/








































































































































































































































































































Текст
Т.Я. КОСОЛАПОВ А
- с.'~ ,
КАРБИДЫ
Т. Я. КОСОЛАПОВА
ИЗДАТЕЛЬСТВО «МЕТАЛЛУРГИЯ»
МОСКВА 1968
УДК 546.261
Карбиды. Коесгламва Т. Я. Изд-во «Метал-
лургия»г;.1968, 300с,
Рассмотрены -все известные в настоящее время
системы металл-— углерод и диаграммы состоя-
ния, дано описание-* физических и -химических
свойств карбидных фаз. Приведены методы по-
лучения-карбидов ^ области их -применения. Рас-
считана ина широкий круг научных, инженерно-
технических •работников' и студентов, интересую-
щихся химией и металлургией тугоплавких соеди-
нений,_Илл. 79, З’абл.’З?. Библ. 920-назв.
3-10-3
60—68
ОГЛАВЛЕНИЕ
Стр.
Предисловие ^г. . л , 4
ГЛАВКА СТРУКТУРА И ФИЗИКО-ХИМИЧЕСКИЕ
СВОЙСТВА КАРБИДОВ
Структуры S' г ч » , у. В
Термодинамические и- тештофизичёские свойства 12
Электрофизические-- иТмагнитные свойства . . 34
Физико-механические ^свойства . . 43
Химические "Свойства L - 45
П
ГЛАВА МЕТОДЫ ПОЛУЧЕНИЯ КАРБИДОВ
IH
ГЛАВА КАРБИДЫ МЕТАЛЛОВ I ГРУППЫ
Карбиды ЩёлочнЫх металлов 65
Карбиды металлов подгруппы меди ; 70
IV
ГЛАВА КАРБИДЫ МЕТАЛЛОВ II ГРУППЫ
Карбиды бериллия, магния и щелочноземельных
металлов1 • s > , . 71
Карбиды 11одгрупгГы -цинка . * - - ~ . 78
V
ГЛАВА КАРБИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Карбиды скандия, иттрия и лантаноидов -- . 79
Карбиды актиноидов . 83
Карбиды металлов-подгруппы IVa . 101
Карбиды металлов подгруппы Va . ~ - - 127
Карбиды "металлов -подгруппы Via * Т 152
Карбиды металлов подгруппы Vila . 172
Карбиды металлов УШ группы _ . - . 175
VI
ГЛАВА КАРБИДЫ ЭЛЕМЕНТОВ ГРУППЫ БОРА
VII
ГЛАВА КАРБИД КРЕМНИЯ
VIII
ГЛАВА ХИМИЧЕСКИЕ СВОЙСТВА КАРБИДОВ
IX
ГЛАВА ПРИМЕНЕНИЕ КАРБИДОВ
Литература v - - 273
ПРЕДИСЛОВИЕ
Кяпбиды относятся к широкому классу соединений,
обладающих рядом ценных свойств, делающих их пер-
спективными материалами для использования в различ-
ных областях новой техники. Карбиды металлов, особен-
но переходных, обладают высокими температурами плав-
ления и твердостью, высокой химической стойкостью,
металлическим характером электро- и теплопроводности,
а также рядом специальных свойств способностью пе-
реходить в сверхпроводящее состояние при относительно
высоких температурах, высокими эмиссионными свойст-
вами и др. Несмотря на большое число работ, посвящен-
ных исследованиям методов получения и свойств карби-
дов, они изучены недостаточно, и часто наблюдаются
большие расхождения между данными разных работ.
Кроме того, эти материалы рассеяны по многочисленным
источникам, что затрудняет возможность подробного оз-
накомления с этим классом соединений..
Всестороннее исследование свойств карбидов пред-
ставляет не только практический, но и научный интерес
с точки зрения выяснения характера химической связи в
карбидах, зависимости свойств от характера химической
связи .и выявления путей для создания материалов с за-
ранее заданными свойствами.
В книге собраны данные по всем известным системам
С, методам получения отдельных фаз, их физиче-
ским и химическим свойствам.
AH^vrr выражает глубокую благодарность чл.-корр.
АН УССР Г. В. Самсонову за ценные указания и по-
стоянную помощь, оказываемую им при работе над ру-
кописью, проф. П. В. Гельду за просмотр рукописи и цен-
ные замечания, а также сотрудникам лаборатории техно-
огии неорганических соединений Института проблем
материаловедения АН УССР, особенно Г. К Макаренко,
плмпшЛ0^0’ Квас и А. В. Ткаченко, оказавшим
Щ при просмотре рукописи и оформлении ее.
4
ГЛАВА I
СТРУКТУРА
И ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА
КАРБИДОВ
СТРУКТУРЫ
Структурные типы карбидов делятся на следующие: с
изолированными атомами углерода, с изолированными
парами атомов углерода, с цепями из атомов углерода и
с сетками из атомов углерода [11].
Щелочные металлы образуют карбиды, в которых при
переходе от карбида лития к карбидам калия, натрия, ру-
бидия и цезия усложняются структурные элементы из
атомов углерода. Так, литий образует один карбид Li2C2
[4], натрий наряду с Na2C2 образует NaC8, NaCie и NaC64
[5]; для калия наиболее характерны карбиды КС8, KCi6,
КС24, КСзб, КС48, КСбо? то же для рубидия и цезия (цезий
карбида Cs2C2 не образует). Эти карбиды характеризу-
ются графитоподобными решетками, в которых между
слоями из атомов углерода расположенье атомы металлов
(рис. 1). Наиболее вероятно расположение атомов ме-
таллов, проекция которого на углеродные сетки дает рас-
пределение их по центрам углеродных тексагонов [895]
(рис. 2). Расстояния между углеродными сетками для
КС8, КС24 и КС36 составляет 0,540, для RbC8, RbC24 и
RbC36 0,565, для NaC64 0,460 и для CsC8 0,594 нм *.
При переходе к карбидам щелочноземельных метал-
лов склонность к образованию сложных анионов из ато-
мов углерода уменьшается и для них характерны карбид-
ные фазы МеС2 со структурами из изолированных пар
атомов углерода. Карбиды СаС2, SrC2 и ВаС2 кристалли-
зуются в тетрагональной ячейке типа СаС2 [896] (рис. 3).
Карбид MgC2 кристаллизуется в тетрагональной ячейке
типа ThC2 (рис. 4).
Лантаноиды и близкие к ним по положению в перио-
дической системе элементов скандий и иттрий образуют
Периоды решетки по всей рукописи даны в нм. 1 нл4=10А.
5
карбидные фазу, соответствующие составам. Л1е3С, МеСг
Мб И^гоиТи ^се -редкоземельные металлы, за исключе-
нием лантана, церия, празеодима и неодима, образуют
карбиды состава Л1<?3С.
-Эти фазы кристалли-
зуются в кубической
структуре типа Fe^N,
близкой к г. ц. к. решет-
ке типа NaCl, но с дефи-
цитом атомов углерода.
Фазы ЛГеС обнару-
Ол
Рис. 1. Структура карбида калия
КС8
жены для иттрия, скан-
дия и церия л кристал-
лизуются в у. ц. к. ре-
шетке типа NaCl [85,'
119, 185, 186, 820].
q. = D,№hm
Рис. 2. Расположение ато-
мов. щелочного металла в
соединении графита с кали-
ем относительно углеродных
гексагональных сеток
Полуторные карбиды Ме2С3 кристаллизуются в боль-
шинстве в о.ц.к. структуре типа PU2C3 (от L&2C3 до
Но2С3), Структуры полуторных карбидов иттрия, эрбия,
туллия и лютеция пока окончательно не определены. Ди-
карбиды образуют иттрий и все лантаноиды. Они кри-
6
Рис. 3. Структурный тип
СаСа
сталлизуются в о.ц. тетрагональной структуре типа СаСо
(рис. з) с двумя атомами в элементарной ячейке. Зави-
симость периодов решетки дикарбидов от порядкового
номера элемента (рис„ 5) свидетельствует об эффекте
лантаноидного сжатияднаблюдаемом и у других кар-
бидных фаз редкоземельных* F
элементов.
Карбиды актиноидов
близки по структурам к кар-
бидам редкоземельных эле-
ментов. Они образуют фазы
МеС, Ме2С3 и МеС2. Моно-
карбиды кристаллизуются в
г. ц. к* решетке типа ЯаС1.
Полуторные карбиды Хза
исключением тория, кото-
рый не образует полутор-
ных карбидов) кристаллизу-
ются в о., ц. к. решетке типа
PU2C3, в которой атомы уг-
лерода образуют парьы Расстояние С—С составляет
0,154 нм. Дикарбиды известны для урана и тория и крис-
таллизуются в г. й,. тетрагональных решетках типа ТЬСг
(см. рис. 4J и СаС2 (UCa).
Карбиды переходных металлов образуют в основном
фазы внедрения или близкие к ним фазы с изолирован-
ными атомами углерода и со .структурными цепями из
атомов углерода [11]. Структуры фаз внедрения, по пра-
вилу Хэгга, образуются при отношении радиуса углеро-
да к радиусу металла, не превышающем 0,59.
Рис. 4. Структурный тип ThC2
7
ДГОМЫ углерод*;ы=и^Тт:м:МГ«?а^И::СпКрИиеэ™Лм
“ Е“раияается ₽ешетка и«°лного.“”лла С °6'
6 а™™ пешеток с высокой симметрией [12].
РЭ’мета" л« подгруппы IVa образуют монокарбиды ко-
торые кристаллизуются в г. ц. к. решетке типа NaCl. Для
Атомный, номер
Рис. 5. Зависимость периодов решетки
дикарбидов от порядкового номера
элемента
С ВО Ф © СВ ~
Рис. 6. Расположение ато-
мов углерода и молибдена
в решетке М02С
этих карбидов характерны широкие области гомоген-
ности. С увеличением содержания углерода в карбидах
значения периодов решетки линейно возрастают.
Металлы подгруппы Va образуют карбидные фазы
и МеСг_х. Фазы Ме2Сг_х кристаллизуются в
гексагональной решетке типа М02С, состоящей из плот-
ноупакованных гексагональных слоев из атомов молиб-
дена и атомов углерода, расположенных в октаэдриче-
ских пустотах (рис. 6) [550]. Монокарбиды кристаллизу-
ются в г.цж. решетке типа NaCl. Области гомогенности
монокарбидов металлов подгруппы Va несколько уже
областей гомогенности монокарбидов металлов подгруп-
пы IVa и шире областей гомогенности полукарбидов ме-
таллов подгруппы Va.
8
Из карбидных фаз хрома (Сг23С6, Сг7С3 и Сг3С2) низ-
ший по содержанию углерода карбид Сг23С6 обладает
структурой с изолированными атомами углерода, кри-
сталлизуется в г. ц. к. решетке и представляет самостоя-
тельный структурный тип. Атомы хрома расположены в
центрах архимедовых кубооктаэдров из атомов углерода
и притупленных тетраэдров: внутри Федоровских кубоок-
Рис. 7. Структурный тип Сг2зС6
таэдров расположены кубы из атомов хрома [896]
(рис. 7). Высшие карбиды хрома образуют структурные
типы с цепями из атомов углерода. Карбид Сг7С3 кри-
сталлизуется в гексагональной решетке типа, близкого
к структурному типу Ru7B3 [897].
Карбид Сг3С2 имеет ромбическую структуру само-
стоятельного типа, в которой каждый из 8 атомов угле-
рода расположен в центре трехгранной призмы. В углах
призмы находятся атомы хрома (рис. 8). Наименьшие
расстояния Сг—Сг 0,2487 нм; Сг—С 0,201 нм [799].
Молибден и вольфрам образуют фазы Ме2С1_х и МеС.
Фазы Мв2С{_х кристаллизуются в гексагональной решет-
2 Косолапова T, Я. 9
ке типа МйС, фазы Л1гС~в гексагональной решетке
т”"Алт'асть гомогенности карбидов подгруппы Via эна-
<же чем у карбидов металлов подгрупп IVa
Гуа У вмших. по содержанию углерода карбидов она
П₽апТПяЧеммалловГподгрупп1>г Vila известны карбидные
фаз^мартнпЖс.^МпЛ. Мп5С2, Мп;Са и МпаС.
Рис, 8, Структура карбида хрома СгзС2
Карбид Мп3С кристаллизуется в ромбической ячейке
структурного типа Fe3C, в котором атомы металла обра-
зуют трехгранные призмы с атомами углерода, располо-
женными в центре (рис. 9) [663], Карбиды Мп23Сб и
Мп7С3 кристаллизуются в кубической и гексагональной
решетках типа Сг23Сб и Сг7С3 соответственно.
Металлы VIII группы образуют карбидные фазы
Ме4С (Fe4C), МегС (Fe3C, Со3С и Ni3C) и Ме2С (Fe2C и
Со2С), Структура карбида Fe4C представлена на рис. 10.
Карбиды 7Ие3С кристаллизуются в ромбической решетке
структурного типа Fe3C (рис. 9)г Л4е2С— в гексагональ-
ной решетке. Области гомогенности карбидов металлов
подгруппы Vila и группы VIII очень узки и составляют
не более 1—2% (ат.) С, Таким образом, при переходе от
карбидов переходных металлов группы IV к карбидам
переходных металлов групп V, VI, VII и VIII ширина
10
области гомогенности уменьшается Cnrnaf.„. «
бот [13, 14, 1721 ширина областил-омХЕХ^??3'
переходных металлов групп IV—VI п₽пиПг,ЮСТа карбидов
мы обусловлена статистиче-ским в^сомРустойчХ* СИСТе’
тронных конфигураций атомов металлами углерода ЭДля
Рис. 9. Структура карбида РезС:
о — 0,45 нм; Ь —• 0,50 нм; с — 0,67 нм
рассматриваемых металлов характерно образование ус
тойчивых d°, d? и d10 электронных конфигурации для
углерода максимально устойчивой является Ф
Ция sp3, образование которой возможно при Р
s-электронов в p-состояние. „Ои0Таппя.
При рассмотрении соединений углерода с
ми наибольший интерес с точки зрения соедин >
2* 11
свойствами, как высокие температуры
дающих такими_с аб вная й химическая стой-
"1аВ'1е^Я,епповодниковые свойства, представляют кар.
бидабор? и кремния, которые и рассматриваются в дан-
ной работе.
Карбиды бора и
В 5 С кристаллизуются в
ромбоэдрической реш®тке Ч»
согласно работе [898], Ь4с
имеет структуру, представ-
ление ю на рис. И- Карбид
Рис. 10. Структура карбида Fe4C
Рис. 11. Структура карбида JB4C
кремния SiC существует в виде Двух основных модифи-
каций: кубический (P-SiC), структурного типа сфалери-
та и a-SiC, который образует большое число структур-
ных типов, имеющих гексагональную и ромбоэдрическую
решетки [816—818].
В табл. 1 приведены структуры и кристаллохимиче-
ские характеристики карбидов.
ТЕРМОДИНАМИЧЕСКИЕ И ТЕПЛОФИЗИЧЕСКИЕ
СВОЙСТВА
Если карбиды щелочных металлов малоустойчивы и
легко разлагаются уже при температурах порядка 800° С,
то при переходе к карбидам металлов подгрупп IVa, Va,
12
н Via наблюдается большой рост температур плавления,
достигающих максимума для карбидов металлов под-
групп IVa и Va (HfC, NbC, ТаС). Температуры плавле-
ния карбидов обычно выше температур плавления соот-
ветствующих металлов. Так, Т^^ТплМес составляет от
0,55 до 0,98.
Исследования испарения дикарбидов редкоземельных
металлов при помощи масс-спектрометрической техники
показали, что в парах над дикарбидами редкоземельных
металлов, кроме атомов металлов, находятся молекулы
дикарбида, а для дикарбидов церия, празеодима и голь-
мия обнаружены и молекулы тетракарбидов [178, 226].
В работе [337] показано, что карбиды титана, цирко-
ния и гафния испаряются без изменения состава в парах.
Состав карбидов ниобия и тантала при испарении в ин-
тервале температур 2770—3300° К меняется -— испаряется
преимущественно углерод до состава МеС01—MeCQTJf
после чего испарение идет с сохранением состава твер-
дой фазы в паре.
Карбиды хрома испаряются ступенчато: Сг23Сб обед-
няется хромом, переходя в Сг7С3, который переходит в
Сг3С2, при испарении выделяющий хром. Анализ имею-
щихся данных по исследованию поведения карбидов пе-
реходных металлов [336, 337, 429, 463, 673, 866] в вакууме
при высоких температурах привел к заключению, что
карбиды металлов подгрупп IVa и Va диссоциируют по
схеме
^ж(тв) = ^б(газ) "Ь Х ^(газ) *
Значения теплот образования карбидов уменьшаются
при переходе от карбидов металлов подгруппы IVa к
подгруппам Va и Via, что свидетельствует об уменьше-
нии энергии связей Me—С, осуществляемых электрон-
ным коллективом и усилением связей Me—Me и С—С
(ковалентных). Такое ослабление энергии связей Me—С,
согласно работе [14], связано с ростом статистического
веса стабильных ^-конфигураций при переходе от карби-
дов металлов подгруппы IVa к карбидам металлов Va и
Via подгрупп, при котором уменьшается вероятность на-
рушения стабильных «реконфигураций углерода и рас-
тет обособленность подрешеток металла и углерода в
карбидах.
13
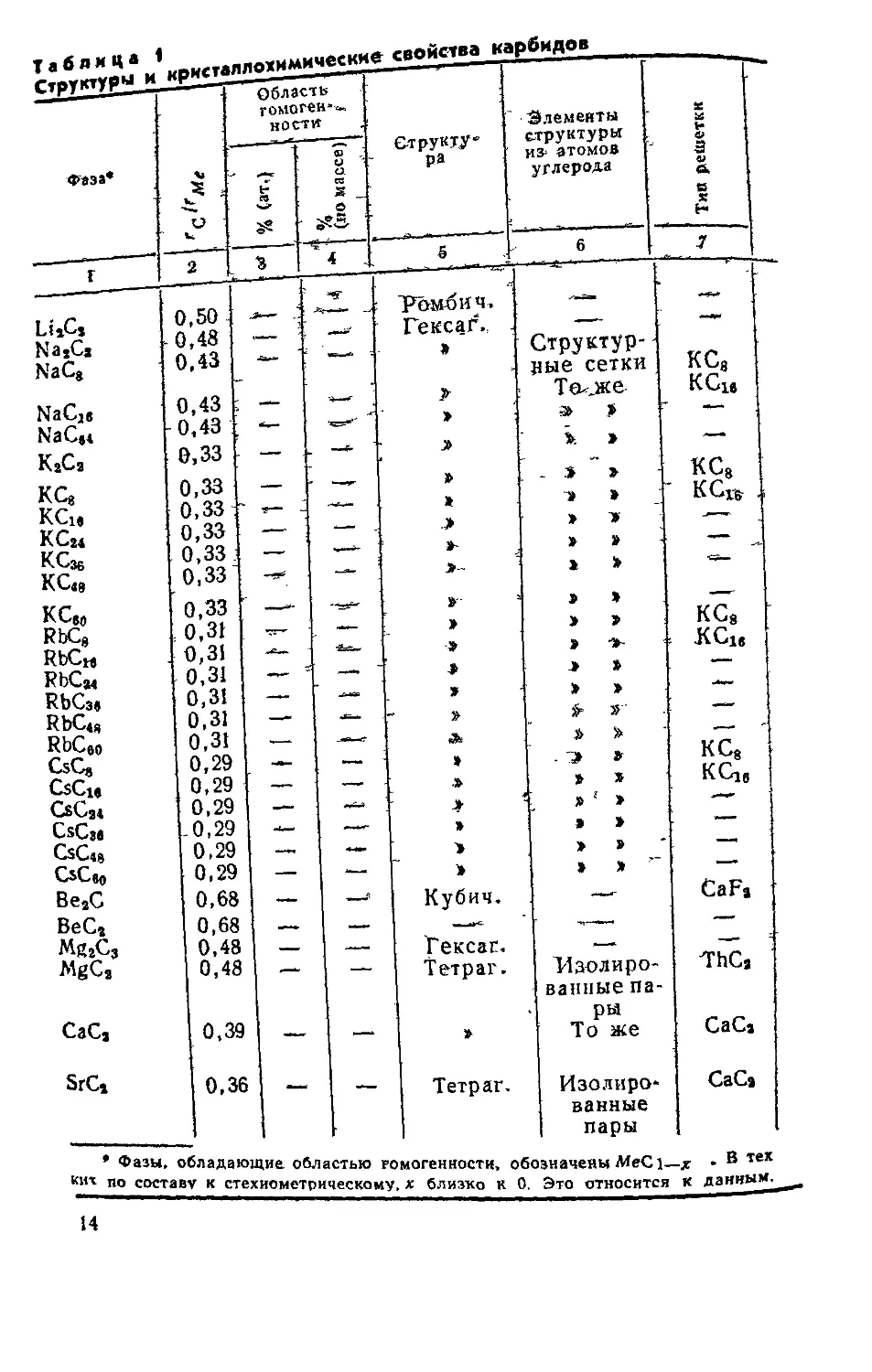
ТаблмЦ* 1
свойства карбидов
«-.ру^туом и христ»™.»™- Структу- ра ~ Элементы структуры из> атомов углерода Тип рефеткк
Фаза* ГЛ< •' чу - - -i- Область гомоген-^ ности
% (ат,) J% (по массе) L_x_i
т 2 3 4 5 6 7
LijCj Na,C» NaC» 0,50 -0,48 0,43 t 1 t Ч11 —а —> - «. •-< - Ромбич» Гексаг», » Структур- • **«* МП»
рые сетки KCg
NaCje 0,43 Те--Же КС1#
-0,43 » >
К2СЯ 0,33 — » — >
KCg 0,33 — - —т- » - » > KCg
KC1, 0,33 - » » KCxg ,
KCm 0,33 ——- > W
КСзв 0,33 » »
kg, 0,33 -я* >- >
кси 0,33 —и— г » » —-r-
RbCg 0,31 » > > KCg
RbCw 0,31 -S— > » = KCle
RbC«M 0,31 * »
RbCat 0,31 1 <1 > » > I.*»»
RbC<g 0,31 Ы&-. & Mia»1
RbCeo 0,31 ч »
CsCg 0,29 » э » KCg
CsCu 0,29 i ' > » KCjg
CSk>2< 0,29 я 1 >
CsCsb -0,29 О 1 Й|Г- > * > -
CsC<8 0,29 4W «мм. > >
CsCeo 0,29 'II Де м » > > ”
BeaC BeCj 0,68 0,68 — < Кубич. CaFg
МЙ2С3 Mgcs 0,48 0,48 —— - Гексаг, Тетраг. "Изолиро- ThCj
ванные па-
CaCj 0,39 ры CaCj
» То же
SrC, 0,36 *—* — Тетраг, Изолиро» CaCj
ванные
пары
кит по состав^к^стехиг^ областью гомогенности, обозначены МеС 1—х - в тех
------~а.Ву к стехиометрическому, х близко к 0. Это относится к данным.
14
Простран- ственная гр) ппа Периоды решетки, ям " Плотность г/см* Литера- турный источник
а * с рентге* невская пикно- метричес- кая
8 9 10 41 12 13 U
0,3655 . 0,6756 9,5440 0,4830 „ 1,2688 Г,91 1,60 1,30 1,58^ 1241] [72]
— III 1| -ШИ »! T-t. . >_S, [5]
W _si. а. г 1 14
0,758 — 1,469 1,62 -М .1 1 г [72]
0,494 2ГГ345Т • ,
0,494 - и 1,745 ' —— - . а.
1 1 1 1 1 1 ||1 — 1 1 - [895,896]
1 —Ч м>* ,,ч'а
- — 0,494 0,494 2,273 1,795 - 1 — - [896]
1 1 — [10.895]
—
— 0,494 * 2,376
0,494 *—* ' 1,851 1 " »!!« и—1 [10,895]
—— —’ —
Fm3m(()5) 0,434 «..А 2,437 2,26 89,93,97]
0,745 ЧР 1,061 — [85]
14 ттт <Dih) 0,555 0,508 2,07 —г [896]
14/ттт (D4h) 0,548 — 0,637 2,21 2,10 (119, 896]
14 ттт (^) 0,582 —-- 0,669 3,26 3,19 [93, 896]
фазах, близ-
помеш₽ии?УЯа состав Фазы определен, приводятся значения х. В
— ным во всех последующих таблицах н тексте.
15
п ПЛ а о Л Ж е н и е таол. х
Структура Элементы структуры из атомон углерода Тия решетки
Фаза* k. к Область гомогенности
% (ат.) % (по массе)
1 2 3 4 5 6 7
ВаСз ScC y3c0>9 YC Y,C3 YCa 1»а2Сз—д> a-LaCj ₽-LaC, CeC Ce^Cj CeCj PraCa 0,35 0,47 0,43 0,43 0,43 0,43 0,41 0,41 0,41 0,42 0,42 0,42 0,42 чан* * 56,2— 60,0 9,98— 11,58 Тетраг. Кубич. » » Тетраг. Кубич. Тетраг. Кубич. » » Тетраг. Кубич. Изолиро- ванные пары Изолиро- ванные атомы То же » Изолиро- ванные пары То же » » » » Изолиро- ванные атомы Изолиро- ванные пары То же » » СаС2 NaCl Fe4N NaCl CaC2 Pu2Cs CaC2 FeS2 NaCl Pu2Cs CaC2 Pu2C3
IS
Простран- ственная группа Периоды решетки, нм Плотность, г/сл3 Литературный источник
а Ь с 1 1 ) рентгеновская пикномет- рическая
8 9 10 11 12 13 14
14 ттт (°Э 0,622 — 0,706 3,72 3,90 - 193,119,896]
1 Fm3tn (°Й 0,499 •г— —— 4,05 3,60 [179—181]
РтЗт М) 0,5068 - 5,41 4,73 17, 864]
Fm3m (ф 0,499 — 5,38 —— [6]
— — — — —— 3,66
14/mmm И) 0,366 -— 0,6169 4,58 3,95 (7,93]
Rm (?1) 0,88185 ххО — — 6,07 —
14/mmm ю 0,3934 — 0,6572 5,35 5,29 (6. 7]
Fm3m № 0,60 «ь 1». — 5,0
Fm3m (°Э 0,513 — — —— [85]
143d М) 0,84476 — 6,969 — [7]
14/mmm Ю 0,3878 — 0,6488 5,56 5,56 (7, 896]
143d (TJ) 0,86072 — — 6,621 [7]
17
Продол жениегабл. 1
1 * Г ** ** ~ Фаза rQ frMe » Область гомогенности Структура Элемент» структуры из атомов углерода Тип решетка
p*«j> % % (тЦ массе}
1 a 3 4 5 в 7
РгСа 0,42 . Тетраг- Изолиро- ванные пары СаС3
NdsC» . 0,42 Кубич., То же Ри2С3
NdCa 0,42 - да Тетраг- * Tf СаС3
Sm&C 0,42 . — Ку бич. Изолиро- ванные атомы JFe4N
Sm2C3 0,42 _ Т1золиро- ' ванные лары PiijCs
SmCi 0,42 sr- Тетр аг. “ То же CaC2
Gd3C 0,38 ——r Кубич. Изолиро- ванные атомы Fe4N
(jdjCg 0,38 э Изолиро7- ванные пары PuaCs
GdC* 0,38 — — Тетраг. То же CaC3
Tb3C 0,44 ~~ Кубич. Изолиро- ванные атомы Fe4N
TbjC3 0,44 — —— » Изолиро- ванные Ри3Сз
TbC» 0,44 — — Тетраг. -пары То же CaCa
Dy3C 18 0,44 4^^» — Кубич. Изолиро- ванные атомы Fe4N
Периоды решетки, нм Плотность, г/сл’
Простран- ственная группа а ь € я й а № © X V S -Л • Г ' пикнометч р^ческая Литературный источник
8 9 10. п 12 - 13 и
1 Мт тт № ьш (7$ 14 ттт № РтЗт (<>1) Ни (Т$) 14/ттт (^) РпгЗт (°1) НЗЗ М) 14/ттт (Dll) РтЗт (01) 143d (71) 14/ттт та РтЗт (01) 0,3855 0,85478 -0,3823 0,5172 0,84257 0,3770 0,5126 0,83407 0,3718 0,5107 0,82617 0,3690 0,5079 0,6434 0^6405 0,6331 0,6275 0,6217 5,-728 6,902 5,970 ' 8,139 7,477° 6,50 _ 8,701 8,024 6,939 8,882 8,335 7,176 9,211 5,58 5,93 ч— 6,421 6,93 7,09 </ [6,7] 1
19
Продол ж е н и е табл. L
а i г ~ - Фа а «и л о и Область гомогенности Структура Элементы структуры из атомов углерода Тип решетки
% <а.т.) % (по массе)
1 2 3 4 5 6 7
DyaCs 0.44 II 1 Кубич. Изолиро- PiijCj
ванные
- пары
DyC2 0,44 Г— Тетраг-. То же СаСа
HosC 0,44 — Кубич. Изолиро- Fe4N
ванные
атомы
НодСз О; 44 » Изолиро- PiijCj
ванные
пары
НоС, 0,44 — Тетраг. То же CaCa
Ег3С 0,44 11 — Кубич. Изолиро- Fe4N
ванные атомы
ЕгС2 0,44 —— — Тетраг. Изолиро- CaC2
ванные
Ти3С пары
0,44 — — Кубич. Изолиро- Fe4N
0,44 ванные атомы
ТиС2 Тетраг, Изолиро- CaC2
ванные
YbaC пары
0,40 — — Кубич. Изолиро- Fe^M
ванные
YbCa атомы
0,40 — Тетраг. Изолиро- CaC2
ванные
LuaC 0,44 — Кубич. пары Изолиро- Fe4N
ванные атомы
простран- стве нав группа Периоды решетки, нм Плотность г! см? Литературный источник
а Ь с рентгеновская пиюномет* рическая
8 9 10 11 12 13 14
143d 01) 0,8198 — —* •*—
14 ттт № 0,3669 — 0,6176 7,450 7,38
РтЗт (о\) 0,5061 — — 9,434.
РтЗт С>1) 0.8176 — — 8,892. —
T13d (^) 0,3643 — - 0,6139 7,701 <7,76
РтЗт (01) 0,5034 — •—- 4,708 — [6,7]
14/mmm (oil) 0,3620 — 0,6094 7,954 7,70
РтЗт (с>1) 0,5016 — 9,901 —
14/mm/n (oil) 0,360 — 0,6047 8,175 —
РтЗт (ol) 0,4993 — 10,26 —
14/ттт. (oil) 0,3637 —— 0,6109 8,097 7,97
РтЗт (ок) 0,4965 — — 10,54 —* J
21
Продолжении^ Область- _геиогеняости Структура Элементы структуры из атомов углерода Тип решетки
Фаза «1 k.
I % — ' г I i (ЭйОэИ ou) L._ .
1 2 ' 3" 4 5 6 7
LuCs ThCx-x 0,05<«<0,1 ThC2— *=0,01 UCp-x UA a-UCj_z 0,41 0,43 0,43 0,50 ' 0,50 ' 0,50 —s. , 1 >.*• . 8— 9 <56 Тетраг. Кубич- " МоНОКЛИНх. ~ Кубич. » Тетраг.. Изолиро- ванные пары Изолиро- ванные атомы Изолиро- ванные пары Изолиро- ванные -атомы Изолиро- ванные пары То же СаС2 NaCl NaCl Ри2Сз СаС3
р-ис3-л 0,50 Кубич,- CaF3
PuCi—x x=0,15 Ри3Сз Ti£i-* *=0,079 ZrCi_x *=0,043 HfC!_x * 0,03 0,48 0,48 0,53 0,48 0,48 46—48 37-50 38,5— 50 36—50 4,04— 4,34 13-20 7,6— 11,64 3,7— 6,3 » Кубич. » » Изолиро- ванные атомы Изолиро- ванные пары Изолиро- ванные атомы То же 4» » NaCl Pu2C3 NaCl NaCl NaCl
22 1
Простран- ственная группа Периоды решетки, ям- -Плотность, а/см3 Литературный, источник
а ь с рентгеновская пикномет - р^ческая
8 9 10 11 12 - 13 14
14/mmm (^) 0,3563 — 0,5964 8,728 —в- 16,7]
/m3m (°л) 0,5335 10,61 1197, 208Г
0,669Г 0,4231 0,6744 9,6 1134, 200, 215] -
ГтЗт (^) 0,4959 *— —ff 13,63 - 12,97 ]199, 264]
нм (rj) 0,8088 — 12,88 12,7 1227]
14 ттт (Ч7) 0,3517 —- 0,5987 11,79 11,28 [259]
Firi3m («а 0,546 — — , —- 1221, 250]
Fm3m W) 0,49582 -— — 13,42 [65 r 265J
им П) 0,81258 —-' 12,70 —S [65, 269]
Fm3m <°а 0,43178 — 4,92 4,93 [286, 335]
Fm3m <°а 0,46828 •— В“— 6,66 6,73 , [335, 374]
FmZtn (°.) 0,46395 .— —<-« 12,67 12,60 [360, 407]
23
Продол ж е н и е табл-. 1
Фаза ' fc /гЛ4е Область гомогенности- Структура Элементы структуры из атомов углерода Тип решетки
% (а?.) % (по массе)
1 2 3 4 5 6 7
VjCi—х ххО 0,57 31 — 33,3 9,9— 40,5 Гексаг. Изолиро- ванные атомы МодС
VC1-X х=0,12 0,57 40—47 13,4^ 17,2 Кубич. То же NaCl
Ьч bjCj—х х*0 0,53 31,0— 33,3 5,8— 6,1 Гексаг, » % МогС
NbCi-x х=0,116 0,53 41,2— 50,0 8,3— 11,4 Кубич. NaCl
Ta<jCi—х х»0 0,53 30,3— 33,3 2,8— 3,2 Гексйг. МогС
TaCi—х х=0,15 0,53 42,2— 49 4г,6— 6,0 Кубич. » » NaCl
СггзСв 0,61 — .— » » > СгззСв
Сг?Сз 0,61 — — Гексаг. Цепи из атомов СгтСз
СГаСд 0,61 — — Ромбич. То же СгзСа
MojCj—х х%0 0,55 31,2— 33,3 5,4— 5,9 Гексаг. Изолиро- ванные атомы Мо2С
WA х xssO 0,55 29,5— 33,3 2,7— 3,2 То же Мо2С
WC 0,55 — » » МоС
МпгзСв 0,59 — Кубич. » » СгазСв
MrixjCi 0,59 —- —— Гексаг.* —
24
Простран- ственная rpjnna Период решетки, нм Плотность, г/см9 Литературный источник
а Ъ - рентгеновская . *4 < Пикномет- рическая
8 8 10 41 12 43
СЗт 0,290 0,4587 - J.75 * 14191
(D3d)
Fm3m 0,4159 — — 5,48 „ 5,36 1419, 864]
(Ф -
СЗт 0,3122 Д),4964 7,85 Т,86 ]442]
№)
Fm3m 0,44691 — — 7,82 7,56 J435 г 442]
(<4)
C3m 0,3104 — 0,4943 15,04 14,8 [466, 468]
(D3d)
Fm3m 0,4456 14,4. 14,3 ]468, 469]
(<4)
Fm3m 1,0638 — -fit 11 6,98 6,97
(Ф
PQtnc 1,398 -III. , 0,4523 6,97 6,92 [371, 490]
(<t)
Pbnm 0,282 , 0,552 1,146 6,74 ' 6,68
C3m 0,3002 1 1 0,4729 9,06 -у— [534]
(*>£)
C3m 0,30008 — 0,47357 17,34 17,2 [65, 152]
M)
C6/n2 0,2903 0,2833 15,77 15,5— [65J
(^) 15,7
Fm3m 1,061 1 7,53 —. [856]
(°л)
—• 0,7492 1,2071 7,30 —— [782, 856]
25
Предо л ж едие табл. I
Фаза as к О ц -Область^ гомогенности 'Структура Элементы структуры на атомов ^углерода Тип решетки
% (ат.) '_х, - ( (аээви би) J
1 2 3 4 S •6 7
МпзС 0,59 — —- 'Ромбич-. Изолиро- ванные атомы* Fe3C
Мп8С» 0,59 —* ' — Моноклин. — IM
Мп7Са 0,59 - —> Гексаг. Цепи из атомов Сг,Сз
Fe»G 0,61 — —ж Ромбич. Изолиро- ванные атомы FeaC
FeaC 0,61 - 7— — Гексаг. То же —
Со8С 0,62 ***> Ромбич. » » Fe3C
Со^С 0,62 » » > ——
Ni3C 0,62 —*— —— № > > Fe3C
А1*С» Вб,эС 0,53 0,85 -—< Ромбо- эдрич. » , ч« < B4C
0,85 17,6— 29,5 19,2— 31,7 —> B4C
₽-SiC 26 0,57 — Кубич. ZnS
Простран- ственная группа Период решетки, мм Платность, «/ел* Литературный ИСТОЧНИК
а & рентгеновская . ч пикномет-* рическая ... —
в 1 9 ' 1b 11 12 - 13 14
РЬтп " 0,4530 0,5080 0,6772 6,89 [11, 856]
—«1 -0,5086 0,4578 , 1,166 (580, 581]
РЬтс 1^330 = 0,454. 3,69 >' [580]
(<&)
РЬпт .0,45235 0,50890 0,67350 7,69 ; 7,07 1856]
-
— 0,2757 ; — 0;4346 7,16 Т152]
РЬпт 0,45а 0,509 0’674 8,07 —л [152, 856]
(»а) - -
i » । 0,2885 <7,4454 0,4364 4 7,6Г~ — [660, 670]
РЬпт 0,2646 - Л, 4329 7,88 7,96 [681]
— 0,853 0,4203 2,584 ; 2,95 2,99 [65, 161]
R3m
0,563 1,219 2,461 [685]
R3tn (D3d) 0,5598 “— 1,212 3,52 3,50 [685, 687]
ТГЗ (т*) 0,4358 — — 3,217 3,211 [856]
27
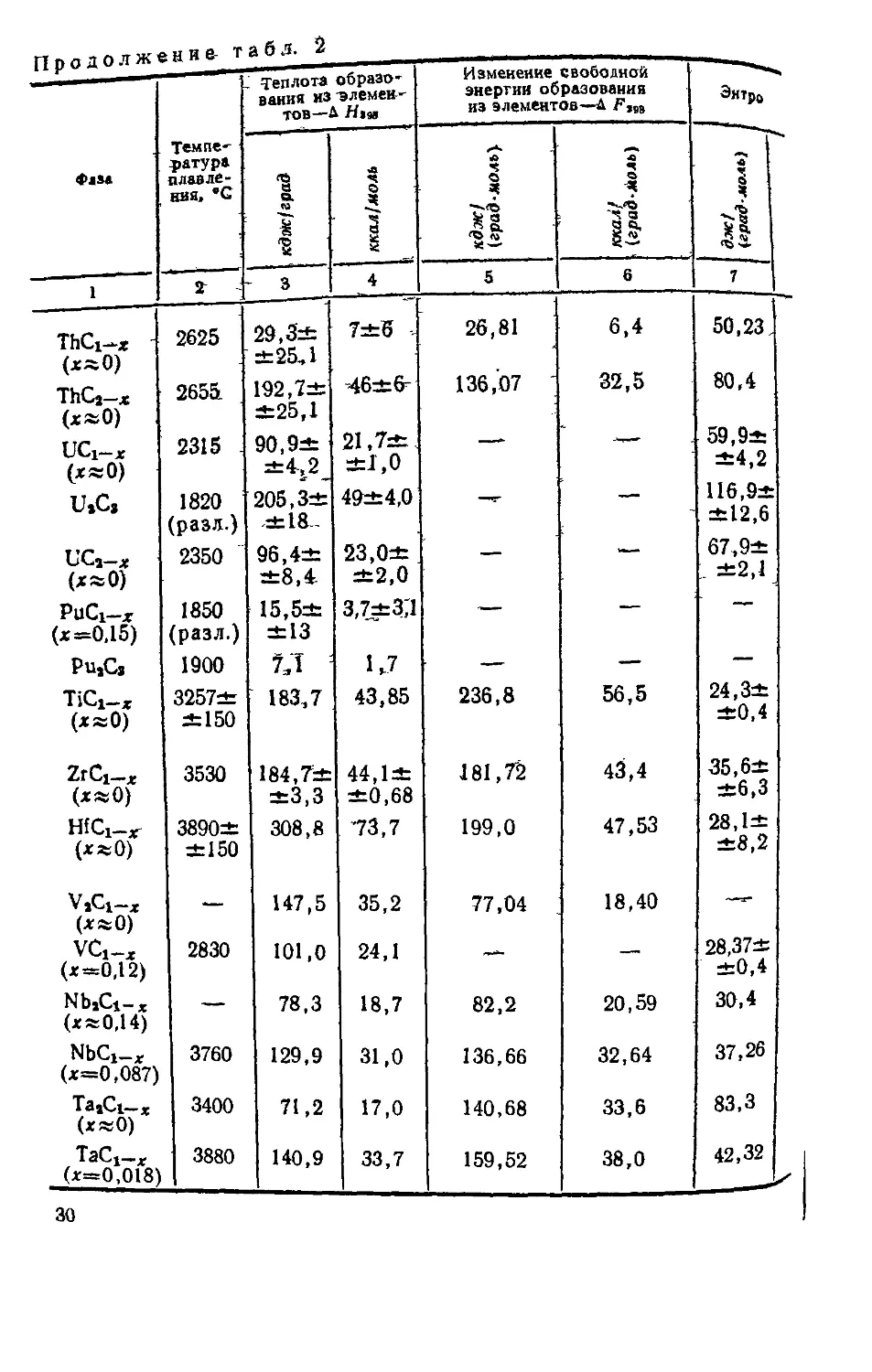
Таблица 2
Термодинамические и теплофизические свойства карбидов
Теплота облазо- Изменение свободной
-вания -иа элем-ен- энергии образования Энтру
тов— А #298 из элементов—Д F+
Фаза Темпе- - ратура "л*
плавле- i ния, °C дж!м.алъ кал(м.ом> О W 2 'В кал/ град мал Ч
к <й^.
1 2 3 4 5 6 7
LI2C2 +59,4 +14,2 — — —
NagCa 800 —17,18 —4,1 —35,558 —8,500 —»
(разл.)
ВегС 2400 91,3± 21,8+5 88,8±21 21,2±5
±21
ВеСг 240,5 57,4 —
MgaCj — 79,6± 19±8,0 —75,36 —18,00 92,1
±33,5 -
MgC3 — 88=5=21 21±5,0 —87,92 —21,0 58,7± ±10,5
СаС2 2300 59,1 + 14,1± 64,306 15,36 70,4±
±8,4 ±2,0 ±2,1
SrC2 1900 (разл.) -— — —
ВаС2 1770— 50,7± 12,1± —, 14,2
2300 (разл.) ±18 ±4,0
ScC 1800 г — — —
YC 1950 —* — - — -' -
Y2C3 1800 — R । » •«нм- -
YCa 2190 1' — —
LaaCg 1425 - -
LaCj 2360 159,1 38,0 - ~
СеаСз 1535 -
CeC2 2290 — - < -
PraCg 1560 —.
PrCa 2120 —
NdC2 2207 — — — — —
28
29
2
П р о д о л ж енне- 1
J Теплота образо- вания из элемен- тов—А Н>и Изменение свободной энергии образования из элементов—A Ftm Энтро
Темпе--
натура плавле- ная, ’G л 5 3
а QU эе 5 §
'S ккал!) кдж} {град ккал/ > {град J дж/ | {град-
1 J - - 3 4 5 6 7
ThC^x 2625 29,3* 7=6 26,81 6,4 50,23.
(х~0) =*25_ 1
ThCi-x 2655 192,7* 46* 6~ 136,07 ' 32,5 80,4
(х«0) =*25,1
UCx-x 2315 - 90,9* 21,7*. —W- 1 1 59,9*
(z«0) =4,2~ *1,0 ^:4,2
и,С, 1820 205,3* 49=4,0 —V 116,9*
(разя.) *18- *12,6
UCj—х 2350 96,4= 23,0= — 67,9*=
(х~0) =8,4 =2,0 . =±=2,1
PuCi—х 1850 15,5*= 3,7*ЗП ч —
(х=0,15) (разя.) *=13
PujCj 1900 U7 —*• —
TiCx-x 3257* 183,7 43,85 236,8 56,5 24,3*
(х«0) =i=150 *=0,4
ZrCi—х 3530 184,7* 44,1* 181,72 43,4 35,6*
(х«0) =3,3 *0,68 *6,3
HfCx-x 3890= 308,8 73,7 199,0 47,53 28,1*=
(х«0) *150 *8,2
VA-x 147,5 35,2 77,04 18,40
(х~0)
VC,-x 2830 101,0 24,1 - — 28,37=
(х=0,12) *0,4
Nb,Ci-x (хл0,14) — 78,3 18,7 82,2 20,59 30,4
NbCx-д. (х=0,087) 3760 129,9 31,0 136,66 32,64 37,26
Ta»Ci—х (х«0) 3400 71,2 17,0 140,68 33,6 83,3
TaCj-х (х=0,018) 3880 140,9 33,7 159,52 38,0 42,32
30
птя Sjm Теплоемкость С Р1М . Теплопроводность Коэффициент терми- ческого расширения —6 1 а»« 10 град . . 1 Литературный источник
кал/ (град моль) 1 дж/ (град-моль) . , ... ,| кал/ (град' мол О 4'1 fl* (QDtk'ITMun 1 кал/ (смгсекград)
8 9 10 11 -» 12 "13 И
12,0 19,2 14,3*0,1 27,9*3,0 16,2^0,5 5,8*0,1 3,5*1,5 6,7*2 ^^045 °o,(fc — 6,77* 2:0,1 7,25 8,9 19,9 Ю,11 ' । »- ” । 1 < 1 12 111» ю 00 <£> S S ’ф сч со 12,1 26,0 . 14,5 12,07 13,05 10,17 25 8,92 7,77 28,9 = 23,9- 23,9 34,3 0,1 Г 20,5 6,3- 24,7 14,2 22,2 0,069 0,057* 0,057 0,082 0,263 (20— J 100 °C) 0,049 0,015 0,059 0,034 0,053 6,53- 8,5 12,4- ' 8,31=Ь *0,68 6,95:2= *0,25 6,06 7,2*0,6 7,0±0,3 6,5 8,29 [65,197, 209—2111 [65,197, 209—211, 213—215] [199,251, 256] [251] [231,2511 1211] 113,317,368, 350,363, 838] [65,152, 368, 397] [65,152, 399, 403] [599] -'[25,173,362 397, 424,599] [152,457, 600, 873] [65, 152, 457, - 459, 873] [152, 315, 467] [152, 315, 368, 467 , 873]
31
Пр дотжЕние табл. 2
Фазе Темпе- ратура плавле- ния, Теплота образо- вания из элемен- Изменение свободной энергии образования из элементов—Д Егт Энтро
тов— Д
। кдж/моль ккал/моль 1 кдж/ (град-моль] ккал/- (град-моль) дж/ (град-моль)
1 2 3 4 5 6 7
СГззСд 1500 68,6 16,38 70,14 16,74 106,0
СГ7С3 1780 178,15 42,51 183,35 43,76 201,1
Сг3С2 1895 88,0 21,01 88,83 21,20 85,5
MojCj—х (х~0) 2690 —17,6 ' —4,2 11,73 2,8 80,0
МоС 2700 —40,6 —9,7 ,— — —
WA-, (х«0) 2730 46,05 1 Г,0 48,98 11,70 —
WC 2870 35,2± ±12,6 8,4± ±3,0 37,60 8,98 35,58
МпазС® 13,8 3,3 — —
МпзС — 15,1 3,6 14,27 3,41 98,9
Fe3C 1650 22,6 —5,4 —19,05 —4,55 101,4
С03С 2300 —33,9 —9,3 —37,7 —9,0 95,7
CojC — —16,8 -4,0 —19,13 —4,57 74,5
Ni3C 2100 —38,5 —9,2 —31,83 —7,6 106,4
В4С1—х (х«0) 2350 57,8 13,8 50,66 12,10 27,1
А14С3 2100 195,7=2= ±41,9 46,7± ±10 150,0 35,8 131,1
SiC 2827 (разл.) 66,2 15,8 51,39 12,27 16,55
32
ПИЯ Заи Теплоемкость С РзвВ Теплопроводность 298 Коэффициент Терми- ческого расширения «2»1()— гРай Литературный источник
кал/ (град'Моль) дж/ (град-моль} кал/ (град-моль} втЦм-град) кал/ (см-секград)
8 9 10 11 12 13 14
25,3 48,0 20,42 19,1 8,5 23,6 24,2 22,9 17,8 25,4 6,47 31,3 3,95 108,5 209,16 98,00 35,7 68,4 0,955 25,89 49,92 23,38 8,53 25,5 12,55 31,4 0,228 6,7 29,0 29,0 8,0 0,076 0,07 ' 0,07 «111 0,02 । м* г-* "Н * * см см ь > । tii । о- 1 II 1 111^- 1 J £368, 483, 492, 497] [60, 103, 152, 553] [65, 152] [65, 553] [103, 553, 900] [587, 589, 596] [65, 589, 593, 594] [86, 152, 664, 900] [60] [65, 103] [152, 368] [152, 684, 697] [103, 152, 174, 900] [103, 152, 742]
3 Косолапова Т. Я.
33
Температуры кипения карбидов очень высоки и со-
ставляют при атмосферном давлении от 2537° С дляВегС
до 6000° С для WC [152].
Величины коэффициентов термического расширения
карбидов переходных металлов такого же порядка, ,как
у металлов, и уменьшаются с ростом порядкового номера
элемента в группе.
Термодинамические и теплофизические свойства кар-
бидов приведены в табл. 2:
ЭЛЕКТРОФИЗИЧЕСКИЕ
И МАГНИТНЫЕ СВОЙСТВА
При рассмотрении "Олектрических свойств карбидов
последние можно разделить на группу карбидов метал-
лов, обладающих металлической проводимостью, и груп-
пу карбидов неметаллов с проводимостью, характерной
для полупроводников. К первой группе относятся в основ-
но I карбиды переходных металлов.
Зависимость удельного электрического сопротивления
карбидов переходных металлов от температуры [903] по-
казана на рис. 12.
п -----------------1_______. . >
7 400 800 1200 1600 2000
Температура, °C
тивлР1н2йа3аЛИС«ИМОСТЬ УДельн°го электрического сопро-
рбидов металлов IVa и Va подгрупп от
температуры
34
Положительные термические коэффициенты удельно-
го электросопротивления, как и абсолютные значения со-
противления, свидетельствуют о металлическом харак-
тере проводимости карбидов.
По работе [397], удельное электросопротивление кар-
бидов титана и циркония, полученных осаждением из та-
зовой фазы, т. е. преимущественно ориентированных и
мало отличающихся от монокристаллов, выше сопротив-
ления этих же карбидов, полученных порошковыми мето-
дами. Так, р для TiC0g2 составляет 200 мком-см, а для
ZrCo.964 N0 002 182 мком • см,.
Систематические исследования удельного электросо-
противления, коэффициентов термо-э.д.с. и эффекта Хол-
та для карбидов переходных металлов IV—VI. групп и
анализ результатов этих исследований [15, 16, 19] позво-
лили оценить величину вклада электронной и дырочной
составляющих в проводимость карбидов.
В случае носителей зарядов двух "знаков
_ 1 — п+ “+
е (га„ и— 4- п.-\- и+У 1
ч
е п— и——|- п-j- и.]-
где R— коэффициент Холла;
Р — удельное электрическое сопротивление;
— п
— п
п_, п+ — концентрации, а и_, и+—подвижность со-
ответственно электронов и дырок.
В работе [19] была вычислена величина
6 == 2L — п и2 — п, и2,
ер® ~ ~ + +
которая характеризует относительный вклад электронов
и дырок в электропроводность. Положительное значение
6 свидетельствует о преимущественной электронной про-
водимости, отрицательное — О дырочной. Для карбидов
величина д положительна и уменьшается при- переходе
от карбидов металлов подгруппы IVa к карбидам метал-
лов подгрупп Va и Via, что свидетельствует о преимуще-
ственно электронной проводимости, доля которой в ряду
MeIVC—MevC—MeViC уменьшается и соответственно
возрастает доля дырочной проводимости. При переходе
з* 35
х .а титана к карбидам циркония и гафния, от
°Т лаР«Иван1дия к карбидам ниобия и тантала абсолют-
КЭР Хение б увеличивается, следовательно, возраста-
вТ ^сследования^электрофизических свойств монокарби-
11^ЯЛВ плпгпупп IVa и Va в области гомогенности
ДОВ металлов подгрупп IVa и
Рис. 13. Зависимость электросопротивления карбидов от
содержания углерода в области гомогенности
[13, 347, 391, 703, 715] показали, что при увеличении содер-
жания углерода коэффициенты термо-э.д.с. и Холла уве-
личиваются (рис. 13, а и б). Отрицательные Значения
термо-э.д.с. и коэффициента .Холла (для TiCj^) свиде-
тельствуют о преимущественно электронном характере
проводимости во всей области гомогенности, причем доля
электронной проводимости увеличивается по мере при-
ближения к стехиометрическому составу. Уменьшение
сопротивления с ростом содержания углерода, по дан-
ным работ [703] и [705], связано с недостаточно высокой
чистотой исследованных карбидов. При исследовании
карбидов повышенной чистоты зависимость прямая.
Высшие по содержанию углерода карбиды редкозе-
мельных металлов (МеС2) также обладают металличе-
ским, преимущественно электронным, характером прово-
димости. С ростом температуры до 800° С сопротивление
П0«ти линейно, в интервале температур 900—
Zn с наблюдается резкий скачок в значениях электро-
ротивления, после чего зависимость снова линейная
36
(рис. 14). Аналогичная аномалия наблюдается и для кап
бида тория, у которого при температуре 141S’r Р‘
ктинная модификация превращается в^кубическуюЭт°о'
позволяет предположить, что у карбидов редкозеХьшых
четатлов происходит фазовое превращение емельнЫх
Изучение электрофизических свойств дикарбидов Г201
позволило вычислить значения концентрации носителей
Рис. 14. Зависимость электросопротивления дикарби-
дов редкоземельных металлов от температуры
тока, которые близки для LaC2, Рг^2 и Nb> 2 капбиды
ляют 0,5 электрона на атом Me. Так как гоЛ0Жа-
кристаллизуются в тетрагональной Решетк ’ й ячей-
щей 4 атома металла и 4 группы С2 в элемен Р
ке, сделано предположение, что 3 группы С2 уЩ У
в виде (С==С)2~, а одна (С = С^ СлелТ%ипет на
12 валентных электронов атомов металлов 10 ид
участие в связи Me—С, а 2 являются электронами про-
водимости. Предположение о такой модели строения Д -
карбидов хорошо согласуется с данными анализа газо-
образных продуктов разложения водой, которые показы-
37
Температура^
Рис. Г5. Зависимость электросопротивления полуторных кар-
бидов редкоземельных металлов от температуры
Рис. 16. Зависимость электросопротивле-
ния технического карбида бора от тем-
пературы
вают, что основные продукты разложения — ацетилен,
этилен и водород. Эта же модель объясняет и высокую
проводимость дикарбидов.
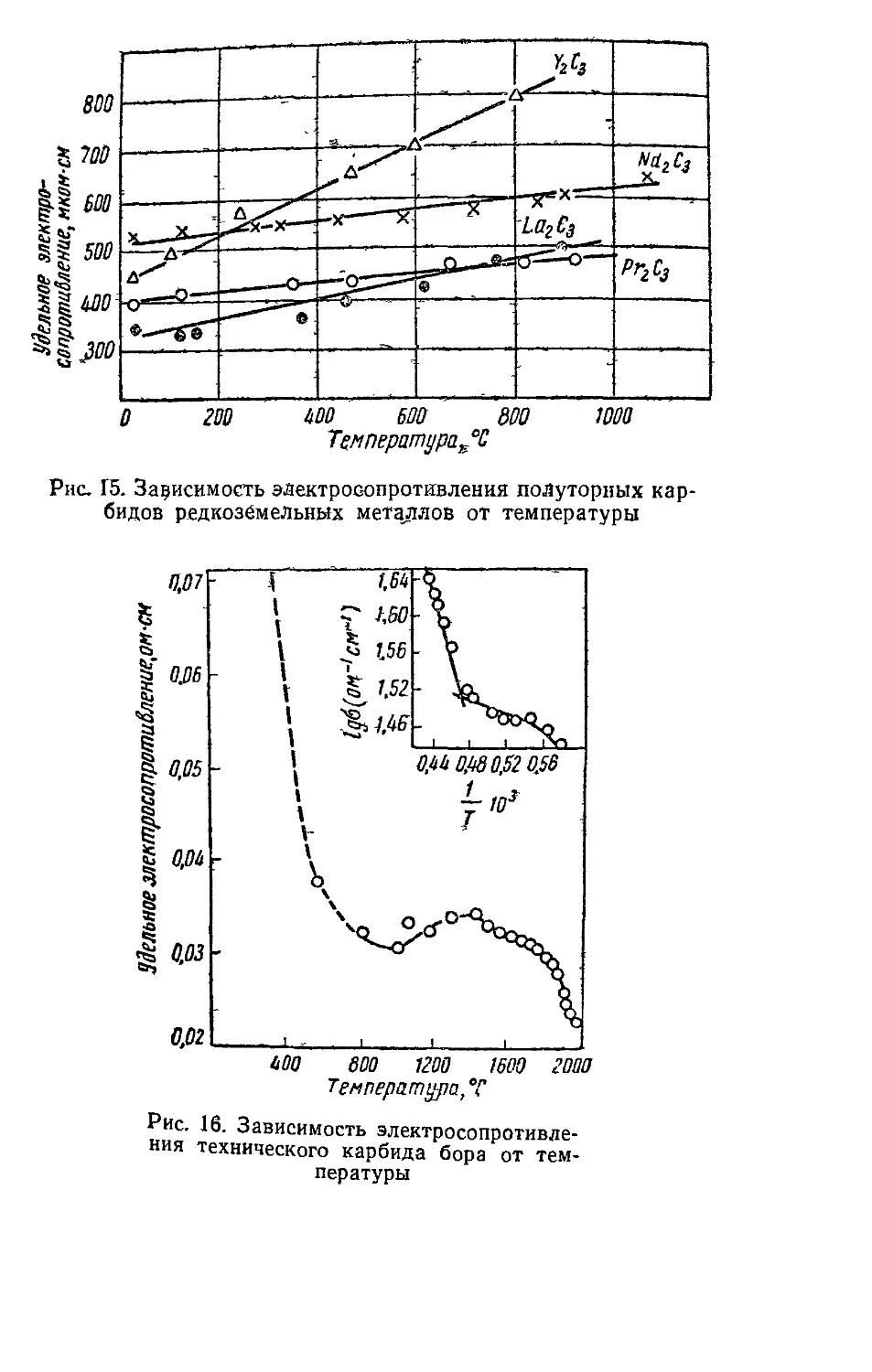
Удельное электросопротивление полуторных карбидов
редкоземельных металлов на порядок выше, чем у дикар-
бидов, и с ростом температуры растет линейно (рис. 15).
Карбиды бора и кремния отличаются полупроводни-
ковым характером проводимости. На рис. 16 представле-
на зависимость удельного электросопротивления техниче-
ского карбида бора- от температуры, “Согласно которой
сопротивление снижается вследствие возбуждения элек-
тронов примесей, после чего несколько повышается из-за
рассеяния на тепловых колебаниях решетки и снова сни-
жается при переходе к собственной проводимости карби-
да [909]. Карбид кремния -- типичный примесный полу-
проводник и сопротивление его существенно зависит от
примесей и дефектов решетки, под которыми понимают
наличие других элементов и отклонения от стехиометри-
ческого состава. Электропроводность технического карби-
да кремния изменяется в пределах 10—10~12 ом^1 -см~1
[711]. Характер температурной зависимости сопротивле-
ния карбида кремния (рис. 17) такой же, как карбида
бора.
Исследование проводимости монокристаллов SiC, по-
лученных осаждением из газовой смеси SiCl4 и толуола,
позволило предположить, что в SiC p-типа носителями то-
ка являются дырки в основной заполненной зоне и элек-
троны в примесной зоне проводимости; и SiC л-типа но-
сителями тока являются электроны основной зоны прово-
димости и электроны примесной зоны [711]. В работе [702]
установлено, что примеси
азота и фосфора сообща-
ют монокристаллам SiC
и-тип проводимости, при-
меси бора и алюминия —
P-тип проводимости. В
первом случае примесь
азота придает кристал-
лам зеленый цвет, во вто-
ром примесь алюминия —
синий или черный цвет.
Магнитны ссвой-
Ства карбидов изучены
Рис. 17. Зависимость электросопро-
тивления карбида кремния от тем-
пературы
Unv для карбидов некоторых переходных ме-
в основном длw у! и редкоземельных металлов. Так’
таЛЛ^пЕ“ переходных металлов в основном являются
как кар6“'}“ ПР„,1Я v которых все внешние электроны
Smho спарены, они слабо парамагнитны. Вычисленные
магнотные моменты дикарбидов редкоземельных эле-
ментов несколько меньше, чем трехвалентных ионов
(кроме YbCs) но близки к ним (рис. 18), что свидетель-
La Ce Pr Nd Pm Sm Ea Gd Tb Uy Ho Er Tu Yb Lit
Металл
Рис. 18. Магнитные моменты редкоземель-
ных металлов и их карбидов?
1 ~~ Me-, 2— МеСг
ствует о малом нарушении 4/-состояний редкоземельных
металлов при образовании карбидов [904].
Сверхпроводимость. Наиболее высокие тем-
пературы перехода в сверхпроводящее состояние наблю-
даются у карбидов ниобия и молибдена, причем у фаз,
содержащих больше углерода, они выше, чем у фаз, со-
держащих меньше углерода. Если для NbC Тк состав-
ляет 14° К, то для Nb2C она только 1,98°К. Значения Тк
для карбидов в области гомогенности падают при умень-
шении содержания углерода и при малых содержаниях
углерода сверхпроводимость практически исчезает.
Термоэмиссионные свойства. Рассматри-
вая термоэмиссионные свойства карбидов, можно заклю-
чить, что для карбидов переходных металлов работа вы-
хода электронов ниже, чем для чистых металлов. Так,
для титана она составляет 4,09 эв, для карбида титана
40
Габлицв 3
* Электрические и магнитные свойства карбидов
1 i • « S е 5 Косолапова Т. Я Удельное электри- ческое сопротив- ление, жком-слс Термический коэффициент электросоп- ротивления а10~3 град—' Коэффи- циент термо- s. д. с., мкв/град
Ве2С 1,1 IO6 —
ScC 274 “““"
YC 4,54.10* -Г— —34,6
y2c3 450 4-0,98 —6,4
yc2 30 4-3,63 —12,0
334 4-0,56 —
LaC2 45 4-4,10 —9,0
Се2С3 383 —‘ —
СеС2 60 4-2,18 —2,4
Pr2C3 396 ±0,24 —г
PrC2 39 4-3,85 —9,1
Nd2C3 500 4-0,26 —
NdCa 44 4-2,65 —10 0
ThCj^ (xsO) 25 — —
ThCo . (x«0) 30 — —
(*~0) 47 *
(x—0,15) 230 1 1 . ——
Постоянная Холла RAO^cm^k Работа выхода Постоянная Ричардсон , а см--град* Температур 1 перехода в 1 сверхпрово- дящее сост - яние, К Литературный источник
10>» дж &в
—4 и — ——* «»»«| 188]
— — | —11,95 1111 ——. 1111 ни 125}
i —4,41 | —5,03 i — i 1 1 И 1 [20]
—2,80 — — <1,28 [152, 20]
1 Г™ ' Г~— — [20]
—5,38 ^л" — — [152, 20]
т-4,38 — » L—. } [20}
—!— — t~r 9rtl [209, 213]
5,6 3,5 550 —— [18, 209]
7,15 4,4-f 4,3-10* <1,20 [152, 264]
— - - ‘ 1 *•— —*• —\ [199]
(£) СП и- - - *4 О ЬО СЛ СЛ Н- 10 4 чСлФ *4 К) N? ЭДЖЛ1+ 1+ г- г. £ & £ Ъл оо сл^ to 00^- Удельное электри- ческое сопротив- ление, । МККМ'СМ i
++ i ‘"F" о 1 с*э о -ч -4* “I- -4“ -Ц ° I -°- 1 isFfffT? - о 1 о О - ° <о w '-) о О О О °0 *° ® ХЛ 00 *4 СТ) СЛ- СЛ СТ) О о »-*- СЛ - - Термический коэффициент электросоп- ротивления а-10 ° град-1
] -<Г 1э , 4- i 1 ] ? -“ ы F 1 I tsg.00 Г|?1+ j? 5я -* 1 = ~ £ о О- >—. <О IT О 1т о о - » W-Д О' О- — 00 СЛ сл со g §^ = 3 >Г у о л. 0у’ •
111 1 F 1 1 Fl 1 Г Т I 1 4 'Joo » >— -* о о ст> 1 F Т 1 И 1+ 1+ 1+ ~ ъ ъ ООО Ю *» ОО SS- Постоянная Холла /?М+4сл3/« L
ел о» о .ы оо сл 1 1 1 1 Г1 Г V J ъ О1^- ьэ л Го ьэ СТ) со ст> м = Работа выхода
, оо" ю со to to со L 1 ”сл 1 I [ | 1~ Г— — го со О г— со со а». со -<л оо сл а
. О ? н- ко 1 Г 1 о! 1 С ( П <=^сл в/ ел ’ Постоянная Ричардсона а/емт-граФ
АЛ ЛАЛ АЛ •—1 >— КОСО-М"—— 1— S ъ k s sf Температура перехода в сверхпрово- дящее состо- яние, °К
•= 72-5 gg?' to—-1°'— ’ —Л сл S3 tsa „ со ” -О Ul“Ul£ м ' ,л S*3 ~£сл СП КО -СЛ gSg^-»» „ з" -“ S-“-Si21!' = е “§ ®s 5 -is § -?.= gT§ •— >—.о - сл » Литературный источник
Продолжение табл.
3 35 эв, для циркония 3,9 эв, для'карбида.циркония 2,18 эв
Максимальные значения работы выхода среди карби-
" _ [ и М0Либ.
карбидов
высокими
„ т. A- [910J-
„Ob'наблюдаются у карбидов титана, -ванадия
пена.
Электрофизические и магнитные- свойства
приведены в табл. 3.
ФИЗИКО-МЕХАНИЧЕСКИЕ СВОЙСТВА
Согласно данным, приведенным в табл. 4,
значениями микрртвердости отличаются карбиды пере-
ходных металлов, высшие по содержанию углероде кар-
бидные фазы редкоземельных металлов и карбиды не-
металлов. Микротвердосты уменьшается при переходе от
карбидов- металлов подгруппы IVa к карбидам металлов
подгрупп Van Via. Данный по микротвердости карбидов
металлов групп VII и VIII пока накоплено мало, но из-
вестно, что твердость их значительно ниже, чем карбидов
металлов подгрупп IVa,JVa и Via. Как показано и рабо-
тах [905, 906], твердость тугоплавких металлоподобных
соединений связана с-энергией межатомного взаимодей-
ствия, т. е. .энергией’кристаллической решетки, которая в
свою очередь определяется электронным строением эле-
ментов; составляющих соединение. Показано [836], что
микротвердость тугоплавких соединений, в частности
карбидов, определяется Статистическим весом стабиль-
ных электронных конфигураций атомов металла и угле-
рода и увеличивается с ростом статистического веса spz
конфигураций углерода.
Исследованиями микротвердости в области гомоген-
ности карбидов [374, 381,473] установлено, что микротвер-
Дость карбидов, циркония и тантала с уменьшением со-
держания углерода линейно падает и при экстраполяции
на нулевое содержание углерода для карбидов металлов
подгруппы IVa значения микротвердости соответствуют
микротвердости чистых металлов. Наиболее высокими
значениями микротвердости обладают карбиды бора и
кремния, характеризующиеся сильными ковалентными
связями между атомами углерода и бора (кремния),
имеющими направленный характер.
Прочностные свойства карбидов изучены еще недо-
статочно. Исследование кратковременной прочности об-
разцов карбидов титана пористостью 20—25% при изги-
4‘ 43
Таблица 4
Механические свойства карбидов
ф Микротвердость Модуль упругости Литературный источник <
Мн/м? Мя!мг кГ/мж’
Ве2С 26270 2690 320000 32000 [88, 1521
S С 26650 2720 [180]
ThC|_х (**0) ThC2_x (х~0) LC,_X 8330 5880 850 600 220000 22050 | [209] [231]
(х~0) и2с, — — —» [220]
—— — —— [231]
(х~0) Т1С,Г 27930+ 39,2 2850+40 315000 32000 [277, 350, 367,
1 S' Н 1 28660+1800 - 2925=±=184 350000 35500 369, 370] [152]
(ХЙО) Н*С1-ж 28540 291 а 350000 35900 [152, 405]
о 7 20520 2094 270000 27600 [152, 424]
(х—0,15) Nb.C,-* 20800+1950 2123+199
(х«0) NbCj х 19220+940 1961+96 — [152]
(х 0,11) ТагС1_х (х»0) СЛ СП О) 00 о О 1+ 1+ 4*. •— ОО СЛ О СП о и— СЛ -ч со СО 4*. 1+ 1+ 4*. *— со СЛ со — — [152, 432]
(х 0,06) Сг2зСо 9500 970 [152]
Сг?Сэ 13030 1330 [152]
Сг Сд 13030 1330 370000 38000 [152, 519]
MwjCj % 14490+2350 1479=1=24 220000 22100 [103, 152 , 908]
(х~0) МоС ^_х 14700 29400 1500 3000 530000 420000 54400 42800 } [152]
(х^О) WC 9040 923 700000 72200 [152, 908]
“6 5*- 54320—56260 5600—5800 — [680]
В4^1_ж йг0) 48450 32400 4950 3340 380000 39400 | [152]
44
Ш081 показало (рис. 19), что с повышением температу-
6е о 1600° С прочность карбида титана изменяется
Ело. С дальнейшим . 1Вв_______________
„иншеиием^мпера- ~ \”
•|
1 5
ZOO 600 1000 1W0 1800 2200
г Температура, °C
Рис. 19. Зависимость прочности
карбида Титана при изгибе от тем-
пературы
туры она резко возрас-
тает, достигая ппи
1900° С 104
(10,4 кГ[мм2),
чего повышение
пературы приводит к
резкому падению проч-
ности. Аналогичен ха-
рактер изменения проч-
ности и у образцов дру-
гих карбидов переход-
ных металлов. Макси-
мум прочности при из-
гибе составляет для образцов карбидов переходных ме-
таллов 0,6
при
Мн/м2
после-
Тем-
g 80
SO
W
20
О
,'1
I
л
ХИМИЧЕСКИЕ СВОЙСТВА
По характеру взаимодействия с водой карбиды мож-
но разделить на две группы: разлагаемые и неразла-
гаемые.
К числу разлагаемых водой относятся карбиды ме-
таллов I и II групп периодической системы, алюминия,
а также карбиды редкоземельных металлов и актинои-
дов. Карбиды щелочных и щелочноземельных металлов
представляют продукты замещения атомов водорода в
ацетилене металлами — ацетилениды и при действии на
них воды и разбавленных минеральных кислот разлага-
ются с выделением ацетилена. Несколько по-иному ведут
себя карбиды бериллия (ВегС) и алюминия, которые при
Действии на них воды разлагаются с выделением метана
и, следовательно, являются продуктами замещения ато-
мов водорода металлами в метане, а также карбид маг-
ния, который разлагается при действии воды с выделе-
нием метилацетилена СНз—С = С—Н. Карбиды редко-
земельных металлов и актиноидов при действии воды
Разлагаются с выделением смеси углеводородов. Соглас-
0 масс-спектрографическим исследованиям [7J, при раз-
45
ложении водой карбидов редкоземельных металлов
Ме^С^ газообразные продукты состоят из метана и во-
дорода.
Продукты разложения фаз Ме3С2 и MeCs-^-ацетилен
и водород, причем количество водорода, выделяющегося
при разложении фазы La2Cs, больше, чем при разложе-
нии ЬаСг-
В литературе имеется много- работ, ^освященных ис--
следованию состава газообразных продуктов разложения
карбидов редкоземельных металлов^и актиноидов водой
[8, 64, 204, 209, 770, 785, 789, 791, 911]. Результаты работ
часто очень расходятся (табл. 5), Это, вероятно, связано
с составами исследованных карбидов, которые часто да-
же не приводятся или не включают в себя данные о со-
держании связанного углерода.
Карбидные фазы редкоземельных металлов, близкие
по составу к стехиометрическим, использовали в работе
[8], в которой разложение проводили обработкой навески
карбида водой в кварцевых реакторах, продуваемых
углекислым газом, с последующим анализом"выделивших-
ся газообразных продуктов хроматографическим мето-
дом [787]. Наиболее надежные данные по составу газооб-
разных продуктов разложения водой карбидов актинои-
дов — результаты работы [209].
Анализ данных по составу газообразных продуктов
разложения водой дикарбидов редкоземельных ме-
таллов приводит к выводу о максимальном выделе-
нии водорода и минимальном ацетилена у дикарбидов
лантана и гадолиния, для которых характерны устой-
чивые состояния 4f° и 4/7 и наличие электронов & d-со-
стоянии.
При переходе от церия к празеодиму, неодиму и т. д.
до гадолиния, согласно правилу Хунда [9], увеличивается
число возможных термов и соответственно уменьшается
вероятность f -> d-электронных переходов. Уменьшение
вероятности участия в связях Me—С d-электронов при-
водит к ослаблению этих связей и увеличению количества
выделяемого при разложении ацетилена. Выделение аце-
тилена и этилена подтверждает предполагаемую на осно-
ании изучения физических свойств модель строения ди-
кароидов редкоземельных металлов, содержащих три
группы (С^С)2" и одну группу (С = С)4-.
46
Таблица 5
Состав газообразных продуктов разложения дикарбидов редкоземельных элементов водой
Фаза Содержание, % — - —. 1 - — - Литератур- ной источник
н» сн, с»н» С.Н. СаЭДб с,н8 С,На и era гомологи
YCa 4,5 71,7 19,0 гД -‘—5 [64}
5,03 15,6 58,6 10,08 8,04 J [8}
LaC2 1,03 27,2 71,75 — 17зб —А [641
10,02 г—Ь 67,19 6,74 . 12,48 1 2 - [789|
15,6 Не обн, 53,0 11?2 1 20,2 1 ". —- 1 И
СеС2 20,27—21,48 75—75 ,‘5 3,52—4,13 ’ 0,96 т“^ [64, 791]
13,4 68,6 5,91 8,81 5,55 [789]
15,9 Не обн. 58,23 10,49 15,35 Не обй. №- [8]
РгС2 зо’,о 66,2 2,50 A. i 1,22 [64 , 791}
6,79 78,16 8,18 10,68 0,75 [789]
7,92 Не обн..- 63,92 Ц.90 16,26 Нё обн. 5 i т—' [8J
NdC2 27,4 66,2 6,34 1Т?9
12,61 67,82 7,61 8,41 1,28 [789]
4,70 Не об и. 67,49 12,50 15,31 Не обн. ь-s- [81
SmC2 21,5 70,65 7,85 ——• [64, 785, 791}
5,1 71,1 7,9 12,2 1,28 2,32 [789]
00
Продолжение табл 5
Фаза н, сн. с,И,
GdCg 17,60 Не обн. 52,8
ThCo.ggOo.c® 9,3 86,4
17,1 29,4 47,7
ThC2_х 59,6 3,1 15,0
16,6 33,2 49,8
ThC1>84O0 об 27,2 2,35 9,5
Содержание. % Литератур- ный источник
с,н. С.Н» с,н8 CeHrt й его гомологи
14,8 13,3 Не обн. — [81
0,75 “— 1,2 2,35—СаН3 1,4—С3Н4 [209]
5,8 — 4— —— [204]
2,8 10,7 Жидкие и твердые углеводо- роды 8,8 [770]
—( — [785]
2,45 1,5 29,1- СаН3 1,5—С3Н4 8,6—С4Н0 Н,3-С4Н8 зи-с^Ню Высшие углеводо- роды 2,0 [209]
К карбидам, не разлагаемым водой, Относятся карби-
ды переходных металлов, бора и кремния.
Карбиды переходных металлов отличаются высокой
химической стойкостью [755, 894] и не разлагаются боль-
шинством минеральных кислот, их смесей и растворов
щелочей (табл. 6 и 7). В ряде случаев химическая стой-
кость карбидов металлов подгруппы Va более высокая,
чем карбидов металлов подгрупп IVa и Via. Это предва-
рительно может быть объяснено тем, чем в карбидах ме-
таллов подгруппы Va, особенно ниобия и тантала, атомы
металла и углерода имеют наиболее высокий, статистиче-
ский вес устойчивых электронных конфигураций при наи-
меньшем статистическом весе' нелокализованных элек-
тронов, которые в основном ответственны за способность
соединения взаимодействовать с реагентами с переходом
к ним электронов. Поэтому карбиды металлов подгруппы
IVa, которые характеризуются высоким статистическим
весом нелокализованных электронов, имеют обычно бо-
лее низкую стойкость, чем карбиды металлов подгруппы
Va. То же наблюдается для карбидов некоторых метал-
лов подгруппы Via, например для карбидов молибдена и
вольфрама, где статистический вес устойчивых ^-конфи-
гураций атомов в металле понижается из-за передачи
части локализованных электронов атомам углерода. Од-
новременно создается повышенная концентрация элект-
ронов, находящихся в свободном состоянии, и возмож-
ность их передачи компонентам реагента с разложением
соответствующего карбида. В то же время карбиды хро-
ма имеют высокую химическую стойкость, которая свя-
зана с тем, что в группировках из атомов хрома и угле-
рода статистический вес устойчивых электронных конфи-
гураций высок и концентрация нелокализованных
электронов между этими группировками мала. Поэтому
вероятность перехода'Этих электронов к атомам реаген-
та существенно уменьшается и карбиды хрома разлага-
ются с трудом. Высказанное предположение требует
экспериментального подтверждения, и работы в этой об-
ласти ведутся.
капУаИ^°^ее Высокой химической стойкостью обладают
Р_иды боРа и кРемния, которые не разлагаются мине-
нлй кислотами и их смесями, включая смесь азот-
вср <РтоРис'гов°Дородной кислот, разлагающую почти
P иды переходных металлов (за исключением кар-
49
Таблица б
Стойкость карбидов металлов подгруппы IVa—Via в различных
Растворитель Нерастворимый'"""’
_ZrCl-x HfC, Д 1^-х МЬС — 1
НС1 (плотность 1,J9) ^9t/100_ 98/98 .. 100/100 100/96
НС1(1:1) 100/97 : "98/95 96/43 100/99
H2SO4(плотность 1,84) 100788 z 97/0 " -100/0 100/0
H2SO4 (1:4) J00/97 98/76 /100/88 100/98
HNOa (1,43) 0/0 - 83/5 60/0 100/гид-
релиз
HNOS (I:l> 0/0 76/6 75/0 —
H3PO4 (1,7) *99/98 98/— 97/0 ЮО/дид-
релиз
H3PO4 (1:3) • 98/99 96/88 98/90 90/99
HCIO* 100/0 - 97/2 —
HCIO4 (1:3) 100/0 . 99/84- — —
H2C2O4 (насыщ. раствор) юо/юа 98/92 97/90 99/99
HC1+HNO8 (3:1) 0/0 14/6 0/0 92/гид-
ролиз
HiSOt+HNO* (1:1) — 2/0 0/0 100/22
HNCM-HF (4:1) 0 0 0 0
HjSCVbHsPCU (1:1) 98/— 97/— - 99/0 91/0
HsSOi+HAO* (1:1) 99/34" 96/0 99/95 100/95
HaSOi+Hi^ (1:1) 99/99- 96/91 98/66 —
H2SO4+H3PO4 (1:1)
NaOH: — -ми —/99
20%-ный раствор 100/100 100/100 100/98 99/100
10%-ный раствор 100/100 - 100/100 100/98 99/—
20%-|-бромная вода 63/86 98/8/ 88/64 100/84
(4:1)
20%+НвОа (4:1) 53/3 34/0 71/88
20%+K3[Fe(CN)fl] 82/37 86/24 —
(10%-ный раствор)
Использованы порошки с размером частиц 40—50 мкм.
В числителе приведен нерастворимый остаток, полученный обработкой кар
Оидов в течение 2 ч при температурах кипения соответствующих растворителем.
50
средах
ост ТОК*
ТЗС!_Х I Сг,С2 "СГуСа СГдзСа Mo2C _ J—x wc Btc, 1—X
100 98 99/— 92,3/— 53/— 89/39 ' 97/48 98/98-
100 98 99/96 99/3,5 —1,3 : ".88/83 : i 96/92 -—/97,6-
100 0 -/- — —' « 89?0 91 II 98/98
99 98 ' 100/68 99/1,6 52/1,3 90/83 = 96/95 /Win
100 99 (H2SCU l-'l) 100/99 (H2SO4 1:1) 100/99 (H2SO4 1:1) 100/98 * S’ : 0/0 - 63/1 97/97
99 98 100/99 — - 0/0 ; -72/10 ’ .—/96,9
98 гид- 100/98 100796 ~ 100М 92/- 91/93 —
ролиз — 98 92/76 96/90 - ——
— — -в— — 73/0 98/40 98/98
—- — 89/58 - 98/93
97 98 100/98 100/96 100/98 89/90 95/95
99/98 98/91 95/94 100/97-. 0/0 28/3 -
91/96 —/84 • -/91 —/97 4/0 . 92/42 -/91,2
0 Не Не Не 0 0 —/90,8
Гидро- раств. —/94 раств, —/4 раств. —/1,4 90/Q 96/0 —w
ЛИЗ 97 97 — —- write. 80/73 95/70 >w
93/98 — T.teBi, 94/95 —
100/99 — — — 92/88 96/93-
99/99 100/99 —/96 0/96 90/94 98/98 -/99,1
99/100 — — — 58/90 97/98 99/99
100 гид- ролиз 62/гид- —/88 -/86 —/86 65/60 70/60 —/99,6
— —"В- w— 31/36 88/87 —/98
ролиз -/61,5 —/53 —с 69/66 68/68 —
бидов в течение 24 ч при температуре 20—25’С, в знаменателе—обработкой кар-
51
Таблица 7
Химическая стойкость карбидов металлов групп IVa и Уа [894]
Нерастворимый остаток, %
Среда Я О о - о© о> / 8
d3 у ц о о" и
ь N К > Z н
NH4F, 5%-ный раствор 9?, 8 76,4 76,9 .—« 92,0 90,3
НгО2, 30%-ная . , . 10,0 97,2 99,6 5,3 0,-0 83,7
(NH4)jSaOg, 25%-ный. 82,2 36,5 0,0 32,5 88,5 91,2
Н^Ог+винная кислота НгОг+лимонная кисло- 3,3 95,6 96,5 64,5 0,0 57,0
та Н2О2+щавелевая кис- 1,2 95,0 97,6 62,7 0,0 57,0
лота 0,0 Гидро- лиз 0,0 59,2 0,0 —
НгОг+насыщ. раствор
комплексона Ш . . . . 12,5 98,0 97,0 0,0 3,5
НгОг+раствор NH4F , 5,3 0,0 0,0 30,0 0,0 3,5
(NH4) 2S2O8+р а створ
комплексона III ... . 97,0 96,4 72,4 97,6
(NH4) 2S2Oe+раствор
nh4f НС14- раствор "79,8 0,7 0,0 80,20 80,0 63,3
(NH4j 282^8 ...... 50,2 44,6 0,0 60,0 97,2 98,4
НС1+НгО2 . . , , HCl+насыщенный рас- 43,8 45,5 2,9 -78,6 99,1
твор^бромной воды , . 15,6 8,5 1,1 45,6 87,5 97,7
, Л??$й4(1 : 4) + раствор (NHO^Oa (25%) . . 70,3 4,3 0,0 15,6 64,0 97,7
+раствор „MW, (25%) 45,3 2,2 0,0 21,8
H2SO4 (1:4)+Н2О2 (30%) 0,25 0,75 0,0 15,4 13,2 —
бидов хрома) (табл. 6). Высокая химическая стойкость
карбидов бора и кремния объясняется сильными кова-
лентными связями между атомами углерода и бора или
кремния и очень низкой концентрацией нелокализован-
ных электронов.
На основании анализа комплекса свойств карбидов,
их кристаллических структур Меерсон [1] предложил
классификацию карбидов, которая несколько уточнена
Самсоновым [2, 17]. Исходя из характера химической свя-
зи, обусловленной особенностями электронного строения,
кристаллических структур, физических и химических
62
св ств карбидов, Самсоновым предложена следующая
1ассификация (рис. 20):
1 Солеобразные или ионные карбиды, образуемые
ei чны& и и щелочноземельными металлами, имеющими
saie тные s-электроны с первым потенциалом ионизации
т 3 до 7 эв.
2. Металлы подгрупп 1б~ и Пб вследствие высоких
низационных потенциалов (7,5— И затрудняющие
П fl
1а
Яо У8 Кб игв
3 p у П _ П Ы
Ш lie"Ine- |Шс
не2
L
E
—I
MJ I
1В 5
|Д|Л
T*|N J
Si “ip45
F 9
S’6
Cln
Ne’“
Аг”
Од
Sc
27
.28
23
Ба31
бе32
AS33
se34
0г35
Кг36
м>3
Sr
,47
Cd'8
SnM
Sb51
Те52
I 53
8а
Rnsf
Pb82
At85
,ао
La
Ro*
Fr
AC
а? ад
Кйалентно- КоШентчсге а кввалентни-
мешяллические ионные
Ро64
Bi83
П81
g- -
Рис, 20. Классификация карбидов
о
3
а
Z/
In*»J
W54
передачу s-электронов на связь с атомами углерода, или
совсем не образуют карбидов, или образуют нестойкие
карбиды. Вследствие этого их можно отнести к первой
группе или выделить в самостоятельную группу кова-
лентно-металлических карбидов.
3- Ковалентные (бора и кремния) и ковалентно-ион-
ные (бериллия и магния) карбиды, образуемые элемен-
тами, имеющими валентные хр-электроны.
По-видимому, способность электронов бериллия к осу-
?™И1° пеРех°Да 1 s22s2 -> ls22s2p приближает кар-
Д бериллия к ковалентным карбидам, образуемым эле-
обТаМИ С внешними P-электронами. В этом карбиде
6рп ЗУЮТСя сильные ковалентные связи между атомами
ериллия. Отвлечение связи от ковалентных между ато-
53
мями углерода яа ковалентные между атомами бериллия
обусловливаем формулу карбида Ве2С в отличие о,
MeCi' для щелочноземельных металлов. Сильная кова-
лентная связь между атомами бериллия при сохранении
доли ковалентной ’связи между атомами -углерода обус-
ловливает высокую асимметрию распределения^электрон-
н ой „плотности и придает карбиду бериллия свойства ион-
ного соединения, т- е. это ковалентно-ионное соединение
с превалирующей долей ковалентной связи.
Карбид магния по составу и структуре близок к кар-
бидамщелочноземельных Металлов, но подхарактеру свя-
зи ближе к -ковалентно-металлическим карбидам. Элек-
троны" атома Магния, как и атома бериллия, способны к
s-> р-переходуг но так как квантовой число его валент-
ных электронов выше, статистический вес ^-конфигура-
ций меньше и они -энергетическимменее устойчивы. В свя-
зи с .этим магний образует карбид MgC2, а не Mg2C, как
бериллий; при этом часть атомов углерода образует проч-
ные ковалентные связи типа С за счет s-электронов
атома магния,- часть их образует более слабые связи
С—С за счет образования sp-конфигураций атомами маг-
ния. Вследствие этого при разложении водой выделяется
не метан, как при~разложении Ве2С, и не ацетилен, как
при разложении, карбидов щелочноземельных металлов,
а метилацетилен СН3 — С С—Н.
4. Металлоподобные карбиды, образуемые ^-пере-
ходными металлами. К этой группе карбидов относятся
карбиды, представляющие фазы внедрения, и карбиды
типа FesC, имеющие структуры, близкие к фазам внедре-
ния: их свойства являются результатом закономерного и
монотонного изменения при переходе от карбидов метал-
лов подгруппы IVa к карбидам металлов подгрупп Va,
Via и VIII группы.
5. Солеобразно-ковалентно-металлические карбиды,
образуемые sdf-переходными металлами. Это карбиды
скандия, иттрия, лантаноидов и актиноидов.
ГЛАВА II
МЕТОДЫ ПОЛУЧЕНИЯ
КАРБИДОВ
Основные методы приготовления карбидов можно раз-
делить на 5 грунт синтез из элементов, восстановление
окислов металлов углеродом, осаждение из -газовой фа-
зы, электролиз расплавленных солеи л химическое выде-
ление. Классификация методов получения карбидов при-
ведена на рис. 21,
Синтез из элементов^ осуществляемый в ду-
говоигэлектрической пеш? прСллавлении, является исто-
рически первым методом, которым: были получены многие
карбиды (21,-22], В настоящее время для этого исполь-
зуют электродуговЫе плавильные установки.
Большинство металлов взаимодействует с углеродом
при температурах ниже температур их плавления, Вслед-
ствие этого более распространен метод получения карби-
дов нагреванием смеси металла и углерода, при темпера-
турах более низких, чем температуры плавления. Этот
метод осуществляется дз среде восстановительного газа
(водород, окись углерода, метан) или в вакууме и ши-
роко используется для- получения карбидов молибдена и
вольфрама. В последнее время- используют метод полу-
чения карбидов синтезом ^из элементов, при котором
смесь порошков металла и углерода нагревают под на-
грузкой,— метод горячего прессования, позволяющий
сразу получать компактные изделия из соответствующих
карбидов. Метод применяют для получения плотных об-
разцов карбидов урана, тантала и ряда других.
Из-за высокой стоимости порошков1 некоторых метал-
лов экономически наиболее эффективный метод получе-
ния карбидов— восстановление окислов со-
ответствующих металлов углеродом или
углеродсодержащими газами. Окислы могут взаимодей-
ствовать с углеродом также в расплаве и при темпера-
Урах ниже температур плавления соответствующих кар-
иДов. Наиболее распространено восстановление окислов
55
Получение карбидов^
Синтез из злементов восста- новление окислов Осаждение из газовой сразы
Химическое
выделение
Электролиз
расплавлен-
ных. солей
Плавление Нагревание при темпе- ратуре ниже f пл Восстанов- ление окислов углеродом
восстанов-
ление
окислов
углеродсо -
держащими
газами
Взаимодей-
ствие метал
лов или окислов
с (.
содержащими
газами
Взаимодей -
ствие гало-
генидов с
углерод- игрерод со-
держащими держащими
материалами
Разложение
карбонилов
вереде
водорода.
Химическое
разложение
Анодное
растворение
В среде
восстанови-
тельного
газа.
В вакууме
Спекание
методом
горячего
прессования
Врасплаве
При темпе-
ратуре
ниже tnn
В восстанови-
тельной среде
в Вакууме
Рис. 21. Классификация методов получения карбидов
«талтов углеродом при температурах ниже температур
шавления карбидов.
Взаимодействие окислов металлов с углеродом осно-
ван на том, что у ряда металлов (особенно переходных
м танов групп IV—VI) сродство их к кислороду мень-
ше, чем сродство углерода к кислороду (рис. 22) и поэто
Рис. 22. Сродство некоторых переходных
металлов к кислороду
му они могут при высоких температурах восстанавли-
ваться углеродом и при достаточно большом сродстве
углерода к металлам образовывать карбиды (рис. 23).
Результаты первых исследований условий получения
карбидов приведены в работах [24—26]. Однако, несмот-
ря на то, что восстановление окислов углеродом — один
из наиболее распространенных типов металлургических
процессов, в течение длительного времени были распро-
странены ошибочные взгляды на его механизм.
Процесс восстановления окислов металлов углеродом
рассматривался как двухстадийный [23, 27], который сво-
дится к диссоциации окисла на металл и кислород и
последующему взаимодействию углерода с кислородом:
57
WOO ?BDO
Температура, °C
Рис 23. Зависимость изобарно-изотермического потенциала
реакций образования карбидов от температуры:
а - карбиды металлов I, П, ИЦ JVa групп и урана;, б-карбиды
металлов Va- н Via подгрупп; в - карбиды металлов VIII группы
и -марганца
2МеО^2Ме + О2;
G Og = СОа.
Этот взгляд да процесс восстановления не'подтвер-
дился экспериментальными данными,, т. е. наблюдаемая
скорость реакций восстановления значительно выше той,
которая должна была бы быть при диссоциации даже
наиболее легкодиссоциирующих окислов.
Другой ошибочный взгляд на процесс восстановления
окислов углеродом развивался в работах [28—32]. Он
сводился к Тому, что реакция протекает при непосред-
ственном взаимодействии между компонентами в твер-
дой фазе, т. е. диффузии углерода в окисел. К такому
заключению авторы пришли "на основе того, что экспе-
риментальные данные хорошо согласуются с законОхМ
Диффузии и2 = £т.
Кроме того, изучая процесс восстановления окислов
меди, кремния, марганца, хрома, железа, никеля и ко-
59
бальта, Баукло с сотрудниками заметили, что газообраз-
ные продукты реакции — это СО и СО2, причем состав
газообразных продуктов постоянен для каждого окисла.
Отсюда делался вывод о зависимости состава газообраз-
ной фазы от упругости диссоциации окисла: чем больше
упругость диссоциации, тем выше содержание СО2 в га-
зовой смеси. Взгляды Там мана и Баукло были подверг-
нуты критике [23, 33—38] и показана их несостоятель-
ность.
Реальная скорость рассматриваемых процессов в сто,
а иногда и в тысячи раз превышает значения скорости,
вычисленной на основе представлений о диффузии в
твердой фазе.
Высокие скорости реальных процессов восстановле-
ния могут быть объяснены лишь решающей ролью газо-
образных продуктов, которые могут образовываться в
процессе реакции.
Неправилен также вывод Баукло о зависимости со-
става газообразных продуктов реакций от упругости
диссоциации исходных окислов, что подтверждается да-
же экспериментами самих авторов.
Так, известно, что Роире,о4>^о2(Сгго,)’ а’ по Данным
авторов, при восстановлении окиси хрома содержание
СО2 в газовой фазе составляет 80%, а при восстановле-
нии РезО4 65%.
Действительный механизм процессов восстановления
окислов металлов углеродом сформулирован Байковым
[33] и сводится к тому, что восстановление происходит за
счет газообразной окиси углерода; при этом соблю-
дается принцип последовательности превращений, со-
гласно которому восстановление идет от высших окис-
лов последовательно через стадии образования низших
окислов.
Образующийся в процессе реакции углекислый газ,
взаимодействуя с углеродом, превращается в окись угле-
рода, т. е. реакция идет по схеме
Л1еО4-СО^Л1е + СО2
С + СО„ 2СО
1 а
Me О -f- С Me + СО
60
Исследования процессов восстановления окиси нио-
б (3 ], окиси ванадия [40], окиси хрома [41], окиси цин-
ка закиси марганца [38] показали, что газообразным
родхктом реакции всегда является СО, а также что ско-
сть процесса зависит от температуры, активности, вос-
став вителя, размеров частиц восстановителя, давления
в системе и не зависит от давления прессования при бри-
кетировании исходных компонентов, т. е. от совершенства
контакта реагирующих веществ. Все это подтверждает
рохождение реакции по указанной схеме.
Расчет энергии активации процесса восстановления
разпичных окислов показал очень близкие друг к другу
значения, причем все эти значения энергии активации
бзизки к энергии активации реакции СО2 + С=2СО. От-
сюда следует, что скорость прохождения реакций вос-
становления окислов металлов углеродом лимитируется
наиболее медленной стадией — регенерацией окиси угле-
рода. Это, однако, нельзя' принять для:, восстановления
всех окислов. При восстановлении -окислов железа^ мар-
ганца, хрома и ряда других труднодиссоциирующих
окиспов энергия активации выше, чем реакции регенера-
ции СО. В этих случаях в начальный период восстанов-
пения лимитирующей стадией является кристаллохими-
ческая перестройка исходных окислов. Затем растет
роль газификации углерода и реакция чувствительна ко
всем параметрам, влияющим на скорость регенерации
СО. К концу процесса, когда поверхность реакционной
зоны сокращается, роль этого фактора снова уменьшает-
ся. Таким образом, процесс восстановления этих окислов
носит автокаталитический характер — скорость восста-
новления в начальный период быстро растет, достигая
максимума, и медленно падает.
Согласно данным работы [26], в среде водорода в пе-
чи водород вступает во взаимодействие с нагретыми
стенками печи по реакциям
nC + m/2H2->CnHw,
МеО + С„НтМеС + Н2 + НаО.
Кроме указанных газообразных продуктов, на ход
восстановления влияет присутствие в газовой фазе хло-
ра, галоидо-водородов, хлорных производных углеводо-
б°идОВ-[?98]ДОбаВКИ УСК°РЯЮТ пР0Цесс образования кар-
61
Пп «АПРИие процесса восстановления окислов угле-
Пр „л получений карбидов в-вакууме исключает воз-
можность Окисления и азотирования, способствует испа-
летучих примесей, увеличивает скорость реакции
S полноту ее прохождения; при этом равновесие реакции
^S+C^C+CO сдвигается вправо [43].
В работе [44] определены температуры начала восста-
новления ряда окислов в вакууме, которые значительно
ниже температур начала восстановления при атмосфер-
ном давлении, а также показано, что процесс проходит
через стадии восстановления до низших окислов. Это
подтверждено манометрическими исследованиями реак-
ций восстановления окислов переходных металлов угле-
родом [44—48].
Метод полученил карбидов взаимодей-
ствием металлов или окислов с углеродсодержащими
газами осуществляется при Достаточно высоком парци-
альном давлении углеводородов, которое может обеспе-
чить необходимое для образования карбида количество
углерода, но препятствует выделению свободного уг-
лерода.
Наиболее подробно взаимодействие металлов с ме-
таном и окисью углерода, а также с другими углеродсо-
держащими газами изучено в работах [49—51]. Резуль-
таты их исследований позволили установить оптималь-
ные температуры некоторых реакций, приведенные ниже:
Мо + Нг4-СН4->МоС + Н+(СН) . г . 700
Мо + На + СН4-МогС + Н2 + (СН) . , . ..... 800
w + Н2 + СЛН*-> W2C у Н2 + (СН) 2100—2400
W2C + H2-HCH) . - . . . . . 2440—2550
+ C-*Hv WC + Н2 + N2 +(СН) , 1000—2200
+ щ + Л . .Л . . . . 1300
Сг + Ha-f-СН4-+Сг3Сг-f-На(СН) 600—800
Этот метод осуществляется при непосредственном
аимодеиствии порошков металлов с углеродсодержа-
ими газами, а также осаждением на металлической
проволоке.
промышленного получения карбидов его исполь-
бмплп -1Л°‘ В основном он применим для получения кар-
_______елеза, никеля и кобальта. Механизм взаимодей-
* СН4, Свн6, СвН5СН3, С2Н2, СО+Н2 и др.
62
ствия л еталлов и .особенно их окислов с углеродсодержа-
щими газами исследован недостаточно.
Осаждение карбидов из гадовой фазы —
дин I з наиболее- перспективных методов получения со-
единении высокой чистоты. Он основан на взаимодей-
ствии между компонентами газообразной смеси, состоя-*
щей из галоидного соединения металла и углеродсодер-
жащих газов [26, 52—54]. Осаждение, происходит на
п верхности раскаленной угольной нити иди нити из туго*
плавких металлов. Водород Способствует развитию реак-
ции и в некоторых случаях значительно понижает тем-
пературу разложения галогениДа. Впервые этот метод
б 1 применен в работах [52, 54] для получения карбида
циркония по реакции
ZrCl4 -f* CHj -4- ZrC 4НС1 -4х
На процесс прохождения реакции взаимодействия
галогенидов металлов „с утлеродсодержащими „тазами
большое влияние оказывают температура нити и соотно-
шение концентраций реагирующих Веществ. Температу-
ра нити в процессе прохождения реакции-должна превы-
шать температуру плавления металлического^ компонен-
та, парциальные давления реагентов'дрлжны исключать
возможность осаждения свободного углерода. Форма
осадка определяется температурой нити.. Чем выше тем-
пература, тем крупнее осаждающиеся кристаллы. При
постоянной температуре Осаждение протекаете образо-
ванием слоя равномерного сечения. Подбирая определен-
ный режим осаждения,, можно получить карбид в виде
монокристалла. Представляется возможным осажде-
ние карбидов из паров соответствующих
карбонильных соединений. Исследование этого
процесса [50] показало, что карбиды молибдена и воль-
фрама могут осаждаться при взаимодействии паров со-
ответствующих карбонилов с водородом при температу-
ре 300—800° С и давлении 13,33—39,09 н/м* (0,1 —
0,3 мм рт. ст.) для МогС и 1333,3 h]mz (10 мм рт. рт.)
Для W2C.
Химическое выделение карбидов основа-
но на различной растворимости Карбидов и ^еталличе-
155~£аЛй?°^’о?тот Метод подробно изучался в работах
об « ’ - ’ карбидные фазы при этом выделяются
работкой различными химическими реагентами, при
63
. п|йталЛы и их сплавы переходят в раствор, а
которой ^т*ся в виде нераСТВорившегося осадка, и
каР™м анодного растворения.
МеМетод приготовления карбидов электролизом
„пиленных солей впервые был использован
Днлпие и Вейсом [63] при приготовлении карбидов мо-
либдена и вольфрама. Он основан на том, что при опре-
Генных условиям при электролизе расплавленных кар-
бматов выделяется осадок аморфного углерода, кото-
вый взаимодействуя с металлом, находящимся в раство-
ре в электролитической ванне, образует карбид. Наи-
более эффективны ванны, содержащие карбонаты, бора-
ты и фторид щелочных металлов и окисел тугоплавкого
металла. Введение в электролит борной кислоты в виде
метабората натрия повышает растворимость окислов
металлов в электролитической ванне.
ГЛАВА III
КАРБИДЫ МЕТАЛЛОВ
1 ГРУППЫ
КАРБИДЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Системна литий— углерод. Изучение системы
литий — карбид лития [4] показало, что существует одна
карбидная фаза Тл2С2, образующая с литием эвтектику,
-с температурой плавления 165° С и содержанием
0^% (ат.ЛС Карбид лития — кристаллическое вещество
белого или сероватого* цвета; кристаллизуется в ромби-
ческой решетке [24.1,.2б2].
Результаты^ термического анализа и рентгенографи-
ческого исследованияГлоказали, что существуют четыре
полиморфные модификации карбида лития с температу-
рами превращения 410, 440- и 560° С [66];
Первый вариант части диаграммы состояния систе-
мы, Li—Li2C2 [4] представлен на рис.24.
Карбид лития L12C2 впервые был получен Муассаном
восстановлением углекислого'лития углем: Ь12СОз4-4С=
= Ы2С24-ЗСС1{64]. Более поздние исследования [212, 241]
показали, Что этот же карбид можно получать нагрева-
нием смеси металла с углем в вакууме при температуре
600—800° С. При более высоких температурах карбид
легко разлагается. При температуре 700—800° С литий
образует карбид при действии на него окиси углерода
и углеводородов.
Нагревание лития в атмосфере этилена приводит к
образованию плавленого серо-белого продукта, соот-
ветствующего, согласно анализу, составу C2H4L16. Так
как образование такого соединения- маловероятно, было
показано, что идет реакция
6L1 + СаН4 =* Li2C2 + 4LiH.
см Разложении продукта реакции водой выделялась
есь ацетилена и водорода в количествах, соответствую-
щих реакции
ЦСа 4- 4LiH + 6НаО = CgHg 4- 4Н2 4- 6LiOH.
5 Косолапова Т. Я. __
Рис. 24. Диаграмма состояния системы
Li—Li2C2 [4]
Карбид лития удалось получить действием ацетилена
на раствор лития в аммиаке [64]. При этом образовыва-
лось двойное соединение ЫгСг’QHa* 2МНз, которое раз-
лагалось в токе водорода при комнатной температуре с
выделением мелкодисперсного белого порошка карбида
лития.
66
. гтема натрий —углерод. При температу-
Сн яоо°с взаимодействия с углеродом не наблюдает-
р х£°641 В работе. [67] показано, что взаимодействие
ся иатоня с углеродом зависит от сорта углеродсо-
пар0Вя1иего материала. Так, с графитом пары натрия
держаш реагИруют, в то время как с сажей реакция
п достаточно интенсивно. В системе натрий — углерод
ляяо\жены три фазы — Na2C2, NaCe и NaCi6 [71], из
°б? оых наиболее изучена фаза Na2C2. Карбид Na2C2
h летавтяет белый порошок, плотность которого при
15° С составляет 1,57 г/см3. При температуре 800° С он
ди социирует [64].
Основной метод получения карбида натриявзаи-
м действие металлического натрия с ацетиленом. При
температурах от температуры плавления натрия до 190° С
в аимодействие идет по реакции
2HC=CH-f-2Na=2NaC s CH + Н2 28,9 ккал (121Д кдж).
При температуре 200° С кислая соль разлагается [68]:
2NaC2H = Na2C2 + СаНа.
При температурах 400—600° С реакция идет сразу с
образованием карбида натрия:
С2Н2 + 2Na = NaaCa + На + 206,6 кдж (49,3 ккал).
Карбид натрия также был получен [69] при пропуска-
нии осушенного ацетилена через раствор натрия в
аммиаке. При этом получается ацетилид натрия:
ЗС2Н2 + 2NH3Na «= 2NaHCa 4- 2NH3 + СаН4,
который при нагревании разлагается с выделени-
ем Na2C2.
п«а^а^ид натрия образуется и при взаимодействии нат-
Р я с органическими соединениями [70].
ты калий— углерод. Исследуя продук-
в атмлгн^ДеЙСТВИЯ калия с углеродом при нагревании
темногп^пплОВ0ДОрОДа’ Деви [64] наблюдал образование
сгопаюшрГА длящего электрический ток порошка, легко
Формуле КрЭ В°ЗДухе- Это соединение соответствовало
леродом^ри^ысокит язаимодействия паров калия с уг-
н д м при высоких температурах [67, 73, 74] было по-
V*
67
l-^йно что графит уже при температуре 50 С раство-
ся в жидком калии, с ростом температуры раство-
^имость увеличивается. При температурах от 300 д0
Ж С пары калия взаимодействуют с углеродом и обра-
™ются продукты, содержащие 4, 8 и 16 молей углерода
на моль калия. Соединение КС8 образует крупные крис-
таллы медно-красного цвета, характеризуется решеткой
графита, у Которого атомы калия расположены в пус-
тотах между слоями. Соединение KCje сине-стального
цвета обладает металлическим блеском. Атомы калия
также расположены в пустотах между слоями. Имеются
данные о существовании карбидов КС24, КСзв, КС48 и
КС60 [Ю].
Наиболее распространенный метод получения карби-
да калия— взаимодействие калия с ацетиленом [75].
В зависимости от температуры может быть получен К2С2
или КНС2, который образуется при температуре 25—
30°Сжли при пропускании ацетилена через раствор ка-
лия в жидком аммиаке. Это бесцветный кристаллический
порошок плотностью 1,37 е/слг3, который при высоких
температурах выделяет ацетилен л превращается в К2С2.
Структура КНС2 близка по типу к СаС2 [72]. Карбид К2С2
получают также взаимодействием ацетилена с калием,
диспергированным в ксилоле при температуре 120—
130° С [5]. После промывки выпадающего при взаимо-
действии объемного осадка сухим бензолом или эфиром
и высушивания получается желтый порошок, содержа-
щий до 70% К2С2.
Карбид калия может быть также получен при тем-
пературе 300° С по реакции
6КОСН, - К2с, + 2KaCOs + 2С + 9На
и
2KCN + 3Mg — К2С2 4- Mg3N2.
„„5ИСТемг РУБИДИЙ — углерод. Карбид руби-
яиикВ“ПеРВЬ1е был получен Действием ацетилена на амми-
типртлЛаСТ°РРУбидия [76]. При этом получалось аце-
нягпрйр соединение Rb2C2*C2H2, которое при быстром
с вылрпрД температуры 300° С в вакууме разлагалось
Ппи мрп М Чистого темно-красного порошка Rb2C2.
углеоолД^мр^я°пМ нагреве его в вакууме образовывались
Пви^лейртпыт Л И ПРОДУКТЫ полимеризации ацетилена.
ри действии паров рубидия на графит, так же как и
68
v калия, образуются соединения, более богатые углеро-
дом—* RbCg, RbC24, RbCss, RbC-a, ЛЬСво [10].
Система цез'ий—-углерод. Карбид цезия по-
лучали при пропускании ацетилена через аммиачный
раствор цезия [76J. При этом раствор быстро обесцвечи-
вался, образовывался моноацетилид цезия в виде белого
кристаллического порошка м выделялся этилен.
Ацетилид Cs2C2'C2H2 яри быстром нагревании в ва-
кууме при температуре 300° С разлагается образова-
*4.20
0 200 ООО 600 * 800 10.00- 1200 М00- 1600 1800
Время, мин
Рис. 25, Зависимость скорости распада CaCs от времени
нием карбида Cs2C2 и выделением ацетилена. Т1ри мед-
ленном нагревании он разлагается на металл, аморфный
углерод и продукты полимеризации ацетилена. При на-
гревании в вакууме CsgCg разлагается на металл и уг-
лерод при температуре-700—800° С [67].
При взаимодействии пиролитического графита с па-
рами цезия при температуре 300° С и давлении 53,3 н/м2
(0,4 мм рт. ст.) получен карбид CsCg[10].
Показано, что при температуре 400° С карбид CsCe
разлагается, причем зависимость скорости разложения
от времени выражается кривой (рис. 25), имеющей два
перегиба: один, соответствующий переходу к фазе CsCje,
и другой — переходу CsCi6 в CsC24-
В работе приводятся данные о существовании кар-
бидов CsCse, CsC48 и CsCgo, свойства которых изучаются.
69
КАРБИДЫ МЕТАЛЛОВ ПОДГРУППЫ МЕДИ
гиетема медь-углер од. Исследование рас.
С углерода в жидкой меди [77] показало, что
творимостиуу р1100оС она составляет 0,0001% и с рос-
пр1! тГипеоатуры несколько увеличивается, достигая при
Lnn° С 0 003 %Р Согласно работе [78], растворимость угле-
™ в меди при температуре 2200-2300’ С составляет
0 024—0,033%. к
Было установлено существование карбидов меди
СщС и Си2С2- _
Рентгеновское Исследование карбида CU2C2 показа-
ло что он обладает структурой, подобной структуре кар-
бида кальция с гранецентрированной тетрагональной ре-
шеткой. Пикнометрическая плотность Си2С2 равна
4,62 г!см3.
При соприкосновении с острыми краями твердых ве-
ществ CU2C2 разлагается со взрывом [80],
При непосредственном взаимодействии меди с угле-
родом получить карбиды не удалось.
При пропускании ацетилена через аммиачный рас-
твор хлорной меди выпадает черный осадок, состав ко-
торого близок к Си2С [66]. При температуре 70—80° С
он разлагается.
Карбид CU2C2—продукт замещения водорода медью
в ацетилене, он образуется при взаимодействии между
аммиачным раствором Cui и КНС2 в соотношении 1:1
или 1:2 [79] в виде темно-красного кристаллического ве-
щества состава Си2С2.
Система серебро — углерод. Растворимость
углерода в серебре составляет [78] в интервале темпера-
тур 1600 1940° С 0,0012—0,0025%. При охлаждении
весь углерод выделяется из раствора в виде графита,
цетилен в аммиачном растворе азотнокислого серебра
гппВЖДает карбид сеРебра серо-желтого цвета. При на-
р ании высушенный Ag2C2 разлагается со взрывом [66].
пяблЛСгяг|Ма золот° —углерод. Золото, согласно
углрплн? 1’ Ра^ТВ0Ряет лишь незначительное количество
натпиЛ.я К-аРбиД Аи2С образуется при взаимодействии
желтого ™0СУльфата золота с ацетиленом [82] в виде
после (Ьипкт™а’ КОТОРЫЙ промывают водой, спиртом и
состоянии Р ия сушат иад сеРН0Й кислотой. В сухом
состоянии — взрывчатое вещество
70
ГЛАВА IV
КАРБИДЫ МЕТАЛЛОВ
П ГРУППЫ
КАРБИДЫ БЕРИЛЛИЯ, МАГНИЯ
И ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
Система бериллий — углерод. Первые иссле-
дования системы Be—С [84—86] показали .наличие в ней
двух карбидов — Ве2С и ВеС2. Однако литературные
данные о высшем карбиде ВеС2 весьма ограничены. Рас-
творимость углерода в бериллии незначительна.
Карбид Ве2С (39,98% С) представляет собой крис-
таллическое вещество красноватого цвета, обладает вы-
сокой твердостью и хрупкостью [89, 93, 94].
Изучение физических свойств горячопрессованных об-
разцов карбида бериллия, содержащих разные количе-
ства графита [88], показало, что добавка Графита, спо-
собствуя получению плотных образцов^не оказывает за-
метного влияния на температурный коэффициент линей-
ного расширения, но влияет на теплопроводность и
удельное электрическое сопротивление, Для каждого со-
става до содержания 50% графита удельное электро-
сопротивление уменьшается с ростом температуры. Со-
став 50% Ве2С с 50% С проявляет свойства, аналогич-
ные графиту.
Изучением равновесия реакции взаимодействия оки-
си бериллия с углеродом ВеОтв-р/2Ств =V2Be2CTB +
гСОг манометрическим методом [91] установлено, что
в интервале температур 1400—1650° С достигается рав-
новесное состояние, соответствующее реакции. При этом
продукты восстановления содержат карбид -бериллия и
непрореагировавшие окись бериллия и углерод.
На рис. 26 приведена кривая зависимости давления
киси углерода от времени при температуре 1890° С.
родукты реакции, соответствующие подъему на кри-
и, содержат Ве2С, ВеО и С. Продукты реакции, соот-
71
Ий конвой; -содержат почти Чистый
ветствуюшие с У а лР^теШ1ераТурах выше 1680° С
род.^ По-видлмому Ркарбйда бериллия: Вё^**
происходит ДИссп0ЦииаяЦаличи/в газовой фазе СО; Вег +
eT??_RpO +cl ЧТО приводит-к осаждению на хо-
лмных частях нагревателя, .кроме углерода, и. окиси бе-
рИ Равновесное давление диссоциации Ве26 по схеме
’2Ве2Ств^Вег + 72Ств, исследованное при темпер ату-
Рис. 26. Зависимость давления -СО: от^вре--
мени взаимодействия па-реакции
ВеО + С~* ВегСМ* СО
рах 1430—1669° К по методу Кнудсена [92], соответству-
ет уравнению
1g р ~ (7,026 ± 0,347) -
Карбид бериллия Ве2С был получен [88] плавлением
бериллия в графитовых тиглях с последующей Обработ-
кой продуктов реакции соляной кислотой^ Этот метод
малопроизводителен и применим лишь для получения
малых количеств карбида.
Второй метод получения карбида бериллия — восста-
новление окиси бериллия углеродом. В работе [87] про-
цесс проводили в вакуумной электрической печи при
температуре 1900° С. При этом выход составлял 60% •
Продукт, содержащий 85—92% Ве2С, получен [89]
прессованием смеси ВеО и древесного угля в брикеты
по^0Сле,?Х!?!?^м нагреванием их в вакууме при темпе-
ратуре 1900° С.
мгплп пРомышленных масштабах Ве2С получали [88],
ьзуя шихту из окиси бериллия и графита, содер-
72
ю 15% избытка графита и 2% парафина. Шихту
жа“у0Вала в брикеты, которые нагревали в графитовом
nPfz в индукционной лечи при; температуре 1950—
С в- течение 8 ч.в атмосфере очищенного водорода.
2 В той же работе исследовали условия получения ком-
пактных образцов карбида бериллия. Спрессованные за-
готовки с-металлическими добавками-V°% Fe, Si)
спекали при температуре,2250—2300°Св защитной атмо-
сфере в течение 30 мин. Полученные образцы были плот-
ны и нехрупки! Плотные и твердые образцы были полу-
чены также методом горячего прессования при темпера-
тур ах'1900—2250° С в атмосфере очищенного водорода.
Опыты,„проводимые в "атмосфере водорода, гелия и
аргона, показали^ что наиболее эффективен водород.
Система магний —углерод. В системе .Mg—-С
известны два карбида^Mg2.Cs (42,56% С) и MgC2
(49,69% С) ,[85].
Получить JVlgCs удалось нагреванием .порошка маг-
ния втоке-ацетилена или других, углеводородов при тем-
пературе 500° С [99[. При действии пентана на металли-
ческий магний при.температуре 656° С образуется Mg2Ca.
Карбид MgC2 при этих,условияг не образуется..
Исследование возможности получения MgC2 взаимо-
действием ацетилена с- этил бромидом магния [100] по-
казало, что образующийся при этом ацетилендибромид
магния при нагревании разлагается на JVIgBr2 и MgC2,
который при нагревании также разлагается по реакции
2MgCa Mg2C3 С.
Разложение MgC2 происходит при температуре 600° С.
При температурах 740—760° С разлагается Mg2C3 по
реакции
MgA 2Mgr 4- зс.
** подробно условия получения карбидов
ёЧг и Mg2C3 исследованы авторами работы [101].
МрсЖЖ взаим°Действие JMgCl2 с СаС2 и смеси
L с СаС2 при температурах 600—700°С в
мечомп 6£е аргона- в результате исследований было за-
тУрах ниже R99° р07}ействие MgCl2 с СаС2 при темпера-
Mgcio — С <темпеРатУРа образования эвтектики
& 2 ^aci2) практически не приводит к образованию
6 Косолапова Т. Я
73
.< г гтпи более высоко» температуре реакция идет че-
п« стадию образования MgCj. Скорость реакции дости-
гает максимума при температуре 600 С. Граница терми-
ческой устойчивости MgaCs соответствует температуре
740° С Это хорошо согласуется е данными работ [99,
100] Роль NaCl в реакции сводится к понижению
температуры плавления MgCl2.
По данным работы [102], образование карбида маг-
ния возможно при электролизе расплавленной смеси со-
лей 20% (мол.) КС1, 32% (мол.) NaCl и 48% (мол.)
MgCl? е применением угольных катодов и молибденовых
анодов. <-
Система кальций — углерод. В системе каль-
ций _ углерод известно существование одной фазы СаС2.
Карбид кальция СаС2 (37,48% С) представляет собой
бесцветные кристаллы плотностью в чистом виде
2,22 г/см3.
Как установлено многочисленными исследованиями
[93, 117—122, 125], карбид кальция существует в четы-
рех модификациях. Наиболее изучены СаС2—I с гране-
центрированной тетрагональной решеткой с четырьмя
молекулами СаС2 в элементарной ячейке, устойчивый
при температурах до 447° С, координационное число Са
по отношению к С равно 10, и СаС2—II, устойчивый при
температурах выше 447° С, кристаллизующийся в три-
клинной сингонии с периодами п=0,842; 6=1,148 и с =
= 0,394 нм- а = 93,4°, ₽-92,5° и у = 89,9°; 2=8.
Карбид СаС2—II был получен нагреванием техниче-
П00°С^П?2] ° и графитом при температуре
СаС2—III, согласно работе [122], обладает моноклин-
ной структурой и СаС2—IV имеет кубическую гранецент-
рированную структуру.
Обычный технический карбид кальция представляет
тетрагональную модификацию СаС2—I.
Электропроводность карбида кальция [124] зависит
от его химического состава и структуры, а также от тер-
мической обработки [123]. Наблюдается анизотропия, во-
никающая в связи с различным режимом охлаждения
тдельных частей слитка. Наружные слои, охлаждаю-
гаппЯ сильнее, имеют более высокое удельное электро-
отивление> чем внутренние, охлаждающиеся мед-
ленно.
74
Отжиг приводит к уменьшению сопротивления что
связано с переходом в другую модификацию
(СаСг—П или СаСг 1П), каждая из которых хаоактр
разуется полимеризованными анионами (С|~)„ или
(C2O*~> п v придающими соединению свойства полуппо-
водников. J у
Теплоту образования карбида кальция определяли в
ряде работ косвенными методами. Так, при определен™
м теплоты растворения CaCs и Са в разбавлений "о
лянои кислоте по реакциям со
СаС24- 2НС1 = СаС12 4- ед 4 Хдж
Са 4 2НС1 СаС12 4 На 4 540,1 дж
СаС2 + Н2 = Са 4ОД ^Х^ 540,1 дж
2С ~i~ Н2 = С2Н2223,8 дж
Са + 2С - СаС2 + {314,3- X) дж
найдена теплота образования карбида кальция,, которая
составляет 59,1 кдж]молъ (14,1—14,2 ккал!моль) [126,
273]. Термодинамические характеристики карбида каль-
ция приведены в работах [60, 103].
При нагревании в вакууме СаСг диссоциирует, что
используется для получения чистого кальция [127, 138].
При температуре 1850° С упругость диссоциации СаСэ
равна 133,3 н/м2 (1 мм рт. ст.), а при 2500°С 101,3 кн!мг
(760 мм рт. ст.}.
Изучая диссоциацию карбида кальция в вакууме,
Гельд и Есин [139] обнаружили, что этот процесс в зна7
чительной степени активируется добавками солей. При
этом роль добавок заключается в том, что анионы адсор-
бированных солей взаимодействуют с кальцием, ослаб-
ляя связь его с углеродом. Так как ионы фтора сильнее
взаимодействуют с кальцием, чем ионы хлора, то и
ускоряющее действие CaFa на процесс диссоциации СаСа
выше, чем СаС12.
По данным работы [104], карбид кальция может быть
получен нагреванием кальция, очищенного дистилляци-
ей в атмосфере аргона при давлении 399,9 н/м2 (3 мм
Tvn С\ еАЛ ^ажей’ прокаленной в вакууме при темпера-
Уре 1500 С, в атмосфере аргона при температуре 1250° С.
слученные таким образом продукты переплавляют в
6*
75
л, па я п гона. Состав окончательных продуктов-
^%СЙРСаС2, до 2% CaQ н да 2% С^.
нЛбочее распространен до настоящего времени Ме.
год получения карбида ^кальция восстановлением окис»
кятьния углем по реакции СаО+ЗС—СдСа+СО притем-
пеоатлрах 1840-2070° С, который- широко описан в ли-
тературе [64, 104—1151-
В качестве углеродистых материалов используют ан-
трацит, кокс или древесный уголь. Основное требование,
предъявляемое к углеродистым материалам, — это ми-
ниматьное содержание-газов и летучих-веществ. Наибо-
лее подходящий в этом отношении- материал — антрацит,
который содержит максимальное'по сравнению е дру-
гими материалами количество углерода и наименьшее
летучих примесей и золы.
Наиболее чистый продукт,, содержащий 99% СаС2,
приготовлен при нагревании смеси цианамида кальция
с углеродом в высоком вакууме при температуре 1100--
1150° С с последующим подъемом температуры до
1350° С. В связи с тем что карбид, кальция легко разла-
гается на воздухе, при получении его ъ процессе охлаж-
дения и дробления он заметно разлагается ХЗ^Ю°/о). В
работе [845] изучали поведение СаС2 в-различных газо-
вых средах с целью выбора среды, препятствующей раз-
ложению. Показано, что при температуре выше 800°С
такие среды, как азот, кислород, воздух, очищенный от
СО2 и влаги, СО, С02, агрессивны и вызывают разло-
жение СаС2. Снижение степени разложения возможно
лишь при интенсификации процесса охлаждения в обла-
сти температуры 1000—1200° С.
Система стронций — углерод. Карбид строн,
ция SrC2 был получен [64] нагреванием смеси окиси или
карбоната стронция с углем.
Рентгенографическое исследование [93] показало, что
обычно получаемый продукт содержит большое количе-
ство примесей в виде SrO и графита.
Известно существование трех модификаций карбида
Ч7П0^ЦИя: кубическая, стабильная при температуре выше
^70 С с периодом решетки а = 0,625 нм; гранецентриро-
-Н тетрагональная с периодами: а = 0,582 и с=
7 шп НМ' с!а~ 1’150» стабильная в интервале температур
( 90) н~( + 370°С) и модификация, устойчивая при
температурах ниже —90° С.
7б
Система, барий—углерод, В системе барий —
роод обнаружено Существование .одной фазы ВаС2, ко-
уГ^яя при комнатной температуре кристаллизуется в те-
агональной решетке типа СаС2 е периодами:. а=0,622,
т2о7О6 «лг, с/а-1,134 [93, 119]. .При температуре выше
?50°С кристаллизуется-в г.ц. к< решетке [85].
Карбид бария, подобно карбидам бериллия и каль-
пия, при нагревании в вакууме при. температуре 1355—
1671° С диссоциирует на металл и "углерод:
ВаС2тв == Ваг SCj-g.
Данные но скорости диссоциации ВаС2 и давлению
диссоциации паров бария над карбидом показали, что
скорость растет с ростом темпер'атурьг.от'4,5-10~4 при
1355°С* до 1,5-1 СП2 -г/(см2 ‘ сек) при'167ГС и-зависи-
мость давления'диссоциации от -температуры выражает-
ся уравнением р 51J
Igp == 3,267 -—7
Карбид бария впервые бйл. получен нагреванием
20%-ной амальгамы бария с угольным "порошком в ат-
мосфере водорода при температуре 700—800° Q [64], Поз-
же при нагревании смеси углекислого-бария е магнием
в присутствии Сажи при температуре 700—800° С получа-
лась смесь карбида бария и окиси магния:
ВаСО3 + 3Mg+ С ВаС2 -р 3MgO.
Содержание карбида бария в смеси составляло не бо-
лее 38%. Отделить же его от окиси магния не представ-
лялось ВОЗМОЖНЫМ-
Исследование реакции восстановления окиси бария
сажей [146, 147] показало, что реакция в вакууме идет
при температуре 1400° С с выделением СО и образовани-
ем карбидно-окисной смеси.
Исследования реакций
и ВаО + ЗС «= ВаС2 + СО
ВаО2 + 4С = ВаС2 -р 2СО
лепппп?Л^14^’ ЧТ0 при Установлении окиси бария уг-
родом в вакууме при температурах 1000—1500° С вслед-
77
нмглкой летучести окислов бария получить карбид
Хктич “ки »е удается. Карбид бария образуется при
Установлении углекислота бария углеродом; при этом
Унует учитывать .то, что упругость диссоциации ВаСО3
температурах 1300-1400° С настолько велика что
он практически полностью разлагается на ВаО и СО2 и
скорость диссоциации ВаСО3 превалирует над скоростью
регенерации СО по реакции Будуара:
СО2 + С = 2СО.
Суммарно процесс идет по реакции
ВаСО3 + ЗС = ВаС2 + СО2 + СО-
Это предположение подтверждено экспериментальными
данными, согласно которым при температуре 1600° С и
использовании шихты, содержащей 5% избытка сажи по
сравнению с расчетной, образуется карбид бария, по со-
ставу близкий к стехиометрическому (Ссвяз =14,0%).
КАРБИДЫ ПОДГРУППЫ ЦИНКА
Система цинк—углерод. Кипящий цинк рас-
творяет лишь незначительное количество углерода {78].
Соединения цинка с углеродом неизвестны. Имеется
ряд работ, посвященных исследованию термодинамики
[154, 155] и и кинетики [156—158] процессов восстановле-
ния окиси цинка углеродом и окисью углерода. В этих
работах показано, что восстановление идет до образова-
ния металлического цинка.
Цинк не образует индивидуальных карбидов, но из-
вестны достаточно устойчивые тройные карбидные спла-
в“' 7L2Z?C,’.,J,r!ZnC'’ Hf2ZnC- NbaZnC, [159, 305[,
^nooZ-rigOv-^O [160].
Кадмий и ртуть, аналогично цинку, не образуют со-
единении с углеродом. г
ГЛАВА V
КАРБИДЫ
ПЕРЕХОДНЫХ МЕТАЛЛОВ
КАРБИДЫ СКАНДИЯ, ИТТРИЯ
И ЛАНТАНОИДОВ
Система скандий—^углерод. Согласно пер-
вым исследованиям, в системе Sc—С [25] существует
один карбид состава, соответствующего формуле Sc4C3.
Однако последующие исследования существование этого
карбида не подтвердили. Получей карбид, соответствую-
щий формуле ScC, кристаллизующийся в кубической ре-
шетке типа NaCl [179], и предположено существование
карбида Sc2C с гексагональной структурой. Виккери с
сотрудниками [180] подтвердили существование карбида
ScC и нашли, что он имеет гексагональную структуру,
что, однако, не подтвердилось в дальнейших исследова-
ниях и, по-видимому, является ошибкой.
В работах [181, 182] показано, что при восстановле-
нии Sc2O3 углеродом при температурах выше 2000° С-в
атмосфере защитного газа продукты реакции наряду с
Sc2O3 и графитом, согласно данным рентгенографическо-
го анализа, содержат кубическую фазу со структурой
NaCl и периодом а —0,447 нм. Сделано предположение,
что образуется либо карбид скандия с дефицитом ато-
мов углерода ScCj^ либо оксикарбид Sc(C, О). Обра-
п°йла1НИД оксикаРбидов подтверждено работами [183] и
[184]. Следовательно, в системе Sc—С существует кар-
ид, близкий по составу к ScC, кристаллизующийся в
кубической структуре, склонный к образованию окси-
карбидов.
Система иттрий — углерод. В системе Y—С
установлено существование четырех фаз: Y3C, YC, Y2C3
и хС2. Низший по содержанию углерода карбид Y3Cj_x
существует в области составов от Y3C0>75 до Y3C12.
79
пи^. -т4екоторы* свойств карбида иттрия со-
ИсслеДОвани &64j докауало> чт0 периоды ре-
ставов УзСо g 1М * о,5О68*±1О~4 и для Y^ п
гт.рткй составляют для ^з^од ’ i и
“ST«дельное, электрическое сопротивление Coot-
О.=» я*, ДА' 16а МЮЯ.С№ И молярная магнитная
ветственно 1^1^ -, 426- КГ4 CGSM.
восприимчивость 400
Монокарбид YC по-
Рис. 27. Диаграмма состояния си-
стемы La—С
лучен при восстанов-
лении окиси иттрия уг-
леродом в вакууме при
Температуре 1900° С.
Исследование условий
получения YC [185, 186]
показало, что при тем-
пературе 1700°С обра-
зуется оксикарбид ит-
трия, с повышением
температуры, кислород
из оксикарбида удаля-
ется в форме СО с об-
разованием при 1900° С
YC.
Выше температуры
1900° С монокарбид
плавится с разложени-
ем на иттрий, частично
испаряющийся в ваку-
уме, и жидкую фазу,
богатую углеродом.
Полуторный карбид
Y2C3 получается при
взаимодействии окиси иттрия с углеродом в вакууме при
температуре 1800° С, При этом, как и в случае получения
монокарбида, процесс идет через стадию образования
оксикарбидов.
Дикарбид YC2 впервые был получен нагреванием тон-
кой смеси иттриевой руды с углем в дуговой печи
188]. Исследование условий получения YC2 восстановле-
нием окиси иттрия углеродом в вакууме [185, 186] по-
казало, что близкий к расчетному составу продукт П®ЛУ'
чается при температуре 1900° С. В отличие от YC и
дикарбид устойчив в вакууме при температуре 1900 С.
80
Система лантан*-углерод. Согласно предло-
иной Г71 диаграмме состояния, в системе La—С (рис.
гстаиовленохуществование двух карбидов: La2C3 и
т яГ из которыхХазС^ плавится инконгруэнтно, a LaC2
^нгоуэнтна-Растворимость углерод* и твердом ланта-
о ппи температуре-6950 С составляет 1,14%. La2C3 гомо-
генен в области содержаний от- 9,98 до 11.58% С (рас-
четное содержание и La2Cg41,48 %).
1а2С3_^ имеет о.ц.к; решетку^ параметр которой
тля стороны, - богатой металлом^ -составляет 0,8803 ±
±0,00004 нмг Для- стЪроньщбогатой углеродом, 0,8185±
±0,00004.нм [7].
Концентрационная область гомогенности LaC2 очень
мала. При7температуре 1800°С LaC2 претерпевает моди-
фикационное' превращение.. Масс-спектрографическое ис-
следование продесса^испарения дикарбида лантана [170]
показало, что прпиемперятуре 2200°С в парах над ди-
карбидом имеются молекулы карбида LaCs и атомы лан-
тан*; отношение Интенсивности пиков -масс -ионов La к
LaCs соответствует 1:16.
Подробное исследование условий получения полутор-
ного карбида Ьа2Сз_х. (196] показало, что при восста-
новлении окиси „лантана углеродом в Вакууме или в
инертной атмосфере (аргон) La2C3_x не образуется,
что вероятно, связано с образованием термодинамически
более устойчивого дикарбида LaC2'l Карбид La2C3_x
получен при взаимодействии дикарбида лантана с ме-
таллическим лантаном при температуре 1400—1500° С в
атмосфере аргона. Однако -этот метод мало технологи-
чен, так как требует предварительного- получения дикар-
ида, весьма неуетойчивого соединения, которое легко
разлагается влагой воздуха.
иппгпСТат0ЧН0 чистый полуторный карбид может быть
У ен синтезом из элементов при дуговой плавке.
во^ЛеД0Вание условий ПолУчения дикарбида лантана
1921пХВоЛеНИеМ окиси лантана углем в вакууме [190-
1500° г Л°’ ЧТ° Реакция начинается при температуре
«нтана со™вХ^85о“Г9Роо^а получения ^карбида
водятсяСТепЯМ™ ерИЙ*~уГлеР°д- В работе [119] при-
СеС Се СД и r’lr0 сУществовании в системе Се—С фаз
» v-,'-2v-'3 и с_ес_2.
81
Рентгеновские исследования этой системы [193] под-
„^ существование только двух карбидов церия:
твердили Уанее описанный Штаккельбергом СеС, по
мнению авторов, является твердым раствором углерода
н пепии Однако в работе [820] при исследовании равно-
весия системы Се-Н2-СН4 обнаружено существование
кавбида СеС, обладающего гранецентрированнои куби-
ческой решеткой. Существование карбида £еС3, описан-
ного Варфом [7], не подтверждено.
Полуторный карбид Се^С3 получен взаимодействием
дикарбида с металлическим церием- и синтезом из эле-
ментов при дуговой плавке [196]. Дикарбид впервые был
получен восстановлением окиси церия ^углеродом [187].
Исследование методов получения и свойств СеС2 описа-
но в работах [64, 180, 192].
Наиболее подробное исследование процесса восста-
новления окиси церия углеродом с целью получения кар-
бида СеСз в вакууме показало, что восстановление на-
чинается при температуре 1300° С, а при 1700—1800° С
образуется продукт, по составу соответствующий карби-
ду СеС2 [192]. Этот продукт представляет собой кристал-
лический порошок золотисто-желтого цвета.
Система празеодим — углерод. В системе
празеодим—углерод на основании данных рентгенов-
ского исследования [7]. было- установлено существование
двух фаз Рг2С3 и РгС2. Существование ранее описанных
фаз Рг3С и РгС не подтверждено.
В работе [820] описано получение монокарбида РгС
при изучении системы Рг—Н2—СН4, однако эти данные
нуждаются в уточнении и проверке.
В работах [7] и [180] карбиды празеодима получали
восстановлением окиси празеодима углеродом. Однако
попытки получения Рг2С3 [196] показали, что получить
однофазный полуторный карбид этим методом не уда-
ется. Поэтому наиболее эффективные методы получения
Полуторного карбида—дуговая плавка смеси празеоди-
ма с углеродом и взаимодействие высшего карбида
ггСг с металлическим празеодимом в печах с индукци-
онным нагревом при температуре 1400—1500° С.
Система неодим —углерод. В системе нео-
дим-углерод обнаружены фазы Nd2C3 и NdC2 [7]. Су-
ществование описанных ранее фаз Nd3C и NdC не под-
тверждено. Условия получения и свойства карбидов нео-
82
аналогичны условиям получения и свойствам
SiWOB празеодима.
2 каобидные фазы самария, европия, гадолиния, тер-
Лпя диспрозия, гольмия, эрбия, тулий, иттербия и лю-
₽пня изучены недостаточно. Известно, что эти металлы
образуют карбиды составов <за исключением
рвоопия) и МеС2. Для некоторых из них установлено су-
ществование фаз УИг2Сз [7, 180].
Почуторные карбиды гольмия, эрбия, тулия и люте-
ция изоструктурны е полуторным карбидом иттрия.
Карбид Но2С3, по данным работы [7], существует в двух
изоструктурных модификациях с решетками Рг2С3 и
УгСз- Карбид Yb2C3 не изоструктурен ни с Рг2С3, ни е
У2С3. В работе [873] был получен карбид Но2С взаимо-
действием гидрида гольмия с углеродом при темпера-
туре 800° С в среде аргона и исследована его структура
методом дифракции нейтронов.
КАРБИДЫ АКТИНОИДОВ
Система торий — углерод. Согласно предло-
женной в работах [197, 198J диаграмме состояния систе-
мы Th—С, в исследуемой системе существуют два сое-
динения: монокарбид с содержанием углерода 4,92%
температурой плавления 2625° С и дикарбид с содержа-
нием углерода 9,37% и температурой плавления 2655°С.
При высоких температурах оба эти карбида дают непре-
рывный ряд твердых растворов; при комнатной темпе-i
ратуре взаимная растворимость их почти полностью
отсутствует.
Более поздние исследования системы Th—G [199] под-
твердили существование кубического гранецентрирован-
ного монокарбида и дикарбида, существующего в виде
моноклинной и кубической (типа CaF2) модификаций.
редложенная диаграмма представлена на рис. 28.
Исследование диффузии углерода в торий [201] опре-
лением градиента концентрации углерода по радиусу
ТОРИЯ ПРИ разных временах диффузионного от-
показало, что коэффициент диффузии при темпе-
So^nJ000,0 составляет 7,4• 10~9 см2!сек, при 1100°С
актИВапИиЛ2/ТЛПри 1200°С 5’7-Ю“8 см2! сек.. Энергия
(38 ««мольД)ИфФузм составляет 159,2 кдж/моль
83
Пои изучении растворимости углерода в тории [202}
«Атолами ренгеновского л металлографического анали-
,пй установлено-,'ЧТО при комнатной температуре раство-
пимость составляет 0,35% (по массе) С, при темпера-
??„е 800«С 0,43^, при 1018° С 0,57% и при 1250»С
=0,91%<»
Монбкарбид. кри-
сталлизуется в г. ц. к.
решетке липа NaCl.
Данные о периоде ре-
шетки, приведенные
разными авторами, не-
сколько' расходятся.
Так, приводятся значе-
ния & = 0,5325 нм [198],
~ 0,529 нм [209] и 0,534 нм
[197].
В работе [197] уста-
новлено, что период ре-
шетки монокарбида го-
• рия изменяется от
0,529 до 0,534 нм в за-
висимости от состава.
Измерение периода ре-
шетки ThC1-:J1. В зави-
симости от состава
[208] показало, что при
периоды решетки не из-
ThC
* wo
Содержание C°L
^0 ЛО 50 50 65
3000
I
ocl
Th+TtlC
ПК/
1^-4-
I
-4
ThCzl
1 1
I 1 TW>ThC2\
гтоо
^zm
1200
SOO
0
ж+тьс
1 -
Д-----
TfifrThCr
10
2 4 6 8
Содержание C, %(no массе)
Рис. 28. Диаграмма состояния си-
стемы Th—С [199]
0<x<0,4, т. e. от Th до ThC0 65
меняются и продукты могут рассматриваться как твер-
дые растворы углерода в тории. При составе ThC0 6? До
ThC egg период решетки монотонно возрастает-от 0,5296
до 0,5335 нм, после чего снова не изменяется.
Дикарбид кристаллизуется в г. ц. тетрагональной ре-
шетке с периодами а = 0,586 и с = 0,529 нм с четырьмя
молекулами в элементарной ячейке [119]. Имеются дан-
ные [209] о том, что он обладает о. ц. тетрагональной
решеткой с периодами а=0,2144 нм и с=0,529 нм с дву-
мя молекулами в элементарной ячейке, а также данные
[204], утверждающие, что ThC2_x—псевдотетрагональ-
ный и кристаллизуется в орторомбической решетке с
периодами а=0,826, 6=1,052 и с=0,422 нм с восемью
молекулами в элементарной ячейке.
84
к паботё [215] показано методами рентгеновского
иза и нейтронной дифракции на монокристаллах,
аЙ ThC кристаллизуется в моноклинной решетке с пе-
"° „н'а-0,653, 6-0,424. с=0,656 ня, ₽=104’ е че-
тырьмя молекулами ТЬС^ в элементарной ячейке. Это
„лпткеождено данными работы fl 34], согласно которым
^пиоды решетки ThC} д9-_ооставляют -а= 0,6691 ± 0,0001,
b=О 4231 ± 0,0001,-с=0,’6744^.0,0002' нм, ₽ =] 03°50'.
В работах [215] и [134] исследования проводили пре-
цизионными методами на .чистых, материалах с учетом
их точного состава,* что позволяет считать их- наиболее
достоверными.
При температуре ШХГС наблюдается переход моно-
клинной модификации ThC2_x в кубическую типа CaF2
с периодом решетки а— 0,5808 нм.
При изучений термодинамических свойств карбидов
тория измеряла^ д, с. ячейки типа Th, ТЬРДСаРгПЬЕ^,
ThCx [200] на -образцах,, в которых отношение С: Th ме-
нялось в интервале ДД—2,8 при, температурах 750—
950° С.
Измерение упругости даров ThC^^. эффузионным
методолг Кнудсена е айализом конденсата активацион-
ным методом [211] дало возможность-вычислить теплоту
образования дикарбида тория (табл. 2).
В работе [214] измеряли- теплопроводность карбидов
тория. Для монокарбида теплопроводность оказалась
равной при температуре 200° С 8,4; при 300° С-7,9; при
400° С 7,9 вт/(м*град) [0,020; 0,019; 0,019 кал[(см -секу.
Хград)]. Для дикарбида теплопроводность равна соот-
ветственно при температуре 200° С 23,4, при 300° С 20,9
и при 400°С 20,5 втКм+град) [0,056; 0,050 и
0,049 кал! (см • сек град) ].
Значения удельного электросопротивления для мо-
нокарбида, приводимые различными авторами [209,
213], значительно расходятся, что можно объяснить раз-
ными составами исследуемых образцов, характеристику
которых авторы приводят недостаточно полно.
С Ростом темРеРатУры удельное электросопротивле-
ние ТпЦ^ линейно возрастает, термо-э. д. с. ТЬС1—Х —
отрицательная и пропорциональна температуре,
тип ^окарбид ^рия получали при нагревании шихты
пиг+ЗС в вакуумной индукционной печи при темпера-
85
1йгПог Г205] Низкая устойчивость ThC^ на воз.
ТУРв не позволяла полностью идентифицировать полу-
?етп«3ПоРбадК™рия впервые был получен при воссга-
дикароид Г углеродом с целью получения
новлении тор ПозжРуэтот же карбид был получен нагре-
Х” смеси окиси тория с углем в виде цилиндров в
луговой электрической печи [203, 2U4J.
АУ В работе [206] дикарбид получали восстановлением
окиси тория сажей при температуре 2130 С. Реакция
при этой температуре в течение 30 мин проходит прак-
тически полностью (99%)/
Исследование радиометрическими методами полно-
ты удаления примесеи из TI1C2_х для его очистки по-
казало, что при температуре- 2130° С такие примеси, как
изотопы Cs, Sr, Be, Ba, Sm, Eu, Ag и Cd, удаляются пол-
ностью.
Исследование условий получения карбидов тория
взаимодействием окиси тория с сажей в вакууме по ре-
акциям
ThOg + 3C = ThC-E2OO,
ThO2 4~ 4C — ThCa 4- 2C0,
проведенное в работе [207], показало, что продукты,
близкие по составу к ThC и ThCs, образуются при тем-
пературах 1800—1900° С из шихт стехиометрического со-
става при давлении 1,33 н/ля2 (~10~2 мм рт. ст.). Полу-
ченные по таким режимам продукты содержат при по-
и 0,1% Ссвоб, при
и 0,3% СсВоб- За-
лучении ThC^ 4,8—4,9% Ссвя'3
получении ThC2 9,2—9,3% Ссвяз
ким образом, приведенная в работе [206] температура
реакции получения ThC2__* 2130° С завышена.
Карбиды тория могут быть получены также синте-
зом из элементов. Спеканием смеси металлического то-
спектРально чистым углем при температуре
1ьии С в среде инертного газа в работе [208] был полу-
чен монокарбид ThC^
каРбида были получены [209] плавкой в дуговой
см₽еиСтЛ2«ЬФРаМ0ВЫМ электР°Дом в атмосфере аргона
фита аллического тория и спектрально чистого гра-
86
fin п тучен Муассаном [217] и ему была приписана
-э ботее соответствовал формуле UC2 [218]. После-
де I сследования подтвердили существование кар^
—; обладающего тетрагональной решеткой
CaC2f и монокарбида UC^ с г.ц. к. решеткой
Гнстема уран —углерод. Карбид урана впер-
р б п п тучен Муассаном [217] и ему была приписана
Апп \ та U2C3. Позже был получен продукт, состав ко-
ор го Сппаа лпп'гпртртйПвал сЬоомуле UC2[2181. После-
д 1—
б" да UC.
т па С.-
типа NaCl [198, 219, 220].
Бы то сделано предположение, что полуторный кар-
бид представляет собой непрерывный ряд твердых ра-
ств р в моно- и дикарбида. Однако рентгеновские ис-
следования [198] показали, что он состоит из одной фазы
о ц к. решеткой. Кроме указанных, других фаз,, в си-
стеме U—С не обнаружено.
Ниже приведены данные о растворимости углерода
в жидком уране при разных температурах [220]г
Тем ера \ра, С. 1350—1400 1450—1500 1550—1600 1650—1700
(помассе) . 0,10 0,17 0,36 0,42
рапра,°С . 1750—1800 1850—1900 1950—2000 2080—213С£
С ( м ссе) . О',51 1,60 1,74 2,92
Впервые диаграмму состояния системы U—С изуча-
т в работе [219] методами рентгеновского и металлогра-
фического исследований и измерения температур плав-
ления. Продолжили исследования в работах [214, 220—
222].
Наиболее достоверны данные, приведенные в работе
[222], на основании которых предложена диаграмма
(рис. 29). Согласно этой диаграмме, в системе U—Q
уще твуют три карбидные фазы:, монокарбид UC^^,
полуторный карбид и2С3_л и дикарбид UC2_^. Дикар-
бид, который кристаллизуется в тетрагональной решетке,
при температурах выше 1800° С претерпевает превраще-
1 ие в кубический типа CaF2.
Монокарбид урана (4,8% С) кристаллизуется в г. ц. к.
решетке типа NaCl. Атомы углерода расположены в ок-
ура^шИЧеСКИХ ПОрах "°ДР^етки, образованной атомамй
Элементарная ячейка UC^ содержит четыре мо-
0е175Лнл ЙОГ"116 U~C составляет °’171 нм’ U~U
0 4^1Лго^пРааетелпР^.водятся значения периода решетки
0,4951 [249, 250], 0,4955 [219], 0,4959 нм [199].
87
По данным работы [221],. период решетки UC^
растет с повышением температуры от 0,49597 при 18~
20° С до 0,5070 при 1800° С.
Монокарбид урана изоморфен мононитриду и моно-
окиси урана и_углерод в нем может замещаться кисло-
родов и азотам. Такое замещение вызывает уменьшение
ою го зо ио 50 55 во 65
Содержание С,°/о( ant)
Рис; 29. Диаграмма состояния системы И—С
[222]
периода решетки UC^. Исследование системы U—
С—О [249] показало, что область одной фазы U(C, О)
соответствует растворимости кислорода 12,5% (ат.).
Период решетки фазы U(C, О) падает с ростом со-
держания кислорода до а = 0,4953±0,0001 нм для соста-
ва UC0 75О0 25. Можно предположить, что значение пе-
риода, равное 0,4959 нм, соответствует карбиду с пре-
дельным содержанием углерода.
Исследование равновесия в системе U—С—О в ин-
тервалах температур 1360—1720 и 1500—1810° С [893]
подтвердило наличие оксикарбида область
существования которога зависит от температуры и отно-
шения ио2/с.
88
Изучение положения атомов углерода в карбидах
урана методом нейтронной дифракции (250] показало,
что связь U—С в значительно слабее, чем в
ис х. и UC2_X, -что объясняет его низкую устойчи-
вость и разложение нВ tJC^ .и UC2_X.
У дикарбида-при повышении температуры от комнат-
ной до 1200” С возрастают оба периода решетки, при
температурах выше .1200° G значение g падает и при
1800° С достигает более низкой, величины, чем при ком-
натной температуре.
Ниже приведены значения пёриодой решетки UC2_Z
при разных температурах [221]:'
Температура, - 25 800 1200
Периоды решетки, ям:
а. -5 . . .. • Й.3516 0,3536 0,3565
с. ...... . 0,5972 0,6013 0,6020
Температура, °C . . . « . 1600 1800 1900—
2200
Периоды решетки, ял:
а. . . 0,3610 0,3660 —
с. . , . . . .. . . v . 0,5995 0,5970 0,535—
0,543
Это обусловлено тем, что при. температурах выше 1800°G
наблюдается переход UC2_r тетрагонального в кубиче-
ский типа CaF2. Расстояние U—С в этой фазе составля-
ет 0,1605, U—-U 0,194 нм [250]._Такое превращение те-
трагонального UC^ в кубический наблюдалось в ра-
ботах [270] и [271].
Теплота перехода карбида состава UClp93 из тетра-
гональной в кубическую модификацию при температуре
1800° С составляет 46,1 кдж{кг (11,0 кал[г~) [263].
По данным рентгенографического исследования спла-
вов системы U—С в сочетании с данными химического
анализа [223], состав монокарбида соответствует форму-
ле исо 32, а дикарбида UCj>91. Однако это противоре-
чит приведенной диаграмме состояния щ не подтвержде-
но многочисленными исследованиями.
Монокарбид представляет собой порошок се-
рого цвета.
Температура плавления, по данным различных авто-
220, 227, 228], колеблется в пределах 2250—
2650 С,
89
По данным работы [251], на основании анализа ре.
"° “в исследований системы U—С скорректированы
основные термодинамические характеристики, которые
также приведены в работах J128, 779].
Исследование поведения монокарбида при нагрева-
нии электронной бомбардировкой в вакууме 133 мкн[м*
НО-6 мм рт. ст,) <г анализом конденсата методами рент-
гене- и электронографии показало, что UC^ испаря-
ется без разложения [211].
Удельное электрическое сопротивление при
комнатной температуре составляет 47 мком-см [213] и
Рис. 30, Зависимость удельного
электрического сопротивления
и коэффициента термо-э. д. с.
UC от температуры:
увеличивается с ростом тем-
пературы до 180 мкОм-см
при температуре 930° С
(рис. 30), ниже 100° р почти
Рис. 31. Зависимость коэффи-
циента термического расшире-
ния от температуры
1 — удельное электросопротивление;
2 — коэффициент термо-э. д. с.
не изменяется с температурой. В интервале температур
1175—2050° К удельное электрическое сопротивление из-
меняется, согласно уравнению [253].
р = 20,4-10-6 -Ь 114,8- 10~9Том см.
Изучение изменения сопротивления при облучении
карбида дозой 1018 нейтр]см3 при температуре 70° С по-
казало, что р при этом увеличивалось на 2,5% [254].
Термо- э. д. с. положительна, достигает мак-
симума при Температуре 200° К (75 мкв/°К), после чего
медленно уменьшается [213] (рис. 30).
Излучательная способность компактного образна
U'-'i—х (при 0,63 мкм) с ростом температуры умень-
шается от 0,67 при 830° С до 0,47 при 1230° С, после чего
90
не изменяется с ростом температуры [255]. По данным
работы [253], в интервале температур 1150—1890° К за-
висимость ее от температуры выражается уравнением
^обБ^^^539-0’02^10-3-
V tvvMKJn
Теплопроводность монокарбида урана в зависимости
от характеристики образцов (содержания углерода и
пористости) приведена в табл. 8 [231].
По данным [224], теплопроводность UCj_^ при тем-
пературе 250° С составляет при содержании углерода
4,5% 0,061, при 4,8% 0,055 и при 5,1% 0,057 кал/(ему
уград-сек) [25, 23 и 24 вт/(м град)], что хорошо согла-
суется С данными, приведенными в табл. 8.
Таблица 8
Теплопроводность образцов UG1_>.
ис Темпера- тура, °C Теплопроводность
содержание С, % плотноеть> % от теорет. вт/(лгграс)) кал/ (смсек.°С)
5,2 — 100 25 0,060
200 23 0,056
300 23 0,054
400 22 0,053
500 23. 0,054
600 24 0,057
700 25 0,060
735 25 0,061
4,8 74,9 60 34 0,080
115 31 0,074
195 25 0,061
265 21 0,050
Зависимость коэффициента термического расширения
от температуры в интервале 150—700° С [235] для спе-
ченных образцов приведена на рис, 31. Изучение тер-
мического коэффициента линейного расширения UCj_z
в области составов от 4,85 до 5,05% С в интервале тем-
ператур 100—2000° С [256] свидетельствует о том, что
коэффициент мало зависит от температуры.
Твердость монокарбида урана, как и ряд других
свойств, зависит от метода приготовления и составляет
По Виккерсу от 550 до 750 HV [231].
91
Максимальная прочность на сжатие при температу-
пр 25*С37,4 М«М2 (33,15 кГ]мм,\ соответствует образ-
Р полученным дуговой плавкой [231}. Исследование
Ц я=гирсти в интервале температур 1200—1425° С при
сжатии [нагрузка. 40 W^2 (4 кГ/мм^)] показало,
что Ярость крипа, при И25°С составляет 0,018%/*
^Исследование.^еханиче&шх свойств образцов, состав
кптооых близок, К расчетному для"монокарбида урана,
в°интервале’^емператур 1600-2300’С [893] показало за-
висимость свойств от содержания, углерода при одина-
ковой тюристосги. Образцы, Содержащие 4,49—4,56% С
(смесь урана.к монокарбида), деформируются под на-
грузкой быстрее, чем образцы, содержащие 4,72—5% С.
Деформация образцов, содержащих 4,6—4,7% С, соот-
ветствует изменению начальной высоты образца от 70
до 20%, а'образцов, содержащих выше 4,7—5,0% С,
20—10%. Такое уменьшение степени деформации связа-
но с наличием^ у образцов, содержащих 4,49—4,56% С,
жидкой фазы.
Монокарбид урана отличается высокой устойчи-
востью при циклической термообработке [239]. Спечен-
ные образцы плотностью 10,2 г/см3 (74,9% от теорети-
ческой), выдерживали 2000 циклов в интервале темпе-
ратур 100—750° С* Согласно данным работы [235],
образцы плотностью 85% от теоретической выдержива-
ли 500 циклов в интервале температур 200—1000° С.
При действии облучения на пластины из монокарби-
да урана, находящиеся в матрице из нержавеющей ста-
ли, при температуре 540—1000° С было обнаружено не-
которое разбухание их, частицы монокарбида имели не-
которую внутреннюю пористость и взаимодействовали со
сталью [231].
При облучении образцов UC^.*. разного состава,
полученных дуговой плавкой в ампулах, заполненных
[уЗК J231]» плотность несколько уменьшалась (на 0,7—
2,5/о) и появлялись трещины. Максимальная темпера-
1лап0 г°ВерХН0СТИ ПРИ 3т0М Достигала 580° С, а в центре
1U00 С. Наиболее стоек против облучения карбид урана
стехиометрического состава.
Дикарбид урана UC2_X—порошок серого цвета.
Термодинамические свойства его приведены в работах
[211, 251,260,261]. Е
92
Теплоемкость продукта, содержащего 05,3% (мол.)
UC2 и 4%- (мол.) UC^b интервале температур (—263) ч-
ч-(+67ГС составляет -60,75 дж! (моль-град)
[14,50 кал! (моль'-град]} [128].
Изучением упругости паров урана над VC$~X мето-
дом Кнудсена [211] с последующим активационным ана-
лизом конденсата установлено, что в интервале темпе-
ратур 2010—2893° С Теплота испарения урана составля-
ет 544,7±2Г кдж]моль (130±50 ккал! моль), что хорошо
согласуется с данными работы Д399], в 'которой в ре-
зультате исследования- испарения дикарбида установ-
лена содержание в газовой фазе молекул урана и
ис^.
Упрутостьдгаров урана выражаетсядуравнением:
lgpu=_—_ + 4f07> а
. 40,4.00 , о по
1g Рис2-Х ~ ~' + 8,38.
Наиболее распространенный метод получения карби-
дов урана — синтез из элементов, взаимодействие урана
с углеродсодержащими тазами и восстановление окис-
лов урана углеродом.
До недавнего времени карбиды урана получали плав-
кой в дуговой электрической печи в атмосфере аргона
и гелия смеси порошка металлического урана с углеро-
дом [224].
Как показано в работе [225], для приготовления кар-
бидов урана порошок металлического урана смешивали
с сахарным углем в различных соотношениях и после
перемешивания смесь смачивали парафином, разбавлен-
ным бензолом, прессовали в брикеты, которые нагрева-
ли в электрической печи* сопротивления с графитовым
нагревателем в среде аргона при температуре 2300° С.
По данным работы [220], температура образования
монокарбида синтезом из элементов составляет 2100° С,
а дикарбида 2400° С.
Получить полуторный карбид синтезом из элементов
не удалось [227, 228]. После охлаждения продукт реак-
ции представлял смесь иС1—х и UC2_X. При нагрева-
нии UCj..* с UC^ в вакууме в интервале температур
1250—1800° С получали U2C3_x[227].
93
Один из наиболее перспективных методов получения
капбидов урана—взаимодействие порошка металличе-
ского урана с углеродсодержащими газами, в частности
с метаном, по реакции
и + СН4 -UC + 2Н2.
Таким образом в работе [219] был получен монокарбид
урана. Над порошком металлического урана пропуска-
ют водород при температуре 225° С. При этом образует-
ся гидрид урана; по завершении процесса гидридсобра-
зования температура увеличивается до 450°С и гидрид
разлагается с образованием высокодисперсного порошка
урана. Такой цикл повторяют трижды, после чего тем-
пература повышается до 600—900° С и над порошком
пропускают метан. При указанных температурах обра^
зуется монокарбид урана, при более высоких — дикар-
бид. Преимущество этого метода сводится к образовав
нию весьма мелкодисперсных порошков карбидов, что
дает возможность в дальнейшем получить из них до-
статочно плотные изделия.
Взаимодействием урана с метаном при температуре
700° С получен карбид с размером частиц около 10 мкм
[199].
Фишер [230] получал UC2^ взаимодействием мел-
коизмельченного урана е углеводородами типа метана
при температурах, несколько более низких, чем темпера-
тура плавления дикарбида.
При использовании пропана или бутана при темпера-
турах 700—750° С получен практически чистый монокар-
бид [216].
При получении карбидов урана восстановлением
окислов углеродом в качестве исходных окислов исполь-
зовали двуокись, окись-закись и трехокись урана.
Двуокись восстанавливается по реакциям
UO2 + ЗС = UC + 2СО,
UOa + 4С = UCa + 2СО,
Реакцию взаимодействия с образованием моно- и
дикарбида изучали в работах [231—235, 237—239].
Впервые этим методом карбиды урана были получе-
ны в работе [232], по которой нагревали смесь UOg с
сахарным углем в вакууме при температуре 2450° С,
94
н [233], в которой изучали равновесие реакции UO2+
+4С=иСг+2СО в интервале температур 1480—1800°С.
Подробное исследование реакции восстановления
UO2 углеродом с целью получения монокарбида прово-
дилось в работа [235]. Манометрическое исследование
реакции показало, что при начальном давлении в систе-
ме 0,13 н/лс2 (Ю~4 ммрт.ст.) и скорости подъема тем-
пературы 50 граЗ[мин реакция с заметной скоростью
начинается при температуре 1300 С.
Оптимальный режим получения UC сводится к на-
греванию шихты UO2 + 3C стехиометрического состава в
вакууме при температуре 1800° С и времени выдержки
после достижения в системе первоначального давления
в течение 10-^30 мин.
При исследовании, восстановления UO2 [199, 237] уг-
леродом с целью получения монокарбида установлено,
что обычно употребляемая при этом окись урана 1Ю2
содержит несколько больше кислорода, чем это соот-»
ветствует формуле UO2, и, кроме того, окись UO2 не-
сколько поглощает влагу. Поглощенная влага меньше
влияет на процесс, чем содержание кислорода в 1Ю2,
которое со временем возрастает, и тем больше, чем
больше поверхность порошка окисла.
В том случае, если используется нестехиометрическая
окись урана, при составлении шихты следует учитывать
ее состав по следующему уравнению:
UO^-t- (3 + х) С -> UC + (2 + х) СО.
Для получения гомогенного карбида реакцию прово-
дят дуговой плавкой брикетов из шихты UO2 и сажи в
атмосфере аргона под давлением 55,65 кн/м2 (0,5 ат).
Плавку проводят в две стадии. При этом в течение часа
может быть получено 100 г царбида в виде брикетов
плотностью 13,58—13,60 г/см? (теорет. 13,66 г!см3)л со-
держащих от 4,75 до 4,88% С. Для получения больших
количеств карбида урана плавкой и литьем используют
специальную установку, на которой можно при силе
тока 4000 а и мощности 100 кет расплавить 2 кг шихты.
При использовании в качестве исходного окисла UgOs
карбиды урана получают по реакциям
U3O8 + 11С = 3UC 4- 8СО;
U3O8 + 14 С = 3UCa + 8СО,
95
к'лтлпие изучались в работах [220; 225, 200]. Первая
реакция идет при температуре 1800 С, вторая —при
2400° С [220, 225].
В работе [238] исследовали две реакции образования
монокарбида урана:
U3O8 + 9С + 2На *= 3UC + 6СО 4 2Н2О;
узО8 +-1 ОС «= 3UC 4~ СО2 + 6СО,
В первом случае шихту нагревали в токе -водорода при
температуре 500° С. При этом U3O8 восстанавливалась
до UO2. Восстановленные брикеты быстро переносили
в вакуумную печь, где при температуре 1200° С проис-
ходила реакция UO2+3C = UC+2CO. Эта реакция за-
вершалась при температуре 1430° Cf затем температуру
поднимали до 1700° С, при которой брикеты спекали в
течение 3 ч.
Продукты реакции представляли однофазный моно-
карбид с некоторым недостатком углерода, что связано
с потерями при взаимодействии углерода шихты с па-
рами воды, образующимися при восстановлении U3Oa
до UO2. Вторую реакцию изучали при нагревании ших-
ты в вакууме. Реакция восстановления U3Os до UO2 на-
чиналась при температуре 620° С, после чего темпера-
туру поднимали до 700° С и восстановление до UO2 за-
вершалось. Затем температуру повышали до 1700° С и
восстанавливали UO2 до UC. При этом содержание уг-
лерода было также ниже соответствующего стехиомег-.
рическому UC.
Для получения UC стехиометрического состава со-
держание углерода в исходной шихте должно на 0,5%
превышать расчетное [200].
Представляет интерес получение монокарбйда урана
с использованием в качестве исходного материала диу-
раната аммония [917].
В работе [243] исследовали условия получения об-
разцов монокарбида урана высокой плотности следую-
щими методами: спеканием UCX_X, спеканием шихты
UO2+C при температурах 1500—1800° С, выдавливани-
ем UCj_x в медной оболочке и горячим прессованием
смеси порошков урана и графита. При применении пер-
ВЫХ25^Х методов полУчены образцы с пористостью вы-
96
Процесс, спекания монокарбида урана с добавками
металлического урана в вакууме изучали в работе [235].
На рис. 32 показана зависимость пористости спеченных
образцов- от температуры и содержания в смеси урана.
При спекании UC^ прй температуре 2200° С в течение
2 ч остаточная пористость- составляет 10%, Добавка
урана понижает пористость и-при содержании его 31,8%
можно достичь пористости 5% спеканием при темпера-
туре 1700° С. Такое уменьшение пористости связано с
Рис. 32. Зависимость пористости спеченных
образцов .UC от температуры и содержания
в смеси урана
наблюдаемым на микроструктуре увеличением размера
зерен UC^ "и изменением формы зерен вследствие пе-
рекристаллизации UCj_x через расплавленный уран.
Исследование, условий, влияющих на плотность спе-
каемых образцов [199, 229, 238], показало, что неболь-
шое загрязнение исходных продуктов кислородом вли-
яет на плотность. При высоких температурах (>1900° С),
при которых происходит спекание за счет кислорода, со-
держащегося в карбиде, протекает реакция
2UC + UO2 = 3U 4- 2СО,
в результате которой спекание происходит в присутст-
вии жидкой фазы, что обеспечивает получение образцов
с высокой плотностью.
При спекании , полученного из смеси метал-
лического урана, обработанного предварительно 25%-
ным раствором HNO3, с графитом в интервале темпера-
ТУРопо/°°~20000 С [199], получены образцы плотностью
Д О /о от теоретической. Однако значительное загряз-
7 Косолапова T. Я. о_
шшие кислородом приводит к резкому уменьшению со-
Н углерода в готовых продуктах. Так, при спе-
S? Хоадкта, «держащего 5% Сг 1,3% О и 0,P3l%N,
при температуре 1900°G-B .образцах содержание угле-
рода составляло 3,87 % •
г При введении-добавок в смесь U+C плотность об-
разцов также повышалась. Так, введением в смесь U+C
10% UFea и спеканием в вакууме при температуре
1200—1450° С достигалась пористость образцов 3,7%.
Добавки- UAh, UBei3 л U3Si2 влияли на спекание мо-
нокарбида урана, интенсифицируя усадку и понижая
температуру спекания [2521.
Более высокой плотности образцов, чем при спека-
ний, можно достичь приготовлением их методом горя-
чего прессования. Исследование1 влияния различных
факторов на плотность образцов, полученных горячим
прессованием [199, 235,^243, 276]^ показало, что этим
методом можно получить образцьГпористостью до 5%.
Образцы с такой пористостью „получены при прессова-
нии порошка UC^, содержащего 0,18% кислорода,
при температуре 1480° С -[199]. Исследование влияния
температуры и давления прессования показало, что ко-
нечная плотность образцов существенно зависит от дав-
ления и мало'зависит от .температуры прессования. Ав-
торы указывают на то, что в процессе горячего прес-
сования UCj_x уплотнение происходит в результате
скольжения по границам зерен; пластическое течение и
объемная диффузия играют второстепенную роль [247].
Введение добавки Mo, W, WC, TiG или SiO2 [248] поз-
волило прессованием при температурах 1000—1200° С
под давлением 19,6 Мн]м2 (200 кГ]см2) получить образ-
цы плотностью 98%.
Наиболее плотные образцы плотностью 98—99,5% с
содержанием 4,75—4,88% С получены плавлением в дуго-
электрической печи в атмосфере аргона или гелия
[1УУ, 231]. При дуговой плавке используют вольфрамо-
вые, графитовые и уранграфитовые электроды. Этот ме-
тод позволяет получать образцы необходимой формы
и размеров. * F
Методом гарниссажной дуговой плавки [272] получе-
ны слитки UC1-JC диаметром 9—25 мм и длиной 150 мм.
лотность слитков составляла 99% от теоретической.
98
Система нептуний—углер од. При нагрева-
нии NpO2 В графитовом-дигле при температуре 2660—
ogQO0 с в течение: 5 мин был. получен дикарбйд NpC2
Т288]. При взаимодействии металлического нептунии с
графитом при температуре 1200° С образуется смесь
карбидов NpsCgiH NpG в соотношении 1:5. Однако дан-
ных о выделении этих фаз в чистом виде нет.
Система плутоний—углерод. В системе
pu—С методом рентгеноструктурного 'анализа иденти-
фицировано два карбида — монокарбид с г. ц. к_. решет-
кой и полуторный карбид с о. ц. к. решеткой [238]. Име-
ются данные о существовании дикарбида РиС2, устойчив
вого при температуре выше 1750° Q
Исследование сплавов Pu—С, полученных дуговой
плавкой [265], показало, что "твердость образцов резко
возрастает при содержании-выше 39,7% (ат.) Су что
свидетельствует о кристаллографическом изменении при
таком составе. Согласно данным рентгенографического
анализа, РиС существует при содержании углерода вы-
ше 39,7%. По данным работы 1236], нижняя граница
фазы РиС соответствует содержанию 42,5% (ат.) С. При
температуре 1000° С эта"'граница сдвигается, принимая
значения 45,5—46,0% (ат.) С. При содержании выше
48% С на рентгенограммах наблюдаются линии PuG и
PU2C3.
Диаграмма состояния системы Pu—С [199] представ-
лена на рис. 33. Согласно этой диаграмме, при содер-
жании 50% (ат.) С имеется смесь РиС и Pu2Cg, откуда
следует, что высшим по содержанию углерода является
монокарбид с предельным содержанием 48% (ат.) С.
Исследование сплавов Pu—-С, содержащих от 38 до
50% (ат.) С, [266] подтвердило, что продукт, содержа-
щий 50% (ат.) С, двухфазный и состоит из РиС1-х и
РщСз. Область устойчивости фазы РиСх_х соответст-
вует содержанию 46—48% С. Появляющаяся на диаг-
рамме £-фаза при содержании 40% (ат.) С предполо-
жительно является соединением, полученным по перитек-
тической реакции Pu+PuC^ при температуре 575° С.
Монокарбид обладает высоким удельным электри-
ческим сопротивлением, мало зависящим от температу-
ры. 1ермо-э. д. с. РиСг__х положительна и растет с рос-
том температуры [213] (рис.34). При температуре ниже
7*
99
^1500
2500
2000
500
О 20 ЬО 60
Содержание С, а/а (ат.)
80 100
Рис. 33. Диаграмма состояния системы Ри—С
Рис. 34. Зависимость удель-
ного электрического сопро-
тивления (/) и коэффициен-
та термо-э. д. с. (2) от тем-
пературы
1730° С монокарбид плутония становится антиферромаг-
нитным. Отношение теплоемкости РиС^ к UCj_^ со-
ставляет 0,2 [2751
Монокарбид плутония получают методами, анало-
гичными методам получения монокарбида урана. Син-
тез из элементов осуществляется дуговой плавкой смеси
плутония и графита, а также непосредственным горячим
прессованием смеси плутония и графита при температу-
ре 1000° С [231]. Взимодействие окиси плутония с угле-
родом по реакциям
Р11О2 4“ (2 х) С РиС 2СО;
2РиО2 4- 7С = Ри2С3 + 4СО
позволяет получить моно- и полуторный карбиды при
температурах 1250—1450° С [199]. В работе [268] уста-
новлено, что взаимодействие РиО2 с углеродом с образо-
ванием монокарбида в вакууме начинается при темпе-
ратуре 1100°С и реакция довольно быстро протекает
при температуре 1350° С. При этом в продуктах реакции
обнаружены два карбида: Ри^^ и PU2C3.
Для получения плотных образцов спекание крабидов
проводят в две стадии: одноразовое спекание монокар-
бидов при температуре от 1250 до 1450° С приводит к
получению образцов с низкой плотностью. После измель-
чения и повторного спекания при тех же температурах
плотность уже достигает 96% от теоретической для
puCj_x и 99% для PU2C3.
КАРБИДЫ МЕТАЛЛОВ
ПОДГРУППЫ IVa
Система титан — углерод. Исследованию систе-
мы титан — углерод посвящено много работ [83, 98, 162,
168, 278—281, 284—290]. Наиболее полный обзор боль-
шей части этих работ приведен в статьях {11, 22, 277].
В результате многочисленных исследований предло-
жена диаграмма состояния системы Ti—С [288] (рис. 35),
согласно которой в системе существует одна карбидная
фаза TiCt_x с широкой областью гомогенности. Ши-
рина области гомогенности, исследуемая Кадоффом и
ильсеном [288], а также в работах [83, 98, 162, 168],
широком интервале составов [до 85% (ат.) С] состав-
101
ляег 37—50% (ат.) с
[13—20% по- массе)]. Су.
ществование карбидов
Ti2C [282] и Т1С2 [291] не
подтвердилось.
„Углерод повышает
.температуру- превраще-
ния п X 0 титана .от 882
до 920° С [288—290]. При
температуре 920° С про-
исходит* перитектическое
-превращение p+TlCX®.
При " этол температуре
растворимость углерода в
а-титане максимальна и
щадает а понижением тем-
пературы. Растворимость
углерода в. 0-титане мак-
симальна при температу-
ре 1750° С-й также пони-
жается с уменьшением
температуры.
Период решетки кар-
бида титана в вависимо-
Рис. 35. Диаграмма состояния сти от его состава из-
системы Ti—С-[288] . меняется от 0,430 до
0,430 нм.
Ниже приведены данные зависимости периода решет-
ки Т1С1-Х от содержания углерода [327, 864], нм:
TiC0>33 . . 0,4313 ' TiC0>5 . . 0,42990^10“4
Т1С0’8Б . . 0,4315* TiC0’& , . 0,43120±5-10-5'
Т1С092 . . 0,4325 [327] TiC0’7 . . олзг^ю*4 [864]
TiC0,96 • * 0,4329 TiC0>8. . . 0,43250±5.10-5
TiCo gg . 0,4320 TiCOjg . , 0,43290=t:5• 10~5
Для TiC стехиометрического состава период [286, 315,
328—333] составляет от 0,4315 до 0,4324 нм.
Расхождения в значении периода связаны с тем, что
карбид всегда несколько загрязнен кислородом, введе-
* Образец содержит 1,53% N и 0,86% О
Ю2
ние которого, как доказано опытами [334], вызывает сжа-
т те решетки и уменьшение периода.
Высокотемпературное рентгенографическое исследо-
вание карбида титана состава ^TiCg^i [335] показало за-
висимость.периода решетки и коэффициента линейного
расширения от температуры, приведенную ниже":
Температура, °C а, ня ”298 граЭ-1 -10-6
23 0,43178=!= ±0_0.0003 —
500 0,43322± ±0,00004 6,99±0,34-
702 0,43395± ±0,00004
848 0,43449± ±0,00004 7,61 ±0,20
Рентгенографическое исследование "распределения элек-
тронной плотности в карбиде титана [240, 245] показало,
что атомы титана в карбиде имеют положительный за-
ряд +4, а атомы углерода — некоторый отрицательный
заряд. Это свидетельствует о том, что в сложной связи
между атомами тйтана и углерода определенная доля
приходится на ионную связь?
Масс-спектрографическое исследование [336] показа-
ло, что при нагревании в вакууме карбид" титана диссо-
циирует на газообразный атомарный титан и углерод.
Скорость испарения и давление паров титана и угле-
рода в зависимости от температуры выражаются данны-
ми, приведенными в табл. 9 [337].
Таблица 9
Скорость испарения карбида титана и давление паров титана
и углерода
Температура, °C Скорость испарения, гЦся2-сек) -10~7 Давление паров Ti-|-C
мкн]м* мм рт. ст.
1800 1,35 2,666 0,020-10—9
1900 4,38 8,931 0,067-10—®
2000 7,78 15,99 0,120-10—8
„Термодинамические свойства карбида титана изуча-
лись в работах [337—342, 673].
103
R паботе [342] изучали зависимости энтальпии карби-
пН пт содержания в них углерода. Образцы готовили
?Хянием спрессованных смесей гидрированного йодид-
кого титана с графитом в вакууме при температурах
«ело_2000°С в течение 3 ч. Анализ полученных продук-
тов свидетельствует об образовании оксикарбидов.
Зависимость энтальпии в интервале составов
_TiCj 0 от содержания углерода линейная. Эн-
тальпия образования оксикарбидов выше, чем это соот-
ветствует экстраполированным значениям карбидов с
со тветствующими содержаниями углерода, причем эн-
тальпия образования оксикарбидов титана аддитивно
складывается из энтальпий углерода и кислорода, при-
соединенных к титану.
Теплоту образования карбида титана из элементов
определяли в работах J336, 338, 339,'673]. Данные, при-
в денные разными авторами, несколько расходятся, что
связано с разным составом образцов, на которых прово-
дились определения. Наиболее вероятны данные работ
[336, 339, 673], которые хорошо совпадают и дают для
карбида, близкого к расчетному составу, значение
д/7298=—183,7± 1,63 кдж/моль (—43,85±0,39ккал/моль).
Теплопроводность TiC^ в интервале температур
500—2100°С составляет 37,7 вт/ (м - град) [0,9 кал/(см*
• сек-град)) при 600° С и 46 вт/(м- град) [0,11 кал!(см •
• сек°К)]при 2000° С [343].
В работе [344] определяли теплопроводность образ-
цов, полученных разными исследователями, и значения
несколько расходились. Так, при температурах 965—
975 С значения теплопроводности образцов соответство-
вали 41 и 30 вт/(м-град) [0,098 и 0,073 кал/(см • сек-
•град)] и при 1415—1425°С 44 и 31 вт/(м-град) [0,0105
и 0,075 кал/(см • сек °К)], что, по-видимому, также опре-
деляется разным составом образцов. Измерения тепло-
проводности TiC0 97 при температурах — 183 и 0°С дали
?ппоеНИЯ’ Равные соответственно 34 и 80 вт/(м • град)
10,082 и 0,19 кал/(см • сек • град)] [349].
Удельное электрическое сопротивление карбида ти-
[345*з4бр°ВЫшением температуры линейно возрастает
удельнЛл°ТЛо^З] изучали температурную зависимость
ктросопротивления и эффекта Холла для
104
препарата TiC Результаты свидетельствуют о росте
удельного электросопротивления и коэффициента Холла
с ростом температуры, что подтверждает металлический
характер проводимости карбида титана.
Изучение температурной зависимости удельного
электрического сопротивления и коэффициента термо-
э. д. с. для карбида титана, нескольких составов в преде-
лах области гомогенности {347], а также коэффициента
Холла и магнитной восприимчивости при комнатной
температуре свидетельствует о том, что удельное сопро-
тивление о увеличением содержания углерода убывает,
а значения коэффициентов термо-э. д. с. увеличиваются.
Аналогичный ход изменения удельного электросопротив-
ления наблюдался в работе [864]. Однако в работах
[703] и [715] показано, что такой ход сопротивления на-
блюдался у образцов невысокой степени чистоты; при
получении карбидов титана из компонентов высокой
чистоты в Глубоком вакууме, зависимость обратная. Из-
менение коэффициента . Холла аналогично изменению
коэффициентов термо-“э. д. с. и совпадает с ними по зна-
ку (табл. 10).
Таблица 10'
Некоторые физические свойства TiCi—х в области гомогенности
при комнатной температуре
Формула соединения Удельное электро " сопротивление, МКОМ'СМ- 'Коэффициент термо- э.д.с., мкв!г-рад Коэффициент Холла, Я-10‘ см[к
^’^0,59 174' —7,7 ±0,2 —4,0
”^*^0,72 131 —8,8 ±0,3 . —4,85
"^’^0,81 104 —10,4±0,3 —6,5
Т* ^0,988 .52,5 —12,5±0,2 —6,7
Отрицательные значения коэффициентов Холла и
термо-э. д. с. свидетельствуют о преимущественно элек-
тронном характере электропроводности карбида титана
в области гомогенности.
Исследование температурной зависимости электросо-
ротивления и термо-э. д. с. на монокристаллах [348] по-
эзало, что с ростом температуры сопротивление моно-
8 Косолапова Т. Я. 105
кписталаов TiC растет; это свидетельствует о металли-
ческом типе их проводимости. Сопротивление при ком-
етной температуре составляет 200 ± 2 мком/см, что хо-
пошо согласуется с данными работы [397], по которой
удельное электросопротивление образца пиролитическо-
го TiC0 92> мало отличающегося от монокристалла, со-
ставляет 200 мком • см.
Определение микротвердости образцов карбида ти-
тана в области гомогенности [333J показало, что изме-
нение микротвердости носит линейный характер и при
экстраполяции до нулевого значения величина микро-
твердости соответствует значению ее для чистого титана.
Механические -свойства образцов из карбида титана
изучались в работах [350—352, 919]. Они зависят от по-
ристости. При переходе от плотных образцов к образцам
с пористостью 5% прочность карбида титана на изгиб
уменьшается почти вдвое. Она значительно возрастает
с уменьшением размеров частиц исходного карбида.
В работе [350] изучали зависимость механических
свойств и электросопротивления образцов карбидов ти-
тана, полученных горячим прессованием, при температу-
ре 3000° С в течение 30 сек, от крупности зерен исходно-
го карбида. Показано, что механическая прочность об-
разцов возрастает с уменьшением зернистости исходного
порошка; электросопротивление при этом падает.
В табл. 11 приведена зависимость предела прочности
на растяжение и сжатие образцов TiCj,^ в зависимости
Таблица и
Предел прочности образцов TiCi—х на растяжение и сжатие
(пористость образцов 1—20%)
Температура, °C SP а сж
Мн/м* кГ/мм1 Мп/м? кГ{мм1
20 800 1000 1200 1300 1400 1600 1800 2000 2200 560—1050 380 280 250 56—105 - 38,0 28,0 0,25 1380 875 521 350 230 310 164 94,5 138 87,5 52,1 35,0 23,0 31,0 16,4 9,45
106
nT температуры [152, 351]. Пористость образцов состав-
ляла от 1 до 20%.
Исследование действия нейтронного облучения на
свойства карбида титана [267] показало, что оно вызы-
вает рост электросопротивления, микротвердости, хруп-
кости и уменьшение размеров зерен; при термическом
отжиге облученных образцов эти свойства возвраща-
Муассан [246, 292] получил карбид, соответствующий
формуле TiC, расплавляя в пламени вольтовой дуги
шихту из двуокиси титана с углем расчетного состава.
Метод приготовления карбида титана восстановлени-
ем окиси титана сажей при более низких температурах
(до плавления шихты) применяли во многих работах
[25, 293—295] с незначительными изменениями и исполь-
зуют для промышленного получения карбида титана.
Однако продукты реакции обычно содержат значитель-
ное количества свободного углерода. Некоторое умень-
шение содержания свободного углерода достигается спе-
циальными методами;
Так, при нагревании карбида титана, содержащего
свободный углерод, с марганцем углерод связывается в
карбид МпзС, который затем удаляется растворением в
разбавленной соляной кислоте. Некоторые авторы [296]
с этой же целью использовали прибавление до 1 % оки-
си хрома. Применяют также метод отделения свобод-
ного углерода флотацией при перемешивании тонкоиз-
мельченного продукта с 0,1—1,5%-ным раствором мыла.
Обработкой карбида титана, содержащего 17,3%
Ссвяз, смесью водорода и метана с парами хлороформа
удалось повысить содержание связанного углерода до
18,6% [298].
Обнаружено также [298], что при введении в газовую
атмосферу печи соединений, содержащих хлор, скорость
реакции образования карбида и полнота прохождения
реакции возрастают. При этом содержащие хлор соеди-
нения, например CCI4, хлорируют окись титана и обра-
зующийся хлористый титан взаимодействует с водоро-
дом и углеродом по уравнению:
TiCl4 4- 2На + С = TiC + 4НС1.
Исследование этой реакции [299] показало, что реакция
11qq?qHT с заметной скоростью при температурах выше
8*
107
Скорость реакции возрастает при замене водорода
е ie омг
TiClJ-H 2Fe + С =^TiC—[- 2FeCl2,
ппи этом реакция идет при более низких температурах.
Р В работе [300] исследовано1 влияние добавок восста-
новленного водородом железа, глинозема, кварцевого
Рис 36. Зависимость- Содержания-- Ссвяз
в продуктах восстановления'ТЮг угле-
родом от времени выдержки (температу-
ра 1900°С> атмосфера—водород):
/ — шихта TiO2 + ламповая сажа; 2 — шихта
TiO2 + газовая сажа; 3 —шихта TiO2 + сахар-
ный кокс; ТПхТ—время первичного подъема
температуры
песка, окиси кальция и металлического кремния на про-
цесс восстановления двуокиси титана углеродом.
Исследования показали, что железо значительно ин-
тенсифицирует процесс восстановления окиси титана и
сдвигает его в сторону более низких температур.
Аналогично добавка AI2O3 в шихту улучшает условия
восстановления TiOg и смещает процесс в область более
низких температур.
Добавки CaO, SiO2 и металлического кремния не
оказывают заметного влияния на процесс.
Наиболее подробно механизм получения карбида ти-
тана восстановлением двуокиси титана углеродом изу-
чали в работах [45—47, 302, 310, 311]. Показано, что про-
цесс восстановления углеродом идет через стадии обра-
зования низших окислов: TI3O5, Ti2O3, TiO и последняя
стадия заключается в восстановлении TiO:
TiO + С — TiC 4- СО.
108
На рис. 36 показана зависимость содержания связан-
ного углерода в продуктах реакции взаимодействия TiO2
с разными сортами восстановителя от времени выдерж-
ки при температуре 1900°С в атмосфере водорода. Из
рисунка следует, что наиболее активный восстановитель
окиси титана —ламповая сажа.'
Нагревание шихты TiO2 с ламповой и газовой сажей
к максимальному содержанию связанного уг-
приводит
лерода в начале процес
са и уменьшению его с
увеличением времени вьь
держки (рис.-37).
Скорость подъема тем-
пературы также сущест-
венно влияет на процесс
образования карбида ги-
тана. Это объясняется
тем, что нагревание ших-
ты при высокой темпера^
туре приводит к росту
размеров кристаллов са-
жи — графитизации её.
В результате этого кон-
центрация ацетилена в
атмосфере печи уменьша-
ется и становится ниже
равновесной, соответст-
вующей максимальному
Рис. 37. Зависимость содержания
Сгряз в продуктах восстановления
TiO2 углеродом от времени вы-
держки при .температуре 1900° С
(атмосфера СО):
1 — Шихта TiO2 4- ламповая сажа;
2 -* шихта- TiO2 + газовая сажа
содержанию связанного углерода в твердой фазе. В свя-
зи с этим содержание связанного углерода уменьшает-
ся— происходит декарбидизация карбида титана. При
^продукты Реакции пРеДставляют твердый раствор
Изучение влияния давления СО на состав продуктов
реакции восстановления TiO^ углеродом [307, 308] в ва-
кууме показало, что максимально насыщенный углеро-
дом карбид титана образуется при давлении 133,3—
1333 н/лх2 (I—ю лхл рт. ст.).
Рассматривая последнюю стадию восстановления
Т10ч C = TiC + CO, Меерсон [312] указывает на то, что
по мере приближения концентрации кислорода в титане
к 0,05% термодинамический потенциал образования свя-
зи Ti О может быть приравнен к потенциалу образо-
109
паиия связи Са — о. Исходя из этого, вычислено значе-
ние термодинамического потенциала для реакции Ti (О) +
4-2C==TiC + CO и значение Кр = РСО для температуры
]300°Cv которое оказалось равным 130,& н/м?
(9 8 мм рт. ст.),, что хорошо согласуется с приведенными
выше экспериментальными данными [307]. При более
высоких температурах наблюдается расхождение между
рассчитанными и экспериментальными значениями рав-
новесных давлений СО, которые-с ростом температуры
увеличиваются и становятся значительно выше получен-
ных экспериментально. Следовательно, по мере повы-
шения температуры энергия связи Ti—О в твердом ра-
створе Ti—С—О становится больше, чем энергия связи
Са—О.
Согласно работе [313], оптимальная температура об-
разования TiC 1800° С, причем при медленном нагреве
реакция идет по схеме TiO^~* TiO -> Ti TiC, при бы-
стром— TiO2~*TiO ->TiC.
Чистый карбид титана, содержащий 20,0% Ссвяз,
был получен в работе [892] нагреванием смеси порошка
чистого титана с ацетиленовой сажей, содержащей 7,5—
10% избытка титана по отношению к расчетному [при
температуре 1747° С й вакууме 266 6 • mkhIm?
(2«10~5 мм рт. ст.)], что связано, по-видимому, с неко-
торой летучестью титана.
Наиболее чистый карбид титана получен осаждением
из газовой фазы. В работе [314] карбид титана осаждали
на вольфрамовой проволоке при температурах 1300—
1700° С из смеси паров четыреххлористого титана, водо-
рода и толуола; согласно работе [287], оптимальная тем-
пература этого процесса 1600—2000° С.
Карбид титана осаждали [315] из смеси паров четы-
реххлористого титана и водорода на угольной нити при
температурах 1530—1830° С'. Полученный продукт со-
держал некоторый избыток титана и соответствовал
формуле Tij j С. После отжига в высоком вакууме при
температуре 1900—2100° С избыток растворенного титана
испарялся и получался карбид, содержащий 19,6% С.
Карбид титана получали [316] осаждением на воль-
фрамовой проволоке при температуре 1300—1700° С из
смеси паров четыреххлористого титана, водорода и уг-
леводородов.
110
Таблица 12
Значение изменения свободной энергии возможных реакций при образовании TiCt «осаждением из
газовой фазы {318]
Реакция AF^. при температуре, °C
900 1050 1300 1600
кдж] моль ккал/моль кдж/моль ккал] моль кдж]моль ] ккал] моль кдж/моль ккал/моль
TiCl4(r)+CH4=TiC4-4HCl +9,6 +2^3 —20,5 —4,9 —75,0 —17,9 —129,5 -30,9
TiCl4(r>4-2H8=Ti+4HCl +210,7 +50,3 +192,3 ' +45,9 +158,0 +37,7 +120,7 +28,8
TiCl4(r)+2H8+C=TiC4-4HCl +39,4 +9,4 +23,0 +5,5 —7,9 -1,9 -34,8 -8,3
TiCl4(r)4-2C-CCl4+TiC +495,2 +118,2 +502,4 + 119,9 +507 +121,0 +521 +124,3
4TiCl3(r)+7C—3CCI4+4TiC +1390 +331 к8 +1404 +335,2 +1452 +346,6 +1536 +366,6
2TiCl2(KOM)+3C=CCl4+2TiC +399 +95,3 +395,5 +94,4 +387Д +92,4 +386,3 +92,2
Tic14(r)+V H2=TiCl3(r)+HCl +40,2 +9,6 +29,3 +7.0 +12,6 +3,0 -5,9 —1,4
TiCl3(r)+4- H3=TiCl2(„M) + +30,2 +7,2 f +39,4 +9,4 +47,8 + 11,4 +60,3 +14,4
+HC1
TiCI2(KoM)+H2^Ti4-2HCl +140,4 +33,5 + 123,6 +29,5 +97,^ +23,3 +66,2
2TiCl2(KOHa)=Tj4-2TiCl3^rj ] +80,0 +19,1 +44,8 +10,7 +2,1 +0,5 —54,5 —13,0
Т1С12(конд)+На-(-С=Т{С4-2НС1 —31,0 —7,4 —45,7 -10,9 —68,3 -16,3 —91,8 —21,9
ЗТ1С12(коиЯ)+С^Т1С+2Т!С13(г) —96,8 —23,1 —107,7 —25,7 —145,4 —34,7 —182,7 —43,6
Аналогично карбид титана получали пропусканием
смеси водорода и метана в соотношении 15:1, насыщен-
ной ларами четыреххлористого титана, над вольфрамо-
вой нитью при температуре 1500° С [317].
Многочисленными исследованиями показано, что
карбид титана осаждается из газовой, фазы только в
присутствии водорода. В*'работе 1318] были проведены
Исходная
Рис. 38. Установка для исследования процесса получения карбида
титана осаждением из тазовой фазы:
а —схема установки: 1—моностаТ; 2 — система очистки; —реометр; 4 —тер-
мостат; б — испаритель; 6 — змеевик; 7 —газометр; Л*- склянки с H2SO4;
S — реометр; 10—сопло Вентури; 11— реакционный сосуд; 12 — испаритель
TiCl,; б—реакционный сосуд
термодинамические расчеты реакций, возможных при
образовании* карбида титана описываемым методом, с
целью выяснения роли водорода (табл. 12)^ Показано,
что при восстановлении ПСЦ водородом выделяется ме-
таллический титан, который взаимодействует с углеро-
дом— продуктом термического распада метана.
Подробнее процесс осаждения карбида титана из га-
зовой фазы [319] исследовали на установке, представ-
ленной на рис. 38. Результаты исследований показыва-
ют, что восстановление паров TiCl4 водородом до титана
при температурах до 1750° С практически не идет.
В присутствии углерода протекает реакция с образова-
на
нием карбида титана, причем роль углерода -сводится к
связыванию титана. „
Выделение углерода из парогазовой смеси J1CI4,
СН4 и Н2 обусловлено распадов метана на поверхности
раскаленной нити. Скорость осаждения карбида титана
(рис. 39) увеличивается.:с ростом парциального давле-
ния СН4 ДО значения lO^2 ат) линейно,
Рис. 39. Зависимость скоро*® Рис. 4(к 'Зависимость плотности об-
еги осаждения карбида ти- разцов карбида титана от температур
тана от парциального дав- ры спекания
ления СН4 б смеси от тем-
пературы, °C:
/- 1320; 2 - 1680;. 3 - 1900 ЗуДЬТат ТОГО, ЧТО В ОбЛЭСТИ
низких, парциальных давле-
ний СН4 лимитирующим процессом является разложе-
ние метана, при более высоком парциальном давлении
лимитирующим становится взаимодействие титана с уг-
леродом.
Разработан метод получения карбида титана взаи-
модействием порошка металлического титана или окиси
титана со смесью паров СС14 или CHCI3. Пары действу-
ют каталитически и, кроме того, являются источником
Углерода [320].
В качестве исходного сырья, подвергающегося вос-
тановлению углеродом, может использоваться вместо
Двуокиси нитрид титана [321] и сульфид титана [322].
*\ароид титана также был получен, нагреванием
Ш
гмеси Фосфата титана с углем в атмосфере аргона
тесном смес“^ е Jюо—16000С или смесй гидрата окиси
Х^Г'мелкоизмельченного угля, и фосфорной кислоты.
Наиболее эффективный и. распространенный в про-
мышленности метод получения карбида титана-вос-
становление двуокиси титана сажей в угольных печах
Рис. 41. Зависимость плотности спеченных об-
разцов от времени спекания и предварительной
обработки порошка:
сплошные линии — предварительно отмытый порошок;
пунктирные линии — порошок неотмытый; а — темпе-
ратура 2600° С; б — температура 2900—2950° С
сопротивления в среде водорода и в трехфазной элект-
родной печи в среде СО, образующейся при реакции
при температуре 2300—2700° С.
Плотные компактные образцы карбида титана, со-
держащего 18,9% Ссвяз и 1,2% Ссвоб, получены [324]
из порошка зернистостью 1 мкм, предварительно обра-
ботанного разбавленной серной кислотой. После такой
обработки порошок высушивали, прессовали под давле-
нием 68,6 Мн!м2 (70 кГ/мм2) и спекали в течение 1 ч в
вакууме 133,3 «М2—533,2 мн/м2 (1—4- 10~4 мм рт. ст.).
Зависимость плотности образцов от температуры спека-
ния приведена на рис. 40. При температуре спекания
Ij’OQC остаточная пористость составляла 5%, при
2050° С 3%.
114
При изучении влияния предварительной очистки И
активации поверхности частиц карбида титана [325] по-
рошок карбида титана, содержащий 18,1% ССВяз и 0,3%
С об, отмывали смесью плавиковой и азотной кислот
вТотичестве, соответствующем растворению 2% от об-
щего количества карбида.
Затем спрессованные бри-
кеты такого порошка спе-
кали при температуре
2600 (рис. 41, а) и 2900—
2950°С (рис. 41,б). Та-
ким образом удалось по-
лучить образцы с остаточ-
ной пористостью 4% при
спекании в течение 7-—
10 мин при температуре
2900° С (рис. 41). Однако
на практике вести спека-
ние при столь высоких
температурах, весьма
близких к температуре
плавления карбида тита-
на, довольно сложно.
Исследование влияния
циклического спекания на
плотность образцов [325]
показало значительное
интенсифицирующее дей-
ствие такого спекания
(рис. 42). Режим цикли-
ческого спекания сводит-
ся к нагреву при максимальной температуре в Течение
3 мин, охлаждению до 400° С и повторному нагреву.
В работе [266] изучали кинетику процесса спекания
пористых изделий из карбида, активированного добавкой
хлористого кобальта; показано, что добавка хлористого
кобальта значительно активизирует процесс.
Система цирконий-=-углерод. Согласно
предложенной в работе [371] диаграмме состояния в си-
nTie£4 0/Zr~ZrC существует один карбид состава ZrC
1 ’ массе)Ь дальнейшем эту систему изучали
в работах [372—375]. В результате всех исследований
енезовский и Руди предложили гипотетическую диаг-
115
Рис. 42. Зависимость плотности об-
разцов TiC от времени спекания,
температуры при циклическом
(сплошные линии) и изотермиче-
ском (пунктирные линии) спекании
.тпания по которой существует один карбид
рамму с°2°ннЫе разных авторов о характеристике об-
ляст7сушествования монокарбида щиркония расходятся
ласти суш нижней границы (табл. 13). Наиболее
достоверны данные работы [859], которые близки к дан-
ны'Т работы [373}.
Таблица 13
Некоторые характеристики областей существования ZrC^-x
по данным разных авторов ____________________,______
Автор и источ- ая* Содержание С (нижняя граница фазы) Период решет- ки, нм Предельное содержание Ссвяз Содержание С {верхняя' граница - фазы) Период решет- ки, нм
(по дос- се) <'1В) % 1 (аэ . 'эви он) , % (ат.) Ч “—тт : (по мас- се) {•IB) %
Уманский [372] 4,8 27,6 0,4656 10,6 47,4 11,62 50 0,4683
Ковальский и Макаренко [373] 7,08 3,5 36,6 0,4652 10,2 40,3 11,62 50 __
Самсонов и Розинова [374] 21,6 0,4582 10,3 45,8 41,62 50 0,4583
Бенезовский и Руди [375] 6,63 35,0 0,4675 if,62 50 0,4694
Сара [859] 7,6 38,5 — —г* — 11,62 50 ——
В работе [859] методами дифференциального терми-
ческого анализа, рентгенографического и металлографи-
ческого анализов изучена диаграмма состояния системы
Zr—С, представленная на рис. 43. Согласно этой диаг-
рамме, подтверждается существование в системе Zr—С
одной фазы ZrCj^, существующей в области содержа-
ния от 38,5 до 50% (ат.) С. Кристаллизуется в г. ц. к. ре-
шетке типа NaCl.
Карбид циркония образует с углеродом эвтектику,
соответствующую содержанию 65% (ат.) С, с темпера-
турой плавления 2850° С [859], что хорошо согласуется
с данными работ [289] и [716].
_3начения пеРиода решетки карбида циркония, приво-
димые многими авторами [314* 315, 329, 383—385],
116
различаются .вследствие разных составов исследуемых
карбидов.
Результаты рентгенографического исследования кар-
бидов циркония разного состава в широкой области тем-
ператур [335t 391] приведены в табл. 14.
ЗБОО
3200
&
%2800
Г
i
$2йОО
2000
О 20 45 БО SO
Содержание С/%>(по массе)
Рис. 43. Диаграмма состояния система
Zr—С£859ф
Рис. 44. Зависимость теплопроводности ZrC от
температуры
Микротвердость фазы ZrC в области гомогенности
рДо6 q° Растет с увеличением содержания углерода
13/0, 374, 386]. Значения микротвердости карбида пре-
117
+12
О о 994 | ] So 1 oil
температуре +1Й г J wo I to tl
S е" м 1 СО + +1Й co - N- О 1
1 о О. cd +1
Ф QO CO -
L в ft о +1 сэ О +1 1 о +1 1Л
C*5 co’
шциента линейного ч с-> О) 0,47146± ±0,00004
и О oj сч о - 1 1 0,47046± i ±0,00004 J
1-Х и коэфЗ гуры [335, 39 «3 Л а> й 2 о» W S а Й СО § +IS 00 о f J t** О f; 1 СЭ О T • * CO • 1 TH о о +1
и jo V* Ыв N « х X X о ь» н ф а х ф ? а* <3 0,46980± ±0,00004 0,47025± ±0,00002 0,46828± ±0,00003 +IS to о 00 о сэ О со - о о +1
о. я
S3
V V X X о |> Зависимость периода расширения от сост< 8 1 0,46961± ±0,00004 0,46957± ±0,00006 1 0,46951± ±0,00001
СО «9 8 L? NJ U5 c? N ъ» 05 c? N сл с? N
118
дельного состава, по
данным различных ав-
торов, составляют
27000—29000 ЖМ2
(2700—2900 кГ1мм).
Зависимость тепло-
проводности карбида
циркония ZrCo.93 в ин-
тервале температур
530—2100° С, согласно
работе [388], выражена
кривой, приведенной
на рис. 44.
Т ер моэ лектрические
свойства карбида цир-
кония изучались в ра-
боте [387]. Изменения
электросопротивления
и термо-э. д. е, в зави-
симости ог температу-
ры свидетельствуют об
электронной проводи-
мости карбида цирко-
ния.
В работах [391, 703]
исследованы удельное
электросопротивление,
коэффициент термо-
э. д. с., теплопровод-
ность и коэффициент
Холла карбида цирко-
ния в области гомоген-
ности от ZrCo.ss ДО
Zr0,96. Показано, что
примесь в образцах
1,4% N оказывает
большое влияние на
свойства карбида цир-
кония. В случае карби-
дов, синтезированных
из чистых циркония и
сажи в глубоком ваку-
уме, обеспечивающем
содержание до 0,09% N, значение удельного электросо-
противления падает с уменьшением содержания углеро-
да. Аналогично меняются термо-э. д. с. и коэффициент
Холта и растет теплопроводность.
Уравнение температурной .зависимости теплоемкости,
выведенное на основе расчета теплоемкости по методу
Дебая в интервале температур 25—2730° С, лриводится
в работах [542] и [844].
В результате исследования процессов испарения кар-
бида циркония [337, 389] вычислена теплота образования
ZrCj_x из элементов, составляющая—199,4± 12,6
дж)моль (—47,6±3 кал/моль) для продукта, 'близкого
по составу к ZrC. В работе [540] методом смешения оп-
ределены значения теплосодержания карбида-ZrC0 9а в
интервале температур 1300—2500° К. и выведена темпе-
ратурная зависимость энтальпии, которая выражается
уравнением
АНгэз = 6,018 Т + 2,192 -10~3Т2 - 381,0.
Восстановлением цирконовой руды сажей в дуговой
электрической печи впервые был получен, расплавленный
продукт, состав которого соответствовал карбиду ZrCj^
[11]. Карбид циркония получали так^е в дуговой печи
независимо от содержания углерода и двуокиси цирко-
ния в шихте, при охлаждении расплава избыточный уг-
лерод выделялся в виде графита [377]; нагреванием сме-
си двуокиси циркония с сажей в среде водорода при тем-
пературе 1900° С [25].
По литературным данным [22], метод получения кар-
бида циркония сводится к нагреву шихты, состоящей из
78,75% двуокиси циркония и 21,25% обугленного сахара
в графитовой печи сопротивления при температуре
2400° С. Продукты реакции содержат Д 1,3% связанного
углерода, 88,92% циркония и следы свободного углеро-
да. Однако обычно продукты восстановления двуокиси
циркония углем не соответствуют составу ZrC, они, как
правило, содержат некоторое количество кислорода и
азота, что связано с образованием устойчивых твердых
растворов ZrC—ZrO—ZrN.
Технически чистый карбид циркония в промышлен-
ных масштабах получают по методу, заключающемуся
в сплавлении цирконового песка, содержащего 67% ZrO2,
119
„ в дуговой печи [378]. Восстановителем являются
5«Л.т гпзЛита (тиглей, электродов).
°ТХ£&сстановление CaO'ZrO2 металлургическим или
«рЛтяным коксом, взятым в небольшом избытке против
стехиометрического, нагреванием .при температурах
1980—2200°С в течение Д ч приводит к образованию
спека который обрабатывают-горячей водой и кислотой.
Нерастворимый остаток представляет практически чис-
тый карбид циркония, загрязненный до 0,2% Са^
Восстановление целесообразно- проводить в вакууме,
что исключает возможность азотирования и, кроме того,
сдвигает равновесие реакции и позволяет-проводить ее
при более низких температурах.
Равновесие в системе ZrO2—С впервые исследовали
в вакууме в интервале температур- 1530—1740° С [380]
при предположении, что реакция идет по уравнению
ZrO2 +30 ZrC4- 2С0.
Однако отсутствие данных полного химического и рент?
геновского анализов продуктов реакции .делает необос-
нованным предположение, что' реакция идет именно до
указанному уравнению^'Исследованием механизма вос-
становления двуокиси циркония углеродом "ц вакууме
0,66—53,2 н/м2 (5-10~4—4« 10"1 мм рт^ ст.), в интерва-
ле температур 800—1950° Q [21] показано, что восста-
новление и науглероживание двуокиси циркония в ваку-
уме проходят через стадии образования низших окислов
и последняя стадия идет по реакции
ZrO + С = ZrC + СО.
Таким образом, результаты исследования, приведен-
ные в работе [380], ошибочны.
В работах [303] и [304] исследовали равновесие в си-
стеме ZrO2—С при высоких температурах.
При температурах 1500—1700° С и давлении 26—
133,3 кн]м2 (150—1000 мм рт. ст?) образуется оксикар-
бид состава ZrC071O0>08 с периодом решетки 0,4665 нм
и уравнение реакции, которому соответствует равновесие
при этих температурах, имеет вид
ZrO2 + 2,62 G Я ZrC0 , О„ л + 1,92 СО.
П„,РРИ темпеРатУре выше 2100° С кислород в карбидах
плпо°НИЯ отсУтствУет» но содержание связанного угле-
рода ниже соответствующего расчетному для ZrC.
Карбид циркония в больших количествах получают
науглероживанием порошка металлического циркония в
вакууме.
Диффузию углерода в цирконии изучали в работах
.[381,. 382, 784}. Определение коэффициента диффузии уг-
лерода в p-Zr в интервалед'емператур 900—1260° С с ис-
пользованием углерода С14 [784] позволило вывести тем-
пературную зависимость коэффициента диффузии, выра-
жающуюся уравнением
D 0,0048 ехр (- 26 700/RT) см2 • сек.
Метод получения карбида циркония осаждением из
газовой-фазы впервые был применен в работе [314]. ZrC
осаждали из газообразной смеси четырекхлористого
циркония, окиси углерода и водорода.
В работе [26] получили карбид циркония, пропуская
смесь метана с водородом через обогреваемую снаружи
колбу, на дне которой находится достаточное количест-
во хлористого циркония, нагреваемого до испарения.
Вверху расположена служащая носителем нить диамет-
ром 0,05 мм. При низких температурах образуются по-
ликристаллический зернистый осадок, при предельно вы-
соких температурах (зависящих от носителя) осадок
монокристаллический.
Металлографическое исследование показало, что все
продукты гомогенны и имеют плотную структуру.
По данным работы [53], при осаждении карбида цир-
кония на вольфрамовой нити с использованием в качест-
ве составляющей СбНдСНз оптимальная температура
нити 1730—2430° С. При этом температура, соответству-
ющая оптимальному давлению пара ZrCl4 300—350° С,
а С6Н5СН3 15° С.
Приведенные данные хорошо согласуются с резуль-
татами работы [50], в которой исследовали осаждение на
высокотемпературных жаропрочных материалах карби-
да циркония. Показано, что ZrC осаждается на вольфра-
мовой нити из паров ZrCl4 + H2 + CxHy (СН4, СбН6,
С6Н5СН3, С2Н2, СО и др.) при, атмосферном давлении
при температурах 1700—2400° С.
Представляет интерес работа [229], в которой иссле-
дованы процессы изготовления тонких пластин из кар-
бида циркония прессованием с дальнейшим активи-
рованным (добавкой NiCOg или Ni2C2O4) спеканием и
121
прокаткой порошка карбида в ленту с последующим спе-
канием. , л
Система г а ф н и и — у г л е р о=д. Согласно гипо-
тетической диаграмме состояния-[371], в системе Hf—C
имеется один «карбид, соответствующий* составу HfC, су-
ществование которого впервые было обнаружено Агте
й Моерсом [53,287].
Наиболее полно диаграмма состояния системы Hf—C
изучена Бенезовским1 и'^Руди 1375], которые на основании
Удержание С, ув(ат.')
Рис. 45. Диаграмма состояния системы Hf—C [375]
результатов рентгеновского и металлографического
анализов, а также измерения температур плавления
предложили диаграмму, приведенную на рис. 457 Однако
измерение температур плавления представляло значи-
тельную трудность и потому не может-считаться совер-
шенно точным.
Согласно полученным данным", -в системе Hf—С име-
ется одна фаза, соответствующая составу Н[С,_Х, с
областью гомогенности от 37 до 50% (ат.) С, Период
решетки HfCt_x линейно увеличивается с ростом со-
держания углерода. Однако данные по абсолютному
значению периодов решетки карбида стехиометрическо-
го состава, приводимые разными авторами [289; 369, 392,
407—409], расходятся и составляют от 0,4614 до
0,4639 нм, что объясняется недостаточно точным опре-
делением состава образцов и учетом примесей. При со-
держании -—70% (ат.) С и температуре 3000° С обра-
зуется эвтектика HfC—С. Согласно данным работы [289],
122
-температура эвтектики составляет 3250±50ff С, что хоро-
шо согласуется с данными работы [716].
Результаты рентгенографического исследования кар-
бидов гафния с разным содержанием углерода- при тем-
пературах ^—190и +26° С приведены в табл. 15 [392].
Таблица 15
Периоды решетки Ли коэффициент термического расширения
карбида тафния
Фаза Я* &М -Да, ил« - СВ НМ 1 1 О <t> Т S' в §•
;26°-С’ —190’ С
0,46382*0,00003 ; ;0,46347*0,00002 О',00035 3,5*0Л
Hf-Сом L 0,46381^0^0004 . 046338±U,00001 л 0,00043, 4,3*0,4
HfC0>?7 0,46395*0,00006' 0,46354*0,00003- 0,00041 4,1*0,7
С jpocTOM температуры коэффициент линейного рас-
ширения увеличивается и в интервале температур 20-=—
612° С составляет 6,59 ±0,04» IO'"6' 12].
Рис. 46. Зависимость удельного элек-
трического сопротивления HfC от тем-
пературы [411]
Температурная зависимость удельного электросопро-
ивления, по данным работы [411], приведена на рис. 46
свидетельствует о металлическом характере проводи-
мости карбида графния.
123
Измерение эмиссии HfCL_x, нанесенного на воль-
флам и тантал, при температурах 1130-1860° С [414] по-
?яРЛло что повышение температуры в указанных преде-
лах приводит к значению работы выхода 3,9 эв (6,24-
10-i9 дж\ которое сохраняется до 2090° С. При на-
гревании катода в течение 3 ч при 2130° С работа выхо-
да увеличивается До 4,1 эв (6,56-10 дж).
Термодинамические характеристики карбида гафния
изучали в работах [402, 403, 540].
Скорость испарения, состав и давление паров карби-
дов гафния изучали на образцах карбида состава
HfC0 951 [337]. Исследования показали, что процесс' испа-
рения идет по схеме
HfC^ = Hfc + сг.
Скорость испарения при температуре 2900° С составляет
(2,5—6,5) X10-5 г! (см2-сек).
Процессы восстановления двуокиси гафния углеро-
дом с целью получения карбида гафйия исследовали в
работах [287, 390, 400—405].
В работах [402, 403] изучали динамическим методом
равновесие реакции восстановления двуокиси гафния уг-
леродом в области температур 1470—1730° С и давлений
933,3—13332 н!м2 (70—1000 мм рт. ст.).
Зависимость 1g р от 1/Т выражается прямой линией,
что свидетельствует о практически постоянном составе
фаз и моновариантности равновесия, которое устанавли-
вается по уравнению
НЮ2 + 2,9 С = Hf<^„ О005 + 1,95 СО.
В более поздней работе [393] определяли теплоты сго-
рания карбидов гафния разного состава и вычисляли
теплоты их образования (табл. 16).
В том случае, если процесс ведут в электрической пе-
чи сопротивления с графитовым нагревателем в среде
в°Дорода, оптимальная температура получения карбида
9^лИокл^ИЗКОГО по С0СтавУ к стехиометрическому,
с nJ С- Полученный при этом продукт содержит
6,26—6,30% С0«щ и до 0,1 % Ссвой,
Исследование условий получения карбида гафния
осстановлением окиси углерода в вакууме [404] показа-
, что полное удаление кислорода при этом возможно
124
Таблица 16
Теплоты сгорания и образования карбидов гафния разного состава
Содержание, % Формула соединения Теплоте сгорания (298а К) Теплота Образования (298° К)
Ш с ^связ ^своб кдж/моль ккал/моль кдж/моль ккал/моль
97,2 2,5 0,1 ^^0,38 —1137 —265,8 —151*3,3 —36,0*0,8
96,15 3,8 0,1 HfQ),59 —1749 —280,4 —172,2*3,8 —41,1*0,9
95,65 4,3 0,1 HfC0>67 —1162 —277,3 —216,6*1,7 —51,7*0,4
94,95 5,0 0,2 HfC078 —1202 —286,7 —220,9—2,1 —52,7*0,5
94,25 5,75 0,05 ^^0,91 —2478 —297,8 —225,02=2,5 -53,7*0,6
93,6 6,05 0,3 ^^0,96 —1269 —303,1 —223,3*4,2 —53,3*1,0
94,0 6,25 0,05 ^^0,99 —1277 —304,9 —227,0*1,2 - 54,2*0,3
93,5 6,3 0,2 HfC —1282 —305,9 —227,0*2,1 —54,22=0,5
^пепатуре около 2000° С-Использование шихты
^Л^етрич^жого состава.-приводит -к образованию
ITXtob содержащих меньше связанного углерода,
SS соответствуем составу HfC, при сохранении ре-
ZrX HfC с уменьшенным периодом, что свидетельст-
вует об образовании-дефектной по углероду решетки.
У Использование шихты, содержащей избыток сажи
(0 4% по отношению к образующемуся карбиду гафния),
приводит к образованию при темпергед>е 2200” С карби-
да с содержанием о,2о /о Шсвяз “ /о ^свбб-
Высокое разрежение приводит к образованию карби-
да, насыщенного углеродом. Сохранение в реакционном
пространстве некоторого остаточного давления СО бла-
гоприятствует полному насыщению карбида тафния уг-
леродом.
Условия прохождения реакции HfO2+3G=HfC-f-2C0
в вакууме исследовали также в работе [405} в интервале
температур 1000—2000* С и давлении 13,33 н/м2
(0,1 мм рт. ст.). Результаты этих исследований, свиде-
тельствуют о том, что при температурах до 2000° С об-
разуется карбид, имеющий дефицит атомов углерода.
Измельчение продуктов, образующихся при темпера-
туре 1900° С, с последующим повторным брикетировани-
ем и нагревом при той же температуре в течение 1 ч поз-
воляет получить карбид, близкий по составу к HfC
(6,31% Ссяз).
Двухстадийный процесс получения карбида гафния
при температуре 1900°С технологически более целесо-
образен, чем выдержка при температуре 2000° С в тече-
ние 1 ч с последующими выдержками при 2400° С в те-
чение 1 ч при 3000° С в течение 5 мин,- предложенные
в работе [406].
Наиболее чистый карбид гафния получен осаждени-
ем на вольфрамовой проволоке диаметром 0,05 мм при
нагревании в атмосфере паров четыреххлористого гаф-
ия, чистого, не содержащего азота водорода и углеводо-
нениюТИПа метана’ Т0лУ0ла> ацетилена [50, 287} по урав-
HfCl4 + СН4 + На = HfC + 4НС1 + Н2.
Tvnnft*P пп осажденного карбида определяется темпера-
250п°с ПРОВОЛОКИ- Оптимальная температура 2100—
с. высокая температура плавления гафния делает
12в
возможным получение жарбиДа Непосредственным на-
углероживанием гафниевой проволоки иди нити, на ко-
торой предварительно осажден гафний, углеводородами
при температурах до 2500° С [54],
Порошок ^карбида гафния легко поддается горячему
прессованию^ образуя плотные компактные образцы с
еерым металлическим блестящим изломом.
В- работе £405]- длГпеследования -некоторых физиче-
ских свойстГмз порошка -карбида' гафния готовили об-
разцы методом горячетсмяпрессования при температуре
около 3000° Of давлении .17,6 .Мн!м? (180 кГ1см?) в тече-
ние 10—15 мин. Лри таком- режиме -получены образцы
г минимальной остаточной пористостью около 6%.
В „работе [410]. исследовали равновесие реакции H1N +
’4-Сг=^ HfC-b^W Показано, что лимйтируется процесс
диффузией частиц углерода в НПФ. Константа равнове-
сия реакции в облает^ температур 1700—2300°С описы-
вается уравнением"
1g Кр = -^+2,00.
Результаты исследований позволяют заключить, что ком-
пактные образцы из карбида гафния можно- получать в
печах сопротивления с. графитовыми нагревателями при
температурах 2500° С в атмосфере азота^При этом мак-
симальное содержание азота не превышает 1 %.
КАРБИДЫ МЕТАЛЛОВ
ПОДГРУППЫ Va
^Система ванадий — углерод. Согласно пер-
вой диаграмме состояния системы ванадий—углерод, в
ней имеется карбид, состав которого изменяется в пре-
делах от VC0>75 до VC [371].
В работе [415] в системе V—С обнаружены две фазы,
соответствующие формулам ViCa (15,01^£) и V5C
(4,5 /оС). V5C^обладает гексагонально^. Длотноупакован-
нои структурой. Как показано в работах [416] и [417], опи-
санная гексагональная структура принадлежит фазеУ2С;
4->з имеет, по данным работы [415], г.ц.к. решетку.
отпсТген^ г^ические'йсслеД°вания сплавов состава
л5по4.а3 Д° [418] показали, что все они идентичны и об-
т г.ц.к. решетко^ типа NaCl. Это позволило пред-
127
„ итп существует фаза VC, имеющая широкую
Т гомогенности, нижний предел которой соответ.
СТВИсТс.“дТомния си’сгемы“-С-[4i9-426] дали несколь.
КО противоречивые данные о предельной составе высше-
го карбида и границах области гомогенности фаз Vf C и
VC Наиболее полное исследование этой системы [421,
Рие. 47. Диаграмма состояния систе-
мы V—С [425]
423] показало, что при взаимодействии трехокиси ванадия
с углеродом образуются следующие фазы:
P-фаза состава VO0 6 Со а —VO0 7 Со 12> обладающая
г.ц.к. решеткой с периодом а = 0,401 нм;
у-фаза состава VC0 33 O0 09 —VC0<50O013, обладающая
гексагональной решеткой с периодами а = 0,2857 -s-
-ь 0,2888 нм, с = 0,4558 -*-0,4567 нм, с/а=\,592 -*- 1,581;
б-фаза, состав которой сильно зависит от условий
опыта, и содержание в ней углерода достигает 13—14%;
она обладает г.ц.к. решеткой с периодом а = 0,4007
-<Ц^4123 нм;
,8 -фаог содержащая от 14 до 18—19% углерода
* *^о,69 * Ц),99' обладающая г. ц. к. решеткой типа NaCl,
с периодом й=0,414^,415 нм.
Г49/ПУ^еСТВ0ВаГИе кубических фаз подтверждено
[424], но интервалы их составов соответствуют VC„
VC0.79hVC0,79-VC„9,.
[42!Я где°ппеплтгТ система исследована в работе
Р Д на диаграмма Состояния, приведенная
128
на оис 47. Установлено, что нижний прёдел области го-
могенности фазы зависит or температуры При 1000 С
он соответствует составу VC^, при 1800 L. и пРи
2165° С VC0j60- Верхний предел соответствует составу
VC 88» чт0 ХОРОШО согласуется с работами [426, 464, 514,
521/538, 555], в которых авторы, обсуждая кристаллохи-
мические особенности кубических карбидов титана, ва-
надия, и ниобия, пришли к выводу, что верхняя граница
области гомогенности соответствует "Составу VC0 875. Бо-
лее полное заполнение подрешетки углерода становится
энергетически невыгодным. При дефектности подрешетки
большей, чем это соответствует составу VC0>667, устойчи-
вость кубического карбида уменьшается и возникают
предпосылки для образования низшего по содержанию
углерода карбида V2C.
Период решетки определяли многие исследователи
[11, 22, 173, 331, 384, 422, 435, 436, 864] и для предельно
насыщенного карбида VCa88 он'равен 0,4159 нм.
Область гомогенности фазы V2Cj_x лежит между
VC0>47 и VC050. В области составов между VCo go и VC0>74
обнаружена новая $ -фаза (при температуре ниже
1334°С), аналогичная найденной в системах Nb—С и
Та-С.
Монокарбид VG образует с углеродом эвтектику, пла-
вящуюся при температуре 2170±50°С [716].
Монокабрид ванадия — серый металлический по-
рошок.
Согласно данным большинства исследований [25, 85,
435], температура плавления карбида ванадия 2830° С.
Карбид ванадия отличается высокой твердостью, цара-
пает корунд.
Результаты измерений удельного электросопротивле-
ния и постоянной Холла [855, 864] свидетельствуют о ме-
таллическом характере проводимости карбида ванадия.
Исследование термоэмиссионных свойств карбида ва-
надия [414] показало, что карбид, содержащий 17,5%
связанного, 0,15% свободного углерода и 80% ванадия,
при температуре катода 1930° С быстро испаряется. При
температурах 1330—4430° С работа выхода карбида, на-
?7лпо г-°Г?тНа тантал» равна 3,9 эв и сохраняется до
ичи с. Повышение температуры до 1860° С повышает
работу выхода до 4 эв.
9 Косолапова Т. Я.
Исследовали процесс испарения карбида ванадия ме-
тодомКиудсеиа [429]. Приведен состав исследуемого кар-
бида VC/Х (18,89%Са1а), «а вызывает сомнение, так
как согласно указанному выше, получить карбид вана-
™я’такого состава невозможно. -
Термодинамические характеристики карбида ванадия
приведены в работах [43, 316, 308, 42У, 48/, 48«, оуу].
Данные о теплоте образования карбида-ванадия из
элементов довольно многочисленны и значительно расхо-
дятся между собой. Так, в работе [429] получено значе-
ние—955 кдж/моль (—22,8 ккал!моль), отнесенное к
составу VCog92; в работе [368]_ —117,3±42 кдж/моль
(—28 0±10 ккал/моль), отнесенное просто к VC пере-
менного состава; в работе [43]—125,7 кдж/моль
(—30,0 ккал/моль), отнесенное к VC0i96j в работе [437]
—171,8 кдж/моль (—43 ккал/моль)_, отнесенное к
VC (91 О004; в работе [599]—101 кдж/моль (—24,1
ккал/моль), отнесенное к карбиду состава"УСб88.
Столь значительные расхождения между приведенны-
1И данными связаны с недостаточной точностью опреде-
ления состава исследуемых образцов. Наиболее преци-
зионные методы использованы в работе [599].
При получении карбида ванадия сырьем могут слу-
жить пятиокись или трехокись ванадия, ванадат аммонии
и металлический ванадий в виде порошка. Исследование
процесса восстановления окиси ванадия углеродом с
цетью получения металлического ванадия в вакууме
описано в работах [41] и [44].
Скорость процесса восстановления V2O3 ламповой са-
жей при температурах до 1000° С весьма незначительна и
быстро растет при температурах 1200—1575° С. Значения
энергии активации рассматриваемого процесса на осно-
?^ПЛрИ1^н\ал^ных данных [4П составляют
4’5~217>5) ’10 дж1м°м>[(48,8—51,9) - Юз кал/молъ\,
СО xP0I¥g совпадает с энергией активации реакции
59 2Jzh72CO7 Равной 184,4-217,9 кдж/моль (44-
зпрнипЛ‘*оль>' Это подтверждает существующую точку
согласно кптппР?ДУ в„осстан°вления окислен углеродом,
ным обоазо °Р°й Действительный восстановитель глав-
ным ооразом газообразная СО [23]
РЫ. размеровНИчастицС VO™ ЛаВления С°' темпе₽атУ
и стиц у2О3, давления прессования и
130
наличия примесей порошкон ниобия, тантала, хрома, мо-
либдена и вольфрама на кинетику процесса восстанов-
ления VsOg углеродом [821фдоказали, что с ростом тем-
п^патуры степень, восстановления V2O3 возрастает от
20% при температуре 1300°С до 68-% пр* 1500° С. Раз-
меры частиц V2O3 и давление прессования не влияют на
степень восстановления. Уменьшение давления СО в-си-
стеме и добавки порошкон тугоплавких металлов способ-
ствуют переходу оксикарбидной б'-фазь! на. основе УС1-Ж
в у'-фазу на ocHOBe VsCi—х,
В первых работах по получению карбида ванадия
V2O5 восстанавливали углеродом [294]. Однако позже, бы-
ло показано [25, 43 Г, 432], Что наиболее эффективно
использование в качестве исходного материала при
получении карбида ванадия трехокиси V2O3, темпе-
ратура плавления которой Составляет 2000° С, в то
время как температура плавления пятиокиси V2O5
675° С.
По данным работы [22], тщательно перемешанную
смесь из 73—75% V2OS и 27—25% ррафита прессуют и
нагревают брикеты в среде водорода при температуре
1800° С» Полученный продукт вторично нагревают и ва-
кууме при температуре^ 1600—1700° С, в результате
чего образуется карбид, содержащий 18,5—19% С общ и
до 0,5% Ссвоб* это значение содержания Ссвоб зани-
жено.
Наиболее подробно изучены условия, получения кар-
бида ванадия в вакууме в работе [434].
Исследование зависимости состава карбида от тем-
пературы показало, что при температуре 1700—1800е С
кислород в продуктах реакции отсутствует при любом
давлении [от 13,33 до 1333,2 н/м2 (от 0,1 до 10 мм рт.ст,)}.
При температуре 1500° С и давлении 13,33 н/л2
t (0,1 мм рт. ст.) для полного удаления кислорода доста-
точно нагрева в течение 2 ч, содержание связанного угле-
1800°Сг°СТаВЛЯеТ —^,3%. Повышение температуры до
также не приводит к увеличению содержания свя-
занного углерода.
maSai7CfiMai7boo/e значение содержания Ссвяз не превы-
Да ван^п^а [433] исследовали условия получения карби-
сырья труи>„ПРИ «пользовании в качестве исходного
9/ тонического ванадата аммония, содержащего
131
56 23%V2O5 (теоретическое содержание 56,2%), по ре-
2NH4VOa 4- 5С 4- 214, “ 2VC-4- ЗСО 4- 2NH3 4* ЗН2О.
Получение карбида ванадия осаждением из четырех-
хлористого ванадия в присутствии нодорода на вольфра-
мовой нити—довольно трудная задача. Ванадий, темпе-
ратура плавления которого 1712° С, при близких к этой
температурах взаимодействует с вольфрамом, что пре-
пятствует образованию карбида. 41оэтому процесс
вначале ведут при более низкой температуре (1400—
1500°С) и лишь после образования пленки карбида на
нити повышают температуру до 2000° С [287].
Аналогичным методом карбид ванадия получали [50],
осаждая его на вольфрамовой нити из смеси четыреххло-
ристого ванадия, водорода и углеводородов.
Система ниобий —углерод, Впервые рентге-
нографическое исследование образцов системы Nb—С,
содержащих от 2,5 до 10,6% ССВязг проводили в работе
[440]. На рентгенограммах образцов, содержащих 37%
(ат.) С, наблюдались линии двух фаз с периодами решет-
ки 0,4425 и 0,4441 нм. Это фаза Nb2G, содержащая
33% (ат.) С, и фаза NbC^, содержащая 41—47% (ат.)С.
Получить карбид с содержанием 50% (ат.) С не удалось.
Предположение о существовании карбида Nb^s было
выдвинуто в работе [441].
Детально система Nb—С исследована в работах
[442—449].
Результаты исследований, по данным работы [443],
приведены в табл. 17.
Позднейшие работы по исследованию системы нио-
бии углерод [446—448] привели к созданию диаграммы
состояния этой системы. В системе подтверждено суще-
ствование трех однофазных областей; а-фаза -— твердый
раствор углерода в ниобии. По предложенной диаграм-
ме с уменьшением температуры растворимость углерода
в ниобии уменьшается; P-фаза —NbaC^, плавящаяся
перитектически при температуре 3090±500° С, с областью
гомогенности NbC0>495-NbC0>500 ниже 2000° С; a-и 0-фазы
2ззч1У9П°гЭВмлоТИКу’г,ЛемпеРатура плавления которой
2335±20 С [448] и 2328±17°С [443]. у-фаза - карбид
с перитектической температурой 3500 ±75° С для
132
Таблица 17
Характеристика образцов в системе Nb—С
Содержание, % (по массе)
Nb собщ ^сноб №+собщ
99,12 0,79 - 99,91
98,15 1,82 — 99,97
96,95 3,00 — 99,95
95,99 4,26 г — 99,95
94,83 5,19 — 100,02
94,04 5,94 — 99,98
93,48 6,45 —— 99,93
92,78 7,26 *—- 100,04
92,34 7,61 *— 99,95
92,03 7,93 99,96
Формула соединения Обнаружен- ные фазы Периоды решетки, нм
a-фа за ₽-фаза — -- f. Т-фаэа
а а с
NbC^ a, p 0,33011 г- —г ч-г-
NbC0>14 a, p 0,33012 *— — г 1
NbCo24 P. a —U 0,3119 0,4957 ? г—
NbC0>34 P, 4 —— 0,3120 0,4957 4—
NbC0_42 P — 0,3121 0,4960 —
NbC049 P —> 0,3123 0,4966 —
NbC0>53 P. 6, (§) —1 0,3128 0,4974 А .
NbCo>6o p.«. Ш — 0,3128 0,4974 0,44317
NbC0>64 3. P. (S) — —— 1—. 0,44300
NbC^ee б, P, (&) —• — — 0,44315
— Продолжение табл. 17
со Содержание, % (по массе) Формула соединения
Nb ‘-'общ Ссвоб №+Собщ
91,72 8,28 100,00 NbC0r70
91,59 8,34 — 99,93 NbC0>71
91,26 8,61 — 99, §7 NbC0(73
90,90 9,12 — 100,02 NbdOi78
90,01 9,95 1 * 99,96 NbCggg
89,80 10,15 — 99,95 NbCf0>8y
89,70 10,24 — 99,04 NbCOt88
89,50 10,51 ' 100,01 ^ьс^91 ^b^o(92
89,29 10,66 Т— 99,95
89,21 10,72 0,02 99,93 NbC0i93 !
88,94 11,01 0,23 99,95 NbCogg
88,57 11,38 0,53 99,95 NbC0,99
1 Обнаружен- ные фазы Дериоды решетки, нм ! . - --> - - - 1 __ . 1
Г ' аЬфазй ₽--фаза J- bi . "' ' '"71 7-фаза
X» h ' .. ? i -П7 г-: Г* Г,. Л
. ц ( и • б V-1 1 Э| ‘ |> 0,4,4^09
б —¥ — 6,44335
б ’ •‘"Г. U—и 0,443^5
б — -1 •Лй-/ * 0t44519
б Hl Hl 0,44618
6 ^-*ч 0,44649
6 — —Хй 0,44660
б •—• Я— ‘ — 0,44690
6 г— Ч— 0,44691
б» графит .•т— *—• 0,44682
б, графит — 0,44692
6, графит —X —, 0,44690
образцов состава Область гомогенности его
NbCon—NbC^ ниже- 2000? CL. Между р- и у-фазой
существует двухфазная область, состоящая из этих
двух фаз.
Предполагаемая: в работе 1443] fe-фаза'в области со-
ставов NbC0>M—NbCOJ0 обнаружена, не была. Граница
фазы NbC—Сбежит в области- NbCfl gi нри температуре
20(10° (L
В работе [449] была исследована система ниобий —
углерод методами рентгеновского и металлографического
Рис. 48. Фазовая диаграмма -состояния систе-
мы Nb—С [449]
анализов сплавов, полученных дуговой плавкой и спека-
нием порошков ниобия с ламповой сажей. Образцы со-
держали от 0,011 до 14,12 % С.
Результаты, представленные в виде фазовой диаграм-
мы (рис. 48), как и предыдущие исследования, свидетель-
ствуют о том, что в системе Nb—С существуют два кар-
ида. гексагональный карбид NbjCj^ с областью гомо-
генности 5,48-5,83% (по массе) С* (NbC0 44— NbC0 48)
135
И г.ц.к. карбид NbC^ с областью гомогенности 8,25—
10,25% (по массе) С (NbG0 70— NbC0>90). Однако грани-
ца фаз гомогенности обоих карбидов, по данным разных
авторов, значительно расходятся.
Наиболее вероятные концентрационные пределы ус-
тойчивости карбидных фаз: для гексагонального карби-
да NbC048 —NbC0>50 и для кубического Nb0>70 —Nb0>99
[444].
Максимальная растворимость углерода в ниобии при
эвтектической температуре. (2230° С) равна 0,8% и
быстро уменьшается с температурой до 0,1% (по массе)
при 1800° С.
Исследование характера взаимодействия монокарби-
да с углеродом [289] показало, что эта система имеет эв-
тектический характер, температура плавления эвтектики
3150±50°С, что хорошо согласуется с данными работы
[716]. Рентгеновский анализ застывших капель эвтектики
показал наличие двух фаз: МЬС^ и графита.
Исследование диффузии углерода в карбид ниобия по-
зволило определить коэффициент диффузии, который При
температуре 2670° С составляет D = 8-10~7 см21сек.
Гельд и Любимов [454] изучали самодиффузию нио-
бия, диффузию углерода в металлической ниобий и само-
диффузию его в карбиде. Самодиффузию ниобия изучали
на образце состава NbC0>98, температурная зависимость
подвижности ниобия описывается уравнение^
D = 5,5- 10~6ехр (—
\ ЦТ )
Энергия активации самодиффузии ниобия в NbC0 98 на
35% меньше, чем в металлическом ниобии.
Температурная зависимость коэффициента диффузии
углерода в ниобий описывается уравнением
D = 1,09-КГ5 ехр(-2^
\ цт }’
пя^^мкг?10 Углерода в каРбиде ниобия изучали на об-
Р NbC0 5, NbC0 75 и NbC0 88, температурные зави-
моапн1ыиаКОЭФФИциентов сам°ДиФфузии описываются
уравнениями
DNbc0. - 5,7- 10~6ехр 29800 \
\ т&Г
136
rye — Я R ICF^ ₽xn 35
Д\ьс0 75" o,o-lu ^ еЛР { ]»
„ 1 nr tn-6~ J "32 300 A
2?NbcrtQ8“l,06'10 expl - 4.
V । “o Vs л 1
Значения энергии активации: процесса самодиффузии
углерода в карбидах ниобия разного состава практически
мало зависят от состава.
Высший карбид/ниобия. NbCf^— серйваТо-коричне-
вый металлический порошок. Расстояние Nb—Ц по дан-^
ным работы [4563, равно- 0,22355 нм, Значения периодов
решетки, приведенные разными^ авторами,, расходятся
[364, 439—444].
Определение периода решетки образцов сплавов
NbCj^ с молярным отношением Nb* С 0,700/0,996 [445]
позволило вывести зависимость периода решетки от ука-
занного отношения, которая выражается уравнением
а = 0,409847 - 0,071820 (C/Nb) - 0,034570.(C/Nb)2,
Значения периодов решетки' карбида ниобия состава
NbC0r884 при высоких температурах [335] й вычисленные
значения коэффициента термического расширения при-
ведены в табл. 18.
Таблица 18
Период решетки и коэффициент линейного расширения NbC0 ^84
Температура, °C
Показатели 20 498 590 700 '720
а, НМ 0,44691± ±0,00003 0,44833± =±=0,00003 0,44895± ±0,00004 —
«298-Ю-S град~1 — 6,65± ±0,28 6,70 —*— 6,76± ±0,23
Продолжение табл. 18
Показатели Температура, °C
848 1000 1500 | 2000 2500
а, нм 0,44943± ±0,00004 —— — -С —
а298'10—*, град— 1 6,84± ±0,21 7,11 8,10 8,27 8,75
10 Косолапова Т. Я.
137
Изучение удельного электрического сопротивления и
постоянной Холла [19, 855] подтверждает металлический
характер проводимости NbCj_^«
Исследование испарения в интервале Температур
2500—2854s С [462] на образцах состава NbC0 976 показа-
ло что диссоциация идет по схеме NbCTB — Nbr + Cr,
’Процесс испарения NbC изучали в работе [463] мето-
дом Ленгмюра в интервале температур 1990—2670 С.
Показано, что при нагревании карбида NbC0>97 в вакууме
при температуре 2670° С отношение C/Nb падает, доходя
до величины, соответствующей составу NbC0 75, после
чего оно не изменяется.
Давление паров углерода над NbCj^, близким по со-
ставу к стехиометрическому, описывается уравнением
. _ опс 32 760
lgpc = 5,296 ---
Вычисленная теплота испарения составляет 628 кдж/моль
(150 ккал!моль).
В результате изучения термодинамических свойств
карбидов ниобия переменного состава [457, 458] выведе-
ны уравнения температурной зависимости теплоемкостей
и изменения энтальпий для карбидов состава NbG^ 749 ,
^ЬСо 8б7 и ^5С100.
Полученная зависимость изменения энтальпии карби-
дов ниобия от состава приведена на рис. 49.
Теплота образования, отнесенная к 1 г-атом, увеличи-
вается с уменьшением содержания углерода, причем за-
висимость хорошо описывается уравнением
^Nbcx= 4,19(48,1 х — 15,9 х2) кдж/моль
[А^ыьс* ~ (48,1 х — 15,9х2) ккал/молъ].
Значения теплот образования карбидов ниобия, при-
веденные в разных работах [339, 457—460, 462, 590, 783],
различаются. В ряде случаев авторы даже не приводят
ja°a испы™ваемых образцов, иногда состав контро-
нир пп™ неД°статочно точно, не определяется содержа-
ствя кя^е»еи’ Наиб°лее полно термодинамические свой-
пичипнЛт А0В ниобия разного состава исследованы пре-
цизионными методами в работах [460, 461].
138
В работе [460] для уточнения термодинамических ха-
рактеристик проведено определение теплоты сгорания
карбидов ниобия разных составов. Использование полу-
ченных данных по теплотам сгорания и данных, получен-
ных авторами работы [461] для теплоты образования
Рис. 49. Зависимость изменения энтальпии карбидов
ниобия разного состава от температуры:
/ —NbCp^o: 2 — НЬСо,749! 3 —NbCo,867: 4 —NbCl.OO
Nb20g, позволило вычислить значения теплот образова-
ния карбидов ниобия разных составов (табл. 19).
Наиболее распространенный метод получения карби-
да ниобия — восстановление окиси ниобия сажей в среде
водорода или в вакууме.
Исследование условий и механизма получения карби-
да ниобия восстановлением Nb2Og углеродом в вакууме
Н6] показало, что оптимальная температура получения
карбида ниобия, близкого по составу к стехиометриче-
скому, 1800° С; при этом для полного завершения реак-
ции время необходимого нагрева составляет 4 ч. При ме-
10*
139
Таблица 19
Значения стандартных теплот сгорания и теплот образования
карбидов ниобия ..— . -..
Карбид Период решетку а, нм Теплота сгорания (298°. К) 'образования (298° К)
кдж[ ! 4. формулу ккал! г. формулу кджТ &. формулу ккал/ г. формулу
NkQ),739 0,4442* *0,0001 —1121,2*: ±1,7 ^-267,6*:= *0,4 —120,2* =*2,1 : —28,7* *0,5
NbCo,783 0,4454* *0,0001 —1137,6* *2,5 —271,5* *0,6 —121,1* *2,9 —28,9* *0,7
Nt^0,838 0,4458* *0,0001 ' —1154.8=!=- *1,7 - = —275,6* *0,4 —125,7* *2,1 —30,0* ±0,5
NbC0gi3 0,4467* *0,0001 —1179,9* ±2,1 —281,6* *0,5 —129,9* ±2,5 -*31,0* *0,6
нее длительном нагреве значительная часть углерода и
ниобия остается в свободном виде.
В работе [450] исследовано равновесие в системе
Nb—С—О и показано, что термодинамически наиболее
вероятно восстановление NbO до высшего карбида NbC,
несколько менее благоприятны условия для восстанов-
ления до Nb2C и наименее благоприятны для образова-
ния металлического ниобия.
Исследование влияния структуры пятиокиси ниобия,
давления, сорта восстановителя и добавок солей щелоч-
ных металлов на процессы углетермического восстанов-
ления пятиокиси ниобия [451] показало, что характер
взаимодействия существенно зависит от метода приготов-
ления Nb2Os. Окислы, полученные Прокаливанием гидро-
окиси ниобия при температурах 800—900° С, в которых
в основном содержится низкотемпературная Т-форма
Nb2Og, реакционно более активны, чем окислы, прокален-
ные при более высоких температурах и содержащие сред-
не- и высокотемпературные М и Н модификации. Это
связано, по-видимому, с рекристаллизацией Nb2Os при
прокаливании и уменьшением реакционной поверхности.
Изучение влияния давления на процесс восстановле-
ния (рис. 50) приводит к заключению, что с повышением
давления от глубокого вакуума до 399,9—533,2 н/м2
140
д ДО 80 120 150 200
Время, мин
Рис, 50. Кинетика восстановления МЬгОй при различных
давлениях газовой фазы, w/jw2; *
1 ~ ,3'33:, 2 ~~ 533,й8: 3 ““ °>13^ 4~13-33 ‘102: 5 — 26>ез 1lfls;
’ — 79,99 • 102; 7 — накопление продуктов реакции
до ра=г79,99 • 102 л/л»2; 8 ~~ 13,33 » 10’
w
Рис, 51, Зависимость стёНени восста-
новлений NbaO? of временй использо-
вания воостановигеля с различной
, Зернистостью:
I сажа; 2—4 Л графит <J размерами ча-
стиц J мм; - Q.10; 3 — 0,25; 4 — 0,5
/3—4 мм рт. СТ.) скорость реакции возрастает, дальней-
шее увеличение давления тормозит реакцию. Это свиде-
тельствует о разном влиянии газовой фазы на характе-
ристику этапов, восстановления и регенерации СО, что
приводит к экстремальному закону зависимости.
F Большое влияние на процесс восстановления оказыва-
ют сорт восстановителя, размеры его частиц (рис. 51) и
добавки солеи щелочных металлов [451, 452]. Из исполь-
зованных в качестве восстановителей ацетиленовой сажи,
графита, каменноугольного пека и продуктов его коксо-
вания наилучшие результаты были получены при работе
с пеком. Это-связано с тем, что при нагревании шихты
происходил пиролиз содержащихся в пеке углеводородов
с образованием сажистого углерода, обладающего высо-
кой удельной поверхностью и большой реакционной спо-
собностью.
Прибавление к шихте 2% (по массе) К2СО3 повысило
степень восстановления МЬгОб при температуре 1150°С
более чем в 2 раза.
Второй метод пригртовления карбида ниобия — синтез
из элементов, осуществляемый при температурах 1700—
1800° С. В качестве исходного сырья служит порошок ме-
таллического ниобия или гидрида ниобия, который при
температуре 800—900° С разлагается с выделением во-
дорода.
Попытка приготовления карбида ниобия осаждением
на вольфрамовой нити из смеси хлорида ниобия, водоро-
да и углеводородов (пары толуола, метан, ацетилен) [50,
287] не дала положительных результатов. При темпера-
турах 900—1000° С на нити интенсивно осаждается ме-
таллический ниобий, Однако при дополнительном нагре-
ве в среде водорода, содержащего углеводороды, при тем-
пературах 1400—1700° С продукты осаждения удалось
полностью перевести в карбид.
Получение изделий различных форм из карбида нио-
оия весьма затруднительно вследствие высокой темпера-
туры спекания заготовок (3000°С). Снижение темпера-
туры спекания достигается при использовании активи-
процесс спекания добавок. В работе [455]
ипл?а00Тан режим активированного спекания карбида
железа и хлористого кобальта, по-
зволяющий получать изделия с пористостью 15—18%
спеканием в течение 1,5 ч при температуре 1500° С. Спе-
142
кание образцов при этих же условиях без активирующих
добавок приводит к получению изделий с пористостью
30—40%,
Система тантал —углербд. Впервые система
Та—С исследована в работе [315], в которой установлено
Рис. 52. Диаграмма состояния системы Та—С [465]
ны две модификации Та2С: a-модификация, кристалли-
зующаяся в гексагональной плотноупакованной решетке
с периодами а = 0,3085±0,0001 нм и с = 0,442± 0,0007 нм,
с/а=1,59, и Р-модификация, кристаллическая структура
которой не определена, но установлено, что рентгено-
грамма ее очень мало отличается от рентгенограммы
а -модификации.
Наиболее полно эта система исследована в работах
[283, 289, 315, 384, 447, 465—469].
Диаграмма состояния система Та—С [465] представ-
лена на рис. 52. Растворимость углерода в тантале при
температуре 1800° С составляет менее 0,1%, при 2000° —
До 0,2% [873]. F
143
п пповодившиеся исследования системы Та С
п-омовном сводились т< рентгенографическим и металле-
Мафическим, данных о химическом составе исследуемых
XSa»™₽«-m прив0± ’jSSSS
состав зависимость периодов решетки от составов и гра
ницы областей гомогенности не могут быть точно оце-
йены-
в -работах [467—469] исследовали систему Та—С с
применением наиболее совершенных методов рентгенов-
ского; химического и термодинамического анализов.
Обнаружены следующие фазы а-фаза кубическая,
в которой углерод практически нерастворим, выражается
составом TaA—TajC^ с периодом решетки от 0,33035
до 0,33066± 0,00002 нм;- Э-фаза^гексагональная, с об-
ластью гомогенности от ТаСр ^^де ДО ^'а^-'о,5О£О,О2 и
периодами решетки от 0=0,31012 и с=0,49372 нм для
ТаС0 gg до а=0,31042. "и д=0,49410 мм для ТаС0>50;
у-фаза г.ц.к. с областью гомогенности ТаС058±0 02 до
TaC0>91±0 Q2 И периодом решетки от 0,44206 до 0,44564 нм.
Карбида состава ТаС авторам получить не удалось.
Существование а-, Р- и у-фаз подтверждено работой
[466]. Области гомогенности и периоды решетки несколь-
ко отличаются от данных работ [467—469].
а-фаза простирается в пределах Та—ТдС0>03 с перио-
дами решетки от 0,33028 до 0,33050 нм. Д-фаза прости-
рается в пределах ТаС0Л1\до TaCas с периодами
0=0,3101 и с=0,4933 нм до а—0,3106 и с=0,4945 нм.
у-фаза простирается в пределах от ТаС0>74 до ТаС1>00.
Кроме того, обнаружена четвертая 4-фаза состава
*аСо>64, не обладающая областью гомогенности и устой-
чивая при температуре ниже 1800° С.
По данным работы [470], максимальное содержание
связанного углерода в карбиде тантала, приготовленно-
го в интервале температур 1500—2500° С, соответствует
формуле TaC0Qfi. J
Имеющиеся расхождения в данных о концентриро-
ванных границах областей гомогенности В*иу-(Ьаз пои-
к еЩе одному исслеД°ванию системы Та—С [1361
и“'те®ва™ установлено, что гексаго-
нальная p-фаза представляет ткрптп.™ icKUdiu
ния на основа Та г „ ^«ляет твердый раствор вычита-
я основе Та,С и область гомогенности его прости-
рается от ТаСмз ТаС^, что хорошб согласуется с
данными работы £466]. Область гомогенности у-фазы со-
ответствует TaQra до ТаС0>96, -что также хорошо согла-
суется с рабртой {466]. Максимальное содержание свя-
занного углерода ву-фазе соответствует составу ТаС09в2.
Период решетки- карбида тантала, .определялся мно-
гими исследователями [294,. 515, 329, 384, 385, 465, 469,
472, 478,479],
В табл. 20 приведены данные-по определению перио-
да решетки и рентгеновской -плотности карбида тантала
в области гомогенности.
Таблица 20
Периоды решетки и плотность карбидов тантала
Формула соединения Период -решетки, нм Объем элементарной, ячейки v—a\ нм3 Рентгеновская йлотность, г/слН
ТаЦ),58 0,4420 0,08635 11,44s
ТаС0>80 .0,4444 0,08776 14,41ff
ТаСо.эо 0,4455 0,08842- 14,39s
Период решетки возрастает с'увеличением содержа-
ния углерода, причем наблюдается линейнад зависи-
мость. Для ТаС предельного состава данные получены
экстраполяцией, так как достичь такого состава не уда-
лось. Сделано предположение, что наиболее часто встре--
чающееся в литературе значение а=0,4455 нм для кар-
бида состава ТаС в действительности соответствует
составам ТаС0 80 и ТаС0 85, которые наиболее легко полу-
чаются. Плотность монокарбида падает с увеличением
содержания углерода и максимальна для состава
TaCo.ss- работе [355] приводятся значения периодов
решетки карбида тантала состава ТаС0 984 и коэффициен-
Ja линейного РасшиРения ПРИ температурах от 23 до
990 С (табл. 21). Эти данные, вероятно, также относятся
к составу с меньшим содержанием углерода, чем
1 а'-0,984*
Значения коэффициента линейного расширения, при-
и[409™е В Та^л’ 21, п°ДтвеРжДены данными работ [301]
145
Таблица 21
Значения периодов решетки и коэффициентов линейного расширения ТаСо>эв4 при разных температурах
Показатели Температура, “С 994
28 498 590 705 848
а, нм 0,44540± ±0,00003 О,44673± ±0,00003 0,44738± ±0,00004 **—1 0,44827± =3=0,00003
а298'^—8’ • град~1 6,29± ±0,29 6,41 . 6,52± ±0,23 6,61 6,64± ±0,14
Температурная зависимость удельного электрическо-
го сопротивления карбида тантала, полученного взаимо-
действием порошка тантала с Ацетиленом в среде водо-
рода и аргона при температуре 2000° С [133], описывает-
ся уравнением
р= [102 + 0,0178 Т (°К)] мком-см,
справедливым в интервале температур 1400—3500° К.
Вычисленная на основании измерения удельного
электрического сопротивления и постоянной Холла [855]
концентрация носителей тока составляет 2,2 электро-
на/формулу.
Термодинамические свойства карбида тантала раз-
личных составов были определены в работах [468,
469], микротвердость и электросопротивление — [480]
(табл. 22, 23).
Имеющиеся в литературе данные по теплотам обра-
зования карбидов тантала весьма многочисленны [129,
337, 339, 447, 468]. Однако в большей части они относят-
ся к продуктам, состав которых, либо совсем не приво-
дится, либо определен с недостаточной точностью. Наи-
более прецизионный метод определения теплот образо-
вания карбидов тантала в зависимости от состава
использован в работах [129]. Образцы подвергали тща-
тельному рентгеновскому и химическому анализу, при-
чем наряду с основными компонентами определяли со-
держание примесей (О, N, Н, Si, Ti, Fe, Mg, Мп, Са, Си,
Г’ N1’ Al’ M°, W, Pb, Bi, Sb и Cd). Калориметриче-
ским методом определяли теплоты сгорания карбидов
пгыЛ’ ГДе Зна„чение * менялось от 0,455 до 0,988, и на
ании найденных величин с учетом значений теплот
146
Таблица 22
Свойства монокарбида тантала в области гомогенности
Формула соедине- ния Д//(ов Л Stag A F,«, Микротвердость Удельное электросопро- тивление, МКОМ/см.
кдж/моль ккал/ моль дж/ (моль-граду кал/ (моль • град) кдж/моль ккал/моль Мн/м? кГ/м/^
ЬСо.60 —78,4 —18,7 —2,01 —0,48 —77,9 — 18,6 11110 НИ 134,0
ТаСов5 — — -г —- —• — 114,0
TaCojo —90,1 —21,5 —1,80 —0,43 т-89,7 —21,4 126,0
ТаСо,75 — — — <— — 13600 1360 110,8
ТаСо.во —106,8 —25,5 —2,10 —0,50 —106,4 —25,4 — —
ТаС0>85 —. — — — —Ь — 14460 1446 41,2
ТаС0 эд —132,8 —31,7 —3,35 —0,80 —132,0 ^-31,5 — —
— — -,-г- —*- — Ц700 1470 34,0
Таблица 23
Некоторые свойства Ta2Ci—х в области гомогенности
Формула соедине- ния Периоды решетки, нм Плот- ность, г/ли3 А /^298 А "$298 А Лае Мийро- твердость j Удельное электросоп- ротивление, МКОМ'см
а Vе кдж/ МОЛЬ ккал/ МОЛЬ дж/ (град/ моль) кал ерад/ моли 'Л — кдж/ моль ккал/ роль Мн/м‘ кГ/ли^
ТаСо.зв 0,31012 0,49372 14,97 ___ . 1. ч. — II 1
^аС-0,40 — —Ч —. —64,9 —15,5 —3,18 —0,76 —64.1 —15,3 7940 810 138,5
ТаЦ),45 —* —ч —- «аж 1 7 8720 890 136,0
Та^о.бо 0,31042 0,49410 15,050 -71,2 -17,0 —2,89 —0,69' —70,4 i —16,8 J1 ' ” 9280 947 80,25
г Таблица 24
00 Теплоты сгорания и образования карбидов тантала
Формула соедине- ния 'l447l k
Теплота сгорания Теплота образования —й7^298
кдж/кг кал/г кдж/моль ккал/моль 1
Та -5627 —1343 -*“V
ТаС0 485 —5975 —1426 95,9*74 23, 9*1,7 А
ТаС(),724 -6277 —1498 113,5*5,4 27,1*1,
Та С 0,749 -6285 —1500 120,2^:2,1 28,7*0,5
ТаСо,802 -6310 —1506 132,0*5,0 31,5*1,2
ТДС 0,821 —6394 —1526 121,5*4,6 £9,03=1,1
ТаСода —6373 -1521 131,0*2,1 31,3*0,5
ТаСо.до* —6465 —-1543 134,9*2,5 32,2*0,6
ТаС о,93б -6469 —1544 144,1*5,4 34,4±1,3
ТаС о, 958 -6566 -1567 132,0*7,1 31,5*1,7
ТаСо,982 -6557 -1565 146,6*2,5 33,8*0,6
Та С 0,988 -6566 -1567 145,0*5,0 34,6*1,2
I783’ Z
Формул^ соедине* НИЯ Теплота сгорания ‘ Г ' Теплота образования *^#298
‘ кдж/кг кд^м^лб (сйал/яоль
—г ТаС07з4 —1200* *6,3 —286,4* *1,5 114,4*6,3 27 Л* 1,5 ।
TaCgjga -*1208* ^3,8 —288,4* *0,9 125,7*4,2' 30,0^1,6
ТаСо,82б —1227*5 ^9.2, —292,8* *2,2 123,6*9,2 29,5^2,2
ТаС0,9б1 —1268* *3,8 —г —302,7* *0,9 135,3±4,2 •WH 32,3*1,0
Та Cq,982 —1270*: *3,8 —303,3* *0,9 141,2*4,2 | 33,7^1,0
образования Та2О5 и С02 вычислили теплоты образова-
ния (табл. 24), которые хорошо согласуются с данными
работы [447].
Процесс испарения карбида тантала изучали на об-
разце состава ТаС059б в интервале температур 2700—
3100° С [337]. Показано, это при нагревании в вакууме
1,33-10' 3 н/м2 (—Ю-5 мм рт. ст.) В указанном интервале
температур отношение. C/Та уменьшается до величины,
соответствующей карбиду состава ТаС0д7, -после чего
оно не меняется.
В работе J469] определяли также шлифспособность
карбидных фаз тантала в зависимости от их состава,
которую сравнивали со шдифспособностью корунда и
карборунда. .Результаты свидетельствуют о росте шлиф*
способности с ростом содержания углерода в карбидной
фазе TaCj_x и о более.высоком значении ее, чему ко-
рунда.
Исследование шлифспособности фазы ТааС^ пока-
зало, что она ниже, чем у монокарбида тантала; в пре-
делах области гомогенности она- растет с ростом содер-
жания углерода.
Карбид тантала был получен нагреванием смеси пя-
тиокиси тантала с сажей при температурах 1200—
1600° С в атмосфере водорода [25, 187, 471).
Оптимальный режим приготовления карбида тантала
методом восстановления Ta2Og сажей [472] — подъем
температуры до 1600° С в течение 1 ч и время выдержки
при этой температуре 2,5 ч в среде водорода.
Результаты опытов по исследованию условий приго-
товления карбида тантала в вакууме свидетельствуют
о том, что при температурах 1400—1500° С и часовой
выдержке в вакууме образуется продукт, близкий по
составу и периоду решетки к стехиометрическому
ТаС.
Метод приготовления карбида тантала синтезом из
элементов был использован многими исследователями
[287, 465, 466, 472]. В работе [472] опыты проводили в
электрической печи сопротивления с графитовыми нагре-
вателями в среде водорода при температурах 1400—
1600° С. Использовали смесь тантала, полученного вос-
становлением фтортанталата калия металлическим нат-
рием, с сажей расчетного состава.
149
Оптимальный режим — нагрев смеси при температу.
ре 1600° С в течение 2,5 ч при предварительном подъеме
температуры в течение 1 ч.
Кроме того, исследовали условия получения ТаС^
из обрезков танталовой жести, которые загружали ~в
угольный патрон. Патрон помещали в угольную лодочку
и засыпали угольной крупкой. Оптимальный режим —
подъем до температуры 1800° С в течение 40 мин и вы-
держка при этой температуре в течение 3 ч.
Получение карбида тантала осаждением из газовой
фазы [287] вызывает затруднения, связанные с тем, что
при температурах 900—1000° С из смеси, содержащей
пары хлористого тантала^ водорода и углеводородов,
осаждается металлический тантал, образование которо-
го подавляет образование карбида.
В работе [315] прокаливали угольную нить в смеси
пятихлористого тантала и водорода. Пятихлористый тан-
тал у поверхности нити восстанавливался до металличе-
ского тантала, который взаимодействовал с образующи-
мися углеводородами с выделением карбида. При тем-
пературах до 2000° С в продуктах реакции имеется
преимущественно тантал, при 2000—2500° С — карбид
тантала.
Чистый карбид тантада осаждали [52, 54] на вольф-
рамовой нити из пятихлористого тантала. Образующую-
ся при осаждении на нити смесь тантала и карбида на-
гревали в среде углеводородов при температуре 2330—
2930° С, в результате чего был получен однофазный
карбид [477].
Изучали [473] условия, при которых осажденный на
нити карбид тантала устойчив. Так как в газовой среде
содержались углеводороды и водород, лампы, в которых
источником света служил монокарбид тантала, работали
более 50 ч при температуре 3200° С.
При высоких температурах из-за высокой концентра-
ции углеводородов выделяется углерод в виде сво-
бодного, равновесная концентрация углеводородов
уменьшается, что вызывает декарбидизацию карбида
тантала.
Прибавление водорода к углеводородной атмосфере
несколько уменьшает интенсивность выделения свобод-
ного углерода. Введение паров 'брома или хлора снижает
необходимое для избежания декарбидизации количество
150
водороДа от давления 26,66 до 6,66 кн/м2 (от 200 до
50 мм Рт- ст’) ' а
Известны методы получения карбида тантала из тан-
„содержащих шлаков. Эти шдаки подвергаются вос-
яновителъной плавке с большим избытком угля в при-
сутствии железа. Образующийся карбид выделяют хи-
ическим путем. Карбид тантала в смеси с карбидом
йремя прессования, мин
Рис. 53. Зависимость плотности образцов от времени и тем-
пературы горячего* прессования
ниобия получают карбидизацией руд 122], ферротант
Карбид тантала Также получали из м^ллическ
тантала и углерода в жидкой алюминиев и ’й в£н_
гретой в графитовом тигле до 2000 С (бй]. л\ид
ной могут служить и металлы группыi железу Получен^
ные слитки обрабатывали кислотой дляt р
разующихся карбидов алюминия, желе й^ни пресс0-
Исследование влияния температурь, Р полу-
вания и добавок металлов на плотное Р п’оказа-
ченных методом горячего прессования L » » темпе_
ло, что уплотнение карбида происход J оатуры на
Ратуре 1400° С, причем ВЛЙЯНЙ®„е чем времени
плотность образцов значительно выше, 1600—
прессования (рис. 53). В интервале температур
' * 1DI
2300° С происходит быстрое увеличение плотности, кота
рая доходит до 95%. Процесс значительно активируетс_
добавками в смесь до 1 % металлов —< никеля, кобальта*
марганца или железа. ’
Образцы, полученные прессованием порошка карбип»
с добавкой 1% Ni при температуре 1600° С и времен?
выдержки в течение 40 60 мин под давлением 30 Мц/мг
(300 кПсм2), имеют остаточную пористость 2—3%; при
прессовании с добавкой 1% Мп при температуре 1800° С
и времени выдержки под тем же давлением 30—40 мин
остаточная пористость составляет 1—2%.
КАРБИДЫ МЕТАЛЛОВ ПОДГРУППЫ Via
Система хро м—у г л е р о д. Исследованию систе-
мы хром—углерод посвящено много работ [483—492].
Одна? из последних работ, позволившая установить
окончательную диаграмму состояния системы Ст—С,—
работа [492]. Согласно предложенной в ней и принятой
в настоящее время диаграмме (рис. 54), в системе Сг—С
существуют три карбида: СгзСа, Сг7Сз и СгазСб, плавя-
щихся инконгруэнтно.
При быстром охлаждении образуется метастабильная
система Сг—Сг7Сз, при медленном — карбид Сг23С6. Это
свидетельствует о том, что координация атомов хрома
И углерода в расплаве ближе к координации, существую-
щей в Сг7Сз, чем в Сг2зСб- Попытки получить карбид СгС
[492],' н§ существование которого указывалось в рабо-
те [483], не увенчались успехом.
Растворимость углерода в хроме исследовалась в ра-
ботах [37], 493—495].
Во всех работйх подтверждена растворимость угле-
рода в хроме, которая достигает максимума 0,3—0,4% С
при температуре около 1500а С.
Электрические свойства карбидов хрома изучались в
работах [518, 843], где показано, что значения коэфф11*
циента Холла и удельного электросопротивления в кар^
оидах монотонно изменяются с ростом содержания угле
рода. г
Небольшие значения коэффициентов термо-э. Д-с->
ложительные значения температурных коэффии1161*
электросопротивления и сравнительно высокое сопро
152
пение свидетельствуют о том, что все три карбида обла-
дают металлической проводимостью.
А у низшего по содержанию углерода карбида Cr23Cs
проводимость преимущественно дырочная. С увеличени-
Рис. 54. Диаграмма состояния системы
Сг—С [492]
Рис. 55. Зависимость удельного электросопро-
тивления карбидов хрома от температуры
содержания углерода в карбидах характер проводи-
ости меняется и она становится преобладающе элект-
ронной.
Измерение температурного коэффициента электриче-
ского сопротивления [902] показало, что у низшего по
153
содержанию углерода карбида Сг23Св электросопротИв.
ление в интервале температур 350—1200 С практически
не изменяется. С ростом содержания углерода в фазе
зависимость его от температуры увеличивается (рис. 55)
Исследование процессов испарения карбидов хрома
[244, 866] показало, что оно носит ступенчатый характер
и идет по схеме
1ls Сг2з^бтп *“ а/э Сг7 С3тв -|- Сгг;
z/s Сг7С3тв 3/б Сг3С2тв + Сгг;
*/3 Сг3С2тв 2/3 Сг + Сгг.
Термодинамические характеристики карбидов хрома
приведены в работах [23, 173, 316, 368, 496 500].
Наиболее распространенный метод получения карби-
дов хрома — взаимодействие окиси с сажей по реакциям
ЗСгаО3 4- 13С = 2Сг3С2 + 9СО;
7CraO3 + 27С = 2Сг7С3 + 21СО;
23CraO3 + 81С = 2Cra3Ce + 69СО.
Равновесие системы Сг2О3—С—СО—Сг^С^ впервые
изучали в работе [233].
При производстве твердых сплавов на основе Сг3С2
[11, 22] брикеты из шихты, состоящей из 74% Сг2О3
и 26% сажи, нагревают при температуре 1600° С в элект-
рической печи сопротивления в среде водорода. Этот ме-
тод приводи? к образованию продуктов смешанного со-
става, содержащих до 30% низших карбидов.
Изучение условий приготовления карбида Сг3С2 опи-
санным методом [501] показало, что карбид, близкий по
составу к Сг3С2, получается в интервале температур
1400—1600° С При температуре 1400° С образуется кар-
бид, содержащий значительное количество примесей
в виде низших карбидов, при температуре выше 1600 С
карбид разлагается и обезуглероживается.
Процессы .получения карбидов хрома восстановлени-
|22]*КИСИ хРома углеродом изучали в работах [502—507,
Подробное исследование условий и механизма полу
чения однофазного карбида Сг3С2 [505, 822] показало,
что процесс образования карбида начинается при тем-
пературе 1150—1200° С и идет через стадии образовайи
низших карбидов. При температурах 1500—1600° С 0
154
разуется однофазный карбид Сг3С2, практически не со-
держащий свободного углерода. Повышение температу-
ры выше 1600 С приводит к получению продуктов со-
держащих значительные количества низшего карбида
Сг?Сз и свободного углерода. Такое явление разуглеро-
живания при высоких температурах связано с тем что
при увеличении температуры выше 1600° С одновременно
со скоростью образования карбида возрастает скорость
графитизации сажи, *в результате чего упругость паров
ацетилена при данной температуре становится ниже рав-
новесной, соответствующей расчетному содержанию свя-
занного в Сг3Сг углерода. Происходит частичное раэ-
углероживание и превращение части Сг3С2 в карбид
СГ7С3 с более низким содержанием углерода.
Исследование влияния температурь^, временй восста-
новления и состава шихты позволило уточнить режим
получения однофазного карбида Сг3С2, который сводит-
ся к тому, что брикеты из шихты расчетного состава с
добавкой 5% раствора декстрина нагревают в печи со-
противления с графитовыми нагревателями при посте-
пенном подъеме температуры до 1500°Q в течение 30—
40 мин и выдержке при этой температуре 1,5—2 ч.
Исследование условий получения карбида Сг7С3 [505,
506, 822] восстановлением окиси хрома сажей по реакции
7Сг2О3 + 27С == 2Сг7С3 + 2 ICO
показало, что оптимальная температура 1200—1300° С
и состав шихты, содержащей 97—98% сажи от расчет-
ного состава.
Целесообразно получать карбид СГ7С3 в две стадии.
Брикеты шихты нагревают при температуре 1200 >
1300° С в течение 1—1,5 ч, затем измельчают и снова
нагревают при той же температуре в течение 20—30 мин.
Получение однофазного низшего по содержанию уг-
лерода карбида Сг23Сб [507] восстановлением Сг2Од угле-
родом затруднено, так как оно тормозится образованием
термодинамически более устойчивых высших карбидов.
Исследования кинетики и механизма процессов вос-
становления окиси хрома в вакууме [41, 508, 865] пока-
зали, что процесс происходит за счет выделяющейся оки-
си углерода и носит автокаталитический характер — ско-
рость восстановления в начальный период быстро растет,
Достигает максимума и медленно уменьшается. На осно-
155
вании этого сделан вывод, что в начальный период вос-
становления окиси хрома лимитирующей стадией являет-
ся кристаллохимическая перестройка окисла в металли-
ческий хром или карбид. Затем растет роль газификации
углерода, и реакция чувствительна ко всем параметрам
влияющим на скорость регенерации окиси углерода’
К концу процесса восстановления, когда поверхность
реакционной зоны уменьшается, роль этого фактора сно-
ва падает.
В работе [497] были получены все три карбида хрома
взаимодействием окиси хрома с сажей в вакууме. Кар-
бид СГ3С2 получали нагреванием шихты Сг2О3 с сажей
в молярном соотношении 3: 13 при температуре 1150—
1200° С, Сг7С3 — нагреванием шихты Сг2О3-ЬС в моляр-
ном соотношении 7 : 27 при 1200—1250° С и Сг23С6 нагре-
ванием СггО3 с сажей в молярном соотношении 2 : 7 при
температуре 1250—1300°
Более поздние исследования условий получения кар-
бидов хрома в вакууме показали, что карбид Сг3С2,
близкий по составу к расчетному, образуется при темпе-
ратуре 1400—1500° С, при температуре 1150—1200° С об-
разуются продукты, содержащие смесь всех карбидов и
окиси хрома.
Получение карбидов хрома в вакууме требует исполь-
зования более сложной аппаратуры, чем получение их
в среде водорода, и более длйтельно, а потому и нецеле-
сообразно для промышленного использования.
Синтезом из элементов карбиды хрома были получе-
ны в работах [25, 483, 487, 488].
Получить чистый однофазный карбид Сг23Сб синтезом
из элементов так же, как и восстановлением окиси хрома
сажей, не удалось. Этот карбид, близкий по составу к
расчетному и не содержащий высших по содержанию уг-
лерода карбидов и окислов, получен при взаимодействии
высшего карбида с окисью хрома по реакции
?200°3 ССГзС2 Сг2зСб+СО в вакууме при температуре
В работе [50] карбид Сг3С2 получен науглероживани-
ем хрома метаном в присутствии водорода при темпера-
туре 600—800° С, согласно реакции
Сг + Н, + СН, Сг3С, + Н, + (СН),
где символом (СН) обозначены продукты разложения
углеводородов,
15S
Очень твердые и износостойкие покрытия на поверх-
ности железа, состоящие в основном из Сг3С2, получены
разложением карбонила хрома в присутствии водорода
при температурах от 250 до 850° С [509].
г В работе [510] получали карбидохромовые покрытия,
для чего образцы армко-железа, стали 18ХНМА и стали
ХГ подвергали электролитическому хромированию в ван-
не, содержащей 150 а СгОз и 1,5 г H2SO4 на 1 л воды
при плотности тока 35 а/дж2, температуре электролита
50° С и продолжительности электролиза 3 ч. Хромиро-
ванные образцы подвергали карбидизации в вертикаль-
ной трубчатой печи при пропускании смеси паров бензи-
на и водорода при температурах 950 и 1050° С в течение
3 и 8 ч.
Покрытия состоят из наружного тонкого слоя карби-
да СгзСг, среднего толстого слоя карбида Сг7С3 и срав-
нительно тонкого слоя карбида Сг23Сб. Под этим слоем
находится слой остаточного непрокарбидизированного
хрома.
Позже исследовали условия получения и свойства
покрытий [511], образующихся при газовой карбидизации
электролитического хрома, осажденного на стали в токе
смеси паров бензина с азотом.
Металлографический и рентгеноструктурный фазовый
анализы подтвердили наличие трех фаз, расположенных
слоями: внешний тонкий слой высшего карбида Сг3С2,
средний толстый слой карбидй Cr7C^ и внутренний тон-
кий слой нитрида Cr2N.
Реакционная диффузия в системе Cr—N—С прохо-
дит как диффузия атомов азота и углерода через обра-
зующийся слой к металлу. Так как атомный радиус азо-
та меньше, чем углерода, диффузионная подвижность его
атомов более высокая, чем атомов углерода, и он прони-
кает в глубь металла с большей скоростью, чем углерод.
Износостойкий слой карбида хрома наносили на по-
верхность стальных изделий осаждением хрома из газо-
образных соединений с последующим взаимодействием
его с углеродом, находящимся в поверхностном слое из-
делия.
Многочисленные исследования посвящены выделению
карбидов из ферросплавов и сталей, которое основано на
разной растворимости карбидов и металлических состав-
ляющих. Найдено [513], что при электролитическом рас-
157
творении хромистой стали в соляной кислоте плотностью
1,02 нерастворимый остаток состоит из смеси карбидов
железа и хрома. *
Из хромистых сталей были выделены карбиды Cr С
и Сг23Св [515], из хромомарганцевых - Сг,С, [516] и
моникелевых — Сг23С6 IP 17J.
Все эти методы, представляя значительный теоретиче.
ский интерес, не применяются в промышленных масщ'.
табах.
Исследование условии приготовления плотных образ-
цов методом горячего прессования [130] позволило полу-
чить образцы плотностью 99—^99,5% при температуре
1750° С и удельном давлении прессования 1500 Мн/м2
(150 кГ/см*). При этом содержание низшего по содержа-
нию углерода карбида Сг7С3 растет от 5% в исходном по-
рошке до 15%.
Система молибден — углерод. Соединение,
содержащее 5,8 % С, впервые было получено в работе
[522] и ему была приписана формула Мо2С. Позже на
основании данных рентгеновского анализа сплавов
Mo—С [522] было показано, что при содержании около
30% (ат.) С образуется твердый раствор углерода в мо-
либдене с плотноупакованной гексагональной решеткой.
С увеличением содержания от 30 до 39% (ат.) С период
решетки а увеличивается от 0,2993 до 0,3004 нм.
Более поздние работы, однако, опровергают это за-
ключение. По данным ряда авторов [523—525], раствори-
мость углерода в молибдене незначительна.
На основании металлографического исследования при-
водятся следующие данные о растворимости углерода в
молибдене [525]:
Температура, °C ... . 1650 1925 2200
Растворимость, % {по
массе) 0,005—0,009 0,012—0,013 0,018—0,022
Это позволяет заключить, что Мо2С — химическое сое-
Область гомогенности при температурах 1400
2200° С от 5,4 до 5,9 % С.
работе [526] при обработке металлического молиб-
дена смесью СО и Н2 получили продукт с г.ц.к. решеткой
периодом 0,414 нм, содержащий от 30 до 35% (ат.) С.
?Т?Му к?РбидУ приписали формулу Мо2С, предположив,
им образом, существование низкотемпературной куби-
158
ческой модификации и высокотемпературной гексаго-
нальной.
При исследовании кристаллической структуры карби-
дов молибдена [11] на рентгенограммах образцов М02С,
нагретых до температуры выше 1400° С, обнаружены но-
вые линии. Сделано предположение, что при высоких тем-
пературах образуется новая фаза Р -М02С, которая имеет
ту же кристаллическую структуру, что низкотемператур-
ная фаза а-МогС, с тем же значением периода а и увели-
ченным периодом с.
Однако существование описанных высокотемператур-
ных модификаций Мо2С не было подтверждено дальней-
шими исследованиями.
При сплавлении молибдена с алюминием и углеродом
в угольном тигле при температуре кипения алюминия
после охлаждения получили, кроме карбида алюминия и
графита, кристаллы соединения, содержащего 11,1 %С и
соответствующего формуле МоС [527]. На существование
этого соединения указывается также в работах [189,
528—530]. Показано [530], что МоС может стабилизиро-
ваться в твердых растворах WC—МоС и NbC—МоС, но
не может быть получен синтезом из элементов или взаи-
модействием молибдена с углеродсодержащими газами.
В работе [51] исследовали возможность- получения
карбидов молибдена взаимодействием металла с мета-
ном. При этом, кроме карбида М02С, получали более
богатый углеродом карбид МоС, который метастабилен
и при высоких температурах разлагался на Мо2С и угле-
род. Данные о структуре монокарбида в работе не при-
водятся.
При рентгенографическом исследовании [531] образ-
цов карбидов молибдена (содержащих около 7,8% свя-
занного углерода) эти образцы представляли смесь из
60% Мо2С и 40% МоС. Кроме того, на рентгенограммах
были обнаружены некоторые линии г.ц.к. решетки с пе-
риодом 0,419 нм, расшифровать которые авторам не уда-
лось.
. При исследовании сплавов молибдена с углеродом
[532] были обнаружены три фазы: а-фаза — твердый рас-
твор углерода в молибдене (предельная растворимость
пРи температуре 1500—2000°С 0,09%), ₽-фаза с содер-
жанием при температуре 1400—2200°С 5,4—6,0%С и
Y-фаза, образующаяся при температуре 2100° С и содер-
159
3000
2660
2600
fz200 У-
Ъ/800
с:
5
,+м
2550 2660 А*™
26)0 А 2600
’ l2S0u^^>&10
М00
I 2200
I
। мо+могс
I
М00 1
I
I
1
I
I
мас*с
ЗС
MDzC+C
WOO
1
БОО
МО МОгС МоС
Рис. 56. Фазовая диаграмма состо-
яния системы Мо—С
L
жащая 12,3— 13,0%С. Однако эта фаза, по данным рент
геноструктурного анализа, несколько отличается от най'
денного раньше монокарбида МоС.
а-фаза образует с 0 -фазой эвтектику при темпера
туре 2200° С, содержащую 1,5 % С. р
Измерения температур плавления сплавов, содержа-
щих от 40 до 75% (ат.) С [533], позволили несколько уточ-
нить диаграмму. Найдена эвтектическая точка МоС__С
соответствующая 700/’
(ат.) [22,5% (по массе)0]
С при температуре 2400° С
(рис. 56).
Более поздние иссле-
дования [534] подтверди-
ли существование двух
карбидных фаз в системе
Mo —С: 0-фаза —Мо2С и
у-фаза — МоС, а также
новой у’-фазы, образую-
щейся при соединении
этих двух фаз.
Науглероживание мо-
либдена при низких тем-
пературах приводит к об-
разованию после 0-фазы
у'-фазьк При нагревании
у'-фазы выше температу-
ры 800° С в атмосфере СО или в вакууме она превраща-
ется в у-фазу, которая при высоких температурах устой-
чивее у'-фазы. При низких температурах у-фаза менее
устойчива и скорость превращения у'-фазы в у-фазу
очень мала, у'-фаза имеет гексагональную решетку, со-
держащую четыре атома молибдена в элементарной
ячейке, которая аналогична ячейке W2B5.
В табл. 25 даны основные рентгенографические пара-
метры всех трех фаз, которые хорошо совпадают с дан-
ными, приведенными в работах [522, 523, 528, 548, 549].
По расположению атомов молибденау'-фаза —проме-
жуточная между 0- и у-фазами. Она выражается также
формулой МоС. Имеются данные о существовании а-Мо
с г.ц.к. решеткой и периодом а = 0,427 нм [398].
Исследование системы V—Mo—С [535] методами рент-
генографического и металлографического анализов пока
160
Таблица 25
Рентгенографические параметры фаз в системе Mo—С
Параметры Фаза
3 V т
а,нм. 0,3002 0,2932 0,2898
с, нм 0,4724 1,097 0,2809
с а Число атомов Мо в элементарной 1,574 3,742 0,969
ячейке 2 4 4
Объем ячейки на атом Мо, ял3 . , 0,01844 0,02042 0,02042
зало, что в системе Мо—С имеются фазы Мо2С и т1-Мо3С2,
кристаллизующиеся в гексагональной решетке. При рез-
ком охлаждении получен карбид а-Мо3С2, кристаллизую-
щийся в кубической решетке с периодом а=0,4281 нм^
Эта фаза устойчива при температуре 1800° С. При мед-
ленном охлаждении и отжиге она переходит в т}-фазу и
распадается на Мо2С и С. Введением в сплавы МоС до
2% В и 1%U или Thri-фаза может быть стабилизиро-
вана.
Предположение о существовании карбида т) -МозС2
подтверждено работами [536] и [537].
Карбид Мо2С (расчетное содержание углерода 5,91 %)
представляет собой металлический порошок темно-серо-
го цвета.
На основании рентгенографического исследования
[553] вычислены термические коэффициенты линейного
расширения карбида Мо2С в направлении оси а и г и ха-
рактеристические температуры 0Д и 0С Показано, что
в интервале температур 17—270° С а(7>ааи аа
= (6,9±0,6) • Ю-6 град-1 и ае = (8,9±0,4) • 10~в град~1.
Различия в характеристических температурах не замеча-
лось = (гпОс) = (21,3±4,0)-Ю~18 г-град2.
Термодинамические свойства карбида Мо2С приведе-
ны в работах [60, 65, 135, 550, 551].
Исследовали [298] процесс восстановления молибде-
нового ангидрида углеродом в атмосфере водорода в при-
сутствии галоидоводородных соединений в качестве ка-
тализаторов.
Результаты опыта, представленные в табл. 26, свиде-
тельствуют о том, что в присутствии галоидоводородных
соединений образуются продукты, близкие по составу к
11 Косолапова Т. Я. 161
Таблица 26
Восстановление МоО3 сажей в различных газовых средах (температура 950° С, время нагрева 0,5 ч)
Содержание ’ %
Среда Чэбщ ^своб С * связ реакцИи %
Вакуум ‘ • 5,6 1,2 4,45 78,5
Водород+1,3% пропана . -. • 5,8 0,73 5,17 87,5
Водород+хлористый водород (6:1,3)+ 1,3% пропана 6,2 0,46 5,77 97,5
То же, е бромистым водородом . », 6,0 0,3 5,72 97,0
То же, е йодистым водородом . . . 5,9 0,15 5,75 98 0
Сгпя, вычислено как
Србщ ^своб
100-Ссвоб
100.
расчетному Мо2С, и процесс восстановления проходит
практически полностью.
Карбид Мо2С получен также восстановлением молиб-
денового блеска, молибдата кальция карбидом каль-
ция [485].
Подробное исследование механизма взаимодействия
молибденового ангидрида с сажей проводили в работе
[541]. Установлено* что при восстановлении молибдено-
вого ангидрида сажей в атмосфере аргона или СО про-
цесс проходит Церез следующие стадии.
1. Восстановление МоО3 до МоО2 при температуре
600—700° С:
2МоО3 4- С — 2МоО2 + СО2.
Восстановление МоО2 до молибдена при темпера-
туре 900—1000° С:
МоО2 4- С=Мо + СО2;
МоО2 + 2СО = Мо + 2СОа;
СО2 + С 2СО.
3. Образование Мо2С по реакции
2Мо+С^ЛЬ2С.
162
Как показано, промежуточные о^ислы между Мо03
и МоО2 и Мо не образуются.
Промышленное производству карбида молибдена [22]
заключается в восстановлении молибденового ангидрида
водородом при температуре 900° С с последующим по-
лучением карбида синтезом из ^металлического порошка
с углеродом в среде водорода дри температуре 1400—
1500° С.
Карбид молибдена впервые получили синтезом из
элементов в работах [25, 527].
Довольно широко распространены методы осаждения
карбида молибдена из газовой фазы. Mo2G получали
[543] науглероживанием порошка модибдена с помощью
СО при температурах 600—1000° С.
Изучение равновесия реакций, 2Мо + СН4=Мо2С + 2Н2
[51, 544] показало, что при температуре 700—850° С
происходит количественное 'разложение метана с об-
разованием карбида Мо2С, содержащего 5,8—
6,0% С.
Карбид молибдена также был получен нагреванием
молибденовой проволойи пропусканием тока в атмосфе-
ре нафталина [545] при использовании смесц паров кар-
бонила молибдена, которые разлагаются при темпера-
турах 300—800° С и давлении 13,33—39,99 (0,1—
3 мм рт. ст.) с водородом, Прц этом в зависимости от
условий проведения опыта получали Мо2С„ или смесь
Мо2С, МоС и Мо.
Значительный интерес для получения *карбидов мо-
либдена представляет электролиз расплавленных солец.
В работах [63, 546] обнаружено, что при электролизе
смесей расплавленных карбоната натрия, метабората
натрия, фторида лития и трехокисй молибдена в разных
соотношениях при температуре 700° С на графитовом
электроде осаждаются карбиды Мо2С, МоС или смесь
обоих карбидов.
Определяющее условие при этом — соотношение
МоОз и СО2. При МоОз: СО2 от 1: 5,5 до 1 г 7 выделяется
карбид Мо2С, содержащий 5,9% С и 94,0% Мо.
При использовании состава ванны с соотношением
М0О3 и СО2 от 1,21 до 1,28 выделяется монокарбид МоС,
содержащий 11,5% С и 87,5% Мо. При соотношении
МоОз и СО2 от 1,5 до 1: 18 можно выделять смесь обоих
карбидов.
11*
163
Пои электролитическом разложении молибдене^
подистых сплавов в разбавленной соляной кислоте (£е
мость 194) при плотности тока от 0,3 до 0,4 a/S°T
осадке остается Мо2С [523]. Кроме того, Мо2С можнп
выделить электролитически из расплавов Ni3Mo°C
Со3Мо3С — Fe3Mo3C, которые мало устойчивы н пГ
охлаждении распадаются с образованием карбида МоС
[547]. „
Система вольфрам углерод. При исслелп
вании системы W — С методами химического и металло'
графического анализов [24] в сплавах, кроме W2C и WC
предположен карбид, соответствующий формуле W3c'
существование которого в дальнейшем не подтвердилось'
В результате рентгеновского анализа и изучения фи^
зических свойств сплавов W — С [430] обнаружена но-
вая фаза, структура которой похожа на структуру w2C.
Эта фаза рассматривается как полнгморфная модифика-
ция p-W2C. Она нестабильна и легко переходит в a-W2C
Таким образом, многочисленные исследования систе-
мы W—С [51, 294, 306, 554, 859, с 250] привели к заклю-
чению, что в этой системе существуют два карбида: W2C
с гексагональной плотноупакованной решеткой, и моно-
карбид WC с простой гексагональной решеткой [259,263,
522, 571]. Монокарбид — наиболее устойчивое соедине-
ние в системе W — С [51]. Максимальная растворимость
углерода в вольфраме [306] составляет 0,3% (ат-) ПРИ
температуре 2400° С и с понижением температуры быст-
ро падает,* составляя 0,05% при 2000° С и следы при бо-
лее низкой температуре.
Твердый раствор, богатый вольфрамом, и низший по
содержанию углерода карбид W2C образуют эвтектику,
н^^1(НС° 2’0% (по массе) С с температурой плавле-
При содержании 3,5% (по массе) С образуется эвтек-
тика W2C WC, плавящаяся при 2760° С.
й wr ТОго> обнаружена вторая модификация карби-
да p-WC, кристаллизующаяся в кубической Решетке п
устойчивая при температурах выше 2525° С [859, с. 25 г
н_ а основании всех имеющихся исследований n0CT£?.ei
ппЛиаграмма состояния системы W—С [859, с. 2514
представленная на рис. 57.
вычисленмЬТ1Те Рентгенографических исследований [
коэффициенты термического расшире
164
вдоль осей а и с и характеристические температуры. По-
казано, что в области температур 22—400° С для моно-
карбида вольфрама, у которого отношение с/а близко к
I, т&1 = (32,4±4,6) • 10~18 град, коэффициенты
линейного расширения ав=3,84*10~® град-1, ас^
3100
3000
2900
у- Z800
2700
?
£ 2600
Содержание С,°/0(по массе)
1 2 3^ 5 6 7 8
0 10 20 30 k0 50 60 90 100
Содержание С, % (ат)
2500
ZWO
Рис. 57. Диаграмма состояния системы W—С [860]
= 3,90*10“® град-1-, для W2C в интервале температур
12—400°С т02 = 29,6 + 6 epad*10“18; mO* =19,3+
±2,9 град*10“18; аа « (6,4±0,16) 10_6 град-1; «с =
= (8,1 ±0,22) • 10~® град-1.
Исследовали [575] влияние высоких давлений и тем-
ператур на свойства карбида вольфрама. Образцы были
получены прессованием порошка WC, содержащего
6,08% С, и спеканием в атмосфере водорода при темпе-
ратуре 2100° С. Спеченные образцы подвергали всесто-
роннему сжатию до 10130 Мн)м2 (100 000 ат) с одно-
временным нагревом до 2100° С. Часть образцов отжи-
гали при температуре 1500° С в течение 1,5 ч.
165
Микротвердость образцов, подвергнутых всесторонне.
mv сжатию, составляет для недожженных 27 400
?1У360 Мн/м2 (2800—3200 кГ!мм2)\ после отжига она
уменьшается до значения, соответствующего образцам
спеченные обычным методом без сжатия.
При изучении адсорбции, миграции и испарения ба-
рия на карбидизированном вольфраме [576] найдено
значение работы выхода W2C, покрытого барием, для по
крытия, соответствующего максимуму автоэлектронного
тока, равное 3,6-10"1а дж (2,25 эв).
Термодинамические свойства карбидов вольфрама
описаны в работах [60, 135, 542, 573, 574].
Карбид вольфрама вначале получали плавлением
вольфрама в дуговой электрической печи [24, 49, 292,
430]. Указанный метод, позволяющий получать низший
по содержанию углерода карбид W2C, близкий по соста-
ву к расчетному, имеет большое промышленное значение
Известны два основных способа приготовления ли-
тых сплавов карбидов вольфрама [556].
Первый заключается в том, что вольфрам, получен-
ный восстановлением вольфрамового ангидрида углеро-
дом, науглероживают и плавят в электрической печи со-
противления с графитовыми нагревателями при темпе-
ратуре 2700° С. Расплав заливают в графитовую форму
при опрокидывании печи.
Второй способ заключается в том, что вольфрам, по-
лученный восстановлением вольфрамового ангидрида во-
дородом, науглероживают и плавят в графитовом тигле
при температуре 3000° С. Расплав отливают с помощью
центробежного устройства в специальные формы.
Высший карбид WC точного состава получить этим
методом нельзя вследствие его перитектического распа-
да на W2C и графит.
Наиболее распространенный метод получения карби-
дов вольфрама в настоящее время — синтез из элемен-
тов в Среде водорода.
наиболее подробно реакции W+C=WC и
=W2С при различных условиях исследованы в
работах [49, 430, 537].
В среде водорода, где образуются углеводороды, они
создают Науглероживающую среду, способствующую по-
лучению карбидов. Это подтверждено более поздними
исследованиями [285], которые показали, что в среде во-
166
дорода в результате взаимодействия сажи и стенок тру-
бы и лодочки с водородом образуется ацетилен. Упру-
гость паров ацетилена над частицами вольфрама ниже,
чем над частицами сажи, вследствие чего атомы угле-
рода переносятся с частиц сажи на частицы вольфрама.
В атмосфере окиси углерода перенос через газовую фазу
происходит по реакции С+СО2 = 2СО, равновесие кото-
рой при температурах выше 800° С полностью сдвигает-
ся в сторону образования СО.
Исследование процесса взаимодействия порошка ме-
таллического вольфрама со смесью окиси углерода и во-
дорода в интервале температур 1000—1100° С при от-
ношениях СО: Н2 от 0,5 до 1 [834] показало, что при этом
образуется карбид вольфрама WC; но полного превра-»
щения вольфрама в WC достичь не удается.
В результате исследования параметров реакционной
диффузии углерода в вольфрам [382] получено значе-
ние энергии активации 14,24 кдж[моль /34000 кал!моль}
17000
и D=l,6- 10-4е т .
В работе [558] подробно исследовали механизм про-
цесса науглероживания вольфрама из твердой и газовой
фаз. В результате были получены образцы, диффузион-
ный слой которых состоял из двух, частей: внешней WC
и внутренней W2C.
Исследование реакционной диффузии углерода в
вольфрам в атмосфере смеси паров бензина с водородом
до температуры 1200° С [559] показало, что заметное вза-
имодействие вольфрама с углеродом начинается при тем-
пературе 1000° С и процесс подчиняется параболическо-
му закону.
Металлографический и рентгеновский анализы образ-
цов подтверждают наличие в диффузионной зоне двух
фаз — внешней WC и внутренней W2C. Изучение реак-
ционной диффузии в тройной системе W—N — С. пока-
зало, что этот процесс также подчиняется параболиче-
скому закону. Диффузионный слой по составу аналоги-
чен слою, образующемуся при диффузии углерода в
вольфрам. Азот замедляет процесс диффузии углерода
в вольфрам. По-видимому, карбиды, образующиеся в
диффузионной зоне, содержат некоторое количество рас-
творенного азота и диффузия углерода через карбидные
фазы, содержащие растворенный азот, затруднена. Об-
167
пазование в данном случае фаз с кристаллическими Ре.
шетками карбидов свидетельствует о том, что при тем-
пературах опытов термодинамическая устойчивость кар-
бидов выше, чем нитридов.
Метод получения монокарбида вольфрама взаимо-
действием порошка вольфрама с сажей в среде водоро.
да широко используют в твердосплавной промышлен-
ности. Процесс ведут в индукционных печах сопротивле-
ния с графитовыми нагревателями. Предложена печь
для карбидизации вольфрама [560] с алундовыми муфе-
лями прямоугольной формы ии молибденовыми нагрева-
телями с рабочей температурой 1450° С. Эта печь харак-
теризуется высокой стабильностью теплового режима,
что обеспечивает высокое качество готового продукта’
Порошок металлического вольфрама получают из
вольфрамового ангидрида, вольфрамовой кислоты или
паравольфрамата аммония [651]. Порошок вольфрама
смешивают с 6,3—6,8% сажи, полученную смесь в виде
порошка или брикетов нагревают при температуре 1400—
1600° С [22].
Проводилось [597] рентгеновское и электронографи-
ческое исследование порошков карбида, синтезированно-
го из вольфрама,, полученного восстановлением вольфра-
мовой кислоты и паравольфрамата. В первом случае по-
рошок карбида более мелкодисперсен, чем во втором.
Однако после 100-ч помола в обоих случаях размер час-
тиц составляет в среднем 0,073 мкм при искажении крис-
таллической решетки 0,16%. Увеличение времени размо-
ла до 300 ч практически не изменяет размера частиц,
искажение кристаллической решетки достигает 5%.
Низший карбид W2C в отличие от всех остальных
тугоплавких соединений получают в промышленных
масштабах плавлением. Смесь технически чистого воль-
фрама с 3% сажи и железа в виде брикетов расплавля-
ют при температуре 3000—3250° С в графитотрубчатых
печах особой конструкции или в высокочастотных печах.
Исследовали [562] условия получения карбида воль-
фрама при непосредственном восстановлении вольфра-
мового ангидрида сажей. Процесс идет эффективно в
среде водорода; при этом содержание углерода в шихте
ТЛВ„ЛЯеТ 12,5% вместо 20,6%, согласно расчету, что
° с Учас™ем в восстановлении ацетилена, обра-
зующегося в реакционной зоне.
168
Влияние примесей различных галоидоводородов на
процесс взаимодействия вольфрамового ангидрида с уг-
леродом (табл. 27) исследовано в работе [298]. Прибав-
ление к газовой смеси галоидоводородных соединений
значительно увеличивает содержание связанного угле-
рода и степень полноты реакции и уменьшает содержа-
ние свободного углерода. Наиболее эффективна при-
месь — йодистый водород.
Таблица 27
Влияние состава газовой атмосферы на состав карбида вольфрама
(соотношение WO3: С в шихте 3,1:1. Температура 1100° С,
время нагрева 1 ч]
А тмосфера Содержание, % Степень полноты реакции %
%бщ С своб С « 1 СВЯЗ I
Водород+0,5 % пропана 6,0 1,85 4,23 69,2
Водород+НС1 на .... (6; 1,3) +0,5% пропа- 6,3 0,66 5,69 92,0
Водород+НВг на ... . (6:1,3) +0,5%. пропа- 6,2 0,45 5,77 92,5
Водород+Ш (6 : 1,3)+0,5% пропана 6,05 0,1 5,95 98,0
Вакуум , . • « W ц • £ » -• 5,8 2,3 3,58 60,4
* г> ^общ ^своб • 100.
~свяа~" 100-сСВ0б 1
Изучали [563] процесс разуглероживания карбида
вольфрама влажным водородом в интервале температур
800—1000° С. Согласно термодинамическим расчетам,
изменение свободной энергии реакции разуглерожива-
ния карбида вольфрама (табл. 28) при использовании
сухого водорода имеет отрицательное значение до тем-
пературы 227° С. Следовательно, при температурах об-
разования карбида сухой водород не может его разугле-
роживать. При использовании влажного водорода раз-
углероживание за счет влаги идет при температурах
выше 1230° С, т. е, при температурах образования кар-
бида.
Исследования показали, что при работе с влажным
водородом наблюдается резкий скачок в скорости реак-
ции разуглероживания при температуре 950° С (рис. 58).
12 Косолапова T. Я.
169
Таблица 28
- свободной энергии реакции разуглерожиаани.
Значения убыли своо д
карбида WC___________-------------- —
Реакция "" дт? ’ —•
27° С 227° С 730° С 1230°^с '
кдж/ моль ккал/ моль кдж/ МОЛЬ ккал] моль кдж/ МОЛЬ ккал) МОЛЬ кдж/ моль ккал/ моль
WC + 2H? = —17.6 — 4,2 — 1,50 — 0,36 +48,8 +11,65 +107,3 +25
= № + <% +82,1 +19.6 +95,1 +22,7 +21,4 + 5.1. — 50,7 12.
Время.ч
Рис. 58. Зависимость скорости разуглероживания кар-
бида вольфрама от времени и температуры
ДЬ1, происходит °зн’ачитА В водороде содержатся пары во-
Раз^лероживание кз^ида06’ растущее с температурой
вого ангидоиТ^1^'1аНИзМг^Восстановления вольфрамо-
ление 1564^ найд^ что восстанов-
стадии образовяииа СИТ от температуры и идет через
Процесс восстановлр»цНИЗШИХ окислов и а-вольфрама
,7П я в челом идет по схеме
I/O
WO^-WQh
WOsr + C,, W„ + cop + GOir;
W„ + WO, -+ wo, •
ТВ 1 Зг 2тв иг»
WOar 4- сгв - WTB + COr;
COa4-C->CO;
WTB + Gra -> WC,..
Известен также метод получения, карбидов вольфра-
ма восстановлением вольфрамата углеродом при тем-
пературе 1400° С [565]. Полученные продукты содержат
смесь WC и W2Ci_x .
Карбиды вольфрама получают и осаждением из га-
зовой фазы. Наиболее распространено получение карби-
дов из карбонила вольфрама.
Так, карбид W2Ci-x получали [526] разложением па-
ра карбонила. Разлагая карбонил при температуре
1000° С [566], получали монокарбид WC,. Карбиды полу-
чали [50] нагреванием смеси карбонила вольфрама с во-
дородом при температурах 300—800° С.
Для получения карбидов карбонил вольфрама под-
вергали [568] термической диссоциации при температу-
рах 650—1000° С. Затем продукты диссоциации нагре-
вали при температурах 1800—1850° С в атмосфере СО.
Кроме того, карбиды вольфрама были получены на-
греванием смеси вольфрамовой кислоты, вольфрамово-
го ангидрида или паравольфрамата аммония с водоро-
дом и метаном при температурах 850—1000° С [569].
При изучении автоэлектронной эмиссии обнаружено
[570] образование карбида вольфрама W2Ci_* цри
взаимодействии вольфрама с углеродом, образующимся
в результате распада молекул углеводорода.
Получение карбидов вольфрама электролизом рас-
плавленных солей было подробно исследовано в рабо-
те [63]. Электролизу подвергали смесь расплавленных
бората натрия, фторида лития и вольфрамового ангид-
рида. Характер образующегося карбида определялся в
основном отношением количества СО2 к WO3 в электро-
лите. При отношении СО2: WO3 от 3,3 до 6 на катоде
осаждается W2Ci—*, при CO2iWOs от 12 до 16—WC;
при CO2:WO3 более 16 карбид не образуется.
12*
171
КАРБИДЫ МЕТАЛЛОВ
ПОДГРУППЫ V»a
Система марганец — углерод. В результат*
изучения системы Мп С была предложена [42, 85, 5771
диаграмму состояния, согласно которой в рассматривае-
мой системе обнаружен один карбид состава МпзС Он
образует с марганцем непрерывный ряд твердых раство
ров в широком интервале температур. Дальнейшие
исследования [578, 579] показали, что этот карбид поли-
морфен и существует в двух формах: а-и Р-Мп3С, из них
а-модификация изоморфна цементиту и образует с ним
непрерывный ряд твердых растворов; ₽-модификация
представляет высокотемпературную форму. Превраще-
ние а в р происходит при температуре 2230° С. Однако
в работе [580] показано, что Мп3С существует лишь в
области температур 950—1050° С, и возможно, что изме-
нение свойств карбида при температурах 1050—1100° С
обусловлено не полиморфным превращением, а разложе-
нием его.
Кроме этого карбида, в системе Мп — С обнаружены
карбиды Мп7С3 и Мп4С, аналогичные соответствующим
карбидам хрома, а также Мп7С2 и МП5С2 [580—584].
Карбид Мп7Сз (8,56% С) изоморфен карбиду хрома
Сг7С3. Атомы углерода в карбиде Мп7Сз связаны между
собой в виде зигзагообразных цепей, параллельных оси
2. Существует, однако, предположение, что Мп7С3 имеет
ромбическую решетку, соответствующую пространствен-
ной группе D^h. Рппга, с периодами а = 0,4546, 6 = 0,6959
и с= 1,1979 нм [857], которое еще недостаточно подтверж-
дено.
Согласно рентгенографическим исследованиям [582],
состав карбида Мп4С более точно выражается формулой
Мп23Сб. Он изоморфен с карбидом Сг2зСб.
Карбид МпдС2 устойчив в широкой области темпера-
тур (до 1050° С) й структурно тождествен с двойным^кар-
бидом (Мп, Fe)5 С2. Карбид Мп7С2 устойчив в узкой об-
ласти температур: 850—1000° С, структура его неиз?гост
на и формула ориентировочна [580]. Предложена [58oJ
диаграмма состояния системы Мп — С, представленная
на рис. 59.
Более поздние исследования системы Мп -— С [782,
856] позволили установить, что содержание углерода в
172
данной фазе составляет 5,48%; кристаллизуется он в
гексагональной решетке с периодами а=0,7492 нм, с~
= 1,2070 нм, с/а= 1,61; плотность 7,30±0,05 г(см\ Все это
приводит к заключению о существовании карбидной фа-
Со держание С, % (по массе)
I 2 3 Ь 5 Б 7 & 9 10
Рис. 59. Диаграмма состояния системы Мп—С
[585]
зы, состав которой выражается формулой МП15С4, а
не Мп7С2.
Наиболее подробно диаграмма состояния системы
Мп — С изучена в работах [584, 586]. Исследования об-
разцов, содержащих от 0 до 8,54% С, приготовленных
в высокочастотной индукционной печи из электролитиче-
ского марганца (99,8% Мп) и электродного графита,
показали, что углерод образует с марганцем твердые
173
оастворы. Высокая растворимость углерода наблюдает,
гя в v-Mn, меньшая в а-» 0- и 6 -Мп. 6-и Р-растворы рас.
палаются перитектически при температурах 1215 и 860° С.
v-oaCTBop распадается эвтектически при температуре
743° С с образованием а-раствора и карбида Мп4С. Кар.
биды МпзС и Мп4С взаимно нерастворимы, в то время
как карбиды Мп7С3 и Мп3С образуют непрерывный ряд
твердых растворов в широкой области составов. Данных
о карбидах Мп5С2 и Мп7С3, упоминающихся в ряде ра-
бот, автор не приводит. Это свидетельствует о том, что
вопрос об окончательной диаграмме состояния системы
Мп с еще полностью не решен.
Наиболее изучены свойства карбидов Мп3С, Мп7С3,
МпгзСб-
Изучение термодинамических свойств карбидов мар-
ганца [173, 587, 589—596] позволило вывести уравнения
температурной зависимости изменения свободной энер-
гии образования соответствующих фаз.
В работе [595] приводятся результаты исследований
методом Кнудсена давления паров марганца над чистым
марганцем и над карбидом марганца в условиях равно-
весия с графитод!. В результате исследований показано,
что в равновесии с графитом и парами марганца в ка-
мере присутствует карбид Мп7С3.
Карбиды марганца можно получать в основном на-
углероживанием твердого марганца, восстановлением
закиси-окиси марганца углем или карбидом каль-
ция.
Для получения карбидов взаимодействием твердого
марганца с углеродом нагревали [588] смеси с разным
содержанием марганца и углерода в графитовой печи
сопротивления в среде аргона при температуре 1250°С
в течение 3 ч. В качестве исходных материалов исполь-
зовали чистый марганец, полученный алюмотермически
из Мп3О4 и очищенный дистилляцией при температуре
1200°С и давлении 1,33 н/м2 (0,01 мм рт. ст.), и графит,
дегазированный нагреванием в вакууме. При нагреве
смеси в вакууме значительные количества марганца те-
ряются вследствие интенсивного испарения его при тем-
пературе выше 1000° С.
Использование в качестве восстановителя сахарного
угля менее эффективно. Так, при температуре 1400 ь
лишь 50% его взаимодействует с металлом.
174
Карбид М23С6 готовили спеканием металлического по-
рошка, содержащего 99,77% Мп, с сажей при темпера-
туре 1050° С в среде аргона в течение суток [587]. Про-
дукт содержал примеси металлического марганца.
Система рений —углерод. Ранними исследо-
ваниями системы Re — С [601] установлено, что при
взаимодействии рения с окисью углерода при темпера-
туре 470—600° С образуется карбид рения. При темпера-
туре 1600° С он разлагается. Более поздние исследова-
ния [602, 603] показали, что диаграмма состояния систе-
мы Re — С является простой эвтектической диаграммой
с эвтектикой, образуемой при температурах 2480—
2500°С с содержанием 16,9% (ат.) С.
Растворимость углерода в рении при температуре
2500° С составляет 11,7% (ат.) и падает до'4,2% при Тем-
пературе 1800° С. Растворимость рения в углероде не об-
наружена. В работе [603] указано, что на рентгенограм-
мах образцов, содержащих КЗ-1—3,7% (ат.) С, рбнаруже-.
ны линии неизвестной фазы. Авторы предполагают
образование карбида, устойчивого при температурах вы-
ше 1900° С.
Исследование тройной системы W—Re—С [604] пока-
зало существование тройного соединения W3Re2C с куби-
ческой структурой. При температуре выше 2500° С обна-
ружен карбид (W, Re) С, обладающий г.ц.к. структурой
типа NaCl.
Технеций — углерод. Систему технеций
углерод исследовали методом рентгеноструктурного ана-
лиза на образцах, полученных нагреванием смеси тех-
неция с сажей, а также технеция в атмосфере смеси
водорода и пара бензола [605]. При температуре 910°С
технеций растворяет до 1 % С. При этом периоды решет-
ки гексагонального технеция растут до а = 0,2892 нм,
с=0,4470 нм, с/а —1,590 (для чистого технеция а =
= 0,2740 нм, с = 0,4398 нм, с/а =1,605)/
При содержаний 1,4—9% С продукты представляют
смесь металлической фазы и карбида ТсС с г.ц.к. решет-
кой и периодом а = 0,3982 нм и плотностью 11,5 г/см3,
КАРБИДЫ МЕТАЛЛОВ
VIII ГРУППЫ
Система железо — углерод. Наиболее пол-
ные обзоры работ по исследованию системы железо—уг-
175
ritsV свойства и-фазы, образующейся при отпуске и пои пластической деформации
Способ получения х -фазы Значе- ние ин- декса X Плот- ность, г/ел3 Значение точки Кюри Намагничеин^Г при температур»
°C °К
0° 20’ к
Отпуск » • • • • 1,91 6,73 265 538 875 800
Деформация . . 2,23 6,96 265 538 985 900
лерод приведены в литературе [85, 86, 606—612]. Исследо-
ваниями этой системы [608—612] было установлено нали-
чие в ней, кроме устойчивого карбида Fe3C (цементит),
нестабильных карбидов Fe2C и Fe4C. Fe2C существует в
виде гексагональной в -фазы [613] и х -фазы другой
структуры, известной под названием карбида Хэгга [611],
которая более точно выражается формулой Fe20Cg. При
температуре 300—400° С гексагональный карбид перехо-
дит в карбид .Хэгга, который переходит в цементит [614,
615].
Неустойчивость е -карбида обусловливается тем, что
межатомное расстояние Fe—Fe [618] составляет 0,270—
0,276 нм, что значительно превышает диаметр атомов
железа.
Электронографическое исследование тонких пленок
карбида железа [619] показало, что превращение в^х
фаз происходит при температуре 380—400° С, а х-* б —
при 550°С. Температура перехода в-карбида в карбид
Хэгга выше 290° С, а при температуре выше 360° С кар-
бид Хэгга переходит в Fe3C [912]. Выше 420° С в-карбид
не существует.
Имеются указания на существование карбидов FeC,
FeC2 и Fe4C3 [616], которые, однако, требуют проверки.
Карбид Fe^C (х-фаза) исследовали методом магнит-
ного анализа [617]. По магнитограммам вычисляли плот-
ность, состав и намагниченность карбида, образующегося
при отпуске стади У11 при температуре 300°С и пласти-
ческой деформации. Вычисленные данные приведены в
табл. 29.
Полученные результаты подтверждают состав х-кар-
бида, соответствующий формуле Fe2C,
Электронографическое изучение карбидов железа, по-
лученных обработкой пленок железа на кристаллах NaCl,
176
со, смесью CO + H2 или ацетиленом [616}, показало на-
личие кубического карбида Fe4C с углеродом в тетраэдре
с четырьмя атомами железа, смещенными вдоль нрост-
Рис. 60. Диаграмма состояния системы Fe—С
ческая координация атомов углерода в решетке и малое
расстояние Fe—С (для аустенита оно равно 0,186 нм)
позволяют предположить, что Данный карбид — химиче-
ское соединение, а не Твердый раствор.
На основании многочисленных исследований
636] приведена диаграмма состояния системы Fe С
177
601 на которой сплошными линиями показана диа
грамма метастабильного равновесия Fe-РезС, а пуик
тарными-диаграмма стабильного равновесия между
расплавом и графитом Наличие метастабильвой системы
наряду со стабильной объясняется [620] разной скоростью
нау/iMJ лйпяЧПКЯНИЯ И ППГТЯ .
Рис. 61. Раствори-
мость углерода в a-Fe
образования и роста зародышей
цементита и соответствующи
графитовых зародышей.
Растворимость углерода в
а -железе при комнатной темпе
ратуре составляет не боле
0,05% [637—639]. Растворимость
при высоких температурах иссл
довали на образцах железа вы
сокой чистоты с применением ме
тодов рентгеновского анализа
[640, 641], метода диффузии [642]
измерения внутреннего трения
твердых растворах углерода в
а -железе [643], измерения релак-
сации упругих напряжений [644],
изучения равновесия реакции
С + СО2^2СО пропусканием СО
над железом, содержащим 0,014
С [645].
На основании изучения рав-
новесия реакции приведена следующая температурная
зависимость растворимости углерода в a-железе до тем-
пературы 728° С [645].
Ig(%C)" = ~- 2,25-10-4Т +3,22.
Исследования с использованием метода меченых атомов
(С14) [648] показали зависимость растворимости углеро-
да в а-железе, приведенную на рис. 61. Экстраполяция
кривой дает растворимость при 728°С0,035%С, что хоро-
шо согласуется с данными работ [645] и [646].
Работами [649, 650] было подтверждено, что мартен-
сит—-твердый раствор углерода в а -железе, а в работах
[651, 652] показана обратимость мартенситных превра-
щений в сплавах Fe—С при нагреве.
Механизм и кинетика превращения аустенита в мар-
тенсит еще не установлены окончательно и имеются зна-
178
чительные расхождения в их объяснении [371, 653—
655].
При изучении системы Fe—С предложен [862] ва-
риант полной диаграммы состояния этой системы
(рис. 62) до 100% С с паровой фазой, согласно которой
карбидов, более богатых углеродом, чем Fe3C, не обна-
ружено. Температура 2380° С —температура кипения
расплава железа,, предельно насыщенного углеродом.
Рис. 62. Полная диаграмма состояния системы Fe — С
Предложено [618] следующее строение карбида Fe3C:
атом углерода образует двойную связь с атомами желе-
за I (расстояние Fe—С 0,185 и 0,189 мм)г а две связи
с атомами железа II (расстояние Fe—С 0,20& и
0,215 нм). Расстояние Fen -“-Fen составляет 0,249 нм.
Карбид Fe2C также кристаллизуется в ромбической
ячейке, периоды которой находятся в простом соотноше-
нии с периодами Fe3C, а именно:
179
aFe,C ~ 2aFe,C '»
^Fe,c ~ 3bFesc;
CFesC ~ lfFe3C *
V ^мнение содержания углерода в ячейке Fe2C по
Уменьшение ^^вызывает заметного сжатия решет-
сра?^пИКзобид FesC- твердое и хрупкое соединение;
К” 1 о» температуре он ферромагнитен и точка
JL ₽гп лежит в пределах 21U ь.
К & пХте 1665] исследовалось удельное электрическое
еопоо?№дание сплавов железо-углерод и установлена
сопротивл v логарифмом удельного электросо-
ShXhh н «держанием Fe3C н сплавах, которая
описывается уравнением:
lg р = 0,90Fe3C4- 0,996.
Содержание С, % . . 0,06 0,17 0,53 0,83 1,12
Удельное электросо- противление, мком-см . 10,45 10,57 11,87 12,94 13,90
Содержание С, % . . 1,23 1,26 3,74 4,33 6,77
Удельное электросо- противление, МКОМ-см 14,43 14,63 31,36 41,17 79,5
хе^забсДса^пи1а РбзС впервые получали плавлением
температуре 7$Х^с7б4?'п^еВаНИеМ "Р"
деожатпрй йот, ч При плавлении смеси, со-
получали пп ТОрый избыток угля, в криптоловой печи
каобил жапа??УКТ’ содеРжа1дий 48% Fe3C. Получали
ления нагпАп^ ПрИ Температуре ниже температуры плав-
леза с сйуяп нием смеси чистого восстановленного же-
ской печи Углем в соотношении 5:1 в электриче-
ние 62 и опРотивления при температуре 920° С в тече-
обпа^штгтЛ Среде В0Д0Р0Да- Содержание карбида в
В обо™Т спекшейся массе составляет 13%
ным раствопр1/^^ ЧИСтый карбид выделяли либо анод-
либо обработкой В раств°Ре 7% -ной соляной кислоты,
трех недель р v Разбавленными кислотами в течение
H2S04 7% Растворителей использовали 2-н.
выделяли чисты?? « 6°^НУЮ СН3СООН. Очищали и
ром уксусной киеппарбид ^656^ обработкой 1-н. раство-
Более распппЛ°ТЫ В течение четырех недель.
железа— взаимп пТ?аНенный мет°Д получения карбида
взаимодействие железа с газообразными угле-
родсодержащими газами. Так, при длительном насыще-
нии листового электролитического железа парами бен-
зина при температуре 700° С получают карбид Fe3C,
который выделяют в чистом виде анодным растворением
в ванне с раствором FeCl2. Полученный крупнокристал-
лический порошок Fe3C промывают разбавленной уксус-
ной кислотой, водой, спиртов и эфиром й сушат в ваку-
уме (82].
В работе [618] чистый карбид Fe3C получали нагре-
ванием восстановленного железа или Fe2O3 в смеси Н2
и СО (15% СО) в течение 16 ч при температуре
500—550° С.
Наиболее распространено получение чистых карби-
дов железа химическим выделением их из сталей и фер-
росплавов. Этот метод можно использовать только для
получения малых количеств карбида для исследований.
Одна группа методов основана на разной раствори-
мости карбида и составляющей металлической основы.
Карбид железа Fe3C был выделен обработкой раз-
бавленными соляной, серной, уксусной, муравьиной кис-
лотами и их смесями [58, 658, 659]. Для предупреждения
окисления выделившегося карбида выделение и сушку
осадка проводили без доступа воздуха. Кислотное выде-
ление дополнялось магнитным отделением примесей.
Наиболее полное выделение наблюдалось при обработке
0,1-н. раствором серной кислоты, но и при этом выход
не превышал 35%.
Электролитическое выделение сводится к тому, что
образец стали служит анодом. В процессе электролиза
металл растворяется, а карбид остается в виде нераство-
рившегося остатка [57, 58, 658, 659, 661, 662].
В качестве электролита используют 0,2-н. раствор
сульфита натрия с добавкой 5% лимоннокислого натрия
[58], 1-н. раствор хлористого калия с добавкой 0,5% ли-
монной кислоты [661], 15%-ный раствор лимоннокислого
натрия с добавкой 1,2% КВг и ОД % KJ [659], 1-н.
раствор соляной кислоты с добавкой 2% Na2S2O3 [57].
Процесс электролиза проводят при охлаждении, необхо-
димая плотность тока составляет 0,02—0,03 а/см2.
Система кобальт — углерод. Первые исследо-
вания системы кобальт — углерод [666] показали, что мак-
симальная растворимость углерода в кобальте составля-
ет 3,9% при температуре 1700° С« При температуре
181
1Чпч6С обнаружена эвтектика [153], содержащая 2чо/ л
Пп1температуре 2100° С состав сплава соотве^’
формуле CosC. Этот карбид существует в расплаве я
охлаждении разлагается на кобальт и графит. АвтЙ
получили сплавы, содержащие и больше углерода
это соответствует Со3С, что позволило предположить Iм
шествование высших карбидов кобальта. У'
Более поздние исследования системы Со—С [667] По
казали, что высокотемпературная область в этой системе
аналогична системе Ni—С. При температуре 1300—
1350° С обнаружена эвтектика, образованная твердым
раствором углерода в кобальте с графитом, содержащая
2,8—3,0% С,, а также карбид Со3С, который несколько
более стабилен, чем Ni3C« Предположение о существо-
вании высшего карбида кобальта подтвердилось работа-
ми [657, 660, 668]. При взаимодействии окиси углерода
с кобальтом при температуре 226—230° С получен кар-
бид Со2С, который изоструктурен с Co2N [669].
В работе [867] исследовали систему кобальт —угле-
род в области высоких содержаний углерода и высоких
температур. Авторы показали, что аналогично системе
железо — углерод на стабильную систему кобальт —
графит накладывается метастабильная система Со—
Со3С, неустойчивость которой обусловлена тем, что кар-
бид СозС легко разлагается при нагревании до
300-350° С (рис. 63).
Исследование равновесия системы метан—углерод
на кобальте, используемом в качестве катализатора
[657] при температурах 350—900° С, показало, что при
350—720° С наряду с реакцией СН4 С+2Н2 образуется
карбид по уравнению
пСо 4- СН4 Со„С 4- 2На -47,26 кдж (11,28 ккал.)
При температуре 428—900° С кобальт насыщается
углеродом при низких концентрациях метана с образова-
нием твердого раствора Со—Со„С. С увеличением со-
рт ъДНИЯ углеР°Да в карбиде соответственно возраста-
максит?^еСНое С0деРжание метана, которое достигав
с и /МЭ ПРИ достижении чистой карбидной фазы.
Ния система КГН ИЛ ге л ь~у г л е Р о д- Первые исследова-
строить [85’ 577’ 667, 671, 672] позволили по-
графическлй ^Ямму состояния этой системы на осН°
интерполяции результатов термическ
182
анализа и определения растворимости углерода в никеле
в жидком состоянии. Согласно этой диаграмме, при тем-
пературе 1314° С образуется эвтектика, соответствующая
содержанию 2,2% (по массе [0,9% (ат.)]С.
Растворимость углерода в никеле в твердом состоя-
нии при температуре 1318° Q составляет 0,55% (по мас-
СО 10 20 30 40 50 80 70 ВО 80 С
Содержание С,%(ат.) (Граорит)
Рис. 63. Диаграмма состояния системы Со—С [867]
се) [2,7% (ат.)] С. Ниже приведены: данные о раствори-»
мости углерода в никеле в твердом состоянии [674]:
Температура,“С , . « .
Растворимость:
% (по массе) . . .
% (ат.)
700 745 925 1055 1170 1265
0,08 0,1 0,2 0,3 0,4 0,5
0,4 0,48 0,95 1,45 1,95 2,4
Растворимость углерода в никеле в жидком состоя-
нии при температуре 2100°С составляет. 6,4%, что соот-
183
ветствует карбиду Ni3C (6,39% С). На основании
жидкий Ni3C кипит и разлагается с выделением графИт'
и паров металла.
При попытке получить карбид никеля с более высп
ким содержанием углерода [676] рентгенографически по-
казано, что во всех продуктах наблюдались только ли-
нии Ni3C.
Подробно сплавы N1—С исследованы в работах [677
678]. Диаграмма состояния системы строилась на осно-
вании металлографического анализа, измерений элек-
тросопротивления в твердом и жидком состоянии и тер-
мического анализа. Результаты позволяют уточнить по-
ложение линий ликвидуса на диаграмме. Эвтектическая
точка, положение которой совпало по результатам тер-
мического анализа и скачку при измерении сопротивле-
ния, соответствует 1,3% С, что расходится с описанными
выше данными работ [577] и [672]. Такое расхождение
объясняется результатами работы [679], в которой пока-
зано, что сплавы никеля с углеродом при больших ско-
ростях охлаждения могут кристаллизоваться по метаста-
бильной диаграмме с выделением карбида Ni3C. При
этом эвтектическая точка сдвинута в сторону высоких
концентраций углерода. Таким образом, в системе Ni—С
обнаружен один карбид Ni3C, который устойчив при
температурах выше 1600° С и ниже 300° С [371].
Систему никель — углерод исследовали [867] в обла-
сти содержаний до 69,2% (ат.) С и температур 3000°С.
Подобно системе Fe—С и Со—С, на стабильную диа-
грамму Ni—С накладывается метастабильная Ni—Ni3C,
которая обусловлена низкой температурой разложения
Ni3C (200-400° С) (рис. 64).
Карбид никеля представляет собой порошок серо-
гается° ^Вета’ ПРИ температуре 370° С полностью разла-
п„Да,^ид никеля Ni3C был получен обработкой никеля
тельнп Углерода [676, 680, 681]. Окись никеля предвари-
320° г останавливали водородом при температуре
nnonveJ3/™М чеРез порошок восстановленного ни_кДл^
влечений ?72кись Углерода при температуре 240—250 С
обпазуюшр&Р М’ Процесс контролировали взвешивание*
Р ующеися углекислоты, которая адсорбировалась
184
едким натром. Вместо СО иногда использовали метан
или ацетилен.
Исследования процессов восстановления окиси нике-
ля твердым углеродом [675] показали, что скорость вос-
становления, которая при температуре 800°С незначи-
тельна, резко возрастает при температуре 900° С.
Рие. 64. Диаграмма состояния системы Ni—С [867]
Система рутений-углерод. Жидкий руте-
ний растворяет углерод, который при охлаждении выде-
ляется в виде графита [66, 85]. Растворимость углерода
в расплавленном рутении увеличивается с ростом темпе-
ратуры и при температуре кипения рутения составля-
Рентгенографическим исследованием продуктов взаи-
модействия рутения с углеродом [771, 772] показано, что
при нагревании смеси рутения с сажей в соотношении
1 -10 в атмосфере гелия при температуре 2600° С в тече-
185
ние 4 ч образуются продукты, на рентгенограммах кота
рых наблюдаются линии новой фазы изоструктур*0;
WC с периодами решетки: а = 0,290785 и с«0,282186 Т?
с плотностью 9,088 г/см3.. Эта фаза идентифицировав ’
как RuC.
Позже была сделана попытка подтвердить существп
вание карбида рутения [819]. Авторы при нагревании
смеси рутения с сажей в соотношении 1 : 10 при темп?
ратуре 2600° С в течение 2 ч получили продукты, на
рентгенограммах которых наблюдались только линии
рутения и графита.
Система родий — углерод. Растворимость
углерода в расплавленном родии составляет 1,42—7,28%
При охлаждении углерод выделяется в форме графита
[66, 85].
Система палладий—углерод. Расплавлен-
ный палладий при нагревании растворяет углерод, кото-
рый при охлаждении выделяется в форме графита.
Растворимость углерода 2,45% [66, 85].
Имеются данные о том, что при действии метана или
смеси метана с водородом на порошок палладия они ак-
тивно взаимодействуют с образованием продукта, содер-
жащего 4,4% С и соответствующего формуле Pd5C2 [773].
Однако эти данные не подтвердились.
Система осмий — углерод. При температуре
кипения осмия в нем растворяется 3,9—4% С [85].
При исследовании продуктов взаимодействия порош-
ка осмия с метаном при температурах 1200 и 2000° С об-
разования карбидов не наблюдалось [601].
Рентгенографическое исследование продуктов взаи-
модействия осмия с сажей в атмосфере гелия в интерва-
ле температур 2200—3000° G показало, что при нагрева-
нии смеси Os—С в соотношении 1:10 при температуре
2600 С в течение 4 ч образуется карбид OsC, обладаю-
щий гексагональной решеткой с периодами а=0,290769,
с=0,282182 нм и плотностью 16,25 г! см3 [771, 772]. Одна-
ко эти Данные не подтвердились работой [819]. Фазовый
состав продуктов взаимодействия осмия с сажей приве
Ден в табл. 30.
иридий — углерод. Расплавленный
темпрпятЛСТВ°рЯет УглеР°Д» количество которого пр
Ппи пу;ЛРе кипения иридия составляет 2,08% [66, 8 ]•
Р аждении углерод выделяется в виде графи
186
Таблица 30
Фазовый состав продуктов взаимодействия осмия с сажей [819]
Молярное отношение Os:С Температура, Врем» ман Фазовый состав по данным рентгеновского анализа
1:10 1:10 1:10 Ta:Os:C 1:10:100 2200 2400 2600 2600 240 240 180 ' 90 Os 4- графит Os -j-графит Os -|- графит Os-j-графит 4-ТаС
Система платина—углерод. Расплавлен-
ная платина растворяет 1,2—1,45% С«. выделяя его прй
охлаждении [66, 85]. При реременном осаждении плата-
ны и углерода на тонкую коллодиевую пленку и нагре-
ве в вакууме полученного продукта при температуре
1100° С образовывалась пленка толщиной 10 нм, кото-
рая, по мнению авторов, является карбидом платины [85].
ГЛ AB A V I
КАРБИДЫ ЭЛЕМЕНТОВ
ГРУППЫ БОРА
Система бор—углерод. Первые исследова-
ния системы бор — углерод [682] установили существова-
ние фазы В4С(В12С3). Многочисленные исследования
этой системы [683—690] позволили предложить оконча-
тельный вид диаграммы, представленный на рис. 65.
Диаграмма в основном подтверждает наличие карбида
В12С3 с большой областью гомогенности [17,6—29,5%
(по массе) С], карбида Bi3C2 и новой фазы.
Металлографическое исследование образцов позволи-
ло предположить наличие в области от 6,4 до 10% С хи-
мического соединения, содержащего около 8% С и соот-
ветствующего составу Bi2C. Рентгенограмма образцов,
близких по составу к Bi2C, имеет индивидуальный ха-
рактер и свидетельствует о сложной структуре этой
фазы.
При исследовании диффузии бора в углерод [696]
установлено, что скорость диффузий в углерод больше,
чем углерода в бор. Температурная зависимость коэф-
фициента диффузии бора в углерод выражается урав-
нением
—28625
D = 3,02e Т :
Карбид бора В4С представляет собой темно-серый
кристаллический порошок, кристаллизующийся в ромбо-
эдрической решетке. Он отличается высокой твердостью,
уступающей только твердости алмаза [48 000 Мн!м
(4950 кГ/мм2)]. Абразивная способность порошка кар-
бида бора (определенная в условных единицах) состав-
ляет для алмаза 0,613, для карбида бора 0,442, для кар-
бида кремния 0,314 [694, 695]. С увеличением содержания
свободного углерода в карбиде бора его абразивная спо
собность резко падает.
188
Карбид В4 Ci-* характеризуется линейной цепочкой
—С—С—С-— и является дырочным полупроводником.
Карбид В13С2 характеризуется линейной цепочкой —С~
—В—С и обнаруживает электронную проводимость.
Измерение удельного электросопротивления и термо-
э. д. с. карбида бора в зависимости от температуры по-
казывает, что до температуры 500° С сопротивление па-
дает почти прямолинейно, при более высоких температу-
Рис. 65. Диаграмма состояния системы В—С [690]
рах снижение замедляется. Ход изменения термо-э.д.с.
обратный. Ширина запрещенной зоны образцов карби-
да, содержащего 78,7% В и 21,7% С, составляет
0,16—0,208 • 10-19 дж (0,1—0,13 эе). Электросопротивле-
ние технического карбида бора почти на порядок ниже
сопротивления чистого карбида.
В работе [379] исследовано испарение карбида бора
и чистого бора в области температур 1750—2500° С и оп-
ределена стандартная теплота реакции 'диссоциации
карбида бора на газообразный бор и графит, которая
составляет 553 кдж (132 ккал). Это хорошо согласуется
с данными работы [376].
189
иМЯ карбида бора синтезом из элемен
метод получения к н ием смеси бора и сажи
псушествляется на Рс^ Этот меТ0Д п03В0ляет Полу
анвс+№ЖеЙ П°РеаММЯ
° 2Во03 + 7Ь 4
„„-ненный метод получения карбида
_ наиболее Р’в^каег в две стадии:
бора. Этот про®» зс0 e 2В + ЗСО,;
4ВН-С==В4С.
При взаимодействий В2О3 + С из расплавленной сме-
си [482] на кусках графита образовывался слой карбида
бора (через который происходит диффузия), образую
щийся при восстановлении В2О3 углеродом бора до пол-
ного превращения графита в карбид.
Температура начала восстановления борного ангид-
рида окисью углерода выше 1320° С. С ростом темпера-
туры скорость реакции восстановления увеличивается
Однако при этом следует учитывать, что карбид бора
при температуре 2250° С перитектически разлагаете .
При этом бор улетает, а оставшийся продукт обогащает-
ся углеродом.
Разработан метод получения карбида бора, по кото-
рому шихта из борной кислоты и сажи в виде брикетов
обезвоживается нагреванием при температуре 800°С;
борный -ангидрид при этом расплавляется и образуется
губка из борного ангидрида, хорошо перемешанного с
еажей. Далее губка нагревается, причем промышленные
методы получения карбида бора отличаются главным
образом по типу печей, в которых ведется процесс. Зто
могут быть керновые, бескерновые, печи типа Таммана,
электродуговые печи. В керновых и бескерновых печа
температура неравномерно распределена по зонам, ши
та" расположена в трех зонах: 1) центральной вь1С° g
температурной, где температура выше 1850° С (в
?оне завершается процесс восстановления борного
гидрида с образованием карбида); 2) промежут° ’
заполненной смесью из недовосстановленного бор_я-
ангидрида, карбида бора и неизрасходованного У.
а) наружной, заполненной в основном исходной Ш
190
Продукты 2-й и 3-й зон используют как полупродукты
для следующей загрузки, а 1-й являются основным про-
дуктом реакции.
Магниетермический метод, получения карбида бора
[692, 693] основан на реакции
2ВаО3 Ч- 6Mg -f G = В4С ф 6MgO.
Он осуществляется нагреванием,, соответствующих шихт
в электрической печи сопротивления с графитовыми на-
гревателями в среде водорода при температуре 1800° С,
Так как продукты реакции содержат окись магния, их
промывают соляной кислотой и остаток прогревают в ва-
кууме при температуре 1800° С для удаления летучих
примесей. Этот способ не нашел широкого промышлен-
ного применения, однако позволяет получать карбид
точного стехиометрического состава.
Хлорид бора восстанавливается водородом в присут-
ствии угля по реакции
4ВС13 + 6Н2 + С = В4С + i 2НС1.
с нагреванием при температуре 600° С Шихты из борного
ангидрида и избытка сажи в токе хлора. При этом обра-
зуется смесь ВС13 и СО. Газообразную смесь ВС1з и CQ
с водородом пропускают над накаленной вольфрамовой
нитью, на которой осаждается слой карбида.
В работе [863] исследовали условия получения моно-
кристаллов карбида бора осаждением из газовой фазы
по реакции
4ВС13 + СС14 4- 8Н2 = В4С 4- 16НС1
на установке; изображенной на рис. 66. Осаждением при
температурах 1550—1650° С, скорости подачи смеси ВС1з
и СС14 г!мин и избытке водорода в течение 4—5 ч полу-
чены монокристаллы длиной 3—4 мм в форме игл и пла-
стин; содержание примесей в полученных продуктах
5-10~4 %. В основном это примеси кремния и магния.
Компактные образцы карбида бора приготовляют в
основном горячим прессованием при температурах выше
2000° С и давлении 3390 Мн/м2 (350 кГ!см2) [692]. Пори-
стость образцов, близкая к нулю, достигается для таких
порошков при температурах 1650—1700° С и давлении
1940 Мн!м2 (200 кГ!см2).
191
Химический состав в процессе горячего прессован
тоже меняется —содержание примесей уменьшается
увеличивается относительное содержание бора и v™
рода. у е'
Система алюминий —углерод. Диаграмм
состояния системы еще полностью не разработана
Часть диаграммы (рис. 67) —лишь гипотетическая [861
Согласно этой диаграмме, в системе А1—С существуют
Рис. 66. Установка для осаждения карбида бора из га-
зовой фазы:
/ — баллон с водородом; 2 —реометр; 3 — еклянка с КОН;
4 — печь; 5 — палладированный асбест; 6 — склянка с КОН;
7 — склянка с Р2О5; 8 — активированный уголь; 9 — сосуд с жид-
ким азотом; 10, 12 — контейнеры е ВС1з и CCL; //—термостат;
13 — медный стержень; 14 — змеевик; 15 — графитовый стержень;
16 — кварцевая труба; 1/ — керамическая прокладка
два карбида: AI3C2 и AI4C3. Сделано предположение
о существовании карбида А13С [85]. Однако многочис-
ленными исследованиями установлено, что единственный
устойчивый карбид в этой системе — AI4C3.
Растворимость углерода в алюминии при температу-
рах 1300—1500°С составляет 0,02—0,04%, при темпера-
турах 1000—1100° С она практически равна нулю [161].
Карбид алюминия AI4C3 — желтый порошок [86].
В вакууме при температурах до 1200° С он устойчив, при
2200° С возгоняется без плавления.
Масс-спектрографическое исследование газообразно-
го карбида алюминия, находящегося в равновесии с па-
рами алюминия и графита [170], показало, что при тем-
пературе 1800°С в парах имеются атомы алюминия, мо
лекулы А1г и AI2C2, высота пиков масс ионов которы
относится как 1050:0,1: 1.
192
Способность карбида алюминия к диссоциации в ва-
кууме использована для полунепрерывного метода полу-
чения чистого алюминия.
Значения теплот образования карбида алюминия, по-
лученные разными авторами [103, 169, 171, 175], значи-
тельно отличаются одно от Другого, что связано с труд-
Рис. 67, Диаграмма состояния системы
Al—С [86]
ностью получения карбида, точно соответствующего со-
ставу А14С3.
В работе [163] описан аморфный карбид алюминия,
образование которого из компонентов наблюдалось на
холодных стенках дуговой печи. Этот карбид термически
менее устойчив, чем получаемый обычно синтезом. Он
разлагается при температуре 1200° С и отличается более
низкой химической устойчивостью. Определение теплоты
образования его показало, что она практически совпада-
ет с теплотой образования кристаллического карбида.
По-видимому, большая реакционная активность этого
карбида связана лишь с большей его дисперсностью.
13 Косолапова Т. Я.
193
В работе [163] исследовано карботермическое восста.
давление окиси алюминия в интервале температип
1400—190Q°C и установлено, что окись алюминия вос-
станавливается углеродом до карбида AI4C3 через про-
межуточные стадии образования оксикарбидов А14О г
и АЬОС.
Карбид алюминия также был получен нагреванием
алюминия с карбидом кальция, восстановлением окиси
алюминия карбидом кальция в дуговой электрической
печи, восстановлением алюмината натрия углеродом
[64], нагреванием смеси алюминия с углем до темпера-
туры 2000° С [164], нагреванием расплавленного в уголь-
ном тигле алюминия до температуры 1700° С [161, 165].
Карбид алюминия образуется в процессах электролиза
криолитглиноземных расплавов [167].
Согласно данным работы [166], существенную роль
при этом играет природа углеродистых материалов, вхо-
дящих в состав подовых блоков и анодной массы. Авто-
ры работы исследовали влияние различных электродных
материалов на продукты взаимодействия их с порошком
алюминия в вакууме при температуре 1000° С. Ниже
приведены результаты исследований, свидетельствую-
щие о влиянии происхождения графита на склонность
его к карбидообразованию:
Графит натуральный серый , . . 43,2 % А14С3
Графит натуральный черный . . .33,6 » »
Графит искусственный..........44,7 » »
Термоантрацит ........ 36,2 » »
Нефтяной кокс пиролизный . . .35,3 » »
Нефтяной кокс крекинговый . . . 33,18 » »
Пековый кокс „ ...... . 44,4 » »
Литейный кокс ........ 25,4 » >
Наиболее склонны к карбидообразованию искусст-
венный графит, наименее — нефтяной и литейный кокс.
Г а л л и й, индий и таллий не образуют со-
единений с углеродом. Известны тройные соединения
Т1з1пС и Ti3TlC [176J, полученные взаимодействием кар-
бида титана с металлическим индием и таллием. Они
кристаллизуются в кубической решетке с периодами
= 0,4199 нм для Ti3InC и а = 0,4209 нм для Ti3TlC. Так-
же получены фазы Zr2TlC, Hf2TlC [177], Ti2TlC, Nb2InC
и Ta2GaC [176]. Эти фазы получены нагреванием смесей
карбидов с металлами при температуре 850° С в тече-
ние 600—900 ч, v г
194
Результаты рентгеновского исследования
ных фаз представлены в табл. 31,
получен-
Таблица 31
Данные рентгеновского анализа
Фаза Природы решетки, нм , с/а - Плотность г/см?
I а С
Ti2InC 0,313а 1,406 4,490 6,19 8,59 8,29 13,01
TiaTlC 0,3158 1,398 4,426
Nb-JnC 0,317а 1,437 4,53n
Ta2GaC 0,3104 1,357 4,37^
Также синтезированы карбидные фазы, содержащие
редкоземельные металлы Nd3InC, Nd3TeC [783]. типа La3InC, Pr3GaC, Nd3GaC,
13*
ГЛАВА VII
КАРБИД КРЕМНИЯ
В системе кремнии-^углерод установлено существо-
вание одного соединения SiC. Имеющиеся в литературе
[698, 699] данные о существовании карбидов Si2C, SiCa
не подтвердились.
Изучение диаграммы состояния системы Si—С весь-
ма затруднительно, что связ.ано с высокой химической
активностью и упругостью паров расплавленного крем-
ния и наличием различных кристаллических модифика-
ций SiC. В работе [700] даны два возможных варианта
диаграммы без учета существования кристаллических
модификаций SiC (рис. 68).
Предельная растворимость углерода в кремнии [701]
составляет 0,6% (по массе) [1,4% (ат.)].
Исследование сплавов в области составов Si—SiC
показало наличие в них двух фаз — твердого раствора
углерода в кремнии и карбида кремния. В образцах со-
ставов SiC—С наблюдается наличие SiC и углерода,
причем длительный отжиг в вакууме при температурах
2000—2100° С приводит к диссоциации карбида крем-
ния, испарению кремния и выделению свободного угле-
рода.
Таким образом, исследования [701, 702] подтвержда-
ют правильность диаграммы, представленной на
рис. 68, а.
Подтверждено [702] существование в системе Si С
только одного соединения SiC, образующегося перитек-
тически при температуре 2830±40°С. Растворимость
углерода в жидком кремнии, по данным авторов, со-
ставляет не более 5 • 10~3% (ат.).
Сгол3лИ£чТем что карбиду кремния посвящено много
нппикт 694» 702, 704—711, 835], приведем кратко лиШЬх_.
овные данные о свойствах и методах получения карой
Да кремния.
196
Карбид кремния существует в виде кубического
0-SiC и гексагонального a-SiC. Температура перехода
одной модификации в другую неопределенна. Некото-
рые считают, что р-SiC неустойчив при температурах
выше 2000° С и переходит в a -SiC [726, 727]. По дан-
ным работы [728], а-SiC может образовываться при тем-
пературах ниже 2100° С’параллельно с P-SiC, в работе
Si+SiC
пар / Пар* С
7 *
t Si+SLC
SIC+G
Si SLC C
a 6
Рис. 68. Фазовая диаграмма состояния системы Si—С
[728] обе модификации были получены при температу-
ре 2700° С.
Однако было установлено [729], что обычно в первую
очередь образуется р -SiC, переход р-модификации в a
происходит при высокой температуре через газовую фа-
зу. Это подтверждено работой [730], в которой вычисле-
ны константы скорости превращения р -SiC в a-SiC при
температурах 2100 и 2300° С, а также энергия актива-
ции процесса превращения, равная 662 кдж/моль
(158 ккал/моль). Это близко к значению теплоты испа-
рения SiC в этом интервале температур [557 кдж/моль
(133 ккал/моль)}. Переход а в р -модификацию обыч-
но не наблюдается. P-SiC имеет одну кристаллическую
форму, обладающую алмазоподобной элементарной,
ячейкой [323, 731].
a -SiC образует много структурных типов с гексаго-
нальной или ромбоэдрической решеткой [816—818].
Слои из атомов кремния и углерода в них смещены раз-
197
личным образом Ьдин относительно другого и образуют
гексагональную упаковку из тетраэдрических структуп
в которой атомы каждой занимают половину пустот дру!
гой. Отдельные тетраэдры из слоев атомов углерода и
кремния расположены параллельно и антипараллель-
но, образуя пачки из одинаково направленных слоев
[732].
Основными структурами гексагонального SiC счита-
ют SiC—II и SiC—III [733, 734]. Все же остальные мо-
дификации представляют различные комбинации этих
типов. Предложены числовые символы. Это последова-
тельные числа, характеризующие число элементарных
слоев с параллельной и антипараллельной ориентацией
тетраэдров, расположенных пачками.
Само возникновение разных форм SiC связано с об-
разованием дефектов на поверхности кристаллов SiC
при их росте. Причина возникновения разных форм SiC
[735] — разная величина частиц карбида в парообразном
состоянии. Эти данные, однако, не подтверждаются дей-
ствительным механизмом образования SiC.
Более вероятно влияние примесей, адсорбированных
растущей поверхностью кристаллов. Так, было обнару-
жено [683], что при наличии примеси бора образуется
SiC—III, при отсутствии его — SiC—II. Химически чис-
тый карбид кремния бесцветен, технический может быть
окрашен в зеленый или черный цвет. Зеленый карбид в
основном состоит из SiC—II, черный — из смеси 60%
SiC—II и 40% SiC—III.
При температуре 2700° С наблюдается диссоциация
карбида кремния [736, 737]. Рентгенографическое иссле-
дование карбида кремния, нагретого в вакууме [738], по-
казало, что при температуре 2050° С a-SiC частично
разлагается с выделением графита. При температуре
2150° С обнаружено полное разложение SiC.
В работах [702, 740] масс-спектрографически опреде-
лены парциальные давления в системах SiC—С и
Si SiC при изучении равновесия системы Si—С методом
Кнудсена и установлен молекулярный состав газовых
продуктов. В них, кроме атомов кремния и молекул
Sig, Sis и Si4, обнаружены молекулы газообразных кар*
бидов SiC, SiC2, Si2C, Si2C3 и Si3C.
Значения теплот образования, энтропий и теплот сго-
рания разных модификаций очень близки [702, 741].
198
Многочисленными исследованиями электрофизиче-
ских свойств карбида кремния, обзор которых приведен
в работах [711, 712], показано, что карбид кремния —
полупроводник примесного типа.
Исследование влияния действия облучения образ-
цов карбида кремния нейтронами, интегральные потоки
которых составляли от 7- 1018 до 1-1021 быстрых
нейтр!см2 [913], показало расширение образцов, макси-
мальная величина которого составляла 2,9%. Период
решетки а увеличивался на 0,89%, с — на 1,09%.
Наиболее распространенный и изученный метод по-
лучения карбида кремния — взаимодействие кремнезе-
ма с углем. Суммарная реакция выражается уравне-
нием:
SiO2 + ЗС = SiC + 2СО.
Она идет в две стадии: восстановление кремнезема уг-
лем до металлического кремния и взаимодействие паров
кремния с углем или с окисью углерода:
SiO 4- 2С = Si + 2СО,
Si 4- С = SiC
или
3Si 4- 2СО = 2SiC 4- SiO2.
Высказано предположение [694, 704, 713], что про-
межуточным продуктом может быть моноокись крем-
ния, которая при температурах промышленных устано-
вок образуется по реакции
SiO2 4- С = SiO 4- СО.
Сырьем для получения карбида кремния служит
кварцевый песок, содержащий не менее 98,5% SiOg и
минимальное количество примесей в виде AI2O3, СаО,
MgO и Ре20з. Это вызвано тем, что примеси глинозема,
окиси магния и кальция могут образовывать силикаты,
способствующие спеканию шихты, окись железа приво-
дит к образованию сплавов железа с кремнием. Угле-
родсодержащее -сырье — малозольный кокс, антрацит и
нефтяной кокс. В шихту добавляют древесные опилки
(для увеличения газопроницаемости) и хлористый нат-
рий (действие которого сводится к рафинированию хло-
рированием примесей).
199
Для приготовления SiC используют электрические
печи сопротивления. Наиболее совершенна в настоящее
время печь, которая по длине разделена на секции, изо-
лированные одна от другой огнеупорным кирпичом.
В торцовых стенках печи закреплены электроды, между
которыми в центре печи расположен угольный сердеч-
ник (три).
Фазовый состав карбида кремния по зонам печи при-
веден в табл. 32.
Таблица 32
Фазовый состав карбида кремния по зонам печи [714]
SiC Место отбора пробы SiC SiC—I SiC—п SiC—III
Зеле- Внутри сердечника 34 0 66 0
ный Средняя зона 29 3 67 0
Зона у поверхности 37 6 52 5
Поверхностный слой 80 Остальное
Чер- Внутри сердечника 28 0 72 0
иый Средняя зона 22 1 72 5
Зона у поверхности 8 5 18 69
Поверхностный слой 23 11 24 42
Описанный метод позволяет получать карбид крем-
ния технического качества, состав которого приведен в
табл. 33.
Высокие полупроводниковые свойства монокристал-
лов карбида кремния вызывают необходимость разра-
ботки производительных методов производства этих мо-
нокристаллов. В работе [702] приводятся следующие
возможные способы получения монокристаллов SiC:
1) кристаллизация из жидкого карбида кремния; 2) кри-
сталлизация из растворов; 3) сублимация; 4) термиче-
ское разложение; 5) термическое восстановление.
Первый метод требует создания установки, в которой
можно достигнуть температуры 2000° С при давлении
сотен тысяч атмосфер, так как в обычных условиях пе-
ревести карбид кремния в жидкое состояние невозможно^
Таблица 33
Состав двух разновидностей карбида кремния
Разновидность Содержание, %
SiC ®*своб ^своб Fe Al CaO SiO2
Зеленый .... 98,8 0,59 0,03 0,11 0,06 0,01 —-
Черный . • •> • 96,2 — 0,13 1,05 —• — 0,94
Второй метод — кристаллизация из растворов — на-
шел довольно широкое применение. Кристаллы SiC п- и
p-типов получали из расплава кремния, в который вво-
дили примеси Li, Mg, В, Al, Ga, Ge, As, Sb, Cu, Fe, Ni и
Au в разных соотношениях при температуре 1600° С. Ис-
следование влияния примесей на кристаллизацию SiC
показало, что примеси стабилизируют a -SiC [702].
Кристаллы SiC получали [717] из кремнистоникеле-
вых расплавов эвтектического состава при температуре
1000° С и из кремнистоалюминиевоцинковых расплавов
при температуре 525° С, Также были получены кристал-
лы SiC из железокремнистых расплавов [718], кремне-
углеродистых расплавов [719], расплавов ферросилико-
хрома [720].
Исследование процесса кристаллизации SiC при
плавке кремния в графитовых тиглях в среде чистого
аргона при высокочастотном нагреве [781] позволило
получить кристаллы кубической структуры размером не
более I мм2 при температуре выше 2000° С. При исполь-
зовании загрязненного аргона получали кристаллы гек-
сагонального SiC.
Метод выращивания монокристаллов карбида крем-
ния сублимацией наиболее распространен. В работе [711]
спекали и затем нагревали в графитово-трубчатой печи
цилиндр из кристаллов SiC. При температуре 2500° С
и выдержке в инертной атмосфере в течение нескольких
часов в полости цилиндра вырастали тонкие пластинки
монокристаллов SiC. При использовании карбида крем-
ния, полученного из смеси кремния и графита высокой
чистоты, выращенные кристаллы были более чистыми
[702]. Но во всех случаях монокристаллы содержали
примесь азота, проникавшего из печи через стенку гра-
фитового реакционного сосуда. Нанесение на стенки ре-
акционного графитового сосуда пирографита пиролизом
14 Косолапова T. Я. 201
метана создавало плотный непроницаемый слой пиро
графита толщиной ДО 50 мкм-
Р Изучен [131] процесс роста кристаллов при сублима-
ции SiC в атмосфере аргона. Исходный порошок SiC по-
ц мещали в пространст
во между графитовым
тиглем и графитовым
цилиндром. Пары кар-
бида кремния поступа-
ли в зону роста крис-
таллов через отверстия
в стенках цилиндра.
Процесс осаждения
Кристаллов регулирова-
ли, изменяя диаметры
отверстий и темпера
туру.
Распространено так-
же получение кристал-
лов карбида кремния
осаждением из газо-
вой фазы термическим
разложением кремнии-
и углеродсодержащих
соединений или восста-
новлением. Так, кар-
бид кремния был полу-
чен [53] из смеси четыреххлористого кремния, толуол
и водорода.
При изучении условий выращивания монокристалло
осаждением из газовой фазы показано {724], что пр
температуре 2000° С из смеси SiCl4, С7Н8 и Н2 при со-
отношений SiCl4 и С7Н8 20 t 1 на угольной нити осаж-
даются кристаллы р-SiC. При отсутствии водорода полу-
чить SiQ не удалось при любых соотношениях газовьх
смесей.
2,0
? о
4
$-2,0
1-4,2
«5
*5
l-v
50
О
<3
*
§
-50
(?)
-100
<з
5S
F
«ь
«5
•9
(О
1400 1600 1800 2000
Температура., °C
Рис. 69. Зависимость изменения изо-
барного потенциала от температуры
для реакций образования SiC из газо-
вой фазы. Цифры на кривых — номе-
ра реакций
-Г5^
§
В работе [780] монокристаллы SiC получали, про-
пуская ток через угольную нить диаметром 0,175мм
ри этом температура нити достигала 2300—2400
днако после 15 мин нить прогорала.
пРовеДен [725] термодинамический анализ пр
Р.5* °бразования каРбида кремния и газовой Фа
тано изменение свободной энергии реакции.
202
(SiCl4)r -h (CCl4)r + 4H2(r) = SiC 4- (8HCl)r; (1)
(SiCI4)r + V6 (CeHe)r + % (H2)r SiC + 4 (HCl)r; (2)
(Sici4)r + v7 (ед)г + 10/7 (H2 )r« Sic + 4 (HCi)r; (3
(SiCI4)r + V7 (ОД)Г - SiC + % (HCl)r +™/7 (Cl2)r. (4)
Результаты, представленные на рис. 69, подтверж-
дают, что при отсутствии водорода реакция образова-
ния SiC невозможна.
Вычислены также изменения свободной энергии ре-
акций образования карбидов возможных примесей:
21FeCl3 + С7Н8 Ч- 27,5Н2 « 7Fe3C + 63НС1; (5)
28А1С13 4- ЗС7Н8 + 15Н2 == 7Al4Cg + 54НС1; (6)
7Т1С14 -4 ед + 10Н2 = 7TiC + 28НС1; (7)
28ВС13 + С7НЙ + 38Н2 =» 7В4С + 84HCI. (8)
Данные расчета показали, что в интервале темпера-
тур, при которых образуется карбид кремния, реакции
(5) — (8) также идут. Вследствие этого для получения
чистого SiC необходимо тщательно очищать исходные
материалы от примесей. Авторы работы исследовали
влияние температуры поверхности осаждения, концентра-
ции реагентов и скорости парогазового потока на ско-
рость осаждения карбида кремния. На основании ис-
следования установлены оптимальные условия получе-
ния карбида кремния из смеси четыреххлористого
кремния, толуола и водорода, которые сводятся к сле-
дующим: температура 1600—1700°С, соотношение SiCl4 :
: С в исходной парогазовой смеси 3: 1, избыток водорода
против расчетного 8—11-кратный, скорость парогазово-
го потока 1,0—1,5 л/мин для реактора диаметром 50 мм.
ГЛАВА V Bel
ХИМИЧЕСКИЕ СВОЙСТВА КАРБИДОВ
Карбид лития легко загорается при комнатной
температуре в атмосфере фтора или хлора с образова-
нием фторида или хлорида лития. С бромом и йодом
реакция идет при нагревании [64]. В атмосфере кисло-
рода, паров серы и селена он сгорает при температурах
700—800° С; с фосфором образует фосфид, который раз-
лагается водой с выделением фосфина.
Водой карбид лития разлагается с выделением аце-
тилена. При комнатной температуре реакция проходит
быстро, при нагревании до 100° С очень бурно. В работе
[4] показано, что чистый карбид лития полностью рас-
творяется в воде с выделением ацетилена в количестве,
точно соответствующем составу Li2C2, Концентрирован-
ные кислоты действуют на карбид медленно; расплавлен-
ная щелочь разлагает его с выделением тепла, расплав-
ленные хлорат, нитрат и перманганат калия быстро раз-
лагают его.
Карбид L12G2 — сильный восстановитель, он восста-
навливает хлорид лития LiCl до Li2Cl с выделением угле-
рода. При высоких температурах происходит разложение
LiaCl—>LiCI 4- Li. Металл при этом снова взаимодейству-
ет с углеродом с образованием Li2C2. При анализе кар-
бида лития [4] содержание углерода определяется изме-
рением объема ацетилена, выделяющегося при его раз-
ложении водой, в градуированной трубке, над слоем
ртути. В растворе содержание лития определяется объ-
емным методом.
Карбид К2С2 под действием паров воды разла-
гается с выделением ацетилена. В среде хлора он заго-
рается при комнатной температуре; с СО при обычной
температуре не взаимодействует.
КНС2 активно взаимодействует с СО2, SO2.
ром [64].
204
Карбид натрия. Сухой воздух не действует на
карбид натрия. Во влажном воздухе он разлагается с вы-
делением ацетилена:
Na2Ca + 2НаО = 2NaOH + СаНа.
Под действием воды разложение происходит со взрывом
с выделением углерода [64].
Хлор и бром также взаимодействуют с Na2C2 со
взрывом с образованием хлористого натрия и выделени-
ем углерода. Реакция с йодом идет менее энергично с
образованием C2J4. При нагревании с фосфором в ат-
мосфере водорода образуется фосфид натрия, который
при действии на него воды выделяет фосфин. В токе хло-
ристого водорода карбид натрия сгорает с образованием
хлористого натрия и выделением углерода и водорода.
Сероводород взаимодействует с Na2C2 при темпера-
туре 150° С; в атмосфере СО2 и SO3 карбид натрия вспы-
хивает уже при комнатной температуре' с выделением
углерода; СО не действует до температуры 250° С.
Такие окислители, как хлораты, нитраты и брома-
ты, образуют с карбидом натрия смеси, которые загора-
ются при ударе и истирании; хлориды и йодиды свинца
и ртути, хлориды алюминия и железа при истирании с
карбидом разлагаются со взрывом.
Спирты разлагают карбид с выделением ацетилена
и образованием соответствующих алкоголятов натрия."
Соединение NaHC2 легко разлагается водой [5]:
NaHC2 + H2O = C2H2-|-NaOH+60,7 дж. В атмосфере хло-
ра и брома NaHC2 разлагается при комнатной темпера-
туре; легко разлагается спиртом с образованием алко->
голята и выделением ацетилена.
Карбид рубидия. При действии воды карбид
рубидия разлагается с выделением ацетилена. Разбав-
ленная и концентрированная соляная кислота легко
разлагает его, азотная кислота взаимодействует е ним со
взрывом.
Карбид рубидия при обычной температуре не окис-
ляется сухим кислородом [64]. В токе фтора, хлора, бро-
ма и йода он загорается при комнатной температуре.
Жидкая сера, фосфор, мышьяк, кремний и бор взаимо-
действуют с Rb2C2 при легком нагревании. Углекислый
и сернистый газ, а также окислы азота разлагают его
лишь при нагревании; СО восстанавливается карбидом
205
рубидия при температурах 700—800° С до атмосферного
углерода. Карбид рубидия восстанавливает окисли, ме-
ди, железа и хрома.
При нагревании смеси карбида рубидия с кальцием
в вакууме выделяется металлический рубидий, который
осаждается на холодных стенках печи в виде блестяще-
го зеркала.
Карбид цезия [64] разлагается водой с образо-
ванием гидроокиси цезия с выделением ацетилена:
Cs2C2 + 2НаО = 2CsOH + С2На.
В концентрированной азотной кислоте загорается.
Галогены взаимодействуют с ним при комнатной
температуре со вспышкой. Аналогично реагирует Cs2C2
с бором и аморфной серой.
Карбид цезия — сильный восстановитель, при не-
большом нагревании он восстанавливает окись железа.
Карбид меди (Си2С2) в воде практически не
растворяется. С раствором КНС2 в жидком аммиаке
взаимодействует по схеме [80]
Cu2C2 + 2КНС2 = 2СиНСа + КаС2;
2СпНС2 + 4КНС2 = 2К2 [Си (НС2)3].
Карбид бериллия Ве2С легко сгорает в токе
фтора с образованием растворимых фторидов берил-
лия [64]. В хлоре он сгорает при температурах 700—
800° С с образованием летучих хлоридов и выделением
графита. Бром взаимодействует с Ве2С лишь при высо-
кой температуре, йод при 800° С не взаимодействует.
Фтористый водород взаимодействует с Ве2С при темпе-
ратуре 450° С. При переходе от фтористого водорода к
хлористому, бромистому и йодистому температура, при
которой идет взаймодействие, возрастает, достигая для
HI 750° С. При этом образуются соответствующие гало-
идные соединения бериллия.
При температурах 700—800° С Ве2С окисляется кис-
лородом; при этом образуется пленка окисла, предохра-
няющая карбиЯ от дальнейшего окисления. При темпе*
ратуре 1000° С пары серы действуют с образованием
сульфида бериллия. В контакте с влажным воздухо
при комнатной температуре [88] Ве2С быстро подвер'
гается гидролизу, образуя Ве(ОН)2.
206
Концентрированные соляная и азотная кислоты
медленно разлагают карбид бериллия при нагревании.
Серную кислоту он восстанавливает до сернистой. Фто-
ристоводородная кислота разлагает его легкр.
Характерна для карбида бериллия его раствори-
мость в воде при комнатной температуре с выделением
метала:
ВеаС 4- 4НаО СН4 4- 2Ве (ОН)2.
Проводилось [95] исследование взаимодействия кар-
бида Ве2С с кремнием и никелем.
Исследование растворимости карбида бериллия в
жидком меднобериллиевом сплаве [96] доказало отсут-
ствие взаимодействия, приводящего к распаду карбида.
При температуре 1700° С становится заметной диссоци-
ация Ве2С по схеме
ВеаСтв <— 2Веж 4- СГраф.
Хенигшмидт [64] анализировал карбид бериллия^ раз-
лагая его при нагревании в токе очищенного й осушен-
ного хлора. При этом выделялся летучий хлорид берил-
лия, по которому определяли содержание бериллия.
Содержание бериллия определяли [96] весовым мето-
дом в виде ВеО после кипячения навески карбида с сер-»
ной кислотой (1 : 1).
Содержание общего углерода определяли [64] после
обработки навески карбида хлором взвешиванием нерас-
творимого остатка. Этот метод малоэффективен, наибо-
лее распространено определение содержания общего уг-
лерода сжиганием навески в токе кислбрбда.
Для определения содержания свободного углерода
[96] сжигали осадок, выделяющийся после разложения
Ве2С длительным кипячением с серной кислотой (1 : 1).
Этот метод может привести к значитёльным ошибкам в
определении содержания Ссвоб, так Как при длительном
кипячении возможно окисление графита серной кисло-
той. Для определения содержания связанного углерода
количество выделяющегося при гидролизе Ве2С метана
определяли сжиганием.
При определении содержания свободного углерода
[765] навеску карбида разлагали серной кислотой (1:1)
при нагревании с обратным холодильником. Нераство-
рившийся остаток сжигали в токе кислорода при темпе-
207
ратуре 900° С в течение 1 ч, поглощая выделяющийся
СО2 аскаритом.
Содержание бериллия определяли в фильтрате осаж-
дением раствором фосфата аммония в виде NH4BePO4
Рис. 70. Зависимость скорости
разложения СаСа водой от
температуры воды и размеров
частиц карбида (цифры на кри-
вых — лии)
воду. При избытке карбида
ция [116].
который после прокали-
вания взвешивали в виде
Be2P20y.
Карбиды магния.
Mg3C2 и MgC2 разлагают
воду, образуя соответст-
венно аллилен и метил-
ацетилен [64]. Содержа-
ние Mg3C2 определяли по
содержанию аллилена,
выделенного при обработ-
ке карбида водой.
Кррбид каль-
ция. СаС2 легко разла-
гается водой с выделени-
ем ацетилена:
СаС2 2Н2О = С2Н2 +
+ Са(ОН)2.
Выделяющийся при этом
ацетилен поглощается ам-
миачным раствором хло-
ристой меди и совершен-
но чист [64]. Указанная
реакция идет при избыт-
ке воды, т. е. когда кар-
бид кальция бросают в
кальция протекает реак-
СаС2 + Са (ОН)2 = 2СаО 4- С2Н2.
Исследовали [743] влияние состава карбида, размера
зерен и температуры воды на скорость разложения кар-
бида. Установлено, что чем ближе карбид к расчетному
составу, тем больше скорость разложения. Увеличение
размеров частиц резко снижает скорость разложения,
что связано с уменьшением поверхности контакта час-
тиц карбида с водой. Повышение температуры воды так-
же увеличивает скорость разложения (рис. 70). Уста-
208
Таблица 34
Зависимость времени разложения карбида от размера частиц
Время, сек Размер частиц карбида, мм
2—4 4—9 8—15 15—25 25—50 50—80
Среднее активного разло- жения 60 99 109 254 810 994
Среднее полного разло- жения ....,, 255 259 304 488 930 1237
Отношение, % . 23,5 38,3 35,9 52 87 80,5
новлено также, что при разложении карбида водой аце-
тилен выделяется неравномерно во времени. Около 97%
карбида разлагается весьма активно, остальные 3%
разлагаются медленно. Отношение времени активного
разложения ко времени полного разложения зависит от
размера частиц карбида (табл. 34).
Пары воды при температурах ниже 250° С взаимодей-
ствуют с СаС2 с выделением С2Н2 и образованием
Са(ОН)2; при температурах выше 250° С образуются
СаО и С2Н2, при температурах 700—800° С — СаО, угле-
род и водород [104].
Изучали [744] взаимодействие карбида кальция с во-
дяным паром методами рентгеноструктурного и химиче-
ского анализов. Показано, что реакционная способность
разных граней кристаллов СаСа по отношению к водяно-
му пару различна и убывает в ряду (100) > (001) >
> (110)> (101) > (lliy.
Разбавленные минеральные кислоты и водные рас-
творы щелочей разлагают СаС2. Дымящая серная кис-
лота действует медленно, концентрированная серная
кислота разлагает его быстрее; дымящая азотная кис-
лота при температуре ее кипения почти не разлагает
карбид кальция.
Сухой водород не реагирует с карбидом кальция до
температуры 2200° С. Выше этой температуры идет ре-
акция
СаС2>к -|- На -+ Саг -|- СаНа.
Азот при нагревании взаимодействует по реакции
СаС2 + N2 - CaCN2 + G,
209
Таблица 35
Константы скорости реакции азотирования карбида кальция
Ускоряющие добавки % от массы шихты К. мкм/мин, при температуре, °C 1050
850 900 950 1000 |
Без доба- вок — — — 0,0055 0,02 0,075
CaCNj+C 10 0,0065 0,025 0,085
CaClj 5 0,04 0,075 0,135 1 '
СаС1я 2,5 0,018 0,043 0,085 0,19 —
CaCI8 1 0,008 0,095 0,21
CaFa 3 — 0,05 0,13 0,265
€aFa 1,5 —’ 0,01 — 0,11 0,275
CaFa 0,5 — — 0,07 0,22
NaF 1 1 0,035 0,12 0,3
BaFa 2,5 0,02 0,045 0,085 0,225
Na2SiFe 1 ?' - 0,01 0,045 0,095 0,26
которую используют в промышленности для получения
цианамида кальция.
Кинетика азотирования порошка СаС2 описывается
линейным уравнением d=kx. Влияние ускоряющих до-
бавок и температуры процесса на константу скорости
реакции приведено в табл. 35 [745—747].
В токе аммиака при температурах выше 650° С идет
также реакция
СаСа -f- 4NH3 ь= CaCNa 4- NH4CN + 4На.
В сухом кислороде СаС2 окисляется при температурах
700—800° С до карбоната кальция. При нагреве в атмос-
фере СО при температура^ выше 1600° С происходит
взаимодействие по реакции
СаС2 + СО = СаО + ЗС.
Сухой хлор взаимодействует с СаС2 при температуре
250° С с образованием СаС12 и выделением углерода;
пары серы образуют сульфид кальция С aS и сероугле-
род, фосфор — фосфид состава СазР2, мышьяк —
СазАз2; в двух последних случаях углерод выделяется в
виде графита.
Газообразный хлористый водород и сероводород так-
же взаимодействуют с карбидом с выделением ацетиле-
на. Все эти реакции протекают с выделением тепла.
Перекись натрия разлагает СаС2 по реакции
210
7NaA + 2CaCa = 2CaO + 4NaaCO3 -f 3Naa.
При взаимодействии карбида кальция с роданидом
калия [748] образовывались сульфид кальция и цианис-
тый калий и выделялся графитг
СаС2 + 5KCNS = CaS5 -Ь 5KCN + 2С.
Органические хлорсодержащие продукты взаимодей-
ствуют при температурах 300—400° С по реакциям
2СаС2 + СС14 = 2СаС12 + 5С;
ЗСаС2 + 2СНС13 = ЗСаС1а + 8С + Н2.
Карбид кальция — сильный восстановителе [749] и
при высоких температурах может полностью восстанав-
ливать сульфаты до сульфидов:
Me SO4 + 4CaG2 = 4СаО -f- Me S -j- 8G;
при этом выделения СО не наблюдается.
Окислы свинца, олова, цинка, железа, марганца, ни-
келя, кобальта, хрома, молибдена и ванадия восстанав-
ливаются с образованием Преимущественно сплавов
кальция. Окислы марганца, хрома, молибдена, вольфра-
ма, титана и кремния при сплавлении с карбидом каль-
ция восстанавливаются до соответствующих карбидов.
При взаимодействии с SiO2 образуются в зависимо-
сти от соотношения компонентов силикаты a-CaSiOs,
P-Ca2SiO4 и y-Ca2SiO4, а также P-SiC. Окись алюминия
восстанавливается карбидом кальция до металлическо-
го алюминия, а при длительном нагревании образует-
ся карбид алюминия [116]. Механизм взаимодействия
карбида кальция с окислами описан в работах
[143—145].
При взаимодействии карбида кальция с ферротита-
ном при температре 1450° С и давлении 66,6 н/м2
(0,5 мм рт. ст.) получен металлический кальций, содер-
жащий менее 1,3% примесей [140].
Опыты по взаимодействию карбида кальция с бором
показали, что при температуре 1300°С карбид кальция
взаимодействует с бором с образованием гексаборида и
борокарбида. При избытке металлического кальция бо-
рокарбид разрушается с образованием СаВ6 и СаС2.
При взаимодействии окиси кальция с борным ангид-
ридом и углеродом при температуре 1900° С [142] обнару-
211
жен борокарбид состава СаС4В2, при взаимодейгт
кальция с углеродом и бором при температуре 1300°г и
борокарбид СаС2В [141], который при обработке соД
кислотой разлагается с выделением углеводородов Ной
При анализе карбида кальция определяют содёп
ние кальция и связанного углерода. Для этого наве^
карбида разлагают водой. В водном растворе опредеп
ют содержание кальция веёовым методом. Содержав
углерода определяют по количеству выделившегося ntt*
разложении карбида ацетилена. ₽и
Давление и объем выделившегося ацетилена [750] и
меряют манометром. Ошибка определения 0,5%. J И3
Предложен экспрессный метод прямого определения
карбида кальция в шлаках [751], который сводится к рас
творению навески в разбавленной азотной кислоте, на
сыщенной селитрой, и сжиганию выделяющегося ацети
лена в печи Марса. Образующаяся при этом углеки -
лота поглощается раствором* щелочи в аппарат
Вюртца-Штролейна. Содержание свободного углерода
определяют следующим методом [752]. Навеску шлака
смачивают в прокаленной фарфоровой лодочке несколь-
кими каплями соляной кислоты (1:1) и высушивают
При этом весь карбид разлагается с выделением ацети-
лена, а в шлаке остается свободный углерод, содержа-
ние которого определяют обычным методом. Содержа-
ние карбида кальция определяют по разности:
%СаС, = (Собщ — Ссвоб)2,67,
(Tie 2,67 коэффициент пересчета содержания С СЕМЗ на
анали3е Карбида кальция определяли [126] с -
держание кальция, связанного в карбид, по количеству
иапХ0,1?3 6 ацетилене, выделяющемся при разложени
ctrpwuki водои- Выход ацетилена из карбида в производ-
ся япр-rJ уСловия* определяют по объему выделяющее -
ся ацетилена по формуле-
—0,5) д.юоо
g
Здесь V — „
ацетилена, выделившегося при анали е»
р = (273 4- 20)
760 (273 + 0 *
212
где t — температура;
Р — атмосферное давление, приведенное к 0° С;
w — упругость водяного пара при температуре /;
g — навеска карбида, г.
Разработан [823] метод полного технического анализа
карбида кальция, по которому определяют содержа-
ние СаС2 и примесей: SiO2, Fe2O3, А12О3, CaO, MgO, S,
Р, CaCN2 и СсВоб- Определение СаС2 основано на из-
мерении объема выделившегося при разложении водой
ацетилена. Шлам, остающийся после определения содер-
жания СаС2, выпаривают с концентрированной соляной
кислотой. Нерастворившийся остаток сплавляют со
смесью соды и буры и в растворе определяют содержа-
ние кремния колориметрическим методом [824]. Из али-
квотных частей фильтрата после отделения нераствори-
мого остатка определяют Fe2O3 колориметрическим
методом с сульфосалициловой кислотой, А12О3 — по реак-
ции с алюминоном, MgO — с красителем титановым
желтым [825]. Содержание СаО определяют комплексо-
нометрически с мурексидом [826].
Определение содержания свободного углерода за-
ключается в сжигании навески карбида в токе кислорода
при температурах 800—900° С с последующим титрова-
нием выделившейся СО2 .раствором Са(ОН)2, При опре-
делении содержания серы и фосфора разлагают навеску
в соляной кислоте с последующим продуванием азотом
H2S и РН3 и определяют серу йодометрическим методом
после улавливания H2S раствором уксуснокислого калия
и фосфора фотоколориметрически после улавливания
РН3 раствором перманганата калия.
Карбид бария. ВаС2 легко разлагается водой
по реакции
ВаС2 + 2НаО = Ва (ОН)2 + Н8С2.
В атмосфере сухого воздуха ВаС2 устойчив до темпе-
ратуры 600—700° С [64, 146]. Галогены легко разлагают
его — пары йода действуют при температуре 122°, пары
брома — при 130° и хлора — при 140°. Фтором ВаС2
разлагается при нагревании с образованием BaF2.
При 700—800° С карбид бария взаимодействует с па-
рами серы с образованием сульфида бария и сероугле-
рода; аналогично взаимодействует селен; пары фосфора
образуют фосфид Ва3Р2, а мышьяк — арсенид.
213
Азот взаимодействует по реакции
ВаСа 4- Na BaNaC + С.
При действии СО
ВаСа + СО = ВаО + ЗС.
Нагревание ВаСа с Ва(ОН)2 приводит к образова-
нию бензола:
ЗВаСа + ЗВа(ОН)а = бВаО + СвНв.
Аналогично кальцию барий при взаимодействии с уг-
леродом и бором при температуре 1300°С образует бо-
рокарбид состава ВаС2В и при взаимодействии окиси
бария е борным ангидридом и углеродом при темпера-
туре 1900° С — борокарбид состава ВаС4В2 [141, 142].
Борокарбиды разлагаются разбавленной соляной кисло-
той с выделением жидких непредельных углеводородов
с открытой цепью, которые полимеризуются в твердое
аморфное вещество.
Анализ карбида бария [64] проводился аналогично
анализу карбида кальция. Карбид разлагали в воде, в
растворе осаждали барий и измеряли количество вы-
делившегося при разложении ацетилена, соответствую-
щего содержанию связанного углерода в карбиде. В Ин-
ституте лроблем материаловедения АН СССР при ана-
лизе карбида бария определяли содержание бария,
связанного и свободного углерода. Содержание бария
определяли после разложения навески карбида в раз-
бавленной (1: 20) соляной кислоте комплексонометри-
ческим методом прибавлением избытка трилона Б и тит-
рованием избытка трилона раствором хлористого никеля
в присутствии мурексида.
.4 Определение содержания связанного углерода осно-
вано на разложении карбида водой в специально при-
способленной для этой цели колбе с последующим сжи-
ганием выделяющихся газов в токе воздуха. Количество
образовавшейся при этом углекислоты определяют обыч-
ным абсорбционно-газообъемным методом.
Для определения содержания свободного углерода
навеску карбида разлагают водой или разбавленной
(1:20) соляной кислотой. Свободный углерод в виде
нерастворившегося остатка отфильтровывают на сл0
прокаленного асбеста, осадок высушивают и сжигают
токе кислорода.
214
Карбид скандия легко разлагается водой, вы-
деляя при этом в виде газообразных продуктов СН4,
С2Нб, а также жидкие и твердые углеводороды. Хи-
мические свойства ScC почти не изучены. В атмосфере
кислорода он окисляется при температурах 700—
800° С.
Карбид иттрия легко разлагается водой с вы-
делением смеси углеводородов и водорода. Основные
продукты разложения YC и У2Сз—метан (95 и 70% со-
ответственно) и YC2 ацетилен.
В концентрированных кислотах карбиды иттрия зна-
чительно устойчивее, чем в разбавленных. В H2SO4 кар-
биды иттрия более устойчивы, чем в НС1 и HNO3.v
Дикарбид иттрия легко взаимодействует с галогена-
ми — с фтором при комнатной температуре, с остальны-
ми — при температурах 700—800° Q легко сгорает в
кислороде, парах серы и селена [64]. При изучении окис-
ления порошков карбидов иттрия {861] показано, что
карбиды иттрия разлагаются на воздухе при комнатной
температуре в основном за счет влаги воздуха, В ат-
мосфере сухого воздуха и сухого кислорода дикарбид
иттрия устойчив при комнатной температуре. Окисление
порошков карбидов иттрия при повышенный температу-
рах кислородом свидетельствует о том, что при 500° С
большая часть карбидов окисляется.
При анализе карбидов иттрия определяют содержа-
ние общего и свободного углерода и иттрия. Содержа-
ние общего углерода определяют обычным методом сжи-
гания в токе кислорода с последующим абсорбционно-
объемным определением выделившегося при сжигании
углекислого газа. Для определения содержания свобод-
ного углерода карбид разлагают в азотной кислоте
(1 : 10) и нерастворившийся остаток сжигают в токе
кислорода. Содержание иттрия определяют или окисле-
нием навески карбида до окиси Y2Oa сжиганием в му-
феле при температурах 900—1000° С, или объемным ме-
тодом — титрованием раствором трилона Б с индикато-
ром арсен азо [788].
Карбиды лантана. Карбиды лантана, так же
как и карбиды всех лантаноидов, взаимодействуют с
водой.
В газообразных продуктах разложения LaC2 обна-
ружены: 53% С2Н2, 15,6% Н2, 11,2% С2Н4 и 20,2° о
215
Таблица 36
Состав газообразных продуктов разложения ЬаС2
Среда Содержание, % ""
СгНг сгн. сн, ~~
НС1 88,8 0,5 6,2
FeCls+^Cl 96,7 0,2 1,2
n
3,4
1,0
С2Н6 [8]. При разложении полуторного карбида основ
ные продукты — метан (75%) и водород (25%).
Исследование газообразных продуктов разложения
дикарбида лантана срляной кислотой и раствором хлор-
лого железа в соляной кислоте [790] показало, что со-
став газообразных продуктов изменяется в зависимости
от среды (табл. 36).
Разбавленные минеральные кислоты [64] легко раз-
лагают дикарбид лантана; концентрированная азотная
кислота не оказывает заметного действия; концентриро-
ванная серная кислота восстанавливается до сернистой.
Исследование поведения дикарбида лантана в раз-
личных средах свидетельствует о практически полной
растворимости LaC2 в минеральных кислотах и устой-
чивости в органических растворах.
При нагревании в токе аммиака LaC2 превращается
в нитрид; легко взаимодействует с галогенами, серой и
селеном с образованием галогенидов, сульфидов и селе-
нидов; при действии газообразного хлористого водорода
LaC2 разлагается с образованием ЬаС1з и выделением
углеводородов; азот и фосфор не взаимодействуют с ним.
Не разлагаясь растворами щелочей, дикарбид легко
разлагается расплавами щелочей, а также такими окис-
лителями, как перманганат, хлорат или нитрат калия.
В сухом воздухе и кислороде дикарбид лантана пр
комнатной температуре практически устойчив. Во вла
ном воздухе быстро разлагается.
Порошок дикарбида лантана в атмосфере кислоро
начинает окисляться при температуре 500° С,
ное окисление происходит при температурах 800—-90 '
Характер окисления LaC2 и La2C3 одинаков, но с
рость окисления La2C3 выше. гс., niwe-
При химическом анализе карбида лантана [64] г
216
Таблица 37
Состав продуктов разложения СеСг соляной кислотой
к раствором FeCls в соляной кислоте
Содержание, %
Среда с,н, с2н. сн« Н,
НС1 74 1,5 — •
FeCls+HCl 94,7 Не обн. 1,6 2,1
деляют содержание общего углерода обычным методом
сжигания, содержание свободного углерода—разложе-
нием карбида разбавленной азотной кислотой и взвеши-
ванием нерастворимого остатка. Содержание лантана
[790] определяли, взвешивая остаток после сжигания
углерода, который представлял Ьа2О3.
Наиболее эффективный метод определения содержа-
ния лантана объемный — титрование раствором трило-
на Б с индикатором арсеназо [788].
Карбиды церия. При разложении дикарбида
церия водой выделяется смесь углеводородов, состав ко-
торых, по данным разных авторов, различен
(см. табл. 5).
Основные продукты разложения полуторного карби-
да Се2Сз водой — метан и водород, кроме того, выделя-
ются этилен, жидкие и твердые углеводороды.
Состав газообразных продуктов взаимодействия ди-
карбида церия с соляной кислотой и раствором хлорно-
го железа в соляной кислоте приведен в табл. 37 [790].
Дикарбид церия разлагается в разбавленных мине-
ральных кислотах; в концентрированной серной кислоте
разлагается при нагревании, восстанавливая H2SOi до
H2SO3. С фтором не взаимодействует при комнатной
температуре, но при нагревании образует летучие фто-
риды, с остальными галогенами при нагревании также
образует соответствующие галогениды церия. При дейст-
вии газообразного хлористого водорода он разлагается
при температуре 650° С с образованием хлорида церия.
Аналогично взаимодействует с Ш. Азот и Ф°СФ°Р
взаимодействуют с СеС2 до температуры 800 С. с с Р
водородом при температуре 600° С образуется см
сульфида церия и графита.
217
Дикарбид церия активно окисляется кислородом
температуре 800" С, полуторный карбид заметно ой
ляется уже при 600 С.
Анализ карбида церия проводят аналогично анализ
карбида лантана.
Карбиды празеодима и неодима. ра,
бавленные кислоты действуют на дикарбиды празеодима
и неодима так же, как и вода. Концентрированные ки
лоты действуют медленнее.
Оба карбида легко взаимодействуют с галогенами с
образованием соответствующих галогенидов, Взаимод й
ствие с фтором медленное при комнатной температуре
с хлором и йодом —при температурах 700 800 С
причем взаимодействие с хлором сопровожда т я
вспышкой. Азот и газообразный аммиак разлагаю
при температуре 1200° С с образованием нитридов. Се
роводород при температурах 700—800° С взаимодеис
вует, образуя сульфиды.
В атмосфере кислорода карбиды празеодима и нео
дима сгорают начиная с температуры 500° С. В атмосф
ре сухого воздуха и кислорода при комнатной темпера-
туре дикарбиды достаточно устойчивы. Анализ карбид
празеодима и неодима проводят так же, как и карбида
лантана.
Карбид самария легко разлагается водой с в -
делением смеси углеводородов. Разбавленные минерал -
ные кислоты разлагают SmC2 тем легче, чем больше о
разбавлены. В токе фтора и хлора SmC2 разлагает
при нагревании с образованием соответствующих гал -
генидов. С бромом и йодом взаимодействие происход
при температуре 700—800° С. В кислороде SmC2 сг р -
ет при температуре 400° С.
Карбиды тория. ThC 1-х и ThC2-x — химическ
малостойкие соединения. При нагревании на возд} е
они легко окисляются до окисла ThO2. Легко разлагают-
ся водой и разбавленными минеральными кислота 1
оставы газообразных продуктов разложения карбид0
тория водой приведены в табл. 5.
рппиДл иДЬ! У р а н а — химически малостойкие с
действии ‘ Наиболее характерно для них разложение _
ются гячп°«Ь1’ ПРИ этом продуктами разложения я
Ды [228] °бразные, жидкие и твердые углевод
218
Монокарбид в виде спеченных образцов легко раз-
рушается в кипящей воде [231]. Изучение влияния при-
месей железа, кремния, вольфрама, азота, кислорода и
водорода на разложение монокарбида водой при темпе-
ратуре 60° С показало, что железо и вольфрам повыша-
ют скорость разложения, в то время как водород и кис-
лород понижают ее. При нагревании с парами воды мо-
нокарбид загорается, превращаясь в окись черного
цвета. При комнатной температуре быстро реагирует с
парами воды. С разбавленными минеральными кислота-
ми (HCk H2SO4, HNO3) легко взаимодействует с выде-
лением водорода, ацетилена, этилена, этана и других
углеводородов. Действие концентрированных кислот бо-
лее слабое. Фосфорная кислота при комнатной темпера-
туре медленно реагирует С монокарбидом, при нагрева-
нии скорость взаимодействия растет.
При действии СО2 монокарбид урана окисляется при
температуре 500° С [231, 774]. Скорость окисления посто-
янна в интервале температур 500—830° С. Сравнение
взаимодействия монокарбида и металлического урана с
СО2 показало, что монокарбид более устойчив. Так, при
температуре 500° С образец окисляется со скоростью
0,6% за 6 ч, а металлический уран—со скоростью 6%.
С водородом монокарбид не взаимодействует до высо-
ких температур.
1}Лонокарбид окисляется углекислым газом по реак-
ции UC + 2CO2 = UO2+C + 2CO. При температурах выше
500° С наблюдается взаимодействие С + СО2 = 2СО и при
1000° С 50% С переходит в газовую фазу в виде СО [774].
Скорость реакции окисленця монокарбида углекислым
газом зависит от плотности образцов. При плотности
80% от теоретической изменение в массе в результате
окисления составляет 140 мг/(см2-ч). Изменение в мас-
се образца плотностью 90% при тех же условиях окис-
ления составляет 10 мг/(см2-ч) [199].
При исследовании взаимодействия монокарбида с СО
(рис. 71) [774] рентгенографически в продуктах реакции
обнаружены дикарбид и двуокись урана. Авторы пред-
полагают, что взаимодействие идет по реакциям
UC + 2СО = UO2 + ЗС;
UC + С = ПСа.
219
_ят«ое мелкодисперсный монокар
монокарбида.
Рис, 71. Зависимость скорости взаимодействия
72. Зависимость скорости окисления монокарбида ура-
на от температуры, °C:
°00* 2 850; 3—700; 4 — 550; Л- — 500; tf_450; 7 —400; в —350
скорой
ратура возгорания возрастала с увеличени в°3'
нагрева и диаметра частиц порошков. Темпер
горания карбида урана на 40 град выше т
возгорания порошка металлического урана.
22Q
Изучение окисления монокарбида урана кислородом
[774] (рис. 72) показало, что конечный продукт окисле-
ния U3O8 и реакция идет по схеме
3UC + 4О2 = U3OS + ЗС.
Параллельно происходит взаимодействие С + О2 = СО2
и 75% С переходит в газовую фазу. '
Данные разных авторов по изучению процесса окис-
ления монокарбида урана [774, 776, 777] значительно
расходятся, что, вероятно, объясняется работой с образ-
цами различных состава и плотности.
Бремя, мин
Рис. 73. Зависимость скорости взаимодействия моно-
карбида урана с азотом от температуры, °C:
/ — 700; 2 — 775; 3 — 850; 4—1000
Исследование окисления образцов монокарбида, при-
готовленных разными методами [777], привело авторов
к заключению, что наилучшие результаты получаются
на образцах, полученных дуговой плавкой. Эти образцы
соответствовали по составу иС1?02 и плотность была
весьма близкой к теоретической. При окислении в чис-
том сухом кислороде в интервале температур 550—
800° С на поверхности образца образовывался легкоотде-
ляемый слой состава U3O8—х- Между этим слоем и по-
верхностью карбида обнаружен слой окисла UOg.
Окисление подчиняется линейному закону.
Энергия активации составляет 90,9±4,2 кдж[моль
(21,7±1 ккал/моль).
Зависимость скорости взаимодействия монокарбида
С азотом приведена на рис. 73.
221
Таблица 38
Взаимодействие монокарбида урана с некоторыми металлами
и карбидами ____________________
Металл или карбид Темпе- ратуре Характер взаимо- действия* Примечание
Натрий . . . . . Натрий т- калий , . Бериллий . .. . . Магний , г • « Кальций , . . » . Олово . . . « . • Свинец . , . * , . Висмут . , . , i Алюминий , . , , Титан Цирконий Л . Ниобий ..... Тантал Хром ...... Молибден . . . s , Железо Нержавеющая сталь . Никель г . . , . , Родий < . г . . | Иридий . . „ | Карбид титана . Карбид циркония . / ’ <500 800 650 870 870 600 600 >1200 1000 1000 1800 1435 750 1170 1098 + + + + + + + 1 + 1 + + ++ I | | | | +11 1,5 Л?н/Л2 (15 кГ/Я10* течение 12 ч ’ В течение 330 ч » > » » Образуется UA13 Образуется эвтекти а содержащая 34±0,5% С Образуется тр йн соединение UCFea Образуется эвтекти а, содержащая 51,5± 5 Fe Образуется эвтектика Образуется соединев е UNis Образуются тро ые соединения типа UjMeC: [920] Образуется раство (10%) карбида титана в монокарбиде урана Образуется непреW ный ряд твердых раст
222
Продолжен не табл. 38
Металл или карбид Темпе-, ратура °C Характер взаимо- действия* Примечание
Карбид гафния . . . 4- То же
Карбид ванадия . . — Наблюдается частич- ное растворение
Карбид ниобия . . • Т— + Образуется непрерыв- ный ряд твердых раство- ров
Карбид хрома (СГ3С2) — + Наблюдается частич- ное растворение
Карбид молибдена — 4- То же
К рбид вольфрама — + Образуется твердый раствор до 10% (мол.) WC в UC
Карбид тория . , , — 4- Образуется непрерыв- ный ряд твердых раство- ров
+ взаимодействует, не взаимодействует.
Монокарбид урана устойчив при взаимодействии с
органическими теплоносителями. После выдержки при
температурах до 350° С в течение 5 ч в дифениле и три-
фениле никакого взаимодействия не обнаружено. При
нагревании в глицерине при температуре 100°С потеря
массы составляла 50 мг!см2.
Результаты исследования взаимодействия монокарби-
да урана с разными металлами приведены в табл. 38
(214, 231, 257, 258].
Полуторный карбид наименее изучен. С водой он при
температуре 750° С заметно не взаимодействует [228].
Энергично вступает в реакцию с соляной кислотой при
температуре 750° С; с серной и азотной кислотами мед-
ленно взаимодействует при температурах до 170° С и
совсем не реагирует при этих температурах с уксусной
кислотой [231]. При нагревании на воздухе до темпера^
тУры 600° С в течение 5,5 ч образец распадается на от-
Дельные гранулы, имеющие металлический блеск. С йо-
Дом полуторный карбид реагирует при 600° С с обра-
зованием йодида урана.
223
Дикарбид медленно разлагается водой при комиа
ной температуре, при нагревании скорость разложеиЗ
значительно возрастает [231]. При отсутствии воздуха *
результате взаимодействия с водой образуется гидпг?
окись зеленого цвета. В присутствии воздуха образуют
ся продукты серовато-черного цвета. В результате об
разуется смесь углеводородов: */з газообразных и 2
жидких и твердых [770]. 3
С водяным паром дикарбид реагирует при темпера
турах 700—800° С с образованием черной двуокиси ура
на; скорость реакции подчиняется линейному зако
ну [231].
Разбавленные минеральные кислоты (НС1, H2SO4 и
HNO3) быстро разлагают дикарбид с образованием рас-
творов солей уранила, окрашенных в желтый цвет [220]
и смеси углеводородов. Концентрированные соляная,
серная и фосфорная кислоты при комнатной темпера-
туре взаимодействуют медленно, при нагревании бы т
ро. Дикарбид легко разлагается щелочами. На в з-
духе или в атмосфере кислорода он воспламени т-
ся при температуре 400° С с выделением СО
и U3O8.
Исследование окисления дикарбида урана [231] при-
вело к заключению, что до температуры 250° С окисл -
ние подчиняется параболическому закону. При темпе-
ратуре 300° С реакция идет с большим выделением т п-
ла, в результате чего температура образца поднимав я
до 1000° С в течение 1 мин.
Взаимодействие с азотом до температуры 700° С под-
чиняется параболическому закону. Весьма интенсивн
реакция идет при температурах выше 1100° С. При
1180° С и выдержке в течение 12 ч карбид полностью
превращается в нитрид. С фтором при комнатной тем-
пературе взаимодействие не наблюдается, но при сла-
бом нагревании реакция идет со взрывом. С хлор м
реакция дикарбида урана протекает при температуре
350° С с образованием летучего хлорида. Смесь UOs и
UCz—х при температуре 600° С частично взаимодействие
с хлором с образованием тетрахлорида урана, а пр*
800—1000° С •—с образованием высших хлоридов. Fe*
акция с бромом начинается при температурах аьш
300° С, а при 900° С образуется UBr4 и выделяется угл
род. Пары йода взаимодействуют при 500° С с обра
224
ванием UI4. В атмосфере сероводорода дикарбид при
температуре 600 С воспламеняется с образованием
сульфида, в атмосфере аммиака разлагается при темпе-
ратурах 700—800° С.
При сплавлении дикарбида урана с нитратом или
перхлоратом калия образуется уранат калия. При на-
гревании смеси UC2-*, UO2 и NaCl до 1000° С образуется
комплексное соединение Na2UO2Cl4.
Анализируя карбиды урана, определяли [225] содер-
жание урана, общего и свободного углерода и азота. Со-
держание урана определяли сжиганием навесок
(100 мг) до образования U3O8 с последующим соответ-
ствующим пересчетом или объемным методом — восста-
новлением урана амальгамой цинка до четырехвалент-
ного состояния и титрованием полученного раствора 0,2-н.
раствором перманганата калия.
Содержание общего углерода определяли сжиганием
навески 0,1 г в токе воздуха с последующей адсорбцией
выделяющегося СО2 натронной известью. Установлено
[227], что при температуре 1260° С навеска полностью
сгорает и нет необходимости в прибавлении флюса.
В работе [778] предложен более точный метод опре-
деления содержания общего углерода. Сжигание прово-
дят при температуре 950—1050° С, в течение 15- мин, про-
пуская смесь аргона и кислорода. Затем постепенно
уменьшают скорость пропускания аргона и соответ-
ственно увеличивают скорость пропускания кислорода,
поддерживая ее постоянной в течение 30 мин. Содержа-
ние выделившегося СО2 определяют по привесу погло-
тительных трубок, содержащих NaOH, нанесенный на
асбест, и слой Mg(C104)2. При содержании 4,64% С и на-
веске в 1,5 г точность определения составляет 0,03%.
Содержание свободного углерода определяли, рас-
творяя навеску в 0,1 г в 50 мл соляной кислоты (1:1)
при нагревании до кипения, после чего к раствору добав-
ляли 1 мл 10%-ного раствора перекиси водорода и ки-
пятили в течение 15 мин. Выделившийся остаток С СВоб-
после охлаждения отфильтровывали, высушивали при
температуре 140—160° С и взвешивали [225]. В работе
[227] содержание свободного углерода определяли сжи-
ганием остатка после обработки навески карбида соля-
ной кислотой 1:1, отфильтрованного на асбестовую про-
кладку, при температуре 1260° С в течение 15 мин.
15 Косолапова Т. Я.
225
Содержание азота определяли [225] по методу дй
МЭ К Vo бидтитана устойчив против действия соля
ной исерной кислот, растворяется в Царской водке ,
смеси азотной и плавиковой кислот [753], Растворимость
карбида титана в минеральных кислотах, их смесях и
растворах щелочей [754, 755] приведена в табл. 7.
Рис. 74. Зависимость толщины пленки от
времени окисления TiC
тана приРтеМператуое 800е Г гт компактныи карбид ти-
при окислении Р к-я800 данным работы [756],
Фаза рутила но обРазУется преимущественно
СЛ°Боле°г 8 ₽аботе в " в“ясненНИКН0ВеНИЯ 5'глерода чере3
карбидов титаня^п°г <)кисление компактных образцов
исследовано в работеНТГ75??ЛерТеМПератур 400-1000°С
приведены на пис 74 та v РезУльтаты исследования
электронографическоо Химическ°е, рентгеновское и
казали, что в пр °е„исследования окисных пленок no-
п. что в первый период окисления на образцах
226
образуется плотная пленка твердого раствора TiC—-ТЮ
вследствие внедрения атомов кислорода в незаполнен-
ные углеродом места в решетке карбида. Затем Кисло-
род диффундирует через слой твердого раствора с обра-
зованием высших окислов Т12О3 и TiOg. Кажущаяся
энергия активации при диффузии ионов кислорода через
слой твердого раствора вычислена авторами и составля-
Рис. 75. Зависимость кон- Рис. *76. Зависимость элек,
станты окисления TiC от трического сопротивления
температуры TiC от температуры окис-
ления
ет 179,7 кдж!моль (42900 кал/моль), Момент прекраще-
ния роста пленки твердого раствора и начало роста слоя
высших окислов фиксируются изменением электросопро-
тивления окисленных образцов (рис. 76).
Устойчивость образцов карбида титана при окисле-
нии на воздухе [350] зависит от содержания углерода и
во всех случаях меньше устойчивости дисилицида мо-
либдена и диборида циркония и выше устойчивости кар-
бида, цементированного металлом (табл. 39).
Порошкообразный карбид титана окисляется с об-
разованием TiO2 и СО2 при температурах 500—600° С.
15* пт
Таблица 39
Устойчивость карбида титана при окислении на воздухе
(температура 1150° С) . ...... .., ,
Материал-
Изменение массы, мг/1см.-сек\
----—1-----—j-------),ДОвРем4^
8
•Т*'*
18,4% Ссвяв,*
1,3%Ссвоб
TlCj_^g 13,3% Ссвяз.о
Й? 0,6% CCBOd. * • f у * * *
4-10% Со ,t 4 >
TiC^ /- 20% Со 5_ * . .Jt? Г J
TiGj_x -4-10 % ЬЙ * к г >
Мо5‘»2,л\ О ’ ’
6
4
10
8
5
2
0,5
7 9 Разру.
шился
5 6 _
Разрушился
18
5
2
Разрушился
5 6
3 3
8
4
При температуре 1200я С и выше газообразным продук
уом реакции является только -СО [758, 759].
Окисление карбида титана в атмосфере воздуха пр
температуре 1200® С и условия создания защищающи
рт окисления покрытий изучали в работе [137], Покр -
тие из окиси алюминия, будучи пористым, оказалось н -
достаточно эффективным/Вследствие этого использо -
ли нанесение тонкослойных покрытий Из полуколлои
ных растворён на окись алюминия. Растворы готовя
смешивая в различных соотношениях водные раствор
борной кислоты и азотнокислого аммония б раств р
кремниевой кислоты, получаемой перемешиванием 100 ял
свежеперегнанного кремниеэтилового -эфира* с 45 мл
85%-ного этилового спирта, 15 мл воды и двумя каплями
серной кислоты,. Раствор наносили краскораспылителе
на поверхность образца карбида титана, n0KPSvL.
Окисью Алюминия, нагретого до температуры бии—
800° С. При попадании на горячую поверхность образ-
ца растворители испарялись и на слое окиси алюмин
оставались SiO2, А12Оа и В2Оа. Пропитанный несколь о
раз образец обжигали при температуре 1400°С в_те-
ние 5—-10 мин., При этом окислы SiOa, А12Оа 9 в
разовывали стекловидную массу к заполняющую _n°Pg
слое А12Оа. Оптимальный состав пропитываемой с
'Соответствует Содержанию 60% SiO<> и 40° о Ь2МЗ
228
Таблица 40
Термодинамические характеристики реакций взаимодействия
карбида титана с парами и газами
Реакция —£#298 A«S«9S йС/1г98
кдж/моль ккал/моль дж/(град-моль} ; । кал/(град-моль) & О 3 лэ кал/(моль град)
TiC+2H8O=TiO3+C+ +2H3 276,9 66,1 84,6 20,2 209,5 5,0
TiC+H2O=TiO4-C4 H3 93,0 22,2 41,9 10,0 104,7 2,5
TiC-}-3 2O2= 1 i03-J-CO 871,9 208,1 89,8 20,0 7,1 1,7
TiC+O2=TiO+CO 446,2 106,5 3,35 —0,8 6,7 1,6
TiC+2CO=TiO2+3C 540,1 128,9 353,6 84,4 —11,7 —2,8
TiC-W3N9=TiN+C 153,3 36,6 84,2 20,1 2,5 0,6
TiC+4HCl=TiCl4+ гНг-рс 209,5 50,0 153,3 36,6 10,9 2,6
Использование такого состава дает алюмооксидное по-
крытие, защищающее карбид титана при окислении на
воздухе при температуре 1200° С в течение 100 ч и выдер-
живающее 100 циклов теплосмен в интервале температур
20—1200—20° С при охлаждении в струе воздуха.
Водяной пар окисляет карбид титана при высоких
температурах [760], вследствие чего использование
влажного водорода при спекании сплавов на основе кар-
бида титана вызывает их разуглероживание.
С азотом карбид титана при температурах 1000—
1200° С взаимодействует медленно, при температурах
выше 1400° С взаимодействие интенсивное [761].
Фтор реагирует с TiC при комнатной температуре,
остальные галоиды — при температурах 400—700° С.
Хлорсодержащие соединения активно взаимодействуют с
ним, образуя хлориды титана, а в присутствии кислоро-
да или серы — хлорокиси или сульфсхлориды1. По отно-
шению к азоту карбид титана устойчив до температуры
1400—1600° С [761, 762]. Выше этой температуры обра-
зуется нитрид [762]. Закись азота быстро окисляет его по
реакции
TiC 4- 3NaO = TiO8 + 3N8 + CO,
229
Таблица 41
Краевые углы смачивания металлов на поверхности Карбида тц.^
Система Температура, °C Краевой угол 6 Среда
Bi—TiC pb—TiC 300—700 400—1000 138—118 152—90 Вакуум Аргон
Cu—TiC Ag—TiC 1100—1300 980 J 08—70 108 Вакуум
Al—TiC 700 118
Zn—TiC 550 120
Окись углерода не реагирует с карбидом [317], Углеки -
лый газ окисляет его по уравнению TiC + 3CO2=TiO2+
4-4СО, скорость которого также описывается уравнени
ем реакции первого порядка.
Исследование устойчивости карбида титана по отно-
шению к газовым средам [763] позволило определить не
которые термодинамические константы для соответству-
ющих реакций (табл. 40).
Такие металлы, как титан, цирконий, ниобий, хром
никель, кобальт и железо, активно взаимодействуют с
карбидом титана [277, 353—358].
При анализе смачиваемости карбида титана разнь-
ми металлами их делят по способности смачивать по-
верхность карбидов на две группы [359]:
1) металлы, слабо взаимодействующие с поверхно-
стью карбидов, они дают большие краевые углы смачи-
вания, и 2) металлы, активно взаимодействующие с по-
верхностью карбидов, они полностью растекаются по п -
верхности, хорошо смачивая каркас из зерен карбида.
Результаты взаимодействия металлов 1-й группы при-
ведены в табл. 41.
Ко второй группе относятся такие металлы, как Fe
Со, Ni, Сг, Nb, Zr, Ti.
В основу описанного деления на группы положено
различие электронных структур металлов, взаимодейст-
вующих с карбидом. Металлы 1-й группы, не смачиваю-
щие карбиды, характеризуются полностью застроенны-
ми «-электронными оболочками, в то время как металль
' ^РУРпь1~это Переходные металлы с недостроенны
и «-оболочками, которые активно взаимодействуют
р идами, хорошо смачивая их поверхность.
230
Характер взаимодействия карбида Титана с тугоплав-
кими металлами и окислами приведен в табл. 42.
При анализе карбида титана обычно определяют со-,
держание титана, общего и свободного углерода [7541
Растворяют навеску в смесях азотной и фтористоводо-
родной кислот, серной и азотной кислот и соляной и
азотной кислот. Содержание титана определяют объем-
ным или весовым методами.
Содержание общего углерода определяют, ожигая
навеску 0,07—0,08 г карбида в печи Марса в токе кисло-
рода при температуре 1100—1200° С с последующим оп-
ределением количества выделившегося СО2 абсорбцион-
но-объемным методом.
Содержание свободного углерода определяют разло-
жением навески карбида в 0,3—0,5 г смесью азотной и
фтористоводородной кислот. При этом карбид растворя-
ется, а свободный углерод остается в виде нераствори-
мого остатка. Его отфильтровывают через слой прока-
ленного асбеста, осадок промывают, переносят в фарфо-
ровую лодочку, сушат и сжигают в печи Марса в токе
кислорода, определяя количество выделившейся СО2
абсорбционно-газообъемным методом.
В некоторых случаях определяют содержание азота
в карбиде титана. В этом случае применяют колоримет-
рический метод г тимолом и гипобромитом [764].
Карбид ц ир к о н и я — химически стойкое соеди-
нение, при комнатной температуре он йрактически не-
растворим в минеральных кислотах. При нагревании он
стоек в соляной кислоте, разбавленной и концентриро-
ванной [25]. В табл. 6 и 7 приведены данные о стойкости
карбида циркония в различных минеральных кислотах,
их смесях и растворах щелочей [754].
С водяным паром карбид циркония не взаимодейст-
вует до температур 600—700° С. Галоиды легко разлага-
ют его: фтор при комнатной температуре, хлор при тем-
пературе 250° С, бром при 300° С и йод около 400° С [64].
^Исследовали [767] взаимодействие карбида циркония
с йодом. При температуре 700—800° С взаимодействие
проходит с достаточно большой скоростью с образовани-
ем Zrl2.
Нагревание компактного образца на воздухе при
температуре 1150° С в течение 48 ч приводит к разруше-
нию образца [350].
231
Таблица 42
Характер взаимодействия карбида титана с некоторыми металлам
и окислами __________________ И
Металл (окисел) Темпе- Характер взаимодействия ЛнтераГ ,тУ₽ный ИСТОЧми
Ниобий 2000 Образуется твердый ра- (363] (3601
Тантал 2000 створ. Энергия активации процесса диффузии ниобия в TiC 352 кдж/моль (84 ккал/моль) Образуется твердый ра-
Хром 2000 створ TiC—ТаС, содержа- щий около 50% (мол.) ТаС Образуется твердый ра- (361]
Молибден 2000 створ, содержащий 7% Сг. Твердость карбида возраста- ет Образуется твердый ра- [361]
Вольфрам 2000 створ, содержащий 20—22% Мо, твердость карбида уменьшается Образуется твердый ра- (361 362
Кобальт Никель Сталь углеродис- та я 1550 1500 1620 створ. Энергия активации процесса диффузии вольф- рама в TiC 481,8±33,5 кдж/моль (115 ± 8 ккал/моль) Взаимодействует [877
Чугун Криолит 1520 1050 Слабо взаимодействует (877
Окись магния 1925 Взаимодействует по реак- (364 365
Окись кремния ции 2MgO+TiC=2Mg + +СО+ТЮ. TiO растворяется в избыт- ке TiC. Возможно образова- ние Mg2TiO4 и частичное восстановление MgO до MgC2, далее идет разложе- ние 3MgC2 Mg3C2+4C= =3Mg+6C Образуется твердый ра- [277
Окись алюминия — створ Ti(C, О) и SiO Не взаимодействует [366
232
Таблица 43
Термодинамические данные реакций взаимодействия карбида
циркония с некоторыми газами___________________________
Реакция —ДНгсв ДС₽2М
кд ж Imo ль ккал) моль дж) (град-моль) § 't е. м X а дж/ (град моль) (wow/pnd?)/vra
ZrC+2H2O=ZrO2+2H2+ +С 473,5 113,0 95,5 22,8 15,6 4,2
ZrC-]-3 / 2O2=ZrO2-|-CU 1068 255 94,7 22,6 3,8 0,9
ZrC-F2CO=ZrO2-|-3CO 736,6 175,8 363,7 86,8 —15,1 —3,6
ZrC+1/2Na=ZrN+C 227,9 54,4 86,7 20,7 —3,8 —0,9
ZrC+4HCl=ZrCl4+2H2+ +c 478,9 114,3 330,2 78,8 31,0 7,4
Выше 1500° С карбид циркония взаимодействует с
азотом с образованием нитрида циркония. Аммиак его
не разлагает.
Некоторые термодинамические данные для реакций
взаимодействия карбида циркония с рядом газов приве-
дены в табл. 43 [769].
Такие окислители, как хлорат, нитрат и перманганат
калия, в расплаве легко разлагают карбид циркония
[766].
Изучение взаимодействия карбида циркония с
NH4HF2 [768] показало, что реакция начинается при ком-
натной температуре и протекает достаточно интенсивно
с образованием (NH4)3ZrF7 и выделением газообразных
продуктов: Н2, NH3, С2Н2 и СН4.
В табл. 44 приведен характер взаимодействия карби-
да циркония с некоторыми металлами и окислами.
При анализе карбида циркония обычно определяют
содержание циркония, общего и свободного углерода.
При определении содержания циркония [303, 304] карбид
сплавляют со смесью КОН и KNO3, выщелачивают
сплав водой, подкисляют раствор соляной кислотой и
осаждают цирконий аммиаком. Вследствие того что кар-
бид циркония растворяется в концентрированной серной
16 Косолапова Т. Я. 233
Таблица 44
Характер взаимодействия карбида циркония с металлами
и окислами
Металл Темпера- тура вза - имодейст- вия, °C Характер взаимодействия Литературный ИСТОЧНИК
Ниобий . , , Тантал f . - < Молибден « Вольфрам < . » Олово, висмут, цинк, свинец, кад- мий Окись магния . . Окись циркония » 1600 2200 1900 2200 350—550 1800 Образуется твердый раствор ZrC—NbC Образуется карбид тантала и раствор цир- кония в тантале, облада- ющий более высокой твердостью, чем тантал Образуется новая фа- за, по твердости близкая к цирконию Образуется новая фа- за, обладающая высокой твердостью, и повышает- ся твердость вольфрама Взаимодействие при контактировании в тече- ние 10 мин не наблюда- лось Частичное восстановле- ние окиси магния с об- разованием карбида маг- ния и твердого раствора Zr(C.O) Образуются оксикарби- ды типа ZrCx Oi_х [360, 394 395] . [360,394] [365]
целесообразно его пя СИ азотной и плавиковой кислот,
растворения пипкпнйСТВ°^ЯТЬ’ а не свлаьлятъ. После
перометрическим пгт И Можно определять объемным, ам-
При₽нагреВаМнии капбиля™ МеТОД0М [754]‘
тал в виде хлпп^.. рбида в токе хлора металл уле-
взвешивали и ₽зЛем°С7й Т0? С0деРжаЩий весь углерод,
кислотой; ппи этой л обрабатывали дымящей азотной
графита [64]. свободный углерод оставался в виде
общего^ТвоХХп Работах £303, 304, 754] содержание
карбиде титана. Углерода определяли так же, как в
234
Определяли [303] также содержание непрореагиро-
вавшей в результате реакции двуокиси циркония. Для
этого навеску обрабатывали смесью 100 мл H2SO4 (1:1)
и 10 мл хромовой кислоты (540 г Сг2О3 в 300 мл Н2О) в
течение часа при температурах 140—155° С. Карбид и
свободный углерод при этом растворялись. Раствор на
75% нейтрализовали едким натром, разбавляли водой
и фильтровали. Фильтр с осадком ZrO2 сжигали и взве-
шивали.
По разности 100— (ZrO6m +Собщ) определяли общее
одержание кислорода; по разности Zro6uI —Zr в ZrO2 —
содержание циркония, связанного в виде Карбида; по
разности Ообщ — О в ZrO2 — колцчество кислорода, вхо-
дящего в карбид.
Карбид гафния стоек против действия соляной,
разбавленной серной и фосфорной ^кислот* растворов ще-
лочей.
Растворимость его в разных средах [755] приведена
в табл. 6 и 7. Характер взаимодействия карбида гафния
с некоторыми металлами и окиСлами приведен в табл* 45.
При анализе карбида гафния определяли содержание
гафния, общего и свободного углерода [734].
Содержание гафния определяли весовым купфероно-f
вым методом или прокаливанием навески при темпера-
туре 900—1000° С.
Определение содержания общего и свободного угле-
рода аналогично определению В карбидах титана и цир-
кония.
Карбид ванадия не разлагается серной й соля-
ной кислотами, в то время как азотнай кислота разлага-
ет его даже при комнатной температуре.
Кислород окисляет его при температуре 800° С; хлор
при температурах выше 500° С взаимодействует с кар-
бидом с образованием летучего жидкого хлорида вана-
дия [64]; аммиак и азот при температурах 700—800° С
образуют нитрид ванадия. Газообразный хлористый во-
дород, пары воды и сероводород при температурах 700—
800° С не разлагают карбид ванадия.
Содержание ванадия определяли сжиганием навески
карбида при температуре 950° С. В связи с тем что пяти-
окись ванадия при температуре 715° С плавится, смачи-
вает стенки лодочки и может перейти на трубку печи
Марса, авторы заполняли лодочку мелкораздробленным
16* 235
Таблица 45
Характер взаимодействия карбида гафния с некоторыми Металлами
М еталл Темпера - тура, °C
Цинк • . • * Кадмий .... 550 450
Алюминий . . • Кремний\. ч - • Олово . * . 1000 1500 350
Свинец «... 450
Ниобий .... 1600
Тантал . . . . 1600
Хром • 1900
Молибден > ч . , =» 1600
Вольфрам . . i 1600
Кобальт . . , . 1550
Никель , . ь , 1500
Окись магния « 2000
Характер взаимодействия литера. тУрный ИСТОЧНИК
Не взаимодействует » » Взаимодействует
» Не взаимодействует » » Образуется твердый раствор HfC—NbC, обла- дающий высокими твер- (877)
достью и температурой плавления Медленно взаимодейст- [360]
вует
Не взаимодействует [877]
Активно взаимодейст- вует с образованием твердого раствора молиб- дена в карбиде Медленно взаимодей- 1
ствует с образованием новой фазы, обладающей твердостью 33320—34300 Мн/м? (3400—3500 кГ/мм2), и увеличением [360]
твердости вольфрама
Не взаимодействует (877
» » [365]
ХХ7повер0хРНо“ь0ква»РИа- Расплав“нный
ке. При темпераrvne ППП'Г л Удерживался в лодоч-
се окисла что rnwTf наблюдалась потеря в мае-
Извест^Г птл Д ТеЛЬСТВует Р его Разложении.
водит к обояаппйиЖИГаНИе каРЬиДа ванадия часто при-
некоторых случяяуИЮ Ьмеси окислов разного состава, в
продукта Наконеп ^ИГание сопровождается выбросом
верхности которого пВВедение кварцевого порошка, на по-
является ИСТ0ЧНйк-тЛа2ИТСЯ °бРазУЮЩИЙСЯ окисел V2O5.
содержания ванадия большой ошибки в определении
этого классический \описа^ным методом. Вследствие
и объемный метод определения вана-
236
дия в карбиде, разложение которого не представляет
труда, безусловно более целесообразен [754, 793, 794].
Был предложен [795, 796] микрометод определения об-
щего углерода, заключающийся в том, что выделяющие-
ся при сжигании карбида в кислороде газы улавливают
в специальной поглощающей промывалке титрованием
раствором Ва(ОН)2, избыток которого оттитровывают
соляной кислотой.
Во избежание возможного воспламенения карбида
ванадия при сгорании в токе кислорода и проскока СО
в конце трубки для сжигания помещают окись меди. До-
статочно удовлетворительные результаты дает определе-
ние содержания общего углерода сжиганием навески в
токе кислорода при температурах 900—1000° С с после-
дующим абсорбционно-газообъемным определением ко-
личества выделившегося СО2. Уточнение методики опре-
деления содержания свободного углерода в карбиде ва-
надия [792] показало, что для растворения его наиболее
целесообразно использовать азотную кислоту концент-
рации 2:1. Навеску 0,2—0,3 г VCi_x растворяют в ней
при нагревании до температуры кипения в течение не-
скольких минут.
Карбид ниобия — химически стойкое соединение,
свойства которого приведены в табл. 6 и 7.
На воздухе карбид ниобия окисляется при температу-
рах 800—900° С, с азотом и аммиаком взаимодействует
при высоких температурах с образованием нитридов [64].
Если химические свойства монокарбида несколько изуче-
ны, то свойства низшего по содержанию углерода кар-
бида Nb2C 1-х практически совершенно неизвестны.
Исследование окисления порошка и кусков размером
до 3 мм Nb2Ci-x на воздухе при температуре 300° С [132]
позволило обнаружить в продуктах реакции новую фа-
зу ромбической структуры, примерный состав которой
соответствует NbC0,4. При этом обнаружено, что яромб =
3 ®гекс> ^ромб^-^гекс И Сромб'"'-'^гекс-
Характер взаимодействия монокарбида ниобия с ту-
гоплавкими металлами и окислами приведен в табл. 46.
Анализ карбида ниобия проводят так же, как кар-
бидов титана и циркония.
Карбид тантала — один из наиболее химически
стойких карбидов. Растворимость его в различных сре-
дах приведена в табл. 6 и 7,
237
Таблица 46
Характер взаимодействия карбида ниобия
с тугоплавкими металлами и окислами
Металл Темпера- тура. °C Характер взаимодействия Ли™РатуРиый ИСТОЧНИК
Ниобий 1600 Образуется Nb2C
Тантал . . • ► 1800 рбразуется слой с мик-
ротвердостью 18620 Мн[м2 (1900 кГ1мм2), что
близко к микротвердости твердого раствора Nb2C—
Та2С
2000 • Образуются два слоя:
прилегающий к танталу с микротвердостью 18530
MhJm2 (1890 кГ[мм2) и внешний слой с микро- твердостью около 34000 [394,360,
Мн/м2 (3470 кг/ми2) 899]
Молибден .... 1800 Взаимодействует с об- разованием пористого слоя, микротвердость ко- торого растет е повыше- нием температуры, дости- гая значений, близких к
микротвердости Мо2С и и МоС. Растет и микро-
твердость молибдена вследствие растворения в нем углерода
Вольфрам , . . 2000 Образуется WC J
Окись магния . , 1800 Активно взаимодейст- [365]
вует с образованием фа- зы с микротвердостью 21440 Мн/м2 (2189 кГ/жи2), предположи- тельно твердого раствора Nb(C,O) и прилегающей к ней мягкой фазы с мик- ротвердостью 1460 Мн/м2
(149 кГ/мм2)
Окись ниобия . Г • 1200 Образуются низшие [918]
окислы и низший карбид Nb2C; при температуре
1600—1700° образуется металлический ниобий
238
Нагревание карбида тантала с водородом [472] при-
водит к его декарбидизации в том случае, если концент-
рация углеродсодержащей фазы (СН4, %) в водороде
ниже следующих значений:
Температура,
°C
2300
2510
2770
3180
В смеси 25%Nj
Я 75% На
V8—-1/*
Va
Азот и смесь 15% N2. и 85% Аг до температуры
2700° С не взаимодействуют с карбидом. Кислород, окис-
ляет его при 800° С до Та2О§.
В табл. 47 приведены термодинамические данные
реакций между карбидом тантала и некоторыми газовы-
ми средами [763].
Таблица 47
Термодинамические данные реакций взаимодействия карбида
тантала с некоторыми газами и парами
Реакция дсп Р298
кдж/моль ккал/моль ч о as aS а о. К Г© кал/ (град мо л ь). дж/{град-моль) ч 1 1
2ТаС+7Н2О=Та2О6 + +2СО+7Н2 251 60,0 —47,3 —11,3 88,4 21,1
2ТаС-р/2О2=Та2О8+ +2СО 1944 464,0 263,5 62,9 18,0 4,3
2TaC+5CO=Ta2Os+ -|-7С 1172 279,8 891,2, 212,7 —26,0 —6,2
Т аС-р/2N2=TaN4-C 90,1 21,5 104,3 24,9 1,26 0,3
TaC+5HCl=TaCls4- +5/2н2+с —61,6 —14,7 533,0 127,2 —18,8 —4,5
Исследование взаимодействия вольфрама Q карби-
дом тантала [362] методом измерения интенсивности из-
лучения нанесенного на шлиф ТаС слоя радиоактивно-
го вольфрама при диффузионном отжиге показало, что
энергия активации процесса диффузии вольфрама со-
239
Таблица 48
Характер взаимодействия карбида тантала
с тугоплавкими металлами и окислами
Металл Темпера- тура, *С Характер взаимодействия Ли„1ХратуРи«й ИСТОЧНИК
Ниобий .... Тантал . . . . • Молибден . . • Вольфрам .... 1200 2200 2200 2200 Образуется твердый раствор Образуется Та2С Образуются новые фа- зы, обладающие высокой твердостью, предположи- тельно тройные фазы Та—Мо—С То же [360, 899]
Окись магния . . Окись тантала . . 2200 Не взаимодействует Образуется металличе- ский тантал [365] [481]
ставляет 595 ±92 кдж/моль (142±22 ккал/моль} и коэф-
ЙТ-™ ДиФФУзии составляют при температуре 2130°С
2,7.10 12 см? сек, при 2170° С 4,2.10~1а СЛР1 сек ппи
СМ2,СеК И при 2300’“’ 1Р7Н
В табл. 48 приведен характер взаимодействия карби-
да тантала с некоторыми тугоплавкими металлами и
окислами.
ла5°держание тантала и свободного углерода опреде-
конияП1? методике’ описанной для карбидов титана, цир-
др.
глпО^?а^°Те £795] предложен микрометод определения
яяк-гтюЛа^Я общего Углерода в карбиде тантала. Метод
ля ппы аТСЯ В ТОМ’ что навескУ сжигают в токе кислоро-
леннпй * мпературе 900—950° С на установке, представ-
fnn°r г,Л рИС' 77’ пРичем температуру поднимают до
nSop пРн отсутствии кислорода, а от 600 до 900-
ппгткш ,Т°/Ке кисл°рода, который пропускают со ско-
ГП мл/мин В течение 30—40 мин. Выделившуюся
пплмыои^6 выключения печи вытесняют кислородом в
твлплм п7^птдК\ЛЯ1^КУ’ наполненную титрованным рас-
(иН)2. Склянку отсоединяют от системы и со-
1 ржв^ е ее титруют соляной кислотой в присутствии
фенолфталеина в токе инертного газа, подающегося для
240
перемешивания раствора. Промывная склянка состо-
ит из отдельных колоколов, насаженных на труб-
ку, подводящую газ, и имеющих в верхней части отвер-
стия для выхода газа. Трубка в нижней части расшире-
ние. 77. Схема установки для определения малых количеств
углерода в карбиде тантала:
1— промывалка е H2SO<; 2 — трубка с ВаО; 3— трубка с Р2О5; 4 — квар-
цевая трубка; 5 — термопара; 6 — печь Марса; 7— защитная кварцевая
трубка; 8 — фарфоровая лодочка; 9 — промывалка е Ва(ОН)2; 10 кон-
трольная склянка е Ва(ОН)2
на и заполнена кольцами из отрезков узкой стеклянной
трубки.
Карбиды хрома отличаются высокой коррози-
онной стойкостью и стойкостью против окисления.
Карбид Сг3С2 стоек против действия всех минераль-
ных кислот, но окисляется при действии расплавленных
селитры и перекиси натрия [483]. При действии кипя-
щей концентрированной хлорной кислоты он разлагается
с выделением ацетилена.
Растворимость карбида Сг7Сз в соляной кислоте
изучали в работах [57, 797, 798]. Показано, что водные
растворы соляной кислоты действуют на карбид слабее,
чем спиртовые, что объясняется некоторой способностью
карбида пассивироваться в водных растворах и терять
пассивность в спиртовых растворах.
На растворимость карбида влияет также концентра-
ция соляной кислоты — в 10%-ном растворе соляном
кислоты растворение проходит полнее, чем в 1%-ном.
Стойкость карбидов хрома при действии на них раз-
личных кислот, их смесей и растворов щелочей увеличи-
вается в ряду Сг23С6—Сг7С3—Сг3С2, что связано с услож-
нением структурных элементов из атомов углерода [805
°06]. Для наименее стойкого карбида Сг23Св характерно
24
изотированное расположение атомов углерода в элеме
тарной ячейке, в то время как для наиболее стойкой"
карбида Сг3С2 характерно наличие зигзагообразных це°
пей из атомов углерода.
В присутствии окислителей карбиды хрома по увели
чивающейся стойкости располагаются в ряд Сг3С
Сг7Сз—СггзСб, что связано с образованием на поверхно
сти пассивной пленки Сг2О3. У низшего карбида Сг23с'
представляющего решетку хрома, в отдельных узлах
которой расположены атомы углерода, эта пленка
наиболее устойчива. Карбиды Сг7С3 и Сг3С2 — более
сложные структуры и по свойствам отличаются от ме-
таллического хрома.
Методом снятия кривых плотность тока — потенциал
электрода изучали [799] электрохимическое поведение
карбидов хрома в растворах серной и соляной кислот.
Результаты исследований показали, что карбид Сг3С2
стоек к окисляющим и восстановительным воздействиям
в кислотах, он имеет высокую анодную поляризуемость.
Согласно многочисленным исследованиям [800—806],
карбиды хрома — наиболее стойкие среди карбидов
переходных металлов IV, V и VI групп периодической
системы элементов против высокотемпературного окис-
ления.
Ниже приведены сравнительные данные о стойкости
карбидов против окисления [800] (температура 900° С,
время 1 ч):
Карбид . . . Сг3С2 Сг7С3 Cr23C6 TiC Nb(Ta)C ZrC VC WC
Потеря в мас-
се, г/ж3 . . 0,66 1,1 1,1 12,1 205 460 435 1140
Исследование окислительной стойкости карбидов
хрома в виде порошков и компактных образцов при тем-
пературах от 400 до 1100° С [802] показало, что окисле-
ние всех трех карбидов хрома в виде порошков начина-
ется при температурах около 700° С,
Компактные образцы Сг3С2 и Сг23Сб до температур
1000° С практически не окисляются. Карбид Сг7С3 в вИ‘
де компактных образцов окисляется заметно уже при
температуре 700° С.
В результате изучения [763] взаимодействия карби-
да Сг3С2 с некоторыми газами и парами вычислены тер-
модинамические характеристики этих реакций (табл.
242
Таблица 49
Термодинамические данные реакций взаимодействия
с некоторыми парами и газами
Реакции —AS298 дсп ”29В
кдж/моль ккал/моль дж/ (моль-град) кал/ (моль/град) дж/' (моль-град) ! кал/ (молъ~град\
2СгзСг4-13Н2О= =ЗСг2Оз4-4СО4* 4-1ЗН2 481,8 115,000 —108,5 —25,9 191,1 45,6
2Сг3С24-1г/9О2= = СгаОз4-4СО 3630 866,400 468,9 111,9 , 60,3 1 14,4
2Сг3Са-|-9СО= =ЗСг2О3-|-13С 2192 523,200 2053 490,1 —21,4 -5,1
СгзСг-Н/гМг3 =3CrN+2C 271,5 64,800 165,5 39,5 41,5 9,9
Сг3Сг4-9НС1= =ЗСгС1з4~2С4- +9 2Н2 370,8 88,500 802,8 191,6 63,3 15,1
При химическом анализе карбидов хрома определяют
содержание общего и свободного углерода и хрома.
Определяли [483] содержание общего углерода после
разложения навески карбида сплавлением с селитрой
или с перекисью натрия. Полученный сплав обрабаты-
вали серной кислотой и выделяющийся при этом угле-
кислый газ поглощали во взвешенных U-образных труб-
ках натронной известью. Сплавление проводили в квар-
цевых колбах, которые выдерживали не более четырех
сплавлений. Этот метод малоэффективен, так как необ-
ходимо учитывать содержание СО2 в перекиси натрия,
которая активно его поглощает, что сложно и связано
с большими ошибками.
Содержание углерода определяли [485] Сжиганием с
прибавлением к навеске карбида перед сжиганием по-
рошка алюминия. Большое количество тепла, выделяю-
щееся при окислении алюминия, способствует полному
разложению карбида. Частично образующийся при этом
карбид алюминия окисляется при прибавлении перекиси
свинца. Таким образом, к навеске карбида 0,4 г прибав-
ляли 2 г смеси, состоящей из 1 ч. алюминиевого порош-
243
ка, 1 ч. окиси алюминия и 10 ч. перекиси свинца с
ние проводили при температуре 750° С. ' ^Жига
Наилучшие условия для полного сжигания капл
хрома в кислороде —сжигание в течение 25—30 ми» Да
температуре 1150—1200° С с использованием в кэчр и
плавня окиси меди [487, 804]. Проверка этого метода е
казала, что ошибка определения составляет около Л?’
Исследование химической стойкости карбида хо* °'
в разных средах показало, что невозможно подобо^
такой реагент, который бы разлагал только карбид ипЬ
только свободный углерод. Поэтому обычно применяв-
шиеся для определения содержания свободного углеоо
да методы в данном случае не могут быть использованы'
При определении содержания свободного углерода
в карбидах хрома использовали [483] разную раствори-
мость их в соляной кислоте. Нерастворившийся в НС1
остаток отфильтровывали, взвешивали и определяли в
нем содержание хрома и углерода. Так как осадок со-
стоит из СГ3С2 и свободного углерода, то по содержанию
хрома определяли количество углерода, связанного с
хромом в виде Сг3Сг. По разности между общим содер-
жанием углерода в нерастворимом остатке и содержани-
ем углерода, связанного с Сг3Сг, определяли содержание
свободного углерода. Этот метод очень длителен и недо-
статочно точен. Определение ;малых количеств свободно-
го углерода этим методом не представляется возмож-
ным.
Содержание свободного углерода определяли [807,
808] сжиганием навески карбида в токе кислорода при
температуре около 600° С. При этом окисления карбида
не наблюдалось, свободный углерод полностью окис-
лялся.
Навеску карбида 0,3—0,5 г сжигали [807] в токе кис-
лорода при температуре 600±20° в течение 30 мин. Ко-
личество выделившегося СОг определяли абсорбционно-
объемным методом.
Навеску 0,5—1 г сжигали [808] в токе кислорода при
температуре 560° С в течение 1 ч. Количество выделив-
шейся СОг определяли по привесу при улавливании в ад-
сорбционных трубках. В работе предложено определять
содержание свободного и связанного углерода из одной
навески. Навеску карбида сжигают при температур
560° С в течение 1 ч, затем меняют трубки, повыша1°
244
температуру до 1300—1450° С и сжигаю?1 в течение
90 мин.
Предложен также метод определения содержания
свободного углерода сжиганием навески карбида при
температуре 540° С и поглощением выделившейся
углекислоты щелочью е кондуктометрическим опре-
делением количества поглощенного углерода [809].
Все эти методы достаточно эффективны, принципи-
ально не различаются и могут с успехом использоваться
при определении содержания свободного углерода в кар-
бидах хрома.
Содержание хрома в карбидах хрома определяли
[489], разлагая навески сплавлением с селитрой или пере-
кисью натрия. Полученный сплав обрабатывали серной
кислотой, образующийся сернокислый раствор бихромата
подщелачивали раствором едкого натра в присутствии
перекиси водорода. Железо и алюминий, содержащиеся
в карбиде в виде примесей, осаждали в виде гидрооки-
сей и отфильтровывали. В фильтрате содержание хрома
определяли йодометрическим методом после подкисле-
ния соляной кислотой [794, 810]. .
Указана возможность определения содержания хро-
ма разложением навески карбидов хрома кипящей хлор-
ной кислотой [497] с последующим окончанием персуль-
фатно-серебряным методом [793, 794].
Проверка данного метода показала, что при такой
обработке навески наблюдаются потери хрома, связан-
ные с летучестью частично образующегося хлористого
хромила.
Наиболее эффективный метод определения содержа-
ния хрома — персульфатно-серебряный метод при пред-
варительном разложении навески сплавлением с пере-
кисью натрия или с едким натром.
Карбиды молибдена. Неокисляющие кислоты
не разлагают МогСх-х. Азотная кислота и смеси ее с
Другими кислотами растворяют его.
В табл. 6 приведены данные о растворимости
Mo2Ci х в разных средах [754]. При действии воды на
поверхности карбида постепенно образуется синий слой
окислов [542].
При нагревании на воздухе порощок начинает окис-
ляться при температуре 500° С и при 800° G полностью
окисляется с образованием МоОз1.
245
Таблица 50 Термодинамические Данные реакции взаимодействия карб молибдена с некоторыми парами и газами
Реакция —Д Наэа
кдж) моль •О О i дж/ (град-моль) (чгоп gods) /vm _ дж/ 1 (град/моль) / (чгомг gvde) а S™
Мо2С4-7НгО= 73,7 17,600 135,7 32,4 71,6 17,1
—2МоОз4-7Нз+СО Мо2С4-?/2О2= =2МоОз4-СО 1586,3 378,600 446,6 106,6 1,26 0,3
МОгС-Н 2^2= =Mo3N4-C 87,1 20,800 85,1 20,3 23,9 5,7
Хлор действует только при высоких температурах,
фтор — при комнатной температуре.
Изучение взаимодействия с некоторыми парами и
газами позволило вычислить термодинамические данные
(табл. 50).
После 15-лшн контакта компактного образца карби-
да с МоО2 при температуре 1700° С, с SiO2 при 1300°С
[539], с жидкой медью при 1200° С, со шлаком мартенов-
ской печи при 1500° С, с Fe2SO4 при 1350° С, с CaSiCb
при 1600° С никакого взаимодействия не наблюдается, в
то время как с жидким никелем, кобальтом, марганцем,
алюминием и медью он взаимодействует [529]. С танта-
лом карбид молибдена вступает в реакцию при темпе-
ратурах 1700—1900° С [552]. Реакция с молибденом идет
с образованием твердого раствора Мо2С—Мо при темпе-
ратуре 1400° С; при взаимодействии с вольфрамом при
Мо^899]Т^Ре 1500°С Образуется твердый раствор WC-
При изучении [811] взаимодействия карбида с щелоч
ным раствором Кз[Ре(СЬ1)6] замечено сильное взаимодем
ствие уже при температуре 20° С. При этом выделяется
Си по реакции:
Мо2С + 14 [Fe (CN)6]3“+ 18 (ОН)"" = 2МоО^“ +
+ 14 [Fe (CN)6] Н- 9Н2О + СО.
246
На основании легкой растворимости карбида молиб-
дена в щелочном растворе K3[Fe(C^)6] и практического
отсутствия растворения свободного углерода в этом рас-
творе автор предложил метод определения связанного
углерода. Он заключается в следующем: навеску кар*
бида растворяют в 10 мл раствора, состоящего из
105 а/л Кз[?е(СМ)б] и 57 г)л NaOH. Раствор разбавляют
водой до 300 мл, к нему прибавляют 30 мл раствора
сульфата цинка (200 г/л) и по каплям соляную кислоту
до достижения pH раствора 1,5. Затем прибавляют 20 мл
раствора KI (50 г/л) и титруют 0,05-н. раствором
МагЗгОз-
Содержание углерода определяют по уравнению
%С^ 4,53X - 60,1,
Е
где V —расход [Fe(CN)6]3r; кг-
Е — навеска, мг,
Определение содержания связанного углерода объ-
емным методом хорошо совпадает С методом сжигания.
Расхождение составляет ±0,06%.
Для определения содержания общего углерода наи-
более распространен метод сжигания,, описанный выше
(см. стр. 231). Содержание свободного углерода опреде-
ляют обычным для большинства карбидов методом —
растворением навески в азотной кислоте (1'1) и сжига-
нием в кислороде нерастворившегося остатка [754].
Предложен колориметрический метод определения
содержания свободного углерода в карбиде молибдена
[812]. Метод основан на том, что карбид молибдена не
адсорбирует из водных растворов такой краситель, как
бромтимоловый синий.
Ниже приведены сравнительные результаты опреде-*
ления содержания свободного углерода колориметриче-
ским и газообъемным методами, которые свидетельству-
ют о хорошей сходимости данных, полученных обоими
методами.
1
Газообъемный метод . . * г 1 .4,18
Колориметрический метод ч .
Относительная ошибка, % . к ч (±0>
Монокарбид вольфрама. В табл. 6 приведено
поведение WC в различных химически агрессивных сре-
дах.
и
2,10
2,09
(-0,5)
247
Таблица 5<
ии9имческие данные реакций взаимодействия
Реакции Д//298 дс;—
кдж/моль 1 ккал/моль дж/ (град-моль) кал/ : (град-моль) 1 ч ч кал} 1 (град-моль) 1
WC4-4H2O= —54,5 —13,000 —15,9 —3,8 62,0 14,8
=WO3-HHa+CO WC+3H2O= =WO34-CH4-J-H2 152,1 36,300 198,6 47,4 15,1 3,6
WC+H2O=W+ —166,8 —39,800 —142,0 —33,9 19,3 4,6
+CO+H2 WC+2O2= =wo3+co 914,2 218,200 161,7 38,6 21,8 5,2
WC4-2H2=W4- +CH4 39,8 9,500 72,9 17,4 —27,6 -6,6
Исследование взаимодействия WC с парами воды,
водородом и кислородом [763] позволило вычислить не-
которые термодинамические характеристики реакций
(табл. 51).
При нагревании на воздухе или в кислороде карбид
вольфрама медленно окисляется до WO3.
Изучение окисления WC [814] показало, что при тем-
пературе 700° С окисление подчиняется линейному зако-
ну и идет полностью по реакции WC + 2,50а=\УОз+СО2.
Окисная пленка содержит только WO3. Степень окисле-
ния WC значительно выше, чем металлического вольфра-
ма, что обусловливается прорывом окисной пленки об-
разовавшимся СО2.
Термогравиметрический анализ показал [813], чт°
тонкий порошок начинает интенсивно окисляться на воз
Духе при температуре 500—520° С, Быстрое окислени
крупнокристаллического WC начинается при температу
ре 565° С,
Фтор взаимодействует с WC при комнатной темпера
туре со взрывом, хлор начинает взаимодействовать ли
при температурах 600—800° С 1754]. йя„а
Исследование электрохимических свойств кар°
248
вольфрама, определение статических потенциалов, изу-
чение зависимости статических потенциалов от pH сре-
ды и от состава электролита проводили [815] компенса-
ционным методом по отношению к насыщенному кало-
мельному электроду сравнения на потенциометре ЛП-58.
Ниже приведена зависимость статического потенциала
WC от среды.
С гатический
Электролит потенциал,
мв
0.001-Н.НС1 (рН = 3)............................ +617
0,001-н.НС1 + 1 % NaCl................. +278
0.001-Н.НС1 + 6,6% NaCl......................... +507
0,001-н.НС1 + 1 % NaCl и 10% лимонной кислоты. . +647
5% NaOH........................... —293
5% NaOH+ 1% NaCl ........................./ . < . —270
5% NaOH+ 10% NaCl ........................... —348
5% NaOH + 1% лимонной кислоты . . j. . , . . . —213
5% NaOH+10% лимонной кислоты . ................. —58
Исследование взаимодействия карбида вольфрама с
молибденом и вольфрамом [849] показало, что в первом
случае при температуре 1850° С образуются две фазы:
Мо2С и твердый раствор Мо — W — С, во втором случае
при 1900° С образуется W2C.
Диффузию вольфрама в карбид WC исследовали
[362] методом измерения интенсивности излучения образ-
цов WC, на которые наносили слой радиоактивного
вольфрама при длительном диффузионном отжиге. От-
жиг проводили при температурах 2130, 2170, 2230 и
2300° С. Энергия активации составляет 578,2 ±
±79,6 кдж]молъ (138±19 ккал/моль^. Коэффициент
диффузии равен при температуре 2130° С 1,9 • 10_12 ,
2170° С 5,4-10~12, 2230° С 7,8-10~12 и 2300° С
16,10~® см21 сек.
Растворение WC в щелочном растворе Кз[Ре(СМ)б]
происходит интенсивно по реакции [811]
WC + 9 [Fe (CN)e]3“ -f- 11 (ОНр = WO1- +
+ 9 [Fe (CN)e]4- + 5,5H2O + J/a CO + Va CO2.
Растворимость WC в растворе Кз[Ре(СМ)в] использо-
вана автором для объемного определения содержания
WC или связанного углерода объемным методом (см.
стр. 247),
249
Содержание WC вычисляют по уравнению
WC — 35,4“ - 245,1%;
Е
С == 2,17-1- - 15,0%,
Е
где расход [Fe(CN)6]3-, мг\
Е — навеска, мг.
Ниже приведено сравнение результатов определения
содержания в WC связанного углерода объемным ме
тодом и методом сжигания, %:
Объемный метод 3,12 6,02 4,87 1 42
Ме?од сжигания 3,27-0,03 5,89-0,04 4,3-0,06 1,5+о,О5
Содержание свободного углерода может быть опр -
делено также колориметрическим методом [812], которы
основан на томк что карбид вольфрама не поглощает я
метиловым оранжевым или метиловым фиолетовым, а
свободный углерод поглощается ими. Методика опр -
деления аналогична описанной для определения сод р
жания свободного углерода в карбиде молибдена. Ре
зультаты определения Ссв0б этим методом приведены в
табл. 52.
Карбиды марганца. Химические свойства кар-
бидов марганца изучены недостаточно.
Карбид Мп3С быстро разлагается водой с выделени-
ем смеси метана и водорода примерного состава 51
СН4 и 49% Нг. Разбавленные минеральные кислоты так-
же легко разлагают его.
Таблица 52
Результаты определения С<.воб, колориметрическим методом
Карбид Ссвоб’ % Относи- тельная ошибка, % Примечание
содержа- ние найдено
WC WC 1,83 1,10 1,76 1,05 —3,7 —4,5 1 Адсорбция метиловых
W2c 1,64 1,56 —4,8 । оранжевым
WC 1,83 1,78 —2,7
WC WC 0,77 1,40 0,74 1,37 —4,0 -^2 I 1 Адсорбция метил вы'
WC 1,76 К 73 —1.7 । фиолетовым
wac 2,13 2,20 +3,2
250
Проводили [588] анализ газообразных продуктов, вы-
деляющихся при обработке образцов марганец-*-угле-
род, содержащих различное количество углерода, соля-
ной кислотой (1:1).
Авторы показали, что при растворении металличес-
кого марганца выделяется только водород; Мп4С при
разложении выделяет, кроме водорода (71%) и метана
(24,2%), до 5% этана, Мп3С выделяет 62% водорода,
27,9% СН4, 9,3% С2Н6 и 2% С3Н8, т. е. с повышением со-
держания углерода возрастает число компонентов газо-
вой смеси, что свидетельствует об усложнении характе-
ра связи.
Фтор действует на Мп зС при комнатной температуре
(образуется фторид марганца), хлор — при нагревании.
В атмосфере кислорода, так же как и в атмосфере
NO и NO2, карбид марганца легко сгорает при неболь-
шом нагревании. В аммиаке при температурах 700—
800° С образуется нитрид марганца [64].
При анализе карбидов марганца [588] определяли со-
держание марганца и связанного углерода. Марганец
осаждали в виде сульфида, пропуская сероводород че-
рез раствор, полученный разложением навески карбида
разбавленной соляной кислотой, и прокаливали осадок
сульфида до образования Мп3О4. Содержание связанно-
го углерода определяли сжиганием газов, выделяющих-
ся при разложении навески разбавленной соляной кис-
лотой.
Карбид железа Fe3C в воде при комнатной тем-»
пературе не растворяется. С ростом температуры выше
150° С растворимость его увеличивается. Разбавленные
минеральные кислоты разлагают Fe3C при температуре
выше 80° С; при этом наряду с газообразными выделя-
ются жидкие и твердые углеводороды, главным образом
пропилен и бутилен. Эти продукты являются результа-
том процессов полимеризации групп СН2 [785, 791].
В атмосфере кислорода мелкодисперсный порошок
Fe3C сгорает при температуре 150° С, в атмосфере хлора
взаимодействие проходит при 100° С, в атмосфере брома
и йода — при температурах 700—800° С, с парами се-
ры—при 500° С.
В литературе в основном приводятся химические
свойства железоуглеродистых сплавов и сталей, свойст-
ва индивидуальных карбидных фаз почти не изучены.
251
Кяобид кобальта, как и карбид Железа пя~
гается при нагревании разбавленными минерал^
^слотам!., продуктами разложения являются пр
полимеризации метилена [785 791]. Содержание общ’“
Хрода в Со3С определяли [153], сжигая навеску
Клплейса в течение 5 ч, содержание свобпп ап'
Жрода^расЛорением в 30%-иой HNO,
щим фильтрованием нерастворившегося свободного уг’.
"^Карбид никеля Ni3C разлагается концентрипп.
ванными и разбавленными минеральными кислотами
Для изучения продуктов разложения карбида нике-
ля соляной кислотой [827] навеску помещали в колбу Для
разложения, которую нагревали на водяной бане. Воз-
дух из колбы вытесняли углекислым газом и кислоту
подавали по каплям. Выделяющиеся газы собирали в
азотометре с концентрированным раствором щелочи. Га-
зообразные продукты разложения состоят в основном из
углеводородов и водорода, причем состав этих продук-
тов непостоянный и меняется. Различный состав реак-
ционных газов, по мнению автора, обусловлен такими
процессами, как образование группы метилена, гидри-
рование метилена до метана, полимеризация группы
метилена до ацетилена, гидрирование этилена до
этана.
Химический анализ карбида никеля проводили, опре-
деляя содержание Ni, Собщ и Ссвоб.
Для определения содержания свободного углерода
навеску карбида разлагали кипячением с концентриро-
ванной соляной кислотой в течение 30 мин. Оставшийся
в виде нерастворимого остатка свободный углерод от-
фильтровывали, высушивали и взвешивали. В растворе
никель осаждали диметилглиоксином. Для определения
содержания общего углерода навеску сжигали в токе
кислорода. Так как навески при загрузке в горячую печь
сразу сгорали, их загружали в холодную печь и посте
пенно поднимали температуру.
Карбид бора — одно из наиболее химически
стойких соединений. По данным работ [682, 722], он
растворяется в минеральных кислотах и их сме^ ’
рацииКЖе В раствоРах Щелочей различной коннен
Приводятся данные [847], согласно которым карбиД
В растворах щелочей различной
252
бора при обработке смесью 2 мл K2SO4 + 0,5 мл НЫО3
полностью растворяется в течение 6 ч.
' Наиболее полное исследование химических свойств
карбида бора проведено в работах [848, 849]. Результаты
сследования представлены в табл. 53 и подтверждают
высокую химическую стойкость В4С.
При использовании карбида бора особое значение
имеет отсутствие в нем большого количества свободного
углерода.
Отделение свободного углерода основано на разных
окислительных свойствах карбида бора и свободного уг-
лерода. В качестве окислителей свободного углерода ис-
пользовали 30%-ный раствор перекиси водорода [846],
раствор хромового ангидрида или бихромата калия в
ерной кислоте [850, 851], смесь концентрированной сер-
ной, азотной и хлорной кислот с бихроматом калия [852].
Наиболее эффективна для окисления сажи смесь,
предложенная в работах [852, 853] (табл. 53 и 54).
Сера, фосфор и азот при температуре 1200° С не взаи-
модействуют с карбидом бора; хлор взаимодействует при
температуре 1000° С с образованием ВС13 и выделением
графита; бром и йод не реагируют.
Кислород до температуры 500° С не окисляет карбид
бора, при 1000° С он сгорает с выделением СО2 и В2О3
[754]. Окисление карбида бора изучали [854] на порошках"
различной зернистости при температурах 820 и 1800° С.
Было установлено, что свободный углерод окисляется зна-
чительно быстрее, чем карбид бора, причем скорость
окисления карбида бора — величина постоянная. На
этом основании авторы разработали метод определения
содержания свободного углерода сжиганием навески в
атмосфере кислорода до достижения постоянного значе-
ния —.
М
Изучение окисления порошка карбида бора [853] по-
казало, что окисление начинается при температуре
600° С, скорость окисления с ростом температуры до
800—1000° С резко возрастает, после чего замедляется и
при 1200—1300°С снова резко увеличивается (рис. 78).
При всех температурах окисление проходит в течение
определенного промежутка времени, после чего прекра-
щается. Это, вероятно, связано с образованием на части-
цах В4С пленки В20з, препятствующей дальнейшему
253
LX- |8№ 84,1
Среда Темпера- тура, °C Время ' обработ- ₽ ки, ч трасте,,, ивший . остаток %
НС1 (конц.) • * НС1 (конц.) • - - м НС1 (1 г О - « ' ’ НС1+НС1О4 ч ‘ ' HZSO4 (конц.) . . л • . - • ' H2SO4 (конц.) H2SO4 (ПЛ < ’ H2SO4+HNO3 » • * ' H2SO4 + HF . ч ' HNO3 (конц.) • ’ • HNO3 (конц.) , . f - * • - • ’ HNO3 (1:1) V - • ‘ HNO3+HF НСЮ4 (конц.) . - НСЮ4 (конц.) . г ’t 1 • " • НСЮ4 (1:1) . . » • * т Ч ' ‘ KJO3 + H3PO4 4 . . ♦ 4 » -и f • H2SO4+KMnO4 H2SO4+К2СГ2О7 Нг5О4+НзРО4+СгОз ..<•••• NaOH 1%-ный ., л , . . - • • NaOH 1%-ный - • • NaOH 1%-ный • л s • • • NaOH 5%-ный NaOH 6%-ный ......... NaOH 8%-ный . • - NaOH 12%-ный ........ z NaOH 12%-ный NaOH 25%-ный •. * . . t . . NaOH 10%+Н2О2 NaOH 10%+Br2 < . . i . f . -> 20 115 110 116 20 280 120 130 280 20 120 110 116 20 120 115 Кипение » » » 20 100 100 20 20 100 20 100 20 100 100 24 1 4 4 24 1 4 4 1 24 1 4 2 24 1 4 3 3 3 3 40 1 2 40 40 2 40 2 40 1 1 98,0 98,0 97,6 97,5 98 0 98,0 97,0 91,2 91 5 97 0 97 0 96.9 90 8 98,0 98,0 97,8 89,7 81,1 100,0 87,8 99,0 99 0 98,5 98,3 98 6 98 5 99,0 99 0 99,1 98 0 99 6
углерод3- *а"
что наиболее
окислению не только карбида бора, но и
я "редположение подтверждается тем,
ггрпат?°е окисление карбида бора наблюдается при тем-
пературах, при которых летучесть В2О3 велика. ЧеМ
ольше содержание углерода в карбиде бора, тем выше
скорость окисления.
взаим°Действии порошка карбида бора с танта
пластинками в интервале температур 1200"”
254
Таблица 54
Результаты обработки В<С смесью FUSCm-HNOa+HClC^+KaCrjO?
(Ю мл: 10 мл: 5 мл : 20 мг|_________________
Время обработки мин Содержание углеро- да. % Содержание бора, % Содержание желе- за, %
до обра- ботки после обработки до обра- ботки после об- работки до обра- ботки после обработки
15 26,3 23,7 70 75 1,1 0,24
23 26,3 22,4 70 77 1,1 1J3
25 26,14 21,4 69,8 77,5 0,23 0,07
1400° С при разрежении 39,9 кн)м2 (З-Ю^лж рт. ст.)
образуется покрытие толщиной до 4 мкм, состоящее из
ТаВ2, ТаВ и Та2В [739].
При анализе карбида бора определяют содержание
бщего и свободного бора, общего и свободного углеро-
да, борного ангидрида [754]. При определении содержа-
ния общего бора навеску карбида разлагают сплавле-
нием со смесью перекиси натрия и едкого натра в желез-
ных тиглях, спеканием с карбонатом бария или окисью
кальция в никелевых
тиглях и сплавлением
с содой в платиновых
тиглях. После переве-
дения навески в рас-
твор содержание бора
определяют объемным
методом — титровани-
ем раствором едкого
натра борноманнитовой
кислоты в присутствии
фенолфталеина. Опре-
деление содержания
свободного бора (вклю-
чая В2О3) основано на
растворении его в сме-
си азотной кислоты с
пергидролем [914], в ко-
торой карбид бора не
разлагается. Анало-
Ш 600 800 1000 1200
температура, °C
Рис. 78. Кривая окисления карбида
бора разного состава:
„ ®связ
А **“-----
связ
255
гично определение содержания борного ангидрида
нано на его разложении водой. В обоих случаях в <Ьи °
рате определяют содержание бора объемным мето?ЬТ
Содержание общего углерода определяют сжигани М
навески в токе кислорода с использованием в качес
плавня окиси меди, окислов свинца или свинца [7211 Ве
температуре 1200° С с последующим абсорбционно-гаРИ
объемным или весовым определением количества вы3°
лившегося СО2. Определение содержания свободного v&'
лерода, как указывалось выше, основано на разных ск
ростях и характере окисления карбида и свободного уг-
лерода [854]. Рассматривая окисление карбида бора и
свободного углерода как суммарный процесс, следует
учитывать, что скорость окисления В4С — величина по
стоянная и что скорость окисления свободно-
го углерода выше скорости окисления карбида бора
Вследствие этого сжигание ведут при температур
1000° С до достижения постоянства отношения ~ . Со-
Д/
держание свободного углерода рассчитывают по фор-
муле
Сс„8 = С-^-«,
д t
где С — общее количество углерода, выгоревшего за
время t.
Карбид алюминия. Характерна для карбида
алюминия реакция разложения его водой с выделениех
метана:
А14С3 + 12НаО = ЗСН4 + 4А1 (ОН)3,
которая протекает медленно при комнатной температуре
Концентрированные соляная и азотная кислоты раз-
лагают его очень медленно, разбавленные — быстро
Согласно данным [164], при обработке соляной кислотой
выделяется метан и до 8% водорода. Концентрирован-
ная серная кислота восстанавливается карбидом ал
миния до SO2. Расплавленные едкие щелочи легко Р
лагают А14С3 при температуре 300° С. тлп—
СЛ0Р°Д окисляет карбид при температурах 7
800 С лишь на поверхности, образующаяся пленка о
256
ла предохраняет его от дальнейшего окисления. Хлор
разлагает при температуре 700—800° С с образованием
хлорида алюминия. Бром взаимодействует при темпера*
туре 700° С, йод при 800—900° С не реагирует. Сера при
температуре 700—800° С разлагает AI4C3 с выделением
тепла и образованием сульфида алюминия. Азот и фос-
фор не действуют на карбид алюминия.
Карбид алюминия — сильный восстановитель, он
взаимодействует при температурах 700—800° С с окисла-
ми меди и свинца, окись железа восстанавливается им.
При высоких температурах наблюдается реакция
А14С3 + А12О3 = 6А1 + ЗСО.
Мелкодисперсный карбид алюминия при нагревании
взаимодействует с КМПО4, К2СГ2О7, РЬО, РЬзО4, KNO3 и
КС1О3.
Анализ карбида алюминия проводили [64] двумя ме-
тодами.
1. Карбид разлагался длительным действием на него
разбавленной соляной кислоты. Раствор, содержащий
после отделения фильтрованием нерастворимого остатка
хлорид алюминия, выпаривали досуха и прокаливали до
AI2O3.
2. Карбид разлагали сплавлением с едкими щелоча-
ми в серебряном тигле, сплав растворяли водой, подкис-
ленной соляной кислотой, и алюминий осаждали гипо-
сульфитом в виде гидрата окиси, который затем
отфильтровывали. Содержание углерода определяли мед-
ленным разложением навески карбида водой с измере-
нием количества выделяющегося метана.
При анализе [169] карбида алюминия также измеря-
ли объем метана, выделяющегося при разложении на-
вески карбида.
Карбид алюминия анализировали [164], разлагая на-
веску 4-н. раствором серной кислоты при нагревании.
Выделяющийся метан сгорал в трубке над окисью меди
и образующаяся углекислота поглощалась в трубках с
едким натром, которые взвешивали. Для поглощения
некоторого количества выделяющихся примесей серо-
водорода и сернистого газа устанавливали трубку с
трехокисью хрома. Нагревание вели в течение 2 ч. Для
надежности после взвешивания нагрев вели еще 1 ч
В нерастворившемся остатке остаются AI2O3 и Ссвоб-
17 Косолапова T. Я. 257
Его отфильтровывали черезг тигель Гуча, высушивали
Лмпеоатуре 300° С и взвешивали. При нераствори-
мом остатке, составлявшем 5,9%. он содержал 8,7%
мом остах , & фильтрате ют нерастворимого
остатка в одной-части определяли удержание алюми-
SТдругой--азота обработкой NaCl и титрованием
пистиллята соляной кислотой.
А К а обид ’кремни а лхимически -стойкое соедине-
ние Он не- разлагается- прйДействии минеральных кис-
лот н их смесей.за исключением фосфорной кислоты, -а
также растворов;, щелочей [754, «28—831). Фосфорная
кислота (плотность IJb-tfcrf). .при температуре 200-
250 °C разлагаетисарбиД. кремния ^^образованием ,£>10*
СО2, Н2 и СН41702].
Данные а растворимости карбидхдфемния в смеси
азотной и фтористоводородной 'кислот .противоречивы.
Указывается [694{на растворимостьвЮвсмесиазотной
и фтористоводородной кислот- ДЭднакр, шэ данным рабо-
ты {832], при шестикратной обработке карбида кремния
смесью HNO3+.HF только. 0,01 переходите раствор.
Эти данные подтверждаются нашими, иссдедованиями
[833]. Такое-расхождение, по-видимому> связансь~-с не-
сколько различной растворимостью отдельных форм
SiC.
В атмосфере водорода -карбид. кремния устойчив до
высоких температур. При взаимодействии с азотом при
температурах, выше 1400°Сообразует литриЫД* [702].
Легко реагирует с фтором^с хлором “при температуре
900—1000° С образует SiCI4^M С; при 1100—1200° (Г
токлоД СС1*; окиси углерода при температур ах» выше
1250 С взаимодействует е SiC, мяшовёрхности которого
при этом выделяется, свободный ^углерод; сера заметно-
действует при температурах выше J10Q°C С парами во-
™п^РчллД начинает-реагировать при темпера-
ЛРп 0 400 С» при 1775—1800° С -активно протека-
ет реакция . F
SiC 4- 2Н2О SiO2 4- СН4...
ппи тр«п6НИе карбида кремния кислородом начинается
TPnln?JuepaType ВЫше 1000°С' В работе {728]проведена
р/зличных^веакпий оценка возможности прохождения
Р И. nniJ Ц При окислении {табл. 55).
окислять SiC еуже при Жомна^н^6^ ЧТ° ХислоРод может
у при жомнатнои температуре^ при этом
258
Таблица 55
Изменение свободной энергии реакций окисления карбида йремйия
Реакция Свободная энергия при температуре, °К
298.16 1000 1500 { ( 2000
кдж/моль, ккал/моль кдж/моль ккал/моль кдж/моль ккал/моль кдж/моль /ждл/маль
SiC+2O2-=SiCH-COa —1172 —279,8 —1049,6 +750.5 —97^,0 —232,0 +892,0 —212,9
SiC+3/A=SiO2+CO —912,2 —217,7 — + +
SiC+Oa=SiO2+C —/79,7 -186,1 —156,8 —576,5 -137^ +-489,0 d16’’1
SiC+8/aOa=SiO+COa —46|,0 —110,1 V *+ '
SiC+O2=SiO+CO —205,3' —.49..0 —З33,9 + 79,1 ,-422.0 M00V7 +^05,3 —126,6
SiC-P/2O2=SiO+C — 70,0 — 16,7 —134,9 1Н32„2 ’М79,3 -т* 42,8 —212*8 50,8
SiC+Oa^Si+COa —345,7 —г 82,5 —351,1 ХЙ8 1+357,9 55,4 J-367.9 M 87,8
SiC-P/aO2=Si+CO — 88,4 ' — 01,1 4-187,5 Д 37 6 —204,9 48,9 +257,3 61j4\
SiC+2CO=SiOa4-3C —509,0 121,5 —258,9 - б|!в + 91,3 2 21,8 4-55^0 f 22,9
SiC+CO=SiO+2C 4* 65,4 f 1М + 64,1 +* 15,3. ,-jt 6М + 151,J f 66,6 f |5,9
SiC+4H2O=SiO2+CO2+4H2 —246,8 pg- 58,9 —282 0 Л 67,^ U316,8 >- 75,6 +s350ttj + 83,7
SiC+3H2O=S iO2+CO+3Ha v-221,2 52,8 . , —sseoj!? —.66,9 ^327^6 ( 'VSjS ^75,4 +89,6
SiC+H2O=Si+CO4-Ha +142,9 4 *34,1 + 64,5 + 8,2 Ц 38,5 ? 9.8 -121 ,l9 4^ 1
SiC+H2Q=SiO+C+Ha f161,3 + 38 j 5 — 57,0 + 13,6 — 13^q ,3^1 + 77,5 ^-+18,5
SiC+1/2SiO2—s/2Si+CO +324,7 +191,5 + 45Д ^02^2 + +* 4*2
SiC-bSiOa=SiO+Si+CQ +621,4 ^148,3 +364,5 М 87,(1 +192,8 + 45^ + 18*8 + 4f5
SiC+SiOa=2&04-q +639,4 +152^7 . 4-3874 4 92,4 +217,9 + $2,0 f &f3. 15Д
«3 Sto-|~C=f=CC)-|-Si + 18,4 “ч 4,4 — 22,6 +* 25,6 - <ч' -+ 44^4 1 <Г^ЛО,6
наиболее вероятны реакции, идущие с образование
SiO». При действии водяного пара возможны реакпи«
выделением SiO2, СО2 и Н2, а также SiO2, СО и нс
Вследствие того что основной продукт окисления кавб 2
да кремния SiO2, объем которой больше чем объем Sir
сплошная пленка SiO2 является защитной, замедляв
щей дальнейшее окисление.
Если сравнить окисляющее действие кислорода па
ров воды и окиси углерода, то видно, что по убывающей
окислительной способности по отношению к SiC их мож-
но расположить в ряд О2—Н2О—СО. Кинетику окисле
ния карбида кремния исследовали [702] термографиче-
ским методом в интервале температур 900—1600° С. Сте
пень окисления рценивали по уравнению
г- 11 пл 2Xq
[1 -(1 -Я)/’]2 = —
го
где Ао — средний диаметр диффундирующей молекуль
г0 —среднее значение радиуса образца;
К' — константа скорости реакции;
t — время;
,3_ г3
----•
rQ
В интервале температур 900—1200° С продуктом
окисления является аморфная окись кремния, теплота
образования которой составляет 83,8 кдж[мол
(20 ккал!моль). При более высоких температурах она
превращается в кристаллическую, теплота образования
ее 69,7 кдж!моль (16,6 ккал)моль}. Пары воды в оки -
лительной атмосфере повышают скорость окисления.
При нагревании образцов SiC при температуре 900°С
и давлении кислорода 13,33 w/л/2 (Ю'1 мм рт. ст.} [702
оорззуется легкая летучая SiO и СО. При температуре
выше Уии С и давлении около 0,1013 Мн/м2 (1 ат) обра-
зуется S1O2. '
Кремния легко взаимодействует с расплава-
таллов °R П°В’ЖФ\ТОВ и гиДРОокисей щелочных ме-
таллов, В2О3, KNO». Na2O2, Na2B4O„ РЬСгО4, КСЮ»
как СяО мепЮ/~К2Сг2О7 + РЬСгО4 с такими окисламн,
[702, 83?]. g ’ U0’ Fe0’ Ni0> M”0’ Сг2°з> Pb0 н Ag2
260
Термодинамический анализ реакций взаимодействия
карбида кремния с окислами кальция и кремния [90] по-
казал, что карбид кремния в присутствии указанных
окислов термодинамически неустойчив в интервале тем-
ператур 1730—2130° С. При этом в системах SiC—SiO
и SiC—SiO2 до температуры 1900° С, наиболее вероятно
образование кремния, а при более высоких температу-
рах—SiO2- Разрушение карбида кремния при темпера-
турах до 1900°С наиболее благоприятно при взаимодей-
ствии с SiO, выше 1900° С — с SiO2. Кроме того, взаимо-
действие в системах SiC—SiO, SiC—SiO2 и SiC—СаО
более благоприятно, чем в системе SiC—SiO2—СаО
(рис. 79).
Обычно в техническом SiC определяют содержание
общего и свободного кремния, кремниевой кислоты, об-
щего и свободного углерода и примесей, в основном же-
леза.
Не останавливаясь на этих методах, которые широ-
ко описаны в литературе [754, 832, 833, 837—839], отме-
тим методы анализа чистого карбида кремния [702,
840—842]. В основном это методы спектрального анали-
за, они сводятся к определению содержания примесей.
В работе [841] приведен спектральный анализ чистого
карбида кремния по методу испарения. Испарение про-
водят при температуре 1850° С в течение 3 мин, фотогра-
фируя спектры на спектрографе ГКП-28. Определяют
содержание меди, алюминия, магния, свинца, сурьмы,
марганца и железа.
Спектральным методом определяют [842] примеси —
медь, кальций, магний, алюминий, титан, ванадий и мар-
ганец в интервале концентрации 10-4—3 • 10~3%.
Примеси меди и железа в карбиде кремния опреде-
ляют [840] полярографическим методом. Для этого на-
веску измельченной и высушенной пробы обрабатывают
смесью серной, фтористоводородной и азотной кислот.
Нерастворившийся остаток обрабатывают соляной кисло-
той, после чего в фильтрате из аликвотных частей опре-
деляют медь и железо. К 50 мл раствора прибавляют
бромфеноловый синий, комплексон III, нейтрализуют
раствором едкого натра, добавляют триэтаноламин и
этилендиамин и полярографируют. £i/2 для Си 0,57 в,
Для Fe 0,99 в.
Содержание примесей никеля, марганца, железа, ме-
18 Косолапова Т.- Я. 261
ди, цинка, сурьмы и молибдена в Si С определяют Метг.
дом нейтронной активации [702]. Навеску об™
чают в реакторе потоком нейтронов плотность^
7-Ю12 нейтр! (см2 • сек) < После облучения навеску Кар
Рис. 79. Зависимость изобарно-изотермического
потенциала от температуры реакций:
1 ~ SiCTB + 23ЮЖ = SiOar + сог; S - SiCTB + SiOr -
= 2Si -4- СО ; 3 - SiC__ + СаО _ = CaSr. + СО_;
Ль 1 ГВ ТВ Л
1 /1 4 \
4 - SiCTB+ —CaSiO3= Са+— Si) +СОГ;
3 \3 5 /ж
5-SiC_„+ — Ca„SiO.= (— Са+ — Si) +СО_
Т 4 2 4 I 2 4 /ж
бида, обрабатывают при температуре 1250°С хлоро
и кислородом. При этом протекают реакции:
SiC 4- 2С12 = SiCl4 + С;
С + Оа = СО2.
Активные примеси отделяют одну от другой и затем и
меряют активность с помощью счетчика Гейгера.
262
ГЛАВА IX
ПРИМЕНЕНИЕ КАРБИДОВ
Свойства карбидов, особенно карбидов переходных
металлов периодической системы элементов Д. Ц. Мен-
делеева, отличающихся высокими температурами плав-
ления и твердостью, коррозионной стойкостью, износо-
стойкостью, металлическим характером проводимости,
определяют возможность широкого применения их в
различных областях техники. Если раньше карбиды
переходных металлов в основном использовались в ка-
честве компонентов твердых сплавов, то за последнее
десятилетие области их применения значительно расши-
рились и в общем виде сводятся к следующим:
1. Огнеупорные материалы.
2. Коррозионностойкие и материалы химической про-
мышленности.
3. Материалы ядерной энергетики.
4. Материалы авиационной и ракетной техники.
5. Материалы электро- и радиотехнического назна-
чения.
6. Высокотвердые и износостойкие материалы.
В табл. 56 приведены основные области применения
отдельных карбидов.
18а Косолапова Тл Я, 263
м Таблица 56
Основные области применения карбидов
Материалы. Фазы Основные свойства Области применения Литератур- ный источник
Огнеупорные TiC (кермет TiC—W) t Устойчив в расплавленных натрии и сплавах натрий — калий при высоких температу- рах (1050° С) и давлении [до 861 кн[м2 (8,5 аг)] Детали насосов для перекач- ки натрия [878]
ZrC, NbC Стойкость против действия расплавленных металлов Тигли и лодочки для плавки металлов, трубы для перекачки расплавленных металлов [152]
NbC, ТаС То же Элементы испарительных ус- тановок для алюминия и цинка
HfC, TaC Высокие температуры плавле- ния, стойкость против действия расплавленных металлов В составе специальных огне- упоров для футеровки тиглей при плавке металлов
Сплавы SiC, Стойкость против действия расплавленных металлов Детали устройств для пере- качки расплавленного алюми- ния, футеровки ванн для полу- чения алюминия электролизом расплавленных солей, тигли для плавки цветных и редких металлов, защитные чехлы тер- мопар [877,922J
Продолжение табл. 56
18а*
Материалы Фазы Основные свойства Области применения» Литературный источник
Коррозионно стойкие мате- риалы химической Разлагается химически свя- занной водой Энергичное водоотнимающее средство 1116]
промышленности Взаимодействие с азотом с образованием цианамида каль- ция Сырье дри промышленном получении цианамида кальция [1161
SrC2, BaCg, Л4еС3 (Me — р.з.э.) Наличие двойных и тройных связей в соединениях Синтез металлоорганических - соединений, содержащих ще- лочноземельные й редкоземель4 ные металлы * |15Ч
CijCa Высокая стойкость по отно- шению к различным Химиче- ским реагентам и стойкость против окисления ФиЛцтры в химической про- мышленностй, электроды (при ^лектроХимийеоких процессах), подшипники и уплотнения в на- сосах, подающих соленую воду( под большим давлением йри промывке нефтяных баков мор- ских танкеров. Детали насосов для перекачки кислот, сопла для агрессивный жидкостей й газов „ ? [884, 886, §87]
СГдСз, Сг МоаО - Каталитические свойства. Катализаторы И процессах органического синтеза ~ - < > ' » — 1889)
тт^4-
to Продолжение табл. 56
Материалы ч Фазы S, . Основные свойства Области применения Литературный источник
SiQ Высока^ стойкость против действия химических реагентов Детали насосов для перекач- ки кислых растворов, насадки сопел для разбрызгивания аг- рессивных жидкостей, холо- дильники и скрубберы, работа- ющие в атмосфере агрессивных {газов
Материалы ядер- иой энергетики Ве2С Высокая температура плавле- ния, жаропрочность, специфи- ческие ядерные свойства Компонент конструкционных материалов, материал замедли- телей, биологической защиты [868, 869]
Добавка Ве2С в металличе- ский бериллий увеличивает длительную прочность бериллия при температурах 650—700° С
UC Высокая температура плавле- ния и теплопроводность, отсут- ствие фазовых превращений, специфические ядерные свойст- ва Сердечники тепловыделяю- щих элементов ядерных реакто- ров, материалы горючего в ре- акторах с натриевым и органи- ческим теплоносителями. Спла- вы LFC—ThC — горючее в реак- торах с газовым охлаждением 1 [231, 242J
Продолжение табл. 56
Материалы Фазы Основные свойства Области применения Литератур- ный источник
ZrC Высокая температура плав- ления, малое поперечное сече- ние захвата нейтронов Покрытие графитовых йат- риц, содержащих карбиды ура- на и тория, являющихся ТВЭЛ- ами в энергетических реакто- рах для газовых турбин с замк- нутым циклом [231]
NbC, ТаС Высокие температуры плав- ления, химическая стойкость • Покрытия дисперсионных ТВЭЛов р матрицей из графита с целью удержания осколков Деления [Ьз1]
В4С Большое поперечное сечение захвата нейтронов Материал поглотителей ре- гулирующих стержней (В4С— А1, В4О-А1203> В4С—ZrBa В4С—циркаллой) [869]
Материалы ави- ационной и ра- кетной техники кэ М BegC Добавка Ве2С к металличе- скому бериллию увеличивает длительную прочность берил- лия, сопротивление разрыву Конструкционный материал, применяемый в авиации [868, 869]
Ч * ' Г1-’ f Г— Материалы ' 1 '”V‘ Фазь! 1 k u Основные свойства ,) Л —p —1» i ; <-4 , Области применения Литератур- ный источник
Ti(f f Высокие температуры плавлен ния^ твердость, жаростойкость жаропрочности “ Г ,1Г| Н 1 > Ч Кбмпонент керметов для из- готовления лопаток тазовых турбин, хорошо выдерживаю- щих жесткие условия работы реактивных авиационных дви- гателей; составной части фрик- ционных дисков Для самолето- строения; защитны^ покрытий элементов ракет (сопел АРД и головных частей) [152, 872, 874, 875]
Материалы элек- тро» и радиотех- нического казна- Mebs (Me — р.З.э.) Высокие температуры Йлавле» ния, низкйя работа выхода В Составе эмиттеров Для тер- моэлектронных преобразова- телей энергии [870]
ThC Высокая плотность тока эмиссии» низкая работа выхода электронов Материал катодов, устойчиво работающих при температуре 1900° С в течение 900 ч [871]
TiC Высокая температура плавле- ния, электропроводность, низ- кая скорость испарения Компонент электродов для подводной электрокислородной резки стали; электроды термо- пар для замера температур до 200° С в восстановительных и нейтральных средах и в вакуу- ме / (876, 877]
Продолжение табл. 56
Материалы Фазы Основные свойства Области применения Литератур- ный источник
Материалы элек- ZrC Высокая температура плавле- Термокатоды Электронных [879, 880]
тро- и радиотех- нического назна- чения NbC, ТаС ния, малая скорость испарения высокая плотность тока эмис- сии Высокие температуры плац- устройств, работающих в раз- борных; установках, в услови- ях неглубокого вакуума и ин- тенсивной полной бомбардиров- ки; компонент катодов (ZrC— UC); термоэлектронных преоб-' разователей тепловой энергии В электрическую "Нагревательные элементы Вы- [152, SlSft
ления, низкая упругость Шаров при высокой температуре, ме- таллический характер проводи- мости, удовлетворительная ме- еСжотемпературных электропе- чей Сопротивления, работаю- щих в восстановительных' ц ней- тральных средах До температу-
ТаС ханичесдая прочность Высокая излучательная спо- ры 3000°С ' ‘ ‘ Световой Источник Высокой [152, 883]
МозС еобность высокая температура плавле- интенсивности!, пригодный дли освещения при киносъемках (1400 ям), Покрытие Да ме-тад - ’ дцчеекие вольфрам д рений для специальных элементов свече- ния, электроламп Припой «яри высокотемпера- [888]
ЬЭ сл к лени» турной пайке в «электронике Л г.р—
« 4—..ц.и.. - /г , « . * .. . 1 — *
to о Продолжение табл. 56
Материалы ,„-7-. . —.... Фазы Основные свойства Области применения Литературный источник
Высокотвердые й. износостойкие Сг2зСв SiC TiC, WC Металлический характер про- водимости, малый температур^ ныц коэффициент электросоп- ротивления ' Высокие электрическое сопро- тивление й химическая стой- кость/ полупроводниковые свой- ства । Высокие температура плавле- ния, твердость, жаростойкость, жаропрочность, способность перекристаллизовываться через расплавленные Металлы Компонент постоянных низ- коомных сопротивлений, рабо- тающих при температурах 300^-400° С с номинальными Значениями сопротивления 5— 50 ом Компонент зажигателен в иг- нитронных выпрямителях, вили- товых сопротивлениях, предназ- наченных для защиты высоко- вольтных Линий электропере- дач, миниатюрных нелинейных сопротивлений, волноводных поглотителей в волноводах, пе- редающих энергию в частотах выше 1000 мгц, -нагревателен электрических печей Компоненты металлокерами- ческих твердых сплавов, обес- печивающих повышение произ- водительности труда в металло- обрабатывающей, горнорудной, угольной* нефтяной промыш- ленности, широко используются твердые сплавы марок ТК, КА, WZ, FS, вк [786] [152, 709, 741] [297 , 567 860, 723, 873, 890, 891, 907, 915, 916] -—i. .
Продолжение табл. 56
Материалы Фазы Основные свойства — - ? . . - Области применения Литературный источник
Высокотвердые и износостойкие ZrC, Сг3Са Высокая абразивная способ- ность Полирующие материалы 1775]
VC, NbC, ТаС Высокие температура плав- ления, твердость, жаростой- кость Легирующие добавки к твер- дым сплавам на основе, TiC и WC. Добавка VC повышает стойкость резцов на 10—20%; добавка 5% ТаС к сплаву В Кб повышает скЬрость обработки чугуна на 10%; добавка 4% ТаС к сплавам маркК ТК повы- шает скорость обработки ста- лей на 20 % [882]
Сг3Са 1"—н— Высокие температура плавле- ния, твердость, сохраняющаяся при высоких температурах, хи- мическая стойкость Компонент наплавочных сплавов, а также твердых спла- вов, используемых для изготов- ления сопел, штампов, высоко- температурных подшипников, пресс-форм для прессования ла- тунных профилей, наконечников пескоструйных аппаратов, вкладышей, в крупногабарит- ные матрицы для протяжки труб [873, 885— 887]
Предолжение табл, 56
Материалы Фазы f Основные свойства Области применения 1 Литературный источник
Покрытия из карбидов тита- на, ванадия, ниобия, хрома на стали повышают износостой- кость от 1,5 до 30 раз, а также значительно повышают жаро- стойкость и коррозионную стой- кость 1321]
i В4С Высокая твердость и зивная^ способность аб^а- Шлифование Й йодирование 'Твердых материалов; заточка и доводка рейдовых пластин из твердых сплавов; режущие эле- менты буровых колонок и инст- рументов для обработки твер- дых материалов, инструмент для правки шлифовальных кру- гов, Пескоструйные сопла, ка- либры, матрицы для протяжки стержней из абразивных мате- риалов 1152, 691, 723)
. w.l ir 1 . .1. SiC Высокая твердость И дивная способность абрат Шлифование, полирование, абразивные изделия [152, 723J
ЛИТЕРАТУРА
1.
Краткая химическая энциклопедия. Изд-всГ «Советская энцикло-
педия*, !964,т. III, е. 424.
Самсонов Г* В. Порошковая металлургия, 1965» № 1, с, 98
педия», 1964.x HI, e. 424.
Самсонов Г* В. Порошковая металлургия, 1965» № 1, с, 98
Самсонов Г. В. В сб... «Редкие щелочные металлы». Новоси-
бирск. Изд-во СО АН СССР, 1965.
Ф ед о р о в П. И., Су М еэ н -ц з э н. Acta Chimica Simca, 1957,
V. 4U, <>= -uu.
5. Алабышев Ал Ф. и др? Натрий и калий. Госхимиздат, 1959
2.
3.
4.
бирск. Изд-во СО АН СССР, 1965.
v_23,№ hp;M
6. Самсонов J. В. Тугоплавкие-соединения редкоземельных* ме-
таллов с неметаллами. Изд-во «Металлургия», 1964.
7. 5 ped ding JP. -a. o.J. Amer. -Chent Soc., 1958, v,: 80, № 17,
8.
9.
10.
таллов с неметаллами. Изд-во «Металлургия», 1964.
р. 3399. л
Косо л а нов аХ. Я. к др^ЖНХГ19б5, r^XOMs 11, с. 243.
Г ер ц б е р т -Г?Атомные реакторы х строение атома. ИЛ» 1948.
Salzano’EJ.fc Arpnson-S. J. Irrorg. Nucl. Chem., 1964
v. 26, p; 1456.
11. С а м сонов X В.,“У манский Я. С. Твердые соединения ту-
гоплавких металлов. Металлургиздат, 1957.
12. Самсонов П В» Изв. ректора физ.-хим. анализа, 1956, т. 27,
с. 97..
13. К у дм а А» Я., С а м с о н о в Г.В. Изв. АН СССР. НеорТаниче-
ские материалы, 1966,/г. II, № 11, с» 1970.
14. Сам сод о в Г. В. Укр. хим» журн., 1965, т. XXI, вып. 12, с. 1233.
15. Самсонов Г, В. 'ЖТФ, J956, т» 26, вып, 4, с. 716.
46. Львов С, Н. и др. Порошковая металлургия, 1962, № 4, с. 3.
17. Сам соя о в Г. В. Укр.-хим, журн., 1965, т. XXXI, вып. 10,
с. 1006.
18. Ф о м е н к о В. С. Эмиссионные свойства элементов и химиче-
ских соединений. Киев. Изд-во «Наукова думка», J964.
19. Л ь в о в С. Н. и др. ДАН СССРГ 1960. т. 135, №. 3, с. 577.
20. Макаренко Г. И и др. Изв. АН СССР. Неорганические ма-
териалы, 1966, т. II, № 4, с. 626„
21. Мее р с он Г. А.» Самсонов Г. В. ЖПХ»1952, т. XXV, вып 7,
с. 744.
22. Киффер Р., Швардкопф П.Твердые сплавы. Металлург-
издат, 1957.
23. Гельд П. В., Вейн О. А. Процессы высокотемпературного
восстановления. Металлургиздат, 1957.
24, Ruff О., W (1 n s ch Р. Z, anorg. Chem., 1914, В. 85, S. 292.
25. Friedrich F,, Sittig L.-Z. anorg. Chem., 1925, B.
S. 163.
26. A g t e M о e r s K. Z. anorg. Chem., 1931» B. 198, S. 233.
27. Tamm an <3., Swofykin A- Z. anorg. Chem., 1928, B.
S. 62.
144,
170,
273
40.
41.
42.
43.
44.
„ /г 7 anorg. Chem., 1923, В. 126; S. 119.
28. Та mm an G. z. a g в s
MBauHoh W, Durrer R. Arch. EisenhuttenWes.r 1930/l93l
D Л. C /1
31 Baukloh W., Zimmerman G. Stahl und Eisen, 1933, B. 53
32 BauUoh W.. Durrer R. Z. anorg Chem., 1935, B. 222, S. 189
r а й к qb А. А. Металлург, 1926, № 3, c. 5.
S’ Гист ^инг A. M. ЖПХ, 1951, т. XXIV, c. 266.
S Гистлйнг А. М.ЖПХ, 1952, т. XXV, c. 277.
36 Позин M. E. и др. ЖПХ, 1954, т. XXVII, № 4, с. 376.
37’ Есин О. А., Гельд П, В. Успехи химии, 1949, т. XVIII, с 63S
38 Гельд П. В. и др. ЖПХ, 1952, т. XXV, № 2, с. 121. ’ Ь58
39. Г е л ь д П. В., Ш в е й к и н Г. П. Изв. АН СССР, ОТН, Метал-
лургия и топливо, 1959; № 1, с. 144.
Карасев Р. А. и др. Изв. АН СССР. Металлургия и топливо
1956 № 4 е. 94.
Самарин А? М., Бертман А. А. Труды Института им
А. А. Байкова. Изд-во АН СССР, 1957, вып. 1, с. 60.
S t a d е1е г A, Metallurgies 1908, v. 5, р. 260.
Самсонов Г. В. Укр. хим. журн., 1957, т. XXIII, вып. 3, с. 287
Kroll W.J., S eh 1 ech t о п A. W. Trans. Electrochem. Soc,
1948„v. 93, p. 274.
45. Меерсан Г. А.гЛиИкес Яи M. ЖПХ, 194Ut. XIV, №3
c. 291.
Е\ А.г Л и it к е с яи М. ЖПХ, 194U т. XIV, № 3
46. M e e p e о н Г. А., Л и и к е с Я- M. ЖПХ, 1945, т. XVIII, № 4/5
с. 251,
47. М е е р с о н Г. А. Изв. сектора физ.-хим. анализа АН СССР,
1943, т. XVI, вып. 1, с. 213.
48. Me ер сон Г,-А и др. Заводская лаборатория, 1953, т. XIX,
№ 10, е. 243.
49. Be eke г К- Z. Elektrochem., 1928, В. 34, S. 640.
50. К a m р b е 11 е J_ а. о. J. Electrochem. Soc, 1949, v. 96, р. 318.
51. Schenk Rw u. a. Z. anorg. Chem., 1931, B. 203, S. 159.
52. V a n A r k e 1 A. E, Physica, 1924, v. 4, p. 286.
53. Moers K. Z. anorg. Chem.., 193Ц B. 198, S. 243.
A. E,„ de Boer J. Z. anorg. Chem., 1925, B. 148,
Ь, 324.
55. McKenna P. Metals Progress, 1939, v. 36, p. 152.
- , лячКо Ю. А., Шапцро M. M. Заводская лаборатория
1950, т, XVI, № IQ, e. 1173.
w Потока H,M. Карбидный, анализ стали. Оборонгиз, 1957.
°8’ 44е<£вед ева С-. Заводская лаборатория, 1947, т. ХП*
№ 12, с 1413.
Щ^ииро Заводская лаборатория, 1947, т. Х1П
№ 12, с. 1426.
-60. Уикс К. Е.,. Блок Ф. Е. Термодинамические -свойства 65 эле-
ментов, их.-окислов, галогенидов, карбидов и нитридов. Изд-во
«Металлургия», 1965.
g 35 К Н* И. и др. Заводская лаборатория, 1956, т. XXII, Xs 1>
м ^P- Заводская лаборатория, 1955, т. XXI, №Д
6 ' П ко/ ’ е U We i s s G. Bull. Soc. Chim. France, 1948. v 15,
Pt ОУ0»
274
64 Honigschmid. Karbide und Silicide, Haale, 1914.
65 Be p яти в У. Д. и др. Термодинамические свойства неоргани.-'
ческих веществ. Справочник. Атомиздат, 1965.
66 . С л а в и н с к и й М. П. Физико-химические свойства элементов.
Металлургиздат, 1952.
67 Fredenh a ge п К-, Cad еп bach G. Z. anorg. Chem., 1926,
В. 158, S. 249.
68 . Matignon H. Compt rend., 1897, w. 124, ’p. 1026.
69* Moissan H., Compt. rend., 1898, v. 127, p. 911.
70. Gmelins Handbuch der anorg. Chem., 1928, № 21.
71. Hoffman U. Naturwiss., 1939, B. 18, S. 229.
72. Fop pl H. Z. angew. Chem., 1952, B. 70, Ke 13, Si 401.
73. Tamman _G., Sworykin A. Z. anorg. Chem., 1927, B. 168,
№2, S. 218.
74 Fredenhagen Such H. Z. anorg. Chem., 1929, B. 178_
S 353.
75 Moissan H. Compt. Tend., 1898, v. 126, p. 303.
76. M о i s s a n H. Compt. rend., 1903, V. 136, p. 1221.
77 Beber M. B., Floe C. F. Trans. AIME, 1946, v. 166, p. 128.
78. Ruff O., Bergd ahi- B. Z. anorg. Chem., 1919, B. 106, S. 79.
79. N a s t R., P f a b W. Chem. Ber., 1956, B. 89, S. 415.
80. N a s t R., P f a b W. Z. anorg. Chem., 1957, B. 292, S. 287„
81. R ri f f O. Z. anorg. Chem., 1917, B. 99, S. 73.
82. Руководство по препаративной неорганической химии, под ред.
Г. Брауера. ИЛ, 1956.
83. Nichimura Н., К i m u г а Н. J. Japan. Inst Metals, 1956,
v. 20, р. 528.
84. S 1 о m а п Н. A. J. Inst, Metals, 1932, v. 49, р. 370.
85 Хансен М., Ан дер к о К, Структура, двойных сплавов. Ме-
таллургиздат, 1962. 4
86. В о л А.. Е. Строение и свойства двойных -.металлических систем.
Физматгнз, 1959.
87. М а 11 е t М, W. а. о. J. Electrochem. Soc., 1954, v. 10, №6, р. 298.
88. Coobs J. H., Koshuba W. J. J. Electrochem. Soc., 1952,
v. 99, № 3, p. 115.
89. Stackelberg M., Q u at г и m F.. J, Phys. Chem., 1934, B. 27,
S. 50.
90. Рябчиков И. В. и др. ЖПХ, 1964, т. XXXVII, № 9, с 2050.
91. Муратов Ф. Ш., Новоселова А. В. ДАН СССР, 1959,
т. 129, № 2, с. 334.
92 Poll ок В. D. J. Phys. Chem., 1959, v. 63, № 4, р. 587.
93. Stackelb^rg М. Z. Electrochem., 1936, В. 37, № 49, S. 542.
94 Neely J. J. а. о. J. Amer. Ceram. Soc., 1950, v. 33, p. 363.
95. Hu men i к M„ Kingery W. J. Amer. Cerani. Soc., 1954,
v. 37, p. 18.
96. Муратов Ф. HI,, Новоселова А, В. ЖНХ, 1961,, т. VI,
е. 1974.
97. Staritzky Е. Anal. Chem.f 1956, v. 28, p. 915.
98. Murakami J. a. o. J. Japan. Inst. Metals, 1957, v. 21, p. 712.
99. Novak J. Z. Physik Chem., 1910, B. 73, S. 513.
100. Rueggeberg W. H. J. Amer. Chem, Soc, 1943, v 65, p. 602.
101. Schneider A., Cordes J, F., Z. anorg. Chem., 1955, B. 279,
275
л о ПоишепчикП. А. Труды Ленингпап
„ амаков Ю. В ’ ПР ститута, 1940, вып. 1, № 4, Рад’
102'сБкого Ивд^ТРИза£?О° Evans Е. Metallurgical thermochemj.
ЮЗ- 5ruvbpeSrgahmeon Press, 1958, chem , 1923 B (3l
S f f o F°erSie
101 f.321. ” д я и др. Сахарная промышленность, 1956, т.
108‘ Мо°53 с 49. я уим поомышленность, 1961, Ns 6, с. 43.
106. В.п»»“ЛАПаг»" е,а И- А' ЖПХ’ 1961' ’ ХХХ1"-№’>.
1 с. 1901- А щварцИ. Д. Общая технология карбида
1°8' и цианамида кальн’строительство, 1934, т. 8, с. 411.
109- Егоров Ф- И. производство карбида кальция. Госхимиз-
110 Куэнецо в а.
дат, 1954. ппоизводство карбида кальция. Госхимиздат,
ill Биагам н. * и
1934. х, дг+а Chem. Scand., 1962, v. 16, № 5, р. 1212.
ц2 V а п n е г b е г g N. Acta1902, B. 31, S. 130.
3 Rothmund M. Z-nf°r|hem 1907, B. 54, S- 170.
114 Rud oil i H. Z. anorg^Chem^ 1QQ7 R xn c ,AI
115? '
116.
117.
118.
119.
120.
121.
122.
Schlumberger K. Z. anorg. Chem., 1927, B. 40, S. 141.
Доронин H. А. Металлургия кальция, Атомиздат, I959.
Frank H. H. u. a. Z. anorg. Chem., 1937, B. 232, S. 61.
В re dig M. A. Z. Phys. Chem., 1942, B. 46, S. 801.
Stackelberg M. Z. Phys. Chem., 1930, B. 9, S. 437.
Bredig M. A. Z. anorg. Chem., 1959, B. 298, № 5—6, S. 255.
Bredig M. A. Z. anorg. Chem., 1961, B. 310, № 4—6, S. 338.
Borchert W4 Rod er M. Z, anorg. Chem., 1959, B. 302
№ 5/6, S. 213.
123‘сГ600.ЬА 11 КологРеева А. Г. ЖПХ, 1948, т. XXI. №6,
125' ?agawmabeHfr%r ?’• Z-panor.^ Chem- В. 39, S. 219.
|,в1а4’Й''? №?и54.1тОГ1 Ka”iL 1 S“
127.' Н о с к 8°п’; eJ«° V Р рУ В‘ Z‘ a"orS- Chem-> 192б> В. 153, S. 17.
р.2969. J’’ PIatzer N- Compt. rend., 1958, v. 246,
129.’ Корни л o'/'a" w °' Trans- Faraday Soc., 1964, v. 60, p. J030.
130. Фет!° Л ЙДР- ЖФХ, 1964, т. XXXVIII, c. 702.
c. 70. Порошковая металлургия, 1961, № 2,
I31f Йис^ллов» и’, Мч Р“жд ест ве н с к а я В. В. В сб. «Рост
J32-Ал ямо в еки йДг°г?аука>>’ Шб4’т-4’с- П7‘
133. EekstelS R мС’ с' И др- ЖНХ> J963> т- vm- с- 200°-
р. 82. Н- Н” F о г гл a n R, J. Appl. Phys., 1962, v. 33,
134. Gantzel Р к- d , ,
9.772. Baldwin N. L. Acta Cryst, 1964, v. И-
,„5’ ?аллуртаяеигомИ” Шва₽цман Л. А. Изв. АН УССР. Ме‘
Д УбровеК дРяН пЯк°’ 1964’ № 2’ с' 18°-
137. Сазонова м Лп Б'Л др’ ФМЛ^, 1964, т. 17, с. 73 м
XXXVII,ВваыПМ4 а н ь к о в с к а я И. Б. ЖПХ, 1^’
• С< f («J.
276
Микулинскйи А. С.» Марон Ф. С. ЖПХ, i960, Т. XXXIII,
' № 4, с. 835.
139 Гельд П. В., Ееин О. А. ДАН СССР, 1950, т. 63, с. 541
14о' Bonnier Е., Poyet Р. Compt. rend. Acad. Sci., 1959, v.248,
‘ № 12, p. Ю48.
141 Ma рко в скин Л. Я.( Векш и н a H. В. ЖПХ, 1961, т, XXXIV,
‘ с. 242.
142 Векшин а Н. В„ М а р к о в с к и й Л. Я, ЖПХ, 1962, т. XXXV,
с. 30. .
143 Me ер сон Г. А. и др. Изв. АН СССР., Металлургия и горное
дело, 1963, № 1, е. 69.
144. Schneider A., Hutt G. Z. anorg. Chem., 1949, В. 257,
S. 289.
145. Меер с он Г. А. и др. Атомная энергия, 1963, т. 14, № 6,с. 563.
146. Gmelins Handbuch der anorg. Chem., 1932, № 30, S. 299.
147. Thomson M. Trans, Electrochem. Soc., 1909, v, 16, p. 197.
148. Самсонов Г. В. и др. ЖОХ, 1962, т. XXXII, вып. 9, с. 2753.
149. Hi el N. A., Cavin О. В. J. Amer. Ceram. Soc., 1964, v. 47,
№ 7, p. 360.
150. Hoch M. J. Appl. Phys., 1958, v. 29, № 11, p. 15—88.
151. De Kay-Thomson M. Trans. Amei\ Electrochem. Soc., 1928,
v. 54, p. 891.
152. Самсонов Г. В. Свойства тугоплавких соединений. Метал-
лургиздат, 1962.
153. Ruff О., Keilig F. Z. anorg. Chem., 1914, В. 88, S. 410.
154. Г е р а с и м о в Я. И., Крестовников А. И. Изв. вузов.
Цветная металлургия, 1958, № 3, с. 54.
155. Horvath Z. Nehezipari musz. egyet kozL, 1960, 21, 277.
156. Есин О. А., Гельд П. В. ЖПХ, 1958, т. XXXI, № 7, с. 986.
157. Чуфаров Г. И. и др. Тр. Института металлургии АН СССР,
Уральский филиал, 1959, № 3, с. 57.
158. Г в е л е с и а н и Г. Г. и др. Изв. АН СССР, ОТН, 1958, № 8,
с. 19.
159. Jeitschko W. u. a. Mh. Chem., 1964, В. 95, S. 156.
160. Myers Н. Can J. Phys., 1957, v. 35, р. 819.
161. Славинский М. П. и др. Металлург, 1934, № 9, е. 12„
162. Murakami J. а. о. J. Japan Inst. Metals, 1957, V. 21, р. 665.
163. Сох J. Н., Р id geon L. М. Canad. J. Chem., 1963, v. 41, №6,
p. 1414.
164. Ruff o., Jellinek E. Z. anorg. Chem., 1916, B. 97, 3. 312.
165. Askenasy P., Lebed eff A. Z. Elektrochem., 1910, B. 16,
S. 559.
166. Беляев А. И. и др. Цветные металлы, 1957, т. 3, с. 45.
icZ’ Б а й м а к о в Ю. В. и др. Легкие металлы, 1937, № 1, с. 22.
168. Murakami J. а. о. New Fac. Engng. Kyoto Univ., 1957, v. 19,
p. 302.
•69- Wohler L., Hofer K. Z. anorg. Chem., 1933, B. 213, S. 249.
Chupka W. A. a. o. J. Physic. Chem., 1958, v. 62, p. 611.
f?,eschi D., Searcy A. J. Phys. Chem., 1959, v. 63, p. 1175.
*<2. Кучма А. Я. Изв. АН СССР. Неорганические материалы,
17о I?67, т- 3- № 5’ с- 884-
Ил Richardsone F. J. Iron. Steel Inst., 1953, v. 175, № 1, p. 33.
74. Campbell C. S. Metallurg. Soc. Conf., 1961, v. 7, p. 412.
175. G r j о t h e i m K. u. a. Z. anorg. Chem., 1964, B. 328, S. 267,
277
47А fpitschko W u. a. Mh. Chem., 1964, B. 95, S. 431, 436.
177. Jeitschko W a. o. L.r.!^t V\7ap’J33'
l7S' Jroidings of a Symposium, Vienna, 1962, 519.
179. Jacobson B., Westgren A- Z, Phys. Chem.„ 1933,
D. D. a. o. Thermodinamics ^of ^Nuclear Materials.
B. 20,
S 361
180. Vickery R. a. a. J. Chem. Soc., 1959, v 159,, p. 503.
ISLAuer-Welsbach H., Nowotny H. Mh. Chem., 1961,
В 92 S 198
182. Nowotny H., Auer-Welsh a ch--H, Mh. Chem., 1961,
B. 92, S. 789.
183. Самсонов Г. В. й др. ДАН СССР, 1962, т. 144, № 5, с. 1062.
184 Самсонов Г. В. и др. В сб. «Редкоземельные элементы»
Изд-во АН СССР, 1963.
185. Самсонов Г... В. и др. ЖНХ, 1962, т, VIII, вып. 5, с. 975.
186. Самсонов Г. В. и др. ЖПХ, 1961, т. XXXIV, № 7, с. 1444.
187. Petterson О. Berichte, 1895, В. 28, S. 2419.
188. Moi ss an Н., Etard Р. Compt. rend., 1897, v. 122, р. 543.
189. Mauzone М. G., Brjgge J. Z. Less-Common alloys of
molybdenum. Climax molybdenum company, 1962.
190. Ковальченко M. С. и др. ЖНХ, 1958, т. XXXI, & 1427.
191. Макаренко Г. Н. ЖПХ, 1963, т. XXXVI, е. 1860.
192. К ос о ла п о в а Т. Я., Макаренко Г. Н. Укр. хим. журн
1964, т. XXX, вып. 8, с. 784.
191
194.
195------------ ------------------------- ,.
196. Косорпова T. Я. а др. ЖПХ, 1966, т. XXXIX, № 11, с.2396.
197. Wil h $1 m H., Chiotti P
198.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
278
В re ver L„ Krikorian O. J. Electrochem. Soc., 1956,
v. 103, p. 38. Л
Moissap H. Compt. rend., 1900, v. 131, p. 595.
Moissan H. Compt. rend., 1J396, v. 123, p. 148.
_______________________ _ . _ Trans. Amer. Soc. Met., 1950,
v. 42, p. 1295.
W i IE e 1 m H. a. o. J. Chem-. Soc.,. 1949, v. 2, p. 318.
Pulvermetallurgie- in der Atomkemtechnik, Metallwerk Plansee
Ag. Reutte/Tirol, 1962.
Techn. Confer, of compounds of Interest in Nucl, Reactor, Colo-
rado, 3—5/VI 11.64.
м®Чг®,0П D' Trans- Am- Soc- Metals^ 1961, v. 53, p. 765.
elson P e te r s о jj 6 Trans. Am,. Soc. Metals, 1958,
v. 56, p. 340.
Troast L. Compt. rend., 1893, v. 116, p. 1229.
Jf°*ssan 4? Etar^ F' Compt. .rend.t 1896, v. 122, p. 573.
Nowotny H. u. a. Planseeber. Pulvermet., 1957, B. 5, № 3,
o. 1UZ,
Scaife D. E., Willie A. W
pos., Sydney, 1958,
Сам co н о в Г, В и дп WTiY
Street R. s„ Wafers TN
gy Author, 1962, № M1114 " ‘
tak^^V1 RA Krikorian
tals, 1961, v, 4, № зг p_ 244 '
Street R. S., Waters т кт
gy Author, 1962,№ Ml U5 ‘
Perr?e tinAmiCSRn-^ar materials. Vienna, 1962, S. 581.
p.740 A” Reithftia“ 1 Helv. СЬЦ Acta, 1943, v. 26,
Austral. Atomic Energy Sym-
1960, т. XXXlII,№ 7, с. 1661.
Res. goup. u^»k. Atomic Ener-
N. Й. J. Less-Common Me-
Res. group u. k. Atomic Ener*
213 Costa P., Lallement R. Physic Letters, 1963, v, 1, Xs 1,
’ p. 21.
214 S h u b b W. Reactor materials, 1963, v. 6, № 2, p. 42.
215 Hund E., Rundle R. J. Amer. Chem, Soc., 1951, v, 73, № 10,
' P- 4777.
216 lonsdall H. K-, Graves J. N. Thermodinamics nuclear
materials, Vienna, 1962, p. 601.
217 Moissan H. Compt. rend., 1896, v. 122, p. 274.
218. Lebeau P. Compt. rend., 1911, v. 152, p. 755.
219. Litz L. a. o. J. Amer. Chem., Soc., 1948, v. 70, p. 718.
220. Кац Дж., Рабинович E. Химия урана. ИЛ, 1954.
221. Wilson W. J. Amer. Ceram. Soc., 1960, v. 45, № 2, p. 77.
222. Chubb W., Phillips W. M. Trans. Amer. Soc, Metals,
1961, v. 53.
223. Tetsuzo A. a. o. Scient Papers, Inst. Phys, and Chem. Res.,
1961, v. 55, № 4, p. 217.
224. S ecrestet A. Preparation and Properties of Uranium Mo-
nocarbide, Castings, BM L-1309, 1959.
225. Esch P., Schneider A. Z. anorg. Chem., 1948, B. 257,
S. 254.
226. De Maria J. a. o. J. Chem. Phys., 1965, v. 43, p. 4449.
227. Mallet M- a. o. J. Electrochem. Soc., 1952, v. 99, p. 197.
228. С и б о p г Г. T., Кац Дж. Химия актинидных элементов.
Атомиздат, 1960.
229. Игнатьев Б. Г. и др. Атомная энергия, 1966, т. 20, с. 489,
230. Fischer R. Chem. Abstr., 1952, v. 46, p. 52786.
231. Strasser A. Nuclear Eng., 1960, v. 5, p. 353.
232. Ruff O., Heinzelmann A. Z. anorg. Chem., 1911, B. 72,
S. 72.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
Heusel O. Z. anorg. Chem., 1926, B. 154, S. 366,
Piarra J., Sinnott M. B. J. Chem. and Engng. Data, 1962,
v. 1, № 4, p. 451.
Meepсон Г. А. и др. Атомная энергия, 1960, № 11, с. 387,
Kruger О. L. J. Amer. Ceram. Soc., 1963, v. 46, p. 80.
H i m m e 1 s t e i n P. u. a. Ber. Deutsch. Keram. Ges., 1963,
B. 40, № 2, S. 153.
New Nucl. Mater. Includ. Non-Metall. Fuels, Vienna, 1963, v. 1,
p. 377.
Craven B., Me. Carthey E. J. Amer. Ceram. Soc., 1961,
v. 44, № 1, p. 12.
Арбузов M. П., X a e н к о Б. В. Порошковая металлур-
гия, 1966, № 3, с. 64.
We hie V. Naturwissenschaft, 1965, В. 52, S. 537.
c ’e r u- a- Planseeber. Pulvermet., 1957, B. 5, № 1,
O. «53.
A., C a ill at R. Powder Met. New York — London,
1961, p. 209.
с 3 P А" С- и ДР- Порошковая металлургия, 1966, № 2,
Арбузов М. П., Хаенко Б. В. Порошковая металлур-
гия, 1966, № 4, с. 74.
М о i s s a n Н. Compt. rend., 1896, v. 122, p. 1297.
Chang R., Rhodes C. G.. J. Amer. Ceram, Sos., 1962, v. 45.
№ 8, p. 379.
279
254.
255.
256.
257.
258.
259 * _. „ „ __, - .4.
260. LeUnaker J. М., Wit tern an W. G. J. Amer. Phvs. IQfio
п Qfi ко, й ч 144.6 J ' ,aw»
it
n Abs’u’/a' Е Acta Cryst.f 1959, v. 12, p. 159.
Techn. Repts. Cer. Internal, Atomic Energy Agency, 1963> * ц
252. JutpeoH Г. А Атомная энергия, Ж т 21, с, б7
253. Grossman L. N. J. Amer, Chem. Soc4 1963, v. 46 м, «
* n 264 * ®*
Griffiths L, B. J. Nucl. Mater., J961, v. 4, Ms 3, n -wR
Novreys 3.7 Wheeler M. Trans. Brit Ceram
v 62, № 2, p. 180, ’ 1963«
M&nder-Penalosa R., Taylor R, J. Amer. Ceram g.
1964", V. 47, № 2, p. 101. S«»
Barta 1 a. P. J. Nucl, Mater., 1961, v. 4, Me 3, n. 222
Toth L, u. eL Mh. Chem., 1961, B. 92, № 3, S. 794
Hagg N. Z, Physik^Chem., J931, B. 12, № 42, S. 42.
В.'зб, № 6, S. 1445. v“" iaw>
261, Levinson L. S. J. Chem. Phys., 1963, B, 38, № 9, S. 2105
262. F 0 p p 1 H. Z, anorg. Chem., 1957, B. 291, S. 12.
263. Rartfee E., Sadag®pan V. Mh. Chem., 1962, B. 93, S 263
264. Pidd R, W. a. o. J. Appl, Phys., 1959, v. 30, № 10, p.1575.
265. Pa dure W. Reactor Materials, 1962, v. 5, № 4, p. 24.
266. Пршедромйрская E. M., Слепцов В. M. Порош о-
вая металлургия, 1966, № 4. с. 11.
267. Кавальченк® М. С., Ог®роднйков В. В. Порош о-
Хая металлургия, 1966, № 10, о- 48.
268. Lied R. W h i t е G. I). J. Amer. Ceram. Soc., 1962, г 45,
p. 149.
269. Zacharjasfen W. Acta Qryst., 1952, v. 5, p. 17.
270. В r e d i g M. A J. Amer. Ceram. Soc., 1960, v. 43, № 9, p. 4
271. Chang R. Acta CiysU 1961, v. 14, № 10, p. 1097.
272. Crauls J. a. o. Trans. Amer. Soc. Metals, 1963, v. 56, p. 7
273, G e i § e 1 e Г G.^ Bileher W. Z, anorg. Chem., 1966, B. 34
№ 5—6, S. 286. s
274. USAEC Report, ANL6573. dune 1962. Reactor materials, 1963,
v. 6, p. 28.
W e h s с h. Prince P. Reactor materials, 1963, v. 6, p 25.
Afeeary A., Blum P. Nucl. Pow., 1960, v. 5, № 50, p. Ig
Титан й его сплавы. Киев, Изд-во АН
vbbF, I960.
Jaffee R. а. о. J. Metals>1950, v. 2,n. 1261
а“7®и@в IAa В л а ты ш e в а Г
Я oUy.
Самсонов Г, в
£ 109, е. 582.
Самсонов
®. 479.
282. Jaeobsoti B.W
Р. 362.
а,яп’,93в-
Х 983Ч^КНЙ С^. Хи декель L. .
275.
276.
277.
278.
279.
280.
281,,
*201. « 9
В. П. ФММ, 1956 t *
В. П. ДАН СССР, 19»
Г7в7‘Эпик А. И. ФММ, 1962, т. 14, выв.Я
е s t g г е n A. Z. Physikal Chem-, 1933, v »
6, v, 28, р. 767. xv
С. С. ЖФХ, 1941. Г л
Латышева
Латышева
280
ой Меерсон Г. А. Редкие металлы, 1935, № 4, с. 4.
9Я6 Ehrlich Р. Z. anorg. Chem., 1949, В. 259, S. 1.
9S7 Agte С., Moers К- Z. anorg. Chem., 1931, В. 198, S. 233.
9Я8 С a d о f f н., Nielsen Р. J. Metals, 1953, v. 5, № 2, p. 248.
289. Портной К. И. и др. Изв. АН СССР, ОТН. Металлургия
и топливо, 1961, № 2, с. 147.
290. Bickerdike R., Hugness G. J. Less-Common Metals, 1959
v. 1, № L P- 42.
291 Roman G., Ramachandran G. N. Current Sci. 1962
‘ v 31, № 8, p. 321.
292 Moissan H. Compt. rend., 1895, v. 120, p. 290.
293. Ruff О., В antzinger H. Z. anorg. Chem., 1923, B. 129,
S. 267.
294. Krainef C., Konopicky R. Berg. u. Hiittenmann. Mh.,
1947, B. 92, S. 166.
295. В г a n 11 e у L-, Beckmann A. J. Amer. Chem. Soc., 1930,
v. 52, p. 3956.
296. Laughein C., Wainer A. Powd. Met. Bull., 1950, v. 5,
p. 24.
297. Norton J. T. Machine Desing. 1956, v. 28, p. 143.
298 Huttig G. a. o. Powd. Met., 1950, v. 5, p. 30.
299. Miinster A., Ruppert W. Z. Elektrochem., 1953, B. 57,
S. 558.
300. Верт ж. Д., Каменцев М. В. ЖНХ, 1959, т. IV, № 1,
а 17.
3 1. Miccioli В. R., Shaffet Р. Т. S. J. Amer. Ceram. Soc.,
1964, v. 47, р. 351.
302. Меер сон Г. А., Липкес Я. М. ЖПХ, 1939, т. XII, с. 1759.
303. К у ц е в В- С. и др. ДАН СССР, 1955, т. 104, с. 567.
304. Желании н В. И. и др. ЖНХ, 1958, т. III, с. 1237.
305. Jaitschko W. u. a. Mh. Chem., 1964, В. 95, S. 1004.
306. Goldschmidt Н. J., Brand J. A. J. Less-Common Me-
307.
308.
309.
310
311.
312.
tals, 1963, V. 5, р. 181.
Меерсон Г. А., Крейн О. Е. ЖПХ, 1952, т. XXV, с. 134.
М е е р с о н Г. А., Крейн О. Е. В сб. «Металлургия цвет-
ных металлов» (МИЦМиЗ). Металлургиздат, 1952, № 22.
Самсонов Г. В. ЖПХ, 1955, т. XXVIII, вып. 9, с. 1018.
Самсонов Г, В. Укр. хим. журн., 1957, т. XXIII, в. 3, с. 287.
Куцев В. С., О р монт Б. Ф, ЖФХ, 1955, т. XXIX, вып. 4,
с. 597.
Меерсон Г. А. Изв. АН СССР, ОТН, Металлургия и топ-
ливо, 1962, № 3, с. 33.
Wagman а. о. Research N. В. S., 1945, V. 34, р. 143.
Van Arkel A. Physika, 1923, В. 3, S. 76.
Г “---- — - ало/ч Г* nie
S. 209"
313.
314.
315.
3,6- Алексеев В. И.
ч №4, с. 545.
• Pollard F., Woodword P.
v.46,p. 190.
°'0- tJiiOTiiH В. П. и др. Изв. вузов. Черная металлургия, 19оо.
,,„№11, с-5-
' ?>ЛЮтии В. П. и др. Изв. вузов. Черная металлургия, 1964,
Wagman a. о. Research N. B. S., 1945, v. 34, p. 143.
Van Arkel A. Physika, 1923, B. 3, S. 76.
cli.IJ’ers W., В as art J. Z. anorg. Chem., 1936, B. 216,
Шварцман Л. А. ФММ, 1961, t. 11,
Trans. Faraday Soc., 1950,
№ 4, с. 545.
V. 46, р. 190.
№ П, с. 5.
№ 3, с. 124.
281
\г The Phycics of powder metallurgy, 1951, p. 295
320. Fattinger V. ^е§пУик д. M. Покрытия из тугоплавки'
32L соединений. Изд^во^ «Металлургия»,^
323^ t Н u^m ^n i k М.‘ Planseeber. Pulvern^.,
324' ?961^.Т№}г-2АР ®0др. Изд. АН СССР, ОТН. Металлургия
325. Меереон Г. А и и2
и топливо, 1УО1> F и лп Физические основы металловедения
326. Умаиский Я. и
ГОНТИ, 1949 Chem., 1957г в. 69 № 9, S. 281,
327. Munster A. Z angUor 1931, В. 77, S. 505.
328. Brantley L. Zj.f ц Д ён, Metallforschung, 1947, В. 2
329. Novotny Н., Ме11
S. 257. с л ъ г я d е г A. Arch. Eisenhiittenwes., 1936/37,
330. Н оf f m a n W„ ь сп 1
B' I0’ -Sk i\v R i x W. Z. anorg. Chem., 1940, B. 244, S. 191.
331. Dawihi w, R‘xrh i960 B. 91, S. 1761.
332. Rudy E. ua MlrChem.3 KQ т г ЖТф>
333. Ковальский A.
1 mа/к”'™pt₽j: ",ys'chera- 1958’ " “•
№ 5, p. 630 Chem., 1958, v. 62, p. 5.
336. C h u p k a W. a. 0. J. Phys. c Испарение тугоплавких
337„фесенко К В-
338.
339.
соединений. Изд-во «Металлургия», 1965.
Humphrey G. J. Amer. Chem. Soc., 1961, v. 73, p. 2261.
Fujishiro S. J. Japan Soc. Powder Met, 1961, v. 8, № 2,
p. 73.
Naylor B. Amer. Chem. Soc,, 1946, v. 68, p. 370.
Kelley K. Bur. Mines Bull., 1949, № 476.
Морозова M. T. и др. ЖОХ, 1962, т. XXXII, № 7, с. 2072.
Taylor R. Е. J. Amer. Ceram. Soc., 1961, v. 44, № 10, p. 525
Hoch M., Vardi J. J. Amer. Ceram. Soc, 1963, v. 46, № 5,
p. 245. c
Glaser F.3 Moskowitz D. Powd. Met. Bull, 1953, v. ,
p. 178.
К о л p м oe ц H. В. и др. ЖТФ, 1958, т. 28, с. 2382.
П ' ' 1 - С. ”. „ мм. ICJU-T, Г. 1О', v' QQC
348. Hollander L. Е. J. Appl. Phys, 1961, v. 32, № К
349. Tsuchido K. a. o. J. Phys. Soc. Japan, 1961, v. 16, P-
Glaser F, Ivanick W. J. Metals, 1956, v. 4, p. « -.ggQ
В e p e й к и н а Л. Л. и др. Заводская лаборатория,
т. XXVI, с. 620.
Bridgman Р. Proc. Amer. Acad.y 1933, v. 66, p. *
Еременко В. H. ЖНХ, 1956, t. I, c. 131. o 1955,
Kin ger у W, Hal den F. Amer. Ceram. Soc. bum,
v. 34, p. 117.
Меереон Г. А. и др. ЖПХ, 1940, т. ХШ, с. 66. у 8,
Blumental Н, Silverman К. J. Metals, 1уэо'
р. 977. мртаЛ^уР’
Еременко В. Н, Лесник Н. Д. Порошковая м
гия, 1961, № 1, с. 43,
340.
341.
342.
343.
344.
345.
346.
£'• М ?1°В С Н' Й ДР’ ЛАН СССР, 1964, Т- 157> № 2> С' 408’
350.
351.
352.
353.
354.
355.
356.
357,
282
358.
359.
60.
361.
362.
363.
364.
365.
366
67.
Paman S., Ramachandran G. N. Current Sci.f 1962.
v 31 № 8, p. 321.
Еременко В. H., Найдич Ю. В. ЖНХ, 1959, т. IV, № 9,
с. 2052.
Самсонов Г. В. и др. Изв. АН СССР, ОТН. Металлургия
и топливо, 1962, № 5, с. 167.
Григорьева В. В., Середа Н. Н. Порошковая метал-
лургия, 1961, № 2, с. 48.
Баски н М. Л. и др. ФММ, 1962, т. XIV, с. 422.
Баскин М. Л. и др. ФММ, 1961, т. XII, № 6, с. 860.
Heltz Н. J. Amer Ceram. Soc., 1950, v. 33, p. 340.
Самсонов Г. В. и др. Изв. АН СССР. Металлургия и гор-
ное дело, 1964, № 4, с. 106.
Barr Н. а. о. Powder Metallurg. Bull., 1950, v. 5, p. 62.
Kieffer R., К о I b i F. Powder Metallurg. Bull., 1949, v. 4,
368 Кубашевский О., Эванс Э. Термохимия в металлургии,
ИЛ, 1954.
69 Rudy Е., Benesowsky F. Planseeber. Pulvermet., 1960,
В 8 S 72.
370 П ад ер и о Ю. Б., Самсонов Г. В. ДАН СССР, 1961,
т. 137, с. 13.
71 Go I'd schmidt М. J. Iron Steel Inst., 1948, v. 160, p. 345.
372. Уманский Я- С. Карбиды твердых сплавов. Металлургиз-
373
374.
375.
дат, 1937.
Ковальский А. Е., Макаренко Т. Г. В сб. «Микро-
твердость». Изд-во АН СССР, 1951.
Самсонов Г, В., Розинова Н. С. Изв. сектора физ.-хим.
анализа. Изд-во АН СССР, 1956, т. 27, с. 126.
Benesowsky F., Rudy Е. Planseeber. Pulvermet., 1960,
В. 8, № 2, S. 66.
Robson H. E., Gieles P. W. J. Phys-. Chem., 1964, v. 68,
p. 983.
Moissan H., Lengfeld M. Compt. rend.,. 1896, v.. 122,
p. 651.
Kroll W. Trans. Electrochem., 1947, v. 92, p. 187.
P- 1367. ~
376.
377
378 ____ _____________________
379. Verhaegen G., Drowart G. J. Chem, Phys., 1962, У. 37,
P- 1367.
380. Prescott С. H. J. Amer. Chem. Soc., 1926, v. 48, p. 2534.
381. Самсонов Г. В., Латышева В. П. ФММ, 1956, т. П,
, с 309.
382. Самсонов Г. В., Эпик А. П. ФММ, 1962, т. XIV, № 3,
с. 479.
383. Norton J., Мои ry A. Trans. AIME, 1949, v. 185, р. 133.
384 Becker К-, Ebert F. Z. Physik, 1925, B. 31 S. 268.
385 Ковальский A. E., Уманский Я. С. ЖФХ, 1946, т. XX,
,8с с. 769.
Ковальский А. Е., Петрова Л. А. В сб. «Микротвер-
,я_ Д°сть»- Изд-во АН СССР, 1951.
38Я Коломоец Н. В. и др. ЖТФ, 1958, т .28, с 2382
Чйа Хау1ог К- Е- J- Amer. Cer. Soc., 1962, v. 45, № 7, p. 353.
^9- Pollack B. D. J. Phys. Chem., 1961, v. 65 p. 751
39?- hotter P., Kohn J. J. Amer. Ceram. Soc., 954, v. 37, p. 415.
691 Нешпор В. С. и др. ЖПХ, 1964, т. XXXVII, с. 2375.
283
392 Krik°rian N- H. a. q. U. Electrochem. Soc., 1963, у
6 p 587 ' ' *
393. Желанкин В. И., Куцев В. С. ЖФХ, 1964, т. XXXVlH
№ 3, с. 562. *>
394 Самсонов Г. В. й др. Огнеупоры, 1961, № 7, с. 335
395’ L i d m a n W., Ham Н. J. Amer. Cer. Soc., 1952, у. 35 n
396 Орм о и т Б. Ф. ЖФХ, 1959, т. XXXIII, с. 1455. р’ 2&
397. Нешпор В. С. и др. Порошковая металлургия, 1967 М> о
с 65 * " **
398. Olougherty Е. V. а. о. Nature, 1961, v. 191, р. Ц94
399. Norman J. Н., W i n е h е I Р. J. Phys. Chem., 1964, v ro
p. 3802. ’ ’
400 Nowotny H. u. a. Planseeber. Pulvermet., 1959, B. 7 № 0
p. 79. ’ * *
401. Kieffer R. u. a. Metall, 1959, B. 18, № 10, S. 919.
402. Желанкин В. И. и др. ЖФХ, 1959, т. 33, № 9, с. 1988.
403. Желанкин В. И. и др. ЖФХ, 1961, т. 35, № 11, с. 2608
404. Меерсон Г. А., Крейн О. Е. ЖНХ, 1960, т. 5 №9
с. 1924. * *
405. Самсонов Г. В., Падерно В. Н. ЖПХ, 1961, т. 34 № 5
е. 963. ' ’
406. Curtis G а. о. J. Amer. Ceram. Soc., 1954, v. 37, № 10, p. 458.
407. Nowotny H. u. a. Mh. Chem., 1959, B. 90, № 5, S. 86.
408. Nowotny H. u. a. Mh. Chem., 1958, B. 89, S. 701.
409. Nowotny H., Laube E. Planseeber. Pulvermet, 1961, B. 9
S. 54.
410. Портной К- И., Левинский Ю. В. ЖФХ, 1963,
т. XXXVII, № 11, с. 2467.
411. Падерно В. Н. Изв. АН СССР^ ОТН. Металлургия и топ-
ливо, 1962, № 6, с. 176.
412. Grisaffe S. J. Amer. Ceram. Soc., 1960, v. 43, № 9, p. 494.
413. Фоменко В. С Радиотехника и электроника, 1961, т. 6,
№ 8, е. 1406.
414. Мацкевич Т. Л. и др. ЖТФ, 1962, т. XXXII, вып. 10, с. 1266.
415. Osawa А., Оу а М. Kinzoka no Kenkyu, 1928, v. 5, р. 34.
416. Westgren A. Metallwirtschaft, 1930, В. 9, S. 921.
417. Н a g g G. Z. physik. Chem., 1933, B. 12, S. 51.
418. Maurer E. u. a. Arch. Eisenhiittenwes., 1939, B. 13, S. 337.
419. Schonberg N. Acta Chem. Scand., 1954, v. 8, № 4, p. 624.
420. Powers K-, Doyle M. Acta metallurg., 1958, v. 6, № Ю.
P. 643. .
421. Гуревич M. А., Ормойт Б. Ф. ДАН СССР, 1954, т. 96,
№6,1165.
422. Желанкин В. И. и др. ЖНХ, 1958, т. III, вып. 5, с. 1237-
423. Гуревич М. А., О р м о я т Б. Ф. ЖНХ, 1958, т. III, с. 403.
424. Алямовский С. И. и др. ЖСХ, 1961, т. 2, № 4, с. 445.
425. Stroms Е. К., М с. Neal R. J. J. Phys. Chem., 19о2, v. 65,
№ 8, р. 1401.
426. Волкова Н. М. и др Изв АН СССР. Металлургия и гор-
ное дело, 1963, № 5, е. 134, г ,«б
*21-£УРевИч М. А., Орм онт Б. Ф. ЖНХ, 1957 т. П, с-1^;
Bitner Н„ Goretzki Н. Mh. Chem., 1962, В. 93, ‘,gg2,
429. Fujishiro S. Qokcen N. A. J. Electrochem., Soc.,
v. 109, Nb 9, p. 835.
284
430. Becker К. Z. Physik, 1928, В. 51, S. 481.
431. Ruff О.» Martin W. Z. angew. Chem., 1912, В 25 № 2
S. 49. ’ ’ ’
432 Беккер К. Тугоплавкие соединения и их использование в
технике. ОНТИ, 1934.
433 Dufek V. Technike spravy, 1959, 6, 13.
434^ Меер сон Г. А., Крейн О. Е. ЖПХ, 1960, т. V с 1934
435. Техника высоких температур, под ред. И. Э. Кемпбелла ИЛ
1959 ' ’
436. Ядерные реакторы, т. III. ИЛ, 1956, "С. 43.
437. Гуревич М. А. ЖНХ, 1963, т. VIII, № 12, с. 2645
438. Алексеев В. И., Шварцмач Л. А. ДАН СССР, I960
т. 133, № 6, с. 1331. ’ ’
439. McKenna Р. Ind. Eng. Chem., 1936, v. 28, p. 767.,
440. Уманский Я. С. ЖФХ, 1940, т. XIV, с. 376.
441. Maurer G. u. a. Arch. Eisenhiittenwes-., 1939—1940 В 13
S. 337. \ ’
442. Brauer G. и. a. Z. anorg. Chem., 1954, В. 277, № 5, S. 249.
443. Brauer G., Lesser R. Z. Metallkunde, 1959, B. 50
S. 487.
444. А л я м о в с к и й С. И. и др. В сб. трудов Уральского поли-
технического ин-та им. С. М. Кирова, 1959, № 92, с. 125.
445. Kemp ter С. а. о. J. Chem. Phys., 4960, v. 33, p. 1873.
446. Stroms E., Krikorian H. J. Phys. Chem.,, 1959, y_ 63.
p. 1749.
447. Nadler M., Kemp ter C. J. Phys. Chem.,. 1960, v. 64, № 10,
p. 1468.
448. Stroms E.r Krikorian H., J. Phys. Chem., 1960, V. 64,
№ 10, p. 1471.
449. Elliot P, Trans. Am. Soc. Met., 1961, v. 53, p. 13.
450. Кусенко Ф. Г„ Гельд П. В. Изв. вузов. Цветная ме-
таллургия, 1961, № 2, с. 43.
451. Гельд П. В., Швейкин Г. П. Изв. АН СССР, ОТН. Ме-
таллургия и топливо, 1959, № 1, с. 144.
452. Гельд П. В. Успехи химии, 1957, т. 29, с. 9.
453. П а д е р н о В. Н., Лапшов Ю. К- Порошковая металлур-
гия, 1963, № 1, с. 75.
454. Гельд П. В., Любимов В. Д. Порошковая металлургия,
1963, № 4, с. 77.
455. Кислый П. С. и др. Порошковая металлургия, 1962, № 2,
с. 38.
456. Stroms Е. а. о. Analyt. Chem., 1960, v. 32, № 12, р. 1873.
457. Гельд П. В., Кусенко Ф. Г. Изв. АН СССР, Металлур-
гия и топливо. 1960, № 2, с. 79.
458. Кусенко Ф. Г., Гельд П. В. Изв. Сибирского отделе-
ния АН СССР, 1960, № 2, с. 46. ' „
459. Мah A., Boyle В. J. Amer. Chem. Soc., 1955, v. 77, № 24,
p. 6512.
460. Корнилов А. И. и др. Вестник МГУ, серия химия, 1962,
№ 6, с. 48.
461. Корнилов А. И. и др. ДАН СССР, 1962, т. 144, с. 355.
462. Самсонов Г. В. и др. Изв. АН СССР, ОТН. Металлургия
и топливо, 1961, № 1, с. 142.
463. Fries R. J. J. Chem. Phys., 1962, v. 37, № 2, p. 320.
19 Косолапова T, Я.
469.
470.
471, .
472. Мальков Л. П
473.
474,
475,
480,
481.
482,
483.
484.
485.
486.
487.
488.
489.
490.
491.
492.
493.
494.
495.
496.
497.
286
4R4 Гельд П. В., НхаЙ В. А. Некоторые вопросы крист
химии кубических окислов и карбидов титана, ванадия
бия Доклад на заседании постоянного межинститутского к«°
локвиума по твердым фазам переменного состава, вЫп. 32 $
465. Е1И пТеАг1F2 Е. Trans. Am. Soc. Metals, 1943, v. 31>
466 Lesser R, Brauer G. Z. Metallkunde, 1958, B. 49, № 12
‘ S, 622.
467. Смирнова
№ 3, c. 557.
468 Смирнова
c. 127.
Смирнова
c. 1327.
KeHeV K- Trans.‘'Am, St. Treat, 1932, v. 19, p.’233.™*'
_ ______ " Ч, Хохлова А, В. Редкие металлы, I935
JMb 1 с. 4.
Cooper P. а. о. XVIII Internal. Congress of Pure and Applied
Chemistry. Montreal, Canada, August, 1961.
Genders K, Harrison R. J. Iron Steel Inst., 1936, № 2,
p. 202.
Naeken M. u. a. Arch. Eisenhiittenwes, 1961, B. 33, № 9,
S. 635.
476 Lersmacher B, Scholz S. Arch. Eisenhiittenwes., 1961,
B. 32, № 6, S. 421.
477. Becker K, Ebert F. Z. Techn. Phys., 1930, B. 148, S. 216.
478. Born R. Nature, 1940, v. 145, p. 741.
479. Norton G, Mowry A. Trans. AIME, 1949, v. 4, p. 387
'*on Самсонов Г. В., Ру кин а В. Б. ДАН УРСР, 1957, № 3,
С. 247.
Колчин О. П., Берлин И. К. Цветные металлы, 1964,
т. 8, е. 67,
Загянский И. Л. и др. ДАН СССР, 1950, т. 74, с. 723.
Ruff Р., Foehr Т. Z, anorg. Chem., 1918, В. 104, S. 27.
Yegesack A. Z. anorg. Chem., 1926, В. 154, S. 30.
Nischk О. Z. Elektrochem, 1923, В. 29, № 5/6, S. 373.
Ruff O. Z. Elektrochem, 1923, B. 29, S. 459. )n,n R
Kraiczek B, Sauerwald F. Z. anorg. Chem, 1930,
185,8. 193.
Friemann E, Sauerwald F. Z. anorg. Chem, w •
B. 203, S. 64.
Westbrook J. J. Metals, 1957, v. 9, № 10, p. 1227. Q
Westgren A, Phragmen G. Z. anorg. Chem,
B. 187, №4, S. 401.
H a ts u t a K. Kinzoku no Kenkyu, 1931, v. 8, p. 88. .
Bloom D, Grant N. Trans. AIME, 1951, v. 188 p. J22
T a m m a n G, S c h б и e r t R. Z. anorg. Chem, 1922, d.
o. 27.
Smith W. J. Metals, 1957, v. 9, № 1, p. 47. _ , 2l.
Sauerwald P. u. a. Z. anorg. Chem, 1933, B. 210, -
Гельд П. В, Есин О. А. ЖПХ, 1950, т. ХХШ, с. и
Kelley К. К. а. о, Technical paper, 1944, № 662.
. -fe 12,
В И, О р о н т Б. Ф. ДАН СССР, 1954, т. 96,
В И ОрмонтБ. Ф. ДАН СССР, 1955, т. юо,
В И., ОРмонт Б' Ф' ЖФХ, 1956, т. XXX,
Z. angew. Chem,_»S8,.„B, 70, № 15.£ 474.
П Хохлова А, В. Редкие металлы, 1935,
jna к ub as chew sky O., Wittig F. 2. Elektrochem., 1941,
498‘ В 47 S. 433.
499 Елютин В. П. и др. Ферросплавы. Металлургиздат, 1951,
nJ нт а йкин Е. 3. ФММ, 1963, т. XVI, № 1, с. 144.
СЛ1 Ито фуракова эдике язико, 1956, т. 13, е. 15 (РЖМ, 1958,
50 №5 реф. 9424).
кл9 Ге ch В. Hutnicke Listy, 1958, v. 13, № 2, р. 113.
wn Cech. В. Hutnicke Listy, 1958, v. 13, № 10, p. 955.
504" Косолапова T. Я-, Самсонов Г. В. ЖПХ, 1959, т. XXXII,
№ 1, с. 55. л
505. Косолапова Т. Я-, Самсонов Г,. В. ЖПХ, 1959, т. XXXII,
’ № 7, с. 1505.
506 Самсонов Г. В., Косолапова Т. Я. Порошковая метал-
лургия в машиностроении. Киев. Изд. НТО Машпром, 1961, с. 28.
507. Косолапова Т. Я., Самсонов Г. В. ЖПХ, 1960, т. XXXII,
' № 8, с. 1704.
508. Вертман А. А., Самарин А. М. Труды совещания по при-
менению вакуума в металлургии, 1958, с. 132.
509 Owen В., Weber R. Am. Inst. Met. Eng., Techn. Publ., 1948,
№ 203.
510. Архаров В. И., Конев В. Н. Вестник машиностроения,
1955, т. 11, с. 55.
511. Конев В. Н. Исследования по жаропрочным сплавам, 1958,
т. 3. с. 415.
512. Мальцева Л. Ф. и др. Порошковая металлургия, 1965, № 11,
с. 87.
513. Arnold A., Read A. J. Iron Steel Inst., 1911, v. 73, p. 249.
514. Цхай В. А, ГельдП. В. ФММ, 1963, т. XVI, с. 493.
515. Grafts W., Lamont I, Trans. AIME, 1950, v. 180, p. 561.
516. Waver F., Koch W. Arch. Eisenhiittenwes., 1950, B. 21, S. 143.
517. Brown J., Clark D. Nature, 1951, v. 167, p. 728.
518. Львов С. H. и др. ФММ, 1961, т. 11, вып. 1, с. 14.
519. Мельничук П. И. и др. ФММ, 1960, т. IX, вып. 6, с. 918.
520. Меньшиков А. 3., Рудомаиов П. Т. Труды Института
физики металлов АН СССР, вып. 22, 1959, с. 69.
521, Цхай В. А., Гельд П. В. Ж- структ. химии, 1964, т. 5,
с. 275.
522- WestgrenA., Phragmen G. Z. anorg. Chem., 1926, B. 156,
Jakei T. Sci. Rep. Tohoku Univ., 1928, № 17, p. 939.
кос S- Trans. Amer. Inst. Min. Met. Engrs, 1930, v. 89, p. 9.
525. Few W., Manning G. Metals, 1952, v. 4, p. 271.
526. Lander J., G a rm er K. Am. Inst. min. Met. Engrs, 1947,
№ 2259.
527. Moi s san H., Hoffman M. Compt rend., 1904, v. 138,
p. 1558.
' Tuttija H. Sci. Pap. Inst. phys. chem. Res. Tokio, 1932, v. 19,
p. 384.
Gmelins Handbuch fur anorg. Chem., 1935, № 53.
530. Dawhil W. Z. anorg. Chem., 1950, B. 262 S. 212
531. Nowotny H., Kieffer R. Z. anorg. Chem., 1952, B. 267,
№ 4_____5 g 261 1
532. Sykes W. P. a. o. Trans. Am. Inst. mln. met Engrs.. 193S,
19. v ll7. P.175. 287
eqo Nowotny Н. u. a. Mh. Chem., 1954, В. 85, S. 255.
534* Kuo К., HaggG. Nature, 1952, v 170, № 4319, p. 245
535. Rudy E-, Benesovsky F. Planseeber. Putvermet
В 10, S. 42. , ’ ’"2,
536 Rudy E. u. a. Z. Metallkunde, 1963, B. 54, S. 345.
лУрР™“.'1"б5? n?ihc: 1 я- m«m.
538 Волкова H. M. и др. ЖНХ, 1965, т. X, с, 1758.
539. Meyer О, Arch. Eisenhiittenwes., 1930/1931, В. 49, S 196
540’ Болгар А. С. и др. Порошковая металлургия, 1967 ’ № 1
с. 40. ’ ’ h
541. Hegedus A. J., Neugebauer J. Z. anorg. Chem 1адп
В. 305, № 3, 4, S. 216. ’’
542. Войтович Р. Ф.„ Шаханова Н. П. Порошковая метал,
лургия, 1967, № 3, с. 75.
543 Meyer О., Ei lender W, Arch. Eisenhiittenwes,, 1938 В 11
S. 545. ’ ’
544. Brewning L., Emmett P. J. Amer. Chem. Soc., 1952 v 14
p. 4773. ’ ‘ ’
545. Pa в дель А. А. Ж. русск. физ.-хим. об-ва, 1930, т 62
с. 1515,
546. Weiss G. An. Chim., 1946, v. I, p. 446.
547. Arnold J., Read A. Proc. -Inst Meeh. Eng., 1914, № 2, p.223.
548. Nowotny H., Kieffer R. Z, Metallforschung, 1947, B. 2,
S 257
549. Fries B., Kemp ter C. Anal. Chem., 1960, v. 32, p. 1898.
550. G1 e i s e r M., Chipman J. J. Phys. Chem., 1962, v. 66, № 8,
p. 1539.
551. Алексеев В. И., Ш в а р ц м а н Л. А. Изв. АН СССР, ОТН,
Металлургия и топливо, 1962, № 6, с. 171.
552. Падерно Ю. Б. и др. Электроника, 1959, № 4, с. 165.
553. Беликов А. М., Умански й Я. С. Кристаллография, 1959,
т. 4, № 5, с. 684.
554. Sykes. Trans Amer. Soc, Steel, Tr., 1930, v. 18, p. 968.
555. Третьяченко Л. А., Еременко В. H. Изв. АН СССР. Не-
органические материалы, 1966, т. II № 9, с. 1568.
556. Воронкова Е. А. В сб. «Твердые сплавы» (ВНИИТС), 1УЬУ,
№ 1, с. 139.
557. S k а и р у F. Z. Elektrochem., 1927, В. 33, S. 487.
558. Креймер Г. С. и др. ЖТФ, 1952, т. 22, с. 848.
559. Герасимов А. Ф. и др. ФММ, 1961, т. XI, е. 596.
560. Карпухин В, В. и др. Цветные металлы, 1957, № 9, с. • _
561. Меереон Г. А. и др. Металлургия редких металлов,
лургиздат, 1955. ч
562. Agte С., Hruska J. HutnickeYisty, 1954, В. 9 S. •
563. Boofi Н. Arch. Eisenhiittenwes., 1959, В. 30, № 12. • •_
564, HegediisA., Gado S. Z. anorg. Chem., i960, B. 3 , -
S. 227.
565. Chretian A. Compt. rend., 1952, v. 234, p. 2608
566. Hurd D. a. o. Ind. Eng. Chem., 1952, v. 44, p. 243 .203-
567. Greenhouse H. M. a. 0. J. Amer. Cer. Soc., 1954,
568. Machin ist, 1954, v. 98, p. 375. _ 1057, v. 79.
569. Newskiz A.j A1 i f e r i s J. J. Amer. Chem. Soc.,
p. 4629,
288
кто Комар А. П Т а л а н и н Ю. Н. Изв. АН СССР, серия фи-
5'и- v logs т. 22, с. 580.
Pfe’v P.> Rix W- Z- Metallkunde, 1954, B. 45, № 3, S 116.
Буторина Л. H. Кристаллография, I960, т. 5, вып. 2, с 234
S Алексеев В. И., Шварцман Л. А. Изв. АН ССР, ОТН:
& Металлургия и горное дело, 1963, № 1, с. 91.
Б74 Алексеев В. И., Шварцман Л. А. ДАН СССР, 1960,
5 ’ т 133, № 6, с. 1331.
К7С Пивоваров Л. X. и др. ФММ, 1964, т. XVII, с. 606.
Б76 Зу бен ко Ю. В. ФТТ. 1961, т. 3, № 2, е. 528.
577 Ruff О., Bormann W. Z. anorg. Chem., 1914, В. 88, S. 365.
578 V о g е 1 R., Doting W. Arch. Eisenhiittenwes^ 1933/36, В. 9,
' S. 247.
579- Southard J. C.f Moore G. E. J. Amen Chem. Soc., 1942,
v. 64, p. 1769.
580 Kuo K-, Pers so m E. J. Iron Steel Inst.t 1954, v. 178, p. 39.
581 Jacobson B., Westgren A. Z. Phys. Chem,, 1933, B. 20,
S. 361.
582. Westgren A. Jernkontorets Ann., 1933, v. 177, p. 501.
583. SchenockR., Meyer K. Z. anorg. Chem., 1938, B.239, S. 161.
584. Тавадзе Ф. H. Труды Института металлов и горного дела.
АН Груз. ССР, 1947, т. 1, с. 197..
585. I s о b е М. Science Repts. Research. Inst. Tohoku Univ., 1951,
v. A3, p. 468.
586. Тавадзе Ф. H. и др. Сообщения АН ГрузССР, 1955, т. 16,
с. 477.
587. Алексеев В. И., Шварцман Л. А. ДАН СССР, т. 141,
с. 346.
Picon М., Flahaut J. Compt. rend., 1957, v. 245, p. 534.
Ulich H., Siemonsen H. Arch. Eisenhiittenwes., 1940, B. 14,
S 27
Ruff O., Gersten E. Chem. Ber., 1913, B. 46, S. 400.
Roth W. A., G r a u R. Arch. Eisenhiittenwes., 1929/30, B. 3,
S. 339.
Карапетьянц M. X. Химическая термодинамика. Госхим-
издат, 1953.
/ к. К«, Moore G. Е. J. Amer. Chem. Soc., 1943,
v. 65, p. 782.
588.
589.
590.
591.
592.
593. Kelley
F. 7^
S’ Sel 1 e У К. К- Bull. Bur. Mines-, 1935, № 407.
ЭД5.M cCabe a. o. J. Metals, 1957, v. 9, № 1, p. 17.
t-Л Алексеев В. И., Ш в a p ц м а н Л. А. ДАН СССР, \960,т. 133'
S’ Sendrus Н- u-a- z- Metallkunde, 1957, В. 48, № 12, S 615.
5ОД LU ] 1S h ’ r ° S- Trans. JaPan Inst Metals, 1960, v. 1, p. 125.
599. Worrel W. L., Chip man J. J. Phys. Chem., 1964, v. 68,
P- 860. E
600. Huber E. J. a. o. J. Phys. Chem., 1961, v. 65, p. 1846.
602 Itze,biatowski W. Z. anorg. Chem., 1937 В 233 S.
rm r Ughes J- Less-Common Metals, 1959, v. 1, № 5, p. 377.
•Евстюхин А. И. и др. Металлургия и металловед
604 w™* Металлов. Госхимиздат, 1963, вып. 4, с. 149. „
с 40ЗЬМЭ Ю- Б’ Н ДР- п°Р°шковая металлургия, 1963, л-
Ь ' а 1 о w s к ( W-, Rubdinski J. Z. Chem., 1962, В.
1ир,
289
606. Epstein S The Alloys of Iron and Carbon, v. I, Constitution
Mew York, 1930-
ОП7 rvwhh К Б. Железоуглеродистые сплавы. Киев. Машги.
ro« filund W u. a. Ber. deutsch. Chem. Ges., 1929, В 62 s’^49-
S Hof fw ann U, c rol 1 E. Z. anorg., Chem., 1930, B. 191 ’Л4?3’
R?o Holed. j. E. a. o. J. Amer. Chem. Soc., 1949, v. I, p ’Jo414-
H fl s S G Z. Kristallogr., 1934, В. A89, S. 92. P’ l89-
Пинск ep 3. Г, Каверин QB. Кристаллография, i957
t. 2, e. 386. ______1OKI .. ло _
606.
617.
618.
619.
620.
621.
622.
623.
624.
625.
626.
627.
628.
629.
630.
631.
632.
633.
634.
635.
636.
637.
638.
639.
640.
641.
642.
643.
644.
645._____
646. Арбузов М. П„”ай
609.
610.
6П.
612.
fill Moreau J. а. о. Rev. metallurgie, 1951, v. 48, р. 486.
614 Ростовцев С. Т. и др. Сталь, 1953, № 8, с. 679.
615 Чуфаров Г. И., Авербух Б Д. ЖФХ, 1934, т. V, с
616.’ П и н с к е р 3. Г., Каверин С. В. Кристаллография,
т 1. вып. 1, с. 66. ’
Ап а ев В. А. ФММ, 1961, т. 12, № 3, с. 409.
Andre М. Bull. Soc. Chem. France, 1961, № 1, p. 143.
Naeakuro S. J. Phys. Soc. Japan, 1959, v. 14, № 2 n iqr
Сирота H. H. ДАН СССР, 1949, т. 64, с. 697. ’ Р’
Reuer R., Kesper R. Ferrum, 1913/14, В. 11, S. 257.
Adcock F. J. Iron Steel Inst., 1937-, v. 135, p. 281.
Antrew J. H., Binnie D. J. Iron Steel Inst., 1929, v 119
p. 309.
Harris G B., Hume-Rothery W. J. Iron Steel Inst., 1953
v. 174, p. 212.
D a r k e n L. S., G u г г у R. W. Trans. AIME, 1951, v. 191, p. 1015.
RuerR., Goernes F. Ferrum, 1916/17, B. 14, S. 161.
Piwowarosky E. Stahl und Eisen, 1934, B. 54, S. 82.
Ruer H., В e r 1 i n J. Z. anorg. Chem., 1920, B. 113, S. 98.
Riimelin G., Mai re R. Ferrum, 1914/15, B. 12, S. 141.
Bardenheuer P. Ferrum, 1916/17, B. 14, S. 129.
Stablein F. Stahl und Eisen, 1926, B. 46, S. 101.
Esser H. Stahl und Eisen, 1927, B. 47, S. 337.
Mehl R. F., Wells C. Trans. AIME, 1937, v. 125, p. 429.
Wark N. J. Stahl und Eisen, 1911, B. 31, S. 2108.
Tchischewsky N., Schulgin N. J. Iron Steel Inst., 1917,
v. 95, p. 189.
Гутовский H. Изв. Томского технол. ин-та, 1914, т. 36, с. 1
Kdster W. Z. Metallkunde, 1930, В. 22, S. 289.
Tamura S. J. Iron Steel Inst., 1927, v. 115, p. 747.
Piding N. P. Trans. Amer. Inst. Min, met. Eng., 1924, v. 43,
p. 145.
Каминский Э. 3., Стеллецкая T. И. Проблемы метал-
ловедения и физики металлов, 1951, № 2, т. 176.
Гриднев В. Н. ЖТФ, 1946, т. XVI, с. 915.
Stanley J. К. J. Metals, 1949, v. 1, р. 752.
We г t С. A. J. Metals, 1950, v. 2, р. 1242.
Lindstrand Е. Acta Metallurg., 1955, v. 3, p. 431.
Петрова E. Ф. и др ДАН СССР, 1958, т. 121, с. 1021.
Арбузе М. П., Ай’зенцок Е. Г. ДАН УССР, 1955, *
6* 1 оО
Migand В., Talbot J. Compt. rend., 1958, v. 245, p.
йо aP V Б‘ И- ДАН СССР, 1959, т. 128, с. 709.
°^ АРбузов М. П. ДАН СССР, 1950, т. 74.
ььо. А п а е в Б. А. ДАН СССР, 1953, т. 93, с. 647.
290
Cl Гриднев В. H. ЖТФ, 1954, т. 24, С. 1812.
6?2 Гриднев В. Н„Трефилов В. И. ДАН СССР, 1954, т. 96
с 741*
553. Штейнберг С. С. Термическая обработка стали. Металлург-
издэт, 1945.
654. Юрьев С Ф. Удельные объемы в мартенситном превращении
аустенита. Металлургиздат, 1950. н и
655 Садовский В. Д. В сб. «Фазовые превращений в железо-
углеродистых сплавах». Машгиз, 1950.
656 Ruff О. Berichte, 1912, В. 45, S. 69.
657 Schenk R. u.a. Z. anorg. Chem., 1927, В. 164, S. 313
658. Гутман С. M. Заводская лаборатория, 1947, № 12, е. 1403.
659 Гудерман и др. Заводская лаборатория, 1946 № Ц________12
с. 900.
660. Hofer J. Е., Р е е b I е s W. С. J. Amer, Chem. Soc, 1947 у 69
№ 4—6, p. 893. ' ’
61. П о п о в a H. M, J* ы б и н а М. Ф. Заводская лаборатория
1947, № 12, с. 1412.
662. Arnold J, Read A. J. Chem. Soc, Ind, 1894, v. 65, p. 788.
663. Meinhardt D, Krisemant O. Arch. Eisenhiittenwes, 1962
B. 33, S. 493.
664. Kubaschewsky O, Gatteral J. A. Thermochemical Data
of Alloys, Pergamon Press, L.—N. Y, 1956.
6 5. R a n d e 1 i f f e S. V, R о 11 a s 0 n E. C, J. Iron Steel Inst, 1958,
v. 189, p. 45.
666 Boecker G. Metallurgie, 1912, B. 9, S. 296.
667. Morrogh H, Williams W. J. J. Iron Steel Inst, 1947,
v. 155, p. 321.
668. Bahr H, Jessen V. Ber. Deutsch. Chem. Ges, 1930, B. 65,
S. 2226.
669. Drain J. Ann. Chim, 1953, v. 8, p. 900.
670. J u z e R, Ruff H. Naturwissenschaften, 1951, № 14, S. 331..
671. Hempel W. Z. anorg. Chem, 1904, B. 17, S. 296.
672. Friedenreich K, Leroux A. Metallurgie, 1910, B. 7, S. 10,
673. F u j i s h i r 0 S, G о c k e n N. A. J. Phys. Chem, 1961, v. 65,
№ 1, p. 161.
674. Lander J. J. a. o. J. Appl. Phys, 1952, v. 23, № 12, p. 1305.
675 Baukloh W, Springorum F Z. anorg. Chem, 1937,
e B. 230, S. 315.
676. Smidt J. Z. anorg. Chem, 1933, B. 216, S. 85.
677. В e p т м а н А. А. н др. Изв. АН СССР, ОТН. Металлургия и
топливо, 1962, № 6, с. 37.
678 Бертман А. А. и др. ДАН СССР, 1963, т. 148, с. 342.
679. Мирошниченко И. С. Изв. вузов. Цветная металлургия,
,Q. 1961, № 1, с. 128.
S’ ?ahr Н. A. Th. Bahr Berichte, 1928, В. 61 S. 2177
681. Jacobson B„ Westgren A. Z. Phys. Chem, 1933, B. M2U,
S. 361.
682. Rid yw ay R. Trans. Amer. Electrochem. Soc, 1934, v. bo-
р. 117,
683. Орм’онт Б Ф. Структуры неорганических веществ. Гостех-
издат
б84. Меереон Г. А, Самсонов Г. В. Изв. сектора физ-хим.
анализа, 1953, т. 22, с. 92.
291
685 Жданов Г. С. и др. ЖФХ, 1954, т. XXVIII, с. 1076
686. Glaser F. а. о. J. Appl. Phys., 1958, v. 24, р. 731.
687. Кудрявцев В. М., Сафронов Г. В. Бюллетень семинар
по жаростойким материалам. Изд-во АН УССР, I960, Вып R
с. 52. °*
688. Самсонов Г. В. и др. ФММ, 1956, т. III, с. 309.
689. С а м е о н о в Г. В., ЖФХ, 1958, т. XXXII, с. 2424.
690. Журавлев Н. Н. и др. Изв. АН СССР, ОТН, Металлуогия
и топливо, 1961, № 1, с. 133.
691. Самсонов Г. В. и др. Бор, его соединения и сплавы Кирп
Изд-во АН УССР, 1960.
692. Dawihl W. Z, Metallkunde, 1952, В. 43, S. 138.
693. Меереон Г. А.; Самсонов Г. В. В сб. «Бор» (Труды
конференции nd химии бора и его соединений). Гостехиздат
1958, с. 52.
694. Марковский Л. Я. и др. Химическая электротермия. Гос-
химиздат, 1952.
695. Алейников Ф. К. ЖФТ, 1953, т. XXVII, с. 567.
696. Кислый П. С., Самсонов Г. В. ЖФТТ, 1960, т. 2, с. 692.
697, Kelley К. К. J. Amer. Chem. Soc., 1941, v. 63, p. 1137.
698. Colson R. Compt. rend., 1882, v. 94, p. 1316.
699. Ta mm an G. Z. anorg. Chem., 1921, B. 115, S. 141.
700. Nowotny N. u. a. Mh. Chem. 1954, B. 85, S. 259.
701. Брохвн И, С., Функе В. Ф. ЖНХ, 1958, т. 3, с. 847.
702. Silicon carbide a high temperature semiconductor, Pergamon
Press, 1960.
703. Августиник А. И. и др. Изв. АН СССР. Неорганические
материалы, 1966, т. II, № 8, с. 1439.
704. Каменцев М. В. Искусственные абразивные материалы,
Машгиз, 1950.
705, Ипполитов Г. М.} Какушадзе Е. П. Производство ко-
рунда и карборунда й их применение. ОНТИ. Машметиздат,
1933.
706. Fitz-Gerald F, A. Carborundum, Halle, 1924.
707. Оршанский Д Л. Электротермия. М.—Д ГОНТИ, 1939.
708. Kiammroth Н. Chemisch Untersuchungen ап Siliziumkarbid-
Heizstaben, Dusseldorf, 1935.
709. Самсонов Г. В. Силициды и их использование в технике.
Киев, Изд-во АН УССР, 1959.
7(0. Бережней А. С. Кремний и его бинарные системы. Киев.
Изд-во АН УССР, 1959.
711. Добролеж С. А. и, др. Карбид кремния. Киев. Гос. изд-во
техн, лит-ры УССР, 1963.
712, Гнесин Г. ДЛеженин Ф. Ф. Порошковая металлургия,
1967. № 2, с, 36.
713. Новиков А. Н. ЖПХ, 1947, т. XX, с. 431.
714. Dietzel A. u. а. Вег. Deutsch. Keram. Ges., 1960, В. 37, S. 524.
715. Нешпор В. С. и др. Изв. АН СССР. Неорганические материа-
лы, 1966, т. II, № 5, с. 855. л « ш
716. Adelshevg L. М. а. о. J. Amer. Cer. Soc., 1966, v. 49, №
Р-573-
717. Bamann Н. N, J. Electrochem. Soc., 1952, v. 99, р. 109.
718, Chipman J. а. о. Acta Metallurgica, 1954, v. 2, p. 439.
719. Hall R. N, J. Appl. Phys., 1958, v. 99, p. 914.
292
728 D i е t z е 1 А. и. а. Вег. deutsch. keram. Ges., 1960, В. 37, S 524’
709 Baumann H. N. J. Electrochem, Soc., 1952, v. 99, n. 109
740 Whitney E. Nature, 1963, v. 199, p. 278.
731.
732.
733.
734.
735.
736.
737.
738. . ..
739. Меер сон Г. А. и др. Изв. АН СССР. Неорганические мате-
740.
741.
742.
7o0 Зарецкая Г. M. и др. Изв. АН СССР, Металлургия и гор-
ное дело, 19о4, т.ч, с. оо.
«' ^*Р/,938И,ЖТслТ"’ Р- ”• Эавод«“ “«о-
да Шзф Р а « И. Г. ЖПХ, 1938, т. 11, № 3. с. 561.
723.' Самсонов Г. В., о р т н о й К- И. Сплавы на основе туго-
плавких соединении. Оборонгиз, 1961. у
774 Kendall Т. J. Chem. Phys., 1953, v. 21, р. 821.
725. М е е р с о н Г. А. и др. Изв. АН СССР. Мет. и горное дело,
1964, № 1, с- 07.
796 Taylor A., Lai die г D. J. Appl. Phys., 1950, v. 1 D 174
727’ Fetterley G. H. J. Electrochem, Soc., 1957, v. 104’ p 322
728 D i e t z e 1 A. u. a. Ber. deutsch. keram. Ges., 1960, B. 37, S 524
729.’ ВаишаппД N. J.,Electrochem. Soc., 1952, v. 99, p. 109.
Ott H. Z. Kristalogr., 1925, B. 61, S. 515,
Белов H. В. Структура ионных кристаллов и металлических
фаз. Изд-во АН СССР, 1947.
Жданов Г. С., Минервйна 3. В. ЖЭТФ, 1947, т. 1/ с 1
Жданов Г. С., МинервинаЗ. В. ЖЭТФ, 1945, т 15
с. 155.
Ramsdell L. S., Kohn J. A. Acta cryst., 1952, v. 5, p. 215.
Ruff O. Trans. Electrochem. Soc., 1935, v. 68, p. 87,
Ruff O., Konchak M. Z. Elektrochem., 1926, B. 32, S. 517.
Bad ami D. V. Nature, 1962, v. 193, № 4815, p. 569.
риалы, 1966, т. II, № 4, c. 604.
Drowart J., de Maria G. Proceedings of the Confer, on <si-
licon carbide, Boston Massachusets Pergamon Press, Oxford, Lon-
don, New York, Paris, 1960.
Rein R. H., C h i p m a n J. J. Phys. Chem., 1963, v. 67, p. 839.
D’Entramont J, C., Chipman J. J. Phys. Chem.,
v. 67, p. 499.
Фалькевич А. Химстрой, 1934, т. 6, e. 442.
if 1, „ Q k i, Fryimori К a n j i- J- Chem.
Japan, 1962, v. 65, p. 167.
' . Знаменский Ю. Д. ЖФХ,
т. XXXVI, c. 2748.
I- HO, c. 1048.
t. 120, c. 140. ..’
1963,
743.
744. T a g a w a Hiroaki.
745. Гольдберг H. ГД.,
т. XXXVI, c. 2748.
746. Гольдберг H. А., Знаменский Ю, Д. ДАН СССР,
т. НО, с. 1048.
747. Гольдберг Н. А., Знаменский Ю. Д. ДАН СССР,
т. 120, с. 140.
Й' Bi es a! ski Е„ Eck Н. Z. anorg. Chem., 1926, В. 156, S. 226.
75п вЬЛг Е-’ B*nder W. Z. anorg. Chem., 1914, B. 88, S. 255.
75?’ Botlle J-< Hinriehsen F. Chem. Ztbl., 1907, B. 78, S. 992.
Богданченко А. Г. Заводская лаборатория, 1949, т. XV,
№ 7, с. 851
521 Б°гДанченко А. Г, Заводская лаборатория, 1963, т. XXIX,
?. с- 163.
754 д’?61’118 Handbuch der anorg. Chemie, 1951, № 41. м
• Анализ тугоплавких соединений. Коллектив авторов. Металлург
издат, 1962.
756 ш0ПЬ1Л0Ва В. П. ЖПХ, 1961, т. XXXIII, № Я е- I936-
Magner F. а. о. Trans. Amer. Soc. Metals, 1956, v. 48, p. 742.
293
Soc.
1962,
1956,
1958,
757. Самсонод Г. В., Голубева Н. К. ЖФХ, 1956, т. ХХх
758. Strikland-Constable. Trans. Faraday Soc., 1944, v 4n
333 *
759. S t r i к 1 a n d - С о n s t a b 1 e. Trans. Faraday Soc., ig47> y 43
760 Bo7o9z H. J. Metall, 1956, B. 10, S. 130.
ТбкЗеликмая A. H., Горовиц H. H. ЖПХ, 1952, T. XXy
762 Monster A. Z. angew. Chem., 1957, B. 69, S. 281.
763i Boor H. J. Metall, 1962, B. 7, S. 40.
764. К л и б у с А.Х., Назарчук Т. Н. ЖАХ, 1961, т. XVI, № i
с 79. ’ *’
765 Reed S. А. а. о. Analyt. Chim. acta, 1954, v. 10, № 5 p 499
766. M e 11 о r J. W. A. comprehensive treatise on inorganic and th₽l
oretical chemictry, 1950.
767. E в с т ю X и н А. И. и др. В сб. «Металлургия и металловеде-
ние чистых металлов», 1959, вып. 1.
768. Niemi ее J. Compt. rend., 1960, v. 251, № 6, р. 875.
769. Tetenbaum М. а. о. Trans. Amer. Nucl. Soc., 1961 v 4
p. 89. ' ’
770. Lebeau P„ Damiens A. Compt. rend., 1913, v. 156, p. 1987
771. Kempter C., Nadler M. J. Chem. Phys., 1960, v. 33, №5
p. 1580.
772. Kempter C. J. Chem. Phys., 1964, v. 41, p. 1515.
773. Gmelins Handbuch fur anorganische Chemie, 1942, № 65.
774. P e a k a 11 К. A., A n t i 11 J. E. J. Less-Common Metals, 1962,
v. 4, p. 426.
775. Артамонов А. Я. и др. Порошковая металлургия, 1967,
№ 2, с. 29.
776. Murbach Е. W. Trans. Met. .Soc. AIME, 1963, v. 227, p. 487.
777. Besson J. a. 0. Compt. rend. Acad. Sci. Paris, 1964, v. 258,
p. 4079.
778. Produet-group. U К Atomic Energy Author Rept., 1963,
№ 444.
779. Combarien A. a. 0. Compt. rend Acad. Sci., 1963, v. 256,
№ 26, p. 5518.
780. W i t z k e H. D., Abhandl. Deutsch. Akad. Wiss. Ber. KI. Nath.,
Phys, und Techn., 1962, № 6, S. 86. „ ,
781, Beckman G. E. J. J. Electrochem. Soc., 1963, v. 110, № L
p. 84.
782. В ouch a u d J. P., Fruchart R. Compt rend. Acad. Sci. Pa*
ris, 1964, v. 258, № 8, p. 3495.
783. H asehke H. u. a. Mh. Chem., 1966, B. 97, № 4, S. 1045.
784. Павлинов Л. В., Быков В Н ФММ, 1965, т. XIX, с.
785. S ch ma hl N. G. Z. Elektrochem., 1934, B. 40, S. 68.
786, Самсонов Г. В. и др. «Электротехнические металлокер
ческие изделия». ВНИИЭМ, 1965.
К а.м i н с и к а О. В. XiMinna промислов!сть, 1962, № 3, с.
Fritz J. S. а. о. Anal. Chem., 1958, V. 30, р. НИ- с 372
Damiens М. A. Bull. Soc. Chim. France, 1914, v. 15, P-
DamiensM. A. Ann. Chem., 1918, v. 9, p. 137.
Schmidt J. Z. Elektrochem., 1934, B. 40, S. 170.
1 у p e в и и M. А. и др. ЖФХ, 1956, т. 11, с. 177.
787.
788.
789.
790.
791.
792.
294
793 Гиллебранд В. и др. Практическое руководство по на-
' органическому анализу. Госхимиздат, 1957.
794. Ды м о в А. М. ТехничеЬкий анализ руд й металлов. Метал-
лургиздат, 1948.
795 С м и р н о в В. И., О р м о и т Б. Ф. ЖАХ, 1954, т. IX, с 359
796 ормонт Б. Ф., Смирнова В. И. ЖФХ, 1957,. т. 69, а 281.'
797 Попова Н. М., Сорокина К. П. Заводская лаборатория
1955, т. XXI, с. 527.
798 П о п о в а Н. М., С о р о к и н а К. П. Заводская лаборатория
1953, т. XIX, № 9, е. 1033. р ’
799 Meinhardt О., К г i s е m е n t О. Z. Naturforsch., 1960, В 15а
S. 880.
800. Leahe J. A. Metallurgia, 1952, v. 45, р. 989.
801. Kennedy J. D. Steel, 1952, v. 181, p. 92.
802. Косолапова T. Я., Самсонов Г, В. ЖФХ, 1961, т. XXXV
с. 363.
803. Hinniiber J., Rfidiger О. Arch. Eisenhiittenwes., 1953,
В. 24, S. 267.
804. Westgren A. a. o. J. Iron Steel Inst., 1928, v. 117, p. 386.
805. Косолапова T. Я., Самсонов Г, В. Труды семинара по
жаростойким материалам. Киев. Изд-во АН УССР, 1961.
806. Косолапова Т. Я., Самсонов Г. В. Укр. хим. журн.,
1962, т. 28, с. 931.
807. Косолапова Т. Я., Р а дзиковекая С. В. Заводская ла-
боратория, 1960, т. 26, № 2, с. 138.
808. DufekV., March Z. Hutn. listy, 1959, В. 14, № 1, S. 909.
809. S t о 1 p e r W. Neue Hfltte, 1964, № 2, S. 110.
810. Кольтгоф И. M., Сейдел Е. Б. Количественный анализ.
ГОНТИ, 1948.
811. Booss Н. Z. anorg. Chem., 1957, В. 292, S. 236.
812. Назарчук Т. Н-, Печентковекая Л. Е. Заводская ла-
боратория, 1961, т. XXVII, с. 256,
813. Newrik A. J. Amer. Chem. Soc., 1955, v. 77, № 17, p. 4521.
814. Webb W. a. o. J. Electrochem. Soc4 1956, v. 103, № 2, p. 112.
815. Машкович Л, А. и др. Заводская лаборатория, 1964, т.XXX.
№ 5, с. 522.
816. Singh G., V е г m а А. Н. Acta cryst., 1964, v. 17, № I, p- 49.
817. Krishna P., Verma A, R. Acta cryst., 1964, v. 17, №2,
p. 51.
818. Gomes de A. H. a. o. Acta cryst., 1964, v. 18, p. 128.
819. Jean tet B., Knap ton A. G. Planseeber. Pulvermet, 1964,
v. 12, p. 12.
820. Dancy E. a. o. Trans, met. Soc. AIME, 1962, v. 224, № 6.
821. Щетников E H., Швейкин Г. П. Порошковая металлур-
гия, 1965, № 4, с. 1.
822. Косолапова Т. Я., Самсонов Г. В. ДАН УССР, 1959,
№ 3, с. 298.
823. Айрапетянц Э Г., Качалова Е. А- Заводская лабора-
тория, 1965, т. XXXI, с. 412. „иэт
°^4. Суворовская Н. А. и др. Технологический анализ в цвет-
ной Металлургии. Металлургиздат, 1957.
295
825. Сен дел Е. Б. Колориметрическое определение следов метал-
826. Пр’ш?б'и°л*рИРКомм4ексоны в химическом анализе. Госхим.
827 ЙтШ J- Z. anorg. Chem., 1933 В. 216, S. 85.
828. Ку кол ев Г. В. Химия кремния и физическая химия силика-
тов. Госстройиздат, 1951.
829 Миклашевский А. И. Заводская лаборатория, 1938, т. VII
№ 2, с. 168. и » v ’
830 Евстропьев К. С., Торопов Н. А. Химия кремния и фи
зическая химия силикатов. Госстройиздат, 1956. *
831. Кайнарский И. С., Дегтярева Э. В. Карборундовые
огнеупоры. Металлургиздат, 1963.
832. Миклашевский А. И. Карборунд, химический анализ и
его свойства. ГОНТИ, 1938.
833 Косолапова Т. Я-, К о т л я р Т. Е. Заводская лаборатория
1958, т. XXIV, № 12, с. 1442. г ’
834. Booss Н. J. Technisch-Wissenschaftliche Abhandlungen der
Osram Gesellschaft Sonderdruck., 1963, 8 B., S. 173.
835. Карбид кремния. Киев. Изд-во «Наукова думка», 1966.
836. Самсонов Г. В.^Стасовская В. В. Порошковая метал-
лургия, 1966, № 12, с. 95.
837. Mflhlhauser О. Z. anorg. Chem., 1894, В. 5, S. 105.
838. Funk Н., Sehauej- R. Chem. Techn., 1954, В. 6, S. 432.
839. Norton. Analyt Chem., 1953, v. 25, p. 1761.
840. A r d e 11 H. W. u. a. Z. anorg. Chem., 1963, B. 3, S. 70.
841. Штейнберг A. H. Tp. Института металлургии АН СССР.
Изд-во АН СССР, 1962, т. II, с. 243.
842. Ehrlich G., Gerdatseh R. Chemia Analiticzna (Polska),
1962, v. 7, № 2, p. 435.
843. Львов С. H. й др. Порошковая металлургия, 1962, № 4, с. 20.
844. Герасимов Я- И. и др. Химическая термодинамика в цвет-
ной металлургии. Металлургиздат, 1963, т. III.
845. Айрапётянц Э. Пи др. ЖПХ, 1967, т. XL, вып. 2, с. 443.
846. Попова Н. М., Засцавская Л. В. Заводская лаборато-
рия, 1955, т. XXI, с. 1285.
847. Шафран И. Г., Павлова М. В. Заводская лаборатория,
1938, т. VII, № JI, с. 1245.
848. Назарчук Т. Н. ЖНХ, 1959, т. IV, № 12, с.2665.
849. Назарчук Т. Н. Тр. семинара по жаростойким материалам.
Киев. Изд-во АН УССР, 196L
850. Самсойов Г. В., К о п н и н а О. И Заводская лаборатория,
1953, т. XIX, fc. 269.
851' ?Л.КОВ А- М.г Самсонов Г. В Порошковая металлургия,
1961, № 5, с. 82. ’
ana’ К. Analyt chim. acta, 1955, v. 13, p. 15.
858, H а з a p ч у к T. H., М е х а н о ш и н а Л. Н. Порошковая ме-
Паллургия> I964- № 2. с. 46.
854‘ V4 е ®pfc0H г- А., Самсонов Г В Заводская лаборатория,
окс 195<?’ т- XVI- с- 1423. 1Й
855‘ Tsu ch i d о a. о. J. Phys. Soc. Japan, 1961, v. !»•
856' №275 paiU579J’ P” F r u c h a r f Bul1- Soc- Chim’ FranCe’ 1964’
296
857. Bouchaud J. P., Fruchart R. Compt rend. Acad. Set Pa*
ris 1964» v. p. lou.
858. Bi’ttner H., Goretzki H Mh. Chem4 1960, B. 91 S 616
859 Sara R. V. J. Amer. Ceram. Soc., 1965, v. 48, № 6 n 243
Й Gurland J. J. Metals, 1957, № 4, p. 152. ' P’ '
861. Кайна pcкий И. С.и др. Огнеупоры, I960, № 4 с. 175
862 Бертман А. А. и др. ДАН СССР, 1964, т. 159 с. 121
863. М i с г z е у е w s k a S.„ N i е m у s Ц Т. J. Less-Common Metals,
1965, v. 8, p. З08.
864. С 0 s t a P., Conte R. R. Compound of Interest in Nucl Rear-
tor Technology, Edit, by I. Waber and P. Chiotti The Metafs
Society AIME, Michigan. 1964, p. 784.
865. Есин О. А., Гельд П. В. ЖПХ, 1958, т. XXXI, с. 1285.
866. F е s е п с о W. W. а. о. Coeepoque intern. Less properties mecha-
niquee et physiko-chimiques des refractores a hatte temperature
Paris, 1965.
867. Бертман А. А. и др. ДАН СССР, 1965, т. 162, № 6 с. 1304
868. Grant N. J., Preston О. J. Metals, 1957, v. 9, p/349.
869. Материаловедение реакторных материалов, под ред. Д. М. Ско-
рова, кн. 3. Атомиздат, 1962. ,
870. Dancy Е. а. о. Trans. Met. Soc. AIME, 1962, v. 224, № 6.
871. Моргулис H. Д. УФН, 1964, т. 43, о. 536.
872. Керметы, под ред. Дж. Р. Тинклпо- и LL В. Крэндалла. ИЛ,
1962.
873. Ogden Н. R. а. о. Trand. Metallurg. Soc. AIME, 1963, v. 227,
№ 6, p. 1458.
874. Munster M., Ruppert W. Z._, Elektrochem.,, 1953, B. 57,
S. 564.
875. H a m i j a n H., Lidman W. J. Metals, 1953, v. 5, № 5, p. 696.
876. Пеньковский В. В., Самсонов Г« В. Автоматическая
сварка, 1962, № 2, с. 39.
877. С амсоновГ. В., Кислый П. С. Высокотемпературные не-
металлические термопары и наконечники, Киев. Изд-во «Нау-
кова думка», 1965.
878. Исследования при высоких температурах, под редакцией
В. А. Кириллина и А. Е. Шейдлина. ИЛ, 1962.
879. Ракитин С. П. и др. Теплофизика высоких температур, 1963,
т. 1, с. 145.
880. М о р г у л и с Н. Д. УФН, 1960, С 70, с. 679.
881. Л и т ц Л. М. Исследования при высоких температурах. ИЛ,
1962.
882. Petrdlick М., Dufex V. Neue Hiitte, 1958, В. 3, S. I.
883. Peek S., Illuminating Eng, 1957, v. 52, p. 96.
884. Bacchela G L a. 0. Bull. Soc. Trans. Miner. Cnst, 1966,
ooc v. LXXXIX, p. 226. л a
885. Григорьева В. В. и др. Технология изготовления и 0,°мп
сти применения карбидохромовых сплавов. ЦИТЭИп, 1» ,
№ М—60—207/3.
886. Kennedy I. Materials and Methods, 1952, v. 38, p- 166.
887. Григор ”e в а В В., Климейко В. М. Сплави на ochobi
карб!ду хрому. Изв. АН УССР, КиГв, 1961.
888. Падерно Ю. Б. и др. Электроника, 1959, № 4, с. 165.
889. Газиев Г. А. и др. ДАН СССР, 1961, т. 140, № 4, с. 863.
297
й в С. и ДР- Производство твердых сплавов.
89°- металлуртиздат, ^бЬ^ pulvermet 1954 В. 2 Si 59.
891. Glaser £ д И- и ДР- ^зв' СССР, Неорганические
892’ материи. 1965 т. ^^^oqu^international. Les properties me-
893. Heiss A-’tDphysrCo chimiques des jeHactanes a hate temperatu-
chamques et pny Juni, j juillet 1965.
re Pans 28 язарчук T. H. Изв. АН СССР. Неоргани-
894 Котляр E- E-’ 1966 P и, № 10- c- 1778-
ческие материалы, 1^’ьюие ф д Графит и его кристалличе-
яак уббелоде А, г•> «Мир», 1965.
8 5’ ческие соединения. Изд во вРкристаллохимию. Изд-во МГУ,
896 Боки й I- ь-
1954’ п В Acta Chem. JScand., 1959, v_13-P- 10°:
897 Aronson В. Асг етьявов н г ЖФХ, 1943, т. 52
898 Ждан0? Г’
с. 326. иа А Л Стр ашинская Л В. Веб. «Высоко-
899. Бурыкина А. ^аничнскяе соедйнения. Киев. Изд-во «Нау-
температурные неиИ
кова думка», I960. КаоаПетянцМ. Л. Таблицы неко-
900 РаЗЛт““ И3*'”
МХТИ, ИМ. Менделеева, 1961.
901. Roberts В. W. Super conductive materials and some of ib;r
properties. New York, 1963. neir
902. Львов С. H, Немченко В. Ф. В сб. «Высокотемператур-
ные неорганические соединения». Изд-во «Наукова думка» 1965
903, Самсонов Г. В., Синельникова В. С. Порошковая
металлургия, 1962, № 4, с. 59.
904. Vickery R. J. J. Chem. Soc., 1959, v. 159, p. 503.
905. Самсонов Г. В. В сб. «Методы испытания на микротвер-
дость, приборы». Изд-во «Наука», 1965.
906. Самсонов Г. В., Нешпор В. С. В сб. «Вопросы порошко-
металлУРгии и прочности материалов». Изд. АН УССР,
1958, вып. 5.
907. Л аз арен ко Б. Р. Вестник АН СССР, 1961, т. 4, с. 83.
уий- 1962 № 2 К °8 В К., С т р у к Л. И. Порошковая металлургия,
909’ ^,McoJ1OB Ё. В., Синельникова В. С. Укр. физ. жури.,
am J?61-т-VI, № 5, с. 687.
qil' Ra м с°но в Г. В. и др, Укр. физ. журн., 1965, т. 10, с. 622.
9 1 В еЛАоп J- а- °- G R. Acad. Sci. Paris, 1965, v. 261, № 8-
P' Jooy,
1966 №^589'5%Г Forscllungsber. Landes Nordrein-Westfalen,
913' Пеньковский В. В. Порошковая металлургия, 1961, № 5
914' cl 207Л Я Р Е- Е ’ н а з а р ч у к Т. Н. ЖАХ, 1960, т. XV, № 2-
915.
916.
917.
Harwood J. J. Metallurgia, 1953, v. 47, P’.ggg v. 38, P- «-c
Knud sen F. P, a. o. J. Amer. Ceram. So •». d-ng non-rne
Якешова Л. New nuclear materials t nna> 1969,
fuels. International atomic energy agency.
p. 3771.
298
»>«. Коеолово»» Т.Й.8 лр. Им. АН СССР. Ли»».-______..
ма«ерямн, 1966. г. 2, № 8, с. 1516. «латесюге
до Струг Л И. Изв. АН ХХСР-. * -
1967. Т. Ш, № 4, с. 624 с ’«Ф^вяшчэме Мак^иалы,
sax Krikorian И. Н. ъ о. J. Nud. Maie^. 1S6X f 51 м> п
921 Jus» Я. « а. anorg. Chem., 1967. КД52, №1/2/58^’
1
КОСОЛАПОВА Татьяна Яковлевна
КАРБИДЫ
Редактор издательства М. С. Архангельская
Технический редактор Г. Н. Каляпина
Переплет художника Т. Л. Сенокосовой
Сдано в производство 18/Х-1967 г.
Подписано в печать 10/IV 1968 г.
Бумага № 2 типографская 84x108732 4,69 бум. л.
9,38 печ. л. 15,76 (усл.) Уч.-изд. л. 15,98. Заказ 1518.
Изд. № 4659. Т-05297. Тираж 2000 экз. Цена 1 р. 14 к.
Издательство «Металлургия», Москва, Г-34,
2-й Обыденский пер., 14
Владимирская типография Главполиграфпрома
Комитета по печати при Совете Министров СССР
Гор. Владимир, ул. Победы, д. 18-6