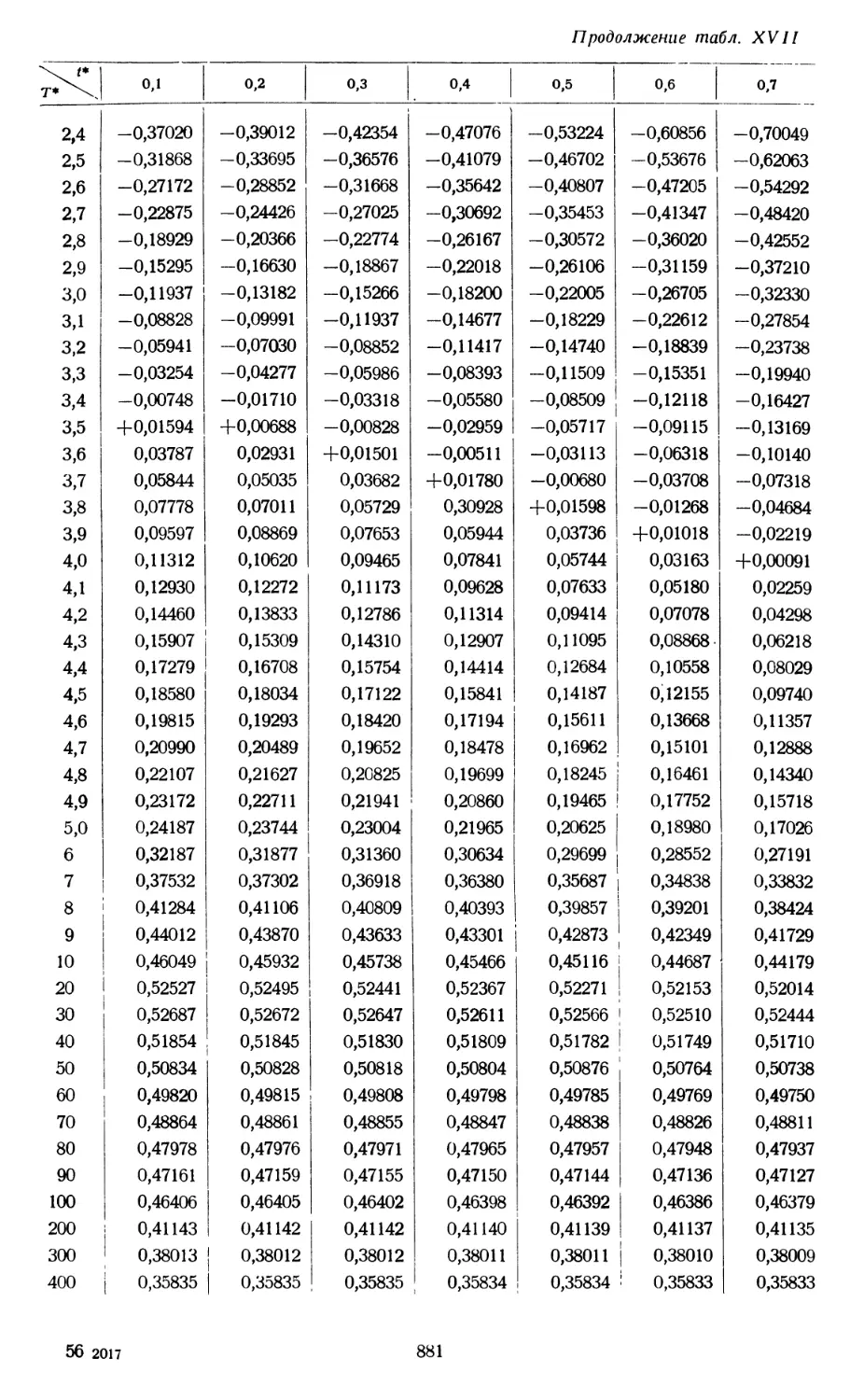
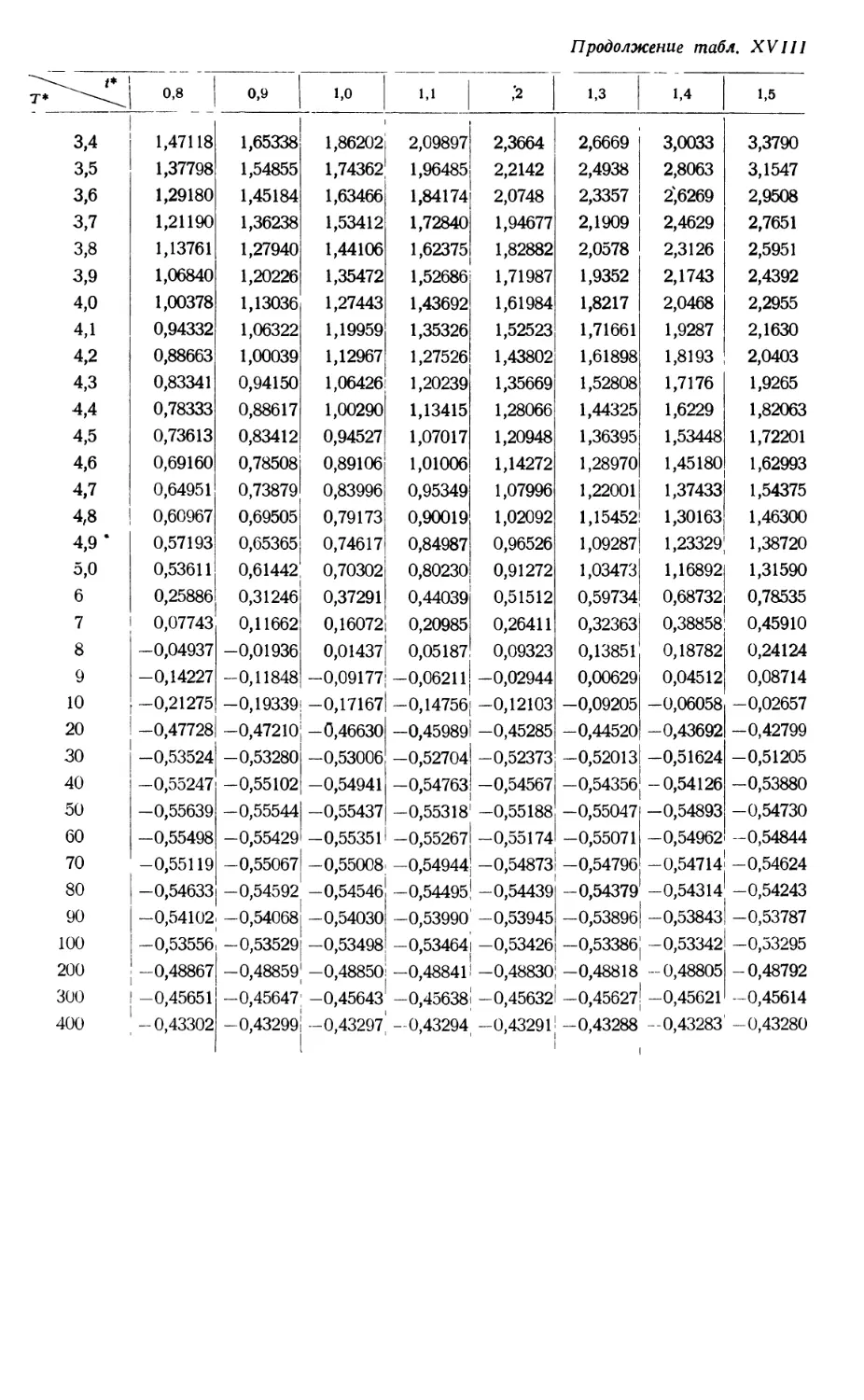
/






















































































































































































































































































































































































































































































































































































































































































































































































































































































































































Автор: Гиршфельдер Дж. Кертисс Ч. Берд Р.
Теги: физика химия молекулярная химия молекулярная физика молекулярная теория
Год: 1961




Текст
Дж. Гиршфельдер, Ч. Кертисс и Р. Берд
МОЛЕКУЛЯРНАЯ ТЕОРИЯ
ГАЗОВ И ЖИДКОСТЕЙ
ПЕРЕВОД С АНГЛИЙСКОГО
ПОД РЕДАКЦИЕЙ
Е. В. СТУПОЧЕНКО
ИЗДАТЕЛЬСТВО ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва 1961
MOLECULAR THEORY OF GASES AND LIQUIDS
J. O. Hirschfelder, Ch. F. Curtiss, R. B. Bird
University of Wisconsin
JOHN WILEY AND SONS, INC., NEW YORK
CHAPMAN AND HALL, LIM., LONDON
19 5 4
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Предлагаемая в русском переводе книга «Молекулярная теория газов и
жидкостей» написана группой известных американских ученых — специали-
стов в области молекулярной физики и смежных областях физической химии.
Ее цели, общий подход к изложению материала и содержание с достаточной
полнотой освещены в предисловии авторов.
Как в советской литературе, так и в зарубежной, до последнего времени
не было книги, в которой столь полно, на основе представлений и методов ста-
тистической физики и теории межмолекулярных сил излагалась бы обширная
область науки о равновесных и неравновесных свойствах жидкостей и газов.
Между тем, потребность в такой книге очень велика. В настоящее время
ряд научных организаций Советского Союза ведут интенсивные работы по
изучению физических и физико-химических свойств жидкостей и газов в широ-
ких интервалах температур и давлений. Следует заметить, что часто для реше-
ния задач, стоящих перед современной техникой, необходимо знать свойства
среды в таких условиях, которые сильно затрудняют или даже делают просто
невозможным при современном уровне экспериментальной техники непосред-
ственное их измерение. В таких случаях очень важно уметь правильно оценить
возможности теоретических методов, опирающихся на доступные экспери-
ментальные данные. С другой стороны, возможность установления связей между
различными свойствами жидкостей и газов не менее важна и при проведении
исследований в экспериментально контролируемых условиях.
С этой точки зрения предлагаемая книга представляет интерес для широ-
кого круга физиков и физико-химиков — научных работников и инженеров.
К тому же она содержит большой табличный материал и примеры численных
расчетов, иллюстрирующие способы вычисления различных величин. Кроме
того, как правильно отмечают авторы, она может служить ценным учебным
пособием для преподавателей, аспирантов и студентов, специализирующихся в
области молекулярной физики и ее приложений.
Основным разделам книги предшествует вводная глава, в которой, не
прибегая к сложному математическому аппарату, авторы ясно изложили физи-
ческие основы более строгих теорий, развиваемых в последующих главах.
Эта глава, а также включенные в текст задачи повышают педагогическую
ценность книги.
При оценке книги, содержащей столь обширный и разнообразный мате-
риал, можно спорить о том, сколь правильно авторы определили роль и удель-
ный вес той или другой части предмета в общем плане изложения. Отметим
лишь, что кинетическая теория ионизованных газов в книге почти полностью
отсутствует. Правда, эти важные вопросы в настоящее время фактически вы-
делились в самостоятельный раздел физики, весьма обширный и интенсивно
разрабатываемый. Однако включение в книгу обзора основных идей и резуль-
татов в этой области представлялось бы естественным.
Авторы широко использовали периодическую литературу и монографии,
опубликованные до 1954 г. (в некоторых случаях включая начало 1954 г.).
Предисловие редактора перевода
Однако списки литературы далеко не охватывают всех опубликованных к
этому времени работ по освещаемым в книге вопросам. Это, впрочем, естест-
венно, поскольку книга не является библиографическим обзором. Поэтому
при переводе редактор счел возможным ограничиться добавлением лишь
отдельных указаний на ряд работ (в частности, позднейших), непосредственно
связанных с текстом. Основные разделы книги дают читателю достаточную
подготовку для изучения позднейшей литературы.
В книге, к сожалению, не учтены многие важные работы советских уче-
ных. Например, во второй части книги при обсуждении проблемы получения
газокинетических уравнений и выводе из них уравнений гидродинамики в
различных приближениях не упоминаются известные работы Н. Н. Боголю-
бова. Это же относится к изложению в первой части книги теории уравнения
состояния, данной на основе работ Урселла, Майера и Кирквуда. Внесенные
редактором примечания не могут, конечно, целиком восполнить этот важный
пробел; для этого потребовалось бы существенно переработать или даже
дописать некоторые параграфы книги.
При переводе книги, охватывающей очень разносторонний материал,
возник ряд терминологических трудностей, решавшихся порой не без извест-
ных колебаний. Например, выражение „peculiar velocity" переведено как
«тепловая скорость», хотя в случае многокомпонентных пространственно неод-
нородных систем среднее значение этой величины для какой-либо одной ком-
поненты, вообще говоря, отлично от нуля.
Английский текст книги содержит большое число опечаток. В русский
текст внесены коррективы на основании списка исправлений, любезно пред-
ставленного в наше распоряжение проф. Дж. О. Гиршфельдером (этот список
содержит около 200 исправлений). Исправлены также и другие замеченные
нами погрешности. Тем не менее следует все же указать на необходимость
известной осторожности при пользовании книгой как справочником.
Отдельные неточности в утверждениях и формулировках отмечены в
примечаниях.
Перевод выполнен В. Б. Леонасом (§4—7 гл. 1; гл. 2 и 3 ; § 9 гл. 9 ; гл. 12);
С. А. Лосевым (§ 1—3 гл. 1 ; гл. 4 ; § 6 и 8 гл. 9); А. И. Осиповым (гл. 5, 6 и
10; § 1—5, 7, 10 гл. 11 ; § 6—9 гл. 13); Е. В. Самуйловым (гл. 7—9 ; § 1—6
гл. 13).
Е. В. Ступоченко
ИЗ ПРЕДИСЛОВИЯ АВТОРОВ
В этой книге описываются свойства газов и жидкостей с «молекулярной»
точки зрения. Эти свойства подразделяются на две группы — равновесные
свойства (такие, как уравнение состояния, коэффициент Джоуля—Томсона
и поверхностное натяжение) и неравновесные (такие, как вязкость, диффузия
и теплопроводность). Выражения для всех этих макросвойств через молеку-
лярные величины и межмолекулярные силы могут быть получены из статисти-
ческой механики. Упомянутые выражения, а также сведения о межмолеку-
лярных взаимодействиях могут быть использованы для предсказания значений
многих физических величин, для которых отсутствуют экспериментальные
данные. Особая целесообразность подхода, основывающегося на статисти-
ческой механике, состоит в том, что равновесные и неравновесные свойства
могут быть взаимно связаны с помощью законов межмолекулярных сил. В прин-
ципе, точное выражение этих законов может быть получено из квантовой меха-
ники молекул. Однако до настоящего времени математические трудности огра-
ничивали успешность этого подхода; тем не менее была получена разумная
качественная картина молекулярных взаимодействий. Поэтому взаимодей-
ствие между двумя молекулами обычно описывают с помощью простых эмпи-
рических функций, форма которых подсказывается квантовой механикой.
Эти функции включают несколько подбираемых постоянных, которые могут
быть определены для различных веществ из анализа экспериментальных изме-
рений макросвойств с помощью соответствующих формул статистической
механики. Следовательно, молекулярный подход дает полное описание макро-
скопического поведения веществ с помощью небольшого числа постоянных,
характеризующих рассматриваемое вещество.
За последнее десятилетие при изучении свойств газов и жидкостей было
получено много теоретических, расчетных и экспериментальных результатов.
Поэтому представлялось желательным дать обзор всей этой области с единой
точки зрения и в единых обозначениях. Мы надеемся, что эта книга будет по-
лезной для нескольких групп читателей. Студенты, специализирующиеся по
химической физике, найдут ее полезной для углубления своих знаний о строении
вещества и для понимания связи между различными разделами статистической
механики. В помощь студентам и преподавателям в конце параграфов пред-
лагаются задачи. Экспериментаторы физико-химики и инженеры найдут в
книге разделы по вычислению величин, характеризующих газы и жидкости.
В книгу включены обширные таблицы и даны примеры численных расчетов,
иллюстрирующие их использование. Лицам, занимающимся статистической
Из предисловия авторов
механикой, и химикам-теоретикам книга будет полезна как справочник. Они
заметят, конечно, что некоторые разделы книги носят незавершенный характер
вследствие отсутствия соответствующих теоретических исследований и пробе-
лов, имеющихся в расчетах. Можно надеяться, что этот недостаток послужит
стимулом для проведения дальнейших исследований.
Обсуждаемый в книге материал естественно делится на три части : равно-
весные свойства, неравновесные свойства и межмолекулярные силы. Основные
проблемы, связанные с каждой из этих областей, обсуждаются в гл. 1.В пос-
ледней части этой главы для справок приведены некоторые результаты клас-
сической и квантовой механики, которые используются во всех разделах книги.
Часть I (гл. 2—6) начинается с обзора статистической механики равно-
весных систем, которая служит основой для теоретического вывода уравнения
состояния. Затем обсуждаются две наиболее важные области приложений
теории — предсказание зависимостей между давлением, объемом и темпера-
турой для газов и жидкостей и анализ экспериментальных данных по урав-
нению состояния с целью получения сведений о межмолекулярных силах.
Часть II (гл. 7—11) посвящена неравновесной статистической механике (ки-
нетической теории и теории явлений переноса). Всякий раз, когда это было воз-
можно, данные этой части сравнивались с результатами, приведенными в части I.
Иными словами, здесь рассматриваются вопросы вычисления макровеличин и
методы получения сведений о межмолекулярных силах. Часть II заканчивается
главой по гидродинамическим приложениям, которая включает общее рас-
смотрение вопросов о распространении звуковых волн, пламен и детонации.
В части III (гл. 12—14) рассматриваются электромагнитная и квантовомеха-
ническая теории сил, действующих между молекулами, атомами и свободными
радикалами. Здесь априорные квантовомеханические вычисления межмоле-
кулярных сил сравниваются с результатами, полученными в первых двух
частях из анализа экспериментальных данных о макросвойствах,
Эта книга возникла на основе работ, проводимых по заказу Артиллерий-
ского бюро военно-морских сил, и является обобщением результатов, полу-
ченных большим числом исследователей. Значительную поддержку в предва-
рительной подготовке различных глав, каждая из которых издавалась как отчет
Исследовательской морской лаборатории Висконсинского университета, ока-
зало Артиллерийское бюро военно-морских сил. Мы особенно благодарны
докторам Гибсону, Эвери и Макклюру (Лаборатория прикладной физики Уни-
верситета Джона Гопкинса), которые сделали возможной организацию под-
готовки книги. Настоящий труд является частичным ответом на запрос Научно-
исследовательского комитета Министерства обороны о современном состоянии
исследований в области уравнения состояния и явлений переноса. Он является
развитием отчета А-116 A942 г.) Национального исследовательского комитета,
озаглавленного «Термодинамические свойства газов, использующихся в дви-
гателях» и написанного Гиршфельдером, Макклюром, Кертиссом и Осборном.
В начальный период работы над рукописью нам любезно оказала помощь
доктор Эллен Спотц. Она принимала участие в написании гл. 3 и 8 и активно
помогала в подготовке, редактировании и чтении корректур других частей
книги. Мы сожалеем, что доктор Спотц не смогла лродолжить работу над
этой книгой до ее завершения.
Из предисловия авторов
Нам хочется также поблагодарить профессора де Бура, директора Ин-
ститута теоретической физики Муниципального университета в Амстердаме,
за предоставление возможности ознакомиться с некоторыми его неопуб-
ликованными исследованиями, за помощь в подготовке глав по квантовым
эффектам (гл. 6 и 10) и очень полезную критику и советы при подготовке дру-
гих разделов книги. В связи с этим один из авторов (Р. Берд) хотел бы выра-
зить свою признательность за предоставленную ему возможность провести год
в институте профессора де Бура в Амстердаме.
Авторы горячо благодарны доктору Чарльзу А. Бойду за подготовку чер-
нового варианта гл. 5, доктору Джону С. Раулинсону за подготовку § 10
гл. 3 и доктору Говарду Б. Палмеру за помощь при написании § 2 гл. 5.
При подготовке этой книги мы использовали большое число научных работ
и поэтому считаем своим долгом поблагодарить всех авторов статей, на которые
мы ссылаемся. Мы благодарим авторов, редакторов и издателей, которые раз-
решили нам перепечатать различные фигуры и таблицы из оригинальных
работ. Несколько источников упоминаются во всей книге особенно часто :
«The Mathematical Theory of Non-uniform Gases», написанная С. Чепменом и
Т. Каулингом (издание Кембриджского университета, 1-е изд. 1939 г., 2-е изд.
1952 г.I) и «Molecular Distribution and Equation of State of Gases», опублико-
ванная Д. де Буром в Reports on Progress in Physics, 12, 305 A949). Эти работы
оказали нам такую помощь при подготовке определенных разделов книги, что
мы желаем их здесь отметить особо.
Висконсинский университет
Февраль 1954.
1) С. Чепмен и Т. Каулинг, Математическая теория неоднородных газов,
ИЛ, I960. — Прим. ред.
ОБОЗНАЧЕНИЯ
Общие
h — постоянная Планка
А = Щ2п
к — постоянная Больцмана
N — число Авогадро
R — газовая постоянная, R = Nk
с — скорость света
е — заряд электрона
а, е — параметры потенциальных функций межмолекулярного взаи-
модействия
А2 == Щ2пткТ
Л*2 = Що*тг
b0 = inNa3 — второй вириальный коэффициент для газа, состоящего из
твердых сфер диаметра а
t* — мера полярности молекул [A0.3) гл.З]
Jn(x) — функции Бесселя
Р?(х) — присоединенные полиномы Лежандра
Yf (д, ф) — сферические функции
S^\x) — полиномы Сонина [C.57) гл. 7]
Г(х) — гамма-функция
Ei(x) — интегральная показательная функция [G.50) гл. 11]
Классическая механика
г,, pi — координаты и импульс i-й частицы в декартовой системе
координат
q,, p/ — обобщенные координаты и импульс
V/ — скорость i-й частицы
mL — масса i-й частицы
Mt — молекулярный вес i-й компоненты
fiy — приведенная масса молекул i и /
Ff — сила, действующая на i-ю частицу
N
/С (pN) = jj? (р}/2/П/) — кинетическая энергия системы из N частиц
Ф(ты) — потенциальная энергия системы из N частиц
Н(гы, ры) — гамильтониан системы из N частиц
12 Обозначения
S — функция Гамильтона—Якоби
5 — «вириал» (гл. 1, §4)
b, g, хфу g) — прицельное расстояние ; начальная относительная скорость ;
угол отклонения в парном столкновении (гл. 1, § 5)
cp{f) — сферически симметричная потенциальная функция меж-
молекулярного взаимодействия
<р(г, в1У 02, ф) — потенциальная функция межмолекулярного взаимодействия,
зависящая от углов
/ — момент инерции
Квантовая механика
/*, = у-gjr —оператор импульса i-й частицы
= JEt ЙГ ("эГ # Эг) — оператор кинетической энергии
= <ЭГ + Ф — оператор Гамильтона
— оператор момента количества движения
Z, я, rjfa) — квантовые числа момента количества движения ; кван-
товые числа энергии; фазовый сдвиг в парном столкно-
вении (гл. 1, § 7)
a(g>%) — угловое распределение рассеянных частиц (гл. 1, §7)
Электродинамика
°У — электростатический потенциал
kj€ — вектор-потенциал
<f — напряженность электрического поля
JZ) — электрическая индукция
J{ — напряженность магнитного поля
е5б — магнитная индукция
6 — поляризация
<^# — намагниченность
С = Ze — общий заряд
fA — вектор дипольного момента
0 — тензор квадрупольного момента
Q — тензор квадрупольного момента, сумма диагональных эле-
ментов которого равна нулю
Q = qe — скалярная величина квадрупольного момента цилиндри-
ческого распределения зарядов
QJ1 — коэффициенты разложения, через которые выражаются муль-
типольные моменты
В1$,пь — коэффициенты «двухцентрового» разложения [A.28) гл. 12]
Х(е) — диэлектрическая восприимчивость
%<ш> — магнитная восприимчивость
е' — диэлектрическая проницаемость
in' — магнитная проницаемость
m — магнитный дипольный момент
q — плотность заряда
1 — сила электрического тока
j — плотность тока
Обозначения 13
/ — плотность энергии
v — частота
г\ — показатель преломления
/, — сила осциллятора
а — поляризуемость
Термодинамика
р — давление
Т — температура
х, — молярная доля i-й компоненты
V ; v — объем ; объем, приходящийся на одну молекулу
U ; и — внутренняя энергия ; внутренняя энергия на одну молекулу
Н ; h — энтальпия ; энтальпия на одну молекулу
Ср9 Cv — удельные теплоемкости при постоянном давлении и постоян-
ном объеме
5 — энтропия
О = н — TS — термодинамический потенциал
А = U — TS — свободная энергия
п — плотность числа частиц (т. е. число частиц в единице объема)
щ — плотность числа частиц i-й компоненты
q — плотность (т. е. масса единицы объема)
г — эффективная плотность числа частиц [B.22) гл. 3]
/г — коэффициент Джоуля—Томсона
6 — химический потенциал i-й компоненты
// — летучесть i-й компоненты
at — активность i-й компоненты
с = КCр/ЭрM — скорость звука
У = cpjcv
Статистическая механика и уравнение состояния
(f р^ ()) — функции распределения1) (классическая статисти-
fN\tMy pN), nN(tN) ] ческая механика)
(п) — матрицы плотности2) (квантовая статистическая
механика)
g(r) — радиальная функция распределения
—«множитель Больцмана» [A.6) гл. 3.]
— «сумма Слетера» [A.13) гл. 6]
Znj ZNq — классическая и квантовая статистические суммы
z — статистическая сумма отдельной молекулы
В(Т), С(Т), D(T)... — второй, третий, четвертый.. .вириальные коэффициенты
Ъ{ — групповые интегралы
Рк — неприводимые интегралы [B.24), B.25) гл. 3]
Vf — свободный объем
а — расстояние между ближайшими соседними молекулами в
кристаллических теориях жидкости
у — поверхностное натяжение
х) Функции распределения обсуждаются в гл. 2, § 1.
2) Матрицы плотности обсуждаются в гл. 2, § 2.
14 Обозначения
Кинетическая теория и явления переноса1)
y)j — сумматорные инварианты
?Р; — обобщенный вектор плотности потока
j — вектор плотности потока массы
р — тензор плотности потока импульса (тензор напряжений)
q — вектор плотности потока тепла
s — часть вектора плотности потока энергии, определяемая
выражением [A.18) гл. И]
б — вектор плотности потока энтропии
г\ — коэффициент сдвиговой вязкости
к — коэффициент объемной вязкости
А — коэффициент теплопроводности
Dy — коэффициент диффузии многокомпонентной смеси
DJ — коэффициенты термодиффузии многокомпонентной смеси
SDy — коэффициенты диффузии бинарной смеси
кТ — термодиффузионное отношение
v — скорость молекулы (гл. 7—10)
v0 — средняя массовая скорость (скорость потока) (гл. 7—10)
v — средняя массовая скорость (гл. 11)
ш — числовая средняя скорость
V ' — тепловая скорость
V — диффузионная скорость
W = f /г/2/cTV— приведенная скорость
g — начальная относительная скорость в парном столкновении
У = ур/2кТ g — приведенная начальная относительная скорость
Q(/)(g) —эффективные сечения переноса [B.2) гл. 8]
QV>S)(T) — интегралы, через которые выражаются коэффициенты пе-
реноса [B.3) гл. 8]
А*, в*, с* — величины, появляющиеся в выражениях для коэффициентов
переноса смесей [B.15) —B.17) гл. 8]
[*7i2 ]i — величина, определяемая выражением B.20) гл. 8
[А12 ]х — величина, определяемая выражением B.34) гл. 8
d, — величина, определяемая выражениями C.27) гл. 7 и B.28)
гл. 11
Ь — величина, определяемая выражением C.28) гл. 7
/,-(г, vh f) — функция распределения скоростей (гл. 7, § 1 и 2)
f\r](t,vht) —члены рядов Энскога [C.11) гл. 7]
ф-1 (r>v/> 0 — функция возмущения [C.21) гл. 7]
Знаки над символами
Статистическое усреднение; парциальная молярная величина обозначается
черточкой (—) над символом
Величина в расчете на моль обозначается тильдой (~) над символом
Величина в расчете на грамм обозначается знаком (/\) над символом
Дифференцирование по времени обозначается точкой над символом %
Определение различных скоростей дано в гл. 7, § 22.
Обозначения 15
Верхние индексы
Комплексно-сопряженная величина (для волновых функций, коэффициентов
разложения и т. п. в квантовомеханических рассмотрениях), а также
величина, приведенная с помощью простейших комбинаций молекуляр-
ных параметров а и е, отмечается звездочкой (*)
Величина, приведенная с помощью комбинации а и е с использованием вели-
чин, связанных с твердыми сферами, отмечается пятиконечной звездоч-
кой (*)
Величина, связанная с активированным состоянием, отмечается значком (X)
Величина, связанная с идеальным газом, отмечена индексом нуль (°)
Величины, связанные с молекулами после парных столкновений, а также
атомы в возбужденных состояниях, отмечены штрихом (')
Нижние индексы
г — величины, приведенные с помощью комбинации критических постоянных
с — значение величины в критической точке
Приведенные величины
Объем V* = VjNo* V* = Vjb0 =
Удельный объем v* = V* = г;/ст3 —
Температура Г* = кТ/е —
Давление р* = por3/e p* = pbJNe
Второй вириальный
коэффициент В* = B/Naz В* = В/Ьо =
Третий вириальный коэффициент С* = C/(/V(t3J С* = С/6О2
Коэффициент вязкости tj* = г\ог\У~хпг г\* =
Коэффициент теплопроводности Я* = Хо%кгху m\z Я* =
Коэффициент диффузии J2)* = «^а"*1 /т/е „2)* =
Термодиффузионное отношение — /сГ* = /ст//ст тв.сф.
Q-интегралы — Qft^* =
Расстояние г* = г/о
Прицельное расстояние Ь* = Ъ\а
Относительная кинети-
ческая энергия стал-
кивающейся пары
молекул с приведенной
массой /г g*2 = (Vj/ig^/e
Потенциальная энергия <р* = q>/e
Дипольный момент /г* = /г/]/ eaz
Поляризуемость а* = а/ст3
Поверхностное натяжение у* = yo2je
Квантовомеханический параметр Л* = hla\ me
16 Обозначения
[А
X
В] -
i
Ах
вх
J
А
By
к
•Аг
ОБОЗНАЧЕНИЕ ВЕКТОРОВ И ТЕНЗОРОВ
Векторы
i, j, к — единичные векторы в направлении х, у, г
А = \АХ + \Ау + kAz — вектор с компонентами АХУ Ау, Az
А = У А\ + А* + А\ — модуль вектора А
(А • В) = ^х^х + АуВу + AZBZ — скалярное произведение А и В
— векторное произведение А и В
[А х [В хС]] = (А • С)В —(А . В)С
г — радиус-вектор с компонентами х, у, z
rf — радиус-вектор i-й частицы
г и = т( — Tj — вектор, направленный от /-й частицы к i-й частице
rv = (rl9 r2, ..., rN) — вектор в SN-мерном пространстве, описывающий по-
ложение N частиц
. p") = ^ (Г/ • Pi)
1 = 1
Тензоры
T — тензор с девятью компонентами, представленными в виде
(т т т \
1 хх 1 ху 1 хг \
Т Т Т
'у* 'уу b*
* ZX * Zy * 22'
Tf — транспонированный тензор Т, полученный путем замены рядов на
столбцы
Т • А)х ^ 1 ХХАХ -\- 1 хуАу -f- 1 XZAZ = 2j ' ха^а
а
(А • Т)х » АХТХХ + АУТУХ + AJn = 2a AJo*
(Т • А) = (А • V)
\ ¦ * ¦ /ху — * хх-* ху "Г ' ху* уу "Г ¦* xz-* zy ~ 2s * ха* ау
(Г: ТО =?2*W=(T':T)
а /3
/1 0 0\
U = 0 1 0 — единичный тензор
\0 0 1/
(U • А) = (А • U) = А
(U : Т) = Тхх + Туу + Та
(U : U) = 3
Обозначения
17
АВ —
Диады
диада1) с девятью компонентами, представленными в виде
(АХВХ АХВУ АХВЛ
\АУВХ АУВУ АуВЛ
\АгВх АгВу АгВг)
(АВ-С) = А(В-С)
(С • АВ) = (С- А)В
СГ:АВ)=((Т-А).В)
(АВ:Т)=(А.(В-Т))
(АВ: CD) = (АС: BD) = (А • (В • CD))
=? (А • (В • C)D) = (А • D)(B • С)
Операция дифференцирования3)
и • 9 • 9 ¦* 9
Эг Эх ' ^ ду * dz
— = i— 4-i— 4-k —
9v 9^x 9^y 9v»
fi
9r
8x
9У
9F = d
92 — graa
ЭО (v) _ | dG
9v "" 9t;x
-э7хА1-
i J k
Эх 9у Эг
4 4 4
"X ^*y •Л12
9Txx i
= rot A
dy
x) Диаду АВ не следует смешивать со скалярным произведением (А • В).
2) Операцию 8/8г часто называют оператором «набла» и обозначают символом v. Обычн
различают 8/8г и 8/8v, записывая Vr и Vv.
18 Обозначения
Операция интегрирования
dz Jdr"=JJ... \dr1dxt... dtN
= JjJ dvx dvy dvz jdt"-* = jj ... JdrA+1 drft+a ...dtN
J F(V)W d\ = lU J F(VOV2dV
J (A-dS)= j (A-n)dS n —нормальный единичный вектор
По поверхности По по^рхности С Внешней СТОрОНЫ ПОВерХНОСТИ
Г (A-ds)= Г (A-t)ds t — тангенциальный единичный
по кривой по кривой • вектор вдоль кривой интегрирования
J (A-dS)= J (99r--A)dr Теорема Грина
кнутой По объему, огранич иному
khjcth этой поверхностью
j (А • ds) = J \^- х AJ • dS j Теорема Стокса
По замкнутой По поверхности,
кривой ограниченной отой
кривой
ГЛАВА 1
ВВЕДЕНИЕ И ОСНОВНЫЕ ПОНЯТИЯ
Уравнение состояния и явления переноса в газах и жидкостях тесно
связаны с силами, действующими между молекулами. Статистическая теория,
связывающая общие свойства с межмолекулярными силами, сейчас хорошо
разработана для разреженных газов и в меньшей степени — для плотных газов
и жидкостей. Измерение макроскопических свойств позволяет в принципе
определить закон, по которому действуют силы между молекулами. Более того,
если вид взаимодействия определен, то становится возможным получить урав-
нение состояния или коэффициенты переноса. Таким образом, описание боль-
шого числа равновесных и неравновесных явлений можно свести к общей
основе и выяснить их взаимосвязь с молекулярной точки зрения.
В этой главе даны наиболее важные понятия и определения, являющиеся
введением в молекулярную теорию газов и жидкостей. Первые три параграфа
посвящены простейшему рассмотрению основных вопросов, обсуждаемых в
этой книге, — уравнению состояния, явлениям переноса и межмолекулярным
силам. Далее следует обсу>одение некоторых важных результатов классиче-
ской механики и их приложений к изучению парных столкновений. Наконец,
дан краткий обзор некоторых важных квантовомеханических понятий, а также
рассмотрена квантовая теория молекулярных столкновений. В этих последних
четырех параграфах приведен ряд важных результатов теоретической физики,
нашедших применения во многих разделах данной книги.
§ 1. УРАВНЕНИЕ СОСТОЯНИЯ. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ
Если рассматриваемый газ состоит из частиц, не имеющих собственного
объема, между которыми не действуют никакие силы, то можно покаЗаТь с
помощью простых кинетических представлений (или с помощью более^строгой
теории), что уравнение состояния такого газа записывается в виде pV = RT.
Как хорошо известно, это соотношение совершенно точно в том случае, когда
газ весьма разрежен, однако при атмосферном давлении отклонения от этого
закона для идеального газа становятся уже ощутимыми. Ван дер Ваальс попы-
тался описать эти отклонения, заменив уравнения идеального газа уравнением,
которое дает достаточно хорошее описание зависимости р от V и Т для газовой
фазы и качественное описание жидкой фазы :
Величина а в уравнении Ван дер Ваальса определяется силами ^притяжения
между молекулами газа (можно видеть, что при постоянных Т и V увеличение
а приводит к уменьшению р); постоянная b определяется собственным объемом
молекул, или, более точно, наличием сильных короткодействующих сил оттал-
кивания между молекулами (увеличение Ъ приводит к увеличению давления
20 Гл. 1. Введение и основные понятия
при постоянных Т и V). Очевидно, что уравнение Ван дер Ваальса является
лишь грубым приближением, так как экспериментально обнаружено, что
молярный объем V в жидкой фазе значительно меньше, чем значение Ь, полу-
ченное из результатов по определению зависимости р от V и Т в газе.
Уравнение Ван дер Ваальса может быть использовано для описания зави-
симости р от V и Т для многих газов в небольших интервалах температур.
Чтобы дать эту зависимость для широкого диапазона изменения переменных,
были предложены многочисленные эмпирические уравнения различной степени
сложности; некоторые из них рассмотрены в гл. 4, § 2. В эти эмпирические
соотношения входят две или более соответствующие постоянные, и они вполне
пригодны для интерполяции существующих результатов. Однако связать
параметры этих уравнений с межмолекулярными силами невозможно, и поэтому
по существу безуспешны попытки установления связи между общим поведе-
нием газа и основными молекулярными взаимодействиями.
В этом параграфе даны простейшие теории уравнения состояния разре-
женных газов и плотных газов (или жидкостей), основанные на модели молекул
в виде твердой сферы. Поведение разреженных газов описывается путем рас-
смотрения отклонений от поведения идеального газа, связанных с молекуляр-
ными взаимодействиями. Поведение плотных газов и жидкостей описывается
путем рассмотрения явлений, происходящих с молекулами в идеальном кри-
сталле, когда они могут покидать свои места в решетке и перемещаться в неко-
тором объеме. Несмотря на то, что используемые здесь доводы достаточно
грубы, результаты упрощенных теорий имеют много общего с результатами
более строгих теорий. В конце настоящего параграфа дано введение к более
строгой статистической теории уравнения состояния.
А. ПРОСТЕЙШАЯ ТЕОРИЯ УРАВНЕНИЯ СОСТОЯНИЯ
РАЗРЕЖЕННЫХ ГАЗОВ
Экспериментальные изотермы в плоскости pV могут быть описаны в очень
широком интервале температуры и давления с помощью соотношения в виде
степенного ряда
lL-\ 4 Ж.
RT ~~ + V + V2 + V3 + ' * '' A# '
которое называется вириальным уравнением состояния. Зависящие от темпера-
туры функции В(Т), С(Т) и т. д. выступают здесь как второй, третий и т. д.
вириальные коэффициенты1).
С помощью элементарных рассуждений получим теперь выражение для
вириальных коэффициентов для газа, состоящего из N молекул, каждую из
которых можно представить в виде твердой сферы с диаметром а. Предположим,
что в некоторый момент времени эти молекулы типа «биллиардных шаров»
помещены в ящик объемом V. Центр первой молекулы, помещенной в ящик,
может двигаться в объеме (К1/з —аK, поскольку он не может приближаться
к стенкам ящика ближе, чем на 1/2а. Центр второй молекулы, добавленной к
первой, ограничен в своем движении объемом (V11* — аK—4/зя<т3> так как он
не может приблизиться к центру первой молекулы ближе, чем на расстояние о.
На фиг. 1 отмечена область, которая ограничивает движение третьей моле-
кулы. Этот процесс можно продолжить до тех пор, пока все N молекул не
будут помещены в ящик. Мы можем тогда сосчитать средний объем, доступный
для центра любой молекулы; очевидно, что
_ of - (A) [|«tf»j „V- ^
г) Слово «вириальный» происходит от латинского vis (множественное число — vires),
что означает «сила». Вириал есть величина, определенная в формуле D.27) и выраженная
через силы, действующие на молекулы. Вириальные коэффициенты определяют отклонения
от идеальности через свойства межмолекулярных сил.
§ 1. Уравнение состояния. Вириальные коэффициенты
21
где предполагается, что размеры ящика велики по сравнению с а. Величина
bQ = 2/3nNa3 является здесь «собственным объемом» молекул и равна учетве-
ренному объему сферических молекул.
Приведенные рассуждения не вполне правильны, так как мы пренеб-
регали тем, что две сферы, указанные на
фиг. 1 пунктирной линией, могут перекры-
ваться ; в результате объем, в котором не
может находиться третья молекула, будет нес-
колько меньше указанного. Следует также
принять во внимание перекрывание сфер, обо-
значенных пунктиром, с пунктирными линиями
на расстоянии 112а от стенок. Это, однако,
связано с поверхностными явлениями и рас-
сматривается отдельно в теории поверхност-
ного натяжения1). Когда принимается во вни-
мание перекрывание двух или более сфер, сред-
ний объем, доступный для центра любой моле-
кулы, записывается в виде
?-*.+-S-+-?-
A.3)
Фиг. 1.
Величины Ь19 Ь2У... — постоянные, зависящие
от геометрии перекрывающихся сфер. Уравне-
ние состояния для твердых сферических молекул тогда запишется в виде
p\v -ьо + ~^- + -~-
= NkT.
A.4)
Разлагая в ряд по степеням 1/К, можно записать это уравнение в вириальной
форме
RT V ч V2
уз
= !+-§-
+ ... =
0,62506?
0,2869 И
A.5)
Численные коэффициенты, фигурирующие во второй строчке этого выражения,
являются результатом точных расчетов второго, третьего и четвертого вириаль-
ных коэффициентов для твердых сферических молекул [1, 2]. Для твердых
молекул вириальные коэффициенты не зависят от температуры2). Значения
второго и третьего вириальных коэффициентов, полученные путем подобных
расчетов для различных веществ, приводятся в табл. 1 ; там же даны соответ-
ствующие экспериментальные значения. Для низких температур эксперимен-
тальные значения второго и третьего вириальных коэффициентов отрицательны,
а для высоких температур положительны. О величине отклонения состояния
газа от идеального при различных температурах и давлениях можно судить
по экспериментальным значениям фактора сжимаемости. Эти отклонения
весьма значительны при низких температурах и высоких давлениях.
Таким образом, уравнение состояния разреженного газа можно записать с
помощью выражений, характеризующих отклонения от уравнения идеального
газа pV = NkT. Очевидно также, что газ ведет себя как идеальный, когда
собственный объем молекул мал по сравнению с объемом сосуда, содержащего
газ.
2) Поверхностное натяжение рассматривается в гл. 5, § 1.
2) Уравнение состояния для твердых сфер, полученное с помощью статистической меха-
ники, приводится в гл. 3, § 5. Вириальные коэффициенты для твердых несферических моле-
кул даны в гл. 3, §8.
22
Гл. 1. Введение и основные понятия
Таблица 1
Некоторые величины, входящие в уравнение состояния,
полученные из экспериментальных измерений и расчетов с помощью модели
твердых сфер
Вещество
Не
Аг
N2
т?к
100
300
500
100
300
500
100
300
500
pV/RT
1 атм
1,0023
1,0005
1,0003
0,9783
0,9999
1,0001
0,990
0,9998
1,0004
50 атм
1,113
1,024
1,013
0,174**
0,971
1,009
0,243**
0,9960
1,0210
1000 атм
1,439
1,242
3,30**
1,675
1,353
3,84**
1,99
1,82
В(Т), смг\молъ
эксперимен-
тальные
значения
9,6
10,8
-178
- 15,2
(8,4)
-149
- 4,4
168
ДЛЯ
твер-
дых
сфер*
21,9
60,9
66,5
С(Т) (см* 1 моль)*
эксперимен-
тальные
значения
F0)***
72
(ПО)
(-1500)
990
710
B080)
1310
640
ДЛЯ
твердых
сфер*
300
2314
2766
¦ Величины для твердых сфер рассчитаны путем использования значений диаметра, полученных из
кинетической теории газов и приведенных в табл. 2 ; Втв сф = Ьо = 7з п N о8 и Ств сф = 6/в Ь^.
•¦ Для жидкости.
**¦ Величины в скобках получены путем экстраполяции экспериментальных данных.
Б. ПРОСТЕЙШАЯ ТЕОРИЯ УРАВНЕНИЯ СОСТОЯНИЯ
ПЛОТНЫХ ГАЗОВ И ЖИДКОСТЕЙ1)
Рассмотрим плотный газ или жидкость, состоящие из N ^по-прежнему
твердых непроницаемых сфер диаметром <т, помещенных в объем V. Представим
себе, что эти молекулы расположены так, что образуют кубическую решетку,
причем расстояние между их центрами
равно (К/ЛI/з. Выберем теперь одну
из этих молекул и предположим, что
она может свобсдно перемещаться от-
носительно своего фиксированного по-
ложения в решетке. Из фиг. 2 можно
видеть, что центр такой «блуждающей»
молекулы может свободно переме-
щаться в объеме, приблизительно рав-
ном кубу с ребром 2(K//VI/a — 2а. Для
молекул с точечной массой (а = 0) этот
объем равен кубу с ребром 2(K//VI/s.
Давление, оказываемое перемеща-
ющейся молекулой на остальные моле-
кулы, равно скорости передачи им-
пульса единице поверхности. Эта ско-
рость прямо пропорциональна частоте, с которой молекула пересекает рассматри-
ваемый объем, и обратно пропорциональна длине ребра указанного куба. Сле-
довательно, давление, создаваемое перемещающейся твердой сферой, относится
к давлению в случае перемещающейся материальной точки как обратное от-
ношение размеров ребер ограничивающих их объемов :
оооо
„Блуждающая " молекула
О Cjj§O О
оооо
Фиг. 2.
RT
2G
2)См. гл. 4, §5, а также [3, 4].
§ 7. Уравнение состояния. Вириальные коэффициенты 23
Это можно переписать в виде
p[V- 0,7816 of V8/»]= RT, A.7)
что является приближенным уравнением состояния для газа из твердых сфери-
ческих молекул при больших плстностях. Численное значение постоянной
зависит от избранного типа упаковки кристалла. Например, для гранецент-
рированной кубической решетки постоянная равна 0,6962, для объемноцентри-
рованной кубической — 0,7163.
В. ВВЕДЕНИЕ В СТРОГУЮ СТАТИСТИЧЕСКУЮ ТЕОРИЮ
УРАВНЕНИЯ СОСТОЯНИЯ
Чтобы получить более строгое уравнение состояния газа или жидкости,
содержащих большое число частиц, необходимо воспользоваться методами
статистической механики равновесных систем. В гл. 2 рассматриваются основ-
ные понятия статистической механики, даются наиболее важные определения,
а также разбираются некоторые свойства идеального газа.
В гл. 3, § 1 показано, что статистическая теория может быть использована
для получения уравнения состояния двояким образом.
1) С помощью статистической суммы ZN:
A.8)
2) С помощью радиальной функции распределения g(rI):
Статистическая сумма ZN является суммой величин ехр (— Е^кТ) по всем
энергетическим состояниям (системы из N молекул), где JE, — энергия /-го
состояния системы. Радиальная функция распределения g(r) определяется так,
что число пар молекул, разделенных друг от друга расстоянием г, равно
В гл. 3 дан строгий вывод классического уравнения состояния при малой
плотности. Показано, что использование как статистической суммы, так и
радиальной функции распределения приводит к уравнению состояния в ви-
риальной форме, представленной в виде соотношения A.2). Таким образом,
статистическая механика позволяет получить выражения для вириальных
коэффициентов в В(Т), С(Т), D(T) и т. д. через силы, действующие между моле-
кулами в газе. Например, если межмолекулярный потенциал <р является функ-
цией только расстояния г между молекулами, то второй вириальный коэф-
фициент можно представить в виде
J
A.10)
Следовательно, для тех газов, для которых межмолекулярные силы известны
как функции расстояния между молекулами, однократное интегрирование
дает значение второго вириального коэффициента как функцию температуры,
что является первой поправкой при учете неидеальности газа. Достаточно точ-
ное измерение коэффициента В(Т) позволяет получить определенные сведения
о характере межмолекулярных сил. Для этого потенциал взаимодействия
предполагается имеющим такой функциональный вид, который качественно
х) Уравнение A.8) может быть использовано в том случае, когда межмолекулярные
силы не зависят от углов, а уравнение A.9) справедливо только для сферически симметрич-
ных потенциалов взаимодействия.
24 Гл. 1. Введение и основные понятия
согласуется с теоретическим рассмотрением и содержит несколько параметров.
Эти параметры затем выбираются такими, чтобы давать наилучшее прибли-
жение к экспериментальным значениям В(Т). Можно также получить теорети-
ческие выражения второго вириального коэффициента для молекул, взаи-
модействие которых описывается потенциалом, зависящим от угла, например
для полярных и длинных молекул. Эти формулы могут также быть исполь-
зованы для анализа экспериментальных данных и получения сведений о меж-
молекулярных силах, зависящих от ориентации молекул. Вычисление вириаль-
ных коэффициентов детально описано в гл. 3. Поскольку в настоящее ^ремя
можно провести оценку только нескольких первых вириальных коэффициентов,
то вириальное уравнение используется лишь только при малых и средних
значениях плотности газа.
В гл. 4 дан вывод уравнения состояния плотных газов и жидкостей. Этот
вывод по-прежнему можно провести либо с помощью статистической суммы,
либо с помощью радиальной функции распределения. В этом случае невоз-
можно, однако, довести до конца вывод уравнения в некотором приближении
без введения ряда упрощающих предположений. Поэтому разные приближения
дают различные результаты. Наибольшее число работ было проделано с по-
мощью метода, заключающегося в использовании статистической суммы.
Первые попытки в этом направлении тесно связаны с простейшей теорией,
изложенной в разд. Б настоящего параграфа, и основаны на использовании
статистической суммы отдельной молекулы, находящейся в ячейке, образо-
ванной ближайшими соседними молекулами. Уточнение этой теории прово-
дится путем рассмотрения поправок на движение молекул в смежных объемах
и наличие «дырок» в структуре решетки. Подробные расчеты проделаны для
различных моделей решеток. В последнее время большое внимание было уде-
лено выводу уравнения состояния плотных газов и жидкостей с помощью ра-
диальной функции распределения. Проведено несколько расчетов, которые
дали многообещающие результаты.
В гл. 5 результаты, полученные в двух предыдущих главах, использованы
для изучения свойств двухфазных систем, в которых пар находится в равно-
весии с жидкой фазой. В гл. 5 включено также рассмотрение поверхностного
натяжения, парахора, критических явлений и явления конденсации.
В гл. 6 уравнение состояния одноатомных и двухатомных газов рассматри-
вается с квантовомеханической точки зрения. Квантовая статистика позволяет
провести описание равновесных свойств вещества в условиях, когда откло-
нения от классических законов значительны. Квантовомеханические эффекты
наблюдаются экспериментально для изотопов водорода и гелия при комнатной
температуре и резко выражены при низких температурах.
§ 2. КИНЕТИЧЕСКАЯ ТЕОРИЯ ГАЗОВ.
КОЭФФИЦИЕНТЫ ПЕРЕНОСА
Явления диффузии, вязкости и теплопроводности физически подобны,
так как они заключаются в переносе некоторых физических величин через газ
или жидкость. Обычная диффузия есть перенос массы из одной области в дру-
гую вследствие наличия градиента концентрации1); вязкость есть перенос
импульса через газ вследствие наличия градиента скорости; теплопровод-
ность есть перенос тепловой энергии в результате наличия градиента темпера-
туры в газе. Этим процессам присвоено название «явления переноса». В настоя-
щем параграфе эти явления описываются с помощью простейшей кинетической
*) Диффузия может также являться результатом наличия градиента температуры (тер-
модиффузия, или эффект Соре), а перенос энергии может быть результатом наличия гра-
диента концентрации (диффузионная теплопроводность, или эффект Дюфура). По этому
поводу см. гл. 7.
§ 2. Кинетическая теория газов. Коэффициенты переноса 25
теории. Несмотря на то, что всюду используются очень грубые допущения,
представляется, однако, возможным получить выражения, описывающие в
основном зависимость коэффициентов переноса от температуры и давления, а
также от массы и размеров молекул газа. Затем кратко дается более строгая
теория и показывается, насколько успешно ее использование.
А. ЭЛЕМЕНТАРНАЯ КИНЕТИЧЕСКАЯ ТЕОРИЯ
РАЗРЕЖЕННЫХ ГАЗОВ
В любом реальном газе молекулы движутся во всех направлениях, и их
скорости имеют очень широкий диапазон значений. Когда две молекулы приб-
лижаются одна к другой, они испытывают очень сложное взаимодействие,
поскольку реальные молекулы притягиваются друг к другу на больших рас-
стояниях и отталкиваются друг от друга, когда межмолекулярное расстояние
становится очень малым. Несмотря на сложность поведения молекул, можно
получить достаточно хорошее описание явлений переноса, если мы рассмотрим
следующую, далекую от реальной модель газа, содержащего п молекул в еди-
нице объема:
I) Молекулы являются твердыми сферами с диаметром а, не притяги-
вающимися друг к другу.
II) Все молекулы движутся с одной и той же скоростью ; наиболее разум-
ным выбором для скорости молекул является использование среднеарифмети-
ческого значения скорости Q = (8/сТ/шяI/2, которое можно вычислить с по-
мощью функции распределения скоростей (см. задачу 4 гл. 2).
III) Все молекулы движутся в направлении, параллельном одной из коор-
динатных осей, так, что шестая часть из них движется в направлении (+ х),
еще шестая часть — в направлении (—х), еще шестая часть— в направлении
(+у)ит.д.
1. Число столкновений молекул в газе. Начнем с рассмотрения зависимо-
сти числа столкновений Г от размеров, плотности числа частиц и средней ско-
рости молекул. Рассмотрим одну молекулу, движущуюся в направлении (+г),
и выясним, с какой частотой она сталкивается с другими молекулами газа.
Очевидно, она не будет испытывать столкновений с другими молекулами,
движущимися в направлении (+ г), так как все они движутся с одинаковой
скоростью Q. По отношению к молекулам, движущимся в направлении (— г),
она имеет, однако, относительную скорость 2Q. Это означает, что за время At
молекулы, центры которых лежат внутри цилиндра с поперечным сечением
жг2 и длиной 2QAt, будут испытывать столкновения с выбранной нами моле-
кулой (предполагая, что последняя не отклоняется при столкновении). По-
скольку число молекул в единице объема равно п и одна шестая всех молекул
движется в направлении (— г), то в единицу времени будет происходить 1l^rtna2Q
столкновений с этими молекулами. Аналогично молекула, движущаяся в
направлении (+ г), обладает скоростью J/2JQ по отношению к молекулам, дви-
жущимся в направлении (+ х); следовательно, в единицу времени будет
происходить 1l6\f^nna2Q столкновений с этими молекулами. Такой же резуль-
тат получается для молекул, движущихся в направлениях (— х), (+ у) и
(—у); таким образом, в целом происходит
Г = S'nTuflQ = S'po*][ *л B.1)
I ГПК 1
столкновений, испытываемых одной молекулой в единицу времени, причем
f = !/8 + 2/3f2. Второе выражение, данное в B.1) для Г, было получено с
помощью выражений р = пкТ (уравнение идеального газа) ий = j/8/сТ/ттг.
Если бы предполагалось, что молекулы движутся во всех направлениях и что
распределение скоростей является максвелловским, то был бы получен тот же
26 Гл. 7. Введение и основные понятия
результат, но мы имели бы ?' = f2, т. е. ?' == 1,414 (по сравнению с прибли-
женным значением 1,276).
2. Средняя длина свободного пробега. Поскольку рассматриваемый нами
газ состоит из непроницаемых упругих сфер, то момент столкновения между
двумя молекулами можно определить достаточно хорошо. Это позволяет ввести
величину, известную как средняя длина свободного пробега, которая является
усредненным расстоянием, проходимым молекулой между столкновениями.
Таким образом, молекула, движущаяся со скоростьюQ в течение достаточно
продолжительного интервала времени At (продолжительность его велика по
сравнению со средним значением промежутка времени между столкновениями),
будет проходить расстояние QAt; если молекула испытывает Г столкновений
в единицу времени, то в течение достаточно продолжительного интервала вре-
мени At молекула будет сталкиваться Г At раз. Отсюда усредненное расстояние,
проходимое молекулой между столкновениями, (или средняя длина свободного
пробега), равно
QAt Q ( .
Подставляя B.1) в B.2), получаем
1 =
где второе выражение получается в результате использования уравнения
идеального газа. Нужно отметить, что при постоянной плотности средняя
длина свободного пробега не зависит от температуры; при постоянном дав-
лении она прямо пропорциональна температуре.
Величина яа2, входящая в знаменатель выражения для средней длины
свободного пробега, является эффективным сечением столкновения для твер-
дой сферической молекулы. Эта величина, входящая во все выражения для
коэффициентов переноса, представляет сечение воображаемой сферы, окру-
жающей молекулу, внутрь которой не может проникнуть центр никакой
другой молекулы.
В этой упрощенной кинетической теории коэффициенты переноса могут
быть выражены через величину /. Поэтому вязкость, диффузию и теплопровод-
ность иногда относят к явлениям, связанным со средней длиной свобоОного
пробега. Однако при более строгом приближении для реальных газов оказы-
вается, что средняя длина свободного пробега не выступает столь естественно
при описании явлений переноса.
3. Потоки молекулярных величин. Коэффициент обычной диффузии числен-
но равен плотности потока молекул сорта i вследствие единичного градиента
плотности числа частиц i; коэффициент вязкости численно равен плотности
потока у-компоненты импульса, создаваемого единичным градиентом у-компо-
ненты скорости; коэффициент теплопроводности численно равен плотности
потока энергии, вызванного единичным градиентом температуры. Во всех трех
случаях векторы плотности потока по направлению противоположны градиен-
там, и это направление определяет направление оси z. Плотность потока
молекул сорта i обозначается через //2. Плотность потока у-компоненты им-
пульса в направлении z записывается через руг = pzyy где р — тензор давления.
Плотность потока энергии обозначается через qz. В силу физического подобия
всех трех явлений описание их может быть дано в общей математической
форме. Для обозначения z-компоненты одного из трех векторов плотности
потока — потока молекул сорта /, потока импульса в направлении у и потока
энергии — будем использовать символ Wp. Символ Р представляет соответ-
ственно плотность числа молекул сорта г, плотность импульса и' плотность
энергии. Эти обозначения иллюстрируются табл. 2.
Рассмотрим поток величины, плотность которой обозначена через Р (т. е.
числа молекул сорта /, импульса в направлении у или энергии), протекаю-
§ 2. Кинетическая теория газов. Коэффициенты переноса
27
щий через плоскость О в направлении (+ z) (фиг. 3). Молекулы, приближаю-
щиеся к плоскости О снизу, испытали последнее столкновение на расстоянии /
ниже плоскости О. Таким образом, они вышли из плоскости А и обладают
значением величины РА, характеризующим эту поверхность. Подобно этому,
молекулы, попадающие в плоскость О после того, как они покинули плоскость
В, обладают значениями Рв, характеризующими эту плоскость. Если величина
Р имеет в плоскости О значение Ро, то мы можем записать
Р =Р -/ —, Р =Р +l-d- B.4)
dz dz
в предположении, что градиент величины Р постоянен на расстоянии порядка
средней длины свободного пробега.
В случае диффузии величина Р равна nh т. е. концентрации числа частиц
сорта /. Значение nL в каждой из плоскостей Л, О, В различно и поэтому
Таблица 2
Диффузия
Вязкость
Теплопроводность
ncvT
Яг
Ф и г. З.
происходит перенос массы. Рассматривая
вязкость, мы предполагаем, что молекулы
в плоскости А движутся в направлении
(— у), молекулы в плоскости О не пере
мещаются вдоль координаты у, а моле-
кулы в плоскости В движутся в напра-
влении (+ у). Такая картина течения
определяется величиной градиента у-ком-
поненты импульса Р = nmvy. При рас-
смотрении теплопроводности плоскости
Л, О и В берут при различных значениях температуры, и величину Р пола-
гают равной плотности энергии ncvT (где cv — удельная теплоемкость на одну
молекулу). Количество величины, плотность которой обозначена через Р, пе-
ресекающее снизу единичную площадь в плоскости О в единицу времени (т. е.
плотность потока этой величины), равно 1IQQPA, где множитель х/в учитывает тот
факт, что только одна шестая всех молекул в плоскости А движется в направ-
лении (+ г). Подобно этому плотность направленного вниз потока той же
величины равна 11вОРв- Следовательно, результирующая плотность потока WP
величины, соответствующей Р, проходящего через плоскость О в направлении
(+ г), равна
где f = 2/3тг, когда ?' взят равным ]/. В частности, значения
Гщ — /ft - - у Ш -j- , (ЛЬ)
представляют величины плотностей потоков молекул сорта i> импульса и
энергии соответственно.
28 Гл. 1. Введение и основные понятия
4. Коэффициенты переноса. Для положения, представленного на фиг. 3,
коэффициенты переноса выражаются через значения плотностей потоков
следующим образом:
*ы = Ы=-Я%; B.9)
^„m,y = /V=->?^. B.10)
»W = *=-*•?; B.11)
здесь о2) — коэффициент диффузии; rj — коэффициент вязкости; X — коэф-
фициент теплопроводности. Сравнивая эти три соотношения с тремя преды-
дущими, находим
2>Л *^
B.13)
B.14)
где q = nm = рт/кТ — плотность газа. Приложение более строгой кинети-
ческой теории (см. гл. 7) к модели газа, состоящего из твердых сфер, позволяет
уточнить приведенные выше выражения для коэффициентов переноса. Строгая
теория для таких молекул показывает, что значение ? различно для каждого
потока. В гл. 8, § 2 показано, что
B.15)
Подставляя эти значения в B.12) — B.14), можно переписать выражения для
коэффициентов переноса в виде
Л) = 2,6280 • Ю-3 ^^- см2/сек, B.16J)
П = 2,6693 • Ю-5 Щ^- г/см ¦ сек, B.17)
А =1,9891- 10т*ЛШ. = J?.JL,7 кал/см-град-сек, B.18)8)
3
8 »
5
16 '
25
32 >
V
Q 3)
tjcv
Хт ~~
Q&Cv
hn
5
6
2
5
12
25
х) Это выражение для ?й справедливо для самодиффузии, т. е. диффузии частиц одина-
ковой массы и одинаковых размеров. Примерами, когда такая формула может употребляться,
являются диффузии орто- и пара-разновидностей газа и диффузия тяжелых изотопов (см.
гл. 8, §2).
*) Для смеси двух химически отличных друг от друга газов коэффициент диффузии для
модели газа из твердых сфер равен
-2,6280• ю- У^СЦ + лу/гльм.
где ог12 = 1/2@4 + <*2).
8) Эта формула для коэффициента теплопроводности приложима только к одноатомным
газам. Для многоатомных газов можно сделать поправку Эйкена (см. гл. 7, § б и гл. 8, § 2);
тогда B.18) заменяется соотношением
15 Я М С„ , 3'
§ 2. Кинетическая теория газов. Коэффициенты переноса
29
где М — молекулярный вес; Т — температура (°К); р — давление (атм)\
а — диаметр молекул (А).
В табл. 3 приведены некоторые значения газокинетических параметров
твердых сфер с целью показать порядки величин, с которыми мы имеем дело.
В таблицу включены также экспериментальные значения, чтобы указать на
недостатки модели газа, состоящего из твердых сфер.
Таблица 3
Некоторые значения газокинетических параметров по упрощенной теории*
при атмосферном давлении**
SO
5S.
с
Il
h
Коэффициент
диффузии 2),
смг»сек~1
||
ев w
о.
111
Коэффициент
ВЯЗКОСТИ 1).
Ю-1 г см-1
сек—1
? 3 •'
Коэффициент
теплопровод-
ности А,
10~7 кал • см~1
сек~1 • град—1
Не
Аг
4,00
39,9
28,02
2,18
3,64
3,75
100
300
500
100
300
500
100
300
500
645
1936
3226
231
694
1157
218
654
1090
11,28
6,51
5,04
9,95
5,75
4,45
12,61
7,28
5,64
7,28
12,60
16,27
2,30
3,99
5,15
2,75
4,76
6,15
0,276
1,437
3,091
0,031
0,163
0,351
0,035
0,183
0,395
@,270)
A,669)
C,890)
0,023
0,186
@,456)
0,027
0,205
@,495)
1123
1946
2512
1273
2204
2846
1005
1740
2247
951
1981
2790
815
2271
3351
698
1663
2657
2093
3625
4679
238
412
531
339
587
766
1744
3583
4844
154
422
631
B32)
F02)
(876)
¦ Значения а взяты у Кеннарда [19].
•• Q =* у8 kTImn ; Г и I определяются соотношениями B.1) и B.3) с учетом $' — К~2~; 3>, у и * следуют
из соотношений B.16)—-B.18).
*** В скобках приведены оценочные величины.
Из вышеприведенных соотношений видно, что коэффициент диффузии
изменяется прямо пропорционально температуре в степени 3/2 и обратно про-
порционально давлению. Коэффициенты вязкости и теплопроводности не за-
висят от давления и растут как квадратные корни из значения температуры.
Все три коэффициента переноса обратно пропорциональны квадрату диаметра
молекул. Эти соотношения будут правильно описывать явления переноса, если
молекулы действительно являются твердыми сферами. Как мы увидим в гл. 8,
эти формулы дают для реальных газов только приближенный вид зависимости
от температуры и давления, так как действительная температурная зависимость
коэффициентов переноса должна включать влияние взаимодействий, которые
имеют место между реальными молекулами. Будет показано, что расчеты, осно-
ванные на более строгой кинетической теории, позволяют предсказывать зна-
чения этих величин с очень большой точностью. Тем не менее следует отметить,
что даже весьма искусственная модель газа, использованная нами, дает хоро-
шие результаты.
Упрощенная кинетическая теория предполагает наличие определенных
отношений молекулярных постоянных, которые не меняются заметно при пере-
ходе от одного вещества к другому и не изменяются заметно с температурой.
Например, комбинация y\Cp\i известна как число Прандтля. Поскольку отно-
шение удельных теплоемкостей у = CpjCv для одноатомных молекул равно
5/э> упрощенная кинетическая теория предсказывает, что число Прандтля
30
Гл. 7. Введение и основные понятия
должно иметь значение 2/3. Подобно этому отношение rj/g ?D известно как число
Шмидта. В соответствии с упрощенной кинетической теорией оно должно
Таблица 4
Экспериментальные значения чисел Шмидта
и Прандтля при 0° С
Газ
лценной кинетической
ИИ
Число
Шмидта
0,73
0,75
0,74
0,70
0,74
0,71
0,73
0,83
Число
Прандтля
0,66
0,67
0,71
0,74
0,72
0,75
0,71
0,67
Ne
Ar
N2
CH4
o2
co2
н2
иметь значение 5/6. В табл. 4 даны значения чисел Шмидта и Прандтля для
нескольких веществ при 0° С, рассчитанные с помощью наилучших экспери-
ментальных значений (см. гл. 8).
Б. ВВЕДЕНИЕ В СТРОГУЮ КИНЕТИЧЕСКУЮ
ТЕОРИЮ ГАЗОВ
Подробное классическое рассмотрение более строгой кинетической теории
разреженных одноатомных газов и смесей (теория Энскога—Чепмена) про-
всдится в гл. 7 (§ 1—4). Поскольку это изложение по необходимости весьма
длинно, то ему для тех читателей, которые в первую очередь интересуются прак-
тическим использованием формул и таблиц гл. 8, полезно предпослать краткое
изложение проблемы и метода рассмотрения, а также суммировать допущения
и предположения, используемые при более строгом выводе выражений для
коэффициентов переноса. Рассматривается также обоснованность полученных
результатов в применении к отдельным типам молекул в определенных
условиях (по температуре и давлению).
1. Краткий обзор вопроса и метода решения. Строгий вывод кинетической
теории газов основан на знании функции распределения /,(r, v?, t). Эта функция
представляет число молекул /-го сорта, которые в момент времени t находятся
в единичном элементе объема вблизи точки г и обладают скоростями внутри
единичного интервала значений вблизи v,. Если в газе нет градиентов кон-
центрации, скорости и температуры, то / (г, vh t) сводится к распределению
Максвелла /,.01 = п^т^лкТK!* ехр (— трЦ2кТ). Когда система не находится
в состоянии равновесия, то функцию распределения можно определить из
интегро-дифференциального уравнения Больцмана. Это уравнение и методы
решения его рассматриваются в гл. 7.
Обычно мы интересуемся свойствами газов, находящихся в условиях, лишь
немного отличающихся от равновесия. Действительно, только в этих условиях,
когда потоки линейны по отношению к производным, можно использовать
обычные определения коэффициентов переноса. В этом ограничении функция
распределения приближается к максвелловской, а уравнение Больцмана можно
решить с помощью метода теории возмущений, развитого Чепменом и Энскогом.
Получающиеся решения затем используются при выводе выражений для по-
токов и для коэффициентов переноса.
§ 2. Кинетическая теория газов. Коэффициенты переноса 3\
Эти выражения показывают, что перенос массы происходит не только
вследствие градиента концентрации, но и при наличии градиента температуры
(термодиффузияI). Подобно этому найдено, что перенос энергии происходит не
только вследствие наличия градиента температуры, но и вследствие градиента
концентрации (эффект ДюфураJ). Эти, а также другие явления второго порядка,
которые не могут быть описаны в рамках упрощенной кинетической теории,
естественно выясняются лишь при более строгом подходе к вопросу.
Конечным результатом теории является то, что мы можем выразить все
коэффициенты переноса через систему интегралов №1>S\ Эти интегралы вклю-
чают достаточно подробно всю динамику столкновения молекулы и, следова-
тельно, закон действия межмолекулярных сил. Неполнота знания в настоя-
щий момент свойств межмолекулярных сил до некоторой степени ограничивает
возможности применения получаемых результатов к практическим Задачам.
С другой стороны, знание зависимости между коэффициентами переноса и
межмолекулярными силами позволяет непосредственно получить некоторые
важные сведения о природе этих сил. Этот вопрос рассматривается подробно
в гл. 8, где проводятся вычисления коэффициентов переноса для реальных
газов при малой плотности.
2. Ограничения теории Энскога—Чепмена. Отмеченная выше кинетиче-
ская теория Энскога—Чепмена основана на различных предположениях,,
которые до некоторой степени ограничивают применимость получаемых резуль-
татов. Рассмотрим каждое ограничение отдельно и обсудим условия, в кото-
рых это ограничение становится существенным.
Так как в теории Энскога—Чепмена рассматриваются только парные
столкновения, то результаты ее не применимы при больших плотностях, когда
тройные столкновения начинают играть значительную роль. В настоящее время
для описания неравновесных свойств плотных газов наиболее употребительна
приближенная теория Энскога. С другой стороны, свойства жидкостей наи-
лучшим образом описываются теорией абсолютных скоростей реакций Эйринга.
Более строгая теория, основанная на использовании радиальной функции рас-
пределения, находится еще в состоянии разЕития. Эта теория, по-видимому,
когда-нибудь заменит теории Энскога и Эйринга. Для иллюстрации степени
влияния давления на коэффициенты переноса рассмотрим коэффициент вязкости.
Согласно теории разреженных газов, вязкость не зависит от давления при по-
стоянной температуре. Этот факт подтверждается экспериментально. Прове-
денные измерения показывают, что вязкость азота при 325° К возрастает в
2,5 раза, если давление увеличивается от 1 до 1ССО атм, тогда как рост состав-
ляет всего лишь 4%, если давление меняется от 1 до 60 атм. Эти результаты
находятся в удивительно хорошем согласии с теорией Энскога. Явления пере-
носа в плотных газах рассматриваются в гл. 9.
Использование классической механики исключает из рассмотрения явления
при низких температурах, когда становятся существенными квантовые эффекты.
Согласно квантовой теории, каждой молекуле можно приписать определенную
длину волны — «длину волны де Бройля» hjmv, — которая обратно пропорцио-
нальна молекулярному весу и скорости молекулы. Длина волны де Бройля,
соответствующая средней арифметической скорости, равна 27,4/У МТ А,
где М — молекулярный вес. При низких температурах средняя скорость моле-
кул мала и соответствующая длина волны де Бройля велика. Когда длина
волны де Бройля имеет порядок размеров молекулы, наблюдаются квантово-
1) Термодиффузия в жидкой фазе обычно называется эффектом Соре. Это явление было
открыто независимо Чепменом и Энскогом в результате точной формулировки кинетической
теории одноатомного газа. Только несколькими годами позже Дутсон дал экспериментальное
подтверждение этого явления [5].
2) Термодиффузия и эффект Дюфура тесно связаны друг с другом в соответствии с соот-
ношениями взаимности Онзагера (см. гл. 11, §2).
32 Гл. 1. Введение и основные понятия
механические «диффракционные эффекты», подобные диффракционным явле-
ниям в оптике. При еще более низких температурах или при больших плот-
ностях, когда длина волны де Бройля становится по порядку величины равной
среднему расстоянию между молекулами, оказываются существенными «стати-
стические эффекты», связанные с принципом Паули. Выше 200° К квантовые
эффекты составляют менее 1% даже для изотопов гелия и водорода. Ниже
этой температуры диффракционные эффекты для легких газов становятся весьма
существенными. Статистические эффекты важны только при температурах
ниже 2° К и проявляются в своеобразном поведении жидкого гелия. Указан-
ные квантовые явления рассмотрены в гл. 10.
Метод решения уравнения Больцмана, развитый Энскогом и Чепменом,
дает приближение к функции распределения в виде ряда. Первое приближение,
которое здесь и рассматривается, справедливо в условиях, когда градиенты
всех физических величин малы. Более высокие приближения дают поправки
для больших градиентов. В первом приближении потоки пропорциональны
первой производной от плотности, скорости и температуры; получающиеся
уравнения, которые описывают изменение плотности, скорости и температуры
со временем (уравнения переноса) называются уравнениями Навье—Стокса.
Высшие приближения дают члены, пропорциональные высшим производным
и степеням менее высоких производных от физических величин. Уравнения
переноса, соответствующие второму приближению, известны как уравнения
рарнетта. Количественно первое приближение справедливо тогда, когда от-
носительные изменения плотности, скорости и температуры на расстоянии
среднего свободного пробега малы по сравнению с единицей [т. е. /(Э In п/Эх)<^ 1
и т. д. ]. В соответствии с табл. 3 средняя длина свободного пробега в Газе при
давлении 1 атм имеет порядок 10~б см. Следовательно, при давлении 1 атм
или выше только в условиях экстремально больших градиентов, вроде тех,
которые имеют место в ударных волнах, появляются отклонения от этого усло-
вия. Проблема ударных волн рассматривается в гл. И. Поскольку средняя
длина свободного пробега обратно пропорциональна давлению, то отклонения
от уравнений Навье—Стокса происходят при низких давлениях даже в усло-
виях менее значительных градиентов. Как указывается в гл. 7, § 5, имеются
значительные сомнения как в сходимости рядов Энскога, так и в справедли-
вости метода. В последнее время были развиты более мощные методы, под-
тверждающие справедливость уравнений Навье—Стокса, но приводящие к
результатам, отличным от уравнений Барнетта и более высоких приближений.
В теории Энскога—Чепмена предполагается, что размеры объема, содер-
жащего газ, а также любых твердых тел внутри этого объема велики по срав-
нению со средней длиной свободного пробега, так что поверхностные слои зани-
мают пренебрежимо малую долю всего объема. При очень малых плотностях
молекулы чаще сталкиваются со стенками содержащего газ объема, чем друг
с другом; следовательно, в этом случае механизм установления локального
равновесия внутри самого газа мало эффективен. В таком газе понятие локаль-
ной плотности, скорости и температуры теряет смысл, и газ более не является
даже приближенно сплошным, а скорее проявляет свойства дискретной среды.
Газ в очень разреженных условиях известен как кнудсеновский газ, и поведение
такого газа достаточно хорошо изучено. Теория газа в диапазоне плотности
между кнудсеновским газом и газом средней плотности, когда применимы
уравнения Навье—Стокса, к настоящему времени развита еще недостаточно.
Строго говоря, кинетическая теория Энскога—Чепмена применима только
к одноатомным газам (к молекулам без внутренних степеней свободы, для
которых потенциал взаимодействия сферически симметричен). Неупругие
столкновения возможны между молекулами, обладающими внутренними сте-
пенями свободы. При этих столкновениях кинетическая энергия уже не сох-
раняется, тогда как количество массы и импульса, очевидно, сохраняется.
Поэтому вязкость и диффузия несущественно зависят от наличия внутренних
степеней свободы, и теория одноатомных газов может успешно применяться
§ 2. Кинетическая теория газов. Коэффициенты переноса 33
к многоатомным молекулами, если молекулы не слишком отличаются от сфе-
рических.
Однако коэффициент теплопроводности сильно зависит от присутствия
внутренних степеней свободы молекул, поскольку поток энергии включает
внутреннюю энергию, так же как и поступательную. С помощью упрощенной
кинетической теории (а также в выводах теории Энскога—Чепмена) мы полу-
чаем соотношение
А = ^> B.19)
связывающее теплопроводность с вязкостью [см. B.14)]. Для одноатомных
газов это соотношение подтверждается экспериментально, но для многоатом-
ных газов наблюдаются значительные отклонения. Для учета влияния внут-
ренних степеней свободы Эйкеном была предложена формула
<2-20>
которая находится в хорошем согласии с большим числом экспериментальных
результатов. Отклонения от этой формулы даются в гл. 7, § 6, и применение
ее обсуждается в гл. 8, § 2.
Когда сталкиваются молекулы, имеющие различную внутреннюю энер-
гию, вероятность перехода в другие внутренние состояния и угловое распре-
деление рассеяния описывается «вероятностью перехода, зависящей от угла».
Кинетическую теорию многоатомных молекул, по форме совершенно подобную
теории для одноатомных молекул, развивали Ван Чанг и Уленбек [б ], а также
де Бур (см. гл. 7, § б). Результаты выражаются через вероятность перехода,
зависящую от угла ; это удобнее, нежели выражать их непосредственно через
потенциальную функцию. Учтя таким образом внутренние переходы, соотно-
шение Эйкена можно получить как предел при больших вероятностях пере-
водов. В настоящее время эти переходы не могут быть рассчитаны теоретически,
за исключением простейших случаев. Однако некоторые сведения о ве-
роятностях переходов были получены из результатов по дисперсии звуковых
волн высокой частоты и в других явлениях, которые зависят от времени релак-
сации [7]. Время релаксации есть характеристическое время установления
равновесия между отдельными внутренними степенями свободы и поступа-
тельным движением. Время релаксации для удобства задается в виде числа
столкновений, необходимых для установления равновесия. Тогда для полу-
чения времени релаксации нужно умножить это число на среднее время между
столкновениями. В водороде, например, для установления равновесия между
вращательными степенями свободы и поступательным движением требуется
около 50 столкновений. Однако последние работы по уширению линий в микро-
волновой области спектра под действием давления, по-видимому, указывают
на то, что это число значительно меньше в случае молекул, обладающих боль-
шим моментом инерции. Обмен колебательной энергией, однако, протекает
значительно труднее. Найдено, что для трехатомных молекул, таких, как
С02, N20, COS и CS2, для установления колебательного равновесия необходимо
от 2500 до 50 000 столкновений.
В. УРАВНЕНИЯ ПЕРЕНОСА И ИХ ПРИМЕНЕНИЯ
Кинетическая теория разреженных и плотных газов позволяет получить
уравнения переноса. Эти уравнения являются дифференциальными уравне-
ниями, описывающими поведение концентрации, скорости потока и темпера-
туры как функции координат и времени. Эти уравнения вместе с термическим
уравнением состояния р = p(V,T) и калорическим уравнением состояния
U = U(V, T) образуют основу для изучения динамики жидкости. Применение
этих уравнений рассматривается в гл. 11. Это рассмотрение включает ряд
34 Гл. 1. Введение и основные понятия
вопросов, представляющих интерес в физике и химии : распространение звука,
распространение волн конечной амплитуды, распространение пламени, удар-
ные волны, детонация и течение газов в ракетах.
§ 3. МЕЖМОЛЕКУЛЯРНЫЕ СИЛЫ И ПОТЕНЦИАЛ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
В предшествующем рассмотрении уравнений состояния и явлений пере-
носа молекулы представлялись в виде твердых сфер. Однако хорошо известно,
что две молекулы притягиваются друг к другу, когда они удалены друг от
друга, и отталкиваются при сближении на достаточно близкое расстояние.
Сила взаимодействия F между двумя сферическими неполярными молекулами
является функцией межмолекулярного расстояния г. В большинстве случаев
удобнее пользоваться потенциальной энергией взаимодействия <р(г), нежели
силой взаимодействия F(r). Эти две функции связаны между собой простыми
соотношениями1)
<p(r)=JF(r)dr. C.1)
Всюду в этой книге мы будем описывать межмолекулярные силы с помощью
таких потенциальных функций.
Для неполярных молекул обычно употребляемой функцией межмолекуляр-
ной потенциальной энергии является потенциал F-12) Леннарда-Джонса
Параметры а и е, имеющие размерности длины и энергии соответственно,
являются постоянными, характеризующими химическую разновидность сталки-
вающихся молекул. На больших расстояниях (г > а) компонента притяжения
с отрицательной шестой степенью превалирует, и молекулы притягиваются
друг к другу с силой, обратно пропорциональной седьмой степени расстояния.
Этот тип сил достаточно точно описывает дипольное взаимодействие между
двумя неполярными молекулами. На малых расстояниях (г <^ а) превалирует
компонента отталкивания, обратно пропорциональная двенадцатой степени,
и молекулы отталкиваются друг от друга с силой, обратно пропорциональной
тринадцатой степени расстояния. Этот тип сил достаточно хорошо описывает
силы отталкивания между большинством типов молекул. При г = о потен-
циальная энергия равна нулю, и поэтому а есть расстояние наибольшего сбли-
жения двух молекул, сталкивающихся друг с другом с нулевой относительной
кинетической энергией. Параметр е является максимумом энергии притяжения
двух молекул, который имеет место на расстоянии г = 2 /«а.
Для полярных молекул наиболее широко используемым видом межмоле-
кулярной потенциальной энергии является потенциал Штокмайера. Эта по-
тенциальная функция межмолекулярного взаимодействия является суммой
функции F-12) Леннарда-Джонса и дополнительного члена, зависящего от угла,
учитывающего электростатическое взаимодействие двух диполей [см. C.33)].
В этой книге мы делаем упор на использование потенциала F-12) Леннарда-
Джонса для неполярных молекул и потенциала Штокмайера для полярных
молекул. С помощью этих двух потенциальных функций межмолекулярного
взаимодействия были рассчитаны многие свойства газов и жидкостей. Это по-
х) Эти соотношения справедливы для таких потенциальных функций и сил, которые
зависят только от межмолекулярного расстояния. Для потенциала, зависящего от углов,
сила, действующая на молекулу, равна — Fa = (д/дга)<раь; кроме того, существует момент,
стремящийся вращать молекулу. Динамика подобных столкновений описывается с помощью-
уравнений Лагранжа (см. § 4 настоящей главы).
§ 3. Межмолекулярные силы и потенциал межмолекулярного взаимодействия 35
зволяет описать большое число равновесных и неравновесных явлений на
общей основе и выяснить взаимосвязь между различными явлениями. Необ-
ходимо, однако, иметь в виду, что эти потенциальные функции идеализируют
действительный вид энергии межмолекулярного взаимодействия.
Потенциал F-12) Леннарда-Джонса и потенциал Штокмайера достаточно
адекватны для большинства простых молекул. Однако взаимодействие длин-
ных молекул, а также молекул в возбужденных состояниях, свободных ради-
калов и ионов нельзя описать с помощью этих двух потенциальных функций.
В гл. 13 и 14 подробно рассматривается природа этих взаимодействий.
В настоящем параграфе мы прежде всего укажем различные источники,
на которых базируются наши знания о межмолекулярных силах. Далее дается
краткий обзор некоторых общих типов взаимодействия, имеющих место между
атомами и молекулами в основных состояниях. Аналитический вид этих типов
взаимодействий дает основу для оценки и интерпретации эмпирических потен-
циальных функций, приведенных в конце настоящего йараграфа. Эти эмпири-
ческие функции используются в различных числовых расчетах во всей книге.
А. ИСТОЧНИКИ СВЕДЕНИЙ О МЕЖМОЛЕКУЛЯРНЫХ СИЛАХ
Природа межмолекулярных сил постигается как из экспериментальных
наблюдений, так и в результате теоретического рассмотрения. По теории пред-
лагается функциональный вид потенциала взаимодействия, а эксперимен-
тальные результаты используются для эмпирического определения соответ-
ствующих параметров потенциальной функции межмолекулярного взаимо-
действия. Эмпирический подход к рассмотрению межмолекулярных сил про-
водится в различных частях книги в связи со свойствами разреженных газов
(вторые вириальные коэффициенты — в гл. 3, коэффициенты переноса — в
гл. 8). Теоретический подход к межмолекулярным силам рассматривается в
трех последних главах.
В гл. 12 проводится обсуждение тех разделов электромагнитной теории,
которые важны для понимания межмолекулярных сил. Классическая электро-
статика позволяет понять взаимодействия между ионами, диполями и более
высокими мультиполями. Она дает также основу для рассмотрения поляри-
зуемости молекул, которая важна при изучении дисперсионных сил, дейст-
вующих между молекулами.
В гл. 13 рассматривается общая теория межмолекулярных сил. Большая
часть этой главы посвящена современному состоянию наших знаний в области
дальнодействующих сил, поскольку представляется возможность дать более
точное описание систем из двух молекул, находящихся на большом расстоянии
друг от друга. Показывается, каким образом дальнодействующие силы могут
быть выражены через дипольные моменты, квадрупольные моменты, поляри-
зуемости, характеристические частоты и вероятности оптических переходов.
В гл. 14 проводятся квантовомеханические расчеты межатомных и меж-
молекулярных сил для ряда простых взаимодействий : Н — Н, Н — Н2,
Н2 — Н2, Н — Не, Не — Не, Ne — Ne, Ar — Аг и некоторые другие. В допол-
нение к имеющимся сведениям о дальнодействующих силах эти расчеты по-
полняют наши теоретические знания о короткодействующих силах.
Для эмпирического определения межмолекулярных сил из макроскопи-
ческих величин необходимо, чтобы экспериментальные измерения были прове-
дены с достаточной точностью, а также чтобы существовало по возможности
полное теоретическое описание этих явлений. В силу последнего ограничения
наиболее полные сведения были получены из свойств разреженных газов и
свойств кристаллов. Зависимость последних от межмолекулярных сил явля-
ется наиболее прямой. Расстояние между сферическими неполярными моле-
кулами в кристалле при абсолютном нуле (исключая небольшие квантовые
поправки) точно равно расстоянию, на котором потенциальная энергия <р(г)
минимальна ; более того, энергия сублимации этих кристаллов при абсолют-
36 Гл. 1. Введение и основные понятия
ном нуле непосредственным образом связана (исключая небольшую поправку
на нулевую энергию кристалла) с минимальным значением ср(г).
Особое значение в этой книге придается определению параметров в раз-
личных эмпирических потенциальных функциях путем анализа свойств раз-
реженных газов. В соответствующих главах даются выражения для второго
вириального коэффициента и вязкости через межмолекулярную потенциальную
функцию <р(г). Если эти величины рассчитаны теоретически для некоторой
эмпирической потенциальной функции, то экспериментальная зависимость
этих величин от температуры может быть использована для расчета указанных
выше параметров. Вообще говоря, параметры, определенные из равновесных
свойств (второй вириальный коэффициент и коэффициент Джоуля—Томсона),
слегка отличаются от значений, определенных из неравновесных свойств
(вязкость, самодиффузия). Это не имело бы место, если бы использовалась
точная функциональная зависимость. Однако, как можно видеть из разных
выражений, различные свойства газа проявляются в выражении для потен-
циала по-разному. Обычно свойства переноса более всего связаны с частью
потенциальной функции, соответствующей отталкиванию, в то время как рав-
новесные свойства более связаны с притяжением.
Б. РАЗЛИЧНЫЕ СОСТАВЛЯЮЩИЕ МЕЖМОЛЕКУЛЯРНЫХ СИЛ1)
Перечислим кратко различные типы взаимодействия, возникающего между
ионами, атомами и молекулами. Эти сведения лягут в основу отдельных моделей
молекул (эмпирических потенциальных функций), которые приведены в конце
настоящего раздела. Это рассмотрение ограничено взаимодействиями между
молекулами в их основных состояниях2).
Удобно, хотя это и несколько произвольно, разделить межмолекулярные
силы на два типа — короткодействующие и дальнодействующие3). Короткодей-
ствующие силы часто называются валентными силами, или химическими силами;
они возникают тогда, когда молекулы настолько сближаются друг с другом,
что их электронные облака перекрываются. Эти силы по своей природе пред-
ставляют собой силы отталкивания и обладают часто ярко выраженным на-
правленным действием. Имеются отдельные экспериментальные указания на
природу короткодействующих межмолекулярных сил из свойств кристалли-
ческой структуры, поверхностных явлений и т. д., однако о межмолекулярных
силах известно гораздо меньше, чем о внутримолекулярных силах. Большая
часть наших сведений об этих силах получена из отдельных квантовомехани-
ческих расчетов, которые были проделаны для ряда молекулярных взаимодей-
ствий. Эти расчеты обычно не обладают достаточно высокой точностью, по-
скольку большинство встречающихся в расчетах интегралов настолько сложно,
что либо они берутся в приближенном виде, либо ими попросту пренебрегают.
С другой стороны, дальнодействующие силы можно рассматривать довольно
строго. К тому же для различных типов взаимодействия легко получаются
функциональные зависимости и формулы, применимые к разным видам моле-
кул. Рассмотрим кратко вначале короткодействующие силы, а затем проведем
более подробное рассмотрение дальнодействующих сил.
Составляющая межмолекулярного потенциала, связанная с коротко-
действующими, или валентными, силами <р<вал>, изменяется экспоненциально
х) Материал, приведенный в настоящем разделе, суммирует результаты, полученные
в гл. 12, § 1 (электростатические силы), в гл. 13, § 3 (дисперсионные силы) и § 5 (индукцион-
ные силы).
2) Взаимодействия между молекулами, находящимися в возбужденных состояниях,
детально рассматриваются в гл. 13, § 6. Там показано, что для такого рода взаимодействий
важную роль в потенциале взаимодействия играет соответствующая «резонансу)) часть <р(рез-).
3) Существуют силы, известные как обменные силы второго порядка, которые важны
для промежуточных расстояний. Эти силы рассматриваются в гл. 14, § 2 в связи с меж-
атомным потенциалом для гелия.
§ J. Межмолекулярные силы и потенциал межмолекулярного взаимодействия 37
в зависимости от расстояния. Действительный вид функции <р<вал-) достаточно
сложен и зависит от типа рассматриваемого взаимодействия. Обычно в боль-
шинстве приложений для приближенного выражения короткодействующей
составляющей используется простейшая форма
в которой а и Ь — постоянные. Постоянная а может быть приближенно выра-
жена в виде [8]
а = ^(ЩЩ+УЁ№), C.4)
где ?7A) и ?7B) — потенциалы ионизации соответственно двух молекул ;
а0 == 0,5292 — радиус первой боровской орбиты ; е — заряд электрона.
Различные составляющие дальнодействующих сил меняются обратно про-
порционально различным степеням межмолекулярного расстояния. Удобно
разделить дальнодействующие составляющие потенциала взаимодействия мо-
лекул на три части : 1) электростатическая составляющая <р<эс>, II) инду-
цированная составляющая ^(инд), HI) дисперсионная составляющая <р<дис>.
Рассмотрим общие свойства этих составляющих и укажем, в каких частях
книги дано более полное описание этих сил. Первые два типа взаимодействия
можно объяснить непосредственно с помощью электростатики, а третий тип —
с помощью квантовой механики.
1. Электростатическая составляющая. Электростатическая составляющая
межмолекулярной потенциальной энергии складывается из взаимодействий
различных мультипольных моментов молекул : зарядов (С), дипольных мо-
ментов (/гI), квадрупольных моментов (QJ) и т. д. Для получения аналитической
записи различных типов электростатического взаимодействия воспользуемся
переменными, определенными так, как это указано на фиг. 4.
Фа>Фь
Фиг. 4.
Точки а и Ь представляют центры двух молекул а и Ь. Пунктирная линия,
проходящая через а, является осью диполя и цилиндрически симметричного
квадруполя молекулы а, а пунктирная линия, проходящая через 6, указывает
на подобную ось второй молекулы. Межмолекулярное расстояние равно г;
углы вш вЪу фа, фь служат для определения ориентации обеих молекул.
г) Мы рассматриваем здесь только «идеальные диполи», т. е. диполи, для которых при
стремлении расстояния между зарядами к нулю величина зарядов возрастает таким образом,
что значение дипольного момента остается постоянным. Аналогичное ограничение имеет
место и в случае квадрупольных моментов. Другими словами, можно сказать, что размеры
мультиполя пренебрежимо малы по сравнению с межмолекулярными расстояниями. Взаи-
модействие между двумя действительными диполями рассматривается в гл. 12, § 1.
2) Квадрупольный момент молекулы в действительности является тензором второго
порядка с девятью компонентами. Если молекула обладает цилиндрической симметрией,
то этот тензор может быть представлен в виде единственной скалярной величины Q (см.
гл. 12, § 1). Формулы, рассматриваемые в этом параграфе, записываются с помощью вели-
чины Q и, следовательно, применимы только для тех молекул, распределение зарядов в
которых обладает цилиндрической симметрией.
38 Гл. 1. Введение и основные понятия
Непосредственное применение закона Кулона для электростатического взаимо-
действия позволяет получить следующие формулы для различных типов взаи-
модействия между молекулами а и Ь :
^ (з.б)
•Q) = + -^ (з cos* eb - i), C.7)
У& "> = - -^ [2 cos 0a cos eb - sin 0a sin eb cos (&- &)], C.8)
<PaVQ) = + -^ [coseeCcos«e, - 1) -
- 2 sin 0a sin db cos 0, cos @e - фъ)), C.9)
<?$'Q) = + -^Tg^- {1-5 cos2 0a - 5 cos2 0ft - 15 cos2 0a cos2 0ft +
+ 2[sin0asin0&cos(^a-^)-4cos0acos^]2}. C.10)
Более полное рассмотрение взаимодействия между зарядами, располо-
женными более сложным образом, дано в гл. 12, § 1. Приведенные формулы
выведены из более общих результатов, полученных в указанном параграфе, и
используются во многих местах этой книги.
Угловая зависимость приведенных выражений до некоторой степени
сложна. Для заданного значения межмолекулярного расстояния существуют
такие относительные ориентации молекул, для которых потенциальная энер-
гия является максимальной (99а&)макс или минимальной (<ра&)мин.. Иногда удобно
пользоваться эффективными сферически симметричными потенциальными функ-
циями1), записываемыми в виде
<РаЬ ехр (— (раь/кТ) do)a dcob
v = U . C-11)
ехр (— (fab/kT) doja dcob
где dco = sin в dQ йф. Здесь rab имеет фиксированное значение, а <раЬ усред-
няется по всем значениям углов. При этом усреднении больцмановский весовой
множитель ехр (—фаь1кТ) включается для того, чтобы учесть тот факт, что
молекулы со статистической точки зрения находятся наибольшую часть вре-
мени в таких положениях, для которых энергия мала. Физически использование
этого эффективного потенциала соответствует предположению, что таЪ несу-
щественно меняется в зависимости от вращения, испытываемого молекулой.
Для малых расстояний или низких температур, когда (<ра&)макс. — (^аь)мин. ^> кТ,
молекулы колеблются около положения минимальной энергии таким образом,
что сраЬ становится почти равным (<ра&)МИн.. Для больших расстояний, когда
(^аь)макс. — (^аь)мин. <^ кТ, больцмановский весовой множитель можно разложить
в ряд по степеням 1//сТ, и мы получаем (см. гл. 13, §5)
^•с> = +^, C.12)
г) Понятие статистического усреднения сраь введено Кеезомом [9], и это усредненное
взаимодействие между двумя диполями или мультиполями известно как усредненная энергия
взаимодействия Кеезома. Усредненная энергия взаимодействия между двумя квадруполями
выдвигалась Кеезомом в качестве возможного объяснения дальнодействующих сил притя-
жения между неполярными молекулами. Этот эффект, однако, пренебрежимо мал по срав-
нению с дисперсионными силами Лондона.
§ 3. Межмолекулярные силы и потенциал межмолекулярного взаимодействия 39
ф(С, Q)
\ _
20/сГ гв '
^(Q, О) _ 7 Qa Qb П]7\
rab — 40/сТ Г10 " \°'1'/
Необходимо отметить, что эти усредненные потенциальные функции, за
исключением $?С), зависят от температуры.
2. Индуцированная составляющая. Когда заряженная частица (например,
ион) взаимодействует с нейтральной молекулой, то заряженная частица а
индуцирует дипольный момент в нейтральной молекуле Ь таким образом,
Фиг. 5. Взаимодействие заряда а с индуцированным дипольным моментом
нейтральной мелекулы /?.
как это показано на фиг. 5. Если поляризуемость молекулы Ь равна аЬу то
дипольный момент, индуцированный в молекуле 6, равен Caabjr2, а энергия
взаимодействия между зарядом и этим наведенным моментом равна (см. гл.
13, §5)
9&HI№II) = -¦%?-¦ C.18)
Подобно этому можно показать, что потенциальная энергия взаимодействия
между точечным диполем и наведенным в нейтральной молекуле диполем
равна
у<?, инд.ц) = _ A*SaftCC0S2ea+l) ^ C.19)
Это является важным вкладом в потенциал взаимодействия между полярной
и неполярной молекулами. Последнее выражение можно усреднить по углам
так, как это было описано выше; в результате получаем
^ ' C.20)
Индуцированные силы подробно рассмотрены в гл. 13, § 5.
3. Дисперсионная составляющая1). Когда взаимодействуют две неполяр-
ные молекулы2), то между ними возникают дальнодействующие силы притя-
жения. Квантовомеханическая теория этих сил рассматривается в гл. 13, § 3.
Допустим, что в некоторый момент времени электроны молекулы а образуют
такое распределение, результатом которого является мгновенный дипольный
х) Этот тип сил отнесен к дисперсионным силам потому, что они могут быть выражены
через величину, называемую силой осциллятора, которая появляется в теории дисперсии
света (см. гл. 12, § 5 и б, и гл. 13, § 3).
2) Дебай [10] полагал, что притяжение двух неполярных молекул можно объяснить,
рассматривая тот факт, что квадрупольный момент одной молекулы индуцирует диполь в
другой молекуле. Это взаимодействие квадруполя с индуцированным диполем соответствует
потенциальной энергии, обратно пропорциональной восьмой степени межмолекулярного
расстояния. Однако было обнаружено, что эта энергия взаимодействия объясняет лишь
пренебрежимо малую долю наблюдаемой энергии. Фалькенгаген [И] применил эффект
индукции к дипольным газам, где это явление весьма существенно.
40
Гл. 1, Введение и основные понятия
момент. Этот мгновенный дипольный момент индуцирует диполь в молекуле Ь.
Индуцированный диполь молекулы Ь взаимодействует затем с мгновенным
диполем молекулы а и вызывает энергию взаимодействия между двумя моле-
кулами безотносительно к ориентации мгновенного диполя. Лондон развил это
представление с квантовомеханической точки зрения и нашел, что это взаи-
модействие, часто называемое индуцированным диполь-дипольным взаимодей-
ствием, приближенно описывается выражением1)
^-. C.21)
У"Ъ - 2\hva+hvb
в котором hva и hvb — характеристическая энергия соответственно двух моле-
кул, приближенно равная их потенциалу ионизации. Можно показать, что
следующие члены дисперсионной энергии — <р(дис'8>, <р(дис.,ю) и т. д. — изменя-
ются соответственно как г~8 (индуцированное диполь-квадрупольное взаи-
модействие), г~10 (индуцированное квадруполь-квадрупольное взаимодействие)
и т. д.
В. ЭМПИРИЧЕСКИЕ ПОТЕНЦИАЛЬНЫЕ ФУНКЦИИ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
При выборе формы межмолекулярного потенциала для расчета различных
величин необходимо иметь в виду два фактора : степень требуемого прибли-
жения к действительности и вычислительные трудности, связанные с исполь-
зованием той или иной функции. В этом разделе приводятся потенциальные
функции межмолекулярного взаимодействия, которые используются затем в
основном изложении, с указанием, какие именно свойства газа или жидкости
рассчитаны с помощью этого потенциала. Вычисления вириальных коэффи-
циентов и коэффициента Джоуля—Томсона описаны в гл. 3 (классические
a Ra
Фиг. б. Графическое представление некоторых сферически симметричных эмпирических
потенциальных функций
а — твердые сферы ; б — точечный центр отталкивания ; в — прямоугольная потенциальная яма ;
г—модель Сюзерленда ; д — потенциал Леннарда-Джонса; е — потенциал Букингема. Эти функции явля-
ются наиболее важными сферически симметричными потенциалами, употребляемыми в книге. Аналити-
ческие выражения для этих потенциалов даны в соотношениях C,22)—C.28).
расчеты) и в гл. 6 (квантовые расчеты). Вычисления коэффициентов переноса
даются в гл. 8 (классическая теория) и в гл. 10 (квантовая теория). Графиче-
ский вид различных потенциальных функций межмолекулярного взаимодей-
ствия приведен на фиг. б.
х) Этот результат] применим только к сферическим молекулам. Дисперсионные силы
между длинными молекулами дают значительно отличное от этого выражение; такие
силы рассмотрены в гл. 13, § 4.
§ 3. Межмолекулярные силы и потенциал межмолекулярного взаимодействия 41
Потенциалы, не зависящие от углов
1. Твердые непроницаемые сферы.
<p(r) = oo, r <а9
C.22)
ср (г) == 0, г > а.
Эта потенциальная функция представляет твердые непроницаемые сферы
диаметром а. Эта модель, часто используемая в вычислениях в силу ее про-
стоты, дает лишь грубое представление о сильных короткодействующих силах
отталкивания (т. е. о том, что молекулы обладают объемом). Для этого потен-
циала первые пять вириальных коэффициентов и коэффициенты переноса рас-
считывались классическим образом (а в некоторых случаях квантовомехани-
чески).
2. Точечный центр отталкивания*
<p(r) = dr6. C.23)
Величина д называется показателем отталкивания ; для большинства моле-
кул этот показатель имеет значение между 9 и 15 (когда 5 = 4, молекулы назы-
ваются максвелловскими). В том случае, когда необходима дифференцируемая
потенциальная функция, эта функция используется в расчетах. Она имеет
также то преимущество, что угол отклонения при столкновении выражается
через единственную переменную. Второй вириальный коэффициент и коэффи-
циенты переноса для этого потенциала были рассчитаны на основе как клас-
сической, так и квантовой теории.
3. Прямоугольная потенциальная яма.
<р(Г) = со, г О,
ср(г) = - е, а < г < Ray C.24)
<р(г) = 0, г > Ra.
Эта модель представляет твердую сферу диаметром <т, окруженную слоем с
полем притяжения величиной е; этот слой простирается до расстояния Ra.
Таким образом, в несколько грубом виде принимаются в расчет как сила оттал-
кивания, так и сила притяжения. Эта модель также используется в расчетах
и дает иногда хорошее согласие для сложных молекул, так как в нее входят
три произвольных параметра. Рассчитаны второй и третий вириальные коэф-
фициенты и коэффициенты переноса для потенциальной прямоугольной ямы.
4. Модель Сюзерленда.
C.25)
ср(г) = — сГ7, г > о.
Этот потенциал описывает твердые сферы диаметром а, которые притяги-
ваются друг к другу по закону обратной пропорциональности некоторому
значению степени расстояния. Это довольно реальная модель, достаточно про-
стая в обращении. Второй вириальный коэффициент и коэффициент переноса
для этой функции вычислены.
5. Потенциал Лен нарда-Джонса.
<Р(г) = 4 с~. C.26)
42 Гл. 1. Введение и основные понятия
Член Щг6 описывает энергию отталкивания, а член —с\гу энергию притя-
жения. В этой книге мы будем пользоваться следующим специальным видом
этого потенциала :
который часто называется потенциалом F-12) Леннарда-Джонса. Величина
а есть значение г, при котором <р(г) = 0. Величина е — максимальное значение
энергии притяжения (или глубина потенциальной ямы), которое достигается
при г = 21/«<7. Притяжение, обратно пропорциональное шестой степени, доста-
точно верно описывает индуцированное диполь-дипольное взаимодействие.
Составляющая потенциальной функции, связанная с отталкиванием, прибли-
женно выражается членом с отрицательным значением степени. Выбор значе-
ния 12 в качестве показателя в этом члене в первую очередь обусловлен мате-
матическими удобствами и не является единственно возможным; для некото-
рых веществ могут оказаться более подходящими другие значения. Эта функция
дает достаточно простое и реальное представление о взаимодействии между
сферическими неполярными молекулами. Многие расчеты были проделаны
с помощью этой потенциальной функции — второй и третий вириальный коэф-
фициенты и коэффициенты переноса в классической теории и обширные вычи-
сления квантовых эффектов.
6. Потенциал Букингема.
<р(г) = be~ar - сг~6 — с'г~8. C.28)
Эта функция с четырьмя параметрами учитывает индуцированное диполь-
дипольное взаимодействие, индуцированное диполь-квадрупольное взаимодей-
ствие, а для составляющей потенциала, связанной с отталкиванием, дает при-
ближение в виде экспоненциального члена. Таким образом, эта модель ближе к
действительности, нежели модель Леннарда-Джонса, но более сложна при
численных расчетах. Некоторые расчеты уравнений состояния проделывались
с помощью этого потенциала, однако для вычислений коэффициентов переноса
он не использовался. Этот потенциал дает нереальные значения вблизи нуля,
где он стремится к—оо. Для описания некоторых свойств газов эта область
задания потенциала не играет существенной роли.
7. Потенциал Букингема—Корнера.
[~4(-^--l)8],r^rm, C.29)
<p(r) = b exp [- a (JL)] - (cr"e + cV~8), r ^ rmy
где
Эта потенциальная функция зависит от четырех параметров: е — глубины
потенциальной ямы ; rm — значения г для минимума энергии ; a — крутизны
экспоненциального отталкивания, чаще всего равной 13,5; /? = c'r^lcr^ —
отношения составляющих, обратно пропорциональных восьмой и шестой сте-
пеням в точке минимума потенциала.
Потенциальная функция Букингема—Корнера учитывает индуцированные
диполь-дипольное и диполь-квадрупольное взаимодействия и описывает от-
талкивание экспоненциального типа. (Дополнительное усложнение было
§ 3. Межмолекулярные силы и потенциал межмолекулярного взаимодействия 43
введено главным образом для компенсации нереальных значений простого по-
тенциала Букингема вблизи начала координат.) Наличие четырех параметров
позволяет получить хорошее согласие с экспериментом; функциональная
запись потенциала, однако, слишком сложна для вычисления. Для этой модели
вычислен второй вириальный коэффициент и первая квантовая поправка к
нему.
8. Модифицированный потенциал F-ехр) Букингема.
^' макс>
C.30)
где Гмакс. —значение г, при котором <р(г), заданное в виде верхнего соотношения,
имеет относительный максимум. Отношение гМакс./гт0ПРеДеляется наименьшим
корнем трансцендентного уравнения
= 1. C.31)
Входящие в выражение для потенциала постоянные е, гт и а имеют то
же значение, что и параметры потенциала Букингема—Корнера. Имеются
числовые таблицы как для второго вириального коэффициента, так и для
интеграла столкновений.
Модифицированный потенциал F-ехр) Букингема несколько более
удобен, нежели потенциал F-12) Леннарда-Джонса, так как он позволяет
варьировать значение диаметра столкновения а (при малой скорости столк-
новения) по сравнению с расстоянием для минимума гт [для потенциала F-12)
Таблица 5
а
12,0
12,5
13,0
13,5
14,0
14,5
15,0
0,87610
0,87983
0,88320
0,88627
0,88910
0,89173
0,89417
гмакс./г»»
0,30247
0,27304
0,24697
0,22382
0,20319
0,18476
0,16825
* Смаке.)/6
1705
• 3 518
7 110
14 115
27 585
53 170
101 222
Леннарда-Джонса о\гт = 0,8909 ]. Энергия относительного максимума <р(гМакс)
обычно настолько велика, что это не приводит к затруднениям. Эти обстоя-
тельства становятся очевидными после рассмотрения табл. 5. Член, учиты-
вающий наведенное диполь-квадрупольное взаимодействие, не входит в эту
потенциальную функцию, так как его влияние может быть достаточно хорошо
учтено небольшим изменением значения параметра а.
Потенциалы, зависящие от углов
9. Твердые эллипсоиды вращения. Эта модель представляет твердый
непроницаемый эллипсоид вращения. Она может быть использована для изу-
чения влияния несферических потенциальных полей на физические свойства
44 Гл. 7. Введение и основные понятия
вещества. Вытянутые эллипсоиды представляют удлиненные молекулы, а
сплющенные эллипсоиды — плоские (дискообразные) молекулы. Для этой
модели рассчитан второй вириальный коэффициент, а также проведены неко-
торые расчеты в кинетической теории эллипсоидальных молекул.
10. Сфероцилиндрические молекулы (Кихара). Длинные молекулы пред-
ставлены для этой модели в виде цилиндров, оканчивающихся полусфе-
рами. Для таких молекул проведены вычисления второго вириального
коэффициента.
11. Твердые сферы, содержащие точечный диполь (Кеезом).
V (г, 0а, вЬу фь -^ = 00, r < а, C.32)
<Кг, вау еь, фь -фа) = - -*^4<ев, оь, фь - фа\
где
g (ва, вь, Фь -Фа) = 2 cos ва cos Ob - sin ва sin вь cos {фь - фа).
Эта модель представляет твердые непроницаемые сферы диаметром а, обла-
дающие точечным диполем с моментом /г. Эта потенциальная функция вклю-
чает короткодействующие силы отталкивания и диполь-дипольное взаимодей-
ствие. Она успешно применяется в расчетах; для этой модели вычислен вто-
рой вириальный коэффициент.
12. Потенциал Штокмайера.
«г, Оау вЬу фь - фа) = 4е [(-f У - [±)] - J±gL g@fl, Ob9 фь - фа\ C.33)
ГДе &Фау Оь> Фь — Фа) — множитель, входящий в выражение C.32) для диполь-
дипольного взаимодействия. Эта потенциальная функция является суперпо-
зицией потенциала F-12) Леннарда-Джонса и взаимодействия двух точечных
диполей. Она достаточно хорошо описывает взаимодействие между такими
полярными молекулами, для которых диполь-квадрупольное взаимодействие
и взаимодействия более высокого порядка не играют существенной роли. Для
этой модели сосчитаны второй и третий вириальные коэффициенты. Однако
расчет коэффициентов переноса для этой потенциальной функции оказался
затруднительным.
§4. КЛАССИЧЕСКАЯ МЕХАНИКА1)
Статистическая механика, как и кинетическая теория газов, является
прямым приложением законов движения. Поэтому большая часть рассужде-
ний в этой книге будет основана на формальных результатах настоящего пара-
графа. Классическая механика обычно дает адекватное описание движения
молекул, за исключением области низких температур, где приобретают важ-
ное значение квантовые эффекты.
Для явлений, обсуждаемых в этой книге, нет необходимости рассматри-
вать релятивистские эффекты. Мы начнем этот параграф с рассмотрения клас-
сических уравнений движения. В заключение будут обсуждены две теоремы,
обычно используемые в статистической механике и кинетической теории, —
теорема Лиувилля и теорема вириала.
1)См. [12—14].
§ 4* Классическая механика 45
А. УРАВНЕНИЯ ДВИЖЕНИЯ КЛАССИЧЕСКОЙ МЕХАНИКИ
Основу классической механики составляет ряд законов Ньютона. Мы
ограничимся здесь рассмотрением второго закона, который утверждает, что1)
F/ = m,-^, D.1)
где F, — сила, действующая 'на r-ю частицу системы ; /я, и г, — соответственно
масса и радиус-вектор (в декартовой системе координат) z-й частицы. В клас-
сической механике динамическое поведение системы частиц вполне опреде-
ляется законами Ньютона, если в некоторый начальный момент времени t0
положение г, и скорости Т) =*= dXi\dt каждой частицы известны. При рассмот-
рении определенных динамических систем и при решении определенных фор-
мальных задач удобно пользоваться законами Ньютона в одном из следующих
видов.
1. Уравнения Лагранжа. Многие задачи значительно удобнее формули-
руются при использовании в качестве переменных углов, или сложных функ-
ций всех координат различных частиц, вместо декартовых координат отдель-
ных частиц. Эти новые координаты называются обобщенными координатами
Як(х1у Yv zi у • • • > хт Уп, 2п) и обычно определяются в виде явных функций от
первоначальных декартовых координат2). Уравнения движения Лагранжа —
это преобразованнв1е к обобщенной системе координат уравнения Ньютона.
Функция Лагранжа, или лагранжиан системы, L(qv ..., q3N; qv ..., qzN)
определяется выражением
L-K — Ф, D.2)
где Ф — потенциальная энергия всей системы3), а К — кинетическая энергия
системы, причем К определяется как сумма кинетических энергий отдельных
частиц выражением
К = Т^т'г1- D.3)
Второй закон Ньютона^ выраженный через функцию Лагранжа, для кон-
сервативных систем4) может быть записан в виде
х) Не обязательно записывать закон Ньютона в виде F/ = -т- (пц —fr\, коль скоро массы
отдельных частиц, за* исключением релятивистских поправок, остаются постоянными.
2) Простым примером обобщенных координат являются сферические координаты г, 0, ц>у
часто используемые вместо декартовых координат х, у, z в задачах, связанных со сферической
симметрией.
3) В задачах, рассматривающих случаи многих частиц, обычно предполагают, что
Это предположение аддитивности, или предположение о парности взаимодействия, обсуж-
дается в гл. 3, § 4.
4) Консервативной системой называется такая система, в которой силы зависят только
от конфигурации системы й выполняется следующее условие: если все частицы, за исклю-
чением одной, закреплены, то интеграл тангенциальной компоненты силы, действующей
на отдельную частицу по замкнутому пути этой частицы, равен нулю. Для таких систем
силы могут быть выражены через единый потенциал всей системы Ф (rw):
F/ - - — Ф
причем N — число частиц в рассматриваемой системе. Часто можно найти функцию Лаг-
ранжа и для неконсервативных систем, так что еще можно применять уравнения Лагранжа.
Например, это возможно в случае движения заряженной частицы в магнитном поле.
46 Гл. 1. Введение и основные понятия
Это — уравнения движенияЛагранжа.Функция дЦд^известткакобобщенный
импульс рк
Р
Легко видеть, что если обобщенными координатами являются декартовы коор-
динаты, то обобщенный импульс сводится к обычному, а уравнение движе-
ния — к уравнению движения Ньютона. Если новыми координатами являются
цилиндрические (q, z, ф\ то рф — обычный момент количества движения1).
Если ими являются, скажем, эйлеровы углы твердого тела, то интерпретация
величины рк уже не столь проста [15]. Уравнения Лагранжа, так же как и
уравнения Ньютона, являются дифференциальными уравнениями второго
порядка ; решения этих уравнений определяются полным набором значений
координат и скоростей в некоторый момент времени.
2. Уравнения Гамильтона. Функция Гамильтона, или гамильтониан
системы, H(qv..., qzN ; pl9..., pzN) определяется соотношением
Важно иметь в виду, что лагранжиан является функцией только координат
и их производных, тогда как гамильтониан — функция координат и соответ-
ствующих импульсов.
В написанное выше выражение гамильтониана переменная qk входит как
явно, так и неявно (через лагранжиан), однако решая выражение D.5), можно
получить qk = qk(q» A)-
Имея это в виду, можно, продифференцировав выражение D.6) по р{ и qh
получить
дН . , v* - Qqi ^ dqj dL
—
Эр/
7 Pj tyi T bpt dqj >
bqi ~ -f- Pi bqt bqt <f dqj dqj '
Эти соотношения можно упростить, воспользовавшись уравнениями Лагранжа
D.4) и определением импульса D.5), и получить
Это—(канонические) уравнения движения Гамильтона. Теперь 3N уравнений
второго порядка Ньютона и Лагранжа преобразованы в 6N дифференциальных
уравнений первого порядка.
Легко можно показать, что гамильтониан системы численно равен полной
нергии. Из определений гамильтониана D.6) и импульса D.5) следует
H = 24,-%-t-L = 24,%-L, D.11)
причем второе выражение правой части D.11) возможно написать потому, что
потенциальная энергия не является функцией скоростей. Поскольку К —
однородная функция второго порядка относительно скоростей, то из теоремы
Эйлера следует2)
241щ: = 2К. D.12)
х) Анализ движения частицы, описываемого сферическими координатами, содержится
в книге Полинга и Вильсона [15].
2) Теорема Эйлера утверждает : если и(хъ х2, ..., хп) — однородная функция степени т
и имеет непрерывные первые производные, то 2х((ди/дх0 = ти. См., например, [17].
§ 4. Классическая механика 47
Таким образом,
И = 2К — L = 2К — (К — Ф) = К + Ф D.13)
и вдоль динамической траектории функция Гамильтона равна константе Е
энергии. Необходимо иметь в виду, что это справедливо только для консер-
вативных систем.
3. Уравнение Гамильтона—Я коби. Можно определить функцию
)
= 2 j Kdt9 D.14)
j
По динамической
траектории
которая является частью действия1), не содержащей явно времени. Из это га
определения вытекает следующее соотношение для обобщенных импульсов и
координат:
Подставляя это выражение в гамильтониан, получаем уравнение Гамильтона—
Якоби
( 1МЬ|) ?- DЛ6)
Это уравнение интересно тем, что оно очень похоже на уравнение Шредин-
гера в квантовой механике.
Б. ТЕОРЕМА ЛИУВИЛЛЯ
Вообразим, что все N частиц образуют отдельную механическую систему.
Для представления изменений состояния системы можно использовать понятие
фазового пространства. Фазовое пространство—это декартово 6ЛГ-мерное про-
странство, координатами которого являются 3N обобщенных конфигурацион-
ных координат qt и 3N соответствующих компонент р{ импульса. Динамиче-
ское состояние системы полностью описывается точкой в фазовом пространстве.
Так как прошлое и будущее системы определяется ее состоянием в настоящий
момент, то точка в фазовом пространстве определяет траекторию однозначно.
С изменением времени изображающая точка системы будет прочерчивать
траекторию в фазовом пространстве. Это движение, называемое естественным
движением фазовой точки, описывается каноническими уравнениями Гамиль-
тона.
Рассмотрим теперь свойство системы F = F(qiy ph t), зависящее от дина-
мического состояния системы и, вообще говоря, явно от времени. Такие свой-
ства системы могут быть интегралами движения, как, например, полная энер-
гия, полное количество движения системы, или полный момент количества
движения. Это могут быть также зависящие от времени характеристики си-
стемы, как, например, количество движения, энергия или положение отдельной
частицы в системе.
г) «Действие» определяется в виде
W @ = JLdt = ^2Kdt-Et.
о о
Реальное движение системы характеризуется тем, что «действие» имеет минимальное зна-
чение. Это положение известно как «принцип наименьшего действия Гамильтона».
48 Гл. 1. Введение и основные понятия
Изменение F во времени, следующее за точкой вдоль естественной траек-
тории, можно представить следующим образом:
DF dF S?(dF • , dF Л /ллн\
+ I9 + g) DЛ7)
?LL+y(LlL-LL\ am
Dt ~ dt tf U/ dpi dPi dqi)' ^'1O'
??-=*?-+ [F,H]. D.19)
В выражении D.18) использованы канонические уравнения Гамильтона, а в
выражении D.19) введено обозначение скобки Пуассона. Интеграл движения
системы является функцией координат и импульсов, которая остается постоян-
ной вдоль каждой естественной траектории в фазовом пространстве. Таким
образом, для интеграла движения а можно написать выражение
[а, Н] = 0. D.20)
Эти представления играют важную роль в постулатах квантовой механики и
используются в статистической механике.
Если мы рассмотрим большое собрание невзаимодействующих систем,
отличающихся друг от друга только их начальными условиями, то состояние
всего ансамбля систем будет изображаться набором точек в фазовом про-
странстве.
При достаточно большом числе систем этот набор изображающих точек
может быть описан непрерывной функцией распределения1) q{qb ph f), назы-
ваемой обычно плотностью в фазовом пространстве. Выбор функции плот-
ности произволен в начальный момент, но в любой последующий момент она
определяется уравнениями движения. Так как отсутствуют «источники» или
«стоки» фазовых точек, то функция распределения удовлетворяет обобщенному
уравнению неразрывности :
D4МО- D21)
Отсюда, используя канонические уравнения Гамильтона и определение скобки
Пуассона, мы получаем уравнение Лиувилля:
Bj-=*L + [6,H]=O. D.22)
Этот результат играет важную роль в статистической механике ; более широко
его применение будет обсуждено в гл. 2.
В. ТЕОРЕМА ВИРИАЛА
Теорема вириала Клаузиуса определяет отношение средней кинетиче-
ской к средней потенциальной энергии механической системы. Эта теорема
справедлива как в квантовой, так и в классической механике. Она оказывается
мощным средством при определении межмолекулярных сил и используется
непосредственно для вывода уравнения состояния.
В классической механике [19, 20] движение частицы массы т{ в направ-
лении оси х под действием силы F, описывается вторым законом Ньютона
D.23)
г) Более подробный вывод уравнения Лиувилля содержится в книге [18].
§ 4, Классическая механика 49
Умножая обе части выражения на х,/2, получаем
1 /с,ч 1 d2Xi I / dxi\2 . 1 d ( dx(
При усреднении по достаточно большому промежутку времени г последний
член исчезает:
d ( dxi\ I f d
О
так как хг(Лс,/<Й) остается ограниченной величиной (если только частица за-
ключена в конечной области пространства), в то время как т становится сколь
угодно большим.
Таким образом, усредняя D.24) по времени, получаем
D.26)
Аналогичные выражения могут быть получены для случаев движения в на-
правлениях у и z. Сложив выражения типа D.26), получим
^s-Vifo-F/^/C,, D.27)
где Kt — среднее значение кинетической энергии частицы. Величина, стоящая
в левой части выражения St = —^OvF/X названа Клаузиусом вириалом
сил. Так как это выражение верно для любой частицы, то оно применимо к
любым системам частиц
ly = ?; D.28)
здесь К = 2 Kt — полная средняя кинетическая энергия, а Я = 2J ~/ — пол"
ный вириал системы.
Если рассматривать только консервативные системы, то теорема вириала
может быть записана в терминах потенциальных энергий. Если, в частности,
потенциальная энергия является однородной функцией координат степени п,
то вириал сил, используя теорему Эйлера1), можно записать в виде
пФ
-1 ^ (г, • F,) = + 4 ^ (*. • -щ *«")) = т пФ. D.29)
Таким образом,
±пФ = К. D.30)
Существуют два особо важных случая потенциалов такого рода. __
1) Простой гармонический осциллятор: ср =х/2/сх2, п = 2 и Ф = К. Это
единственный случай, когда энергия равномерно распределена между кине-
тической и потенциальной. _ __
2) Кулоновское взаимодействие2): ц> = e2fr, п = — 1 и Ф = —2К. Для
молекул, описываемых потенциалом взаимодействия типа Леннарда-Джонса
г) См. примечание к выражению D.12), стр. 46.
2) Этот случай подробно рассмотрел Шоттки [21].
50 Гл. 1. Введение и основные понятия
где
из теоремы вириала следует
В последнем параграфе этой главы будет рассмотрен вывод теоремы вириала
в квантовой механике. В гл. 3 классическая теорема вириала будет исполь-
зован^ для получения уравнения состояния в вириальной форме.
§ 5. СТОЛКНОВЕНИЯ МОЛЕКУЛ В КЛАССИЧЕСКОЙ МЕХАНИКЕ
(ОДНОАТОМНЫЕ МОЛЕКУЛЫ)
Как уже упоминалось, строгое развитие кинетической теории зависит
от знания природы процесса столкновения, имеющего место при взаимодей-
ствии двух одноатомных молекул. В этом параграфе будут рассматриваться
адиабатические столкновения двух частиц, потенциал взаимодействия которых
является функцией только расстояния между ними1).
Рассмотрим вначале такие характеристики столкновения, которые связаны
с движением системы двух частиц как целого. Такое рассмотрение не требует
детального описания движения отдельных частиц. Затем будут рассмотрены
траектории отдельных молекул и выведено выражение для угла отклонения
налетающей частицы. Угол отклонения — это единственная характеристика
процесса столкновения, которая используется при расчетах явления переноса.
А. СУММАТОРНЫЕ ИНВАРИАНТЫ СТОЛКНОВЕНИЯ
Предположим, что сталкивающаяся система состоит из двух молекул; одна —¦
сорта i с массой т„ а другая — сорта / с массой т;. Эти частицы действуют
друг на друга с силой, зависящей только от расстояния между ними. Пусть
скорости молекул перед столкновением (т. е. перед тем, как взаимодействие
сталкивающихся частиц станет значительным) будут соответственно V, и vy.
Соответствующие скорости после столкновения обозначим через vj и yj. За-
коны сохранения массы, импульса и энергии системы как целого определяют
величины, известные как сумматорные инварианты; они будут использоваться
в гл. 7.
Если при столкновении молекул i и / не происходит химической реакции,
то, конечно, в любой момент во время столкновения массы индивидуальных
частиц не изменяются. В частности, это справедливо до и после столкновения ;
можно записать
rrii = m'i9 ruj = m'j. E.1)
Таким образом,
mt + nij = m't + m'j. E.2)
Выражения E.1) и E.2) являются следствиями закона сохранения массы как
для отдельных частиц, так и для системы в целом.
*) Для каждого /с-го электронного состояния системы двух сталкивающихся молекул
существует своя потенциальная функция <рк(г). Адиабатическое столкновение — это такое
столкновение, при котором электронное состояние остается неизменным. Неадиабатическими
называют такие столкновения, при которых система с начальным электронным состоянием
и потенциалом <рк(г) в результате столкновения переходит в другое электронное состояние
с соответственно изме*яющейся потенциальной функцией. Такие неадиабатические столк-
новения происходят между молекулами, находящимися в возбужденных состояниях, или
при столкновении очень быстрых частиц. Этот вопрос обсуждается в гл. 13, § 1.
§ 5. Столкновения молекул в классической механике (одноатомные молекулы) 51
Если система сталкивающихся частиц в целом не подвергается действию
внешних сил, то можно путем интегрирования выражения второго закона Нью-
тона по времени показать, что импульс системы постоянен. Это означает, что в
любой момент во время столкновения сумма количества движения отдельных
частиц остается постоянной. В частности, поскольку это справедливо до и
после столкновения, то можно записать
т. V/ + mj \j = mi v{ + rrij v). E.3)
Это выражение является следствием закона сохранения импульса для
системы, рассматриваемой как целое.
Если система не подвергается действию внешних сил, то ее полная энергия
остается постоянной. Следовательно, в любой момент столкновения сумма
кинетической и потенциальной энергии сталкивающихся частиц и энергии,
обусловленной взаимодействием частиц, остается постоянной. До и после
столкновения энергия системы равна просто сумме кинетической и потенциаль-
ной энергии отдельных частиц. Следовательно, можно записать
i- m, vf +1 mj v] = ±mt v? + \ rrij vf, E.4)
что является выражением закона сохранения кинетической энергии.
Все приведенные выше выражения законов сохранения для системы стал-
кивающихся частиц записаны в следующей форме :
Wi + V] = Vi + Vy > E-5)
где \pt — соответственно mh компоненты myt и х/а т$. Более того, можно пока-
зать, что любая функция скоростей, удовлетворяющая соотношению E.5),
является линейной комбинацией этих величин. Величины у,- называют сум-
маторными инвариантами.
Б. ТРАЕКТОРИИ ЧАСТИЦ ПРИ СТОЛКНОВЕНИИ
Рассмотрим теперь динамическую задачу о столкновении двух частиц в
трехмерном пространстве. Проведем это рассмотрение следующим образом :
сначала сведем задачу к плоской задаче двух тел, затем к плоской задаче о
движении отдельного тела и в конце концов к задаче об одномерном движении
отдельной гипотетической частицы. Каждое последовательное упрощение ведет
к менее точному описанию движения. Однако в целях достижения более про-
стых средств наглядного представления задачи вполне удобно поступиться
полнотой описания.
Построить в нашем воображении или на страницах книги траектории
сталкивающихся частиц при произвольном потенциале взаимодействия не-
сколько затруднительно. Поэтому целью настоящего обсуждения является
достижение лучшего понимания процесса столкновения в классической
механике и получение формулы для «угла отклонения», входящего в формулы
для коэффициентов переноса в гл. 7 и 8.
Начнем описание динамики столкновения с записи закона движения
для обеих частиц:
^ E.6)
; E.7)
В этих выражениях F, и Fy — силы, действующие соответственно на молекулы
/и/1; г, и tj — радиусы-векторы, определяющие положение этих молекул.
Равенство Fy = —F, в выражении E.7) указывает на то, что единственная
сила, действующая на молекулу г, обусловлена присутствием молекулы /, и
наоборот. Если E.6) умножить на mjy а E.7) — на т, и вычесть их друг из друга,
52
Гл. 1. Введение и основные понятия
то получим
E.8)
где /г — приведенная масса для сталкивающихся молекул, определяемая
выражением1)
Если обе части E.8) векторно умножить на (г, — гу), то правая часть выпадает
(поскольку предполагается, что межмолекулярные силы действуют вдоль
линии центров2), а векторное произведение коллинеарных векторов равно
нулю) и, следовательно,
^ ] E.10)
/-',) х ^ (г,-- г,)] =0.
Последнее выражение можно записать в виде
-[-ar<r'-|>) х
<5Л1>
Второй член обращается в нуль в силу коллинеарности векторов ; интегрируя
оставшийся член по времени, получаем
[(г, - г,) х ± (г, - tj)] = [(г, - tj) х (V,- - v,)] = К, E.12)
где К — постоянный вектор (не зависящий от времени), перпендикулярный
плоскости, в которой лежат векторы (г,- — гу) и (v,—у/). В каждый момент
времени обе частицы и центр их масс находится в плоскости, перпендикуляр-
ной вектору К. На фиг. 7 графически изображено столкновение двух частиц.
Молекула i
W
Фиг. 7. Схематическое пространственное изображение парного столкновения.
Видно, что сталкивающиеся частицы все время расположены в плоскости (ориентация которой в простран-
стве не изменяется), движущейся с постоянной скоростью центра масс. Изображенные плоскости являются
как бы мгновенными снимками частиц, сделанными через равные промежутки времени. Молекула / дви-
жется вначале со скоростью vt в отрицательном направлении оси у. Молекула j вначале движется со ско-
ростью Vj вдоль линии W, лежащей в плоскости XY. Проекция траектории центра масс на плоскости XY
представлена линией СС. Если все изображенные плоскости совместить, то получим фиг. 8.
2) Необходимо заметить, что для сталкивающихся частиц одинаковой массы т при-
веденная масса равна (т/2).
2) Для дипольных, сигарообразных молекул, а также молекул в возбужденных со-
стояниях силы не будут действовать вдоль линии центров. Настоящий анализ применим
только в случае неполярных сферических молекул, для которых потенциал взаимодейст-
вия зависит только от расстояния между ними.
§ 5. Столкновения молекул в классической механике (одноатомные молекулы) 53
Из фигуры видно, что обе частицы все время находятся в плоскости, ориен-
тация которой в пространстве остается неизменной и движущейся с постоянной
скоростью центра масс.
Тот факт, что столкновение происходит в плоскости, движущейся вместе
с центром тяжести системы, позволяет свести задачу к двухмерной. Наглядно
это можно представить совмещением всех плоскостей фиг. 7 в одну плоскость.
Это и представлено на фиг. 8. Аналитически мы просто выбираем систему коор-
динат, движущуюся вместе с центром тяжести и осью z, направленной по К.
Для удобства начало координат может быть помещено в центр масс системы.
I Молекула J*^-~ ^Г^-'Д -~'
-Молекула i
Плоскость XY
v
Фиг. 8. Схематическое плоское изображение парного столкновения.
Это изображение получено путем совмещения всех плоскостей фиг. 7. Здесь г — расстояние между сталкиваю-
щимися молекулами ; 0 — угол, определяющий направление (полярный угол); вт — угол, соответствую-
щий наибольшему сближению частиц; b — прицельное расстояние ; % — угол отклонения.
В такой системе координат кинетическая энергия системы двух частиц
относительно центра их масс может быть записана в виде
К = \
у]).
E.13)
Это выражение можно переписать, используя переменные г и 0, где г — меж-
молекулярное расстояние, а в — угол наклона по отношению к положительной
оси х (в изменяется от О до 2тг, а г — от О до оо). Так как начало координат со-
впадает с центром масс, то координаты xb yn xJy yj можно выразить через гид
следующим образом :
E.14)
E.15)
E.16)
у, = ^—г sine. E.17)
'J mi -f- mj v
Подстановка этих выражений в выражение E.13) приводит к следующему:
E.18)
54
Гл. 7. Введение и основные понятия
Этого соотношения и знания потенциальной энергии взаимодействия <р(г) до-
статочно для изучения задачи о столкновении. Интересно заметить, что точно
такая же информация потребовалась бы для решения задачи о двухмерном
движении одной частицы массы /г в сферически симметричном потенциальном
Фиг. 9. Схематическое изображение, соответствующее задаче о движении тела в потен-
циальном поле, эквивалентной в динамическом отношении задаче о столкновении двух частиц.
Величины г, в, ет, b и х имеют тот же смысл, что и на фиг. 8.
поле (р(г). Поэтому проблема столкновения обычно обсуждается в форме экви-
валентной задачи о движении одного тела. На фиг. 9 дано схематическое изо-
бражение соответствующего явления. Для получения уравнений движения в
эквивалентном случае движения одного тела в потенциальном поле (как и в
случае задачи двух тел) мы просто запишем уравнения сохранения энергии и
импульса. Приравнивая импульс и энергию, соответствующие величине г,
большей эффективного радиуса действия потенциала, их значениям в произ-
вольной точке траектории, получаем два соотношения:
iir*Q E.19)
у №2 = у М'2 + г2 в2) + <р(г).
E.20)
Здесь b — прицельное расстояние (расстояние наибольшего сближения при
отсутствии взаимодействия), g — от-
Полная энергия носительная начальная скорость стал-
сталкивающейсягшры^Чгрд*_ кивающихся молекул. Соотношения
^ -- E.19) и E.20) представляют собой
уравнения движения системы и дают
полное описание движения в терми-
нах потенциала взаимодействия и
двух характеризующих столкновение
параметров.
Графическое представление про-
цесса столкновения может быть еще
более упрощено. Если величину q вы-
ражения E.19) подставить в E.20), то
мы получим следующее соотношение
для г как функции времени :
1 2 _ 1
2 2
Так как E.21) не содержит переменной
0, то это соотношение можно рас-
сматривать как описание одномерного движения частицы массы /г с полной
Фиг. 10. Эквивалентная задача о
движении одного тела.
При рассмотрении динамики столкновений бывает
удобно изучать эффективные потенциальные кривые
для эквивалентной одномерной задачи о движении
одного тела. В гл. 7 кривые такого типа обсуждаются
в связи с рассмотрением столкновений молекул с
потенциалом взаимодействия Леннарда-Джонса.
~^2^+<р(г). E.21)
§ 5. Столкновения молекул в классической механике (одноатомные молекулы) 55
энергией 1l2/*g2j движущейся в неком эффективном потенциальном пом
= <p(r) + \tig*^ry E.22)
где V^g^/f2) — так называемый центробежный потенциал. Типичная кривая
эффективного потенциала для двух взаимодействующих молекул представлена
на фиг. 10. В любой точке на этой кривой эффективная потенциальная энергия
системы в сумме с кинетической энергией равна полной энергии V^g2- Оче-
видно, имеется целое семейство кривых <рЭфф.(О> зависящих от значения вели-
чины 1l2pg2b2.
В. УГОЛ ОТКЛОНЕНИЯ ПРИ СТОЛКНОВЕНИИ
Единственной величиной, характеризующей столкновение и входящей в
формулы для коэффициентов переноса, является угол отклонения % (угол
между векторами относительной скорости сталкивающихся частиц до и после
столкновения). Этот угол измеряется в системе координат, связанной с центром
масс системы (см. фиг. 8 и 9). Угол % связан простым соотношением с углом
0т, определяемым как значение переменной 0, соответствующее минимальному
расстоянию г между молекулами :
Х = п-2вт. E.23)
Это расстояние тт называется расстоянием наибольшего сближения. В прин-
ципе можно исключить время из уравнений движения и получить г как функцию
0, т. е. получить аналитическое выражение для траектории. Полагая в этом
выражении drjdd равным нулю, получим значения гт и 0Ш. Однако вт проще
может быть получено следующим образом. Выражение для производной dr/dd
может быть найдено, если учесть, что г и 0 являются известными функциями
времени E.21) и E.19). Поэтому
dd "~ dd/dt "~ Ь
Знак минус взят потому, что вдоль траектории при возрастании г величина в
уменьшается. Таким образом, интегрируя E.24), мы получаем выражение
для вт
1/ т _ уС) _ _*1
Комбинируя это выражение с выражением E.23), получаем окончательную
формулу для угла отклонения :
№) = *-2b( ¦. dZ .. E-26)
rJ
Эта формула верна для любых сферически симметричных потенциалов взаи-
модействия <р(г). Значение гт может быть получено путем приравнивания нулю
производной йг\йв в выражении E.24). Если можно ограничиться только пар-
ными столкновениями, то через угол отклонения может быть выражен целый
56 Гл. 1. Введение и основные понятия
ряд свойств разреженных газов. Например, вязкость rj(T) и второй вириаль-
ный коэффициент В(Т) выражаются в виде
RT =4^кя
о
е у2 уЧ \ sin* х db* dy, E.27)
B(T) = -| N Ще-v* у* jX db* dy} E.28I)
о i-o J
где у2 = 1l<j*g2lkT. Соответствующие выражения для всех коэффициентов пере-
носа приводятся в гл. 7 ; в гл. 8 для различных потенциалов взаимодействия
проводится вычисление угла отклонения и величин коэффициентов переноса.
§ 6. КВАНТОВАЯ МЕХАНИКА2)
Теория межмолекулярных сил, излагаемая в гл. 13 и 14, и поведение лег-
ких газов при низких температурах, рассматриваемое в гл. б и 10, могут
быть описаны лишь на основе квантовомеханических представлений, так как
классическая механика неприменима к описанию поведения частиц с малой
массой и малой энергией. В этом параграфе мы сначала обсудим экспери-
ментальные результаты, которые привели к формулированию квантовой ме-
ханики, а затем изложим некоторые фундаментальные принципы квантовой
механики. Фундаментальным уравнением, в известном смысле соответствую-
щим второму закону Ньютона классической механики, является уравнение
Шредингера. Это дифференциальное уравнение второго порядка может быть
решено в нескольких случаях, однако для большинства обсуждаемых проблем
решения можно получить, только используя приближенные методы.
В заключение параграфа будет дан вывод теоремы вириала в квантовой
механике.
А. ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ,
О «НЕКЛАССИЧЕСКИХ» СВОЙСТВАХ СИСТЕМ
Развитие квантовой механики в значительной мере было вызвано рядом
искусных и важных экспериментов, которые не оставили сомнения в том, что
классическая механика не способна точно описать атомные явления. Квантовая
природа излучения, существование систем в стационарных энергетических
состояниях, дуализм излучения и вещества и существование электронного и
ядерного спинов были подтверждены следующими экспериментами.
1. Квантовая природа излучения. 1) Планк [23] счел необходимым для
объяснения наблюдаемого распределения в спектре излучения черного тела
предположить существование фотонов с энергией hv.
2) Эйнштейн [24] предсказал, а опыты Лоуренса и Бимса [25] подтвер-
дили, что при падении света на металлическую поверхность максимальная
скорость испущенных электронов никогда не превышает hv. Кроме того, число
х) В § 1 настоящей главы дана простая формула, выражающая В(Т) через <р(г). Соотно-
шение E.28) приводится для иллюстрации того, что В(Т) может быть выражено через %. Эта
формула справедлива только для монотонно убывающих потенциальных функций.
2) Для читателей, не знакомых с основами квантовой механики, можно рекомендовать
книгу Гайтлера [22]. Более полное изложение можно найти в книге Полинга и Виль-
сона [15].
§ 6. Квантовая механика 57
испускаемых в единицу времени электронов пропорционально интенсивности
падающего света1).
3) Комптон [26] показал, что столкновение фотона с электроном описы-
вается законами упругого удара, причем энергия фотона изменяется от hv до
W в соответствии с законами сохранения энергии и количества движения.
2. Стационарные энергетические состояния. 1) Ритц [27, 28] сформули-
ровал комбинационный принцип, согласно которому частота света, испускае-
мого или поглощаемого атомом или молекулой, может быть представлена как
разность двух «термов», которые Эйнштейн интерпретировал как характеристи-
ческие уровни энергии, деленные на постоянную Планка ft.
2) Франк и Герц [29] показали, что при прохождении электронов в газе
молекулы его имеют тенденцию поглощать только определенные порции энер-
гии, соответствующие величинам термов, измеренным спектроскопически.
3) Критический потенциал ионизации атома, измеренный непосредственно
при столкновении электронов с атомами, соответствует разности спектральных
термов, соответствующих нижнему и предельному верхнему состояниям.
3. Волновая природа вещества. 1) Дэвисон и Джермер[30], Рапп [31, 32],
Кикучи [33, 34 ] и другие показали, что пучок электронов может дать диф-
фракционную картину, аналогичную получаемой для рентгеновских лучей,
причем длина волны равна приблизительно Я = ][ 150/? А (где Е — энергия
электрона в вольтах). Такие пучки электронов ведут себя подобно волнам и
могут контролироваться с большой точностью. Это привело к развитию элек-
тронной оптики и электронной микроскопии.
2) Демпстер [35] показал, что а-частицы могут давать диффракционную
картину.
3) Джонсон [36], Эллет, Ольсон, Цаль [37] и другие показали, что пучки
атомов могут также давать диффракционную картину.
4. Электронный и ядерный спины. Штерн и Герлах [38—40] показали,
что электроны и ядра обладают спином, что приводит их к отклонению в ту
или иную сторону при движении во внешнем неоднородном магнитном поле.
Отношение магнитного момента к моменту количества движения, обусловлен-
ному спином, оказывается аномальным.
Б. КВАНТОВОМЕХАНИЧЕСКОЕ ОПИСАНИЕ СИСТЕМ
Квантовая механика развивалась одновременно по двум направлениям :
во-первых, формулировка волновой механики Шредингера [41, 42] и, во-
вторых, матричная механика Гейзенберга [43] и Борна и Иордана [44]. Шре-
дингером [45] и Экартом [46] независимо было показано, что обе формули-
ровки теории (матричная и волновая) эквивалентны. В некоторых случаях
удобнее пользоваться матричной формулировкой, однако для изучения про-
цессов столкновений, межмолекулярных сил и получения численных резуль-
татов чаще используется формулировка Шредингера. Поэтому далее мы
рассмотрим тодько теорию Шредингера.
1. Уравнение Шредингера. В квантовой механике состояние системы N
частиц описывается волновой функцией xF(tNy t); эта функция имеет тот смысл,
что | ^(r^V) |2 dtN выражает вероятность того, что в некоторый момент времени t
координаты системы заключены внутри области dtM около точки rN. Чтобы W(tN, t)
могла интерпретироваться таким образом, необходимо, чтобы она обладала
г) Пропорциональность фототока интенсивности излучения установлена А. Г. Столе-
товым [см. ЖРФХО, 21, 159 A889)]. — Прим. ред.
58 Гл. 1. Введение и основные понятия
интегрируемым квадратом, т. е. для системы N тождественных частиц
V* (rN, t) V(rN, t) drN=l. F.1)
По всему
пространству
В случае пучка, содержащего N тождественных частиц в единице объема,
удобно нормировать волновую функцию таким образом, чтобы
Г У* (г", 0 ?(rN, t) drN = N. F.2)
_ J
По единичному
объему
Динамика системы полностью описывается квантовомеханическим гамиль-
тонианом системы
^{4), F.3)
который аналогично классическому является суммой операторов кинетической
и потенциальной энергии. (Некоторые волновомеханические операторы обсуж-
даются ниже, в разд. В.) Изменение состояния системы во времени тогда
определяется волновым уравнением Шредингера:
jrV(r»,t)=--^V(r»,t). F.4)
Имеется набор функций ^(r^, t), характеризующих «стационарные состоя-
ния» системы. Если система находится в одном из этих стационарных состояний,
то физические свойства системы не изменяются со временем. В этом случае
можно определить полную энергию системы Ej с помощью выражения
.^У=Е^, = _А-^.. F.5)
Для этих стационарных состояний волновая функция может быть разделена
на две части — зависящую только от времени и зависящую только от коор-
динат y)j(rN):
(if) F.6)
Часть волновой функции, зависящая только от координат, удовлетворяет урав-
нению
F.7)
известному как волновое уравнение Шредингера для стационарных состояний.
В условиях, когда поведение системы близко к классическому, это уравнение
приводится к уравнению Гамильтона—Якоби классической механики. В разд. Г
это будет обсуждаться в связи с решением уравнения Шредингера прибли-
женным методом Вентцеля—Крамерса—Бриллюэна.
2. Нормировка и условия ортогональности. Стационарные состояния
системы описываются волновыми функциями, являющимися решением урав-
нения Шредингера F.7); эти функции непрерывны, имеют непрерывные пер-
вые производные и везде конечны. Для ограниченных систем можно (и обычно
это бывает удобно) нормировать волновые функции так, чтобы они удовле-
творяли соотношению F.1) или F.2). Часто волновое уравнение обладает упо-
мянутыми решениями только для ряда дискретных стационарных энергети-
ческих уровней Е = Ev ?2,...
§ 6. Квантовая механика 59
Волновая функция W(rN, f), описывающая любое состояние системы,
может быть записана в виде линейной комбинации волновых функций щ(ты)
стационарных состояний, т. е.
У(гаг,0 = 21су@уЮ- F.8)
В этом смысле любое состояние системы является «смесью» стационарных со-
стояний1). Этот принцип суперпозиции волновых амплитуд приводит к диффрак-
ционным явлениям, характерным для квантовой механики.
Собственные функции обладают и другим свойством, часто используемым
в квантовомеханических выкладках. Если функции щ и \pj являются собствен-
ными функциями, соответствующими различным собственным значениям, то
J
; у, *" = (). F.9)
Это — так называемое условие ортогональности. Если ipt и y)j соответствуют
одному и тому же собственному значению, то могут быть найдены такие их
линейные комбинации, которые удовлетворяют соотношению F.9).
3. Плотность тока вероятности. Мы можем определить плотность тока
вероятности I как произведение плотности вероятности q = W*W и средней
скорости частиц2). Можно показать, что справедливо соотношение
Если W действительна, то I = 0 и поток частиц через некоторую площадку
невозможен. Плотность тока вероятности удовлетворяет уравнению неразрыв-
ности
tllH- . <6Л1>
Ток вероятности—удобное понятие, связанное не только с квантовомеханическим
потоком, но и с проблемами резонанса.
В. ОПЕРАТОРЫ В ВОЛНОВОЙ МЕХАНИКЕ
Как мы видели, динамическое состояние системы определяется волновой
функцией, а ее поведение во времени описывается волновым уравнением Шре-
дингера. Теперь коротко обсудим измеряемые динамические величины, которые
представляются в квантовой механике операторами. Вначале дадим несколько
специфических примеров квантовомеханических операторов, а затем рассмо-
трим некоторые операции с ними.
1. Некоторые квантовомеханические операторы. Каждой измеряемой
динамической величине классической механики соответствует квантовомеха-
нический оператор. Например, координате х в классической механике соответ-
ствует оператор х в квантовой механике3). Действие этого оператора на волно-
вую функцию ip(x) эквивалентно простому ее умножению на х. Это же справед-
г) Употребление в указанном смысле термина «смесь» нельзя считать удачным. Выра-
жение F.8) представляет собой так называемый чистый случай (reine Fall), или чистое со-
стояние. Термин смесь состояний относится к случаю неполного квантовомеханического
описания системы с помощью матрицы плотности (см., например, В. Паули, Общие прин-
ципы волновой механики, М., 1947 или Л. Ландау и Е. Лифшиц. Статистическая
физика, М., 1951). — Прим. ред.
2) Приведенное определение не является строгим, поскольку в силу соотношения не-
определенности нельзя говорить о значении скорости частиц в данной точке пространства.
Однако если вектор плотности тока вероятности мало зависит от г, указанное истолкование
вектора I становится возможным, так как в этом ^лучае можно с достаточной точностью
определить скорость частицы. — Прим. ред.
3) Здесь мы ограничимся обсуждением метода Шредингера. Возможно использование
совершенно другого набора операторов, как это, например, делается в методе импульса [47 ].
60 Гл. 7. Введение и основные понятия
ливо для любой функции координат, определяющих положение в пространстве;
таким образом, действие оператора потенциальной энергии в (б.З) на волновую
функцию ip(tN) эквивалентно произведению <P(tN)y)(tN). Оператор, соответ-
ствующий компоненте классического импульса рх, — это дифференциальный
оператор
Следовательно, оператор кинетической энергии для системы частиц выражается
в соответствии с (б.З) следующим образом :
М _ v & ( э э \
Два последних выражения справедливы только для декартовой системы коор-
динат; они, вообще говоря, не справедливы в других системах координат [48,49].
Квантовомеханические операторы момента количества движения могут
быть выражены через операторы импульса точно так же, как это делается в
классической механике :
= 1 (У/ fizi - Zt fly<) = — 2' [УI -^ - Zt -^rj , F.14)
F.15)
(бЛб)
В дополнение к этим трем операторам можно написать также не имеющие клас-
сического аналога операторы спинового момента количества движения. Необ-
ходимость введения понятия спина показали Уленбек и Гаудсмит [50] и Паули
[51], разработавшие формальные метсды квантовомеханического описания
спина. Позже Дирак [52, 53], изучая поведение электрона в релятивистской
квантовой механике, показал, что результаты релятивистской теории авто-
матически приводят к свойствам электрона, которые прежде приписывались
гипотетическому вращательному движению электрона [53, 47]. Магнитный
момент, связанный со спином электрона, оказался в два раза больше величины,
которую следовало ожидать, если бы спин был подобен другим формам момента
количества движения. Известны спины практически для всех ядер.
2. Собственные функции и собственные значения операторов. Для каждого
оператора *j? могут быть найдены такие функции ф^ что в результате действия
на них оператора, мы получаем первоначальные функции, умноженные на
некоторую постоянную:
ф А^ F.17)
Если при этом функции ф] удовлетворяют определенным граничным условиям
и некоторым другим требованиям, то это так называемые собственные, или
характеристические, функции оператора «^f, a Aj — соответствующие соб-
ственные значения. Уравнение F.17) может быть дифференциальным или инте-
гральным, и могут существовать только дискретные значения Ajy для которых
существуют физически допустимые решения ф^ [16, 54]. Собственными функ-
циями оператора импульса пх являются ф{х) = А ехр (— грхх\Щ \ собствен-
ными функциями с^#2 = с^#| + <^#| + J$\ являются шаровые функции.
Мы будем рассматривать только эрмитовы операторы, т. е. такие опера-
торы, для которых
J>J", F.18)
где f(rN) и g(rN) — произвольные функции. Важное свойство эрмитовых опера-
торов заключается в том, что они имеют только действительные собственные
§ 6. Квантовая механика 61
значения. Если система находится в состоянии, характеризуемом собственной
функцией оператора, соответствующего динамической переменной, то значе-
ние этой переменной в точности равно собственному значению. Именно поэтому
мы рассматриваем только эрмитовы операторы.
Если система находится не в собственном состоянии ф^ а в состоянии,
характеризуемом некоторой функцией У7^, t) [которую можно разложить по
собственным функциям типа разложения F.8)], то математическое ожидание
величины, соответствующей оператору <^f, дается выражением
А = (V* ^Фdr = 2Т cf Cj\ф?\А0jdrN' = 2rk/|2 At. F.19)
•* ij J i
В этом выражении использованы свойства ортогональности и нормированности
собственных функций ф^
3. Коммутативность операторов. Если некоторые квантовомеханические
операторы Л и & коммутируют, то это означает, что действие оператора Л&
совпадает с действием оператора *5&^б\ Однако вообще
= ih [Л,Щ , F.20)
где {Л& - &Л) — коммутатор операторов Л и *$, а [Л,Щ - оператор, соот-
ветствующий классической скобке Пуассона от Л и «jg. С помощью соотношения
F.20) можно для упоминавшихся ранее операторов написать следующие соот-
ношения :
хпх — ахх = ih9 F.21)
F.22)
o^f2 <^f 2 — <J?Z <^f2 = 0, F.23)
^#2 ЛГ — JTo^f2 = 0. F.24)
Некоторая функция может быть одновременно собственной функцией обоих
операторов только в том случае, если они коммутируют. Таким образом, из
F.21) видно, что невозможно сдновременно знать положение и сопряженный
импульс системы. Если Л— некоторый оператор, явно не содержащий времени,
то зависимость его от времени дается уравнением
F.25)
из которого можно видеть, что если оператор Л коммутирует с гамильто-
нианом, то динамическая переменная, соответствующая оператору^, является
интегралом движения.
Г. НЕРАЗЛИЧИМОСТЬ ТОЖДЕСТВЕННЫХ ЧАСТИЦ
В классической теории движение группы тождественных частиц вполне
определяется вторым законом Ньютона; в любой момент времени можно раз-
личать частицы. В квантовой теории принимается, что тождественные частицы
не могут быть различимы. Это ведет к интересным явлениям; некоторые из
них будут обсуждены в этой книге.
Рассмотрим систему N неразличимых частиц, пренебрежимо слабо взаи-
модействующих д[ уг с другом. В этом случае гамильтониан системы можно
написать в виде суммы гамильтонианов отдельных частиц :
ЛГ - .3fx + JT2 + ... + JFN, F.26)
где >Ж/ зависит только от координат /-й частицы. Вся система частиц подчиняется
волновому уравнению Шредингера
F.27)
62 Гл. 1. Введение и основные понятия
Согласно F.26), уравнение Шредингера распадается на N уравнений :
а(г1) = аафа(т1),
Фъ{Ч) = «ъФъ(?ъ), F.28)
«а + «&+ ... +ап = ек. F.29)
Величину ф](хт) можно интерпретировать как функцию, описывающую т-ю
частицу в /-М квантовом состоянии с энергией а;. С математической точки зре-
ния собственной функцией системы является произведение собственных функ-
ций отдельных частиц. Однако физические системы ведут себя таким образом,
что полную волновую функцию можно образовать только из определенных
линейных комбинаций волновых функций, причем допустимы только такие
комбинации, которые симметричны или антисимметричны относительно пере-
становок любых двух частиц.
1. Антисимметричные волновые функции; статистика Ферми—Дирака.
Если мы потребуем, чтобы полная волновая функция была симметрична, то
можно записать искомую комбинацию волновых функций отдельных частиц
в виде детерминанта
F.30)
Это выражение можно символически представить также в виде
W = 24- l)pP{^i)^(r2)...}. F.31)
Сумма берется по всем перестановкам Р алфавитных индексов F.31). Символ
р (четность перестановки) является четным или нечетным числом в зависимости
от того, четно или нечетно число перестановок пар букв а, &, с,..., необходимое
для превращения некоторого произведения, например фс(г^) 0а(г2)..., в произ-
ведение с нормальным порядком индексов фа (г2) фь (г2)... Легко видеть, что
волновая функция F.30) обладает требуемыми свойствами симметрии, так как
перестановка двух столбцов детерминанта, соответствующая перестановке двух
частиц, изменяет знак полной волновой функции. Кроме того, необходимо за-
метить, что если две частицы находятся на одном и том же энергетическом
уровне (что соответствует замене индекса Ь на а), то две строки детерминанта
становятся одинаковыми и полная волновая функция обращается в нуль. По-
этому мы заключаем, что две частицы не могут одновременно находиться в од-
ном и том же квантовом состоянии (принцип Паули). О частицах, волновые
функции которых обладают таким свойством симметрии, говорят, что они под-
чиняются статистике Ферми—Дирака. Статистика такого типа необходима для
описания всех элементарных частиц и всех атомов и молекул, содержащих
нечетное число элементарных частиц1). Следствие этих ограничений симметрии
будет рассмотрено в гл. 2, 6 и 10.
*) Имеются в виду нуклоны (протоны и нейтроны), входящие в состав ядер, и электроны,
входящие в состав (электронных оболочек) атомов и молекул. Известны элементарные
частицы, подчиняющиеся статистике Бозе—Эйнштейна; таковы все частицы с целым
(или нулевым) спином. Статистике Ферми—Дирака подчиняются частицы с полуцелым
спином. — Прим. ред.
§ 6. Квантовая механика 63
2. Симметричные волновые функции; статистика Бозе—Эйнштейна. Если
частицы неразличимы и если потребовать, чтобы полная волновая функция
была симметрична относительно перестановки двух каких-либо частиц, то
последняя может быть записана в виде
F.32)
Сумма берется по всем перестановкам алфавитных индексов. Статистика, кото-
рой подчиняются системы с волновыми функциями такого типа, называется
статистикой Бозе—Эйнштейна. Для таких систем нет ограничений на
число частиц, находящихся в различных возможных квантовых состояниях
а, Ь}..., п. Этими свойствами симметрии обладают системы молекул и атомов,
содержащие четное число элементарных частиц.
Д. ПРИБЛИЖЕННЫЕ МЕТОДЫ РЕШЕНИЯ
УРАВНЕНИЯ ШРЕДИНГЕРА1)
Поскольку уравнение Шредингера только для простейших систем допу-
скает решение в замкнутой форме, то большую важность имеют приближенные
методы решения. Обсудим кратко три метода. В вариационном методе мы
строим приближенную собственную функцию подходящего вида, содержащую
несколько переменных параметров ; эти параметры подбираются таким обра-
зом, чтобы получить лучшее значение энергии. В методе возмущений мы должны
вначале знать решение волнового уравнения, отличающееся от истинного только
отсутствием некоторых членов, влияние которых на систему мало. Затем мы
исправляем решение, с тем чтобы учесть дополнительный возмущающий член
в потенциале. В методе Вентцеля—Крамерса—Бриллюэна (ВКБ) мы прини-
маем классическое решение в качестве первого приближения и вычисляем затем
квантовые отклонения от классического поведения. Все эти методы играют
важную роль в задачах, связанных с молекулярным взаимодействием, и будут
использованы ниже.
1. Вариационный метод. Пусть функции щ представляют полный набор
собственных функций гамильтониана системы -3f\ Е{ — соответствующий набор
собственных значений, а ? — произвольное пробное решение. Тогда, поскольку
ipi образуют полную ортонормированную систему, можно разложить произ-
вольную функцию в ряд по функциям щ:
? = 24w F.33)
Отсюда
Jre = 2EiatVi- F.34)
Умножим оба эти выражения на ?* и проинтегрируем по всему пространству ;
воспользовавшись свойством ортонормированности собственных функций, по-
лучим
J F.35)
Таким образом,
где Ео — наинизшее собственное значение гамильтониана.
Так как Е( — Ео > 0, то
1
О См. [56].
F.38)
64 Гл. 7. Введение и основные понятия
и равенство возможно только в том случае, если ? будет точной собственной
функцией, соответствующей наинизшему состоянию. Этот результат является
основой приближенного метода решения. Произвольная пробная функция ?
может содержать любое необходимое число параметров. Можно вычислить
величину отношения интегралов и путем подбора параметров сделать ее мини-
мальной. Полученное таким образом отношение является лучшим приближе-
нием к энергии (наинизшего состояния), которое может быть получено с проб-
ной функцией такого вида.
Легко видеть, что если пробная функция и собственная функция, соответ-
ствующая наинизшему собственному значению, ортогональны, то отношение
интегралов будет большим или равным Ev Аналогично, если пробная функция
и собственные функции, соответствующие двум наинизшим собственным зна-
чениям, ортогональны, то отношение будет равным или большим Е2 и т. д.
Последние результаты ценны в тех случаях, когда на основе симметрии пробная
функция может быть выбрана ортогональной к основному состоянию.
В качестве примера рассмотрим пробную функцию ?, являющуюся линей-
ной комбинацией функций /,. Ряд может быть конечным или бесконечным и
необязательно ортонормированным. Таким образом,
F.39)
Для удобства воспользуемся следующими сокращенными обозначениями :
dt» = JT% F.40)
l*. F.41I)
В соответствии с равенством F.38) приближенное значение энергии дается
выражением
• F.42)
Е = .. . •
2 bi Dj Aji
Лучшее приближение для Е получим, минимизируя это выражение относи-
тельно всех входящих в него параметров. В результате получим
р/ЛГу-ЕЛу) = 0. F.43)
Этот ряд уравнений имеет нетривиальное решение только в том случае, если
удовлетворяется уравнение
|ЛГу-ЕЛу| = 0. F.44)
Наименьший корень этого секулярного уравнения является, очевидно,
приближенным наинизшим собственным значением гамильтониана *Ж'. Боль-
шие корни являются приближениями к высшим собственным значениям.
2. Метод возмущений. Часто оказывается, что гамильтониан системы
может быть записан в виде суммы двух членов
ЛГ = ,jr<°> + Х-ЭРЫ, F.45)
где ,Jf@) — главный член; А^A)— малое возмущение (Я—малая величина,
которая может быть переменной). Например, возмущенный член может опи-
сывать влияние внешнего электрического или магнитного поля. Приближенное
решение можно получить, используя в качестве пробной функции ^0) линей-
ную комбинацию собственных функций невозмущенного гамильтониана ^@)
1) Если ft ортонормированы, то Дц =
§ 6. Квантовая механика 65
Тогда если ?(/° — невозмущенная энергия, то из уравнений F.40) и F.41)
следует
0Г0 = J xpf)*:Jtvf> dtN = E<f» ди + Х,Ж<$, F.46)
где
ЛГ $ = J ^°>* .ЭРЫ ^@) *" F.47)
4у=«/у F.48)
Ряд энергетических уровней возмущенной системы можно получить, решая
секулярное уравнение F.44), приобретающее вид
| (JEJ» - Е) ди + hTf |=0. F.49)
Если это уравнение будет решено точно, то полученные решения будут со-
ответствовать энергетическим уровням гамильтониана .Ж. Обычно этого
сделать не удается. Точность приближенных решений зависит от характера
возмущения и от порядка, до которого уравнения решаются. Исследовать
сходимость трудно, поэтому сложно определить ошибку. Решение секуляр-
ного уравнения предполагается близким к одному из значений Е\°\ и, следо-
вательно, его можно разложить по степеням А:
Ек = Е^ + АЕ?> + V Ер + ... F.50)
Таким образом, с точностью до первых степеней,
и) | = 0. F.51)
Раскрывая детерминант и удерживая только члены первой степени относи-
тельно А, находим, что если начальный ряд собственных значений не вырожден
(т. е. ?<?> ф Д°> при i ф /с), то
?W=JTW F.52)
так что
E = ?<°> + A.jr#>. F.53)
Если исходный ряд энергетических уровней вырожден, то получение той
же точности достигается путем пренебрежения матричными элементами JT$}
для состояний, принадлежащих различным вырожденным группам. В этом
случае детерминант превращается в произведение детерминантов. Ряд возму-
щенных уровней, возникающих от одной вырожденной группы, может быть
получен из решения уравнения
|(Jrg>-?p«i;)|=O. F.54}
Строчки и столбцы этого детерминанта соответствуют начальному ряду
вырожденных состояний.
Удерживая более высокие степени А, можно получить более точные резуль-
таты. Для случая невырожденных уровней теория возмущений второго порядка
приводит к выражению для энергии
Е = ЕР + ДЛГЙ» - Я-
Многочисленные применения метода возмущений к решению простых
задач можно найти в любом учебнике по квантовой механике.
3. Метод Вентцеля—Крамерса—Бриллюэна (ВКБI). Метод ВКБ очень
удобен для апроксимирования решений одномерных квантовомеханических
х) Краткое описание этого метода есть в работах [47, 48 ]. Более подробно см. работы
[57, 58].
Гл. 7. Введение и основные понятия
задач. Этот метод позволяет получить поправки к классическому поведению,
выраженные через ряд по степеням постоянной Планка. Рассмотрим одномер-
ную задачу — случай, когда потенциальная энергия Ф(х) и полная энергия
системы Е представляются фиг. 11.
В классической точке поворота
х0 имеем Ф(х0) = Е, так что класси-
ческая кинетическая энергия в этой
точке равна нулю. Из фиг. 11 ясно,
что для значений х, больших х0, ки-
нетическая энергия Е — Ф(х) поло-
жительна и эта область классически
7
К
! Классическая
область
Классическая
(ip=A exp iSJfi)
достижима. Однако для х, меньших
х0, кинетическая энергия Е — Ф(х)
отрицательна и эта область не может
быть достигнута классической сис-
темой. Это различие проявляется
в разных формах решения для
областей х <^ х0 и х ^> х0.
Уравнение Шредингера
рассматриваемой одномерной
дачи можно записать в виде1)
для
за-
х
Квантовая
(точное ф)
Тогда S(x) удовлетворяет диф-
Фиг. п. Схематическое изображение амплитуд ференциальному уравнению вида
вероятности, рассчитанных классически, кванто-
вомеханически и по методу В КБ. ;* d $
Определим новую функцию S(x)
следующим образом :
F.57)
-2т[Е-Ф(х)] =0. F.58)
За исключением члена /A(rf2S/rfx2), это уравнение в точности совпадает с клас-
сическим уравнением Гамильтона—Якоби D.16). Разложив S(x) в ряд по
степеням л
S(x) = S0(x) + AS^x) + A2S2(x) + ... F.59)
p подставив его в уравнение F.58), а затем, приравняв члены одинаковой сте-
пени относительно А, получим ряд уравнений. Первые два из них будут иметь
вид
F.60)
F.61)
. d*S0
1
dS0 dS1 _
dx dx -
Уравнение F.60) для нулевого приближения S0(x) в точности совпадает с урав-
нением Гамильтона—Якоби. В этом выражается соответствие между кван-
товой и классической механикой. Интегрирование F.60) дает
So=± J рт[Е-Ф{х)\йх;
F.62)
г) При рассмотрении решений радиального волнового уравнения х заменяется на г,
а Ф(х) на Ф(г) + А2/(/ + \)/2тг2; у) равна радиальной волновой функции, умноженной на г.
§ в. Квантовая механика 67
подставляя это выражение в F.61), получаем
S ТЖ ( Ш) U'»{2m [Е ~ Ф(х)]}. F.63)
Следовательно,
Sl = -L\n{^[E-0(x)}\ F.64)
и для первого приближения волновой функции у(х) получаем выражение
**> - ( тТЁ^ФЩ-Т' К е*Р (т 1 B m Р - «W1 >"• Ас) +
X
+ А2 ехр (- {- J {2ш [? - Ф(х)] J1'» dx )]. F.65)
Для неклассической области, где ? — Ф(х) отрицательно, удобнее записать
волновую функцию в эквивалентном виде :
**> = (шщ=*\Iи [*>• ехР (J- J V т t0^ -?1 >1/2 dx) +
х
Хо
+ Вг ехр (- 1 J {2ш [Ф(х) -?] }V. dx)], (б.бб)
В общем случае, когда имеется более одной классической точки поворота,
связь между А19 А2 и til9 B2 затруднительно записать в явном виде. Однако в
рассматриваемом случае, когда Ф(х) > Ь, для всех значений х < х0, очевидно,
Вг = 0. Можно показать, что решение волнового уравнения в классической
оЬласти имеет вид
* cos [- т + т ] ^т t? " Ф^ У1' dx\ • <6-
**> =2 в
Это выражение можно использовать для вывода квантовомеханических по-
правок ко второму вириальному коэффициенту и коэффициентам переноса
При высоких температурах (см. гл. 6 и 10).
Е. ТЕОРЕМА ВИРИАЛА В КВАНТОВОЙ МЕХАНИКЕ
Как было показано [59, 60], теорема вириала и ее применение справед-
ливы в квантовой области так же, как и в классической, если средние по вре-
мени заменить на средние по пространству. Ниже приводятся доказательство
справедливости квантовомеханической теоремы вириала, ее приложения к
теории уравнения состояния рассмотрены в гл. 6, § 2, а приложения к теории
молекулярной структуры и взаимодействия — в гл. 13, § 1.
Для системы N частиц стационарные уравнения Шредингера для функции
у) и ее комплексно-сопряженной у* имеют вид
Продифференцировав первое из них по х7 и умножив результат на x/tp*, получим
68 Гл. 1. Введение и основные понятия
Подставляя (Ф — ?)^* из уравнения F.69) в F.70) и суммируя по всем /,
получаем
Первый член можно преобразовать, воспользовавшись соотношением
V Эх? Эху Эх? Эх,;~ ^ Щ
Тогда, интегрируя F.71) по xh получаем выражение
f Э2у>* Ь
J 22 xj (v*
_ CD *
= -2 )V wdx' +1 <y*J ^r^*-12 x>-w\l • FJ3)
Если система ограничена, то член в скобках при подстановке обоих пре-
делов обращается в нуль. Таким образом, окончательно получаем
[к2' - я? щ) »*" - - i .к (- f * f) ""rN ¦
это — выражение квантовомеханической теоремы вириала. Левый член пред-
ставляет собой ожидаемую величину кинетической энергии ; член справа —'
квантовомеханический аналог вириала. Если потенциал Ф — электростати-
ческого происхождения, т. е. 2 xj(®&lQxj) = —Ф>то уравнение F.?4) сводится к
, F.75)
что находится в соответствии с результатом классической теории К = —112Ф-
§ 7. СТОЛКНОВЕНИЯ МОЛЕКУЛ В КВАНТОВОЙ
МЕХАНИКЕ1)
Развитие квантовомеханической теории уравнения состояния и явлений
переноса требует знания квантовой теории столкновений. Мы здесь рассмотрим
только столкновения таких двух частиц, для которых потенциал взаимодей-
ствия не зависит от угла.
Мы уже упоминали выше, что в классической теории свойства разрежен-
ного газа (второй вириальный коэффициент и коэффициенты переноса) выра-
жаются через угол отклонения %{by g) вектора относительной скорости двух
сталкивающихся частиц. В квантовой теории, однако, нельзя одновременно
точно определить положение и скорость молекулы (принцип неопределенности
Гейзенберга). Следовательно, нельзя точно определить и угол отклонения.
В лучшем случае можно исследовать, какова вероятность отклонения на угол
%. Эта вероятность выражается через фазовый сдвиг г\{(к) радиальной волно-
вой функции. Фактически фазовый сдвиг является единственной характери-
стикой столкновения, которая входит в окончательные формулы для второго
вириального коэффициента и коэффициентов переноса в разреженном газе. В
классическом пределе фазовый сдвиг просто связан с углом отклонения.
. [611.
§ 7. Столкновения молекул в квантовой механике
69
А. ВЗАИМОДЕЙСТВИЕ ДВУХ ЧАСТИЦ;
ФАЗОВЫЕ СДВИГИ /?Д*)
При обсуждении классической теории столкновений (см. § 5) было отме-
чено, что задача двух тел (включающая 6 степеней свободы), если выделить
поступательное движение центра масс системы двух частиц, может быть легко
сведена к эквивалентной задаче одного тела (включающей 3 степени свободы).
Аналогичное упрощение может быть проделано и в квантовой теории ; при
этом результирующее уравнение Шредингера дня двух частиц с потенциалом
взаимодействия ср{г) может быть записано в виде
_ Л_1 {JL И\ 4- М — F П 1)
здесь Е — полная энергия системы. Если потенциал взаимодействия имеет
минимум, то существуют два типа собственных функций <р(г) этого уравнения
(фиг. 12). Если эти две частицы связаны, то система может находиться в одном
Ф и г. 12. Типичная кривая потенциальной энергии для системы двух частиц.
Показаны энергетический уровень в континууме и дискретный уровень.
или нескольких дискретных состояниях с отрицательной энергией1) и ? = ?"•
Если частицы испытывают столкновения, то их энергия будет в области конти-
нуума и
? =i- 2
где g — относительная скорость частиц перед столкновением. Именно эту
последнюю возможность мы и хотим рассмотреть. Если ввести величину х,
определяемую выражением hx = /*g, то уравнение Шредингера принимает вид
' = 0. G.2)
Это уравнение может быть решено методом разделения переменных, подста-
новкой у) = y)xl (r) Yf (в, ф\ где Yf @, ф) — шаровые функции2), а грх1 (г) удов-
летворяет радиальному волновому уравнению
(г Vw
~//@)(г^/) = 0,
G.3)
где
/,(/•) =
1) Принимаем, что нуль энергии и потенциала соответствует удаленным друг от друга
частицам.
2) Определение шаровых функций Y?@, 0) дается в приложении Б гл. 12.
70 Гл. 1. Введение и основные понятия
В случае «идеальных» частиц, для которых <р(г) = 0, это уравнение превра-
щается в
-?? (г У?/) + (*2 - ^г11) (г^) = 0. G.4)
Индексом 0 обозначены величины, соответствующие случаю «идеальных» частиц.
При вычислении второго вириального коэффициента в квантовой механике
необходимо знать дискретные энергетические уровни системы взаимодей-
ствующих частиц и сплошной спектр энергетических уровней, точнее плот-
ность энергетических состояний в континууме. Для вычисления плотности
состояний в положительной области спектра, даваемой G.3) и G.4), удобно
сначала превратить энергетический спектр в дискретный [62 ]. Это связано с
введением искусственного граничного условия, согласно которому волновая
функция относительного движения должна обращаться в нуль при расстоянии
г между молекулами, большем некоторого значения г = L. Впоследствии L
полагается равным бесконечности.
Общее решение G.4) может быть записано в_виде суммы двух функций
Бесселя J 1+Уя (xr) nj _,_y2(*r), умноженной на ][хг [63]. Но так как послед-
няя функция обращается в бесконечность при г = 0, то ее следует исключить.
Таким образом, решение уравнения G.4) мы записываем в виде
; G.5)
это выражение для больших значений г асимптотически стремится к
г yg, = A sin Г xr — -=- 1ти (для больших г). G.6)
Используя искусственное граничное условие, можно следующим образом
определить х:
xL-^Ы^пл, п = 0,1,2,... G.7)
С уравнением G.3) можно поступить аналогичным образом. Для значений г
при которых <р(г) обращается в нуль, волновая функция может быть записана
в виде
г ч>ш = Afa) [" ^-f Ji+v* (* г) + Bfyc) [n f-] * J_,_,, (х г). G.8)
Для больших г она асимптотически переходит в
г грх1 = [А%х) + В)(х)] к sin [и г - -i- In + rj^x)] (для больших г), G.9)
где
^ G.10)
Тогда искусственное граничное условие дает
*L-yte + 4,(x) = /w, л = 0,1,2, ... G.11)
Таким образом, мы видим, что асимптотические решения для реальных (взаи-
модействующих) и «идеальных» (невзаимодействующих) молекул имеют сину-
соидальный характер и отличаются только по фазе синусоидальной функции.
Это различие и является фазовым сдвигом rjt(x). Фазовый сдвиг зависит от
орбитального квантового числа/[или момента количества движения А У 1A +1)]
и от орбитального квантового числа х (или импульса относительного
движения hx). Это напоминает классический случай, когда угол отклонения %
вычислялся как функция параметра столкновения Ь (или момента количества
движения pbg) и относительной скорости g (или импульса относительного
движения )
§ 7. Столкновения молекул в квантовой механике 71
Плотность состояний q°(x) = (An/AxH и q(x) = AnjAx можно теперь
определить из соотношений G.7) и G.11) соответственно. Дифференцирование
зтих соотношений приводит к
{Ап)« = ±(Ах)\ G.12)
Ап^Ан + Щ-Ах. G.13)
Таким образом, мы видим, что при Ах -> О разность плотностей энергии, выра-
женная через х1 дается
[<**)-<№\=^ik' G.14)
Этот результат используется при выводе формулы для второго вириального
коэффициента в области низких температур.
Фазовый сдвиг для квазиклассического столкновения может быть вычи-
слен методом ВКБ, описанным в § б. В первом приближении находим, что для
монотонно убывающего потенциала фазовый сдвиг просто связан с углом от-
клонения
где g = hxlfi и Ь = У 1A + 1)/и. Более высокие приближения дают отклог
нения от классического поведения. В гл. 10 описано разложение фазовых
сдвигов по методу ВКБ. Эти результаты могут затем быть использованы для
вычисления квантовых поправок ко второму вириальному коэффициенту и
коэффициентам переноса.
Б. ВЕРОЯТНОСТЬ ОТКЛОНЕНИЯ НА УГОЛ %
Продолжим обсуждение эквивалентной задачи одного тела, описываемой
уравнением G.2), и изучим рассеяние системы с приведенной массой /г при ее
приближении к центру рассеяния, расположенному в начале системы коор-
динат, в отрицательном направлении вдоль оси (+ г). Наиболее общее решение
уравнения G.2) в соответствии с цилиндрической симметрией задачи и физиче-
ским условием, что волновые функции конечны при г — О, может быть записано
в виде
V = ic/VAi(r)P/(cosz), G.15)
z=o
где С/ — постоянная.
Для изучения угловой зависимости рассеяния разделим у) на две части :
\р!у представляющую падающую волну, и ^s, представляющую рассеянную
компоненту.
Для падающей волны (система приближается вдоль оси z) имеем1)
y)i = ехр(— ixz) = exp(*'*rcos#). G.16)
Выражение G.16) может быть разложено в ряд по полиномам Лежандра ;
коэффициенты разложения содержат функции Бесселя [б]
*>' = 2 B/ + J>il У^тТ ¦/<+* <* г> р< (cos *)• <7Л7>
Этот ряд для больших г можно записать в виде
^ = 7^-^B/+Oi'sin^r-^/Jp^cos^). G.18)
1) Это выражение может быть видоизменено в случае столкновения двух одинаковых
(неразличимых) частиц; необходимая симметризация будет рассмотрена позже.
72 Гл. 7. Введение и основные понятия
Выражение G.18) дает асимптотическое поведение падающей волны. Рассмот-
рим теперь рассеянную волну, которая описывается выражением G.15), и,
следовательно, ее асимптотический вид
Vs = у *'"/(*), G.19)
гДе fix) — произвольная функция угла #. Полная волновая функция, являю-
щаяся суммой этой функции и уI [даваемой G.18)], представляет собой реше-
ние полного волнового уравнения, содержащего потенциал взаимодействия.
Чтобы оно было справедливым, функция /(#) должна быть выбрана так, чтобы
полная волновая функция была непрерывна и имела непрерывные производ-
ные. Это связано с таким выбором /(#), чтобы гр1 + tps асимптотически совпа-
дало при некотором выборе постоянных С/ с выражением, даваемым реше-
нием G.15).
Вводя ?>/ — коэффициенты разложения функции /(#) по полиномам Ле-
жандра — можно уравнение G.19) переписать в виде
vs = ±e'*rvD/P/(cos;0. G.20)
Поскольку, с одной стороны, асимптотическое решение волнового уравнения
G2) складывается из двух асимптотических функций гр1 и ^, а с другой сто-
роны, дается полной волновой функцией у G.15), то можно записать
2 |^B/ + l)8in [кг - \
1 a t ч> (-21>
= 2 {у С, №) + ВДГ/2sin (>.г— -^-Z — 4z(«)j}Pi(cosz).
Приравнивая коэффициенты при Pz(cos%), получаем
-1B/ + 1) f'sin(кг - у/) + Dtliw -
= С/ [АЦн) + BK«)]l/« sin [кг - 11 + v] , G.22)
что является тождеством относительно г. Следовательно, приравнивая коэф-
фициенты при С/ и D/, получаем связь
Д = -1гB/+1)(^-1)> G.23)
и, окончательно,
V>S = -?^2Bl+ IK*2'41- l)Pi(cosx). G.24)
Доля частиц, рассеянных в данном направлении, дается пределом отно-
шения j ips |2/| уI12 для больших расстояний.# После достаточного удаления
молекул друг от друга, так что они больше «не чувствуют» потенциала <р(г),
они разлетаются с относительной скоростью g. Следовательно, число молекул,
пересекающих сферу радиуса г в единицу времени, для скоростей, заключен-
ных в телесном углу dco и направленных под углом со, равно
G.25)
откуда
? 2 B/ +J) (e2im -J)pi (cos xJ.
Это выражение дает распределение рассеянных частиц в предположении, что
Задачи TS
частицы различимы; т. е. эта формула справедлива для статистики Больц-
мана. Если частицы неразличимы, то волновая функция должна быть симмет-
ризована и, следовательно, гр1 = еЫг должна быть заменена на
y>i = B)-K[eixZ±e-ixz],
где знак плюс соответствует статистике Бозе—Эйнштейна, а минус — стати-
стике Ферми—Дирака.
Для рассеяния одинаковых частиц в случае статистики Бозе—Эйнштейна
(Б. Э.) получаем
W& X)]б.э. = -? 2 Bl + J) (e2im ~ J)pi <cos X) \ G.27)
/ четное
а в случае статистики Ферми—Дирака (Ф. Д.)
2 («* x) \ G.28>
/ нечетное
Выражения G.27) и G.28) справедливы только для частиц с нулевым спином.
Для Б. Э. и Ф. Д. газов с частицами, обладающими спином s, имеем соответ-
ственно
{§&] «(в°к + [^ «$>д., G.29)
[] ^ • <7-30>
Индекс 0 обозначает величину, описываемую соотношениями G.27) и G.28),
соответствующую спину, равному нулю. Этот анализ применим к случаю столк-
новений частиц, обладающих ядерным спином s. При столкновениях молекул
потенциал не является сферически симметричным и необходимо более детальное
рассмотрение.
Задачи
К Используя выражение A.10), вычислить второй вириальный коэффициент для
случаев :
а) модели твердой сферы,
б) прямоугольной потенциальной ямы.
2. Для двухатомной молекулы при Cv = */%R вычислить величину поправки Эйкена
к коэффициенту теплопроводности.
3. Какова максимальная энергия притяжения для потенциала Букингема?
4. Вычислить среднюю по времени кинетическую и потенциальную энергии для одно-
мерного гармонического осциллятора и сравнить результат с результатом, предсказываемым
теоремой вириала.
5. Вычислить угол отклонения для случаев :
а) модели твердой сферы,
б) кулоновского потенциала.
6» Найти волновую функцию и энергетические уровни в случае частицы, заключенной
в одномерном потенциальном ящике.
7* Найти фазовые сдвиги для модели твердой сферы.
в. Используя E.27) и E.28) и результаты задачи 5, вычислить для модели твердой сферы
второй вириальный коэффициент и коэффициент вязкости.
9. Разложить уравнение Ван дер Ваальса по степеням величины 1/V и выразить второй
вириальный коэффициент через величины а и Ь.
10. Рассмотреть взаимодействие двух молекул для потенциала F-12) Л еннарда-Джойса.
Какова величина силы взаимодействия в момент, когда расстояние между ними
г = 2<т и г = 0,9 а?
74 Гл. 7. Введение и основные понятия
ЛИТЕР АТУРА
1. Н а р р е 1 Н., Ann. d. Phys., 4, 21, 342 A906).
2. М a j u m d a r R., Bull. Calcutta Math. Soc, 21, 107 A929).
3. E у r i n g H., H i r s с h f e 1 d e r J. O., Journ. Phys. Chem., 41, 249 A937).
4. H i r s с h f e 1 d e r J. O., Journ. Chem. Ed., 16, 540 A939).
5. Chapman S., Dootson F. W., Phil. Mag., 33, 248 A917).
6. Wang Chang C.S., Uhlenbeck G. E., Transport Properties in Polyatomic Gases,
CM-681, Project No. 7924 University of Michigan A951).
7. Gay don A. G., Spectroscopy and Combustion Theory, New York, 2d Ed., 1948 (см.
перевод: Гейдон А., Спектроскопия и теория горения, ИЛ, 1950).
8. Zener С, Phys. Rev., 37, 556 A931).
9. Keesom W. H., Phys. Zs., 22, 129 A921).
10. Debye P., Phys. Zs., 21, 178 A920).
11. Fa Ik en hag en H., Phys. Zs., 23, 87 A922).
12. Jo os G., Theoretical Physics, New York, 1932.
13. С о r b e n H. C, Stehle P., Classical Mechanics, New York, 1950.
14. To 1 man К. С, Principles of Statistical Mechanics, Oxford, 1938.
15. P a u 1 i n g L., Wilson E. В., Introduction to Quantum Mechanics, New York, 1935.
16. M a r g e n a u H., Murphy G. M., Mathematics of Physics and Chemistry, New York,
1943.
17. S о k о 1 n i k о f f I. S., S о k о 1 n i k о f f E. S., Higher Mathematics for Engineers and
Physicists, New York, 1941.
18. Хин чин А. Я., Математические основания статистической механики, М.—Л., 1943.
19. Ken na r d E. H., Kinetic Theory of Gases, New York, 1938.
20. Tolman R. C, Statistical Mechanics, New York, 1927.
21. Schottky W., Phys. Zs., 21, 232 A920).
22. He i tier W., Elementary Wave Mechanics, Oxford, 1946. (см. перевод: Гайтлер В.,
Элементарная квантовая механика, ИЛ, 1948).
23. Planck M., Ann. d. Phys., 4, 553 A901).
24. Einstein A., Ann. d. Phys., 17, 132 A905).
25. Lawrence E. O., Beams J. W., Phys. Rev., 32, 478 A928).
26. Co mp ton A. H., Phys. Rev., 21, 207, 483, 715 A923).
27. Ritz W., Phys. Zs., 4, 406 A903).
28. Ritz W., Ann. d. Phys., 12, 264 A903).
29. Franck J., Hertz G., Verhandl. Deutsch. Phys. Ges., 16, 457, 512 A914).
30. Da vis son С J., Germer L. H., Phys. Rev., 30, 705 A927).
31. Rupp E., Zs. Phys., 95, 801 A935).
32. Rupp E., Ann. d. Phys., 9, 458 A931).
33. Kikuchi S., Proc. Imp. Acad. Tokyo, 4, 271, 275, 354, 471 A928).
34. К i k u с h i S., Phys. Zs., 31, 777 A930).
35. Dempster A. J., Phil. Mag., 3, 115 A927).
36. Johnson Т. Н., Phys. Rev., 37, 847 A931).
37. El let t A., Olson H. F., Zahl H. A., Phys. Rev., 34, 493 A929).
38. Stern O., Zs. Phys., 7, 249 A921).
39. Gerlach W., Stern 0., Zs. Phys., 8, 110 A922).
40. Gerlach W., Stern 0., Zs. Phys., 9, 349 A922).
41. Sc h г о d i n g e r E., Ann. d. Phys., 79, 361 A926).
42. Schr o"d i nger E., Collected Papers on Wave Mechanics, London, 1928.
43. H e i s e n b e r g W., Zs. Phys., 33, 879 A925).
44. В о r n M., Jordan P., Zs. Phys., 34, 858 A925).
45. S с h r 6 d i n g e r E., Ann. d. Phys., 79, 734 A926).
46. Eckart C, Phys. Rev., 28, 711 A926).
47. R о j a n s k у V., Introductory Quantum Mechanics, New York, 1938.
48. Kemble E. C, Fundamental Principles of Quantum Mechanics, New York, 1937.
49. Pauli W., Handb. d. Phys., 2 Aufl., 24, 1 Teil.
50. Uhlenbeck G. E., Goudsmit S., Naturwiss., 13, 953 A925).
Литература 75
51. Pauli W., Zs. Phys., 43, 601 A925).
52. Dirac P. A. M., Proc. Roy. Soc, A117, 610 A928).
53. Dirac P. A. M., The Principles of Quantum Mechanics, 3rd Ed., Oxford, 1947 (см.
перевод 3-го издания: Дирак П. А. М., Принципы квантовой механики, М.—Л., I960).
54. Murnaghan F. D., Introduction to Applied Mathematics, New York, 19^8.
55. G о m b a s P., Mehrteilchenproblem der Wellenmechanik, Basel, 1950 (см- перевод:
Гомб аш П., Проблема многих частиц в квантовой механике, ИЛ, 1952).
56. Е у г i n g H., Walter J., К i m b а 11 G. E., Quantum Chemistry, New York, 1944
(см. перевод: Эйринг Г., Уолтер Дж., Кимбалл Дж., Квантовая химия, ЙЛ,
1948).
57. Schiff L. I., Quantum Mechanics, New York—Toronto—London, 1955 (см. перевод:
Шифф Л., Квантовая механика, ИЛ, 1959).
58. Dunham J. L., Phys. Rev., 41, 713, 721 A932).
59. Slater J. С, Journ. Chem. Phys., 1, 687 A933).
60. Born M., Hei sen berg W., Jordan P., Zs. Phys., 35, 557 A925, 1926).
61. Mot t N. F., Masse у H. S., The Theory of Atomic Collisions, Oxford, 1949 (см.
перевод : М о т т Н., М е с с и Г., Теория атомных столкновений, ИЛ, 1951).
62. Kahn В., Диссертация, Utrecht, 1938.
63. J a h n k e E., E m d e F., Funktionentafeln, Leipzig, 1945 (см. перевод : Я н к е Е.,
Эмде Ф., Таблицы функций, М.—Л., 1948).
64. Watson G. N., Bessel Functions, Cambridge, 1922 (см. перевод 2-го издания:
В а т с о н Г. Н., Теория бесселевых функций, ИЛ, 1949).
ПУСТАЯ СТРАНИЦА
Часть I
РАВНОВЕСНЫЕ СВОЙСТВА
ПУСТАЯ СТРАНИЦА
ГЛАВА 2
СТАТИСТИЧЕСКАЯ МЕХАНИКА
Методы классической и квантовой механики очень эффективны при
рассмотрении поведения простых систем, обладающих только несколькими
степенями свободы. Однако очевидно, что невозможно применять эти метсды
для изучения поведения очень сложных систем, например объема газа, содер-
жащего 6,023 • It26 молекул. Более того, знание поведения микроскопических
элементов системы, полученное таким образом, не представляет особого инте-
реса. Макросостояние системы и его изменение во времени могут быть пред-
сказаны статистической механикой, если сведения о начальном состоянии
системы ограничиваются результатами измерений макросвойств.
Статистическую механику можно представить состоящей из двух важных
разделов : механики равновесных и неравновесных состояний.
Первый раздел хорошо понятен с точки зрения основных принципов и
формальных методов теории. Основные принципы и важнейшие результаты
теории будут изложены в настоящей главе. В последующих четырех главах
будет обсуждаться применение результатов этой главы к некоторым важным
задачам: уравнению состояния разреженных газов, свойствам сжатых газов
и жидкостей, критическим явлениям, квантовым эффектам и явлениям при
низких температурах. Хотя прогресс в этом направлении до сих пор сильно
тормозился трудностями численных расчетов, все же известно многое о свой-
ствах вещества и их зависимости от сил, действующих между молекулами.
Неравновесная статистическая механика развивалась в более позднее
время и является сейчас сдним из передовых участков фронта исследователь-
ских работ. Около ста лет назад Максвелл, Больцман и другие рассмотрели
случай разреженного газа, использовав специальные методы. С этими методами
обычно связывается термин «кинетическая теория». Общее обсуждение фор-
мального метода статистической механики неравновесных систем дается в
гл. 7, в последующих главах обсуждаются его применения к гидродинамике
и явлениям переноса.
§ 1. ОПИСАНИЕ СТАТИСТИЧЕСКИХ АНСАМБЛЕЙ
В КЛАССИЧЕСКОЙ МЕХАНИКЕ
В классической статистической механике микросостояние системы описы-
вается заданием положений и импульсов всех частиц системы. В этой главе
мы рассмотрим сложные системы типа газа или жидкости, содержащие N мо-
лекул с / степенями свободы1). Этот параграф начинается с рассмотрения раз-
х) Число степеней свободы молекул — это число координат, необходимых для описа-
ния ее геометрического положения и ориентации. Обычно пользуются представлением о со-
ставляющих молекулу атомах, как о точечных частицах (пренебрегая ядерным и электрон-
ным движением). Тогда одноатомная молекула имеет 3 степени свободы, твердая двухатом-
ная молекула (способная вращаться, но не колебаться) — 5, нетвердая двухатомная моле-
кула — б, нетвердая п-атомная молекула — Зп степеней свободы.
-80 Гл. 2. Статистическая механика
личных способов описания динамического микросостояния системы. Вводятся
понятия гиперпространств различных типов, что дает удобный метод сумми-
рования сведений о состоянии системы.
В целях статистического изучения системы мы рассмотрим не одну си-
стему, а большой набор Г (ансамбль) таких систем, где Г — число систем.
Необходимо помнить, что эти системы тождественны и состоят из того же
числа молекул одного типа, заключенных в одинаковых резервуарах. Они
отличаются, однако, тем, что представляют систему в различных динамических
состояниях, появляющихся вследствие различных начальных условий. Поня-
тия ансамбля и функции распределения будут обсуждгны подробно. Этот
параграф ограничивается описанием классического состояния системы; ана-
логичное квантовомеханическое рассмотрение дается в § 2.
А. КОНФИГУРАЦИОННОЕ ПРОСТРАНСТВО,
ПРОСТРАНСТВО ИМПУЛЬСОВ И ФАЗОВОЕ ПРОСТРАНСТВО
Для изображения геометрии и динамики сложной системы используются
гииерпространства нескольких видов. Здесь мы дадим сводку всех обозначений,
необходимых для описания системы (газ, жидкость или твердое тело), содержа-
щей N подсистем (одноатомных молекул) с 3 степенями свободы каждая. Об-
суждаемые здесь понятия легко могут быть обобщены на случай многоатомных
молекул. В дальнейшем будет использоваться декартова система координат,
но с тем же успехом можно пользоваться обобщенными координатами и импуль-
сами.
Положение отдельной одноатомной молекулы может быть задано тремя
координатами х, у, г, т. е. молекулы мы изображаем точкой в трехмерном кон-
фигурационном пространстве молекулы. В этом пространстве система N моле-
кул изображается N точками. Таким образом, задавая векторы положения
rv r2,..., rN в конфигурационном пространстве молекулы, мы получаем конфи-
гурацию всей системы. Это обычный способ представления газовых систем в
трехмерном пространстве. Однако иногда удобнее рассматривать гиперпро-
странство 3N измерений (т. е. пространство с 3N взаимно ортогональными
осями). В этом конфигурационном пространстве газа одна точка служит для
задания конфигурации всей системы, т. е. можно считать, что геометрия сис-
темы определяется вектором1) rN е= гх, г2, ..., rN с 3N компонентами.
Аналогично импульс отдельной одноатомной молекулы может быть пред-
ставлен точкой в трехмерном пространстве импульсов молекулы. В этом про-
странстве импульсы молекул представляются N точками. С другой стороны,
можно рассмотреть Замерное пространство импульсов газа, в котором компо-
ненты вектора pN = pv p2,..., pN определяют 3 компоненты импульса для
всех N молекул.
В гл. 1 уже говорилось, что знания всех импульсов и координат системы
достаточно для полного динамического описания системы. Поэтому часто исполь-
зуется понятие фазового пространства. Фазовым пространством молекулы, или
/^-пространством, как его обычно называют (/г вместо молекулы), является соеди-
нение конфигурационного пространства и пространства импульсов молекулы,
т.е. шестимерное пространство в случае одноатомной молекулы. Отдельная точка
этого пространства описывает положение и импульс молекулы. Динамическое
состояние системы N молекул дается соответственно N точками в //-пространстве.
Фазовое пространство газа, обычно называемое ^пространством (у вме-
сто газа), получаем при соединении конфигурационного пространства и про-
странства импульсов газа, и, таким образом, это пространство имеет 6N изме-
рений. Полное динамическое состояние системы в этом случае описывается
точкой в у-пространстве. Движение этой точки подчиняется законам движения
Ньютона или эквивалентным уравнениям Гамильтона.
Векторные обозначения см. в начале книги.
§ 1. Описание статистических ансамблей в классической механике 81
Важно ясно понять связь /л- и у-пространств. Динамическое состояние
системы N молекул можно представить либо одной точкой в у-пространстве,
либо облаком точек в /г-пространстве. Если все молекулы тождественны, то
при замене одних частиц другими облако точек ^-пространства будет представ-
лять одно и то же состояние системы. Таким образом, имеется N! таких пере-
становок молекул, которые соответствуют одному и тому же облаку точек в
/г-пространстве(т. е.тому же расположению пронумерованных точекI). Каждая
такая перестановка изображается новой точкой в у-пространстве. Таким
образом, в у-пространстве имеется N\ различных точек, соответствующих
облаку N точек //-пространства.
Для молекул с / степенями свободы конфигурационное пространство и
пространство импульсов молекулы имеют каждое / измерений; таким образом,
/г-пространство — 2/-мерное, а у-пространство имеет 2N/ измерений. Может
возникнуть вопрос, почему при построении этих фазовых пространств исполь-
зуются импульсы, а не скорости. Удобство такого выбора станет ясным в связи
с теоремой Лиувилля.
Б. АНСАМБЛИ И ФУНКЦИИ РАСПРЕДЕЛЕНИЯ2)
Сведения, касающиеся макроскопических свойств системы, можно полу-
чить путем статистического изучения большого числа систем, или ансамбля
динамически подобных систем. Например, если мы хотим предсказать поведе-
ние моля газа, заключенного в сосуде кубической формы, то представим себе,
что мы имеем большое число экземпляров таких систем, состоящих из того
же числа частиц того же сорта и заключенных в одинаковые сосуды. Каждый
из экземпляров совершенно независим от других, и они отличаются друг от
друга только тем, что фазы движения в различных сосудах различны.
Мгновенное динамическое состояние каждой системы ансамбля можно
описать соответствующей точкой в у-пространстве. Если ансамбль состоит из
множества систем, то точки систем образуют в у-пространстве нечто подобное
облаку. В этом случае можно ввести функцию q = q(tn} pN) плотности точек в
у-пространстве, так что q dtM dpN равно числу точек в элементе объема drN dpN.
Не теряя общности, такая функция может быть введена для дискретного облака
точек, так как можно представить q в виде набора б-функций Дирака. Иногда
удобно пользоваться нормированной функцией
pn{tn пт ^
где Г — число систем ансамбля. Тогда Р<^>(г^, р^) — функция плотности
вероятности. Вероятность нахождения выбранной случайным образом системы
ансамбля в состоянии со значениями tN pN, лежащими в пределах от г^ до
tn + dtN и от р" до р" + dpN, равна Р<">(г", р^) • dt"dp".
Для использования при выводе уравнения состояния в следующей главе
удобно ввести понятия функции полотности вероятности низшего порядка.
Выберем группу h молекул z, к,..., А и обозначим эту группу через {rj).
Теперь найдем вероятность3) P(h) (r*7, р^) того, что группа молекул {rj} находится
в состоянии со значениями тп, р*7, лежащими в пределах от т*1 до drn и от р^
до dpny причем г^ и р^ — это векторы в ЗЛ-мерном конфигурационном
подпространстве и подпространстве импульсов набора молекул {rj}, a
drv dpv = dt{ dtx... drkdpt dpx ... dpk. Очевидно,
pc>(r», p») = JJ P<N>(rN, p") dr"-* dpN~\ A.2)
x) Речь идет о нумерации состояний, по которым распределяются молекулы. — Прим. ред.
2) Обозначения различных видов функций распределения в этой главе совпадают с обо-
значениями, использованными в работе [1].
3) Если в группу {*7}входят молекулы 1, 2, 3, ..., Л, то мы используем обозначение
82 Гл. 2. Статистическая механика
где dr"1 dpN~n —элемент объема в 3(N — й)-мерном подпространстве у-про-
странства, соответствующего не принадлежащим группе {rj} молекулам.
Заметим, что в обозначении Р(/1) (г**, р^) символ Р указывает на число рассмат-
риваемых молекул, а г и р указывают на то, какие именно молекулы рассматри-
ваются.
Интегрирование Р<^>(г^, р^) или Pt^f1, pv) по пространству импульсов
приводит к
Р<")(Г") =.Jp<")(r", p") dp" A.3)
и
р<>0(гч) = [рС)^», pn) dp»7 = |ж")(г") drN~\ A.4)
Эти выражения дают соответственно вероятности того, что одна случайно выб-
ранная система ансамбля находится в положении, соответствующем хы или гп.
Величина P(N) (rN, pN) dtN dpN определилась как вероятность нахождения
случайно выбранной системы ансамбля в элементе объема dtN dpN около
значений tM, pN у-пространства. Для //-пространства вероятность нахождения
точек, соответствующих молекулам системы в элементе объема dr1dp1,...,
dxN dpN около значений tjpv..., rN pN с учетом всех возможных перестановок
молекул, будет в N! раз больше
/<N>(rN, pN)dxx rfpx... drNdpN = N ! P<N>(rN, p^)dtNdp". A.5)
Аналогично, поскольку h объектов из общего числа N могут быть выбраны
[N!/(N — Л)!] способами, функции плотности нижнего порядка для //-про-
странства записываются в виде
/(^ А = [-<л^ JJ/(N)(rN, pN)dr"-»dpN-\ A.6)
Функция наинизшего порядка имеет вид /A)(гх, р±) = NPA\rv рг). Именно эта
функция появляется в уравнении Больцмана (см. гл. 7, § 1) и из нее выводятся
формулы для коэффициентов переноса. Интегрирование /(l)(ri> Pi) по Pi Дает
плотность числа частиц газа n(rj. В кинетической теории обычно используется
не функция импульсов, а функция скоростей J(tv у2). В соответствии с этим мы
используем f(tv vx) в выводах кинетической теории. [Заметим, что эта функция
отличается от f^^i, Pi) отсутствием индекса A).]
Вероятности некоторой конфигурации по аналогии с A.3) и A.4) даются
выражениями
n(N)(rN) = N ! P<">(rN) A.7)
A.8)
Функция низшего порядка л^г^ есть просто плотность числа частиц п(гх),
а пB)(Г/, гу) — бинарная функция плотности вероятности, важная функция,
используемая в теории газов и жидкостей. Величина пB)(г/, ry) dtt dtj дает ве-
роятность нахождения молекул в элементах объема dth dtj около точек г, и
Гу соответственно.
Если N велико по сравнению с единицей, то иногда бывает удобно ввести
понятие радиальной функции распределения g(rh ry/), определяемой выражением
пB)(г„ tj) = л(г,) n(rj) g(rh tjd. A.9)
Функция g(rh Tji) стремится к единице при стремлении гп = \г] — г,| к беско-
нечности, а отклонения от единицы являются мерой корреляции положений
пары молекул. В условиях однородности в изотропной среде g(th х]г) есть
функция только rJh a 27r/22Vg(r/l) r2jt dr^ — плотность числа пар молекул,
расстояние между которыми заключено в интервале от rjt до гд + dr^.
§ 7. Описание статистических ансамблей в классической механике 83
Функции Р(л)(г^, р") и Р(л) (гп) называют видовыми функциями распреде-*
ления, а функции fhKfr}> Pv) и л(/1)(гП) — родовыми функциями распределения1).*
Последние имеют то преимущество, что множители N1 и (N — ft)! не появ-
ляются в формулах, в которых используются эти функции. Все эти функции
распределения вообще являются функциями времени, хотя зависимость от,
времени в приводившихся выражениях явно не выражена.
В. ИЗМЕНЕНИЕ ПЛОТНОСТИ ВЕРОЯТНОСТИ ВО ВРЕМЕНИ
В гл. 1, § 4 показано, что изменение во времени функции типа плотности
вероятности PlN)(tN, pM, t) описывается уравнением Лиувилля
= [Н,Р] - (# • S3 -(# • Sfl . <UO)
Как показано в гл. 1, § 4, уравнение Лиувилля является уравнением
неразрывности потока изображающих точек в фазовом пространстве. Так как
источники и стоки отсутствуют, то уравнение может быть записано в виде
0, A.11)
где DjDt — субстанциональная производная.
Обращение субстанциональной производной в нуль указывает на то, что
при движении любой точки в у-пространстве не происходит изменения во вре-
мени плотности точек, лежащих в ближайшей окрестности этой точки. Это
следствие теоремы Лиувилля известно как сохранение фазовой плотности.
Может быть также обсуждено второе следствие о сохранении фазового объема.,
Это следствие утверждает, что объем, занимаемый данным рядом точек, не
изменяется во времени при движении, хотя форма его непрерывно изменяется.
Рассмотрим такую малую область фазового пространства Ах, чтобы плотность
точек была существенно постоянной внутри этой области. Число точек, изо-
бражающих системы, в этой области дается выражением
Ап = Р^Ах. A.12)
Если поверхность элемента объема Ах в любой момент времени определяется,
точками, вначале* лежавшими на ней, то мы сможем следить за поведением
элемента объема во времени. В силу единственности механического движения
ни одна точка внутри области, ограниченной поверхностью, не сможет поки-
нуть объема ; источники и стоки точек также отсутствуют. Следовательно,
D(An) _
Dt ~ Dt nx^^ Dt —и- \iA6)
Член DP(N)/Dt обращается в нуль в силу A.11), так что
¦Ш(^) = О. A.14)
Справедливость последнего соотношения для фазового объема любых размеров
можно установить, проводя суммирование этого результата по всем малым
элементам объема.
Г. АНСАМБЛИ, ПРЕДСТАВЛЯЮЩИЕ ЗАМКНУТЫЕ СИСТЕМЫ
В СОСТОЯНИИ РАВНОВЕСИЯ
Чтобы ансамбль описывал систему в равновесии, плотность точек в фазо-*
вом пространстве должна оставаться постоянной во времени. Это означает, что
*) В оригинале „specific distribution functions" и „generic distribution functions".
Терминология «видовых определений» и «родовых определений* введена Гиббсом (см.
Дж. В. Г и б б с, Основные принципы статистической механики, М.—Л., 1946). — Прим. рео.
84 Гл. 2. Статистическая механика
в соответствии с A.10) мы должны потребовать выполнения следующего соот-
ношения :
^,p^,0 = [^,P<N>] =0. A.15)
Оно удовлетворяется, если Р<^> — постоянная относительно координат
г", р" или если PiN) = P(N)(a), где а = а(ты} pN)—интеграл движения. Инте-
гралы движения —это такие величины систем, которые не изменяются со вре-
менем (если система изолирована). Известными примерами интегралов дви-
жения являются энергия, импульс, момент количества движения. Здесь
будет рассматриваться только первый из этих интегралов, поскольку второй и
третий интегралы, соответствующие поступательным и вращательным дви-
жениям системы, обычно не существенны в термодинамическом отношении.
Для интегралов движения имеет место
[Я,а]=0; A.16)
следовательно, если P(N) — некоторая функция интеграла движения, то
[#,f*N>] =——[Я, а] =0. A-17)
Так что в соответствии с A.10)
Поэтому если pw= Р<лО(а), то ансамбль представляет установившееся, или
равновесное, распределение. Существуют две важные функции распределения
для равновесного состояния, имеющие свои названия. Сейчас мы просто дадим
их определение; позже будет показана полезность этих представлений и их
основания.
1*. Микроканонический ансамбль. Микроканоническим, или «ансамблем
энергетического слоя», является однопараметрический ряд распределений,
используемых для изучения изолированных систем, когда известны объем и
энергия системы. Функция распределения определяется равномерным распре-
делением изображающих систему точек в области между двумя соседними
поверхностями постоянной энергии фазового пространства ; вне этой области
распределение точек отсутствует. Математически ансамбль описывается так:
P(N) = PQ (постоянная) для значений энергии H(rN, pN),
лежащих в пределах от Е до Е + АЕ; A.19)
p(N) = 0 вне этой области.
Полагая АЕ -> 0, получаем поверхностный ансамбль, для которого все системы
обладают точно энергией Е.
2. Канонический ансамбль. Канонический ансамбль
Ж">(г", р") = [ZNNl Л*»] -i exp [- ^g^-] A.20)
также является однопараметрической функцией распределения для различных
значений Т. Здесь k — постоянная Больцмана, а Т позже будет отождествлена
с температурой; ZN — так называемая статистическая сумма, или сумма по
состояниям (или Zustandsumme), a Z^1 — нормировочный множитель. Вклю-
чение множителей N! и №м обсуждается в § 4.
Канонический ансамбль используется для изучения систем, температура и
объем которых заданы1). Для системы с большим числом степеней свободы он
х) Это показано в § 3.
§ 1. Описание статистических ансамблей в классической механике 85
близко апроксимирует микроканоническое распределение с энергией, соответ-
ствующей наиболее вероятному значению.
Таким образом, распределение по энергиям, представляемое каноническим
ансамблем, имеет очень острый максимум. Плотность точек для других значе-
ний энергии пренебрежимо мала1). Это легко показать. Число изображающих
систем — точек в заданном объеме — определяется выражением
pw dtNdpN = [ZN N! Л3"]-1
Очевидно, что это распределение имеет острый максимум, так как drjdH возра-
стает [как И в степени CN — 2)/2 \ в то время как экспонента быстро умень-
шается (см. задачу 2 в конце этой главы).
Средние значения, получаемые с помощью канонического ансамбля, в
точности совпадают со значениями, вычисляемыми с помощью микроканони-
ческого ансамбля. Однако при изучении флуктуации мы должны проявить
осторожность, выбирая подходящий ансамбль.
Д. АНСАМБЛИ, ПРЕДСТАВЛЯЮЩИЕ ОТКРЫТЫЕ СИСТЕМЫ
В СОСТОЯНИИ РАВНОВЕСИЯ2)
До сих пор рассматривались только системы с фиксированным числом
молекул. Они называются замкнутыми системами. Системы, в которых проис-
ходят химические реакции или изменения физического состояния3), называются
открытыми системами4). В этих случаях можно представить себе ансамбль,
различные системы которого имеют различное число v подсистем (молекул).
Можно показать, что ансамбль, представляющий открытые системы при по-
стоянной температуре Т в данном объеме V, является большим каноническим
ансамблем
/*> (г*, р") = [Z*v\ АЗ']-* ехр [Щ exp [- ^ХЦ . A.22)
Нормировочная постоянная ZB- — большая статистическая сумма системы ;
/г — химический потенциал ; P(v)(rv, pv) dtv dpv — вероятность того, что случайно
выбранная из ансамбля система состоит из v молекул и имеет координаты и
импульсы, заключенные в области dtv dpv около точки т% р\ Подобные ан-
самбли могут быть построены для открытых систем и при других условиях,
например система при постоянном давлении и температуре5).
х) Здесь имеется в виду тот факт, что плотность точек рассчитывается на единицу интер-
вала энергии. Плотность точек в фазовом пространстве максимальна при минимальных зна-
чениях энергии. — Прим. ред.
2) Более детальное обсуждение большого канонического ансамбля и его применения
см. в работе [2].
3) Имеются в виду изменения агрегатных состояний или фазовые превращения. —
Прим. ред.
4) Обычно открытыми системами называются системы, обменивающиеся веществом
с окружающей средой (см., например, С. д е Гроот, Термодинамика необратимых про-
цессов, М., 1956). Химические к фазовые превращения могут протекать и в закрытых системах.
Однако при подобных превращениях число частиц определенного вида (т. е. число частиц
данной химической компоненты или число частиц вещества в данном агрегатном состоянии)
может рассматриваться как переменное. Поэтому такие системы независимо от того, явля-
ются ли они открытыми или закрытыми, описываются с помощью большого канонического
ансамбля Гиббса. — Прим. ред.
5) Более подробное обсуждение большого канонического ансамбля и его применений
дано в работе [2].
86 Гл. 2. Статистическая механика
§ 2. ОПИСАНИЕ СТАТИСТИЧЕСКИХ АНСАМБЛЕЙ
В КВАНТОВОЙ МЕХАНИКЕ1)
До сих пор мы рассматривали только такие системы, которые адекватно
описываются в рамках классической механики. Однако при рассмотрении
систем, состоящих из атомов и молекул, необходимо использовать квантово-
механические представления. Так как существенной особенностью последних
является невозможность полного определения значений всех координат и
импульсов, то здесь должен быть использован другой подход. Прежде чем
применять квантовую механику к ансамблям систем, будут изложены некото-
рые основные идеи и элементы обозначений, относящихся к квантовоме^ани-
ческому описанию отдельных систем.
А. КВАНТОВОМЕХАНИЧЕСКОЕ ОПИСАНИЕ ОТДЕЛЬНЫХ
СИСТЕМ
Пусть состояние отдельной квантовой системы дается нормированной
волновой функцией 4f(tNy f). Эта функция имеет тот смысл, что ] W(tNyt)\2utN
является вероятностью нахождения в момент / координат системы в пределах
от г* до г^ + й*ы-
Динймика системы полностью определяется квантовомеханическим га-
мильтонианом ЛГ, а изменения состояния системы во времени описываются
вол: овым уравнением Шредингера [см. гл. 1, F.4)]. Волновая функция
системы ?P(rN, t) может быть «разложена» с помощью полного ортонормиро-
ванного ряда функций фп(ты) с рядом квантовых чисел {q} = qv q2,. .., q3N :
^(rN,O = 2^e(O^e(rN) B.1)
Q
[например, волновую функцию можно разложить по собственным функциям
энергии w (rN) с рядом квантовых чисел {а} = alf <т2,..., <r3N ]; ce(f) назы-
вают «амплитудой вероятности», а | cQ(t) |2 представляет собой вероятность
нахождения системы в состоянии {#}. Условие 2 \ cQ(t) |2 = 1 сразу следует
Q
из ортонормированности функций ^е(г^). Зависимость cQ(t) от времени дается
«обобщенным» (или «преобразованным») уравнением Шредингера
-TlF = ^<V@^Ve, B.2)
где
J г») d r». B.3)
= J &*(г")
Состояние системы, таким образом, может быть одинаково хорошо описано как
функцией W(tN, t), так и рядом амплитуд вероятности cq(t), а характер изменений
состояния системы во времени может быть описан одинаково хорошо как урав-
нением F.4) гл. 1, так и уравнением B.2) настоящей главы. Описание системы
в терминах амплитуд вероятности cQ(t) позволяет рассматривать, системы в
общем представлении, в котором координаты не играют особой роли. Действи-
тельные значения коэффициентов вероятности, конечно, зависят от частного
выбора ортонормированных функций в разложении B.1). Предположим, на-
пример, что W(tNy t) разложена также в ряд по функциям ^т(г^):
AtN). B.4)
J) Настоящий параграф написан совместно с профессором Амстердамского университета
И. де Буром. Более подробное рассмотрение обсуждаемых вопросов дано в работе [3].
§ 2. Описание статистических ансамблей в квантовой механике 87
Если %х(тм) и фп(тм) связаны унитарной матрицей1) преобразования afiT, то
Хт(т") = 2 VФ1*"\ или Ф1*ы) = 2<Zx(rN). B.5)
Q х
Тогда
?е = 2 V dt, или dT = 2 «7,1 се B.6)
Г Q
дает связь между амплитудами вероятности cQ(f) и df(f). Матричные элементы
*3f^ преобразуются следующим образом:
или 6 Q B.7)
Эти различные способы описания квантовомеханических систем будут исполь-
зованы в последующих параграфах, а также в гл. 6 при выводе квантового
уравнения состояния.
Б. ОПРЕДЕЛЕНИЕ МАТРИЦЫ ПЛОТНОСТИ ВЕРОЯТНОСТИ
До сих пор мы ограничивались случаем простой квантовомеханической
системы. Рассмотрим теперь ансамбль Г тождественных систем; состояние
/с-й системы будет определяться функцией W(k\tM, t) или рядом амплитуд веро-
ятности 4к)@> выражаемых аналогично B.1) следующим образом:
V«KtN,t) = 2c«Xt)te(r»). B.8)
Q
Определим теперь квантовомеханическую матрицу2) плотности вероятности
следующими двумя способами :
f f)f B.9)8)
B.Ю)
/c-1
Во втором выражении матрица «^$Р зависит от определенного ряда волновых
функций ^e(rN), выбираемого для описания системы. Переход к новой системе
собственных функций #T(rN), даваемой выражением B.5), позволяет, используя
выражение B.7), ввести матрицу плотности вероятности S5^, связанную с
&§) соотношением
или B.11)
т т*
Необходимо отметить, что матрицу ^ы\гм9 г/дг) можно рассматривать как
специальный вид обобщенной матрицы с^ее/. В этом случае для представления
х) Унитарная матрица — это такая матрица, для которой комплексно-сопряженная
транспонированная матрица равна обратной матрице, т. е.
атп = апт •
2) Нейман [4, 5 ] и Дирак [б, 7 ] показали, что величина, определяемая выражением
B.9), является квантовым аналогом классической вероятности в у-пространстве.
3) Заметим, что г2^ и t* — два различных вектора в ЗМ-мерном пространстве так же,
как q и q' — два различных квантовых числа.
88
Гл. 2. Статистическая механика
системы выбираются «собственные функции координат» (т. е. б-функции
ДиракаI)
V(b)(»t) $V(k\"N)d(''N-x«)dx"N. B.12)
Матрицей преобразования [соответствующей матрице <v в выражении B.5)>
связывающем функции 0Q(xN) и д(г"м — тм) ] является фХг"ы)> так что можно
записать выражения, аналогичные выражениям B.5) — B.7):
или
или
0Q(tN) = J ^(r"N)
cf\t) = J <pQ(r"N) W«Xr"N, t) d r"",
B.13)
B.14)
г*; r'") =
или
т'") d x'" dx"N.
Соответственно выражение
^N\r"; г'") =
или
W
B.15)
B.16)
^ B.17)
дает связь между матрицами <i#(N)(rN; х'н) и с^^.
Удобно ввести оператор вероятности ^N\ определяемый следующим
образом:
B.19)
Тм) d xN. B.20)
Выбор этого оператора вероятности ^<лг) [и> следовательно, матриц
^(N)(rN j r'N) и ^(^)] зависит от системы, которая изображается ансамблем.
Мы покажем вскоре, что для равновесного ансамбля оператор должен
являться функцией оператора, соответствующего некоторому интегралу дви-
жения системы.
Это позволяет выражения B.16) и B.17) записать в виде
В. ФИЗИЧЕСКИЙ СМЫСЛ МАТРИЦЫ ПЛОТНОСТИ
ВЕРОЯТНОСТИ
В классической статистической механике плотность вероятности
; pN) является функцией N векторов положения хы и N векторов
импульса pN.
Мы говорили выше, что квантовый ее аналог — функция Sfi(N\xM ; x'N),
являющаяся функцией двух рядов N векторов г^ и х'ы (или в общем
случае функция <?^\ являющаяся функцией двух рядов 3N квантовых
чисел {q} и {q'}). Фактически для матриц ^(м\хм, г/дг) и ^<^> невозможно
х) E-функция Дирака обладает тем свойством, что она равна нулю для всех значений
аргумента, кроме значения |r"w — rw| = 0. В этой точке она имеет столь большое значение,
что интеграл от функции по всему пространству координат равен единице. О других свой-
ствах E-функции см. в работе Шредингера[8].
§ 2. Описание статистических ансамблей в квантовой механике 89
дать четкую физическую картину, но, как обычно в квантовой механике,
удобно приписать физический смысл диагональным элементам матрицы. В
классической теории вероятность нахождения произвольно выбранной из
ансамбля системы в положении г^ дается выражением
Ny pN) d pN# B.21)
В квантовой механике эту вероятность получаем, беря диагональный элемент
^ы\гы, гы). Таким образом, с этой величиной связывается вероятность нахож-
дения произвольно выбранной из ансамбля системы в положении хы. Анало-
гично с &vp связывается вероятность пребывания системы в состоянии с соот-
ветствующим набором квантовых чисел {д}. Особенно важен случай, когда
разложение волновой функции системы B.1) производится в ряд по собствен-
ным функциям энергии ^(rN), так что <&$) представляет вероятность нахож-
дения системы в энергетическом состоянии {а}.
По аналогии с нормировкой классической функции плотности вероятности
мы определим квантовомеханическую матрицу плотности вероятности так,
чтобы
j^N\tN; г") d г» = 2 ^й° = 1 • B.22)
Классической операции интегрирования функции плотности вероятности по
всему у-пространству в квантовом случае соответствует след матрицы плот-
ности вероятности.
Г. ДРУГИЕ МАТРИЦЫ ПЛОТНОСТИ ВЕРОЯТНОСТИ
Можно ввести несколько других матриц плотности вероятности, просто
получаемых из матрицы ^(ы\тм; r'N). Использование этих матриц плотности
упрощает многие формулы, которые мы встретим при обсуждении квантового
уравнения состояния. Во-первых, мы определим видовую матрицу плотности
вероятности <i#(/l)(rn ; г'1») следующим образом :
г*"» ; г"» г""") dt-Nf>. B.23)
Эта матрица получается из диагональных элементов, соответствующих моле-
кулам, не входящим в группу h молекул i, к,..., A = {i?}, и последующим
интегрированием по всем векторам положения (N — К) молекул. Тогда диа-
гональный элемент ^h)(tv; г**) дает вероятность того, что в любой произвольно
выбранной системе группа молекул {rj} будет находиться в положении г*7.
Определим еще родовую матрицу плотности вероятности
здесь множитель [NlfcN — ft)! ] — число возможных способов выбора ft объек-
тов из общего числа N объектов. Диагональные элементы этой матрицы явля-
ются квантовыми аналогами родовой конфигурационной плотности вероят-
ности, даваемой выражением A.8) этой главы.
Д. ЗАВИСИМОСТЬ МАТРИЦЫ ПЛОТНОСТИ ОТ ВРЕМЕНИ ;
АНСАМБЛИ ДЛЯ ЗАМКНУТЫХ СИСТЕМ В СОСТОЯНИИ РАВНОВЕСИЯ
Комбинируя выражение B.9) и формулу F.4) гл. 1 или выражения
B.10) и B.2), можно сразу получить уравнения
—- ^(N)(rN . f'N) = _ !_ (,3f?5№ _ ^j(N) , Jf ) (fN . r'N)? B.25I)
~—&5(N) = — (.J^^(N) ^3(N) <Jf\ ' . B.26)
r; r'N) означает r^ ; т'я — элемент матрицы (<Т&>Ш —,
90 Гл. 2. Статистическая механика
Эти уравнения аналогичны уравнению Лиувилля в классической статистиче-
ской механике.
Распределение для стационарного состояния (для которого д^ыIЫ = 0)
определяется выражением {>У?<&>'Ы) — <9>{ы)<Ж) = 0. Обращение в нуль этого
коммутатора может иметь место только в том случае, если матрица плотности
вероятности является функцией некоторой матрицы, представляющей интеграл
движения. В случае двух типов равновесных ансамблей, используемых для
представления замкнутых систем и обсуждавшихся в предыдущем разделе,
матрицы могут быть записаны в явном виде1).
1. Микроканонический ансамбль.
— Сд<т<т' в малом интервале значений энергии Ео ^ Ev ^ Ео + дЕ, B.27)
= 0 вне этого интервала.
2. Канонический ансамбль.
=ZjU exp (-^f?1)- B.28)
Здесь нормировочным множителем Zn9 является квантовомеханическая
статистическая сумма. Микроканонический ансамбль, очевидно, используется
для представления изолированной, замкнутой системы при постоянной энергии.
Позже будет показано, что каноническим ансамблем представляются изолиро-
ванные, замкнутые системы при постоянной температуре. Оба ансамбля дают
одинаковые результаты при вычислении средних значений (см. § 3), и, следо-
вательно, для этих целей можно одинаково успешно пользоваться обоими
ансамблями. Поскольку микроканонический ансамбль не является непрерыв-
ной функцией энергии, а канонический ансамбль является непрерывной функ-
цией, то последний значительно более удобен с математической точки зрения.
Е. АНСАМБЛИ ДЛЯ ОТКРЫТЫХ СИСТЕМ
(БОЛЬШОЙ КАНОНИЧЕСКИЙ АНСАМБЛЬ) В СОСТОЯНИИ РАВНОВЕСИЯ
Для рассмотрения открытых систем, заключенных в данном объеме V при
постоянной температуре Г, оператор вероятности может быть представлен в
виде
jew = (ZB)-i exp (¦?) ехр (- *?) , B.29)
где Zf — нормировочная постоянная (большая квантовомеханическая стати-
стическая сумма); /г — химический потенциал; >Ж^ — гамильтониан системы
v молекул. Число частиц v представляющего ансамбля выражается здесь обыч-
ным числом. В действительности, строго говоря, v должно быть заменено опе-
ратором в основном таким же образом, как представляется оператором и энер-
гия2).
Используя выражения B.19) и B.20), можно получить
J*w (г; г) = (Zf)-i ехр (?) 2 Ф*« (П ехр (- ^г) Ф* (г") > B-30)
<*&> = (ZB)-i exp (i?) ехр (- ^). B.31)
представляющие вероятности того, что случайно выбранная из ансамбля си-
стема будет системой v молекул с положением г" и системой v молекул с энер-
гией Е^ соответственно.
1) Матрица плотности в энергетическом представлении называется обычно статисти-
ческой матрицей ; соответствующий оператор — статистическим оператором. — Прим. ред
2) Подробное обсуждение этого вопроса см. в работе К. Хушими [9], особенно в ч. II,
§6, 11 и 12.
§ 3. Основы статистической механики 91
§ 3. ОСНОВЫ СТАТИСТИЧЕСКОЙ МЕХАНИКИ
Выше было проведено классическое и квантовое рассмотрение сложных
систем и введена терминология, необходимая для статистического рассмотре-
ния подобных систем. Теперь уже без использования статистических методов
будет весьма трудно продвинуться дальше в описании свойств таких систем.
Это можно было бы сделать с помощью быстродействующих вычислительных
устройств. Однако полученные при этом сведения о детальном микроповедении
имели бы небольшую ценность, если бы они не выражались в терминах макро-
свойств. Как известно, при эмпирическом или термодинамическом рассмо-
трении макросостояние системы определяется небольшим числом «параметров
состояния», таких, например, как температура и давление. С макроскопи-
ческой точки зрения полный набор параметров состояния полностью опре-
деляет прошлое и будущее системы.
А. ОБОСНОВАНИЕ МИКРОКАНОНИЧЕСКОГО АНСАМБЛЯ
Макроскопические свойства равновесной системы не изменяются во вре-
мени. Напротив, микроскопическое динамическое состояние системы изме-
няется со временем, и в соответствии с представлениями классической меха-
ники изображающая точка прочерчивает траекторию в фазовом пространстве.
Наблюдаемые свойства системы являются средними во времени вдоль траек-
тории. Из важной теоремы, доказанной Биркгофом (см. [10,11]), следует,
что почти для всех траекторий (за исключением множества меры нуль) эти
средние по времени существуют в том смысле, что существуют предельные их
значения для больших интервалов времени.
Вторая часть теоремы Биркгофа утверждает, что вдоль траектории среднее
по времени от произвольной функции координат и.импульсов системы равно
среднему по некоторому подпространству фазового пространства. Это под-
пространство является той частью фазового пространства, для которой полный
набор интегралов движения имеет определенный заданный ряд значений.
«Эргодическая» система — это такая система, для которой единственными инте-
гралами движения являются энергия и компоненты импульса и момента коли-
чества движения. Весьма вероятно, что большинство интересующих нас физи-
ческих систем являются эргодическими. Следовательно, предположив, что мы
рассматриваем эргодическую систему, можно представить ее ансамблем, в кото-
ром изображающие точки равномерно распределены в той области фазового
пространства, где энергия, импульсы и момент количества движения имеют
заданные значения. Статистическая механика обычно применяется к изучению
невращающихся систем, находящихся в покое относительно выбранной си-
стемы координат1). Следовательно, члены ансамбля обладают равными нулю
импульсом и моментом количества движения и заданной энергией. Это не что
иное, как микроканонический ансамбль, определенный в § 1. Этот ансамбль
представляет собой систему с заданной энергией, т. е. изолированную систему.
Как правило, нас интересуют не свойства изолированной системы, а поведе-
ние системы, находящейся в термостате при известной температуре. Как мы
вскоре увидим, канонический ансамбль представляет собой такую систему.
Б. ИСПОЛЬЗОВАНИЕ МИКРОКАНОНИЧЕСКОГО АНСАМБЛЯ
ДЛЯ ПОЛУЧЕНИЯ ВЫРАЖЕНИЯ РАСПРЕДЕЛЕНИЯ ЭНЕРГИИ
ПО МАКРОСКОПИЧЕСКИМ ПОДСИСТЕМАМ
Рассмотрим изолированную систему, состоящую из большого числа N
одинаковых подсистем. Подсистемы предполагаются макроскопическими по
1) Вращающиеся системы могут быть рассмотрены этим методом путем введения фик-
тивных внешних сил или путем рассмотрения нового ансамбля. Последний случай рас-
смотрен Толманом [3].
92 Гл. 2. Статистическая механика
размеру (так что они могут быть пронумерованы) и отделенными друг от друга
таким образом, что от одной системы к другой возможна только передача энер-
гии, но не массы. Таким образом, каждая подсистема фактически помещена в
термостат, образованный конечным числом одинаковых членов ансамбля.
В этом случае может быть использована терминология классической меха-
ники. Результаты квантового рассмотрения будут даны позже. Обозначим
фазовое пространство всей системы как у-пространство, а для отдельной под-
системы как /г-пространство ; таким образом, микроскопическое динамиче-
ское состояние отдельной системы может быть изображено точкой в у-про-
странстве или облаком пронумерованных точек в /г-пространстве.
Для вычислительных целей удобно разделить у-пространство на большое
число малых ячеек, каждая объемом Агу. Пронумеруем затем эти ячейки, после
чего состояние системы может быть задано координатами точки, изображающей
систему, или номером ячейки, в которой эта точка расположена. Конечно, вто-
рой метод описания динамического состояния системы несколько менее точен.
Однако при выборе ячеек достаточно малыми состояние системы может быть
задано с любой степенью точности.
Теперь каждая ячейка у-пространства соответствует набору N ячеек в
^-пространстве, соответствующих N одинаковым подсистемам. Если мы гово-
рим, в какой ячейке у-пространства находится изображающая точка си-
стемы, то можно сказать, в каких ячейках /г-пространства лежат изобра-
жающие точки подсистем. Вследствие идентичности подсистем при рас-
смотрении макросвойств единственно важным является вопрос о том, сколь-
ко изображающих точек (а не какие именно изображающие точки) подсистем
находится в каждой ячейке ^-пространства.
Таким образом, мы определяем макросостояние системы посредством
ряда чисел, указывающих, сколько изображающих точек подсистем лежит в
каждой ячейке /г-пространства. Вообще, одному и тому же макросостоянию
соответствует несколько микросостояний. Пусть п( — число изображающих
подсистемы точек, находящихся в i-й ячейке /г-пространства. Величину nh
характеризующую макросостояние, мы назовем числом заполнения. Количество
ячеек в у-пространстве (т. е. число состояний системы), соответствующее дан-
ному макросостоянию, дается выражением
пг\ п2! л8!... 1°л'
Как указывалось выше, наблюдаемые свойства системы являются средними
по времени вдоль естественной траектории в у-пространстве. Это среднее по
времени, однако, является тем же, что и «фазовое среднее» вдоль «эргодической
поверхности». Для большинства (если не для всех физических систем) эта
эргодическая поверхность — «поверхность» постоянной энергии в у-простран-
стве1). Поэтому для вычисления вероятного поведения отдельной системы мы
представляем ее микроканоническим ансамблем, т. е. утверждаем, что все
состояния системы с данной энергией равновероятны2). Следовательно, вероят-
ность W нахождения системы в определенном макросостоянии, характеризуе-
мом числами заполнения п19 п2, п3,..., выражается в виде
^=4w- C-2>
3
где С — нормировочный множитель.
х) Более строгое рассмотрение см. в книге [10].
2) Здесь необходимо использовать представление об объеме фазовой ячейки, так как
распределение изображающих точек на поверхности постоянной энергии не равномерно, а
зависит от скорости изменения величины H(r*% p*) с расстоянием в направлении, перпенди-
кулярном поверхности. Этот факт связан с понятиями «теории меры», используемой при
доказательстве теоремы Биркгофа.
§ 3. Основы статистической механики 93
Ясно, что наиболее вероятное макросостояние системы описывается набо-
ром чисел заполнения, соответствующим максимальной величине W. Однако
нельзя независимо изменять все числа заполнения, так как они должны удо-
влетворять следующим двум условиям :
^nj = N, C.3)
^njEj = E, C.4)
где Е — полная энергия системы, а ?у — энергия подсистемы, изображающая
точка которой расположена в /-й ячейке.
Таким образом, для определения наиболее вероятного макросостояния
системы мы должны найти максимум выражения W относительно всех вариа-
ций чисел п, с учетом условий C.3) и C.4). Предположим, что числа л, доста-
точно велики, так что справедливо приближение Стирлинга, а также допустим,
что числа щ могут рассматриваться как непрерывные переменные. Эти допу-
щения не вносят заметной ошибки. Пусть 6njy 6lnWf 6N и ЬЕ — малые изме-
нения величин njy \nW, N и Е соответственно. Тогда для экстремальных зна-
чений W имеем
д In W = - 2 (In Tij + l)dtij = 0 C.5)
наряду со следующими условиями :
= O, C.6)
ij = O. C.7)
Воспользовавшись методом множителей Лагранжа1), можно написать выра-
жение
«In W-(a- l)dN-PdE = O, C.8)
где а и р — произвольные постоянные, которые позже будут выражены через
N и ?.
Используя выражения C.5)—C.7), можно переписать последнее выра-
жение в виде
2 (In tij + a + pEj) dnj = O, C.9)
где а и р — неопределенные постоянные. Так как <5пу можно теперь рассматри-
вать как произвольную вариацию, то коэффициенты при каждом бпу должны
быть равны нулю. Таким образом,
j pj ,
или C.10)
fij = ехр [-а - PEj],
где черточкой обозначено наиболее вероятное значение Щ (пу — значения
чи^ел заполнения, дающие максимальное значение вероятности W). Этот резуль-
тат дает наиболее вероятное макросостояние системы или наиболее вероятное
распределение подсистем по ячейкам //-пространства.
В. СРЕДНИЕ ПО АНСАМБЛЮ И ФЛУКТУАЦИИ
Обычно имеется конечная вероятность W(nvn2,...) нахождения системы
в определенном макросостоянии, отличном от наиболее вероятного. Однако
если система содержит большое число подсистем, то величина W(nv п2,...),
См., например, книгу [12].
94 Гл. 2. Статистическая механика
отличная от максимального значения W = W(nv п2У...), стремится к нулю
при больших N. Этот результат становится точным при N -> со. Для конечных
значений N имеются небольшие флуктуации около наиболее вероятного рас-
пределения.
Рассмотрим для системы динамическую переменную X(tN, pN).
Здесь Х(ю — значение динамической переменной для системы в fc-м со-
стоянии (т. е. когда изображающая точка системы находится в fc-й ячейке
у-пространства). Среднее по ансамблю для величины X выражается через
У — У Р ХМ v И 11 ^
у\ — ^j * к "^ > \*^ /
где Рк — нормированная вероятность нахождения системы в fc-м состоянии.
Таким образом, ряд значений Распределяет ансамбль. В этом случае, поскольку
ансамбль —микроканонический, Рк = 0, если ячейка не соответствует энергии Е.
В последнем случае величина Рк равна постоянной, определяемой из условий
нормировки.
Вследствие тождественности подсистем величина Х(Л) имеет одно и то же
значение для всех микросостояний, соответствующих одному макросостоянию.
Следовательно, сумма в выражении C.11) может быть переписана в виде суммы
не по микросостояниям, а по макросостояниям
Х~ V II/ V(O СХ 1 0\
i
где Wi — вероятность нахождения системы в макросостоянии /, определяемом
набором чисел заполнения п,-. Значения величины Wt даются выражением C.2).
Максимальное значение величины Wiy обозначенное W, соответствует наи-
более вероятным значениям чисел заполнения и дается выражением C.10).
Рассмотрим вероятность W + 6W макросостояния, очень мало отличаю-
щегося от наиболее вероятного. Как было показано, отношение величины
W + 6W к величине W для наиболее вероятного макросостояния становится
очень малым при увеличении числа N подсистем. Пусть лу — числа заполне-
ния, определяющие наиболее вероятное макросостояние, а /2у + <5пу — числа
заполнения для слегка отличного макросостояния. Тогда из выражения C.2)
следует
w + sw П ml
C.13)
Теперь, используя приближение Стирлинга и тот факт, что величины Пу и воз-
мущенные величины Яу + drij удовлетворяют условиям C.3) и C.4), можно
преобразовать C.13) и получить выражение для величины In [(W + dW)/W]
с точностью до второго порядка вариации Ьщ
Ч"^?^\ = -2*"№-т2^+--- C.14)
Поскольку щ — числа заполнения, соответствующие максимуму величины
Wif то первая вариация равна нулю и мы получаем выражение для «кривизны»
(ЗЛ5)
где at — n,/N и <5а,- = <5n,/N — наиболее вероятное значение относительных
чисел заполнения и соответствующая вариация. При возрастании числа N
наиболее вероятное значение относительных чисел заполнения остается по-
стоянным. Следовательно, величину отношения вероятности любого опреде-
ленного макросостояния к вероятности наиболее вероятного макросостояния
можно сделать как угодно малой, если в достаточной степени увеличить полное
число подсистем N.
§ 3. Основы статистической механики 95
Чтобы показать, что при предельно больших N среднее значение вели-
чины X определяется только наиболее вероятным макросостоянием, рассмо-
трим выражение C.12). Для больших значений N суммирование можно заме-
нить интегрированием, т. е.
Г... [xwdnxdn2...
X = у-7 C.16)
... XWdrixdnt...
Хотя здесь знаменатель равен единице в силу нормированности, удобно запи-
сать это выражение именно в этой форме. Интегрирование ведется по всей
гиперповерхности с учетом условий C.3) и C.4). Теперь, разделив числитель
и знаменатель на N в одной и той же степени и воспользовавшись для W выра-
жением C.15), получаем
Г... Гх ехр I- ^ 2 («* - <№] <tai <*«¦-..
X = f г / N х, f , (ЗЛ7)
J ... J ехр |- ^- Z («* - vJm dax da%...
где вместо вариации &at написано (а( — я,). Величина X зависит только от
относительных чисел заполнения, и X может быть разложена в ряд Тэйлора
вблизи значений, соответствующих наиболее вероятному макросостоянию.
Разложение можно записать в виде
4^)..., C.18)
где индексом О обозначены значения, соответствующие а(. При интегрировании
учтем, что Хо, (ЭХ/Эа,H (Э2Х/Эа/ЭауH,... являются постоянными величинами.
Первый член в выражении C.17) соответствует вкладу величины Хо в значение
X. Другие члены дают такие добавки, которые при увеличении N могут быть
сделаны сколь угодно малыми. Сделав простую замену переменных, можно
увидеть, что величина X, в которую входит s-я производная от X, стремится
к нулю при больших значениях N как N~s/2. Таким образом, для больших
значений N имеем
X = Хо. C.19)
Этот результат указывает на то, что для систем, содержащих большое число
подсистем, только наиболее вероятное макросостояние определяет среднее
значение. Более того, в пределе при бесконечно больших N флуктуации состоя-
ния системы становятся равными нулю.
Г. РАСПРЕДЕЛЕНИЕ ЭНЕРГИИ МЕЖДУ МОЛЕКУЛАМИ
В ГАЗЕ
Применим только что рассмотренный метод к случаю классического иде-
ального газа, в котором отсутствует взаимодействие между молекулами. В этом
случае системой является данный объем газа, а подсистемами — молекулы
газа. Полная энергия системы Е является суммой энергий отдельных моле-
кул Ej. В свою очередь энергия ?/ есть сумма кинетической энергии и энергии
внутренних степеней свободы молекулы. Величина Ej зависит от координат1)
/-й ячейки (q, p) и определяется функцией Гамильтона #(q, p) для отдельной
молекулы.
Так как РA)(Ч, Р), по определению, есть нормированная функция распре-
деления изображающих точек в //-пространстве, то числа заполнения могут
х) Здесь q представляет набор обобщенных координат, описывающих положение и кон-
фигурацию отдельной молекулы, а р соответствует набору сопряженных импульсов. Таким
образом, р и q — это векторы в /-мерных пространствах координат и импульсов соответст-
венно.
96 Гл. 2. Статистическая механика
быть выражены в виде
л, = JV J PA) (q, P) dq dp. C.20)
По у-й ячейке
Следовательно, если ячейки выбраны достаточно малыми, то наиболее вероят-
ная функция распределения имеет вид
), C.21)
где zhf — соответствующая нормировочная постоянная. В гл. 4, § 2, будет
показано, что множитель Лагранжа /? имеет физический смысл величины 1//сТ.
Так как газ состоит из конечного числа молекул, то имеется конечная, хотя
и малая вероятность пребывания газа в другом макросостоянии, когда газ
описывается другой функцией распределения. Для одноатомного газа (без вне-
шних и внутренних сил) гамильтониан отдельной молекулы имеет вид
"('.»)="?•• C-22)
Таким образом, выражение C.21) превращается в
P<i> (г, р) = -L BлткТ)-г!* е-Рг12ткт. C.23)
Эта функция нормирована так, чтобы при ее интегрировании по всему //-про-
странству получить единицу. Для некоторых целей (и, в частности, при обсуж-
дении кинетической теории) удобнее ввести функцию распределения /A)(г, Р),
нормированную так, что интегрирование по всему пространству импульсов
молекулы дает величину п (плотность числа частиц). Эта функция имеет вид
/а) (г, р) = п BлткТ)-г1> е-Рг12ткТ, C.24)
или, выражая через молекулярную скорость V, получаем
, v) = n {-~f е-™ч™ C.25)
— так называемую «функцию распределения Максвелла—Больцмана»1). В кине-
тической теории этот же результат получается как равновесное решение урав-
нения Больцмана. Можно показать, что функция Максвелла—Больцмана
C.25) описывает распределение скоростей молекул также и в неидеальном
газе (в газе взаимодействующих молекул) и газе многоатомных молекул. При-
меры применения выражений C.21) и C.25) даны в задачах в конце этой главы.
Д. ОБОСНОВАНИЕ КАНОНИЧЕСКОГО АНСАМБЛЯ
Методы, описанные в предыдущем разделе, неприменимы при рассмот-
рении неидеальных газов, жидкостей или твердых тел. Для таких систем пол-
ная энергия является суммой энергий отдельных молекул и энергии, связан-
ной с межмолекулярными взаимодействиями. Следовательно, необходимо
использовать более мощный метод статистической механики, который может
быть развит путем другой интерпретации рассмотренного в гл. 1, § 3 общего
формализма.
При изучении свойств идеального газа отдельные его молекулы рассматри-
вались как подсистемы, а находящийся в сосуде газ — как система. При изу-
чении реальных веществ, молекулы которых взаимодействуют, в качестве под-
системы мы выберем содержащийся в сосуде неидеальный газ (жидкость или
твердое тело) N молекул. В качестве системы мы выберем большое собрание Г
таких сосудов с газом, каждый из которых имеет жесткие стенки, и располо-
женных так, что между ними имеется тепловой контакт. Таким образом, между
сосудами возможна передача энергии, но нет переноса массы. Можно пред-
ставить себе, что каждый из этих сосудов помещен в большой термостат, обра-
х) В советской литературе распределение C.25) обычно называется распределением
Максвелла. — Прим. ред.
§ 3. Основы статистической механики 97
зованный из собрания подобных сосудов. В этом случае выражение (ЗЛО)
можно интерпретировать как дающее вероятность нахождения изображающей
сосуд точки в /-й ячейке фазового пространства с энергией Ej. Энергия Ej
зависит от координат /-й ячейки (rN, pN) и является суммой кинетических
энергий всех молекул в сосуде и энергии, связанной с внутренними степенями
свободы и межмолекулярными силами. Таким образом, Ej дается функцией
Гамильтона H(tN, рм). Так как, по определению, P(N\tN, ры) — нормирован-
ная функция распределения в фазовом пространстве, соответствующем N мо-
лекулам газа, то числа заполнения пу можно выразить через интеграл по /-й
ячейке этого фазового пространства
я, = Г J Р<"> (г", р") drN dp". C.26)
По у-й ячейке
Следовательно, если ячейки выбраны достаточно малыми, то функция, пред-
ставляющая наиболее вероятное распределение фазовых точек (соответствую-
щих сосудам с газом), может быть записана в виде
" р~) =
где N\h3NZN — нормировочный множитель, а /?, как будет показано дальше,
равна 1/fcT. В разд. В было показано, что если предположить, что система
состоит из большого числа Г сосудов с газом, то в пределе при Г, стремящемся
к бесконечности, вероятность нахождения собрания сосудов в макросостоянии,
отличном от описываемого выражением C.27), равна нулю.
Большое число одинаковых сосудов с газом, окружающих рассматри-
ваемый сосуд и имеющих с ним тепловой контакт, образует идеальный
термостат. Однако свойства газа в равновесии не зависят от материала или
конструкции термостата. Это можно показать статистическим путем, при-
меняя обсуждавшиеся выше аргументы к сосуду, в котором молекулы типа
А и В находятся по разные стороны непроницаемой теплопроводящей пере-
городки. Выше мы нашли, что функции распределения для двух типов газов
характеризуются одним и тем же значением величины /?. Теперь, переходя
последовательно к более сложным системам, в которых сосуд А окружен боль-
шим количеством сосудов В, находящихся в тепловом контакте с ним, мы
найдем, что значение величины /? остается неизменным, а распределение по-
прежнему дается выражением C.27).
Ансамбль, описываемый функцией распределения C.27), известен как
канонический ансамбль и представляет систему известной температуры или
систему, помещенную в термостат. Как было показано в § 1, если N велико,
эта функция распределения приводит к имеющему очень острый максимум
распределению энергии. Поэтому канонический ансамбль можно рассматри-
вать как удобное математическое приближение к микроканоническому ансам-
блю. В самом деле, коль скоро рассматриваются только усредненные свойства
системы, оба ансамбля приводят к одинаковым результатам. Однако при изу-
чении флуктуационных явлений важно, чтобы рассматриваемая система была
представлена соответствующим ансамблем.
Функция распределения Р^\гы9 pN) дает более полное описание состоя-
ния системы, чем описание, даваемое простой функцией Максвелла—Больц-
мана P(l)(q, p) (полученной в предыдущем параграфе). Функция P^\t^f p^)
дает сведения о вероятности относительного положения молекул, т. е. о веро-
ятности появления пар и групп молекул. Функция распределения P^q, p)
может быть получена из Р^(ты9 рм) интегрированием по всем координатам
и импульсам, за исключением координат и импульсов одной молекулы. Функ-
ция P(l)(q, p), полученная таким образом, подтверждает справедливость при-
менения распределения Максвелла—Больцмана к случаю неидеальных газов,
жидкостей и твердых тел. [Необходимо напомнить, что функция PA)(q, p)
получена в разд. Г только для идеальных газов. ]
Гл. 2. Статистическая механика
Е. ВЫЧИСЛЕНИЕ СРЕДНИХ ПО АНСАМБЛЮ
В начале этого параграфа было показано, что наблюдаемые на опыте вели-
чины могут быть вычислены как средние значения по ансамблю. В частности,
для величины X(rN9 pN) среднее (наблюдаемое) значение дае'Гся выражением
Х=\^Х (г", pN) Р<"> (г", р") dtN dpN. C.28)
Функция распределения для микроканонического ансамбля дается выраже-
нием A.19); для канонического ансамбля соответствующая функция дается
выражением C.27) и
z^ Я х <r" р"> е~т (г*л drN d*N
выражает среднее значение динамической переменной X. Рассмотрение, про-
водимое здесь, строго применимо только к классическим системам. Аналогич-
ные результаты могут быть получены при использовании методов квантовой
механики. Квантовомеханическое среднее значение динамической переменной
Х(ты, pN) (представляемой оператором Об) для fc-й системы ансамбля, находя-
щейся в состоянии, характеризуемом волновой функцией Ч*к\ выражается
следующим образом:
А'<*> = J У<*>* (fN) ^ У< ) (fN) dTN # C.30)
Среднее значение динамической переменной по ансамблю является просто сред-
ним арифметическим
2
к
= г-*222#>• @4k)@ ,,
/с е с'
где Г — число систем в ансамбле. Последнее выражение записано через коэф-
фициенты разложения c(k\t) [см. B.1)]. Его можно записать также через ста-
тистический оператор & :
X = f (^ Об) (г"; г") dr» = 2(& Ж)ее. C.32)
J Q
Статистический оператор для микроканонического ансамбля дается выраже-
нием B.27), а для канонического — выражением B.28).
В последнем случае для среднего значения динамической переменной,
соответствующей квантовомеханическому оператору Об, мы имеем
(з.ЗЗ>
±-
§ 4. ОСНОВЫ СТАТИСТИЧЕСКОЙ ТЕРМОДИНАМИКИ
Чтобы результаты, полученные в предыдущих параграфах, могли быть
использованы, необходимо выравить их в терминах макроскопически изме-
римых величин. В настоящем параграфе мы покажем, каким образом статисти-
ческая механика может быть использована для определения термодинами-
ческих величин — внутренней энергии, энтропии и температуры. Более того,.
представляет сокращенную запись интеграла
§4. Основы статистической термодинамики 99
из статистических принципов могут быть выведены все три основных закона
термодинамики. Связь статистической механики и термодинамики обычно
обсуждается посредством рассмотрения так называемой статистической суммы.
Поэтому мы предпошлем рассмотрению статистической термодинамики краткое
обсуждение статистической суммы в квантовой и классической статистике.
А. СТАТИСТИЧЕСКАЯ СУММА
Квантовомеханическая статистическая сумма1) системы N молекул ZNq уже
была определена выше как нормировочный множитель для квантовой кано-
нической функции распределения. Статистическая сумма (называемая иногда
суммой по состояниям, или Zustandssumme) определяется двумя эквивалент-
ными соотношениями
7 = у р-рЕ) — V a.p-PEj (Л\\
Сумма по всем Сумма по всем
энергетическим энергетическим
состояниям уровням
В первом соотношении ?, дает энергию системы в квантовом состоянии с кван-
товыми числами { /} = jv /2... С другой стороны, во втором соотношении Е}
соответствует энергии /-го уровня, а вырожденность уровня указывается сим-
волом gj (т. е. /*-й уровень состоит из gy состояний). В литературе обычно исполь-
зуются оба выражения статистической суммы; в этой книге будет исполь-
зоваться то или другое выражение в зависимости от удобства. Статистическая
сумма является функцией величины C (позже будет показано, что /? = 1/fcT),
а также некоторых внешних параметров системы, влияющих на энергетические
уровни. Обычно единственным учитываемым механическим параметром явля-
ется объем. Характер зависимости Е1 от объема будет обсуждаться в § 5 в связи
с практическими термодинамическими расчетами.
Квантовомеханическая статистическая сумма может быть записана также
в виде интеграла по конфигурационному пространству аналогично выражению
B.19) для &N(rN,rM)
ZNq = I 2 Ф*{*") е ~ р<* фе(г") dtN. D.2)
J Q
Для статистической суммы, записанной таким образом, зависимость от объема
выражается через пределы интегрирования по конфигурационному прост-
ранству, а также через граничные условия, которым должна удовлетворять
функция разложения фг В предельном случае классического поведения
системы можно показать2), что статистическая сумма, выражаемая формулой
D.2), является интегралом по классическому фазовому пространству
ZN = [N! ft3"] \\e~№N> p*> dr"dp". D.3)
Это выражение классической3) статистической суммы справедливо только для
случая системы тождественных частиц.
Появление в знаменателе множителя N1 обусловлено принципом Паули.
Вследствие тождественности частиц определенные области фазового прост-
х) Как классическая, так и квантовомеханическая статистические суммы обозначаются
символом Zn. Чтобы различить их, мы будем квантовомеханическую статистическую сумму
обозначать индексом q(Zsq).
2) В гл. 6 детально рассматривается квантовоклассическое соответствие плотности ве-
роятности. Точное доказательство того, что в пределе функция ZNq выражения D.2) стре-
мится к функции Zn выражения D.3), может быть проведено аналогичным образом и поэтому
здесь не приводится.
3) Классическую статистическую сумму иногда называют фазовым интегралом. Этот
термин не следует путать с фазовым интегралом &pdq Зоммерфельда—Вильсона старой
квантовой теории.
100 Г л, 2. Статистическая механика
ранства эквивалентны в том отношении, что они соответствуют простой перену-
мерации частиц. Так как в наборе N частиц возможны N! перестановок, то
множитель A/N!) вносит «поправку» на неразличимость частиц. Такие поправ-
ки должны всегда вводиться в классические формулы. Множитель Л3^ в выра-
жении для классической статистической суммы можно интерпретировать как
объем ячейки в у-пространстве. Ниже в этом параграфе будет использована
квантовомеханическая форма статистической суммы.
Б. ВНУТРЕННЯЯ ЭНЕРГИЯ И ПЕРВЫЙ ЗАКОН
ТЕРМОДИНАМИКИ
Используя уже полученную формулу для квантовомеханической стати-
стической суммы и выражение для среднего по ансамблю C.33), можно запи-
сать следующее выражение для внутренней энергии и системы, помещенной в
термостат1) :
U = -L D.4)
Внутренняя энергия может быть также выражена непосредственно как произ
водная от логарифма статистической суммы
91nZ"
dp
где индекс V указывает на то, что дифференцирование ведется при постоянных
значениях внешних параметров системы (таких, например, как объем). В диф-
ференциальной форме первый закон термодинамики может быть записан в
виде
dU = dq- dw, D.6)
где bq — малое количество тепла, поглощенное системой ; bw — работа, совер-
шенная системой во время малого изменения состояния. Здесь dU — полный
дифференциал, тогда как 6q и bw не являются таковыми. Можно также за-
писать выражение для dU через энергетические уровни системы.
Полагая aj = Zu1 ехр (—fiEj), можно выражение D.4) написать в виде2)
U = f*jEj, D.7)
откуда следует
dU = 2 Ej duj + 2 *j dEj. D.8)
Первый член в правой части представляет собой изменение энергии, обу-
словленное перераспределением полной энергии по различным квантовым
состояниям системы. Второй член дает изменение энергии, возникающее вслед-
ствие смещения энергетических состояний системы, вызванного изменением
объема (или других внешних параметров). Последний можно, очевидно, отож-
дествить с Sw, а первый — с bq; таким образом,
jdaj D.9)
dw = -2ejdEj. D.10)
Тогда соотношение D.8) можно рассматривать как микроинтерпретацию пер-
вого закона термодинамики.
х) В соответствии с обозначениями выражения C.33) необходимо обозначить эту вели-
чину через Ё. Однако мы предпочитаем пользоваться символом U.
2) dj = Fij/N — относительные числа заполнения} введенные в § 3 и эквивалентные
величине &у из § 2. Ввиду двойных индексов последним обозначением пользоваться не-
удобно.
§4. Основы статистической термодинамики 101
В. ТЕМПЕРАТУРА, ЭНТРОПИЯ И ВТОРОЙ ЗАКОН
ТЕРМОДИНАМИКИ
Теперь необходимо ввести понятия температуры и энтропии. В аксиома-
тической термодинамике [13] величина, обратная температуре, определяется
как интегрирующий множитель для сообщаемого системе количества тепла,
а энтропия — как получаемый при этом полный дифференциал. Статистическое
определение энтропии основано на аналогии с этим. Выше были получены вы-
ражения, которые мы интуитивно связывали с понятиями бесконечно малого
количества работы и тепла. Величина, обратная температуре, определяется
как интегрирующий множитель для этого бесконечно малого количества тепла,
а дифференциал энтропии — как получаемый при этом полный дифференциал.
Определим функцию S (энтропию) выражением
где к — постоянная Больцмана, a So — постоянная. Теперь полный дифферен-
циал dS энтропии можно выразить через
D.12)
Используя определение статистической суммы и выражения D.9) и D.10)
для bq и Sw, получаем
^ kUdp =
= kfi [dU -2ujd?j] = kpdq= ±dq. D.13)
Этот результат показывает, что величина &/?, или A/Т) является интегрирующим
множителем для количества тепла. Таким образом, умножение неполного диф-
ференциала bq на A/Т) приводит к появлению полного дифференциала dS
энтропии.
Однако таким путем температура определяется не единственным образом.
Если в дополнение к этому потребовать, чтобы энтропия была экстенсивным
свойством, то можно показать, что выбор единственен. При этом не прини-
мается во внимание мультипликативная постоянная (фактор шкалы). Эмпири-
чески температура часто определяется с помощью термометра идеального газа.
Эта температура связана с уравнением состояния, выражаемым формулой
pV = NkT. Термодинамическая температура, введенная выше, тождественна
с этой температурой, так как в гл. 3, § 1, будет показано, что она приводит к
тому же уравнению состояния.
Следовательно, выражение для энтропии можно переписать в виде
S0. D.14)
При таком способе мы вводим понятие температуры и энтропии методом, ана-
логичным термодинамическому. Ясно, что S и Т —функции состояния. В за-
ключение статистического доказательства второго закона термодинамики необ-
ходимо показать, что энтропия изолированной системы никогда не уменьшается.
Это утверждение является содержанием известной Я-теоремы. Общее доказа-
тельство этой теоремы слишком длинно, чтобы приводить его здесь, и может
быть найдено в других учебниках1). Однако доказательство, применимое к спе-
циальному случаю идеального газа, обсуждается в гл. 7, § 3.
1) См., например, книгу Толмана [83], где даны доказательства этой теоремы в класси-
ческой и квантовой статистике.
102 Гл. 2. Статистическая механика
Г. ЭНТРОПИЯ ПРИ АБСОЛЮТНОМ НУЛЕ И ТРЕТИЙ ЗАКОН
ТЕРМОДИНАМИКИ
Постоянная So, появившаяся при определении энтропии, не зависит ни
от температуры, ни от механических параметров системы. В пределе, когда
температура стремится к абсолютному нулю,
limS = limok[lnZgje-^T + J-—- ]+ Sq = k]ngo + s&> D.15)
где g0 — вырожденность основного еостояния системы.
Рассмотрим теперь два состояния одной и той же системы: в одном слу-
чае вещество АВ находится в сосуде при абсолютном нуле температуры; в
другом случае мы имеем два сосуда, в каждом из которых находятся вещества
Аи В при абсолютном нуле. Можно представить себе обратимый процесс, в
котором система может быть переведена из одного состояния в другое. Следо-
вательно, два состояния АВ и А + В являются двумя действительными состо-
яниями одной и той же системы, отличающимися только описывающими состо-
яние внешними параметрами. Так как So — одна и та же величина для обоих
состояний, то разность энтропии в двух состояниях дается выражением
D.16)
Если все три вещества А, В и АВ таковы, что их основные состояния не вырож-
дены, т. е. gA = gB = gAB= 1, то
AS = SAB-(SA + SB) = O. D.17)
Для многих систем нижнее состояние обычно считается многократно выро-
жденным. Однако обычно осуществляется небольшое разделение энергетических
уровней, вызванное малыми возмущениями, которыми, как правило, пренеб-
регают. При приближении кТ к нулю это очень малое разделение уровней ста-
новится большим по сравнению с кТ. Следовательно, строго говоря, имеется
вероятность того, что основное состояние всегда будет невырожденным, т. е.
выражение D.17) останется справедливым. Так как постоянная So никогда
не принимается во внимание, то удобно, приняв ее равной нулю, определить
энтропию выражением
S = k\nZN + -jr. D.18)
В соответствии с этим определением третий закон термодинамики принимает
вид
D.19)
т—о
а если основное состояние системы невырождено, то
lim S = 0. D.20)
Т-+О
Величина g0 выражает истинную вырожденность нижнего квантового состоя-
ния системы. Однако при сравнении статистически рассчитанных значений эн-
тропии со значениями, измеренными калориметрически, может оказаться необ-
ходимым несколько изменить смысл этой величины. При расстоянии между
нижними энергетическими уровнями, малыми по сравнению с кТ0, где То — ве-
личина минимальной измеримой температуры, на кривой зависимости удель-
ной теплоемкости от температуры в области низких температур появится
небольшой горб. При обработке экспериментальных данных о величине Ср/Т
изменением энтропии, вызванным за счет этого, пренебрегают. Для этой цели
необходимо рассматривать как вырожденные группу уровней, разделенных
энергиями, малыми по сравнению с величиной кТ0.
§ 5. Вычисление термодинамических свойств идеальных газов 103
Д. ВЫРАЖЕНИЕ ТЕРМОДИНАМИЧЕСКИХ ВЕЛИЧИН
ЧЕРЕЗ СТАТИСТИЧЕСКУЮ СУММУ
Мы уже видели, каким образом основные термодинамические функции —
энергия и энтропия — могут быть выражены через статистическую сумму,
которая, в свою очередь, зависит от энергетических уровней системы. Поэтому
если доступна подробная информация об энергетических уровнях системы, то
с помощью статистической суммы могут быть вычислены все термодинамиче-
ские величины. Соотношения, необходимые для этого, таковы :
-y-, D.23)
А = U - TS = - kT\nZN. D.24)
Для систем, единственным внешним параметром которых является объем V,
можно записать дополнительно соотношения
--(—) -кт( -.)
~- {dV)T~~Ki[ dV )ту
^) + kTV (-^-)T, D.26)
G = H-TS=- kTlnZN + kTv\j^~^T. D.27)
Первое из этих соотношений используется в статистической механике при
выводе уравнения состояния.
§ 5. ВЫЧИСЛЕНИЕ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ИДЕАЛЬНЫХ ГАЗОВ
В этом параграфе будет подробно обсуждено важное применение принци-
пов статистической термодинамики — вычисление термодинамических свойств
газов при достаточно низких давлениях, при которых газы можно считать
идеальными. Во-первых, мы покажем, что ограничения симметрии, наклады-
ваемые на волновые функции систем, образованных из тождественных частиц,
приводят к двум типам распределений энергии между молекулами в газе :
одно — распределение для газа Ферми—Дирака, а другое — для газа Бозе—
Эйнштейна. При высоких температурах обе функции распределения совпадают
и термодинамические свойства газов обоих типов становятся одинаковыми.
Эта предельная форма статистик упомянутых двух типов называется статисти-
кой Больцмана. Во второй части параграфа вычисляются термодинамические
свойства газа Больцмана и рассматривается вклад, обусловленный внутрен-
ними степенями свободы.
А. СТАТИСТИЧЕСКАЯ СУММА ИДЕАЛЬНОГО ГАЗА
Если плотность газовой системы N молекул достаточно низка и если меж-
молекулярные силы короткодействующие (в сравнении с кулоновскими), то
время, в течение которого молекула испытывает столкновение, пренебрежимо
мало по сравнению со временем, протекающим между столкновениями. В этих
условиях^ можно утверждать, что потенциальная энергия межмолекулярного
взаимодействия пренебрежимо мала по сравнению с полной энергией отдель-
ных молекул. Каждая из молекул может находиться в любом квантовом
104 Гл. 2. Статистическая механика
состоянии свободной молекулы. Однако, поскольку молекулы тождественны,
состояние газа определяется числом молекул в каждом состоянии и бессмы-
сленно определять, какие именно молекулы находятся в том или другом со-
стоянии. Следовательно, будет удобно характеризовать состояние газа набором
чисел заполнения nf, дающих число молекул в /-м квантовом состоянии моле-
кулы, если газ находится в /с-м состоянии с полной энергией Ек. Пусть энергия
молекулы в /-М состоянии будет bj. Тогда, поскольку энергия газа есть сумма
энергий отдельных молекул, энергию газа в Л-м состоянии можно выразить
в виде
Ek = 2n}ej9 E.1)
j
где п) удовлетворяет соотношению
N = 2nf. E.2)
Кроме того, для величины п) имеется ограничение, обусловленное типом ста-
тистики. Как указывалось в гл. 1, § 6, для статистики Ферми—Дирака числа
заполнения заключены между значениями 0 и 1. Однако для частиц Бозе—
Эйнштейна требования симметрии волновой функции газа не ограничивают
числа молекул в любом состоянии. Статистическую сумму для всего газа
можно записать в виде
2 w? w? u>?... , E 3)
к к к V ' '
где
Wj = e*ikT. E.4)
Подразумевается, что сумма по к берется только по тем наборам чисел п), кото-
рые совместимы с определенной статистикой молекул и ограничением E.2).
Введем теперь производящую функцию /(С), определяемую выражением
4 E.5)
В этом и последующих выражениях верхний знак соответствует статистике
Ферми—Дирака, а нижний — статистике Бозе—Эйнштейна. Статистическая
сумма ZN является просто коэффициентом при члене CN разложения функции
/(?)¦ Если в качестве С взять комплексное переменное, то, разделив функцию
/(?) на fto+i) и воспользовавшись методом теории вычетов (или теоремой Коши),
можно получить выражение для ZN:
E.6)
Замкнутый контур С интегрирования в комплексной области должен быть
выбран так, чтобы была охвачена точка С = 0. Этот интеграл может быть
вычислен методом «наиболее крутого спуска», и мы получаем
lnZN = - Nlnfo ± vin(l ± Zoe-e*lkT). E.7)
Для определения параметра ?0, являющегося положительным и действитель-
ным числом, служит вспомогательное соотношение
l]-1. E.8)
2
Б. РАСПРЕДЕЛЕНИЕ ЭНЕРГИИ МЕЖДУ МОЛЕКУЛАМИ
ИДЕАЛЬНОГО ГАЗА
В § 3 было получено распределение энергии между молекулами в класси-
ческом (больцмановском) газе. Теперь мы хотим найти это распределение для
газов Ферми—Дирака и Бозе—Эйнштейна. Как уже было показано, канони-
§ 5. Вычисление термодинамических свойств идеальных гаъов 105
ческий ансамбль представляет систему с заданными объемом и температурой.
Соответственно среднее значение любой динамической величины дается выра-
жением C.33). Используя эту формулу, мы найдем, что математическое ожи-
дание величины rij дается выражением1)
s; пь е-ЕШ/кт
к
к
или, если выразить Ек с помощью соотношения E.1), выражением
zN• <5Л0>
Окончательно получаем
- кТ (dZN\ /^ 11Ч
Так как Со является функцией ej9 необходимо проявить осторожность
при получении производной CZN/3ey) из выражения E.7). Продифференцировав,
получим2)
nJ=lCj1e-*kT±l]-1. E.12)
Это выражение описывает распределение энергии между отдельными моле-
кулами газов Ферми—Дирака и Бозе—Эйнштейна. Можно показать, что пара-
метр Со связан со свободной энергией выражением G = NkT In ?0.
Для большинства реальных систем Со1 > 1 ; основными исключениями
являются электроны в металле, газы при очень низких температурах и очень
плотные газы. Если применить условие Со1 > 1, то можно написать
0-* V (МТ)\
где V — объем (смэ1моль), а М — молекулярный вес. Для идеального газа при
нормальных условиях
Со1 = 31,590 Af/«.
Системы, для которых величина Со1 мала, так что величиной ± 1 пренебречь
нельзя, иногда называют вырожденными системами или говорят, что они нахо-
дятся в состоянии вырождения. В большинстве приложений можно пользо-
ваться распределением
rij^Coe^ikT. E.13)
Таким образом, мы видим, что распределения Ферми—Дирака и Бозе—Эйн-
штейна в пределе приближаются к полученному в § 3 распределению Макс-
велла—Больцмана. Более того, в пределе статистическая сумма, выражаемая
E.7), превращается в
ZN = ^, E.14)
где z — статистическая сумма отдельной молекулы в сосуде
z^jje-iw. E.15)
j
Величина N!, появляющаяся в выражении E.14), обусловлена принципиаль-
ной неразличимостью молекул. О системах, статистические суммы которых
г) Здесь черточкой обозначено среднее значение числа заполнения, тогда как в выра-
жении C.10) черточкой обозначалось наиболее вероятное значение. Для систем, содержащих
большое число молекул, эти величины по существу одинаковы и отличаются только вслед-
ствие пренебрежимо" малых флуктуации.
2) Функции распределения для газов Ферми—Дирака и Бозе—Эйнштейна можно полу-
чить также методом комбинаторного анализа, как это сделано в §3. См., например, [3].
106 Гл. 2. Статистическая механика
даются выражением E.14), говорят, что они подчиняются статистике Больц-
мана. Именно эта статистика будет использоваться в оставшейся части этой
главы. Проблема вычисления термодинамических свойств для газа Больцмана
сводится к вычислению статистической суммы отдельной молекулы в сосуде.
Вычисление статистической суммы для отдельной молекулы будет рассмотрено
в оставшейся части этого параграфа.
В. ВЛИЯНИЕ ПОСТУПАТЕЛЬНОГО И ВНУТРЕННЕГО ДВИЖЕНИЯ
МОЛЕКУЛЫ НА ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА
Для точного вычисления статистической суммы для сложных молекул
необходимо знать все энергетические уровни системы. Вследствие большого
числа степеней свободы это весьма затруднительно и обычно многое из необ-
ходимых сведений недоступно. Однако для большинства практических целей
оказывается достаточным идеализировать положение, пренебрегая слабым вза-
имовлиянием различных степеней свободы. Статистическая термодинамика
идеального газа излагается во многих учебниках, поэтому здесь будет прове-
дено лишь краткое обсуждение.
Пусть *jfr — квантовомеханический гамильтониан сложной молекулы;
y)j к ej — волновая функция и энергия, соответствующие /-му квантовому
состоянию, так что
Л Sjipj. E.16)
Для обсуждаемых здесь целей ряд энергетических уровней еу отсчитывается
от основного состояния, принятого за нуль. При рассмотрении смесей различ-
ных химических компонент необходимо относить энергии основных состояний
химических компонент к соответствующему ряду стандартных состояний, выб-
ранных для элементов. Это будет обсуждено ниже. Движения, совершаемые
многоатомными молекулами, крайне сложны. Однако можно рассматривать
движение состоящим из шести разных видов движения : поступательного
движения как целого (пост.); различных валентных и деформационных коле-
баний (кол.); вращательного движения молекулы как целого (вр.); различ-
ных вращений или ограниченных вращений групп внутри молекулы (вн. вр.);
движения электронов (элн.); ядерного спина (яд.). Эти типы движения в той
или иной степени зависимы друг от друга. Например, вращательное движение
почти не зависит от движения электронов, но сильно связано с колебательным
движением. Тем не менее в качестве первого приближения можно предполо-
жить, что все эти типы движения независимы, так что можно написать выра-
жения
= Jf (пост') + ЛГСвр') + 2" Jr(K0Jb) + 2 ЛГ(вн#вр") + ,JT(9JIH#) + ^Г(яд) E.17)
В выражении E.18) индексом (вн.) обозначены различные типы внутренних
(непоступательных) движений. Такое рассмотрение позволяет нам разделить
уравнение Шредингера E.16) на две составные части :
= ?(пост.)
(вн.) ^(вн.) = ?(вн.) ^
Решение этих уравнений дает нам энергетические уровни, необходимые для
получения статистических сумм для различных типов движения:
E.22)
§ 5. Вычисление термодинамических свойств идеальных газов 107
Исходя из определения статистической суммы молекулы, статистических сумм
для различных видов движения и того, что энергия молекулы в любом состоянии
принимается равной сумме энергий, соответствующих различным типам дви-
жения, можно записать выражение
Теперь в соответствии с выражением E.14) статистическая сумма для всего
газа N молекул записывается в виде
ZN [2(nocT.)]N j
Поскольку в выражения для термодинамических функций всегда входит
величина In ZN, то они могут быть представлены в виде суммы членов, выра-
жающих вклад поступательного и внутренних движений :
X = Х<пост-> + 2 лу*.>, E.25)
где X может быть и внутренней энергией, и удельной теплоемкостью, и сво-
бодной энергией и т. п. Обычно принято включать величину N\ в член, соответ-
ствующий вкладу поступательного движения ; это и сделано в выражении
E.25). Этот член во многих случаях имеет преобладающее значение. Соответ-
ственно выражения для вкладов различных типов движения можно записать
в виде1)
Nk [ln (*=р>.) + i] , s<bh.) = I*™ + Nik in 20-);
Д(пост.) = _ NkT ln p^-) - N/cT, A<8H-> = - NkT In z<BH->; E.26)
^(пост.) = (у(пост.) _j_ jy^J ^ #<BH-> = C/(bh.) .
(-(пост.) = Д(пост.) ^_ ^y^j- ? Q(bh.) = Д(вн.).
^(пост.) _ ( дЩпост.) -| (BH#) _ f Э^/(вн.) -j #
С(пост.) = С(пост.) + N& ? С(вн.) = ^вн.)^
Таким образом, при вычислении термодинамических функций идеального газа
мы просто складываем выражения, соответствующие различным типам дви-
жения, происходящим внутри молекулы. Полное описание методов вычисления
выражений для различных типов движения в случае сложной молекулы тре-
бует много времени и включения в текст множества табулированных функций.
Такое полное рассмотрение вопроса можно найти в ряде книг (см.,например [14]),
поэтому мы ограничимся некоторыми простыми вычислениями для случая
одноатомных и двухатомных молекул.
1. Вклад поступательного движения. Если газ не находится при темпера-
туре, близкой к абсолютному нулю, то вклад поступательного движения в тер-
модинамические свойства может быть вычислен с помощью классической ста-
тистической суммы. Принимается, что молекула заключена в объеме Vy а ее
классический гамильтониан — это просто выражение (р? + р* + р|)/2/п.
*) В выражениях для членов, соответствующих поступательному движению, исполь-
зована формула Стирлинга In N\ ^ N In N — N.
108 Гл. 2. Статистическая механика
Тогда фазовый интеграл для поступательного движения можно записать в виде
и V V V - р*+ру+р%
= ^. j J j e 2тит dpxdpydpz, E.27)
— 00 — 00 —0
где множитель V — результат интегрирования по пространственным коорди-
натам х, у, z. Интегрирование по пространству импульсов непосредственно
приводит к
2<пост.) = JJ- B яткТуь = VX- з, E.28)
где А = Л/У 2ntnkT — длина волны де Бройля для среднеарифметической
скорости молекулы (см. гл. 1, § 2), умноженная на 2/я. Из этого выражения
отчетливо видно, как статистическая сумма может зависеть от определенных
внешних параметров системы — в данном случае от объема. Из формул, связы-
вающих статистическую сумму с термодинамическими функциями, можно
легко получить известные выражения для термодинамических величин, при-
ходящихся на моль:
0<посг.) = _|_ RTf ?(пост.) = _|_ Ry ?(пост.) = А ## E.29)
Можно показать также, что энтропия, приходящаяся на моль вещества, выра-
жается следующим образом:
^ A in Г + In к) + 2,6546 =
= R (-|- In М + -A In T - In p] - 2,3141, E.30)
где числовые постоянные являются комбинациями различных универсальных
постоянных1). Это выражение для энтропии есть так называемое уравнение
Саккур—Тетроде. Для одноатомного газа вклад поступательного движения в
термодинамические свойства дается нижней строчкой выражения E.30). Для
газов, состоящих из многоатомных молекул, поступательное движение дает
основной вклад в термодинамические свойства.
2. Вклад вращательного движения двухатомных молекул. Если пред-
ставить двухатомную молекулу в виде жесткой гантели с моментом инерции /,
вращающейся в пространстве трех измерений, то решение уравнения Шредин-
гера для этой системы дает энергию и вырожденность J-ro уровня, где J —
вращательное квантовое число
2J+l. E.32)
Таким образом, статистическая сумма имеет вид
z<bp.) =У B!+1)е 21кТ
Величина момента инерции может быть получена из спектроскопических дан-
ных. Для вычисления выражений типа E.33) удобно воспользоваться форму-
^Эти значения постоянных справедливы, если R выразить в кал/моль-град, Т — в %
V — в литрах, ар — в атм. В в соответствии с рекомендациями Национального бюро
стандартов мы будем пользоваться для калории величиной 4,1833 международных джоуля и
4,18401 абсолютных джоуля. Для величины R принято значение R = 1,98718 кал/моль-град.
§ 5. Вычисление термодинамических свойств идеальных газов 109
лой Эйлера—Маклорена :
Д /(У)=j/U)</ + -т\1Ш + f(M +
Здесь Bk — числа Бернулли1).
Подставляя вместо f(J) величину Bу+1)ехр[—h2J(J -f \)\2IkT) и
полагая Jo = О, a Jx = оо, получаем
Для двухатомных молекул при нормальных температурах отношение 2/fcT/A2
значительно больше единицы и поэтому существенен только первый член.
Обычно в знаменатель статистической суммы вводят число симметрии а, которое
равно единице для двухатомных молекул с различными ядрами и двум — для
молекул с одинаковыми ядрами. Этот множитель связан с ограничениями,
накладываемыми на число допустимых квантовых состояний принципом
Паули. Из выражения B.25), включив в него число симметрии и пренебрегая
членами высших порядков, мы получаем выражения для термодинамических
величин на моль
0
E.37)
E.38)
3. Вклад колебательного движения двухатомных молекул. Если пред-
положить, что растяжение связи в двухатомной молекуле подчиняется закону
Гука (т. е. сила, стремящаяся вернуть молекулу в равновесное положение,
прямо пропорциональна величине деформации связи), то задача сводится к
решению уравнения Шредингера для одномерного простого гармонического
осциллятора. Энергия п-го колебательного состояния дается выражением
E.39)
Эти колебательные состояния невырождены. Колебательная статистическая
сумма обычно выражается через энергии, отсчитываемые от нулевого уровня,
так что
7(кол.) __ у p-nhvlkT _ * /к ЛС\\
ZK ?ое ™ \-e-MkT • У°Аи)
Последнее выражение получено путем использования формулы суммы гео-
метрической прогрессии. Соответствующий вклад в термодинамические свой-
ства выражается следующим образом:
(/(кол.) = ?(кол.) = ffhv(eMkT _ 1)- 1^ E.41)
р _ Rln (j __ е-*/*т)# E.42)
Частота v — это собственная частота колебаний молекулы и может быть полу-
чена из колебательного спектра молекулы. При высоких температурах необ-
ходимо применять поправку на возрастающую ангармоничность колебаний.
х) Несколько первых чисел Бернулли имеют следующие значения: Вх = Ve» ^2 = Узо*
В3 = у42, В4 = Узо* Вб = б/вв и т. д. Разложение Эйлера—Маклорена выводится и обсу-
ждается во многих математических книгах (см., например, [15]).
ПО Гл. 2. Статистическая механика
4. Вклад электронного движения двухатомных молекул. Большинству
молекул для возбуждения высших электронных уровней необходима довольно
высокая температура. Соответственно во многих случаях г(элн) — это просто
вырожденность основного электронного состояния. Электронное вырождение
для одно-, двух- и многоатомных молекул можно получить из величин «спек-
тральных термов» следующим образом.
а) Одноатомные молекулы. В символах термов, обозначающих электронное
состояние атома (например, 3Р3, г82, 4F0), правый индекс указывает на величину
полного (орбитальный + спиновый) момента количества движения электронов
в атоме. Электронное вырождение выражается так:
г(элн.)==2у+ 1. E.43)
б) Двухатомные молекулы. В символах термов, обозначающих электронное
состояние двухатомной молекулы (например, 32^, 2ПШ гА^, левый индекс
указывает на величину «мультиплетности спина» —2$+ 1. Электронное вы-
рождение тогда имеет вид
2 S + 1 для состояния 2
2BS + 1)для состояний П}Л...
в) Многоатомные молекулы. Для многоатомных молекул с насыщенными
валентностями g*3™) = 1 или 2 в зависимости от четности или нечетности
числа электронов (так как для четного числа электронов результирующий
спиновой момент количества движения равен нулю, а для нечетного — резуль-
тирующий спин равен половине).
5. Вклад ядерного спина. Статистическая сумма для ядерного спина
равна просто произведению величин мультиплетности ядерного спина всех
атомов молекулы:
<) 1), E.44)
где 5уяд) — ядерный спин /-го атома. Однако поскольку этот вклад отражается
только на величине всегда отбрасываемой аддитивной постоянной в выражении
для энтропии, то им можно пренебречь (за исключением случая процессов,
включающих превращения элементов). В результате мы получаем скорее
«возможное», чем «абсолютное» значение энтропии.
Г. СМЕСИ ИДЕАЛЬНЫХ ГАЗОВ
Можно показать, что для смеси идеальных газов, содержащей NA моле-
кул вещества А и NB молекул вещества В, статистическая сумма выражается
в виде
ZNA 2NB HA(s0)A + N?(e0)?
2»=^7^ге~ kT ¦ E-45)
Здесь zA и zB — введенные выше статистические суммы молекул, для которых
все энергии отнесены к основным состояниям соответствующих молекул.
Множители NA! и NBl появляются в силу неразличимости молекул одного
сорта точно так же, как появляется величина JV! в соответствующем выражении
для чистого вещества. Члены (ео)А и (ео)в — энергии основных состояний
молекул Аи В, отнесенные к соответствующему ряду стандартных состояний
элементов. Таким образом, (ео)А — это энергия образования молекулы А в
основном состоянии из соответствующих элементов в собственных стандартных
состояниях. За стандартные состояния элементов обычно принимаются те,
которые соответствуют нулю градусов по Цельсию и давлению в 1 атм. Из
выражения статистической суммы E.45) можно вычислить различные термоди-
§ 6. Теория флуктуации 111
намические величины для смеси газов. Таким образом, найдем
O Oj, E.46)
2
XjHj, E.47)
E.48)
В этих выражениях Uj и Hj — не просто величины, вычисленные описанным
методом. Это величины, к которым для учета изменения нуля энергии при-
бавлено N(eo)j. Последнее выражение справедливо только для случая нереаги-
рующих смесей. Энтропия смеси газов не равна простой сумме соответствую-
щих вкладов отдельных компонент. Подстановка статистической суммы для
смеси газов в выражение D.24) показывает, что энтропия смеси выражается
следующим образом:
5см. = 2 *j Sj~R2 *j In Xj. E.49)
j j
Величина —R2X]\riXj называется энтропией смешения. Аналогичные доба-
вочные члены, обусловленные смешиванием, появляются в выражениях для
свободной энергии Гиббса 6 и свободной энергии Гельмгольца Л.
При рассмотрении температурной зависимости термодинамических вели-
чин для смесей необходимо внимательно учитывать возможность пренебре-
жения химическими реакциями и возможность поддержания химического
равновесия определенных молекулярных компонент. В первом случае числа
NA и NB остаются постоянными, тогда как во втором случае они меняются
в соответствии с постоянными равновесия.
§ 6. ТЕОРИЯ ФЛУКТУАЦИИ
До сих пор наше обсуждение ограничивалось описанием равновесных
свойств, которые могут быть вычислены как наиболее вероятные величины
или как средние по ансамблю. Именно эти равновесные свойства подчиняются
законам термодинамики. Однако вследствие молекулярной природы вещества
существует конечная вероятность отклонений от этого среднего поведения.
Методы статистической механики дают не только описание среднего поведе-
ния, но и способ вычисления вероятности флуктуации любой величины около
ее среднего значения. Вследствие большого числа механических степеней
свободы системы типа газа или жидкости макроскопические флуктуации обычно
крайне малы и появляются редко. Можно рассматривать флуктуации любых
макроскопических или термодинамических свойств методами общего форма-
лизма [16—19]. Мы не будем здесь излагать этого общего формального рас-
смотрения, а дадим детальное обсуждение флуктуации внутренней энергии
и плотности, поскольку эти величины важны при изучении критической об-
ласти (см. гл. 5). Флуктуации плотности приводят к рассеянию света (см. гл.7,
§ 12). Флуктуации внутренней энергии приводят вблизи критической точки
к аномально высоким значениям удельной теплоемкости при постоянном
объеме. Мы не рассматриваем флуктуации химического состава многокомпо-
нентной системы, так как это потребовало бы подробного анализа, который
не найдет прямого применения в настоящей книге1). Очень простой пример
2) Флуктуации состава многокомпонентной системы при наличии и отсутствии хими-
ческих реакций изучались Фаулером [17]. Эти флуктуации проявляются в мутности рас-
творов. Недавно в работах [20, 21] были рассмотрены флуктуации с учетом высших прибли-
жений и членов, которыми ранее пренебрегали, но без учета химических реакций.
112 Гл. 2. Статистическая механика
флуктуации термодинамических величин дает внутренняя энергия U. Среднее
значение внутренней энергии Uy выраженное через энергетические состояния
Ej для системы, находящейся в равновесии с термостатом при температуре Т,
дается выражением D.4). Среднеквадратичное отклонение внутренней энергии
может быть записано через среднее значение U и среднее значение U2:
(U - U)* = IP - 2A/ U) + (UY = (Uf - (U)\ F.1)
В соответствии с C.33) можно написать
Л^ 1к^) р^)\ F.2)
) р
Т
Из сравнения двух последних соотношений с выражениями D.21) и D.22)
следует _______
(U-UJ = kT*Cv. F.3)
Следовательно, в соответствии с этим соотношением удельная теплоемкость
при постоянном объеме является мерой флуктуации внутренней энергии.
А. ФЛУКТУАЦИИ ПЛОТНОСТИ,
ВЫРАЖЕННЫЕ ЧЕРЕЗ ТЕРМОДИНАМИЧЕСКИЕ ВЕЛИЧИНЫ
Рассмотрим сосуд объема V, в котором находится N молекул одного и
того же сорта. Представим далее, что объем V разделяется воображаемыми
перегородками на множество малых элементов объема Vs. Тогда молекулы в
сосуде могут свободно перемещаться из одного элемента объема в другой.
В среднем в любом элементарном объеме Vs содержится v = N(VsjV) молекул.
Однако вследствие флуктуации имеется конечная вероятность того, что число
молекул в одном из элементов объема отличается от этой величины. Будем
рассматривать элемент объема Vs как «систему», а сосуд объема V — как «ан-
самбль» систем.
Поскольку и вещество и энергия могут перетекать из одной системы в
другую, то целесообразно представлять любую данную равновесную систему
большим каноническим ансамблем. В соответствии с выражением B.31) для
большого канонического ансамбля вероятность того, что сисггема содержит v
молекул и находится в /-м квантовом состоянии, можно записать в виде
Т (б-4)
здесь E\v) — энергия /-го состояния системы v молекул. Величина /г — химиче-
ский потенциал системы, который может быть выражен через термодинами-
ческий потенциал А и свободную энергию G:
Символ ZB означает большую статистическую сумму и является нормировоч-
ным множителем для величины Щ:
ZB = 2 ev^lkT е~ Е^!кТ. F.6)
Удобно ввести величину A(v), являющуюся свободной энергией Гельмгольца
для системы v молекул и связанную следующим соотношением со статистиче-
ской суммой Zv системы:
Z, = e-AWkT = 2e~ E<iv)lkT. F.7)
§ 6. Теория флуктуации 113
Тогда вероятность того, что система содержит v молекул (и находится в любом
состоянии), можно выразить в виде
^ F.8)
Большая статистическая сумма может быть записана в виде
ZB = 2 ^lkT e~AWkT. F.9)
Введение термодинамической величины A(v) позволяет вычислять флуктуации
через непосредственно измеримые величины. Среднеквадратичное отклонение
величины v от ее среднего значения выражается разностью средней величины
v2 и квадрата средней величины v
(у - VJ = ^ - 2Щ + (^ = И - (^. F.10)
Однако обычно удобнее рассматривать относительную флуктуацию 6, выра-
жаемую в виде
Из F.8) для вероятности того, что система содержит v молекул, следует
? (у _
Если v достаточно велико, то распределение, выраженное формулой F.8), по
существу совпадает с распределением Гаусса и выражение F.12) может быть
легко вычислено. Это можно показать следующим образом. Используя F.5),
можно разложение величины A(v) в ряд Тэйлора вблизи v представить в виде
f ... F.13)
Подстановка этого выражения в F.12) приводит к
F.14)
Экспонента представляет распределение Гаусса.
Рассмотрим изменения размера указанной области, т. е. изменения вели-
чины Vs и соответствующие изменения величины v. Поскольку величина /л
не зависит от размера области, то величина экспоненты пропорциональна Vs
или v. Таким образом, для больших значений v (т. е. для областей больших
размеров) функция распределения имеет очень острый максимум и в выражении
суммы существенны только члены, соответствующие v, близким к v. Кроме
того, поскольку члены высших степеней в разложении величины A(v) в F.13)
еще более высокого порядка, то мы имеем право пренебречь ими. Тогда, за-
меняя в выражении F.14) суммирование интегрированием, мы приходим к
следующему выражению для относительной флуктуации плотности1):
х) Второе выражение в формуле F.15) можно получить следующим образом. Учтем,
что А как функция v и Vs при постоянной Т является однородной функцией первого порядка
относительно переменных. Следовательно, по теореме Эйлера А — Vs(dA/dVs) + v(dA/dv).
Дифференцируя это выражение по Vs, получаем C2A/dvdVs) = —(Vs/v)(8'i.4/9Vr|). Анало-
гично, дифференцируя по v, получаем {d2A/dvdVs) = — (v/Vs) (ЭМ/Э?2). Следовательно,
<8M/92) = (Vt/v*)(d2A/dVs)' Таким образом, так как /г = (ЭД/Э*>) ир=- ЪА/dVs, получаем
dv
Это соотношение и использовано в выражении F.15).
114 Гл. 2. Статистическая механика
кТ кТ
8 ^ (v
W(dp/dvs)T ' FЛ5)
Поскольку объем есть экстенсивная величина, то это выражение указывает
на то, что флуктуация плотности имеет порядок величины 1/v, т. е.
<бЛ6>
где величина в квадратных скобках зависит только от состояния жидкости и
не зависит от объема Vs рассматриваемой области. В случае идеального газа
это выражение дает для относительной флуктуации плотности
«=4- <6Л7>
Это выражение показывает, что если размеры области выбраны достаточно
малыми, то число молекул в этой области флуктуирует. Этого можно ожидать,
если область будет порядка молекулярных размеров. Однако если размеры
области достаточно велики, так что в среднем область содержит много молекул,
то вероятность заметных отклонений от среднего мала. Тем не менее даже эта
мгновенная неоднородность плотности приводит к макроскопически наблю-
даемому явлению рассеяния света атмосферой и броуновскому движению.
Рассеяние света рассмотрено в гл. 12, § 7.
В критической точке величина (др]дп)т равна нулю и из выражения F.16)
следует, что флуктуация бесконечно велика. Однако этот результат неправилен,
так как в выражении F.14) мы пренебрегли членами высших порядков разло-
жения A(v). Продолжая выкладки, проделанные в примечании к выражению
F.15), находим следующие термодинамические соотношения :
9»;* _ Vl (Wp\ >oVl(dp\ -
( (
vi (эзр ч 8 v\ (d*
Таким образом, для критической точки, где (dpldV)T и (d2pl3V2)T равны нулю
первая и вторая производные от /г и соответствующие члены в разложении вели-
чины A(v) равны нулю. Так как (Э3р/Э V3)T не равно нулю1) в критической точке,
то третья производная от /г тоже не равна нулю и в области критической точки
в разложении A(v) должны удерживаться члены вплоть до этого порядка.
Рассмотрение флуктуации в окрестности критической точки усложнено, так как
необходимо рассматривать полную последовательность членов разложения.
Однако мы можем очень легко вычислить флуктуацию в критической точке.
В критической точке (т. е. при v и Т, соответствующих критической точке)
разложение A(v) имеет вид
±^\+ ... F.20)
Таким образом, из распределения F.8) следует, что относительная флуктуация
может быть выражена следующим образом :
<*,=
F.21)
х) К этому приводит классическая интерпретация критической точки. Однако точная
природа критической точки все еще недостаточно понятна (см. гл. 5, § 2).
§ 6. Теория флуктуации 115
Тогда, заменяя (на тех же основаниях, что и выше) суммирование интегриро-
ванием, получаем
А1/*)
iy2
где Г (х/4) = 3,6256 и ГC/4) = 1,2254 — гамма-функции. Соотношение F.19)
можно использовать для выражения флуктуации через величины, получаемые
из уравнения состояния, и для флуктуации плотности получаем
с А74) L п2(эзр/9п3)
Величина в квадратных скобках зависит только от характера уравнения со-
стояния в окрестности критической точки. Так как флуктуация имеет вели-
чину порядка (l/vI/2, то полученные значения много больше величин, давае-
мых апроксимацией, обсуждавшейся ранее. Однако необходимо отметить, что
этот результат дает конечную величину флуктуации плотности в критической
точке. Если для жидкости справедливо уравнение Ван дер Ваальса [выра-
жение B.1) гл. 4], то из выражения для относительной флуктуации в крити-
ческой точке F.23) следует
8с=^ F.24)
Уравнение состояния Дитеричи [см. уравнение B.3) гл. 4] приводит к числен-
ному множителю, равному 1,59 вместо 0,90. Для любого уравнения состояния
можно ожидать, что этот численный множитель будет порядка единицы.
Б. ФЛУКТУАЦИИ ПЛОТНОСТИ,
ВЫРАЖЕННЫЕ ЧЕРЕЗ РАДИАЛЬНУЮ ФУНКЦИЮ РАСПРЕДЕЛЕНИЯ1)
Можно также получить выражение для флуктуации плотности через
радиальную функцию распределения. Это выражение может быть выведено
из обсуждавшихся выше результатов. Однако несколько удобнее вывести это
выражение непосредственно из общих представлений.
Рассмотрим сосуд с газом объема V, содержащий N молекул. Состояние
газа описывается функцией распределения /(N) в фазовом пространстве системы.
Эта функция и ее нормировка подробно обсуждались в § 1. Из определения
функции fN) ясно, что вероятность нахождения z-й молекулы в точке г, а /-й
в точке г' выражается следующим образом:
-L Г Г д(Г,. - Г) Щ - Г') /<»>(*">, pN) dtN dpN
Рассмотрим теперь малую область объема Vs внутри сосуда с газом. Вероятное
значение квадрата числа молекул в этой области можно получить из выраже-
ния F.25) суммированием по всем i и / и интегрированием г и г' по объему Vs:
f J \d(Tt - r)d
= Jn2 J dT\i8(Ti ~ r)/<N>(r^
1 v
J \drdr' J J"*r< - r
1
2
lJv, v,
v,
F.26)
l) Радиальная функция распределения g(r) определяется выражением A.9;. Приме-
нение ее к описанию уравнения состояния обсуждается в гл. 3, § 1, и гл. 4, § 9.
116 Гл. 2. Статистическая механика
Второе выражение в этом соотношении написано таким образом, что выделен
член, соответствующий i = /. Это соотношение можно упростить, вводя обыч-
ную плотность п(г) и бинарную функцию распределения пB)(г, г'), определен-
ную в § 1. Если все молекулы одинаковы, то соотношение F.26) приводится к
виду
гГг = [ n(t)dr + J J n<2> (r, r') dr df. F.27)
Первый член этого соотношения дает вероятное число v молекул в объеме Vs.
Второй член можно выразить через бинарную функцию распределения g(r).
Таким образом, F.27) можно переписать в виде
J \
или
= V+ J \'nW"W &>r' - r)dTdT'>
n(r) n(r>) [g(r, T' - r) - 1] dr df. F.29)
В равновесных условиях, когда жидкость макроскопически однородна, п(г)
не зависит от г, a g(r, г' — г) зависит только от расстояния | г'—г|. Кроме
того, как показано в гл. 4, § 9, g(r) равна единице для значений г, больших
по сравнению с молекулярными размерами. Таким образом, если объем Vs
велик по сравнению с молекулярными размерами, то
гГг _ (уу = v + 4nn2Vs\ [g(r) - 1] r2dr. F.30)
И выражение для относительной флуктуации плотности через радиальную
функцию распределения имеет вид
J- l]r*dr]. ' F.31)
Мы получили два выражения для относительной флуктуации : одно — выра-
женное непосредственно через уравнение состояния и второе — выраженное
через радиальную функцию распределения. Сравнение обоих результатов —
выражений F.16) и F.31) — приводит к
^ = 1+ Аш J [g(r) -1] г* dr. F.32)
Это выражение весьма интересно и ценно. Другой вывод [1 ] этого выражения
можно дать, используя вириальное разложение уравнения состояния и
радиальной функции распределения. Вириальное разложение подробно обсуж-
дается в гл. 3.
Задачи
1. Рассмотреть графически и аналитически классическое движение следующих систем :
а) Материальная точка в потенциальном ящике.
б) Несжимаемая материальная точка, движущаяся с постоянным ускорением.
в) Одномерный простой гармонический осциллятор.
г) Двумерный жесткий ротатор.
д) Простой маятник в двумерном случае.
2. Показать, что для системы, гамильтониан которой не содержит члена, соответствую-
щего потенциальной энергии, канонический ансамбль может быть весьма близким к микро-
каноническому ансамблю с энеогиейЕ. [Указание: гамильтониан системы при использовании
радиальной координаты R =У?Тр*/2т) и 3iV—1 углов в ЗЛГ-мерном гиперпространстве
может быть записан в «полярных координатах». Тогда Н = R2. Число систем, заключенных в
слое толщины dR, для случая микроканонического ансамбля может быть выражено сле-
дующим образом :
P(N) ах = Дехр (- р Н) R™~ * dR.
Задачи 117
Далее можно найти число систем в слое толщины dH и подобрать его таким, чтобы оно весьма
близко совпало с микроканоническим ансамблем. Можно показать, что при увеличении числа
систем в ансамбле до бесконечности каноническое распределение сильно сужается, прибли-
жаясь, таким образом, к микроканоническому распределению.]
3. Вывести следующие выражения распределения Максвелла—Больцмана :
а) Выражение для числа молекул, скорости которых заключены в пределе от и до v+dv:
п (v) dv = 4 лы( 2^г )в/" e-mvil2kT v* dv.
б) Выражение для числа молекул, поступательная энергия которых заключена в
предел от е до е + de :
п (е) de = 2nN ()tU
Изобразить графически распределения а и б. Применимы ли эти выражения в случае
многоатомных молекул ?
4. Обычно пользуются несколькими значениями «средней» скорости молекул в газе,
которые получаются из закона распределения задачи За. Показать, что
а) среднеарифметическая скорость Ъ равна
б) среднеквадратичная скорость равна
в) наиболее вероятная скорость Ь равна
Ъ = ][
Ш = 12,900 1/-^- см/сек.
т
Последняя скорость соответствует максимуму кривой распределения n(v). Вычислить
среднеквадратичную скорость для мЪлекул Н2 и SFe при температуре 0 и 400° К. Какая
доля (в процентах) молекул имеет скорости, большие некоторой данной величины v? Какая
доля молекул водорода имеет скорости, превышающие 104 см/сек?
5. Для многих случаев найдено, что полная энергия системы может рассматриваться
равномерно распределенной по различным степеням свободы системы (теорема о равно-
мерном распределении энергии по степеням свободы). Важно отметить, что равномерное
распределение не является общим следствием статистической механики. Скорее это результат
особой формы зависимости энергии от импульса и координат. Несколько примеров поясняют
это положение:
а) Показать, что для разреженного одноатомного газа, состоящего из N молекул,
средняя энергия, связанная с движением /-й молекулы в направлении х, равна ё/х = Va kT.
Доказать далее, что средняя энергия всего газа может быть получена суммированием энер-
гий, соответствующих всем степеням свободы
N 1
Е =, 2<ъ +**. + h) = -jNkTm
б) Для разреженного двухатомного газа (предполагая, что молекулы—жесткие рота-
торы) показать, что средняя энергия, соответствующая каждой степени свободы, равна г/2 кТ.
Проверить справедливость закона равномерного распределения в этом случае и показать,
что средняя энергия всего газа равна
в) Классический гамильтониан для кристалла, выраженный через нормальные
координаты (qt, p{) системы, имеет вид
Щри Яд = 2 (у^. +4 я2 "< m q\],
118 Гл. 2. Статистическая механика
где vt — частоты нормальных колебаний. Проверить справедливость закона равномерного
распределения в этом случае. Заметим, что во всех трех случаях гамильтониан системы
выражается через координаты и импульсы одинаковым образом:
6. Закон распределения Максвелла—Больцмана может применяться в самых различ-
ных задачах. Используя этот закон, найти выражения :
а) для изменения плотности с высотой в изотермической атмосфере;
б) для изменения плотности коллоидных частиц с высотой в коллоидальной суспензии ;
в) для изменения плотности коллоидных частиц с расстоянием от оси вращения при
вращении суспензии с помощью центрифуги;
г) для распределения по углам осей магнитных диполей в магнитном поле. [Указание:
энергия магнитного диполя с моментом т в магнитном поле напряженности JT выражается
так;
е(в) = — тЖ cos 0,
где в — угол между направлением поля и диполем.]
7. Показать, что вклад вращательных степеней в статистическую сумму молекулы
может быть помимо использования числа симметрии получен из классического фазового
интеграла.
8. Вычислить термодинамические функции О, Д, С, Я, S, Си и Ср для аргона и азота
при температурах 300 и 1000° К. Ядерным спином и движением электронов пренебречь. В
случае азота при вычислении вклада вращательных и колебательных степеней свободы вос-
пользоваться значениями hv/k = 3336,6 °К и А/2/А: = 2,847 °К.
9. Используя литературные данные, вычислить термодинамические функции задачи
8 для атомарного водорода и брома при температурах 300 и 1000 °К.
10. Вывести уравнение состояния идеального газа с помощью статистической суммы.
11. Энергия системы в классической статистике может быть выражена как среднее от
гамильтониана H(rw, p/v) по каноническому ансамблю. Показать, что энтропия может быть
выражена как среднее от величины —k In [ЛМ h™PW(rNf pvj], т. е. что выражение
TS=-kT jj Р(") (г", р") In Р(Ю (г", р") dr" dp" - кТ In (N\ h3N) =
(rN) drN + _|
справедливо в классической статистике. Написать аналогичное выражение квантовой ста-
тистической механики.
ЛИТЕРАТУРА
1. de Boer J., Rep. Progr. Phys., 12, 305 A949).
2. F о w 1 e г R. H., Guggenheim E. A., Statistical Thermodynamics, Cambridge, 1939
(см. перевод: Фаулер Р., Гуггенгейм Э., Статистическая термодинамика,
ИЛ, 1949).
3. То 1 man R. С, The Principles of Statistical Mechanics, Oxford, 1938.
4. N e u m a n n J., Gott. Nachr., 245 A927).
5. N e u m a n n J., Math. Grundlagen der Quantenmechanik, Berlin, 1929.
6. D i г а с P. A. M., Proc. Cambr. Phil. Soc, 25, 62 A929) ; 26, 240 ; 27, 376 A930).
7. Dirac P. A. M., The Principles of Quantum Mechanics, Oxford, 1935 (см. перевод:
3-го издания Дирак П. А. М., Принципы квантовой механики, М.—Л., 1960.
8. S с h i f f L. I., Quantum Mechanics, New York, 1949 (см. перевод Ш и ф ф Л., Кван-
товая механика, ИЛ, 1957).
9. Husimi K-, Proc. Phys. Mat. Soc, Japan, Ser. 3, 22, 264 A940).
10. Хинчин А. Я., Математические основания квантовой статистики, М.—Л., 1951.
11. Grad H., Kinetic Theory and Statistical Mechanics, New York, 1950.
12. S о k о 1 n i k о f f I. S., S о k о 1 n i k о f f E. S., Higher Mathematics for Engineers and
Physicists, New York, 1941.
Литература 119
13. С h a n d r a s е к h а г S., An Introduction to the Study of Stellar Structure, Chicago, 1939
(см. перевод: Чандрасекар С, Введение в учение о строении звезд, ИЛ,
1950).
14. Mayer J. E., Mayer M. G., Statistical Mechanics, New York, 1940 (см. перевод:
М а й е р Дж., Г е п п е р т-М а й е р М., Статистическая механика, ИЛ, 1952).
15. В u r i n g t о n R. S., Тоггапсе С. С, Higher Mathematics, New York, 1939.
16. Klein M. J., Tisza L., Phys. Rev., 76, 1861 A949).
17. Fowler R. H., Statistical Mechanics, Cambridge, 1929.
18. Einstein A., Ann. d. Phys., 33, 1275 A910).
19. S m о 1 u с h о w s k i M., Ann. d. Phys., 25, 225 A908).
20. К i r k w о о d J. G., Goldberg R. J., Journ. Chem. Phys., 18, 54 A950).
21. Stockmayer W. H., Journ. Chem. Phys., 18, 58 A950).
ГЛАВА 3
УРАВНЕНИЕ СОСТОЯНИЯ ДЛЯ ГАЗОВ
МАЛОЙ И УМЕРЕННОЙ ПЛОТНОСТИ1)
Наблюдаемые экспериментально отклонения от уравнения состояния
идеального газа были качественно объяснены Ван дер Ваальсом [1 ], предпо-
ложившим, что молекулы притягиваются друг к другу на больших расстояниях
и отталкиваются при очень малых расстояниях между ними. Это объяснение
привело Ван дер Ваальса к формулированию его известного уравнения состоя-
ния, содержащего две постоянные. В последующие годы значительные усилия
были направлены на нахождение эмпирического уравнения состояния, которое
хорошо согласовывалось бы с экспериментальными данными. Некоторые наи-
более важные эмпирические соотношения, такие, как уравнения Дитеричи,
Вертело и Битти—Бриджмена, будут обсуждены в следующей главе.
В этой главе мы займемся виршльным уравнением состояния, записывае-
мым в общем виде2)
Коэффициенты В(Т), С(Т)У D(T) и т. д. называются соответственно вторым, тре-
тьим, четвертым и т. д. вириальными коэффициентами. С помощью статисти-
ческой механики эти вириальные коэффициенты можно выразить через потен-
циал межмолекулярного взаимодействия. Далее, с помощью межмолекуляр-
ных сил можно получить количественную интерпретацию отклонений от урав-
нения состояния идеального газа. И, действительно, многие ценные.сведения
о межмолекулярных взаимодействиях получены при анализе данных о зависи-
мости ру V и Т.
Из выражений для вириальных коэффициентов, даваемых статистической
механикой, становится очевидным, что второй, третий, четвертый и т. д. вири-
альные коэффициенты дают отклонения от поведения идеального газа, обуслов-
ленные парными, тройными, четверными и т. д. столкновениями. Следовательно,
при малых плотностях отклонения от идеальности адекватно описываются
вторым вириальным коэффициентом, тогда как при более высоких плотностях
х) Эта глава подготовлена совместно с доктором Эллен Спотц, Висконсинский универ-
ситет.
2) Вириальное разложение фактора сжимаемости впервые использовал Камерлинг Оннес
[2] как способ представления экспериментальных данных. Некоторые ученые предпочи-
тали выражать полученные экспериментальные данные путем разложения фактора сжи-
маемости в ряд по степеням давления:
Коэффициенты этого разложения просто связаны с коэффициентами уравнения @.1):
в-- в с- с-вг
В настоящей книге мы обычно пользуемся вириальными коэффициентами, опреде-
ляемыми уравнением @.1). Обозначения вириальных коэффициентов для разложения по
давлению отличаются от коэффициентов, соответствующих уравнению @.1), штрихом.
§ 7. Формальный вывод уравнения состояния в статистической механике 121
необходимо пользоваться большим числом вириальных коэффициентов. На-
пример, для азота при 0° С численная величина добавок к фактору сжимае-
мости за счет вириальных коэффициентов выражается следующим образом:
1 атм pV/RT = 1 - 0,0005 + 0,000003 + ...,
10 атм pV/RT = 1 - 0,005 + 0,0003 + ...,
100 атм pV/RT = 1 - 0,05 + 0,03 + ...
Область справедливости вириального разложения ограничивается сходи-
мостью рядов. Ряды расходятся при плотностях, соответствующих жидкости.
Поэтому основным применением вириального уравнения состояния является
изучение газов малой и умеренной плотности.
В первой половине этой главы дается вывод вириального уравнения со-
стояния. Получаются явные выражения для вириальных коэффициентов через
межмолекулярные силы. Вторая половина главы посвящена численным расче-
там вириальных коэффициентов для потенциалов различных видов. Вычи-
сляются все функции, необходимые для расчетов, и результаты представляются
в форме таблиц, удобных для расчетов. Результаты этой главы не вполне
справедливы в случае изотопов водорода и гелия при температуре ниже ком-
натной и в случае газов при очень низких температурах, так как для них суще-
ственны квантовые эффекты. Квантовая теория уравнения состояния и явление
равновесия при низких температурах рассмотрены в гл. 6.
§ 1. ФОРМАЛЬНЫЙ ВЫВОД УРАВНЕНИЯ СОСТОЯНИЯ
В СТАТИСТИЧЕСКОЙ МЕХАНИКЕ
При изучении свойств газов и жидкостей используются два вида урав-
нения состояния. В этом параграфе, во-первых, будет показано, как урав-
нение состояния1) может быть выведено из соотношения статистической термо-
динамики, связывающего давление и статистическую сумму. Затем обсуждается
другой вывод, при котором уравнение состояния получается из теоремы ви-
риала классической механики с помощью найденного среднего для pV по
каноническому ансамблю. Этот метод приводит к уравнению состояния, выра-
женному через радиальную функцию распределения. Это уравнение состоя-
ния имеет вид, сильно отличающийся от получаемого из статистической суммы,
но эквивалентность обоих уравнений может быть легко показана.
А. МЕТОД СТАТИСТИЧЕСКОЙ СУММЫ
При обсуждении в гл. 2, § 4, статистической термодинамики было показано,,
что статистическая сумма и давление в системе связаны соотношением2)
A.1)
В классической статистике статистическая сумма системы N одинаковых
молекул выражается в виде
W JJ> A.2)
x) Некоторые авторы соотношение р — p(V, T) называют термическим уравнением со-
стояния. Они пользуются этой терминологией для различения его от калорического уравнения
состояния, дающего выражение для внутренней энергии U = U(V, T). Вывод последнего
подобен выводу термического уравнения состояния и поэтому здесь не приводится (см. за-
дачу 1 в конце этой главы).
2) Можно показать [3, 19] также, что давление может быть выражено и через большую-
статистическую сумму (см. гл. 2, § 1).
pV = /cTlnZB-
122 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
где гамильтониан Н(тм, рм) дается выражением
N п*
A.3)
1=1
В случае идеального газа гамильтониан содержит только член кинетической
энергии, и классическую статистическую сумму можно проинтегрировать в
замкнутой форме, получив
yN
Z° A4)
где А2 = Щ2пткТ. Подставляя эту статистическую сумму в выражение для
давления, получаем уравнение состояния идеального газа pV = NkT.
В случае неидеального газа можно провести интегрирование статистиче-
ской суммы по импульсу, получив для величины ZN выражение
"!*-' A.5)
где WN(tN) — множитель Больцмана
Qn — конфигурационный интеграл1)
A.7)
Вычислению этого интеграла посвящен § 2. В изложенном там выводе не де-
лается никаких предположений относительно характера величины Ф(г^), т. е.
не вводится предположение о парном взаимодействии.
Б. МЕТОД, ОСНОВАННЫЙ НА ТЕОРЕМЕ ВИРИАЛА
Теорема вириала классической механики для системы частиц дается вы-
ражением D.28) гл. 1: В настоящем обсуждении мы рассмотрим в качестве
«частиц» системы молекулы (массы т) газа или жидкости, пренебрегая на время
внутренними степенями свободы молекул2). Теорема вириала утверждает, что
•средняя полная кинетическая энергия молекул К =1/22 /nt?? равна величине
i
вириала 3 = —^^(VF/)- Каждая молекула газа испытывает силы, дейст-
вующие со стороны всех других молекул, а также действие удерживающей
силы резервуара. В соответствии с этим существует вириал межмолекулярных
сил St и вириал внешних сил 2е.
Вириал межмолекулярных сил дается выражением
Чтобы получить триал внешних сил, рассмотрим силу, действующую со сто-
роны молекулы на стенку и равную, но противоположно направленную силе,
действующей со стороны стенки на молекулу. Рассмотрим элементарную пло-
щадку ndS стенки резервуара, где п — единичный вектор, направленный по
нормали снаружи. Средняя по времени сила, действующая со стороны нале-
г) Этот интеграл иногда называют конфигурационной статистической суммой.
2) В гл. 4, § 3, излагается другая точка зрения. Там рассматриваемая система (газ или
жидкость) принимается состоящей из совокупности ядер и электронов. Такая точка зрения
лозволяет получить сведения о кинетической энергии электронов при высоких плотностях.
§ 1. Формальный вывод уравнения состояния в статистической механике 123
тающих молекул на эту элементарную площадку, есть р п d S, а сила, дейст-
вующая на молекулы со стороны стенки равна — рп dS. Если г — вектор
положения элемента поверхности, то вклад этого элемента в вириал равен
1/2р(г • n) dS. Интегрирование по поверхности резервуара дает вириал внеш-
мих сил
Зе = ±р J (r.n)dS = i-p J (?•') = |pV. A.9)
По поверхности По объему
резервуара резервуара
Здесь в соответствии с теоремой Грина поверхностный интеграл преобразован
в интеграл по объему1).
Тогда теорема вириала принимает вид
= Si+Se = ^-y[rr^-0(TN)\ +^-pV, A.10)
из которого следует выражение pV через полную кинетическую энергию и
лотенциал межмолекулярного взаимодействия молекул
N
Черта над членами этого выражения указывает на то, что эти величины —
средние по времени. В соответствии со статистической механикой эти средние
по времени величины могут быть заменены средними по ансамблю. Восполь-
зовавшись выражением для классической плотности вероятности соответст-
вующей каноническому ансамблю, получим уравнение состояния
Во втором выражении этого соотношения проведено интегрирование по им-
лульсам. Из этого результата ясно видна роль межмолекулярных сил. Если
межмолекулярные силы отсутствуют, то интеграл по конфигурационному про-
странству обращается в нуль и мы получаем уравнение состояния идеального
газа pV = NkT.
Для неидеального газа полученное выражение может быть упрощено
если предположить, что полная потенциальная энергия системы является
суммой потенциальных энергий пар молекул
Тогда уравнение состояния принимает вид
= NkT- *»%-* Jg@(f)r3dr, A.13)
где nB\tv r2) и g(r) — бинарные функции распределения, определяемые выра-
жением
r*> = ov-2)lQN f WN(rN)dT»-2 = NiNvT1) i(r12). A.14)
См. замечание об обозначениях векторов и тензоров в начале книги.
124 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Появление бинарных функций распределения и отсутствие функций высшего
порядка является следствием сделанного допущения об аддитивности потен-
циальной энергии системы. Уравнение состояния может быть преобразовано
к вириальному виду путем разложения бинарной функции распределения
в ряд по степеням плотности. Этот вывод дан в § 3.
В. ЭКВИВАЛЕНТНОСТЬ УРАВНЕНИЯ СОСТОЯНИЯ,
ПОЛУЧЕННОГО МЕТОДОМ СТАТИСТИЧЕСКОЙ СУММЫ
И С ПОМОЩЬЮ ТЕОРЕМЫ ВИРИАЛА
Тождественность выражений уравнения состояния, даваемых соотноше-
ниями A.1) и A.12), была показана Борном и Грином [4, 5]. В первом из этих
соотношений необходимо продифференцировать статистическую сумму по
объему. Зависимость классической статистической суммы от объема прояв-
ляется только через определяемые размерами сосуда пределы интегрирования
по конфигурационному пространству.
Статистическая сумма может быть преобразована в интеграл, для которого
размеры сосуда уже не будут фигурировать в пределах интегрирования. Это
достигается введением характеристической макроскопической длины L. Эта
длина — порядка размеров сосуда и, таким образом, объем резервуара пропор-
ционален L3. Если приведенные координаты fy = ry/L ввести в интеграл клас-
сической статистической суммы, то он принимает вид
. A.15)
Тогда уравнение состояния выражается в виде
_кТ L dzN _
kTL™ Г Г щп* р*IкТ ГдгкШ
р*IкТ Гдг к Ш.~\ dtNdbN (I 16>
L 3kT dL}U "P ' U# '
Дифференцирование гамильтониана приводит к выражению
Подставляя его в уравнение состояния A.16) и возвращаясь к первоначальным
пространственным координатам, получаем выражение
pV = NkT -1
эквивалентное выражению A.12).
§ 2. ВЫВОД ВИРИАЛЬНОГО УРАВНЕНИЯ СОСТОЯНИЯ
С ПОМОЩЬЮ СТАТИСТИЧЕСКОЙ СУММЫ1)
В этом параграфе2) мы продолжим вывод уравнения состояния уже описан-
ным методом статистической суммы. Воспользуемся введенным Юрселлом [6 J.
разложением интеграла по конфигурационному пространству по «[/-функциям»
и получим статистическую сумму для газа в виде суммы произведений интег-
ралов Ь/. Эти интегралы называются групповыми интегралами, так как они
х) В обозначениях и содержании этого и следующего параграфов мы будем следовать
де Буру [5].
2) См. также монографию Боголюбова (Н. Н. Боголюбов, Проблемы динамиче-
ской теории в статистической физике, М., 1946), в которой развит метод получения степен-
ных разложений для уравнения состояния на основе канонического распределения Гиббса. —
Прим. ред.
§2. Вывод вириального уравнения состояния с помощью статистической суммы 125
включают группы из 1, 2, 3,..., N молекул. Вириальные коэффициенты выра-
жаются непосредственно через эти групповые интегралы; /-й вириальный
коэффициент выражается комбинацией групповых интегралов b1,b2,...ybj.
Окончательные выражения вириальных коэффициентов могут быть несколько
упрощены, если привлечь к рассмотрению силы парного взаимодействия. Вве-
дение этих сил отложено до § 4.
А. «17-ФУНКЦИИ»
Юрселл [б] показал, что множитель Больцмана WN(rN), появляющийся
в интеграле по конфигурационному пространству, может быть представлен
в виде суммы произведений функций [//(гА). Эти U-функции1) выражаются через
комбинации множителей Больцмана, соответствующие малому числу молекул :
U2 (г„ г,) = W2 (TifTj) - Wx (г,) Wx (г,),
Ut (г* rj9 rk) = W, (г„ TJt rk) - W2 (г„ rj) Wx (tk) -
- W2 (i>, rk) Wx (r) - W2 (rft, r,) W± (tj) +
+ 2W1(ti)W1(rJ)W1(rk). B.1)
Эта схема должна учитывать все возможные распределения молекул в группы
и дать сумму произведений {/-функций, соответствующих этим распределениям.
Необходимо заметить, что при этом перестановки молекул внутри группы не
производятся. Коэффициентами, стоящими перед различными членами, явля-
ются выражения вида (— l)""^ — 1)!, где п —число групп в соответствующем
члене. Определенные таким образом [/-функции обладают следующим важным
свойством : функция ?//(гА) равна нулю для «разделенных» конфигураций, в
которых молекулы в ряду {А} разделяются на две или больше групп, удален-
ных одна от другой на такие расстояния, что можно пренебречь взаимодей-
ствием между группами, ^то свойство [/-функций может быть легко прове-
рено для нескольких первых [/-функций.
Множители Больцмана для случая нахождения в объеме V одной, двух,
трех и т. д. частиц могут быть записаны через [/-функции
W2 (г„ tj) = U2 (г„ гу) + ^(г,), иг (г,),
(г„ г„ г,) = U3 (г„ г„ г,) + U2 (г,, Tj) иг (rk) +
+ U2 (гу, г,) иг (г,) + U2 (tk, г,-) U, (Tj) +
rk). B.2)
Таким образом, выражения для [/-функций, представленных через множители
Больцмана, совпадают с видом выражений для множителей Больцмана, пред-
ставленных через [/-функции, за исключением последнего, в котором для всех
членов коэффициенты равны + 1. Множитель Больцмана для системы N
молекул Wn(tn) можно, таким образом, символически записать в виде
WN(t») = Z 77 ад*). B.3)
(ZlmN)
Здесь суммирование должно быть распространено по всем возможным распре-
делениям N молекул в т групп по одной, т2 групп по две, ..., т{ групп по /
молекул с учетом условия 2 1Щ = N.
Эти [/-функции используются по следующей причине. Скажем для разре-
женного газа, в котором парные столкновения часты, а тройные — редки,
х) Как и в гл. 2, § 1, индекс / означает число молекул, а Я указывает, какие именно
молекулы рассматриваются.
126 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
функции иг(т{, Гу, rft) равны нулю, так же как и все {/-функции более высоких
порядков. Выражение для WN(rN) тогда будет состоять из сумм произведений
{/-функций отдельных молекул и пар. Если мы интересуемся лишь вторым
вириальным коэффициентом, который обусловлен только парными взаимодей-
ствиями, то можно отбросить {/-функции для групп из трех и более молекул.
Б. ГРУППОВЫЕ ИНТЕГРАЛЫ Ь/
Выразив в соответствии с выражением B.3) множитель Больцмана для
системы N молекул суммой произведений {/-функций, мы можем теперь фор-
мально взять конфигурационный интеграл. В результате получаем1) выра-
жение
где суммирование проводится по всем наборам значений mly удовлетворяющих
условию 21Щ = N. Величины bt—это так называемые групповые интегралы,
определяемые выражением
bi = (Vll)-1 J Ut (rlt r2, ..., г,) йтг dr2 ... tf,, B.5)
где пределы интегрирования соответствуют физическим размерам резервуара
объема V. Групповые интегралы для малых значений I почти не зависят от объе-
ма. Это можно показать, проводя (приближенно) интегрирование по коорди-
натам одной молекулы группы для получения множителя V. Этот множитель
может быть сокращен с V, входящим в выражение, определяющее bt. Прибли-
жение может быть проиллюстрировано путем рассмотрения группового инте-
грала Ь2, записываемого в специальном случае двух частиц, находящихся в
одномерном ящике размером L:
L L
ь* =ж\ $и* (*i> xJtoi&b' B.6)
О О
Поскольку {/2 является функцией только относительного положения двух
частиц, то можно провести замену переменных
L L-Xx
ь*=4т$ J U*(x*-xd*ib-xdtei- B.7>
О -xt
Функция {/2(x2 — xx) ={ exp [—q>(x2 — хгIкТ]— 1 } не равна нулю только
в очень узком интервале значений переменной х2 — х1У так как межмолекуляр-
ным взаимодействием можно пренебречь на расстояниях, ббльших нескольких
ангстрем. Если длина L — макроскопических размеров, то значение Ьг не
х) Этот ро/льтат получен следующим образом. Определенному ряду значений mi соот-
ветствует много членов разложения B.3), так как возможны различные способы распреде-
ления N частиц по группам. Все эти члены дают после интегрирования тот же результат,
а именно
N
Число этих членов дается выражением
так как перестановка частиц внутри группы и перестановка групп одного и того же размера
не приводят к появлению новых членов. Перемножая выражения (а) и (б) и суммируя по
всем значениям mi, получаем выражение B.4).
§ 2. Вывод вириального уравнения состояния с помощью статистической суммы 12Т
изменится, если пределы интегрирования для х2 — хх положить бесконечными
L +со
b* = 4r\ J U2(x2-x1)d(x2-x1)dx1. B.8)
Это позволяет провести интегрирование по хг и получить выражение
+ 00
»W*(b. B-9)
где х21 — положение молекулы 2 относительно молекулы 1.
Точное значение bz в трехмерном случае может быть апроксимировано
способом, аналогичным только что описанному для одномерного интеграла Ъ2.
Так как XJ{ для «разделенной» конфигурации равна нулю, то можно провести
интегрирование по координатам одной молекулы. Таким образом, групповые
интегралы B.5) могут быть представлены в виде
1, ...-rlJdT21...drlv B.10>
где пределы интегрирования бесконечны, а Гд — координаты, отсчитываемые
относительно положения молекулы 1. Это не зависящее от объема приближе-
ние для групповых интегралов отлично оправдывается для случая интегралов
низших порядков. Следовательно, использование выражения B.10) не вносит
дополнительных ограничений в теорию газов. При больших значениях вели-
чины I приближение становится менее точным, так как большие группы зани-
мают заметную долю объема сосуда. Поэтому выражение B.10) не может исполь-
зоваться для жидкостей, в которых преобладают большие группы. В соответ-
ствии с выражениями B.5) и B.10) первыми групповыми интегралами являются
следующие :
= 27 JJ [
= -±- A* [2! Z2Z\] = 4"J [e
о
[w* ^T»Гз) - w^ r*>w^ -
- W2(r2, r3) Wx (г,) - W2(r3, rj W&J +
+ 2W1{r1)W1{r2)WM\dr1dr2drz = ^
= -L Г Г [е-ф(Тги Г.,)/ЛТ _ ?-|>(r81) + V(r81) + <p(rM)]lkT j ^f2i ^f3i _[_
1] dr21 drn +
4"[ J
B.1
В последней формуле Ф(г21, г31) — потенциальная энергия системы трех моле-
кул, а г21 и г31 — координаты молекул 2 и 3 соответственно, отсчитываемые
относительно молекулы 1. Если принять, что потенциальная энергия системы
трех молекул равна <р(г12) + <р(г23) + <р(г1з)> то первый интеграл обращается а
нуль.
128 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
В. ВЫЧИСЛЕНИЕ СТАТИСТИЧЕСКОЙ СУММЫ
Для вычисления конфигурационного интеграла выражения B.4) было
предложено несколько методов. Здесь мы рассмотрим метод «наиболее крутого
спуска», введенный в статистическую механику Дарвином и Фаулером [7] и
использованный для решения этой задачи Борном и Фухсом [8].
Введем производящую функцию
(
B.12)
где С — комплексное переменное. Если разложить эту функцию в ряд по сте-
пеням С, то коэффициент при CN будет равен QN. Умножая производящую
функцию на f-tf-i и применяя теорему вычета Коши1), мы получаем следующее
выражение для конфигурационного интеграла :
В качестве контура интегрирования С выбирается окружность с центром,
лежащим в плоскости С. Вводя в это выражение С = г ехр ((ф) и dC = it йф,
получаем
QN = J-{ ехр [2 Vbfce'*I - Шф - N In r] йф. B.14)
Подынтегральное выражение имеет минимум для значения г = г, лежащего
на положительной оси ф = 0 и получаемого из условия
~ [/-N^6,^ = о, или N = 2 Vlb^. B.15)
Вследствие того что в подынтегральном выражении содержатся взаимно
компенсирующиеся высокие степени г при г = г, этот минимум оказывается
очень острым. С другой стороны, модуль подынтегрального выражения имеет
острый максимум в направлении, перпендикулярном действительной положи-
тельной оси. Эта точка С = г, таким образом, может быть названа «седловидной
точкой». Экспоненты подынтегрального выражения B.14) можно теперь раз-
ложить в ряд по степеням ф вдоль контура | С | = z:
2 Vbiize**)l— N In (zeW) = v у Ъ{ zl- N In z - 4" ф2 2 VI2 Ь{ zl + О(ф*). B.16)
Хорошее приближение получается при использовании ряда Тэйлора, если от-
брасывать члены, следующие за ф2. Интегрирование выражения B.14) можно,
таким образом, провести с исчезающе малой ошибкой. Поэтому, учитывая,
что область изменения ф простирается от — оо до + ©о, получаем
B.17)
Окончательно для величины In ZN, необходимой при термодинамических рас-
четах, . получаем
In ZN = - N In z Я3 + 2Vbi zl. B.18)
x) Теорема вычета Коши гласит: если /(С) аналитична внутри и на контуре С, за исклю-
чением точки С = 0, в которой находится полюс, то интеграл от /(С) по контуру равен умно-
женному на 2ni коэффициенту при члене С-1 в разложении /(?) в ряд в точке С = 0.
§ 2. Вывод вириального уравнения состояния с помощью статистической суммы 129
Это выражение получено при пренебрежении квадратным корнем в знамена-
теле соотношения B.17); это допустимо, поскольку 2УЬ&1 и 2 PVbtzl пропор-
ци'ональны N.
До сих пор параметр z являлся просто некоторой величиной, определяе-
мой выражением B.15). Его физический смысл можно вскрыть, если восполь-
зоваться выражением статистической суммы B.18) для получения свободной
энергии G
G = NkT In A3 + NkT In z. B.19)
Но из термодинамики известно, что величина G связана с летучестью / и актив-
ностью газа а отношением
G = G° + NkT \n-t- = G° + NkT In a. B.20)
Здесь индексом 0 отмечены величины, соответствующие стандартному состоя-
нию, которое выбирается как состояние идеального газа при такой же темпера-
туре и удельном объеме. Тогда G0 может быть вычислено методом, описанным
в гл. 2, § 5, и мы получим G° = NkT In А%/У/У), так что выражение B.20) сво-
дится к
G = NkT In A3 + NkT In ^~-. B.21)
Из сравнения этого выражения с B.19) следует, что параметр z связан с актив-
ностью а:
z = -^- = an, B.22)
где п = N1V — плотность числа частиц. Параметр z имеет размерность концент-
рации и при очень низких плотностях просто совпадает с плотностью числа
частиц. В этом можно убедиться, рассмотрев выражение B.15). Параметр z
можно представлять себе как активную плотность числа частиц1).
Г. УРАВНЕНИЕ СОСТОЯНИЯ В ВИРИАЛЬНОЙ ФОРМЕ
Подстановка выражения статистической суммы B.18) в выражение A.1)
для давления приводит к уравнению состояния, записанному через активную
плотность числа частиц z
pV = kTV(~^^)T = kT2 VbjZ1. B.23)
Для выражения pV в виде ряда по степеням плотности числа частиц п необ-
ходимо знать, как z выражается через п.
Мейер [9] и Каан [10] показали, что уравнение B.15) может быть решено,
если п выражается числа через z
z = nexp(-2Pknk)y B.24)
k=l
где fik — различные комбинации групповых интегралов :
р2 = З&з - 661, B.25)
0з = 464 - 24&2&з + -у-*| ит. д.
Выражения для высших рк могут быть получены разложением z в выражении
B.24) в ряд по степенями, подстановкой его в уравнение B.15) и приравнива-
нием коэффициентов при одинаковых степенях п.
х) Параметр z Гуггенгеймом был назван активностью, а Мейером — летучестью. Тер-
мин активная плотность числа частиц представляется более подходящим.*
130 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Подставляя выражение B.24) в формулу B.23) для pV, получаем урав-
нение состояния в вириальной форме типа выражения @.1)
РУ _ 1 v kh ( N )k B 26)
Таким образом, видно, что величины Aк непосредственно связаны с вириальными
коэффициентами
4
B.27)
Явные выражения для вириальных коэффициентов будут даны в § 4.
§ 3. ВЫВОД ВИРИАЛЬНОГО УРАВНЕНИЯ СОСТОЯНИЯ
С ПОМОЩЬЮ ТЕОРЕМЫ ВИРИАЛА1)
Продолжая обсуждение, начатое в § 1, мы покажем здесь, как уравнение
состояния может быть выведено с помощью теоремьк вириала. Уравнение
состояния будет выражено через бинарные функции распределения пB) (tvt2).
Это выражение уравнения состояния может быть преобразовано к вириальной
форме путем разложения функции распределения в ряд по степеням плотности.
Эта процедура проводится посредством модифицирования метода, изложен-
ного в предыдущем параграфе, так как функции распределения n(h\th) явля-
ются интегралами от множителей Больцмана по всем, кроме первых ft, моле-
кулам :
^J <зл>
Вначале множитель Больцмана записывается в виде суммы произведений
модифицированных U-функций, затем проводится интегрирование модифици-
рованных U-функций по ограниченному чи'слу координат, что дает повод ввести
модифицированные групповые интегралы. Именно через эти интегралы выра-
жается функция распределения п(/1)(гл). Вириальное уравнение состояния мо-
жет быть получено из разложения бинарной функции распределения по сте-
пеням плотности.
А. «МОДИФИЦИРОВАННЫЕ 17-ФУНКЦИИ»
Для последующего вывода удобно формально принять группу молекул
{ft} = 1, 2, ..., ft за отдельную частицу с координатами rh = rv г2, ..., г.
Не содержащие молекул {ft} {/-функции определяются точно соотношением
B.1). Для групп молекул, включающих группу {Л}, модифицированные
U-функции определяются следующим образом :
rj)- Щг")
- W(t\ тк) W1 (tj) - W(rJf тк) Wx (f) + 2W(r") W,(tj) Wi (rk). C.2)
лГ\5, 11].
§ 3. Вывод вириального уравнения состояния с помощью теоремы вириала 131
Тогда множители Больцмана выражаются через суммы произведений
[/'-функций:
= U(th,rJyTk)
U(r*)U1(rJ)U1(rk). C.3)
Эти два ряда выражений тождественны с выражениями B.1) и B.2), если в них
г, заменить на гл.
Б. «МОДИФИЦИРОВАННЫЕ ГРУППОВЫЕ ИНТЕГРАЛЫ»
Итак, функция Больцмана WN(tN) = W(rh, г^"л) может быть записана
в виде суммы произведений [/-функций и в каждом произведении появляется
модифицированная [/-функция типа C.2). Можно показать, что WN(rM) может
быть представлена окончательно в виде
WN (г") = U (г») WN-h (г"-*) + V; U (г*, г,) Wn-ь-! (г""*) +
+ SjShUP,*j,**) Wn-m^"*-f) + . • • C.4)
Подстановка этой функции в выражение C.1) для n(h\rh) дает
C.5)
где Ьф — модифицированные групповые интегралы, определяемые в виде
&<"> (г*) = TiV J • • • J U (**> г*+ь ¦ ¦ ¦ , ^+i) *л+ь • . . , **+/ ~
^ -i-J ... | (/(г*, гл+1, ... , rh+l_Jdrh+1, ... , *л+/в1. C.6)
Для интегрирования по объему координаты относят к положению 1-й моле-
кулы. После интегрирования система координат смещается таким образом, что
Гу соответствуют положению относительно некоторой молекулы группы {Л}.
Таким образом, модифицированные групповые интегралы являются функциями
относительных координат г\ Они зависят от объема таким же образом, как и
обычные групповые интегралы. Фактически групповые интегралы являются
частным случаем модифицированных интегралов Щ> для Л, равного единице.
В. ВЫРАЖЕНИЕ БИНАРНОЙ ФУНКЦИИ РАСПРЕДЕЛЕНИЯ
ЧЕРЕЗ ПЛОТНОСТЬ
Для вычисления отношения конфигурационных интегралов функция
распределения C.5) при помощи выражения B.18) может быть записана через
активную плотность частиц г. В результате для бинарной функции распре-
деления (Л = 2) получаем
^Ы^'ЯЫ^1. C.7)
Используя выражение B.24), связывающее z и п, можно записать бинарную
функцию распределения в виде ряда по степеням плотности числа частиц
C.8)
132 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
здесь ск(г12) — различные комбинации модифицированных групповых инте-
гралов
ф12) = 2bf{r12) - 4ft2 ft?)(r12), C.9)
<2) - \2b2 bp(r12) + Dfti - 6ft3) »?(g и т. д.
Аналогичные разложения функций распределения по степеням плотности
числа частиц можно написать и для функций распределения высших поряд-
ков1). Функция распределения нетождественно равна плотности числа частиц
п = NJV.
Г. УРАВНЕНИЕ СОСТОЯНИЯ В ВИРИАЛЬНОЙ ФОРМЕ
Если предположить, что полная потенциальная энергия является суммой
потенциальных энергий молекулярных пар, то функция распределения C.8)
может быть подставлена в выражение A.13) для величины pV. Тогда урав-
нение состояния принимает вид
Вириальные коэффициенты при этом даются выражениями
В(Т) = - Щ | Cl(r12) r?2 ^ drn, C.11)
С(Г)= -|^-fc2(r12)r?2^-dr12 ит.д. C.12)
Явные выражения вириальных коэффициентов через силы межмолекуляр-
ного взаимодействия будут даны в следующем параграфе.
§ 4. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ
Вывод с помощью статистической суммы (см. § 2) уравнения состояния в
вириальной форме и разложение функций распределения лл(гл) в ряд по сте-
пеням плотности проводились без предположения об аддитивности межмолеку-
лярных сил. В настоящем параграфе будет сделано это предположение и будут
описаны связанные с ним упрощения. Затем будут даны явные выражения
вириальных коэффициентов через силы межмолекулярного взаимодействия,
полученные как с помощью статистической суммы, так и с помощью теоремы
вириала. Дается распространение этих результатов на случаи многоатомных
молекул и газовых смесей.
А. ПРЕДПОЛОЖЕНИЕ ОБ АДДИТИВНОСТИ
МЕЖМОЛЕКУЛЯРНЫХ СИЛ
Если сила взаимодействия двух произвольных молекул не зависит от рас-
положения всех других молекул, то полная потенциальная энергия системы
х) Выражения для функций распределения лл(гл) описанным выше методом получили
также Мейер и Монтроль [12]. Они с самого начала предполагали аддитивность межмо-
лекулярных сил, тогда как при выводе выражения C.7) и формул для функций распреде-
ления высших порядков это предположение не делалось. Преимущество изложенного метода
заключается в том, что он может быть полностью перенесен в квантовую механику, если
дать другую интерпретацию величине \Уы(гн). Множите ли Больцмана классической теории
в квантовой статистической механике (см. гл. б) переходят в суммы Слетера.
§ 4. Вириальные коэффициенты
133
может быть записана в виде Ф(тм) = 1/2У У <Ри9г№<Ри—потенциал взаимо-
i J
действия для изолированной пары молекул. Вообще это предположение спра-
ведливо1), за исключением случаев молекул, имеющих тенденцию к ассоцииро-
ванию (например, молекул с гидроксильными или аминогруппами), и моле-
кул, обладающих водородной связью.
Фиг. 13. Вид апроксимированной функции /(г)
Мейер [14] и его сотрудники при выводе уравнения состояния исполь-
зовали функцию
Мги)=[е-^т-\]. D.1)
Эта функция обладает тем свойством, что она отлична он нуля только в том
случае, если две рассматриваемые молекулы сблизились настолько, что энер-
гия взаимодействия заметно отличается от нуля. Приближенный ход функции
fu для реальных мопекул представлен на фиг. 13. Предположение аддитивности
позволяет выразить {/-функции через функцию //у:
D-2)
Г2> Гз) = /l2 /
23
/l2 /23 + /23 /13 + /12 /l3 •
В общем случае функция Uk(rk) содержит все произведения функций fu так,
что все молекулы группы {к} как бы «связаны» по отдельности. Четыре
комбинщии функций fij} образующие U3(tvr2,T3)y изображены на фиг. 14.
^2*23 'tf;23
Фиг. 14. Четыре комбинации функций ///, учитываемые в выражении D.2) для
Основное свойство [/-функций — стремление к нулю для «разделенных» кон-
фигураций — может быть легко понято, если воспользоваться функциями //у.
х) Оно почти справедливо для дисперсионных сил Лондона, но не выполняется для
сил отталкивания или химических сил первого порядка (см. гл. 12). В работе [13] вычисля-
лась ошибка в возмущении третьего порядка, изменяющаяся по закону 1/г9. Однако эта
ошибка имеет очень малое значение при обычных газокинетических столкновениях.
134 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
В этом случае по крайней мере один из множителей /,7, описывающих связь мо-
лекулы одной группы с молекулой другой группы, должен быть равен нулю.
Первые несколько групповых интегралов выражаются через функции //;:
D.3)
где bt — неприводимые интегралы, в которых некоторые из членов могут быть
представлены в виде произведения интегралов. Например, первый член в
выражении для й8 может быть записан в виде
JJJ/ii /u*i*«*. = V\f12dt2 J/13dr3. D.4)
Такое преобразование возможно только тогда, когда размеры резервуара
велики по сравнению с радиусом действия межмолекулярных сил. Интегралы
(}кУ определяемые выражением B.25), не могут быть преобразованы таким же
образом. Поэтому мы назовем их неприводимыми интегралами.
Б. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ ДЛЯ НЕ ЗАВИСЯЩИХ
ОТ УГЛОВ ПОТЕНЦИАЛОВ
По методу статистической суммы (см. § 2) для первых трех вириальных
коэффициентов получены выражения
00
J [e-«'»kT - 1 ] r* dr, D.5)
f&i> гз) + W2(r2, г8) - 1] йгг dt2 dr3 =
N2 \0VW\7 *\7 7 Л- 7*\ ГК7№('?7 72^21 —
= - 3V" Jj J /l2 /23 /l3 *1 ^2 d4 =
$л2 N2 С С i* ...
lilt f f Y Г Г /if* ПГ ilf
— ~ з J J J '12 '13 '23 12 13 23 12 13 23 ~~
(По всем rltr«srM, образующим треугольник^
(По т^м гхвг18гц, образующим треугольник, для которых гм ^ г„ и г18 ^ г18).
м + б/12/13/14/2з/84 +
x dr2 dr3 drt. D.7)
Формулы, выраженные через функции /,7, справедливы только в том случае,
если выполняется предположение об аддитивности; справедливость формул,
§ 4. Вириальные коэффициенты 135
выраженных через статистические суммы или множители Больцмана, не зависит
от этого предположения. Кроме того, эти формулы выведены для случая одно-
атомных молекул со сферически симметричным потенциалом взаимодействия
и взаимодействующих по законам классической механики. Первое из этих пред-
положений не играет существенной роли в случае симметричных молекул,
так как внутренние степени свободы слабо влияют на уравнение состояния1);
поэтому необходимо рассматривать только поступательное движение. Если
молекулы несферичны, то полученные выше результаты должны быть обоб-
щены на случай потенциала, зависящего от угла. При выводе используются
представления классической механики, поэтому полученные формулы не могут
применяться ни при очень низких температурах, ни даже при комнатной тем-
пературе для изотопов водорода и гелия. Формулы, справедливые для легких
газов и при низких температурах, даются в гл. 6, где обсуждается квантово-
механическое уравнение состояния.
Методом теоремы вириала, описанным в § 3, для второго и третьего вириаль-
ных коэффициентов получаем выражения
В(Т) = - ^f- J г» А е-Фтг dr D8)
О
и
^ JJJ (l+fi2)f2zf^n2r23rls^dr12dr2Zdrlz. D.9)
JJ ^
(По всем г18 гх, гм,
образующим треугольник)
Эквивалентность выражения D.8) и последнего выражения формулы D.5)
может быть легко проверена интегрированием по частям. Аналогичным образом
можно показать эквивалентность соответствующих формул для третьего вири-
ального коэффициента.
В. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ ДЛЯ ПОТЕНЦИАЛОВ,
ЗАВИСЯЩИХ ОТ УГЛОВ2)
До сих пор рассмотрение относилось только к случаю одноатомных моле-
кул, обладающих тремя поступательными степенями свободы каждая. При
этом подразумевалось, что потенциал взаимодействия зависит только от рас-
стояния между молекулами. Рассмотрим теперь молекулы, которые можно
представить в виде жесткого ротатора с тремя поступательными и двумя вра-
щательными степенями свободы. Можно предположить, что потенциал взаи-
модействия таких молекул является функцией межмолекулярного расстояния
и трех углов, описывающих взаимную ориентацию молекул. Эта модель исполь-
зуется при изучении свойств двухатомных и других линейных молекул и при-
менима в случае молекул, обладающих дипольным моментом. Формулы для
вириальных коэффициентов газа, состоящего из жестких ротаторов, легко
можно получить из формул для одноатомного газа, если дать другую интер-
претацию некоторым обозначениям, использованным при их выводе.
Гамильтониан для случая газа, состоящего из N жестких ротаторов массы
m с моментом инерции /, можно записать в виде
Ж) > <4Л°)
г) Это справедливо до тех пор, пока межмолекулярные силы не зависят от ориентации
и внутренней конфигурации молекул. На языке квантовой механики это требование озна-
чает независимость потенциала межмолекулярного взаимодействия от квантовых состояний
молекул.
2)См. [15].
136 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
где q — набор из пяти координат (х, у, z, 0, ф), необходимых для задания поло-
жения и ориентации отдельной молекулы; рв и рф — компоненты импульса,
соответствующие координатам в и ф. Тогда статистическая сумма системы
имеет вид
Zn = дда-J • • • \e-^N^^dpxldpyldpzldpeidp01 х
х йх1йу1йг1йв1йф1... D.11)
Интегрирование по импульсам приводит к выражению
Zn= з^ А2ы | • • • JV*«.W {sin^ sin в,...} х
iV * Лпост. лвр. ' J
х йв1йф1йх1йу1йг1... йвыйфыйх^уыйгы, D.12)
где А^ост = Щ2пткТ и A*p = Щ2л1кТ. Это выражение можно записать в виде
,
где WN(qN) — классический множитель Больцмана, определяемый выраже-
нием A.6), а
dq( = —— dxi dyt dzt sin 0, dO( dфi. D.14)
С этого момента дальнейший вывод можно провести в соответствии со схемой,
описанной в § 2. Единственное отличие состоит в замене элемента объема rfr,
более общим элементом dq(. Для удобства в определении dqt введен множитель
4л. В предельном случае, когда потенциал взаимодействия не зависит от угло-
вых координат, интегрирование dq, по углам просто дает rfrz. В этом смысле
множитель 4 л является нормировочным множителем. Необходимость этой
нормировки видна из формального рассмотрения выражения B.15). Элемент
объема dqt следует выбрать так, чтобы Ьг было равно единице, иначе выражение
B.24) и последующие не будут выполняться.
Окончательные формулы для второго и третьего вириальных коэффи-
циентов в случае потенциалов, зависящих от углов, имеют вид
_ »2яя л
= ~~ Т I* J J f /l2 sin 6l ddl sin в*dd* d&* ~ ^ r™ dri2' D'15)
' 0 0 0 0
D.16)
В этих формулах, как и прежде, //у = ехр (—Фи/кТ)—1, за исключением
того, что теперь //у — это функции не только межмолекулярного расстояния,
но и трех углов, необходимых для задания взаимной ориентации двух молекул.
Эти формулы будут использованы в § 8 и 10 при получении уравнения состоя-
ния для газа длинных и полярных молекул.
Г. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ ДЛЯ СМЕСЕЙ
Вириальное уравнение состояния смеси может быть получено путем за-
писи статистической суммы газовой смеси и использования метода, изложен-
ного в § 2 [16]. Таким образом, найдено, что уравнение состояния смеси v
§ 4. Вириальные коэффициенты 137
компонент описывается выражением @.1), а соответствующие вириальные
коэффициенты даются выражениями
?(Г)см. =| Д Вар(Т) ха хр, D.17)
С(Г)см. = 222 Са/>у(Т)хаХрХу. D.18)
а=1 /Зя»1 у-=1
Здесь ха — молярная доля а-компоненты в газовой смеси ; В^Т) — второй
вириальный коэффициент для чистой компоненты а ; Вар(Т) — второй вириаль-
ный коэффициент, вычисленный для потенциала (рар(г12)У описывающего взаи-
модействие между молекулами компонент а и /3:
B*f>(T) = -Yv /ia2<Mr2- D.19)
Функция fif = exp (—<paiflkT)—1 определяется выражением D.1). Индексы
a и ft указывают на принадлежность молекул i и / химическим компонентам
а и /?. Аналогичным образом выражается третий вириальный коэффициент
fr2dr3; D.20)
Сару(Т) остается неизменным для любых перестановок индексов, а Сааа(Т) —
это третий вириальный коэффициент для чистой компоненты а.
Д. ОПРЕДЕЛЕНИЕ ВИРИАЛЬНЫХ КОЭФФИЦИЕНТОВ
ИЗ ДАННЫХ ЗАВИСИМОСТИ МЕЖДУ Р, V и Т
Вириальные коэффициенты могут быть получены из данных зависимости
между р, V и Т несколькими различными методами. В одном из методов урав-
нение состояния, включающее третий вириальный коэффициент, преобразовы-
вается к виду
^=(^-1)? = В+ -?+... D.21)
Отсюда можно видеть, что второй вириальный коэффициент дается выражением
В(Т) = lim [j^r - 1J V = lim Л. D.22)
Третий вириальный коэффициент равен производной от ^ при давлении,
равном нулю
С{Т) = lim (u*-B)V. D.23)
0
Этот метод получения вириальных коэффициентов В и С требует очень
точных данных относительно величины сжимаемости при очень низких плот-
ностях.
Другой метод основан на представлении фактора сжимаемости в виде
конечного полинома относительно экспериментальных значений величины 1/V
PV _ , , Аг(Т) ЩТ) Ап(Т) (А 2Д.
rt ~ * v V2 ''' 9п V****)
В этом случае при различных температурах берется ряд значений
постоянных Aj. Эти значения определяются методом наименьших квадратов
или другим методом подбора выражения для кривой, описывающей экспери-
ментальные данные. Величины Aj(T) принимаются непосредственно за вириаль-
ные коэффициенты. Однако при этом необходимо соблюдать осторожность,
138 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
так как величины Aj(T) зависят как от интервала давлений, при которых
велись измерения, так и от степени полинома, используемого для построения
кривой, охватывающей экспериментальные точки. Поэтому, если эксперимен-
тальные данные не соответствуют области достаточно низких давлений, функ-
ция АХ(Т) не будет обязательно вторым вириальным коэффициентом. Боль-
шинство тщательных измерений фактора сжимаемости газа проводилось при
умеренных и высоких давлениях, так что получить из них надежные значения
вириальных коэффициентов затруднительно. Коэффициент А2(Т) и последу-
ющие коэффициенты крайне чувствительны к показателю степени выбираемого
полинома ; поэтому при вычислении третьего вириального коэффициента по
экспериментальным данным мы также сталкиваемся со значительными труд-
ностями.
Часто экспериментаторы выражают, свои результаты в виде полинома,
отличающегося от выражения D.24). Датские ученые (Михель и сотрудники в
Амстердаме и Камерлинг Оннес с сотрудниками в Лейдене) в качестве единицы
давления используют международное определение атмосферы. Используемая
при этом единица объема, известная как единица «амага», соответствует моляр-
ному объему рассматриваемого газа при давлении в 1 международную атмо-
сферу и температуре 0° С. Эта стандартная единица молярного объема несколько
различна для разных газов и вследствие небольших отклонений свойств газов
от идеальности не равна 22,414 см31моль даже при атмосферном давлении. Мо-
лярный объем газа при произвольном давлении и температуре определяется
отношением истинного значения к величине стандартной единицы молярного
объема :
»« = -?¦; D.25)
здесь va — молярный объем в единицах «амага». Аналогично плотность газа в
единицах «амага» выражается так:
Обычно удобно записывать уравнение состояния в виде
pva = а + bga + CQ2a + dQ*+ ... D.27)
и выражать экспериментальные результаты для различных температур, под-
бирая значения коэффициентов а, Ь, с... (при этом используются конечные
ряды). Сравнение этого уравнения состояния с вириальной формой @.1) по-
казывает, что коэффициенты В, С, D,... можно выразить через а, 6, с, d,...
следующим образом :
В V
D.28)
D = — Kf и т. д.
где Vs = RTja. Так как эти соотношения справедливы только при бесконеч-
ности ряда D.27), то при вычислении вириальных коэффициентов ими можно
пользоваться только как приближенными.
Немецкие ученые (в частности, Гольборн и Отто в Берлине) пользуются в
качестве единицы давления давлением, соответствующим 1 м pm. ст. В каче-
стве единицы молярного объема принимается объем моля газа при давлении
1 м рт. ст. и температуре 0° С. Уравнение состояния записывается в виде
ру = А + Вр + Ср2+ ... D.29)
§ 5. Вириальные коэффициенты в случае простых потенциалов взаимодействия 139
и Л, В, С,... относят к различным температурам. Вириальные коэффициенты
уравнения @.1) выражаются через эти величины
В = 0,76 RTj,
С = @,76 RT)*^ +В*. D.30)
Эти выражения так же точны только в случае бесконечности ряда D.29).
§ 5. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ
В СЛУЧАЕ ПРОСТЫХ ПОТЕНЦИАЛОВ ВЗАИМОДЕЙСТВИЯ,
НЕ ЗАВИСЯЩИХ ОТ УГЛОВ
В этом параграфе результаты вириального разложения используются
для вычисления уравнения состояния в случае нескольких простейших моде-
лей молекулы. Для каждой такой простой потенциальной функции можно в
аналитическом виде взять интегралы и получить вириальные коэффициенты.
Эти простые аналитические выражения удобны для представления экспери-
ментальных данных, соответствующих узкому интервалу температур. При
представлении данных для более широких интервалов необходимо пользо-
ваться более реалистическими потенциальными функциями. Кроме того, более
реалистическими потенциальными функциями необходимо пользоваться и для
получения силовых постоянных, которые в свою очередь могут использоваться
для вычисления уравнения состояния и коэффициентов переноса. Вириальные
коэффициенты для таких улучшенных потенциалов даются в последующих
параграфах.
А. МОДЕЛЬ ТВЕРДЫХ СФЕР
Модель твердых сфер в связи с ее простотой нашла широкое использование
в ранних работах по изучению уравнения состояния и при расчетах высших
вириальных коэффициентов. Второй и третий вириальные коэффициенты могут
быть легко вычислены по формулам предыдущего параграфа. Однако четвер-
тый [17], пятый1) и высшие вириальные коэффициенты вычислить значительно
труднее.
Вириальные коэффициенты от второго до пятого для газа, состоящего из
твердых сфер с диаметром столкновения <х, выражаются следующим образом:
С="8"Ьо, E.1)
D = 0,2869 ftg,
? = @,115 ±0,005)ftg.
Вириальные коэффициенты для модели твердых сфер не завися! от температуры
и, вероятно, все положительны. Поэтому коэффициент сжатия pVjRT всегда
больше единицы и является функцией плотности.
Приближение типа твердых сфер хорошо оправдывается при очень высо-
ких температурах, когда взаимное притяжение молекул становится несуще-
1) Величина пятого вириального коэффициента была вычислена в Лос-Аламосе (частное
сообщение Розенблюта) на счетной машине IBM 701 методом Монте-Карло.
140 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
ственным. Например, эта модель успешно использовалась в задачах внутренней
баллистики для апроксимирования уравнения состояния горячих пороховых
газов. Задавая энергию реального взаимодействия между двумя молекулами,
бывает очень трудно точно подобрать подходящую для модели твердых сфер
величину диаметра столкновений1). Одним из разумных определений величины
может являться расстояние наибольшего сближения двух сталкивающихся
молекул с энергией кТ. Часто величина а вычисляется путем приравнивания
величины Ьо собственному объему молекулы b в уравнении Ван дер Ваальса.
Однако величина ван-дер-ваальсовой постоянной Ь определяется не единст-
венным образом. Другой обычный метод выбора а связан с величиной вязкости
(см. гл. 8). Для уравнения состояния высокотемпературного порохового газа2)
оказывается, что величина о равна 0,81 величины силовой постоянной о потен-
циала Леннарда-Джонса (см. § 6).
Б. МОДЕЛЬ ТОЧЕЧНЫХ ЦЕНТРОВ ОТТАЛКИВАНИЯ
Подстановка потенциальной функции <p(r) = dr6 [выражение 3.23) гл. 1]
в формулу второго вириального коэффициента D.8) приводит [22 ] к
= - |#[г-(- Mr--*)ехр (^) dr =
0 E.2)
Поскольку интеграл расходится на верхнем пределе, если не выполняется
условие д > 3, то этот результат не приложим ни к электронному, ни к иони-
зованному газам. Второй вириальный коэффициент всегда положителен для
газа, молекулы которого рассматриваются как точечные центры отталкивания.
В. МОДЕЛЬ СЮЗЕРЛЕНДА
В этой модели молекулы представляются твердыми сферами диаметра о,
окруженными потенциальным полем <р(г) = — сг~у [см. выражение C.25)
гл. 1 ]. Подстановка этой потенциальной функции в формулу второго вириаль-
ного коэффициента приводит к [23]
^ /! iy - 3
Интегрирование можно провести, разложив экспоненту в ряд Тэйлора. Мо-
дель Сюзерленда является специальным случаем потенциала Леннарда-Джонса
при бесконечно слабом отталкивании. При низких температурах модель при-
водит к отрицательной величине второго вириального коэффициента. Это обу-
словлено важностью члена отталкивания потенциальной функции при столк-
новениях медленных молекул.
х) В книге Стюарта [20 ] даны диаметры столкновений для модели твердых сфер, опре-
деленные по различного рода экспериментальным данным.
2) Высокотемпературная область для пороховых газов оказывается близкой к области
температур, в которой потенциал Леннарда-Джонса приводит к максимальному значению
второго вириального коэффициента, равному приблизительно 0,52 Ьо [21 ].
§ 5. Вириальные коэффициенты в случае простых потенциалов взаимодействия
141
Г. МОДЕЛЬ ПРЯМОУГОЛЬНОЙ ПОТЕНЦИАЛЬНОЙ ЯМЫ
Для прямоугольной потенциальной ямы имеем выражение C.24) гл. 1
<р(г) = со, 0<г<о,
<р(г) = - е, а < г < /?<т,
9<г) = 0, г>/?ог.
Выражения, приведенные в § 4, разд. Б могут быть использованы для вычисле-
ния второго и третьего вириальных коэффициентов. В результате получаем [24 ]
E.4)
E.5)
ЦТ) = i-ftg [5 - (Я6 - 18/?4 + 32/?3 - 15)d -
- B/?6 - Зб/?4 + 32/?3 + 18/?2 - 16) А* -
- (б/?6 - 18/?4 + 18/?2 - б) А% R < 2,
С(Г) = ^/>§ [5 - ПА - (- 32/?3 + 18/?2 + 48) zl2 -
- E/?6 - 32/?3 + 18/?2 + 26) J3], /?>2,
где А = ехр (е/кТ) — 1, а Ьо = 2/3 tzNo3. Значения параметров Ьо, е//с и R полу-
чены для большого числа молекул [25 ] путем сопоставления данных* по вто-
рому вириальному коэффициенту с формулой E.4). Полная сводка этих пара-
метров, включая значения для многих органических молекул типа углеводо-
родов и фреонов, дана в табл. 6.
Таблица 6
Силовые постоянные для потенциала прямоугольной потенциальной ямы,
полученные по значениям второго вириального коэффициента*
Газ
с,\
Ь.Л,
см* 1 моль
Литература
Неон
Аргон
Криптон
Азот
Двуокись азота
Этан
Пропан
п-Бутан
п-Гептан
Этан
Пропен
Пропадиен
1-Бутен
2-Метилпропен
шранс-2-Бутен .
цис-2-Бутен ...
CC13F
CHC12F
CC12F2
CC12F-CC1F2
Метилхлорид ..
Аммиак
Пар
2,382
3,162
3,362
3,299
3,917
17,05
39,87
47,93
45,29
75,79
1,87
1,85
1,85
1,87
1,83
19,5
69,4
98,3
53,7
119
27
27
28]
27
[30
3,535
4,418
4,812
6,397
3,347
4,316
4,511
5,592
5,570
5,276
5,747
4,534
2,797
4,812
3,697
4,294
2,902
2,606
55,72
108,8
140,6
330,3
47,28
101,4
115,8
220,6
218,0
185,3
239,5
117,6
27,60
140,6
63,73
99,90
30,83
22,33
1,652
1,464
1,476
1,314
1,677
1,460
1,373
1,249
1,254
1,324
1,215
1,545
2,321
1,394
2,075
1,337
1,268
1,199
244
347
387
629
222
339
382
492
490
465
537
399
306
345
335
469
692
1260
[31]
[32]
[33]
[34]
[35]
[36]
[37]
[38]
[39]
[40]
[41]
[42]
• Постоянные для сложных молекул взяты из работы [25].
142 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Модель прямоугольной потенциальной ямы особенно удобна при напи-
сании уравнения состояния газов, образованных из сложных молекул, так как
в потенциальную функцию входят три подбираемые постоянные. Результаты,
полученные для этой модели, не применимы к высокотемпературным газам,
так как этот потенциал нельзя использовать для описания столкновений моле-
кул с большими энергиями — столкновений, играющих решающую роль при
повышенных температурах.
Было найдено, что второй вириальный коэффициент для модели прямо-
угольной потенциальной ямы апроксимирует коэффициент, соответствующий
потенциалу F-12) Леннарда-Джонса [выражение C.27) гл. 1 ], если R принять
равным 1,8, значение диаметра столкновения а равным значению а для по-
тенциала F-12) Леннарда-Джонса, а глубину потенциальной ямы принять
равной 0,56 величины максимальной энергии притяжения для потенциала
Леннар да-Джонса. При таком частном выборе параметров выражения для
второго и третьего вириальных коэффициентов имеют вид
= 1 -4,832 А, E.6)
= 0,6250 - 2,085/1 + 10,118Л2 - 8,430/I3. E.7)
Хотя первое из этих выражений приводит к совпадению с вычислениями для
случая потенциала F-12) Леннарда-Джонса, значение третьего вириального
коэффициента E.7) отличается в 2—3 раза от соответствующего потенциалу
Леннарда-Джонса. Это является результатом того, что второй вириальный
коэффициент относительно нечувствителен к виду потенциальной функции
межмолекулярного взаимодействия, тогда как высшие вириальные коэффи-
циенты значительно более чувствительны.
Модель прямоугольной потенциальной ямы может быть использована [25 ]
при вычислении второго и третьего вириальных коэффициентов для газовых
смесей. Потенциал взаимодействия z-й и /-й молекул компонент а и /? дается
выражениями
<^(г/7)=оо, <Уар>Гф
Ч>* (ги) = - e^ aap < ru < Rap aapj E.8)
^Ы = 0, ru>Rapaafi.
Тогда, полагая фо)ар = у п N о*р и zJa/5 = exp (-^-)— 1, можно написать сле-
дующие соотношения дня Вар{Т) и Сар(Т):
= /@) - [/A,1L,, + /0,2) 4^, +
+ [/B,1L,КУ + 1B,2L„Д* +
-[IC)AapAayAfiy], E.10)
где
/@) = J(Oap, °ay, Ору),
/A,2) = Лаф Rayaay, apy) - /@),
/A,3) = J(oafi,aaY,RfvaltY) - 1@),
1B,2) = J(Raftoap, aay, Rpy о,у) - [/@) + /A,1) + /A,3)],
/B,3) = J(aafi, Ray aay, Rfiy apy) - [/@) + /A,2) + /A,3)],
/C) =J(Rapoap,Rayaay,Rpyafiy)-\l@)+ v/(l,fc)
i
ft=l
§ 6. Вириальные коэффициенты для потенциала F-12) Леннарда-Джонса 143
Функция J(a, Ь, с) в формулах для величины / имеет вид
при
при
^ при с^а+Ь, E.12)
j(a,b, с) «= ^ [а6 + Ь6 + с* + 18а2Ь2с2 + 1б(?»3с3 + с3а3 + а
- 9{а*(&2 + с2) + Ь\с* + а2) + с\а* + Ь2)}]
Прямоугольная потенциальная яма является наиболее реалистичным
потенциалом, для которого может быть точно вычислен третий вириальный
коэффициент смеси.
§ 6. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ ДЛЯ ПОТЕНЦИАЛА
F-12) ЛЕННАРДА-ДЖОНСА1)
Потенциал F-12) Леннарда-Джонса [см. C.27) гл. 1 ]
широко используется при вычислениях свойств газов, жидкостей и твердых
тел. Поэтому здесь подробно рассматривается вычисление вириальных коэф-
фициентов для этой важной модели. Показано также, что параметры а и е,
входящие в потенциальную функцию, могут быть определены из значений вто-
рого и третьего вириальных коэффициентов. В заключение показано, что экспе-
риментальные значения коэффициента Джоуля—Томсона также могут быть
использованы для получения сведений о силах, действующих между молеку-
лами. В последующем обсуждении многие формулы принимают более простой
вид, если воспользоваться следующими обозначениями2) приведенных величин:
F.,)
— * Jт*^ » ^"к '
Величины В? и CJ - безразмерные; через них могут быть выражены
юдинамические свойства.
Величины В? С?
термодинамические свойства.
х) Некоторые расчеты вириальных коэффициентов были предприняты для потенциалов
Леннарда-Джонса с не равным двенадцати показателем степени члена отталкивания. По-
скольку найдено, что двенадцатая степень приводит для многих веществ к лучшему со-
гласию с экспериментальными значениями второго вириального коэффициента и коэффи-
циента Джоуля—Томсона, то в этом параграфе будут даны результаты только для потен-
циала F-12) Леннарда-Джонса. Однако для молекул гелия, ртути и некоторых других может
оказаться несколько лучшим потенциал F-9) Леннарда-Джонса. Многочисленные расчеты
второго вириального коэффициента методом перфорированных карт для потенциала
F-9) Леннарда-Джонса в интервале значений Т* от 0,5 до 100 были сделаны в работе [43].
Третий вириальный коэффициент для потенциала F-9) рассчитан в работе [57].
2) В литературе используются два способа приведения величин типа второго вириального
коэффициента. Пользуются выражением В* = B/iVa8, и В* = В/ВТв.сф. = В/Ьо. Оба спо-
соба имеют свои преимущества. Мы будем обозначать значком (*) приведение величин путем
простейших комбинаций параметров а и е, а значком (*)—приведение относительно ком-
бинаций величин а и е, используемых для модели твердых сфер. Оба типа приведенных
величин, конечно, связаны просто.
144 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
А. ВЫЧИСЛЕНИЕ ВТОРОГО ВИРИАЛЬНОГО
КОЭФФИЦИЕНТА
При использовании потенциала Леннарда-Джонса [44] интеграл для
второго вириального коэффициента может быть взят в аналитическом виде.
Подстановка потенциала F-12) Леннарда-Джонса [см. C.27) гл. 1 ] в формулу
D.8) для второго вириального коэффициента приводит к
F.2)
Если ехр [D/Т*)г*~6 ] разложить в бесконечный ряд, то интегрирование можно
провести в аналитическом виде. Приведенный1) второй вириальный коэффи-
циент может быть записан в виде
В(Т) = Ьо В*(Т*), В*(Г*) = 2 #ЛГ*-<?'+1>'4. F.3)
Коэффициенты2) Ьи) даются выражением
^) F.4)
и сведены в табл. IV3).
Сумма в выражении F.3) расходится очень быстро при значениях приве-
денных температур, больших 4. Для меньших значений Т* сумма расходится
не так быстро : при Т* = 0,30 для получения точности до пятой значащей
цифры необходимо удерживать около 30 членов ряда. Функция В* вычисля-
лась [45] методом перфорированных карт в интервале значений Т* от 0,3
до 400. Эти значения приведены в табл. II (стр. 854). Там же приводятся зна-
чения функции В?(Г*), входящей в выражения для термодинамических вели-
чин. Таблица различных термодинамических величин, выраженных через
вириальные коэффициенты, приведена в приложении Б в конце настоящей
главы (см. стр. 190).
Рассмотрим свойства функции В*(Т*). Как видно из фиг. 15, при низких
температурах В* отрицательна, а выше температуры Бойля*) Т% — 3,42 она
положительна и имеет максимум вблизи Г* = 25. Поведение функции В*
можно понять, исходя из характера потенциальной функции. При низких
температурах средняя энергия молекул того же порядка, что и глубина потен-
циальной ямы. Поэтому сталкивающиеся молекулы проводят значительную
часть времени взаимодействия в области притяжения потенциальной кривой.
Эта особая роль притяжения между молекулами приводит к понижению дав-
ления, и, таким образом, второй вириальный коэффициент становится отрица-
тельным. При высоких температурах средняя энергия молекул велика по срав-
нению с максимальной энергией притяжения. Молекулы как бы «не замечают»
падения потенциальной кривой, так что решающий вклад в вириальный коэф-
фициент обусловлен членом отталкивания потенциальной функции. Особое
значение взаимного отталкивания молекул приводит к увеличению давления,
х) Для общего вида потенциала Леннарда-Джонса [см. C.26) гл. 1] аналогичный вывод
приводит к формуле для В(Т):
В (Т) = - 2*L (?\ У у*«б-у) 2 W (V, б) yJ, F.3a)
6 ycj у-о
2) Коэффициенты b(J) не следует путать с интегралами bi (§ 2).
3) Таблицы, пронумерованные римскими цифрами, помещены в Приложении в конце
книги (стр. 849).
4) См. задачу 4 в конце главы.
§ 6. Вириальные коэффициенты для потенциала F-12) Леннарда-Джонса
145
и, таким образом, второй вириальныи коэффициент становится положительным.
При очень высоких температурах особое значение имеют столкновения, при
которых молекулы в значительной степени взаимопроникают друг в друга,
что приводит как бы к уменьшению объема молекул. Поэтому величина В*
достигает максимума и затем начинает уменьшаться. Этот максимум наблю-
дался у гелия [27].
1ft
0,5
О
-0,5\-
-2,0
-2,5
-з,о
-3,5
-4,0
1
-
/it
— II
- / / /
' 1
1 1
-с
1 1
i 1 1
-
• -Аг
o-N2
e-CH4
«-He
I i i
5 10
20
50 то
Фиг. 15. Приведенный второй вириальныи коэффициент для потенциала Леннарда-Джонса.
а кривая для Не, рассчитанная квантовомеханическим методом ; b — кривая для Н2, рассчитанная кван-
товомеханическим методом (см. гл. 5, § 5); с — кривая, рассчитанная классическим методом. Класси-
ческую кривую для В*(Т*)можно сравнить с экспериментальными данными для различных газов. Отчет-
ливо видно, в какой области температур для Не и Н, существенны квантовые отклонения [26].
Чтобы вычислить второй вириальныи коэффициент с помощью табл. II
(стр. 854) нужны только значения параметров а и е потенциала Леннарда-
Джонса. Их получают из экспериментальных значений второго вириального
коэффициента (это будет обсуждаться в следующем разделе), коэффи-
циентов переноса (см. гл. 8, § 4), постоянных, характеризующих критическую
точку, точку плавления или точку кипения (см. гл. 4, § 1), или из значений
Таблица 7
Силовые постоянные потенциала F-12) Леннарда-Джонса,
полученные из значений вторых вириальных коэффициентов*
Газ
Ne
Аг
Кг
Хе
N2
о2
сн4
со2
в/к, °К
34,9
119,8
171
221
95,05
118
148,2
189
<т, А.
2,78
3,405
3,60
4,10
3,698
3,46
3,817
4,486
д0, см3/моль
27,10
49,80
58,86
86,94
63,78
52,26
70,16
113,9
Литература
[27]
[46]
[28]
[47]
[48]
[49]
[50]
[51]
• Более подробная таблица силовых постоянных потенциала F-12)
Леннарда-Джонса приведена на стр. 851. Там же приведены силовые постоя-
нные, вычисленные из данных по вязкости.
146 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
других величин. Значения этих силовых постоянных даны в табл. I (стр. 851)
для большого числа молекул. В табл. 7 приведены некоторые из этих парамет-
ров. Если силовые постоянные получены из экспериментальных данных раз-
личного типа, то при вычислении свойств, связанных с уравнением состояния,
обычно более целесообразно пользоваться значениями, полученными с по-
мощью второго вириального коэффициента.
Для ряда газов было получено несколько наборов силовых постоянных
в соответствии с рассмотрением различных выражений для уравнения состоя-
ния. Рассмотрим числовой пример, поясняющий применение табулированных
величин и формул, связанных с вычислением уравнения состояния.
ПРИМЕР
Задача. Вычислить фактор сжимаемости для аргона при давлении 5 атм и темпера-
турах 15 и 250° С.
Решение. Из табл. 7 получаем постоянные для Аг: е//с=П9,8°К и Ьо=49,80 см3/моль.
Тогда Т = 15 + 273,2 = 288,2 °К, а Т* = кТ/е = 288,2/119,8 = 2,406. Соответствующее
этому Г* значение величины В* из табл. II находим равным —0,3605. Второй вириаль-
ный коэффициент В тогда равен В = Ь0В* = D9,80) (—0,3605) = 17,95 смг/моль. Тогда
вириальный коэффициент для уравнения состояния в виде ряда по степеням давления [см.
примечание к выражению @.1) на стр. 120) равен В' = B/RT = (—17,95^/(82,06) B88,2) =
= —0,00076 атм-1. Окончательно для фактора сжимаемости имеем
РУ = 1 + В'(Т)р + • • • = 1 - @,00076) E) + • • • = 0,9962.
NkT
Аналогичным образом для Г = 250° С В* =0,1702, а фактор сжимаемости равен 1,0010.
Б. ОПРЕДЕЛЕНИЕ МЕЖМОЛЕКУЛЯРНЫХ СИЛ ПО ЗНАЧЕНИЯМ
ВТОРОГО ВИРИАЛЬНОГО КОЭФФИЦИЕНТА
Определив экспериментально значения второго вириального коэффициента
одним из методов, изложенных в § 4, можно использовать их для получения
значений параметров любой потенциальной функции взаимодействия. Два
подбираемых параметра потенциала Леннарда-Джонса могут быть определены
из экспериментальных значений вириального коэффициента В(Т) для двух
температур. Можно воспользоваться следующей процедурой. Во-первых, введем
величину
— I В(Т ) э п ^
Тогда е/к можно определить, решая уравнение
F.6)
методом повторных ошибок.
В качестве первого приближения величины е\к можно использовать зна-
чение, вычисленное по температурам кипения, плавления или критической
температуре с помощью выражений, приведенных в гл. 4, § 1. Диаметр столк-
новений получаем из выражения1)
Ьо= 1,2615 a--[J^T], F.7)
где Т( равно либо Т19 либо Т2. Несколько отличный набор постоянных может
быть получен при выборе других значений температур Тх и Т2. Однако это
расхождение мало и обусловлено тем, что потенциальная функция Леннарда-
В этом соотношении величина Ьо выражена в см3/моль, а диаметр столкновений
А
§ 6. Вириальные коэффициенты для потенциала F-12) Леннарда-Джонса
147
Джонса является эмпирической функцией и не дает точного описания зависи-
мости межмолекулярных сил от расстояния. Для иллюстрации способа опре-
деления силовых постоянных из значений второго вириального коэффициента
здесь приводится числовой пример.
ПРИМЕР
Задача. Используя экспериментальные данные [47] табл. 7, определить для ксенона
силовые постоянные потенциала Леннарда-Джонса. Критическая температура ксенона
равна 289,8 °К.
Решение. Так как для большого числа газов кТс/е = 1,3, то приближенное значение
е/к равно 223 °К- Теперь решаем уравнение (б.б) для трех комбинаций температур 7\ и Г2.
Результаты решения методом последовательных ошибок можно представить табл. 8.
Таблица 8
Тг
298,2
348,2
373,2
498,2
548,3
573,3
кв
0,3003
0,2963
0,2894
Пробные
значения
e/fc
223
220
219
223
220
221
220
221
222
B*(Tj)
в*(тЬ
0,3060
0,3010
0,2993
0,3009
0,2940
0,2964
0,2839
0,2867
0,2895
Наиболее подходящие значения величины е\к для каждой пары темпера-
тур подчеркнуты. Совпадение значений, полученных для широкого темпера-
турного интервала, показывает точность потенциала Леннарда-Джонса. Для
elk = 221° К диаметр столкновений в соответствии с выражением F.7) равен
а = 4,10 А.
В табл. 9 приведены значения В(Т), вычисленные с помощью табл. II
(стр. 854), и для сравнения их измеренные значения.
Таблица 9
Измеренные и вычисленные значения второго вириаль-
ного коэффициента В(Т) для ксенона
Температура,
°к
298,2
348,2
373,2
498,2
548,3
573,3
В{Т), см*/моль
измеренный
-130,2
-94,5
-81,2
-39,1
-28,0
-23,5
вычисленный
-128,4
-94,5
-81,5
-38,9
-28,0
-23,4
Силовые постоянные потенциала Леннарда-Джонса, определенные таким
образом, даны в табл. 7. Более полные данные можно найти в табл. I (стр. 851).
Если доступные экспериментальные данные были получены двумя раз-
личными группами ученых, то вычислялись два набора силовых постоянных.
148
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Представленные в табл. I силовые постоянные для водорода и гелия
были получены как уже описанным методом, так и аналогичным методом,
в котором учитывались квантовые эффекты (см. гл. б, § 5). Кривая второго
вириального коэффициента В*(Г*) для потенциала Леннарда-Джонса пред-
ставлена на фиг. 15. С помощью потенциала Леннарда-Джонса были обрабо-
таны экспериментальные данные для большого числа газов ; результаты также
показаны на фиг. 15. Видно, что эта потенциальная функция довольно хорошо
описывает температурную зависимость второго вириального коэффициента.
Это является отличной иллюстрацией применимости закона соответственных
состояний, подробно обсуждаемого в следующей главе.
До сих пор вычисление силовых постоянных ограничивалось рассмотре-
нием взаимодействия молекул одного и того же вещества. В принципе возможно
определить закон взаимодействия молекул различных веществ из значений
второго вириального коэффициента для смеси, выражаемого соотношением
D.17). Однако вириальные коэффициенты для смесей зависят не только от сил,
действующих между неодинаковыми молекулами, но (и очень сильно) от взаи-
модействия одинаковых молекул. Поэтому для определения сил взаимодействия
неодинаковых молекул необходимо иметь точные экспериментальные значения
второго вириального коэффициента для чистых компонент и для смеси. Такие
данные обычно недоступны. Другая возможность [52 ] заключается в исполь-
зовании явления изменения объема при смешении двух газов. В этом случае,
как и прежде, зависимость от сил взаимодействия неодинаковых молекул в
определенной степени маскируется зависимостью от взаимодействия одинако-
вых молекул. Измерения коэффициента диффузии и отношения постоянных-
термодиффузии являются лучшим источником информации о силах взаимо-
действия между неодинаковыми молекулами1).
Таблица 10
Экспериментальные и
Газовая смесь
Ha-Na
на-соа
N2-He
Na-Os
Na-COa j
N2-QH,
N,-CHSF
CO2-He
CO,-Oa j
СОа-С,Н4
CO2-CH,F
Темпера-
тура,
°C
25
25
25
25
30
25
30
25
25
25
25
30
25
25
вычисленные значения
Вц(Т), СМ^МОЛЬ
Экспериментальные значения,
полученные в работах
[52]
13,5
-41,3
-39,0
12,5
—
-50,4
-54,8
—
-40,2
-58,3
-128
—
[53]
12,8
—
- 1Д
—-
—
-42,6
—
-36
—
I
—
-149
[54]
(ИД)*
—
—
—
- 9,7
-40,6
—
—
—
-41,5
—
—
Вычислен-
ные
значения
11,5
0,8
3,0
17,7
-10,1
-41,1
-49,1
-44,2
-40,6
21,3
-52,1
-49,9
-133
-105
• Этот результат получили Михель и Вассенаар [55].
1) В табл. 90 (стр. 461) сравниваются силовые постоянные, полученные из эксперимен-
тальных диффузионных и термодиффузионных данных, со значениями, вычисленными с
помощью эмпирического правила комбинирования.
§ 6. Вириальные коэффициенты для потенциала F-12) Леннарда-Джонса 149
Когда другие сведения отсутствуют, необходимо пользоваться эмпири-
ческим правилом комбинирования, связывающим силовые постоянные между
неодинаковыми молекулами с их значениями для одинаковых молекул :
««* = («««*)*. F.9)
Соотношение F.8), очевидно, является точным в случае модели твердых сфери-
ческих молекул. Соотношение F.9) следует из простой интерпретации диспер-
сионных сил через поляризуемость отдельных молекул (см. гл. 12, § 3). Силовые
постоянные, полученные с помощью этого правила комбинирования, по-види-
мому, приводят к хорошим результатам при вычислениях для смесей. В табл.
10 сравниваются экспериментальные значения В12(Т), полученные из данных
по изменению объема при смешении, со значениями, вычисленными с помощью
эмпирического правила комбинирования.
ПРИМЕР
Задача. Вычислить второй вириальный коэффициент для смеси, состоящей из 25%
N2 и 75% СН4, при температуре 50° С C23,2 °" К) (концентрация в молярных долях).
Решение. Из табл. I (стр. 851) получаем следующие значения параметров:
N2 b0 = 63,78 см*/моль, ~ = 95,05° К,
СН4 Ьо = 70,16 см*/моль, ~ = 148,2а К.
/с
Значения параметров для взаимодействия молекул N2 и СН4 получаем из выражений F.8)
и F.9)
—СН4) = -i-{[60(N2)]1/e + [bo(CHjfl*Y = 66,96 см*/молъ,
± (N2 - СН4) = |± N2 ± СН4|/2 = 118,7 ° К.
Используя эти значения, получаем
Na Г* = 3,400, В* = — 0,0043, В = — 0,2743 см*/моль,
СН4 Г* = 2,181, В* = — 0,4942, В = — 34,67 см*/молъ,
N2—СН4 Г* = 2,723, В* = — 0,2143, В = — 14,34 см*/моль.
Затем из выражения D.17) для второго вириального коэффициента получаем
В = @,25)*(— 0,2743) + 2 @,25) @,75) (— 14,35) +
+ @,75J (— 34,67) = — 24,90 см*/моль.
В. ВЫЧИСЛЕНИЕ ТРЕТЬЕГО ВИРИАЛЬНОГО КОЭФФИЦИЕНТА
Интеграл для третьего вириального коэффициента может быть вычислен
методом, аналогичным использовавшемуся для вычисления второго вириаль-»
ного коэффициента [56]. Для потенциала F-12) Леннарда-Джонса получаем
С(Т) = Ь20С*(Т*), С*(Г*) = ZcMT*~(J+W. F.10)
Это выражение весьма похоже на выражение F.3) для второго вириального
коэффициента, но если коэффициенты b(j) в выражении F.3) являются гамма-
функциями, то коэффициенты разложения c(j) есть сложные интегралы. Зна-
чения си\ вычисленные Кихарой1), даны в табл. V (стр. 858). Метод получения
г) В работе [57] постоянные сО) представляют собой исправленные постоянные,
приведенные в работе [56].
150
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
значений коэффициентов с0) описан в приложении А в конце настоящей главы.
Третий вириальный коэффициент вычислялся также путем прямого вычисления
неприводимого интеграла /32 методом перфорированных карт. Результаты этих
вычислений1) даны в табл. II (стр. 854) для интервала приведенных темпера-
тур Г* от 0,70 до 400. Протабулированы также функции С?(Г*), используемые
при расчетах термодинамических свойств. Вычисления третьего вириального
коэффициента в ограниченном интервале температур проводились также двумя
другими группами ученых [59,60].
0,7
0,6
0,5
0,4
? °Л
0.1
О
-0,1
-0.2
I
/
/
_ /
/
/
/
_/
1 1
1
\ ©э
ф ^Sj®^
фф ax,j3
фДд ^
ф
ф
ф
QyJ
О
О
О
1
вб
I
1 1
• -Аг
®-СН4
Д-С2Н4
ф-С02
1 1
a-D2
е-Н2
в-Не
o-N2
©в
-
10
20
50
Фиг. 16. Приведенный третий вириальный коэффициент для потенциала Л еннарда-Джонса.
Кривая получена вычислением неприводимого интеграла рг методом перфорированных карт. Эксперимен-
тальные точки соответствуют различным газам. Отклонения от теоретической кривой отчетливо видны в
случае несферических молекул. Различное поведение легких газов (водород, дейтерий, гелий) является
следствием квантовых эффектов [58].
На фиг. 16 показан ход С*(Т*), полученный из вычислений по методу пер-
форированных карт [56 ]; приводится также ряд экспериментальных точек.
Эти экспериментальные значения были обработаны с использованием силовых
постоянных, полученных из значения второго вириального коэффициента.
В целом совпадения кривой и экспериментальных точек нет. Все точки, соот-
ветствующие простым газам, лежат выше вычисленной кривой. Несовпадение,
вероятно, обусловлено в некоторой степени способом определения значений
третьего вириального коэффициента по данным о сжимаемости (см. §4). Откло-
нений следует также ожидать вследствие эмпирического характера потенциала
Леннарда-Джонса. Для третьего вириального коэффициента существенны
иные области потенциальной кривой, нежели для второго вириального коэф-
фициента. Кихара [57] нашел, что несколько лучшее совпадение получается
для потенциала F-9) Леннарда-Джонса, но это не значит, что потенциал F-9)
Леннарда-Джонса имеет преимущества по сравнению с потенциалом F-12)
Леннарда-Джонса. Значение 12 для показателя степени силы отталкивания
выбрано потому, что оно обеспечивает лучшее соответствие с эксперименталь-
ными значениями второго вириального коэффициента и коэффициента Джоуля—
Томсона для большого числа газов. Кроме того, для потенциала F-12) Леннарда-
Джонса наблюдается исключительно хорошее совпадение для коэффициентов
переноса (см. гл. 8).
г) Пересмотренные вычисления Кихары [57 ] находятся в хорошем согласии с ними.
§ 6. Вириальные коэффициенты для потенциала F-12) Леннарда-Джонса 151
Из этих результатов можно сделать единственное заключение, что если
сведения о межмолекулярных силах получены из измерений различных физи-
ческих величин, то необходимо выбирать потенциальную функцию, более
реалистическую, чем потенциал Леннарда-Джонса.
Исключительно плохое согласие экспериментальных и рассчитанных зна-
чений третьего вириального коэффициента наблюдается для удлиненных моле-
кул. Даже для второго вириального коэффициента, относительно не чувстви-
тельного к виду потенциала, несимметричность молекул проявляется в том,
что ход кривой для В(Т) отличается от хода, соответствующего потенциалу
Леннарда-Джонса. В § 8 будет показано, что очень хорошее согласие с экспери-
ментальными значениями В(Т) получается, когда вычисления проводятся с
потенциальной функцией, учитывающей зависимость от угла. До сих пор не
проделаны вычисления третьего вириального коэффициента для удлиненных
молекул.
Очень большие отклонения от теоретической кривой получены в случае
изотопов водорода и гелия. Эти отклонения, обусловленные квантовыми эффек-
тами, обсуждаются в гл. 6. Для третьего вириального коэффициента никаких
вычислений величины квантовых отклонений не проводилось и поэтому ни-
какого количественного объяснения этих эффектов дать нельзя.
Ввиду упоминавшихся выше отклонений таблица приведенных значений
третьего вириального коэффициента имеет несколько более узкие пределы
применимости, чем в случае второго вириального коэффициента. Таблица зна-
чений С*(Т) должна давать очень хорошие результаты для сферических непо-
лярных молекул при высоких температурах и при таких плотностях, что по-
правка фактора сжимаемости, обусловленная третьим вириальным коэффи-
циентом, мала по сравнению с поправкой, обусловленной вторым вириальным
коэффициентом. Для иллюстрации способа пользования таблицами мы дадим
здесь числовой пример.
ПРИМЕР
Задача. Вычислить третий вириальный коэффициент для криптона при температуре
250° К.
Решение. Из табл. 7 для силовых постоянных взаимодействия получаем е/k — 171° К
и Ьо = 58,86 см?/моль. Приведенная температура Т* равна Т/(е/к) = 250/171 = 1,46. Для
этого значения Т* из табл. III (стр. 856) получаем для С* значение 0,55357. Тогда третий
вириальный коэффициент равен С(Т) = Ь20С*(Т*) = E8,86J @,55357) = 1918 (емумольJ.
До сих пор наше обсуждение ограничивалось третьим вириальным коэф-
фициентом для чистых веществ. Еще не было проделано вычислений значений
Сару(Т)ддя потенциала Леннарда-Джонса в соответствии с выражением D.18).
Однако эти значения могут быть найдены, если воспользоваться тем, что тре-
тий вириальный коэффициент для смеси получен в аналитическом виде для
модели прямоугольной потенциальной ямы, в частности для случая двух по-
стоянных [см. E.10)]. Определим функцию А(Т*) таким образом, что ее куб
равен отношению значений величины С(Т), вычисленной для чистой компо-
ненты а и потенциала Леннарда-Джонса, и величины С(Т), вычисленной для
модели прямоугольной ямы с двумя постоянными [выражение E.7) ] :
[CUT)]л.-д. = [Сааа(Г)]пр.я. [А(Га)]*. F.11)
Функция А слабо зависит от Г* и табулирована в табл. IV (стр. 857). Пред-
ставляется разумным выразить вклады СаРу(Т) в третий вириальный коэф-
фициент для смеси компонент а, /?, у в виде
[СаРу(Т)]л.-д. - [Са/>у(Г)]пр.я. [А(Га) А(Т;) А(ГУ)]; F.12)
[Сару(Т) ]пр.я. дается выражением E.10), где Rafi = R^ = Rya = 1,8, а глу-
бина трех прямоугольных потенциальных ям принята равной 0,56
глубины, соответствующей случаю потенциала F-12) Леннарда-Джонса.
152
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Справедливость выражения F.12) не может быть проверена до тех пор,
пока не будет получено хорошее эмпирическое уравнение состояния для смеси
газов, содержащей существенно сферические молекулы. До тех пор, пока соот-
ветствующие данные не доступны, выражение F.12) должно давать разумный
метод нахождения величины фактора сжимаемости для смесей при высоких
температурах.
Г. КОЭФФИЦИЕНТ ДЖОУЛЯ—ТОМСОНА
Коэффициент Джоуля—Томсона /г, определяемый как скорость изменения
температуры с давлением при изэнтальпическом расширении, связан с уравне-
нием состояния соотношением
v)h = c?[t(if)p-v]- <бЛЗ>
Правую часть этого соотношения можно разложить в ряд1) по степеням 1/V,
и тогда коэффициент Джоуля—Томсона можно выразить через приведенные
второй и третий вириальные коэффициенты и их производные
-..., <6Л4)
где С°р — значение молярной теплоемкости при давлении, равном нулю, и рас-
сматриваемой температуре2). Первый член разложения равен коэффициенту
Джоуля—Томсона /г° при давлении, равном нулю. Коэффициент при члене
1/К, деленный на RT, дает наклон касательной к кривой зависимости /* от дав-
ления в точке р = 0.
0
-1
1
I
«л
I I
I I I
b
-, /
i i 1
• -Аг
®-сн4
©-Не
-
, „, -
г 1 1
10 20
50 100 200
500
Фиг. 17. Приведенный коэффициент Джоуля—Томсона в случае потенциала
Леннарда-Джонса.
о- — кривая, рассчитанная классическим методом ; Ь — кривая для Не, рассчитанная квантовомеханическим
методом. Классическая кривая (Вх*—В*) сравнивается с соответствующими экспериментальными дан-
ными. Показаны также экспериментальные данные, полученные для гелия, для которого заметно проявление
квантовых эффектов [61 ].
В табл. 11 (стр. 854) даны значения функции В?—В*, необходимые для
вычисления коэффициента Джоуля—Томсона при давлении, равном нулю.
В графической форме эта функция представлена на фиг. 17. На той же фигуре
х) Разложения в ряд удельной теплоемкости и других термодинамических величин даны
в приложении Б в конце главы.
2) Значение Ср может быть вычислено по методу, изложенному в гл. 2, § 5.
Л 6. Вириалъные коэффициенты для потенциала F-12) Леннарда-Джонса 153.
показаны экспериментальные значения /*°С°/&0. Силовые постоянные, полу-
чаемые из значений второго вириального коэффициента, используются для
обработки экспериментальных данных. Согласие экспериментальных и теоре-
тических значений очень хорошее. Это сравнение является убедительной иллю-
страцией эффективности использования данных о межмолекулярных силах,
получаемых из измерений какой-либо термодинамической величины, для вы-
числения другой величины, измеряемой другим способом. Необходимо заме-
тить, что величина JBf—В* положительна при низких температурах (охлаж-
дение при расширении) и отрицательна (нагревание путем расширения) при
температурах, превышающих температуру инверсии Т) = 6,47 (см. задачу
5 в конце главы).
Очевидно, таблицу значений Bf—В* можно использовать для вычисле-
ния силовых постоянных потенциала Леннарда-Джонса. Значения а и е,
полученные таким образом, по точности сравнимы со значениями, полу-
ченными из второго вириального коэффициента. Эти силовые постоянные
могут затем быть использованы для вычисления второго вириального коэф-
фициента. Таким образом, используя результаты статистической механики,
можно определить значения второго вириального коэффициента по экспери-
ментальным значениям коэффициента Джоуля—Томсона. Термодинамика не
предоставляет такой возможности, так как ее результаты содержат неиз-
вестные постоянные интегрирования.
пример
Задача, Найти температуры инверсии для ксенона и неона.
Решение. Из табл. II видно, что функция В* — В* меняет знак при Т\ = 6,47. Так как
в/k для ксенона равно 221° К, то температура инверсии Т\ = F,47) B21) = 1430° К = 1157° С.
Для неона в/к = 34,9° К, так что Т, = F,47) C4,9) = 226° К = — 47° С.
Пользуясь табл. II (стр. 854), можно также вычислять коэффициенты
Джоуля—Томсона для смесей. Удельную теплоемкость Cg смеси идеальных
газов можно получить из выражений E.48) гл. 2, а выражение D.17) настоя-
щей главы дает выражение для второго вириального коэффициента смеси.
Для коэффициента Джоуля—Томсона при давлении, равном нулю, для двух-
компонентной смеси получаем формулу
„о _ *i (c°p)i А + А (с°рJ А + 2х, х2 (ьоI2 [(в*I2 - (в*I2] ,
* см- ~~ Xi(Cj)i K (ЬЛ5>
На фиг. 18 представлены вычисленные1) и экспериментальные значения
коэффициента Джоуля—Томсона при давлении, равном нулю, для смеси
Не — N2[61]. Эмпирические правила комбинирования [см. F.8) и F.9)] исполь-
зовались для вычисления величины (Ь0I2 [(Bf I2 — (В*I2 ]. Принципиальна
возможно определить силы взаимодействия одинаковых молекул из измерений
коэффициента Джоуля—Томсона для смесей. Однако имеющиеся данные не
позволяют сделать этого единственным образом. На фиг. 19 наклон кривой за-
висимости [I от р, выражаемый соотношением F.14), сравнивается с соответ-
ствующими экспериментальными значениями, где величина (/^"^(Э/г/ЭрH дается
для различных газов как функция температуры. Для аргона вычисленная кри-
вая проходит определенно выше экспериментальных точек. Это соответствует
тому, что в указанном интервале температур С?ыч. <С?Сп. и С^ыч. > CitKCn.
(что видно из фиг. 16). Ошибка складывается из двух эффектов. Однако для
двуокиси углерода и азота С?,ч. > С?сп. и С^ыч.4 >С*ЭКСП., и эти два проти-
воположных эффекта стремятся взаимно компенсировать друг друга. В самом
х) Вычисление проводилось на основе использования «эффективных» силовых постоян-
ных для гелия. Таким образом, экспериментальные значения сравнивались со значениями,,
полученными по классическим формулам. Полученные постоянные не учитывают квантовых
эффектов.
154 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
деле, для этих газов экспериментальные точки не располагаются ниже теорети-
ческой кривой, как в случае аргона. Таким образом, расхождения измеренных
и вычисленных значений коэффициента Джоуля—Томсона, по-видимому, суще-
ственно связаны с различиями вычисленных и измеренных значений третьего
вириального коэффициента.
о/ю
0,30
0,25
| 0,20
- 0,10
0,05
О
-0,05
-0,70
7
\
1 1
1
-—-*—
1
1 1 1
а-16,6% Не
Ь-33,2% Не
с-51,0%Не
d-75,5%Hz
I l I
0
-700 -50
50 700
150 200 250
Фиг. 18. Коэффициент Джоуля—Томсона смеси гелий—азот, соответствующий нулевому
давлению [61 ].
В заключение этого параграфа рассмотрим приложимость полученных
результатов и покажем, каким образом можно достичь дальнейших успехов
в установлении связи межмолекулярных сил и уравнения состояния. Для
-0,004{-
400 0 100 200 300 400
Температура, °С
О
-0,074
-100 0 100 200 300 400
Температура, °С
0,001
0,002
0,003
0,004
0,005
0,006
0,007
0,008
-0,018
- -0,00$ -
-100 0 100 200 300 400
Температура, °С
-от,
400 0 100 ZOO 300 400
Температура j °C
Фиг. 19. Сравнение рассчитанных (сплошная кривая) и экспериментальных значений
(точки) величины (^y^dft/dpH для различных газов [58].
благородных газов и почти сферически симметричных молекул потенциал Лен-
нарда-Джонса в широком интервале температур дает отличное совпадение с
измерениями для второго вириального коэффициента и коэффициента Джоуля—
§ 7. Второй вириальный коэффициент для более сложных потенциалов 155
Томсона, экстраполированному к нулевому давлению. Для третьего вириаль-
ного коэффициента и величины наклона кривой зависимости /г от р при дав-
лении, равном нулю, совпадение не столь хорошее. Кроме того, несколько от-
личные значения силовых постоянных получаются при вычислении из значе-
ний коэффициентов переноса и свойств кристалла. Следовательно, можно
заключить, что, хотя потенциал Леннарда-Джонса имеет большую ценность
при изучении связей между различными свойствами вещества, более точные
сведения о межмолекулярных силах можно получить, только пользуясь более
сложным законом взаимодействия. В § 7 мы изложим результаты, достигнутые
в решении задачи построения потенциальных функций взаимодействия для
сферических неполярных молекул.
Для всех обсуждавшихся в настоящем параграфе величин в случае суще-
ственно несферических молекул наблюдаются несколько большие расхождения
между вычисленными и измеренными значениями. Для этих молекул при вы-
числении вириальных коэффициентов необходимо пользоваться потенциалом
взаимодействия, зависящим от углов. Вириальные коэффициенты для случая
несферических молекул рассмотрены в § 8. Полярные молекулы рассматри-
ваются в последнем параграфе настоящей главы.
§ 7. ВТОРОЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ
ДЛЯ БОЛЕЕ СЛОЖНЫХ СФЕРИЧЕСКИ СИММЕТРИЧНЫХ
ПОТЕНЦИАЛОВ
Вычисления второго вириального коэффициента будут проведены для
нескольких функций межмолекулярного взаимодействия типа Букингема.
Эти функции имеют член отталкивания в виде экспоненты и содержат более
двух подбираемых постоянных. В этом параграфе коротко указаны возможные
вычисления для таких потенциальных функций и способы определения под-
бираемых силовых постоянных.
А. ПОТЕНЦИАЛ БУКИНГЕМА—КОРНЕРА
Довольно.большие расчеты второго вириального коэффициента (как клас-
сического, так и квантового) были проделаны с применением потенциала Бу-
кингема—Корнера [см. C.29) гл. 1 ]:
_
Этот потенциал содержит четыре постоянные : е — глубина потенциальной
ямы; гт — значение г в минимуме потенциальной кривой ; a — крутизна
экспоненциального отталкивания ; C = c'fcr^. Эта потенциальная функция
более реалистична, чем потенциал Леннарда-Джонса. Она описывает силу
отталкивания экспонентой и в ее общем виде содержит члены притяжения,
пропорциональные как г~8, так и г.
Букингем и Корнер [63] провели численное интегрирование выражения
второго вириального коэффициента с использованием потенциала C.29) гл. 1.
Свои результаты для второго вириального коэффициента и коэффициента
156
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Джоуля—Томсона они выразили в виде
G.1)
G.2)
где T* = kTje. Функции Fo и Go табулированы в табл. XXIV и XXV (стр. 894-896)
в интервалах значений C от О до 0,2, а от 12,5 до 14,5 с шагом 0,5 и Т* от 1,25
до 100. Максимальная ошибка вычислений составляет менее 0,1% значений,
приведенных в таблицах. Функции Fo и Go, очевидно, имеют тот же характер-
ный ход, что и функции В* и Bf — В* для потенциала Леннарда-Джонса.
Была вычислена также первая квантовая поправка к величинам второго вири-
ального коэффициента и коэффициента Джоуля—Томсона (см. гл. 6, § 5). Од-
нако до сих пор для этой потенциальной функции не были проделаны вычи-
сления коэффициентов переноса.
Результаты расчетов Букингема и Корнера не использовались столь широко
для определения межмолекулярных сил по данным зависимости между р, V и Т.
Параметры потенциала Букингема—Корнера для двух благородных газов —
аргона и неона — приведены в табл. И. Эти постоянные получены Корнером с
учетом малыхквантовыхэффектов.Значения второго вириального коэффициента,
вычисленные при использовании этих постоянных с точностью до 0,5%, со-
гласуются с экспериментальными значениями [27] в интервале температур до 600°.
Аналогичное совпадение имеет место и для коэффициента Джоуля—Томсона.
Параметры потенциала Букингема—Корнера
Таблица 11
Газ
Неон [64]
Аргон [64]
Р ,
0,00
0,15
0,20
0,00
0,14
0,20
а
13,6
13,3
13,2
13,9
13,5
13,4
elk
37,1
37,4
37,5
123
125
125
Гт
3,16
3,16
3,16
3,87
3,87
3,87
/о
3,20 [65]
3,81 [67]
589 [66]
2030 [68]
а, р, е/к, тт — параметры, входящие в потенциальную функцию ; /0 — расстояние (в А) между бли-
жайшими соседями кристаллической решетки при температуре О* К; Ло — теплота (кал\молъ) сублимации
кристалла при 0° К.
Одинаково хорошее согласие как для значения /? = О, так и для ft = 0,2,
по-видимому, указывает на то, что второй вириальный коэффициент недоста-
точно чувствителен к форме потенциальной кривой и поэтому не дает сведений
о степени относительной важности членов г~6 и г~8. В соответствии с квантово-
механическими расчетами Маргенау (см. гл. 14, § 2) величина /3 должна быть
порядка 0,15. Недавние измерения квадрупольного момента методом уширения
радиоволн в микроволновом диапазоне (см. гл. 13, § 7) позволяют точно опре-
делить величину ft для различных молекул. Полученные таким образом значения
сильно отличаются от полученных Маргенау и сильно зависят от точной элек-
тронной конфигурации.
Б. МОДИФИЦИРОВАННЫЙ ПОТЕНЦИАЛ
F-ехр) БУКИНГЕМА
Недавно Рейс и Гиршфельдер [69 ] подготовили таблицы значений второго
вириального коэффициента, вычисленного на основе модифицированного по-
тенциала (б-ехр) Букингема [см. C.30) гл. 1 ]:
ср{г) =
'макс, у
Т <С ''макс. •
§ 8. Второй вириальный коэффициент для несферических молекул
157
В этом выражении гмакс. — значение г, при котором <р(г) имеет фиктивный мак-
симум. Три постоянные е, гт и а имеют тот же смысл, что и в случае потенциала
Букингема—Корнера.
Для модифицированного потенциала (б-ехр) Букингема второй вириаль-
ный коэффициент можно записать в виде
= ЬтВ*(а;Т*), G.3)
где bm =2I^Nr3m и Т* =/сГ/е. Значения функции В*(а; Г*) табулированы в
табл. XXVIII (стр. 902), а значения параметров е, rm и а для различных газов
даны в табл. 12. Эти параметры определялись с помощью коэффициента вяз-
кости, второго вириального коэффициента и свойств кристалла. Литературные
ссылки на работы, использованные для вычисления параметров, даны в послед-
них трех столбцах табл. 12. Для случая модифицированного потенциала
(б-ехр) Букингема были вычислены интегралы, необходимые для вычисления
коэффициентов переноса. Эти расчеты описаны в гл. 8, § 4.
Таблица 12
Параметры модифицированного потенциала F-ехр) Букингема [70]
Вещество
н2
Не
Ne
Аг
Кг
Хе
СН4
Na
СО
Гт,А
3,337
3,135
3,147
3,866
4,056
4,450
4,206
4,040
4,011
<4,099
3,937
Параметры
efk, °K
37,3
9,16
38,0
123,2
158,3
231,2
152,8
113,5
101,2
> 132,0
119,1
а
14,0
12,4
14,5
14,0
12,3
13,0
14,0
16,2
17,0
>17
17,0
Экспериментальные данные, использованные
для определения параметров
коэффициенты
вязкости
[71-93]
[71,77,78,81,84-86,
90,91,100-103]
[77,78,85,88,90,91,
110,111]
[71,76-78,91,103,
115,116]
—
—
[71,78,80,89,90,
120, 121 ]
—
[73, 76-78,89, 90,
92,102,132 — 134]
—
[73,76,77,
91, 103,132]
вторые
вириальные
коэффициенты
[94-99]
[95, 98,
104-109]
[95,112,113]
[95,117-119]
[28]
[47]
[122,123]
[48,128-131]
—
[135-138]
—
кристалли-
ческие
свойства
_
—
[114]
[114]
[114]
[114]
[124-127]
[124-127]
—
[124-127]
—
§ 8. ВТОРОЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ
ДЛЯ НЕСФЕРИЧЕСКИХ МОЛЕКУЛ
Точно так же, как для случая сферических молекул простейшей моделью
является модель твердых непроницаемых шаров, в случае несферических
молекул можно рассматривать модели твердых непроницаемых объектов раз-
личной формы. Поэтому мы начнем это обсуждение с изложения работы Иши-
хары, показавшего, как можно рассчитывать второй вириальный коэффициент
в случае любой твердой выпуклой молекулы. Кихара продолжил эту работу,
рассмотрев модели удлиненных и сплюснутых молекул, взаимодействующих
согласно потенциалу Леннарда-Джонса, в котором за расстояние между моле-
кулами принято кратчайшее расстояние между «сердечниками» этих молекул.
158
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
В заключение параграфа обсуждается ранняя работа Корнера, вычислявшего
второй вириальный коэффициент для модели цилиндрических молекул. Он
представлял цилиндрическую молекулу в виде трех или четырех распреде-
ленных вдоль оси центров сил. В этом случае потенциал межмолекулярного
взаимодействия выражался суммой взаимодействий различных центров двух
молекул.
А. РАССМОТРЕНИЕ МОДЕЛИ ТВЕРДЫХ ВЫПУКЛЫХ МОЛЕКУЛ
МЕТОДОМ ИШИХАРЫ
В § 5 было показано, что второй вириальный коэффициент для модели
твердых шаров равен учетверенному объему молекул газа. Для модели твер-
дых несферических молекул второй вириальный коэффициент можно записать
в виде
B = 4Nvmf, (8.1)
где vm — объем отдельной молекулы; / — множитель (всегда больший еди-
ницы), указывающий отклонение от поведения для модели твердых шаров.
Ишихара [139, 141 ] показал, каким образом, используя теорию групп и пред-
ставления дифференциальной геометрии, может быть вычислен множитель /
для любых твердых выпуклых молекул. Результаты его строгой математиче-
ской работы крайне просты по виду. Для газа, содержащего молекулы одного
сорта, множитель / дается выражением
(8.2)
где sm — площадь поверхности молекулы, a R — среднее значение радиуса
кривизны. Последний выражается следующим образом :
(8.3)
где Rt и R2 — основные радиусы кривизны, a dco — элемент поверхности на
сфере единичного радиуса.
Таблица 13
Множитель / для различных несферических моделей [139, 141]*
Форма молекул
Размер
Шар
Куб
Правильный тетраэдр
Правильный октаэдр
Цилиндр
Радиус = 1
Ребро = 1
Ребро = 1
Ребро = 1
Длина = /;
радиус=а
4я
6
Уз
1
3/4
2ла (а+1)
1
11/8
1,926
1,330
п а2 + (я + 3) al + /*
8а/
• Для эллипсоидальных и сфероцилиндрических молекул / дается выражениями (8.4)—(8.7).
Эти результаты были обобщены на случай моделей негладких молекул
(например, твердый четырехгранник) и смесей молекул различных форм. Тот
факт, что / всегда больше единицы, указывает на то, что для несферических
молекул второй вириальный коэффициент всегда превышает коэффициент,
соответствующий модели сферических молекул того же объема. Второй вириаль-
ный коэффициент для всех твердых моделей не зависит от температуры. Методы,
использованные в этом анализе, могут найти применение в других геометри-
чески трудных задачах.
§ 8. Второй вириальный коэффициент для несферических молекул
159
Множитель / вычислялся для ряда моделей несферических молекул [139]
и результаты этих вычислений приведены в табл. 13. Были также сделаны
вычисления для модели эллипсоида вращения (как для вытянутого, так и для
сплюснутого) и получено выражение
(8.4)
где е0 — эксцентриситет, определяемый соотношением е§ = (а2 —
(а и Ь — большая и малая полуоси образующих эллипса соответственно,
фиг. 20). Если эксцентриситет мал по сравнению с единицей, то выражение
для / можно записать в виде ряда по степеням г§
-J5"
15
175
31
525
(8.5I)
На фиг. 21 показано изменение функции / в зависимости от отношения а\Ъ.
Этот результат будет полезен при определении формы молекул по данным об
осмотическом давлении в растворах высокополимеров [139].
2а
* 2Ь
Фиг. 20. Эллипсоидальная молекула.
вращение вокруг оси а а' приводит к вытя-
нутому сигарообразному эллипсоиду; вра-
щение вокруг оси ВВ' приводит к сплюсну-
тому (блинообразному)эллипсоиду ; аи b —
большая и малая полуоси эллипсоида соот-
ветственно ; / — расстояние между
фокусами /ь /,.
12 3 4 5 6
Отношение величии полуосей а/Ь
Фиг. 21. Изменение величины
/ в зависимости от отношения
величин Полуосей ЭЛЛИПСОИДаль-
пыл.
Второй вириальный коэффициент для модели сфероцилиндрических молекул
был вычислен Кихарой [29]2). Вытянутые сфероцилиндры образуются (фиг. 22)
при вращении молекул вокруг оси АА\ а сплюснутые — при вращении вокруг
оси ВВГ. Для таких молекул / дается выражениями
[О -I |
?2 _|_ 4| (вытянутая молекула),
/=!+¦
(сплюснутая молекула),
(8.6)
(8.7)
г) Подобное решение в виде ряда получено ранее [15] и в неопубликованной работе
Котани. Коэффициент при члене ej дан в работах [139, 141 ] равным 37/60, что является
ошибочным.
2) Авторы благодарны за предоставление возможности ознакомиться с этой работой,
до ее опубликования в журнале.
160 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
где ? = Qmjl. Кихара использовал эти выражения для моделей твердых эллип-
соидальной и сфероцилиндрической молекул и получил выражения второго
вириального коэффициента при потенциале Леннарда-Джонса, зависящем от
углов. Эта работа будет рассмотрена в следующем разделе.
\А'
Фиг. 22. Сфероцилиндрическая молекула.
Вращение вокруг оси А А' приводит к вытягиванию сфероцилиндра (капсюлеобразная молекула); вра-
щение вокруг оси В В' приводит к сплющиванию сфероцилиндра (пилюлеобразная молекула).
Б. ОБОБЩЕННАЯ МОДЕЛЬ СФЕРОЦИЛИНДРИЧЕСКИХ МОЛЕКУЛ
КИХАРЫ
Кихара [29] показал, как можно обобщить сфероцилиндрическую модель
для включения сил отталкивания и притяжения, соответствующих взаимо-
действию типа Леннарда-Джонса. Обобщение проводится таким образом,
чтобы сохранить интегрируемость выражения для второго вириального коэф-
фициента. Результаты применимы как к длинным, так и к плоским молекулам.
Во-первых, необходимо определить, что означает термин сердечник моле-
кулы. Для гомополярной двухатомной молекулы за сердечник принимается
линейный отрезок, соединяющий их ядра. Сердечник других молекул выпуклой
формы может быть определен аналогичным образом относительно остова моле-
кулы. Например, расстояние О — О в молекуле СО2 и расстояние С — С в
молекулах С2Н2, С2Н4 или С2Н6 образуют сердечники. Длина сердечника /,
определенного таким образом для нескольких молекул, приводится в табл. 14.
Потенциал взаимодействия <р(е) двух таких длинных молекул принимается
в форме потенциала F-12) Леннарда-Джонса [выражение C.27) гл. 1 ]. Однако
аргумент функции q определяется не как расстояние между центрами молекул,
а как кратчайшее расстояние между их сердечниками. Физическая реальность
подобного потенциала сомнительна, в частности при столкновениях, в которых
концы сердечников молекул находятся в пределах расстояния взаимодействия.
Тем не менее при этом подходе вводятся основные понятия взаимодействия
несферических молекул и получено хорошее совпадение теории с эксперимен-
том. Метод Корнера, описанный в разд. Г, в связи с этим оказывается более
реалистическим; однако этот метод значительно сложнее в случае длинных
молекул и вообще неприменим в случае плоских молекул.
За сердечник плоских молекул принимается тонкий круглый диск. На-
пример, для бензола за сердечник выбирается круглый диск, окруженный
шестью атомами углерода. При этом межмолекулярное расстояние определяется
§ 8. Второй вириальный коэффициент для несферических молекул 161
как кратчайшее расстояние между сердечниками. При необходимости разли-
чить сердечники плоских и длинных молекул их называют соответственно
дисковыми сердечниками и стержневыми сердечниками. Длина стержневого и
диаметр дискового сердечников обозначаются символом /. Эта величина может
быть легко получена из известных в литературе данных о внутримолеку-
лярных расстояниях. Таким образом, потенциал межмолекулярного взаимо-
действия будет характеризоваться тремя величинами : длиной I и параметрами
а и е потенциала F-12) Леннарда-Джонса. В действительности Кихара в каче-
стве одного из используемых параметров вместо диаметра выбрал величину
расстояния, соответствующего минимуму потенциальной кривой дт = 21/в<т.
Расстояние qm отождествляется также с наименьшим размером («малая ось»)
сфероцилиндра, как показано на фиг. 22. При / = 0 потенциал межмолекуляр-
ного взаимодействия сводится к обычному сферически симметричному случаю
потенциала F-12) Леннарда-Джонса. При е = О и замене показателя степени
силы отталкивания 12 на ©о модель сводится к модели твердых сфероцилинд-
рических молекул, показанных на фиг. 22.
Для сферически симметричных молекул второй вириальный коэффициент
может быть записан в виде
Г=оо
В(Т) = J [1 - *-*'>/*т] dbo(r), (8.8)
г=О
где bo(r) — второй вириальный коэффициент для модели твердых шаров с
диаметром г; bo(r) = 2nNr3j3.Аналогично для обобщенной сфероцилиндри-
ческой модели второй вириальный коэффициент выражается1) в виде
В(Т) = [ [1 — е-?<е)/«т] db{o) + 6,@). (8.9)
е=О
В этом выражении b^g) — второй вириальный коэффициент для твердой
сфероцилиндрической модели с сердечником длины / и малой осью q
где / дается выражениями (8.6) и (8.7) для вытянутых и сплюснутых сфероци-
линдров соответственно.
Замена потенциала Леннарда-Джонса потенциалом ср(о) и подстановка
выражения bfa) (8.9) приводит к интегрируемому в аналитическом виде выра-
жению для второго вириального коэффициента. Метод интегрирования тот же,
что использовался при интегрировании для второго вириального коэффициента
для модели Леннарда-Джонса в F.2). Окончательно получаем
Н ^р— F± r^fj (вытянутые сфероцилиндры), (8.10)
х)Это можно показать следующим образом. Выражение D.15) для второго вириального
коэффициента записывается через расстояние между центрами молекул г. Расстояние q между
концами сердечников является тогда функцией г и трех углов, определяющих взаимную
ориентацию молекул в19 02> #• Эта функция может быть преобразована к виду
г = r(g, д19 02, 0). Тогда выражение D.15) можно записать в виде
N ГС ГС ,ч . ^ dr
В (Т) =» -г |М|A —e-?te>/*T) [г (е, 01Э 02 0)]2 -т— dg sin 0г sin 02 ddx dd2 d&.
4 jJJJ UQ
Так как потенциальная функция <p(g) для модели Кихары не зависит от углов, то интегри-
рование по углам приводит к выражению
00
4 J
о
где Y(q) — одна и та же функция для любых потенциалов взаимодействия (р(д). Тождест-
венность этого результата и выражения (8.9) очевидна.
162
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
В(Т)
n*fJQ*ml
Гft It^T
+
+ -§¦] Qml2 Pi \jjf) H fg— (сплюснутые сфероцилиндры), (8.11)
где
к
12
2/ ( в
7Т Ы
6l-k
—12—
J
(8Л
Значения функций Fk табулированы в табл. XXIII (стр. 893). Эти резуль-
таты могут быть легко обобщены на случай, когда показатели степени сил
взаимодействия отличаются от значений 6 и 12.
Таблица 14
Параметры для обобщенной модели сфероцилиндрических и эллипсоидальных
молекул Кихары [29], определенные из значений второго
вириального коэффициента
Газ
N,
сн, = сн,
со,
'Л
0,74
1,10
1,34
2,2
Сфероцилиндрические
молекулы
em, A
2,81
3,47
4,0
3,7
•/fc, °К
39,4
124,0
266,0
279,0
Эллипсоидальные
молекулы
em, A
3,24
4,09
4,8
4,6
е/к, °К
32,4
101,0
216,0
224,0
В табл. 14 даны значения длин сердечников для четырех молекул, а также
значения параметров qm и е, полученные из данных по второму вириальному
коэффициенту. На фиг. 23 проводится сравнение вычисленных и измеренных
значений второго вириального коэффициента для этих газов.
50
чоо
I II I I I Г I I
I Г
I I I I I I I 1
Q5 1,0 1,5
lgkT/c
Фиг. 23. Сравнение экспериментальных и теоретических значений второго вириального
коэффициента, полученных для обобщенной модели сфероцилиндрической молекулы
Кихары [29].
§ 8. Второй вириальный коэффициент для несферических молекул 163
В, ОБОБЩЕННАЯ МОДЕЛЬ ЭЛЛИПСОИДАЛЬНЫХ МОЛЕКУЛ
НИХ АРЫ [29]
Модель твердого эллипсоида для учета межмолекулярных сил отталки-
вания и притяжения может быть обобщена методом, аналогичным описанному
в предыдущем разделе для сфероцилиндрической модели. За стержневый
сердечник длинной молекулы принимается линия, соединяющая фокусы удли-
ненных эллипсоидов вращения. Межмолекулярное расстояние q определяется
следующим образом. Мы строим на обоих сердечниках конгруэнтные эллип-
соиды такой величины, что они касаются друг друга. Тогда суммарная длина
малых полуосей, касающихся эллипсоидов, принимается за расстояние q, a
потенциал <р(д) межмолекулярного взаимодействия принимается соответствую-
щим взаимодействию по Леннарду-Джонсу. Аналогично для случая плоских
молекул за межмолекулярное расстояние q принимается суммарная длина
малых полуосей, касающихся сплюснутых сфероидов, построенных на диско-
вых сердечниках.
Эта модель межмолекулярного взаимодействия сводится к модели Лен-
нарда-Джонса в случае совпадения фокусов и к модели твердых эллипсоидов
при отсутствии притяжения и равном бесконечности показателе степени силы
отталкивания в потенциале Леннарда-Джонса.
Выражение (8.9) можно теперь использовать при вычислении второго
вириального коэффициента для обобщенной модели эллипсоидальных молекул.
Результат может быть выражен в виде ряда по степеням величины lJQm
В(Т) = |*
Коэффициенты kj для удлиненных эллипсоидов выражаются в виде
к -- *---^- к --^- к - 12Q1 к - 1643
*i ~ 2 > Л2 — 120 ' 3 — 240 ' 4 — 67 200 ' *б ~~ 134 400 ' *' ' >
а для сплюснутых
ъ 1 v _i_ k О k 2 . 2
*l — 1у К2 — 15 ' 3 "" ' 4 "~ ~~ 25"» б "~ ^25"' " # '
Функции Fk(ejkT) определяются выражением (8.12) и приведены в
табл. XXIII. Поскольку выражение (8.13) не сходится при I, большем или
равном gmy то иногда удобно выражать второй вириальный коэффициент
степенным рядом относительно величины эксцентриситета1) е0. В этом случае
получаем
J) ()
где коэффициенты Kj выражаются следующим образом:
Выбор длины сердечника (т. е. межфокального расстояния) / делается так
же, как указано выше. Параметры е и Qm получаются тогда из значений вто-
рого вириального коэффицента и выражений (8.13) и (8.14). Значения пара-
метров, найденных таким образом, приведены в табл. 14, а кривые для изме-
ренного и вычисленного второго вириального коэффициента — на фиг. 24.
Различие между величинами большой и малой полуосей,эллипсоидальных
молекул значительно меньше соответствующей величины для сфероцилинд-
рических молекул. Только по одним значениям второго вириального коэффи-
х) Эксцентриситет е0 = 1/YP+ q%i- .
164 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
циента невозможно с определенностью заключить о том, какая модель более
реалистична. Однако для молекул водорода эллипсоидальная модель подходит
значительно хуже сфероцилиндрической. Для последней модели длины большой
Ф и i. 24. Сравнение экспериментальных и теоретических значений второго вириального
коэффициента, полученных для обобщенной модели эллипсоидальной молекулы Кихары [29].
и малой полуосей только на несколько процентов меньше соответствующих
величин для потенциальных функций, вычислявшихся квантовомеханическим
путем [142—145] (см. гл. 14, §4).
Г. ЧЕТЫРЕХЦЕНТРОВАЯ МОДЕЛЬ ДЛИННЫХ МОЛЕКУЛ КОРНЕРА
Для нахождения второго вириального коэффициента длинных молекул
Корнер [146] применил четырехцентровую модель, в которой молекула пред-
ставляется четырьмя центрами сил, распределенными равномерно на длине,
равной 26W (фиг. 25). Энергия взаимодействия двух таких молекул предпо-
лагается равной
Где rij — расстояние между точечными центрами / и / первой и второй молекул
соответственно. Суммирование ведется по шестнадцати возможным взаимо-
действиям силовых центров1). Индекс с под силовыми постоянными указывает
на то, что они относятся к случаю взаимодействия между силовыми центрами,
а не между самими молекулами.
Это выражение для потенциала межмолекулярного взаимодействия можно
записать в виде
{(^Г{^)'1 (8-16)
х) Лондон [147] критиковал использование модели точечных центров для представле-
ния дисперсионных сил, так как она не учитывает тензорный характер поляризуемости
молекул (см. гл. 13, § 4). При вычислениях второго вириального коэффициента этим эффек-
том, однако, можно, безусловно, пренебречь.
§ 8. Второй вириальный коэффициент Оля несферических молекул
165
где теперь силовые постоянные зависят от ориентации со = { в1У 02, ф }. Кор-
нер получил формулы для зависимости постоянных а(со) и е(со) от углов, вы-
числив <р(г) с помощью (8.15) для большого числа ориентации и подобрав кри-
вую, описывающую полученные результаты. В его выражения входит пара-
метр Цас и они хорошо выполняются в интервале значений Цас от 0 до 0,75.
Зависящий от углов потенциал взаимодействия (8.16) затем подставляется в
интеграл для второго вириального коэффициента D.15). Интегрирование по г
может быть проведено аналитически, если использовать метод, примененный
Фиг. 25. Схематическое изображение двух взаимодействующих четырехцентровых молекул»
к вычислению второго вириального коэффициента для потенциала Леннарда-
Джонса [см. F.2) ]. После этого можно интегрировать по углам. Тогда во вто-
рой вириальный коэффициент войдут три параметра ос> I и ё, причем послед-
ний1) соответствует некоторой средней глубине ямы для потенциала Леннарда-
Джонса. Окончательный вид выражения для В(Т) таков:
Щ}] (8.17)
где В*, Б* и В2* — функции, определяемые соотношением (б.З) и табулирован-
ные в табл. II (стр. 854). Величины а и /? являются функциями Цас; а выра-
жается формулой
а значения /? приведены в табл. 15.
Таблица 15
0,00
0,15
0,30
0,45
0,60
0,75
Р
0,0000
0,0024
0,0084
0,0143
0,0159
0,0129
! Здесь е — следующее среднее значение е(со) по ориентациям со
- _ Г Ш Vе (щ) [° (ft>I8 sin flj sin в2 ddx dd2 d @2 — 0
6 " L Ш1^Й]3 sin 0X sin 02 d0x rf02 d @2 - 0x)
166
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Малость значений /? указывает на то, что при отклонениях от сферичности
молекул происходит незначительное изменение температурной зависимости
второго вириального коэффициента. И действительно, второй вириальный коэф-
фициент для несферических молекул, очевидно, очень близок к коэффициенту
для сферических молекул с диаметром столкновений, равным <хса1/а.
Значения параметров /, aCi e для нескольких типичных молекул приве-
дены в табл. 16. Там же даны значения приведенных критических постоянных
кТе(ё и VclQ[2a*a.l0™).
Таблица 16
Параметры для четырехцентровой модели Корнера
и значения приведенных критических постоянных [146]
Газ
ае,к | е,°К | kTeJe \ Ve/(]/2 о*а • 10")
Азот
Окись углерода
Углекислый газ
Метан
Этилен
Этан
Пропан
Пропилен
/пранс-2-бутен .
л-Бутан
0,15
0,14
0,29
0,00
0,21
0,19
0,25
0,26
0,38
0,39
3,37
3,60
3,57
3,79
4,1
4,5
4,9
4,9
4,9
4,9
95
98
198
148
192
206
228
220
281
254
1,33
1,37
1,54
1,29
1,47
1,48
1,62
1,66
1,53
1,68
1,29
1,07
0,90
1,28
0,92
0,82
0,78
0,70
0,75
0,77
Если молекулы имеют сферическую форму, то значения приведенных кри-
тических постоянных не будут отличаться для различных молекул. Заметно,
что с увеличением длины молекулы значения постоянных возрастают. Оче-
видно, что приведенная критическая температура почти не чувствительна
к удлинению молекул, но критический объем при этом заметно уменьшается.
Возможность упаковки длинных молекул рядом друг с другом делает такую
тенденцию, по-видимому, приемлемой. Графическое сравнение эксперимен-
тальных и вычисленных для различных потенциальных функций результатов
для нескольких газов проводится в следующем параграфе.
§9. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ,
ПОЛУЧЕННЫХ ДЛЯ НЕСКОЛЬКИХ ПОТЕНЦИАЛЬНЫХ
ФУНКЦИЙ МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
НЕПОЛЯРНЫХ МОЛЕКУЛ
Четыре предыдущих параграфа были посвящены обсуждению вычислений
второго вириального коэффициента для веществ, состоящих из неполярных
молекул. Перед тем как приступить к изучению полярных молекул, мы сумми-
руем сведения по второму вириальному коэффициенту и межмолекулярным
силам взаимодействия, полученные для неполярных молекул. В этом параграфе
сравниваются различные потенциальные функции и значения получаемого
для этих функций второго вириального коэффициента. Сначала мы рассмотрим
в качестве примера сферических молекул некоторые благородные газы, а затем
в качестве примера несферических молекул — азот и углекислый газ.
А. СФЕРИЧЕСКИЕ МОЛЕКУЛЫ
Экспериментальные и теоретические результаты, полученные для гелия,
неона, аргона и криптона, приведены на фиг. 26—29 соответственно. Для каж-
дого из этих газов приведены экспериментальные значения В(Т), а также
§ 9. Обсуждение результатов, полученных для нескольких потенциальных функций 167
вычисленные значения второго вириального коэффициента для нескольких
эмпирических потенциальных функций. Для сравнения показаны также кри-
вые различных потенциальных функций. Рассмотрим каждый из этих газов
отдельно.
1. Гелий. При температурах выше 400° К для описания второго вириаль-
ного коэффициента может использоваться классическая теория. При комнатной
Фиг. 26. Второй вириальный коэффициент В(Т), рассчитанный для различных
молекулярных моделей гелия.
Показаны также потенциальные функции, рассчитанные по экспериментальным значениям В(Т), взятым
из работ [27, 155].
а — потенциал Леннарда-Джонса ; b — потенциал Интемы и Шнейдера и экспериментальные данные рабо-
ты [155J; с — данные работы [27]; d — потенциал Мэзона и Раиса; е — потенциал Месси, Букингема
и Гамильтона.
температуре квантовые эффекты малы, но могут быть обнаружены экспери-
ментально. Квантовые отклонения хорошо описываются теорией; соответ-
ствующие вычисления проделаны в гл. 6, § 5. При температурах ниже 20° К
квантовые эффекты становятся ощутимыми. Теория и соответствующие вычи-
сления в области низких температур излагаются в гл. 6, § 4.
Имеется большое количество экспериментальных данных, которые могут
быть использованы для определения энергии взаимодействия двух атомов гелия.
Для описания экспериментальных данных использовались четыре потенциаль-
168 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
ные кривые. Месси, Букингем и Гамильтон [148—151 ] пользовались для низ-
котемпературной области потенциалом Букингема; де Бур и Михельс [152]
и Лунбек [26 J1) пользовались потенциалом F-12) Леннарда-Джонса как для
области низких, так и для области высоких температур; Интема и Шнейдер
[155] в области высоких температур пользовались потенциалом Букингема ;
Мэзон и Райе [156] использовали модифицированный потенциал F-ехр) Бу-
кингема для обработки данных как для второго вириального коэффициента,
так и для коэффициента вязкости. Вычисленные де Буром и Михельсом, Лун-
беком, Интемой и Шнейдером, Мэзоном и Райсом значения второго вириаль-
ного коэффициента сравниваются на фиг. 26 с экспериментальными данными
Гольборна и Отто [27], Интемы, Шнейдера и Даффи [155—157].
К сожалению, до сих пор не проводилось вычислений второго вириаль-
ного коэффициента для области высоких температур с использованием потен-
циала Месси—Букингема—Гамильтона. Однако из фиг. 26 можно видеть, что
кривая для этой функции по форме почти совпадает с кривой для потенциала
де Бура—Михельса—Лунбека, так что результаты должны были бы быть
очень близкими.
Месси, Букингем и Гамильтон [148—151 ], принимая во внимание кванто-
вые эффекты, проделали тщательный анализ данных по второму вириальному
коэффициенту для гелия в области низких температур. При обработке экс-
периментальных данных использовалась потенциальная функция, соответ-
ствующая двум участкам кривой потенциала Букингема [выражение C.28)
гл. 1 ]. Для г ^2,61 А (при г = 2,61 А потенциал равен нулю) они пользова-
лись теоретическим соотношением Слетера—Кирквуда (см. гл. 14, § 2). Для
г ^ 2,61 А постоянная а принималась соответствующей соотношению Слетера—
Кирквуда, а отношение величин с и с' — равным значению, предсказанному
Маргенау (см. гл. 14, § 2); остающиеся две постоянные определялись из экспе-
риментальных данных. Полное выражение для потенциала имеет вид
<р(г) = [770е-4*60' - ^-\ • Ю-12 эрг, г<2,61 А; (9.1а)
10-12 эрг, r>2,6lA. (9.16)
Де Бур, Михельс [152] и Лунбек [26] нашли параметры в случае потен-
циала F-12) Леннарда-Джонса по экспериментальным значениям второго
вириального коэффициента в интервале температур от 20 до 400° К; способ
расчета описан в гл. 6, §5. В этих вычислениях в указанном интервале темпера-
тур учитывались также квантовые эффекты. Таким образом было найдено
[26, 152], что а = 2,556 А, а е/к = 10,22° К. Эти значения параметров позво-
ляют с помощью теории, изложенной в гл. 6, §4, [159], определить второй
вириальный коэффициент для гелия вплоть до температуры 1°К. Коэффициенты
вязкости и теплопроводности газообразного гелия рассчитывались с помощью
потенциала F-12) Леннарда-Джонса с этими значениями параметров. Вы-
числения проводились для области очень низких температур (от 1 до 15° К
[160]) с использованием квантовомеханических расчетов гл. 10, §2, а для
области высоких температур [161 ] (примерно от 100 до 500° К) —с использо-
ванием классической теории. Очень хорошее совпадение получено для всего
интервала температур. Поскольку упомянутые значения параметров приводят
к удовлетворительному согласию как для равновесных, так и для неравновес-
ных свойств, то можно с уверенностью сказать, что это наиболее подходящие
значения для использования в случае потенциала F-12) Леннарда-Джонса.
х) Значения силовых постоянных потенциала F-12) Леннарда-Джонса были получены
таким путем де Буром и Михельсом [152 ]. Приводимые здесь значения взяты из работы Лун-
бека, исправившего ранее полученные значения с помощью новейших значений основных
физических постоянных kt N и h, взятых из работы Дю Монда и Коэна [153, 154].
§ 0. Обсуждение результатов, полученных для нескольких потенциальных функций 16$
Интема и Шнейдер [155] для обработки своих экспериментальных данных
по второму вириальному коэффициенту в интервале температур от 0 до 1200° С
пользовались потенциалом Букингема. Для постоянной с бралось значение,
следующее из простого теоретического анализа Лондона, а для отношения
сие' — значение, получаемое при идеализировании атома простым гармони-
ческим осциллятором1)^, гл. 14, §2). Для наилучшего выражения потенциала
ими было получено выражение
<р(г) = [1200 <?-4>72 г - 1^- _ i^-J. 10-й эрг (г _ в д)#
Обработка экспериментальных данных с помощью потенциала Леннарда-
Джонса не дала хорошего результата.
Мззон и Райе [156] пользовались как значениями второго вириального
коэффициента в интервале температур от 0 до 1200° К, так и значением коэф-
фициента вязкости в интервале от 200 до 1100° К и модифицированным потен-
циалом (б-ехр) Букингема [см. C.30) гл. 1 ]. Они нашли а = 12,4, ejk = 9,16° К,
гт = 3,135А. Отклонения были в пределах экспериментальной ошибки для
всего температурного интервала, и это определенно говорит в пользу приме-
нения потенциала F-12) Леннарда-Джонса.
Из фиг. 26 видно, что потенциал Интемы—Шнейдера (кривая Ь) имеет
минимум, превышающий примерно на 0,3 • Ю-15 эрг минимум потенциала
де Бура—Михельса—Лунбека и Месси—Букингема—Гамильтона. Минимум
потенциала Мэзона—Раиса расположен между двумя другими кривыми. Для
малых расстояний между молекулами кривые потенциала Интемы—Шнейдера
(Ь) и Мэзона—Раиса (d) пересекают кривую потенциала Леннарда-Джонса
(а), что указывает на более слабое отталкивание. В гл. 14, § 2 будет показано,
что кривая теоретического потенциала Слетера—Кирквуда располагается
между этими двумя кривыми. Потенциал Розена—Маргенау—Пейджа (пред-
ставляющий в настоящее время результат наиболее тщательного теоретиче-
ского анализа) имеет минимум, превышающий примерно на 0,3 • 10~15 эрг
минимум, соответствующий потенциалу Интемы—Шнейдера.
2. Неон. Букингем [148] и Корнер [64] получили выражения для потен-
циала взаимодействия двух атомов неона (фиг. 27) на основании измерения
второго вириального коэффициента в интервале температур от 65 до 673° К
и значений минимального расстояния между соседними атомами в кристалле и
теплоты сублимации кристалла, соответствующих температуре 0° К. Букин-
гем предположил, что для значений г, больших примерно 4,4 А, потенциальная
функция имеет вид
<Р(г) = - [^- + Щ- Ю-12 эрг (г - в А). (9.3)
Далее он нашел, что для значений г между 2,6 и 3 А
(р(г) = [2570е-4>26г - -?-]• Ю-12 эрг (г — в А). (9.4)
Корнер использовал потенциал Букингема—Корнера [см. C.29) гл. 1 ]. Резуль-
таты уже обсуждались в § 7 и приведены в табл. 11. Мэзон и Раис [156], поль-
зуясь той же кристаллической структурой и значениями второго вириального
коэффициента и коэффициента вязкости (в интервале от 80 до 1100 °К), искали
постоянные для модифицированного потенциала Букингема ; полученные ими
результаты приведены в табл. 12. Эти результаты находятся в хорошем согласии
с результатами Корнера.
г) Лучшими теоретическими значениями для постоянных сие' следует считать
с = 1,39 • Ю-12 эрг А11 и с' = 3,0 • Ю-12 эрг А14[162]. Однако Интема и Шнейдер не могли
воспользоваться этими значениями постоянных при обработке своих данных.
170 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
15
10
с
0
-5
-10
-15
-20
-25
-30
-35
-40
-45
-50
-55
СП
ьи
-65
-75
0
I I , I ' L-li-i-lAi
- /Г :*
- If . °-d
- v
/7
. !
- I
i_L i I .
5 -
4 -
3 -
<v> 2 -
ч П -
I"-
-2 -
-3 -
-4 -
WO 200 300
r
3 4 5 6 7 8 9
i i i i i i
/
/^
1
МГ~с
•Фиг. 27. Второй вириальный коэффициент B(T)f рассчитанный для различных молекуляр-
ных моделей неона.
Показаны также потенциальные функции, рассчитанные по экспериментальным значениям В(Т), взятым
из работы [27].
¦а — прямоугольная потенциальная яма, b — потенциал Лен нарда-Джонса; с — потенциал Букингема и
Корнера ; d — данные работы [27 ].
Корнер использовал также для обработки данных потенциал типа Лен-
нарда-Джонса [см. C.27) гл. 1 ] с показателем степени силы притяжения у,
равным 6, и различными показателями Ь сил отталкивания. Он нашел, что
лучшее совпадение, даже для случая твердого тела, получается при значении
Ъ = 12. Для других постоянных были найдены следующие значения:
в = 5,01 • Ю-15 эрг, или - = 36,3° К,
а = 2,82А.
(9.5)
Почти те же значения постоянных были найдены по температурной зависи-
мости коэффициента вязкости (см. табл. I, стр. 851).
3. Аргон. Букингем [148] и Корнер [64] изучали также потенциал взаи-
модействия двух атомов аргона (фиг. 28). Букингем использовал параметры
кристалла и значения второго вириального коэффициента в интервале темпера-
тур от 173 до 673° К. Корнер пользовался, кроме этих данных, измеренными
значениями коэффициента Джоуля—Томсона при нулевом давлении примерно
в том же интервале температур. Потенциал Букингема имеет вид
~12 эрг, г>5,3А; (9.6а)
- 1>Q2 J0- эрг, 3,4 А < г < 3,6 А. (9.66)
Значения постоянных, полученные Корнером, даны в табл. 11. Мэзон и Райе
[156] рассмотрели те же данные в интервале температур от 55 до 1900° К для
9. Обсуждение результатов, полученных для нескольких потенциальных функций 171
получения постоянных модифицированного потенциала Букингема, которые
приведены в табл. 12. Их потенциал весьма близок к потенциалу Корнера.
Фиг. 28. Второй вириальный коэффициент В(Т), рассчитанный для различных молекуляр-
ных моделей аргона.
Показаны также потенциальные функции, рассчитанные по экспериментальным значениям В(Т), взятым
из работ [27, 46].
п — прямоугольная потенциальная яма; b — потенциал Леннарда-Джонса ; с — потенциал Букингема
и Корнера; d — данные работы [27]; е — данные работы [46].
Корнер проделал также анализ для потенциала Леннарда-Джонса. Лучшее
значение показателя силы отталкивания для аргона было найдено равным
12,3. Однако если выбрать для показателя значение 12, то силовые постоянные
имеют значения1)
е = 1,647-Ю-14 эрг, или у=119,3°К;
а = 3,45 А.
(9.7)
Эти значения очень близки к полученным из данных по коэффициенту вязкости
(см. табл. I).
Райе [163] получил потенциал межмолекулярного взаимодействия путем
более детального анализа данных для твердого тела и использования значений
второго вириального коэффициента. Он предположил, что на больших расстоя-
ниях взаимодействие описывается потенциалом Букингема [см. (9.6а)]. В этом
значения
х) Лунбек [26 ] в соответствии с данными Михельса и др. [46 ] приводит следующие
ния постоянных в интервале температур от 0 до 150 °С : а = 3,405 А, е/k = 119,8 °К.
172
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
случае он получил два выражения для потенциала в области минимума :
(р(г) = [_ 1,923 + 5,344 (г - 3,8267J - 6,726 (г - 3,8267K +
+ 2,043 (г - 3,8267L] • 104 эрг, (9.8)
<р(г) = [_ 1,945 + 5,430(г - 3,8608J - 4,885 (г - 3,8608K] • Ю4 эрг*
Эти два выражения для потенциала получены на основе двух различных пред-
положений относительно разупорядоченности в твердом поле.
4. Криптон. Совпадение экспериментальных значений второго вириаль-
ного коэффициента, полученных Битти и др. [28], и значений, вычисленных
на основании потенциала F-12) Леннарда-Джонса, показано на фиг. 29.
5
о
-5
40
-15
-20
-25
-30
?-35
^ -40
-45
-50
-55
-60
-65
-70
-75
-80
200 300 400 500
7,°К
Фиг. 29. Второй вириальный коэффициент В(Т), рассчитанный для различных молекуляр-
ных моделей криптона.
Показаны также потенциальные функции, рассчитанные по экспериментальным значениям В(Т), взятым
из работы [28].
а — прямоугольная потенциальная яма ; b — потенциал Леннарда-Джонса ; с — данные
работы [28].
1 1 ' I
-
1 1 1
/
I t
: i
, 1 , 1
1 ' 1
/
20
8. Ю
й
^ n
-10
-20
I
--a
- A
" II
f
4
1
r
5 6
f
la
i
-
-
7 a
i i
Это типичное совпадение, которого можно ожидать для сферических молекул,
когда отсутствуют какие-либо другие силы, кроме короткодействующих валент-
ных и дальнодействующих дисперсионных сил.
Б. НЕСФЕРИЧЕСКИЕ МОЛЕКУЛЫ
Мы дадим два примера молекул N2 и СО2, являющихся несколько асиммет-
ричными. Поскольку эти молекулы почти сферические, то зависимость второго
вириального коэффициента от температуры очень хорошо описывается с по-
мощью потенциала F-12) Леннарда-Джонса. Второй вириальный коэффи-
циент не чувствителен к форме молекулы, тогда как эффект асимметрии прояв-
ляется в температурной зависимости третьего вириального коэффициента.
Однако весьма затруднительно отличить эти отклонения для третьего вириаль-
ного коэффициента, вызванные асимметрией, от отклонений, обусловленных
неточным знанием радиальной зависимости для сферического потенциала.
§ 9. Обсуждение результатов, полученных для нескольких потенциальных функций 173
1. Азот. Превосходные измерения величин ру V и Т для азота проде-
лали Гольборн и Отто [27], а также Михельс, Воутерс и де Бур [48]. Первые
авторы осуществили измерения в широком интервале температур. На фиг. 30
сравниваются экспериментальные и теоретические значения второго вириаль-
ного коэффициента, полученные а) для потенциала F-12) Леннарда-Джонса
Фиг. 30. Второй вириальный коэффициент В(Т)> рассчитанный для различных молекуляр-
ных моделей азота.
Показаны также потенциальные функции, рассчитанные по экспериментальным значениям В(Т), взятым
из работ [27, 48].
а — прямоугольная потенциальная яма ; Ъ — потенциал Леннарда-Джонса ; с — четырехцентровая модель
Корнера ; d — данные работы [27 ]; е — данные работы [48 ]; / — модель Корнера (потенциал взаимо-
действия двух центров сил).
с силовыми постоянными, подобранными по данным Гольборна и Отто; б) для
сфероцилиндрической и обобщенной эллипсоидальной модели Кихары (см. § 8)
и в) для четырехцентровой модели Корнера (см. § 8). Во всех случаях сов*
падение хорошее. Из табл. 14 видно, что отношение длины к ширине для моле-
кулы азота равно приблизительно 1,3, так что эта молекула только слегка асим-
метрична.
2. Углекислый газ. Кривые температурной зависимости второго вириаль*
ного коэффициента, полученные с помощью обсуждающихся выше потенциалов,
представлены на фиг. 31. Эти кривые сравниваются с прекрасными эксперт-
174 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
ментальными значениями, полученными А. Михельсом и К. Михельсом [51J
и Мак-Кормаком и Шнейдером [30 ]. Молекула углекислого газа значительно
более асимметрична, чем молекула азота, и имеет отношение длины к ширине,
20
15
10
5
0
-5
40
45
-20
-25
-30
-35
-40
-45
-50
-55
-60
-65
-10
-75
-80
1 i • i ¦ i • i ' i ¦ i ¦ i • i
a S,
ь /^
- -с /jr
- +-9 //
/
.III. I
25
20
15
10
й л
Ь °
к-5
^/5
-20
-25
-30
и
-
-
г
и?»
//
//
*——
300 400 500
- Гк
600
Фиг. 31. Второй вириальный коэффициент В(Т), рассчитанный для различных
молекулярных моделей углекислого газа.
Показаны также потенциальные функции, рассчитанные по экспериментальным значениям В(Т), взятым
из работ [30, 51].
а — прямоугольная потенциальная яма ; b — потенциал Леннарда-Джонса ; с — эллипсоидальная модель
Кихары ; d — сфероцилиндрическая модель Кихары ; е — четырехцентровая модель Корнера ; / — дан-
ные работы [51 ]; g — данные работы [30J; h — модель Корнера (потенциал взаимодействия двух центров сил)
равное приблизительно 1,6. Однако вследствие нечувствительности второго
вириального коэффициента к форме молекулы для всех типов потенциальных
функций имеется хорошее согласие с экспериментом.
В. СРАВНЕНИЕ РАЗЛИЧНЫХ ПОТЕНЦИАЛЬНЫХ ФУНКЦИЙ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
На фиг. 26—31, где сравнивались значения второго вириального коэф-
фициента для благородных газов, азота и углекислого газа, построены также
кривые потенциальных функций различных типов, параметры которых под-
бирались так, чтобы получилось наилучшее совпадение с экспериментальной
температурной зависимостью второго вириального коэффициента. Эти раз-
личные потенциалы имеют совершенно отличные формы и глубину минимума
и тем не менее приводят к довольно хорошему совпадению с экспериментом в
широком температурном интервале. Проведенное графическое сравнение под-
черкивает тот факт, что температурная зависимость В(Т) не чувствительна к
форме функции, выбранной для межмолекулярного потенциала. Единственное
свойство, по-видимому, присущее всем этим кривым потенциальных функций,
заключается в приблизительном равенстве площадей, охваченных кривой. Эта
о»
площадь f q>(r) dr для потенциала F-12) Леннарда-Джонса равна B4/55)есг, а для
а
прямоугольной потенциальной ямы равна (R—1)ео. Отношение площадей,
§ 0. Обсуждение результатов, полученных для нескольких потенциальных функций 175
соответствующих потенциалу F-12) Леннарда-Джонса и прямоугольной потен-
циальной яме, для пяти рассмотренных газов приведено в табл. 17. Как видно,
это отношение очень близко к единице.
Таблица 17
Молекула
Ne
Аг
Кг
N2
со2
(Я— 1)(еа)потя#
B4/55)> о)л.-д.
0,96
1,03
1,05
0,99
1,06
Хилл [164] подобрал параметры потенциала Букингема для получения
лучшего совпадения с экспериментальными значениями второго вириального
коэффициента для гелия, неона и аргона
v = be-*-±-. (9.9)
Он сравнил эти потенциалы с потенциалами F-12) Леннарда-Джонса, силовые
постоянные которых также подбирались по тем же значениям второго вириаль-
ного коэффициента, и выразил постоянные потенциала Букингема через по-
стоянные потенциала Леннарда-Джонса. Он нашел
b = 828 000 г, а = 0,0826 а, с = 4,50 е о*.
(9.10)
Минимум потенциала Букингема соответствует межмолекулярному расстоя-
нию, равному 1,1310а (по сравнению с 1,-2616а для потенциала Леннарда-
Джонса), а минимальное значение потенциала равно —1,210 е. Потенциал
Букингема обращается в нуль при расстоянии между молекулами 1,003 а.
Потенциал Букингема [экспоненциальная сила отталкивания и показа-
тель степени силы притяжения, равный 6 (или 8) ] страдает тем недостатком^
что при очень малых расстояниях между молекулами гмакс потенциал имеет
максимум, а при г->0 потенциал становится отрицательным и стремится к —оо„
В табл. 18 даны значения величин межмолекулярных расстояний и энергий,,
соответствующих максимуму, для нескольких газов. Видно, что энергия мак-
симума крайне велика с точки зрения энергии тепловых столкновений. Нет
большой необходимости разделять потенциал на две части, как, например, в
случае потенциала Букингема—Корнера. Проще положить потенциал равным
бесконечности для всех расстояний г, меньших гмакс., и пользоваться потенциа-
лом Букингема для всех расстояний г, больших Гмакс..
Таблица 18
Фиктивный максимум потенциала Букингема
Вещество
Не
Ne
Аг
Тип потенциала
Потенциал Интема—Шнейдера (9.2)
Потенциал Слетера—Кирквуда (9.1а)
Потенциал Корнера табл. A1)
Потенциал Корнера табл. A1)
гмакс.» ^
0,82
0,65
0,73
0,78
Р(гмакс.)
10-" эрг
Н,7
19,0
55,2
520
ккал/моль
168
270
795
7490
Проверкой качества потенциальной функции является ее способность
описывать как равновесные, так и неравновесные свойства вещества в твердой>
176
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
жидкой и газообразной фазах. Параметры потенциала прямоугольной потен-
циальной ямы для трех газов, найденные по значениям второго вириального
коэффициента и по измерениям коэффициента вязкости (см. гл. 8, § 3), пред-
ставлены в табл. 19. Очевидно, что эти параметры, вычисленные по значениям
одной величины, не могут использоваться для вычисления другой макроскопи-
ческой величины. Аналогичное сравнение сделано и для потенциала Леннарда-
Джонса. В силу большей реалистичности потенциала Леннарда-Джонса в этом
случае совпадение значительно лучше, чем для прямоугольной потенциальной
ямы.
Таблица 19
Сравнение параметров потенциалов, вычисленных по значениям второго
вириального коэффициента и коэффициента вязкости
Газ
Ne
Ar
N2
Силовые
постоянные
а
е/к
R
а
е/к
R
а
elk
R
Прямоугольная потенциальная яма
по значениям
В(Т)
2,38
19,5
1,87
3,16
69,4
1,85
ЗД)
53,7
1,87
по значениям
V(T)
2,38
101,0
1,54
2,98
167,0
1,96
3,36
80,0
2,08
Потенциал Леннарда-Джонса
по значениям
В(Т)
2,74
35,7
3,405
119,75
3,698
95,05
по значениям
*П(Т)
2,80
35,7
3,418
124,0
3,681
91,46
§ 10. ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ ДЛЯ ГАЗОВ,
состоящих из полярных молекул1)
Выше были описаны вычисления второго вириального коэффициента для
большого числа потенциальных функций, однако до сих пор обсуждение огра-
ничивалось только рассмотрением неполярных молекул. Взаимодействие двух
сложных полярных молекул носит весьма запутанный характер и поэтому
иногда удобно рассматривать полный межмолекулярный потенциал взаимо-
действия в виде суммы следующих отдельных членов : 1) валентного члена
<р(вал-), связанного с короткодействующим отталкиванием; 2) лондоновских
дисперсионных членов, соответствующих г~6, г~8 и т. д. и слабо зависящих от
ориентации молекул <р<дисп->; 3) зависящего от углов электростатического
члена, обусловленного взаимодействием мультиполей, причем наиболее важ-
ными являются диполь-дипольное взаимодействие <p(w\. пропорциональное
г, и диполь-квадрупольное взаимодействие <pifQ\ пропорциональное г~4;
4) индукционного члена, обусловленного индуцированным взаимодействием.
Здесь наиболее важным является диполь-дипольный индуцированный потен-
циал ср^ инд- *>, пропорциональный г~6. Эти различные типы взаимодействия
коротко обсуждались в гл. 1, § 3, и подробно будут рассмотрены в гл. 13, § 5.
В настоящем параграфе мы сначала покажем, как можно вычислить вто-
рой вириальный коэффициент для простейшей модели полярной молекулы,
а именно твердой сферы с помещенным внутри точечным диполем. Эта модель
включает два наиболее важных члена потенциала взаимодействия — корот-
)
Англия.
Этот параграф написан совместно с Дж. Раулинсоном, Манчестерский университет,
§ 10. Вириальные коэффициенты для газов, состоящих из полярных молекул 177
кодействующие силы отталкивания и диполь-дипольное взаимодействие. Затем
будут обсуждены расчеты, проделанные для потенциала Штокмайера. Этот
потенциал включает члены г2 и г, соответствующие силе отталкивания и
диполь-дипольному индуцированному притяжению, а также включает диполь-
дипольное взаимодействие. Для этого потенциала были подготовлены объе-
мистые таблицы, позволяющие вычислить для полярных молекул значения
второго и третьего вириальных коэффициентов и коэффициента Джоуля—
Томсона. В общем получено хорошее согласие измеренных и вычисленных зна-
чений. Лучшее согласие можно, несомненно, получить при учете важного
квадруполь-дипольного члена потенциальной функции. Некоторые расчеты
были проделаны в этом направлении и они обсуждаются в конце параграфа.
А. МОДЕЛЬ' ТВЕРДЫХ СФЕР, СОДЕРЖАЩИХ
ТОЧЕЧНЫЕ ДИПОЛИ
Для твердых сфер диаметра а, в центре которых расположены точечные
диполи с дипольным моментом /г, потенциал взаимодействия дается выражением
C.32) гл. 1
<Р(г, в19 02, ф2 - &) = - ? Z(ev К
ё(в1> 92> Фг — 0l) = 2 C0S в1 C0S 02 — Si
Переменные г, 0г, 02, фх и ф2 представлены на фиг. 32. Второй вириальный
коэффициент вычисляется [165] путем подстановки этого потенциала в выра-
жение D.15). Для значений г < о интегрирование приводит для второго вири-
ального коэффициента к выражению Bя/^(т3/3), соответствующему модели твер-
дых сфер. Для значений г><т величина ехр (—<р//сТ) может быть разложена
Фиг. 32. Координаты, описывающие взаимную ориентацию двух полярных молекул
в ряд по степеням A/fcT), и тогда легко провести интегрирование по г. После-
дующее интегрирование по углам приводит к выпадению нечетных степеней
A//сТ), и окончательный результат имеет вид
A0.1)
55 125 {^ЧсТ) —•••]•
Величины Gk появляются в результате интегрирования по углам и определя-
ются выражением
2л п п
G* = тМ Г f [g @i, в2, ф2 — фЛ] *к sin вг sin 02 йвг йд2 й(фг — фг). A0.2)
О 00
178 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Кеезом [166] вычислил также величину второго вириального коэффициента
для случая молекул, обладающих квадрупольными моментами.
Б. ВТОРОЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ ДЛЯ ПОТЕНЦИАЛА
ШТОКМАЙЕРА
Ббльшая часть вычислений второго вириального коэффициента для по-
лярных молекул основана на использовании потенциала Штокмайера [167]
C.33) гл. 1
4<г, вх, в2, ф2, - фх) = 4е [(-f I2 - ф§] - ? g(eif в2,ф2- фг)у
гДе gWv ^2> Фч — Фг) — функция, определенная выше. Параметры а и е, ин-
терпретация которых несколько отличается от соответствующей потенциалу
Леннарда-Джонса, можно использовать для определения величин1)
(Ю.З)
Если подставить потенциал Штокмайера в выражение D.15) для второго ви-
риального коэффициента, то интегрирование по г можно провести аналогично
случаю потенциала Леннарда-Джонса и получить результат в виде ряда гамма-
функций. Интегрирование по углам, как следует из разд. А, приводит к появ-
лению коэффициентов Gk> определяемых выражением A0.2). Окончательно
получаем
X
л=1 fc—O
Следовательно, второй вириальный коэффициент для потенциала Штокмайера
может быть записан в виде
;/г*). A0.5)
Это выражение аналогично полученному для потенциала Леннарда-Джонса
F.3), за исключением того, что приведенный здесь второй вириальный коэф-
фициент зависит не только от приведенной температуры, но параметрически
зависит также и от приведенного дипольного момента /г*. Эта последняя зави-
симость графически представлена на фиг. 33, где В* нанесена как функция
от Г* для нескольких значений параметра /¦ = 8гу* /г*2. Из того, что кривая
В*(Т*) для полярных молекул располагается ниже кривой, соответствующей
неполярным молекулам, следует ббльшая величина фактора сжимаемости
неполярных газов по сравнению с полярными (при тех же значениях постоян-
ных а и е). Это результат дополнительного сжатия за счет сил притяжения
двух молекул, обладающих электрическим моментом2).
Кривые фиг. 33 построены на основании данных табл. XVII3) (стр. 880).
С помощью этих данных и значений силовых постоянных, приведенных
^Величина/* введена Раулинсоном [170]; множитель/8 вводится в некоторые фор-
мулы для нахождения более простого математического выражения.
^Табулирование значений дипольных моментов было проделано в работе [168].
8) Эта таблица подготовлена Раулинсоном по данным, полученным методом перфориро-
ванных карт [169]. Такой стсоб представления более удобен для интерпретации экспери-
ментальных данных, чем способ, использовавшийся ранее, и впервые предложен Раулин-
соном [170]. Менее полные таблицы приводятся также в работах [25, 171].
§ 10. Вириальные коэффициенты для газов, состоящих из полярных молекул 179
в табл. 20, можно вычислить значения второго вириального коэффициента для
полярных молекул. Определение силовых постоянных по экспериментальным
данным обсуждается в следующем разделе.
I I I I ГТТТТ
1 I I I ММ
I I I I I 1 1 I
I Mill
2 3 4 5 6 8 Ю ZV 30 40 ВО ВОЛЮ
Г*
Фиг. 33. Зависимость приведенного второго вириального коэффициента В* от Т* и t*
в случае потенциала Штокмайера.
Таблица 20
Силовые постоянные потенциала Штокмайера [170,171]
Ьо (смЧмоль) = 2я No»/3 = 1,2615 [<r(A)F; t* - 8-Vi/<»«, м*
Вещество
CHC18
CHC12F*
С2НбС1
CH3CI
СН3СОСН3
СН3ОН
NH3
CH3F
H2O
CHgCN
CH3CHO
//, дебаи
1,05
1,29
2,02
1,89
2,74
1,66
1,47
1,82
1,83
3,5
2,7
t*
0,1
0,1
0,2
0,6
0,7
0,8
1,0
1,07
1,2
1,2
1,4
e/kt °K
1060
381
320
380
520
630
320
207
380
400
270
a, A
2,98
4,82
5,41
3,43
3,76
2,40
2,60
3,36
2,65
4,02
3,68
CM* j МОЛЬ
33,45
141,0
199,7
50,73
66,87
17,48
22,12
47,85
23,42
82,04
62,75
Литература
[176]
[37]
[76]
[40]
[176]
[176]
[177-179]"
•••
[180, 181]"'
[176]
[182]
* Силовые постоянные для СНС1 F вычислены в примере, приведенном в разделе В»
** Данные работы были коррелированы в работе [177].
••• Данные Михельса и Виссера (готовятся к печати).
ПРИМЕР
Задача, а) Вычислить фактор сжимаемости (pV/RT) для водяного пара при давлении
15 атм и температуре 300 °С.
б) Какова температура Бойля для пара?
Решение, а) Из табл. 20 находим Ьо = 23,42 см*/моль, e/k=38Q °K, t*= 1,2. Приведенная
температура равна Г* = кТ/в = 573/380= 1,51. Из табл. XVII (стр.880) находим В•(Т* = 1,51,
t* = 1,2) = — 4,750. Тогда В(Т) = b0 B*(T*, t*) = B3,42) (— 4,750) = — 111,2 см*/моль.
180 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Эта значение второго вириального коэффициента соответствует разложению по степеням
1/V. Соответствующее значение для разложения по степеням давления
b
Фактор сжимаемости тогда равен -?=- = 1 + В'р + ... = 1 + (—0,002365) A5) + ... =
= 1—0,03548 = 0,9645.
б) Температура Бойля — температура, для которой B*(T*f t*) = 0. Из табл. XVII
можно найти Г&.= 4,-876, следовательно, Гб.= Tg. -|- = D,876) C80) = 1853 *К= 1580 °С.
В. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ ПОТЕНЦИАЛА ШТОКМАЙЕРА
ПО ЗНАЧЕНИЯМ ВТОРОГО ВИРИАЛЬНОГО КОЭФФИЦИЕНТА
Если известен дипольный момент молекул газа, то можно воспользоваться
значениями второго вириального коэффициента для нахождения значений
а и е, а также совместимого с ними значения параметра /¦ = 8~у*(/л21 ео*).
Поскольку необходимо определять значения двух параметров, то для этого
нужно использовать также две независимо измеренные величины. В качестве
таковых мы выберем второй вириальный коэффициент и величину наклона
кривой зависимости В от Г при нескольких определенных значениях тем-
пературы1).
Во-первых, введем величину у, в которую входят только измеряемые на
опыте величины :
У
твЩ
V8
3231/*2
V '
Во-вторых, введем «видимый температурный коэффициент» s, определяемый
следующим образом:
В(Т) = ATS, A0.7)
где А — постоянная. Величина 5 — тангенс наклона касательной2) к кривой
зависимости In | В \ от In | Т \ — определяется графически путем использо-
вания этих двух величин, что соответствует знанию второго вириального коэф-
фициента и его производной по температуре при некоторой температуре Т.
Воспользовавшись приведенными величинами,, определяемыми выражением
/10.3), величины у и s можно написать в виде
у = ^В* (Т*, *•) = у (Т*, *•), A0.8)
y(s - 1) =.^ (Bf - В*) = zGV*). A0.9)
Эти даа уравнения могут быть разрешены численно относительно /* и Т*
(следовательно, и г/к) с помощью таблиц для функций (Т*//*)В* и В* — В*,
приведенных в Приложении (см.табл. XVIII и XIX, стр. 884—888). Тогда значе-
ния Ьо (и а) получаются из выражения Ьо = В/В*. Этот метод иллюстрируется
следующим примером.
ПРИМЕР
Задача. При температуре 283° К второй вириальный коэффициент для CHC12F равен
— 616 см3/моль. Показатель степени температуры s выражения A0.7) равен — 1,72. Диполь-
лый момент /* = 1,29 дебая. Найти значения постоянных а и е потенциала Штокмайера.
Проверить совместимость полученных значений ау е и t*.
х) Описываемый здесь метод несколько отличается от процедуры, использованной
Раулинсоном [174].
2) Величина /л выражается в дебаях A дебай = 1 • Ю-18 эл. стат. ед.).
§ 10, Вириальные коэффициенты для газов, состоящих из полярных молекул 181
Решение. Сначала следует вычислить величину у выражения A0.6). При температуре
283° К она равна
ТВ _ B83) (-616) _
У 3231/г2 C231) A,29J " *****
y(s— 1) = (— 32,44) (— 2,72) = + 88,24.
Теперь, подставляя t* и Г*, необходимо одновременно решать уравнения
у(Т*,П =-32,44, (а)
z(T*, t*) = + 88,24 ; (б)
функции у и z даются выражениями A0,8) и A0.9) соответственно. Значения функции
y(T*,t*) приведены в табл. XIX (стр. 888), а функции (В* —В*) = (t*/T*) z(T*f t*) в табл.
XX (стр. 890).
Подбор начнем со значения t* = 0,3. Пользуясь табл. XIX, можно численно решить
уравнение (а) и получить Г* = 0,399. По этим значениям Г* и /* из табл. XVIII для z(T*, /*)
получается значение z = 136. Оно слишком велико, так как z = 88,24 в соответствии с выра-
жением (б). Таким образом, повторяем эту операцию при других значениях t*, пока не будут
найдены значения Г* и /*, удовлетворяющие выражению (б). Результаты можно представить
в виде табл. 21.
Таблица 21
t+
0,3
0,2
0,1
т*
0,399
0,427
0,742
?<**-**>
136
115
go^y(s-i)
Отсюда видно, что с точностью до ошибки эксперимента значения t* = 0,1 и Т* — 0,742
удовлетворяют как выражению (а), так и выражению (б). Используя эти значения, получаем
т
т*
в
~в*
283
0,472
= 381° К,
- 4,3765
===- = 141 см9/моль,
Совместимость можно проверить, вычисляя значение
Теперь для проверки применимости этих значений вычислим значения В(Т) и сравним их
с измеренными значениями табл. 20.
Таблица 22
т
239
250
283
311
339
366
394
422
450
т*
0,627
0,656
0,742
0,816
0,890
0,960
1,034
1,107
1,181
я*
-5,942
-5,455
-4,377
-3,704
-3,180
-2,784
-2,443
-2,163
-1,925
см*1молъ
-838
-769
-616
-522
-448
-393
-345
-305
-271
см*1молъ
-766
-734
-616
-528
-446
-403
-354
-310
-271
^выч.—^эксп.
?выч.
+8,6
+4,6
0,0
-м
+0,4
-2,5
-2,6
-1,6
0,0
Таким образом, видим, что параметры t* = 0,1, b0 ^ 141 смь/моль и е/k = 381° К при-
водят к значениям второго вириального коэффициента В(Г), совпадающим с точностью до
ошибки эксперимента с измеренными значениями.
182
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Раулинсон [174] проанализировал данные для девяти газов полярных
молекул и получил значения параметров, представленные в табл. 20. Как это
видно из табл. 23, значения второго вириального коэффициента, вычисленные
с использованием этих значений параметров, согласуются с экспериментом с
ошибкой в 2—3%. Приведенные в табл. 20 данные для фтористого метила по-
лучены Лунбеком и Сельдамом [171 ]. В таблице приведены также соответ-
ствующие ссылки на литературу.
Таблица 23
Сравнение значений В(Т), измеренных и вычисленных для случая потенциала
Штокмайера [17]
Газ
Ацетонитрил \ R
. (Изм.
АцеТ0Н (выч.
„ (Изм.
Хлористый этил <
(Изм.
Хлороформ |Выч
—В(Т), смЧмоль при температуре (°С)
50
4000
3760
1560
1530
580
560
1000
1010
70
2840
2750
1280
1230
510
500
840
850
90
2110
2160
1040
1010
450
450
730
730
110
1690
1740
850
850
390
410
630
630
130
1330
1410
700
730
350
370
Г. КОЭФФИЦИЕНТ ДЖОУЛЯ—ТОМ СО НА ДЛЯ ПОТЕНЦИАЛА
ШТОКМАЙЕРА
Коэффициент Джоуля—Томсона при нулевом давлении для случая по-
тенциала Штокмайера выражается аналогично случаю потенциала Леннарда-
Джонса F.14)
/*° = -й- № <г*> '*) - в* (г*> '*)] • A0Л°)
ср
Значения функции (Bf — В*) для полярных молекул приведены в табл. XVIII
(стр. 884) для различных значений параметра /¦ = /**2/]Г8. Таким образом,
если параметры для потенциала Штокмайера известны, то легко вычислить
величину коэффициента Джоуля—Томсона, экстраполированного к нулевому
давлению.
Значения коэффициента Джоуля—Томсона могут использоваться для
определения параметров а и е таким же путем, как использовались значения
параметров состояния. Это было сделано для пара [172]; полученные значе-
ния силовых постоянных приведены в табл. 24. Сравнение вычисленных и
измеренных значений, приведенное в табл. 25, показывает отличное совпадение.
Таким образом, мы видим, что коэффициент Джоуля—Томсона является удоб-
ным альтернативным источником сведений^ о межмолекулярных силах для
полярных молекул.
Таблица 24
Силовые постоянные
По значениям В(Т)*
По значениям /л°С°р**
t*
1,2
1,2
для Н2О
е{к, °K
380
373
[172]
а, А
2,65
2,68
К
см*1моль
23,42
24,30
• Литературу см. в табл. 20, стр. 179.
••Данные по коэффициенту Джоуля—Томсона заимствованы из работ[179,
180,. Данные теплоемкости по спектроскопическим данным заимствованы из
работы [182J ; они были исправлены в работе [184J.
§ 10. Вириальные коэффициенты для газов, состоящих из полярных молекул 183
Таблица 25
Измеренные и вычисленные значения коэффициента
Джоуля—Томсона для Н2О [172]
г, °с
39
59
80
100
125
166
Значение и0 Ср
измерен-
ное
5170
3640
2710
2110
1590
987.
вычислен-
ное
5150
3620
2700
2080
1570
980
Т, °С
196
225
260
300
347
Значение ^ Ср
измерен-
ное
780
646
507
400
322
вычислен-
ное
780
640
510
400
320
Д. ТРЕТИЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ ДЛЯ ПОТЕНЦИАЛА
ШТОКМАЙЕРА
Раулинсон [173] показал, каким образом можно численно проинтегриро-
вать выражение для третьего вириального коэффициента. При этом применя-
лись замена переменных (см. § 6), пред-
ложенная Кихарой при вычислении
третьего вириального коэффициента в
случае потенциала Леннарда-Джонса,
и метод, использованный Штокмайером
при интегрировании выражения для
второго вириального коэффициента для
полярных молекул. Результат имеет
вид двойной суммы по различным сте-
пеням величин /¦ и Г*, коэффициенты
которой — сложные восьмикратные
интегралы; здесь они не приводятся.
Окончательно третий вириальный
коэффициент для полярных молекул
можно записать в виде
A0.11)
Таким образом, приведенный третий
вириальный коэффициент является
функцией приведенной температуры и
параметрически зависит от приведен-
ного дипольного момента. Значения
C*(T*f t*) табулированы в интервале
значений Г* от 0,70 до 400 для ряда
значений t* (см. табл. XX, стр. 890).
Графически зависимость функции С*
от Г* представлена на фиг. 34 для не-
скольких значений /*. Необходимо отме-
тить, что отличия от поведения непо-
лярных молекул для третьего вириаль- Фиг. 34. Зависимость приведенного тре-
тьего вириального коэффициента С* от Г* и
6 8 ю
вириального коэффициента i
/* в случае потенциала Штокмайера.
ного коэффициента носят иной харак-
тер по сравнению с отличиями для
второго вириального коэффициента.
Однако Раулинсон установил, что кривая С*, соответствующая <* = 1,
входит в отрицательную полуплоскость приблизительно при Г* = 1, и что
кривые для полярных молекул расположены в этой области под кривыми,
соответствующими неполярным молекулам. Этот результат показан на фиг. 35.
184
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
Сравнение вычисленных и измеренных значений проводится на фиг. 36
только для трех газов : Н2О, NH3 и CH3F. Для получения приведенных экспе-
риментальных значений использованы значения параметров табл. 20. Для
аммиака и фтористого метила со-
гласие достаточно хорошее. Для обоих
этих газов экспериментальные точки
располагаются несколько выше и кри-
вые проходят менее круто. Эти откло-
нения носят тот же характер и имеют
примерно тот же порядок величины,
что и отклонения, обнаруженные
Фиг. 35. Кривые
С*(Т*) для газа полярных
молекул [173].
а — неполярные молекулы (t* = 0) ;
b — полярные молекулы (t* = 1).
-2
-4
-6,
Ш3(зксп)
CH3F (эксп.)
Н20 (оыч., t*=1,2)
СН3?(выч,г*=1,07)-
№3(выч.,1*=!,0)-
Для неполярных молекул (t*=0)
1 1 L
7,2 1,3 Ifl 1,5
7,7 1,8 1,9 2,0
для третьего вириального коэф-
фициента в случае потенциала
F-12) Леннарда- Джонса (см.
§ б). Явное расхождение вы-
численных и измеренных зна-
чений для Н2О, несомненно,
обусловлено пренебрежением
взаимодействием высших муль-
типолей (в частности, диполь-
квадрупольным взаимодействием)
в выражении для потенциала
Штокмайера. Даже для вто-
рого вириального коэффициента
влияние этих взаимодействий
в случае воды в несколько раз
больше, чем в случае аммиака,
молекула которого симметрична [174]. По-видимому, это различие значительно
сильнее отражается на величине третьего вириального коэффициента.
В результате молекулы воды объединяются в группы больше, чем ожидалось,
и коэффициент С* становится отрицательным при температуре ниже Т* = 1,8,
а не при температуре порядка Т* = 1,0. Геометрия молекул воды является,
до-видимому, фактором, способствующим такому объединению, так как она
допускает образование решетки типа вюрцита. В такой решетке каждая
молекула притягивает всех своих соседей одинаково, что невозможно ни для
дипольных молекул, ни для молекул аммиака. Более полное обсуждение
явлений взаимодействия высших мультиполей проводится в разд. Ж.
Фиг. 36. Сравнение экспериментальных и
теоретических значений третьего вириального
коэффициента для аммиака, фтористого ме-
тила и паров воды.
Е. ВЫЧИСЛЕНИЕ ДЛЯ СМЕСЕЙ
Значения вириальных коэффициентов и коэффициента Джоуля—Томсона
для смесей можно получить из выражений D.17), D.18) и F.15). Для прове-
дения любых расчетов необходимо располагать значениями силовых постоян-
ных парного взаимодействия одинаковых и неодинаковых молекул газовой
§ 10. Вириальные коэффициенты для газов, состоящих из полярных молекул 185
смеси. Силовые постоянные парного взаимодействия одинаковых полярных
молекул даны в табл. 20. Параметры потенциала взаимодействия неодинаковых
молекул в принципе можно получить из величины вириального коэффициента
для смеси или из величины объема смешения. Трудности, с которыми сталки-
ваются на этом пути, уже обсуждались в § б, разд. В. Соответственно самое
целесообразное — использовать эмпирическое правило комбинирования, свя-
зывающее силовые постоянные неодинаковых молекул с постоянными для
одинаковых молекул. Для взаимодействий двух полярных молекул сортов
1 и 2 можно использовать соотношения
*и = у <*! + *«>' (Ю.12)
(l0J4>
Данные, позволяющие проверить применимость этих соотношений, отсутст-
вуют.
Для смесей, состоящих как из полярных, так и неполярных молекул,
необходимо определить взаимодействие между полярными и неполярными
молекулами. Можно показать (см. гл. 13, § 5), что эффективная полная энергия
взаимодействия полярной и неполярной молекул имеет тот же вид, что и соот-
ветствующее выражение для неполярных молекул. Следовательно, при вы-
числениях Вц для смеси можно использовать таблицы значений В*(Т*) для
случая потенциала Леннарда-Джонса. Силовые постоянные взаимодействия
полярной (обозначим их индексом р) и неполярной (индекс п) молекул можно
получить с помощью эмпирического правила комбинирования
°пР = \(°п + ор)Г\ A0.15)
A0.16)
Множитель ? дается выражением
где ап — поляризуемость неполярной молекулы в А3; ап — диаметр столк-
новений неполярной молекулы в А ; (Ь0)п выражено в смг/моль. Использование
различных комбинационных формул иллюстрируется числовым примером.
пример
Задача. Вычислить второй вириальный коэффициент при температуре 400° К для
смеси 50 мол.% воздуха, 45 мол.% CHC12F и 5 мол.% паров воды. Предполагается, что воздух
состоит на 4/б из азота и на уб из кислорода.
Решение. Второй вириальный коэффициент для четырехкомпонентной смеси имеет вид
В(Т) = х?Вп + х*В22 + х§В3
+ 2ххх2В12 + 2
Поэтому мы должны вычислить величины членов, обусловленных имеющими место
десятью различными типами взаимодействия. Прежде всего мы должны вычислить силовые
постоянные для различных типов взаимодействия. Затем, используя эти параметры, можно
вычислить значения различных Bif с помощью таблиц. В табл. 26 для иллюстрации способа
проведения вычислений сведены все расчеты. Некоторые детали расчета указаны в приме-
чаниях.
186
Гл. 3, Уравнение состояния для газов малой и умеренной плотности
Таблица 26
Вычисление второго вириального коэффициента В(Т) для
четырехкомпонентной смеси полярных и неполярных молекул
Компоненты Молярные доли Поляризуемости0
ах - 1,76 А»
а8 = 1,60 Ав
1
2
3
4
- N,
- О,
- СНС1, F
- н,о
Xi
х?
- х,
х*
- 0,40
- 0,10
= 0,45
= 0,05
I
1
1
1
1
2
2
2
3
3
4
I
1
2
3
4
2
3
4
3
4
4
3,698*
3,58г
4,26*
3,14е
3,46*
4,14е
3,03е
4,82е
3,74™
2,65е
(Ъ W
см3/моль
63,78*
57,88д
97,53d
39,05д
52,26*
89,51д
35,09д
141е
66,0д
23,42е
Hilk, °K
95,05*
105,9*
192е
213е
118*
214е
237е
381е
380™
380е
С/
—
—
—
—
—
—
0,1е
0,35™
1,2е
g
4,208
3,777
2,083
1,878
3,389
1,869
1,688
1,050
1,053
1,053
• и
ви
+ 0,1478
+ 0,0759
- 0,5637
- 0,7350
- 0,0070
- 0,7435
- 0,9385
- 2,3750
- 2,9017
-14,2
В(Т) = ]?У
и
вн
+ 9,427
+ 4,393
- 54,98
- 28,70
- 0,366
- 66,55
- 32,93
-334,9
-191,5
-332,6
с х В.=
j и
Вклад в
В(Т), обу-
словлен-
ный
взаимо-
действием
+ 1,508
+ 0,351
- 19,79
- 1,148
- 0,004
- 5,990
- 0,329
- 67,82
- 8,618
— 0,832
-102,7
смь1моль
а Из работы [20] ; см. также табл. 115.
б Табл. 7.
в Табл. 20.
г Следует использовать соотношения F.8) и F.9).
f
е Следует использовать соотношения A0.15)—A0.17).
ж Следует использовать соотношения A0.12)—A0.14).
, т* кТ 400
И " е,, = ty/fc *
и Для взаимодействия неполярных молекул с неполярными и полярных молекул с неполярнымй
второй вириальный коэффициент В*(Т*) следует вычислять с помощью табл. II (стр. 854). Для взаимодей-
ствия полярных молекул с полярными при вычислении В *(Т*,^*) нужно воспользоваться табл. XVII (стр.880).
л Для взаимодействия Ы соответствующий вклад имеет вид XfB«, а для взаимодействия i-j вклад
имеет вид 2xixfBii. Тогда [В(Т)]СМ - ?* х/ Вц.
Ж. ДИПОЛЬ-КВАДРУПОЛЬНОЕ ВЗАИМОДЕЙСТВИЕ
СЛОЖНЫХ МОЛЕКУЛ
Если молекула имеет сложным образом распределенный заряд, то может
оказаться необходимым включить в потенциальную функцию члены, учиты-
вающие взаимодействия высших мультиполей. Эти члены, вероятно, особенно
важны в случае взаимодействия молекул, имеющих тенденцию образовывать
водородные связи. Наиболее важным вкладом от взаимодействия высших
мультиполей является член q>W))9 пропорциональный г~4 и обусловленный
взаимодействием диполя одного распределения заряда с квадруполем другого
распределения заряда. Следующий член, пропорциональный г~б, является
суммой квадруполь-квадрупольного и диполь-октупольного взаимодействий.
Этим членом обычно пренебрегают даже в случае сложных молекул. Подробное
рассмотрение взаимодействий двух общих распределений заряда проводится
в гл. 12, § 1.
Рассмотрим влияние диполь-квадрупольного взаимодействия на величину
второго вириального коэффициента. Для случая этого взаимодействия выра-
жение потенциала будет значительно сложнее, чем в случае диполь-дипольного
взаимодействия, так как'квадруполь является тензором и вообще имеет более
§ 10. Вириальные коэффициенты для газов, состоящих из полярных молекул 187
чем одну неравную нулю компоненту. Наиболее важными случаями являются
квадруполи, обладающие цилиндрической симметрией относительно оси ди-
поля. Таким квадруполям соответствует распределение вдоль линии трех
или более зарядов или распределение типа, показанного на фиг. 37, где кру-
говое кольцо положительного заряда расположено в плоскости, лежащей над
отрицательным зарядом. Для молекул, об-
ладающих таким типом симметрии, можно
потенциал взаимодействия представить мо-
дифицированным потенциалом Штокмайера,
включающим член, пропорциональный г,
и учитывающим энергию диполь-квадру-
польного взаимодействия :
Кольцо
положительного
заряда
Здесь /г — дипольный момент, a Q — ци-
линдрически симметричный квадрупольный
момент молекулы1). Функция g определя-
ется выражением C.32) гл. 1, a h имеет вид
Л («1, К Фг - Фг) = у (cos 0i - cos 9*> х
X [2 sin Вг sin 02 cos (ф2 — фг) —
-Scosflicosej— 1]. A0.19)
Функция h представляет угловую зависи-
мость взаимодействия диполя «1» и квад-
руполя «2» плюс взаимодействие квадру-
поля «1» с диполем «2». Маргенау и Майерс
[175] при вычислениях второго вириаль-
ного коэффициента для паров воды пред-
положили, что угловая зависимость описывается членом г~4 аналогично
случаю диполь-дипольного взаимодействия.
Для потенциала A0.18) Раулинсон [174] получил выражение второго
вириального коэффициента. Его результат можно представить в виде поправки,
которую необходимо добавить к результату A0.4), полученному для потенциала
Штокмайера :
^42 «Ю (п\(к\ пГ 6П-3/С + 2/-3
Отрицательный
^ заряд
Фиг. 37. Распределение заряда, ис-
пользовавшееся в § 10 при рассмот-
рении важности диполь-квадруполь-
ного взаимодействия полярных
молекул.
п=2к=2 1=1
12
X
A0.20)
Коэффициенты ИкЬ появляющиеся в результате интегрирования по углам'
имеют вид
2л л п
Hkl = ^ J J j ^^
0 0 0
a u* = C/f32) (iMQ/e^). Хотя это выражение справедливо только в случае
цилиндрического распределения заряда, оно может быть легко обобщено
также для описания более сложных взаимодействий. Число членов, которые
Определение величины Q дано в гл. 12, § 1.
188
Гл. 3. Уравнение состояния для газов малой и умеренной плотности
необходимо учитывать при суммировании по I в выражении для Л В*, зависит
от величины рассматриваемых значений Г*, /* и и*. Для паров воды при тем-
пературе выше 100° С члены при I > 1 пренебрежимо малы и, следовательно,
необходимы только коэффициенты Нк1. Значения нескольких первых из этих
коэффициентов приведены в табл. 27 [174 ]. Используя эти значения, Раулинсон
Таблица 27
Значения коэффициентов разложения
к
2
0,8889
3
-0,2844
4
1,0464
5
-1,088
б
2,283
вычислил для паров воды параметры потенциальной функции A0.18). Значения
этих параметров вместе со значениями, соответствующими потенциалу Шток-
майера, приведены в табл. 28.
Таблица 28
Силовые постоянные для паров воды
Параметр
t*
и*
Потенциал
Штокмайера
2,65
380°
1,2
—
Потенциал
A0.18)
2,725
356°
1,2
0,654
Сравнение измеренных значений и значений второго вириального коэф-
фициента, вычисленных с использованием постоянных табл. 28, проводится в
табл. 29.
Таблица 29
Вычисленные и измеренные значения второго вириального
коэффициента для паров воды
Т °С
100
150
200
300
400
—В(Т)
измеренные
[179, 180]
450
284
197
112
72
вычисленные для
потенциала
Штокмайера
(см. [170])
460
290
202
115
74
вычисленные для
потенциала в
A0.1в) [1/4]
450
281
205
123
80
Как видно, совпадение не удается улучшить и при учете диполь-квадру-
польного члена, особенно при высоких температурах. Однако с определен-
ностью можно считать, что в случае учета диполь-квадрупольного взаимодей-
ствия полученные значения параметров более правдоподобны. В частности,
значения а, определенные из A0.18), более близки к величине межмолекуляр-
ного расстояния для льда. Вычисления Раулинсона показывают, что вклад
диполь-квадрупольного взаимодействия в величину второго вириального
коэффициента для паров воды в интервале температур от 100 до 400° С будет
порядка 25% величины, соответствующей вкладу диполь-дипольного взаи-
модействия. Поэтому диполь-квадрупольным взаимодействием нельзя прене-
брегать при вычислении второго вириального коэффициента сложных поляр-
ных молекул.
Приложение А
Приложение А
РАЗЛОЖЕНИЕ КИХАРЫ ТРЕТЬЕГО ВИРИАЛЬНОГО
КОЭФФИЦИЕНТА
Последнее соотношение в выражении D.6) для третьего вириального
коэффициента можно легко преобразовать [56 ] к виду
V4 У^У* СО
C(T) = -4^J J [[/12/i3/23/-f2^i2]^d(y2). (АЛ)
Новые переменные х, у определены на фиг. 38 и связаны следующими соотно-
шениями с г23 и г13:
) (А.2)
Сначала можно проинтегрировать (АЛ) по г12, так как г12 не входит в пределы
интеграла. Интеграл по г12 интегрированием по
частям можно преобразовать к виду
9(/i2 /13 /23)
(А.З)
Произведение / функций можно записать в виде
суммы
где
= Г> - 2 /(fc) +2 2
А=1 к ]>к
(А.4)
/(О) = ехр [_
Фиг. 38. Определение
переменных х и у.
(А.5) Лопатообразная область, ограниченная
сплошными линиями, — область, в кото-
рой движение третьей молекулы при
данном значении величины r1Jf совмести-
мом с условием г13^ г1г и г28 ^ rlf,
запрещено.
За потенциальную функцию ц> принимается общее выражение потенциала
Леннарда-Джонса [см. C.19) гл. 1 ], где п и т — показатели степени силы
отталкивания и силы притяжения соотзетственно. Вначале полагают т > б,
затем рассматривается значение т -> б. ,
После подстановки выражений (А.4) и (А.5) в (А.З) можно провести интег-
рирование таким же способом, как для выражения F.2). В результате разло-
жение квадратной скобки выражения (АЛ) имеет вид
2
где t == Cw//cT) {kT\v)mln. Коэффициенты А, являются функциями переменных
х и у и даются следующим выражением :
Aj = A + |« + ,-)*/" [(-^^
/ J
п)т-„- J —
4- я»
A"
)m/n J
4е),
(А.7)
190 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
где
f-i = у*2 + у»; а ir1 = КA - хJ + У2-
Подстановка выражения (А.6) в (АЛ) и последующее интегрирование Д,- по
х и у дает1)
Д , я) (^ -^-)[Л"-т) + Г (А.8)
где коэффициенты сС»(т, п) выражаются через двойные интегралы:
1 J "'«"О* <A.9>
Для вычисления со)(т, п) можно воспользоваться численным интегрированием,
хотя для больших значений / возможно аналитическое интегрирование по
методу наиболее крутого спуска. В результате получаются простые асимптоти-
ческие формулы. В коэффициент с'^б, п) входит Г [(т — 6)/п ] при т = 6,
Однако Кихара показал, что
Для потенциала F-12) Леннарда-Джонса выражение для С(Т) имеет вид F.10),
а коэффициенты разложения с0) F, 12) приведены в табл. V (стр. 858). Кихара
[57 ] вычислил также значения коэффициентов разложения с(у) F, 9).
Приложение Б
ВЫРАЖЕНИЕ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ЧЕРЕЗ УРАВНЕНИЕ СОСТОЯНИЯ
И ВИРИАЛЬНЫЕ КОЭФФИЦИЕНТЫ
Отклонения термодинамических функций от значений, соответствующих
идеальному газу, можно выразить через параметры уравнения состояния и
вириальные коэффициенты. В этом приложении мы дадим сводку выражений
для различных термодинамических функций непосредственно через параметры
уравнения состояния, вириальные коэффициенты, а также через приведенные
вириальные коэффициенты и приведенный молярный объем V* = Vjb0.
1. Внутренняя энергия
We = " Г* (~V*~ + " (V*f +•••)• (БЛ)
г) bo = 2liriNaby а а есть значение г, при котором <р(г) — 0; е — максимальная энергия
притяжения.
Приложение Б
191
2. Энтальпия
Н-Н»
Ne
= 7*
в*-в* . c*_v2d*
V*
(V*J
3. Энтропия1)
?
*-*•--«'»'+*»?-
ino ! i
R - mP \V* +
4. Теплоемкость при постоянном объеме
CV-C%
Г2 d2B
2Cf + C
Г» <PC
_
R V* 2(V*J "f "
5. Теплоемкость при постоянном давлении
{dp/dV)T
d2B \ dTj ^ dT 2 dT*
ср-с°Р
v* ^
V2
1
Г • • •
6. Коэффициент Джоуля—Томсона
rp UP n j 1__
2B2 — 2TB-^- — 2C + Г
, /?Г2 d2B Г^ dB
= Вх* - В* + -1- [2 (В*)8 - 2В* Bf - 2С* + Cf +
+ *Bt(Bt-B*)]+...
LP J
Величина 5° определяется как предельное значение § при р -»• О
(Б.2)
(Б.З)
(Б.4)
, (Б.5)
192 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
7. Скорость звука
,2 _ __ J^L (ОЦ
с°-~ м [dvjT'
где y = Cp/Cv.
Задачи
1. Вывести калорическое уравнение состояния (см. примечание 1 на стр. 121) двумя
способами :
а) используя соотношение, связывающее внутреннюю энергию и функцию распре-
деления ;
б) используя тот факт, что внутренняя энергия газа или жидкости при данной тем-
пературе и удельном объеме есть среднее значение классического гамильтониана по канони-
ческому ансамблю; показать, что оба вывода приводят к одинаковому результату.
2. Предполагая в § 2, что все Ui для / ^ 3 равны нулю, учесть это при выводе урав-
нения состояния и получить выражение для второго вириального коэффициента.
3. Получить выражение для изменения объема при смешении а, молей сорта Аг с а2
молями сорта А2 и упростить результат путем его разложения в ряд и удержания только
членов первого порядка малости. Из этого результата увидим, что изменение объема зависит
как от сил взаимодействия между молекулами одного сорта, так и от сил взаимодействия
между молекулами разных сортов.
4. При низких температурах кривая зависимости pV от р имеет минимум, называемый
точкой Бойля. С ростом температуры точка Бойля смещается в сторону меньших значений
давления. В конце концов при р = 0 и некоторой температуре (температуре Бойля) появ-
ляется точка, где касательная имеет нулевой наклон. Выше этой температуры изотермы не
имеют точек с нулевым наклоном касательных. Показать, что при температуре Бойля
В(Т) = 0. Далее, используя это, найти для потенциала F-12) Леннарда-Джонса значение
е/k для следующих газов с соответствующими температурами Бойля:
Воздух, 74° С Аргон, 137° С
Азот, 50° С Кислород, 150° С
5. При низких температурах коэффициент Джоуля—Томсона положителен ; это озна-
чает, что охлаждение будет сопровождаться изэнтальпическим расширением. При высоких
температурах коэффициент Джоуля—Томсона отрицателен, что соответствует нагреванию
при дросселировании. Для каждой величины давления существует некоторая температура,
при которой коэффициент Джоуля—Томсона меняет знак (точка инверсии). Температура,
при которой коэффициент Джоуля—Томсона, соответствующий р = 0, меняет знак, назы-
вается температурой инверсии. Показать, что при этой температуре инверсии d(B/T)/dT = 0.
Можно ли ожидать наличия температуры инверсии в случае молекул, ведущих себя как то-
чечные центры отталкивания?
6. Используя таблицы для потенциала Л еннарда-Джонса, вычислить :
а) изменение объема при смешении 4 л N2 и 1 л Ог; смесь приготовляется при
давлении 10 атм и температуре 300° К ;
б) второй вириальный коэффициент для смеси молярной концентрации 10% СН4,
50% СО2, 40% N2 при температуре —50° С.
7. Используя таблицы для потенциала, Штокмайера, вычислить:
а) величину фактора сжимаемости pV/RT для пара при температуре 600° С и давле-
нии 50 атм;
б) изменение объема при смешении 10 л аргона и 5 л аммония ; смесь приготовля-
ется при температуре 500е С и давлении 50 атм;
в) коэффициент Джоуля—Томсоча, соответствующий нулевому давлению при тем-
пературе 400° К для смеси молярной концентрации 25% воздуха и 75% фтористого метила.
8. Построить графики следующих потенциальных функций :
<р(г) = ©о , г < а,
<р(г) = - пев (-^-J In (-—-J, r > а.
Литература 193
Каков физический смысл параметров п, е и <х? Показать, что второй вириальный коэф-
фициент для этого потенциала дается выражением
9. В случае несферических молекул может использоваться потенциал вида
<Р (г, 0Ъ 02, 02 - 0х) = 9>i (г) + 9>2 (г) [cos2 0Х + cos2 0a].
Показать, что для этого потенциала классический второй вириальный коэффициент
дается выражением
= - 2я N ( f^-e-^"- ff erf j/^)' - l]
О
В (Г) = - 2я N ( f^-e-^"- ff erf j/^) - l] r» dr.
О
10. Используя потенциал взаимодействия двух атомов гелия типа Интема—Шнейдера
(9.2), путем численного интегрирования найти второй вириальный коэффициент при тем-
пературе 300° К. Сравнить эту величину с величиной, соответствующей потенциалу F-12)
Леннарда-Джонса.
ЛИТ ЕР АТУР А
1. Van der Waals J. D. Sr., Диссертация, Leiden, 1873.
2. К a m e r 1 i n g h О n-n e s H., Comm. Phys. Lab., Leiden, No. 71 A901).
3. К r a m e г s H., Proc. Roy. Acad., Amsterdam, 41, 10 A938).
4. Born M., Green H. S., Proc. Roy. Soc, A191, 168 A947).
5. d e В о e r J., Rep. Progr. Phys., 12, 305 A949).
6. U r se 11 H. D., Proc. Cambr. Phil. Soc, 23, 685 A927).
7. F о w 1 e r R. H., Statistical Mechanics, 2nd ed., Cambridge, 1936.
8. В о r n M., F u с h s K., Proc. Roy. Soc, A166, 391 A938).
9. Mayer J. E., Journ. Chem. Phys., 5, 67 A937).
10. К a h n В. Диссертация, Utrecht, 193«.
11. de Boer J., Physica, 15, 680 A949).
12. M а у e r J. E., M о n t г о 11 E., Journ. Chem. Phys., 9, 2 A941).
13. Axil rod В. М., Teller E., Journ. Chem. Phys., 11, 299 A943).
14. Mayer J. E., Mayer M. Q., Statistical Mechanics, New York, 1940. (см.перевод:
M а й е р Дж., Гепперт-Майер М., Статистическая механика, ИЛ, 1952).
15. Wang Chang С. S., Диссертация, University of Michigan, 1944.
16. Mayer J. E., Journ. Phys. Chem., 43, 71 A939).
17. H a p p e 1 H., Ann. d. Phys., 30, 246 A906).
18. Majumdar R., Bull. Calcutta Math. Soc, 21, 107 A929).
19. F о w 1 e r R. H., Proc. Cambr. Phil. Soc, 34, 382 A938).
20. Stuart H. A., Molekulstruktur, Berlin, 1934.
21. Hirschfelder J. O., McClure F. Т., Curtiss С F., О s borne D. W.,
Thermodynamic Properties of Propellant Gases, NDRC Report, A-116, November, 1942,
22. Jeans J., Dynamical Theory of Gases, Cambridge, 1925.
23. К e e s о m W. H., Comm. Phys. Lab. Leiden, Suppl., 24B, 32 A912).
24. К i ha г а Т., Nippon-Sugaku-Buturigakukaisi, 17, 11 A943).
25. H i г s с h f e 1 d e r J. 0., McClure F. Т., Weeks I. F., Journ. Chem. Phys., 10,
201 A942).
26. L u n b e с k R. J., Диссертация, Amsterdam, 1950.
27. Hoi born L., Otto J., Zs. Phys., 33, 1 A925).
28. В e a 11 i e J. А., В r i e r 1 e у J. S., В a r r i a u 11 R. J., Journ. Chem. Phys., 20, 1613
A952).
29. Kihara Т., Journ. Phys. Soc. Japan, 6, 289 A951).
30. M а с С о r m а с к К. Е., Schneider W. G., Journ. Chem. Phys., 18, 1269 A950).
31. В e a 11 i e J. A., H a d 1 о с к, Р о f f e n b e r g e r, Journ. Chem. Phys., 3, 93 A935).
32. В e a 11 i e J. A., Kay, К a m i n s к у, Journ. Am. Chem. Soc, 59, 1589 A937).
194 Гл. 3 Уравнение состояния для газов малой и умеренной плотности
33. В eat tie J. A., S i m a r d, S u, Journ. Am. Chem. Soc, 61, 26 A939).
34. Smith, В eat tie J. A., Kay, Journ. Am. Chem. Soc, 59, 1587 A937).
35. R о p e r E. E., Journ. Phys. Chem., 44, 835 A940).
36. Thermodynamic Properties of Trichloromonofluoromethane, Kinetic Chemicals, Inc., 1938.
37. Thermodynamic Properties of Dichloromonofluoromethane, Kinetic Chemicals, Inc., 1939.
38. Thermodynamic Properties of Dichlorodifluoromethane, Circular 12, Am. Soc. Refr. Eng.,
1931.
39. Thermodynamic Properties of Trichlorotrifluoroethane, Kinetic Chemicals, Inc., 1938.
40. Methyl Chloride, E. I. du Pont de Nemours & Co., the R. and H. Chemicals Department,
5th Ed., 1940.
41. Meyers С H., Jessup R. S., Refr. Eng., 11, 345 A925).
42. К e e n a n J. H., Keyes F. G., Thermodynamic Properties of Steam, New York, 1936.
43. E p s t e i n L. F., H i b b e r t С J., Journ. Chem. Phys., 20, 752 A952).
44. L e n n a r d - J о n e s J. E., Proc. Roy. Soc, A106, 463 A924).
45. Bird R. В., Spotz E. L., University of Wisconsin, CM 599 (Project No. 9938),
May 10, 1950.
46. Michel s A., Wijker Hub., Wijker Hk., Physica, 15, 627 A949).
47. В e a 11 i e J. А., В a r r i a u 11 R. J., В r i e r 1 e у J. S., Journ. Chem. Phys., 19, 1222
A951).
48. Michels A., Wouters H., de Boer J., Physica, 1, 587 A934).
49. Hoi born L., Otto J., Zs. Phys., 10, 367 A922).
50. Michels A., Nederbragt G. W., Physica, 3, 569 A936).
51. Michels A., Michels C, Proc. Roy. Soc, A153, 201- A936).
52. E d w a r d s A. E., Roseveare W. E., Journ. Am. Chem. Soc, 64, 2816 A942).
53. L u n b e с к R. J., Boerboom A. J. H., Physica, 17, 76 A951).
54. G о r s к i R. A., Miller J. G., Journ. Am. Chem. Soc, 75, 550 A953).
55. Michels A., W a s s e n a a r Т., Appl. Sci. Res., Al, 258 A949)".
56. К i h a r а Т., Journ. Phys. Soc. Japan, 3, 265 A948).
57. К i h a r а Т., Journ. Phys. Soc. Japan, 6, 184 A951).
58. В i r d R. В., SpotzE. L, H i r s с h f e 1 d e r J. O., Journ. Chem. Phys., 18, 1395
A950).
59. M о n t г о 11 E. W., MayerJ. E., Journ. Chem. Phys., 9, 626 A941).
60. d e В о e r J., M i с h e 1 s A., Physica, 6, 97 A939).
61. H i r s с h f e 1 d e r J. O., Roseveare W. E., Journ. Phys. Chem., 43, 15 A939).
62. R о e b u с к J. R., OsterbergH., Journ. Am. Chem. Soc, 60, 341 A938).
63. В u с к i n g h a m R. А., С о r n e r J., Proc Roy. Soc, A189, 118 A947).
64. С о r n e r J., Trans. Farad. Soc, 44, 914 A948).
65. d e S m e 11 J., KeesomW.H, M о о i j W. H., Leiden. Comm., 203e, 1930.
66. KeesomW. H., H a a n t j e s, Physica, 2, 460 A935).
67. Simon F., Simson C, Zs. Phys., 25, 160 A924).
68. Born M., Ann. d. Phys., 69, 473 A922).
69. Rice W. E., H i r s с h f e 1 d e r J. O., Journ. Chem. Phys., 22 A954).
70. Mason E. A., Rice W. E., Journ. Chem. Phys., 22 A954).
71. I s h i d a Y., Phys. Rev., 21, 550 A923).
72. Guenther P., Zs. Phys. Chem. 110, 626 (924).
73. T r a u t z M., В a u m a n n P. В., Ann. d. Phys., 2, 733 A929).
74. TrautzM., Stauff F. W., Ann. Phys., 2, 737 A929).
75. T r a u t z M., К i p p h a n K. F. Ann. d. Phys., 2, 743 A929).
76. T r a u t z M., L u d e w i g s W., Ann. d. Phys., 3, 409 A929).
77. Trautz M., Binkele H. E., Ann. d. Phys., 5, 561 A930).
77a. Trautz M., Melster A., Ann. d. Phys., 7, 409 A930).
78. Trautz M., Zink R., Ann. d, Phys., 7, 427 A930).
79. Trautz M., Kurz F., Ann. d. Phys., 9, 981 A931).
80. Trautz M., Sorg K. G., Ann. d. Phys. 10, 81 A931).
81. Trautz M., H e b e r 1 i n g R., Ann. d. Phys., 10, 155 A931).
82. S u t h e r 1 a n d B. P., M a a s O., Canad. Journ. Res., 6, 428 A932).
83. T r a u t z M., H e b e r 1 i n g R., Ann. d. Phys., 20, 118 A934).
Литература 195
84. Trautz M., Husseini I., Ann. d. Phys., 20, 121 A934).
85. Trautz M., Zimmermann H., Ann. d. Phys., 22, 189 A935).
86. v a n 111 e r b e e к А., С 1 a e s A., Nature, 142, 793 A938).
87. van Itterbeek A., Claes A., Physica, 5, 938 A938).
88. van Itterbeek A., van Paemel. O., Physica, 7, 265 A940).
89. J о h n s t о n H. L., M с С 1 о s к е у К. Е, Journ. Phys. Chem., 44, 1038 A940).
90. Wobser R., Mueller F., Kolloid-Beihefte, 52, 165A941).
91. van Itterbeek A., van Paemel O., van Lierde, Physica, 13, 88 A947).
92.de Troyer, van Itterbeek A., Rietveld, Physica, 17, 938 A951).
93. Bud den berg J. W., Wilke С R., Journ. Phys. Coll. Chem., 55, 1491 A951).
94. Hoi born L., Otto J., Ann. d. Phys., 63, 674 A920).
95. H о 1 b о r n L., Otto J., Zs. Phys., 23, 77 A924).
96. Hoi born L., Otto J., Zs. Phys., 38, 359 A926).
97. Nijhoff G. P., Keesom W. H., Leiden Comm., 188b, 188d A927).
98. G i b b y, Tanner, M a s s о n, Proc. Roy. Soc, A122, 283 A929).
99. M i с h e 1 s A., G о u d e к е t M., Physica, 8, 347 A941).
100. States M. N., Phys. Rev., 21, 662 A923).
101. Nasini A. G., Rossi C, Gazz. chim. ital., 58, 433, 898 A928).
102. van Itterbeek A., Keesom W. H., Physica, 5, 257 A938).
103. Johnston H. L., G r i 11 у Е. R., Journ. Phys. Chem., 46 948 1942).
104. Boks J. D. A., Kamerlingh Onnes H., Leiden Comm., 170» A924).
105. Hoi born L., Otto J., Zs. Phys., 10, 367 A922).
106. N i j h о f f G. P., Keesom W. H., 11 i i n, Leiden Comm., 188c A927).
107. Michel s A., Wouters H., Physica, 8, 923 A941).
108. Schneider W. G., Duff ie J. A. H., Journ. Chem. Phys., 17, 751 A949).
109. Yntema J. L., Schneider W. G., Journ. Chem. Phys., 18, 641 A950).
110. Edwards R. S., Proc. Roy. Soc, A119, 578 A92g).
111. Buddenberg J. W., Wilke C. R., Journ. Coll. Chem. 55, 1491 A951).
112. Ka merli ng h Onnes H., С г о m e 1 i n С A., Leiden Comm., 147d A915).
113. Crommelin C. A., Martinez, Kamerlingh Onnes H., Leiden Comm.,
154a A919).
114. Kane G., Journ. Chem. Phys., 7, 603 A939).
115. van Itterbeek A., van Paemel O. Physica, 5, 1009 A938).
116. Vasilesco V., Ann. de phys., 20, 137, 292, A945).
117. Kamerlingh Onnes H., Crommelin С A., Leiden Comm., 118b A910).
118. Hoi born L., Otto J., Zs. Phys. 30, 320 A924).
119. Michel s A., Wijker Hub., Wijker Hk., Physica, 15, 627 A949).
120. Rankine A. O., Smith С J., Phil. Mag., 42, 615 A921).
121. Jung G., Schmick H., Zs. Phys. Chem., B7, 130 A930).
122. Freeth F. A., Ver-schoyle T. T. H., Proc. Roy. Soc, A130, 453 A931).
123. M i с h e 1 s A., N e d e r b r a g t G. W., Physica, 2, 1000 A935).
124. Landolt-Bornstein. Pysikalisch-Chemische Tabellen. (см. перевод: Ландолбт-
Бе рнштейн, Физико-химические таблицы, М. — Л., 1939).
125. С 1 u s i u s К., Zs. Phys. Chem., B3, 41 A929).
126. Clayton J. 0., Giauque., W. F., Journ. Am. Chem. Soc, 54, 2610 A932).
187. Clayton J. 0., Giauque W. F., Journ. Am. Chem. Soc, 55, 4875, 5071 A933).
128. Kamerlingh Onnes H., van Urk А. Т., Leiden Comm., 169d, 169e A924).
129. Kamerlingh Onnes H., v a n U r k А. Т., Zs. Phys., 10, 367 A922); 23, 77 A924)
130. Kamerlingh Onnes, van Urk А. Т., Zs. Phys., 30, 320 A924).
131. Kamerlingh Onnes H., van Urk А. Т., Zs. Phys., 33, 1 A925).
132. Smith С J., Proc. Phys. Soc, 34, 155 A922).
133. Trautz M., Gabriel E., Ann. d. Phys., 11, 606 A931).
134. Rig den P. J., Phil. Mag., 25, 961 A938).
135. Verschoyle Т. Т. Н., Proc. Roy. Soc, Alll, 552 A926).
136. Scott G. A., Proc. Roy. Soc, A125, 330 A929).
137. Town end D. T. A., Bhatt L. A., Proc. Roy. Soc, A134, 502 A931).
138. Michel s, Lupton, Wassenaar, de Graaf, Physica, 18, 121 A52).
196 Гл. 3. Уравнение состояния для газов малой и умеренной плотности
139. I s i h а г а А., Н а у a s h i d а Т., Journ. Phys. Soc. Japan., 6, 40 A951).
140. К i h a r а Т., Journ. Phys. Soc. Japan., 8 A933).
141. Isihara A., Journ. Chem. Phys., 18, 1446 A950).
142. Margenau H., Phys. Rev., 63, 385 A943).
143. Margenau H., Phys. Rev., 64, 131 A943).
144. Evett A. A., Margenau H., Journ. Chem. Phys., 21, 958 A953).
145. d e Boer J., Physica, 9, 363 A942).
146. Corner J., Proc. Roy. Soc, A192, 275 A948).
147. London F., Journ. Phys. Chem., 46, 305 A942).
148. Buckingham R. A., Proc. Roy. Soc, A168, 264 A938).
149. M a s s e у H. S. W., В u с к i n g h a m R. A., Proc Roy. Soc, A168, 378 A938).
150. M a s s e у H. S. W., Buckingham R. A., Proc. Roy. Soc, A169, 205 A939).
151. Buckingham R. A., Hamilton J., M a s s e у H. S. W., Proc. Roy. Soc, A179,
103 A941).
152. de Boer J., Michels A., Physica, 5, 945 A938).
153. du Mond J. W. M., Cohen E. R., Rev. Mod. Phys., 20, 82 A948).
154. du Mond J. W. M., Cohen E. R., Rev. Mod. Phys., 21, 651 A949).
155. Yntema J. L., Schneider W. G., Journ. Cherm. Phys. Japan, 18, 641 A950).
156. Mason E. A., Rice W. E., Journ. Chem. Phys., 22 A954).
157. Schneider W. G., Duffie J. A. H., Journ. Chem. Phys., 17, 751 A949).
158. H о 1 b о r n L., О 11 о J., Zs. Phys., 33, 1 A925).
159.de Boer J., Michels A., Physica, 6, 409 A939).
160. d e В о e r J., P h у s i с a, 10, 348 A943).
161.de Boer J., van Kranendonk J., Physica, 14, 442 A948).
162. Margenau H., Phys. Rev., 56, 1000 A939).
163. Rice О. К., Journ. Am. Chem. Soc, 63, 3 A941).
164. Hill T. L., Journ. Chem. Phys., 16, 399 A948).
165. Keesom W. H., Comm. Phys. Lab. Leiden, Suppl. 24b, Section 6 A912).
166. Keesom W. H., Comm. Phys. Lab. Leiden, Suppl. 39a A915).
167. Stockmayer W. H., Journ. Chem. Phys., 9, 398 A941).
168. Wesson L. G., Table of Electric Dipole Moments, M. I. T., The Technology Press, 1948.
169. Bird R. В., Диссертация, University of Wisconsin, 1950.
170. R о w 1 i n s о n J. S., Trans. Farad. Soc, 45, 974 A949).
171. Lun beck R. J., ten Seldam С A., Physica, 17, 788 A951).
172. R о w 1 i n s о n J. S., Диссертация, Oxford, 1950.
173. Rowlinson J. S., Journ. Chem. Phys., 19, 827 A951).
174. Rowlinson J. S., Trans. Farad. Soc, 47, 120 A951).
175. Margenau H., Myers V. W., Phys. Rev., 66, 307 A944).
176. .Lambert, Roberts, Rowlinson, Wilkinson, Proc. Roy. Soc, A196, 113
A949).
177. Keyes F. G., Journ. Am. Chem. Soc, 60, 1761 A938).
178. Lawrence С. К., Journ. Am. Chem. Soc, 52, 6 A930).
179. Keyes F. G., Smith L. В., Gerry H. Т., Proc. Am. Acad. Arts Sci., 70, 319 A936).
180. С о 11 i n s C, Keyes F. G., Proc. Am. Acad. Arts. Sci., 72, 283 A938).
181. Alexander, Lambert, Trans. Farad. Soc, 37, 421 A941).
182. Gordon A. R., Journ. Chem. Phys., 2, 65 A934).
183. W i 1 s о n E. В., Journ. Chem. Phys., 4, 526 A936).
ГЛАВА
УРАВНЕНИЕ СОСТОЯНИЯ ПЛОТНЫХ ГАЗОВ
И ЖИДКОСТЕЙ
В предыдущей главе было показано, что уравнение состояния может быть
получено двумя способами : по методу статистических сумм и по методу бинар-
ной функции распределения. Показано также, что оба метода приводят к выра-
жению для фактора сжимаемости в виде ряда по степеням плотности. Такое
вириальное представление успешно используется для количественного изу-
чения равновесных свойств газов при малых и средних значениях плотности.
Оно также может быть использовано для качественного объяснения конден-
сации и критических явлений; этот вопрос рассматривается в гл. 5.
В этой главе мы рассмотрим применение указанных двух методов в области
высоких плотностей. Метод статистических сумм используется в виде раз-
личных приближений к действительной статистической сумме для N молекул.
Эти приближения образуют основу для так называемой теории решеток, или
теории свободного объема. Теория решеток, в свою очередь, разделяется на
две общие категории : «теорию ячеек», в которой движение каждой молекулы
ограничивается некоторой ячейкой, но не принимается в расчет тот факт, что
некоторые ячейки в действительности пусты, и «теорию дырок», которая учиты-
вает наличие свободных ячеек. С помощью этих теорий проведены обширные
вычисления.
Метод бинарной функции распределения можно применить в области
высоких плотностей, получая решения приближенного интегрального урав-
нения для бинарной функции распределения. Немногие полученные до сих
пор численные результаты находятся в разумном согласии с эксперименталь-
ными значениями; этот метод, по-видимому, имеет большое значение.
Несмотря на то, что в настоящее время многие вопросы подверглись изу-
чению в связи с развитием указанных выше теорий, тем не менее для удовле-
творительного описания свойств плотных газов и жидкостей потребуется еще
большая работа как теоретическая, так и вычислительная. Имея в виду недо-
статочность существующих теорий, начнем эту главу с обсуждения закона
соответственных состояний. Этот закон устанавливает связь между различ-
ными свойствами разных веществ и является основой для построения обобщен-
ной диаграммы сжимаемости Хогена и Ватсона [1 ]. Дадим также краткий
обзор ряда эмпирических уравнений состояния.
§ 1. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ1)
Закон соответственных состояний успешно применяется для оценки пове-
дения плотных газов и жидкостей. Этот закон в том виде, в котором он был
первоначально сформулирован Ван дер Ваальсом, основан на выражении пере-
менных в их отношении к значениям в критической точке. Это является основой
для построения обобщенной диаграммы сжимаемости Хогена и Ватсона.
*) Рассмотрение закона соответственных состояний ограничено здесь соотношениями
между р, V и Г. Некоторые из развиваемых здесь положений могут применяться также для
изучения явлений переноса (см. гл. 9, § 1)
198 Гл. 4. Уравнение состояния плотных газов и жидкостей
В другом виде закона соответственных состояний для получения приведенных
величин используются молекулярные постоянные. Тогда становится возмож-
ным ввести дополнительные параметры, позволяющие теоретически обосно-
ванно применять этот закон к различным типам молекул —длинным, полярным,
а также легким молекулам, для которых квантовые эффекты играют значи-
тельную роль1).
А. ЭМПИРИЧЕСКИЙ ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ
Критическая точка определяется как точка, в которой как (Эр/ЭУ)г, так
и (&p]dV2)T обращаются в нуль; значения давления, объемами температуры
в этой точке (критические постоянные) обозначаются через рс, Vc и Тс соответ-
ственно. Эти постоянные можно использовать для определения следующих
приведенных переменных:
Эмпирический закон соответственных состояний может быть сформули-
рован следующим образом: «Все вещества подчиняются одному уравнению
состояния, если это уравнение выражено через приведенные переменные».
Вообще говоря, состояние системы может быть описано любыми двумя из трех
переменных : давление, объем и температура. Таким образом, согласно закону
соответственных состояний, любая безразмерная комбинация есть универсаль-
ная функция двух из трех приведенных переменных. В частности, фактор
сжимаемости pV/RT можно записать в виде универсальной функции приве-
денного объема и приведенной температуры:
^r = F{Vry Tn). A.2)
Для реальных систем обычно до некоторой степени удобнее в качестве пара-
метров использовать приведенные значения давления и температуры. В этом
случае
-щг = г(рпТг). A.3)
Согласно закону соответственных состояний, как F, так и z являются универ
сальными функциями, одинаковыми для всех веществ.
Для приведенных параметров критическая точка2) определяется значе-
ниями W = 1 и Тг = 1. Из A.2) замечаем, что в критической точке
^ ). A.4)
Таким образом, значение этого критического коэффициента (фактор сжимае-
мости в критической точке) будет являться универсальной постоянной. Зна-
чения факторов сжимаемости в критической точке для ряда веществ приве-
дены в табл. 30. Для простых сферических и неполярных молекул значение
критического коэффициента приближается к 0,292. Следовательно, представ-
х) Закон соответственных состояний в квантовой механике рассматривается в гл. 6,
§ 6 (термодинамические свойства) и в гл. 10, § 4 (явления переноса).
2) Из A.2) следует, что
. RTc
— F (Vr, Tr)
Используя затем определение критической точки, мы получаем из этого соотношения два
других соотношения, включающих только V>, Tr, функцию F и ее первые две производные.
Эти уравнения представляют условия, налагаемые на функцию в точке A,1). Они удовле-
творяются, если указанная функция действительно представляет экспериментально полу-
ьенную сжимаемость.
§ 7. Закон соответственных состояний
199
ляется вероятным, что газ, состоящий из этих молекул, будет подчиняться
единому уравнению состояния в приведенных переменных.
В табл. 30 приведены также значения критического коэффициента для
некоторых углеводородов. Для этих веществ значение pcVc\RTc приближается
к 0,267. Следовательно, можно было бы предполагать, что углеводороды под-
чиняются единому приведенному уравнению состояния, отличному, однако,
Таблица 30
Фактор сжимаемости pV/RT в критической точке
Простые почти сферические неполярные молекулы
Вещество
Не
н2
Ne
Аг
PcVJRTc 1
1
0,300
0,304
0,296
0,291
Вещество
)
Хе
N2
о2
сн4
со2
PeVelRTc
0,293
0,292
0,292
0,290
0,287
Углеводороды
Вещество
Этан
Пропан
Изобутан
п-Бутан
Изопентан
п-Пентан
п-Гексан
п-Гептан
п-Октан
PeVcIRT, 1
0,267
0,270
0,276
0,257
0,268
0,266
0,260
0,258
0,258
Вещество
Бензол
Циклогексан
Диизопропил
Диизобутил
Этиловый эфир
Этилен
Пропилен
Ацетилен
PeVJRTe
0,265
0,276
0,266
0,262
0,262
0,291
0,273
0,275
Полярные молекулы
Вещество
CH3CN
Н2О
NH3
СН3ОН
CH3CI
С2Н5С1
PcVc/RTc
0,181
0,224
0,238
0,220
0,258
0,269
V в а» К8 j
1,2
1,2
1,0
0,8
0,6
0,2
от уравнения для сферических молекул. Большинство молекул углеводородов
имеет форму, близкую к эллипсоиду. Камерлинг Оннес уже давно обнаружил,
что вещества, состоящие из молекул некоторой одинаковой формы, будут
обладать одинаковыми объемными свойствами. Он назвал это принципом меха-
нического подобия.
Некоторые полярные газы также включены в табл. 30. Вместе с величиной
критического коэффициента для них дано значение величины t* = (^2/ea3f 8),
являющейся характеристическим параметром для полярных молекул
(см.гл. 3, § 10). Можно видеть,что критический коэффициент изменяется от 0,269
до 0,181, тогда как параметр f* увеличивается. Поэтому очевидно, что единый
200
Гл. 4. Уравнение состояния плотных газов и жидкостей
закон соответственных состояний можно применять ко всем веществам, неза-
висимо от формы, массы и дипольного момента молекул, только в весьма при-
ближенном смысле.
Из закона соответственных состояний, записанного в виде A.2), следует
Тг). A.5)
Поскольку подобное выражение предполагает, что критический коэффициент
(pcVJRTc) есть универсальная постоянная, то отсюда следует, что рг — универ-
сальная функция Vr и Тг:
pr = G(VnTr). A.6)
Вид этой зависимости показан на фиг. 39.
Закон соответственных состояний дает удобное, но приближенное сред-
ство для определения свойств плотного газа или жидкости. Единственно необ-
ходимым для нас является знание величин двух критических постоянных рас-
сматриваемого вещества. Критический объем
чрезвычайно трудно измерять даже с неболь-
шой точностью, поэтому обычно удобнее поль-
зоваться выражением для сжимаемости через
рг и Тг A.3), нежели через VT и Тг A.2). Зна-
чения критических постоянных могут быть, в
свою очередь, получены с помощью более
доступных величин, таких, как точка кипе-
ния, точка плавления, парахор и плотность
жидкости [1 ]. Следовательно, для получения
уравнения состояния, описывающего поведение
вещества с точностью 10—15%, требуется
очень немного данных.
Опытным путем найдено, что1)
Шидкая
фаза
[3,4],
A.7)
vr=v/vf
иг. 39. Характерное расположени
изотерм.
Vlb = 0,376 Vc
V°m = 0,321 Vc
[5-7], A.8)
[8-10], A.9)
[8-10]. A.10)
Первое из этих соотношений, известное как правило Гульдберга (и предло-
женное независимо Гюи), устанавливает, что при атмосферном давлении тем-
пература кипения по абсолютной шкале равна приблизительно 2/8 значения
критической температуры. В действительности из закона соответственных
состояний следует, что при фиксированном приведенном давлении (т. е. при
некотором определенном значении доли критического давления) точка кипения
во всех случаях является одинаковой частью критической температуры. Таким
образом, сравнение различных значений точек кипения при атмосферном дав-
лении, а не при некотором частном значении приведенного давления р\рс
может быть оправдано только в том отношении, что критические давления
большинства веществ имеют одинаковый порядок величины, а точка кипения
(по абсолютной шкале) относительно мало чувствительна к изменению дав-
ления. Для гомологических рядов наблюдается рост величины отношения
ТЬ\ТС с увеличением молекулярного веса ; это можно ожидать в силу соот-
*) Подробное обсуждение эмпирических соотношений такого типа проведено Партит-
тоном [2].
§ 7. Закон соответственных состояний 201
ветствующей тенденции критического давления. Однако нельзя предполагать,
что аналогичные соотношения могут иметь место с достаточной степенью точ-
ности и для точки плавления по следующим причинам : некоторые молекулы
вращаются в жидкости, но не вращаются в кристалле. Иногда процесс плав-
ления связан с изменением структуры решетки (числа ближайших соседних
молекул и типа симметрии). Кроме того, квантовые эффекты могут быть важны
в кристалле и несущественны в жидкости. По этим причинам плавление как
таковое может быть совершенно различным для разных веществ, и закон соот-
ветственных состояний в этом случае может оказаться неприменимым. Тем
не менее приведенные выше соотношения могут быть использованы для гпубых
оценок.
Б. ОБОБЩЕННЫЕ ДИАГРАММЫ СЖИМАЕМОСТИ
И ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ ПЛОТНЫХ ГАЗОВ
И ЖИДКОСТИ (ПО ХОГЕНУ И ВАТСОНУ)
Практическое значение закона соответственных состояний возросло после
опубликования Хогеном и Ватсоном[1,11] диаграмм обобщенной сжимаемости
и термодинамических свойств газов и жидкостей1). Эти величины выражаются
в виде функций от приведенного давления pr = pjpc для различных значений
приведенной температуры Tr = TjTc. Диаграммы были получены путем усред-
нения значений для следующих семи газов : Н2, N2, C02, NH3, CH4, С3Н8г
С5Н12. Поскольку эта группа состоит из различных типов молекул, то нельзя
ожидать, что результирующая кривая будет достаточно точно давать значения
рассматриваемых величин для каждого из этих газов. Тем не менее эти диа-
граммы весьма полезны для проведения приближенных расчетов вследствие
быстроты и легкости, с которыми могут быть получены необходимые резуль-
таты. Для более полного знакомства с диаграммами следует обратиться к пер-
воисточнику [1 ]. Эти диаграммы даны на фиг. 40—43. На практике расчеты
лучше всего проводить с помощью более полных диаграмм, опубликованных
отдельно [1, 11, 12]. Они дают обобщенный фактор сжимаемости и поправки
на неидеальность для энтальпии, энтропии и теплоемкости плотных газов.
Диаграммы, на которых даны зависящие от давления поправки к различным
термодинамическим величинам, получены с помощью обобщенного фактора
сжимаемости вместе с обычными термодинамическими соотношениями (см.
приложение Б к гл. 3). Величины с индексом 0 относятся к идеальным газам
(т. е. к значениям в условиях предельно низких давлений), а величины без
индекса — к реальным газам. Другие диаграммы Хогена и Ватсона дают сле-
дующие величины :
Газы: летучесть, вязкость.
Жидкости: поправки для энтальпии, энтропии, теплоемкости, коэффи-
циент теплового расширения и фактор сжимаемости.
Указанные диаграммы часто используются в задачах, связанных со сме-
сями. Установлено, что с достаточно хорошим приближением смесь ведет себя
как одно вещество с критическими постоянными, усредненными в соответствии
с молярными концентрациями компонент,
A.11)
A.12)
Здесь X/ — молярная доля i-й хи шческой компоненты. Однако такое рассмо-
трение смесей возможно только в парообразной фазе и неприменимо внутри
или вблизи пограничной кривой для смеси.
г) Обширные диаграммы соответственных состояний для легких, средних и тяжелых
углеводородов, подобные диаграммам Хогена и Ватсона, были даны Максвеллом [12].
2,0
I,U
0,8
0,6
0,5
0,3
0,2
01
Щ
1
¦
0,10 ^
i
- 1
——
^^
о&Г*
*ч
с
Ш
ч
ч
|
ч:
ч
5
ч
ч
\1
09
¦9
=
V
i
ч,
?5
ч
\
\
\
===
- ¦¦
4S
\
\ \ \
\\ \ \
\\ \ >
|\ '
I— -I
=
—i
ч
\
-1,0
--пр
uet
=
ч
ч,
?de
¦•».
ч
/;
/
ш
[—2,0
I
1,2.
1 74 >
шят
—--•
^г
^Л7/7
ера
—-¦
—
V
777I/
—-¦
.—-
^/
=
/
Е =
^ i»j
/
0
и
с
1риведенная тем
9-
*•*
9
G50^s
в —
7
—(
III'
/^
А-
^*»
.«—
»—¦
——
-——
0
ч^
— —
\
— —
ч.
Ч
¦—¦—
"Ч,
>>
1
0 0,1 0,2 0,3 0,4
Интервал низких давлении, рг
20 25 30 40 50
0,1 0,2 0,25 Q3 Ofl 0,5 0,6 0,8 1,0 2 3 4 5 6 7 8910
Приведенное давление, рг
Фиг. 40. Фактор сжимаемости как функция приведенного давления и приведенной температуры [1 ].
10
8
6
5
4
о*
•О
i
га:
0,8
¦0,6
. 0,5
0,4
аз
0,2
0,1
/
А
у
у
/
/ /
у
у
у
у
/
/
/
у
у
/
/
/
A
\J
/
/
/
У
у
у
У
у
/
7
f у
у
" У
/
/
у
у
А
/
/
/
у
У
/
к
у
А
/
у
/
/
/
А
у
у
у
у
/
/
/
/
/
У
/
aS
у
\
/
/
у
У
/
/
/
/
у
гу
/
/
у
у
у
у
У4
/у
у
/
/
/
/
у
А
/ f
/у
/
у
А
*
А
/
у
*
у
А
л
/
/
/
/
/
У
у
у
у
У/
у
у
/у
У/
-л
/
у
у
у
У/
у
у
у
Уу
/
у
у
/
у
V,
/
К
/
У
у^
/
/
у
/
у
у
у^
/
/
/
i
/
>
у
/
/
/
/
у
/
у
//
уу
у i
/у
'/у
уу
у у
У4
//
/
f
у
/
л
//
У.
у
/
У/
у/
Уу
у
У
//
/
/
~у_
у
А
У
у
у
у
Уу
у
'у
V
/
/
У
у/
у
/(
/
у
У
/
у
у
/,
у
/
/
rs^J
/
/
/
г
у
у
~У
У
у
у
У
/
/
/
/\
/
ft
/
у \
/<
у
у
у\
/а
/
у
1
/
j
' /
' /
у
/
V
Ф А
/
у
л л
у
/
/
/
/
V
/
/
/
7
/
У/
у
у
у
У
1
/
/
ff
/
/
/
/
/
у
у
г
У
у
у
у
(
1
f
/
/
/
/
у
у
у
у
у*
у
/
и
\
'/
У /
/
л
у
У
у_
у
у
у
У
/
/
*
У
V
у .
'У
у\
у
у
У
у
Уу
^ У
-f
/
/
у
у
у
у
у
J
у
у
С
~у_
у
у
у
у
у
у
у
у
у
у
/
/
/
у
У
у
/
у
у
у
у
I
1
*0*У
А
У
А
А
~у
у
у
Ж/
у
/у
^у
уу
У
уУ
А
у
< у
у
УЛ
у
**-
у
>
у
У
У\ л
<\
у
У
'\
у
у4
у
У
у
У
у
у
>
^у
>
^-*
у
у ^
у1
у ^
^^
¦ вва
f^
я* ——
T-=i - Приведенная
г Тс температуре
р =1L - Приведенное
Рс давление
т Критическая
'с температур
р Критическое
Гс давление
Д°- Энтальпия идеаль
*, ного газа
И — Энтальпия реаль-
ного газа
7
1
0,01 0,02 0,03 0.04 0.06 0,08 0,1
3 4 5 6 8
Фиг. 41. Поправка на неидеальность для
0,2 0,3 0,4 0,5 0,6 0,8 1,0 2
Приведенное давление, рГ
энтальпии как функция приведенного давления и приведенной температуры
70
[1 ].
/0
8
6
5
4
3
сз
^ Up
о Ofi
о 05
0,3
0,2
ni
AT
J
у
'-
у
У
у
/
/
у
f
У
А
г
у
/
/
W
У
У
у
/
у
у
/
у
/
/
/
/
/
у
У
у
А
у
А
у
у
А
А
у
у
у
у
А
у
У,
у
у
у
У
у
у
у
А
А
у
J
/*
л
/
/
/
у
/
/
/
J
/
/
/
/
А
г
J
2_
>
у
г
У
л
У
/
у
J
J
J
/
/
/
У
г
У
У
у
У
у
у
J
1
Тс "
Рс -
5°-
/
/
у
А
г
у
у
у
/л
'/<
_ <
/
/
/
/
/
/
У
г
с
0
'с
/
У
>
/
/
/
у
/
/
/
i i i
- Приведенная
температура
_ Приведенное
давление
Критическая
температура
Критическое
давление
Энтропия идеального
газа
Энтропия реального
газа
1—
°-<о
Л*
у
у
У
у
А
у
У
у
у
А
у
/
У
У
у
у
//
/
/
7
У
у
у
у
у
А
г
У
/
/
г
7
у.
у
?
2
У
/у
у_
у
у
/
У
/
/
у
А
/
У
У
у
J
у
Уа
У/,
//
У
у
'у
У у
/
У
/
/
/
/
у
/у
у
у
'у
у
(
J
f
у
/
/
у
У
у
/
А
у
у
у
\/
Уу
>
/
У
/
у*
J
У,
/
/
у
у
/
/
J
/
У
/
у
у1
\/
/
/
У
у
/
/
/
/
/
у
>
f
/
/
/
у'
/
/
У
t
/
1
/
/
А
/
у
У*
/
/
/
Г А
1
1
/
J
/
/
* /
ф У
1У
/
у
f
У/
/ У
/
А
'/,
-к
\j
1
J
/
J
/
/t
~y_
A
A
f
у
У
у
1
/
г
/
/
/
у
у
/
/
У
У,
'А
г
У
/
Г
/ /
/
1
г
у
у_
+1
у
2
у
у
/
/
у
У
у
\ /
/
/
/
\7
у
Z
у
у
у
у
у
Уу
у
Уу
{ /
'у
'у
//,
У
1
/
I
у
У
/
А
/
' у
/
/
Ух
у
у
/
/
у
у
у
у
у
/
/
у
у
/
г
/
~у
у
у
у
у
/
У
/
У
* j
/
у
у
у
у
у
?
у*
у
/
у
у
А
'у
у
А
Уу
уу
у
у
А
У/
'А
^^
у
у
у
у
/,
у
у
у
/у
'у
у
у
у
у
у
УЛ
у
у
У
у
у
у
у
У
у
у
у
у
у
А
Лу
У
1
¦1,0-
¦1,1-
¦1,2-
-/
,3'
,f
,6
W-
',2-
'¦4':
>
л*,
'Q01 0,02 0,03 0,04 0,06 0,08 0,1 0,2 0,3 0,4 0,5 0,6 0,8 1.0 2 3 4 5 6
Приведенное давление, рг
Фиг. 42. Поправка на неидеальность для энтропии как.функция приведенного давления и приведенной температуры [1].
§ 1. Закон соответственных состояний
205
'0,01 0,02 0,030,04 0,060,080,1 0,2 0,3 0,4 0,6 0,810
Приведенное давление, рг
2 3 4 5 67
Ф и г. 43. Поправка на неидеальность для теплоемкости как функция приведенного
давления и приведенной температуры [1 ].
В. ТЕОРЕТИЧЕСКИЙ ВЫВОД ЗАКОНА
СООТВЕТСТВЕННЫХ СОСТОЯНИЙ ДЛЯ ГАЗОВ,
СОСТОЯЩИХ ИЗ СФЕРИЧЕСКИХ НЕПОЛЯРНЫХ МОЛЕКУЛ
Существование зависимостей (связывающих соответственные состояния)
для газов, состоящих из сферических неполярных молекул, может быть пока-
зано и теоретическим путем. Для таких газов потенциальная энергия системы
предполагается состоящей из суммы членов ср(г0), каждый из которых пред-
ставляет энергию взаимодействия между парой молекул. Кроме того, вполне
приемлемо предположить, что потенциал <р(г) можно представить в виде неко-
торой универсальной функции /, причем в выражение для потенциала войдут
две скалярные величины а и е, характеризующие молекулу:
A.13)
Потенциал Леннарда-Джонса q>(r) = 4е [(с/гI2 — (tf/rN] имеет две постоянные
и является некоторым приближением к универсальной функции.
В соответствии с соотношением A.1) гл. 3, уравнение состояния газа (в
котором движение молекул подчиняется законам классической механики)
206
Гл. 4. Уравнение состояния плотных газов и жидкостей
может быть записано в виде
A.14)
Следовательно, давление зависит только от кТ, объема, двух скалярных
величин ей а, входящих в потенциальную функцию, и вида универсальной
функции /. Величины е и а можно использовать для определения приведен-
ных переменных1)
т* =
кТ
е
A.15)
Отсюда, а также из анализа размерностей следует, что приведенное дав-
ление можно представить как некую универсальную функцию приведенного
объема и приведенной температуры :
р* а p*(V*, Т*).
A.16)
Вид этой функции зависит только от вида потенциальной функции /.
Из такой записи закона соответственных состояний и определения крити-
ческой точки следует, что критические постоянные р*, V* и Г*, выраженные
через молекулярные постоянные, являются универсальными постоянными.
Значения, приведенные в табл. 31, однако, показывают, что существует зна-
чительный разброс по величине. Отклонения значений для Не и Н2 объясня-
ются влиянием квантовых эффектов, рассматриваемых в гл. 6, § 6.
Таблица 31
Критические постоянные некоторых почти сферических полярных молекул,
полученные с помощью силовых постоянных потенциала F-12) Леннарда-Джонса
Газ
Не
н2
Ne
Аг
Хе
N2
о2
сн4
тс,°к
5,3
33,3
44,5
151
289,81
126,1
154,4
190,7
Ve, CM*
57,8
65,0
41,7
75,2
120,2
90,1
74,4
99,0
Ре»
атпм
2,26
12,8
25,9
48
57,89
33,5
49,7
45,8
т:
0,52
0,90
1,25
1,26
1,31
1,33
1,31
1,29
V*
2,75
2,05
1,59
1,51
1,38
1,41
1,28
1,41
К
5,75
4,30
3,33
3,16
2,90
2,96
2,69
2,96
р.*
0,057
0,134
0,232
0,243
0,276
0,274
0,297
0,264
Р:
0,027
0,064
0,111
0,116
0,132
0,131
0,142
0,126
PcVJRT*
0,300
0,304
0,296
0,291
0,293
0,292
0,292
0,290
Приближение, при котором р*, V* и Т* считаются универсальными по-
стоянными, приводит непосредственно к простым методам оценки значений
величин, входящих в выражения для межмолекулярных сил. Таким образом,
для сферических неполярных молекул, подчиняющихся потенциалу F-12)
Леннарда-Джонса, получаются приближенные соотношения
-f = 0,77 7c
Ьо = 0,75 Vc= 18,4 -?.
A.17)
A.18)
Рассмотренные выше соотношения между значениями Т и V в точке плавления,
д) Величины р* = pbJNe и V* = V/b0 также могут быть использованы для определения
приведенного давления и приведенного объема.
§ 7. Закон соответственных состояний
207
точке кипения при нормальном давлении и в критической точке приводят к
следующим приближенным зависимостям:
A.19)
A.20)
A.21)
A.22)
A.23)
sm.
bo = 2,3Vs
Экспериментально обнаружено, что
Ьо =2,293 V8,,
где Vq — молярный объем вещества в твердом состоянии при 0° К. Из таблицы
для второго вириального коэффициента (табл. II, стр. 854) видно, что [13]
= 0,292 7Б,
A.24)
где Тб — температура Бойля. Эти выражения дают простой метод для оценки,
межмолекулярных потенциалов. Для получения результатов большей точ-
ности, относящихся к какому-либо отдельному типу молекул, можно приме-
нить закон соответственных состояний к молекулам, обладающим аналогичной
0,6
0.5
-Аргон [14]
-Кислород [/5,16]
-Азот [11]
[18]
10
15 20 25
V*=V/b0
_1_
30 35
Фиг. 44. Сжимаемость некоторых газов, состоящих из сферических неполярных молекул,
как функция приведенного объема V* для отдельных значений приведенной температуры Г*.
208 Гл. 4. Уравнение состояния плотных газов и жидкостей
структурой. Результаты таких вычислений должны рассматриваться, однако,
в качестве грубых оценок и использоваться только в том случае, когда более
точные методы невозможны.
Из закона соответственных состояний, записанного в виде A.16), с очевид-
ностью следует, что фактор сжимаемости является универсальной функцией
V* и Т*, т. е.
?ф$ A.25)
Результаты экспериментальной проверки этих соотношений приведены
на фиг. 44. Поскольку приведенные параметры в критической точке V* и Т*
имеют всегда одинаковые значения, то из этого выражения непосредственно
следует эмпирический закон соответственных состояний A.2), так же как и
формула A.3).
Г. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ
ДЛЯ МНОГОАТОМНЫХ МОЛЕКУЛ
Для иллюстрации способа обобщения закона соответственных состояний
на случай многоатомных молекул можно дать несколько отдельных примеров.
В этих примерах для описания внутренней структуры молекул вводятся до-
полнительные параметры. Они появляются в выражении для приведенного
уравнения состояния и связаны с идеей механического подобия Камерлинг
Оннеса.
В гл. 3, § 8 рассматривалась четырехцентровая модель цилиндрических
молекул Корнера. В этой модели предполагается, что длинные (сигарообразные)
молекулы могут быть представлены как четыре силовых центра, расположен-
ных вдоль одной линии. Расстояние между центрами сил в молекуле опреде-
ляется значением /. Суммарное значение межмолекулярной силы определяется
результатом взаимодействий между четырьмя центрами, причем каждое взаи-
модействие следует выражению A.13) с параметрами е и а. Из вышеприведен-
ных рассуждений ясно, что уравнение состояния для такой модели имеет вид
p=p(V,kTye,oyl). A.26)
Введение приведенных переменных, данных в A.15), и приведенного расстоя-
ния /* = Цо дает выражение
Р* = Р*(У*,Г*;/*). A.27)
Следовательно, для цилиндрических молекул с одинаковым значением отно-
шения /¦ приведенное давление является универсальной функцией V* и Т*.
«Круглые» многоатомные молекулы типа ХУп (такие, как NH3, CBr4, SF6)
можно представить в виде жесткой системы, состоящей из п идентичных сило-
вых центров, расположенных на одинаковом растоянии а от центра молекулы.
Тогда, так же как и в модели Корнера, можно предполагать, что энергия
взаимодействия между двумя молекулами1) складывается из суммы взаимодей-
ствий между указанными силовыми центрами (центральный атом в этих взаи-
модействиях не учитывается). Предполагается, что потенциал, описывающий
взаимодействие между силовыми центрами, представлен в виде A.13). Тогда
уравнение состояния такой модели запишется в виде
kTye,oyayn). A.28)
Вводя приведенное расстояние а* = а\о и приведенные переменные, заданные
г) Возражения, возникающие при введении предположения о существовании взаимо-
действия указанного типа, указаны в примечании на стр. 164.
§ 7. Закон соответственных состояний
209
в A.15), из соображений размерности получаем
р* = р*( V*, Т*, а*, л). A.29)
Это — другой пример более обобщенного вида связей между соответствен-
ными состояниями. Уравнение A.29) можно также получить, подставив выра-
жение для межмолекулярного потенциала в классический интеграл состояний
[19].
Д. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ
ДЛЯ ПОЛЯРНЫХ МОЛЕКУЛ
Простое соотношение, связывающее соответственные состояния, можно
получить и для полярных молекул, если предположить, что потенциал взаи-
модействия имеет вид
Ж) = */ (~) - 4- g{K 02, Фг—&), О -3°)
где /(г/а) — универсальная функция приведенного расстояния г\о, а выражение
g@i> #2> Ф% — Фл) описывает угловую зависимость взаимодействия между двумя
а 0,4
g1
| I 0,2
. . . . 1
г
. . , , 1
о
о
' 1 '
¦
1 1 I
1 ' 1
0,5 г Ю
, . . . I i . . i • . .
Неполярные газы Полярные газы
• Аргон в Клористый этил ¦ Аммиак
^ Неон а Хлористый метил и Вода
О Азот в Ацетон и Ацетонитрил
е Водород а Уксусный альдегид
Фиг. 45. Приведенные критические постоянные полярных газов.
Даны изменения различных приведенных критических величин в зависимости от параметра t* = a«*b/V8.
а — отношение peVJRTe для ряда полярных газов ; б — р* = Pebo/Ne; в — kTeje = Т* ; г — значения
приведенного критического объема V^ = Ve/fee. Все эти величины изменяются (не сильно) с изменением
параметра f*. Силовые постоянные, использованные для приведения критических величин, получены для
потенциала Штокмейера из экспериментальных значений второго вириального коэффициента [20]. Когда
значения для неполярных газов лежат слишком близко друг к другу, приведено значение только для аргона.
точечными диполями. Уравнение состояния вещества, составленного из мо-
лекул, подчиняющихся такому типу потенциала, записывается в виде
tfT9e,o,p). A.31)
210 Гл. 4. Уравнение состояния плотных газов и жидкостей
Скалярные величины е и а можно использовать для определения приведенных
переменных так же, как это сделано в A.15), и ввести приведенный дипольный
момент /г* = /г/ У еа3. Тогда с помощью анализа размерностей приходим к
соотношению между приведенными давлением, объемом и температурой
p*=p*(V»,T* ;/*•). A.32)
Это объясняет тот факт, что различные равновесные свойства полярных ве-
ществ, записанные в безразмерных величинах, дают систематические откло-
нения от свойств неполярных веществ в соответствии со значением /г*, харак-
теризующим это вещество. В гл. 3 достаточно отчетливо показана зависимость
вириальных коэффициентов от /г* для молекул, подчиняющихся потенциалу
Штокмайера. Аналогичная картина наблюдается для кривой насыщающих
паров, линии плавления и критических параметров. Изменение значения при-
веденных критических постоянных в зависимости от значения приведенного
дипольного момента показано на фиг. 45 [20 J1).
§ 2. ЭМПИРИЧЕСКИЕ УРАВНЕНИЯ СОСТОЯНИЯ
В качестве уравнений состояния газов и жидкостей предложены много-
численные эмпирические соотношения, каждое из которых предназначено для
применения в том или другом специальном случае2). Некоторые из этих урав-
нений достаточно точны лишь в узкой области значений температуры и плот-
ности, другие применимы к описанию как газообразной, так и жидкой фазы.
Вначале рассматриваются уравнения состояния с двумя постоянными (урав-
нения Ван дер Ваальса, Дитеричи и Вертело). Эти уравнения очень удобны,
так как имеют простой аналитический вид. Более громоздкими, но значительно
более подходящими для точных расчетов являются уравнения Битти—Бридж-
мена (с пятью произвольными постоянными) и Бенедикта—Вебба—Рубина
(с восьмью параметрами). Параграф заканчивается обзором полуэмпирических
уравнений, которые используются для описания жидкостей.
А. УРАВНЕНИЯ СОСТОЯНИЯ С ДВУМЯ ПОСТОЯННЫМИ
Для полуколичественного рассмотрения основных особенностей урав-
нения состояния используются следующие уравнения:
Ван дер Ваальса
(р + -fr) <? -b) = RT- <2Л>
Вертело
[P+^-)(9-b) = RT> B-2>
Дитеричи
B.3)
Постоянная а является мерой сил сцепления между молекулами, постоянная Ь
пропорциональна объему молекул. Все эти уравнения позволяют предсказать
*) В качестве дополнительной литературы, посвященной рассмотренным в этом
параграфе вопросам, можно указать книгу A. L. Lydersen, R. A. Greenkorn,
О. А. Н о u g e n, Generalised Thermodynamic Properties of Pure Fluids, Wisconsin, 1955,
а также работу J. О. H i r s с h f e 1 d e r, R. J. В u с h 1 e y, H. A. M с С e e, J. R. S u 11 о п„
Industr. and Eng. Chem., 53, 386 A958). — Прим. ред.
2) Хороший обзор эмпирических уравнений состояния можно найти у Тэйлора и Глее-
стона [21], а также у Партингтона [2]. Весьма полная сводка уравнений состояния дана в
книге М. П. В у к а л о в и ч, И. И. Новиков, Уравнение состояния реальных газов,,
М, 1948. — Прим. ред.
§ 2. Эмпирические уравнения состояния
211
температуру Бойля и критическую изотерму. Ни одно из этих уравнений не
дает значения максимума второго вириального коэффициента как функции тем-
пературы; в первых двух уравнениях третий вириальный коэффициент не
Таблица 32
Значения ряда величин для уравнений состояния, включающих
две постоянные
Величины
Ре
Vc
Тс
PcVclRTc
В(Т)
С(Т)
Уравнения
Ван дер Ваальса
Вертело
Дитеричи
27 Ь%
ЪЪ
8а
27ЬЯ
0,375
а
b-
RT
Ь*
216 б8
3b
27bR
0,375
a
~~RT*
29,56 6*
2b
a
4bR
0,2706
RT
ь*
RT
а'
1.0
0,5
зависит от температуры. Критические постоянные и вириальные коэффициенты
для этих трех простых уравнений состояния собраны в табл. 32. Уравнение
Дитеричи дает значение pcVJRTa
которое находится в превосходном
согласии со средним значением этой
величины для 25 неполярных газов,
—0,272 [22]. Следовательно, уравне-
ние Дитеричи замечательно точно
выполняется вблизи критической
точки и является вообще наилучшим
уравнением состояния, зависящим от
двух постоянных.
Интересно сравнить значение вто- -0,5
рого вириального коэффициента, полу-
ченное из уравнений Ван дер Ваальса в
или Дитеричи, со значением, полу- -j
ченным с помощью потенциала Лен-
нарда-Джонса, который, как было
показано, достаточно точно описывает
поведение разреженного газа. В соот-
ветствии с уравнением Ван дер
Ваальса второй вириальный коэффи-
циент изменяется линейно по отно-
шению к ЦТ. Из фиг. 46 можно ви-
деть, что В(Т) для потенциала Лен-
нарда-Джонса значительно отклоня-
ется от прямой линии. Это поясняет
тот факт, почему постоянные а и Ь}
входящие в уравнение Ван дер Ваальса,
существенно зависят от температуры
или от того, каким образом они определены. На кривой для второго вириаль-
ного коэффициента, полученной из потенциала Леннарда-Джонса, наблю-
дается максимум. Он лежит в области между Г* = 10 и Т* t= 100; этот
-/,5-
0
Ф и г. 46. Зависимосгь В* от 1/7* для
потенциала Леннарда-Джонса.
212 Гл. 4. Уравнение состояния плотных газов и жидкостей
интервал температур весьма важен в горении и задачах внутренней баллис-
тики. В этой области второй вириальный коэффициент почти не зависит от
температуры и может быть приближенно положен равным Ьо/2.
Вводя небольшую поправку на неидеальность разреженного газа, Вертело
предложил следующее выражение для второго вириального коэффициента :
Это выражение широко используется [2] для определения молекулярного
веса по плотности газа и величины скрытой теплоты парообразования по дав-
лению паров ; в обоих случаях учитываются небольшие поправки на неидеаль-
ность газа. Использование выражения B.4) вполне оправдано, поскольку оно
дает хорошее согласие с температурной зависимостью В(Т), полученной из
потенциала Леннарда-Джонса в диапазоне между Г* = 0,3 и Т* = 2 [23].
Все три вышеприведенных уравнения с двумя постоянными могут при-
меняться к смесям. Параметры аи b изменяются в зависимости от состава
а = ап х\ + 2а12 хг х2 + а22 xi, B.5)
Ь = Ьц А + 2Ь12 хг х2 + Ь22 х\. B.6)
Величины xt — молярные доли компонент, а постоянные аи и ЬИ — просто по-
стоянные чистых компонент. Величины а12 и Ь12 являются постоянными, харак-
теризующими столкновения между разнородными молекулами. Если экспери-
ментальные данные по величине этих постоянных отсутствуют, то они могут
быть грубо оценены с помощью следующих эмпирических соотношений :
fe4fe feb B-7)
Эти соотношения соответствуют выражениям F.8) и F.9) гл. 3. Соотношения
B.5) и B.6) были впервые получены Ван дер Ваальсом [24]. В некоторых
случаях они достаточно точно подтверждаются экспериментальными резуль-
татами.
Б. УРАВНЕНИЕ СОСТОЯНИЯ БИТТИ—БРИДЖМЕНА1)
Уравнение Битти—Бриджмена является одним из лучших эмпирических
Представлений зависимости между р, V и Т для газов при давлениях до зна-
чений порядка 250 атм; оно имеет вид
^)() B.9)
Значения пяти эмпирических ^параметров Ао, Во, a, b и с определены для боль-
шого числа газов и приведены в табл. 33.
При очень больших давлениях уравнение Битти—Бриджмена выпол-
няется неудовлетворительно. Выполнимость уравнения при очень низких дав-
лениях можно исследовать, представив его в вириальной форме. Второй ви-
риальный коэффициент записывается в виде
В(Т) = В0--^-^. B.10)
Это выражение согласуется с выражением для второго вириального коэф-
фициента, полученного с помощью потенциала Леннарда-Джонса в широкой
области температур, что обеспечивается должным выбором постоянной с.
1) См. [25, 26].
§ 2. Эмпирические уравнения состояния
213
Таблица 33
Значения постоянных, входящих в уравнение состояния Битти—Бриджмена,
для различных газов [21]
р = [ЯГA — <5)/V«] [V + В] — A/V\
А - А,A — a/VO, В = Be(l — bfV), <* = с/ VT*
Размерность : атм, л/моль, °К
Газ
Не
Ne
Аг
Кг [28]
Хе [29]
н2
№
о2
Воздух
J2
со2
NH,
СН4
С2Н4
с2н.
с3н8
1-С4Н8
изо-С4Н8
л-С4Н10
изс-С4Н]0
п-С6Н12
нео-С6Н1а
л-С,Н1в
СН3ОН
(С2Н6JО
Л.
0,0216
0,2125
1,2907
2,4230
4,6715
0,1975
1,3445
1,4911
1,3012
17,0
5,0065
2,3930
2,2769
6,1520
5,8800
11,9200
16,6979
16,9600
17,7940
16,6037
28,2600
23,3300
54,520
33,309
31,278
а
0,05984
0,02196
0,02328
0,02865
0,03311
-0,00506
0,02617
0,02562
0,01931
0,0
0,07132
0,17031
0,01855
0,04964
0,05861
0,07321
0,41988
0,10860
0,12161
0,11171
0,15099
0,15174
0,20066
0,09246
0,12426
в9
0,01400
0,02060
0,03931
0,05261
0,07503
0,02096
0,05046
0,04624
0,04611
0,325
0,10476
0,03415
0,05587
0,12156
0,09400
0,18100
0,24046
0,24200
0,24620
0,23540
0,39400
0,33560
0,70816
0,60362
0,45446
ь
0,0
0,0
0,0
0,0
0,0
-0,04359
-0,00691
0,004208
-0,01101
0,0
0,07235
0,019112
-0,15870
0,03597
0,01915
0,04293
0,10690
0,08750 '
0,09423
0,07697
0,13960
0,13358
0,19179
0,09929
0,11954
с 10-*
0,0040
0,101
5,99
14,89
30,02
0,504
4,20
4,80
4,34
4000
66,00
476,87
12,83
22,68
90,00
120,00
300,00
250,00
350,00
300,00
400,00
400,00
400,00
32,03
33,33
Чтобы получить согласие в наиболее важной в физическом отношении
области температур, нами [27 ] выбрано следующее значение для с:
тогда
с = 0,0236 -j^j;
в = 0,04127 -jfe- -
bo= 1,249 Bo.
Ю-15,
B.11)
B.12)
B.13)
Первое из этих соотношений может быть использовано как критерий возмож-
ности применимости потенциала Леннарда-Джонса. В табл. 34 сравниваются
значения е и Ьо, рассчитанные по соотношениям B.12) и B.13), со значениями,
определенными непосредственно из второго вириала или результатов по вяз-
кости. В последних двух колонках значение с, рассчитанное по B.11), сравни-
вается с экспериментально наблюдаемым значением. Согласие получается
хорошее в тех случаях, когда другие обстоятельства указывают на примени-
мость в этом случае потенциала Леннарда-Джонса. По-видимому, это является
достаточно тонким критерием применимости потенциала Леннарда-Джонса.
Внутренняя энергия U является одной из наиболее важных термодинами-
ческих величин, связанных с уравнением состояния. Ее можно определить
214
Гл. 4. Уравнение состояния плотных газов и жидкостей
Таблица 34
Газ
Не
Ne
Аг
Н,
N2
02
Воздух
со2
NH3
СН4
С2Н4
сгн.
с,н8
1-С4Н8
ызо-С4Н8
л-С4Н10
ызо-С4Н10
л-с6н12
нео-С6Н12
п-СтН1в "
СН,ОН
Ь„,см
получено
из урав-
нения
Битти—
Бридж-
мена
17,49
25,73
49,10
26,18
63,02
57,75
57,59
405,9
130,85
42,65
69,78
151,8
117,4
226,1
300,3
302,3
307,5
294,0
492,1
419,2
884,5
753,9
567,6
Значения bQ e и
*/моль
получено
из потен-
циалаЛен-
нарда-
Джонса
21,17*
25,95
49,80
31,41*
63,78
57,75
55,11
113,9
70,16
78
226
155
884
8, 10-
получено
из уравне-
ния Битти-
Бридж-
мена
0,776
5,189
16,02
4,739
13,40
16,22
14,19
26,30
24,03
35,25
20,49
25,45
31,46
33,12
34,92
35,25
36,35
35,47
36,07
34,96
38,72
27,75
34,61
с [25,26]
16, эрг
получено
из потен-
циала
Лен-
нарда-
Джонса
1,402
4,928
16,529
5,075
13,12
16,22
13,69
26,09
20,46
33,54
33,40
40,99
38,92
с - 10~"*, л/град
получено из соот-
ношения B.11)
0,00022
0,0966
5,942
0,07488
4,077
6,625
4,425
198,2
48,84
50,19
16,15
67,30
98,26
220,8
343,9
355,7
397,0
352,8
620,9
481,5
1380
433200
632,7
• МОМ
наблю-
даемое
0,0040
0,101
5,99
0,0504
4,20
4,80
4,34
4000
66,00
476,87
12,83
22,68
90,00
120,00
300,00
250,00
350,00
300,00
400,00
400,00
400,00
32,03
33,33
•)Эти силовые постоянные были получены путем согласования экспериментальных результатов с тео-
ретическим выражением, учитывающим квантовые эффекты.
(с точностью до аддитивной функции температуры) из термодинамического соот-
ношения
Внутреннее «Тепловое» Внешнее
давление давление давление
Внутреннее давление CUldV)T является разностью между тепловым и внеш-
ним давлением. Интегрирование (BUjBV)T по объему (при постоянной темпера-
туре) дает значение внутренней энергии. Таким образом, для уравнений со-
стояния
Ван дер Ваальса
-4г, B-15)
Дитеричи1)
B.16)
l)Ei(x) — экспоненциальная интегральная функция.
§ 2. Эмпирические уравнения состояния
215
Битти—Бриджмена
о =
-пА-
-Z-w- <217)
На фиг. 47 проводится сравнение этих рассчитанных значений внутренней
энергии с экспериментальными значениями для аргона при 0° С. Постоянные,
ill i I i
Эксперимент _
ВандерВаальс
I I I I I I I I I I I I I
0 40 80120160200240280320360400440480520560600640-р,единицыАмага
">56ШJ80187140 112933801O0,062256/M0,946,74$40,037?3Sfl - V, СМЪ/тль
О 318 752 111 146 182 220 263 311367 435 519 622 753 91911281393-p, атм
Ф и г. 47. Сравнение экспериментальных зйачений внутренней энергии аргона при 0° С
с величинами, рассчитанными по эмпирическим уравнениям состояния.
входящие в уравнение Битти—Бриджмена, взяты из табл. 33. Постоянные а и Ь,
необходимые для использования в выражениях, полученных из уравнений Ван
дер Ваальса и Дитеричи, получаются, если положить Ъ — a/RT равным вто-
рому вириальному коэффициенту Битти—Бриджмена, a ajRT2 — произбодной
по температуре от второго вириального коэффициента Битти—Бриджмена.
Выражение Ван дер Ваальса для внутренней энергии оказалось неожиданно
точным. Выражение Битти—Бриджмена случайным образом отклоняется в
противоположном направлении. Выражение Дитеричи совершенно неудовле-
творительно при высоких значениях плотности, когда V становится меньше
или сравнимо с Ь. Это очевидно из B.16), так как Ei@) = — «>. Обычно выше
некоторого значения давления уравнение Битти—Бриджмена дает совершенно
ошибочные значения внутренней энергии. Например, для углекислого газа
при Т = 150° С уравнение Битти—Бриджмена плохо воспроизводит значения
внутренней энергии при плотностях, превышающих критическое значение
плотности. Это показано на фиг. 48.
216
Гл. 4. Уравнение состояния плотных газов и жидкостей
-500
4000
« -1500
1 -2000
I
^-2500
-3000
-3500
-4000
i—i—I—i—i—г
С02 при 150°С
Битти-Бриджмен
т—i—г
Эксперимент
Дитеричи
Ван дер Ваальс
О 40 80 120 160 200 240 280 320 360 400 440 480 520 560 600-р, единицы Амага
I I I I I I I I I I I I I I I I „
оо556,6278186139 111 92,8 79,569,661,855,7 50,6 46,4 42,8 39,8 37J-V, см3/моль
I I I I I I I I I I I I I I I I
О 511106,0149,9192 235 284 344423 531 686 908122716812320 3201-р} атм
Фиг. 48. Сравнение экспериментальных значений внутренней энергии С02 при 150° С
с величинами, рассчитанными по эмпирическим уравнениям состояния.
В. УРАВНЕНИЕ СОСТОЯНИЯ БЕНЕДИКТА—ВЕББА—РУБИНА
Бенедикт, Вебб и Рубин [30] провели обобщение уравнения Битти—Бридж-
мена как для чистых веществ, так и для смесей. Они нашли, что для описания
наблюдаемой зависимости между р, V и Т для углеводородов до значений
плотностей, вдвое превышающих критическую плотность, необходимо исполь-
зовать уравнение
B.18)
содержащее восемь параметров. Предполагается, что для смесей эти восемь
параметров примут вид
B.19)
§ 2. Эмпирические уравнения состояния
217
Величины xt — молярные доли различных компонент, индексы / и / при по-
стоянных Ло, Во, ... означают величины, относящиеся к чистым компонентам.
Таблица 35
Эмпирические постоянные уравнения Бенедикта—Вебба—Рубина [31]*
Размерность : атм, л/моль, °К; газовые постоянные : R — 0,08207, Г = 273,13 + f(°C)
Газ
Азот
Метан
Этилен
Этан
Пропилен
Пропан
шо-Бутан
изо-
Бутилен
н-Бутан
U30-
Пентан
н-Пентан
я-Гексан
н-Гептан
Ао
1,19250
Во
0,0458000
1,85500,0,0426000
3,33958 0,0556833
4,15556 0,0627724
с„ • ю-6
0,00588907
0,0225700
0,131140
0,179592
6,11220 0,0850647 0,439182
6,87225 0,0973130 0,508256
10,23264 0,137544
8,95325 0,116025
10,0847
12,7959
12,1794
14,4373
17,5206
0,124361
0,160053
0,156751
0,177813
0,199005
0,849943
0,927280
0,992830
1,74632
2,12121
3,31935
4,74574
а |
0,0149000
0,494000
0,259000
0,345160
0,774056
0,947700
1,93763
1,69270
1,88231
3,75620
4,07480
7,11671
10,36475
ь
0,00198154
0,00338004
0,0086000
0,0111220
0,0187059
0,0225000
0,0424352
0,0348156
0,0399983
0,0668120
0,0668120
0,109131
0,151954
е9 • 10—°
0,000548064
0,00254500
0,021120
0,0327670
0,102611
0,129000
0,286010
0,274920
0,316400
0,695000
0,824170
1,51276
2,47000
а- 108
0,291545
0,124359
0,178000
0,243389
0,455696
0,607175
1,07408
0,910889
1,10132
1,70000
1,81000
2,81086
4,35611
7- 10»
0,750000
0,60000
0,923000
1,18000
1,82900
2,20000
3,40000
2,95945
3,40000
4,63000
4,75000
6,66849
9,00000
* Значения постоянных для азота взяты из частного сообщения Бенедикта.
Значения постоянных для азота и некоторых углеводородов приведены в
табл. 35. Выражения для термодинамических величин, соответствующие урав-
нению состояния Бенедикта—Вебба—Рубина, записываются в виде
Летучесть:
RT In /, = RT In
± j-L
2c
(yi
(т.
Энтальпия:
ye
-y/v
y/V2
2V2
B.20)
~^'"w"e+^e"^
Энтропия:
B-22)
.218 Гл. 4. Уравнение состояния плотных газов и жидкостей
Эти соотношения справедливы для большого числа углеводородных смесей
з широкой области значений от жидкости до газа, включая критическую об-
ласть.
Г. ЭМПИРИЧЕСКИЕ ЗАВИСИМОСТИ ДЛЯ ЖИДКОСТЕЙ
Уравнение Тайта [32] и его видоизменение, сделанное Кирквудом, пред-
ставляют собой лучшие эмпирические выражения уравнения состояния
для жидкостей. Уравнение Тайта, предназначавшееся сначала для описания
сжимаемости воды, записывается в виде
dV к
~~dp ~ L + P '
где К — постоянная; L — функция температуры. В интегральной форме урав-
нение представляется как
здесь pQ — некоторое стандартное значение давления ; Vo — соответствующий
объем жидкости. В настоящее время нет еще достаточного теоретического
обоснования этого уравнения. Тем не менее изучение свойств большого числа
жидкостей [33,34] показывает, что уравнение B.24) дает почти полное согласие
с экспериментальными наблюдениями. Это уравнение оказывается примени-
мым к различным жидкостям (производные бензола, растворы солей и др.) и
годным в широкой области значений температуры и давления (до 1000 атм).
В условиях очень высоких давлений уравнение Тайта требует некоторого
видоизменения, так как левая часть выражения B.24) стремится к бесконеч-
ности, тогда как правая часть не может превышать значение п. Поэтому было
предложено следующее видоизменение этого уравнения [35, 36 ]:
In (Р±А<Щ -п\п
ln { A(S) ) - пln
В этом соотношении в качестве независимой переменной взята не температура
а энтропия ; приращение объема заменено логарифмом отношения величины
объема при нулевом давлении У@, S) к величине объема при некотором дав-
лении р. Ричардсон и др. [37 ] показали, что это соотношение дает хорошее
выражение для уравнения состояния воды до 25 000 атм.
Уравнение B.25) можно записать в виде, напоминающем выражение для
адиабаты идеального газа :
[p+A(S)]Vn = F(S), B.26)
где F(S) = A(S) [V@, S)]n. Тогда величину внутренней энергии можно полу-
чить из (ЭС//Э V)s = — р, которое после интегрирования принимает вид
)-V). B.27)
Первый член pV/(n — 1) является основным. Второй член дает поправку,
обусловленную приращением объема [У@, S)— V]. До тех пор пока послед-
ний член мал, ясно, что выражения B.27) и B.25) будут соответствовать экс-
периментальным результатам. Однако для больших изменений объема в выра-
жение для внутренней энергии необходимо ввести дополнительные члены,
пропорциональные более высоким степеням приращения объема. Интересно
отметить, что п равно отношению удельных теплоемкостей у только в случае
идеального газа, когда pV = RT и A(S) = 0.
Приближенной, но удобной формой записи уравнения адиабаты для
жидкости является
= F(S). B.28)
§ 3, Газы при очень высоком давлении 219
Это соответствует тому случаю, когда п = 3, а поправочный член, содержащий
A(S), считается пренебрежимо малым. Для этого уравнения как скорость
звука, так и римановы характеристики (см гл. 11, § 5) пропорциональны дав-
лению.
§ 3. ГАЗЫ ПРИ ОЧЕНЬ ВЫСОКОМ ДАВЛЕНИИ
В этом параграфе рассматриваются вопросы, связанные с изучением горе-
ния пороховых газов, детонации и других процессов, в которых участвует газ
при очень высоком давлении. Вначале рассматриваются некоторые эмпири-
ческие зависимости, используемые для описания термодинамических свойств
газов при очень высоком давлении. Затем мы рассмотрим изменение струк-
туры молекул при высоких давлениях. При этом для объяснения роста кине-
тической энергии электронов при большом сжатии привлекается теорема о
вириале.
А. УРАВНЕНИЕ СОСТОЯНИЯ ПОРОХОЕЫХ ГАЗОВ
Для газа при умеренно высоких давлениях можно рекомендовать [27, 38 J
уравнение
pV _ . В(Т± О&ЯЬ* , 0,2869 6» , 0,1928b\ /Q n
где В(Т) — второй вириальный коэффициент; Ьо — обычная постоянная Ван
дер Ваальса. Третий и четвертый вириальные коэффициенты справедливы
для твердых сфер с объемом, равным fto/47V. Пятый вириальный коэффициент
выбран таким, чтобы согласовать величину сжимаемости с уравнением Эйринга
E.3) в диапазоне плотностей, соответствующих жидкому состоянию.
Для пороховых газов при температуре около 2500° С второй вириальный
коэффициент почти не зависит от температуры. Это можно видеть из фиг. 46
или из значений В(Т) для потенциала Леннарда-Джонса, приведенных в
табл. II (стр. 854). Значение В(Т) очень мало изменяется в диапазоне между
Г* = кТ\г = 10 и Т*= 100. Максимальное значение В(Т), равное 0,527 Ьо, дости-
гается вблизи Г* = 30, что соответствует высокой температуре, наблюдаемой
при горении пороховых газов. Таким образом, в этих задачах В(Т) полагается
равным bf а значение Ь выбирается равным 0,527 Ьо. Значение Ьо для смеси поро-
хового газа можно оценить с помощью правила сложения (простого, но не-
достаточно точного)
0Л2(Ь& C.2)
где Х( — молярная доля i-й компоненты; (ЬД — значение Ьо, вычисленное по
потенциалу Леннарда-Джонса для отдельных компонент. Вследствие наличия
максимума второго вириального коэффициента в этом интервале температур
вполне успешно решаются задачи внутренней баллистики, в которых моле-
кулы рассматриваются как твердые сферы с описанным выше собственным
объемом.
Б. УРАВНЕНИЕ СОСТОЯНИЯ ГАЗА ПРИ ДЕТОНАЦИИ
В задачах по детонации, когда давление может достигать 200000 атм,
может показаться, что наиболее точным является уравнение Леннарда-Джонса
и Девоншайра (см. § 7). Однако в этих случаях большое преимущество имеет
использование уравнения Халфорда—Кистиаковского—Вильсона1)
C.3)
*) Это видоизменение уравнения Беккера было сформулировано Халфордом и изло-
жено . в работе [39 ] (см.. также [40 ]).
220 Гл. 4. Уравнение состояния плотных газов и жидкостей
Значение постоянной К определяется из выражения
K = ?ntKi9 C.4)
где щ — число молей i-й компоненты газа в 1 см3 смеси; К( — эмпирические
постоянные, характеризующие каждую из химических компонент и в 5,5 раза
превышающие значения Ь0У полученные с помощью потенциала Леннарда-
Джонса для рассматриваемой компоненты. Для воды и аммиака К, равны 108
и 164 см3/моль соответственно. Успешное применение этого уравнения в боль-
шой мере обязано тому, что описание большинства детонационных явлений
мало зависит от вида уравнения состояния.
|В. ИСПОЛЬЗОВАНИЕ ТЕОРЕМЫ О ВИРИАЛЕ ДЛЯ ИЗУЧЕНИЯ
ДЕФОРМАЦИЙ МОЛЕКУЛ В УСЛОВИЯХ ЭКСТРЕМАЛЬНО ВЫСОКИХ ДАВЛЕНИЙ
Когда вещество подвергается действию экстремально высоких давлений,
строение молекул претерпевает изменения : электронные уровни энергии сме-
щаются, а химические связи ослабевают или разрушаются, существенные изме-
нения, происходящие в свойствах вещества под действием высоких давлений,
могут быть проиллюстрированы на примере электропроводности : при низком
давлении газ является изолятором, при повышенном — превращается в полу-
проводник и, наконец, при экстремально высоком давлении он становится
металлическим проводником. В настоящее время обнаружено, что такое метал-
лическое состояние достигается при давлении порядка 1 000 000 атм в зави-
симости от вещества. Необходимо провести еще огромное число эксперимен-
тальных исследований в области очень высоких давлений. Многие интересные
результаты могут быть предсказаны. Например, некоторые химические реакции,
происходящие при атмосферном давлении лишь в условиях чрезвычайно высо-
кой температуры, могут протекать при высоком давлении и при комнатной
температуре.
При увеличении давления квантовомеханические уровни молекул сдви-
гаются и кинетическая энергия системы возрастает. Это явление можно объя-
снить с помощью теоремы о вириале. Величину изменения внутренней энергии
и изменения кинетической энергии системы можно выразить через эмпириче-
ское уравнение состояния. Наиболее непосредственную информацию могла
бы дать абсорбционная спектроскопия. Однако в настоящее время в этом на-
правлении еще нет экспериментальных результатов1). В принципе изменение
диэлектрической постоянной или показателя преломления могло бы указать
на степень сжатия молекул, однако в настоящий момент теоретическое пони-
мание этих свойств (см. гл. 12, § 2) еще недостаточно хорошо развито.
В гл. 3, § 1 теорема о вириале используется для вывода формального вы-
ражения уравнения состояния через межмолекулярные силы. В этом выводе
предполагается, что газ состоит из молекул ; внутренняя структура молекул
не рассматривается. В этом параграфе мы применяем теорему о вириале к урав-
нению состояния в предположении, что газ составлен из ядер и электронов.
В соответствии с A.10) гл.З, средняя кинетическая энергия частиц может быть
записана в виде суммы вириала, определяемого внешними силами (т. е. влия-
нием стенок сосуда), */2pV и вириала, определяемого силами взаимодействия
между частицами
К ^\ .). C.5)
*) Экспериментальное исследование поглощения рентгеновских лучей при высоких
давлениях проводилось в работах : J. Saurel, R. Bergeon, P. Johannin, J. Dap-
signy, J. Kieffer, B. Vodar, Disc. Faraday Soc, No. 22, 64A956); J. Dapsigny,
J. Kieffer, B. Vodar, Journ. de phys. et le Radium., 17, 606 A956). — Прим. ред.
§ 3. Газы при очень высоком давлении 221
Здесь (F,)bh. — сила, действующая на i-ю частицу (ядро или электрон) со сто-
роны всех других частиц. Поскольку ядра и электроны взаимодействуют друг
с другом по закону Кулона, то вириал сил взаимодействия между частицами
может быть выражен через Ф — полную потенциальную энергию системы
[см. D.29) гл. 1]
|^ВД = |Ф, C.6)
Следовательно, C.5) можно переписать в виде
Сумма средней кинетической энергии и средней потенциальной энергии как
раз и является в термодинамике внутренней энергией U
и = К + Ф. C.8)
Последние два соотношения дают выражения произведения pV и внутденне^
энергии через К и Ф. Эти соотношения можно решить относительно К и Ф,
которые тогда будут выражены через экспериментально измеримые термо-
динамические величины __
C.9)
C.10)
Дифференцирование этих функций (известных как соотношения Шоттки [41])
с использованием обычных термодинамических формул1) дает [42, 43]
?p (%)T]p, C.11)
- <* = & (w), ~ 2CJ dT + l3V + 2T Шр + »P (?)J <P- C-12)
Полная кинетическая энергия К является суммо_й кинетической энергии
атомных ядер Ка и кинетической энергии электронов Ке. Если поступательные
и колебательные движения атомных ядер описываются классически, то Ка
будет являться функцией только температуры и не зависеть от давления. В этом
случае изменение Ке в зависимости от давления будет точно равно изменению
К. Хотя это и не совсем правильно, однако очевидно, что_изменение величины
Ке является существенной частью изменения значения К.
Бриджмен [42, 43] использовал формулу C.11) для получения зависи-
мости К от давления в твердых телах. Из его экспериментальных результатов
следует, что рост кинетической энергии при 19 000 атм в литии составляет
18 ккол/моль, в висмуте — 30, в алюминии — 14 и в железе — 9 ккаа/моль.
Для газов Михельс с сотрудниками показал [44—46 ], что изменение кине-
тической энергии пропорционально давлению. На фиг. 49 показано, что в СО2
кинетическая энергия увеличивается на 8 тал/моль при давлении 2800 атм;
на фиг. 50 показано, что этот рост для азота составляет 15 ккал/моль при
7800 атм. В этилене кинетическая энергия растет на 9 ккал/моль при росте
давления до 2500 атм\47, 48], а в аргоне — на 4 ккал/моль при росте давления
до 2000 атм [49, 50].
Вначале Михельс и др. предполагали, что большому изменению кинети-
ческой энергии с давлением соответствует большое изменение полной (кинети-
ческая плюс потенциальная) энергии электронов. Если бы это имело место, то
химические связи ослабевали бы и разрушались при таких сравнительно
*) Для определения dU использовалось соотношение
222
Гл. 4. Уравнение состояния плотных газов и жидкостей
низких давлениях, как 50000 атм. Однако Коттрел [51,52] применил теорему о
вириале и показал, что полная энергия электронов меняется лишь приблизи-
тельно на 1/2) изменения кинетической энергии. Поэтому для разрушения хими-
ческих связей потребуется давление порядка 1 000 000 атм.
ю
V
О 500 W00 1500 2000 2500 3000 3500
р, атм
Фиг. 49. Изменение кинетической энергии в углекислом газе как функция давления [45].
/
4
А
/
0
л
(<;
/
/
/
1000 2000 3000 4000 5000 6000 ЩО 8000
р, атм
Фиг. 50. Изменение кинетической энергии в азоте как функция давления [46J.
Коттрел предполагал, что газ состоит из двухатомных молекул. Допустим,
что функции потенциальной энергии [например, кривые Морзе Фп(г)] для
этих молекул известны как функции расстояния между двумя ядрами. Тогда
в соответствии с развитым Слетером методом применения теоремы о вириале
к молекулярным задачам (см. гл. 13, § 1) средняя кинетическая энергия элект-
ронов является известной функцией от г
Ке(г)=-Фп(г)~Г^, C.13)
или если г0 — расстояние между ядрами в равновесии, то изменение электрон-
ной кинетической энергии, когда молекула сжата от г0 к г, составляет
ЛКе = ТС.(г) - Ке(г0) = Ф„(г0) - <Р„(г) -
C.14)
§ 3. Газы при очень высоком давлении 223-
Подобное сжатие приводит к изменению средней полной электронной энергии
АФе = Фе(г) - Фе(г0) = Фп(г) - Фп(г0). C.15)
Из экспериментального измерения зависимостей р от V и Г, таких, как изме-
рения Бриджмена и Михельса, для отдельных давлений известна величина
изменения электронной кинетической энергии ЛКе для данного вещества. Из
C.14) можно найти соответствующее значение г. Затем значение г можно под-
ставить в C.15) для получения величины изменения полной электронной энер-
гии АРе, соответствующей экспериментально наблюдаемому изменению кине-
тической энергии АКе.
Для проверки щ авильности этого вывода Коттрел рассуждал следующим
образом. Мы знаем, что для получения определенного значения г требуется
сила, сжимающая молекулу до этого межъядерного расстояния, F = —d»n\dr*
Следовательно, для каждого давления р можно определить силу F, действую-
щую на молекулу. Пусть эта сила выражается произведением эффективной
площади поперечного сечения молекулы Д умноженной на давление. Тогда
из измерений зависиуости р от V и Т можно определить А для различных
молекул. Если А остается постоянным, в то время как давление растет, то из
рассуждений, обратных приведенным, можно определить изменение кинети-
ческой энергии с давлением при давлениях, намного превышающих те, при
которых проводились измерения. Таким образом, из работы Михельса и др. [44 ]
можно найти, что для N2 эффективная площадь поперечного сечения молекулы
равна А = 145 А и остается постоянной до 30С0 атм — предела известных экс-
периментальных результатов. Если эта площадь поперечного сечения остается
постоянной до 100 0С0 атм, то средняя электронная кинетическая энергия
будет испытывать изменение на 180 ккал/моль, в то время как полная энергия
электронов Фп(г) должна была бы возрасти на 4 тал/моль. На самом деле для
увеличения Фп{т) до значения энергии диссоциации требуется давление порядка
1000 000 атм. Задолго до того как будет достигнуто давление в 1 000 0С0 атм,
соседние молекулы азота будут испытывать сильные искажения вследствие
взаимодействий друг с другом, так что смысл экстраполяции Коттрела к очень-
высоким значениям давления не ясен. Тем не менее большая величина отно-
шения изменения кинетической энергии к изменению полной электронной
энергии должна служить предупреждением тому, кто полагает, что ослабление
химических связей и другие изменения в электронной структуре могут иметь
место при сравнительно низких давлениях.
Г. КВАНТОВОМЕХАНИЧЕСКОЕ РАССМОТРЕНИЕ
ДЕФОРМАЦИИ МОЛЕКУЛ ПРИ ВЫСОКОМ ДАВЛЕНИИ
Другим способом изучения влияния очень высоких давлений на вещество*
является использование квантовой механики для теоретического определения
эффекта сжатия отдельного атома. Рассмотрены две задачи подобного типа г
атом водорода, находящийся в твердом сферическом объеме [53 ], и ион моле-
кулы4 водорода (Hg) — в шаровом объеме [51]. Мы рассмотрим здесь только вы-
вод для атома водорода, проделанный де Гроотом и Селдамом1).
Свободный атом водорода имеет бесконечное число связанных отрицатель-
ных состояний энергии и континуум положительных уровней энергии. Если
атом заключен в большой сферический объем, то континуум становится дискрет-
ным, а число связанных состояний — конечным. При уменьшении радиуса г^
этого сферического объема уровни энергии, соответствующие связанным со-
стояниям, постепенно поднимаются и, таким образом, на вершине потенциаль-
ной ямы становятся положительными, т. е. электрон в этих энергетических
*) Недавно Селдам [54] суммировал свои квантовомеханические расчеты в доктор-
ской диссертации. Селдам и де Гроот [55 ] проделали также квантовомеханические рас-
четы свойств сжатого гелия и аргона.
224
Гл. 4. Уравнение состояния плотных газов и жидкостей
состояниях стремится быть свободным. Состояние 2s имеет равную нулю энер-
гию при г0 = 6,153 а0, 2р-состояние — при г0 = 5,086 а0, а ls-состояние — при
го= 1,835 а0. На фиг. 51 показаны полная энергия ?, средняя кинетическая
Is 2р 2s
3765 ЩЗг ТТПП
2510 108,8
1255 54t4
05
4255 -54,4
-25W 408,8
- I
1s 2p 2s
i i i i i iii
2
1,06
4
2,12
6 8 a0
3,18 4,23 A
Фиг. 51. Кинетическая, потенциальная и полная энергии атома водорода, заключенного
в сферический объем радиусом г0 [53].
15
I'0
I
р, атм
1500 3000 4500 6000
0,1
0,2
П1
\
4
Фиг. 53. Относительное изменение поляри-
зуемости атома водорода, заключенного в сфе-
рический объем, как функция давления [53].
0 2500 5000
р,атм
Ф]иг. 52. Изменение кинети-
ческой энергии как функция
давления для атома водорода,
заключенного в сферический
объем [53].
энергия электронов К и средняя потенциальная энергия электронов Ф для
различных значений радиуса г0 ограничивающего объема для Is-, 2s- и
2р-состояний. На фиг. 52 показано изменение электронной кинетической энергии
как функции давления, оказываемого атомом на стенки ограничивающего его
объема. Удивительно, что при давлении 2700 атм изменение электронной кине-
тической энергии составляет 8 ккал\моль. Эти данные находятся в хорошем
согласии с наблюдаемыми экспериментальными' значениями для СО2 и N2.
Вероятно, одним из наиболее значительных признаков сжатия молекул явля-
ется изменение поляризуемости с давлением. Это видно из расчетов для атомар-
ного водорода (фиг. 53).
§ 4. Некоторые общие положения метода ячеек 225
Другой подход к рассматриваемой задаче имеется у Вигнера и Хантинг-
тона [56 ], которые рассматривали возможность наличия металлической моди-
фикации водорода. Они рассчитали физические свойства объемноцентриро-
ванной кубической решетки, составленной из атомов водорода. Ими найдено,
что водород можно сжать до состояния металлического кристалла с плот-
ностью 0,80 (величина обычной плотности твердого водорода 0,087). В дейст-
вительности, при некотором давлении металлическая модификация может
оказаться нестабильной. Тем не менее Вигнер и Хантингтон утверждают, что
легко рассчитать, что даже в условиях наиболее благоприятной сжимаемости
при высоком давлении величина давления, необходимого для осуществления
этого перехода, составляет 250 000 атм. К аналогичному выводу пришли
Крониг и др. [57].
Д. ОПТИЧЕСКИЕ И ЭЛЕКТРИЧЕСКИЕ МЕТОДЫ ИЗУЧЕНИЯ
ДЕФОРМАЦИИ МОЛЕКУЛ ПРИ ВЫСОКИХ ДАВЛЕНИЯХ1)
Изменение показателя преломления г\ или диэлектрической постоянной
е' с давлением позволяет использовать простые экспериментальные методы для
изучения деформации молекул при высоких давлениях. В первом прибли-
жении функция Клаузиуса—Мосотти A/е) (е' — 1)/(е' + 2) и функция Ло-
рентц—Лоренца (l/eHtj8— l)/(?f + 2) зависят только от поляризуемости мо-
лекул. Поэтому изменение значений величин, входящих в эти функции, дает
возможность экспериментально определить изменение поляризуемости при
деформации молекул. В гл. 12, § 2 показано, что усовершенствование теории
диэлектрической поляризуемости приводит при больших плотностях к по-
правкам к функциям Клаузиуса—Мосотти и Лорентц—Лоренца вследствие
наложения электрического поля наведенных диполей на электрическое поле,
являющееся результатом действия окружающих молекул. Беря эти поправки
в расчет, Бётчер [58 ]2) рассмотрел экспериментальные результаты Михельса
и др. [44] по показателю преломления углекислого газа в виде функции плот-
ности и нашел, что поляризуемость СО2 падает на 10% при давлении лишь
100—200 атм и на 20% при давлении порядка 2000 атм. Это значение не-
сколько больше значения, получаемого из теоретических расчетов для сжатого
атома водорода, но, однако, находится еще в согласии с этими расчетами.
Следует усовершенствовать теоретическое рассмотрение диэлектрической
поляризуемости, прежде чем можно будет с хорошей точностью определить
изменения поляризуемости молекул с давлением.
§ 4. НЕКОТОРЫЕ ОБЩИЕ ПОЛОЖЕНИЯ МЕТОДА ЯЧЕЕК
До сих пор обсуждение было ограничено эмпирическим рассмотрением
и общими результатами, полученными из закона соответственных состояний.
Теперь мы вернемся к теоретическому исследованию поведения жидкостей и
плотных газов. Жидкость или плотный газ можно представить либо как весьма
неидеальный газ, в котором часты многократные столкновения, либо как иска-
женный кристалл, в котором утрачен дальний порядок3).
Первый случай является развитием вириального представления уравне-
ния состояния, которое было описано в гл. 3. Получаемые из этой теории
результаты вполне удовлетворительны с теоретической точки зрения, за исклю-
1)См. [54, 55].
2) См. также |9 ].
3) О соотношении между твердым и жидким и между газообразным и жидким состоя-
ниями подробно изложено в книге Я. И. Френкель, Кинетическая теория жидкостей,
М., 1945. — Прим. ред.
226 Гл. 4. Уравнение состояния плотных газов и жидкостей
чением возможных затруднений со сходимостью в жидкой фазе. Эта теория
используется при изучении явлений конденсации (см. гл. 5, § 2). Однако ви-
риальное уравнение состояния в настоящее время не имеет значения для про-
ведения практических численных расчетов в области высоких значений плот-
ности.
С другой стороны, кристаллическое приближение не привело еще к фор-
мальным решениям. Тем не менее оно оказалось ценным, поскольку приводит
к ряду приближенных способов рассмотрения, которые можно использовать
для получения численных результатов. Существует два главных типа таких
приближений : 1) теория ячеек, в которой жидкость рассматривается в виде
деформированного кристалла с молекулами, локализованными вблизи или
непосредственно в узлах решетки, и 2) теория дырок, в которой считается, что
жидкости отличаются от кристаллов тем, что некоторые узлы решетки свободны.
В этом и последующих трех параграфах проводится подробное рассмотрение
теории ячеек. В § 8 суммированы расчеты, проведенные на основе теории дырок.
А. КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА КАК ОСНОВА
МЕТОДА ЯЧЕЕК
Твердое тело, состоящее из молекул без внутренних степеней свободы,
представляется в виде системы частиц, совершающих малые колебания около
положения равновесия. Статистическая сумма подобной системы является
произведением статистических сумм гармонических осцилляторов. Если бы
было возможно разложить эти колебания на нормальные, то можно было бы
написать точное выражение статистической суммы системы. Однако в прием-
лемом приближении (приближение Эйнштейна или Грюнайзена) мы можем
считать, что каждая молекула колеблется независимо друг от друга в
таком поле, которое получается тогда, когда все соседние молекулы на-
ходятся в положении равновесия. В таком приближении все частоты коле-
баний одинаковы и статистическая сумма для системы из N частиц попросту
равна произведению N одинаковых сомножителей1).
Для жидкости, в которой плотность частиц немного меньше, чем в твердом
теле, амплитуда движения молекул больше. Представление о малых колеба-
ниях в жидкости несправедливо. Однако удельная теплоемкость вещества в
жидкой фазе непосредственно вблизи точки плавления почти такая же, как и
теплоемкость твердой фазы. Это позволяет нам обратиться к представлению о
молекуле, движущейся в таком силовом поле, которое существовало бы при
том условии, что все соседние молекулы находятся в своих средних положе-
ниях. Таково существо «метода ячеек», который используется для получения
уравнения состояния. Все вещество представляется в виде регулярной
решетки, состоящей из ячеек, в каждой из которых находится по одной моле-*
куле. Совершенно очевидно, что подобная модель вещества в жидком состоянии
страдает следующими двумя недостатками:
1) Отсутствует связь между движением молекул в соседних ячейках.
Ввиду отсутствия в жидкости или плотном газе дальнего порядка, пренебре-
жение связью между движением соседних молекул является, вероятно, луч-
шим приближением, нежели приближение Эйнштейна для твердого тела.
2) Свободный обмен молекулами между ячейками полностью исключается.
Это ограничение приводит к ошибке в величине энтропии системы, которая
обычно исправляется с помощью слагаемого, называемого коллективной энтро-
пией2). Вопрос о коллективной энтропии обсуждается в следующем разделе.
В развитии теории ячеек существует два основных направления: Эйринга
и его сотрудников и Леннарда-Джонса и Девоншайра. Обе эти группы иссле-
дователей основывают свои теории жидкого состояния на достаточно обосно-
х) По вопросу о статистической теории кристаллов см., например, [60] или [61].
2) В оригинале: „communal entropy". — Прим. ред.
§ 4. Некоторые общие положения метода ячеек
227
ванной физической интуиции. Основные соотношения,- являвшиеся исходными
точками в этих исследованиях, недавно были установлены Кирквудом [62],
который тщательно разобрал предположения теорий Эйринга и Леннарда-
Джонса и Девоншайра. Поэтому мы отказываемся от хронологической послв-
довательности изложения и начинаем с результатов, полученных Кирквудом.
Б. ПОНЯТИЕ КОЛЛЕКТИВНОЙ ЭНТРОПИИ
Смысл коллективной энтропии можно выяснить, рассматривая идеализи-
рованную систему, состоящую из N невзаимодействующих частиц. Рассчитаем
разность энтропии двух состояний этой системы, изображенных на фиг. 54.
о
D
О
О
о
х>
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
О
о
о
о
о
о
о о
о
о
о
Q
о
о
О
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
О
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
о
О
о
о
О
о
о
о
.о
о
о
о
О
о
о
о
:о
о
о
о
о
о
о
о
о
Фиг. 54. Иллюстрация понятия о коллективной энтропии.
I идеальный газ, состоящий из N молекул в объеме V; II — N молекул, каждая из которых находится
в ячейке объема V/N.
В состоянии I все молекулы ограничены полным объемом V. Классическая
статистическая сумма для этого состояния такая же, как и для идеального
газа, состоящего из N молекул
*"•"-??-, DЛ)
где А2 = Щ2пткТ. В состоянии II, когда каждая молекула ограничена соб-
ственной ячейкой с объемом VJN, статистическая сумма записывается в виде
D.2)
В последнем выражении N! отсутствует вследствие возможной различимости
частиц. Статистические суммы Z$ и Zft!) дают совершенно одинаковые выра-
жения для внутренней энергии и давления. Следовательно, наличие пере-
городок не сказывается на величине внутренней энергии и давления, если
частицы являются невзаимодействующими. Однако эти статистические суммы
дают различные значения для энтропии S = к\п ZN + U\T. Разность значений
энтропии между двумя состояниями равна
- S<»> = к In (J^) = к Ш (^J-j = Aft.
D.3)
Здесь использована формула Стирлинга для In N!
Последний результат означает, что для газа в условиях предельно малой
плотности модель ячеек дает заниженную на величину Nfc энтропию и преуве-
личенную на NkT свободную энергию. Этот недостаток можно поправить,
введя в выражение для статистической суммы Z^ множитель ем. Этот прием,
228 Гл. 4. Уравнение состояния плотных газов и жидкостей
однако, не вполне удовлетворителен : множитель eN необходим лишь в пре-
дельном случае идеального газа, но не для вещества, находящегося в кристал-
лическом состоянии. Где-нибудь между этими двумя предельными случаями
учет «коллективной энтропии» становится эффективным. Одно из предполо-
жений [38] заключается в том, что эта «коллективная энтропия» появляется
скачкообразно в точке плавления, когда она дает дополнительный вклад в
антропию величиной R (на 1 моль) для жидкости. Этот взгляд, однако, под-
кергся критике со стороны Раиса [63], который утверждает, что множитель
еы в статистической сумме появляется постепенно. Это привело бы к тому,
что «коллективная энтропия» стала бы функцией объема, так же как и темпера-
туры, и, таким образом, дала бы вклад в уравнение состояния.
Более полное понимание вопроса о коллективной энтропии можно полу-
чить в результате следующего более формального объяснения, в котором рас-
сматривается система из N одинаковых взаимодействующих частиц. В гл. 3,
§ 1 было показано, что уравнение состояния может быть записано через стати-
стическую сумму
ZN = (N! ft3") JJexp [- H(r^pN)] dr»dp» =
X-+NQN, D.4)
Где qn — так называемый конфигурационный интеграл. Поделим теперь про-
извольным образом все пространство координат на N ячеек Al9 A2i..., AN.
Тогда конфигурационный интеграл можно записать в виде суммы членов, в
которых значения координат каждой молекулы ограничены отдельной ячей-
кой. Если имеется N молекул и N ячеек, то QN содержит NM членов, соответ-
ствующих NN способам размещения N молекул в N ячейках
2 .-• 2 J / ... J ехрГ-^ldr^...dr*. D.5)
L J
2
Так как молекулы неразличимы, т^ при перестановке молекул в ячейках вели-
чина выражения не меняется.
Обозначим через QN(mv /п2,.. ., mN) различные члены в D.5), соответст-
вующие такому случаю, когда тг молекул находится в ячейке 1, т2 молекул —
в ячейке 2, a mN молекул — в ячейке N. Вследствие возможности перестановок
N молекул имеется ЛМ/Дап,! способов получения отдельных групп
{/л} = т19 /п2,..., Шдг. Следовательно, полный конфигурационный интеграл
получается в виде
Qn = (N!)-1^ тЙл Qn(mlym2,
{m}
_ v
{m} Ul mX
Суммирование ведется по всем группам чисел {т} с тем ограничением, что
2 Щ = N.
1 Последнее выражение может быть записано формально в виде
Qn^^QW, D.7)
где
!,...,!), D.8)
. D.9)
{«}
При достаточно высоких плотностях силы отталкивания между молекулами
§ 4. Некоторые общие положения метода ячеек 229
препятствуют многократному заполнению ячеек; тогда все QN(mv т2,..., mN),
исключая Q$, равны нулю, так что
а = 1 (предельное значение при высокой плотности). D.10)
В случае экстремально разреженного газа взаимодействие между молекулами
практически отсутствует и, таким образом, все QN(mv т2,..., mN) равны между
собой и дают1)
а =? е (предельное значение при малой плотности). D.11)
Оба эти результата находятся в согласии с результатами описанной выше
упрощенной картины.
В общем случае значение а лежит между 1 и е9 и изменение величины а
с плотностью указывает на появление коллективной энтропии. При рассмот-
рении теории свободного объема в последующих двух разделах предполагается,
что д = е. Это может явиться источником ошибок в получаемых уравнениях
состояния, поскольку производная от а по удельному объему дает вклад в
значение давления. Приближенную оценку а можно получить при рассмот-
рении эффекта многократного заполнения ячеек (см. § б и 7). Можно избежать
необходимость решения этой задачи, если допустить существование дырок в
структуре решетки (см. § 8).
В. КОНЦЕПЦИЯ СВОБОДНОГО ОБЪЕМА2)
Теперь можно показать, что результат, представленный в соотношении
D.7), можно использовать для получения исходных выражений для метода
ячеек, описанного в следующем параграфе. Этот вывод поясняет допущения,
вводимые в методе ячеек.
Начнем с определения плотностей вероятности Р[К)(т^} pN) и P[n\tn)
с помощью
<NV ^ exp [— ЯУ°] D.12)
p("> (г") = [N! Х*»г®]-1 exp [- ^p-]. D.13)
Для системы из N взаимодействующих молекул, подчиненных условию, по
которому они должны оставаться в своих отдельных ячейках (по молекуле в
каждой ячейке), статистическая сумма записывается в виде Z$=(A3^ NlJ-^Qft*.
Эти функции распределения отличаются от выражений A.1) и A.3) гл. 2
учетом того фактора, что молекулы не могут двигаться через границы своих
собственных ячеек, хотя они еще могут взаимодействовать с молекулами, на-
ходящимися в дгугих ячейках. Введем теперь приближение, согласно кото-
рому плотность вероятности в конфигурационном пространстве может быть
выражена в виде произведения функций, каждая из которых является функ-
цией координат только одной молекулы3)
D.14)
х) Для этого случая соотношение D.9) упрощается и записывается в виде
так что а как раз равно числу членов в исходной сумме D.6), деленному на N1 Таким образом,
в соответствии с формулой Стирлинга d^ = N*/N \ = е*.
2)См. [62]. '
8) Это приближение подобно тому, которое использовано Хартри при решении урав-
нения Шредингера в случае проблемы многих тел.
230 Гл. 4. Уравнение состояния плотных газов и жидкостей
Функция s(r) нормирована на одну ячейку
= 1. D.15)
Вектор г является вектором, указывающим положение молекулы в ячейке
относительно некоторого удобного начала координат в ячейке. Наилучший
выбор функции s(r) получается из условий минимума свободной энергии
А = U — TS при постоянных Т и А, полученных с учетом условий норми-
ровки для s(r).
Свободная энергия задается в виде
А = - kTlnZN = - kTIn(l~*NQN) =
= _ NkT In a - kT In (A-** Q(i)) = D.16)
= - NkT\na + AW - kTlnN !.
Величина A^ = Z7(l)— TS(l) есть работа системы N взаимодействующих частиц,
размещенных по ячейкам. Величины ?7A) и TS(l) записываются следующим
образом1):
jjpW (tN} pN) H (TNf pN) dfN dfN = JpJJV) (fN) ф (fN) dfN+ |. NfcT> D 1?)
A A A
N - AT In (N ! h™) =
= - kTJP<N>(r^)InP[N)(rN)drN + ^-NkT-kT'In(N ! A3^) DЛ8)
Величина A(l), выраженная через функции s(r), соответственно равна
AV/NkT = J 5(r) In s(r) dt + -^ [J ?(r - r') s(r) s(r') dr rff +
J AA D.19)
+ -^ln(N!A3N).
В этом выражении величина Е(т) определяется соотношением
S(r)=jM|Ri/-r!) D.20)
и является составной частью полной потенциальной энергии взаимодействия
частицы 1 в положении г по отношению к началу координат своей ячейки с
частицами 2, 3, ..., N, расположенными в начале координат своих соответ-
ствующих ячеек. Вектор R17 является вектором, характеризующим положение
начала координат /-й ячейки по отношению к началу координат ячейки 1.
Рассматривая теперь А как функцию от s(r), получаем условия экстремума
дА = J{/cT lns(r) +\E(r - r')s(T')dt'}dsdr = 0 D.21)
А
вдоль границ
Используя множители Лагранжа, из D.21) и D.22) можно получить интеграль-
ное уравнение для s(r). Предпочтительнее, однако, записать это в виде интег-
См. задачу 11 гл. 2.
§ 4. Некоторые общие положения метода ячеек 231
рального уравнения для у(г), которое дает для s(r) соотношения
D.23)
А
Тогда интегральное уравнение для у)(т) записывается в виде
Y>(r) = I w(r - г') ехр |"^т^) 1 df, D.25)
л
где
u,(r) = F(r)-?0, D.26)
Ео = Jj?(r - r') s(r) 5(f) dr Л*. D.27)
Решение интегрального уравнения D.25) определяет наилучшее приближение
к Р[Ы)(тм), взятое в виде произведения функций s(r).
Предыдущий вывод позволяет записать статистическую сумму ZN через
статистическую сумму z для отдельной молекулы
ZN = z» = [A-»*tyexp (- ^-)]N, D.28)
где vj — свободный объем, определяемый соотношением
0/=J ехр [--*&]*. D.29)
Л
Из этих выражений можно получить другие термодинамические величины и
уравнение состояния.
В качестве нулевого приближения можно предположить, что молекулы
расположены в центре соответствующих ячеек. Если s(r) выражается через
б-функцию Дирака1), то в первом приближении2) статистическая сумма моле-
кулы и свободный объем будут равны соответственно
z = *-»**, ехр [—fPJ D.30)
г, D.31)
где Е(т) задается соотношением D.20), а А2 = Щ2пткТ. Эти выражения вместе
с соотношением
являются исходным пунктом теории ячеек, излагаемой в последующих трех
параграфах. Таким образом, мы рассмотрели приближения, легшие в основу
метода ячеек. Несмотря на то, что метод ячеек является значительным дости-
жением теории жидкостей и плотных газов, очевидно, что возможности этого
метода ограничены трудностью точной оценки величины а. Теория дырок,
рассматриваемая в § 8, в этом отношении превосходит теорию ячеек.
*) Недавно Карери [66] рассмотрел использование в этой связи функций Гаусса.
2) Подстановка нулевого приближения для у>(г) в правую часть уравнения D.25) дает
y>i(r) [первое приближение к хр(т) ]. В свою очередь, хрх можно подставить в интеграл и полу-
чить следующее приближение у^1")- Ценность полученных результатов сомнительна ввиду
ошибки, вносимой за счет неточностей в оценке значения а.
232
Гл. 4. Уравнение состояния плотных газов и жидкостей
§ 5. УПРОЩЕННАЯ МОДЕЛЬ ЯЧЕЕК
ДЛЯ ЖИДКОСТЕЙ И ПЛОТНЫХ ГАЗОВ1)
С помощью последних соотношений предыдущего параграфа можно раз-
вить достаточно хорошую простую теорию жидкостей, если сделать некоторые
приближения для свободного объема vf и «энергии решетки» N?@)/2. Резуль-
таты, полученные с помощью этой упрощенной модели, сравниваются затем
с экспериментальными и расчетными значениями для различных термодина-
мических величин. Этот метод позволяет получить также правило Трутона и
правило Гильдебранда для давления насыщенных паров.
А. ПРИБЛИЖЕННЫЕ ВЫРАЖЕНИЯ
ДЛЯ СВОБОДНОГО ОБЪЕМА И ЭНЕРГИИ РЕШЕТКИ
Для оценки величины свободного объема предположим, что молекулы
представляют собой твердые сферы с диаметром а. Все молекулы, кроме одной
(«блуждающей»), закрепляются в положении равновесия в правильной куби-
ческой решетке. Блуждающая молекула свободно движется в ячейке, образо-
ванной «ближайшими соседями». Этот свободный объем имеет очень сложную
геометрическую форму2). Однако в случае этой упрощенной теории значение
свободного объема рассчитывается следующим образом. Рассмотрим ряд
молекул, расположенных вдоль оси х в решетке так, как это показано на
фиг. 55. Центр блуждающей (заштрихованной) молекулы может перемещаться
Фиг. 55. Приближенное значение свободного объема в случае простой кубической упаковки
молекул.
вдоль оси х на расстояние 2 [г;1'» — а ]; аналогично он может двигаться вдоль
осей у и г. Тогда свободный объем по упрощенной теории приближенно запи-
сывается в виде
где мы ввели Ъ = 27iNosj3 — величину, входящую в уравнение Ван дер Ваальса.
Рассмотрим теперь энергию реиетки д/2/ ?@), отнесенную к 1 молю.
Достаточно хорошее приближение для энергии решетки можно получить, по-
ложив ее равной отрицательному значению внутренней энергии парообразо-
вания в расчете на 1 моль, AUnap.. Гильдебранд[67] показал, что AUnax>. можно
записать в виде функции типа a(T)jVn. В табл. 36 показано, что для большого
числа веществ значение п = 1 является вполне удовлетворительным. Взяв
это значение п, для энергии решетки по упрощенной теории можно записать
E.2)
ЧСм. [64, 65].
2) Точная форма рассматривается в § 6.
§ 5. Упрощенная модель ячеек для жидкостей и плотных газов
233
Таблица 36
Внутренняя энергия парообразования [67]
Экспериментально полученные значения л в формуле ^^Пар. = a(T)jVn
Вещество
Вещество
я-Гептан
Четыреххлористый кремний
Четыреххлористый углерод ,
Бензол
Четырехбромистый кремний,
Хлористое олово
Четыреххлористый титан ...
1,09
1,09
1,07
1,05
1,04
1,04
1,02
Хлороформ
Этиловый эфир .
Ацетон
Сероуглерод
Метиловый спирт
Ртуть
1,02
1,01
0,89
0,89
0,34
0,33
В дальнейшем будет показано, что функция а(Т) является величиной а
Ван дер Ваальса.
Строго говоря, энергия решетки и свободный объем зависят как от сил
притяжения, так и от сил отталкивания, действующих между молекулами. Этот
упрощенный подход основан, однако, на зависимости свободного объема глав-
ным образом от сил отталкивания (величина Ь Ван дер Ваальса) и на зависи-
мости энергии решетки главным образом от сил притяжения (величина а Ван
дер Ваальса).
Б. УРАВНЕНИЕ СОСТОЯНИЯ ЭЙРИНГА
Подстановка этих упрощенных выражений для свободного объема и энер-
гии решетки в выражение для статистической суммы D.30) и использование
D.32) дает
~"" Ч| (у _ о,7816 ft1/. И») = /?Т, E.3I)
что и является уравнением состояния Эйринга. Постоянная 0,7816 соответ-
ствует простой кубической решетке. Для объемноцентрированной кубической
решетки значение постоянной равно 0,7163, а для гранецентрированной —
0,6962. Это уравнение подобно уравнению Ван дер Ваальса, за исключением
того, что собственный объем здесь не остается постоянным, а меняется как 2/3
объема.
Фактически уравнение Эйринга можно рассматривать как предельную
форму уравнения Ван дер Ваальса, когда последнее исправляется на случай
перекрытия твердых сфер. Необходимость такого шага была отмечена еще
Ван дер Ваальсом и Больцманом. Они записывали уравнение состояния в виде
бесконечного ряда типа
-т1[1 + т+°-625 (т)'+°-2869 [т
I1
Больцман и Егер высчитали член 0,625 (bfVJ, рассматривая одновременные
столкновения трех молекул. Хаппель [68] и Маюмдар [69] рассчитали коэф-
фициент при следующем члене, учитывая одновременные столкновения четырех
х) Чтобы показать зависимость от vt в явном виде, это соотношение можно записать
и иначе:
' в (Т) \ ( 1 1/ »/Л
V2 J {2 ' J (Э.за)
где vf дается соотношением E.1).
234
Гл. 4. Уравнение состояния плотных газов и жидкостей
молекул. Чтобы согласовать уравнение E.4) с уравнением Эйринга в области
жидкого состояния [27, 38], указанный ряд можно ограничить членом
0,1928 {bjvy. Поскольку этот член имеет тот же порядок величины, что и другие
члены, то представляется, что уравнение Элринга является логическим продол-
жением уравнения Ван дер Ваальса в область более плотной упаковки.
Можно составить таблицы, показывающие отличное численное согласие
между величинами а и Ь, определенными из жидкого и газового состояний.
Однако различные методы расчета этих величин, исходя из данных по газу,
дают настолько различные значения, что это табличное согласие кажется иллю-
зорным. Более надежной мерой справедливости уравнения состояния является
сравнение расчетных и экспериментальных значений фактора сжимаемости и
коэффициента теплового расширения. Примером полученного соответствия
служат результаты, приведенные в табл. 37.
Таблица 37
Фактор сжимаемости и коэффициент теплового расширения
жидкостей
Фактор сжимаемости /3 = V— (9/p)y
Коэффициент теплового расширения а = V—1(QV/6T)P
Вещество
(QH^o
CC14
CHCl,
од.
/3- 10*
расчетное*
2,12
1,07
1,03
0,85
атм~х
наблюдаемое
1,29
1,05
1,00
0,95
а- 10»,
расчетное*
1,68
1,14
1,31
1,12
град—1
наблюдаемое
1,58
1,23
1,27
1,24
* По уравнению E.3).
В. ДАВЛЕНИЕ НАСЫЩАЮЩИХ ПАРОВ,
ПРАВИЛО ГИЛЬДЕБРАНДА И ПРАВИЛО ТРУТОНА1)
Выражение для давления насыщающих паров можно получить, прирав-
нивая потенциал Гиббса G моля жидкости той же величине для 1 моля газа.
Предположим, что газ идеальный и, более того, величиной произведения pV
для жидкости можно пренебречь по сравнению со значением pV для газа. Эти
предположения позволяют нам записать потенциал Гиббса для 1 моля в виде
п пт
Gnap. = — RT
J ,
= - ЯГ In [ir*avf exp f- fg-)],
E.5)
E.6)
где А2 == Щ2пткТ. Приравнивая оба значения потенциала в двух фазах, полу-
чаем
E.7)
Таким образом, мы видим, что а появляется в выражении для давления
паров. В этой упрощенной теории мы принимаем, что в твердой фазе д = 1 ;
в точке плавления он скачком переходит к значению е. В соответствии с этим
в выводе условий равновесия жидкость — пар а берется равным е.
. [67].
§ 5. Упрощенная модель ячеек для жидкостей и плотных газов
235
Если предположим, что энергия решетки равна отрицательному значе-
нию внутренней энергии парообразования, то выражение для давления паров
можно записать в виде
f (^) E.8)
<59>
где АНпар. — молярная энтальпия парообразования. Используя уравнение
состояния Эйринга в виде E.3а), можно записать выражение для свободного
объема в форме
Во второй записи предполагалось, что давлением р можно пренебречь по срав-
нению с внутренним давлением а/1/2, которое можно записать через молярную
теплоту парообразования в соответствии с E.2). Подстановка этого выражения
для свободного объема в выражение для давления паров дает
ij exp у
RT
j
E.10)
Мейер [70, 71] показал, что для очень многих веществ величина
является универсальной функцией от AHmpJRT; это утверждение находится
в соответствии с последним соотношением. Такой результат позволяет получить
правило Гильдебранда для давления насыщающих паров. Концентрация пара
спар. = PnapJRT является универсальной функцией от AHnapJRT. Согласно
E.10), это утверждение справедливо, за исключением множителя V. Однако
изменение этого множителя от вещества к веществу незначительно в сравнении
с сильно выраженной температурной зависимостью
в виде экспоненциального множителя.
Правило Трутона можно получить, положив
RT/8V приближенно равным некоторому универ-
сальному числу. Для Т = 300 °К и V = 82 см*/моль
/W = 35 (i|p. - l)8 exp (-
E.11)
В точке кипения при нормальных условиях, ха-
рактеризуемых Т = Ть и р — 1 агпм, из этого
соотношения для теплоты парообразования можно
получить
i|n^_ = 10>4. E.12)
Это и есть правило Трутона с наилучшим значением
численной постоянной. Эта постоянная совсем не
зависит от значения числа, взятого для RT/8 V:
когда последнее изменяется в 2 раза, постоянная
Трутона меняется лишь на несколько процентов.
Для благородных газов, которые имеют очень низкие
точки кипения и малые молярные объемы, соотно-
шение E.10) дает значение постоянной Трутона,
почти равное 12; подобная величина находится в
согласии с экспериментом.
Г. ТЕПЛОЕМКОСТЬ
в
Фиг. 56. Потенциальная
энергия молекулы, находя-
щейся в ячейке.
a — упрощенное представление для
жидкости ; б — твердое тело ;
в — потенциальная кривая для
молекулы жидкости, дающая
исправленное значение
теплоемкости.
Прежде чем мы сможем рассчитывать тепло-
емкость, потребуются некоторые поправки к упро-
щенной теории жидкости. До сих пор мы предполагали, что молекулы дви-
жутся в потенциальном поле в виде ящика, подобно тому, как это изобра-
жено на фиг. 56, а. Перпендикулярные стенки соответствуют твердым, упру-
236
Гл. 4. Уравнение состояния плотных газов и жидкостей
гим молекулам. Мы знаем, однако, что в твердом теле, где молекулы почти
всегда находятся в состоянии столкновения со своими соседями, потенциаль-
ное поле имеет почти параболическую форму (см. фиг. 56, б) и кривизну,
связанную с характеристической температурой. Рассматривая потенциаль-
ное поле между молекулами в жидкости, мы предполагаем следующее : 1) как
и раньше, в промежутках между столкновениями молекулы движутся в про-
странстве, свободном от сил ; 2) во время столкновения молекулы отталки-
ваются от своих соседей точно таким же образом, как и в твердом теле.
Таким образом, стенки потенциальной ямы в жидкости имеют ту же кри-
визну, что и потенциальная кривая в твердом теле. Улучшенный свободный
объем образуется путем разделения потенциала для твердого тела по пунктирной
линии, делящей кривую на две части (фиг. 56, б), и включеш я области, свобод-
ной от сил, так, как это показано на фиг. 56, в. Используя этот метод, Кинкейд
и Эйринг [72, 73 ] рассчитали теплоемкость и большое число других термоди-
намических показателей жидкой ртути с точностью до ошибок эксперимента.
Конечно, некоторые постоянные, входящие в их соотношения, были опреде-
лены из экспериментальных данных, однако в силу согласия с экспериментом
полученные теоретические соотношения по крайней мере можно рассматривать
в качестве хороших эмпирических интерполяционных формул.
Д. ИЗМЕНЕНИЕ ЭНТРОПИИ ПРИ ПЛАВЛЕНИИ
Уже отмечалось, что в этой упрощенной теории предполагается, что 6
меняется в точке плавления скачкообразно от 1 к е. Этот разрыв величины а
соответствует энтропии плавления R в расчете на 1 моль. Эта «коллективная
энтропия» уже рассматривалась в § 4. В соответствии с этой теорией весьма
обоснованным было бы полагать, что энтропия плавления составляет около
2 кал\моль • град. Экспериментально было обнаружено, что среднее значение
энтропии плавления для большого числа одноатомных веществ составляет
2,2 кал/моль • град.
Определение энтропии плавления для многоатомных молекул усложняется
наличием колебаний в твердой фазе. Величину энтропии колебаний Sv можно
оценить, если из спектров комбинационного рассеяния или других данных
известны частоты колебаний. Здесь для численных расчетов в соответствии с
наблюдениями Полинга [74] используется значение 50 см*1. Энтропию вра-
щения Sr в жидкой фазе можно сосчитать, исходя из величины момента инер-
ции. Тогда энтропия плавления Sf составит
Sf = R + Sr-Sv. E.13)
В табл. 38 приведены данные, иллюстрирующие соответствие между расчет-
ными и экспериментальными значениями. В общем случае можно полагать,
Таблица 38
Вещество
С1а
Вг,
J2
cs2
Sr
13,6
16,1
18,3
14,5
13,4
s.
8,8
9,9
10,7
7,2
7,7
(ЗДрасч.
6,8
8,2
9,6
9,3
7,7
E/)набл.
6,8
9,5
9,5
8,4
8,6
что S/ многоатомных молекул приближенно равна 2 +3D кал/моль-град,
где D — число степеней свободы молекул, приходящееся на колебания, кото-
рые в процессе плавления переходят к вращению.
§ 6. Уравнение состояния для твердых сфер, не обладающих силами притяжения 237
Несмотря на то, что описанная здесь картина процесса плавления пред-
ставляется довольно убедительной и согласие предсказанных величин с экс-
периментальными достаточно удовлетворительно, однако не следует принимать
эту концепцию слишком всерьез. Как было показано в § 4, веские теоретические
соображения приводят к тому, что параметр а изменяется непрерывно от точки
плавления до точки кипения.
Леннард-Джонс и Девоншайр [75, 76] принимают, что в процессе плав-
ления появляется некоторое число новых положений решеток (названных
^-положениями), равное числу исходных положений (а-положений). Перерас-
пределение атомов между старыми а-положениями и новыми ^-положениями
дает прирост энтропии, энергии и объема; с помощью подходящего и обосно-
ванного выбора параметров эти авторы получили превосходное согласие
с экспериментом.
§ 6. УРАВНЕНИЕ СОСТОЯНИЯ ДЛЯ ТВЕРДЫХ СФЕР,
НЕ ОБЛАДАЮЩИХ СИЛАМИ ПРИТЯЖЕНИЯ
В предыдущем параграфе была описана упрощенная модель ячеек
жидкостей и плотных газов. В этой теории были сделаны такие предположения
о свободном объеме и энергии решетки, для которых затруднительно указать
границы их справедливости. В настоящем параграфе рассматриваются твердые
сферические молекулы и не вводится никаких других предположений, кроме
тех, которые обсуждались в § 4. Прежде всего проводится расчет точной вели-
чины свободного объема и ошибок за счет «сглаживания» свободного объема.
Рассматривается также влияние положения узлов в ячейке, отличного от того
случая, когда центральная точка является «началом координат» в ячейке.
Найдено, что путем соответствующего выбора начала координат можно полу-
чить уравнение состояния для плотных газов, которое при малых плотностях
дает правильный второй вириальный коэффициент.
А. ТОЧНЫЙ И «СГЛАЖЕННЫЙ» СВОБОДНЫЙ ОБЪЕМ
В этом выводе мы рассматриваем твердые сферы диаметра а, занимающие
объем V и образующие в совокупности гранецентрированную кубическую
решетку. Каждая молекула в такой решетке имеет 12 ближайших соседей на
расстоянии а от нее. Лучше всего это можно изобразить так, как на фиг. 57,
полагая, что ближайшие соседние молекулы занимают средние точки на всех
12 ребрах воображаемого куба, построенного около данной молекулы. Объем,
приходящийся на одну молекулу, v = V/N, связан с расстоянием между бли-
жайшими соседними молекулами соотношением
Как можно видеть из фиг. 57, ячейка, соответствующая этому объему, имеет
форму додекаэдра. Тогда полный объем V может быть представлен в виде
объема, составленного из этих ячеек—додекаэдров, содержащих по одной моле-
куле в каждой ячейке. Эти ячейки подобны тем, которые были рассмотрены в §4.
Назовем теперь центральную молекулу (фиг. 57) «блуждающей». Соседние
с ней молекулы остаются закрепленными на своих местах в решетке, тогда
как она может свободно перемещаться до тех пор, пока v > <х3/)/2 (удельный
объем, соответствующий наиболее плотной возможной упаковке). В условиях
высокой плотности(г;<2<х3) «блуждающая» молекула не может выйти из «клетки»,
образованной ближайшими соседними молекулами. В таких условиях величина
свободного объема, определенного в соответствии с D.31), как раз равна объему,
доступному центру «блуждающей» молекулы. В условиях умеренно низкой
238
Гл. 4. Уравнение состояния плотных газов и жидкостей
плотности (г; > 2<х3) эта молекула уже не ограничивается объемом, образован-
ным ближайшими соседними молекулами. Однако свободный объем все еще
определяется соотношением D.31). В соответствии с этим определением движе-
ние «блуждающей» молекулы ограничено отчасти ближайшими соседними
Фиг. 57. Додекаэдр, образованный плоскостями, делящими пространство между молекулой
в центре и ее ближайшими 12 соседями [77].
молекулами, а отчасти воображаемыми границами ячейки—додекаэдра. При
очень низкой плотности (г; > 41/2<т3) «блуждающая» молекула не испытывает
столкновений с ближайшими соседними молекулами и свободно движется по
всей ячейке (т. е. vf = v). Вводя приведенный удельный объем v* = г;/а3, ска-
занное выше можно суммировать следующим образом :
1
v* = -j=r наиболее плотная упаковка,
V* = 2 «блуждающая» молекула может выйти из «клетки»,
#*^4|/2 «блуждающей» молекуле доступна вся ячейка.
На фиг. 58 приведены примеры точной фермы свободного объема для
рассматриваемой модели. Геометрия модели и аналитическое выражение точ-
ного свободного объема очень сложны и здесь не приводятся [77 ].
Вследствие большой сложности выражения для точного свободного объема
(даже для твердых сфер) обычно при расчетах свободного объема используется
представление о «сглаженном», или «приведенном к форме сферы», свободном
объеме. Это достигается заменой зависящей от угла величины [E(t) — Е@)]
в соотношении D.31) приближенно эквивалентной сферически симметричной
функцией. Сначала проводится усреднение по углам функции взаимодействия
<р{\ а + г |) между рассматриваемой молекулой (находящейся в точке г своей
ячейки) и одной из ближайших соседних молекул (находящейся в точке а по
отношению к центру рассматриваемой ячейки). Обозначим результат этого
усреднения через <р(|а + г|). Тогда полное взаимодействие рассматриваемой
молекулы со всеми ее с соседними молекулами берется равным с у(\ а + г |).
Таким образом,
F.2)
§ 6. Уравнение состояния для твердых сфер, не обладающих силами притяжения 23Э
что дает
F.3)
Приведение к форме сферы особенно просто в случае рассматриваемой решетки,
составленной из твердых сфер. Сглаженный свободный объем является сферой
v*=4VZ
v*=3V3j2
V*=16/3V3
Фиг. 58. Точная форма свободного объема (вверху) и соответствующие
«сглаженные» свободные объемы (внизу) [77].
наибольшего диаметра, которую можно вписать в рассмотренный выше точ-
ный свободный объем
vf==*r = -i7Ch-1) • F-4)
Тогда уравнение состояния запишется в виде
NkT ~~ L а
F.5)
Величина свободного объема и значения, получаемые из уравнения состояния
для этой модели, приведены в табл. 39 вместе с аналогичными результатами
Таблица 39
Сравнение величины точного и сглаженного свободного объема
V*
0,7071
0,8104
0,8779
1,0240
1,1854
1,5573
2,0000
При
гочном
расчете
•;
0,0000
0,0006
0,0025
0,0131
0,0407
0,1767
0,4912
pVINkT
оо
23,0
14,0
9,04
6,48
4,53
3,77
Для объема,
приведенного к сфере
•;
0,0000
0,0004
0,0018
0,0095
0,0275
0,1143
0,2977
p\INkT
оо
22,50
14,37
8,61
6,32
4,32
3,41
точных расчетов. Это сравнение показывает, что ошибки в уравнении состояния,
полученные после сглаживания, несущественны, если диапазон плотности
таков, что молекула действительно ограничена ячейкой, образованной ближай-
шими соседними молекулами.
240
Гл. 4. Уравнение состояния плотных газов и жидкостей
Б. УРАВНЕНИЕ СОСТОЯНИЯ ПРИ МАЛОЙ ПЛОТНОСТИ ;
СТРОГО И НЕСТРОГО ЦЕНТРИРОВАННЫЕ СВОБОДНЫЕ ОБЪЕМЫ
В выводах § 4 предполагалось, что в нулевом приближении молекулы
расположены в центре своих ячеек. До тех пор пока плотность достаточно
велика, движение молекулы ограничено некоторым объемом; в этом случае
вполне обоснованно полагать, что равновесное положение молекул совпадает
с центрами соответствующих ячеек. Однако в условиях малой плотности огра-
ничение движения молекулы рамками своей ячейки будет искусственным.
Поэтому с помощью метода ячеек в условиях предельно малой плотности едва
ли можно получить уравнение состояния идеального газа. Покажем теперь,
каким образом можно видоизменить метод ячеек, чтобы получить для малой
плотности уравнение состояния, справедливое вплоть до второго вириального
коэффициента.
Рассмотрим несколько способов, с помощью которых можно построить
ячеистую решетку из двумерного гексагонального расположения N молекул.
На фиг. 59, а представлен обычный тип решетки, когда молекулы расположены
в центре своих ячеек. Очевидно, что при малых плотностях парные столкно-
вения исключены. Однако ячейки можно расположить так, чтобы каждая
молекула находилась на одной из сторон своей ячейки ; в этом случае могут
происходить парные столкновения (фиг. 59, б). Другой способ получения
такого же эффекта заключается в замене вертикальных перегородок новыми,
на которых расположены молекулы так, как это указано на фиг. 59, в. Обна-
ружено, что для трехмерной гранецентрированной кубической решетки про-
ще сделать выбор модели, аналогичной той, которая дана на фиг. 59, в. Обе
последние модели (фиг. 59, б и в) дают правильное значение величины второго
вириального коэффициента. Однако общее уравнение состояния для этих двух
случаев различно. Отсюда следует, что требование определения правильного
значения второго вириального коэффициента не является однозначным усло-
вием для выбора типа ячейки.
Рассмотрим расположение, представленное на фиг. 59, в. Когда блуждаю-
щая молекула находится в левой половине своей ячейки, она ведет себя так,
Фиг. 59. Некоторые способы построения ячеек в гексагональной решетке.
Зачерненная молекула — «блуждающая»; все другие молекулы считаются неподвижными.
как если бы она находилась одна в ячейке типа, представленного на фиг. 59, а,
с 12 ближайшими соседними молекулами. С другой стороны, когда рассматри-
ваемая молекула находится в правой половине ячейки, она ведет себя, как если
бы она находилась в гексагональной ячейке вместе с другой молекулой, поме-
щенной в центре ячейки с 11 ближайшими соседними молекулами. Следова-
тельно, в этом приближении свободный объем можно представить как средне-
арифметическое двух типов свободного объема. Мы полагаем, таким образом,
что этот усредненный свободный объем составлен из суммы нестрого центри-
рованного свободного объема (когда рассматриваемая блуждающая молекула
находится в ячейке одна) и строго центрированного свободного объема (когда
§ 6. Уравнение состояния для твердых сфер, не обладающих силами притяжения 241
рассматриваемая блуждающая молекула находится в ячейке вместе с другой
молекулой в центре ячейки, окруженные 11 ближайшими соседними молеку-
лами). Сказанное иллюстрируется фиг. 60.
Фиг. 60. Замена свободного объема «усредненным свободным объемом» vja.
vft и
— «нестрого центрированный» и «строго центрированный» свободные объемы; зачерненная
молекула — «блуждающая».
Величина свободного объема и уравнение состояния для гранецентриро-
ванной кубической решетки, составленной из твердых сферических молекул,
уже были рассчитаны с использованием «нестрого центрированной модели»
(это соответствует обычному методу центрирования ячеек в решетках, как это
описывалось в предыдущем разделе); выше описано также получение «усред-
ненного» свободного объема. В условиях большой плотности оба метода дают
одинаковые результаты. Для малой плотности нестрого центрированная модель
дает уравнение состояния идеального газа с нулевым вторым вириальным
Фиг. 61. Сравнение значений фактора сжимаемости, полученных с помощью «нестрого
центрированной» модели (кривая а) и по методу «усредненного свободного объема»
(кривая Ь) [77].
коэффициентом, тогда как модель с усредненным свободным объемом дает
правильное значение второго вириального коэффициента для твердых сфер
RT~
Полученный таким образом третий вириальный коэффициент уже неверен
и равен bl вместо 6/8&q. Фактор сжимаемости для обоих случаев дан графически
на фиг. 61.
Попл [78 J1) несколько подробнее рассмотрел некоторые стороны выводов
Кирквуда, описанных в § 4. Сделав некоторые приближения, Попл получил
выражение для д. В этом выражении учитывается влияние многократного
*) Содержание работы Попла суммировано в конце § 7 настоящей главы.
242 Гл. 4. Уравнение состояния плотных газов и жидкостей
заполнения ячеек. Если рассматривать только двукратное заполнение, то
результат Попла может быть записан через определенные выше значения
нестрого и строго центрированных свободных объемов
= v/s + f2 vfs vfh . F.7)
Это приводит к уравнению состояния, несколько отличному от тех, которые
до сих пор описывались. Второй вириальный коэффициент в этом уравнении
составляет 58,6% правильного значения.
§ 7. УРАВНЕНИЕ СОСТОЯНИЯ ЛЕННАРДА-ДЖОНСА
И ДЕВОНШАЙРА1)
Как и в предыдущих двух параграфах, в качестве исходной точки2) мы
берем три последних соотношения § 4. На этот раз, однако, мы будем пользо-
ваться в расчетах более реальной потенциальной функцией — потенциалом
F-12) Леннарда-Джонса
Параметры а и е, входящие в эту функцию, можно использовать для опреде-
ления следующих приведенных переменных:
т* кТ
/ * = -j- приведенная температура ;
* г
Г* = — приведенное расстояние;
а
а
п* = — приведенное расстояние между ближайшими
а соседними молекулами;
V* = —j- приведенный размер ячейки, равный ——-, V • /
о у2
и приведенный объем, приходящийся на одну молекулу ;
V* = -~ приведенный свободный объем ;
р* = —^— приведенное давление.
В дополнение к ним обычно вводят две следующие безразмерные величины:
Ц G.2)
Vf Vf
% = 2 я а» = 2тГа**'
использование которых упрощает некоторые формулы.
Поскольку потенциальная функция Леннарда-Джонса дает вполне удов-
летворительное уравнение состояния и коэффициенты переноса в разреженных
газах, то ее использование в методе ячеек должно дать достаточно хороший
!) См. [79].
2) Как упоминалось в § 4, материал излагается не в хронологическом порядке. Вывод,
описанный в этом параграфе, вначале был получен полуинтуитив шм образом. Киркву-
довское подтверждение этого метода было получено значительно позже.
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра
243
критерий пригодности этого метода. Как видно из предыдущего рассмотрения,
хорошее согласие с экспериментом получается в случае жидкости ; для крити-
ческой плотности и ниже ее получаются неудовлетворительные результаты.
Расчеты Попла [78], Янсенса и Пригожина [80] показывают, что рассматри-
ваемая модель может быть несколько улучшена, если учесть двойное запол-
нение.
А. ВЫВОД УРАВНЕНИЯ СОСТОЯНИЯ
Возьмем снова для нашей модели гранецентрированную кубическую решет-
ку. В действительности значение потенциальной функции внутри ячейки
естественно зависит от угла. Для упрощения расчетов используется эквива-
лентный ей сферически симметричный потенциал. Вид потенциального поля
внутри ячеек различных размеров показан на фиг. 62. Картина, соответствующая
случаю, когда плотность газа очень мала, показана на фиг. 62, а. Потенциал
внутри ячейки имеет постоянное значение, за исключением области низкого
12с
а ~ 1,65 а
V*~3,18
0 0,5
J2C
а~1,37а
v*~1,82
0,5
-
~1,195а
v*~l,21
0
\J
0,5
а 6 в
Фиг. 62. Потенциальное поле внутри ячейки для различных ее размеров [79].
потенциала вблизи границы; стороны ячейки являются как бы адсорбирую-
щими поверхностями для молекулы, находящейся внутри. При обычных зна-
чениях плотности жидкости области пониженного значения потенциала час-
тично перекрываются с образованием между ними энергетического барьера,
как указано на фиг. 62, б. При повышении плотности до значения, соответ-
ствующего твердому состоянию, создается такое положение, при котором этот
барьер исчезает и оба минимума сливаются (фиг. 62, в). С дальнейшим ростом
плотности перекрытие полей отталкивания ведет к увеличению минимума.
Допустим на мгновение, что рассматриваемая нами молекула не взаимо-
действует с молекулами, удаленными от нее на расстояние, превышающее
расстояние до оболочки, образуемой 12 соседними ближайшими молекулами.
В соответствии с этим мы заменяем величину [Е(г) — Е@) ] в соотношении
D.31) сглаженной функцией, описанной в § 6. В результате для потенциала
Леннарда-Джонса получаем
[Е(г) — Е@)] ^ с
J J
где
1(у) = A + 12у + 25,2у2 + 12уЗ +
+ г2 - 2 ar cos в) sin вйвйф- <р(а) =
- у)-ю _ 1,
G.3)
G.4)
Подставляя это выражение для [Е(г) — Е@)] в D.31), получаем величину сво-
бодного объема. Тогда из соотношений D.30) и D.32) можно получить урав-
244 Гл. 4. Уравнение состояния плотных газов и жидкостей
нение состояния ; предполагая, что а не зависит от объема, получаем1)
Величина g является безразмерным свободным объемом, определенным в G.2);
g, и gm— комбинации2) производных от g соответственно по Г* и t>*. Эти вели-
чины записываются в виде следующих интегралов3) :
У> <7'6>
= J КУ) Гу ехр {- ? [Ж. _ 2 «g> ]) dy, G.7)
ехр {-?[-Ж._ 2^]}<fy. G.8)
Верхний предел интегрирования в этих интегралах для случая гранецентри-
рованной кубической решетки равен
уо = (A ][2f = 0,30544. G.9-
Уравнение G.5) является уравнением состояния Леннарда-Джонса и Девон
шайра. Оно выражает фактор сжимаемости pV/NkT через приведенную темпера-
туру Т* и приведенный удельный объем (т. е. приведенный размер ячейки) е?*.
Это вполне согласуется с законом соответственных состояний, рассмотренным
в § 1. Очевидно, что фактор сжимаемости приближается в пределе к единице
для очень высоких температур и (или) низкой плотности. Получающиеся из
уравнения критическая изотерма и кривые (р, V) при более низких темпера-
турах имеют S-образную форму, подобно изотермам уравнения Ван дер Ваальса.
Тот факт, что изотермы не имеют горизонтального участка, соответствующего
совместному существованию пара и жидкости, является результатом введенных
в теорию приближений. Как говорится в гл. 5, используя обычные термодина-
мические соображения, можно ввести горизонтальный участок и рассмотреть
равновесие жидкость—пар.
Интегралы g, gh gm были вычислены Леннардом-Джонсом и Девоншайром
[79 ], Пригожиным и Ролье [82 ], Пригожиным и Гарикьяном [83 ] и Хиллом
[84]. Результаты этих расчетов находятся в полном согласии между собой.
Пригожий и Гарикьян пользовались в расчетах потенциалом F-12) Леннарда-
Джонса, а также моделью Сюзерленда. Они нашли, что термодинамические
величины, вычисленные с помощью метода ячеек, относительно мало зависят
от вида потенциала. Следовательно, едва ли имеет смысл для уравнения со-
стояния Леннарда-Джонса и Девоншайра проводить вычисления с использо-
ванием более сложных межмолекулярных потенциальных функций. Кванто-
вые поправки к уравнению Леннарда-Джонса и Девоншайра были вычислены
Лунбеком [85 ]; эти расчеты рассматриваются в гл. 6, § б.
х) В своем первоначальном рассмотрении Леннард-Джонс и Девоншайр в качестве
коэффициента при \/v*2 использовали величину [1,2 + Bgm/g)] вместо [1,0 + Bgm/g)]. Это
объясняется влиянием второй и третьей оболочек на энергию решетки, а не на величину-
свободного объема.
2) В частности,
*• JL = JL [Л ?"L| т* —Z- =
dv* T* [v** v**y дТ*
Т*
8) Предельные значения интегралов gf gi и gm при очень высоких и очень низких тем-
пературах вычислены Раулинсоном и Кертиссом [81 ].
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра
245
Б. МОДИФИКАЦИЯ УРАВНЕНИЯ СОСТОЯНИЯ ЛЕННАРДА-ДЖОНСА И
ДЕВОНШАЙРА, УЧИТЫВАЮЩАЯ ВЗАИМОДЕЙСТВИЕ С ТРЕМЯ ОБОЛОЧКАМИ
Наиболее обширные вычисления из числа проделанных для уравнения
состояния Леннарда-Джонса и Девоншайра были выполнены Венторфом,
Бюлером, Гиршфельдером и Кертиссом [86]. В этих вычислениях проведен
учет взаимодействия рассматриваемой молекулы с первыми тремя ближай-
шими окружающими эту молекулу оболочками. (Первая оболочка содержит
12 молекул и находится на расстоянии а, вторая — 6 молекул, на расстоянии
аУ2 и третья—24 молекулы, на расстоянии а |^3.) С учетом этой модификации
уравнение состояния запишется в виде
= 1+-^
)--^A,2045+^)]. G.10)
Величины G} GL и GM являются интегралами, подобными интегралам
и gm, за исключением того, что функции /(у) и т(у) заменяются функциями
и М(у)у имеющими вид
±т D-у) +±т (i-у) .
G.11)
На фиг. 63 величина р* представлена как функция v* и Т*9 высчитанная в
соответствии с G.10). Фактор сжимаемости, pVjNkT, также рассчитанный с
помощью этого уравнения, дан графически на фиг. 64. Те же самые результаты
изложены в виде таблиц (табл. VIII, стр. 861). Влияние на фактор сжи-
маемости учета взаимодействия рассматриваемой молекулы с различным чис-
лом ближайших молекул показано в табл. 40.
Таблица 40
Влияние соседних молекул на величину сжимаемости,
вычисленную с помощью модели Леннарда-Джонса и Девоншайра
___
V*
1,195
1,414
1,826
2,575
а
b
с
а
b
с
а
b
с
а
b
с
т*
0,7
3,607
-1,161
-3,292
0,018
-3,395
-3,566
0,071
-1,998
-2,142
0,099
-0,951
-0,923
1,0
3,764
0,459
-0,344
1,935
-0,453
-0,852
1,194
-0,255
-0,325
0,888
-0,255
-0,325
1,7
2,000
0,053
2,194
2,994
1,588
1,515
2,181
1,329
1,300
1,667
1,235
1,200
a) Вычисления с учетом только одной оболочки соседних молекул, осно-
ванные на использовании g-функций Пригожина и Гарикьяна [83] и выра-
жения G.5).
b) Вычисления с учетом одной оболочки соседних молекул при расчете
свободного объема и трех оболочек при расчете энергии решетки, основан-
ные на g-функциях Прчгожина и Гарикьяна для одной оболочки [83] и вы-
ражения G.5) с коэффициентом 1,2; см. примечание 1 на стр. 2i4.
c) Вычисления с учетом трех оболочек соседних молекул при расчете
как свободного объема, так и энергии решетки, основанные на G-функциях
Венторфа и др. [86] для трех оболочек и выражения G.10).
2,0
18
IS
12
1.0
0,8
0,6
0,4
0,2
n
0,2
J
w
1
I
\
1
\
\\
1 \
1
\
\
\
N..
V
N
—X
——
— —
Критическая точна
/
г
/
-
- —
^Идеальный газ
-—,
—-—
——
—.а
__ _
——.,
__ _
•^.
^Равновесие тидкость-пар
— —
г
1,2
1,3
1,2
10 И 18 22
ЗА 38 42 46
26 30
Фиг. 63. Приведенное давление рщ = ро*/екак функция
приведенного объема.
Кривые — изотермы соответствуют определенным значениям
приведенной температуры Т* [86].
300
200
100
80
60
АО
20
Л
' 7V
\
¦^—
d
^-*
— -¦
—
ч
\
—-
\
\
4
1
1
i
/
1
f
jL
i
1
/
0
/A
/)
V\
/;
Y/
у
/
/
/y
* /
z
/
у
op
1,0
1,2
is
2,0
3,0
5,0
7,0
i
i
6 -t—
W
0,8
0,6
0,4
0,1
Фиг. 64. Фактор сжимаемости как функция приведенной
плотности.
Кривые — изотермы соответствуют определенным значениям
приведенной температуры Т* [86].
0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра
247
Разность между значениями термодинамических величин для неидеаль-
ного вещества и соответствующими значениями для идеального газа при оди-
наковой температуре обозначена штрихами
G.12)
S\v, T) = S(v, T) - S\T) + R In p(v, T) ;
здесь U°(T) и C%(T) — значения внутренней энергии и удельной теплоемкости
при температуре Т и нулевом давлении; S°(T) —предел величины (S + R In р)
«0
2,4
1,8
12
Ofi
О
-0,6
со
2,4
:э .
з,о
-3,6
-«2
-4,8
5,4
-6,0
-6,6
-V-
400\,50
W
/
///
///
/,
ч/
If/
*-
к
4
4J
\
^s
S^1
\
/
ч
/20
**+
4;
4
sV
l>
•"^
4.,,
4
ч
ч
ч
4
^\
^S
\
s
s
л
\
4
\
\
^««
*^
\
ч
ч
\
N
/
•>•».
4
4.
\
s
/
/
'
—-
/
/
/
s
У
/
I
/
/
/
J
1
1
1
j
1
/
J
/
/
у
I
1
у
7~
~7
i~
1
//
1
J_
f /
/\
7
у
/
/
4
t
1
1
1
I
A
7
/
/
n
/I
7
/j
/
IJ
t
/
г
7
/ /
1
>
4
Q
\l\
/ Us
( ii
111
щи
/hi
л
Ш
I
In
/II
'I
7
11
0,4 0t5 0t6 0,7 0,8 0,9 1ft 1,1 1,2 /,3 1,4 1,5
1/v*
Фиг. 65. Поправка на неидеальность для приведенной внутренней энергии газа U'/Ne
как функция приведенной плотности.
Кривые — изотермы соответствуют определенным значениям приведенной температуры Т* [86].
при р -> 0. На фиг. 65—67 представлены значения U'y C'v и S' как функции
от Т* и t?*, рассчитанные из интегралов G, GL и GM. Аналогичные сведения
можно найти в приложении (табл. IX—XI, стр. 863—867).
2.1
IS
1,3
V
0,9
0,1
0,5
0.3-
0,1
0,8 ~
4 7/
тру-
ч
/
/
г
У
/
//
У
/
//
/
У/
//
у
У
/
' У
//
V
/
/
'/
\/
/
/
/
/
/
У
/Г
/
у
/
у
у
У
у
у
^У
у
у*
1
у
у
У
10,8
/
/
/1,5
^2,0
У
^10
^200
yoofl
03 пЗ
l/v*
Фиг. 66. Поправка на неидеальность для приведенной
теплоемкости Cv/Nk как функция приведенной плотности.
Кривые — изотермы соответствуют определенным значениям
приведенной,температуры Т* [86].
0,3
0
'0,6
-1,2
>°
-2,4
'3,0
'3,6
-4,2
-48
-5,4
-6fl
-6,6
-7,2
-7,8
-8,4
-зд.
V
ч
\
ч
\
¦**
ч
ч
\
\\
\^
*v\
•«^
ч^
ч
\
ч
ч
\
ч
\
\
V
\
\
ч
у
\
\
\
ч^
\
ч
ч
\
ч
\
\
s
\
\
\
ч^
\
\
\
\
V
S
ч
S,
\
s
ч
ч^
N
\
г S
\
ч
ч
чч
\
ч
ч
\
ч^
ч
ч
s
Г
J00
sJOO
1
0
±20
^^
0
Ч 5
3
2
13
К07
0,2 0,4 0,6 0,8 1,0 7,2 1,4 1,6 1,8 20
1/V*
Фиг. 67. Поправка на неидеальность для
приведенной энтропии S'/Nk как функция
приведенной плотности.
Кривые — изотермы соответствуют определенным
значениям приведенной температуры TV186].
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра
В. СРАВНЕНИЕ С ЭКСПЕРИМЕНТАЛЬНЫМИ РЕЗУЛЬТАТАМИ
Чувствительным критерием правильности уравнения состояния, получен-
ного теоретическим путем, является описание критических явлений. Изтабл.,У1П
(стр. 861) можно найти, что
7* _ кТе _ ] on
1 с — е — 1 >^и>
ст8
= 1,77,
G.13)
Эти значения можно сравнить с соответствующими экспериментальными значе-
ниями, приведенными в табл. 31 (стр. 2сбI). Видно, что метод Леннарда-Джонса
и Девоншайра очень хорошо предсказывает величину Т% однако теоретические
значения р?, v* и pcvc\kTc ошибочны. Пригожий и Гарикьян [83] показали,
что величина pcvclkfc относительно мало зависит от вида потенциальной функ-
ции межмолекулярного взаимодействия и, следовательно, маловероятно, чтобы
этот метод мог дать удовлетворительное количественное описание критической
области. В следующем параграфе показано, что введение понятия «дырок» в
теорию жидкостей позволяет получить намного лучшее значение критического
коэффициента.
В табл. 41 сравниваются теоретические и экспериментальные значения
объема жидкости, находящейся в равновесии со своим паром, при различны*
Таблица 41
Сравнение расчетных и экспериментальных значений удельных объемов жидкостей,
находящихся в равновесии со своим паром [86]
Газ
Азот
Неон
Аргон
Метан
г, °к
77
27,26
90
111
122
111,6
133
153
191,05
т*
0,844
0,764
0,726
0,9026
0,9871
0,818
0,976
1,122
1,400
Эксперимен-
тальное значение
плотности
жидкости, г/см*
0,804
1,204
1,374
1,224
1,138
0,4245
0,3916
0,3547
0,1615
Давление,
атпм
1
1
1,5
7,4
13,7
1
4,38
11,84
45,8
V
экспери-
менталь-
ное
1,16
1,27
1,209
1,357
1,459
1,07
1,16
1,28
2,82
*
i
расчетное
1,09
1,07
1,05
1,11
1,17
1,08
1,15
1,27
1,77
температурах. Согласие вполне хорошее, так же как и общий ход изменения
величин. Это сравнение, однако, не может служить достаточно хорошим кри-
терием вследствие быстрого изменения давления в зависимости от объема в
рассматриваемой области.
х) В этой таблице приведены данные для Не и Н2. Вследствие квантовых эффектов
приведенные критические постоянные этих газов сильно отличаются от значений для
тяжелых газов. Этот вопрос подробно рассмотрен в гл. 6.
Таблица 42
Сравнение экспериментальных и расчетных значений фактора сжимаемости
для различных веществ
Аргон*
Плотность
еди-
ницы
Амага
316,6
419,9
503,6
626,3
г/см»
0,5642
0,7483
0,8975
1,1161
т,°с
т*
V*
2,944
2,219
1,851
1,488
0
2,203
р,атм
(эксп.)
[50]
310
470
900
1330
р /кТ
(эксп.)
[50]
0,97
1,13
1,38
2,07
P'lkT
(расч.)
1,45
1,61
1,77
2,10
75
2,808
р, атм
(эксп.)
[60],
450
710
1090
1820
pvfkT
(эксп.)
[50]
1,14
1,36
1,64
2,34
plkT
(расч.)
1,64
1,87
2,09
2,51
150
3,413
р, атм
(эксп.)
[50]
610
980
1400
2250
pv/kT
(эксп.)
[50]
1,25
1,50
1,80
2,32
pvlkT
(расч.)
1,76
2,02
2,28
2,74
Азот*»
Плотность
еди-
ницы
Амага
186,6
233,6
315,1
415,9
578,5
г/см*
0,2332
0,2795
0,3938
0,5198
0,7231
Т,°С
Т*
V*
3,998
3,337
2,368
1,794
1,290
0
2,987
р, атм
(эксп.)
[91]
190
240
390
680
1750
pvlkT
(эксп.)
[91]
1,03
1,07
1,25
1,63
3,03
pvlkT
(расч.)
1,41
1,62
1,87
2,21
3,07
50
3,533
р, атм
(эксп.)
[91]
250
310
510
880
2170
pvfkT
(эксп.)
[91]
ода
R:.i,i6.
1,37
1,79
3,17
pv/kT
(расч.)
1,44
1,70
1,98
2,36
3,28
100
4,080
р, атм
(эксп.)
[91]
300
370
630
1070
2570
pvfkT
(эксп.)
[91]
1,16
1,22
1,46
1,89
3,25
pvlkT
(расч.)
1,46
1,75
2,06
2,46
3,40
Водород»'
Плотность
еди-
ницы
Амага
939,0
758,1
675,5
583,1
483,3
391,7
348,8
г/см»
0,08441
0,06815
0,06072
0,05241
0,04345
0,03516
0,03135
Г,°С
г*
V*
1,516
1,878
2,108
2,442
2,946
3,640
4,082
0
8,20
р, атм
(эксп.)
[90]
2540
1610
1290
995
735
539
461
pvlkT
(эксп.)
[90]
2,707
2,124
1,910
1,706
1,520
1,378
1,321
pvlkT
(расч.)
••••
3,126
2,612
2,415
2,215
1,977.
1,593
1,342
50
9,70
р, атм
(эксп.)
[90]
2932
1882
1516
1175
872
641
548
pvlkT
(эксп.)
[90]
*
)
]
>,640
>,098
1,900
1,704
1,525
1,386
1,329
pvfkT
(расч.)
•¦••
3,122
2,630
2,434
2,234
1,976
1,542
1,313
150
12,71
р, атм
(эксп.)
[90]
2406
1952
1525
1140
842
721
pvlkT
(эксп.)
[90]
2,049
1,866
1,688
1,522
1,389
1,334
pvlkT
(расч.)
3,118
2,619
2,429
2,224
1,933
1,498
1,275
•Критическая плотность 0,531 г\смг\ плотность в жидком состоянии (90 ° К) 1,41 г/см»; плотность
в твердом состоянии D0 °К) 1,65 г/см*; удельный объем при нормальных условиях 22 390 см*1г - моль.
•• Критическая плотность 0,311 г/см»; плотность в жидком состоянии G7 °К) 0,804 г/см9; плотность
в твердом состоянии B0 °К) 1,03 г(смг; удельный объем при нормальных условиях 22 407 см*1г • моль.
••• Критическая плотность 0,03172 г/см9; плотность в жидком состоянии B0 °К) 0,070 г{смг; плотность
в твердом состоянии A3 °К) 0,076 г1смг; удельный объем при нормальных условиях 22 427 см*1г • моль.
•••• Вычисления основаны на классической теории.
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра
251
Экспериментальные и вычисленные значения фактора сжимаемости для
аргона, азота и водорода сравниваются в табл. 42. Очевидно, что рассматривае-
мая теория наиболее удовлетворительна при низких температурах и высоких
плотностях. В этих условиях движение молекул почти ограничено ячейками
так, как это предполагается в теории. При низких плотностях и высоких тем-
пературах рост молекулярного движения делает ячеистую картину строения
жидкости значительно менее привлекательной.
Для иллюстрации точности, которую мы можем ожидать при расчете тер-
модинамических величин по методу Леннарда-Джонса и Девоншайра, вычи-
сленные и экспериментальные значения поправок на неидеальность для внут-
ренней энергии, теплоемкости и энтропии приводятся в табл. 43—45 соответ-
ственно.
«Экспериментальные» значения получены с помощью численного
анализа результатов по сжимаемости. Можно видеть, что значения внутренней
энергии согласуются более удовлетворительно для аргона, нежели для азота.
Таблица 43
Сравнение экспериментальных и расчетных значений поправки на неидеальность
для внутренней энергии V (кал/моль)
Аргон
Плотность
единицы
Амага
280
400
520
640
г/слс»
0,4995
0,7136
0,9277
1,1418
Г,°С
т*
1*
3,325
2,3275
1,7904
1,4547
(
)
2,203
эксп. [49]
-429
-600,9
-769,3
-923,8
расч.
-445,1
-634,2
-825,7
-1035,3
75
2,808
эксп. [49]
-401,5
-563,8
-717,8
-851,0
расч.
-415,4
-592,0
-768,5
-956,2
150
3,413
эксп. [49]
-385,6
-539,0
-679,5
-971,3
расч.
-386,2
-549,8
-711,8
-879,4
Азот
Плотность
единицы
Амага
200
240
280
320
400
480
г (см9
0,2500'
0,3000
0,3500
0,4000
0,5000
0,6000
т, °с
т*
V*
3,731
3,109
2,665
2,332
1,865
1,554
0
2,987
эксп. [92]
-302
-360
-418
-475
-588
-693
расч.
-267
-316
-370
-423
-531
-640
50
3,533
эксп. [92]
-285
-340
-395
-449
-554
-650
расч.
-252
-295
-346
-395
-495
-594
150
4,627
эксп. [92]
-270
-312
-372
-422
-514
-594
расч.
1 1 1 1 1 1
Это можно связать с тем фактом, что двухатомные молекулы азота обладают
большим квадрупольным моментом и не имеют сферически симметричного по-
тенциала. Для удельной теплоемкости расхождение между вычисленными и
экспериментальными значениями заметно растет с ростом температуры. Харак-
тер расхождения для энтропии указывает на то, что теория приписывает рас-
сматриваемой системе больше порядка, чем он в действительности имеет место
при низкой плотности, т. е. в рассматриваемой модели молекулы значительно
сильнее связаны со своими ячейками, нежели в действительности.
Таблица 44
Сравнение экспериментальных и расчетных значений поправки на неидеальность
для теплоемкости C'v (кал/моль • град)
Аргон
Плотность
единицы
Амага
280
400
520
640
г/слс»
0,4995
0,7136
0,9277
1,1418
т,°с
т*
г*
3,325
2,3275
1,7904
1,4547
0
2>203
эксп. [49]
0,42
0,55
0,74
1,02
расч.
0,411
0,558
0,765
1,069
75 | 150
2,808
эксп. [49]
0,30
0,42
0,62
0,91
расч.
0,395
0,562
0,761 ,
1,039
3,413
эксп. [49]
0,16
0,26
0,43
0,69
расч.
0,379
0,564
0,755
1,015
Азот
Плотнл/""Г1~
единицы
Амага
200
240
280
320
400
480
г/слс8
0,2500
0,3000
0,3500
0,4000
0,5000
0,6000
т,°с
т*
3,731
3,109
2,665
2,332
1,865
1,554
0
2,987
эксп. [92]
0,40
0,45
0,49
0,56
0,71
0,91
1
расч.
0,31
0,42
0,49
0,55
0,72
0,93
50
3,533
эксп. [92]
0,27
0,33
0,38
0,45
0,60
0,78
расч.
0,29
0,41
0,49
0,55
0,72
0,92
150
4,627
эксп. [92]
0,06
0,07
0,10
0,14
0,22
0,33
расч.
0,24
0,40
0,49
0,55
0,71
0,89
* Таблица 45
Сравнение экспериментальных и расчетных значений поправки на неидеальность
для энтропии S' (кал/моль-град)
Аргон
Плотность
единицы
Амага
280
400
520
640
г/слс8
0,4995
0,7136
0,9277
1,1418
т, °с
т*
V*
3,325
2,3275
1,7904
'1,4547
0 | 75 | 150
2,203
эксп. [49]
-1,234
-1,861
-2,600
-3,480
расч.
-0,6^35
-1,699
-2,754
-3,924
2,808
эксп. [49]
-1,147
-1,741
-2,433
-3,244
расч.
-0,5365
-1,561
-2,567
-3,666
3,413
эксп. [49]
-1,101
-1,671
-2,329
-3,085
расч.
-0,4773
-1,450
-2,418
-3,463
Азот
единицы
Амага
200
240
280
320
400
480
г/с*8
0,2500
0,3000
0,3500
0,4000
0,5000
0,6000
т, °с
т*
V*
3,731
3,109
2,665
2,332
1,865
1,554
0
2,987
эксп. [92]
-1,05
-1,29
-1,54
-1,80
-2,38
-3,05
1
расч.
-0,28
-0,66
-1,08
-1,49
-2,34
-3,21
50
3,533
эксп. [92]
-1,00
-1,22
-1,46
-1,72
-2,28
-2,91
1
1
расч.
-0,22
-0,59
-1,00
-1,40
-2,22
-3,05
150
4,627
эксп. [92]
-0,95
-1,16
-1,39
-1,63
-2,16
-2,74
расч.
-0,16
-0,48
-0,87
-1,25
-2,03
-2,82
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра 253
Г. МОДИФИКАЦИЯ УРАВНЕНИЯ СОСТОЯНИЯ ЛЕННАРДА-ДЖОНСА
И ДЕВОНШАЙРА, УЧИТЫВАЮЩАЯ ДВОЙНОЕ ЗАПОЛНЕНИЕ ЯЧЕЕК
В условиях малой плотности уравнение состояния Леннарда-Джонса и
Девоншайра неудовлетворительно с физической точки зрения, так как оно
предполагает отсутствие переходов молекул от одной ячейки к другой. Учет
возможности таких переходов приводит к тому, что некоторые ячейки будут
заполнены многократно, тогда как некоторые ячейки будут свободными.
Влияние многократного заполнения было недавно независимо и почти
одинаковым образом изучено Янсенсом и Пригожиным [80] и Поплом [78].
Ограничимся здесь рассмотрением лишь двойного заполнения, т. е. допустим,
что ячейки могут содержать 0, 1 или 2 молекулы и не более. Резуль-
таты подобного рассмотрения совершенно аналогичны результатам Леннарда-
Джонса и Девоншайра, за исключением того, что величина а (которая обычно
в выводах Леннарда-Джонса и Девоншайра полагалась равной е) берется те-
перь равной
" = l+-(iyf2. G.14)
Величины г//} и v{f равны значениям свободного объема для случаев, когде
в ячейке, образованной ближайшими соседними молекулами, находится одни
и две частицы соответственно :
G15)
v(f = [\\е- Ы1нте«т№те«тгЯктdTidrjK f
л
где через со(г) сокращенно обозначена величина с [<р(а + г) — ср(а) ]. Предель-
ными значениями а, как видно из G.14), являются единица в случае очень
высокой плотности и 1 + 1/ 2 в случае очень низкой плотности. Предельное
значение при малой плотности должно быть равно е ; это указывает на то, что
при малой плотности 88% коллективной энтропии обязано двойному запол-
нению ячеек.
Значения свободного объема v(f и v*f можно [78] связать с величинами
строго и нестрого центрированных свободных объемов vfh и vfs, о которых шла
речь в предыдущем параграфе,
v<p = vfs, G.17)
G.18)
Именно с помощью этих соотношений было получено уравнение (б.б). Следо-
вательно, рассмотрение строго и нестрого центрированных свободных объемов
почти эквивалентно учету двойного заполнения.
Модификация, учитывающая двойное заполнение ячеек, при больших
плотностях дает результаты, совпадающие с результатами первоначального
вывода Леннарда-Джонса и Девоншайра ; однако при малых плотностях, как
и предполагалось, наблюдаются большие расхождения. Для этой модели было
проделано лишь ограниченное число расчетов, и они показывают, что крити-
ческие постоянные аргона можно вычислить с большей точностью, нежели в
том случае, когда учитывается лишь однократное заполнение [80].
254 Гл. 4. Уравнение состояния плотных газов и жидкостей
§ 8. ТЕОРИЯ ДЫРОК ДЛЯ ЖИДКОСТИ И ПЛОТНОГО ГАЗА1)
В предыдущем параграфе указывается, что одна из трудностей теории
Леннарда-Джонса и Девоншайра заключается в том, что она не учитывает
наличия свободных мест в решетке. Давно установлено, что в решетчатой струк-
туре жидкостей имеется много «дырок» (около 0,5% в нормальной жидкости и
около 50% вблизи критической точки). Различные теории ставят целью учет
этого обстоятельства. В этих теориях отсутствует фактор а, характеризующий
коллективную энтропию. Мы начнем с рассмотрения того, как из представления
о наличии в решетке свободных мест получить теорию, описывающую поведение
жидкости. Затем рассматриваются четыре метода расчета величины свобод-
ного объема.
А. ОБЩАЯ ТЕОРИЯ ДЫРОК ДЛЯ ЖИДКОСТЕЙ2)
Как и при выводе общей теории ячеек в § 4, исходным пунктом в этом
случае также является классическая статистическая сумма ZN или конфигу-
рационный интеграл QN ансамбля, состоящего из N одноатомных молекул
ZN = A-3N qn = _!_ J е-ф(
N f
где А2 = Щ2птпкТ и интегрирование проводится по всему объему V сосуда,
содержащего жидкость. Разделим теперь этот объем на некоторое число ячеек.
Чтобы допустить возможность существования дырок в структуре жидкости,
выберем число ячеек L несколько большим числа молекул N. Тогда размер
ячейки q и объем v, приходящийся на молекулу, будут отличаться друг от
друга3):
? "
Далее предполагается, что ячейки достаточно малы, так что вероятность пре-
бывания двух молекул в одной ячейке пренебрежимо мала, и достаточно
велики, так что межмолекулярные силы между смежными ячейками не играют
роли. Оба эти условия могут выполняться только в том случае, когда меж-
молекулярные силы являются короткодействующими. Это, однако, имеет место
лишь в случае взаимодействия между неполярными нейтральными молекулами»
Выбор точных размеров ячеек будет проведен ниже.
Конфигурационный интеграл можно записать в виде суммы интегралов
по всем ячейкам :
?" = ЖГ 2 2 • • • 2 J dri J *i • • • J drNe-*Nw . (8.3)
Сумма состоит из LN членов, соответствующих L различным ячейкам, в
которых можно разместить N различных молекул. Так как ячейки выбраны
такими, что вероятность пребывания двух молекул в одной и той же ячейке
пренебрежимо мала, то члены суммы (8.3), соответствующие многократному
заполнению ячеек, можно отбросить. Тогда сумма будет состоять только из
Llj(L — JV)! оставшихся членов, соответствующих полному числу способов,
которыми N молекул можно разместить в L ячейках таким образом,
чтобы в каждой ячейке было не более одной молекулы. Эти члены составляют
) [81].
2) Этот вывод приближается к выводу, проделанному Оно [87]. (См. Я. И. Френкель,
Кинетическая теория жидкостей, М., 1945. — Прим. ред.)
8) В предыдущем параграфе размер ячейки и объем, приходящийся на одну молекулу,
совпадали и обозначались через v.
§ 8. Теория дырок для жидкости и плотного газа 255
LljNl(L — N)\ групп из JV! членов, каждый из которых отличается друг от
друга только нумерацией молекул. Так как все молекулы одинаковы, то эти
члены имеют одинаковое численное значение. Следовательно, конфигурацион-
ный интеграл можно переписать в виде
Qn = 2 j *i I * 1 • • • J drN e-«rN>i*T, (8.4)
где суммирование ведется по всем тем размещениям N молекул в L ячейках,
которые отличаются друг от друга не только перестановкой (или нумерацией)
молекул и при которых в каждой ячейке находится не более одной молекулы.
Как и в рассмотренном выше методе ячеек, оценка интеграла зависит от
выбора системы решетки и формы и ориентации ячеек. Рассмотрим такую
систему решетки, когда в любом из узлов решетки имеется с ближайших моле-
кул, расположенных на расстоянии а. (Для гранецентрированной кубической
упаковки с = 12 и а3 = q ]Г2.) Для любой заданной конфигурации можно
определить
СО[ = доля ближайших соседних узлов i-й молекулы, которые свободны,
Если все молекулы находятся в центре соответствующих ячеек, то потенциаль-
ная энергия i-й молекулы равна сA —<*>i)(p(a). Соответственно полная потен-
циальная энергия системы равна
Ф(г") = ± (N - Q) <р (а) (8.6)
(молекулы в центре ячеек).
Когда молекулы расположены не в центре ячеек, для потенциальной энергии
системы можно получить приближенное выражение путем, подобным тому,
который использовался в теории ячеек. Сначала мы рассмотрим взаимодействие
«блуждающей» молекулы с одной из ближайших соседних молекул, когда послед-
няя расположена в центре своей ячейки. Когда блуждающая молекула нахо-
дится в точке г своей ячейки, она обладает запасом потенциальной энергии, на
[<р(а + г) — ср(а) ] выше того значения, которое получается, когда эта моле-
кула находится в центре ячейки. Аналогичная разность значений потенциаль-
ной энергии получается при наличии A — ы)с ближайших соседних молекул
путем применения процесса «сглаживания» так, как это было описано в § 6.
Таким образом, мы предполагаем, что потенциальная энергия рассматриваемой
молекулы зависит не от положения своих соседей, а только от их количества.
Следовательно, предполагается, что полная потенциальная энергия системы
может быть с достаточной точностью записана в виде
Ф(г") = | (N - О) ср{а) + 2 с A - со, Й
(молекулы не находятся в центре ячеек).
В действительности, конечно, зависимость энергии от оу{ не столь проста ;
энергия зависит от расположения свободных узлов решетки так же, как и от
их количества.
Статистическую сумму ZN можно теперь получить, подставляя этот резуль-
тат в (8.4),
ZN = k^Ne-Nc^'2kT2{i(^i)](^) • • • /(a>N)}eOe«a)pkT, (8.8
где
J[i^^TF)-9(fl)) Am*dr. (8.9>
256
Гл. 4. Уравнение состояния плотных газов и жидкостей
Функция ](со) является обобщенным свободным объемом, в котором учиты-
вается наличие свободных соседних узлов решетки. Когда таких узлов нет,
о = 0 и i(co) становится в точности свободным объемом, приведенным к сфери-
ческой форме и определенным в F.3). Когда все ближайшие соседние молекулы
отсутствуют, со = 1 и /(со) равна размеру ячейки.
Из (8.8) можно получить предельные выражения
для высокой плотности
ZN = A-3N [j @)] N e-Nc<p(a)l2kT ^
ДЛЯ НИЗКОЙ ПЛОТНОСТИ
L1
N!(L-N)!
)! e
(8.10)
(8.11)
Первое из этих соотношений является приближением Эйнштейна для статисти-
ческой суммы кристалла. Второе выражение содержит множитель еы, кото-
рый необходим в предельном случае для идеального газа (см. § 4). Таким обра-
зом, мы видим, что при выводе соотношения (8.8) можно обойтись без всякого
упоминания о сомнительной «коллективной энтропии»; эта зависимость по-
зволяет делать плавный переход от самых высоких до самых низких плотно-
стей.
Б. ЛИНЕЙНОЕ ПРИБЛИЖЕНИЕ ЛОГАРИФМА ВЕЛИЧИНЫ
СВОБОДНОГО ОБЪЕМА
Для доведения до конца суммирования в (8.8) необходимо провести неко-
торое упрощение функциональной зависимости для ](со). Точная запись выра-
жения зависимости от со, как видно из (8.9),
слишком сложна. Сравнение выражения (8.9)
с F.3) показывает, что /(со) при температуре
Т такова же, как и vf при температуре
7/A — о>) для тех же размеров ячейки.
Следовательно, функциональную зависимость
j(co) можно получить из функции g(v*, Т*),
определенной с помощью соотношений G.2)
и G.6)
(^) (8.12)
1
In(a3/Y2) >
/1
/ 1
/ 1
/ 1
/ 1
/ I
ь/ /
/ /
/ / ^
/ ^/
4n[2Jta3g(v*T*)]
С
а
W
где q* — приведенный размер ячейки q\o*. На
фиг. 68 приведен график зависимости in f(co)
от со9 полученный из этого соотношения и
таблиц для g{v*y T*) описанных в § 7.
До сих пор расчеты, проведенные на
основе теории дырок, опирались на допу-
Фиг. 68 Действительная зависи- щение 0 том, что In/(со) есть линейная функ-
мость обобщенного свободного объе- ГГ ' 1Х ' ту
ма In/ (со) от со согласно (8.12) Ция от ^
{сплошная кривая) и линейные приб-
лижения к In/ (со), используемые
различными исследователями в рас-
четах (пунктир). где точки j и j ограничивают отрезок прямой
и — усредненное значение со для тем- Tnm/.QV Л1 _ П и л» — 1 Ня гЬмг ««пт/ячянм
пературы, плотности и размера ячейки, В ТОЧКаХ СО — U И СО — 1. Па фИГ. 00 ПОКазаНЫ
для которых проводилась кривая d [8i]. сделанные четыре линейных приближения к
In/(со): а — приближения Чернуши и Эйринга
[88]; b —Оно [87]; с — Пика и Хилла [89]; d — Раулинсона и Кертисса
[81]. Очевидно, что прямые линии недостаточно хорошо описывают зависи-
мость In j(co) от со.
(8.13)
§ 8. Теория дырок для жидкости и плотного газа 257
Если линейное приближение в виде (8.13) подставить в (8.8), то для стати-
стической суммы получим выражение
ZN = А-алг e-Nc<Ka)!2kT{jN % да, L, Q) еп«кТ}, (8.14)
где
i [±) (8.15)
Величина K(N, L, Q) есть число конфигураций с заданным значением Qy когда
N молекул распределяются по L ячейкам. Нельзя дать точного значения этой
функции, однако ее можно оценить с помощью «квазихимического приближе-
ния»1). Это позволяет нам записать статистическую сумму в окончательном виде
здесь
где
Величина х относится к среднему числу дырок около какой-либо одной моле-
кулы сед так, что
При высоких плотностях число ячеек почти равно числу молекул, х при-
ближается к единице, а со — к нулю, т. е. в среднем отсутствуют свободные
узлы решетки, ближайшие к данному. При низких плотностях число ячеек
намного больше числа молекул, х стремится к нулю, со — к единице, т. е.
в среднем все ближайшие узлы решетки не заняты молекулами. Показано,
что в критической точке приблизительно половина всех узлов решетки сво-
бодна.
Из выражения для статистической суммы в виде (8.16) можно получить
уравнение состояния и выражения для различных термодинамических вели-
чин2). Эти величины нельзя вычислить достаточно точно, пока не определены
размер ячейки и параметры /0 и /г Когда это сделано, термодинамические вели-
чины получаются как функции температуры и плотности. Параметры /0 и /,
можно получить различным образом; ниже рассматриваются случаи, пред-
ставленные на фиг. 68. Размер ячейки можно положить равным некоторому
произвольному постоянному значению, не зависящему от плотности и тем-
пературы ; например, размер ячейки можно взять равным объему, приходя-
щемуся на одну молекулу в кристалле при абсолютном нуле. Он может быть
также выбран таким, чтобы при каждом значении температуры и плотности
величина свободной энергии А = U — TS была минимальна. Последнее опре-
деление теоретически более обосновано. Однако первое имеет то преимущество,
что оно иногда позволяет получить для критических постоянных подробные
выражения.
*) Этот метод разработан в различных формах Гутгенгеймом и Бете [61 ].
2) Подробная запись этих выражений, полученных с помощью ряда теории дырок, дана
Раулинсоном и Кертиссом [81 ].
258
Гл. 4. Уравнение состояния плотных газов и жидкостей
В. СРАВНЕНИЕ ВЫЧИСЛЕНИЙ, ПРОВЕДЕННЫХ НА ОСНОВЕ
ТЕОРИИ ДЫРОК, С ЭКСПЕРИМЕНТОМ
Разочаровывающим является то обстоятельство, что указанные выше
четыре теории дырок имеют мало преимуществ по сравнению с выводами Лен-
нарда-Джонса и Девоншайра. Особенно существенно то, что приближение
Раулинсона и Кертисса (линия d на фиг. 68) не дает лучшего описания плот-
ных газов и жидкостей. Это приближение в определенном отношении является
наилучшим из всех четырех, представленных на фиг. 68, поскольку оно дает
правильное значение как величины In /(со), так и ее первой производной при
со = со. Тот факт, что этот выбор линейного приближения кажется лишь не-
многим лучше остальных, по-видимому, указывает на то, что нельзя добиться,
решительного улучшения теории без отказа от некоторых из допущений, вве-
денных в основы теории1).
Мы заканчиваем этот раздел сравнением результатов вычислений, прове-
денных на основе указанных теорий дырок, друг с другом и с известными экспе-
риментальными данными.
1. Предел при малых плотностях. Второй вириальный коэффициент.
В первую очередь эти теории предназначены для получения уравнения состоя-
00,4 Q6 0,8 1
40 60 80100
Фиг. 69. Расчетное значение второго вириального коэффициента как функция приведенной
температуры в соответствии с различными теориями.
а — Чернуши и Эйринг [88]; Ь — Оно [87]; с — Пика и Хилла [89]; d — Раулинсона и Кертисса [81 ].
Внизу справа показано изменение размера ячейки в зависимости от приведенной температуры в соответствии
с кривой d [81].
ния жидкостей. Простым критерием удовлетворительности получаемого урав-
нения являются значения второго вириального коэффициента. С другой
стороны, имеются основания полагать, что если какая-либо модель не дает
г) Из позднейших работ, посвященных теории дырок, можно указать М с L е 11 а п
A. G., Phil. Mag., 3, 707 A958); Hill Т. L., Journ. Chem. Phys., 28, 1179 A958). —
д
Прим. ред.
§ 8. Теория дырок для жидкости и плотного газа
259
приемлемого значения второго вириального коэффициента, то она не даст и
удовлетворительного значения критического коэффициента pcVcIRTc.
Теория Леннарда-Джонса и Девоншайра для второго вириального коэф-
фициента дает нуль, поскольку первый член поправки на неидеальность про-
порционален v*. Значения второго вириального коэффициента, получаемые
из теорий дырок, даны на фиг. 69 вместе с кривой В*(Т*) для потенциала
F-12) Леннарда-Джонса, рассчитанной по формуле F.3) гл. 3. Кривая d явля-
ется наилучшим приближением ко второму вириальному коэффициенту. Инте-
ресным обстоятельством является то, что приближения Ьу с и dy когда размеры
ячеек определяются условием минимума для А = U — TS, дают при высоких
температурах
В*(Г*) = const x
(8.19)
Как можно видеть из соотношения F.3) гл. 3, эта зависимость от Т* для пре-
дельно высоких температур является правильной ; постоянная пропорцио-
нальности оказывается неожиданно хорошей, будучи лишь на 15% больше.
2. Фактор сжимаемости плотного газа. Казалось бы, что полученные
удовлетворительные значения второго вириального коэффициента позволяют
надеяться на получение на основе теории дырок уравнения состояния, хорошо
согласующегося с экспериментом при высоких плотностях. Однако сравнение
2JS-
2ft-
Щ
1 i 1
1 1
Теория
Роулинсона-^ ,
и Кертисса^'
Теория s у
Леннарда-Джонса ,' ,
и Девоншайра ч^' /
У
_ У
У
У
- •
/
V
^у^Энсперимк
\ |
s у
' S -
у
гнт
-
1
100 200 300 400 500 600
Плотность, единицы Амага
700
Фиг. 70. Уравнение состояния водорода при 0°С в соответствии с теорией Леннарда
Джонса и Девоншайра и теорией Раулинсона и Кертисса.
Экспериментальная кривая получена Михельсом и др. [90].
кривых, приведенных на фиг. 70, показывает, что это не так. Проводится срав-
нение между результатами измерений для водорода при высокой плотности и
расчетов, проделанных как Леннардом-Джонсом и Девоншайром, так и Раулин-
соном и Кертиссом. Обе расчетные кривые приближаются друг к другу при
ш -+ 0, но ни одна из них не совпадает с экспериментальными результатами.
3. Критическая точка. В табл. 46 проводится сравнение результатов рас-
четов критических постоянных, проведенных с помощью различных теорий.
Ни одна из теорий не дает хорошего набора значений критических постоянных.
Значения, получаемые с помощью метода Раулинсона и Кертисса, не приве-
дены, так как в критической точке, по-видимому, размер ячеек приближается
к 5, что выходит из области затабулированных значений g, gz и gm. Это само
260
Гл. 4. Уравнение состояния плотных газов и жидкостей
Таблица 46
Средние значения для
Ne, Ar, N2
Леннард-Джонс и Девоншайр
Чернуши и Эйринг [88 ]
Оно [87]
Пик и Хилл [89]
PeVe/RT,
0,293
0,591
0,342
0,342
0,719
1,28
1,30
2,74
0,75
1,18
0,119
0,434
0,469
0,128
0,261
3,15
1,77
2,00
2,00
3,25
0,000
0,455
0,455
0,175
по себе является слабой стороной теории, одно из условий которой заклю-
чается в том, что размер ячеек должен быть настолько малым, чтобы возмож-
ность пребывания более чем одной молекулы в любой ячейке была прене-
брежимо мала.
4. Равновесие жидкость—пар. Проведено сравнение [81 ] эксперимен-
тальных значений давления насыщенного пара и результатов расчетов, проде-
ланных с помощью теории Леннарда-Джонса и Девоншайра и теории Оно.
Расчетные кривые указывают на линейное изменение рпар. в зависимости от
1/7, характерное и для экспериментальных кривых. Выводы Леннарда-Джонса
и Девоншайра дают несколько лучшее согласие с экспериментом, нежели
теория Оно.
5. Точка кипения. Точка кипения жидкости при 1 атм является величи-
ной, которую нельзя вычислить из уравнений типа уравнений «соответственных
состояний». Однако если предполагать, что наибольшие критические давления
приближаются к 40 атму то за точку кипения можно брать такую температуру,
при которой Рпар./Рс = 1/40. Экспериментальное значение Т% равно прибли-
зительно 0,87. Теория Леннарда-Джонса и Девоншайра дает значение, равное
примерно 0,80, а теория Оно — около 0,51. Тем не менее отношение значений
Т% и Т* весьма постоянно:
Эксперимент
0,67
Теория Леннарда-Джонса и Девоншайра
0,62
Теория Оно
т*
= 0,68
Эти результаты находятся в хорошем согласии со старым эмпирическим пра-
вилом Гульдберга и Гюи (см. § 1).
6. Энтропия испарения в точке кипения. Экспериментальное значение
энтропии испарения при температуре кипения равно 16,8 кал/моль для инерт-
ных газов и около 21 кал/моль для высоко кипящих неполярных компонент.
Теория Леннарда-Джонса и Девоншайра и теория Оно дают 20,3 и 23,2 кал)моль
соответственно.
Из результатов этого и предыдущего разделов со всей очевидностью сле-
дует, что различные теории, использующие представления о решетках, содер-
жат предположения, существенно ограничивающие применимость получаемых
результатов. В настоящее время представляется, что более точные результаты
можно получить с помощью метода, опирающегося на радиальную функцию
распределения; этот метод рассматривается в следующем параграфе.
§ 9. Выражение уравнения состояния через бинарную функцию распределения 261
§ 9. ВЫРАЖЕНИЕ УРАВНЕНИЯ СОСТОЯНИЯ
ЧЕРЕЗ БИНАРНУЮ ФУНКЦИЮ РАСПРЕДЕЛЕНИЯ1)
Различные решеточные теории уравнения состояния, рассмотренные в
предыдущем параграфе, основаны на различных апроксимациях статистиче-
ской суммы N частиц. Эти приближения соответствуют очень высоким плот-
ностям, когда система имеет структуру, более или менее отвечающую строению
решетки. Для умеренных значений плотности удовлетворительных резуль-
татов пока не получено.
Наиболее обещающий альтернативный подход к решению этой проблемы
основан на выражении уравнения состояния через бинарную функцию распре-
деления пB\г12) или радиальную функцию распределения2) g{r12)y которые опре-
деляются соотношением A.14) гл. 3. В гл. 3, § 1, было показано, что уравнение
состояния можно записать в виде A.13)
pV = NkT-\
или
Здесь введены предположения о том, что: 1) межмолекулярные силы явля-
ются аддитивными (т. е. силы, действующие между любой парой молекул, не
зависят от присутствия любых других молекул); 2) межмолекулярный потен-
циал <р(г) не зависит от углов; 3) для описания системы применяется классиче-
ская механика.
Тот факт, что уравнение состояния зависит только от бинарной функции
распределения, а не от функций распределения более высокого порядка явля-
ется следствием первого предположения об аддитивности сил.
Сначала мы рассматриваем свойства бинарной функции распределения и ее
связь с потенциалом усредненной силы. Затем выводим различными путями
точные интегральные уравнения для лB)(г12). Встречающееся здесь затруднение
заключается в том, что в этих уравнениях содержится также nC)(ri> Г2> гз) —
функция распределения для трех молекул. Следовательно, должны быть
сделаны упрощения, исключающие из уравнений эти функции ; достигается
это путем так называемого суперпозиционного приближения. Необходимо
провести еще большую расчетную работу, чтобы оценить пригодность этих
методов.
А. СВОЙСТВА БИНАРНОЙ ФУНКЦИИ РАСПРЕДЕЛЕНИЯ3)
Бинарная функция распределения пB)(г1} г2) и радиальная функция рас-
пределения g(r12) определялись соотношением A.14)
i, r2) = n*g(r12) = [(N - 2)!
Функция g(r) стремится к единице, когда г возрастает до бесконечности ; величи-
на 1/2^л?(г) АпгЧг есть число пар молекул, расстояние г между которыми лежит
внутри интервала г и г + dr. В гл. 3, § 3 показано, что радиальная функция
распределения имеет следующий вид в пределе при низких плотностях:
^ (9.1)
ЧСм. [93].
2) В жидкости g(r) обычно называют функцией распределения Дебая—Менке.
8) О различных типах функций распределения в классической статистической механике
см. гл. 2, § 1.
262
Гл. 4. Уравнение состояния плотных газов и жидкостей
В условиях умеренных значений плотности радиальную функцию распреде-
ления можно разложить в степенной ряд по плотности числа частиц в единице
объема так, как это показано в выражении C.8) гл. 3. Если предполагается,
что межмолекулярные силы аддитивны, то это соотношение при умеренных
значениях плотности принимает вид
gM = e~^r^kT [ 1 + п J (*-«*'»>/*т - 1) (*-*'«>/«• — l)dr3 + О(п2)]. (9.2)
Сравнивая это выражение с C.8) и (ЗЛО) гл. 3, видим, что член, стоящий внутри
скобок и пропорциональный п, относится ктретьему вириальному коэффициенту.
Вычисления1) величины g(r12)
были проделаны для потенциала
Леннарда-Джонса. На фиг. 71 по-
казаны результаты, соответствую-
щие выражению (9.1) для различ1
ных значений приведенной темпе-
ратуры 7* в условиях предельно
малой плотности. Функция рас-
пределения имеет единственный
максимум, соответствующий тому,
что вероятность пребывания пары .
молекул на таком расстоянии друг
от друга, при котором потенциаль-
ная энергия минимальна (г = 21/«а),
превышает вероятность пребывания
на любом другом расстоянии.
Уменьшение величины максимума
с ростом Т* соответствует увели-
чению беспорядка, наступающего
с ростом теплового движения.
* На фиг. 72 даны кривые g(r*)
во втором приближекии, заданном
ь ¦
1 1
Фиг. 71. Бинарная функция распре-
деления в первом приближении, полу-
ченная из соотношения (9.1).
Для получения этих кривых использовался
потенциал F-12) Леннарда-Джонса [93].
Фиг. 72. Бинарная функция распределения
во втором приближении, полученная с
помощью (9.2).
Для получения кривых использовался потенциал
F-12) Леннарда-Джонса [93 ].
Соотношением (9.2) с использованием потенциала Леннарда-Джонса [94, 95 ]. На
фиг. 72, а показано изменение g(r*) с плотностью при постоянной температуре
а на фцг. 72, б дано изменение g(r*) с температурой при постоянной плотности.
г) Оценка величины бинарной функции распределения выполнена де Буром и Михель-
сом [94], а также Монтроллом и Майером [95]. Результаты этих двух работ находятся между
собой в хорошем согласии.
§ 9. Выражение уравнения состояния через бинарную функцию распределения 263
Эти кривые имеют два максимума, т. е. у любой заданной молекулы сущест-
вует две окружающие ее оболочки, на которых относительно наиболее веро-
ятно найти другую молекулу. Третье, четвертое и т. д. приближения для g(r*)
приводят к появлению на кривой третьего, четвертого и т. д. максимума. Это
соответствует росту «ближнего порядка» с ростом плотности.
Кривые g(r*) можно вычислить из опытов по рассеянию рентгеновских
лучей [96, 102 ]; полученные таким образом кривые имеют тот же вид, что и
теоретические [102]. Можно удивляться тому, что результаты по рассеянию
рентгеновских лучей не использованы для расчета уравнения состояния. К не-
счастью, в настоящее время в большинстве случаев это невозможно потому,
что: 1) требуются исключительно точные экспериментальные измерения; 2) необ-
ходимо провести эксперименты в широком диапазоне температуры и плотности;
3) должны быть более точно известны показатели рассеяния рентгеновских
лучей, на атомах; 4) в многоатомных молекулах существуют эффекты интер-
ференции вследствие рассеяния различными атомами в молекуле.
Б. «ПОТЕНЦИАЛ УСРЕДНЕННОЙ СИЛЫ»
Другим способом представления бинарной функции распределения явля-
ется выражение ее через усредненную силу, действующую на одну из двух
молекул. Рассмотрим пару молекул 1 и 2 и будем интересоваться средней силой,
действующей на молекулу 1 со стороны всех других молекул, как функцией
межмолекулярного расстояния г12. Ее можно получить в результате соответ-
ствующего усреднения величины F^r^) = — ЭФ^^/Э^ по каноническому
ансамблю, представляющему систему молекул
J J ?-
= [(N - 2)! Q^J-i J(- *L) e-*W*r dr*-2. (9.3)
Из определения пB)(гх, г2) очевидно, что F^, r2) имеет следующую связь с
бинарной функцией распределения :
F (г г ^ - dW{Tl>Га) - кТ Э1п"B)(г1>г2)
*Wi, ч) — ъг— - Kl о^— •
Здесь мы ввели также потенциал усредненной силы ^Р(т1} г2). В соответ-
ствии с этим бинарная функция распределения запишется в виде
л<2)(Г1, г2) = п2 e~w^ т^кТ. (9.5)
Множитель п2, появляющийся в связи с произвольной постоянной интегриро-
вания, выбран таким, чтобы ^(r^rj) -> 0 при г12 -> «э. Очевидно, что в пределе
при малой плотности потенциал усредненной силы приближается к межмоле-
кулярной потенциальной функции <р(г12).
Величины, определяемые последними двумя соотношениями, могут быть
обобщены :
(9.7)
Здесь F^r*) — сила, действующая со стороны других молекул на молекулу 1
как функция расположения группы молекул {ft} = 1, 2, 3, ..., ft.
264 Гл. 4. Уравнение состояния плотных газов и жидкостей
В. ВЫВОД ИНТЕГРАЛЬНЫХ УРАВНЕНИЙ ДЛЯ БИНАРНОЙ
ФУНКЦИИ РАСПРЕДЕЛЕНИЯ1)
Как было показано, уравнение состояния можно выразить через бинар-
ную функцию распределения г№. Рассмотрим теперь вывод интегрального
уравнения для этой функции. Получающееся уравнение содержит не только
п'2\ но и пC). Подобно этому, уравнение для лC) содержит функцию распре-
деления, на один порядок более высокую и т. д. Следовательно, чтобы получить
точное решение для лB), необходимо решить систему из N связанных уравнений.
Мы рассмотрим здесь три различных вывода, которые дают подобную
систему из N уравнений. В следующем разделе обсуждаются методы прибли-
женного решения этих уравнений.
1. Вывод с помощью потенциала усредненной силы. Понятие о потенциале
усредненной силы можно использовать в качестве исходного пункта для вы-
вода уравнения для бинарной функции распределения. Если межмолекуляр-
ные силы являются аддитивными, то
- F>=ж ф^ = J ?*'«>¦ <9-8>
Используя это соотношение и выражение для ?lt записанное в виде (9.3) вместе
с определением лC), получаем
uJ " J ^
я(«)(Г1,г2,г3)
F ^-
tl - ~ ЭГ1
Первый член в правой части этого равенства дает непосредственно силу,
действующую на молекулу 1 со стороны молекулы 2. Второй член дает вклад
других молекул в усредненную силу ; величина лC)/лB) есть вероятность найти
третью молекулу в точке г3, когда положение молекул 1 и 2 фиксировано. При-
равнивая это выражение выражению для ?г в виде (9.4), получаем
. т Э In яB) (г1? г2) _ Эу(г12) Г Эу(г1э) яC>(Г1, г2, г3) - ,Q * nv
KI дГг "" frl J ~Ш[ Щ^^Г *' ( }
Подобным образом можно получить уравнения для других л(л), в которых
фигурируют также функции более высокого порядка л(/1+1).
Полученные результаты можно несколько упростить, учитывая, что функ-
ции /1(л) являются функциями только относительных координат2)
г2)= п\гп)^п<*1
1, Г2, Г3) = ПC)(Г12, Г23, Г13) = /2<i>3 .
Умножая обе части уравнения (9.10) на (г12/г12) = (гх — г2)/г12, получаем
Эг12 "- Эг1а J
Эг18 пB)
J
^7 J J b
О |Гм-Гм|
Последняя форма уравнения представлена в «биоцентрических» координатах.
2. Вывод из уравнения Лиувилля. Функции распределения л<л)(гл) можно
получить из функций распределения fN\rN9 pN) интегрированием по pN и
г) По этому вопросу см. книгу : Н. Н. Боголюбов, Проблемы динамической теории
в статистической физике, М., 1946. — Прим. ред.
2) Эти соотношения справедливы только в условиях равновесия.
§ Р. Выражение уравнения состояния через бинарную функцию распределения 265
г^~л, как это описано в гл. 2, § 1. Зависимость fN\rMy ры) от времени описы-
вается уравнением Лиувилля A.10) гл. 2. Поэтому можно получить уравнение
для nSh\rh) путем интегрирования уравнения Лиувилля по р^ и rN~h9 а также
используя то обстоятельство, что мы рассматриваем условия равновесия.
Уравнение Лиувилля для fM)(rM, ры) можно записать в виде
* <9ЛЗ>
где ?к — сила, действующая на fc-ю молекулу со стороны всех других молекул.
Уравнение для функции распределения более низкого порядка /(л) для набора
молекул {Л} = 1, 2, 3, ..., ft можно получить путем интегрирования этого
уравнения по остающимся N — h координатам
-«^-.)j =0. (9.Н,
Для получения этого уравнения используется теорема Грина и предполагается,
что fN) достаточно быстро стремится к нулю при | рк | -> оо и | qk | -> оо. Если
предполагается, что в системе действуют только парные силы1), то для преоб-
разования уравнения (9.14) можно воспользоваться соотношениями типа (9.8)
Таким образом, единственное уравнение для функции fN) преобразовано в
систему связанных уравнений для функций распределения различных поряд-
ков. Уравнение для каждой /(Л) включает функцию следующего более высокого
порядка /(й+1). Вот почему появление в этих уравнениях функций распреде-
ления двух различных порядков является следствием предположения о на-
личии парных взаимодействий.
Поскольку полученная система связанных уравнений содержит 6N пере-
менных, то она не имеет большего практического значения, чем исходное урав-
нение Лиувилля, пока не сделаны допущения, позволяющие ее замкнуть. По-
следовательность уравнений можно ограничить, если /(/1+1) приближенно
выразить через функции распределения более низкого порядка. Например,
если -предполагать, что система приближается к идеальному газу, так что
играют роль лишь парные столкновения, или если предполагается наличие
«молекулярного хаоса», то представляется возможным достаточно точно выра-
зить /B^ через /A). Это приближение приводит в кинетической теории к обычному
уравнению Больцмана (см. гл. 7, § 1).
Свойства плотных газов можно выразить через f*\tl9 r2, рх, р2). В действи-
тельности, в условиях равновесия необходима лишь функция пB\г1} г2). Урав-
нение для этой функции можно получить, умножая (9.15) на р, и интегрируя
по пространству импульсов рл. Нами также используются соотношения
Jp,/Wrfp* = O, / = 1,2,3,..., Л, (9.16)
=4*r, 1 = 1,2,3,..., Л. (9.17)
Первое соотношение содержит утверждение о том, что средний момент коли-
чества движения равен нулю, а второе в кинетической теории служит опре-
*) Имеется в виду сформулированное в начале этого параграфа предположение об адди-
тивности межмолекулярных сил. — Прим. ред.
266 Гл. 4. Уравнение состояния плотных газов и жидкостей
делением температуры (см. гл. 7, § 2). Тогда уравнение для л(Л)(гЛ) легко полу-
чается в виде
1X1 "~Э7Г~ ~ ~ "Э7Г (Г ' J 8г, ~n<*>(?)—UIM-i- ^1U'
Если Л = 2, то получается тот же результат, что в уравнении (9.10).
3. Вывод с помощью введения параметра связи1). Другим способом решения
задачи является представление полной потенциальной энергии системы Ф(ты)
в таком виде, который включает параметр связи ?,
N N N
Ф(г^) = ?2 <рг] + 2Т 2 Vjk • (9.19)
У=2 j = 2k=j+l
Действительное положение, конечно, описывается значением 1=1.
Когда f = 0, полная потенциальная энергия не учитывает взаимодействия
молекулы 1 с другими молекулами, т. е. молекула 1 является «несвязанной».
Если функцию распределения записать с помощью потенциала (9.19), то она
также будет функцией параметра f.
Подстановка приведенного выше выражения для Ф(гы) в формулу для
/zB) и соответствующее дифференцирование по параметру связи дает
= -ср(г ) - [Ыг ) ГпC)(гьг2>гз) _ "B>(г1,г3)-| df =
(9.20)
J
О | г1Ш-г1Я |
Этот прием позволяет нам получить систему связанных интегральных урав-
нений в форме, отличной, но эквивалентной той, которая была получена с
помощью двух других описанных методов.
Г. РЕШЕНИЕ ИНТЕГРАЛЬНОГО УРАВНЕНИЯ
ДЛЯ БИНАРНОЙ ФУНКЦИИ РАСПРЕДЕЛЕНИЯ.
СУПЕРПОЗИЦИОННОЕ ПРИБЛИЖЕНИЕ
Из интегральных уравнений (9.12) и (9.20) нельзя найти лB) до тех пор,
пока не сделаны какие-либо предположения о записи лC) через лB). Для этой
цели предложен так называемый метод суперпозиционного приближения.
Границы справедливости этого метода неизвестны.
Обычно предполагают, что потенциальную энергию системы, состоящей
из трех молекул, можно записать как
Ф(Гь г2, гз) = <р(г12) + ср(гп) + <р{г^). (9.21)
Если то же соотношение записать для потенциала усредненной силы
Пгг, Ч, Гз) = У(д + УМ + УУ, (9-22)
го из (9.7) следует
. [103].
§ Р. Выражение уравнения состояния через бинарную функцию распределения 267
Это «суперпозиционное приближение»1) является, очевидно, достаточно точным
в условиях предельно низкой плотности. При высоких плотностях это при-
ближение не полностью оправдано2).
Суперпозиционное приближение было использовано для исключения лC)
из двух интегральных уравнений для г№. Так как соотношение (9.23) не вполне
точно, то его использование в уравнениях (9.12) и (9.20) приводит к двум раз-
личным приближенным интегральным уравнениям для бинарной функции
распределения — уравнению Борна—Грина—Айвена и уравнению Кирквуда.
Решения этих двух уравнений различны, и это различие является мерой закон-
ности суперпозиционного приближения. Поскольку принцип суперпозиции
дает точное значение лC) для предельно малых плотностей, то получающееся
решение для г№ в виде степенных рядов по л правильно до второго члена.
Поэтому получающееся уравнение состояния имеет правильный третий ви-
риальный коэффициент. Выражения для последующих вириальных коэффициен-
тов, однако, являются только приближенными3).
1. Приближенное интегральное уравнение Борна—Грина—Айвена4). Рас-
смотрим теперь первое интегральное уравнение для г№ (9.12) и используем
суперпозиционное приближение. Это уравнение можно полностью проин-
тегрировать по г12 от произвольного значения г12 = г до г12 = °°. Мы будем
использовать следующие предельные значения : лB) -> л2 и <р -> 0 при г12 -> оо.
Чтобы вычислить интеграл по г12, г23, г13, необходимо изменить порядок интег-
рирования. Однако в этом частном случае сходимость интегралов такова, что
не позволяет делать простого изменения порядка интегрирования. Непосред-
ственные, но очень длинные преобразования дают следующее уравнение для
g(r) = ^lfi
kT\ng{r)=-<p{r) + 2nn\ J -Ig-frsfeg, ~ 1)['" ~ Bгг3" r)i]r*3dri3dr13. (9.24)
О r-ria
Здесь у и g с отрицательными аргументами обозначены через <р{— г) = <р(г)
и g(— г) = g(r)y а также gtj — g{ru). Это приближенное уравнение было полу-
чено независимо Борном и Грином [109], а также Айвеном [108]. Грин [110]
добился успеха в решении этого интегрального уравнения, сделав ряд даль-
нейших допущений, анализ которых несколько сложен. Из решения были по-
лучены приближенные интегралы для вириальных коэффициентов ; эти инте-
гралы значительно проще в обращении, нежели соответствующие точные
интегралы, рассмотренные в гл. 3. С помощью уравнения (9.24) Кирквуд и
Боггс [111] получили уравнение состояния для твердых сфер.
2. Приближенные интегральные уравнения Кирквуда6). Рассмотрим теперь
применение принципа суперпозиции ко второму интегральному уравнению
х) Это суперпозиционное приближение может быть представлено как аналогия при-
ближению Хартри—Фока в квантовой механике, примененному к функции пC) в конфигу-
рационном пространстве трех молекул в виде произведения функций лB) в конфигурационном
пространстве молекулярных пар. Соответствующее приближение типа Хартри—Фока в
конфигурационном пространстве единичной молекулы приводит к теории свободного объема
плотных газов и жидкостей (см. § 4). [Условия применимости суперпозиционного прибли-
жения рассмотрены в работе В. В. Ватолло, ЖФХ, 33, №11, 2393A959). — Прим. ред.]
2) Нийбур и Ван Хоув [104, 105] недавно предложили другое приближение, которое
дает для твердых сфер лучшие результаты, нежели суперпозиционное приближение.
8) Для твердых сфер точное значение четвертого вириального коэффициента равно
D = 0,2869 Ь*. Согласно принципу суперпозиции и приближенному интегральному урав-
нению Айвена, Борна и Грина, D = 0,2252 bl. Это значение недавно получили Харт, Валлис
и Под [106], а также Рашбрук и Скойнс [107]. В соответствии с приближением Нийбура и
Ван Хоува [104, 105] D = 0,2500 Ь%.
4)См. [108, 109].
5)См. [103].
268
Гл. 4. Уравнение состояния плотных газов и жидкостей
для лB) (9.20). Интегрируя это уравнение по
что g -> 1 при ? -> 0, можно получить
• * (г+г18)
J
+1
J
и используя то обстоятельство,
(9.25)
О О | г-г» |
Это и есть приближенное интегральное уравнение, полученное Кирквудом
[103]. Недавно оно было использовано для вычисления бинарной функции
распределения для твердых сфер [112]1).
3. Сравнение двух приближенных интегральных уравнений. Оба описан-
ных приближенных интегральных уравнения можно записать в обобщенном
виде. Введем параметр связи ? в
91—i -т ,_ уравнения Борна и Грина и для
этих двух случаев определим
ядро K(f, f)
\t\
(Борн—Грин—Айвен), (9.26)
(9.27)
о \t\
(Кирквуд).
С помощью этих функций оба
приближенных интегральных
уравнения записываются в виде
+ 2яп х
х f[K(r-r23, Z)-K(r + r^,I)] x
"о
-l)-^-dre. (9.28)
4. Численное решение при-
ближенных интегральных урав-
нений. Интегральные уравнения
для радиальной функции рас-
пределения были решены как для
твердых сферических молекул
[112], так и для молекул, взаи-
модействующих друг с другом
в соответствии с модифициро-
ванным потенциалом Леннарда-
Джонса [114]. На фиг. 73 срав-
нивается фактор сжимаемости,
вычисленный из уравнения Борна—Грина—Айвена и уравнения Кирквуда для
твердых сферических молекул. Приводится также соответствующая кривая,
полученная из теории приведенного к сфере свободного объема для твердых
Фиг. 73. Фактор сжимаемости pv/kT как функ-
ция приведенного объема v* для твердых сфери-
ческих молекул диаметром с.
а — по приближенному интегральному уравнению Киркву-
да (9.25); Ь — по приближенному интегральному уравне-
нию Борна—Грина—Айвена (9.24); с — по теории свобод-
ного объема, приведенного к форме сферы F.5) [114].
г) Результаты Кирквуда, Мауна и Олдера [112] недавно были исправлены и дополнены
Мак-Лелланом [113].
§ 9. Выражение уравнения состояния через бинарную функцию распределения 269
сфер [соотношение F.5)]. Из этих графиков можно видеть, что уравнение
Борна—Грина—Айвена дает значение pVjNkT, для предельно низких плотно-
стей почти совпадающее с результатами, полученными из уравнения Кирк-
вуда. Для более высоких плотностей согласие также достаточно хорошее.
Модифицированный потенциал F-12) Леннарда-Джонса, используемый
при решении уравнений для радиальной функции распределения, является
обычным потенциалом F-12) Леннарда-Джонса для г > сг, а для г < а он имеет
бесконечно большое значение. Фактор сжимаемости и поправки к величине
внутренней энергии и энтропии, вычисленные из уравнений Борна—Грина—
Айвена для отдельных значений приведенных объема и температуры, даны
в табл. 47—49. Значения приведенных критических постоянных равны
pl = 0,199, VI = 2,585,
Гс = 1,433, PcVc/RTc = 0,358.
Таблица 47
Фактор сжимаемости,
полученный с помощью радиальной функции распределения [114]
13,82
3,632
2,260
1,483
1,222
0,833
-0,594
-1,445
-2,433
-2,829
1,000
0,629
-0,156
-0,734
-1,268
-1,382
1,250
0,768
0,264
-0,038
-0,115
0,052
1,677
0,883
0,670
0,649
1,018
1,467
2,500
1,064
1,326
2,139
2,856
5,000
1,456
1,998
3,242
4,223
00
1,167
1,833
2,667
4,333
5,567
Приведенное «избыточное» значение внутренней энергии [114]
и' и— и»
Таблица 48
Ne
^\^^ Т*
V* ^"\^
13,82
3,632
2,260
1,483
1,222
0,833
2,280
3,370
5,073
6,313
1,000
0,741
2,148
3,277
5,024
6,280
1,250
0,621
2,035
3,192
4,974
6,232
1,667
0,537
1,939
3,118
4,925
6,181
2,500
1,856
3,050
4,873
6,125
5,000
1,787
2,990
4,822
6,066
Приведенное «избыточное» значение энтропии [114]
S' S— S*+Rlnp
Таблица 49*
^\^^ т*
V* ^^\^
13,82
3,632
2,260
1,483
1,222
1,000
-0,74
R
1,250
-0,50
-1,24
-6,31
1,667
-0,33
-1,39
-1,99
-2,55
-2,97
2,500
-0,99
-1,36
-1,90
-2,39
5,000
-0,73
-1,00
-1,54
-2,06
* Эта таблица отличается от таблицы, приведенной в работе [114], различием в опреде-
лении «избытка» энтропии (см. § 7 настоящей главы).
270
Гл. 4. Уравнение состояния плотных газов и жидкостей
Сравнение теоретической и экспериментальной радиальных функций рас-
пределения для жидкого аргона при 91,8 °К и 1,8 атмпроводится на фиг. 74.
Величина а в потенциальной функции межмолекулярного взаимодействия
была при этом взята такой, чтобы первые макси-
мумы обеих кривых совпадали. Видно, что со-
гласие между этими кривыми хорошее. На фиг. 75
экспериментальные результаты измерения фак-
тора сжимаемости аргона при 273 °К как функ-
ции плотности сравниваются с теоретическими
Фиг. 74. Сравнение экспериментальной
и теоретической радиальной функции
распределения для жидкого аргона при
91,8° К и 1,8 атм.
Экспериментальные значения взяты из
работы [100].
'200 400 600 S00
Плотность, единицы Амага
Фиг. 75 Сравнение экспери-
ментальных и теоретических
значений фактора сжимаемости
аргона при 273 °К.
а — экспериментальные значения
[50 ]; Ь — теоретические значения,
полученные с помощью радиальной
функции распределения ; с — теоре-
тические значения, полученные по
методу ячеек [ 114 ].
значениями, полученными по методу ячеек Леннарда-Джонса и Девоншайра
(см. § 7 настоящей главы).
Задачи
1. Рассчитать радиальную функцию распределения для твердых сферических молекул
с диаметром <г, используя соотношение (9.2). Пусть п* = паг будет приведенной плот-
ностью числа частиц, г* = г/а — приведенное межмолекулярное расстояние, так что потен-
циал равен
9>(г*) = оо, г*<1,
г*>1.
Покажите, что [115]
g(r*) = 1 + -~- [l - -J r* + -^-
г*<1,
К г* < 2,
2<г*.
Сравните эти результаты с результатами, полученными для потенциала Леннарда-
Джонса.
2. Вычислите потенциал усредненной cnnbiW(r*2) для п* = 1 для твердой сферической
модели. Нанесите W(r*) и <р(г*) на один и тот же график. Заметьте, что при г* < 2 с умень-
шением расстояния уменьшается ^(г*), что соответствует силе притяжения. Эта усреднен-
ная сила, действующая на молекулу 1 в направлении молекулы 2, может быть интерпрети-
рована следующим образом : молекула 2 экранирует молекулу 1 от столкновений с осталь-
ными N — 2 молекулами, и, следовательно, для г* < 2 это результирующая сила, дей-
ствующая на молекулу 1 в направлении молекулы 2.
Литература 271
3. Используя закон соответственных состояний, оценить фактор сжимаемости, энталь-
пию, энтропию и Ср для N2 при 60 атм и 100 °К.
4. Используя уравнения состояния Ван дер Ваальса, Вертело, Дейтеричи, Битти—
Бриджмена и Бенедикта—Вебба—Рубина, оценить фактор сжимаемости N2 при 60 атм
и 100 °К.
5. Используя теорию Леннарда-Джонса и Девоншайра, вычислить фактор сжимае-
мости, энтальпию, энтропию и удельную теплоемкость N2 при 60 °К и 1 атм.
6. Недавно было предложено [116] следующее уравнение состояния с двумя постоян-
ными :
_ RT а__
Р v-.bV-xl* р§/.г'/."
Покажите, что а = 0,9099 RTbJ*V2cl* и Ь = 0,1567 У\1г и уравнение состояния дает значение
V/RT равное 0,276, что находится в хорошем согласии с экспериментом.
ЛИТЕРАТУРА
. 1. Н о u g e n О. A., Watson К. М., Chemical Process Principles, Part II, New York, 1947.
2. Partington J. R., Advanced Treatise of Physical Chemistry, Vol. I, New York, 1949.
3. G u 1 d b e r g, Zs. Phys. Chem., 5, 374 A890).
4. Guye, Bull. Soc. Chim., 4, 262 A890).
5. Clark, Am. Chem. Journ., 18, 618 A896); Zs. Phys. Chem., 21, 183 A896).
6. van Laar, Ann. Phys. Boltzmann Festschr., 316 A904).
7. Lorenz, Herz, Zs. anorg. Chem., 116, 103 A921).
8. Lorenz, Zs. anorg. Chem. u. allgem. Chem., 94, 240 A916).
9. Herz, Zs. Elektrochem., 25, 215 A919). .
10. Lorenz, Herz, Zs. anorg. Chem. u. allgem. Chem., 138, 331 A924).
11. H о u g e n O. A., Watson К. М., Chemical Process Principles Charts, Madison, Wiscon-
sin, 1946.
12. Maxwell J. В., Data Book on Hydrocarbons, New York, 1951.
13. L e n n а г d - J о n e s J. E., Physica, 4, 941 A937).
14. Kamerlingh Onnes H., Crommelin C. A., Commn. Phys. Lab. Leiden,
118b A910).
15. N i j h о f f G. P., К e e s о m W. H., Commn. Phys. Lab. Leiden, 179b A925).
16. v a n U r k A. Th., N i j h о f f G. P., Commn. Phys. Lab. Leiden, 169c A924).
17. Kamerlingh Onnes H., van Urk A. Th.,* Commn. Phys. Lab. Leiden, 169d
A924).
18. Kamerlingh Onnes H., Crommelin С A., Commn. Phys. Lab. Leiden,
147d A915).
19. T r a p p e n i e r s N., Physica, 17, 501 A951).
20. Rowlinson J. S., Journ. Chem. Phys., 19, 831 A951).
21. Taylor H. S., Glass tone S., States of Matter, New York, 1951.
22. В eat tie J. A., Stockmayer W. H., Rep. Progr. Phys., 7, 195 A940).
23. L a m b e r t J. D., Roberts G. A. H., Rowlinson J. S., Wilkinson V. J.,
Proc. Roy. Soc, A196, 113 A949).
24. van d e г W a a 1 s J. D., Die Continuity des gasf6rmigen u. flussigen Zustandes, Leipzig,
1899, 1900.
25. В e a 11 i e J. А., В r i d g e m a n О. С, Journ. Am. Chem. Soc, 49, 1665 A927).
26. В e a 11 i e J. A., Bridgeman О. С, Proc. Am. Acad. Arts Sci., 63, 229 A928).
27. Hi rschf elder J. O., Roseveare W. E., Journ. Phys. Chem., 43, 15 A939).
28. В e a 11 i e J. А., В r i e г 1 e у J. S., В a r r i a u 11 R. A., Journ. Chem. Phys., 20, 1613
A952).
29. В eat tie J. A., Barriault R. А., В r i e r 1 e у J. S., Journ. Chem. Phys., 19,
1219 A951).
30. Benedict M., Webb G. В., Rubin L. C, Journ. Chem. Phys., 8, 334 A940) ;
10, 747 A942).
272 Гл. 4. Уравнение состояния плотных газоз и жидкостей
31. Benedict M., Webb G. В., Rubin L. С, Chem. Eng. Progr., 47, 419 A951).
32. N е w i 11 D. M., High Pressure Plant and the Properties of Fluids at High Pressures,
Oxford, 1940.
33. G i b s о n R. E., L о e f f 1 e г О. Н., Ann. N. Y. Acad. Sci., 51, 727 A949).
34. Gibson R. E., Loef f ler О. Н., Journ. Am. Chem. Soc, 61, 2515 A939).
35. К i г к w о о d J. G., В e t h e H., The Pressure Wave Produced by an Underwater Explo-
sion, Part I, OSRD, Report No. 588.
36. К i г к w о о d J. G., Richardson J. M., The Pressure Wave Produced by an Under-
water Explosion, Part III, OSRD, Report No. 813.
37. R i с h a r d s о n J. M., A r о n s А. В., H a 1 v e г s о n R. R., Journ. Chem. Phys., 15,
785 A947).
38. Hirschfelder J. O., Stevenson D. P., Eyring H., Journ. Chem. Phys.,
5, 896 A937).
39. К i s t i а к о w s к у G., Wilson E. В., Jr., Div. B. NDRC Report Serial No. 52 (OSRD
114).
40. Kirkwood J., Brinkley S., Richardson J., Div. B. NDRC (OSRD 2022)
p. 113; Final Report NDRC, Div. 8.
41. Schottky W., Phys. Zs., 21, 232 A920).
42. Bridgman P. W., Rev. Mod. Phys., 7, 1 A935).
43. В r i d g m a n P. W., Phys. Rev., 46, 930 A934).
44. Mich els A., de Boer J., Bijl A., Physica, 4, 981 A937).
45. Michels A., de Groot S. R., Nederland. Tijdschr. Natuurk., 12, 77 A946).
46. Michel s A., Lunbeck R. J., Wolkers G. J., Physica, 17, 801 A951).
47. M i с h e 1 s A., d e Groot S. R., G e 1 d e r m a n s M., Appl. Sci. Res., Al, 55 A947).
48. Michels A., de Groot S. R., Geldermans M., Physica, 12, 105 A946).
49. Michels A., Lunbeck R. J., Wolkers G. J., Physica, 15, 689 A949).
5O.Michels A., Wijker Hub., Wijker Hk., Physica, 15, 627 A949).
51. Cottrell T. L., Trans. Farad. Soc, 47, 337 A951).
52. Co tt re 11 T. L., Journ. Chem. Phys., 18, 1117 A950).
53. de Groot S. R., ten Seldam С A., Physica, 12, 669 A946).
54. t e n Seldam С. А., Диссертация, University of Utrecht, 1953.
55. ten Seldam C. A., de Groot S. R., Physica, 18, 891 A952).
56. W i g n e г E. P., H u n t i n g t о п Н., Journ. Chem. Phys., 3, 764 A935).
57. Kronig R., de Boer J., Korringa J., Physica, 12, 245 A946).
58. BOttcher C. J. F., Physica, 9, 937, 945 A942).
59. d e Groot S. R., ten Seldam C. A., Physica, 13, 47 A947).
60. Mayer J. E., Mayer M. G., Statistical Mechanics, New York, 1940 (см. перевод:
M а й е р Дж., Гепперт-Майер М., Статистическая механика, ИЛ, 1952).
61. F о w I e r R. H., Guggenheim E. A., Statistical Thermodynamics, Cambridge, 1939
(см. перевод: Фаулер Р., Гуггенгейм Э., Статистическая термодинамика,
ИЛ, 1949).
62. Kirkwood J. G., Journ. Chem. Phys., 18, 380 A950).
63. R i с е О. К., Journ. Chem. Phys., 6, 476 A938).
64. E у r i n g H., H i r s с h f e 1 d e r J. O., Journ. Phys. Chem., 41, 249 A937).
65. Hirschfelder J. O., Journ. Chem. Ed., 16, 540 A939).
66. Care ri G., Journ. Chem. Phys., 20, 1001 A952).
67. H i 1 d e b r a n d J. L., Scott R. L., Solubility of Non-Electrolytes, 3rd. Ed., New York,
1950.
68. H a p p e 1 H., Ann. d. Phys., 21, 342 A906).
69. Majumdar R., Bull. Calcutta Math. Soc, 21, 107 A929).
70. Mayer J. E., Journ. Chem. Phys., 5, 67 A937).
71. Mayer J. E., Ackermann P. G., Journ. Chem. Phys., 5, 74 A937).
72. Kincaid J. F., Eyring H., Journ. Chem. Phys., 5, 587 A937); 6, 620 A938).
73. Kincaid J. F., Eyring H., Journ. Phys. Chem., 43, 37 A939).
74. Pauling L., Phys. Rev., 36, 430 A930).
75. L e n n a r d - J о n e s J. E., Devonshire A. F., Proc. Roy. Soc, A169, 317 A939);
A170, 464 A939).
Литература 273
76. L e n n a r d - J о n e s J. E., Proc. Phys. Soc, 52, 38 A940).
77. Buehler R. J., Wentorf R.H., Hirschfelder J. 0., Curtiss C. F.,
Journ. Chem. Phys., 19, 61 A951).
78. Pople J., Phil. Mag., 42, 459 A951).
79. L e n n a r d - J о n e s J. E., Devonshire A. F., Proc. Roy. Soc, A163, 53 A937).
80. J a n 8 s e n s P., P r i g о g i n e I., Physica, 16, 851 A950).
81. R о w 1 i n s о n J. S., Curtiss С F., Journ. Chem. Physique, 19, 1519 A951).
82. Prigogine I., Raulier S., Physica, 9, 396 A942).
83. P r i g о g i n e I., G а г i к i a n G., Journ. Chim. Phys., 45, 273 A948).
84. Hill T. L., Journ. Phys. Colloid Chem., 51, 1219 A947).
85. Lunbeck R. J., Диссертация, Amsterdam, 1951.
86. Wentorf R. H., Buehler R. J., Hirschf elder J. O., Curtiss С F.,
Journ. Chem. Phys., 18, 1484 A950).
87. О n о S., Memoirs of the Faculty of Engineering, Kyushu University, Japan, 10, 190, No. 4
A947).
88. С e r n u s с h i F., E у г i n g H., Journ. Chem. Phys., 7, 547 A939).
89. P e e к H. M., Hill T. L., Journ. Chem. Phys., 18, 1252 A950).
90. M i с h e 1 s A., G о u d e к е t M., Physica, 8, 347 A941).
91. Mich els A., Wouters H., de Boer J., Physica, 3, 585 A936).
92. M i с h e 1 s A., Wouters H., d e Boer J., Physica, 3, 597 A936).
93. d e В о e r J., Rep. Progr. Phys., 12, 354 A949).
94. d e В о е г J., M i с h e 1 s A., Physica, 6, 97 A939).
95. M о n t г о 11 E., Mayer J. E., Journ. Chem. Phys., 9, 626 A941).
96. M e n к е Н., Phys. Zs., 33, 593 A932).
97. Debye P., Menke H., Phys. Zs., 31, 797 A930).
98. D e b у e P., Menke H., Ergeb. der Techn. Rontgenkunde, II A937).
99. Zernike F., Prins J. A., Zs. Phys., 41, 184 A927).
100. E i s e n s t e i n A., Gingrich N. S., Phys. Rev., 58, 307 A940); 62, 261 A942).
101. Warren В. Е., Journ. Appl. Phys., 8, 645 A937).
102. M о r r e 11 W. E., H i 1 d e b r a n d J. H., Journ. Chem. Phys., 4, 224 A936).
103. Kirkwood J. G., Journ. Chem. Phys., 3, 300 A935).
104. N i j b о e r B, R. A., van Hove L., Koninklijke Nederlandse Akademie van Weten-
schappen, 54, 256 A951).
105. N i j b о e r B. R. A., van Hove L., Phys. Rev., 85, 777 A952).
106. Hart R. W., Wall is R., Pode L., Journ. Chem. Phys., 19, 139 A951).
107. Rushbrooke G. S., S с о i n s H. I., Nature, 167, 366 A951).
108. Y v о n J., Actualites scientifiques et industrielles, Paris, 1935.
109. Born M., Green H. S., Proc. Roy. Soc, A188, 10 A946).
110. Green H. S., Proc. Roy. Soc, A189, 103 A947).
111. Kirkwood J. G., Boggs E. M., Journ. Chem. Phys., 10, 394 A942).
112. К i г к w о о d J. G., Maun E. K., Alder B. J., Journ. Chem. Phys., 18, 1040 A950).
113. Me Lei Ian A. G., Proc. Roy. Soc, A210, 509 A952).
114. К i г к w о о d J. G., L e w i n s о n V. A., Alder B. J., Journ. Chem. Phys., 20, 929
A952).
115. de Boer J., Rep. Progr. Phys., 12, 305 A949).
116. Benson S. W., Golding R. A., Journ. Chem. Phys., 19, 1413 A951).
ГЛАВА 5
РАВНОВЕСИЕ ЖИДКОСТЬ—ПАР И КРИТИЧЕСКИЕ
ЯВЛЕНИЯ1)
В предыдущих двух главах были изучены равновесные свойства жидких
и газообразных систем в отдельности. Настоящая глава посвящена исследо-
ванию систем, состоящих одновременно из жидкой и газообразной фазы и нахо-
дящихся в равновесии.
. Сначала будет изучена промежуточная область между жидкой и газо-
образной фазой. Термодинамический подход к изучению промежуточной об-
ласти приводит к установлению связи между поверхностным натяжением и
другими термодинамическими свойствами поверхности. С другой стороны,
статистический подход позволяет понять микроструктуру переходной области.
Установлено, что ширина переходной зоны, являющейся поверхностным слоем
между жидкой и газообраз: ой фазой, прогрессивно увеличивается с ростом
температуры. При приближении к критической температуре вдоль всего сосуда
.под влиянием, силы тяжести возникает градиент плотности. С этого момента
исчезает различие между жидкостью и паром. Один из методов определения
критической точки основан на явлении исчезновения мениска при возрастании
температуры. Однако в настоящее время не ясно, будет ли температура, при
которой исчезает мениск, совпадать с критической температурой, определенной
из изучения геометрических свс йств изотерм. Экспериментальные измерения
показали, что разность между критическими температурами, определенными
этими двумя методами, не превосходит нескольких сотых градуса.
Затем будет изложена классическая теория фазовых переходов и крити-
ческого состояния. Эта теория основана на искусственном предположении,
согласно которому вплоть до критической точки существуют две отчетливо
выраженные, полностью гомогенные фазы, отделенные четким мениском. Такая
теория позволяет правильно объяснить многие явления, связанные с фазовыми
переходами и критическим состоянием. Существуют, однако, такие явления,
как градиенты плотности в жидкости и эффект гистерезиса, которые не укла-
дываются в рамки классической теории и требуют для своего объяснения зна-
чительно более подробного изучения проблемы. Эти «аномалии» с большим или
меньшим успехом можно объяснить, учитывая действие силы тяжести и флук-
туации плотности. Но, чтобы до конца понять эти явления, необходимо еще
проделать большую теоретическую и экспериментальную работу.
В заключение сделана попытка суммировать результаты проведенных
исследований и показано, как можно применить результаты двух предыдущих
глав для интерпретации некоторых явлений, имеющих место в двухфазных
системах.
Глава заканчивается обсуждением некоторых интересных явлений, наблю-
даемых при фазовых переходах в бинарных системах. Явления обратной кон-
денсации, имеющие большое значение при перегонке нефти, обсуждаются с
помощью фазовых диаграмм. Поведение бинарных двухфазных систем также
рассматривается с термодинамической точки зрения.
Глава написана в сотрудничестве с профессором Чарльзом Бойдом.
§ 1. Промежуточная область между жидкостью и паром 275
§ 1. ПРОМЕЖУТОЧНАЯ ОБЛАСТЬ МЕЖДУ ЖИДКОСТЬЮ
И ПАРОМ
Теоретическое рассмотрение двухфазных систем должно включать рас-
смотрение промежуточной области и изучение природы поверхностного натя-
жения. При теоретическом изучении поверхностного натяжения можно при-
менить те же два метода, которые были использованы при изучении равновес-
ных свойств жидкостей1), т. е. метод свободного объема и метод радиальной
функции распределения. Первый метод использовали Леннард-Джонс и Корнер,
второй метод — Кирквуд и Бафф*). Здесь будут изложены работы этих двух
групп исследователей, а затем будет рассмотрено поверхностное натяжение
капель и соотношение между парахором и молекулярными силами.
А ОПРЕДЕЛЕНИЕ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
Двухфазная система, находящаяся в вытянутом объеме, продольная ось
которого совпадает с осью z, показана на фиг. 76, а; зависимость плотности
от расстояния z изображена на фиг. 76, б. В жидкой фазе плотность равна п@
и не зависит от расстояния г, в газообразной фазе плотность равна постоянной
величине пD С макроскопической точки зрения плотность скачкообразно
меняет свое значение при переходе через поверхность раздела жидкость—пар.
При формулировке микроскопической теории поверхностного натяжения
необходимо учитывать, что в действительности никакого разрыва непрерыв-
ности не существует. Плотность меняется непрерывно при переходе через по-
верхность раздела, как указано на фиг. 773). Другие равновесные характеристики
вещества изменяются аналогичным образом. Тензор давления, например, равен
p0U в жидкости и также выражается в газовой .фазе (р0 — статическое давление).
В переходной области между двумя фазами тензор давления p(z) плавно ме-
няется от значений, равных значению в жидкости, до значений, равных зна-
чению в газе.
Гиббс [4 ] показал, что с математической точки зрения удобно ввести «раз-
деляющую поверхность» между жидкой и газообразной фазами. Эта разделяю-
щая поверхность нормальна к градиенту плотности. Положение такой поверх-
ности до некоторой степени произвольно. Удобно выбрать его, потребовав,
чтобы 2 = 0 (координата поверхности) удовлетворяло условию
О +z<'>
J [п«> - n(z)] dz = J [n(z) - лс>] dz, A.1)
@ О
x) Кроме этих двух методов, для определения поверхностного натяжения жидкостей
применяется теория дырок жидкостей, изложенная в гл. 4, §.8 [1 ]. Эта теория приводит к
зависимости плотности от расстояния, аналогичной той, которая приведена на фиг. 77.
2) В работе [2 ] проводится сравнение и обобщение этих двух методов.
8) Хилл [3 ] развил простой, но грубый метод расчета поверхностного натяжения. Он
потребовал, чтобы химический потенциал имел одну и ту же величину не только в гомоген-
ных жидкой и газообразной фазах, но и во всех промежуточных точках внутри поверхности
раздела. Значение химического потенциала определялось из величины свободного объема и
энергии взаимодействия молекул. Величина свободного объема как функция плотности опре-
делялась из модифицированного уравнения Ван дер Ваальса. Молекулярная плотность
имела вид «пятен» в том смысле, что радиальная функция распределения выбиралась так,
что она была равна нулю вплоть до расстояний, равных эффективному диаметру столкно-
вений, и равна единице для всех ббльших расстояний. Таким образом легко вычислить
энергию взаимодействия молекулы как функцию изменения плотности с расстоянием г.
Равенство химических потенциалов при всех высотах приводит к интегральному урав-
нению для определения функции распределения плотности. Как только определена функция
распределения плотностей, легко находится напряжение как функция расстояния г, а сле-
довательно, и поверхностное натяжение. Такие расчеты выполнены для молекул, являю-
щихся твердыми шариками диаметра ст, которые на больших расстояниях притягиваются
друг к другу с силой, потенциал которой обратно пропорционален шестой степени расстояния.
276
Гл. 5. Равновесие жидкость—пар и критические явления
что иллюстрируется фиг. 76. Назовем такую разделяющую поверхность экви-
молекулярной поверхностью раздела. При таком выборе поверхности раздела
гипотетическая система, в которой плотность на разделяющей поверхности
изменяется скачком от значения, равного значению в жидкой фазе, до значения,
равного значению в газовой фазе, будет содержать такое же число молекул,
как и истинная система.
Пар(и)
Поверхност-
ная — •(
пленка (s)
Жидкость (I)
\~z ¦"
L J
-ПB)
Фиг. 76. Схематическое изоГ ражение двухфазной системы, состоящей из жидкой (/) и
газообразной (v) фаз, разделенных поверхностной пленкой (s).
Положение «эквимолекулярной Поверхности раздела» (соответствующей координате z = О) определяется
уравнением A.1). В соответствии скатим уравнением области А и В должны быть равны.
0,8
0,6
0,4
0,2
-5-4-3-2-101234561
г/а
Фиг. 77. Зависимость плотности от давления в переходной области [3].
Представим себе, что заштрихованная стенка объема переместилась обра-
тимо и изотермически в направлении оси х на расстояние их, как показано на
фиг. 76. При таком перемещении система совершает работу
A.2)
Единственной компонентой тензора давления, которую необходимо при этом
рассматривать, является компонента хх9 описывающая силы, действующие в
§ 1. Промежуточная область между жидкостью и паром 277
направлении оси х на элемент поверхности, перпендикулярный к оси х. Пусть
далее дно и крышка контейнера будут перемещаться обратимо и изотермически
так, что объем системы примет свое первоначальное значение. При таком пере-
мещении система совершает работу
2
f podz. A.3)
После всех этих операций система перейдет в состояние, в котором объем,
давление и температура равны своим первоначальным значениям. Распреде-
ление молекул между частями объема, находящимися по обе стороны от раз-
деляющей поверхности, также не изменится в результате совершенных про-
цессов1). Единственная разница заключается в увеличении площади поверхност-
ной пленки. Работа, которую необходимо затратить, чтобы увеличить площадь
пленки на единицу, обозначается через у и называется поверхностным натя-
жением. В соответствии с этим при растяжении пленки на их (площадь пленки
увеличивается на величину да = Ьдх) затрачивается работа
у 8а = _ 8w = 8aj(Po _ РххB)) йг . A.4)
Таким образом, поверхностное натяжение действует в направлении оси х на
единичную площадку, расположенную в плоскости уг, и аддитивно склады-
вается с обычным нормальным давлением, действующим на обе фазы.
Рассмотрим теперь вклад поверхностного слоя в термодинамические функ-
ции двухфазной системы. Любой термодинамический параметр, относящийся
ко всей системе в целом, можно представить в виде суммы трех членов, обязан-
ных своим происхождением жидкости, газу и поверхностному слою. Например,
свободная энергия двухфазной системы имеет вид
А = ДО + AW + Д<5>. A.5)
В этой формуле А@ — свободная энергия единицы объема гомогенной
жидкости (т. е. объема, находящегося внутри жидкости), умноженная на
объем системы, находящейся ниже эквимолекулярной поверхности раздела;
A{v) определяется аналогично для газовой фазы. Поскольку свободная энергия
всей системы А хорошо известна, формула A.5) может служить для опреде-
ления A(s\
Изучим теперь термодинамику двухфазной системы. С помощью первого
закона термодинамики элемент работы dw выражается следующим образом :
dw = pdV — yda. A.6)
х) По определению эквимолекулярной поверхности раздела число молекул над поверх-
ностью (обозначаемое как число молекул пара) равно abz(v)n(v). Условие равенства числа
молекул пара до и после этих процессов записывается в виде
Точно так же в жидкой фазе
abz«) п @ = (а + д х) te@' л(О.
В этих формулах г(?У и z<0' — расстояния от разделяющей поверхности до концов контейнера
после всех процессов. Поскольку
zW z<0' a
z(v) z(O а + дх'
то объемы жидкой и газообразной фаз не изменяются в результате совершенных процессов
Поэтому перечисленные процессы являются процессами при постоянном объеме (в то же
время и при постоянном давлении, так как давление пара остается постоянным при изотер-
мических процессах), при которых объемы двух фаз не меняются. Подчеркнем, что этот резуль-
тат получается как прямое следствие определения эквимолекулярной поверхности раздела
и будет неверен при других определениях разделяющей поверхности.
278 Гл. 5. Равновесие жидкость—пар и критические явления
Учитывая эту формулу, получаем для дифференциала свободной энергии вы-
ражение
dA = dU - d(TS) = -SdT- pdV + yda . A.7)
Аналогично для жидкой и газообразной фазы
= - Sfb dT - pdV&, A.8)
= - S<*>df - pdVW . A.9)
Комбинируя два последних соотношения и используя тот факт, что
+ Viv) = V, получаем выражение
= - S<s> dT + yda. A.10)
Вследствие аддитивности объемов1) A(s) = G(s), поэтому поверхностное натя-
жение можно записать в в де2)
Все остальные термодинамические величины поверхностной пленки можно
выразить посредством у и ее производных. Например, из уравнения Гиббса—
Гельмгольца можно найти внутреннюю энергию поверхностной пленки
A.12)
Эту величину можно получить непосредственно из измерений поверхност-
ного натяжения, поэтому она часто применяется для сравнения теоретических
результатов с экспериментальными. Изложенные термодинамические рассуж-
дения ограничены однокомпонентными системами с эквимолекулярной поверх-
ностью раздела.
Поверхностное натяжение можно также рассматривать с точки зрения
закона соответственных состояний, изложенного в гл. 4, § 1. Для приведения
переменных используются критические постоянные. Можно ожидать, что при-
веденное поверхностное натяжение yr = yV^kT^r1 будет универсальной
функцией приведенной температуры Тг = Т/Тс. Ьсли для приведения перемен-
ных используются молекулярные единицы (например, постоянные а и е
в потенциале Леннагда-Джонса), то приведенное поверхностное натяжение
у* = ya2je будет универсальной функцией приведенной температуры Т* •= kTje.
Закон соответственных состояний можно использовать для установления
связи между существующими экспериментальными данными и предсказаниями
величины поверхностного натяжения у таких веществ, которые еще не изучены
экспериментально, поскольку не существует строгих теорий в тех областях,
которые подвергаются наиболее интенсивному экспериментальному изучению.
Б. РАСЧЕТ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ МЕТОДОМ
СВОБОДНОГО ОБЪЕМА
Леннард-Джонс и Корнер [9, 10] развили теорию поверхностного натя-
жения, ведущую свое начало с уравнения A.11). Их работа является обобще-
нием теории свободного объема жидкостей Леннарда-Джонса и Девоншайра,
учитывающим существование поверхностного слоя. При таком подходе они
г) Равенство A(s) = G(s) справедливо лишь в том случае, когда за поверхность раздела
жидкость—пар принята эквимолекулярная поверхность раздела. При других определениях
поверхности раздела жидкость—пар, например если поверхность раздела определить как
«поверхность натяжения», о которой будет говориться дальше в связи с кривизной поверх-
ности, это равенство уже не будет иметь места.
2) В работах Фаулера [5, 6] показано, что поверхностное натяжение не увеличивает
потенциальную энергию поверхности, как ранее предполагали Бредли [7 ] и Маргенау [8 ].
§ U Промежуточная область между жидкостью и паром 279
смогли вычислить свободную энергию единицы поверхности. Их расчет основан
на следующих предположениях :
1) Плотность пара столь мала, что при расчете поверхностного натяжения
можно пренебречь существованием молекул в газовой фазе.
2) Жидкость и ее поверхность образуют полу бесконечную колонну. Струк-
тура жидкости подобна структуре кристаллической решетки, узлы которой
являются положениями равновесие молекул. Поверхностное натяжение в этом
случае будет зависеть от того, какая кристаллическая плоскость рассматри-
вается в качестве свободной поверхности. Расчеты поверхностного натяжения
выполнены для различных кристаллических плоскостей. Истинное поверх-
ностное натяжение, очевидно, должно быть минимальным.
3) Узлы решетки в поверхностном слое слегка сдвинуты по направлению
внутрь жидкости так, что потенциальная энергия системы, состоящей из всех
молекул, расположенных в своих узлах, в полубесконечной колонне достигает
минимума. За исключением поверхностного слоя, узлы решетки расположены
так, как если бы вообще не существовало поверхности.
4) Свободные объемы молекул внутри жидкости не изменяются вследствие
наличия поверхности. При расчете свободного объема молекул, находящихся
в поверхностном слое жидкости, все соседние молекулы точно расположены
в своих узлах, а рассматриваемая молекула «блуждает» в результирующем
потенциальном поле.
Для расчета свободной энергии поверхностной пленки Леннард-Джонс и
Корнер рассчитали свободную энергию 1 моля жидкости, ограниченной поверх-
ностной пленкой площади а, и вычли из полученного результата свободную
энергию 1 моля жидкости внутри жидкой фазы. Последняя величина, которая
не меняется в присутствии поверхностной пленки, определяется теорией
Леннарда-Джонса и Девоншайра
д@ = ф<0 _ №Т In А-3 — NkT In vf - NkT, A.13)
где А2 = h2j2nmkT ; индекс (l) указывает, что данная величина вычислена для
элемента жидкости, находящегося внутри жидкости ; Ф@ — общая потен-
циальная энергия N молекул, расположенных в узлах решетки.
Общая потенциальная энергия молекул 1 моля жидкости, окруженного
поверхностной пленкой, обозначается Ф(/)(г). Эта энергия зависит от расстояния
z, на которое поверхностный слой молекул смещен внутрь жидкости. Можно
найти величину смещения г0, при котором Ф(/)(г) минимальна1); эта величина
в дальнейшем и будет определять положение узлов решетки в поверхностном
слое. Таким образом, свободная энергия 1 моля жидкости, окруженной поверх-
ностной пленкой, имеет вид
= Ф(П(г0) - NkT In A-3 - (N - N<s>) kT In vf - N<s> kT In г>^> — NkT. A.14)
Индекс (/') относится к характеристикам жидкости, окруженной поверхностной
пленкой ; N(s) — число молекул в поверхностной пленке ; v(p — свободный
объем молекулы, находящейся на поверхности. Поверхностное натяжение в
этом случае в соответствии с формулой A.11) определяется выражением2)
х) За расстояние z0 при более строгом расчете следовало бы брать расстояние, при кото
ром минимальна свободная энергия или поверхностное натяжение. Однако такой способ рас-
чета значительно труднее, а получаемые результаты незначительно отличаются от преды-
дущих.
2) Из этого уравнения непосредственно следует закон Этвеша
-Г), A.15а)
где К и То — постоянные. Величина К обычно принимается равной 2,1 эрг/град, а То прибли-
280
Гл. 5. Равновесие жидкость—пар и критические явления
Корнер [10 ] применил этот результат для расчета поверхностного натяже-
ния жидкости, состоящей из сферических неполярных молекул, взаимодействие
которых определяется потенциалом Леннарда-Джонса, причем эти молекулы
образуют гранецентрированную кубическую решетку. Схема его расчета сле-
дующая. Сначала выбирается рассматриваемая поверхность. Это должна быть
поверхность, соответствующая минимальному значению свободной энергии
поверхности, т. е. минимальному значению поверхностного натяжения. Из
трех главных плоскостей такой решетки плоскость A00) можно исключить
сразу1), поскольку она соответствует разрыву четырех ближайших соседних
связей на площади а2 (где а — расстояние между ближайшими соседями),
тогда как плоскость (ПО) соответствует разрыву 3,54, а плоскость A11) — раз-
рыву 3,56. Разрыв ближайших соседних связей требует значительной энергии,
поэтому наиболее целесообразно рассматривать плоскости (ПО) и A11). Де-
тальные расчеты показали, что плоскость A11) несколько предпочтительнее,
поэтому в дальнейшем она только и будет рассматриваться.
Некоторые результаты этих расчетов приведены в табл. 50. Сначала ука-
зана величина смещения поверхностного слоя внутрь жидкости z0. Затем дано
значение потенциальной энергии молекулы на поверхности %(z0) при смещении
поверхностного слоя на расстояние z0, а затем та же величина при отсутствии
смещения поверхностного слоя #@). В последней колонке приводятся значения
разности [Ф(п(го) — Ф(/)] в формуле A.15), которые как раз равны увеличению
потенциальной энергии идеальной решетки при бесконечно малом удалении двух
полубесконечных блоков жидкости с плоской поверхностью. Эти величины
Таблица 50
Сводка некоторых промежуточных результатов при вычислении
поверхностного натяжения по методу свободного объема
е,а — постоянные в потенциале Леннарда-Джонса ; а
ближайшими соседями; a I' 3/2 — расстояние между
. 0,7
0,8
0,9
1,0
"о
(аКЗ/2)
0,075
0,054
0,036
0,020
*<*о)
в
10,6524
11,1648
11,5496
11,8077
*@)
в
10,1361
10,8585
11,3994
11,7589
— расстояние между
' ПЛОСКОСТЯМИ A11)
[Ф(*>Bо) — ФЩ <**
а 8
1,550
1,990
2,377
2,703
были получены тщательным суммированием вкладов более чем от 400 узлов
решетки и приблизительным суммированием вкладов от молекул, находящихся
от поверхности на расстоянии более 5а.
Свободный объем tfif молекул в поверхностном слое вычислен с
помощью модифицированного метода Леннарда-Джонса и Девоншайра.
зительно соответствует критической температуре. Чтобы получить это уравнение, обычно
предполагают, что отношение свободных объемов [vty/vty] не зависит от температуры. Оче-
видно, что \Wl'Xzo) — ФМ] и N(s> пропорциональны числу молекул на единице поверх-
ности, которое в свою очередь пропорционально V*/». Несколько лучшее эмпирическое
соотношение для поверхностного натяжения простых молекул было найдено Гуггенгеймом
[11, 12]; оно имеет вид
A.156)
где у0 — то же самое, что и у0У*с3/Тс = 4,4 эрг • град-1 моль-*!*.
х) В работе [13] определена разность [W)(z0) — 0КО], но, к сожалению, только для
плоскости A00).
§ 7. Промежуточная область между жидкостью и паром
281
Симметрия гексагональной упаковки в поверхностном слое позволяет
с большой степенью точности считать, что потенциал обладает цилиндрической
симметрией. Потенциал необходимо рас-
считывать для каждого отдельного случая,
один из примеров которого показан на
фиг. 78.
Чтобы проделать численные расчеты
поверхностного натяжения, необходимо
знать плотность жидкости, находящейся в
равновесии со своим паром, как функцию
температуры. Для простых жидкостей
Гуггенгейм [11] эмпирически нашел эту
зависимость, которая в приведенных пере-
менных Vr = V/Ve, Tr = TjTc имеет вид
п
-f(l-TrL'. A.16)
-//€ "
-/2€
Положительная ось направлена по нормали
К "'«Р^иДР^проведена „ля
V 0,2 о,з о,4
Ф и г. 78. Потенциальная энергия мо-
Корнер Г101 использовал эмпирические лекулы, расположенной на поверхности,
соотношения Wy.~l.29 и VJNo* ~ 3,05 ГоТлГре^ГиТ^ГниХТрГд:
для получения из формулы A.16) значения нег0 расстояния между плоскостями
v* как функции Г*. Вычисленные таким A11).
ПУТеМ Значения Т* Приведены В Табл. 51.
Приведены ТаКЖе Некоторые реЗуЛЬ-
таты расчетов величины свободного объема.
Интересно отметить, что при температуре больше 0,62 Тс свободный объем
поверхностных молекул фактически становится меньше свободного объема
молекул, находящихся внутри жидкости. В жидком аргоне при температуре
90° К (Т1Те = 0,60) поверхностное натяжение равно 1,63 е/а3 = 23,1 дин/см.
Экспериментальное значение равно 11,9 дин/см [14, 15]. Расчет по методу
функции распределения, выполненный Кирквудом и Баффом и изложенный
в следующем разделе, приводит к величине 14,9 дин/сму которая значительна
лучше согласуется с экспериментальными данными.
Таблица 51
Сводка результатов расчета поверхностного натяжения
по методу свободного объема
1
v*2
0,7
0,8
0,9 t
1,0
Tr
0,620
0,521
0,421
0,319
l
T*
1,2495
1,4871
1,8432
2,4280
vf
0,009
0,153
0,295
0,427
1,545
1,902
2,236
2,542
Теория поверхностного натяжения, основанная на методе свободного
объема, по существу является лишь интересной точкой зрения, позволяющей
нарисовать простую качественную картину поверхностного явления. Для
точных численных расчетов эта теория оказывается слишком грубой. Более
того, она совершенно не пригодна при рассмотрении влияния кривизны поверх-
ности на величину поверхностного натяжения. В настоящее время теории,
основанные на методе радиальной функции распределения (изложенном r
следующем разделе), кажутся более многообещающими.
282 Гл. 5. Равновесие жидкость—пар и критические явления
В. РАСЧЕГ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
МЕТОДОМ РАДИАЛЬНОЙ ФУНКЦИИ РАСПРЕДЕЛЕНИЯ1)
Давление в гомогенной фазе, выраженное через радиальную функцию
распределения [формула A.13) гл. 3], имеет вид
A.17)
В гл. 9, § 4, показано, что в общем виде тензор давления выражается через
радиальную функцию распределения
U - 1л(Г1)J/i(r2)g(r1(Гг1) -?-^drn. A.18)
В этом случае радиальная функция распределения параметрически зависит от
координаты молекулы 1. Величина n(r2)g(rlf r21)dr21 равна числу молекул,
находящихся в объеме dr21 вокруг молекулы в гг Вектор гх отсчитывается от
некоторого фиксированного начала, тогда какг21 измеряется от конца вектора гг
Это выражение применимо и к негомогенной области перехода между двумя
гомогенными фазами. (Столь же хорошо оно применимо и к неравновесным
состояниям.)
Из формулы A.18) можно найти выражение для pxx(z), которое затем сле-
дует подставить в выражение для поверхностного натяжения [формула A.4)].
Таким путем получаем
У = _ кт Г<1) + ± J Г™(г21) ;г^ -g- *n. A.19)
Величины Г^ и Г?2) называются соответственно поверхностной плотностью
числа молекул и поверхностной плотностью пар и являются частными случа-
ями функций
О со
Г}1)= J z{ [n(Zl) - n<b] dzx + \ zi[nB,)-/!<•)]&! A.20)
— ОЭ 0
и
0
f = J *i f^fe) "te) gfe, r2i) - n<z>2 g(/)(r2i)] ^1 +
00
— 00
здесь g@ и g(t>) — обычные радиальные функции распределения в жидкой и
газообразной фазах вдали от разделяющей поверхности.
Поверхностное натяжение многокомпонентной системы можно, обобщив
формулу A.19), написать в виде
у = - кТ2{Г^\ + 4 V (Г&\г21))и^р- — dr^- A.22)
Здесь (Г^)^ — поверхностная плотность молекул i-й компоненты, определен-
ная выражением, аналогичным формуле A.20); (Г^2)),у — поверхностная плот-
ность пар молекул /-го и /-го сорта, определенная выражением, аналогичным
формуле A.21), в которой теперь первый член относится к молекулам г-го
сорта, а второй — к молекулам /-го сорта ; <Ри(г21) — энергия взаимодействия
молекул 1 и 2, относящихся к *'-му и /-му сортам.
Вплоть до этого момента ничего не говорилось о положении разделяющей
поверхности между жидкостью и паром, которая находится в начале z-коор-
!) СМ. [16].
§ 7. Промежуточная область между жидкостью и паром 283
динаты. Совершенно ясно, что расчетная величина поверхностного натяжения
не зависит от выбора разделяющей поверхности. В случае эквимолекулярной
поверхности раздела, определенной формулой A.1) (просто утверждающей,
что Г^> = 0), выражение для поверхностного натяжения упрощается и
принимает вид
Написанная формула является точным выражением поверхностного натя-
жения, не зависящим от частных предположений, которые делаются в методе
свободного объема. Трудность, конечно, заключается в том, что наши знания
о плотности и бинарной функции распределения в промежуточной области
неполны. Тем не менее этот подход кажется многообещающим для изучения
поверхностных явлений. В разд. Д будут приведены некоторые простые
результаты, полученные этим методом.
Г. ВЛИЯНИЕ РАДИУСА КРИВИЗНЫ НА ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
Термодинамическая теория, учитывающая влияние кривизны поверхности
на величину поверхностного натяжения, создана Толменом [17] и обобщена
впоследствии Кирквудом и Баффом [16, 18, 19]. Эта теория описывает термо-
динамические свойства очень малых капелек или групп, которые появляются
при сгущении пересыщенного пара. В будущем эта теория может оказаться по-
лезной при объяснении критических явлений, когда преобладают большие
группы.
Для капелек понятие эквимолекулярной поверхности раздела перестает
быть столь удобным. Гиббс [4 ] показал, что существует разделяющая поверх-
ность, называемая поверхностью натяжения1), которая при вычислении термо-
динамических свойств в случае капелек приводит к наиболее простым резуль-
татам2).
Для сферических поверхностей Бафф и Кирквуд [16, 18] нашли, что по-
верхность натяжения имеет радиус, меньший на г0 радиуса эквимолекулярной
поверхности раздела, причем г0 определяется формулой
-f с
J
A.24)
Используя формулу A.19) для поверхностного натяжения и определение
функций Г^р и Г<?>, можно получить выражение для г0
В гл. 4, § 9, были рписаны приближенные интегро-дифференциальные урав-
*) Точное определение поверхности натяжения связано с длинными термодинамиче-
скими рассуждениями. Мы не будем давать здесь этого определения, а отошлем читателя к
работе Толмена [17].
2) Работа образования сферической капли является величиной определенной. Она равна
произведению поверхностного натяжения на площадь поверхности
W = -АА = 4пг*у.
В этом параграфе г означает радиус кривизны поверхности натяжения, а у — поверх-
ностное натяжение, определенное относительно этой разделяющей поверхности. Для любой
другой разделяющей поверхности, имеющей радиус кривизны г' и площадь поверхности
4лг'2, поверхностное натяжение равно у*. С хорошим приближением можно написать
Ал г2 у = 4л г'2 у'.
Значения у и/ сильно различаются лишь для очень малых капелек.
284 Гл. 5. Равновесие жидкость—пар и критические явления
нения Кирквуда и Борна—Грина—Айвена, устанавливающие связь между
бинарной функцией распределения и плотностью. Из этих уравнений следует
[16, 18]:
W
- G+W
Подставляя этот результат при / = 1 в предыдущее уравнение, получаем
Эти выражения для z0, строго говоря, применимы лишь в случае плоских по-
верхностей. Для кривых поверхностей в выражениях для Г<р и Г<?> интегри-
рование вдоль декартовой координаты должно быть заменено соответствующим
интегрированием вдоль радиальной координаты. Этим эффектом пренебрегли, и
вплоть до настоящего времени зависимость z0 от радиуса кривизны никем не
учитывалась.
Однако при вычислении поверхностной плотности Г(^> пренебрегать кри-
визной уже нельзя. Для капелек, поверхность натяжения которых имеет
радиус кривизны г, поверхностная плотность Г<?>, равная разности, отнесенной
к единице площади разделяющей поверхности, фактического количества веще-
ства двухфазной системы и того количества вещества, которое имелось бы, если
бы жидкая и газообразная фазы имели 'однородные плотности вплоть до раз-
деляющей поверхности, равна
П1} = J Иг) - ifi>] (l + уJdz + j [n(z) - /ЮО] (l + -fJ dz. (i.28)
-г О
Если поверхность натяжения берется на расстоянии z0 ниже поверхности, для
которой Г^> = 0, то относительно поверхности натяжения, принятой теперь
за начало отсчета, поверхностную плотность можно записать в виде
= г* = z0 [яЛ - лО] [l + i +1 (^J]. A.29)
Гиббс получил уравнение
dy=-r*df*, A.30)
связывающее дифференциал поверхностного натяжения с химическим потен-
циалом /л. В изотермических процессах, при которых равновесие между двумя
фазами сохраняется,
^ = ^</г=-4т; о-31)
здесь p{v) — давление в газе, а рA) — давление в жидкости или в капельке.
Избыток давления в жидкости над давлением в паре дается известным урав-
нением Гиббса—Кельвина
pib-jfi>) = MLm A.32)
Дифференцируя последнее выражение и используя три предыдущих уравнения,
можно получить соотношение Гиббса—Толмена—Кенига [20 ]:
Г йУ
Интегрируя уравнение A.33) от г = оо (соответствующее плоской поверхности)
§ 7. Промежуточная область между жидкостью и паром
до любого значения радиуса кривизны капельки г, получаем
285
A.34)
здесь у0 — поверхностное натяжение плоской поверхности. В табл. 52 при-
ведены значения отношения (у/у0) для малых радиусов кривизны.
Таблица 52
Зависимость поверхностного натяжения от радиуса
кривизны поверхности натяжения
2./Г
0,00
0,01
0,02
0,05
0,10
0,20
0,30
Y/Yo 1
1,00
0,98
0,96
0,91
0,83
0,70
0,60
| Zjr
0,40
0,50
0,60
0,70
0,80
0,90
1,00
Y/Y.
0,52
0,46
0,41
0,36
0,33
0,30
0,28
В первом приближении
A.35)
Сомнительно, чтобы величина z0 оставалась постоянной и не зависела от радиуса
кривизны для таких малых капелек, где г становится порядка г0. В связи с этим
численные результаты, приведенные в табл. 52, нельзя считать строго устано-
вленными. Поверхностное натяжение очень малых капель, содержащих не-
сколько сотен молекул, представляет значительный интерес в связи с теориями
образования зародышей. Однако прежде, чем теория примет удовлетворитель-
ный вид, необходимо еще проделать огромную работу по выяснению влияния
размеров капель на величину поверхностного натяжения.
Д. РАСЧЕТ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
В ПЕРВОМ ПРИБЛИЖЕНИИ
Для расчета поверхностного натяжения необходимо знать плотность как
функцию координат л(г) и радиальную функцию распределения g(rlf г21) вблизи
поверхности. В принципе эти функции, как указали Кирквуд и Бафф [16, 18],
можно найти с помощью теории жидкости Борна и Грина [21, 22]. Здесь,
однако, мы ограничимся рассмотрением двух очень упрощенных моделей.
1. Жидкость с нормальной плотностью вплоть до поверхности раздела
Гиббса в контакте с вакуумом. В этом случае
A.36)
Единственная часть ГB), отличная от нуля, возникает при интегрировании от
zx = — z21 до Zjl = 0 при условии, что zM положительно. Таким образом,
286 Гл. 5. Равновесие жидкость—пар и критические явления
If> = (_iy+ii|in@-g('>(r21), z21>0, (
rf=O, z21<0.
Подставляя эти выражения в формулы A.23) и A.27), получаем величину по-
верхностного натяжения и координату поверхности в виде
о
Легко показать, что внутренняя энергия поверхности U<s) выражается сле-
дующим образом:
Ц)йЧ1. A.40)
(здесь Г(§) относится к эквимолекулярной поверхности раздела), поэтому
Кирквуд и Бафф [16, 18] применили эти уравнения для расчета поверх-
ностных свойств жидкого аргона. Они использовали также потенциал Лен-
нарда-Джонса и экспериментальные значения для кривой g(l\r)f полученные
Эйзенштейном и Гингричом [23] при 90 °К :
у = 14,9 дин/см {экспериментальное значение 11,9)
5) = 27,2 дин/см (экспериментальное значение 35),
Г* = 4,99 • Ю-8 г/см2,
z0 = 3,63 • Ю-8 см.
Приведенные в скобках экспериментальные значения получены Бали
и Доннаном [14, 15]. Согласие между данными удивительно хорошее. Напом-
ним, что Корнер [10], применяя ячеечную модель, в этом случае получил
7 = 23,1 dunfCM. Таким образом, метод радиальной функции распределения
оказывается значительно более многообещающим применительно к расчету
поверхностного натяжения.
2. Жидкость с нормальной плотностью вплоть до эквимолекулярной
поверхности раздела в контакте с газом постоянной плотности. В этом случае
n(Zl) = n«>, Zl<0,
2l>0,
и
zx>0 и 21 + 221>0,
2l<0 и + >0
или гх>0 и
§ 7. Промежуточная область между жидкостью и паром 287
Ясно, что ^1\г21) и g(t>)(r2j) есть обычные радиальные функции распреде-
ления, относящиеся к элементу, находящемуся в объеме жидкости или
газа соответственно. Функция g(/>r)(r21) является некоторым видом корреляции
между молекулами жидкости и молекулами газа. Вид этой функции в настоя-
щем грубом приближении не может быть точно вычислен.
Из уравнения A.21) следует
<o(Гя) _ „со„« gcw (ГяI, *21 > о,
(L44>
Zfi- [п пg«>> (г21) - л<*g<»> (г21)], г21 < 0 .
Используя далее полярные координаты для вычисления угловых частей трой-
ного интеграла по г21, получаем из формул A.23) и A.40) следующие выражения
для определения поверхностного натяжения и внутренней энергии поверх-
ности :
00
W I1** 8е0 W - 2) "W ^ <г) + n<t>)' ^ Wl dr > У -45>
о
00
(/<*> = - |- J г* ?> (г) [n<v g<o (г) - 2л«> л<•> g<W (г) + ФУ ?<•> (г)] dr . A.46)
о
Точно такие же результаты были получены Фаулером [5, б ], который произвел
непосредственный расчет этой модели. Уравнение A.46) служит основой теорий
парахор Фаулера, к которой мы и перейдем сейчас.
Е. УРАВНЕНИЕ МАК-ЛЕО ДА И ПАРАХОР
Мак-Леод [24 ] предложил следующее эмпирическое уравнение для опре-
деления поверхностного натяжения1):
у = const (q0) - e(v)L, A.47)
где q1) и q{v) — истинные значения плотности жидкости и пара. Сагден [25
записал его результат в следующем виде :
= - Ухи . П48>
е() Q() j.10 — /.(•) ^ лЧ°'
Постоянная Р называется парахором. Парахор с большой степенью точности
можно представить в виде суммы постоянных, относящихся к отдельным ато-
мам или группам атомов в молекуле. Сагден считал, что парахор связан с
объемом молекулы. В любом случае парахор данного вещества почти не зависит
от температуры в довольно широкой области температур.
Фаулер [5, 6] получил уравнение Мак-Леода путем следующих теорети-
ческих рассуждений. Оэщее уравнение состояния вблизи критической точки
можно представить в виде ряда
A.49)
Л) Это уравнение известно в русской литературе под названием уравнения Бачинского.
Оно было впервые предложено А. И. Бачинским в статье «О формулах поверхностного натя-
жения>, Изв. физич. инст. МНИ, 2, 60 A922). — Прим. ред.
288 Гл. 5. Равновесие жидкость—пар и критические явления
Опущенные члены пренебрежимо малы по сравнению с записанными. Урав-
нение A.49) является кубическим уравнением относительно объема, поэтому
любому заданному давлению соответствуют три значения К, удовлетворяющие
уравнению. В двухфазной области действительны все три корня l/(/), Viv) и
посторонний третий объем, появляющийся вследствие S-образной формы изо-
терм. Давление пара при данной температуре (см. § 2) определяется с помощью
термодинамического соотношения
С помощью этого соотношения Фаулер показал, что вблизи критической точки
в первом приближении выполняется равенство
[nU-tfi»Y=$rnUTe-T), [A.51)
где Vc — молярный объем в критической точке; пс— плотность в критической
точке; а = (Эр/Э7)с; 0 = (Э3р/Эуз)с.
Далее, радиальные функции распределения для жидкости, газа и проме-
жуточной области в критической точке совпадают, поэтому их можно запи-
сать в виде
(г) = gc (г) + р^Г) Gff..) (г)
A.52)
О функциях G(r) нам почти ничего не известно.
Подставляя эти выражения в формулу A.46), получаем величину поверх-
ностной энергии вблизи критической точки
f n*f гз <p(r) [G«\r) + G^(r) - 2 GF*. v\r)] dr. A.53)
о
Заменяя в последнем выражении [п(/) — n(v)] по формуле A.51), находим
У<5> = В(ТС — Т), A.54)
где
^Д f r*<P(r) [GQ\r) + G(v\r) - 2G(/' v\r)] dr. A.55)
Тс J
О
Подставляя C/(s) [формула A.54)] в уравнение Гиббса—Гельмгольца и интег-
рируя от Т до ТСУ получаем поверхностное натяжение. Разлагая поверхностное
натяжение вблизи критической точки в ряд, получаем
?\ A.56)
§ 7. Промежуточная область между жидкостью и паром
289
или, согласно формуле A.51),
Таким образом, парахор имеет вид
A.57)
A.58)
Значения парахора, определенные Сагденом для некоторых веществ, приве-
дены в табл. 53.
Таблица 53
Выражение парахора через физические постоянные
Вещество
Аг
Ne
N2
СО
сн4
р
54
25
60,4
61,6
73,2
/р- ю-"\
[ o'fie1!* J
7,0
7,6
7,0
6,8
7,1
Среднее
7,1
Леннард-Джонс и Корнер [9 ] показали (см. табл. 53), что в случае простых
молекул парахор с точностью до 3,6% можно выразить эмпирическим соотно-
шением
p = G,1.1023)?W/. A.59)
где а и е — постоянные в потенциале F-12) Леннарда-Джонса (в см и эрг
соответственно). Основываясь на этом соотношении, эти авторы предложили
чувствительный метод определения диаметра молекулярных столкновений а
по значению парахора (е1^ обычно легко определяется). Тот факт, что Р почти
пропорционально а3, согласуется с предположением Сагдена о пропорциональ-
ности парахора объему молекулы. Фаулер рассчитал значения парахора для
некоторых примеров, используя ряд упрощающих предположений, в резуль-
тате которых полученные им данные носят полуэмпирический характер. В на-
стоящее время удовлетворительного объяснения независимости парахора от
температуры не существует.
§ 2. ФАЗОВЫЕ ПЕРЕХОДЫ В ОДНОКОМПОНЕНТНЫХ
СИСТЕМАХ
Большая часть этого параграфа посвящена изложению макроскопической
теории фазовых переходов и критического состояния однокомпонентных
веществ. Метод исследования заключается в изучении геометрических свойств
поверхности р = p(V, T), изображающей уравнение состояния. Исследуются
как экспериментальные поверхности, так и поверхности, описываемые про-
стыми аналитическими уравнениями. Молекулярный подход к изучению кри-
тического состояния осуществляется с помощью методов статистической меха-
ники. Излагаемая теория является дальнейшим развитием теории уравнения
состояния гл. 3, § 2. В заключительной части параграфа дан краткий обзор
современного состояния теории.
290
Гл. 5. Равновесие жидкость—пар и критические явления
А. МЕТОДЫ ОПРЕДЕЛЕНИЯ КРИТИЧЕСКОЙ ТОЧКИ
Уравнение состояния однокомпонентной системы можно представить
геометрически в виде поверхности р = p(V, Т). Схематически эта поверхность
изображена на фиг. 79, представляющей диаграмму зависимости между
р, V и Т. Кривая ADCEB называется линией сосуществования. Точка этой
линии, в которой температура максимальна, называется критической точкой.
Фиг. 79. Диаграмма зависимости между р, V и Т для жидкой и газообразной фаз
однокомпонентной системы.
В центре — поверхность р = p(V,T)\ в правой верхней проекции — различные изотермы на диаграмме
р — V; в левой верхней проекции — различные изохоры на диаграмме р — Т; на нижней проекции —
различные изобары на диаграмме Т — V.
Часть кривой сосуществования ADC} расположенная слева от критической
точки, называется «линией жидкости», а часть справа от критической точки —
«линией пара». Языкообразная сбласть, ограниченная линией сосущество-
вания, соответствует условиям, при которых система распадается на жидкую
и парообразную фазы, находящиеся в равновесии.
Поскольку трехмерным представлением диаграммы зависимости между р, V
и Т пользоваться не совсем удобно, то для изображения этой диаграммы обычно
применяются три двухмерные диаграммы, показанные на фиг. 79. На верхней
правой проекции изображены обычные изотермы на диаграмме р — V. Они
получены проектированием на плоскость р — V кривых (изотерм), образую-
щихся при сечении поверхности p = p(Vy T) плоскостями, перпендикулярными
оси Т. Изотерма, проходящая через точку С, называется «критической изо-
термой» ; в критической точке (С) на этой изотерме выполняются условия
§ 2. Фазовые переходы в однокомпонентных системах
291
= (92p/9V2)T = 0. В левой верхней проекции изображены изохоры
на диаграмме р — Т. Они получены проектированием на плоскость р — Т
кривых, образующихся при сечении поверхности р = /?(/, Т) плоскостями,
перпендикулярными оси V. Кривые (А, В), (D, ?), (С), пересекающиеся с С,
являются проекцией языкообразной области сосуществования на плоскость
р — Т и подобны кривой^давления пара. Нижняя проекция изображает изо-
бары на диаграмме Т — V и получается путем проектирования на ^плоскость
Т — V кривых, образующихся при сечении поверхности р = p(V, T) пло-
скостями, перпендикулярными
ОСИ р. ПО ЛЮбОМу ИЗ ЭТИХ Трех 75,4
графиков можно восстановить
первоначальный вид поверхности
р = р(у} Т) в трех измерениях.
Методы определения критических
постоянных основаны на ана-
лизе этих трех проекций.
1. Определение критической
точки из изотерм. Анализируя
поведение изотерм, можно опре-
делить критическую температуру
с точностью до нескольких сотых
градуса. Этот метод иллюстри-
руется фиг. 80, на которой при-
ведены экспериментальные дан-
ные Михельса и др. [26 ] для СО2.
Последние измерения Венторфа
и Бойда [27, 28], показанные на
фиг. 81, оченб близко подходят к
критической точке.
7О?
35 39 43 47 51 55 59 63
va х W4 - объем, единицы Амага х Ю4
Фиг. 80. Изотермы углекислого газа в
критической области [26],
67
2. Определение критической
точки по изохорам. Существуют
два метода изучения критичес-
кого состояния, основанные на
изучении поведения определен-
ного количества веществ в сосуде
постоянного объема при изменении температуры. В первом методе измеряется
давление системы, т. е. непосредственно определяются изохоры [29—31].,
Во втором методе изучается поведение мениска [32, 33] при изменении тем-,
пературы.
4550,0
5540,0
5530,0 -
5522,5
-
ч
CV
Ok,
—
¦
>—
а».
31
31,
31,1О°С
зш°с
М06°С
02°
--
с-
га
р
1,0 с
°С
=
-tr
-¦¦»
——с
—о
——.
-о—
__
——
¦—.
—
—
>
0**
0
о
1825
2,0
2,5
22 2,3
1/р, см%
Фи г. 81. Изотермы углекислого газа в критической области [27].
292
Гл. 5. Равновесие жидкость—пар и критические явления
Первый метод сводится к измерению изохор, показанных на фиг. 79, по-
этаму основная задача в нем заключается в определении предельного значения
плотности, при котором кривая р как функция Т не испытывает разрыва. Из
фигуры видно, что изохоры ведут себя совершенно различно в зависимости от
того, будет ли общая плотность q (средняя плотность пара и жидкости) меньше,
больше или равна критической плотности &.. На фиг. 79 приведены следующие
примеры этих трех случаев :
Если :
Q>Qc
то система при изменении
своего состояния переме-
щается вдоль следующей
кривой р как функции Т :
(Л, В) ЕЕ'
(А, В) DD'
(A,B)(D,E)C
Последний случай соответствует предельному положению кривой и позволяет
определить величину критической плотности.
Во втором методе вместо измерения давления системы измеряется зависи-
мость высоты мениска, разделяющего жидкую и газообразную фазы от темпера-
туры [33]. Так же, как и раньше, существует три направления изменения
системы при повышении температуры в зависимости от величины среднего
значения общей плотности :
Если:
Q < Qc, то мениск опускается до тех пор, пока в системе
останется только газообразная фаза;
Q > Qc, то мениск поднимается до тех пор, пока в системе
останется только жидкая фаза;
q = QCt то мениск постепенно исчезает, находясь все время
в середине сосуда.
Исчезновение мениска может служить критерием, определяющим критиче-
скую точку.
Поведение мениска в этих трех случаях можно объяснить следующим
образом. Пусть g — средняя плотность вещества в сосуде, a g(v) и qA) — плот-
ности жидкой и газообразной фаз, находящихся в равновесии. В этом случае
часть объема, занятая жидкой фазой, равна (q — e(t>))/(e(/)— Q(v))- Если средняя
плотность вещества в сосуде ? > ес, to при нагревании сосуда достигается
такая температура, при которой q=qP\ и система целиком становится жидкой.
Аналогичным образом, если ё<ес, то при нагревании сосуда достигается
такая температура, при которой g = е(% и система целиком становится
газообразной. Если средняя плотность выбрана таким образом, что q = Qc,
то при нагревании сосуда достигается такая температура, при которой
g = qc = gW = ^># при этом значении температуры мениск исчезает при-
близительно на середине сосуда1).
2. Определение критической точки по плотности жидкой и газообразной
фаз (закон Нальете и Матиаса). Следующий метод определения критической
точки основан на свойствах диаграммы Т — V, изображенной на фиг. 79.
Привычнее изображать температуру как функцию плотности, нежели как
х) Вблизи критической точки 112(<№) + #@)
исчезает, находясь как раз на середине сосуда
ре + а(Т>— Тс). Если а = 0, то мениск
§ 2. Фазовые переходы в однокомпонентных системах
293
функцию объема. Для определения критического значения плотности Кальете
и Матиас предложили вычертить среднюю плотность жидкой и газообразней
фаз, находящихся в равновесии. Средние значения плотности Vafe^ + Q(l))
ложатся на прямую линию, известную под названием прямолинейного диа-
метра1). Точка пересечения прямолинейного диаметра с кривой плотностей
определяет критическое значение плотности и температуры. Этот метод иллюст-
рируется на фиг. 82 на примере водорода.
0,08
0,01
0,06
0,05
0,04
0,03
0,02
0,01
-258 -256 -254 -252 -250 -243 -246 -244 -242 -2W
Т9°С
Фиг. 82. Определение критической точки из измерений плотности жидкой g@ и газооб-
разной #00 фаз по методу Кальете и Матиаса.
Приведенные данные относятся к водороду [36]. Определенные таким способом критические аначения тем-
пературы и плотности находятся в превосходном согласии с результатами, полученными путем визуального
определения мениска [32].
ft—
-Л—
(
)(V)
л
<
(D
о»
—о
^—
\
¦О—
Критическая точка-"
/
\
¦о-
/
р
-
Б. СТАБИЛЬНЫЕ И МЕТАСТАБИЛЬНЫЕ СОСТОЯНИЯ
Языкообразная область на фиг. 79, ограниченная кривой сосуществования,
требует несколько более подробного рассмотрения. Истинных равновесных
условий, описываемых кривыми, не всегда удается получить экспериментально.
Например, будем сжимать пар. При этом может возникнуть состояние переча-
сыщешя, при котором существует одна гомогенная парообразная фаза, хотя в
соответствии с фазовой диаграммой часть пара должна была бы конденсиро-
ваться в жидкость, находящуюся в равновесии с оставшимся паром. Состояние
перенасыщения образует метастабильное состояние, которое, однако, может
существовать в течение значительного времени. Для образования жидкой фазы
необходима конденсация вещества на ядрах конденсации. Ядра конденсации
могут возникать спонтанно; ими могут служить также ионы (например, в камере
Вильсона), или обычно присутствующие частички пыли, или другие малые
частички твердых примесей [38—41 ]. После того как началась конденсация,
равновесие жидкость — пар устанавливается очень быстро.
Другой тип метастабильного состояния наблюдается при перерасшире-
нии жидкостей. Представим себе, что гомогенная жидкость находится под
большим давлением в цилиндре. Если поршень цилиндра движется так, что
х) Исчерпывающее изложение закона прямолинейного диаметра дано в работе [37 J.
Закон Кальете и Матиаса утверждает, что средняя плотность не зависит от температуры и
равна критической плотности. Этот закон верен лишь приблизительно. В действительности
средняя плотность изменяется линейно или квадратично с температурой.
294
Гл. 5. Равновесие жидкость—пар и критические явления
жидкость расширяется, то при давлении, меньшем давления насыщения, часть
жидкости должна превращаться в пар. Вместо этого, однако, гомогенная жид-
кая фаза может существовать, не испаряясь, в течение длительного промежутка
времени, даже если давление отрицательное (в смысле стремления разорвать
жидкость). Для испарения жидкости необходимо, чтобы молекулы имели воз-
можность испаряться в пузырьки или пустоты. Эти пустоты могут образовы-
ваться спонтанно, но чаще они образуются газами, растворенными в жидкости,
или на поверхности обычно присутствующих твердых примесей [42, 43].
Рассмотрим теперь изотермы этих метастабильных состояний на диаграмме
р V. Это удобнее сделать, воспользовавшись каким-либо простым аналити-
ческим уравнением состояния, например уравнением Ван дер Ваальса. Изотер-
мы этого уравнения состояния ниже критической точки не имеют горизонталь-
ного отрезка, соответствующего равновесию жидкость—пар, а обладают
S-образной формой, показанной на фиг. 83. Например, температуре Т2 соответ-
ствует теоретическая изотерма DQRSE, тогда как истинное равновесие на
Фиг. 83. Изотермы на диаграмме р — V, соответствующие приближенному простому
аналитическому уравнению состояния типа уравнения Ван дер Ваальса.
Равновесные состояния соответствуют линиям DE и Л В. Отрезки кривой DQ и АХ изображают состояние
жидкости при перерасширении, а отрезки SE и ZB — состояние пересыщения пара. Отрезки QRS и XYZ
соответствуют физически нереализуемым состояниям.
диаграмме р — V изображается пунктирной линией DE. Эта линия описывает
давление пара при температуре Т2. Максвелл [44] первый предложил изме-
рять давление пара (т. е. положение пунктирной линии DE) из изотерм с по-
мощью простых аналитических уравнений. При переходе 1 моля жидкости в
газообразное состояние термодинамический потенциал G=H—TS изменяется
на величину
J B.1)
Интегрирование происходит вдоль S-образной части изотермы (линии DQRSE).
Поскольку в равновесии G(/) равно G(t°, то интеграл равен нулю. Это значит,
что отрезок DE необходимо выбирать таким образом, чтобы область, ограничен-
ная линией VR и кривой DQR, была равна области, ограниченной линиями
RE и RSE. Подчеркнем, что в этом выводе не используется какой-либо частной
интерпретации различных областей петли (за исключением того, что они и
физически реализуемые отрезки изотерм описываются одним аналитическим
выражением).
Истинное равновесие описывается соединяющим отрезком, таким, как
DE. Отрезки S-образной аналитической кривой DQ и SE можно интерпретиро-
вать с помощью экспериментально реализуемых метастабильных состояний.
Сегмент SE изотермы Т2 соответствует состояниям перенасыщения, а сегмент
§ 2. Фазовые переходы в од покомпонентных системах
295
DQ — состояниям пере расширения. Интересно отметить, что даже отрицательные
давления, имеющиеся на метастабильной части изотермы Тг для жидкости
(фиг. 83), не лишены смысла. Эти метастабильные состояния наблюдались в
лаборатории [40 ] и представляют значительный интерес при изучении кави-
тации в жидкостях [41 ]. Части изотерм QRS и XYZ соответствуют физически
нереализуемым состояниям. Более полная интерпретация различных частей
S-образных изотерм может быть получена с помощью нижеследующего термо-
динамического рассмотрения.
Рассмотрим изотермическую вариацию свободной энергии (А = V — TS)
при изменении объема для гипотетической системы, показанную на фиг. 84.
В соответствии со вторым законом термодинамики устойчивое состояние будет
характеризоваться минимальной величиной А относительно вариаций при
постоянных температуре и давлении. Рассмотрим систему в состоянии, изобра-
жаемом точкой а. Можно изменить состояние системы, сжав часть системы до
&
03
Объем V
Фиг. 84. Зависимость свободной энергии А от объема для гипотетической устойчивой
системы при постоянной температуре.
состояния Ьу а оставшуюся часть расширив до состояния с так, чтобы общий
объем системы не изменился. Очевидно, что величина Д соответствующая
новому состоянию системы, будет обозначаться на диаграмме точкой d. Следо-
вательно, если кривая обращена вогнутостью вверх, то новому состоянию
системы соответствует большая величина свободной энергии и это состояние
неустойчиво. И наоборот, если кривая обращена вогнутостью вниз, то новому
состоянию системы соответствует меньшее значение свободной энергии, по-
этому оно более устойчиво, чем первоначальное. В последнем случае система
будет изменять свое состояние до тех пор, пока ее свободная энергия не достиг-
нет минимума.
Поскольку кривая на фиг. 84 обращена вогнутостью вверх, то
B.2)
и она соответствует устойчивым состояниям. Поскольку давление связано со
свободной энергией
B.3)
то уравнение состояния может соответствовать серии устойчивых состояний
лишь при условии
B.4)
Если это условие не выполняется, то система неустойчива по отношению к
бесконечно малым флуктуациям плотности.
296
Гл. 5. Равновесие жидкость—пар и критические явления
Изменение свободной энергии А вдоль S-образной изотермы р — V пока-
зано на фиг. 85. Согласно B.3), давление в любой точке равно тангенсу угла
наклона, взятого с обратным знаком. Кроме того, из соотношения
B.5)
следует, что отрезок, отсекаемый на вертикальной оси касательной в какой-
либо точке, равен термодинамическому потенциалу в этой точке. Поскольку
при равновесии давление и термодинамический потенциал G одинаковы в жидкой
и газообразной фазах, то касательные к кривой в точках, соответствующих
равновесному существованию жидкой или газообразной фазы в отдельности,
совпадают. Это показано на фиг. 85 пунктирной линией, которая проходит через
точки D и Е.
I
1
«5
Объем V
Фиг. 85. Зависимость свободной энергии от объема для ван-дер-ваальсова газа ниже
критической точки при постоянной температуре.
Точки D, Qt R, S, Е те же, что и изображенные на фиг. 83 на изотерме Т,.
Рассмотрим теперь различные части кривой DQRSE на фиг. 83 (этой кри-
вой соответствует кривая DQRSE на фиг. 85). Части кривой DQ и SE> тангенс
угла наклона которых отрицателен, удовлетворяют, уравнению B.4) и поэтому
соответствуют устойчивым по отношению к малым флуктуациям плотности
состояниям системы. Поскольку эти части кривой лежат внутри соединяющего
отрезка, то они изображают состояния системы, неустойчивые по отношению
к фазовым переходам, включающим конечные флуктуации плотности. Эти от-
резки соответствуют метастабильным состояниям перенасыщения и перерасши-
рения. Часть изотермы QRS на фиг. 83 не удовлетворяет условию B.4), поэтому
состояния, описываемые этим сегментом, даже неметастабильны и совершенно
недостижимы.
В. ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА КРИТИЧЕСКОЙ
ОБЛАСТИ
Рассмотрим термодинамические свойства однокомпонентной системы вблизи
критической точки. Теплоемкость при постоянном давлении Ср в критической
точке равна бесконечности, однако теплоемкость при постоянном объеме Сг,
скорость звука с и коэффициент Джоуля—Томсона /л являются конечными
величинами.
1. Теплоемкость при постоянном объеме. Теплоемкость при постоянном
объеме можно вычислить как сумму теплоемкости идеального газа С? и инте-
§ 2. Фазовые переходы в однокомпонентных системах
297
грала от второй производной давления по температуре
J
B.6)
Второй член можно рассчитать, воспользовавшись экспериментальными дан-
ными о сжимаемости. Результаты таких расчетов для СО2 вблизи критической
точки [45 ] приведены на фиг. 86. Существование отчетливого максимума вблизи
критической точки очевидно. Этот максимум можно объяснить флуктуациями.
100 200 300 400 500
ра-плотность, единицы Амага
600
Фиг. 86. Зависимость Cv от плотности при различных температурах для углекислого газа,
определенная из данных о сжимаемости (критическая плотность 237 единиц Амага) [45].
В гл. 6, §2, показано, что теплоемкость при постоянном объеме пропорцио-
нальна среднеквадратичному отклонению внутренней энергии :
B.7)
В связи с этим аномально большие значения удельной теплоемкости вблизи
критической точки объясняются большими флуктуациями и образованием
молекулярных групп в этой области.
При нагревании смеси жидкости и пара в сосуде постоянного давления
потребляемую энергию можно представить в виде суммы энергий, необходимых
для нагревания жидкой и газообразной фаз в отдельности, плюс энергия, необ-
ходимая для испарения некоторой части жидкости, чтобы поддержать фазовое
равновесие при высокой температуре. Если средняя плотность системы больше
или меньше критической плотности, то кривая средней удельной теплоемкости
как функция температуры имеет разрыв в точке, соответствующей переходу
системы в однофазное состояние. Если, с другой стороны, средняя плотность
равна критической, то средняя теплоемкость остается непрерывной. Послед-
ние измерения теплоемкости в углекислом газе [46 ], выполненные по этой мето-
дике, привели к величине удельной теплоемкости, более чем вдвое превосхо-
дящей результаты, полученные из диаграммы зависимости между ру V и Т.
Причина расхождения до сих пор еще остается невыясненной.
298 Гл. 5. Равновесие жидкость—пар и критические явления
2. Теплоемкость при постоянном давлении. Теплоемкость при постоянном
давлении можно рассматривать с помощью сосуда, снабженного поршнем, дви-
жущимся без трения и поддерживающим давление в системе на постоянном
уровне. Начальная температура системы достаточно мала, так что система
целиком состоит только из жидкой фазы. При легком нагревании системы тем-
пература возрастает, объем увеличивается и величина Ср остается конечной.
Аналогичные рассуждения применимы и при достаточно высоких температу-
рах, когда система полностью газообразна. Если, однако, температура системы
возрастает до такой величины, что давление в системе станет равным равно-
весному давлению пара, то начинается образование пара. Дальнейшее легкое
нагревание не будет сопровождаться увеличением температуры, а подводи-
мая теплота будет потребляться как скрытая теплота парообразования. Сле-
довательно, в этой точке величина Ср становится бесконечной и остается тако-
вой до тех пор, пока система целиком не перейдет в результате фазового пере-
хода в газообразное состояние. Таким образом, при любом давлении, меньшем
критического, существует область объемов (или средних плотностей), выше
которых Ср бесконечна. В предельном случае критического давления рс об-
ласть объемов (или плотностей) охватывает только одну точку — критический
объем (или критическую плотность). Таким образом, при приближении системы
к критической точке со стороны однофазной области величина Ср приближается
к бесконечности.
3. Скорость звука. Скорость звука при нулевой частоте1) равна
<2-8>
где М — молекулярный вес. Вторая форма этого уравнения становится неопре-
деленной в критической точке, так как отношение у = CV\CV стремится к беско-
нечности, а (9р/9К)г стремится к нулю. Однако простыми термодинамическими
преобразованиями [47] уравнение B.8) можно привести к
c-v( T V/2№) [l- AiM^
Так как (Зр/ЗК)т в критической точке равно нулю, а вторая величина остается
конечной, то
ШШ <210)
Значение (9p/3T)v в критический точке можно получить'либо из обычной диа-
граммы зависимости между р, V и Т, либо из кривой давления пара. Вследствие
непрерывности фаз кривая давления пара идентична с критической изохорой, а
(Эр/ЭТ)у равно тангенсу угла наклона этой кривой.
В настоящее время, к сожалению, не существует экспериментальных
данных о величине скорости звука в критической области при частотах доста-
точно низких, чтобы можно было считать с ^ с0. Следовательно, в настоящее
время невозможно экспериментально проверить формулу B.10J). Поведение
г) Следует различать скорость звука при нулевой частоте с0, которая является термо-
динамически определенной величиной, и скорость звука при любой другой частоте с, которая
зависит от времени релаксации и других характеристик процесса (см. гл. 11, §4).
2) Шнайдер (частное сообщение, март 1952 г.) измерил скорость и дисперсию звука в
ксеноне в критической области на частотах 250, 750, 1250, 1750 и 2250 кгц. С уменьшением
частоты скорость становилась меньше. Экстраполяция к нулевой частоте, конечно, очень
груба, однако при этом скорость приближается к значению, предсказываемому формулой
B.10).
§ 2. Фазовке переходы в однокомпонентных системах
299
320
20 40 60 80
Давление р, атм
Фиг. 87. Зависимость скорости звука в СО2 от давления вблизи критической точки
при частоте 260 кгц.
Минимальное значение скорости достигается на критической изотерме [48].
100
90
5? 80
-о
I
Г"
60
50
41
1 1
1 1 1
Спорость
звука
43
Коэффициент
поглощения
44 45 46 47
Температура, °С
0,30
0,25
0,20 |
0,15 1
а/о
0,05s
48 49
Фиг. 88. Скорость звука и коэффициент поглощения (частота 600 кгц) в SFe в критической
области.
Измерения проводились в системе, начальная средняя плотность которой равнялась критической. Коэф-
фициент поглощения а определялся уравнением А = А9е~аХ' , где А и Ао — амплитуды а Я длина
волны [49, 50].
скорости звука при конечных частотах в критической области показано на
фиг. 87, где нанесены результаты измерений в CO.J48]1). Последние измерения
Шнайдера в SFe показаны на фиг. 88.
4. Коэффициент Джоуля—Томсона. Коэффициент Джоуля—Томсона мож-
но записать в виде
х) В этой работе имеются также данные для С2Н4.
300
Гл. 5. Равновесие жидкость—пар и критические явления
Эта формула не определена в критической точке. Термодинамическими
преобразованиями это выражение можно привести [47 ] к виду
г • B-12)
(др/ъщ [1 - (с./-
В критической точке это выражение принимает очень простой вид
1
(др;дТу •
B.13)
Таким образом, коэффициент Джоуля—Томсона в критической точке равен
обратной величине тангенса угла наклона кривой давления как функции тем-
пературы.
-50
50 100 150 200 250 300
Т.'С
Фиг. 89. Коэффициент Джоуля—Томсона для СО2 в критической области][51].]
На фиг. 89 приведены экспериментальные значения коэффициента Джоуля—
Томсона в СО2 как функции температуры при различных давлениях [51].
Г. ПОВЕДЕНИЕ ВЕЩЕСТВА В КРИТИЧЕСКОЙ ОБЛАСТИ1)
В предыдущих разделах рассматривалась природа фазовых переходов
и критического состояния. Это рассмотрение основывалось на предположении
об однородности каждой фаны во всех свойствах, а также о существовании рез-
кой границы между фазами. Рассмотрим сейчас подробно свойства жидкости
непосредственно вблизи критической точки. Сначала перечислим эффекты,
которые становятся особенно важными вблизи критической точки, а затем,
учитывая эти эффекты, изучим различные наблюдаемые явления.
Поверхность, или переходная область, между двумя фазами не имеет отчет-
ливых границ вблизи критической точки. Структура переходной области под-
робно обсуждалась в § 1. При приближении к критической точке толщина пере-
ходного слоя резко увеличивается, а разница в плотности между двумя фазами
резко уменьшается. Размазывание границы между жидкой и газообразной
Раздел написан совместно с Г. Пальмером.
§ 2. Фазовые переходы в однокомпонентных системах 301
фазами усиливается эффектами гравитации и тем фактом, что (Зр/31/)т мало.
В сосуде, содержащем жидкость, давление медленно меняется с высотой вслед-
ствие гидростатического давления верхних слоев жидкости. Изменение давле-
ния с высотой, а также тот факт, что (Зр/ЭК)т мало, приводит к зависи-
мости плотности от высоты. Эти два эффекта — размазанность поверхности
и влияние гравитации — экспериментально неразличимы и приводят к появ-
лению макроскопических градиентов плотности.
Статистическая теория флуктуации изложена в гл. 2, § 6. Там показано,
что флуктуации плотности становятся особенно большими вблизи критиче-
ской точки. сИю значит, что существуют большие микроскопические градиенты
плотности в случайных направлениях, накладывающиеся на макроскопические
градиенты плотности в вертикальном направлении. В гл. 12, § 7, показано, что
эти микроскопические флуктуации плотности приводят к рассеянию света.
Вблизи критической точки рассеяние особенно велико и известно под названием
«критической опалесценции».
Экспериментально установлено, что вблизи критической точки трудно
получить равновесные условия на протяжении всей системы. Это является источ-
ником многочисленных интересных явлений, которые можно иллюстрировать
в экспериментах с перевернутой U-образной трубкой. Если жидкость поме-
щена в один из рукавов перевернутой U-образной трубки и система нагревается
несколько выше критической точки, то при охлаждении жидкость стремится
реконденсироваться в том рукаве, который первоначально содержал жидкость.
Макроскопические градиенты плотности в жидкости вблизи критической
точки наблюдались многими исследователями [52, 55 ]. Маасе с сотрудниками
[53, 54 ] применили метод, в котором плотность жидкости в различных точках
камеры определялась по плавучести маленького поплавка. Типичная кривая
градиента плотности, полученная этим методом, приведена на фиг. 90 [56].
Совсем недавно Пальмер измерил градиенты плотности оптическим методом
Шлирена [55]. Его результат приведен на фиг. 91. Результаты обоих методов
показывают, что градиенты плотности зависят не только от температуры, но
и от предыстории системы. Эти «гистерезисные» эффекты будут изложены далее.
Градиенты плотности, возникающие в жидкости под действием сил грави-
тации, можно связать с уравнением состояния [57 ]. Давление жидкости изме-
няется с высотой под влиянием веса верхних слоев жидкости по закону
-t- = -<?g. B-14)
Температура постоянна на протяжении всей жидкости, поэтому производная
плотности по высоте имеет вид
9?
_Й__ 9?
dz~- (др/дд)т ' ^Л0)
Поскольку вблизи критической точки (9р/Э^)т мало, то это и объясняет сущест-
вование больших градиентов плотности.
Уравнение B.15) определяет градиент плотности как функцию макроско-
пических переменных. Функцию распределения плотности можно выразить
также через микроскопические переменные, воспользовавшись довольно гру-
бым статистическим рассмотрением. Систему, функция распределения плот-
ности которой изображена на фиг. 90, можно представить в виде жидкой фазы
плотности q°\ отделенной от газообразной фазы плотности g{v) довольно широ-
кой переходной областью. Такая система состоит из большого числа молеку-
лярных групп различных размеров. Для простоты будем рассматривать идеаль-
ную систему, жидкая фаза которой состоит из групп объема vKC) и плотности
(fc\ а газообразная фаза представляет собой гомогенную среду плотности еD
В переходной области между этими фазами молекулярные группы распреде-
лены по барометрическому закону. В дальнейшем будет предполагаться, что
302
Гл. 5. Равновесие -жидкость—пар и критические явления
жидкая фаза состоит из молекулярных групп сферической формы, образующих
плотную упаковку. В этом случае д(с) = [3 |/2/я:]е(/) = 1,ЗЕ05 р(/). Если начало
оси z выбрано на поверхности жидкости, то плотность как функция z выра-
жается в виде
Q = jW + (,«> - ^) ехр [- Mzl??>2!«*] . B.16)
Величина v(c) в этой формуле является эмпирической постоянной, имеющей
смысл среднего размера группы. Ее можно определить, подставляя в B.16) экс-
периментальные значения плотности. Сплошная кривая для метилового эфира
W
0,21 0,22 0,23
р -плотность, г/см3
о.™
U,ZJUU
0,2200
0,2100
0,2000
0,1900
0,1800
0}1700
I i i l l
^*^\\
: U
Средняя плотность \
_ 0,207г/см3 \
Высота контейнера \
5,96 см \
1 1 1 I i
0 12 3 4 5 6
Высота, см
Фиг. 91. Плотность этана как
функция расстояния при различных
температурах выше критической [55].
Ф и г. 90. Плотность метилового эфчра
как функция расстояния в сосуде.
Экспериментальные данные взяты из работы
[56]. Сплошная линия соответствует кривой,
вычисленной по формуле B.16) в предполо-
жении, что в молекулярных группах содер-
жится 1,8 10 молекул.
на фиг. 90 построена с помощью B.16) при v(c) = 6- Ю"5 смг. Группы такого
размера имеют молекулярный вес 8,5- 108 и содержат 2- Ю7 молекул мети-
лового эфира. Диаметр такой группы приблизительно равен 270 молекулярных
диаметров. Размеры групп по порядку величины совпадают с размерами, опре-
деленными из экспериментов по рассеянию света в этилене в критической об-
ласти [58]1).
Поскольку для установления равновесия в такой жидкости требуется
значительный промежуток времени, то очень трудно определить термодинами-
О рассеянии света см. гл. 12, § 7.
§ 2. Фазовые переходы в однокомпонентных системах
303
чески устойчивые градиенты плотности в этой жидкости. На фиг. 90 изображена
типичная кривая плотности как функция высоты в объеме. Рассмотрим неко-
торую точку, лежащую в нижней половине объема в области жидкой фазы.
На фиг. 92 показано, как меняется плотность в этой точке при повышении или
понижении температуры (со скоростью нагревания около 0,01 град/час). Верх-
няя кривая описывает поведение
плотности при увеличении темпера-
туры, а нижняя — при уменьшении.
Ни одна из этих кривых не обнару-
живает признаков нестабильности.
После встряски в системе снова уста- |
навливается прежнее распределение §
плотности, которое в дальнейшем |
заметным образом не меняется в те- |
чение нескольких дней. Этот «гисте- ^
резис» в поведении градиента плот-
ности, по всей видимости, обязан тому
факту, что в условиях равновесия с
ростом температуры размеры молеку-
лярных групп уменьшаются, тогда как
при понижении температуры ОНИ уве- Фиг- ^2• Схематическое изображение зави-
личиваются. При возрастании темпе- ^П
ратуры молекулярные группы стре-
мятся уменьшиться, причем скорость
уменьшения их достаточно велика, поэтому процесс установления равновесия
протекает довольно быстро. С другой стороны, при уменьшении температуры
молекулярные группы растут, причем скорость роста настолько мала, что
равновесие не достигается в течение нескольких дней. Таким образом, часть
кривой на фиг. 92, соответствующая возрастанию температуры, вероятно,
изображает равновесное состояние системы, а кривая, полученная при пони-
жении температуры, соответствует неравновесным состояниям1).
Т-температура
Д. СТАТИСТИЧЕСКАЯ ТЕОРИЯ КОНДЕНСАЦИИ
До сих пор изложение ограничивалось термодинамикой и простейшими
эмпирическими рассуждениями. Явление фазового перехода и критическое
состояние должны также получаться при строгом статистическом рассмотрении
уравнения состояния жидкости. Некоторые успехи в этом направлении уже
сделаны, однако потребуется еще много работы, чтобы придать теории закон-
ченную форму. Первая теория конденсации, основанная на теории групповых
интегралов неидеальных газов, изложенной в гл. 3, § 2, предложена Майером
[60]. Псзднее им и его сотрудниками [61—63] было высказано утверждение о
существовании аномальной области выше обычно наблюдаемой критической
температуры. В частности, было предсказано существование конечного тем-
пературного интервала выше температуры исчезновения мениска, в котором
существуют изотермы, не изменяющиеся с давлением внутри конечного интер-
вала плотностей, но имеющие при всех давлениях непрерывные производные
по плотности. Эта теория конденсации вызывает различные возражения, в
частности существование аномальной области вблизи критической точки явля-
ется интересным вопросом, требующим, однако, экспериментальной проверки.
*) В работе [59] методом радиоактивных индикаторов измерено относительное распре-
деление плотности в зависимости от высоты в ксеноне в критической области. Распределение
плотности при температурах выше критической в основном совпадает с предсказанным клас-
сической теорией критическим состоянием и некоторыми последними данными об уравнении
состояния в критической области. Интересно отметить, что гистерезиса в поведении плот-
ности не обнаружено.
304 Гл. 5. Равновесие жидкость—пар и критические явления
Для систем, заключенных в конечный объем, изотермы являются непре-
рывными функциями плотности. Разрыв производных, указывающий на фазовый
переход, появляется в предельном случае, когда объем и число молекул велики,
а объем, приходящийся на одну молекулу, остается постоянным. Существо-
вание такого предела молчаливо предполагается в развиваемой теории урав-
нения состояния.
В гл. 3, § 2, показано [см. B.23), стр. 129], что
где z — активная плотность, a bt — групповые интегралы, определенные фор-
мулой B.5) гл. 3. Групповые интегралы являются интегралами по всем коор-
динатам / молекул, заключенных в объеме Vy от выражения, зависящего от
потенциальной энергии и всех координат I молекул. При малых значениях I
групповые интегралы bt почти не зависят от V. При больших, но конечных
значениях I групповые интегралы bt стремятся к своим пределам b*f\ соответ-
ствующим бесконечным значениям V при условии, что межмолекулярный по-
тенциал достаточно быстро стремится к нулю с увеличением расстояния.
Поскольку в формуле B.23) гл. 3 берется сумма конечного числа членов,
то изотерма, изображаемая этим уравнением, является аналитической кривой,
не имеющей разрывных производных. Разрыв первых производных, указываю-
щий на существование фазового перехода, появляется лишь в пределе при
стремлении V и N к бесконечности, причем V/N = v (постоянная). Поэтому
давление ps, при котором начинается конденсация, можно связать с первой син-
гулярностью (т. е. с сингулярностью, соответствующей наименьшей величине z)
в уравнении
V/v
p = kT\\m 2btzl. B.17)
Майер предположил, что эта сингулярность совпадает с первой сингулярностью
уравнения
p = kT2bfj. B.18)
Кацура и Фуджита [64 ] указали, что ввиду зависимости активной плотности
z от V (а также от 7, так как v = V/N) это совпадение будет необязательным.
Они считают, что первые сингулярности уравнений B.17) и B.18) будут раз-
личны.
Чисто формальными преобразованиями уравнение B.18) можно привести
к вириальной форме :
[]Jf] B.19)
рк определены формулой B.25) гл. 3 посредством интегралов Ьф для I <? к + 1.
Из этого выражения непосредственно получается производная изотермы, кото-
рая имеет вид
(w)r=^[1-i^"k]- <2-20>
В работе [65] показано, что первая сингулярность в уравнении B.18)
соответствует наименьшему значению л среди тех, для которых выражение
2кркп* B.21)
имеет сингулярность или при которых
i*&n*=l. B.22)
§ 2. Фазовые переходы в однокомпонентных системах
305
Майер определяет /?0(Т) как предел выражения1)
= lim [&]»"<. B.23)
В этом случае конденсация появляется при л = 1//?0, если уравнению B.22)
не удовлетворяют меньшие значения плотности. В последних случаях конден-
сация появляется при меньших значениях плотности. Поэтому в пределе в
точке конденсации (Зр/3л)т > О появляется разрыв в наклоне изотерм. Это
соответствует обычным представлениям о конденсации ниже критической точки.
Если существует какой-либо конечный температурный интервал, где урав-
нение B.22) определяет величину плотности, при которой начинается конден-
сация, то из формулы B.20) следует, что (Зр/Эл)т стремится к нулю при прибли-
жении к этому значению плотности ; поэтому эти изотермы непрерывны.
Такой температурный интервал и аномальная область существуют, если
fik таково, что выполняются одновременно следующие четыре условия :
1) 2 кркпк сингулярно ;
2) сингулярная точка является точкой ветвления ;
3) 2 к$кпк -> 1 при приближении к сингулярной точке ;
к
4J
к
О при приближении к сингулярной точке.
Последнее условие эквивалентно утверждению, что вторая производная
давления по плотности вдоль изотермы равна нулю. Убедительность аргумен-
тации Майера вызывает сомнение прежде
всего потому, что требование одновремен-
ного выполнения четырех условий является
слишком жестким. Если аномальная область
существует, то изотермы имеют вид, изобра-
женный на фиг. 93. Ниже температуры Тс
изотермы имеют разрывные производные в
точке конденсации; выше этой температуры,
но ниже температуры Т'с изотермы имеют
непрерывный наклон, но существует по
крайней мере одна точка, в которой наклон
равен нулю. Внутри заштрихованной об-
ласти, связанной как раз с этими изотер-
мами, вещество будет обладать совершенно
своеобразными свойствами. Майер утвер-
ждает, что в этой области не существует
поверхности натяжения между двумя фа-
зами. С точки зрения влияния гравитации
на распределение плотности внутри объема,
рассмотренного в § 2, можно с уверен-
ностью сказать, что существует широкая переходная область между двумя
фазами без видимого разрыва. Кроме того, согласно теории флуктуации, изло-
женной в гл. 2, § б, в этой области можно ожидать больших флуктуации плот-
ности. Действительно, если изотермы имеют плоскую часть, на которой все
производные равны нулю, то флуктуации по существу будут бесконечными (за
исключением того, что общий объем системы будет конечным).
Теория конденсации Майера подвергалась критике со стороны различных
авторов по разным причинам. Кан и Уленбек [67, 68] выдвинули некоторые
возражения против теории Майера и предложили альтернативную теорию
конденсации. Майер [69] не считает эти возражения правильными.
Объем —»-
изотерм вблизи крити-
соответствии с теорией
Майера [66].
г) Подчеркнем, что при"конечных значениях / интеграл bi, как показано, приближается
к конечному пределу Ьф. Поскольку /?л определены через b*f\ может оказаться, что вели-
чина ро(Т) зависит от порядка, в каком вычисляется предел.
306 Гл. 5. Равновесие жидкость—пар и критические явления
Цимм показал [70], что «принцип суперпозиции» потенциалов средних
сил (см. гл. 4, § 9) приводит к тому, что критическая точка является сингуляр-
ной точкой изотермы. В этой сингулярной точке производные всех порядков
от давления по плотности равны нулю, тогда как обычно предполагается, что
только две первые производные равны нулю. Кроме того, Цимм показал, что
если критическая точка является сингулярной точкой, то теория конденсации
Майера приводит к единственной критической температуре и не предсказы-
вает существования аномальной области. Майер указал [69], что единственное
заключение, которое можно сделать из замечаний Цимма, сводится к несов-
местимости существования аномальной области с принципом суперпозиции.
Совершенно ясно, что вопрос о существовании аномальной области является
дискуссионным как с точки зрения теории, так и с точки зрения эксперимента.
§3. ФАЗОВЫЕ ПЕРЕХОДЫ В ДВУХКОМПОНЕНТНЫХ
СИСТЕМАХ1)
В однокомпонентных системах конденсация возникала при понижении
температуры и повышении давления, а испарение — при повышении тем-
пературы и понижении давления. Поведение двухкомпонентной системы при
фазовых переходах значительно сложнее вследствие обратного явления. Если
сжимать смесь газов, то при некотором давлении появляется жидкая фаза.
Однако при определенных температуре и составе дальнейшее возрастание
давления может привести к уменьшению и даже полному исчезновению
жидкой фазы. Это явление впервые наблюдал Кальете [73 ] при сжижении смеси
воздуха и углекислого газа и назвал его обратной конденсацией первого рода.
Аналогичное явление возникает при изобарическом изменении температуры
системы. Оно известно под названием обратной конденсации второго рода. Эти
обратные явления можно объяснить, анализируя диаграмму зависимости
между р, VyT их бинарной смеси. В конце этого параграфа будет дан общий
термодинамический анализ двухфазных систем.
А. КРИТИЧЕСКАЯ ТОЧКА И ОБРАТНОЕ ПОВЕДЕНИЕ
В § 2 было показано, как можно интерпретировать уравнение состояния
чистых веществ с помощью трех двухмерных проекций поверхности р = p(V, T),
изображенных на фиг. 79. Если система находится в состоянии, в котором
одновременно существуют две фазы, находящиеся в равновесии, то на фазовой
диаграмме этому состоянию соответствует точка, лежащая в языкообразной
области (включая и линию сосуществования ADCEB). Проекцией языкообраз-
ной области на плоскость р — Т является обычная кривая давления пара
Рп&р(Т), кончающаяся в критической точке.
Возможные состояния бинарной системы изображаются поверхностью
(в пространстве четырех измерений) р = p(V, Т> х), где х — молярная доля
первой или второй компоненты. Эту поверхность можно изучить с помощью
различных трехмерных проекций. В частности, для изучения равновесия
жидкость—пар удобно подробно исследовать трехмерную поверхность, по-
лучающуюся проектированием части поверхности р = p(V, T, х), описываю-
щей совместное сосуществование фаз в равновесии. Эта поверхность pnapG\ x)
показана на фиг.942) и является двухкомпонентным аналогом кривой давления
пара для чистого вещества3). Кривая КгСг является кривой давления пара для
Ч [71, 72].
2) Это чертеж трехмерной модели, сделанной Бойдом [27 ] и Экштейном.
3) Необходимо заметить, что поверхность pnap(Tf х) не является функцией р = p(VfT, x)
при постоянном V, так же как /?пар(Г) не является функцией р = p(V, T) при постоян-
ном V. С целью подчеркнуть это различие употребляется индекс «пар».
§ 3. Фазовые переходы в двухкомпонентных системах
307
чистой компоненты 1, оканчивающейся в точке Clf которая является крити-
ческой точкой для этой компоненты. Аналогичным образом К2С2, оканчиваю-
щаяся в точкеС2, является кривой давления пара для чистой компоненты 2. Через
любую точку (р, х, Т), находящуюся внутри поверхности, можно провести
прямую, параллельную оси х. Эта линия пересечет поверхность в двух точках,
определяющих состав жидкой и газообразной фаз в равновесии при заданных
величинах р и Т. Давление, температура (и состав), при которых эти две точки
совпадают, определяют критическую точку смеси1). Кривая СгС0С2 является
геометрическим местом критических точек смесей различного состава. Эта
кривая делит поверхность на две части, соответствующие жидкому и газооб-
разному состояниям.
-50
О 10 20 30 40 50 60 70 80 90 100
х-процентное содержание молей этана
Фиг. 94. Трехмерное представление поверхности рпарG\х) для системы этан—гептан.
Экспериментальные данные взяты из работы [74]. Сг и С, — соответственно критические точки для гептана
и этана ; кривые К^ и К%С% — соответствующие кривые давления пара ; кривая CtC9Ct — геометрическое
место критических точек для смесей различного состава. А — линии относятся к стороне поверхности,
обращенной к жидкости ; В — линии относятся к стороне поверхности, обращенной к пару.
Чтобы облегчить понимание трехмерной поверхности, изображенной на
фиг. 94, ее удобно представить в виде трех двухмерных проекций, аналогич-
ных проекциям, изображенным на фиг. 79. Эти проекции показаны на
фиг. 95-^97. С помощью этих диаграмм рассмотрим равновесие жидкость—пар
в двухфазной системе и явление обратной конденсации.
На фиг. 95 изображены изобары в плоскости Т — х. Они получаются
проектированием на плоскость Т — х кривых, образующихся при сечении
поверхности рпар = Pnap(T, х) плоскостями, параллельными плоскости Т — х.
Это обычные кривые равновесия жидкость—пар, которые часто употребля-
ются для определения поведения бинарных систем при фазовых переходах.
С помощью этой диаграммы рассмотрим некоторые возможные варианты по-
ведения системы при фазовом переходе.
х) Критическую точку смеси по аналогии с однокомпонентной системой можно опре-
делить как точку, в которой исчезает различие между двумя фазами. Заметим, однако, что
только в случае чистых веществ критическая точка соответствует наивысшим значениям
температуры и давления, при которых одновременно существуют две фазы.
308
Гл. 5. Равновесие жидкость—пар и критические явления
300
Рассмотрим сначала систему, состояние которой изображается точкой
А на диаграмме Т — х; пусть давление в этой системе равно 14,06 кг/см*.
Повышаем температуру системы таким образом, чтобы объем системы увели-
чивался, а давление оставалось постоянным. Точка, изображающая состояние
системы в этом процессе, будет перемещаться вверх по пунктирной прямой
до тех пор, пока не достигнет положения В. При этой температуре появляется
газообразная фаза (т. е. фаза меньшей плотности), изображаемая точкой В\
которая существует в равновесии с жидкостью. При дальнейшем увеличении
температуры объем, занимаемый газообразной фазой, увеличивается, а объем
жидкой фазы уменьшается, пока сис-
тема не дойдет до точки D, в которой
исчезают последние следы жидкости,
имеющей состав, определяемый точкой D'.
С увеличением давления погранич-
ная кривая жидкость—пар сдвигается
в сторону более высоких температур. Из-
менение точек кипения чистых ком-
понент (х = 0 и х = 1) с давлением сле-
дует вдоль кривой давления пара для
двух чистых веществ. С увеличением дав-
ления температуры кипения чистых ком-
понент непрерывно возрастают, пока не
достигнут критической температуры
одной из компонент. Для системы этан—
гептан при давлении, несколько мень-
шем 28,12 кг/см2, это будет температура
гептана. Соответствующая критичес-
кая температура равна ~ 250° С. На-
чиная с этой температуры и выше,
в системе выше некоторого предельного
состава, определяемого давлением, не
могут одновременно существовать две
жидкие фазы.
Исследуем теперь поведение системы
при давлении 42,18 кг/см2. Гетерогенная
область, ограниченная кривыми жид-
кости и пара, не простирается налево далее х = 0,18. Следовательно, если
изобарически нагревать смесь, состав которой х = 0,15, то фазового разде-
ления, которое наблюдалось при давлении 14,06 /сг/сл*2, не происходит. Ли-
ния жидкости изобары 42,18 кг)см2 при возрастании температуры, прежде чем
соединиться с линией пара в точке с, проходит через минимум относительно
содержания этана. Точка с, являющаяся точкой пограничной кривой, наклон
которой равен нулю, представляет критическую точку для смеси, состоящей
из 0,22 молярных долей этана. Только в этой точке жидкая и газообразная
фазы неразличимы по плотности и составу.
Наличие минимума по составу на жидкой границе приводит к возникно-
вению одного из видов обратной конденсации. Рассмотрим смесь при давлении
42,18 кг/см2, состояние которой на фиг. 95 изображается точкой а. При нагре-
вании смеси таким путем, что ее давление и состав не меняются, точка, изоб-
ражающая состояние системы, будет перемещаться от а к Ь, где существуют
условия, когда возникает газообразная фаза, состав которой определяется
точкой Ъ*. Если происходит дальнейшее увеличение температуры, то объем,
занимаемый жидкой фазой, уменьшается, достигая минимума, а затем снова
начинает возрастать вплоть до точки d, когда исчезают последние следы газо-
образной фазы, состав которой определяется точкой d', и система снова пере-
ходит в однофазное жидкое состояние. Таким образом, при нагревании системы
процесс парообразования сменяется в дальнейшем процессом конденсации.
0,20 0,40 0,60 0,80 {00
х-молярные доли этана
Фиг. 95.Изобары на плоскости Т — х
для системы этан—гептан.
График получен проектированием на плоскость
Т — х кривых, образующихся при сечении по-
верхности Рпар(Т,х) плоскостями, перпенди-
кулярным к оси р. Этот график использован
для иллюстрации обратной конденсации второго
рода в процессах, которым соответствует линия
ае для изобары 42,18 кг/см* [71 ].
§ 3. Фазовые переходы в двухкомпонентных системах
309
Это явление известно под названием обратной конденсации второго рода
[75, 76 Р). Описываемое явление наблюдается, конечно, если точка а лежит
слева от критической точки с и справа от самой левой точки линии жидкости.
Если критическая температура второй компоненты превосходит первую, то
система при нагревании ведет себя аналогичным образом, только на этот раз
возникает и исчезает жидкая фаза.
На фиг. 96 изображены изотермы в плоскости р — л. Они получаются
проектированием на плоскость р— х кривых, образующихся при сечении
поверхности, изображенной на фиг. 94, плоскостями, перпендикулярными
оси Т. Кривые такого типа могут служить для иллюстрации явления, извест-
ного под названием обратной конденсации первого рода [73, 75, 76 ]. Рассмотрим
84,36 -
О 20 40 60 80
х-молярные доли этана
Фиг. 96. Изотермы на плоскости р — х
для системы этан—гептан.
Получены проектированием на плоскость р — х
кривых, образующихся при сечении поверхности
рпар(Т,х) плоскостями, перпендикулярными
оси Т. Пунктирные линии иллюстрируют
обратную конденсацию первого рода при 150°С.
Кривая "
'давления
компоненты 1
50 100 150 200 250
Т-температура, °С
300
Фиг. 97. Линии постоянного состава
на плоскости р — Т.
Получаются проектированием на плоскость р—Т
кривых, образующихся при сечении поверхности,
изображенной на фиг. 94, плоскостями, перпен-
дикулярными оси х. Пунктирные линии ае и
ае — пути, на которых происходит обратная
конденсация первого и второго рода. Молярная
доля этана в этой смеси х = 0,265 [71 ].
систему при температуре 150 °К, состояние которой на фиг. 96 изображается
точкой а. Поскольку эта точка не лежит внутри гетерогенной области, то она
соответствует только однофазному состоянию системы. При изотермическом
увеличении давления изображающая точка будет двигаться вверх по линии ае.
В точке р появляется жидкая фаза, состав которой определяется точкой /Г.
При дальнейшем увеличении давления объем, занимаемый жидкой фазой,
сначала увеличивается, а затем уменьшается. В точке д исчезают последние
следы жидкой фазы, состав которой определялся точкой 6'. Такое обратное
поведение не наблюдается, конечно, в смесях, содержание этана в которых
меньше, чем в критических смесях при этом давлении (отмеченном точкой С),
или в смесях, содержащих этана больше, чем в состоянии, соответствующем
крайней правой точке на линии пара.
В общем случае обратное поведение свойственно таким процессам, при
которых изображающая точка на трехмерной диаграмме входит или выходит
из гетерогенного объема на одной и той же фазовой поверхности независимо
от того, соответствует ли она жидкой или газообразной фазе. Ее путь не обяза-
тельно параллелен оси температуры или давления, а может сопровождаться
одновременным изменением давления и температуры.
г) Обратной конденсацией первого рода называется аналогичное явление, которое возни-
кает при изотермическом изменении давления.
310
Гл. 5. Равновесие жидкость—пар и критические явления
На фиг. 97 изображены линии постоянного состава на диаграмме р — Т.
Они получены проектированием на плоскость р — Т кривых, образующихся
при сечении поверхности, изображенной на фиг. 97, плоскостями, перпенди-
кулярными оси х. Часть кривой, образующейся при пересечении плоскости
с поверхностью, соответствующей жидкости, обычно называется линией точек
кипения, а с поверхностью, соответствующей газообразному состоянию, —
линией точек росы. Критической точкой смеси данного состава является точка
соединения линии росы с линией кипения. Если система изменяется так, что
ее изображающая точка движется в двухфазную область, пересекая линию
точек росы, то в системе возникает жидкая фаза, по составу отличающаяся от
газообразной. Если изображающая точка пересекает линию точек кипения,
то появляется газообразная фаза. Эти два типа обратной конденсации пока-
заны на фиг. 97 линиями ае и ае.
В последнее время интерес к проблеме обратной конденсации значительно
усилился благодаря той важной роли, которую играет дистилляция углево-
дородов под большим давлением в нефтяной промышленности. Кроме- того,
эти явления постоянно используются в целях увеличения добычи нефти из
скважин. Поддерживая умеренное давление в скважине, удается избежать
потери высококипящих фракций, происходящей при понижении давления.
I
Жидкая фаза
Газообразная N
фаза
Температура
исчезновения
Фиг.!
Температура
, Зависимость электропроводности растворов NH4C1 в СН8ОН от температуры вблизи
критической точки [80 ].
Физические свойства растворов нелетучих компонент вблизи критической
области растворителя также представляют значительный интерес.
В этой области наблюдаются следующие явления :
1) В алкоголе выше критической точки существуют обычные ионные усло-
вия в жидкости [77, 78 ]. Это доказывается путем изучения спектров погло-
щения раствора кобальт—хлорид алкоголя выше и ниже критической тем-
пературы. Спектры поглощения ионизованных солей остаются одинаковыми.
2) Молекулы йода остаются сольватированными молекулами карбон-
тетрахлорида при прохождении такого раствора через критическую точку
растворителя [79]. Это доказывается тем фактом, что спектр поглощения
йода скорее напоминает спектр йода, растворенного в жидкости, чем спектр
газообразного йода. Таким путем доказывается существование влияния раст-
ворителя выше критической точки.
3) Электропроводность растворов метилалкоголя либо в KJ, либо в NH4C1
как функция температуры обладает непрерывными производными в крити-
ческой области [80 ]. Из фиг. 98 видно, что вблизи критической точки электро-
проводность газа увеличивается, а жидкости — уменьшается.
§ 3. Фазовые переходы в двухкомпонентных системах 311
4) В разведенных растворах критическая температура пропорциональна
молярной концентрации разведенного вещества [81 ]. Например, при повы-
шении молярной концентрации жидкого СО2 на 1% критическая температура
повышается на 8,8° С, для жидкого нашатырного спирта — на 13,0° С.
Б. ТЕРМОДИНАМИЧЕСКОЕ РАССМОТРЕНИЕ
Для изучения состояния смеси при фазовом переходе можно использовать
эмпирическое уравнение состояния смеси. В § 2 было показано, что давление
пара в однокомпонентной смеси можно найти, воспользовавшись эмпирическим
уравнением состояния и проведя горизонтальную линию на диаграмме р — V
так, чтобы области, заключенные между изотермой и линией по обе стороны
последней, были равны друг другу. Обобщение этой идеи на случай бинарной
смеси — не простая задача.
Если различные фазы многокомпонентной смеси находятся в равновесии,
то в соответствии с термодинамическими требованиями химический потенциал
(т. е. парциальный молярный термодинамический потенциал) i-й химической
компоненты должен быть одинаков во всех фазах. В частном случае равновесия
жидкость—пар в бинарной системе это условие сводится к требованию
/*<$ = № /*$ = №, C.1)
где верхние индексы (I) и (v) относятся к величинам в жидкой и газообразной
фазах, а нижние индексы 1 и 2 — к компонентам.
Химический потенциал f-й компоненты смеси идеальных газов имеет вид
МТ, Р) = 1*ЩТ) + RT\np+RT In Xi, C.2)
где Xi — молярная доля i-й компоненты, а /*<?>(Г) — химический потенциал
или термодинамический потенциал, отнесенный к 1 молю чистой i-й компоненты
при температуре смеси и единичной летучести. Разность между химическим
потенциалом i-й компоненты в реальном газе и этим выражением можно
записать с помощью уравнения состояния смеси. Из термодинамики известно,
что
где V( — парциальный молярный объем i'-й компоненты. Таким образом, в
смеси реальных газов химический потенциал i-й компоненты имеет вид
р) = №(Т) + RTIn(pXi) + J [Vt ~-y-]dp. C.4)
Интеграл берется при постоянной температуре.
Состав бинарной смеси определяется молярной долей одной из компонент.
Будем считать, что хг известно, и определим выражение
C.5)
Поскольку парциальный молярный объем V( определяется из уравнения
состояния смеси, то функция /,(Т, р, хх) считается известной, если известно
уравнение состояния. В § 2 было показано, что эмпирические уравнения со-
стояния таковы, что некоторые изотермь! имеют S-образную форму. Таким
312
Гл. 5. Равновесие 'жидкость—пар и критические явления
образом, при определенных условиях на изотерме существуют три точки,
соответствующие определенному давлению, а следовательно, /?-(Т, р, хг) явля-
ется трехзначной функцией. Средняя точка лишена физического смысла, а что
касается крайних точек, то необходимо различать величины, связанные с изо-
термами пара f?\T, p, хг), и величины, от-
носящиеся к изотермам жидкости /ф(Т, р, хх).
Условия равновесия двух фаз в бинарной
смеси [формула C.1) ], выраженные с помощью
этих функций, имеют вид
C.6)
При фиксированных значениях темпера-
туры и давления эти уравнения определяют
хф и х^, являющиеся молярными долями
компоненты 1 в жидкой и парообразной фазах.
В общем случае фазы обладают различными
составами. Критический состав при заданных
температурах и давлении определяется вели-
чиной xlf удовлетворяющей равенству хф =х({К
Ниже критической температуры это равенство
уже не выполняется. Как указывалось ранее,
при более высоких температурах может суще-
ствовать 0, 1 или 2 такие точки.
Аналитическое выражение функции //(T,p,Xj)
для смеси, подчиняющейся уравнению Ван
дер Ваальса, приведено в приложении А.
Вид этих функций изображен на фиг. 99.
Эти кривые даны с характеристическими постоянными системы этан—гептан
для температуры 400° К и давления 20 атм. Пунктирные отрезки кривой
обязаны своим происхождением недостижимым центральным частям изотерм
Ван дер Ваальса. Решение уравнения C.6) приводит к значениям x{f = 0,30
и хф = 0,75. Из фиг. 94 очевидно, что при некоторых значениях температуры
и давления уравнения C.6) вообще не имеют решения.
Фиг. 99. Вид функций ff* и
определяющих поверхность сосу-
ществования двухкомпонентных
смесей.
Прило жен ие А
ХИМИЧЕСКИЙ ПОТЕНЦИАЛ СМЕСИ ГАЗОВ, ПОДЧИНЯЮЩЕЙСЯ
УРАВНЕНИЮ ВАН ДЕР ВААЛЬСА
Уравнение Ван дер Ваальса имеет вид
R Т а / а 1 \
P=^—j-Jf (АЛ>
Хотя первоначально это уравнение было предложено как уравнение состоя-
ния р — V — Т для однокомпонентных газов, Ван дер Ваальс применил его
к смесям. В бинарных смесях постоянные а и Ь будут зависеть от состава в
соответствии с формулами B.5) и B.6) гл. 4.
Используя это уравнение состояния, можно найти функции /,G, р, Xj),
определенные формулой C.5). Они имеют следующий вид:
Ji{T, P, хг) — in -j^—: b)
(A.2)
Литература 313
Величины At и Bt определяются выражениями
А1 = 2(х1ап — 2х2а12),
&22).
Аналогичным образом определяются Л2 и Ва.
Задачи
К С помощью уравнения Ван дер Ваальса найти удельный объем пара, удельный объем
жидкости, а также давление пара в однокомпонентной системе при температуре Va^c-
2. Предположим, что вблизи критической точки путем экспериментальных измерений
давления и температуры при различных значениях плотности в контейнере высоты h опре-
делен ряд изотерм. Как необходимо исправить изотермы, если принять во внимание изме-
нение давления и плотности в контейнере, вызванное силами притяжения [27 ]?
3. Определить состав и удельный объем жидкой и газообразной фазы смеси, состоящей
из равного числа молей этана и гептана при 100° С. Применить уравнение Ван дер Ваальса
совместно с правилом, определяющим постоянные а и Ь смеси [см. формулы B.5) и B.6)
гл. 4]. Примем
=$-?* и я = з<>с??
([72]); экспериментальные данные для системы этан—гептан приведены в работе [74].
ЛИТЕРАТУРА
1. О п о S., Memoirs of the Faculty of Engineering, Kyushu University, 10, 195 A947).
2. MacLellan A. G., Proc. Roy. Soc, A213, 274 A952).
3. H i 11 T. L., Journ. Chem. Phys., 19, 261 1203 A951); 20, 141 A952).
4. G i b b s J. W., The Collected Works, Vol. I, p. 219, London A928).
5. Fowler R. H., Proc. Roy. Soc, A159, 229 A937).
6. Fowler R. H., Physica, 5, 39 A938).
7. Bradley, Phil. Mag., 11, 846 A936).
8. Margenau, Phys. Rev., 38, 365 A931).
9. Lennard-Jones J. E., Corner J., Trans. Farad. Soc, 36, 1156 A940).
10. С о r n e r J., Trans. Farad. Soc, 44, 1036 A948).
11. Guggenheim E. A., Journ. Chem. Phys., 13, 253 A945).
12. Guggenhe i m E. A., Thermodynamics, 2nd ed., Amsterdam, 1950.
13. Ha ra si ma A., Proc Phys. Math. Soc Japan, 22, 825 A940).
14. В a 1 y, D о n n a n, Journ. Chem. Soc, 81, 907 A902).
15. Rudorf G., Ann. d. Phys., 29, 751 A909).
16. К irk wood J. G., Buff F. P., Journ. Chem. Phys., 17, 338 A949).
17. T о 1 m a n R. C, Journ. Chem. Phys., 16, 758 A948); 17, 118, 333 A949).
18. Buff F. P., Kirk wood J. G., Journ. Chem. Phys., 18, 991 A950).
19. Buff F. P., Journ. Chem. Phys., 19, 1591 A951).
20. Кое nig F. 0., Journ. Chem. Phys., 18, 449 A950).
21. В о г n M., Green H. S., Proc. Roy. Soc, A188, 10 A946).
22. Green H. S., Proc Roy. Soc, A189, 103 A947).
23. Eisenstein A., Gingrich N. H., Phys. Rev., 62, 261 A942).
24. McLeod, Trans. Farad. Soc, 19, 38 A923).
25. S u g d e n S., The Parachor and Valency, London, 1930.
26. Michels A., Blaisse В., Mi с he Is C, Proc. Roy. Soc, A160, 358 A937).
27. W e n t о r f R. H., В о у d С. A., University of Wisconsin Naval Research Laboratory
Report, CM-724, May 1952.
28. Wentorf R. H., Диссертация; University of Wisconsin, December 1951.
29. S a j о t s с h e w s k i W., Ann. Phys. Beibl., 3, 741 A879).
314 Гл. 5. Равновесие жидкость — пар и критические явления
30. Cailletet L., Colardeau, Compt. rend., 112, 563 A891).
31. I p a t i e f f, Monroe, Ind. Eng. Chem. (Anal. Ed.), 14, 171 A942).
32. White D., Friedman A., Johnston H., Journ. Am. Chem. Soc, 72, 3565 A950).
33. Cagnaird de la Tour, Ann. chim. (Ser. 2), 21, 127 A922).
34. С a i 11 e t e t L., M a t h i a s E., Compt. rend., 102, 1202 A886); 104, 1563 A887).
35. С a i 11 e t e t L., M a t h i a s E., Journ. Phys., 5, 549 A886).
36. Mathias E., Crommelin С A., Kamerlingh Onnes H., Comm. Phys.
Lab. Leiden, 154b. A921).
37. Par ting ton J. R., Treatise on Physical Chemistry, Vol. I, London, 1949.
38. Becker R., During W., Ann. d. Phys. E), 24, 719 A935).
39. Becker R., Trans. Farad. Soc, 45, 55 A949).
40. Reiss H., Journ. Chem. Phys., 18, 529 A950).
41. T u r n b u 11 D., F i s h e r J., Journ. Chem. Phys., 17, 71 A949).
42. Scott A. F., Shoemaker D. P., Tanner K. N., Wendel J. G., Journ.
Chem. Phys., 16, 495 A948).
43. Pies set M. S., Journ. Appl. Mech., 16, 277 A949).
44. The Scientific Papers of James C. Maxwell, Cambridge, 1890.
45. Mich els A., Bijl A., Michels C, Proc. Roy. Soc, A160, 376 A937).
46. M i с h e 1 s A., S t r i j 1 a n d J. C, Physica, 15, 813 A950).
47. С u r t i s s C.F., В о у d С. A., Palmer H. В., Journ. Chem. Phys., 19, 801 A951).
48. Herget С. М., Journ. Chem. Phys., 8, 537 A940).
49. S с h n e i d e r W. G., Journ. Chem. Phys., 18, 1300 A950).
50. Schneider W. G., Canad. Journ. Chem., 29, 243 A951).
51. Roebuck J. R., Murrell T. A., Miller E. E., Journ. Am. Chem. Soc, 64,
400 A942).
52. He in P., Zs. phys. Chem., 86, 385 A914).
53. M a a s s O., Chem. Rev., 23, 17 A938).
54. M с I n t о s h R. L., Dacey J. R., M a a s s O., Canad. Journ. Res., 17B, 241 A939).
55. Palmer H. В., Диссертация, University of Wisconsin, 1952.
56. Winkle г С A., Maass O., Canad. Journ. Res., 9, 613 A933).
57. Yvon J., Actualites scientifiques et industrielles, Paris, 1937.
58. С a t a 1 d i H. A., D г i с к a m e r H. G., Journ. Chem. Phys., 18, 650 A950).
59. Weinberger M. A., Schneider W. G., Canad. Journ. Chem., 30, 422 A952).
60. Mayer J. E., Journ. Chem. Phys., 5, 67 A937).
61. Mayer J. E., Ackermann P. G., Journ. Chem. Phys., 5, 74 A937).
62. Mayer J. E., Harrison S. F., Journ. Chem. Phys., 6, 87 A938).
63. Harrison S. F., Mayer J. E., Journ. Chem. Phys., 6, 101 A938).
64. Katsura S., Fujita H., Journ. Chem. Phys., 19, 795 A951).
65. Born M., Fuchs K-, Proc. Roy. Soc, A166, 391 A938).
66. Mayer J. E., Mayer M. G., Statistical Mechanics, New York, 1940. (см. перевод:
Май ер Дж., Гепперт-Майер М., Статистическая механика, ИЛ, 1952).
67. Капп В., Uhlenbeck G. E., Physica, 5, 399 A938).
68. К a h n В., Диссертация, Utrecht, 1938.
69. Mayer J. E., Journ. Chem. Phys., 19, 1024 A951).
70. Zimm В. Н., Journ. Chem. Phys., 19, 1019 A951).
71. Hougen O. A., Watson KM., Chemical Process Principles, Part II, New York,
1947.
72. В о у d С. A., Journ. Phys. Colloid. Chem., 54, 1347 A950).
73. С a i 11 e t e t L., Compt. rend., 90, 210 A880).
74. Kay W. В., Ind. Eng. Chem., 30, 459 A938).
75. К u en en J. P., Arch. Neerland. Sci., 26, 354 A893).
76. Katz D. L., Kurata F., Ind. Eng. Chem., 32, 817 A946).
77. H a n n а у V. В., Proc. Roy. Soc, 30, 478, 484 A880).
78. H a n n а у V. В., Hogarth J., Proc. Roy. Soc, 29, 324 A879); 30, 178 A880).
79. Villa rd P., Compt. rend., 120, 182 A895).
80. Kraus С A., Phys. Rev., 18, 40, 89 A904).
81. Booth H. S., Bi dwell R. M., Chem. Rev., 44, 477 A949).
ГЛАВА б
КВАНТОВАЯ ТЕОРИЯ УРАВНЕНИЯ СОСТОЯНИЯ1)
В предыдущих главах изучались равновесные свойства таких газов и в
таких условиях, в которых классическая механика адекватно описывает по-
ведение системы. Существуют, однако, два типа квантовых эффектов, которые
при определенных условиях необходимо принимать во внимание : 1) диффрак-
ционные эффекты, объясняющиеся волновой природой молекул и проявляю-
щиеся в условиях, когда длина дебройлевской волны молекул порядка моле-
кулярного диаметра ; 2) эффекты симметрии, связанные со статистикой частиц
и проявляющиеся в условиях, когда длина дебройлевской волны молекул
порядка среднего расстояния между молекулами в газе.
При комнатных температурах диффракционные эффекты достигают изме-
римой величины у гелия и водорода и не существенны для тяжелых газов.
При низких температурах квантовые поправки, обусловленные этими эф-
фектами, становятся существенными для гелия и водорода, а также вполне
заметными для тяжелых газов. Эффекты симметрии сказываются лишь при
очень больших плотностях или очень низких температурах.
Настоящая глава посвящена расчету этих квантовых эффектов и, исклю-
чая последний параграф, в ней обсуждаются одноатомные газы. После крат-
кого изложения теории канонического ансамбля в квантовой механике выве-
дено квантовое уравнение состояния двумя общими методами, описанными
в гл. 3 в связи с классическим уравнением состояния.
С помощью этих результатов может быть найдено уравнение состояния
идеального газа, равновесные свойства которого могут быть выражены в
аналитическом виде. Это позволит получить сведения об упомянутом выше
«эффекте симметрии», а также о явлении конденсации Бозе—Эйнштейна.
Затем подробно рассмотрен квантовомеханический расчет второго вириального
коэффициента и коэффициента Джоуля—Томсона при давлении, равном
нулю. В тех случаях, когда это возможно, произведены сравнения вычи-
сленных и экспериментальных данных.
В начале гл. 4 обсуждался закон соответственных состояний в класси-
ческой механике. Аналогичный закон в квантовой механике имеет вид
р*= р*(У*, Т* ; Л*). Как видно из этой формулы, степень отклонения системы
от классической определяется величиной Л* — h/оУ те. Эта величина равна
отношению длины дебройлевской волны (соответствующей системе с приве-
денной массой ц — т\2 и энергией е) к диаметру столкновений а и харак-
терна для каждого вещества. В этой главе закон соответственных состояний
применяется при изложении квантовых эффектов в жидкостях и плотных
газах.
*) Написана Дж. де Буром и Р. Байрон Бердом.
316 Гл. 6. Квантовая теория уравнения состояния
§ 1. ПРЕДВАРИТЕЛЬНЫЕ СВЕДЕНИЯ ИЗ СТАТИСТИЧЕСКОЙ
МЕХАНИКИ
Прежде чем обсуждать вывод уравнения состояния, коротко суммируем
основные сведения о каноническом ансамбле в квантовой механике. Сначала
обсудим некоторые формы матрицы плотности вероятности, затем статисти-
ческую сумму, средние величины и «сумму Слетера». Сумма Слетера является
квантовомеханическим аналогом множителя Больцмана. Показано, что сумма
Слетера переходит в множитель Больцмана в классическом пределе.
А. МАТРИЦА ПЛОТНОСТИ ВЕРОЯТНОСТИ ДЛЯ КАНОНИЧЕСКОГО
АНСАМБЛЯ
В замкнутой системе, находящейся в равновесии при температуре Т,
оператор плотности вероятности имеет вид
&№=Zjher*ikT, A.1)
где <Э? — квантовомеханический оператор Гамильтона системы N частиц, а
оператор ехр (—<Э?\кТ) по определению равен 2(—<Э?\кТУ\\\. Множитель
Zfc — нормировочный. J
В соответствии с формулой B.19) гл. 2 матрица плотности вероятности
K ; r'N) в гильбертовом пространстве имеет вид
; г'") =
Диагональный элемент
; гаг) = Zrfq 2 Ф\ (г") *-ЛГ'*т ф6 (г") A.3)
Q
является квантовомеханическим выражением плотности вероятности в конфи-
гурационном пространстве rN, a ZNq — квантовомеханическая статистическая
сумма, получаемая из условия B.22) гл. 2 и определяемая формулой D.2) гл. 2.
Функции фй(тм) образуют полный ортонормированный ряд собственных функ-
ций, симметризованных соответствующим образом. Два последних выражения
матрицы плотности обладают тем важным свойством, что они не зависят от
ряда ортонормированных функций фй{хы)у выбранных в качестве базисной
системы при разложении волновой функции (см. гл. 2, § 2). В дальнейших
расчетах будут использованы два ряда функций.
1. Собственные функции оператора энергии. Волновая функция системы
^(гА0 [формула B.1) гл. 2] может быть разложена по собственным функциям
w(rN) оператора Гамильтона >Ж. В этом случае матрица плотности вероятности
принимает вид
(о . гаг) =Zjj\2\ V>* (*N) I2 е-в*1кТ ; 0.4)
здесь ехр (—EffjkT) — вероятность энергетического состояния {ст}, а | ipJrN) |2 —
плотность вероятности в этом состоянии. В такой форме интерпретация
^<дг)(гаг;Гдг) как вероятность в конфигурационном пространстве rN очевидна.
Ниже будет показано, что это выражение стремится к классическому пределу
р(лг)(глг) _ Гр<лг)(гл^ pM)dpM при высоких температурах, когда система мало
отклоняется от классической.
2. Собственные функции оператора импульса. Волновая функция системы
^(rN) [формула B.1) гл. 2] может быть разложена по собственным функциям
оператора импульса ехр [—(i/A) (p^ • rN) ]. Если система заключена в куб
со стороной L, то значения, которые принимает р;-, ограничены некоторой сово-
купностью дискретных значений. Поэтому удобно ввести безразмерный вектор
§ 1. Предварительные сведения из статистической механики 317
6N — (Ljh)pMt допуская в дальнейшем, что он может принимать все положи-
тельные или отрицательные значения. Ряд функций, используемых в качестве
базисной системы при разложении волновой функции, должен, конечно,
соответствовать симметрии системы. Таким образом, ряд симметризованных
собственных функций оператора импульса, которые будут использованы в
дальнейшем, имеют вид
N ж J 2
1)* Z <
где P — оператор перестановки (порядка pY)N частиц, соответствующий любым
перестановкам индексов г^ = гь г2,..., rN. Знак плюс соответствует статистике
Бозе—Эйнштейна, а знак минус — статистике Ферми—Дирака. В этом случае
матрица плотности [формула A.2)] принимает вид
doN =
N ! ^
Второе из приведенных здесь выражений для ^ы\ты; r'N) допустимо
лишь при инвариантности оператора Гамильтона относительно операции пере-
становок Р. Третье выражение получается в результате обратной замены 6N
на р^. Суммирование по g в формуле A.2), которое проводится для всех раз-
личных наборов квантовых чисел, здесь заменено интегрированием по 6N,
причем это интегрирование проводится независимо по всем возможным компо-
нентам импульса от— оо до + оо. Следовательно, дополнительный множитель
AM, появляющийся в формуле A.6), возникает в результате того обстоятельства,
что вследствие тождественности частиц все состояния импульса, получаю-
щиеся простой перестановкой частиц, содержатся в интегрировании.
Матрица плотности вероятности может быть также записана в виде фор-
мулы B.20) гл. 2
^#> = -LJ фп(г")е-Ж1ьтф^(rN)dTN . A.7)
Эта матрица зависит от системы ортонормированных функций, выбранных в
качестве базиса. Способ перехода к другой системе указан формулой B.11) гл.2.
Если, в частности, выбраны собственные волновые функции оператора энер-
гии <3fy)a = ?л\г> то ^e?} принимает диагональный вид
^-Ео1хту A#8)
причем *?#(?? — вероятность того, что система находится в собственном энер-
гетическом состоянии {а} с энергией Еа, a ZNq — квантовомеханическая
статистическая сумма, определяемая из условий нормировки формулы B.22)
гл. 2 и выражаемая формулой D.1) гл. 2.
Б. МАТРИЦА ПЛОТНОСТИ ВЕРОЯТНОСТИ В КЛАССИЧЕСКОМ
ПРЕДЕЛЕ
Диагональный элемент квантовомеханической матрицы плотности и соот-
ветствующее классическое выражение равны
[2Ф1(гы)е-*''*т 0в(г")] A.9)
х) Здесь р — число перестановок пар молекул, необходимых для восстановления перво-
начального порядка.
318 Гл. 6. Квантовая теория уравнения состояния
и1)
* ^ A.10)
где Ф(ты) — потенциальная энергия системы, а А2 = Щ2пткТ. Множитель
д-заг появляется вследствие интегрирования по всем импульсам. Чтобы до-
биться сходства между классической и квантовой формулировками, эти два
выражения необходимо записать в виде
\ wW <u2>
ZNNU3N vv
Величина ^N(rN) известна как сумма Слетера2), а ее классический аналог
WN(r*) обычно называется множителем Больцмана. Эти две величины опре-
деляются следующим образом :
A.13)
A.14)
Покажем3), что сумма Слетера переходит в множитель Больцмана в класси-
ческом пределе в области высоких температур, где система мало отличается
от классической.
Для доказательства в качестве базисных функций ф(*ы) удобно выбрать
соответствующим образом симметризованные собственные функции импульса,
данные формулой A.5). В соответствии с этим сумма Слетера может быть запи-
сана в виде
. A.15)
Применение оператора е~^^кТ к стоящей за ним функции в этой формуле не-
сколько затруднено тем обстоятельством, что операторы кинетической и потен-
циальной энергий не коммутируют друг с другом. В связи с этим для расчета
функции
A.16)
входящей в интеграл, будет применен другой метод. Можно показать, что эта
функция удовлетворяет дифференциальному уравнению Блоха
р";Т) A.17)
и должна иметь асимптотическое решение при 7, стремящемся к бесконечности
ввидеХ = exp [O'/A)(pN • г^)].При Т <оо решение ищут в следующей форме :
X = ени*№-.г*)е-зг(гх.рх)/кт у(ты,ры;Т). A.18)
Подставляя это выражение в дифференциальное уравнение Блоха, получаем
дифференциальное уравнение для определения У, которое должно быть решено
с учетом граничного условия : Y -> 1 при Т -> с». Решение этого уравнения
х) Это следует из уравнения A.3) и A.20) гл. 2.
2) Суммы Слетера для нескольких простых систем вычислены в задаче 1 в конце этой
главы.
8) Доказательство взято у Кирквуда [1 ].
§ 1. Предварительные сведения из статистической механики 319
ищется в виде ряда1)
*). A.19)
Неизвестные величины у7- определяются уравнениями, получающимися после
подстановки выражения A.19) в дифференциальное уравнение для Y и прирав-
нивания коэффициентов при одинаковых степенях постоянной Планка. После
этого выражение для Y подставляется обратно в формулу A.15) и интегри-
руется по всем импульсам. Во время последней операции члены, содержащие
нечетные степени постоянной Планка, исчезают. Окончательное выражение
для суммы Слетера имеет вид2)
N (гаг) = Wn (г") [{1 + Я* 2 и>Ф + А4 2 *>Ц +
+ Члены более высокого порядка Л } ± / * О()\з\
4
j k
+ {Другие члены, учитывающие перестановки более высокого порядка}] у
где А2 = Щ2пт кТ и
±{4 y
1 j
с-22)
Формула A.20) может быть применена для нахождения квантовых попра-
вок к классической функции Больцмана при условии, что температура системы
настолько велика, что квантовые эффекты малы; при низких температурах
ряд по степеням постоянной Планка расходится. Члены, учитывающие пере-
становки высших порядков, необходимо учитывать при рассмотрении третьего
и более высших вириальных коэффициентов ; на величину второго вириаль-
ного коэффициента они не влияют.
Из приведенного выше рассмотрения ясно, что в предельном случае высо-
ких температур сумма Слетера Ж*и(ты) стремится к множителю Больцмана
WN(rN), а квантовомеханическая статистическая сумма [формула D.1) или
D.2) гл. 2] переходит в классическую статистическую сумму [формула D.3)
гл. 2]. Как указывалось в вводной части к этой главе, поведение системы отли-
чается от классического вследствие волновой природы молекул («диффрак-
ционные эффекты») и статистик молекул («эффекты симметрии»). Вклад этих
эффектов ясно виден в формуле A.20). Члены, стоящие в первом ряду скобок,
возникают исключительно вследствие диффракционных эффектов, остальные
члены появляются вследствие требований симметрии, налагаемых на вол-
новую функцию системы из N тождественных частиц.
^Имеются и другие способы решения этого уравнения. См., например, [2, 3].
2) 27 ? означает суммирование по всем / и к> исключая члены, для которых / = к,
j к
8) Чтобы понять смысл членов в этом уравнении, рассмотрим случай N = 3. Тогда
в формуле A.15) суммирование по всем перестановкам представляется в виде
2 (± 1)Р = Pl2S ± (i°218 + ^132 + ЯШ) + (Р« +/>312).
Члены в первых фигурных скобках формулы A.20) возникают вследствие перестановки,
аналогичной Р128. Следующие три оператора перестановки ответственны за появление вто-
рых фигурных скобок в формуле A.20), учитывающей перестановку двух молекул. Третьи
фигурные скобки, соответствующие двум последним перестановкам, учитывают перестановки
третьего порядка.
320 Гл. 6. Квантовая теория уравнения состояния
§ 2. ВЫВОД УРАВНЕНИЯ СОСТОЯНИЯ ИЗ СТАТИСТИЧЕСКОЙ
МЕХАНИКИ
По содержанию этот параграф тесно примыкает к гл. 3, § 1, где выводится
уравнение состояния для классических систем. В этом параграфе будет пока-
зано, что уравнение состояния1) может быть получено как из квантовомехани-
ческой статистической суммы, так и с помощью квантовомеханической теоремы
вириала, причем результаты оказываются одинаковыми. Вывод вириальной
формы уравнения состояния излагаться детально не будет (как это сделано в
гл. 3, § 2 и 3), поскольку он почти одинаков с классическим. Однако будет
показано, как необходимо изменить классические результаты гл. 3, § 4, чтобы
они удовлетворяли квантовым статистикам.
А. МЕТОД СТАТИСТИЧЕСКОЙ СУММЫ
Как показано в гл. 2, произведение pV связано со статистической суммой
соотношением
(°^). B.1)
Квантовомеханическая статистическая сумма ZNq может быть записана
в виде D.1) гл. 2
а
или в виде D.2) гл. 2
ZNq = Г [2 Ф\ (*N) е-ЛПитф (г*)] dr«,
J Q
где Jf=Jf + Ф — сумма операторов кинетической и потенциальной энергии.
Первое выражение является обычной формой «суммы по состояниям» для
статистической суммы. В этой форме зависимость ZNq от объема содержится
в уровнях энергии ЕаУ которые сложным образом зависят от внешних пара-
метров системы. Второе выражение ZNq является такой формой записи, при
которой зависимость от объема содержится в пределах интегрирования, а
также в граничных условиях, которым должны удовлетворять функции фг{ты).
Используя вторую формулировку для статистической суммы, уравнение со-
стояния можно написать в виде
Б. МЕТОД, ОСНОВАННЫЙ НА КВАНТОВОМЕХАНИЧЕСКОЙ
ТЕОРЕМЕ ВИРИАЛА
Произведение pV может быть также найдено из среднего по ансамблю в
соответствии с формулой C.33) гл. 2. Чтобы применить эту формулу, необхо-
димо знать вид оператора pV. С помощью квантовомеханической теоремы
вириала, изложенной в гл. 1, § б, получается выражение для оператора pV,
аналогичное формуле A.11) гл. 3,
х) Здесь, как и в гл. 3, имеется в виду термическое уравнение состояния[р = p(V, T)].
Калорическое уравнение состояния [U = U(V, T)] будет обсуждено в задаче 2 в конце этой
главы.
§ 2. Вывод уравнения состояния из статистической механики 321
где ЗГ и Et — соответственно оператор кинетической энергии и оператор, со-
ответствующий вириалу межмолекулярных сил системы. Произведение pV
в таком случае может быть записано в виде
B.4)
Если учитывать только парное взаимодействие, то предыдущую формулу
можно записать в виде
Jl$^ B.5)
что в точности соответствует формуле A.13) гл. 3, причем классическая
бинарная функция распределения п'2) заменена квантовомеханической ХB),
определяемой формулой B.24) гл. 2.
В. ЭКВИВАЛЕНТНОСТЬ УРАВНЕНИЙ СОСТОЯНИЯ,
ПОЛУЧЕННЫХ ИЗ СТАТИСТИЧЕСКОЙ СУММЫ
И С ПОМОЩЬЮ ТЕОРЕМЫ ВИРИАЛА1)
Если в формулу B.2) подставить собственные функции импульса [формула
A.5)], то выражение для произведения pV примет вид
PV= NkfZs •^^ul\2{± l)Pe-2niW'p^e-^lkTe+2n«k«'r«Hk". dr" . B.6)
В этой формуле вместо импульсов р, стоят волновые числа k; = py/ft и положено
V = L3 [так что V(d/dV) =1/зД9/9^)]- Чтобы преобразовать дифференциро-
вание2), введем новые переменные f, = r;/L и k; = k;/L. Тогда последнее выра-
жение принимает вид3)
х [~ Т Гаг) e~'r'kT] е+2л1Fлг •rlr)
Оператор дифференцирования внесен внутрь интеграла, так как от L зависит
лишь оператор Гамильтона
Легко показать, что производная по L равна
4^яЬ B-9)
Подставляя этот результат в формулу B.7) и возвращаясь к первоначальным
переменным, получаем в точности формулу B.4), выведенную из теоремы
вириала.
Г. ОКОНЧАТЕЛЬНОЕ ВЫРАЖЕНИЕ КВАНТОВОМЕХАНИЧЕСКОГО
УРАВНЕНИЯ СОСТОЯНИЯ
Метод статистической суммы может быть применен для получения урав-
нения состояния в вириальной форме таким же образом, как это сделано в
гл. 3, § 2. Единственное отличие заключается в том, что ZN и WN(rN) всюду
х) Приводимое здесь доказательство взято из работы де Бура [4 ]. Аналогичное доказа-
тельство, основанное на представлении статистической суммы в форме «суммы по состояниям»,
дал Айвон [5].
2) Этот метод был использован Грином 16 ] в аналогичном классическом доказательстве
(см. гл.3,§ 1.)
3) Здесь использован тот факт, что, хотя операторы dJ^/dL и ехр(— JP/kT) в общем случае
не кохммутируют, порядок их безразличен при вычислении следа (см. [4]).
322 Гл. 6. Квантовая теория уравнения состояния
должны быть заменены на ZNq и Ж*ц (гы). Конечные выражения для второго
и третьего вириальных коэффициентов, приведенные в формулах D.5) и D.6)
гл. 3, справедливы также и в квантовой механике при условии, что в них про-
изведена такая же замена (в квантовой механике, однако, не применимы фор-
мулы, в которые входят /,у). Так же как и в классическом случае (см. гл. 3, § 3),
можно развить метод, основанный на теореме вириала. Степенное разложение
для пB), данное формулой C.8) гл. 3, применимо и для jfB). Единственное из-
менение заключается в том, что во всех формулах множитель Больцмана дол-
жен быть заменен суммой Слетера [7]1).
§ 3. СВОЙСТВА ИДЕАЛЬНОГО ГАЗА
Рассмотрим специальный случай идеального газа, в котором энергия
взаимодействия молекул равна нулю2) [Ф(гы) =0]. Неразличимость молекул,
как уже говорилось в гл. 1, § б, налагает определенные ограничения симметрии
на форму волновой функции системы. В системах, состоящих из молекул с
нечетным числом нуклонов и электронов, волновая функция должна быть
антисимметричной относительно перестановки любых двух молекул. Вол-
новые функции, описывающие системы, состоящие из молекул с четным числом
нуклонов и электронов, должны быть симметричными [8]. Большинство мо-
лекул относится к последней категории, известными исключениями являются
Не3 и HD.
При вычислении суммы Слетера и уравнения состояния идеального газа
необходимо заметить, что, хотя между молекулами идеального газа не действует
никаких сил, газ Ферми—Дирака будет отличаться от классического идеаль-
ного газа в известном смысле так, как если бы он состоял из молекул, отталки-
вающихся друг от друга. Противоположными свойствами обладает газ Бозе—
Эйнштейна. В связи с этим удобно говорить о кажущемся отталкивании
между молекулами газа, имеющего антисимметричные волновые функции (т. е.
удовлетворяющие принципу Паули), и кажущемся притяжении между моле-
кулами газа, имеющего симметричные волновые функции. Последний случай
тесно связан с явлением конденсации Бозе—Эйнштейна.
А. СУММА СЛЕТЕРА ИДЕАЛЬНОГО ГАЗА, СОСТОЯЩЕГО ИЗ МОЛЕКУЛ
СО СПИНОМ, РАВНЫМ НУЛЮ
Оператор Гамильтона идеального газа совпадает с оператором кинети-
ческой энергии i3T = — Щ2т JV [C/8f;) • (Э/Эг,)]. Поскольку экспоненциальные
функции в формуле A.15) являются собственным функциями оператора им-
пульса, то оператор <Ж = ЭГ можно заменить его собственным значением
2 (р*12т). После интегрирования сумма Слетера принимает вид3)
Г) = ? (± \)р e-(n!»)Z(rk-rPky C.1)
р
(верхний знак соответствует статистике Бозе—Эйнштейна, нижний — Ферми—
Дирака). Здесь rPk означает положение частицы, получающееся после приме-
нения перестановки Р к fc-й частице. При тождественных перестановках аргу-
мент экспоненты равен нулю. Следовательно, в отсутствие эффектов симметрии
х) См. задачу 3 в конце главы.
2) Величины, относящиеся к такой системе, будут обозначаться индексом 0.
8) Интегрирование сводится к вычислению интеграла
eibx е-а* X* fa — г71 е-
а
§ 3. Свойства идеального газа 323
() = 1,т.е. в точности равно величине классического множителя Больц-
мана при Ф(гм) = 0.
В случае системы двух частиц формула C.1) принимает вид
ЗГ™ (т19 г2) = 1 ± г-2«тУ» C.2)
(верхний знак соответствует статистике Бозе—Эйнштейна, нижний — Ферми—
Дирака).
Знак плюс соответствует волновым функциям, симметричным относительно
перестановки двух частиц, а знак минус — антисимметричным волновым функ-
циям. Для симметричных волновых функций вероятность нахождения двух
молекул на расстоянии г12 становится больше единицы1) при уменьшении г12 и
достигает двух, когда г12 приближается к нулю. Это пример «кажущегося при-
тяжения» между молекулами в системе Бозе—Эйнштейна. Для антисимметрич-
ных волновых функций вероятность нахождения двух молекул на расстоянии
г12 уменьшается с уменьшением расстояния и приближается к нулю, когда
г12->0. Таким образом, вероятность нахождения двух молекул Ферми—Ди-
рака в одной и той же точке пространства равна нулю, хотя в случае идеаль-
ного газа молекулы не обладают протяженностью в пространстве (т. е. нет
межмолекулярных сил). Это пример «кажущегося отталкивания» между моле-
кулами газа, имеющего антисимметричные волновые функции (т. е. удовлет-
воряющие принципу Паули).
В обоих случаях расстояние, на котором сказывается влияние молекул
друг на друга в соответствии с требованиями симметрии, будет порядка
А = hl\2nmkT. Это расстояние с точностью до числового множителя порядка
единицы равно дебройлевской длине волны молекул, имеющих температуру Т.
Если среднее расстояние между молекулами порядка дебройлевской длины
волны, то «эффекты симметрии» начинают играть важную роль и о системе
говорят как о «вырожденной» или находящейся «в состоянии вырождения».
Б. СУММА СЛЕТЕРА ИДЕАЛЬНОГО ГАЗА, СОСТОЯЩЕГО ИЗ МОЛЕКУЛ
СО СПИНОМ, НЕ РАВНЫМ НУЛЮ
До сих пор атомы рассматривались как материальные точки. Часто, од-
нако, оказывается, что атомы обладают собственным постоянным моментом
количества движения. Этот «ядерный спин» влияет на статистические веса и
свойства симметрии различных состояний.
Для простоты рассмотрим систему двух тождественных частиц, каждая
из которых обладает собственным моментом количества движения sh. Такой
спин может быть ориентирован Bs + 1) разными способами по отношению к
фиксированной оси, и все эти ориентации обладают одинаковой энергией.
В статистике Болыдмана спиновые состояния независимы, поэтому наличие спина
приведет лишь к тому, что каждое состояние системы из двух частиц будет
обладать Bs + IJ степенью вырождения и, следовательно, статистическая
сумма должна быть просто умножена на этот множитель. Практически, однако,
удобнее рассматривать статистическую сумму (и сумму Слетера), деленную
на этот множитель. Поэтому этот множитель не появляется ни в одной из фор-
мул. Наличие спина не влияет на термодинамические свойства системы. Однако
в случае статистик Бозе—Эйнштейна и Ферми—Дирака влиянием спина нельзя
пренебрегать.
Суммы Слетера для молекул со спином Ж^^\т1У г2) можно выразить через
суммы Слетера для молекул без спина 2t^<°\rv r2) следующим образом. Рассмот-
рим снова систему, состоящую из двух частиц, но теперь уже подчиняющихся
статистике Бозе—Эйнштейна. Общая волновая функция системы должна
быть симметрична относительно перестановки двух частиц. Если частицы не
г) Речь идет о ненормированных вероятностях. — Прим. ред.
324 Гл. 6. Квантовая теория уравнения состояния
обладают собственным спином, то пространственные волновые функции долж-
ны быть симметричны. Однако если частицы обладают спином, то симметрич-
ными должны быть уже произведения пространственных и спиновых волно-
вых функций. Из Bs + IJ различных спиновых функций можно образовать
E + 1) B5 + 1) симметричных и sBs + 1) антисимметричных комбинаций.
Чтобы получить симметричную общую волновую функцию, последние спиновые
функции необходимо умножить на антисимметричные пространственные вол-
новые функции, а первые — на симметричные. Следовательно, в выражение
для суммы Слетера состояния с симметричными пространственными функциями
входят с весом (s + l)/Bs + 1), а состояния с антисимметричными простран-
ственными функциями — с весом sjBs + 1). Таким образом, для частиц Бозе—
Эйнштейна сумма Слетера принимает вид
«"I, Га) = ("ИЛ) Г®э. (г», г2) +
C.3)
где, верхний индекс у Ж^(г1У г2) означает величину спинового квантового числа 5.
Аналогично для частиц Ферми—Дирака получаем
)<в%.(г1)г2). C.4)
Заметим, что если спиновое число очень велико, то Ж^дХ1^ т2) и
Р>э (rlf г2) смешаны в почти одинаковых пропорциях, и сумма Слетера в
этом случае очень близка к соответствующей сумме в статистике Больцмана.
Качественно можно сказать, что наличие спина, отличного от нуля, в этом
случае как бы компенсирует действие принципа Паули.
В. УРАВНЕНИЕ СОСТОЯНИЯ ИДЕАЛЬНОГО ГАЗА1)
Групповые интегралы для идеального газа можно получить из формулы
B.10) гл. 3; подставляя вместо У(о) выражение C.1). В результате получаем
&? = (± 1)'-Ч-6/.ЛЗ('-о. C.5)
Из этих групповых интегралов находятся давление и плотность в виде ряда
по стейеням «активной плотности числа частиц» z, определенной в гл. 3, § 2
-Ч-1/.[Я82]/> C.6)
-Ч-*1*[ЛЧ]1. C.7)
В этих уравнениях верхний знак соответствует симметричным собственным
функциям (статистика Бозе—Эйнштейна), а нижний знак — антисимметрич-
ным собственным функциям (статистика Ферми—Дирака).
Особый интерес представляет второй вириальный коэффициент
-^ = TN2-v.L|— f C.8)
(верхний знак соответствует статистике Бозе—Эйнштейна, нижний — Ферми—
Дирака).
Видно, что второй вириальный коэффициент пропорционален (тТ)"9!;
поэтому он оказывается существенным лишь при низких температурах для
частиц малых масс. В статистике Бозе—Эйнштейна В°(Т) отрицателен, а в
статистике Ферми—Дирака — положителен. Это согласуется с представ-
лением о кажущемся притяжении и отталкивании.
*)См. [9, 10].
§ 3. Свойства идеального газа
325
Г. КОНДЕНСАЦИЯ БОЗЕ—ЭЙНШТЕЙНА1)
Исследуем формулы C.6) и C.7) применительно к статистике Бозе—Эйн-
штейна и изучим их поведение при возрастании плотности газа. При достаточно
низких плотностях произведение 1ъг меньше единицы, поэтому вопроса о схо-
димости этих двух рядов не возникает. При возрастании плотности произ-
ведение Л32 становится равным единице и эти уравнения принимают вид
-/. = 1,34 А-»,
C.9)
(ЗЛО)
При дальнейшем возрастании плотности произведение А3г становится
больше единицы и ряд расходится. Согласно представлению Эйнштейна, кото-
рый первый изучал это явление, п°с есть максимально достижимая плотность
при данной температуре Т. При дальнейшем сжатии газа молекулы «конден-
сируются» в состояние с нулевой энергией и не вносят вклада ни в давление,
ни в плотность системы, оставляя давление на уровне р°. Таким образом, на-
блюдается явление, похожее на конденсацию : при всех температурах суще-
ствует плотность п?, при которой начинает появляться «конденсация» и свя-
занное с ней давление насыщения p°s. Из формул C.9) и C.10) видно, что не
существует такой температуры, выше которой не наблюдалось бы явление
конденсации. Это значит, что не существует критической точки в том смысле,
как это имеет место при конденсации реальных газов.
При конденсации реального газа производная (dpjdV)T претерпевает раз-
рыв в точке перехода. Из формулы C.6) и C.7) видно, что в случае идеального
газа Бозе—Эйнштейна эта производная равна
[27/-V.(A»z)']
(ЗЛ1)
и она непрерывна в точке конденсации. При A3z^> 1 производная равна нулю,
так как ряд Z /-к расходится. На основании этих результатов можно начер-
тить изотермы для идеального газа Бозе—Эйнштейна, которые и показаны на
фиг. 100. Из диаграммы зависимости между р, V и Т видно, что конденсиро-
ванная фаза будет существовать при нулевом объеме.
ю
м
'«2
1 1 1 1
|\
\ \
\ \
\ \
\ \
- \
\
-
1 1 1 1
V
ч -~~——
1 1 1
1 1 1 1 1 1
-
—
-
-
" з°
2°
1 1 1 1 1 1
50
V, СМ *
100
150
Фиг. 100. Изотермы Не4 для идеального газа Бозе—Эйнштейна [10].
г) См. [9—13]. Работа [12] дает хороший обзор этого вопроса; кроме того, в ней приво-
дится довольно полный список работ, посвященных теории и применениям конденсации
Бозе—Эйнштейна.
326 Гл. 6. Квантовая теория уравнения состояния
Исследуем конденсированную систему с термодинамической точки зрения.
Напишем сначала выражения для термодинамических параметров при зна-
чении Л32 = 1:
до =
G° =/?Т In (zA*)-> 0,
eg = ^
(^
Разность между значениями термодинамических параметров в газообразной и
<окидкой» фазах можно^ получить из последней группы формул, положив соот-
ветственно V = Ve и V = 0 ; отсюда видно прежде всего, что AG0 = 0.
В соответствии с термодинамикой это условие означает, что жидкая и
газообразная фазы находятся в равновесии. Из формул C.9) следует
dp°s _ 5 p°s _ AS» m (~ Пч
"SfTT" Ay°'J K }
первая часть этого выражения совпадает с разностью энтропии и объема между
жидкой и газообразной фазами, т. е. удовлетворяется уравнение Клапейрона.
Таким образом, термодинамические условия, которым должно удовлет-
ворять явление конденсации, выполняются. Но мы видели, что
1) объем конденсированной фазы равен нулю;
2) критической температуры не существует;
3) (9р/ЗК)г непрерывна при V = Vc;
4) Cv непрерывна при V = Vc.
Б.Э
Фиг. 101. Удельная теплоемкость при постоянном объеме как функция температуры.
Пунктирная кривая — классический предел для одноатомных газов [14].
В этих пунктах изученное явление отличается от конденсации реальных
газов. Газ Ферми—Дирака не конденсируется. На фиг. 101 сравниваются
кривые зависимости С„ от Т для идеальных газов Ферми—Дирака й Бозе—
Эйнштейна.
§4. ВТОРОЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ РЕАЛЬНЫХ ГАЗОВ
ПРИ ОЧЕНЬ НИЗКИХ ТЕМПЕРАТУРАХ1)
Второй вириальный коэффициент в квантовой механике зависит от стати-
стической суммы двух взаимодействующих молекул. Энергетические уровни,
используемые при вычислении статистической суммы, получаются из решения
х) Для понимания этого параграфа необходимо знание квантовой механики системы,
состоящей из двух тел, а также квантовой теории парных столкновений. Необходимые
сведения об этом были даны в гл. 1, § 7.
§ 4. Второй вириалъный коэффициент при очень низких температурах 327
волнового уравнения Шредингера для двух частиц. Существуют два типа
решений : решения, соответствующие паре молекул, пойманных й потенциаль-
ной яме, так называемые «двойные молекулы», и решения, соответствующие
двум сталкивающимся молекулам. Эти два типа решений обсуждены в
гл. 1, § 7, и изображены на фиг. 12, стр. 69.
В этом параграфе будет показано, как можно выразить статистическую
сумму (а следовательно, и второй вириальный коэффициент) через стационар-
ные энергетические уровни (соответствующие двойным молекулам) и «фазовые
сдвиги» в радиальном волновом уравнении (соответствующем столкновениям).
Коротко будут изложены расчеты фазовых сдвигов для некоторых простых
типов потенциала, а также для потенциала Леннарда-Джонса. В конце пара-
графа будут приведены результаты расчетов В(Т) для изотопов гелия и водо-
рода при низких температурах.
А. ВЫРАЖЕНИЕ ВТОРОГО ВИРИАЛЬНОГО КОЭФФИЦИЕНТА
ЧЕРЕЗ ФАЗОВЫЕ СДВИГИ И ЭНЕРГИЮ СТАЦИОНАРНЫХ СОСТОЯНИЙ
Второй вириальный коэффициент в классической статистической механике
выражается с помощью классической статистической суммы по формуле D.5)
гл. 3. Как уже указывалось, эта формула справедлива также и в квантовой
теории уравнения состояния при условии, что классические статистические
суммы Zx и Z2 заменены квантовомеханическими статистическими суммами
Zlq и Z29. В соответствии с этим для неидеального и идеального (Ф(гм) = 0)
газов можно написать следующие выражения :
Неидеальный газ
Идеальный газ
В°(Т) = --?yW [2Z§? - (*?,)¦], D2)
где А2 = Щ2птпкТ; Zlq и Z\q — статистические суммы для одной молекулы;
Z2q — статистическая сумма для двух взаимодействующих молекул ; Z% —
статистическая сумма для двух невзаимодействующих молекул. Разность
между выражениями D.1) и D.2) равна
В(Т) - В°(Т) = - -f
= - 4- W [2е-Е°1кТ -2е-Е>т]} D.3)
у
где Еа — собственные значения энергии системы, состоящей из двух молекул
взаимодействующих по закону <р(г); Е°а — собственные значения системы двух
невзаимодействующих молекул. Каждое собственное значение энергии харак-
теризуется набором шести квантовых чисел {а}.
Как показано в гл. 1, § 7, волновая функция системы двух частиц может
быть записана в виде произведения волновой функции поступательного дви-
жения, изображающей перемещение системы как целое, и волновой функции
относительного движения
Ф(Г1у г2) = у(пост.) (Гс) ^(отн.) (fia) = ^(пост.) (Гс) ^/(ri2) ym ф) я
Здесь гс = 112(т1 + г2) — координата центра масс; г12 = гх — г2 — координата
относительного движения. Волновая функция относительного движения мо-
жет быть разложена на радиальную волновую функцию грп1 и угловую часть
У[", представляющую собой шаровые функции. Произведению волновых функ-
ций соответствует сумма собственных значений энергий
Е = ?(пост.) _|_ ?<отн.)#
328 Гл. 6. Квантовая теория уравнения состояния
Следовательно, статистическая сумма в выражении D.3) может быть записана
в виде
Z2q = 22<пост-> 2 е-Етт1кт = у/, у Я-8 2 B / + 1) 2 е~Е^кТу D.6)
n,l,m I n
Z% = Z<n°CT-> 2 e-E^kT = 2s/° V A-» 2 B1 + 1) 2 <rES''fcT. D.7)
n,l,m I n
Здесь учтено значение статистической суммы поступательного движения, при-
веденное в формуле E.28) гл. 2. Множитель 28/* появлчется вследствие того,
что суммарная масса системы равна 2т. Энергетические уровни относительного
движения не зависят от квантового числа т, но существование этих уровней
необходимо принимать во внимание при вычислении статистической суммы,
поскольку каждый из этих уровней обладает мультиплетностью B1 + 1), что
и сделано в двух последних формулах. Таким образом, состояние системы
двух молекул задается двумя квантовыми числами ли/, которые определяют
энергию и момент количества движения системы.
Подставляя выражения для статистических сумм в формулу для второго
вириального коэффициента, получаем
[В(Т) - В°(Т)] = - N 28/« Яз 2 B1 + 1) [2 е~Е«*1кТ - 2 е~ЕЫкТ]. D.8)
/ п п
Суммирование по всем п может быть разбито на две части — суммирование
по отрицательным энергетическим состояниям и суммирование по состояниям
с положительным значением энергии. Отрицательный энергетический спектр
является дискретным и соответствует «связанному состоянию молекул» с дис-
кретными уровнями. В этом случае, конечно, нереагирующие молекулы не вно-
сят соответствующего вклада, поэтому вторую сумму по л в формуле D.8)
можно не рассматривать. Сумма по л в этих условиях принимает вид
2e-EnilkT. D.9)
п
Величины отрицательных дискретных уровней энергии E~t получаются из
решения радиального волнового уравнения для уп/(г12). Положительный энер-
гетический спектр непрерывен и появляется вследствие кратковременных
столкновений между двумя молекулами. Энергия относительного движения
равна относительной кинетической энергии частицы приведенной массы р :
Е = р2/2/л = h2x2j2[x. Суммирование по л в этом случае должно быть заменено
интегрированием по и, поэтому члены, стоящие в правой части формулы
D.8) в квадратных скобках, принимают вид
Г*йч- Dло)
При введении фазовых сдвигов rj^x), которые определяются из решения ра-
диального волнового уравнения для положительных значений энергии, было
использовано уравнение G.14) гл. 1.
Окончательное выражение второго вириального коэффициента в кванто-
вой механике получается в результате комбинирования последних трех формул
- N2+V.Яз2B1+ 1) 2 e-^ix7 -
I п
^ N2^P 2 B1 + l)je-ht^l^kT[^-^^]d>c. D.11)
Здесь использовано явное выражение В°(Т), приведенное в формуле C.8).
Знак минус соответствует статистике Бозе—Эйнштейна, а знак плюс — ста-
тистике Ферми—Дирака. При суммировании по / необходимо учитывать только
§4. Второй вириальный коэффициент при очень низких температурах 329
четные значения I в случае статистики Бозе—Эйнштейна и нечетные в случае
статистики Ферми—Дирака1).
Из формулы D.11) видно, что выражение для В(Т) состоит из трех частей,
каждая из которых имеет определенный физический смысл. Первый член по-
казывает, как «кажущееся притяжение» (или отталкивание) уменьшает (или
увеличивает) давление в газе. Второй член показывает, как уменьшается дав-
ление при образовании связанных молекул. Интеграл по х учитывает откло-
нение газа от идеальности, возникающее вследствие учета парных столкно-
вений в газе.
Приведенные результаты справедливы для молекул с нулевым спином.
Для молекул со спином, равным s, можно написать [аналогично формулам
C.3) и C.4).]
[Й] Ы] DЛ2)
D.13)
причем В(о) соответствует молекулам с нулевым спином и вычисляется по фор-
муле D.11).
В разд. В изложен расчет второго вириального коэффициента для Не3.
Поскольку этот атом имеет нечетное число нуклонов и спин, равный х/2,
то для расчетов необходимо применять формулу D.13).
Б. ВЫЧИСЛЕНИЕ ФАЗОВЫХ СДВИГОВ ДЛЯ НЕКОТОРЫХ
ПРОСТЫХ ПОТЕНЦИАЛОВ
Фазовые сдвиги, которые необходимо знать для вычисления второго вири-
ального коэффициента, формально определены формулой G.10) гл. 1. Чтобы
вычислить фазовые сдвиги, необходимо задать межмолекулярную потенциаль-
ную функцию в радиальном волновом уравнении. В общем виде невозможно
получить аналитическое выражение для фазовых сдвигов как функции меж-
молекулярного потенциала. Однако можно получить решение в замкнутой
форме в случае модели твердых сфер (для всех I) и прямоугольной потенциаль-
ной ямы (для I = 0). На основе результатов, полученных с помощью простых
моделей потенциалов, можно попытаться понять природу фазовых сдвигов.
1. Модель твердых сфер. Поскольку для твердых сфер диаметра а потен-
циал равен нулю при г > <т, то радиальную волновую функцию в этой области
можно принять равной выражению G.8) гл. 1. Отношение [Bl{x)jAl (x)]
в этом случае определяется из граничного условия о\р{о) = 0. В соответ-
ствии с формулой G.10) гл. 1 фазовые сдвиги получаются в виде
nix) = arctg [(- 1)'+' J0^) ]; D.14)
при I = О имеем щ(х) = — ха. Фазовые сдвиги для твердых сфер графи-
чески изображены на фиг. 102. То обстоятельство, что фазовые сдвиги в случае
модели твердых сфер являются величинами отрицательными, легко можно
понять, рассмотрев их геометрический смысл. Поскольку drjjjdx также
всюду отрицательно, то плотность энергетических состояний в статистической
сумме двух взаимодействующих молекул будет везде меньше аналогичной
величины для невзаимодействующих молекул. Это приводит к тому, что вклад
г) Причина этого заключается в следующем: при суммировании по состояниям относи-
тельного движения в формуле D.8) учитываются только такие состояния, волновые функции
которых симметричны или антисимметричны относительно перестановки двух частиц. Пере-
становка частиц означает замену 0 на 0 + п и в на (п — 0). Угловая часть волновой функции
Yf1 не меняется при такой замене в случае четных / и меняет знак на противоположный в
случае нечетных /. Следовательно, для частиц Бозе—Эйнштейна необходимо суммировать
по четным состояниям 1У а для частиц Ферми—Дирака — по нечетным.
330
Гл. 6. Квантовая теория уравнения состояния
от взаимодействия во второй вириальный коэффициент, как видно из формул
D.10) и D.11), положительный. Подставляя формулу D.14) в D.11), получаем
для приведенных вириальных коэффициентов В* = В/2/3я:Мг3 соответственно
Фиг. 102. Фазовые сдвиги для модели твердых сфер [15].
для статистик Бозе—Эйнштейна и Ферми—Дирака [15] следующие выра-
жения :
D.16)
Из этих выражений видно, что при дебройлевской длине волны (приблизительно
А = ft/ Y2nmkf), большей диаметра молекул, основной вклад происходит от
эффектов статистики.
2. Прямоугольная потенциальная яма. Рассмотрим прямоугольную по-
тенциальную яму, определенную формулой C.24) гл. 1, при jR = 3/2. Для этого
потенциала найдем решение радиального волнового уравнения [формула
G.3) гл. 1 ], описывающего относительное движение двух частиц с относительной
энергией ? .и 1 = 0. Поскольку при г = о волновая функция обращается в
нуль, решение можно записать в виде
DЛ7)
D.18)
где
D.19)
е — глубина прямоугольной потенциальной ямы. Требование непрерывности
гу(г) и d(r\p{r))jdr в точке г = 3/2<т непосредственно приводит к
ггр(г) = A sin к'(г — о),
гу)(г) = В sin (кг + %), г > -~ау
А* к2 = 2[лЕ} А2 к'* = 2[л(Е + е);
%(«) = arctg [^ tg •=?] - -f ~.
Зависимости фазовых сдвигов от ко изображены графически на фиг. 103 при
различных значениях глубины потенциальной ямы.
Из фиг. 103 можно определить, как меняются фазы при возрастании глу-
бины ямы. Если глубина ямы е меньше У^то2, то кривые фазовых сдвигов
берут свое начало в точке нуля при на = 0. Если глубина ямы лежит между
ЦьЩто* и 9/4Л2/шст2, то кривые фазовых сдвигов принимают значение, равное п
при ко = 0. В общем случае величина фазовых сдвигов скачкообразно меня-
ется на п при ко = 0, если [tgK'<x/2 ]Е=0 = с» или е = (л —1l2Jh2jma2.
§4. Второй вириальный коэффициент при очень низких температурах 331
Можно показать, что при этих значениях е на краю прямоугольной ямы
(Е = 0) как раз существует дискретный уровень. Следовательно, по поведению
фаз при ко = О можно судить о том, существует ли один или несколько дискрет-
ных уровней для сталкивающейся пары молекул.
Анализируя фиг. 103, можно получить и другой интересный вывод. Видно,
что с ростом глубины потенциальной ямы фазы становятся все более поло-
жительными при столкновении молекул с небольшой относительной энергией.
Фиг. 103. Фазовые сдвиги для прямоугольной потенциальной ямы (R = 8/2) для / = О
при различных значениях глубины е (в единицах h2/mo2).
е = О соответствует модели твердых сфер [15].
Это значит, что при подобных предположениях поведение сталкивающихся
молекул сильно отличается от случая модели твердых сфер, поскольку взаи-
модействующая система чувствует наличие ямы на потенциале. При столкно-
вениях с большой относительной энергией система оказывается не особенно
чувствительной к деталям потенциала и фазовые сдвиги приближаются, как и
следовало ожидать, к соответствующим сдвигам в случае модели твердых
сфер.
В. ВЫЧИСЛЕНИЯ ДЛЯ ПОТЕНЦИАЛА ЛЕН НАР ДА-ДЖОНСА
Для других потенциальных функций, ближе стоящих к действительности,
чем рассмотренные выше модели, фазовые сдвиги могут быть определены числен-
ным интегрированием радиального волнового уравнения [формула G.3) гл. 1 ]
с граничным условием г \р{г) = О при г = 0. Интегрирование можно провести
также в области, где потенциальная энергия равна нулю. В этой области вол-
новые функции известны [см. формулу G.8) гл. 1 ]. Сшивая волновые функции,
полученные численным интегрированием, с волновыми функциями, данными
формулой G.8) гл. 1, можно найти коэффициенты At{x) и Bt(x). С помощью
этих коэффициентов можно определить величины фазовых сдвигов по формуле
G.10) гл. 1, причем для каждого значения I эту операцию необходимо прово-
дить при многих значениях к.
Для потенциала F-12) Леннарда-Джонса радиальное волновое уравнение
[формула G.3) гл. 1) может быть записано в приведенной форме :
- М (г*У) = 0, D.21)
где г* = г/<т, х* = ко и Л* =hl<s)ft№.
332
Гл. 6. Квантовая теория уравнения состояния
Это уравнение описывает столкновения между молекулами любых газов.
Величина Л* меняется, конечно, в зависимости от природы газа, что согла-
суется с законом соответственных состояний, изложенным в § 6. Метод опре-
деления параметра Л* дается в § 5, а табулированные значения Л* для раз-
личных веществ приведены в табл. 57 (стр. 337).
Многочисленные вычисления, фазовых сдвигов проделаны для Не3 в случае
потенциала Леннарда-Джонса [16], а также для Не4 в случае нескольких
потенциальных полей [17—19]1). По-
скольку Не4 состоит из четного числа нук-
лонов и электронов и его спин равен нулю,
то необходимо вычислять лишь фазовые
сдвиги для четных значений I. Для Не3,
состоящего из нечетного числа нукло-
нов и электронов и имеющего спин,
равный г/2, необходимо знать фазовые
сдвиги как для четных, так и для не-
четных значений Z. Фазовые сдвиги для
изотопов гелия приведены на фиг. 104, а
полный перечень вычисленных резуль-
татов дан в табл. VI и VII (стр. 857 и 860).
Эти результаты можно теперь рас-
смотреть с точки зрения результатов,
полученных для модели твердых сфер
и прямоугольной потенциальной ямы.
Как для Не3, так и для Не4 фазовые сдви-
ги напоминают фазовые сдвиги в случае
модели твердых сфер при больших зна-
чениях энергии. Однако они не прибли-
жаются асимптотически к фазовым сдви-
гам для твердых сфер, поскольку при
больших относительных энергиях час-
тицы в какой-то степени проникают
внутрь потенциала Леннарда-Джонса.
Положительные фазовые сдвиги появ-
ляются в результате действия притяги-
вающей части потенциала. Из поведения
фазовых сдвигов для Не3 при ко, стре-
ГелГя SS [ТГн^Щ, ™°е„°Г «хся к нулю, видно, что для двух
для потенциала F-12) Леннарда-Джонса. взаимодействующих атомов Не3 стацио-
а — фазовые сдвиги для Не*; б — фазовые НарНЫХ СОСТОЯНИЙ Не Существует. С Дру-
сдвиги для не». гой СТОрОНЫ> для Не4 трудно решить,
существует или не существует дискретный
уровень вблизи Е = 0. По всей видимости, при некотором определенном выборе
потенциала дискретного уровня не существует, однако небольшое изменение
формы этого потенциала сможет привести к появлению такого уровня2).
С помощью этих фазовых сдвигов были вычислены вторые вириальные
коэффициенты изотопов гелия при очень низких температурах. Лишь при тем-
пературах ниже 7* = 1 сумма по I в формуле D.11) сходится достаточно
быстро, чтобы сделать ряд пригодным для численного расчета. Для расчета
Не4, являющегося газом Бозе—Эйнштейна со спином, равным нулю, применя-
ется формула D.11). Для Не3, являющегося газом Ферми—Дирака со спином,
равным 112, применяется формула D.13). Полученные с потенциалом Леннарда-
г) В работе [17] расчеты основаны на потенциале F-12) Леннарда-Джонса, в работе
[18] — на потенциале Букингема, а в работе [19] — на потенциале Букингема—Корнера.
2) Существование дискретных уровней для потенциала F-12) Леннарда-Джонса обсуж-
дается в работе [20].
§ 4. Второй вириальный коэффициент при очень низких температурах 333
Джонса результаты для обоих газов приведены в табл. 54. Те же результаты
изображены графически на фиг. 105, там же нанесены соответствующие экспе-
риментальные данные для Не4.
Таблица 54
Значения второго вириального коэффициента для изотопов
гелия при очень низкой температуре [15]
Потенциал F-12) Леннарда-Джонса
т, °к
0,51
1,02
2,04
3,07
В{Т)
Не8
-293,0
-199,3
-113,5
- 76,52
Не*
-1066
-389
-164
-101
т, °к
4,09
5,11
6,13
7,15
В(Т)
Не8
—56,70
-44,24
-35,67
-29,35
Не*
-71,6
-54,1
-42,4
-34,2
На фиг. 106 представлены результаты,
полученные различными авторами для Не4.
Анализируя эти результаты, можно сделать
некоторые выводы о чувствительности В(Т)
при низких температурах к форме межмо-
лекулярной потенциальной функции.
- Чтобы можно было сравнить резуль-
таты, полученные с помощью различных
потенциальных кривых, к каждой кривой
подбирается потенциал F-12) Леннарда-
Джонса так, чтобы эти потенциальные
кривые возможно теснее примыкали друг
к другу. Значения постоянных а и е, соот-
ветствующие этим потенциальным кривым,
приведены в табл. 55 [24 ], где сравниваются
характеристики этих потенциальных функ-
ций. На основании приведенных значений
силовых постоянных можно вычислить Л*.
В табл. 55 приводятся также данные об
энергии стационарных состояний, найден-
ных с помощью различных потенциальных
функций, и их вклад в вириальные коэф-
фициенты при 1 и 7° К- Из анализа при-
веденных данных видно, что для получения
хорошего согласия вычисленных с помощью
потенциала Леннарда-Джонса результатов с экспериментальными данными
в этой температурной области необходимо уменьшить Л* для Не4 на ~5%.
Таблица 55
Значения второго вириального коэффициента при низких температурах,
вычисленные с помощью различных потенциальных функций [24]
Фиг. 105. Квантовомеханические зна-
чения второго вириального коэффи-
циента для Не4 и Не3 при низких
температурах.
Показаны вклады Бозе—Эйнштейна и Фер-
ми—Дирака во второй вириальный коэффи-
циент для Не8. / — данные Кистемейкера и
Кеезома [21J; 2 — данные Кеезома и Крааке
122 ; 3 — данные Кеезома и Вальстра [231;
[16].
Расчетные кривые взяты
и
из
работы
Кривые на
фиг. 106
i
5
А
ВМ
В
С
Энергия
дискретного уровня
E~je
-0,0058
-0,0004
0
—
—
t\k, °К
11,84
10,64
10,24
8,87
7,10
а, А
2,62
2,62
2,56
2,62
2,62
л*
2,42
2,56
2,67
2,80
3,14
Вклад от
дискретного
уровня в
В(Т), %
при
1°К
12
1
—
—
—
при
7 °К
1,3
0,1
—
—
334
Гл. 6. Квантовая теория уравнения состояния
Расчеты второго вириального коэффициента при низких температурах для
водорода и дейтерия выполнил Мияко [25]. Его расчеты основаны на ранее
изложенной теории одноатомного газа и, следовательно, не учитывают наличия
квантованных вращательных состояний. Однако Мияко принимал во внимание
существование в водороде орто- и лара-состояний. Это значит, что газ рас-
сматривался как бинарная смесь двух типов молекул водорода и, следовательно,
требования симметрии применялись лишь к одной части общего числа столкно-
вений [26]. Найденные величины не очень хорошо согласуются с эксперимен-
тальными данными, очевидно, потому, что не учитывались вращательные кван-
товые эффекты. Краткое изложение этих вращательных квантовых эффектов
дано в § 7.
Кривая S
Фиг. 106. Второй вириальный коэффициент для Не4 при низких температурах по данным
различных авторов [24].
/ — данные Кеезома и Крааке [22]; 2 — данные Кеезома и Вальстра [23]; 3 — данные Кистемейкера и
Кеезома [21].
Кривая ВМ (по расчетам де Бура и Михельса[17]). Потенциал F-12) Леннарда-Джонса с параметрами вид,
определяемыми из условия совпадения с экспериментальными значениями второго вириаль-
ного коэффициента в области высоких температур, где квантовые поправки малы,
(по расчетам Месси и Букингема [18]). Потенциальная функция в 1,3 больше вычисленной
Слетером берется в виде
<р(г) - Ь ехр (— аг) -в
где Ь = 10-» эрг, с = 1,91 • Ю-11 эрг А', а - 4,60 а-1.
Кривая А (по расчетам Букингема, Гамильтона и Месси [19]). Потенциальная функция с членом, учиты-
вающим наведенное диполь—квадрупольное взаимодействие, имеет вид
<р(г) — Ь ехр (— аг) — сг — * — с' г—•,
где Ь - 9,77 • 10-10 эрг, с - 1,50 • 10-" эрг А«, с' = 2,51 • 10-" эрг А8 и а = 4,60 А-1.
Кривая В (по расчетам Букингема, Гамильтона и Месси [19]). Потенциальная функция — такая же, как
и в случае А, но умноженная на постоянный множитель 0,83. Это изменяет глубину потен-
циальной ямы не меняя положение минимума.
Кривая С (по расчетам Букингема, Гамильтона и Месси [19]). Потенциальная функция такая же, как и
в случае А, но умноженная на постоянный множитель 0,67.
§5. ВТОРОЙ ВИРИАЛЬНЫЙ КОЭФФИЦИЕНТ РЕАЛЬНЫХ ГАЗОВ
ПРИ «ПРОМЕЖУТОЧНЫХ ТЕМПЕРАТУРАХ»
В предыдущем параграфе было показано, как с помощью квантовомехани-
ческого выражения второго вириального коэффициента [формула D.11)]
можно получить уравнение состояния при очень низких температурах. В гл. 3
было получено классическое выражение для второго вириального коэффи-
циента, справедливое при высоких температурах. В этом параграфе будет иссле-
§ 5. Второй вириалъный коэффициент при промежуточных температурах 335
довано поведение уравнения состояния в промежуточной температурной
области, где система мало отличается от классической. Будет показано, что
квантовомеханический второй вириальный коэффициент может быть представ-
лен в виде классического выражения плюс поправочный член.
В гл. 1, § б, было показано, что в случае систем, мало отличающихся от
классических, удобно применять метод ВКЕ. Первое приближение метода
ВКБ дает в точности классический результат, второе приближение — первую
квантовую поправку, и т.д. Кан [10] с помощью этого метода рассчитал отри-
цательные энергетические уровни дискретных состояний Е~ь а также фазовые
сдвиги rjfa) в формуле D.11). Таким путем он получил правильное класси-
ческое предельное выражение для второго вириального коэффициента, а также
первую квантовую поправку. В этом параграфе его подход обсуждаться не
будет, поскольку то же самое выражение для квантовой поправки к В(Т)
можно получить более простым путем из формулы A.2С) для суммы Слетера.
К тому же техника применения метода ВКБ подробно описана в гл. 10 в связи
с квантовым выводом коэффициентов переноса.
А. ОБЩИЕ КВАНТОВЫЕ ВЫРАЖЕНИЯ ВТОРОГО ВИРИАЛЬНОГО
КОЭФФИЦИЕНТА1)
Второй вириальный коэффициент в классической статистической меха-
нике определяется посредством множителя Больцмана W2(rv r2) по формуле
D.5) гл. 3. Это выражение остается справедливым и в квантовой теории урав-
нения состояния, если множитель Больцмана заменить его квантовым анало-
гом — суммой Слетера Ж^2{г1у г2). В соответствии с этим можно написать
для неидеального газа
B(T) = --^r\\[^2(rlyT2)-l]dr1dT2f E.1а)
для идеального газа
4
Вычитая второе уравнение из первого и интегрируя по координатам одной
из молекул, получаем
\В(Т) - В\Т)\ = - 4 J [^«('и) - *4(rii)№ii. E-2)
Если написать сумму Слетера [формула A.20)] для двух частиц и подставить
ее в последнюю формулу, то получается следующее выражение для второго
вириального коэффициента2):
где
00
| = - 2п N J [e-wikT - 1 ] г2 dr, E.4)
Вп(Т) = - 2nN A92OJft*;r«)f<r^>/*r X
О
х [^+*г^+-?гЛг?*--^^уЛг. E.б)
х) См. [27].
2) При этой подстановке используются лишь члены первого ряда скобок в формуле A.20).
336
Гл. 6. Квантовая теория уравнения состояния
Здесь ВКЛ{Т) — обычное классическое выражение второго вириального коэф-
фициента В°(Т) = (Л2//п)8/« В0(Т) — второй вириальный коэффициент идеаль-
ного газа, определенный формулой C.8).
Б. ПРЕДСТАВЛЕНИЕ КВАНТОВОЙ ПОПРАВКИ К В (Г) В ВИДЕ РЯДА
(ПОТЕНЦИАЛ ЛЕННАРДА-ДЖОНСАI)
В гл. 3, § б было показано, что выражение ВКЛ.(Т) в случае потенциала
Леннарда-Джонса может быть представлено в виде быстро сходящегося беско-
нечного ряда гамма-функций. Подобное выражение может быть получено для
функций ВХ{Т) и Ви(Т). Используя выражения ejk и bo = 2/z7iNa* для при-
ведения к безразмерному виду температуры и В(Т), получаем для потенциала
Леннар да-Джонса
в* = 1Вк
А*2 В*
В*,
где
EJ)
2
7=0
I
/-о
в*(т*) = ? ьф т*-<23+б;>/12,
11 7=0 1
Коэффициенты разложения b(j\ бф и b\§ имеют вид [28]
E.8)
E.9)
E.10)
E.11)
E.12)
E.13)
491520я* А /! Г I 12 Г ^Л^
Величины b(i\ 6</> и Ь$ табулированы в табл. V (стр. 858). Функции
п., Bf<, Bf[ и В? приведены в табл. 56 [29 ]2).
Таблица 56
Величина классической и квантовомеханической доли в приведенном втором
вириальном коэффициенте [28]
в* = [ ВЮ1. + А*' в* + Л*' ви + ¦ ¦ О Т А*'В?
МП - - (Ш + 472S/+3024/2 n ,21+*1.Л г ( 6/ + 1 ~|
ч I 491520л* ){ /1 ) { 12 )'
т*
1
2
4
6
9
12
25
100
я*
-2,538
-0,6274
+0,1154
0,3229
0,4406
0,4759
0,5290
0,4640
в*
0,4145
0,08203
0,03247
0,01826
0,01057
0,007324
0,003025
0,0006126
в*
-0,09463
-0,01095
-0,001176
-0,4227 • 10-3
-0,1372 • Ю-8
-0,6761 • 10-*
-0,117 • 10-*
—
R*
DO
0,005359
0,001895
0,6699 • Ю-8
0,3646 • 10~3
0,1985 • Ю-3
0,1289 • Ю-3
0,4287- 10-*
0,5359 • 10-6
1)См. [15, 28].
2) Величины Вкл., Bf, Bf\ связаны с соответствующими величинами Б^., В* , В*\у при-
мененными де Буром и Лунбеком, равенством В* = C/2л) В*, как это объяснено в списке обо-
значений.
§ 5. Второй вириальный коэффициент при промежуточных температурах 337
Величина Л* в помещенных выше формулах равна дебройлевской длине
волны относительного движения двух молекул с относительной кинетической
энергией е, деленной на диаметр столкновений а
E.15)
Отсюда видно, что приведенный второй вириальный коэффициент зависит не
только от приведенной температуры 7*, но и от значения квантовомеханичес-
кого параметра Л*. Это согласуется с квантовомеханическим законом соответ-
ственных состояний, который, подробно обсуждается в следующем разделе.
Ббльшая величина Л*, например, соответствует большей квантовой поправке
в макросвойствах.
В. ВЫЧИСЛЕНИЯ ДЛЯ ПОТЕНЦИАЛА ЛЕН НАР ДА-ДЖОНСА
Чтобы использовать результаты последнего раздела, необходимо знать
«силовые постоянные» а и ejk для рассматриваемых молекул. Для точного
определения этих параметров, необходимых при квантовомеханических
расчетах, опишем экспериментальные данные о величине второго вириального
коэффициента (или коэффициента Джоуля—Томсона) формулой E.7). Это
достигается путем последовательных приближений : сначала определяются
значения параметров а и ejk, для чего используется лишь классическое выра-
жение второго вириального коэффициента. С помощью этих параметров опре-
деляется величина Л* в первом приближении. Затем, используя найденное
значение Л*, можно получить новые значения параметров а и ejk по формуле
E.7) с помощью экспериментальных данных. Этот процесс повторяется до тех
пор, пока не получается согласующийся ряд значений с, ejk и Л*. Полученные
таким образом величины для большого числа различных газов приведены
в табл. 57 [29 J1).
Как только определены эти параметры, второй вириальный коэффициент
и коэффициент Джоуля—Томсона при нулевом давлении могут быть легко
Таблица 57
Значение квантовомеханического
параметра Л*
Благородные
газы
Не8
Не*
Ne
Аг
Кг
Хе
3,08
2,67
0,593
0,186
0,102
0,064
Изотопы
водорода
н2
HD
D2
НТ
DT
т2
1,729
1,412
1,223
1,223
1,095
1,00
Другие
газы
сн4
N2
СО
о2
0,239
0,226
0,220
0,201
вычислены. Ряд по степеням постоянной Планка [формула E.3)] сходится
тем медленнее, чем больше система отклоняется от классической. Это значит,
что формула E.3) не может быть использована при температурах ниже 40 °К
для гелия и ниже 75 и 45 °К для водорода и дейтерия соответственно.
В табл. 58 приведены числовые значения различных членов в разложении
второго вириального коэффициента для четырех газов.
г) Если определены or, е и Л* для одного из изотопов молекулы, то значение Л* легко
вычисляется для любого другого изотопа, поскольку а и е одинаковы для всех изотопов
вещества. Таким путем были определены параметры * для Не3 и некоторых изотопов водо-
рода.
338
Гл. 6. Квантовая теория уравнения состояния
Таблица 58
Порядок величин различных членов, входящих в В(Т) [28]
Газ
Не*
н2
Ne
г,°к
27,3
83,5
256,0
49,2
182,8
592,0
37,0
182,8
592,0
35,6
95,0
392,0
^кл.
- 4,87
8,87
11,13
-47,1
7,55
15,7
-78,94
7,55
15,7
-66,2
- 6,23
12,1
в°
0,50
0,093
0,017
0,57
0,080
0,014
0,31
0,029
0,004
0,03
0,007
0,0008
Wm)Bi
9,16
1,82
0,48
20,68
2,26
0,49
19,60
1,13
0,25
3,80
0,55
0,07
(h*fm)*Bu
-4,05
-0,19
-0,01
-8,63
-0,19
-0,01
-10,21
-0,05
0
-0,47
-0,01
0
На фиг. 15 (стр. 145) показаны расчетные и экспериментальные значения
второго вириального коэффициента. Как видно, теория хорошо предсказывает
квантовые поправки в уравнении состояния при низкой плотности. Подобное
сравнение проведено на фиг. 17 (стр. 152) для коэффициента Джоуля—Томсона
при нулевом давлении, который в квантовой механике имеет вид
+ №MW*- EЛ6)
аналогичный формуле E.3).
В табл.1 (стр.851) приведены два ряда силовых постоянных для легких
газов. Параметры, обозначенные через Qu, получены из экспериментальных
данных по формуле E.7). Можно, однако, определить экспериментальные дан-
ные и с помощью классической формулы F.3) гл. 3. Таким путем были
определены параметры для С1. Эти величины, однако, должны рассматриваться
как «эффективные» силовые постоянные. Для точных расчетов необходимо
использовать высокотемпературную экстраполяцию и значения параметров Qu,
найденные при очень низких температурах.
Г. ВЫЧИСЛЕНИЯ ДЛЯ ПОТЕНЦИАЛА БУКИНГЕМА—КОРНЕРА1)
Букингем и Корнер [30 ] вычислили значение второго вириального коэф-
фициента и коэффициента Джоуля—Томсона, используя потенциальную функ-
цию, определенную формулой C.29). Их результаты имеют вид
. С°р =
0(а,/?; Г*),
^(а,/?; Т*),
д «о К/5; Т*),
Gx(а,/?; Г*).
E.17)
E.18)
Значения функций Fo, G0J Fv Gx приведены в табл. I—IV (стр. 851—857). Эти
таблицы охватывают широкую температурную область и вычислены при пяти
значениях а и двух значениях /?. Они не применялись для вычисления силовых
постоянных потенциала Букингема—Корнера, учитывающего квантовые эф-
фекты в легких газах.
Применяемые здесь обозначения определены в гл. 3, § 7.
§ 6. Закон соответственных состояний в квантовой механике 339
§6. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ
В КВАНТОВОЙ МЕХАНИКЕ
Настоящее изложение очень близко к изложению закона соответственных
состояний для классических систем, данное в гл. 4, § 1. Здесь будет сформули-
рован аналогичный закон для квантовых систем. Затем будет показано, как этот
закон можно применить для выяснения природы квантовых эффектов в жидком
состоянии и предсказания свойств некоторых изотопов водорода и гелия. Обсуж-
дение коэффициентов переноса с точки зрения квантовомеханического закона
соответственных состояний будет дано в гл. 10, § 4.
А. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ В КВАНТОВОЙ МЕХАНИКЕ
Закон соответственных состояний выводится в квантовой механике так
же, как и в классической теории: сначала переходят к переменным, приведен-
ным с помощью параметров критического состояния (макросвойства), а затем
с помощью параметров межмолекулярного потенциала (молекулярные свойства).
Впервые это было предложено Буком [31 ], который нашел квантовую
поправку к уравнению состояния при высоких давлениях и низких темпера-
турах из закона соответственных состояний. Он предложил приведенное урав-
нение состояния [формула A.6) гл. 4] записать таким путем, чтобы постоянная
Планка входила в него в безразмерном виде. Следуя Буку, уравнение состояния
можно написать в виде
Pr = Pr(Vr,Tr; Лг), F.1)
где ЛТ — безразмерная комбинация
л h //* л\
Г~ (VJNI!* (rnkTI!* в V '
Параметр Лг характеризует степень отклонения от классического закона соот-
ветственных состояний. Вообще говоря, этот параметр не особенно хорошо
подходит для изучения квантовых эффектов^ поскольку сам параметр Лг за-
висит еще от квантовых эффектов через Vc и Тс. Тем не менее видно, что кван-
товые поправки будут велики для легких газов с малыми критическими тем-
пературами и объемами. Квантовомеханическое уравнение состояния можно
записать аналогично классической формуле A.16) гл. 4 в виде функции от
параметров, приведенных к безразмерному виду с помощью молекулярных
постоянных [32—34]. В общем случае такое уравнение состояния имеет вид
р = р^,кТ; Л,т,<г,г) F.3)
при условии, что межмолекулярная потенциальная функция дается формулой
A.13) гл. 4. Если для приведения переменных использовать молекулярные
параметры а и е, то уравнение состояния в приведенной форме можно записать
в виде
F.4)
Приведенное давление р* = pa*je может зависеть лишь от безразмерных вели-
чин V* = V/No* и Т* = кТ/е, а также от безразмерной комбинации Л*, опре-
деляемой следующим образом:
4
а уте
поскольку V*, Т* и Л* — единственные ^безразмерные параметры, которые
можно образовать, комбинируя величины V/N, кТ} Л, ш, е и а.
Формула F.4) выражает закон соответственных состояний в квантовой
механике применительно к уравнению состояния. Полученный результат
340 Гл. 6. Квантовая теория уравнения состояния
выведен без каких-либо ограничений на плотность или температуру системы,
поэтому он справедлив для газов, жидкостей и твердых тел.
Таким образом, приведенное давление в общем виде является функцией
приведенного объема и приведенной температуры и зависит от параметра Л*.
В дальнейшем будет показано, что эта функция имеет различный вид для мо-
лекул, подчиняющихся статистике Бозе—Эйнштейна и статистике Ферми—
Дирака. Это связано с тем обстоятельством, что при суммировании по всем
квантовым состояниям в статистической сумме учитываются лишь симмет-
ричные состояния в первом случае и антисимметричные во втором. Однако
для температур и плотностей, при которых дебройлевская длина волны мала
по сравнению со средним расстоянием между молекулами, различие между
статистическими суммами по симметричным и антисимметричным состояниям
исчезает и, следовательно, оба приведенных уравнения состояния переходят
в одно приведенное квантовомеханическое уравнение состояния.
Закон соответственных состояний уже был проиллюстрирован на примере
поведения некоторых равновесных свойств газа. На фиг. 15 (стр. 145) приведены
кривые зависимости В* от Т* в классическом случае. Проведено также срав-
нение вычисленных кривых В* с экспериментальными данными для некото-
рых легких газов. Совершенно ясно, что квантовые поправки больше для
веществ с большим значением Л*. Аналогично ведет себя третий вириальный
коэффициент (см. фиг. 16, стр. 150), а также коэффициент Джоуля—Томсона
при нулевом давлении (фиг. 17, стр. 152).
Б. КВАНТОВЫЕ ЭФФЕКТЫ В ЖИДКОЙ ФАЗЕ1)
Теоретическое изучение квантовых поправок к величинам, характери-
зующим свойства газов, приводит к удовлетворительному количественному
описанию наблюдаемых явлений. Однако такие чисто теоретические резуль-
таты для жидкости еще не получены, поэтому здесь придется ограничиться
полуэмпирическим исследованием квантовых эффектов, основанным на законе
соответственных состояний. Закон соответственных состояний будет взят в
форме уравнения F.4), причем для параметра Л* взяты значения, полученные с
помощью потенциала F-12) Леннарда-Джонса и приведенные в табл. 48 (стр. 269).
Для иллюстрации общих свойств квантовых поправок будут коротко рассмот-
рены экспериментальные данные о свойствах вещества в критическом состо-
янии, а также молярные объемы жидкости, равновесие жидкость—пар,
пар—твердое тело и поверхностное натяжение.
1. Свойства вещества в критическом состоянии. На фиг. 107 показана
зависимость приведенных критических постоянных благородных газов и водо-
рода от величины параметра Л*. Из этого графика находятся значения крити-
ческих постоянных в предельном случае Л* = 0:
Г* =^=1,26,
F.6)
Для легких газов величины критических параметров сильно отличаются от
своих классических значений, причем степень отклонения увеличивается с
ростом Л*.
. [33, 34, 29].
§ 6. Закон соответственных состояний в квантовой механике
341
Не4 Не3
Фиг. 107. Критические параметры как функции Л*.
Экстраполяционные величины параметров в критическом состоянии при Л* = О (квантовые эффекты отсут-
ствуют) даны в формулах F.6). Критические параметры для Не* предсказаны путем графической экстра-
поляции трех кривых. Это хорошая иллюстрация применения закона соответственных состояний
в квантовой механике [29].
2. Объемы жидкости. Молярный объем жидкости очень чувствителен к
квантовым эффектам. Как видно из фиг. 108, отклонения приведенного моляр-
ного объема от классического предельного значения для не слишком боль-
ших величин Л* пропорциональны Л*2. Эта пропорциональность была иссле-
дована теоретически [34]1) с помощью уравнения состояния Леннарда-Джонса
Фиг. 108. Приведенные молярные объемы одноатомных жидкостей и изотопов водорода
при различных приведенных температурах.
Видно, что приведенный молярный объем жидкостей является почти линейной функцией Л*2 [34]
и Девоншаира для жидкости. В этой теории статистическая сумма одной моле-
кулы в жидкости, опуская не зависящий от объема множитель, имеет вид
(б.7;
здесь (о(г) — среднее потенциальное поле молекулы в ячейке объема Л. Чтобы
можно было применять теорию Леннарда-Джонса и Девоншаира в квантовой
механике, необходимо заменить множитель Больцмана ехр (—ео(г)//сГ) его
г) Квантовые эффекты в жидкой фазе изучены в работах [35, 36 ].
342
Гл. 6. Квантовая теория уравнения состояния
квантовомеханическим аналогом — суммой Слетера. Сумму Слетера можно
представить в виде ряда в соответствии с формулой A.20). В результате по-
лучаем следующее выражение для статистической суммы одной молекулы:
г,Т)+ ...]Л\ F.8)
Подставляя это выражение в формулу D.32) гл. 4, получаем окончатель-
ное выражение для приведенного уравнения состояния в форме
Р* = Pin + Л*2Р1 +
+ ...
F.9)
Из этого выражения видно, что квантовые поправки для различных свойств
жидкости прямо пропорциональны Л*2 при малых значениях Л*. Величина
8г#/ЭЛ*2, найденная с помощью теории де Бура [34], отличается от эксперимен-
тального наклона кривой tf как функции Л*2 (см. фиг. 108) на множитель
порядка 2. Учитывая приближенный характер теории Леннарда-Джонса и
Девоншайра, это совпадение нельзя считать плохим.
3. Равновесие жидкость—пар и пар—твердое тело. Приведенное давле-
ние пара в соответствии с классической теорией соответственных состояний
является универсальной функцией приведенной температуры. Для тяжелых
благородных газов (Аг, Кг, Хе) это действительно подтверждается последними
экспериментальными данными1), приведенными на фиг. 109. На той же фигуре
Линия критических точек
Классические
пррдрл
Фиг. 109. Приведенное давление пара при различных температурах.
Видно, что квантовыми поправками нельзя пренебрегать в случае легких газов Не, Н„ Ne и что квантовые
эффекты особенно заметны при больших л* [29].
изображены кривые давления пара для легких благородных газов [40, 41 ] и
трех изотопов водорода [42 ]. Как видно из фигуры, квантовые поправки для
этих газов оказываются заметными даже для неона. Кроме того, кривые дав-
ления пара для трех изотопов водорода явно отличаются друг от друга. Этот
эффект классическая теория вообще не может предсказать. Из фигуры видно,
что бблыыим значениям Л* соответствуют ббльшие отклонения от класси-
ческих значений. Интерпретация этого явления может быть дана с помощью
теории твердого тела Дебая и теории жидкости Леннарда-Джонса и Девон-
шайра [29].
х) См. для ксенона работу [37], для аргона—работу [38] и для криптона—работу [39].
§ 6. Закон соответственных состояний в квантовой механике
343
Теоретические выводы о равновесии пар—твердое тело находятся в удов-
летворительном согласии с экспериментальными данными прежде всего по-
тому, что твердая и газообразная фазы достаточно хорошо изучены. Количест-
венные заключения о равновесии жидкость—пар, конечно, менее удовлетвори-
тельны, поскольку наших знаний о жидком состоянии еще недостаточно.
4. Поверхностное натяжение. Классическая теория поверхностного натя-
жения изложена в гл. 5, § 1, однако квантовые поправки теоретически еще не
изучены. Из фиг. 110 видно, что квантовые эффекты очень сильно влияют на
величину поверхностного натяжения легких веществ. Согласно классическому
.11
0,9
0,8
0,7
0,6
.0,5
0,3
0,2
o,iV
О
\
Х- точка
л \
\ \ \
\
\ \
\\
0,1 0,2 0,3 04 05 06 Ц7 08 09 ^5 Tti} 7,2 И/3
r* _ *3 *4 Т^кТ/е а 4 ¦DTt'Nel'
7с "^Не Не Н2 HD D2 T2 Аг
Фиг. 110. Зависимость приведенного поверхностного натяжения у* от приведенной тем-
пературы Т* и квантовомеханического параметра Л*.
Стрелки над символами атомов и молекул указывают на значение приведенной критической температуры
каждого из веществ.
принципу соответственных состояний можно ожидать, что приведенное по-
верхностное натяжение у* = уо*\е будет универсальной функцией приведен-
ной температуры Т* = kTje для веществ, молекулярное взаимодействие кото-
рых адекватно описывается потенциалом Леннарда-Джонса. Имеющиеся
экспериментальные данные указывают, что у* зависит, кроме того, от значения
квантовомеханического параметра Л*.
В. СВОЙСТВА ИЗОТОПОВ ГЕЛИЯ И ВОДОРОДА
В настоящее время большой интерес представляет вопрос о разделении и
свойствах изотопов. К числу наиболее интересных и малоизученных изотопов
принадлежат два изотопа гелия и шесть изотопов молекулярного водорода.
В этом разделе будет показано, как можно определить некоторые свойства
Не3, D2, T2, HD, HT и DT.
1. Свойства Не3 [34]. В последнее время Не3 стал привлекать к себе вни-
мание тем, что он не обладает своеобразными свойствами Не11- Поскольку а и е
одинаковы для обоих изотопов, а масса Не3 равна 3/4 массы Не4, то Л* для Не3
составляет У*[гЛ* для Не4, или 3,08. Таким образом, получается довольно
большое численное значение для Л*.
Если известна зависимость каких-либо физических свойств от величины
Л* и других приведенных параметров, то эту зависимость можно экстраполи-
ровать к значениям Л* = 3,08 и таким образом определить свойства Не3 с
344
Гл. 6. Квантовая теория уравнения состояния
точностью до 10—20%. Таким путем в 1948 г. были предсказаны [34] крити-
ческие постоянные Не3 (см. фиг. 107). В следующем году эти постоянные были
измерены [43]. Экспериментальные результаты и результаты, предсказанные
с помощью квантовомеханического закона соответственных состояний, срав-
ниваются в табл. 59.
Таблица 59
Критические постоянные изотопов гелия
Газ
Не*
(экстр. [34]
"е(эксп. [43]
те,°к
5,20
3,37
3,34
ре, атм
2,26
1,12
1,15
Ve, см*/моль
57,76
70,0
72,0
т*
0,509
0,33
РГ
0,0271
0,0135
К
5,742
7,0
Таким же путем в 1948 г. [34] были вычислены кривые давления пара
Не3. Спустя два года эти величины были экспериментально измерены [44].
Сравнение экспериментальных и теоретических данных, проведенное в табл. 60,.
указывает на очень хорошее согласие.
Таблица 60
Давление пара изотопов гелия (смрт.ст.)
Газ
Не4
Не3
^\
(экстр.
(ЭКСП. •
т*
\.
[34]
[44]
1,21
0,07
2,07
1,33
0,14
3,10
1,52
0,39
5,35
1,63
0,64
7,04
1,8
1,23
2,0
2,34
14,0
15,2
2,4
6,26
27,0
29,1
2,8
13,27
48,0
49,11
3,0
18,12
59,0
61,79
3,2
24,05
75,0
76,40
3,3
27,48
82,0
84,47
С помощью закона соответственных состояний была установлена также
величина поверхностного натяжения для Не3. Это было сделано путем графи-
ческого представления данных о величине поверхностного натяжения для
Н2, Не4, Ne и Аг с помощью уравнения
9- F.10)
Зависимость величины /} от Л* изображена в верхнем правом углу фиг. 110;
отсюда путем экстраполяции для Не3 получается значение р = 0,56. С помощью
найденной величины /? и значения Т? = 0,32 можно построить кривую поверх-
ностного натяжения. Экспериментальных данных о величине поверхностного
натяжения в Не3 до сих пор еще нет.
2. Свойства изотопов Н2. Свойства Не3 определяются экстраполяцией;
для определения свойств Н2 необходимо воспользоваться процессом интерпо-
ляции. При этом необходимо сделать два не совсем очевидных допущения ;
во-первых, считать, что изотопы Н2, представляющие собой двухатомные
молекулы, являются благородными газами и, во-вторых, что межмолекулярное
взаимодействие различных изотопов Н2 описывается одним и тем же потен-
циалом. Молекулы водорода являются вращающимися двухатомными молеку-
лами, поэтому можно ожидать отклонения от закона соответственных состоя-
ний, поскольку в рассматриваемой форме он был выведен только для сфери-
ческих молекул. Более того, существует несколько причин, вследствие
которых второе предположение, справедливое для изотопов гелия, может не
§ 6. Закон соответственных состояний в квантовой механике
345>
выполняться для изотопов водорода. Это, во-первых, небольшое различие в нуле-
вой энергии, которое приводит к некоторому различию силовых полей Н2, D2
и Т2 [45 ]. Во-вторых, и это является более серьезной причиной, HD, НТ и DT
вращаются вокруг центра тяжести, расположенного не на середине расстояния
между атомами, как это имеет место в случае молекул Н2, D2 и Т2. Экспери-
менты покажут, будут ли эти причины влиять на согласие экспериментальных
и расчетных данных, основанных на указанных выше двух предположениях.
С помощью интерполяции кривых, описывающих физические свойства
как функции Л*2, можно найти значения этих свойств для различных изото-
пов водорода [29, 47]. В табл. 61 и 62 приведены вычисленные таким образом.
Таблица 61
Критические постоянные изотопов водорода
Вещество
Л*
г?
V*c
ТсСК)
pdamM)
Vc
(смг/моль)
(теор. [29]
(эксп.
(теор. [29]
(эксп.
(теор. [29]
(эксп.
н,
1,729
0,897
6,46
4,42
C3,18)
33,18
A2,98)
12,98
F6,95)
66,95
HD
1,412
1,03
7,8
4,1
38,0
35,9 [47]
15,5
14,6 [47]
62,0
62,8 [47]
Dt; HT
1,223
1,10
8,6
3,9
41,0
38,3 [47,48]
17,5
16,3 [47,48]
59,0
60,3 [47]
DT
1,095
1,14
9,2
3,8
42,0
—
18,5
—
57,0
—
т,
1,00
1,17
9,6
3,7
43,0
—
19,0
—
56,0
—
критические постоянные и кривые давления пара. Там же приведены сущест-
вующие экспериментальные данные о критических постоянных [48, 49 ]. Точ-
ность процесса экстраполяции не может быть больше 5%, поэтому имеющиеся
данные не могут дать однозначного ответа о том, существуют ли отклонения
от интерполяционных величин. Вместе с тем это позволяет утверждать, что
перечисленные выше причины не приводят к существенному отклонению от
интерполяционных величин. Аналогичные вычисления проделаны и для
некоторых других параметров [29].
Таблица 62
Давление пара изотопов водорода [29] (мм pnt. cm.)
Т,вК
10
12
14
16
18
20
25
30
35
н,
1Дз
12,7
55,4
153,3
345,9
675,7
2525,0
6200,0
—
HD
0,28
2,94
16,8
65,2
176,4
382,8
1630,0
4415,0
8060,0
D.; HT
0,05
0,73
5,44
25,4
87,2
219,9
1040,0
3180,0
6650,0
DT
0,21
1,85
11,5
43,5
130,0
770,0
2540,0
5790,0
Т,
0,09
0,98
7,43
31,6
106,0
700,0
2345,0
5400,0
346 Гл. 6. Квантовая теория уравнения состояния
§7. КВАНТОВЫЕ ЭФФЕКТЫ В ДВУХАТОМНЫХ ГАЗАХ
Приведенные до сих пор теоретические рассмотрения в этой главе ябля-
ются строгими только для одноатомных газов. Следует сказать, что изложение
теории уравнения состояния водорода при очень низких температурах, а также
вывод квантовых поправок к уравнению состояния водорода и дейтерия при
промежуточных температурах проводился на основе результатов, полученных
в теории одноатомных газов. В настоящем параграфе будут непосредственно
изучены квантовомеханические эффекты, связанные с вращательным движением
этих молекул. Сначала будет изложена общая постановка задачи, а затем будет
определена величина вращательного квантового эффекта в Н2. Расчеты прове-
дены для области промежуточных температур (аналогично § 5) и не сделаны
для области очень низких температур (аналогично § 4).
А. КРАТКАЯ СВОДКА ТЕОРЕТИЧЕСКИХ РЕЗУЛЬТАТОВ
Квантовую статистическую теорию равновесных объемных свойств раз-
вила Ван Чанг [49 ]. Она исходила из предположения, что двухатомные моле-
кулы можно представить в виде модели жестких трехмерных ротаторов массы
т и момента инерции / с пятью степенями свободы — тремя поступательными
и двумя вращательными. Оператор Гамильтона системы N таких молекул
является квантовомеханическим аналогом классического гамильтониана [фор-
мула D.10) гл. 3]. Квантовомеханическая статистическая сумма такой системы
также похожа на классическую статистическую сумму [формула D.13)] при
условии, конечно, что множитель Больцмана заменен суммой Слетера. Ван
Чанг показала, что сумму Слетера можно выразить через [/-функции точно
так же, как это сделано в гл. 3, § 2. Таким путем были получены выражения
для вириальных коэффициентов.
Ван Чанг показала также, что выражения для вириальных коэффициен-
тов, выведенные таким путем, сводятся к аналогичным выражениям [формулы
{4.15) и D.16) гл. 3], полученным для вращающихся двухатомных молекул в
классической теории. Затем она получила квантовые поправки ко второму
вириальному коэффициенту по схеме § 5 для многоатомных газов. Оказалось,
что существуют два типа квантовых поправок: поправки, пропорциональные
последовательным степеням (h2jm) и связанные с поступательным движением,
и поправки, пропорциональные последовательным степеням (Л2//) и связанные
ч: вращательным движением.
Окончательный результат, аналогичный формуле E.3), можно записать
в виде
в(Т) = Вкл. + [(!-) Мпост) + (-?-)'
где
^Ц G.2)
G3)
Ш+(*
+ cosec2 0Х [-^f + cosec2 02 [-^f] r* dr dQ. G.4)
§ 7. Квантовые эффекты в двухатомных газах
347
В этих выражениях
J
2л п я
exsinв2йв2й(ф2 —фг).
G.5)
0 0 0
Поправки более высокого порядка (пропорциональные ft4) также найдены.
Они не приводятся здесь ввиду их громоздкости, а также потому, что они
везде опубликованы [49]. Выражения, даваемые формулами G.1)—G.5), не
являются еще наиболее общими выражениями второго вириального коэффи-
циента. Ван Чанг получила более общие выражения, учитывающие наличие
0р/л0-/шра-состояний у гомеополярных двухатомных газов. Здесь эти формулы
не приводятся, однако полученные с их помощью результаты будут кратко
обсуждены.
Б. СРАВНЕНИЕ ТЕОРЕТИЧЕСКИХ И ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ
ДЛЯ ВОДОРОДА
Для расчета вращательных квантовых поправок в водороде Ван Чанг пред-
положила, что межмолекулярный потенциал водорода приближенно можно
представить в виде
9 (г, 0i, КФ) = - ¦?¦ + 7^- + ju (cos* вг + cos* 02).
G.6)
Постоянные а, бис выбираются из условия наилучшего совпадения с разно-
образными квантовомеханическими расчетами, проведенными для несфери-
ческого потенциального поля водорода [50, 51J1).
На основе потенциала G.6) проведены расчеты второго вириального коэф-
фициента. Результаты расчета приведены в табл. 63 совместно с первыми кван-
товыми поправками. Зависимость В от Г лучше согласуется с эксперименталь-
ными данными при низких температурах, где наклон dBjdT велик. Это связано
с тем обстоятельством, что поправки второго порядка всегда отрицательны, и
при низких температурах они не малы, как видно из табл. 64.
Таблица 63
Вклад различных членов, входящих во второй вириальный коэффициент для
На (см*/моль)
-В
4- * В(П0СТ-> 4- Н%
т. *к
123
173
223
323
373
423
Вкл.
-3,06
+5,68
9,49
13,15
14,06
15,22
(/i./m)B<nocT->
4,23
2,58
1,76
1,13
0,97
0,64
1,00
0,46
0,28
0,15
0,12
0,08
*
^выч.
2,17
8,72
11,53
14,43
15,15
15,94
^эксп.
2,56
9,14
12,08
15,14
15,55
15,68
1) Найденные величины равны а ¦¦
с = 4,784- 10-8 эрг- А12.
1,25 • Ю-11 эрг . Ae ; b = 4,438 • 10-» эрг . А12;
348
Гл.6. Квантовая теория уравнения состояния
Вращательные квантовые поправки второго порядка имеют знак, про-
тивоположный поправкам первого порядка ; аналогичное положение имеет
место и для поступательных квантовых поправок (см. табл. 58). Из анализа
порядка величины В<вр> и fJftp-> видно, что сходимость рядов ухудшается с
уменьшением температуры и, следовательно, разложение в ряд по степеням
ft2// справедливо лишь при относительно высоких температурах.
Таблица 64
Вращательные квантовые поправки первого и второго
порядка (см*/моль)
Т, °К
123
173
223
323
(Л«//)в<вр0
1,00
0,46
0,28
0,15
(*/|)и4вр->
-0,42
-0,14
-0,07
-0,02
Ван Чанг [49] рассчитала разность между вторыми вириальными коэф-
фициентами нормального и пара-водорода при 173 и 223° К. При этих темпера-
турах разность ВНа— Впара равна соответственно 0,19 и 0,11 смг\моль. Эти
величины показывают, что разность (ВНа — Впара) уже при 173° К меньше
экспериментальной ошибки.
Задачи
1. Найти сумму Слетера для следующих простых систем [10]:
а) Свободно движущаяся материальная точка в одномерном потенциальном ящике
длиной L :
б) Линейный гармонический осциллятор1) с частотой v:
., где У = ъ
в) Две невзаимодействующие материальные точки в одномерном потенциальном ящике
длиной L ; рассмотреть случаи статистики Больцмана, Бозе—Эйнштейна и Ферми—Дирака :
. (х) = A -
Б. Э. (X) I = с^^
^Ф.Д. (х) J
Обсудить поведение суммы Слетера при переходе к классическому пределу.
2. Вывести следующие калорические уравнения состояния и доказать их эквивалент-
ность :
U = -¦
При выводе необходимо воспользоваться соотношением
где Нп(у) — эрмитовы полиномы. Это соотношение приведено в работе [52 ].
Литература 349
3. Вывести следующее квантовомеханическое выражение для бинарной функции рас-
тления в идеальном газе:
пределения
Дать интерпретацию этого выражения с помощью представления о кажущемся отталкивании
и притяжении молекул.
ЛИТЕРАТУРА
1. Kirkwood J. G., Phys. Rev., 44, 31 A933).
2. Н u s i m i K., Proc. Phys.-Math. Soc. Japan, Ser., 3, 22, 264 A940).
3. Mayer J. E., Band W., Journ. Chem. Phys., 15, 141 A947).
4. d e Boer J., Physica, 15, 843 A949).
5. Y v о n С R., Compt. rend., 227, 763 A948).
6. G r e e n H. S., Proc. Roy. Soc, A189, 103 A947).
7. d e Boer J., Nuovo Cimento, Suppl. Vol. VI, Ser. IX A949).
8. Ehrenfest P., Oppenheimer J. R., Phys. Rev., 37, 333 A931).
9. К a h n В., U h 1 e n b e с k G. E., Physica, 5, 399 A938).
10. К a h n В., Диссертация, Utrecht, 1938.
11. London F., Phys. Rev., 54, 948 A938).
12. L о n d о n F., International Conference Report, Vol. II, London, 1947.
13. Einstein A., Bed. Ber., 261 A924); 3 A925).
14. d e Boer J., Nederlands Tijdschrift voor Natuurkunde, 16, 89 A950).
15. d e Boer J., Диссертация, University of Amsterdam, 1940.
16.de Boer J., van Kranendonk J., Compaan K., Physica, 16, 545
A950)
17. d e Boer J., M i с h e 1 s A., Physica, 6, 409 A939).
18. M a s s e у H. S. W., Buckingham R. A., Proc. Roy. Soc, A168, 378 A938); A169,
205 A939).
19. Buckingham R. A., Hamilton J., M a s s e у H. S. W., Proc. Roy. Soc, A179,
103 A941).
20. К i 1 p a t r i с k J. E., К i 1 p a t r i с k M. F., Journ. Chem. Phys., 19, 930 A951).
21. К i s t e m a k e г J., Keesom W. H., Physica, 12, 227 A946).
22. Keesom W. H., Kraak H. H., Physica, 2, 37 A935).
23. Keesom W. H., Walstra W. K., Physica, 6, 1146 A939).
24. de Boer J., Rep. Progr. Phys., 12, 305 A949).
25. Mi yak о R., Proc. Phys. Math. Soc. Japan, Ser. 3, 24, 852 A942).
26. Halpern 0., Gwathmey E., Phys. Rev., 52, 944 A937).
27. U h 1 e n b e с k G. E., В e t h E., Physica, 3, 729 A936).
28. de Boer J., Michels A., Physica, 5, 945 A938).
29. L u n b e с k R. J., Диссертация, Amsterdam, 1951.
30. В u с k i n g h a m R. A., Corner J., Proc Roy. Soc, A189, 118 A947).
31. Byk A., Ann. d. Phys., 66, 157 A921); 69, 161 A922).
32. d e Boer J., Physica, 14, 139 A948).
33. d e Boer J., В 1 a i s s e B. S., Physica, 14, 149 A948).
34. de Boer J., Lunbeck R. J., Physica, 14, 520 A948).
35. H a m a n n S. D., Trans. Farad. Soc, 48, 303 A952).
36. David H. G., Hamann S. D., Trans. Farad. Soc, 49, 711 A953).
37. M i с h e 1 s A., W a s s e n a a r Т., Physica, 16, 253 A950).
38. Clark A.M., Din F., Robb J., Michels A., Wassenaar Т., Zwiete-
, ring Th., Physica, 17, 876 A951).
39. Michels A., Wassenaar Т., Zwieterling Th., Physica, 18, 63 A952).
40. D i t с h b u r n R. W., G i 1 m о u r J. C, Rev. Mod. Phys., 13, 310 A941).
41. Landolt-Bdrnstein, Physikalisch-Chemische Tabellen, Berlin, 1936 (см. перевод:
Ландольт-Бернштейн, Физико-химические таблицы, М.—Л., 1939).
350 Гл. 6. Квантовая теория уравнения состояния
42. Woolley H. W., Scott R. В., Brickwedde F. G., Journ. Res. Natl. Bur.
Stand. 41, 379 A948).
43. Sydoriak S. G., Grill у E. R., Hammel E. F., Phys. Rev., 75, 303, 1103
A949).
44. Abraham B. M., Os borne D. W., We ins toe к В., Phys. Rev., 80, 366
A950).
45. В e 11 R. P., Proc. Roy. Soc, A174, 504 A940).
46. H a m m e 1 E. F., Journ. Chem. Phys., 18, 228 A950).
47. A rn о 1 d R. D., Hoge H. J., Journ. Chem. Phys., 18, 1295 A950).
48. Friedman A. S., White D., Johnston H. L., Journ. Chem., Phys., 19, 126
A951).
49. Wang Chang C.S., Диссертация, University of Michigan, 1944.
50. d e Boer J., Physica, 9, 363 A943).
.51. Marge паи Н., Phys. Rev., 64, 131 A943).
52. U hie n beck G. E., Ornstein L. S., Phys. Rev., 36, 823 A930).
Часть II
НЕРАВНОВЕСНЫЕ СВОЙСТВА
ПУСТАЯ СТРАНИЦА
ГЛАВА 7
КИНЕТИЧЕСКАЯ ТЕОРИЯ РАЗРЕЖЕННЫХ ГАЗОВ
В предыдущих главах были рассмотрены свойства систем, находящихся
в равновесии. В настоящей и последующих главах статистические методы
используются для описания неравновесных свойств, и в частности для описа-
ния явлений переноса. Элементарная теория явлений переноса изложена в
гл. 1, § 2. Там же дано краткое изложение подхода, основывающегося на стро-
гой кинетической теории и используемого в этой главе, а также рассмотрены
предположения, используемые в теории, и границы ее применимости. В настоя-
щей главе получены гидродинамические уравнения и формулы для коэффициен-
тов переноса для малых плотностей. В гл. 8 проводятся оценки и практические
расчеты коэффициентов переноса. Состояние теории явлений переноса при
высоких давлениях рассмотрено в гл. 9. Квантовая теория явлений переноса
обсуждается в гл. 10. В гл. И рассматриваются гидродинамические уравнения,
а также подробно обсуждаются некоторые приложения этих уравнений.
Большая часть всей теории, излагаемой в упомянутых выше главах, ограничи-
вается случаем одноатомных газов, однако в настоящей главе отмечается,
что внутренние степени свободы оказывают очень небольшое влияние на
коэффициенты диффузии и сдвиговой вязкости. Поэтому некоторые из ре-
зультатов, полученные для одноатомных газов, практически имеют более
широкую область применимости, чем это следует из теории. Напротив,
коэффициент теплопроводности существенно зависит от обмена энергией
между поступательными и внутренними степенями свободы. Теория явлений
переноса в газах, состоящих из молекул, обладающих внутренними степенями
свободы, кратко изложена в § б этой главы.
Мы начнем с того, что покажем, как свойства разреженного газа полностью
могут быть выражены через функцию распределения /(г, v, f). Эта функция
распределения является решением интегро-дифференциального уравнения
Больцмана. Уравнение Больцмана справедливо лишь для достаточно малых
плотностей газа, когда влиянием столкновений более чем двух молекул можно
пренебречь. Если длина среднего свободного пробега молекулы в газе мала по
сравнению с макроскопическими размерами, то газ ведет себя как сплошная
среда. Для таких случаев из уравнения Больцмана можно получить гидро-
динамические уравнения Навье-Стокса и выражения для векторов потоков.
Коэффициенты переноса определяются векторами потоков и выражаются через
интегралы ?(/'s), значение которых зависит от вида потенциальной функции
межмолекулярного взаимодействия.
§ 1. ФУНКЦИИ РАСПРЕДЕЛЕНИЯ КИНЕТИЧЕСКОЙ ТЕОРИИ1)
В этой главе кинетическая теория основывается на уравнении Больцмана
для функции распределения /(г, v, /). Мы изложим здесь два вывода уравнения
Больцмана. Первый, простой физический вывод дает физическую интерпре-
тацию различных членов уравнения. Второй, более строгий вывод основан на
Используемые здесь функции распределения определены в гл. 2, § 1.
354 Гл. 7. Кинетическая теория разреженных газов
интегрировании уравнения Лиувилля. Перед тем как изложить эти выводы,
мы обсудим некоторые свойства различных функций распределения кинетиче-
ской теории и их использование для описания неравновесных систем.
А. ФИЗИЧЕСКОЕ ОПИСАНИЕ НЕРАВНОВЕСНЫХ СИСТЕМ1)
Динамическое состояние системы частиц совершенно точно определяется
значениями полного набора координат и импульсов всех частиц. Законы клас-
сической механики позволяют по известному динамическому состоянию си-
стемы в начальный момент времени точно предсказать е есостояние в любой
последующий момент времени.
Однако фактически невозможно дать полного описания состояния слож-
ной макроскопической системы. Поэтому мы должны довольствоваться значи-
тельно менее полным ее описанием. Проблема предсказания вероятного пове-
дения системы по неполным данным о системе в некоторый определенный
момент времени является статистической проблемой. При статистическом опи-
сании системы полезно использовать метод описания системы с помощью ан-
самбля, состоящего из большого числа систем, тождественных изучаемой.
Состояние такого ансамбля определяется функцией распределения fN(rN, ры, t),
задаваемой в фазовом пространстве отдельной системы. Функция распре-
деления выбирается таким образом, что описание системы с помощью средних
по ансамблю величин в точности соответствует неполному макроскопическому
описанию состояния системы в некоторый определенный момент времени.
При этом принимается, что вероятное поведение системы в последующие момен-
ты времени является средним поведением систем представляющего ансамбля.
Ансамбль можно образовать различными способами. Вследствие этого функция
распределения определяется неоднозначно. Для равновесного состояния ан-
самбль определяется эргодической теоремой Биркгоффа. Для случая нерав-
новесного состояния аналогичная теорема еще не рассматривалась. Однако
последнее не является источником непреодолимых трудностей, так как в ста-
тистической механике неравновесных систем рассматриваются главным обра-
зом функции распределения низшего порядка /A) и /B). Уравнения для функций
распределения низшего порядка выводятся путем использования различных
ограничений, таких, как концепция «молекулярного хаоса», которая ограничи-
вает набор рассматриваемых типов функций распределения fiN).
Изменение функции распределения ры)(тыу pN, t) в зависимости от времени
описывается уравнением Лиувилля, которое было получено в гл. 1, § 4. Найти
решение этого уравнения трудно, поскольку в него входят 6N переменных.
Однако обычно мы не интересуемся таким полным описанием системы,
которое можно получить с помощью функции /м, а можем ограничиться менее
полным описанием, даваемым одной из функций распределения fh(rhy рл, t)
низшего порядка. Эти функции получаются интегрированием fN) по ко-
ординатам и импульсам N — h молекул, исключая группу 1, 2, 3, ..., Л,
как указано в гл.2, § I2). Особый интерес представляют функции распределе-
ния с Л = 1 и h = 2.
В настоящей главе мы рассматриваем главным образом функцию /(l)(r, p, f),
которая дает вероятность обнаружения отдельной молекулы с данными
значениями координат и импульса. Координаты и импульсы других N — 1
молекул остаются неопределенными. При рассмотрении систем из одинаковых
молекул функцию распределения fN) можно считать симметричной по коор-
динатам всех молекул, так как в физических экспериментах молекулы такой
системы не отличаются друг от друга. Следовательно, для получения /A) не-
важно, какая именно молекула рассматривается. Очевидно, что р& достаточна
для описания тех свойств газа, которые не зависят от относительного поло-
г) Данное здесь обсуждение аналогично обсуждению, проведенному Трэдом [1].
2) По поводу определения и нормировки этих функций см. гл. 2, § 1.
§ 7. Функции распределения кинетической теории 355
жения двух или большего числа молекул. Это значит, что объем сведений о
системе, доставляемых нам функцией /A), достаточен для изучения поведения
умеренно разреженных газов.
Для газов более высокой плотности необходимо знать функции распре-
деления более высокого порядка. Однако если можно предположить, что меж-
молекулярные силы являются парными силами, т. е. что Ф(гм) = 1f2 2 q>ij9
ij
то для определения всех макроскопических свойств системы достаточно
знать функцию распределения порядка h = 2. Функция распределения
f*XTv r2> Pi» P2> 0 задается в фазовом пространстве пары молекул. Использо-
вание бинарной функции распределения при изучении поведения плотных
газов обсуждается в гл. 9, § 4. Напомним, что, как показано в гл. 3, § 1, в
результате предположения об аддитивности сил равновесные свойства плотных
газов оказалось возможным выразить только через бинарную функцию распре-
деления равновесной системы.
Рассмотрим теперь зависимость различных функций распределения от
времени. Как было отмечено выше, зависимость функции fN) от времени дается
уравнением Лиувилля. Иначе говоря, каждой функции fN\ заданной в началь-
ный момент времени /0, однозначно соответствует функция fN) в любой более
поздний момент времени tv Однако для функций распределения низшего по-
рядка, зная функцию fh)(t0), невозможно однозначно предсказать /(/1)(^).
Например, возможно, что для момента времени t0 имеется ряд функций
f(MXQ> /(А/)'('о)> fN)"(Q и т- Д-> которые при интегрировании по координатам
и импульсам N — 1 молекул все дают одну и ту же функцию рг)(г0). В более
поздний момент времени ряд функций iV-ro порядка переходит в функции
/(N)d)> fN)'(ti)> fN)"(ti) и т- Д- Этим функциям соответствуют функции распре-
деления первого порядка f1^), /^'('iX f^"D) и т- Д-> которые, вообще говоря,
различаются между собой. Это значит, что для функций распределения /Г1) не
существует единственного интегро-дифференциального уравнения. Чтобы
исключить эту неоднозначность, необходимо использовать дополнительные
условия, которые ограничивают возможный выбор функций /(^>. Таким
условием, используемым при выводе уравнения Больцмана для рх), является
условие молекулярного хаоса1).
Б. ФИЗИЧЕСКИЙ ВЫВОД УРАВНЕНИЯ БОЛЬЦМАНА
Рассмотрим смесь одноатомных газов, находящихся в неравновесном со-
стоянии. Предположим, что газ достаточно разрежен, так что для описания
свойств такого газа необходимо учитывать парные, а не тройные столкновения
молекул. Для общности предположим, что на молекулы /-го сорта действует
внешняя сила X,. Будем полагать, что X, — функция координат и времени,
не зависящая от скорости. Влияние сил, зависящих от скорости, рассматри-
вается несколько иным способом [2]. Предполагается, что внешние силы много
меньше сил, действующих на молекулы во время столкновения. Межмолеку-
лярные силы значительно превосходят силы тяжести и действуют только в
течение момента столкновения.
Как указывалось выше, свойства газа в неравновесном состоянии могут
быть выражены через функцию распределения, заданную в фазовом простран-
стве одной молекулы (/г-пространство). Каждая компонента смеси имеет функ-
цию распределения /ф(г, р„ f), определяемую таким образом, что вероятное
число молекул i-го сорта в элементе объема их около точки г, обладающих
импульсами в элементе dp, около р„ равно /ф(г, р„ ()йхд$[.
В гл. 2, посвященной статистической механике равновесных систем, по-
казано, что функция распределения fip для таких систем не зависит от времени
г) Более общий и строгий метод вывода газокинетических уравнений развит Н. Н.Бого-
любовым в работе «Проблемы динамической теории в статистической физике», М.—Л.,
1946. — Прим. ред.
356 Гл. 7. Кинетическая теория разреженных газов
и пространственной координаты и является функцией распределения Макс-
велла. Теперь нашей задачей является определение зависимости /()}(r, р, f)
от тех же переменных для неравновесных систем. Иными словами, мы хотим
определить характер потока фазовых точек в шестимерном фазовом простран-
стве, где каждая фазовая точка представляет одну молекулу и молекулы взаи-
модействуют между собой. Если бы взаимодействие между молекулами отсут-
ствовало, то поведение функции f}\r, р„ t) определялось уравнением Лиувилля
для точек в ^-пространстве. Мы покажем, что при учете взаимодействия в
уравнении Лиувилля появляется дополнительный член, который учитывает
столкновения молекул.
Выделим элемент объема dt dp,, расположенный около точки (г, р,). В этом
элементе объема имеется ffdr dp, фазовых точек, представляющих частицы
z-ro сорта. В отсутствие столкновений молекулы, соответствующие этим фазо-
вым точкам, перемещаются таким образом, что к моменту времени t + dt их
радиусы-векторы1) будут равны [г + (Р,М) dt ], а векторы импульса равны
[р, + X/ dt]. При отсутствии столкновений другие фазовые точки в эту область
попасть не могут, и поэтому
^] ) A.1)
Однако вследствие столкновений в газе не все фазовые точки по прошествии
времени dt из (г, р,) попадают в {[г + (Р,М) dt ], (р, + X, dt)}} так как
импульсы молекул, фазовые точки которых испытывают отклонения за счет
столкновений, изменяются. Некоторые фазовые точки, первоначально не на-
ходившиеся в (г, р,), попадают в {[г -f (P,M) dt], (p, + X,d/)} в результате
столкновений с другими молекулами. Обозначим число молекул i'-го сорта,
находящихся в элементе объема dx около точки г и выбывающих из области
значений импульсов от р, до р, + dp, вследствие столкновений с молекулами
/-го сорта в течение времени dt, через F^j/dr dp,- dt, а число молекул i'-го сорта,
которые вследствие столкновений с молекулами /-го сорта в течение времени
dt присоедийяются к группе точек, начавших свое движение из (г, р,), через
Г<#> dx dp, dt. Если в уравнении для потока фазовых точек учесть влияние столк-
новений, то оно приобретает вид
+ $[ dt}> № + Х< *]' I' + d']) dr*' =
= /р>(г, pz, t)dr dp, + 2 [ЛЯ - ЛЯ1 drdVtdt. A.2)
Левая часть полученного уравнения может быть разложена в ряд Тэйлора
около точки (г, р„ 0:
ik dt]> №'+x<d'J> р+dt])
t + ...jdrdp,. A.3)
Пользуясь уравнениями A.2) и A.3), получаем уравнение Больцмана
которое описывает скорость изменения во времени функции /ф. Полученное
уравнение имеет ту же форму, что и уравнение Лиувилля, за исключением
дополнительного члена столкновений в правой части.
Следует отметить, что в величинах Ffy и Fty не учитываются столкнове-
ния молекул со стенками. Как показано в гл. 11, столкновения молекул со стен-
ками учитываются в граничных условиях для гидродинамических уравнений.
г) Мы используем декартову систему координат.
§ 1. Функции распределения кинетической теории
357
Точное выражение для членов правой части уравнения Больцмана, назы-
ваемых интегралами столкновений, может быть найдено следующим образом.
Рассмотрим член Ftfdrdpidt, представляющий собой число молекул /-го
сорта, выбывающих из элемента фазового объема dr dp,- в течение времени dt
за счет столкновений с молекулами /-го сорта. Рассмотрим молекулу /-го сорта,
расположенную в точке г и имеющую импульс рг Нам необходимо найти
вероятность того, что эта молекула за время dt столкнется с молекулой /-го
сорта при прицельном расстоянии1) &, находящемся в интервале от Ъ до Ъ + db.
Если молекулу / рассматривать как неподвижную, то молекула / будет при-
ближаться к ней с относительным импульсом (ру — р,). Такой способ рассмот-
рения столкновения изображен на фиг. 111. Предполагается, что межмолеку-
лярные силы пренебрежимо малы на расстоянии, превышающем А, которое
мало по сравнению с длиной среднего свободного пробега молекулы.
Фиг. 111. Столкновение молекул /-го сорта с одной молекулой /-го сорта при параметре
столкновения, равном Ь.
Расстояние А равно такому межмолекулярному расстоянию, на котором практически необходимо учиты-
вать действие межмолекулярного потенциала. Любая молекула /-го сорта, находящаяся в начальный
момент времени между цилиндрами (с радиусами Ь и Ь + db и высотой gif dt), столкнется с молекулой / в
течение малого интервала времени dt.
Из фиг. Ill ясно, что любая молекула /-го сорта, находящаяся в секторе
цилиндрической оболочки, испытывает в течение времени dt столкновение с
молекулой I, характеризуемое прицельным расстоянием b и начальной относи-
тельной скоростью
A.5)
mj
mi
Вероятное число молекул /-го сорта, находящихся в этом секторе, равно
/j1}(r>Py>0 gijbdbdedt, A.6)
где
&] = &л = \%л\- A.7)
Общее число столкновений, испытываемое молекулой i'-го сорта с любыми
молекулами /-го сорта, можно получить суммированием столкновений, харак-
теризуемых любыми значениями b и е и любыми значениями относительных
скоростей g;7. В результате получим
A.8)
^Геометрия столкновения рассматривается в гл. 1, §5.
358 Гл. 7. Кинетическая теория разреженных газов
Так как вероятное число молекул r-го сорта в элементе объема dx около *
точки г, обладающих импульсами в пределах от р, до р, + dp,, равно
/ф(г, piyi)dxdpb то
Г^drdp, df = dxdP/ Л JJJ/j1}(r, P;, 0 #>(r, p,, t) glJ b db de dpj. A.9)
Отсюда
) jjjWW A.10)
дает вклад в Э/ф/Э/, учитывающий выбывание молекул из группы / за счет
столкновений с молекулами /'. В последнем выражении через /ф и /<j> со-
кращенно обозначены /ф(г, р„ t) и /ф(г, ру, /) соответственно.
Другая часть интеграла столкновений Г<^> может быть найдена аналогич-
ным образом. Рассмотрим столкновение молекул, обладающих импульсами
р, и ру, характеризуемые прицельным расстоянием Ь. Импульсы молекул после
столкновения обозначим через р- и р}. Величины импульсов после столкновения
определяются величинами импульсов до столкновения, параметром столкно-
вения и характером межмолекулярных сил. Для сферически симметричной
потенциальной функции из закона сохранения энергии следует, что абсолют-
ные величины относительной скорости до и после столкновения равны, т. е\
Из закона сохранения момента количества движения следует, что равны
также прицельные расстояния до и после столкновения, т. е.
b = b'. A.12)
Кроме того, вследствие симметрии уравнений динамики можно сказать,
что в результате столкновения, характеризуемого прицельным расстоянием Ь,
молекулы, обладающие импульсами р\ и р;', приобретают импульсы р, и ру.
Методами, аналогичными использованным выше, можно показать, что
вероятное число столкновений в элементе объема dx около точки г за время
dty в результате которых молекулы приобретают импульсы в пределах от р;
до pi + dph равно
г(;> dx dP/ dt = dx dp] dt J J J>(r, p;, о ft» (г, р;, о gffj v db' de dp). (i. 13)
Здесь величины со штрихами являются функциями величин без штрихов.
Вид этих функций определяется характером межмолекулярных сил. Прямым
следствием теоремы Лиувилля является условие1)
dPidp;=dpidPj. A.14)
Используя это условие, а также равенства A.11) и A.12), получаем
A.15)
где через /ф' и /(}>' обозначены /ф(г, pj., t) и fj\xy py, t) соответственно. Величина
Гф дает вклад в Э/ф/сЗ'» учитывающий увеличение числа молекул рассматри-
ваемой группы за счет столкновений.
Подставляя выражение для Г<?> и Fty в уравнение Больцмана, записан-
ное в виде A.4), получаем уравнение для /ф(г, р„ f)
= Sjjj (/}»' /jiv - № /j4) gtj b db de dpj. A.16)
x) Преобразование, переводящее одну точку фазового пространства в другую, находя-
щуюся на той же траектории, что и первая точка, и удаленную от нее на расстояние, для
прохождения которого необходим фиксированный интервал времени, является контактным
преобразованием. Теорема сохранения объема в фазовом пространстве, обсуждаемая в
гл.2. § 1, тесно связана с теоремой, которая устанавливает, что якобиан контактного преоб-
разования равен единице. Так как предполагается, что векторы положения двух молекул
не изменяются при столкновении, то преобразование (р/, pj) -> (р{, pj) является контактным
и его якобиан равен единице.
§ 1. Функции распределения кинетической теории 359
Это важное уравнение называется интегро-дифференциальным уравнением
Больцмана для функции распределения. Аналогичные уравнения могут быть
написаны для всех компонент газовой смеси. В каждом из этих уравнений в
правые части под знак интеграла входят функции распределения всех
компонент. Следует помнить, что эти интегралы неявным образом зависят от
законов сил, действующих между молекулами. Функции /<}>' и /<}>' являются
функциями величин р- и pj, которые могут быть найдены на основе законов
динамики при известных р,, py, Ь и при известной потенциальной энергии
межмолекулярного взаимодействия.
Изложенный вывод уравнения Больцмана прост и дает непосредственно
физическую интерпретацию членов уравнения. Однако в нем имеются логи-
ческие трудности, связанные с конечностью размеров молекул и конечностью
времени столкновения1). Кирквуд [4] вывел уравнение Больцмана более стро-
гим путем непосредственно из теоремы Лиувилля2).
В. ВЫВОД УРАВНЕНИЯ БОЛЬЦМАНА ИЗ ТЕОРЕМЫ
ЛИУВИЛЛЯ
Газ, состоящий из N молекул, может быть представлен ансамблем, описы-
ваемым функцией распределения fN) (гы, рм, t) в у-пространстве 6N измерений.
Согласно уравнению Лиувилля, полученному в гл. 1, § 4, изменение функции
распределения fN) со временем определяется уравнением
Э/<'У) ( дН 8/(N) Ч Г дН df(N) \ __
di •" у Э»рлг * 8rN J (~ЭглГ # ~~8pv~~J (*•!¦/
или
8/(w) , у ( 1 ( 9/(v) ч / Э/(лО ^ _
где тк — масса молекулы к ; ?к — сила, действующая на молекулу к со сто-
роны всех остальных молекул ; Хк — сила внешнего поля, действующая на
молекулу к.
Как было сказано выше, макроскопическое поведение газа обычно описы-
вается с достаточной точностью с помощью функций распределения низшего
порядка. Например, макроскопическое поведение газа достаточно малой плот-
ности описывается набором функций распределения /ф. Эти функции опреде-
ляются как интегралы от fN) по координатам и импульсам всех молекул, за
исключением одной. Благодаря симметрии fN) функция /(р зависит только от
сорта молекул, отмечаемого индексом /, и не зависит от того, какую именно
молекулу данного сорта мы выбираем.
Уравнение для /ф может быть получено из уравнения Лиувилля A.18)
путем интегрирования его по координатам и импульсам (N — 1) молекулы.
Выполняя такое интегрирование и используя тот факт, что /ф исчезает,
когда | р^ | -> оо, а также на стенках сосуда, получаем
о-м>
Это уравнение само по себе не определяет поведения функции /ф. Как
указывалось выше, при любом уменьшении подробности описания системы
г) См. [3, стр. 218].
2) Авторы книги, по-видимому, не зна комы с более ранней работой Н. Н. Боголюбова
(«Проблемы динамической теории в статистической физике», М.—Л., 1946), в которой вопрос
о выводе уравнения Больцмана рассмотрен наиболее строго. — Прим. ред.
+ i kT) + («."?) - -ЯК ?)*.* 0.20)
360 Гл. 7. Кинетическая теория разреженных газов
необходимо вводить условия, ограничивающие характер рассматриваемой
системы. В данном случае для получения уравнения Больцмана необходимо
ввести концепцию «молекулярного хаоса»1).
Кирквуд [4 ] показал, что при выводе уравнения Больцмана неявно исполь-
зуется предположение о том, что функция распределения f()\t) не изменяется
заметно в течение столкновения. Это может быть показано следующим образом.
Для газа, состоящего из одной компоненты, межмолекулярные силы которого
являются парными, уравнение A.19) приводится к виду
Т
Если межмолекулярные силы короткодействующие, то диаметр столкновения
г0 можно определить, используя условие, что F12 = 0, когда \тг — r21 ^> г0.
При этом весь вклад в интеграл уравнения A.20) вносится областью, где
I ri — r21 < го- Принцип молекулярного хаоса предполагает, что за пределами
сферы взаимодействия
m) = A1\t)f(i\t), |г1-г,|>г0. A.21)
Но внутри сферы взаимодействия точный вид бинарной функции распре-
деления fH(t) неизвестен. Рассмотрим способ, с помощью которого можно в
принципе вычислить /<8@-
Если пренебречь тройными столкновениями, то в фазовом пространстве
двух частиц через точку (гъ рх; г2, р2) проходит только одна траектория. Если
система в момент времени t находится в точке (т19 рх; г2, р2), то, следуя по траек-
тории в обратном направлении, можно установить, что в момент времени t — dt
система была в определенной точке (ri, r2; pi, p2). Определим интервал времени
Щги Г2; Pi, Рг 5 0 таким образом, что | ri — г21 = г0. В момент времени t — dt
/(Ж r2; pi, p2, t - dt) = /^(ri, pi, t - dt) f?\t'2, p2, t - dt).
Отсюда следует, что
h; Pi, P2, 0 = /^(rl, Pi, * - K) /<№, p2, / - dt). A.22)
Таким образом, для каждого значения (г2, р2) под интегралом уравнения
A.20) бинарной функции распределения соответствует функция распределения
для одной частицы в момент времени t — dt. Для каждой точки (г2, р2) интер-
валы времени dt, имеющие порядок времени столкновения и малые по срав-
нению с интервалами времени, характерными для макроскопических измере-
ний, различны.
Кирквуд устранил разницу в dt для различных точек (г2, р2), усреднив
уравнение A.19) по интервалу времени, несколько большему времени столк-
новения. Усреднение по времени ниже отмечается чертой над усредняемой
величиной, так что усредненная по времени функция распределения обозна-
чается в виде /ф. Усредняя таким образом уравнение A.20) по времени, Кирк-
вуд получил
mi\ri \ Ьт ;^Г' Эр,-
О-
Это уравнение совпадало бы с уравнением Больцмана, полученным в
предыдущем разделе, если бы /ф/ф =¦ /(Р/(Р- Последнее выполняется при
условии, что функции распределения не изменяются заметным образом на
"""" г) См. примечание на стр. 355. — Прим. ред.
§ 2. Уравнения переноса 361
интервале времени т0, сравнимом с временем столкновения, по которому произ-
ведено усреднение. В этом можно убедиться следующим путем :
Wfp=± / т + г) ш
} ) (JL J
-т,/2
/}« fjD = (-f } f\4t + г) их) (JL J #)(, +
° /2 °/2
4
Vv "t" ~24~ ^П 0/2 Г /; Qt2 J \l^HJ
В обычном выводе уравнения Больцмана величиной Ы пренебрегают и
аргументируют это тем, что ошибка, вносимая таким пренебрежением, срав-
нима с ошибкой вследствие пренебрежения в функции распределения величи-
ной расстояния между центрами сталкивающихся молекул. Ошибками, вно-
симыми обоими упомянутыми пренебрежениями, можно пренебречь при усло-
вии, что функции распределения не изменяются заметным образом за время
порядка времени столкновения или на расстояниях порядка молекулярного
диаметра.
Г. РАСПРЕДЕЛЕНИЕ ПО СКОРОСТЯМ
При решении большинства проблем кинетической теории в качестве пере-
менной удобнее пользоваться скоростью, чем импульсом.
Ниже в этой главе вместо функции распределения /(}}(г, р„ t) в фазовом
пространстве мы будем пользоваться функцией распределения в пространстве
скоростей /,(r, V/, t). Уравнение Больцмана для функции распределения в
пространстве скоростей имеет вид1)
t + (V<- ТЕ") + i(X<- щ) = fJlJirirj-Wujbdbdstoj. A.25)
Это уравнение является основой для анализа явлений переноса в газах.
§ 2. УРАВНЕНИЯ ПЕРЕНОСА
Гидродинамические уравнения переноса — уравнения сохранения массы»
импульса и энергии — могут быть получены непосредственно из уравнения
Больцмана. При выводе этих уравнений появляются определенные величины,
выражающиеся через функцию распределения. Можно показать, что они пред-
ставляют собой потоки массы, импульса, энергии и непосредственно связаны
с диффузионной скоростью, тензором давлений и потоком тепла. В настоящем
параграфе выводятся соотношения между указанными величинами. Прибли-
женное решение уравнения Больцмана и вычисление потоков проводится в
следующих параграфах. Мы начнем этот параграф с ряда определений раз-
личных скоростей, необходимых при обсуждении гидродинамических урав-
нений.
х) Мы опускаем индекс 1 везде, где функция распределения выражается в виде функции
от скорости, но не от импульса.
352
Гл. 7. Кинетическая теория разреженных газов
А. СКОРОСТИ МОЛЕКУЛ И СКОРОСТИ ПОТОКА1)
Линейную скорость молекулы /-го сорта относительно неподвижной сис-
темы координат обозначим через v;, а компоненты этой скорости — через
vJX, Vjy, vJz. Абсолютную величину линейной скорости vy- обозначим через vj.
Среднюю скорость молекулы Vy компоненты /, имеющей плотность числа
частиц njy определим следующим образом :
B.1)
Средняя скорость является функцией координат и времени и представляет
собой макроскопическую скорость потока компоненты /'. Черта сверху озна-
чает среднюю величину функции скорости. Например,
B.2)
есть среднее значение величины a(v;).
Средняя массовая скорость2) определяется следующим образом :
где e(r, t) — суммарная плотность газа в некоторой точке
B.4)
Средняя массовая скорость является взвешенной средней величиной, так
как каждая молекула дает вклад в эту скорость, пропорциональный ее массе.
Импульс единицы объема газа оказывается тем же самым, как если бы все
молекулы этого объема перемещались со средней массовой скоростью v0. Эта
скорость обычно называется скоростью потока или скоростью течения.
Тепловая скорость молекулы /-го сорта определяется как скорость моле-
кулы относительно системы координат, движущейся со средней массовой ско-
ростью v0
V,.(v,,r,0 = v;.-vo. B.5)
*) Поскольку обозначения для скоростей, используемые в настоящей книге, отличаются
от обозначений, используемых в книге Чепмена и Каулинга [2], мы приводим здесь список
этих обо значений:
Скорость
Скорость молекулы
Компоненты скорости молекулы
Средняя массовая скорость
Средняя числовая скорость
Тепловая скорость молекулы
Диффузионная скорость
Приведенная скорость
Начальная относительная скорость ...
Приведенная начальная относительная
скорость •.
По Чепмену
и Каулингу
с
u,v,w
со
с
С
Использован-
ные в настоя-
щей книге
vxtvytvz
ш
V
V
W
g
2) В гл. 11, где излагается гидродинамика жидкостей, индекс 0 у символа v0 для средней
массовой скорости опущен. Это не может привести к недоразумениям, так как в гл. 11 ско-
рость молекулы v не употребляется.
§ 2. Уравнения переноса 363
Диффузионная скорость химической компоненты / есть скорость потока
молекул / относительно системы координат, движущейся со средней массовой
скоростью газа
Vy(r,0 = Vy-vo. B.6)
Очевидно, что диффузионная скорость есть средняя тепловая скорость и на
основе определения vy и Vy может быть записана в виде
B.7)
Отметим, что v0 является функцией положения и времени, но не Vy. Из опре-
деления диффузионной скорости и средней массовой скорости следует
2 rij raj Vy = 2 nj Щ (Vy — v0) = 0. B.8)
Иногда используется иная средняя скорость — числовая средняя скорость.
Она определяется следующим образом :
uj Сг ft = "V tx • v • B Q)
П у
где п = 2 nj— общая плотность числа частиц смеси. Легко показать, что эта
скорость просто связана со средней массовой скоростью, причем
j
Для упрощения записи некоторых выражений в последующих параграфах
часто используется приведенная скорость Wy. Она определяется следующим
образом :
w/=^Vy, B.11)
Модуль Wy обозначается через Wy.
Помимо определенных здесь скоростей, в последующих параграфах упо-
требляются еще две другие скорости — начальная относительная скорость g/y
двух сталкивающихся молекул, определенная по A.5), и приведенная началь-
ная относительная скорость у/у
yif/. <2Л2>
где fXtj — приведенная масса двух сталкивающихся молекул.
Температуру в кинетической теории мы определим через усредненную
по всем молекулам кинетическую энергию теплового движения1):
B.13)
Понятие температуры вводится в гл. 2, § 4 на основе термодинамических
соображений. Данное там термодинамическое определение температуры имеет
смысл только для равновесных систем. Настоящее более общее определение
температуры согласуется с термодинамическим в этом предельном случае.
х) Кинетическая энергия молекул вычисляется в системе координат, движущейся со
скоростью v0. — Прим. ред.
354
Гл. 7. Кинетическая теория разреженных газов
Б. ВЕКТОРЫ ПОТОКОВ1)
В газе, находящемся в неравновесном состоянии, могут существовать гра-
диенты величин, характеризующих макроскопические физические свойства
системы : градиенты концентрации, средней массовой скорости и температуры.
Наличие этих градиентов является причиной молекулярного переноса в газе
массы rrij, импульса ffiyVy и кинетической энергии х/2 rnjVj. Так как механизмы
переноса каждой из этих молекулярных величин могут быть рассмотрены с
единой точки зрения, обозначим эти величины, как это было сделано в гл. 1,
§ 2, через тру. Напомним, что эти величины уже рассматривались в гл. 1, § 5
b связи с их важностью как сумматорных инвариантов столкновений.
Рассмотрим перенос величин xpj с микроскопической точки зрения. Выде-
лим в газе малый элемент поверхности dS, перемещающийся со скоростью,
равной средней массовой скорости газа v0. Ориентацию элемента поверхности
будем характеризовать единичным вектором п, направленным по нормали к
поверхности. В соответствии с определением B.5)
скорость молекул /-го сорта относительно элемента
поверхности dS равна Vy. Все молекулы /-го сорта,
имеющие скорость2) Vy и пересекающие dS в течение
времени dt, должны первоначально находиться в
цилиндре с основанием dS и образующей, парал-
лельной Vy и равной | Vy | dt (фиг. 112). Этот
цилиндр имеет объем (п • Vy) dS dt. Число молекул
в единице объема, имеющих скорость Vy, равно
/у dVj. Отсюда число молекул, которые пересекают
поверхность dS в течение времени dt> равно
fj(n-Vj)dVjdSdt. B.14)
Если с каждой молекулой через dS перено-
сится величина y>j, зависящая от Vy, то
Vjfj(n-Vj)dVjdSdt B.15)
представляет собой количество этой величины,
переносимое через dS в течение времени dt всеми
молекулами, имеющими скорости в области dVj
около Vy. Плотность потока величины ipj (т. е. общее
количество y)j, которое переносится молекулами
выделенной группы через единицу поверхности в
единицу времени) равна
B.16)
Фиг. 112. Цилиндр, содер-
жащий все молекулы /-го
сорта, обладающие скоро-
стью Vy, которые в течение
времени dt пересекут
поверхность dS.
Плотность общего потока через элементарную поверхность можно получить
путем интегрирования B.16) по всем значениям скоростей. В результате получим
\vjfj(nj *Y/)rfV; = (n ' f VjfjVjdVj) = (n# T/)» B-l7)
где
Vj = \y>jfjVjdVj B.18)
называется вектором плотности потока величины уу. Физический смысл
вектора плотности потока состоит в том, что компонента его в любом направ-
лении п представляет собой плотность потока соответствующей физической
величины через поверхность, нормальную к п.
Рассмотрим векторы плотностей потоков, характеризующие перенос
массы, импульса и кинетической энергии.
х) Элементарное изложение вопроса о векторах потоков дано в гл. 1, § 2, в связи с изло-
женной там элементарной кинетической теорией газов.
2) Или, более точно, молекулы, имеющие скорости в области dWj около Vy.
§ 2. Уравнения переноса 365
1. Перенос массы. Если у;- = Щ то1)
1 dV> = ^-шу V,- = jy B.19)
представляет собой вектор плотности потока массы.
2. Перенос импульса. Если уу = mj Vjx, то
*у = Щ J Vyx Vy /у dVy = лу my V^Vj B.20)
означает вектор плотности потока, характеризующий перенос х-компоненты
импульса (относительно v0). Этот вектор имеет компоненты, пропорциональные
VjxVjx, VjxVjy и VJXVjZ. Аналогичные векторы потоков могут быть получены
для у- и г-компонент импульса. Указанные три вектора потока полностью
характеризуют перенос импульса. Девять компонент этих трех векторов обра-
зуют симметричный тензор р второго ранга :
= mjjfj VJx VJxdVj =nj
(Pj)xy = (Pj)yx = Щ Щ Vjx VJy. B.21)
Символически тензор ру, характеризующий парциальное давление /-й химиче-
ской составляющей газа, можно записать в виде
Pj = njmjVjVj. B.22)
Сумма тензоров парциальных давлений по всем сортам молекул газа образует
тензор давлений смеси2):
j J
Физический смысл тензора давлений состоит в том, что он представляет
собой поток импульса в газе. Отдельные компоненты тензора давлений имеют
следующее значение : диагональные элементы рхх, рууу ргг равны нормальным
напряжениям ; иначе говоря, рхх равно силе, действующей на единицу пло-
щади поверхности, нормальной к направлению х, в направлении х. Недиаго-
нальные элементы тензора давлений представляют собой сдвиговые напря-
жения, например рух равно силе, действующей на единицу площади поверх-
ности, перпендикулярной направлению у, в направлении х. Компоненты
тензора давлений рух, р^у р^, в сумме дающие результирующую силу ру,
действующую на единицу поверхности, перпендикулярной направлению у,
показаны на фиг. 113. В тензор давлений в качестве компонент входят напря-
жения или давления, которые могут быть измерены приборами, перемещаю-
щимися в потоке со скоростью v0. Давление, измеренное в потоке с помощью
неподвижного манометра, зависит от v0 и от ориентации манометра.
В § 3 показано, что в системе, находящейся в равновесии, сдвиговые
напряжения равны нулю, а нормальные напряжения равны между собой.
В этом случае сила, действующая на любой выделенный в газе элемент поверх-
ности, постоянна и независимо от ориентации поверхности перпендикулярна
к ней, т. е.
Рхх = Руу = Pzz = Р>
Рху = Pyz = • •. = 0, B.24)
где р — гидростатическое давление для равновесной системы.
г) Необходимо отметить, что при вычислении средних величин интегрирование по V/
эквивалентно интегрированию по V/, так как V/ отличается от V/ на постоянную величину и
интегрирование ведется по всем значениям скоростей.
2) Введенный здесь тензор давлений отличается от обычно рассматриваемого в механике
сплошных сред тензора напряжений только знаком. — Прим. ред.
366
Гл. 7. Кинетическая теория разреженных газов
Z Ру2
L ¦'
pf-v
A
'fll
Фиг. 113. Значение компонент тензора давлений.
Показан малый элемент поверхности, нормаль к которой совпадает с направлением оси Y. На единицу
площади выделенного в газе элемента поверхности действует общая сила ру. Компонентами этой силы явля-
ются рп (нормальное напряжение) и рух и руг (сдвиговые напряжения). Девять компонент такого типа
образуют тензор давлений.
3. Перенос кинетической энергии. Если y>j =ll2mjVj> T0
= 4j B.25)
есть вектор потока, характеризующий перенос кинетической энергии молеку-
лами /-го сорта. Сумма таких векторов по всем компонентам смеси газа равна
вектору плотности потока тепла:
B.26)
Физический смысл компонент qX} qy, qz вектора плотности потока тепла q за-
ключается в том, что они представляют собой плотности потоков кинетической
энергии в направлениях х, у и z соответственно.
Необходимо отметить, что приведенные здесь выражения для векторов
потоков справедливы только для разреженных газов. Для более плотных
газов в этих выражениях появляются дополнительные члены, учитывающие
перенос энергии и импульса за счет столкновений. При столкновении двух
молекул от одной молекулы к другой почти мгновенно передается некоторое
количество энергии и импульса. При этом если центры двух сталкивающихся
молекул находятся на противоположных сторонах от элемента поверхности
dSy то результирующий перенос импульса или энергии через поверхность dS
за счет столкновений будет вносить дополнительный вклад в потоки, который
не учитывался в приведенных выше выражениях1).
Указанные ограничения применимости выражений для потоков не сущест-
венны для настоящей главы, так как влияние переноса импульса или энергии
за счет столкновений в условиях применимости используемого нами уравнения
Больцмана пренебрежимо мало.
В. ОБЩЕЕ УРАВНЕНИЕ ПЕРЕНОСА2)
Основные гидродинамические уравнения непрерывности, движения и
баланса энергии могут быть получены из уравнения Больцмана без определе-
ния вида функций распределения /,. Умножая уравнение Больцмана для /-й
г) Детальный анализ этого вопроса проведен Кирквудом [5 ] (см. гл. 9, § 3 и 4).
2) Здесь приведен вывод уравнений переноса для случая разреженного газа, однако
вид этих уравнений сохраняется также для плотных газов и жидкостей (см. гл. 9, § 4).
§ 2. Уравнения переноса 367
компоненты [уравнение A.25)] на величину %ри соответствующую молекулам
1-го сорта, и интегрируя результат по всем значениям у,-, получаем
= 2 \\\\ Wiif'i // - /, /,) gij b db ds dv, dvj. B.27)
Три члена в левой части этого уравнения могут быть просто преобразованы :
B.28)
B.29)
Последние два равенства записаны только для х-компонент1). В равенстве
B.30) первый член, появляющийся при интегрировании по частям, исчезает,
так как предполагается, что /,у,- достаточно быстро стремится к нулю при воз-
растании v(. Пользуясь B.28) — B.30), получаем
Эг i l lJ l\dt
Х'
=^ ЯЯ
Это уравнение известно, как общее уравнение переноса Энскога для
величины y>if соответствующей молекулам i-го сорта. Суммируя уравнения
B.31) по всем /, получаем уравнение переноса \р для смеси.
Г. ИСЧЕЗНОВЕНИЕ ИНТЕГРАЛОВ СТОЛКНОВЕНИЙ ДЛЯ СУММАТОРНЫХ
ИНВАРИАНТОВ
Уравнения типа B.31) для любых %pt мало полезны вследствие наличия в
правой части этих уравнений очень сложных интегралов. Однако если щ —
масса 1-й молекулы, то, используя тот факт, что масса отдельной молекулы
не изменяется при столкновении, можно показать, что
ЯЯ ш<</'< Л - h /у) ft;b <Н> ** <*i <*j = 0- B.32)
Далее, исходя из того, что масса, импульс и энергия молекул сохраняются
при столкновении, можно показать, что при y>if равном ть m,V, или х/2 т№>
(fl fj ~ ft fj) iu b <*b d? &i d*J = 0. B.33)
г) Величина щ может зависеть от г и t через vo(r, /)• Величины г, v, и / являются незави-
симыми переменными.
368 Гл. 7. Кинетическая теория разреженных газов
Эти тождества дают возможность упростить общее уравнение переноса.
Справедливость последних двух тождеств может быть установлена сле-
дующим образом. Интеграл
ШЬ(/; Я - Ш Subdbdedv,dwj B.34)
равен интегралу
JJJJ ?>Шу - /; fj) gij V dV de dv\ dv;y B.35)
написанному для обратных столкновений. В § 1 показано, что
gtj = Su> b = b'> rfv- dvJ = Щ rfvyt B.36)
поэтому интеграл B.35) может быть запии" н клле
,. B.37)
Так как интегралы B.34) и B.37) равны, то каждый и^ них равен полусумме
обоих интегралов, т. е.
Vj. B.38)
Для случая щ = т( из B.38) немедленно следует справедливость B.32).
Равенство щ — у\ = 0 выражает тот факт, что массы отдельных молекул не
изменяются во время столкновений.
Суммируя B.38) по i и / и пользуясь тем, что немые индексы взаимоза-
меняемы, получаем
= 4 2 ЯЯ ^ ~ **> W К - fi ^ «/у * ^ de dv, rfyy. B.39)
Таким образом,
Wj. B.40)
По определению инвариантов столкновений [см. выражение E.5) гл. 1]
сумма (tpi + tpj — \p'i — tp'j) равна нулю. Следовательно, тождество B.33)
соблюдается при y>h равном miy т(\( и 1/гш^?-
Д. ЧАСТНЫЙ ВИД УРАВНЕНИЙ ПЕРЕНОСА
После того как была доказана справедливость тождеств B.32) и B.33),
можно вывести уравнение переноса для конкретных молекулярных величин.
Это можно сделать, полагая в уравнении B.31) \pt равным miy гп,У, и 1l2^iV}
и затем суммируя полученные уравнения по всем значениям /. Подставляя в
B.31) у)( = mi и пользуясь тем, что интеграл столкновений, как было показано
выше, при этом равен нулю, получаем
§. 2. Уравнения переноса 369
или, переходя к диффузионным скоростям,
)О. B.42),
Последние два уравнения представляют собой две формы уравнения не-
прерывности для *-й компоненты. Если каждое из_уравнений B.42) умножить
на т( и затем сложить их, то, поскольку 2 п1тУг = °> будем иметь
О. 2.43)
Это — уравнение непрерывности смеси газов.
Если подставить в B.31) \pt = /n,-V/ и просуммировать полученные урав-
нения по всем значениям /, то интеграл столкновений в правой части уравнения
исчезнет и в результате мы получим
a B.44)
Это уравнение можно упростить, используя соотношение между диффу-
зионными скоростями B.8) и определение тензора давлений B.23). Следуег
отметить, что при дифференцировании г и у,- рассматриваются как независимые
переменные. После некоторых преобразований получим уравнение движения
газа
?( N H )i <2-45>
Подставляя в общее уравнение переноса V/^Vi771/^ и выполняя
преобразования, аналогичные проделанным при выводе предыдущих урав-
нений, получаем
Уравнение B.46) может быть преобразовано такими же способами, какие
были применены при выводе уравнения движения. Вводя тензор давлений,
определяемый выражением B.23), а также вектор плотности потока тепла,
определяемый выражением B.26), получаем уравнение баланса энергии?
: i Vo) - 2 ni № • Vi) = 0, B.47)
где (J^oct.) — внутренняя энергия газа на единицу массы, равная энергии
поступательного движения молекул1)
#<пост.) = ±2>± ntmt Vf. B.48)
Эта величина равна полной энергии единицы массы газа в системе коор-
динат, движущейся со средней массовой скоростью v0 (кинетическая энергия
*) Для молекул, не обладающих, внутренними степенями свободы, т. е. для молекул
рассматриваемого типа, эта энергия представляет собой единственный вклад во внутреннюю
энергию. В общем случае, однако, в выражение для внутренней энергии входят еще и другие
члены. Этот вопрос рассматривается в § б настоящей главы.
370 Гл. 7. Кинетическая теория разреженных газов
потока в эту энергию не входит), исключая потенциальную энергию газа во
внешнем поле1). Используя уравнение непрерывности B.43), уравнение баланса
нергии B.47) можно записать в другом виде :
э л
0$(пост.) / 80(пост.) \ f д \ ( д \ —
+ e(v^VJ) №«) (Р: ) + ^(XV) B.49)
Используя определения #(пост-) и Г, это уравнение можно преобразовать к виду
+ 2 Пг (X, • V,) + 4 кТ (-?- • 2 ni V/)- B.50)
Последние два уравнения представляют собой два вида уравнения баланса
энергии. Как показано в § б настоящей главы, уравнение B.49) является общим
и если в нем О(пост) заменить на О, то оно справедливо даже для реагирующих:
смесей многоатомных молекул. Уравнение B.50), однако, справедливо только
для нереагирующих смесей, которые состоят из молекул, не обладающих внут-
ренними степенями свободы.
§ 3. РЕШЕНИЕ УРАВНЕНИЯ БОЛЬЦМАНА
МЕТОДОМ ЭНСКОГА
Вопросу получения приближенного решения уравнения Больцмана посвя-
щен ряд работ. Мы рассмотрим здесь метод возмущений Энскога, представляю-
щий собой видоизменение метода Гильберта. Рассматриваемый метод последо-
вательных приближений в принципе может быть распространен на системы,
в которых градиенты термодинамических величин велики. В нулевом прибли-
жении функцией распределения является локальная функция распределения
Максвелла, а получаемые при этом уравнения переноса являются уравнениями
Эйлера. В следующем, первом приближении получаются уравнения Навье—
Стокса2), во втором приближении — уравнения Барнетта. В более высоких
приближениях получаются более сложные уравнения, в которые входят век-
торы потоков, зависящие от высших производных, от термодинамических
величин и от высших степеней низших производных. Результаты, получаемые
в более высоких приближениях, используются редко.
Газ, находящийся в любом начальном состоянии и не подвергающийся в
течение достаточно продолжительного времени внешним возмущающим воз-
действиям, в конце концов достигает некоторого стационарного состояния.
Если система адиабатически изолирована и не подвергается воздействию внеш-
них сил, то таким стационарным состоянием является однородное состояние,
в котором все функции распределения являются максвелловскими. Доказа-
тельство того, что равновесными функциями распределения являются макс-
велловские функции (Н-теорема), приводится в разд. А. Остальные разделы
настоящего параграфа посвящены изложению решения уравнения Больц-
мана методом Энскога; а также подробному обсуждению результатов, полу-
х) Некоторые авторы записывают уравнение баланса энергии таким образом, что в
энергию входят дополнительно либо кинетическая энергия, связанная с движением газа
со средней массовой скоростью v0, либо потенциальная энергия газа во внешнем поле сил,
либо и то и другое. Определение, данное в настоящем параграфе, будет использоваться далее
во всей книге.
2) Уравнения Навье—Стокса применимы к системам с малыми градиентами величин,
характеризующих макроскопические свойства системы, т. е. для систем, в которых эти вели-
чины не изменяются заметно на расстоянии, равном средней длине свободного пробега мо-
лекулы.
§ 3. Решение уравнения Больцмана методом Энскога 371
ченных по методу Энскога в первом приближении. Появляющаяся при исполь-
зовании этого метода система интегральных уравнений решается вариацион-
ным методом, что приводит к получению быстросходящихся рядов.
Недавно Грэд развил новый метод решения уравнения Больцмана, который
является более мощным методом изучения систем, находящихся в состояниях,
далеких от равновесного. В первом приближении его результаты совпадают
с результатами Энскога, однако в высших приближениях появляются расхож-
дения. Метод Грэда излагается в § 5.
А. Я ТЕОРЕМА (РЕШЕНИЕ ДЛЯ СЛУЧАЯ РАВНОВЕСНОГО
СОСТОЯНИЯ СИСТЕМЫ)
Функции распределения, описывающие поведение газовой смеси, являются
решением системы уравнений Больцмана для каждой компоненты смеси. Из
уравнений, полученных в § 1, приведенных в конечном счете к виду A.25),
найдем, что в отсутствие внешних сил для системы, находящейся в однородном
состоянии, скорость изменения функций распределения /, со временем дается
уравнениями
Ж =^Jtt(f'tfj-fifj)gubdbdedVj. C.1)
То, что функции распределения /, стремятся в пределе к максвелловским функ-
циям распределения, очевидно из статистической механики равновесных сис-
тем, изложенной в гл. 2. Однако интересно показать, что уравнение Больцмана
приводит к тем же функциям распределения равновесных систем.
Для анализа решения для случая равновесия удобно ввести функцию
H(f), определяемую следующим образом:
Я (/) = 2 J h (v,, 0 In /, (у,, 0 dvt. C.2)
Эта функция является обобщением энтропии, определенной в гл. 2 (см. задачу
12 в конце главы). Данное здесь доказательство Н-теоремы представляет собой
частный случай общего доказательства (проводимого методами статистической
механики) второго закона термодинамики. Дифференцируя H(t) и используя
уравнение Больцмана для dfjdt, получаем
vy. C.3)
Интеграл в правой части этого уравнения может быть симметризован методами,
описанными в § 2, что дает
¦Ж = -4 ЯЯ МЙ^'Я"t^Sijbdbdedv.dyj. C.4)
Каждая из подынтегральных функций имеет вид (х — у) In (х/у), где х = /{/J
и у = /,/у. Если х>у, то (х—у) и In (х/у) положительны; если х<у, то (х — у)
и In (х/у) отрицательны. Поэтому подынтегральная функция каждого из ин-
тегралов в правой части уравнения C.4) всегда положительна или равна
нулю. Отсюда dHjdt отрицательна или равна нулю, так что H(t) никогда не
может увеличиваться. Из определения C.2) следует, что H(t) ограничена и
приближается к определенному пределу1) для больших значений /. В этом
*) Для простоты вместо смеси рассмотрим однокомпонентную систему. Функция Я может
быть равна —«э, только если интеграл (а) = — / In / dv расходится. Если / убывает быстрее,
чем e-1l^v^ikri то очевидно, что интеграл (а) сходится. Если / убывает медленнее, чем
e-y2mv*/kTf T0 интеграл (а) все же сходится по следующей причине. Подынтегральная функ-
ция — / In / в последнем случае в пределе меньше 1/гпн?1. Однако в любом случае интеграл
(b) = !/2 mtPfdv, соответствующий полной кинетической энергии молекул, должен схо-
диться. Чепмен и Каулинг рассматривают этот вопрос более подробно [2, стр. 70 ].
372 Гл. 7. Кинетическая теория разреженных газов
пределе функции распределения таковы, что подынтегральная функция каж-
дого из интегралов в правой части уравнения C.4) тождественно равна нулю,
т. е. при равновесии
l/ l/ ln/J + ln/J. C.5)
Поэтому логарифмы функций распределения являются сумматорными инва-
риантами молекулярных столкновений.
Можно показать, что любой сумматорный инвариант является линейной
комбинацией трех инвариантов столкновений, рассмотренных в гл. 1, § 5 :
массы ть импульса тр{ и кинетической энергии 112гпрЬ Таким образом, в рав-
новесии наиболее общее выражение для In/, имеет вид
In /, = at m, + (b • (m, v,)) + с [±- mt vf), C.6)
где ah b, с — постоянные, зависящие (через начальные функции распределения)
от полного числа молекул /-го сорта полного импульса и полной энергии систе-
мы. Удобно записать это выражение через физические параметры : плотность
числа частиц щ молекул /-го сорта, среднюю массовую скорость v0, опреде-
ляемую выражением B.3), и температуру Т, определяемую выражением B.13).
При этом функция распределения для системы, находящейся в равновесии,
приобретает вид
где V/ = (v, — v0) — тепловая скорость, определяемая по B.5).
Для систем, находящихся в равновесном состоянии, /, не зависит от вре-
мени, вследствие чего правая часть уравнения C.1) равна нулю. Равенство
нулю правой части уравнения C.1) выражает собой состояние равновесия
в процессах столкновений, которое заключается в том, что число молекул
1-го сорта, выбывающих за счет столкновений из определенного интервала
скоростей, точно компенсируется числом молекул, прибывающих в этот интер-
вал за счет столкновений. Помимо этого, Я-теоремой доказывается, что реше-
ние уравнений Больцмана для случая равновесия1) таково, что не только правая
часть уравнения C.1) равна нулю, но тождественно равна нулю также подын-
тегральная функция каждого интеграла. Это является доказательством стати-
стического принципа детального равновесия, который состоит в том, что в рав-
новесии число молекул /-го сорта, выбывающих из определенного интервала
скоростей за счет столкновений определенного рода с молекулами /-го сорта,
точно балансируется числом прибывающих в этот интервал молекул за счет
обратных столкновений.
Б. РАЗЛОЖЕНИЕ ФУНКЦИЙ РАСПРЕДЕЛЕНИЯ В РЯДЫ
ПО МЕТОДУ ЭНСКОГА2)
Уравнение Больцмана A.25) может быть записано в виде
t
t
гДе J(fb tj) — билинейная форма:
y(//,/y) = JJJ(/?/; - fifj)g>jbdbdedvj, C.9)
обозначающая интегралы столкновений.
Решение уравнения Больцмана в виде ряда может быть получено путем
введения в уравнение Больцмана параметра возмущения е таким образом,
х) Доказательство этого утверждения по существу основывается на доказательстве того,
что сумматорные инварианты являются линейными комбинациями упомянутых выше пяти
скаляров.
2) См. [6].
§ 3. Решение уравнения Больцмана методом Энскога 373
чтобы при произвольном изменении частоты столкновений относительное число
столкновений определенного вида оставалось неизменным. Мы рассмотрим
некоторую гипотетическую задачу, в которой уравнение Больцмана имеет вид
а 1/е есть мера частоты столкновений. При малом е столкновения происходят
очень часто, вследствие чего газ можно рассматривать как сплошную среду,
в которой в каждой точке достигается локальное равновесие. Функцию распре-
деления при этом можно разложить в ряд1) по параметру е
fl = flo) + efM + e*fp+... . C.11)
Если этот ряд подставить в преобразованное уравнение Больцмана (ЗЛО)
и приравнять коэффициенты при одинаковых степенях е, то мы получим сле-
дующую систему уравнений для функций ff\ /[,1], /[,-2] и т. д.:
2
тг) тйг ( i?rJ
= 2 У № №+J №]> tf11) + J №> №1 •
Из уравнений C.12) для f\r^ однозначно определяются функции распределения.
Первое уравнение из системы уравнений C.12) состоит из нескольких
интегральных уравнений, рассмотренных при решении уравнения Больцмана
для случая равновесия и при доказательстве Я-теоремы (см. разд. А). На осно-
вании анализа, приведенного в разд. А, можно сказать, что наиболее общее
решение рассматриваемых уравнений имеет вид
Л11 = "' (штТЛе~"к(л^ркГ- (злз)
Величины
л, = л,(г,0; vo = vo(r,0; т = т(г,о C.14)
при решении этих уравнений остаются произвольными функциями координат
и времени. Чтобы эти величины соответствовали локальным значениям плот-
ности числа частиц, средней массовой скорости и температуре, необходимо,
чтобы решения остальных уравнений системы C.12) были бы такими, чтобы
выполнялись условия
J7v,= n,, C.15)
tfiur, = eirt, C.16)
-^¦2m' f <v< - voJ/<dv< = -тпкТ- (ЗЛ7>
2
Можно показать [2], что из остающихся уравнений системы C.12), используя
дополнительные условия
J O, г=\, 2, 3,..., C.18)
0, г = 1, 2, 3,..., C.19)
=0' r=l> 2> 3'"" C'20)
!) Не следует путать обозначения /С1), /(*) и т .д., где индексом в круглых скобках отме-
чен порядок функции распределения, с обозначениями /М, /121 и т.д., где индексами в квад-
ратных скобках отмечены порядки приближений.
374 Гл. 7. Кинетическая теория разреженных газов
можно однозначно определить все функции /[г]. Так как определенные таким
образом функции распределения удовлетворяют условиям C.15) — C.17), то
они могут быть использованы как решение уравнения Больцмана.
В. РЕШЕНИЕ УРАВНЕНИЯ БОЛЬЦМАНА В ПЕРВОМ ПРИБЛИЖЕНИИ
Уравнение для fp может быть записано через функцию возмущения ф(9
определяемую следующим образом:
t\1] (Г, V,, 0 = ДО (Г, V,, 0 Фг (Г, V,, О- C.21)
Подставляя C.21) во второе уравнение системы C.12), получаем
C-22
Дополнительные условия C.18) — C.20) через ф, запишутся следующим
образом:
J>A<*Vi = O, C.23)
$г, = 0, C.24)
ir2mt J(v, - vo)*/F,01 ФМ = 0. C.25)
Как было сказано выше, этой системы уравнений достаточно для однознач-
ного определения функций возмущения ф{.
Могут быть вычислены производные от функции fo(\ входящие в уравнение
C.22). В окончательные выражения для этих производных входят производные
по координатам и по времени от функций nf(r, t\ vo(r, /) и T(r, /). Производные
по времени исключаются с помощью уравнений переноса B.42), B.45), B.50).
В рассматриваемом приближении в интегралах для векторов плотностей пото-
ков, которые входят в уравнения переноса, ft заменяется на /[0Д так что j, = О,
р = pU, q = 0. В результате уравнение для функции возмущения ф{ приобре-
тает вид
Н <3-26)
Величины d/ и Ь, выражаются следующим образом :
Ь, = 2 [w, Wy - 4" W*i U] • C*28)
Безразмерная скорость W,- определяется по B.11).
Г. ИНТЕГРАЛЬНЫЕ УРАВНЕНИЯ
Функция возмущения ф{ зависит от координат и времени только через
величины пь Vp, Г и их пространственные производные. Из вида интегрального
уравнения для ф{ следует, что функция возмущения линейно зависит от произ-
водных и имеет вид
^ y). C.29)
§ 3. Решение уравнения Больцмана методом Энскога 375
где А,, В/ и Сф — функции безразмерной скорости W,, локального состава
смеси и локальной температуры. Если смесь состоит из v компонент, то неза-
висимыми являются только 1у — 1) векторов Ль так как из определения d,
следует, что
^«, = 0. C.30)
Это дает возможность для каждого i один из коэффициентов Сф считать равным
нулю. В целях симметризации выражений будем полагать
0'> = О. C.31)
Подставляя выражение для ф{ C.29) в уравнение C.26) и приравнивая
коэффициенты при одинаковых градиентах, получаем интегральные уравне-
ния1) для функций Сф, В, и Af:
у>' - c\ky - cj* - - cp> - су» + c\k + с}*>} х
C.32
Л01 Ь, == -2J№ {В',+ В; - В, - uj}pt0]t[j>]gijbdbdedvJ} C.33)
Л01 D - W<) v* e ^JJJ {А'< + Ai -А/ - *}jfW]gijbdbdedvj. C.34)
В эти уравнения в качестве переменных входят л,(г, t), Г(г, /) и Vf(r, t)
[вместо Vf(r, 0 фактически мы будем использовать W,(r, t) ]. Пространственные
координаты (или производные по пространственным координатам) явно в
полученные уравнения не входят. Следовательно, Ah В/ и Сф зависят от прост-
ранственных координат только через упомянутые выше переменные. Можно
показать, что любая векторная функция от W,- равна самому вектору, умно-
женному на некоторую скалярную функцию от абсолютной величины вектора
W/. Поэтому А,, и Сф имеют вид
C.35)
C.36
где Сф и Д- — функции Wh которые параметрически зависят от Г и от всех rij.
Можно также показать, что тензор В, в соответствии с видом интегрального
уравнения для Bf C.33) должен иметь вид
в, = {w, w, - 4-w*и} в> W^ • <3-37)
x) Уравнение C.32) получается следующим образом. В результате подстановки C.29)
в C.26) имеем
JL /JO] (V/. ||) = 2h fff {{ Ф' + Cf - CW - Cf>}. йь) № /JO] gU bdbdsd vj.
Используя условие ? d/ = 0 и делая некоторые алгебраические преобразования, получаем
• = 2 <»* • 2 J J J [Ф' + tfy - Ф' -«$*'- Ф - Ф + Ф + Ф] t\01 t[j0] iub db de d*J-
Левую часть полученного уравнения можно переписать в виде
t\4fi 2 (fia,-
Яг К
Приравнивая коэффициенты при одинаковых йд, получаем C.32).
376 Гл. 7. Кинетическая теория разреженных газов
Интегральные уравнения для А,, В,- и Сф [уравнения C.32)—C.34) ]
должны решаться с учетом дополнительных условий, выражаемых уравнениями
C.23)—C.25). Эти дополнительные условия при подстановке в них А„ В,
и Сф приобретают вид
j O, C.38)
C.39)
Дополнительного условия, аналогичного двум приведенным выше, для В,
не имеется, так как функции вида C.37) автоматически удовлетворяют
условиям C.23)—C.25) при любом произвольном виде функции B^Wi).
Решения интегральных уравнений C.32)—C.34) могут быть получены
двумя эквивалентными методами: методом, разработанным Чепменом и Каулин-
гом [2], и вариационным методом [7]. В обоих методах скалярные функции
Сф(И^), Bj(Wi) и A(W,) разлагаются в ряды по полиномам Сонина1). Чепмен
и Каулинг использовали бесконечные ряды из этих полиномов, в результате
чего коэффициенты переноса у них выражаются через отношения детерминантов
бесконечного порядка.
Для получения численных величин коэффициентов переноса при этом
необходимо учитывать только несколько элементов этих детерминантов, так
как при последовательном добавлении столбцов и колонок отношение детер-
минантов быстро сходится. Здесь мы рассмотрим решение интегральных урав-
нений вариационным методом.
Так как интегральные уравнения C.32)—C.34) все имеют одинаковый вид,
можно написать одно общее интегральное уравнение для тензора ТЦ*к\ заменя-
ющее все три уравнения2)
JJf } ff"gubdbdedvj. C.40)
Символы R(,M> и T\htk) для трех рассматриваемых уравнений имеют зна-
чения, приводимые ниже:
Уравнение R\htk) l\h*k
C.32) -i/pi (dih - д(к)\( <#> - С}*>
C.33) - 2 fW (\у, w, - -~ W? U) В,
C.34) /l.o](-|--W?)v, A,
Необходимо иметь в виду, что индекс i при символе Т(?>Л) указывает на то,
что тензор является функцией вектора скорости W/. Индексы при других сим-
волах R, А, В, С имеют то же значение. Мы будем искать приближенное реше-
ние интегрального уравнения для Т^ C.40) вариационным методом. Из этого
уравнения вместе с дополнительным условием
2V^ij^k)'^dfi0] Лг, = О, C.41)
которое эквивалентно уравнениям C.38) и C.39), ТЦ>к) определяется одно*
значно.
х) Определение полиномов Сонина дано формулой C.57).
*)Два из этих уравнений являются уравнениями относительно векторных величин
(тензоров первого порядка) At и С*?* — С*?*-
§ 3. Решение уравнения Больцмана методом Энскога 377
Д. НЕКОТОРЫЕ ВАЖНЫЕ ИНТЕГРАЛЬНЫЕ ТЕОРЕМЫ
В последующих рассуждениях полезно использовать некоторые сокращен-
ные обозначения. Обозначим через G,y и Н/у какие-либо два тензора, вообще
являющиеся функциями W, и Wy. Через [Gl7; Н^]^1) обозначим величину
[Gl7; H,7],y= -7^tftf(G<7:(Htf- "JWtrgijbdbdsdVidvj. C.42)
Из соображений симметрии, аналогичных соображениям, рассмотренным в
§ 2, получим
[G,,; На]и = -^ JjJJ((G;y - G,7):(H;y - Hl7))/[»l/foigybdbded^dvj. C.43)
Отсюда видно, что выражение [G,y; Н/у ],у симметрично относительно переста-
новки Gu и Иф а также относительно перестановки индексов i и / у скобки,
[Gu; "ij)ij = [Hu; Gu]u = [Gl7; Hu]jt = [Hu; G/;]y/. C.44)
Оператор [ ] является линейным. Если G/y и Н/7 имеют вид
Gu^Kt + Lj; H/y=M, + Ny, C.45)
где К/ и М, зависят только от W/, a Ly и Ny — только от Wy, то
- [К*; Ща +[КГ, Щц +[lj; M,]u + [L,; Щи. C.46)
Необходимо отметить, что индексы при символах тензоров в скобках указы-
вают на функциональную зависимость тензоров от скоростей W/ и Wy. Индекс
ij при скобке указывает на то, что интеграл вычисляется для столкновений
между молекулами i и /.
Рассмотрим два ряда тензорных функций К, и L, и определим через эти
функции величину
{KL} ^[K Ky;L/ + Ly]/y. C.47)
Выражение в фигурных скобках удовлетворяет соотношениям
{K;L} = {L;K} C.48)
{К; L + М} = {К; L} + {К; М}. C.49)
Так как {К; К} выражается через сумму интегралов, подынтегральные
функции каждого из которых неотрицательны, то
{К;К}>0. C.50)
Можно легко показать, что {К ; К} исчезает в том случае, и только в том,
когда К/ является линейной комбинацией сумматорных инвариантов. Линейная
комбинация сумматорных инвариантов, удовлетворяющая дополнительным
условиям C.41), тождественно равна нулю. Отсюда, если ограничиться рас-
смотрением ряда тензорных функций К„ которые удовлетворяют дополнитель-
ным условиям, то {К ; К} исчезает в том случае, и только в том случае, когда
каждое из К, тождественно равно нулю.
х) Индексы // у скобок указывают на интегрирование по всем значениям V/ и у/.
378 Гл. 7. Кинетическая теория разреженных газов
Е. ВАРИАЦИОННЫЙ ПРИНЦИП
Применим вариационный принцип для получения приближенного решения
интегральных уравнений C.40). В качестве пробных функций используем ряд
функций 1(?»Л), которые удовлетворяют уравнениям
>: Rp>)rfv, = -2nt щ [!<"•*>;фк >+1^% C.51)
и включают столько произвольных параметров, сколько необходимо.
Умножая бискалярно интегральное уравнение для Т(,М) C.40) на t(,M)
и интегрируя полученное произведение по всем v,, получаем
2щщ№ ; f %, C.52)
где \ую — пробные функции, а Т(?>/с) — точные решения интегральных урав-
нений. Приравнивая правые части последних двух уравнений и суммируя их
по всем /, а также используя свойства симметрии C.44) и определение C.47),
получаем
{(M) т<л*>} 0<л*> <"*>} C.53)
Поскольку выражение в фигурных скобках для двух одинаковых функций
неотрицательно C.50), то
{j(h,k) _ t(/a>. у<м> _ \(h,k)}^ о. C.54)
Далее, пользуясь свойством линейности оператора { } C.49) и соотно-
шением между пробными функциями и точными решениями C.53), находим
{1<м>; **•*>}<{Пй-*>; Т<Л>*>}. C.55)
Условие C.55) является формулировкой вариационного метода1) для полу-
чения приближенных решений Т(,М). Подробнее вариационный метод решения
состоит в следующем. Выбирается ряд пробных функций t(?>/?), которые включают
некоторое число произвольных параметров. Далее, если рассматриваются
только те пробные функции, которые удовлетворяют дополнительным усло-
виям C.41), то знак равенства в C.55) имеет место только тогда, когда T(?>fc)
и t<^*> совпадают. Иными словами, наилучшее приближение к точному решению
интегрального уравнения получается при значениях произвольных параметров,
соответствующих максимуму левой части C.55), т. е. в наилучшем прибли-
жении
f C.56)
Вариационное условие C.56) совместно с уравнениями C.51), которые
ограничивают выбор пробных функций, образуют основу вариационного метода
решения интегральных уравнений C.40).
Ж. ПРИЛОЖЕНИЕ ВАРИАЦИОННОГО ПРИНЦИПА
(РАЗЛОЖЕНИЕ ПО ПОЛИНОМАМ СОНИНА)
Рассмотрим обсужденный выше вариационный метод в случае, когда проб-
ными функциями являются конечные ряды из полиномов по квадрату ско-
рости W*. В качестве таких полиномов используем полиномы Сонина S^\
определяемые следующим образом2):
qm /Y\ ^ (— !)¦/ (т + П)\ ] /о су\
°П VV ^ /п _L Al lm _ A! rl Л ' V '
г) Впервые это было сделано Хелландом и Улингом [8].
2) За исключением нормировки, эти полиномы совпадают с соответствующими полино-
мами Лагерра. Первые два полинома Сонина имеют вид
— х.
§ 3. Решение уравнения Больцмана методом Энскога
379
Эти полиномы удовлетворяют условиям ортогональности
>(x) dX - -&±«Й1 am
C.58)
и удобны вследствие того, что для них соблюдаются следующие два частных
случая условий ортогональности:
ЗгцкТ
$
C.59)
C.60)
В качестве пробных функций t(?»fc> мы возьмем конечные линейные комби-
нации из полиномов Сонина S' v
пи
m—0
C.61)
в которых индекс п и тензоры W/ могут иметь следующие значения (табл. 65).
Таблица 65
При Т^'1*'
равном
А/
В/
Индекс
п равен
•/¦
•/•
•/•
Wi равен
W/
W/Wf-VtW'U
W/
Коэффициенты ^^разложе-
ния функций tj***' по полино-
мам Сонина обозначаются
следующим образом
вы (в
По причинам, которые будут выяснены позднее, нами указана зависимость
коэффициентов разложения от числа членов ? конечных рядов, представляю-
щих пробные функции. Здесь мы только отметим, что коэффициенты разложе-
ния зависят от числа используемых членов разложения t(,M), так как они не
являются коэффициентами бесконечных рядов разложения. Введем следующие
определения :
/?&'*> = J (b(ihik): W,) Sg"> (W*) d\t C.62)
и
ь — & 4m *\im • \O.\jOj
Уравнения C.51), накладывающие определенное ограничение на пробные
функции, выражаются через эти величины следующим образом :
w,("'*> = 0, C.64)
где
2
т—О
'i1 *
i 2
m-Om'-O
а формулировка вариационного критерия C.56) приобретает вид
C.65)
C.66)
380 Гл. 7. Кинетическая теория разреженных газов
Таким образом, проблема определения коэффициентов разложения проб-
ных функций состоит в нахождении экстремума g(M) при ограничивающих
условиях C.64). Экстремум g(M) в таком случае может быть найден с помощью
метода множителей Лагранжа. Обозначим эти множители через А<^*). Урав-
нение C.64), а также уравнения
= 0 C.67)
образуют систему уравнений, из которой можно определить №}>*> и коэффи-
циенты разложения f(*/?}(f)- Дифференцируя g(M) и w^k) no №\$у из C.67)
получаем
[I+№•»]#№> + 2*2 щщх
j m'-0
[W, S<T> (W?); W, SS»'> (WPU = 0, C.68)
/=1,2,...,»,
Решением этой системы уравнений, совместным с уравнениями C.64), будет
Xf,k) = 1 f i« 1, 2, 3,...,». C.69)
При этом fllilPiS) — постоянные, определяемые из уравнений C.68), в которых
д(й,а) = 1ф Уравнения для определения коэффициентов №$ могут быть запи-
саны в виде
2 21 ОТГ № @ = - R№, C.70)
j m'— 0
где
Q7" = f ^ щ {ди [ W, Sg»>(lVf); W, Sg»')
+ *ji [W, SW(lVf); W? S<["'>(W?)]f/}. C.71)
При T(fM), равных В,- (и t(f}^\ равных W\$\ уравнения C.70) линейно
независимы и отсюда все lft\$ могут быть определены из этих уравнений.
Однако когда Т(/Л>л) есть А, или (С(Р — C(f), можно показать, что при m = О
одно из уравнений системы линейно зависимо. В этих случаях необходимо
использовать дополнительные условия C.41), которые выражаются через fi}$
следующим образом:
2 %У™№ k) (f) J W*i ЭД^') Л01 Wi = 0. C.72)
Вследствие условий ортогональности C.59) члены этой суммы с m ф 0 равны
нулю. Поэтому дополнительные условия приобретают вид
0. C.73)
Следовательно, когда Т(*м) есть А, или (Cf — Cf), пробные фикции должны
быть выбраны такими, чтобы
УШ; = 0. C.74)
Уравнение C.74) является дополнительным уравнением, необходимым
для определения всех коэффициентов /^(f). Используя C.74), можно
§ 4. Формулы для вычисления коэффициентов переноса 381
уравнение C.70) преобразовать к виду
2 21 $?Г t%m = - RW\ 3.75)
j /л'—О
где
(ОТ', когда ffaP = bJm.,
j
QT' = jQmm' _ *Ш QT> дт0 8т>0> КОГда ffcp = ajm, или ф*>. <3J6)
Из уравнений C.75) можно получить ^rfP(f), через которые выражаются
функции А,, В* и Сф. Через последние в свою очередь выражаются функция
ф{ и функция распределения /, с учетом поправочного члена первого порядка.
Зная функцию распределения /, неравновесных систем, можно вычислить
коэффициенты переноса. При вычислении коэффициентов переноса в разло-
жении по полиномам Сонина оказывается необходимым учитывать лишь
небольшое число членов. Для коэффициентов вязкости и диффузии хорошее
приближение можно получить, учитывая один первый член разложения. При
учете двух первых членов разложения значения перечисленных коэффициентов
отличаются от значений, полученных с учетом одного члена, только на несколько
процентов. При учете только первого члена разложения коэффициент диф-
фузии оказывается не зависящим от концентрации. Слабая зависимость коэффи-
циента диффузии от концентрации получается лишь при учете двух членов
разложения по полиномам Сонина. Перенос массы вследствие наличия гра-
диента температуры, т. е. термодиффузия, в выражении для диффузионной ско-
рости не появляется вовсе, если учитывать в разложении по полиномам Сонина
только один член. Для получения первого приближения коэффициента тепло-
проводности в разложении по полиномам Сонина необходимо также учитывать
два члена.
§ 4. ФОРМУЛЫ ДЛЯ ВЫЧИСЛЕНИЯ КОЭФФИЦИЕНТОВ
ПЕРЕНОСА
Выражения для векторов потоков массы, импульса и энергии выведены
в § 2. Для вычисления интегралов, входящих в это выражение, необходимо
знать функцию распределения /,. Приближенное выражение для функции
распределения в виде /, = /[?JA + Фд было получено в предыдущем параграфе.
Если в интегралах, через которые выражаются векторы потоков, использовать
функцию фи даваемую вьфажением C.29), то диффузионная скорость, тензор
давления и вектор теплового потока выразятся через интегралы от функций
Ai(Wi), B((Wi) и Cty(Wt). Получению и обсуждению именно этих выражений
для векторов потоков и посвящается настоящий параграф.
А. ВЫРАЖЕНИЕ КОЭФФИЦИЕНТОВ ДИФФУЗИИ И ТЕРМОДИФФУЗИИ
ЧЕРЕЗ КОЭФФИЦИЕНТ РАЗЛОЖЕНИЯ ПО ПОЛИНОМАМ СОНИНА
Интеграл для диффузионной скорости B.7), записанный через функцию ф1У
имеет вид
^N iJ D.1)
Используя для ф{ выражение C.29) и замечая, что член, включающий В,, при
интегрировании исчезает, можно записать V/ в виде
D.2)
382 Гл. 7. Кинетическая теория разреженных газов
Используя выражения C.35) и C.36), дающие вид функций А,- и Сф, получаем1)
В этом выражения uj имеет вид C.27), a D(J и Df определены следующим обра-
зом :
DJ = J« |f^ J АДГ) WT Л« dV,. D.5)
Здесь Dtj и Df называются коэффициентами диффузии и коэффициентами
термодиффузии многокомпонентных смесей соответственно*).
Таким образом, мы видим, что диффузионная скорость V/ включает члены,
пропорциональные градиенту концентрации, градиенту давления, разности
внешних сил, действующих на различные сорта молекул, и член, пропорцио-
нальный градиенту температуры. До работы Чепмена и Энскога термодиффузия
в газовой фазе не была известна теоретически и не наблюдалась эксперимен-
тально. Последующие эксперименты Чепмена и Дутсона [9] показали, что
теоретическое предсказание явления термодиффузии совершенно точно. Это
один из интересных примеров предсказания экспериментально наблюдаемого
явления на основе строгого теоретического анализа. Эксперименты по измере-
нию Du обычно проводятся таким образом, что вклады в диффузионную ско-
рость за счет градиента давления, внешних сил и градиента температуры оказы-
ваются пренебрежимо малыми.
Используя разложения Сф и А, по полиномам Сонина C.61), выражения
для коэффициентов диффузии можно привести к виду
mi V /.(/,0m Г 1/2 Q(m)/u/?\ *[0] ИМ D.6)
где ? равно числу членов, удерживаемых при разложении Ду(?) и Df(?) в
ряды по полиномам Сонина. Вычисляя с помощью C.59) интегралы, входящие
в выражения D.6) и D.7), получаем
D.9)
Таким образом, мы выразили DtJ и DJ только через нулевые коэффициенты
разложения по полиномам Сонина. Независимо от того, сколько членов исполь-
зуется при разложении (т. е. независимо от ?), после интегрирования остаются
г) Здесь нами использована теорема, которая может быть доказана, исходя из сообра-
жений симметрии. Если F(r) — любая функция от абсолютной величины г, то
J F(r) n dt = у U J F(r) r2 dr.
2) Разные авторы различным образом определяют эти величины. Определенный здесь
коэффициент Dij таков, что для двухкомпонентной смеси Dy сводится к обычному коэффи»
циенту диффузии Л)/у бинарных смесей. Коэффициент Df ранее определен не был. В случае
двух компонент определенный здесь коэффициент D/ не сводится к коэффициенту термодиф-
фузии для бинарных смесей, введенному Чепменом и Каулингом. Соотношения между
различными коэффициентами обсуждаются в гл. 8, § 1.
§ 4. Формулы для вычисления коэффициентов переноса 383
только нулевые коэффициенты. Однако величины коэффициентов сЦ$(?) и
а,0(?), которые могут быть найдены из системы уравнений C.75), зависят от
числа членов разложения.
При f = 1 для Du получаются достаточно хорошие результаты. Полагая
f = 2, мы получаем дополнительно небольшой поправочный член. За исклю-
чением особых случаев, обычно не возникает необходимости использовать любые
приближения с величиной $ > 2. Коэффициенты Df при ? = 1 тождественно
равны нулю. Это получается вследствие того, что при R(,M) = /l?J [б/2 — W? ] V/
и при л = 3/г интеграл C.62) исчезает, так как функция (б/2 — WJ) равна
S§l(Wfy и поэтому ортогональна Sift(WJ). Для вычисления коэффициента тер-
модиффузии в разложении по полиномам Сонина необходимо учитывать по
крайней мере два члена, т. е. полагать ? = 2. Для реальных потенциальных
функций такое приближение приводит к ошибке от 5 до 10%.
Б. ВЫРАЖЕНИЕ КОЭФФИЦИЕНТА ВЯЗКОСТИ ЧЕРЕЗ КОЭФФИЦИЕНТЫ
РАЗЛОЖЕНИЯ ПО ПОЛИНОМАМ СОНИНА
Интеграл для тензора давлений B.23) выражается через функцию возму-
щения ф1 следующим образом:
D.10)
D.11)
- pU + 2mjjVj Vy/y» fjdVj, D.12)
где p = nkT D.13)
— равновесное гидростатическое давление при локальной температуре и плот-
ности. Используя выражение C.29) для функции возмущения фь находим
D.14)
Можно показать, что из соображений симметрии члены с А, и Сф обращаются
в нуль. Используя для тензора В,- выражение C.37) и снова применяя сообра-
жения симметрии, получаем
р = pU - \-*r2J±jBj(Wj) V*j /jridVy) S, D.15)
где S — тензор скорости сдвига, определяемый выражением
<4Лб>
Коэффициент вязкости г\ определяется соотношением
p = pll-2*?S. D.17)
Из выражения D.15) следует
f
ч - -m2-efjBi(wj) WV D.18)
Используя разложение By по полиномам Сонина C.61), получаем
2Ь**®№<Ю)Щ*Щ. D19)
т—О
Здесь снова аргумент у т?(?) указывает на порядок приближения. Вычи-
сляя интегралы в D.19) с учетом условий ортогональности C.60), находим, что
384 Гл. 7. Кинетическая теория разреженных газов
коэффициент вязкости смеси выражается через коэффициенты bj0(?) разло-
жения по полиномам Сонина
kT2nbJO(i;). D.20)
Как и в случае диффузии, первое приближение здесь дает главный вклад в зна-
чение г}. Вычисления показывают, что при учете следующих членов разло-
жения коэффициент rj изменяется очень мало.
В. ВЫРАЖЕНИЕ КОЭФФИЦИЕНТА ТЕПЛОПРОВОДНОСТИ
ЧЕРЕЗ КОЭФФИЦИЕНТЫ РАЗЛОЖЕНИЯ ПО ПОЛИНОМАМ СОНИНА
Интеграл для вектора плотности потока энергии q B.26) следующим обра-
зом выражается через функцию возмущения ф/.
4-21)
При подстановке в D.21) выражения для ф] C.29)] член, включающий Ву,
исчезает, в результате чего имеем
^^j D.22)
Поток энергии может быть подразделен на две части: на часть, учиты-
вающую поток энергии за счет движения молекул относительно средней массо-
вой скорости, и на часть, учитывающую поток энергии за счет других причин.
Подставляя в D.22) выражение для А, C.35) и Сф C.36), получаем
К
-^)у^01^- С4-23)
Используя, как и выше, соображение симметрии, находим
Ц-п(т'*2Ь2у=№<Рй(тг-wi)w2j№dVJ> <4-24)
где
V^yj2S ?J) m D.25)
Первый член представляет собой плотность потока энергии за счет пере-
носа массы, второй — за счет градиента температуры, а третий учитывает допол-
нительный перенос энергии под действием градиента концентрации. Последний
член аналогичен члену, учитывающему влияние градиента температуры на
диффузию, т. е. члену, учитывающему термодиффузию.
Выражение для q может быть преобразовано следующим образом. Из
интегрального уравнения для А, C.34) можно получить соотношение
D j) w
Я( j, D.26)
которое может быть использовано, чтобы переписать выражение для q D.24).
§ 4. Формулы для вычисления коэффициентов переноса 385
Далее, «симметризуя» сумму по i и / и используя условие 2 Лк = О, получаем
к
22
+ Cf> - Ci"> - Cj">])/J°] /J»l gub db de dV, d\}. D.27)
Из интегрального уравнения для С(*} C.32) следует
(<5,ft 6ik)
= 2jjjj(A, ¦ № + Cf)' - C<*>' - Cj*>' -
- 0") - C?1) + 0ft> + Cj")]) fp ff* gu b db de dVt dVj. D.28)
Суммируя D.28) no i и «симметризуя» сумму по i и /, находим
X fi0] flj0] gu b db de dV, dVj. D.29)
Используя определение Dj D.5), а также условие 2 йк = О, плотность потока
энергии можно записать в виде *
i^^ D.30)
Коэффициент А' не равен обычно определяемому коэффициенту теплопровод-
ности. Общепринято исключать градиенты dy из выражения для q с помощью
выражения для диффузионных скоростей D.3). В результате такого исклю-
чения плотность потока энергии выражается через диффузионные скорости и
градиент температуры. Вследствие того, что в выражение для диффузионной
скорости входит термодиффузионный член, к А' добавляется малый член, кото-
рый в сумме с А' дает обычный коэффициент теплопроводности А. Окончатель-
ное выражение для А D.65) будет получено в этом параграфе ниже.
Если функции Ai(Wi) записать в виде рядов по полиномам Сонина, как это
было сделано выше C.61), то выражение для А' D.25) приобретает вид
Цх
x aJm(S)fsW(Wj)(-^--WJ)w]№dVj. D.31)
Далее, поскольку
s$0^) = -§--W?, D-32)
из условий ортогональности полиномов C.58) получим
Л' (f) = - 4 к 2 nj f Щ- aix (]¦)< D.33)
386 Гл. 7. Кинетическая теория разреженных газов
Г. ИНТЕГРАЛЫ №>*)
Выше коэффициенты переноса были выражены через коэффициенты разло-
жения по полиномам Сонина. Эти коэффициенты разложения, как нетрудно
вспомнить, получаются путем решения системы совместных уравнений C.75).
Из этой системы видно, что коэффициенты разложения tfy *>({) являются слож-
ными комбинациями «скобочных» интегралов, которые определяются по C.42).
Чепмен и Каулинг [2] показали, что последние могут быть представлены в
виде линейных комбинаций набора интегралов Q(l>s) [2]1). Для столкновений
между молекулами 1-го и /-го сорта интегралы ??(/'.s) определяются следую-
щим образом:
Q(i)S) = ]l^f.[ J «"'& П+3(! - cos '*) * db dYiS. D.34)
6 6
В этих интегралах /ли — приведенная масса сталкивающихся молекул i и /
[определение /ли дано формулой E.9) гл. 1]; ytj — приведенная начальная
относительная скорость сталкивающихся молекул B.12); # — угол отклонения
молекул в системе координат центра тяжести; Ь — прицельное расстояние
(Ь и % показаны на фиг. 9, стр. 54). Таблица наиболее часто встречающихся
«скобочных» выражений, записанных через интегралы &ls\ дана в прило-
жении А (стр. 406). С помощью этой таблицы и системы уравнений C.75) можно
вычислить любые коэффициенты разложения по полиномам Сонина t% k).
Динамика столкновений входит в коэффициенты переноса через интегралы
столкновений D.34). Для вычисления ??(/>s) необходимо знать % как функцию
начальной относительной скорости g(j и прицельного расстояния Ь. Зависи-
мость % от gu и b дается выражением E.26) гл. 1. В это выражение явно входит
потенциальная энергия взаимодействия <р(г). Таким образом, задавая потенциал
взаимодействия, можно вычислить интегралы f2(/>5) и отсюда коэффициенты
разложения $пЛ)(?).
Формулы для коэффициентов переноса, выражаемых через интегралы f3(/»s),
приводятся в следующих разделах настоящего параграфа. Окончательное
вычисление углов %(g, ft), интегралов ??(/|S) и коэффициентов переноса для
некоторых законов межмолекулярных сил производится в гл. 8. Наиболее
удовлетворительными и удобными для использования являются вычисления,
выполненные на основе либо потенциала F-12) Леннарда-Джонса, либо моди-
фицированного потенциала Букингема. Эти потенциалы хорошо описывают
взаимодействие между сферическими неполярными молекулами. Формулы и
таблицы, приводимые в гл. 8, дают возможность практически использовать
теорию. В этой же главе показано, что согласие между вычисленными и экспе-
риментальными результатами вполне удовлетворительное.
Д. ТОЧНЫЕ ФОРМУЛЫ ДЛЯ КОЭФФИЦИЕНТОВ ПЕРЕНОСА, ВЫРАЖЕННЫЕ
ЧЕРЕЗ ИНТЕГРАЛЫ ?<*>«)
Здесь, так же как и в предыдущих разделах, обсуждается метод получения
выражений для коэффициентов переноса через величины, характеризующие
законы межмолекулярных сил и динамику парны;, столкновений. Детали
алгебраических преобразований, приводящих к представлению этих выражений
через интегралы Q{U s\ очень громоздки и вследствие этого опускаются, за исклю-
чением кратких указаний на то, каким образом полученные до сих пор резуль-
таты могут быть использованы для получения низших приближений различных
коэффициентов переноса.
х) Интегралы Q(l>s) в настоящей книге совпадают с интегралами Q(l)(s) по книге Чепмена
и Каулинга [2] и отличаются от интегралов Q(l>s) по книге Энскога [6 ].
§ 4. Формулы для вычисления коэффициентов переноса
387
1. Коэффициент диффузии. Коэффициент диффузии многокомпонентной
смеси может быть получен в очень хорошем приближения при учете в разло-
жении по полиномам Сонина только одного члена. Уравнения, из которых
определяются с$'Л)A) C.75), при этом приобретают вид
= -/?$'*>. D.35)
Записанные через интегралы ??(/» s\ эти уравнения выглядят следующим обра-
зом :
¦Ли
[щ т{ди - дЛ) -
tl)) Q\}* =
D.36)
Для бинарной смеси из уравнений D.36) немедленно можно получить
— th h \
Таким образом, в первом приближении коэффициент диффузии1) бинарной
смеси [из D.8) ] имеет вид
Ф /i\_ 3(mt+mj) kT ( .
По определению [или на основании вида выражения D.37)],
#« = 48Л-#Л- D-39>
Используя этот результат, а также выражение для диффузионных постоянных
D.37) и соотношение для диффузионных постоянных бинарных смесей D.38),
из D.36) можно получить систему уравнений для обобщенных диффузионных
постоянных
2 Fij{mhDJh(l) - mk DJk(l)} = Fih - dik), D.40)
где
D.41)
Легко может быть получено формальное решение этой системы уравнений.
Обозначил через Fij адьюнкту элемента F(j детерминанта |F|, составленного
из Fij} т. е.
u ... Flt;_i F^y+i ... Flv
= (_ iy+J
i-l, J-l Fi-l, j+l
Fi+м • • •
D.42)
Решая далее уравнение D.40), получаем
mhDih{\)-
D.43)
x) Для бинарной смеси мы обозначаем D// через
388 Гл. 7. Кинетическая теория разреженных газов
Так как Dti з= 0, то из D.43) следует
Соотношения D.44) связывают обобщенные коэффициенты диффузии смеси
с коэффициентами диффузии бинарных смесей для различных пар компонент
смеси. Однако результаты в виде D.44) обычно трудно использовать при реше-
нии конкретных задач. Поэтому часто выгодна иная формулировка задачи.
Из D.3) найдем
D.45)
Это выражение может быть значительно упрощено путем использования осо-
бого вида уравнений D.40). Дополнительные условия C.74), которым должны
удовлетворять коэффициенты с%> к\ могут быть записаны через коэффициенты
диффузии. В результате такого преобразования получим
2{ЩmhDih{\) - mtmkDik{\)} = 0. D.46)
С учетом D.46) уравнения D.40) преобразуются к виду1)
К mh Ddl) - nt mk Djk(l) - njmh Dih(\) +
2
+ njmkDik{\)) = {dih-dik)Q. D.47)
Уравнения этой системы линейно зависимы и поэтому не могут быть исполь-
зованы для получения коэффициентов без учета дополнительных условий
D.46), хотя и являются справедливыми. Пользуясь уравнениями D.47) и усло-
вием 2&к =0 и предполагая, что Djk может быть заменено первым приближе-
нием, т. е. Djk(l), уравнения D.45) можно преобразовать к виду
l) l Эг Г n^ij(
Эта система из (v — 1) независимых уравнений часто употребляется при реше-
нии гидродинамических задач.
2. Коэффициент термодиффузии. Чтобы получить коэффициент термодиф-
фузии, необходимо вычислить функции Д-. При этом в разложении по поли-
номам Сонина должны быть учтены два члена. Использование только одного
члена приводит к нулевым значениям коэффициента термодиффузии ; по этой
причине о термодиффузии часто говорят как об эффекте второго порядка. В рас-
сматриваемом' случае уравнения C.75) имеют вид
2 2 QT'ajm>B) = -Rim. D.49)
/ m'=O
Через интегралы QP>s) величины Q1]m' выражаются следующим образом :
> D.50)
J) Эта система уравнений представляет собой частный случай более общей системы
C.70).
§ 4* Формулы для вычисления коэффициентов переноса
389
=-8
/8 -у Щ
4-
D.51)
D.52)
*«ву-дд)х
D.53)
15
2АГ
D.54)
Система линейных уравнений D.49) для коэффициентов а]0 и а]х может
быть решена по правилу Крамерса1). Далее из D.9) получим выражение для
коэффициента термодиффузии
Лоо
VU
О00
V21
Л10
V12
Vrl Vr2
«5,
,2
Ой QU
Qll Qll
®l
Q
Qll Qll
Qll 0
Qll 0
<?? 0
11 Л11
11 Vl2
Q,1? ОЙ Ой
<*,„ о о
о о
Qll
ll
k
OO
I
k
Qll
k
6n
. D.55)
3. Коэффициент вязкости. Для v-компонентной смеси коэффициент вязкости
в соответствии с формулой D.20) в первом приближении выражается следующим
образом:
± D.56)
г) Правило Крамерса состоит в том, что решение системы линейных уравнений может
быть выражено через отношение детерминантов из коэффициентов системы.
390
Гл. 7. Кинетическая теория разреженных газов
Коэффициенты bj0(l) определяются из v уравнений C.75), которые в рассматри-
ваемом случае имеют вид
=-1, /-1,2,3,...,,,
; I Rio >
Q4! = 2 nt n,{a,7[W,, W,],, + <5«[W,-; W,],,}.
В этом случае W,- и Ri0 определяются из соотношений
/?,о = /2 (W,: W,) /f,oi dvt = - 4 J Wf № dV, = - 5л,,
D.57)
D.58)
D.59)
D.60)
Из уравнений D.57) мы, конечно, можем по правилу Крамерса найти коэф-
фициенты bjo(l) в виде отношений детерминантов v-to порядка. Однако вслед-
ствие того, что коэффициент вязкости выражается через 2! пр]0(\), его лучше
представить как отношение двух детерминантов — детерминанта (v + 1)-го
порядка в числителе к детерминанту v-го порядка в знаменателе, т. е. в виде
4A) =
н,
'11
21
H
lv
щ/п
г w тт
nVl nv2
njn п2/п
Н„ nv\n
nv/n 0
где
\НЦ\
D.61)
D.62)
Величины
могут быть выражены через Q^s):
я/ гщ ^, тц mi
и 32
15 n2mjkT —*
D.63)
Полученные результаты легко могут быть распространены на случай, когда
в разложении по полиномам Сонина учитывается большее число членов. Фор-
мальные результаты могут быть значительно упрощены для случаев бинарных
смесей и чистых газов (см. гл. 8, § 2).
4. Коэффициент теплопроводности. Выражение для плотности потока
энергии обычно записывается через диффузионные скорости и градиент темпе-
ратуры. Используя выражение для плотности теплового потока D.30) и урав-
нения D.48), получаем
В этом выражении
2л Jj Щ A) L m mt nj mi J
— обычный коэффициент теплопроводности. Величина А' выражается через
§ 5. Решение уравнения Больцмана методом Грэда
391
коэффициенты разложения по полиномам Сонина в виде D.33), откуда с помощью
методов и результатов, описанных выше, получим
А'B) = --?
9??
9fi О
93
«а
о
99
98
98
•
•
«S
1
1
О
9Я
9Й
9Й
9»
D.66)
9Й
Здесь
m' _
D.67)
где Q"' выражается по формулам D.50)—D.53).
§ 5. РЕШЕНИЕ УРАВНЕНИЯ БОЛЬЦМАНА МЕТОДОМ ГРЭДА
Как упоминалось в § 1, состояние газа может быть описано с различной
степенью полноты. Полнота сведений о системе, которые дает нам функция
распределения /(г, v, /) одной молекулы, позволяет полностью характеризовать
поведение газов малой плотности, где необходимо учитывать только парные
столкновения, а столкновениями более высокого порядка можно пренебречь.
Изменение функции распределения /(г, V, /) со временем дается уравнением
Больцмана. Оно не может быть решено точно, и поэтому в большинстве
случаев мы удовлетворяемся гидродинамическим описанием состояния газа
через макроскопические переменные — плотность, скорость и температуру.
Уравнениями, описывающими изменение этих величин со временем, являются
уравнения переноса, полученные в § 2. При выводе этих уравнений были вве-
дены определенные дополнительные величины (векторы плотностей и потоков) :
диффузионные скорости, тензор давлений и вектор плотности теплового потока.
Значения этих величин зависят от степени отклонения функции распределения
системы от максвелловской функции распределения равновесных систем.
Решение уравнения Больцмана методом Энскога приводит к выражению
векторов плотностей потоков через пространственные производные от макроско-
пических величин. В первом приближении, которое детально обсуждалось в
предыдущих параграфах, векторы плотностей потоков выражаются через пер-
вые производные. В следующих приближениях векторы плотностей потоков
392 Гл. 7. Кинетическая теория разреженных газов
выражаются через высшие производные и через производные низшего порядка
в различных степенях.
Знание макроскопических переменных (плотности, средней массовой
скорости и температуры) эквивалентно знанию первых трех моментов ско-
ростей функции распределения. Решение уравнения Больцмана методом
Энскога дает полное описание поведения газа в любой момент времени, если
для начального момента времени в каждой точке пространства известны первые
три момента функции распределения. Очевидно, что ряд функций /(г, v, <),
имеющих одинаковые первые моменты в начальный момент времени, дает раз-
личные распределения, соответствующие различным наборам первых трех
моментов в последующие моменты времени. Следовательно, метод Энскога не
дает общего решния, а некоторым способом ограничивает класс рассматривае-
мых функций распределения. Этим характеризуется процесс понижения пол-
ноты описания состояния системы. Всякий раз, когда мы понижаем степень
подробности описания системы, необходимо вводить условия, ограничивающие
область рассматриваемых возможных состояний системы. В случае решения
уравнения методом Энскога не ясно, каким образом накладываются эти ограни-
чения. Не ясно также и то, является ли полученное таким образом решение
истинным решением, так как сходимость рядов Энскога часто подвергается
сомнению.
Грэд [1 ] развил метод решения уравнения Больцмана, в котором полу-
чается первое приближение Энскога и есть возможность получать описание
системы с любой полнотой, начиная с описания через функцию распределения
/(г, v, t) и кончая гидродинамическим приближением. Метод Грэда весьма ценен
при изучении газов достаточно малой плотности, когда газ нельзя рассматривать
как непрерывную среду. Метод позволяет использовать более общие граничные
условия при решении гидродинамических задач. Этот метод решения кратко
излагается ниже. Для простоты мы ограничиваемся случаем однокомпонент-
ного газа, когда имеется только одна функция распределения, одно уравнение
Больцмана и не возникают диффузионные проблемы.
А. УРАВНЕНИЯ МОМЕНТОВ
Рассмотрим тензор п-го порядка, образованный из компонент скорости v:
0$L = «W«>ft--' EЛ)
Здесь индексами внизу обозначены компоненты скорости v. Каждому тензору
скоростей может быть сопоставлен тензор моментов функции распределения
п-го порядка
\ E.2)
Очевидно, что нулевой момент равен плотности числа частиц л. Первый момент
равен макроскопической плотности потока п v0. Макроскопическая темпера-
тура выражается через свернутый второй момент1):
\nkT =±nmV* ~2S® -^nmvl. E.3)
i
Тензор давлений выражается через полный второй момент
ри = mSff — nmvoi vOJ. E.4)
Вектор плотности потока энергии выражается через свернутый третий момент
2
x) Свернутый тензор есть тензор, ранг которого ниже исходного тензора на два ранга.
Свернутый тензор образуется из компонент исходного тензора (в которых два индекса оди-
наковы) путем суммирования по этим индексам.
§ 5. Решение уравнения Больцмана методом Грэда 393
Если уравнение Больцмана умножить на величины v(n) и затем проинтегри-
ровать по всем значениям скоростей, то мы получим уравнения моментов.
В отсутствие внешних сил эти уравнения имеют вид
E.6)
Уравнения для моментов первых двух порядков и уравнение для свернутого
момента третьего порядка представляют собой обычные уравнения переноса,
полученные в § 2. В этих уравнениях правая часть, учитывающая роль столк-
новений, вследствие свойств сумматорных инвариантов равна нулю. Вообще
же, как можно видеть, в уравнение для момента определенного порядка входит
момент, порядок которого на единицу выше порядка рассматриваемого момента.
Помимо этого, в уравнение через интеграл столкновений входит полная
функция распределения. Система уравнений для моментов эквивалентна
исходному уравнению Больцмана.
Метод решения состоит в следующем. Искомая функция, представляющая
функцию распределения, выражается через v произвольных параметров. Эти
параметры могут быть записаны через скалярные моменты первых v порядков.
Остающиеся моменты выражаются через моменты низшего порядка. Скалярные
уравнения первых v порядков системы уравнений для моментов E.6) описывают
изменение параметров со временем, а, следовательно, также и поведение газа.
Грэд в качестве искомой функции использовал функцию распределения
Максвелла, умноженную на конечные ряды из многомерных полиномов Эрмита.
Он показал, что ряды, получающиеся при учете все большего и большего числа
членов в рядах из полиномов Эрмита, сходятся к истинному решению уравне-
ния Больцмана.
Б. «ТРИНАДЦАТИМОМЕНТНОЕ» ПРИБЛИЖЕНИЕ
Рассмотрим детально «тринадцатимоментное» приближение. В этом прибли-
жении образуется функция распределения, зависящая от тринадцати скалярных
параметров. Тринадцать параметров выражаются через тринадцать независи-
мых скаляров, определяемых моментами S(o), S(l), S& и свернутым моментом
третьего порядка 2 S\J]. Зависимость тринадцати параметров от времени
можно получить, используя уравнения моментов, т. е. уравнения E.6) с
л =0, 1,2, и сумму уравнений, получающихся при л = 3. Если учесть приве-
денные выше соотношения между моментами и величинами плотности, скорости,
температуры, тензора давлений и плотности потока тепла, то можно убедиться,
что при л = 0, 1 уравнения моментов, а также сумма «диагональных» уравнений
моментов при л = 2, представляют собой соответственно уравнение непрерыв-
ности, уравнение движения и уравнение баланса энергии. В описываемом
приближении тензор давлений и вектор плотности потока тепла являются
независимыми функциями, удовлетворяющими своим дифференциальным урав-
нениям — остающимся уравнениям моментов. Этими уравнениями являются
ЭР// , ^ Э
Эх*
v 3 . v 5 рк дТ кТ ^ 9 р
f "ш*?*qi) + T~srw + иг 2. э^г"* +
7 к _ дТ 1 _Э 9
к j) к к
394 Гл. 7. Кинетическая теория разреженных газов
В уравнениях E.7) и E.8) символ { } означает следующее :
{Аи} = i-(Аи + AJt) - 4*и2 Аыс- E-9)
Тензор Р определяется следующим образом :
P = p-pU, E.10)
^ и Я — интегральные выражения, формально соответствующие коэффициентам
вязкости и теплопроводности, полученным в предыдущем параграфе.
Уравнения E.7) и E.8) наряду с обычными уравнениями переноса образуют
систему уравнений для макроскопических переменных л, v0 и Т, а также р
и q. Дл? решения этих уравнений требуются более полные граничные условия,
чем для решения уравнения Навье—Стокса, и поэтому с помощью полученных
уравнений могут быть решены более общие задачи. Можно показать, что если
поток таков, что свойства малого элемента газа не изменяются заметно в течение
времени столкновения (т. е. в течение среднего времени между столкновениями),
то за время нескольких столкновений р и q достигают значений, даваемых
уравнениями
j^4f{M <5Л1>
<5-12>
Эти уравнения справедливы во многих случаях, хотя они не применимы в таком
особом случае, как прохождение газа через фронт ударной волны.
Если известна зависимость скорости и температуры от положения, то
уравнения E.11) и E.12) образуют систему из двух уравнений для q и Р. Члены,
взаимосвязывающие эти уравнения (вторые члены в правой части каждого из
уравнений), выражаются через вторые производные. Если скорость и темпера-
тура газа не изменяются заметно на длине среднего свободного пробега, то эти
взаимосвязывающие члены оказываются пренебрежимо малыми, и уравнения
E.11) и E.12) сводятся к соотношениям Навье—Стокса. Дополнительные
члены важны при изучении газов малой плотности, где средняя длина свобод-
ного пробега велика и газ нельзя рассматривать как сплошную среду.
§ 6. ВЛИЯНИЕ ХИМИЧЕСКИХ РЕАКЦИЙ И ВНУТРЕННИХ СТЕПЕНЕЙ
СВОБОДЫ МОЛЕКУЛ
До сих пор в настоящей главе обсуждалась кинетическая теория нереаги-
рующих смесей газов, состоящих из простых молекул. Предполагалось, что
молекулы сферически симметричны и не обладают внутренними степенями
свободы. Мы начнем этот параграф с обсуждения изменений, которые необхо-
димо внести в уравнения переноса, когда в смеси многоатомных газов проис-
ходят химические реакции1). Затем мы изложим общую теорию явлений пере-
носа в газах, молекулы которых обладают внутренними степенями свободы.
В связи с этим будет дана оценка поправочного члена Эйкена. Для много-
атомных молекул не было сделано обширных расчетов. Однако материал, при-
водимый в этом параграфе, полезен с точки зрения указания путей, по которым
может далее развиваться теория многоатомных газов.
х) Диффузия возбужденных или ионизованных молекул относительно молекул того же
сорта, находящихся в основном состоянии, является аномальной вследствие сильных резо-
нансных сил [101.
§ б. Влияние химических реакций и внутренних степеней свободы молекул 395
А. УРАВНЕНИЯ ПЕРЕНОСА ДЛЯ РЕАГИРУЮЩЕЙ
ГАЗОВОЙ СМЕСИ
В § 1 было показано, что правая часть уравнения Больцмана пропорцио-
нальна числу молекул определенного сорта, приобретаемых «группой» молекул
в фазовом пространстве за счет столкновений. Следовательно, если столкнове-
ния приводят к химической реакции, то правая часть уравнения Больцмана
должна быть видоизменена. Это изменение уравнения Больцмана, очевидно,
повлияет на интегральные свойства, используемые при выводе уравнений пере-
носа. Интеграл
J-JVHv, F.1)
равен общей скорости увеличения молекул f-ro сорта за счет столкновений с
молекулами /-го сорта в единице объема. (При отсутствии химических реакций .
этот интеграл, конечно, равен нулю.) Отсюда для реагирующей газовой смеси
общая скорость увеличения (в единице объема) числа молекул i за счет столкно-
вений со всеми типами молекул, включая /-й сорт, будет
\ ь F.2)
где К/ — общее увеличение числа молекул /-го сорта в единице объема за еди-
ницу времени за счет химических реакций. Так как при химической реакции
общая масса сохраняется, то
2*т,А:, = а (б.З)
i
Условие F.2) при выводе уравнения непрерывности заменяет собой условие
B.32). В столкновениях, сопровождающихся химическими реакциями, масса,
импульс и энергия сохраняются. Поэтому B.33) остается неизменным, за
исключением того факта, что сумматорными инвариантами теперь являются
mh т(\( и x\^n{V\ + u<*H->, где upH-> — энергия внутренних степеней свободы
молекулы f-го сорта.
Используя условие F.2), получаем уравнение непрерывности для f-й ком-
поненты газа :
^ { )Ki, F-4)
которое заменяет уравнение B.42). Член К/ представляет собой интенсивность
источника образования молекул /-го сорта за счет химических реакций. Урав-
нение непрерывности полной системы [уравнение B.43) ]
F.5)
остается неизменным (так как т{ — сумматорный инвариант). Уравнение
F.5) может быть также получено из уравнений F.4) путем умножения каждого
из них на т{ и последующего суммирования по всем i.
Импульс системы из двух сталкивающихся молекул сохраняется в любых
столкновениях, в которых происходят химические реакции. Следовательно,
как и прежде, для уравнения движения мы имеем
^**- F.6)
Уравнение баланса энергии может быть получено способом, совершенно ана-
логичным способу, описанному в § 2. Для многоатомных молекул необходимо
учесть только то, что величина \рь входящая в общее уравнение переноса, равна
теперь не поступательной энергии молекулы, а общей ее энергии — поступатель-
ной плюс внутренней. Это изменение приводит к тому, что в уравнении B.49)
396 Гл. 7. Кинетическая теория разреженных газов
?)(пост.) заменяется на О, в результате чего уравнение баланса энергии приобре-
теат вид
.(XI..VI.), F.7)
где О — термодинамическая внутренняя энергия на 1 г смеси
0 = ±-2п(т@(; F.8)
здесь 0t — энергия на 1 г каждой составляющей.
Уравнение баланса энергии можно записать также через температуру,
как это было сделано при получении уравнения B.50). При этом окончательное
уравнение имеет вид, несколько отличный от вида уравнения для одноатомных
газов. Используя F.4) для исключения Щ/Ы, появляющегося при дифферен-
цировании О, получаем
+ 2 Щ (V, • X,) - 2 т, К О, + 2 О, (¦? • щ т, V,). F.9)
В этом уравнении
есть средняя теплоемкость на 1 г при постоянном объеме.
Выражения для потоков через функцию распределения при наличии в
газовой смеси химических реакций не изменяются. В результате столкновений,
сопровождающихся химическими реакциями, в неравновесных системах проис-
ходит воЗхмущение функции распределения. Однако этот эффект мал и в боль-
шинстве случаев им можно пренебречь [11—13].
Б. ВЛИЯНИЕ ВНУТРЕННИХ СТЕПЕНЕЙ СВОБОДЫ МОЛЕКУЛ
НА ЯВЛЕНИЯ ПЕРЕНОСА (ПОПРАВКА ЭЙКЕНА)
Наличие внутренних степеней свободы у молекул не изменяет выражений
для коэффициентов диффузии и вязкости, записанных через функцию распре-
деления. Выражение для вектора плотности теплового потока, очевидно, видо-
изменяется, так как при выводе уравнения баланса энергии поступательная
энергия заменяется на величину 1l2rniV2i + u(BH->. Отсюда для многоатомных
молекул выражение B.26) должно быть заменено выражением
Здесь вектор q — общая плотность потока энергии. Прямым следствием видо-
изменения выражения для q, если учесть, что в хорошем приближении и(вн->
не зависит от V,-, является то, что второй член F.11) просто добавляется ко
второму члену выражения для плотности теплового потока D.64), вследствие
чего
где ht — энтальпия на одну молекулу i-го сорта
Л. = -|-/сТ + и<вн>. F.13)
Однако наличие у молекул внутренних степеней свободы, помимо отмеченного
эффекта, может влиять на величину потока за счет запаздывания перехода
энергии от поступательных степеней срободы к внутренним степеням свободы.
§ 6. Влияние химических реакций и внутренних степеней свободы молекул 397
Вследствие возможности неупругих столкновений коэффициент тепло-
проводности оказывается чувствительным к виду внутренних степеней свободы
молекул. Точное выражение для коэффициента теплопроводности зависит от
вероятности перехода энергии от одних степеней свободы к другим и особенно
от вероятности передачи энергии от поступательных степеней свободы к внут-
ренним степеням свободы. Вследствие того, что точное рассмотрение проблемы
сложно, мы вначале коротко обсудим «предположение Эйкена», которое поз-
воляет быстро оценить влияние внутренних степеней свободы на коэффициент
теплопроводности.
Эйкен [14] использовал предположение,, применимое, если скорость пере-
ходов энергии между молекулами велика, или, иными словами, если скорость
переходов энергии такова, что распределение молекул по различным состояниям
является равновесным распределением, соответствующим локальной темпера-
туре1). Рассмотрим газ, состоящий из молекул одного сорта. В соответствии с
формулами B.10) и B.11) гл.8, если газ состоит из молекул одного сорта, не
обладающих внутренними степенями свободы, коэффициент теплопроводности
в первом приближении выражается следующим образом:
1 = \пСщ. F.14)
Молекулы газа могут находиться в различных возбужденных квантовых состоя-
ниях2). Эти различные квантовые состояния молекул можно рассматривать как
различные составляющие газа. В такой интерпретации газ является хими-
чески реагирующей смесью с большим числом компонент, каждая из которых
не обладает внутренними степенями свободы. Неупругие столкновения рассмат-
риваются как химические реакции. Массы всех молекул равны. В хорошем
приближении одинаковы также и все потенциалы взаимодействия.
Для смеси молекул с одинаковыми массами и с одинаковыми законами
взаимодействия между любыми двумя молекулами коэффициент термодиффузии
равен нулю. Отсюда по F.12) вектор плотности потока энергии для такой смеси
имеет вид
q = _ тП_ + ^щ^-кТ + ир»->) V,. F.15)
Здесь ufBH-> — энергия внутренних степеней свободы молекулы в квантовом
состоянии i, а А(о) — коэффициент теплопроводности системы, рассматриваемой
как смесь. Из F.14), учитывая, что теплоемкость Cv на 1 г равна Щ2гп, получаем
Am x
Скорость диффузии определяется формулой D.3), которая для рассматри-
ваемой простой смеси приобретает вид
Здесь ?D — коэффициент само диффузии. Величина п^п имеет значение доли
молекул в квантовом состоянии i в смеси. Вообще эта величина сложным
образом зависит от всех градиентов. Чтобы получить приближение Эйкена,
предположим, что доля молекул в состоянии i определяется равновесным
х) В гл. 13, § 7 показано на основании экспериментальных данных по влиянию давления
на распространение микроволн в газах, что для большинства молекул скорость передачи
вращательной энергии при столкновениях сравнима со скоростью передачи поступательной
энергии. Предположение Эйкена разумно для вращательных степеней свободы, но оно не
справедливо для колебательных степеней свободы, где, как известно, для передачи энергии
требуется большое число столкновений (от 50 до 5000).
2) Концепция квантовых состояний введена здесь не ради точности, а для удобства об-
суждения вопроса.
398 Гл. 7. Кинетическая теория разреженных газов
(максвелловским) распределением при соответствующей локальной температуре.
Далее
э (пЛ Г d (niW дТ /r ia\
Подставляя это выражение в формулу для диффузионной скорости F.17)
и используя тот факт, что 2 пУ( = 0 (когда все mt равны), из F.15) для вектора
плотности потока энергии получаем
Ч№)]г FЛ9)
Здесь OjBH> = ujBHY"J — энергия внутренних степеней свободы на единицу
массы молекулы в состоянии /.
Введем 0<BH> — среднюю энергию внутренних степеней свободы на 1 г
и <?<""•> — теплоемкость внутренних степеней свободы на 1 г
F.20)
2,
Lv ~С» 2т ~ 2, Ui W Ц
Используя эти выражения, вектор плотности потока энергии и коэффициент
теплопроводности системы можно представить в виде
4 = -*-wT> <6-22)
vV. F.23)
В последнем выражении для коэффициента теплопроводности введено отно-
шение теплоемкостей : у — CpjCv = 1 + klCvm.
Безразмерное отношение q JZ)\r) является функцией температуры и имеет
значение порядка единицы. Полагая это отношение точно равным единице,
коэффициент теплопроводности можно записать следующим образом :
* = -^(Vy-5)Cvri. F.24)
Множитель (9 у — 5)/4 хорошо известен как поправочный множитель Эйкена
для теплопроводности многоатомных молекул. Первоначально Эйкен получил
его, исходя из простых предположений. Вследствие того что переход энергии
от поступательных степеней свободы к внутренним степеням свободы затруднен,
выражения F.23) и F.24) для А недостаточно хорошо согласуются с экспери-
ментальными данными при обычных температурах. Однако при достаточно
высоких температурах формула F.23) дает более точное совпадение с экспери-
ментом.
В. ФОРМАЛЬНАЯ КИНЕТИЧЕСКАЯ ТЕОРИЯ МНОГОАТОМНЫХ МОЛЕКУЛ1)
Кинетическая теория многоатомных газов была создана путем приложения
формальных методов обычной кинетической теории к случаю многоатомных
молекул или молекул, обладающих внутренними степенями свободы. В кине-
тической теории многоатомных молекул поступательные степени свободы
^Теория, развитая в этой главе, является итогом работы Ванг Чанг и Уленбека[15].
Простое изложение теории было также независимо дано Дж. де Буром (не опубликовано).
[См. также работу J. S. Dahler, Journ. Chem. Phys., 30, 1447 A959). — Прим. ред.]
§ 6. Влияние химических реакций и внутренних степеней свободы молекул 399
удобно рассматривать классически, а внутренние степени свободы — квантово-
механически. Введем функции распределения //(г, v, /) для каждого квантового
состояния и Индексом i обозначается ряд квантовых чисел {/}= il9 i2, *3> • • •>
характеризующих внутренние состояния молекулы1). Каждая функция рас-
пределения удовлетворяет собственному уравнению Больцмана.
Интегралы столкновений можно записать через 1%(е9О9ф) — дифферен-
циальные эффективные сечения рассеяния на углы 0, фу где индексами /, /
обозначен^ состояния молекул до столкновения, а индексами fc, I — состояния
молекул после столкновения ; через g обозначена абсолютная величина началь-
ной асимптотической относительной скорости двух сталкивающихся молекул.
Направление начальной относительной скорости выбрано в качестве оси
полярной системы координат. Далее, используя те же соображения, что и в
§ 1, получаем более общее уравнение Больцмана. Число молекул, теряемое
группой молекул в состоянии i за счет столкновений с молекулами в состоянии
/, можно записать через эффективное сечение Щ [по аналогии с A.10)] в виде
, = 2 WItttjgtjtSunOdOdtdvtdvj. F.25)
к, iJJJ
Аналогично число молекул, приобретаемых группой молекул в -состоянии i
за счет столкновений, в которых образуются молекулы в состоянии /, выра-
жается следующим образом :
2 ttgt F.26)
k, lJJJ
Здесь V/,7 и V/ — скорости молекул перед такими столкновениями, в которых
конечными скоростями являются v, и Vy. Пользуясь фактом существования
обратных столкновений2), можно записать
. F.27)
Таким образом, обобщенное уравнение Больцмана имеет вид
- 2 fff(M-/,/,)fo/?j<S,9,f*)sine*)<fj*<!v,. F.28)
Это обобщенное уравнение Больцмана было решено методом возмущений в
двух предельных случаях. В одном случае предполагается, что переходы энер-
гии между поступательным и внутренним движением осуществляются легко,
в другом —эти переходы затруднены. Теория и результаты решения уравнения
Больцмана в рассматриваемом случае очень похожи на случай одноатомных
газов. Ниже обсуждаются некоторые результаты этой теории для газа, состоя-
щего из одной компоненты.
1. Обмен энергией между поступательным и внутренним движениями не
затруднен. Рассмотрим случай, когда переходы энергии осуществляются легко.
Вид уравнений переноса при этом сохраняется таким же, каким он был для
одноатомного газа. Тензор давлений, однако, выражается следующим образом :
, F.29)
где S —тензор скорости сдвига, определенный формулой D.16).
г) В предыдущих параграфах через //(г, v, t) обозначалась функция распределения i
составляющей смеси. Так как сейчас мы ограничиваемся случаем газа, состоящего из одной
компоненты, то во избежание недоразумений следует помнить, что здесь индексом / обозна-
чены различные квантовые состояния молекул.
2) Заметим, что gki ifa йчн dv\ = gij /{/ tfv/ dvj. Последнее было доказано, однако, только
для случая невырожденных состояний внутреннего движения.
400 Гл. 7. Кинетическая теория разреженных газов
Коэффициент г] аналогичен коэффициенту вязкости, появляющемуся в
кинетической теории одноатомных молекул, и называется коэффициентом
сдвиговой вязкости. Коэффициент к — дополнительный коэффициент переноса,
называемый коэффициентом объемной вязкости1). Этот коэффициент тесно связан
с «временем релаксации», характеризующим время, необходимое для перехода
энергии от поступательных степеней свободы к внутренним.
Удобно рассматривать каждый вид внутреннего движения или внутренних
степеней свободы отдельно и определять время релаксации хь характеризую-
щее эти каждые виды. Можно показать, что объемная вязкость связана с вре-
менами релаксации т, следующим образом :
где сф — вклад внутренних степеней свободы в удельную теплоемкость на одну
молекулу cv.
Выражение для плотности потока энергии совпадает с выражением, полу-
ченным в простом случае. Однако коэффициент теплопроводности удобно рас-
сматривать как сумму двух членов, один из которых учитывает влияние посту-
пательного движения, а другой — внутренних степеней свободы
Ч==-(А(пост.) + Я(вн.))_^.. F.31)
Подробные выражения для коэффициентов переноса имеют следующий вид:
1) Коэффициент сдвиговой вязкости
^' ' kj
х JJJ [у4 sin2 в + -| у2 Ле (l - -| sin2 в)] у3 е~+ Ц (g, 0, ф) sin в dd dф dy; F.32)
здесь
*< = -§-> F.33)
где Е( — энергия f-ro состояния
Ле = ek + et — ei — г;. F.34)
Приведенная относительная скорость определена формулой B.12).
2) Коэффициент теплопроводности
*~»—
__15 ,
4
т Г4»
2/п | ас - Ь2
Зс<вн->
Я(вн.) = _ ^1__ f?— * L (б.Зб)
где
а =
2 Г пт
X JJJ{3 (Ае)* + 2 [(в, - ej) у - (ек - в,) у']} X
X у3 «г?1 /gf (g, 0, ^) sin 0 dB йф dy, F.37)
Иногда он называется коэффициентом вязкости расширения.
§ 6. Влияние химических реакций и внутренних степеней свободы молекул 401
с = 4
х JJJy» е-У It) (g, в, ф) sin 0 йв йф йу, F.38)
W 1 ^. „, _
nm
х JJJ {у4 sin2 в + у* (Ле) (j + cos2 0]J у8 er* Ifj (g, в, ф) sin в йв йф йу, F.39)
где
у'2 = у2 - Ле F.40)
и 4ВН) — вклад внутренних степеней свободы в теплоемкость на одну молекулу.
3) Время релаксации
2№ X
х JJJr3 г-v /f/(g, 0, ^) sin 0d0 d^ ^. F.41)
2. Обмен энергией между поступательным и внутренним движениями
затруднен. Результаты теории для случая, в котором переходы энергии относи-
тельно редки, выглядят отчасти по-иному. Для простоты сосредоточим наше
внимание на молекулах, которые имеют только один вид внутренних степеней
свободы. Все рассуждения легко могут быть перенесены на молекулы, обладаю-
щие несколькими видами внутренних степеней свободы. Вследствие того, что
равновесие устанавливается медленно, можно рассматривать две темпера-
туры — температуру, характеризующую поступательную энергию Т<пост->,
и температуру, характеризующую внутреннюю энергию Т<вн->.
Обе температуры связаны уравнением, которое дополняет полученные
ранее уравнения переноса. Это уравнение имеет вид
(б.42)
Здесь, как и ранее, 4ВН) — вклад в теплоемкость внутренних степеней свободы;
х — время релаксации; Л<вн-> — коэффициент теплопроводности, характеризую-
щий перенос энергии внутренними степенями свободы. Если отвлечься от влия-
ния члена, учитывающего теплопроводность, то из уравнения F.42) следует,
что температура Т<вн-) экспоненциально стремится достигнуть поступательной
температуры ТЧП0СТ) с характерным временем г.
Обычные уравнения переноса остаются неизменными. В данном случае
тензор давлений не включает члена с объемной вязкостью. Плотность потока
энергии равна сумме двух членов :
q = - Л<пост-> А Т<пост-> - А<вн-> А Г<вн->. F.43)
Подробные выражения для коэффициентов переноса имеют следующий вид:
1) Коэффициента сдвиговой вязкости
1 8 ]
X
X JJjV <rv% /Упр. (g, 0, ф) sin3 в dd йф dy, F.44)
где /упр. —эффективное сечение упругих столкновений. В данном случае на
величину коэффициента вязкости влияют только упругие столкновения.
402 Гл. 7. Кинетическая теория разреженных газов
2) Коэффициент теплопроводности
1_ =__32 1_ у.
Жпост.) "~ 75 УятРТ (-Г6-*J Xt
I k,l
X JJJV етУ /упр. (g, 0, ф) sin3 в йв йф dy, F.45)
8{
к, I
X JJj> е^1 /упр. (g, 0, ф) sin 0 d0 4* dy. F.46)
3) Время релаксации
X JjjV8 г-у% 1% (g, 0, ф) sin 0 d0 rf^ dy. F.47)
Чтобы использовать приведенные здесь формулы, а также формулы F.32) и
F.41), необходимо знать дифференциальные эффективные сечения выбираемых
моделей. Следует отметить, что последние для реальных моделей вычислены
еще не были.
Г. НЕКОТОРЫЕ СПЕЦИАЛЬНЫЕ МОДЕЛИ
(ТВЕРДЫЕ ОВАЛОИДЫ, ШЕРОХОВАТЫЕ СФЕРЫ, НАГРУЖЕННЫЕ СФЕРЫ)
Развитию общей формальной теории многоатомных молекул, в общих
чертах изложенной выше, предшествовали различные исследования явлений
переноса для некоторых специальных моделей молекул, обладающих внутрен-
ними степенями свободы. Вследствие того, что из формальной теории еще не
были получены численные результаты, мы изложим здесь некоторые результаты
для твердых овалоидов, совершенно шероховатых сфер и нагруженных сфер
1. Твердые овалоиды. Ишида1) развил кинетическую теорию газа, состоя-
щего из твердых, несферических молекул, не обладающих, помимо отмеченных
свойств, какими-либо другими внутренними степенями свободы. Он получил
обобщенное уравнение Больцмана, которое приложимо к описанию этого типа
молекул. Из видоизмененного таким образом уравнения Больцмана получаются
обычные уравнения переноса, а также дополнительное уравнение, выражающее
закон сохранения момента количества движения, имеющее вид
М • {и? + (уо • Ww«)| + 4-W * Ишо • М) + пЩТЩ) - 6 = 0; F.48)')
здесь G — внешний момент сил, действующий на отдельные молекулы; М —
тензор момента инерции; ш0—средняя угловая скорость молекул; Q = ш — ш0 —
относительная угловая скорость молекул. Если молекулы сферические, то
дополнительное уравнение не представляет интереса, так как все моменты коли-
чества движения при этом характеризуют макроскопическое движение газа
(или, наконец, в таком газе отсутствует возможность обмена между макроскопи-
ческим моментом количества движения и моментом, связанным с вращением
молекул). Однако если молекулы несферические, то возможность обмена момен-
тами количества движения приводит к некоторым интересным следствиям,
каковыми в данном случае является возникновение дополнительных явлений
г) Формально результаты Ишида [16] правильны, однако используемые им удельные
переменные, а также якобианы, встречающиеся при интегрировании, требуют дальнейшего
уточнения.
2)Это уравнение, как показал Кертисс [21, 22], неверно.
§ 6. Влияние химических реакций и внутренних степеней свободы молекул 4Q34
переноса, связанных с появлением времени релаксации. Описываемая модель
не была изучена в такой степени, чтобы на основе теории Ишида можно было
бы получить какие-либо численные результаты для коэффициентов переноса.
2. Совершенно шероховатые сферические молекулы1). Одной из простейших
идеализированных моделей, для которой были получены численные результаты,
является модель совершенно шероховатых твердых сфер, предложенная Брай-:
ном [17]. Изложенный в настоящей главе метод Чепмена и Энскога был рас-1
пространен на эту модель Пиддаком [18, 2 ]. В модели совершенно шероховатых
сфер молекулы при столкновениях не скользят одна относительно другой:
Более точно, модель шероховатых сфер такова, что относительная скорость в
точке соприкосновения при столкновении меняется на обратную. Сталкиваю-
щиеся молекулы могут обмениваться моментами количества движения как
между собой, так и с поступательным движением. Иными словами, действующие
между молекулами силы не строго центральны. Тем не менее теория значи-;
тельно упрощается вследствие того, что в данном случае нет необходимости;
вводить дополнительные координаты, описывающие ориентацию молекулы.
Функция распределения /(г, v, ш, t) зависит от радиуса вектора, линейной ско-1
рости молекулы и угловой скорости молекулы ш.
Решение уравнения Больцмана для функции /(г, v, ш, t) может быть полу-
чено тем же методом, который использовался для решения аналогичной пробг;
лемы в случае точечных молекул, взаимодействующих по законам центральных»
сил. Результаты могут быть выражены через величину а — безразмерный)
радиус вращения
« = -^7 <б-49>
где т, а и / — соответственно масса, диаметр и Момент Инерции шероховаты?
сферических молекул. Величина а изменяется от1 нуля, когда вся масса cocpeL
доточена в центре сферы, до 2/з> когда вся масса распределена по поверхности
сферы. Для сферы однородной плотности а — 2/в.
Резюмируем результаты кинетической теории для шероховатых' сфер;,
выписав первые приближения коэффициентов переноса. Чтобы сравнить их
с результатами для гладких сфер, выражения для коэффициентов переноса
представим в виде произведений значений коэффициентов для гладких сфер2}
(в квадратных скобках) на поправочные множители (в фигурных скобкахK)
О) (\\ __ Г 3 1/ kT I \mi qi + "*2 а2 + (mx f m2) аг a2 \
•^eW - L 8"^ |f 2я/л12 J W a, + m,a2 \- 2(тг'+ m.) a, aJ'
75 M1^] / 12Q + «J C7+151a + 50a2)
64? Г Hm\ \ l»02+5a+101a« + 102a»)
г) Эта модель может быть изучена с помощью метода (разработанного Ванг Чанг и^
Уленбеком), который изложен в разд. В. Результаты при этом совпадают с приведенными-
здесь.
2) Коэффициенты переноса для гладких сфер получены в гл. 8, § 3.
8) Величины al2 и ^2 — соответственно диаметр столкновения и приведенная масса {
двух сталкивающихся молекул : al2 = *ki?i + ?г) и (l//*i2) = 0/^) + A/пц)
404
Гл. 7. Кинетическая теория разреженных газов
Для иллюстрации хараюгера отклонения значений коэффициентов для
шероховатых молекул от их значений для гладких сфер на фиг. 114 даны попра-
вочные множители в функции от а для коэффициентов самодиффузии (Ь) и
вязкости (а). Следует отметить, что на основании выписанных формул величина
отношения тЛB)/кг)A) для шероховатых сфер при а = 0 равна 5,55, в то
время как для гладких сфер эта величина равна 15/4 = 3,75. Это есть следствие
того факта, что модель шероховатых сфер допускает обмен между вращатель-
ной и поступательной энергиями.
Для иллюстрации связи коэффициента теплопроводности с коэффициентом
вязкости выражение для АB) можно записать в виде
CF + 13а) C7 + 151а + 50а2) \
10A2+75а + 101а2 + 102а3)}'
F.54)
Интересно сравнить это выражение с формулой F.23), написанной на
основе предположения о быстрых переходах энергии. Если в F.23) мы примем
Cv = Зк/пг и используем в качестве q ?D\r\ величину, полученную на основе
формул F.51) и F.52) для шероховатых сфер, то коэффициент теплопроводности
будет иметь вид
к п(\\\ 3C7+101а + 50а2)
4A) { -^6ТТ
Отношение значений коэффициента теплопроводности, рассчитанного по фор-
муле F.54) для шероховатых сфер, к значениям, рассчитанным по формуле
F.55), представлено на фиг. 115. Из графика видно, что для а = О отношение
О Щ 02 0,3 Ofl 0,5 Q.6 0.7
а
Фиг. 114. Отношения коэффициентов
вязкости (а) и самодиффузии (b) для
шеррховатых сфер к тем же коэффи-
циентам для гладких сфер.
О 0,1 0,2 0,3 0,4 0,5 0,6 Q7
а
Фиг. 115. Отношение коэффициента
теплопроводности для шероховатых
сфер к коэффициенту, вычисленному
с помои»ью видоизмененного прибли-
жения Эйкена.
равно единице ; это получается вследствие того, что в данном случае переходы
энергии осуществляются легко й равновесие устанавливается очень быстро. Для
других значений а отклонение отношения от единицы характеризует влияние
релаксации на теплопроводность. Из графика следует также, что поправка
Эйкена дает удовлетворительные результаты с ошибкой в пределах от 10°/0 и
меньше. Последнее подтверждается также вычислениями коэффициента
теплопроводности, приведенными в гл. 8.
§ 6. Влияние химических реакций и внутренних степеней свободы молекул 405
3. Нагруженные сферы. Для газа, состоящего из шероховатых сферических
молекул, время релаксации, т. е. время, потребное для установления равно-
весия между поступательной и внутренней энергиями молекул, еще не вычис-
лено. Однако соответствующие расчеты были выполнены Джинсом [19] для
газа, состоящего из молекул, которые можно назвать «нагруженными сферами».
В этой модели молекулы — сферические с диаметром а и массой т. Центр
тяжести молекулы находится на малом расстоянии д от геометрического центра.
Предполагается, что Ма и что распределение массы имеет ось симметрии.
Молекула имеет момент инерции / относительно оси, проходящей через центр
масс перпендикулярно оси симметрии. В работе Джинса используется раннее
максвелловское изложение кинетической теории, поэтому результаты Джинса
непосредственно не сравнимы с результатами, обсуждаемыми здесь. Тем не
менее некоторые его результаты представляют интерес.
Нагруженные сферические молекулы при движении колеблются и пере-
дают вращательную энергию при столкновениях. Отсюда можно выяснить,
как устанавливается равновесное распределение энергии, а также найти время,
необходимое для установления этого равновесия. Во-первых, необходимо отме-
тить, что столкновения не могут влиять на вращение молекул относительно их
оси симметрии. Поэтому если рассматривать только этот тип вращения, то меха-
низм для достижения равновесия отсутствует. Энергия вращения около других
двух осей может переходить в поступательную энергию и при этом может уста-
навливаться равновесие. Эта картина отчасти, но не вполне, похожа на дву-
мерное вращение двухатомных молекул.
Обозначим через и<пост-> и н<вр> соответственно среднюю поступательную
и среднюю вращательную энергии на молекулу (в последней исключена энергия
вращения около оси симметрии). Джине показал, что в однородном газе с плот-
ностью числа частиц п
Н<вр.) _ 2 ц(пост#) | _ 5 пр yjfi^. F.5б)
3
здесь р — постоянная, зависящая только от структуры молекулы,
р=1щ+_ущя (б57)
Из уравнения F.56) следует, что |u<bp-> — 2/3и<пост->[ — монотонная функция*
спадающая к нулю. Это согласуется с известными равновесными значениями
и(пост.) = zj^j и и(вР.) _, iff ддд двумерного ротатора. Уравнение F.56) также
показывает, что система приближается к равновесию. Величина |ц<вр-> —*/8и<пост-)|
экспоненциально убывает со временем, уменьшаясь в е раз в течение времени
(«времени релаксации»)
т = -3 F.58)
5рп Уи(пост.) • х
В этом выражении использована формула F.57) для р.
В соответствии с формулой B.1) гл. 1 среднее время между столкновениями
твердых сферических молекул равно
F.59)
При нормальных условиях это число имеет порядок 10~10 сек. Время релак-
сации
F.60)
Отсюда следует, что если момент инерции мал, то энергия передается очень
легко и время релаксации мало. Однако если момент инерции велик, то для
передачи энергии необходимо большое число столкновений и время, потребное
406 Гл. 7. Кинетическая теория разреженных газов
для достижения равновесия, становится большим. Увеличение асимметрии
молекулы, характеризуемое величиной <5, облегчает передачу энергии ; умень-
шение д увеличивает время релаксации. Следует, однако, отметить, что резуль-
таты, полученные на основе рассматриваемой модели, сомнительны, так как в
большинстве случаев передача энергии происходит вероятнее не вследствие
'асимметричного распределения массы, а вследствие несферичности потенциала
взаимодействия.
Методом, аналогичным изложенному в гл. 11, § 4, Джине рассмотрел также
распространение звука в газе, состоящем из нагруженных сферических молекул.
Он показал, что для частот °vy малых по сравнению с 1/т, скорость распростра-
нения звука в разреженном газе характеризуется отношением удельных тепло-
емкостей' у ч= 7/б- Такое отношение удельных теплоемкостей имеет газ, состоя-
щий из нагруженных сфер. Таким образом, при низких частотах вращательное
движение согласуется с возмущениями, создаваемыми проходящей волной.
•При высоких частотах v ^> 1/т скорость распространения характеризуется
,у = б/3. Такое значение у имеет газ, молекулы которого обладают только посту-
пательной энергией. Отсюда при частотах, больших по сравнению с обратной
величиной времени релаксации, вращательное движение не согласуется с вол-
новыми возмущениями. При частотах порядка 1/т происходит резонансное
поглощение звука. Аналогичные расчеты могут быть сделаны для шерохова-
тых сфер.
Приложение А
ЗНАЧЕНИЯ «СКОБОЧНЫХ» ВЫРАЖЕНИЙ,
ИСПОЛЬЗУЕМЫХ ПРИ ВЫВОДЕ КОЭФФИЦИЕНТОВ ПЕРЕНОСА
ЗАПИСАННЫЕ ЧЕРЕЗ ИНТЕГРАЛЫ ?№»
(А.2)
(A.3)
miimtmjfl* Г 595 о/1д) ^89
(nn+mj)* [ 16 Ь 8
) ^
16 Ь 8
^] (A.4)
+ щу\ (а.5)
(A.6)
[W,; S^{Wb W,],7 - - 8 _4__{^}.*> _ |fl||4)j, (A.7)
' !) Формулы (A.I)—(A.3), (A.6), (A.7), (A.9), (A.12), (A.13) даны по Чепменуи Kay
лингу [2]. Формулы (A.4), (А.5), (А.8), (A. 10), (A.ll) взяты у Мэзона [20].
Литература 407
[W,; Sft (Wf) Vf,\u = 4 -fc&p [f a$# - ICQ* + Щ*], (A.8)
= 8
,; ft
- Ът) Offt* + m) Q\)*> + 2m, my ?}?•«], (A.9)
X
x [-ff A2m? + 5mJ) Oft* - ^- Dm? + 5m,2) Д^>
+ -^- mf й\}* - 4 /п)Щ* + 7m,-m,^'» - 2m,-
= 8 i^w [SD0m'+168 m'm'
- -|- my2 (84/n? + 35m|) ^}-2> + -i- m2 A08m? + 133 mf) fl^«> -
- 14m, mj i^2-3) + 2m, mj ?$•*> + 2m? mf fl^«l, (A.I 1)
и если W, = W, VJi--~-WJ U, to
|W,; Wy],7 = - 4
; W'b/" "Г 7JH7W H^ + 4"т№»}'
1. Получить явное выражение для коэффициента диффузии тройной смеси.
2. Получить явное выражение для коэффициента теплопроводности бинарной смеси.
3. Вычислить коэффициенты переноса газа из шероховатых сферических молекул од-
нородной плотности.
ЛИТЕРАТУРА
1. Grad H., Communications on Pure and Applied Mathematics, 2, 331 A949).
2. С h a p m a n S., Cowling T. G., The Mathematical Theory of Non-uniform Gases,
Cambridge, 1939 (см. перевод: С. Чепмен, Т. Каулинг, Математическая теория
неоднородных газов, ИЛ, I960).
3. G г е е n H. S., Molecular Theory of Fluids, New York, 1952.
4. К i r k w о о d J. G., Journ. Chem. Phys., 15, 72 A947).
5. К irk wood J. G., Journ. Chem. Phys., 18, 817 A950).
6. E n s k о g D., Archiv fOr Mathematik, Astronomi och Fysik, 16, § 16A922); Диссертация,
Upsala, 1917.
7. С u г t i s s С F., H i r s с h f e 1 d e r J. O., Journ. Chem. Phys., 17, 550 A949).
8. Hellund E. J., U eh ling E. A., Phys. Rev., 56, 818 A939).
9. С h a p m a n S., D о о t s о n F. W., Phil. Mag., 33, 248 A917).
10. Buckingham R., Dalgarno A., Proc. Roy. Soc, A213, 506 A952).
11. Prigogine I., Xhrouet E., Physica, 15, 913 A949).
12. Prigogine I., Ma hie u M., Physica, 16, 51 A950).
408 Гл. 7. Кинетическая теория разреженных газов
13. С и г t i s s C. F., Диссертация, University of Wisconsin, 1948 (опубликована в виде
научного отчета по военно-морским проблемам СМ-476 Висконсинского университета).
14. Eucken A., Phys. Zs., 14, 324 A913).
15. W a n g - С h a n g С. S., U h 1 e n b e с k G. E., Transport Phenomena in Polyatomic
Molecules, University of Michigan Publication, CM-681, 1951.
16. I shi da Y., Phys. Rev., 10, 305 A917).
17. Bryan G. H., Brit. Assoc. Rep., p. 83 A894).
18. P i d d u с k Ft В., Proc. Roy. Soc, A101 A922).
19. J e a n s J. H., Dynamical Theory of Gases, Cambridge, 1904.
20. Ma sen E. A., Journ. Chem. Phys., 22 A954).
21. Curtiss С F., Journ. Chem. Phys., 24, 225 A956).
22. Curtiss С F., Journ. Chem. Phys., 26, 1619 A957).
ГЛАВА 8
ЯВЛЕНИЯ ПЕРЕНОСА В РАЗРЕЖЕННЫХ ГАЗАХ*)
В гл. 7 была детально изложена строгая кинетическая теория одноатомных
газов2). Конечными результатами этой теории являются формулы для различных
коэффициентов переноса, выраженные через интегралы щ*s), которые, в свою
очередь, зависят от законов сил межмолекулярного взаимодействия. В этой
главе будут даны оценки этих интегралов, а также расчеты коэффициентов
переноса для пяти различных сферически симметричных потенциальных функ-
ций. Наиболее полезной потенциальной функцией, для которой были сделаны
такие вычисления, является потенциал F-12) Леннарда-Джонса. Значительная
часть настоящей главы посвящается обсуждению 'расчетов для этого потен-
циала, примерам расчетов практических задач, а также определению пара-
метров межмолекулярного потенциала из экспериментальных измерений коэф-
фициентов переноса. Вообще можно сказать, что вопрос о явлениях переноса
в газах, состоящих из молекул со сферически симметричными потенциалами
взаимодействия, разработан достаточно хорошо.
Однако вопрос о явлениях переноса в газах, состоящих из молекул с несим-
метричными потенциалами взаимодействия, еще требует своего решения. Как
указывалось в конце предыдущей главы, некоторые расчеты были сделаны
лишь для специальных моделей — твердых овалоидов, шероховатых сфер и
нагруженных сфер. Была также разработана общая теория газов, молекулы кото-
рых обладают внутренними степенями СЕободы (см. гл. 7, § 6). Однако в настоя-
щее время эта теория еще не использовалась для изучения явлений переноса
в газах, состоящих из длинных молекул или из полярных молекул. Можно
сделать лишь приближенные расчеты коэффициентов переноса полярных газов
и смесей, которые включают одну полярную компоненту. Эти расчеты обсуж-
даются в конце настоящей главы.
§ 1. ВЕКТОРЫ ПЛОТНОСТЕЙ ПОТОКОВ И КОЭФФИЦИЕНТЫ
ПЕРЕНОСА
В гл. 1, § 2 было показано, что в элементарной кинетической теории кине-
тические коэффициенты определяются через потоки и градиенты различных
физических величин. Аналогичные результаты были получены в предыдущей
главе на основе строгой математической кинетической теории газов. Перед
анализом различных расчетов коэффициентов переноса целесообразно подвести
общий итог проделанной работы по определению коэффициентов переноса на
основе строгой теории, а также дополнительно рассмотреть некоторые вопросы,
1)Эта глава была подготовлена при участии доктора Висконсинского университета
Эллен Л. Спотц.
2) В гл. 1, § 2 дана элементарная теория явлений переноса. Там же обсуждены ограни-
чения и предположения теории Чепмена—Энскога, обсужденной в гл. 7. Настоящая глава
основывается на результатах теории одноатомных газов, изложенной в гл. 7.
410
Гл. 8. Явления переноса в разреженных газах
представляющие практический интерес. Следует отметить, что выражения,
данные здесь для векторов плотностей потоков, включают только первые
пространственные производные различных физических величин —температуры,
концентрации и средней массовой скорости. Вообще векторы плотностей потоков
включают высшие производные, и поэтому результаты, приводимые в настоящей
главе, применимы только в том случае, если градиенты перечисленных физи-
ческих величин малы. Приведенные в настоящей главе выражения для векто-
ров плотностей потоков и коэффициентов переноса справедливы только при
достаточно низких плотностях, когда можно пренебречь тройными столкно-
вениями. Помимо этого, обсуждаемые выражения для векторов плотностей
потоков, строго говоря, справедливы только для одноатомных газов и содер-
жат небольшую ошибку, если их применять к многоатомным газам. Внутренние
степени свободы не оказывают заметного влияния на поток массы (диффузию)
и поток импульса (вязкость). Поэтому коэффициенты диффузии и вязкости газов
из многоатомных молекул адекватно описываются приведенными в настоящей
главе формулами. Что же касается плотности потока энергии (теплопровод-
ности), то он включает как поступательную энергию, так и энергию внутренних
степеней свободы. Поэтому формула для коэффициента теплопроводности видо-
изменяется так, чтобы это обстоятельство принималось во внимание (поправка
Эйкена).
Существует принципиальное различие, которое следует отметить между
приведенными здесь выражениями для векторов плотностей потоков и выра-
жениями, данными в гл. 1, § 2. По элементарной кинетической теории перенос
массы возникает под действием градиента концентрации, а перенос энергии —
под действием градиента температуры. По строгой кинетической теории, помимо
отмеченных причин, перенос массы происходит под действием градиента темпе-
ратуры (эффект Соре, или термодиффузия), а перенос энергии — под действием
градиента концентраций (эффект Дюфура, или диффузионный термоэффект).
Эти дополнительные эффекты усиленно изучались с точки зрения так называе-
мых соотношений взаимности Онзагера, которые обсуждаются в гл. 11, § 2.
Выражения для плотности потока импульса и коэффициента вязкости, давае-
мые строгой кинетической теорией, по существу те же, что и выражения, давае-
мые элементарной теорией в гл. 1, § 2.
Этот параграф посвящен обсуждению выражений для векторов плотностей
потоков, полученных из строгой кинетической теории, аналогичных элементар-
ным выражениям B.9)—B.11) гл. 1. Обозначения, использованные в гл.7 для
обсуждаемых плотностей потоков, можно представить в следующем виде:
Плотность
потока *Pt
Статистическое
выражение для
плотности
потока
щ 1Л( V/ V/
112 Щ Tti[ Vх V/
Обозначе-
ние
1/
Р/
Я/
i = o
р
я
Плотности потоков обозначаются символом У; в скобках дано обозначение
физической величины, для которой написан символ плотности потока; индек-
сом i указана компонента смеси. Символами ],, р, и q, обозначены соответственно
вектор плотности потока массы, тензор давлений и вектор плотности потока
тепла 1-й составляющей многокомпонентной смеси. Соответствующими симво-
лами без индексов обозначены плотности потоков смеси. Вектор плотности
потока массы смеси тождественно равен нулю, так как диффузионная скорость
§ 7. Векторы плотностей потоков и коэффициенты переноса 411
V/ определена как скорость потока молекул r-го сорта относительно средней
массовой скорости газа1).
А. ПЕРЕНОС МАССЫ И КОЭФФИЦИЕНТЫ ДИФФУЗИИ
В элементарной кинетической теории, как следует из формулы B.9) гл. 1,
коэффициент диффузии определяется через поток молекул, возникающий под
действием градиента концентрации. В строгой кинетической теории ^компо-
нентной газовой смеси аналогичное выражение для вектора плотности потока
массы выглядит значительно сложнее :
^j^uj-DT^* /=1, 2, 3,...,v, A.1)
где uj выражается через градиенты молярных долей и давления, а также вклю-
чает члены, учитывающие влияние внешних сил Х^, действующих на молекулы
i
здесь пЛ — плотность числа частиц (т. е. число молекул в 1 см3) различных ком-
понент; п и q — соответственно плотность числа частиц и массовая плотность
смеси; Вц и DJ—^соответственно коэффициенты диффузии2) и термодиффузии
многокомпонентной смеси.
Из формулы A.1) следует, что поток массы может возникать по четырем
причинам : ,1) под действием градиента концентрации, как в элементарной кине-
тической теории ; 2) под действием градиента давления, как, например, во вра-
щающемся газе, где тяжелые молекулы выталкиваются в наиболее удаленную
от оси вращения часть объема3); 3) под действием внешних сил, которые, напри-
мер, приводят к появлению диффузии электрически заряженных частиц в
ионизованном газе под влиянием электрического поля4); 4) под действием гра-
диента температуры, приводящего к возникновению термодиффузии. При экс-
периментальном определении коэффициента диффузии используется первый
способ возбуждения диффузии. Коэффициент термо диффузии обычно измеряется
при наличии градиента концентрации и градиента температуры, когда в системе
достигается равновесное состояние.
Для гидродинамических приложений иногда удобнее заменять v формул
A.1) на v — 1 независимых соотношений6)
81пГ ^
d -
Здесь о2)/у — обычный коэффициент диффузии бинарных смесей.
1) Различные виды молекулярных скоростей, используемые в кинетической теории,
обсуждаемой в настоящей книге, определены в гл. 7, § 2. Там же дано определение тензора
давлений, вектора плотности потока тепла и температуры в кинетической теории. Особенно
следует быть осторожным в связи с определением диффузионной скорости. Некоторые авторы
предпочитают определять эту величину не относительно средней массовой скорости v0, а от-
носительно средней числовой скорости ш. В настоящей книге использовано первое опреде-
ление.
2) Коэффициент Dfj связан с обычным коэффициентом диффузии бинарной смеси Я)у
соотношением D.44) гл. 7. Для трехкомпонентных смесей соотношение D.44) гл. 7 пере-
ходит в B.41) гл. П.
8) Гравитационное ускорение приводит к диффузии под действием градиента давления,
но не к диффузии под действием внешних сил. Сила, действующая на молекулу при ускоре-
нии а, равна X/ = т/а. Подстановка этой силы в выражение A.2) приводит к тому, что
член, обязанный своим происхождением внешним силам, исчезает.
4) См. Чепмен и Каулинг [1 ], гл. 18, а также Герцфельд [2].
б) Вывод этих соотношений дан в гл. 7, § 4. Относительно обозначения см. примечание 1
на стр. 417.
412 Гл. 8. Явления переноса в разреженных газах
Во многих лабораторных экспериментах, проводимых с целью изучения
диффузии в нереагирующих газовых смесях, положение значительно упро-
щается. Когда в системе достигается постоянная температура и давление при
отсутствии внешних сил Хк, действующих на молекулы, диффузия происходит
только в одном направлении. В таких условиях соотношение A.1) можно ком-
бинировать с уравнениями непрерывности для смеси газов, чтобы получить
для концентраций дифференциальное уравнение второго порядка по коорди-
натам и первого порядка по времени. Если уравнения непрерывности B.42)
гл. 7 просуммировать по всем компонентам, то результирующее уравнение
может быть значительно упрощено путем использования того, что п—^щ
является величиной постоянной вследствие постоянства давления и темпе-
ратуры. Если диффузия происходит только в направлении х, то получаем
?2 n<("o + V,) =1-*>(*,/) = 0 . A.4)
Таким образом, средняя числовая скорость о>(х, /) = A/л) 2J л,(г>0 + Vt) не зави-
2J
сит от положения и является только функцией времени1). Уравнения непрерыв-
ности B.42) гл. 7 могут быть записаны через среднюю числовую скорость
Подставляя выражения для диффузионной скорости из формулы A.1) в послед-
нее уравнение, получаем
Это уравнение выражает собой обобщенный второй закон диффузии Фиш.
До сих пор все результаты относились к многокомпонентным системам.
Для двухкомпонентных систем формулы могут быть значительно упрощены.
Выражение A.1) для компоненты 1 из двухкомпонентной смеси можно записать
в виде
i1 = n1m1W1 = ^-m1m2^12u2-Dl^; A.7)
поскольку dx = — d2 и \г = — j2, то
-Z>i2 = -Z>21 Dl = -Dl. A.8)
Из формулы A.7) можно получить аналог общего уравнения A.3) для двух-
компонентных систем
Здесь мы ввели величину feT, определенную следующим образом:
г) Если диффузия происходит в некотором произвольном направлении, то уравнение
A.4) может быть записано в виде
Важно отметить, что если скорость не вращательная, то из последнего уравнения не
следует, что средняя числовая скорость независима от пространственной координаты.
§ 7. Векторы плотностей потоков и коэффициенты переноса 413
Эта величина, являющаяся мерой относительной значимости термодиффузии
и обычной диффузии, известна как термодиффузионное отношение. Величина
кТ определена так, что когда кт > 0, то компонента 1 перемещается в холодную
область, а когда кт < 0, то компонента 1 перемещается в нагретую область.
Рассмотрим обычную диффузию при отсутствии температурных градиентов.
Написав уравнение A.6) для двух компонент и учтя, что Dn = D22 = 0, получим
два уравнения, имеющие следующий вид:
э/ " эх Н^2 эх J WW эх • <1Л1'
Это уравнение справедливо для любой нереагирующей бинарной смеси газов
при постоянных температуре и давлении и при наличии диффузии только в
направлении х. Для системы, находящейся в покоящемся закрытом сосуде,
средняя числовая скорость со (t) равна нулю. Поэтому уравнение A.11) упро-
щается и приобретает вид
Э/ — Эх \-"ъ Эх J ' l1'1^
Это хорошо известный второй закон диффузии Фика1). Уравнение A.12) исполь-
зуется для анализа данных2) по диффузии в закрытых сосудах для определения
JDl2. Однако существует много практических случаев, когда имеет место испа-
рение, в которых co(t) не равно нулю, и должно использоваться более общее
уравнение A.11). Рассмотрим термодиффузию в бинарной смеси газов при отсут-
ствии внешних сил и градиентов давлений. Уравнение A.9) при этом имеет вид
Vi - V, — й? -2>и {I- ft) + *r I in Т). A.13)
Когда достигается равновесие, диффузионные скорости обращаются в нуль,
вследствие чего уравнение A.13) упрощается
Это уравнение может быть проинтегрировано, пссле чего получаем
Вследствие того, что кт не только функция температуры, но и функция состава,
полученный результат не является точным решением уравнения A.14). Однако
практически состав обычно изменяется так слабо, что изменением кт вследствие
изменения состава можно пренебречь, тогда как изменением кт вследствие
изменения температуры пренебрегать нельзя. Термодиффузионное отношение
сложным образом зависит от температуры, поэтому во избежание интегрирова-
ния при вычислении интеграла A.15) пользуются теоремой о среднем значении
интеграла. Это дает возможность написать
(l/r2^ n ifi^
здесь температура Т равна некоторой средней температуре3), лежащей между
Т и Г.
х) В первом приближении коэффициент диффузии не зависит от концентрации и урав-
нение поэтому сводится к
2)ГМ., например. [3].
8) Броун [4] предложил следующее соотношение для Т:
~ [т'-т)
1п
414 Гл. 8. Явления переноса в разреженных газах
Обычно уравнением A.16) пользуются при анализе экспериментов по
термодиффузии. Величина кт находится по равновесному составу в двух сооб-
щающихся сосудах, в одном из которых поддерживается температура Т, а в
другом — более высокая температура Т'.
Экспериментальные данные обычно выражаются через величины, обозна-
чаемые1) к\ или а и определяемые следующим образом:
#,* (/Сг)эксп. (кт)жсп. п 17Ч
Для табулирования экспериментальных значений к? необходимо пред-
варительно выбрать произвольно диаметр столкновения твердых сфер. Оче-
видно, что при сравнении экспериментальных и вычисленных величин к$ сле-
дует позаботиться о том, чтобы при приведении экспериментальных и вычислен-
ных величин кт использовались одинаковые диаметры столкновений. Величина
к* вводится с целью получения величины, слабо зависящей от температуры и
концентрации. Величина а используется потому, что она почти не зависит от
концентрации.
Б. ПЕРЕНОС ИМПУЛЬСА И КОЭФФИЦИЕНТ ВЯЗКОСТИ
В элементарной кинетической теории выражение для плотности потока
импульса и определение коэффициента вязкости дается формулой B.10) гл. 1.
Выражение для тензора давлений, получаемое в строгой кинетической теории,
имеет вид2)
A.18)
здесь г) — коэффициент вязкости, a S — тензор скорости сдвига, определяемый
следующим образом:
Символ f обозначает транспонированный тензор, который получается заменой
строк столбцами. Типичные диагональные и недиагональные элементы тензора
давлений равны
Физический смысл компонент тензора давлений показан на фиг. 113 (стр. 366).
Определение плотности потока импульса и коэффициента вязкости, давае-
мое формулой A.18), является непосредственным обобщением определения тех
же величин, данного в гл. 1, § 2, в элементарной кинетической теории. Для
х) В литературе к % обычно обозначается через Rt. Мы предпочитаем символ к*, так как
он согласуется с обозначениями приведенных величин, используемых в настоящей книге.
2) Более общее выражение для тензора давлений, которое справедливо для плотных
газов и жидкостей, дается в гл. 11. Там показано, что тензор давлений состоит из трех членов:
гидростатического давления, члена, включающего сдвиговую вязкость r\t и члена, включаю-
щего объемную вязкость х. Для разреженных одноатомных газов х = 0, вследствие чего
выражение A.18) является обоснованным. Для плотных одноатомных газов х ф 0. Для
многоатомных газов объемная вязкость х вообще мала, но не равна нулю и зависит от вре-
мен релаксации (гл. 7, §6).
§ 1. Векторы плотностей потоков и коэффициенты переноса
415
случая, представленного на фиг. 116, тензор давлений имеет упрощенный вид:
р о -v^w-
Op О
A.21)
Элемент тензора давлений рхх равен х-компоненте силы, действующей на еди-
ницу площади поверхности, перпендикулярной г-направлению. Отсюда из
Скорость и
Сила F
' Площадь А
Фиг. 116. Физическая картина, соответствующая тензору напряжений A.21).
выражения A.21) может быть получено обычное элементарное определение
вязкости :
Здесь 5 — расстояние между пластинами, имеющими площадь А и параллель-
ными плоскости XY, одна из которых перемещается под действием силы F со
скоростью и.
В. ПЕРЕНОС ЭНЕРГИИ И КОЭФФИЦИЕНТ
ТЕПЛОПРОВОДНОСТИ
Определение коэффициента теплопроводности А через плотность потока
энергии в элементарной кинетической теории дается формулой B.11) гл.1.
Аналогичное выражение для v-компонентной смеси одноатомных газов, полу-
ченное методами строгой кинетической теории в гл. 7, имеет вид1)
A.23)
В соответствии с этим выражением существуют три механизма передачи энергии
через многокомпонентный газ : 1) под действием градиента температуры, как
это объясняется элементарной кинетической теорией ; 2) потоком 2! n<V* моле-
г) Это выражение получено для газов из одноатомных молекул. В гл. 7, § б показано,
что для газов из многоатомных молекул вектор плотности потока тепла имеет тот же общий
вид. Разница заключается лишь в том, что член ъ/2кТ27л^У/ заменяется членом 27n,7z,V/,
в котором hi — энтальпия на молекулу /-й компоненты, равная (mid + kT). В некоторых
задачах в выражении для вектора плотности потока тепла необходимо учитывать тепло,
теряемое или приобретаемое за счет излучения. Этот вопрос рассматривается в гл. 11, § 3#
416 Гл. 8. Явления переноса в разреженных газах
кул в единицу времени относительно массовой скорости, причем каждая моле-
кула переносит с собой в среднем ь\гкТ тепловой энергии ; 3) «обратным про-
цессом» — термодиффузией, известной как эффект Дюфура. В лабораторных
экспериментах обычно измеряется поток, обязанный своим происхождением
только первому механизму.
Экспериментально теплопроводность определяется в системах, находя-
щихся в стационарном состоянии, в которых все V/ равны нулю и в которых
поток тепла имеет место только в одном направлении. В таких условиях выра-
жение A.23) упрощается и приобретает вид
) 0-24)
Здесь Т и Т — температуры двух граничных поверхностей, а (х' — х) — рас-
стояние между ними. В статических системах, в которых все скорости равны
нулю, в соответствии с уравнением F.7) гл. 7 и приведенным выше выражением
A.23) имеем
Если теплоемкость cv постоянна, как, например, в случае газа из одноатомных
молекул, где cv = */2ky то О = сТ//п, так что
Между этим уравнением и уравнением A.12) имеется значительная аналогия.
§ 2. СВОДКА ФОРМУЛ КИНЕТИЧЕСКОЙ ТЕОРИИ
ДЛЯ ЧИСТЫХ ГАЗОВ И МНОГОКОМПОНЕНТНЫХ СМЕСЕЙ
В предыдущей главе было показано, что коэффициенты переноса могут
быть выражены через ряд интегралов ??(/«s), массы молекул /л, и плотности
числа частиц щ различных компонент. В настоящем параграфе мы снова
займемся формулами для коэффициентов переноса и представим их в наиболее
удобном для непосредственных практических расчетов виде. Эти результаты
используются в следующих параграфах главы в расчетах коэффициентов переноса
при различных потенциальных функциях.Формулы для коэффициентов переноса
в настоящем параграфе мы запишем через молекулярные веса Мь молярные
доли xt и ряд величин ??(/'s)*, равных интегралам f3(/jS) для рассматриваемого
потенциала, деленным на аналогичные интегралы для твердых сферических
молекул. Все формулы для коэффициентов переноса даются лишь в первсм
приближении ; точные выражения для вторых приближений можно найти в
приложении А в конце настоящей главы.
А. ВЕЛИЧИНЫ Qds)*
Чтобы рассчитать коэффициенты переноса для какой-либо потенциальной
функции ф(г), сначала необходимо по формуле E.26) гл. 1 вычислить угол
отклонения %х) при столкновении. Для твердых сферических молекул легко
написать аналитическое выражение для %, но для любой реальной потенциаль-
ной функции необходимо использовать численные методы расчета. Имея од-
нажды рассчитанные значения % для большого числа различных g и ft, можно
х) В этой главе используются результаты анализа динамики столкновения двух тел, а
также различные обозначения, употребляемые при описании парных столкновений, данные
в гл. 1, §5.
§ 2. Сводка формул кинетической теории для чистых газов
417
вычислить двойные интегралы в формулах D.34) гл. 7 и получить таким
образом fl(/»s). Далее коэффициенты переноса могут быть рассчитаны
как функции температуры. Для вычисления коэффициентов переноса в первом
и втором1)приближениях необходимы интегралы i3(/|S), приведенные ниже:
Коэффи-
циент
п
А
3)
кт
i
Чистый
газ A-е
прибли-
жение)
/2B.2)
/2B'2)
/2G.1)
Смесь
A-е приближение)
/2B,2), ?A,1)
/2B.2), ?U,S)
s= 1,2,3
Ж11)
/2B.2), /2<М)
s = 1, 2, 3
Чистый газ
B-е приближение)
/2B.)
s l?B i
s = 2, 3, 4
/2B,2), ?(l,s)
s= 1,2,3
/2B,0 /2(!,s), /2C,3)
s= 1,2, 3,4, 5
/ = 2, 3,4
Точные формулы для коэффициентов переноса, записанные через f2<'» s\ при-
водятся ниже.
Поскольку выражения для % E.26) гл. 1 и для интегралов i3(/»s> D.34) гл. 7
являются исходными для всех последующих вычислений коэффициентов пере-
носа, проводимых в этой главе, то для удобства мы выпишем их здесь снова,
но в несколько измененном виде, а также определим дополнительно эффек-
тивные сечения Q(/):
dr/r*
B.1)
1 _ *L _ gfr)
г2 V^g1
-cos
B.2J)
B.3K)
1) В предыдущей главе различные порядки приближения для разных коэффициентов
переноса отмечались указанием числа членов разложения по полиномам Сонина. Например,.,
через ri(g) обозначался коэффициент вязкости, вычисляемый с учетом ? членов разложения
по полиномам Сонина. В этих обозначениях низшие приближения коэффициентов вязкости,
теплопроводности, диффузии и термодиффузии записываются следующим образом:
т?A), ЛB),Я)A),?гB). Это говорит о том, что для получения низших приближений коэффициен-
тов теплопроводности и термодиффузии в разложении по полиномам Сонина C.61) гл. 7
необходимо учитывать два члена ряда.
Из соображений удобства мы используем в этой главе обозначения, принятые Чепме-
ном и Каулингом [1 ]. Эти авторы обозначают первое отличное от нуля приближение, как
первое приближение. Упомянутым символам в обозначениях гл. 7 в этой главе соответствуют
обозначения Чепмена и Каулинга : [r)]lf [X]ly [?>]1у [кт]г. Высшие приближения обозна-
чаются следующим образом : [т?]2 = т?B), [А]2 = АC) и т. д.
2) По Чепмену и Каулингу, величина Q(O(g) равна функции 0</)(gp), умноженной на
2 njg. Мы предпочитаем использовать QW(g), так как она имеет размерность эффективного
сечения. По Чепмену и Каулингу, величины Q(l>s) совпадают с величинами Q(l)(s), однако
их не следует путать с величинами Q(i»s) по Энскогу (см. [1, гл. 9]).
8) Заметим, что
418 Гл. 8. Явления переноса в разреженных газах
Здесь у2 = 1121&?21кТ и [л — приведенная масса двух сталкивающихся молекул.
Если рассматриваются столкновения двух молекул /-го и /-го сортов, то в
качестве потенциальной функции следует использовать потенциальную функ-
цию (ри, характеризующую столкновения двух различных молекул. В этом
случае приведенная масса определяется по формуле A/fty) = A/гп,) + A/гпу).
Чтобы отметить, что интегралы Q(l>s) (T) соответствуют взаимодействию между
частицами /-го и /-го сорта, будем записывать их в виде Q\]> S\T). Для столкно-
вения между молекулами одного сорта все перечисленные величины обозна-
чаются соответственно как cpiiy Ри^^Щ и?2$'8) (Т).В расчетах коэффициентов
переноса для чистых газов, где имеют место только столкновения типа i — /,
нет необходимости отмечать индексами сорт молекул, но следует помнить о том,
что в формулах должна использоваться приведенная масса. В расчетах коэф-
фициентов переноса для многокомпонентных смесей, где случаются столкно-
вения обоих типов i — i и / — /, особое внимание должно быть уделено
точному указанию того, какое именно взаимодействие учитывается.
Для твердых сферических молекул1) диаметра а формулы B.2) и B.3)
упрощаются и приобретают вид
[QCOW = [l - 1 ^Т^] по\ B.4)
*>]тв.сф. = Щ^ -Ц^11- [Q«W • B-5)
Для этой элементарной модели эффективные сечения не зависят от g. Для
диффузии эффективным сечением, соответствующим модели твердых сфер,
является QA) = до*2, для вязкости и теплопроводности должно использоваться
2<2>2/2
Все потенциальные функции, обсуждаемые в этой главе, могут быть оха-
рактеризованы параметрами а и е. С помощью этих параметров потенциальные
функции могут быть записаны в виде
B.6)
где предполагается, что /(г/ст) имеет одинаковый вид для любых газов. Различ-
ные переменные кинетической теории можно привести к безразмерному виду
путем деления их на подходящую комбинацию параметров, входящих в потен-
циальные функции. Таким путем мы .определим следующие приведенные пере-
менные :
г*= — приведенное межмолекулярное расстояние,
b
b* = — приведенное прицельное расстояние,
Ф* = — приведенная потенциальная энергия межмолекулярного
е взаимодействия,
кТ
Т* = приведенная температура,
g*2 = -у /*g2/e приведенная относительная кинетическая энергия.
г) Модель твердых сфер подробно описывается в § 3. Мы приводим здесь результаты
этого параграфа с целью использования их при получении приведенных интегралов Q(l>s).
Только для твердых сфер y2nju/kTQ(l*s) пропорционально Q@.
§2. Сводка формул кинетической теории для чистых газов 419
Удобно определить приведенные величины для Q@(g) и ??(/>s)(T), разделив
их на соответствующие величины для твердых сфер1), т. е. записать
по)* _ _[&_
_
]тв,ф. - 1/а(в
Физический смысл ?№s>* и Q(/)* состоит в том, что они указывают на отличие
какой-либо модели молекулы от идеализированной модели твердых сфер.
В первом приближении формулы строгой кинетической теории для коэффи-
циентов переноса через упомянутые приведенные величины запишутся в виде2)
M-Ji — 2 U^2^<2'2)* )~m ' ( '
Эти результаты можно сравнить с результатами элементарной кинетической
теории [см. B.12) —B.14) гл. 1].
Основные исходные формулы для вычисления коэффициентов переноса
B.1) —B.3) могут быть теперь записаны в приведенных единицах
, ft*) = я - 2b* f , -*!^1 , B.12)
' J 1/ b*2 q?*(r*) v '
00
| A - cos'X)b*db*, B.13)
- /i 1 + / J о
B.14)
В выражения для коэффициентов переноса для чистых газов и смесей
часто входят три комбинации из ?№•>*, для которых мы вводим специальные
обозначения3). Этими комбинациями являются следующие:
г) Это согласуется с использованным в настоящей книге определением приведенного
второго вириального коэффициента, т. е.
в*(г*)= В(Т) =
2) При соответствующей интерпретации величин, входящих в формулу для Л), она ока"
жется справедливой также для коэффициента диффузии бинарных систем. Формулы для
г) и А не могут быть таким простым способом обобщены на случай бинарных смесей.
8) Приведенные здесь величины А*, в* и с* равны величинам а, в и с по Чепмену и
Каулингу [1, стр. 164], деленным на их значения для твердых сфер, т. е.
420 Гл. 8. Явления переноса в разреженных газах
Все эти три величины близки к единице.
Б. КОЭФФИЦИЕНТ ВЯЗКОСТИ
Коэффициент вязкости для чистых газов или смесей в любом приближении
может быть получен из выражения D.20) гл. 7. Сначала мы обсудим формулу
для коэффициента вязкости чистого газа, а затем для бинарной смеси и фор-
мулу для вязкости многокомпонентных смесей.
1. Коэффициент вязкости чистого газа. В первом приближении коэффи-
циент вязкости чистого газа равен
= 266,93 ^~ЩТ*) , ( 2.18)
где г} — вязкость, г/см-сек; Т —температура, °К; Т*—приведенная темпе-
ратура = kTje'y M — молекулярный вес; а — диаметр столкновения, А; е/к —
параметр потенциальной функции межмолекулярного взаимодействия, °К.
В fc-м приближении коэффициент вязкости равен
M* = Mi/?°. B.19)
Функция fff очень медленно изменяется в зависимости от Т* и только
слегка отличается от единицы3). Явное выражение этой функции через ?№*)*
при к = 3 дано в приложении А в конце главы.
Вязкость как функция температуры при атмосферном давлении была
измерена для большого числа газов. Из этих данных можно получить опреде-
ленные сведения относительно межмолекулярных сил. С этой целью по B.6)
выбирается вид потенциальной функции, а затем вычисляется Й<2'2>*(Т*).
Наряду с экспериментальными данными по вязкости для определения пара-
метров а и е потенциальной функции используется уравнение B.19). Пример
определения межмолекулярных сил из данных по вязкости при использовании
потенциала F-12) Леннарда-Джонса дается в гл. 8, § 4.
2. Коэффициент вязкости бинарных смесей. При анализе вязкости бинар-
ных смесей удобно пользоваться величиной
*) Величина А* непосредственно связана с отношением q[^]i/[v]i [cm. B.48)].
2) Величина с* непосредственно связана [19] с производной по температуре при по-
стоянном давлении от [Я) ]х:
2 б[
L " х ' 3 d In Г* 2 б I Э In Г
Это соотношение легко может быть выведено из рекуррентной формулы для /Ж«), данной в
примечании к формуле B.3). Величина в* может быть выражена через первую и вторую
производные от коэффициента диффузии.
8) Для потенциала Леннарда-Джонса функция /(з) дана в табл. XIV (стр. 871) и отличается
от единицы менее чем на 0,8%.
§ 2. Сводка формул кинетической теории для чистых газов 421
или в практических единицах
[4i.li- Ю7 = 266,93 JY2m7m2ti(m1±aQ)
где Т — температура, °К ; РЬ = kTje12— приведенная температура; М19
М2 — молекулярные веса компонент 1 и 2 ; ст12, е12/к — параметры потенциаль-
ной функции, характеризующей взаимодействие 1—2 (А и °К соответственно).
Эту величину можно рассматривать как коэффициент вязкости гипотети-
ческого чистого газа, молекулы которого имеют молекулярный вес, равный
2 M1M2I(M1-{'M2)J и взаимодействуют по потенциальной кривой, определяемой
параметрами взаимодействия ai2 и е12. (Аналогичную искусственную величину В12
мы определили в связи с вычислением второго вириального коэффициента.)
Если в формуле B.21) индекс 2 заменить индексом 1, то мы получим формулу,
аналогичную формуле B.18) для чистого газа из молекул сорта 1.
Теперь мы можем записать вязкость бинарной смеси в соответствии с
определениями B.18) и B.21):
- А4 v J'
Ха
Шг ~*~ [ih.li ~*~ h.li ' •
2х1хЛ ((Мг + М^ЛГ [у12]1 \ . х\ (М%
" I JlJ" I
h.li J
где Xj, x2 — молярные доли компонент 1 и 2; М1У М2 — молекулярные веса
компонент 1 и 2; а^ — функция от /сТ/е12, определенная формулой B.15).
Вообще величина [A + Уч/Хч)/A + Zv)] заметно отличается от единицы
и может вносить изменение в окончательный результат, достигающее 50%.
Величина Xv вносит основной вклад в значение коэффициента вязкости, если
молекулярные веса компонент 1 и 2 не сильно отличаются друг от друга и
если силы, действующие между различными и одинаковыми парами молекул,
почти одинаковы. Для бинарной смеси тяжелых изотопов коэффициент &яз-
кости в хорошем приближении можно вычислять по формуле
B.23)
Следует отметить, что величина rj12 непосредственно связана с коэффициен-
том диффузии
г ! _ 5 mxm2 п[ДIа]1 _ 5 МгМ2
Wi2Ji~ з (mx + m2) a* " 3 (Мг+М2) а*
Если имеются данные по диффузии, то для получения величины [rj12 ]г вместо
формул B.20) или B.21) лучше пользоваться формулой B.24). Ввиду того
что в настоящее время этих данных недостаточно, формулу B.22), по-видимому,
лучше записывать через величину [rj12 ]v Формула для коэффициента вязкости,
записанная через [rj12]v имеет то преимущество, что она более удобна для
вычислений.
В настоящее время накоплен значительный объем экспериментальных
данных по-вязкости смесей из двух газов. С помощью этих данных и формулы
B.22) в принципе возможно определить параметры потенциала парного взаи-
модействия между неодинаковыми молекулами. Однако практически такое
определение оказывается невозможным, так как парное взаимодействие между
неодинаковыми молекулами вуалируется парным взаимодействием между
одинаковыми молекулами. Аналогично, как отмечается в гл. 3, § б, для опре-
деления законов парных сил, действующих между неодинаковыми молекулами,
недостаточно измерений второго вириального коэффициента бинарных смесей.
422
Гл. 8. Явления переноса в разреженных газах
3. Коэффициент вязкости многокомпонентных смесей. На основании стро-
гой кинетической теории многокомпонентных газовых смесей коэффициент вяз-
кости v-компонентной смеси определяется выражением D.61) гл. 7. Для
расчетных целей это выражение может быть записано в виде
tfu Я
г г г г
^12 #2
#1Я #
12
23
#23
#яя
#1„
#2*
#2„ #я
о
#12
#**
#]
Я,
Я9о Я
Ни
я2.
B.25)
'23
зз
я,
Элементы Я,7 определителей могут быть выражены через fr/yh или
через коэффициенты диффузии [j2),y ]x:
„ = х? , ^ 2х/х/с MjMk \ 5^ АЛ' п
RT
/с=1
Mk)
3 Мк А*
B,26)
Н = - 2%iXj MiMj Г 5 — l] / -? /
/?Г
B.27)
гДе [Vi]i — коэффициент вязкости, определяемый по формуле B.18); [п^ —
величина, определяемая по формуле B.20); [-?)//<]! — коэффициент диффузии,
определяемый по формуле B.44); а$—функция, определяемая по формуле
B.15); xh M( — молярная доля и молекулярный вес i-й компоненты.
Отношение детерминантов в формуле B.25) может быть представлено в
виде следующего разложения :
4f ~ m <U V <U w w V W
- ... B.28)
1 — 1 У—1 А:=1
Поскольку недиагональные элементы Ни малы по сравнению с диагональными
элементами Иш то основной вклад в коэффициент вязкости многокомпонентной
смеси вносит первый член разложения B.28). Чтобы недиагональные элементы
точно были равны нулю, необходимо предположить, что а* == б/3. Если это же
самое предположение использовать для диагональных элементов, то B.28)
приобретает вид
" хг х?
г- ' " ^ ' " ^ —^ -^Г~ ' B-29)
1-1
Mi
§ 2. Сводка формул кинетической теории для чистых газов 423
Вследствие того, что при получении этой формулы мы пренебрегли высшими
членами разложения B.28) и для величины а* было выбрано нереальное зна-
чение, формула B.29) не очень хорошо соответствует действительным значе-
ниям вязкости. Однако в результате широкого изучения экспериментальных
данных Будденберг и Вилке [6 ] показали, что формула B.29) дает значения
вязкости, хорошо совпадающие с действительными значениями, если множи-
тель 2 заменить эмпирической величиной, равной 1,385, Таким образом, в очень
хорошем приближении формула B.25) может быть заменена формулой
1?^ B.30)
1>385^Х/Х*
^
Это соотношение позволяет очень просто вычислить вязкость многокомпонент-
ной смеси по данным вязкости чистых компонент и по данным о коэффициентах
диффузии различных пар химических компонент смеси. При отсутствии экспе-
риментальных данных [fj^ может быть вычислено по формуле B.18), a [jZ)/fc]j —
по формуле B.44). Формула B.30) впервые была получена Будденбергом и
Вилке [6] не из строгой кинетической теории многокомпонентных газов. При
выводе этой формулы они исходили из формулы, данной ранее Сюзерлендом[7],
Тиссеном [8] и Шуделем [9]. Будденберг и Вилке установили, что для учета
парных столкновений между неодинаковыми молекулами в формулу необ-
ходимо ввести коэффициент диффузии, после чего они на основе тщательного
анализа экспериментальных данных вычислили постоянную, равную 1,385х).
В. КОЭФФИЦИЕНТ ТЕПЛОПРОВОДНОСТИ
Коэффициент теплопроводности для чистых одноатомных газов или для
смесей одноатомных газов может быть получен в любом приближении из фор-
мул D.65) и D.66) гл. 7. Для многоатомных газов эти формулы не могут быть
использованы вследствие значительного влияния на теплопроводность внутрен-
них степеней свободы молекул. В настоящее время лучшим способом исправ-
ления формул для одноатомных газов с целью распространения их на много-
атомные газы является использование поправки Эйкена, полученной в гл. 7, § 6.
Формулы этого параграфа для одноатомных газов получены на основе стро-
гой кинетической теории, а формулы для многоатомных газов должны рассмат-
риваться как приближенные.
1. Коэффициент теплопроводности чистого газа. Коэффициент теплопро-
водности чистого газа в первом приближении равен
[Я],. 10' = 1989,1 Щ
где А — коэффициент теплопроводности, кал1см-сек-°К'> Т—температура, °К;
Т* = kTje — приведенная температура; М — молекулярный вес; ст, г/к — па-
раметры потенциальной функции, А и °К соответственно.
Таким образом, в первом приближении коэффициент теплопроводности
пропорционален коэффициенту вязкости. Этот вывод согласуется с результа-
тами элементарной кинетической теории.
Высшие приближения коэффициента теплопроводности можно вычислить
по форхмуле
И* = Mi № = -г ~йГ Щ ' B-32>
Функции f# и /W мало отличаются от единицы и медленно изменяются с 7*.
См. также К. S. Brokaw, Journ. Chem. Phys., 29, 391 A958) — Прим. ред.
424 • Гл. 8. Явления переноса в разреженных газах
Точные выражения ff и /<*> через интегралы ?№s>* даны в приложении А в
конце главы.
Для многоатомных молекул формула B.31) должна быть заменена таким
соотношением, в котором приближенно учитывалась бы передача энергии между
поступательными и внутренними степенями свободы молекул [см. F.24)],
т. е. соотношением вида
№йкен = -т!МгD4 + 4)- B-33>
Для одноатомных газов, для которых теплоемкость С„ = 3/?/2 на моль, формула
B.33) упрощается и переходит в B.31).
2. Коэффициент теплопроводности бинарных смесей. Для изучения тепло-
проводности бинарных смесей удобно определить величину [Л12]1У аналогич-
ную величине [rj12]v определенную выражением B.20):
25 ( У("*1 + т2) п кТ12тг т2
\ ( 3 Л _ 15
) V2 *J " ~^
или в практических единицах
+ М2O2М1 м2
где Т — температура, °К : Т{2 = кТ\е12 — приведенная температура;
М19 М2 — молекулярные веса компонент 1 и 2 ; ст12, ei2jk — параметры потен-
циальной функции, характеризующей взаимодействие между молекулами сорта
1 и 2, А и °К соответственно.
Через эту величину и теплопроводность чистых компонент теплопровод-
ность смеси одноатомных газов может быть записана в виде
T+ZT - л*V^brzrJ • B.36)
[А2]
_ Xl TJA) I 2xlX2 rj(Y) i ^2 г гB)
Zx = xf иы + 2хгх2 G<z> + x
15 A« 12 I 5 12+ M Afx + 2
15 Ai2 I 4M1M2 J ТВДпГ Т2 l"T Bl2
_ j_ _
32a*4 5 12
да - •] -i№ ¦»+о ¦
где xx, x2 — молярные доли компонент 1 и 2 ; Mv M2 — молекулярные веса
компонент 1 и 2 ; а&, в^ —функции от Т}2 = /сГ/е12, определенные форму-
лами B.15) и B.16).
Для смесей тяжелых изотопов в хорошем приближении выражение B.36)
сводится к
1 *i , х, B.37)
§ 2. Сводка формул кинетической теории Оля чистых газов 425
Величина [А12]х непосредственно связана с коэффициентом диффузии [«2I23i
25
При наличии данных о диффузии для вычисления А для бинарных и многоком-
понентных смесей лучше пользоваться этими данными. Только потому, что к
настоящему времени произведено еще очень мало измерений по диффузии, мы
записали формулу B.36) через величины [А12]г
В настоящее время не разработан строгий метод вычисления теплопровод-
ности смесей многоатомных газов. Однако теплопроводность смесей многоатом-
ных газов может быть оценена с помощью следующего эмпирического метода1).
Предполагая, что молекулы одноатомны, вычислим величину, определяемую
формулой B.36). Результат обозначим через Аодн.. При известных экспери-
ментальных значениях А для чистых газов для каждой компоненты смеси можно
определить отношение
(^) (Я/)эКСП.
1 (А/)од
На основе этих предварительных определений эмпирическое выражение для
теплопроводности смеси записывается в виде
v м -|Эйкен ri 1
[Яс„. ]Эйкен = [Асм. \ + У х,- Щ —1& , B.40)
где [Асм. ]v [А,- ]Эйкен и ^ ]1 — величины, определяемые формулами B.42),
B.33), B.31) соответственно. Это выражение получено путем обобщения вывода
формулы F.23) гл. 7.
Для чистых газов теплопроводность пропорциональна вязкости. Вычисле-
ние теплопроводности смесей значительно облегчилось бы, если бы теплопро-
водность смесей оставалась также пропорциональной вязкости с легко вычи-
сляемым коэффициентом пропорциональности. Лучшим эмпирическим соотно-
шением такого типа является формула
1/ v-2 О v v v-2 \
[Асм. ]i [^см. ]эксп,
где о, = (А^Джсп.. Однако по расхождениям экспериментальных значений и
значений, вычисленных на основе этой формулы, можно сказать, что соотно-
шение B.41) дает менее надежные результаты, чем вычисляемые по формуле
B.39).
3. Коэффициент теплопроводности многокомпонентных смесей. Коэффи-
циент теплопроводности многокомпонентной смеси вычисляется по формуле
D.65), которая может быть записана в виде
ГА 1 - ГА' 1 1 R V ^ RT Xi XJ \ [°?]l [DjT]l Г
[Асм. ]г - [А см. ]х - тR 2 2 ЛЩ~г ixJW ~~ ~^Mj\ '
г) Имеется альтернативный эмпирический метод, который также приводит к хорошему
согласию с экспериментальными значениями коэффициента теплопроводности бинарных
смесей многоатомных газов. Если из данных по вязкости известны силовые постоянные
чистых компонент и известны экспериментальные значения А для чистых газов, то можно
провести следующие расчеты. Для каждой составляющей смеси из данных по вязкости исполь-
зуем только параметр et/k. Параметр о\ получим из формулы B.31), подставляя в нее необ-
ходимые значения Г* и экспериментальные значения А/. Используя среднегеометрическое
параметров etfk и среднеарифметическое подбираемых параметров aj, по формуле B.35)
вычислим [Хи]г. Используя подбираемый параметр а\ или экспериментальные значения
А/ вместо [А/]ь находим, что формула B.36) хорошо воспроизводит концентрационную зави-
симость коэффициента теплопроводности.
426
Гл. 8. Явления переноса в разреженных газах
В хорошем приближении с точностью до 1% это выражение может быть заме-
нено выражением
141
Lll xv
xv 0
B.42a)
L}\ ... Lll
В выражениях B.42) и B.42a) [a2)ij]l — первое приближение коэффициента
диффузии бинарных систем, определяемое по формуле B.44), [DJ] ^ — термо-
диффузионная постоянная, определяемая по формуле B.53), и
«.]! = 4
L??
US
L\l
0
О
О ... О хг ... xv О
/00 /00 /01
^11 • • • L*\v *-ll
L11 / 11
11 • • • иЪ
Luvv i-ryl . . .
B.43)
где
2xt xj
v +
2x]XkMj
16 Г Г x,xy
25p L[]
41
8Г
= -bxix1
M
(Mi +
§ 2. Сводка формул кинетической теории для чистых газов 427
- bp **J (Mi
_ Mi r oi
2XiхьЩ-Nft+^-Ml-ZM\ в^ + 4MiMk
/11 **Л1 X*
Г. КОЭФФИЦИЕНТ ДИФФУЗИИ
Здесь мы начнем изложение с того, что дадим выражение для коэффициента
диффузии бинарной смеси газов. Затем мы покажем, что при определенных
условиях может быть получен предельный вид коэффициента диффузии, кото-
рый называется «коэффициентом самодиффузии». Наконец, мы выведем выра-
жение для коэффициента диффузии многокомпонентных газовых систем.
1. Коэффициент диффузии бинарной смеси. Коэффициент диффузии бинар-
ной смеси может быть получен из формулы D.4) гл. 7. В первом приближении
имеем
l = 0,00262801Жр|рЖ , B.44)
где „2I2 — коэффициент диффузии, см2/сек ; р — давление в атм ; Т — тем-
пература, °К ; Tf2 = кТ/е12; Mv М2 — молекулярные веса компонент 1 и 2 ;
^12» ei2l^ — параметры потенциальной энергии молекул, характеризующие
взаимодействие молекул сорта 1 и 2, А и °К соответственно.
Высшие приближения коэффициента диффузии определяются по формуле
[.2yfc = [.?>u]i/&. B-45)'
Функция /(||, является функцией молекулярного веса, молярных долей,
вязкости двух компонент и температуры. Явное выражение для этой функции,
которая только слегка отлична от единицы1), дано в приложении А к настоящей
главе.
Заметим, что в первом приближении коэффициент диффузии зависит только
от взаимодействий между неодинаковыми молекулами. Силы, действующие
между одинаковыми молекулами, влияют на коэффициент диффузии лишь во
втором приближении. Это значит, что измерения коэффициента диффузии как
функции температуры являются превосходным методом определения законов
сил, действующих между неодинаковыми молекулами. К сожалению, таких
экспериментальных данных в настоящее время еще мало.
х) Для потенциала Леннарда-Джонса эта функция для большинства бинарных смесей
имеет значение от 1,00 до 1,03.
428 Гл. 8. Явления переноса в разреженных газах
2. Коэффициент самодиффузии. Если выражение B.44) записать для одной
компоненты, то мы получим формулу для коэффициента самодиффузии
Шг = 0,0026280-^5?—, B.46)
где Л) — коэффициент само диффузии, см2/сек ; р — давление, атм ; Т — тем-
пература ; Т* — приведенная температура, равная kTle; M — молекулярный
вес; a, ejk — параметры потенциальной функции, А и °К соответственно.
Высшие приближения для коэффициента самодиффузии вычисляются по фор-
муле
[2>L UZ>]/^. B.47)
Поправочный множитель fi?\ являющийся функцией только приведенной тем-
пературы, дан в приложении А в конце главы.
Выясним теперь, каков смысл коэффициента самодиффузии. Очевидно,
что если все молекулы газа физически идентичны, то измерение их взаимной
диффузии невозможно. Однако можно экспериментально измерить величины,
очень близкие к коэффициентам само диффузии.
1) Взаимодиффузия изотопов. При диффузии одного изотопа некоторого
газа в другом изотопе процесс диффузии можно исследовать с помощью извест-
ных методов определения концентрации изотопов. Так как число нейтронов в
атомном ядре не оказывает существенного влияния на межмолекулярные
силы, то а12 = аг = о2 и е12 = ег — е2. Кроме того, если молекулы достаточно
велики, то отношение 2М1М21(М1 + М2) очень близко к Мг или М2. Отсюда
формула B.44) упрощается и приводится к виду B.46). С увеличением доступ-
ности получения изотопов изучение диффузии изотопов могло бы стать интерес-
ным методом получения сведений о межмолекулярных силах.
2) Взаимодиффузия орто- и пара-состояний. Наряду со взаимодиффузией
изотопов может быть также исследован процесс диффузии молекул в орто-
состоянии через газ из молекул в пара-состоянии1). Вследствие того, чтс орто- и
пара-состояния газа экспериментально могут быть различимы только для
легких молекул, такой метод может быть использован только при изучении
диффузии изотопов водорода. Для молекул водорода взаимодействия между
орто- и орто-, орто- и пара-, пара- и пара-состояниями могут рассматриваться
как взаимодействия, характеризующиеся одним и тем же набором силовых
постоянных, хотя, конечно, взаимодействие слегка зависит от вращательного
состояния молекулы. Помимо этого, BМ1М2)/(М1 + М2) = М} = М2. Следо-
вательно, формула B.44) снова приводится к виду B.46).
Очевидно, что коэффициент самодиффузии должен рассматриваться как
некая искусственная величина. Правильнее считать его просто предельной
формой коэффициента диффузии бинарных смесей. Интересно отметить, что в
пределе при равенстве масс и межмолекуляриых сил формула B.24) упро-
щается и приводится к виду
1Ш. = |а*. B.48)
Функция а*, определенная соотношением B.15), очень медленно изменяется
при изменении 7*, и поэтому отношение q \JD у [rj ]г очень близко к постоянной.
3. Коэффициент диффузии многокомпонентных газовых систем. Коэффи-
циенты диффузии многокомпонентных смесей могут быть найдены по формуле
D.44) гл. 7, которую можно записать в виде
gTZrL> B-49)
г) Экспериментально это может быть сделано путем измерения электрического сопро-
тивления нагретой проволоки, теплоотвод которой в атмосфере орто- или пара-газа раз-
личен.
§ 2. Сводка формул кинетической теории для чистых газов
429
Xi
Xk
где Кц = 0 ;
К | — детерминант из К/у; KJi — миноры :
О ... Klt ,-х /Сх t i+1
= (- I)'4'
К/-1Д
V, 1
Kv
Д. КОЭФФИЦИЕНТ ТЕРМОДИФФУЗИИ
Коэффициент термодиффузии D? для многокомпонентных систем может
быть получен из строгой кинетической теории и имеет вид D.55) гл. 7. Из стро-
гой кинетической теории может быть получено также термодиффузионное
отношение кт для двухкомпонентных систем. Ниже будет дано в наиболее
общей форме выражение для вычисления термодиффузионного отношения
цля любой пары газов и выражение специального вида, предназначенное для
расчетов термодиффузии изотопов.
1. Термодиффузионное отношение для бинарных газовых смесей. В пер-
вом приближении термодиффузионное отношение кт выражается следующим
образом :
15' Г М2 -
l
2М2
2 - МП _ t
2Afx J 1у
B.50)
15
2M1 [A2]i 4 a* I 2M2
где xv x2 —молярные доли компонент 1 и 2 ; М19 М2 — молекулярные веса
компонент 1 и 2 ; [Ах ]L, [А2 ]L — к оэффициенты теплопроводности, вычисляемые
по формуле B.31); [Я12]1 — величина, определяемая по формуле B.34); а&
сй — функции от Т*12 = кТ/е12у определяемые по формулам B.15) и B.17);
Хь и Yk — функции, определяем ые по формуле B.36).
Термодиффузионное отношение является очень сложной функцией тем-
пературы, концентраций и молекулярных весов и зависит параметрически
от законов межмолекулярных сил . Зависимость от концентрации в основном
определяется множителем ххх2 и в меньшей мере множителем (S^l)xx — SB)x2).
Главная зависимость от масс моле кул дается величинами S(l) и SB), принци-
пиальная зависимость от температуры — множителем (бс^ — 5). Термодиф-
фузионное отношение может быть положительным или отрицательным. Поло-
жительное значение кт означает, что компонента 1 диффундирует в холодную
область, а компонента 2 — в более нагретую область1). Температура, при кото-
рой термодиффузионное отношение меняет знак, называется инверсионной тем-
пературой.
Следующие высшие приближения для термодиффузионного отношения
могут быть получены по формуле, приведенной в приложении А в конце главы.
х) Значение кт зависит от определения этой величины. В настоящей книге принято
то же определение кт, которое использовалось Чепменом и Каулингом [1].
430
Гл. 8. Явления переноса в разреженных газах
Из небольшого числа расчетов, проведенных для [кт ]2, можно сделать вывод,
что ошибка в первом приближении для кт больше, чем для любого другого
коэффициента переноса1).
2. Термодиффузионное отношение для бинарных смесей изотопов [11]
В специальном случае тяжелых изотопов выражение B.50) может быть значи-
тельно- упрощено. Разлагая выражения B.50) в ряд по степеням отношения
(М1 — М2I(М1 + М2), для кт в результате получим формулу
r. 1 _ 15Bа* + 5)Fс*-5) (Мг-М2)
l"rji — 2А* A6 а* - 12 в* + 55) (Мг + М2) Л1
B.51)
В литературе2) часто используется величина /с$, определяемая по формуле
A.17). В первом приближении эта величина выражается следующим образом :
\к 1* = [kT]l =Ъ9 Bа*+5)Fс*-5)
1 rJl 1>тТГв-сФ- 7 а* A6 а* -12 в* + 55) >
[кт!
B.52)
и является универсальной функцией Т*. Так было показано Винтером [12];
это рыражение справедливо, если (Мх — М2)/(Мг + 7И2) не превышает 0,15.
3. Коэффициент термодиффузии для многокомпонентных газовых смесей»
Исходным выражением для коэффициента термодиффузии многокомпонентных
смесей является выражение D.55) гл. 7. Это выражение можно записать в виде
8Affc
5Я
Lu ...
1я ...
Li? ...
& :::
L?s ...
jl :::
i« ...
/ 00
/ 00
in
Ll
LJ?
/ 01
J 01
L« ...
0
LSi -
L?J ...
L\\ ...
Lg ...
/01
f 11
o"
L?J
Lg
L,V
0
0
0
B.53)
где Ц\т' определяются по формулам B.43).
г) См. табл. 67, в которой даны итоговые результаты для твердых сфер. Кроме того,
см. недавно выполненные Мэзоном [10] расчеты [кт\ для модифицированного потенциала
Букингема.
2) Величина к г в литературе обычно обозначается через Rt. Мы используем обозначение
кт> чтобы согласовать его с принятым в этой книге обозначением, которое состоит в том,
что величины, деленные на их значения для твердых сфер, отмечаются пятиконечной звез-
дочкой.
§ 3. Вычисление коэффициентов переноса для простых потенциалов 431
§3. ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ ПЕРЕНОСА ДЛЯ ПРОСТЫХ
ПОТЕНЦИАЛОВ
В гл. 3 было показано, что для ряда простых потенциалов вторые вириаль-
ные коэффициенты могут быть получены в аналитическом виде. Однако сделать
это для коэффициентов переноса невозможно. Аналитически эффективное сече-
ние столкновения можно получить для потенциала твердых сфер, но для дру-
гих простых потенциальных функций, обсуждаемых в этом параграфе, исполь-
зуется численное интегрирование. Здесь приводятся результаты этих числен-
ных расчетов, так как они могут быть полезны для грубых вычислений и для
интерполирования данных в пределах небольшого температурного интервала.
Удобна модель обратной степенной зависимости ср{г) — йгд> так как ее можно
дифференцировать, и двойной интеграл по Ь и g для этой модели может быть
сведен к однократному интегралу. В прошлом для установления связи с экс-
периментальными данными широко использовалась модель Сюзерленда (твер-
дые сферы, окруженные полем притяжения). Для модели Сюзерленда в литера-
туре приводятся «постоянные Сюзерленда» для многих веществ. Для сложных
молекул может быть использована модель прямоугольной ямы, так как она
включает три подбираемые постоянные. При проведении точных расчетных
работ, основывающихся на реальном потенциале взаимодействия, следует
иметь в виду, что в настоящее время лучшими и наиболее доступными являются
вычисления для потенциала F-12) Леннарда-Джонса и для модифицирован-
ного потенциала (б-ехр) Букингема, который обсуждается в § 4. Все эти потен-
циальные функции имеют вид формулы B.6).
А. ТВЕРДЫЕ УПРУГИЕ СФЕРЫ
Результаты вычислений для твердых сфер использовались в предыдущих
параграфах для приведения эффективных сечений столкновений. Для модели
твердых сфер [ср(г) = оо для г < о ; ср(г) = 0 для г > а ] интеграл по углам
отклонения может быть легко вычислен. Расстояние наибольшего сближения
двух сталкивающихся молекул равно гт = оу если Ца,игт = 6 для Ь ^> <т.
Отсюда из B.1) немедленно получим
os(^) &<ег,
Эти результаты легко могут быть получены также из простых геометрических
соображений. Как и следовало ожидать, для непроницаемых сфер угол откло-
нения не зависит от относительной кинетической энергии. Подстановка только
что полученных значений % в B.2) и B.3) приводит к выражениям для Q(/) и
q«,s) в виде ^2.4) и B.5). Приведенные величины Q<*>* ий(/'5)* определены таким
образом, что для модели твердых сфер они точно равны единице. Это значит, что
все формулы для коэффициентов переноса, данные в § 2, могут быть исполь-
зованы для твердых сфер, если все значения ??(/»s)* положить равными единице.
Выражения для вязкости и самодиффузии B.18) и B.46) с &1>S)*, рав-
ными единице, могут быть использованы при анализе экспериментальных
данных для получения молекулярных диаметров а или молекулярных эффек-
тивных сечений па2. В табл. 66 сравниваются молекулярные диаметры, вычи-
сленные на основе этих двух формул. Из таблицы следует, что значения диа-
метров столкновения твердых сфер, полученные из данных о вязкости, выше
значений, полученных из данных о самодиффузии. Из этого можно сделать
вывод, что для реальных молекул ??B»2)* немного больше ??AД)*. Вычисления
эффективных сечений столкновений для более реальных потенциалов подтвер-
ждают этот факт.
432
Гл. 8. Явления переноса в разреженных газах
На основе модели твердых сфер были получены важные результаты о
быстроте сходимости разложений по полиномам Сонина, используемых при
выводе выражений для коэффициентов переноса. Эти результаты приводятся
Таблица 66
Молекулярные диаметры твердых сфер, вычисленные из данных
о вязкости* и из данных о само диффузии [15], измеренных
при О °С и 1 атм
10» - 266,93
\МТ
h = 0,0026280 '
Газ
Аг
Ne
N2
о2
сн4
со2
г/слс • сек
2099
2967
1663
1918
1030
1366
см*{сек
0,156
0,452
0,185
0,187
0,206
0,0974
о,
из данных о
вязкости
3,64
2,58
3,75
3,61
4,14
4,63
А
из данных о
самодиффузии
3,47
2,42
3,48
3,35
3,79
4,28
* Для Ne и Аг данные о вязкости взяты из измерений Джонстона и Макк-
лоскей [13], для N2, O2, СН4и СО2 — из измерений Джонстона и Грилли [14].
в табл. 67, где даны отношения fc-x приближений коэффициентов переноса к
их действительным значениям. Ошибки для термодиффузионного отношения
значительно выше, чем для любого другого коэффициента. Вообще говоря,
ошибки в первом приближении для твердых сфер больше, чем для более реаль-
ных молекулярных моделей.
Таблица 67
Доли k-х приближений коэффициентов переноса
относительно их действительных значений
(на основании модели твердых сфер)*
[Р]к—к-е приближение коэффициента Р,
[Р] — действительное значение коэффициента Р.
р
V
X
?0
кт
[Р]г![Р]
0,984
0,976
0,883
0,77
[P]J[P]
0,999
0,998
0,957
0,88
[Р]8/[Р]
0,999+
0,999+
0,978
—
• См. [1, стр. 169 и 196]
Б. ТОЧЕЧНЫЕ ЦЕНТРЫ ОТТАЛКИВАНИЯ
При очень высоких температурах силы отталкивания играют более важную
роль, чем силы притяжения. При этом молекулы можно представить как точеч-
ные центры сил отталкивания с потенциальной энергией взаимодействия
cp(r)'= dr~d [см. формулу C.23) гл. 1 ]. Показатель д является мерой жесткости
или мягкости молекул. При д = ©о эта модель сводится к модели твердых сфер.
Модель с конечным 6 описывает естественную «сжимаемость» молекул. Для
большинства молекул 6 имеет значение между 9 («мягкие» молекулы) и 15
(«жесткие» молекулы).
Одно из преимуществ этой потенциальной функции заключается в том,
что угол отклонения, являющийся функцией Ъ и g, может быть выражен через
§ 3. Вычисление коэффициентов переноса для простых потенциалов
433
единственную комбинацию этих двух переменных. Если мы следующим обра-
зом определим величины у, ут и у0:
==ol dd
то угол отклонения можно записать в виде
Ут(Уо)
—2 J [1-у-4ШГ>-
C.2)
C.3)
Предел ут соответствует такому значению величины у, для которой корень в
знаменателе интегрируемой функции обращается в нуль. Угол отклонения как
функция у0 для 6 = 12 приведен на фиг. 117. Значения ут(у0) и %(у0) даны в
Фиг. 117. Угол отклонения как функция одного параметра у0 для потенциала
взаимодействия 0(г) = dr-12.
По неопубликованным вычислениям Берда.
табл. XXI (стр. 891). Эти величины были получены численным методом. В ре-
зультате подстановки угла отклонения в соотношении B.2) и B.3) получим
следующие формулы для эффективных сечений столкновений:
Г
2л
Jl«-
cos'
dd
C.4)
C.5)
Значения интегралов Л(/)(й) приведены в табл. 68.
Используя найденное выше выражение C.5) для QV*S\ получаем сле-
дующие соотношения для коэффициентов переноса:
C.6)
Эти соотношения полезны для получения данных о коэффициентах переноса
при высоких температурах, где вклад в значения коэффициентов переноса за
счет сил отталкивания более важен, чем за счет сил притяжения.
434
Гл. 8. Явления переноса в разреженных газах
Особого внимания заслуживают два специальных случая взаимодействия
рассматриваемого типа. В одном из них молекулы взаимодействуют по закону
отталкивания с д = 4 и известны как максвелловские молекулы. С точки зрения
истории вопроса интересно отметить, что Максвелл задолго до работ Энскога
и Чемпена показал, что для такого типа молекул можно развить строгую кине-
тическую теорию, не решая интегрального уравнения, из которого определяется
Таблица 68
Значения А^1\б) для точечного центра отталкивания*
д I А^\д) I
1 i
4
6
8
10
12
14
20
24
0,298
0,306
0,321
0,333
0,346
0,356
0,500
0,308
0,283
0,279
0,278
0,279
0,280
0,286
0,289
0,333
•См. [1, стр. 172]. Величины Д<*>(<5) связаны с величи-
нами Ai{6), протабулированными Чепменом и Каулингом,
соотношением
= АЦЪ + 1)/2'
функция Больцмана. Однако хотя максвелловские молекулы очень удобны с
математической точки зрения, они слишком далеки от реальности, чтобы быть
важными с точки зрения физики. Для таких молекул коэффициент термодиф-
фузии тождественно равен нулю [1, стр. 169 и 196].
Во втором специальном случае д = 1 и d = еге2. Этот случай соответствует
кулоновскому взаимодействую1) между двумя зарядами е1 и е2. Для кулонов-
ского потенциала угол отклонения может быть получен в явном виде
x(yQ) = 2 arc s!n
Vl+4"yJ
C.7)
Подставляя выражение C.7) в формулу C.4), находим, что интеграл при верх-
нем пределе расходится. Физическое объяснение возникающего математического
затруднения заключается в том, что поле сил спадает настолько медленно, что
становится неясным, как следует определить парные столкновения. Очевидно,
все столкновения в этом случае следует рассматривать в известном смысле
как многократные. В разреженных газах, однако, игнорируя многократные
столкновения, происходящие на далеких расстояниях, можно получить при-
ближенные выражения для коэффициентов переноса.
В разреженных газах величины Ь, при которых происходит отклонение
на заметный угол, значительно меньше среднего расстояния D между части-
цами в газе. В таких условиях кажется разумным в качестве верхнего предела
выбрать b = D или у0 = DA/2ftg2lele2) =y'o. При таких значениях верхнего
предела можно получить следующие выражения для эффективных сечений :
C.8)
C.9)
\- = Ц г,' '2 1пA+4уо2)- . ,7 , .
*) Явления переноса в ионизованных газах исследовал Кихара [16]. (См. также
[86—89]. — Прим. ред.)
§ 3. Вычисление коэффициентов переноса для простых потенциалов 435
Чтобы получить й(/»s), эти выражения должны быть проинтегрированы по всем
значениям относительной скорости g, которая входит в эффективные сечения
в явном виде как g~4 и неявно через у'о. Вследствие того, что Q(/) есть медленно
меняющаяся функция у'о, удобно ввести дополнительное упрощающее предпо-
ложение, заключающееся в том, что %ug2 заменяется своим средним по всевоз-
можным типам столкновений значением, которое [1, стр. 93] равно 2/сГ.
В результате в первом приближении коэффициенты переноса для газа, состоя-
щего из одной компоненты, приобретают вид
. = 4 PiL ("'Я Ш' / "" С + 4«*>Ь <зло>
где yn0 =
В. МОДЕЛЬ СЮЗЕРЛЕНДА1)
В модели Сюзерленда молекулы рассматриваются как твердые сферы
диаметра о, окруженные полем сил притяжения, обратно пропорциональным
некоторой степени расстояния при максимальном значении энергии притя-
жения, равном е. Потенциал взаимодействия имеет вид C.25) гл. 1
?(') = °°> г<а>
Точный расчет углов отклонения и эффективных сечений столкновения
требует трудоемкой вычислительной работы. Однако если предположить, что
притяжение относительно слабо, то высшими степенями г, которые появляются
при точном подходе к проблеме, можно пренебречь. В результате получим
формулы Сюзерленда для вязкости и диффузии, широко используемые для
получения кривых, соответствующих экспериментальным данным
Ыг = 1г)—*SK C.13)
Постоянные Сюзерленда Sv и S^, пропорциональные энергии взаимодействия
двух молекул при их соприкосновении, выражаются следующим образом :
Sa=/A)(y)y, C.15)
5,= ^(у)у. C.16)
Постоянные пропорциональности №(у) для некоторых показателей притя-
жения у приведены в табл. 69. Результаты такого приближенного подхода
справедливы только при очень высоких температурах, когда можно ожидать,
что силы притяжения играют малую роль в молекулярном взаимодействий.
х)См. [17] и [1, стр. 182].
436
Гл. 8. Явления переноса в разреженных газах
Если энергия притяжения изменяется, как 1/г6, то потенциальная функция
имеет вид
(р(г) = оо Г < G,
(ЗЛ7)
где снова е равно максимальному значению энергии притяжения, a a — диа-
метр твердого ядра.
Таблица 69
Постоянные пропорциональности для постоянных
Сюзерленда
7
2
3
4
6
8
*A)G)
0,2662
0,2276
0,2010
0,1667
0,1444
i<«>(y)
0,2336
0,2118
0,1956
0,1736
0,1556
Полный расчет углов отклонения и эффективных сечений столкновения
для такого потенциала, без каких-либо предположений о малости сил притя-
жения, выполнил Котани [17 ]. Значения вычисленных им интегралов ??(/'s)*(T*)
даны в табл. XXIII (стр. 893). С помощью этой таблицы по формулам, приве-
денным в § 2, можно вычислить первое и второе приближения коэффициентов
переноса чистых газов и их смесей. Силовые постоянные для неона и азота,
определенные изданных измерений вязкости и диффузии, и силовые постоянные
для двуокиси углерода, полученные из данных о вязкости, приведены в табл. 70.
Таблица 70
Силовые постоянные для потенциала Сюзерленда, полученные из
данных вязкости [13, 14] и самодиффузии [15] при температурах
между 200 и 300° К
Газ
Ne
N2
СОа [18]
Из измерений
вязкости
<г,А
•/*, °К
2,33
3,07
3,43
192
416
638
Из измерений
самодиффузии
«/*. °К
2,20
3,17
196
202
Г. ПРЯМОУГОЛЬНАЯ ПОТЕНЦИАЛЬНАЯ ЯМА1)
Простейшим потенциалом с тремя постоянными, для которого были
вычислены коэффициенты переноса [см. гл. 1, формулы C.24)], является пря-
моугольная потенциальная яма
<р(Г) = оо , Г<(Х,
<р(г) = — е, а<г</?а,
у(г) = О, r>Ro.
Углы отклонения и эффективные сечения столкновения для этой потенциальной
функции были вычислены численно. Полученные таким образом интегралы
. [19].
§ 4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 437
?>('»s)* даны в табл. XXII (стр.892). Параметры потенциальных функций (опре-
деленные из вязкости), необходимые для использования табл. XXII, приво-
дятся в табл. 71. При использовании упомянутых параметров и табулирован-
ных функций хорошо воспроизводятся экспериментальные данные. По таб-
лицам параметров и табулированных функций могут быть вычислены также
другие коэффициенты переноса.
Таблица 71
Силовые постоянные для прямоугольной потенциальной ямы,
полученные из данных о вязкости [19]
Вещество
Не
Ne
Аг
н2
о2
232
101
167
94
94
а, к
1,90
2,38
2,98
2,57
3,16
1
R
0,720
0,650
0,510
0,700
0,470
Вещество
N2
СО
со2
сн4
Воздух
80
91
200
174
87
в, А
3,36
3,29
3,46
3,35
3,30
7
R~
0,480
0,440
0,450
0,510
0,480
Однако вследствие того, что форма обсуждаемой потенциальной функции
далека от реальной, параметры, полученные из данных о коэффициентах пере-
носа, нельзя использовать для вычисления вторых вириальных коэффициентов.
Таблица 72
Сравнение силовых постоянных для прямоугольной потенциальной ямы,
полученных из данных о вязкости [19] и из данных о втором вириальном
коэффициенте [20]
Вещество
Этан ...
Пропан
л-Бутан
Этен ...
Пропен
elk, °K
Из данных о
вязкости
°, А
Из данных о
втором вириальном
коэффициенте
<г,А
244
347
387
222
339
3,81
4,21
4,50
3,70
4,02
0,437
0,480
0,460
0,440
0,450
3,54
4,42
4,82
3,35
4,32
0,606
0,683
0,678
0,597
0,685
В табл. 72 сравниваются силовые постоянные, полученные из данных о вяз-
кости и из данных о втором вириальном коэффициенте. Значительное расхож-
дение между двумя рядами постоянных объясняется тем, что вязкость в боль-
шей степени, чем второй вириальный коэффициент, зависит от частей потен-
циальной функции, соответствующих силам отталкивания.
§ 4. ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ ПЕРЕНОСА
ДЛЯ ПОТЕНЦИАЛА F-12) ЛЕННАРДА-ДЖОНСА1)
Четыре простые потенциальные функции, обсуждаемые в предыдущих
параграфах, дают довольно хорошие результаты для коэффициентов переноса
в ограниченном интервале температур и указывают на основную температур-
ную зависимость этих коэффициентов. Более реальной потенциальной функ-
г) Вязкость для потенциала D-8) Леннарда-Джонса вычислили Хассе и Кук [21, 22].
Их результаты для изучения термодиффузии были применены Джонсом [23].
438 Гл. 8. Явления переноса в разреженных газах
цией, для которой для неполярных молекул были вычислены коэффициенты
переноса, является потенциал F-12) Леннарда-Джонса
здесь е — глубина потенциальной ямы (максимум энергии притяжения), а
а — диаметр столкновений молекул с малой энергией [т. е. значение г, для
которого <р(г) = 0 ]. Расчеты для этой потенциальной функции независимо были
выполнены авторами из четырех различных стран [24—27, 33]. Поскольку
результаты всех этих расчетов превосходно согласуются между собой, можно
с уверенностью сказать, что в табулированных функциях вычислительная
ошибка мала. Здесь мы приведем лишь наиболее важные результаты вычисле-
ний, что же касается деталей, то их можно найти в оригинальных работах.
Сначала мы изложим вопрос о вычислении углов отклонения и эффективных
сечений столкновения. Далее мы проведем вычисление различных коэффициен-
тов переноса и сравним экспериментальные и расчетные результаты.
Недавно Мэзон [10], используя методы, аналогичные методам, описанным
в настоящем параграфе, вычислил ряд интегралов ?(/» s)* для модифицирован-
ного потенциала Букингема. Таблицы этих интегралов и связанных с ними
величин даны в табл. XXVIII—XXXII (стр. 902—911). Работа была выполнена
столь недавно, что к настоящему времени определены силовые постоянные
лишь для нескольких видов молекул (см. табл. 12, стр. 157). В других случаях
потенциал еще не применялся.
А. ДИНАМИКА ПАРНЫХ СТОЛКНОВЕНИЙ1) И ВЫЧИСЛЕНИЕ
ЭФФЕКТИВНОГО СЕЧЕНИЯ СТОЛКНОВЕНИЯ
В гл. 1, § 5 было отмечено, что динамика парных столкновений может
быть изучена с помощью эффективной потенциальной энергии <рзфф.(О> которая
определяется по формуле E.22) гл. 1 как сумма потенциальной энергии межмо-
лекулярного взаимодействия и центробежной потенциальной энергии. Для
потенциала F-12) Леннарда-Джонса функция эффективней потенциальной
энергии выражается через приведенные величины следующим образом :
%* ?. D.2)
Приведенная эффективная потенциальная энергия является функцией г* и
параметрически зависит от величины g*2&*2. На фиг. 118 приведены кривые
(р*ФФ. для нескольких величин g*2fe*2. Горбы на кривых грубо расположены в
точках, где силы притяжения Ван дер Ваальса балансируются центробежным
отталкиванием. При критическом значении g*2b*2 = -^ E1/з) имеется точка
перегиба с приведенной энергией 0,8. Выше этого критического значения кри-
вые эффективной потенциальной энергии монотонно спадают.
Кривые на фиг. 118 можно использовать для анализа динамики столкно-
вений. Если начальная приведенная кинетическая энергия пары сталкиваю-
щихся молекул g*2 < 0,8, то в зависимости от значения величины g*2fc*2, харак-
теризующей столкновение, динамически возможны несколько типов столкно-
вений. При g*2 я^ 0,2 можно проиллюстрировать несколько возможных слу-
чаев : а) Если Ь* таково, что g*2fc*2 = 10, то молекулы постепенно отклоняются
одна относительно другой. Это столкновение можно назвать «касательным»
столкновением. Влияние отталкивающей части потенциала в таком столкновении
пренебрежимо мало, б) Если fc* таково, что g*2fc*2 = 0,008, т.е. если происходит
х) Классическая динамика молекулярных столкновений подробно изложена в гл. 1, § 5.
Характерные для потенциала Леннарда-Джонса результаты, обсуждаемые в настоящем
параграфе, основываются на этом изложении.
§4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 430
почти «лобовое» столкновение, то на траекторию движения на далеких расстоя-
ниях влияют силы притяжения, а на близких — силы отталкивания, в) Если
А* таково, что система не может перейти совсем за горб, то наблюдается явление
образования «квазисвязанного состояния»1). Молекулы длительное время нахо-
дятся на расстоянии, близком к
положению горба, вращаясь одна
относительно другой2) с угловой
скоростью, определяемой выраже-
нием E.19) гл. 1. Три описанных
типа столкновений изображены на
фиг. 119.
Углы отклонения могут быть
найдены путем вычисления интег-
рала в выражении B.12) числен-
ными методами3). В табулированном
виде [26, 27, 33 ] результаты даны
в табл. XVI (стр. 873) и графи-
чески представлены на фиг. 120, где
A — cos x) построена как функция
Ь*2 для четырех значений g*2. Пло-
щади под этими кривыми равны
эффективным сечениям Q(l)*. Для
небольших значений относительной
кинетической энергии g*2 кривые
быстро осциллируют, что соответ-
ствует образованию квазисвязанных
состояний. Для столкновений с
g*2*C0,8 для каждого g* имеется
одно значение &*, при котором
энергия вершины горба точно сов-
падает с энергией системы. При
таких значениях Ь* = Ь% угол отклонения %, как можно видеть из табл. XVI,
равен бесконечности.
Горб кривой A — cos %) постепенно исчезает с увеличением энергии столк-
новения, так как на сталкивающиеся системы все в меньшей и меньшей степени
влияет углубление на кривой потенциальной энергии. При g*2 = 10 горб,
появляющийся вследствие сил притяжения, почти полностью пропадает.
+1,2
+1,0
+0,8
+0,6
+0,4
+0,2
-0,2
-0,4
-0,6
-0,8
-in
l \ ¦
" Л \ -
-
-
-
-
\ .2.2 \
P^—;
g*2h*z=0fl08
-
. i . i . i . i . i . i .- i . '
0123456789
r*= r/a
Фиг. 118. Приведенный эффективный потенциал
D.2) для потенциала F-12) Леннарда-Джонса
при некоторых значениях g*2b*2.
х) В оригинале : the phenomenon of «orbiting». — Прим. ред.
2) Столкновения, приводящие к образованию «квазисвязанного состояния», не следует
•смешивать со «связанными молекулами» на периодических орбитах, которые рассматривал
Кирквуд [28]. Последние представляют собой попавшие в потенциальную яму две молекулы,
вращающиеся одна относительно другой и находящиеся друг от друга на расстоянии порядка
размеров молекулы. Если предположить, что в газе имеют место парные столкновения, но
столкновения более высокого порядка не происходят, то «связанные молекулы» существо-
вать не могут, так как для образования или разрушения этого состояния требуются тройные
столкновения. Уравнение Больцмана, на основании которого была развита кинетическая
теория, не учитывает влияния тройных столкновений. Кирквуд показал, что в пределе для
малых плотностей (т. е. в рассматриваемом здесь приближении) связанные молекулы не
влияют на коэффициенты переноса. Однако при высоких плотностях такое влияние имеется.
Этому вопросу в дальнейшем уделяется внимание во введении к гл. 9. Столкновения, при-
водящие к образованию квазисвязанных состояний, дают небольшой вклад в эффективные
сечения. Однако при высоких температурах их влияние совсем мало и практически пренеб-
режимо. В связи с обсуждением вопроса о связанных молекулах следует отметить, что эти
состояния учтены во втором вириальном коэффициенте. Это является следствием того факта,
что в статистических суммах учитывается возможность попадания пар молекул в потен-
циальные ямы.
8) Для вычисления углов отклонений было использовано несколько методов. Один из
них состоит в подборе кривой для медленно меняющейся части подынтегральной функции,
которая позволяет далее провести интегрирование аналитически [26, 27, 33]. Другой, так
называемый гониометрический метод недавно использовали де Бур и Кранендонк [25].
440
Гл. 8. Явления переноса в разреженных газах
При этом кривая A — cos %) для потенциала Леннарда-Джонса очень близка к
кривой для твердых сфер и проходит лишь немного ниже нее, потому что столк-
новения молекул Леннарда-Джонса при высоких энергиях происходят по
закону отталкивания г*12. Иными словами, молекулы Леннарда-Джонса для
Фиг. 119. Различные типы столкновений
молекул Леннарда-Джонса.
о- — касательное столкновение, наиболее важны
силы притяжения ; б — почти лобовое столкно-
вение, вступают в игру большие силы отталки-
вания ; в — случай образования квазисвязанного
состояния, возможен вследствие наличия горба на
кривой эффективной потенциальной энергии, ко-
торый появляется при взаимном уравновешивании
силы притяжения и центробежной силы.
Фиг. 120. Угол отклонения для потенциала
Леннарда-Джонса.
Представлена зависимость A—cos*) от Ь*ш для
различных значений приведенной кинетической
энергии я*1. Около точки ftj1 функция A — cos x>
бесконечное число раз осциллирует между •
значениями О и 2.
столкновений с большой энергией имеют немного меньшие эффективные сече-
ния, чем твердые сферы (имеющие то же а)у в то время как для столкновений с
.низкой энергией их эффективные сечения больше.
По углам отклонения, являющимся функциями fc* и g*, из выражений
B.13) и B.14) путем численного интегрирования были получены эффективные
сечения Q<*>*(gr*2) и величины Q«>S)*(T*). Зависимости ?<'>• от приведенной
относительной кинетической энергии g*2 и ?<''*>* от приведенной температуры
доказаны на фиг. 121 и 122 соответственно. По определению, для модели твер-
дых сфер все эти величины равны единице. Как было отмечено выше, откло-
нение величин (?('>• и i№ *>* от единицы указывает на отклонение поведения
любой другой модели от модели твердых сфер. Из фиг. 121 ясно, что эффектив-
ные сечения для молекул Леннарда-Джонса при высоких энергиях вследствие
«мягкости» отталкивательной части потенциала меньше эффективных сечений
твердых сфер и, напротив, для малых энергий вследствие влияния сил притя-
жения эффективные сечения молекул Леннарда-Джонса в несколько раз
больше эффективных сечений твердых сфер. Поведение {2<''s>*, показанное на
§4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 441
Ii miii
I I Ml Г
фиг. 122, может быть объяснено тем же путем, так как процесс усреднения
усиливает роль столкновений с низкой энергией при низких температурах и
столкновений с высокой энергией при высоких температурах.
С целью облегчения вычислений коэффициентов переноса были составлены
обширные таблицы эффективных сечений и связанных с ними величин. В табл.
XII—XIV (стр. 868—871) [26, 33]
следующие величины табулированы
как функции Т* от 7* = 0,30 до
Т* =. 400: все ?2(/>s)*, необходимые
для вычисления третьего прибли-
жения вязкости и теплопроводности,
второго приближения коэффициента
диффузии и первого приближения
термодиффузионного отношения;
величины а*, в*, с*, определен-
ные формулами B.15) — B.17), и
-,#)*
I I Г I I I 1
0,1 Ц2 Ofl 0,6 1ft
2 4 6 810 20 40 60 100
9*2
Фиг. 121. Зависимость Q@* от приведенной
кинетической энергии относительного
движения g*2.
QW* —эффективное сечение для диффузии; Q<2> *" — для
вязкости и теплопроводности; Q(*)* появляется в
выражениях для третьего приближения коэффициентов
вязкости и теплопроводности.
2 3 4 56 810 2030406080100 200 400*
Т*=кТ/е
Фиг. 122. Зависимость •?(/,«)*
от температуры.
факторы Д3), #3> и /(J>, определяемые формулами B.19), B.32) и B.47). С помощью
этих таблиц и формул, приведенных в § 2, могут быть вычислены коэффициен-
ты переноса для потенциала F-12) Леннарда-Джонса. Следующие параграфы
настоящей главы посвящены обсуждению различных коэффициентов переноса
как с точки зрения их вычисления по межмолекулярным силам, так и с точки
зрения их экспериментальных значений. Даются также иллюстративные при-
меры, демонстрирующие использование табулированных функций.
Б. КОЭФФИЦИЕНТ ВЯЗКОСТИ ЧИСТЫХ ГАЗОВ
Чтобы вычислить для какого-нибудь чистого газа коэффициент вязкостц
по формулам B.18) или B.19), необходимо энать силовые постоянные в и а.
Они могут быть получены из анализа экспериментальных данных о вязкости
и самодиффузии, как описано ниже, или из данных о втором вириальном
коэффициенте и о коэффициентах Джоуля—Томсона, как описано в гл. 3, §6.
Грубые оценки могут быть получены из постоянных, характеризующих
критическую точку или точки плавления и кипения, как сказано в гл. 4, § L
Силовые постоянные некоторых газов, определенные из данных измерения
442
Гл. 8. Явления переноса в разреженных газах
вязкости, приведены в табл. 73. Более полные сведения по силовым постоянным
можно найти в табл. I (стр. 851). Силовые постоянные, приведенные в этой
таблице, получены рядом исследователей из различных измерений. Для вычи-
сления различных коэффициентов переноса желательно, если это возможно1
использовать параметры, определенные из вязкости.
Таблица 73
Силовые постоянные для потенциала F-12) Леннарда-
Джонса, определенные из данных о коэффициенте
вязкости41
Газ
е/к, °К
о, А
Эксперимен-
тальные
данные
Ne
Аг
Кг
Хе
N2
о2
сн4
со2
35,7
124
190
229
91,5
113
137
190
2,789
3,418
3,61
4,055
3,681
3,433
3,882
3,996
[14]
[28]
[29]
[13]
• Более полные таблицы силовых, постоянных для потенциала
Леннарда-Джонса даны в табл. I. В этой же таблице приведены силовые
постоянные, определенные из второго вириального коэффициента.
На фиг. 123 показана кривая вязкости ряда газов в приведенных едини-
цах. На этой же фигуре изображена теоретическая кривая для потенциала
F-12) Леннарда-Джонса. Для иллюстрации превосходного согласия между
-0,4
-0,6-
-1,0
-1,1
1
-
i
1
з
/ <
1 1 1 1—
Теоретическая кривая
для потенциала F-12) —. ©
Леннарда -Джонса "\.^- -
Р ©« V^""
^г## А - Неон
© <$* л
-Jy^ # • - яргон
/ © в - Водород
/-§- < - Дейтерий
в ®/^ © - Гелий
5/ -о- -Ксенон
i i i i
-1,0
-0,5
0,5
1,0
Ф и г. 123. Сравнение теоретической кривой \g(r)*/VT*) с экспериментальными данными.
Экспериментальные данные пересчитаны в приведенные величины по формуле -q* = ^/VmT, где в
качестве с и а использованы величины табл. I. Кривые для Неуеор# [74] и для Не^еор# [80] вычислены
на основе квантовомеханической теории (см. гл. 10, § 2). Экспериментальные данные: для неона и ар-
гона [72], для водорода и дейтерия [81], для гелия [82], для ксенона [30].
экспериментальными и вычисленными величинами в табл. 74 приведено их
численное сравнение. Рассмотрим теперь пример использования таблиц flW)*
и формулы для вязкости B.19).
§4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 443
ПРИМЕР
Задача. Вычислить вязкость СН4 при температуре 450 °К. Какова вязкость CD4 при той
же температуре?
Решение. Из табл. 73 найдем, что силовые постоянные метана равны е/k = 137 °К и
с = 3,882 А. Приведенная темпепатура Г* = кТ/е = 450/137 = 3,28. Из табл. XII (стр. 8С8я
найдем, что для Т* = 3,28 ?B,2)* = 1,016, а из табл. XIV (стр. 871) /<з) = 1,004. Молеку)
лярный вес СН4 равен 16,04. Отсюда по формуле B.19) найдем п
СН4:
= 266,93 _
- 1,004 = 1487 г/см. сек.
Для тяжелого изотопа метана CD4 (молекулярный вес 20,07)
самые, что и для легкой молекулы. Отсюда вязкость CD4 можн
силовые постоянные те ж-
, можно найти, умножая значение
вязкости СН4 на корень квадратный из отношения молекулярных весов:
CD4: D?]s • 107 = 1487 i/ -tJ^t- = 1663 г/см. сек.
Таблица 74
г,°к
80
100
120
140
160
180
200
220
240
260
280
300
400
500
800
1000
1200
1500
Сравнение экспериментальных и вычисленных значений вязкости*
[для потенциала F-12) Леннарда-Джонса]
rj • 10 г/см • сек
Аг
эксп. теор.
688
649
839 814
993 979
1146
1298
1447
1594
1739
1878
2014
2145
2270
4621
5302
5947
6778
1142
1300
1454
1601
1744
1882
2014
2143
2269
2839
3347
4641
5391
6083
6983
Ne
N.
эксп. теор. эксп. ! теор.
1198
1435
1646
1841
2026
2204
2376
2544
2708
2867
3021
3173
5918
6800
1212
1451
1665
1867
2054
2231
2396
2558
2713
2862'
3008
3149
3812
4383
5945
6872
г
698
826
948
1068
1183
1295
1403
1505
1603
1696
1786
3493
4011
4452
5050
i
687
820
947
1070
1186
1296
1402
1503
1600
1693
1785
2202
2570
3528
4068
4554
5268
сн4
эксп. теор. эксп. теор,
I
403
478
560
629
703
778
850
919
986
1053
1116
I
393
472
553
630
707
780
852
921
987
1052
1116
1405
1661
2312
2687
3034
3498
768
917
1061
1202
1341
1476
1604
1728
1845
1958
2071
4115
4720
!
757
910
1059
1203
1342
1474
1602
1726
1845
1959
2070
2578
3031
4183
4853
5457
6264
со,
эксп. теор.
1015
1112
1209
1303
1400
1495
3391
3935
4453
5139
1014
1114
1212
1308
1402
1495
1923
2309
3285
3839
4348
5052
• Силовые постоянные и указания на литературу об экспериментальных данных приведены в табл. 73
Два подбираемых параметра потенциала Леннарда-Джонса могут быть
получены из данных о ьязкости методом, аналогичным тому, который был
использован в гл. 3, § б для анализа данных о втором вириальном коэффициенте.
Для определения о и е необходимо иметь экспериментальные значения вяз-
кости при двух температурах Тг и Т2. Процедура определения состоит в сле-
дующем. Сначала определяется величина кп из соотношения
затем с помощью формулы B.19) составляется уравнение
k =[Q(*.*)*(T*)
V I Ж2' 2)* (Т*)
D.3)
D.4)
444
Гл. 8. Явления переноса в разреженных газах
из которого методом подбора или каким-либо другим приближенным методом
определяется величина е/к. [В уравнении D.4) TJ = кТ(/е.\ В качестве пер-
вого приближения е/к при решении уравнения D.4) можно использовать вели-
чины, вычисленные по формулам, приведенным в гл. 4, § 1, из данных о тем-
пературе кипения или о критической температуре. После того как величина
е/к определена, по формуле
^ 266,93 ЩТг /<з)(?7) /А -.
-2- D.5)
можно определить диаметр столкновения. В формуле D.5) Г, равно либо Tv
либо Т2. Параметры, определенные при различных 7\ и Т2, могут несколько
отличаться друг от друга, так как потенциал F-12) Леннарда-Джонса является
эмпирической функцией и не дает точного описания зависимости межмолекуляр-
ных сил от расстояния между молекулами. Для иллюстрации определения
силовых постоянных из измерений вязкости ниже приведен численный пример.
ПРИМЕР
Задача. В табл. 76 приведены экспериментальные данные о вязкости для ксенона. Из
этих данных получить параметры е и а для потенциала F-12) Леннарда-Джонса. Сравнить
полученные таким образом величины силовых постоянных с силовыми постоянными, полу-
ченными из второго вириального коэффициента в гл. 3, § 6. (Критическая температура Хе
равна 289,8 °К.)
Решение. Полагая Тг = 293 ° К и Т2 = 500 °К, по формуле D.3) найдем/^ = 1,2370.
Так как кТс/е^ 1,3 и Тс = 289,8 °К, в первом приближении для е/к получим 223 °К.
Значения кч для этой величины е/к и для следующих приближений е/к, вычисленные по фор-
муле D.4), приводятся в табл. 75.
Таблица 75
«//с, °К
223
230
[229]
228
т;
1,3139
1,2739
1,2795
1,2851
а(М)*(г?)
1,3923
1,4121
1,4093
1,4065
1,0001
1,0001
1,0001
1,0001
т*
S
2,2422
2,1739
2,1834
2,1930
Я(М)*(Г)
1,1312
1,1427
1,1410
1,1393
(8) ( Л
1,0019
1,0018
1,0018
1,0018
1,2330
1,2379
1,2373
1,2366
Наилучшее согласие вычисленного значения кп с экспериментальным значением полу-
чается при е/к = 229 °К- Далее по формуле D.5) для любой из температур 7\ или Т2 найдем
0е = 16,440 и а = 4,055 А. Из измерений второго вириального коэффициента Хе, проведен-
ных Битти, Баррио и Брайерли, мы нашли, что а = 4,10 А и е/к = 221 °К.
Таблица 76
Экспериментальные [30] и вычисленные величины
г)(Т) для ксенона
Температура,
107, г/см • сек
289,7
293
400
450
500
550
эксп.
2235
2260
3009
3351
3652
3954
выч.
2236
2260
3016
3341
3653
3951
В табл. I (стр. 851) приведены силовые постоянные некоторых газов,
молекулы которых не попадают в категорию сферических или полярных. Для
таких газов экспериментальные данные удалось согласовать с теоретическими
результатами для потенциала Леннарда-Джонса вследствие того, что вяз-
кость слабо зависит от внутренних степеней свободы. Табулирование силовых
§4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 445
постоянных для упомянутых газов кажется полезным по той простой причине,
что по таким данным можно экстраполировать известные значения вязкости в
области высоких или низких температур. Однако наряду с этим было устано-
влено, что для некоторых газов температурная зависимость вязкости значи-
тельно отличается от предсказанной для потенциала Леннарда-Джонса. Для
таких веществ невозможно подобрать параметры для потенциала F-12) Леннар-
да-Джонса. Ниже обсуждаются эти аномальные случаи и объясняется, почему
свойства этих газов проявляют такие отклонения1).
1. Полярные молекулы: Н2О, NH3, HBr, HCN, HJ, HgCl2. Энергия взаи-
модействия полярных молекул значительно отличается от энергии взаимодей-
ствия неполярных молекул, так как потенциальная функция полярных молекул
включает член с сильной угловой зависимостью, обратно пропорциональной
третьей степени расстояния между молекулами. Поэтому неудивительно ано-
мальное поведение таких молекул относительно потенциала F-12) Леннарда-
Джонса.
2) Длинные молекулы : л-гептан. Коэффициенты переноса и, в частности,
вязкости не очень чувствительны к форме молекул. Однако если отношение
длины молекулы к ее диаметру становится слишком большим, то форма молекул
влияет на все физические свойства газа. Например, температурная зависимость
вязкости п-гептана сильнее той, которую можно было бы ожидать для сфери-
ческих молекул.
3) Свободные радикалы или молекулы в возбужденных состояниях. Как
объясняется в гл. 13, молекулы с насыщенными валентными связями ведут
себя совершенно иным образом, чем молекулы, в которых общий результирую-
щий спин электронов равен нулю. На больших расстояниях, когда пере-
крытие электронных волновых функций двух молекул несущественно, моле-
кулы взаимодействуют по обычному закону притяжения Ван дер Ваальса, в
котором силы взаимодействия обратно пропорциональны седьмой степени рас-
стояния. Когда же молекулы сближаются на малые расстояния, потен-
циальная кривая может принимать различные формы в зависимости от того,
оказывается ли сталкивающийся комплекс, образованный из двух молекул,
в состоянии притяжения или в состоянии отталкивания. Эти типы взаимо-
действия являются следствием принципа Паули, который накладывает опре-
деленные ограничения на свойства симметрии электронных и спиновых
волновых функций. Квантовая механика позволяет вычислить вероятность
того, в каком состоянии — отталкивания или притяжения — оказывается
комплекс молекул при столкновении.
В развитие только что сказанного рассмотрим простейший случай двух
сталкивающихся атомов водорода. На больших расстояниях два атома притя-
гиваются друг к другу по закону 1/г6. При тесном сближении атомов требо-
вания симметрии, накладываемые на общую волновую функцию, приводят к
тому, что могут образовываться1^- или ^'-состояния. Синглетное состояние
(состояние притяжения) соответствует обычной гомеополярной связи и имеет
силовые постоянные, равные а = 0,5 А и ejk = 51 000 °К. Одно столкновение
из каждых четырех происходит по потенциальной кривой, характеризуемой
этими постоянными. Триплетное состояние соответствует первому возбужден-
ному состоянию молекулы водорода (в этом состоянии, исключая большие
расстояния, молекулы отталкиваются) и имеет силовые постоянные2), равные
а = 3,5 А и elk = 3,8°K. Из каждых четырех столкновений по закону, харак-
теризуемому этими постоянными, происходят три столкновения.
Так как коэффициенты переноса этих газов не могут быть описаны с по-
мощью потенциала F-12) Леннарда-Джонса, рассматриваемый случай удобно
обсудить на основе результатов для простых потенциалов. В § 3 было показано,
^Эпштейну и Мариону [31 ] удалось объяснить температурную зависимость вязкости
паров ртути и металлов ра основе потенциала F-9) Леннарда-Джонса.
*) По неопубликованным грубым квантовомеханическим расчетам Гиршфельдера.
446
Гл. 8. Явления переноса в разреженных газах
что для молекул, которые отталкиваются одна от другой по закону обратной
степенной зависимости, коэффициент вязкости имеет простой вид
V = k'Ts. D.6)
Постоянные к' и s («температурный показатель») определяются из экспери-
ментальных данных. Установлено также,
что для молекул, взаимодействующих сог-
ласно модели Сюзерленда, вязкость прибли-
женно выражается следующим образом:
D.7)
где ksn S (постоянная Сюзерленда) — под-
бираемые параметры. Значения к' и s, a
также ks и S для веществ, для которых
невозможно подобрать параметры потен-
циала Леннарда-Джонса, даны в табл. 77.
Наилучшее представление о формулах
D.6) и D.7) можно получить на основе
сравнения их с результатами, даваемыми
потенциалом Леннарда-Джонса. Таким спо-
собом можно получить зависимость s и S
от приведенной температуры Т*. Резуль-
таты сравнения приведены на фиг. 124. Диапазон изменения «постоянных» s
и S с температурой определенно указывает на то, что простые формулы D.6)
и D.7) не следует использовать более широко, чем для интерполяции в пре-
делах небольшого интервала температур, особенно если для рассматриваемых
газов известны силовые постоянные потенциала F-12) Леннарда-Джонса.
2,0 10.0
Т*=кТ/е
Фиг. 124. Температурная зависимость
постоянной Сюзерленда S и темпера-
турного показателя s.
Таблица 77
Вязкость аномальных газов
Ю7 =
1 +(
Газ
НаО
NH8
HJ
НВг
HCN
HgCla
л-С7Н1в
Интервал
температур.
300-400
500-600
600-700
300-400
500-600
600-700
300-400
400-500
300-400
300-400
500-600
500-600
600-700
750-850
350-450
450-550
к'
2,039
1,227
1,598
1,203
2,576
5,207
6,889
10,42
5,004
0,7443
1,131
1,521
4,841
3,406
2,163
0,6715
s
1,079
1,164
1,123
1,181
1,053
0,9427
0,9837
0,9152
1,040
1,215
1,144
1,180
1,000
1,057
0,9789
1,172
к8
140,2
235,8
244,4
2С2,7
189,4
164,7
221,9
229,4
245,3
166,3
185,4
351,6
248,1
314,1
72,73
133,1
s
459,4
1051,0
1108,0
740,7
684,3
518,7
312,9
334,4
376,2
836,2
999,1
1191,0
656,5
982,8
363,2
1022,0
S*
т
1,313
1,911
1,705
2,116
1,244
0,7980
0,8940
0,7431
1,075
2,389
1,817
2,165
1,010
1,229
0,9080
2,044
Литература
[32]
[32]
[28]
[32]
[32]
[32]
[28]
¦ Постоянная Сюзерленда поделена на среднюю температуру из указанного интервала температур.
§ 4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 447
Ясно также, что если S/X > 1,1 или s> 1,01, то невозможно выбирать такое
е/ку которое может быть использовано в потенциале F-12) Леннарда-Джонса.
Известен ряд таких газов. Некоторые из них перечислены в табл. 77.
в. коэффициент вязкости смесей
Для вычисления вязкости бинарных смесей по формуле B.22) необходима
знать силовые постоянные сие, описывающие взаимодействия между двумя
одинаковыми и двумя неодинаковыми молекулами. Силовые постоянные для
взаимодействия между одинакорыми молекулами могут быть получены иа
экспериментальных данных о вязкости чистых газов. Для вычисления силовых
постоянных, характеризующих взаимодействие между неодинаковыми моле-
кулами, в принципе могут быть использованы данные о вязкости бинарных
смесей. Однако при этом/как и в случае определения силовых постоянных из
вторых вириальных коэффициентов смесей, необходимо иметь чрезвычайн0
Вязкость бинарных газовых смесей*
rj • 1О7 г/см • сек
Таблица 78
Аргон—неон
г,вк
293,16
373,16 .
473,16
Данные
ЭКСП. [34]
выч.
ЭКСП.
выч.
ЭКСП.
выч.
0,0
3092
3070
3623
3566
4220
4170
26,80
2808
2795
3313
3281
3890
3847
Аг, %
60,91
2504
2493
2990
2963
3529
3495
74,20
2401
2390
2885
2853
3413
3375
100,0
2213
2208
2693
2660
3222
3164
Кислород—окись углерода
т,°к
300,06
400,06
500,06
Данные
ЭКСП. [35]
выч.
ЭКСП.
выч.
ЭКСП.
выч.
о„ %
0,0
1776
1771
2183
2178
2548
2539
23,37
1841
1840
2268
2263
2650
2638
42,01
1900
1896
2343
2331
2741
2717
77,33
1998
2003
2482
2464
2908
2871
100,0
2057
2073
2568
2550
3017
2972
* Широкое сравнение экспериментальных и вычисленных результатов было проведено Гиршфельдером
Бердом и Спотц [33].
точные данные как для чистых компонент, так и для их смеси. Наиболее просто
и точно силовые постоянные, характеризующие взаимодействие между двумя
неодинаковыми молекулами, можно получить из измерений коэффициента
диффузии как функции температуры. Однако практически измерения такого
рода всегда отсутствуют, поэтому обычно используются эмпирические «комби-
национные правила», которые связывают силовые постоянные одинаковых моле-
кул с силовыми постоянными неодинаковых молекул. Эти правила имеют вид
еи =
D.9)
448
Гл. 8. Явления переноса в разреженных газах
Соотношения D.8) и D.9) обсуждались в гл. 3, § б в связи с вычислением вто-
рого вириального коэффициента смесей. Для иллюстрации точности, которую
можно ожидать при вычислении вязкости смесей по силовым постоянным, по-
лученным из комбинационных правил, в табл. 78 сравтваются вычисленные
и экспериментальные значения вязкости для двух газовых смесей.
Ниже приводится пример численного расчета вязкости бинарных смесей.
ПРИМЕР
Задача. Вычислить вязкость смеси, состоящей по объему из 23,37% N2 и 76,63% СО,
при температуре 226,9 °С.
Решение. Силовые постоянные чистых газов выберем по табл. I (стр. 851). Силовые
постоянные, характеризующие взаимодействие между двумя неодинаковыми молекулами,
найдем по эмпирическим комбинационным правилам D.8) и D.9). Выпишем найденные сило-
вые постоянные, необходимые для расчетов:
N2: аг = 3,681 А, ег /к = 91,5 °К,
СО : <т2 = 3,706 А, е2 /к = 88,0 °К,
Na —СО: <т12=3,694А, е12/к = 89,7 °К.
Для Т = 226,9 °С = 500,1 °К приведенные температуры равны Т\ = 5,466, Т% = 5,683,
^12 = 5,575. По табл. XII и ХШ (стр. 868 и 870) табулированных функций для потенциала
F-12) Леннарда-Джонса найдем
М2|2)* E,466) = 0,9127,
?<2'2)* E,683) = 0,9060,
По этим данным можно вычислить величины
[77ili- 107 = 2555 [по
[*fe1i'10? = 2539 Епо
[г?12]1.107 = 2546 [по
А*
E,575) = 0,9093,
E,575) = 1,102.
эрмуле B.18)],
эрмуле B.18)],
эрмуле B.21)].
Концентрационную зависимость вязкости смеси N2 — СО при 226,9 °С найдем по формуле
[*?cm.]i- Ю7 = —5ДJ3 х\ + 13,0455 хг х2 + 6,5423 х\ '
Для хг = 0,2337 и х2 = 0,7663 найдем, что [^см.]1 • 107 = 2542 г/см • сек. Экспериментальное
значение вязкости по Л андольту-Бернштейну " [29 ] равно ^•107 = 2550 г/см-сек.
Таблица 79
т °к
193
373
473
Ne
55,76
31,93
21,66
21,89
55,76
31,93
21,66
21,89
55,76
31,93
21,66
21,89
Вязкость тройных
Состав (в % по объему)
Аг
26,70
32,13
58,51
23,82
26,70
32,13
58,51
23,82
26,70
32,13
58,51
23,82
смесей газов
Не
17,54
35,94
19,83
54,29
17,54
35,94
19,83
54,29
17,54
35,94
19,83
54,29
fob-
выч.*
2718
2562
2429
2500
3205
3025
2895
2938
3752
3551
3425
3449
1О7 г/см • сек
эксп. [36]
2740
2569
2411
2504
3237
3044
2886
2957
3790
3574
3415
3470
* Вычисления выполнены по формуле B.25).
Вязкость многокомпонентной смеси может быть найдена по формуле
B.25). Для использования этой формулы необходимо знать величины й<'»в>*
и а*, а также силовые постоянные, характеризующие взаимодействия между
любыми двумя одинаковыми и любыми двумя неодинаковыми молекулами.
Таблица 80
Вязкость многокомпонентных смесей промышленных газов
со,
8,6
13,3
6,2
10,4
10,80
6,70
6,40
6,0
10,6
8,9
8,7
3,70
1,7
2,1
2,0
3,3
2,2
2,2
2,5
4,8
3,5
3,1
о,
2,3
3,9
10,7
2,00
0,10
3,00
0,10
0,30
0,9
0,9
1,4
0,6
0,6
1,0
0,8
0,3
0,3
0,5
Соста!
СО
28,5
7,80
0,30
25,70
29,8
30,7
32,8
27,10
6,0
5,7
4,6
3,8
4,1
4,0
14,9
26,4
27,3
28,6
J (В % ПО
н,
1,6
2,20
2,20
0,70
11,50
3,9
3,3
1,5
9,50
57,5
53,0
54,9
51,3
53,1
52,3
53,0
17,2
14,4
17,7
объему)
СН4
0,3
0,4
0,2
1,60
24,0
24,3
23,5
29,6
29,5
29,9
18,1
2,6
3,7
4,2
N.
89,1
82,8
83,1
59,5
85,00
83,20
89,60
56,70
55,4
56,7
56,8
57,80
7,8
ПД
11,6
10,0
9,2
9,4
9,1
48,2
50,0
45,0
тяже-
лые
угле-
водо-
роды
2,1
2,3
2,0
1,4
1,3
1,2
1,6
0,5
0,8
0,9
Темпера-
ч%>
293
293
293
293
300,5
524,5
973
1279
307,5
519
975
1285
314
518
974,5
1287
302
526
976
1283
293
293
293
300,5
565,5
981
1282
293
293
293
293
293
293
293
293
293
293
т} • 107 г/см сек
эксп.
1756
1749
1793
1738
1827
2715
4117
4856
1842
2655
4048
4808
1904
2706
4113
4895
1823
2686
4041
4777
1743
1747
1749
1815
2819
4045
4792
1262
1304
1310
1332
1306
1307
1355
1714
1712
1715
выч.*
1761
1765
1789
1798
1792
2661
4008
4753
1835
2653
4019
4783
1856
2644
4017
4777
1829
2696
4042
4821
1794
1797
1802
1816
2823
4041
4803
1254
1290
1398
1269
1254
1261
1373
1743
1732
1719
Лите-
ратура
|[37]
)
[38]
[38]
[38]
[38]
[37]
)
[38]
[37]
* Вычислено по формуле B.25).
450
Гл. 8. Явления переноса в разреженных газах
В табл. 79 сравниваются вычисленные и экспериментальные значения вяз-
кости тройных смесей газов, и можно убедиться, что между вычисленными и
экспериментальными значениями имеется согласие в пределах 1%. В табл. 80
такое же сравнение проводится для некоторых смесей промышленных газов.
Более чем в 50% рассмотренных случаев согласие превосходное. Для некото-
рых смесей имеющиеся расхождения, возможно, являются следствием ошибок
в газовом анализе. Вычисленные величины для этих многокомпонентных газо-
вых систем отличаются от экспериментальных приблизительно в пределах 1%.
ПРИМЕР
Задача. Вычислить коэффициент вязкости тройной газовой смеси, состоящей из
50%СН4,35% N2 и 15% О2, при давлении, равном 1 атм,и температуре 380 аК. Использо-
вать уравнения B.25) и B.30) и сравнить результаты, получаемые по этим двум формулам.
Решение, а) Чтобы использовать формулу B.25), необходимо вычислить элементы
определителя Htj по формулам B.26) и B.27). Для этого в свою очередь необходимо знать
силовые постоянные шести возможных типов взаимодействий между тремя различными
химическими сортами молекул. В табл. 81 выписаны необходимые для расчета силовые
постоянные, табулированные функции и некоторые величины, получающиеся в процессе
расчета. Благодаря этой таблице ясно, в каком порядке удобнее проводить вычисления.
Вычислив Я/у, найдем отношение детерминантов [?7CMt]t • 107 = 1762 г/см»сек.
Таблица 81
Расчет вязкости тройной газовой смеси по формуле B.25) для потенциала F-12)
Леннарда-Джонса
Компоненты Молярные доли Молекулярные веса
1—СН4
2 — N.
3 —О,
х, - 0,50
х, - 0,35
х, - 0,15
Мх - 16,04
М, - 28,02
М, - 32,00
1
1
1
1
2
2
3
j
1
2
3
2
3
3
3,882я
3,782*
3,658*
3,681е
3,557*
3,433a
41 our
Л f *
136,5*
111,7*
124,3*
91,46*
101,8*
113,2я
" ^п
2,784
3,402
3,057
4,155
3,733
3,357
1,060
1,007
1,034
0,9622
0,9851
1,010
2MiMie
Л* +Mi
16,04
20,40
21,37
28,02
29,88
32,00
\/2MiMfTd
V Mi+м,
78,07
88,05
90,11
103,2
106,6
110,3
[Vif]\m\07t
г)см • сек
1305е
1632™
1738™
2113е
2283™
2473е
А*з
1,096
1,095
—
1,097
—
Mi
Mi
1,747
1,995
—
1,142
—
Mi Mi
(Mi + Mfy
315
0,2224
—
0,2489
—
+4213"
- 259*
- 100*
+ 1923"
- 59*
+ 754"
а По табл. I стр. 851 ; силовые постоянные из данных о вязкости.
б Использованы эмпирические комбинационные правила D.8) и D.9).
в По табл. XII, стр. 868.
г Равна М< при i — j.
д Равна УТЙ^Гпри / - j.
«По формуле B.18).
ж По формуле B.21).
з По табл. XIII, стр. 870.
и По формуле
к По формуле
б) Данные, содержащиеся в табл. 81, также могут быть использованы для вычисления
вязкости по формуле B.30), которую мы перепишем в виде
х?
2 А*[ад]1
В рассматриваемом случае v = 3. Из этой формулы найдем
[^см.]1-Ю7= 1731 г/см-сек.
§ 4. Вычисление коэффициентов переноса Оля потенциала F-12) Леннарда-Джонса 451
Г. КОЭФФИЦИЕНТ ТЕПЛОПРОВОДНОСТИ
Коэффициент теплопроводности для одноатомных газов в первом и третьем
приближениях может быть вычислен по формулам B.31) и B.32) соответственно.
Поправочный множитель /(я3) для потенциала F-12) Леннарда-Джонса приведен
в табл. XVI (стр. 873).
Коэффициент [А]3 легко может быть найден, если известны параметры а и е.
В табл. 82 сравниваются значения коэффициентов теплопроводности, получен-
ные экспериментальйо, со значениями, вычисленными теоретически, с исполь-
зованием силовых постоянных, найденных из данных о вязкости (см. табл. I).
Таблица 82
Теплопроводность одноатомных газов. Сравнение экспериментальных значений
с вычисленными на основе потенциала F-12) Леннарда-Джонса
А- 1О\ кал/см сек • °К
T, eK
90,2
194,7
273,2
373,2
491,2
579,1
Не
выч.* |
1711
2817
3507
4296
5135
5711
эксп.**
1655
2706
3390 [40]
3406
3438 [41]
3510 [42]
4165
4947
5504
Ne
выч.
498
878
1105
1356
1608
1791
эксп.
489
876
1092 [28]
1110
1357
1595
1789
Аг
выч.
137
292
392
504
619
696
эксп.
141
293
385[41]
390[43]
394
506
614
685
Кг
выч.
68
148
206
273
343
390
эксп.
152
[ 190[28]
1
( 208
272
340
388
Хе
выч.
40
85
120
161
204
234
эксп.
91
123
124 [28]
168
208
237
* Значение [А ], вычислено по формуле B.32). Силовые постоянные взяты из данных о вязкости по
табл. I, стр. 851.
** Если отсутствует указание на источник, то эти величины взяты из работы [39].
Для многоатомных газов наилучшей формулой для расчета [А]х является
формула B.33), включающая множитель(-^--^-+у|, приближенно учитываю-
щий переходы энергии между поступательными и внутренними степенями
свободы. В табл. 83 сравниваются экспериментальные значения X для много-
атомных газов с вычисленными [А]х по формуле B.33). Силовые постоянные
для чистых газов взяты из^ данных о вязкости. В таблице приводятся также
(А. Г* *\ \
15* ~i~ ~*~ т) *
Из табл. 83 следует, что если известна теплоемкость как функция темпера-
туры, то коэффициент теплопроводности можно получить в достаточно хорошем
приближении, используя формулу B.33). Ниже приводится пример расчета
коэффициента теплопроводности чистого вещества.
ПРИМЕР
Задача. Вычислить коэффициент теплопроводности : а) чистого аргона; б) чистого
водорода при Т = 273,16 °К.
Решение, а) По табл. I найдем силовые постоянные для аргона:
а = 3,418 А, у = 124°К.
Далее вычислим приведенную температуру Т* = 273,16/124 = 2,203. По табулированным
функциям для потенциала F-12) Леннарда-Джонса найдем
?<2,2)* B,203) =1,138,
ГУ B,203) = 1,003,
452
Гл. 8. Явления переноса в разреженных газах
И, наконец, по формулам B.31), B.32) вычислим [А]3 • 107 = 393 кал/см • сек - °К.
Вычисленное значение [А]3 хорошо согласуется с величинами, данными в табл. 82.
б) Силовые постоянные для Н2 равны
а = 2,968 A, y = 33,3°К.
Далее, Т* = 273,16/33,3 = 8,203, откуда из табулированных функций найдем
?B,2)* (8,203) = 0,8508.
Затем по формуле B.31) вычислим [Я]х- 107 = 3090 кал/см • сгк .°К.
Эта величина должна быть умножена на поправочный множитель Эйкена
_4_ _С*_ j_ JT| _ Г 4 4,883 , 3
_ 15 R
В результате найдем
L 15 R + 5 J ~~ L 15
1,987
[А]Эйкен . ю? = 3878 кал/см. сек .°К.
Сравнение этой величины с экспериментальными данными приведено в табл. 83.
Таблица 83
Теплопроводность многоатомных газов. Сравнение экспериментальных
значений с вычисленными для потенциала F-12) Леннарда-Джонса
(с поправкой Эйкена)*
Газ
н2
О*
со2
сн4
N0
100
200
273
300
100
200
273
300
200
273
300
100
200
273
300
150
200
273
300
А • 1О7, кал/см - сек *К
выч.**
1618
3053
3878
4140
224
436
568
615
235
345
386
247
493
676
741
347
445
576
619
эксп.
1625
3064
J39651
\4040J
4227
216
438
Г5841
\577J
635
227
Г3491
\360J
398
254
522
Г7341
1720J
1 У
819
321
425
567
619
4 Cv 3
15 Я 5
1,057***1
1,217***]
1,255
1,1259
1,267 1
1,268/
1,273
1,278
1,370
1,487
1,527
1,400 1
1,407/
1,460
1,479
1,333
1,310
1,297
1,293
Ссылка на источник
экспериментальных
данных
[40]
[40,43]
[40]
[40]
[40,45]
[40]
[40]
[40,43]
[40]
[40]
[40,45]
[40]
[40]
• Величины Cv, необходимые для вычисления поправочного множителя Эйкена (-- * + -,. ), взяты из
работы [44'. Яйкен *
¦• Вычисленные величины равны [Я]^*1 пс" и найдены по формуле B.33). Силовые постоянные, необхо-
димые для расчетов, взяты из табл. I, стр. 851, полученной на основе данных о вязкости.
•¦¦ Значения С„ для Н, при 100 и 200е К взяты для определенного отношения концентраций орто- и
л ара-модификаций, равного 3 : 1, а не из работы [44].
Таблица 84
Сравнение вычисленных и экспериментальных бинарных газовых смесей*
Одноатомные газы
Газовая смесь
(Г, °К)
Ne—Аг
B73,16)
Процент
легкой
компоненты
смеси
0,0
27,04
45,37
84,68
94,61
100,00
v • ю7,
г/сл* • сек
эксп.
2089 [34]
2173
2206
2118
2017
1923
А • 107
кал/см • сек • °К
эксп.
389 [46]
742
1077
2320
2939
3386
вычислено
по формуле
B.36)
391
755
1102
2439
3046
3466
х »?эксп. ' 10?
390
536
695
1660
2530
3580
Многоатомные газы
Гяяовая смесь
* aouoa/i wiviccd
(Г, °К)
Н2-СО2
B73,16)
Н2~СО
B73,16)
H2-N2
B73,16)
H2-N2O
B73,16)
Н2-О2
B95)
Процент
легкой
компоненты
0,0
10,0
14,2
25,0
35,5
50,0
75,0
90,1
100,0
0,0
16,3
27,2
56,6
63,4
79,4
100,0
0,0
15,9
39,0
65,2
79,5
80,3
100,0
0,0
7,5
20,9
38,6
59,9
81,2
100,0
0,0
3,36
25,0
50,0
75,0
94,74
100,0
-п • Ю7,
г/см • сек
эксп.
1369 [47]
1386
1392
1406
1415
1417
1341
1163
854
1706 [48]
1715
1649
1538
1488
1303
853
1688 [48]
1670
1600
1449
1285
1274
853
1358 [47]
1366
1379
1388
1369
1273
854
2045 [48]
2044
1994
1855
1588
1088
887
эксп.
360 [43]
510
570
770
1000
1350
2270
3150
4040
530 [43]
800
1030
1800
2090
2700
4040
550 [43]
800
1270
1940
2520
2570
4040
380 [43]
480
710
1070
1700
2720
4040
625 [49]
651
1112
1827
2749
3744
4180
А • 10',
кал/см • сек • К
вычислено
B.40)
360
515
586
785
1007
1377
2301
3191
4040
530
770
965
1701
1934
2633
4040
550
786
1241
2023
2655
2996
4040
380
502
745
1131
1748
2671
4040
625
676
1060
1675
2606
3765
4180
по формуле
B,41)
360
427
461
565
701
977
1936
3154
4040
530
690
805
1425
1656
2416
4040
550
698
1028
1753
2449
2498
4040
380
430
545
775
1282
2397
4040
625
657
920
1441
2496
3850
4180
454
Гл. 8. Явления переноса в разреженных газах
Продолжение табл. 84
Гя*човяя смрск
1 CtoUDd/1 CIVIC С О
(Т, °К)
Н2-Аг
B73,16)
N2-Ar
B73,16)
Процент
легкой
1/пмппиритм
l\UiVlI lUnCn 1 хзй
0,0
9,0
18,0
40,0
60,0
80,2
100,0
0,0
20,38
35,87
61,08
78,04
100,00
v • Ю7,
г/см • сек
эксп.
2135 [28]
2126
2111
2020
1863
1480
854
2089**
2937
3064
3043
2863
1659
€КСП.
390 [43]
550
730
1260
1870
2700
4040
385 [41 ]
417
444
490
524
566
А • 107,
кал jсм ¦ сек • ° К
вычислено i
B.40)
390
509
647
1087
1676
2588
4040
385
440
476
524
• 547
566
ю формуле
B.41)
390
451
527
802
1272
2148
4040
385
605
690
798
839
566
¦ Силовые постоянные во всех расчетах взяты по табл. I, стр. 851.
•* Величины вязкостей смесей N, — Аг были вычислены по формуле B.22).
Теплопроводность смеси двух одноатомных газов может быть найдена по
формуле B.36). Если силовые постоянные для чистых газов, полученные из
данных о вязкости, известны, то силовые постоянные, характеризующие взаи-
модействие между неодинаковыми молекулами, могут быть приближенно
вычислены по комбинационным правилам D.8) и D.9). В табл. 84 приведены
экспериментальные значения А для смесей аргон—гелий. Там же даны зна-
чения А, вычисленные по формуле B.36). Силовые постоянные определены из
данных о вязкости (взяты из табл. I) и по формулам D.8) и D.9). Согласие полу-
чается достаточно хорошее. В таблице приведены также экспериментальные
15 R
значения вязкости смеси и табулирована величина -г- —п,-т—tj" *7эксп.
Сравнение последней величины с измеренными значениями коэффициента теп-
лопроводности показывает, что теплопроводность смеси не пропорциональна
ее вязкости.
В настоящее время лучшим методом вычисления коэффициента тепло-
проводности бинарных смесей многоатомных газов является метод вычисления
его по формуле B.40), т. е. вычисления Асм. по формуле для одноатомных газов
и последующего умножения Асм. на поправочный множитель. Другой менее
удовлетворительный метод состоит в умножении коэффициента вязкости смеси
на некоторый множитель в соответствии с формулой B.41). В табл. 84 сравни-
ваются экспериментальные данные с результатами вычисления теплопровод-
ности обоими методами.
Д. КОЭФФИЦИЕНТ ДИФФУЗИИ1)
Для вычисления коэффициента диффузии JD12 бинарных смесей газов по
формуле B.44) необходимо знать силовые постоянные ст12 и е12, характеризую-
щие взаимодействия между двумя неодинаковыми молекулами. В настоящее
время в наилучшем приближении эти постоянные можно найти по комбина-
ционным правилам D.8) и D.9). В табл. 85 сравниваются вычисленные и экспе-
риментальные значения коэффициентов диффузии.
х) Наиболее полное изложение современного состояния вопроса об обычной диффузии
и термодиффузии в различных типах систем, находящихся в газообразном и жидком состоя-
ниях, как с экспериментальной, так и с теоретической точек зрения, дано в монографии
Иоста [50].
Таблица 85
Сравнение вычисленных и измеренных коэффициентов диффузии
Газовая смесь
Аг-Не
Аг-Н2
Ar-N2
Ar—O2
Аг-СО2
N2-H2
N2-O2
N2-CO
N2-CO2
N2-C2H4
N.-C.H.
N2-nC4H10
N2-u30-C4H10
Na—(/ис-бутен-2
Ha-O2
H2-CO
H2-COa
Ha-N2O
H2-SFe
Ha-CH4
H2-C2H4
H2-C2He
H2—(/ис-бутен-2
co-o2
coa-oa
CO2-CO
CC^-N.O
CO2-CH4
3,059
3,193
3,550
3,426
3,707
3,325
3,557
3,636
3,839
3,957
4,050
4,339
4,511
4,467
3,201
3,279
3,482
3,424
3,922*
3,425
3,600
3,693
4,111"
3,512
3,715
3,793
3,938
3,939
21,3
64,3
106
118
153
55,2
102
100
132
137
145
194
169
188
61,4
60,6
79,5
85,6
89,Г
67,4
82,6
87,5
113**
112
147
145
204
161
T, °K
273,2
293,2
293,2
293,2
293,2
273,2
288,2
293,2
273,2
293,2
273,2
273,2
288,2
293,2
298,2
298,2
298,2
298,2
298,2
298,2
273,2
273,2
273,2
288,2
293,2
298,2
273,2
298,2
273,2
298,2
298,2
298,2
298,2
273,2
273,2
293,2
273,2
273,2
273,2
Вычисление
значения
слР/сек
0,653
0,770
0,188
0,188
0,136
0,656
0,718
0,739
0,175
0,199
0,174
0,130
0,143
0,147
0,152
0,156
0,144
0,0986
0,0970
0,0947
0,689
0,661
0,544
0,597
0,616
0,634
0,552
0,473
0,607
0,705
0,595
0,556
0,413
0,175
0,128
0,146
0,128
0,092
0,138
Эксперимен-
тальные
значения
см} 1 сек
0,641
0,77
0,20
0,20
0,14
0,674
0,743
0,76
0,181
0,22
0,192
0,144
0,158
0,16
0,165
0,163
0,148
0,0960
0,0908
0,095
0,697
0,651
0,550
0,619
0,60
0,646
0,535
0,420
0,625
0,726
0,602
0,537
0,378
0,185
0,139
0,16
0,137
0,096
0,153
Лите-
ратура
[1]
TQ А 1
[84]
[1]
[85]
[84]
[1]
[84]
[1]
[1]
[85]
[84]
[3]
)
[1]
J
[85]
[84]
[3]
[1]
[3]
[1]
[3]
[84]
)
[1]
Замечание. Приведенные в таблице величины а1% и elt/k вычислены по комбинационным правилам
D.8) и D.9). Силовые постоянные для чистых компонент получены из экспериментальных данных о вязкости.
Для вычисления [^)isb была использована формула B.44).
• Силовые постоянные чистого SF, оценены по критическим величинам.
** Силовые постоянные чистого цнс-бутена-2 оценены по параметрам точки кипения и по второму
вириальному коэффициенту.
456
Гл. 8. Явления переноса в разреженных газах
Если учесть, что при расчетах были использованы приближенные значе-
ния силовых постоянных, полученные из комбинационных правил, и что рас-
четы были проведены по формулам первого приближения, то согласие вычис-
ленных величин с экспериментальными значениями коэффициента диффузии
можно считать в общем вполне удовлетворительным.
Для вычисления коэффициента диффузии во втором приближении необ-
ходимо использовать формулу B.45). Для потенциала Леннарда-Джонса для
-0,4
-0,5
V -0,6
*^-0,1
~-0,8
-0,9
-ho
п 1 1 1 1 1 г
Вычисленная классическая кривая
для потенциала F-12)
Леннарда-Джонса
-0,8 -0,4 0 0,4 0,8 1,2 1,6 2,0 2,4 2,8
ЦТ*
Ю/а
Фиг. 125. Коэффициент самодиффузии в приведенных единицах Ю* У
Кривая вычислена по соотношению 2>*р*Т*—*'* = C/8 Уп) (l/fl*1»1)*). Отдельные точки нанесены по
экспериментальным данным в приведенных единицах. Указания на источники экспериментальных
данных см. в табл. 86.
большинства бинарных смесей коэффициент диффузии во втором приближении
отличается от его значения в первом приближении не более, чем на 3%. Во
втором приближении коэффициент диффузии слегка зависит от концентрации
(см. приложение А в конце главы). Однако достаточно точные эксперименталь-
ные данные, позволяющие проверить эту концентрационную зависимость, в
настоящее время отсутствуют.
В первом приближении коэффициент диффузии зависит от взаимодействий
только между неодинаковыми молекулами. Это значит, что измерение коэффи-
циента диффузии как функции температуры является лучшим методом полу-
чения силовых постоянных а12 и е12у характеризующих взаимодействия между
неодинаковыми молекулами. Однако сейчас таких экспериментальных данных,
к сожалению, еще нет. При наличии измерений коэффициента диффузии законы
сил, действующих между неодинаковыми молекулами, могут быть найдены из
экспериментальных данных методом, аналогичным методу, примененному при
определении законов сил, действующих между одинаковыми молекулами, из
измерений вязкости (см. разд. Б).
ПРИМЕР
Задача. Используя формулу первого приближения для ЯI2, вычислить коэффициент
диффузии для смеси Ne — Хе при температуре 320° К и давлении 2 атм.
Решение. Обозначим Ne индексом 1, а Хе индексом 2. По табл. I (стр. 851) найдем
ejk = 35,7 °К, ах = 2,789 A, ejk = 229° К, <т2 = 4,055 А. По формулам D.8) и D.9) найдем
el2/k = 90,42 °К и <г12 = 3,422 А. Для температуры Т = 320°К, Т*% = 3,539. Молекулярные
веса неона и ксенона равны Мх = 20,183, М2 = 131,30. По табл. XII (стр. 868) найдем
ЛС1'1)* C,539) = 0,9096. Далее, пользуясь формулой B.44), для первого приближения Ю12
получаем ЯI2 = 0,119 см2/сек.
§4. Вычисление коэффициентов переноса для потенциала F-72) Леннарда-Джонса 457
Коэффициент самодиффузии JZ) при известных силовых постоянных а и е,
характеризующих взаимодействие между одинаковыми молекулами, может
быть вычислен по формуле B.46). В табл. 86 сравниваются экспериментальные
значения коэффициентов самодиффузии со значениями, вычисленными по фор-
муле B.46). В расчетах были использованы силовые постоянные, найденные
из экспериментальных значений вязкости. Почти во всех случаях совпадение
достаточно хорошее. Последнее служит прекрасной иллюстрацией того, как по
силовым постоянным, определенным из данных об одном свойстве, можно рас-
считать другое свойство. На фиг. 125 наряду с экспериментальными точками
для некоторых веществ приведена вычисленная кривая зависимости lg o2)*p*T*~3/a
от lg T*. Поправочный множитель /§}, входящий в формулу B.47) для опреде-
ления высших приближений JZ), приведен в табл. XIV, стр. 871.
Экспериментальные данные по коэффициентам самодиффузии в зависимо-
сти от температуры, так же как и измерения других коэффициентов переноса,
Таблица 86
Сравнение вычисленных и обследованных коэффициентов самодиффузии
при давлении 1 атпм
Газ
Ne
Аг
Кг
Хе
н2
N2
о2
сн4
со2
НС1
т,°к
353,2
273,2
77,7
353,2
273,2
273,2
77,7
293,0
300,5
273,0
85,0
20,4
353,2
273,2
273,2
77,7
353,2
273,2
273,2
77,7
353,2
273,2
90,2
362,6
312,8
273,2
194,8
295,0
смш1сек*
0,669
0,435
0,0491
0,245
0,154
0,154
0,0133
0,093
0,0571
1,243
0,167
0,0104
0,273
0,174
0,174
0,0161
0,279
0,176
0,176
0,0154
0,302
0,189
0,0215
0,157
0,119
0,0920
0,0474
0,127
Экспериментальные
значения 2>,
см*1сек
0,703 ±0,005
0,452 ±0,003
0,0492 ±0,0004
0,249 ±0,003
@,156 ±0,002
( 0,158 ±0,002
0,0134 ±0,0002
0,093 ±0,0045
0,0576 ±0,0009
1,285 ±0,002
0,172 ±0,008
0,00816±0,0002
0,287 ±0,009
{0,185 ±0,006
@,172 ±0,002
0,0168 ±0,0003
0,301 ±0,004
@,187 ±0,003
@,175 ±0,001
0,0153 ±0,0002
0,318 ±0,006
0,206 ±0,005
0,0266 ±0,0023
0,1644
0,1248
0,0970
0,0516
0,1246
Литература
} [15]
[15]
[15]
[51]
[15]
[52]
[53]
[54]
[15]
[55]
[15]
} [15]
[55]
[15]
[15]
[56]
[57]
1 Вычислено по формуле B.46). Силовые постоянные получены из данных о вязкости.
458
Гл. 8. Явления переноса в разреженных газах
могут быть использованы для определения силовых постоянных. Однако в на-
стоящее время для этого удобнее использовать более простые и до некоторой
степени более точные измерения вязкости.
Е. ТЕРМОДИФФУЗИОННОЕ ОТНОШЕНИЕ
Термодиффузионное отношение в первом приближении может быть вы-
числено по формуле B.50). Для случая термодиффузии тяжелых изотопов эта
формула может быть переписана в виде B.51). Величина к$у определяемая в
общем случае по формуле A.17), а в случае тяжелых изотопов по формуле
B.52), табулирована как функция Т* в табл. XV, стр. 872. Расчеты термодиф-
фузии во втором приближении для потенциала Леннарда-Джонса еще не сде-
ланы. Даже в первом приближении термодиффузионное отношение является
сложной функцией концентраций и температуры и зависит от сил, действую-
щих между двумя одинаковыми и неодинаковыми молекулами. При б с* — 5,
равном нулю, термо диффузионное отношение меняет знак. Из табл. XIII,
стр. 870, видно, что для потенциала F-12) Леннарда-Джонса температурная
инверсия имеет место при Т* = 0,4 и 7* = 0,95. Недавно наблюдалась
инверсия при Г* = 0,95 [58 ] при изучении термодиффузии в смесях Н2 — D2.
Сравнение некоторых экспериментальных значений кт с вычисленными
для потенциала F-12) Леннарда-Джонса дано в табл. 87. Как видно из этого
сравнения, согласие вычисленных и экспериментальных величин не столь
Таблица 87
Сравнение экспериментальных значений кт с вычисленными для
потенциала F-12) Леннарда-Джонса [12]*
Газовая
смесь
He-Ne
He-Ar
Не-На
Процент
легкой
составляющей
53,8
53,8
53,8
20
30
40
50
60
51,2
51,2
51,2
10
20
30
40
50
22,2
81
81
50
60
70
80
90
кт • 10е
эксп.
7,65
7,82
7,83
5,31
7,24
8,64
9,70
10,04
9,10
9,20
9,56
2,50
4,76
6,60
8,10
9,31
2,39
2,03
1,79
4,81
4,42
3,50
2,84
1,32
выч.**
8,15
8,28
8,39
4,53
6,21
7,45
8,15
8,26
9,44
9,57
9,61
2,59
4,94
6,98
8,93
9,79
1,98
2,38
2,31
3,74
3,56
3,08
2,31
1,29
т °к
л t «х
205
330
365
330
330
330
330
330
179
205
365
330
330
330
330
330
46
118
358
330
330
330
330
330
[60]
[61]
[60]
[61]
[62]
[63]
[64]
§4. Вычисление коэффициентов переноса для потенциала F-72) Леннарда-Джонса 459
Продолжение табл. 87.
Газовая
смесь
He-N2 '
Ar-Ne
Аг-Н2
Ar-N2
H2-N2
на-со
Н2-СО2
Процент
составляющей
34,5
53,1
34,5
53,1
51,2
51,2
51,2
51,2
20
30
40
50
60
47
55,6
47
55,6
46
62,5
70
46
62,5
70
29,4
42,0
77,5
29,4
42,0
77,5
24
53
24
53
53
53
эксп.
7,42
9,42
8,31
10,7
3,50
3,80
4,15
4,77
2,33
3,39
4,07
4,57
4,67
5,14
5,73
6,35
7,12
1,01
0,842
0,83
1,82
1,70
1,53
3,95
5,21
4,84
5,48
7,49
6,63
3,76
5,83
4,45
7,38
6,89
8,99
выч.**
7,12
9,23
7,65
9,92
3,47
3,85
4,25
4,90
2,57
3,53
4,24
4,63
4,68
5,89
6,25
7,71
8,17
0,624
0,596
0,536
1,32
1,28
1,16
3,97
5,01
4,44
5,90
7,37
6,36
3,21
5,08
4,81
7,66
8,39
9,60
т °к
i , «\
145
145
261
261
179
205
261
406
324
324
324
324
324
167
167
258
258
157
157
157
252
252
252
143
143
143
264
264
264
142
142
246
246
300
370
Лит^плтл/пя
v aril CJJA1 У Р**
[65]
[60]
L J
[61]
\ [66]
[66]
Looj
i [66J
J
j [67]
• Т - ТТ//(Т/ — Т) In (Г7Г), где Т и Т* — температуры холодного и нагретого баллонов.
•• Величины [kT]i вычислены по формуле B.50).
хорошее, как для других коэффициентов переноса. Это, по-видимому, есть
результат пренебрежения высшими приближениями в кт, а также того, что
наши знания о силах, действующих между неодинаковыми молекулами, недо-
статочно точны. В табл. 88 и 89 аналогичное сравнение делается для изотропной
термодиффузии. Здесь совпадение вычисленных и экспериментальных значений
кт несколько лучше, по-видимому, вследствие того, что обе взаимодиффунди-
рующие компоненты взаимодействуют по одинаковым законам сил.
Данные о термодиффузии вместе с данными об обычной диффузии можно
использовать для определения а12 и е12 в потенциале F-12) Леннарда-Джонса,
который описывает взаимодействие между двумя неодинаковыми молекулами.
Таким методом Сривастава и Мадан [59 ] определили силовые постоянные для
460
Гл. 8. Явления переноса в разреженных газах
Таблица 88
Концентрационная зависимость кт при изотопной
термодиффузии в водорододейтериевых смесях при
Т = 327 °К
Н о/
"г, /в
10
20
30
40
50
60
70
80
90
кт • Ю1
эксп. [64]
1,45
2,65
3,56
4,16
4,32
4,16
3,62
2,81
1,66
выч.
1,48
2,66
3,54
4,12
4,34
4,22
3,76
2,91
1,67
Таблица 89
Температурная зависимость изотопной термодиффузии*
? = <ад/<*г>гв. сф.
Взаимодиффундирую-
щие компоненты
Ne^-Ne22
Ar"-Ar*«
О1вО1в—О1вО18
Легкий
изотоп, %
90
0,307
97,5
эксп.
0,382
0,550
0,713
0,816
0,0673
0,151
0,312
0,466
0,534
0,367
0,475
0,538
•
выч.**
0,480
0,587
0,618
0,625
0,030
0,096
0,359
0,528
0,572
0,371
0,468
0,499
т, °к
129
238
432
712
129
154
300
555
720
284
386
443
Литература
(экспери-
ментальные
данные)
1 [69]
)
\
< [69]
I
\ [70]
* Настоящая таблица дана по Винтеру [12]. Об определении Т см. § 1. См. также табл. 87.
** к т вычислено по данным табл. XV (стр. 872) для различных величин Т* = kTfe.
пяти различных пар молекул1). Их результаты приведены в табл. 90. и 91. Там
же дано сравнение параметров, которые получили Сривастава и Мадан, с сило-
выми постоянными, вычисленными по эмпирическим комбинационным прави-
лам D.8) и D.9). Достаточно хорошее совпадение постоянных, полученных двумя
методами, дополнительно подтверждает справедливость комбинационных пра-
вил.
х) Силовые постоянные, которые определили Сривастава и Мадан, содержат небольшую
ошибку, так как при расчетах авторы пренебрегали вторым приближением термодиффузион-
ного отношения, выражение для которого дано в приложении А в конце главы.
§ 4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-Джонса 461
ПРИМЕР
Задача. В эксперименте по изучению термодиффузии двухбалонным методом газовая
смесь состоит из 35,6% Н2 и 64,4% Ne. Вычислить изменение концентрации, когда темпера-
тура одного баллона достигает 290,4°К, а другого — 90,2°К.
Решение. Изменение концентрации может быть найдено по формуле A.16) или вычи-
слено численным интегрированием соотношения A.15). Проведем расчет обоими методами.
а) В соответствии с формулой A.15) имеем
Ахг - кт (Т) In -у-.
Здесь 7* — средняя температура, которая может быть приближенно найдена из соотношения
В нашей задаче Т= 90,2°К, а? = 290,4°К- Отсюда средняя температура Т = 153°К. Первая
часть расчетов может быть выполнена в последовательности, представленной в табл. 91.
Далее, по формуле B.50) можно вычислить кт(Т). Промежуточные величины имеют значения
ХХ + Yx = 3,030 • Ю-3,
SO-) = -15,94,
SB) = 12,75.
Термодиффузионное отношение равно кт(Т) = — 0,0607. Пользуясь найденным значением
кт(Т), находим разность концентраций Н2 в двух баллонах
Ахг = хг(Т) — хх(Т) = — 0,0710.
б) Для получения более точного результата необходимо выполнить численное интег-
рирование формулы A.15). Сначала вычислим кт в ряде точек температурного интервала
от Т = 90,2°К до Т' = 290,2°К. Для этого можно использовать только что изложенный
метод. В результате получим
т, °к
90,2
115,2
140,2
кТ
-0,0444
-0,0532
-0,0589
165,2
190,2
215,2
кт
-0,0626
-0,0653
-0,0672
т, °к
240,2
265,2
290,2
кТ
-0,0685
-0,0694
-0,0699
На основании этих результатов по правилу Симпсона1) численно вычислим интеграл от
кт/Т в интервале температур от 90,2 до 290,2 °К. После этого найдем Ахх = — 0,0707. Экспе-
риментальное значение [61 ] Ахг = — 0,069.
Таблица 90
Силовые постоянные, определенные из данных о термодиффузии
и из данных об обычной диффузии
Смесь газов
Не-Аг
Н2-Аг
H2-N2
Н2-О2
Н2-СО
эксп.
26,65
56,41
46,53
59,81
52,02
геометри-
ческое
среднее
27,45
64,47
53,86
60,67
57,55
<*12,А
эксп.
3,04
3,24
3,34
3,19
3,36
арифмети-
ческое
среднее
3,06
3,20
3,33
3,21
3,31
О методах численного интегрирования см., например, [68].
462
Гл. 8. Явления переноса в разреженных газах
Таблица 91
Расчеты изменения концентраций при термодиффузии
Молекулярные веса
Мг - 2,016
М% - 20,18
Компоненты
1-
2-
-н,
-Ne
Молярные доли
хг = 0,356
х, - 0,644
i
j
Оф А
еф, °К
1
1
2,968а
33,За
4,595
0,9424е
2
2
2,789*
35,7а
4,286
0,9560
1
2
2,878*
34,5*
4,435
0,9492
i
J
Au
в*
с*
кал/см- сек -°К
1
1
...
2087д
2
2
1,099г
1,093г
0,928*
737 д
1
2
1634е
а По табл. I, стр. 851.
б По формулам D.8) и D.9).
в По табл. XII, стр. 868.
г По табл. XIII, стр. 870.
д По формуле B.31).
е По формуле B.35).
ПРИМЕР
Задача. Вычислить kf для изотопной термодиффузии Агзв и Аг40 при температуре 300°К-
Решение. По табл. I (стр. 851) найдем для аргона г/k = 124°К. Отсюда для Т = 300°К
получим Т* = 300/124 = 2,42. Далее по табл. XV (стр. 872) найдем, что для потенциала
F-12) Леннарда-Джонса kf B,42) равно 0,359.
§ 5. СРАВНЕНИЕ РЕЗУЛЬТАТОВ ВЫЧИСЛЕНИЯ КОЭФФИЦИЕНТОВ
ПЕРЕНОСА ДЛЯ НЕКОТОРЫХ СФЕРИЧЕСКИ СИММЕТРИЧНЫХ
ПОТЕНЦИАЛЬНЫХ ФУНКЦИЙ, ОПИСЫВАЮЩИХ ВЗАИМОДЕЙСТВИЕ
МЕЖДУ НЕПОЛЯРНЫМИ МОЛЕКУЛАМИ
В двух предшествующих параграфах мы рассмотрели расчеты коэффициен-
тов переноса для различных молекулярных моделей. Расчеты для простейших
потенциалов, таких, как твердые упругие сферы, точечный центр отталкивания,
модель Сюзерленда, прямоугольная потенциальная яма, рассмотрены в § 3.
Расчетам для потенциала F-12) Леннарда-Джонса полностью посвящен § 4.
В настоящем параграфе мы сравним результаты расчетов для перечисленных
потенциальных функций (за исключением точечного центра отталкивания) с
экспериментальными данными по коэффициентам вязкости и само диффузии.
Проведенное здесь сравнение аналогично сравнению для второго вириального
коэффициента в гл. 3, § 9.
Экспериментальные1) и теоретические значения ^/Тх/» для неона, аргона,
азота и двуокиси углерода представлены на фиг. 126—129. Теоретические зна-
чения рассчитаны по формулам B.18) и B.19). Справа на фигурах даны потен-
циальные функции, полученные на основе экспериментальных данных о вяз-
кости. Из графиков видно, что температурная зависимость ^/Тх/» наилучшим
образом воспроизводится с помощью потенциала F-12) Леннарда-Джонса.
Поскольку Траутц с сотрудниками получил экспериментальные данные
для значительно более высоких температур, чем Джонстон, то имеется не-
которое различие между силовыми постоянными, найденными из этих двух
серий экспериментов. Тем не менее с помощью обеих потенциальных функций
х) Для Ne и Аг — данные Джонстона и Макклоскей [13 ]; для N2 и СО2 — Джонстона и
Грилли Г14]; для Ne и Аг'— Траутца и Винкеле [34]; для Ne, Ar, N2, СО2 — Траутца и
Цинке [71 ]; для Ne — Траутца и Циммермана [72] ; для N2 — Траутца и Баумана [73]
и Траутца и Мелстера [35]; для СО2 — Траутца и Курца [47] и Василеско [18].
Потенциальные
I функции, получен:
ные из экспери-
ментальных
данных
О 200 400 600 800 1000 1200 1400 1600
Г К
Фиг. 126. Коэффициент вязкости неона, вычисленный
для некоторых моделей молекул.
а — потенциал F-12) Леннарда-Джонса; Ь — модель прямоугольной
потенциальной ямы; с — модель Сюзерленда ; d — модель твердых
сфер; е — данные работы [14]; / — данные работ [34, 35, 72].
О 200 400 600 800 1000 1200 1400 1600
Г/К
Фиг. 127. Коэффициент вязкости аргона, вычисленный
для некоторых моделей молекул.
а — потенциал F-12) Леннарда-Джонса [14] ; Ь — то же [34, 35];
с — модель прямоугольной потенциальной ямы; d — модель
твердых сфер; е — данные работы [14];/—данные работ [34,35];
g — потенциал Леннарда-Джонса.
/50
140
130
120
110
• 90
- 80
W
60
50
40
30
Потенциальные
функции, получен-
ные из экспери- _
ментальных
данных
0 200 400 600 800 1000 1200 1400 1600
Г К
Фиг. 128. Коэффициент вязкости азота, вычисленный
для некоторых моделей молекул.
а — модель прямоугольной потенциальной ямы; Ь — потенциал F-12)
Леннарда-Джонса [13] ; с — потенциал F-12) Леннарда-Джонса [73];
d — модель Сюзерленда ; е — модель твердых сфер ; / — данные
работы [13]; g — данные работ [35, 73].
140
130
120
ПО
100
' 50
.80
10
60
50
40
30
1
-
-
— i X
It
9
1
1
1
" ;
- /
-
l
1
/
7
f
i
i
V
/
эрг
X
1
1
у'
20
0
-20
-40
-60
-80
i
i
—
/ 2
i [
-
-
-
i
д
о
0
.1
!
| !|
i
ii
j,-
1
•i
1
1
1
- b
- С
- d
- e
- f
-9
- h
4\5Г6 7 8
| Q 1 \ 1 —
L/i-
Потенциальные"
функции, получен-
ные из экспери-
ментальных -
данных
i
0 200 400 600
800 WOO 1200 1400 1600
Г к
Фиг. 129. Коэффициент вязкости двуокиси углерода,
вычисленный для некоторых моделей молекул.
а — потенциал F-12) Леннарда-Джонса [13]; Ь — то же [35, 47];
с — модель твердых сфер ; d — модель прямоугольной потенциальной
ямы ; е — модель Сюзерленда; / — данные работ [35, 47 ]; g — данные
работы [13]; Л —данные работы [18 ; п — потенциал F-12) Леннарда-
Джонса.
§ 5. Сравнение результатов вычисления коэффициентов переноса
465
1000
250
температурная зависимость rj/T11* воспроизводится одинаково хорошо во
всем интервале температур, для которого известны экспериментальные данные.
Для потенциала Сюзерленда хоро-
шее совпадение с экспериментом
получается только на сравнительно
малом температурном интервале.
Это ясно из фиг. 126 и 128. По этой
причине модель Сюзерленда не сле-
дует использовать для слишком
далекой экстраполяции. Очень хо-
рошее согласие между наблюден-
ными и вычисленными величинами
получается для прямоугольной по-
тенциальной ямы во всем интервале,
для которого были вычислены интег-
ралы столкновений для этого потен-
циала. Такое совпадение отчасти
является следствием того, чю
прямоугольная потенциальная яма
включает три подбираемых пара-
метра.
На фиг. 130—133 аналогичное
сравнение между эксперименталь-
ными1) и теоретическими результа-
700
650
-« 1 1 L
ЮО 150 Z00 250 300 350~
Т,°К
вычисленный для некоторых моделей молекул,
а—^данные работы [74]; Ь — данные работы [15];
с — потенциал Леннарда-Джонса ; d — модель пря-
моугольной "потенциальной ямы; е — модель Сюзер-
ленда ; / — модель твердых сфер.
тами проводится для JDIT*1*. Тео- ,огч _. АА
ретические данные вычислены по фиг* 130. Коэффициент сомодиффузии неона,
формуле B.46) с помощью силовых
постоянных, полученных из данных
о вязкости для каждой потенциаль-
ной функции2). Это позволяет видеть
результаты использования потен-
циальных функций, полученных на основе измерений одних коэффициентов, для
вычисления других коэффициентов. К сожалению, сейчас отсутствуют данные
400
Фиг. 131. Коэффициент самодиффузии аргона, вычисленный для некоторых моделей молекул.
а — данные работы [51]; Ь — данные работы [15]; с — потенциал Леннарда-Джонса ; d— модель твер-
дых сфер; е — модель прямоугольной потенциальной ямы.
!)См. [\5, 74, 51, 55].
2) Силовые постоянные для потенциала F-12) Леннарда-Джонса взяты из работ [13, 14],
поскольку здесь нас интересует область низких температур (см. замечание к табл. I)i
466
Гл. 8. Явления переноса в разреженных газах
о самодиффузии в таком широком интервале температур и имеющие такую же
точность, как данные о вязкости. Вследствие этого проверка потенциальных
функций не может быть сделана в достаточно широком интервале температур,
как для вязкости. Для Ne, Ar и N2 потенциал F-12) Леннарда-Джонса дает
to и
400
300
250
-
-
(/
1
1 1 1
e
© $
ф ^^^*"~ ~~
© — a
в — b
f
i i i i i i i i
100
150
200
250
300
350
Фиг. 132. Коэффициент самодиффузии азота, вычисленный для некоторых моделей молекул.
а — данные работы [15]; Ь — данные работы [55]; с — потенциал F-12) Леннарда-Джонса ; d — модель
твердых сфер ; е — модель прямоугольной потенциальной ямы ; / — модель Сюзерленда.
250
225
200 -
175
• - a
© - b
в - с
e
_ _ f
: ^
I i i 1
Ф
! f ! 1
150
200
250
300
350
Фиг. 133. Коэффициент самодиффузии двуокиси углерода, вычисленный для некоторых
моделей молекул.
а данные работы [56]; b — данные работы [15]; с — данные работы [55]; d — потенциал F-12) Лен-
нарда-Джонса ; е — модель твердых сфер ; / — модель потенциальной прямоугольной ямы ; g — модель
Сюзерленда.
намного лучшее воспроизведение температурной зависимости JD\T*1*, чем
другие потенциалы. Для СО2 модель Союзерленда и прямоугольная потен-
циальная яма дают лучшее совпадение для ряда экспериментальных точек,
расположенных выше, а потенциал Леннарда-Джонса дает несколько луч-
шее согласие с экспериментальными точками, расположенными ниже.
§ 6. Вычисление коэффициентов переноса чистых газов и газовых смесей 467
§ 6. ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ ПЕРЕНОСА ЧИСТЫХ ГАЗОВ
И ГАЗОВЫХ СМЕСЕЙ, СОДЕРЖАЩИХ ОДНУ ПОЛЯРНУЮ
КОМПОНЕНТУ (ПРИБЛИЖЕННОЕ РАССМОТРЕНИЕ)
Теория явлений переноса для молекул, обладающих внутренними степе-
нями свободы, не была еще применена к полярным газам. В настоящее время
для полярных газов сделаны расчеты только для эффективного сферически
симметричного потенциального поля. Эти расчеты, которым посвящается на-
стоящий параграф, дают превосходный способ описания экспериментальных
данных и вполне приемлемы для экстраполяции в область высоких температур.
При использовании сферически симметричного потенциала для расчетов эффек-
тивных сечений следует иметь в виду, что эти результаты имеют небольшое
значение для интерпретации природы межмолекулярных сил, которые сильно
зависят от углов. Таким образом, обсуждаемые здесь результаты не дают
нам возможности использовать данные о вязкости для предсказания сжи-
маемости газа, тогда как для сферических неполярных молекул это может
быть сделано с помощью обширных вычислений, проделанных для потенциала
F-12) Леннар да-Джонса.
В гл. 3, § 10 показано, как может быть вычислен второй вириальный коэф-
фициент смесей полярных и неполярных газов. В настоящем параграфе мы
покажем, как могут быть проделаны аналогичные вычисления для коэффициен-
тов переноса, если в смеси имеется только одна полярная компонента.
Хотя описываемые .здесь способы вычисления коэффициентов переноса
полярных газов и их смесей не являются строгими, тем не менее они лучше
других известных способов. Изложение метода вычислений коэффициентов с
помощью эффективного сферически симметричного потенциала кажется вполне
разумным, так как этот метод полезен для вычисления свойств промышленных
газов, которые часто включают полярную компоненту.
А. ВЯЗКОСТЬ ЧИСТЫХ ГАЗОВ1)
В гл. 3, § 10 изложен метод расчета вириальных коэффициентов и коэффи-
циента Джоуля—Томсона для полярных молекул. При этом для описания
взаимодействия между двумя полярными молекулами используется потен-
циал Штокмайера [C.33) гл. 1 ]
V (г, 0i, 0„ Л- А) = 4г [(^) - (?)•] - ? g (в19 в2, фъ-фд,
S (ev К Фч — Ф-д = 2 cos 01cos 02 — sin 0isin 02 cos @2 — Фг)-
Зависящий от углов член, пропорциональный г~3, описывает взаимодействие
двух «точечных диполей» (см. гл. 12, § 1). Для столкновений с большой энергией
более важны силы отталкивания, чем силы притяжения. Поэтому в этом случае
в очень хорошем приближении член, зависящий от углов, можно заменить чле-
ном, соответствующим взаимодействию двух точечных диполей, расположен-
ных вдоль одной линии. Это предположение позволяет записать приближенный,
не зависящий от углов потенциал в виде
1) См. [83].
2) Необходимо отметить, что второй вириальный коэффициент для этого потенциала
не может быть вычислен, так как без множителя, зависящего от углов, интеграл для В(Т)
не сходится. Поэтому результаты расчетов, основанные на потенциальной функции F.1),
не могут сравниваться с соответствующими результатами статистической механики равно-
весных систем.
468
Гл. 8. Явления переноса в разреженных газах
Для столкновений с низкой энергией силы притяжения обычно весьма важны,
и потенциальная функция F.1) оказывается плохим приближением к функции,
описывающей реальное взаимодействие.
Коэффициенты переноса, вычисленные на основе не зависящей от углов
потенциальной функции F.1), зависят от параметров а и г и от безразмерной
величины
F.2)
Эта величина (аналогичная величине /*, определенной в связи с использованием
потенциала Штокмайера в гл. 3, § 10) является мерой степени отклонения по-
ведения данного вещества от неполярного.
Для функции потенциальной энергии F.1) были вычислены [83] приве-
денные эффективные сечения столкновений ??<2»2)*, характеризующие вязкость
и теплопроводность. Эффективные сечения ?К2»2>*для этой функции приведены
в табл. XXXIV (стр. 914) в очень широком интервале приведенных температур Т*.
С помощью этой таблицы были определены параметры потенциальной функ-
ции F.1) для двенадцати газов. Вычисленные параметры приводятся в табл. 92.
Таблица 92
Параметры приближенного, не зависящего от углов потенциала
для полярных молекул* [функция F.1)]
Газ
N0
СО
СНС13
H2S
НС1
so2
N0C1
СН3С1
CH3OH
СН3СОСН8
NH3
н2о
//, дабан**
0,0672
0,1172
1,05
0,931
1,034
1,611
1,83
1,861
1,680
2,85
1,437
1,831
112,4
109,9
415,2
221,1
218,0
191,4
205,2
243,6
194,9
158,4
146,8
230,9
•.А
3,508
3,585
5,117
3,733
3,506
4,341
4,332
4,076
4,082
5,485
3,441
2,824
д*
0,003371
0,009830
0,07177
0,2739
0,4123
0,5995
0,7272
0,7606
0,7715
1,1253
1,2499
2,333
•Данные по силовым постоянным для.полярных газов собраны Кригером [83].
•• I дебай = 10—18 эл. стат. ед.
На фиг. 134 показана кривая вязкости неполярных газов в приведенных
единицах. На этом же графике приведены вычисленные кривые для некоторых
полярных газов. С помощью графика можно сделать заключение о величине и
направлении отклонения поведения полярных газов от неполярных. На график
также нанесены экспериментальные точки для четырех газов. Вычисленные
кривые хорошо ложатся на экспериментальные точки.
§ 6. Вычисление коэффициентов переноса чистых газов и газовых смесей
-V
О 0,1 0,2 0,3 0,4 0,5 0,6 0,7
08
Фиг. 134. Сравнение экспериментальных данных с вычисленными на основе потенциальной
функции Кригера F.1).
Отклонения от поведения неполярных газов наибольшие для больших величин <5*, что согласуется с законом
соответственных состояний для полярных молекул.
ПРИМЕР
Задача. Вычислить вязкость NH3 при 800°К.
Решение. На основании табл. 92 силовые постоянные NHa имеют значения
а = 3,441 А,
б* = 1,2499.
Отсюда Г* = 5,4496. По табл. XXXIV (стр. 914) ?B,2)* (Т*, д*) = 0,9388, М = 17,032.
По формуле B.18) найдем
,. 10' = 266,93
Sy =2Жг/см.сек.
Б. КОЭФФИЦИЕНТЫ ВЯЗКОСТИ И ДИФФУЗИИ СМЕСЕЙ,
СОДЕРЖАЩИХ ОДНУ ПОЛЯРНУЮ КОМПОНЕНТУ
В гл. 3, § 10 дан метод расчета вириальных коэффициентов смесей, вклю-
чающих полярные и неполярные газы. Тот же метод может быть применен
к расчету коэффициентов переноса.
Рассмотрим сначала диффузию полярного газа в неполярном. В этом случае
для расчетов необходимо знать только силы, действующие между неодинако-
выми молекулами, т. е. между полярными и неполярными молекулами.
В гл. 13, §5 показано, что эффективная потенциальная энергия взаимодействия
полярной и неполярной молекул имеет тот же вид, который она имеет для двух
неполярных молекул. Следовательно, для описания взаимодействия полярной
и неполярной молекул может быть использован потенциал Леннарда-Джонса.
Можно показать, что силовые постоянные, характеризующие это взаимодей-
470
Гл. 8. Явления переноса в разреженных газах
ствие, могут быть найдены по комбинационным правилам
апР =
в которых
F.3)
F.4)
F.5)
где апу еп — параметры потенциала Леннарда-Джонса для неполярнь!х молекул
(см. табл. I, стр. 851); ор, ер — параметры потенциала Штокмайера для по-
лярных молекул (см. табл. 20); а* — приведенная поляризуемость неполярной
молекулы а* = ап\о\ ; /лр — приведенный дипольный момент полярной моле-
кулы /лр = /Лр1У'еро* ; t*p = /л*р21У8 (приведена в табл. 20).
Используя эти силовые постоянные по формуле B.44) с помощью таблиц
для flO»1)* (табл. XII, стр. 868), можно вычислить первое приближение коэф-
фициента диффузии. В табл. 93 сравниваются вычисленные и эксперименталь-
ные значения коэффициентов диффузии паров воды в некоторых неполярных
газах. Согласие в общем удовлетворительное.
Таблица 93
Коэффициенты диффузии для паров воды и некоторых
неполярных газов
Взаимодействующие
компоненты
Н2О-Н2
Н2О-СО2
Н2О-Не
H2O-N2
т, °с
34,4
55,5
79,2
34,4
55,5
79,2
34,0
55,3
79,3
34,4
55,4
79,0
¦Ф», см*/сек
эксп. [75]
1,02
1,12
1,20
0,202
0,211
0,241
0,90
1,01
1Д2
0,256
0,303
0,359
эксп. [76, 77]
0,91
0,99
1,10
0,174
0,192
0,215
выч.
0,95
1,07
1,21
0,183
0,208
0,239
0,95
1,07
1,21
0,255
0,289
0,329
ПРИМЕР
Задача. Оценить коэффициент диффузии для пары газов NH3 — СН4 при давлении
5 атм и температуре 500°К.
Решение. Силопые постоянные имеют значения :
СН4 : 2L = 136,5°К, ап = 3,882 А
NH3: 4г = 320 Ж, ор = 2,60 А
к
t*P = 1,0,
а„ • 1024 - 2,61 см*,
опр = 3,216 А
= 0,04461
= 229,7°К
(табл. I, стр. 851),
(табл. 20, стр. 179),
[по формуле F.5)],
[по формуле F.3)],
[по формуле F.4)].
Используя эти силовые постоянные, найдем Т%р = кТ/епр = 2,177. Для потенциала
Леннарда-Джонса по табл. XII (стр. 868) найдем ^1»1)* (Т%р) = 1,045. Используя далее
формулу <2.44), получаем [Sbnp] i = 0,134 см2/сек.
Таблица 94
Расчет вязкости тройной смеси, включающей одну полярную компоненту
Компоненты
1 — СН4 (неполярная)
2 — N. (неполярная)
3 — NH3 (полярная)
Молярные доли Молекулярные веса Поляризуемости
хх = 0,50
х2 = 0,35
х8 = 0,15
Mt = 16,04
М2 = 28,02
М3 = 17,03
at = 2,61 А3
а, = 1,76 А8
I
1
1
1
2
2
3
C)
J
1
2
3
2
3
3
C)
ai>, A
3,882а
3,7825
3,216е
3,681а
3,117е
3,44И
2,60д
«<//*» °К
13б,5а
111,7^
229,7е
91,46а
187,5е
14б,8г
320д
Параметры
полярной
молекулы
—
—
-
-
-
t*=\fid
Т* - кТ/ц,
2,784
3,402
1,654
4,155
2,027
2,589
—
1,060е
1,007е
1,263е
0,9622е
1,170е
1,357™
—
2 Mi М^
(Mt+MfK
16,04
20,40
16,52
28,02
21,18
17,03
—
]/2MiMjT
\ (М,+М})и
78,07
88,05
79,23
103,2
89,71
80,45
—
г/см • сек
1305"
1632Л
1619я
2113*
2Ю7Л
1337*
—
А*
—
1,096*
1,096*
—
1,094*
—
—
Mj
ж
—
1,747
1,062
—
0,608
—
—
MtMj
{Mi + MjY
—
0,2315
0,2498
—
0,2351
—
—
+4145«
- 259°
- 121°
+ 1872«
- 61°
+ 1122"
—
а Силовые постоянные для потенциала F-12) Леннарда-Джонса (табл. I, стр. 851).
б Для определения силовых постоянных, характеризующих взаимодействие неполярных молекул, использованы эмпирические комбинационные правила D.8) и D.9).
в Для определения силовых постоянных, характеризующих взаимодействие полярной и неполярной молекул, использованы эмпирические правила F.3) и F.4); для
полярных компонент использован потенциал Штокмайера.
г Силовые постоянные для потенциала Григера [выражение F.1)], табл.92.
д Силовые постоянные для потенциала Штокмайера [выражение C.33) гл. 1], табл. 20.
е По таблице для потенциала F-12) Леннарда-Джонса (табл. XII, стр. 868).
ж По таблице для потенциала Кригера (табл. XXXIV, стр. 914).
з Величина равна М< при i = j.
и Величина равна \ М»Т при i = j.
к По формуле B.18).
л По формуле B.21).
м По таблице для потенциала F-12) Леннарда-Джонса (табл. XIII, стр. 870).
н По формуле B.26).
о По формуле B.27).
472
Гл. 8. Явления переноса в разреженных газах
Этот же метод может быть использован для вычисления вязкости смеси
газов, включающих одну полярную компоненту. Для расчетов могут быть
использованы формулы B.22) или B.25). При этом сначала необходимо вычи-
слить величины {пп]ь {nnp]i> lvP]i и а*р. Первая из этих величин (вязкость
чистой неполярной компоненты) может быть вычислена (с помощью силовых
постоянных, определенных из данных вязкости) по формуле B.18). Вязкость
чистой полярной компоненты [г)р]г может быть вычислена по методу, изло-
женному в разд. А, или может быть использовано ее экспериментальное
значение. Величины [т)пр ]х и а*р, зависящие от взаимодействий между поляр-
ными и неполярными молекулами, могут быть вычислены с помощью таблиц
для потенциала F-12) Леннарда-Джонса и комбинационных правил F.3)—
F.5). Ниже мы приводим численный пример, чтобы пояснить детали расчетов
ПРИМЕР
Задача. Вычислить коэффициент вязкости тройной смеси, состоящей из 50% СН4.
35% N2 и 15% NH8 при давлении 1 атм и температуре Т = 380°К. Использовать формулы
B.25) и B.30) и сравнить результаты расчетов по этим двум формулам.
Решение, а) Коэффициент вязкости трехкомпонентной смеси может быть вычислен
по формуле B.25;. Для этого сначала необходимо найти элементы детерминантов #//, что
можно сделать, рассмотрев предварительно шесть типов взаимодействий, которые имеют
место между тремя составляющими. Силовые постоянные этих взаимодействий, табулиро-
ванные функции, необходимые для расчетов, и некоторые промежуточные результаты расче-
тов даны в табл. 94. После того как мы нашли Н#, можно вычислить детерминанты в фор-
муле B.25).
Окончательно получим
[?7см.]1-1О7= 1672 г/см-сек.
6) Промежуточные результаты табл. 94 могут быть использованы для вычисления
вязкости по формуле B.30), записанной в виде, который использовался в связи с примером
к табл. 81. В данном случае результат расчета по этой формуле с точностью до четырех зна-
чащих цифр совпадает с результатом, полученным по формуле B.25). Обычно оба метода
дают результаты, различающиеся не более чем на 2%.
Вязкость влажного воздуха
Таблица 95
Ю7 г/см -сек
Температура, °К
Процент влажности
0,0*
0,5
5,0
100
150
200
273,16
300
400
500
600
800
1000
1200
1400
1600
2000
3000
5000
702 G03)
1038 A036)
1337 A335)
1724 A717)
1851 A843)
2290 B276)
2678 B659)
3034
3680
4257
4761
5251
5735
6680
8685
12070
701
1035
1335
1722
1849
2289
2678
3034
3680
4260
4765
5253
5739
6685
8689
12090
7Q0
1033
1333
1720
1847
2288
2677
3035
3681
4263
4768
5257
5744
6690
8691
12100
688
1016
1315
1704
1832
2278
2671
3033
3687
4274
4786
5280
5770
6720
8730
12160
• Значения вязкости чистого воздуха, данные в скобках, были вычислены
в предположении, что воздух является многокомпонентной смесью.
Приложение А 473
В табл. 95 приведены результаты расчета (по изложенному выше методу)
вязкости влажного воздуха. В расчетах предполагалось, что воздух является
чистым газом и имеет силовые постоянные, приведенные в табл. I, стр. 851.
Интересно отметить, что в температурном интервале от 100 до 500 °К вязкость
уменьшается с увеличением влажности, тогда как при температуре выше
800 °К имеет место обратная зависимость. В настоящее время еще нет экспери-
ментальных данных, подтверждающих эти результаты.
Приложение А
ФОРМУЛЫ ДЛЯ ВЫСШИХ ПРИБЛИЖЕНИЙ КОЭФФИЦИЕНТОВ
ПЕРЕНОСА
Известны два метода получения высших приближений коэффициентов
переноса. Один из них — метод Чепмена и Каулинга — обсужден в гл. 6, § 3,
другой — новый метод, который развил Кихара1). Метод Чепмена и Каулинга
дает следующие результаты:
1) Коэффициент вязкости чистых газов [см. формулу B.19)]
/C) = 1 J&
"*" Фп Ьгг ~ bU) (Ьгг Ь22 Ъъъ + 2b12 bls Ь2ь - Ь\2 bzz - Ь\, Ьг1 - Ь\ъ Ь22)
где
J25
2) Коэффициент теплопроводности чистых газов [см. формулу B.32)]
Qii(fli2fl23-~ Q22fliaJ /д 2)
(an а22 - а\2) (аи а22 azz + 2а12 als a2Z - en a\z - а22 a\z - а3з «ii) '
х) Метод, который развил Кихара [78 ], впервые был опубликован в Японии (Asakura
Bookstore, Tokyo, 1949) и переведен на английский язык Военно-воздушной базой Райт-Пат-
терсон США.
474
Гл. 8. Явления переноса в разреженных газах
где
bu
77
256
012 =
'13 J
20fi<2'4>*,
где
3) Коэффициент самодиффузии [см. формулу B.47)]
fB)=_L_
Fс*-5J
(А.З)
E5-
функции а*, в*, с* определены соотношениями B.15) —B.17).
4) Коэффициент диффузии бинарных смесей [см. формулу B.45)]
где
60 (Хя+Уя)
¦IV;
функция с* определена соотношением B.17); Хк и Уя определены формулой
B.36); W выражается следующим образом :
15
"Г 8А* МгМ2
где х, и 7W, — молярные доли и молекулярные веса соответственно; функция
а* определяется по B.15); [А,^ — теплопроводность, выражающаяся по
B.31); [Я12]х — вспомогательная величина, определяемая B.35).
5) Термодиффузионное отношение бинарной смеси [10]
(А.5)
где Soo, S0] и 50_х — детерминанты:
5-2-2
5-1-2
51-2
52-2
5-1-
1-1
5-21
5-п
5ц
5—22
5-]2
52-
2-i
5-2-2
5-1-2
«1-2
52-2
5-2-2
5-1-2
5l-2
52-2
Приложение
5_2-i
5-i-i
«i-i
52-i
5-20
5-ю
5io
«20
A
5-20
5-ю
5io
«20
5-21
«-11
Sll
«21
5—22
5—12
Sl2
«22
5—22
«-12
512
522
475
Элементы для этих детерминантов выражаются следующим образом
«00 = 8-77^; "
(мг
5
4
35
12
2
1
J '
01
20
М2
02'
7Afj Af,
Ш2
«1-1 — «—и —
«1-2 = «-21 = ~~
Ш" ["Ж D0 м*+т Ml Ml + 35 Mf) Q<&»* -
i2 >* + -| Mf A08 Mf + 133 Ml) Q{
- 105 M! Q&V* + 45 M| ^(Л'5>* + -I- M, M2 DM? + 7M1) fi
- 56 MxMli2<22'3)* + 40M1M|.G&4>* + 12 M\ M
476 Гл. 8. Явления переноса в разреженных газах
8(М,М2)^ Г 8505^,^ 2499
^ )* + 45 Q<&V* - -^-
56?<|3>* -
Остальные элементы могут быть получены, если учесть, что stJ = s_,_y, когда
индексы, отмечающие одну компоненту, заменены индексом второй компоненты,
и наоборот (это правило не может быть использовано, если один из индексов i
или / равен нулю). Величины ст, появляющиеся в выражениях для элементов
детерминантов, представляют собой характеристические длины, которые
используются при определении приведенных интегралов столкновений fl^»*)*.
Кихара [78] получил несколько более простые выражения для высших
приближений коэффициентов переноса, причем сделал это следующим способом.
Согласно Чепмену и Каулингу [1, стр. 149], коэффициенты переноса выражаются
в виде отношений детерминантов бесконечного порядка. При этом различные
порядки приближений соответствуют замене детерминантов бесконечного по-
рядка детерминантами соответствующих конечных порядков. Кихара заметил,
что недиагональные элементы детерминантов малы по сравнению с диагональ-
ными элементами, и использовал это. Его метод состоит в разложении беско-
нечных детерминантов по степеням недиагональных элементов. С помощью
разложения с точностью до второй степени недиагональных элементов полу-
чаются значительно более простые результаты, чем результаты второго при-
ближения для вязкости, теплопроводности и диффузии и первого приближе-
ния для термодиффузии [1]. Метод Кихара дает точные результаты как в
случае газа Лорентца (Мг <^ М2 и хг <4 х2), так и в случае газа Максвелла
(ср = dr~4). Кихара получил следующие результаты :
для чистых газов
для бинарных смесей
где
2М* У/а ( ^i2>2)* 1 ( а1 \ _ А М1^2 А* _ ЗМ2(М2-Мг) .
г + М2) [ ^Ь1>* J 1 о\2 ) 5 (Мг + M2f (Мг + М2J '
9 A (-±
°А 5 I M
S2 = S± при условии, что индекс 1, обозначающий первую компоненту, заменен
индексом 2, обозначающим вторую компоненту, и наоборот
S И
Литература 477
Q2 = Q± при условии, что индекс 1, обозначающий первую компоненту, заменен
индексом 2, обозначающим вторую компоненту, и наоборот
32М1М2 .
А "Г"
^ (М
8 (M
5 (
Вычисление ^ихаРа> много легче, чем вычисление [кТ]2, так как /стКиха:>
включает меньше интегралов. Для бинарных смесей тяжелых изотопов вместо
формулы B.52) Кихара получил
Щ6^^) (ДЛ0)
Задачи
1. Показать, что для потенциала F-12) Леннарда-Джонса произведение j
является универсальной функцией приведенной температуры Т* = кТ/е. (В — второй ви-
риальный коэффициент ; rj — коэффициент вязкости ; Т — абсолютная температура.)
2. Используя табулированные функции для потенциала F-12) Леннарда-Джонса,
вычислить :
а) коэффициент вязкости SFe при 500°К и давлении 2 атм;
б) коэффициент вязкости смеси С12 и Вг2 в соотношении 1 : 1 при 450°К и давлении
1 атм;
в) коэффициент диффузии смеси Н2О — СН4 при давлении 5 атм и 500°К ;
г) безразмерное отношение q?>/t] для Ne при 250°К ;
д) величину кр для термодиффузии изотопов С12 при комнатной температуре и давле -
нии 1 атму например для С135С135 в С137С187.
3. Сделать оценки:
а) коэффициента теплопроводности СН3СОСН3 при 450°К ;
б) термодиффузионного отношения смеси из 85% СН4 и 15% NH3 при 500бК ;
в) коэффициента вязкости смеси из 80% N2, 15% О2 и 5% СН3ОН при 800°К и 5 атм.
4. Получить силовые постоянные потенциала F-12) Леннарда-Джонса для N2 и Аг
из экспериментальных данных о коэффициентах самодиффузии, приведенных в табл. 86.
Сравнить результаты с параметрами, полученными из анализа вязкости и данных о сжимае-
мости.
5. Использовать результаты этой главы для анализа относительной важности частей
межмолекулярного потенциала, характеризующих притяжение и отталкивание, определяю-
щих коэффициент вязкости чистого газа. Вычислить/2B,2)• для Рлд = 4е ["(ст/гI2—(^А*)в]>
а такжедля99=4К<т/гI2 при Т*=кТ/е=\, 10, 100, 400. Показать, что для Т* выше 10 вклад
в г}(Т) или?B>2)* за счет притяжения меньше 10%. Использовать результаты гл. 3 для
аналогичного анализа по второму вириальному коэффициенту, для которого силы притя-
жения при Т* = 10 играют чрезвычайно важную роль.
ЛИТЕРАТУРА
1. Chapman S., Cowling T. A., The Mathematical Theory of Non-uniform Gases,
Cambridge, 1939 (см. перевод: Чепмен С, Каулинг Т., Математическая теория
неоднородный газов, ИЛ, I960).
2. Herzfeld К. F., Freie Wegla"nge und Transporterscheinungen in Gasen, Hand- und
Jahrbuch der Chemischen Physik, Band 3, Teil 2, Abschnitt IV, Leipzig, 1939.
3. В о у d С. A., Stein N., S t e i n g r i m s s о n V., Rumpel W. F., Journ. Chem.
Phys., 19, 548 A951)
4. Brown H., Phys. Rev., 58, 661 A940).
5. С u r t i s s C- F., H i r s с h f e 1 d e r J. O., Journ. Chem. Phys., 17, 550 A949).
6. Bud den berg J. W., Wilke С R., Ind. Eng. Chem., 41, 1345 A949).
7. Sutherland W., Phil. Mag., 40, 421 A895).
8. Thiesen M., Verhandl. deutsch. phys. Gesellschaft, 4, 348 A902).
9. Schudel W., Schweiz. Ver. Gas- und Wasserfach. Monats-Bull., 22, 112, 131 A942).
478 Гл. 8. Явления переноса в разреженных газах
10. Mason E. A., Journ. Chem. Phys., 22 A954).
11. Jones Clark R., Phys. Rev., 58, 111 A940).
12. Winter E. R. S., Trans. Farad. Soc, 46, 81 A950).
13. J о h n s t о n H. L., M с С 1 о s к е у Е. S., Journ. Chem., 44, 1038 A940).
14. Johnston H. L., Grill у Е. R., Journ. Chem. Phys., 46, 948 A942).
15. Win n E. В., Phys. Rev., 80, 1024 A950).
16. Kihara Т., Rev. Mod. Phys., 24, 45 A952).
17. Kotani M., Proc. Phys.-Math. Soc, Japan, 24, 76 A942).
18. V a s i 1 e s с о V., Ann. de phys. A1), 20, 292 A945).
19. Ho lie ran E. M., Hulburt H., Journ. Chem. Phys., 19, 232 A951).
20. H i r s с h f e 1 d e r J. О., М с С 1 u r e F. Т., Weeks I. F., Journ. Chem. Phys., 10, 201
A942),
21. Ha see H. R., Cook W. R., Phil. Mag., 3, 977 A927).
22. Hasse H. R., Cook W. R., Proc. Roy. Soc, 125, 196 A929).
23. Jones R. C, Phys. Rev., 59, 1019 A941).
24. Kihara Т., Kotani M., Proc Phys.-Math. Soc, Japan, 25, 602 A943).
25. de Boer J., van KranendonkJ., Physica, 14, 442 A948).
26. Hirschf elder J. O., Bird R. В., Spotz E. L., Journ. Chem. Phys., 16, 968
A948).
27. Hirschf elder J. O., Bird R. В., Spotz E. L., Journ. Chem. Phys., 17, 149
A950).
28. Kir к wood J. G., Journ. Chem. Phys., 15, 72 A947).
29. Landolt-Bornstein, Physikalisch-Chemische Tabellen, Berlin (см. перевод:
Ландольт-Бернштейн, Физико-химические таблицы, М.—Л., 1939).
30. Trautz M., Heberling R., Ann. d. Phys. E), 20, 118 A934).
31. Epstein L., Marion M., Journ. Phys. and Collod. Chem., 57, 336 A953).
32. Braune H., Linke R., Zs. phys. Chem., A148, 195 A930).
33. Hirschf elder J. O., Bird R. В., Spotz E. L., Chem. Rev., 44, 205 A949).
34. T r a u t z M., В i n k e 1 e H. E., Ann. d. Phys. E), 5, 561 A930).
35. Trautz M., Melster A., Ann. d. Phys. E), 7, 409 A930).
36. TrautzM., К i p p h a n K. F., Ann. d. Phys., 2, 746 A929).
37. H e r n i n g F., Z i p p e r e r L., Gas- und Wasserfach. Monats.-Bull., 79, 49, 69 A936).
38. Schmid, Gas- und Wasserfach, Monats.-Bull., 85, 92 A942).
39. Kannuluik W. G., Carman E. H., Proc. Phys. Soc, 65B, 701 A952).
40. Johnston H. L., Grilly E. R., Journ. Chem. Phys., 14, 233 A946).
41. Weber S., Ann. d. Phys. D), 54, 325 A917).
42. Dickins B. G., Proc. Roy. Soc, A143, 517 A934).
43. Ibbs T. L., Hirst A. A., Proc Roy. Soc, A123, 134 A929).
44. National Bureau of Standards Tables, American Petroleum Institute Research Project
No. 44, June 30, 1949.
45. Weber S., Ann. d. Phys. D), 54, 437 A917).
46. Wachsmith J., Phys. Zs., 9, 235 A908).
47. Trautz M., Kurz F., Ann. d. Phys. E), 9, 981 A931).
48. Van Itterbeek A., van Paemel O., van Lierde J., Physica, 13, 88 A947).
49. Васильева A., Phys. Zs., 5, 737 A904).
50. J о s t W., Diffusion in Solids, Liquids and Gases, New York, 1952.
51. Hutch in son F., Journ. Chem. Phys., 17, 1081 A949).
52. Groth W., Harteck P., Zs. Elektrochem., 47, 167 A941).
53. Visner S., Rep. K-688 (May 1951) of K-25 Plant, Carbide and Carbon Chemicals Co.
54. Harteck P., Schmidt H. W., Zs. Phys. Chem., 21B, 447 A933).
55. Winter E. R. S., Trans. Farad. Soc, 47, 342 A951).
56. Amdur I., Irvine J. W., Jr., Mason E. A., Ross J., Journ. Chem. Phys.,
20, 436 A952).
57. Braune H., Zehle F., Zs. Phys. Chem., 49B, 247 A941).
58. d e T г о у e r A., van Itterbeek A., R i e t v e 1 d A. O., Physica, 17, 938 A951).
59. Srivastava B. N., Madan M. P., Proc Phys. Soc, 66, 27B A953).
60. Grew К. Е., Proc Roy. Soc, A189, 402 A947).
Литература 479
61. Atkins В. Е., Ва stick R. E., Ibbs Т. L., Proc. Roy. Soc, A172, 142 A939).
62. van Itterbeek A., van Paemel O., van Lierde J., Physica, 13, 231 A947).
63. Murphey B. F., Phys. Rev., 72, 834 A947).
64. Heath H. R., Ibbs T. L., Wild N. E., Proc. Roy. Soc, A178, 380 A941).
65. Ibbs T. L., Grew К. Е., Proc. Roy. Soc, 43, 142 A931).
66. Ibbs T. L., Grew К. Е., Hirst A. A., Proc. Roy. Soc, 41, 456 A929).
67. В a stick R. E., Heath H. R., Ibbs T. L., Proc Roy. Soc, A173, 543 A939).
68. M i 1 n e W. E., Numerical Calculus, Princeton, 1949.
69. Stier L. G., Phys. Rev., 62, 548 A942).
70. Whalley E., Winter E. R. S., Trans. Farad. Soc, 45, 1091 A949).
71. Trautz M., Zink R., Ann. d. Phys. E), 7, 427 A930).
72. T r a u t z M., Zimmerman H., Ann. d. Phys. E), 22, 189 A935).
73. Trautz M., Baumann P. В., Ann. d. Phys. E), 2, 733 A929).
74. Groth W., Sussner E., Zs. Phys. Chem., 193, 296 A944).
75. Schwertz F. A., Brow J. E., Journ. Chem. Phys., 19, 640 A951).
76. Winkelman A., Wied. Ann., 22, 152 A884).
77. W i n к e 1 m a n A., Wied. Ann., 36, 93 A889).
78. К i ha г а Т., Imperfect Gases, Tokyo, 1949.
79. d e Boer J., Physica, 10, 348 A943).
80. С о h e n E. G. D., Physica, 17, 993 A951).
81. v a n Itterbeek А., С1 a e s A., Physica, 5, 938 A938).
82. v a n Itterbeek A. Keesom W. H., Physica, 5, 257 A938).
83. К r i e g e г F. J., The Viscosity of Polar Gases, Proj. RAND Report, RM-646, July 1, 1951.
84.* Board man L. E., Wild N. E., Proc. Roy. Soc, A162, 511 A937).
85.* Waldman L., Naturwissenschaften, 32, 223 A944).
86.* Cohen R. S., Spitzer L., Jr., Routly P. M., Phys. Rev., 80, 230 A956)
[см. перевод: Коэн Р., Спитцер Л., Роутли П., Проблемы современной
физики, ИЛ, № 2 A956)].
87.* S p i t z e r L., Jr., H a r m R., Phys. Rev., 89, 977 A953) [см. перевод: С п и т ц е р Л.,
Хэрм Р., Проблемы современной физики, ИЛ, № 2A956)].
88.* Cowling Т. G., Proc Roy. Soc, A183, No 995 A945) (см. перевод : К а у л и н г Т.,
Современные проблемы астрофизики и физики солнца, ИЛ, 1951).
89.* Landshof i R., Phys. Rev., 82, 442 A951).
* Звездочкой отмечена литература, добавленная при переводе. — Прим. ред.
ГЛАВА 9
ЯВЛЕНИЯ ПЕРЕНОСА В ПЛОТНЫХ ГАЗАХ
И ЖИДКОСТЯХ
В двух предыдущих главах рассмотрена кинетическая теория разрежен-
ных газов и использование этой теории для вычисления коэффициентов пере-
носа. Упомянутая теория основывается на интегро-дифференциальном урав-
нении Больцмана для функции распределения /(г, v, t). В этом уравнении учи-
тываются только парные столкновения молекул и предполагается, что раз-
меры молекул малы по сравнению со средним расстоянием между молекулами.
Поэтому результаты этой теории не приложимы к плотным газам и жидкостям.
В настоящей главе мы рассмотрим четыре различных подхода к изучению явле-
ний переноса в плотных средах и изложим их в порядке возрастающей мате-
матической сложности и строгости.
В § 1 рассматривается возможность установления связи между зависимо-
стью коэффициентов переноса от температуры и зависимостью от давления с
помощью закона соответственных состояний в его различных формах. К на-
стоящему времени на основе этого закона составлены обобщенные диаграммы
для коэффициента вязкости, по которым можно получить наилучшие оценки
вязкости плотных газов. К сожалению, недостаток экспериментальных данных
не позволяет составить диаграммы соответственных состояний для других
коэффициентов переноса.
В § 2 обсуждается приложение теории абсолютных скоростей реакций
Эйринга к анализу различных явлений переноса1). Здесь энергия и энтропия
активации, появляющиеся в общем определении различных коэффициентов
переноса, вычисляются путем сравнения теоретических результатов с экспери-
ментальными данными и затем интерпретируются с помощью простых моделей
жидкости или плотного газа. Теория Эйринга особенно полезна в приложениях
к пластическому течению. Она, по-видимому, хороша для описания явлений
переноса в жидкостях, но не подходит для газовых систем.
Изложению теории плотных газов Энскога посвящен § 3. Эта теория,
предшествующая работе Эйринга, является развитием теории газов малой
плотности, изложенной в гл. 7, и создана только для газов, состоящих из твер-
дых сфер. Последнее ограничение сделано потому, что при использовании его
нет необходимости рассматривать многократные столкновения. При развитии
кинетической теории разреженных газов с целью применения ее к плотным
газам в нее вносятся поправки, учитывающие тот факт, что в плотных газах и
жидкостях молекулярные диаметры не малы по сравнению со средними меж-
молекулярными расстояниями. Вследствие упомянутого факта необходимо
принимать во внимание перенос импульса и кинетической энергии, возникаю-
щий в результате мгновенного обмена этими величинами между центрами двух
твердых сферических молекул при столкновении. «Перенос столкновениями»
является главным механизмом переноса при высоких плотностях2).
х) Основы теории абсолютных скоростей реакций изложены в приложении в конце
главы.
2) Включение в теорию плотных газов и жидкостей механизма переноса импульса столк-
новениями позволяет объяснить один момент из теории разреженных газов, который на пер-
вый взгляд кажется парадоксальным. В статистической механике равновесных систем учет
парных столкновений приводит к тому, что в уравнении состояния идеального газа появля-
ется поправочный член, выражающийся через второй вириальный коэффициент. В статисти-
ческой механике неравновесных систем (кинетической теории) в интегро-дифференциальном
уравнении Больцмана также учитываются парные столкновения. Однако в этой теории полу-
§ 7. Закон соответственных состояний 481
Хотя теория Энскога была создана для твердых сфер, тем не менее ее
результаты могут быть использованы для реальных газов. Коэффициенты пере-
носа плотных газов могут быть оценены в последнем случае на основе сведений
о коэффициентах переноса разреженных газов, данных о сжимаемости плотных
газов и по эффективным диаметрам столкновений для молекул. Если эффек-
тивный диаметр столкновений подбирается таким образом, что вычисленные
значения коэффициентов переноса при высоких плотностях согласуются с
экспериментальными, то получается превосходное совпадение с эксперимен-
том в широком интервале плотностей.
В § 4 кратко излагается строгая кинетическая теория плотных газов, с
которой связаны имена Кирквуда, Борна и Грина. Показано, что методами
статистической механики, исходя из уравнения Лиувилля, можно получить
точное выражение для векторов потоков через неравновесную радиальную функ-
цию распределения. В таком подходе именно отыскание неравновесной ради-
альной функции распределения является основной проблемой. Для решения
этой задачи были предложены несколько приближенных методов, однако к
настоящему времени вполне удовлетворительный метод еще не создан. Подход
к проблеме, основывающийся на статистической механике, в конечном счете
должен приводить к кинетической теории плотных газов, по своей точности
сравнимой с кинетической теорией разреженных газов, обсуждаемой в гл. 7.
Однако в настоящее время теория еще не столь совершенна и имеются только
весьма скромные результаты.
§ 1. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ
Исследованию зависимости коэффициентов переноса от давления до сих
пор экспериментаторы уделяли очень мало внимания , и поэтому имеются
лишь весьма скромные экспериментальные результаты [1 ]. Эти сведения
могут быть объединены и сопоставлены с помощью закона соответствен-
ных состояний. Приложение закона соответственных состояний к уравнению
состояния плотных газов и жидкостей обсуждено в гл. 4, § 1. Там показано,
что закон соответственных состояний дает практический метод использования
измеренных свойств одного вещества для предсказания свойств другого, для
которого нет экспериментальных данных и не существует удовлетворительных
теоретических методов определения его свойств. Аналогичный метод может
быть использован в теории явлений переноса.
А. ОБЗОР ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ О КОЭФФИЦИЕНТАХ
ПЕРЕНОСА ПРИ ВЫСОКИХ ПЛОТНОСТЯХ
Этот параграф мы начнем с подведения итогов по результатам эксперимен-
тальных исследований явлений переноса при высоких плотностях. Это об-
суждение даст грубое количественное представление о влиянии давления
на коэффициенты переноса и укажет на поведение этих коэффициентов при
сжатии газа до достижения им жидкого состояния.
чается тензор давлений, который в случае равновесия сводится к уравнению состояния
идеального газа. Очевидное расхождение между двумя теориями, основывающимися на учете
парных столкновений, является следствием того факта, что в кинетической теории разре-
женных газов парные столкновения в действительности рассматриваются недостаточно
полно. Когда к уравнению Больцмана добавляется член, учитывающий перенос столкно-
вениями, последующий анализ совершенно естественно приводит к тензору давлений, кото-
рый включает второй вириальный коэффициент.
[Кинетическое уравнение, получаемое в теории Н. Н. Боголюбова (см. «Проблемы дина-
мической теории в статистической физике», М.—Л., 1946), отличается от обычного уравнения
Больцмана и приводит к поправкам в уравнении состояния и в выражениях коэффициентов
переноса. Это связано с более строгим и полным учетом парных столкновений. — Прим. ред. ]
482
Гл. 9. Явления переноса в плотных газах и жидкостях
1. Вязкость1). На фиг. 135 даны экспериментальные изотермы для коэф-
фициента вязкости СО2 как функции давления, полученные Филлипсом [11].
Для этого газа были проведены измерения, простирающиеся в критическую
область, вследствие чего на их основе можно получить хорошее представление
о поведении вязкости как в газовой, так и в жидкой фазах. Ниже критической
температуры в изотермах вязкости имеются разрывы. По аналогии с изотер-
мами р — V можно начертить S-образные кривые, чтобы связать изотермы
в точках разрыва. На фиг. 135 эти участки изотерм проведены пунктиром.
20
40
_J L.
0,5
60 р,атм*°
¦ v I i
1,0
100
-J I L.
120
/.5
Рг=Р/Рс
Фиг. 135. Вязкость СО2 как функция давления при различных температурах.
Участок А А кривой может быть интерпретирован как вязкость перегретой
жидкости, участок ВВ' — как вязкость переохлажденного пара. Отметим,
что при переходе системы из газовой фазы в жидкую наблюдается пересечение
изотерм, так как с увеличением температуры вязкость жидкости уменьшается,
а вязкость газа увеличивается. Пересечение в действительности происходит до
того, как газ сжижается.
2. Теплопроводность. Измерений теплопроводности газов при высоких
давлениях сделано недостаточно2). На фиг. 136 представлен коэффициент теп-
лопроводности Аг [15] вблизи критической области. Некоторые эксперимен-
тальные измерения были сделаны в этой же области для N2 и СО2 [13, 14]. Для
давлений от 50 до 760 мм рт. ст. измерения теплопроводности были сделаны
в воздухе, двуокиси углерода и для некоторых органических паров [16]. В этой
области давлений обычные молекулы оказывают малое влияние на теплопровод-
ность газов. Однако для таких веществ, которые стремятся образовать связанные
водородом димеры, обнаружено, что теплопроводность быстро увеличивается с
давлением в этой области.
г) Широкий обзор измерений вязкости при высоких давлениях, проведенных до 1944 г.,
дали Каминг, Мейланд и Игли [2]. Полный обзор данных по коэффициенту объемной (или
второй) вязкости газов и жидкостей дали Карим и Розенхед [3 ]. Экспериментальные изме-
рения этого коэффициента выполнил Либерманн [4—6].
Экспериментальные данные о вязкости можно найти у Ньюитта [7]. О вязкости
см. также в работе Хоутена и Ватсона [8 ]. В недавно вышедшей книге Максвелла [9 ] приве-
дены карты соответственных состояний, аналогичные картам Хоугена и Ватсона для коэф-
фициента вязкости, фактора сжимаемости и всех других термодинамических свойств для
углеводородов с малым и большим молекулярным весом.
Измерение вязкости N2 с большой точностью провели Михельс и Гибсон [10].
*) Сводка об экспериментальных измерениях теплопроводности дана Франком [12].
§ 7. Закон соответственных состояний
483
II
з,
2,0
1,0
0
3
*20
, кал
0
80
у
' р-24атл!
-
i I
10L
i
0,6
^\
Df=l
)
o;i
N
р=36атм pr=jy/
р=48атм
р=42атм рг
р=24атм pr=j-
i I
120
1 L_
0,8 0,9
I—
4
Pr=1
_l
140
Г/К
•96атм
о \
/
_J
1,0
1 г
Pr=2
\
\
=====
50
V
1—
1—
\
¦^^D-64 о.
180
1,2
r
—' 1
-
-
-
«Д.-Г
-
-
200
13
Tr=T/Tc
Фиг. 136. Теплопроводность аргона как функция температуры при различных давлениях.
3. Диффузия. Согласно кине-
тической теории разреженных газов,
коэффициент диффузии обратно про-
порционален полной плотности сме-
си. Поэтому при определении влия-
ния давления на коэффициент диффу-
зии следует изучать зависимость от
давления произведения^^. Одна-
ко, за исключением недавних изме-
рений1) коэффициентов самодиффу-
зии СО2, CS2, CH4 и коэффициентов
обычной диффузии некоторых жид-
ких смесей, практически данных о
зависимости qJD12 от давления, к
сожалению, не имеется. Для само-
диффузии СО2 при 42 °С произведе-
ние qJD уменьшается до половины
того значения, которое оно имеет
при низких давлениях и плотности
около ^ = 0,7 г/сл*3.
4. Термодиффузия. Вследствие
особого значения термодиффузии
как процесса, приводящего к раз-
делению, за последнее время боль-
шое внимание уделяется его экспе-
риментальному изучению2).
р, атм
Фиг. 137. Зависимость от давления величины
а = кт/х^у характеризующей термодиф-
фузию, при Т = 427° К.
Средние молярные доли компонент имеют значения
около 0,5.
г) Самодиффузию в СО2 изучали и Теммерхауз и Дрикеймер[17, 18], Робб и Дрикеймер
[ 19]; самодиффузию в CS2 — Колер и Дрикеймер [20]; в СН4 — Джефрис и Дрикеймер
(работа опубликована после 1954 г.); диффузию в жидкой фазе в J2 изучали Кова и Дрикей-
мер (работа опубликована после 1954 г.); в SnJ4 — СС14 — Доане и Дрикеймер (работа опуб-
ликована после 1954 г.); в CS2 — органические смеси — Колер и Дрикеймер [21]; в воде и
сульфатных растворах — Коддебак, Колер и Дрикеймер [22]; в воде и спиртовых рас-
творах — Коддебак и Дрикеймер [23 ].
2) Беккер [24,25] изучал термодиффузию в системах СО2—Н2, Н2—N2, СО2—N2,CO2—СН4,
СО2—Аг и N2—СН4 двухбалонным методом. Дрикеймер с сотрудниками для изучения влия-
ния давления на термодиффузию использовал разделительную колонну (см. СО2—С3На [26],
Аг—Ne [27], Хе—СН4 [28], Хе—С2Н2 [29, 30]).
484 Гл. Р. Явления переноса в плотных газах и жидкостях
Для иллюстрации влияния давления на термодиффузионное отношение на
фиг. 137 представлены экспериментальные результаты для некоторых бинарных
смесей газов [24]. Очевидно, что для некоторых газовых систем значительно
большее разделение может быть достигнуто при высоких давлениях.
Б. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ ДЛЯ СФЕРИЧЕСКИХ
НЕПОЛЯРНЫХ МОЛЕКУЛ
Закон соответственных состояний может быть выведен с помощью анализа
размерностей. Рассмотрим газ, состоящий из сферических неполярных молекул.
Предположим, что межмолекулярная потенциальная энергия может быть оха-
рактеризована двумя параметрами; характеристической длиной а и харак-
теристической энергией е. Это соответствует действительности, например
если силы, действующие между молекулами, являются парными силами и если
потенциал взаимодействия является потенциалом типа Леннарда-Джонса.
Свойства молекул вещества полностью определяются массой т и двумя постоян-
ными а и е. Макросвойства, такие, как коэффициенты переноса, являются функ-
циями шести величин : г>, Т, т, к, о и е. Как и в случае уравнения состояния,
температуру и объем можно выразить через безразмерные комбинации упомя-
нутых величин Т* и г>*
T*=*L, V*=JL = 4-- A.1)
Аналогично, сами коэффициенты переноса могут быть записаны через
безразмерные комбинации JZ)*, rj* и А*
т * п* ^ Ло* т (] 2)
В том, что эти величины действительно безразмерны, можно убедиться по фор-
мулам B.9)—B.11) гл. 8 для коэффициентов переноса в газах малой плотности.
Безразмерные комбинации „2)*, rj* и А* являются функциями 7*, г?*, /с, ту а и г.
Первые две переменные безразмерны, тогда как остальные четыре имеют опре-
деленные размерности. Очевидно, что любая безразмерная функция может
быть записана через безразмерные аргументы. Поэтому, поскольку безраз-
мерных комбинаций из /с, т, а и е не существует, следует, что JZ)*, rj* и А* от
этих параметров не зависят1). Таким образом,
?)* = ?)* (г,*, Т*), rj* = f* (*>*, Г*), А* = А* (*;*, Г*). A.3)
Функциональный вид этих приведенных коэффициентов переноса зависит
только от вида предполагаемой функции межмолекулярной потенциальной
энергии. Из формул, приведенных в гл. 8, § 2, следует, что в предельном случае
малых плотностей приведенные коэффициенты переноса имеют вид
?D* (г?*, Т*) = — ~——— v* у
Г7> О'4)
A* (v*, Т*) = 75
64 fit ?B>2)*(T*)
При конечных давлениях функциональный вид более сложен и еще не
получен теоретически. Ввиду этого закон соответственных состояний может
иметь большое значение для предсказания коэффициентов переноса при
высоких плотностях.
х) О методе размерностей см. [31] или [32]. [См. также Л. И. Седов, Методы по-
добия и размерности в механике, М.—Л., 1951. — Прим. ред.]
§ 7. Закон соответственных состояний 485
В. ПРИЛОЖЕНИЯ ЗАКОНА СООТВЕТСТВЕНЫХ СОСТОЯНИЙ
Из всех коэффициентов переноса только коэффициент вязкости широко
изучен с помощью закона соответственных состояний. Этот закон может быть
применен различными способами. Эти способы мы коротко описываем ниже.
1. Приведение с помощью молекулярных параметров. Соотношение A.3)
можно непосредственно применить для изучения коэффициентов переноса
при высоких давлениях. Однако этот метод не используется. Он менее целе-
сообразен, чем остальные, так как может быть успешно применен только к тем
газам, которые удовлетворяют основному предположению метода, т. е. пред-
положению о том, что молекулы являются сферическими и неполярными.
С помощью соотношений A.3) могут быть изучены благородные газы, а также
другие группы газов, молекулы которых химически подобны.
2. Приведение с помощью критических постоянных рс и Тс. Согласно
закону соответственных состояний, примененному к уравнению состояния,
величина е ?авна универсальной постоянной, умноженной на ТСУ а а пропор-
циональна К1/3. Следовательно, коэффициенты переноса, приведенные с по-
мощью различных комбинаций Vc и ТСУ являются функциями приведенной
температуры Тг = Т1ТС и приведенного объема Vr = V/Ve. Этот метод приве-
дения вязкости впервые предложил и использовал Камерлинг Оннес [33].
Так как критический объем точно измерить трудно, указанное приведение
удобнее проделать с помощью критического давления, т. е. можно написать
= Л)п (рп Тг),
= rjR(PryTr)y A.5)
=XR(pr,Tr).
Эти соотношения вообще более применимы, чем A.3).
3. Приведение с помощью значений коэффициентов переноса в критиче-
ских точках. Коэффициенты переноса могут быть приведены путем деления
их на значения коэффициентов переноса в критической точке. Поступая таким
образом, получим следующие универсальные соотношения :
J7)r = -§; = JZ)r(pnTr)y rjr = ^- = rjr(pnTr)f Xr = ^ = Xr(PnTr)y A.6)
в которых Тг = TjTc и рг = plpc. Очевидно, что Л)п уг и К — постоянные крат-
ные величинам JZ)Ry rjR, XR соответственно. На фиг. 138 показаны кривые «об-
общенных коэффициентов вязкости» rjr(pn Tr)y подготовленные Уехара и Ватсо-
ном [66]. Конечно, коэффициенты переноса не могут быть легко измерены в
критической точке. Однако эти критические значения могут быть получены с
помощью подгонки экспериментальных данных к обобщенной кривой в любой
другой области. Таким образом, этот метод предусматривает метод экстрапо-
ляции измеренных величин и необходимость точной подгонки обследованной
области. В этом отношении этот метод предпочтительнее, чем предыдущий.
Однако он неудобен по той причине, что для его использования необходимы
экспериментальные значения коэффициентов переноса. Значение вязкости в
критической точке можно оценить с помощью соотношения [7]
A.7)
Это соотношение согласуется с изложенным в предыдущем пункте. Аналогич-
ные выражения могут быть получены для других коэффициентов переноса.
'Ц4 0,5 Ofi Ofi 1tO • 2,0 3 4 5 6
Приведенная температура ТГ=Т/ТС
Ю
Фиг. 138. Приведенная вязкость rjr = v/rjc как функция приведенной температуры
при различных значениях приведенного давления рг = р/рс.
и
9
8
7
Vе'
I5
§4
СО
со
¦ HI
у
««•
е-
У
—
#
Т1
-J—
Л 1
/\ '
/
У
Х'
/
1
/-
(
у
i
/
*
-—
У
/
,/
/
/
1
- •
- -
ty
. —
¦ е
^*
_/
/
_ _ - - -
Л + *
у'
11111 >Н
- -7-
\
у*—
уУ*
У
^т —'
¦ —
Г
*>У
^*
.——
h
У
т —
а
<i
—-
—
»—-
^"
0
Гз
-1
***
У
у
п
у*-
00
*
у
у
У
у
У*
у
У
у
у*
у*
¦^
+**
у*
у*
у
У
«г:
у р
У
у
у *
у
у-
у '
0,2 0,3 0,4 0,5 0,6 0,7 0,8 Q91,0 2
Приведенное давление рг
4 5 6 7 8 910
Фиг. 139. Вязкость, приведенная с помощью значений вязкости при нулевом давлении,
гак фун1>т(и«1 прнрр^рнного ц'ячениа и темпер'-п рм [21
488 Гл. 9. Явления переноса в плотных газах и жидкостях
Очевидно, что использование таких выражений для коэффициентов в крити-
ческой точке сводит обсуждаемый метод к методу, изложенному в предыдущем
пункте.
4. Приведение с помощью значений коэффициентов переноса при предельно
малых плотностях. Другой тип обобщенных диаграмм может быть получен
путем приведения коэффициентов переноса с помощью их предельных значений
при малых плотностях. В этом случае приведенная вязкость определяется
в виде
(Для большинства газов в качестве предельного значения вязкости можно
взять значение вязкости при 1 атм, так как коэффициент вязкости почти не
зависит от давления при малых плотностях.) По закону соответственных состоя-
ний эта приведенная вязкость есть функция только приведенного давления
Рг = PlPc и приведенной температуры Tr = TjTc. Использование закона соот-
ветственных состояний в таком виде имеет то преимущество, что результаты
подгоняются к величинам при нулевом давлении и, таким образом, приведе-
ние фактически проводится в этом пределе. Вязкость при малых плотностях
известна для большого числа веществ. Ее можно также оценить с помощью
методов, изложенных в предыдущей главе. Диаграмма обобщенной вязкости [1],
соответствующая обсуждаемому способу приведения, показана на фиг. 139 [2].
Г. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ ДЛЯ ПОЛЯРНЫХ
МОЛЕКУЛ1)
Обсужденные выше приложения закона соответственных состояний строго
пригодны только для сферических неполярных молекул. Там предполагалось,
что потенциальные функции, описывающие взаимодействие между двумя
различными молекулами, характеризуются параметрами а и е. Как отмечалось
в гл. 3, § 10, силы взаимодействия между полярными молекулами, помимо
а и е, зависят также от величины дипольного момента р. Поэтому соотношения
A.3) для полярных газов приобретают вид
\те
,»•;/«•), A.9)
IT*,v*;h*),
где /** = /i/yест3 — приведенный дипольный момент.
Как указывалось в гл. 8, §6, для полярных молекул кинетическая теория
еще не была настолько развита, чтобы на основе ее можно было строго уста-
новить вид функции rj*(T*, v* ; ^*) даже для предельно малых плотностей.
Рассмотрим полярные молекулы, для которых измерены вязкость и второй
вириальный коэффициент. Как отмечалось в гл. 3, § 10, второй вириальный
коэффициент может быть использован для оценки молекулярных силовых
постоянных и /л*. Эти постоянные совместно с данными о вязкости при нулевом
давлении были использованы для вычисления приведенной вязкости
Уте
!) См. [34].
§ 2. Теория Эйринга явлений переноса в газах и жидкостях
489
CJ
J*=0,8
+ -сн3сл г- п,в
ч - СН3ОСН3 t*-0,7
а- СН3ОН Г 0,8
0,25
0,50
0,75
1,00 1,25
T*=kT/e
150
/,75
2,00
Фиг. 140. Экспериментальные значения ?B»2)* для некоторых полярных газов.
Кривые соответствуют экспериментальным значениям Я*1-1)* (для некоторых полярных газов), вычисленным
по формуле [OA'f)]3Kcn. ™ 266,93 fy МТ1о*(г) • 107)]. В качестве величин айв использованы величины,
вычисленные Раулинсоном по второму вириальному коэффициенту (см. гл. 3, § 10). Параметр /* просто
связан с приведенным дипольным моментом /л* соотношением /* = ju**/K 8 = fi*jV~Sea*, В соответствии с
законом соответственных состояний, отклонения от кривой для неполярных газов больше для больших
значений t*.
На фиг. 140 показаны кривые, характеризующие rj*} а также теоретически
вычисленные [по A.4) гл. 9] кривые для ^* = 0. Влияние тех членов межмо-
лекулярных потенциалов, которые учитывают полярность молекул, грубо
характеризуется зависимостью кривых от /г*.
§ 2. ТЕОРИЯ ЭЙРИНГА ЯВЛЕНИЙ ПЕРЕНОСА В ГАЗАХ
И ЖИДКОСТЯХ1)
Прежде чем изложить метод изучения процессов переноса в плотных
газах и жидкостях с помощью строгой кинетической теории, рассмотрим инте-
ресный подход к этой проблеме, основывающийся на теории абсолютных ско-
ростей реакций. Теория абсолютных скоростей реакций довольно успешно
применяется для вычисления коэффициентов переноса в жидкой фазе и дает
простое и наглядное истолкование процессов переноса. Основным уравнением
теории является уравнение Эйринга для удельной константы скорости к'
[уравнение (А.11) гл. 9]
B.1)
г) Краткий вывод уравнения скорости процесса Эйринга дан в приложении в конце
главы. Более полное изложение этого вопроса можно найти в работе Глесстона, Лайдлера
и Эйринга [35].
Впервые приложение теории абсолютных скоростей реакций к явлениям переноса
было сделано Эйрингом [36]. Обзор работ по этому вопросу дали Пауэлл, Розевир и Эйринг
[37 ], а также Кинкайд, Эйринг и Штерн [38 ]. Обзоры очень кратки. В обеих статьях под-
водятся итоги по приложению теории к расчету вязкости (с учетом влияния температуры
и давления), диффузии, ионной проводимости и теплопроводности. В статье [38] рассматри-
ваются также специальные задачи, возникающие при описании движения больших молекул.
Эти задачи представляют интерес при рассмотрении пластического течения. Обзор прило-
жений теории скоростей реакций к явлениям переноса дали также Глесстон, Лайдлер и
Эйринг [35, гл. IX].
490
Гл. 9. Явления переноса в плотных газах иг жидкостях
где zJG+, АЙ+ и AS+ — свободная энергия Гиббса, энтальпия и энтропия акти-
вации соответственно. Множитель х называется коэффициентом прохождения
и равен вероятности того, что процесс действительно происходит, если система
находится в активированном состоянии. Для большинства процессов к изме-
няется от х/2 до 1. В настоящем обсуждении мы будем полагать его равным
единице.
Очевидно, что для того, чтобы при изучении диффузии, вязкости и тепло-
проводности применить эту теорию, необходимо предположить, что в среде
действует какой-то тип механизма, который ответствен за поток молекул и в
котором имеется медленная стадия, отождествляемая с «активированным
состоянием». Нужно также знать число молекул, участвующих в элемен-
тарном процессе потока, но в точном определении самого механизма нет
необходимости. Однако априори невозможно установить, сколько молекул
(одна, две или большее число) участвует в элементарном процессе потока. Эй-
ринг предположил, что в поток вовлекается обычно только одна молекула,
так что процесс является одномолекулярным. Результаты расчетов хорошо
согласуются с экспериментом. Получающиеся значения энергии и энтропии
активации могут быть объяснены на основе простой одномолекулярнои модели.
А. КОЭФФИЦИЕНТ ВЯЗКОСТИ
Чтобы описать процесс вязкого течения, рассмотрим модель жидкости,
изображенную на фиг. 141. Будем полагать, что число молекул в единице объема
жидкости равно п. Вообразим (какэто было сделано в гл. 4, § 4 при обсуждении
уравнения состояния для больших плотностей), что жидкость имеет решетчатую
©00
Активированное состояние.
Без сдвиговой силы
При наличии сдвиговой силы F
Расстояние
Фиг. 141. Эффективное сечение идеализированной жидкости, иллюстрирующее основной
скоростной процесс в вязком потоке.
Чтобы попасть в свободную от молекул полость, молекула должна пройти через горловину.
структуру и что каждая молекула под действием соседних молекул заключена
в свою ячейку. Расстояние между узлами решетки вдоль потока обозначим
через а, а расстояние между соседними плоскостями — через 6. Основным
процессом, который мы здесь рассматриваем, является движение молекулы
к расположенной около нее свободной ячейке. Чтобы пройти через «горловину»,
образуемую ближайшими соседями, блуждающая молекула должна пройти,
как показано на фиг. 141, через область высокой потенциальной энергии.
§ 2. Теория Эйринга явлений переноса в газах и жидкостях 491
Если на систему не действуют внешние силы, то потенциальная кривая симметр ична
относительно «горловины». Если же на единичную площадку действует внешняя
сила F, стремящаяся сместить один слой молекул относительно другого, то
кривая потенциальной энергии искажается так, что молекула, перемещающаяся
в направлении силы, спускается «под гору», а молекула, перемещающаяся в
обратном направлении, поднимается «в гору».
Процесс движения молекулы в полость по направлению приложенной
силы будем называть прямым процессом, а процесс движения молекулы в
полость в направлении, противоположном действующей силе, — обратным
процессом. Через к} и к'г обозначим частоты прямого и обратного процессов.
При этом общая скорость потока молекул относительно узлов решетки в на-
правлении приложенной силы будет равна
vo = a(k'f — К).
Частоты к} и к'Г определяются по формулам B.1) с учетом того обстоятель-
ства, что свободная энергия активации для двух процессов различна, так как
прямой процесс осуществляется легче, чем обратный. Сила, действующая на
отдельную молекулу и заставляющая перемещаться молекулу вперед, равна
силе F, действующей на единицу площади и умноженной на площадь, зани-
маемую одной молекулой 1/пй. Сила F/nd уменьшает свободную энергию акти-
вации, необходимую для перескока молекулы в направлении силы, и увеличи-
вает свободную энергию активации, необходимую для перескока молекулы
в противоположном направлении. Если активированное состояние находится
на полпути между узлами решетки, то работа, производимая внешней силой
при перемещении молекулы на расстояние х/2 а в активированное состояние,
равна ^aF/nd. Эта работа представляет собой то количество энергии, на которое
изменяется свободная энергия активации в двух направлениях. Таким обра-
зом, частоты к'/ и к'г можно записать в виде
B.2)
B.3)
где fco равна величинам к) и к'Т в отсутствие внешней силы F. Общая скорость
потока одного слоя относительно слоя, следующего за ним, равна
v0 = a (fcj - К) = 2ак'о sh {^щг) ' B.4)
Из этого результата, а также с помощью определения коэффициента вязкости,
данного выражением A.22) гл. 8, получим
*i — JL Fd
vo/6
Из этого соотношения следует, что вязкость зависит от внешней силы, т. е.
поток не является ньютоновским1). Следует отметить, что из всех существую-
щих молекулярных теорий явлений переноса только теория Эйринга объясняет
такое поведение вязкости. Для обычных жидкостей и разумно малых значений
внешней силы aFfidnkT очень мало по сравнению с единицей. Отсюда
sh (aFj2bnkT) может быть заменен первым членом разложения Тэйлора,
который равен aF/2dnkT} после чего выражение для коэффициента вязкости
приобретает вид
HiritiI^- <2в>
В этом случае поток является ньютоновским в том смысле, что вязкость не за-
висит от приложенной силы. Часто для упрощения полагают, что расстояние
между молекулярными слоями д равно расстоянию между узлами решетки а,
*) По поводу потоков, не являющихся ньютоновскими, см. Рейнер [39], Филиппов
[40] и Штейнкопф (литографированное издание 1942 г.).
492
Гл. 9. Явления переноса в плотных газах и жидкостях
т. е. полагают, что множитель (д/аJ равен единице. В пластических потоках
прилагаемые силы достаточно велики, так что аргумент гиперболического
синуса велик по сравнению с единицей и вязкость зависит от величины прило-
женных сил.
В соотношениях B.5) и B.6) вязкость выражается через свободную энергию
активации AG*, которая получается эмпирически. Так как связи между сосед-
ними молекулами, которые должны быть преодолены для образования полости,
те же, что и преодолеваемые в процессе испарения, то энергия, необходимая
2 4 6 8^ 10 11 14 16 18
AUV> ккал
Фиг. 142.. График, иллюстрирующий справедливость соотношения B.7).
Квадратиками отмечены «идеальные газы»; треугольниками — водородосвязывающие жидкости ;
кружками — остальные жидкости [38].
для образования полости (размером, равным характерному объему молекулы),
является энергией испарения AUV. Поэтому мы можем предположить, что
энергия активации пропорциональна энергии испарения.
Энтальпия испарения AHV = AUV + RT до некоторой степени более при-
вычная величина, чем AUV. Эмпирически найдено, что
AG+ =
2,45
Справедливость этого соотношения показана на фиг. 142, на которой полу-
ченная эмпирически (из данных по измерениям вязкости большого числа раз-
личных жидкостей) свободная энергия активации сравнивается с измеренными
значениями энергии ^испарения [38].
Тот факт, что AG+ = AU„/2,45, по-видимому, указывает на то, что размер
полости, необходимой для объяснения процесса вязкого потока, является
только частью характерного объема молекулы. Возможный механизм [41 ]
вязкого потока, согласующийся с этим результатом, показан на фиг. 143.
Здесь пара молекул сжимается, поворачивается приблизительно на 90° и затем
разделяется. При этом приходящийся на одну молекулу объем приблизительно
равен х/3 части объема молекулы.
Энергия испарения может быть оценена по правилу Трутона
АО% = АН, - RTb = 9,4 RTb, B.8)
где Ть — температура кипения при 1 атм. Используя это соотношение, вяз-
кость жидкости можно записать в виде
rj = hne3>8T>iT. B.9)
Последнее соотношение полезно для грубых оценок.
§ 2. Теория Эйринга явлений переноса в газах и жидкостях
493
Из закона соответственных состояний все вещества при температуре плав-
ления Тт должны иметь одинаковую величину AUvIRTm. Таким образом,
AG+IRTm и г\\п в точке плавления
для всех веществ должны быть оди-
наковы. Для молекул средних раз-
меров величины л для различных
жидкостей мало отличаются друг от
друга. В точке плавления вязкость
для жидкостей из таких молекул
приблизительно равна 0,02 пуаза.
Рассмотрим теперь, какие све-
дения могут быть получены из экспе-
риментальных измерений вязкости.
По температурной зависимости
вязкости при постоянном давлении
можно определить энтальпию ак-
тивации. Для большинства веществ
она изменяется от х/3 до х/4 энергии
испарения. Для ассоциированных
жидкостей она много больше, чем
для нормальных жидкостей. Для
жидких металлов теплота испаре-
ния очень мала и имеет величину,
согласующуюся с представлением о
том, что единицей потока является
ион металла, а электроны прово-
димости внешних оболочек в поток
не вовлекаются. Для длинных мо-
лекул-полимеров теплота активации
найдена равной около 10 ккал/моль
и не зависит от длины цепи. Это
соответствует единице потока, рав-
ной от 20 до 40 атомов углерода в
молекуле. Другими словами, длинные молекулы, по-видимому, перемещаются
змеевидным образом — по звену за раз.
С помощью измерения температурной зависимости вязкости при постоян-
ном объеме могут быть получены некоторые сведения о роли полостей в про-
цессе потока. Можно думать, что число полостей в жидкости почти не зависит
от температуры при постоянном объеме. Если теплота активации зависит
исключительно от образования полостей, то зависимость вязкости от темпера-
туры при постоянном объеме может быть действительно очень малой. Для
нормальных жидкостей это соответствует действительности, но для ассоцииро-
ванных жидкостей наблюдается значительное влияние температуры. Таким
образом, в ассоциированных жидкостях часть теплоты активации, очевидно,
идет на разрыв связей между ассоциированными молекулами, что приводит к
образованию малых единиц, которые могут облегчить течение.
По зависимости вязкости от давления при постоянной температуре может
быть определен объем активации или добавочный объем, необходимый для
активированного состояния1). Для нормальных жидкостей он приблизительно
составляет х/в объема, приходящегося на одну молекулу.
Наилучшим эмпирическим соотношением для зависимости вязкости от
состава, которое смогли получить Эйринг и его коллеги, является соотношение
Фиг. 143. Вязкий поток, осуществляющийся с
помощью вращения двойных молекул.
Две молекулы сталкиваются и мгновенно образуют двой-
ную молекулу. Если около нее достаточно свободного
пространства, то двойная молекула может вращаться
и затем диссоциировать. С помощью последователь-
ности таких процессов один слой жидкости может
перемещаться относительно другого.
In г) = хг In г)г + х2 In rj2;
B.10)
г) Фриш, Эйринг и Кинкайд [42] показали, что при увеличении давления энергия акти-
вации для вязког потока увеличивается как pv'. Из экспериментальных данных найдено,
что v' обычно составляет ув объема, приходящегося в жидкости на одну молекулу.
494 Гл. 9. Явления переноса в плотных газах и жидкостях
здесь ^и?|2 — вязкости двух чистых компонент, а хг и х2 — молярные доли
компонент.
Недавно Мур, Гиббс и Эйринг [43 ] распространили теорию на углеводо-
роды. Они нашли резкие переходные точки, в которых возбуждаются новые
типы движения. При низких температурах движение молекул преимущественно
поступательное. При промежуточных температурах становится существенным
колебательное движение и, наконец, при высоких температурах возбуждается
свободное вращение молекул около их длинных осей. При низких температурах
течение осуществляется с помощью объединений двух или большего числа
молекул. При высоких температурах течение является одномолекулярным
процессом.
Найдено, что в точке кипения для большинства алифатических и ароматиче-
ских углеводородов [вместо B.7)] AG+ = —450 (кал/моль) + (AUvll,87)
Вязкость смазочных масел может быть объяснена на основе настоящей теории.
Однако это чрезвычайно сложная задача.
Б. КОЭФФИЦИЕНТ ДИФФУЗИИ
Теория диффузии легко получается из теории вязкости. Рассмотрим, во-
первых, самодиффузию в системе, где числовая плотность л является функцией
координаты х. Если концентрация в некотором слое молекул равна л, то кон-
центрация в соседнем слое, находящемся от предыдущего на расстоянии 6,
равна л + b{dnjdx). Число молекул, переходящих с 1 см2 первого слоя ко вто-
рому слою за 1 сек, равно Ьпк'о; число молекул, проходящих в обратном на-
правлении, равно дк'о [п + 6(dn/dx) ]. Разность между первым и вторым пото-
ками, взятая с обратным знаком, равна коэффициенту самодиффузии JZ), умно-
женному на градиент концентрации. Отсюда коэффициент самодиффузии выра-
жается в виде
B.11I)
здесь к'о — частота переходов в отсутствие внешней силы. С помощью формулы
B.6) эта величина может быть выражена через коэффициент вязкости, в резуль-
тате чего получим
(J^ B.12)
Если считать, что 5 = а = гг% то выражение B.12) может быть упрощено :
!?- = п1ЬкТ. B.13)
При диффузии в смесях движущей силой является градиент активности, а не
градиент концентрации. Чтобы учесть градиент активности аг вместо градиента
концентрации п19 можно преобразовать соотношение B.11). В результате по-
лучим формулу
Ч
г) В формулах B.11)—B.13) 2) отличаются от используемых в гл. 11, §2, на множитель
Q/nkT. Чаще употребляются 2> из этих формул.
2) Вектор потока массы в двухкомпонентной неидеальной изотермической системе
равен [см. B.28), B.29) гл. 11]
Коэффициент диффузии ЯI2 определяется следующим образом:
§2. Теория Эйринга явлений переноса в газах и жидкостях
495
Множитель dlnajd In пг может быть вычислен с помощью данных о давлении
паров. Правило для оценки концентрационной зависимости коэффициента
диффузии может быть установлено на основе эмпирически найденного факта,
заключающегося в том, что отношение
tiJZ}'12l(d In ajd In пг) изменяется линейно в
зависимости от концентрации. Этот факт*
иллюстрируется фиг. 144 [37 ].
Согласие между вычисленными и изме-
ренными коэффициентами диффузии очень
плохое. Для ряда пар жидкостей вычислен-
ные величины либо слишком велики, либо
слишком малы, причем одни величины могут
превосходить другие в 4 раза. Возможно,
что механизм диффузии каким-то образом
отличается от механизма вязкого течения.
Например, диффузия может осуществляться
с помощью бимолекулярного процесса, как
это предполагается в механизме потока,
изображенном на фиг. 143.
В. КОЭФФИЦИЕНТ ТЕПЛОПРОВОДНОСТИ
Теплопроводность плотных газов или
жидкостей зависит от очень быстрой пере-
дачи энергии через сами молекулы1). Этот
механизм лучше всего можно понять, рас-
смотрев вопрос о скорости распространения
звука в жидкостях. Найдено, что скорость
звука в жидкости в 5—10 раз больше ско-
рости самих молекул по кинетической
теории. Скорость звука в жидкости с1 мо-
жет быть найдена в предположении, что при
столкновении двух молекул энергия мгно-
венно передается от центра одной моле-
кулы к центру другой. На этой основе
HiI"
cg,
B.15)
W
Ацетон
где v = л — объем, приходящийся на одну
молекулу в жидкости, a vf — ее «свободный ' хлороформ
объем» (см. гл.4, § 4). Свободный объем Фиг. 144. Влияние активности на
жидкости может быть вычислен по давлению произведение коэффициента диффузии
пара или из уравнения состояния. Найдено, и вя31<ости.
что соотношение B.15) дает превосходное
согласие с измеренными значениями скорости звука для большого числа
жидкостей. Эйринг и его сотрудники настолько уверены в справедливости соот-
ношения B.15), что используют его для оценок свободных объемов жидкостей
из данных по измерениям скорости звука.
На основании грубых соображений кинетической теории теплопровод-
ность одноатомного газа дается формулой B.14) гл. 1, т. е. имеет вид
№ = -^-ncvQl; B.16)
здесь п = У ЪкТ\пт — средняя скорость молекул по кинетической теории ;
г) Этот процесс известен как процесс переноса энергии столкновениями. Более детально
он обсуждается в § 3 и 4.
496 Гл. Р. Явления переноса в плотных газах и жидкостях
I — средний свободный путь ; cv — удельная теплоемкость на молекулу при
постоянном объеме. Для жидкости это выражение должно быть преобразовано
следующим образом. Удельная теплоемкость cv для жидкости очень близка
к 3/с (вместо З/с/2 для газа), так как потенциал, в котором движутся молекулы,
может быть апроксимирован простым трехмерным гармоническим потенциалом.
Средний свободный путь можно заменить »*/• — расстоянием, на которое пере-
дается энергия при одном столкновении. Далее, в согласии с представлениями
о процессе распространения звука, скорость, с которой перемещается энергия
в жидкости, отличается от скорости, с которой она перемещается в газе в
(vjVfI!* раз. Таким образом, грубые соображения кинетической теории при-
водят к следующему выражению для теплопроводности одноатомных жидко-
стей :
Для исключения свободного объема из полученного выражения может быть
использовано соотношение B.15). Полагая с8 = УукТ/т} получаем
B.18)
где у = cpjcv. Эта формула пригодна только для одноатомных жидкостей.
Теплопроводность многоатомных молекул может быть получена умно-
жением предыдущего результата на поправку Эйкена (9 у— 5)/ [15 (у— 1)],
которая учитывает внутренние степени свободы молекул. Чтобы согласовать
окончательное соотношение с очень удовлетворительным эмпирическим соот-
ношением Бриджмена [44, 45 ], Эйринг предположил, что в переносе энергии
участвуют только вращательные и поступательные степени свободы, так что
подходящей величиной у в поправке Эйкена обычно является величина у = 4/3.
Таким образом, для многоатомных жидкостей можно написать
к1 = 2,80 кп2'* у-1'* с1. B.19)
Здесь отношение удельных теплоемкостей у следует брать равным величине,
получаемой из измерений скорости звука (по-видимому, оно отлично от зна-
чения 4/3, используемого в попаравке Эйкена). Эта формула чрезвычайно хоро-
шо (со средним отклонением в 10%) выражает теплопроводность большого
числа жидкостей. Бриджмен указал, что формула B.19) дает правильную
температурную зависимость теплопроводности жидкости при 1 атм. Однако
для большинства жидкостей теплопроводность увеличивается в 2 раза, когда
давление поднимается до 12 000 атм, тогда как из уравнения B.19) следует,
что теплопроводность увеличивается в 4 раза.
§ 3. ТЕОРИЯ ЭНСКОГА ЯВЛЕНИЙ ПЕРЕНОСА
В ПЛОТНЫХ ГАЗАХ И ЖИДКОСТЯХ1)
Строгая кинетическая теория газов, изложенная в гл. 7, основывается на
уравнении Больцмана A.25) гл. 7. При выводе этого уравнения (гл. 7, § 1)
учитывались только парные столкновения и предполагалось, что молекулы не
имеют протяженности в пространстве (или, более точно, молекулярный диа-
метр а мал по сравнению со средним расстоянием между молекулами). Оба
эти предположения несомненно справедливы для разреженных газов. Однако
для плотных газов эти два предположения должны быть пересмотрены.
г) См. работу Энскога [46]. Краткое изложение ее дано Чепменом и Каулингом [47,
гл. 16]. Недавно Кертисс распространил теорию Энскога на реальные газы. Работа Кертисса
изложена в отчете OOR-3 A953) Военно-морской научно-исследовательской лаборатории
Висконсинского университета. [См. также работу R. F. S u i d е г, С. F. С и г t i s s, Phys.
Fluid, I, № 2 (Ш58). — Прим. ред.]
§ 3. Теория Энскога явлений переноса в плотных газах и жидкостях 497
Энског был первым, кто продвинулся в этом направлении. Он развил
кинетическую теорию газов, состоящих из твердых сферических молекул диа-
метра <х. Для этой специальной модели несущественны тройные столкновения
и столкновения более высокого порядка. Рассматривая только парные столк-
новения и принимая во внимание конечные размеры молекул, Энског смог
изложить теорию плотных газов на основе развитой ранее теории разреженных
газов. Вследствие того что молекулы обладают некоторым объемом, при сжатии
газа становятся важными два эффекта : 1) перенос импульса и энергии столк-
новениями — при столкновении двух твердых сферических молекул происхо-
дит мгновенный перенос импульса и энергии от центра одной молекулы к центру
другой ; 2) изменение числа столкновений в секунду — частота столкновений
увеличивается, так как а непренебрежимо мало по сравнению со средним
расстоянием между молекулами, и уменьшается, так как молекулы настолько
близки друг к другу, что они защищают друг друга от налетающих молекул.
Таким образом, частота столкновений в газе из твердых сфер отличается в Y
раз от частоты столкновений в газе, состоящем из точечных частиц1). Как будет
показано позднее [см. C.32)], уравнение состояния для твердых сфер, запи-
санное через У, имеет вид
^г=1+^У. C-1)
где Ьо = 2/3 nNo* — второй вириальный коэффициент. Величина Y уже была
получена в связи с вириальным разложением уравнения состояния [см. E.1)
гл. 3]; она имеет вид
Y = 1 + 0,6250-^- + 0,2869 №-\* + 0,115 (-^)\ .. C.2)
Решение, данное в этом параграфе (только для чистых газов), очень близко к
решению, изложенному в гл. 7. Обозначения, используемые здесь, совпадают
с обозначениями гл. 7. После изложения теории мы даем краткий обзор резуль-
татов, полученных для твердых сферических молекул. Далее мы обсуждаем,
каким образом Энског использовал эти результаты, чтобы связать данные о
коэффициентах переноса при высоких давлениях с данными уравнения состоя-
ния. Завершается это вычислением на основе измерений, относящихся к урав-
нению состояния, множителя Y путем замены внешнего давления р в C.1)
суммой внешнего и внутреннего давлений. Сумма давлений равна «тепловому
давлению»2)
Показано, что этот метод выбора У физически разумен и согласуется с точкой
зрения, принятой Эйрингом и Хилдебрандом при изучении жидкой фазы.
А. ВИДОИЗМЕНЕНИЕ УРАВНЕНИЯ БОЛЬЦМАНА
ДЛЯ ПЛОТНЫХ ГАЗОВ
В гл. 7, § 1 уравнение Больцмана для функции распределения /(г, v, t)
получается на основе физических и геометрических соображений. Аналогичный
способ может быть использован для вывода интегро-дифференциального урав-
нения для функции распределения в плотных газах. Очевидно, что левая часть
(«потоковые члены») уравнения Больцмана остается неизменной в плотных
газах, но интеграл в правой части уравнения Больцмана видоизменяется, так
как молекулы имеют конечные размеры.
г) Энског обозначил этот множитель через #. Мы используем символ У, так как % исполь-
зовано для обозначения угла отклонения.
2) Для твердых сфер тепловое давление равно внешнему давлению, так что соотношения
C.1) и C.3) эквивалентны.
498 Гл. Р. Явления переноса в плотных газах и жидкостях
В гл. 7, § 1 показано, что частота столкновений в элементе объема dx около
точки г, при которых молекулы обладают скоростями в области ds около v и в
dvt около vx и параметры столкновений находятся в интервале db около & и в
интервале de около е, равна1)
/ (г, v) /x (r, v,) gb db de dv dw1 dr. C.4)
Чтобы найти частоту столкновений молекул в плотных газах из твердых
сфер, в это выражение необходимо ввести два изменения :
1) Вследствие того что сталкивающиеся молекулы обладают конечными
размерами, центры двух молекул не находятся в одной и той же точке. Если
одна молекула находится в точке г, то центр другой молекулы находится в
точке (г — <rk), где к — единичный вектор, направленный от центра второй
молекулы к центру первой молекулы в момент соприкосновения молекул при
столкновении. Соответственно функция распределения /х(г, ух) должна в дейст-
вительности вычисляться в точке (г — ok).
2) В плотных газах объем, приходящийся на одну молекулу vy имеет тот
же порядок, что и объем молекулы ^l^ltfJ- Поэтому объем, в котором может
находиться центр какой-либо одной молекулы, уменьшается, а вероятность
столкновения возрастает. Таким образом, частота столкновений C.4) увели-
чивается в Y раз, причем для твердых сфер Y зависит от плотности числа частиц
л(г, /). Функция Y вычисляется в точке соприкосновения двух сферических
молекул, т. е. в точке (г—112аЮ- Следовательно, при рассмотрении плотных
газов вместо выражения C.4) должно использоваться выражение
r. C.5)
С помощью этого видоизмененного выражения для частоты столкновений,
используя методы вывода уравнения Больцмана, аналогичные изложенным
в гл. 7, § 1, можно получить интегро-дифференциальное уравнение для функции
распределения
_Y(r--^ok)f(r,v)f1(r-ak,v1)]gbdbdedv1. C.6)
Предполагается, что состояние газа медленно изменяется в пространстве.
Отсюда функции /(г + <xk, v, t) и Y(r + 112<*К t) могут быть разложены в ряды
Тэйлора, после чего уравнение C.6) может быть записано в виде
где
*) В гл. 7 рассматривались многокомпонентные газы и столкновения между различными
составляющими. Поэтому две сталкивающиеся молекулы отмечались индексами i и /, обозна-
чающими тип молекулы. Для чистых газов необходимо делать различие между двумя моле-
кулами в столкновении: это делается с помощью индекса 1.
При обсуждении теории разреженных газов угол е не необходим вследствие цилинд-
рической симметрии. Этот угол, изменяющийся от О до 2щ определен на фиг. 111, стр. 357.
§ 3. Теория Энскога явлений переноса в плотных газах и жидкостях 499
Из этого уравнения может быть определена функция распределения плотных
газов, состоящих из твердых сферических молекул. Перед тем как обсудить
решение полученного уравнения, рассмотрим векторы плотности потоков и
уравнения переноса.
Б. ВЕКТОРЫ ПЛОТНОСТИ ПОТОКОВ
В гл. 7, § 2 были рассмотрены потоки различных молекулярных свойств —
массы, импульса и кинетической энергии — через элемент поверхности в
разреженных газах. Поскольку молекулы рассматривались там как точечные
частицы, то общее выражение для векторов плотности потоков B.18) гл. 7 пред-
ставляет собой перенос молекулярных свойств за счет потока молекул через
элемент поверхности. Теперь рассмотрим дополнительный перенос молекуляр-
ных свойств за счет столкновений, в которых центры сталкивающихся молекул
находятся на противоположных сторонах элемента поверхности. В таких
столкновениях происходит мгновенная передача импульса и энергии от центра
одной молекулы к центру другой, т. е. имеет место так называемый перенос
столкновениями, о котором мы уже упоминали выше.
Используя фиг. 112 (стр. 364), рассмотрим механизм переноса столкнове-
ниями. Вектор п — единичный вектор, нормальный к элементу поверхности dS}
направленный от отрицательной стороны поверхности к положительной. При
столкновении двух молекул, в котором первая молекула находится на поло-
жительной стороне поверхности, а вторая — на отрицательной, произведение
(к • п) положительно (так как к — единичный вектор, направленный от второй
молекулы к первой). В этих столкновениях линия, соединяющая центры
молекул, пересекает элемент поверхности dS в момент столкновения. Поэтому
центр первой молекулы находится в цилиндре, основанием которого служит
dS, а образующая параллельна к и имеет длину а. Объем этого цилиндра равен
ст(к • n) dS. Средними положениями центров двух молекул в момент столкно-
вения являются (т + 1/2аЮ и (г—ViA)» а точка соприкосновения в среднем
находится в г. Отсюда по аналогии с C.5) вероятное число таких столкновений
в единицу времени, в которых скорости молекул лежат в областях dv и dvx
около v и Vj и параметры столкновений находятся в интервале db около b и
de около е, равно
^{] C.8)
Так как в каждом столкновении такого сорта через поверхность передается
количество свойства у, равное (гр' — гр), то общая скорость переноса свойства у)
столкновениями в единицу времени через единицу площади равна
(п УФ) = oY (r) JJJJ (у/ - V)f[r + -±-ok,v}t) x
х 'i (r ~~ 4"аk> vi'') <к ' п> %b db de
Интегрирование проводится по всем переменным так, что (к • п) остается поло-
жительным.
Ограничения, накладываемые на область интегрирования, неудобны. Воз-
никающие при этом трудности могут быть обойдены следующим образом.
Сначала представим себе, что две сталкивающиеся молекулы поменялись ро-
лями, т. е. переменим индексы у переменных интегрирования в C.9).
500 Гл. 9. Явления переноса в плотных газах и жидкостях.
Затем заменим к на —к и (гр' — гр) на (гр[ — у)г). Так как гр — сумматор-
ный инварианту то (гр'г — грг) = — (гр' — гр). Следовательно, подынтегральная
функция в C.9) от этой замены переменных интегрирования не изменяется.
Отличие состоит только в том, что интегрирование по всем переменным про-
водится так, что (к • п) остается отрицательным. Отсюда C.9) может быть
переписано в виде
х
xkgbdbdedvdvv (ЗЛО)
где интегрирование производится по всем значениям переменных.
Таким образом, мы показали, что вклад переноса столкновениями в плот-
ность потока свойства гр равен
х kgbdbdedwdw^ C.11)
Как и при выводе уравнения C.7), функции распределения могут быть разло-
жены в ряды Тэйлора, в результате чего Ч*ф можно переписать в виде
... . C.12)
В дополнение к переносу \р столкновениями в поток входит также перенос гр
за счет потока молекул через поверхность. Эта часть плотности потока дается
выражением B.18) гл. 7
?/j>/d C.13)
Общий вектор плотности потока свойства гр является суммой двух плотно-
стей потоков
у = ФК + Ч*ф. C.14)
Индексами К и Ф обозначены потоки, дающие вклады в общий поток за счет
кинетической энергии (поток молекул) и за счет потенциальной энергии (пере-
нос столкновениями вследствие конечности размеров молекул) молекул соот-
ветственно. Член, учитывающий поток за счет кинетической энергии, вносит
главный вклад в общий поток при низких плотностях, а член, учитывающий
поток за счет потенциальной энергии, является главным при высоких плот-
ностях.
В. УРАВНЕНИЯ ПЕРЕНОСА
Уравнения переноса могут быть выведены способом, во многом аналогич-
ным способу, примененному в теории разреженных газов. Умножая видоиз-
мененное уравнение Больцмана C.7) на сумматорный инвариант и интегрируя
его по всем v, получаем
где
rv. C.15a)
§ 3. Теория Энасога явлений переноса в плотных газах и жидкостях 501
Можно показать, что после сложных преобразований1) интегралы /, приоб-
ретают вид
= -ТаУШ{v~ ?) (к" W
-Ta2Y JIfl> - ?\ (кк: l{ffiw]nT
/, = 0. C.16)
Легко видеть, что из этих выражений следует
}) C.17)
1}). C.18)
6
Сравнение этих результатов с C.12) показывает, что 2 А в точности равна
JL . Фф\у так что C.15) можно переписать в виде
Это уравнение есть общее уравнение переноса.
г) Исчезновение /х доказано в гл. 7, § 2. Те же соображения приложимы к доказатель-
ству /в = 0. Преобразования для /2 выглядят следующим образом. Соотношением C.15а) /2
определяется в виде
Согласно доводам, приведенным в гл. 7, § 2,
/2 = - aY j(JJ* (к • j/' -|l + / -^-j) g& db de dv dvx. F)
Отсюда
4JJjJ{}({§ ^j) (в)
Аналогичными способами можно показать, что
V aY Ш1(V " V^ (k' rlf)
Отсюда следует
jJJJ( ^J (д)
Теперь пусть молекулы поменяются ролями. При этом
[как это имело место в связи с получением C.9) и C.10)]. Следовательно,
/2 = aY Jj| j (V - у') (k. U -|-] ^ db de dw ^
Комбинируя последние два выражения, получаем для /2 выражение C.16). Выражения,
данные там для других //, получаются путем аналогичных преобразований.
502 Гл. 9. Явления переноса в плотных газах и жидкостях
Полагая снова гр равным т, тМ и 1l2mV2 (какэто было сделано в гл. 7, § 2),
получаем обычное уравнение непрерывности, уравнение движения и урав-
нение баланса энергии. Различие между уравнениями гл. 7, § 2 и уравнениями,
полученными здесь, состоит только в том, что тензор давлений р = рк + рф
и вектор плотности потока тепла q = qk + qo теперь складываются, как ука-
зано в C.14), из двух членов — кинетического и потенциального.
Г. РЕШЕНИЕ ВИДОИЗМЕНЕННОГО УРАВНЕНИЯ БОЛЬЦМАНА
Видоизмененное для высоких плотностей уравнение Больцмана C.7) мо-
жет быть решено методом, совершенно аналогичным методу, использованному
для решения обычного уравнения Больцмана (для малых плотностей) и изло-
женному в гл. 7, § 3. Функция распределения может быть разложена в ряд,
как указано в гл. 7, § 3 [см. разложение C.11I. Однако мы рассмотрим только
возмущение первого порядка и просто запишем
[l+fJ(r,v,0], C.20)
где /f°] — невозмущенная функция распределения
= п (w Г e-m(v-v')w- C.21)
Далее всюду функция возмущения ф и производные от п, v0 и Т рассматри-
ваются как малые величины.
Подставляя выражение C.20) для /(г, v, t) в уравнение C.7), получаем
интегро-дифференциальное уравнение для функции возмущения ф. Члены
левой части уравнения («потоковые» члены) и первый член в правой части ]х
совпадают с полученными в теории разреженных газов. В рассматриваемом
нами приближении функция возмущения не появляется в интегралах J2 и уз>
так как эти члены уже включают производные макроскопических величин.
Аналогично в этом приближении J4, J5, у6все равны нулю, так как в них входят
вторые производные макроскопических переменных или квадраты первых
производных. В результате интегрирования1) J2 и у3 получим
л - - -f^3 Yfm [2 (vt)+D-W2 - 4) (
+ МУГ»:ъ*) + [-т*Л-1)(?-.,.)], C22)
Уз =-4 f)
где, как и в гл. 7, безразмерная скорость W определена следующим образом :
Используя эти результаты наряду с результатами гл. 7, § 3, получаем урав-
нение для функции возмущения ф
= 2«JJ/i«/t01 {Ф' + Ф'х-Ф- Фдi*^Фгг, C.23)
в котором, как ив гл. 7, Ь = 2 (WW — 7з W2 UJ. При получении этого уравне-
ния для исключения производных по времени от макроскопических переменных
') Детали алгебраических преобразований, используемых при интегрировании и в
остальном анализе, см. [47, стр. 278].
§ 3. Теория Энскога явлений переноса в плотных газах и жидкостях 503
были использованы уравнения переноса. Уравнение C.23) для плотных газов
аналогично уравнению C.26) гл. 7 для разреженных газов. Два уравнения
совпадают, за исключением того, что в уравнении для плотных газов
(9/3r)v0 дополнительно умножается на A/У) (l +~^nn<fi y), а Э1пГ/Эг—на
(l/Y)(l +-^-nnasY). Поэтому решение уравнения C.23) имеет вид
)(:i4 C.24)
где функции А и В определены и вычислены в гл. 7, § 3.
Д. КОЭФФИЦИЕНТЫ ПЕРЕНОСА
Теперь мы имеем все необходимое для вычисления коэффициентов пере-
носа : выражения для плотностей потоков C.12)—C.14) в виде интегралов
от функции распределения /, функцию распределения / C.20), выраженную
через функцию возмущения, и, наконец, выражение C.24) для самой функции
возмущения. Сначала рассмотрим вклады рк и qK, вносимые в тензор давлений
и вектор плотности потока тепла за счет кинетической энергии молекул. Затем
получим выражения для рф и яФ, учитывающие в тензоре давлений и векторе
плотности потока тепла вклады, возникающие за счет переноса столкновениями,
и, наконец, получим точные выражения для коэффициентов объемной и сдви-
говой вязкости и для коэффициента теплопроводности.
Вклад, вносимый в тензор давлений и вектор плотности потока тепла за
счет кинетической энергии молекул, может быть получен с помощью выра-
жения C.13). Это выражение формально совпадает с выражением, использо-
ванным в кинетической теории разреженных газов. Однако выражение для ф,
которое мы используем теперь, отличается от выражения, полученного в гл. 7
в коэффициентах при А и В, которые описаны в связи с выводом уравнения
C.23) и выражения C.24). Поэтому мы можем немедленно написать1)
рк = nkT U - 4" (l + 4 */ю» Y) rj» S , C.25)
Як = ~ 4" A + -g-™*" Y) А° 1Г <3-26)
Здесь rj° и Л° — коэффициенты вязкости и теплопроводности в разреженных
газах ; S — тензор скорости сдвига, определенный по D.16) гл. 7.
1. Коэффициенты сдвиговой и объемной вязкости. Вклад в тензор давле-
ний за счет переноса столкновениями может быть получен из C.12), если гр
принять равным ту. Первый интеграл в этом выражении может быть вычислен.
Этот интеграл равен
±пп<Р(птУ) (VV + -±-V2U) = -^ппв* Y (pK +~-nkT и) =
= nkT [-|-ппо* У) U - -yjrтгпо* [l + -^rnno* y) rf>S. C.27)
Данная здесь вторая форма записи получена путем использования выражения
C.13) для рК и определения температуры в кинетической теории. Третья
форма записи получена с помощью C.25).
*) Соотношение C.25) отличается от D.17) гл. 7 не только тем, что в него входит допол-
нительный множитель, введенный в C.24), но и тем, что мы вместо статического давления р
написали nkT. Это сделано потому, что в р# имеется дополнительный член, который добав-
ляется к упомянутому члену, образуя вместе с ним истинное уравнение состояния.
504 Гл. 9. Явления переноса в плотных газах и жидкостях
Во второй интеграл выражения C.12) входят производные функции рас-
пределения, и поэтому этот интеграл не включает функцию возмущения в том
приближении, которое мы рассматриваем. Следовательно, интеграл может
бьгп> вычислен непосредственно. Результат имеет вид
C.28)
где
C.29)
Величина х, которая, как показано ниже, является коэффициентом объемной
вязкости, может быть записана через коэффициент сдвиговой вязкости B.10)
для нулевого давления1)
х = 1,002 (-|- wo*J Yrj°. C.30)
Общий вклад в плотность потока импульса за счет переноса импульса столкно-
вениями равен сумме выражений C.27) и C.28). Добавляя к этой сумме вклад,
определяющийся переносом импульса за счет кинетической энергии молекул,
получаем общий тензор давлений
Сравнение этого выражения с F.29) гл. 7 показывает, что к есть коэффициент
объемной вязкости. Из C.31) следует, что кинетическая теория приводит к
выражению для статического давления
р = nkT (l + -§- пп& Y). C.32)
Поскольку известно статическое давление из уравнения состояния для твердых
сфер, то из C.32) можно вычислить величину Y. Сравнивая C.31) с соотно-
шением F.29) гл. 7, являющимся определением коэффициента сдвиговой вяз-
кости % получаем
=4- (i+4-ша3 Y+°>761 Dлпа3 YT) v°- <3-33)
Это выражение обсуждается в разд. Е.
2. Коэффициент теплопроводности. Вклад в вектор плотности потока тепла
за счет переноса столкновениями можно получить из C.12), полагая в этом
выражении у) равными y2mv2. Первый интеграл в этом выражении равен
0^-- C.34)
Второй интеграл выражается следующим образом :
Зк х дт
^Числовой множитель 1,002 равен 16 @,984)/5л. Величина0,984является поправкой к
коэффициенту вязкости при нулевом давлении за счет высших приближений в рядах из
полиномов Сонина (см. табл. 67, стр. 432).
§ 3. Теория Энскога явлений переноса в плотных газах и жидкостях 505
где к — коэффициент объемной вязкости, выражение для которого дано выше.
Складывая полученные два члена с вкладом, вносимым в вектор плотности
потока тепла за счет кинетической энергии и выражающимся в виде C.26),
получаем выражение для полного вектора плотности потока тепла
* + ?»}?¦ C.36)
Отсюда коэффициент теплопроводности имеет вид1)
= 4~ (l + 4"яда* Y + °>755 ("I"
4"
Эту формулу мы рассмотрим еще в разд. Е.
Формула C.37) справедлива, конечно, только для одноатомных молекул.
Для плотных многоатомных газов эта формула может быть видоизменена на
основе идей, которые приводят к поправке Эйкена в разреженных газах
(см. гл. 7, § 6). В результате получим
= 4-(-г+4-)-т^+4-лпа*[4-+°>755D-лпа3) Y]4r^ <3-37a>
Однако вследствие того, что вращение молекул при высоких плотностях затруд-
нено, нельзя ожидать, что эта формула приложима выше области средних плот-
ностей.
3, Коэффициент диффузии. Обсуждавшиеся до сих пор теория и прило-
жения ограничивались случаем чистых газов. Теория Энскога плотных газов
распространена на случай бинарных смесей2). Результаты для вязкости, тепло-
проводности и термодиффузии выглядят очень сложно и не приводятся здесь.
Однако коэффициент диффузии для плотных газов просто связан с его величиной
при малых плотностях3)
Величина У12 является обобщением функции Y, введенной ранее, и зависит
от диаметров молекул обоих сортов ах и <т2
(?±) ... . C.39)
В этом параграфе мы убедились, что плотность потока импульса (тензор давле-
ний) и плотность потока энергии (вектор плотности потока тепла) состоят из
двух частей : одна возникает вследствие потока молекул, другая — из-за пере-
носа столкновениями. Вектор плотности потока массы j, связанный с коэффи-
циентом диффузии, отличается от этих двух векторов тем, что он (и это совер-
шенно ясно) не зависит от переноса столкновениями.
^Численная постоянная 0,755 дает поправку за счет высших приближений (см. табл.
67, стр. 432).
2) Это обобщение было проделано Торном в университете Сиднея [47, стр. 292 ].
3)^ фф фф б й й
щ рд р ур д [, р ]
)i2 — коэффициент диффузии бинарных смесей, вычисленный на основе кинетической
теории для малых плотностей для газа, температура и плотность которого равны темпера-
туре и плотности плотного газа.
4) ^12 B формуле C.38) не совпадает с ^I2 в гл. 11, § 2. Это различие обязано тому, что
d12 по Чепмену и Каулингу [47, стр. 293] не совпадает с частным случаем йг по B.28) гл. 11.
506 Гл. 9. Явления переноса в плотных газах и жидкостях
Е. КРАТКИЙ ОБЗОР РЕЗУЛЬТАТОВ ТЕОРИИ ПЛОТНЫХ ГАЗОВ
ЭНСКОГА ДЛЯ ТВЕРДЫХ СФЕР
Суммируем теперь результаты теории Энскога коэффициентов переноса в
плотных газах из твердых сферических молекул. Коэффициенты переноса, делен-
ные на их значения при нулевом давлении (отмеченные индексом 0) и умно-
женные на приведенный объем, могут быть записаны через безразмерную вели-
чину
()() C.40)
Коэффициенты переноса через у выражаются следующим образом :
Сдвиговая вязкость -уг- = —- + 0,8 + 0761 у,
Объемная вязкость —^- = 1,002у,
C.41)
Теплопроводность1) ~- = -~ + 1,2 + 0,755 у,
?DV 1
Само диффузия2)
Приведенная сдвиговая вязкость, умноженная на приведенный объем как
функции параметра у, имеет минимум. То же справедливо для приведенной
теплопроводности, умноженной на приведенный объем. Эти минимумы равны
о-='.'*>.
<342)
Изменение различных коэффициентов переноса в зависимости от у графически
представлено на фиг. 145.
Для твердых сферических молекул параметр у равен фактору сжимаемости
минус единица
4
В областях низких и средних плотностей величина у может быть записана в
вириальной форме
у= (-^)+0,6250 (Aj2 + 0,2869 (Aj3 +0,115 (АL+... C.44)
В области высоких плотностей лучшее уравнение состояния для твердых сфер
получили с помощью радиальной функции распределения Кирквуд, Мон и
Алдер [48 ]3). Их результаты даны в табл. 96.
х)Это соотношение справедливо для одноатомных газов. Для многоатомных молекул
нужно использовать формулу C.37а).
а) ?>° может быть вычислено из Л)° = М?>(р = \)IqRT, где Л)(р = 1) — коэффициент
самодиффузии при давлении в 1 атм, вычисляемый по методам гл. 8.
8) См. также гл. 4, § 9. Кирквуд, Мон и Алдер [48 ] для плотностей, превышающих
bo/V = 1,6, получили меньшие значения у, чем можно получить из уравнения состояния
Эйринга [из уравнения E.3) гл. 4, используя постоянную 0,6962] в V/0,6962 b0 раз. Эти
результаты недавно были подтверждены и развиты Мак-Лелланом [49]. Однако недавние
неопубликованные расчеты уравнения состояния (по методу Монте-Карло), выполненные
Розенблатом, показывают, что предпочтительнее уравнение Эйринга, полученное на основе
представлений о свободном объеме.
§ 3. Теория Энскога явлений переноса в плотных газах и жидкостях
507
Таблица 96
Величина у как функция bo/V для очень плотных газов из твердых сфер
PV
У
RT
— 1
0,3535
0,6250
0,8511
1,047
1,224
у
0,44
0,91
. 1,39
1,89
2,40
UV
! 1,377
1,527
1,664
1,805
1,934
У bJV
2,91
3,43
3,93
4,44
4,95
2,058
2,160
2,278
2,387
2,962
у
5,46
5,99
6,50
6,93
со
Зависимость у от bojV для твердых сфер показана на фиг. 146 [48]. Этот
график, а также графики, представленные на фиг. 145, дают полную сводку
результатов теории Энскога явлений
6,0
5,0
а - (х/х°) (v/b0)
Ь -(rj/rj°)(V/b0)
С -(x/v°)(V/b0)
d - @/0°)(V/bo)
у р
переноса в плотных газах и жид-
костях, состоящих из твердых сфер.
Фиг. 145. Коэффициенты переноса газа,
состоящего из твердых сферических
молекул (по теории Энскога).
8,0
1.0
6,0
5,0
У
4,0
3,0
2,0
W
0
Фиг
i i i i i i i i i i
:
-
-
. /
: /
1,0 U
bo/V
. 146. Величина у==
как функция bJV
твердых сфер.
1 f
/ I
/ г
/ f
/ L
г
|_
1
г
L
. i • . 1
> 3,0
(pV/RT)-
для
Кривая асимптотически приближается
к вертикальной пунктирной линии.
Ж. ПРИЛОЖЕНИЕ РЕЗУЛЬТАТОВ К РЕАЛЬНЫМ ГАЗАМ
Хотя приведенные выше формулы C.4) для коэффициентов переноса полу-
чены для газов, состоящих из твердых сферических молекул, Энског [46,47]
показал, что они могут использоваться для реальных газов, причем получаются
вполне приемлемые результаты. Чтобы использовать приведенные выше фор-
мулы, необходимо определить Ьо и у. Энског предположил, что давление в
C.43) может быть заменено «термическим давлением» Т(Эр/ЭГ)у, так что вели-
чина у может быть определена из экспериментальных данных по сжимаемости
с помощью соотношения
C.45)
508
Гл. 9. Явления переноса в плотных газах и жидкостях
Легко показать, что для твердых сфер это соотношение и соотношение C.43)
совпадают. Однако для случая реальных газов этого не происходит. Энског
предположил также, что Ьо вычисляется1) по минимуму на кривой (rj/rj0) V как
функции у2).
Подтверждение метода Энскога может быть получено из следующих сообра-
жений. Давление, испытываемое одной молекулой, состоит из двух компонент :
внешнего давления р вследствие сдерживающего действия стенок сосуда и
«внутреннего давления» (Э?//ЭК)Г, которое объясняется наличием сил сцепле-
ния молекул. Сумма внешнего и внутреннего давлений называется «термическим
давлением» и термодинамически равна Т(др/дТ)у. Когда Энског предположил,
что величина у может быть определена из экспериментальных данных по сжи-
маемости с помощью C.45), он полагал, что реальный газ эквивалентен газу из
твердых сфер, в котором внешнее давление может быть заменено «термическим
давлением». Это предположение является основой теории растворимости неэлек-
тролитов в жидкостях Гильдебранда и основой теории уравнения состояний
жидкостей Эйринга (см. гл. 4, § 5). На основании того, что теории Гильдебранда
и Эйринга хорошо согласуются с экспериментом, можно заключить, что упомя-
нутая выше модель удовлетворительна для жидкостей и плотных газов. Однако
она не подходит для разреженных газов и все менее подходит с понижением
температуры. В таких условиях молекулы реального газа имеют тенденцию
образовывать небольшие скопления, что не может произойти с твердыми
сферическими молекулами.
Таблица 97
Вязкость азота при температуре 50° С как функция давления41
Р t QfflM
15,37
57,60
104,5
212,4
320,4
430,2
541,7
630,4
742,1
854,1
965,8
Qt с/СМ
0,01623
0,06049
0,1083
0,2067
0,2875
0,3528
0,4053
0,4409
0,4786
0,5117
0,5404
(|)ЭКСПЮ%
\ Q /ЭКСП.
см*/сек
117900
32 740
19280
11480
9520
8870
8660
8590
8700
8890 *
9090
17 • Ю7, г/см • сек
ЭКСП.***
1913
1981
2088
2373
2737
3129
3509
3786
4163
4550
4913
выч.
1810
1900
2050
2240
2660
3080
3480
3800
4180
4550
4920
* Сравнение взято из работы [47, стр. 289].
** Данные о сжимаемости из работы [50].
*** Данные о вязкости из работы [10].
В табл. 97 показана применимость полуэмпирического обобщения Энско-
га теории плотных газов из твердых сфер . В этой таблице сравниваются вычис-
ленные и экспериментальные значения вязкости N2 при 50° С до давлений
1000 атпм. Очевидно, что согласие вполне удовлетворительное. В табл. 98 [46]
г) Полученные таким путем величины Ьо имеют тот же порядок, какой получается из
коэффициентов вязкости разреженных газов для модели твердых сфер. Однако согласие не
особенно хорошее. Другие методы определения Ьо появятся после того, как будет накоплено
больше экспериментальных данных.
2) Фактически величина Ьо не может быть задана произвольно. Чтобы г)°, А0 и Л>° были
предельными значениями соответствующих коэффициентов переноса для низких плотностей,
необходимо иметь уV -> Ьо, когда q -> 0. Если у определяется с помощью C.45), то следует,
что Ьо = В + T(dB/dT), где В — второй вириальный коэффициент.
§4. Рассмотрение явлений переноса в плотных газах и жидкостях
509
Вязкость
45,3
64,3
75,9
82,7
86,8
89,2
91,7
94,9
101,6
114,6
* двуокиси
Q. г/см*
0,100
0,170
0,240
0,310
0,380
0,450
0,520
0,590
0,660
0,730
углерода при
давления
(—) •1о:'
\ о /чксп
смг/сек
18000
11500
9080
7840
7240
7020
7040
7220
7560
7950
40,3° С как
t] • 10:,
эксп.
1800
1960
2180
2430
2750
3160
3660
4260
4990
5800
Таолица 98
функция
г/см • сек
выч.
1910
1990
2160
2400
2730
3150
3670
4270
4980
5780
* Экспериментальные данные о сжимаемости и вязкости взяты из работы [50].
аналогичное сравнение проводится для СО2. Данные здесь два численных
сравнения служат довольно убедительным доказательством того, что метод
Энскога приложения к реальным газам результатов, полученных для твердых
сфер, некоторым образом учитывает основные особенности явлений переноса
в плотных газах.
§ 4. РАССМОТРЕНИЕ ЯВЛЕНИЙ ПЕРЕНОСА В ПЛОТНЫХ ГАЗАХ
И ЖИДКОСТЯХ МЕТОДАМИ СТАТИСТИЧЕСКОЙ МЕХАНИКИ
НЕРАВНОВЕСНЫХ СИСТЕМ1)
В гл. 7 развита теория явлений переноса в разреженных газах на основе
принципов статистической механики неравновесных систем. Путем решения
уравнения Больцмана для функции распределения рг\т, р, t) там получены
точные выражения для коэффициентов переноса через законы межмолекуляр-
ных сил. В этом параграфе на основе уравнения Лиувилля развита строгая
кинетическая теория плотных газов. Если предположить, что силы, действую-
щие между молекулами, являются парными силами, то векторы плотности
потоков могут быть выражены через бинарную функцию распределения нерав-
новесных систем. Таким образом, главной задачей кинетической теории плотных
газов и жидкостей является задача вычисления этой функции распределения
неравновесных систем. (В гл.4, § 9 показано, что проблема вычисления бинарной
функции распределения равновесных систем является ключом к пониманию
уравнения состояния плотных газов и жидкостей.) Несомненно, что этот подход
к изучению явлений переноса с теоретической точки зрения более удовлетвори-
телен, чем подходы, развитые в § 2 и 3. Однако в настоящее время проделаны
лишь очень ограниченные вычисления, так что этот метод еще не может быть
использован практически. Мы представим здесь основные черты этого направ-
ления теории, так как подход, основывающийся на методах статистической
механики, весьма перспективен и некоторые элементы теории дают возмож-
ность лучше уяснить смысл теорий Эйринга и Энскога.
Приведенное здесь обсуждение основывается на классической статисти-
ческой механике. Рассматривается система, состоящая из N одинаковых моле-
кул2). Для простоты предполагается, что эти молекулы являются точечными
ЧСм. [51].'
2) Ирвинг и Кирквуд [51] создали теорию смесей, состоящих из нескольких компонент.
510 Гл. 9. Явления переноса в плотных газах и жидкостях
частицами, не обладающими внутренними степенями свободы. Однако многие
результаты носят более общий характер. В дальнейшем предполагается, что
между молекулами действуют парные силы, так что потенциальная энергия
системы Ф(гы) может быть записана в виде
±22
где <р(г^) — потенциальная энергия взаимодействия между молекулами i и /,
когда они находятся друг от друга на расстоянии rfj.
А. УРАВНЕНИЕ ЛИУВИЛЛЯ И ОБЩЕЕ УРАВНЕНИЕ ПЕРЕНОСА1)
Для рассматриваемой системы, состоящей из N молекул, может быть
образован ансамбль динамически подобных систем, как описано в гл 7, § 1.
Состояние отдельной системы в ансамбле определяется одной точкой в 6N-Mep-
ном фазовом пространстве (у-пространстве); состояние представляющего
ансамбля описывается большим числом точек в том же фазовом простран-
стве. Функция распределения fN\ представляющая распределение этого облака
точек в фазовом пространстве, удовлетворяет уравнению Лиувилля
9 _ piv.e (L . « D 1)
dt ~ т [v дг")^[дгм 8pN J • V4-1'
Как отмечено в гл. 2, § 1, функция распределения нормирована к N!
= AM D.2)
Пусть а^, рм) — некая динамическая переменная, не зависящая от
времени. Тогда в соответствии с основными принципами статистической меха-
ники неравновесных систем (см. гл. 7, § 1) ожидаемое значение а является
средним значением
5=АЯа (г">ры) /(А0 <г"р" *>drM dfM • D-3)
Именно это значение а можно ожидать при измерении. На основании опреде-
ления D.3) и уравнения Лиувилля скорость изменения ожидаемого значения а
выражается в виде
т 1Р дг") [дг"
D 4)
Переход от второго выражения к третьему сделан с помощью теоремы Грина
и предположения о том, что fN) спадает достаточно быстро, когда г^ -> оо и
ры _+ оо# уравнение D.4) известно как «общее уравнение переноса».
Б. МАКРОСКОПИЧЕСКИЕ ПЕРЕМЕННЫЕ
Три основных уравнения переноса могут быть получены из общего урав-
нения переноса, если в качестве динамической переменной использовать дина-
х) В этом разделе используются функции распределения классической статистической
механики. Полный обзор этих функций, их определения и взаимосвязь даны в гл. 2, § 1.
§4. Рассмотрение явлений переноса в плотных газах и жидкостях 511
мические переменные, связанные с плотностями в обычном трехмерном про-
странстве массы, импульса и энергии.
Вероятность на единицу объема, что молекула /с находится в точке г,
равна
Щ^Т Я D-5>
где д(гк — г) — функция Дирака. Следовательно, (вероятная) общая плотность
числа частиц в точке г равна
N .
п(г,0= 2 8(гк-г), D.6)
а общая плотность массы равна
N
Q(t,t)= Д/ЛE(г,-г). D.7)
Аналогично вклад молекулы к в импульс на единицу объема в точке г
равен
и соответственно общая плотность импульса равна
N
D-8>
Q (г, 0 v0 (r, t)= 2Vh* (г* - г); D.9)
vo(r, f) — средняя массовая скорость молекул, т. е. макроскопическая скорость
течения.
Во внутреннюю энергию газа входят кинетическая энергия хаотического
движения молекул (т. е. движения относительно v0) и потенциальная энергия,
связанная с межмолекулярными силами1). Кинетическая энергия на единицу
объема в точке г равна
- DЛ0>
Однако эта величина включает кинетическую энергию г/2 q v$, являющуюся
энергией макроскопического движения жидкости. Поэтому плотность кинети-
ческой энергии хаотического движения молекул равна
где uK(r, t) — вклад во внутреннюю энергию на одну молекулу за счет кинети-
ческой энергии.
Плотности массы, импульса и кинетической энергии могут быть определены
прямым способом, так как масса, импульс и кинетическая энергия молекулы
могут рассматриваться как величины, локализованные в молекуле. В противо-
положность этому потенциальная энергия у(гу), связанная с силами, действую-
щими между молекулами i и /, зависит от положения обеих молекул и поэтому
*) Ирвинг и Кирквуд [51 ] определяют внутреннюю энергию как суммУ общей кинети-
ческой энергии, потенциальной энергии межмолекулярного взаимодействия и потенциаль-
н ой энергии, связанной с внешними силами. Иначе говоря, эти авторы включили в допол-
нение к учтенным здесь членам потенциальную энергию системы в поле внешних сил и кине-
тическую энергию, связанную с макроскопическим течением. Различие в определениях при-
водит к небольшим изменениям в уравнении энергии, но, конечно, не влияет на определения
векторов плотностей потоков.
512 Гл. 9. Явления переноса в плотных газах и жидкостях
в действительности не локализуема. Область действия межмолекулярных сил
мала по сравнению с макроскопическими размерами, поэтому в этой проблеме
с макроскопической точки зрения не возникает трудностей. В качестве разум-
ного приема можно формально принять, что в каждой молекуле локализовано
по половине потенциальной энергии <р{гг]). Такое определение сохраняет естест-
венную симметрию и дает возможность написать следующее выражение для
общей плотности потенциальной энергии:
п (г, 0 иф (г, /) = \ 2 2 9иЩ-*); D.12)
здесь иф(г, t) — вклад потенциальной энергии во внутреннюю энергию, при-
ходящуюся на одну молекулу. В результате полная внутренняя энергия на одну
молекулу равна
u(r,0 = MM) + MM). D.13)
Таким образом, нам удалось записать плотности массы, импульса и внутренней
энергии через средние величины или, иными словами, через интегралы от
/<">(г, р, О-
В. МАКРОСКОПИЧЕСКИЕ ПЕРЕМЕННЫЕ, ВЫРАЖЕННЫЕ ЧЕРЕЗ ФУНКЦИИ
РАСПРЕДЕЛЕНИЯ НИЗШИХ ПОРЯДКОВ
Запишем теперь полученные результаты через интегралы от функций рас-
пределения низшего порядка. Функция распределения fu в фазовом простран-
стве одной молекулы равна интегралу от ры) по координатам положения и
импульсам всех молекул, за исключением одной. Во избежание выделения какой-
либо одной молекулы в процессе интегрирования функцию f1} можно записать
в симметричном виде
I
г,Р,0 = I 4h-r).6(fik-p). D.14)
Аналогично функцию распределения в пространстве пар молекул можно запи-
сать в виде
/W (г, г', р, р', 0 = 11 6(rj-r)d(tk-r')d(Pj-p)d(pk-tf). D.15)
Числовая плотность, средняя массовая скорость, кинетическая и потенциаль-
ная энергии на одну молекулу через эти функции могут быть записаны следую-
щим образом:
n(r,O=J/<1>(r,p,Odp, D.16)
D.17)
D.19)
Первые три соотношения соответствуют определениям, данным в гл. 7, § 2.
Для рыводов, которые следуют ниже, полезны несколько других функций
распределения. Вероятность того, что одна молекула находится в точке г,
а другая — в точке г', дается функцией пB)(г, г', t). В соответствии с A.8) гл. 2
она определяется следующим образом :
п<2> (г, г', 0 = |Т/B) (г, г', р, р', 0 dp dp'. D.20)
§4. Рассмотрение явлений переноса в плотных газах и жидкостях 513
Удобно выразить <|ункцкю пB) через обычную плотность числа частиц
и «радиальную функцию распределения неравновесных систем» g(r, R, t)
л<*> (г, г + R, 0 = п (г, 0 п (г + R, 0 g (r, R, 0; D.21)
здесь мы вместо (г' — г) написали R. Эта радиальная функция распределения
зависит макроскопически от г и микроскопически от R. Для R, больших по
сравнению с областью действия межмолекулярных сил, g(r, R, t) приближается
к единице. В статистической механике равновесных систем радиальная функция
распределения не зависит от г и t и сводится просто к g(R).
Выражение D.19) для нф(г, t) теперь может быть записано через радиаль-
ную функцию распределения неравновесных систем
D.22)
Поскольку плотность числа частиц п(г, /) зависит от г только макроскопически
и область действия межмолекулярных сил мала, то это выражение с небольшой
ошибкой может быть записано в виде
j D.23)
Другой функцией распределения, которая будет использована ниже,
является функция /<2), определяемая следующим образом:
P(r,r',0 = jj(p 0P')/B)(r,r',p,p',Odpdp'. D.24)
Эта функция представляет собой плотность вероятности импульса в конфигу-
рационном пространстве пар молекул, а (р © р') является шестикомпонентным
вектором, первые три компоненты которого равны р, а вторые три компоненты
равны р'.
Г. ВЕКТОРЫ ПЛОТНОСТИ ПОТОКОВ И УРАВНЕНИЯ
ПЕРЕНОСА
В гл. 7, § 2 были выведены три уравнения переноса для разреженных газов.
Аналогичные уравнения могут быть выведены для плотных газов путем под-
становки различных динамических переменных а в D.4). Единственное различие
в результатах состоит в том, что для векторов плотностей потоков получаются
более общие выражения. Вывод этих выражений очень длинен [51 ], и поэтому
мы здесь представим лишь результаты этих выводов.
Уравнение непрерывности может быть получено, если в D.4) принять
а = т 2 d(Tk-t). D.25)
Так как скорость изменения ожидаемого значения а является ожидаемым
значением динамической переменной
D.26)
то из определений плотности и средней массовой скорости [см. D.7) и D.9)]
следует
9г
Это уравнение совпадает с уравнением непрерывности, полученным в гл. 7, § 2*
514 Гл. Р. Явления переноса в плотных газах и жидкостях
Уравнение движения выводится аналогичным способом. Его можно полу-
чить, используя в качестве динамической переменной в общем уравнении пере-
носа переменную ее, определяемую следующим образом :
"=2fP*<4r*-r). D.28)
После сложных преобразований [51 ] получим то же уравнение движения,
которое мы вывели для разреженных газов, т. е.
) D.29)
Однако здесь тензор давлений состоит из двух членов
Р = Рк + Рф> D.30)
где
рк (г, 0 = т J (-?- - V,) (-?- - v0) /<*> (г, р) dp, D.31)
Рф(г,О = -4Hr>0]2J^g(r,R,0^r<iR. D.32)
Первый член рк учитывает влияние кинетической энергии хаотического дви-
жения молекул и является преобладающим членом в газах1). Второй член рф.
учитывает влияние потенциальной энергии межмолекулярного взаимодействия
и является преобладающим в жидкостях. Этот член описывает перенос импульса
столкновениями или обмен импульсами между центрами сталкивающихся
молекул, который происходит почти мгновенно в процессе столкновения2).
В условиях равновесия тензор давления р диагоналей, причем каждый диаго-
нальный элемент его равен
\^^ D.33>
Это соотношение является уравнением состояния, выведенным в гл. 3, § 1
на основе теоремы вириала.
Уравнение баланса энергии может быть получено из общего уравнения
переноса аналогичным способом. Результат имеет вид
). D.34>
Это уравнение совпадает с уравнением, полученным для разреженных газов,
за исключением того, что вектор плотности потока тепла q теперь имеет более
общий вид
Я = Як + Яф. D.35)
где
L$\\2() D.36).
т v0 (r, 0 лB> (г, г + R, 0] <*R ; D.37)
х) Член р^ совпадает с выражением для тензора давлений в разреженных газах, который
выражается в виде B.23) гл. 7 ; член I——vol равен тепловой скорости молекул, определя-
емой соотношением B.5) гл. 7.
2) Хотя аналогия и не очевидна, тем не менее эти члены и соответствующие члены в
потоке энергии являются обобщением соответствующих чле! оз, входящих в теорию Энскога
(СМ. §3).
§ 4. Рассмотрение явлений переноса в плотных газах и жидкостях 515
здесь U — единичный тензор, а Ц — первые три компоненты jB). Первый член
qK учитывает влияние кинетической энергии хаотического движения и явля-
ется преобладающим в газах1). Второй член цф учитывает влияние потенциаль-
ной энергии межмолекулярного взаимодействия, появляется вследствие пере-
носа энергии столкновениями и является преобладающим в жидкостях.
Д. ВЫЧИСЛЕНИЕ КОЭФФИЦИЕНТОВ ПЕРЕНОСА
Таким образом, мы убедились, что обычные уравнения переноса прило-
жимы к плотным газам и жидкостям и получили выражения для векторов
плотностей потоков через бинарную функцию распределения и интегралы от
нее. Эти выражения являются основными из тех, которые должны быть вычис-
лены, чтобы развить теорию явлений переноса в плотных газах и жидкостях.
Бинарная функция распределения может быть получена строго либо
путем решения уравнения Лиувилля, либо путем решения эквивалентного
ряда связанных уравнений для функций распределения низшего порядка
[см. (9.15) гл. 4]. Однако практически это полностью осуществить невозможно
и необходимо использовать некоторые приближенные методы, ограничивающие
последовательность уравнений. В этих приближенных методах используется
некоторый вид суперпозиционного приближения, обсужденного в гл. 4, § 9.
Борн и Грин [52, 53 ] использовали один вид суперпозиционного прибли-
жения, чтобы получить интегральное уравнение для радиальной функции рас-
пределения. Это уравнение было решено методом возмущений, применимым
в условиях, когда система только слегка удалена от состояния равновесия.
Решение далее использовалось для получения векторов плотностей потоков
и величин коэффициентов переноса для плотных газов и жидкостей. Хотя
формальное решение было получено, тем не менее полный численный расчет
коэффициентов переноса еще не был сделан. Согласно Борну и Грину, сравнение
с экспериментальными данными показывает, что результаты их расчетов дают
правильный порядок величин и имеют качественно правильную зависимость
от плотности и температуры.
Справедливость принципа суперпозиции анализировалась Клейном и
Пригожиным [58] путем рассмотрения гипотетического одномерного газа.
Они нашли, что результаты, основывающиеся на приближениях, не могут
описывать диссипативных эффектов.
Кирквуд [54] вычислил коэффициент трения без использования супер-
позиционного приближения. Решение Кирквуда методом возмущений приводит
к теории коэффициента трения, аналогичной той, которую развил Чандрасекар
[55 ] на основе феноменологической теории броуновского движения. Частицы,
движущиеся в жидкости, испытывают вязкое сопротивление. Если частицы
представляют собой большие сферы радиуса г, то жидкость воздейстьует на
них как непрерывная среда и постоянная трения при этом равна стоксовской
величине бтггуг. Кирквуд обобщил понятие коэффициента трения, чтобы исполь-
зовать его для мельчайших частиц или молекул. Отношения двух коэффициентов
вязкости к коэффициенту трения были выражены через интегралы от меж-
молекулярного потенциала и функции распределения равновесных систем.
Кирквуд, Бафф и Грин [56 ] применили эти результаты для вычисления отноше-
ния коэффициента сдвиговой вязкости к постоянной трения для жидкого аргона
при 89° К, используя потенциал Леннарда-Джонса и экспериментально най-
денную радиальную функцию распределения Эйзенштейна и Гингрича [57].
Теория постоянной трения вполне закончена, но пока еще невозможно вычи-
слить постоянную точно. Грубые оценки постоянной трения приводят к зна-
чениям сдвиговой вязкости аргона, находящимся в вполне приемлемом согла-
сии с экспериментом.
Член q^ совпадает с выражением B.26) гл. 7.
516 Гл. 9. Явления переноса в плотных газах и жидкостях
Приложение А
ТЕОРИЯ СКОРОСТЕЙ РЕАКЦИЙ ЭЙРЙНГА1)
Теория абсолютных скоростей реакций первоначально была создана
Эйрингом [59] с целью объяснения скоростей химических реакций. Однако
вскоре выяснилось, что эта теория с успехом может быть применена к изучению
скоростей многих других процессов. Приложение теории Эйринга к теории
явлений переноса в плотных газах и жидкостях обсуждено в § 2. Здесь будут
изложены основные идеи теории абсолютных скоростей реакций на основе
рассмотрения частного случая химической реакции.
Рассмотрим гипотетическую реакцию вида
А + ВС^АВ + С. (АЛ)
В соответствии с принципами химической кинетики скорость R реакции про-
порциональна концентрациям пА и пвс:
R = k'nAnBC. (A.2)
Величина /с' называется «удельной скоростью реакции».
Прежде в химической кинетике удельная скорость реакции обычно выра-
жалась в форме, которую предложил Аррениус,
tikT; (А.З)
здесь Е^ — энергия активации ; Г — число столкновений в единице объема
в единицу времени между молекулами типа А и молекулами типа ВС ; sF —
«частотный коэффициент». Произведение Г e~E+ikT равно числу столкновений
в единице объема в единицу времени между молекулами А и ВС, кинетическая
энергия которых, соответствующая компоненте относительной скорости, на-
правленной вдоль линии, соединяющей центры молекул при столкновении,
больше энергии активации. Числом s определяется вероятность того, что гео-
метрические и внутренние соотношения благоприятствуют реакции. Соотно-
шение Аррениуса неудобно потому, что величина s не может быть предсказана.
В некоторых случаях s очень мала по сравнению с единицей, и в некоторых —
очень велика по сравнению с единицей. Когда s больше единицы или когда
удельная скорость уменьшается с температурой, простое объяснение скорости
реакции на основе представлений о столкновениях непригодно и необходимо
более точное рассмотрение, основывающееся на статистической механике.
Рассмотрим подробно реакцию (А. 1) с микроскопической точки зрения.
Если предположить, что реакция между атомом и двухатомной молекулой
происходит таким образом, что А, В и С все время остаются на прямой линии2),
то за течением реакции можно проследить по значениям двух межатомных
расстояний гАВ и гвс. Перед столкновением между Л и ВС расстояние гАВ
велико, а расстояние гвс мало. Если в течение столкновения происходит хими-
ческая реакция, то после того, как продукты реакции удаляются друг от друга,
малым будет расстояние гАВ и большим гвс. Если происходит такая хими-
ческая реакция, то в течение столкновения образуется одно состояние, в кото-
!) См. [35, 36, 38, 59, 60 (гл. XVI)].
2) Предположение о том, что три атома в течение столкновения находятся на одной линии,
вводится для упрощения изложенного здесь обсуждения, поскольку оно устраняет необхо-
димость введения третьей динамической переменной — угла между Л, В и С. В реальной
химической реакции траектория наименьшей энергии часто соответствует расположению
атомов на одной линии. Отклонения от этой линии принимаются во внимание путем учета в
статистической сумме активированного комплекса связанных колебаний.
Приложение Л. Теория скоростей реакций Эйринга
517
ром все три атома очень близко расположены друг от друга. Об этой соединен-
ной молекуле обычно говорят, как о «комплексе столкновения». Обозначим
этот комплекс химическим символом (АВС)+ и перепишем реакцию (А. 1) в
виде
A + BC^(ABC)i -+АВ + С.
Мы записали первый этап процесса в виде обратимого процесса, отмечая этим
тот факт, что комплекс, будучи однажды образован, может затем развалиться
на составные части таким путем, что в результате образуются либо компоненты
А и ВС, либо компоненты АВ и С.
Траектория
реакции
Фиг. 147. Поверхность потенциальной
энергии для реакции А + ВС, проис-
ходящей в одном измерении.
Пунктирная линия — «траектория реакции»;
(s) и (а) — симметричные и антисимметричные
виды нормальных колебаний активированного
комплекса соответственно.
cs:
:з
со
3-
сз
нциальи
/
/
/
/
/
/
/
/
А+ВС/
т
(А1
\ -
Е*
Е
\
\
\
t
1
1
1
\
\
\
\
\
\
\
\ABiC /
п
Координата реакции
Фиг. 148. Потенциальная энергия как
функция расстояния вдоль «траектории
реакции».
Вершина этой кривой соответствует точке седла
на фиг. 147.
За течением реакции можно проследить на поверхности потенциальной
энергии вида, показанного1) на фиг. 147. Конфигурационное пространство может
быть разделено на три области ; верхний левый угол соответствует начальному
состоянию А+ВС) нижний правый угол соответствует конечному состоянию
АВ + С; полоса посередине соответствует «активированному состоянию)»,
в котором образуется комплекс столкновения. Пунктирной линией отмечена
траектория, по которой стал бы скатываться тяжелый шар, если его поместить
в наинизшую точку седла и предоставить ему возможность перемещаться вниз
по любой стороне энергетического холма. Эта траектория известна как «траек-
тория реакции». Эффективное сечение поверхности энергии вдоль траектории
реакции показано на фиг. 148.
Рассмотрим теперь расчет скорости реакции R и удельной скорости реак-
ции к'. Скорость реакции равна произведению трех сомножителей: 1) среднего
числа молекул в активированном состоянии п+; 2) частоты, с которой моле-
г) Необходимо отметить, что поверхности потенциальной энергии вида, показанного
на фиг. 147, имеют косоугольную систему координат и шкалы по осям абсцисс и ординат
не всегда совпадают. Это делается для упрощения описания динамики процессов столкно-
вения. В косоугольной системе координат градиент на поверхности энергии в любой точке
пропорционален силе, действующей на систему, и кинетическая энергия пропорциональна
сумме квадратов скоростей изменения этих новых координат. Траектория столкновения на
поверхности энергии совпадает с траекторией движения центра масс на этой поверхности
энергии [35, стр. 100].
518 Гл. 9. Явления переноса в плотных газах и жидкостях
кулы проходят через активированное состояние, т. е. средней скорости е>,
деленной на ширину барьера w; 3) «коэффициента прохождения» я, равного
вероятности того, что произойдет химическая реакция после достижения систе-
мой активированного состояния. Таким образом, можно записать выражение
для скорости реакции
D) (А.5)
Коэффициент прохождения неизвестен, но обычно полагается равным единице1).
Ширина области активированного состояния является наиболее плохо опре-
деляемой величиной, но в данном случае она исключается. Средняя скорость у,
с которой система пересекает активированное состояние2), равна |/ kT\2n m+
где ш+ — эффективная масса, которую имеет активированный комплекс на
траектории реакции.
Величина rv- может быть выражена через постоянную равновесия К+ ,
описывающую равновесие между исходными компонентами А и ВС и активи-
рованным комплексом (АВС)+ ,
^ 4-; (A.6)
пА пВС
здесь zA и zBC — статистические суммы для атома А и молекулы ВС; z+ — ста-
тистическая сумма для активированного комплекса. Чтобы найти z+, сначала
следует изучить поведение комплекса столкновения (АВС)+. Когда система
(s)
В
(а)
ABC
Фиг. 149. Симметричный и антисимметричный виды колебаний активированного комплекса.
находится в состоянии А + ВС, то она обладает двумя поступательными степе-
нями свободы и одной колебательной степенью свободы; то же самое справе-
дливо для случая, когда система находится в состоянии АВ + С. Когда сис-
тема находится в состоянии в виде ко мплекса (АВС)+, она обладает .одной
г) Многочисленные расчеты для различных гипотетических случаев [61,62] показывают,
что коэффициент прохождения имеет порядок единицы или лежит между 7г и 1 Для тех реак-
ций, которые ведут себя классически. Однако если реакция приводит к квантовомеханиче-
скому пересечению двух поверхностей потенциальной энергии, то коэффициент прохождения
может быть чрезвычайно малым — порядка Ю-6.
2) Скорость v определяется следующим образом:
00
Приложение А Ттеория скоростей реакций Эйринга 519
поступательной степенью свободы и двумя колебательными степенями свободы.
Нормальные типы колебаний показаны на фиг. 146. Двумя типами нормальных
колебаний являются симметричные колебания (s), перпендикулярные траек-
тории реакции, и антисимметричные колебания (а), происходящие вдоль траек-
тории реакции. Эти колебания изображены на фиг. 149. Симметричные коле-
бания являются подлинными колебаниями, антисимметричные же колебания
ло существу не являются колебаниями, так как потенциальная поверхность в
направлении этих «колебаний» почти плоская. Ввиду этого предполагается, что
такой тип «колебаний» может быть заменен поступательным типом движения
и что комплекс столкновения ограничен в своем движении одномерным ящиком
длины W. Физически это соответствует предположению о том, что вершина
барьера очень близка к плоскости1). Исходя из упомянутого предположения,
статистическую сумму г+ для активированного комплекса можно записать
в виде
(A.7)
здесь Z+' — статистическая сумма идеализированного активированного ком-
плекса, в котором антисимметричные колебательные степени свободы опущены;
у B
л m+kT/h2) w— статистическая сумма, соответствующая ограниченному
поступательному движению вдоль траектории реакции на вершине барьера.
Далее мы можем определить постоянную равновесия К
Это позволяет записать (А. 5) в виде
[) (А.9)
откуда получается постоянная удельной скорости реакции
' <АЛ0>
Универсальный множитель kTjh равен 6,25. Ю^Г/ЗОО) сек. Обычно К удобно
записать через свободную энергию активации AG+. При этом (АЛО) приобре-
тает вид
К = я [^ е-жг1*т = я (*Г| е-ий*-тА§]1«г- (А.П)
Это и есть формула Эйринга для постоянной удельной скорости реакции2).
х) Вигнер [63] получил поправки к значению скорости реакции для кривых энергети-
ческих поверхностей вблизи активированного состояния [35, стр. 191 ].
2) Когда течение процессов во времени обычно, т. е. когда квантовыми эффектами можно
пренебречь, х изменяется между г/2и 1. Однако экспериментально трудно отличить влияние
& от А§+. Поэтому для некоторых целей удобно определить эффективную энтропию акти-
вации /dS+эфф., которая учитывает влияние х, и переписать (А. 11) в виде
= (_кТ_\ e-AG^JRT = (кТ\
где
+ + + + +
а у ^Scaa = AS + R In x.
520 Гл. 9. Явления переноса в плотных газах и жидкостях
Полученные результаты позволяют интерпретировать частотный коэффи-
циент в формуле Аррениуса. В соответствии с формулами (А.З) и (А.11) этот
коэффициент s Г равен
sr = F,25.
Для большого числа реакций энтропия активации может быть оценена, исходя
из простых термодинамических соображений или на основе статистической
механики. Можно ожидать, что для одномолекулярных реакций энтропия
активации A 5f мала, так что частотный множитель может иметь значение
порядка Ю13. Для бимолекулярных реакций можно ожидать отрицательной
энтропии активации вследствие потери трех поступательных степеней свободы,
когда две реагирующие молекулы сталкиваются и образуют один активиро-
ванный комплекс. Это количественно подтверждается экспериментом. В неко-
торых активированных комплексах пропадают вращательные степени свободы,
которыми обладают молекулы перед образованием комплекса. Это приводит
к тому, что скорость реакции становится необычно медленной. В других акти-
вированных состояниях возбуждается вращение, которое было невозможно
в исходном состоянии. Это приводит к необычно быстрым реакциям. Реакция
между (C2H5JS и С2Н5Вг
(C2H6JS + С2НбВг -> (С2НбK SBr
случается в 100 раз чаще, чем это следует на основании расчетов частоты
столкновений между такими молекулами на основе кинетической теории. Это
может быть объяснено, исходя из того, что в активированном состоянии возбуж-
даются вращательные степени свободы. Реакция окисления окиси азота имеет
отрицательный температурный коэффициент вследствие того, что у этой реак-
ции отсутствует энергия активации и вращение возбуждается только в активи-
рованном состоянии. Такое объяснение подтверждается количественными рас-
четами для наблюдаемых аномальных результатов [64].
Теория абсолютных скоростей реакций может быть применена к хими-
ческим реакциям в жидкой фазе [65 ] в той же мере, как и для реакций в газо-
вой фазе. В этих приложениях числовые плотности заменяются активностями.
Выражение Эйринга для скорости реакции сводится к выражению Бронстеда,
и различные коэффициенты активности позволяют учесть влияние окружающей
среды на удельную скорость реакции. Для обобщенной химической реакции,
имеющей следующий стехиометрический вид:
аА + ЬВ + ... 71 (аА - ЬВ - .. .)* -*Продукты, (А.13>
скорость реакции в плотных газах или в жидкой фазе равна
аАаВ- • • ,
\КпП*Апъв. (А. 14)
Здесь коэффициенты активности а% аьв, ... и а+ исходных молекул и активи-
рованного комплекса определены по отношению к разреженному газу, в кото-
ром коэффициенты активности равны единице. Удельная скорость реакции
fcras равна скорости данной реакции в разреженном газе. Независимо от порядка
реакции удельная скорость реакции в разреженных газах выражается по
(А.11).
Хотя соотношение (А.11) выведено для специального случая химической
реакции, тем не менее его можно применять к большому числу различных проб-
лем, связанных с изучением скоростей процессов, в которых система проходит
через какой-нибудь вид активированного состояния. В гл. 9, § 2 мы обсудили
приложения этой теории к явлениям переноса в плотных газах и жидкостях»
Литература 521
Задачи
1. Используя табл. 97 и закон соответственных состояний, вычислить вязкость аргона
как функцию давления при постоянной температуре.
2. Используя закон соответственных состояний, оценить вязкость и теплопроводность
N2 при 60 атм и 100°К.
3. Используя теорию Эйринга явлений переноса, оценить вязкость жидкого N2 в нор-
мальной точке кипения.
4. Объяснить, почему вязкость газа возрастает с ростом температуры, а вязкость жид-
кости уменьшается с ростом температуры.
ЛИТЕР АТУРА
1. Р а г t i n g t о n J. R., Advanced Treatise on Physical Chemistry, New York, 1950.
2. Comings E. W., May land B. J., Egly R. S., The Viscosity of Gases at High
Pressures, Engineering Experimental Station Bulletin Ser. 354, Vol. 42, No. 15, Uni-
versity of Illinois, November 28 A944).
3. Karim S. M., Rosen head L., Rev. Mod. Phys., 24, 108 A952).
4. L i e b e r m a n n F., Phys. Rev., 73, 537 (A) A948).
5. Lie be r man n F., Journ. Acoust. Soc. Am., 20, 868 A948).
6. Liebermann F., Phys. Rev., 75, 1415 A949).
7. N e w i 11 D. M., The Design of High Pressure Plant and the Properties of Fluids at High
Pressures, Oxford, 1940.
8. Hougen O. A., Watson К. М., Chemical Process Principles, New York, 1947.
9. Maxwell J. В., Data Book on Hydrocarbons, New York, 1951.
10. Michels A., Gibson A. O., Proc. Roy. Soc, A134, 288 A931).
11. Phillips P., Proc. Roy. Soc, A87, 48 A912).
12. Fran ck E. U., Chemie-Ingenieur Technik, 25, 238 A953).
13. Боровик Е., Journ. of Phys. (СССР), 11, 149 A947).
14. S e 11 s с h о р р, Forsch. Gebiete Ingenieurw., 5, 162 A934).
15. U h 1 i r A., Jr., Journ. Chem. Phys., 20, 463 A952).
16. L a m b e r t J. D., S t a i n e s E. N., Woods S. D., Proc Roy. Soc. (London), A200,
262 A950).
17. T i m m e r h a u s K. D., D r i с k a m e r H. G., Journ. Chem. Phys., 19, 1242 A951),
18. Tim me rha us K. D., Drickamer H. G., Journ. Chem. Phys., 20, 981 A952).
19. R о b b W. L., Drickamer H. G., Journ. Chem. Phys., 19, 1504 A951).
20. К о e 11 e r R. C, Drickamer H. G., Journ. Chem. Phys., 21, 267 A953).
21. Кое Her R. C, Drickamer H. G., Journ. Chem. Phys., 21, 575 A953).
22. С u d d e b а с k R. В., К о е 11 e r R. C, Drickamer H. G., Journ. Chem. Phys.*
21, 589 A953).
23. Cud de back R. В., Drickamer H. G., Journ. Chem. Phys., 21, 597 A953).
24. Becker E. W., Journ. Chem. Phys., 19, 131 A951).
25. В е с k e r E. W., Zs. Naturforsch., 5a, 457 A950).
26. Drickamer H. G., O'Brien V. J., В res ее J. С, Ockert С. Е., Journ.
Chem. Phys., 16, 122 A948).
27. D r i с k a m e г H. G., Mellow E. W., Tung L. H., Journ. Chem. Phys., 18, 945
A950).
28. Tung L. H., Drickamer H. G., Journ. Chem. Phys., 18, 1031 A950).
29. Pierce N. C, Duffield R. В., Drickamer H. G., Journ. Chem. Phys., 18,
950 A950).
30. G i 11 e г Е. В., D u f f i e 1 d R. В., Drickamer H. G., Journ. Chem. Phys., 18„
1027 A950).
31. Bridgman P. W., Dimensional Analysis, Yale, 2nd ed., 1931. (см. перевод:
БриджменП., Анализ размерностей, М.-Л., 1934).
32. Perry J. H., Chemical Engineer's Handbook, New York, 3rd ed., 1950.
33. Kamerlingh Onnes H., Verhand. Akad. Amsterdam, 21, 2 Stuk, 8 A881).
34. Rowlinson J. S., Town ley J. R., Trans. Farad. Soc, 49, 1 A953).
322 Гл. 9. Явления переноса в плотных газах и жидкостях
35. Glass tone S., Lai die г К. J., Eyring H., Theory of Rate Processes, 1941
(см. перевод: Глесстон С, Лейдлер К., Эйринг Г., Теория абсолютных
скоростей реакций, ИЛ, 1948).
36. Eyring H., Journ. Chem. Phys., 4, 283 A936).
37. Powell R. E., Roseveare W. E., Eyring H., Ind. and Eng. Chem., 33, 430
A941).
38. Kincaid J. F., Eyring H., Stearn A. E., Chem. Rev., 28, 301 A941).
39. R e i n e r M., Deformation and Flow, London, 1949.
40. P h i 1 i p p о f f W., Viskositat der Kolloide, Berlin, 1942.
41. H i r sch f e 1 d e r J. O., Stevenson D., Eyring H., Journ. Chem. Phys., 5,
896 A937).
42. Frisch D., Eyring H., Kincaid J., Journ. Appl. Phys., 11, 75 A940).
43. Moore R. J., Gibbs P., Eyring H., Journ. Phys. Chem., 57, 172 A953).
44. Bridgman P. W., Proc. Am. Acad. Arts Sci., 59, 109 A923).
45. Bridgman P. W., The Physics of High Pressures, London, 1931.
46; Enskog D., Kungliga Svenska Vetenskapsakademiens Handlingar, 63, No. 4 A922).
47. Chapman S., Cowling T. G., The Mathematical Theory of Non-uniform Gases,
Cambridge, 1939 (см. перевод: Чепмен С, Каулинг Т., Математическая тео-
рия неоднородных газов, ИЛ, I960).
48. К i r k w о о d J. G., Maun E. К., Alder В. J., Journ. Chem. Phys., 18, 1040 A950).
49. McLellan A. G., Proc. Roy. Soc. (London), A210, 509 A952).
50. Warburg E., v. Babo L., Wied. Ann., 17, 390 A882).
51. Irving J. R, К irk wood J. G., Journ. Chem. Phys., 18, 817 A950).
52. Born M., Green H. S., A General Kinetic Theory of Liquids, Cambridge, 1949.
53. Green H. S., Molecular Theory of Fluids, New York, 1952.
54. К i г к w о о d J. G., Journ. Chem. Phys., 14, 180 A946).
55. Chandrasekhar S., Rev. Mod. Phys., 15, 1 A945).
56. К i г к w о о d J. G., В u f f F. P., GreenM.S, Journ. Chem. Phys., 17, 988 A949).
57. Eisenstein A., Gingrich N. S., Phys. Rev., 62, 261 A942).
58. Klein G., Prigogine I., Physica, 19 A953).
-59. Eyring H., Journ. Chem. Phys., 3, 107 A935).
60. Eyring H., Walter J., Kim ball G. E., Quantum Chemistry, New York, 1944
(см. перевод: Эйринг Г., Уолтер Д., Кимбалл Д., Квантовая химия, ИЛ,
1948).
61. Hirschfelder J. О., Wigner E., Journ. Chem. Phys., 7, 616 A939).
62. H u 1 b u r t H. M., H i r s с h f e 1 d e r J. O., Journ. Chem. Phys., 11, 276 A943).
63. Wigner E. P., Zs. phys. Chem., 1319, 903 A932).
64. G e r s h i n о w i t z H., Eyring H., Journ. Am. Chem. Soc, 57, 985 A935).
«65. W у n n e - J о n e s W. F. K., Eyring H., Journ. Chem. Phys., 3, 107 A935).
«66. Uyehara, Watson, National Petroleum News, Technical Section 36, 764 (Oct. 4,
1944).
ГЛАВА Ю1)
КВАНТОВАЯ ТЕОРИЯ И ЯВЛЕНИЯ ПЕРЕНОСА
Три предыдущие главы были посвящены классической теории переноса
в плотных и разреженных газах. В этой главе будут рассмотрены те изменения
в теории переноса, к которым приводит квантовая механика.
В первом параграфе дана общая формулировка теории переноса на основе
квантовой статистической механики неравновесных систем. Показано, что
лолучаемые при этом результаты формально аналогичны результатам класси-
ческой теории. Далее получено уравнение Больцмана, соответствующее кван-
товомеханическому описанию разреженных газов. Это уравнение отличается
от соответствующего классического уравнения тем, что принимаются во вни-
мание квантовые эффекты, вызванные волновой природой молекул (диффрак-
ционные эффекты), а также статистикой частиц (эффекты симметрии). Диф-
фракционные эффекты существенны в условиях, когда дебройлевская длина
волны молекул — порядка размера молекул. Эффекты симметрии становятся
важными в условиях, когда дебройлевская длина волны молекул — порядка
среднего расстояния между молекулами в газе.
Квантовая теория явлений переноса в разреженных газах может быть
развита двумя методами. При «очень низких» температурах, когда квантовые
эффекты играют первостепенную роль, коэффициенты переноса могут быть
выражены посредством «фазовых сдвигов». При «промежуточных температурах»,
когда система мало отличается от классической, фазовые сдвиги можно полу-
чить методом ВКБ. В этом случае коэффициенты переноса получаются в виде
ряда по степеням постоянной Планка. Эти два подхода, которые обсуждены в
§ 2 и 3, аналогичны двум квантовомеханическим формулировкам второго вири-
ального коэффициента (см. гл. 6, § 4 и 5).
В настоящее время очень мало сделано в квантовой теории переноса плот-
ных газов и жидкостей. Некоторые сведения можно получить, анализируя
экспериментальные данные с позиций закона соответственных состояний в
квантовой механике. Этот вопрос вкратце рассматривается в последнем пара-
графе.
§1. КВАНТОВАЯ СТАТИСТИЧЕСКАЯ МЕХАНИКА
НЕРАВНОВЕСНЫХ СИСТЕМ
В гл. 9, § 4 изложена классическая статистическая механика плотных
газов и жидкостей. Здесь будет указано, как можно обобщить эту теорию на
случай жидкости, в которой существенны квантовые эффекты. Кроме того,
будет показано, что обычные уравнения гидродинамики могут быть получены
из квантовомеханического уравнения Лиувилля2). Эти результаты справедливы
для плотных газов и жидкостей. В предельном случае малых плотностей при
таком подходе можно получить квантовомеханическое уравнение Больцмана.
х) Глава написана Дж. де Буром и Р. Байроном.
2) Задача о выводе уравнений гидродинамики для квантовомеханической системы рас-
смотрена в работах К. П. Гурова [ЖЭТФ, 18, ПО A948); 20, 279 A950)]. — Прим. ред
524 Гл. 10. Квантовая теория и явления переноса
А. ОБЩИЕ ПОЛОЖЕНИЯ СТАТИСТИЧЕСКОЙ МЕХАНИКИ1)
Классическая статистическая механика основана на концепции ансамблей
и функции распределения2) в фазовом пространстве р**\гы, pNy f) (см. гл. 2Г
§ 1). Скорость изменения со временем этой функции распределения определяется
уравнением Лиувилля.Функции распределения низших порядков определяются
как интегралы от fN\tNy pN, t), и скорость изменения со временем этих функций
может быть получена соответствующим интегрированием уравнения Лиувилля*
Среднее значение физической величины X(tNy рм) получается путем
интегрирования по всему фазовому пространству функции Х(г^, р^), умно-
женной на функцию распределения. Эти обозначения применялись при
изложении классической теории явлений переноса в плотных газах и жид-
костях (см. гл. 9, § 4).
При формулировке квантовой статистической механики концепция функ-
ции распределения не может быть непосредственно перенесена из классической
теории, поскольку в соответствии с принципом неопределенности Гейзенберга
невозможно точно определить положение системы в фазовом пространстве.
Существуют два различных способа определения функции распределения в
квантовой теории. Один из них был отмечен в гл. 2, § 2, где показано, что при
матричной формулировке квантовой механики возможно построить «матрицу
плотности вероятности», которую можно интерпретировать как аналог класси-
ческой функции распределения в фазовом пространстве. Временная зависи-
мость матрицы плотности определяется "квантовомеханическим аналогом урав-
нения Лиувилля. Матрицы плотности низшего порядка определяются как неко-
торые интегралы от первоначальной матрицы плотности. Среднее значение физи-
ческой величины X(tM, pN) определяется следом матрицы, которая является
произведением матрицы плотности и матрицы, соответствующей физической
величине Х(тм9 pN). Эта формулировка квантовой статистической механики
применялась в гл. 6, где излагалась квантовая теория уравнений состояния.
Существует второй подход при формулировке квантовой статистической
механики, щ который был предложен Вигнером [2 ]. В этом методе строится
«функция распределения» ^ы\гы9 ры, f), которая не имеет простой интерпрета-
ции в терминах концепции вероятностей, но может быть непосредственно
применена для вычисления средних величин по формулам, совершенно анало-
гичным классическим формулам для средних величин. Этот метод и будет
положен в основу при дальнейшем изложении.
Функция распределения Вигнера может быть определена с помощью
матрицы плотности в координатном или импульсном представлении3):
ы} t) = -^ J«*w <**«*> jrw (г" - 4" г'">fN + 4" г'">') dt'N =
)
p'"- A-1)
*)См. [1].
2) Различные функции распределения приведены в гл. 2. При изложении статистической
механики равновесных систем удобно применять функцию распределения P(W)(r^ pNy /^
определенную в гл. 2 [формула A.1)]. При изложении кинетической теория многие авторы
предпочитают применять функцию распределения в виде /(аг)(глг, pw, t), которая определена в
формуле A.5) гл. 2. Последняя имеет то преимущество, что функция наинизшего порядка
/<х)(г, р, t) как раз является обычной (унарной) функцией распределения, применяемой в кине-
тической теории разреженных газов.
8) Матрица плотности c/fXiV) (r^, r/jV, t) определена формулой B.24) гл. 2 и отличается от
матрицы «#W(rN,r/N, t) множителем N1 Такое же различие существует и между функциями
/(N)(rW, pN, t) и P(N) (nN), pN, t) в классической статистической механике.
Величина of"(N)(pN, p/N, t) есть *лГ-матрица в импульсном представлении. Она связана с
)(ты, r/N, t), поскольку преобразование Фурье устанавливает зависимость между коорди-
натным и импульсным представлениями волновой функции. В этом пункте заключено основ-
ное различие между квантовой и классической формулировками статистической механики.
В последнем случае плотности вероятности в конфигурационном пространстве и простран-
стве импульсов являются независимыми, поэтому классическая функция распределения
для канонического ансамбля может быть записана в виде P(N)(rN, pN) = RN)(rN) P(N)(pN).
§ 7. Квантовая статистическая механика неравновесных систем 525
Функция распределения ^{ы\гы, pN, t) всюду вещественна, хотя и не
обязательно положительна. Интегрирование этой функции по всему простран-
ству импуЛьсов дает диагональные элементы матрицы плотности в конфигура-
ционном пространстве. Точно так же интегрирование по всему координатному
пространству дает диагональные элементы матрицы плотности в представлении
импульсов
J = Jfto) (fNf tNf t) , A.2)
J $Пы) (Г
= JfiN) (pMy pNy f). A#3)
Особое преимущество функции распределения Вигнера заключается в том,
что вычисление средних значений величин происходит точно так же, как и в
классической статистической механике, т. е. прямым интегрированием по всему
фазовому пространству
^ A.4)
Таким образом можно избежать техники вычислений, описанной в гл. 2, § 2.
Имея в виду выражение для средних значений [формула A.4)], можно
перенести многие результаты классической статистической механики в кван-
товую статистическую механику простой заменой fM)(rN,pNyf) на ^ы)(гм,ры, t).
В частности, можно перенести в квантовую механику выражения векторов
плотности потоков и уравнения переноса, выведенные в гл. 9, § 4. Таким путем
устанавливается, что обычные уравнения гидродинамики применимы в кван-
товой теории столь же хорошо, как и в классической [1 ].
Б. УРАВНЕНИЕ БОЛЬЦМАНА ДЛЯ РАЗРЕЖЕННОЙ СМЕСИ
ГАЗОВ
В гл. 7, § 1 было показано, что классическое уравнение Больцмана для
разреженных газов может быть получено из уравнения Лиувилля для функции
распределения fN)(tM, ры, t). Аналогично этому квантовомеханическое урав-
нение Больцмана для разреженных газов можно получить из дифференциаль-
ного уравнения для функции распределения Вигнера ^(м\г^у р^, t). Таким
образом, можно показать, что уравнение Больцмана для обычной (унарной)
функции распределения /,(г, v,, t) /-го вещества r-компонентной газовой смеси
в квантовой механике имеет следующий вид1):
= 2лДJJ[/;/;O +e,/,)(i +0//,)-Ш1 +0,/',)(i +еМ х
; A.5)
здесь 0, = (ft//n/KE/Gz), где Q( — статистический вес i-й молекулы, а
й = —1,0,1 соответственно для статистик Ферми—Дирака, Больцмана и
Бозе—Эйнштейна. Величины gtj и % — соответственно начальная относитель-
ная скорость и угол отклонения траектории двух взаимодействующих частиц ;
a(giJf%) — сечение рассеяния, определенное в гл. 1, § 7. В левой части уравне-
ния Больцмана «гидродинамические члены» такие же, как и в классической
теории2), однако интеграл столкновений в правой части отличается от соответ-
ствующего классического выражения в двух отношениях : 1) заменой gtj bdb
на a(gijy x) sin %d% вследствие диффракционных эффектов; 2) введением множи-
х) Уравнение Больцмана в такой форме впервые было получено Улингом и Уленбеком.
[4], которые построили первую полную квантовую теорию явлений переноса.
2) Доказательство этого утверждения содержится в работе Нордгейма и Кикучи [5].
526 Гл. 10. Квантовая теория и явления переноса
теля A + 0,/,), учитывающего «эффект симметрии». Обсудим коротко различия
между классической и квантовомеханической формулировками уравнения
Больцмана.
Первое отличие возникает вследствие существенно волновой природы моле-
кул. В классической теории столкновений угол отклонения % можно определить
точно при любом законе взаимодействия. Число столкновений молекул в еди-
нице объема за единицу времени, скорости которых vh Vj и Ь лежат в области
dvh d\j и db, можно Еыразить следующим образом :
fifJguBnbdb)dvidvJ. A.6)
С другой стороны, в волновой механике в соответствии с принципом
неопределенности Гейзенберга невозможно одновременно определить поло-
жение и скорость молекулы. Следовательно, невозможно определить точно и
угол отклонения % после столкновения двух молекул. Рассмотрим, однако,
поток молекул массы ть скорости v, и плотности числа частиц пь который
взаимодействует с потоком молекул массы mjf скорости v7 и плотности числа
частиц rij. В этом случае можно определить вероятное число столкновений моле-
кул в единице объема за единицу времени, относительные скорости которых
после столкновенияg'tj лежат в телесном угле dw = 2^sin%rf% около поляр-
ной оси gtj. Совершенно ясно, что это вероятное число столкновений пропор-
ционально плотности числа частиц в обоих пучках, а также множителю, завися-
щему от % и ?/;• Следовательно, вероятное число столкновений равно
ninj a(Su> x)d w- Этим выражением определяется величина a(gijy %). Пусть
теперь в первом потоке скорости лежат в интервале dv( около \h а во втором — в
dVj около Vj. Тогда число столкновений в единице объема за единицу времени,,
подсчитанное по формулам квантовой механики, равно
// fj«(ёФ *) Bл sin х d%) dvt dVj. A.7)
Следовательно, заменяя gtfidb на a(gijy x)sm%dx B классических выражениях
гл. 7, можно изменить их так, чтобы они учитывали волновую природу молекул.
Примеры квантовомеханических расчетов a(gijy x) даны в гл. 1, § 7.
Второе отличие состоит во введении множителей A + 0,/,), которые появля-
ются исключительно вследствие «статистики» молекул. Рассмотрим однокомпо-
нентный газ, заключенный в объеме V, молекулы или частицы которого под-
чиняются статистике Ферми—Дирака (справедлив принцип Паули). В этом
случае известно, что в области скоростей dv не может находиться больше чем
VG(mltiK dv частиц ; G — «статистический вес» частицы, равный, по определе-
нию, числу независимых квантовых состояний, в которых может находиться
частица с той же самой внутренней энергией. Например, для электрона G = 2,
так как есть два различных спиновых состояния. Если Vf(v) dv молекул имеют
скорости, лежащие в области dv около V, то вероятность столкновений, в резуль-
тате которых в этой области скоростей появляются дополнительные молекулы,
уменьшится в отношении |l—/(v)(—] /GJ. Этот результат связан с кон-
цепцией «кажущегося отталкивания» между частицами Ферми—Дирака, о кото-
рой говорилось в гл. 6, § 3. Последнее утверждение эквивалентно положению,
согласно которому, если в области скоростей dv находится VG(mlhK dv частиц,
то все другие столкновения, переводящие частицы в эту же область, просто
не будут происходить. Для частиц, подчиняющихся статистике Бозе—Эйн-
штейна, необходимо включить множитель Tl +f(v){-m~f/G]> соответствующий
«кажущемуся притяжению» между молекулами. В статистике Больцмана раз-
решены все типы столкновений, поэтому в( = 0.
Если квантовые поправки, вызванные эффектами статистики, становятся
существенными, то о газе говорят как о «вырожденном». Такая терминология
применялась и при изложении квантовой теории уравнений состояния. Для
определения условий, при которых учет эффектов симметрии становится необ-
§ 2. Явления переноса при очень низких температурах 527
ходимым, нужно учесть, что число частиц в единице объема в области скоростей
dv не может превосходить G(m/ftK dv. В случае максвелловского распределения
скоростей число частиц в единице обьема равно
причем наибольшая плотность достигается при v = 0, где fdv = п (-о^г)
Тогда при нормальных условиях
и заселенность уровней настолько мала, что вырождение не появляется.
В противоположном предельном случае больших плотностей или низких тем-
ператур
bSf(^ A.10)
плотность частиц много больше плотности состояний. В этом случае вырож-
дение, связанное со статистиками Ферми—Дирака или Бозе—Эйнштейна, при-
водит к отклонению от максвелловского распределения. Следуя Зоммерфельду
[б]1), введем такой «параметр вырождения» A3n/G, что
№n\G <^ 1 соответствует слабому вырождению,
а
А3 п/О 1 соответствует сильному вырождению.
Здесь А2 = Щ2пткТ.
Из рассмотренного видно, что вырождение будет наибольшим для легких
частиц или молекул при больших плотностях и низких температурах. Это
отчетливо проявляется в случае электронного газа. Однако даже для самых
легких атомных или молекулярных газов этим эффектом можно пренебречь,
исключая случай очень низких температур, например для Не — около 1° К-
Таким образом, можно сделать вывод, что наиболее важными квантовыми
эффектами являются те, в которых проявляется волновая природа молекул,
т. е. диффракционные эффекты.
Прежде чем обсуждать подробно эти два эффекта, подчеркнем основную
разницу между ними. Диффракционные эффекты начинают играть важную
роль при таких достаточно низких температурах, когда дебройлевская длина
волны — порядка размеров молекул. Чтобы эффекты симметрии стали сущест-
венны, кроме очень низкой температуры, необходима еще и очень высокая
плотность, при которой дебройлевская длина волны стала бы порядка величины
среднего расстояния между молекулами в газе. Следовательно, величина эффек-
тов симметрии в основном зависит от плотности, тогда как диффракционные
эффекты от плотности не зависят.
§2. ЯВЛЕНИЯ ПЕРЕНОСА ПРИ ОЧЕНЬ НИЗКИХ
ТЕМПЕРАТУРАХ
Квантовомеханическое уравнение Больцмана [формула A.5)], так же как
и классическое уравнение гл. 7, может быть рассмотрено с разных точек зрения.
Можно показать, не решая уравнения Больцмана, что гидродинамические
уравнения переноса получаются из уравнения A.5). Решение уравнения,
необходимое для получения явных выражений потоков и коэффициентов пере-
носа, можно получить таким же путем, как это сделано в гл. 7 для класси-
См. также гл. 2, § 5.
528 Гл. 10. Квантовая теория и явления переноса
ческой теории. В связи с этим в дальнейшем будут приведены только конечные
результаты.
Сначала рассмотрим результаты, полученные при обсуждении только
диффракционных эффектов. Затем будет показано, как можно обобщить эти
результаты, чтобы учесть эффекты симметрии. В заключение будут суммиро-
ваны результаты различных квантовомеханических расчетов, сделанных в
случае низких температур.
А. ДИФФРАКЦИОННЫЕ ЭФФЕКТЫ
Мы подчеркивали, что диффракционные эффекты можно учесть, заменив
gbdb в классических выражениях величиной a(g, %) sin % d х- В соответствии
¦с этим выражение для сечения Q(n)(g), даваемое формулой B.2) гл. 8 в класси-
ческой теории, в квантовой механике принимает вид
B.1)
Величина a(g, %) представляет собой вероятность отклонения на угол %. Для
этой величины с помощью квантовой теории'столкновений можно получить
явные выражения в случае статистик Больцмана, Бозе—Эйнштейна и Ферми—
Дирака [см. формулы G.26)—G.28) гл. 1 ].
Исследуем сначала выражения для Q(l) и QB) в случае статистики Больц-
мана.
Квантовомеханическое сечение в случае статистики Больцмана после
подстановки a{g,%) в формулу B.1) и проведения длинных алгебраических
лреобразований принимает вид1)
/=0,1,2,...
2) .;n2/
0B) _ B*} $ ('+!)(/ + 2) .
Q ~Wdf (/ + 3/2) s
Здесь I и х — квантовые числа, характеризующие момент количества движения
и относительную кинетическую энергию при парных столкновениях, а rjfa) —
фазовый сдвиг в радиальных волновых функциях при парных столкновениях
{см. гл. 1, § 7). Интересно сравнить эти результаты с аналогичными формулами
классической кинетической теории, которые имеют вид
B.3)
При высоких температурах отклонения от классической теории пренебре-
жимо малы, поэтому квантовомеханические выражения переходят в класси-
ческие с учетом соответствия
), B.4)
х) Это те же формулы, которые приводятся в обычных учебниках по квантовой теории
явлений переноса, за исключением того, что они обычно пишутся в более сложной форме.
Приведенная здесь сокращенная запись впервые была предложена профессором Крамерсом
из Лейденского университета. Эти формулы проще с точки зрения вычислений, и в них очень
хорошо проявляется аналогия с классическими формулами.
§ 2. Явления переноса при очень низких температурах 529
где ii — приведенная масса сталкивающейся пары молекул. Это соответствие
подробно обсуждается в гл. 10, § 3.
В гл. 8 было показано, что первостепенной задачей, возникающей при
расчетах явлений переноса, является определение угла отклонения #(&, g)
как функции параметра столкновения Ь и начальной относительной скорости g.
Другими словами, необходимо найти угол отклонения как функцию момента
количества движения pbg и относительной кинетической энергии %/*g2.
Аналогично этому в квантовой теории явлений переноса основной проблемой
является определение фазового сдвига ^/(и), который получается из решений
радиального волнового уравнения для сталкивающихся частиц, как функция
квантовых чисел I и к или функция момента количества движения Aj/7(/ + 1)
и относительной кинетической энергии Л2 х2/2 /л. В классической теории сечение
получается путем интегрирования по всем Ь, аналогично этому в квантовых
формулах производится суммирование по всем I. Как в классической, так и в
кв.штовой теориях интегралы Q(n& находятся из выражения
**L Qln,t) = J ?-v y2t+3 qw dyt B.5)
0
В классической теории у2 = jLtg2l2kT, в квантовой теории у2 = Л2 я2/2 /г кТ.
Если с помощью классического или квантовомеханического сечения вычислено
?Кл»'>, то коэффициенты переноса можно найти по формулам, приведенным в
гл. 8, § 2.
Таким образом удается выразить коэффициенты переноса, а следова-
тельно, и свойства самого переноса через фазовые сдвиги. Методы вычисле-
ний фазового сдвига описаны в гл. 6, § 4 в связи с расчетом второго вири-
ального коэффициента при очень низких температурах. Непосредственное
вычисление коэффициентов переноса при низких температурах проведено в
разд. В.
Б. ЭФФЕКТЫ СИММЕТРИИ
До сих пор все рассуждения ограничивались статистикой Больцмана.
Если считать частицы неразличимыми и принимать во внимание симметрию
волновых функций, то a(g, %) необходимо находить по формулам G.27) и
G.28) гл. 1, выведенным соответственно для случая статистик Бозе—Эйнштейна
и Ферми—Дирака. Эффективные сечения в случае статистики Бозе—Эйн-
штейна имеют вид
= 4 (-^Л jj? B1 + 1) sin2 rji,
B.6)
"Ь 1) (/ + 2) .
.— sin2 (rji «|_2 — "Пи у
а в случае статистики Ферми—Дирака
B.7)
i
I—1,3,э, .«.
Эти выражения справедливы для частиц со спином, равным нулю. Для частиц
530 Гл. 10. Квантовая теория и явления переноса
со спином s ф 0 сечения имеют вид
) Ы) . B-8)
• B-9)
Величины Q$3 и ф^д. без индекса s представляют сечения для частиц со
спином, равным нулю, и вычисляются по формулам B.6) и B.7). Формула B.9)
применяется для вычисления сечения и коэффициентов переноса Не3, который
содержит нечетное число частиц и имеет спин s = г/2. Для вычисления коэффи-
циентов переноса выражения B.6)—B.9) можно непосредственно подставить
в формулу B.5) и получить интегралы Q(n>fy>.
Второй эффект, который необходимо рассмотреть после того, как принята
во внимание симметризация волновых функций, заключается в определении
влияния члена 6/, включенного в квантовомеханическое уравнение Больц-
мана. Эта поправка, которая, как часто говорят, присуща «эффекту симметрии»,
рассматривается ниже.
Основная проблема в теории явлений переноса, как было показано в гл. 7,
заключается в решении ряда интегральных уравнений для функций At(Wi),
utiWi), и CftWt), которые дают соответственно коэффициенты теплопроводности,
вязкости и диффузии. Простая замена gb db на a (g, %) s\n%d%y какмы видим, поз-
воляет сейчас же получить правильные квантовомеханические выражения для
коэффициентов переноса, которые учитывают диффракционные эффекты и пер-
вый эффект «статистики», указанный выше. Для получения коэффициентов
переноса в квантовой механике, учитывающих как «диффракционные» эффекты,
так и эффекты «симметрии», необходимо заново решить ряд указанных выше
интегральных уравнений, учитывающих множитель A + 0,/,). Уравнения эти
решены и формальные результаты можно найти всюду как для чистых газов [4 ],
так и для газовых смесей [9 ].
Для вязкости и теплопроводности чистых газов приводятся следующие
явные выражения [7, 8], справедливые во втором приближении при решении
интегральных уравнений :
SS^[ ^\ BЛ1)
Знак плюс соответствует статистике Бозе—Эйнштейна, а знак минус —
статистике Ферми—Дирака. Величины №2>2\ /?2) и /(jp должны вычисляться
квантовомеханическим путем с помощью сечений по формулам B.6)—B.9).
Множители /(|> и /(|> очень близки к единице1) и представляют следующие высшие
члены в разложении по полиномам Сонина в гл. 7, но не высшие члены в методе
решения уравнения Больцмана, развитом Энскогом, которые включают произ-
водные высших порядков от градиентов температуры, концентрации и давления.
Уравнения B.10) и B.11) без скобок представляют собой квантовомехани-
ческие выражения для коэффициентов переноса, учитывающие, конечно, сим-
метризацию эффективного сечения, как указано выше в формулах B.6) и B.7)-
Наличие скобок указывает на «эффект симметрии», вызванный корректировкой
1)Эти множители определены в классической теории в формулах B.19) и B.32) гл. 8-
Для потенциала отталкивания, обратно пропорционального двенадцатой степени расстоя-
ния, /^ и fty близки к 1,016 и 1,025 соответственно при Г, близком к абсолютному нулю.
Расчеты этих величин для потенциала F-12) Леннарда-Джонса показывают, что поправка
существенна лишь при очень низких температурах [103].
§ 2. Явления переноса при очень низких температурах 531
0/, рассмотренной здесь. Функции 6(^ и йф, появляющиеся в первом прибли-
жении, имеют вид
7 - J* r(>3) + 6rB>2)] Г2 13)
/ 3з/2 9^B|2) j. </л^
Величины Х<п>'> представляют собой интегралы flC11»'), определенные формулой
B.5), в которых ехр (—у2) заменено на ехр(—4/3у2). Члены 6B) и 6<f>, появляю-
щиеся во втором приближении, содержат QW> и Г<л»'> с п = 2, 4 и t = 2, 3, 4, 5.
Эти члены малы по сравнению с fity и <5(р и поэтому в дальнейшем не рассматри-
ваются.
Таким образом, включение множителя A + 0,/,) в уравнение Больцмана
приводит к появлению дополнительного вклада, зависящего от плотности и
вызванного эффектами симметрии. Непосредственное вычисление [9] этой
поправки для газа Не4 при температурах около 1° К показывает, что она такого
же порядка величины, как и поправка Энскога для плотных газов1), которая
приближенно учитывает зависимость коэффициентов переноса от плотности
в классической теории. Изменения величины коэффициента теплопроводности
такого порядка наблюдал Уббинк [10]. Однако в настоящее время невозможно
анализировать эти данные, поскольку неизвестно влияние одновременных
столкновений нескольких частиц на зависимость коэффициента переноса от
плотности.
Поскольку в этом разделе рассматривается лишь не зависящая от плотности
часть коэффициентов переноса, то этот второй эффект симметрии в дальнейшем
не будет приниматься во внимание. Однако эффекты статистики, влияющие на
размеры сечения, существенны уже при умеренно низких температурах, поэтому
они должны приниматься во внимание.
В. ВЫЧИСЛЕНИЯ ДЛЯ СЛУЧАЯ ОЧЕНЬ НИЗКИХ ТЕМПЕРАТУР
Вязкость гелия и водорода вычислена по формуле B.6) для модели твердых
сфер [7, 11 ], для которой все фазовые сдвиги можно найти в аналитическом
виде (см. гл. 6, § 4). Найдено, что в таком простом потенциальном поле квантово-
механические результаты значительно лучше согласуются с эксперименталь-
ными данными, чем расчеты, основанные на классической теории.
Вязкость Не4 рассчитана [12] на основе межатомного потенциала Сле-
тера2). Согласие между вычисленными и экспериментальными результатами не
особенно хорошее, хотя форма кривой вязкости как функция температуры в
общем соответствует действительности. Необходимо, однако, помнить, что
потенциал Слетера выведен из чисто теоретических соображений квантово-
механическим путем и не содержит постоянных, определяемых из эксперимента.
В связи с этим расчеты, основанные на этом потенциале, не должны считаться
неудовлетворительными. Результаты расчетов, «основанные на потенциале Сле-
тера и модели твердых сфер, сравниваются с соответствующими эксперимен-
тальными данными на фиг. 150.
Наиболее многочисленные расчеты параметров переноса при низких тем-
пературах были выполнены с потенциалом F-12) Леннарда-Джонса. Метод
расчета фазовых сдвигов для этого потенциала описан в гл. 6, § 4; из таблиц
фазовых сдвигов найдена вязкость изотопов гелия [12, 14] и изотопов водо-
рода [14]. В качестве иллюстрации рассмотрим результаты, полученные для
Не3 и Не4. Совсем недавно были вычислены коэффициенты самодиффузии для
изотопов гелия [16].
х) Теория Энскога о зависимости коэффициентов переноса от плотности для газов,
состоящих из твердых сферических молекул, изложена в гл. 9, § 3.
2)См. формулу B.13) гл. 14.
532
Гл. 10. Квантовая теория и явления переноса
Исходной точкой для квантовомеханического расчета параметров пере-
носа в плотных газах является решение радиального волнового уравнения
для пары взаимодействующих мо-
лекул. Это уравнение может быть
записано в приведенной форме с
помощью параметров потенциала
F-12) Леннарда-Джонса. Радиаль-
ное волновое уравнение в таком
случае имеет вид
2000-
dr*2
Л*2
где г* == г/а, a я* = и а.
B.14)
Из этого уравнения видно, что
характеристика вещества входит в
уравнение лишь через квантовоме-
ханический параметр Л* = ft/a f/л е.
Метод получения фазовых сдвигов
из предыдущего уравнения описан
в гл. б, § 4, где приводятся числен-
ные результаты. Величина кванто-
вомеханического параметра Л* для
различных веществ дана в табл. 57
(стр. 337).
После того как из радиального
уравнения найдены фазовые сдвиги,
можно рассчитать сечение Q(l) для
диффузии и QB) для вязкости и теп-
лопроводности. Для Не4, спин ко-
торого равен нулю, сечение Q^3
необходимо рассчитывать по формуле B.6). Для Не3, спин которого равен
г/2, необходимо найти как Q$Rm, так и ф^э. и применить формулу B.9) для
Фиг. 150. Сравнение вычисленных и экспе-
риментальных данных о вязкости для Не4.
а — экспериментальные данные ; Ь — вычисленные на
основе модели твердых сфер в классической теории;
с — вычисленные на основе модели твердых сфер в кван-
товой теории (Улинг); d — вычисленные с помощью
потенциала Слетера в квантовой теории. Цифровые дан-
ные взяты из работы [19].
Фиг. 151. Зависимость квантовомеханического эффективного сечения Q<?) от энергии.
Величина Q<«>* —эффективное сечение, деленное на соответствующую величину в случае модели твердых
сфер; х* = хаА где х — волновое число сталкивающихся частиц [16].
§ 2. Явления переноса при очень низких температурах
533
\ I I I I I I I I
па)+
Q классическое
I
0,02 0,05 0,1 0,2 0,5 1
10 20
Фиг. 152. Сравнение эффективных сечений QC1)* и QB)*, вычисленных
с помощью классической и квантовой теорий.
Квантовомеханические кривые рассчитаны на основе статистики Болыдмана.
150'
1/00
50
Не
вычисления сечения. Рассчитанные величины сечений для изотопов гелия
представлены на фиг. 151. Эта фигура хорошо иллюстрирует диффракцион-
ные эффекты, встречающиеся в явлениях переноса. На фиг. 152 сравниваются
классические кривые для QA) и Q'2) с
аналогичными квантовомеханическими
кривыми, рассчитанными с помоицю
статистики Больцмана для величины
Л*, относящейся к легкому изотопу ге-
лия. Из этой фигуры видно, что вычис-
ленное с помощью квантовой теории се-
чение столкновений с малыми энергиями
значительно меньше соответствующей ве-
личины, полученной с помощью класси-
ческой теории.
Значения вязкости, рассчитанные с
помощью сечения, показанного на
фиг. 151, приводятся в табл. 99 и изобра-
жены графически на фиг. 153. Как
видно, вязкость Не3 значительно больше
вязкости Не4. Более того, при очень
низких температурах вязкость тяжелого
изотопа пропорциональна температуре в
степени 3/2, тогда как для легкого изотопа
она пропорциональна корню квадрат-
ному из температуры. Такое различие в температурной зависимости объясня-
ется тем фактом, что для пары атомов Не3 не существует стационарного состоя-
ния, в то время как для Не4 существует либо реальный, либо виртуальный
уровень вблизи края энергетической ямы.
Дальнейшее графическое сравнение полученных результатов можно найти
на фиг. 123 (стр.442), на которой представлены значения вязкости, полученные
с помощью классической и квантовой теорий.
При вычислении второго вириального коэффициента указывалось, что
расчеты свойств газов методом фазовых сдвигов ограничены областью очень
низких температур, поскольку для более высоких температур необходимо
2,0
4,0
Фиг. 153. Зависимость вязкости Не3 и
Не4 от температуры, рассчитанная кван-
товомеханическим путем* для потенциала
Леннарда-Джонса [16].
534
Гл. 10. Квантовая теория и явления переноса
Таблица 99
Вязкость и теплопроводность Не3 и Яе4, рассчитанная квантовомеханическим
путем с потенциалом F-12) Л еннар да-Джонса [9,14]
Не»
Г,°К
0,0
0,51
1,02
2,04
3,07
4,09
5,11
г/см • сек
0,0
37,7
72,0
113,0
124,0
131,0
140,0
А • 107,
кал/см • сек °К
0,0
93,1
178,0
279,0
306,0
324,0
346,0
Не*
Т, °К
0,091
0,183
0,303
0,366
0,457
0,640
0,915
1,280
1,634
1,828
2,285
2,740
V W7,
г/см • сен
0,7
2,4
8,0
12,6
20,1
30,9
33,8
36,1
41,2
44,6
55,6
67,3
; • ю7,
кал/см • сек К
1,3
4,6
14,9
23,6
37,8
57,9
63,4
67,6
77,2
83,7
104,1
127,1
находить фазовые сдвиги для значительно большего числа значений квантовых
чисел I и я. Для высоких температур, при которых система не сильно отличается
от классической, решение можно получить в виде ряда по степеням постоянной
Планка аналогично тому, как это сделано для второго вириального коэффи-
циента в гл. 6, § 5. Это приближение обсуждается в следующем параграфе.
§3. ЯВЛЕНИЯ ПЕРЕНОСА ПРИ ПРОМЕЖУТОЧНЫХ
ТЕМПЕРАТУРАХ
В предыдущем параграфе было показано, что коэффициенты переноса
можно вычислить при очень низкой температуре с помощью квантовой статисти-
ческой механики. В гл. 8 приводятся классические формулы для коэффициен-
тов переноса, на основании которых "можно определять свойства газа при
высоких температурах. В этом параграфе будет изложена теория явлений пере-
носа в «промежуточной температурной» области, в которой система мало
отличается от классической.
В гл. 6, § 5 изложен метод вычисления второго вириального коэффициента
в этой промежуточной температурной области. Показано, что квантовая по-
правка для второго вириального коэффициента может быть представлена в
виде ряда по степеням А2. Это разложение в ряд может быть получено двумя
методами. Первый метод заключается в решении дифференциального уравне-
ния Блоха. Этот метод применен в гл. 6, § 5. Во втором методе фазовые сдвиги
определяются методом ВКБ. Последний метод, развитый Каном [18], может
быть также применен для вычисления коэффициентов переноса. Оказывается,
что квантовая поправка к коэффициентам переноса может быть записана также
в виде ряда по степеням Л2.
А. ОПРЕДЕЛЕНИЕ ФАЗОВЫХ СДВИГОВ МЕТОДОМ ВКБ1)
Фазовые сдвиги для парных столкновений получаются из решения радиаль-
ного волнового уравнения G.3) и G.4) гл. 1. Первое из этих уравнений является
дифференциальным уравнением для (ггрх) и описывает относительное радиаль-
х) Краткое описание метода ВКБ дается в гл. 1, § б. Более подробное объяснение
приводится в книге Шиффа [19], и, наконец, очень подробное изложение имеется в работе
[20]. Квантовая динамика парных столкновений изложена в гл. 1, §7. Полученные там
результаты применены в теории фазовых сдвигов.
§ 3. Явления переноса при промежуточных температурах 535
ное движение двух частиц, энергия взаимодействия которых дается потенциаль-
ной функцией ср(г). Второе из этих уравнений является дифференциальным
уравнением для (гу#/) и описывает относительное движение двух невзаимодей-
ствующих частиц. Решение первого из этих двух уравнений может быть запи-
сано в виде
(г^) = ехрр^]. C.1)
В соответствии с методом ВКБ функция их1(г) ищется в виде ряда
и* (г) = «Й> + (у) и<? + (у)' и9> + • •. C.2)
Функции и$(г) определяются уравнениями, получающимися путем подста-
новки этого выражения в формулу G.3) гл. 1 и приравнивания коэффициентов
при одинаковых степенях постоянной Планка1). При этом решение получается
в виде
где I — орбитальное квантовое число ; к — волновое число, характеризующее
общую относительную энергию сталкивающейся пары молекул. Величина
//(г) равна [1A + 1I г2] + [2 /a (p(r)\h2 ], а гт — классическая точка поворота,
определяемая уравнением
//(г) = *2.
Дальнейшие рассуждения будут ограничиваться такими потенциалами взаи-
модействия, для которых имеется только одна классическая точка поворота —
расстояние наибольшего сближения двух молекул.
Из двух решений C.3) необходимо составить две линейные комбинации :
одну для «классической области», где /z < я2, а другую — для «неклассической
области», для которой /z > и2. Для сшивания этих двух решений в класси-
ческой точке поворота воспользуемся формулами сшивания Крамерса [19, 20].
Если найдено решение для (ripxl) методом ВКБ, то легко найти и фазовые
сдвиги. Они могут быть также разложены в ряд
т (*) = *г!1] (*) + ч!11 (*) + v\z] (*) + ..., C.4)
причем следующие один за другим члены возникают из различных членов
экспоненты C.3). Дальнейшие обсуждения будут ограничены первыми двумя
приближениями в методе ВКБ. Первое приближение дает правильную класси-
ческую формулу для параметров переноса, второе приближение дает квантовую
поправку, пропорциональную Л2. С учетом формул сшивания Крамерса вол-
новые функции в первом приближении метода ВКБ в «классической области»
имеют вид
cos (- тг
Гт
Для больших г это упрощается :
г
(ryj) ~ cos(*r - хгт - -J- + {[К*1-//(г)- *] dr). C.6)
х) При выводе этих выражений учитывается тот факт, что Л появляется также и в кван-
товомеханическом выражении центробежного потенциала [Л2/(/ + 1)/2/гг2].
536
Гл. 10. Квантовая теория и явления переноса
Полученное решение является решением радиального волнового уравнения
G.3) гл. 1 для потенциала взаимодействия <р(г), согласно первому приб-
лижению метода ВКБ. Чтобы получить решение уравнения G.4) гл. 1
в том же приближении, необходимо в формуле C.6) заменить //(г) на 1A + 1)/г2
и произвести указанное интегрирование. Таким образом находится радиальная
волновая функция двух невзаимодействующих частиц на большом расстоя-
нии г
(пр2/) ~ cos [кг — -^ %- Vl(l+l)]• C.7)
Фазовый сдвиг определяется как разность фаз радиальных волновых функций
на больших расстояниях с учетом и без учета взаимодействия <р(г). Из формул
C.6) и C.7) фазовый сдвиг в первом приближении ВКБ получается в виде
- Ь (г) -к) dr.
C.8)
Аналогичным образом получается и во втором приближении метода ВКБ
C.9)
16 У/ (/+1) 32 J («2-//M^
ftn
Интеграл от этого выражения расходится. Исследование формулы связи Кра-
мерса (см. [18]) показывает, однако, что этот интеграл должен рассматриваться
как половина интеграла по контуру, показанному на фиг. 154 в комплексной
¦е
Фиг. 154. Путь интегрирования в комплексной плоскости г, использованный
при вычислении интеграла C.9) [20].
плоскости г. Как показал Хадамард [21], этот интеграл в комплексной пло-
скости гможет быть переписан в виде главной части интеграла вдоль действи-
тельной оси г. Это значит, что мы должны интегрировать по частям C.9) до тех
пор, пока не останется сходящийся остаток, а все бесконечные члены, появля-
ющиеся в результате интегрирования по частям, должны быть отброшены.
Произведя такую операцию, получим
_J_ Г
24 J
(fnti) -
y«i-/f(r)
dr.
C.10)
Таково выражение фазового сдвига во втором приближении метода ВКБ.
§ 3. Явления переноса при промежуточных температурах 537
Б. ВЫРАЖЕНИЕ СЕЧЕНИЯ В ВИДЕ РЯДА ПО СТЕПЕНЯМ
ПОСТОЯННОЙ ПЛАНКА1)
Сечение как функция фазовых сдвигов в статистике Больцмана определя-
ется формулой B.2). Подставляя фазовые сдвиги rf}](x) и rf^(x) в выражение
для сечения, получаем ряд, члены которого пропорциональны последователь-
ным степеням А.
Разность фазовых сдвигов [Vi+n — Ч/], появляющуюся в формуле B.2),
можно записать через производные, что соответствует разложению в ряд Тзй-
лора x\i+n как функции /
Удобнее, однако, ввести переменную Ь, определяемую равенством
Щ + 1) = ngb, и выразить разность фазовых сдвигов через производные по Ь.
Кроме того, заменим х эквивалентным выражением pg/h. Используя выра-
жения rf-}\x) и rff](x), полученные по методу ВКБ, находим
4>й(?Г- <злз>
Величины х и %р определяются с помощью первого и второго приближений к
фазовому сдвигу и имеют вид
C.15)
Используя явные выражения для фазовых сдвигов C.8) и C.10), полученные
в первом и втором приближениях, имеем следующие интегральные выражения
для х и V '•
] C.16)
C.17)
Гт
В этих выражениях величина F(r) является «эффективным потенциалом»,
выраженным в единицах начальной кинетической энергии относительного дви-
жения Va ^g2,
^+f C.18)
Штрих при F(r) означает дифференцирование по г. Величина %(&, g) — класси-
ческий угол отклонения, определенный формулой E.26) гл. 1, следовательно,
введенный нами параметр b является прицельным расстоянием, определенным
в гл. 1, § 5. Из уравнения C.13) следует
х) См. [22].
538 Гл. 10. Квантовая теория и явления переноса
Это выражение устанавливает соответствие1) фазовых сдвигов и угла откло-
нения.
Подставляя выражения C.12) и C.13) в формулы для Q(l) и QB), которые
в случае статистики Больцмана имеют вид B.2), можно определить эффектив-
ные сечения. Величины sin (??/+„ — *?/) можно представить в виде ряда по
степеням A//xg&, а суммирование по I можно заменить интегрированием по пере-
менным Ь
№)[ 4Ш' ]• C-20)
Таким путем можно получить эффективное сечение в виде ряда, члены кото-
рого содержат возрастающие степени постоянной Планка. Первый член, про-
порциональный А0, представляет собой классическое сечение Q$ или Q{$,
определенное формулой B.3). Можно показать, что члены, пропорциональные
нечетным степеням А, будут исчезать при интегрировании по частям. Поэтому
окончательное выражение имеет вид
?)' Q(,n) + (•?)* Q(,1> + • • •; C.21)
здесь Q({!) — первая квантовая поправка к эффективному сечению, которая
имеет вид
00
» = 2я[±[ (si
6
[ (sin *)(*-
6
= 2* [т
О
Явное выражение для Qfr> еще не получено.
Подставляя эти выражения в формулу B.5) для 42(л> *\ получаем
[-?-Hj».O + (JL| flft.O+..., C.23)
где
= С е-у у2,+з Qjn> g-2ф, > (з.24)
3) Это соответствие установлено другим методом в работе Мотта и Месси [23]. Приведен-
ный здесь метод не позволяет непосредственно получить квантовые поправки к классической
величине.
§ 3. Явления переноса при промежуточных температурах 539
Переменная у определяется следующим образом: у2 = г/2 !*g*/kT. Таким обра-
зом, получены выражения для квантовых поправок к классическим величинам
Q(n>f\ а следовательно, и ко всем коэффициентам переноса.
В. ВЫЧИСЛЕНИЯ ДЛЯ ПОТЕНЦИАЛА ОТТАЛКИВАНИЯ,
ОБРАТНО ПРОПОРЦИОНАЛЬНОГО ДВЕНАДЦАТОЙ
СТЕПЕНИ РАССТОЯНИЯ
Рассчитать квантовые поправки к коэффициентам переноса по приведен-
ным выше формулам в случае модели твердых сфер не просто, поскольку в
выражения для % и у входят производные потенциальной функции. Простей-
шей моделью, для которой удается провести такие вычисления, является
модель отталкивающихся молекул. В дальнейшем будет рассматриваться
потенциальная функция
(^I2 C.25)
представляющая часть потенциала F-12) Леннарда-Джонса, соответствующего
силам отталкивания. Классические расчеты коэффициентов переноса с этим
потенциалом изложены в гл. 8, § 3. Основное преимущество этой модели заклю-
чается в том, что величины % и у можно записать как функции одной перемен-
ной у0 и, более того, интегрирование по у в формуле C.24) можно провести
аналитически.
Удобно ввести новые переменные, которые определяются выражениями
ь by
Ут
Ут C.26)
lVl2_ ( ь Л ( У2^2 У/»
где гт — классическое расстояние наибольшего сближения молекул, опреде-
ленное формулой E.26), а уту являющееся функцией у0, определяется как поло-
жительный корень уравнения
&Г-0- <3-27>
В новых переменных интегралы % и у принимают вид, удобный для вычислений *
где
^(^f]n, C.30)
C.31)
В табл. XXI (стр. 891) приведены значения ym, #, у) и /==d^/dy0 как
функции уо-
Зная х и Ъ можно вычислить классические сечения [формула B.3)] и
первую квантовую поправку [формула C.22)].
540 Гл. 10. Квантовая теория и явления переноса
В результате получаем
lf <3-32>
Величины Л(п) и В(н) являются интегралами пр у0 и не зависят от g. Численные
значения их равны
> = + 0,3458, ВA> = - 0,2680,
=:+ 0,2787 , #2> = + 0,5251.
Интегралы Q("'*> можно записать в виде
до, 0 ^ л W |/^ (.?.) V. „* г (t + f) [l + К, (л, 0 ^- + ... ] , C.33)
где
л* - h т* - -*L
Ki (п, 1) = Г^-1 G,706 -10-3) ?
C.34)
/Ci (п, 2) = [-1^-] E,439 -10-»).
В соответствии с расчетами первой квантовой поправки к коэффициентам
переноса квантовые выражения бязкости, теплопроводности и самодиффузии
для Не и Н2 уменьшаются не более чем на 0,5% при комнатных температурах.
Величина второй квантовой поправки до сих пор еще не найдена. Имеющиеся
в настоящее время экспериментальные данные недостаточно хороши для точ-
ного сравнения вычисленных и наблюденных результатов.
§ 4. ЗАКОН СООТВЕТСТВЕННЫХ СОСТОЯНИЙ
В КВАНТОВОЙ МЕХАНИКЕ1)
Все рассуждения в предыдущих двух параграфах относились к разрежен-
ным газам, когда можно было пренебречь тройными столкновениями. Однако
теорию коэффициентов переноса можно сформулировать с помощью закона
соответственных состояний, который не ограничен областью малых плотностей,
а справедлив при всех значениях плотности, в частности и в жидком состоянии.
Закон соответственных состояний может быть выведен с помощью соображений
теории размерности, как это сделано в гл. 9, § 1. Пусть потенциал взаимодейст-
вия между двумя молекулами характеризуется двумя параметрами — харак-
теристической длиной а и характеристической энергией е. Тогда квантовая тео-
рия объемных свойств, таких, как коэффициенты переноса, будет функцией
г) Изложение закона соответственных состояний можно найти в разных местах этой
книги : в гл. 4, § 1 излагается классическая форма этого закона применительно к уравнению
состояния, в гл. 6, § 6 излагается квантовая форма применительно к уравнению состоя-
ния, а в гл. 9, § 1 — классическая форма применительно к явлениям переноса.
§ 4. Закон соответственных состояний в квантовой механике 541
семи переменных: v,T,m, fc, or, ей ft. Анализ размерности показывает, что при-
веденные коэффициенты переноса, определенные формулой A.2) гл. 9, могут
быть записаны в виде
?)* = ?)*(v*,T*; Л*),
fl*=tp<p*,T*; Л*), D.1)
А* = Л* (г;*, Т* ; Л*).
Переменные г;* и Г* — приведенный объем и температура, определенные фор-
мулой A.1) гл. 9, а Л* — квантовомеханический параметр
± D.2)
Смысл и интерпретация этого параметра даны в гл. б, § б, где закон соответ-
ственных состояний применен для определения равновесных свойств1). Вели-
чина Л* для различных веществ приведена в табл. 57 (стр. 337).
Закон соответственных состояний можно сформулировать в переменных,
приведенных к критическим постоянным. Например, можно написать
?)r = ?)r(pnTr; Лг),
Чг = Уг(Рг,Тг; Лг), D.3)
К=К(Рг,Тг; Лг),
где приведенные коэффициенты переноса, приведенное давление и приведенная
температура определены формулой A.6) гл. 9. Величина Лг является безраз-
мерной комбинацией
Лг = v
которая применялась в первых теориях, посвященных выводу квантовых от-
клонений от классического уравнения состояния. Однако, как показано в
гл. 6, § б, величина Лг неудовлетворительна в том смысле, что она сама
зависит от квантовых эффектов через vc и Тс. Параметр Л* свободен от этих
недостатков.
В настоящее время нет достаточного количества данных о вязкости, тепло-
проводности и диффузии, чтобы можно было применять закон соответственных
состояний в квантовой механике. После того как такие данные накопятся, этот
закон позволит установить связь между экспериментальными фактами и
предсказать квантовые свойства жидкостей.
Задача
1. Найти квантовую поправку к вязкости для неона при ЗОО°К.
2. Справедливо ли соотношение
[см. формулу B.31) гл. 8] в квантовой механике?
См. гл. 6, § б, а также работы Байка [ 24 ].
542 Гл. 10. Квантовая теория и явления переноса
ЛИТЕРАТУРА
1. Irving J. H., Zwanzig R. W., Journ. Chem. Phys., 19, 1173 A951).
2. Wig пег Е. P., Phys. Rev., 40, 479 A932).
3. M о r i H., О п о S., Prog. Theor. Phys., 8, 327 A952).
4. Uehling E. A., Uhlenbeck G. E., Phys. Rev., 43, 552 A933).
5. N о r d he i m, К i к u с h i, Zs. Phys.; 60, 652 A930).
6. Sommerfeld A., Zs. Phys., 47, 1, 43 A928).
7. Uehling E. A., Phys. Rev., 46, 9Г7 A934).
8. H e 11 u n d E. J., Uehling E. A., Phys. Rev., 56, 818 A939).
9. de Boer J., Physica, 10, 348 A943).
10. U b b i n к J. В., Диссертация, Leiden, 1945.
11. Masse у H. S. W., Mohr С. В. О., Proc. Roy. Soc, A141, 434 A933).
12. Masse у H. S. W., Mohr С. В. О., Proc. Roy. Soc, A144, 188 A934).
13. Slater J. C, Phys. Rev., 32, 349 A928).
14. de Boer J., Cohen E. G. D., Physica, 17, 993 A951).
15. Mi yak о R., Proc. Phys.-Math. Soc, Japan (Ser. 3), 24, 852 A942).
16.'Cohen E. G. D., Offerhaus M. J., de Boer J., Physica (в печати).
17. С h a p m a n S., Cowling T. G., The Mathematical Theory of Non-uniform Gases,
Cambridge, 1939 (см. перевод: Чепмен С, Каулинг Т., Математическая
теория неоднородных газов, ИЛ, I960).
18. К a h n В., Диссертация, Utrecht, 1938.
19. Schiff L. I., Quantum Mechanics, New York, 1949 (см. леревод: Шифф Л.,
Квантовая механика, ИЛ, 1957).
20. Kemble E. С, Fundamental Principles of Quantum Mechanics, New York, 1937.
21. H a d a m a r d J., Le probleme de Cauchy, Paris, 1932.
22. de Boer J., Bird R. В., Phys. Rev., 83, 1259 A951).
23. Mott N. F., Massey H. S. W., The Theory of Atomic Collisions, Oxford, 1949
(см. перевод : Мотт Н., Месси Г., Теория атомных столкновений, ИЛ, 1951).
24. Byk A., Ann. d. Phys., 66, 157 A921); 69, 161 A922).
ГЛАВА 11
ГИДРОДИНАМИЧЕСКИЕ ПРИМЕНЕНИЯ УРАВНЕНИЙ
ПЕРЕНОСА
В гл. 2—б изучались свойства газов и жидкостей в равновесии, а в
гл. 7—10 — их свойства в неравновесном состоянии. В настоящей главе мы при-
меним полученные результаты к вопросам гидродинамики. Для решения любой
гидродинамической проблемы необходимы следующие основные уравнения :
1) термическое уравнение состояния р = p(V, T),
2) калорическое уравнение состояния U = U(V, T),
3) уравнение непрерывности,
4) уравнение движения,
5) уравнение энергии.
Эти пять уравнений совместно с уравнениями, описывающими зависимость
коэффициентов переноса от температуры и плотности, образуют исходную
систему уравнений для решения гидродинамических задач. При решении
практических гидродинамических задач необходимо, чтобы решения этих
уравнений удовлетворяли определенным начальным и граничным условиям.
С помощью этой системы уравнений и соответствующих граничных условий
можно описать множество различных явлений. В качестве иллюстрации при-
менения этих уравнений в настоящей главе будет рассмотрено распространение
волн бесконечно малой амплитуды (звуковых волн), волн конечной амплитуды
и ударных волн. Мы покажем также, как, явно учитывая химические реакции,
можно описать пламена и детонацию. В заключение мы обсудим течения в
соплах, применяемых в ракетных двигателях. Для простоты мы ограничимся
здесь лишь обсуждением ламинарных течений.
Уравнения переноса (уравнения 3, 4, 5) содержат4вектор плотности потока
масс j,, тензор плотности потока импульса р и плотность потока тепла q. При
конкретном применении уравнений переноса к гидродинамическим проблемам
необходимо выразить потоки какфункции коэффициентов переноса и градиентов
физических характеристик жидкости. Уравнения, связывающие потоки, могут
быть получены из термодинамики необратимых процессов и соотношения
Онзагера. Мы покажем здесь, как можно включить перенос энергии излучением
в общий вектор плотности потока энергии.
§ 1. УРАВНЕНИЯ ГИДРОДИНАМИКИ
В основе динамики жидкости лежат уравнения переноса. С их помощью
можно найти макроскопические свойства жидкостей (такие, как локальная
плотность числа частиц, скорость течения и температура) как функцию потока
масс (скоростей диффузии), потока импульса (тензора давления), потока энер-
гии и скоростей химических реакций.
Уравнения переноса выведены классическим путем (см. гл. 9, § 4) и с по-
мощью квантовой механики (см. гл. 10, § 1) при очень общих предположениях.
В этом параграфе мы суммируем полученные результаты и обсудим границы
применимости этих уравнений. Мы дадим также вывод уравнения для изме-
нения энтропии, которое пригодится впоследствии в параграфе, посвященном
теории распространения звуковых волн, волн конечной амплитуды, детонации
и течению в соплах.
544 Гл. 11. Гидродинамические применения уравнений переноса
А. ПРИМЕНИМОСТЬ УРАВНЕНИЙ ПЕРЕНОСА
Вообще говоря, уравнения переноса применимы к любой жидкости, однако
они дают физически осмысленные результаты лишь в том случае, когда можно
говорить о локальных свойствах жидкости. Локальная плотность, скорость и
температура могут быть определены формально, но эти определения разумны
лишь при условиях, в которых жидкость ведет себя как сплошная среда. Если
макроскопические свойства жидкости заметно меняются на расстояниях по-
рядка длины свободного пробега, то распределение скоростей уже не будет
максвелловским. В этих условиях плотности потоков уже не могут быть выра-
жены как функции локальной плотности, скорости, температуры и их первых
производных. Приведем два хорошо известных примера :
1) сильно разрежённый газ [1J1) (или «газ Кнудсена»), в котором размеры
сосуда или размеры погруженного в него тела порядка средней длины свобод-
ного пробега ;
2) газ в ударной волне, в которой макроскопические свойства как функция
расстояния скачкообразно изменяются на расстоянии порядка нескольких
длин свободного пробега.
В обоих этих случаях говорить о значениях макроскопических величин в
точке не имеет смысла.
Можно спросить также о применимости уравнений переноса для описания
турбулентного движения. Хорошо известно, что поток жидкости в трубах может
быть как ламинарным, так и турбулентным. Ламинарный поток, появляющийся
при малых скоростях движения жидкости2), характеризуется плавным изме-
нением линий тока. Однако с ростом скорости поток становится турбулентным :
появляются малые завихрения и на весь поток накладываются более или менее
случайные движения. Размеры этих вихрей велики по сравнению со средней
длиной свободного пробега, поэтому турбулентное движение является скорее
макроскопическим, чем молекулярным. Так, при турбулентном движении
жидкость тоже можно рассматривать как континуум и применять к ней обыч-
ные уравнения переноса. В этих уравнениях переменные являются функциями
мгновенных значений величин в некоторой точке. Однако практически интерес
представляют значения переменных, усредненные по промежутку времени,
большему по сравнению с периодом флуктуации. Следовательно, в турбулент-
ном потоке для получения соотношений между средними значениями макро-
скопических переменных уравнения переноса должны быть усреднены по
времени. Уравнения, приведенные в этой главе, получены на основе теории
турбулентности, которая в дальнейшем больше обсуждаться не будет3).
Рассмотрим теперь более подробно уравнение энергии. Передача энергии
практически4) происходит одним или несколькими способами из четырех теоре-
тически возможных : теплопроводности5), диффузии, конвекции и излучения.
Рассмотрим здесь кратко явления передачи энергии двумя типами конвекции—
вынужденной и естественной. При вынужденной конвекции среда описыва-
ется непосредственно уравнениями гидродинамики и граничными условиями
конкретной задачи. При естественной конвекции в потоке образуется градиент
плотности вследствие действия теплопередачи и гравитационного притяжения.
*)См. гл. 1, §2 и гл. 7, §5.
2) Характер течения в трубах крупного сечения определяется числом Рейнольдса
Dqv/г), где D — диаметр трубы, q — плотность, v — скорость, а г\ — коэффициент вязкости.
При малых числах Рейнольдса поток ламинарный, при больших числах — обычно турбу-
лентный.
8) Для дальнейшего изучения теории турбулентности см. работы [2—7 ]. (См. также
работы советских ученых по теории турбулентности, изложенные, например, в следующих
книгах : Л. Ландау, Е. Л и ф ш и ц, Механика сплошных сред, М.—Л., 1953;
Л. Седов, Методы подобия и размерности в механике, М.—Л., 1957. — Прим. ред.)
4) Общее рассмотрение практической теплопередачи в газах и жидкостях дано в работах
[8-10].
б) Математическая теория теплопроводности в твердых телах (без потока) дана в работах
[И, 12].
§ 1. Уравнения гидродинамики 545
Среди всех возможных состояний среды стационарное состояние, осуществляе-
мое в природе, является наиболее устойчивым по отношению к возмущениям.
Хотя естественная конвекция принадлежит к одному из наиболее трудных
типов гидродинамических краевых задач, существует небольшое число част-
ных примеров, для которых удается найти удовлетворительное теоретическое
решение задачи. Наиболее известными среди них являются следующие:
1) конвективные токи в термодиффузионной колонне Клузиуса—Дикеля [13]
(газ или жидкость заключены в кольцевую область между двумя концентри-
ческими вертикальными трубками, поддерживающимися при разных темпера-
турах) ; 2) ячеечная конвекция в газе или жидкости, заключенных между
двумя большими горизонтальными плоскими пластинами, в которых поддер-
живается различная температура1). Конвекция играет очень важную роль в
диффузионных методах разделения смесей [22]. Естественная конвекция,
подобно турбулентности, остается вне сферы этой книги.
Если энергия передается путем излучения, плотность потока энергии q
в уравнениях переноса необходимо заменить суммой (q + qR) тепловых потоков
за счет теплопроводности и диффузии, а также за счет излучения2). Происхож-
дение и величина потока излучения подробно рассматриваются в § 3. В пламе-
нах или других системах, имеющих высокую температуру, излучение может
являться одним из основных механизмов передачи энергии. Вообще говоря,
давление излучения pR аддитивно добавляется к гидродинамическому тензору
давления, а плотность энергии излучения OR — к внутренней энергии системы.
Однако лишь в астрономических условиях или в условиях, соответствующих
атомному взрыву, энергия излучения3) или давление излучения4) становятся
заметными. В связи с этим последние два эффекта излучения здесь не рассма-
триваются.
Б. СВОДКА УРАВНЕНИЙ ПЕРЕНОСА
Состояние системы описывается, как известно, следующими макроскопи-
ческими переменными : плотностью числа частиц различных компонент nh
средней массовой скоростью или скоростью потока5) и температурой Т. Эти
переменные связаны с величинами, сохраняющимися при молекулярном столк-
новении : массой, импульсом и энергией сталкивающихся молекул. Соответ-
ственно для каждого из этих «сумматорных инвариантов» можно вывести и
уравнение переноса: уравнение непрерывности каждой компоненты, уравнение
движения и уравнение энергии6).
1) (а) уравнение непрерывности i-й компоненты
(б) общее уравнение непрерывности
*)См. работы [14—17].Экспериментальная проверка теории дана в работах [18, 19].
2) Подробное изложение см. в работах [23—25].
3) Что энергия излучения обычно пренебрежимо мала по сравнению с внутренней энер-
гией, можно показать следующим образом. Энергия излучения черного тела при температуре
Т равна Or = gaT* = 1,833 • 10-22g Г4 кал/г, где а — постоянная Стефана—Больцмана. Если
плотность q порядка единицы, а Т = 100 000°К, то Or = 0,02 кал/г.
4) В равновесных условиях давление излучения равно pR — аТА = 2,523 • Ю-21 Г4 апгм.
Если Г=10 000°К, PR = 2,5- 10~б атм; если Г=100 000°К, Pr= 0,25 атм. Таким
образом, для всех обычных применений давление излучения пренебрежимо мало.
ь) В гл. 7—10 средняя массовая скорость обозначалась v0. В гидродинамических прило-
жениях, описанных ниже, 0 в индексе оказывается излишним, поэтому он будет опущен.
в) Сохранение момента импульса при столкновении приводит еще к одному уравнению
переноса, которое устанавливает связь момента импульса — макроскопического движения
газа с вращательным движением молекул. Это уравнение, вид которого, вообще говоря,
неизвестен, обсуждалось в гл. 7, § 6.
546 Гл. 11. Гидродинамические применения уравнений переноса
2) уравнение движения
дй ( а п\ Ив , л
3) уравнение энергии
dO дй
Здесь е — плотность потока, а G — внутренняя энергия, отнесенная к 1 г.
Величины, приводящие к необратимости течения, следующие : К( — скорость
образования i-й компоненты в единице объема; V/ — скорость диффузии /-й
компоненты; р — тензор давления; q + qR—тепловой поток. Выражения
для этих величин в разреженном газе в отсутствие излучения приводятся в
гл. 8, § 1. Для получения выражений плотности потоков, справедливых в более
общих условиях, в § 2 использована термодинамика необратимых процессов.
В § 3 рассмотрена доля энергии излучения в общем потоке энергии.
Субстанциональная производная DjDt, появляющаяся в уравнениях
A.1) — A.4), описывает скорость изменения в системе координат, связанной с
элементом среды, движущимся со скоростью v. В соответствии с уравнением
A.1) плотность числа частиц i-й компоненты меняется под действием трех
причин : расширения среды (первый член в правой части), процессов диффузии
(второй член) и возникновения i-й компоненты в результате химической реакции
(третий член). Общая плотность среды, как показывает уравнение A.2), изме-
няется лишь в результате расширения среды, описываемого членом в правой
части уравнения. Скорость элемента среды в соответствии с уравнением A.3)
изменяется под действием градиента давления внешних сил, различных для
каждой компоненты. Тензор давления можно записать в виде суммы двух
членов
p = pU + P. A.5)
Первый член соответствует статическому давлению, а второй член учитывает
эффекты вязкости. Внутренняя энергия, как показывает уравнение A.4), изме-
няется под действием трех причин : 1) существования потока энергии, учиты-
вающего также и излучение (первый член в правой части); 2) работы pV и
эффектов вязкости (второй член); 3) работы диффузионных сил против внеш-
них сил.
В некоторых случаях удобнее пользоваться уравнениями переноса в пере-
менных Тир. Для однокомпонентной системы эти уравнения, являющиеся
частным случаем вышеприведенных, имеют вид
<¦¦*>
§ 1. Уравнения гидродинамики 547
Аналогичные уравнения в общем случае многокомпонентной системы сложнее,
поскольку парциальные молярные внутренние энергии являются функциями
не только температуры, но также плотности и химического состава.
В. УРАВНЕНИЕ ЭНТРОПИИ
Приведенные выше уравнения позволяют полностью описать гидро-
динамику среды. Однако оказывается весьма полезным, в частности для адиа-
батически изолированных систем, ввести еще одно уравнение — уравнение,
описывающее скорость изменения энтропии. Это уравнение записывается в
форме, учитывающей возникновение энтропии в каждом из процессов, идущих
в газе ; выражения для плотности потоков могут быть при этом найдены
с помощью методов термодинамики необратимых процессов. Эти методы
позволяют определить выражения плотности потоков и в жидком состоянии,
что особенно ценно, поскольку другие методы для этой цели неизвестны.
Рассмотрим изменение энтропии внутри конечной области пространства Л.
Область ограничена поверхностью, каждая точка которой движется вместе
с потоком с локальной скоростью v. Энтропия внутри области меняется по двум
причинам : 1) вследствие обратимого потока энтропии через ограничивающую
поверхность и 2) вследствие возникновения энтропии в результате необратимых
процессов. Пусть 5—энтропия единицы массы, б — плотность обратимого
потока энтропии, a g — скорость возникновения энтропии в необратимых
процессах1). В таком случае можно написать
Jdr; A.8)
здесь интеграл Г d S означает интегрирование по всей поверхности рассматри-
л
ваемого объема Л. Это — уравнение энтропии в интегральной форме.
Внося операцию дифференцирования под знак интеграла, это уравнение
можно переписать в виде
JJ A.9)
л л л л
Применяя теорему Гаусса, получаем
ЬН -«]dt=°- AЛ0>
Поскольку это выражение справедливо для любой области пространства, то
можно написать
*. (Ml)
Используя уравнение непрерывности [формула A.2)], получаем
Это—уравнение энтропии в дифференциальной форме.
х) Следует иметь в виду, что «обратимый» поток энтропии с связан с необратимыми про-
цессами — теплопроводностью, диффузией и т. д. [см., например, выражения A.16)—A.19)].
Термин «обратимый» в данном случае имеет тот смысл, что первый член в правой части равен-
ства A.8) может быть как положительным, так и отрицательным, тогда как второй член,
связанный с необратимыми процессами внутри объема А, всегда положителен. — Прим. ред.
548 Гл. 11. Гидродинамические применения уравнений переноса
Удобно записать уравнение непрерывности с помощью субстанциональной
производной DjDt. В таком случае уравнение A.12I) принимает вид
Уравнение энтропии в такой форме может быть также получено из урав-
нений переноса, приведенных в разд. Б. Сравнение двух уравнений позволяет
найти явное выражение для скорости возникновения энтропии g.
В равновесных условиях энтропия зависит лишь от состояния системы
в целом. В неравновесных условиях локальное значение удельной энтропии
принимается такой же функцией температуры, плотности и состава. Дифферен-
циал энтропии равен
J*>d ; A.14)
t Q
здесь ц{ — химический потенциал. Для систем, находящихся в равновесии,
р — давление жидкости. В неравновесных системах р принимается такой же
функцией температуры, плотности и состава. Как будет показано в § 2, только
при определенных условиях х/3 суммы диагональных элементов тензора давле-
ния будет равна р. Из уравнения A.14) ел едут
С помощью формул разд. Б можно исключить производную по времени в
правой части этого уравнения. В результате получаем уравнение, совпадаю-
щее по форме с A.13), причем d и g выражаются следующим образом:
6 = 4- [{ч + ч*} - 2 ni п V<] > 0 • 1б)
Запишем выражения 6 и g в несколько более удобном виде.
Вектор 6 представляет плотность обратимого потока энтропии. Это легко
увидеть из следующих рассуждений. Общий поток энергии q + q^ можно за-
писать в виде суммы двух членов
q + qR = e + 2n.HiVi, A.18)
где Н( — парциальная молярная энтальпия. Второй член соответствует по-
току энергии вследствие процессов диффузии, тогда как в (которое определяется
этим уравнением) является плотностью потока тепла. Из уравнений A.16) и
A.18) ел едут
где St — парциальная молярная энтропия. В такой форме видно, что б явля-
ется суммой двух членов. Первый член соответствует плотности потока энтро-
пии, возникающего под действием теплового потока ; второй член обязан
процессам диффузии.
*) Необходимо отметить, что при выводе этого уравнения не были использованы специ-
фические свойства энтропии 5, а следовательно, подобное уравнение можно написать для
любого экстенсивного параметра газа.
§ 7. Уравнения гидродинамики 549
Величина g может быть записана через скорости возникновения молекул
различного типа Kf. Химические процессы, протекающие в реагирующей
смеси, можно описать уравнениями реакции, которые символически записы-
ваются следующим образом :
АЛП + АЛ2] + • • • -%y[i] +%Л2] + • • •; A.20)
здесь Ри и г\ц — целые числа, а [/ ] — концентрация /-и компоненты. Пусть
постоянная скорости для /-и прямой реакции равна fey, а для/-й обратимой
реакции равна fej, тогда скорость прямой реакции равна
kj№№..., A.21)
где //—текущая концентрация i-й компоненты. Подобное выражение можно
написать и для обратной реакции. Общая скорость образования 1-х молекул в
результате всех химических реакций равна
К, = 2{r,u-h) [<!<№№ • • •) - (kjfl1Jffj ...)]• A-22)
Назовем химическим сродством Уу и скоростью rj /-й реакции выражения
0-23)
.). A.24)
Тогда в соответствии с формулой A.17) вклад химических реакций в общую
скорость возникновения энтропии можно записать в виде
--f-^'K'e-F-?r'K'- A-25)
i i
Если это выражение, а также выражение для потока энергии [формула
A.18)] подставить в формулу A.17), то получим выражение
где j, = nitntfi — плотность потока массы /-й компоненты, определенной фор-
мулой B.19) гл. 7, а А/ равно1)
Л — L^ l*LL
4
mi эг * mt dr mi
mi Д {щ)т,р,хк эг "^ m/ эг rrn Л/#
г) Вторая форма этого уравнения получается в результате использования того факта,
что химический потенциал /*/ зависит от состояния системы и, в частности, может рассматри-
ваться как функция Г, р и xj (/ ф i). Частные производные по Г и р исключаются с помощью
термодинамических соотношений
Заметим, что V/ — парциальный молярный объем, который нельзя путать с V/, обозначаю-
щей скорость диффузии.
550 Гл. 11. Гидродинамические применения уравнений переноса
Таким образом, формула A.26) выражает скорость возникновения энтро-
пии в необратимых процессах. Первый член соответствует вкладу процесса
диффузии, второй— переносу импульса, третий — переносу энергии и, наконец,
последний — химическим реакциям. Эта формула написана в таком виде, что
выражения плотности потоков, входящие в нее, могут быть определены с по-
мощью термодинамики необратимых процессов. Прежде чем рассматривать
применения этого уравнения, изложим коротко основные принципы термо-
динамики необратимых процессов.
§ 2. ТЕРМОДИНАМИКА НЕОБРАТИМЫХ ПРОЦЕССОВ1)
До сих пор явления переноса изучались с точки зрения статистической меха-
ники неравновесных систем или кинетической теории. Как известно, в урав-
нения переноса входят потоки, которые можно выразить через интегралы от
функции распределения. В конечном счете наша цель — выразить эти потоки
через свойства отдельных молекул. На основе такой молекулярной теории
можно предсказывать свойства переноса в условиях, где экспериментальные
данные отсутствуют. Такая молекулярная теория развита лишь для некоторых
определенных моделей молекул и пригодна лишь в определенных условиях.
Например, в гл. 7 выведены классические выражения2) плотности потоков и
коэффициенты переноса в разреженном одноатомном газе. Эти же выражения
в гл. 10 получены квантовомеханическим путем.
В гл. 7, § б дано обобщение этой теории на случай разреженного много-
атомного газа, однако численные результаты до сих пор не получены. Явления
переноса в плотных одноатомных газах обсуждаются в гл. 9 в рамках недавно
развитой статистической механики неравновесных систем. Хотя формально
результаты получены, необходимо еще выполнить много расчетной работы,
чтобы придать этим результатам практическую ценность.
Для жидкости, состоящей из произвольных молекул и находящейся при
обычных температуре и давлении, в настоящее время с помощью статистической
механики невозможно получить выражения потоков и найти коэффициенты
переноса как функцию межмолекулярных сил. Для такой жидкости в общем
смысле слова можно получить некоторые сведения с помощью термодинамики
необратимых процессов. Можно получить выражения потоков и с помощью
«соотношения взаимности» Онзагера найти связь между коэффициентами пере-
носа. Однако чисто термодинамическим путем невозможно получить коэффи-
циенты переноса как функции молекулярных свойств.
А. СООТНОШЕНИЕ ВЗАИМНОСТИ ОНЗАГЕРА
В предыдущих главах было показано, что потоки, приводящие к возник-
новению необратимых процессов, зависят от градиентов температуры и концен-
трации, а также от внешних сил. Причины, вызывающие потоки, обычно
называются обобщенными силами или сродством и обозначаются Х(; с каждой
обобщенной силой связан поток Jt. Обобщенные силы и потоки могут быть
скалярными, векторными и тензорными величинами. Однако при абстрактном
рассмотрении удобнее всего считать их все скалярными. Вообще говоря, любая
обобщенная сила может вызвать появление любого потока. Например, если в
системе существуют градиенты плотности и температуры, то возникают сле-
дующие потоки :
1) поток массы, вызванный градиентом плотности (обычная диффузия);
*) См. [26,27].
2) Сводка этих результатов дана в гл. 8, § 1.
§ 2. Термодинамика необратимых, процессов 551
2) поток энергии вследствие температурного градиента (теплопровод-
ность) ;
3) поток массы вследствие температурного градиента (термическая диф-
фузия или «эффект Соре»);
4) поток энергии вследствие градиента плотности («эффект Дюфура»).
Отсюда видно, что имеются два типа эффектов — прямые эффекты, такие,
как 1 и 2, и перекрестные эффекты, такие, как 3 и 4.
В качестве постулата термодинамика необратимых процессов прини-
мает, что в состояниях, близких к положению равновесия, потоки можно пред-
ставить в виде
Ji-^ijXj. B.1)
Эта формула показывает, что потоки являются линейными функциями обоб-
щенных сил.
Величины at] называются феноменологическими коэффициентами. Диаго-
нальные коэффициенты ап являются коэффициентами, представляющими
прямые эффекты; недиагональные коэффициенты а/;(/^=/) являются фено-
менологическими коэффициентами перекрестных эффектов. Выражения пото-
ков, полученные при рассмотрении явлений переноса в предыдущих четырех
главах, имеют тот же общий вид. Такие же «линейные» соотношения получаются
и во многих других физических или химических процессах, например в термо-
магнитных, в термогальванических, в электрокинетических и т. д. [26, 27 ].
Линейные соотношения применимы в условиях, достаточно близких к поло-
жению равновесия ; таким образом, формула B.1) определяет область приме-
нимости термодинамики необратимых процессов.
Основная теорема термодинамики необратимых процессов была доказана
Онзагером [28 ]. Эта теорема, доказательство которой будет дано ниже, утвер-
ждает, что при «соответствующем выборе» потоков и обобщенных сил феноме-
нологические коэффициенты симметричны:
а,7 = ау/. B.2)
Эти уравнения обычно называются «соотношениями взаимности». Термин
«соответствующий выбор» потоков или сил можно объяснить следующим обра-
зом. Рассмотрим изолированную систему. Состояние системы описывается
набором переменных wvw2,w9,... Этими переменными могут являться, например,
локальные значения температуры, плотности и концентрации. В этом случае
благодаря непрерывному изменению свойств переменные образуют бесконеч-
ный ряд. Для удобства переменные выбираются таким образом, чтобы в состоя-
нии термодинамического равновесия они равнялись нулю. Тогда, учитывая,
что энтропия в состоянии равновесия имеет максимум, отклонение энтропии от
своего максимального значения в первом приближении можно записать в виде
квадратичной формы по этим переменным1)
Теперь можно пояснить термин «соответствующий выбор» потоков и сил.
Под «соответствующим выбором» потоков и сил подразумевайся потоки и
силы, определяемые через переменные состояния и их производные по вре-
мени2)
У, = */, B.4)
^uWj. B.5)
х) Коэффициенты gy как смешанные вторые производные, появляющиеся при разло-
жении в ряд Тэйлора, являются симметричными.
2) Величина Т вводится в Я",- для удобства. Это согласуется с обычным определением AS
552 Гл. 11. Гидродинамические применения уравнений переноса
Основная теорема Онзагера [формула B.2)] справедлива для потоков и сил,
определенных таким образом.
Доказательство соотношений взаимности Онзагера основывается на очень
общих положениях статистической механики и на концепции микроскопической
обратимости. Принцип микроскопической обратимости означает симметрию
механических уравнений движения относительно времени или, другими сло-
вами, инвариантность уравнений движения относительно преобразования
t -> —t. Доказательство основано на формальном рассмотрении кинетики необ-
ратимых процессов и не зависит от конкретной природы рассматриваемых
процессов.
Предполагается, что скорость макроскопического необратимого процесса
совпадает со средней скоростью затухания статистических флуктуации в сис-
теме. Рассмотрим систему, находящуюся в некотором неравновесном состоянии.
Определим скорость приближения системы к равновесию. Для этого рассмот-
рим ту же систему в состоянии равновесия. Благодаря статистическим флук-
туациям имеется малая, но конечная вероятность нахождения системы в любом
микроскопическом состоянии. Подождем до тех пор, пока в результате флук-
туации система не перейдет в рассматриваемое нами неравновесное состояние.
Спустя некоторое время t система может находиться в любом другом микро-
скопическом состоянии. Однако среди всех возможных классов неравновесных
состояний один наиболее вероятен и соответствует новому неравновесному
состоянию системы. Таким путем на основе теории флуктуации можно про-
следить приближение нашей системы к состоянию равновесия.
Благодаря большому числу степеней свободы макроскопической системы
вероятность флуктуации мала и поэтому система с подавляющей вероятностью
будет находиться в микроскопическом состоянии, соответствующем непосред-
ственно равновесному состоянию. Этим объясняется плодотворность статисти-
ческого метода. По этой же причине путь затухания флуктуации достаточно
хорошо определен, вероятность отклонения от этого пути будет величиной
значительно меньшего порядка, чем вероятность следования по этому пути.
Физически это соответствует тому факту, что путь необратимого процесса
хорошо определен.
С помощью этих аргументов Онзагер развил формальную теорию скоро-
стей необратимых процессов. Предполагая далее, что потоки связаны с обоб-
щенными силами по линейному закону, он получил выражения для коэффи-
циентов. Принцип микроскопической обратимости в этом случае приводит к
соотношениям взаимности.
Если продифференцировать формулу B.3) по времени, то получается вы-
ражение, содержащее у, и Х{ в том виде, как они определены двумя послед-
ними формулами
4 ±i- B-б>
Это выражение определяет скорость возникновения энтропии. Формула A.2б>
определяет величину g. Она представляет собой скорость необратимого возник-
новения энтропии в малом элементе объема жидкости, движущемся с потоком
со скоростью v. [Действительная величина изменения энтропии элемента
объема, являющегося, конечно, неизолированной системой DS/Dt, представ-
ляет сумму g и члена (З/Зг - б), изображающего обратимое изменение энтропии
вследствие потока тепла.] Выражение для g [формула A.26)], как легко ви-
деть, имеет ту же форму, что и написанное выше равенство. Интуитивно можно
сравнить эти две формулы и получить выражение для J{ и Х(. Однако при
этом остаются неопределенными, во-первых, числовой множитель, а, во-вто-
рых, сами потоки, поскольку можно взять любую линейно независимую ком-
бинацию у,. Тем не менее соотношения взаимности справедливы при любом
выборе потоков и обобщенных сил, лишь бы комбинация последних в B.6)
§ 2. Термодинамика необратимых процессов 553
приводила к скорости возникновения энтропии. Однако необходимо заметить,
что потоки Ji9 определенные по этому методу с помощью формулы A.26),
не совпадают с потоками, фигурирующими в основной теореме Онзагера и
определенными формулой B.4). Например, поток тепла е, как известно, не
может быть выражен в виде производной по времени от какой-либо макроско-
пической переменной состояния. Применение соотношений взаимности к пото-
кам и обобщенным силам в том виде, как они обычно определяются, требует
дальнейшего обоснования. Доказательство применимости соотношений Он-
загера к различным частным случаям даны в работе [26 ].
Макроскопические переменные системы определяются с помощью микро-
скопических переменных, а именно с помощью координат и импульсов всех
частиц. Определение обычно дается в форме средней величины или интеграла.
При выводе соотношения взаимностей Онзагер молчаливо предполагал, что
все макроскопические переменные wt являются четными функциями скорости
частицы, т. е. они не меняют знака при инверсии времени. Казимир и Теллеген
[26 ] рассмотрели более общий случай, в котором считается, что некоторые
переменные являются нечетными функциями. Они показали, что аи симмет-
ричны по индексам, если оба индекса i и / относятся одновременно к четным
или одновременно к нечетным переменным, в противоположном случае ai}
антисимметричны по индексам.
В применениях соотношений взаимности, которые будут рассмотрены в
этом параграфе, в качестве переменных фигурируют лишь концентрации п1У
средняя массовая скорость v и температура Т. Эти величины связаны с микро-
скопическими переменными формуламиC.15)—C.17) гл.7. Вблизи положения
равновесия функция распределения симметрична по скоростям, поэтому щ и Т
будут четными функциями молекулярных скоростей, a v — нечетной функцией.
Следовательно, аи (связывающие процессы диффузии и теплопроводности)
будут симметричны относительно i и /, тогда как коэффициенты, связываю-
щие эффекты вязкости с любыми другими процессами, будут антисимметричны.
Однако, как будет показано в разд. Б, в изотропной среде не существует
связи между эффектами вязкости и теплопроводностью или диффузией.
Б. ПРИМЕНЕНИЕ ТЕРМОДИНАМИКИ НЕОБРАТИМЫХ ПРОЦЕССОВ
К ЯВЛЕНИЯМ ПЕРЕНОСА1)
Применим соотношения взаимности Онзагера для изучения явлений пере-
носа в жидкостях, в которых пренебрегается переносом энергии путем излу-
чения. Скорость возникновения энтропии в потоке под действием необратимых
Таблица 100
Потоки и обобщенные силы в движущейся жидкости
Поток, Ji Обобщенная сила (сродство), Xt
m пц \i
e = q - Г
п [определена формулой
A.24)]
— Af [определена формулой A.27)]
_ j_
T dr
Yi [определена формулой A.23)]
процессов дается формулой A.26). Сравнение этой формулы с формулой B.6)
позволяет произвести соответствующий выбор выражений для потоков и обоб-
щенных сил. Эти величины представлены в табл. 100, где для удобства введен
1)См. [29].
554 Гл. 77. Гидродинамические применения уравнений переноса
тензор Р, определяемый как тензор давления без гидростатического члена
P = p-pU. B.7)
Предположим, что система находится вблизи равновесия, так что к этому выра-
жению можно применить линейный закон. Это приводит к выражениям плот-
ности потоков, эквивалентных первому приближению Энскога, т. е. выраже-
ния плотности потоков линейны относительно первых производных макро-
скопических переменных и не содержат высшие производные или степени
первой производной. Линейный закон можно также использовать при получении
предельной формы выражений для скоростей химических реакций вблизи сос-
тояния химического равновесия. Однако, поскольку последние результаты имеют
ограниченную область применимости, они здесь обсуждаться не будут.
Если рассматриваемая система находится в состоянии, близком к поло-
жению равновесия, то в соответствии с линейным законом [формула B.1)]
каждая компонента потоков может быть записана в виде линейной ком-
бинации всех компонент обобщенных сил. Однако можно показать, что в случае
изотропных сред члены, соответствующие связи тензоров, порядок которых
отличается на нечетное число, будут отсутствовать. Например, отсутствует
сеязь между j/ или е (тензор первого ранга) и Р (тензор второго ранга) или rt
{тензор нулевого ранга). Однако между потоком энергии е и потоком масс j,
связь существует и она приводит к появлению эффектов Соре и Дюфура. Суще-
ствует также связь между потоком импульса и скоростями химических реакций,
поскольку описывающие их тензоры отличаются на два ранга. Эта связь про-
порциональна следу обобщенной силы —(Э/Эг)у. Такой эффект не изучен экспе-
риментально, и в дальнейшем он обсуждаться не будет.
В следующем разделе будет показано, как можно применить термодина-
мику необратимых процессов для изучения потока импульса в жидкости, в
которой отсутствуют химические реакции. Будет показано также, как можно
вывести тензор давления и как можно вычислить вклад эффектов вязкости в
скорость возникновения энтропии. В дальнейшем будут изучены потоки масс
и энергии и определен вклад процессов диффузии и теплопроводности в выра-
жение для скорости возникновения энтропии.
В. ПРИМЕНЕНИЕ ТЕРМОДИНАМИКИ НЕОБРАТИМЫХ ПРОЦЕССОВ
К ПЕРЕНОСУ КОЛИЧЕСТВА ДВИЖЕНИЯ
Применение линейного закона [формула B.1)] к компонентам потока Р
и обобщенным силам —C/Эг)у приводит к следующим результатам :
здесь t?!, v2, и t;3 — компоненты v вдоль осей х, у, г соответственно. Совокупность
феноменологических коэффициентов afj образует матрицу, состоящую из 81 эле-
мента. Однако не все эти элементы независимы. Например, из соотношений
взаимности Онзагера следует, что
o!fj = ag. B.9)
Кроме того, при статистическом выводе уравнений переноса тензор давления
определяется как симметричный. Такое определение необходимо, пос-
кольку в противном случае на каждый малый элемент объема будет дейст-
вовать момент кручения. В соответствии с этим а$ должны удовлетворять
следующим требованиям :
*% = "$. B.10)
§ 2. Термодинамика необратимых процессов 555
Комбинируя два последних результата, получаем
< = <• B.11)
Теперь можно записать формулу B.8) в более простом виде
1 I 9**Л ^ /О 1О\
+ -&[) • BЛ2>
Благодаря найденным соотношениям симметрии существует лишь 21 неза-
висимый коэффициент а$. Это «упругие постоянные» среды. Эти же условия
симметрии можно получить из услтеия, что Р тождественно равно нулю для
движений, соответствующих перемещению или вращению тела как целого.
Следовательно, в этом случае соотношения взаимностей Онзагера имеют про-
стую физическую интерпретацию. Если среда обладает каким-либо элементом
симметрии, то число независимых упругих постоянных значительно меньше 21.
В частности, для изотропной среды связь между Р и [(d/9r)v + (8/9r)vt] инва-
риантна относительно вращения координатной системы. В этом случае можно
показать, что а)] имеют такую форму, что1)
] + D)?-«)D-.v)u> B.13)
где г] и х — независимые коэффициенты, известные под названием соответ-
ственно коэффициентов сдвиговой и объемной вязкости2). Эта форма следует
непосредственно из условий изотропии, поскольку единствеными тензорами,
от которых может зависеть Р, являются U, C/8r)v и транспонированная вели-
чина [(9/3r)v]t. Следовательно, Р должно быть линейной комбинацией этих
трех тензоров. Поскольку антисимметричная комбинация [(9/3r)v] и [C/3r)v]t
запрещена условиями симметрии, изложенными в предыдущем параграфе,
то единственная линейная комбинация этих тензоров имеет вид B.13). Таким
образом, в изотропной среде существуют, вообще говоря, два коэффициента
вязкости. Сдвиговая вязкость rj играет важную роль в потоках, в которых
последовательные плоские слои жидкости движутся с различными скоростями.
Коэффициент объемной вязкости к оказывается существенным при процессах
расширения среды [33].
Из A.26) и B.13) следует, что вклад эффектов вязкости в выражение для
скорости возникновения энтропии равен
где S—тензор «сдвиговой скорости», определенный формулой D.16) гл. 7.
Это выражение можно преобразовать к виду
)\ B.15)
Множители, стоящие при rj и и, являются суммами квадратов и, следовательно,
оба положительны. В соответствии со вторым законом
Sv>0 B.16)
при всех (Э/Эг)у. Из условия того, что один из множителей может быть равен
нулю, тогда как другой не равен нулю, следует, что
к>0. B.17)
х) Эта трактовка по существу совпадает с рассмотрением Сен-Венана [31] и Стокса [32].
Интересно отметить, что при этом делается лишь одно предположение, что нормальные и
сдвиговые напряжения являются линейными функциями скоростей деформации.
2) Коэффициент объемной вязкости обсуждается в гл. 7, § б в связи с кинетической
теорией многоатомных молекул и в гл. 9, § 3 в связи с теорией плотных газов.
556 Гл. 11. Гидродинамические применения уравнений переноса
Кинетическая теория разреженных газов в приближении Навье—Стокса
(как изложено в гл. 7) приводит к выражению для тензора давления, полностью
согласующемуся с формулой B.13). В этом пределе, однако, коэффициент
объемной вязкости х равен нулю. Интегральные выражения коэффициента
сдвиговой вязкости идеального газа, данные в гл. 7, приводят к положительным
значениям коэффициента вязкости в соответствии с формулой B.17).
Г. ПРИМЕНЕНИЕ ТЕРМОДИНАМИКИ НЕОБРАТИМЫХ ПРОЦЕССОВ
К ЯВЛЕНИЯМ ПЕРЕНОСА МАССЫ И ЭНЕРГИИ
Рассмотрим теперь применение линейного закона и соотношений взаим-
ности Онзагера к процессам диффузии и теплопроводности в среде, состоящей
из v компонент. Если рассматривать только эти члены, то скорость возникно-
вения энтропии в соответствии с формулой A.26) равна
BЛ8>
Положение несколько усложняется тем обстоятельством, что не все потоки \t
линейно независимы. Действительно, по определению, скорости диффузии1),
V
2'j/ = 0. Следовательно, одно из \t можно исключить из формулы B.18).
В частности, исключим поток масс для fc-й компоненты \к. Тогда выражение для
g может быть записано в виде
Применяя линейный закон, можно получить следующие выражения для пото-
ков е и j,:
е =
B.20)
\фк
Постоянные a}j являются феноменологическими коэффициентами Онзагера,
удовлетворяющими соотношениям взаимности2). Индекс 0 относится к пере-
менной, обозначающей температуру; остальные индексы характеризуют ком-
поненты смеси. Пока феноменологические коэффициенты с произвольным
индексом к не определены.
Выражения для е и j, можно записать в другой форме так, чтобы к-я ком-
V
понента не играла особой роли. Из формулы B.21) и из того факта, что 2 h = О,
следует ых
^& B.22)
yi/I
]Фк i^k
х) Скорость диффузии определена в гл. 7, § 2.
2) В общем виде феноменологические коэффициенты являются тензорами. Соотношения
взаимностей Онзагера в этом случае приводят к выражениям вида
где значок f означает операцию транспонирования. Для изотропной системы коэффициенты
являются скаляром, умноженным на единичный тензор. Только последний случай и рас-
сматривается здесь.
§ 2. Термодинамика необратимых процессов 557
Уравнение B.21) будет формально справедливо и для i = k, если определить
«*; = -2*и, / = 0,1,2,3, ... , v . B.23)
Определим ajk так, чтобы выполнялись соотношения взаимностей
*jk = *ki=-2*n> / = 0,1,2,3,... ,v. B.24)
Используя эти выражения, формулы B.20) и B.21) можно записать в упрощен-
ном виде
^^ B.25)
2
Постоянные а^ симметричны и удовлетворяют линейным соотношениям [фор-
мулы B.23) и B.24)], которые можно записать в виде
t?iaij=s?iaji=l0- B-27)
Удобно выражения для в и j, записать с помощью величин d,, тесно свя-
занных с Л,. Эти величины d, являются обобщением тех величин йь которые
определены формулой C.27) гл. 7 в кинетической теории плотных газов. Опре-
делим обобщенные величины d, следующим образом1):
d = лит д _ лит ?р + лит у nXj. B.28)
' Р QP 9r QP •— ' J
Исключая из этой формулы Л, [см. формулу A.27)], получаем2)
„ у "'
П/ /Л/ Г e v ^4? n Y
— A; — > '*/ A/
Применяя соотношение Гиббса—Дюгема и используя свойства парциальных
молярных величин, легко можно показать, что ? <*/ = 0- Принимая во вни-
мание выражение для dz-, а также соотношения, данные формулой B.27), можно
х) Другое определение d/ получается путем умножения правой части формулы B.28)
на р/пкТ. Если, помимо этого, D// определить иначе, умножив его на пкТ/р, то выражение
B.33) остается неизменным. Эти определения обычно используются при изучении плотных
газов и жидкостей.
2) В случае смеси идеальных газов
V- = -1- (*&-) = кТ
1 п ' UxJt• р,хк XI •
Подставляя эти величины в формулу B.29), получаем выражение для d/, в точности совпа-
дающее с формулой C.27) гл. 7.
558 Гл. 11. Гидродинамические применения уравнений переноса
получить следующие выражения для потоков :
Г Эг Р2L п!Щ J
7 = 1
Эти выражения эквивалентны ранее полученным и имеют формальное сходство
с соответствующими выражениями, получаемыми в кинетической теории иде-
ального газа.
Кинетическая теория разреженных газов, изложенная в гл. 7, приводит
к следующим выражениям для плотностей потоков энергии и диффузии :
° = -*'llF-P2-$y*J> B-32)
*' = —ТГ IF + -T2mimJDu*j- B-33)
Величина А' тесно связана с коэффициентом термодиффузии [см. формулу D.65)
гл. 7]. Величины Dtj — обобщенные коэффициенты диффузии, a DJ — обоб-
щенные коэффициенты термодиффузии (эти величины определены в гл. 7, § 4).
Поскольку не все uj линейно независимы, то D/; связаны с atj не простым обра-
зом. В гл. 7 Dtj были определены условием
Du = 0. B.34)
В случае а/у однозначность достигается совершенно другим путем — с помощью
соотношений взаимности.
Установим связь между результатами кинетической теории разреженных
газов и термодинамики необратимых процессов. Совершенно ясно, что выпол-
няется следующий ряд соотношений взаимности :
<*/о = «о, = ?>Г. B.35)
Связь остальных коэффициентов не так ясна. Исключая «диагональные» эле-
менты из B.31) с помощью условия 21 d,- = 0 и сравнивая полученный резуль-
тат с формулой B.33), можно найти, что коэффициент Du связан с аи следую-
щим образом :
Это уравнение можно решить относительно аи
] B-37)
i
Отсюда видно, что Вц должны быть такими, чтобы эта линейная комбинация
была симметричной1). В случае двухкомпонентной смеси формула B.37) пере-
ходит в формулу
? B-38)
2) Для смеси, состоящей из v компонент, существует 1/2v(v—1) соотношений взаимности
и, следовательно, v(v—1) величин Ду могут быть выражены через x/2v{y—1) величин. На-
пример, величины D(j в кинетической теории разреженных газов могут быть выражены
через x/#(v—1) величин постоянных бинарной диффузии Щ для различных пар компонент
[см. D.44) гл. 7].
§ 2. Термодинамика необратимых процессов 559
и, следовательно, соотношения симметрии будут иметь вид
Dti = Dn. B.39)
В этом простом случае результаты тривиальны и их можно получить непосред-
ственно из формулы B.33), принимая во внимание условие JJ d, = 0. Однако в
общем случае соотношения взаимности налагают довольно серьезные огра-
ничения.
Для трехкомпонентной смеси формула B.37) переходит в формулу
. B.40)
Кинетическая теория, изложенная в гл. 7, приводит к следующему соотно-
шению между D12 и постоянными бинарной диффузии Л)ц:
Аналогичные выражения получаются и для других пи. Таким образом, для
разреженного газа
„ п2 пг па т1 т2
^ ®гг + п2 лI3 + л3 ЛI2) х B.42)
х [п3 /п| ^ -2Jз — /л, (в — п2 m2) „2I2 „2J3 -m1(Q- пг
Это выражение симметрично относительно индексов 1 и 2, и, следовательно, в
этом случае результаты кинетической теории согласуются с соотношениями
взаимности. Можно показать, что это согласование осуществляется в общем
виде в случае многокомпонентной смеси.
Скорость возникновения энтропии вследствие процессов теплопроводности
и диффузии дается формулой B.18). Вводя в эту формулу d, и используя соотно-
шение 2! d, = 0, получаем следующее выражение для g:
Тогда, согласно формуле B.32), а также формулам D.48) и D.65) гл. 7, получаем
для идеального газа
Так как каждый член этой формулы может в отдельности быть равен нулю, то,
принимая во внимание второй закон термодинамики, который утверждает, что
B.45)
получаем
Я>0 и -2>,7>0. B.46)
Д. СВОДКА РЕЗУЛЬТАТОВ
Выше в общем виде были получены выражения для плотности потоков r
среде. Суммируем полученные результаты,
а) Тензор давления [см. B.13)]
t4D)]D). B.47>
560 Гл. 11. Гидродинамические применения уравнений переноса
Коэффициенты rj и к — соответственно коэффициенты сдвиговой и объемной
вязкости. В разреженном газе, состоящем из молекул, лишенных внутренних
степеней свободы, коэффициент объемной вязкости я = 0 и статическое
давление соответствует 2/3 следа тензора давления. В гл. 7, § б было показано,
что при наличии внутренних степеней свободы объемная вязкость конечна,
а в гл. 9 показано, что объемная вязкость также не равна нулю в случае плот-
ных газов или жидкостей. Тем не менее в общем случае влияние объемной
вязкости мало, и с большой степенью точности этими членами можно пренеб-
речь.
б) Плотность потока энергии q определяется формулами A.18) и B.32).
Для однокомпонентной жидкости ее можно записать в виде
« = -*¦?> B-48)
где А — коэффициент теплопроводности. В случае смеси газов q с большой
•степенью точности можно записать в виде
Ч = -*%г + 2 л,"Д., B.49)
тде V/ — скорость диффузии компоненты i. Дополнительный член соответ-
ствует потоку энергии в результате процессов диффузии. Это выражение для q
«аписано с точностью до членов, соответствующих эффекту Дюфура, прямому
влиянию градиентов концентрации и давления на поток энергии. Эффект
Дюфура, являющийся процессом, обратным термодиффузии, вообще говоря,
мал и им обычно перенебрегают.
в) Скорость диффузии /-й компоненты [формула B.33)], выраженная
•через dz [формула B.29)], имеет вид
< = 7i77 2 mJDu*J --ЩЩ-5Г- <2-50>
Последний член в этом выражении соответствует прямому эффекту темпера-
турного градиента. Этот эффект называется термодиффузионным эффектом или,
применительно к жидкости, эффектом Соре. Вклад от такого эффекта обычно
мал и в большинстве случаев им можно пренебречь. Оставшиеся члены зависят
от dy, определенных формулой B.29). Первый член в uj изображает прямой
эффект градиента концентрации. В разреженном газе этот член просто равен
градиенту молярной доли /-й компоненты. Дополнительные члены соответ-
ствуют градиенту давления (диффузионное давление) и внешним силам. В фор-
муле B.50) Ьи, определенные условием Du = 0, известны под названием коэф-
фициентов многокомпонентной диффузии. Эти коэффициенты зависят от состава
газа или жидкости. В частном случае бинарной смеси удобно применять JZ512
для обозначения D12 и определить
jj- B-51)
Если использовать это определение, то выражение для скорости диффузии
[формула B.50)] в частном случае бинарной смеси примет вид
V, = -?¦ т2 JZI2 [d, - kT *$L]. B.52)
Поскольку коэффициенты бинарной диффузии симметричны, JZI2 = JD21y а ско-
рости диффузии определены так, что выполняется условие 2 nimiSi = 0, то
можно написать
?ф ЗД B-53)
§ 2. Термодинамика необратимых процессов 561
В случае разреженных газов этот результат можно обобщить, получив фор-
мулу D.48) гл. 7 для скоростей диффузии в многокомпонентной смеси
^ щщ ГУ У)й д1пТ V ЩП! { dTj D^)
п.т.) •
Эмпирически установлено, что эти результаты также хорошо применимы к
плотным газам и жидкостям.
Гидродинамические уравнения Навье—Стокса для многокомпонентной
смеси с учетом химических реакций определяются формулами A.1) — A.4),
причем тензор давления дается формулой B.47), плотность потока энергии —
формулой B.49), а скорости диффузии — формулой B.50). Эти выражения
плотности потоков, а следовательно, и уравнения Навье—Стокса применимы
одинаково хорошо как к плотным газам или жидкостям, так и к разреженным
газам при условии, что градиенты скорости, концентрации, температуры и
давления умеренно малы.
При решении задач диффузии1) удобно выражать уравнения непрерывности
через среднюю по компонентам скорость
«=v + 4^nyVy. B.55)
Заменяя с помощью этой формулы v в формуле A.1), получаем уравнения
непрерывности в форме
Если все эти уравнения сложить, то получается уравнение непрерывности
Из формулы B.57) видно, что ш — постоянная, не зависящая от координат и
времени, которую легко определить из граничных условий, если воспользо-
ваться следующими условиями :
1) химические реакции не изменяют общего числа молекул в системе
2 Kj = 0 ;
i 2) температура и давление в системе постоянны, так что п — постоянная ;
3) поток безвихревой, т. е. [(З/Эг) хш]=0.
Подставляя явные выражения для скоростей диффузии [формула B.52),
а также A.2) гл. 8 ] в формулу B.56), можно в случае разреженного газа, состоя-
щего из двух компонент, получить следующее выражение для К:
Э1"Р
Г
^щ Х2 - maXl) + kT !?L}) • B.58)
Легко видеть, что формула B.58) сводится к обычному выражению закона
диффузии Фика [формула A.12) гл. 8] для двухкомпонентного разреженного
нереагирующего газа, находящегося в замкнутом сосуде при постоянных тем-
пературе и давлении.
х) Решение многих задач диффузии дано в книге Иоста [34]. Решения некоторых част-
ных задач диффузии в условиях, когда коэффициент диффузии зависит от концентрации»
приведены в работах [35, 36].
562 Гл. 11. Гидродинамические применения уравнений переноса
§ 3. ПЕРЕДАЧА ЭНЕРГИИ ИЗЛУЧЕНИЕМ1)
Передача энергии излучением существенна в системах с высокими тем-
пературами. Если среда достаточно прозрачная, то энергия в виде излучения
может передаваться на большие расстояния. Примером этого может служить
нагрев комнат радиаторами или электрическими нагревательными при-
борами. Если среда не прозрачная, то излучение пройдет небольшой
случайный путь с момента испускания до момента поглощения. Среднее рас-
стояние между этими точками известно под названием «средней длины свобод-
ного пробега» излучения, поэтому теория передачи энергии излучением в не-
прозрачной среде по терминологии очень похожа на кинетическую теорию
диффузии. Именно такова природа поведения излучения в расплавленном
железе в плавильных печах. Другим примером может служить излучение в
горячих газах в двигателе Дизеля.
В гл. 12, § б развита теория испускания и поглощения излучения. В при-
сутствии излучения молекула может с определенной вероятностью испустить
или поглотить свет некоторой характеристической частоты, переходя при этом
из одного квантового состояния в другое. Молекула в возбужденном состоя-
нии имеет определенную вероятность спонтанно излучить свет такой же харак-
теристической частоты [38, 39]. Поэтому каждое вещество обладает своим
спектром поглощения, который можно охарактеризовать с помощью коэффи-
циента поглощения /*„, являющегося функцией частоты. Поскольку спектр
поглощения зависит от распределения молекул по различным квантовым со-
стояниям, то /Ltv зависит также от температуры вещества.
В гл. 13, § 7 показано, что спектральные линии изолированных молекул
имеют естественную ширину (благодаря спонтанному излучению света) и
вследствие взаимодействия молекул эти линии уширяются (уширение вслед-
ствие давления) и смещаются (вследствие деформаций самих молекул). Таким
• образом, коэффициент поглощения зависит от плотности системы или от ее
агрегатного состояния. Теория функций /^(Т, q) изложена в гл. 12 и 13. Однако
теоретический расчет этих функций очень труден2), и в настоящее время лучшие
выражения для этой функции, за исключением немногих астрономиче-
ских примеров [23, 24], получены эмпирически. После того как излучение
много раз поглотится и снова испустится, интенсивность излучения как функ-
ция частоты будет характеризовать скорее температуру тела, чем его состав.
Такое тепловое излучение известно под названием излучения абсолютно чер-
ного тела3). Таким образом, можно измерить температуру по интенсивности излу-
чения — чем тело менее прозрачно, тем меньший оптический путь необходим,
чтобы получить такое излучение.
Из простых рассуждений ясно, что поток излучения в любой точке гх
можно выразить в виде интеграла по всему пространству. Излучение, прихо-
дящее в эту точку, может быть испущено веществом в другой точке г2. Поток
света может попасть прямо в некоторую точку из источника излучения, а мо-
жет по дороге и рассеяться4). Обычно в большинстве интересующих riac задач
*) См. [23—25]; большая часть материала этого параграфа взята из работы [37].
2) Приближенный расчет спектров излучения двухатомных газов был проделан Пенне-
ром [40].
3) Излучение абсолютно черного тела подробно рассматривается во многих монографиях,
например ^38, 41 ].
4) Рассеяние сильно усложняет расчет передачи энергии путем излучения. В работе
[23] показано, что общее сечение рассеяния света на молекулах равно as = 512л7а2/ЗА4 см2}
где а — поляризуемость молекул, выраженная в cmz, а X — длина волны, выраженная в см.
Для видимого света А — порядка Ю-4 см ; для средней молекулы a — порядка Ю-23 смъ.
Таким образом, as — порядка 5 • Ю-26 см2. Средняя длина свободного пробега рассеяния равна
\/nas. Рассеянием можно пренебречь, если средняя длина свободного пробега рассеяния
велика по сравнению с размерами системы или велика по сравнению со средней длиной сво-
бодного пробега излучения. В газе, плотность которого равна п = 1019 молекул в 1сл*3, сред-
няя длина свободного пробега рассеяния равна 2 • 10* см. Рассеяние может играть важную
роль в мутных средах (см. гл. 12, § 7) или средах, содержащих дым, пыль или другие твердые
частицы в суспензии.
§ 3. Передача энергий излучением 563
рассеяние не играет заметной роли, поэтому в дальнейшем оно не будет рас-
сматриваться.
Если А — величина поглощенной энергии излучения в единице объема в
единицу времени, a R — величина испущенной энергии излучения в единице
объема в единицу времени, то дивергенция потока излучения q^ равна
A- C.1)
Обозначим коэффициент поглощения для света частоты v через рр(см2/г), а
величину испущенной энергии в 1 см*/сек в единичном интервале частоты около
v — через Rv. Пусть также qv(r) будет обозначать общий поток энергии в этой
области частот. В таком случае общую величину испущенной энергии R и об-
щий поток излучения q^ можно записать в виде
00
R(r)=JRv(r)dv, C.2)
<fr. C.3)
Путем геометрических рассуждений можно показать, что qv выражается через
Rv и nv :
1^) -^ ехр (- f ^ dl) dt,. C.4)
Здесь г12 = | тг — г21; интеграл Г /лр dl берется вдоль линии, соединяющей эти
о
две точки, a q — плотность вещества.
Если рассматривается одномерная задача в том смысле, что Rvupv — функ-
ции только х, то компоненты потока излучения в направлении осей у и z равну
нулю, а х-компонента ^ может быть выражена в виде одномерного интеграла1)
Ы,, C.5)
— «• X
где
xt
C.7)
Символ Ei (— т„(х2, х2)) обозначает интегральный логарифм.
А. ПОТОК ИЗЛУЧЕНИЯ В ДВУХ ЧАСТНЫХ СЛУЧАЯХ
Существуют два частных случая, для которых поток излучения можно
выразить достаточно просто : 1) непрозрачные среды, в которых поглощение
излучения настолько сильно, что излучение находится в локальном термо-
Я(*) [ехр ( т„ (х2, хд) + т„ (х,, Xi) Ei (- т„ (хг, х^)] C.6)
и
г) Декартовы координаты в формуле C.4) заменяются цилиндрическими координатами,
@2> пг> хг)- Интегрирование по всем 02 выполняется сразу и приводит к появлению множи-
теля 2л. Исключая далее д2 с помощью новой переменной s = (х\ + е!)^2» получаем двойной
интеграл qvx(xx) = — у2 J dx2 J Rv(x2){xJs2)qx^[—tv(x2, x^s/x^ds. Интегрирование по s
-« о
приводит к формуле C.5).
564 Гл. 11. Гидродинамические применения уравнений переноса
динамическом равновесии с веществом; 2) прозрачные среды с небольшим
поглощением энергии.
1. Интенсивное поглощение излучения и метод Россе ланд а. В неограни-
ченной среде, находящейся при постоянной температуре, плотность энергии
излучения ev (в единичной области частот) будет характерной для черного тела
при температуре Т среды, с которой излучение находится в равновесии,
Поскольку скорость света равна с, а коэффициент поглощения вещества равен
fivy то поглощенная вэтой области частот энергия равна сед/лр (на 1 см*)сек). В силу
условий равновесия испущенная энергия равна поглощенной энергии, так что
Rv = cevQfiv. C.9)
Таким образом, уравнение C.4) можно записать в виде
(ЗЛО)
Рассмотрим теперь систему, в которой температура медленно меняется на
расстоянии (в/*,) для любых частот. Тогда в первом приближении Rv опре-
деляется формулой C.9), а е„(г2) зависит от локальной температуры T(t^.
Тогда ev в точке г2, расположенной вблизи г1У можно разложить в ряд Тэйлора
и ограничиться первыми двумя членами
еХч) = еМ + [т? • (г, - О] • C.11)
Коэффициент поглощения меняется достаточно медленно, поэтому его можно
считать локальной постоянной.
Интегрирование первого члена ev(rr) в формуле C.10) дает нуль, а интег-
рирование второго члена приводит к выражению1)
Иначе говоря, поскольку ev есть функция лишь локальной температуры,
то это выражение можно переписать в виде
с dev дТ /Q 1О\
Общий поток энергии, равный в соответствии с формулой C.3) интегралу от
qv по всем частотам, можно записать в виде
с Гр 1 dep А 1 дТ
L0 J
Росселанд [25] определяет среднюю длину свободного пробега для теплового
излучения следующим образом :
_ aR d(aT*) дт m<tt
Это интегрирование легко выполняется в сферических координатах.
§ 3. Передача энергии излучением 565
Здесь а — постоянная Стефана—Больцмана G,67 • Ю-15 эрг • см~* • град-4);
аТ4 — плотность энергии излучения черного тела, равная
00
aT*=fevdv. C.16)
о
Сравнение формул C.14) и C.15) приводит к выражению1)
Определение длины свободного пробега, данное Росселандом, основано на
физической картине движения фотонов со скоростью света в случайных направ-
лениях со средним шагом XR. С этой точки зрения формула C.15) — простое
выражение закона диффузии фотонов, которое можно получить с помощью
кинетической теории.
2. Слабое поглощение излучения (несветящиеся пламена и т. д.). Сущест-
вует много случаев (например несветящиеся пламена [42]), когда величина
поглощаемой энергии А мала по сравнению с величиной испускаемой энер-
гии R. В этих условиях величиной А можно пренебречь ; тогда формула C.1)
принимает вид
(^) C.18)
причем формула C.4) принимает вид
k^. C.19)
Существуют два типа процессов испускания — тепловое излучение и хемилю-
минисценция. Величина энергии теплового излучения Rv выражается формулой
C.9) как функция /у> и плотности излучения черного тела ev. Излучение хеми-
люминисценции зависит от кинетики фотохимических реакций, и каждый от-
дельный случай должен рассматриваться особо. Согласно экспериментальным
данным, излучение пламен в основном является комбинацией хемилюминис-
ценции и теплового излучения, причем хемилюминисценция играет преобла-
дающую роль2). Изменения удельной теплоемкости, связанные с релаксацион-
ным процессом перехода энергии между поступательными и внутренними сте-
пенями свободы [44]3), приводит к появлению нетеплового излучения, которое
может быть поставлено в один ряд с хемилюминисценцией.
В случае теплового излучения поток излучения можно определить с по-
мощью коэффициента поглощения /г(Г). Однако в инженерной практике дан-
ные по излучению выражаются с помощью эффективной испускательнои спо-
собности е, которая зависит от размеров экспериментальной аппаратуры, а
также плотности (или давления) вещества.
Рассмотрим излучение среды, заключенной в полусфере радиуса L, темпера-
тура и плотность которой постоянны. В той части, которая касается излуче-
ния, вещество вне оболочки полусферы может не приниматься во внимание.
г) В таком изложении пренебрегают вынужденным излучением. Учет этого эффекта
приведет к появлению множителя [1 —ехр(—hv/kT)] у коэффициента поглощения в фор-
муле C.17). См. работы [23—25].
2) См. книгу Гейдона [44], которая содержит наиболее полный анализ различных меха-
низмов излучения, встречающихся в пламенах, а также многочисленные ссылки на другие
работы.
3) Теоретическое исследование времени молекулярной релаксации дано в работах
[45, 46]. Экспериментальные результаты изложены в работах [47—49].
566 Гл. 17. Гидродинамические применения уравнений переноса
В этих условиях формулу C.4) можно проинтегрировать (в сферических коор-
динатах), причем получаемая аксиальная компонента потока в центре полу-
сферы имеет вид
1'=т?A-*г**)- <3-20)
Поскольку излучение является тепловым, то R9 определяется формулой
C.9); поэтому
C.21)
Общая аксиальная компонента потока излучения в этой точке равна
1r = jq, dv = \ аТ* 1 - -^ J e,er«^ dv . C.22)
Множитель саТ4/4 представляет собой поток излучения, испускаемый абсолют-
но черным телом при температуре Т. Поэтому второй Множитель можно интер-
претировать как эффективную испускательную способность
eG\ QL) = 1 -^eve~^dv. C.23)
о
Величины е(Г, qL) табулированы для небольшого числа веществ1). Эти испу-
скательные способности соответствуют эффективным коэффициентам погло-
щения /гG\ qL), определяемым отношением
f ev e-QpvL dv
o__ . C.24)
I evdv
о
Эффективный коэффициент поглощения можно выразить с помощью испуска-
тельной способности посредством формулы
/• =--^111A^6G,8^). C.25)
Для определения потока излучения необходимо во всех предыдущих
формулах /lcv заменить на /хG, qL). При этом, конечно, возникают ошибки, во-
первых, вследствие того, что не принимается во внимание частотная зависи-
мость коэффициента поглощения, а, во-вторых, потому, что в практических
задачах со сложной геометрией неизвестно, какое значение необходимо брать
для qL.
Б. СТАЦИОНАРНЫЙ ФРОНТ ИЗЛУЧЕНИЯ
Если в движущемся теле поддерживается высокая температура, то окру-
жающая среда постепенно прогревается и температурное распределение
в ней стремится к некоторому стационарному состоянию.
Рассмотрим простейший пример стационарного фронта излучения. Пред-
положим, что бесконечно большая плоская пластина, имеющая постоянную
температуру 70, движется со скоростью v в однородой среде с постоянной
удельной теплоемкостью С„ перпендикулярно своей плоскости. Если прене-
х) См. работы [23—25], а также [50, 51 ]. В работе [48 ] определены е для С02, Н20 вплоть
до 2000° С.
§ 4. Теория распространения звука 567
бречь кинетической энергией и теплопроводностью жидкости, а также считать
внешние силы равными нулю, то уравнение энергии [формула A.4)] в стацио-
нарном случае (dO/dt = 0) принимает вид
здесь х—расстояние от движущейся стенки, a qRx — компонента плотности
потока излучения, перпендикулярного стенке. Формулу C.26) можно сразу
проинтегрировать; при этом получается следующий результат:
qvCv (Т - Т.) = - Яях + (ft*)- • C.27)
Индекс оо соответствует х = оо. Предположим, что величинами Т«, и (qRx)<o
можно пренебречь. Если поглощение настолько велико, что плотность потока
излучения можно выразить через среднюю длину свободного пробега излу-
чения Росселанда, то формула C.27) принимает вид
с Т cXr d(aT*) dT __ 0 п Mv
Если средняя длина свободного пробега зависит от температуры
C.29)
где Ао — постоянная, не зависящая от температуры, то формулу C.28) можно
проинтегрировать, причем получается
fj^ = 0 . C.30)
Если положить
то формулу C.30) можно записать в виде
либо в виде
(^I/(s+3) C.33)
Постоянная х0 соответствует расстоянию от плоскости пластины х = 0, под-
держиваемой при температуре Го, на котором температура падает до нуля.
Излучение, * сопровождающее ударные волны или волны детонации, имеет
много общего с рассмотренным примером.
§ 4. ТЕОРИЯ РАСПРОСТРАНЕНИЯ ЗВУКА
b оставшейся части этой главы будут рассмотрены некоторые неравно-
весные процессы течения. Исходными уравнениями для анализа этих процес-
сов будут служить уравнения переноса (см. в § 1 и 2). В этом параграфе будет
рассмотрена теория распространения звуковых волн в жидкостях. В случае
бесконечно малых возмущений уравнения переноса можно упростить, прене-
брегая квадратами и более высокими степенями малых величин, так что эти
уравнения становятся линейными дифференциальными уравнениями.
Начнем с того, что пренебрежем «диссипативными членами», учитывающими
диффузию, вязкость, теплопроводность и химическую кинетику. Линеаризо-
ванные дифференциальные уравнения, получаемые в этом случае, приводят к
решениям в виде незатухающих волн, причем решение применимо к любой
568 Гл. 11. Гидродинамические применения уравнений переноса
жидкости. Если учесть диссипативные члены, то линеаризованные дифференциаль-
ные уравнения приведут к решениям в виде распространяющихся волн с по-
глощением. Коэффициент поглощения зависит от коэффициентов переноса,
химических реакций и любых других релаксационных процессов, протекаю-
щих в системе; получаемые при этом результаты применимы, строго говоря,
лишь в случае разреженных газов.
А. РАСПРОСТРАНЕНИЕ БЕЗ ПОГЛОЩЕНИЯ
При изучении распространения звука вместо уравнения энергии удобнее
рассматривать в качестве одного из уравнений переноса уравнение энтропии.
Если пренебречь диссипативными членами, приводящими к поглощению, то
уравнение энтропии [формула A.11)] принимает вид
В этом приближении энтропия элемента газа, движущегося со скоростью по-
тока v, остается постоянной. Для простоты предположим, что невозмущенная
система однородна и находится в покое, т. е., исключая возмущения в системе,
возникающие вследствие прохождения ударной волны, скорость потока по-
всюду равна нулю, а плотность и давление всюду постоянны и равны соответ-
ственно р0 и #0.
Пренебрегая диссипативными членами, уравнение движения [формула
A.3)] можно записать в виде
Это уравнение можно линеаризовать следующим путем. Во-первых, массовая
скорость v, появляющаяся вследствие прохождения звуковой волны, является
бесконечно малой величиной. Поэтому член [ev-(9v/9r)], включающий квадрат
малой величины, можно опустить. Далее, в члене e(9v/9/) плотность g можно
заменить равновесной величиной g0J так как сама производная dv/dt является
малой величиной. Таким образом, линеаризованное уравнение движения имеет
вид
Запишем это уравнение, вводя вместо давления р плотность д. Давление явля-
ется функцией § и q, но вследствие сохранения энтропии при прохождении
звуковой волны [формула D.1)] р является функцией только д и уравнение
можно записать в виде
? = <*?; <4-4>
здесь с0 равно1)
с ==
в условиях равновесия2). Величина с является термодинамической характери-
стикой системы. Из дальнейшего станет ясно, что с0 — скорость распростра-
нения звуковых волн. Таким образом, формула D.3) принимает вид
х) Для идеального газа р = дкТ/т и с = ] /у -Е- = / у —, где у = Cp/Cv =
У Q Г т
= 1 + (R/Cv). В реальных средах с слабо зависит от плотности.
2) Последнее выражение в формуле D.5) следует непосредственно из общих принципов
термодинамики.
$ 4. Теория распространения звука 569
Рассмотрим уравнение непрерывности A.2). Член [v-(8^/9r)] является малой
величиной второго порядка, поэтому линеаризованное уравнение непрерыв-
ности можно записать в виде
(|)O. D.7)
Уравнения D.1), D.6) и D.7) являются исходными в теории распростра-
нения звука без затухания. Из этих трех уравнений можно получить дифферен-
циальное уравнение второго порядка для плотности, решение которого позво-
ляет описать волновое движение. Если уравнение непрерывности продиффе-
ренцировать по времени, то получается
~W~ +
С помощью уравнения движения D.6) из формулы D.8) можно исключить v:
Решение этого уравнения имеет вид
^] D.10)
Это решение описывает гармоническую волну длины А и амплитуды д0 ф0У
бегущую в направлении z со скоростью с0. Общее решение записывается в виде
суперпозиции таких волн, каждая из которых характеризуется своим напра-
влением и длиной волны.
Подставляя формулу D.10) в уравнение движения D.6), получаем ком-
поненты скорости
^ = ^ = 0'. D.11)
Интегрирование по t приводит к1)
v2 = Cousin Ц- (z - с00; »х = vy = 0 • D.12)
Из этой формулы видно, что волна скорости с амплитудой фосо находится в
фазе с волной плотности. Поскольку плотность и давление связаны уравнением
адиабаты [формула D.4)], волны плотности и давления находятся в фазе,
причем амплитуда волны давления равна ^
Б. РАСПРОСТРАНЕНИЕ С ПОГЛОЩЕНИЕМ2)
Рассмотрим теперь роль диссипативных членов в уравнениях переноса.
В любой точке пространства, через которую проходит звуковая волна, газ
будет попеременно сжиматься и расширяться. Необратимость этого цикла,
приводит к поглощению энергии. Необратимость и, как следствие поглощения,
дисперсия вызваны следующими причинами : 1) вязкостью, 2) теплопровод-
ностью, 3) диффузией, 4) химическими реакциями и 5) конечностью времени
обмена энергией между различными степенями свободы молекул. Обмен энер-
гией математически может рассматриваться как частный случай химической
реакции.
Систему линейных дифференциальных уравнений, описывающих распрост-
ранение звуковой волны (с поглощением), можно получить следующим образом.
г) Аддитивная постоянная полагается равной нулю, так как считается, что до прохож-
дения звуковой волны жидкость находится в покое.
2)См. [52, 53].
570 Гл. 11. Гидродинамические применения уравнений переноса
Звуковая волна приводит к появлению малых возмущений. В соответствии с
этим каждая макроскопическая величина может быть записана в виде суммы
двух членов, один из которых соответствует невозмущенной величине, а дру-
гой — возмущению. Например, плотность можно записать в виде
e = a>[i+fl. D.13)
Если выражения такого типа подставить в уравнения переноса и отбросить
члены, являющиеся произведениями возмущений (как малые второго порядка),
и члены, содержащие лишь невозмущенные величины переменных (поскольку
эти величины сами удовлетворяют уравнениям переноса), то получится одно-
родная линейная система дифференциальных уравнений для возмущений.
Решение такой однородной системы линейных дифференциальных урав-
нений можно записать в виде суперпозиции гармонических волн различных
частот v. Например, возмущение плотности имеет вид
ф = ф0 е«а^-2ш)у a=z*L + iH. D.14)
Возмущения других величин имеют такую же форму. Величина ф0 — ампли-
туда, а а — комплексное Еолновое число. Действительная часть а равна произ-
ведению 2тг на обратную длину волны, а мнимая часть — коэффициенту погло-
щения к.
Если выражения для возмущений типа формулы D.14) подставить в сис-
тему линеаризованных уравнений переноса, то получим однородную систему
линейных алгебраических уравнений для амплитуд. Эта система уравнений
имеет нетривиальное решение, если детерминант, составленный из ее коэффи-
циентов, равен нулю. Из последнего условия находим а как функцию v. Если
в этом уравнении опустить диссипативные члены, то для а получим только
действительные значения. Это значит, что в отсутствие поглощения решение
совпадает с решением, приведенным в разд. А. Включение диссипативных
членов приводит к комплексной величине а, действительная часть которой
27г/Я очень просто связана со скоростью распространения, а мнимая часть к
является коэффициентом поглощения.
Поскольку поглощение звука вследствие процессов переноса, т. е. в отсут-
ствие химических реакций и релаксационных явлений, вообще говоря, мало,
величину к удобно разложить в ряд, сохраняя при этом лишь члены первого
порядка. В этом приближении к можно представить в виде суммы трех членов
x = xv + xx + xD. D.15)
Каждый член соответствует одному из процессов переноса. Вклад процессов
вязкости, теплопроводности и диффузии в многокомпонентной смеси можно
представить соответственно в виде
_ 4и»(Г-Р' "' Я „ .«
J J
yn -f 4>jk[njmj H^
Основной член в последней формуле можно преобразовать к виду
4512п1пк(Щ - mk)(mjDtj- mkDik). D.19)
4
§4. Теория распространения звука 571
Физически он соответствует диффузии вследствие градиента давления. Для
бинарной смеси1) xD можно записать в более простом виде посредством термо-
диффузионного отношения, определенного в формуле A.10) гл. 8:
D.20)
Из написанных выше выражений для к^ и яя видно, что эти величины
положительны. Кроме того, из последней формулы видно, что в случае бинар-
ной смеси kd положительно. Последнее утверждение, по-видимому, справед-
ливо и в общем случае, поэтому к всегда положительно.
В рассматриваемом до сих пор линейном приближении скорость распрост-
ранения звука считалась равной с0. Процессы переноса незначительно меняют
скорость звуковой волны (появляются члены второго и более высокого поряд-
ков, связанные с коэффициентами переноса).
Рассмотрим теперь влияние химических реакций на процесс поглощения.
В большинстве случаев оказывается, что химические реакции приводят к зна-
чительному увеличению поглощения и дисперсии, причем выражение для к
получается очень сложным [55, 56 ]. Однако если поглощение звука вследствие
химических реакций мало, то выражение для к можно разложить в ряд и огра-
ничиться линейным относительно постоянной скорости членом. Вклад в коэф-
фициент поглощения в результате химических реакций будет иметь вид
к. = }v 7, Щ У [(Аи); — (rjj — Bf) cv Г]2kf nbпЬк .. . D.21)
Здесь (Au)j — изменение внутренней энергии под действием /*-й реакции;
с9 — теплоемкость смеси в расчете на одну молекулу; Ду-— число молекул /-го
сорта, участвующих в /*-й реакции; (rjj — /fy) — прирост числа молекул в резуль-
тате реакции.
Переход поступательной энергии в энергию внутренних степеней свободы
молекулы можно рассматривать как частный случай химической реакции.
При этом предполагается, что газ состоит из молекул двух типов : невозбуж-
денных молекул А и молекул А*, обладающих некоторым избыточным запасом
энергии, сосредоточенным во внутренних степенях свободы. «Химическая»
реакция в этом случае записывается в виде
М + А^М + А*, D.22)
где Л4.может быть как молекулой Д так и молекулой А*. Этот тип химической
реакции был изучен Кнезером [57]. Скорость такой реакции характеризуется
временем релаксации т, определяемым следующим образом :
+ k'a)]-\ D.23)
где ка и к'а — постоянные скорости прямой и обратной реакций2). Пусть са —
часть теплоемкости при постоянном объеме на одну молекулу, связанная
с нерелаксирующими степенями свободы (или для нереагирующей смеси,
см. гл. 2, § 5). Добавка к теплоемкости при постоянном объеме на одну моле-
кулу вследствие релаксирующих степеней свободы или благодаря реакции
(в условиях полного равновесия) имеет вид
сг = к \fjrj е*кТ A + ef/"T)-2. D.24)
Пусть
cv = ca + Cl D.25)
будет удельной теплоемкостью смеси в условиях полного равновесия. Кнезер
1) Выражения для бинарной смеси получены Колером [54 ].
2) Более общее выражение для времени релаксации получено в работе [45 ].
572 Гл. 11. Гидродинамические применения уравнений переноса
показал, что в таком случае скорость звука выражается следующим образом :
а коэффициент поглощения имеет вид
н= kClWv*T
С [С* (CV+к)+ Са(Са +к) Bnvr)*] • V***1'
Скорость звука как функция частоты, даваемая формулой D.26), является
ступенчатой функцией и изображена на фиг. 155. Из этого выражения видно,
что в области низких частот по сравнению с характеристической частотой т
скорость звука дается обычным выражением с = VykT/m, где у— отношение
удельных теплоемкостей в реагирующей смеси.
В этом случае при прохождении звуковых
волн состав смеси меняется таким образом, что
в каждой точке, в каждый данный момент сос-
тав смеси находится в равновесии с локаль-
ными значениями температуры и плотности.
В области высоких частот в выражение для
скорости звука входит величина у для не-
реагирующей смеси, так как в этом случае сое-
In v тав смеси не успевает следовать за колебания-
ми звуковой волны и остается постоянным.
Кривая с2 как функция In v имеет точку пере-
гиба при значении частоты, равном
* —^ ^ 1 с
|;=_i_iL. D.28)
Фиг. 155. Зависимость скорости
звука и коэффициента поглощения Коэффициент поглощения на длину волны
от частоты. равен xc\v. Следует ожидать, что эта величи-
на будет мала как в области низких, так и
в области высоких частот. Эта кривая изображена на фиг. 155. Максимум
этой кривой соответствует точке
v = -!_ ]/*(* + *> , D.29)
2лт у са (са + к)
которая приближенно совпадает со значением частоты в точке перегиба для
верхней кривой фиг. 155. При са того же порядка, что и сю полуширина кривой
поглощения приблизительно равна 3,8 октав.
Интересные осложнения получаются при изучении поглощения высоко-
частотного звука. Если длина волны звука приближается к; длине среднего
свободного пробега молекул, то газ перестает вести себя как континуум и урав-
нения Навье—Стокса становятся несправедливы. Как указывалось в гл. 7, § 3,
решение уравнения Больцмана, данное Энскогом, является разложением
по степеням величины, которая в основном определяется отношением длины
среднего свободного пробега к характерной длине, на которой макроскопиче-
ские свойства заметно меняются. Уравнения Навье—Стокса получены из пер-
вого приближения Энскога. Приближение второго порядка соответствует
уравнению Барнетта. Для изучения поглощения коротковолнового звука
необходимо использовать высшие приближения.
Примаков [58 ], а позднее Цин и Шамберг [59 ] применили второе прибли-
жение, или уравнения Барнетта, для получения поправок к коэффициенту
поглощения, вызванных эффектами, связанными с приближением длины волны
к длине свободного пробега. Ван Чанг и Уленбек [60, 61 ] применили эти резуль-
таты для расчета дисперсии звука в гелии. Поправки к коэффициенту погло-
щения в высшем приближении становятся заметными в области коротких
длин волн. Трудности, возникающие вследствие применения высших прибли-
жений, оказываются настолько большими, что практически нецелесообразно
. § о. Распространение волн конечной амплитуды в одном измерении
573
идти дальше результатов, полученных Ван Чанг и Уленбеком. Кроме того, по
всей видимости, ряды Энскога обладают лишь асимптотической сходимостью.
Следовательно, для исследования свойств газа с большими макроскопическими
градиентами необходимо искать какие-то другие методы.
Мотт-Смит1) получил выражение для коэффициента поглощения звука
малой длины волны, используя уравнение Больцмана и не вводя уравнения
переноса. Если предположить, что функция распределения молекул по скоро-
стям лишь слегка отличается от функции Максвелла, то непосредственно из
уравнения Больцмана можно получить линейное уравнение, описывающее
распространение звуковых волн. Такой подход позволяет избежать трудностей,
присущих предыдущим методам, и не зависит от величины отношения длины
среднего свободного пробега к длине волны2). Оказалось, что коэффициент
поглощения в области высоких частот меньше, чем следовало ожидать, исходя
из уравнений Навье—Стокса, однако уравнения Барнетта приводят к правиль-
ному результату.
§ 5. РАСПРОСТРАНЕНИЕ ВОЛН КОНЕЧНОЙ АМПЛИТУДЫ
В ОДНОМ ИЗМЕРЕНИИ3)
Перейдем от рассмотрения волн бесконечно малой амплитуды к волнам
конечной амплитуды. Распространение волн конечной амплитуды в одном
измерении рассмотрим с помощью метода характеристик Римана, пренеб-
регая при этом эффектами вязкости,
теплопроводности и диффузии4). Как
следствие такого пренебрежения,
поток можно считать изэнтропичес-
ким. Однако в уравнении переноса
члены второго порядка не будут
отбрасываться.
Скорость звука с определяется
формулой D.5) как термодина-
. мическая величина. Можно по-
казать, что для любой реальной
жидкости скорость звука растет с
увеличением давления вдоль адиа-
баты. Тот факт, что скорость звука
больше в области сжатия и объя-
сняет образование ударных волн.
Рассмотрим синусоидальную волну,
распространяющуюся в положи-
тельном направлении оси х, как
показано на фиг. 156, а. В соответ-
ствии с теорией, развитой в преды-
дущем параграфе, любые малые
возмущения распространяются С Фиг. 156. Схемы образования ударной волны,
местной скоростью звука. Таким
образом; гребень волны распространяется быстрее, чем впадина, поэтому
спустя некоторое время волна примет вид, показанный на фиг. 156,6. В конце
в
г) Частное сообщение.
2) В аналогичной постановке вопрос о дисперсии и поглощении звука малых длин волн
теоретически исследовался в работах И. И. Ольховского [ЖЭТФ, 31, 238 A956); ДАН
СССР, 118, 468; 123, № 2; 123, 821 A958); Научн. Докл. Высш. школы, серия физ.-мат.
наук, № 4, 143, 181 A958)]. — Прим. ред.
3)См. [62].
4) Оригинальный метод ограничивается одномерным случаем в изэнтропическом при-
ближении. Однако, как будет показано дальше, этот метод с небольшими изменениями может
быть применен для численного решения трехмерных задач с учетом явлений переноса.
574 Гл. 11. Гидродинамические применения уравнений переноса
концов, спустя большой промежуток времени горб догонит впадину и волна
по виду станет похожа на зубья пилы, как показано на фиг. 156, в. Имеющий
место разрыв непрерывности во фронте такого зуба называется ударной
волной. Поведение ударных волн обсуждается в § 8.
А. МЕТОД ХАРАКТЕРИСТИК РИМАНА
Так же как и в предыдущем параграфе, исходными для построения теории
являются уравнения переноса. Как и раньше, удобно применять уравнение
энтропии [формула A.11)] вместо уравнения энергии [формула A.4)]. Пренеб-
регая диссипативными процессами, уравнение энтропии, уравнение непрерыв-
ности и уравнение движения можно записать соответственно в виде1)
Ы +v dz — e ~dz~
Таковы основные уравнения для развиваемой в дальнейшем теории.
Последние два уравнения определяют q и v как функции координат и
времени. Эти уравнения могут быть решены следующим методом Римана.
Рассмотрим некоторую термодинамическую величину o(q, S), характеризу-
ющую среду, которая будет определена в дальнейшем. Применяя оператор
[Э/Э* + г;(Э/Эг)] к cf(q, S) и используя уравнение непрерывности E.2), получаем
( 9 . д
Ы+vi*
а=
В последней формуле в соответствии с уравнением энтропии E.1) считается»
что энтропия элемента среды, движущегося со скоростью v, постоянна. Доба-
вляя к последнему уравнению уравнение движения E.3), получаем
Добавляя к обеим частям последнего равенства член c[(dvjdz) + (9o/9z)],
получаем
Аналогичным путем получаем выражение
Важным моментом метода является определение величины a(qy §). Эта
термодинамическая величина, называемая характеристикой Римана, опре-
деляется следующим образом :
a(QfS)= I — \do • E.8)
J \Q )
Qo{S)
*) Уравнение движения [формула E.3) ] в этой форме получается путем комбинации
формулы D.2) в одномерном случае с формулой E.1). Величина с определяется из термоди-
формулы D.2) в одномерно
намической формулы D.5).
§ 5. Распространение волн конечной амплитуды в одном измерении 575
Интегрирование в этой формуле ведется вдоль адиабаты, a qq(§) — любое фик-
сированное значение плотности. Величина Q0(S) не влияет на расчеты, поскольку
она не меняется в процессе расчета. Широко применяются следующие два опре-
деления qo(§) : 1) в качестве до(§) берется Плотность жидкости при стандарт-
ных условиях температуры и давления (не зависящая поэтому от S); 2) в ка-
честве до(§) берется плотность жидкости, удельная энтропия которой при стан-
дартном давлении равна S. Выбор того или иного значения qo(S) обычно опре-
деляется условиями задачи.
В соответствии с определением в(ду §) производная ее по плотности равна
Уравнения E.7) в таком случае упрощаются :
= 0> / = » + <>> E.10)
Из уравнения E.10) следует, что величина / = v + а остается постоянной
вблизи точки, движущейся со скоростью (я + с). Аналогично из уравнения
E.11) вытекает, что величина g = v — а остается постоянной около точки,
движущейся со скоростью (г; — с). Эти уравнения совместно с уравнением
энтропии полностью определяют гидродинамику газа. Уравнение энтропии
утверждает, что энтропия вблизи точки, движущейся со скоростью vy не зависит
от времени.
Предположим, что в некоторый начальный момент времени /0 скорость
v(z), плотность g(z) и удельная энтропия §(z) являются известными функциями
координат. Зная термодинамические свойства системы, можно найти величины
c(z) и o(z), а следовательно, f(z) и g(z). С помощью метода, изложенного в § 4,
можно найти значения величин /(г), g(z) и §(z) в некоторый более поздний
момент времени t0 + At. Зная эти величины, по следующим формулам можно
легко найти v и q
у [/(*) + ?(*)]> EЛ2)
^[f(z)-S(z)]. E.13)
Отсюда с помощью термодинамических соотношений можно найти c(z)
[и если желательно q(z), p(z) и T{z) ]. Этот процесс можно повторять до тех пор,
пока не будет получено полное решение гидродинамической задачи.
Метод Римана применим до тех пор, пока «характеристики» J(z) = (v + a)
и g{z) = (г; — о) остаются однозначно определенными. Однако в общем случае,
как будет видно из дальнейшего, возможно, что две различные /-характерис-
тики — А и /2 (начинающиеся в начальный момент времени соответственно в
точках zx и z2) — в дальнейшем пересекутся. При этих условиях скорость, плот-
ность и другие гидродинамические переменные не определены однозначно в
точке пересечения, поэтому точку пересечения следует считать местом, где
образуется ударная волна. Конечно, такого положения в действительности
не бывает. При приближении к точке пересечения характеристик градиенты
плотности, температуры и давления становятся большими. На некотором
расстоянии от точки пересечения градиенты становятся настолько большими,
что пренебрежение членами, учитывающими вязкость и теплопроводность,
становится незаконным. Тем не менее по пересечению характеристик можно
судить о месте и времени образования ударной волны.
Этот «метод характеристик» может быть применен для решения трехмер-
ных задач и задач, учитывающих вязкость, теплопроводность, диффузию и
внешние силы. В этих задачах основные уравнения, т. е. уравнения энтропии и
576 Гл. 11. Гидродинамические применения уравнений переноса
дифференциальные уравнения для / и g, имеют не равные нулю правые части.
Следовательно, /, g и <§ медленно меняются вдоль соответствующих кривых,
вместо того чтобы оставаться постоянными. Это, однако, не является серьезным,
препятствием для численных расчетов.
Б. ПРИМЕНЕНИЕ МЕТОДА ХАРАКТЕРИСТИК К СЛУЧАЮ
ИДЕАЛЬНОГО ГАЗА
Некоторое количество качественных сведений о распространении волн
конечной амплитуды можно получить, рассматривая простой случай идеаль-
ного газа с постоянной удельной теплоемкостью. Для такого газа можно напи-
сать следующие уравнения :
Уравнение состояния
p
Уравнение адиабаты
Скорость звука
Характеристика Римана
ff=7=Tc< EЛ7)
При определении характеристики Римана фиксированное значение плот-
ности полагается равным нулю. Обсудим теперь решение для идеального
газа с у = 3 или, более общее,
2/л + 1 ч
у = 2т_ y (ш ~"целое)-
1. у = 3 или т = 1. Решение гидродинамических уравнений особенно
просто в частном случае, когда у = 3. Эти решения грубо описывают распрост-
ранение волн конечной амплитуды в твердых телах, поскольку большинство
твердых тел по своему поведению при адиабатическом сжатии приближенно
соответствует идеальному газу с у = 3, т. е. удовлетворяют уравнению
(plp0) == (qIq0)*. (Это значение у нельзя путать с истинной величиной отношения
удельных теплоемкостей, которая в случае твердых тел лежит между 1 и 5/3.)
В этом случае скорость звука пропорциональна плотности, и характеристика
Римана в точности равна скорости звука
E.18)
Следовательно, / = (г; + с) постоянна вдоль кривой z = z{t\ наклон которой
равен (v + с), a g = (г; — с) постоянна вдоль кривой, наклон которой равен
(г; — с). В связи с этим / постоянна вдоль линии
z = a(f)+ft, E.19)
a g постоянна вдоль линии
Функции a(f) и b(g) определяются из начальных условий. Решая уравнения
E.19) и E.20) совместно, получаем
_ b(g) - a(j)
l~ t-g '
г = fb(g) - gfl(/> . E.22)
/ о
§ 5. Распространение волн конечной амплитуды в одном измерении 577
Вспоминая определения / и g, можно написать выражения
^0=y(/ + g), E.23)
Ф> 0 =!(/-?)• E.24)
Последние четыре выражения являются общим решением гидродинами-
ческих уравнений, записанным в параметрической форме. Зная скорость и
плотность как функции координат в начальный момент времени, можно найти
скорость звука c(z, 0), а также /(z, 0) и g(z, 0). Последние функции определяют
соответственно вид функций a(f) и b(g). Зная a(f) и b(g), по формулам E.21) и
E.22) можно определить значения z и t при любых желаемых / и g. С помощью
формул E.23) и E.24) можно найти соответственно v(zy t) и c(z, t). Таким образом,
учитывая, что с = co(qIqo), можно определить все характеристики движущейся
среды.
2. у = j:m+| (m — целое). Дарбу [63] получил решение гидродинами-
C.TYI 1 2/77 -4- 1
ческих уравнений в аналитическом виде для всех значений у = 2т _ ^ , где
т — целое число. Случай у = 3(т = 1) уже рассмотрен. Практический инте-
рес представляют еще два частных случая : т = 2, соответствующий идеаль-
ному одноатомному газу (у = 1,677), и т = 3, соответствующий воздуху
(у = 1,400). Решение Дарбу получается из решения следующих параметри-
ческих уравнений :
/ iyn-1 Г 8(m-l) f a(f) + b(g)
E.25)
)\v >
где a(/), &(g) и Z — параметры. Гидродинамические уравнения решаются таким
же образом, как и в случае у = 3.
В. ОБРАЗОВАНИЕ УДАРНЫХ ВОЛН В ИДЕАЛЬНОМ ГАЗЕ
Ударная волна, как указывалось выше, образуется при пересечении харак-
теристик, соответствующих различным значениям v + а. Приведенный ниже
пример показывает, когда могут возникать такие условия.
Рассмотрим идеальный газ, находящийся в момент времени / = 0 в сле-
дующих условиях :
О < г < оо газ находится в покое ; температура,
плотность и давление одинаковы;
— оо < z < 0 газ произвольно неоднороден.
Характеристики / и g изображены схематически на фиг. 157 как функции вре-
мени. На этой фигуре можно выделить три области :
/. Газ остается невозмущенным. Все характеристики — прямые линии.
Эта область ограничена прямой линией с наклоном (dz/dt) = с0, где с0 — ско-
рость звука в однородной среде.
II. Газ остается произвольно неоднородным. Эта область ограничена вол-
нистой линией с наклоном (dz/dt) = —с, где с — локальная скорость звука в
этих условиях.
III. Между областями I и II /-характеристики являются прямыми линия-
ми, g-характеристикам, пересекающимся с / (в силу того, что они возникают
в области /), соответствует одна и та же величина g = —2со/(у— 1). Наклон
578
Гл. 11. Гидродинамические применения уравнений переноса
/-характеристик, равный (v + с), определяется формулой
^ E.28)
и, следовательно, постоянен для каждой данной величины /.
Поскольку /-характеристики в области III — прямые линии с наклоном,
являющимся функцией самой величины /, то вполне вероятно, что две различ-
ные /-характеристики могут пересечься в точке S, как показано на фиг. 157.
Как раз в этой точке и образуется ударная волна.
Ф и г. 157. Поведение характеристик / и g в общем случае.
Для иллюстрации образования ударной волны рассмотрим поведение газа,
заключенного в цилиндр с поршнем, который может двигаться произвольным
образом. Положение поршня в любой момент времени характеризуется выра-
жениями
= W(t),
E.29)
Пусть движение поршня ограничено дозвуковыми скоростями.
На фиг. 158 /-характеристики с наклоном (v + с) отходят от поверхности
поршня. Предположим, что газ удовлетворяет уравнению адиабаты для идеаль-
Нет газа
Фиг. 158. Схема образования ударной волны.
§ 5. Распространение волн конечной амплитуды в одном измерении
579
ного газа, т. е. а = 2с/(у— 1). Так же как и в предыдущей задаче, проведем
линию с наклоном dzfdt = с0. Слева от этой линии (область /) газ остается не-
возмущенным. В любой точке на поверхности поршня (я — <х) = —2со/(У— 1)
[так как линии, соответствующие постоянным значениям величины (v — а),
возникают в невозмущенной части газа, как и в предыдущей задаче ]. Таким
образом, поскольку на поверхности поршня v = dW/dt, то в этой точке
_ 2с„
у-1
сг =
dW
dt
Учитывая, что с= [{у—1)/2) ] or и v = dW/dt, получаем
с = с0 Н 2 w >
-Г I
— <г0Н
dW
IF
E.30)
E.31)
E.32)
Из выражения E.31) видно, что если dWjdt положительно, то с>с0 и плот-
ность газа вблизи поверхности поршня возрастает. Если dW/dt отрицательно,
то вблизи поверхности поршня происходит расширение газа. Линия, начинаю-
щаяся в точке CvfJ, где dWjdt — малая положительная величина, будет иметь
Фиг. 159. Несколько примеров образования ударных волн.
а — поршень быстро вдвигается в газ, ударная волна образуется непосредственно у стенки ; б — поршень
постепенно вдвигается в газ, ударная волна образуется позднее ; в — поршень быстро удаляется из газа,
ударная волна не образуется.
меньший наклон, чем линия, начинающаяся в точке (z2, f2), где dWfdt — боль-
шая величина. Таким образом, если z2> zv то эти линии пересекаются в неко-
торой точке. Совершенно ясно, что чем меньше ускорение поршня, тем меньше
разность в наклонах линий, соответствующих постоянным значениям величины
(v + а), и тем больше время, проходящее до момента пересечения характери-
стик, т. е. до момента образования ударной волны. Таким образом:
1) если поршень движется ускоренно, то линии, соответствующие по-
стоянным (v + or), сходятся, приводя к образованию ударной волны;
2) если поршень движется замедленно, то линии, соответствующие постоян-
ным (я + а), расходятся и ударная волна не образуется.
Это два частных случая более общей теоремы, согласно которой ударная
волна всегда образуется при наличии достаточного количества времени, если
газ сжимать, и не образуется, если газ расширять. Некоторые примеры, иллюст-
рирующие эту теорему, изображены на фиг. 159.
О превращении синусоидальной волны в волну пилообразной формы гово-
рилось в начале этого параграфа. Расстояние, на котором происходит полное
превращение волны, можно определить следующим образом. Рассмотрим
сначала газ, находящийся в сосуде с поршнем. Предположим, что поршень
движется по синусоидальному закону с частотой v и малой амплитудой, гене-
рируя при этом звуковые волны: На достаточно большом расстоянии от поршня
580 Гл. 11. Гидродинамические применения уравнений переноса
звуковая волна примет пилообразную форму. Для определения расстояния,
необходимого для такого преобразования волны, найдем место пересечения
двух соседних характеристик. Рассмотрим две /-характеристики, выходящие
с поверхности поршня, как показано на фиг. 160. Точки (zly tL) и (z2, t2) нахо-
дятся рядом. Наклон т /-характеристики в соответствии с формулой E.32)
равен
т = (v + с) = с0 + ^±1 V, E.33)
где V — скорость поршня. Наклон характеристики,
выходящей из точки (z2, t2), равен
Ж Л " «} • E.34)
Используя эти соотношения, можно показать, что
расстояние до точки пересечения соседних характеристик имеет вид
¦»¦-„
= Ш ~ у + 1 dV
dt 2 dt
Предположим теперь, что закон движения поршня задается формулой
V = Vo sin 2nvt, V0<zc0. E.36)
В таком случае
или, выражая через длину волны А = cojv,
— - ( 2 1 с»
здесь Vo — амплитуда звуковой волны. Однако амплитуду чаще выражают
через перепад давления, который имеет вид
Р-Ро = УРо^- E-39)
Таким образом, расстояние до места образования ударной волны, выраженное
в единицах длины волны, как функция перепада давления в звуковой волне
имеет вид
^ - ( 2у 1 [ Ро
В качестве примера рассмотрим звуковую волну в воздухе (у = 1,4) при
давлении 1 атм и перепаде давления р — р0 = 0,001 атм. В этом случае волна
будет принимать пилообразную форму на расстоянии Лг = 186 длин волн.
Это значит, что все звуковые волны вырождаются в ударные волны малого
периода, поэтому упрощенная теория распространения звука не применима на
больших расстояниях. Кроме того, в изложенной теории пренебрегается вяз-
костью и теплопроводностью. Учет этих факторов приводит к поглощению
звуковой волны. Вязкость и теплопроводность еще имеют тенденцию размывать
разрыв непрерывности в ударной волне и, следовательно, тормозят процесс
§ 6. Одномерные уравнения переноса в стационарном случае 581
образования ударной волны. Действительное изменение формы волны проис-
ходит под действием двух конкурирующих процессов — процесса превраще-
ния синусоидальной волны в волну пилообразной формы и процесса погло-
щения.
§ 6. ОДНОМЕРНЫЕ УРАВНЕНИЯ ПЕРЕНОСА
В СТАЦИОНАРНОМ СЛУЧАЕ
В трех последующих параграфах проведено теоретическое рассмотрение
пламен, ударных волн и детонации ; все эти явления можно описать как уста-
новившиеся. В соответствии с этим математическое описание указанных трех
явлений имеет очень много общего. Поэтому целью настоящего параграфа
является обсуждение некоторых общих закономерностей установившихся
явлений и вывод основных уравнений, которые являются отправными для
выводов, проводимых в последующих трех параграфах. Мы получим здесь
также линеаризованные уравнения, описывающие поведение системы, когда
ее состояние лишь немного отклоняется от состояния химического и тепло-
вого равновесия.
Для простоты рассмотрение ограничено одномерным случаем систем в
стационарном состоянии, т. -е. систем, в которых свойства среды одинаковы
в параллельных друг другу плоскостях и являются функциями единственной
координаты z. В теории пламен система координат связана с горелкой. В теории
ударных волн и детонации система координат связана с движущейся волной.
Тогда значение z соответствует некоторому положению относительно волны,
и свойства в этой точке не зависят от времени. Большие положительные зна-
чения z соответствуют тем точкам, в которых газ уже прошел через волну и
достиг химического и теплового равновесия. Свойства потока при z = оо обоз-
начаются значком оо. Большинство соотношений, рассматриваемых в этом пара-
графе, приложимо к любым газам, при любых плотностях и любых температурах.
В ряде случаев даются отдельные формулы, которые, строго говоря, справед-
ливы только для разреженного газа, т. е. такого, который подчиняется урав-
нению идеального газа (р = пкТ) и явления переноса в котором описываются
теорией Энскога—Чепмена (см. гл. 7). В тексте ясно указывается, какие урав-
нения принадлежат к этой последней категории и в какой степени они могут
быть применимы к случаю, когда газ неразрежен. Предполагается, что поток
является реагирующей многокомпонентной смесью. Внешние сцлы и излучение
не рассматриваются, поскольку их включение усложнило бы выводы. Они
обычно не играют значительной роли в определении гидродинамики среды.
А. ОСНОВНЫЕ УРАВНЕНИЯ СИСТЕМЫ В ОБЩЕМ СЛУЧАЕ
Основные уравнения гидродинамики даны в § 1. Там уравнения непре-
рывности, движения энергии выражены через .плотности потоков: скорости
диффузии V,- (или плотности потоков массы j,), тензор давления р и вектор плот-
ности потока тепла q. Эти величины можно записать через коэффициенты пере-
носа и производные от переменных так, как это сделано в § 2. Следовательно,
можно переписать уравнения гидродинамики, использовав коэффициенты пере-
носа. В проведенном в последующих трех параграфах рассмотрении оказалось,
однако, более удобным сделать следующее. Мы записываем уравнение движения
через коэффициент вязкости и уравнение энергии через коэффициент тепло-
проводности. Однако уравнение непрерывности мы оставляем в первоначаль-
ной форме — выраженным через скорости диффузии. Затем в дополнение к
уравнениям переноса мы включаем соотношения, которые связывают скорости
диффузии с коэффициентами диффузии. Эти соотношения мы назвали «уравне-
ниями диффузии». Наконец, для описания поведения среды необходимо опреде-
582 Гл. 11. Гидродинамические применения уравнений переноса
лить уравнение состояния. Следовательно, основными уравнениями, которые
мы рассматриваем, являются : 1) уравнение непрерывности; 2) уравнение
движения, 3) уравнение энергии, 4) уравнение диффузии и 5) уравнение состоя-
ния.
1. Уравнение непрерывности. Полное уравнение непрерывности для од-
номерной системы в установившемся состоянии получается из уравнения A.2),
если положить производную по времени равной нулю. Так как никакие свой-
ства газа не зависят ни от х, ни от у, то мы получаем
¦йг^-о. (ел)
Это уравнение можно сразу проинтегрировать:
M = qv. F.2)
Определенная таким образом постоянная интегрирования М является плот-
ностью потока массы (на единицу поперечного сечения в плоскости ху). Вели-
чина М не зависит от координаты z.
Подобно этому уравнения непрерывности для отдельных химических ком-
понент [уравнения A.1)] запишутся в виде
4- [n((v+ V,)] = K(. F.3)
Удобно в качестве новой независимой переменной ввести величину Gh которая
является долей плотности потока массы, вносимой 1-й компонентой,
о, = тиа&
Выражая уравнения непрерывности через Gh получаем
Af-g*-=*!,*,. F.5)
Рассмотрим некоторое число линейно независимых Qt для системы, которая
состоит из 5 химических разновидностей и в которой протекает Г химических
реакций (обозначенных индексом /); пусть стехиометрическая запись этих
реакций имеет вид
... F.6)
Если kj и kj— постоянные скоростей прямой и обратной реакций соот-
ветственно, то результирующая скорость /-й реакции равна
О = ЧТ) пхг* п2%> • • • ~ */'0О п\ч п J; • • •, F.7а)
г, = kj(T) (JL)?" х\ч ф ... - к'(Т) (¦?)?" х\ч х%>... F.76)
Тогда величины Kt задаются в виде
Поскольку rjij и pij — постоянные, то отсюда следует, что наибольшее число
линейно независимых величин К/ не превышает V в соответствии с числом V
функций Гу. В некоторых случаях не все Гу линейно независимы; тогда число
линейно независимых К, может быть меньше Г. Например, если присутствует
а различных видов атомов, a vik — число атомов fc-го вида в молекуле z-ro
сорта, то обязательно существует а соотношений типа
24^ = 0, к =1,2,..., а. (б9)
§ 6. Одномерные уравнения переноса в стационарном случае 583
Соотношение* F.9) выражает тот факт, что при химических реакциях не проис-
ходит ни распада, ни образования атомов. Однако не все а соотношений в F.9)
в действительности линейно независимы, если в ходе химических реакций не-
которые группы молекул останутся неизменными. В этом случае число неза-
висимых соотношений F.9) равно числу независимых компонент (в смысле
правила фаз) а! <^ а. Таким образом, число I линейно независимых К( равно
либо (s — a'\ либо Г, в зависимости от того, что меньше. Из уравнения F.5)
ясно, что число линейно независимых величин О( то же, что и число линейно
независимых величин К*.
2. Уравнение движения. Уравнение движения выражено в A.3) через
тензор давления. В § 2 показано, что вид тензора давления может быть получен
в самой общей форме с помощью термодинамики необратимых процессов. Для
одномерного потока гг-компонента тензора давления записывается в виде
FЛ°)
Подстановка этого выражения в уравнение движения, записанного для уста-
новившегося состояния в одном измерении, дает
+ Ц^^Ы0 FЛ1)
Поскольку qv = М является постоянной величиной, то это уравнение можно
сразу проинтегрировать
(± )<? = B; F.12)
здесь В — постоянная интегрирования, которая зависит только от граничных
условий. Если в точке z = оо достигается полное химическое и тепловое равно-
весие, то (dvjdz)* = 0 и В = Mvm + р». Следовательно, для уравнения дви-
жения мы получаем окончательно
3. Уравнение энергии. Уравнение энергии выражено в A.4) через вектор
плотности потока тепла. Если пренебречь внешними силами и излучением,
то в одномерном установившемся случае это уравнение запишется в виде
Это выражение сразу интегрируется и дает
)?]=L' <бЛ5)
где L — постоянная интегрирования. Если в точке z = с» достигается полное
химическое и тепловое равновесие, то L = М(О + (р/о) + х/2 ^2)«- Величина
(О + (P/q)) является энтальпией единицы массы газа
/7 + ! = Д = -1^/пД.. FЛ6)
Величина Й((Т)—энтальпия на 1 г чистой компоненты L
Выражение для вектора плотности потока тепла через коэффициенты
переноса было получено в кинетической теории разреженных газов [см. соот-
584 Гл. 11. Гидродинамические применения уравнений переноса
ношение D.64) гл. 7]. Это выражение записывается в виде
Vk-Vj). F.17)
Строго говоря, этот результат применим только в случае разреженного газа-
Однако в соответствии с рассмотрением, проведенным в § 2, подобная запись
применима также и в случае жидкостей, за тем исключением, что' при этом
энтальпии, входящие во второй член, должны интерпретироваться как пар-
циальные молярные величины, а термодиффузионный член, который обычно
мал, должен быть несколько видоизменен. Подставляя выражение для вектора
плотности потока тепла в F.14) и используя уравнение движения F.13), в
конце концов получаем
- р V
4. Уравнение диффузии. Кинетическая теория разреженных газов дает
выражения, связывающие векторы плотности потоков массы (или скорости
диффузии) с коэффициентами диффузии и термодиффузии [см. соотношение
D.48) гл. 7 ]. Эти уравнения в одномерной задаче можно записать через вели-
чины Gt:
dz п ^
_ Л _ птЛ d\np M
Ч Q J dz '
1 din Г
+ n dz *2
Как было сказано в § 2, несмотря на то, что этот результат достаточно строго
применим лишь в случае разреженного газа, в некоторых случаях его при-
менение эмпирически обосновано и для описания процессов в жидкостях.
5. Уравнение состояния. Ради простоты мы часто предполагаем, что урав-
нение состояния можно представить в виде уравнения состояния идеального
газа
р = пкТ. F.20)
Давление р замеряется по отношению к движущемуся потоку газа.
Эта система из пяти уравнений определяет переменные р, xh Gh v и Т
как функции координаты z, если заданы граничные условия. В точке z = оо,
где достигается полное химическое и тепловое равновесие, производные от
каждой из этих переменных по z равны нулю. Из уравнений диффузии, а также
из условия, что все величины (dXtldz)* обращаются в нуль, следует
Из уравнений непрерывности и требования, чтобы {dGildz)» обращалось в
нуль, следует
К/(ху.>Т.) = 0. F.22)
Определение температуры при z = оо требует знания некоторых дополнитель-
ных граничных условий, например знания величин температуры при z = О
или z = — ©о.'
§ 6. Одномерные уравнения переноса в стационарном случае 585
Б. ОСНОВНЫЕ УРАВНЕНИЯ ДЛЯ СИСТЕМЫ ВБЛИЗИ
РАВНОВЕСИЯ1)
Рассмотрим основные уравнения, соответствующие установившемуся со-
стоянию, когда система лишь немного отклоняется от состояния химического
и теплового равновесия. Когда z приближается к ©о, уравнения становятся
линейными и выражаются через разность текущих значений переменных и их
предельных значений при г=°о.Эти линейные соотношения легко решаются и
дают асимптотические решения общих уравнений гидродинамики. В рассмат-
риваемых здесь уравнениях символ <5 обозначает разность между текущим
значением переменной и предельным значением; например, 6Т = Т — Тю.
Понятно также, что все полученные из уравнений величины выражаются
через их значения при z =00. Если имеется I независимых величин G, и s хими-
ческих компонент, то всего получается (I + s + 2) линейно независимых
функций. Мы возьмем в качестве независимых функций первые I из Gb первые
(s — 1) из xf и переменные Т, v и р. Остающиеся Qt связаны линейными комби-
нациями с первыми I:
Qt = 2xauQj + bi9 «' = (/+1), (Z + 2),...,s. F.23)
Постоянные a(j определяются соотношениями между К,, а постоянные bt
подбираются такими, чтобы получить правильное предельное значение Gt в
соответствии с F.21). Подобно этому, xs связано с первыми (s— 1) величинами
из
xs = 1 - 2xj. F.24)
Разложим теперь в ряды Тэйлора различные части уравнений переноса и огра-
ничимся членами первого порядка от дТ, 6vy dp, dG( и дх(. В таком приближении
уравнения переноса примут следующий вид:
1) Уравнение непрерывности.
(а) Сохранение общей массы
? (mi - т8) dxt
4
2* miXi
f-1
(б) Непрерывность отдельных компонент
¦ I
^ = Ао<ТдТ + Аа,рбр + $2 AG{X,dXj, i = 1,2,3, ...,/. F.26)
2) Уравнение движения
3) Уравнение энергии
Ш1 = Атт ЬТ + ATp dp + 2 ATXf dxj + 2 ATG, dGj. F.28)
4) Уравнения диффузии
dT + Bv ~W~ +BiT ~dF~ = 2 Ax* ** °XJ + 2,
F.29)
i= 1,2,3, ...,s-l
x) Это рассмотрение справедливо только для разреженного газа.
586 Гл. 11. Гидродинамические применения уравнений переноса
Коэффициенты А и В задаются соотношениями
л Ш{ 8 Ki
L
МкТ «ГсГ D,T x,dJ,xidT
Ду+ ? Й,аи]-
t-i+i J
М кТ Г «
ху ту .ф/у
ЛИ
м ~др~'
mt
F.33)
лГ Г^Г ~ "^rJ' F-32)
F-35)
F.366)
F.37)
F.38)
Уравнения F.25) — F.29) образуют тогда систему (/ + s + 1) линейных
дифференциальных уравнений первого порядка и одного линейного алгебраи-
ческого уравнения для того же числа зависимых переменных. Решения этих
переменных имеют вид
37 2f'(k)
- exp (a<*> z), F.41)
г) Частные производные от Kt по молярным долям можно получить из соотношений
(б.76) и F.8), если все s молярных долей будут линейно независимы.
§ 6. Одномерные уравнения переноса в стационарном случае 587
здесь f(k) — постоянные, определяемые из граничных условий. Если все fk\
кроме одной, приравнять нулю и полученные выражения подставить в выше-
приведенные соотношения, то мы получим систему линейных алгебраических
уравнений для а(к) и постоянных [x(fk), [Gtfk\ [pfk) и [vfk\ Этими урав-
нениями являются
8-1
пц-т8)[хг](к)
=0, F.42)
Ао,т + AalP [p](fc> + 'z Ао,м [*,]<*> - «<*> [О;]« = 0, / = 1,2,...,/, F.43)
1 = 1
+ [М - (j ri + *) а(к))М(Л) = 0, F.44)
jT - а<*> BJp [р]Ы + 2? (AXfXi - а« E/у) [*,-]<*> +
i/A[GJ(k) = O, /=l,2,...,s-l, F.45)
(Атт - а<^>) + ЛГ/7 [р]*> + 2? Лтх< [xj» +
+ iiWG/](-0 = 0. F.46)
Эти линейные уравнения можно решить с помощью обычных алгебраических
методов с использованием (или без использования) детерминантов.
Величины a(fc) тогда получаются как корни векового уравнения1)
11 1 (Ttl-i — Ttl$ Л (Tfis—i — Ms \ r\ r\
• -=r — — I —= 1... I = I U ••• U
T . p v ' \ m ) \ m )
0 1 M-(%r) + x)a.W 0 ... 0 0 ... 0
rp 0 AtXi ••• Дтх,-! Атох •• Atq,
0 (A*x.—ctk))...
0 Ax,_tXl ...
0 AQlXl ... Доххм — a(k) -. 0
= 0.
AQlT AGlP 0 Ло^ ...
F.47)
Отсюда получается (I + s + 1) корней a(k). Если полное химическое и тепло-
вое равновесие достигается лишь при г, приближающемся к бесконечности,
то физический смысл имеют только отрицательные значения a(k), имеющие
отличные от нуля значения №.
В вековом уравнении для удобства введено обозначение
8
588
Гл. 11. Гидродинамические применения уравнений переноса
§ 7. ТЕОРИЯ РАСПРОСТРАНЕНИЯ ПЛАМЕНИ1)
Пламя представляет собой тепловую волну, сопровождаемую2) экзотер-
мическими химическими реакциями и распространяющуюся с дозвуковыми
скоростями. Детонационная волна в отличие от пламени расйространяется со
сверхзвуковыми скоростями относительно вещества перед фронтом волны, т. е.
относительно непрореагировавшего вещества. Скорость детонационной волны
определяется уравнениями сохранения массы, импульса и энергии (совместно
с условием Чепмена—Жуге), тогда как скорость распространения пламени
зависит от характерных особенностей химической кинетики, а также от коэф-
фициентов диффузии и теплопроводности. В этом параграфе будет рассмотрена
одномерная теория стационарных ламинарных пламен в гомогенных газовых
смесях. В большинстве пламен падение давления во фронте пламени мало,
поэтому эффекты вязкости можно не принимать во внимание. Кроме того, кине-
тическая энергия движущихся газов мала по сравнению с энергией, выделяе-
мой при реакции. Сначала будут рассмотрены граничные условия и общие
уравнения, описывающие распространение пламени. Затем в качестве примера
будет подробно изучено строение пламени, в котором происходит лишь одна
мономолекулярная реакция.
Стационарность пламени должна
обеспечиваться стабилизатором3). Пламя
распространяющееся вниз по трубке
большого диаметра, не имеет плоского
фронта, обычно является турбулентным
и не достигает стационарного состоя-
ния4). Скорость распространения такого
пламени зависит от материала и диа-
метра трубки, причем при достаточной
длине трубки пламя либо гаснет, либо
переходит в детонационную волну5).
¦Зона пламени
Зона охлаждения
Смесительная
I камера]
Топливо
Окислитель
А. КАЧЕСТВЕННАЯ ТЕОРИЯ ПЛАМЕНИ
ГОРЕЛКИ БУНЗЕНА
Фиг. 161. Линии тока в зоне пламени (или
конуса) горелки Бунзена.
Пламя Бунзена является хоро-
шим примером стационарной системы.
В горелке Бунзена, как показано на
фиг. 161, топливо и окислитель вводятся
раздельно на дне смесительной камеры. Смесительная камера делается доста-
точно длинной, чтобы обеспечить равномерное смешение. Зона пламени пред-
1)См. [64—69].
2) См. также : В. Н. Кондратьев, Кинетика химических газовых реакций, АН
СССР, 1958 ; Л. Н. X и т р и н, Физика горения и взрыва, Изд. Московского университета,
1957. — Прим. ред.
3) Хотя необходимость стабилизатора ясна из математических соображений, однако
возникает вопрос, необходим ли он в реальных физических условиях. Маркштейну [70]
удалось получить стационарное пламя в цилиндре из пирекса диаметром 10 см. Геометрия
этих экспериментов аналогична изображенной на фиг. 163, только стабилизатор отсутствует.
В этом случае теплопередача к стенкам достаточна для стабилизации пламени.
4) Полное описание экспериментов, связанных с пламенами, дано в работах [71, 72].
Очень хорошее изложение первых теорий распространения пламени имеется в работе Эванса
[73]. Другой хороший обзор дан в работе М.ак-Клура и Берла [74].
б) Теоретические и экспериментальные данные о распространяющихся пламенах под-
робно изложены в работах [71, 72]. Вследствие поверхностного давления и теплопередачи к
стенке фронт пламени искривлен. Благодаря увеличению поверхности вследствие искривления
кажущаяся скорость пламени в трубках большого диаметра более чем вдвое превосходит
скорость пламени в горелке Бунзена. Гидродинамическая устойчивость фронтов пламени
рассмотрена теоретически Эйнбиндером [75].
§ 7. Теория распространения пламени 589
ставляет собой круговой конус1), отделенный от выходного отверстия трубки
зоной охлаждения2). Угол раствора конуса зависит от природы газов и скорости
течения.
Пусть е0 — плотность холодных газов, v0 — компонента скорости3) холод-
ных газов, нормальная к фронту пламени4), a Qm и va — соответствующие вели-
чины с нагретой стороны фронта. Величина v0, известная как скорость пламени,
является характеристикой газовой смеси и не зависит от конструкции горелки
или скорости течения газов. Скорость пламени v0 определяется теоретически
из решения уравнений переноса.
Экспериментально скорость пламени можно определить по углу раствора
конуса на горелке Бунзена. Рассмотрим подробно течение газов в зоне пламени.
Вследствие цилиндрической симметрии целесообразно рассматривать двумер-
ную картину, изображающую течение в плоскости, проходящей через ось
симметрии трубки. Пусть 0 — угол между образующей конуса и осью трубки,
как показано на фиг. 161. В таком случае компоненты скорости холодных газов
v(o), перпендикулярные и параллельные горящей поверхности, имеют вид
= »о, GЛ)
G.2
Пусть ф —угол между образующей конуса и направлением течения горячих
газов, как показано на фиг. 161. В таком случае компоненты скорости газов с
нагретой стороны v(a)) имеют вид
v(<o) = p(«) cos ф # ^7.4)
Компоненты скорости, параллельные фронту пламени, не изменяются при
переходе через фронт
г?<0) cos в = г?<°°> cos ф . G.5)
Выражая последнюю формулу через величины vQ и vm> получаем
-*—**- G.6)
Поскольку вследствие уравнения непрерывности qqv0 = ^<»г?«,,то этот результат
можно записать в виде
Qo tg0 •
На фиг. 162 показаны действительные линии тока в пламени горелки Бун-
зена. Как видно из формулы G.1) [79], зная v(o) и измеряя угол 0, который об-
разует горящая поверхность с вертикалью, можно определить скорость пла-
мени. В соответствии с формулой G.7), измеряя углы отклонения линий тока,
можно определить отношение плотностей горячих и холодных газов. Такие
наблюдения позволяют судить о степени достижения горящими газами полного
химического равновесия. Этот метод анализа вместе с микроскопическим изу-
чением пленок, изображенных на фиг. 162, является основным при экспери-
х) Зона пламени (исключая самую вершину и углы) является правильным прямым кру-
говым конусом при условии, что скорость газа в трубке не зависит от расстояния от центра
трубки. Постоянство скорости по профилю можно обеспечить соответствующим выбором
формы выходной части трубки [76, 77].
2) Длина зоны охлаждения определяется величиной теплового потока от пламени к вы-
ходному отверстию трубки [78].
3) В этой главе символ v означает среднюю массовую скорость, которая в гл. 7 обозна-
чалась через v0. Последнюю не следует путать с величиной г>0, определяемой здесь.
4) В советской литературе v0 называется нормальной скоростью распространения пла-
мени. — Прим. ред.
590
Гл: 17. Гидродинамические применения уравнений переноса
ментальном определении температуры как функции расстояния внутри фронта
горения [79 ].
Обычно скорость пламени и другие характеристики горения не зависят за-
метно от выбранной линии тока (исключая самую вершину и углы), хотя кривизна
конического фронта пламени значительно меняется от линии к линии. Это про-
исходит потому, что радиус кривизны обычно велик по сравнению с толщиной
Фиг. 162. Фотография линий тока в конусе горелки Бунзена.
Выходное отверстие трубки спроектировано так, чтобы получился постоянный профиль скорости [79].
зоны горения (по порядку равной миллиметру). Поскольку характеристики
пламени не чувствительны к величине радиуса кривизны, рассмотрим предель-
ный случай — плоскую горящую поверхность, соответствующую бесконечному
радиусу кривизны. В таком пламени температура и химический состав зави-
сят лишь от расстояния, перпендикулярного этой поверхности. Результаты
такой одномерной теории можно применить к трехмерным горелкам, если
предположить, что температура, химический состав и компонента скорости,
нормальная к поверхности пламени, изменяются с расстоянием от поверхности
пламени в горелке по такому же закону, как и в гипотетическом одномерном
случае, и что компонента скорости газов, параллельная поверхности пламени,
не изменяется при прохождении через фронт пламени.
§ 7. Теория распространения пламени
591
Хотя зона пламени настолько тонка, что можно применять одномерную
теорию, тем не менее она достаточно толста, чтобы можно было применять
внутри нее макроскопические понятия континуума. Поскольку толщина зоны
составляет несколько сот длин свободного пробега, то к ней можно применять
понятия локальной температуры, давления, плотности и химического состава
и пользоваться обычными уравнениями гидродинамики для определения этих
величин.
В некоторых пламенах [44] обмен энергией между колебательными и
поступательными степенями свободы происходит настолько медленно, что
наряду с поступательной температурой необходимо определять и колебатель-
ную. В других системах существуют метастабильные возбужденные электрон-
ные состояния, которые необходимо рассматривать как независимые молекуляр-
ные компоненты. Эти явления делают недействительным понятие локальной
температуры и приводят к тому, что наблюдаемые в пламенах скорости хими-
ческих реакций отличаются от результатов, полученных в статической системе.
Однако в настоящем изложении, чтобы не затемнять основные законы распро-
странения пламени, эта неполнота теплового равновесия не будет рассматри-
ваться явно.
Излучение играет важную роль при распространении пламени, однако
детальное рассмотрение изучения и процессов охлаждения необходимо прово-
дить в каждом конкретном случае отдельно [44 ]. Учитывая результаты, полу-
ченные в § 3, можно включить передачу энергии излучением в рассматриваемую
здесь теорию пламен [80 ]. Экспериментальное определение состава излучения
показывает, что оно состоит из хемилюминисценции и теплового излучения,
причем хемилюминисценция обычно преобладает. Очень яркое пламя испу-
скает менее 0,5% своей химической энергии в форме видимого света. Основ-
ная часть излучения испускается в форме инфракрасных полос излучения.
Все молекулы, возникающие в процессе экзотермической химической реакции,
первоначально находятся либо в возбужденном электронном, либо в возбуж-
денном колебательном состояниях. Предоставленные самим себе, такие моле-
кулы отдают эту энергию в виде излучения. Однако при столкновении молекул
эта энергия может передаваться другим молекулам в форме поступательной
или тепловой энергии. Обычно необходимо очень большое ч^сло столкновений,
чтобы произошла такая передача
I:
1 -
О —
1
i pa»
"о
Смесительная
камера
•—Зона пламени
Зона охлаждения
Пористая
*- перегороди —
стабилизатор
энергии. Число требуемых столк-
новений очень сильно зависит от
молекулярного состава. Некоторые
вещества в высшей степени эффек-
тивны для передачи энергии и по-
этому они тушат излучение, нап-
ример при горении окиси углерода
небольшая примесь водяных паров
[81 ] уменьшает долю химической
энергии, испускаемой в форме из-
лучения с 24 до 3%. В этом случае
скорость пламени чрезвычайно чув-
ствительна к присутствию водяных
паров.
Б. ОДНОМЕРНАЯ СТАЦИОНАРНАЯ
ТЕОРИЯ РАСПРОСТРАНЕНИЯ
ПЛАМЕНИ Фиг. 163. Схема горелки, моделирующей
одномерные пламена.
Одномерное стационарное пламя
является идеализацией поведения
газов в малом сегменте зоны пламени горелки Бунзена. На фиг. 163 показана
рассматриваемая нами гипотетическая горелка. Нижняя часть ее очень похожа
Топливо-
-+— Окислитель
592 Гл. 11. Гидродинамические применения уравнений переноса
на горелку Бунзена в той части, которая является впускным устройством и
смесительной камерой. Однако вблизи выходного отверстия трубки располо-
жена пористая перегородка, которая играет роль «стабилизатора». Над пористой
перегородкой прямые стороны горелки продолжены дальше, чтобы линии
тока не расходились. Горелка считается достаточно большой, чтобы можно
было не учитывать роль стенок и считать зону пламени плоской по крайней
мере вдали от стенок.
Стабилизатор необходим, чтобы процессы в такой горелке описывались
стационарным решением уравнений гидродинамики. Без стабилизатора воз-
никают следующие трудности, усложняющие математическое решение задачи.
Во-первых, продукты горения начнут диффундировать назад через зону пламени
в непрореагировавший газ, препятствуя тем самым установлению стационарного
состояния. Во-вторых, скорости обычных химических реакций таковы, что
реакция происходит, хотя и медленно, уже во впускном устройстве. Вслед-
ствие этих причин1) для поддержания стационарного режима в одномерной
задаче необходимо вводить стабилизатор, который обладал бы следующими
двумя свойствами :
1) Стабилизатор должен представлять полупроницаемую пористую пере-
городку, свободно пропускающую молекулу окислителя и топлива и препят-
ствующую прохождению молекул продуктов горения, образующихся в резуль-
тате химических реакций в пламени. Тем самым обратная диффузия в смеси-
тельную камеру, не совместимая со стационарным состоянием, запрещена.
2) Стабилизатор должен служить тепловой губкой, забирающей тепло из
пламени [см. формулу B.49) ]:
() + BП;Й( Vdo- G.8)
В этой формуле z означает расстояние от стабилизатора, а индекс 0 относится
к величинам на поверхности стабилизатора.
Температура, плотность и массовые скорости течения различных компо-
нент в смесительной камере известны, те же значения они имеют и на холодной
границе z = 0. Теплопередачу к стабилизатору необходимо определять. Со
стороны горячей границы (z = оо) имеет место полное тепловое и химическое
равновесие, так что Тту е* и (х,)*, известны.
Уравнения, описывающие распространение пламени, являются одно-
мерными стационарными уравнениями переноса и уравнениями диффузии,
выведенными ранее. Эти уравнения совместно с граничными условиями допу-
скают только одно решение для величины массовой скорости течения М.
Это решение описывает структуру фронта пламени. В качестве граничных
условий берутся условия при z = ©о (горячая граница) и условия при z = О
(холодная граница) на стабилизаторе.
Граничные условия на горячей границе, где существует химическое и теп-
ловое равновесие, определены в § б, разд. Б. Граничные условия на холодной
границе зависят от свойств стабилизатора. Вследствие диффузии продуктов
горения из пламени назад к стабилизатору химический состав газов на
поверхности стабилизатора, т. е. (Х/)о, не определен. Однако доли (G^, вносимые
различными химическими компонентами в массовую скорость течения на
поверхности, стабилизатора, известны, поскольку они определяются через
химический состав холодных газов в смесительной камере :
(G,)o =
) В смесительной . G.9)
камере
Поскольку (Х/)о не определены, то проинтегрировать уравнения переноса в
области между холодной и горячей границами простым путем невозможно.
х) В трехмерной задаче горения в бунзеновском конусе пламя стабилизируется концами
трубки.
§ 7. Теория распространения пламени 593
Величины То и q0, являющиеся температурой и потоком тепла к стабилизатору,
полностью определяются условиями на холодной границе. Поскольку (х,H
не определены, численные решения1) уравнений переноса обычно ищут, начи-
ная от температуры пламени Т« по направлению к низшим температурам.
Исходные условия на горячей границе определены в § б, разд. Б. Если имеется
несколько линейно независимых величин Gh то существует несколько отрица-
тельных корней а(Л) [см. формулу F.47) ] и задача определения скорости пла-
мени становится задачей многих собственных значений. При фиксированном
значении скорости пламени уравнения можно проинтегрировать в напра-
влении холодной границы, не требуя знания величин То или q0. Наименьшая
возможная температура стабилизатора, согласующаяся с законом сохранения
энергии, обозначается символом Т?. Эта температура соответствует случаю
<7о = О.
В настоящее время имеются лишь три примера, для которых можно срав-
нить теоретические и экспериментальные результаты.
а) Разложение окиси этилена. Эта реакция считается мономолекулярной
[83]
(v0) эксп. = 12,5 см)сек,
(г;0) выч. = 9,6 CMJceK .
б) Разложение гидразина. Реакция считается мономолекулярной, постоян-
ная скорости берется из экспериментов Сцварка [84]. Адаме и Стоке [85]
считают, что можно получить лучшее согласие теории с экспериментом, если
рассматривать эту реакцию как цепную. При начальной температуре 150° С и
давлении 1 атм
Ы эксп. = 200 см/сек [8б]2),
= 2&4 см(сек [68]8).
в) Разложение озона. Это первый случай применения теории к цепным
реакциям
Для смеси, состоящей из 25% (молярных) озона и 75% кислорода, при началь-
ной температуре 300° К и давлении 624 мм рт. ст. имеем:
55 см/сек [87]*)
47 см/сек (используя наилучшую постоянную диффузии) [68],
51 см/сек (используя коэффициенты диффузии, меньшие на 20%) [68],
85 см/сек (коэффициенты диффузии считаются равными нулю) [6S],
255 см/сек (коэффициенты диффузии полагаются равными нулю и считается,
что концентрация атомов кислорода соответствует состоянию
псевдостационарного равновесия с озоном) [87].
(г>0)выч.:
Влияние давления на распространение пламени можно понять, анализируя
зависимость различных величин от давления. Коэффициенты диффузии &ц
изменяются обратно пропорционально первой степени давления ; коэффициент
теплопроводности А не зависит от давления ; скорости химических реакций
х) Трудности в получении численных решений чаще всего связаны либо с «неустойчи-
востью*, либо с «переустойчивостью» семейства решений дифференциальных уравнений.
Численная процедура, наиболее плодотьорная в этом случае, изложена в работе [82].
2) Вследствие типографской ошибки приводимая ими величина Л уменьшена в 10 раз.
3) Приведенная величина исправлена в соответствии с предыдущим замечанием.
*) Поскольку здесь наблюдались сферически расширяющиеся пламена, то (г>0)Эксп.
будет несколько меньше тех значений, которые должны наблюдаться в случае плоского
фронта пламени.
594 Гл. 11. Гидродинамические применения уравнений переноса
изменяются, как р^, где /?—порядок химических реакций. В большинстве обыч-
ных пламен можно пренебречь кинетической энергией газов, термодиффузией
и потоком излучения. Если все химические реакции имеют один и тот же по-
рядок /?, то уравнения переноса можно записать в виде
Oj Gj
х' mj xJl^
1 dx, , 1 t
где индексом 1 обозначены величины, относящиеся к атмосферному давлению.
Поскольку правые части этих уравнений не зависят от давления, то все урав-
нения, так же как и граничные условия, инвариантны относительно преобразо-
вания
G.13)
причем одновременно все величины z, определенные при давлении 1 атм, умно-
жаются на р-Р12. Поскольку скорость пламени пропорциональна М/р, то для
мономолекулярных реакций v0 ~ р~^2, для реакций второго порядка v0 не
зависит от р, для реакций третьего порядка v0 ~ рУ*. В настоящих пламенах
с цепными реакциями механизм инициирования цепей — либо первого, либо
второго порядка, реакции развития цепей — второго порядка, а реакции обрыва
цепи — либо второго, либо третьего порядка. При высоких давлениях реакции
инициирования цепей становятся менее важными, чем реакции обрыва. Это
приводит к выводу, основанному на экспериментальных исследованиях, что
эффективная величина /? будет меньше 2. Однако поскольку большинство заро-
дышей цепей образуется в горячей части пламени и затем диффундирует назад,
то кинетика инициирования цепей не имеет большого значения, поэтому прак-
тически /? очень близко к 2.
В. ПРОСТОЙ ПРИМЕР ПЛАМЕНИ :
МОНОМОЛЕКУЛЯРНАЯ ОБРАТИМАЯ РЕАКЦИЯ
Решение основных уравнений, описывающих реальное пламя, связано с
большими вычислительными трудностями. Трудности появляются в первую
очередь вследствие сложности кинетики протекающих реакций, а также боль-
шого числа переменных, которые необходимо рассматривать. Для выяснения
основного характера решения, не осложненного присутствием многочисленных
химических реакций, будет рассмотрен простой частный случай. Рассмотрим
пламя, в котором происходит одна-единственная обратимая мономолекулярная
реакция. Реакция состоит в том, что молекулы топлива А реагируют, пре-
вращаясь в молекулы продуктов В, освобождая при этом энергию, равную
Q кал/моль,
А^В. GЛ4)
Эта система достаточно проста, чтобы можно было исследовать основные черты
решения гидродинамических уравнений, определить смысл граничных условий
и найти зависимость скорости пламени и структуры зоны горения от значений
различных параметров.
Поскольку одна молекула топлива превращается в одну молекулу продук-
тов сгорания, то тА = тв = т. Для простоты считаем, что удельная тепло-
емкость при постоянном давлении не зависит от температуры и одинакова для
обеих компонент : (СР)А = (Ср)в = Ср. Кроме того, в этом примере имеется
лишь одна независимая молярная доля (поскольку хА + хв = 1) и одна неза-
§ 7. Теория распространения пламени 595
висимая часть массовой скорости течения (поскольку GA + Ов = 1). Для
упрощения обозначений будем считать, что хА = х, a GA = О. Скорость обра-
зования молекул А (в молекулах на 1 см3 в 1 сек) равна
К = - nxk'e-E++]kT + /7A - x)k'e'iEt+Q)lkT. G.15)
Предполагается, что стерический фактор к1 одинаков для прямой и обратной
+
реакций, а Е+ —энергия активации прямой реакции, отнесенная к одной моле-
куле. В этом примере удобно ввести в уравнения переноса безразмерные пере-
менные. Определим безразмерные переменные — температуру, скорость и рас-
стояние :
кТ
г = приведенная температура, G.16)
11 = приведенная скорость, G.17)
2
С = МСр\ -i- приведенное расстояние (от стабилизатора). G.18)
о
Если уравнения переноса записать с помощью этих переменных, то получаются
уравнения для определения х, и и т как функции С. При этом в уравнениях
появляются определенные безразмерные параметры. Эти параметры следующие :
1) Приведенная массовая скорость течения
G.19)
заменяющая величину М и являющаяся собственным значением задачи.
2) Число Маха на горячей границе
G.20)
где с = (у'/сГ/ш)^2 — скорость звука, а у' — отношение удельных теплоемко-
стей для реагирующей смеси газов. Последнюю величину можно получить из
формулы D.26)
У' = 1+
здесь у — отношение удельных теплоемкостей смеси газов, в которой не проис-
ходит химических реакций. Если рассматриваемая реакция не имеет обратной
реакции, то отношение удельных теплоемкостей у' равно у.
3) Приведенная энергия реакции
А = -4". G.22)
4) Приведенный коэффициент диффузии
равный отношению числа Прандтля к числу Шмидта (см. гл. 1, § 2). Численная
величина этого параметра обычно лежит около значения д = 4у/(8у — 5),
равного приблизительно единице.
5) Приведенный коэффициент вязкости
G.24)
596 Гл. 11. Гидродинамические применения уравнений переноса
равный числу Прандтля (см. гл. 1, § 2), умноженному на множитель 4/3. Прини-
мая во внимание поправку Эйкена (см. гл. 7, § б и гл. 8, § 4), со можно записать
в виде-у g-^-g-, причем последнее выражение равно примерно единице.
Уравнения переноса, описывающие распространение пламени и выражен-
ные через безразмерные переменные и параметры, имеют следующий вид.
Уравнение непрерывности
Ж = jtf (*>*)• G-25)
Уравнение движения
JL = ±h(u,r). G.26)
Уравнение сохранения энергии
-g- = g(",*,G). G.27)
Уравнение диффузии
i| = ±(x-G). G.28)
В этих уравнениях функции /, g и h равны
/ (х, т) = <?-•/* [ _ х + A - х) е-*'), G.29)
h (a,т) = («- i) + _sLr(-2_-i)> G.30)
-T)—Zfy'T.(u-iy]. G.31)
Граничные условия на горячей границе таковы, что производные в формулах
G.25) — G.28) равны нулю при С = <». Выражая эти условия через т«, получаем
*- = 1Т^Г' G*32)
ц. = 1, G.33)
G. =х« . G.34)
Связь т. с х0 обсуждалась ранее.
Граничные условия на холодной границе определяются свойствами стаби-
лизатора и топлива. Для простоты предположим, что топливо состоит из чистого
газа Л, т. е.
G0=l. G.35)
Вследствие обратной диффузии к стабилизатору величина х^ не может быть
определена. Величина g на холодной границе, выраженная через поток тепла к
стабилизатору q0, имеет вид
= (Е* мСР\ _ лк*. A - хь). G.36)
V W I*0 m
Кроме того, на поверхности стабилизатора {dvldz\ = 0, так что
Ло = й(Ио1*о) = О. G.37)
Это окончательное выражение граничных условий.
§ 7. Теория распространения пламени 597
Граничные условия можно теперь переписать в несколько более удобной
форме. Поскольку h = 0 на холодной границе, то уравнение, определяющее
h(u, t) [формула G.30) ], превращается в квадратное уравнение относительно
и0, являющейся функцией т0. Решение этого уравнения имеет вид
Используя это решение в уравнении для g(u, т, G) [формула G.31) ], из формулы
G.36) можно найти связь между т0, т«, q0 и х0. Физически начальная темпера-
тура газа т0 и градиент температуры или g0 являются величинами определен-
ными. Следовательно, этот результат можно применить для определения т*
через т0. Действительно, поскольку численное интегрирование обычно прово-
дится в направлении от горячей границы к холодной, то часто удобнее задать
величину То, и, используя написанное выше уравнение, определить т0 с по-
мощью г». В результате получаем выражение для т0, которое зависит от потока
тепла q0. Наименьшая возможная величина т0 получается при q0 = 0. Эта вели-
чина обозначается как т'о.
Для большинства пламен число Маха ««,, относящееся к высокой темпера-
туре, мало по сравнению с единицей. Таким пламенам соответствует нижний
знак в формуле G.38). Если это выражение разложить, то получается простая
связь между щ и т0
Поскольку массовая скорость течения М постоянна, то, используя определение
и и уравнение состояния идеального газа, получаем
"=-?-^-т = 1г^г- GЖ))
Следовательно, формула G.39) определяет величину отношения давлений
Если в это выражение подставить типичные значения параметров, то оказы-
вается, что для обычных пламен отношение давлений Рсо/р0^ 0,998. В связи с
этим в обычных пламенах эффектом вязкости можно пренебречь.
Если пренебречь вязкостью газа, то уравнение движения G.26) упрощается,
превращаясь в Л(и, т) = 0. Таким путем получается квадратное уравнение для
и как функции т, решение которого имеет вид
<7-42)
Эту формулу можно использовать для исключения и из уравнения энергии.
В таком случае остаются только три уравнения, описывающие распростра-
нение пламени и определяющие х,тиб как функции ?
В оставшихся разделах этого параграфа будет получено и обсуждено реше-
ние этих уравнений, при условии пренебрежения вязкостью. Как указыва-
лось выше, кинетической энергией движущегося газа обычно можно прене-
бречь. В связи с этим первоначально будет рассмотрено решение уравнений
распространения пламени в этом приближении, т. е., пренебрегая кинетической
энергией движущегося газа, сначала без учета диффузии (й = 0), а затем с
учетом диффузии. В заключение параграфа будет рассмотрена роль кинетиче-
ской энергии, которой нельзя пренебрегать в случае высокоскоростных пламен.
598
Гл. 11. Гидродинамические применения уравнений переноса
Г. ПРОСТОЙ ПРИМЕР ПЛАМЕНИ (КИНЕТИЧЕСКОЙ ЭНЕРГИЕЙ
И ДИФФУЗИЕЙ ПРЕНЕБРЕГАЕТСЯ)
Простейший пример пламени можно получить, если пренебречь кинети-
ческой энергией газов (что справедливо для большинства обычных пламен) и
положить коэффициент диффузии равным нулю. В этом случае уравнение диф-
фузии G.28) принимает вид G = х. Действительная диффузия мало влияет на
величину Си сильно — на величину х. Поэтому характер поведениях, определен-
ный при дальнейшем анализе, подобен поведению G в пламени с диффузией.
Поделив уравнение непрерывности G.25) на уравнение энергии G.27), получим
дифференциальное уравнение, определяющее х как функцию температуры,
их
йт
G.43)
где /(х, т) определяется формулой G.29), a g(x, т) — формулой G.31), в которой
пренебрегается членом, описывающим кинетическую энергию (т. е. последним
членом),
g (х, т) = (i^-L) fi (x - х J - (т. - т). G.44)
Следовательно, в этом случае поведение пламени описывается только одним
обыкновенным дифференциальным уравнением.
Исходя из физических соображений, для этого уравнения можно написать
два граничных условия. На горячей границе т = г., а х определяется форму-
лой G.32). На холодной границе х = 1, а т = т0. Величина т0 находится из
формулы G.36). Величина т0 как функция Tq, определяемой по формуле
- *«), G.45)
выражается следующим образом:
*о = К ~ Яо
Е+МСг
G.46)
Поскольку граничные условия переопределены, то удовлетворительное реше-
ние ураьнения G.43) можно получить лишь для одной величины (или,
возможно, для небольшого числа величин) безразмерного значения массовой
скорости fi.
Фиг. 164. Схематическое изображение решений в случае простых пламен.
Диффузией и кинетической энергией пренебрегается.
§ 7. Теория распространения пламени
5Э9
Решение уравнения G.43) схематически показано на фиг. 164. В точке
(Хоо, Та,) система находится в состоянии полного химического и теплового равно-
весия, так что как /, так и g равны нулю. Величина хт определяется как точка
пересечения двух кривых / = 0 и g = 0. Фактически эти пересекающиеся кри-
вые делят плоскость хт на четыре области, имеющие различные знаки / и g и
характеризующиеся своим знаком dxjdt. Решение, имеющее физический смысл,
получено лишь в области /. Из дифференциального уравнения [формула
G.43) ] видно, что любое решение пересекает кривую / = 0 под углом наклона,
равным нулю относительно оси абсцисс (это показано на кривых d и е). Анало-
гичным образом любое решение пересекает кривую g = 0 под прямым углом
относительно оси абсцисс (это показано на кривых с и /). Кривые, проходящие
через сингулярную точку (*«,, г»), представляют исключение из этого пра-
вила. В этом случае производная dxfdx в сингулярной точке не определена.
Эту неопределенность можно раскрыть по правилу Лопиталя, рассмотрев
частное от деления производной числителя на производную знаменателя. Такая
процедура приводит к квадратному уравнению относительно (dxidx)my которое
определяет два решения (кривые а и ft), проходящие через точку (х*,, т*,). Совер-
шенно ясно, что кривая b не имеет физического смысла, поскольку ее наклон
всюду положителен и она не удовлетворяет условиям на холодной границе.
Таким образом, кривая а является единственным решением, удовлетворяющим
условиям на горячей границе, которое при соответствующей величине /х может
также удовлетворять условиям на холодной границе. Следовательно, только
это решение имеет физический смысл и лишь оно будет обсуждаться в даль-
нейшем. Начальный наклон кривой а равен
Aг). - -
BY + Щ,
G.47)
где
е
•-l/Too
п _^_
Чем больше предполагаемая величина /г (или М), тем меньше начальный на-
клон. Влияние /г (или М) на сингулярное решение а показано на фиг. 165.
Совершенно ясно, что кривая а должна
соответствовать очень малым значениям /г,
чтобы удовлетворить условиям на холодной
Фиг. 165. Влияние [л (или М) на
сингулярное решение.
Фиг. 166. Графическое изображение ft
как функции т0, иллюстрирующее
зависимость скорости пламени от
мощности теплового фильтра.
границе. С другой стороны, кривые end соответствуют очень большим вели-
чинам /л. Кривая Ь будет соответствовать действительности, если т0 достаточно
велико по сравнению с т'о.
600
Гл. 11. Гидродинамические применения уравнений переноса
Сингулярное решение при любом значении /г может быть истинным реше-
нием лишь при некоторых значениях q0. Температура т0, при которой справед-
ливо найденное решение, определяется по точке пересечения кривой решения
с линией х = 1. Зависимость т0 от /л изображена на фиг. 166. Для т0, несколько
превышающего Tq, /г почти не зависит от т0. Действительно, То должно возрасти
на величину порядка 1000°, чтобы /а изменилось заметным образом при условии,
конечно, что для параметров пламени берутся приемлемые значения.
Происхождение плато на кривой /г как функции т можно понять, изучая
характер решения уравнения G.43) вблизи х= 1. В области холодных тем-
ператур обратной реакцией можно пренебречь. Если к тому же в правой части
уравнения G.43) положить х = 1, то получается легко интегрируемое урав-
нение
dX -4гт^- G-48)
dr
Решение этого уравнения записывается в виде
G.49)
где е — постоянная интегрирования, численную величину которой необходимо
выбирать очень малой, чтобы удовлетворить условию х(т0) = 1. Функция
F(r, т0) определяется следующим образом :
-^Ei(JL-4-)' G.50)
где Ei — интегральный логарифм1). Функциональная зависимость F(r, to) от г
показана на фиг. 167 при tq = 0,02. Эта величина х'о соответствует приемлемым
значениям Т? = 300° К и Ei =30 000 кал/моль. Как видно из фиг. 167, я,
-/5
0,02 Q04 0,06 0,08 0,10 0,12 0,14
т
Фиг. 167. Функциональная зависимость F(t,Tq) от т при т'о =¦ 0,02.
величина х остается практически постоянной вплоть до значения г = 0,08,
или Т = 1200° К, при котором начинается реакция. Из этой фигуры, вообще
говоря, не видно, что F(t, т'о) становится положительной и даже бесконечной
при очень малых значениях разности т—т©. Чтобы показать это, часть фиг. 167,а
представлена на фиг. 167, б в увеличенном масштабе. Характер решения
в этой области до некоторой степени искажен приближениями, сделанными
§ 7. Теория распространения пламени 601
при выводе уравнения G.48). Однако некоторые результаты не вызывают сом-
нения. Величина F(ry х'о) изменяется от 10~21 до оов узкой области т — т^ = 10,
или Т — Т'о = 0,015 °К. Поскольку 1//г2 имеет порядок 10G0, то оказывается,
что экстремально малое значение теплового потока (—q0), приводящее к тому,
что Го только на 0,015 °К выше Гд, достаточно, чтобы сделать вычисленную
величину х(Т0) практически независимой от То в очень большом интервале
То; этим и объясняется наличие плато на кривой fi как функции т. Величина
/ll по существу не зависит от потока тепла при условии, что т0 больше т'о + 10"в
и меньше 0,08. В пламенах с диффузией точная величина т0 оказывает неболь-
шое влияние на G и сильно влияет на х вблизи холодной границы.
Д. ПРОСТОЙ ПРИМЕР ПЛАМЕНИ (КИНЕТИЧЕСКОЙ ЭНЕРГИЕЙ
ПРЕНЕБРЕГАЕТСЯ, А ДИФФУЗИЯ УЧИТЫВАЕТСЯ)
Диффузия в обычных пламенах играет важную роль. Если диффузией
не пренебрегать, то величины х и G перестают быть равными.
Тремя уравнениями, определяющими х, т и G как функции С, являются
уравнение непрерывности G.25), уравнение сохранения энергии G.27) и урав-
нение диффузии G.28). Функция /(х, т) определяется формулой G.29), а функ-
ция g{t>G)— формулой G.31), в которой членом, описывающим кинетическую
энергию, т. е. последним членом, пренебрегается. Можно получить два связан-
ных дифференциальных уравнения для xuG как функций т, поделив уравнение
сохранения энергии на два оставшихся уравнения.
Точка (Go, Xoo, Too) является сингулярной точкой уравнений распростра-
нения пламени, поэтому в ее окрестности необходимо дополнительно исследо-
вать характер решений. Если /(х, т) разложить в ряд Тэйлора в окрестности
этой точки, то вблизи горячей границы уравнения распространения пламени
будут линейны относительно переменных (G — (?«), (х — х») и (г — тю):
. (о - GJ + (г - О , G.51)
^^-O, G.52)
(x-xJ. G.53)
Параметры А и В определяются формулами G.47). Наиболее общее решение
этой системы уравнений имеет вид
G-GO0=blw1 еа* + b2 w2 ea*< + й3 iv3 ea*c, G.54)
x-xm=b1s1 e«t + b2 s2 ea*< + ?>3 s3 ea*cy G.55)
t-r^i?! ea* + b2 ea* + b3 ea*<. G.56)
Уравнение для щ получается путем подстановки этих выражений в формулы
G.51) — G.53) и приравнивания нулю детерминанта, составленного из коэф-
фициентов этих трех алгебраических уравнений,
-<5а3 + (<5+ 1)а2-(-В + 1 - дА)а -(А + В) = 0. G.57)
Решение алгебраических уравнений приводит к следующим результатам для
W,- и s( как функций корней а( кубического уравнения :
_ У
\-щб)
602
Гл. 11. Гидродинамические применения уравнений переноса
В соответствии с физическим смыслом параметров а( существует один от-
рицательный корень уравнения G.57), который будет обозначаться как аг.
Поскольку в теории пламени лишь одни отрицательные значения а имеют
смысл, то единственным физически приемлемым начальным условием будет
условие Ь2 = Ь3 = 0. С хорошим приближением величину отрицательного
корня можно написать в виде
в
G.60)
Если обратная реакция при мономолекулярном превращении отсутствует, то
А = 0. Поскольку величина А обычно очень мала, то единственным послед-
ствием, связанным с полным пренебрежением обратной реакцией, будет очень
небольшое изменение значения корня ; характер решения при этом не изме-
нится.
Если начальные условия на горячей границе определены, т. е. известны
(dG/dr)а, = wx и (dxjdt)* = sv то интегрирование уравнений распространения
пламени оказывается несложной численной задачей. Однако если постоянная
д мала, то уравнение диффузии «усложняется» и поэтому требуется применение
особых методов [82]. Влияние диффузии на функциональные зависимости
li и х от г, G от г и х от С показаны1) на фиг. 168—170. Отметим, что диффузия
мало влияет на зависимость G от т. С хорошим приближением G п х можно
выразить в форме
G.61)
ar2 e-*
G.62)
здесь а и b — соответствующим образом подобранные постоянные.
0,04
0,02
Фиг. 168. Зависимость безразмерной массовой скорости течения ft от безразмерного
коэффициента диффузии д.
Имеются два приближенных метода определения скорости и структуры
пламени, применяющиеся наиболее часто ; один из них развит Бойсом и
Корнером [88, 89], другой — Адамсом [90]. Основные схемы этих решений
г) Эти кривые и числовые примеры в оставшейся части этого параграфа рассчитаны при
следующих значениях параметров:
Too = 3000 °К, П = 300 °К,
Е+ = 30 000 кал/моль, х« = 0,00439,
Р
0,2,
1,0848,
= 0,02,
у =1,2.
§ 7. Теория распространения пламени
603
следующие. В уравнении распростра-
нения пламени
dG
dr
величины х и О в правой части
апроксимируются выражениями
X = X0.+51(T-Te), G.64)
G = Goo + w1(t — т J. G.65)
В этом случае правая часть уравнения .
G.63) становится функцией только г, |
поэтому уравнение может быть решено xtG
простым интегрированием. Справед-
ливость такой апроксимации опреде-
ляется тем фактом, что она обоснована
в окрестности точки (Goo, xe, тт), где
правильное решение лежит вблизи
кривой g(G, т) = 0. Уравнение распро-
странения пламени интегрируется в
этой области и далее предполагается,
что f(x, т) и g(G, т) повсюду не очень
чувствительны к величинам х и G. При
таком рассмотрении удается получить
правильный порядок для величины
скорости распространения пламени в
таких простых случаях, как моно-
молекулярные превращения, при ус-
ловии, что коэффициентдиффузиинеоченьмал. Для пламен с цепными реакциями
такая апроксимация может оказаться очень плохой.
Фиг. 169. Влияние коэффициента
диффузии д на величины х(т) и G(r).
о,го
0,18
0,16
0,14
0,12
Г
0,10
0,08
0,06
8
0
€,25
0,50
0,75
1,00
0,03849
0,03102
0,02673
0,02357
0,02132
-4,0 -3,5 -3,0
0,5
Фиг. 170. Зависимость температуры от расстояния.
Ci — значение С в точке преломления.
604
Гл. 11. Гидродинамические применения уравнений переноса
Е. ПРОСТОЙ ПРИМЕР ПЛАМЕНИ (ДИФФУЗИЕЙ ПРЕНЕБРЕГАЕТСЯ,
КИНЕТИЧЕСКАЯ ЭНЕРГИЯ УЧИТЫВАЕТСЯ)
Обычные пламена распространяются со скоростью порядка 100 см/сек,
скорость пламени в горелках порядка 1000 см/сек. Скорость звука в нагретых
газах порядка 100 000 cMJceic, поэтому число Маха имеет порядок хл =0,01.
В этих условиях можно пренебречь кинетической энергией газов и поэтому
ранее рассмотренное приближение является удовлетворительным. Однако при
изменении давления и состава смеси скорость распространения пламени может
сделаться настолько большой, что
кинетической энергией газов уже
нельзя будет пренебрегать. Для про-
стоты предположим, что коэффициенты
диффузии и вязкости равны нулю.
Более полное рассмотрение, учиты-
вающее влияние вязкости, дано в со-
кращенном виде в § 9 в связи с теорией
детонации.
Уравнения, описывающие пламена,
представляют теперь уравнение непре-
рывности G.25) и уравнение сохране-
ния энергии G.27). Функция /(х, т)
дается выражением G.29), а функция
g(x, r) — выражением G.31), в котором
G заменено х, а и — выражением
G.42). Таким образом, функция х(т)
описывается дифференциальным урав-
нением G.43), в котором функция
g(x, т) теперь содержит члены, описы-
вающие кинетическую энергию.
Характер решений дифференциаль-
ного уравнения можно описать с по-
мощью диаграммы, аналогично изобра-
женной на фиг. 164. Учет кинети-
ческой энергии в первую очередь
2 Z* ПРИ различных приводит к тому, что прямые линии
g(x, г) = 0 в плоскости хх переходят
в параболы. На фиг. 171 изображены
кривые g(x, т) = 0 при различных значениях х9. При хш = 1/f/ производная в
точке тв бесконечна. При хя= 1 производная кривой g = 0 равна производной
кривой / = 0. При «а, > 1 параболы g = 0 пересекают кривую / = 0 в точках
выше г», обозначаемых как т«. Тщательное изучение показывает, что реше-
ние, начинающееся в т« при ««> 1, не имеет физического смысла1). Это значит,
что хж = 1 соответствует наибольшей величине числа Маха, относящегося к
высокой температуре, при котором еще возможно стационарное распростра-
нение пламени. Как будет показано в § 9, оно соответствует «нижней точке
Чепмена—Жуге» и может быть предсказано путем рассмотрения законов
сохранения массы, количества движения и энергии.
При и., лежащем между 1/У/ и единицей, температура на кривой g = О
достигает максимального значения
Фиг. 171. Кривые р
G.66)
г) Верхняя точка пересечения rm представляет хорошую начальную точку для расчета
пламени, однако относительно этой новой температуры новое число Маха хв будет меньше
единицы.
§ 7. Теория распространения пламени
605
В этой же точке скорость, давление и плотность имеют вид
PS__ 1+*2оо/
" 2
G.67)
G.68)
На фиг. 172 показаны решения уравнений распространения пламени в области
между ts и г». Любое физически приемлемое решение должно проходить через
точку (xs, ts). Независимо от величины
li существует такое решение, прохо-
дящее через горячую границу (х«,,Тсо)
и точку (xs,ts)f причем это решение f
единственное. В связи с этим решение х
уравнения, удовлетворяющее условиям
как на горячей, так и на холодной
границе, можно получить численным
интегрированием от точки (х5, т8) в
сторону стабилизатора. Величина р
при некотором определенном выборе яв
определяется из требования, чтобы
решение удовлетворяло условию на
холодной границе.
Напомним, что при рассмотрении
быстрых пламен с изменением я« ме-
няются либо т0, либо г». С практи-
ческой точки зрения удобнее считать
т0 постоянным при изменении я.,
меняя при этом соответственно т«.
Результаты ряда вычислений, проде-
ланных при значении т0 = 0,02, при-
ведены в табл. 101.
Фиг. 172. Графическое изображение решений
уравнений распространения пламени при
Кривая а изображает единственное решение,
удовлетворяющее граничным условиям.
Таблица 101
Зависимость параметров пламени от величины кинетической энергии газов
0
0,527
0,907
0,922
1,000
10,000
9,738
9,257
9,173
9,110
Ро
Роо
1,0000
1,3064
1,9295
1,9621
2,1349
0,1000
0,0786
0,0560
0,0556
0,0514
J/ Ср
' А/С'ЛЕ+
оо
0,03525
0,02576
0,02537
0,02535
Л Хк' тпп
0,03858
0,03525
0,03079
0,03043
0,03043
г* |/ m E+ КкТ0)
0,0000
1,0000
1,195
1,200
1,200
Анализируя эту таблицу, можно сделать следующие выводы:
1) Существует критическое давление холодного газа, ниже которого не-
возможно установление стационарного режима горения. В рассматриваемом
примере это давление имеет вид р0 = 0,02535 yx'AnE+jCp. В случае бимоле-
кулярной реакции в противоположность мономолекулярной реакции критерий
образования стационарного режима горения не зависит от начального давле-
ния ; вместо этого существует критическая температура т0, ниже которой горе-
ние невозможно.
606
Гл. 11. Гидродинамические применения уравнений переноса
2) При очень высоких начальных давлениях кинетическая энергия газов
пренебрежимо мала ; параметр fi постоянен, а скорость распространения
пламени меняется как ро1/2.
3) При уменьшении начального давления скорость пламени возрастает,
пока при критическом начальном давлении число Маха горячего газа не станет
равным единице.
4) Массовая скорость течения практически не меняется с давлением.
При критическом начальном давлении /л составляет 0,7887 своей величины при
очень высоких начальных давлениях.
§8. ТЕОРИЯ РАСПРОСТРАНЕНИЯ УДАРНЫХ ВОЛН
Ударная волна — это такая волна, в которой на небольшом расстоянии
резко меняются свойства газа. Стремление волны сжатия в газе перейти к удар-
ной волне рассматривалось в § 5. Если в гидродинамических уравнениях пре-
небречь членами, включающими вязкость, теплопроводность и диффузию, то
ударные волны проявляются как математическая разрывность решений.
Когда учитываются эти диссипативные процессы, то разрыв превращается в
несколько более постепенный переход, имеющий место на расстоянии нескольких
длин свободного пробега (например, приблизительно 10~5 см в газе при 1 атм
и комнатной температуре). Вследствие наличия в ударной волне очень сильных
градиентов макроскопических величин, часто уравнений Навье—Стокса недо-
статочно для описания структуры и толщины ударной волны. В конце настоя-
щего параграфа рассмотрены попытки более точного анализа структуры удар-
ной волны.
На фиг. 173 представлена ударная волна, образованная поршнем, движу-
щимся с постоянной скоростью (v0— г?оо). В этом случае установившееся со-
стояние обеспечивается наличием
давления р0 на одной стороне удар-
ной волны и роэ — на другой сто-
роне. Это положение совершенно
отличается от того, когда ударная
волна образуется при взрыве или в
ударной трубе, что не является
действительно установившимся про-
цессом. При взрыве или в ударной
трубе после прохождения ударной
волны давление падает (иногда дав-
ление в волне разрежения р« ста-
новится меньше р0 или колеблется)
и в конце концов возвращается к
значению р0. Однако даже в таких
распространяющихся ударных вол-
нах поведение и структуру ударного
фронта можно описать для любого
момента времени с помощью теории
с установившегося состояния, так как
Фиг. 173. Значения скорости газа по отно- толщина ударного фронта (рассто-
шению к газу, неподвижному при z = — оо (а) и яние, на котором имеет место рост
значения скорост*, газа по отношению к непо- давления> равНое примерно 10~> см)
Газ неподвижен
Ро
- Поршень
б
+ J
Ро
Ударная волна
неподвижна
С ДЛИНОЙ
движной ударной волне (б).
у* F у ' очень мала в сравнении
всей волны.
Рассмотрим плоские ударные волны [62, 91]. В системе координат, движу-
щейся с ударной волной так, как показано на фиг. 173, распределение пара-
метров получается из установившегося решения гидродинамических уравнений.
§ 8. Теория распространения уйарных волн 607
(Формирование такой волны, т. е. преобразование решения, зависящего от
времени, в установившееся решение, рассмотрено в § 5.) Условия, связывающие
значения параметров газа по обе стороны ударного разрыва, можно легко
получить из гидродинамических уравнений (см. § б). Предполагается, что
не только в пределе для х->оо (обозначенного индексом оо), но и для предель-
ных условий при х->— оо (обозначенного индексом 0) все производные равны
нулю. Скорость ударной волны по отношению к газу при —со тогда будет равна
v0. В этом случае уравнения переноса дают соотношения между значениями
параметров по обе стороны ударного разрыва. При этом предполагается, что
существуют решения дифференциальных уравнений, удовлетворяющие этим
условиям. Что это всегда справедливо, показывается далее в настоящем
параграфе, а также в следующем параграфе в связи с рассмотрением вопроса
детонации.
Сначала рассмотрим соотношения, связывающие значения параметров по
обе стороны ударной волны. Несмотря на то, что эти соотношения справедливы
даже с учетом явлений переноса, они не дают представления о детальной
структуре ударной волны. Подробное строение ударной волны, описываемое
уравнениями переноса, обсуждается в последнем разделе этого параграфа.
Показано, что толщина ударной волны зависит от величины коэффициентов
переноса. Решения уравнений переноса становятся разрывными в пределе,
когда rj и А стремятся к нулю.
А. СООТНОШЕНИЯ ГЮГОНИО
Уравнения переноса для установившейся одномерной задачи приведены
в § 6. Если предполагается, что производные при х = —со равны нулю, то
эти уравнения F.2), F.13), F.15) дают следующие соотношения между пере-
менными по обе стороны ударной волны :
= ?«»•, (8.1)
f Poo, (8.2)
Йо + -i-t>g = Й„+±1%. (8.3)
Эти соотношения, известные как соотношения Гюгонио, связывают восемь вели-
чин: #0, v0, р0, Йо, Qmy vо,, ро» и Йт. Однако если мы используем термодинами-
ческую зависимость п от q и р, то число переменных может быть сведено к
шести. Следовательно, этих соотношений достаточно для определения трех
переменных через остающиеся три. Обычно задается плотность и давление в
газе низкого давления (д0 и р0), а также дополнительный параметр, характе-
ризующий силу ударного разрыва, например давление рт в газе высокого
давления. Для нахождения значений оставшихся величин vOyvm и Qm решаются
соотношения Гюгонио. Значение v0 соответствует скорости притока холодного
газа к ударному разрыву или скорости распространения ударной волны в
неподвижном газе, находящемся под низким давлением. Величина г>« является
скоростью движения горячих газов от фронта ударной волны.
Удобно представить соотношения Гюгонио в несколько иной форме.
Из уравнений непрерывности и движения (8.1) и (8.2) можно получить
М =
Поскольку г/2 (г? — v%) = г/2 (г;то — г;0) (г^ + v0), то из той же] пары уравнений
следует
4" К - »о2) = - (P-"^; + ft) • (8-5)
608 Гл. 77. Гидродинамические применения уравнений переноса
Далее, комбинируя этот результат с уравнением энергии (8.3) и соотношением
Й = и + (plo), получаем зависимость
/ГУ П\— (Р°° + А>) (goo - g0) # /о дч
(U Uo) - 2^ (8.6)
Последние три зависимости являются соотношениями Гюгонио, записан-
ными в несколько более удобной форме. Эти соотношения нельзя окончательно
решить, если неизвестны уравнение состояния и термодинамические свойства
газа. В реальном газе расчеты могут оказаться несколько громоздкими. Однако
можно получить качественное описание явления, рассматривая распростра-
нение ударных волн в идеальном газе с постоянной удельной теплоемкостью.
Б. ПРИМЕНЕНИЕ СООТНОШЕНИЙ ГЮГОНИО
К ИДЕАЛЬНОМУ ГАЗУ
Для идеального газа с постоянной удельной теплоемкостью внутренняя
энергия 1 г вещества по отношению к 0° К составляет
О = ЬТ=-р*т>-в~ (8-7)
Используя это выражение для внутренней энергии, можно решить уравнение
энергии (8.6) и получить выражение для плотности #« газа высокого давления
через ео> А, и Р~
Л —Л (У + *) Роо + (У - *) Ро
Эту зависимость можно удобно записать через отношение плотности ы0 и отно-
шение давления ? на скачке, определенные как
Тогда
„ _ (У- 0
Это соотношение часто называют ударной адиабатой Гюгонио1), несмотря на
то, что это неверно, так как энтропия газа при переходе через скачок меняет
свое значение2).
Если ударная волна слаба, то ? приближается к единице и, разлагая
ударную адиабату Гюгонио (8.10) в ряд, получаем
«o=l+-f(f-l)--2^(y-l)(f-lJ + 4^(>'-1J^-1K+..- (8.11)
Это разложение можно сравнить с разложением соотношения, описывающего
изэнтропическое сжатие газа,
Интересно отметить, что первые три члена разложения ударной адиабаты
Гюгонио и соотношения для изэнтропического сжатия одинаковы. Это выра-
жает тот факт, что в слабых разрывах наблюдается очень малый скачок энтро-
пии.
х) Это соотношение, конечно, не применимо к изменению давления с плотностью в удар-
ной волне [см. (8.43)].
2) Следует иметь в виду, что при необратимом адиабатическом процессе энтропия воз-
растает. — Прим. ред.
§ 8. Теория распространения ударных волн 609
Для сильных разрывов ? велико, и из ударной адиабаты Гюгонио (8.10)
следует, что отношение плотности и0 приближается к предельному значению
в ?. (8.13)
Например, для воздуха у = 1,4, и предельное значение и0 равно 6.
Из уравнения состояния идеального газа
* оо Poo Qo S /О 1 Л\
В сильных разрывах f велико, тогда как и0 приближается к некоторому пре-
делу. Следовательно, при сильных разрывах Тт настолько велико, что ударные
волны могут даже светиться.
Ударную адиабату Гюгонио (8.10) можно переписать в форме, известной
как соотношение Ретина—Гюгонио
Рсо-Ро Рсо + Ро. (815)
Qoo-Qo Qoo + Po v '
Эта форма записи особенно интересна с точки зрения аналогии с соотношением
которое дает наклон касательной к кривой адиабаты идеального газа.
Выражения для скоростей v0 и t?« газа по обе стороны волны можно полу-
чить из (8.1) и (8.4):
-ш Г ~, Г"
(8.17)
...у
Qoo
Qo
Qo
(Poo
(Qoo
(Poo
-Po)
-Qo)
-Po)
Из этих выражений и соотношения Ренкина—Гюгонио (8.15) можно получить
значения чисел Маха для потока по обе стороны ударного фронта, выраженные
через п0 и |, и скорость звука, определенную соотношением D.5),
<8Л9>
Ниже показано, что физически реализуется лишь тот случай, когда ио^>1.
Из ударной адиабаты Гюгонио (8.10) следует, что если и0 ^ 1, то f ^ и0. Следо-
вательно, из последних двух выражений имеем
ко>1 или г>0>с0, (8.21)
*ж<1 или г;да<с<о. (8.22)
Эти результаты говорят о том, что газ низкого давления перемещается со сверх-
звуковой скоростью по отношению к ударному фронту, а скорость нагретого
сжатого газа является дозвуковой по отношению к фронту. Если ударная
волна распространяется в неподвижном газе, то она набегает на неподвижный
газ со сверхзвуковой скоростью v0.
Из соотношений (8.19) и (8.20) ясно, что в предельном случае слабых удар-
ных волн, когда и0 и f равны единице и свойства газа по обе стороны разрыва
одинаковы, скорость распространения волны равна скорости зрука. Действи-
тельно, в этом предельном случае нет различия между слабой ударной волной
и звуковой волной.
610 Гл. 11. Гидродинамические применения уравнений переноса
Физический смысл имеют только те случаи, когда ио^> 1, так как это усло-
вие необходимо для того, чтобы изменение энтропии газа при переходе через
ударную волну было положительным. Энтропия 1 г идеального газа с постоян-
ной удельной теплоемкостью равна
5 = Ср In Т - -^- In р + const = Cv In (pQ-r) - Ср In (A.) + const. (8.23)
Таким образом, разность значений энтропии газа по обе стороны фронта соста-
вляет
(^?)Cvln^Uoy). (8.24)
Поскольку за положительное направление z нами выбрано такое, когда газ течет
из области 0 в область °о, то из второго начала термодинамики следует
?ыоу>1. (8.25)
Рассматривая это неравенство вместе с ударной адиабатой Гюгонио (8.10),
можно показать, что из второго начала следует f ;> 1 и щ ;> 1. Следовательно,
в ударной волне газ течет со стороны низкого давления к высокому давлению
(или от низкой плотности к высокой плотности). Противоположный поток будет
противоречить второму началу термодинамики.
Использование интегральных уравнений — соотношений Гюгонио — ско-
рее делает необходимым привлечение дополнительных аргументов, основанных
на втором начале, чем в случае дифференциальных уравнений гидродинамики
с членами, представляющими диссипативные процессы. В действительности,
уравнения гидродинамики включают уравнение изменения энтропии и второе
начало и, следовательно, нет необходимости дополнительно учитывать эти
соотношения. Можно показать, что дифференциальные уравнения не имеют
решений, представляющих термодинамически невозможные ударные волны
разрежения. Нереальность ударных волн разрежения была также показана
при рассмотрении процесса образования ударных волн в § 4 настоящей главы.
Величина изменения энтропии 1 г газа при переходе через ударную волну,
выраженная через отношение давлений, может быть получена из ударной адиа-
баты Гюгонио (8.10) и соотношения (8.24). Развертывая это выражение, для
слабых ударных волн получаем
5. - §0 =-|^ (У* - 1) (^ ~ О3 + ... (8.26)
Поскольку первый член в этом выражении-является кубическим, то этот резуль-
тат указывает на то, что в слабых разрывах наблюдается небольшое изменение
энтропии.
После прохождения ударной волны от взрыва или в ударной трубе газ
адиабатически расширяется до своего первоначального давления р0. Вследствие
роста энтропии газа после расширения газа до первоначального давления р0
остаточная температура Т'т несколько выше начальной температуры. Этот рост
температуры связан с ростом энтропии соотношением
S.-S0 = (?pln-5f. (8.27)
Это соотношение вместе с предыдущим позволяет получить общее выражение
для роста температуры. Для слабых разрывов это выражение записывается в
виде разложения
Т'„ - То = То (-^-) (I - IK + • ••• (8.28)
В качестве примера точного расчета рассмотрим прохождение ударной волны
в воздухе, для которого у = 1,4 при температуре Гв = 273 °К. Зависимость
§ 8. Теория распространения ударных волн
611
роста температуры от силы ударной волны (отношения давлений) показана
в табл. 102. Из таблицы видно, что диссипация энергии мала, если р<*1р0 меньше
2 или 3, но становится большой для более сильных разрывов. В сравнении с
газами прохождение ударных волн в жидкости дает пренебрежимо малый
нагрев. Например, для ударных волн с давлением приблизительно 1000 атм,
распространяющихся в воде, Т'„ — То = 0,025° К.
Таблица 102
Рост температуры в ударной волне в воздухе
Роо
Ро
(т1-го),°к
2
2,5
3
10,3
5
31
10
97
50
579
100
1065
500
1417
Помимо отношения давлений роо/р0, существуют две другие важные меры
силы ударной волны:
1) Положительный импульс /, определенный в виде
(8.29)
Интегрирование здесь проводится по промежутку времени, за который дав-
ление в ударной волне возвращается к р0.
2) Энергия ударной волны Е (на единицу поверхности ударного фронта)
задается в виде
E=f(p-po)vdt.
(8.30)
Для слабых разрывов v ^ (р — po)j!qoco, так что
(8.31)
Поскольку энергия диссипирует путем нагрева газа, через который проходит
ударная волна, то полная энергия ударной волны падает согласно зависимости
dE
(8.22)
На очень больших расстояниях давление в ударной волне уменьшается
как z~v*. В трехмерных сферических ударных волнах на расстоянии R от
источника давление падает как R^RJu
В. СТРУКТУРА И ТОЛЩИНА УДАРНОЙ ВОЛНЫ
В ИДЕАЛЬНОМ ГАЗЕ1)
Структура ударной волны описывается решением одномерных уравнений
гидродинамики для стационарного случая. В качестве примера рассмотрим
распространение ударной волны в идеальном газе, состоящем только из одной
химической компоненты с постоянной удельной теплоемкостью2). Для такого
газа одномерные уравнения гидродинамики в установившемся виде такие же,
1)См. [92—95].
2) Каулинг [96] рассмотрел влияние диффузии на структуру ударной волны. Он пока-
зал, что диффузия по существу оказывает такое же влияние, как и вязкость и теплопровод-
ность. Скорость диффузии часто составляет существенную долю скорости звука.
612 Гл. 11. Гидродинамические применения уравнений переноса
как и в § 6, но записанные для однокомпонентной системы :
е» = е.». = л«, (8.33)
±% + pa, (8.34)
Уравнение состояния идеального газа может быть использовано для
исключения Т из уравнений гидродинамики. Уравнения тогда можно объеди-
нить для получения дифференциального уравнения второго порядка для v
как функции z
М(^ОК~^) = 0, (8.36)
где
^4 j ^ ^l (8.37)
Здесь v0 — скорость в пределе для z -> — оо. Эта зависимость, конечно, содер-
жится в соотношениях Гюгонио, рассмотренных в предыдущем разделе. Диф-
ференциальное уравнение можно удобно записать в безразмерной форме с
помощью безразмерных величин и, С и со, определенных соотношениями G.17),
G.18) и G.24):
dul . y+\ da
^и) = О. (8.38)
Решение этого дифференциального уравнения второго порядка для и(?)
для произвольных значений параметров затруднительно. Однако, когда вели-
чина со (составляющая 4/з числа Прандтля) раина единице, решение можно
получить в простой форме [93—95 ]. В соответствии с табл. 2, в которой при-
ведены экспериментальные значения числа Прандтля для ряда газов, значение
3/4 П1ля числа Прандтля (или со = 1) является вполне обоснованной величиной.
Поэтому в дальнейших выводах по структуре ударных волн мы рассматриваем
только случай со = 1.
Для со = 1 общим решением уравнения (8.38) будет
" Ж + Чг (и - 1) (Ц, - и) = &, (8.39)
где С — произвольная постоянная. Так как предполагается, что в пределе при
z -> оо как vy так и dvjdz остаются конечными, то постоянная С берется равной
нулю. Таким образом, единственное решение, имеющее физический интерес,
запишется в виде
Общее решение этого уравнения имеет вид
гл'г 2У tf0 in | и0 — и | — in 1 ц —
где Со — произвольная постоянная. Это соотношение дает в неявном виде
скорость как функцию положения. Типичные кривые приведены на фиг. 174.
Полное решение задачи, т. е. оценку других переменных, можно получить пара-
§ 8. Теория распространения ударных волн
613
метрически через значение безразмерной скорости и. Величина и0 связана с
исходным числом Маха я0 соотношением (8.19).
Если уравнение движения (8.34) записать через безразмерные переменные
и полученное выражение решить относительно давления р, то найдем
щ-уи + усо^. (8.42)
ур
Для со = 1 параметрическое уравнение для давления можно получить заменой
-8-6-4-20246
Фиг. 174. Структура ударных волн различной интенсивности.
производной в этом уравнении выражением (8.40). Тогда если воспользоваться
(8.37), то
У + 1 «о
Р = У- 1
— и
(8.43)
у-1
Для того чтобы давление при и = и0 (z -> — ©о) было положительным, необхо-
димо, чтобы величина ии была меньше (у + \)j(y— 1). Это—результат (8.13),
непосредственно полученный из соотношений Гюгонио.
Толщину ударной волны можно определить как расстояние, на котором
наблюдается основная часть изменения скорости. Определим толщину как
расстояние между точкой, в которой и = и0— е(и0— 1), и точкой, в которой
и = 1 + е(и0— 1), где е — произвольно малое число. Из соотношения (8.41)
следует, что это расстояние в безразмерных единицах равно
(8.44)
~* y+iUo-
Если коэффициент теплопроводности А постоянен, то это выражение дает для
толщины
Приближенное выражение для теплопроводности через среднюю длину сво-
бодного пробега I дано в соотношении B.14) гл. 1. Если с = У у кТ\т — ско-
рость звука, то из выражения для коэффициента теплопроводности находим
Am = 2_
6ск 3(у- 1) У уп
А/.
(8.46)
614
Гл. 11. Гидродинамические применения уравнений переноса
Как видно из соотношений (8.21) и (8.22), скорость газа внутри ударной волны
равна приблизительно локальной скорости звука. Следовательно, это выра-
жение можно использовать для получения выражения для толщины через
среднюю длину свободного пробега для газа внутри ударной волны. Это число
средних длин свободного пробега равно
I
ул
е )
(8.47)
2 3
Число Маха
На фиг. 175 представлено отношение средней длины свободного пробега
к толщине ударной волны как функция начального числа Маха. Кривая а
получена Томасом [97], который учитывал влияние изменения тепло-
проводности и вязкости с температурой.
Видно, что умеренно сильные ударные
волны занимают расстояние, включающее
лишь несколько длин свободного пробега.
Так как градиенты макроскопических
величин весьма значительны, то использо-
вание уравнений Навье—Стокса не оправ-
дано. Эти уравнения основаны на первом
приближении Энскога—Чепмена. В гл. 7,
§ 3 было отмечено, что первое приближение
справедливо лишь тогда, когда изменения
макроскопических величин на длине сво-
бодного пробега несущественны.
Ван Чанг [98] провела тщательное
изучение структуры ударных волн, осно-
ванное на решении уравнения Больцмана
по методу Энскога. Она рассматривала вли-
яние следующих двух приближений, помимо
Фиг. 175. Толщина ударной волны которое давало уравнение Навье—
в ««-«i™^^ (а) Сто^са РезРультАаТы говорят о том, что ряды
сходятся очень медленно, если исходное
число Маха значительно превышает еди-
ницу. Это указывает на то, что при наличии больших градиентов, которые
имеют место в сильных ударных волнах, понятие о локальных значениях мак-
роскопических величин и понятие о континууме теряют смысл. Для изучения
структуры сильных ударных волн необходимо пересмотреть уравнение Больц-
мана, а также рассматривать другие виды приближенных решений.
Недавно Мотт-Смит [99] сделал приближение, основанное непосредственно
на уравнении Больцмана, избегая какого-либо использования гидродинами-
ческих уравнений или понятия о векторах потоков. Он получил приближенное
решение уравнения Больцмана, описывающее структуру ударной волны. При
обычном рассмотрении предполагается, что функция, описывающая распреде-
ление скоростей молекул, является почти максвелловской. Мотт-Смит пред-
положил, что функция распределения является суммой двух точно максвеллов-
ских членов. С физической стороны это дает картину смеси двух газов различной
температуры и скоростей движения. В качестве уравнения для параметров,
входящих в выбранную функцию распределения, было использовано уравнение
Больцмана. Теория является приближенной вследствие предположений о
функции распределения. Однако показано, что предполагаемый вид функции
распределения не меняется заметно со временем, так что решение оказы-
вается почти установившимся. Эти результаты вместе с результатами Томаса
показаны на фиг. 175.
Хорниг, Коуэн и Грин [100—103] использовали оптические методы для
определения толщины ударных волн в азоте, аргоне, водороде и кислороде.
Эти методы основаны на изменении отражательной способности ударной волны
§ 9. Теория детонации
615
с длиной волны света, что использовалось для измерения профиля плотности
в ударной волне. Теория Томаса позволяет предполагать, что показатель пре-
ломления как функция расстояния внутри ударной волны записывается в виде
=1 +
— Уо
(8.48)
Здесь параметр L можно использовать как меру толщины волны. Эксперимен-
тальные результаты сравниваются с предсказаниями теории Томаса в табл. 103.
Видно, что экспериментальные результаты примерно вдвое превышают вели-
чины, предсказываемые теорией Томаса. Сравнение с фиг. 175 показывает, что
эти результаты приближаются к кривой Мотт-Смита.
Таблица 103
Сравнение экспериментальных и
теоретических значений толщины
ударной волны в N2 при 25° С
р
р.
1,71
1,71
1,71
Рв
42
68
85
L- 10
эксп.
3,2
2,0
1,8
', см
теор.
2,0
1,3
1,0
Из результатов по изучению ударной волны [103] следует, что для уста-
новления равновесия между вращательной и поступательной энергиями в водо-
роде требуется 150 столкновений; в азоте и кислороде равновесие наступает
значительно быстрее.
§9. ТЕОРИЯ ДЕТОНАЦИИ
Детонационная волна — это устойчивая волна, в которой стационарные
условия поддерживаются за счет поступления энергии экзотермической хими-
ческой реакции. Эту волну можно представлять как фронт пламени, пред-
шествуемый ударной волной, предварительно нагревающей вещество. Детона-
ция отличается от обычного распространения пламени тем, что ее скорость
превышает скорость звука.
Как и в теории ударных волн, обсуждавшейся в предыдущем параграфе,
резкое возрастание давления, плотности и температуры в детонационной волне
описывается уравнениями сохранения массы, количества движения и энергии.
Скорость распространения устойчивой детонации определяется условием
Чепмена—Жуге. Это условие, которое мы сейчас рассмотрим, позволяет выбрать
из всех возможных решений уравнений переноса наиболее устойчивое решение.
Это решение соответствует минимальной возможной скорости распространения.
Таким образом, все характеристики детонационной волны полностью опре-
деляются уравнением состояния и уравнением теплового баланса и не зависят
от химической кинетики или явлений переноса. Структура детонационной волны,
однако, определяется решением дифференциальных уравнений переноса. Это
те же уравнения, что были использованы в двух предыдущих параграфах для
описания структуры фронта пламени и ударных волн. Детонационное решение
вблизи холодной границы подобно решению для ударной волны, а вблизи горячей
границы подобно решению для пламени.
616 Гл. 11. Гидродинамические применения уравнений переноса
А. СООТНОШЕНИЯ ПОГОННО И УСЛОВИЕ ЧЕПМЕНА—ЖУГЕ
Как и при обсуждении ударных волн и пламен, мы будем пользоваться
системой координат, движущейся вместе с волной. В этой системе картина
стационарна. Холодная граница соответствует z = — ©о, а горячая z = °о.
Условия, соответствующие холодной границе, мы обозначим индексом 0, а на
горячей границе индексом ©о. Вещество движется в положительном направле-
нии и скорость при z = — ©о, обозначенная v0, является скоростью распростра-
нения детонационной волны в непрореагировавшем веществе. Поэтому v0
названа скоростью детонации.
Соотношения, связывающие значения переменных по обе стороны дето-
национной волны, аналогичны тем, что применялись для ударных волн
[см. (8.1)—(8.3)]. По-прежнему удобно преобразовать эти выражения для
получения соотношений типа (8.4)—(8.6)
(9.2)
(9.3)
Хотя они и справедливы для значений по обе стороны детонационной волны,
однако сама возможность появления детонационной волны зависит от существо-
вания и устойчивости решений дифференциальных уравнений, описывающих
структуру детонационной волны.
Начальные значения концентрации х/0, температуры То, давления р0 и
плотности q0 предполагаются известными, так что
s _
2 */о Vi (То> Ро> х/о)
^0 = -^—1 (9.4)
Различие уравнений для детонационной и ударной волн выражается в конкрет-
ном виде функции От. Для ударной волны химический состав остается неиз-
менным, тогда как в детонационной волне химический состав изменяется так,
что при прохождении газа через детонационную волну выделяется энергия и
устанавливается химическое равновесие. Требование химического равновесия
при z = ©о означает, что скорости всех реакций в выражении F.8) равны
нулю :
К{(хУв, Т., р J = 0, i = 1,2, ..., 5. (9.5)
Из этих выражений можно определить равновесный химический состав
xja>(Tm, p.») как функцию температуры и давления. Для выражения темпера-
туры через давление, плотность и химический состав можно воспользоваться
уравнением состояния
Tw = f(poo,Qw,xjoo). (9.6)
Используя эту форму уравнения состояния, можно определить равновесный
химический состав в виде функции давления и плотности худа(р00, ?«,). Тогда
если парциальные молярные внутренние энергии и{(Тту pmj xja>) известны,
то известна и внутренняя энергия О» на 1 г вещества как функция р» и ?«
(p., Qj = -^ 5 (9.7)
2 х<» Mi
§ Р. Теория детонации
617
Точка
Чепмена-Щге
Если использовать это соотношение и подставить его в (9.3), то получим соотно-
шение между р« и q».
На фиг. 176 представлена кривая Гюгонио DBCB', полученная таким
способом. Нижняя кривая ANA'O соответствует случаю ударной волны с
теми же условиями перед фронтом, за которой начальный химический состав
остается неизменным. Все уравнения переноса (9.1)—(9.3) применяются к
конечному состоянию газа, соответствующему точке только на верхней кривой.
Однако единственная реальная ус-
тойчивая детонационная волна —
это волна, конечному состоянию
которой соответствует точка Чеп-
мена—Жуге С. Эта точка характе-
ризуется тем, что прямая линия,
проходящая через точку начального
состояния О и точку С, касается
кривой Гюгонио при давлении, |
большем, чем1) р0. Выбор единст- '
венной точки является результатом
условия Чепмена—Жуге, дополни-
тельного к условиям сохранения.
Мы дадим доказательство справед-
ливости условия Чепмена—Жуге,
но сначала рассмотрим некоторые
свойства этого особого решения.
В соответствии с (9.1) взятый
с обратным знаком тангенс угла
наклона прямой, соединяющей на
фиг. 176 точку начального состо- ]/р—*- ]/9о
ЯНИЯ О С ТОЧКОЙ КОНечНОГО СОСТО- фиг 1?б Кривая Ре„кина-Гюгонио, на
ЯНИЯ — ТОЧКОЙ кривой ГЮГОНИО которой показана точка Чепмена—Жуге.
DBCB', равен квадрату величины
массовой скорости потока М. Таким
образом, условие Чепмена—Жуге из всех возможных решений выбирает ре-
шение, соответствующее минимальной скорости распространения.
Решение Чепмена—Жуге — это решение, для которого скорость горячего
газа относительно фронта*г>« точно равна местной скорости звука, что можно
показать следующим образом. Поскольку точки кривой Гюгонио изображают
состояния термодинамического равновесия, то производная внутренней энергии
О *> вдоль этой кривой выражается в виде
RlQ —производная энтропии вдоль кривой Гюгонио. [Это непосредствен-
но следует из термодинамических соотношений (ЗО/З^ = p/g2 hCO/3SN= Т.]
Но поскольку внутренняя энергия вдоль кривой выражается непосред-
ственно соотношением (9.3), то производную можно записать также в виде
g0O "
dg«
Приравнивая два последних выражения, получаем выражение для производ-
ной энтропии вдоль кривой Гюгонио
TdSpo goo — gp Г Poo ~ Ро
-— d-
(9.10)
x) Имеется и другая линия, для которой точка касания соответствует давлению, мень-
шему /?0. Это—«нижняяточка Чепмена—Жуге», и она соответствует максимальной возможной
скорости пламени (см. § 7). Эта точка здесь не рассматривается.
618 Гл. 11. Гидродинамические применения уравнений переноса
Таким образом, ясно, что условие Чепмена—Жуге соответствует точке С,
в которой dSco/d Q* = 0. Производная равна нулю также при ?« = q0. Можно
показать [62], что энтропия имеет максимум в точке q0 и минимум в точке
Чепмена—Жуге С. Так как в точке Чепмена—Жуге энтропия экстремальна,
то тангенс угла наклона в этой точке имеет вид
^!г—*(?k-HrzV- <9И)
Но (Эроо/З^сэ)^ — это квадрат скорости звука; таким образом, из (9.1) следует
v* = c~. (9.12)
Иначе говоря, скорость горячего газа относительно фронта равна местной
скорости звука.
Условие Чепмена—Жуге приводит к наименьшей скорости детонации,
возможной при том условии, что химическая реакция завершается пол-
ностью. Меньшая скорость детонации соответствовала быпрямой на диаграмме
Ренкина—Гюгонио (прямая О А' на фиг. 176), не пересекающей кривую Гюгонио
конечного состояния. Очевидно, что в случае реальной детонации возможны
химические реакции, завершающиеся только частично. Но поскольку скорости
реакции всегда конечны, то рассмотренная картина уже не сможет быть описана
математически правильным стационарным состоянием, для которого горячая
граница бесконечно далека от фронта волны. Если скорость больше скорости
Чепмена—Жуге, то конечное состояние будет изображаться точками типа В
или В'. Рассмотрим подробно эту возможность.
Нейман [104], Деринг [105] и Зельдович [106] независимо пришли к
выводу, что детонация — это процесс горения, вызываемый ударной волной.
Они предположили, что время, необходимое для прохождения реакции, велико
по сравнению с временем прохождения газа через ударную волну. Таким
образом, на диаграмме Ренкина—Гюгонио (см. фиг. 176) детонация может быть
представлена ударной волной, в которой состояние изменяется от О до точек
N или А, затем следует превращение в зоне реакции или во фронте пламени,
в результате чего достигается точка на кривой Гюгонио конечного состояния.
Из соотношения (9.1) следует, что если в действительности ударная волна и
химическая реакция существуют порознь, то эти три точки лежат на прямой
линии.
Рассмотрим волну, для которой точка конечного состояния лежит, как и
точка В, выше точки Чепмена—Жуге С. Поток газа за таким фронтом детонации
будет иметь относительно фронта дозвуковую скорость. Таким образом, волна
разрежения может догнать детонационную волну и ослабить ее так, что точка
конечного состояния будет двигаться вниз по кривой Гюгонио. Как только
будет достигнута точка С, скорость потока горячего газа станет равной ско-
рости звука и волны разрежения не смогут уже больше догнать детонационную
волну.
Возможность существования детонационной волны, в которой ударная
волна сопровождается расширением до состояния типа точки В', лежащей
ниже точки Чепмена—Жуге С, может быть легко установлена. Такая волна
будет состоять из ударной волны, переводящей газ в состояние Д следующего
за ней фронта пламени, переводящего газ в состояние В, и «ударной волны
разрежения», в которой газ приходит в состояние В'. Однако ударные волны
разрежения неустойчивы и распадаются на ряд слабых разрывов. (На самом
деле не существует стационарных решений уравнений переноса, соответствую-
щих ударной волне разрежения, см. § 8.) Например, ударная волна разрежения
может распасться на две ударные волны : одну, соответствующую переходу из
состояния В в С, и другую — из состояния С в В'. Вторая ударная волна рас-
пространяется с меньшей скоростью и отстает. Первая распространяется быстрее
первоначальной волны и, как было показано выше, догоняет детонационную
волну и ослабляет ее так, что точка конечного состояния достигает точки С.
§ 9. Теория детонации
619
-*• с
t
г или
Возможность решения, для которого будет иметь место непосредственно
переход из точки О в точку, расположенную ниже точки Чепмена—Жуге
(например, в точку В'), не может быть показана только на основе гидродинами-
ческих рассуждений. В такой волне
химические реакции должны завер-
шаться в заметной степени в газе,
состояние которого близко к началь- f
ному и который не был нагрет и р
сжат ударной волной.
Эйринг, Пауэлл, Дюфей и Пар- ^
лин [107] распространили теорию on гили
Неймана—Деринга—Зельдовича на а
задачи, включающие искривленные 1
фронты детонации и неустойчивые
состояния.
Качественная картина дето-
нации, соответствующая теории и n t z или
Неймана — Деринга — Зельдовича,
представлена на фиг. 177. Головная Т
часть детонационной волны (извест- v
ная как пик Неймана) является
почти идеальной ударной волной, в
которой химическая реакция почти
не происходит. Давление на пике '
(точка N на фиг. 177) равно при-
близительно удвоенному значению
pte, тогда как температура равна
примерно половине То,. Это резкое | и п 2 или
возрастание давления и темпера- х
туры происходит на длине порядка
Ю см. Вторая фаза детонационной
волны сопровождается постепенным
падением давления, возрастанием и п гили{
температуры и завершением хими-
ческих реакций. Эта вторая фаза Фиг. 177. Схематическое представление детона-
протекает на длине порядка 1 см. ционной волны.
Длину этой зоны реакции / (рас-
стояние от точки О до С на фиг. 177)
можно определить экспериментально по значению критического диаметра
стержня взрывчатого вещества, в котором уже не распространяется устойчивая
детонация, по изменению скорости детонации при окружении стержня
взрывчатого вещества слоем инертного вещества различной толщины или по
уменьшению скорости детонации при прохождении искривления известного
радиуса кривизны. В табл. 104 приводятся измеренные значения длины зоны
реакции детонационной волны для ряда взрывчатых веществ [107].
Таблица 104
Измеренная длина зоны реакции для различных
взрывчатых веществ
0 N
-J L.
О N
О N
О N
-*. С
Взрывчатое вещество
тнт
Гексоген
Пикриновая кислота
60-40 аматол
1,СМ
0,076
0,083
0,22
0,398
Взрывчатое вещество
Минол-2
Нитрогуанидин
2 СО+О2 (газ) .
U см
0,538
0,88
1,1
620 Гл. 11. Гидродинамические применения уравнений переноса
Б. ПРИМЕНЕНИЕ ТЕОРИИ К СЛУЧАЮ ДЕТОНАЦИИ
ИДЕАЛЬНОГО ГАЗА
Свойства детонационной волны определяются уравнением состояния
и уравнением теплового баланса. Детонация в твердых взрывчатых ве-
ществах очень сильно зависит от уравнения состояния твердого тела. Од-
нако, чтобы иллюстрировать характер решений, рассмотрим детонацию для
системы, подчиняющейся уравнению состояния идеального газа, при постоян-
ном значении отношения удельных теплоемкостей у.
Если 0 — количество энергии, освобождаемое 1 г вещества в ходе хими-
ческих реакций, то для системы, являющейся идеальным газом с постоянным
значением у, можно записать
Это выражение можно подставить в (9.3) и разрешить его относительно
В результате получаем
У + 1 ! 2<$go g0
У 1 Р g
У— 1 goo
Это выражение есть обобщение выражения (8.10), применявшегося к ударным
волнам. Оно описывает кривую Гюгонио для конечного состояния (кривая
DBCB' на фиг. 176) и, если положить 0 = 0, описывает кривую Гюгонио для
начального состояния AN АО.
Условие Чепмеиа—Жуге требует, чтобы производная dp«>/d(l/?co), опре-
деляемая из выражения (9.14), была равна (рга — Ро)/ [(V^~) — О/бо)]- ТогДа
эта величина, взятая с обратным знаком, будет равна М2. Проводя необ-
ходимые операции, находим
^tb+v^t- (9Л5)
+^i-^. (9.16)
Величина отношения давлений роо/р0 получается подстановкой выражении
(9.16) в выражение (9.14). Верхний знак применяется для детонационного реше-
ния. Знак минус соответствует максимальной возможной скорости распростра-
нения пламени и не будет рассматриваться. С помощью полученных резуль-
татов легко можно показать, что для идеального газа скорость горячих про-
дуктов относительно фронта детонации равна местной скорости звука
^ = свв. (9.17)
Этот результат, как было показано выше, справедлив для любого газа.
Во многих случаях при детонации начальное давление бывает порядка
1 атм, тогда как давление за волной — порядка 200 000 атм. Поэтому инте-
ресно рассмотреть предельный случай р0 <^ р« и соответствующий вид полу-
ченных выше результатов. Тогда скорость распространения детонации равна
(9.18)
Остальные величины даются следующими простыми выражениями :
f=^-, (9.19)
?=. = 2(у-1)-^- (9.20)
Эти соотношения полностью описывают эффекты, сопровождающие детонацию.
§ 9. Теория детонации
621
Таблица 105
Сравнение вычисленных и измеренных значений скорости детонации в газах [108]*
ро = 1 атм, То - 291 °К
Взрывчатая смесь
B^ + 0^
BН2 + О2) + 5О2
BH2 + O2) + 5N2
BН2 + О2) + 4Н2
BН2 + О2) + 5Не
BН2 + О2) + 5Аг
Роо> атМ
18,05
14,13
14,39
15,97
16,32
16,32
Т, °К
3583
2620
2685
2976
3097
3097
Скорость детонации,
м/сек
выч.
2806
1732
1850
3627
3613
1762
измер.
2819
1700
1822
3527
3160
1700
• Берете, Грин и Кистяковский [109] повторили опыты Эльбе и Фриофа [108] и проделали вычисления
с уточненными термохимическими данными. Их результаты очень хорошо совпали с данными Эльбе и Фриофа.
а точность их вычислений была достаточна, чтобы установить расхождение между теорией и экспериментом.
В табл. 105 сравниваются измеренные и вычисленные значения скорости
детонации1) смесей водород—кислород с инертными добавками и без них. Вели-
чина 0 вычислялась на основе
предположения о полном термо- Ю000
динамическом равновесии в точке 8000
Чепмена—Жуте (равновесная 60п00
смесь Н2О, Н2О2, Н, О и ОН), так то
что величина 0 несколько отли-
чалась для каждого опыта2). Зна-
чение у равно его значению для
реагирующей смеси [см. выра-
жение G.21)]. Согласие вычис-
ленных и измеренных значений
очень хорошее, за исключением
систем, находящихся вблизи пре-
делов воспламенения, для кото-
рых имеются заметные расхо-
ждения.
Существование пределов вос-
пламенения зависит как от раз-
меров сосуда (и свойств его по-
верхности), так и от химической
кинетики системы. Для газов, в
которых химические реакции
включают разветвленную или
энергетическую цепь, всегда
имеются два, а иногда три пре-
дела воспламенения. На фиг. 178
показаны пределы воспламенения
стехиометрической смеси кисло-
род—водород [71]. Кривая А соот-
ветствует нижнему пределу вос-
пламенения ; В — кривая верх-
него предела; С —третий предел
воспламенения. Устойчивая детонация возможна только в пределах «языко-
образного полуострова воспламенения», ограниченного кривыми А и В, или
420 440 460 480 500 520 540 560 580
Температура, X
Фиг. 178. Пределы воспламенения стехиометри-
ческой смеси кислород—водород.
х) Подробное обсуждение явления детонации в газах дано в работах [71, 72].
2) Подробности расчетов даны в работе [110].
622 Гл. 11. Гидродинамические применения уравнений переноса
в области высоких давлений над кривой С. В области очень малых давлений,
ниже кривой нижнего предела, детонация невозможна, так как слишком
большая доля свободных радикалов поглощается и разрушается на поверхности
сосуда или взрывной бомбы. Верхний предел воспламенения обусловлен гомо-
генной рекомбинацией свободных радикалов в газовой фазе при тройных
соударениях. В смеси кислород—водород верхний предел обусловлен реакцией
Н2 + О3 + Третья молекула-> НО2 + Третья молекула.
Соединение НО2 сравнительно инертно и обычно будет диффундировать на
стенку, где и распадется. Третий предел воспламенения, по-видимому, присущ
системе Н2 + О2, так как он зависит от давления в системе, когда оно стано-
вится достаточно высоким, чтобы НО2, распадаясь в газовой фазе, возвращало
атомы Н быстрее, чем они диффундировали бы к стенке.
В. СТРУКТУРА ДЕТОНАЦИОННОЙ ВОЛНЫ
Структура детонационной волны описывается решением одномерных ста-
ционарных уравнений переноса. Это те же уравнения, что использовались при
изучении распространения пламен в § 7. Существенное отличие состоит в том,
что при изучении детонации мы ищем решений, для которых скорость волны
относительно холодного газа больше скорости звука.
Как при обсуждении распространения пламени, рассмотрим в качестве
примера одномерную устойчивую детонационную волну в газе, энергия в кото-
рой выделяется в результате единственной мономолекулярной реакции :
А -+ В. (9.21)
Однако при обсуждении детонации значительное упрощение связано с возмож-
ностью пренебрежения обратной реакцией. Обратная реакция играет очень
малую роль и не меняет существенно свойства решений. Дальнейшее упро-
щение можно получить, пренебрегая диффузионными эффектами (т. е. полагая
коэффициент диффузии равным нулю). Однако вязкость играет важную роль
при определении структуры как детонационной, так и ударной волны. Таким
образом, нельзя пренебречь эффектами вязкости, как это делалось при изучении
распространения пламен. Однако фактически упрощение может быть достиг-
нуто, если коэффициент вязкости таков, что постоянная со [определяемая
выражением G.24)] равна единице. Как показано в § 8, такой выбор величины со
физически разумен и дает возможность получить аналитическое решение
уравнений, описывающих структуру ударной волны без реакции.
Уравнения переноса, описывающие структуру такой детонационной
волны, — это уравнения G.25)—G.27). Но поскольку пренебрегаем обратной
реакцией, то у' = у и из выражения, определяющего функцию /(х, т), выпа-
дает второй член. Как указывалось выше, мы пренебрегаем диффузией,
поэтому G = х. Однако при рассмотрении детонации проще принять в
качестве начальных переменных вместо т, и и х величиныр/р*,, и = Qmjq и х,
молярную долю компоненты А. В соответствии с уравнением состояния легко
преобразовать переменные
j_ = JL*l = uJ-. (9.22)
Tee Poo Q р« V '
Таким образом, уравнения переноса, описывающие структуру детонационной
волны, принимают вид
(9.23)
(9.24)
(9.25)
§ 9. Теория детонации 623
где ? — приведенная координата, определяемая выражением G.18)
а /, guh—функции, определяемые выражениями G.29)—G.31). Поскольку для
детонационной волны справедливо условие Чепмена—Жуге, то к„ = 1. Тогда
выражения разд. Б определяют М, е»/бо и Р»/Ро (а также т»/т0). Явный
вид функций /, h и /, соответствующих детонационной волне, для которой
х. = 1 и со = 1, дается выражениями
(9.27)
<9'28>
Постоянная /л определяется из выражения G.19)
, (9.30)
где к' — стерический фактор в выражении для скорости химической реакции
[см. выражение G.15)]. Если в теории пламен величина /л являлась собствен-
ным значением, определяемым детальными решениями уравнений переноса,
то в теории детонации величина /л известна априори в силу условия Чепмена—
Жуге.
Характер решений этих уравнений может быть представлен с помощью
диаграммы Ренкина—Гюгонио (см. фиг. 176). На фиг. 179 построены соответ-
ствующие кривые1). Сначала соотношение (9.14) используется для получения
кривой Гюгонио конечного состояния. Тогда точка Чепмена—Жуге С опре-
деляется из (9.15) или путем проведения прямой линии из точки начального
состояния О, касающейся кривой Гюгонио конечного состояния. Затем для
построения кривой Гюгонио (ударной адиабаты) начального состояния исполь-
зуется выражение (9.14), где Q приравнивается нулю. Линия, проходящая
через точку О и точку Чепмена—Жуге С, пересекает ударную адиабату в точке
N, известной как «пик Неймана». Пику Неймана соответствуют значения плот-
ности и давления, равные значениям за ударной волной, распространяющейся
со скоростью детонации. Однако если газ переводится ударным сжатием из
состояния О в состояние JV, то давление как функция плотности уже будет
описываться не ударной адиабатой, а решением уравнения переноса (8.43).
Кривая этого решения приведена на фиг. 179.
На фиг. 180 показана зависимость Т/То от qJq вдоль кривой решения и
прямой линки OCN. На этой фигуре видно, что температура в пике Неймана N
равна несколько менее половины значения температуры в конечном состоя-
нии — точке Чепмена—Жуге С. Так как в области между О и N температура
мала, то нельзя ожидать, что химические реакции завершатся заметно за корот-
кое время сжатия газа в ударной волне. В действительности температура в
точке N настолько мала, что во многих практических случаях можно ожидать
появления временной задержки или зоны затухания, прежде чем начнутся
реакции.
Ряд качественных свойств решений можно получить путем рассмотрения
характера сингулярности точки С и рассмотрения поверхностей, на которых
2) Значения постоянных, используемых в примерах этого параграфа, совпадают со зна-
чениями, использованными при обсуждении пламен. См. примечание на стр. 602.
624
Гл. 11. Гидродинамические применения уравнений переноса
производные обращаются в нуль. На фиг. 179 прямая OCN связывает значения
давления и плотности, получаемые при Л = 0 (эта связь не зависит от пере-
менной х). Из уравнения (9.24) ясно, что решения «вертикальны», т. е.
d(PlPo)ld(OolQ) = °°> когда они пересекаются прямой OCN. Кривые постоянного
Q2 0,4 0,6 as 1,0 1,2
Ро/р
Фиг. 179. Диаграмма Ренкина—Гюгонио
для ударной волны, иллюстрирующая
решение уравнения переноса (Д), кривые
Гюгонио для начального (С) и конечного
(В) состояний, линии Л = 0 и / = 0.
13
12
П
10
9
8
6
5
4
3
2
1
N
Решение'
для ударной
волны
i i i I I t I I 1 L.
О 0t2 Oft 0,6 0,8 W
Ро/Р
Фиг. 180. Зависимость Т/Тоотqo/q.
Показан ход температурной кривой для
ударной волны и кривая Л = 0.
значения х (кривые х = xj) являются замкнутыми кривыми, определяемыми
условием / = 0. Из уравнения (9.25) следует, что решения «горизонтальны»,
т. е. d(pjpo)ld(QoJQ) = 0, если они пересекаются с кривой / = 0, т. е. если зна-
чение х на кривой соответствующего решения равно Xj.
Удобно разделить плоскость [(р/ро)> (QoIq)] фиг. 179 на две области : область
I — слева от линии h = 0 и область II — справа от этой линии. Из выражения
(9.28) видно, что ft, а следовательно, и d(gole)ldC отрицательны в области I и
положительны в области II. Точно так же из (9.29) следует, что величины /',
а следовательно, и d(plpo)ld? положительны при значениях х, больших х7,
и отрицательны при значениях, меньших Xj. Таким образом,
Область I :
d(Q0lQ)
d (P/Po)
1Ж
(9.31a)
§ 9. Теория детонации 625
Область II: dK°) >0> есЛИ
(9.316)
d(plPo) ^q х<Х
Горячая граница (точка С) является сингулярной точкой ряда дифферен-
циальных уравнений. Рассмотрим характер этой сингулярности. По причинам,
которые будут сейчас изложены, удобно ввести параметр нЛ и рассматривать
детонационные волны, необязательно удовлетворяющие условию Чепмена—
Жуге. Иначе говоря, мы рассматриваем случай детонации, для которого конеч-
ное состояние лежит на кривой Гюгонио фиг. 179 выше точки С. В области
вблизи горячей границы функции правой части уравнений переноса (9.23)—
(9.25) линейны по отношению к отклонениям переменных от предельных зна-
чений. Эти «линеаризованные» уравнения имеют вид
(934)
Общее решение ряда этих уравнений можно записать в виде
х = к Mi е°'с + U М, е°* + tt [х], е^, (9.35)
u-l=t1 ^ + tz e< +13 e<*, (9.36)
?=^ til ^+*• Ш. ^+ь Ш. ^ • ^9-37>
Постоянные tv t2, t3 произвольны и определяются граничными условиями
задачи. Величины а,, [х], и [р/р«],- определяются подстановкой этих выра-
жений в дифференциальные уравнения (9.32)—(9.34). Легко показать, что
«i=--^r-(l-*i), (9.38)
[x]i = 0, (9.39)
(9.40)
а, = --!-*->/'-, (9.41)
= узт^ [^ е-'/г" - 0 ~ «2)] [!+¦?¦ ^1/г~] 0.42)
(9.44)
(9.45)
И.-0'
Для детонационного решения переменные стремятся к конечным пределам
при С, стремящемся к бесконечности. Таким образом, поскольку а3 положи-
Гл. 11. Гидродинамические применения уравнений переноса
тельно, то ta должно быть принято равным нулю и асимптотическое решение
имеет вид
. х = Шге°л, (9.47)
р _
(9.48)
(9-49)
^ ч Пламя
где постоянные tx и t2 определяются из условий на холодной границе. Мо-
лярная доля х должна быть всегда поло-
жительна. Таким образом, поскольку [х]2
положительно, то ясно, что и t2 должно-
быть выбрано положительным. Постоян-
ная tx может быть положительной, отри-
цательной или равной нулю. Решения
трех типов показаны схематически на
фиг. 181 и обозначены: НДЗ, 2 и 3.
Однопараметрическое множество решений
типа НДЗ и 3 соответствует различ-
ным возможным значениям отношения
tjt2. Кривая, обозначенная НДЗ пред-
ставляет типичное решение, следующее
из теории Неймана—Деринга—Зельдо-
вича. Это решение начинается с реше-
ния дифференциальных уравнений для
ударной волны без химической реакции.
Затем по мере возрастания температуры
и появления химических реакций реше-
ние отклоняется от хода кривой для
ударной волны. Точка максимального дав-
ления достигается при х = Xj. Затем ре-
шение НДЗ идет круто вниз, достигая
точки С по кривой, обозначенной «пламя».
Эта кривая графически представляет ре-
шение уравнения, когда начальному со-
стоянию соответствует точка N, а конеч-
ному — точка С. Два других совмес-
тимых типа решений детонационных урав-
нений представлены кривыми 2 и 3.
Кривые этих решений идут от точки О
Фиг. 181. Схематическое представление к с, не достигая максимума по давлению,
трех возможных типов решения дето- реп1ение о ттогтигярт тпш™ Г ппппи гмн
няпипнныу лгпяпнйний гешение z достигает точки с вдоль син-
гулярного решения при f, = 0, t2 ф 0, тогда
как 3 достигает С по другому сингуляр-
ному решению. Возможность и физическая реальность решений этого типа
являются интересным вопросом.
50
40
30
20
10
Решение
ударной волны
национных уравнений.
§ 10. ПОТОК ТОПЛИВНЫХ ГАЗОВ В РАКЕТНЫХ
ДВИГАТЕЛЯХ1)
В качестве другого примера применения гидродинамических уравнений
можно рассмотреть течение топливных газов в ракетных соплах. Внутренняя
баллистика ракеты зависит от скорости возникновения топливных газов и от
течения этих газов через сопло. В ракетах твердого топлива скорость возникно-
[111].
§ 10. Поток топливных газов в ракетных двигателях 627
вения топливных газов определяется скоростью горения твердого заряда ;
чтобы поддерживать давление в камере сгорания постоянным в течение вре-
мени горения, необходимо контролировать скорость горения. Скорость горения
можно контролировать, регулируя состав твердых компонент или изменяя
их геометрическую конфигурацию. В идеальном случае удается поддерживать
стационарное состояние почти в течение всего процесса горения. В моторах
жидкого топлива скорость возникновения газов контролируется регулировкой
скоростей двигательного потока.
В этом параграфе будут рассмотрены стационарные задачи при условии,
что скорость потока одинакова на протяжении любого сечения и что тепло-
проводностью и торможением у стенок можно пренебречь. Чтобы учесть эти
факторы, необходимо дальнейшее углубление в теорию пограничного слоя,
а также в аэродинамику и теорию турбулентности. Во многих практических
случаях, однако, рассматриваемая идеализация является хорошим прибли-
жением, подтверждающимся экспериментально. Стационарность задачи поз-
воляет значительно упростить теоретические расчеты условий на выходе ракеты.
Зная геометрию сопла, в любой его части можно найти давление, температуру
и скорость газа, а также результирующую тягу, ускоряющую ракету.
А. УРАВНЕНИЯ ГИДРОДИНАМИКИ
Течение газа в сопле описывается уравнениями гидродинамики, выведен-
ными в § 1. В этом параграфе за независимые уравнения удобно выбрать урав-
нения непрерывности, уравнение энергии и уравнение энтропии. Предположим,
что существует стационарное состояние, и рассмотрим решение уравнений, не
зависящих от времени. Не зависящие от времени уравнения, описывающие
течение через геометрическую систему с изменяющимся сечением, можно
свести к одномерным уравнениям.
Пусть ось z совпадает с направлением оси ракеты (фиг. 182), а /(х, у) = z
— уравнение граничной поверхности, т. е. внутренней поверхности камеры и
Сечение At
Внешнее давление р0 \ Сечение Ае
Сопло
Головной конец *-Камера Хвостовой конец
Фиг. 182. Схематическое изображение камеры сгорания и сопла ракетного
двигателя.
сопла. Обычно эта поверхность обладает цилиндрической симметрией относи-
тельно оси z. Рассмотрим плоскость, нормальную оси z, и пусть S будет часть
плоскости, отсекаемая поверхностью /(х, у) = z. Вывод одномерных уравнений,
применяемых в настоящей задаче, основан на следующей простой теореме.
Если F(r) описывает поле векторов, касательных к поверхности /(х, у) = z,
то выполняется равенство
?) sJJ <1ол>
S
где Fz — z-компонента F.
628 Гл. 11. Гидродинамические применения уравнений переноса
Уравнение непрерывности в трехмерном случае для стационарного сос-
тояния дается формулой A.2)
(l) (Ю.2)
Поскольку скорость газа на внутренней поверхности камеры сгорания и сопла
либо равна нулю, либо касательна к стенке, то можно применить теорему,
приводящую к равенству A0.1). Таким путем получаем
^ O. A0.3)
Интегрируя затем по z, находим
J M, A0.4)
где М—постоянная1). По смыслу М—масса, переносимая потоком через любое
поперечное сечение в 1 сек. Условия в сопле близки к однородным на протя-
жении любого сечения. Следовательно, предыдущую формулу можно записать
в виде
q(z)v(z)A(z) = M, A0.5)
где A(z) — поперечное сечение как функция z, a q(z) и v(z) — средние значения
плотности и скорости, определенные таким образом, чтобы было справедливо
уравнение A0.4).
Уравнение сохранения энергии может быть выведено аналогичным образом.
Если уравнение движения A.3) умножить на q\ и сложить с уравнением энер-
гии A.4), то получим
Принимая во внимание уравнение непрерывности A0.2), находим
o. (ю.7)
Это уравнение можно проинтегрировать во всей плоскости, нормальной оси z.
Если предположить, что давление на стенке равно нулю, а теплообмен со стен-
кой отсутствует, то можно воспользоваться формулой A0.1). В результате
получаем
JJ [О + у *>2) Qvzdxdy + ^ (p-v)zdxdy + ^ qzdxdy = const. A0.8)
Если пренебречь эффектами вязкости и теплопроводности, то это уравнение
можно апроксимировать следующим одномерным уравнением :
(О + \ г;2) qvA +pvA = const. A0.9)
Поделив на постоянный множитель gvAy получим уравнение сохранения
энергии
Й + 4rv* = const, A0.10)
где Й = О + (pIq) —энтальпия единицы массы. В этом уравнении постоянную
удобно ЕЫражать, используя условия в камере сгорания, т. е. в точке, находя-
щейся далеко (в отрицательном направлении оси) от сопла. Обозначая условия
в этой точке индексом с, получаем
* (^_*,2) = #c--#. A0.11)
г) Символ М использовался в предыдущих параграфах для обозначения несколько дру-
гой величины — потока массы через единичное сечение.
§ 10. Поток топливных газов в ракетных двигателях 629
Если точка с выбирается на стенке камеры сгорания, то скорость vc равна нулю.
Практически условия внутри камеры сгорания ракет близки к однородным,
а кинетической энергией потока в любой точке камеры можно пренебречь.
В этом случае уравнение энергии [см. формулу A0.40) в практических едини-
цах] принимает вид
Vtf-tf). A0.12)
Если пренебречь эффектами диффузии, вязкости, теплопроводности и откло-
нением от химического равновесия, то в соответствии с результатами § 1 поток
можно считать изэнтропическим. Это условие используется в качестве третьего
уравнения гидродинамики. Последнее уравнение совместно с уравнением непре-
рывности A0.5) и уравнением энергии A0.12) полностью описывает течение газа
через сопло или насадок.
Уравнение энергии A0.12) позволяет выразить скорость газа как функцию
энтальпии. Конкретный вид этого уравнения определяется видом уравнения
состояния и термохимией процессов в рассматриваемом газе. Практически эти
расчеты оказываются довольно сложными. Необходимые числовые результаты
будут приведены в конце этого параграфа. Однако для получения качествен-
ных результатов сначала будет рассмотрен идеальный газ с постоянной удель-
ной теплоемкостью.
Б. ТЕЧЕНИЕ ИДЕАЛЬНОГО ГАЗА В СОПЛЕ
При постоянной удельной теплоемкости газа уравнение энергии A0.12)
можно записать в виде
T)^(Tc-T). A0.13)
Эта формула выражает зависимость скорости течения газа от температуры.
Вследствие изэнтропичноаи течения связь между температурой и давлением
в движущемся газе выражается адиабатическим уравнением состояния. Для
идеального газа эта связь, выраженная через параметры газа в камере сгорания,
имеет вид
С помощью этих формул уравнение A0.13) записывается следующим образом :
Полученный результат можно выразить через скорость звука [определенную
формулой D.5) ] в камере сгорания
Как видно из формулы, максимальная скорость достигается при полном рас
ширении, когда р/рс = 0. Эта максимальная скорость равна
][ Т у/
пп 17\
• (ЮЛ7)
В качестве примера типичных условий, встречающихся в ракетах, возьмем
следующие : температура в камере сгорания 3500° К, средний молекулярный
вес 25, отношение удельных теплоемкостей у = 1,20 [т. е. (г>т/сс) = УЩу—1)^3].
Поскольку {кТс\т) = 1,1 • Ю10 см2/сек2, то максимально достижимая скорость
истечения в ракетах, использующих это топливо, составляет ~ 3,6 • 10б см/сек,
или 12 000 фут/сек. В действительности полное расширение никогда не
достигается, поэтому скорость истечения будет приблизительно в 2 раза
меньше скорости звука в камере, т. е. порядка 2 • 105 см/сек, или 7000 фут\сек.
630
Гл. 11. Гидродинамические применения уравнений переноса
Параметры газа внутри сопла как функции площади сечения получаются
из уравнения непрерывности A0.5). Исключая из уравнения непрерывности
плотность с помощью уравнения адиабаты A0.14) и скорость с помощью фор-
мулы A0.16), получаем выражение
(у- 1 у/2 M
{ 2 j QcCc
W"
A0.18)
Эта формула выражает давление через сечение ; вид функциональной зависи-
мости изображен на фиг. 183.
Кривая, выражающая зависимость А от р, имеет минимум. Значения
величин в этой точке будем обозначать индексом t. Простым дифференцирова-
нием можно показать, что pt и At имеют вид
Pt = ( 2 \
Рс { У + 1 J
A0.19)
A0.20)
С помощью уравнения адиабаты A0.14)
и формулы для скорости можно найти
выражения для ft и vt, которые имеют вид
2 у/<у-0
Фиг. 183. Зависимость давления в сопле
от площади поперечного сечения сопла.
Aа22)
К(щбинируя последнюю формулу с выра-
жением для скорости звука [формула D.5) ],
а также с формулами A0.19) и A0.21), можно написать
vt = ct; (Ю.23)
это значит, что скорость потока в минимальном сечении равна местной скорости
зрука.
Для типичной ракеты, рассмотренной выше, сТс = 3500° К, у = 1,2 и
молекулярным весом 25, найдено, что ptjpc = 0,56, a vt = 110 000 см\сек. Если
давление в камере рс равно 200 атм, то (М/Д) = ft vt = 1,200 г/см2-сек.
Рассмотрим теперь поток газа, вытекающий из камеры сгорания через
сопло наружу. Предположим, что давление в камере сгорания рс, а давление
окружающей йнешней среды р0. Пусть At — сечение сопла или насадка, а
Ае — это же сечение на конце сопла. Из фиг. 183 видно, что на конце сопла
давление может быть равно либо реу либо р'е. В связи с этим могут существовать
три типа течения :
1) Простое течение Бернулли, появляющееся при
Ро>Ре-
A0.24)
В этих условиях давление нигде не достигает значения ри а критическое
сечение не соответствует минимуму, изображенному на фиг. 183. Скорость газа
остается дозвуковой на протяжении всего потока. Скорость звука возрастает,
достигая максимума, а давление падает до минимума в узком- сечении.
2) Обычное течение в ракетах, появляющееся при
А><Л- (Ю.25
§ 10. Поток топливных газов в ракетных двигателях 631
В этих условиях критическое сечение совпадает с точкой, обозначаемой индек-
сом t. Скорость газа равна местной скорости звука в критическом сечении и
непрерывно возрастает за ним. Давление вдоль расширяющейся части сопла
монотонно падает.
3) Течение с ударными волнами в соплах с перерасширением, появляю-
щееся при
Р'е>Ро>Ре. (Ю.26)
В этих условиях не может осуществиться изэнтропическое течение и в сопле
образуются ударные волны. Вверх по течению от ударной волны поток такой
же, как в обычных ракетных соплах.
Результирующая сила или тяга возникает под действием неуравновешен-
ных сил в камере сгорания. Если камера сгорания и сопло обладают цилиндри-
ческой симметрией, то результирующая тяга приложена в направлении, про-
тивоположном соплу. Как видно из фиг. 182, тягу можно рассматривать как
результирующую четырех сил
F = F, + F2 + Fz + Ft, A0.27)
где F2 — сила давления газа на переднюю стенку камеры сгорания ; F2 — сила
давления газа на тыльную стенку камеры сгорания ; F3 — сила давления газа
в расширяющейся части сопла; F4 — неуравновешенная сила, возникающая
в результате давления внешней атмосферы р0 на всю камеру сгорания, за исклю-
чением области выходного отверстия Ае.
Эти силы можно записать в виде
F1 = pcACJ F4 = — А> Ае у
Ас А,
F2 = - J pdA, F3 = J pdA. A0.28)
At At
Давление в подынтегральных выражениях является функцией сечения, как это
в'идно из формулы A0.18) или из фиг. 183. В выражении для F2 интегрирование
осуществляется вдоль правой ветви кривой на фиг. 183, а в F3 — вдоль левой
ветви.
Интегралы, входящие в выражение для тяги, берутся непосредственно.
Результат, выраженный через скорость истечения газа, даваемую формулой
A0.16), и расход массы М можно записать в виде
F = Mve + (pe-p0)Ae. A0.29)
Таким образом, общая тяга ракеты равна скорости изменения импульса двига-
тельных газов в выходном сечении сопла плюс избыточное давление газов на
выходное сечение.
В. ТЕРМОХИМИЯ СМЕСЕЙ РЕАЛЬНЫХ ГАЗОВ
В предыдущем разделе была изложена теория течения идеального газа в
ракетном сопле. Однако характеристики реальных ракет зависят от свойств
топливных газов, т. е. от свойств горячих химически реагирующих смесей.
В общем случае свойства газа и динамика потока зависят от кинетики хими-
ческих реакций. Однако при высоких температурах, существующих в камерах
сгорания ракет, скорости реакций настолько велики, что состав газа с хорошим
приближением можно считать термодинамически равновесным. Если предпо-
ложить, что это условие выполняется, то свойства смеси полностью описываются
законами обычной термодинамики.
При достаточно высоких температурах предположение о термодинами-
ческом равновесии выполняется хорошо, поскольку все химические реакции
в этом случае протекают очень быстро. Если, однако, газы расширяются и
охлаждаются, то состав смеси может оказаться «замороженным». Это происходит
632
Гл. 11. Гидродинамические применения уравнений переноса
в том случае, когда скорости химических реакций меньше скорости охлаждения.
Рассмотрим, например, реакцию азота с кислородом с образованием окиси
азота, которая происходит при нагревании воздуха :
N2 + O2^2NO. A0.30)
Время, необходимое для образования половины равновесной концентрации
окиси азота, приведено в табл. 106 [112]. При температурах выше 2300° К
равновесное состояние достигается в течение малых долей секунды, тогда как
при температуре 1500 °К для достижения равновесия требуются дни.
Таблица 106
Время, необходимое для образования половины равновесной
концентрации окиси азота в воздухе при атмосферном давлении
г,°к
1500
1700
1900
1,09
3,54
1,25
сек
• 10*
• 103
• 102
Г, °К
1 2100
2300
2500
5,06
2,25
1,06
, сек
• ю-1
• ю-2
т, °к
2700
2900
3100
U/t, сек
5,25-
3,45
1,86
10-*
ю-б
ю-6
Для мономолекулярной реакции с энергией активации Е+ время релакса-
ции1) реакции (время образования половины равновесной концентрации) —
порядка секунды при E+/RT = 30. Если ?+ = 40000 кал/моль, то при темпе-
ратурах выше 700° К время релаксации меньше 1 сек. Для реакций с энергией
активации 100 000 кал (соответствующей разрыву углеродно-водородной связи)
время релаксации меньше 1 сек уже при температурах выше 1700° К.
Существенным облегчением для вычислительной работы является то обсто-
ятельство, что процесс горения в большинстве орудий и ракет происходит при
таких высоких температурах, при которых термодинамическое равновесие
осуществляется с большой степенью точности. Однако при этих температурах
молекулы диссоциируют, поэтому необходимо рассматривать большое число
молекулярных компонент. Даже небольшая примесь свободных радикалов или
атомов может оказывать заметное влияние на внутреннюю энергию и другие
термодинамические свойства.
Равновесный состав можно получить путем совместного решения уравне-
ний сохранения массы и уравнений химического равновесия (равенство хими-
ческих потенциалов). Поскольку в химических реакциях не возникают или
уничтожаются атомы, то общее число атомов каждой компоненты остается
постоянным. Таким образом, если в 1 г смеси всего содержится gj атомов /-го
сорта и если молекула /-го сорта содержит vJt атомов /-го сорта, a fit — число
молекул /-й химической компоненты в 1 г смеси, то
iNi = ij, /=1,2,...,s. A0.31)
Для каждой из 5 атомных компонент существует уравнение такого типа.
Если имеются / химических реакций, которые можно записать в стехио-
метрической форме в виде
Аа [1] + Ая И + .. •
[1]
+
A0.32)
нием
) При нормальной величине стерического фактора скорость реакции дается выраже-
k = lO^e-E+IRTceK-1, к = 1 сек-1 при E+/RT = 30.
§ 10. Поток топливных газов в ракетных двигателях 633
и если /*, — химический потенциал i-й компоненты, то в условиях химического
равновесия
2 [Рш ~ Чш\ Л = 0, * = 1,2, ... , I. A0.33)
Химический потенциал /*,, выраженный через свойства чистой компоненты i
и уравнение состояния смеси, дается формулой C.4) гл. 5.
Уравнения A0.31) и A0.33) совместно с уравнением состояния определяют
равновесный состав. Точный метод решения зависит во многом как от специфики
проблемы, так и от наличия быстродействующих счетных машин. Бинкли [ИЗ]
рекомендует получать первое приближение для состава, используя законы
идеального газа, затем с помощью первого приближения для состава получить
уравнение состояния в первом приближении и вычислить интеграл в фор-
муле C.4) гл. 5. Таким путем методом последовательных приближений можно
найти истинный состав смеси. В работах Корнера [114] и Гиршфельдера, Мак-
Клура, Кертисса и Осборна [115] приведен подробный расчет состава порохо-
вых газов. При высоких температурах (не выше 3000° К) реакции диссоциации
несущественны, необходимо лишь рассматривать реакцию с участием водяного
пара :
СО + Н2О ^СОа + Н2. A0.34)
Для пороховых газов в области температур от 2000 до 2500° К в равновесном
состоянии на каждые 77 молей СО приблизительно приходится 23 моля СО2.
Принимая во внимание этот факт, можно развить [115] упрощенный метод
расчета термохимических параметров пороховых газов, в котором температура
пламени, удельная теплоемкость и число молей в 1 г выражаются аддитивно
через первоначальный состав пороха, т. е. в виде суммы молярных долей хими-
ческих компонент в порохе, каждая из которых умножена на характеристи-
ческую постоянную.
Температура пламени топлива определяется просто из закона сохранения
энергии. Пусть Of — энергия, которая выделится, если 1 г топлива разложить
на элементы в их стандартном состоянии и эту смесь охладить до абсолютного
нуля. Эта энергия равна энергии, необходимой для образования результирую-
щей смеси газов и нагрева смеси до температуры пламени, а также некоторой
работе, связанной с расширением газа. Таким образом, уравнение сохранения
энергии имеет вид
Of = O+JpdV, A0.35)
где V = 1/е — удельный объем, а О — внутренняя энергия смеси, отнесенная
к 1 г.
Рассмотрим два частных случая. Для получения изохорической темпера-
туры пламени будем считать V постоянным. В этом случае интеграл равен
нулю и
Of=O (ПРИ изохорическом процессе). A0.36)
Для получения изобарической температуры пламени будем считать р постоян-
ным. В таком случае
j A0.37)
где Vf — удельный объем топлива. Уравнение сохранения энергии имеет вид
O + pV, A0.38)
или
Й* = Й (при изобарическом процессе). A0.39)
Расчет термодинамических свойств смеси, находящейся в равновесии,
позволяет получить сведения, необходимые для применения формул разд. А
к течению смеси реальных газов в сопле.
634
Гл. 11. Гидродинамические применения уравнений переноса
Г. ТЕЧЕНИЕ РЕАЛЬНОГО ГАЗА В СОПЛЕ
Для применения формул, описывающих течение газа в сопле, удобно сум-
мировать свойства газа, что и сделано на фиг. 184 и 185, на которых графи-
чески изображены свойства газа, возникающего при горении типичного ракет-
ного твердого топлива. Фиг. 184 представляет собой диаграмму Молье, где
давление и температура газа изображены как функции удельной энтальпии Й
и удельной энтропии 5. Изобарическая адиабатическая температура пламени
нанесена на графике так, что, зная давление в камере сгорания рс, можно по
Н
1500
то
1300
1200
1100
1000
900
800
100
600
500
2,0
Адиабатическая тртерптщ
У/ju пламени
р - Давление, атм
Т - Температура, °К
Н - Энтальпия, кал/г
S - Энтропия, кал/град г
/У/ /van/ /
///Awn/ /
\/ / /
/ / /
/
/У/ /won/ ~7
////won/ / У j
/У/ /пор/ У /' У / /
/У/ Ушо/ У У У \ У У
J_
2,1
2,1
2,5
2,6
Фиг. 184. Диаграмма Молье, описывающая термохимические свойства газов, образую-
щихся при горении типичного ракетного пороха.
точке пересечения линии этого давления с линией адиабатической температуры
пламени найти начальную энтальпию Йс и начальную энтропию §с пороховых
газов. На фиг. 185 изображена зависимость р/Тд от Т при постоянном давле-
нии. Этот график позволяет найти уравнение состояния в форме, наиболее удоб-
ной для рассматриваемых применений. Зная давление и температуру, можно
довольно просто определить соответствующее значение плотности q. (Диа-
граммы давления как функции плотности для различных значений температуры
недостаточно точны.) В соответствии с разд. А, в котором обсуждались урав-
нения переноса, течение газа в сопле является изэнтропическим. Таким образом,
условия вдоль трубки тока в сопле, соответствуют на диаграмме Молье усло-
виям вдоль вертикальной линии (см. фиг. 184). Как видно из формулы A0.12),
увеличение кинетической энергии газа равносильно уменьшению энтальпии.
Если энтальпию выражать в кал\г, то эту'зависимость можно написать в виде
v = ЗQ0t2Vйc-Й фут/сек = 9150 Ыс - Й см/сек. A0.40)
С помощью этой формулы и диаграммы Молье можно найти скорость газа как
функцию давления в камере сгорания и текущего давления в сопле, т. е.
v = v(pc, p). Подобным же образом комбинация диаграммы Молье с диаграм-
мой уравнения состояния позволяет определить плотность пороховых газов
в сопле как функцию начального давления в камере сгорания и текущего дав-
ления, т. е. q = д(рсу р).
Изменения свойств газа вдоль сопла можно получить, используя урав-
нение непрерывности, утверждающее, что М = gvA — постоянная. Для газа,
§ 10. Поток топливных газов в ракетных двигателях
635
входящего в сопло, v мало, a q велико. Для выходящего газа q становится малым,
а г? велико. Величина qv имеет максимум, который равен M/At. Следовательно, в
любой точке А\АХ как функция р равна максимальному значению qv, деленному
на текущую величину qv. Эту кривую можно получить графически с помощью
функций, описанных в предыдущем параграфе. Затем с помощью графического
метода пересечений можно получить q, v или любые другие переменные как
функции AjAt.
3,50
р - Давление, атм
Т - Температура, °К
р - Плотность, г/см3
1000 1500 2000 2500 3000 3500
Фиг. 185. Уравнение состояния для типичного порохового газа.
Действительная характеристика ракеты определяется тягой ракеты F.
Можно показать [116], что выражение
+ (pe-p0)Ae) A0.41)
выведенное в частном случае идеального газа [формула A0.29)], сохраняет
свою силу и в общем случае. Поскольку указанным методом можно вычислить
ve и ре как функции AeJAh то тяга на единицу площади выходного сечения
F/Afy а следовательно, и характеристика ракеты определяются термодинами-
ческими свойствами пороховых газов.
Рассмотрим некоторый частный пример. Предположим, что в камере сго-
рания ракеты находится типичный ракетный порох, о котором шла речь раньше;
пусть также отношение площади поверхности пороха к критическому сечению
выбрано так, чтобы было обеспечено стационарное давление в камере сгорания,
равное 250 атм. Это отношение определяется скорее из характеристической
скорости горения пороха, чем из термодинамических соображений. Из диа-
граммы Молье для этого пороха (см. фиг. 184) видно, что при рс = 250 атм
изобарическая адиабатическая температура пламени равна 3420° К. Удельная
энтальпия Йс в этих условиях равна 1476 кал/г, а удельная энтропия §с равна
2,391 кал\г° К. Если сопло хорошо рассчитано и в нем отсутствуют тепловые
потери, то газ расширяется в сопле без изменения энтропии так, что давление
и температура в процессе расширения изменяются вдоль пунктирной линии.
Для составления табл. 107 из диаграммы Молье были взяты величины
Т и Й, соответствующие различным значениям давления, и с их помощью
по формуле A0.40) были найдены значения скорости v. Из фиг. 185 для опре-
деления g были взяты значения pjTq при каждом полученном значении темпе-
ратуры. Затем с помощью формулы A0.5) были найдены значения qv= M/A.
636
Гл. 11. Гидродинамические применения уравнений переноса
Таблица 107
Параметры типичного порохового газа в сопле при давлении в камере сгорания
250 атм
Р/Ре
1
0,9
0,8
0,6
0,4
0,2
од
0,04
0,02
0,01
0,004
Р,
атм
250
225
200
150
100
50
25
10
5
2,5
1
г, °к
3419
3373
3323
3180
2982
2654
2361
1986
1742
1530
1295
Йс—Й,
кал/г
0
26
55
135
237
397
531
688
790
878
970
V
см/сек
0
46 800
67 800
106300
140 800
182 300
210 800
239900
257 100
271000
284 900
фут(сек
0
1535
2224
3488
4619
5981
6916
7871
8435
8891
9347
PlTg,
атм • см*
г • град
3,379
3,368
3,357
3,335
3,307
3,278
3,263
3,259
3,257
3,255
3,254
Q, г/см*
0,02164
0,01981
0,1793
0,01414
0,01014
0,005747
0,003245
0,001545
0,0008813
0,0005020
0,0002373
Ml А,
г\смгсек
0
927
1216
1503
1428
1048
684,0
370,6
226,6
136,0
67,6
A/At
оо
1,632
1,244
1,007
1,060
1,444
2,212
4,082
6,677
11,12
22,38
1750
1500
Г-—i г 1 1 1 г
Критическое сечение
Изобразив графически величину этого отношения как функцию давления
(фиг. 186), можно определить, что максимальная величина отношения
1513 г-см сек достигается при давлении р= 137 атм. Это значит, что
М\Аг = 1513 г-см сек при давлении в камере сгорания, равном 250 атм.
(Каждому значению давления в
камере сгорания соответствует
характеристическая величина
M/At.) Для поддержания рав-
новесного давления в камере
сгорания на уровне 250 атм
необходимо, чтобы в 1 сек сгорало
1513Д г пороху. Значения A\At
получаются делением числа
1513 г-см сек на величину
М\А. Функциональная зависи-
мость скорости от величины AjAt
2оо 225 250 графически представлена на
фиг. J.87.
Фиг. 186. Зависимость массовой скорости течения
пороховых газов от давления.
Давление в камере сгорания 250 атм.
25 50 75 100 125 150
р, атм
Для расчета тяги при любой
величине отношения Ae]At7 опре-
деляемой конструкцией сопла,
воспользуемся формулой A0.41).
помощью табл. 107 найдем соответствующие значения ve и ре.
Таблица 108
Тяга ракетного двигателя на пороховом топливе в зависимости от величины
отношения
1,000
1,060
1,444
2,212
4,082
6,672
11,12
22,38
ре, атм
137
100
50
25
10
5
2,5
1,0
Mve/At
г(см*
177 700
217 200
281300
325 200
370100
396 700
418100
439600
фунт/дм*
2527
3089
4001
4625
5264
5642
5947
6253
AiiPe-
атм
136
104,9
70,7
53,1
36,7
26,7
16,7
0
-Ро)/Л
фунт/дм*
1999
1542
1039
780
539
392
245
0
фунт/дм*
4526
4631
5040
5405
5803
6034
6192
6253
Задачи
637
Подставляя эти величины в формулу A0.41), определяем отношение тяги к пло-
щади критического сечения при различных значениях отношения AeIAt.
Полученные результаты приведены
в табл. 108 для р0 = 1 атм.
Как видно из таблицы, при даЕ-
лении на выходе, равном 1 атм, отно-
чшение выходного сечения к крити-
ческому сечению равно 22,38. Для
большинства ракет эта величина зна-
чительно меньше приведенной здесь.
Обычно отношение выходного сечения
к критическому сечению заключено в
пределах от 2 до 4, т. е. давление на
выходе лежит между 25 и 10 атм.
Из табл. 108 видно, что тяга в таких
соплах составляет от 86 до 93% тяги
в соплах с тем же самым критическим
сечением и с давлением на выходе,
о,з
1,8 2,1 2,4 2J 3,0
равным 1 атм.
0,6 0,9 1,2 1,5
Скорость
Фиг. 187. Зависимость скорости пороховых
газов A03 фут/сек) от площади поперечного
сечения сопла.
Начальное давление 250 атм.
Задачи
1. Найдено [117], что в углекислом газе при 3600°F испускательная способность равна
0,085 при L — 2 фут и р = 1 атм. Чему равна величина /л? Чему равен поток энергии, испу-
щенный сферой углекислого газа радиусом 1 дюйм при давлении 24 атм и температуре
3600 °F в вакуум?
2. Найти скорость звука в N2 и в Не при комнатных температурах и давлении. Рассчи-
тать также эту величину для смеси N2 и Не в отношении 1 : 1 в тех же условиях. Расчет
произвести в двух случаях: предполагая газ идеальным и принимая во внимание второй
вириальный коэффициент. Сравнить эти скорости со средними квадратичными скоростями
молекул. Найти частоту, при которой длина волны равна средней длине свободного пробега.
3. Исследовать поведение волны разрежения методом характеристик Римана. В на-
чальный момент времени t = 0 считается, что
v(z) = О,
7B) = Го (постоянная),
71Z
р (z) = р0 — рг cos -у— ; —
— < z < у
Предполагается, что газ идеальный, а отношение удельных теплоемкостей у является по-
стоянным.
4. Рассмотреть ударную волну в азоте при комнатной температуре и атмосферном
давлении, предполагая, что отношение давлений р»/р0 = 10, и считая газ идеальным. Найти
скачок плотности, температуры и скорости в ударной волне. Определить скачок энтропии в
ударной волне, а также конечную температуру при адиабатическом расширении газа до
начального давления. Пролелать те же вычисления для случая p<*>/Pq ~ 2100.
5. На основе термодинамических данных, приведенных в таблицах [Бюро стандартов
(U. S. Bureau of Standards Tables (Circular C461) „Selected Values of Properties of Hydro-
carbons") или в любых других источниках ], рассчитать скорость детонации и давление
Чепмена—Жуге при взрыве стехиометрической смеси водорода и кислорода, первоначально
находящейся при давлении 1 атм и Т = 300°К.
6. Рассчитать изохорическую и изобарическую температуры пламени в стехиометри-
ческой смеси водорода и кислорода, первоначально находящейся при давлении 1 атм при
Т = 300°К.
а) Считать, что концентрация гидроксила равна равновесной при температуре
пламени.
б) Считать, что при температуре пламени концентрация гидроксила равна нулю.
638 Гл. 11. Гидродинамические применения уравнений переноса
ЛИТЕР АТУРА
1. Knudsen M., Kinetic Theory of Gases, London—New York, 1950.
2. Goldstein S., Modern Developments in Fluid Dynamics, Vol. 1, 2, Oxford, 1938.
3. D г у d e n H. L., Sr., Quart. Appl. Math., 1, 7 A943).
4. Batchelor G. K., The Theory of Homogeneous Turbulence, Cambridge, 1953.
5. G о 1 d s t e i n S., Statistical Theory of Turbulence, Lecture Series No. 6, Institute for
Fluid Dynamics and Applied Mathematics, University of Maryland, 1950.
6. F r e n к i e 1 F. N., Introduction to Some Topics in Turbulence, Lecture Series No. 3,
Institute for Fluid Dynamics and Applied Mathematics, University of Maryland, 1950.
7. Schlichti n g H., Grenzschicht-theorie, Karlsruhe, 1951.
8. M с A d a m s W. H., Heat Transmission, New York, 1942.
9. J а к о b M., Heat Transfer, Vol. I, New York, 1949.
10. P e г г у J. H., Chemical Engineers Handbook, New York, 3rd Ed., 1950.
11. Car slaw H. S., Jaeger J. C, Conduction of Heat in Solids, Oxford, 1947.
12. Inge r so 11 L. R., Zobel O. J., Ingersoll A. C, Heat Conduction, New York,
1948.
13. Jones R. C, Furry W. H., Rev. Mod. Phys., 18, 151 A946) (см. перевод:
Джонс К., Ферри В., Разделение изотопов методом термодиффузии, ИЛ, 1947).
14. Lord Rayleigh, Phil. Mag., 32, 529 A916).
15. J e f f г е у s H., Phil. Mag., 2, 833 A926).
16. Jeff reys H., Proc. Roy. Soc, A118, 195 A928).
17. Jeff re ys H., Quart. Journ. Mech. Appl. Math., IV C), September A951).
18. Chandra K-, Proc. Roy. Soc, A164, 231 A938).
19. С hand rase к ha r S., Proc. Roy. Soc, A210, 26 A951).
20. Benedict M., Boas A., Chem. Eng. Progr., 47, 51 A951).
21. В e n e d i с t M., Diffusion Separation Methods, Encyclopedia Chem. Techn., 5,76 A950).
22. Ruhemann M., Separation of Gases, Oxford, 2nd. Ed., 1949.
23. Chandrasekhar S., Radiative Transfer, Oxford, 1950.
24. Chandrasekhar S., An Introduction to Stellar Structure, Chicago, 1939 (см. пе-
ревод : Чандрасекар С, Введение в учение о строении звезд, ИЛ, 1950).
25. R о s s e I a n d S., Theoretical Astrophysics, Oxford, 1936.
26. De Groot S. R., Thermodynamics of Irreversible Processes, Amsterdam, 1951.
27. Prigogine I., Etude Thermodynamique des Processus Irreversibles, Диссертация,
Paris, 1947.
28. О n s a g e r L., Phys. Rev., 37, 405 A931).
29. О n s a g e r L., Phys. Rev., 38, 2265 A931).
30. Onsager L., Ann. N. Y. Acad. Sci, 46, 241 A945).
31. Venant St., Compt. rend., 17, 1240 A843).
32. Stokes, Trans. Cambridge Phil. Soc, 8 A845).
33. Karim S. M., Rosenhead L., Rev. Mod. Phys., 24, 108 A952).
34. J о s t W., Diffusion in Solids, Liquids, Gases, New York, 1952.
35. Hwang J. L., Journ. Chem. Phys., 20, 1320 A952).
36. Stokes R. H., Trans. Farad. Soc, 48, 887 A952).
37. M a g e e J. L., The Effect of Radiation in Flames, CM-627,.University of Wisconsin Naval
Research Laboratory, August 1950.
38. Ruark A. E., Urey H. C, Atoms, Molecules and Quanta, New York, 1930.
39. E у r i n g H., W a 11 e r J., К i m b a 11 G. E., Quantum Chemistry, New York, 1944
(см. перевод : Эйринг Г., Уолтер Дж., К и м б а л л Дж., Квантовая химия,
ИЛ, 1948).
40. Penner S. S., Journ. Appl. Phys., 21, 685 A950).
41. GALCIT (Guggenheim Aeronautical Laboratory, California Institute of Technology),
Progress Report 9—38, 1949.
42.. В r i 11 о u i n L., Die Quantenstatistik, Berlin, 1931 (см. перевод: БриллюэнЛ.,
Квантовая статистика. Харьков—Киев, 1934).
43. На slam R. Т., Love 11 W. G., Hunneman R. D., Ind. Eng. Chem., 17, 272
A925).
Литература 639
44. G а у d о n A. G., Spectroscopy and Combustion Theory, New York, 2nd Ed., 1948
(см. перевод: Гейдон А., Спектроскопия и теория горения, ИЛ, 1950).
45. Ландау Л., Те л л ер Е., Phys. Zs. Sowjetunion, 10, 34 A936).
46. Schwartz R. N., Slawsky Z. I., Herzfeld K. F., Journ. Chem. Phys., 20,
1591 A952).
47. Меттер И. М., Phys. Zs. Sowjetunion, 12, 232 A937).
48. Kantrowitz A., Journ. Chem. Phys., 14, 150 A946).
49. Ha be r P. W., Kantrowitz A., Journ. Chem. Phys., 15, 275 A947).
50. H о 11 e 1 H. C, Trans Am. Inst. Chem. Eng., 38, 531 A942).
51. Sc hack A., Arch, fur Eisenhtitten, 19, 11 A948).
52. M a r k h a m J. J., Beyer R. Т., Lindsay R. В., Rev. Mod. Phys., 23, 353 A951).
53. В e r g m a n n L., Der Ultraschall, Zurich, 1949 (см. перевод с 2-го изд.: Бергман Л.,
Ультразвук, ИЛ, 1954).
54. К о h I e r M., Ann. d. Phys., 39, 209 A941).
55. Da mk о hie r G., Zs. Electrochem.,'48, No. 2, 3 A942).
56. Herzfeld K., Journ. Chem. Phys., 9, 513 A941).
57. Kneser M. O., Ann. d. Phys., 2, 761 A931).
58. Primakoff H., Journ. Acoust. Soc. Am., 13, 14 A942).
59. Tsien H. S., Sc ham berg R., Journ. Acoust. Soc. Am., 18, 334 A946).
60. Wa n g Chang С S., On the Dispersion of Sound in Helium, University of Michigan,
Department of Engineering Research Report UMH-3-F, APL/JHU CM-467, May 1948.
61. Wang Chang C.S., U h 1 e n b e с k G. E., On the Propagation of Sound in Monatomic
Gases, University of Michigan, Department of Engineering Research Report to ONR,
October 1952.
62. С о u r a n t R., F r i e d e r i с h s К. О., Supersonic Flow and Shock Waves, New York,
1948 (см. перевод: Курант Р., Фридрихе К., Сверхзвуковое течение и удар-
ные волны, ИЛ, 1950).
63. Hadamard J., Lemons sur la propagation des ondes, Paris, 1902.
64. Hirschfelder J. O., Curtiss С F., Journ. Chem. Phys., 17, 1076 A949).
65. Hirschfelder J. O., Curtiss С F., Journ. Phys. and Colloid. Chem., 55, 744
A951).
66. Curtiss С F., Hirschfelder J. 0., Campbell D. E., Theory of Flame
Propagation and Detonation, III, University of Wisconsin Naval Research Laboratory
CM-690, 1952.
67. Curtiss С F., Hirschfelder J. O., Campbell D. E., Fourth International
Symposium on Flames and Combustion, New York, 1953.
68. H i r s с h f e 1 d e r J. O., Curtiss C.F., Campbell D. E., Journ. Phys. Chem.,
57, 403 A953).
69. Hirschfelder J. O., Campbell E. S., Analytical (Power Series) Solutions of the
Flame Equations, University of Wisconsin Naval Research Laboratory CM-784, 1953.
70. Markstein G. H., Journ. Aeronaut. Sci., 18, 199 A951).
71. L e w i s В., Von Elbe G., Combustion, Flames and Explosions, New York, 1951
(см. перевод 1-го изд.: Льюис Б., Эльбе Г., Горение, пламя и взрывы в газах,
ИЛ, 1948).
72. J о s t W., Explosion and Combustion Processes in Gases, New York, 1946 (см. пере-
вод : И о с т В., Взрывы и горение в газах, ИЛ, 1952).
73. Evans M. W., Theoretical Concepts of Flame Propagation, Chem. Rev., 53,363A952).
74. McClure F. Т., Berl W. G., Ind. and Eng. Chem., 45, 1415 A953).
75. Einbinder H., Journ. Chem. Phys., 21, 480 A953).
76. Mac he H., He bra A., Akad. Wiss. Abteilung Wien (lla), 150, 157 A941).
77. Andersen J. W., Rev. Sci. Instr., 19, 822 A948).
78. F r i e d m a n R., Third Symposium on Combustion, Flames and Explosion Phenomena,
New York, 1948.
79. Andersen J. W., Fein R. S., Journ. Chem. Phys., 17, 1268 A949).
80. M a g e e J. L., The Effect of Radiation in Flames, University of Wisconsin Naval Research
Laboratory CM-627 A950).
81. Hall D. A., Tawada K., Trans. Farad. Soc, 26, 600 A930).
640 Гл. 11. Гидродинамические применения уравнений переноса
82. С u r t i s s С. F., H i r s с h f e 1 d e r J. О., Integration of Stiff Equations, Proc. Natl.
Acad. Sci., 38, 235 A952).
83. G e r s t e i n M., McDonald G. E., S с h a 11 a R. L., Fourth Symposium on Flames,
Combustion and Detonations, Boston, 1952.
84. S z w a r с М., Journ. Chem. Phys., 17, 505 A949).
85. A d a m s G. K., Stocks G. W., Fourth Symposium on Flames, Combustion, and Deto-
nations, Boston, 1952.
86. Murray R. C, Hall A. R., Trans Farad. Soc, 47, 743 A951).
87. Lewis В., Von Elbe G., Journ. Chem. Phys., 2, 283, 537 A934).
88. В о у s S. F., Corner J., Proc. Roy, Soc, A197, 90 A949).
89. С о r n e r J., Proc. Roy. Soc, A198, 388 A949).
90. H e n к e 1 M. J., S p a u 1 d i n g W. P., H i r s с h f e 1 d e r J. O., Third Symposium
on Combustion, New York, 1949.
91. Cole R. H., Underwater Explosions, Princeton, 1948 (см. перевод: Коул Р., Под-
водные взрывы, ИЛ, 1950).
92. Taylor G. I., M а с с о 11 J. W., «The Mechanics of Compressible Fluids» в книге Aero-
dynamic Theory, ed. by W. F. Durand, Pasedena, 1943.
93. Becker R., Zs. Phys., 8, 321 (Ш22).
94. M о r d u с h о w M., L i b b у P. A., Journ. Aeronaut. Sci., 16, 674 A949).
95. Grad H., Comm. Pure and Appl. Math., 5, 257 A932).
96. С о w 1 i n g T. G., Phil. Mag., 33, 61 A942).
97. T h о m a s L. H., Journ. Chem. Phys., 12, 449 A944).
98. Wang Chang C.S., On the Theory of Thickness of Weak Shocks, University of Michigan
Department of Engineering Report UMH-3-F (APL/JHU CM-503), August 19, 1948.
99. M о t t-S m i t h H. M., Phys. Rev., 82, 885 A951).
100. Cowan G. R., Hornig D. F., Journ. Chem. Phys., 18, 1008 A950).
101. Greene E. F., Cowan G. R., Hornig D. F., Journ. Chem. Phys., 19, 427 A951).
102. Greene E.. F., Hornig D. F., Technical Report No. 4, Office of Naval Research
Contract N. 7, Brown University, August 1, 1952.
103. Greene E. F., Hornig D. F., Journ. Chem. Phys., 21, 617 A953).
104. von Neumann J., O. S. R. D., Rep. No. 549 A942); Ball. Res. Lab. File No. X-122.
105. D 0 r i n g W., Ann. d. Phys., 43, 421 A943).
106. Зельдович Ю. Б., ЖЭТФ, 10,542A940).
107. Ey r ing H., Powell R., Duffey G., Parlin R., Chem. Rev., 45, 69 A949).
108. Lewis В., Friauf J., Journ. Am. Chem. Soc, 52, 3905 A930).
109. Berets D. J., Greene E. F., К i s t i a k о w s k у G. В., Journ. Am. Chem. Soc,
72, 1080 A950).
110. Lewis В., Von Elbe G., Phil. Mag. G), 20, 44 A935).
111. Liepmann H. W., P u с k e 11 A. E., Introduction to Aerodynamics of a Compressible
Fluid, New York, 1947.
112. J e 11 i n e k K., Zs. anorg. Chem., 49, 229 A906).
113. Brinkley S. R., Journ. Chem. Phys., 14, 563 A946); 15, 107 A947).
114. Corner J., Theory of the Interior Ballistics of Guns, New York, 1950.
115. Hirschfelder J. O., McClure F. Т., Curtiss С F., Osborne D. W.,
Nat. Def. Res. Com. Report A-116, November 1942.
116. Hirschfelder J. 0., Sherman J., Nat. Def. Res. Com. Report A-101, October
1942, Nat. Def. Res. Com. Armor and Ord ance Memoranda A-67 M—A-70 M, March 1943.
117. Mali na F. J., Journ. Franklin Inst, 230, 433 A930).
118. Perry J. H., Chemical Engineers Handbook, New York, 1950.
Часть III
МЕЖМОЛЕКУЛЯРНЫЕ СИЛЫ
ПУСТАЯ СТРАНИЦА
ГЛАВА 12
ЭЛЕКТРОМАГНИТНЫЕ ОСНОВЫ ТЕОРИИ
МЕЖМОЛЕКУЛЯРНЫХ СИЛ
В настоящей главе изложены основы электромагнитной теории, необходи-
мые для полного изучения межмолекулярных сил. Дальнодействующие меж-
молекулярные силы имеют электромагнитное происхождение. Существуют три
общих типа сил : силы чисто электростатического происхождения (напри-
мер, силы взаимодействия между диполями и высшими мультиполями), индук-
ционные силы и «дисперсионные» силы, связанные с электромагнитным явле-
нием индуцированного поглощения и излучения света.
Можно предполагать существование взаимодействия между магнитными
моментами молекул, но эти силы редко имеют существенное значение. Коротко-
действующие силы, возникающие в результате перекрывания электронных
облаков молекул, должны рассматриваться исключительно на основе кванто-
вой механики.
В § 1 настоящей главы рассмотрено электростатическое взаимодействие
двух общих распределений заряда. Потенциал этого взаимодействия выражается
через мультипольные моменты распределений. Недавно разработанное «двух-
центровое» разложение величины 1/г12 позволило упростить решение задачи
изучения взаимодействия сложных распределений заряда, даже когда распре-
деления перекрываются. Математический подход такого рода весьма ценен
при вычислении кулоновских интегралов в квантовомеханическом рассмотре-
нии короткодействующих межмолекулярных сил. В § 2 рассматривается связь
макроскопических электрических свойств вещества со свойствами отдельных
молекул. Например, дипольный момент и поляризуемость молекулы можно
определить из температурной зависимости диэлектрической восприимчивости.
Дисперсионные силы могут быть приближенно выражены через поляризуемости
молекул. Из зависимости диэлектрической восприимчивости от давления можно
получить подтверждение образования дамеров или ориентирования соседних
молекул в плотном газе или жидкости.
До этого момента все рассуждения основаны на законе Кулона. Для
описания магнитных и дисперсионных сил необходимо ввести электромагнит-
ные уравнения Максвелла. Эти дифференциальные уравнения образуют постуля-
тивную основу электромагнитной теории. Из этих уравнений получают силы,
действующие между двумя группами движущихся зарядов. При движении
зарядов внутри молекулы возникают магнитное поле и силы. Если молекулы
обладают постоянными магнитными моментами, т. е. вещество парамагнитно,
то величины магнитных моментов играют важную роль при определении меж-
молекулярных сил. Например (см. § б, разд. Д), силы, действующие между
двумя атомами, ни один из которых не находится в S-состоянии, сильно зави-
сят от магнитных моментов (или вращательных квантовых чисел).
Последние два параграфа посвящены теории излучения и поглощения
света. Эти явления тесно связаны с зависимостью поляризуемости от
частоты. Поляризуемость, в свою очередь, может быть выражена через эле-
менты матрицы дипольного момента. Эти выводы позволяют связать макро-
скопические электромагнитные свойства с внутренними свойствами отдель-
ных молекул.
644 Гл. 12. Электромагнитные основы теории межмолекулярных сил
§ 1. ЭЛЕКТРОСТАТИКА
Из основного выражения для электростатического взаимодействия (закон
Кулона) можно получить электростатический потенциал <2? распределения
заряда и потенциальную энергию q>ab взаимодействия двух распределений заряда.
Обе эти задачи рассмотрены как для дискретного, так и для непрерывного рас-
пределений заряда. Выражение потенциала V получается в виде «одноцентро-
вого» разложения (Неймана) по сферическим функциям. Аналогичное выраже-
ние для потенциала взаимодействия q>ab получено в виде двухцентрового раз-
ложения по сферическим функциям. В обоих этих выражениях величины1)
Q™ близко связаны с мультипольными моментами, появляющимися в коэффи-
циентах разложения.
В заключение описано приложение этих результатов к частному случаю
дипольного распределения заряда. В последующем обсуждении рассмотрены
как «дискретные», так и «непрерывные» распределения заряда. В первом случае
распределение заряда считается образованным рядом дискретных точек, заряд
в /-й точке равен е(. В последнем случае полный заряд внутри элементар-
ного объема drt в точке г, принимается равным #(г,) йгг. .
А. КУЛОНОВСКОБ ВЗАИМОДЕЙСТВИЕ ЗАРЯДОВ
И РАСПРЕДЕЛЕНИЙ ЗАРЯДА
Потенциальная энергия взаимодействия двух дискретных зарядов ег и ej
или двух элементов непрерывного заряда о(т() dxt и q(tj) йу есть энергия куло-
новского взаимодействия
*и = *%ч 0-1а)
ridtJ. A.16)
С этим потенциалом связана сила, действующая на 1-й заряд со стороны /-го
заряда,
здесь ги — радиус-вектор, направленный из /-го заряда в i'-й заряд, так что
Рассмотрим теперь два сложных распределения заряда (которые могут
являться, например, молекулами), обозначив их через а и Ь. Полную потен-
циальную энергию взаимодействия <р этой системы получим путем суммиро-
вания потенциальных энергий взаимодействия всех возможных пар зарядов
обеих молекул а и ft. Удобно разделить полную энергию на три части
V = ?a + Vb+9t*, 0-3)
где <ра и <рь — собственные энергии взаимодействия распределений заряда а и Ъ
соответственно ; у^—энергия взаимодействия2) зарядов молекул а и Ь. Если
заряды, принадлежащие распределению а, обозначить индексами i и V, а рас-
пределению b — индексами / и /', то отдельные члены выражения полной потен-
г) Эти величины Qff не следует путать с присоединенными функциями Лежандра второго
рода; в этой книге они нигде не используются.
2) Если аи b молекулы, то <раь совпадает с потенциалом межмолекулярного взаимодей-
ствия, за исключением членов, описывающих возмущение одного распределения заряда
вследствие присутствия другого распределения.
§ 7. Электростатика 645
циальной энергии системы двух взаимодействующих распределений заряда
даются в виде
22W
^- *,*,, A.46)
A.56)
22
^*!*,. 0.66)
Полная сила, действующая на заряды а и обусловленная присутствием зарядов
Ь, связана с потенциальной энергией у<й
*°ь = 22*4 = 22^*4' 0-7а)
j$^&A.76)
В разд. Д приведено выражение для потенциальной энергии в виде двухцен-
трового разложения по сферическим функциям.
Б. ЭЛЕКТРОСТАТИЧЕСКИЙ ПОТЕНЦИАЛ И НАПРЯЖЕННОСТЬ
ЭЛЕКТРИЧЕСКОГО ПОЛЯ
Рассмотрим заряд еа, помещенный в точке а вблизи непрерывного распре-
деления заряда. Сила, действующая на этот заряд и обусловленная близостью
распределения заряда, равна
2
Принято определять напряженность электрического поля &, являющуюся силой,
действующей на единичный заряд со стороны распределения заряда, в виде
С этой величиной связан электростатический потенциал У (потенциальная^
энергия единицы заряда), определяемый соответственно в виде1)
ff = _ j^ A Ю)
8г
Тогда электростатический потенциал в точке а, создаваемый совокупностью
зарядов еь выражается в виде
i a
Как &, так и Т, конечно, являются функциями положения точки а.
Если распределение заряда b просто является зарядом еа, то ^ = фаь/еа.
646 Гл. 12. Электромагнитные основы теории межмолекулярных сил
Полную силу, действующую на распределение а со стороны распределе-
ния Ь9 можно теперь записать в виде
A.12)
здесь ff( — напряженность электрического поля, созданного распределением b
в точке расположения заряда i распределения а. Так как сумма сил, действую-
щих со стороны различных зарядов одного распределения друг на друга, равна
нулю, то fft можно считать напряженностью поля, созданного зарядами обоих
распределений.
Формулы для напряженности электрического поля и электростатического
потенциала могут быть также записаны и для непрерывных распределений
заряда. В разд. Г электростатический потенциал ^ выражается в виде
одноцентрового разложения (Неймана) по сферическим функциям.
В. МУЛЬТИПОЛЬНЫЕ МОМЕНТЫ
С каждым распределением заряда связан ряд величин, известных под на-
званием мультипольных моментов, играющих важную роль в электростатике.
Двумя первыми моментами являются полный заряд С и дипольный момент р.
Эти величины определяются следующим образом :
с = 2'i, 0.13)
i
tL=2*th* 0-14)
Вообще все мультипольные моменты, за исключением первого момента, не рав-
ного нулю, зависят от положения начала координат. Обычно начало координат
выбирается либо в центре тяжести, либо в геометрическом центре распределе-
ния зарядов.
Большие затруднения вызывает определение квадрупольного момента.
Прежде обычно определяли квадрупольный момент в форме, аналогичной при-
веденным выше С и ^, т. е.
е=2ем. A.15)
Однако во многих случаях удобнее определять квадрупольный момент в
виде тензора, след которого равен нулю
A.16)
Это определение квадрупольного момента, используемое в настоящей книге.
Для распределения заряда (линейного вдоль оси z или цилиндрически
симметричного1) относительно оси г) недиагональные элементы Q равны нулю,
a Qxx и Qyy равны —V2Qzz- Поэтому для такого распределения заряда тензор
квадрупольного момента полностью определяется скалярной величиной, напри-
мер Qa. Скаляр Qa = Q мы определим как квадрупольный момент.
При рассмотрении молекулярных задач удобно ввести величину q9 также
определяемую нами как скалярная величина квадрупольного момента, т. е. [1 ]
где е — абсолютная величина заряда электрона (q имеет размерность эффектив-
ного сечения). Необходимо заметить, что величиной 1/2Q2Z = && — @хх иногда
пользуются как скалярной величиной квадрупольного момента [2, 3 ]. Некото-
рые величины квадрупольных моментов в литературе относятся к вращаю-
2) Дискретное распределение заряда с осью симметрии, имеющей по меньшей мере трой-
ную симметрию, обладает аналогичными свойствами.
§ 7. Электростатика 647
щимся молекулам. Эти величины являются средними от квадрупольных момен-
тов для фиксированной в пространстве системы координат. Они зависят от
точного вращательного состояния молекулы, но обычно равны приблизитель-
но половине значения для невращающихся молекул.
При разложении <раЬ и W в ряд по сферическим функциям в коэффициентах
разложения появляется ряд величин Q™, связанных с определенными выше
квадрупольными моментами. Эти величины определяются для дискретного или
непрерывного распределения заряда в виде1)
A.18а)
A.186)
здесь Р*? (cos в) — соответствующие полиномы Лежандра (см. приложение Б
в конце главы).
Уравнение A.186) может быть записано в более простом виде, если разло-
жить плотность распределения заряда в бесконечный ряд по сферическим
функциям:
Функции Qntm(r) выражаются через плотность распределения заряда с помощью
соотношения
A.20)
Тогда выражение A.186) принимает вид
A.21)
получаемый подстановкой A.19) в A.186) и использованием условий ортого-
нальности сферических функций [см. выражение (Б.4), стр. 696 J.
Мультипольные моменты, определяемые выражениями A.13), A.14) и
A.16), могут быть выражены через величины QJ?
С = Q8, A.22)
0-23)
0-24)
x) Величины ОЯ1 зависят от положения и ориентации системы координат. Обычно их
выбирают так, чтобы учесть преимущество естественной симметрии, имеющейся в задаче.
Везде далее в книге используется равенство
648
Гл. 12. Электромагнитные основы теории межмолекулярных сил
Г. ОДНОЦЕНТРОВОБ РАЗЛОЖЕНИЕ ЭЛЕКТРОСТАТИЧЕСКОГО
ПОТЕНЦИАЛА РАСПРЕДЕЛЕНИЯ ЗАРЯДА
Выражение электростатического потенциала для дискретного распределе-
ния заряда имеет вид A.11). В этом и аналогичном выражениях для непрерыв-
ного распределения заряда появляется величина 1/г?а, где ria — расстояние
между f-м зарядом, расположенным в точке rh вь фь и некоторой точкой поля
а с координатами г, 0, ф (фиг. 188). Если символами г< и г> обозначить соот-
Ф и г. 188. Система координат, использованная при рассмотрении вклада заряда,
находящегося в точке /, в электростатический потенциал У? в точке а.
ветственно меньшее и большее значения г{ и г, то величину 1/г/а можно пред-
ставить в виде одноцентрового разложения, или разложения Неймана [4, 5]1)
7Г=2 2 %; 111 >! ¦?
п-От—п к ' '' >
e> g<m@<~0) ¦
Подстановка выражения для 1/г,а в формулы электростатического потен-
циала дискретного или непрерывного распределения заряда дает
» +П /я_1т|\| _ .^Г1 ^Г» ъ , л ч / ,*
Tf* I/ '_ Dm /рЛС Й\ Р—1ТП0 I > а гП P"i (рлс Я Л pimfpi к
—гт-г Гп (COS 0> е ^ ,, ^ tt ft Гп (tOb V() t -f
2 2
n-Om—n
2 2 fe
n=Om--n v
2 ^-фгрп
здесь Qnymif) — коэффициенты разложения, определяемые выражением A.20).
В точке поля а, расположенной вне распределения заряда, для всех зарядов
r>rh и выражение A.26) упрощается :
=± 2 % +
n=0m—n \ •
A.27)
Величина 1/па может быть разложена с помощью эллипсоидальных координат [б].
§ 1. Электростатика
649
Величины Q™ определяются выражениями A.18) и A.21) для дискретного и
непрерывного распределений заряда соответственно. Этим выражением электро-
статического потенциала в виде ряда по степеням 1/г особенно удобно пользо-
ваться в случае больших расстояний от распределения заряда.
Д. ДВУХЦЕНТРОВОЕ РАЗЛОЖЕНИЕ ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ
ВЗАИМОДЕЙСТВИЯ ДВУХ РАСПРЕДЕЛЕНИЙ ЗАРЯДА
Выражения A.6) представляют потенциальную энергию взаимодействия
двух распределений заряда для дискретного и непрерывного распределений.
В эти выражения входит величина Гц — расстояние между i'-м зарядом первого
Фиг. 189. Системы координат, использовавшиеся при обсуждении двухцентрового
разложения.
Точка а — начало системы координат, расположенной в распределении заряда а; точка i — положение
заряда этого распределения; точка Ь — начало системы координат, расположенной в распределении за-
ряда Ь ; точка j — положение заряда этого распределения.
распределения и /-м зарядом второго. На фиг. 189 изображено это расстояние
и другие координаты, используемые в дальнейшем обсуждении. Обратная вели-
чина 1/Г/у может быть представлена в виде двухцентрового разложения^1)
здесь мы пользуемся символом п< для обозначения наименьших значений па
и пь. Коэффициенты В^ь представляют собой сложные функции rh rj и rQb.
Формулы и численные значения этих коэффициентов приводятся в конце главы
в приложении А. Углы 0, и 6j отсчитываются относительно центров а и & соот-
ветственно, как это показано на фиг. 189.
Подстановка этого двухцентрового разложения в выражение A.6) дает
для потенциальной энергии взаимодействия двух распределений заряда
9*= 2 2
O O
<раь= 2 2 2 ГГ
п.-0п»-0т--п< J J
x e( ej P%a (cos 0,) P^ (cos 0y) eimWi~
(ri f П J rab) Qna,m (г() Qnbtm (Г/) Г? (Щ Г) dfj
A.29а)
A.296)
Звездочка над Qna>m указывает на комплексно сопряженную величину. Эти
выражения справедливы для любых двух перекрывающихся и неперекрываю-
щихся распределений заряда. Функции &п$т(г) определяются выражением A.20).
х) Выражения для некоторых простейших коэффициентов В были даны несколько раньше
в работе [8].
650
Гл. 12. Электромагнитные основы теории межмолекулярных сил
Если два распределения заряда не перекрываются, то гаЬ > (г,- + о).
В этом случае (обозначен как область I в приложении А, стр. 690) коэффи-
циенты В1&1 имеют вид
/ 1\ и. i •*,/,» I м\| «Па Jit
A.30)
Потенциальная энергия взаимодействия двух распределений заряда выражается
рядом по степеням ljrab
<РаЬ= 2, 2 2
Пав0 nj«=O /П = —Л<
Члены этого разложения описывают взаимодействия различных мультиполей.
Последнее выражение для додано не в наиболее удобной форме. Величины
Qna определяются выражением A.18) как для дискретного, так и для непрерыв-
ного распределений заряда и применяются в изображенном на фиг. 189 случае
Фиг. 190. Системы координат, использовавшиеся при нахождении (?5? и QЯ1.
распределения заряда вокруг точки а. Аналогичным образом величины Q™
определяются и для распределения вокруг точки Ь. В системе координат,
используемой при вычислении величин Q™ и (#?, ось направлена вдоль линии,
соединяющей эти точки (центры распределения).
Обычно удобнее вычислять величины Q? в системе координат, фиксирован-
ной некоторым образом относительно самих распределений заряда. Системы
координат, фиксированные относительно распределений заряда, представлены
на фиг. 190. Величины, вычисляемые в случае этих вращающихся систем коор-
динат, обозначаются черточками над соответствующими символами. Когда
потенциал сраЬ выражается через величины Q^ вместо Q#, то, очевидно, зависи-
мость энергии взаимодействия от относительной ориентации распределений
заряда становится ясной.
Для выражения сраЬ через Q™ необходимо знать соотношение между этими
величинами nQ% Это соотношение может быть получено методами теории групп.
Из определения (ЭД следует, что относительно вращения она преобразовывается
аналогично сферической функции я-го порядка. Сферические функции состав-
ляют базисное представление трехмерной группы вращения (см. приложение Б,
стр. 695). Пусть Sa — вращение, преобразующее систему координат ха> уа> za
в систему хв> уш za> a Sb — аналогичное вращение для распределения заряда
относительно точки Ь. Тогда можно показать, что справедливо выражение вида
(Па-\та\)\(Па+\т\)\
§ 1. Электростатика 651
и аналогичное выражение для распределения заряда Ь. Коэффициенты Dne(Sa)mem
являются коэффициентами представления трехмерной группы вращения. В при-
ложении Б (стр. 695) даны формулы для этих величин и обсуждаются некоторые
их свойства.
Выражение потенциала сраЬ взаимодействия двух распределений заряда
через QJ?, вычисляемых для систем координат, помещенных внутрь распреде-
лений, находим из A.31) и A.32)
— | /л |) I (ла + ! /па |)! (пь +1 m |)! (пь— |m
X
Dn*(Sa)*mamDnb(Sb)mm 1 A.33)
Это выражение отчетливо указывает на зависимость энергии взаимодействия
двух распределений заряда от расстояния гаЬ и относительных ориентации.
Ориентации распределений заряда выражаются вращениями Sa и Sb.
Из выражения A.33) и свойств коэффициентов представления следует, что
если.^аь усреднено по всем ориентациям двух распределений заряда а и ft
(по отдельным вращениям), то все члены с па и пЬ} не равными нулю, выпадают.
Тогда эта средняя величина q>ab равна Qoo(a) Qw>(b)lrab- Это—потенциал взаимодей-
ствия двух точечных зарядов, величины которых равны общим зарядам обоих
распределений.
Е. СВОЙСТВА ЭЛЕКТРИЧЕСКОГО ДИПОЛЯ
Вернемся опять к простейшему примеру дискретного распределения
заряда — диполю. Диполь состоит из двух равных зарядов противоположных
знаков —е и +е, находящихся на расстоянии /. В соответствии с A.14) диполь-
ный момент такой системы равен
H = e\. A.34)
Векторы fe и 1 направлены от отрицательного к положительному заряду. Для
реального диполя расстояние / конечно. Однако на расстояниях, больших по
сравнению с /, напряженность электрического поля зависит в основном от
величины дипольного момента, а не от величин е и / по отдельности. Учитывая
это, определим идеальный диполь (или точечный диполь) как такой, для кото-
рого е -> оо при / -> 0, так что их произведение остается постоянным.
Из определений идеального диполя и вектора напряженности электричес-
кого поля легко получить формулы, описывающие их взаимодействие :
Потенциальная энергия диполя в электрическом поле
?=-(fi.tf). (Ь35)
Сила, действующая на диполь в электрическом поле,
A.36)
Вращающий момент, действующий на диполь в электрическом поле,
L=[iix&]. A.37)
Заметим, что вращающий момент стремится повернуть диполь так, чтобы он
был ориентирован по направлению поля.
Покажем теперь, как результаты, полученные в первой части этого раз-
дела, могут быть применены к реальным и идеальным диполям. Сначала рас-
смотрим одноцентровое разложение электростатического потенциала отдельного
диполя, обсуждавшееся в разд. Г. Затем рассмотрим диполь-дипольное взаимо-
действие и выразим потенциальную энергию через двухцентровое разложение,
описанное в разд. Д.
652 Гл. 12. Электромагнитные основы теории межмолекулярных сил
1. Электростатический потенциал диполя. Электростатический потенциал
реального диполя, конечно, может быть выражен в виде суммы членов, соот-
ветствующих каждому из зарядов. Однако часто бывает удобно пользоваться
Фиг. 191. Система координат и символы, использовавшиеся при нахождении
электростатического потенциала диполя.
разложением A.27). Для случая системы координат, изображенной на фиг. 191,
величины Q?J, описывающие реальный диполь, имеют вид
, A-38)
здесь Q? — дипольный момент /г. Для реального диполя не равны нулю только
величины мультиполей с нечетными п. Из этого выражения для Q% и выра-
жения электростатического потенциала распределения заряда A.27) мы полу-
чаем выражение электростатического потенциала (в точке а фиг. 191) реального
диполя (при г > V2O
A.39)
Отсюда очевидно, что для г t> l поле реального диполя совпадает с полем
идеального, и единственным членом потенциала является «дипольный член»
общего выражения A.27).
Дифференцирование выражения A.39) дает напряженность электрического
поля диполя
A.40)
ЭГ
где
A.41)
а ег и е# — единичные векторы, направленные в сторону возрастания гиб.
2. Потенциальная энергия взаимодействия двух диполей. Системы коор-
динат, которые могут использоваться при описании взаимодействия двух дипо-
лей, представлены на фиг. 192. Общее выражение для потенциальной энергии
взаимодействия двух дискретных распределений заряда A.29) упрощается для
диполь-дипольного взаимодействия
оо <» т=Bл< + 1) /л i 1 л i I \
42 2 2 B^^^lLVhir^y
2 2 2
#=9 л»=0 т=-Bл< + 1)
х Р1ЯА, (cos 0а) Р{^„ (cos в
§ 7. Электростатика
653
Это выражение справедливо для любых взаимных расстояний и ориентации
двух диполей.
Когда расстояние между центрами диполей гаЬ превышает величину
1/2Aа + W>T0 распределения «не перекрываются» и следует пользоваться выра-
жением A.33). Величины Q™, используемые в этом выражении, вычисляются
для осей za и zb, изображенных на фиг. 192. Эти величины почти совпадают с
{?? выражения A.38) и не равны нулю только при m = 0. Коэффициенты
Dn(S)*m B A.33) просто выражаются через сферические функции
A.43)
Углы 0 и ф описывают ориентацию фиксированной оси z в системе (xyz) коорди-
нат, ось z которой проходит вдоль линии центров. Пользуясь этими соотноше-
Ф и г. 192. Система координат и символы, использовавшиеся при обсуждении
диполь-дипольного взаимодействия.
Диполи о и Ь имеют соответственно длины 1а и /».
ниями1), находим потенциальную энергию взаимодействия двух реальных
диполей
- (-1L (Яд + Щ)\ A
/2 lb)nb
Па
X
Рпа (cos 0в) Р% (cos вь)
A.44)
ab
Поскольку реальные диполи обладают только мультипольными моментами с
нечетными номерами, то выражение энергии взаимодействия можно представить
рядом, содержащим члены \\rlby l/rgb, ljrlb, ...
Определяющим членОхМ этого ряда для сраЬ является
- 2cos0acos0b + sinваsinвьcos(фь - фа)].
A.45)
Это выражение представляет энергию взаимодействия двух идеальных диполей.
х) Имеется альтернативная возможность использования формулы A.31) заменяя Q21
на (?™. В системе xyz фиг. 192 Q% выражается в виде
Q« = [1 - (_ l)«](l/2 l)n epm (cos в) е1т0л
654
Гл. 12. Электромагнитные основы теории межмолекулярных сил
Именно эта функция входит в потенциал взаимодействия полярных молекул
Штокмайера [см. C.33), гл. 1]. Если таЬ и / сравнимы по величине, то реальные
и идеальные диполи существенно отличаются. Сравнение потенциалов взаимо-
действия (раЬ реальных и идеальных диполей проводится на фиг. 193 для четырех
Фиг. 193. Влияние конечных размеров на величины энергии взаимодействия двух
реальных диполей.
различных относительных ориентации. При (rabjl) < 2 отклонение свойств реаль-
ного диполя от идеального существенно.
Иногда удобно выражать энергию взаимодействия двух идеальных диполей
в векторной форме
<Раъ = -^ [(!"«• ъ) - 3
rab
1.46)
Если (вга}
%, Ф'ъ) и @abf ф'аЬ) — направляющие углы трех векторов /иа
§ 2. Поляризация вещества и диэлектрическая восприимчивость 655
и f^ в обычной системе координат, то соотношение A.46) принимает вид
Ч>аЪ = ГЗГ*- lC0S д'а C0S дЬ + Sin O'a Sill O'b COS @ft' - ^ - 3 (COS O'a COS Q'ab +
A.47)
sin 6'a sin 0дЬ cos (ф'аЬ — fQ) (cos 0» cos d'ab + sin Si sin в'аЬ cos (&ь —
Эти выражения используются при рассмотрении межмолекулярных сил по-
лярных молекул в гл, 13, § 5 и 6 и обусловленного столкновениями полярных
молекул уширения радиоволн вследствие давления в гл. 13, § 7.
§ 2. ПОЛЯРИЗАЦИЯ ВЕЩЕСТВА И ДИЭЛЕКТРИЧЕСКАЯ
ВОСПРИИМЧИВОСТЬ1)
Вещество состоит из двух видов заряда — свободного и связанного. Свобод-
ные заряды могут проходить значительные расстояния, а связанные заряды,
как, например, электроны в атомах, могут сдвигаться от своего среднего поло-
жения только на расстояния, малые по сравнению с размерами атома, т. е.
на малые доли ангстрема. Электрон в атоме сохраняет стационарную орбиту
за счет уравновешивания силы притяжения к ядру и центробежной силы,
связанной с этим движением. Внутренние электронные силы в атомах и моле-
кулах очень велики по сравнению с силами, связанными с внешними электри-
ческими полями. В проводниках часть электронов может свободно перемещаться
из одного места в другое. Эти свободные электроны распределяются так, чтобы,
обеспечивалось постоянство электрического потенциала внутри проводника.
Любое другое распределение свободных электронов требует более высокой
энергии. В изоляторе электроны плотно связаны с отдельными атомами, моле-
кулами или ионами. Когда такой изолятор помещается в электрическое поле,
электроны будут несколько сдвинуты в направлении, противоположном направ-
лению сдвига ядер. Это относительное смещение зарядов в веществе мы назовем
поляризацией.
Когда две молекулы приближаются друг к другу, имеет место взаимная
поляризация. Эта поляризация молекул может внести существенный вклад
в потенциал межмолекулярного взаимодействия. Степень поляризации можно
найти, изучая диэлектрическую восприимчивость вещества.
При изучении электростатических явлений в диэлектриках существенны три
вектора: ff, 3) и йГ<Л0К->. Напряженность электрического поля 8 является
мерой силы, действующей на макроскопический пробный заряд в диэлектрике.
Вектор электрической индукции Л) описывает поле, источником которого явля-
ются макроскопические заряды. Разница между 8 и 3) связана с поляри-
зацией вещества. Вектор локальной напряженности поля ^<лок-) является ме-
рой силы, действующей на молекулы в диэлектрике, и характеризует поле,
вызывающее поляризацию молекул и ориентацию постоянных диполей.
А. ПОЛЯРИЗУЕМОСТЬ И ПОЛЯРИЗАЦИЯ
Рассмотрим совокупность связанных зарядов во внешнем поле. Как ска-
зано выше, внешнее поле вызывает смещение зарядов. Разности между мульти-
польными моментами возмущенного и невозмущенного распределений заряда
известны как индуцированные мультипольные моменты. Для слабых внешних
полей индуцированный дипольный момент может быть записан в виде
а^; B.1)
. [9].
656 Гл. 12. Электромагнитные основы теории межмолекулярных сил
здесь а — поляризуемость объекта. Энергия индуцированного диполя в электри-
ческом поле & равна
<р= _J (р<инд.>.Л j) =-^ag%. B.2а)
В общем случае поляризуемость а является тензором, т. е, индуцированные
дипольные моменты необязательно располагаются по направлению приложен-
ного электрического хполя. Тогда /и<инд-> = (а<&) и энергия индуцированного
диполя в электрическом поле <f выражается в виде
(р =—2~ (#* •«• #0 . B.26)
Смысл понятия поляризуемости может быть иллюстрирован на простом
примере. Рассмотрим проводящую сферу радиусом /?, помещенную в однородное
электрическое поле напряженностью &9 направленное по оси z. Электростати-
ческий потенциал вне сферы равен [10]
B.3)
Но этот же потенциал соответствует также точечному диполю с моментом
R3 ffy помещенному в центр сферы, и его потенциал накладывается на внешнее
поле. Поэтому поляризуемость проводящей сферы равна
а = #3 . B.4)
Для проводящего эллипсоида поляризуемость является тензором, главные оси
которого совпадают с главными осями эллипсоида.
Поляризация вещества, обусловленная влиянием внешнего электрического
поля, связана с двумя явлениями : 1) поляризуемостью отдельных молекул и
2) частичным ориентированием постоянных диполей молекул. Средний диполь-
ный момент, рассчитанный на единицу объема, обусловленный этими двумя
явлениями, называется поляризацией и обозначается символом &.
Б. ВЕКТОРЫ & И 8
Электрическое поле в вакууме полностью обусловлено присутствием мак-
роскопических заряженных тел. Если диэлектрик (т. е. изолятор) поместить
в это поле, то поле внутри диэлектрика будет создаваться электрическим полем
макроскопических заряженных тел и полем, связанным с поляризацией отдель-
ных молекул и постоянными моментами молекул диэлектрика. Однако такое
раздельное рассмотрение полей редко представляет интерес. Вместо этого
подойдем к задаче со статистической точки зрения.
Этот подход связан с введением двух векторов J3) и ff. Макроскопические
заряженные тела представляют собой источники вектора электрической индук-
ции .2). Таким образом, интеграл от вектора & по замкнутой поверхности равен
охваченному поверхностью заряду, умноженному на 4 л,
B.5)
Вектор напряженности электрического поля ff является мерой силы, действую-
щей в диэлектрике на макроскопическое заояженное тело. Поскольку это поле
консервативно, то полная работа, произведенная при переносе пробного заряда
по замкнутому пути, равна нулю. Таким образом, интеграл по замкнутому
контуру имеет вид
<$(#\ds) = O. B.6)
Два последних соотношения служат определением векторов j?) и ff и исполь-
зуются для получения дифференциальных соотношений для векторов и их
§ 2. Поляризация вещества и диэлектрическая восприимчивость 657
поведения на границе. Применяя теорему Грина к выражению B.5) и теорему
Стокса к выражению B.6), можно получить следующие дифференциальные
соотношения для векторов ?fr и g :
) B.7
Эти уравнения применимы внутри диэлектрика.
На границе двух диэлектриков 1 и 2 (или диэлектрика и вакуума) может
быть поверхностная плотность реальных зарядов #(s). С помощью приведенных
выше интегральных соотношений можно показать, что на границе двух диэлек-
триков векторы электрической индукции Л) и напряженности электрического
поля g удовлетворяют соотношениям
(п • {&<* - ?)&}) = 4я^>, B.9)
0, B.10)
где п — единичный вектор, нормальный к поверхности и направленный из среды
1 в среду 2.
Для нахождения связи между векторами JD и g рассмотрим следующий
эксперимент. Сначала приложим напряжение к двум параллельным пластинам
так, чтобы в вакууме электрическое поле было направлено по оси z. Напряжен-
ностью поля может быть как вектор j?), так и вектор &у поскольку в вакууме
он и совпадают. Теперь заменим вакуум изотропным однородным диэлектриком.
В то время как вектор jZ) останется неизменным, напряженность электрического
поля к изменится вследствие поляризации среды.
Выделим внутри диэлектрика малый элемент объема, размеры которого
велики по сравнению с расстоянием между молекулами. Теперь можно спро-
сить, каков должен быть заряд на поверхности пустой полости, чтобы силы,
действующие на пробный заряд в полости, были равны силам, действующим
на пробный заряд в однородном диэлектрике. Дипольный момент этих поверх-
ностных зарядов равен полному дипольному моменту выделенного элемента
вещества.
Для удобства выберем в качестве такой полости цилиндр, торцы которого
параллельны пластинам конденсатора. Примем радиус цилиндра г значительно
превышающим высоту, так что краевыми эффектами можно пренебречь. Рас-
смотрим две торцевые поверхности, перпендикулярные оси z. Из выражения
B.9) следует
{2)(?2)ф} 4^> B.11)
где e(s) — плотность заряда на поверхности (для обеих поверхностей плотности
равны и имеют противоположные знаки зарядов), а индексы о и i обозначают
положение вне и внутри цилиндра соответственно. Так как внутри полости
вещества нет, то
J0<0 = <?@, B.12)
и поскольку сила, действующая на пробный заряд внутри полости, равна силе
перед удалением этого малого цилиндра, то j?)@ и #@ равны напряженности
электрического поля & перед удалением полости. Ввиду определенного выбора
формы полости вектор электрической индукции не зависит от присутствия
полости и
4яес> = ^I-гГ2, B.13)
где 3)z — значение вектора индукции перед удалением полости. Поэтому можно
сделать заключение, что удаленное из полости вещество обладает дипольным
658 Гл. 12. Электромагнитные основы теории межмолекулярных сил
моментом, z-компонента которого равна
V ^-#2). B.14)
Поляризация &* определяется как полный дипольный момент единицы объема
вещества. В соответствии с этим г-компонента вектора поляризации & равна
^ B.15)
Тогда, поскольку другие компоненты векторов J#, Л> и 9 равны нулю,
B.16)
Можно показать, что этот результат приложим как к анизотропной, так и к
изотропной средам.
В. ВЕКТОР НАПРЯЖЕННОСТИ ЛОКАЛЬНОГО ПОЛЯ ?<Л01<)
Выше напряженность электрического поля g была выражена через электри-
ческую индукцию Л) и поляризацию ч&. Необходимо помнить, что вектор &
связан с полем сил, действующих на макроскопический пробный заряд. Рас-
смотрим теперь вектор #<лок->, дающий силу, действующую на отдельную моле-
кулу. Это поле является суммой поля Л) внешних зарядов и поля, обусловлен-
ного поляризацией всех других молекул (за исключением данной молекулы).
В то время как вывод выражения для g был проведен с помощью макроскопи-
ческих аргументов, вывод выражения для &(ЛОК-) требует микроскопической
точки зрения.
Рассмотрим конденсатор с круговыми пластинами радиуса R, верхняя
и нижняя пластины которого параллельны плоскости ху и пересекают ось z
в точках +L и —L. Примем R настолько бблыиим L, что поле вблизи центра
цилиндрической области между пластинами конденсатора существенно парал-
лельно оси г. Если на верхней и нижней пластинах поверхностная плотность
заряда, приходящегося на единицу площади, равна #(s), то вектор j?) обладает
только z-компонентой, которая равна 4 nQ(s).
Пусть теперь между пластинами конденсатора помещается изотропный
диэлектрик, на единицу объема которого приходится п молекул, каждая из
которых обладает идеальным дипольным моментом fe. Полный дипольный
момент |и<полн-) молекулы является суммой постоянного дипольного момента /и
и индуцированного дипольного момента /и<инд->. Средняя по времени величина
полного дипольного момента j?(noJIH-) является вектором, направленным вдоль
оси г, т^е. по направлению электрического поля. Тогда вектор поляризации*^
равен п |?<полн.)#
Выясним теперь, какова напряженность поля ^(лок), действующего на
молекулу, помещенную в центр цилиндрической области. Сначала заметим,
что электрический потенциал в точке расположения этой «центральной моле-
кулы», созданный диполем с моментом fi(n0JlH-\ находящимся в точке г, равен
, B.17)
а г— компонента напряженности электрического поля, соответствующей этому
потенциалу, равна
?(*==^) <2Л8>
Плотность числа молекул или диполей в окрестности точки г равна ng(r\
гАе ё(г) — радиальная функция распределения, подробно обсуждавшаяся в
гл. 4, § 9. Поэтому полная напряженность поля всех диполей, действующего
на центральную молекулу, имеет только г-компоненту, величина которой дается
§ 2. Поляризация вещества и диэлектрическая восприимчивость 659
умноженным на весовой множитель интегралом радиальной функции распре-
деления по цилиндрической области. Для получения выражения ^(лок-) к нему
должен быть прибавлен вектор Л):
B.19)
цил.обл.
где %\ дается выражением B.18). Выражение B.19) может быть записано через
производную g'(r) = dg\dr от радиальной функции распределения
J
г
цил. обл. О
~-2>, + nJV(s)[ |>J*]&. B.20)
О V(s)
Для изменения порядка интегрирования и получения другого вида этого выра-
жения необходимо предположить, что g'(r) отлична от нуля только для зна-
чений 5, малых по сравнению с линейными размерами конденсатора. Объем
V(s) — это объем, ограниченный снаружи поверхностью цилиндра, а изнутри
поверхностью сферы радиуса 5.
Можно показать [11 ], что интеграл по г не зависит от 5 и имеет величину
f = 4л р<п°лн.) Г- -| + у L 1 ^ - -^/*»"».> для R > L . B.21)
Условие того, что /? значительно больше L, физически соответствует требованию
однородности поля в направлении оси г. Подставляя полученный результат
в выражение B.20) и интегрируя по s (необходимо воспользоваться тем, что
g(oo) = 1, a g@) = 0), окончательно получаем
#(лок.) = & _ Щ. п^(попн.) =&-*?-&. B.22)
Этот результат может быть также записан через вектор & в виде
Это выражение не зависит от вида радиальной функции распределения, но
отражает тот факт, что радиальная функция распределения g(r) не зависит от
угла. Таким образом, результат несколько отличен в случае среды, имеющей
локальную кристаллическую структуру. Именно локальное поле #<лок-> воз-
действует на отдельную молекулу и стремится поляризовать ее и расположить
в направлении поля.
Г. ВЫРАЖЕНИЕ ДИЭЛЕКТРИЧЕСКОЙ ВОСПРИИМЧИВОСТИ
ЧЕРЕЗ ДИЭЛЕКТРИЧЕСКУЮ ПОСТОЯННУЮ
Поляризация &> средний дипольный момент р(полн.) и напряженность локаль-
ного электрического поля #(лок-> могут быть связаны друг с другом выражением,
подобным B.1),
^5 = п |1<полн> = л х(е) гГ<Л0К->. B.24)
Величина %(е) называется диэлектрической восприимчивостью,' рассчитанной
на одну молекулу [восприимчивость, рассчитанная на моль, равна N х(е) ]•
Она аналогична поляризации в выражении B.1). В самом деле, если молекулы
не обладают постоянным дипольным моментом, то х(е) совпадает с а. С другой
стороны, если молекулы обладают постоянным дипольным моментом, то в выра-
жение для %(е) входит другой член, зависящий от температуры. Этот член обус-
ловлен ориентированием диполей в электрическом поле.
660 Гл. 12, Электромагнитные основы теории межмолекулярных сил
Диэлектрическую восприимчивость можно связать с диэлектрической
постоянной е', определяемой в виде
?) = е'&. B.25)
Диэлектрическая постоянная легко находится экспериментально. В неизотроп-
ной среде е' является тензором, так как направления векторов «2) и 8 не сов-
падают. С помощью величины е' выражения B.16) и B.23) могут быть пред-
ставлены в виде
i B.26)
0Члок.) = _*_ (е' + 2) g . B.27)
Из этих соотношений и выражения, определяющего восприимчивость, рассчи-
танную на одну молекулу, следует
или, вводя плотность вещества q, молекулярный вес М и число Авогадро N,
<2-29>
Это выражение известно как формула Клаузиуса—Мосотти1). Можно показать,
что, за исключением случаев высоких плотностей и напряженности электри-
ческого поля, восприимчивость смеси аддитивна, т. е.
х<* = 2*1*9, B.30)
где х( — молярная доля компоненты i, а х\е) — восприимчивость i-й чистой
компоненты.
Формулой Клаузиуса—Мосотти можно пользоваться для измерения вос-
приимчивости х(е) и для предсказания зависимости диэлектрической постоянной
от плотности. В следующем разделе показано, что %(б) является функцией тем-
пературы и параметрически зависит от двух молекулярных величин — поляри-
зуемости и постоянного дипольного момента. Изменение %(е) с плотностью может
быть использовано для обнаружения изменений молекулярной структуры, как,
например, образования димеров.
Д. ВЫРАЖЕНИЕ ДИЭЛЕКТРИЧЕСКОЙ ВОСПРИИМЧИВОСТИ
ЧЕРЕЗ МОЛЕКУЛЯРНЫЕ ВЕЛИЧИНЫ
В выражении B.24) диэлектрическая восприимчивость определялась через
величины ]й(полн) и ^(лок-). Рассмотрим теперь точное вычисление величины
^(полн.)> являющейся суммой среднего постоянного дипольного момента и инду-
цированного момента молекулы диэлектрика в электрическом поле. Эта вели-
чина может быть выражена через поляризуемость и постоянный дипольный
!) Поправки к формуле Клаузиуса—Мосотти при высоких плотностях или напряжен-
ностях электрического поля обсуждались в работах [9, 12—15]. Бус показал, что при высо-
ких напряженностях электрического поля
«' = V2 + [Y*n(if + 2)/л/ё ] Цр(г? + 2) ftg/kT),
где г\ — коэффициент преломления света; /л — дипольный момент молекулы; L(x) — функ-
ция Ланжевена, рассматриваемая ниже в § 2; у и р — числовые постоянные, значения
которых должны быть найдены двумя различными методами. Онзагер нашел, что у = 4/в и
ft = 7з- Кирквуд получил у = 28/3 У73 и /3 = ^73/6. Как будет показано в гл. 13, при взаи-
модействии двух молекул воды роль квадрупольного взаимодействия очень велика. Таки
образом, для молекул воды необходимо несколько видоизменить выражение, полученное
Бусом.
§ 2. Поляризация вещества и оиэлектрическая восприимчивость 661
момент молекул, а тем самым диэлектрическая восприимчивость может быть
выражена через молекулярные величины.
В качестве примера диэлектрика рассмотрим идеальный многоатомный
газ. В гл. 2, § 3 показано, что в такой системе энергия распределена среди мо-
лекул в соответствии с распределением Больцмана. Среднее значение любой
молекулярной величины может быть соответственно выражено в виде сред-
него по распределению Больцмана. Здесь нас интересует молярная величи-
на, которой является дипольный момент, зависящий, конечно, от квантового
состояния молекул. Если мы обозначим через ^полн> квантовомеханическое
среднее полного (постоянный плюс индуцированный) дипольного момента
молекулы в /-м квантовом состоянии, то получим
здесь индекс / заменяет всю совокупность квантовых чисел, описывающих
состояние молекулы, a ej —энергия соответствующего состояния.
Это выражение для jU(noJ1H-> можно упростить, если сделать некоторые
предположения, которые обычно справедливы: 1) все молекулы находятся
в основных электронных состояниях ; 2) вращательная энергия не зависит от
колебательного квантового числа; 3) дипольный момент ^олн) не зависит от
колебательного квантового числа ; 4) разность энергий вращательных уровней
мала по сравнению с величиной fcT, так iro вращение можно рассматривать
классически. Если сделать эти предположения, то можно показать [16], что
B.31) сводится к классическому среднему
Г ,|<полн.) (R) е-<КЮ1кТ dR
J* *. /2.32)
здесь через R обозначены эйлеровы углы1), определяющие ориентацию молекулы,
<p(R) — потенциальная энергия молекулы для определенной ориентации, а
dR = sin в йв йф dy>- Таким образом, средний дипольный момент равен просто
усредненной по всем ориентациям величине fe<n0J1H>, если весовым множителем
является множитель Больцмана ехр (—cpfkT).
Для нахождения |й<полн> необходимо в интегралы предыдущего выражения
подставить |и<полн> и <р, выраженные через углы. Пусть вектор напряженности
{f электрического поля направлен вдоль оси z фиксированной системы коорди-
нат. В изотропном веществе вектор ^(лок> имеет то же направление. Тогда пол-
ный дипольный момент и потенциал молекулы в поле ^<лок) даются выражениями
Н-> = /, sinfl cos ф,
/4П0ЛН-> = /г sinfl sin ф, B.33)
q> = — fz <f <лок> cos в — у а <f (Л01<>2. B.34)
Интегралы в выражении B.32) для х- и у-компонент ^полн-> равны нулю. Сле-
довательно, направление рЕ<полн-> совпадает с осью z и имеет величину
к
_ ag(лок.) _j_ О_
> cos в] е-мё(Л0К) cos щт sin Odd
е-м^ЛОКа)соь eikT sin BdB
B.35)
kT
г) Определение эйлеровых углов дано в приложении Б настоящей главы.
662
Гл. 12. Электромагнитные основы теории межмолекулярных сил
где Цх) — функция Ланжевена1)
+х
= cth x —
45
B.36)
Рассмотрим теперь два предельных случая этого выражения. В случае
сильных электрических полей или низких температур [когда х = ^Г<>Т
велико и L(x) стремится к единице ] ^(по™.) стремится к величине
» == a g<лок-> + /г . B.37)
Эта величина соответствует «насыщению», т. е. идеальной ориентации всех
молекул. В случае слабых электрических полей или высоких температур р(полн>
выражается в виде
л „ ^IJtnie I v 4 , ta I ЛПК. 143
B.38)
Здесь использовано разложение функции Ланжевена в ряд Маклорена. Срав-
нение этого последнего выражения с определением диэлектрической восприим-
чивости показывает, что при дос-
таточно малых напряженностях поля
или высоких температурах
B.39)
0,002 0,003
0,004 0,005
Фиг. 194. Экспериментальные кривые,
изображающие температурную зависимость
диэлектрической восприимчивости.
Тангенс угла наклона равен постоянному диполь-
ному моменту молекулы, а ордината точки пересе-
чения с осью *<•>— величине поляризуемости [33].
Это соотношение вместе с формулой
Клаузиуса—Мосотти позволяет нахо-
дить постоянный дипольный момент и
поляризуемость молекул по экспери-
ментальным измерениям зависимости
диэлектрической постоянной от тем-
пературы. График зависимости ди-
электрической восприимчивости от
величины 1/7 для некоторых веществ
представлен на фиг. 194. Прямые линии
получены теоретически с помощью
выражения B.39). Тангенс угла на-
клона этих прямых является мерой
величины дипольного момента, а от-
резок, отсекаемый на вертикальной
оси, — мерой поляризуемости. Знание этих молекулярных величин имеет
большое значение при изучении взаимодействия полярных молекул с поляр-
ными и неполярными молекулами. Этот вопрос широко обсуждается в следую-
щей главе.
§ 3. ЭЛЕКТРОМАГНИТНЫЕ УРАВНЕНИЯ МАКСВЕЛЛА
В предыдущих параграфах обсуждались свойства статических электри-
ческих зарядов. При рассмотрении электродинамики движущихся зарядов
необходимо ввести несколько новых величин для описания протекания электри-
ческого тока и сил, связанных с движущимися зарядами.
г) При низких температурах функция Ланжевена для учета дискретной природы вра-
щательных энергетических состояний должна быть заменена функцией Бриллюэна [171.
jf 3. Электромагнитные уравнения Максвелла 663
Сила электрического тока i определяется как произведение величины
линейной плотности зарядов а на их скорость v
i = ov. C.1)
Другой используемой величиной является плотность тока, которая определя-
ется в виде
J = 0V. C.2)
Эта величина характеризует поток заряда, т. е. величину заряда, протекающего
через единицу площади поперечного сечения в единицу времени. Она удовлет-
воряет уравнению
выражающему закон сохранения заряда.
В § 1 было показано, что сила, действующая на статический заряд, выра-
жается через напряженность электрического поля 8. На заряженную движу-
щуюся частицу действуют добавочные силы и для их описания удобно ввести
понятие вектора магнитной индукции. Вектор магнитной индукции & выра-
жается через силу F, действующую на движущийся со скоростью v заряд е,
уравнением вида
F-e*+i-[vxJBl; C.4)
здесь с — скорость света в вакууме.
Многочисленные опыты с электрическими и магнитными полями привели
к формулированию системы дифференциальных уравнений — уравнений Макс-
велла, — определяющих свойства векторов g и Л?, связанных с движущимися
в вакууме зарядами. Эти уравнения подытоживают экспериментальные резуль-
таты и в настоящее время образуют постулятивную основу электродинамики.
В действительности невозможно каким-либо образом «вывести» эти уравнения,
так же как нельзя вывести основные постулаты квантовой механики. По-
этому мы начнем этот параграф с перечисления четырех основных уравне-
ний, а затем выясним происхождение некоторых членов, входящих в них.
Из уравнений Максвелла можно получить закон кулоновского взаимодей-
ствия, составляющий основу обсуждения, проводившегося в двух предыдущих
параграфах.
. В § 2 показано, что в материальной среде необходимо рассматривать два
вектора ^и.2), которые могут быть связаны друг с другом с помощью диэлектри-
ческой постоянной е' или диэлектрической восприимчивости %(е). Аналогичными
величинами в случае магнитного поля являются векторы & и JC, которые
могут быть связаны друг с другом с помощью магнитной проницаемости /*'
или магнитной восприимчивости %(т). В заключение параграфа мы запишем
уравнения Максвелла для материальной среды.
А. УРАВНЕНИЯ МАКСВЕЛЛА В ВАКУУМЕ
Следующие четыре ^дифференциальных уравнения описывают свойства
электрического и магнитного полей в вакууме:
() C.5)
(З.б)
664 Гл. 12. Электромагнитные основы теории межмолекулярных сил
Эти уравнения вместе с граничными условиями служат для однозначного
нахождения векторов ff и J@.
Два первых уравнения описывают «источники» полей. Связь дивергенции
напряженности электрического поля & с плотностью электрического заряда
выражает закон Кулона, использовавшийся в § 1 настоящей главы. Тот факт,
что магнитная индукция Л подчиняется соотношению, аналогичному C.6),
бьщ показан экспериментально. Это соотношение можно интерпретировать как
утверждение, что изолированные магнитные полюсы (заряды) не существуют.
Третье из приведенных уравнений связано с именем Фарадея, показавшим,
что изменение магнитного поля всегда связано с появлением электрического
поля. Именно этот закон используется в электрических двигателях и генера-
торах.
Четвертое уравнение появилось впервые в виде закона Ампера. Если по
круговому витку радиуса R протекает ток силы г, то в центре витка создается
магнитное поле, перпендикулярное плоскости витка и имеющее величину
индукции, равную
^ C.9)
Ампер истолковал это явление и результаты изучения величины полной маг-
нитной индукции &, показав, что она равна сумме вкладов отдельных элемен-
тов, образующих виток провода, величина которых ds выражается в виде
Ul**].; (ЗЛО)
здесь г — радиус-вектор, идущий из элемента длины ds в данную точку поля.
Из этого соотношения может быть выведен закон Ампера
<ЗЛ1>
описывающий магнитное поле постоянного тока.
Максвелл с помощью излагаемых ниже аргументов показал, что в общем
случае это выражение несовместимо с уравнением, описывающим сохранение
заряда C.3). Беря дивергенцию от обеих частей уравнения C.11), получаем
в-**])-* №¦••)• «"?>
Поскольку дивергенция ротора всегда равна нулю, то отсюда и из C.3) следует,
что (Bg/dt) = 0. Это справедливо только в стационарном, но не в общем случае.
Поэтому Максвелл пересмотрел закон Ампера и, добавив член A/с) (df/df),
называемый «током смещения», заменил уравнение C.11) уравнением C.8).
Совместимость этого уравнения C.8) с уравнением сохранения заряда можно
показать следующим образом. Взяв дивергенцию от обеих частей уравнения
C.8), получаем
Комбинируя это выражение с уравнением C.5), получаем уравнение сохранения
заряда
Справедливость уравнения Максвелла с членом Г^Х^! и током смещения
хорошо подтверждается прямыми наблюдениями магнитных полей, создаваемых
движущимися зарядами. Токи смещения имеют особое значение в задачах,
связанных с высокочастотными переменными токами.
§ 3. Электромагнитные уравнения Максвелла 665
Б. СКАЛЯРНЫЙ И ВЕКТОРНЫЙ ПОТЕНЦИАЛЫ ;
МАГНИТНЫЕ МУЛЬТИПОЛИ
В § 1 было отмечено, что удобно ввести понятие электростатического потен-
циала, связанного с напряженностью электрического поля. Уравнения Макс-
велла могут быть упрощены путем введения двух потенциалов — скалярного
потенциала V и векторного потенциала %j€. Так как «Ф является вектором,
дивергенция которого равна нулю, то он может быть выражен через ротор
другого вектора. Поэтому векторный потенциал определяется так, что
C.15)
Можно показать, что в общем случае g легко выразить через ^ и скалярный
потенциал в виде
Очевидно, определение потенциалов <j? и V неоднозначны. К первому можно
прибавить градиент скалярной функции, не меняя при этом величины &*
Тогда если соответственно изменить и потенциал ^, то величина 8 также не
изменится. Неопределенность ддя^ и ^"удобно устранить, потребовав, чтобы
они удовлетворяли соотношению
Тогда прямая подстановка этих выражений в уравнения Максвелла приводит к
Решения уравнений для %j? и Т могут быть выражены в виде
Интегрирование ведется по всему распределению заряда : г, — координаты
1-го заряда, a ria — вектор, направленный из точки поля в r'-й заряд. Заметим
здесь «эффект запаздывания», выражающийся в том, что величинами, опре-
деляющими значение потенциала в момент t = t0, являются коо рдинаты и ско-
рости в момент t = t0 — (r/ct/c).
Вследствие подобия выражений для потенциалов *j? и Т во многих случаях
обе величины могут рассматриваться аналогичным образом1). Рассмотрим сна-
чала векторный потенциал в точке на некотором расстоянии от совокупности
движущихся зарядов. Этот потенциал может быть разложен в ряд, аналогичный
использованному в § 1:
{ ^^иЦ_^; C.22)
здесь г, — радиус-вектор, соединяющий начало системы координат с зарядом eh
движущимся со скоростью vf; г—радиус-вектор, соединяющий начало с про-
х) Иногда оС и c?f объединяют в виде четырехм ерного потенциала, особенно удобно га
при релятивистском рассмотрении взаимодействия излучения и вещества [18].
$66 Гл. 12. Электромагнитные основы теории межмолекулярных сил
нзвольной точкой поля. «Электростатическое» выражение этого разложения
имеет вид
где для мультипольных моментов введено запаздывание в виде
C.24)
C.25)
-,- -'*-*.- I*f-' C.26)
Эти величины соответствуют полному заряду, дипольному и квадрупольному
моментам распределения заряда.
После многочисленных преобразований с использованием уравнения сохра-
нения заряда выражение C.22) можно записать в виде [17]
здесь m — магнитный дипольный момент набора зарядов, определяемый выра-
жением
m = 4r2le* h х V'1W -^> C-28)
а /и и © — определенные выше дипольный и квадрупольный моменты распре-
деления заряда. В разложение дляс^б, конечно, входят магнитные мультиполи
высших порядков, аналогичные электрическим мультиполям высших порядков.
В стационарном состоянии первый и третий члены разложения C.27) равны
нулю и ряд для %j€ становится подобным ряду для электростатического потен-
циала.
Магнитное поле совокупности зарядов записывается через ротор от вектор-
ного потенциала, выражаемого C.27). В соответствии с этим выражением основ-
ной вклад в магнитную индукцию S8 дают три члена : 1) магнитное поле осцил-
лирующего электрического диполя ; 2) поле магнитного диполя; 3) поле
осциллирующего электрического квадруполя. Для стационарных токов эффект
запаздывания равен нулю. В этом случае производные по времени от электри-
ческих моментов равны нулю и выражение для ЛЭ сводится к виду
^© = 3 (тг;г) Г — -^ (стационарный ток). C.29)
Магнитный момент, входящий в это выражение, является частным случаем
выражения C.28). Если поле создается стационарным током силы i, то момент
магнитного диполя равен
Ш = -я— $ [г X ds] (стационарный ток). C.30)
Аналогично моменту количества движения частицы L, равному т [г х v ],
магнитный момент частицы, даваемый выражением C.28), может быть (пре-
небрегая запаздыванием) записан в виде
. C.31)
Пропорциональность m и L используется при нахождении связи магнитного
момента с динамикой молекулы.
§ 3. Электромагнитные уравнения Максвелла 667
В. НАМАГНИЧИВАНИЕ ВЕЩЕСТВА
Вследствие явления поляризации вещества (обсуждавшегося в § 2) и ана-
логичного явления — намагничивания вещества — электрические и магнитные
поля в веществе не описываются уравнениями Максвелла в вакууме. Молекулы
среды обладают движущимися заряженными частицами, взаимодействующими
с наложенными электрическими и магнитными полями. Силы, действующие на
молекулы со стороны внешнего поля, стремятся ориентировать и деформировать
молекулы, а движение заряженных частиц (молекул) создает дополнительные
электрические и магнитные поля, которые складываются с внешними полями.
Рассмотрим магнитное поле тока, текущего по витку в вакууме. Можно
измерить силу, действующую на движущийся заряд или малый пробный контур
(маленький виток, несущий ток). Было замечено, что эта сила изменяется при
заполнении пространства веществом и что она зависит от природы среды. Этот
опыт аналогичен опыту, проводившемуся в электростатике, где было замечено,
что величина силы, действующей на заряд, помещенный между пластинами
конденсатора, зависит от природы диэлектрика. Магнитное поле заставляет
вещество само создавать дополнительное поле, которое складывается с полем
внешних источников.
Аналогично случаю электростатической индукции j2) обозначим вклад
внешних постоянных магнитор и токов в полное магнитное поле через J?. Сила,
действующая на пробный контур, определяется полем, являющимся суммой X
и поля, связанного с намагничиванием вещества. Это магнитное поле, опре-
деляющее силу, действующую на микроскопическое тело, назовем по аналогии
с ff магнитной индукцией J8. Величину, известную как намагниченность,
можно определить следующим образом :
-4я<лГ = К-ЛВ. C.32)
Как показано в § 4, намагниченность соответствует магнитному моменту еди-
ницы объема. Это совпадает с определением поляризации &
+ 4n& = m2)-ff. C.33)
Отрицательный знак является несколько неожиданным, но, как показано в
следующем параграфе, он имеет важное значение при физической интерпрета-
ции величин о/# и чФ.
Можно также ввести величину магнитной проницаемости ц':
C.34)
Это определение аналогично определению диэлектрической постоянной е', кото-
рая выражается в виде
& = e'ff. C.35)
Как /#', так и в' в действительности являются тензорами, однако в изотропных
средах они могут считаться скалярными величинами. Они тесно связаны с
величинами xJt и & и являются постоянными материальной среды. Как /*',
так и г' для вакуума равны единице.
Вопросы, связанные с намагничиванием вещества и магнитной восприим-
чивостью, широко обсуждаются в следующем параграфе. Основным поводом
для введения здесь векторов JC и «Я? является возможность написания урав-
нений Максвелла для материальной среды.
Г. УРАВНЕНИЯ МАКСВЕЛЛА ДЛЯ МАТЕРИАЛЬНОЙ СРЕДЫ
Рассмотренные выше уравнения Максвелла приложимы только к полям
в вакууме. Рассматриваемым ниже методом можно показать, что поля в мате-
риальной среде удовлетворяют аналогичной системе уравнений
C.36)
668 Гл. 12. Электромагнитные основы теории межмолекулярных сил
(¦!¦•<•)-О, C.37)
C.39)
Эти уравнения наряду с уравнениями C.34) и C.35) служат для однозначного
определения векторов 3>, ff, J? и &, если, кроме того, потребовать, чтобы е' и /г'
были независимы от координат и времени, исключая границы раздела диэлек-
триков.
Мы можем теперь ввести скалярный и векторный потенциалы, определяе-
мые выражениями C.15) и C.16). Если *j€ и V связаны соотношением
<3-40>
то прямая их подстановка в уравнения Максвелла для материальной среды
приводит к уравнениям для потенциалов :
Из уравнений Максвелла можно получить следующее соотношение, выра-
жающее сохранение электромагнитной энергии:
*>. C-43)
Здесь / — плотность энергии электромагнитного поля, a S — вектор Пойн-
тинга, или поток электромагнитной энергии, определяемый в виде
' = -5г{(*-Х)+ (-»•*)}> C.44)
8 = ?[*ХЯ]. C-45>
Член (gv-j?) соответствует работе, производимой в единицу времени полем,
действующим на заряды в единице объема. Уравнение C.43) приводит к поня-
тиям электромагнитного импульса и давления излучения.
Для области пространства, свободной от макроскопических зарядов и
токов, уравнения Максвелла могут быть преобразованы к виду волновых
уравнений
)* C-46)
Эта форма уравнений ведет непосредственно к свойствам электромагнитных
волн. Так как коэффициент преломления ц определяется отношением скорости
света в вакууме к скорости света в веществе, то из этих двух уравнений прямо
следует
г) = YJT* . C.48)
Обычно /л' очень близко к единице, так что r}^^]fef. Интенсивность электро-
магнитной волны определяется обычно как абсолютная величина усреднен-
ного по времени вектора Пойнтинга.
При изучении свойств электрических и магнитных полей в материальной
среде можно использовать два подхода. В микроскопическом рассмотрении
используются уравнения Максвелла для вакуума, а различные заряды, обра-
§4. Намагничивание вещества и магнитная восприимчивость 669
зующие среду, рассматриваются по отдельности. Поляризация и намагничи-
вание вещества объясняются с микроскопической точки зрения. Если мы знаем
координаты и скорости всех элементарных зарядов в среде, то этот подход пол-
ностью дает детальную структуру электрических и магнитных полей. Величины
этих полей будут сильно меняться от точки к точке и во времени и ро многих
приложениях будут иметь малую ценность. С другой стороны, при макроско-
пическом рассмотрении используются ураьнения Максвелла для материальной
среды; поляризация и намагничивание вещества учитываются макроско-
пически через величины /*' и е'. При использовании этого подхода мы учиты-
ваем только макроскопические заряженные тела и юки; результирующее
описание полей также макроскопично.
Рассмотрим теперь связь между решениями, получаемыми в ДЕух этих
подходах, и интерпретацию введенных выше векторов *ЛГ и &. Решение для
обоих случаев может быть записано через векторный и скалярный потенциалы.
Рассмотрим сначала решение микроскопической задачи. Потенциалы могут
быть усреднены по малой области пространства, т. е< малой относительно
любых макроскопических вариаций, но достаточно большой, чтобы содержать
много молекул. В результате такого сглаживания появляется новая функция,
которая выявляет только макроскопические изменения. Можно показать [17],
что эта новая функция совпадает с функцией, получаемой при прямом решении
макроскопических уравнений Максвелла. При этом процессе усреднения ве-
личины*^ и xjf могут быть отождествлены с микроскопическими свойствами
вещества. Показано, что *#* есть полный электрический дипольный момент
молекул единицы объема, a *At — полный магнитный дипольный момент моле-
кул единицы объема. Как показано в следующем параграфе, величина *ЛГ
может быть связана с магнитным моментом молекулы тем же методом, который
использовался в § 2 при рассмотрении аналогичных задач электростатики. Эти
результаты являются подтверждением уравнений Максвелла для материаль-
ной среды.
§ 4. НАМАГНИЧИВАНИЕ ВЕЩЕСТВА И МАГНИТНАЯ
ВОСПРИИМЧИВОСТЬ
В этом параграфе мы рассмотрим магнитную аналогию обсуждения, про-
веденного в § 2 для электростатики. Рассмотрим поведение векторов напря-
женности поля Ж и магнитной индукции & на границе раздела двух сред и
покажем, как эти векторы связаны с вектором намагниченности %Jf. Затем мы
рассмотрим локальный вектор магнитной индукции «$(лок->, который соответ-
ствует силе, испытываемой отдельной молекулой среды. В заключение обсудим
магнитную восприимчивость.
Магнитной восприимчивостью удобно пользоваться при определении маг-
нитных моментов индивидуальных молекул. Эти моменты являются ключом
к изучению структуры молекул. Если вещество парамагнитно, т. е. молекулы
обладают постоянными магнитными моментами, то силы взаимодействия маг-
нитных диполей могут играть важную роль в межмолекулярных силах.
А. ВЕКТОРЫЦС И &
Дифференциальными уравнениями для векторов J? и & являются урав-
нения Максвелла для материальной среды C.37) и C.39). В стационарном
состоянии «током смещения» можно пренебречь, и соотношения принимают
вид
О, D.1)
*!• D.2)
Эти соотношения справедливы внутри вещества.
670
Гл. 12. Электромагнитные основы теории межмолекулярных сил
Применяя теорему Грина к первому из этих соотношений, а теорему Стокса
ко второму, можно получить следующие интегральные выражения :
dS) = O, D.3)
-?¦', <4-4>
где i — полный ток, текущий по поверхности, ограниченной контуром, вдоль
которого ведется интегрирование. На границе двух сред 1 и 2 (или на границе
среды и вакуума) может существовать поверхностная плотность тока j(s) (или
тока на единицу длины нормально к потоку). С помощью написанных выше
интегральных соотношений можно показать, что на
границе двух сред выполняются следующие условия
для векторов «Я? и }? :
= 0, D.5>
~j(s\ D-6>
где п — единичный вектор, нормальный к поверх-
ности и направленный из среды 1 в среду 2. Эти
выражения аналогичны выражениям B.9) и B.10).
Для получения соотношения между векторами
Ку & и %Jt рассмотрим простой опыт, подобный опыту,
приведенному в § 2, разд. Б. Рассмотрим длинный ци-
линдр из диэлектрика, окруженный катушкой, по
которой протекает электрический ток (более идеализи-
рованный случай — цилиндр охвачен поверхностным
Ф и I. i \)o. аил иьдрическая током, текущим по внешней поверхности). Простоты
полость в материальной ради рассмотрим изотропную среду, так что векторы
среде. Л и JsB параллельны. Удалим теперь вещество из по-
лости, ограниченной цилиндром длиной L и радиусом /?,
ось которого совпадает с направлением поля. Предполагается, что линейные
размеры этого цилиндра велики по сравнению с молекулярными размерами.
Вид полости представлен на фиг. 195. Выясним, какова должна быть вели-
чина поверхностных токов, чтобы сила, действующая на макроскопический
пробный контур тока внутри полости, была равна силе, действовавшей да
удаления вещества из этой полости. Магнитный дипольный момент этих
токов равен моменту удаленного вещества. Применяя к полости выражения
D.5) и D.6), получаем
jq(o) _ ^g@ = о
(торец цилиндра),
= — /E) (боковая поверхность цилиндра),
D.7)
D.8)
где индексами i и о обозначены значения внутри и вне полости соответственно,
a /(s) — величина поверхностного тока, циркулирующего вокруг цилиндра в
направлении, указанном на фиг. 195.
Так как предполагается, что сила, действующая на движущийся заряд в
цилиндрической полости, равна силе, действовавшей до образования полости,
и поскольку в полости вещество отсутствует, то
ЯЮ-ХЮ»*, D.9)
где«5Э — величина магнитной индукции до образования полости. Кроме того,
вследствие выбора формы полости
К@) = ЗС; DЛ0>
§ 4. Намагничивание вещества и магнитная восприимчивость 671
здесь К — величина напряженности до образования полости. Следовательно,
необходимо ввести поверхностный ток, который равен
Поскольку полный ток i вдоль поверхности равен L/(s), то в соответствии
с выражением C.30) магнитный момент этого тока равен
^*\ D.12).
но направлен против поля. Тогда вектор намагниченности <Jt, являющийся
полным магнитным моментом единицы объема, получается из двух последних
выражений и связан с JC и S8 следующим образом:
Это выражение совпадает с выражением C.32), и оно показывает, что данная
в § 3 интерпретация вектора <Jt правильна.
Б. ЛОКАЛЬНЫЙ ВЕКТОР МАГНИТНОЙ ИНДУКЦИИ *(лок.)
В проведенном выше обсуждении была найдена связь между векторами:
J& и вектором намагниченности*лГ. Необходимо иметь в виду, что вектор &г
описанный выше, соответствует силе, действующей на макроскопический проб-
ный контур. Рассмотрим теперь силу, действующую на отдельную молекулу1).
По аналогии с соответствующим электрическим вектором &(лок-) введем локаль-
ный вектор магнитной индукции ^б<лок>.
Для определения силы, действующей на некоторую молекулу, рассмотрим
длинную цилиндрическую катушку (длиной L и радиусом /?), по которой проте-
кает ток. Силовые линии магнитного поля внутри цилиндра параллельны при
условии, что L^> R. Внутри этого цилиндра поместим некоторое количества
вещества, молекулы которого являются постоянными магнитными диполями с
моментом т. Полный магнитный дипольный момент молекулы т(полн>является
суммой постоянного момента m и индуцированного момента т<инд>. Усреднен-
ный по времени полный магнитный дипольный момент т(полн> является векто-
ром, направленным по направлению или против оси z, т. е. по (или против)
внешнему полю. Вектор намагниченности J# равен nmSnojlH).
Действие магнитного поля на молекулу внутри катушки складывается иа
действия внешнего магнитного поля JC и действия поля, обусловленного на-
магниченностью вещества внутри цилиндра. Рассмотрим опять изотропное
вещество так, чтобы вектор намагниченности был параллелен направлению
поля. Здесь ситуация очень сходна с имевшей место в § 2, разд. В при рассмотре-
нии аналогичной задачи электростатики. Найдем начальное поле магнитных
диполей, помещенных в центр молекул, образующих вещество. В результате
получим выражение, очень похожее на B.19):
J g(r)JTldr9 D.14)
б
цил. обл.
где g(r) — радиальная функция распределения ; п — плотность числа единиц ;
3f\ — начальная напряженность поля магнитных диполей
<4Л5>
Вычисление величины лб?лок;> может быть осуществлено методом, аналогичным
использованному при вычислении соответствующего интеграла для ^>
х) Другой вывод и хорошее обсуждение локального вектора магнитной индукции ^(л
имеется у Френкеля [19].
672 Гл. 12. Электромагнитные основы теории межмолекулярных сил
Однако, получив выражение, аналогичное B.21), необходимо выбрать другое
предельное значение. Для получения внутри катушки параллельного поля и
исключения краевых эффектов необходимо принять L^> R (вместо L <^ R в
случае электростатики). Это приводит к отличному результату
J^ D.16)
V(s)
так что окончательные выражения для величины «$<лок-) имеют вид
ДОюк.) = К + |лШ(полн.) = Ж + Щ-^у D.17)
|L D.18)
Вывод выражения D.17) аналогичен выводу выражения B.22), а D.18) анало-
гичен B.23). Происхождение этих двух пар выражений совершенно различно
вследствие того, что граничные условия, применяемые в обоих случаях, раз-
личны. Таким образом, мы приходим к неожиданному результату, что если
рассматривать локальные векторы, то ?В и SD, по-видимому, будут играть экви-
валентную роль, тогда как с точки зрения физического смысла полей экви-
валентную роль играют векторы 38 и ff.
В. ВЫРАЖЕНИЕ МАГНИТНОЙ ВОСПРИИМЧИВОСТИ
ЧЕРЕЗ МАГНИТНУЮ ПРОНИЦАЕМОСТЬ
Магнитная восприимчивость, рассчитанная на одну молекулу, и магнит-
ная проницаемость определяются аналогично диэлектрической постоянной
Щ(полн.) = х(т) ^лок.)^ D.19)
.0 = jw'J{. D.20)
В неизотропной среде направления векторов <$6 и J? не совпадают, и поэтому
/*' и х(т) являются тензорами. Если п — плотность числа частиц (молекул),
то магнитная восприимчивость, рассчитанная на одну молекулу, связана с
магнитной проницаемостью
Х ~ Ann U'+
Это — магнитный аналог формулы Клаузиуса—Мосотти. Восприимчивость,
рассчитанная на 1 моль, равна 7V%(m).
Существует важное различие между диэлектрической и магнитной воспри-
имчивостями, состоящее в том, что е' всегда больше единицы (а %{е) всегда поло-
жительна), тогда как /г' может быть больше или меньше единицы (а %(т) может
быть как отрицательна, так и положительна). Вещества, для которых /г' меньше
единицы, называются диамагнетиками. Для этих веществ значение /г' столь
близко к единице, что величина Й(лок<) очень близка к «$??, и их едва ли следует
различать. В этом случае восприимчивость диамагнитных веществ, рассчитан-
ная на одну молекулу, дается выражением вида
^-1)- D.22)
Восприимчивость диамагнитных веществ отрицательна потому, что вследствие
ларморовой прецессии магнитное поле индуцирует в молекуле момент, на-
правленный против поля.
С другой стороны, если молекулы обладают постоянным магнитным диполь-
ным моментом, то поле стремится ориентировать молекулы по направлению
поля, так что %(т) положительна, а /г' больше единицы. Этот случай соответ-
ствует парамагнитному веществу.
§ 4. Намагничивание вещества и магнитная восприимчивость 673
Средний магнитный дипольный момент молекулы и магнитная воспри-
имчивость могут быть вычислены с помощью молекулярных величин в основ-
ном так же, как и для электростатики, обсуждавшейся в § 2. Легко видеть, что
по аналогии с выражением B.35) средний магнитный дипольный момент, рас-
считанный на одну молекулу, совпадает с направлением оси z и имеет величину
/й<полн.) = ш(инд.) + mL
здесь Цх) — функция Ланжевена1). Если молекулы не обладают постоянным
магнитным дипольным моментом, то второй член равен нулю и средний момент
просто равен индуцированному моменту. Это приводит к отрицательной вели-
чине магнитной восприимчивости диамагнитных веществ, так как индуциро-
ванный момент отрицателен, т. е. направлен против поля.
Если молекулы являются постоянными магнитными диполями, то первый
член обычно пренебрежимо мал и средний момент в основном зависит только
от ориентирования постоянных диполей. Для малых значений аргумента
функция Ланжевена может быть разложена в ряд, и мы получаем из выражения
D.23) для слабых полей или высоких температур
/л<поли> = з^ ле(ло4 D.24)
Таким образом, для слабых полей или высоких температур восприимчивость
пропорциональна 1/Г и множитель пропорциональности является мерой вели-
чины постоянного магнитного дипольного момента. Используя выражение
вектора ^(JI0K) из D.17) и D.25), находим
где величина
0 = f^- D.27)
известна как температура Кюри,
Теория ферромагнетизма Вейсса [20] основана на выражении D.23).
Выше температуры Кюри в вещество, молекулы которого обладают постоян-
ным моментом, парамагнитно. Ниже температуры Кюри существует самопроиз-
вольное намагничивание, т. е. намагничивание не равно нулю в отсутствие
поля. Это состояние назовем ферромагнетизмом. Если J? равно нулю, то выра-
жение D.23) может быть записано в виде
-Т^ЗЦа), D.28)
где
!==(т\
т
Но 2L{a)ja монотонно спадает от единицы при а = 0 до нуля при а = оо.
Поэтому если Т < 0, то имеется не равное нулю значение а, а следовательно, и
ш<полн.) не раВно нулю, так что уравнение D.28) удовлетворяется. Однако если
Т > 0, то единственным решением уравнения D.28) является а = 0 (следо-
вательно, /п(полн-> = 0) и поэтому самопроизвольное намагничивание невоз-
можно. Хотя классическая теория и дает качественное описание явления фер-
ромагнетизма, однако количественные результаты зависят от аномального по-
ведения электронных спинов и их взаимодействия. Интересно отметить, что
х) При низких температурах для учета дискретной природы вращательных энергети-
ческих состояний функция Ланжевена должна быть заменена функцией Бриллюэна [17]
674 Гл. 12, Электромагнитные основы теории межмолекулярных сил
существование температуры Кюри зависит от знака члена, прибавляемого в
выражении D.17) к J{ для получения локального вектора *5Э(Л0К). Знак анало-
гичного члена в электростатике противоположен и, следовательно, электри-
ческой аналогии ферромагнетизма не существует.
§5. КЛАССИЧЕСКАЯ ТЕОРИЯ ПОГЛОЩЕНИЯ СВЕТА
И КОЭФФИЦИЕНТА ПРЕЛОМЛЕНИЯ
Существуют два типа дальнодействующих межмолекулярных сил : элек-
тростатические силы (такие, как сила диполь-дипольного взаимодействия) и
«дисперсионные», или лондоновские силы (такие, как сила притяжения непо-
лярных молекул, обратно пропорциональная шестой степени расстояния).
Эти дисперсионные силы тесно связаны с испусканием и поглощением света
молекулами. В этом и следующем параграфах излагается классическая
и квантовая теория поляризуемости и коэффициента преломления. Результаты
выражаются в терминах «силы осциллятора»//и«характеристическихчастот»^.
В § 3 дисперсионные силы выражаются через эти величины. Величины /, и vt
могут быть определены при изучении зависимости коэффициента прелом-
ления от частоты. Эти величины, в свою очередь, используются для предска-
зания сил взаимодействия неполярных молекул. Настоящий параграф начи-
нается с обсуждения электрических и магнитных полей, создаваемых вибра-
тором Герца (или осциллирующим диполем), представляющим идеализирован-
ную антенну или источник'света. Затем обсуждается движение заряженной
частицы в электромагнитном поле. Обсуждение этих двух вопросов дает необ-
ходимый фундаментальный материал для классического и квантового рассмо-
трения взаимодействия света и вещества.
А. ОСЦИЛЛИРУЮЩИЙ ДИПОЛЬ (ВИБРАТОР ГЕРЦА)
Рассмотрим излучение, создаваемое в вакууме зарядом + е, колеблющимся
относительно (неподвижного) заряда —е. Пусть колебания происходят вдоль
линии и имеют амплитуду А и частоту v = с/А. Этот осциллирующий диполь
является простейшим примером электромагнитного излучателя и представ-
ляет радиоантенну или источник света. Электромагнитные свойства этой систе-
мы могут быть получены из решений уравнений C.18) и C.19). Мы здесь не
будем подробно обсуждать этих решений, но коротко рассмотрим результаты
в двух предельных случаях г <^ А и г^>Х.
Можно показать1), что в непосредственной близости к осциллятору (г <^ А)
напряженность & ранна напряженности статического диполя с моментом
fi = eA cos Bтгс*/А). Соответственно напряженность магнитного поля JC совпа-
дает с напряженностью поля, создаваемого элементом тока,
Вдали от осциллятора (г > А) компоненты векторов & и JC выражаются
в виде
*• = - "W sin в cos -X <r - d>' E.1а)
sin в C0S "Т <Г ~ ^ ' E.16)
г) См., например, [21 ] и [22 ]. Прекрасное изложение электромагнитной теории света и
оптики содержится в книге Борна [23].
§ 5. Классическая теория поглощения света и коэффициента преломления 675
Так как %% = <J^0, а все другие компоненты векторов <f и Л равны нулю, то
векторы напряженности магнитного и электрического полей взаимно перпен-
дикулярны и равны по величине. Кроме того, отсутствуют компоненты векторов
<f или Л в направлении распространения излучения г. Поэтому на достаточно
большом расстоянии от вибратора Герца векторы & и Л образуют плоскую
бегущую электромагнитную волну. Можно показать, что электромагнитное поле
на большом расстоянии от любой совокупности движущихся зарядов (суммар-
ный заряд которого не равен нулю) может быть представлено в виде набора
вибраторов Герца, обладающих различными частотами и направлениями коле-
баний. Поэтому электромагнитное излучение в пространстве, свободном от
поля, состоит из ряда распространяющихся в разные стороны налагающихся
волн различной частоты и поляризации. Для каждой из этих волн векторы
<f и Л равны по величине и взаимно перпендикулярны.
Для удобства поляризация света определяется через плоскости, в которых
расположены векторы <f и г. Свет плоско поляризован, если эта плоскость остается
инвариантной. Свет эллиптически поляризован, если <fx = Я^х sin [Btt/A)(z — ct)]
и &у = &2 cos [Bтг/А) (z — ct) ]. Круговая поляризация является частным слу-
чаем эллиптической поляризации, когда <?х = ^Г2.
Позже мы дадим описание поведения молекулы в электромагнитном поле.
Предполагается, что длина световой волны велика по сравнению с молеку-
лярными размерами и что молекула находится на расстоянии многих длин
волн от источника. В этих условиях напряженность электрического поля с
хорошим приближением однородна в пределах длины молекулярных разме-
ров. Для света, поляризованного в направлении оси х и распространяющегося
в направлении оси г, единственными не обращающимися в нуль компонентами
являются х-компонента вектора напряженности электрического поля & и
у-компонента вектора напряженности магнитного поля Л- Величины нгпря-
женностей в области порядка молекулярных размеров в этом случае равны
E.2а)
E.26)
Плотность энергии электромагнитного излучения / в соответствии с выра-
жением C.44) раьна среднему значению величины
Так как средние значения ^Г2 и <Жг равны
т
lim 05 — sin2 —=— ^=-o^0) E.4)
^ Т J Л ?*
О
где ^0 — амплитуда колебаний электрического и магнитного векторов, то
усредненная плотность энергии равна
/ = i^o- E.5)
В этом случае интенсивность электромагнитной волны или поток энергии
равен с/.
Б. УРАВНЕНИЕ ДВИЖЕНИЯ ЗАРЯЖЕННЫХ ЧАСТИЦ
Ргссмотрим молекулы газа, достаточно разреженного, чтобы можно было
пренебречь поляризацией, т. е. #<лок-> = g = Л) и &<лок-> = «$б = Л •
На заряженные частицы (электроны и ядро) молекулы действуют внешние
поля и силы со стороны других частиц молекулы. Пусть Tt и Лег — соответ-
ственно скалярный и векторный потенциалы в точке, где находится 1-я
частица. Предположим далее, что сила, действующая на заряженную частицу
676 Гл. 12. Электромагнитные основы теории межмолекулярных сил
со стороны других частиц молекулы, обусловлена исключительно электроста-
тическим (кулоновским) взаимодействием1). Следовательно, эта сила равна
отрицательной величине градиента потенциала Ф (потенциальной энергии
молекулы). Сила, действующая на /-ю частицу с зарядом eiy выраженная через
эти потенциалы, равна
Хотя это поле сил неконсервативно, тем не менее можно написать лагран-
жиан так, что уравнения движения смогут быть представлены в виде рыра-
жения D.4). Этот лагранжиан [17 ] имеет вид
и приводит к обобщенным импульсам [см. D.5) гл. 1 ], отличным от обычных
величин. Например, в декартовой системе координат
p/ = m,v, + -2-i*/. E.8)
С помощью лагранжиана можно в соответствии с D.6) построить гамильто-
ниан. В декартовой системе координат эта функция имеет вид
н -= 2-Ш1 (р. - т •*')' + ф + ^ Ti • E-9)
Гамильтониан вместо V и ^€можно выразить через веюоры# и 3?. Тогда, если
система координат (необязательно декартора) определена независимо от век-
торов g и ЗС и если оба этих вектора постоянны в пространстве и времени, то
можно показать [24], что компоненты дипольных моментов р и m в напраг-
лении приложенного поля выражаются в виде
Эти соотношения используются при нахождении электрических и магнитных
моментов молекулы путем вычисления энергии молекулы во внешнем поле.
Для упрощения выражения E.6) силы, действующей на заряженную
частицу, можно воспользоваться результатами рассмотрения вибратора Герца.
Как было показано, на больших расстояниях от вибратора Герца векторы
напряженности электрического и магнитного полей равны по величине. Из
этого, а также из выражения C.4) следует, что магнитная сила меньше электри-
ческой в отношении v/c. В атомах и молекулах скорости электронов порядка
1/137 скорости света. Соответственно влиянием магнитного поля Л в этих
случаях можно пренебречь и выражение E.6) ci едется к виду
Это ьыражение будет использовано в примерах, рассмотренных в этом и сле-
дующем параграфах.
х) Предположение о наличии только кулоновского взаимодействия эквивалентно прене-
брежению слабым магнитным взаимодействием электронов в молекуле, обусловленным их
FT T>tV4lAAt f ft Л ll W vsO nrttitfvAtf f\wmw m ws'v r%
р
движением и наличием спинов.
§ 5. Классическая теория поглощения света и коэффициента преломления 677
В. КОЭФФИЦИЕНТ ПРЕЛОМЛЕНИЯ (ТЕОРИЯ ДРУДЕI)
Классическая теория коэффициента преломления света была разработана
Друде. Эта теория была перенесена в квантовую механику лишь с небольшими
изменениями, обсуждаемыми в следующем параграфе. Молекулярная модель,
испольгоранная в теории Друде, состоит в следующем. Молекула рассматри-
вается как совокупность частиц с зарядом et и массой /л,. Связь каждой из этих
частиц со своим положением равновесия изотропна и подчиняется закону упру-
гих сил. Силовая постоянная срязи для /-й частицы равна kiy частота ее колебаний
vt = A/2тг) Vkjlmh а смещение от положения равновесия равно <$г, = г, — г/0).
Тогда потенциальная энергия молекулы равна Ф = */2 2 k№rif-
Представим теперь, что молекула помещается в пучок' света (переменное
электромагнитное поле) с частотой v0, достаточно низкой, чтобы длина волны
До = c/v0 была велика по сравнению с молекулярными размерами. Внешнее
электриче кое поле тогда будет существенно однородным на протяжении
молекулярных размеров, а напряженность его равна <f = ff0 cos 2л vot. Сила,
действующая на r'-ю частицу молекулы, дается выражением E.11)
F,- = — ki д г, + е( &о cos 2tzv0 t. E.12)
В соответствии со вторым законом Ньютона уравнение движения /-й частицы
имеет вид
Щ •§***( + kiд г/ = et &о cos 2nv01. E.13)
После достаточно длительного промежутка времени (такого, что переходные
процессы успевают затухнуть) решение этого уравнения не будет зависеть от
начальных условий и примет вид2)
Оно дает величину смещения /-й частицы молекулы, обусло ленного действием
внешнего переменного электрического поля.
Из этого последнего результата можно получить выражение для зависи-
мости величины индуцированного диполыюго момента от времени
„(„„д.)= »J!
Так как индуцироганный дипольный момент напрарлен по полю, то поляри-
зуемость а, определяемая выражением B.1), является скаляром, равным
/*<инд->/У. Следовательно, мы выразили поляризуемость молекулы через свой-
ства образующих ее частиц :
"w-^r-?^F- <5Л6а)
Таким образом, поляризуемость является функцией частоты приходящего
излучения. Поляризуемость молекулы в электрическом поле, не меняющемся
во времени, выражается в виде
Поляризуемость часто выражают через силу осциллятора fb определяе-
мую соотношением
4
е2/т '
^См. [24].
2) Обсуждение уравнений такого типа см., например, в книге [22].
678 Гл. 12. Электромагнитные основы теории межмолекулярных сил
где е и т —заряд и масса электрона. В классической интерпретации значение
/ дает эффективное число электронов, колеблющихся с характеристической
частотой. Поляризуемость молекулы, выраженная через эти величины, имеет
вид
?^Г E18)
Если мслекулы не обладают постоянным дипольным моментом, то поля-
ризуемость совпадает с электрической восприимчивостью. Диэлектрическая
постоянная, экстраполированная к нулевой частоте, может быть получена из
формулы Клаузиуса—Мосотти B.28)
В связи с выражениями C.46) и C.47) необходимо заметить, что коэффициент
преломления г\ очень близок к величине f е'. Таким образом, коэффициент
преломления света частоты v0 дается выражением
S-j^t- E-20)
Во многих случаях эти простые выражения очень хорошо воспроизводят
экспериментальные дисперсионные кривые. Как показано в § б, квантовоме-
хгнический вывод дает для величины a(vQ), e' и г\ выражения того же вида.
Силы осциллятора в этом случае будут определенными матричными элементами,
связанными с дипольными моментами.
§ 6. КВАНТОВОМЕХАНИЧЕСКАЯ ТЕОРИЯ ПОГЛОЩЕНИЯ СВЕТА
И КОЭФФИЦИЕНТА ПРЕЛОМЛЕНИЯ
В этом параграфе мы получим квантовомеханические выражения для
поляризуемости и коэффициента преломления. Эти выражения имеют тот же
общий вид, что и полученные в предыдущем параграфе с помощью класси-
ческих представлений. Поэтому мы получим возможность дать квантовую
интерпретацию силы осциллятора через элементы матрицы дипольного мо-
мента, а интерпретацию характеристических частот — через разности двух
энергетических уровней молекулы. Вывод этих выражений сделан в терминах
вероятностей переходоь | alk |2 для индуцированного поглощения или излуче-
ния света. В связи с этим параграф начинается с обсуждения упрощенной
квантовомеханической теории поглощения света.
А. ВЕРОЯТНОСТИ ПЕРЕХОДОВ МОЛЕКУЛЫ В ЭЛЕКТРОМАГНИТНОМ
ПОЛЕ
Рассмотрим молекулу, невозмущенный гамильтониан которой <^@). Соот-
ветственно невозмущенные нестационарные волновые функции удовлетворяют
уравнению Шредингера
» = ?, Wf = A ^L, (б. 1)
где Et—энергетические уровни. Волновая функция может быть разбита на
множители
F.2)
где ipty — стационарные волновые функции невозмущенной молекулы.
$ б. Квантовомеханическая теория поглощения света и коэффициента преломления 679
Рассмотрим теперь молекулу в поле электромагнитного излучения частоты
v0 = с/Я0, падающего по направлению оси z и поляризованного в плоскости xzy
т. е. вектор & направлен вдоль оси х. Как и в предыдущем параграфе, длина
волны излучения предполагается большой по сравнению с молекулярными
размерами, т. е. электромагнитное поле может считаться в пределах молекуляр-
ных размеров постоянным. Далее, поскольку скорость электроноь мала по
сравнению со скоростью света, то влиянием напряженности магнитного поля
светового луча можно пренебречь. Следовательно, при этих предположениях
единственной компонентой электромагнитного поля, подлежащей рассмотре-
нию, является %?х = lf0 sin 2% vot. Поэтому полный гамильтониан молекулы в
поле излучения имеет вид
- (Уо sin 2лг01) 2 et xt, F.3)
где et и xt — заряд и координата г-й частицы молекулы.
Поэтому перЕым шагом в квантовомеханическом изучении поглощения
света будет запись уравнения Шредингера для возмущенной системы
- (вг0 sin 2w0 0 2 et xt) Wt = ^ ^f • F.4)
Все величины, соответствующие невозмущенной молекуле, обозначим
индексом 0. Волновая функция возмущенной системы в 1-м состоянии может
быть разложена в ряд по невозмущенным волновым функциям
§М0Т (б-5)
В общем случае волновые функции комплексны и мы будем обозначать комп-
лексно сопряженные волновые функции звездочкой. Подставляя это выраже-
ние в уравнение F.4), умножая его на ^>* и интегрируя по всему простран-
ству, получаем следующие дифференциальные уравнения для коэффициентов
разложения а1т:
ifl F.6)
здесь через (f*x)mj обозначен интеграл
F.7)
Эти интегралы являются матричными элементами х-компоненты дипольного
момента.
Предположим, что в момент t = 0 молекула находится в /-м состоянии,
так что пц@) = 1, а все другие alm@) равны нулю. Мы не определяем, является
ли /-состояние верхним или нижним энергетическим состоянием. Рассмотрим
вероятность переходов из этого состояния в другие состояния, вызванных воз-
мущениями электромагнитным полем. Исследуем поведение коэффициентов
alm(t) после короткого промежутка времени. Вероятность того, что переход
произойдет в течение этого короткого промежутка времени, мала, так что^
alv 0/2, a/3,... пренебрежимо малы по сравнению с величиной аш которая почти
в точности равна единице. Тогда выражение (б.б) приводится к виду
^ ]. F.8)
Прямое интегрирование этого выражения дает
alm =-ёГ0 {1Хх)т1 ^ [ Щ-Ет + Ь. Ei-Em-hb J '
680 Гл. 12. Электромагнитные основы теории межмолекулярных сил
Из этого выражения можно получить величину | а1т |2 = а*т а1т
\п 12 - л- е*\(Г*)«*\ш Г2-2cos [(Я/-Ед+ *»,)*/*]
I «*m I t" 4 [ (Ei-Em+hvoy "T"
2 - 2 cos [(?/ - Em - hv0) t/h] __ 2 + 2 cos 4яу0 t - 2 cos [(?/ - Em + ftv0) г/Л]
"+" (Я/ - Ят - Ю2 (Я/ - Emy - (/boI
_ 2cos[(?/ — ECT - Л
Это выражение дает вероятность перехода молекулы из /-состояния в т-состоя-
ние в течение короткого промежутка времени t под влиянием поляризован-
ного срета частоты v0.
Б. ИНДУЦИРОВАННОЕ ПОГЛОЩЕНИЕ И ИСПУСКАНИЕ СВЕТА
Предположим, что \Ег — Em\^hv0. Этот случай соответствует задаче
резонансного поглощения или испускания света. Если Ег < EmJ то переход
Ее^ет к поглощению сг.ета молекулой, если же ?, > Ет, то молекула допол-
нительно испускает свет частоты v0. Выражение F.10) получено в предполо-
жении, что падающий на молекулу сгет монохроматичен. Обычно это не вы-
полняется, и для наших целей мы предположим, что частоты падающего излу-
чения заключены в узкой полосе частот, включающей \Et — Em \jh и ограни-
ченной значениями vx и v2. В этом случае мы заменим %\ в выражении F.10)
на вя/, где / — плотность энергии излучения, рассчитанная на единицу длины
частотного интервала. В этом случае вероятность перехода а\т получается
путем интегрирования выражения F.10) от v± до v2, так что, пренебрегая ма-
лыми членами, имеем
(б.п)
где переменной интегрирования является f = (| Ет — Ег\ — hv)t/h. Если
теперь полоса частот широка по сравнению с естественной шириной линии
испускания1), то замена f (vx) на — оо и f (vj) на + оо не даст большой ошибки.
Тогда после интегрирования получаем2)
FЛ2)
Таким образом, вероятное^ перехода пропорциональна интенсирности срета час-
тоты | Ех—Ет |/Л, квадрату компоненты матрицы дипольного момента и времени.
В приложении В (стр. 700) приведены величины этих матричных компонент
г) Эта линия испускания никогда не бывает идеально тонкой. Если молекула находится
в вакууме, то естественная ширина линии обусловлена силой реакции испущенного излу-
чения на излучающий электрон. При обычных давлениях ширина линии обусловлена расши-
рением вследствие давления, т. е. тем, что энергетические уровни молекул слегка смещены
в результате ван-дер-ваальсова взаимодействия с другими молекулами (см. гл. 13, §7).
2) Это соотношение может быть получено в предположении, что свет поляризован.
В Случае изотропного излучения можно показать, что
2 2л it
где
§ 6. Квантовомеханическая теория поглощения света и коэффициента преломления 681
для многих переходов ?тома водорода и для возбуждения других атомов из
основного состояния (s-состояния) в низшее Р-состояние. В случае водорода
видно, что все компоненты матрицы дипольного момента обращаются в нуль,
если азимутальное квантовое число изменяется не на ± 1, а магнитное кван-
товое число не на 1, 0 или —1.
В соответствии с выражением F.12), если компоненты матрицы диполь-
ного момента обращаются в нуль, переходы невозможны. В действитель-
ности такие запрещенные переходы происходят, однако интенсивность спект-
ральной линии в 100 и даже 1000 раз слабее интенсивности линий, соответ-
ствующих разрешенным переходам. Метастабильными являются такие состоя-
ния, из которых обычно правила отбора запрещают переходы в нижние энерге-
тические состояния. Запрещенные переходы играют большую роль в системах
при низких давлениях, когда запрещенные переходы с испусканием происхо-
дят значительно чаще, чем превращения электронной энергии в поступатель-
ную в процессах столкновений. Например, свечение ночного неба в основном
создается излучением, возникающим в результате таких запрещенных пере-
ходов [25].
Другой пример запрещенных переходов дают инфракрасные спектры
двухатомных молекул. При элементарном рассмотрении вообще утверждается,
что газы, состоящие из гомоядерных двухатомных молекул, не могут давать
вращательных и колебательных спектров, так как при любых расстояниях
между ядрами молекулы не обладают постоянными дипольными моментами.
Это правило следует из обращения в нуль элементов квантовомеханической
матрицы дипольного момента в силу выражения F.12). Эти матричные элементы
вычисляются путем использования волновых функций невозмущенной моле-
кулы и даются выражением F.7). Небольшое поглощение в газе двухатомных
гомоядерных молекул, обусловленное вращательными и колебательными пере-
ходами, тем не менее может осуществляться и за счет других механизмов.
1) Изотопический эффект. В обычных газах всегда присутствуют неболь-
шие количества молекул изотопов, таких, как HD или О1вО18, не являющихся
строго гомополярными.
2) Электрический квадрупольный эффект. Если квадрупольный момент
свободной молекулы изменяется при изменении межъядерного расстояния, то
имеет место некоторое поглощение. Член, описывающий это поглощение, не
представлен в выражении F.7), так как при его получении предполагалось,
что поле однородно в пределах молекулярных размеров. Этот член зависит от
первой производной напряженности поля.
3) Магнитный дипольный эффект. Если магнитный дипольный момент
свободной молекулы изменяется с изменением межъядерного расстояния, то
в результате появляется некоторое поглощение. Этот член выпал вследствие
пренебрежения в возмущенном гамильтониане членом, соответствующим
магнитному полю, и изменением поля поперек молекулы.
4) Эффект электрической деформации [26, 27]. Если молекулы помещают-
ся в однородное внешнее поле, то они деформируются и у них индуцируется
дипольный момент, так что в этом случае возможно поглощение.
5) Эффект, вызванный давлением [28, 29]. В плотных газах и жидкостях
деформация молекул при столкновениях может вызвать появление погло-
щения.
Три первых эффекта имеют место для изолированных молекул, второй и
третий можно получить, если отказаться от ограничений, введенных в начале
этого параграфа1), четвертый и пятый из перечисленных эффектов обусловлены
деформациями, вызванными внешними силами, и являются примерами «вынуж-
денного дипольного излучения». В первых четырех случаях поглощение про-
порционально плотности поглощающей среды ; однако величина поглощения,
вызванного давлением, пропорциональна квадрату плотности.
., например, [30, 4].
682 Гл. 12. Электромагнитные основы теории межмолекулярных сил
В. КОЭФФИЦИЕНТ ПРЕЛОМЛЕНИЯ
Квантовая формула для коэффициента преломления может бьпь получена
методом, совершенно аналогичным описанному в § 5, разд. В методу клас-
сической теории. Если молекулы не обладают постоянным дипольным момен-
том, то компонента электрического дипольного момента, индуцированного
световым пучком, имеет вид
= J Yf Betх) У,dr" . F.13)
Этот интеграл отличается от интеграла F.7) тем, что здесь использованы воз-
мущенные волновые функции У,. Подставляя в это выражение W{ = 2аЧ/(\
интегрируя и учитывая, что аи ^ 1, а все другие alk малы, получаем k
F.14)
к
Теперь, воспользовавшись выражением для амплитуд F.9), найдем
)ч _
<инд.)ч _ %> у 2 | ([лх)щ I2 (Ек -
2
к
*P у 21 Ыт 1* hv0 sin [(Ei - Ек) t/h]
+ ^°^ (Ei-Ek)*-(hv0)* • (бЛ5>
Поляризуемость определяется как усредненный по времени индуцированный
дипольный момент. Таким образом, второй член этого выражения не даст
вклада в поляризуемость, так как в течение первой половины периода он сов-
падает с фазой приложенного электрического поля, а в течение второй поло-
вины противоположен этой фазе.
Таким образом, если ось х является главной осью тензора поляризуемости,
то для этого направления компонента поляризуемости выражается в виде
(п /v\\ __ у 2 1 ([AX)ik I2 (Ек - Ej) {R\fa\
(«ххЫ)и - 2 (Ei-Ek)*-(hvoy > <б- 16а>
а усредненная по всем направлениям поляризуемость — в виде
(« Ы)и = у Ка** W)// + (Оуу Ы)и + («« Ы)и] • F.166)
Это выражение является квантовомеханическим аналогом классического вы-
ражения Друде E.16).
Если все молекулы находятся в /-состоянии, то коэффициент преломления
будет выражаться формулой Клаузиуса—Мосотти E.20), аналогичной класси-
ческой формуле
(U FЛ7)
Здесь п — число молекул в 1 см3. Если распределение молекул по энергети-
ческим состояниям типично для состояния термодинамического равновесия,
то вероятность нахождения молекулы на 1-м уровне равна
§ 6, Квантовомеханическая теория поглощения света и коэффициента преломления 683
где g, — статистический вес /-го состояния. Соответственно коэффициент пре-
ломления выражается в виде
Г П2 ~ П _
U2 + 2J ~
Эта формула впервые была получена Крамерсом [31, 17 ] на основе закона соот-
ветственных состояний. Отрицательные члены приводят к «отрицательной
дисперсии» или отрицательным значениям силы осциллятора /, что трудно
объяснить классически.
Интересно сравнить эту квантовую формулу с классической формулой
Друде, обсуждавшейся в предыдущем параграфе. Прежде всего видно, что
зависимость поляризуемости и коэффициента преломления от частоты излу-
чения одинакова в обеих формулах. Квантовомеханическое рассмотрение дает
возможность лучшей интерпретации постоянных. Собственные характеристи-
ческие частоты Друде, являющиеся характеристическими частотами испуска-
ния или поглощения, равны
vlk = Щ^-, F.20)
где Ег и Ек —энергии состояний / и к молекулы соответственно. Тогда кванто-
вомеханический аналог выражения E.18) имеет вид
Здесь flk— сила осциллятора, соответствующая переходу из /-состояния в
/с-состояние, определяемая выражением вида
, _ 2 hvik I m I2
где
Сила осциллятора, определяемая выражением F.22), относится к отдель-
ным состояниям молекулы. Если молекула обладает вырожденными состоя-
ниями, то удобно специально указывать на вырожденность квантового числа.
Например, энергетические уровни атома вырождены по магнитному кванто-
вому числу m (если только нет внешнего поля, как в задачах, связанных с
эффектом Зеемана). В этом случае состояния обозначаются двойным индексом
lm, где / заменяет совокупность всех квантовых чисел, отличных от /л, а сила
осциллятора //ш; кш> соответствует переходу из Z/п-состояния в fc/л'-состояние.
Из выражения F.21) видно, что поляризуемость молекулы в Z/л-состоянии выра-
жается в виде
4l-|^- F-23)
Однако обычно интерес представляет усредненная величина поляризуемости
молекул в состояниях с вырожденными уровнями. Эта величина имеет вид
(«М« = -2ЩГТ 2 (« WU ш = -?ж2 tAi > <6-24>
m к № О
где flk — усредненная сила осциллятора, определяемая выражением
flk = 2 Ц + 1 ^ Щ flm' kmt' F.25)
т т'
684 Гл. 12. Электромагнитные основы теории межмолекулярных сил
a BL, + 1) — вырождение 1-го уровня. Сумма сил осцилляторов flk по всем
дискретным и непрерывным энергетическим состояниям к равна полному
числу электронов системы. Это — так называемое правило /-суммы Рейхе—
Куна—Томаса [32, 54, 25].
Применение дисперсионной формулы связано с одной трудностью, состоя-
щей в том, что суммирование должно вестись как для переходов в дискретные,
так и для переходов в непрерывные энергетические состояния. Влияние пере-
ходов в континууме часто бывает важным, и иногда оно трудно вычисляется [34].
Дисперсионные соотношения и силы осцилляторов, обсуждавшиеся здесь,
играют очень важную роль при определении дальнодействующих межмоле-
кулярных сил (см. гл. 13, § 3).
§7. РАССЕЯНИЕ ЭЛЕКТРОМАГНИТНЫХ ВОЛН1)
Рассеяние электромагнитного излучения может происходить как с изме-
нением, так и без изменения частоты. Если частота не меняется, то рассеяние
называется релеевским рассеянием. Если происходит изменение частоты, то
явление называют римановским рассеянием. Мы рассмотрим здесь только реле-
евское рассеяние. Оно обусловлено рассеянием отдельными молекулами и интер-
ференционными явлениями, возникающими в результате флуктуации плот-
ности рассеивающей среды2). Теория, излагаемая ниже, приложима как к види-
мому свету, так и к рентгеновскому излучению. Рассеяние видимого света
Фиг. 196. Разность фаз для волн, рассеянных молекулами i и /.
приводит к явлению опалесценции, наблюдаемому ьблизи критической точки.
Рассеяние рентгеновских лучей дает возможность по экспериментальным
данным вычислить радиальную функцию распределения3) g(r).
Рассмотрим рассеяние электромагнитных волн в среде, содержащей N моле-
кул, достаточно слабое, так что необходимо рассматривать только одно-
кратное рассеяние. В этом случае результат может быть выражен через радиаль-
ную функцию распределения g(r). Рассмотрение многократного рассеяния
связано с введением функций распределения высших порядков. Пусть к0 —
волновой вектор электромагнитной волны перед рассеянием, а к—тот же
ЧСм. [35—39, 23].
2) Флуктуации плотности подробно рассматривались в гл. 2, § 6.
8) Подробное обсуждение радиальной функции g(r) распределения проводилось в гл. 4,
§ 9 в связи с уравнением состояния при высоких плотностях.
§ 7. Рассеяние электромагнитных волн 685
вектор после рассеяния. Кроме того, поскольку настоящее обсуждение ограни-
чено только случаем упругого рассеяния, то fc0 = k = 2я/Л, где А — длина
волны. Направления векторов к^ и к указывают направления падающего и
рассеянного пучков соответственно.
Разность фаз для волн, рассеянных в определенном направлении молеку-
лами i и /, графически изображена на фиг. 196. Длина оптического пути луча,
проходящего через точку /, больше соответствующей длины для точки i на вели-
чину, равную сумме расстояний а и Ъ. Угол между ко и к равен в. Угол rj0 равен
углу между векторами к0 и (г/ — г,), а угол rj равен углу между векторами
к и (Tj — г,). Расстояние а, очевидно, равно Гц cos rj0J а расстояние Ь равно
Гц COS (<П — Г}) ИЛИ — Гц COS Г/.
Таким образом, разность фаз двух волн, равная сумме расстояний, умно-
женной на волновое число fc, выражается в виде
к0 Гц cos % — кГц cos rj или (k^ — k) • (гу — г,). G.1)
Пусть А — амплитуда электромагнитной волны, уходящей после рассеяния на
отдельной молекуле. Тогда величина электрического вектора в точке г в ре-
зультате рассеяния на молекуле / равна
А ).(f/-n)| G.2)
I г — r/1
где д( — фаза падающей на молекулу i электромагнитной волны. Рассмотрим
теперь излучение на расстоянии R от рассеивающих молекул, большем по
сравнению с линейными размерами объема, содержащего молекулы (т. е.
R > г, для всех /). Тогда электрический вектор рассеянной волны равен
«>•«». G.3)
-1
Плотность энергии рассеянного пучка можно получить непосредственно
из соотношения
1 = 4- 1#\12= -т^г У >'cos[(ko-k) • г/1 .
Сначала проведем суммирование по отдельности для i = / и i Ф /; тогда
получим выражение вида
rN + у у cos [(ко - к) • Тц]]. G.5)
Если рассеивающие молекулы образуют макроскопически однородную
фазу объема V, то плотность энергии рассеянного света можно выразить
через радиальную функцию распределения g(r). В элементе объема dtj (внутри
объема V) на расстоянии Гц от молекулы /содержится ng(ru) молекул. В силу
аргументов, аналогичных использовавшимся в гл. 2, § 6, суммирование в выра-
жении G.5) может быть заменено интегрированием по объему, занимаемому
веществом,
7 =
\N + Л i <г<;)cos №о - ^ • тц] dtt dTj ]. G.6)
vv
Кроме того, интеграл по объему можно разбить на две части, прибавляя и от-
нимая величину этого интеграла, получаемую для g(r(j) равной единице. Таким
образом, плотность энергии рассеянного света можно записать в виде
/ = /w + /. + //f G.7)
Гл. 12. Электромагнитные основы теории межмолекулярных сил
где
j7k) ' ГЛ*'*У> G.9)
vv
- !>cos кк° - k> • глdr<
Эти члены имеют следующий смысл :
1) Im — плотность энергии для света, рассеянного отдельными молеку-
лами в отсутствие интерференции. В этом смысле 1т дает вклад молекул в
рассеяние.
2) /s — плотность энергии для света, рассеянного при наличии интер-
ференции, связанной с геометрией поверхности, охватывающей вещество.
Именно это рассеяние в основном ответственно за диффракционную картину,
возникающую при падении света на каплю [40]. Если вещество сконцентри-
ровано в сфере радиуса /?0, то интеграл можно взять и получить в результате
Is = *%?- [sin (sR0) - (sR0) cos (s/?0)P, G.11)
где
s=\ko-k\ = 2ksm^ = ^-sm^. G.12)
3) 11 — плотность энергии для света, рассеянного возможными микро-
скопическими неоднородностями или в мутной среде. Так как [g(fy) — 1 ]
существенно отличается от нуля только для значений г/7, малых по сравнению
с макроскопическими размерами среды, то увеличение пределов интегриро-
вания в выражении G.10) с тем, чтобы допустимо было считать ги бесконечно
большим, приводит к небольшой ошибке. В этом случае выражение для /, при-
водится к виду
оо
In П
rbdr'J \d*u ffeOtf) - 1] cos (srl7 cos 0,7) sin0,7d0,7 =
0 0 0
п^* <7J3)
Эти формулы могут использоваться для описания рассеяния электромаг-
нитного излучения двух типов : видимого света, длина волны которого А велика
по сравнению с размерами молекулярных скоплений, и рентгеновских лучей,
длина волны которых порядка величины молекулярных размеров. В обоих,
случаях рассеяние может быть описано выражениями G.7) — G.9) и G.13),
задавая амплитуду А, зависящую от характера электромагнитных волн. В
дальнейшем мы пренебрежем поверхностным рассеянием, так что / = 1т + /,-.
А. РАССЕЯНИЕ ВИДИМОГО СВЕТА
Для световых волн в видимой части спектра, где длина волны велика по
сравнению с молекулярными размерами, можно показать, что
A = fcVHH*> sin у, G.14)
где /г<инд-> — дипольный момент молекулы, индуцированный электромагнит-
ной волной, а у — угол между направлением индуцированного дипольного
§ 7. Рассеяние электромагнитных волн 687
момента и направлением распространения рассеянной волны. Для сферически
симметричных молекул направление индуцированного дипольного момента
совпадает с направлением электрического вектора в падающем световом пучке.
Рассмотрим полярную систему координат, направление полярной оси которой
совпадает с направлением электрического вектора в падающем световом пучке
(т. е. с направлением волнового вектора 1ц,). Тогда направление электрического
вектора в падающем световом пучке определяется полярным углом я/2 и ази-
мутальным углом ф0. Пусть направление распространения рассеянного пучка
(т. е. направление волнового вектора к) дается углами 0 и ф. Тогда угол у
выражается в виде
cos у = sin 0 cos (ф — фо). G.15)
Электрический вектор рассеянной волны перпендикулярен направлению
волнового вектора к и расположен в плоскости, в которой лежат волновой
вектор и вектор индуцированного дипольного момента.
Индуцированный дипольный момент |и<инд) просто связан с поляризуе-
мостью а молекулы и локальной напряженностью поля &(лок-):
|и(инд.) = а^(лок.) в G.16)
Локальная напряженность поля обсуждалась в §2, разд. В. С хорошим при-
ближением справедливо выражение вида
!* = ±а(е + 2)&, G.17)
где е — диэлектрическая постоянная среда, a g—электрический вектор па-
дающей волны. Таким образом, окончательно для амплитуды электрического
вектора рассеянной световой волны получаем
А = ±к*а(е + 2)% [1 - sin20cos2(^ - фо)]У>. G.18)
Плотность энергии рассеянного света в видимой области спектра дается
соотношением G.7), в которое должна быть подставлена амплитуда из выра-
жения G.18). Для света этих длин волн расстояние корреляции1) положений
молекул вещества мала по сравнению с длиной волны А. Следовательно, вели-
чина sr мала для всех значений г, при которых радиальная функция распре-
деления g(r) существенно отлична от единицы. Таким образом, величину sin (sr)
можно разложить в ряд и получить для плотности энергии рассеянного света
выражение в виде
00 00
j^^ ...]. G.19)
Сравнение выражений G.19) и F.31) показывает, что первые два члена тесно
связаны с флуктуациями плотности. В действительности в пределе для боль-
ших длин волн (s -* 0) высшие члены ряда равны нулю и интенсивность явно
связана с флуктуациями плотности. В этом приближении, введенном Релеем,
предполагается, что рассеяние происходит на группах, размеры которых малы
по сравнению с длиной волны. Добавочные члены этого ряда важны только в
случае малых длин волн (большие s) или если расстояние корреляции велико,
так что g(r) (радиальная функция распределения) медленно приближается к
единице.
г) Расстояние корреляции равно расстоянию между молекулами, на котором еще
заметна корреляция между их положениями. — Прим. ред.
688 Гл. 12. Электромагнитные основы теории межмолекулярных сил
Введем теперь параметр f с помощью соотношения вида
G.20)
Этот параметр является мерой расстояния корреляции пар молекул, т. е. мерой
приблизительной реличины имеющихся групп. С помощью этой величины и
выражения F.32) плотность энергии, даваемая выражением G.19), приводится
к виду
Тогда, используя выражение для амплитуды рассеянной волны G.18) и опре-
деление величины s G.12), это выражение можно переписать в виде
С помощью выражения E.5) соотношение G.22) можно записать через плотность
энергии в падающем пучке /0 = A/4я) | W |2, так что доля рассеянного света
равна
Если падающий свет поляризован, то рассеяние для определенных значений
0 максимально в плоскости ф = ф0 + (я/2), перпендикулярной направлению
электрического вектора падающего пучка. Для этой плоскости интенсивность
не зависит от полярного угла 0, за исключением членов второго порядка.
В плоскости электрического вектора ф = ф0, интенсивности для прямого @=0)
и обратного направлений @ = п) равны, а рассеяние в перпендикулярном
направлении (для 0 = я/2) равно нулю.
Интенсивность рассеянного света для неполяризованного пучка получим,
усреднив выражение G.23) по всем значениям углов ф0. В результате имеем
± _ 8я<_ N д«(« + 2)« п 2fll Г кТ
т;~"9 ж —а*—ll +cos ™ 1
Это распределение, очевидно, цилиндрически симметрично. Интенсивности
света, рассеянного в прямом и обратном направлениях, равны. При в = я/2
интенсивность равна половине величины интенсивности для 0 = 0 или 0 = п.
Голубой свет неба обусловлен флуктуациями плотности воздуха [41, 42].
Величина А4 в знаменателе выражения G.24) приводит к сильной зависимости
от длины волн, так что коротковолновая часть солнечного спектра рассеивается
сильнее. Красный цвет закатов и зори обусловлены тем, что длинноволновое
излучение относительно меньше рассеивается. Другим фактором, определяю-
щим цвет неба, является, конечно, рассеяние, обусловленное частицами пыли
и дыма [43].
Выражения для интенсивности рассеянного света G.23) и G.24) несправед-
ливы вблизи критической точки. Эти выражения предсказывают бесконечно
большое рассеяние в критической точке. Очевидно, это неправильно, хотя рас-
сеяние и становится большим. Орнштейн и Цернике [44]1) пытались видоизме-
См. также [37, 45 ].
§ 7. Рассеяние электромагнитных волн 689
нить вывод выражений для рассеяния, с тем чтобы получить выражение, спра-
ведливое в критической точке. Видоизменение, являющееся приближенным,
основано на представлении о корреляции флуктуации плотности в соседних
объемах. Интенсивность рассеянного света для неполяризованного пучка
можно выразить через величину а, интерпретируемую как средний диаметр
групп молекул или центров рассеяния :
/ -?- -S" °2 («+ 2)М1 + cos2 в)кТ
т = -^ г • G-25)
т г
0 А* (9/?/8л)г + 2яа <та еЯ2 кТ sin2 -|.
В состояниях, далеких от критической точки, это выражение сводится к G.24).
Вблизи критической точки [когда (Эр/9п)г = 0] из этой теории следует1), что
интенсивность рассеянного света изменяется пропорционально А~2, а не про-
порционально А. Выражение G.25) используется для получения приближен-
ных размеров групп молекул в области критической точки по данным экспери-
ментального наблюдения рассеяния света [47]. Размеры групп, полученные
этим методом, находятся в разумном согласии со значениями, получаемыми из
интерпретации результатов измерения вариаций плотности с высотой
(см. гл. 5, § 2). Трудность в этой теории связана с тем, что в критической точке
интеграл от интенсивности по всем телесным углам бесконечен. Более поздняя
теория Рокарда обходит эту трудность, однако в ней не содержится в явном
виде диаметр групп [48]. По-видимому, потребуется дальнейшая работа,
прежде чем будет создана удовлетворительная теория.
Б. РАССЕЯНИЕ РЕНТГЕНОВСКИХ ЛУЧЕЙ
Длина волны рентгеновских лучей имеет порядок молекулярных размеров
и поэтому каждый электрон рассеивает независимо от других. Можно показать,
что амплитуда А рассеянной отдельным электроном электромагнитной волны
выражается в виде
A = -m^ffsinV> <7-26)
где, как и выше, у — угол между электрическим вектором и направлением
распространения рассеянного пучка. Если электромагнитная волна рассеи-
вается атомом, обладающим несколькими электронами, то выражение для
амплитуды рассеянной волны усложняется вследствие интерференции волн,
рассеянных на различных электронах. Кроме того, в жидкости, состоящей из
многоатомных молекул, выражение для амплитуды рассеянной волны услож-
няется также вследствие интерференции волн, рассеянных на отдельных ато-
мах, образующих молекулу. Амплитуду А обычно нельзя получить теорети-
чески, однако она может быть вычислена из экспериментальных данных по
рассеянию рентгеновских лучей в разреженном газе.
Применим выражение G.7) к рассеянию рентгеновских лучей. Вместо
величины А мы будем пользоваться величиной /т, так как она определяется
из эксперимента. Тогда выражение G.7) для интенсивности излучения дает
G.27)
Рассмотрим теперь рассеяние неполяризованного пучка рентгеновских лучей.
Интенсивность рассеянного излучения зависит только от полярного угла 0,
который можно заменить параметром s, определяемым выражением G.12) и
г) Баттачария [46 ] нашел, что рассеяние всегда пропорционально А-1. Рокарду и Понту
[37] не удалось подтвердить закон А-2,
690
Гл. 72. Электромагнитные основы теории межмолекулярных сил
равным усредненному по азимутальным углам ф значению интенсивности
выражения G.27). Так как угол ф входит только в 1т, то результат имеет
аналогичный вид, где Im(s) зависит
только от усредненного по
ф значения величины | А |2.
Жидкий аргон
Кривая 7 54,4°К
Кривая 1 5/,8°К
Кривая 3 126,7°К
Кривая 4 144,1° К
Кривая 5 149,3°К
Фиг. 197. Экспериментальные кривые для ра-
диальной функции распределения g(r).
Кривые представляют зависимость от приведенного рас-
стояния г* — rja, где а = 3,42 А. Данные заимствованы
из работы [35] и основаны на результатах работы [57].
углам
Вели-
чина Im(s) является молекулярной
характеристикой и не зависит от
плотности. В соответствии с выра-
жением (9.2) гл. 4 радиальная функ-
ция распределения в предельном
случае низких плотностей стре-
мится к виду ехр(<р(г)//сГ). Следо-
вательно, в предельном случае низ-
ких плотностей I(s) линейно зависит
от плотности для всех значений s.
Иначе говоря, вблизи нулевой плот-
ности рассеяние будет исключитель-
но молекулярным явлением, а интер-
ференцией волн от различных мо-
лекул можно пренебречь. Этот факт
лежит в основе метода экспери-
ментального определения величины
/m(s). Величина Im(s) представляет
интерес и сама по себе, так как
она тесно связана с молекулярной
структурой.
Экспериментальные значения
величины Im(s) (интенсивности
рассеянного излучения), экстрапо-
лированной к нулевой плотности, и величины I(s) (то же при конечной
плотности) можно подставить в выражение G.27) для получения радиальной
функции распределения. Воспользовавшись преобразованием Фурье, выра-
жение G.27) приведем к виду
G.28)
Полученные таким путем экспериментальные кривые радиальной функции
распределения g(r) для жидкого аргона представлены на фиг. 197.
Приложение А
КОЭФФИЦИЕНТЫ ДВУХЦЕНТРОВОГО РАЗЛОЖЕНИЯ
Аналитические выражения коэффициентов разложения B\^b (rh г;; rab)
будут получены для четырех областей значений переменных гь г^ и гаЬ. Эти
четыре области представлены на фиг, 198. Для изучения взаимодействия зна-
чительно удаленных друг от друга распределений заряда необходимо рассмат-
ривать коэффициенты разложения в области I. Когда два распределения заряда
перекрываются, необходимо пользоваться коэффициентами разложения для
областей II, III и IV.
[7].
Приложение А.
691
III
Область I
Область II
Область III
Область IV
/V
Фиг. 198. Четыре области, использовавшиеся при нахождении коэффициентов
двухцентрового разложения.
Область 1 [raft> r( + rj\
па,пь v I > 'y > 'aft/ —
Область 11 [rab + rt
Область III [rab + гу
'ab
О
(А.1)
(А.2)
(пд -
Область IV [|г, - гу
(Па — ПЬ)\
0
г
ТаЪ
) —
/С,/
rk+l—na—nj—l
(А.З)
(А.4)
Значения коэффициентов A,{e%' и D,!^ для na, пь =0, 1, 2, 3 и соответ-
ствующие значения т приведены в табл. 109. Значения функций, для которых
па > пЬу не приводятся, так как они должны определяться путем перестановки
индексов, в соответствии с правилом
В\ т\ (г г • г \ — ( 1 \п»+пь R\ т I (г, г • г \ (А *Л
па,пъ У i 9'] у ' аЬ/ — \ */ °Пь,пЛ\г j > ' i y'ab/ * \™"^/
Для значений па или пь, не приведенных в табл. 109, может использоваться
следующая общая формула для коэффициента В]п%&г(, rj; rab) в области IV:
2т па— \т\ пь—\т\
u=O v«O w«O
; (А.6)
здесь / = и + v + w и
Таблица 109
Коэффициенты разложения для области IV
гЛ-Лв-1 т1—пь-\
"ПшП* аЪ
Замечание. Пользуясь этой формулой и таблицей, можно вычислить значения коэффициентов разложения
ВТ^лПь в области IV для значений индексов, меньших или равных 3. Суммирование ведется по всем значениям
таблицы. Максимальное значение величины {к + 1) — Bл« + щ + !)•
пь
Па
m
Dm
к 1
0 0
1
2
3
4
6
8
1 0
1
3
5
7
2 0
2
4
6
3 0
3
5
4 0
2
4
6 0
2
8 0
0
0
0
4
-1
2
1
0
0
0
0
2
2
0
0
0
-1
0
0
0
0
0
0
0
0
0
0
0
0
1
0
0
16
3
0
-6
0
3
0
0
о
0
-8
0
0
6
6
0
0
0
0
0
1
0
0
0
0
0
1
1
0
16
2
0
-9
8
0
-1
0
0
0
0
0
0
g
0
9
0
8
-16
0
0
9
0
-1
0
0
1
1
1
64
1
0
g
16
-9
1
0
0
0
0
0
0
-9
18
-9
0
16
16
0
-9
-9
0
Г
0
0
2
0
0
32
-5
0
5
0
5
-5
0
16
0
0
16
0
-15
-10
-15
0
0
0
0
5
5
0
-1
0
0
2
1
0
256
-25
0
60
0
-30
-20
15
0
0
0
0
0
180
-60
60
-180
-256
0
384
90
-60
-270
20
60
-9
2
1
1
512
-5
0
20
0
-30
20
-5
0
0
0
0
0
60
-60
-60
60
-128
0
-128
90
60
90
-20
-20
3
3
0
0
256
35
0
-28
0
-14
-28
35
-128
0
0
0
-128
140
84
84
140
0
0
0
-70
-84
-70
28
28
-5
692
Продолжение табл. 109
пь
Па
т
ипапь
к 1
2
2
0 .
256
2
2
1
1536
2
2
2
12 288
3
1
0
256
Ат
3
1
1
1024
Лк>1)
3
2
0
3072
3
2
1
12 288
3
2
2
49152
0 0
2
4
5
6
8
10
12
2 0
2
4
6
8
10
3 0
7
4 0
2
4
6
8
5 0
5
7
6 0
2
4
6
8 0
2
4
10 0
2
12 0
-19
75
-150
128
-50
25
-9
0
75
-100
50
-100
75
0
0
0
-150
50
150
-450
0
128
768
0
-50
-100
-450
0
25
75
0
-9
0
0
-9
50
-150
128
0
-25
. 6
0
50
-100
0
100
-50
0
0
0
-150
0
-150
300
0
128
-512
0
0
100
300
0
-25
-50
0
6
0
0
-3
25
—150
256
-150
25
_з
0
25
-100
150
-100
25
0
0
0
-150
150
150
-150
0
256
256
0
-150
-100
-150
0
25
25
0
-3
0
0
21
-42
14
0
0
21
-14
0
-210
84
0
-84
210
0
384
-512
-210
0
126
420
0
0
0
0
0
-84
-140
0
21
42
0
-6
0
N 0
7
-21
14
0
14
-21
7
0
-105
84
42
84
-105
0
256
256
-210
-126
-126
-210
0
0
0
0
70
84
70
0
-21
-21
0
3
0
0
189
-630
735
0
-420
315
-294
105
-1050
1470
-420
-420
1470
-1050
0
0
3675
-1260
-630
-2940
7875
-4608
0
-15360
2100
1260
2940
10500
-525
-1470
-2625
294
630
-75
49
-210
315
0
-140
-105
126
-35
-350
630
-140
140
-630
350
0
0
1575
-420
210
1260
-2625
-2048
0
5120
700
-420
-1260
-3500
175
630
875
-126
-210
25
7
-42
105
0
-140
105
-42
7
-70
210
-140
-140
210
-70
0
0
525
-420
-210
-420
525
-1024
0
-1024
700
420
420
700
-175
-210
-175
42
42
-5
693
Продолжение табл. 70
пь
Па
т
Dm
к 1
0 0
2
4
6
7
8
10
12
14
2 0
2
4
6
8
10
12
4 0
2
4
6
8
10
6 0
2
4
6
8
7 0
7
8 0
2
4
6
10 0
2
4
12 0
2
3
3
0
10 240
3
3
1
49152
3
3
2
245 760
3
3
3
2 949 120
520
-2450
4900
-7350
5120
0
-1470
980
-250
-2450
5880
-4410
0
4410
—5880
2450
4900
-4410
0
-2940
14 700
-12 250
-7350
0
-2940
-19 600
61250
5120
-102400
0
4410
14 700
61250
-1470
-5880
-12 250
980
2450
131
-735
1715
-2695
2048
-735
539
—343
75
-735
2058
-1617
588
-1617
2058
-735
1715
-1617
294
1078
-5145
3675
-2695
588
1078
6860
-18 375
2048
30 720
-735
-1617
-5145
-18 375
539
2058
3675
-343
-735
20
-147
490
-1225
1024
0
-245
98
-15
-147
588
-735
0
735
—588
147
490
-735
0
-490
1470
-735
-1225
0
-490
-1960
3675
1024
-6144
0
735
1470
3675
-245
-588
-735
98
147
5
-49
245
-1225
2048
-1225
-245
-49
5
-49
294
-735
980
-735
294
-49
245
-735
490
490
-735
245
-1225
980
490
980
-1225
2048
2048
-1225
-735
-735
-1225
245
294
245
-49
-49
14
-250
75
-15
694
Приложение Б 695
| /л |)! (п» + | m
1 Г, _ fi?»±
2t-2na+lll [ г,
fi - f
«(|/n|)l W (-l)*Bfc)l
u! -4 А! BЛ-и)! (т - Л)! I 2re»ry J '
x
= у или iL±i, (A.9)
- 1)п.-|т|-„-р B |щ| + 2v + 2р)\ [{rjb -
= па — \т\ — 2v, когда v<V2(na — 1Ш I) ИЛИ 1k(na — \т\ — О> или же рх =
U/|ml Г^ = {~1Г V
Л (nb-q)\(nb-\m\—2q-w)\q\
q=° (AM)
^^VjK-I/hI-w) или 1l2(nb-\m\-w-l).
Индексы суммирования предполагают только интегральные величины, так
что выбор kD рх и q2 однозначен. Нахождение коэффициентов A{n%l(k,t) и
Dn?nl в явном виде связано с дальнейшим разложением Т, С/, V и W'.
Приложение Б
КОЭФФИЦИЕНТЫ ПРЕДСТАВЛЕНИЯ ТРЕХМЕРНОЙ
ГРУППЫ ВРАЩЕНИЯ
В этом приложении мы рассмотрим явный вид и некоторые свойства коэф-
фициентов представления трехмерной группы вращения. Так как коэффициенты
представления тесно связаны со сферическими функциями, то следует начать
это обсуждение с точного определения сферических функций, используемых
в настоящей книге.
Обычные полиномы Лежандра определяются выражением
для любых целых чисел /1>0, югда как присоединенные функции Лежандра
определяются выражением1)
~Р,(х) (Б.2)
для любых целых чисел т.
х) Обозначения, применяемые здесь, совпадают с обозначениями, принятыми в работе
[49]. Другие авторы пользуются другими обозначениями.
696 Гл. 12. Электромагнитные основы теории межмолекулярных сил
Из этих выражений следует, что функции тождественно равны нулю
для всех значений т, не удовлетворяющих неравенству — l^^1
Несколько первых полиномов имеют вид
= 1, РЦх) = 15A -х*)х,
= х, Pf(x)=15(l-x2L
Pi (х) = A - х2I/', ^ ^ = р^ ^ =_1_ C5 х* _ 30 ха + 3),
Р\ (х) = 4 A - *2I/s G х3 - Зх),
Р|(х) = 3A-х2), Р42(х)=-?A-х2)Gх2-1),
ро(х) = Р3(х) = 4"Eх8 - Зх), Р!(х) = 105A - х2)''«х,
Р\ (х) = -|- A - х2I^ Eх2 - 1), Р|(х) = 105 A - х2J.
Нормированные сферические функции выражаются через приведенные выше
функции
Y? (в, ф) = i""'-
Эти функции ортонормированы в том смысле, что имеет место выражение вида
J J УГ (^ Л* У?' (б, Л sin 0d6<# = ди. 6тт.. (Б.4)
о о
Знак нормировочного множителя, используемый здесь, следующий:
У? (в, Ф) = (- 1)т УТт (О, ФУ ¦ (Б.5)
При таком определенном выборе знака нормировочного множителя сфери-
ческие функции выражаются рекуррентными соотношениями:
УA + т+1)A-т) УГ1 F,0) = [т ctg в - ±] Y?(в,0), (Б.б)
УA + т)A-т+1)У?-1 (в,0) = [т ctg в + -?\ Yf (в,0), (Б.7)
2Г+1 cos в Yf (в, 0) = y<'-g>g + m> YfLi(в,0) +
«+ij Kjni@>O)) (Б.8)
/27ТТ sin 0 К? @,0) = y<f + "»-_M + w> ym-! @> 0) _
w + g уд-»@>0)$
sin в Yf (в, 0) = - y</-"Otfj«-*) к«+х(в, О)
Приложение Б
697
Рассмотрим две декартовые системы координат с
щающиеся одна относительно другой. Относительная
координат может быть описана с помощью эйлеровых
Будем обозначать координаты первоначальной
сверху, а новой системы — символами с черточками,
можно описать, рассматривая следующий
набор вращений. Первоначальная сис-
тема координат поворачивается на угол
а вокруг оси z в направлении, противо-
положном часовой стрелке (таким обра-
зом, что ось х движется в сторону оси у),
затем система координат наклоняется на
угол /? относительно новой оси у' так, что
ось z движется в новой плоскости xz в
направлении, соответствующем положи-
тельному направлению оси х. Теперь сис-
тема координат поворачивается на угол
у относительно новой оси z в направ-
лении против часовой стрелки. Оконча-
тельное положение системы координат
соответствует осям с подчеркнутыми
сверху символами.
Расположение системы координат
с подчеркнутыми сверху символами,
повернутой на углы а, (}, у относительно
положения, соответствующего непод-
черкнутым символам, представлено на
фиг. 199.
Пусть 0 и ф — полярные коорди-
наты точки на единичной сфере в старой
системе координат, а в и ф — в новой
системе.
Тогда можно показать, что
общим началом, но вра-
ориентация двух систем
углов1) а, 0, у.
системы без черточек
Смысл эйлеровых углов
Фиг. 199.
Описание эйлеровых углов
а, Р и у.
хУ?(в,ф). (Б.11)
m
Коэффициенты разложения Dt(R)mn зависят от вращения R = (а, /?, у),
которое переводит неподчеркнутые координаты в подчеркнутые. Коэффициенты
Dl(R)mn при фиксированном значении / образуют B1 + 1)-мерную матрицу.
Можно показать, что матрицы мультипликативны так же, как и сами вра-
щения, т. е.
2d Ч \*\)тп^ \Р)пт' — *** \К^)тт' у yD.lZ)
п
где RS — общий результат вращения S, за которым следует вращение R.
Таким образом, матрицы образуют представление трехмерной группы вра-
щения [52]. Сферические функции образуют базисное представление. Коэф-
фициенты представления имеют вид
где Z — положительные целые числа или нуль. Величины dl((})nm, за исклю-
чением нормировочного множителя, являются полиномами Якоби с аргумен-
х) Углы a, ft, у часто обозначают 0, в и % (см., например, [51]). Углы в и 0 — это обыч-
ные углы, используемые в сферической системе координат для задания направления новой
оси г. Угол х является мерой вращения системы координат или объекта относительно новой
оси г.
698 Гл. 12. Электромагнитные основы теории межмолекулярных сил
том х = cos /?. Явное выражение для d'(/3)nm имеет вид при п > 0, т > О
Выражения для отрицательных значений индексов даются соотношениями
л>0, т>0, (Б.15)
п>0, т>0, (Б. 16)
, /п>0. (Б.17)
Величины d'(/S)nm могут быть записаны1) несколькими эквивалентными
выражениями (дли всех пат)
-(- IV"]l(/-n)\(f-m)\ х
л)!(/+ т)!
n)\ (/ - яI {f + т) I (/ - т)!
х cos2/-n+m-2s-|-sin"-m+2s4-- (БЛ9)
Коэффициенты представления ортогональны в том смысле, что
J Dl (#)*m & (R)n.m. sin fidadfidy = д1Г дпп, дтт, J*^-. (Б.20)
Из вида выражений для коэффициентов представления (Б. 14) и опреде-
ления сферических функций непосредственно следует
= f-jj^j- ^Г (/5, «).
j- ^Г (/5, «). (Б.22)
Произведение двух коэффициентов представления может быть выражено
через суммы коэффициентов представления
D'(R)nm D1' (R)n.m. = 2 s'Un- s'Um- DL (R)n+n., m+m-. (Б.23)
L,
Явное выражение для коэффициентов $?„,„', известных как коэффициенты
Вигнера или Клебша—Жордана, было дано Вигнером2),а таблицы значений
*) Коэффициенты представления определяются свойствами группы вращения только в
пределах преобразования подобия. Коэффициенты, однако, полностью определяются опре-
делением базиса, т. е. нормировкой сферических функций. Коэффициенты dl(f$)nm, введенные
здесь, отличаются от аналогичных величин Вигнера [52] множителем (—1)п+т. Однако
сравнивать коэффициенты представления Dl(R)nm трудно вследствие отличия в определении
эйлеровых углов, что ведет к различиям в параметризации группы. Наши выражения для
коэффициентов представления совпадают с выражениями, используемыми Маргенау и
Мэрфи [49].
2) Коэффициенты s^'n^' обозначаются Кондоном и Шортли [25] в виде
(/, /', л, п' | /, /', L, п + п')- Коэффициенты Рака, связанные с коэффициентами Вигнера, обсуж-
даются в работе [53]. Отличие наших выражений для коэффициентов представления от
коэффициентов Вигнера (см. предыдущее примечание) не отражается на значениях s^>ntfl/.
Приложение Б
699
этих коэффициентов для случая /', равного 1 или 2, приводятся Вигнером [52 ]
и Кондоном и Шортли [25].
Коэффициенты $?„,„/ обладают следующим свойством симметрии :
bL,n\n —
2ZT+1
и удовлетворяют соотношениям ортогональности
bL' — °LL'
(Б.25)
(Б.26)
L
Воспользовавшись предыдущими выражениями, можно легко показать,
что трехмерное представление (I = 1) имеет вид
— е-*
2 е
cosp)e~ia -^-^-/ysini
cosp
— cos
Oia
4r-eiy A — cos P) е~(а —-^
Базисное представление имеет вид
cosp)eia
. (Б.27)
(Б.28)
(Б.29)
(Б.ЗО)
Выражение (Б.27) отличается от матрицы вращения R преобразованием подо-
бия, базисом которого являются х/г, у/г и г/г.
Можно использовать специальный случай соотношения (Б.23), когда
V = 1, и получить обобщенные рекуррентные выражения для соотношений
(Б.8) — (Б. 10), которые могут применяться для коэффициентов предствления
+ ") (* — "О (/+ ffl) nl-i(O\
1B1+ 1) \К)п
nm
ГI (f?\ -4-
sin
sin
- -I-
—I
- n+ l)(l + n+ l)(l- m+ 1){1 - m + 2)
sin
/B/+1)
/(/+ 1)
T
-n+
m+
m + 2)
(/ + 1) {21 + 1)
1(R)n>m+i* (Б.ЗЗ)
700
Гл. 72. Электромагнитные основы теории межмолекулярных сил
Приложение В
МАТРИЧНЫЕ КОМПОНЕНТЫ ДИПОЛЬНОГО МОМЕНТА
ДЛЯ ОПТИЧЕСКИХ ПЕРЕХОДОВ
Состояние атома водорода характеризуется четырьмя квантовыми числами :
п — главное квантовое число ; I — азимутальное квантовое число ; т1 (иногда
просто т без индекса)—магнитное квантовое число или проекция орбитального
момента количества движения на ось z; ms — спиновое квантовое число.
Состояния с различными азимутальными квантовыми числами имеют специаль-
ные обозначения : состояние с / = О называют s-ссстоянием ; состояние с
/ = 1 называют р-состоянием; состояние с / = 2 называют d-состоянием ;
состояние с / = 3 называют /-состоянием. Пространственная часть волновой
функции атома водорода может быть записана в виде1)
= /?„,/(г)
(В.1)
здесь У7@, ф) — нормированные сферические функции, определенные выра-
жением (Б.З), a Rn,i(r) — нормированные радиальные функции распределения,
выражаемые через присоединенные функции Лягерра.
Таблица 110
Значения величины G(n, I; n\l— 1) для атома водорода
Is
2s
3s
4s
5s
2P
3p
4/>
bp
3d
4d
5d
2p
1,66479
27,00
0,88
0,15
0,052
3p
6,267
9,18
162,0
6,0
0,9
3d
22,52
101,2
1J
0,23
4/>
0,093
1,64
29,9
540,0
21,2
Ad
2,92
57,2
432,0
4/
104,6
252,0
2,75
5p
0,044
0,60
5,1
72,6
1125,0
5d
0,95
8,8
121,9
1181,25
5/
n,o
197,8
900,0
Воспользовавшись выражениями (Б.8) — (Б. 10), можно показать, что
квадраты абсолютных величин компонент матрицы дипольного момента [опре-
деляемых выражением F.7) ] для оптических переходов атома водорода выра-
жаются в виде
/, /п | ^ | п% / -
тj G (п,
(В.2)
[51, стр. 132].
Приложение В
701
(л, /, т | fix | л', / - 1, mOI2 = \(п, I, т | /*у | п', I - 1, m')l2 =
(/ - m - 1) (/ - m) + EOT_ljm. (/ + /п - 1) (I + m)] x
(В.З)
здесь функции G(n, I; n',1 — 1) интегралы1) вида
л',/- l) =
(B.4)
Значения G(n, 1; n\ I — 1) приведены в табл. 110. Видно, что во всех слу-
чаях наибольшие значения имеют место при п = п' и значения становятся
очень малыми, если \ п — п' \ будет большим.
Значения компонент матрицы дипольного момента для ряда различных
элементов приводятся в табл. 111 [55]. Основным уровнем для всех элементов
в этой таблице является S-состояние. Оптические переходы, для которых
Таблица 111
Дипольные моменты перехода и значения силы осциллятора [55]
Атом
Н
Li
Na
К
Rb
Энергия
перехода
S-+P
0,7500
0,1358
0,1546
0,1184
0,1154
Сила осцил-
лятора f8P
0,416
0,7500
0,9755
-0,98
—0,98
\\иг)8Р\*
* 0,555
5,52
6,31
8,28
8,47
Атом
Cs
Zn
Cd
Hg
Энергия
перехода
S-+P
' 0,1019
0,4261
0,3981
0,4926
Сила осцил-
лятора fSp
0,98
-1,2
1,20
1,19
\\1*м)8Р\
9,62
2,82
3,01
2,42
приведено значение дипольного момента, являются переходами из основного
состояния в первое возбужденное Р-состояние. Приводятся также значения
энергии и силы осциллятора для этого перехода.
Под величиной | (pz)Sp |2 подразумевается
1
2
Сила осциллятора
Ep-Es \Q*z)sp\*
(В.5)
(В.б)
Значения дипольных моментов для переходов из основного ^-состояния
гелия в конфигурации гР, (Is) (np) приведены в табл. 112.
х) Бете [54] приводит весь ряд формул для G(n, I; п',1—1), численные значения
G(n, I; п'}1 — 1) и силы осцилляторов для всех переходов, включая переходы в непрерывные
энергетические состояния. Кондон и Шортли [25] приводят формулы и численные значения
G(n,l; n\l—1) для большого числа переходов. Заметим, что величина R(n, [) у Бете,
а также у Кондона и Шортли для обозначений, используемых в этой книге и Полингом и
Вильсоном [51 ], равна rR(n, /).
702
Гл. 12. Электромагнитные основы теории межмолекулярных сил
Таблица 112
Дипольные моменты для переходов из
основного состояния гелия [56]
п
2
3
4
0,224
0,0547
0,0204
n
5
6
7
0,0100
0,00588
0,00351
Полная сила осциллятора / для перехода из основного состояния в конти-
нуум равна для водорода 0,437, для гелия 1,58, для лития 0,24 и для натрия
ОДШ1).
Задачи
1. Рассмотреть две молекулы воды, центры тяжести которых удалены на 4А. Найти
величину энергии и относительную ориентацию, соответствующую минимальной энергии.
Использовать представленную на фиг. 220 (стр. 788) и табл. 136 (стр. 789) модель молекулы
воды Попла.
2. Показать, что для любых распределений заряда взаимодействие двух неперекры-
вающихся распределений заряда не может быть записано в виде <р = be-ar.
3. Вычислить коэффициент преломления как функцию частоты для атома водорода в
основном состоянии, а также величину поляризуемости, экстраполированной к нулевой
частоте. Значение силы осциллятора приведены в табл. 110.
ЛИТЕРАТУРА
1. Ramsey N. F., Phys. Rev., 78, 221 A950).
2. Green how С, Smith W. V., Journ. Chem. Phys., 19, 1298 A951).
3. S1 a t e r J., Frank N., Electromagnetism, New York, 1947.
4. Eyring R, Walter J., Kim ball G. E., Quantum Chemistry, New York, 1944
(см. перевод : Эйринг Г., Уолтер Дж., К и м б а л л Дж., Квантовая химия,
ИЛ, 1948).
5. Mac Robert Т. М., Spherical Harmonics, New York, 1927.
6. Rude n berg K., Journ. Chem. Phys., 19, 1459 A951).
7. В u e h 1 e r R. J., H i r s с h f e 1 d e r J. 0., Phys. Rev., 83, 628 A951); 85, 149 A952).
8. С a r 1 s о n B. C, R u s h b г о о k e G. S., Proc. Cambr. Phil. Soc, 46, 626 A950).
9. F г б 1 i с h H., Theory of Dielectrics, Oxford, 1950.
10. Page L., Introduction to Theoretical Physics, New York, 1935.
11. Jaf f e G., Journ. Chem. Phys., 8, 879 A940).
12. Booth F., Journ. Chem. Phys., 19, 391 A951).
13. О n s a g e r L., Journ. Am. Chem. Soc, 58, I486 A936).
14. К irk wood J., Journ. Chem. Phys., 7, 911 A936).
15. А н с е л ь м А., ЖЭТФ, 14, 364 A945).
16. R u s h b г о о k e G. S., Introduction to Statistical Mechanics, Oxford, 1949.
17. V a n V1 e с k J. H., The Theory of Electric and Magnetic Susceptibilities, Oxford, 1932.
18. Frank P., von Mises R., Riemann-Webers Differentialgleichungen in der Physik,
Braunschweig, 1927, 7 Aufl., Bd. II, Teil IV (A. Sommerfeld).
19. Френкель Я., Lehrbuch der Elektrodynamik, Berlin, Bd. II, 1928.
20. S t о n e r E. C, Magnetism, London, 1946.
21. Abraham M., Becker R., Electricity and Magnetism, London, 1947.
22. J о о s G., Theoretical Physics, New York, 1934.
См. работу E i s e n s t e i n A.; Gingrich N. S., Phys. Rev., 62, 261 A942). — Прим. ред.
Литература 703
23. В о г n M., Optik, London, 1943.
24. D r u d e P. К. L., The Theory of Optics, London, 1933.
25. С о n d о n E. U., S h о r 11 e у G., Theory of Atomic Spectra, Cambridge, 1935 (см.
перевод: Кон дон Е., Шорт л и Г., Теория атомных спектров, ИЛ, 1949).
26. С о n d о n E. U., Phys. Rev., 41, 759 A932).
27. Grawford M. F., Dagg I. R., Phys. Rev., 91, 1569 A953).
28. van Kranendonk J., Bird R. В., Physica, 17, 953, 968 A951).
29. van Kranendonk J., Диссертация, Amsterdam, 1952.
30. К e m b 1 e E. C, Fundamental Principles of Quantum Mechanics, New York, 1937.
31. Kramers H. A., Nature, 113, 673 A924); 114, 310 A924).
32. Kuhn W., Zs. Phys., 33, 408 A925).
33. L e F 6 v r e R. J. W., Dipole Moments, London, 2nd Ed., 1948.
34. Wheeler J. A., Phys. Rev., 43, 258 A933).
35. d e Boer J., Rep. Progr. Phys., 12, 305 A949).
36. Lord R а у 1 e i g h, Phil. Mag., 41, 447 A871).
37. С a b a n n e s J., Diffusion moleculaire de la lumiere, Paris, 1929.
38. Oster G., Chem. Rev., 43, 319 A948).
39. Chandrasekhar S., Radiative Transfer, Oxford, 1950.
40. Brillouin L., Journ. Appl. Phys. 20, 1110 A949).
41. Lord Rayleigh, Collected Works.
42. Chandrasekhar S., Elbert D., Nature, 167, 51 A951).
43. К u i p e r G. P., Atmosphere of Planets, Chicago, 1948.
44. Ornstein L. S., Zernike F., Proc. Acad. Sci., Amsterdam, 17, 793 A914); Phys.
Zs., 27, 761 A926).
45. R о s e n f e 1 d L., Theory of Electrons, Amsterdam, 1951.
46. Bhattacharyya D. K., Proc. Indian Assoc. Cultural Sci., 8 A923).
47. Cataldi H., Drickamer H. G., Journ. Chem. Phys., 18, 650 A950).
48. R о с a r d Y., Compt. rend. 195, 771 A932); Journ. de phys. et le Radium, 4,165 A933).
49. M a r g e n a u H., Murphy G. M., The Mathematics of Physics and Chemistry,
New York, 1943.
50. J a h n k e E., E m d e F., Tables of Functions, 1945 (см. перевод : Янке Е,
Эмде Ф., Таблицы функций, М.—Л., 1948).
51. Pauling L., Wilson E. В., Jr., Introduction to Quantum Mechanics, New York, 1935.
52. W i g n e r E. P., Gruppentheorie und ihre Anwendung auf die Quantenmechanik der
Atomspektren, London, 1944.
53. В i e d e n h а г n L. G., В 1 a 11 J. M., Rose M. E., Rev. Mod. Phys., 24, 249 A952).
54. В e t h e H. A., Handbuch der Physik, XXIV/1, Berlin, 1933, S, 440.
55. King G. W., Van Vleck J. H., Phys. Rev., 55, 1165 A939).
56. Vinti J. P., Phys. Rev., 42, 632 A932).
ГЛАВА 13
ТЕОРИЯ МЕЖМОЛЕКУЛЯНЫХ СИЛ
Большая часть настоящей книги посвящена вычислению свойств газов и
жидкостей. Было показано, что статистическая механика дает способ получе-
ния теоретических выражений для различных равновесных и неравновесных
свойств веществ через по*енциальную энергию взаимодействия пары молекул.
Для практических расчетов на базе этих теоретических выводов необходимо
сделать предположение об аналитической форме потенциала взаимодействия.
Сводка различных эмпирических потенциальных функций взаимодействия,
используемых в настоящей книге, дана в гл. 1, § 3. Целью этой и следующей
главы являетря обсуждение состояния наших теоретических знаний о меж-
молекулярных силах1), которые образуют основу для этих идеализированных
моделей.
До сих пор предполагалось, что энергию взаимодействия всегда возможно
представить как функцию межмолекулярного расстояния и взаимной ориента-
ции взаимодействующей пары. В § 1 этой главы мы обсудим применимость
концепции межмолекулярного потенциала с помощью подробного анализа
процесса столкновения молекул. Показано, что концепция функции потен-
циальной энергии полезна в пределе, когда две молекулы сближаются мед-
ленно. Эта концепция несправедлива, если энергия взаимодействия на рассто-
янии одной волны де-Бройля [1,2] изменяется на величину, сравнимую с
расстоянием между соседними поверхностями потенциальной энергии. Напри-
мер, быстрые столкновения, встречающиеся в химии горячих атомов или в
молекулярных пучках, не могут быть поняты с помощью одной функции потен-
циальной энергии. Помимо этого, имеется несколько особых случаев, в кото-
рых две поверхности потенциальной энергии очень близко подходят одна к
другой, так что даже тепловые столкновения не следуют одной кривой потен-
циальной энергии.
Межмолекулярные силы удобно—хотя это и искусственный прием— под-
разделить на два типа : дальнодействующие (ван-дер-ваальсовы) силы и корот-
кодействующие (валентные или химические) силы. Эта глава в основном посвя-
щена* общему теоретическому рассмотрению дальнодействующих сил. Эти силы
могут быть строго описаны через физические свойства отдельных молекул.
Строгая трактовка короткодействующих сил не может быть дана с помощью
свойств отдельных молекул. Здесь необходимо рассматривать каждую пару
мслекул как особый случай. Вычисление межмолекулярных, сил для значи-
тельного числа различных молекулярных пар обсуждается в гл. 14. В этих
последних двух главах очень мало говорится о магнитных взаимодействиях
между молекулами, обладающими магнитными моментами, например о свобод-
ных радикалах и молекулах в возбужденных состояниях. При изучении столк-
новений таких молекул в гамильтониане, описывающем молекулярное взаи-
модействие, необходимо учитывать дополнительные члены. Вид гамильтониана,
в который включены эти магнитные взаимодействия, обсуждается в прило-
жении А (стр. 798).
При изучении дальнодействующих сил обычно рассматривают четыре типа
сил : 1) электростатические силы ; 2) индукционные силы ; 3) дисперсионные
Введение в теорию межмолекулярных сил дано в гл. 1, § 3.
Гл. 13. Теория межмолекулярных сил 705
силы (силы Лондона); 4) резонансные силы. Силы, действующие между двумя
неполярными молекулами, которые не имеют результирующего момента коли-
чества движения, являются силами дисперсионного типа. При взаимодействии
неполярной молекулы с полярной молекулой между молекулами действуют
дисперсионные и индукционные силы. При столкновении двух полярных моле-
кул вступают также в действие электростатические силы. Резонансные силы
действуют между двумя одинаковыми молекулами, когда квантовые числа
двух молекул таковы, что оптические правила отбора разрешают свободный
обмен фотонами между молекулами. Электростатические и индукционные силы
могут быть поняты с помощью классической теории. Напротив, дисперсионные
силы Лондона и резонансные силы по своему происхождению являются строго
квантовэмеханическими.
Подробное рассмотрение дисперсионных сил проведено в § 3 и 4, резонанс-
ных сил — в § 6. С точки зрения теории возмущений, химические (коротко-
действующие), электростатические, индукционные и резонансные силы все
вместе появляются в выражении для энергии как члены возмущения первого
порядка. Дисперсионные же силы появляются в выражении для энергии как
члены возмущения второго порядка. В вариационной трактовке все эффекты
объединяются при условии, что пробная волновая функция является доста-
точно подходящей. Таким образом, различие между разными типами сил до
некоторой степени фиктивное и в действительности приводит к серьезным
трудностям, когда мы пытаемся получить общее выражение для силы, комби-
нируя короткодействующие и дальнодействующие силы, как это делается в
гл. 14.
Дальнодействующие силы, действующие между асимметричными неполяр-
ными молекулами, рассмотрены в § 4. Когда молекулы находятся на расстоянии
в пределах двух диаметров столкновения, межмолекулярные силы становятся
существенно отличными от сил, действующих между сферическими молекулами.
Энергия не изменяется более как 1/г?& и при некоторых ориентациях она имеет
максимум или минимум по трудно устанавливаемым причинам. Для выяснения
природы таких сил необходимо проделать значительную работу.
К настоящему времени роль диполей в межмолекулярных силах понята
довольно хорошо. Однако роли квадруполей до недавнего времени уделялось
значительно меньшее внимание. Причина этого заключалась в том, что только
в течение последних нескольких лет в распоряжении экспериментаторов появи-
лись хорошие способы (микроволновые спектры) для измерения квадрупольных
моментов. К тому же квантовая механика молекул только теперь раз-
вилась настолько, что стало возможным вычислять квадрупольные моменты
теоретически (см. § 8 этой главы и гл. 14, § 4). Взаимодействие между
электрическими мультиполями обсуждается в § 5 и 6. Использование микро-
волновых спектров для получения важных сведений о молекулах описано в § 7.
Определение межмолекулярных сил из свойств кристаллов при темпера-
турах, близких к абсолютному нулю, обсуждается в § 9. Тип кристаллической
решетки, расстояние между молекулами, теплота сублимации, теплоемкость
или дебаевская характеристическая температура могут быть найдены путем
использования любого предполагаемого вида потенциальной энергии взаимодей-
ствия между парами молекул. Согласие между экспериментальными и вычисля-
емыми величинами является чувствительным методом апробации потенциальной
функции. До настоящего времени эти свойства кристаллов широко использо-
вались для определения сил, действующих между молекулами благородных
газов. Однако при изучении сил, действующих между сложными многоатом-
ными молекулами, свойства кристаллов могут оказаться даже более полезными.
Результаты этой главы дают теоретическую основу для некоторых эмпири-
ческих функций потенциальной энергии, которые использовались в связи с
уравнением состояния или в связи с вычислением свойств переноса. Многие
результаты, данные здесь, слишком сложны для использования в предшествую-
щих выводах. Однако они будут необходимы, когда мы будем больше знать
706 Гл. 13. Теория межмолекулярных сил
об отношении макроскопических свойств вещества к свойствам составляющих
его молекул. Например, при вычислении свойств переноса веществ, состоящих
из многоатомных молекул (см. гл. 7, § 6), величинами, которые должны быть
определены, являются дифференциальные эффективные сечения столкновений Щ.
Эти величины хорошо описывают как рассеяние, так и изменение моле-
кулярных квантовых чисел при столкновении. К сожалению, пока нет случаев,
где бы эти важные величины были определены. Трудности, встречающиеся
при их вычислении, обсуждены в § б в связи с описанием взаимодействия между
двумя симметричными полярными молекулами.
§ 1. ФУНКЦИИ ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ МЕЖМОЛЕКУЛЯРНОГО
ВЗАИМОДЕЙСТВИЯ
Этот параграф мы начнем с качественного обсуждения применимости и
ограничений концепции функции потенциальной энергии межмолекулярного
взаимодействия. Затем мы перейдем к формулировке проблемы молекулярных
взаимодействий на языке квантовой механики и обсудим приближекия, которые
вводятся при разделении электронного и ядерного движений пары сталкиваю-
щихся молекул. Это позволит яснее понять обоснованность концепции
функции потенциальной энергии межмолекулярного взаимодействия. Далее
будет показано, что теорема вириала дает важные сведения о природе как меж-
молекулярных, так и внутримолекулярных сил. Затем мы обсудим важную
теорему квантовой механики, из которой следует, что сила, действующая на
любое ядро в системе ядер и электронов, является в точности классическим
элеюростатическим воздействием на рассматриваемое ядро со стороны других
ядер и распределения электронных зарядов, вычисляемого из уравнения Шре-
дингера. Наконец, будет показано, каким образом с помощью квантовой меха-
ники может быть вычислена функция потенциальной энергии межмолекуляр-
ного взаимодействия.
А. КОНЦЕПЦИЯ ФУНКЦИИ ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
В гл. 12, § 1 были подробно обсуждены электростатические силы, действую-
щие между рядом зарядов или распределений зарядов. В настоящем параграфе
мы намерены продолжить это обсуждение и рассмотреть далее специальный
случай взаимодействия двух молекул. С этой целью мы рассмотрим молекулы
а и Ьу которые вместе состоят из v ядер и п электронов. Каждый электрон
несет заряд—е, а каждое ядро несет заряд Zae. Конфигурацию ядер и электро-
нов символически запишем в виде
г" = г 1, т2У... , га,.. • , г„ и г" = тъ г2,... , г,-,... гл
соответственно. Обозначения г* и тп* используем для записи конфигурации va
ядер и па электронов молекулы а и аналогичные обозначения — для молекулы Ъ
(греческие индексы для ядер и латинские для электронов).
В формулировке проблемы взаимодействия двух молекул в результате
разделения различных видов движения возникает несколько разных видов
потенциальных функций. Прежде всего мы запишем полную потенциальную
энергию системы ядер и электронов, которую мы будем использовать в гамиль-
тониане, описывающем полную систему. Второй вид возникает при выделении
электронных степеней свободы с целью получения потенциальной энергии для
движений ядер. В большинстве задач маловероятно изменение колебательных
§ 1. Функции потенциальной энергии межмолекулярного взаимодействия 707
квантовых чисел при столкновении [3—7 ]3) и поэтому удобно выделить коле-
бательный вид движения, чтобы образовать третий вид потенциала, который
является функцией только межмолекулярного расстояния и относительной
ориентации молекул. Четвертый вид образуется в результате выделения как
вращательного вида движения, так и колебательного. Однако практически это
бесполезно, так как изменение вращательных квантовых чисел при столкно-
вении очень вероятно. Наиболее распространенными формами потенциальной
энергии являются: второй вид для взаимодействия атомов и третий вид для
взаилодействия молекул. Чтобы показать соотношение между различными
видами и уточнить используемые обозначения,' мы даем следующую сводку.
1. Электронное и ядерное движения не разделяются. Пара молекул может
рассматриваться как система ядер и электронов, взаимодействующих по куло-
новскому закону сил. В соответствии с выражением A.1) гл. 12 общая потен-
циальная энергия системы равна
^±^^ ±1^' (l.i)
(Индекс е добавлен, чтобы отметить, что этот потенциал именно тот, который
используется для получения электронной волновой функции в разд. Б.) Эта
потеадытькдя функцич обращается в нуль, когда расстояния между всеми
ядрами и электронами велики. Когда рассматривается система из двух молекул
(двух раздельных наборов зарядов), удобно, как указано в соотношении A.3)
гл. 12, разделить общую потенциальную энергию на три части : собственную
энергию молекулы а, Фа(г'«, гп«), собственную энергию молекулы Ь, Ф6(Г», гл»)
и энергию взаимодействия cpab(r\ гл). Собственные энергии определяются выра-
жениями, аналогичными выражению, данному выше для Фе. Энергия взаимо-
действия определяется как энергия, равная
щ = <pQb (г", г") = Фе (Г, г") - Фа (г*, г"*) - Фь (г», I*). A.2)
Эта функция потенциальной энергии определена так, что она равна нулю,
когда обе молекулы достаточно далеко удалены одна от другой.
2. Электронное и ядерное движения разделяются (разделение Борна—
Оппенгеймера). Если электронное и ядерное движения рассматриваются раз-
деляющимися, то можно получить волновое уравнение, которое описывает
электронное движение относительно ядер, находящихся в фиксированном поло-
жении. При этом оказывается полезным ввести символ Фт обозначающий потен-
циальную энергию системы при определенной конфигурации ядер2):
A.3)
х) Вероятное число столкновений, необходимое для перехода энергии от поступатель-
ного движения к колебательному движению и увеличения на единицу колебательного кван-
тового числа, может быть установлено из измерений дисперсии и абсорбции звука. Дисперсия
и абсорбция звука в результате таких «релаксационных явлений» обсуждается в гл. 11, § 4.
Вероятное число столкновений есть экспериментально найденное время релаксации, делен-
ное на среднее время между столкновениями. Экспериментально обследовано [3, 4], что при
20° С число столчновений, необходимое для перехода энергии в колебательный вид, равно
34 000 для С12, 7500 для N2O и 50 000 для СО2. Для других молекул найдено, что, если колеба-
тельный вид движения является «либрацией» (заторможенным вращением), число столкно-
вений может быть меньшим и равным 50. Теория взаимных переходов между колебательной
. и поступательной энергиями была рассмотрена Зенером [6, 7 ].
2) В этой и следующих формулах вертикальная черта отделяет квантовые числа от пере-
менных.
708 Гл. 13. Теория межмолекулярных сил
Эта функция зависит от квантовых чисел электронов системы, которые мы
здесь обозначаем символически буквой к. Если рассматриваемая система состоит
из двух молекул, то, как и прежде, удобно ввести функцию-потенциальной
энергии, которая стремится к нулю, когда молекулы удаляются одна от другой.
Соответственно мы определим функцию <pQb{rv)
?аь(Г>)^<раЬ(каУкь\Г) = Фп(к,Г*)-ФМ^ A.4)
в которой через ка и кь обозначены наборы квантовых чисел электронов, описы-
вающих молекулы а и Ь. Отсюда следует, что для каждого электронного состо-
яния ка и кь существуют различные функции потенциальной энергии межмоле-
кулярного взаимодействия.
3. Электронное и колебательное движения отделяются от других дви-
жений. Если колебательное движение рассматривается независимо от других
ядерных движений, то можно говорить о потенциальной функции межмоле-
кулярного взаимодействия пары молекул, находящихся в определенных элек-
тронных и колебательных состояниях. В таком случае мы используем функ-
цию потенциальной энергии
<Раъ (г, соау соь) = сраЬ {кау va ; kb, vb | г, coa, щ). A.5)
Эта функция потенциальной энергии зависит от межмолекулярного расстоя-
ния г, от ориентации молекул сош соЬу а также, конечно, от квантовых чисел,
характеризующих электронное (ка, kb) и колебательное (vay vb) движения. Иногда
оказывается удобным усреднить функцию потенциальной энергии, определя-
емую по A.5), по всем ориентациям молекул, используя в качестве весового
множителя множитель Больцмана,
=<раЬ(ка, va; kb,vb \г,Т)= J ге-9л(г,ш..«>тмть 0-6)
Следует отметить, что полученный усредненный потенциал зависит от темпе-
ратуры.
4. Все типы движений отделяются один от другого. Если электронное
колебательное, вращательное и поступательное движения возможно рассмат-
ривать раздельно, то удобно использовать потенциальную функцию
<РаЬ (Г) = <Pab (ka, Vm jaf ТПа J kbf Vbf jb, Шь\г) , A.7)
в которой maumb — магнитные квантовые числа ; ja и jb — вращательные кван-
товые числа. Эта потенциальная функция зависит от межмолекулярного рас-
стояния и параметрически от электронных, колебательных и вращательных
квантовых чисел. Иногда удобно работать с усредненной потенциальной функ-
цией, в которой усреднение проведено по магнитным квантовым числам :
2 2 щъ (тпу ть \ г)
(<раьу = <<рпЬ(kQ,va,и; кь,vb,jb|г)> = т'ть 22Х A.8)
Этот средний потенциал не зависит от температуры. По аналогии с усреднением
A.6) можно определить зависящую от температуры среднюю величину, беря
больцмановское среднее значение <jpab> по другим квантовым состояниям.
В качестве конкретного примера рассмотрим взаимодействие между двумя
атомами, имеющими атомные номера Za и Zb и имеющие па и пъ электронов
соответственно. Для удобства обозначим электроны атома а индексом i, а элект-
роны атома b индексом /. Геометрия расположения ядер и электронов пред-
§ 7. Функции потенциальной энергии межмолекулярного взаимодействия 709
ставлена на фиг. 189. Рассматривая только электростатические взаимодействия
между различными электронами и ядрами, потенциальную энергию можно
записать в виде
Так как волновые функции, используемые в квантовой механике, обычно выра-
жаются через небольшое число сферических функций, то удобно выразить
различные члены сре через сферические функции. Для рыражения второго члена
через сферические функции используется двухцентровое разложение A.28)
гл. 12. Третий и четвертый члены выражаются через одноцентровое разло-
жение A.25) гл. 12. Если межъядерное расстояние гаЬ больше (г,- + rj) для всех
электронов, то разложение может быть записано в виде
где
/п — (z<* ~ ла) № - пь) е* ^ bk+i
re — - г ^ "JmT >.
Tab Л= j Таь
bk+1 =-e* [Zb2г>[Рк(cos0t) + (- l)kZahfPk(cos0,)] +
4- р* V V V ^_ {-^)m+kbk\rkrkbrf
^Огп±к<^П{к^кЬ+\т^^кЬ+\т\)'
X PV-кь (COS в,) Р^ (COS ву) *""<*-*>. A.11)
Здесь fc< меньше кь и к — кь.
Для взаимодействия нейтральных атомов Ь2 = 0 и первые несколько членов
этого разложения, записанные в декартовых координатах, имеют вид
A.12а)
Bх,ху + 2у,у;-3ziZj)(Zj-г,)], A.126)
Эта форма ^ используется в расчетах дальнодействующих сил, рассматриваемых
в настоящей главе. Разложение A.10) справедливо только в том случае, если
для всех электронов соблюдается условие таЪ > (г, + rj). Там, где это условие
не выполняется, <ре может быть представлено в виде ряда по обратным сте-
пеням г,- и Г/. При очень большом расстоянии между атомами дальнодействую-
щие силы могут быть строго представлены в виде ряда по обратным степеням rab.
Однако при меньшем расстоянии между атомами (из-за членов с обратными
степенями г,- и rj) силы включают экспоненциальные функции, зависящие от
расстояния между ядрами. Поэтому силы очень трудно вычисли1ъ для проме-
жуточных значений межъядерного расстояния. Метод расчета сил для этих
промежуточных межъядерных расстояний подробно рассматривается в гл. 14,
§ 1, где обсуждается взаимодействие двух атомов водорода.
Рассмотрим взаимодействие двух атомов с точки зрения соотношения
A.4). Для такой системы ядерная конфигурация определяется расстоянием
710
Гл. 13. Теория межмолекулярных сил
между атомами. Поэтому поверхности потенциальной энергии оказываются
одномерными, как показано схематически на фиг. 2С0. Эти функции
электронной энергии служат в качестве функций потенциальной энергии
межмолекулярного взаимодействия при движении ядер. Обычно за ну-
левую энергию удобно принимать потенциальную энергию межмолекуляр-
ного взаимодействия, соответствующую электронной энергии атомов, удален-
ных один от другого на большое расстояние. На фиг. 200 изображено несколько
типов поверхностей потенциальной энергии. Кривые а, с и е имеют глубокий
минимум, так что атомы притяги-
ваются, когда они достигают межъ-
ядерного расстояния, соответству-
ющего минимуму, после чего они
отталкиваются. Кривые b и / соот-
ветствуют отталкиванию при любых
расстояниях между атомами.
Вопрос о том, полезны ли по-
верхности межмолекулярной потен-
циальной энергии в описании про-
цессов столкновений, зависит от воз-
можности разделения электронного
и ядерного видов движения. При
описании медленных столкновений
такое разделение является хорошим
предположением; для быстрых стол-
кновений оно непригодно. В бы-
стрых столкновениях взаимодей-
ствие между электронным и ядер-
ным движениями ведет к переходам
от одной поверхности потенциаль-
ной энергии к другой. Например,
для потенциальных кривых, изо-
браженных на фиг. 200, вероятность
того, что столкновение, вначале
А Bр) + BBs)
ABs) + BBp)
ABs) + BBs)
Фиг. 200. Гипотетическая энергия
взаимодействия двух атомов А и В.
происходящее по кривой а, может в
конце идти по кривой /, мала. Однако, когда две поверхности потенциальной
энергии, такие, как е и d> почти пересекаются, вероятность того, что столкно-
вение, начавшееся вдоль кривой е, в конце будет происходить по кривой d,
оказывается очень большой.
Б. ПРИБЛИЖЕНИЯ, ВВОДИМЫЕ РАЗДЕЛЕНИЕМ ЭЛЕКТРОННОГО
И ЯДЕРНОГО ДВИЖЕНИЙ (РАЗДЕЛЕНИЕ БОРНА— ОППЕНГЕЙМЕРА)
Возможность использования при описании процессов столкновений меж-
молекулярных потенциальных функций основывается на возможности разде-
ления электронного и ядерного движений. В сьязи с этим мы проанализируем
ошибки, которые вносятся предположением о разделимости [1, 8, 9].
Начнем с гамильтониана для объединенной системы из электронов и ядер.
В приложении А (стр. 798) дано полное выражение для гамильтониана, вклю-
чающего влияние внешних электрических и магнитных полей. Если молекулы
имеют результирующий электронный спин или орбитальный момент коли-
чества движения, то некоторые из членов, описывающих магнитное взаимодей-
ствие, в этом выражении могут оказаться важными с точки зрения определения
межмолекулярных сил. В противном случае, достаточно рассмотреть только
электростатическое взаимодействие между частицами. Это приближение мы
здесь и рассмотрим.
В большинстве задач о столкновениях удобно пользоваться системой коор-
динат, связанной с центром тяжести. С этой целью мы вычтем оператор, пред-
§ 7. Функции потенциальной энергии межмолекулярного взаимодействия 711
ставляющий кинетическую энергию центра тяжести системы, из полного опера-
тора Гамильтона. Это приводит к следующему гамильтониану для относитель-
ного движения1):
A.13)
где
A.14)
здесь индексы а и /? относятся к различным ядрам; индексы / и / — к различным
электронам ; та — массы ядер ; т — масса электрона ; М — общая масса
молекуляной системы. Потенциальная энергия Фе является величиной, опре-
деляемой выражением A.1). Волновое уравнение для объединенного электрон-
ного и ядерного движений имеет вид
JTi2(r,r")= -4"?"> <1Л5)
где Q(r, rn) — полная волновая функция системы.
Раздельное рассмотрение ядерных и электронных координат может быть
проведено на основе теории возмущений. Невозмущенная электронная вол-
новая функция получается путем рассмотрения определенной фиксированной
конфигурации ядер Г. (Это представляется вполне разумным, так как класси-
чески электронное движение значительно быстрее ядерного.) Электронная
волновая функция у>к (rv, rn) такой гипотетической системы удовлетворяет
уравнению
JT. п (Г, *") = фп (к | Г) у>к (г", г"); A.16)
здесь Фп(к\тр) — энергетические уровни системы, соответствующие электрон-
ным состояниям, характеризующимся набором квантовых чисел электронов,
обозначаемым символом к. В соответствии с соотношением A.4) величина
Фп(к | Г) тесно связана с функцией потенциальной энергии межмолекулярного
взаимодействия сраЬ. Функции ^(г', гп) для какой-либо фиксированной ядер-
ной конфигурации образуют полный набор ортогональных функции в коорди-
натном пространстве электронов. Иными словами, любая функция электрон-
ных координат может быть выражена в виде линейной комбинации функций
\рк(т\ гп). ТакихМ образом, не теряя общности, волновую функцию Х2(г', гп) объе-
диненного электронного и ядерного движений исходной задачи можно запи-
сать в виде
Q (г^ гп) = ^ Хк (г0 У>к (г">г") • (LI7)
к
Подставляя функцию fi(r, rn) в таком виде в уравнение A.15), умножая обе
стороны уравнения на y>f(rvf тп) и интегрируя по всему пространству электрон-
ных координат, получаем
= -±%- A.18)
1 °1
г) Уравнение A.13) симметрично относительно всех N частиц. Кембл[10, стр. 63] выра-
зил Ж через 3N — 3 независимых координат, например координат относительно некоторой
частицы.
712 Гл. 13. Теория межмолекулярных сил
Величины Alk> Blk и Clka выражаются следующим образом:
A»-?J4?? +.?-?),,*", A.19)
*.--.2 ?/*(?•?)**¦ +
& ??i 2k%) 0.20)
Дифференциальное урарнение для функции %k(r) может быть в принципе
решено после того, как будут получены величины Ф„(/|г") из уравнения A.16).
Решение уравнения A.18) дает описание движения ядер.
Выражения для величин А1Ь Вш и Cika могут быть упрощены следующим
образом. Так как потенциальная энергия системы, а также кинетическая энер-
гия движения системы относительно центра тяжести не зависит от переноса
системы в целом, то отсюда следует, что функции \pk{xv, гп) зависят только от
относительного расположения различных частиц. Рассматривая влияние на
электронную волновую функцию изменений каждой из ядерных координат на
величину А г и влияние на электронные волновые функции изменений каж-
дой из электронных координат на величину —Л г, приходим к следующему
выводу:
( + Аг,г") = у>к(т%г" - Аг"). A.22)
Разлагая обе функции в ряды Тэйлора, получаем
^ч* 9ул >? ®У*к /1 оо\
у^. -г = — s'. —is— ( 1 .ZOI
a 8г<* i ЪЪ
И
^_^_J_v,/,==^JrJrV,fc. A.24)
Используя этот результат, из A.19) убедимся, что А,л — нуль и что второй инте?-
грал в выражении A.20) исчезает, так что
A.25)
Интегралы СШа не упрощаются.
' Члены, в которые входят величины В1к и С^а, обычно малы и ими можно
пренебречь. Из уравнений A.23) и A.24) следует, что производные по ядерным
координатам грубо имеют тот же порядок величины, что и производные по
электронным координатам. В таком случае легко показать, что члены, в которые
входят величины В1к, имеют порядок (mlM)Kef где Ке — кинетическая энергия
электронов. Аналогично члены, в которые входят С/Ла, имеют порядок величины
\(mjM) КеКп, где Кп — кинетическая энергия ядер. Так как т/М может изме-
няться в пределах от 0,0005 до 0,000002, то ошибка, вносимая пренебрежением
этими членами в обычных задачах молекулярной физики, очень мала. Напри-
мер, ошибка в энергии основного состояния молекулы водорода примерно равна
0,005 ккал/моль. Таким образом, уравнение A.18) обычно может быть упро-
щено путем отбрасывания членов, включающих Blk и С1каУ в результате чего
ядерное движение определяется решением уравнения
_Э_ Э ^ Л2 у, (_д д_\
= -А ЗД A.26)
§ 7. Функции потенциальной энергии межмолекулярного взаимодействия
713
Отсюда ясно, что ФпA\т*), определяемые из уравнения A.16), являются функ-
циями потенциальной энергии ядерного движения.
Эйринг и Полани [И]3) показали, что путем перехода от координат га
к линейной комбинации та из уравнения A.26) можно исключить перекрестные
члены (-р- • -р-1 xi и представить уравнение для ядерного движения в виде
уравнения Шредингера в пространстве 3 v—3 измерений, где v — общее число
ядер. Чтобы представить ядерное движение через 3v—6 относительных коор-
динат ядер, необходимо выделить вращательное движение молекулярной
системы. Это было сделано [13—15 ], но результаты имеют весьма сложный вид..
Фактически Blk и Zlka определяют связь между различными электронными
состояниями, которая обычно слаба. При столкновениях, при которых моле-
кулы находятся вначале в электронных состояниях, характеризующихся набо-
ром квантовых чисел Z, они могут при разлете оказаться в других электронных
состояниях, характеризующихся набором квантовых чисел к. Переход от сос-
тояния I к состоянию к обычно случается, когда ядерная конфигурация такова,
что между энергиями ФпA\тг) и Фп(к\Г) имеется лишь очень малое различие.
Теория таких неадиабатических столкновений обсуждалась Колманом и Лон-
доном [16]2), Райсом [18], Ландау [19], Зенером [20, 21 ]. Большой процент
столкновений молекул в возбужденных состояниях является неадиабатическим
в таком смысле. Вопрос об уменьшении энергии возбуждения за счет переходов
энергии при столкновении является предметом значительного количества экспе-
риментальных и теоретических исследований3). К электронным переходам могут
также приводить некоторые столкновения молекул, находящихся в основных
состояниях и обладающих большой энергией4).
Фиг. 201. Слева — две почти пересекающиеся функции потенциальной энергии;
справа — увеличенная часть области, где Фл@ и &n(k) близки одна к другой.
Приближенное выражение для вероятности неадиабатического столкно-
вения двух атомов можно получить следующим образом. Если две функции
потенциальной энергии одинаковой симметрии ФпA\г) и Фп(к\г) имеют почти
одинаковые значения вблизи точки г0, как показано на фиг. 201, то можно про-
вести две асимптотические линии Ах и Ак, которые соединяют ФпA) и Фп(к)
в точках, расположенных после и перед точкой их наибольшего сближения.
Определим далее следующие величины : г0 — расстояние, при котором раз-
ность ФпA) — Фп(к) имеет наименьшую величину 2еA, к); st = (dA^dr)^ и
sk = (dAkldr)r0 — наклон асимптот, a v(r0) = (drldt)ro — относительная скорость
столкновения, когда молекулы находятся на расстоянии г0. Вероятность Р,
^См. также [12].
2) Приложения вариационных принципов к проблемам столкновений получили недавно
важное развитие в работе Алтшулера [17].
3) Удачное изложение проблемы неадиабатических столкновений дано Эйрингом, Уолте-
ром и Кимбаллом [22].
*)См. [23], а также [24].
714 Гл. 13. Теория межмолекулярных сил
что система перейдет из состояния ФпA) в состояние Фп(к) (или наоборот) во
время столкновения, выражается приблизительно следующим образом [22 ]:
CXPL hv(ro)\si-sk\}>
где ft — постоянная Планка. Таким образом, увеличение сближения кривых
потенциальной энергии, повышение кинетической энергии столкновения, уве-
личение угла между двумя кривыми приводит к увеличению вероятности пере-
хода.
Когда заметная доля столкновений происходит неадиабатически, класси-
ческое представление о межмолекулярных силах теряет свое значение, но
результаты процессов столкновений все еще могут описываться на статисти-
ческой основе.'Таким образом, концепцию межмолекулярного потенциала сле-
дует подвергнуть тщательной проверке в каждом частном случае.
В. СВЕДЕНИЯ О МЕЖМОЛЕКУЛЯРНЫХ ПОТЕНЦИАЛАХ, ПОЛУЧАЕМЫЕ
С ПОМОЩЬЮ ТЕОРЕМЫ ВИРИАЛА
Слетер [25]1) показал, что для определения отношения средней кинети-
ческой энергии электронов к средней потенциальной энергии электронов моле-
кулярной системы может быть использована теорема вириала. Это позволяет
лучше понять природу как межмолекулярных, так и внутримолекулярных
сил, а также приводит к большому числу интересных приложений теоремы
вириала. Например, Коттрелл воспользовался результами Слетера для опре-
деления смещения уровней электронной энергии молекул из эксперименталь-
ных данных завт самости между р, V и Т, когда газ находится под очень
высоким давлением (см. гл. 4, § 3). Другое приложение теоремы вириала —
приближенное квантовомеханическое вычисление энергий связи. В таких
вычислениях в приближенных волновых функциях вводится масштабный
множитель, который подбирается так, чтобы получить наименьшую энергию
(см. приложение Б, стр. 799). Подбор масштабной постоянной автоматически
обеспечивает получение правильного отношения средней кинетической энергии
электронов к средней потенциальной энергии электронов.
Рассмотрим молекулярную систему, состоящую из п электронов и v ядер,
которые притягиваются или отталкиваются по закону Кулона (магнитными
взаимодействиями мы пренебрегаем). Когда система находится в состоянии
равновесия, то в соответствии с теоремой вириала средняя потенциальная
энергия равна удвоенной средней кинетической энергии, взятой с отрицатель-
ным знаком. Теорема вириала может быть применена даже тогда, когда ядра
находятся в фиксированном неравновесном положении. Вследствие раздели-
мости электронных и ядерных координат теорема вириала дает очень мало
сведений о неравновесных случаях.
Потенциальная энергия рассматриваемой молекулярной системы записы-
вается в виде A.1). Поскольку эта функция потенциальной энергии является
однородной функцией координат в степени —1, то в соответствии с уравнением
D.29) гл. 1 можно написать
где F/ и Fa — силы, действующие на z'-й электрон и a-е ядро соответственно.
Вириал сил, действующих на элеюроны в соответствии с D.28) гл. 1, равен
средней величине кинетической энергии электронов Ке:
. также [26].
§ 7. Функции потенциальной энергии межмолекулярного взаимодействия 715
Электроны движутся значительно быстрее ядер. Поэтому до тех пор, пока
рассматривается движение электронов, ядра находятся в стационарном состоя-
нии. Исходя из этого допущения, соотношения A.28) с помощью A.29) можно
записать в виде
Ке=-\фе + -^2^?-)' С1-30)
Сила, действующая на а'-е ядро, выражается следующим образом :
*.= -?,*«. 0-31)
где Фп — потенциальная энергия системы для определенной ядерной конфи-
гурации. В уравнении Шредингера для движения электронов A.16) Фп играет
роль общей энергии .электронов, так что для фиксированной конфигурации
ядер имеем
Ке + Фе = Фп.~ A.32)
Подстановка A.31) р A.30) дает
Если потенциальная энергия Фп является функцией только расстояний га/3
между ядрами, то
У(т .А-ф)-А-У Уг i?l. П34^
Отсюда соотношение A.33) может быть записано в виде
*1?7- С-35)
Далее, комбинируя полученный результат с соотношением A-32), получаем
соотношения
Таким образом, мы получили выражение для средней кинетической и потен-
циальной энергий электронов через Фп. Соотношения A.36) и A-37) очень
полезны для понимания изменений, происшедших ь электронных состояниях
молекул под влиянием очень высокого давления (см. гл. 4, § 3).
Рассмотрим столкновение двух атомов с заполненными оболочками, на-
пример атомов аргона. Если предположить, что взаимодействие атомов аргона
подчиняется потенциалу F-12) Лениарда-Джонса, то
. A.38)
716 Гл. 13. Теория межмолекулярных сил
Следовательно, средняя кинетическая энергия электронов равна
Ke=--L0e+l2e[2(^-(^f]. (U9)
Таким образом, для таких атмов
A.40)
±-Феу
где г = 21/вог — равновесное расстояние между атомами.
Г. ЭКВИВАЛЕНТНОСТЬ МЕЖМОЛЕКУЛЯРНЫХ СИЛ, ВЫЧИСЛЕННЫХ
НА ОСНОВЕ КЛАССИЧЕСКОЙ И КВАНТОВОЙ МЕХАНИКИ
Геллман [27, стр. 285] и Фейнман [28] доказали, что силы, действующие
на ядра, определенные по квантовомеханическим поверхностям потенциальной
энергии, в точности совпадают с силами, рассчитываемыми на основе класси-
ческой электростатики и знания плотности вероятности распределения элек-
тронов. Другими словами, как только из решения уравнения Шредингера
определено распределение электронного облака, то силы, действующие на ядра,
могут быть вычислены с помощью электростатической формулы A.7) гл. 121).
Эта теорема, называемая теоремой Геллмана—Фейнмана, очень полезна для
установления качественной связи между распределением электронного заряда
в молекулах и межмолекулярными силами. Она также значительно упрощает
наше изложение вопроса о природе межмолекулярных сил. Следует отметить,
что спиновый и обменный эффекты играют важную роль при расчете электрон-
ного облака, но как только распределение электронов фиксировано, то силы
являются принципиально классическими электростатическими силами. Перво-
начально имелась тенденция полагать, что спиновые и обменные эффекты
играют скорее более непосредственную роль, а не роль вторичного фактора в
определении этих сил.
Рассмотрим систему из v ядер и п электронов, в которой ядерное и элек-
тронное движения разделимы. Нормированные электронные волновые функции
ip==y)k(rvy rn) удовлетворяют уравнению Шредингера A.16); Фп = Фп(к | г1')
— энергетические уровни системы. Последние могут быть выражены в виде
A.41)
Подставляя это выражение в соотношение A.31), получаем следующее выра-
жение для силы, действующей на а-е ядро в х-направлении :
-^,^^df. A.42)
Выражение A.42) может быть упрощено, если принять во внимание, что опе-
ратор .3ft является эрмитовым, а также если учесть свойства волновых функций.
х) Теорема Геллмана—Фейнмана доказана с помощью обычного гамильтониана с учетом
только электростатических взаимодействий между частицами. Если принять во внимание
малые спиновые и орбитальные связывающие члены в полном гамильтониане, приведенном
в приложении А (стр. 798), то можно найти небольшие отклонения от результатов, получен-
ных Геллманом и Фейнманом.
§ 7. Функции потенциальной энергии межмолекулярного взаимодействия 717
В результате такого упрощения получим1)
Fax= -JV^A»- -j>?„А« A.43)
Далее, в соответствии с определением Ф,, A.1) можно записать
ОФе 8 /- у ZjZf* \ v7 . 9
Последний член тесно связан с электрическим полем в х-направлении, созда-
ваемым а-м ядром в точке, где находится i-й электрон. Это поле равно
<р (г\ ? о 9 [ ^ \ ^а в (Ха — Xj) /1 у| к\
гГ^От) -2ае-д^(—J ^ A.45)
Отсюда A.43) можно записать в виде
*
Чй
-- -Щ Чй + •?' «/*•
Дальнейшее упрощение можно сделать, заметив, что плотность заряда ?х
электрона 1 равна
01 = ejy>*y>drzdrs... drn. A.47)
Вследствие тождественности электронов системы выражение для плотности
заряда всех п электронов должно быть одинаковым. Отсюда общая плотность
заряда электронов g в точке равна щг. Окончательно выражение для х-компо-
ненты силы, действующей на а-е ядро, записывается в виде
Это и является доказательством теоремы Геллмана—Фейнмана.
Этот важный результат означает, что сила, действующая на какое-либо
ядро в фиксированном положении в любой системе из ядер и электронов, сов-
падает с классической электростатической силой, действующей на рассматри-
ваемое ядро со стороны других ядер и распределения заряда, создаваемого
всеми электронами. Иначе говоря, сила, действующая на ядро, равна заряду
этого ядра, умноженному на электрическое поле, слагающееся из поля, созда-
ваемого в точке, где расположено рассматриваемое ядро, другими ядрами, и из
электрического поля, вычисляемого классически из распределения заряда
электронов. Если волновая функция вычислена для определенной ядерной
конфигурации, то теорема Геллмана—Фейнмана позволяет провести пря-
мое вычисление всех ядерных сил. Это значительно сокращает работу по срав-
Поскольку JTе — эрмитов оператор, то
Далее у)* удовлетворяет уравнению Шредингера
Поэтому последние два члена выражения A.42) могут быть записаны в виде
откуда следует A.43). Так как часть JFe, определяемая кинетической энергией, не зависит
от ха, то
д<Же = дФе
дха ~ дХа '
718
Гл. 13. Теория межмолекулярных сил
нению с обычной схемой вычисления энергии системы для большого числа
ядерных конфигураций и намного увеличивает точность результатов.
Из теоремы Геллмана—Фейндоана ясно, что для того, чтобы два ядра двух
атомов притягивались, между ними должна быть некоторая концентрация
электронного заряда, которая притягивала бы оба ядра. В значительной мере
ядро движется вследствие его притяжения к собственному электронному
облаку. В антисимметричном 32'-состоянии молекулы Н2 узел в плотности
заряда электронов находится посередине между ядрами. Поэтому плотность
электронов между ядрами уменьшается и атомы отталкиваются. Аналогично
силы притяжения Ван дер Ваальса могут быть интерпретированы как такое
искажение распределения электронов р молекулах, при котором плотность
электронов становится большей на стороне, обращенной к налетающей моле-
куле, i
Очень интересное наглядное истолкование смысла теоремы Геллмана—
Фейнмана было дано Берлином [29]. Дело в том, что одн i облает 1 электронного
облака способструют связыванию ядер, другие — разрыхлению. Рассмотрим, на-
пример, двухатомную молекулу, изображенную на фиг. 202. Расстояние вдоль оси,
соединяющей ядра, измеряемое от средней точки этой оси, обозначим через х,
а расстояние точки от оси х— через у. Ядра удалены одно от другого на рас-
стояние г. На фиг. 202,а отмечена точка электронного облака (хг, уг), которая
способствует свя?ыванию ядер. Иначе говоря, элемент отрицательного заряда,
расположенный в точке (xv ух), приводит к возникновению действующей на
а
V
I
/
Фиг. 202. Связывание (а) и разрыхление (б) в двухатомной молекуле, состоящей из ядер
а и b с зарядами Za и Z&.
ядро а кулоновской силы, которая имеет составляющую в х-направлении, и
притягивает ядро а к ядру Ь. Аналогично кулоновская сила, действующая со
стороны отрицательного заряда на ядро &, имеет составляющую в х-направ-
лении, которая также притягивает ядро а к ядру Ь. Таким образом, отрица-
тельный заряд в точке (xv уг) способствует объединению ядер в молекулу аЬ.
Рассмотрим далее точку (х2, у2) на фиг. 202, б. Отрицательный заряд, располо-
женный в этой точке, приводит к возникновению кулоновских сил, действую-
щих на ядра а и b в х-направлении. Если составляющая силы Fbx, действую-
щей на ядро а в х-направлении, больше составляющей силы Fax> действующей
на ядро b в х-направлении, то сила, равная разности Fbx — Faxy является
той силой, которая приводит к взаимному отталкиванию двух ядер. Иначе
говоря, отрицательный заряд, расположенный в точке (х2, у2), в рассматривае-
хмых условиях является разрыхляющим. Поверхность, разделяющая связываю-
щую и разрыхляющую зоны электронной плотности, определяется условием
Fa*=Fbxy A.49)
§ 1. Функции потенциальной энергии межмолекулярного взаимодействия
719
или, если использовать закон Кулона, условием
Za [х + -J-] [(* + -J-J + y-f' = Z. [* --? ] [(х - ^J
A.50)
Для молекулы, состоящей из одинаковых ядер (с Za = Z&), уравнение гра-
ничной поверхности имеет вид
у -
Граничная кривая, приведенная на фиг. 203, похожа на гиперболу и имеет
асимптоты — прямые линии, пересекающиеся под углом 109°28'. Разрыхляю-
щая зона электронной плотности заштри-
хована. С увеличением отношения ZJZb
одна из поверхностей становится более
плоской и приближается к поверхности
х = — г/2, другая — свертывается и обра-
зует замкнутую область, которая стяги-
вается в точку (х = г/2, у = 0).
Для хлористого водорода, имеющего
ZJZb = 17, небольшая разрыхляющая об-
ласть, как показано на фиг. 2С4, прости-
рается по оси х от ядра водорода в точке
х = г/2, до х = 0,82г, и по оси у от
у= +0,19г до v = —0,19г. Для хлори-
стого натрия, имеющего ZJZb = 17/11, как
показано на фиг. 2С5, поверхности более
похожи на случай молекулы из одина-
ковых ядер, когда поверхность, проходя-
щая через ядро С1, немного уплсщена, а
поверхность, проходящая через ядро Na,
более вогнута.
С помощью аналогичного анализа можно определить, в каких областях
пространства электронное облако способствует притяжению молекул, а в
фиг- З03* Молекула, состоящая из
одинаковых ядер, Za = lb.
Ф и г. 204. Молекула
хлористого водорода,
Za/Zb = 17.
Фиг. 205. Молекула хлористого
натрия, Za/Zb = 17/11.
каких оно природит к возрастанию отталкивания между молекулами. В любом
случае часть электронного облака, находящаяся непосредственно между двумя
молекулами, всегда яьляется связывающей.
720 Гл. 13. Теория межмолекулярных сил
Д. КВАНТОВОМЕХАНИЧЕСКИЙ РАСЧЕТ ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
Квантовомеханический расчет энергии взаимодействия двух молекул, по
существу, аналогичен расчету энергии связи отдельной молекулы. Много при-
меров подробно изложено в гл. 14. Двумя главными методами расчета энергии
взаимодействия являются вариационный метод и метод возмущений (см. гл. 1,
§ бI). Помимо этого, недавно развит обобщенный метод самосогласованного
поля, который мы здесь обсудим. Вариационный метод имеет то преиму-
щество, что энергия, вычисленная с помощью этого метода, всегда слишком
высока, так что добавление большего числа членов или большего числа под-
бираемых параметров при апроксимации волновой функции приводит к умень-
шению ошибки. Вариационный метод может быть применен для любых расче-
тов межмолекулярного потенциала, как в случае близко расположенных мо-
лекул, так и в случае значительного их удаления друг от друга. Вариационный
метод и метод возмущений правильнее применять к расчетам взаимодействия
двух молекул, чем к расчетам энергии связи отдельной молекулы, так как энер-
гия взаимодействия действителы о мала по сравнению с потенциалами иони-
зации и другими мерами молекулярной энергии. Первое приближение метода
возмущений применяется для расчетов химических сил или сил отталкива-
ния, которые возникают, когда молекулы находятся близко одна от другой
и их электронные облака перекрываются. Его можно также использовать
для вычисления дальнодействующих сил, действующих между молекулами,
обладающими постоянными дипольными или мультипольными моментами
(см. § 5 и 6). Для вычисления дальнодействующих сил, которые возникают
вследствие индуцирования электрических моментов у одной молекулы под
действием электрического поля другой молекулы, необходимо использовать
второе приближение метода возмущений (см. § 3 и 4).
Вычисление химических сил или сил отталкивания с помощью первого
приближения метода возмущений особенно просто в случае, когда структура
связи двух молекул определена, так что нет необходимости рассматривать
резонанс. Если электроны в молекулах а и Ь находятся на обобщенных одно-
электронных орбиталях ахA), а2A),... и &хB), &2B),... соответственно (индек-
сом внизу отмечены орбитали, числами в скобках обозначены электроны),
то общая энергия системы из двух взаимодействующих молекул выражается
следующим образом2):
—2~J%Lab. A.52)
х) Прекрасное обсуждение этих методов дано Гомбашом [30], Моттом и Снеддоном [31 ]
а также Эйрингом, Уолтером и Кимбаллом [22].
2) В квантовой механике молекул принята следующая терминология:
Интегралы кулоновской энергии. Кулоновская энергия системы с четырьмя элек-
тронами равна
J ах A)* а2 B)* Ьх C)* Ьг {А)*Жах A) а2 B) Ъг C) Ь2 D) dr*.
Когда гамильтониан <ЗР выражается в виде суммы членов, то интеграл кулоновской энергии
переходит в сумму интегралов, каждый из которых называется «кулоновским интегралом».
Однократно обменные интегралы энергии. Однократно обменные интегралы энергии
имеют вид
| ах C)* а2 B)* Ъх («« Ь, D)*Жах A) а2 B) Ъх C) Ьг D) dr«.
Эти интегралы можно привести к виду
M3)*MD*-?- at A)^C)*г*,.
Многократно обменные интегралы энергии. Многократно обменные интегралы
энергии отличаются от однократно обменных интегралов энергии тем, что в исходном и ко-
§ 7. Функции потенциальной энергии межмолекулярного взаимодействия 721
Здесь 2 Lab — сумма обменных интегралов между парами, образующими обоб-
р
щенные электронные связи ; 2 Lab — сумма обменных интегралов между всеми
и
непарными электронными орбиталями ; Q — сумма энергий всех кулоновских
взаимодействий плюс сумма энергий взаимодействия, обусловленных дально-
действующими силами — дисперсионными, индукционными и дипольными.
Часть Q, возникающая за счет кулоновских взаимодействий, Qc, выражается
следующим образом :
Qc= 2 *?f-+ 2 Ja+ 2 Kab, A.53)
а>/3 'ар По всем По всем
По всем орбиталям парам
парам ядер
где
tf?r1dr2, A.54)
A.55)
Обменные интегралы в выражении A.52) имеют вид
Lab = jja A)* Ъ B)* ? Ь A) а B) ЛггЛгш. A.56)
В выражении A.52) мы пренебрегли многократно обменными интегралами
энергии и интегралами перекрытия.
Энергия взаимодействия пары молекул вычисляется следующим образом.
Сначала вычисляется энергия ?Прибл. системы из двух молекул при столк-
новении, затем вычисляется знергия двух молекул, удаленных одна от другой
на очень большое расстояние. Разность этих двух энергий и есть энергия взаи-
модействия. Энергии взаимодействия, вычисленные таким способом, получаются
достаточно точными.
Для расчета межмолекулярной энергии очень полезен недавно развитый
Слетером [32—34] обобщенный метод самосогласованного поля1). Отдельно
рассматривается каждая из молекул. Затем рассматривается объединенная
система. Пусть of$(rj, a(f(h)> aB}(ri)- • • образуют полный набор ортогональных
одноэлектронных молекулярных орбиталей для электрона 1 в молекуле а.
Различные приближенные волновые функции молекулы а могут быть образо-
ваны путем составления различных линейных комбинаций из детерминантов
Слетера, образованных из этих орбиталей. Составляя линейные комбинации
из бесконечного числа детерминантов Слетера, можно получить достаточно
точную волновую функцию. Однако практически найдено, что очень хорошая
функция получается с помощью только нескольких детерминантов Слетера,
соответствующих взаимодействию между несколькими важными конфигура-
циями. Пусть приближенная волновая функция ^прибл. представляет собой
некоторую частную линейную комбинацию детерминантов Слетера, выражен -
кечном произведениях орбиталей меняют места более чем одна пара электронов. Примером
такого типа интегралов является интеграл
«i C)* а2 D)* Ьх A)* Ь2 B)* -?- ах A) а2 B) Ьх C) Ь2 D) dr«.
Интегралы перекрытия. Термин интеграл перекрытия может быть использован
либо в общем смысле для интеграла Jy>* щ drN, чтобы отметить факт неортогональности двух
волновых функций, либо для того, чтобы отметить неортогональность двух орбиталей ах и
о19 т. е. для интеграла §,агA)* ЬгA) dr^
*) Полное изложение стандартных методов самосогласованного поля Хартри и Хартри—
Фока дано Гомбашом [30].
722 Гл. 13. Теория межмолекулярных сил
ных через набор орбиталей а^\ аA\ а^\... С помощью этой функции можно
однозначно определить эффективное поле потенциальной энергии Ф0^), в кото-
ром движется какой-нибудь из электронов (например, электрон 1),
Функция Ф*0^) представляет собой энергию взаимодействия электрона 1 со
всеми остальными электронами, усредненную по всем движениям этих элект-
ронов и их спингм, в предположении, что ядра остаются в фиксированных
положениях. Это следует из того факта, что
¦ ¦ ¦ *п A.58)
есть вероятность нахождения электрона 1 в области drv усредненная по всем
движениям и спинам всех остальных электронов. Соответственно Ф°\т^) —
потенциальная энергия взаимодействия электрона (с определенным спином),
расположенного в точке гх с распределением электронного заряда
0-59)
Ч5
. Сил. *«*••
при фиксированных положениях ядер. Используя функцию Ф*0^) в качестве
потенциала в уравнении Шредингера для одного электрона, получаем новый
набор молекулярных орбиталей ^C) ^iV)
здесь ек—энергия электрона на fe-й орбитали. Новые орбитали аA)(Гх) служат
для определения нового одноэлектронного потенциального поля #(l)(ri)« Этот
процесс повторяется до тех пор, пока он не сойдется к самосогласованному
потенциальному полю. При определении общей энергии объединенной системы
из двух сталкивающихся молекул будем исходить из приближенной волновой
функции упривл., образованной путем антисимметризации произведений приб-
лиженных волновых функций отдельных молекул. С помощью этой функции
образуем Ф°\т^). Из одноэлектронного потенциала получим набор молекуляр-
ных орбиталей для объединенной системы. Далее, повторяя процесс до тех
пор, пека он не сойдется, получаем общую энергию объединенной системы и
отсюда энергию взаимодействия.
Этот процесс нахождения энергии взаимодействия, по-видимому, особенно
удобно использовать при расчетах на быстродействующих вычислительных
машинах. Меклер [35, стр. 62 ; 36, стр. 19] показал, что в случае молекуляр-
ного кислорода процесс сходится легко и быстро. Общая энергия молекулы,
соответствующая потенциалу &°\т^), дает возмущения первого порядка, а
энергия, соответствующая {P^fo), дает возмущения второго порядка. Так как
^@)(ri)> ^(lVi) и т- Д- вообще имеют ту же симметрию, что и молекула, то опре-
деление орбиталей можно значительно упростить, используя теорию групп.
§ 2. ПОЛЯРИЗУЕМОСТЬ МОЛЕКУЛ1)
Поляризуемость молекул играет очень важную роль в некоторых теориях
дальнодействующих межмолекулярных сил, обсуждаемых в последующих
параграфах. Поэтому настоящий параграф мы посвятим обсуждению методов
расчета поляризуемостей. Сначала излагается общая, квантовомеханическая
^Поляризуемость вещества обсуждается в гл. 12, §2.
§ 2. Поляризуемость молекул 72 3
теория и ее приложение к расчету поляризуемости молекулярного водорода.
Затем обсуждается принцип аддитивности поляризуемостей. И, наконец, рас-
сматривается использование экранировочных постоянных для расчета поля-
ризуемостей и других свойств молекул.
В молекуле, помещенной в электрическое поле, происходит смещение заря-
дов, вследствие чего индуцируется дипольный момент. Величина этого инду-
цированного дипольного момента равна
рС-нд.) = («.*), B.1)
где а — тензор поляризуемости молекулы. Если молекула имеет постоянный
дипольный момент ц*, то энергия молекулы в электрическом поле минус энергия
вне поля может быть получена из выражений A.35) и B.2) гл. 12 :
Е-Е0=-(р.г0--1(гГ.а.*), B.2)
где ?0 — энергия невозмущенной молекулы, a E—энергия молекулы, на-
ходящейся в электрическом поле 8'.
Следует отметить, что соотношение B.1) справедливо только для малых
напряженностей поля. При столкновениях молекул напряженности электри-
ческих полей столь велики, что возникает вопрос [37], пригодны ли в этом
случае обычные поляризуемости, подходящие для полей малых напряжен-
ностей. На расстоянии 3 А от электрического заряда напряженность поля
имеет значение 5,3-Ю5 эл.-ст. ед/см, или 160 млн. в/см. На расстоянии ЗА
от молекулы, имеющей дипольный момент 3 дебая1) и расстояние между
зарядами 2А, электрическое поле равно примерно 75 млн. в/см. Коулсон,
Макколл и Саттон развили теорию, из которой следует, что поляризуемость
бензола и других родственных и ароматических молекул, у которых про-
странственные и электрические свойства очень различны в разных направ-
лениях, значительно увеличивается в полях с напряженностью порядка
100 млн. в/см.
А. ПРИМЕНЕНИЕ ВАРИАЦИОННОГО МЕТОДА2) ДЛЯ РАСЧЕТА
ПОЛЯРИЗУЕМОСТЕЙ
Поляризуемость молекулы может быть найдена с помощью простой вариа-
ционной процедуры, которую предложили Хиллераас [38] и Хааз [39, 40].
Рассмотрим невозмущенную молекулу, имеющую п электронов и v ядер, кван-
товомеханический оператор Гамильтона которой равен <Ж$. Энергию молекулы
в основном состоянии обозначим через ?0. Соответствующая этой энергии
волновая функция Wo удовлетворяет уравнению Шредингера
.y0V0 = E0V0 B.3)
и условию нормировки
rW*WQdxn= 1. B.4)
Когда молекула испытывает внешнее возмущающее влияние (например,
влияние электрического поля), в гамильтониане появляется дополнительный
член Л?^, обусловленный этим возмущением. Общий гамильтониан возму-
щенной молекулы при этом имеет вид
jr = ^fo + '^i- B.5)
1 дебай = Ю-18 эл.-cm. ед.
а; 1 оеоаи = ш-хо эл.-ст. ео.
2) Вариационный метод как способ приближенного решения уравнения Шредингера
обсуждался в гл. 1, § 6.
724 Гл. 13. Теория межмолекулярных сил
Этому гамильтониану соответствует волновая функция W и энергия Е. Возму-
щенная волновая функция может быть приблизительно записана в виде1)
V = (l + AJrjY09 B.6)
где А — варьируемый парамерт. Этот параметр вычисляется из условия, чтобы
приближенная величина энергии
была минимальной.
Подставляя гамильтониан B.5) и волновую функцию W B.6) в выражение
для энергии B.7), получаем
Используя свойства невозмущенной волновой функции B.3) и B.4), полученное
выражение можно привести к виду
Р _ р _ \Уг + (Qx + 2W2) А + (Qa + W>) Л»
где
Если постоянная А выбирается из условия минимальности величины Е,
то выражение B.9) дает смещение энергетического уровня за счет влияния
возмущения. Применим полученный результат к специальному случаю моле-
кулы, помещенной в электрическое поле.
Для определения поляризуемости нейтральной молекулы в качестве воз-
мущения выберем однородное электрическое поле, имеющее напряженность
(fx в х-направлении. Возмущающий потенциал при этом имеет вид
^ 2
1=1 a=l
Здесь е — абсолютная величина заряда электрона; х( и ха — координаты элек-
тронов и ядер соответственно ; Za — атомные номера ядер. Теперь х-компо-
нента постоянного дипольного момента невозмущенной молекулы равна
) B.11)
где чертой отмечено квантовомеханическое усреднение по невозмущенной
волновой функции. Так как все электроны эквивалентны, то можно опустить
х) Мы предполагаем, что молекула не перемещается относительно электрического поля.
В таком случае волновая функция стационарного состояния может быть выбрана действи-
тельной. Поэтому в последующем обсуждении Wo полагается действительной функцией.
Член JFj чаще является функцией, чем обобщенным оператором. Последующий анализ без
этих допущений был бы несколько более длинным, но результаты оказываются очень близ-
кими.
§ 2. Поляризуемость молекул 725
индекс у х, и называть х средней х-координатой электрона. Таким образом,
можно видеть, что, когда интеграл Wly определенный выше, вычисляется для
возмущающего потенциала B.10), результат можно записать в виде
Wi—Ac**. B.12)
Выражение для смещения уровня энергии B.9) при этом принимает вид
Р р у A(Ql + 2Wt-
Это выражение для смещения уровня энергии имеет тот же вид, что и B.2).
Отсюда вычисление интегралов Qt и Wt дает возможность вычислить коэффи-
циент при %\, который просто связан с поляризуемостью.
Отметим, что из определения Wt следует, что обе величины — как Wz,
так и Wejtx&x — пропорциональны |Г?. Поэтому эти члены не дают вклада в
поляризуемость и далее не будут рассматриваться. Можно показать, что интег-
рал Qx равен нулю, а другие интегралы в выражении B.13) имеют значения1)
B.14)
х) Выражение для W2 получается путем подстановки Л?1У имеющего вид B 10), в опре-
деление W2, имеющее вид B.9), и использования выражения B.11):
- 2 B Za хо) B xi
2 (xi-xi)(xj-
Используя тот факт, что х одинаково для всех п электронов (так как | ^ol2 симметрична
относительно перестановки любой пары электронов), получаем выражение B.14). Чтобы
вычислить <?, и Q2, необходимо рассмотреть (JT0— Е0)Л^г^0. Некоммутирующими частями
операторов J?o и <WX являются член кинетической энергии в Жь и электронный член в Jpx.
В частности,
(JT,-
Отсюда интеграл Q2 приобретает вид
Здесь был использован факт тождественности электронов. Интегрируя и используя тот факт,
что ^2 = 0 при хх = ± оо, получаем 0х = 0.
Интеграл 02 имеет вид
Члены с / т* 1 не дают вклада в интеграл, поэтому
Интегрированием по частям легко показать, что
"¦«¦ ^f **--¦?•
так что
726 Гл. 73. Теория межмолекулярных сил
где п — число электронов в молекуле ; а0 = (k2lme2) = 0,5292 А — боровский
радиус; (х± — хJ— среднеквадратичное отклонение электрона в х-направ-
лении от его среднего положения ; (хх — х) (х2 — х) — средняя корреляция
между мгновенными х-координатами двух различных электронов. Эта корре-
ляция должна быть равна нулю для молекулярной орбитальной волновой
функции. Однако для многоатомных молекул эта корреляция весьма велика,
если волновая функция является функцией типа описывающей химическую
связь с использованием атомных орбиталей или функцией типа коррелиро-
ванной молекулярной орбитали.
Варьируемый параметр А может быть определен путем приравнивания
нулю производной (ЗЕ/дА) и решения получающегося уравнения относительно А.
Можно легко показать, что в предельном случае малой напряженности элек-
трического поля (т. е. в случае, когда %?х -+ 0) параметр А изменяется, как
$1, и имеет вид
A = <**-W*\ B.16)
откуда
F F и Г (f2xt2x-W2J @ 17.
Ь — Ьо= — [*хбх ^ • \*Л1)
Сравнивая этот результат с выражением B.2) и используя выражения B.14)
и B.15) для Q2 и W2, получаем следующую формулу для хх-компоненты поля-
ризуемости :
ахх = ± [<х~=г)« - (л - 1) (х1-х)(х2-х)]К B.18а)
Этот результат, справедливый для молекул, является обобщением выражения
для поляризуемости атомов, которое было получено Кирквудом [41 ].
Поляризуемость атома изотропна. Если волновая функция атома апрок-
симируется орбитальной волновой функцией атома, то второй член в формуле
B.18а) исчезает. Далее, для атома х = 0их| = у| = if = Vs'i"- Поэтому фор-
мула B.18а) для атома приобретает вид
^ BЛ86)
Эта формула дает простой приближенный метод оценки поляризуемости. Для
атома водорода из формулы B.186) получаем а = 4а§, что хорошо согла-
суется с экспериментальным значением а = 4,5ag. Для атома гелия формула
B.186) дает а= 1,0 ag (для простейшей приближенной волновой функции,
выражающейся в виде произведения двух водородных орбиталей). Экспери-
ментальное значение поляризуемости для гелия равно 1,37 ag. Хорошие
результаты получаются для молекулярного водорода [42], если использовать
формулу B.18а).
Б. ПОЛЯРИЗУЕМОСТЬ МОЛЕКУЛЯРНОГО ВОДОРОДА
Очень точные значения поляризуемости молекулярного водорода и его
изотопов вычислили Ишигуро, Араи, Котани и Мицушима [43 ]. Они использо-
вали одиннадцатичленную волновую функцию Джеймса и Кулиджа [44 ] для
нормальной невозмущенной молекулы водорода и предположили, что влияние
внешнего электрического поля на волновую функцию можно учесть путем
добавления десяти подбираемых постоянных, умноженных на члены типа
Джеймса и Кулиджа. Эти десять постоянных выбирались затем такими, которые
приводили к наименьшему значению энергии. Таким путем они получили ре-
зультаты, приведенные в табл. 113. Экспериментальные значения диэлектри-
ческой постоянной молекулярного водорода при 0° С и 1 атм колеблются в
пределах от 1,000259 до 1,000273, так что теоретическое значение, равное
1,000267, вполне удовлетворительно. Экспериментальные значения поляри-
§ 2. Поляризуемость молекул
727
зуемостей имеют значительное расхождение. Наиболее аккуратные измерения
были приведены Фолькманом [45]. Данные его экспериментов приведены в
табл. 113.
Таблица 113
Теоретические и экспериментальные значения поляризуемостей
и диэлектрической постоянной е' молекулярного водорода
(для нулевой частоты)
а у — поляризуемость (А*) в направлении, параллельном связи ; а± — поля-
ризуемость (А*) в направлении, перпендикулярном связи ; а — среднее зна-
чение поляризуемости, равное V» (а ц + 2 а^)
а||
а±
а
е'(О°С,
1 атм)
Эксперимен-
тальные
значения [45]
н,
0,934
0,718
0,790
1,000259
до 1,000273
Теоретические значения [43]
н,
0,9746
0,6968
0,7894
1,0002666
HD
0,9651
0,6917
0,7829
1,0002643
0,9537
0,6856
0,7749
1,0002616
В. АДДИТИВНОСТЬ ПОЛЯРИЗУЕМОСТЕЙ СВЯЗЕЙ
Поляризуемость сложной молекулы просто можно оценить суммированием
вкладов, вносимых в поляризуемость различными составляющими связями.
Справедливость этого метода может был установлена с помощью теоретиче-
ских расчетов, развитых в разд. А.
Предположим, что электроны в п-электронной молекуле могут быть под-
разделены на некоторое число групп Д В, С,..., включающих пА, пв, пс,...
электронов соответственно. Предполагается, что эти группы более или менее
независимы одна от другой, так что волновая функция (перед тем, как она
будет антисимметризована) может быть представлена в виде произведения
ряда функций, каждая из которых представляет одну из групп электронов.
Мы используем обозначение Ха{ для координаты /-го электрона группы А
Средняя величина хА есть величина Хд„ усредненная по невозмущенной вол-
новой функции группы А. Суммы, фигурирующие в разд. А, могут быть
записаны в виде
1-1
22
/-U-1
22
i j
Z2
i J
I j
B.19)
?2
i j
вследствие чего
п п
2 2{
i j
22(*
i J
Но если волновая функция действительно может быть представлена в виде
произведения волновых функций различных групп электронов, то статисти-
728
Гл. 13. Теория межмомкулярных сил
ческая корреляция движений электронов двух различных групп должна отсут-
ствовать, т. е. должно соблюдаться условие
-хв) = 0. B.20)
Таким образом, поляризуемость может быть представлена в виде суммы
поляризуемостей составляющих групп
OL = (Хд -\- CLq -f- OLq -p . . . \Z./,l)
Таким образом, аддитивность поляризуемостей является мерой разделимости
молекулы на группы электронов, такие, как внутренние оболочки электронов
или электроны химических связей.
Таблица 114
Поляризуемости связей [46]
Связи
С—С (алифатическая)
С—С (ароматическая)
С=С
С=С
С—Н (алифатическая)
С-С1
С-Вг
ав • 10«5,
см*
18,8
22,5-
28,6
35,4
7,9
36,7
50,4
а±. 10",
см*
0,2
4,8
10,6
12,7
5,8
20,8
28,8
Связи
С=О (карбонил)
С=О (СО2)
C=S (CS2)
C=N (HCN)
N-H (NH8)
S-H (H2S)
a H ' 10«5.
CM3
19,9
20,5
75,7
31
5,8
23,0
aj_ • 10«s,
CM*
7,5
9,6
27,7
14
8,4
17,2
В табл. 114 приводятся поляризуемости некоторого числа связей, оценен-
ные Денбигом [46]. Отметим, что поляризуемость связи аи в направлении,
параллельном связи, отличается от поляризуемости ах в направлении, перпен-
дикулярном связи. Если электрическое поле направлено под углом в к хими-
ческой связи, то поляризуемость ае вычисляется по формуле1)
ае = а „ cos2 в + ах sin2 в. B.22)
Усредняя U0 по всем ориентациям (по всевозможным телесным углам), полу-
чаем среднее значение поляризуемости связи
а = -j- (а и + 2а j_). B.23)
Средняя поляризуемость молекулы является суммой средних поляризуемостей
ее связей.
Эти поляризуемости связей очень полезны для расчетов энергии протя-
жения неполярных или полярных многоатомных молекул, пропорциональной
обратной шестой степени расстояния между молекулами (дисперсионных сил
Лондона). Этот вопрос обсуждается в § 3. Описанные выше простейшие правила
получения поляризуемости молекул непригодны для молекул, в которых
имеется резонанс между структурами друх или большего числа связей.
2) В декартовых координатах с осью г, направленной вдоль связи, тензор поляризуе-
мости связи имеет вид
Yaj. О 0 \
a = 10 ах О
\0 О aj
В формуле B.22) а$ является гг-компонентой поляризуемости в новой системе координат,
повернутой на угол в относительно системы координат с осью z, направленной вдоль связи.
Таблица 115
Поляризуемость молекул1)
Молекула
н2
N2
о2
С12
HF
НС1
НВг
HJ
N2O
СО
СО2
soa
H2S
cs2
NH3
(CNJ
HCN
CH4
C2He
CH2 = CH2
CH s CH
C8H8
CH8C1
CHaCl2
CHC18
CC14
CH3OH
a- 10»»,
CM'
7,9
17,6
16,0
46,1
24,6
26,3
36,1
54,4
30,0
19,5
26,5
37,2
37,8
87,4
22,6
50,1
25,9
26,0
44,7
42,6
33,3
62,9
103,2
45,6
64,8
82,3
105,0
32,3
a, • 10",
CM*
9,3
23,8
23,5
66,0
(9,6)
31,3
42,2
65,8
48,6
26,0
40,1-41,0
54^
40,4
151,4
24,2
77,6
39,2
26,0
54,8
E6,1)
51,2
50,1
123,Г
54,2
50,2
66,8
105,0
40,0
a, 10«\
CM*
7,1
14,5
12,1
36,2
G,2)
23,9
33,1
48,9
20,7
16,25
19,7-19,3
27,2
34,4
55,4
21,8
36,4
19,2
26,0 '
39,7
C5,9)
24,3
69,3
63,5
41,4
84,7
90,1
105,0
25,6
a, • 10«s,
CM*
7,1
14,5
12,1
36,2
G,2)
23,9
33,1
48,9
20,7
16,25
19,7-19,3
24,9
40,1
55,4
21,8
36,4
19,2
26,0
39,7
C5,9)
24,3
69,3
123,1
41,4
59,6
90,1
105,0
31,4
Положение главных осей и направле-
ние дипольного момента; структура
молекулы
ах ось симметрии
ах то же
«1
«1
<*i
ах ось симметрии
ах то же
«1
ах ось симметрии; молекула ли-
нейна и не симметрична.
N = N = 0
а, ось симметрии
ах симметричная, линейная
А* = Hz, «2i плоскости OSO
/и = Рг> «21 плоскости HSH
ах ось симметрии; линейная
молекула
ах ось симметрии ; /г = цх
пирамидальная
ах ось симметрии, линейная
ах - то же
Правильный тетраэдр
ах ось симметрии
аг то же
ах ось симметрии, линейная
au плоскости ССС
au плоскости кольца
1* = Н>\> ai ось симметрии
[л = ^3; au плоскости C1QC1
ах ось симметрии ; fi = /их
Правильный тетраэдр
Вычислено для /г_1_0С;
Z(ax, /г) = 700
*) Поляризуемости, приведенные в таблице, взяты из справочника Ландольта—Бернштейна [94].
730 Гл. 13. Теория межмолекулярных сил
Экспериментальные значения поляризуемостей приведены в табл. 115.
Величины а1У а2, а3 являются тремя главными компонентами поляризуемости.
Средняя поляризуемость а равна а = х/8 (ai + а2 + аз)-
ПРИМЕР
Задача. Вычислить ахх, ауу, azz и а для молекулы этилена.
Решение. Выберем систему координат с осями, направленными вдоль главных осей
молекулы
С помощью формулы B.22) и табл. 114 получим поляризуемость вдоль оси С = С
azz = (а„)с=с + 4(а„)с-н cos2 56° + 4(ах)с-н sin2 56°
= 28,6 + 4 G,9) @,5592J + 4 E,8) @,8290J
= 28,6 + 9,9 + 15,9 = 54,4 • Ю-25 см*.
Поляризуемость в направлении, перпендикулярном оси С = С в плоскости молекулы, равна
<*хх = (ai)c=c + 4(an)c-H cos2 34° + 4(ai)c-H sin2 34°
= 10,6 + 4 G,9) @,8290J + 4 E,8) @,5592J
= 10,6 + 21,7 + 7,3 = 39,6 • Ю-26 см\
Поляризуемость в направлении, перпендикулярном плоскости молекулы, равна
ауу = (ai)c=c + 4(а1)с-н = Ю,б + 4 E,8)
= 10,6 + 23,2 = 33,8 • 102« см*.
По формуле B.23) и с помощью табл. 114 получим усредненную по углам поляризуе-
мость молекулы:
(а)Сгн4= Е «= Г 7»(а|! + 2а±)
по всем по всем
связям связям
а для связи С=С : 4" B8>6 + B) A0>6)) = 16>6'
а для связи С—Н : 4" G>9 + B) E>8)) = 6,5.
Отсюда (а)с2н4 = A6,6) + 4F,5) = 42,6-10-26 см*. Следует отметить, что это то же самое,
что и a = 73 (ахх + ауу + аи).
Г. ОЦЕНКА ПОЛЯРИЗУЕМОСТИ И ДРУГИХ СВОЙСТВ АТОМОВ
ПУТЕМ ИСПОЛЬЗОВАНИЯ ЭКРАНИРОВОЧНЫ* ПОСТОЯННЫХ
Поляризуемость и многие другие физические свойства атомов, такие, как раз-
меры, диамагнитная восприимчивость, электрическая восприимчивость и иони-
зационный потенциал, могут быть просто и довольно точно оценены с помощью
«экранировочных постоянных». Основой этого метода является тот факт, что
электрон в атоме не «чувствует» полного воздействия ядра, так как электрон
экранируется от ядра другими электронами. Поэтому хотя заряд ядра и равен
Ze, 1ем не менее электрон в атоме ведет себя так, как если бы. он двигался в
поле ядра заряда (Z—S)e. Величина S называется экранировочной постоянной.
Из одного свойства атома можно определить ряд экранировочных постоянных
§ 2. Поляризуемость молекул
731
и затем использовать их для вычисления других свойств. Из ряда способов,
предложенных для эмпирического определения атомных свойств [47, 48],
наиболее удовлетворительным является способ Слетера [49 ], который позднее
обосновали теоретически Гомбаш и Гаспар [50].
Предположим, что волновые функции каждого электрона в атоме можно
записать в виде
.t lt m = Rn. (г) YT@, ф); /?„* (г) =
B.24)
здесь У™@, ф) — нормированные сферические функции, определенные выра-
жением (Б.З) гл. 12; г измеряется в единицах боровского радиуса
ао=0,5292 А. На больших расстояниях эта функция ведет себя, как водородо-
подобная волновая функция с главным квантовым числом п* и эффективным
зарядом ядра (Z — S). Эффективное главное квантовое число п* и эффективный
заряд ядра (Z — S) для различных электронов в атоме определяются из сле-
дующих эмпирических правил:
1) Если главное квантовое число электрона п, то величина п* определяется
из следующих данных :
п
п*
1
1
2
2
3
3
4
3,7
5
4,0
б
4,2
2) Для определения (Z — S) электроны подразделяются на следующие
группы, каждая из которых имеет различные экранировочные постоянные :
is
2s
2р
3s
Ър
3d
4s
Ар
Ad
4/
5s
Ър
Ы
Иными словами, электроны s и р для одинакового п группируются вместе, но
электроны d и / рассматриваются раздельно. Группы образуются, начиная от
внутренней, в порядке, приведенном выше, считая Is самой внутренней.
3) Экранировочная постоянная S составляется для любой группы элект-
ронов из следующих вкладов :
а) нуль от любой оболочки, являющейся внешней, по отношению к рас-
сматриваемой оболочке ;
б) 0,35 от каждого электрона рассматриваемой группы (исключая группу
Is, где используется величина 0,30);
в) 0,85 от каждого электрона с квантовым числом, на единицу меньшим
квантового числа рассматриваемой оболочки, если таковой является оболочка
s или р ; 1,00 от каждого электрона, квантовое число которого отличается от
квантового числа рассматриваемой оболочки больше, чем на единицу;
г) если рассматриваемой оболочкой является оболочка d или /, то 1,00 от
каждого электрона внутренних оболочек.
Ниже приводится несколько примеров, показывающих, как определяются
величины п* и S.
примеры
Задача. Найти эффективные ядерные заряды (Z — S) и эффективные главные кванто-
вые числа л* для : а) нормального углерода C(ls2, 2s2, 2p2) и б) однократно ионизованного
углерода C(ls2, 2р2, 2р). Для углерода Z = 6.
Решение.
а) (Z — SIS =6 — 0,30 = 5,70,
(Z - 5JS, 2Р = б — C) @,35) - B) @,85) = 3,25,
б) (Z — SIS =6 — 0,30 = 5,70,
(Z - SJS, 2P = б - B) @,35) - B) @,85) = 3,60,
= 2,
= 1,
= 2.
732 Гл. 13. Теория межмолекулярных сил
Задача. Найти эффективные ядерные заряды (Z — S) и я* для 26 электронов ь нормаль-
ном железе Fe (Is2, 2s2, 2р«, 3s2, 3pe, 3d6, 4s2).
Решение.
(Z — S)is = 26 — 0,30 = 25,70, n\ = 1,00,
(Z — SJS,2p = 26 — G) @,35) - B) @,85) = 21,85, n* = 2,00,
(Z - S)8s,8p = 26 — G) @,35) - (8) @,85) - B) A,00) = 14,75,) n* - 4 00
(Z — S)zd = 26 — E) @,35) — A8) A,00) = 6,25, 1 п» " 6>Ш>
(Z — S)iS = 26 — A) @,35) — A4) @,85) — A0) A,00) = 3,75. n* = 3,70.
Для вычисления поляризуемости и диамагнитной восприимчивости атомов1)
необходимо знать г?. Для волновой функции B.24) средняя величина Л-й сте-
пени радиуса для /-го электрона равна
_ j
г? = -
/с
[// B п? + /)] 4. B.25)
Следовательно, зная экранировочные постоянные и эффективные квантовые
числа, можно вычислить атомные поляризуемости. Кроме того, эти же эмпири-
чески определяемые постоянные могут быть использованы для вычисления
энергий ионизации и атомных радиусов. В качестве иллюстрации полезности
метода экранировочных постоянных приводятся следующие примеры.
ПРИМЕРЫ
Задача. Вычислить поляризуемость и диамагнитную восприимчивость атомного водо-
рода в его основном состоянии.
Решение. В данном случае (Z — S) = 1 и п* = 1, так что
? = [(z
= [2(z-sj]Bл*+!) Bп*+ 2) а*= Ш*
В соответствии с формулой B.186) поляризуемость равна ___ (г2J = 4а». Диамагнит-
ная восприимчивость равна 9а0
fjx(m) = — 7,92 • Ю-7 -~- = - 2,38 • 10-« см*/моль.
Задача. Вычислить поляризуемость и диамагнитную восприимчивость атомов углерода
в их основных состояниях.
Решение.
? []2 C) D) aS = °'°923 al
E) (б) а% = 2,8402 a*.
Поляризуемость тогда равна 14,35 я» а диамагнитная восприимчивость равна
Ntfjn) = —9,14 • 10-* смг\молъ.
Задача. Энергия, необходимая для удаления всех электронов от v-электронного
атома в определенном состоянии, равна
Е = - 13,603 ^f-^A)%e, B.26)
где 13,603 — величина е*/2а0 в эв. Вычислить энергию нормального и однократно ионизован-
ного углерода и затем энергию ионизации углерода.
х) Для атомов диамагнитная восприимчивость на 1 моль равна
См. [51, стр. 91].
§ 3. Дисперсионные силы Лондона, действующие между симметричными молекулами 733
Решение. Для нормального углерода C(ls2, 2s2, 2ра)
Е = - 13,603 |^2 E,70J + 4 (~"J] = — 1027,5 эв.
Для однократно ионизованного углерода C(ls2,2s2,2p)
Е' = - 13,603 Г2 E,70J + 3 (^-J] = - Ю16,1 эв.
Энергия ионизации равна
Яион. = ?' — Е = - 1016,1 + 1027,5 = 11,4 эв.
Задача. Радиус определенной электронной оболочки определяется как расстояние, на
котором плотность электронного заряда е \ у> \* имеет максимум. Это расстояние равно
(Этот атомный радиус полезен для оценки межъядерных расстояний в валентных объедине-
ниях. Длина связи равна сумме двух атомных радиусов двух атомов или ионов, образующих
связь.) Вычислить атомный радиус для 4s электронов в железе.
Решение.
§3. ДИСПЕРСИОННЫЕ СИЛЫ ЛОНДОНА, ДЕЙСТВУЮЩИЕ МЕЖДУ
СИММЕТРИЧНЫМИ МОЛЕКУЛАМИ1)
В квантовомеханических расчетах энергии взаимодействия двух молекул
дисперсионные силы Лондона появляются при учете членов возмущения вто-
рого порядка. Дисперсионными они называются потому, что эти члены возму-
щения выражаются через те же силы осцилляторов //ь которые появляются
в формулах для дисперсии света (см. гл. 12, § 6).
Классическое объяснение дисперсионных сил, действующих между двумя
молекулами а и Ь, следующее. В любсй момент электроны в молекуле а имеют
определенную конфигурацию — такую, что молекула имеет мгновенный
дипольный момент (даже если она не обладает постоянным электрическим
моментом). Этот мгновенный диполь молекулы а индуцирует диполь в молекуле ft.
Взаимодействие между этими двумя диполями приводит к возникновению
сил притяжения между двумя молекулами. Тогда дисперсионная сила равна
этой мгновенной силе притяжения, усредненной по всем мгновенным конфи-
гурациям электронов в молекуле а. Это объяснение выглядит качественно пра-
вильным и вполне правдоподобным. Однако этот механизм не очевиден с точки
зрения квантовомеханических расчетов. В этом отношении лучше не делать
упора на классическом истолковании этих сил.
Мы начнем с простой трактовки дисперсионных сил, основанной на модели
Друде (см. гл. 12, § 5), в которой молекула представляет собой трехмерный
осциллятор. Затем мы обсудим возмущения второго порядка. Ослабление дис-
персионных сил на очень больших расстояниях вследствие эффектов запазды-
вания (конечности скорости распространения электромагнитных волн) обсу-
ждается в конце настоящего параграфа.
А. УПРОЩЕННАЯ ТЕОРИЯ ДИСПЕРСИОННЫХ СИЛ, ОСНОВАННАЯ
НА МОДЕЛИ ДРУДЕ
В гл. 12, § 5 обсуждена простая теория дисперсии света, основанная на
модели молекулы, предложенной Друде. Здесь мы используем эту модель для
описания дисперсионных сил, действующих между молекулами, чтобы полу-
См. [52, 54].
734
Гл. 13. Теория межмолекулярных сил
чить выражения для сил через поляризуемости молекул. В модели Друде
молекула а состоит из пары гармонически и изотропно связанных частиц,
обладающих равными и противоположными по знаку зарядами еа и имеющих
приведенную массу та. (В гл. 12, § 5 молекула состояла из ряда гармонически
связанных частиц. Здесь для простоты мы рассматриваем только одну пару
частиц.) Силовые постоянные обозначаются через ка, так что частота колебаний
va = A/2я) Ука1та. Аналогичные соображения применимы к молекуле Ь.
Рассмотрим сначала уравнения движения пары молекул, находящихся
одна от другой на расстоянии rab. Система из двух молекул имеет шесть частот
колебаний. Если молекулы находятся на достаточно большом расстоянии, то
набор частот колебаний состоит из трижды вырожденной частоты va (соответ-
ствующей невозмущенным колебаниям молекулы а) и трижды вырожденной
частоты vb (соответствующей колебаниям молекулы Ь). Когда молекулы сбли-
жаются, частоты колебаний возмущаются. Энергия взаимодействия молекул
получается из кванторомеханических соображений. Если молекулы по всем
шести колебательным степеням свободы находятся в основных состояниях, то
энергия взаимодействия равна разности между нулевой энергией возбужденной
системы и нулевой энергией двух разделенных молекул. Очевидно, что метод
позволяет также найти энергию взаимодействия молекул в возбужденных
колебательных состояниях, но эти взаимодействия трудно интерпретировать.
Пусть дга и дгь — смещения вибраторов а и & из их положения равновесия.
Потенциальная энергия системы составляется из вкладов, обуслоьленных сме-
щениями осцилляторов
и члена, представляющего электростатическое взаимодействие. Если молекулы
удалены одна от другой на достаточно большое расстояние (так что применима
концепция идеального диполя), то энергия взаимодействия дается формулой
A.45) гл. 12 или членом Ьг\г\ь выражения A.10). В декартовой системе коорди-
нат энергия равна
<РаЪ = ^~ [Ь*а д*Ь + дУа fyb ~ 2<fefl dzb] , C.2)
где ось z направлена вдоль оси, соединяющей ядра.
Сила, действующая на выбратор а, равна градиенту потенциальной энергии
системы по дгау взятому с обратным знаком. Таким образом, в соответствии со
вторым законом Ньютона уравнения движения системы приобретают вид трех
пар связанных уравнений :
d2 (dXg)
d2 (дхь)
пгп
= — kh dxh —
~dt2
I **^Ь ал —
еаеь
rib
еаеь
rib
еаеь
еаеь
rib
ext
b у
C.3)
тпа
Решения этих уравнений зависят от начальных условий. Однако по истечении
очень короткого промежутка времени неустановившаяся часть решения зату-
§ 3. Дисперсионные силы Лондона, действующие между симметричными молекулами 735
хает и решения оказываются периодическими. Если решения вида
6ха = Ахе^ы, дуа = Ауё**ы и т. д. подставить в дифференциальные урав-
нения, то мы получим набор линейных однородных уравнений для амплитуд.
Эти уравнения имеют решение, только если детерминант из коэффициентов
уравнений равен нулю. Это приводит к «секулярному», или детерминантному,
уравнению для частот колебаний. Детерминант может быть раскрыт, в резуль-
тате чего получим два одинаковых уравнения ьида
(ка - та 4л> »*) (къ - ть 4л» *) = J™ C.4)
и одно уравнение вида
(ка - та 4я* **) (кь - тъ 4тг* v*) = ^f?-. C.5)
'ab
Рассматриваемая система имеет шесть колебательных степеней свободы и
шесть фундаментальных частот колебаний. Однако вследствие цилиндрической
симметрии (оси х и у эквивалентны) имеются две пары дважды вырожденных
частот. Эти частоты определяются как корни уравнения C.4). Две невырожден-
ные частоты являются корнями уравнения C.5). Уравнения для частот колеба-
ний могут быть записаны через частоты колебаний отдельных молекул и поля-
ризуемости. В соответствии с изложенным в гл. 12, § 5, поляризуемость (модели
молекулы Друде) в стационарном электрическом поле (поле нулевой частоты)
выражается в виде
Таким образом, уравнения для частот колебаний могут быть переписаны в виде
C.7)
М-ОМ-^ = 4*2»*-^. C.8)
'ab
С точки зрения квантовой механики энергия рассматриваемой системы
равна сумме шести членов — каждой нормальной частоте соответствует один
член. Иными словами, энергия выражается следующим образом :
Общая энергия = ^hvt In, + -у I, C.9)
1=1
где vt — классические частоты колебаний, вычисляемые только что описанным
способом ; щ — колебательные квантовые числа соответствующей нормальной
частоты. Энергия взаимодействия равна разности между общей энергией систе-
мы и общей энергией разделенных молекул. Энергия взаимодействия двух
молекул, находящихся в низших колебательных состояниях, равна
б \
f лисп ^ *^ ^^Г* о о I /*^. Л С\\
УаЪ ' ~ ~2~ J-iVi — ^ Va — ^ vb\ ' (О. IV)
Очевидно, что существование энергии взаимодействия зависит от существо-
вания нулевой энергии, что является чисто квантовомеханической концепцией.
Явные выражения для фундаментальных частот vty, vty и т. д. из урав-
нений C.7) и C.8) могут быть получены в двух специальных случаях:
1) Системы, в которых возмущения малы по сравнению с разностью между
частотами разделенных молекул, т. е.
\va — Vb\
ГаЬ
736 Гл. 13. Теория межмолекулярных сил
В этом случае
() _ v B) __
x -У у -
2) Системы, в которых обе молекулы подобны и возмущения малы, т. е.
va = vb = v0,
<*a = ab = a C.12)
В этом случае
C.13)
Рассмотрим.теперь общую энергию системы, состоящую из двух взаимо-
действующих вибраторов, находящихся в осноьном состоянии. Иначе говоря,
рассмотрим столкновение двух молекул, каждая из которых находится в основ-
ном состоянии, и предположим, что во время столкновения не происходит изме-
нения колебательных квантовых чисел. Если возмущение мало по сравнению
с разностью между частотами разделенных молекул (случай 1), то энергия
взаимодействия (т. е. разность между общей энергией системы и энергией раз-
деленных атомов) равна
Если молекулы подобны и возмущение мало (случай 2), то найдем, что энергия
взаимодействия равна
Следует отметить, что это выражение является специальным случаем выра-
жения C.14).
Таким образом, мы получили выражение для потенциала взаимодействия
между неполярными молекулами—потенциала «дисперсионных сил». Выра-
жения получены с помощью сильно идеализированной модели, но результаты
интересны. Зависимость потенциала от поляризуемостей отдельных молекул и
зависимость от межмолекулярного расстояния по закону обратной шестой
степени согласуются с результатами более точной трактовки.
§ 3. Дисперсионные силы Лондона, действующие между симметричными молекулами 737
Б. ДИСПЕРСИОННЫЕ СИЛЫ КАК ВОЗМУЩЕНИЯ
ВТОРОГО ПОРЯДКА
Более точная теория дисперсионных сил, действующих между реальными
молекулами, может быть основана либо на вариационном решении квантово-
механической проблемы, либо на решении методом возмущений. Для очень
больших расстояний между молекулами обменные интегралы убывают по
экспоненциальному закону и становятся пренебрежимо малыми по сравнению
с кулоновскими интегралами. Поэтому нет необходимости антисимметризовать
волновые функции относительно координат электронов, и в качестве волновой
функции системы в нулевом приближении можно выбирать простое произве-
дение волновых функций отдельных молекул.
Подробные расчеты энергии взаимодействия двух атомов водорода вариа-
ционным методом приводятся в гл. 14, § 1. Здесь мы рассмотрим приложение
результатов второго приближения теории возмущений к общей теории дис-
персионных сил.
В «сферических» молекулах эффект всех ядер таков, как будто они распо-
ложены в одном центре, так что при рассмотрении далеких взаимодействий
можно считать, что молекулы ведут себя, как отдельные атомы. В соответствии
с изложенным в § 1 квантовомеханическая проблема расчета потенциальной
энергии межмолекулярного взаимодействия является проблемой определения
электронной энергии для частной конфигурации. Общая потенциальная энер-
гия системы Фе определяется выражением A.1). Часть, представляющая взаи-
модействие между двумя молекулами <ре [выражение A.2)], действует как
возмущающий потенциал.
Возмущающий потенциал сре в выражении A.10) разложен по обратным
степеням расстояния между молекулами. Чтобы получить главный член в раз-
ложении дисперсионной энергии, необходимо рассмотреть только первый не-
исчезающий член в разложении (ре> т. е. член bsjr%b. В первом приближении
энергия взаимодействия, равная значению q>e, усредненному по квадрату невоз-
мущенной волновой функции, равна нулю, если атомы (или молекулы) не имеют
результирующего момента количества движения (например, атомы в S-состоя-
ниях).
Поэтому необходимо рассмотреть возмущения второго порядка. Если
атомы (или молекулы) имеют результирующий момент количества движения,
то возмущение первого порядка не равно нулю. Эффекты возмущения первого
порядка рассматриваются в § б как мультиполь-мультипольные электростати-
ческие взаимодействия. Они накладываются на дисперсионные силы (возму-
щения второго порядка), обсуждаемые в настоящем параграфе.
Решение квантовомеханических задач методом возмущений обсуждалось
в гл. 1, § б. Охарактеризуем состояние изолированной молекулы а магнитным
квантовым числом та и числом ка> символически обозначающим все остальные
квантовые числа. Аналогичные обозначения используем для молекулы Ь. Оче-
видно, что энергии отдельных молекул вырождены по магнитному кванто-
вому числу. Обозначим эти энергии через Ека и Екь. Волновую функцию
нулевого порядка сложной системы (т. е. пары сталкивающихся молекул)
запишем в виде произведения двух волновых функций изолированных
молекул :
У>аъ (ка та кь ть | г") = гра (ка та | г"«) уь (кь ть | г"»). C Л б)
Выражение F.55) гл. 1 для энергии возмущения во втором приближении
строго применимо только к невырожденным системам. Однако, как хорошо
известно, это выражение может быть использовано для вырожденных систем,
если используются «правильные» вырожденные волновые функции. Отсюда
найдем, что энергия возмущения системы во втором приближении, т. е. диспер-
738 Гл. 13. Теория межмолекулярных сил
сионная энергия пары молекул1) равна
„(дисп.)/ь т k т \г \ — X" 1 (fa та kb ть \ я>е \ ка та кь ть) j2 n m
<Раь Ука та кь ть | г at) - - 2 Ей + ЕК-Еь-Еь ' {ЗЛ7)
к'Ьу ть
Суммирование распространяется на все квантовые числа, за исключением чле-
нов, имеющих к'а = ка и к'ъ — кь, которые опускаются. Матричные элементы
числителя определяются следующим образом:
(катакътъ\<ре\кат'ак'ьть) = (jyg(ката|г"-)ygf(kbтъ\гп») х
х ч>е Wa № та \ г"-) уь (к'ь т!ь \ г"») Л* dr«b. C.18)
Если мы примем <рс = b3jrlb и используем явное выражение для Ь^ давае-
мое формулой A.12а), то выражение C.17) для дисперсионной энергии примет
вид
= г%^ ^* Ект + Ёкь — Ек'а^Ёк'ь ^а Ша ' Х<2^а та^ ^ъ Ш& I X& I kb т^ ~^~
к* т^
Н Щ
+ (kama\ya\kfam'a)(khmb\yb\kfbmfb)-2(kama\za\kamfa)(kbm C.19)
Обычно мы интересуемся только средним потенциалом межмолекулярного взаи-
модействия для столкновений молекул в состояниях, характеризуемых всеми
значениями магнитных квантовых чисел. Если полученное выражение для
дисперсионной энергии усреднить по всем магнитным квантовым числам обеих
молекул, то мы получим выражение, которое просто может быть записано через
«квантовомеханические силы осцилляторов», определяемые выражением F.22)
гл. 12:
Ч>аЪ \Ка Ч | Гаь) ~ { 2л ) г\Ъ Т
fa (ка , k'g) fb (kb , kb)
fat [Ekb - Ец\ [Ek. + Ekb - Ек> - Ец] '
где т—масса электрона.
Следует отметить, что метод возмущений полезен (т. е. дает хорошее при-
ближение), если только возмущения малы по сравнению с разностью между
энергетическими уровнями или, более точно, если неравенство
75" \(ка та | xj К т'а) {кьть \ хь \ к'ът'ь) | < | Ека + Екь - ЕК - Ец\ C.21)
удовлетворяется для всех значений квантовых чисел и если аналогичные усло-
вия удовлетворяются для у- и z-направлений. Энергию систем, для которых
эти условия не соблюдаются, следует находить вариационным методом (см.,
например, § 6).
Из определения сил осцилляторов [F.22) гл. 12] ьидно, что fa(ka, k'a)
имеет тот же знак, что и (Ека, — Ека). Отсюда из выражения для дисперсионной
энергии непосредственно следует, что для молекул в основных состояниях (р^сп)
отрицательно и силы являются силами притяжения. Молекулы в возбужден-
ных состояниях могут притягиваться или отталкиваться одна от другой.
г) Более подробная и употребительная трактовка дисперсионных сил, действующих
между молекулами, находящимися в вырожденных состояниях, дана Дал ером и Гиршфель-
дером [95].
§ 3. Дисперсионные силы Лондона, действующие между симметричными молекулами 731
Определим постоянную с так, чтобы
<№*¦>=—&' C>22)
Эйзеншитц и Лондон [55] вычислили сумму. Они нашли, что для взаимодей-
ствия между двумя нормальными атомами водорода
с = 6,47 е* а%, C.23)
где % — радиус первой боровской орбиты, равный 0,5292 А. Эта величина срав-
нима с более точным значением
с = 6,5Ое2я|5, C.24)
полученным вариационным методом [56 ]. Интересно отметить, что по расчетам
методом возмущений заметная доля дисперсионной энергии получается за счет
состояний непрерывного спектра энергий.
Дисперсионная энергия C.20) выражена через силы осцилляторов. Оценка
этих величин может быть получена из измерений изменения показателя прелом-
ления света с частотой. Из выражений F.17) и F.22) гл. 12 найдем
у fa (kg, kg)
где т — масса электрона. Милликен [57, 58] показал, что в действительности
по существу очень часто происходит только один переход из основного состоя-
ния, который имеет заметный матричный элемент дипольного момента. В этих
случаях важна только одна величина fa(ka, k'a), и изменение показателя прелом-
ления с частотой может быть апроксимировано одним членом, т. е. выражением
где А и Ej—эмпирические постоянные. Постоянная ?7 интерпретируется как
средняя, или эффективная, разность энергий. Постоянная А пропорциональна
силе осциллятора и отсюда пропорциональна статической поляризуемости,
которая, согласно формуле F.21) гл. 12, имеет вид
*Д . /о 97^
Исходя из этих положений, приближенное выражение для дисперсионной
энергии можно записать через статические поляризуемости отдельных молекул
и эмпирические постоянные Е1а и EIb:
„,<дисп.)_ L ( Я/«Я/» ) а°аь . И ЭЯ\
таЬ — о I R 4- Е ) ""г* \у.^дэ/
Часто обнаруживается, что величины ?7 приблизительно равны энергии иони-
зации. Приближенное выражение C.28) для уЙГ011'* тесно связано с результатами
разд. А, полученными с помощью идеализированной модели Друде. Так как
потенциалы ионизации большинства молекул имеют одинаковый порядок вели-
чины, то из последнего результата следует, что в хорошем приближении по-
стоянная взаимодействия пары неодинаковых молекул связана с постоянными
взаимодействия между одинаковыми молекулами
саЬ = У саа сьь. C.29)
Это соотношение является основой одного из «комбинационных правил» для
силовых постоянных, которое обсуждалось в гл. 3 и 8 [формулы F.9) гл. 3 и
D.9) гл. 8].
740 Гл. 13. Теория межмолекулярных сил
Отметим еще два других приближенных выражения для дисперсионной
энергии. Леннард-Джонс [59 J1) с помощью приближенных квантовомеханиче-
ских расчетов получил выражение2)
саЬ = б /га? К й») + ab rfft, C.30)
где т — масса электрона ; с — скорость света ; %<т) — магнитная восприим-
чивость. Из других соображений Слетер и Кирквуд [60, 41 ] получили прибли-
женное соотношение
где /?2 и п? — числа электронов во внешних оболочках двух молекул; Oq — ра-
диус первой боровской орбиты, равный 0,5292 А. Это соотношение может быть
подвергнуто критике в том отношении, что при его выводе не учитывались
электроны внутренних оболочек. Помимо этого, если молекулы не одноатомны,
к выражению C.31) следует добавить дополнительные члены.
В. ЧЛЕНЫ ВЫСШИХ ПРИБЛИЖЕНИЙ В ВЫРАЖЕНИИ
ДЛЯ ДИСПЕРСИОННОЙ ЭНЕРГИИ
При получении выражений для дисперсионной энергии в разд. А и Б в
разложении сре учитывался только первый неисчезающий член. Если в разложении
уе учесть высшие члены (т. е. bJribJ b5jiiby...), то в выражении для дисперсион-
ной энергии мы получим члены, пропорциональные обратной шестой, восьмой,
десятой и т. д. степеням расстояния между молекулами, т. е.
(дисп.) L с ь /о оо\
таЬ — гв г8 г10 * * * \О«о&)
В разд. Б показано, как можно вычислить коэффициент с для атомов любого
сорта. Расчеты следующих коэффициентов с" и с" для реальных атомов или
молекул были выполнены только в двух случаях — в случае взаимодействия
двух атомов водорода (см. гл. 14, § 1) и в случае взаимодействия двух атомов
гелия (см. гл. 14, § 2). Однако для модели Друде расчеты могут быть выпол-
нены просто.
Предположим, как и в разд. А, что молекулы можно рассматривать как
трехмерные простые гармонические осцилляторы [61 ], обладающие единст-
венной классической частотой v0. Подтверждением возможности использования
только одной частоты могут служить экспериментальные наблюдения Милли-
кена [57, 58 ], из которых следует, что в передаче заряда от атома к атому эффек-
тивен переход только одного типа. В этом случае волновая функция отдельной
молекулы имеет вид [62, §11]
V(nx, пуу пг) = НПх(УРх)НПу(УРу)НпЛГР2)е~^> C.33)
где пх, пу, пг — колебательные квантоБые числа для колебаний р трех направ-
лениях ; Нп — полиномы Эрмита. Величина 0 равна
^ = ЖЛ"о = ^-, C.34)
х) Выражение C.30) может быть получено из выражения C.28) путем использования
экранировочных постоянных и приближенных соотношений для а, #(, а также соотно-
шений для энергии ионизации, приведенных в примере § 2.
2) По формуле C.30) саь получается примерно в 4 раза больше, чем по несколько лучшей
формуле, данной Маргенау [61 ]:
бтеаааь
Саь= -
— Прим. ред.
§ 3. Дисперсионные силы Лондона, действующие между симметричными молекулами 741
где т — приведенная масса осциллятора. Вторая форма записи получена с
помощью формулы (З.б), которая дает выражение а через /п.
Предположим, что волновые функции отдельных молекул а и b имеют вид
C.33), но с различными величинами /?, соответствующими различным фунда-
ментальным частотам va и vb и различным поляризуемостям аа и аь. Удержим
в выражении для <р€ члены, пропорциональные третьей, четвертой и пятой
обратным степеням расстояния гаЬу т. е. запишем q>€ в виде
C.35)
где &3> &4, h — определяются выражениями A.12). Чтобы получить выражение
для дисперсионной энергии, выражения A.12) необходимо подставить в C.17).
Если обе молекулы находятся в основных состояниях (все квантовые числа
равны нулю), то можно показать, что [63]
C.36)
C.37)
C.38)
L —
г»"
3
2
45
8
hva vb aa аь
(va + Vb) '
fl2 va vb aa аь Г
e2 L
) h*vlv2ba2aa2b
aava
2va + Vb
va+ 2vb J
16"
Если обе молекулы одинаковы, то эти выражения сводятся к выражениям,
полученным Маргенау [61 ]. Они дают лучший способ для оценок вкладов,
вносимых в дисперсионную энергию высшими приближениями.
Можно было бы предположить, что возмущения третьего порядка вносят
в ^Й"^ член, изменяющийся, как \\г%ь. Однако вследствие симметрии волно-
вых функций простых гармонических осцилляторов C.33), т. е. вследствие
правил отбора, этот член равен нулю.
Таблица 116
Сравнение теоретических и эмпирических расчетов дисперсионной энергии
Газ
Не
Ne
Аг
Кг
Хе
N2
о2
С12
сн4
со2
НС1
Квантовомеханические расчеты
с - 1Ово,
эрг • см*
11,4
1,23
4,67
55,4
107,0
233,0
57,2
39,8
321,0
112,0
152,0
111,0
с1 • 10™
'о
эрг - см9
31,0
1,89
6,9
120,0
275,0
710,0
120,0
96,0
1000,0
310,0
410,0
320,0
с" • 10й,
эрг - см10
45,0
1,65
5,3
136,0
370,0
1120,0
130,0
120,0
1630,0
440,0
590,0
480,0
[61]
<ввыч/ 1Ов°>
эрг • см*
15,5
1,52
5,64
66,7
130/)
280,0
66,8
48,8
386,0
135,0
180,0
144,0
Эмпири-
ческие
расчеты
*°вязк. • 1°в0>
эрг • см*
12,57
1,29
9,50
109,2
232,0
561,2
125,6
102,3
957,0
257,9
427,1
259,1
Сравнение
6°вязк.
гОвыч.
0,811
0,849
1,68
1,64
1,79
2,00
1,88
2,10
2,48
1,91
2,37
1,80
С помощью приведенных выше формул постоянные с, с', с" были вычислены
для ряда простых веществ1). Эти величины даны в табл. 116. В этой же таблице
приведена вычисленная из данных о вязкости величина сЛязк. = 4сог«,
г) Для Hg вычисленное значение с равно 255* Ю-60 эрг-см6 [54].
742 Гл. 13. Теория межмолекулярных сил
являющаяся коэффициентом при члене 1/г6 в потенциале Леннарда-Джонса.
Эту величину скорее следует сравнивать не с с [выражения C.32)], а с вели-
чиной Свыч., равной
с' с"
Свыч.— с 4" ~ъ Н~ -^4~ • C.39)
В табл. 116 мы приводим величину с?ыч. для сравнения с СвЯЗк. = 4есгв. Для
Н2 и Не имеется вполне удовлетворительное согласие между &ыч. и CbW, но
во всех других случаях сВЯзк. в 1,5—2,5 раза больше &ыч.. Вообще с' и с" вносят
в Свыч. вклад, равный приблизительно 20%. Причины отмеченного расхож-
дения неясны. Возможно, что это указывает на более быстрое убывание корот-
кодействующих сил в потенциале F-12) Леннарда-Джонса, нежели убывание
члена 1/г12, так что силы притяжения для получения того же значения потен-
циала могут не быть такими большими, как в потенциале F-12) Леннарда-
Джонса.
Г. ВЛИЯНИЕ «ЗАПАЗДЫВАНИЯ» НА ДИСПЕРСИОННЫЕ СИЛЫ
НА БОЛЬШИХ РАССТОЯНИЯХ
Если две нейтральные, неполярные и невозбужденные молекулы удалены
одна от другой на очень большое расстояние, то дисперсионная энергия оказы-
вается значительно меньше энергии, предсказываемой выражениями, полу-
ченными выше в этом параграфе. Причиной этого является «запаздывание»,
т. е. влияние конечности скорости распространения электромагнитных волн.
Уменьшение сил притяжения может быть качественно описано следующим
образом. Мгновенный диполь первой молекулы порождает электромагнитное
поле. По истечении времени таЬ\с (с — скорость света) это поле индуцирует
электрический момент во второй молекуле, и к моменту времени 2гаЬ\с электри-
ческое поле от индуцированного диполя достигнет первой молекулы. К этому
моменту времени мгновенный диполь первой молекулы изменится как по вели-
чине, так и по направлению, так что энергия взаимодейстгия несколько умень-
шится. Такая причина уменьшения энергии взаимодействия была предло-
жена [64] для объяснения стабильности лиофобных коллоидов, в которых
дальнодействующие силы оказываются несколько меньшими, чем можно было
ожидать.
Казимир и Полдер [65] рассмотрели проблему эффектов запаздывания с
точки зрения квантовой электродинамики. Так как их расчеты весьма сложны,
мы обсудим только результаты. Сначала они рассмотрели взаимодействие ней-
тральной молекулы с проводящей стенкой. Для малых расстояний между
молекулой и стенкой они нашли обычное выражение Лондона для энергии
взаимодействия
*=-^. г^Ь C-40)
где с — скорость света, a v0 — наиболее важная частота в выражениях для
показателя преломления, поляризуемости и дисперсионной энергии. Иначе
говоря, v0 является частотой, связанной с энергией Ег в выражении C.26).
Как отмечалось выше в связи с обсуждением выражения C.26), hv0 во многих
случаях приблизительно равна энергии ионизации.
Для расстояний, больших по сравнению с характеристической длиной,
К=~ • C.41)
Казимир и Полдер [65 ] нашли, что энергия взаимодействия между нейтраль-
ной молекулой и стенкой равна
ЗЛса с /л io\
r^> <3-42)
§ 4. Дисперсионные силы, действукщие между асимметричными молекулами 743
т. е. энергия взаимодействия на больших расстояниях вследствие эффектов
запаздывания уменьшается и имеет вид энергии взаимодействия для малых
расстояний, уменьшенной на величину ЗА0/2л;2г. На фиг. 206 показано, как этот
множитель изменяется с расстоянием.
Если hv0 выразить в эв, то Ло = 12 394//iv0. Потенциал ионизации — по-
рядка 10 эв. Отсюда характеристическая длина Ао — порядка 1200 А. Исполь-
зуя этот результат, по фиг. 206 можно установить, что около 400 А эффекты
запаздывания уменьшают энергию в 2 раза.
Значительно труднее, чем в случае взаимодействия молекулы со стенкой,
получить выражение для энергии взаимодействия двух нейтральных неполяр-
ных молекул, удаленных одна от другой на расстояние г. С этой целью были
рассмотрены первый, второй, третий и четвертый порядки возмущений. В ре-
зультате было найдено [65], что энергия взаимодействия выражается следую-
щим образом:
C.43)
где g— множитель, изменяющийся в пределах от единицы (для г <^ Ао) до
23А0/оя2г (для г ^> Яо). Для промежуточных расстояний зависимость g от г/А0
показана на фиг. 207. Поскольку Ао ^ 1200 А, влияние запаздывания стано-
вится важным на расстояниях порядка 200—300 А.
0,25 0,50 0,75 1,00
Фо
Фиг. 206. Поправочный множотель,
определяющийся эффектами запаз-
дывания, для взаимодействия ней-
трального атома с проводящей
стенкой.
Энергия взаимодействия равна
/(ft/8»)
Фиг. 207. Поправочный множитель,
определяющийся эффектами запаз-
дывания, для взаимодействия двух
нейтральных неполярных молекул.
Этот множитель входит в выра-
жение C.43).
Таким образом, эффектом запаздывания можно пренебречь в случае взаи-
модействия небольших молекул. Однако он становится важным во взаимодей-
ствиях высоких полимеров и других больших молекул.
§ 4. ДИСПЕРСИОННЫЕ СИЛЫ, ДЕЙСТВУЮЩИЕ МЕЖДУ
АСИММЕТРИЧНЫМИ МОЛЕКУЛАМИ
Дисперсионные силы, действующие между асимметричными молекулами,
отличаются от сил, действующих между сферически симметричными молеку-
лами в друх отношениях : 1) на больших расстояниях энергия взаимодействия
изменяется, как 1/г^, но ее величина зависит от относительной ориентации
молекул ; 2) на промежуточных расстояниях закон изменения дисперсионной
энергии отклоняется от l/rgb. (Дисперсионная энергия становится значительно
744 Гл. 13. Теория межмолекулярных сил
меньше той, которую можно было бы ожидать на основе экстраполяции выра-
жения для энергии, справедливого для больших расстояний в область проме-
жуточных расстояний.) Помимо этого, дисперсионная энергия становится
чрезвывайно чувствительной к относительной ориентации двух молекул.
В этом параграфе мы сначала рассмотрим дисперсионные силы на больших
расстояниях, затем изложим результаты общей теории дисперсионных сил
Лондона [66 ] для средних расстояний и, наконец, обсудим расчеты дисперсион-
ных сил Дэвиса и Коулсона [67 ] для частной модели, удобной для описания
сопряженной двойной связи или ароматических молекул.
В системах с сопряженными двойными связями электрический заряд
свободно может перемещаться от одного конца молекулы к другому. Инду-
цированный диполь в такой молекуле обладает очень большим расстоянием
между его положительным и отрицательным зарядами. По этой причине со-
пряженная двойная связь и ароматические молекулы имеют аномальную
дисперсионную энергию. Кроме того, энергия может иметь некоторое число
максимумов и минимумов при изменении расстояния между молекулами и
изменении их относительных ориентации, что представляет собой весьма инте-
ресный результат квантовомеханических расчетов.
А. ДИСПЕРСИОННЫЕ СИЛЫ, ДЕЙСТВУЮЩИЕ МЕЖДУ
АСИММЕТРИЧНЫМИ МОЛЕКУЛАМИ НА БОЛЬШИХ РАССТОЯНИЯХ
Для вычисления дисперсионных сил, действующих между двумя асим-
метричными молекулами на больших расстояниях, Лондон [66] использовал
основы теории дисперсионных сил, обсужденные в § 3. Рассмотрим две моле-
кулы а и ft, обладающие аксиальной симметрией. Вследствие этой симметрии
каждая молекула имеет две компоненты поляризуемости вдоль главных осей
(см. § 2): поляризуемости вдоль оси, соединяющей молекулы аи(а) и <*„(&),
и поляризуемости вдоль оси, перпендикулярной оси, соединяющей молекулы
aL{a) и а±(Ь). Предполагается, что молекулы представляют собой анизотропные
трехмерные осцилляторы, обладающие фундаментальными частотами vn(a) и
vn{b) для колебаний вдоль оси и дважды вырожденными частотами vL(a) и
v±(b) для колебаний, перпендикулярных оси. Ориентации двух молекул харак-
теризуем углами вау фау вЬу фь (по поводу определения этих углов см. фиг. 192,
стр. 653). Можно показать [66, 96], что для асимметричных молекул, обладаю-
щих описанными свойствами, обобщением выражения C.14) является
= С Fд,
fab
где с (ва, фа; 06, фь) — коэффициент, зависящий от углов,
С (ва, фа ; КФь) =
= (А - В - В' + С) [sin ва sin 0„ cos (фь -фа)-2 cos 6a cos Qbf +
+ 3 (В - С) cos2 ва + 3 (В' - С) cos2 бь + (В + В' + 4С). D.2)
Величины А, В, В' и С определяются следующим образом:
§4. Дисперсионные силы, действующие между асимметричными молекулами 745
В этом параграфе для упрощения обозначений у д>(дисп<) опускается индекс ab-
Дисперсионная энергия D.1), усредненная по всем ориентациям двух осцил-
ляторов, равна
9>(дисп) = - ~ [ Л + 2 (В + В') + 4 С]. D.7)
Выражения D.1) и D.7) сводятся к результатам, полученным для изотропных
молекул, когда поляризуемости и частоты колебаний вдоль различных осей
равны между собой (А = В = В' = С). Поляризуемости вдоль оси, параллель-
ной и перпендикулярной оси, соединяющей молекулы, могут быть оценены
с помощью методов и таблиц, приведенных в § 2. Частоты нетрудно оценить,
так как маловероятно, чтобы vxx и vL сильно отличались одна от другой. Таким
образом, при предположении, что, как и в случае сферических молекул,
vn = vL = v и что hv = I (I—потенциал ионизации), в выражение для диспер-
сионной энергии вносится лишь небольшая ошибка1). Изложенный способ
описания дисперсионных сил, действующих между асимметричными моле-
кулами, полезен при рассмотрении взаимодействия между двумя молекулами
водорода (см. гл. 14, § 4).
Б. ДИСПЕРСИОННЫЕ СИЛЫ, ДЕЙСТВУЮЩИЕ МЕЖДУ АСИММЕТРИЧНЫМИ
МОЛЕКУЛАМИ НА СРЕДНИХ РАССТОЯНИЯХ
Простая теория дисперсионных сил, действующих между асимметричными
молекулами, находящимися в основных состояниях, на средних расстояниях
была развита Лондоном [66]. В соответствии с выражением C.17) дисперсион-
ная энергия асимметричных молекул, так же как и симметричных, находя-
щихся в основных состояниях, во втором приближении теории возмущений
записывается в виде
X" 1@>0; Q,Q\<Pe\k'atm'a\ k'b,m'b)\2
къ\ ть'
Разложение <ре по степеням A/га&), приведенное в A.10), непригодно для много-
атомных молекул. Поэтому мы должны использовать для <ре исходное выра-
жение A.2).
Некоторые приближения, использованные при расчетах взаимодействия
симметричных молекул, могут быть использованы при расчетах взаимодействия
между асимметричными молекулами. Во-первых, из выражения C.20) ясно, что
в главный член ^Сдисп.) (т. е. член, пропорциональный 1/rgJ дают вклад только
те переходы, в которых квантовые состояния молекул а и b изменяются одно-
временно. Во-вторых, следует заметить, что с каждым из переходов, который
дает вклад в главный член, связан дипольный момент [см. F.7) гл. 12].
И, в-третьих, для передачи заряда между атомахми важен только один частный
переход. Вследствие этого сумму в выражении D.8) оказывается возможным
свести к одному члену. Если к'а Ф 0 и к'ь Ф 0, то члены ZaZbe*\raby 2' Zbe2lrb. и
>! Zae2jraf, входящие в q>e, не дают вклада в матричные элементы. Соответственно
мы можем апроксимировать D.8) выражением
= _ 1@,0; o>O|ye|^,o;
(ЕЕ) + (Е
Де Бур [68] независимо получил D.1), предполагая, что уц = v±,
746
Гл. 13. Теория межмолекулярных сил
в котором
@,0; 0,
D.10)
где па и пь — числа электронов в молекулах а и Ь соответственно.
Определим электронную плотность Qa, связанную с переходом @,0)->(fca, 0)
в молекуле а :
=Я *''
... drna.
D.11)
Определим также Ra(—) как «центр тяжести» области, где да отрицательна,
а Ra(+) как «центр тяжести» области, где Qa положительна :
D.13)
Теперь электрический момент, связанный с переходом и выражающийся фор-
мулой F.7) гл. 12, можно записать в виде
-R«(-)K- D.14)
Вторая форма записи (ia является определением заряда еа, участвующего в
переходе. Аналогично могут быть определены величины eby R&(+) и R6(—).
Можно показать, что выражение D.9) через эти величины записывается в виде
^(ДИСП.) _- _
el el
h(va
vb) Г
\т +
I2.
J '
D.15)
здесь hva и hvb соответственно равны (Ека — ЕОа) и (Екь — ЕОь).
В качестве иллюстрации зависимости дисперсионной энергии от взаимной
ориентации пары молекул рассмотрим два крайних случая — ориентацию А
и ориентацию JB, изображенные на фиг. 208. В ориентации А обе молекулы
Я?1
'ab
Ориентация В
Ориентация ?
Фиг. 208. Для ориентации А : | #<,(+) — /&(+) | = | Ra(+) — Rb(—) И Ra{—) - Rb(+) !=
= | Ra(—) — Rb(—) 1 и <р(дисп.) =0;
для ориентации В: Ra(+) и Rb(—) столь близки одна к другой,
что ^(дисп.) выражается в виде D.17).
§ 4. Дисперсионные силы, действующие между асимметричными молекулами 747
перпендикулярны одна другой. В ориентации В обе молекулы располагаются
вдоль одной линии, причем их одноименные дипольные концы обращены в
одинаковые стороны. Для этих частных ориентации выражение D.15) переходит
в следующие:
Ориентация А
<р(ДИСП.):=0, DЛ6)
Ориентация В
ф(дисп.) = Ъ*Ь 1 /л \
9 h(va + vb) |Ra(+)-R*(-)|2 V
По-видимому, простейшим путем, дающим возможность понять взаимо-
действие между асимметричными молекулами, является путь, в котором моле-
кулы рассматриваются как реальные диполи с расстоянием между зарядами,
равным R(+) — R(—). Геометрия взаимодействующих молекул представлена
на фиг. 192 (стр. 653). Если D определить как отношение энергии взаимодей-
ствия реальных диполей к энергии взаимодействия идеальных диполей, то
. диполь-диполь
9>идеал. диполь-диполь
1
= ll (l l)[ l()
| Ra (+) - Ra (-) | | Rb (+) — R» (—) | [— 2 COS da COS 6b + Sin Oa Sin 0b COS @b - 0a)]
D.18)
Величина отношения D для четырех ориентации диполей, в которых
/ = j Ra(+) — Ra(—) | = | Rb(+) — Rb(—) \, изображена на фиг. 193 (стр. 654).
Выражение D.15) можно записать через D
> к'ь> °I2 D2[-2 cos ва cos вь + sin да sin вь cos (Фь - 0a)Y и
*r — h(va + ) %
D.19)
Как видно из фиг. 193, для большинства ориентации пары молекул D -> 1,
когда гаЬ становится большим. Отсюда 9?(дисп) для больших значений расстоя-
ния между молекулами изменяется, как 1/г^, и сильно зависит от взаимной
ориентации пары молекул. Можно также видеть, что это предельное поведение
потенциала практически достигается, когда rab^2ly где в качестве / выби-
рается наибольшая из величин | Ra(+) — Ra(—) I и I Rft(+) — R*(—) I-
В. ДИСПЕРСИОННАЯ ЭНЕРГИЯ ВЗАИМОДЕЙСТВИЯ ДЛИННЫХ
АРОМАТИЧЕСКИХ МОЛЕКУЛ С СОПРЯЖЕННЫМИ ДВОЙНЫМИ СВЯЗЯМИ
Дэвис и Коулсон [67] получили ряд волновых функций молекул, пред-
ставляющих различные электронные квантовые состояния длинных аромати-
ческих молекул с сопряженными двойными связями. Они использовали эти
волновые функции для прямых расчетов дисперсионной энергии для значи-
тельного числа относительных ориентации молекул и расстояний между ними.
Существенное отличие их трактовки от обсужденной в предыдущих пунктах
состоит в том, что в рассуждениях Лондона предполагается, что заметный вклад
в дисперсионную энергию дает переход только к одному из возбужденных
электронных состояний. Лондон не дает также какого-либо указания о том,
которое из возбужденных состояний является этим состоянием. Дэвис и
Коулсон рассмотрели все возбужденные состояния для частной модели
молекулы и нашли, что наиболее важный вклад дают переходы, которые соот-
ветствуют перескоку одного электрона в каждой молекуле с наименее связы-
вающей (или из состояния с высшей энергией) из связывающих молекулярных
748
Гл. 13. Теория межмолекулярных сил
орбиталей в состояние с низшей энергией несвязывающих молекулярных
орбиталей. Для больших расстояний между молекулами (см.табл. 118, стр. 753)
всеми другими переходами можно перенебречь.
При сближении молекул с сопряжейными двойными связями (или аро-
матических молекул) силы приобретают характер существенно направленных
сил, вследствие чего молекулы сближаются преимущественно под такими
углами, что они находятся в различных плоскостях. Тот факт, что почти во
всех органических молекулярных кристаллах, таких, как нафталины и антра-
цены, плоскости соседних молекул непараллельны, указывает на то, что это
заключение правильно. Чтобы внести полную ясность в эту проблему, тре-
буется проделать еще много работы.
Коулсон1) предложил следующее объяснение направленных сил. Пред-
ставим себе классический электрический осциллятор, в котором имеется опре-
деленное число стоячих волн для каждого вида колебаний. Электрический
потенциал таких осцилляторов или антенных систем имеет сильные направ-
ленные свойства. Поскольку при переходе от классической к квантовой теории
стоячие волны заменяются волнами де Бройля, то для нормальных видов дви-
жения электронов можно ожидать тех же направленных свойств. Это точно
совпадает с тем, что было получено Дэвисом и Коулсоном из подробных расче-
тов. Рассмотрим теперь коротко их метод и результаты.
Молекула а
Молекула b
Фиг. 209. Геометрия, которую Дэвис и Коулсон использовали для описания взаимодействия
двух линейных молекул.
Рассмотрим линейную молекулу, состоящую из 2/с подобных атомов оди-
наковых размеров с расстоянием / между соседними атомами, изображенную
на фиг. 209. Чтобы представить полиэновую цепь, во всех численных расчетах I
полагается равным 1,4 А, что соответствует нормальному разделению С—С.
Если в качестве атомного радиуса выбрать величину, равную J/2, и прибавить
ее к обоим концам молекулы, то общая длина молекулы окажется равной
L = 2Ы. Предположим, что каждый атом дает один электрон в общее число
подвижных электронов, или электронов, подобных тем, которые имеются в
металле. Свойства молекул как протяженных осцилляторов определяются
распределением электронов по молекулярным орбиталям, которые прости-
раются на всю длину молекулы. В рассматриваемых расчетах удобно считать
эти электроны принадлежащими 2рл атомным орбиталям2).
Пус1ь ось z направлена по перпендикуляру к оси молекулы. Явное выра-
жение для нормализованной 2рл орбитали, связанной с а-м ядром, занятым
*) Частное сообщение, декабрь 1951 г.
2) По поводу атомных орбиталей, детерминантов Слетера и т. д. см., например, F9]
или [70].
§ 4. Дисперсионные силы, действующие между асимметричными молекулами 749
/-м электроном, будет следующим:
ta(i) = y4-zaje-««; D.20)
здесь с — подбираемая экранировочная постоянная. В численных расчетах с
выбирается равной 1,625/а0, что соответствует 2рл орбитали углерода. Вслед-
ствие того, что орбитали ра(/) предполагаются взаимно ортогональными, интег-
ралами перекрытия можно пренебречь.
Из 2fc атомных орбиталей можно образовать 2fc линейных комбинаций,
известных как молекулярные орбитали,
1 f 2?ТТ sin BЕТГ) **•«>. D.21)
Если отрицательная величина E — резонансный интеграл [69, стр. 76] для
соседних атомов, то энергия электрона на /-й молекулярной орбитали, отсчи-
тываемая от его энергии на орбитали изолированного атома, равна
D-22)
Приемлемое значение ?, равное —40 ккал/моль, было предложено Милликеном,
Рике и Броуном [71 ], использовавшими данные спектроскопии и горения.
Так как /? отрицательно, то меньшему / соответствует низшая энергия. Когда
1 ^ / <; k, Sj отрицательно и молекулярные орбитали являются связывающими.
Когда к + 1 < / <; 2fc, ej положительно и молекулярные орбитали являются
разрыхляющими.
Основное состояние молекулы получается путем заполнения каждой из
низших к молекулярных орбиталей (/ = 1, 2, ..., к) в соответствии с принци-
пом Паули. Волновая функция основного состояния представляет собой детер-
минант Слетера [69 ], диагональный элемент которого имеет вид
Zi(l)ZiB)*.<3)ztD) • • • Z*<2* - i)xkBk); D.23)
здесь черта над буквой обозначает одно спиновое состояние, отсутствие черты —
другое. Возбужденные состояния представляют собой конфигурацию, в
которых один или большее число электронов находятся на разрыхляющих
орбиталях. В настоящей проблеме необходимо рассмотреть только синглетные
возбужденные состояния, в которых один или более электронов переходят
на разрыхляющие орбитали. Состояние, в котором электроны с /?-й связываю-
щей молекулярной орбитали переходят на q-ю разрыхляющую орбиталь,
известно как состояние (pq). Волновая функция этого состояния равна сумме
двух детерминантов Слетера, диагональные элементы которых имеют вид
D.24)
Рассмотрим теперь взаимодействие двух таких молекул. Дисперсионная
энергия имеет вид D.8). В сумме выражения D.8) наиболее важное возбужден-
ное состояние характеризуется (кау ка + 1 ; кЬу кь + 1). Это состояние является
тем состоянием, в котором один электрон каждой молекулы переходит с выс-
шего связывающего состояния на низшее возбужденное состояние.
При рычислении матричных интегралов всеми трех- и четырехцентровыми
интегралами можно пренебречь. Так как рассматриваются только сравнительно
большие расстояния между молекулами, то вклад дают только кулоновские
интегралы. Расчетные трудности возникают при рассмотрении ориентации, в
которых две молекулы непараллельны, так что направление тг-орбиталей двух
молекул различно. Интегралы, появляющиеся в расчетах, связаны с интег-
750
Гл. 13. Теория межмолекулярных сил
ралами, которые вычислил Бартлетт [72 ]. Вычисление интегралов значительно
упрощается в предположении, что на рассматриваемых расстояниях можно
пренебречь экспоненциальными членами. Число встречающихся интегралов
таково, что практически для всех возбужденных состояний необходимо вычи-
сление 2fc = 6 матричных элементов. Однако из расчета матричного элемента,
соответствующего переходу из основного состояния в состояние (ка, ка + 1 ;
hi К + 0> а также на основе сведений об относительной важности других
состояний с 2fc = 2, 4 и 6, можно сделать хорошие оценки энергий взаимодей-
ствуя реальных длинных молекул.
X'
э-
Фиг. 210. Координаты, использованные для описания положения одной молекулы
относительно другой.
Для описания положения одной (второй) молекулы относительно другой
(первой) необходимо задание четырех координат—х, у, в и ф. Эти координаты
изображены на фиг. 210 и могут быть определены следующим образом. Осью х
является ось первой молекулы. Ось у перпендикулярна оси х, проходит через
Моле пула
в плоскости ху
Фиг. 211. Три ориентации, рассмотренные при изучении взаимодействия двух длинных
молекул.
центр первой молекулы и находится в плоскости, на которой расположен центр
второй молекулы. Относительное положение центра второй молекулы описы-
вается заданием значений хиу. Оси х' и у' проходят через центр второй моле-
кулы и параллельны осям хиу. Угол в есть угол между осью второй молекулы
и осью х1. Плоскость, образуемая осью х' и осью второй молекулы, образует
угол ф с плоскостью ху.
§ 4. Дисперсионные силы, действующие между асимметричными молекулами 751
Обсудим теперь результаты, которые получаются для трех различных
ориентации двух молекул, показанных на фиг. 211.
Ориентация I. Молекулы параллельны одна другой и перпендику-
лярны межмомкулярной оси (х = в = ф = 0). Результаты расчетов для этой
ориентации, изображенной на фиг. 211, приведены в табл. 117 [67]. Когда
y\L > 1,8, энергия изменяется, как обратная шестая степень у, что можно ожи-
дать из теории Лондона. Для меньших расстояний между молекулами энергия
Значения энергии взаимодействия —
Таблица 117
для ориентации I (кал/моль)
2
4
6
10
20
• у. А
4
121,0
935,0
2620,0
7950,0
—
8
2,33
34,1
140,0
668,0
—
15
0,627
1,06
5,58
39,6
382,0
20
0,0567
0,199
1,12
9,10
116,0
30
0,06507
0,07343
0,1084
0,996
16,9
50
0,1134
0,09050
0,05965
0,0517
1,16
взаимодействия много меньше той, которую можно было ожидать на основе
распространения на эту область закона обратной шестой степени. Для малых
значений y\L энергия пропорциональна 2fc + 1 ^ L, для больших расстояний
между молекулами она пропорциональна Bfc + IN ^ L5. Для значений y/L,
больших 1,8, энергия взаимодействия приблизительно выражается следующим
образом :
?(дисп.) _
D.25)
Так как Дэвис [73] показал, что поляризуемость длинной молекулы изме-
няется, как L3, а потенциал ионизации изменяется, как L, теория Лондона
также ведет к пропорциональности 9?(дисп) величине L5: однако численные
коэффициенты несколько различны. Дэвис и Коулсон [67] нашли, что для
больших молекул вклад 2ра электронов в дисперсионную энергию много мень-
ше 2рп электронов, и поэтому они полагают, что настоящая модель весьма точна.
В табл. 118 для гексатриена СН2 = СН — СН = СН — СН = СН2, имею-
щего 2fe = б, даны вклады в дисперсионную энергию, вносимые различными
переходами. Очевидно, что наибольший вклад вносит переход C3; 44). Это
переход, в котором электрон в каждой молекуле перескакивает со связываю-
щей молекулярной орбитали с наивысшей энергией (или с последней связываю-
щей орбитали) на разрыхляющую орбиталь с низшей энергией. При расстоянии
между молекулами в 4 А заметный вклад дает переход C3 ; 55). Однако на
больших расстояниях дисперсионная энергия может быть вычислена с неболь-
шой ошибкой при учете только перехода C3 ; 44) и при пренебрежении всеми
остальными переходами.
Ориентация II. Молекулы смещены и параллельны (изменяются х и
у при в = ф — 0). Изменение энергии межмолекулярного взаимодействия при
изменении координаты х двух параллельных молекул (см. ориентацию II на
фиг. 211) представлено на фиг. 212. Для каждого значения у энергия притяжения
— ^(дисп.) максимальна при отсутствии смещения по х(х= 0) и проходит через
минимум для некоторого значения смещения хо(у). Положение этого минимума
трудно объяснить, так как вклад каждого возбужденного состояния в — ^(wcn>
обладает тем же свойством и имеет минимум при различных значениях х. Эта
сложная картина аналогична взаимодействию двух излучающих антенн. На
фиг. 213 показано изменение энергии взаимодействия в зависимости от х для
10000
1000 ~
о
Фиг,
JUU
100
10
W
0,1
-
л
i
\2к=6
\
\2к=4
\V
\
\2к=2
\ /
I
2к=Ю
V
^—
^—•
I
I
у=8А
^ ^^
I
10
Х,А
15
20
500-
400-
300 -
1
200-
/00 -
i I
/ /Вклад cm перехода
/1
C4; 34)
Вклад от перехода^
C5; 35)
_
у
/
/
уу
\ /
\ /
\ V
\ л
/ \ / -
/ N /
Вклад от w
перекода р- —
C4; 35) у \
/ \
/ v —
/ \
* \
30 60
в, град
90
212. Дисперсионная энергия
для ориентации II [67].
Фиг. 213. Зависимость дисперсионной
энергии от длины молекулы для ориен-
тации II при у = 8 А [67].
Фиг. 214. Дисперсионная энергия для
гексатриена Bк = 6) для ориентации III
при у = 7А.
Показаны вклады от различных членов [671.
§4. Дисперсионные силы, действующие между асимметричными молекулами 753
различных размеров молекулы при у = 8 А. Из изложенного становится ясно,
что при взаимодействии молекул действуют значительные силы, стремящиеся
удержать молекулы параллельными и обратньши одна другой.
Таблица 118
Относительная роль различных переходов для ориентации I гексатриена B1с=6)
(Энергия выражена в кал/моль)
Переход
C3; 44)
C3; 55)
C3; 46)
C2; 45)
C2; 56)
C2; 65)
C3; 66)
B2; 55)
C1; 46)
B2; 66)
C1; 66)
B1; 56)
(П; 66)
Вырождение
или
эквивалентное
число термов
1
4
4
2
8
4
4
1
2
4
4
2
1
Энергия,
необходимая
Л ПО ПАПАУЛПЭ
дл>1 Перехода
71000
135 000
125 000
135 000
190000
190 000
180 000
200000
180000
244 000
234 000
244 000
288 000
Дисперсионная энергия равна сумме
всех вкладов
Вклады в
У = 4А,
2205,0
366,0
6,5
3,7
0,04
14,8
17,1
3,7
0,03
0,8
0,03
6,005
0,001
2620,0
__9(nHcn.) от
переходов
у = 8А
135,5
4Д
0,09
0,04
0,03
0,02
0,02
0,005
0,0008
0,0004
0,00009
0,00001
0,000005
140
различных
у = 15 А
5,50
0,02
0,03
0,03
0,004
0,0001
0,00009
0,00004
0,0003
0,000002
0,000002
0,000001
0,000000003
5,58
Энергия притяжения —^(дисп*) максимальна, когда две молекулы парал-
лельны одна другой и перпендикулярны межъядерным осям. При некотором
угле у0 = arctg(x/y) энергия притяжения проходит через минимум; при
некотором угле у2 она имеет вторичный максимум. Значения у0 и у2 даны в
табл. 119 [67].
Таблица 119
Величины у0 и уа как функции расстояния у
Молекула
2/с=6
Длинная цепь
Bк велико)
Расстоя-
ние у, А
3
8
10
15
y/L>l
град
66
44
42
39
35
град
69
56
56
53
51
Для молекул, имеющих вид длинной цепи Bfc велико), для больших рас-
стояний между молекулами имеем
= _ 16
Bk + IM cos2 у C sin2 у - IJ.
D26)
754 Гл. 13. Теория межмолекулярных сил
Ориентация III. Вращение второй молекулы в плоскости двух моле-
кул (изменяются в и у при х = ф = 0). Изменение в показывает, что энергия
взаимодействия —у№сп-) ддя больших молекул максимальна при 0, имеющем
значение где-то между 60 и 90°. Это наиболее удивительный результат расчетов
Дэвиса и Коулсона. В случае 2fc = 2 энергия минимальна, когда в = 0, и равна
нулю, когда в = 90°. Таким образом, для 2/с = 2 разность q^HP — 9>(™о?'}
раьна 5 кал/моль, когда у = 7 А, и 120 кал\мольу когда у = 4 А. На фиг. 214
показано изменение —<р(дисп.) для молекулы 2к = 6 при у = 7 А. Из фигуры
ясно, что энергетически угол в пределах от 60 до 90° предпочтительнее и что
при энергии около 240 кал/моль имеется энергетический барьер, препятстпую-
щий вращению. Аналогичным образом бутадиен B/с = 4) имеет энергетиче-
ский барьер при энергии около 860 кал/моль, когда у = 4,2 А. Такие же зна-
чения предполагаемых углов, по-видимому, имеют место во взаимодействиях
нафталиновых, антраценовых и других ароматических молекул. Из фиг. 214
можно видеть, что результаты очень чувствительны к способу описания вы-
сших возбужденных состояний.
§ 5. СИЛЫ, ДЕЙСТВУЮЩИЕ МЕЖДУ МОЛЕКУЛАМИ,
ИМЕЮЩИМИ ПОСТОЯННЫЙ ЭЛЕКТРИЧЕСКИЙ МОМЕНТ1)
Рассмотрим энергию далеких взаимодейстрий друх реальных молекул,
имеющих постоянные электрические моменты. Эта энергия состоит из трех
членов2)
Вклад, вносимый в общую энергию величиной qt9*-), определяется чисто элект-
ростатическим взаимодействием между постоянными распределениями заря-
дов двух молекул ; <р<инд-) — энергия индукции Дебая—Фалкенхагена — пред-
ставляет собой энергию взаимодействий между постоянным распределением
заряда одной молекулы и моментами, индуцируемыми в другой молекуле.
Член у<д*сп-> соответствует дисперсионной энергии Лондона и представляет собой
энергию взаимодействия двух индуцированных распределений зарядов.
Вопрос об электростатической энергии взаимодействия двух постоянных
распределений зарядов обсуждался в гл. 12, § 1, как вопрос об энергии взаи-
модействия мультипольных моментов. Дисперсионная энергия рассматри-
валась в § 3 и 4. В этом параграфе рассматривается энергия индукции. Во
многих случаях используется потенциал межмолекулярного взаимодействия,
усредненный по всем ориентациям двух взаимодействующих молекул. Здесь
обсуждается усредненный потенциал и анализируется относительное значение
различных вкладов, из которых составляется усредненный потенциал.
А. ЭНЕРГИЯ ИНДУКЦИИ
Энергия индукции возникает вследствие индуцирования моментов в рас-
пределении заряда под действием электрического поля, создаваемого другим рас-
пределением заряда. Энергия молекулы, обладающей поляризуемостью ее в
электрическом поле напряженности ff, выражается в соответствии с формулой
B.2) гл. 12 в виде —^^-cc-ff). Таким образом, если gQ(xb) — поле, созда-
ваемое у молекулы Ь молекулой а [аналогичный смысл имеет ffb(ra)l T0
вклад индукционных эффектов в потенциальную энергию межмолекулярного
ЧСм. [53,54].
2) Резонансные силы рассматриваются отдельно в § о.
§ 5. Силы, действующие между молекулами, имеющими постоянный электр. момент 755
взаимодействия определяется следующим образом :
= —J- (ye (tb) .<*b.ffa (r,)) —J- (ffb (ra) -*a.gb (ra)). E.2)
Электрические поля, выраженные через электростатические потенциалы, имеют
вид, даваемый формулой A.10) гл. 12. Электростатические потенциалы в свою
очередь выражаются через мультипол! ные моменты распределений заряда и
определяются по формуле A.27) гл. 12.
Мы рассмотрим специальный случай изотропной поляризуемости моле-
кул, когда тензоры аа и щ могут быть заменены скалярами аа и аь. Далее мы
предположим, что распределения зарядов обладают цилиндрической симмет-
рией, так что квадрупольные моменты могут быть охарактеризованы скаляр-
ными величинами Qa и Qb [см. A.17) гл. 12 ]. В этом случае индукционная энер-
гия равна
/ИНД|) __ Cl аь + Сь аа __ 2Cfl fig ab cos 0g + 2Cb f*b eg cos 0»
_ А аь C cos» 0fl + 1) + ц\ aa C cos» 0b + 1)
3СQgcu>Ccos20e- \) + 3CbQbaaCcos*0b- 1)
б (ла Qa аь cos8 вд + 6/ль Qb aa Cos8 0O ,- оч
где Ca и Cb —заряды молекул а и Ь ; ва и вь — углы между осями симметрии
молекул и линией, соединяющей центры молекул. Члены в <р<инд>, изменяю-
щиеся, как 1/г4 и 1/гб, важны во взаимодействии иона с нейтральной молекулой.
Член, изменяющийся, как 1/г6, играет главную роль во взаимодейстгии нейт-
ральных молекул, обладающих дипольными моментами. Однако в случае
уширения линий микроволнового спектра аммония под действием давления,
создаваемого благородными газами, как отмечается в § 7, важен член [74],
изменяющийся, как 1/г7.
Б. ПОТЕНЦИАЛЬНАЯ ЭНЕРГИЯ ВЗАИМОДЕЙСТВИЯ,
УСРЕДНЕННАЯ ПО ВСЕМ ОРИЕНТАЦИЯМ МОЛЕКУЛ
Потенциальная энергия взаимодействия двух несферических молекул
зависит от ориентации молекул. Однако во многих приложениях необходима
потенциальная энергия, усредненная по всем ориентациям двух молекул.
В разреженном газе в случае равновесия вероятность обнаружения двух
молекул, удаленных одна от другой на расстоянии г и ориентированных
некоторым частным образом, пропорциональна множителю Больцмана
ехр{— (p(raby0)a)(ob)lkT}. Таким образом, средняя потенциальнаяэнергия, которую
мы рассматриваем, получается путем такого усреднения по всем ориентациям,
в котором в качестве весового множителя берется множитель Больцмана
JJV (ГаЬ, ft)a, (Ob) е~Я<г*ь><»а,ъI d0)a d(Ob
9 {ГаЬ' Т) = Я «-*-*.-Wdmda» E'4)
Следует отметить, что этот средний потенциал зависит от температуры.
Выражения для <р обычно выглядят весьма сложно, так что вычислить
интегралы E.4) трудно. Однако для тех значений га&, для которых для всех
ориентации \ q> — ^| мало по сравнению с кТ, экспоненту удобно разложить
в ряд и представить у в виде
_ _ SS<pe-(<P-v)lkTda)ada>b _ Я <Р V - (? ~ ЩТ + - ¦. ] (tog dm
Л e-to-№T dcoa da>b "" Л [1 - (<р - 9>)/кТ + ...] dcoa da>b
756 Гл. 13. Теория межмолекулярных сил
Таким образом получаем1) разложение среднего потенциала по степеням XjkT
E-6)
Средний потенциал ф(гаЬу Т) E.6) выражается через интегралы от завися-
щего от углов потенциала cp(rab, coa9 cob), который, как следует из E.4), равен
сумме трех членов. Подробный анализ выражений E.6) и E.1) показывает, что
в хорошем приближении перекрестными членами можно пренебречь [52, 53]
и записать ^ в виде
у = ^(эл.) -[- ^(инд.) _[_ ^(дисп.) . /5.7)
здесь ^<эл> имеет вид, аналогичный выражению E.6), в котором <р заменено на
^(эл.)ф j0 же справедливо и в отношении ^<инд-) и y(**cn.)t Рассмотрим теперь
каждую из этих составляющих отдельно и выясним их основную зависимость
от межмолекулярного расстояния.
Средняя энергия электростатического взаимодействия может быть вычи-
слена путем рассмотрения молекул, имеющих распределение зарядов, обла-
дающих общими мультиролями. Подставляя общее выражение A.33) гл. 12
для 9><эл> в E.6) и пользуясь условием ортогональности (Б.20) гл. 12 коэффициен-
тов представления, находим
<±С^__!_ v (Hi - I пи 1) I (ла - | ш, 1I1^ + па) 1]»
г кТ ? (n1 + \m\)\(n1-\ A
m
х _
А (п, + | ш, |) ! B щ + 1) Bла + 1) Г2(п1+п,+1)
Член, в котором nL и п2 равны нулю, не входит в сумму. Далее, поскольку
У 1 B^ + 2^1
получаем
-(эл) _ Caa _ J_ v (ni - I mil) I (n, - | m8|)lBn1+ 2n,)l
^ ~ г кТ ^ (п1+|яц|I(п + |т|)!Bв1)!Bв)!Bв1+1)Bв+1)
Интересно отметить, что в величину <р<9Л), полученную путем непосредственного
усреднения уС*"-), т. е. в интеграл \\ (р^-Ысо^со^ дает вклад только кулоновское
взаимодействие общих зарядов Са и Сь двух молекул. Если молекулы ведут
себя как нейтральные идеальные диполи, то в ?<эл*> дает вклад только тот член
суммы, у которого пг = п2 = 1 иш1 = /п2 = 0.В этом случае
При вычислении средних по пространству удобно использовать результаты :
cos2 в == Vs» sin2 0 = 2/з» cos2 @a - 0&) = г/2 >
COS* в = Ve > Sin2 в COS2 0 - 2/16, Sin* в = 8/M, COS4 @a — 0b) = 3/8 •
§ 5. Силы, действующие между молекулами, имеющими постоянный электр. момент 757
Энергия индукции в специальном случае цилиндрически симметричных
молекул выражается в виде E.3). Если это выражение подставить в формулу
E.6), то найдем, что средняя энергия индукции не зависит от Т вплоть до чле-
нов, пропорциональных 1/г?ь, и имеет вид
E.12)
Из этого выражения следует, что потенциальная энергия взаимодействия
нейтральной неполярной молекулы а и нейтральной полярной молекулы Ь
включает член —а^Цг6. Этот член учитывается при выводе1) комбинационных
правилA0.15) и A0.16) гл.З и F.3) и F.4) гл.8. Использование средних по углам
в этом выводе оправдано, так как вклад за счет индукционных сил мал по срав-
нению с вкладом, даваемым дисперсионными силами.
Для симметричных молекул дисперсионная энергия не зависит от ориен-
тации молекул, так чю из C.32) имеем
^(Дисп.) = ^(дисп.) = _ JL _ _?_. _ JjL _ m m e EЛ 3)
Первый член объясняется взаимодействием взаимноиндуцируемых диполей, а
последующие члены описывают взаимодействия между высшими индуцируе-
мыми моментами.
В. ОТНОСИТЕЛЬНЫЕ ЗНАЧЕНИЯ ВКЛАДОВ В МЕЖМОЛЕКУЛЯРНЫЙ
ПОТЕНЦИАЛ
Для взаимодействий подобных симметричных молекул главный член в
выражениях для каждого из трех вкладов —за счет электростатических, индук-
ционных и дисперсионных сил — изменяется, как 1/г2ь- Коэффициентами про-
порциональности соответственно являются —2/г4/3/сТ, —2/ьс2а и —*Ua*hv0.
Отсюда, чтобы сравнить относительные значения вкладов, необходимо срав-
нить только эти три коэффициента. В табл. 120 приводятся значения коэффициен-
тов для некоторых типичных молекул [53]. Очевидно, что влияние индукцион-
ных сил не вел^ ко во взаимодействии между нейтральными молекулами. Од-
нако влияние дисперсионных сил важно даже во взаимодействии молекул
с большими дипольными моментами.
х) Если предположить, что вклады валентных и дисперсионных сил в межмолекуляр-
ный потенциал <р(г) определяются потенциалом F-12) Леннарда-Джонса, то взаимодействие
полярной и неполярной молекул описывается потенциалом
Это выражение приводится к виду потенциала F-12) Леннарда-Джонса с параметрами о'аъ
и е'аъ, имеющими вид
J_
2
где
L 4 Sab Oab J
4Gla еьь Gib r eaa
Предполагается, что
*i
оаа <*bb-
758
Гл. 13. Теория межмолекулярных сил
Таблица 120
Сводка вкладов, вносимых во взаимодействие между подобными полярными
молекулами различными типами взаимодействий [53]
Молекулы
СО
HJ
НВг
НС1
NH8
нао
1* • 10",
эл.'ст. ед
0,12
0,38
0,78
1,03
1,5
1,84
а- 10м,
СМ*
1,99
5,4
3,58
2,63
2,21
1,48
14,3
12
13,3
13,7
16
18
Электростати-
ческий ф(ЭЛ.)г1
(Т - 2^3 °К)
B) "' 10"
\з) 293k 10 •
10~ee эрг - ем9
0,0034
0,35
6,2
18,6
84
190
Дисперсионный
^(дисп.) г«
-j a* h v0 • 10вв,
КГ"*0 эрг • см*
67,5
382
176
105
93
47
Индукционный
ф(инд.) ri
2 fx* а • 10е»,
10 ~м эрг • см*
0,057
1,68
4,05
5,4
10
10
Энергия взаимодействия неполярной молекулы а с ионом Ь, имеющим-
заряд Cbf равна
Первый член представляет собой энергию индукционного взаимодействия за-
ряда с индуцированным диполем, второй член — лондоновскую дисперсион-
ную энергию взаимодействия двух взаимноиндуцированных диполей. Чтобы
проиллюстрировать относительную роль двух членов, рассмотрим их значе-
ния на расстоянии, равном «диаметру столкновения кинетической теории».
На таком расстоянии общий потенциал (включая отталкивательные члены)
приблизительно равен нулю. Энергии взаимодействия на таких расстояниях
приведены [75] в табл. 121. Очевидно, что член ион-дипольного взаимодействия
велик, однако и дисперсионная энергия имеет значения, которыми нельзя
пренебречь.
Таблица 121
Энергия взаимодействия на расстоянии «диаметра столкновения
Молекулы
Не
Ne
Аг
Кг
Хе
Na
оа
со2
Ы
у(инд.)
- 202
- 298
- 781
- 931
-1200
- 429
- 658
- 728
-1000
+
^(дисп.)
- 46
- 58
-142
-165
-209
- 94
-100
-133
-179
Na+
9(инд.)
-134
-202
-557
-668
-883
-301
-472
-520
-730
ф(дисп.)
- 37
- 50
-116
-130
-162
- 71
- 83
-104
-144
К+
у(инд.)
- 71
-111
-322
-402
-538
-173
-283
-303
-441
у(дисп.)
- 37
- 53
-114
-125
-148
- 62
- 86
- 97
-137
кинетической теории»
Rb+
^(инд.)
- 55
- 87
-257
-323
-437
-137
-228
-242
-359
ф(Дисп.)
- 35
- 52
-114'
-123
-143
- 60
- 88
- 95
-138
Cs+
ф("НД.)
- 43
- 69
-210
-266
-363
-111
-188
-196
-293
у(дисп.)
- 41
- 62
-136
-145
-169
- 68
-104
-ПО
-163
Замечание. В первых колонках даны энергии ион-дипольного взаимодействия (^1/ri4), во вторых -
лондоновские дисперсионные энергии взаимодейстия \~11Г1Ь)- Все энергии даны в 10~15 эрг.
Г. ВОДОРОДНЫЕ СВЯЗИ КАК ЭЛЕКТРОСТАТИЧЕСКИЕ СИЛЫ
Термин «водородная связь» был введен Латимером и Родебушом [70],
чтобы отметить большую энергию взаимодействия между группами ОН или
NH и атомами О, N, F или С1. Одно время полагали, что сила водородных связей
§ б.Квантовомеханическая трактовка резонансных и электростатических сил 759
объясняется некоторым видом квантовомеханического резонанса. Однако все
современные данные указывают на то, что водородные связи представляют собой
обычные электростатические взаимодействия между соответствующими груп-
пами1). Важно знать точное расположение электрических зарядов, ибо группы
могут сближаться достаточно тесно, так что концепция идеальных диполь-
ных сил оказывается несправедливой. Хорошим примером, который обсуж-
дается в § 8, является пример взаимодействия молекул воды. Дэвис [781 про-
вел очень тщательное изучение физических аспектов водородных связей. Он
дал следующие энергии связи для водородных связей димеров и полимеров в
газовой фазе2):
АН
6HF q± (HF)e 6800 кал/моль
2НСООН ^± (НСООН)а 7060
2СН8СООН ^± (СН8СООН)а 9430-4,88 Т
2CH8COOD ^± (CH8COODJ 7950
гС^СООН ^± (С^СООН),, 9200
ЗСаНбСООН ^±1 (QiH6COOH)8 8000
2(C8H6COOD) ^± (C^COOD), 7040
Вследствие больших энергий взаимодействия между этими молекулами
только небольшая доля их существует в газовой фазе в виде мономеров. Следо-
вательно, их вириальные коэффициенты [79, 80] и коэффициенты переноса8)
соответственно аномальны.
§ 6. КВАНТОВОМЕХАНИЧЕСКАЯ ТРАКТОВКА РЕЗОНАНСНЫХ
И ЭЛЕКТРОСТАТИЧЕСКИХ СИЛ
Рассмотрим взаимодействия двух молекул с чисто квантовомеханиче-
ской точки зрения. Здесь ориентации, а также электронное и колебательное
движения неявно характеризуются квантовыми числами в статистическом
смысле. Энергия взаимодействия соответствует типу IV, обсужденному в § 1.
Квантовомеханическэя трактовка межмолекулярных сил отличается от
классической трактовки в двух отношениях. Во-первых, в кваитовомеха-
нической трактовке часто возможны резонансные силы, которые не имеют
классической аналогии. Во-вторых, ориентации молекул характеризуются
квантовыми числами только частично. Квантовомеханическое взаимодействие
двух молекул, находящихся в определенных квантовых состояниях, можно
срарнить с классическим взаимодействием, усредненным по всем ориентациям,
когда в качестве весового множителя берется квадрат волновой функции.
Резонанс иногда играет важную роль во взаимодействии двух неполярных
молекул (обычно одинаковых) Помимо этого, резонанс часто усложняет элек-
тростатическое взаимодействие полярных молекул. В случае резонанса энергия
взаимодействия аномально велика, иногда как энергия притяжения, а иногда
как энергия отталкивания.
Мы начнем настоящий параграф с рассмотрения типов резонансных сил,
которые могут иметь место при взаимодействии молекул. Затем мы обсудим
*) При тесном сближении групп, т. е. очень коротких водородных связях, имеющих,
например, место в кристаллах солей кислот, таких, как КН2РО4, характер взаимодействия
изменяется от электростатического до гомеополярного [77].
а) Слабая тенденция ацетальдегида образовывать димеры
[2СН8СНО ^ (СН8СНО)„ /\Н = 2610 ккал/моль
рассматривалась как пример водородной связи. Однако этот пример не служит показателем
отличия ацетальдегида от ацетона и полярных газов, которые не могут образовывать водо-
родные связи.
8) О вязкости см. работу [82], а о теплопроводности — работу [83].
760 Гл. 13. Теория межмолекулярных сил
квантовомеханическое взаимодействие между двумя идеальными диполями,
вставленными в линейные или аксиально симметричные молекулы; это даст
пример чисто электростатического взаимодействия, усложненного в некото-
рых случаях резонансом. Далее мы обсудим два характерных примера взаи-
модействия иона и нейтральной молекулы : взаимодействие протона с возбуж-
денным атомом водорода и взаимодействие протона с возбужденным атомом
гелия. Первый пример иллюстрирует случай сильного резонанса, второй —
случай, близкий к резонансу. Заканчивается параграф изложением первого
приближении квадруполь-квадрупольного взаимодействия атомов (находя-
щихся в S-состояниях). Это до известной степени сложно, поскольку трактовка
вопроса зависит от того, насколько мало или велико расстояние между сосед-
ними энергетическими уровнями по сравнению с кТ. Во взаимодействиях, в
которых участвуют свободные радикалы или молекулы в возбужденных состоя-
ниях, возникают силы, аналогичные рассматриваемым в настоящем параграфе.
А. ПРИРОДА РЕЗОНАНСНЫХ СИЛ
Возможность возникновения резонансных сил появляется всякий раз,
когда взаимодействие снимает вырождение волновых функций. Иными сло-
вами, резонанс связан с возможностью гибридизации волновых функций.
В далеких взаимодействиях между молекулами такая гибридизация эффек-
тивна для возникновения резонанса только тогда, когда состояния, образую-
щие гибрид, имеют очень близкие энергии на больших расстояниях. Если
состояния имеют точно одинаковую энергию, то возникает очень острый резо-
нанс. Если состояния имеют близкие значения энергии, то резонанс не эффекти-
вен до тех пор, пока не будут достигнуты несколько меньшие расстояния.
Имеются два типа резонанса : резонанс между молекулами и резонанс в
пределах одной молекулы. Резонанс между молекулами может иметь место в
столкновениях одинаковых молекул, обладающих такими квантовыми числами,
что молекула из нижнего энергетического состояния может при поглощении
фотона, не нарушая правил отбора для дипольного излучения, перейти в состоя-
ние с квантовыми числами, которыми обладает другая молекула. Если эти
условия для квантовых чисел не удовлетроряются, то эффективность резонанса
в изменении потенциала взаимодействия значительно уменьшается. Резонанс
в пределах одной молекулы иллюстрируется на примере взаимодействия про-
тона с возбужденным атомом водорода. В этом взаимодействии азимутальное
квантовое число не является больше «хорошим» квантовым числом и волновая
функция представляет собой гибридизированную сумму всех орбиталей атома
водорода, имеющих одинаковые значения главного и магнитного квантовых
чисел, но различные значения азимутального квантового числа.
Вследствие резонанса происходит расщепление поверхностей потенциаль-
ной энергии. Иначе говоря, существует определенная вероятность того, что в
процессе столкновения молекулы попадут на любую из ряда поверхностей
потенциальной энергии. Эта вероятность пропорциональна статистическому
вырождению определенной энергетической поверхности. Резонансная энергия
может быть положительной или отрицательной. На больших расстояниях
сумма резонансных энергий, умноженных на их априорные вероятности,
равна нулю.
Рассмотрим теперь оба типа резонансных явлений : резонанс между моле-
кулами и резонанс в пределах одной молекулы.
1. Резонанс между молекулами. Состояние резонанса между двумя моле-
кулами а и Ь существует тогда, ко^да квант энергии может быть легко испу-
щен молекулой а и поглощен молекулой б1). Согласно квантовой механике, в
систехме молекул невозможно точно определить состояние молекулы а и моле-
*) На существование резонансных явлений первым указали Эйзеншитц и Лондон [551.
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 761
кулы Ь раздельно. Если yja(i) — волновая функция молекулы а в /-состоянии,
то волновой функцией системы из двух молекул может быть либо функция
У (g) = Va (О П (/) + Wa (/) V» @ , FЛ)
либо
*Чи) =У>а(!)М!) - V>a(i)Vb(i). F.2)
Поэтому столкновение двух молекул может происходить по поверхностям
потенциальной энергии, характеризуемыми ^-состояниями, или по поверх-
ностям, характеризуемым (и)-состояниями. Теория возмущений в первом
приближении дает следующие выражения для энергии взаимодействия системы
молекул, находящихся в одном из двух упомянутых состояний:
(б.з)
F.4)
Поскольку нас интересуют только члены, изменяющиеся, как \\г\ь, т.е. дающие
наибольший вклад в энергию резонансного взаимодействия, то достаточно
удержать в разложении сре в ряд [см. A.10) ] только главный член. Подставляя
главный член разложения q>e в F.3) и F.4), получаем
[| Wy | + l(fy)i. |2 _ 2 | (^),7 |i] . F.5)
Здесь (рх)ф (ру)ф (pz)ij — компоненты дипольного момента, связанного с
переходом отдельной молекулы из r-состояния в /-состояние [см. F.7) гл. 12].
Ось z направлена вдоль линии, соединяющей ядра. Таким образом, сильное
резонансное взаимодействие возможно только при неисчезающем дипольном
моменте, связанном с этим переходом.
Для резонансного взаимодействия необходимо, чтобы одна из молекул
находилась в возбужденном состоянии. Например, если при взаимодействии
двух атомов водорода в основных состояниях резонансные силы не прояв-
ляются, то при взаимодействии атома водорода в ls-состоянии с атомом водо-
рода в 2р-состоянии такие силы имеют место. Из приложения В гл. 12 можно
видеть, что неисчезающими матричными элементами дипольного момента для
переходов атома водорода из ls-состояния в различные 2р-состояния являются
только матричные элементы
I (/О* 2р,|2 = Ш», %; I2 = IGfyb. 2р, I2 = 0,555 е* а%.
Таким образом, резонансная энергия может иметь четыре различных
значения :
или ±0,555-^^ F.6)
В одной шестой части столкновений пары атомов водорода, один из которых
находится в ls-состоянии, а другой — в 2р-состоянии, энергия резонансного
взаимодействия равна 1,110 еЧЦгЪь; в одной шестой столкновений она равна
— 1,110 е2аЦг%ь; в одной третьей столкновений энергия равна 0,555 е2аЦг1ь,
а еще в одной третьей о ш равна —0,555 е2аЦг1ь.
Этот результат, выражаемый формулой (б.б), может быть легко обобщен
на случай резонансных сил, возникающих при столкновении любого атома в
S-состоянии с таким же атомом в Р-состоянии [84]. Во всех этих случаях,
взаимодействующая система может попасть в следующие классы состояний :
2е>2юПшП§. Каждый из этих классов состояний имеет свою функцию потен-
762
Гл. 13. Теория межмолекулярных сил
циальной энергии. Кинг и Ван Флек показали, что резонансная энергия,
соответствующая каждому из этих классов, может быть записана в виде
+2^
-2
rib '
rib '
\(t*z)sp\f
rb
ng,
П,.
F.7)
В табл. Ill (стр. 701) даны значения | (/*z)Sp |2 для различных атомов для пере-
ходов и^ основного S-состояния в первое возбужденное Р-состояние. Сравнивая
формулы F.6) с формулами F.7), мы видим, что при столкновении атома водо-
рода в ls-состоянии с атомом водорода в 2р-состоянии д>(рез<) = -J2 1,10 (e2a%lr%b)
для -Г-состояний и 9?(рез) = ±0,555 (еЧЦг^ для/7-состояний. Если рассматри-
ваются далекие взаимодействия, энергия Kotopbix изменяется, как 1/г2&, то
синглетные и триплетные состояния имеют одинаковые энергии. Поскольку
при образовании синглетных и триплетных g- и и-состояний должен соблю-
даться принцип запрета Паули, то
/7-состояния имеют удвоенную ве-
роятность по сравнению с 27-сос-
тояниями.
Несмотря на то, что резонан-
сная энергия Zg- и /7и-состояний
для Н, Li, Na, К, Rb, Cd, Hg на
больших расстояниях положите-
льна, из эксперимента известно, что
на малых расстояниях эти состоя-
ния соответствуют стабильным мо-
лекулам. На фиг. 215 показана
форма кривых потенциальной энер-
гии этих состояний. Для натрия
максимум кривой потенциальной
энергии соответствует энергии около
0,7 ккал/моль; для лития он достигает значения 2,8 ккал/моль. Во всех этих
случаях резонансная энергия велика и соответствует очень большим рассто-
яниям между молекулами. Резонансная энергия равна кТ при комнатной тем-
пературе @,6 ккал/моль) при расстоянии между молекулами, изменяющимися
в зависимости от сорта атомов от 6 до 20 А.
2. Резонанс в пределах одной молекулы. Некоторые молекулы обладают
вырожденными состояниями, которые под действием возмущения, возникаю-
щего при взаимодействии с другой молекулой, расщепляются. В таких случаях
появляется возможность возникновения резонансных сил. Простым примером
явления такого типа может служить взаимодействие протона с возбужденным
атомом водорода.
На первый взгляд кажется, что следовало бы рассмотреть возможность
колебаний электрона между центрами а и Ь и записать волновую функцию в
виде ?/=?/аA)+ 1^A), где УаA) — волновая функция атома а плюс протон ft,
a ^Vl) — волновая функция протона а плюс атом Ь. Однако легко показать,
что точно такое же значение энергии на больших расстояниях получается,
если волновую функцию представить в виде У=УаA), опустив У&A). В резуль-
тате включения в волновую функцию как ФаA)9 так и !РЬA) в выражении для
•энергии появляются члены, экспоненциально убывающие с расстоянием и
§ б. Квантовомеханическая трактовка резонансных и электростатических сил 763
имеющие заметную величину только на малых расстояниях. Таким образом,
протон рассматривается скорее не как частица, а как источник электри-
ческого потенциала. Рассмотрим случай, изображенный на фиг. 216. Электрон
Фиг. 216. Координаты, использованные при обсуждении взаимодействия протона
и атома водорода.
находится на расстоянии га1 от центра атома. Угол между осью, соединяющей
ядра, и радиусом-вектором, направленным от а к электрону, обозначим через
6av Энергия возмущения атома а, возникающая вследствие присутствия про-
тона, равна
SS[()fe)i] <6-8>
В настоящей задаче мы интересуемся только определением члена резонан-
сной энергии <р<Рез->, который изменяется как l/rf&. Появление членов, пропор-
циональных \)гЪь% удивительно, так, как подобную зависимость от расстояния(
можно ожидать для электрических заряд-дипольных сил, а атом водорода не
имеет диполя. Как мы сейчас покажем, гибридизация волновой функции при-
водит к полярному распределению заряда в атоме и, в результате, — к появ-
лению упомянутого члена.
В качестве примера рассмотрим состояние, в котором протон и атом водо-
рода, имеющий главное квантовое число п = 2, находятся на большом расстоя-
нии один от другого. Энергия атома водорода одинакова для состояния с ази-
мутальным квантовым числом I = 0 B$-состояние) и состояний с / = 1
Bр-состояния). Под влиянием электрического поля протона азимутальное
квантовое число перестает быть «хорошим» квантовым числом.
Таким образом, волновая функция является гибридом из функций
2s и 2р:
F.9)
Поскольку на магнитное квантовое число возмущение не оказывает влия-
ния, то неортогональными орбиталями будут те, которые имеют одинаковые
значения магнитных квантовых чисел [69, стр. 71 ]. В нашем случае магнитное
квантовое число может быть только нулем (единственное значение магнитного
квантового числа в 2$-состоянии). Физически равенство нулю магнитного
квантового числа означает, что момент количества движения не имеет компо-
ненты в направлении оси, соединяющей ядра.
Резонансная энергия получается при решении уравнения первого при-
ближения теории возмущений, т. е. уравнения F.49) гл. 1. Линейная комбина-
ция волновых функций F.9) используется как волновая функция нулевого
приближения, а главный член <ре [см. A.10)] используется в качестве возму-
щающего потенциала. В обозначениях, принятых в уравнении F.49) гл. 1,
25-состояние обозначается индексом 1, а 2р-состояние — индексом 2; пара-
метр А равен —е\г\ь. В-соответствии с этим запишем
= - ега1 Рг (cos 0al), (б. 10)
Е - ?<0) = Е _ ?<0) =9
764 Гл. 13. Теория межмолекулярных сил
Далее, секулярное уравнение может быть представлено в виде
= 0. F.12)
Матричные элементы возмущения равны
s = 0, F.13)
„2/7, = О, F.14)
Значения матричного элемента (ftz)is, ?р. даны в приложении В гл. 12.
Таким образом, резонансная энергия выражается следующим образом :
^^ F.16)
Вследствие обратной квадратичной зависимости от расстояния д>(рез>>
является энергией, имеющей заметные значения на более далеких расстояниях,
чем те расстояния, на которых имеет заметное значение энергия, изменяющаяся,
как 1/Гаь> и обычно ожидаемая при взаимодействии иона и неполярной моле-
кулы. В столкновениях протона с атомом водорода, имеющим главное кванто-
вое число п = 2, вероятности того, что энергия взаимодействия будет положи-
тельной или отрицательной, равны. Полная энергия взаимодействия протона
с атомом водорода равна сумме энергий резонансного и электростатического
взаимодействий (это обсуждается в разд. Г).
Б. КВАНТОВОМЕХАНИЧЕСКОЕ ВЗАИМОДЕЙСТВИЕ ДВУХ ИДЕАЛЬНЫХ
ДИПОЛЕЙ, ВСТАВЛЕННЫХ В ЛИНЕЙНЫЕ МОЛЕКУЛЫ
Для понимания поведения коэффициентов переноса полярных газов и
для понимания явления уширения линий микроволнового спектра при повы-
шенном давлении (см. § 7) очень важно иметь подробные сведения о взаимо-
действии диполей. Квантовомеханический расчет энергии двух взаимодейст-
вующих дипольных молекул может быть проделан с помощью первого и вто-
рого приближений теории возмущений. Электрическое взаимодействие первого
порядка (с энергией, изменяющейся, как \lrlb) исчезает, за исключением случая
резонанса. Причиной такого поведения является квантовомеханическое опре-
деление ориентации диполей. Линейный диполь характеризуется азимуталь-
ным квантовым числом / и магнитным квантовым числом т. Числа / и т фикси-
руют ось вращения молекулы в определенных пределах.
Дипольный момент молекулы всегда перпендикулярен оси вращения.
В результате, если оси вращения двух молекул фиксированы, то для ориен-
тации диполей равновероятны четыре конфигурации, изображенные на
фиг. 217, вследствие чего усредненная по волновым функциям двух молекул
электростатическая энергия в первом приближении исчезает.
Волновые функции изолированной молекулы, представляют собой норма-
лизованные сферические функции Yf{dy ф\ данные в (Б.З) гл. 12. Индексами
/ и т соответственно обозначены азимутальное и магнитное квантовые числа.
Энергия молекулы в /-м состоянии равна
8яЧ
где / — момент инерции молекулы.
Теперь рассмотрим взаимодействие двух таких молекул а и ft. На очень
больших расстояниях, где <р€ = 0, волновая функция системы представляет
собой произведение волновых функций изолированных молекул. Предполо-
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 765
'аЬ
Фиксированная
ось вращения \а
молекулы а ^-
/
/
4 Фиксированная
rab Ь/ ось вращения
—' молекулы о
[ab
\ |
Фиг. 217. Равновероятные ориентации двух диполей с фиксированными осями вращения.
жим, что молекулы первоначально находятся в состояниях /а, та и jb> ть соот-
ветственно. При этом невозмущенная волновая функция системы имеет вид
V0 (/а, гпа; /ь, mb) = Yp @а, фа) Yff (вь, фь). (б. 18)
Произведение b.Jrlb на уР является линейной комбинацией самое большее
двенадцати величин уH
F.19)
^ V0 На, та; /», ть) = ^ -2"»,-
Здесь индексом / отмечены следующие состояния:
+l, ma; jb+hmb),
- 1; /»+ l,m»+ 1),
; /» - 1, mb),
+ i; л- i,m»- 1),
= V° (/в - 1,
F.20)
; /ft - l,m6 - 1),
= f°(J'a-I, ma; jb+l,mb),
-1, Ша+l; jb+l,mb-\),
Значения численных постоянных », легко определяются из уравнений
(Б.8) — (Б. 10) гл. 12. Величины vt равны нулю для тех состояний (они могут
не учитываться), которые имеют либо отрицательное значение /, либо значение
| т \, большее соответствующего /. Мы будем отличать первоначальное состоя-
ние индексом 0.
766 Гл. 13. Теория межмолекулярных сил
Для решения, рассматриваемой проблемы Лондон [85, 52] использовал
метод возмущений (который он назвал методом неострого резонанса). Он
образовал приближенную волновую функцию в виде линейной комбинации из
функции у? и функций состояний (самое большее двенадцати), с которыми она
взаимодействует в соответствии с уравнением F.19). Наиболее точное значение
энергии может быть получено с помощью такой линейной комбинации из
решения секулярного уравнения. Это секулярное уравнение имеет особенно
простой вид, так как состояния с if большим нуля, не взаимодействуют с любым
другим состоянием, за исключением состояния 0. К тому же диагональные
элементы 1, 2 и 3; 4, 5 и 6 ; 7, 8 и 9, а также 10, И и 12 образуют группы из
трех элементов. Таким образом, секулярное уравнение имеет нулевые элементы
вдоль нулевой строки, внизу нулевой колонки и вдоль диагонали. Выполняя
указанные перемножения и делая некоторые преобразования, получаем (в об-
щем случае) для д>(/а, та ; jb9 ть \ rab) уравнение пятого порядка
здесь е, выражаются в виде
. _ *' Г(/«+1) , (ft+ 1I
8l ~ ~4я* L~77~ + ~~Ть~\
~ 4л* L a \
F.22)
~ 4я* [~Га 7TJ'
Величины a%i равны
а\ = v\ + v I + tj =
[4 [(/« + 1)» - ml] [(/»+ I)» - ml] +
+ 54 Ua
+ 'A Ua
+ XA (fa +ma-l)<Ja + ma)(jb - ть - 1)Ць - ть)]
<4/«-1><4/,-1> (б2^
+ lA (Ja + Ша + 1) (/a + /Ла + 2) (/& + /Ш — 1) (jb + Ш&) +
+ У' ija-ma)(]a - ma - 1) (/ft - fflft + 1) (/ft - ПЦ + 2)]
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 767
Для больших расстояний (за исключением случаев резонанса, которые мы
обсудим ниже) ср оказывается очень малым по сравнению с eh вследствие чего
члены в правой части уравнения F.21) могут быть разложены в ряды по сте-
пеням q>
<6-21a>
Рассматривая в правой части уравнения F.21а) только первые три члена,
получаем для <р квадратное уравнение. Решение этого уравнения, имеющее
физический смысл, выражается в виде ряда ;
^^f^ ...> F.24)
где В; имеет вид
Bt=2x^r> F.25)
здесь е1к — /-я степень ек.
Рассмотрим столкновения между одинаковыми молекулами. В этом случае
/ла = [ль = ii и моменты инерции 1а = 1Ь = /. Определим параметр z, пропор-
циональный шестой степени расстояния, и приведенный потенциал взаимодей-
ствия <р':
Далее, уравнение F.21) может быть записано в виде
+
<p'+(fa
Приведенный потенциал взаимодействия, не считая частных молекуляр-
ных постоянных, является функцией только z и параметрически зависит от
вращательных квантовых чисел взаимодействующих молекул.
Из уравнения F.27) ясно, что в тех случаях, когда | /а — jb \ = 1, знаме-
натель третьего или четвертого членов становится равным q>'. Покажем, что
энергия взаимодействия на больших расстояниях становится пропорциональной
г|ь вместо г1ъ и что существует резонансное вырождение состояний с ]а = jb + 1
и /& = /а+1. Рассмотрим случай ja = jb+\. При этом уравнение F.27)
может быть (после умножения на q>) записано в виде
Для достаточно больших расстояний ср' становится малым по сравнению
с B/а + 1) или 2. Таким образом, для больших расстояний уравнение F.28)
может быть разложено в ряд по степеням q>'. Удерживая первые три члена
разложения, получаем квадратное уравнение для <р'
(z + DJv'2 + D2<p' — al = 0, F.29)
где
Di = B/a+l)a ~" B/fl-iJ + "'
F-30>
n _ a* fl[_ , ^L
^2 B/e+l) B/a-l) ^ 2 #
768
Гл. 13. Теория межмолекулярных сил
Решения уравнения F.29) имеют вид
Разлагая эти решения в ряд по степеням l/zy», получаем два асимптоти-
ческих решения, пригодных для больших расстояний,
г1/.
D2
2г
J
2г
J>1
F.32)
F.33)
Таким образом, при наличии резонанса существуют два потенциала взаи-
модействия на больших расстояниях : один положительный, а другой отрица-
тельный. Энергия взаимодействия на больших расстояниях изменяется, как
2ГУ* ИЛИ IfГ^.
Значения величин Blt В2 и В3 для нерезонансных систем приведены в
табл. 122.
Таблица 122
Важные параметры, характеризующие взаимодействие двух одинаковых
линейных молекул
(Нерезонансный случай)
fa =
/a =
la ==
la =
/„ =
о,
о,
i,
i,
2,
h
ib
h
h
h
= 0
Q
= 1
= 3
2
0
0
0
0
0
0
1
1
0
0
0
0
1
0
0
0
1
1
; 1
1
2
2
2
ТПь
0
0
1
2
0
1
1
-1
0
1
2
3
з
-2
-1
0
1
2
3
0
1
2
2
-1
1
2
2
-1
2
0,33333333
-0,28571429
-0,22619048
-0,04761905
0,58000000
0,41000000
0,32000000
0,07000000
-0,22567901
-0,20074074
-0,12592593
-0,00123457
-0,01790124
-0,10370370
-0,16074074
-0,18901235
-0,18851852
-0,15925926
-0,10123457
0,57482993
0,50170068
0,28231293
0,17687075
0,39965986
0,48299320
0,34353741
0,02721088
0,17687075
0,36054422
Bt
0,16666667
0,42857143
0,36011905
0,15476190
0,84500000
0,35250000
0,28000000
0,09250000
0,37691358
0,34703704
0,25740741
0,10802469
0,04552469
0,14907407
0,22092593
0,26108025
0,26953704
0,24629630
0,19135802
0,62159864
0,53103741
0,25935374
0,16638322
0,42077664
0,49716553
0,31916100
0,04024943
0,16638322
0,34580499
Вз
0,08333333
-0,39285714
-0,32663690
-0,12797619
0,66125000
0,33812500
0,27000000
0,03562500
-0,29983882
-0,27532922
-0,20180041
-0,07925240
-0,02936814
-0,13289609
-0,20407922
-0,24291752
-0,24941101
-0,22355967
-0,16536351
0,59119898
0,50641298
0,25205499
0,15421863
0,39825444
0,47869426
0,31509826
0,02158919
0,15421863
0,34334845
§ б.Квантовомеханическая трактовка резонансных и электростатических сил 769
Во многих случаях достаточно знать tp(jv /g)—энергию взаимодействия
двух диполей, усредненную (алгебраически) по всем магнитным квантовым
состояниям. Это усреднение по магнитным квантовым состояниям соответ-
ствует классическому пространственному усреднению по всем ориентациям
осей вращения диполей (при постоянном моменте количества движения отно-
сительно этих осей). Результат, изменяющийся, как l/rg&, не имеет простой
классической аналогии. Лондон [85] нашел, что на больших расстояниях
vtiv /2) может быть выражено в виде
/2); F.34)
здесь G(jly /2) — числа, приведенные в табл. 123.
Значения G(JU j2)
Таблица 123
0
1
2
3
4
о
-0,5
-0,Ш
0,25
0,100
0,0555
1
-0,111
-0,5
-0,0678
0,194
0,0786
0,25
-0,0678
-0,5
-0,0645
0,181
3
0,100
0,194
-0,0645
-0,5
-0,0336
4
0,0555
0,0786
0,181
-0,0336
-0,5
Если | ]\ — /21 велико, то
О (Я, /2) -> -от 7^—п~ • F.35)
Z l[Ji—]2) — 1J '
Для случая резонанса, когда j\ = / и /2 = / — 1 или ]\ = / — 1 и /2 = /,
F.36)
Такое детальное описание взаимодействия диполей необходимо для точ-
ной интерпретации свойств переноса полярных молекул и интерпретации уши-
рения линий микроволнового спектра под действием давления, рассматри-
ваемого в § 7.
В. КВАНТОВОМЕХАНИЧЕСКОЕ ВЗАИМОДЕЙСТВИЕ ДВУХ ИДЕАЛЬНЫХ
ДИПОЛЕЙ, ВСТАВЛЕННЫХ В СИММЕТРИЧНЫЕ ВОЛЧКИ
Квантовомеханическое взаимодействие двух диполей можно также рас-
смотреть в случае, если диполи вставлены в сферы (молекулы, у которых равны
главные моменты инерции) или симметричные волчки (молекулы, у которых
равны два главных момента инерции). Это реалистичные модели полярных
молекул. Расчеты очень схожи с расчетами для линейных диполей.
Волновая функция молекулы типа симметричного волчка со вставленным
идеальным диполем зависит от трех квантовых чисел fc, которые определяют
компоненты момента количества движения в направлении дипольного мо-
мента. Если к = 0, то положение во многом совпадает со случаем линейного
диполя. Однако если к не равно нулю, то в направлении межъядерно$ оси име-
ется результирующий электрический момент. Это можно видеть из фиг. 218.
Если предположить, что ось вращения фиксирована заданными квантовыми
числами / и т (как с линейным диполем), а ориентация диполя относительно
этой оси фиксирована заданным квантовым числом к, то диполь вынужден
осциллировать между двумя положениями, показанными на фиг. 218.
Таким образом, если квантовые числа ка и кь двух молекул отличны от
770 Гл. 73. Теория межмолекулярных сил
нуля, между молекулами имеет место электростатическое взаимодействие
первого приближения, и энергия взаимодействия изменяется, как 1/г^.
Волновые функции пары изолированных симметричных волчков [62, стр.
275 ; 86, 87 ] равны произведениям коэффициентов представления, определяе-
мых соотношением (Б.13) гл. 12
F.37)
Квантовые числа имеют следующий смысл : (Л2/4тг2)/(/ +1) — квадрат общего
момента количества движения ; (й/2тг)/с — компонента момента количества
Ось z
Ось вращения
молекулы
Фиг. 218. Возможные ориентации диполя, если ось вращения молекулы не совпадает с
направлением диполя.
Помимо показанного движения ось вращения молекулы вращается около оси г.
движения вдоль оси симметрии молекулы ; (й/2тг)/п — компонента момента
количества движения вдоль некоторого фиксированного произвольного на-
правления в пространстве. Квантовые числа могут иметь следующие значения :
/с =-/,-/+ 1, ... , 0,...,/- 1,/,
т =-/,-/+ 1, ... ,0, ...,/'- 1,/,
/= 0,1,2,...
Энергия пары изолированных молекул равна
та; jb, кЬу ть) = -^2~[]±
где /3 — момент инерции относительно оси симметрии; /х — два других равных
момента инерции.
Как и в случае линейных дипольных молекул [см. F.19)], произведение
потенциала взаимодействия и невозмущенной волновой функции может быть
представлено в виде линейной комбинации большого числа (до 27) невозму-
щенных волновых функций. Это непосредственно следует из соотношений
(Б.31) — (Б.ЗЗ) гл. 12 между коэффициентами представления. В результате,
если мы запишем произведение потенциала взаимодействия на невозмущенную
волновую функцию в виде
К> та; А, кь, ть)= 2 vt V>°(ja, ft«, /ni ; j'b, К, m'b), F.39)
Tab
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 771
то коэффициенты разложения (или матричные элементы) vt будут отличны от
нуля только при условии, что k'a = ka> k'b = kb, rn'a + m'b = ma + mb9 j'a = ja
или (/a ± 1), ]'ь = h или (/ft + 1) и ma = ma или (ma ± 1). В общем случае в вы-
ражение F.39) войдут 27 волновых функций, с которыми может взаимодейство-
вать любая частная волновая функция. Вследствие того, что в проблеме сим-
метричного волчка имеют место взаимодействия, определяющиеся как первыми,
так и вторыми порядками теории возмущений, эта проблема значительно более
сложна, чем в случае линейных дипольных молекул. Взаимодействия, в кото-
рых / и к для друх молекул остаются неизменными, рассматриваются как взаи-
модействия первого порядка. На больших расстояниях они дают наиболее
важный вклад в энергию взаимодействия. Члены первого порядка входят в
выражение для энергии только тогда, когда ка и кь отличны от нуля.
Маргенау и Уоррен [88] рассмотрели взаимодействия первого порядка.
Однако даже в этом случае проблема сложна. Для каждого значения /а, ка;
]ь, кь энергия системы расщепляется на большое число энергетических уровней.
Все состояния, для которых сумма та -f ть имеет одинаковые значения,
взаимодействуют и образуют свое собственное секулярное уравнение. Когда
/а = jb = ka = kb=l, секулярному уравнению удовлетворяют следующие шесть
видов энергии взаимодействия :
— 2 А з ¦ (дважды вырождена),
ДД3Ь (дважды вырождена),
^' ab
A - Щ-
F.40)
(дважды вырождена),
A+Кз)
Случай диполя, вставленного в симметричный волчок, отличается от случая
диполя, вставленного в линейную молекулу, тем, что среднее значение <раЬ
не исчезает в первом порядке. Это согласуется с тем фактом, что для симметрич-
ных дипольных молекул имеет место эффект Штарка первого порядка.
Кэррол [89] вычислил взаимодействие второго порядка, усредненное по
всем магнитным квантовым числам. Вычисление энергии взаимодействия с
учетом членов второго порядка в том виде, как это сделал Лондон для линей-
ных дипольных молекул, вследствие большого числа состояний, которые при
этом следует одновременно рассматривать, сложно (но не невозможно).
Г. ДАЛЕКИЕ ВЗАИМОДЕЙСТВИЯ МЕЖДУ ПРОТОНОМ^ И АТОМОМ
ВОДОРОДА ИЛИ АТОМОМ ГЕЛИЯ
Взаимодействие протона с атомом водорода или атомом гелия, находя-
щихся в основных состояниях, типично для взаимодействия иона с нейтральной
неполярной молекулой. При взаимодействии протона с возбужденным атомом
водорода появляются два дополнительных эффекта : 1) возбужденный атом
водорода имеет (за исключением s-состояний) квадрупольные моменты, так что
при этом возникают электростатические силы, и 2) вырождение возбужденных
состояний приводит к возникновению значительных резонансных сил, обсуж-
денных в разд. А. При взаимодействии протона с возбужденным атомом гелия
возникают электростатические силы того же сорта. Однако вследствие того,
Гл. 13. Теория межмолекулярных сил
что в данном случае возбужденные состояния лишь близки к вырождению,
резонансные силы оказываются значительно менее важными.
Легко показать, что атом, имеющий азимутальное квантовое число L,
обладает мультиполями порядка п = 0, 2,..., 2L. Таким образом, атом в
S-состоянии с L = 0 не имеет мультипольных моментов ; атом в Р-состоянии
с L = 1 имеет квадрупольный момент; атом в D-состоянии с L = 2 имеет
квадрупольный и шестнадцатипольныймоменты, и т. д. (по поводу определения
моментов см. гл. 12, § 1).
Таким образом, между протоном, и атомом в S-состоянии нельзя ожидать
прямого электростатического взаимодействия (здесь будут действовать лишь
индуцированные дипольные силы). Однако между протоном и атомом во всех
других состояниях будут действовать заряд-квадрупольные силы. Так как
основными состояниями атомов водорода и гелия являются S-состояния, то
такие силы будут иметь место лишь в случае, если атомы находятся в возбуж-
денных состояниях. Заряд-квадрупольные силы изменяются, как 1/г^,, вслед-
ствие чего они являются более дальнодействующими, чем силы, действующие
между зарядом и индуцированным диполем, изменяющиеся, как l/r?ft. Однако
для некоторых значений магнитного квантового числа заряд-квадрупольные
силы являются силами притяжения, для других — силами отталкивания.
Энергия взаимодействия заряда и квадруполя, усредненная по магнитному
квантовому числу (что соответствует классическому пространственному усред-
нению по всем ориентациям атома), исчезает.
Энергия взаимодействия между протоном и атомом водорода или атомом
гелия в основных состояниях, обычно имеющая характер энергии взаимодей-
ствия заряда и индуцированного диполя, равна <р(инД> = — ае2/2г^ь и не услож-
няется резонансными или электростатическими взаимодействиями. Однако
злектростатические взаимодействия между протонбм и возбужденным атомом
водорода или возбужденным атомом гелия являются примерами, в которых
резонанс в пределах одной молекулы влияет на природу дальнодействующих
межмолекулярных сил. В столкновении протона с возбужденным атомом
водорода резонанс приводит к появлению в выражении для энергии взаимодей-
ствия, как объяснялось в разд. А, членов, изменяющихся, как 1/г^,. Этот резо-
нанс возникает вследствие того, что все состояния атома водорода с одинако-
вым главным квантовым числом п имеют точно одинаковые энергии. Для атома
гелия состояния с одинаковыми главными квантовыми числами для двух
электронов имеют только небольшое различие в энергиях для различных зна-
чений азимутальных квантовых чисел. Таким образом, при взаимодействии
протона с возбужденным атомом гелия резонанс уже не столь резок и не при-
водит ни к появлению члена, изменяющегося, как \jrlby ни к влиянию на обыч-
ный член заряд-квадрупольного взаимодействия, изменяющегося, как l/rjj&;
однако он вызывает появление дополнительного члена, изменяющегося, как
V^iby который добавляется к энергии взаимодействия заряда с индуцирован-
ным диполем..
1. Взаимодействие протона с атомом водорода. Проблема взаимодействия
протона с атомом водорода является одной из немногих проблем молекулярной
квантовой механики, которая может быть решена точно. Для больших расстоя-
ний энергия взаимодействия протона с атомом водорода может быть точно опре-
делена [90, 91 ] в виде ряда по обратным степеням расстояния между прото-
ном и атомом
здесь каждое из Е( точно выражается через Е{ с меньшим значением и Ана-
логично может быть разложена в ряд по степеням ЦгаЬ волновая функция
y) = y)o + 7^y)l + 7hyJ+ ... F.42)
§6. Квантовомеханическая трактовка резонансных и электростатических сил 773
Пусть i3f0 — гамильтониан невозмущенного атома водорода, а Ео — энергия
частного состояния невозбужденного атома водорода. Возмущающий потенциал,
создаваемый полем протона, выражается в виде F.8), Подставляя степенное
разложение возмущающего потенциала, энергии взаимодействия и волновой
функции в уравнение Шредингера и приравнивая член с одинаковыми степе-
нями \/rab9 получаем следующую систему совместных уравнений :
-». F.43)
- га1Рг(соьва1)щ =
= Е2у>г + ?8 у>0,
Вследствие того что все уравнения разделяются в параболических коорди-
натах, решение их значительно упрощается, если вместо сферических исполь-
зовать параболические координаты
Для взаимодействия протона с атомом водорода в основном состоянии
Коулсон [90 ] получил следующее выражение для энергии взаимодействия :
213/4
1773/2 ' 86049/16]
ЫЫ* + (К)" J #
Крогдал [92] решила уравнения для взаимодействия протон^ с атомом
водорода в возбуждённом состоянии и получила результаты, приведенные в
табл. 124. В таблице п, /, т — главное, азимутальное и магнитное квантовые
числа соответственно ; Ео — энергия невозмущенного атома водорода. Энергии,
полученные этим способом, включают индукционную энергию и дают общую
энергию взаимодействия. Следует отметить, что функции потенциальной энер-
гии расщеплены. Например, атом, имеющий главное квантовое число п = 3
и магнитное квантовое число т = 0, может взаимодействовать с протоном по
любой из трех потенциальных функций :
F.45)
72 I
I2 (ГаьМ'У
Коулсон и Гиллам [91 ] показали, как обобщить результаты, которые
получила Крогдал, чтобы определить энергию взаимодействия с точностью
до любой степени l/raft.
Энергия взаимодействия протона с атомом водорода в любом состоянии
на малых и средних расстояниях вычисляется в гл. 14, § 5. Система протона и
атома водорода охватывает случай двухатомного иона водорода Щ> изучен-
ный весьма подробно, поскольку проблема взаимодействия протона с атомом
водорода является одной из немногих проблем молекулярной квантовой меха-
ники, которая может быть решена точно.
2. Взаимодействие протона с возбужденным атомом гелия. Взаимодействие
протона с возбужденным атомом гелия может быть изучено с помощью мето-
дов, во многом совпадающих с использованными для изучения взаимодействия
774
Гл. 13. Теория межмолекулярных сил
Таблица 124
Энергия взаимодействия протона с атомом водорода [92] в единицах е2/а0
п
1
2
3
4
5
М
0
1
0
2
1
0
3
2
1
0
4
3
2
1
0
/
0
1
0,1
2
1,2
0,1,2
3
2,3
1,2,3
0,1,2,3
4
3,4
2,3,4
1,2,3,4
0,1,2,3,4
Е.
-1/2
-1/8
-1/8
-1/18 |
-1/18
-1/18
-1/32
-1/32
-1/32
1/32
-1/50
-1/50
1/50
-1/50
-1/50
0
0
± з
0
± 9/2
0
± 9
0
± 6
0
±12
± б
±18
0
±15/2
0
±15
±15/2
±45/2
0
±15
±30
Et
0
+ 6
- б
+ 36
+ 9
+ 36
- 72
+ 120
+ 72
+ 120
- 72
+ 72
-312
+300
+225
+300
0
+225
-375
+300
0
-900
- 9/4
- 78
-1377
-3360
-46875/4
протона с атомом водорода. Существенное отличие первого случая от второго
заключается в том, что состояния с одинаковым главным квантовым числом,
но различными азимутальными квантовыми числами имеют слегка различные
энергии. В результате резонанс оказывается не «острым» и энергия взаимодей-
ствия имеет вид [93]
w — JL Г Mflo> _ (аУдо> + 1 • (б 46)
здесь ^ — квадрупольный момент частного состояния изолированного атома
гелия. Поляризуемость а' отличается от поляризуемости изолированного
атома вследствие близости системы к резонансу ; а' была вычислена на основе
предположения, что волновую функцию атома гелия, возмущенного протоном,
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 775
можно представить в виде линейной комбинации волновых функций состояний,
близких к резонансу. Энергия уаЬ была определена путем решения соот-
ветствующих секулярных уравнений. Поляризуемость а' просто связана с
коэффициентом при \jr%b в разложении q>ab в степенной ряд. Значения величин
qjal и a'/aj) приведены в табл. 1251).
Таблица 125
Квадрупольный момент q и величина а' для атома гелия в различных состояниях
п
2
2
2
3
3
3
3
3
3
4
4
4
4
4
4
4
4
4
5
5
5
О
*1
1
0
1
2
1
2
2
О
1
2
3
1
2
3
2
3
О
1
2
m
О
0
1
О
О
О
1
1
2
О
0
О
О
1
1
1
2
2
О
1
О
я
<
О
-24
12
0
-144
-72
72
-36
72
О
-480
-288
-192
240
-144
-144
288
0
0
-1200
-5400/7
о!
<
+428
-428
-20362
+42 429
-22 067
+85 964
+ 136 366
+3357 782
-3580110
л
5
5
5
5
5
5
5
5
5
6
6
6
6
6
6
6
6
6
б
б
6
б
6
6
/
3
4
1
2
3
4
2
3
4
0
1
2
3
4
5
1
2
3
4
5
2
3
4
5
т
0
0
1
1
1
1
2
2
2
0
0
0
0
0
0
1
1
1
1
1
2
2
2
2
я
<
-600
-3000/7
600
-2700/7
- 450
-2550/7
5400/7
0
-1200/7
0
-2520
-11736/7
-1392
-7920/7
- 840
-1260
-5868/7
-1044
-6732/7
- 756
11736/7
0
-3168/7
- 504
В каждой конфигурации один ид электронов находится в ls-состояш.и, другой электрон — в сос-
тоянии, характеризуемом квантовыми числами п, I, т.
Д. СИЛЫ КВАДРУПОЛЬ-КВАДРУПОЛЬНОГО ВЗАИМОДЕЙСТВИЯ АТОМОВ,
НИ ОДИН ИЗ КОТОРЫХ НЕ НАХОДИТСЯ В ^-СОСТОЯНИИ
Взаимодействие двух атомов, ни один из которых не находится в S-cocto-
янии, является примером квадруполь-квадрупольного взаимодействия. Инте-
ресно отметить, что существует несколько различных потенциалов взаимодей-
ствия, характерных для сталкивающейся системы, причем каждый из этих
потенциалов соответствует своему набору «молекулярных» квантовых чисел,
описывающих объединенную систему двух атомов. Эти энергии взаимодействия
(изменяющиеся, как 1/г^) намного больше дисперсионной энергии. Например,
энергия притяжения двух атомов кислорода, находящихся на расстоянии
3,0 А, составляет 0,6 ккал/моль; для двух атомов бора такая же энергия притя-
жения будет на расстоянии 5,1 А, а для атома углерода, взаимодействующего
с атомом кислорода, такая же энергия достигается на расстоянии 3,2 А. Можно
х) Значения величин, приведенные в таблице, легко получаются из величин, табулиро-
ванных в [93 ].
776 Гл. 13. Теория межмолекулярных сил
ожидать, что аналогичные силы проявляются при взаимодействии свободных
радикалов или других молекул, обладающих отличным от нуля суммарным
моментом количества движения электронов. Учет этих сил позволит объяснить
некоторые аномалии химических и физических свойств, обнаруженные у таких
молекул.
Книпп [97 ] изучил энергию взаимодействия двух атомов, ни один из кото-
рых не находится в S-состоянии. В этом случае энергия квадруполь-квадру-
польного взаимодействия появляется в первом порядке. В зависимости от
величин магнитных квантовых чисел некоторые атомы притягиваются, а другие
отталкиваются при взаимодействии с выбранным атомом.
Система из двух атомов образует «двухатомную молекулу». На больших
расстояниях, где взаимодействием можно пренебречь, волновые функции со-
стояний, характеризующихся определенными молекулярными квантовыми
числами, являются линейными комбинациями произведений атомных волно-
вых функций [98—101 ]. Если спины двух атомов равны соответственно S9l и S2,
то полный спин «молекулы» может принимать одно из следующих значений :
S^S^S,, Sl + S1-l,...f|Sl-S1|. F.47)
Для наших целей важны квантовые числа ДйиГ:
Л = | mLl + mL21 — квантовое число, характеризующее абсолютное
значение проекции орбитального момента количества движения на межъядер-
ную ось; Л может принимать любые целые значения от 0 до Lx + L2, где
Lx и L2 — орбитальные моменты количества движения атомов. Как было
ранее условлено, состояниям, обозначаемым 27, П, А, Ф,..., соответствуют
значения Л = 0, 1, 2, 3,... Поскольку Л равно абсолютному значению
mLl + mL2, то состояния П,А,Ф,... — двукратно вырожденные, так как проек-
ция орбитального момента количества движения на межъядерную ось может
быть либо положительной, либо отрицательной, а состояния Z не являются
вырожденными.
Q = | Щд -f mj21 — квантовое число, характеризующее абсолютное зна-
чение проекции полного момента количества движения на межъядерную ось.
Г = | mjx + mLl | — особое квантовое число, являющееся смешанной
характеристикой.
Расчет энергии взаимодействия атомов на больших расстояниях услож-
няется тем фактом, что расщепление вследствие атомных спин-орбитальных
взаимодействий может быть величиной того же порядка. Спин-орбитальное
расщепление изменяется от 0,0018 эв для основного состояния бора до 0,94 эв
для наинизшего терма таллия. Если спин-орбитальное расщепление мало по
сравнению с энергией взаимодействия порядка кТ, то имеет смысл рассматри-
вать орбитальное квантовое число атома L. Если спин-орбитальное расщепле-
ние велико по сравнению с кТ, то имеет смысл рассматривать полный момент
количества движения атома J. По этим причинам рассмотрим столкновения
трех типов:
A) Спин-орбитальное расщепление обоих атомов 1 и 2 мало по сравнению с кТ.
Б) Спин-орбитальное расщепление атома 1 велико по сравнению с кТ,
а атома 2 мало по сравнению с кТ.
B) Спин-орбитальное расщепление обоих атомов 1 и 2 велико по срав-
нению с кТ.
Книпп [97] нашел, что энергия взаимодействия, получаемая в первом
порядке теории возмущений, в этих трех случаях может быть выражена сле-
дующим образом:
A) 9 = ^[r{nylf]1[r{nylf]2B1{L^Bz{L2)X{Ll) L2; Л),
Б) 9> = -? [г (п, /J]i [ФЖ D, (Л) В2 (L2) А (Л; L2; Г), F.48)
B) <Р = 4 [г (л, 1)*]г [г (п, /)*]2 D, (Л) D2 (Л) Я (Л; J2; ii).
Таблица 126
Элемент
В*
А1*
Ga
In
Tl
С*
Si*
Qe
Sn
Pb
0*
s
Se
Те
F
Cl
Br
J
Атомы с незаполненными
Основной
уровень
2p
3p
4p
5p
6p
2p*
3p*
4p«
5p*
6p*
2p*
3p*
4p«
5p*
2p«
3p«
4p*
5p»
• n
)
\sP
1 2
j
)
1 a o
1 8/>Vf
j
B(L)
-0,6325
-0,6325
-0,6325
-0,6325
-0,6325
0,6325
0,6325
0,6325
0,6325
0,6325
-0,6325
-0,6325
-0,6325
-0,6325
0,6325
0,6325
0,6325
0,6325
0
0
0
0
0
0
0
0
0
0
-0,3743
-0,3743
-0,3743
-0,3743
0,4472
0,4472
0,4472
0,4472
р-оболочками
Радиус м
ной плотнс
внешней
в ед
метод
экраниро-
вочных
постоян-
ных
Слетера
1,54
2,57
2,74
3,20
3,53
1,23
2,17
2,42
2,83
3,12
0,88
1,65
1,97
2,30
0,77
1,48
1,80
2,11
аКСИМоЛЬ"
>сти заряда
орбитали
.а.
метод
самосо-
гласован-
ного
поля
1,80
1,26
0,85
0,73
[г(л,
О']
в ед. а*
метод
экраниро-
вочных
постоян-
ных
Слетера
4,4
10
11
14
17
2,8
7,3
8,5
И
14
1,4
4,2
5,6
7,5
1Д
3.4
4,7
6,2
0
метод
самосо-
гласован-
ного
поля
9,12
4,882
2,440
1,82
Звездочка у названия элемента означает, что спин-орбитальное расщепление терма, соответствующего
наинизшей энергии, составляет менее 0,036 эв, или 0,6 ккал/моль.
Таблица 127
Элемент
Sc*
Y
La
Ti
Zr
Hf
V
w
Fe
Co
Ni
ir
•
Атомы с i
Основной уровень
3</4s2
4</5s2
5</6s2
Зс/Мя2
4gP5s2
ЗсГЧз8
5d«5s2
3de4s2
3d4s*
3d4^
5d*
)
j
Ц
J
•F.,,
6^o
tF'l.
1езаполненными d-оболочками
B{L)
•
-0,5345
-0,5345
-0,5345
-0,2213
-0,2213
-0,2213
0,2213
0,5345
-0,5345
-0,2213
0,2213
0,5345
D(J)
-0,4472
-0,4472
-0,4472
-0,1833
-0,1833
-0,1833
0,1533
0
-0,4006
-0,1935
0,2003
0,4782
Метод экрани
постоянных
радиус макси-
мальной плотно-
сти заряда внеш-
ней орбитали в ед.
а0
3,00D,55)
4,55E,33)
5,33E,88)
2,47 D,35)
3,75 E,08)
4,38E,60)
2,09D,15)
3,23E,11)
1,44C,65)
1,32C,51)
1,21 C,38)
2,58
ровочных
Слетера
Ил, 0*1
в ед. а0
14
30
40
9,5
20
27
6,8
15
3,2
2,7
2,3
9,4
Числа в скобках означают положения максимума радиальной плотности двух электронов в s-оболочке,
лежащей выше незаполненной rf-оболочки. Положение этих электронов определяет радиус атомов.
Величины A(J*i> h2f or) для целых значений д
Таблица 128
д
0
1
2
3
4
5
h, - 1
Л2 - 1
0 еа
3,6/6
0 /Ь
0 г
-2,4/
0,6/
2
1
-1,014 а
2,324 Ь
-0,295 Ь
2,456
0,047
-1,489
0,356
-2,891
1,014
3
1
-1,225 а
2,096 Ъ
-0,382 Ъ
2,061
-0,170
-1,401
2,199
0,230
-1,939
0,646
-3,096
1,225
4
1
-1,305 а
2,013 Ъ
-0,418 Ъ
1,972
-0,289
-1,392
1,367
0,347
-1,424
2,032
-0,032
-1,710
0,853
-3,222
1,354
1,5
1,5
2,4 еа
0 еа
3,2 fb
-0,8 fb
-1,6 е
1,2/
-0,8/
0,4 е
-2,8/
1,2
2
2
2,774 еа
-1,060 еа
3,261 /&
1,508 fb
-0,484 /ft
1,202 е
-0,917 е
1,127 /
-1,413 /
-2,000 е
0,857 /
-1,857 /
0,857 е
-2,571 /
1,714 /
Таблица 129
Величины Wlyh2,6) для полуцелых значений д
б
0,5
1,5
2,5
3,5
4,5
5,5
ht = 1,5
Л, - 1
2,746
0
-1,049
0,176
-2,722
0,848
2
1,5
2,864
1,551
-0,377
-1,168
0,916
-1,195
. -1,872
0,619
-2,770
1,434
4,5
1
1,977
-0,399
-1,345
1,916
-0,206
-1,476
1,883
0,125
-1,774
2,054
0,518
-2,339
0,934
-3,270
1,401
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 779
Значения B(L), D(J) и [r(n, If ] приведены в табл. 126 и 127 [97 ] для основ-
ных состояний многих элементов. Там же приведены значения среднего квад-
рата радиуса электрона [г(п, IJ ], полученные как с помощью метода экрани-
ровочных постоянных Слетера, так и по методу самосогласованного поля. Эти
величины очень чувствительны к распределению заряда. Коэффициенты А при-
ведены в табл. 128 и 129 [97 ].
В табл. 128 и 129 каждому набору аргументов соответствует несколько
состояний. Спектроскопические обозначения состояний с помощью букв пони-
маются следующим образом. Определим с = wtw2(—l)h*+h*9 где w — число
электронных состояний. Спектроскопические обозначения состояний, соответ-
ствующие различным энергиям, получаются при этом по следующим пра-
вилам.
Если атомы различны, то а соответствует 2>-состоянию при с = — 1
или ^'"-состоянию при с = + 1, а & соответствует -^-состоянию при с = + 1
или ^--состоянию при с = — 1. Если атомы одинаковы, то е соответствует
g-состоянию при S нечетном и u-состоянию при S четном ; ^-состояния от-
рицательны ; / соответствует g-состоянию при S четном и u-состоянию при S
нечетном ; ^-состояния положительны.
Для других случаев спектроскопические обозначения состояний ясны и
символ a, b, e, f можно не рассматривать, однако энергии взаимодействия по-
прежнему определяются вышеуказанными выражениями. В табл. 130 [97]
приведены значения Я, полученные для некоторых типичных случаев.
Таблица 130
Значения А для некоторых типичных молекул
Молекула
F2 или В2
С2 или О2
СО
А
+3,6
+0,6
0,0
-2,4
+3,6
+0,6
0,0
-2,4
+3,6
+0,6
0,0
-2,4
Состояния
1Г» + ЗУ>
^gy ^и
xAg,*Au
Ит **gy ^gy 2*\лу ^g
1Ilg, 3/7u (нижележащие состояния)
1Г+ Зу+ 5у +
xAg^AmbAg
^пу sZg-y 5Г~, гЕ+у 3Г^, bZg
1/7 3/7 5Г7
1ЛУ §У U
xngi 3/7u, 5llg (нижележащие состояния)
1Z+,3Z+y5Z+ (нижележащие состояния)
11 у— 3 у — 5У—
\%*пУп+'1П'ЗП'ЬП
Из примеров, приведенных в табл. 131 [97] видно, что силы квадруполь-
квадрупольного взаимодействия достигают большой величины. Расстояния,
приведенные в таблице, соответствуют примерно удвоенным равновесным
расстояниям для нормальной двухатомной молекулы. При любых других
расстояниях энергия изменяется обратно пропорционально пятой степени
отношения расстояний.
780
Гл. 13. Теория межмолекулярных сил
. Таблица 131
Энергии притяжения на расстояниях, равных удвоенной сумме
атомных радиусов
ва
во
с2
CF
о2
F2
ВС
BF
СО
OF
Известные состояния
двухатомных молекул
22+92П
8/7и?
_ в
Tab, A
3,80
2,80
2,66
2,10
1,80
1,54
3,23
2,67
2,23
1,67
fab, Эв
-0,112
-0,139
-0,190
-0,231
-0,341
-0,406
-0,202
-0,195
-0,347
-0,549
Состояния, дающие
наибольшую энергию
притяжения на больших
расстояниях
гП8*Пи
2П, 4/7
ingf *пш *ng
>
1Яг,зЛ„,«Яг
Wg, 8Я„
*?-,*?-
12>+' 3U+, 6Z+ -
Замечание. Атомные радиусы определены как расстояния, соответствую-
щие положениям последнего максимума на функции электронной плотности.
Спин-орбитальным взаимодействием пренебрегается и значения г1 берутся
из последней колонки табл. 126 и 127. Последние четыре молекулы имеют
при этих расстояниях дополнительно к указанным еще ^-состояния, соот-
ветствующие притяжению с энергиями, равными Ve энергий 27-состояний и
с той же мультиплетностью (пренебрегая другими силами). В таблице
приведены известные наблюдавшиеся состояния, молекулы из которых
диссоциируют на нормальные атомы.
На фиг. 219 изображены энергии взаимодействия атомов углерода и кис-
орода, находящихся в основных состояниях, на больших расстояниях. Четыре
0,016
Фиг. 219. Энергия взаимодействия атома углерода с атомом кислорода, находящихся
в основных состояниях.
Ьривая а —энергия в состояниях 127+, *П, 1П ; кривая Ь — в состояниях ХА, *А, *А ; кривая с — в со-
стояниях ЧГ—, *?—, *?—, 127+, »Г+, ХП, *П, *П; кривая d - б состояниях хП, *Я, *П. Эти кри-
вые справедливы лишь при расстояниях, больших 3 А.
потенциальные кривые на больших расстояниях расщепляются на 18 отдель-
ных потенциальных кривых при малых расстояниях. Основное состояние
молекулы окиси углерода обозначается как 1Г+. Таким образом, продолжение
§ 7. Определение межмолекулярных сил по уширению микроволнового спектра 781
потенциальной кривой основного состояния молекулы окиси углерода на
больших расстояниях должно в соответствии с табл. 130 слиться с потенциаль-
ной кривой наинизшего значения энергии, показанной на фиг. 219, так как две
потенциальные кривые, соответствующие состояниям с одной и той же симмет-
рией, не могут пересечься.
Обсуждаемое здесь квадруполь-квадрупольное взаимодействие, по всей
видимости, играет большую роль при взаимодействии и рекомбинации свобод-
ных радикалов. Эти силы важны также при определении физических свойств
веществ, содержащих большое число возбужденных молекул, таких, например,
как горячие газы или системы, в которых происходят быстрые химические
реакции.
§ 7. ОПРЕДЕЛЕНИЕ МЕЖМОЛЕКУЛЯРНЫХ СИЛ
ИЗ ДАННЫХ ПО УШИРЕНИЮ МИКРОВОЛНОВОГО СПЕКТРА
ВСЛЕДСТВИЕ ДАВЛЕНИЯ
Уширение линий микроволнового спектра вследствие давления является
важным источником сведений о мультипольных моментах молекул и дально-
действующих силах межмолекулярного взаимодействия. В настоящее время
в высокоразрешающих микроволновых спектрометрах фактически устранены
ошибки, связанные с «шириной щели», что позволяет получать эксперименталь-
ные данные с большой точностью. Однако, вследствие того, что теоретический
анализ данных находится еще в начальной стадии, интерпретация эксперимен-
тальных данных довольно грубая. В этом параграфе сначала будут изложены
некоторые важные факты, касающиеся уширения спектральных линий, а за-
тем будет указано, какие сведения можно получить из теоретического анализа
данных по уширению вследствие давления.
А. УШИРЕНИЕ ЛИНИЙ В МИКРОВОЛНОВОМ СПЕКТРЕ
Ширины спектральных линий обусловливаются тремя факторами:
1) естественная ширина линий AvN, обязанная «радиационному трению»;
2) допплеровское уширение AvD, связанное со скоростью молекул ; 3) уширение
вследствие давления AvPBy обязанное межмолекулярным силам. При микровол-
новых частотах (от 5 • Ю4 до 5 • Ю10 сек*1) уширение вследствие радиационного
трения и эффекта Допплера мало по сравнению с уширением вследствие
давления. При давлениях порядка 1 мм рт. ст. уширение линий вследствие
давления превосходит допплеровское уширение от 20 до 100 раз, а последнее
значительно больше уширения вследствие радиационного трения. Для объя-
снения уширения вследствие давления необходимо принимать во внимание
дипольные и квадрупольные моменты, а также поляризуемость молекул.
Очень часто по ширине линий можно определять вероятность перехода посту-
пательной энергии во вращательную при столкновении. В этих процессах
особенно важную роль играют дальнодействующие силы, поскольку в неко-
торых случаях наблюдаемые сечения перехода поступательной энергии во
вращательную фактически больше газокинетических сечений.
1. Естественная ширина линий,вызванная радиационным трением1). Рас-
смотрим переход между двумя уровнями энергии Ег и Е2. Поскольку эти уровни
обладают конечными ширинами АЕХ и АЕ2У то спектральная линия, связанная
с переходом между двумя уровнями, также будет обладать естественной полу-
шириной AvN. Если молекулы находятся в среднем в течение промежутка вре-
мени 1Л в состоянии 1 с энергией Е1} то в соответствии с принципом неопреде-
ленности
h^E1 = k. G.1)
»)См. [102, 103].
782 Гл. 13. Теория межмолекулярных сил
Кроме того, 1/*! можно интерпретировать как скорость спонтанного перехода
из ?гсостояния во все fc-состояния, для которых Ek < Ev Выражая 1/^ через
«силы осцилляторов», определенные уравнением F.22), получаем
где vlk и flk — частоты и силы осцилляторов, связанные с переходом вниз
из 1 в к. Таким путем Вайскопф и Вигнер [104 ] и Хойт [105 ] нашли, что интен-
сивность линии определяется выражением
где
Av 4$
4$
+ 2 Л ft] G.4)
является полушириной линии. В этой формуле fc-состояния соответствуют
энергиям, лежащим ниже уровня Е19 а /-состояния —энергиям, меньшим Е2.
Величина в скобках обладает максимальным значением — порядка г^/3.
Таким образом, максимальное значение естественной полуширины линии со-
ставляет Ю А и не зависит от длины волны.
2. Допплеровское уширение спектральных линий. Молекула, движу-
щаяся со скоростью v в направлении распространения света, поглощает час-
тоту
(f) G.5)
где v0 — частота, соответствующая скорости, равной нулю. Если принять во
внимание функцию распределения тепловых скоростей молекул в газе, то
можно показать, что допплеровская полуширина равна
An = i
3. Уширение микроволновых линий вследствие давления. Очень подроб-
ная теория уширения вследствие давления была развита Андерсоном [106]1),
а упрощенный вариант ее — Смитом и Ховардом [108]. Существенные члены
в теории уширения микроволнового спектра вследствие давления совершенно
отличаются от членов, играющих важную роль при изучении уширения опти-
ческих спектров вследствие давления [102]. Так же как и в случае естествен-
ной ширины линии, можно применить принцип неопределенности для Еыра-
жения полуширины линий AvPB через характеристическое время т:
xh AvPB = h , или AvPB = JL • G.7)
Время т является средним интервалом времени, в течение которого процессы
излучения не прерываются столкновениями. Для молекул в виде твердых сфер
диаметром аРВ, движущихся со средней скоростью Q, время т является сред-
ним временем между двумя столкновениями2)
Т== y2nUQ '' G>8)
здесь п — число молекул в 1 см*. Комбинируя два последних соотношения,
можно определить диаметр столкновения аРВУ связанный с уширением микро-
х) См. также работу [107].
2) Уравнение G.8) согласуется со средним временем между двумя столкновениями,
определенным формулой B.1) гл. 1 при f = fl.
§ 7. Определение межмолекулярных сил по уширению микроволнового спектра 783
волнового спектра вследствие давления, через наблюдаемую ширину линий:
B. G.9)
В газе, состоящем из двух сортов молекул а и Ь, уширение линий газа а вслед-
ствие давления определяется выражением
2 AvPB = 1/2 [па Qa (аРВIа + пь Qab (oPB)$b] ; G.10)
здесь Qab = У {Ql + Qb)/2 — средняя скорость ; {оРВ)аЬ — диаметр столкнове-
ния при смешанных столкновениях, связанный с уширением вследствие дав-
ления.
Эти соотношения всегда применяются для определения микроволнового
диаметра столкновения, независимо от закона сил, действующих между моле-
кулами. Однако значение аРв зависит от закона сил и характера столкновений.
Время х всегда есть время между столкновениями. Существует два предельных
случая столкновений: адиабатические столкновения, в которых фаза испущен-
ного излучения значительно меняется, и неадиабатические столкновения, при
которых происходит скачкообразное изменение квантового состояния, т. е.
обмен энергией между рассматриваемой и налетающей молекулами. В случае
оптического уширения вследствие давления важны все адиабатические столк-
новения, тогда как неадиабатическими столкновениями можно пренебречь.
С другой стороны, в теории уширения микроволнового спектра вследствие
давления адиабатическими столкнорениями обычно пренебрегают. Пусть
вероятность перехода молекулы а из вращательного состояния ja в состояние
ja равна | а^уа |2. Вероятность перехода по истечении времени столкновения
равна | aiai'a{t — °° ) |2. В соответствии со строгой теорией Андерсона все неадиа-
батические столкновения, для которых | aja^(t = ©о) |2 ^> i/4, эффективны в
уширении вследствие давления в том смысле, что они определяют эффективный
жесткий диаметр аРВ и время т, используемое в уравнениях G.7) — G.10).
В этом случае величина аРВ непосредственно определяет меру вероятности
перехода энергии обычно из поступательных степеней свободы во вращатель-
ные в процессе столкновений.
Вероятности перехода можно получить с помощью теории возмущения,
зависящей от времени [109]. Пусть <Жа — квантовомеханический оператор
Гамильтона для изолированной молекулы а. При столкновении с другой моле-
кулой Ь к этому гамильтониану необходимо добавить возмущение, возникаю-
щее вследствие взаимодействия двух молекул. Запишем зависящее от времени
возмущение в виде
<Р @ = ч>аь На, та ; jb9 ть \ гаЬ it)]. G.11)
Это значит, что потенциал возмущения явно зависит от квантовых состояний
двух взаимодействующих молекул и является неявной функцией времени ввиду
зависимости от межмолекулярного расстояния rab(t). Зависимость гаЬ от /
определяется траекторией сталкивающейся пары. Используя этот потенциал
возмущения, уравнение Шредингера можно записать в виде
МГ« + v(t)] у (г-, о - - 4™ <7Л2>
Волновую функцию можно записать в виде линейной комбинации волновых
функций начального /а-состояния и конечного /д-состояния :
в/.у.(ОУу. + а/.л(О*гл. G.13)
До столкновения (t = — °о) молекула находится в /а-состоянии, так что
вМ.(-~)=1, влл(-~) = 0. G.14)
784 Гл. 13. Теория межмолекулярных сил
После столкновения (t = оо) вероятность перехода из /а- в /д-состояние опре-
деляется квадратом абсолютной величины выражения
л*в- GЛ5)
Диаметр столкновения огРВ является расстоянием наибольшего сближения1)
(если траектория — прямая линия) для критического столкновения, при кото-
ром | ajaj'a(t = оо) |2 = !/4. Если оРВ велико по сравнению с газокинетическим
поперечником молекул, что встречается довольно часто, то критическая траек-
тория является почти прямым путем и с хорошим приближением
rlb(t) = o%B + v4\ G.16)
где v — относительная скорость двух молекул в отсутствие взаимодействия.
Кроме того, Андерсон показал, что если hv\aPB меньше {Eja — Е/а)у как это
обычно бывает, то пренебрежение экспонентой в формуле G.15) приводит к
ошибке порядка нескольких процентов.
Б. СВЕДЕНИЯ О ДАЛЬНОДЕЙСТВУЮЩИХ СИЛАХ ИЗ ДАННЫХ
ПО УШИРЕНИЮ ВСЛЕДСТВИЕ ДАВЛЕНИЯ
Проиллюстрируем, как можно использовать указанные выше результаты
для получения некоторых сведений о дальнодействующих межмолекулярных
силах. Рассмотрим различные формы нотенциала межмолекулярного взаи-
модействия cp(t) в уравнении G.11).
1. Диполь-дипольное взаимодействие. Энергия взаимодействия двух дипо-
лей определяется формулой A.47) гл. 12. Пусть критическая траектория явля-
ется прямой линией с расстоянием наибольшего сближения, равным аРв.
Направление радиуса-вектора от молекулы а к молекуле Ь, находящейся на
расстоянии наибольшего сближения, примем за направление полярной оси,
по отношению к которой будем измерять углы в'ау, Q'b и В'аЬ. Остальные углы
Фа, Фь и фаь измеряются относительно плоскости, в которой происходит
столкновение. Для критического столкновения вдоль прямой линии rab(f)
определяется уравнением G.16); в этом случае cos 6'аЬ = Ъ\таЬ [где Ь при-
цельное расстояние, определенное в связи с уравнением E.2С) гл. 1 ], а
sin Q'ab = vtjrab и фаЬ = 0. Если мы пренебрежем экспонентой в формуле G.15)
и произведем интегрирование по времени до вычисления матричных элементов
вращательных переходов, то получим
Г fab @ dt = -^g?- (-COS в'а COS в'„ + Sin в'а Sitl в'ь Sitl ф'а sitl ф'ь). G.17)
— 00
В этом случае вероятности переходов получаются умножением уравнения
G.17) на волновые функции начального и конечного состояний и .интегриро-
ванием по всем координатам молекулы. Результаты до некоторой степени услож-
няются резонансным взаимодействием, обсужденным в § б, разд. Б и В. Андер-
сон [106] и Мицушима [107] вывели выражение для уширения вследствие
давления в случае молекул типа симметричного волчка и линейных- полярных
молекул, а также для аммиака. Молекулы аммиака принадлежат к молекулам
типа симметричного волчка с той лишь разницей, что они имеют дополнитель-
ные степени свободы, соответствующие возможности инверсии.
Фактически самоуширение спектра аммиака представляет особенно про-
стой пример, потому что вращательные энергии достаточно велики, и необ-
Автор пользуется здесь непереводимым, термином «miss distance». — Прим. ред.
§ 7. Определение межмолекулярных сил по уширению микроволнового спектра 785
ходимо рассматривать лишь несколько переходов или несколько матричных
элементов. Теоретические данные Андерсона сравниваются с эксперименталь-
ными результатами в табл. 132. Поскольку средняя скорость столкновений
пропорциональна корню квадратному из температуры, то аРВ пропорцио-
нально Г/*. Эта температурная зависимость подтверждена в работе Ховарда
и Смита [108].
Таблица 132
Ширина линий Av для спектра инверсии NH8 при комнатной температуре*
J
2
3
3
3
4
5
5
5
5
к
1
1
2
3
4
1
2
3
5
Avt мггц/мм рт. ст.
эксп.
15,5
14,5
19
27
27
11
15,5
20
28
теор.
15,5
14
20
27
27,5
11
15
20
29
орв, А
эксп.
10,4
Ю,1
11,6
13,8
13,8
8,8
10,4
11,9
14,1
J
6
6
i 6
7
7
8
10
11
к
3
4
6
5
6
7
9
9
Av, мггц/мм рт. ст.
эксп.
18,5
21,5
28
24,5
23,5
24,5
25
17,5
теор.
17
21
29
22
26
26
27
24,5
орв, А
эксп.
11,4
15,2
14,1
13,1
12,7
13,1
13,3
11,1
* Таблица взята у Андерсона [106J и изменена в соответствии с данными Ховарда и Смита [108]
В настоящее время самоуширение спектров линейных полярных молекул
несколько менее хорошо изучено, чем самоуширение в случае молекул сим-
метричного волчка. В связи с этим можно думать, что уширение вследствие
давления в газовых смесях интерпретировать будет даже труднее. Однако это
не так. Экспериментальные эффективные сечения молекул типа симметричных
волчков (СН8С1 и СНС13), относящиеся к уширению линии аммиака 3, 3,
могут быть объяснены тем же способом, что и самоуширение аммиака. Экспе-
риментальные результаты для этих молекул приведены в табл. 133 [108].
Таблица 133
Величины аРВ (для столкновений с NH3) и дипольные моменты из данных по
уширению вследствие давления линии аммиака j = 3 и к — Зпри столкновении
с другими газами
Налетающая
молекула
NH8
COS
HCN
C1CN
~ A
<*PB, A
13,8
7,56
10,0
11,9
fi • 10",
эл.-ст. ед.
1,44
0,720
2,96
2,80
Налетающая
молекула
СН3С1
СН2С12
СНС18
so,
_, А
°РВр А
11,3
10,3
13,7
10,4
ц •10»,
эл.-ст. ед.
1,87
1,59
0,95
1,7
2. Диполь-квадрупольное взаимодействие1). Рассмотрим взаимодействие
молекулы с дипольным моментом /га, сталкивающейся с молекулой, имеющей
квадрупольный момент Qb. В этом случае потенциал возмущения, связанный с
взаимодействием, имеет вид
9ф = 4~
- 3cos20;) + 2cosflisinв'ьsinд'аcos(ф'а - ф'ь)\, G.18)
м. [108].
786
Гл. 13. Теория межмолекулярных сил
где 6'ау О'ь, фа и ф'ь — полярные и азимутальные углы диполя и квадруполя
относительно межъядерной оси. Подставляя это выражение в формулу G.15),
интегрируя и усредняя по всем углам, получаем
ре —
0,84 [Xa
G.19)
В табл. 134 [103] приведены величины квадрупольных моментов, определен-
ные из данных по уширению вследствие давления линии NH3 с / = 3, к = 3
другими молекулами. В этой таблице q = Qje в соответствии с формулой A.17)
гл. 12.
Таблица 134
Квадрупольные моменты молекул, полученные с помощью микроволновых
диаметров столкновения
Молекула
оа
N0
СО
со2
COS
csa
<т,А
по кине-
тической
теории
4,09
4,02
3,90
3,96
4,46
из уширения
линии
3,3 NH3
5,54
3,86
5,64
5,97
7,59
7,56
7,72
?вр., А' а
0,31*
| 0,W
0,29
0,33
0,64
0,60
0,64
Молекула
N2O
HCN
C1CN
C2H2
C2H4
с2нв
н2
по кине-
тической
теории
4,35
4,79
4,86
из уширения
линии
3,3 NH,
7,32
10,0
11,9
8,79
6,67
5,64
Явр., А* °
0,91б
1,60
2,39
1,10
0,48
<0,27
4=0,220
а Величины <7вр. относятся к вращающимся молекулам.
* Использованы средние экспериментальные значения для расчета ?Вр
6 Для получения верхнего предела двр# использовано значение а = 4,18 А, которое считается наи-
более правдоподобным.
г Андерсон, Смит и Горди [111] получили это значение из измерений ширины линий тонкой структуры
микроволнового спектра кислорода. Точность этого числа, однако, вызывает сомнение.
д Для Н, величина q скорее связана с межъядерной осью, чем представляет собой величину для вра-
щающихся молекул. Квадрупольный момент измерили Харрик и Рамзей [112J, применившие микровол-
новый резонансный метод молекулярного пучка [113] для измерения вращательного магнитного момента.
Наш квадрупольный момент молекулы q равен (г^ — Qe), где г^ = 0,742 А — межъядерное расстояние,
a Q0 = 0,330 А* — величина, которую Харрик и Рамзей определили как «квадрупольный момент электрон-
ного распределения». Этот результат хорошо согласуется с теоретическим значением q = 0,248 • 10~1в см*,
полученным Джеймсом и Кулиджем[114]. Величина N(r), определенная Джеймсом и Кулиджем, равна поло-
вине нашей величины q.
3. Взаимодействие дипольных молекул с неполярными молекулами, не
обладающими квадрупольными моментами. Первоначально Андерсон [106]
предположил, что уширение вследствие давления спектра полярной молекулы
а происходит в результате столкновений с неполярной молекулой ft, не имею-
щей квадрупольного момента, и обязано наведенному диполь-дипольному взаи-
модействию. В этом случае потенциал возмущения определяется формулой
E.3)
^ G.20)
Член cos2 в приводит к появлению небольшой вероятности перехода, значение
которой было рассчитано Андерсоном с помощью методов теории групп, раз-
витых Рака [НО]. Однако рассчитанная ширина линий оказалась слишком
малой. В связи с этим Андерсон высказал предположение, не является ли
квадрупольный момент полярной молекулы Qb тем источником, который при-
водит к уширению вследствие давления. В этом случае следующий член в у
§ 8. Определение квадрупольного момента молекулы воды 787
определяется формулой E.3)
G.21)
Эта часть взаимодействия приводит к значительно большему коэффициенту
перехода, чем выражение G.20).
Для линии аммиака / = 3, к = 3
. G.22)
Квадрупольный момент аммиака равен q = 0,2759 А2. Результаты расчетов
уширения вследствие давления для линии NH3 / = 3, к — 3 приведены в
табл. 135. Расхождение данных в случае Аг еще не объяснено. Наличие квад-
рупольного момента в случае О2 является причиной расхождения экспери-
ментальных и теоретических значений.
Таблица 135
Величина сРВ9 определенная из уширения вследствие
давления линии NH3;
j = 3, h = 3
Налетающие
молекулы
Не
Аг
эксперимен-
тальные
значения
[уравнение
G.9)]
2,71
2,00
3,73
3,86
теоретические
значения
[уравнение
G.22)]
2,57
2,15
3,42
3,35
4. Квадруполь-квадрупольное взаимодействие. Мицушима [107] изучил
квадруполь-квадрупольное взаимодействие и успешно рассчитал самоуши-
рение спектра О2, учитывающее температурную зависимость.
§ 8. ОПРЕДЕЛЕНИЕ КВАДРУПОЛЬНОГО МОМЕНТА
МОЛЕКУЛЫ ВОДЫ
В § 4 было показано, что межмолекулярные силы для асимметричных
молекул сильно-зависят от положения электрических зарядов внутри молеку-
лы. Для изучения взаимодействия двух молекул воды необходима подробная
модель, в которой была бы строго определена пространственная локализаций
каждого из шести различных электрических зарядов. Такие модели опреде-
ляются независимо теоретическими и эмпирическими путями. Получаемые
таким образом модели в основном согласуются друг с другом. Используя эти
модели, можно довольно точно объяснить физические свойства льда, воды
и пара.
Для объяснения некоторых физических свойств газовой фазы оказывается
достаточным описывать электрические свойства молекул, представляя их в
виде идеальных диполей и квадруполей. В этом параграфе вычислены квадру-
польные моменты воды. Эти результаты являются добавлением к квадруполь-
ным моментам, вычисленным в предыдущем параграфе на основе анализа
микроволновых спектров.
788
Гл. 13. Теория межмолекулярных сил
А. ТЕОРЕТИЧЕСКОЕ ОПРЕДЕЛЕНИЕ1)
В первых теоретических расчетах молекулы Н2О предполагалось, что
электроны в атоме кислорода расположены на орбитах (IsJBsJBpzJBpx)Bpy).
В соответствии с этой картиной электроны 2рх и 2ру могут образовать связь
с двумя атомами водорода. Попл [115] нашел, что точнее считать орбиты
2s и 2р «гибридизированными»2), чтобы получить тетраэдрическую симметрию,
как в случае одинарных валентных орбиталей углерода. Две из этих орби-
талей имеют по одному электрону, которые могут образовать связь с ато-
мами водорода. Чтобы получить правильную величину дипольного момента
(^= 1,84-Ю8 эл.-ст. ед.) и правильную величину угла Н-О-Н A05°),
необходимо предположить, что угол между двумя разрыхляющими орбита-
лями равен 120,2°.
Фиг. 220. Теоретическая модель молекулы воды, предложенная Дунканом и Поплом [116].
Электроны на разрыхляющих орбиталях известны под названием «неподе
ленной пары электронов», поскольку их электронные функции распределения
имеют наибольшие значения вблизи атома кислорода, вдали от атома водорода.
Эти неподеленные пары ответственны за вурцитную структуру льда. На
фиг. 220 показано расположение ядра кислорода и двух внутренних электронов,
двух протонов Н и Н', центров электронных функций распределения сие'
для пар связывающих электронов и центров электронных функций распре-
деления а и а' для неподеленных пар. Координаты этих точек приведены в
табл. 136 [116]. Дипольный момент воды направлен вдоль оси z. В табл. 137
[116] приведены значения различных вкладов в z-компоненту дипольного
момента (относительно системы координат с началом в ядре кислорода). Эта
таблица содержит также значения 0ХХ, Оуу и О^ (относительно той же системы
координат), связанные выражением A.15) гл. 12 с квадрупольными моментами.
Дункан и Попл рассчитали эти величины, определяя как среднеарифметиче-
ские, так и среднеквадратичные значения координат, соответствующих различ-
х) См. [116]. Дункан и Попл [22] обобщили теоретическую трактовку Н2О и проделали
аналогичные расчеты для NH3 и HF.
2) Обсуждение «гибридизации» дано, например, в монографиях [67, 117].
§ 8. Определение квадрутыльного момента молекулы воды
789
Таблица 136
Координаты зарядов для теоретической модели Дункана и Попла молекулы Н2О
(Все расстояния даны в А)
Частицы
Неподеленные пары электронов ...
Ядро кислорода и два Is электрона
Связывающие электроны
Протоны
Символы*
а
а1
О
сг
с
Н'
Н
Заряд
-2е
-2е
бе
-2е
-2е
е
е
г
-0,158
-0,158
0,0
0,355
0,355
0,586
0,586
У
0,0
0,0
0,0
0,463
-0,463
0,764
—0,764
0,275
-0,275
0,0
0,0
0,0
0,0
0,0
* Символы пояснены на фиг. 220.
ным компонентам электронной функции распределения. Тот факт, что эти сред-
ние совершенно различны, затрудняет достаточно точное объяснение электри-
ческих свойств молекул в рамках модели точечных зарядов. Из табл. 137 видно,
что неподеленная пара электронов играет важную роль в определении электри-
ческих моментов молекулы.
Таблица 137
Теоретические вклады в дипольный и квадрупольный моменты молекулы Н2О
Частицы
1(Уиэл.-ст.ед.) A0-«вэл.-ст.ед.)к\Ог"эл.-ст.ед.) A0-м эл7-ст.ед.)
Неподеленные пары электронов
Связывающие электроны
Протоны
Полный момент относительно системы
координат с центром в атоме кисло-
рода
Полный момент относительно системы
координат с началом в центре масс
+3,03
-6,82
+5,63
+ 1,84
+ 1,84
-3,463
-3,084
0,000
-6,547
-6,547
-1,906
-9,080
+5,506
-5,480
-5,480
-2,422
-6,697
+3,318
-5,801
-5,951
Во многих случаях желательно определить квадрупольные моменты в
системе координат с началом в центре масс. Перемена начала координат не
сказывается на величинах Охх и 0ууу но слегка меняет &zz. Из уравнения A.16)
гл. 12 следует, что квадрупольные моменты относительно центра масс имеют
следующие значения :
Qxx = — 1,663 • 10-2в эл. - ст. ед.,
Qyy = + 1,538 • Ю-26 эл. - ст. ед.у
Qzz = + 0,125- 10-2вэл. -ал. ед.
(8.1)
Путем рассмотрения, аналогичного анализу молекулы воды, Дункан и
Попл [116] показали, что в случае аммиака вклад моментов относительно
системы координат с центром в атоме азота (ось z совпадает с направлением
дипольного момента) равен значениям, приведенным в табл. 138.
790
Гл. 13. Теория межмолекулярных сил
Таблица 138
Теоретические вклады в дипольный и квадрупольный моменты молекулы NH8
Частицы
A0—"эл.-ст. ед.)
вы - вуу
A0-" эл.-ст. ед.)
A0-««эл.-ст. ед.)
Неподеленные пары
Связывающие электроны
Протоны
Полный момент относительно системы
координат с центром в атоме азота
+3,67
-7,68
+5,48
+ 1,46
-1,335
-13,851
+6,361
-8,825
-2,631
-8,411
+2,080
-8,962
Б. ЭМПИРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ1)
Квадрупольный момент воды определен эмпирически Раулинсоном, кото-
рый сравнил экспериментальные значения второго вириального коэффициента
паров воды с теоретическими величинами, рассчитанными с помощью потен-
циала межмолекулярного взаимодействия воды
(8.2)
где gFv 02, ф) и h@v 02, ф) — функции трех углов, описывающих взаимную
ориентацию двух молекул воды. Явный вид функций определяется формулой
C.32) гл. 1 и A0.19) гл. 3 соответственно2). Параметры t* и и* связаны простыми
соотношениями с дипольным и квадрупольным моментами молекул
= 7^"^, U* =
еет*
(8.3)
Раулинсон предположил, что молекулы паров воды быстро вращаются вокруг
своей оси симметрии (оси z), так что в среднем молекулы обладают цилиндри-
ческой симметрией и Q = Qz2. После вычисления второго вириального коэффи-
циента с помощью этого потенциала и сравнения полученных результатов с
экспериментальными значениями для воды было найдено, что параметры потен-
циала равны следующим значения :
= 2,725 А, е = 707 кал/моль, t*=l,2, и* = 0,654.
(8.4)
Раулинсон предположил, что молекулу Н2О можно апроксимировать
четырехзарядной моделью, изображенной на фиг. 221. В таком случае доста-
точно дипольного и квадрупольного моментов для определения координат
зарядов. Эти сведения приведены в табл. 139 [ 117 ]3). С помощью этой таблицы
получены следующие значения для компонент тензора квадрупольного момента
х) См. [117—119]. При обсуждении расчетов Раулинсона [117] о влиянии эффектов
диполь-квадрупольного взаимодействия на второй вириальный коэффициент см. также
гл. 3, § 10.
2) Фактически для описания взаимной ориентации двух молекул воды необходимы лишь
пять углов. Приведенная потенциальная функция описывает взаимодействие двух цилинд-
рически симметричных зарядов, изображенных на фиг. 37 и получаемых путем усредне-
ния распределения заряда в молекулах воды, вращающихся вокруг своих диполь-
ных осей.
8) Имеется также частное сообщение Раулинсона A951 г.).
§ 8. Определение квадрупольного момента молекулы воды
791
в электростатических единицах (относительно системы координат с началом
в атоме кислорода):
Qxx = — 3,30 • Ю-26 эл.-ст. ед.,
Qyy = + 2,85 • Ю-26 ЭЛ.-СП1. ед.,
Qzz = + 0,45 • Ю-26 эл.-ст. ед.
(8.5)
Между значением квадрупольного момента, полученным Раулинсоном [117] и
Поплом [116], имеется существенная разница. Значения Раулинсона, по всей
вероятности, ненадежны, поскольку
модель точечных зарядов недооце-
нивает вклад внешних областей
молекул в квадрупольный момент;
кроме того, при изучении паров
Раулинсон идеализировал квадру-
польные силы. Тем не менее эмпири-
ческое согласие получено хорошее.
Такое распределение зарядов
приводит к теплоте сублимации,
равной 11,10 шал/моль при 0°К,
что согласуется с эксперименталь-
ным значением 11,32. Предсказан-
ное значение расстояния между
ближайшими соседями 2,73 А
также хорошо согласуется с экспе-
риментальной величиной 2,72 А.
Другая интересная особенность
заключается в том, что эта модель
приводит к энергиям, более предпо-
чтительным для Еурцитной струк-
туры, нежели для решетки типа
алмаза, и допускает случайные ориентации при комнатной температуре. Эти
результаты хорошо согласуются с экспериментальными данными. Однако
Фиг. 221. Эмпирическая модель молекулы
воды, предложенная Раулинсоном [117].
Таблица 139
Координаты
Частицы
Связывающие
электроны ...
Протоны
зарядов в эмпирической модели Раулинсона
молекулы Н2О
Символы*
а
а'
Н'
н
Заряд
-0,32 е
-0,32 е
0,32 г
0,32 е
Z
0,0
0,0
0,584
0,584
У
0,0
0,0
0,762
-0,762
0,51
-0,51
0,0
0,0
* Символы пояснены на фиг. 221.
Кэмпбел [120] исследовал различные предложенные модели Н2О с точеч-
ными зарядами и показал, что в случае льда мультипольные моменты выс-
ших порядков, включая и пятый, играют важную роль. Кроме того, энергия
взаимодействия зависит от предполагаемой ориентации не только соседних,
но и более удаленных молекул.
792 Гл. 13. Теория межмолекулярных сил
§9. ОПРЕДЕЛЕНИЕ МЕЖМОЛЕКУЛЯРНЫХ СИЛ ИЗ СВОЙСТВ
КРИСТАЛЛОВ ПРИ АБСОЛЮТНОМ НУЛЕ
Существуют три характеристики кристалла1), которые можно использовать
для определения межмолекулярных сил : энтальпия сублимации при 0°К
на молекулу й<су6л-> ; расстояние между ближайшими соседями при 0°К d0;
дебаевская характеристическая температура 6D. Дополнительно к этим
характеристикам иногда тип кристаллической решетки позволяет полу-
чить довольно чувствительный критерий для формы функции потенциальной
энергии.
Полная энергия кристаллической решетки при абсолютном нуле Uo равна
сумме потенциальной энергии решетки Фо и нулевой энергии Ко, связанной с
нулевыми колебаниями молекул решетки
Ко = Nhgy°»-K (9.1)
Нулевая энергия определяется с помощью дебаевской характеристической
температуры по формуле (см., например, [124, гл. 11])
K0 = -^-NkeD. (9.2)
При расчетах полной потенциальной энергии решетки обычно предполагается,
что межмолекулярные силы аддитивны2); это значит, что можно написать
0O = ±NE(O; d0), (9.3)
где N — число молекул решетки : E(O;do)—энергия взаимодействия одной
молекулы со всеми соседними в предположении, что расстояние между бли-
жайшими соседями равно d0.
Расстояние между ближайшими соседями в кристалле при абсолютном
нуле отличается от расстояния Гмин., при котором межмолекулярная потен-
циальная функция q>(r) имеет минимум вследствие нулевой энергии и притя-
жения молекул, расположенных позади ближайших соседей. Эффект нулевой
энергии сказывается на расширении решетки [125] (для аргона это расшире-
ние равно приблизительно 0,05 А на d0J а для неона — около 0,15 А). Притя-
жение ближайших соседей стремится сократить размеры решетки [126]. Для
гранецентрированных кубических кристаллов, взаимодействие молекул кото-
рых описывается потенциалом F-12) Леннарда-Джонса, линейные размеры
решетки вследствие этого эффекта уменьшаются приблизительно на 3%.
Для неона (rf0 — гмин.) = 3,20 — 3,16 = 0,04 А, а для аргона (d0—Гмин.) =
= 3,81—3,87 = —0,06 А. Наиболее полное и общее теоретическое рассмотре-
ние было проведено для потенциальной функции Леннарда-Джонса. Однако
много частных расчетов проделано для потенциала Букингема и Букингема—
Корнера, а также для других межмолекулярных потенциальных функций
(см. гл. 3, § 7 и 9).
*) Эти характеристики кристалла, так же как и линия плавления твердых тел, были
изучены де Буром с сотрудниками с точки зрения квантовомеханического закона соответ-
ственных состояний [121—123].
2) Очевидно, что силы внутри кристалла не совсем аддитивны (см. гл. 3, § 4) вслед-
ствие важности интегралов перекрывания и обмена. Отсутствие аддитивности приводит к
отклонению от условий Коши, связывающих коэффициенты упругости кристалла.
§ Р. Определение межмолекулярных сил из свойств кристаллов при абсолютном нуле 793
А. ПОТЕНЦИАЛЬНАЯ ЭНЕРГИЯ КРИСТАЛЛИЧЕСКОЙ РЕШЕТКИ
ПРИ АБСОЛЮТНОМ НУЛЕ
Для дальнейшего удобно записать величину ?@ ; d0) [определенную фор-
мулой D.20) гл. 4] в виде
?@; do) = 2ni<P(ri), (9.4)
где Г/ — расстояние от избранной молекулы до точек /-го рода решетки, а
п{ — число молекул, принадлежащих этим точкам решетки. В табл. 140 при-
ведены величины г, и щ для пяти типов кристаллических решеток, а также
соотношения между d0 (расстоянием между ближайшими соседями) и v0 (удель-
ным объемом кристалла). С помощью табл. 140 можно определить ?@; d0)
для любой частной потенциальной функции межмолекулярного взаимодей-
ствия. Например, для потенциальной функции отталкивающего взаимодей-
ствия, изменяющейся обратно пропорционально s-й степени расстояния
[ (r) = ft/r5], получаем
do) = -^L. (9.5)
Значения постоянных Cs приведены в табл. 141 для четырех типов кристаллов.
Для потенциальной функции (t, s) Леннарда-Джонса [q>(r) = br~s — cr~' ]
потенциальную энергию кристалла при абсолютном нуле можно выразить
в виде
E(O;do) = ^— -§-, (9.6)
"о о
где постоянные Cs и Ct те же, что и в случае отталкивающего потенциала,
рассмотренного раньше. Их значения приведены в табл. 141. Атомы инертного
газа при кристаллизации образуют гранецентрированную кубическую решетку.
Кихара и Коба [127 ], однако, указали, что при t = 6 и любом значении s опре-
деляемое формулой (9.4) ?@) имеет в случае гексагональной плотно упако-
ванной структуры более низкое значение, чем в случае гранецентрированной
кубической структуры. Этот результат можно согласовать с предыдущим
утверждением, учитывая тот факт, что истинный межмолекулярный потен-
циал отличается от потенциала Леннарда-Джонса, и, кроме того, принимая
во внимание эффекты нулевой энергии.
Б. НУЛЕВАЯ ЭНЕРГИЯ КРИСТАЛЛИЧЕСКОЙ РЕШЕТКИ
ПРИ АБСОЛЮТНОМ НУЛЕ
Дебаевскую характеристическую температуру можно выразить через
увеличение потенциальной энергии кристалла, вызванное смещением какой-
либо одной молекулы на расстояние г от своего равновесного значения
[см. уравнение D.20) гл. 4]. Однако для расчета дебаевской температуры нет
необходимости знать увеличение потенциальной энергии кристалла для каждой
ориентации смещения, достаточно лишь знать величину, усредненную по всем
ориентациям смещения1)
я
Е (г; d0) - Е @; d0) = 2 щ 1J [9 (J^ + г* - 2 г г, cos 0) - <р (г,)] sin в dO. (9.7)z>
О
Если ограничиться взаимодействием ближайших соседей, то (9.7) переходит
в формулу F.2) гл. 4. При очень низких температурах смещения г малы по
сравнению с расстоянием между узлами решетки, поэтому межмолекуляр-
ный потенциал можно разложить в ряд по степеням г в окрестности точки г,-.
г) См. работу Раиса [130]; формулы (9.8) и (9.9) являются обобщениями соответствую-
щих формул Раиса.
а) Формула (9.7) переходит в формулу G.3) гл. 4 в случае гранецентрированной куби-
ческой решетки с потенциалом F-12) Леннарда-Джонса и при условии, что учитывается
только взаимодействие между ближайшими соседями.
Таблица 140
Расстояние между ближайшими молекулами в кристалле [127]
Гранецентрированный кубический (кубическая плотная упаковка) кристалл
1
2
3
4
5
б
7
8
9
10
11
12
13
15
16
щ
12
6
24
12
24
8
48
6
36
24
24
24
72
48
12
do
r%ildl
17
18
19
20
21
22
23
24
25
26
27
28
29
31
32
- 2V. rV.
щ
48
30
72
24
48
24
48
8
84
24
96
48
24
96
6
= 1,1224,/,
33
34
35
36
37
38
39
40
41
42
43
44
45
47
48
ni
96
48
48
36
120
24
48
24
48
48
120
24
120
96
24
11*1
49
50
51
52
53
54
55
57
58
59
60
61
63
64
65
108
30
48
72
72
32
144
96
72
72
48
120
144
12
48
б) Гексагональный плотно упакованный кристалл [127]
rfe = 21/. JU «= 1,1224 vj/з
i
2
2!
з3
4
5
5?
6
%
в?
7
71
'8
8j
9
%'
10
ю|
10.1
и
И;
И*
12
121
щ
12
6
2
18
12
6
12
12
6
6
12
24
6
12
12
24
12
12
2
12
6
24
6
12
r%ildl
13
13?
14?
14?
15
15?
15?
16
16?
17
17?
18
18а1
183-
19
19?
20|
21
213?
22
22?
22|
23
238-
24
12
6
24
12
12
24
6
12
24
24
18
12
12
24
12
12
36
24
12
18
12
24
12
у*:
238-
24
25
26
26^
26?
27
27|
271
28
2831
29
298а
Ч
30*-
31
31а1
31?
328-
33
зз8?
34
35
Збз*
48
2
36
24
12
12
42
6
12
24
12
12
36
12
24
72
12
24
12
48
24
24
24
12
36
36?
37
37?
38
38i
38:-
39
393-
39?
40
408-
41
41?
42
42?
42:-
43
43?
4з:-
45
453а
461
18
24
72
12
24
12
36
36
6
24
12
12
24
48
36
12
2
72
12
12
60
36
6
24
Продолжение табл. 140
47
47i
47|
48
48j
49
49?
50
50*
Щ
48
12
24
6
12
60
48
30
24
r%*ldl
«*
51
511
52
52,'
53
53|
54
54|
12
36
48
36
12
36
24
18
6
ч?
55
55J
™\
568-
57
58
58J
58?
щ
36
48
18
24
24
48
36
24
24
59
593-
591-
60
61
615
36
6
60
12
24
72
24
в) Объемноцентрированная кубическая решетка [128]
d0 = 2~V» 3V2 „V. = 1,0911 rV«
1
la1
2*
31
п,
8
6
12
24
4
5j
6j
6*
-
8
6
24
24
8
9
IDS
24
32
12
48
12
30
г) Простая кубическая решетка [128 ]
d, = rV.
1
2
3
4
6
12
8
6
5
6
8
9
24
24
12
30
10
11
12
13
24
24
8
24
14
48
д) Структура алмаза [128]
de = 2~V» 3V1 vxl* - 0,8660 i V»
1
28?
4
12
12
6
бз1
8
9
12
24
16
12
и?
13s1
145
16
24
24
12
8
17
24
796
Гл. 13. Теория межмолекулярных сил
Таблица 141
Постоянные Cs в потенциальной энергии кристалла
S
4
5
6
7
8
9
10
И
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
Гранецентри-
рованная
(плотно упа-
кованная)
кубическая
[127, 129]
25,33830
16,9675
14,45392
13,35939
12,80194
12,49255
12,31125
12,2009
12,13188
12,08772
12,05899
12,04002
12,02736
12,0198
12,01300
12,009353
12,006280
12,004496
12,00306
12,00218
12,001511
12,001075
12,000748
12,000531
12,00037
12,000263
12,000185
Гексагональ-
ная плотно
упакованная
[127, 129]
14,45489
13,36035
12,80282
12,49332
12,31190
12,13229
12,05923
12,02748
12,01306
Объемно-
центри-
рованная
кубическая
[129]
22,63872
14,7585
12,2533
11,05424
10,355
9,8945
9,564
9,31326
9,11418
8,95180
8,8167
8,70298
8,60625
8,52363
8,45250
8,39138
8,33860
8,29306
8,253675
8,219626
8,19015
8,16465
8,1425
8,123469
8,106921
8,092593
8,080186
Простая
кубическая
[124]
16,5323
10,3775
8,40192
7,4670
6,94580
6,6288
6,4261
6,29229
6,2021
6,140
6,09818
6,06876
6,04826
6,0339
6,02388
6,01682
6,011863
6,008369
6,00590
6,004170
6,002945
6,00208
6,001470
6,001040
6,000734
6,000519
6,000367
В этом случае интегралы в формуле (9.7) можно вычислить явно и результат
принимает вид
E(r; do)-?(O; d0) = 2 Щ [±- fa)'
(9-8)
Для малых колебаний членами, содержащими г в степени выше второй, можно
пренебречь, и квадрат основной частоты v(d0) молекулы массы та) движущейся
в такой трехмерной потенциальной яме, становится равным
Величины v2, определенные равенством (9.9), приближенно являются
средним значением квадратов частот в кристалле [130]. Если взять дебаев-
скую функцию распределения частот, то максимум, или (дебаевская) частота,
будет равен (б/з)*М^о)> а дебаевская характеристическая температура равна
5У/..*Ш (9Л0)
В этом случае нулевая энергия кристалла при абсолютном нуле Ко опреде-
ляется по формуле (9.2).
§ 9. Определение межмолекулярных сил из свойств кристаллов при абсолютном нуле 797
Райе [130] произвел тщательное изучение температурной зависимости
внутренней энергии кристаллов благородных газов вблизи абсолютного нуля.
При более высоких температурах (например, вблизи 73°К для аргона) в перио-
дической структуре кристаллов появляются нарушения, которые приводят
к важным отклонениям в функции распределения частот. Однако при достаточно
низких температурах предположение о выполнимости дебаевской функции
распределения является удовлетворительным.
Равновесное расстояние при абсолютном нуле можно определить теоре-
тически, минимизируя сумму потенциальной и нулевой энергий
(9.11)
d(d0)
<*№)
[4- NE @; ^) + -|- Nk0o (d0)] = 0.
Таким образом, при любой форме потенциальной функции парного взаимо-
действия можно вычислить все свойства кристаллов при абсолютном нуле.
Или, наоборот, экспериментальные данные можно использовать для опреде-
ления потенциальной функции парного взаимодействия.
В. ОПРЕДЕЛЕНИЕ/СИЛ ВЗАИМОДЕЙСТВИЯ МЕЖДУ АТОМАМИ
БЛАГОРОДНОГО ГАЗА
Применяя методы, изложенные выше, Райе [130] определил энергию
взаимодействия двух атомов аргона, даваемую формулой (9.8) гл. 3. Аналогич-
ным образом Корнер [125] определил потенциал Букингема—Корнера для
неона и аргона [см. табл.. 11 (стр. 156)], и недавно Мэзон и Райе приспособили
модифицированный потенциал Букингема к Ne, Ar, Кг и Хе (см. гл. 3, § 7).
Канн [132] с помощью экспериментальных данных о свойствах кристалла
при низких температурах определил постоянные в потенциале Букингема
?>(r) = fcexp(~(zr)--^-^- (9.12)
для аргона, криптона и ксенона. Его работа может служить иллюстрацией
такого рода расчетов. Он проверил для а две величины : аг = 4,782 и а2 = 2,899.
Блейк и Майер теоретически нашли (см. гл. 14, § 2), что величина аг справед-
лива для взаимодействия двух атомов неона, Борн и Майер [133] нашли, что
а2 пригодно для описания взаимодействия галогенидов- щелочных металлов.
Для неона, аргона и ксенона Канн принял с' равным нулю; для криптона он
взял величину с', определенную теоретически Букингемом [134]. Значения
b и с были определены из условия совпадения рассчитанных величин с экспе-
Таблща 142
Определение потенциальной функции межмолекулярного взаимодействия с помощью
экспериментальных данных о твердом состоянии
Неон
Аргон
Криптон
Ксенон
Экспериментальные значения*
do, A
3,20
3,83
3,95
4,34
(субл.)
кал( моль
448
1850
2678
3778
64
80
63
55
Расчетные значения
а, А-1
4,782
2,899
4,782
2,899
4,782
2,899
2,899
4,782
2,899
ь, ю-1»,
эрг
10 250
97
830000
1600
2180000
3135
3550
18400000
11730
с , 10-»,
эрг-А*
8,07
14,0
87,4
125
148
209
190
354
477
с', 10-",
эрг.А»
0
0
0
0
0
0
587
0
0
Od, °K
60
53
105
81
91
68
70
87
65
• Здесь Ло у л*'— экспериментальные значения теплоты сублимации при 0°К, не исправленные на
нулевую энергию.
798 Гл. 13. Теория межмолекулярных сил
риментальными данными для теплоты сублимации и для расстояния между
соседями при абсолютном нуле. Выбор величины ах или а2 в этом случае опре-
делялся сравнением рассчитанных частот с наблюдаемыми дебаевскими часто-
тами. Результаты расчетов Канна приведены в табл. 142 [132]. По всей види-
мости, расчеты Корнера и Мэзона и Раиса более точны.
До настоящего времени экспериментальные данные о свойствах кристалла
при.низких температурах применялись лишь для определения потенциалов
межмолекулярного взаимодействия сферически симметричных молекул. Воз-
можно, что этот метод окажется очень полезным в случае сложных несиммет-
ричных молекул.
Приложение А
ПОЛНЫЙ ГАМИЛЬТОНИАН СИСТЕМЫ ВО ВНЕШНЕМ
ЭЛЕКТРИЧЕСКОМ И МАГНИТНОМ ПОЛЯХ1)
Обычно при рассмотрении энергии молекулярной системы (с фиксирован-
ными ядрами) предполагается, что гамильтониан электронной системы равен
Jte. Однако полный гамильтониан должен содержать, кроме <3fe, еще дополни-
тельные члены, учитывающие магнитные взаимодействия и релятивистские
эффекты. Эти поправочные члены могут оказаться важными при определении :
1) вероятности перехода из одного квантового состояния в другое в случае
быстрых столкновений, представляющих интерес в химии «горячих атомов»
или в экспериментах с молекулярными пучками ; 2) потенциальной энергии
в случае столкновений тяжелых частиц, таких, как J2 с малыми скоростями,
в результате которых может разрываться связь между спиновым и орбитальным
моментами молекул ; 3) межмолекулярных сил между свободными радика-
лами, электронно-возбужденными молекулами или очень длинными молеку-
лами, представляющими интерес в биологии. Пусть fft — напряженность
внешнего электрического поля, }?{ — напряженность внешнего магнитного
поля, а к>Е[ — вектор-потенциал (внешнего магнитного поля), все относя-
щиеся к /-му электрону. Кроме того, пусть операторы спина и импульса z-ro
электрона будут соответственно st и р, = (А/*) (З/Эг) + (е/с) xj€(. В таком
случае гамильтониан с учетом членов, пропорциональных A/сJ, где с — ско-
рость света, может быть записан в виде2)
(A.I)
где <3fe определяется формулой A.4), a
- Д4" [(r, - tj) x p,] • s,. + 2^ ([(r, - i» x pj] • s,)}, (A.2)
2 jr [rfcfor- sj) - 3 (s, • (r,- - r,)) (sr (r,,- r,)) ] -
2
- ~^22(srSj)S(ri - tj), (A.3)
Ц (A.4)
x) Авторы благодарны Сесслеру, Ван Флеку, Клейнеру, Бете, Слетеру и Паули за по-
мощь в подготовке этого приложения. Сесслеру принадлежит идея введения второго члена
в Жг и обсуждение Жг. Приближенный гамильтониан [уравнение (А.8)] и обсуждение
3?ь принадлежат Ван Флеку. Клейнер исправил ошибку, допущенную ранее авторами
при вычислении &х.
2) См. [135]. Несколько другое обсуждение гамильтониана дано в работах [136—141].
Приложение Б 799
= - tS^t 2 [^ (Р, -9j> + -fc ((r, - tj) • р,) ((г,- - г,) • р,)}, (А.5)
(A.6)
«,)• (А.7)
2
pf, (A.6)
Здесь различные члены имеют следующий смысл:
'3?! состоит из двух частей : первый и наибольший член описывает спин-
орбитальное взаимодействие спина электрона с магнитным моментом, обуслов-
ленным движением электрона. Оставшийся член в <ЖХ дает взаимодействие
«спин-другая орбита», являющееся взаимодействием спина одного электрона
с магнитным моментом, обусловленным движением другого электрона.
<Жг описывает спин-спиновое магнитное взаимодействие, которое часто
очень значительно1). Второй член в <Жг с дельта-функцией Дирака <5(г, — г7)
должен быть введен, чтобы принять во внимание тот факт, что первый член в
Жг плохо определен для неперекрывающихся электронных распределений,
поскольку радиальное среднее от 1/г?у равно бесконечности, тогда как много-
кратные угловые интегралы дают нуль. Поэтому первый член в <_^*2 необходимо
вычислять вне области малой сферы вокруг каждого электрона или, что экви-
валентно, полагая все промежуточные величины равными нулю. Это соотно-
шение для ,J^% приводит к формуле Ферми для сверхтонкой структуры S-состоя-
ния, если его применять к электрону и протону.
<Ж.6 содержит поправочные члены, введенные Дираком для учета электрон-
ного спина. Эти члены присутствуют даже для одного электрона.
<Ж± описывает магнитное взаимодействие электронов типа орбита—орбита.
JT5 является релятивистской поправкой к кинетической энергии, обуслов-
ленной зависимостью массы электрона от скорости.
<J^% учитывает влияние внешнего электрического и магнитного полей.
Первые два члена являются релятивистскими поправками к эффектам электри-
ческого поля и ими обычно пренебрегают. Магнитный член важен лишь по-
стольку, поскольку он связан со сверхтонкой структурой S-уровней, для
которых электронные волновые функции велики вблизи ядра. В этом случае
электрическое поле у ядра играет роль «внешнего» поля.
На больших расстояниях, при которых не происходит перекрывания
электронных распределений, члены от <J^X до,Ж% можно заменить членом, опи-
сывающим обычное взаимодействие между двумя диполями. Это значит, что
если а,{ — оператор орбитального момента количества движения z-го электрона,
то полный гамильтониан можно записать в виде
Ш^х + */) • («у
- 3 [(«, + 2S/) • (гГ- г,)] [(aj + 2sj) • (г, - г,)]}. (А.8)
Прило жение Б
ОТНОШЕНИЕ КИНЕТИЧЕСКОЙ ЭНЕРГИИ ЭЛЕКТРОНОВ
К ПОТЕНЦИАЛЬНОЙ ЭНЕРГИИ В МОЛЕКУЛЯРНОЙ СИСТЕМЕ
Для улучшения приближенных молекулярных волновых функций можно
применить теорему вириала. Фок [143 ] показал, что если ввести масштабный
множитель в приближенную функцию электронного заряда и изменять его
так, чтобы получилось наименьшее значение энергии, то теорема вириала будет
автоматически выполняться в любой квантовомеханической системе, потен-
Второй член среден Ферми [142].
800 Гл. 13. Теория межмолекулярных сил
циал которой является однородной функцией координат. Фок придал этой
теореме форму, удобную для применения к атомным системам.
Эта теорема обобщена [144] на случай молекул или металлов, в которых
межъядерные расстояния могут рассматриваться как параметры. Пусть г" и rv
относятся соответственно к электронным и ядерным координатам, a ^(rV )
соответствующим образом силшетризованная приближенная волновая функция
либо основного состояния, либо наинизшего энергетического состояния. В этом
случае можно образовать новую приближенную волновую функцию у*(гП> г")>
вводя масштабный множитель s таким образом, чтобы электронные расстояния
увеличивались в s раз, тогда.как межатомные координаты оставались бы неиз-
менными. Таким путем, учитывая условия нормировки, получаем
% (Г", TV) = S<3"/2) Wi EГ*, Г) . (Б Л}
Из соображений размерности следует, что кинетическая энергия электронов
К^г"), соответствующая новой волновой функции, простым образом связана
с K(l\rv) кинетической энергии, соответствующей старой волновой функции
). (Б.2)
Аналогично потенциальная энергия электронов, соответствующая новой функ-
ции, связана с потенциальной энергией, соответствующей старой функции, со-
отношением
0P(rv) = s0p(stv). (Б.З)
Полная энергия электронов в молекуле является суммой кинетической и потен-
циальной энергий
Ф<?> (Г) = К<*> (Г) + Ф<*> (Г) = s2 К?> (sr) + s ФЫ (sr). (Б.4)
В случае равновесия
] 0
Наилучшее значение энергии получается при масштабном множителе, удов-
летворяющем уравнению
Комбинируя два последних уравнения, находим наилучшее значение s при
равновесном расстоянии
_ -УгФ^Eг>)
5 —w^r' (Б-7)
которому соответствует наилучшее или наименьшее значение полной энергии
- (Б-8)
Теорема вириала автоматически выполняется при этой волновой функции, так
как для этой величины s
Ф?} (rv) = - K<s> (Г) = 4 Ф?> (г). (Б.9)
Это значит, что если приближенную функцию электронного распределения
растягивать или сжимать, то размеры, соответствующие наинизшей энергии,
Приложение Б 801
приводят к правильному значению отношения потенциальной энергии к кине-
тической. При неравновесных расстояниях правильная величина отношения
потенциальной энергии к кинетической определяется уравнениями A.36) и
A.37). Для выполнения этого отношения необходимо, чтобы 5 было равно
~ [ФО> <**)]¦' dr J'
что соответствует энергии
(Б.11)
Обычно эта величина соответствует наинизшей энергии, наблюдаемой при
неравновесных расстояниях, и поэтому величина s должна быть выбрана путем
сравнения с непосредственными рассчитанными значениями энергии для не-
равновесных расстояний.
Задачи
1. Рассмотрим две частицы, взаимодействующие в соответствии с двумя потенциаль-
ными кривыми. Кривые «основного состояния» и «возбужденного состояния» имеют вид
<р0 = 100 [еJЩкал/моль,
9l = 50 [*-8<r-l,5)__ 2*-4<г-!,5)] + 50,5 ккал/моль;
здесь г — расстояние в А.
Рассчитать вероятность того, что при столкновении двух одинаковых частиц в нор-
мальном состоянии с относительной скоростью v(r) произойдет переход в возбужденное
состояние. Изобразить вероятность перехода как функцию v(r).
2* Рассчитать поляризуемость в направлениях z и х атома водорода с электроном,
находящимся на орбите 2pz,
rcosflexp(-r/2fl0)
3. Рассчитать расстояние для каждой из следующих пар молекул, энергия притяжения
которых равна 0,6 ккал/моль (кТ при комнатной температуре), предполагая, что наиболее
благоприятные ориентации следующие:
а) Аг + Аг,
б) Аг + НС1,
в) НС1 + НС1,
г) С + О (в основных состояниях),
д) Н (Is) + протон,
е) Н Bр) + протон.
4. Дано кубическое уравнение для энергии взаимодействия протона с атомом гелия
О + 9* {+ 0,0155023 + -Ив) + 9 C,19474. 10-* + ±
- + *-
1,150106» Ю-3 0,3524904
и три его корня с точностью до 1/г3
?! = 0,0130552 + ?L+...f
<р2 = 0,00024471-^- + -^- + ...,
36 , а3 ,
Найти коэффициенты аъ а2 и а3 У членов 1/г4.
5. Рассчитать квадруполь-квадрупольную энергию взаимодействия двух атомов С1,
находящихся в основных состояниях. Какая при этом возникает потенциальная кривая и как
она связана с основным состоянием молекулы С12?
802 Гл. 13. Теория межмолекулярных сил
ЛИТЕР АТУР А
1. Pelzer H., Wigner E. P., Zs. phys. Chem., B15, 445 A932).
2. Hirschfelder J. О., Wigner E. P., Journ. Chem. Phys., 7, 616 A939).
3. Eucken A., Osterr. Chem.-Ztg., 20, 1 A935).
4. H e г z f e 1 d K. F., Freie WeglSnge und Transporterscheinungen in Gasen, Hand- und
Jahrbuch der Chemischen Physik, 3, 97 Leipzig, 1939.
5. Lambert J. D., R owl ins on J. S., Proc. Roy. Soc, A204, 424 A950).
6. Zener C, Phys. Rev., 37, 556 A931).
7. Zener C, Proc. Cambr. Phil. Soc, 29, 136 A933).
8. Born M., Oppenheimer J. R., Ann. d. Phys., 84, 457 A927).
9. В a t e s D. R., F u n d a m i n s к у A., M a s s e у H. S. W., Leech J. W., Phil.
Trans. Roy. Soc, A, No. 860, 93 A950).
10. К e m b 1 e E. C, Fundamental Principles of Quantum Mechanics, New York, 1937.
if. Eyring H., Polanyi M., Zs. phys. Chem., B12, 279 A931).
12. Hirschfelder J. О., Диссертация, Princeton University, 1936.
13. Curtiss С F., Hirschfelder J. O., Adler F. Т., Journ. Chem. Phys., 18,
1638 A950).
14. H i r s с h f e 1 d e r J. O., Wigner E. P., Proc. Nat. Acad. Sci., 21, 113 A935).
15. Curtiss С F., Journ. Chem. Phys., 21, 1199 A953).
16. К a 11 m a n, London, Zs. phys. Chem., B2, 207 A929).
17. Altshuler S., Phys. Rev., 89, 1278 A953).
18. Rice O. K., Proc. Nati. Acad. Sci., 17, 34 A931).
19. Ландау Л. Д., Phys. Zs. d. Sowjetunion, 2, 46 A932).
20. Zener СМ., Proc. Roy. Soc, A137, 696 A933).
21. Zener С M., Proc. Roy. Soc, A140, 66Q A933).
22. E у г i n g H., W a 11 e г J., К i m b a 11 G. E., Quantum Chemistry, New York, 1944.
(см. перевод: Эйринг Г., Уолтер Д., Кимбалл Г., Квантовая химия, ИЛ,
1948).
23. М о 11 N. F., M a s s е у Н. S. W., Theory of Atomic Collisions, Oxford, 2nd Ed., 1949
(см. перевод: Мотт H., Месси Г., Теория атомных столкновений, ИЛ, 1951).
24. Phelps А. V., Molnar J. P., Phys. Rev., 89, 1202 A953).
25. Slater J. С, Journ. Chem. Phys., 1, 687 A933).
26. Born M., He i sen berg W., Jordan P., Zs. Phys., 35, 557 A925—1926).
27. H e 11 m a n n H., Quantenchemie, Leipzig, 1937.
28. Fey n man R. P., Phys. Rev., 56, 340 A939).
29. Berlin Т., Journ. Chem. Phys., 19, 208 A951).
30. G о m b k s P., Theorie und Lo'sungsmethoden des Mehrteilchenproblems der Wellen-
mechanik, Basel, 1950 (см. перевод: Г о м б а ш П., Проблема многих частиц в
квантовой механике, ИЛ, 1952, 1953).
31. Mott N. F., Sneddon I. N., Wave Mechanics and Its Applications, Oxford, 1948.
32. Slater J. C, Phys. Rev., 91, 528 A953).
33. Slater J. C, Phys. Rev., 81, 385 A951).
34. Electronic Structure of Atoms and Molecules, Solid State and Molecular Theory Group
of Massachusetts Institute of Technology, Technical Report No. 3, February 1953.
35. M e с k 1 e r A., Solid State and Molecular Theory Group of Massachusetts Institute of
Technology, Quarterly Progress Report, July 1952.
36. M e с k 1 e r A., Solid S'tate and Molecular Theory Group of Massachusetts Institute of
Technology, Quarterly Progress Report, October 1952.
37. Coulson С A., Maccoll A., Sutton L., Trans. Farad. Soc, 48, 106 A952).
38. H у 11 e r a a s E., Zs. Phys., 65, 209 A930).
39. H a s s ё Н. R., Proc. Cambr. Phil. Soc, 26, 542 A930).
40. Hasse H. R., Proc Cambr. Phil. Soc, 27, 66 A931).
41. Kirk wood J. G., Phys. Zs., 33, 57 A932).
42. H i r s с h f e 1 d e r J. O., Journ. Chem. Phys., 3, 555 A935).
43. Ishiguro E», Arai Т., Kotani M., Mizushima M., Journ. Phys. Soc,
A65, 178 A952).
Литература 8G3
44. James H. M., Coolidge A. S., Journ. Chem. Phys., 1, 825 A933).
45. V о 1 к m a n n H., Ann. d. Phys., 24, 457 A935).
46. D e n b i g h K. G., Trans. Farad. Soc, 36, 936 A940).
47. Pauling L., Sherman J., Zs. Krist., 81, 1 A932).
48. Pauling L., Proc. Roy. Soc, A114, 181 A927).
49. Slater J. C, Phys. Rev., 36, 57 A930).
50. Go mb as P., Gaspar R., Acta Phys. Sci. Hung., 3, 317 A952).
51. Van Vleck J. H., Electric and Magnetic Susceptibilities, Oxford, 1932.
52. Margenau H., Rev. Mod. Phys., 11, 1 A939).
53. L о n d о n F., Trans. Farad. Soc., 33, 222 A930).
54. London F., Zs. phys. Chem., Bll, 222 A930).
55. Eisenschitz R., London F., Zs. Phys., 60, 491 A930).
56. Pauling L., Beach J. Y., Phys. Rev., 47, 686 A935).
57. Mull ike n R. S., Journ. Chem. Phys., 7, 14, 20, 121, 339, 353, 364, 570 A939).
58. Mull ike n R. S., Journ. Chem. Phys., 8, 234, 382 A940).
59. Lenna r d-J о nes J. E., Interatomic Forces, Indian Association for Cultivation of
Science, Calcutta, 1939.
60. Slater J. С, К irk wood J. G., Phys. Rev., 37, 682 A931).
61. Margenau H., Journ. Chem. Phys., 6, 897 A938).
62. Pauling L., Wilson E. В., Jr., Introduction to Quantum Mechanics, New York,
1935.
63. Hornig J. F., Hirschfelder J. O., Journ. Chem. Phys., 20, 1812A952).
64. Ve г we у E. J. W., Over bee к J. Т. G., van Ness K., Theory of Stability of
Lyophobic Colloids, Amsterdam, 1948.
65. Casimir H. B. G., Polder D., Phys. Rev., 73, 360 A938).
66. London F., Journ. Phys. Chem., 46, 305 A942).
67. Da vies P. L., Coulson С A., Trans. Farad. Soc., 48, 777 A952).
68. d e Boer J., Physica, 9, 363 A942).
69. Coulson С A., Valence, Oxford, 1952.
70. Pitzer K. S., Quantum Chemistry, New York, 1953.
71. Mul liken R., Rieke C, Brown W., Journ. Am. Chem. Sci., 63, 41 A941).
72. Bartlett J. H., Phys. Rev., 37, 507 A931).
73. Da vies P. L., Trans. Farad. Soc, 48, 789 A952).
74. A n d e r s о n P. W., Phys. Rev., 80, 511 A950).
75. M a r g e n a u H., Philosophy of Science, 8, 603 A941).
76. La time г W. M., Rode bush W. H., Journ. Am. Chem. Soc, 42, 1419 A920).
77. U b b e 1 о h d e A. R., Journ. Chem. Phys., 46, 429 A949).
78. Da vies M., Annual Reports of the Chemical Societies for 1946,43, 5, London A947).
79. Alexander A. E., Lambert J. D., Trans. Farad. Soc, 37, 421 A941).
80. Lambert J. D., Roberts G. A. H., R о w 1 i n s о n J. S., Wilkinson V. J.,
Proc. Roy. Soc, A196, 113 A949).
81. Lambert J. D., Strong E. D. Т., Proc. Roy. Soc, A200, 566 A950).
82. Craven P. M., Lambert J. D., Proc Roy. Soc, A205, 439 A951).
83. Lambert J. D., S t a n i e s E. N., Woods S. D., Proc. Roy. Soc, A200, 262 A950).
84. King G. W., Van Vleck J. H., Phys. Rev., 55, 1165 A935).
85. London F., Zs. Phys., 63, 245 A930).
86. Dennison D. M., Rev. Mod. Phys., 3, 280 A931).
87. I n f e 1 d L., Hull Т. Е., Rev. Mod. Phys., 23, 32 A951).
88. Margenau H., Warren D. Т., Phys. Rev., 51, 748 A937).
89. Carroll K., Phys. Rev., 53, 310 A938).
90. Coulson С A., Proc Roy. Soc, Edinburgh, A61, 20 A941).
91. Coulson С A., Gil lam СМ., Proc Roy. Soc, Edinburgh, A62, 360 A948).
92. Krogdahl M. K., Astrophys. Journ., 100, 311 A944).
93. К г о g d a h 1 M. K., Astrophys. Journ., 100, 333 A944).
94. Landolt-Bornstein, Zahlenwerte und Funktionen, Berlin, 1951.
95. Da hie r J. S., Hirschf elder J. O., Journ. Chem. Phys., 25 A956).
96. Coulson С A., Proc Roy. Soc, 169A, 413 A939).
804 Гл. 13. Теория межмолекулярных сил
97. К n i p p J. К., Phys. Rev., 53, 734 A938).
98. Wigner E., Wit me r E., Zs. Phys., 51, 859 A928).
99. Mul liken R. S., Phys. Rev., 36, 1440 A930).
100. H u n d F., Zs. Phys., 63, 723 A930).
101. S p о n e r H., Molekulspektren, Berlin, 1936.
102. Margenau H., Watson W. W., Rev. Mod. Phys., 8, 22 A936).
103. G о r d у W., Smith W. V., T r a m b а г u 1 о R., Microwave Spectroscopy, New York,
1953 (см. перевод : Г о р д и В., С м и т В., Т р а м б а р у л о Р., Радиоспектроскопия,
М.—Л., 1955).
104. Weisskopf V., Wigner E., Zs. Phys., 63, 54 A930); 65, 18 A931).
105. Hoyt F., Phys. Rev., 36, 860 A931).
106. A n d e r s о n P. W., Phys. Rev., 76, 647 A949).
107. Mizushima M., Phys. Rev., 83, 94 A951).
108. Smith W. V., Howard R., Phys. Rev., 79, 132 A950).
109. F о 1 e у Н. M., Phys. Rev., 69, 616 A946).
110. Racah G., Phys. Rev., 62, 438 A942).
111. Anderson R. S., Smith W. V., Gordy W., Phys. Rev., 82, 264 A951),
112. Ha r rick J., Ramsey N. F., Phys. Rev., 88, 228 A952).
113. Ramsey N. F., Phys. Rev., 78, 221 A950).
114. James H. M., Coolidge A. S., Astrophys. Journ., 87, 447 A938).
115. Pople J. A., Proc. Roy. Soc, A202, 323 A950).
116. Duncan A., Pople J., Trans. Farad. Soc, 49, 217 A953).
117. Rowlinson J. S., Trans. Farad. Soc, 47, 120 A951).
118. Bjerrum N., Kgl. Danske Videnskab. Selskabs (Math.-Phys.-Medd., 27, 1 A951).
119. Bjerrum N., Science, 115, 385 A952).
120. Campbell E. S., Journ. Chem. Phys., 20, 1411 A952).
121. d e Boer J., Physica, 14, 139 A948).
122. d e Boer J., В 1 a i s s e B. S., Physica, 14, 149 A948).
123. Lunbeck R. J., Диссертация, Amsterdam, 1951.
124. Mayer J. E., Mayer M. G., Statistical Mechanics, New York, 1940 (см. перевод
M а й е р Дж., Гепперт-Майер М., Статистическая механика, ИЛ, 1952).
125. Corner J., Trans. Farad. Soc, 35, 711 A939); 44, 914 A948).
126. L e n n a r d - J о n e s J. E., Physica, 4, 941 A937).
127. Kihara Т., Koba S., Journ. Phys. Soc, Japan, 7, 348 A952).
128. Prins J., Peter sen H., Physica, 3, 147A936).
129. Lennard-Jones J. E., Ingham A. E., Proc. Roy. Soc, A107, 636 A952).
130. Rice О. К., Journ. Am. Chem. Soc, 63, 3 A941).
131. Buckingham R., Proc. Roy. Soc, A168, 264 A938).
132. Kane G., Journ. Chem. Phys., 7, 603 A939).
133. Born M., Mayer J., Zs. Phys., 75, 1 A932).
134. Buckingham R., Proc. Roy. Soc, A160, 94 A937).
135. В e t h e H. A., Handbuch der Physik, 24, 1, Berlin, 2 Verlag, 1933.
136. В г е i t G., Phys. Rev., 39, 616 A932).
137. Be the H. A., Fermi E., Zs. Phys., 77, 296 A932).
138. Condon E. U., S h о r 11 e у G. H., The Theory of Atomic Spectra, Cambridge, 1935
(см. перевод: Кондон Е., Шортли Г., Теория атомных спектров, ИЛ, 1949).
139. В г е i t G., М е у е г о 11 R. E., Phys. Rev., 72, 1023 A947); 75, 1447 A948).
140. В re it G., Brown G. E., Arfken G., Phys. Rev., 76, 1299 A949).
141. Allen L. H. Ufford С W., Van Vleck J. H. Astrophys. Journ., 109,42 A949).
142. Fermi E., Zs. Phys., 60, 320 A930).
143. Фок В., Zs. Phys., 63, 855 A930).
144. H i r s с h f e 1 d e г J. О., К i n с a i d J. F., Phys. Rev., 52, 658 A937).
ГЛАВА 14
КВАНТОВОМЕХАНИЧЕСКИЕ ВЫЧИСЛЕНИЯ
МЕЖМОЛЕКУЛЯРНЫХ СИЛ
Наши знания о межмолекулярных силах являются результатом сочетания
экспериментальных наблюдений и общих теоретических рассмотрений. Теория
приводит к общим функциональным выражениям, а эксперимент служит для
определения числовых постоянных. Наиболее важно то обстоятельство, что
теория позволяет делать качественные предсказания о межмолекулярных
силах, что, в свою очередь, позволяет непосредственно интерпретировать
экспериментальные данные и ставить новые эксперименты для выявления
характерных особенностей. В настоящее время качественная теория межмоле-
кулярного взаимодействия разработана только для простых молекул, причем
обычно предполагается, что молекулы находятся в основном состоянии. Для
определения сил между другими типами молекул, а также для выяснения
влияния этих сил на физические и химические свойства системы потребуется
еще очень много работы. В некоторых простейших случаях, как, например,
в случае взаимодействия между двумя атомами водорода или гелия, силы
межмолекулярного взаимодействия удается рассчитать строгим квантовомеха-
ническим путем. Однако вплоть до настоящего времени не существует строгих
квантовомеханических расчетов, которые бы с достаточной степенью точности
позволили выразить энергию межмолекулярного взаимодействия при всех
расстояниях—малых, промежуточных и больших. Поясним, что под боль-
шими расстояниями подразумеваются такие, на которых необходимо учиты-
вать лишь дальнодействующие силы, рассмотренные в гл. 13; под малыми
расстояниями подразумеваются такие, в которых основную роль играют хими-
ческие силы, и, наконец, под промежуточными расстояниями — такие, где
области короткодействующих и дальнодействующих сил переходят одна в
другую.
В гл. 13 дально действующие силы выражены через физические харак-
теристики изолированных молекул. Для объяснения дальнодействующих
взаимодействий достаточны первый и второй порядок теории возмущений,
поскольку возмущения действительно малы. Более того, нет необходимости
антисимметризовать волновые функции, так как все обменные интегралы и
интегралы перекрывания пренебрежимо малы. Эти упрощения перестают быть
справедливыми, если взаимодействующие молекулы находятся на малых или
промежуточных расстояниях друг от друга.
Вплоть до настоящего времени большинство расчетов межмолекулярных
энергий проделано для очень малых расстояний, представляющих интерес не
только с точки зрения кинетики химических реакций, но и с точки зрения
спектроскопии. Однако изучению межмолекулярного взаимодействия на про-
межуточных расстояниях, которым соответствуют равновесные расстояния
между соседними молекулами в кристалле, уделено очень мало внимания ;
энергия взаимодействия при таких промежуточных расстояниях определяет
физические свойства газа, жидкости и твердого состояния. Расчеты энергии
при промежуточных расстояниях обсуждаются на примере взаимодействия
двух атомов водорода (см. § 1). С одной стороны, расчеты этой энергии более
трудны, а с другой — легче по сравнению с определением энергии при мень-
ших расстояниях. Они более трудны в том смысле, что необходимо полнее
знать функциональный вид волновой функции, которая приводит к удовлет-
806 Гл. 14. Квантовомеханияеские расчеты межмолекулярных сил
верительным значениям дисперсионной энергии при больших расстояниях и
энергии химических связей при малых расстояниях. Но расчеты при проме-
жуточных расстояниях оказываются более легкими в том смысле, что много-
центровые кулоновские и обменные интегралы становятся численно менее
важными и их значение найти значительно легче.
Расчеты межмолекулярных сил при малых и промежуточных расстояниях
требуют применения вариационного принципа совместно с приближенными
волновыми функциями со многими экспериментально определяемыми парамет-
рами1). При этом необходима очень большая числовая точность, так как полная
энергия бимолекулярной системы, которая вычисляется непосредственно,
обычно значительно превосходит 100 эв, а для получения энергии взаимодей-
ствия, по порядку составляющей десятую или сотую долю 1 эв, необходимо
из полной энергии взаимодействия вычесть энергию изолированных молекул.
Только после появления быстродействующих вычислительных машин стало
возможно провести вычисление большого числа необходимых интегралов
(см. приложение А, стр. 842).
Последние успехи в технике квантовомеханического расчета молекул
[2—8]2) (см. в гл. 13, § 1 о методе обобщенного самосогласованного поля
Слетера) позволяют надеяться, что в течение нескольких лет удастся рассчи-
тать теоретические кривые потенциальной энергии с точностью, сравнимой с
экспериментальной.
Можно надеяться, что в конце концов межмолекулярные силы при малых
и промежуточных расстояниях удастся выразить через физические харак-
теристики изолированных молекул точно так же, как в настоящее время даль-
нодействующие силы выражаются посредством потенциалов ионизации, поля-
ризуемости и электрических моментов. Чтобы проверить справедливость любых
таких выражений, необходимо все еще сравнивать эти результаты с очень точ-
ными вариационными расчетами, проделанными для небольшого числа частных
случаев.
В этой главе современное состояние теории межмолекулярных сил рас-
сматривается на небольшом числе частных примеров, для которых существует
строгий теоретический расчет энергии взаимодействия3):
1) Н + Н, 5) Н + Н2,
2) Не + Не, 6) Н2 + Н2,
3) Ne + Ne, 7) Н или Н2 и различные поло жительные.
и отрицательные ионы водорода,
4) Аг + Аг, 8) Не + Н+ и Не+ + Не'.
Во всех этих расчетах функциональная форма приближенной волновой
функции ^прибл. выбиралась из условия компромисса между точной функцией
и функцией, допускающей интегрирование. Приближенная энергия системы
из двух молекул определялась из соотношения4)
Е_ II ^прибл. <%? ^прибл. dr"* drnb /n t v
прибл. — ——— • VV#1/
II ^прибл. ^прибл. йтпайтпь
К сожалению, во многих случаях при интегрировании допускаются различные
приближения, так что ошибка в ?прибл. возникает не только вследствие неточ-
г) См. работу Ван Флека и Шермана [1 ], которая до сих пор является лучшей по изло-
жению основных методов квантовой механики молекул.
а) Одним из наиболее интересных последних достижений является обобщение статисти-
ческой модели атома Томаса—Ферми, проведенное Амальди, Дираком, Иенсеном, Гомбашом,
Фейнманом, Геллером и другими, которые учитывали электронные спины, обменные силы и
корреляцию между движением различных электронов. См. [7, гл. 9]. Применение этого
метода к молекулам обсуждается в работе [8, гл. 7 ].
а) В этой главе штрих сверху указывает на возбужденное состояние.
4) Как и в гл. 13, па и щ означают число электронов в молекулах а несоответственно.
Гл. 14. Квантовомеханические расчеты межмолекулярных сил 807
ности приближенной волновой функции, не ив результате сделанных при
интегрировании приближений. Численное значение ?ПрИбл. минимизировано
путем варьирования по возможности наибольшего числа параметров. Если
Еа и Еь — соответственно полные энергии изолированных молекул а и b (элек-
трона и ядра полностью разделены), то энергия взаимодействая равна
ФаЬ == ^прибл. — Еа — Еь . @.2)
Для расчета всей потенциальной кривой, начиная от больших и кончая
малыми расстояниями, необходимо найти Еа и Еъ,' используя те же прибли-
женные волновые функции и те же приближения при интегрировании, что и
при расчетах системы из двух молекул. В противном случае, q>ab не будет стре-
миться к нулю при бесконечном разделении молекул. Даже если при интег-
рировании не вводится дополнительно никаких ошибок, вариационный прин-
цип не может непосредственно применяться для нахождения сраЬ, так как нет
гарантий, что ошибка, возникающая при расчетах энергии комбинированной
системы, будет больше или меньше ошибок, возникающих при расчетах энергии
изолированных молекул. По всей видимости, одна из наибольших трудностей
при теоретических расчетах межмолекулярных сил заключается в операции
сшивания энергии химического взаимодействия, рассчитанной при малых
расстояниях, с дисперсионной энергией, рассчитанной при больших расстоя-
ниях. Обычно химическая и дисперсионная энергии рассчитываются совер-
шенно различными методами. Если просто прибавить дисперсионную энергию
к химической энергии, то получаются совершенно различные результаты, за-
висящие от того, включаются ли члены, обратно пропорционально восьмой,
десятой и т. д. степени расстояния. Принимая это во внимание, трудно
оценить теоретическую точность полученных таким образом потенциальных
функций.
Перечислим работы, посвященные атомным взаимодействиям, которые
не рассматриваются в этой главе.
1) Н + Не [9]. (К сожалению, Джентил не смог вычислить многие необходимые инте-
гралы, поэтому полученный им результат не имеет большой ценности. В настоящее время
все эти интегралы могут быть вычислены и эту задачу следует рассмотреть заново.)
2) N + N [10]. (Это довольно полное рассмотрение молекулы N2 вблизи равновесных
расстояний, непосредственно учитывается взаимодействие 10 электронов.)
3) Be + Be' [11]. (Это довольно полное рассмотрение взаимодействия нормального
BsJ атома Be с возбужденным 2s 2р атомом Be по методу Гайтлера—Лондона, в котором
учитываются все получающиеся состояния энергии и рассчитываются все интегралы. В работе
приведена таблица всех использованных интегралов.)
4) Li + Li [12—14]. (Бартлетт и Фарри [14] получили превосходное согласие с
экспериментом для энергии диссоциации Li2, рассчитанной по методу Гайтлера—Лондона
с некоторыми дополнительными приближениями. Джеймс [12] показал, что это согласие
нарушается и результаты становятся неудовлетворительными, если не делать этих прибли-
жений. По его мнению, причина такого расхождения заключена во влиянии электронов
внутренних оболочек. Коулсон и Дункансон [13] применили метод молекулярных орбит
для расчета энергии связи молекулы в нормальном состоянии ; однако они не достигли точ-
ности расчетов Джеймса.)
5) Ве+ Н и Ве'+ Н [15]. (Айрлэнд применил метод Гайтлера—Лондона для рас-
чета целого ряда кривых потенциальной энергии, возникающих при взаимодействии атома Н
либо с нормальным BsJ, либо с возбужденным 2s2p атомом Be.)
6) Li + Н и Li+ + Н [16,17]. (Книпп [16] применил одиннадцатичленную волновую
функцию Джеймса и Кулиджа, чтобы показать, что LiH+ имеет наинизшее ^-состояние,
энергия отталкивания которого составляет 0,181 эв при расстоянии За0 относительно
системы Li+ + Н. Расчеты Книппа, воспользовавшегося техникой Джеймса и Кулиджа,
привели к значительно более низким и менее удовлетворительным энергиям диссоциации,
чем расчеты Хатчисона и Маската [17], применявших метод Гайтлера—Лондона.)
7) Li + Li+ [18]. (Джеймс, используя волновые функции типа Джеймса и Кулиджа,
нашел, что при расстоянии в 2,98 а0 энергия связи Li+ равна 1,243 эв.)
8) К + Н [19]. (Геллман развил свою систему приближений, считая, что электроны
внутренних оболочек удовлетворяют условиям Томаса—Ферми, а внешние электроны под-
чиняются модифицированному уравнению Шредингера. В случае К + Н его результаты,
по общему признанию, достаточно плохи. Однако метод Геллмана может оказаться полез-
ным при расчетах межмолекулярных сил на больших расстояниях.)
808 Гл. 74. Квантовомеханические расчеты межмолекулярных сил
9) Na + Na и К + К [20, 21 ]. (Модифицированный метод Гайтлера—Лондона приме-
нен для расчета энергии взаимодействия двух атомов, имеющих один s электрон во внешней
оболочке. Рассчитаны все интегралы, описывающие взаимодействие внешних электронов.
Получено блестящее согласие энергии связи, равновесного расстояния и основных частот
для нормальных двухатомных молекул с экспериментальными данными.)
§ 1. ВЗАИМОДЕЙСТВИЕ ДВУХ АТОМОВ ВОДОРОДА
Поскольку взаимодействие двух атомов водорода представляет собой
простейший пример молекулярного взаимодействия, оказалось возможным
выполнить для этой системы более подробные и точные вычисления, чем для
какой-либо другой системы1) Чтобы выбрать наилучшие кривые потенциальной
энергии, необходимо использовать различную информацию. Если два атома в
ls-состояниях сталкиваются с относительно малыми значениями кинетической
энергии, то в одном случае из четырех они пойдут по энергетической кривой
*?, соответствующей нормальной молекуле Н2, а в трех случаях — по энер-
гетической кривой 327, соответствующей наинизшему состоянию отталкивания
Н22). Высокоэнергетические столкновения, представляющие интерес в химии
«горячих атомов», могут, конечно, сопровождаться ионизацией или возбужде-
нием других состояний. Потенциальная кривая синглетного состояния нормаль-
ной молекулы водорода имеет диаметр столкновения, соответствующий нулевой
энергии, а =0,5 А, и минимальную энергию 103 ккал\моль G160-10~15 эрг
на 1 молекулу) при г = 0,74 А. Триплетное состояние обладает диаметром
столкновения а = 4,0 А и минимальной энергией при г = 4,5 А, равной
0,0074 ккал/моль @,514- Ю6 эрг на 1 молекулу).
Аналитический вид волновых функций, пригодных для расчетов энергии
взаимодействия при всех расстояниях, должен удовлетворять следующим тре-
бованиям : 1) волновые функции должны приводить к хорошим значениям
энергии связи на малых расстояниях ; 2) они должны приводить к хорошим
значениям дисперсионной энергии на больших расстояниях; 3) они должны
быть достаточно простыми, чтобы можно было без большого труда вычислить
обменные и кулоновские интегралы при разных межъядерных расстояниях.
Большинство приближенных волновых функций, приводящих к удовлет-
ворительным значениям энергии химической связи при малых расстояниях,
не дают нужного значения дисперсионной энергии на больших расстояниях.
Например, волновые функции типа Гайтлера—Лондона
^=^A)^B) + ^B)^A) A.1)
не удовлетворяют этим требованиям, так как они приводят к нулевым значе-
ниям дисперсионной энергии независимо от формы одноэлектронных орбиталей
уаA) и %A). Здесь индекс 1 относится к электрону. Ни одна из двух других
стандартных форм волновых функций, таких, как функция Розена [26] или
х) В работах Кембла и Ценера[22, 23] проведены довольно грубые расчеты энергии
взаимодействия тех состояний, которые образуются при столкновении атома водорода в
ls-состоянии с другим атомом водорода, первоначально находящимся либо в 25-состоянии,
либо в 2р-состоянии. При таких столкновениях возникает восемь -Г-состояний и четыре
/7-состояния. Два из этих /7-состояниЙ устойчивы. Одно из них, называемое ^-состоянием,
обладает энергией диссоциации, равной 3,37 эв, тогда как расчеты Кембла и Ценера дают
лишь величину 2,64 эв. Наиболее точно кривая потенциальной энергии для состояний, обра-
зующихся при столкновении Is- и 2р-атомов водорода, определена в работе [24]. С помощью
двенадцатичленной волновой функции в работе [25 ] проделаны очень точные (с точностью
до 6 ккал/моль) расчеты энергии отталкивающего \so2pa*Zu- и притягивающего \sd2so*Zg-
состояний водорода при расстояниях от 1,3 а0 до 1,9 а0.
2) При больших расстояниях энергии xi7- и ^-состояний приблизительно равны, по-
этому относительная вероятность нахождения атомов водорода в том или ином состоянии
просто равна отношению чисел вырождения (в данном случае, спиновых мультиплетностей)
одному к трем.
§ 7. Взаимодействие^ двух атомов водорода
809
Вейнбаума [27], не удовлетворяет предъявленным требованиям. Функция
Розена принимает во внимание поляризацию, а функция Вейнбаума учитывает
как ионные, так и гомополярные члены. Действительно, чтобы рассчитать диспер-
сионную энергию, необходимо принять во внимание статистическую корреля-
цию между координатами двух электронов точно так же, как в классической
теории дисперсионной энергии учитывается, что при любом мгновенном поло-
жении электрона и протона атома а в атоме b путем изменения статистического
распределения электронного заряда наводится дипольный момент; взаимо-
действие между мгновенным и наведенным дипольными моментами и приводит
к появлению энергии притяжения.
Простейший вид волновой функции как для Х27-, так и для ^-состояний,
удовлетворяющий перечисленным выше требованиям, следующий :
+ yalyb2)
PZ*(zalzb2» ±
pZ*(za2zbl)]
ab
A.2)
Знак плюс соответствует ^-состоянию, а знак минус— ^-состоянию. Ось z
направлена вдоль линии, соединяющей ядра, а оси х и у — перпендикулярно
этой линии. Через zal обозначается
разность z-координат электрона 1 и
ядра а. Используемые обозначения
показаны на фиг. 222. В уравнении
A.2) 0^A) и &0A) являются водородо-
подобными волновыми функциями
атомных орбит Is, центры которых
расположены соответственно в атомах
а и 6, а эффективные заряды ядра
равны Z:
= DТ ехр (- Zral),
]L A-3)
exP ( Zrbi) . Фиг. 222. Схематическое изображение
расстояний в молекуле водорода.
В формулах A.3) га1 и rbl выра-
жены в единицах а0. В квантовой
механике атомов или молекул удобно расстояния выражать в единицах боров-
ского радиуса а0 = 0,5292 А, а энергию измерять в «атомных единицах»
<?Х = 627,46 ккал/моль = 27,206 эв = 4,3584- 10-и эрг.
Параметры в формуле A.2) имеют следующий смысл :
1) Параметр Z называется эффективным зарядом ядра, так как волновые
функции атомных орбит аоA) и ЬоA) являются решениями уравнения Шредин-
гера для электрона в кулоновском поле ядра заряда Ze. Увеличение параметра
Z приводит к уменьшению размеров орбиты. При фиксированном межъядерном
расстоянии увеличение Z приводит к уменьшению отношения средней потен-
циальной к средней кинетической энергии электронов. Однако, как показано
в приложении Б гл. 13 (стр. 799), на равновесных расстояниях при варьиро-
вании Z с целью получения наинизшего значения энергии автоматически уста-
навливается правильная величина отношения потенциальной энергии к кине-
тической.
2) Параметр у определяет вклад ионных членов или членов Вейнбаума [27 ]
в приближенную волновую функцию. Для волновой функции молекулярной
орбиты у ч= 1. Благодаря взаимному отталкиванию электроны преимущественно
находятся вблизи различных ядер. Это приводит к тому, что величина у стано-
вится значительно меньше единицы. Вследствие симметрии задачи величина у
в 327-состоянии равна нулю.
Гл. 14. Квантовомеханическае расчеты межмолекулярных сил
3) Параметр а устанавливает корреляцию между мгновенными положе-
ниями двух электронов по отношению к осям, перпендикулярным линии ядер.
Величина а для xi7- и ^-состояний отрицательна при всех расстояниях. Это
значит, что распределение заряда будет больше в том случае, когда знаки коор-
динат х или у у обоих электронов будут различными. Таким образом, отри-
цательное значение а есть простое следствие взаимного отталкивания элект-
ронов.
4) Параметр /? устанавливает корреляцию между мгновенными положе-
ниями двух электронов по отношению к расстояниям вдоль линии ядер. Поло-
жительная величина /? соответствует максимально возможному расстоянию
между электронами. Для 12f- и ^-состояний при больших межъядерных рас-
стояниях величина /? положительна. Тот факт, что величина /? положительна
при больших расстояниях, во многом определяет дисперсионную энергию.
При малых расстояниях /? остается положительной для ^-состояния и ста-
новится отрицательной для ^-состояния. Отрицательная величина /J соответ-
ствует концентрации отрицательных зарядов в области между ядрами. Как
показано в гл. 13, § 1, концентрация отрицательного заряда между ядрами
приводит к появлению сил притяжения между атомами. Герни и Маги [29]
показали, что волновые функции Гайтлера—Лондона или Ванга1) можно значи-
тельно улучшить, если считать, что центры электронных орбит несколько ближе
друг к другу, чем ядра.
При очень больших расстояниях как для ^-состояния, так и для
^-состояния у становится равной нулю, Z = 1, а а принимает вид
Такая волновая функция есть довольно грубая апроксимация волновых
функций, использованных Хассе [31] и Кирквудом [32] для вариационных
расчетов дисперсионной энергии. Волновая функция Гиршфельдера—Линнета
приводит к следующему выражению для энергии взаимодействия на очень
больших расстояниях :
Приведенный результат согласуется с результатами Полинга и Бича [33 J2)
9*=-$ [6,49903 (¦?)' + 124,399 (?-)' + 1135,21 (¦?)"] . A.6)
Полинг и Бич получили свой довольно точный результат, применяя вариацион-
ный метод и решая секулярное уравнение 26-го порядка для определения
численного коэффициента при члене, обратно пропорциональном шестой сте-
пени расстояния, секулярное уравнение 17-го порядка для получения члена,
обратно пропорционального восьмой степени расстояния, и секулярное урав-
нение 2б-го порядка для определения члена, обратно пропорционального деся-
той степени расстояния. К счастью, большинство коэффициентов в секулярном
уравнении исчезает, однако все же остается достаточное количество нену-
левых членов, поэтому решение уравнений связано с очень большой затра-
той труда. В течение последних двадцати лет очень развилась техника тео-
ретических расчетов, которая может в значительной степени уменьшить затрату
труда, связанную с этими расчетами.
х) См. любую книгу по квантовой механике молекул, например [30].
2) В работе [34] показано, что числовой коэффициент у члена, обратно пропорциональ-
ного шестой степени расстояния, должен быть равен или больше 6,4976.
§ 7. Взаимодействие двух атомов водорода
811
А. СОСТОЯНИЕ 1?9 СООТВЕТСТВУЮЩЕЕ НОРМАЛЬНОЙ
МОЛЕКУЛЕ Н2
Теоретические расчеты, использующие волновую функцию, определяемую
уравнением (L2), были проделаны, начиная от самых малых и кончая наиболь-
шими расстояниями. Таким путем получена единственная кривая потенциальной
энергии, которая на больших расстояниях дает дисперсионную энергию, а на
малых—энергию химической связи. Однако на очень больших расстояниях эта
волновая функция не дает дисперсионной энергии с той точностью, как в тео-
ретической работе Полинга и Бича [33 ], а при малых расстояниях вычисленные
значения энергии не столь точны, как величины, получаемые из эксперимен-
тальных значений уровней колебательной энергии молекулы водорода. «Наи-
лучшая» или «наиболее вероятная» кривая потенциальной энергии показана на
фиг. 223; относящиеся к ней числовые значения приведены втабл. 143. Расчеты
Гиршфельдера—Линнета правильно описывают лишь промежуточный отрезок
«наилучшей» или «наиболее вероятной» кривой потенциальной энергии.
120
WO
80
60
40
20
| о
-20
-40
-60
-80
-too
U,UIO
0,0/0
0,005
0
|-0,005
1
-0,0/0
-0,0/5
-0,020
-0025
1 I 1
\
¦ r"
il , ,
1
-
• -
_
—
-
-
1
8
11
9 10
ГаЬ/<*0
Фиг. 224. Ван-дер-ваальсова энергия
взаимодействия двух Is атомов водорода.
7 — расчет Гиршфельдера—Линнета ;
2 — расчеты Полинга и Бича [28].
Фиг. 223. Энергия взаимодействия двух Is
атомов водорода как функция межъядерного
расстояния.
7 — расчеты Гиршфельдера—Линнета ; 2 — расчеты
Джеймса, Кулиджа и Презента ; 3 — кривая Морзе ;
4— кривая, определенная Ридбергом и Бейтлером из
экспериментальных значений колебательной
энергии [28J.
Наиболее правдоподобные экспериментальные данные для нормальной
молекулы Н2 на расстояниях, не превосходящих 3,7а0, были получены Рид-
бергом [35 J и Бейтлером [36], проанализировавшими по методу Клейна
первые 10 колебательных уровней молекулярного водорода. Их результаты
приведены в табл. 144 (кривая 4 на фиг. 223). Межъядерные расстояния
определены с точностью 0,005 А или 0,01а0. Из фиг. 223 видно, что теорети-
ческая кривая Гиршфельдера—Линнета переходит в экспериментальную кривую
Ридберга на расстояниях около 5а0. Поэтому теоретическая кривая, по всей
видимости, достаточно точна в области от 5а0 до таких расстояний, на ко-
торых дисперсионные силы начинают играть заметную роль.
Таблица 143
Наиболее вероятная потенциальная энергия взаимодействия нормальных
атомов водорода (ХЕ)
во
0,78
0,79
0,81
0,83
0,85
0,88
0,92
0,96
1,01
1,09
1,23
1,40
1,68
1,94
2,15
2,32
2,49
2,66
2,84
3,05
3,28
3,51
3,73
4,00
4,5
5,0
6,0
7,0
8,0
9,0
10,0
11,0
12,0
Энергия
связи,
ккал/моль
14,38
20,14
26,68
33,91
41,84
50,46
59,70
69,57
80,13
91,32
103,20
109,32*
103,20
91,32
80,13
69,57
59,70
50,46
41,84
33,91
26,68
20,14
14,38
10,00
3,983
1,858
0,3995
0,0919
0,0251
0,0097
0,0044
0,0027
0,0013
Источник расчетов
Экспериментальные значения Ридберга, определен-
1 ные из данных об уровнях колебательной
энергии
Промежуточное значение, определенное из условий
достаточно гладкого сшивания
• Расчеты Гиршфельдера—Линнета
Расчеты Полинга и Бича [формула A.6)]
* Максимальная энергия связи.
§ 7. Взаимодействие двух атомов водорода
813
ГДС
Представляет интерес сравнить экспериментальную кривую Ридберга с
обычной кривой Морзе [37, 30 ], которая очень часто применяется благодаря
своей простоте:
109,46 [ — 2 ехр (— х) + ехр (— 2х)] ккал\моль, A.7)
х = 1,0298 [^ - 1,401). A.8)
Кривая Морзе хорошо согласуется с экспериментальной кривой Ридберга
вплоть до расстояний гаЬ1а^ = 2,5, однако при больших расстояниях она при-
водит к слишком завышенным энергиям связи.
Из фиг. 224 видно, что энергия притяжения, рассчитанная Гиршфельдером
и Линнетом, больше энергии, рассчитанной Полингом и Бичем на расстояниях,
меньших 90q. Поскольку в расчетах Полинга эффекты обмена не принимались
во внимание, то точка пересечения двух рассчитанных кривых является точкой,
начиная с которой обменные или валентные силы становятся важными.
Таблица 144
Экспериментальная кривая потенциальной энергии Ридберга
для нормальной молекулы водорода [35]
Колебатель-
ный уровень
0
1
2
3
4
5
б
7
8
9
10
оо
Энергия
связи,
ккал/молъ
103,20
91,32
80,13
69,57
59,70
50,46
41,84
33,91
26,68
20,14
14,38
0
Минимальное
расстояние
в
А
0,652
0,576
0,536
0,506
0,486
0,466
0,451
0,441
0,431
0,416
0,411
атомные
единицы а.
1,23
1,09
1,01
0,96
0,92
0,88
0,85
0,83
0,82
0,79
0,78
Максимальное
расстояние
в
А
0,887
1,028
1,138
1,228
1,318
1,409
1,504
1,614
1,734
1,855
1,975
атомные
единицы а.
1,68
1,94
2,15
2,32
2,49
2,66
2,84
3,05
3,28
3,51
3,73
Б. СОСТОЯНИЕ *29 СООТВЕТСТВУЮЩЕЕ ОТТАЛКИВАНИЮ ДВУХ
АТОМОВ ВОДОРОДА, НАХОДЯЩИХСЯ В ls-СОСТОЯНИИ
«Наилучшая» или «наиболее вероятная» кривая потенциальной энергии
для ^-состояния получается путем комбинации рассчитанных Полингом и
Бичем [33 ] значений дисперсионной энергии при больших расстояниях, рассчи-
танных значений Гиршфельдера—Линнета при промежуточных расстояниях,
и очень точных теоретических расчетов Джеймса, Кулиджа и Презента при
малых расстояниях. «Наиболее вероятная» энергия взаимддействия изображена
на фиг. 223 и 224, соответствующие численные значения приведены в табл. 145.
Кривая потенциальной энергии для ^-состояния обсуждается в работе
Джеймса, Кулиджа и Презента [38]. Используя удобную вариационную функ-
цию и применяя определенные поправки, они рассчитали, что при
гаь1^о = 1Д 1,6 и 1,87 энергии отталкивания равны соответственно 119,3, 105,6 и
77,9 ккал/моль. Аналитическая формула, описывающая их результаты, имеет вид
A.9)
9>д.к.п. = 349,7 (-^-)~4/* ехр [- 0,357 (-J-)] ккал/моль.
Теоретическая кривая Гиршфельдера—Линнета приводит к значениям, пре-
восходящим рассчитанные Джеймсом, Кулиджем и Презентом соответственно
814
Гл. 14. Квантовомеханические расчеты межмолекулярных сил
на 33, 24 и 10 ккал/моль. Схематически это изображено на фиг. 223. Рассчитан-
ная Гиршфельдером и Линнетом энергия превосходит энергию, рассчитанную
Джеймсом, Кул и джем и Презентом в точке 1,5я0 на 28%, в точке 1,6а0 на 23%,
в точке 1,87а0 на 13%. Как процентное, так и абсолютное значения разности
между результатами этих двух расчетов уменьшаются при увеличении rab.
Поскольку энергии, рассчитанные Джеймсом, Кулиджем и Презентом, были
получены с помощью значительно более правдоподобной вариационной функ-
ции, то, без сомнения, они более реальны. Оказывается, что расчеты Гиршфель-
дера—Линнета хорошо апроксимируют потенциальную кривую гЕ на расстоя-
ниях, больших 2,5а0, и что в целом наилучшая кривая проходит через три точки,
определенные Джеймсом, Кулиджем и Презентом, и далее при rab, бблыиих
2,5а0, переходит в потенциальную кривую Гиршфельдера—Линнета. Значения
этой наиболее правдоподобной потенциальной энергии приведены в табл. 145.
Из фиг. 224 видно, что ^-состояние обладает минимальной потенциальной
энергией —0,0074 ккал/моль при rab/a0 = 8,45, энергией взаимодействия, рав-
ной нулю при rabla0 = 7,51, и положительной энергией при меньших расстоя-
ниях. •
Таблица 145
Наиболее вероятные значения потенциальной энергии для возбужденной
молекулы водорода [28]
Tab
1,0
1,25
1,5
1,75
2,0
2,5
3,0
3,5
4,0
4,5
5,0
6,0
7,0
7,51
8,0
8,45
10,0
12,0
Энергия
отталкивания,
тал/моль
245
166
119
88,8
68,0
30,45
16,35
8,45
4,300
2,008
0,9074
0,1558
0,01393
0,00000
-0,00586
-0,0074*
-0,0038
-0,0013
Источник расчетов
Из квантовомеханических расчетов Джеймса, Кулиджа
и Презента [38 ]. Цифровые значения получены из
уравнения A.9)
> По расчетам Гиршфельдера—Линнета [28]
По расчетам Полинга и Бича [33] [уравнение A.6)].
Рекомендуется при гаь/а0 > 12
* Минимум энергии.
§ 2. ЭНЕРГИЯ ВЗАИМОДЕЙСТВИЯ АТОМОВ БЛАГОРОДНОГО ГАЗА,
НАХОДЯЩИХСЯ В ОСНОВНЫХ СОСТОЯНИЯХ
Взаимодействие атомов благородного газа, находящихся в основных состоя-
ниях, позволяет легко сравнивать теорию с эксперимеетом. Выражение энергии
взаимодействия имеет простой вид вследствие того, что внешняя электронная
оболочка атомов благородного газа заполнена. Существующие теоретические
расчеты приводят к правдоподобным потенциальным кривым, однако «наилуч-
шая», потенциальная кривая получается из экспериментальных данных.
§ 2. Энергия взаимодействия атомов благородного газа 815
А. ВЗАИМОДЕЙСТВИЕ ДВУХ АТОМОВ ГЕЛИЯ
Энергия взаимодействия двух атомов гелия может быть выражена в виде
суммы четырех членов
ф == ^(вал.) _|_ ^(об.,2) _j_ ^(дисп.,6) _|_ ^(дисп.,8) . ^2.1)
здесь 9?(BaJIt) — валентная, или химическая, энергия притяжения, получаемая
в первом порядке теории возмущений ; ^(дисп.,6) и ^(дисп.,8) — дисперсионная
энергия, обратно пропорциональная соответственно шестой и восьмой степени
расстояния ; g>(o6*>2) — обменная энергия второго порядка, которая будет вскоре
обсуждаться. Член д>(об) не сказывается на дальнодействующих силах и несу-
ществен на малых расстояниях ; ето необходимо учитывать в промежуточной
области, Где дальнодействующие силы сливаются с короткодействующими.
Рассмотрим каждый из этих членов отдельно.
1. Валентное взаимодействие. П. Розен [39] применил теорию возмущений
в первом порядке для расчета валентной энергии. Его расчет отличается от
предыдущих вычислений Слетера [40], Джентила [9] и Н. Розена [20] тем,
что он вычислил все интегралы, тогда как в предыдущих расчетах одними
интегралами пренебрегалось, а другие апроксимировались. П. Розен исполь-
зовал пространственно несимметризованную волновую функцию для невозму-
щенного атома гелия1) в виде
^""^11") B.2)
и получил результат, который можно представить в виде
где г — в А. Этот результат находится в удовлетворительном согласии с широко
применяемым выражением валентной энергии, полученным Слетером,
4вал> = 7,7• Ю-10ехр(-4,60г) эрг. B.4)
Энергия взаимодействия по Слетерув2,1 раза меньше энергии взаимодействия
П. Розена при г = 5а0.
2. Обменная энергия второго порядка. Так же как и в теории возмущения
первого порядка, где валентная энергия получается в виде суммы кулоновского
и обменного членов, теория возмущений во втором порядке приводит к появле-
нию кулоновского и обменного членов. Кулоновские члены второго порядка
дают обычную дисперсионную энергию, рассмотренную в гл. 13, § 3. Обменные
члены второго порядка не рассматривались в гл. 13, § 3, так как они с увели-
чением расстояния экспоненциально падают и поэтому не вносят заметного
вклада в дальнодействующие силы. Однако они становятся важными при про-
межуточных расстояниях (особенно для молекул, у которых внутренние элект-
ронные оболочки отсутствуют). Обменные члены второго порядка получаются
так же, как и обычные дисперсионные члены при вычислении энергии возму-
щения во втором порядке с волновыми функциями, антисимметричными по
отношению к перестановке любых двух электронов.
Эйзеншитц и Лондон [41 ] изучали обменные члены второго порядка, соот-
ветствующие взаимодействию двух атомов водорода, а Маргенау [42 ] обобщил
их метод на случай взаимодействия двух атомов гелия.
х) В работе [30] подробно обсуждаются свойства этой функции. Расчеты энергии невоз-
мущенного атома, выполненные с помощью этой функции, приводят к значениям, превосхо-
дящим правильную величину на 0,76 эв.
816 Гл. 14. Квантовомеханические расчеты межмолекулярных сил
Существуют два метода, с помощью которых можно рассчитать диспер-
сионные силы, —теория возмущений во втором порядке и вариационный метод.
Обменная энергия второго порядка появляется при любом из этих двух рас-
смотрений. Однако практически для грубой оценки у(о6) лучше применить
вариационный метод в простейшем виде. Возьмем в качестве варьируемой
функции следующую:
(lb) B.5)
где щ — антисимметричная невозмущенная волновая функция ; <ре — потен-
циал возмущения, возникающий вследствие взаимодействия двух атомов;
А — вариационный параметр. Для расчета энергии необходимо вычислить
интеграл
JVoЧ>\ VodhdTt dr3dr4 . B.6)
Для системы, состоящей из двух атомов гелия, невозмущенная волновая
функция имеет вид
2)bC4)- *C2)Л<14)-
1,2),
где %аA, 2) и %ьC, 4) — волновые функции основного состояния изолированных
атомов гелия. Обменный член второго порядка получается в виде
. B.8)
Интегралы такого типа исчезают на больших расстояниях, где перекрывание
мало. Маргенау проделал подробные расчеты, учитывающие все обменные
интегралы, и нашел, что для системы, состоящей из двух атомов гелия, член,
описывающий взаимодействие (без учета валентной энергии, получающейся
в первом порядке теории возмущений, и дальнодействующих дисперсионных
сил), имеет вид
^<об.,2) = _ 5,60 • Ю-10 ехр (- 5,33 г) эрг, B.9)
где г — в А.
3. Дисперсионная энергия, обратно пропорциональная шестой степени
расстояния. Маргенау [42 ] проделал блестящие расчеты дисперсионной энер-
гии, обратно пропорциональной шестой степени расстояния. Он использовал
все значения сил осциллятора /01- для переходов между основным уровнем и
всеми возбужденными состояниями гелия, включая и континуум, как это
сделали Винти [44 ] и Уиллер [43 ]. Он несколько изменил теоретические значе-
ния /0/, с тем чтобы получить согласие между расчетной величиной поляризуе-
мости гелия и наилучшим экспериментальным значением. Подставляя эти силы
осцилляторов и энергии, соответствующие различным переходам, в уравнение
для наведенного диполь-дипольного взаимодействия [уравнение C.20) гл.13],
Маргенау получил
139
где г — в А. Наилучшие расчеты были проделаны Слетером и Кирквудом [34] :
^ B.11)
Значение энергии, полученное Слетером и Кирквудом [42 ], было найдено
вариационным методом, который, как известно, также приводит к завышенным
значениям и в случае других пар молекул [42 ].
4. Дисперсионная энергия, обратно пропорциональная восьмой степени
расстояния. Пейдж применил вариационный метод для расчета дисперсионной
§ 2. Энергия взаимодействия атомов благородного газа
817
энергии, обратно пропорциональной восьмой степени расстояния. Он получил
B.12)
где г— в А. Числовой множитель хорошо согласуется со значением, полученным
Букингемом [46] (числовая постоянная 3,53), и с предыдущими расчетами
Маргенау [47].
5. Полная энергия взаимодействия и сравнение с экспериментом. Мар-
генау [42] тщательно исследовал ошибки, возникающие при представлении
полной энергии в виде аддитивной суммы валентной энергии, обменной энергии
второго порядка и дисперсионной энергии. Он пришел к выводу, что при взаи-
модействии двух атомов гелия возникает небольшая ошибка при расстояниях,
представляющих интерес для столкновений с относительно малой энергией.
Таким образом, «наилучшая» теоретическая потенциальная кривая Розена—
Маргенау—Пейджа имеет вид
(теор.)
Ф.М.П.
где г —в А. Приведенное выра-
жение находится в удовлетвори-
тельном согласии с более ранним
потенциалом Слетера—Кирквуда,
имеющим широкое применение,
(вал.) , (дисп.,6)
<Рск = Vc + 9?ск =
Ю-12 эрг. B.14)
Межмолекулярная потенциаль-
ная функция для гелия опреде-
лена тремя эмпирическими мето-
дами, которые приводят к [резуль-
татам, хорошо согласующимся друг
с другом:
1) С помощью анализа данных,
относящихся ко второму вириаль-
ному коэффициенту в области тем-
ператур от 30 до 400° К, выполнен-
ного де Буром, Михельсом и Лунбе-
ком с потенциалом F-12) Леннарда-
Джонса в соответствии с методом,
описанным в гл. б, § 5, где показано,
как можно принять во внимание
квантовые поправки [48—50 ]. Опре-
деленная таким образом потенциаль-
ная функция описывает экспери-
ментальные данные о вязкости ге-
лия [51] с точностью до нескольких
процентов.
2) С помощью анализа данных,
относящихся к высокотемператур*
ному второму вириальному коэф-
фициенту в области от 0 до 1200° С
эрг 9
.13)
-2
о
г. А
Фиг. 225. Потенциальные кривые, описываю-
щие взаимодействие двух атомов гелия.
Кривые, построенные по данным теории: 1 — Слетера—
Кирквуда [34]; 2—Розена—Маргенау—Пейджа [39,
42, 45, 47]. Кривые построены по экспериментальным
данным: 3 — потенциал F-12) Леннарда-Джонса, опре-
деленный с помощью значений второго вириального
коэффициента при низких температурах D8, 49];
4 — модифицированный потенциал (б-ехр) Букингема,
определенный из значений второго вириального коэф-
фициента при высоких температурах и коэффициента'
вязкости [52]; 5 — потенциал Букингема—Корнера,
определенный с помощью значений второго вириаль-
ного коэффициента при низких температурах и коэф-
фициента вязкости [53].
818 Гл. 74. Квантовомеханические расчеты межмолекулярных сил
и к коэффициенту вязкости в области от 200 до 1100° К, выполненного Мэзоном
и Райсом [52 ] с модифицированным потенциалом (б-ехр) Букингема (см. гл. 3, § 9);
при этих температурах квантовыми эффектами можно полностью пренебречь.
3) С помощью анализа второго вириального коэффициента и коэффициента
вязкости при очень низких температурах, проделанного Букингемом, Гамиль-
тоном и Месси [53 ] для определения потенциальной функции межмолекуляр-
ного взаимодействия гелия (см. гл. 3, § 9); эти три экспериментальные кривые
совместно с теоретическими кривыми Слетера—Кирквуда и Розена—Марге-
нау—Пейджа показаны на фиг. 225.
Два атома Не4 могут находиться в связанном дискретном колебательном
квантовом состоянии (см. табл. 55, стр. 333). Совсем недавно Килпатрик1) пока-
зал, что для потенциала (б-ехр) Букингема, модифицированного Мэзоном и
Райсом, энергия такого дискретного уровня будет равна Е = —4,677 • Ю8 эрг,,
или Е\е = —0,003699. К сожалению, экспериментальные данные недостаточно
точны для однозначного определения потенциала.
Б. ВЗАИМОДЕЙСТВИЕ ДВУХ АТОМОВ НЕОНА
Блейк и Майер [54 ] рассчитали энергию отталкивания двух атомов с за-
полненными электронными оболочками, применяя теорию возмущения Гайт-
лера—Лондона в первом порядке. Они воспользовались волновыми функциями
одноэлектронных орбит, рассчитанными Броуном [55 ] по методу самосогласо-
ванного поля Хартри. Пусть ipai соответствуют орбитам с центром в атоме ау
а индекс i относится к одной из пяти орбит — Is, 2s, 2p_, 2p0 и 2р+. Волновая
функция ipbj определяется аналогичным образом. Энергия взаимодействия выра-
жается через величины Lih sijy S и Uy. Обменные интегралы L,7 равны
^*,. B.15)
Интегралы перекрывания равны
Stj=*jrtiVbj*r. B.16)
Суммарное перекрывание равно
^ B.17)
Пусть д><эл-ст-) — классическая электростатическая энергия взаимодействия
двух пространственных зарядов, определенная в уравнении A.66) гл. 12 через
плотности зарядов Qa и gb9 относящиеся соответственно к атомам а и ft, и пусть
электростатические потенциалы атомов а и Ь будут равны Та и ОУЪ. В таком
случае
j jdr #
Аналогичным образом можно определить
Uu = ~ е*J (Уа + Tbj)tfiVbjdr, B.19)
гДе Tat — потенциал атома а, в котором отсутствует один электрон на /-ft
орбите; Tbj определяется аналогичным образом. Используя эти величины,
энергию взаимодействия двух атомов неона можно записать в виде
2 E.. Uu +LiJ) . B.20>
*) Частное сообщение, сентябрь 1953 г. Килпатрик произвел расчеты на вычислитель-
ной машине MANIAC в Лос-Аламосе. Он вычислил также фазовые сдвиги при различных
значениях квантового углового числа в довольно широкой области кинетических энергий.
Полученные им результаты для потенциала Букингема значительно отличаются от резуль-
татов, основанных на потенциале де Бура—Михельса—Лунбека.
§ 2. Энергия взаимодействия атомов благородного газа
819
Значения потенциальной энергии, рассчитанные при трех различных межъядер-
ных расстояниях, приведены в табл. 146.
Таблица 146
Энергия взаимодействия двух атомов неона [54]
'.А '
1,8
2,3
3,2
0,0756
0,0115
0,000271
Ю-14 эрг
-729
-79,7
-1,19
\О~иэрг
283
35,3
0,663
р(ЭЛ. CT.)f
10—м эрг
-102
-9,45
-0,101
9,10-" эрг
344
34,9
0,426
Рассчитанные значения энергии могут быть описаны аналитическим выра-
жением
2,97.10-е
эрг>
B.21)
где г — расстояние в А. Это выражение согласуется с расчетами при г = 1,8
и 3,2 А, но приводит к значению 19,8-Ю4 эрг (вместо 34,9- \0~и эрг)
при г = 2,3 А. Лучшее согласие получается, если воспользоваться экспо-
ненциальной формой потенциала 6
?>=1,9.10-8ехр(-4,782г)эрг, B.22)
где г — расстояние в А. Это выра-
жение согласуется с расчетами при
г = 1,8 и 3,2 А и приводит к зна-
чению 31,5- Ю4 эрг при г — 2,3 А.
Последнее значение с точностью до
10% совпадает с рассчитанным.
Таким образом, экспоненциальная
форма потенциала приводит к более
точному описанию. \
Маргенау [42] исследовал выра-
жение для обменной энергии вто-
рого порядка двух взаимодейству-
ющих атомов неона и нашел, что она
будет пренебрежимо мала. Таким
образом, полная энергия взаимо-
действия с большой точностью
равна сумме валентной энергии,
соответствующей короткодействую-
щим силам, и дисперсионной энер-
гии, соответствующей дальнодейс-
твующим силам. Определение потен-
циала межмолекулярного взаимо-
действия неона с помощью экспери-
ментальных данных, относящихся к
уравнению состояния, обсуждается
в гл. 3, § 9. Ввиду отсутствия луч-
шей информации предполагается,
что первый порядок теории возму-
щений дает отталкивающую часть
4
3
2
5 О
-/
-2
-3
-5 -
I L
* 3 4 5 . 6 1 8
г, А
Фиг. 226. Три потенциальные кривые для
взаимодействия двух атомов неона.
7 — экспериментальная кривая потенциала F-12)
Леннарда-Джонса, определенная из данных по вязкости;
2 — экспериментальная кривая модифицированного
потенциала (б-ехр) Букингема, определенная с помо-
щью данных о втором вириальном коэффициенте, ко-
эффициенте вязкости и свойствах кристалла [52];
3 — теоретическая кривая Блейка и Майера [54] с
учетом дисперсионной энергии.
820
Гл. 14. Квантовомеханические расчеты межмолекулярных сил
потенциала межмолекулярного взаимодействия, тогда как во втором порядке
теории возмущений получается притягивающая часть. Полная потенциальная
кривая соответствует сумме этих членов. «Наилучшая» потенциальная кривая
получается сложением притягивающей части потенциала F-12) Леннарда-
Джонса с аналитическим выражением, описывающим теоретические резуль-
таты Блейка и Майера [уравнение B.22) ]; таким образом,
Ртеор. = 1,9- 10-8ехр(- 4,782 г) - 9^г6ш эрг . B.23)
Сравнение этого потенциала с экспериментальными функциями проведено
на фиг. 226. 1) Постоянные в потенциале F-12) Леннарда-Джонса определены
из данных о коэффициенте вязкости [56 ], приведенных в табл. 73, стр. 442. Эти
постоянные очень близки к соответствующим постоянным, определенным из
кривой, описывающей второй вириальный коэффициент. 2) Параметры в моди-
фицированном потенциале (б-ехр) Букингема совпадают со значениями Мэзона
и Раиса [52 ]2), найденными из данных о втором вириальном коэффициенте,
коэффициенте вязкости и свойствах кристалла, приведенных в табл. 12, стр. 157.
Из фиг. 226 видно, что имеется очень хорошее согласие экспериментальных и
теоретических функций.
В. ВЗАИМОДЕЙСТВИЕ ДВУХ АТОМОВ АРГОНА
Кунимуне [58 ] применил метод, развитый Блейком и Майером для неона,
к расчету валентного взаимодействия двух атомов аргона. Результаты расчетов
Кунимуне представлены в табл. 147, в которой применены те же обозначения,
что и в табл. 146.
Таблица 147
г А
2,12
2,64
3,70
Энергия взаимодействия двух атомов аргона [58]
s
0,300
0,0656
0,00232
10-м эрг
-2750
-400
-7,63
Ю-14 эрг
271
28,2
0,447
^(эл. ст.),
\О-иэрг
-410
-44,0
-0,98
<р(вал.),
Ю-14 эрг
2090
328
6,20
Теоретические значения, приведенные в этой таблице, можно описать сте-
пенной зависимостью
B.24)
г, эрг
или экспоненциальной функцией
<Р<вал.) = 5,15 • Ю-8 ехр (- 3,68 г) эрг,
B.25)
где г — расстояние в А. Выражение, обратно пропорциональное расстоянию
в степени 10,5, приводит к лучшему согласию, чем любая другая степень.
Однако математически удобнее пользоваться выражением, обратно пропор-
циональным двенадцатой степени расстояния. Наилучшее совпадение расчет-
ных данных с теоретическим потенциалом, обратным двенадцатой степени рас-
стояния, получается при
Щ?1 B.26)
х) Эти потенциалы для Ne и Аг хорошо согласуются с потенциалом Букингема—Корнера,
определенным в работе [57].
§ 3. Взаимодействие атома водорода с молекулой водорода
821
Маргенау [42] исследовал выражение для обменной энергии второго
порядка при взаимодействии двух атомов аргона и нашел, что она будет пре-
небрежимо мала. Таким образом, полную энергию взаимодействия с большой
степенью точности можно пред-
ставить в виде суммы валентной
энергии, соответствующей корот-
кодействующим силам, и диспер-
сионной энергии, соответствую-
щей да л ьно действующим силам.
«Наилучшая» теоретическая кри-
вая получается при сложении
притягивающей части экспери-
ментального потенциала F-12)
Леннарда-Джонса с теоретичес-
ким значением валентной энергии
•3,68 г)- «
эрг, B.27)
1,092-Ю-
для
где г — в А. На фиг. 227 теорети-
ческая кривая изображена сов-
местно с тремя эмпирически
определенными потенциалами:
1) потенциал F-12) Леннарда-
Джонса с параметрами, опреде-
ленными из данных по вязкости
[56], приведенных в табл. 73,
стр.442. Параметры потенциала,
определенные из данных по вяз-
кости, хорошо согласуются с
соответствующими параметрами,
определенными из данных о вто-
ром вириальном коэффициенте;
2) модифицированный потенциал
(б-ехр) Букингема, постоянные
в котором определены Мэзоном и
В. Райсом [52 ] и приведены в табл. 12, стр. 157 ; 3) потенциал О. Раиса, опре-
деленный выражением (9.8) гл. 3 и полученный из рассмотрения свойств кри-
сталла и второго вириальногй коэффициента. Теоретическая и эксперименталь-
ные кривые хорошо согласуются между собой.
Ф и г. 227. Четыре потенциальные кривые
взаимодействия двух атомов аргона.
1 —экспериментальная кривая F-12) Леннарда-Джонса,
определенная из данных по вязкости; 2 — эксперимен-
тальная кривая модифицированного потенциала F-ехр)
Букингема, определенная с помощью данных о втором
вириальном коэффициенте и свойствах кристалла [52 ];
3 — экспериментальная кривая О. Раиса; 4 — теорети-
ческая кривая Кунимуне [58], учитывающая дисперси-
онную энергию.
§ 3. ВЗАИМОДЕЙСТВИЕ АТОМА ВОДОРОДА С МОЛЕКУЛОЙ
ВОДОРОДА
Расчетам энергии взаимодействия атома водорода с молекулой уделялось
много внимания. Если столкновение атома водорода с молекулой водорода
происходит с относительно малой кинетической энергией выше 7 ккал/моль,
то с большой вероятностью осуществляется простейшая из всех возможных
химических реакций:
Н + Н2->На + Н. C.1)
К этому типу часто принадлежат также 0р/Л0-пара-превращение водорода и
реакции между легким и тяжелым водородом. Столкновения с относительно
малой кинетической энергией встречаются в пламенах, где происходит
822 Гл. 14. Квантовомеханические расчеты межмолекулярных сил
диффузия атомов водорода (или других свободных радикалов) через газ с насы-
щенными валентностями, например такой, как Н2. Эффективное сечение столк-
новения атомов водорода, имеющих очень большую кинетическую энергию,
с молекулами водорода измерено Амдуром [60] по методу атомных пучков.
С теоретической точки зрения, вариационный расчет энергии системы, состоя-
щей из трех атомов водорода, представляет труднейшую задачу молекулярной
физики, которая когда-либо исследовалась, если не пренебрегать некоторыми
важными интегралами.
А. ПОЛУЭМПИРИЧЕСКИЙ МЕТОД ЭЙРИНГА
Качественную картину столкновения легче всего понять, используя «полу-
эмпирический» метод Эйринга, применяемый для расчета поверхности потен-
циальной энергии [61—63] системы, состоящей из трех атомов водорода:
а, Ь и с. Полуэмпирическая схема представляет дальнейшую апроксимацию
теории возмущений в первом порядке, рассмотренную в гл. 13, § 1, где энергия
системы, состоящей из многих атомов, выражена через энергии связи каждой
пары атомов, рассматриваемых как изолированные двухатомные молекулы.
Из уравнения A.53) гл. 13 следует, что кулоновская энергия системы Qc выра-
жается в виде суммы членов СаЬ (называемых кулоновской энергией двухатом-
ной молекулы а — b) и членов Еа (называемых энергией изолированного атома а)
Qc= 2 Cab+ 2 Еа; C.2)
Все Все
пары атомы
атомов
здесь
в атоме Ъ Орбитали
в атоме а
2
J" JJa,(ir^B)*-^-a,(lN;B)dr1dra, C.3
2
Орбитали Орбитали
в а в Ь
а Еа — сумма всех членов в Qc, которые не включают какие-либо орбитали или
ядра, не принадлежащие атому а. Представление Qc в виде суммы членов СаЬ
и Еа не требует каких-либо приближений. Однако отождествление СаЬ с куло-
новской энергией двухатомной молекулы предполагает, что орбитали двухатом-
ной молекулы не меняются в присутствии других атомов; отождествление Еа
с полной энергией атома а предполагает так же, как и в случае двухатомных
молекул, что орбитали изолщ ованных атомов не меняются в присутствии дру-
гих атомов. Отсюда ясно, что от полуэмпирической схемы нельзя ожидать коли-
чественной точности, однако с качественной точки зрения она очень полезна.
В системе, состоящей из трех атомов водорода, с каждым атомом связан
лишь один электрон. Следовательно, существует лишь один обменный интеграл
Lab, связанный с каждой парой атомов, т. е. с каждой двухатомной молекулой,
и определенный формулой A.56) гл. 13. В соответствии с первым порядком
теории возмущений [уравнение A.52) гл. 13] энергия связи молекулы водорода
а — ft, выраженная как функция межъядерного расстояния rab, имеет вид
<РаЬ (Tab) = Cab Ы + ^ab fob) • C-4)
Для удобства, а также ввиду отсутствия лучшей информации функция (раь(гаь)
обычно берется в форме потенциальной кривой Морзе [30, 37 ] для двухатомной
молекулы а — Ь. Для молекулы водорода потенциальная кривая Морзе
дается формулой A.7).
§ 3. Взаимодействие атома водорода с молекулой водорода
823
В полуэмпирической схеме дополнительно предполагается, что СаЬ и Lab
пропорциональны <раЬ:
Cab (fab) = П <раЬ (ГаЪ) , Lab (ГаЬ) = A - Л) <раЬ (ГаЬ) J
C.5)
здесь я, по определению,—«доля кулоновской энергии», которая обычно считается
постоянной, не зависящей от rab. Для Н2 Сугиура [64] непосредственно рассчи-
тал интегралы и нашел, что п довольно сильно зависит от межъядерного рас-
стояния, как показано на фиг. 228. В довольно широкой области расстояний,
представляющих интерес для многих
задач теории столкновений, п^О, 14. 0,16
По аналогии с этим химики считают,
что для всех молекул п = 0,14.
В действительности для получения °>12
согласия между рассчитанным и
наблюденным значением энергии ак-
тивации реакции
р
Н2->Н2
0,08
0,04
I I
1 I I \
Равновесны расстояние
между атомами водорода
1 \ f \ I I I
О 0,5 1,0 7,5
2,5 3,0 3,5 4,0
Г, А
с учетом поправки на нулевое зна-
чение энергии необходимо выбрать
л = 0,20 [63 ]. Представление о
постоянстве п приводит к появлению
ложного минимума [65 ] на энерге-
тической потенциальной поверх-
ности трех атомов, и, несомненно,
это предположение совершенно не-
верно для межъядерных расстояний,
сравнимых с равновесными рассто-
яниями в двухатомных молеку-
лах. Ошибочность предположения
л = 0,14 подтверждена в работе
Розена и Икехары [21 ], проделав-
ших расчеты для щелочных метал-
лов, аналогичные расчетам Гайт-
лера—Лондона и Сугиуры для водорода. Они нашли, что п = 0,23 для Li2,
п = 0,32 для Na2 и п = 0,38 для К2. Аналогичным образом Фог [66 ] показал,
что для связи углерод—водород h = 0,63.
При .рассмотрении трехатомной системы с одним электроном в каждом
атоме по методу теории возмущений в первом порядке химические связи могут
существовать лишь между а и Ь или Ь и с (волновая функция, описывающая
связь между а и с, является линейной комбинацией волновых функций двух
других связывающих состояний и поэтому не представляет что-нибудь новое).
Если принять во внимание взаимодействие двух связывающих состояний, то
полная энергия системы с учетом возмущения первого порядка будет иметь вид
Фиг. 228. Доли кулоновской энергии в соот-
ветствии с приближением Гайтлера—Лондона—
Сугиуры [63].
Кулоновская энергия
п "** Полная энергия связи '
Г)
прибл. — Vc
Ц
е
— LabLbc — Lab Lac — Lbc L
abLbc
bc Lac
C.6)
Если в это выражение подставить полуэмпирические значения кулоновских
и обменных энергий, то энергия взаимодействия cpafic атома водорода а с моле-
кулой водорода be (относительно состояния, соответствующего бесконечному
разделению атома и молекулы) принимает вид
<Ра,Ъс = DHt + П [(рнг (ГаЬ) + №. {he) + 4>Ht (Гас)] Т
Т A - Л) [рн. Ы2 + 9НШ (he)* + <PHt (Гас)* - УН. Ы УН, (ГЬС) -
— УН, (Гаь) <PHt (ГаС) ~ УН, (гЬс) Ун, (Гас)\1% \
C.7)
824
Гл. 14. Квантовомеханичесте расчеты межмолекулярных сил
здесь Dh, —энергия диссоциации молекул водорода без учета нулевой энергии,
т. е. DHf = Ю8,8 ккал/моль. На фиг. 229 схематически изображен потенциал
взаимодействия при столкновении молекулы с атомом водорода. Предполагается,
что атомы в молекуле водорода расположены на равновесном межъядерном
расстоянии. Сплошные кривые соответствуют линиям постоянной потенциаль-
ной энергии при различных положениях атома. На больших расстояниях атом
Уровень реакции
Область высокой
энергии > 15ккал
^Уровень кинетической энергии
Боровская орбита 0,53А-
Фиг. 229. Потенциал взаимодействия атома Н с молекулой Н2 [62].
притягивается под действием слабых дисперсионных сил. На малых расстоя-
ниях притяжение сменяется отталкиванием. При тепловых скоростях, соот-
ветствующих средней комнатной температуре, атом начинает отталкиваться,
прежде чем достигнет первой сплошной линии. На больших расстояниях сплош-
ные линии очень близки к окружностям, однако атомы, обладающие большой
кинетической энергией, при столкновениях, происходящих параллельно линии
ядер, могут проникать глубже внутрь молекулы, чем при столкновениях,
происходящих перпендикулярно линии ядер. Если атом обладает достаточным
запасом энергии и приближается параллельно линии ядер, то он пересекает
значение энергии, отмеченное как уровень реакции. С этого момента атом
начинает притягиваться. Если двухатомная молекула свободна, она слегка
расширяется при приближении атома, и после того, как атом достигнет уровня
реакции, все три атома начинают сильно колебаться. В результате один иа
внешних атомов отрывается. Если оторвавшийся атом ранее принадлежал
молекуле, то происходит химическая реакция, иначе в результате столкнове-
ния происходит лишь переход кинетической энергии во внутреннюю энергию.
Одна из принципиальных трудностей, возникающая при применении полу-
эмпирической схемы, заключается в том, что нам неизвестна энергия связи
нормальной двухатомной молекулы при больших межъядерных расстояниях.
Кривые Морзе представляют довольно плохую апроксимацию на больших
расстояниях [28, 67]1), и это делает полуэмпирическую систему бесполезной
для количественных расчетов.
См. также § 1,
3.§ Взаимодействие атома водорода с молекулой водорода
825
Б. ПРЯМОЙ РАСЧЕТ ДИСПЕРСИОННОЙ ЭНЕРГИИ В ПЕРВОМ
ПОРЯДКЕ ТЕОРИИ ВОЗМУЩЕНИЙ
Прямой расчет энергии взаимодействия атома и молекулы водорода в пер-
вом порядке теории возмущений приводит к гораздо более удовлетворительным
результатам, чем полуэмпирический расчет. Применяя новую технику интегри-
рования, можно без труда вычислить все необходимые трехцентровые интег-
ралы [68], поэтому задача оказывается вполне разрешимой. Такие расчеты
уже проделаны для трех атомов водорода, расположенных симметрично вдоль
прямой или образующих равносторонний треугольник [69, 73 ], однако эти
результаты не представляют большого интереса с точки зрения кинетической
теории столкновений между атомами и молекулами водорода.
Маргенау [74] довольно грубо апроксимировал трехцентровые обменные
интегралы. Это позволило ему рассчитать отталкивающую часть потенциала
межмолекулярного взаимодействия в первом приближении теории возмущений.
Используя волновую функцию типа Гайтлера—Лондона или типа Ванга с эффек-
тивным зарядом ядра Z = 1,20 (были рассмотрены также две другие величины
Z, одна больше, а другая меньше предыдущей, однако они не привели к ра-
зумным результатам), Маргенау рассмотрел две конфигурации :
А) Атом приближается к молекуле перпендикулярно линии ядер
г
и
Здесь ге = 0,73 А при равновесном разделении атомов в Н2.
В) Атом приближается вдоль линии ядер
Результаты расчетов Маргенау приведены в табл. 148.
Таблица 148
Расстоя-
ние
г, А
2,205
2,646
3,087
Энергия отталкива-
ния в первом порядке
теории возмущений,
10-" эрг
конфигу-
рация А
173,6
39,8
8,87
конфигу-
рация В
231,2
55,96
12,45
Дисперсион-
ная энергия
или энергия
притяжения.
10-" эрг
141,0
39,94
14,10
Расстоя-
ние
г, А
3,528
3,969
4,410
Энергия отталкива-
ния в первом порядке
теории возмущений,
10-" эрг
конфигу-
рация А
1,69
0,333
0,066
конфигу-
рация В
2,59
0,495
0,096
Дисперсион-
ная энергия
или энергия
притяжения,
10-" эрг
5,82
2,701
1,370
Притягивающая часть потенциальной энергии (дисперсионная энергия)
была рассчитана по методу, указанному в гл. 13, § 3. Для атома водорода
Маргенау применил силу осциллятора / = 1 и соответствующую резонанс-
ную энергию, равную 0,487 е2/а0 (это значение приводит к правильному чис-
ловому коэффициенту при г~6 для взаимодействия двух атомов водорода).
826
Гл. 14. Квантовомеханические расчеты межмолекулярных сил
Для молекулы водорода он взял два дисперсионных члена (эти члены
были введены Вольфом и Герцфельдом [75 ] для получения особо точных резуль-
татов об изменении показателя преломления с частотой). Их значения сил осцил-
ляторов и резонансных энергий
равны
= 0,69, Ег = 0,630 *>/Оо,
= 0,84, Е2 = 0,492 е*/а0 .
C.8)
В этом случае дисперсионная энер-
гия имеет вид
C.9)
Гн,н, Гн,н,
где г — в А. В этих расчетах асим-
метрией молекулы Н2 пренебрега-'
лось. Однако зависимость дисперси-
онной энергии от ориентации можно
определить по методу гл. 13, § 4,
принимая во внимание различие в
поперечной и продольной поляри-
зуемости.
На фиг. 230 изображен полный
потенциал, полученный путем ком-
бинации притягивающей и оттал-
кивающей частей потенциала
(В первом порядке
Фиг. 230. Потенциал взаимодействия атома ,~ ir\\
водорода с молекулой водорода для конфи- W* *Ч>
гураци и . для К0НфИГурацИИ ^ диаметр
столкновения а = 2,85 А, а глубина
потенциала е = 5 • Ю6 эрг; для конфигурации В диаметр столкновения
о = 3,03 А, а глубина потенциала е = 3,5- Ю5 эрг. Если учесть асимметрию
поляризуемости, то значение е слегка возрастает для конфигурации В и умень-
шается для конфигурации А.
§ 4. ВЗАИМОДЕЙСТВИЕ ДВУХ МОЛЕКУЛ ВОДОРОДА
Взаимодействие двух молекул водорода представляет один из наиболее
сложных примеров, для которых проделаны подробные квантовомеханические
вычисления. С помощью ранее выведенной грубой апроксимации можно полу-
чить теоретический потенциал, хорошо согласующийся с энергией взаимодей-
ствия, выведенной из экспериментов. Энергию взаимодействия можно пред-
ставить в виде суммы четырех членов
<рнг н, = <р(вал>) + <p(Q>Q) + <p<wcn-'6> + <р(дисп"8); D.1)
здесь 9><вал-> — химическая, или валентная, энергия, возникающая вследствие
перекрывания волновых функций ; <р<^> — электростатическая квадруполь^
квадрупольная энергия взаимодействия; <р(дисп.,б> — часть дисперсионной
б й (дасп8)
дру р р
энергии, обратно пропорциональная шестой степени расстояния; ^
часть дисперсионной энергии, обратно пропорциональная восьмой степени
расстояния. Точные расчеты энергии взаимодействия в первом приближении
теории возмущений позволяют определить как квадруполь-квадрупольную,
так и валентную энергию. Однако вследствие приближений, допускаемых в
§ 4. Взаимодействие двух молекул водорода 827
настоящем рассмотрении, квадруполь-квадрупольное взаимодействие требует
отдельного рассмотрения. Проанализируем вычисления, приводящие к различ-
ным типам энергии взаимодействия.
А. ХИМИЧЕСКАЯ, ИЛИ ВАЛЕНТНАЯ, ЭНЕРГИЯ
Вычисление химической, или валентной, энергии представляет наиболее
трудную часть расчета энергии взаимодействия двух молекул водорода. Как
при вариационном расчете, так и при расчете в первом порядке теории возму-
щений появляется много трех- и четырехцентровых многократно обменных
интегралов. Сейчас уже существуют формулы для вычисления всех этих интег-
ралов [76 J1). Для единственного примера, в котором четыре атома водорода
расположены вдоль прямой линии на расстоянии 1,4 а0 один от другого, Тэйлор
оценил все эти интегралы и определил энергию взаимодействия [68]. Несом-
ненно, что подобные вычисления вскоре будут проделаны для различных кон-
фигураций двух молекул водорода. Однако до последнего времени вычисления
этих интегралов были трудно преодолимой задачей. Численно они настолько
велики, что любая попытка их апроксимировать должна быть сделана очень
тщательно. Но если все многократно обменные (как энергии, так и перекры-
вания) интегралы положить равными нулю, то во многих задачах значения
энергии взаимодействия, вычисленные в первом порядке теории возмущений,
оказываются более точными по сравнению с энергией, вычисленной с учетом
обменных интегралов [63, 77, 78 ]. Причина этого кажется неясной, но воз-
можное объяснение должно исходить из того, что происходит компенсация
ошибок следующим образом: энергию взаимодействия, вычисленную в первом
порядке теории возмущений, можно выразить в виде отношения суммы интегра-
лов энергии к сумме интегралов перекрывания. Каждому интегралу энергии
в числителе соответствует интеграл перекрывания в знаменателе. Если прибли-
женные волновые функции достаточно точны, то отношение любой пары этих
интегралов не зависит от выбора пары и в точности равно энергии взаимодей-
ствия системы, поэтому пренебрежение какой-либо парой многократно обмен-
ных интегралов, одного в числителе и одного в знаменателе, не вносит ошибок.
Де Бур [79 ] применил теорию возмущений в первом порядке, или метод
Гайтлера—Лондона для расчета энергии взаимодействия двух молекул 'водо-
рода. Он использовал водородоподобные атомные функции с постоянной экра-
нирования, близкой к единице. При расчетах он положил все многократно
обменные интегралы равными нулю и пренебрег всеми интегралами, учитываю-
щими влияние электронного обмена внутри молекул на кулоновское взаимо*
действие между молекулами. Эветт и Маргенау [80, 81 ]2) недавно проделали
подобные вычисления с учетом всех тех интегралов, которые отбросил де Бур.
Трудно сказать, какие из этих вычислений приводят к более правдоподобным
результатам. Однако результаты, полученные де Буром, удобнее, поскольку
они выражены в замкнутой форме, с явной зависимостью от ориентации. По
этой причине в оставшейся части этого параграфа будет рассмотрена лишь
трактовка де Бура.
Де Бур нашел, что в области межмолекулярных расстояний от 4а0 до
10а0 для двух молекул водорода а — & и с — d валентная энергия может быть
*) См. также приложение А, ссылки на работы [11, 12].
2) В работах [80, 81 ] использовалось значение эффективного заряда ядра Z = 1,166,
которое приводит к наилучшему значению энергии молекулы водорода в основном состоянии
при равновесном расстоянии между атомами. Для четырех положений, обсуждаемых в
этом параграфе, результаты Эветта и Маргенау, обозначаемые индексом Э. М., отличаются от
результатов де Бура следующими числовыми множителями:
Положение abed
4BM>(iBaJb) 0,3 1,0 1,8 1,8
Оба ряда валентных энергий одинаково зависят от межмолекулярного расстояния. В работах
Эветта и Маргенау [80, 81] исправлена допущенная в работах Маргенау [82] числовая
ошибка, которая приводит к неправильным результатам.
Таблица 149
Теоретические значения энергии взаимодействия двух молекул водорода
Положение а
d/J^ &ab — Ocd ea 0
по
5,0
5,5
6,0
6,5
7,0
7,5
8,0
8,5
9,0
9,5
10,0
А
2,65
2,91
3,18
3,44
3,70
3,97
4,23
4,50
4,76
5,03
5,29
,(»¦"•>
289,0
113,5
44,55
17,49
6,87
2,70
1,058
0,415
0,163
0,064
0,025
285,5
78,7
24,68
8,63
3,30
1,37
0,604
0,283
0,139
0,072
0,038
16,3
10,2
6,57
4,40
3,04
2,15
1,559
1,151
0,865
0,660
0,511
^(дисп., 6)
-38,8
-21,9
-12,99
-8,04
-5,15
-3,41
-2,312
-1,607
-1,140
-0,824
-0,606
^(дисп., 8)
-12,9
-6,0
-3,01
-1,59
-0,877
-0,505
-0,301
-0,186
-0,117
-0,076
-0,051
*1
253,6
95,8
35,12
12,26
3,88
0,94
0,004
-0,227
-0,229
-0,176
-0,121
250,1
61,0
15,25
3,40 ,
0,31
-0,40
-0,450
-0,359
-0,253
-0Д6&
-одоа
Положение Ь
0, 0cdt = у ,
7
по
5,0
5,5
6,0
6,5
7,0
7,5
8,0
8,5
9,0
9,5
10,0
А
2,65
2,91
3,18
3,44
3,70
3,97
4,23
4,50
4,76
5,03
5,29
75,3
29,9
11,83
4,68
1,85
0,729
0,288
0,113
0,045
0,018
0,007
102,2
31,8
11,07
4,23
1,75
0,775
0,364
0,179
0,092
0,049
0,027
-8,2
-5,1
-3,29
-2,20
-1,52
-1,076
-0,780
-0,576
-0,433
-0,330
-0,255
^(дисп., 6)
-31,6
-17,8
-10,57
-6,54
-4,19
-2,770
-1,880
-1,307
-0,928
-0,671
-0,493
^(дисп., 8)
-12,9
-6,0
-3,01
-1,59
-0,877
-0,505
-0,301
-0,186
-0,117
-0,076
-0,051
*1
22,6
1,0
-5,04
-5,65
-4,74
-3,622
-2,673
-1,956
-1,433
-1,059
-0,792
9ц
49,5
2,9
-5,8
-6,10
-4,84 •
-3,576
-2,597
-1,890
-1,386
-1,028
-0,772
828
Продолжение табл. 149
Положение
во* — ecd = -5-» 0ab — 0л* ¦* 0
а0
5,0
5,5
6,0
6,5
7,0
7,5
8,0"
3,5
9,0
9,5
10,0
А
2,65
2,91
3,18
3,44
3,70
3,97
4,23
4,50
4,76
5,03
5,29
„(вал.)
35,8
14,2
5,65
2,24
0,887
0,351
0,139
0,054
0,022
0,009
0,003
„(вал.)
36,0
12,9
5,06
2,13
0,956
0,453
0,225
0,117
0,063
0,035
0,020
„«?> <?>
6,1
3,8
2,46
1,65
1,140
0,807
0,584
0,432
0,324
0,248
0,192
^(дисп., 6)
-27,5
-15,5
-9,20
-5,69
-3,647
-2,411
-1,637
-1,138
-0,807
-0,584
-0,429
^(дисп., 8)
-12,9
-6,0
-3,01
-1,59
-0,877
-0,505
-0,301
-0,186
-0,117
-0,076
-0,051
v\
1,5
-3,5
-4,10
-3,39
-2,497
-1,758
-1,215
-0,838
-0,578
-0,403
-0,285
*н
1,7
-48
-4,69
-3,50
-2,428
-1,656
-1,129
-0,775
-0,537
-0,377
-0,268
Положение d
1
а*
5,0
5,5
6,0
6,5
7,0
7,5
в,о
8,5
9,0
9,5
10,0
А
2,65
2,91
3,18
3,44
3,70
3,97
4,23
4,50
4,76
5,03
5,29
„(вал.)
35,2
14,0
5,58
2,22
0,879
0,348
0,138
0,055
0,021
0,008
0,003
34,8
12,6
4,98
2,11
0,949
0,451
0,224
0,116
0,062
0,035
0,020
„(<?> Q)
2,0
1,3
0,82
0,55
0,380
0,269
0,195
0,144
0,108
0,083
0,064
^Дисп., 6)
-26,8
-15,1
-8,99
-5,56
-3,564
-2,356
-1,599
-1,112
-0,789
-0,570
-0,419
^(дисп., 8)
-12,9
-6,0
-3,01
-1,59
-0,877
-0,505
-0,301
-0,186
-0,117
-0,076
-0,051
*1
-2,5
-5,8
-5,60
-4,38
-3,182
-2,244
-1,567
-1,099
-0,777
-0,555
-0,403
<рп
-2,9
-7,2
-6,20
-4,49
-3,112
-2,141
-1,481
-1,038
-0,736
-0,528
-0,386
829
830 Гл. 14. Квантовомеханические расчеты межмолекулярных сил
представлена в виде
2Д8 il fe-1,87 гас/ао _|_ е-1,87 г*/* -f ?^Х'87 '•»/* + е~!'87 ™/*о] . D.2)
Однако это уравнение применять не всегда удобно, поскольку зависимость
у(вал.) от уГЛ0В выражена не явно через четыре межъядерных расстояния. При-
нимая это во внимание, де Бур предложил для формулы D.2) апроксимирующее
выражение, справедливое в области межмолекулярных расстояний от 6а0 до 8а0.
Эта область расстояний представляет наибольший интерес для столкнове-
ний с относительно малой кинетической энергией, которые определяют коэф-
фициенты переноса и вириальные коэффициенты. Апроксимирующее выраже-
ние де Бура для формулы D.2) имеет вид
ал.) = j^_ j4800 ^у + 129 3б0 JAJ13 [ 13 cos2 ваЬ + 13 cos2 6cd - 2] +
+ 412 010(^-I5A - 15cos20aft- 15cos20cd- 195 cos2 0a6 cos2 0cd +
+ 2 [sin ваЬ sin dcd cos (фаЬ - фсй) - 14 cos ваЬ cos 0cd]2) +
+ 68640 [?ff F - 90 cos2 ваЬ - 90 cos2 dcd + 255 cos4 ваЬ + 255 cos4 0cd)}. D.3)
В этой формуле углы 6ab и 0cd соответствуют углам ва и вь на фиг. 192 (стр. 653),
а г — расстояние между центрами двух молекул. К сожалению, зависимость
формулы D.3) от углов очень сложная.
Чтобы получить грубую картину зависимости энергии взаимодействия от
ориентации молекул, рассмотрим четыре основные ориентации молекул.
В табл. 149 приведены значения (рA\ал) и g>(Fiaj° для этих четырех конфигураций.
Производя усреднение <р<вал-> по всем возможным пространственным ориен-
тациям двух молекул (см. гл. 13, § 5), получаем
-(вал.) = ^ |-48 000 (-^)U + 862 400 (-^I8 + 8 788 200 f^)"
Членом, содержащим 1/fcT, можно пренебречь, так как прямые вычисления
показывают, что его величина составляет менее 2% от первого члена.
Б. ЭНЕРГИЯ ВЗАИМОДЕЙСТВИЯ НА БОЛЬШИХ РАССТОЯНИЯХ
Энергия взаимодействия двух молекул водорода на больших расстояниях
равна сумме трех членов yWiQ), <р(дисп) и <р(дисп.,8)#
1. Квадруполь-квадрупольное взаимодействие. Энергия квадруполь-квад-
рупольного взаимодействия определяется формулой C.10) гл. 1 через квадру-
польные моменты нормальных молекул водорода
^н, = г^н-47| + 47?; D.5)
здесь г| — средний квадрат z-координаты (отсчитываемой от центра молекулы
вдоль оси) одного из электронов молекулы, усредненной с помощью функции
распределения электронной плотности. Аналогичным образом xf — средний
квадрат координаты одного из электронов в направлении, перпендикулярном
межъядерной оси. Расстояние между двумя атомами водорода обозначается
через гнн. Очень точное вычисление qHt было сделано Джеймсом и Кули джем
[83 ], которые применили свою тринадцатичленную волновую функцию. Полу-
ченный ими результат имеет вид
qHt = [0,248 + 0,172 [^ - 1,4) —0,115(-^ - 1,4)*]. 10-16о<2. D.6)
§ 4. Взаимодействие двух молекул водорода 831
Поскольку равновесное расстояние Н — Н в молекуле водорода равно 1,4а0,
то теоретическое значение квадрупольного момента для водорода равно
qHt= 0,248-10"6 см2. Эта величина согласуется с экспериментальным значением
Харрика и Рамзея (см. примечание к табл. 134, стр. 786) </н, =? 0,220- 10~1в см2.
Кроме того, расчеты для #н, были проделаны Коулсоном [84 ], использо-
вавшим для расчета Н2 метод самосогласованного поля, и Месси и Букингемом
[85 ], применившими собственные функции типа Ванга. Они получили соответ-
ственно значения 0,325-Ю6 и 0,141-Ю6 см2. Большая . разность между
различными рассчитанными значениями показывает, что величина квадруполь-
ного момента очень чувствительна к распределению электронной плотности.
Значения <p((?>Q) для четырех различных ориентации молекул, вычисленные
с помощью квадрупольного момента, найденного Джеймсом и Кулиджем, при-
ведены в табл. 149. Если <p(Q,Q) усреднить по всем возможным пространственным
рриентациям молекул, то, как объяснено в гл. 13, § 5, величина оказывается
равной нулю.
2. Дисперсионная энергия, обратно пропорциональная шестой степени
расстояния. Вычисление дисперсионной энергии, обратно пропорциональной
шестой степени расстояния, усложняется вследствие малой несферичности
молекул водорода. Для решения этой задачи де Бур [79 ] использовал выра-
жение D.2) гл. 13, предположив, что основные колебательные частоты, соотбет-
ствующие перпендикулярным и параллельным относительно молекулярной
оси колебаниям, равны1). С помощью этого выражения дисперсионную энергию
^(дисп.,6) можно определить через среднюю по ориентациям энергию ^(дисп.,6)
и анизотропию поляризуемости
Jb*L D.7)
При вычислении табл. 149 принималось, что « = 0,1173. Такая вели-
чина к следует из теоретических расчетов Ишигуро, Араи, Котани и
Мицушимы (см. гл. 13, § 2). Средняя дисперсионная энергия представляет
собой энергию взаимодействия, как если бы молекулы были сферическими.
Наиболее надежно вычисление средней дисперсионной энергии выполнено
Маргенау [80, 81 ]. Он идеализировал задачу, предположив, что только два
перехода определяют дисперсионную энергию. Это хорошее приближение,
поскольку Вольф и Герцфельд [75 ] показали, что экспериментальную зависи-
мость показателя преломления водорода от частоты можно с большой степенью
точности объяснить, используя двухчленную дисперсионную формулу. Марге*
нау воспользовался значениями сил осциллятора и энергий, связанных с силами
осциллятора ' [формула C.8) ], эмпирически найденными Вольфом и Герц-
фельдом. Подставляя эти значения сил осцилляторов и энергий в уравне-
ние C.20) гл. 13, Маргенау получил следующее выражение для средней диспер-
сионной энергии:
il |^je D.8)
Числовой множитель 10,9 сравним со значением 14,2, найденным де Буром с
помощью одночленного приближения [уравнение C.28) гл. 13 ]. В табл. 149
х) Месси и Букингем [85 ] рассчитали дисперсионную энергию для молекулы водорода
вариационным методом с помощью волновых функций типа Ванга. Рассчитанная ими вели-
чина больше предлагаемой нами от 1,4 до 1,7 раза. Маргенау [80,81 ] обратил внимание, что
вариационный метод Хассе, использованный Месси и Букингемом, обычно приводит к слиш-
ком большим энергиям взаимодействия. Зависимость потенциала Месси и Букингема от углов
значительно отличается от соответствующей зависимости в выражении D.2) гл. 13. Букингем
(частное сообщение, ноябрь 1950 г.) указал на небольшую ошибку в числовых коэффициен-
тах в выражении для дисперсионной энергии, полученной Месси и Букингемом с помо-
щью функций типа Ванга (но не Коулсона).*
832
Гл. 14. Квантовомеханические расчеты межмолекулярных сил
приведены значения ^(дисп.,6) ддЯ четырех основных ориентации двух молекул,
а в табл. 150 — значения средней дисперсионной энергии.
Таблица 150
Экспериментальные и теоретические значения энергии взаимодействия
двух молекул водорода
(Энергии выражены в единицах 10—" эрг)
1
во
5,0
5,5
6,0
6,5
7,0
7,5
8,0
8,5
9,0
9,5
* 10,0
А
2,65
2,91
3,18
3,44
3,70
3,97
4,23
4,50
4,76
5,03
5,29
86,2
26,9
9,46
3,65
1,53
0,682
0,323
0,160
0,083
0,045
0,025
-(дисп., б)
-30,4
-17,2
-10,18
-6,30
-4,04
-2,669
-1,812
-1,259
-0,894
-^0,646
-0,475
-(дисп., 8)
-12,9
-6,0
-3,01
-1,59
-0,877
-0,505
-0,301
-0,186
-0,117
-0,076
-0,051
-(полн.)
42,9
3,7
-3,73
-4,24
-3,39
-2,492
-1,790
-1,285
-0,928
-0,677
-0,501
-(полн.)
экс.
31,4
0,8
-4,84
-4,81
-3,77
-2,762
-1,991
-1,436
-1,043
-0,766
-0,569
3. Дисперсионная энергия, обратно пропорциональная восьмой степени
расстояния. Доля дисперсионной энергии в полной энергии взаимодействия
двух молекул водорода настолько мала, что нет необходимости определять
зависимость ее от ориентации молекул. Маргенау [80, 81 ] применил значения
сил осциллятора и энергий, найденных Вольфом и Герцфельдом [75 ] для рас-
чета дисперсионной энергии, обратно пропорциональной восьмой степени рас-
стояния.
Однако он не применял специальной двухчленной формулы, а, взяв средние
значения от /х и /2 и Ег и Е2У воспользовался приближением, соответствующим
одному осциллятору [формула C.37) гл. 13], и получил
D.9)
(дисп.,8) = _ 116 il
ф
В. ПОЛНАЯ ЭНЕРГИЯ ВЗАИМОДЕЙСТВИЯ И СРАВНЕНИЕ
С ЭКСПЕРИМЕНТОМ
Полная энергия взаимодействия для четырех основных ориентации моле-
кул дана в табл. 149. В соответствии с двумя теоретическими выражениями
для валентной энергии [формулы D.2) и D.3) ] получаются два ряда значений
полной энергии. Поскольку формула D.3) была определена де Буром из усло-
вия лучшего совпадения с кривой <р(валЛ при расстояниях г от ба0 до 8а0, то
полная энергия q>Y будет более надежна, чем q>u. Эти потенциальные кривые
изображены на фиг. 231. Полные энергии хорошо согласуются друг с другом
в случае ориентации Ь, с и d и не согласуются в случае ориентации а. Принци-
пиальное преимущество выражения сри для полной энергии заключается в том,
что в этом случае полная энергия выражается в виде степеней синусов и коси-
нусов различных углов1). Для потенциала в такой форме легко рассчитать
среднее по ориентации значение дисперсионной энергии; результаты такого
х) Для некоторых теоретических целей желательно найти энергию взаимодействия в
виде простой функции от ориентации молекул. Принимая это во внимание, Ван Чанг [86]
предложила следующую апроксимацию потенциала де Бура:
где
<р(полн.) = [а + Р (COS2 Sab + COS2 вы)] Г2 - СГ-*,
а = 4,438-10-9эрг. А12; Р = 4,784- 10-»эрг. А12; с = 1,25- К)-11 эрг. А«.
§4. Взаимодействие двух молекул водорода
833
расчета приведены в табл. 150. Труднее рассчитать среднее значение (рг.
Из табл. 149 легко определить относительную долю различных типов энергий
взаимодействия. Из та блицы видно, что как квадруполь-квадрупольное взаимо-
4
4,76
Фиг. 231. Сравнение теоретических потенциалов <pt и ^ для различных ориентации,
а также <рд (теор.) с экспериментальной энергией взаимодействия двух молекул водорода.
действие, так и дисперсионная энергия, обратно пропорциональная восьмой сте-
пени расстояния, вносят малый, но вполне заметный вклад в полную энергию
взаимодействия. При очень больших расстояниях (г> 1 \а0) энергия квадруполь-
квадрупольного взаимодействия становится преобладающей, и полная энергия
для одних ориентации будет положительная, а для других — отрицательная.
Надежное экспериментальное значение энергии взаимодействия полу-
чается из сравнения потенциала Леннарда-Джонса с экспериментальными дан-
ными по сжимаемости с учетом поправки на квантовые эффекты [87 ]
(см. гл. 6, § 5). Таким путем найдено, что постоянные в потенциале F-12)
Леннарда-Джонса равные = 5,107 • 10~15 эрг и а = 2,928 А. Экспериментальные
и теоретические потенциалы сравниваются в табл. 150 и на фиг. 231. Согла-
сие очень хорошее.
834
. Гл. 14. Квантовомеханические расчеты межмолекулярных сил
§ 5. ВЗАИМОДЕЙСТВИЕ АТОМОВ ИЛИ МОЛЕКУЛ ВОДОРОДА
С РАЗЛИЧНЫМИ ИОНАМИ ВОДОРОДА
Взаимодействие атомов или молекул водорода с различными ионами слу-
жит иллюстрацией сил взаимодействия между ионами и нейтральными моле-
кулами. В этом параграфе рассматривается взаимодействие Н или Н2 с Н+
или Н". Дополнительно к этому обсуждаются столкновения Н2 с Н и вероят-
ность образования ионной группы.
А. ВЗАИМОДЕЙСТВИЕ Н + Н+
Взаимодействие протона с атомом водорода с образованием Щ является
одной из немногочисленных проблем квантовой механики молекул, которая
допускает точное решение. Уравнение Шредингера разделяется как в эллипти-
ческих, так и в параболических координатах; получающиеся при этом одно-
мерные линейные обыкновенные дифференциальные уравнения могут быть
решены методом последовательных приближений с любой желаемой степенью
точности. Наиболее точные вычисления для основного состояния были выпол-
нены Хиллераасом [88], Сандеманом [89], Хеллмигом [90] и Джонсоном [91 ].
Энергии многих возбужденных состояний были вычислены Теллером [92].
8 .9
Фиг. 232. Энергия основного состояния (\sa) иона Н\, рассчитанная Хиллераасом [88],
и энергия возбужденных состояний, рассчитанная Теллером [92].
На фиг. 232 изображены потенциальная кривая основного состояния (lscr),
рассчитанная Хиллераасом, и кривые возбужденных состояний, рассчитанные
Теллером. Спектр HJ не наблюдался, но Бейтлер и Юнгер [93] определили
энергию диссоциации основного состояния Н? по энергии связи и потенциалу
ионизации молекулы водорода. Найденное ими экспериментальное значение
§ 5. Взаимодействие атомов или молекул водорода с различными ионами водорода 835
61,07 ккал/моль очень хорошо согласуется с теоретическим значением энергии1
диссоциации 61,01 ккал/моль. Равновесное расстояние между атомами в Н?
равно 1,06 А.
Если довольно медленный протон сталкивается с атомом водорода, находя-
щимся в нормальном состоянии Is, то с равной 'вероятностью может образо-
ваться как состояние lscrBZ^), так и состояние 2poBZ+). Состояние 2ра на
очень больших расстояниях обладает отрицательной энергией, соответствую-
щей притяжению, однако при расстояниях, меньших 5,9 А, энергия этого
состояния положительна. Энергия взаимодействия протонов с атомами водо-
рода, находящимися в различных состояниях, для частного случая больших
расстояний рассмотрена в гл. 13, § 6.
Б. ВЗАИМОДЕЙСТВИЕ Н + Н~
В результате взаимодействия атома водорода с Н~ может образоваться
ион Н^. Однако ион Н^ никогда еще не наблюдался, и кажется невероятным,
чтобы он когда-нибудь наблюдался, Q
ибо по расчетам Эйринга, Гирш-
фельдера и Тэйлора [94] ион Н^г 2д
обладает минимальной энергией
—37,4 ккал/моль при межъядерном g 40
расстоянии 1,8А. Однако потенциал ь- |
ная кривая для Н2 плюс электрон ^ $0
пересекает кривую для Н^" как раз <|
вблизи этой точки (фиг. 233). Таким § 80
образом, столкновение между Н~ и °*
атомом водорода приводит к образо- юо
ванию молекулы
электрона.
Н» и свободного
г
\
\
/
г-
с
1нг+
„—'
у
элею
прон
— ¦
— -
В. ВЗАИМОДЕЙСТВИЯ
Н + Щ, Н2 + Н+ и Н2 + Щ
120
Q5 40 15 2fl 2,5 3ft 3,5 4ft 4,5
Ядерное расстояние, А
Фиг. 233. Потенциальные кривые Наи Н^ [94].
При взаимодействиях Н + Н?, Н2+ Н+ и Н2 + НJ образуется ион Щ. Этот
трехатомный ион водорода наблюдается при масс-спектроскопических иссле-
дованиях, если водородный газ ионизуется при давлениях, достаточно высоких,
чтобы произошли столкновения Щ с молекулами водорода [95—100]. Факти-
чески оказывается, что почти при каждом столкновении происходит реакция
H+lUvU+lU /С 1 \
2 + П2-*Н3 + Н • (Э.1)
Ион Нд может также образоваться и при столкновении Н+ с На или
Н а с Н. Попытки обнаружить спектр Нд до сих пор не увенчались успехом.
Квантовомеханические вычисления энергии Н? были выполнены с помощью
водородоподобных волновых функций ls-состояния, взятых во многих комби-
нациях и с варьирующимися параметрами [69, 71—73, 101 ]. Иой Н? очень
устойчив и обладает энергией, лежащей более чем на 184 ккал/моль ниже
уровня энергии, соответствующего двум разделенным атомам водорода и
протону. Таким образом, химическая реакция E.1), несомненно, изотерми-
ческая и сопровождается выделением более 11 ккал/моль, а вероятнее всего
38 ккал/моль1). Трехатомный ион водорода обладает устойчивой конфигурацией,
х) Величина 11 ккал/моль получается при сравнении рассчитанной энергии Н^" сточным
значением энергии Н2 и Н. Поскольку расчеты Н+ выполнены вариационным методом, то
значение 11 ккал/моль должно быть нижним пределом. Величина 38 ккал/моль получается
при сравнении рассчитанной энергии Н8+ со значением энергии Н2 и Н, полученным с по-
мощью тех же самых приближенных волновых функций, что и для Н^. Поскольку не из-
вестно, превышает ли ошибка при вычислении энергии Н2 ошибку при вычислении Н+,
то не известно, занижена или завышена величина 38 ккал/моль.
836
Гл. 14. Квантовомеханические расчеты межмолекулярных сил
в которой атомы расположены приблизительно по углам равностороннего
треугольника, а расстояние между атомами приблизительно равно 1,79 А.
По-видимому, две колебательные частоты лежат в инфракрасном спектре в
области волнового числа 1100 см*1 и могут экспериментально наблюдаться.
Энергия притяжения протона и молекулы водорода схематически изобра-
жена на фиг. 234. Сплошные линии дают энергию системы, когда положение
молекулы водорода фиксировано (атомы водорода закреплены на равновесном
-во
ккал
-60
-40
Стабильная
конфигурация
н3+
Фиг. 234. Энергия взаимодействия протона и молекулы водорода [72].
расстоянии), а протон проходит все положения. Черные круги, размеры кото-
рых — порядка боровского радиуса, обозначают положение молекулы водо-
рода. Заштрихованная область соответствует энергетически труднодостижи-
мым конфигурациям. На больших расстояниях энергия притяжения протона
и молекулы водорода определяется дисперсионной формулой вида
E.2)
здесь ан, — поляризуемость молекулы Н2, числовые значения которой в соот-
ветствии с табл.113 (стр. 727) заключены в пределах от 0,6968-Ю4 смг
(когда протон движется перпендикулярно к оси молекулы) до 0,9746- Ю4 см*
(когда протон движется параллельно оси молекулы); гн+,н2 — расстояние
между протоном и центром тяжести молекулы водорода. Величины чистого
электростатического взаимодействия, определяемые формулой E.2), соответ-
ствуют линиям —10, —20 и —40 жал/моль. При больших сближениях стано-
вятся важными химические силы.
Г. ВЗАИМОДЕЙСТВИЕ Н2 + Н~
Можно ожидать, что при взаимодействии молекулы водорода с ионом
Н~ образуется ион Н^. Однако ион Н^ еще никогда не наблюдался. С помощью
вариационного квантовомеханического расчета [71 ] показано, что на больших
расстояниях ион Н~ притягивается к Н2, а на малых расстояниях наблюдается
отталкивание, так как ион Н^ диссоциирует на Н2 + Н~* и при этом освобож-
§ 5. Взаимодействие атомов или молекул водорода с различными ионами водорода 837
дается энергия, равная 65,0 ккал\моль. Таким образом, ион Нз по своему пове-
дению очень похож на ион Н^. Рассмотрение, основанное на концепции энтро-
пии, также приводит к необходимости разложения Н^. Таким образом (пред-
полагая, что частота колебаний Н2 относительно Н"~ в волновых числах состав-
ляет 100 см'1 и в равновесии расстояние между Н" и Н2 равно 2,8 А), можно
показать [71 ], что равновесная концентрация Нд определяется выражением
= 0,2 рн.. E.3)
Квадратные скобки означают концентрацию, а рн, — давление водорода в ат-
мосфере.
Д. ГРУППА ИОНОВ1)
Основную проблему группирования ионов можно сформулировать в тер-
минах равновесной реакции
А + В+ 2 ЛВ+ , E.4)
для которой постоянная равновесия К может быть записана с помощью пар-
циальных функций z для различных компонент
[АВ+] _к__
Если г — расстояние между ионами В+ и молекулой, то
где
здесь а — поляризуемость; /л — дипольный момент молекулы А (см. гл. 13,
§ 5). Метод расчета парциальных функций изложен в гл. 2, § 5. Парциальная
функция группы равна
2 кТ )
za zb W2 Г 2 кТ ) L Л5 J 9
здесь САВ+ = r2mAmBl(mA + mB); vAB+ — основная колебательная частота
молекулы АВ+ (рассматриваемой как двухатомная молекула). Из уравнений
E.5), E.7) и уравнений для определения парциальных функций Аи В+ можно
показать
К = 3,822 ГМ* + М*1* *""%гг молъ1см* . E.8)
' L MaMb J sh(hvAB+/2kT) ' v '
В этом выражении МА и Мв — молекулярные веса ; а выражено в А3; г — рас-
стояние в А. Для частоты vAB+ наиболее вероятное значение равно 100 смг1,
поэтому при 300° К sh(ft vAB+/2kT) = 0,2403. Если парциальное давление А
равно рА атму то концентрация А при 300° К равна 4,0600- 10~б рА моль/см3.
Таким образом, считая, что vAB+ = 100 слг\ а температура равна 300° К,
уравнение E.8) можно записать в виде
. [94].
838 Гл. 14. Квантовомеханические расчеты межмолекуяярных сил
Для ионных групп водорода в предположении, что г = 2,8 А и а = 0,8, из
уравнения E.9) следует
[Н+]2 =0,186 рн. атм, E.10)
Г""+1 E.11)
Дополнительные расчеты показывают, что при атмосферном давлении
вероятность образования группы из двух молекул и иона водорода приблизи-
тельно равна квадрату вероятности образования группы из одной молекулы
и иона.
§ 6. ВЗАИМОДЕЙСТВИЕ АТОМА ГЕЛИЯ С ВОЗБУЖДЕННЫМ АТОМОМ
ГЕЛИЯ ИЛИ G ПРОТОНОМ
В результате взаимодействия нормального атома гелия с возбужденным
атомом гелия или с протоном образуются двухатомные молекулы. В этом
параграфе показано, как «инертные газы» могут образовывать химические ком-
бинации. Двухатомные молекулы и ионы, обсуждаемые здесь, обладают боль-
шим временем жизни и их необходимо учитывать в теории электрических раз-
рядов или других мощных способов возбуждения.
А. ВЗАИМОДЕЙСТВИЕ НОРМАЛЬНОГО И МЕТАСТАБИЛЬНОГО
АТОМОВ ГЕЛИЯ
В результате взаимодействия нормального атома гелия с возбужденным
атомом гелия, находящимся в первом триплетном или синглетном метастабиль-
ном состоянии, образуется сильносвязанная двухатомная молекула. Этот тип
связи часто называют «ван-дер-ваальсовой», или «поляризационной», связью
[102], хотя это, как будет сейчас показано, и не совсем правильно. Энергию
взаимодействия можно представить в виде суммы короткодействующей «валент-
ной энергии» д>вал- и дисперсионной энергии д>дисп-.
Букингем и Далгарно [78 ] по методу Гайтлера—Лондона рассчитали в
первом порядке теории возмущений валентную энергию взаимодействия в
области расстояния от а0 до 12а0. Для изолированных атомов гелия они
приняли аналитические волновые функции типа Морзе—Юнга—Гаурвица
[103]. Таким образом, координатная часть волновой функции нормального
атома гелия бралась в виде
(i|^ ) F.1)
а координатная часть волновых функций триплетного и синглетного состояний
возбужденного атома — в виде
<W».2) = Мч~~ 2'93415)ехр (" 1'22^~-°'61^)> <6-2>
где Nu и N12 — нормировочные множители. Возбужденными состояниями
атома гелия, соответствующими конфигурациям Is2p, пренебрегалось, так как
эти энергетические состояния лежат более чем на 1 эв выше 1$2$-состояний.
Букингем и Далгарно вычислили все интегралы, появляющиеся в первом
порядке теории возбуждений. Полученные ими результаты приведены в
табл. 151.
§6. Взаимодействие атома гелия с возбужденным атомом гелия или с протоном 839
Таблица 151
Энергия взаимодействия между нормальным (lsls) атомом гелия и метаста-
бильным (\s2s) возбужденным синглетным или трип летным атомом гелия
Tab
во
1.0
2,0
2,5
3,0
4,0
5,0
6,0
7,0
8,0
9,0
10,0
11,0
12,0
'Zu
+29800,0
-1120,0
-847,3
-61,0
+451,1
+266,3
+ 130,8
+58,0
+24,8
+ 10,0
+3,5
+1,3
+0,4
„(вал.) t
10-"эрг
67600,0
10070,0
4385,0
2310,0
920,9
351,3
144,7
60,6
25,3
10,0
3,5
1,3
0,4
+26800,0
-1918,0
-1440,0
-308,6
+400,1
+259,8
+ 130,8
+58,4
+24,8
+ 10,0
+3,5
+ 1,3
+0,4
51100,0
9819,0
4472,0
2521,0
977,6
359,1
145,1
60,1
25,3
10,4
3,5
1,3
0,4
10-w эрг
-202,1
-53,0
-17,7
-7,0
-3,14
-1,57
-0,83
-0,48
-0,27
Дисперсионная энергия была вычислена по формуле Букингема [46 ]
9
F.3)
здесь г? — средний квадрат радиуса z'-ro электрона атома a; rj — средний
квадрат радиуса /-го электрона атома Ь.
Усреднение проведено по всем соответствующим распределениям электрон-
ной плотности. Применяя для атома гелия волновые функции Морзе—Юнга—
Гаурвица, Букингем и Далгарно нашли, что дисперсионная энергия имеет вид
?• (б-4)
Они вычислили также дисперсионную энергию, применяя самосогласованное
поле для распределения электронной плотности в нормальном и возбужденном
атомах, как это сделано Вильсоном и Линдсеем [104].
Вычисления по методу самосогласованного поля приводят к дисперсион-
ным энергиям, превосходящим в 1,5 раза значения энергии, полученные с
помощью волновых аналитических функций, определяемых выражением B.7).
Однако Букингем и Далгарно не считают вычисления по методу самосогласо-
ванного поля строгими, так как этот метод приводит к слишком большому
растяжению 2$-орбиты.
При расстояниях, меньших 4а0, дисперсионная энергия составляет малую
часть полной энергии, поэтому Букингем и Далгарно произвольно полагают
ее равной нулю. При расстояниях, больших 4а0, для получения полной энергии
взаимодействия они складывают валентную и дисперсионную энергии
ф = у>(вал.) _J_ ^(дисп.) ^ F.5)
Результаты показаны на фиг. 235. Состояния г?и и гЕи обладают мини-
мумами энергии при 2,1а0 и положительными максимумами соответственно в
6,7 и 6,0 ккал/моль при расстояниях около 4а0. Энергия диссоциации, вычис-
ленная для 327„-состояния, заключена приблизительно в пределах от 23 до
28 ккал/моль. Это значение энергии сравнимо с величиной 51,5 ккал/моль при
расстоянии 2,0а0, определенной Мюлликеном из экспериментальных данных.
Вычисленное значение энергии диссоциации для ^„-состояния заключено в
840
Гл. 14. Квантовомеханические расчеты межмолекулярных сил
пределах от 37 до 39 икал/моль, но не существует экспериментальных данных,
с которыми можно было бы сравнить это значение. Энергия 3?g- и ^-состояний
положительна при всех расстояниях, меньших 12а0. На достаточно больших
расстояниях энергия этих g-состояний обладает небольшим минимумом, однако
эта область лежит за пределами настоящих расчетов.
Горб на потенциальных кривых 327„- и ^-состояний напоминает кривые
потенциальной энергии, возникающие при резонансном взаимодействии, рас-
смотренном в гл. 13, § 6. Однако взаимодействие нормального атома гелия
с возбужденным (Is2s) атомом гелия не включает резонанса. Появление горба
должно быть связано с положительной валентной энергией при промежуточных
расстояниях.
5000
-2000 -
-зооо t
Фиг. 235. Кривые хЕи- и ^-состояний, образующихся при столкновении нормального
(lsls) атома гелия с метастабильным (Is2s) возбужденным синглетным атомом гелия.
Кривые %Еи - и *2?0-состояний образуются при столкновении нормального атома гелия с метастабильным
(Is2s) возбужденным триплетным атомом гелия [78].
Используя потенциальные кривые, изображенные на фиг. 235, и метод Месси
и Смита [106 ], Букингем и Далгарно [107 ] вычислили фазовые сдвиги, полное
эффективное сечение упругого рассеяния и эффективное сечение диффузии для
столкновения триплетного (Is2s) мета стабильного атома гелия с нормальным.
Эти вычисления приводят к заключению, что коэффициент диффузии триплет-
ных метастабильных атомов, проходящих через нормальный гелий при полном
давлении 1 мм рт. ст., равен ?D = 370 см2/сек при 300° К и Л) = 130 см2/сек
при 77° К. Впоследствии Фелпс и Молнар [108] измерили коэффициенты диф-
фузии при этих двух условиях и получили соответственно значения 410 и
130 см2/сек, что находится в очень хорошем согласии с теорией. Букингем и
Далгарно показали, что коэффициент диффузии для синглетных (Is2s) метаста-
бильных атомов, проходящих через нормальный гелий при температурах,
меньших 650° К, приблизительно равен коэффициенту диффузии для триплет-
ных атомов. При более высоких температурах синглетные атомы будут обла-
дать несколько меньшим коэффициентом диффузии. Эти расчеты являются
иллюстрацией того, как можно применять сложные потенциальные кривые,
обсуждаемые в этой главе, для определения макроскопических физических
свойств газов.
§ 6. Взаимодействие атома гелия с возбужденным атомом гелия или с протоном 841
Б. ВЗАИМОДЕЙСТВИЕ НОРМАЛЬНОГО АТОМА ГЕЛИЯ
С ПРОТОНОМ
В гл. 13, § б рассматривались дальнодействующие силы взаимодействия
атома гелия с протоном. Исследуем теперь короткодействующие силы взаи-
модействия. Энергия взаимодействия обладает глубоким минимумом, харак-
терным для стабильного двухатомного иона. Система НеН+ изоэлектронна с
молекулой водорода, однако благодаря разным зарядам двух ядер она более
сложная. Коулсон и Дункансон [109 ] провели довольно интенсивные изучения
основного состояния двухатомного иона. Они вычислили энергию связи с помо-
щью пятичленной волновой функции Джеймса и Кулиджа. Однако наинизшее
значение энергии они получили с помощью волновых ионо-полярных функций
типа Ванга. Пусть а соответствует атому гелия, а Ь — протону, тогда орбитали
электрона 1 в атоме гелия имеют вид
*'He'ei, (б.б)
^He гм 9 (б.7)
а орбиталь электрона вблизи протона имеет вид
; F.8)
здесь Zne, 2не и Zh — три различных эффективных ядерных заряда.
Поэтому пространственная часть ионо-полярной волновой функции Ванга
может быть выражена в виде
хР==Ца0A)Ь0B) + а0B)Ь0A)]+,га0A)а0B) +
+ v[ao(l)acB) + aoB)ac(l)]. F.9)
Первый член в формуле F.9) появляется вследствие гомеополярного взаимодей-
ствия электрона в атоме гелия с электроном в атоме водорода ; второй член
соответствует чисто ионному вкладу, когда оба электрона находятся в наиниз-
ших состояниях около атома гелия ; третий член соответствует состоянию,
в котором оба электрона находятся около атома гелия, причем один из них
поляризован в возбужденное состояние. Тот факт, что эффективные ядерные
заряды не равны действительным зарядам ядер, превращает выражение F.9)
в функцию типа Ванга. Особенность рассмотрения Коулсона—Дункансона
заключается в том, чю они вычислили все интегралы, появляющиеся в этой
задаче1). Они получили следующие значения энергии взаимодействия протона
и нормального атома гелия:
Расстояние, взаиТод^твия,
0 ккал/моль
1,25 —47,9
1,50 —53,3
2,00 —37,5
1,446 (равновесное) —53,6
При равновесном расстоянии постоянные в потенциальной функции равны
следующим значениям:
А = 0,71730, ГНе = 1,8900,
[л = 0,24843, ГНе= 1,5586,
v = 0,02979, Z'H = 1,5226.
х) Бич [ПО] вычислил энергию взаимодействия атома гелия с протоном, однако он не
достиг такой точности, как Коулсон и Дункансон, так как не вычислял обменные и кулонов-
ские интегралы с неравными эффективными ядерными зарядами.
842 Гл. 14. Квантовомеханические расчеты межмолекулярных сил
Энергия нулевых колебаний для НеН+ вблизи положения равновесия состав-
ляет 4,8 ккал1молъ. В случае взаимодействия протона с атомом гелия «на две
трети устойчивость двухатомного иона достигается за счет образования кова-
лентной связи и на одну треть — за счет поляризации атома Не» [109,110].
Коулсон и Дункансон провели также точное вычисление энергии взаимо-
действия Не+ с протоном. Энергия взаимодействия в этом случае всегда поло-
жительна, поэтому задача имеет незначительный физический интерес.
Приложение А
ИНТЕГРАЛЫ, ПРИМЕНЯЕМЫЕ ПРИ РАСЧЕТАХ ЭНЕРГИЙ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
Имеется много работ, в которых различные интегралы, применяемые при
вычислении энергий межмолекулярного взаимодействия, представлены в виде
функций от межатомных расстояний или табулированы при некоторых частных
значениях межатомного расстояния и других параметров. Ниже приводится
список некоторых из этих работ.
Одноцентровые и двухцентровые интегралы
1. Root ha an С. С, «Study of Two-Center Integrals Useful in Calculations on Molecular
Structure, b, Journ. Chem. Phys., 19, 1445 A951) (формулы).
2. RQdenbergK., «A Study of Two-Center Integrals Useful in Calculations on Molecular
Structure, II. The Two-Center Exchange Integrals», Journ. Chem. Phys., 19, 1459 A951)
(формулы).
3. С о u 1 s о n С. A., «Two-Center Integrals Occurring in the Theory of Molecular Structure»,
Proc. Cambr. Phil. Soc, 38 (Pt. II), 210 A941) (формулы).
4. P a r r R. G., С r a w f о r d B. L., «On Certain Integrals Useful in Molecular Orbital Cal-
culations*, Journ. Chem. Phys., 16, 1049 A948) (формулы).
5. H e 11 m a n n G., Einfuhrung in die Quantenchemie, Berlin, 1937 (формулы).
6. С о u 1 s о n С A. «Evaluation of Certain Integrals Occurring in Studies of Molecular Struc-
ture», Proc. Cambr. Phil. Soc, 33 (Pt. I), 104 A937) (теория).
7. С о ц 1 s о n С. A., D u n с a n s о n W. E., «Wave Functions for HeH++ and HeH+», Proc.
R6y. Soc, A165, 90 A938) (формулы для вычисления обменных интегралов в случае
разных зарядов).
Трехцентровые и четырехцентровые интегралы
8. Barnett М. Р., С о u I s о п С. A., «The Evaluation of Integrals Occurring in the Theory
of Molecular Structure. Parts I and II», Phil. Trans. Roy. Soc, A243, 221 A951) (вычи-
слены одно- и двухцентровые интегралы, а также трех- и четырехцентровые интегралы ;
изложена теория и приведены формулы).
9. Н i r s с h f e I d e r J. О., W е у g a n d t С. N., «Integrals Required for Computing the
Energy of H8 and H,+», Journ. Chem. Phys., 6, 795 A938) (таблицы).
10. Hirschfelder J. O., Eyring H., Rosen N., «I. Calculation of Energy of H3
Molecule», Journ. Chem. Phys., 4, 121 A936) (формулы).
11. Lundquist S. O., L б w d i n P. O., «On the Calculation of Certain Integrals Occurring
in the Theory of Molecules Especially Three-Center and Four-Center Integrals», Arkiv
fOr Fysik (Stockholm), 3, 147 A951) (теория и формулы).
12. R u d e n b e r g K., «On the Three- and Four-Center Integrals in Molecular Quantum
Mechanics», Journ. Chem. Phys., 19, 1433 A951) (теория).
13. Hirschf elder J. О., Диссертация, Princeton University A936) (табулированы
трехцентровые интегралы для трех атомов водорода, расположенных вдоль прямой
на равных расстояниях).
14. Н i r s с h f е 1 d e r J. О., Diamond Н., Е у г i n g H., «III. Calculation of the Energy
of H8 and H+», Journ. Chem. Phys., 5, 695 A937) (табулированы трехцентровые
интегралы для трех атомов водорода, расположенных антисимметрично относительно
некоторой линии).
Значения некоторых одно- и двухцентровых интегралов, наиболее часто
встречающихся при расчетах энергий межмолекулярного взаимодействия, при-
ведены в табл. 152 [28].
Таблица 152
q^zrat
0,0
1,6
1,5
1,75
2,0
2,5
3,0
3,5
4,0
4,5
5,0
6,0
7,0
8,0
10,0
12,0
Q-Zrab
0,0
1,0
1,5
1,75
2,0
2,5
3,0
3,5
4,0
4,5
5,0
6,0
7,0
8,0
10,0
12,0
0,0
1,0
1,5
1,75
2,0
2,5
и.
1,000000
0,858385
0,725173
0,6552725786
0,586453
0,458308
0,348509
0,259194
0,189262
0,136085
0,096577238
0,047096292
0,02218913
0,0101756997
L
0,002012730219
0,0003747969535
Z—Юи
1,000000
0,894694
0,804017
0,7583760654
0,714289
0,633281
0,563267
0,503922
0,453937
0,411796
0,3760937
0,3194558
0,2769694
0,244140805
0,197000003
0,164930556
0,391406
0,37433
0,360048
0,352943470
0,345879
0,331183
/cc
1,000000
0,735759
0,482519
0,3514849529
0,225559
0,005130
-0,159319
-0,264856
-0,318692
-0,332576
-0,3189295
-0,2503540
-0,1714338
-6,107191492
-0,034912546
-0,009652558
Z~lJo*
1,000000
0,735759
0,557826
0,4778783446
0,406006
0,287298
0,199148
0,135888
0,091578
0,061100
0,04042768
0,01735126
0,007295056
0,003019163651
0,0004993992274
0,00007987476059
0,825000
0,773402
0,719081
0,688560847
0,657139
0,594686
/и
2,000000
1,646880
1,308467
1,133534347
0,965275
0,668203
0,438045
0,274051
0,164675
0,095545
0,05376360
0,01574933
0,004235616
0,0010632933
0,0000570751761
0,0000026064767
±-Чсе
0,500000
0,306566
0,153402
0,08371921757
0,022556
-0,0701144
-0,124468
-0,147841
-0,149578
-0,138168
-0,1201601
-0,08055943
-0,04848173
-0,027116562
-0,007316955
-0,001729596
0,145833
0,123914
0,103043
0,0922190597
0,081725
0,062773
0,042188
0,01898
0,008325
0,00317153
-0,001432
-0,008268
1,000000
0,729329
0,583688
0,5239755402
0,472527
0,390567
0,330028
0,284542
0,249581
0,222071
0,19994552
0,16665949
0,142856193
0,124999873
0,0999999977
0,083333333
2~lJu
1,000000
0,778930
0,586554
0,4933150199
0,407421
0,264909
0,163102
0,095946
0,054308
0,029744
0,01583449
0,004179292
0,001021136
0,000234625836
0,0000107521224
0,000000427866198
Z-lKoe. t
0,072916
0,042717
0,018936
5 0,0086000949
-0,000030
-0,011818
0,500000
0,548950
0,536304
0,5172387306
0,492661
0,435666
0,378482
0,327590
0,284908
0,250068
0,22185876
0,180181700
0,151539775
0,1308487433
0,1029997191
0,0850694374
0,625000
0,554521
0,490338
0,4576042080
0,425974
0,368388
0,319804
0,279944
0,247554
0,221192
0,19956908
0,16659267
0,142844724
0,124997956
0,099999472
0,083333332
z-*L-t „
0,625000
0,436651
0,296835
0,23611543
0,184156
0,106622
843
Продолжение табл. 152
q^Zrab
3,0
3,5
4,0
4,5
5,0
6,0
7,0
8,0
10,0
12,0
q=Zrab
0,0
1,0
1,5
1,75
2,0
2,5
3,0
3,5
4,0
4,5
5,0
6,0
7,0
8,0
10,0
12,0
q=Zrab
0,0
1,0
1,5
1,75
2,0
2,5
3,0
3,5
4,0
4,5
5,0
6,0
7,0
8,0
10,0
12,0
0,314537
0,295358
0,274176
0,252151
0,2305073
0,1917270
0,1610067
0,137725185
0,106504961
0,087020932
0,536225
0,483922
0,438333
0,399107
0,365497910
0,311778878
0,271296778
0,239903208
0,194539121
0,163411431
Z~xLee се
0,391406
0,2035
0,093613
0,0675754
0,04395
0,043284
0,061445
0,075931
0,078505
0,070531
0,0569410
0,0294102
0,0121820
0,0043184
0,000392491
0,0000265330
Z-^o.. о.
0,047309
0,035359
0,026430
0,019880
0,01511295
.0,009097311
0,005801824
0,0039011236
0,0019998488
0,0011574032
0,825000
0,66567
0,51691
0,441865
0,37095
0,24907
0,15813
0,095715
0,055647
0,031249
0,017028
0,004688
0,001188
0,000283
0,0000137
0,000000568
Z—lL(a0, а0; а0, Ьо)
0,625
0,50704485
0,40536896
0,3552642088
0,30803646
0,22559548
0,16074246
0,1121558092
0,07698167
0,05215030051
0,034953043
0,015311456
0,006537867888
0,0027387379
0,0004610930366
0,000074658581
-0,011760
-0,012569
-0,011690
-0,010003
-0,008107280
-0,004846741
-0,002741192
-0,001544918
-0,000535527
-0,000216840
z-*LoifOt
0,145833
0,11119
0,08153
0,0674753
0,054758
0,034269
0,020260
0,011433
0,006206
0,003261
0,00166632
0,00040649
0,000092185
0,00001973
0,0000007952
0,00000002822
t-Wco
0,0
0,187988301
0,1816793613
0,1689187589
0,1538428958
0,1232108094
0,0967894319
0,07602178750
0,06029852650
0,04851960921
0,03966222452
0,02772640534
0,02040042496
0,01562384476
0,009999974833
0,00694444390^
-0,017301
-0,018497
-0,017316
-0,015112
-0,01268642
-0,008510270
-0,005669731
-0,003873036
-0,001998731
-0,001157364
0,042188
0,00845
-0,01550
-0,0246928
-0,03114
-0,03629
-0,03367
-0,02715
-0,01987
-0,01350
-0,008656
-0,003105
-0,0009711
-0,000275
-0,0000175
-0,00000089
Z-*L(a0, ае; а0, Ье)
0,07291666667
0,04063749
0,01559917
0,004775861
-0,004239108
-0,016473472
-0,021943231
-0,022678274
-0,020684210
-0,0174718272
-0,0140023272
-0,0080856288
-0,00424690746
-0,00209458666
-0,000450608960
-0,0000872422396
Z-Чоо оо
0,058508
0,030766
0,015627
0,007714
0,00371704
0,000814027
0,000167600
0,0000328960
0,00000113835
0,0000000354839
г-Ч-осое
2'-
0,072916
0,04625
-0,114956
-0,1348339
-0,144307
-0,138115
-0,112495
-0,082014
-0,055077
-0,034701
-0,0208652
-0,0066051
-0,0018605
-0,00047860
-0,000025982
-0,00000116873
-1Ца0, а,; по, ЬЛ)
0,145833
0,122314
0,10013
0,0887086
0,077650
0,057715
0,041534
0,029146
0,020042
0,010373
0,009057
0,003921
0,001653
0,000673
0,000108
0,000018
844
Задачи 845
Интегралы определяются посредством следующих функций :
(A.2)
at(l) bsB)= (^-)ralrft2sin0alsin0&2cos(^2-^)e-^, (А.З)
где ао{\), ac(l), as(\) - водородоподобные волновые функции с центром в атоме а;
1 относится к электрону. Аналогичные волновые функции можно написать
для атома Ь или электрона 2. Считая, что / или / означают о} с или s, интересую-
щие нас интегралы можно записать в виде
t (A.4)
(A.5)
(Ae6)
2O^rfrxft1, (A.7)
2)^dr1dr2, (A.8)
L(aifaJfakfbl)=\\ai(l)aJ(l)akB)blB)^-drldr29 (A.9)
— — uoc = J —— at1. (A.
Задачи
1. Вычислить энергию взаимодействия атома водорода с протоном, находящимся на
расстоянии 5 боровских радиусов, используя наилучшие волновые функции, для которых зна-
чения всех необходимых интегралов приведены в табл. 152.
2. Используя интегралы табл. 152, рассчитать энергию взаимодействия атома водорода
с атомом гелия как функцию расстояния. Применить водородоподобные атомные функции с
одинаковым эффективным зарядом на двух центрах.
846 Гл. 14. Квантовомеханические расчеты межмолекулярных сил
ЛИТЕР АТУРА
1. Van VI ее к J. H., Sherman A., Rev. Mod. Phys., 7, 167 A935).
2. L е п п а г d - J о п е s J. E., P о p 1 e J. A., Proc. Roy. Soc, 210A, 190 A952).
3. Lennard-Jones J. E., Journ. Chem. Phys., 20, 1024 A952).
4. Roothaan С. С, Rev. Mod. Phys., 23, 69 A951).
5. M о n t e t G. L., Keller S. P., Mayer J. E., Journ. Chem. Phys., 20, 1057 A952).
6. Per-Olov L6wdin, Journ. Chem. Phys., 19, 1570, 1579 A951).
7. G о m b a s P., Theorie und Lflsungsmethoden des Mehrteilchenproblems der Wellen-
mechanik, Basel, 1950.
8. G о m b a s P., Die Statistische Theorie des Atoms und ihre Anvendungen, Wien, 1949
(см. перевод: Гомбаш П., Статистическая теория атома и ее применения, ИЛ,
1951).
9. Gentile G., Zs. Phys., 63, 795 A930).
10. К о р i n е с k H., Zs. Naturforsch., 7a, 22, 314 A952).
И. Furry W., Bartlett J., Phys. Rev., 39, 210 A932).
12. J a m e s H., Journ. Chem. Phys., 2, 794 A934); Phys. Rev., 43, 589 A933).
13. С о u 1 s о n C, D u n с a n s о n W., Proc. Roy. Soc, A181, 378 A943).
14. Bartlett J., Furry W., Phys. Rev., 38, 1615 A931).
15. I r e 1 a n d C, Phys. Rev., 43, 329 A933).
16. Knipp J., Journ. Chem. Phys., 4, 300 A936).
17. Hutchisson E., Muskat M., Phys. Rev., 40, 340 A932).
18. J a m e s H., Journ. Chem. Phys., 3, 9 A935).
19. H e 11 m a n n H., Journ. Chem. Phys., 3, 61 A935) ; Acta Physicochimica (СССР), 1
913 A935).
20. Rosen N., Phys. Rev., 38, 255 A931).
21. Rosen N., Ikehara S., Phys. Rev., 43, 5 A933).
22. Kemble B.C., Zener С. М., Phys. Rev., 33, 512 A929).
23. Zener СМ., Kemble E. C, Phys. Rev., 34, 999 A929).
24. King G. W., Van Vleck J. H., Phys. Rev., 55, 1165 A939).
25. Co'olidge A. S., James H. M., Journ. Chem. Phys., 6, 730 A938).
26. Rosen N., Phys. Rev., 38, 2099 A931).
27. We in ba urn S., Journ. Chem. Phys., 1, 593 A933).
28. Hirschfelder J. O., Linnett J. W., Journ. Chem. Phys., 18, 130 A950).
29. Gurnee E. F., Magee J. L., Journ. Chem. Phys., 18, 142 A950).
30. P a u 1 i n g L., Wilson E. В., Introduction to Quantum Mechanics, New York, 1935.
31. Hasse H., Proc. Cambr. Phil. Soc, 26, 542 A930); 27, 66 A931).
32. К i r k w о о d J. G., Phys. Zs., 33, 57 A932).
33. P a u 1 i n g L., В е а с h J. Y., Phys. Rev., 47, 686 A935).
34. Slater J. C, Kirk wood J. G., Phys. Rev., 37, 682 A931).
35. Rydberg R., Zs. Phys., 73, 376 A931).
36. В e u 11 e r H., Zs. phys. Chem., B27, 287 A934).
37. M о г s e P., Phys. Rev., 34, 57 A929).
38. J a m e s H. M., С о о 1 i d g e A. S., Present R. D., Journ. Chem. Phys., 4, 187, 193,
A936).
39. Rosen P., Journ. Chem. Phys., 18, 1182 A950).
40. S1 a t e r J. C, Phys. Rev., 32, 349 A928).
41. Eisenschitz R., London F., Zs. Phys., 60, 491 A930).
42. M a r g e n a u H., Phys. Rev., 56, 1000 A939).
43. W h e e 1 e r J. A., Phys. Rev., 43, 258 A933).
44. Vinti J. P., Phys. Rev., 42, 632 A932).
45. Page С. Н., Phys. Rev., 53, 426 A938).
46. Buckingham R. A., Proc. Roy. Soc, A160, 94 A937).
47. Margenau R, Rev. Mod. Phys., 11, 1 A939).
48. de Boer J., Michels A., Physica, 5, 945 A938).
49.de Boer J., L u n b e с k R. J., Physica, 14, 510 A948)
50. Lunbeck R. J., Диссертация, Amsterdam, 1951.
Литература 847
51. de Boer J., van Kranendonk J., Physica, 14,442A948).
52. M a s о n E. A., Rice W. E., Journ. Chem. Phys., 22, January A954).
53. В u с к i n g h a m R. A., H a m i 11 о n J., M a s s e у H. S. W., Proc. Roy. Soc, A179,
103 A941).
54. Bleick W. E., Mayer J. E., Journ. Chem. Phys., 2, 252 A934).
55. В г о w n F. W., Phys. Rev., 44, 214 A933).
56. H i r s с h f e 1 d e r J. O., Bird R. В., S p о t z E. L., Journ. Chem. Phys., 16, 963
A948).
57. С о r n e r J., Trans. Farad. Soc, 44, 914 A948).
58. К u n i m u n e M., Progr. Theor. Phys. (Japan), 5, 412 A950).
59. Farkas A., Farkas L., Orthohydrogen, Parahydrogen and Heavy Hydrogen,
Cambridge, 1935.
60. A m d u r I., Journ. Chem. Phys., 11, 157 A934).
61. E у r i n g H., P о 1 a n у i M., Zs. phys. Chem., B12, 279 A931).
62. Hirschfelder J. 0., Eyring H., To pie у В., Journ. Chem. Phys., 4, 170
A936).
63. Hirschfelder J. 0., Journ. Chem. Phys., 9, 645 A941).
64. S u g i u r a Y., Zs. Phys., 45, 484 A927).
65. К a s s e 1 L. S., Kinetics of Homogeneous Gas Reactions, New York, 1932,
66. Voge H., Journ. Chem. Phys., 4, 581 A936).
67. Hulburt H. M., Hirschfelder J. O., Journ. Chem. Phys., 9, 61 A941).
68. Taylor R., .Proc. Phys. Soc, A64, 249 A951).
69. H i r s с h f e 1 d e r J. 0., Eyring H., Rosen N., Journ. Chem. Phys., 4, 121;
4, 130 A936).
70. Hirschfelder J. O., Stevenson D. P., Eyring H., Journ. Chem. Phys.,
5, 896 A937).
71. Stevenson D. P., Hirschfelder J. O., Journ. Chem. Phys., 5, 933 A937).
72. H i r s с h f e 1 d e r J, O., Journ. Chem. Phys., 6, 795 A938).
73. Hirschfelder J. O., Weygandt С N., Journ. Chem. Phys., 6, 806 A938).
74. Margenau H., Phys. Rev., 66, 303 A944); 64, 131 A943).
75. W о 1 f K. L., H e r z f e 1 d K. F., Handbuch der Physik, Vol. 20, 1928.
76. В a r n e 11 M. P., С о u 1 s о n С A., Phil. Trans. Roy. Soc, A243, 221 A951).
77. С о u 1 s о n С A., Valence, Oxford, 1952.
78. Buckingham R., Dalgarno A., Proc Roy. Soc, A213, 327 A952).
79. d e Boer J., Physica, 9, 363 A942).
80. Evett A. A., Margenau H., Phys. Rev., 90, 1021 A953).
81. Evett A. A., Margenau H., Journ. Chem. Phys., 21, 958 A953).
82. Margenau H., Phys. Rev., 63, 131, 385 A943).
83. J a m e s H. M., С о о 1 i d g e A. S., Astrophys. Journ., 87, 438 A938).
84. С о u 1 s о n С A., Proc. Cambr. Phil. Soc, 34, 204 A938).
85. Masse у H., Buckingham R., Proc Roy. Irisch Acad., A45, 31 A938).
86. W a n g - С h a n g, Диссертация, University of Michigan, 1944.
87. й e Boer J., Physica, 14, 139 A948).
88. Hylleraas E. A., Zs. Phys., 71, 739 A931).
89. S a n d e m a n J., Proc. Roy. Soc Edinburgh, 55, 72 A935).
90. H e 11 m i g E., Zs. Phys., 104, 694 A937).
91. J о h n s о n V. A., Phys. Rev., 60, 373 A941).
92. T e 11 e r E., Zs. Phys., 61, 458 A930).
93. В e u 11 e r H., J u n g e r H. O., Zs. Phys., 100, 80 A936).
94. Eyring H., Hirschfelder J. O., Taylor H.S., Journ. Chem. Phys., 4, 479
A936).
95. Smyth H. D., Rev. Mod. Phys., 3, 347 A931).
96. В 1 e а к n e у W., Phys. Rev., 40, 496 A932).
97. Bleakney W., Phys. Rev., 35, 1180 A930).
98. H о g n e s s T. R., L u n n E. G., Phys. Rev., 26, 44 A925).
99. В r a s e f i e 1 d С J., Phys. Rev., 31, 52 A928).
100. Dorse h K. E., Kallmann H., Zs. Phys., 53, 80 A929).
848 Литература
101. Н i r s с h f e 1 d e r J. О., Diamond Н., Е у г i n g H., Journ. Chem. Phys., 5, 695
A937).
102. Herzberg G., Spectra of Diatomic Molecules, New York, 2nd ed. 1950 (см. перевод:
1-го изд.: Герцберг Г., Спектры и строение двухатомных молекул, ИЛ, 1949).
103. Morse P. M., Young L. A., H a u r w i t z E. S., Phys. Rev., 48, 948 A935).
104. Wilson E. В., Lindsay R. В., Phys. Rev., 47, 681 A935).
105. M и 11 i k e n R. S., Rev. Mod. Phys., 4, 1 A932).
106. Masse у H. S. W., Smith R. A., Proc. Roy. Soc., A142, 142 A933).
107. Buckingham R. A., Dalgarno A., Proc. Roy. Soc, A213, 506 A952).
108. P h e 1 p s A. V., M о 1 n a r J. P., Phys. Rev., 89, 1202 A953).
109. С о и 1 s о n С A., D и n с a n s о n W. E., Proc. Roy. Soc, A165, 90 A938).
110. Beach J. Y., Journ. Chem. Phys., 4, 353 A936).
ПРИЛОЖЕНИЕ
ПУСТАЯ СТРАНИЦА
Таблица I
Постоянные сил взаимодействия для потенциала F-12) Леннарда-Джонса
В тех случаях, когда это возможно, результаты, полученные из измерений величины вязкости, исполь-
зуются для проведения вычислений коэффициентов переноса, а экспериментальные значения второго вириаль-
ного коэффициента — для вычисления уравнения состояния и термодинамических величин.
Для легких газов приведены два ряда значений постоянных сил взаимодействия : отмеченные сим-
волом (кл.) были получены с помощью классических формул (которые не являются вполне справедливыми),
а отмеченные символом (кв.) — из квантовомеханических формул. Последние дают действительное значение
постоянных сил взаимодействия, тогда как первые — только «эффективное» значение постоянных сил взаи-
модействия. Для точных вычислений и экстраполяции в область высоких температур необходимо использо-
вать значения с символом (кв.) — вместе с квантовомеханическими формулами и затабулированными функ-
циями. Значения, отмеченные символом (кл.), пригодны для грубых расчетов вместе с классическими форму-
лами и затабулированными функциями.
Здесь приведены также постоянные сил взаимодействия для некоторого числа веществ, состоящих
из полярных и (или) несферических молекул, которые не описываются потенциалом Леннарда-Джонса.
Однако эти постоянные, а также затабулированные функции, основанные на потенциале Леннарда-Джонса,
могут быть полезны для расчетов, пока не будет развита теория, необходимая для описания сложных молекул.
В том случае, когда приводятся два значения постоянных сил взаимодействия, полученных из резуль-
татов по измерению вязкости, следует иметь в виду, что интервал температуры для результатов работ
[1,3] заключен между 80 и 300° К, а для результатов работы [4]—обычно между 300 и 1000° К.
Газ
Легкие газы
Не (кв.)
Не (кл.)
Н2 (кв.)
Н2 (кл.)
D2 (кв.)
D2 (кл.)
Инертные газы
Ne
Аг
Кг
Хе
Простейшие
многоатомные
газы
Воздух
о2
Постоянные сил
результатов по измерению
вязкости
elk, °K
10,22
33,3
38,0
39,3
35,7
27,5
124,0
116,0
190,0
229,0
97,0
84,0
91,5
79,8
113,0
88,0
а, А
2,576
2,968
2,915
2,948
2,789
2,858
3,418
3,465
3,61
4,055
3,617
3,689
3,681
3,749
3,433
3,541
Литера- 1
тура
[1,2]
[2,3]
[4,5,6,
7,8]
[9]
[1]
[4,5,6]
[1]
[4,5]
[2]
[10]
[3]
[4,5,11]
[3]
[4,7]
[3]
[4]
взаимодействия, полученные из
второго вириального коэффициента
в/k, °К
10,22
6,03
37,00
29,2
37,00
31,1
35,60
34,9
119,8
122,0
171,0
158,0
221,0
217,0
99,2
102,0
95,05
95,9
117,5
118,0
а, А
2,556
2,63
2,928
2,87
2,928
2,87
2,749
2,78
3,405
3,40
3,60
3,597
4,100
3,963
3,522
3,62
3,698
3,71
3,58
3,46
2
Ьо - ^ п N а\
СМ* /МОЛЬ
21,07
22,84
31,67
29,76
31,67
29,77
26,21
27,10
49,80
49,58
• 58,86
58,7
86,94
78,5
55,11
60,34
63,78
64,42
57,75
52,26
Литера-
тура
[21]
[21]
[22]
[22]
[23]
[23]
[24]
[25]
[26]
[25]
[27]
[28]
[29]
[28]
[30]
[31]
[32]
[25]
[28]
[31]
54*
851
Продолжение табл. I
Газ
СО
соа
N0
N20
СН4
CF4
СС14
soa
SFe
Fa
Cla
Bra
J2
Другие неоргани-
ческие вещества
(пары)
НС1
HJ
AsH8
HgJa
HgBra
SnBr4
SnCl4
Hg
Углеводороды
CH = CH
CH2= CH2
C2H,
с3н8
л-С4Н10
'-C4H10
n-QHu
л-С,Н14
n-C,Hw
л-С8Н18
л-С,Нзд
Циклогексан
с.н,
Постоянные сил взаимодействия, полученные из
результатов по измерению
вязкости
e/fc, #K
110,0
88,0
190,0
213,0
119,0
91,0
220,0
237,0
137,0
144,0
327,0
252,0
112,0
357,0
257,0
520,0
550,0
360,0
324,0
281,0
698,0
530,0
465,0
1550,0
851,0
185,0
205,0
230,0
254,0
410,0
313,0
345,0
413,0
320,0
240,0
324,0
440,0
а, А
3,590
3,706
3,996
3,897
3,470
3,599
3,879
3,816
' 3,822
3,796
5,881
4,290
3,653
4,115
4,400
4,268
4,982
3,305
4,123
4,06
5,625
5,414
6,666
4,540
2,898
4,221
4,232
4,418
5,061
4,997
5,341
5,769
5,909
7,451
8,448
6,093
5,270
Литера-
тура
[3]
[4,7]
[3]
[4,8]
[3]
[10]
[3]
[8]
[3]
[4,12]
[2]
[4]
[13]
[14]
[10]
[15,16]
[14]
[17]
[10]
[2]
[14]
[14]
[14]
[14]
[18]
[19]
[2]
[2]
[2]
[19]
[19]
[20]
[20]
[2]
[2]
[20]
[2]
второго вириального коэффициента
elk *К
100,2
189,0
205,0
131,0
189,0
148,2
152,5
200,9
199,2
243,0
242,0
297,0
282,0
а, А
3,763
4,486
4,07
3,17
4,59
3,817
4,70
5,51
4,523
3,954
5,637
4,971
8,88
Ьо = |- п N а»,
смг\молъ
67,22
113,9
85,05
40,0
122,0
70,16
131,0
211,1
116,7
78,0
226,0
155,0
884,0
Литера-
тура
[33]
[34]
[35]
[36]
[36]
[37]
[38]
[38]
[39]
[28]
[28]
[28]
[28]
852
Продолжение табл. I
Газ
Другие органиче-
ские вещества
(пары)
СН3ОН
Одрн
СН3С1
CHaClj
CHCI3
C*N2
COS
CS2
Постоянные сил взаимодействия, полученные из
результатов по измерению
вязкости
507,0
391,0
855,0
406,0
327,0
339,0
335,0
488,0
о, А
3,585
4,455
3,375
4,759
5,430
4,38
4,13
4,438
Литера-
тура
[20]
[20]
[14]
[14]
[2]
[2]
[2]
[20]
второго вириального коэффициента
e/v, °K
°, А
Ь. - 4 п N а»,
см*1молъ
Литера-
тура
Таблица II
Второй вириальный коэффициент и коэффициент Джоуля—Томсона при
нулевом давлении для потенциала F-12) Леннарда-Джонса [40]
В(Т) = Ь9В*(Т*)
Т* = кТ/е
Ьо = ^ п No*
В8*
*>,>№* - В*)
T*(dB*ldT*)
T**(d*B*ldT**)
г- |
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,00
2,10
2,20
2,30
2,40
2,50
2,60
2,70
2,80
2,90
в*
-27,880581
-18,754895
-13,798835
-10,754975
- 8,720205
- 7,2740858
- 6,1979708
- 5,3681918
- 4,7100370
- 4,1759283
- 3,7342254
- 3,3631193
- 3,0471143
- 2,7749102
- 2,5380814
- 2,3302208
- 2,1463742
- 1,9826492
- 1,8359492
- 1,7037784
- 1,5841047
- 1,4752571
- 1,3758479
- 1,2847160
- 1,2008832
- 1,1235183
- 1,0519115
- 0,98545337
- 0,92361639
- 0,86594279
- 0,81203328
- 0,76153734
- 0,71414733
- 0,66959030
- 0,62762535
- 0,55063308
- 0,48170997
- 0,41967761
- 0,36357566
- 0,31261340
- 0,26613345
- 0,22358626
- 0,18450728
- 0,14850215
76,607256
45,247713
30,267080
21,989482
16,923690
13,582156
11,248849
9,5455096
8,2571145
7,2540135
6,4541400
5,8034061
5,2649184
4,8127607
4,4282616
4,0976659
3,8106421
3,5592925
3,3374893
3,1404074
2,9642040
2,8057826
2,6626207
2,5326459
2,4141403
2,3056683
2,2060215
2,1141772
2,0292621
1,9505276
1,8773287
1,8091057
1,7453722
1,6857016
1,6297207
1,5275444
1,4366294
1,3552188
1,2819016
1,2155320
1,1551691
1,1000353
1,0494802
1,0029572
в,*
-356,87679
-189,46536
-116,36604
- 78,87795
- 57,33952
- 43,88245
- 34,91869
- 28,64050
- 24,06266
- 20,61311
- 17,94190
- 15,82546
- 14,11557
- 12,71081
- 11,53985
- 10,55133
- 9,70744
- 8,97985
- 8,34700
- 7,79217
- 7,30227
- 6,86692
- 6,47777
- 6,12805
- 5,81225
- 5,52578
- 5,26485
- 5,02628
- 4,80738
- 4,60587
- 4,41980
- 4,24750
- 4,08753
- 3,93863
- 3,79972
- 3,54814
- 3,32647
- 3,12974
- 2,95401
- 2,79614
- 2,65355
- 2,52416
- 2,40623
- 2,29831
вх* — в*
104,488
64,003
44,066
32,744
25,644
20,8563
17,4468
14,9137
12,9672
11,4299
10,1884
9,1665
8,3120
7,5877
6,9663
6,4279
5,9570
5,5419
5,1734
4,8442
4,5483
4,2810
4,0385
3,8174
3,6150
3,4292
3,2579
3,0996
2,9529
2,8165
2,6894
2,5706
2,4595
2,3553
2,2573
2,0782
]
]
1
1,9183
1,7749
1,6455
1,5281
1,4213
1,3236
1,2340
i
1,1515
854
Продолжение табл. II
Т*
3,00
3,10
3,20
3,30
3,40
3,50
3,60
3,70
3,80
3,90
4,00
4,10
4,20
4,30
4,40
4,50
4,60
4,70
4,80
4,90
5,0
6,0
7,0
8,0
9,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
100,0
200,0
300,0
400,0
в*
-0,11523390
-0,08441245
-0,05578696
-0,02913997
-0,00428086
0,01895684
0,04072012
0,06113882
0,08032793
0,09839014
0,11541691
0,13149021
0,14668372
0,16106381
0,17469039
0,18761774
0,19989511
0,21156728
0,22267507
0,23325577
0,24334351
0,32290437
,0,37608846
0,41343396
0,44059784
0,46087529
0,52537420
0,52692546
0,51857502
0,50836143
0,49821261
0,48865069
0,47979009
0,47161504
0,46406948
0,41143168
0,38012787
0,35835117
я*
0,9600031
0,9202229
0,8832774
0,8488746
0,8167606
0,7867145
0,7585430
0,7320758
0,7071630
0,6836715
0,6614830
0,6404922
0,6206045
0,6017352
0,5838082
0,5667545
0,5505118
0,5350237
0,5202387
0,5061101
0,4925951
0,3839722
0,3082566
0,2524801
0,2097011
0,1758670
0,0286638
-0,0174929
-0,0393115
-0,0516478
-0,0593621
-0,0645039
-0,0680819
-0,0706470
-0,0725244
-0,0775400
-0,0765245
-0,0747534
о*
в2
-2,19920
-2,10785
-2,02340
-1,94511
-1,87231
-1,80447
-1,74108
-1,68174
-1,62605
-1,57371
-1,52441
-1,47789
-1,43394
-1,39234
-1,35291
-1,31548
-1,27991
-1,24606
-1,21381
-1,18305
-1,15367
-0,919393
-0,757930
-0,639879
-0,549792
-0,478779
-0,170403
-0,072012
-0,024109
0,003927
0,022147
0,034817
0,044056
0,051031
0,056441
0,077296
0,081397
0,082055
в? — в*
1,0752
1,0046
0,93906
0,87802
0,82104
0,76776
0,71782
0,67094
0,62684
0,58528
0,54607
0,50900
0,47392
0,44067
0,40912
0,37914
0,35062
0,32346
0,29756
0,27285
0,24925
0,06107
-0,06783
-0,16095
-0,23090
-0,28501
-0,49671
-0,54442
-0,55789
-0,56001
-0,55758
-0,55316
-0,54787
-0,54226
-0,53659
-0,48897
-0,45665
-0,43310
Таблица III
Третий вириальный коэффициент и его производные для потенциала F-12)
Леннарда-Джонса [41]
Т* -
= J я N а9
С* -
Сх* = (/)
С,* - T*«(d«C*/dT*«)
т*
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,00
2,10
2,20
2,30
2,40
2,50
2,60
2,70
2,80
2,90
С* '
-3,37664
-1,79197
-0,84953
-0,27657
+0,07650
0,29509
0,42966
0,51080
0,55762
0,58223
0,59240
0,59326
0,58815
0,57933
0,56831
0,55611
0,54339
0,53059
0,51803
0,50587
0,49425
0,48320
0,47277
0,46296
0,45376
0,44515
0,43710
0,42260
0,40999
0,39900
0,38943
0,38108
0,37378
0,36737
0,36173
0,35675
С,*
28,68
18,05
11,60
7,561
4,953
3,234
2,078
1,292
0,7507
0,3760
+0,1159
-0,0646
-0,1889
-0,2731
-0,3288
-0,3641
-0,3845
-0,3943
-0,3963
-0,3929
-0,3858
-0,3759
-0,3643
-0,3516
-0,3382
-0,3245
-0,3109
-0,2840
-0,2588
-0,2355
-0,2142
-0,1950
-0,1777
-0,1621
-0,1482
-0,1358
С,*
-220,0
-140,0
-92,1
-62,1
-42,7
-29,8
-21,0
-14,9
-10,6
-7,52
-5,29
-3,66
-2,46
-1,57
-0,910
-0,420
-0,050
+0,224
0,427
0,572
0,680
0,755
0,806
0,837
0,854
0,859
0,856
0,830
0,794
0,749
0,700
0,651
0,602
0,557
0,514
0,473
г* |
3,00
3,10
3,20
3,30
3,40
3,50
3,60
3,70
3,80
3,90
4,00
4,10
4,20
4,30
4,40
4,50
4,60
4,70
4,80
4,90
5,0
6,0
7,0
8,0
9,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
100,0
200,0
300,0
400,0
с*
0,35234
0,34842
0,34491
0,34177
0,33894
0,33638
0,33407
0,33196
0,33002
0,32825
0,32662
0,32510
0,32369
0,32238
0,32115
0,32000
0,31891
0,31788
0,31690
0,31596
0,31508
0,30771
0,30166
0,29618
0,29103
0,28610
0,24643
0,21954
0,20012
0,18529
0,17347
0,16376
0,15560
0,14860
0,14251
0,10679
0,08943
0,07862
С,*
-0,1247
-0,1148
-0,1060
-0,09826
-0,09133
-0,08510
-0,07963
-0,07462
-0,07024
-0,06634
-0,06286
-0,05989
-0,05709
-0,05458
-0,05237
-0,05040
-0,04865
-0,04712
-0,04579
-0,04461
-0,04359
-0,03893
-0,03989
-0,04231
-0,04529
-0,04825
-0,06437
-0,06753
-0,06714
-0,06566
-0,06388
-0,06203
-0,06025
-0,05857
-0,05700
-0,04599
-0,03970
-0,03551
с,*
0,439
0,400
0,369
0,340
0,313
0,288
0,266
0,246
0,227
0,210
0,194
0,183
0,169
0,156
0,145
0,134
0,125
0,116
0,108
0,100
0,0934
0,0449
0,0258
0,0192
0,0183
0,0199
0,0502
0,0654
0,0717
0,0742
0,0750
0,0748
0,0741
0,0732
0,0722
0,0619
0,0547
0,0496
856
Таблица /V
Функция Л(Т*) для оценочных значений третьего вириального
коэффициента для газовых смесей [41]
(с</*)пот. ящ.
т*
1,00
1,05
1,10
1Д5
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75-
1,80
1,85
1,90
1,95
2,00
2,10
2,20
2,30
2,40
2,50
2,60
2,70
2,80
2,90
3,00
3,10
3,20
3,30
3,40
А(Т*)
0,71099
0,74603
0,76746
0,78196
0,79246
0,80047
0,80684
0,81206
0,81649
0,82032
0,82369
0,82671
0,82947
0,83199
0,83432
0,83648
0,83851
0,84041
0,84219
0,84388
0,84545
0,84834
0,85091
0,85315
0,85510
0,85678
0,85823
0,85944
0,86043
0,86123
0,86185
0,86231
0,86261
0,86278
0,86283
г*
3,50
3,60
3,70
3,80
3,90
4,00
4,10
4,20
4,30
4,40
4,50
4,60
4,70
4,80
4,90
5,00
6,00
7,00
8,00
9,00
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0 •
90,0
100,0
200,0
300,0
400,0
А(Т*)
0,86275
0,86259
0,86233
0,86198
0,86157
0,86109
0,86055
0,85996
0,85931
0,85863
0,85792
0,85717
0,85640
0,85560
0,85478
0,85394
0,84502
0,83580
0,82692
0,81857
0,81075
0,75449
0,71957
0,69443
0,67488
0,65893
0,64549
0,63391
0,62374
0,61468
0,55663
0,52415
0,50186
857
Таблица V
Коэффициенты разложения для второго и третьего вириальных коэффициентов (и квантовые
поправки ко второму вириальному коэффициенту) для потенциала F-12) Леннарда-Джонса
Второй вириальный коэффициент (включая квантовые поправки):
В* - В*л + Л*гВ$ + Л*'ВЙ +...,
У=0
* 2 ьФт*
7=0
4ft T
Третий вириальный коэффициент в классическом представлении:
СКЛ. у = 0 сПТ*
j
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
+ 1,7330010
-2,5636934
-8,6650050 (-1)
-4,2728224 (-1)
—2,1662512 (-1)
-1,0682056 (-1)
-5,0545862 (-2)
-2,2890120 (-2)
-9,9286513 (-3)
-4,1329383 (-3)
-1,6547753 (-3)
-6,3872683 (-4)
-2,3818733 (-4)
-8,5982461 (-5)
-3,0100597 (-5)
-1,0236007 (-5)
-3,3872440 (-6)
-1,0913390 (-6)
-3,4305829 (-7)
-1,0530464 (-7)
-3,1597475 (-8)
*Р
8,297 (-2)
6,И (-2)
7,65 (-2)
6,79 (-2)
5,903 (-2)
3,362 (-2)
2,01 (-2)
1,10 (-2)
5,66 (-3)
2,692 (-3)
1,237 (-3)
5,39 (-3)
2,234 (-3)
8,90 (-4)
-2,630 (-3)
-9,037 (-3)
-2,549 (-2)
-1,494 (-2)
-1,40 (-2)
-1,12 (-2)
-6,26 (-3)
-5,02 (-3)
-2,941 (-3)
-1,584 (-3)
-8,03 (-4)
-3,839 (-4)
-1,74 (-4)
-7,53 (-5)
1
+ 1,729
-3,203
+ 1,519
+0,958
+0,429
+0,059
-0,140
-0,210
-0,205
-0,168
-0,123
—0,084
-0,059
-0,035
-0,020
-0,011
-0,006
-0,004
21
22
23
24
25
26
27
28
29
30
31
32
! 33
i 34
35
36
37
38
39
40
-9,2768372 (-9)
-2,6673193 (-9)
-7,5168046 (-10)
-2,0778030 (-10)
-5,6376036 (-11)
-1,5024114 (-11)
-3,9350796 (-12)
-1,0135315 (-12)
-2,5684633 (-13)
-6,4073832 (-14)
-1,5742194 (-14)
-3,8108431 (-15)
-9,0935023 (-16)
-2,1397782 (-16)
-4,9670392 (-17)
-1,1378186 (-17)
-2,5730157 (-18)
-5,7457408 (-19)
-1,2674099 (-19)
-2,7623753 (-20)
Табулированные здесь величины взяты из следующих источников :
fctf) : Берд и Спотц [40],
b\j\ Ь$: де Бур [42],
с<*>: Кихара [43].
Примечание. Числа в скобках указывают значения степени от 10, на которые нужно умножить
соответствующее число.
858
Таблица VI
Фазовые сдвиги г?/ (х&) для Не4, рассчитанные с помощью
потенциала F-12) Леннарда-Джонса [44]
хо
0,25
0,50
0,75
1,00
1,50
2,00
2,50
3,00
3,50
4,00
/ = о
1,180
0,850
0,510
0,205
-0,355
-0,890
-1,410
-1,913
-2,375 .
-2,800
/ =- 2
0,010
0,050
0,137
0,415
0,510
0,390
0,105
-0,220
-0,550
/ = 4
0,013
0,078
0,205
0,325
0,385
0,375
/ = 6
0,012
0,055
0,113
0,182
859
Таблица VII
Фазовые сдвиги B1+ l)'j/(xa) для Не8, рассчитанные с помощью потенциала F-12)
Леннарда-Джонса [45]
на
0,0
0,2
0,4
0,6
0,8
1,0
1.2
1,4
1,6
1,8
2,0
2,2
2,4
2,6
2,8
3,0
3,2
3,4
3,6
3,8
4,0
ха
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
2,0
2,2
2,4
2,6
2,8
3,0
3,2
3,4
3,6
3,8
4,0
l = 1
0,00
0,03
0,20
0,54
0,88
1,08
1,08
0,92
0,65
0,30
-0,09
-0,51
-0,95
-1,40
-1,87
-2,34
-2,82
-3,29
-3,77
-4,24
-4,72
/ = о
0,00
0,34
0,39
0,27
0,11
-0,07
-0,26
-0,46
-0,66
-0,86
-1,05
-1,25
-1,44
-1,63
-1,81
-1,99
-2,17
-2,34
-2,51
-2,67
-2,83
/ = з
0,01
0,04
0,11
0,22
0,39
0,65
0,98
1,32
1,66
1,97
2,18
2,29
2,23
2,06
1,82
1,53
1,21
0,82
/ = 2
0,02
0,10
0,29
0,58
0,93
1,27
1,56
1,66
1,55
1,29
0,92
0,49
0,02
-0,46
-0,96
-1,48
-2,00
-2,53
/ =5
0,01
0,01
0,03
0,05
0,10
0,17
0,26
0,40
0,55
0,73
0,94
1,18
1,48
1,86
2,25
2,64
3,00
/ = 4
0,02
0,04
0,09
0,18
0,29
0,44
0,62
0,86
1,13
1,44
1,77
2,11
2,45
2,78
3,02
3,13
3,09
/ - 7
0,01
0,01
0,02
0,04
0,05
0,08
0,11
0,15
0,21
0,27
0,35
0,44
0,55
0,68
0,82
/ = б
0,01
0,02
0,03
0,05
0,08
0,12
0,18
0,25
0,33
0,43
0,55
0,70
0,87
1,07
1,30
1,54
/ - 9
0,01
0,01
0,02
0,03
0,04
0,06
0,08
0,10
0,13
0,17
0,21
0,26
0,32
/ - 8
0,01
0,02
0,02
0,03
0,04
0,06
0,09
0,12
0,16
0,21
0,27
0,34
0,41
0,49
/ = и
0,01
0,01
0,02
0,03
0,04
0,05
0,06
0,08
0,10
0,12
0,15
/ - 10
0,01
0,01
0,02
0,02
0,03
0,04
0,05
0,06
0,08
0,11
0,14
• 0,18
0,21
/ - 13
0,01
0,01
0,01
0,02
0,02
0,03
0,04
0,05
0,06
0,08
/ - 12
0,01
0,01
0,02
0,02
0,02
0,03
0,03
0,04
0,05
0,07
0,09
0,11
/ = 15
0,01
0,01
0,01
0,01
0,02
0,02
0,03
0,03
0,04
/ - 14
0,01
0,01
0,02
0,02
0,03
0,03
0,04
0,04
0,05
1 - 17
0,01
0,01
0,01
0,01
0,01
0,02
0,02
0,03
/ = 16
0,01
0,01
0,02
0,02
0,03
0,03
1 = 19
0,01
0,01
0,01
0,01
0,01
0,02
1 = 18
0,01
0,02
0,02
860
Таблица VIII
Фактор сжимаемости pV/NkT, основанный на модели Леннарда-Джонса и Девоншайра
с учетом трех оболочек [46]
Ч а*8
Г*\
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,1
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
400
0,8
215,9
202,2
190,2
179,6
170,13
161,66
154,02
147,10
140,80
129,74
120,38
111,33
99,23
89,03
80,866
66,149
56,330
49,300
44,015
36,676
28,074
21,584
13,735
8,717
6,733
4,476
0,9
117,27
109,97
103,57
97,92
92,92
88,40
84,35
80,68
77,355
71,525
66,593
62,365
55,492
50,142
45,856
38,120
33,020
29,262
26,438
22,471
17,897
14,307
9,998
6,969
5,630
3,962
г* =
1,0
64,01
60,26
56,97
54,07
51,49
49,18
47,10
45,21
43,50
40,50
37,953
35,768
32,205
28,521
27,258
22,172
20,435
18,468
16,982
14,809
12,312
10,373
7,866
5,910
4,902
3,592
kTJe
1,2
17,650
17,142
16,528
15,984
15,500
15,065
14,672
14,316
13,991
13,419
12,932
12,510
11,816
11,266
10,817
9,885
9,284
8,838
8,488
7,963
7,281
6,659
5,688
4,624
4,000
3,096
v* = г /а» « (а/а)8/К2 = а*3/К2
1,4
2,358
2,695
2,987
3,240
3,462
3,657
3,830
3,886
4,018
4,245
4,432
4,587
4,826
4,998
5,123
5,310
5,395
5,427
5,430
5,393
5,259
5,087
4,576
3,905
3,462
2,777
1,5
-0,8136
-0,3818
+0,0812
0,4862
0,8429
1,1593
1,4411
1,6935
1,9205
2,311
2,634
2,904
3,326
3,636
3,870
4,248
4,458
4,579
4,648
4,705
4,720
4,591
4,211
3,652
3,267
2,655
1,6
-2,642
-1,999
-1,442
-0,955
-0,5276
-0,1491
+0,1881
0,4897
0,7608
1,2272
1,6128
1,9355
2,4417
2,8163
3,1010
3,570
3,841
4,006
4,110
4,253
4,283
4,212
3,920
3,444
3,104
2,543
1,8
-3,738
-3,059
-2,472
-1,960
-1,511
-1,113
-0,759
-0,443
-0,1585
+0,3309
0,7357
1,0750
1,6086
2,0054
2,3090
2,816
3,149
3,336
3,457
3,595
3,684
3,673
3,486
3,122
2,849
2,319
2,0
-3,566
-2,948
-2,413
-1,947
-1,537
-1,1743
-0,8515
-0,5627
-0,3030
+0,1444
0,5151
0,8262
1,3168
1,7135
1,9919
2,460
2,741
2,922
3,043
3,185
3,290
3,305
3,175
2,885
2,657
2,082
2,2
-3,066
-2,528
-2,062
-1,654
-1,296
-0,9789
-0,6961
-0,4427
-0,191
+0,203
0,528
0,803
1,234
1,557
1,807
2,230
2,486
2,653
2,766
2,901
3,009
3,035
2,942
2,702
2,499
1,856
861
Продолжение табл, VIII
2,4
2,5
2,6
2,8
3,0
3,5
4,0
5,0
6,0
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1Д
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
400
-2,535
-2,070
-1,667
-1K15
-1,004
-0,729
-0,483
-0,2624
-0,0634!
+0,2803
0,5663
0,8067
1,1887
1,476
1,698
2,077
2,310
2,461
2,565
2,691
2,797
2,829
2,760
2,556
2,348
1,661
-2,310
-1,877
-1,501
-1,172
-0,8808
-0,6232
-0,3932
-0,1865
+0,0002
0,322:
0,5908
0,8173
1,177
1,448
1,658
2,017
2,238
2,383
2,483
2,605
2,708
2,743
2,683
2,492
2,270
1,579
-2,107
-1,701
-1,349
-1,0407
-0,7681
-0,5265
-0,3105
-0,1162
+0,0590
0,3623
0,6151
0,8287
1,1684
1,425
1,624
1,965
2,176
2,314
2,410
2,529
2,630
2,666
2,614
2,431
2,188
1,506
-1,760
-1,402
-1,091
-0,817
-0,575
-0,360
-0,168
+0,005
0,161
0,432
0,659
0,850
1,156
1,387
1,568
1,878
2,070
2,198
2,287
2,398
2,495
2,534
2,494
2,308
2,020
1,387
-1,486
-1,165
-0,885
-0,6395
-0,4219
-0,2282
-0,0547
+0,1015
0,2427
0,4880
0,6933
0,8673
1,1453
1,357
1,522
1,806
1,985
2,104
2,187
2,291
2,385
2,424
2,394
2,178
1,856
1,296
-1,0215
-0,7626
-0,5366
-0,3376
-0,1613
-0,0040
+0,1371
0,2645
0,3798
0,5804
0,7489
0,8922
1,122
1,297
1,435
1,675
1,826
1,928
2,001
2,093
2,178
2,217
2,166
1,829
1,520
1,156
-0,7382
-0,5168
-0,3234
-0,1531
-0,0021
h0,1329
0,2541
0,3634
0,4626
0,6353
0,7806
0,9044
1,1035
1,256
1,376
1,586
1,719
1,810
1,874
1,957
2,031
2,048
1,901
1,531
1,305
1,085
-0,399
-0,221
-0,0661
+0,0708
0,1924
-0,1841
-0,0329
+0,0990
0,2149
0,3173
0,3009! 0,4084
0,3985
0,4865
0,5664
0,7057
0,8230
0,9230
1,084
1,207
1,304
1,471
1,571
1,633
1,670
1,700
1,682
1,606
1,402
1,191
1,101
1,026
0,4897
0,5676
0,6282
0,7411
0,8340
0,9113
1,030
1,115
1,161
1,266
1,305
1,321
1,323
1,311
1,271
1,219
1,128
1,056
1,029
1,007
Значение поправки на неидеальность газа для приведенной внутренней
основанное на модели Леннарда-Джонса и Девоншайра с учетом трех
Таблица IX
энергии й'/Ке,
оболочек [46]
Г*\
0,70
0,75
0,80
0,85!
0,90
0,95
1,00
1,05
1,1
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
400
0,8
14,94
15,05
15,16
15,26
15,36
15,45
15,54
15,63
15,71
15,88
16,04
16,19
16,49
16,79
17,08
17,80
18,52
19,23
19,92
21,38
23,95
27,73
39,08
69,86
114,29
311,2
0,9
2,375
2,451
2,525
2,598
2,675
2,742
2,814
2,885
2,957
3,100
3,244
3,387
3,674
3,958
4,240
4,934
5,666
6,310
6,949
8,214
10,687
14,05
24,79
52,31
91,08
264,5
1,0
-3,590
-3,517
-3,444
-3,372
-3,300
-3,228
-3,157
-3,086
-3,015
-2,874
-2,734
-2,596
-2,323
-2,015
-1,758
-1,120
-0,4897
+0,1330
0,7476
1,876
4,124
7,372
17,21
42,11
76,05
231,2
1,2
-7,374
-7,284
-7,220
-7,156
-7,093
-7,030
-6,967
-6,904
-6,841
-6,717
-6,593
-6,470
-6,226
-5,986
-5,749
-5,217
-4,668
-4,130
-3,602
-2,576
-0,6285
+2,095
10,28
30,14
57,82
186,7
1,4
-7,505
-7,446
-7,387
-7,329
-7,271
-7,214
-7,157
-7,120
-7,066
-6,958
-6,851
-6,745
-6,536
-6,331
-6,129
-5,638
-5,165
-4,707
-4,262
-3,405
-1,792
+0,5218
7,150
23,75
47,18
158,3
1,5
-7,177
-7,137
-7,084
-7,032
-6,979
-6,927
-6,876
-6,825
-6,774
-6,673
-6,573
-6,475
-6,282
-6,093
-5,908
-5,459
-5,028
-4,612
-4,207
-3,428
-1,912
+0,1349
6,213
21,55
43,36
147,4
1,6
-6,785
-6,735
-6,686
-6,637
-6,509
-6,541
-6,494
-6,447
-6,400
-6,308
-6,217
-6,127
-5,951
-5,779
—5,611
-5,204
-4,813
-4,434
-4,066
-3,324
-1,982
-0,1051
+5,503
19,78
40,21
137,45
1,8
-5,947
-5,906
-5,866
-5,827
-5,788
-5,749
-5,710
-5,672
-5,634
-5,559
-5,485
-5,412
-5,269
-5,129
-4,991
-4,656
-4,317
-4,002
-3,964
-3,097
-1,961
-0,3553
+4,504
17,08
35,30
117,14
2,0
-5,220
-5,189
-5,157
-5,126
-5,095
-5,064
-5,033
-5,002
-4,972
-4,912
-4,852
-4,792
-4,675
-4,553
-4,438
-4,158
-3,886
-3,618
-3,356
-2,845
-1,861
-0,4584
+3,841
15,13
31,65
95,25
2,2
-4,638
-4,612
-4,587
-4,562
-4,537
-4,512
-4,487
-4,462
-4,435
-4,385
-4,336
-4,286
-4,188
-4,090
-3,994
-3,755
-3,520
-3,289
-3,061
-2,613
-1,745
-0,4962
+3,375
13,66
28,64
74,18
Продолжение табл. IX
\. а**
Г* \.
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1Д
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3
3,5
4
5
7
10
20
50
100
400
2,4
-4,177
-4,156
-4,135
-4,114
-4,093
-4,072
-4,051
-4,030
-4,009
-3,967
-3,925
-3,883
-3,799
-3,715
-3,631
-3,423
-3,216
-3,012
-2,810
-2,411
-1,631
-0,5013
+3,033
12,51
25,66
56,17
2,5
-3,984
-3,964
-3,945
-3,926
-3,966
-3,887
-3,868
-3,848
-3,828
-3,789
-3,750
-3,711
-3,632
-3,553
-3,475
-3,279
-3,084
-2,891
-2,699
-2,320
-1,577
-0,4968
+2,895
12,00
24,06
48,57
2,6
-3,810
-3,792
-3,774
-3,756
-3,738
-3,720
-3,702
-3,684
-3,665
-3,629
-3,592
-3,555
-3,481
-3,407
-3,333
-3,147
-2,903
-2,779
-2,596
-2,235
-1,524
-0,489
+2,774
11,50
22,36
41,88
2,8
-3,5129
-3,4968
-3,4807
-3,4656
-3,4484
-3,4321
-3,4158
-3,3994
-3,3830
-3,3501
-3,3171
-3,2840
-3,2174
-3,1505
-3,0835
-2,9155
-2,7475
-2,5799
-2,4128
-2,0808
-1,4261
-0,4670
+2,5735
10,457
18,81
30,99
3,0
-3,2666
-3,2517
-3,2368
-3,2219
-3,2070
-3,1920
-3,1770
-3,162
-3,1469
-3,1168
-3,0864
-3,0561
-2,9951
-2,9338
-2,8722
-2,7177
-2,5628
-2,4078
-2,2532
-1,9451
-1,3358
-0,4396
+2,4091
9,263
15,321
22,834
3,5
-2,7997
-2,7862
-2,7729
-2,7596
-2,7464
-2,7332
-2,7201
-2,7070
-2,6940
-2,6679
-2,6418
-2,6158
-2,5636
-2,5113
-2,4589
-2,3270
-2,1953
-2,0630
-1,9306
-1,6660
-1,1399
-0,3641
+ 1,9559
5,879
8,208
10,470
4,0
-2,4644
-2,4514
-2,4386
-2,4260
-2,4136
-2,4013
-2,3891
-2,3770
-2,3649
-2,3410
-2,3173
-2,2937
-2,2467
-2,1998
-2,1530
-2,0359
-1,9186
-1,8010
-1,6834
-1,4485
-0,9874
-0,3456
+ 1,1918
2,9761
3,804
4,5205
5,0
-2,0005
-1,9878
-1,9756
-1,9636
-1,9519
-1,9404
-1,9292
-1,9181
-1,9071
-1,8856
-1,8645
-1,8436
-1,8027
-1,7623
-1,7226
-1,6255
-1,5325
-1,4446
-1,3623
-1,2153
-0,9839
-0,7479
-0,3793
-0,0967
+0,00948
0,09335
6,0
-1,6861
-1,6738
-1,6620
-1,6506
-1,0396
-1,6290
-1,6188
-1,6088
-1,5992
-1,5807
-1,5632
-1,5466
-1,5159
-1,4882
-1,4631
-1,4099
-1,3674
-1,3330
-1,3045
-1,2606
-1,2036
-1,1556
-1,0931
-1,0520
-1,0376
-1,0266
Таблица X
Значение поправки на неидеальность газа для приведенной теплоемкости &/Nk,
основанное на модели Леннарда-Джонса и Девоншайра с учетом трех оболочек [46]
N. а**
Т* N^
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1Д
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
0,8
2,17
2,07
1,99
1,92
1,85
1,79
1,74
1,69
1,617
1,567
1,529
1,493
1,470
1,460
1,440
1,422
1,415
1,410
1,400
1,275
1,231
1,108
0,974
0,855
0,9
1,494
1,466
1,450
1,443
1,440
1,435
1,435
1,435
1,435
1,435
1,430
1,427
1,417
1,404
1,391
1,376
1,283
1,274
1,256
1,190
i,iio
1,035
0,864
0,747
1,0
1,452
1,447
1,442
1,438
1,432
1,425
1,421
1,416
1,402
1,390
1,380
1,345
1,310
1,283
1,268
1,253
1,237
1Д96
1,127
1,108
1,060
0,946
0,773
0,656
1,545
1,272
1,268
1,265
1,261
1,258
1,254
1,250
1,242
1,234
1,226
1,210
1,194
1,150
1,096
1,087
1,067
1,046
1,008
0,947
0,887
0,779
0,621
0,536
1,4
1,177
1,168
1,159
1,150
1,142
1,120
1,100
1,080
1,073
1,065
1,060
1,037
1,019
1,000
0,964
•
0,931
0,903
0,879
0,840
0,792
0,746
0,635
0,521
0,455
1,6
0,934
1,054
1,047
1,041
1,034
1,028
1,022
1,015
1,002
0,990
0,977
0,956
0,935
0,918
0,879
0,848
0,821
0,799
0,772
0,728
0,665
0,584
0,483
0,423
1,6
0,989
0,980
0,971
0,961
0,953
0,946
0,937
0,930
0,916
0,903
0,891
0,869
0,850
0,833
0,798
0,770
0,746
0,738
0,719
0,653
0,611
0,540
0,451
0,397
1,8
0,804
0,795
0,787
0,780
0,773
0,767
0,760
0,755
0,744
0,734
0,726
0,710
0,696
0,684
0,674
0,654
0,623
0,610
0,587
0,555
0,524
0,469
0,399
0,351
2,0
0,632
0,628
0,624
0,621
0,617
0,614
0,610
0,607
0,602
0,596
0,591
0,590
0,580
0,568
0,553
0,540
0,529
0,520
0,505
0,482
0,459
0,417
0,359
0,313
2,2
0,504
0,503
0,502
0,501
0,500
0,500
0,500
0,500
0,497
0,495
0,493
0,489
0,485
0,482
0,473
0,466
0,460
0,454
0,443
0,427
0,410
0,376
0,327
0,278
55 2017
865
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,1
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
2,4
0,417
0,418
0,419
0,419
0,420
0,420
0,421
0,421
0,421
0,421
0,421
0,420
0,419
0,418
0,415
0,410
0,406
0,403
0,396
0,384
0,371
0,344
0,296
0,240
2,5
0,385
0,386
0,387
0,388
0,389
0,390
0,391
0,391
0,392
0,393
0,393
0,393
0,393
0,393
0,391
0,388
0,385
0,382
0,377
0,367
0,355
0,330
0,280
0,218
2,6
0,359
0,360
0,362
0,363
0,364
0,365
0,366
0,366
0,368
0,369
0,369
0,370
0,371
0,371
0,370
0,368
0,366
0,364
0,360
0,351
0,341
0,317
0,263
0,195
2,8
0,321
0,323
0,324
0,325
0,326
0,327
0,328
0,328
0,330
0,331
0,332
0,333
0,335
0,335
0,336
0,336
0,335
0,333
0,330
0,324
0,316
0,294
0,227
* 0,149
3,0
0,297
0,298
0,298
0,299
0,300
0,300
0,301
0,302
0,303
0,304
0,304
0,306
0,307
0,308
0,309
0,310
0,310
0,309
0,307
0,302
0,296
0,271
0,188
0,107
3,6
0,268
0,266
0,265
0,264
0,262
0,262
0,262
0,261
0,261
0,261
0,261
0,261
0,262
0,262
0,263
0,264
0,265
0,265
0,264
0,261
0,252
0,207
0,099
0,041
Продолжение
4,0
0,258
0,254
0,250
0,247
0,245
0,243
0,241
0,240
0,238
0,237
0,236
0,235
0,234
0,234
0,234
0,235
0,235
0,235
0,233
0,224
0,200
0,130
0,0434
0,0145
5,0
0,249
0,242
0,237
0,232
0,227
0,224
0,220
0,218
0,213
0,210
0,207
0,203
0,200
0,198
0,190
0,181
0,170
0,159
0,137
0,101
0,069
0,030
0,0067
0,0019
табл, X
6.0
0,241
0,232
0,223
0,216
0,209
0,202
0,196
0,190
0,180
0,170
0,162
0,146
0,132
0,120
0,0957
0,0770
0,0629
0,0525
0,0388
0,0235
0,0138
0,0050
0,00097
0,00025
866
Таблица XI
Значение поправки на неидеальность газа для приведенной энтропии S'/Nk,
оенованное на модели Леннарда-Джонса и Девоншайра с учетом трех оболочек [46]
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1>1
1,2
1,3
1.4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
400
0,8
-8,816
-8,643
-8,491
-8,359
-8,239
-8,132
-8,036
-7,948
-7,866
-7,723
-7,5965
-7,4834
-7,2854
-7,1145
-6,9628
-6,6442
-6,3855
-6,1697
-5,9854
-5,6719
-5,2402
-4,7916
-3,9568
-3,0070
-2,3994
-1,4539
Т* ^^\
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,1
1,2
1,3
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
7
10
20
50
100
400
0,9
-7,7406
-7,6333
-7,5360
-7,4465
-7,3612
-7,2852
-7,2116
-7,1417
-7,0752
-6,9488
-6,8372
-6,7317
-6,5422
-6,3759
-6,2284
-5,9209
-5,6645
-5,4465
-5,2962
-5,0146
-4,5988
-4,1602
-3,4121
-2,5594
-2,0251
-1,1926
2,4
-1,9982
-1,9695
-1,9426
-1,9172
-1,8933
-1,8705
-1.8490
-1,8285
-1,8089
1,0
-7,0399
-6,9413
-6,8490
-6,7627
-6,6814
-6,6047
-6,5322
-6,4634
-6,3979
-6,2763
-6,1652
-6,0632
-5,8813
-5,7108
1,2
-5,8313
-5,7235
-5,6415
-5,5647
-5,4925
-5,4244
-5,3600
-5,2989
-5,2408
-5,1327
-5,0338
-4,9428
-4,7804
-4,6390
-5,5756 ! -4,5140
-5,2917
-5,0623
-4,8707
-4,7068
-4,4138
-4,0361
-3,6502
-2,9641
-2,2027
-4,2383
-4,0386
-3,8729
-3,7320
-3,5032
-3,1757
-2,8508
-2,2900
-1,6735
-1,7277 -1,2864
-0,9841
2,5
-1,8619
-1,8354
-1,8105
-1,7871
-1,7650
-1,7439
-1,7239
-1,7049
-1,6867
-1,7723 | - 1,6526
-1,7386 | -1,6212
-1,7074
-1,6512
-1,6017
-1,5576
-1,4647
-1,3897
-1,3265
-1,2724
-1,1833
-1,0520
-0,9176
-0,6714
-0,3790
-0,1939
-0,0236
-1,5921
-1,5396
-1,4932
-1,4518
-1,3644
-1,2934
-1,2338
-1,1825
-1,0978
-0,9728
-0,8422
-0,6080
-0,3270
-0,1565
-0,0174
-0,6703
2,6
-1,7402
-1,7155
-1,6923
-1,6704
-1,6497
- 1,6301
-1,6113
-1,5936
1,4
-4,8227
-4,7413
-4,6657
-4,5952
-4,5293
-4,4674
-4,4091
-4,3340
-4,2838
-4,1904
-4,1050
-4,0266
-3,8873
-3,7664
-3,6602
-3,4413
-3,2688
-3,1277
-3,0088
1.5
-4,3804
-4,2865
-4,2185
-4,1549
-4,0953
-4 0393
-3,9865
-3,9366
-3,8893
-3,8018
-3,7221
-3,6493
-3,5205
-3,4092
-3,3117
-3,1116
-2,9544
-2,8259
-2,7179
-2,8175 ! -2,5439
-2,5456 | -2,2976
-2,2792
-1,8159
-1,3014
-0,9738
-0,4437
2,8
-1,5323
-1,5101
-1,4894
-1,4698
-1,4512
-1,4336
-1,4169
-1,4010
-1,5765 -1,3857
-1,5446 - 1,3571
-1,5151
-1,3306
-1,4877 -1,3061
-1 4383
-1,3947
- 1,3556
-1,2729
-1,2056
-1,1489
-1,1002
-1,0194
-0,8999
-0,7766
-0,5494
-0,2796
-0,1253
-0,0128
-1,2616
-1,2222
-1,1870
-1,1120
-1,0507
-0,9990
-0,9544
-0,8803
-0,7701
-0,6560
-0,4444
-0,1989
-0,0788
-0,0071
-2,0538
-1,6290
- 1,1538
-0,8490
-0,3526
1,6
-3,9672
-3,8988
-3,8354
-3,7764
-3,7212
-3,6695
-3,6209
-3,5750
-3,5316
-3,4514
-3,3786
-3,3122
-3,1948
-3,0936
-3,0050
-2,8233
-2,6805
-2,5638
-2,4655
-2,3083
-2,0823
-1,8587
-1,4670
-1,0250
-0,7395
-0,2744
3,0 , 3,5
-1,3611
-1,3406
-1,3214
-1,3033
-1,2862
-1,2701
-1,2547
-1,2400
- 1,2260
-1,1998
-1,1754
-1,1530
-1,1123
-1,0762
-1,0437
-0,9748
-0,9183
-0,8705
-0,8293
-0,7605
-0,6579
-0 5513
-0,3530
-0,1369
-0,0490
-0,0040
-1,0393
-1,0207
-1,0035
-0,9874
-0,9723
-0,9581
-0,9446
-0,9319
-0,9197
-0,8970
-0,8761
1,8
-3,2834
-3,2277
-3,1761
-3,1281
-3,0834
-3,0414
-3,0020
-2,9647
-2,9295
-2,8643
-2,8051
-2,7511
-2,6553
-2,5727
-2,5000
-2,3504
-2,2339
-2,1369
-2,0545
- 1,9215
-1,7299
- 1,5388
-1,1996
-0,8107
-0,5502
-0,1362
4,0
-0,8104
-0,7924
-0,7759
-0,7606
-0,7464
-0,7331
-0,7206
-0,7088
-0,6976
-0,6768
-0,6578
-0,8568 -0,6403
-0,8220
-0,7912
-0,7636
-0,7049
-0 6568
-и,6160
-0,5806
-0,5216
-0,4330
-0,3407
-0,1779
-0,0493
-0,0149
-0,0011
-0,6089
-0,5813
-0,5567
-0,5044
-0,4616
-0,4254
- 0,3940
-0,3415
-0.2639
-0,1873
-0,0772
-0,0171
-0,0048
-0,0004
2,0
-2,7417
-2,6979
-2,6572
-2,6193
-2,5807
-2,5502
-2,5187
-2,4888
-2,4605
-2,4079
-2,3600
-2,3160
-2,2376
-2,1717
-2,1116
-1,9865
-1,8869
-1,8046
-1,7345
-1,6203
-1,4546
-1,2876
-0,9877
-0,6389
-0,4084
-0,0841
5,0
-0,4954
-0,4780
-0,4621
-0,4476
-0,4342
-0,4218
-0,4103
-0,3995
-0,3893
-0,3705
-0,3536
-0,3382
-0,3108
-0,2871
-0,2661
-0,2228
-0,1888
-0,1617
-0,1397
-0,1068
-0,0676
-0,0391
-0,0119
-0,0021
-0,0006
-0,00008
2,2
-2,3220
-2,2872
-2,2547
-2,2242
-2,1955
-2,1684
-2,1428
-2,1184
-2,0977
-2,0545
-2,0148
-1,9781
-1,9126
-1,8551
-1,8042
-1,6977
-1,6120
-1,5408
-1,4798
-1,3798
-1,2336
-1,0849
-0,8152
-0,4976
-0,2881
-0,0445
6,0
-0,2812
-0,2642
-0,2489
-0,2352
-0,2226
-0,2112
-0,2006
-0,1909
-0,1820
-0,1658
-0,1518
-0,1395
-0,1190
-0,1027
- 0,0894
-0,0656
-0,0501
-0,0394
-0,0318
-0,0220
-0,0122
-0,0064
-0,0018
-0,0003
-0,00013
+ 0,00002
55*
867
Таблица XII
Значение интегралов ?'Л»)* для вычисления коэффициентов переноса с помощью
потенциала F-12) Леннарда-Джонса
7'*
2,662
2,476
2,318
2,184
2,066
1,966
1,877
1,798
1,729
1,667
1,612
1,562
1,517
1,476
1,439
1,406
1,375
1,346
1,320
1,296
1,273
1,253
1,233
1,215
1,198
1,182
1,167
1,153
1,140
1,128
1,116
1,105
1,094
1,084
1,075
1,057
1,041
1,026
1,012
0,9996
0,9878
0,9770
0,9672
0,9576
0,9490
0,9406
0,9328
0,9256
0,9186
0,9120
0,9058
1
2,256
2,078
1,931
1,808
1,705
1,618
1,543
1,479
1,423
1,375
1,332
1,295
1,261
1,231
1,204
1,179
1,157
1,137
1,119
1,102
1,086
1,072
1,059
1,046
1,034
1,023
1,013
1,004
0,9947
0,9860
0,9780
0,9707
0,9633
0,9567
0,9500
0,9380
0,9267
0,9167
0,9073
0,8987
0,8907
0,8833
0,8767
0,8700
0,8640
0,8580
0,8520
0,8473
0,8420
0,8373
0,8327
1,962
1,795
1,663
1,556
1,468
1,396
1,336
1,285
1,242
1,205
1,172
1,144
1,119
1,096
1,076
1,058
1,041
1,027
1,013
1,000
0,9887
0,9780
0,9680
0,9588
0,9502
0,9420
0,9345
0,9272
0,9205
0,9142
0,9082
0,9023
0,8968
0,8917
0,8867
0,8775
0,8688
0,8612
0,8538
0,8470
0,8407
0,8347
0,8290
0,8237
0,8187
0,8138
0,8093
0,8048
0,8007
0,7967
0,7928
2,785
2,628
2,492
2,368
2,257
2,156
2,065
1,982
1,908
1,841
1,780
1,725
1,675
1,629
1,587
1,549
1,514
1,482
1,452
1,424
1,399
1,375
1,353
1,333
1,314
1,296
1,279
1,264
1,248
1,234
1,221
1,209
1,197
1,186
1,175
1,156
1,138
1,122
1,107
1,093
1,081
1,069
1,058
1,048
1,039
1,030
1,022
1,014
1,007
0,9999
0,9932
2,535
2,375
2,232
2,105
1,992
1,893
1,806
1,729
1,661
1,602
1,549
1,502
1,460
1,422
1,388
1,357
1,329
1,304
1,280
1,259
1,239
1,221
1,205
1,189
1,175
1,162
1,149
1,137
1,126
1,116
1,106
1,097
1,088
1,080
1,073
1,055
1,045
1,033
1,022
1,012
1,002
0,9935
0,9855
0,9780
0,9708
0,9643
0,9578
0,9518
0,9463
0,9408
0,9358
2,333
2,163
2,016
1,889
1,781
1,689
1,610
1,542
1,484
1,434
1,389
1,350
1,316
1,286
1,258
1,234
1,212
1,192
1,174
1,157
1,142
1,128
1,115
1,103
1,092
1,081
1,072
1,063
1,054
1,046
1,038
1,031
1,024
1,018
1,012
1,000
0,9895
0,9800
0,9710
0,9630
0,9555
0,9485
0,9415
0,9355
0,9295
0,9240
0,9185
0,9135
0,9085
0,9040
0,8995
2,152
1,978
1,833
1,713
1,614
1,532
1,463
1,406
1,357
1,315
1,278
1,247
1,219
1,194
1,172
1,152
1,135
1,119
1,104
1,091
1,078
1,067
1,057
1,047
1,037
1,029
1,022
1,014
1,007
1,000
0,9942
0,9875
0,9825
0,9767
0,9717
0,9617
0,9525
0,9450
0,9375
0,9300
0,9233
0,9175
0,9117
0,9058
0,9008
0,8958
0,8908
0,8867
0,8825
0,8783
0,8742
1,990
1,819
1,682
1,574
1,486
1,415
1,356
1,307
1,267
1,231
1,201
1,175
1,152
1,131
1,113
1,097
1,082
1,068
1,056
1,045
1,035
1,025
1,016
1,008
1,000
0,9929
0,9860
0,9795
0,9735
0,9677
0,9623
0,9569
0,9520
0,9473
0,9427
0,9343
0,9261
0,9190
0,9120
0,9058
0,8996
0,8940
0,8887
0,8836
0,8788
0,8742
0,8698
0,8656
0,8617
0,8577
0,8539
2,557
2,378
2,223
2,090
1,975
1,875
1,788
1,712
1,645
1,587
1,535
1,488
1,447
1,410
1,377
1,347
1,319
1,294
1,272
1,251
1,232
1,215
1,198
1,183
1,169
1,156
1,144
1,133
1,122
1,112
1,103
1,094
1,085
1,078
1,070
1,056
1,043
1,032
1,021
1,012
1,003
0,9942
0,9863
0,9792
0,9721
0,9658
0,9596
0,9538
0,9483
0,9433
0,9383
868
Продолжение табл. XII
т*
3,7
3,8
3,9
4,0
4,1
4,2
4,3
4,4
4,5
4,6
4,7
4,8
4,9
5
6
7
8
9
10
20
30
40
50
60
70
80
90
100
200
300
400
Жы>*
0,8998
0,8942
0,8888
0,8836
0,8788
0,8740
0,8694
0,8652
0,8610
0,13568
0,8530
0,8492
0,8456
0,8422
0,8124
0,7896
0,7712
0,7556
0,7424
0,6640
0,6232
0,5960
0,5756
0,5596
0,5464
0,5352
0,5256
0,5130
0,4644
0,4360
0,4170
0,8287
0,8240
0,8200
0,8167
0,8127
0,8093
0,8060
0,8027
0,7993
0,7960
0,7933
0,7907
0,7873
0,7847
0,7607
0,7420
0,7260
0,7127
0,7013
0,6293
0,5909
0,5651
0,5459
0,5307
0,5181
0,5075
0,4984
0,4903
0,4403
0,4135
0,3955
0A.8)*
0,7892
0,7857
0,7822
0,7790
0,7758
0,7727
0,7697
0,7668
0,7640
0,7613
0,7585
0,7560
0,7535
0,7510
0,7295
0,7120
0,6973
0,6847
0,6735
0,6048
0,5680
0,5432
0,5248
0,5100
0,4980
0,4878
0,4790
0,4713
0,4233
0,3975
0,3802
#(«.?)*
0,9870
0,9811
0,9755
0,9700
0,9649
0,9600
0,9553
0,9507
0,9464
0,9422
0,9382
0,9343
0,9305
0,9269
0,8963
0,8727
0,8538
0,8379
0,8242
0,7432
0,7005
0,6718
0,6504
0,6335
0,6194
0,6076
0,5973
0,5882
0,5320
0,5016
0,4811
flM*
0,9308
0,9263
0,9218
0,9175
0,9133
0,9093
0,9055
0,9018
0,8985
0,8950
0,8918
0,8885
0,8855
0,8823
0,8565
0,8360
0,8193
0,8048
0,7923
0,7160
0,6750
0,6475
0,6268
0,6105
0,5970
0,5855
0,5755
0,5670
0,5128
0,4835
0,4638
#(«,«)*
0,8955
0,8915
0,8875
0,8840
0,8805
0,8770
0,8735
0,8705
0,8670
0,8640
0,8610
0,8585
0,8555
0,8530
0,8295
0,8105
0,7945
0,7810
0,7690
0,6950
0,6555
0,6285
0,6085
0,5930
0,5795
0,5685
0,5590
0,5505
0,4978
0,4694
0,4502
до*
0,8700
0,8667
0,8633
0,8592
0,8558
0,8533
0,8500
0,8467
0,8442
0,8408
0,8383
0,8358
0,8332
0,8307
0,8083
0,7902
0,7749
0,7617
0,7501
0,6783
0,6396
0,6135
0,5940
0,5784
0,5657
0,5548
0,5454
0,5371
0,4857
0,4580
0,4393
1
1
0,8504
0,8469
0,8436
0,8404
0,8371
0,8342
0,8312
0,8283
0,8256
0,8229
0,8202
0,8176
0,8152
0,8127
0,7912
0,7736
0,7587
0,7458
0,7345
0,6643
0,6264
0,6007
0,5817
0,5664
0,5539
0,5433
0,5342
0,5261
0,4757
0,4486
0,4302
0,9333
0,9288
0,9246
0,9204
0,9167
0,9125
0,9088
0,9054
0,9021
0,8988
0,8954
0,8925
0,8892
0,8863
0,8613
0,8413
0,8246
0,8108
0,7988
0,7242
0,6842
0,6571
0,6367
0,6208
0,6075
0,5963
0,5867
0,5779
0,5246
0,4954
0,4758
869
Таблица XIII
Величины а*, в* и с* Для вычисления коэффициентов переноса в смесях
с помощью потенциала F-12) Л еннарда-Джонса
т*
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,00
2,10
2,20
2,30
2,40
2,50
2,60
А* | В*
1,046
1,062
1,075
1,084
1,093
1,097
1,101
1,102
1,104
1,105
1,105
1,105
1,104
1,103
1,103
1,102
1,102
1,101
1,100
1,099
1,099
1,098
1,097
1,097
1,097
1,096
1,096
1,096
1,095
1,094
1,094
1,094
1,094
1,094
1,094
1,094
1,094
1,094
1,094
1,094
1,094
1,289
1,296
1,296
1,289
1,284
1,275
1,263
1,254
1,24*
1,233
1,223
1,216
1,206
1,200
1,192
1,183
1,179
1,172
1,169
1,165
1,159
1,156
1,154
1,148
1,143
1,140
1,137
1,137
1,133
1,129
]
1,127
,126
,124
,122
1,119
1,116
1,113
1,110
1,108
1,106
1,104
с*
0,8475
0,8392
0,8329
0,8278
0,8251
0,8231
0,8224
0,8223
0,8233
0,8247
0,8265
0,8289
0,8315
0,8342
0,8367
0,8392
0,8417
0,8450
0,8475
0,8508
0,8525
0,8558
0,8583
0,8608
0,8633
0,8658
0,8683
0,8708
0,8725
0,8742
0,8767
0,8783
0,8800
0,8825
0,8842
0,8875
0,8908
0,8933
0,8967
0,8992
0,9017
т*
2,70
2,80
2,90
3,00
3,10
3,20
3,30
3,40
3,50
3,60
3,70
3,80
3,90
4,00
4,10
4,20
4,30
4,40
4,50
4,60
4,70
4,80
4,90
5,00
6,00
7,00
8,00
9,00
10,00
20,00
30,00
40,00
50,00
60,00
70,00
80,00
90,00
100,00
200,00
300,00
400,00
А*
1,094
1,094
1,095
1,095
1,095
1,096
1,096
1,096
1,097
1,097
1,097
1,097
1,097
1,098
1,098
1,098
1,099
1,099
1,099
1,100
1,100
1,100
1,101
1,101
1,103
1,105
1,107
1,109
1,110
1,119
1,124
1,127
1,130
1,132
1,134
1,135
1,137
1,138
1,146
1,151
1,154
в*
1,103
1,104
1,102
1,101
1,100
1,096
1,099
1,096
1,096
1,095
1,097
1,093
1,093
1,095
1,093
1,093
1,094
1,094
1,092
1,091
1,093
1,095
1,091
1,092
1,090
1,092
1,090
1,091
1,094
1,095
1,095
1,095
1,095
1,096
1,095
1,095
1,096
1,095
1,095
1,095
1,095
С*
0,9042
0,9067
0,9083
0,9108
0,9125
0,9133
0,9158
0,9167
0,9183
0,9192
0,9208
0,9217
0,9225
0,9242
0,9250
0,9258
0,9267
0,9275
0,9283
0,9292
0,9300
0,9308
0,9308
0,9317
0,9375
0,9400
0,9425
0,9433
0,9450
0,9475
0,9483
0,9483
0,9483
0,9483
0,9483
0,9483
0,9483
0,9483
0,9483
0,9483
0,9483
870
Таблица XIV
Функции для вычисления высших приближений
для коэффициентов переноса
Г*
0,30
0,50
0,75
1,00
1,25
1,5
2,0
2,5
3,0
4,0
5,0
10,0
50,0
100,0
400,0
1,0014
1,0002
1,0000
1,0000
1,0001
1,0004
1,0014
1,0025
1,0034
1,0049
1,0058
1,0075
1,0079
1,0080
1,0080
1,0022
1,0003
1,0000
1,0001
1,0002
1,0006
1,0021
1,0038
1,0052
1,0076
1,0090
1,0116
1,0124
1,0125
1,0125
1,0001
1,0000
1,0000
1,0000
1,0002
1,0006
1,0016
1,0026
1,0037
1,0050
1,0059
1,0076
1,0080
1,0080
1,0080
871
Таблица XV
Функция [кт]* для вычисления изотопического коэффициента диффузии
[**]* -
.-Д. F-12)
Г* = кТ/ш
т*
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1;50
1,55
1,60
[кт]*
0,086
0,035
-0,002
-0,032
-0,048
-0,059
-0,063
-0,063
-0,057
-0,049
-0,039
-0,025
-0,010
+0,005
0,019
0,033
0,047
0,066
0,080
0,099
0,108
0,127
0,141
0,155
0,169
0,183
0,197
г*
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,0
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
3,0
3,1
3,2
3,3
3,4
3,5
3,6
3,7
3,8
3,9
0,211
0,221
0,230
0,244
0,253
0,263
0,277
0,286
0,305
0,323
0,337
0,356
0,370
0,383
0,397
0,411
0,420
0,434
0,443
0,447
0,461
0,466
0,475
0,479
0,489
0,493
0,497
г*
4,0
4,1
4,2
4,3
4,4
4,5
4,6
4,7
4,8
4,9
5,0
6,0
7,0
8,0
9,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
100,0
200,0
300,0
400,0
[ад*
0,507
0,511
0,516
0,520
0,525
0,529
0,533
0,538
0,543
0,542
0,547
0,578
0,591
0,604
0,607
0,616
0,625
0,627
0,625
0,623
0,622
0,621
0,621
0,620
0,619
0,615-
0,612
0,611
872
Таблица XVГ
Расстояние наибольшего сближения и угол отклонения
X « х{Ь*, g*) - угол отклонения
Ь* = Ь/а — приведенные прицельные расстояния
g** -. i де*/е - приведенная начальная кинетическая энергия
г?п(Ь*,8*) = гт1° — приведенное расстояние наибольшего сближения
1 + У\ +
2
0,0025
0,0040
0,0050
0,0063
0,0075
0,0100
0,0110
0,0120
0,0126
0,833
0,835
0,840
ь*
2,838
2,696
2,643
2,598
2,572
2,544
2,539
2,538
2,537
2,516
2,503
2,470
*
г
я»
2,704
2,500
2,409
2,318
2,251
2,146
2,112
2,082
2,065
1,027
1,026
1,025
X
-0,323
-0,543
-0,706
-0,945
-1,205
-1,998
-2,576
-3,937
— оо
-5,117
-4,339
-3,416
-0,1
2
0,842
0,850
0,860
0,880
0,900
0,912
0,925
0,938
0,950
0,970
1,000
ь*
2,456
2,400
2,328
2,171
1,996
1,881
1,744
. 1,593
1,436
1,1195
0
1,025
1,023
1,021
1,017
1,014
1,011
1,009
1,007
1,005
1,001
0,9960
X
-3,177
-2,509
-1,943
-1,124
-0,491
-0,150
0,201
0,542
0,862
1,437
п
0,0050
0,0075
0,0100
0,0150
0,0170
0,0180
0,0200
0,0220
0,0240
0,0250
0,0254
0,0255
0,0256
0,730
0,735
2,522
2,411
2,350
2,287
2,274
2,269
2,262
2,257
2,256
2,255
2,255
2,255
2,255
2,242
2,226
2,400
2,243
2,138
1,998
1,957
1,938
1,905
1,875
1,848
1,835
1,830
1,829
1,828
1,046
1,045
-0,329
-0,514
-0,718
-1,219
-1,477
-1,628
-1,997
-2,538
-3,660
-5,552
-9,248
-13,592
— оо
-4,140
-4,032
.0,2
i
0,740
0,745
0,750
0,755
0,760
0,780
0,800
0,820
0,850
0,860
0,880
0,900
0,930
0,950
1,000
2,209
2,192
2,175
2,158
2,140
2,065
1,984
1,897
1,751
1,697
1,583
1,455
1,230
1,046
0
1,043
1,042
1,041
1,040
1,039
1,034
1,030
1,026
1,020
1,018
1,014
1,010
1,004
1,001
0,9923
-3,753
-3,472
-3,209
-2,969
-2,751
-2,022
-1,452
-0,982
-0,381
-0,198
0,151
0,49а
1,008
1,377
п
-0,4
0,008
0,010
0,015
0,020
0,025
0,030
0,035
2,297
2,235
2,138
2,083
2,050
2,029
2,015
2,204
2,123
1,984
1,892
1,823
1,768
1,723
-0,269
-0,341
-0,530
-0,737
-0,969
-1,235
-1,553
0,040
0,048
0,049
0,050
0,05366
0,565
2,006
2,002
2,000
1,999
1,999
1,988
1,685
1,635
1,629
1,624
1,605
1,084
-1,959
-3,127
-3,259
-3,777
— оо
-5,121
Параметр z применялся в первоначальном численном определении x(b, g) [47]. Значения пара-
метра z приведены здесь для облегчения интерполяции в таблице.
873
Подолжение табл. XV Г
Z
0,570
0,575
0,583
0,590
0,600
0,610
0,650
0,700
0,750
0,800
0,840
0,850
ь*
1,981
1,974
1,963
1,952
1,938
1,922
1,852
1,750
1,629
1,485
1,347
1,308
г*
1,082
1,081
1,078
1,076
1,073
1,070
1,059
1,046
1,034
1,023
1,015
1,013
X
-4,394
-3,954
-3,514
-3,229
-2,920
-2,656
-1,896
-1,180
-0,586
-0,045
0,384
0,492
0,4
z
0,875
0,900
0,913
0,925
0,938
0,950
0,960
0,970
0,980
0,990
1,000
ь*
1,204
1,086
1,017
0,948
0,866
0,780
0,699
0,608
0,498
0,353
0
г*
т
1,008
1,003
1,001
0,9984
0,9961
0,9940
0,9922
0,9905
0,9888
0,9857
0,9855
• X
0,768
1,055
1,213
1,365
1,539
1,713
1,872
2,048
2,253
2,517
л
- 0,6
0,010
0,015
0,020
0,030
0,040
0,050
0,060
0,065
0,070
0,080
0,088304
0,407
0,410
0,420
0,440
0,450
0,475
-0,230
-0,360
-0,478
-0,752
-1,062
-1,428
-1,887
-2,153
-2,531
-3,746
— оо
-3,235
-3,561
-3,719
-3,353
-3,164
-2,732
0,500
0,550
0,600
0,650
0,700
0,725
0,750
0,775
0,800
0,825
0,850
0,875
0,900
0,925
0,950
0,975
1,00
1,785
1,733
1,670
1,594
1,505
1,454
1,399
1,339
1,273
1,201
1,121
1,031
0,930
0,811
0,667
0,475
0
1,099
1,082
1,067
1,052
1,039
1,033
1,028
1,022
1,017
1,011
1,006
1,002
0,9968
0,9923
0,9879
0,9836
0,9795
-2,381
-1,789
-1,290
-0,832
-0,362
-0,193
0,007
0,208
0,413
0,622
0,838
1,064
1,305
1,569
1,871
2,253
л
- 0,8
0,015
0,020
0,025
0,050
0,075
0,100
0,150
0,1708
0,250
0,275
0,290
0,300
0,335
0,350
0,380
2,044
1,974
1,925
1,812
1,775
1,761
1,755
1,754
1,751
1,747
1,744
1,741
1,730
1,725
1,711
1,961
1,870
1,801
1,605
1,500
1,430
1,336
1,308
1,227
1,208
1,197
1,191
1,169
1,160
1,145
-0,267
-0,360
-0,456
-0,984
-1,639
-2,535
-7,817
-6,091
-4,736
-4,188
-3,891
-3,111
-2,863
-2,518
0,400
0,450
0,500
0,550
0,600
0,650
0,700
0,750
0,800
0,850
0,870
0,900
0,920
0,950
0,970
1,000
1,700
1,667
1,627
1,577
1,517
1,446
1,363
1,266
1,151
1,012
0,949
0,840
0,755
0,602
0,469
0
1,135
1,113
1,093
1,076
1,061
1,047
1,034
1,022
1,011
1,001
0,9969
0,9913
0,9877
0,9824
0,9790
0,9741
-2,340
-1,800
-1,430
-1,072
-0,715
-0,375
-0,040
0,299
0,650
1,025
1,186
1,446
1,635
1,963
2,235
л
874
Продолжение табл. XVI
Z
0,015
0,025
0,050
0,075
0,100
0,110
0,120
0,130
0,150
0,160
0,170
0,180
0,190
0,200
0,220
0,230
0,250
0,270
0,300
b*
2,031
1,908
1,789
1,745
1,726
1,722
]
1
]
1
1
1
]
1
]
1,719
1,716
1,713
i,711
1,710
L,709
1,708
1,707
1,705
1,703
1,700
1,696
1,688
1,956
1,797
1,601
1,496
1,426
1,404
1,383
1,365
1,333
1,319
1,305
1,293
1,281
1,270
1,250
1,241
1,224
1,208
1,187
X
-0,240
-0,408
-0,861
-1,385
-2,005
-2,284
-2,580
-2,889
-3,496
-3,757
-3,964
-4,108
-4,195
-4,235
-4,231
-4,197
-4,074
-3,877
-3,484
.0,9
z
0,350
0,400
0,450
0,500
0,550
0,600
0,650
0,675
0,700
0,725
0,750
0,800
0,850
0,900
0,925
0,950
0,975
1,000
b*
1,669
1,643
1,611
1,569
1,520
1,462
1,392
1,354
1,312
1,267
1,218
1,107
0,974
0,807
0,704
0,579
0,412
0
r*
1,157
1,132
1,110
1,091
1,073
1,058
1,044
1,037
1,031
1,025
1,019
1,008
0,9982
0,9988
0,9842
0,9799
0,9756
0,9715
X
-2,787
-2,197
-1,715
-1,304
-0,936
-0,593
-0,262
-0,099
0,064
0,228
0,393
0,733
1,095
1,500
1,732
2,000
2,341
Л
0,015
0,025
0,050
0,075
0,100
0,150
0,200
0,210
0,220
0,230
0,240
0,250
0,300
0,350
0,400
2,025
1,901
1,778
1,732
1,711
1,695
1,687
1,685
1,683
1,682
1,680
1,678
1,664
1,645
1,619
1,954
1,794
1,599
1,494
1,424
• 1,331
1,269
1,259
1,249
1,240
1,231
1,223
1,186
1,156
1,130
g*. =
-0,229
-0,387
-0,812
-1,289
-1,830
-3,000
-3,608
-3,631
-3,631
-3,612
-3,576
-3,524
-3,084
-2,529
-2,021
0,95
0,450
0,500
0,550
0,600
0,650
0,700
0,750
0,800
0,850
0,900
0,925
0,950
0,975
1,000
1,586
1,545
1,495
1,437
1,369
1,290
1,198
1,088
0,957
0,7925
0,6912
0,5682
0,4045"
0
1,108
1,089
1,072
1,057
1,043
1,030
1,018
1,007
0,9970
0,9875
0,9830
0,9786
0,9744
0,9703
-1,584
-1,201
-0,852
-0,522
-0,202
0,115
0,436
0,768
1,123
1,521
1,750
2,014
2,351
Л
0,025
0,050
0,075
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
1,894
1,768
1,721
1,698
1,678
1,668
1,658
1,643
1,623
1,596
1,563
1,792
1,597
1,492
1,422
1,330
1,267
1,221
1,184
1,154
1,129
1,107
«*«
-0,369
-0,768
-1,207
-1,689
-2,664
-3,178
-3,135
-2,784
-2,320
-1,871
-1,470
= l
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,522
1,473
1,415
1,348
1,270
1,178
1,070
0,9410
0,7795
0,5588
0
1,088
1,071
1,055
1,041
1,028
1,017
1,006
0,9957
0,9863
0,9774
0,9691
-1,109
-0,775
-0,458
-0,148
0,161
0,474
0,800
1,149
1,541
2,027
Л
875
Продолжение табл. XVГ
г
0,025
0,050
0,075
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
ъ*
1,871
1,737
1,681
1,653
1,624
1,607
1,591
1,572
1,550
1,522
1,487
С
1,784
1,589
1,485
1,416
1,323
1,261
1,215
1,179
1,149
1,124
1,102
8*
X
-0,635
-1,313
-1,922
-2,253
-2,261
-2,053
-1,743
-1,411
-1,091
-1,2
z
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
ь*
1,446
1,398
1,342
1,277
1,202
1,115
1,012
0,8893
0,7362
0,5276
0
г*
1,083
1,066
1,050
1,036
1,024
1,012
1,001
0,9910
0,9817
0,9728
0,9646
х
-0,792
-0,507
-0,232
0,042
0,319
0,604
0,905
1,232
1,604
2,069
я
1,4
0,025
0,050
0,075
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
1,853
1,712
1,651
1,618
1,582
1,560
1,540
,519
,494
,465
1,430
1,776
1,582
1,479
1,410
1,318
1,256
1,210
1,174
1,144
1,119
.1,097
-0,546
-1,089
-1,540
-1,779
-1,791
-1,642
-1,407
-1,140
-0,870
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,389
1,342
1,287
1,223
1,151
1,066
0,9678
0,8499
0,7033
0,5038
0
1,078
1,061
1,046
1,032
1,019
1,008
0,9968
0,9867
0,9774
0,9686
0,9604
-0,605
-0,346
-0,091
0,165
0,426
0,697
0,984
1,297
1,654
2,102
л
0,025
0,050
0,075
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
1,837
1,692
1,627
1,591
1,549
1,523
1,500
1,477
1,450
1,420
1,385
1,769
1,576
1,473
1,404
1,312
1,251
1,205
1,169
1,139
1,114
1,093
g*% =
-0,481
-0,938
-1,297
-1,485
-1,494
-1,373
-1,177
-0,947
-0,706
. 1,6
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,344
1,297
1,243
1,181
1,110
1,028
0,9326
0,8186
0,6771
0,4849
0
1,074
1,057
1,041
1,028
1,015
1,003
0,9927
0,9827
0,9734
0,9647
0,9565
-0,465
-0,224
0,016
0,259
0,508
0,768
1,045
1,347
1,693
2,129
я
0,025
0,050
0,075
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
1,824
1,675
1,607
1,568
1,522
1,493
1,468
1,442
1,415
1,383
1,347
1,762
1,570
1,467
1,399
1,307
1,246
1,200
1,165
1,135
1,110
1,088
в**
-0,432
-0,827
-1,128
-1,281
-1,285
-1,180
-1,007
-0,801
-0,580
-1,8
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,307
1,260
1,207
1,146
1,077
0,9968
0,9038
0,7930
0,6558
0,4694
0
1,069
1,053
1,037
1,024
1,011
0,9996
0,9889
0,9789
0,9697
0,9610
0,9528
-0,355
-0,128
0,101
0,334
0,575
0,826
1,094
1,388
1,726
2,151
Л
876
Продолжение табл. XVI
= 2,0
z
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
b*
1,661
1,549
1,500
1,467
1,440
1,413
1,385
1,353
1,317
1,276
r*
m
1,564
1,393
1,302
1,241
]
1,196
1,160
1,131
1,106
,084
1,066
X
-0,393
-0,743
-1,001
-1,130
-1,126
-1,033
-0,876
-0,686
-0,480
-0,267
z
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
b*
1,229
1,177
1,117
1,049
0,9706
0,8797
0,7716
0,6379
0,4565
0
r*
m
1,049
1,034
1,020
1,007
0,9960
0,9853
0,9754
0,9662
0,9575
0,9493
X
-0,050
0,170
0,396
0,629
0,874
1,135
1,422
1,752
2,169
я
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1,632
1,512
1,456
1,419
1,388
1,358
1,327
1,294
1,257
1,216
1,551
1,382
1,292
1,231
1,186
1,151
1,122
1,097
1,076
1,057
g*% •¦
-0,323
-0,597
-0,789
-0,877
-0,868
-0,783
-0,647
-0,481
-0,298
-0,105
= 2,5
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,170
1,119
1,061
0,9953
0,9204
0,8335
0,7306
0,6035
0,4316
0
1,040
1,025
1,012
0,9992
0,9878
0,9772
0,9674
0,9582
0,9496
0,9415
0,094
0,299
0,511
0,731
0,963
1,212
1,486
1,803
2,203
л
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1,610
1,484
1,424
1,383
1,349
1,317
1,285
1,251
1,214
1,173
1,540
1,372
1,282
1
1
1
]
1,222
1,178
1,142
1,113
1,089
1,068
1,049
g*% ¦
-0,277
-0,504
-0,656
-0,720
-0,703
-0,622
-0,497
-0,345
-0,175
0,006
= 3,0
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,128
1,077
1,020
0,9566
0,8839
0,7999
0,7008
0,5786
0,4136
0
1,033
1,018
1,004
0,9919
0,9806
0,9701
0,9603
0,9512
0,9427
0,9347
0,194
0,389
0,591
0,802
1,026
1,266
1,532
1,839
2,228
я
- 4,0
1,576
1,444
1,378
1,332
1,295
1,261
1,227
1,192
1,154
1,113
1
]
]
1,521
1,355
1,266
1,207
1,163
1,128
1,099
1,075
1,054
1,036
-0,219
-0,390
-0,496
-0,531
-0,503
-0,425
-0,311
-0,172
-0,017
0,149
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,068
1,020
0,9643
0,9029
0,8334
0,7536
0,6596
0,5442
0,3887
0
1,020
1,005
0,9916
0,9794
0,9683
0,9579
0,9483
0,9393
0,9308
0,9229
0,324
0,507
0,697
0,898
1,110
1,339
1,593
1,887
2,262
877
Продолжение табл. XVГ
Z
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
ь*
1,551
1,415
1,345
1,298
1,258
1,222
1,187
1,151
1,113
1,073
1,504
1,340
1,253
1,194
1,151
1,116
1,088
1,064
1,043
1,025
X
-0,184
-0,321
-0,401
-0,420
-0,385
-0,307
-0,198
-0,066
0,082
0,240
= 5,0
2
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
ь*
1,028
0,9799
0,9263
0,8666
0,7994
0,7223
0,6318
0,5209
0,3719
0
Г*
m
1,009
0,9943
0,9811
0,9691
0,9580
0,9478
0,9382
.0,9293
0,9210
0,9132
X
0,407
0,582
0,766
0,959
1,164
1,387
1,633
1,919
2,284
я
= 10
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1,477
1,334
1,259
1,205
1,162
1,123
1,086
1,049
1,011
0,9704
1,449
1,291
1,207
1,150
1,108
1,075
1,048
1,025
1,005
0,9874
-0,110
-0,179
-0,206
—0,192
-0,141
-0,060
0,043
0,163
0,297
0,440
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
0,9277
0,8816
0,8315
0,7762
0,7145
0,6444
0,5627
0,4632
0,3302
0
0,9718
0,9578
0,9451
0,9335
0,9229
0,9130
0,9038
0,8953
0,8872
0,8797
0,592
0,752
0,920
1,099
1,289
1,496
1,726
1,993
2,335
я
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1,435
1,291
1,214
1,159
1,115
1,075
1,037
1,000
0,9620
0,9225
1,414
1,260
1,178
1,122
1,081
1,049
1,023
1,000
0,9806
0,9635
g** -
-0,082
-0,127
-0,136
-0,109
-0,051
0,031
0,133
0,250
0,379
0,518
= 15
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
0,8807
0,8360
0,7875
0,7344
0,6754
0,6086
0,5310
0,4368
0,3111
0
0,9483
0,9347
0,9223
0,9110
0,9005
0,8909
0,8819
0,8736
0,8657
0,8584
0,664
0,819
0,982
1,155
1,339
1,540
1,763
2,023
2,356
я
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1,405
1,262
1,184
1,129
1,084
1,044
1,006
0,9690
0,9312
0,8922
1,388
1,237
1,156
1,102
1,062
1,030
1,004
0,9818
0,9627
0,9460
g*%
-0,066
-0,099
-0,098
-0,064
-0,003
0,081
0,183
0,299
0,425
0,561
= 20
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
0,8511
0,8073
0,7600
0,7083
0,6510
0,5863
0,5113
0,4204
0,2993
0
0,9311
0,9176
0,9055
0,8944
0,8841
0,8747
0,8659
0,8577
0,8500
0,8428
0,705
0,857
1,017
1,186
1,368
1,565
1,785
2,040
2,368
я
878
Продолжение табл. XV Г
z
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
ъ*
1,312
1,173
1,096
1,041
0,9964
0,9567
0,9196
0,8834
0,8472
0,8100
1,304
1,162
1,086
1,035
0,9971
0,9672
0,9427
0,9220
0,9040
0,8883
8**
X
-0,034
-0,040
-0,017
0,030
0,100
0,187
0,289
0,402
0,525
0,656
-50
z
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
ь*
0,7712
0,7303
0,6864
0,6387
0,5862
0,5273
0,4593
0,3772
0,2683
0
г*
0,8743
0,8617
0,8503
0,8399
0,8303
0,8214
0,8131
0,8054
0,7982
0,7914
X
0,794
0,940
1,093
1,256
1,431
1,621
1,832
2,079
2,395
п
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1,244
1,109
1,035
0,9812
0,9376
0,8990
0,8630
0,8280
0,7931
0,7576
1,239
1,104
1,032
0,9835
0,9476
0,9192
0,8959
0,8762
0,8592
0,8442
g*t e
-0,020
-0,013
0,018
0,072
0,145
0,234
0,336
0,449
0,570
0,699
¦• 100
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
0,7206
0,6818
0,6403
0,5954
0,5461
0,4908
0,4272
0,3507
0,2493
0
0,8309
0,8190
0,8081
0,7982
0,7891
0,7806
0,7728
0,7654
0,7586
0,7521
0,835
0,978
1,129
1,289
1,460
1,646
1,854
2,097
2,407
Л
879
Таблица XVII
Значения второго вириального коэффициента для газа полярных молекул
(потенциал Штокмайера)
В{Т) - Ъ0В*(Т*; *¦) Т* - *Т/е
8 *(Т* Г»)
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
•0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,00
2,1
2,2
2,3
0,1
-31,129
-20,355
-14,717
-11,339
-9,1199
-7,5631
-6,4159
-5,5381
-4,8460
-4,2871
-3,8268
-3,4414
-3,1142
-2,8330
-2,5889
-2,3750
-2,1862
-2,0183
-1,8680
-1,7328
-1,6105
-1,4994
-1,3980
-1,3051
-1,2197
-1,1410
-1,0681
-1,0006
-0,93775
-0,87917
-0,82445
-0,77322
-0,72516
-0,67998
-0,63745
-0,55947
-0,48969
-0,42693
0,2
-42,968
-25,879
-17,777
-13,241
-10,401
-8,4786
-7,1001
-6,0677
-5,2675
-4,6304
-4,1116
-3,6815
-3,3193
-3,0103
-2,7437
-2,5114
-2,3072
-2,1265
-1,9653
-1,8208
-1,6905
-1,5724
-1,4649
-1,3667
-1,2766
-1,1937
-1,1171
-1,0462
-0,98038
-0,91908
-0,86190
-0,80844
-0,75834
-0,71130
-0,66707
-0,58607
-0,51373
-0,44877
0,3
-72,01
-38,07
-24,090
-16,985
-12,841
-10,181
-8,3495
-7,0213
-6,0183
-5,2364
-4,6110
-4,1000
-3,6758
-3,3166
-3,0102
-2,7455
-2,5145
-2,3113
-2,1312
-1,9706
-1,8264
-1,6963
-1,5784
-1,4710
-1,3728
-1,2827
-1,1998
-1,1232
-1,0523
-0,98633
-0,92498
-0,86772
-0,81417
-0,76398
-0,71686
-0,63076
-0,55409
-0,48540
0,4
-64,11
-36,28
-23,733
-17,026
-12,996
-10,360
-8,5234
-7,1813
-6,1627
-5,3659
-4,7271
-4,2045
-3,7695
-3,4021
-3,0881
-2,8167
-2,5799
-2,3716
-2,1870
-2,0223
-1,8746
-1,7413
-1,6205
-1,5106
-1,4101
-1,3179
-1,2330
-1,1547
-1,0821
-1,0147
-0,95197
-0,89345
-0,83873
-0,78747
-0,69408
-0,61121
-0,53721
0,6
-60,4
-35,92
-24,11
-17,53
-13,477
-10,789
-8,8965
-7,5043
-6,4433
-5,6113
-4,9432
-4,3961
-3,9406
-3,5559
-3,2271
-2,9430
-2,6952
-2,4773
-2,2844
-2,1124
-1,9581
-1,8190
-1,6931
-1,5785
-1,4738
-1,3778
-1,2896
-1,2079
-1,1325
-1,0625
-0,99736
-0,93662
-0,87985
-0,77679
-0,68573
-0,60472
0,6
-58,8
-36,36
-24,91
-18,33
-14,185
-11,394
-9,413
-7,9476
-6,8267
-5,9457
-5,2373
-4,6567
-4,1732
-3,7649
-3,4160
-3,1146
-2,8519
-2,6211
-2,4168
-2,2348
-2,0717
-1,9247
-1,7917
-1,6708
-1,5604
-1,4594
-1,3662
-1,2804
-1,2011
-1,1275
-1,0590
-0,99526
-0,87993
-0,77850
-0,68865
0,7
-59,0
-37,3
-26,0
-19,34
-15,05
-12,13
-10,040
-8,486
-7,292
-6,3523
-5,5953
-4,9744
-4,4572
-4,0203
-3,6471
-3,3249
-3,0442
-2,7976
-2,5795
-2,3854
-2,2115
-2,0549
-1,9133
-1,7846
-1,6674
-1,5597
-1,4610
-1,3699
-1,2858
-1,2078
-1,1353
-1,0048
-0,89060
-0,78989
Таблица составлена Раулинсоном [48 ].
880
Продолжение табл. XVII
2,4
2,5
2,6
2,7
2,8
2,9
3,0
3,1
3,2
3,3
3,4
3,5
3,6
3,7
3,8
3,9
4,0
4,1
4,2
4,3
4,4
4,5
4,6
4,7
4,8
4,9
5,0
6
7
8
9
10
20
30
40
50
60
70
80
90
100
200
300
400
0,1
-0,37020
-0,31868
-0,27172
-0,22875
-0,18929
-0,15295
-0,11937
-0,08828
-0,05941
-0,03254
-0,00748
+0,01594
0,03787
0,05844
0,07778
0,09597
0,11312
0,12930
0,14460
0,15907
0,17279
0,18580
0,19815
0,20990
0,22107
0,23172
0,24187
0,32187
0,37532
0,41284
0,44012
0,46049
0,52527
0,52687
0,51854
0,50834
0,49820
0,48864
0,47978
0,47161
0,46406
0,41143
0,38013
0,35835
0,2
-0,39012
-0,33695
-0,28852
-0,24426
-0,20366
-0,16630
-0,13182
-0,09991
-0,07030
-0,04277
-0,01710
+0,00688
0,02931
0,05035
0,07011
0,08869
0,10620
0,12272
0,13833
0,15309
0,16708
0,18034
0,19293
0,20489
0,21627
0,22711
0,23744
0,31877
0,37302
0,41106
0,43870
0,45932
0,52495
0,52672
0,51845
0,50828
0,49815
0,48861
0,47976
0,47159
0,46405
0,41142
0,38012
1 0,35835
о,з
-0,42354
-0,36576
-0,31668
-0,27025
-0,22774
-0,18867
-0,15266
-0,11937
-0,08852
-0,05986
-0,03318
-0,00828
+0,01501
0,03682
0,05729
0,07653
0,09465
0,11173
0,12786
0,14310
0,15754
0,17122
0,18420
0,19652
0,20825
0,21941
0,23004
0,31360
0,36918
0,40809
0,43633
0,45738
0,52441
0,52647
0,51830
0,50818
0,49808
0,48855
0,47971
0,47155
0,46402
0,41142
0,38012
0,35835
0,4
-0,47076
-0,41079
-0,35642
-0,30692
-0,26167
-0,22018
-0,18200
-0,14677
-0,11417
-0,08393
-0,05580
-0,02959
-0,00511
+0,01780
0,30928
0,05944
0,07841
0,09628
0,11314
0,12907
0,14414
0,15841
0,17194
0,18478
0,19699
0,20860
0,21965
0,30634
0,36380
0,40393
0,43301
0,45466
0,52367
0,52611
0,51809
0,50804
0,49798
0,48847
0,47965
0,47150
0,46398
0,41140
0,38011
0,35834
0,5
-0,53224
-0,46702
-0,40807
-0,35453
-0,30572
-0,26106
-0,22005
-0,18229
-0,14740
-0,11509
-0,08509
-0,05717
-0,03113
-0,00680
+0,01598
0,03736
0,05744
0,07633
0,09414
0,11095
0,12684
0,14187
0,15611
0,16962
0,18245
0,19465
0,20625
0,29699
0,35687
0,39857
0,42873
0,45116
0,52271
0,52566
0,51782
0,50876
0,49785
0,48838
0,47957
0,47144
0,46392
0,41139
0,38011
0,35834
о,б
-0,60856
-0,53676
-0,47205
-0,41347
-0,36020
-0,31159
-0,26705
-0,22612
-0,18839
-0,15351
-0,12118
-0,09115
-0,06318
-0,03708
-0,01268
+0,01018
0,03163
0,05180
0,07078
0,08868 •
0,10558
0,12155
0,13668
0,15101
0,16461
0,17752
0,18980
0,28552
0,34838
0,39201
0,42349
0,44687
0,52153
0,52510
0,51749
0,50764
0,49769
0,48826
0,47948
0,47136
0,46386
0,41137
0,38010
: 0,35833
0,7
-0,70049
-0,62063
-0,54292
-0,48420
-0,42552
-0,37210
-0,32330
-0,27854
-0,23738
-0,19940
-0,16427
-0,13169
-0,10140
-0,07318
-0,04684
-0,02219
+0,00091
0,02259
0,04298
0,06218
0,08029
0,09740
0,11357
0,12888
0,14340
0,15718
0,17026
0,27191
0,33832
0,38424
0,41729
0,44179
0,52014
0,52444
0,51710
0,50738
0,49750
0,48811
0,47937
0,47127
0,46379
0,41135
0,38009
0,35833
56 2017
881
Продолжение табл. XVИ'
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,00
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
3,0
3,1
3,2
3,3
3,4
i
0,8
-58,8
-38,5
-27,3
-20,50
-16,05
-12,973
-10,759
-9,103
-7,828
-6,820
-6,008
-5,3413
-4,7855
-4,3161
-3,9152
-3,5690
-3,2676
-3,0030
-2,7691
-2,5609
-2,3745
-2,2069
-2,0554
-1,9180
-1,7923
-1,6775
-1,5720
-1,4749
-1,3852
-1,3021
-1,1531
-1,0234
-0,90957
-0,80895
-0,71944
-0,63934
-0,56729
-0,50216
-0,44304
-0,38917
-0,33989
-0,29466
-0,25302
-0,21459'
0,9
1
| -59,7
-40,0
-28,8
-21,78
-17,14
-13,90
-11,612
-9,790
-8,440
-7,342
-6,4696
-5,7520
-5,1537
-4,6483
-4,2164
-3,8438
-3,5193
-3,2345
-2,9829
-2,7591
-2,5589
-2,3788
-2,2165
-2,0685
-1,9340
-1,8Н0
-1,6981
-1,5941
-1,4982
-1,3268
-1,1785
-1,0490
-0,93509
-0,83413
-0,74412i
-0,66342
-0,59072
-0,52492
-0,46511
-0,41054
-0,36057
-0,31468
-0,27239'
1,0
i
1
-41,8
-30,4
-23,2
-18,4
-14,92
-12,42
-10,54
-9,08
-7,915
-6,976
-6,203
-5,559
-5,014
-4,5484
-4,1466
-3,7970
-3,4903
-3,2192
-2,9783
-2,7629
-2,5697
-2,3943
-2,2357
-2,0912
-1,9591
-1,8380
-1,7265
-1,5285
-1,3580
-1,2100
-1,0803
-0,96584
-0,86421
-0,77343
-0,69191
-0,61833
-0,55165
-0,49096
-0,43552;
-0,38472,
-0,33800!
!
! -43,5
-32,0
-24,7
-19,61
-16,00
-13,36
-11,35
-9,780
-8,534
-7,524
-6,693
-5,998
-5,4111
-4,9092
-4,4762
-4,0994
-3,7688
-3,4768
-3,2173
-2,9860
-2,7770
-2,5889
-2,4183
-2,2630
-2,1211
-1,9910
-1,7610
-1,5643
-1,3943
-1,2461
-1,1159
-1,0008
-0,89828
-0,80654
-0,72400
-0,64940
-0,58168
-0,51998
-0,46355
-0,41179
1,2
i
1
I
I
-45,8
-34,4
-26,3
-21,0
-17,2
-14,3
-12,21
-10,53
-9,20
-8,11
-7,21
-6,471
-5,839
-5,298
-4,831
-4,426
-4,069
-3,7548
-3,4760
-3,2256
-3,0014
-2,7990
-2,6156
-2,4487
-2,2963
-2,0282
-1,8002
-1,6044
-1,4345
-1,2860
-1,1551
-1,0391
-0,93559
-0,84275
-0,75908,
-0,68333
-0,61449
-0,55167
-0,49417
1 1.3
i
1
-47,0
-35,7
-27,9
-22,3
-18,4
-15,42
-13,13
-11,34
-9,90
-8,74
-7,780
-6,976
-6,296
-5,714
-5,212
-4,774
-4,3909
-4,0532
-3,7517
-3,4831
-3,2419
-3,0244
-2,8273
-2,6479
-2,3342
-2,0693
-1,8431
-1,6479
-1,4780
-1,3289
-1,1972
-1,0802
-0,97554
-0,88152
-0,79663
-0,71967
-0,64962
-0,58562
1,4
-50,3
-37,8
-29,8
-24,3
-19,6
-16,5
-14,1
-12,2
-10,7
-9,41
-8,38
-7,51
-6,78
-6,15
-5,612
-5,145
-4,735
-4,370
-4,046
-3,758
-3,498
-3,265
-3,053
-2,6846
-2,3758
-2,1138
-1,8889
-1,6940
-1,5239
-1,3742
-1,2416
-1,1235
-1,0177
-0,92240
-0,83625
-0,75801
-0,68671
-52,0
-40,0
-31,0
-25,4
-21,1
-17,7
-15,1
-13,1
-11,45
-10,12
-9,00
-8,08
-7,29
-6,62
-6,045
-5,541
-5,097
-4,706
-4,359
-4,049
-3,771
-3,520
-3,0857
-2,7248
-2,4204
-2,1606
-1,9368
-1,7423
-1,5719
-1,4215
-1,2879
-1,1687
-1,0616
-0,96505
-0,87758
-0,79805
882
Продолжение табл. XVII
3,5
3,6
3,7
3,8
3,9
4,0
4,1
4,2
4,3
4,4
4,5
4.6
4.7
4.8
4,9
5.0
6
7
8
9
10
20
30
40
50
60
70
80
90
100
200
300
400
0,8
-0,17900
-0,14598
-0,11527
-0,08664
-0,05989
-0,03486
-0,01140
+0,01064
0,03137
0,05089
0,06932
0,08672
0,10318
0,11877
0,13355
0,14758
0,25614
0,32667
0,37524
0,41012
0,43593
0,51854
0,52367
0,51665
0,50707
0,49729
0,48795
0,47924
0,47117
0,46370
! 0,41132
0,38008
0,35832
0,9
-0,23332
-0,19713
-0,16352
-0,13225
-0,10308
-0,07582
-0,05030
-0,02636
-0,00387
+0,01729
0,03723
0,05605
0,07383
0,09065
0,10659
0,12170
0,23818
0,31341
0,36502
0,40197
0,42927
0,51672
0,52281
0,51613
0,50673
0,49704
0,48776
0,47909
0,47105
0,46360
0,41129
1 0,38006
0,35831
1,0
-0,29493
-0,25510
-0,21818
-0,18388
-0,15193
-0,12213
-0,09426
-0,06815
-0,04366
-0,02064
+0,00103
0,02145
0,04073
0,05896
0,07620
0,09255
0,21799
0,29853
0,35355
0,39283
0,42180
0,51468
0,52184
0,51556
0,50635
0,49676
0,48755
0,47893
0,47091
0,46349
0,41126
0,38005
0,35830
1,1
-0,36415
-0,32018
-0,27949
-0,24176
-0,20667
-0,17397
-0,14345
-0,11490
-0,08814
-0,06302
-0,03941
-0,01717
+0,00380
0,02360
0,04232
0,06004
0,19554
0,28199
0,34082
0,38270
0,41353
0,51243
0,52077
0,51493
0,50593
0,49646
0,48732
0,47874
0,47077
0,46337
0,41123
0,38003
0,35829
1,2
-0,44135
-0,39270
-0,34776
-0,30615
-0,26752
-0,23158
-0,19807
-0,16677
-0,13747
-0,11000
-0,08421
-0,05995
-0,03709
-0,01554
+0,00483
0,02409
0,17077
0,26379
0,32682
0,37157
0,40444
0,50996
0,51960
0,51423
0,50546
0,49613
0,48707
0,47854
0,47061
0,46323
0,41119
0,38001
0,35828
,,з !
-0,52697
-0,47305
-0,42333
-0,37736
-0,33476
-0,29519
-0,25834
-0,22397
-0,19184
-0,16175
-0,13353
-0,10702
-0,08207
-0,05856
-0,03637
-0,01540
+0,14364
0,24389
0,31154
0,35942
0,39453
0,50728
0,51833
0,51348
0,50496
0,49576
0,48679
0,47833
0,47043
0,46309
0,41115
1 0,37999
0,35827
1,4
-0,62149
-0,56164
-0,50657
-0,45574
-0,40870
-0,36507
-0,32451
-0,28673
-0,25145
-0,21846
-0,18755
-0,15854
-0,13127
-0,10560
-0,08140
-0,05855
+0,11409
0,22225
0,29494
0,34625
0,38379
0,50438
0,51695
0,51266
0,50441
0,49537
0,48650
0,47810
0,47024
0,46293
0,41110
0,37997
0,35825
1,5
-0,72546
-0,65899
-0,59792
-0,54166
-0,48968
-0,44155
-0,39687
-0,35530
-0,31654
-0,28033
-0,24645
-0,21469
-0,18486
-0,15681
-0,13039
-0,10548
+0,08206
0,19885
0,27702
0,33203
0,37221
0,50125
0,51547
0,51179
0,50383
0,49495
0,48618
0,47784
0,47004
0,46276
0,41105
0,37994
0,35824
Таблица XVIII
Коэффициент Джоуля—Томсона для полярных газов (потенциал Штокмайера)
^*
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,0
2Д
2,2
2,3
о,,
123,498
72,564
48,640
35,492
27,438
22,1035
18,3565
15,6028
13,5052
11,8606
10,5403
9,4591
8,5590
7,7987
7,1487
6,5870
6,0970
5,6661
5,2843
4,9437
4,6382
4,3627
4,1129
3,8855
3,6776
3,4870
'3,3113
3,1492
2,9990
2,85950
2,72960
2,60837
2,49495
2,38863
2,28878
2,10628
1,94360
1,79774
/"°Ср = Ьо [В* — В*1 Т* = кТ/е
Ьо = | л Л? а8 * = 8-1
В*(Т*,/*) — В* (Т*;
0,2
197,638
103,764
64,580
44,773
33,366
0,3
402,4
179,79
100,272
64,351
45,340
26,1573 34,084
21,2763
17,7928
15,2019
13,2102
11,6374
10,3674
9,3226
8,4494
7,7095
7,0753
6,5258
6,0457
5,6227
5,2473
4,9121
4,6109
4,3390
4,0924
3,8676
3,6620
3,4732
3,2993
3,13866
2,98972
2,85133
2,72242
2,60204
2,48938
2,38375
2,19109
2,01983
1,86666
26,8448
21,8888
0,4
364,1
177,22
103,156
67,640
48,166
36,383
28,7059
18,3263 23,4079
15,6644
13,6121
11,9883
10,6764
9,5953
8,6921
7,9269
7,2710
6,7030
6,2609
5,7700
5,3824
5,0365
4,7260
4,4457
4,1916
3,9602
3,7486
3,5545
3,3757
3,21054
3,05758
2,91549
2,78319
2,65970
2,54419
2,33421
2,14835
1,98271
19,5818
16,7158
14,5035
12,7530
11,3386
10,1751
9,2036
8,3816
7,6778
7,0694
6,5385
6,0716
5,6582
5,2896
4,9591
4,6613
4,3916
4,1463
3,9222
3,7168
3,5279
3,3536
3,19224
3,04254
2,90328
2,77342
2,53836
2,33137
2,14779
¦)
0,5
349,5
181,58
109,34
72,96
52,405
39,738
31,3874
25,5836
21,3750
18,2157
15,7751
13,8439
12,2845
11,0032
9,9346
9,0318
8,2601
7,5939
7,0137
6,5043
6,0538
5,6528
5,2940
4,9712
4,6791
4,4138
4,1720
3,9503
3,7469
3,5595
3,38617
3,22556
3,07632
2,80747
2,57215
2,36456
0,6
347,1
189,8
117,58
79,68
57,691
43,904
34,718
28,2907
23,6106
20,0896
17,3667
15,2118
13,4727
12,0450
10,8556
9,8520
8,9955
8,2573
7,6153
7,0525
6,5557
6,1141
5,7196
5,3651
5,0449
4,7547
4,4900
4,2482
4,0265^
3,8224*
3,6339
3,45947
3,14689
2,87504
2,63666
0,7
352,0
200,6
127,3
87,51
63,83
48,76
38,611
31,464
26,238
22,2972
19,2456
16,8297
14,8801
13,2805
11,9493
10,8272
9,8708
9,0475
8,3325
7,7068
7,1550
6,6655
6,2285
5,8365
5,4832
5,1627
4,8715
4,6055
4,3620
4,1380
3,9314
3,5635
3,24571
2,96873
"таблица рассчитана Раулинсоном по данным, полученным методом перфорированных карт [48, 49].
884
Пподолжение табл. XVIII
т*\
2,4
2,5
2,6
2,7
2,8
2,9
3,0
3,1
3,2
3,3
3,4
3,5
3,6
3,7
3,8
3,9
4,0
4,1
4,2
4,3
4,4
4,5
4,6
4,7
4,8
4,9
5,0
6,0
7,0
8,0
9,0
10,0
20
30
40
50
60
70
80
90
100
200
300
400
0,1
1,66625
1,54710
1,43869
1,33963
1,24877
1,16516
1,08796
1,01650
0,95015
0,88839
0,83078
0,77692
0,72646
0,67910
0,63454
0,59258
0,55299
0,51559
0,48018
0,44664
0,41481
0,38457
0,35582
0,32843
0,30234
0,27742
0,25364
0,06411
-0,06559
-0,15923
-0,22952
-0,28389
-0,49641
-0,54427
-0,55780
-0,55995
-0,55754
-0,55312
-0,54785
-0,54224
-0,53657
-0,48897
-0,45665
-0,43310
0,2
1,72885
1,60426
1,49107
1,38782
1,29327
1,20638
1,12627
1,05219
0,98349
0,91962
0,86009
0,80448
0,75243
0,70361
/0,65773
0,61455
0,57383
0,53538
0,49901
0,46458
0,43192
0,40091
0,37143
0,34338
0,31665
0,29115
0,26682
0,07325
-0,05885
-0,15404
-0,22540
-0,28052
-0,49550
-0,54384
-0,55754
-0,55978
-0,55741
-0,55304
-0,54778
-0,54219
-0,53653
-0,48895
-0,45664
-0,43310
0,3
1,83421
1,70036
1,57911
1,46879
1,36801
1,27558
1,19055
1,11205
1,03939
0,97194
0,90919
0,85065
0,79592
0,74465
0,69654
0,65130
0,60869
0,56849
0,53051
0,49458
0,46053
0,42823
0,39754
0,36837
0,34058
0,31410
0,28884
0,08850
-0,04761
-0,14539
-0,21852
-0,27491
-0,49398
-0,54313
-0,55712
-0,55950
-0,55721
-0,55288
-0,54765
-0,54208
-0,53645
-0,48893
-0,45663
-0,43310
0,4
1,98389
1,83675
1,70393
1,58350
1,47379
1,37347
1,28140
1,19663
1,11834
1,04582
0,97846
0,91575
0,85724
0,80251
0,75122
0,70308
0,65779
0,61512
0,57486
0,53680
0,50079
0,46666
0,43427
0,40351
0,37424
0,34637
0,31981
0,10993
-0,03184
-0,13326
-0,20887
-0,26704
-0,49186
-0,54212
-0,55653
-0,55911
-0,55693
-0,55266
-0,54748
-0,54195
-0,53634
-0,48889
-0,45661
-0,43308
0,5
2,18016
2,01533
1,86717
1,73333
1,61183
1,50109
1,39976
1,30675
1,22105
1,14185
1,06843
1,00031
0,93682
0,87758
0,82214
0,77018
0,72140
0,67551
0,63227
0,59147
0,55289
0,51639
0,48180
0,44896
0,41776
0,38809
0,35983
0,13758
-0,01150
-0,11762
-0,19644
-0,25690
-0,48913
-0,54084
-0,55577
-0,55860
-0,55656
-0,55239
,-0,54726
-0,54177
-0,53619
-0,48886
-0,45660
-0,43307
0,6
2,42601
2,23865
2,07097
1,92014
1,78373
1,65983
1,54684
1,44343
1,34843
1,26088
1,17996
1,10496
1,03527
0,97037
0,90976
0,85308
0,79994
0,75004
0,70310
0,65887
0,61712
0,57767
0,54032
0,50492
0,47134
0,43943
0,40909
0,17155
0,01345
-0,09846
-0,18121
-0,24448
-0,48579
-0,53926
-0,55484
-0,55798
-0,55611
-0,55205
-0,54700
-0,54156
-0,53601
-0,48880
-0,45657
-0,43305
0,7
2,72533
2,50995
2,31811
2,14628
1,99151
1,85143
1,72415
1,60802
1,50166
1,40391
1,31382
1,23052
1,15329
1,08154
1,01468
0,95227
0,89387
0,83913
0,78772
0,73938
0,69380
0,65078
0,61014
0,57167
0,53522
0,50062
0,46776
0,21194
0,04307
-0,07572
-0,16317
-0,22977
-0,48183
-0,53740
-0,55374
-0,55725
-0,55558
-0,55165
-0,54669
-0,54130
-0,53581
-0,48874
-0,45654
-0,43304
885
Продолжение табл. XVIII
г* \-^
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,0
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
3,0
3,1
3,2
3,3
0,8
360,0
213,0
138,2
96,3
70,72
54,24
43,02
35,071
29,234
24,818
21,395
18,6830
16,4942
14,6991
13,2061
11,9486
10,8780
9,9574
9,1590
8,4609
7,8463
7,3015
6,8161
6,3813
5,9891
5,6346
5,3125
5,0189
4,7502
4,5034
4,0662
3,6913
3,3666
3,08298
2,83332
2,61200
2,41467
2,23766
2,07805
1,93355
; i,802ie
1,68218
1,57224
0,9
372,0
227,0
150,3
105,82
78,31
60,29
47,981
39,097
32,600
27,645
23,808
20,766
18,3101
16,2961
14,6216
13,2126
12,0135
10,9836
10,0913
9,3120
8,6265
8,0198
7,4800
6,9960
6,5610
6,1679
5,8113
5,4863
5,1893
4,6661
4,2207
3,8376
3,5050
3,2138
2,9569
, 2,72887
2,52523
2,34232
2,17732
2,02781
; 1,89171
[ 1,7673'
1,0
242,0
163,2
116,3
86,6
66,93
53,31
43,54
36,29
30,776
26,485
23,078
20,327
18,071
18,1956
14,6177
13,2764
12,1252
11,1284
10,2587
9,4946
8,8194
8,2178
7,6804
7,1974
6,7614
6,3661
: 6,0064
5,3769
4,8450
4,3908
. 3,9987
1 3,6573
1 3,3578
3,0932
2,8579
1 2,6474
\ 2,458 И
I 2,2873:
2,13231
1 1,9911
258,4
176,8
127,3
95,50
74,12
59,20
48,40
40,350
34,213
29,427
25,623
" 22,548
20,026
17,930
16,1663
14,6681
13,3829
12,2709
11,3016
; 10,4514
9,6990
9,0309
8,4339
7,8981
; 7,4147
' 6,9770
1 6,2158
5,5779
5,0369
4,5729
4,1714
3,8210
, 3,5128
I 3,2400
2,9970
) 2,7794
\ 2,5835
2,4064
1 2,2455'
1,2
i
i
1
1
276,0
192,0
139,2
105,1
81,9
65,5
53,67
44,77
37,97
32,64
28,39
24,980
22,167
19,828
17,861
16,192
14,759
13,521
12,4433
11,4964
10,6612
1 9,9194
9,2575
8,6636
8,1284
7,2043
6,4361
5,7895
5,2387
4,7650
4,3537
3,9939
3,6769
3,3955
3,1447
2,9197
' 2,7169
1 2,5332
1,3
293,0
206,6
151,6
115,2
90,2
72,42
59,37
49,57
42,03
36,13
31,436
27,626
24,498
21,895
19,708
17,849
16,257
14,8825
13,6838
12,6337
11,7072
10,8855
10,1524
9,4952
8,3684
7;4402
6,6650
6,0093
5,4488
4,7650
4,5439
4,1746
3,8483
3,5585
3,2996
3,0669
2,8571
1,4
i
i
313,0
223,0
165,0
126,0
99,0
79,8
65,5
54,7
46,5
39,93
34,72
30,49
27,02
24,14
21,71
19,65
17,884
16,358
15,029
13,866
12,840
11,932
11,121
9,7415
8,6158
7,6835
6,9006
6,2357
5,6653
5,1715
4,7406
4,3616
4,0263
3,7278
3,4606
3,2204
1,5
332,0
239,0
178,0
137,5
108,7
87,6
72,0
60,4
51,21
44,02
38,26
33,59
29,76
26,56
23,88
21,595
19,640
17,951
16,482
15,196
14,064
13,060
11,3642
9,9944
8,8694
7,9319
7,1414
6,4675
5,8873
5,3836
4,9425
4,5540
4,2096
3,9024
3,6271
886
Продолжение табл. XVIII
3,4
3,5
3,6
3,7
3,8
3,9
4,0
4,1
4,2
4,3
4,4
4,5
4,6
4,7
4,8
4,9 е
5,0
6
7
8
9
10
20
30
40
50
60
70
80
90
100
200
300
400
0,8
1,47118
1,37798
1,29180
1,21190
1,13761
1,06840
1,00378
0,94332
0,88663
0,83341
0,78333
0,73613
0,69160
0,64951
0,60967
0,57193
0,53611
0,25886
0,07743
-0,04937
-0,14227
-0,21275
-0,47728
-0,53524
-0,55247
-0,55639
-0,55498
-0,55119
-0,54633
-0,54102
-0,53556
-0,48867
-0,45651
-0,43302
0,9
1,65338
1,54855
1,45184
1,36238
1,27940
1,20226
1,13036
1,06322
1,00039
0,94150
0,88617
0,83412
0,78508
0,73879
0,69505
0,65365
0,61442
0,31246
0,11662
-0,01936
-0,11848
-0,19339
-0,47210
-0,53280
-0,55102
-0,55544
-0,55429
-0,55067
-0,54592
-0,54068
-0,53529
-0,48859
-0,45647
-0,43299
1,0
1,86202
1,74362
1,63466
1,53412
1,44106
1,35472
1,27443
1,19959
1,12967
1,06426
1,00290
0,94527
0,89106
0,83996
0,79173
0,74617
0,70302
0,37291
0,16072
0,01437
-0,09177
-0,17167
-0,46630
-0,53006
-0,54941
-0,55437
-0,55351
-0,55008
-0,54546
-0,54030
-0,53498
-0,48850
-0,45643
-0,43297
1,1
2,09897
1,96485
1,84174
1,72840
1,62375
1,52686
1,43692
1,35326
1,27526
1,20239
1,13415
1,07017
1,01006
0,95349
0,90019
0,84987
0,80230
0,44039
0,20985
0,05187
-0,06211
-0,14756
-0,45989
-0,52704
-0,54763
-0,55318
-0,55267
-0,54944
-0,54495
-0,53990
-0,53464
-0,48841
-0,45638
-0,43294
,'2
2,3664
2,2142
2,0748
1,94677
1,82882
1,71987
1,61984
1,52523
1,43802
1,35669
1,28066
1,20948
1,14272
1,07996
1,02092
0,96526
0,91272
0,51512
0,26411
0,09323
-0,02944
-0,12103
-0,45285
-0,52373
-0,54567
-0,55188
-0,55174
-0,54873
-0,54439
-0,53945
-0,53426
-0,48830
-0,45632
-0,43291
1,3
2,6669
2,4938
2,3357
2,1909
2,0578
1,9352
1,8217
1,71661
1,61898
1,52808
1,44325
1,36395
1,28970
1,22001
1,15452
1,09287
1,03473
0,59734
0,32363
0,13851
0,00629
-0,09205
-0,44520
-0,52013
-0,54356
-0,55047
-0,55071
-0,54796
-0,54379
-0,53896
-0,53386
-0,48818
-0,45627
-0,43288
1,4
3,0033
2,8063
2Ч,6269
2,4629
2,3126
2,1743
2,0468
1,9287
1,8193
1,7176
1,6229
1,53448
1,45180
1,37433
1,30163
1,23329
1,16892
0,68732
0,38858
0,18782
0,04512
-0,06058
-0,43692
-0,51624
-0,54126
-0,54893
-0,54962
-0,54714
-0,54314
-0,53843
-0,53342
-0,48805
-0,45621
-0,43283
1,5
3,3790
3,1547
2,9508
2,7651
2,5951
2,4392
2,2955
2,1630
2,0403
1,9265
1,82063
1,72201
1,62993
1,54375
1,46300
1,38720
1,31590
0,78535
0,45910
0,24124
0,08714
-0,02657
-0,42799
-0,51205
-0,53880
-0,54730
-0,54844
-0,54624
-0,54243
-0,53787
-0,53295
-0,48792
-0,45614
-0,43280
1>ункци
Таблица XjX
[Я (Г*//*) В*, применяемая для определения параметров в потенциале 1
по экспериментальным значениям второго вириального i
Г^\| 0,1
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1
,10
,15
,20
[,25
,30
,35
,40
,45
,50
,55
1,60
,65
,70
1,75
1,80
1,85
1,90
1,95
2,0
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
3,0
-93,387
-71,243 !
-58,868
-51,026
-45,600
-41,597
-38,495
-35,998
-33,922
-32,153
-30,614
-29,252
-28,028
-26,914
-25,899
-24,938
-24,048
-23,210
-22,416
-21,660
-20,937
-20,242
-19,572
-18,924
-18,296
-17,686
-17,090
-16,510
-15,942
-15,385
-14,840
-14,305
-13,778
-13,260
-12,749
-11,749
-10,773
-9,8194
-8,8848
-7,9670
-7,0647
-6,1763
-5,3001
-4,4356
-3,5811
0,2
-64,452
-45,288
-35,554
-29,792
-26,003
-23,316
-21,300
-19,720
-18,436
-17,364
-16,446
-15,646
-14,937
-14,299
-13,718
-13,185
-12,690
-12,227
-11,792
-11,380
-10,988
-10,614
-10,254
-9,9086
-9,5745
-9,2512
-8,9368
-8,6312
-8,8333
-8,0420
-7,7571
-7,4781
-7,2042
-6,9352
-6,6707
-6,1537
-5,6510
-5,1609
-4,6814
-4,2119
-3,7508
-3,2975
-2,8512
-2,4114
-1,9773
0,3
-72,01
-44,42
-32,120
-25,478
-21,402
-18,665
-16,699
-15,213
-14,043
-13,091
-12,296
-11,617
-11,027
-10,503
-10,034
-9,6093
-9,2198
-8,8600
-8,5248
-8,2108
-7,9144
-7,6334
-7,3659
-7,1098
-6,8640
-6,6273
-6,3989
-6,1776
-5,9630
-5,7536
-5,5499
-5,3509
-5,1564
-4,9659
-4,7791
-4,4153
-4,0633
-3,7214
-3,3883
-3,0630
-2,7446
-2,4323
-2,1256
-1,8238
-1,5266
0,4
-56,10
-36,28
-26,700
-21,283
-17,870
-15,540
-13,851
-12,567
-11,555
-10,732
-10,045
-9,4601
-8,9526
-8,5053
-8,1603
-7,7459
-7,4172
-7,1148
-6,8344
-6,5725
-6,3268
-6,0946
-5,8743
-5,6648
-5,4641
-5,2716
-5,0861
-4,9075
-4,7342
-4,5662
-4,4029
-4,2439
-4,0888
-3,9374
-3,6439
-3,3617
-3,0890
-2,8246
-2,5674
-2,3167
-2,0717
-1,8317
-1,5963
-1,3650
0,5
-48,3
-32,33 '
-24,11
-19,28
-16,172
-14,026
-12,455
-11,256
-10,309
-9,539
-8,8978
-8,3526
-7,8812
-7,4674
-7,0996
-6,7689
-6,4685
-6,1933
-5,9394
-5,7035
-5,4827
-5,2751
-5,0793
-4,8934
-4,7162
-4,5467
-4,3846
-4,2277
-4,0770
-3,9313
-3,7900
-3,6528
-3,5194
-3,2625
-3,0172
-2,7817
-2,5548
-2,3351
-2,1220
-1,9145
-1,7120
-1,5141
-1,3203
Цтокмайерг
<оэффициента
0,6
-44,1
-30,30
-22,83
-18,33
-15,367
-13,293
-11,77
-10,597
-9,6712
-8,9186
-8,2924
-7,7612
-7,3031
-6,9023
-6,5473
-6,2292
-5,9415
-5,6791
-5,4378
-5,2145
-5,0066
-4,8118
-4,6286
-4,4555
-4,2911
-4,1350
-3,9848
-3,8412
-3,7034
-3,5704
-3,4418
-3,3175
-3,0798
-2,8545
-2,6398
-2,4342
-2,2365
-2,0456
-1,8606
-1,6809
-1,5060
-1,3353
0,7
-42,0
-29,3
-22,3
-17,96
-15,05
-13,00
-11,474
-10,30
-9,375
-8,6210
-7,9933
-7,4616
-7,0042
-6,6048
-6,2522
-5,9373
— 5,6535
-5,3954
-5,1590
-4,9412
-4,7389
-4,5501
-4,3733
-4,2066
-4,0494
-3,8993
-3,7569
-3,6205
-3,4900
-3,3646
-3,2437
-3,0144
-2,7990
-2,5954
-2,4017
-2,2165
-2,0388
-1,8676
-1,7021
— 1,5410
-1,3856
Таблица рассчитана Раулинсоном по данным, полученным методом перфорированных карт [48,49].
888
Продолжение табл. XIX
^--. t*
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25 •
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,0
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
3,0
0,8
-40,4
-28,9
-22,2
-17,94
-15,05
-12,973
-11,431
-10,24
-9,296
-8,525
-7,886
-7,3443
-6,8792
-6,4742
-6,1175
-5,7996
-5,5141
-5,2553
-5,0190
-4,8017
-4,6006
-4,4138
-4,2393
-4,0758
-3,9207
-3,7744
-3,6353
-3,5029
-3,3764
-3,2553
-3,0269
-2,8144
-2,6150
-2,4269
-2,2483
-2,0779
-1,9146
-1,7576
-1,6060
-1,4594
0,9
-39,8
-28,9
-22,4
-18,15
-15,24
-13,13
-11,612
-10,33
-9,378
-8,566
-7,9073
-7,3498
-6,8716
-6,4560
-6,0904
-5,7657
-5,4745
-5,2111
-4,9715
-4,7518
-4,5492
-4,3611
-4,1867
-4,0221
-3,8680
-3,7226
-3,5849
-3,4539
-3,3293
-3,0959
-2,8808
-2,6808
-2,4936
-2,3170
-2,1497
-1,9903
-1,8378
-1,6914
-1,5504
1,0
-29,3
-22,8
-18,6
-15,6
-13,43
-11,80
-10,54
-9,53
-8,707
-8,022
-7,444
-6,949
-6,518
-6,1403
-5,8052
-5,5057
-5,2355
-4,9898
-4,7653
-4,5588
-4,3685
-4,1900
-4,0243
-3,8687
-3,7223
-3,5841
-3,4530
-3,2099
-2,9876
-2,7830
-2,5927
-2,4146
-2,2469
-2,0883
' -1,9373
-1,7932
-1,6550
1,1
-29,7
-23,3
-19,1
-16,04
-13,82
-12,15
-10,83
-9,780
-8,922
-8,208
-7,606
-7,089
-6,6409
-6,2481
-5,9004
-5,5901
-5,3106
-5,0572
-4,8260
-4,6147
-4,4180
-4,2364
-4,0671
-3,9088
-3,7601
-3,6200
-3,3619
-3,1286
-2,9154
-2,7188
-2,5361
-2,3655
-2,2049
-2,0530
-1,9087
-1,7711
1,2
-30,5
-24,4
-19,7
-16,6
-14,3
-12,5
-11,19
-10,09
-9,20
-8,45
-7,81
-7,280
-6,812
-6,402
-6,039
-5,717
-5,425
-5,1629
-4,9243
-4,7040
-4,5021
-4,3151
-4,1414
-3,9791
-3,8272
-3,5494
-3,3004
-3,0751
-2,8690
-2,6792
-2,5027
! -2,3380
-2,1830
-2,0366
! -1,8977
1,з !
-31,0
-24,7
-20,4
-17,2
-14,9
-13,05
-11,62
-10,47
-9,52
-8,74
-8,079
-7,513
-7,022
-6,593
-6,214
-5,876
-5,5731
-5,3003
-5,0504
-4,8228
-4,6135
-4,4203
-4,2410
-4,0737
-3,7706
! -3,5019
-3,2609
-3,0423
-2,8423
-2,6578
-2,4865
-2,3266
-2,1762
-2,0343
1,4
-32,3
-25,7
-21,3
-18,2
-15,4
-13,6
-12,1
-10,9
Q ОЛ.
-9,07
-8,38
-7,78
-7,26
-6,81
-6,414
-6,064
-5,750
-5,463
-5,202
-4,966
-4,747
-4,548
-4,361
-4,0269
-3,7334
-3,4727
-3,2381
| -3,0250
-2,8301
-2,6502
-2,4832
-2,3273
-2,1808
1,5
-33,0
-27,0
-22,0
-18,6
-16,2
-14,2
-12,6
-11,4
-10,31
-9,445
-8,70
-8,С8
-7,53
-7,06
-6;650
-6,280
-5,947
-5,647
-5,376
-5,129
-4,902
-4,693
- 4,32Ш
'' -3,9904
1 -3,7113
-3,4570
1 -3,2280
-3,0200
-2,8294
-2,6535
-2,4899
-2,3374
Таблица XX
Третий вириальный коэффициент для полярных газов (потенциал Штокмайера) [51]
С(Т)
г* ^--^
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
4,0
6,0
8,0*
10,0
= ь*с*
0,0
0,4297
0,5924
0,5683
0,5180
0,4728
0,4371
0,3811
0,3523
0,3266
0,3077
0,2962
0,2861
G-;/*)
0,1
0,4440
0,6177
0,5900
0,5351
0,4861
0,4476
0,3873
0,3563
0,3286
0,3085
0,2966
0,2863
т*
0,2
0,5304
0,7162
0,6679
0,5940
0,5311
0,4826
0,4076
0,3692
0,3350
0,3109
0,2978
0,2871
= кТ/в
0,3
0,740
0,9216
0,8221
0,7075
0,^161
0,5478
0,4445
0,3924
0,3463
0,3151
0,3000
0,2884
ь.
С*(Т*;
0,4
1,268
1,075
0,8899
0,7507
0,6496
0,5195
0,4275
0,3630
0,3213
0,3031
0,2902
0,5
1,78
1,451
1,158
0,9455
0,7957
0,5807
0,4761
0,3859
0,3295
0,3072
0,2926
0,6
2,5
2,0
1,53
1,214
0,995
0,6871
0,5403
0,4156
0,3401
0,3124
0,2957
0,7
3,4
2,7
2,03
1,572
1,257
0,825
0,6223
0,4529
0,3532
0,3188
0,2995
/*2 = 8-
0,8
4,6
3,7
2,69
2,03
1,595
0,999
0,7248
0,4986
0,3?90
0,3265
0,3039
1,0
7,0
6,3
4,72
3,36
2,46
1,482
1,002
0,6194
0,4095
0,3459
0,3151
1,2
9,0
7,0
5,2
4,0
2,19
1,401
0,7857
0,4640
0,3715
0,3297
890
Таблица XXI
Расстояние наибольшего сближения и угол отражения для отталкивающего
потенциала9 обратно пропорционального двенадцатой степени расстояния
b
Ут
It)"
48ea" J
(yf = dx/dy и v — выражения, необходимые для расчета квантовых выражений коэффи-
циентов переноса)
Уо
Ут
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
0,00000
0,06148
0,12285
0,18398
0,24475
0,30503
0,36466
0,42349
0,48134
0,53799
0,59321
0,64668
0,69808
0,74697
0,79284
0,83509
0,87305
0,90599
0,93332
0,95474
0,97048
0,98132
0,98839
0,99283
0,99555
1,25
1,30
1,35
1,40
1,45
1,50
0,99723
0,99825
0,99888
0,99927
0,99952
0,99968
3,1416
3,0049
2,8679
2,7303
2;5917
2,4519
2,3105
2,1673
2,0217
1,8736
1,7225
1,5684
1,4109
1,2504
1,0871
0,9228
0,7596
0,6020
0,4559
0,3282
0,2249
0,1477
0,0943
0,0592
0,0371
0,0232
0,0148
0,0093
0,0061
0,0041
0,0026
-2,731
-2,736
-2,744
-2,761
-2,783
-3,070
-2,770
-2,328
-1,804
-1,287
-0,859
-0,546
-0,340
-0,210
-0,131
-0,083
-0,048
-0,031
-0,025
3,1416
2,553
1,958
1,356
0,743
2,811
2,844
2,886
2,936
2,991
3,051
3,116
3,182
3,243
3,286
3,292
3,234
0,116
-0,529
-1,195
-1,887
-2,611
-3,388
-4,127
-4,993
-5,847
-6,668
-7,341
-7,847
-8,056
-7,840
-7,048
-5,799
-4,335
-3,085
-2,087
-1,358
-0,874
-0,565
-0,362
-0,243
-0,157
-0,104
891
Таблица XXII
Интегралы ?(/s'*, применяемые при расчете коэффициентов переноса и вычисленные
для модели потенциальной ямы [51]
Т* = кТ/е
ЦТ*
0,4
0,6
0,8
1,1
1,2
1,3
2,2
1,1
1,2
1,3
2,2
1,1
1,2
1,3
2,2
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
5,00
0,2
1,1513
1,0925
1,0654
1,1833
1,0911
1,0581
1,0418
1,0996
1,0491
1,0315
1,0217
1,0573
2,500
1,66/
1,250
0,833 ' 0,500
| 0,4
0,6
0,8
1,0
1,2
1,3388
1,2113
1,1483
1,4333
1,1894
1,1257
1,0916
1,2335
1,5263
1,3421
1,2431
1,7100
1,2794
1,1944
1,1440
1,3687
1,0947 | 1,1281
1,0688
1,0515
1,1259
1,1001
1,0792
1,1823
1,7038
1,4725
1,3429
1,9817
1,3578
1,2593
1,1966
1,4933
1,1519
1,1244
1,1029
1,2231
1
I
1,8713
1,5996
1,4418
2,2417
1,4267
1,3191
1,2467
1,6052
1,1700
1,1432
1,1222
1,2520
2,0300
1,7233
1,5396
2,4908
1,4872
1,3746
1,2944
1,7057
1,1844
1,1583
1,1379
1,2723
2,0
2,5638
2,1804
1,9183
3,3688
1,6578
1,5535
1,4629
2,0037
1,2234
1,1977
1,1796
1,3093
892
Таблица XXIII
Интегралы Q(l>s>>*, применяемые при расчете коэффициентов переноса и вычисленные для
потенциала Сюзерленда с притягивающим членом, обратно пропорциональным шестой
степени расстояния
с
0,00
0,01
0,02
0,03
0,04
0,05
0,075
0,100
0,125
0,150
0,175
0,200
0,225
0,250
0,375
0,500
0,625
0,750
0,875
1,000
1,125
1,250
1,375
1,500
1,625
1,750
1,875
2,00
т*
оо
50
25
16,67
12,50
10,00
6,67
5,00
4,00
3,33
2,86
2,50
2,22
2,00
1,33
1,00
0,800
0,667
0,571
0,500
0,444
0,400
0,364
0,333
0,308
0,286
! 0,267
0,250
i
1,0000
1,0034
1,0072
1,0112
1,0154
1,0198
1,0318
1,0450
1,0592
1,0738
1,0000
1,0023
1,0045
1,0068.
1,0091*
1,0115
1,0179
1,0246
1,0319
1,0397
1,0888 1,0479
1,1042
1,1348
1,2096
1,2802
1,3466
1,4090
1,4676
1,5228
1,5748
1,6238
1,6702
1,7142
1,7560
1,7962
1,8344
1,8714
1,0566
1,0751
1,1248
1,1765
1,2284
1,2793
1,3286
1,3757
1,4205
1,4630
1,5034
1,5417
1,5782
1,6131
1,6465
1,6789
Котани [52] табулировал функцию FJ(C),
шением :
1,0000
1,0017
1,0034
1,0052
1,0070
1,0088
1,0137
1,0188
1,0243
1,0302
1,0366
1,0432
1,0576
1,0972
1,1379
1,1776
1,2163
1,2542
1,2915
1,3280
1,3635
1,3980
1,4313
1,4634
1,4944
1,5242
1,5529
Д(»,2)*
1,00С0
1,0036
1,0074
1,0116
1,0159
1,0205
1,0330
1,0468
1,0617
1,0774
1,0939
1,1115
1,1285
1,1464
1,2376
1,3264
1,4090
1,4846
1,5537
1,6167
1,6753
1,7294
1,7800
1,8275
1,8725
1,9153
1,9560
1,9950
я<«.»>*
1,00С0
1,0027
1,0055
1,0084
1,0114
1,0146
1,0232 *
1,0325
1,0425
1,0532
1,0645
1,0763
1,1014
1,1694
1,2404
1,3101
1,3763
1,4383
1,4957
1,5491
1,5988
1,6452
1,6889
1,7301
1,7693
1,8066
1,8422
#(М)*
1,0000
1,0021
1,0043
1,0066
1,0092
1,0114
1,0177
1,0248
1,0322
1,0400
1,0483
1,0570
1,0752
1,1272
1,1837
1,2419
1,2993
1,3547
1,407]
1,4565
1,4980
1,5463
1,5872
1,6258
1,6631
1,6973
1,7305
которая связана с функцией &№к следующим соотно
#(/,«)* :
+ 1)!
4
в
2кТ
[¦
i 1 + (-1)'
2 1 + /
1
2 7* *
893
Таблица XXIV
Второй вириальный коэффициент, рассчитанный с помощью потенциала
Букингема—Корнера
В(Т) - 2 я Яг? F0(a, /3; Т*)
Т* = кТ/е
1/Т*
1/Т*
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
т*
100,0
50,0
33,33
25,00
20,00
16,67
14,29
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,539
1,429
1,333
1,250
F0(a, /*; Г*) для fi
a - 12,5
0,0885
0,1012
0,1062
0,1080
0,1079
0,1066
0,1044
0,1016
0,0982
0,0945
0,0716
0,0444
0,0144
-0,0176
-0,0514
-0,0866
-0,1233
-0,1614
-0,2008
-0,2413
-0,2836
-0,3270
-0,3718
-0,4180
13
0,0922
0,1050
0,1102
0,1121
0,1121
0,1109
0,1088
0,1061
0,1029
0,0993
0,0771
0,0506
0,02J3
-0,0100
-0,0429
-0,0774
-0,1132
-0,1504
-0,1889
-0,2287
-0,2697
-0,3121
-0,3559
-0,4010
-0
13,5
0,0958
0,1087
0,1140
0,1160
0,1161
0,1150
0,1130
0,1104
0,1073
0,1038
0,0821
0,0562
0,0276
-6,0030
-0,0352
-0,0689
-0,1040
-0,1404
-0,1781
-0,2170
-0,2572
-0,2987
-0,3416
-0,3857
14
0,0993
0,1123
0,1177
0,1197
0,1199
0,1189
0,1170
0,1144
0,1114
0,1080
0,0868
0,0615
0,0335
0,0035
-0,0282
-0,0612
-0,0956
-0,1313
-0,1682
-0,2064
-0,2458
-0,2865
-0,3285
-0,3718
14,5
0,1028
0,1158
0,1212
0,1233
0,1236
0,1226
0,1207
0,1183
0,1153
0,1120
0,0913
0,0664
0,0389
0,0094
-0,0216
-0,0541
' -0,0879
-0,1229
-0,1592
-0,1967
-0,2354
-0,2753
-0,3165
-0,3590
F0(a, Р; Т*) для Р = 0,2
a = 12,5
0,01
0,02
0,03
0,04
0,05
0,06
0,07
100,0
50,0
33,33
25,00
20,00
16,67
14,29
0,0905
0,1034
0,1086
0,1105
0,1106
0,1094
0,1074
13
13,5
14
0,0942
0,1072
0,1126
0,1147
0,1149
0,1138
0,1120
0,0979
0,1110
0,1165
0,1186
0,1189
0,1180
0,1163
0,1015
0,1146
0,1202
0,1224
0,1228
0,1220
0,1203
14,5
0,1050
0,1182
0,1238
0,1261
0,1265
0,12^7
0,1242
См. [53]; имеется также частное сообщение R. A. Buckingham, октябрь, 1952.
894
Продолжение табл. XXIV
F0(a, 0; Т*) для /9 = 0,2
1/7*
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
т*
и - 12,5
13
13,5
14
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,539
1,429
1,333
1,250
0,1047
0,1016
0,0981
0,0762
0,0501
0,0212
-0,0097
-0,0424
-0,0764
-0,1119
-0,1488
-0,1869
-0,2264
-0,2672
-0,3093
-0,3527
-0,3975
I _
0,1094
0,1064
0,1030
0,0819
0,0565
0,0284
0,0017
0,0336
0,0668
0,1014
0,1373
0,1745
0,2130
0,2527
0,2938
0,3361
0,3798
0,1138
6,1109
0,1076
0,0871
0,0623
0,0349
0,0055
-0,0256
-0,0580
-0,0919
-0,1269
-0,1633
-0,2009
-0,2397
-0,2798
-0,3212
-0,3639
0,1180
0,1152
0,1120
0,0919
0,0677
0,0409
0,0121
-0,0183
-0,0501
-0,0832
-0,1175
-0,1531
-0,1899
-0,2280
-0,2672
-0,3078
-0,3496 |
14,5
0,1219
0,1192
0,1160
0,0964
0,0727
0,0464
0,0182
-0,0116
-0,0427
-0,0753
-0,1089
-0,1439
-0,1799
-0,2172.
-0,2557
-0,2955
-0,336S
Таблица XXV
Коэффициент Джоуля—Томсона при нулевом давлении, рассчитанный с помощью
потенциала Букингема—Корнера3"
1/Г*
0,01
0,02
0,03
0,04
0,05
0,06
0,07
/,*Ср
a,P; T*)
Т* = кТ/е
1/Т*
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
т*
100,0
50,0
33,33
25,00
20,00
16,67
14,29
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,539
1,429
1,333
1,250
G0(a,
a = 12,5
-0,1090
-0,1166
-0,1156
-0,1110
-0,1040
-0,0958
-0,0866
-0,0767
-0,0662
-0,0552
+0,0047
0,0706
0,1409
0,2152
0,2931
0,3747
0,4598
0,5487
0,6415
0,7382
0,8391
0,9443
1,0541
1,1686
/3; Т*) для /3 =
,3
-0,1128
-0,1206
-0,1199
-0,1155
-0,1087
-0,1008
-0,0919
-0,0822
-0,0720
-0,0613
-0,0028
+0,0616
0,1303
0,2029
0,2790
0,3587
0,4419
0,5287
0,6193
0,7138
0,8124
0,9152
1,0224
1,1342
= 0
13,5
-0,1165
-0,1245
-0,1239
-0,1198
-0,1132
-0,1054
-0,0967
-0,0873
-0,0773
-0,0669
-0,0096
+0,0534
0,1207
0,1917
0,2663
0,3442
0,4257
0,5107
0,5994
0,6919
0,7883
0,8889
0,9938
1,1031
14
-0,1201
-0,1283
-0,1278
-0,1238
-0,1173
-0,1098
-0,1013
-0,0920
-0,0822
-0,0720
-0,0159
+0,0459
0,1119
0,1816
0,2547
0,3311
0,4110
0,4944
0,5813
0,6720
0,7665
0,8651
0,9679
1,0752
14,5
-0,1237
-0,1318
-0,1315
-0,1275
-0,1213
-0,1139
-0,1055
-0,0965
-0,0869
-0,0768
-0,0217
+0,0390
0,1038
0,1723
0,2441
0,3192
0,3976
0,4794
0,5648
0,6538
0,7466
0,8434
0,9443
1,0496
G0(a, /3; Т*) для /3 = 0,2
Т*
12,5
См.
100,0
50,0
33,33
25,00
20,00
16,67
14,29
[53] ; имеется
-0,1109
-0,1190
-0,1182
-0,1139
-0,1075
-0,0997
-.0,0909
13
-0,1149
-0,1232
-0,1227
-0,1186
-0,1124
-0,1049
-0,0964
13,5
14
14,5
-0,1187
-0,1272
-0,1269
-0,1230
-0,1170
-0,1097
-0,1014
также частное сообщение R. А.
-0,1224
-0,1310
-0,1308
-0,1271
-0,1213
-0,1142
-0,1061
1. ОКТЯбОЬ.
-0,1260
-0,1346
-0,1345
-0,1309
-0,1253
-0,1183
-0,1105
1952.
896
Продолжение табл. XXV
1/Г*
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
7*
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,539
1,429
1,333
1,250
Go(a
a = 12,5
-0,0814
-0,0714
-0,0608
-0,0031
+0,0606
0,1287
0,2005
0,2760
0,3550
0,4376
0,5238
0,6138
0,7077
0,8057
0,9080
1,0146
1,1260
, 0; T*) для 0 =
13
-0,0872
-0,0774
-0,0671
-0,0109
+0,0512
0,1176
0,1876
0,2613
0,3383
0,4188
0,5029
0,5907
0,6823
0,7778
0,8775
0,9815
1,0901
= 0,2
13,5
-0,0924
-0,0829
-0,0729
-0,0180
+0,0427
0,1076
0,1761
0,2480
0,3233
0,4020
0,4842
0,5700
0,6595
0,7529
0,8503
0,9520
1,0580
14 | 14,5
-0,0973
-0,0880
-0,0782
-0,0244
+0,0350
0,0985
0,1656
0,2361
0,3098
0,3869
0,4674
0,5513
0,6389
0,7304
0,8257
0,9252
1,0290
-0,1018
-0,0927
-0,0831
-0,0304
+0,0279
0,0903
0,1560
0,2252
0,2975
0,3731
0,4520
0,5344
0,6203
0,7099
0,8034
0,9009
1,0027
Таблица XXVI
Первая квантовая поправка для второго вириального коэффициента,
рассчитанная с помощью потенциала Букингема—Корнера
(tfrW/сГ) F^a, /3; Г»)
Т* = ЛГ/е
Fx(a, /5; Т*) для /5 - О
0,01
0,02
0,03
0,04
0,0#5
0,06
0,07
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
г*
100,0
50,0
33,33
25,00
20,00 .
16,67
14,29
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,538
1,429
1,333
1,250
a r= 12,5
0,065
0,081
0,091
0,100
0,107
0,113
0,119
0,124
0,129
0,134
0,157
0,179
0,201
0,224
0,247
0,272
0,299
0,326
0,356
0,388
0,422
0,458
0,497
0,539
13
0,069
0,085
0,096
0,104
0,111
0,118
0,124
0,129
0,135
0,140
0,164
0,186
0,209
0,232
0,257
0,282
0,310
0,339
0,370
0,402
0,438
0,475
0,515
0,559
13,5
0,074
0,090
0,100
0,109
0,116
0,123
0,129
0,135
0,140
0,145
0,170
0,193
0,217
0,241'
0,266
0,293
0,321
0,351
0,383
0,417
0,453
0,493
0,534
0,579
14
0,078
0,094
0,105
0,114
0,121
0,128
0,134
0,140
0,146
0,151
0,176
0,200
0,225
0,250
0,276
0,303
0,332
0,363
0,396
0,432
0,470
0,510
0,554
0,600
14,5
0,0ЙЗ
0,099
0,110
0,119
0,127
0,134
0,140
0,146
0,152
0,157
0,183
0,208
0,233
0,25а
0,285
0,314
0,344
0,376
0,410
0,447
0,486
0,528
0,573
0,621
ЦТ*
0,01
0,02
0,03
0,04
0,05
0,06
0,07
г*
100,0
50,0
33,33
25,00
20,00
16,67
14,29
а = 12,5
0,068
0,084
0,094
0,103
0,110
0,116
0,122
р; Г*) для р «
.3
0,072
0,088
0,099
0,107
0,114
0,121
0,127
- 0,2
13,5
0,076
0,093
0,103
0,112
0,119
0,126
0,132
,4
0,081
0,097
0,108
0,117
0,124
0,131
0,137
14,5
0,085
0,102
0,113
0,122
0,129
0,136
0,143
См. [53]; имеется также частное сообщение R. A. Buckingham, октябрь 1952.
898
Продолжение табл. XXVI
P; Г*) для 0 - 0,2
1/Т*
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
т*
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,538
1,429
1,333
1,250
а = 12,5
0,127
0,132
0,137
0,160
0,182
0,203
0,225
0,248
0,272
0,296
0,323
0,351
0,380
0,411
0,445
0,481
0,518
13
0,132
0,137
0,142
0,166
0,189
0,211
0,234
0,257
0,282
0,307
0,335
0,363
0,394
0,426
0,460
0,496
0,536
13,5
0,138
0,143
0,148
0,172
0,196
0,219
0,242
0,266
0,292
0,318
0,346
0,376
0,407
0,441
0,476
0,513
0,553
14
0,143
0,149
0,154
0,179
0,203
0,227
0,251
0,275
0,302
0,329
0,358
0,388
0,421
0,455
0,491
0,529
0,571
14,5
0,149
0,155
0,160
0,186
0,210
0,234
0,259
0,285
0,312
0,340
0,370
0,401
0,434
0,469
0,506
0,545
0,588
899
Таблица XXVII
Первая квантовая поправка для коэффициента Джоуля—Томсона при нулевом
давлении, рассчитанная с помощью потенциала Букингема—Корнера
CJ - (tirJkT) Qx{a, /5; Г*)
Г* - kTfe
Ох(а, # Г*) для /3 = О
ЦТ*
0,01
0,02
0,03
0,04
0,05
0,06
0,07
0,68
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
г*
100,0
50,0
33,33
25,00
20,00
16,67
14,29
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,538
1,429
1,333
1,250
а = 12,5
0,151
0,186
0,210
0,230
0,247
0,262
0,277
0,291
0,304
0,317
0,381
0,446
0,514
0,586
0,664
0,749
0,841
0,941
1,050
1,169
1,298
1,440
1,595
1,762
13
0,160
0,195
0,219
0,239
0,257
0,273
0,288
0,302
0,316
0,330
0,396
0,463
0,533
0,608
0,689
0,776
0,872
0,976
1,089
1,213
1,348
1,495
1,656
1,830
13,5
0,169
0,204
0,229
0,249
0,267
0,284
0,299
0,314
0,328
0,342
0,411
0,480
0,553
0,630
0,714
0,805
0,903
1,011
1,129
1,257
1,398
1,551
1,718
1,900
14
0,178
0,214
0,239
0,260
0,278
0,295
0,311
0,326
0,341
0,355
0,426
0,497
0,572
0,652
0,739
0,833
0,935
1,047
1,169
1,303
1,449
1,609
1,782
1,971
14,5
0,188
0,224
0,250
0,271
0,290
0,307
0,323
0,339
0,354
0,369
0,441
0,514
0,592
0,674
0,764
0,861
0,967
1,083
1,210
1,349
1,500
1,665
1,845
2,040
ЦТ*
0,01
0,02
0,03
0,04
0,05
0,06
0,07
г*
100,0
50,0
33,33
25,00
20,00
16,67
14,29
--ОЛ а, /5; Г*) для р
а = 12,5
0,157
0,193
0,216
0,235
0,252
0,268
0,282
13
0,166
0,201
0,225
0,245
0,262
0,278
0,293
-0,2
13,5
0,174
0,210
0,235
0,255
0,273
0,289
0,305
14
0,183
0,219
0,245
0,265
0,284
0,301
0,316
14,5
0,193
0,229
0,255
0,276
0,295
0,312
0,329
См. [53]; имеется также частное сообщение R. A. Buckingham, октябрь, 1952.
900
Продолжение табл. XXVII
ЦТ*
0,08
0,09
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
Т*
12,50
11,11
10,00
6,667
5,000
4,000
3,333
2,857
2,500
2,222
2,000
1,818
1,667
1,538
1,429
1,333
1,250
— GX°, Р, Т*) для Р
а - 12,5
0,296
0,309
0,322
0,385
0,449
0,515
0,584
0,659
0,738
0,824
0,916
1,016
1,125
1,242
1,370
1,507
1,654
13
0,307
0,321
0,335
0,400
0,466
0,534
0,606
0,682
0,764
0,853
0,949
1,052
1,164
1,285
1,416
1,557
1,709
= 0,2
13,5
0,319
0,334
0,348
0,415
0,483
0,553
0,627
0,706
0,791
0,882
0,981
1,088
1,203
1,327
1,461
1,606
1,762
14
0,332
0,346
0,361
0,430
0,500
0,572
0,648
0,729
0,817
0,911
1,013
1,123
1,241
1,368
1,506
1,654
1,815
14,5
0,344
0,359
0,374
0,445
0,517
0,591
0,669
0,752
0,842
0,940
1,045
1,157
1,277
1,408
1,549
1,702
1,867
901
Таблица XXVIII
Второй вириальный Коэффициент, рассчитанный с помощью модифицированного
потенциала F-ехр) Букингема [54]
В(Г) = Ьт В*(а, Г»)
Г* = kTje
"\ а
Т* \^
0,40
0,45
0,50
0,55
0,60
0,65
0,70
0,75
0,80
0,85
0,90
0,95
1,00
1,05
1,10
1,15
1,20
1,25
1,30
1,35
1,40
1,45
1,50
1,55
1,60
1,65
1,70
1,75
1,80
1,85
1,90
1,95
2,0
2,1
2,2
2,3
2,4
2,5
2,6
2,7
2,8
2,9
3,0
3,1
3,2
3,3
12,0
-10,347
-8,060
-6,536
-5,455
-4,653
-4,035
-3,546
-3,149
-2,822
-2,547
-2,313
-2,111
-1,9362
-1,7827
-1,6470
-1,5263
-1,4181
-1,3208
-1,2327
-1,1526
-1,0795
-1,0125
-0,9509
-0,8941
-0,8416
-0,7928
-0,7475
-0,7052
-0,6657
-0,6287
-0,5940
-0,5614
-0,5307
-0,4744
-0,4241
-0,3788
-0,3379
-0,3007
-0,2669
-0,2359
-0,2075
-0,1813
-0,1572
-0,1348
-0,1141
-0,0948
12,5
-10,052
-7,826
-6,342
-5,290
-4,508
-3,906
-3,430
-3,043
-2,725
-2,457
-2,229
-2,033
-1,8623
-1,7127
-1,5805
-1,4628
-1,3574
-1,2625
-1,1767
-1,0986
-1,0274
-0,9621
-0,9020
-0,8467
-0,7954
-0,7479
-0,7037
-0,6625
-0,6240
-0,5880
-0,5542
-0,5224
-0,4924
-0,4376
-0,3885
-0,3443
-0,3045
-0,2683
-0,2353
-0,2051
-0,1774
-0,1519
-0,1283
-0,1066
-0,0863
-0,0675
i
13,0
-9,787
-7,616
-6,168
-5,141
-4,378
-3,791
-3,326
-2,949
-2,638
-2,377
-2,154
-1,963
-1,7960
-1,6499
-1,5208
-1,4059
-1,3029
-1,2103
-1,1264
-1,0502
-0,9806
-0,9168
-0,8582
-0,8041
-0,7541
-0,7076
-0,6645
-0,6242
-0,5866
-0,5514
-0,5184
-0,4873
-0,4581
-0,4045
-0,3565
-0,3134
-0,2744
-0,2391
-0,2068
-0,1774
-0,1503
-0,1254
-0,1024
-0,0811
-0,0614
-0,0430
13,5
-9,548
-7,427
-6,012
-5,008
-4,262
-3,688
-3,233
-2,864
-2,560
-2,304
-2,087
-1,899
-1,7363
— ]
—;
1,5934
1,4671
1,3546
1,2539
1,1632
1,0812
-1,0066
-0,9385
-0,8761
-0,8187
-0,7657
-0,7168
-0,6713
-0,6291
-0,5897
-0,5529
-0,5184
-0,4861
-0,4557
-0,4271
-0,3746
-0,3276
-0,2854
-0,2473
-0,2127
-0,1811
-0,1523
-0,1258
-0,1014
-0,0789
-0,0580
-0,0387
-0,0207
14,0
-9,332
-7,256
-5,871
-4,887
-4,157
-3,595
-3,149
-2,788
-2,490
-2,239
-2,026
-1,842
-1,6823
-1,5422
-1,4184
-1,3082
-1,2095
-1,1206
-1,0401
-0,9670
-0,9002
-0,8391
-0,7828
-0,7309
-0,6829
-0,6383
-0,5969
-0,5583
-0,5222
-0,4884
-0,4567
-0,4269
-0,3988
-0,3474
-0,3013
-0,2660
-0,2225
-0,1886
-0,1577
-0,1294
-0,1034
-0,0795
-0,0574
-0,0370
-0,0180
-0,0004
14,5
-9,134
-7,099
-5,741
-4,777
-4,061
-3,509
-3,072
-2,718
-2,425
-2,179
-1,970
-1,790
-1,6330
-1,4955
-1,3740
-1,2658
-1,1689
-1,0816
-1,0027
-0,9309
-0,8653
-0,8052
-0,7500
-0,6990
-0,6519
-0,6082
-0,5675
-0,5296
-0,4941
-0,4609
-0,4298
-0,4005
-0,3730
-0,3224
-0,2772
-0,2366
-0,1999
-0,1665
-0,1361
-0,1083
-0,0828
-0,0593
-0,0377
-0,0176
0,0010
0,0184
15,0
-8,953
-6,956
-5,623
-4,676
-3,973
-3,431
-3,002
-2,654
-2,366
-2,125
-1,919
-1,742
-1,5877
-1,4526
-1,3332
-1,2269
-1,1316
-1,0459
-0,9682
-0,8976
-0,8332
-0,7742
-0,7199
-0,6698
-0,6234
-0,5804
-0,5404
-0,5031
-0,4683
-0,4357
-0,4050
-0,3763
-0,3492
-0,2995
-0,2550
-0,2151
-0,1789
-0,1461
-0,1163
-0,0889
-0,0638
-0,0407
-0,0194
0,0003
0,0186
0,0357
902
Продолжение табл. XXVIII
N. а
пЧ
3,4
3,5
3,6
3,7
3,8
3,9
4,0
4,1
4,2
4,3
4,4
4,5
4,6
4,7
4,8
4,9
5,0
6,0
7,0
8,0
9,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
100,0
200,0
300,0
400,0
12,0
-0,0768
-0,0600
-0,0442
-0,0295
-0,0156
-0,0026
0,0097
0,0212
0,0322
0,0425
0,0523
0,0616
0,0704
0,0787
0,0867
0,0943
0,1015
0,1581
0,1955
0,2215
0,2403
0,2540
0,2937
0,2903
0,2811
0,2712
0,2619
0,2533
0,2455
0,2384
0,2319
0,1876
0,1624
0,1444
12,5
-0,0500
-0,0336
-0,0183
-0,0039
0,0096
0,0223
0,0343
0,0455
0,0562
0,0663
0,0758
0,0848
0,0934
0,1016
0,1С93
0,1167
0,1237
0,1788
0,2152
0,2406
0,2587
0,2721
0,3102
0,3065
0,2971
0,2872
0,2778
0,2692
0,2613
0,2542
0,2477
0,2033
0,1780
0,1599
13,0
-0,0258
-0,0098
0,0051
0,0192
0,0324
0,0448
0,0564
0,0674
0,0779
0,0877
0,0970
0,1058
0,1142
0,1222
0,1297
0,1369
0,1438
0,1976
0,2331
0,2578
0,2755
0,2885
0,3252
0,3213
0,3118
0,3018
0,2924
0,2837
0,2759
0,2687
0,2622
0,2177
0,1921
0,1739
13,5
-0,0039
0,0117
0,0264
0,0401
0,0530
0,0652
0,0766
0,0874
0,0976
0,1072
0,1163
0,1249
0,1331
0,1409
0,1483
0,1554
0,1621
0,2147
0,2495
0,2736
0,2909
0,3036
0,3392
0,3349
0,3254
0,3154
0,3059
0,2973
0,2895
0,2823
0,2758
0,2311
0,2053
0,1870
14,0
0,0161
0,0314
0,0458
0,0593
0,0719
0,0838
0,0951
0,1056
0,1156
0,1250
0,1340
0,1425
0,1505
0,1581
0,1654
0,1723
0,1789
0,2305
0,2645
0,2882
0,3051
0,3175
0,3522
0,3477
0,3381
0,3281
0,3187
0,3101
0,3022
0,2951
0,2885
0,2438
0,2178
0,1994
14,5
0,0345
0,0496
0,0637
0,0770
0,0894
0,1011
0,1121
0,1225
0,1323
0,1415
0,1503
0,1586
0,1665
0,1740
0,1811
0,1879
0,1944
0,2451
0,2785
0,3017
0,3183
0,3305
0,3643
0,3598
0,3502
0,3402
0,3308
0,3222
0,3143
0,3072
0,3007
0,2559
0,2298
0,2112
15,0
0,0516
0,0664
0,0803
0,0933
0,1055
0,1170
0,1279
0,1381
0,1477
0,1568
0,1655
0,1736
0,1814
0,1888
0,1958
0,2025
0,2088
0,2586
0,2916
0,3144
0,3307
0,3427
0,3758
0,3712
0,3616
0,3516
0,3423
0,3337
0,3259
0,3188
0,3123
0,2675
0,2413
0,2227
903
Таблица XXIX
Интегральные функции столкновения, применяемые при расчетах коэффициентов переноса
и вычисленные для модифицированного потенциала F-ехр) Букингема [55]
Z«.*> (Г*)= [Г*A -г б/а)]1/» О<*-*>* (Г*)
Т* - кТ/е
а =» 12
Г*
7*
C»3>
1,1870
1,1911
1,1662
1,1243
1,0750,
1,0282
0,9873
0,9530
0,9248
0,9016
0,8825
0,8541
0,8350
0,8221
0,8135
0,8080
0,8023
0,8031!
0,8070
0,8126
0,8253
0,8384
1,0551 0,9672 0,9027 0,8572
1,0523 0,9584 0,8883 0,8320
1,0142 0,0925 0,8113,0,7338
0,9515
0,8900
0,8402
0,8232(
0,7597
0,7160
0,8025' 0,6870
0,7745 0,6679
0,7538 0,6555
0,7387
0,7272
0,7130
0,7058
0,7028
0,7025
0,7037
0,7104
0,7195
0,7288
0,7239
0,6667
0,6328
0,6131
0,6019
0,6475
0,6005
1,1178
1,1079
1,0704
1,0161
0,9641
0,6388| 0,5985
0,6414
0,6451
0,6496
0,5764 0,9194
0,5643 0,8828
0,5589 0,8536
0,5959,0,5572 0,8305
0,5930,0,557610,8125
0,5923| 0,5593 0,7985
0,7793
0,7684
0,7625
0,7601
0,6475
0,6425
0,6385| 0,5942 0,5646
0,6039
0,5711
0,5779
0,6096 0,5845
0,6155' 0,5910 0,7600
0,6616 0,6296 0,6059,0,7654
0,6735 0,6425 0,6180| 0,7746
0,6847 0,6541 0,6305J 0,7853
0,7382| 0,6950 0,6644 0,6404 0,7963
0,7556|0,7129 0,6819 0,6572 0,8174
0,7709 0,7280 0,6962! 0,6705
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
0,8364
0,8509,0,7842 0,7409 0,7081
0,7961 0,7520 0,7185
0,8623
0,8730
0,8829
0,9002
0,9150
0,9280
0,9394!
0,9497
0,9710
0,9885
1,0032
1,0157
1,0270
1,0371
1,0549
1,0700
1,0834
1,0959
1,1071
1,1906
0,8067
0,8160
0,6817 0,8533
0,6912 0,8680
0,6993 0,8813
0,7066 0,8929
0,7189 0,9133
0,8457,0,7969| 0,7594' 0,7292 0,9301
0,7380 0,9444
0,7618 0,7272
0,7702 0,7351
0,8322 0,7848 0,7485
ll
0,8572 0,8074| 0,7690
0,8674 0,8164.0,7773
0,8762
0,8951
0,9103
0,9232
0,9346
0,9446
0,9538
0,9701
0,9846
0,8245
0,8415
0,8555
0,8674
0,8781
0,8876
0,8965
0,7846
0,8007
0,8138
0,7459' 0,9568
0,7530 0,9680
0,7682 0,9910
0,7808
0,8253! 0,7921
0,8355 0,8021
0,8450
0,8535
0,9123,0,8691
0,8114
0,8199
0,8355
0,9264 0,8827 0,8495
0,9973,' 0^9388' 0,8957,' 0,8622
1,0091
1,0201
1,1038
0,9504 0,9074 0,8743
1,0094
1,0250
1,0385
1,0508
1,0614
1,0807
1,0978
1,1129
1,1265
0,9615 0,9184 0,8853! 1,1391
1,0462 1,0049 0,9729 1,2356
12
Z(M)
1,1947 1,0951
1,1983 1,1002
1,2098! 1,1094
1,2065
1,1799
1,1421
1,1028
1,0667
1,0352
1,0084
1,0936
1,0471
0,9974
0,9538
0,9185
0,8907
0,8692
1,0221 0,9706'0,9193
1,0291 0,97500,9323
1,0401 0,9770! 0,9188
0,9978
0,9367
0,8835
0,9129 0,8381
0,8453 0,7711
0,7953 0,7280
0,8428' 0,7614 0,7017
0,8133 0,7391 0,6862
0,7923 0,7249' 0,6775
0,7775 0,7160 0,6731
0,9859,0,8528
0,9520 0,8309| 0,7561
0,7673
0,9290
0,9137
0,8189
0,8130
0,9039 0,8111
0,7525
0,7531
0,7560
0,7108
0,6716
0,7075 0,6736
0,7093 0,6790
0,7139 0,6860
0,7198 0,6936
0,8978 0,8116
0,8930] 0,8189 0,7742
0,8963 0,8300 0,7891
0,9033' 0,8423 0,8035
0,9122! 0,8546 0,8171
0,7604 0,7263
0,9313 0,8778,
0,9503 0,8987
0,8415
0,8623
0,7435
0,7600
0,7014
0,7202
0,7374
0,7752 0,7529
O,7892j 0,7668
0,8135! 0,7906
0,8339 0,8105
1,0927
1,1062
1,1119
1,0843
1,0360
0,9870
0,9450
0,9111
0,8847
0,8642
0,8488
0,8282
0,8170
0,8120
0,8107
0,8119
0,8207
0,8331
0,8466
0,8599
0,8849
0,9072
T* I Z<"»«>
Z(M>
Z<«'e> I Z<4'4>
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
0,9681
0,9845
0,9995
1,0133
1,0381
1,0598
1,0788
1,0958
1,1113
1,1448
1,1729
1,1976
1,2193
1,2393
1,2573
1,2901
1,3189
1,3451
1,3690
1,3915
1,5600
0,917210,8806 0,8516' 0,8275;
0,9339'0,8967 0,8670,0,8424
0,9488 0,9110 0,8808 0,8559
0,9624' 0,9241 0,8935j 0,8678
0,9862*0,9469 0,9153 0,8891
,0068 0,9664j 0,934l] 0,9073
,0248 0,98380,9508 0,9236
,0409
,0557
,0875
1,1145
1,1382
1,1593
,1786
,1962
,2283
,2571
,2828
0,9993! 0,9658
1,0134
1,0441
1,0703
0,9796
1,0095
0,9383
0,9516
0,9812
1,0353] 1,0067
1,09351 1,0582! 1,0294
1,1143
1,1332
1,1509
1,1826
1,2113
1,2370
,3068 1,2609
,3292 1,2835
,4988 1,4547
1,0787
1,0976
1,1152
1,1469
1,1756
1,2014
1,2257
1,2481
1,4208
1,0499
1,0688
1,0863
1,1180
1,1468
1,1730
1,1973
1,2198
1,3939
0,9269
0,9445
0,9602
0,9745
0,9998
1,0213
1,0404
1,0575
1,0731
1,1070
1,1357
1,1608
1,1832
1,2041
1,2228
1,2572
1,2878
1,3157
1,3413
1,3649
1,5470
Величина Qll-s) с помощью параметра rm стала безразмерной.
904
Продолжение табл. XXIX
» 13
1,1870 1,0551 0,9672'
1,1813 1,0447] 0,9528
1Д6181 1,0150,0,9084
1,1275; 0,9619,0,8389
1,0851| 0,9071
0,8572 1,1178
1,0436
1,0068
0,9756
0,9498
0,9285
0,9112
0,8854
0,8617
0,8268
0,9027
0,8846
0,8217 0,7479| 1,0718
0,7429
0,7807' 0,6983,
0,8305 1,1020
«1:
0,7400 0,6575;
0,7129
0,8009 0,6953
0,7818 0,6841
0,7678
0,7577
0,7454
0,6772
0,6732
0,6710
0,8684 0,740010,6729
0,8573 0,7386 0,6770,
0,8503
0,8461
0,8436
0,7396
0,7421
0,7519
0,8472' 0,7637
0,8535'0,7757
0,8614] 0,7874
0,87820,8091
0,8950 0,8282 0,7862,
0,6823
0,6881'
0,7032'
0,7181
0,7320
0,7446
0,7671!
0,6393!
0,6294'
0,6244
0,622б[
0,6229
0,6266
0,6326
0,6395
0,6468
0,6542
0,6715!
0,6875!
0,7017;
0,7145
0,7365
0,7547
0,6681 1,0250
0,623б| 0,9787
0,6011| 0,9383
0,5904 0,9048
0,5862 0,8780
0,5855 0,8569
0,5871 j 0,8405
0,5897 0,8279
0,5970
0,6052
0,6136
0,8111
0,8022
0,7981
0,6219'0,7974
0,6299| 0,7987
0,6483,0,8073
0,6643 0,8193
0,6785] 0,8323
0,6910 0,8454
0,7121 0,8700
0,7296 0,8922
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
0,9107
0,9253'
0,9389'
0,9514]
0,9736
0,9927
1,0096
1,0247
1,0384
1,0674'
1,0913
1,1118
1,1296
1,1456
1,1602
1,1861
1,2087
1,2287
1,2469
1,2639
1,3892
0,8451
0,8601
0,8734
0,8855
0,9065
0,9245
0,9399
0,9537J
0,9659
0,9922
1,0139
1,0328
1,0492
1,0643
1,0778
1,1023
1,1239
1,1431
1,1608
1,1774
1,3027
0,8024
0,8166
0,8292
0,8404 0,8059
0,8599
0,8765
0,8907
0,9034
0,9147
0,7702'0,7441
0,7836J 0,7568
0,7954; 0,7678
0,7777
0,7948
0,8094
0,9392 0,8987
0,9596
0,9775
0,9933
1,0076
1,0208
1,0447
1,0659
1,0849
1,1025
1,1193
1,2456
0,8240
0,8394
0,8528 0,8223
0,8648! 0,8337
0,8754
0,9184
0,9357
0,9510
0,9651
0,9783
1,0017
1,0230
1,0422
1,0598
1,0762
1,2042
0,8440
0,8664
0,8855
0,9027
0,9179
0,9318
0,9447
0,9682
0,9894
1,0089
1,0267
1,0434
1,1723
0,9118
0,9294
0,9453
0,9596
0,9847
1,0063
1,0250
1,0417
1,0569
1,0895
1,1166
1,1403
1,1616
1,1806
1,1983
1,2297
1,2577
1,2830
1,3063
1,3277
1,4915
13
Г*
1,1947
1,1985
1,2052
1,2056
1,1862
1,1549
1,1207
1,0882
1,0951
1,0994
1,1090
1,0977
1,0602
1,0164
0,9766
0,9436
1,0221 0,9706] 0,9193 1,0927
1,0269 0,9709 0,9265) 1,0986
1,0376 0,9783 0,9247 1,1116
1,0077] 0,9278 0,8562 1,0944
0,9549 0,8664 0,7934 1,0526
0,9
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
6
1,0594 0,9175
1,0346'0,8972
1,0138] 0,8820
0,8618
0,8514
0,8471
0,8465
0,8484
0,8590
0,8731
0,9824
0,9613
0,9475
0,9391
0,9344
0,9327
0,9388
0,9059
0,8676
0,8395
0,8196
0,8057
0,7965
0,8190
0,7866
0,7653
0,7519
0,7267
0,7120
0,9485 0,8880
0,9597
0,9831
1,0058
0,9027
0,9303
0,9548
0,7519 0,7043
1,0076
0,9681
0,9360
0,9110
0,7439 0,7009 0,8917
0,7398 0,7005] 0,8775
0,7870| 0,7382] 0,7044 0,8590
0,7850
0,7871
0,7916
0,7976
0,8148
0,8326
0,8497
0,8658
0,7418 0,7118J 0,8499
0,7481 0,7206 0,8466
0,7557 0,7299 0,8469
0,7638 0,7392
0,7844 0,7615
0,8040 0,7817
0,8219 0,7998
0,8381! 0,8161
0,8496
0,8617
0,8771
0,8931
0,9087
0,8944 0,8668 0,8443 0,9378
0,9191 0,8911 0,8679 0,9637
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
l,O27o'0,9769 0,9408 0,9121
1,0464 0,9964' 0,9600 0,9309
1,0643] 1,0143] 0,9773 0,9477;
1,0809 1,0306 0,9929 0,9628
1,1105
1,1363
1,1595
1,1801
1,1991
1,2406
1,2758
1,3066
1,3344
1,3595
1,3826
1,4243
1,4615
1,4951
1,5262
,0592
,0843
,1064
,1264
,1448
1,1847
1,2187
1,2486
1,2757
1,3004
1,3232
1,3644
1,4015
1,4348
1,4656
1,554611,4942
1,7718 1,7128
1,0207
1,0449
1,0662
1,0857
1,1033
1,1423
1,1757
1,2052
1,2320
1,2565
1,2792
1,3202
1,3569
1,3906
1,4215
1,4500
1,6699
0,9898
1,0133
1,0342
1,0530
1,0704
1,1085
1,1416
1,1709
1,1975
1,2217
1,2444
1,2854
1,3224
1,3559
1,3869
1,4161
1,6376
0,8885'
0,9066
0,9230
0,9377
0,9641
0,9871
1,0076
1,0262
1,0432
1,0812
1,1138
1,1430
1,1694
1,1937
1,2165
1,2577
1,2945
1,3282
1,3595
1,3885
1,6114
0,9867
1,0072
1,0257
1,0425
1,0725
1,0984
1,1216
1,1422
1,1611
1,2027
1,2382
1,2694
1,2974
1,3230
1,3466
1,3896
1,4280
1,4628
1,4948
1,5248
1,7518
905
Продолжение табл. XXIX
а= 14
•Л A.1)
г*
1,1870
1,1727
1,1575
1,1286
1,0914
1,0542
1,0208
0,9924
0,9688
0,9495
0,9337
0,9106
0f8956
0,8863
0,8808
0,8780
0,8784
0,8843
0,8928
0,9025
0,9228
0,9426
1,0551 0,9672
1,0382 0,9481
1,0146 0,9115
0,9684 0,8497
0,9191
0,8776
0,8455
0,8217
0,8042
0,7917
0,7827
0,7724
0,7686
0,7687
0,7710
0,7747
0,7871
0,8011
0,8152
0,8288
0,7963
0,7585
0,7336
0,7176
0,7076
0,7018
0,6989
0,6984
0,7019
0,7074
0,7139
0,7208
0,7853
0,7557
0,7716
0,7863'
0,8539 0,8125'
0,8764 0,8349
0,9027
0,8814
0,8281
0,7566
0,7069
0,6773
0,6608
0,6521
0,6483
0,6476
0,6488
0,6542
0,6617
0,6700
0,6784
0,6868
0,7068
0,7252
0,7416
0,7565
0,7824
0,8042
0,8572
0,8287
0,7571
0,6836
0,6420
0,6214
0,6121
0,6091
0,6096
0,6121
0,6157
0,6246
0,6342
0,6440
0,6534
0,6625
0,6836
0,7022|
0,7188
0,7335
0,7587
0,7796
1,1178
1,0987
1,0729
1,0308
0,9891
0,9524
0,9218
0,8974
0,8781
0,8633
0,8519
0,8373
0,8300
0,8273
0,8277
0,8300
0,8406
0,8545
0,8692
0,8840
0,9121
0,9376
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
0,9612
0,9784
0,9944
0,8962
0,8543
0,9140 0,8715
0,9300
1,0092 0,9445
1,0358
1,0588
,0794
,0978
,1143
,1501
,1802
1,2060
,2286
,2491
,2678
,3008
,3297
,3555
,3789
0,9701
0,9918
1,0110
1,0280
1,0436
1,0766
1,1044
1,1281
1,1494
1,1685
1,1862
1,2176
1,2455
1,2708
1,2938]
0,8867
0,8229
0,8391
0,8536
0,9005 0,8666
0,9245 0,8893
,4009; 1,3150
,5626 1,4758
0,9450
0,9629
0,9789
0,9931
1,0244
1,0507
1,0736
1,0940
1,1125
1,1296
1,1604
1,1877
1,2125
1,2354
1,2568
1,4185
0,9086
0,9255
0,9408
0,9546
0,9844
1,0097
1,0320,
1,0520
1,0703
1,0871
1,1179
1,1450
1,1699
1,1926
1,2141
1,3768
0,7974
0,8129
0,8266
0,8390
0,8606
0,8792
0,8956
0,9101
0,9235
0,9526
0,9772
0,9992
1,0191
1,0372
1,0538
1,0844
1,1115
1,1363
1,1595
1,1809
1,3452
0,9605
0,9812
0,9999
1,0169
1,0468
1,0726
1,0953
1,1157
1,1339
1,1737
1,2070
1,2359
1,2615
1,2848
1,3063
1,3446
1,3789
1,4095
1,4377
1,4637
1,6611
I
14
T*
Z<2»e>
1,1947
1,2017
1,2090
1,2109
1,1947
1,1670
1,1359
1,1060
1,0794
1,0564
1,0370
1,0078
0,9885
0,9761
0,9688
0,9649
0,9651
0,9729
0,9844
0,9973
1,0244
1,0507
1,0951
1,1027
1,1128
1,1046
1,0716
1,0317
0,9948
0,9641
0,9397
0,9209
0,9066
0,8882
1,0221
1,0301
1,0416
1,0168
0,9690
0,9706 0,9193
0,9742 0,9296
0,9831
0,9391
0,8825
0,9235 0,8382
0,8878
0,8615
0,8428
0,8300
0,8215
0,8132
0,8790 0,8122
0,8756
0,8759
0,8785
0,8910
0,9069
0,8151
0,8205
0,8271
0,8464'
0,8076
0,7877
0,7754
0,7682
0,7647
0,7641
0,7686
0,7756
0,9239 0,8862
0,7930
0,8162
0,8666 \),8385
0,8594
0,9309
0,8695
0,8109
0,7718
0,7481
0,7346
0,7277
0,7249
0,7250
0,7299
0,7380
0,7476
0,7840 0,7580
0,9409 0,9048 0,8785
0,9729 0,9384; 0,9121
1,0015 0,9675 0,9408
0,7683
0,7937
0,8172
0,8384
0,8575
0,8908
0,9188
1,0927
1,0990
1,1098
1,0965
1,0607
1,0206
0,9846
0,9549
0,9315
0,9135
0,9001
0,8829
0,8746
0,8720
0,8731
0,8764
0,8905
0,9081
0,9267
0,9449
0,9790
1,0092
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
1,0755
1,098
1,1192
1,1385
1,1731
1,2034
1,2302
1,2540
1,2758
1,3229
1,3622
1,3963
1,4266
1,4543
1,4793
1,5239
1,5636
1,5990
1,6314
1,6612
1,8858
1,0274
1,0504
1,0712
1,0902
1,1237
1,1526
1,1781
1,2007
1,2213
1,2659
1,3034
1,3363
1,3653
1,3917
1,4161
1,4596
1,4983
1,5332
1,5656
1,5950
1,8198
0,9882|
1,0154,
1,0357
1,0539
1,0859
1,1136
1,1379
1,1596
1,1792
1,2222
1,2583
1,2902
1,3185
1,3445
1,3684
1,4115
1,4497
1,4846
1,5165
1,5461
1,7713
0,9656 0,9428
0,9875 0,9639
1,0068 0,9827
1,0244
1,0554
1,0818
1,1051
1,1261
1,1452
1,1868
1,2222
1,2535
1,2816
1,3070
1,3308
1,3735
1,4118
1,4463
1,4782
1,5079
1,7344
0,9996
1,0293
1,0550
1,0777
1,0980
1,1168
1,1576
1,1925
1,2231
1,2510
1,2766
1,2999
1,3427
1,3806
1,4156
1,4474
1,4771
1,7043
1,0361
1,0600
1,0816
1,1013
1,1359
1,1656
1,1917
1,2153
1,2364
1,2826
1,3217
1,3559
1,3863
1,4141
1,4396
1,4856
1,5263
1,5633
1,5972
1,6289
1,8674
906
Продолжение табл. XXIX
» 15
T*
1,1870
1,1722
1,1577
1,1320
1,0984
1,0645
1,0337
1,0075
0,9857
0,9678
0,9533
0,9323
0,9191
0,9112
0,9070
0,9054
0,9083
0,9167
0,9273
0,9389
0,9627
0,9856
1,0551 0,9672 0,9027
1,0378 0,9477
1,0163 0,9153
0,9750
0,9300
0,8916
0,8618
0,8397
0,8235
0,8121
0,7954
0,7931
0,7944
0,7979
0,8028
0,8594
0,8099
0,7747
0,7514
0,8810
0,8343
0,7687
0,7221
0,8572
0,8285
0,7655
0,6970
0,6579
0,6944 0,6388
0,6792
0,7367 0,6715
0,7278
0,7230
0,8040 0,7208
0,7218
0,7266
0,7335
0,7412
0,7494
0,6687
0,6687
0,6707
0,6777
0,6867
0,6964
0,6306
0,6285
0,6299
1,1178
1,1007
1,0765
1,0392
1,001:
0,9672
0,9387
0,9158
0,8978
0,6333 0,8840
0,6378
0,6484
0,6596
0,6708
0,7063| 0,6817
0,7159,0,6921
0,8179 0,7699 0,7389
0,8344 0,7896.'0,7597
0,8507 0,8078,'0,7783
0,7160
0,7372
0,8735
0,8605
0,8548
0,8535
0,8554
0,8593
0,8734
0,8903
0,8662
0,8951
0,9203
0,8245
0,8541
0,8796
0,7558 0,9077
0,7953 0,7725 0,9245
0,8245 0,8012 0,9558
0,8494] 0,8250 0,9836
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
1,0069
1,0268'
1,0449
1,0619
1,0922
1,1188
1,1426
1,1640
1,1834
1,2257
1,2614
1,2923
1,3199
1,3446
1,3675
1,4083
1,4443
1,4766
1,5059
1,5331!
1,7372
0,9017 0,8708'
0,9214 0,8895'(
0,9816' 0,9390 0,9063|(
0,9430
0,9632
0,8458 1,0086
0,8638 1,0308
0,8800| 1,0514
0,998110,9549,' 0,921б| 0,8946,1,0699
0,9205 1,1032
1,0277
1,0530
1,0756
1,095'
1,1140
1,1537
1,187:
1,2167
1,2429
1,2666
1,2886
1,3280
1,3630
1,3945
1,4235
1,4505
1,6538
0,9829
1,0070
1,0284
1,0475
1,0648
1,1029
1,1353
1,1637
1,1890
1,2123
1,2339
1,2726
1,3073
1,3389
1,3675
1,3945
1,5991
0,9483
0,971
0,9921
1,0104
1,0273
1,0642
1,0957
1,1234
1,1486
1,1718
1,1929
1,2316
1,2660
1,2974
1,3267
1,3534
1,5596
0,9428
0,9628
0,9805
0,9970
1,0333
1,064!
1,0920
1,1169
1,1397
1,1609
1,1996
1,2340
1,2654
1,2946
1,3217
1,5291
1,1132
1,1579
1,1812
1,2026
1,2494
1,2897
1,3254
1,3574
1,3869
1,4141
1,4631
1,5072
1,5471
1,5837
1,6180
1,8773
а= 15
I
1,1947
1,2016
1,2081
1,2107
1,1978
1,1739
1,1462
1,1191
1,0945
1,0734
1,0555
1,0285
1,0110
1,0003
0,9944
0,9920
0,9953
1,0059
1,0196
1,0345
1,0651
1,0943
Z(M>
1,0951
1,1025
1,1114
1,1061
1,0783
1,0426
1,0088
0,9803
0,9575J
0,9400!
0,9268
0,9102
0,9027
0,9009
0,9027
0,9067
0,9223
0,9409
0,9602
0,9790
1,0139
1,0452
1,0221
1,0306
l,0403|
1,0211
0,9791
0,9706 0,9193
0,9736 0,9290
0,9828 0,9325
0,9465J 0,8796
0,8952
0,9374 0,8536
0,8252
0,7879
0,7654
0,7529
0,7470
0,7883' 0,7454
0,8423' 0,7858' 0,7466
0,9039;
0,8791
0,8617
0,8498
0,8247
0,8060
0,7946
0,8359' 0,7872
0,8365 0,7936
0,8412
0,8481
0,8562
0,8786
0,9013
0,9229
0,9431
0,9796
1,0111
0,8024
0,7535
0,7637
0,7751
0,8124 0,7868
0,8227 0,7986
0,8488,0,8266
0,8734
0,8960
0,9169
0,9533
0,9844
0,8521
0,8750
0,8958
0,9318
0,9623
1,0927
1,1061
1,1150
1,1029
1,0705
1,0333
0,9996
0,9718
0,9499
0,9331
0,9209
0,9057
0,8992
0,8982
0,9007
0,9054
0,9222
0,9419
0,9622
0,9819
1,0187
1,0514
T*
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
z(*»f>
1,1213
1,1462
1,1691
1,1904
1,2283
1,2616
1,2911
1,3179
1,3423|
1,3954'
1,4406
1,4802
1,5152
1,5474
1,5769
1,6298
1,6768
1,7190
1,7580
1,7938
2,0647
1,0733
1,0985
1,1212]
1,1421
1,1791
1,2111
1,2397
1,2655
1,2887
1,3399
1,3835
1,4219
1,4561
1,4874
1,5163
1,5684
1,6150
1,6569
1,6952
1,7312
2,0026
1,0389
1,0634
1,0856
1,1059
1,1416
1,1727
1,2002
1,2250
1,2477
1,2975
1,3400
1,3775
1,4114
1,4424
1,4710
1,5225
1,5687
1,6107
1,6491
1,6850
1,9572
1,0115
1,0356
1,0570
1,0766
1,1115
1,1416
1,1683
1,1925
1,2148
1,2634
1,3054
1,3425
1,3759
1,4067
1,4352
1,4865
1,5326
1,5747
1,6132
1,6489
1,9227
0,9887
,0121
,0332
,0523
,0860
,1154
,1417
,1656
,1873
,2353
,2768
,3138
,3471
,3776
,4060
,4757
,5034
,5453
,5841
,6200
,8945
1,0805
1,1064
1,1300
1,1515
1,1895
1,2225
1,2518
1,2781
1,3020
1,3544
1,3992
1,4382
1,4737
1,5057
1,5353
1,5889
1,6365
1,6797
1,7194
1,7562
2,0351
907
Таблица XXX
Функции для вычисления высших приближений коэффициентов переноса [55]
для видоизмененного потенциала F-ехр) Букингема
Функция /
г*
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
-3,5
4
5
6
а=12
1,0020
1,0017
1,0010
1,0002
1,0001
1,0002
1,0003
1,0003
1,0002
1,0002
1,0001
1,0001
1,0001
1,0003
1,0006
1,0009
1,0017
1,0024
1,0030
1,0035
1,0042
1,0047
а = 13
1,0020
1,0017
1,0012
1,0004
1,0001
1,0001
1,0001
1,0001
1,0001
1,0001
1,0000
1,0001
1,0003
1,0005
1,0008
1,0012
1,0020
1,0028
1,0035
1,0041
1,0049
1,0053
а=14
1,0020
1,0018
1,0013
1,0005
1,0001
1,0000
1,0000
1,0000
1,0000
1,0000
1,0000
1,0002
1,0004
1,0007
1,0011
1,0015
1,0024
1,0032
1,0039
1,0045
1,0053
1,0059
а =15
1,0020
1,0018
1,0014
1,0006
1,0002
1,0000
1,0000
1,0000
1,0000
1,0000
1,0001
1,0003
1,0006
1,0009
1,0013
1,0017
1,0028
1,0037
1,0044
1,0050
1,0059
1,0064
г*
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
а=12
1,0049
1,0051
1,0052
1,0053
1,0053
1,0053
1,0052
1,0052
1,0051
1,0050
1,0049
1,0048
1,0048
1,0047
1,0047
1,0047
1,0047
1,0047
1,0047
1,0048
1,0053
а=13
1,0057
1,0059
1,0060
1,0061
1,0061
1,0062
1,0061
1,0061
1,0060
1,0059
1,0058
1,0058
1,0058
1,0058
1,0058
1,0058
1,0058
1,0059
1,0059
1,0060
1,0067
а=14
1,0062
1,0065
1,0066
1,0067
1,0068
1,0069
1,0068
1,0068
1,0068
1,0068
1,0067
1,0066
1,0067
1,0066
1,0067
1,0067
1,0068
1,0069
1,0070
1,0070
1,0077
а=15
1,0068
1,0070
1,0072
1,0073
1,0074
1,0075
1,0075
1,0075
1,0075
1,0075
1,0075
1,0075
1,0075
1,0075
1,0076
1,0077
1,0077
1,0078
1,0079
1,0081
1,0088
Функция
т*
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
а = 12
1,0017
1,0019
1,0017
1,0012
1,0003
1,0001
1,0001
1,0002
1,0002
1,0002
1,0001
1,0001
1,0001
1,0003
1,0005
а = 13
1,0017
1,0018
1,0021
1,0014
1,0005
1,0001
1,0000
1,0001
1,0001
1,0001
1,0001
1,0001
1,0002
1,0004
1,0007
а = 14
1,0017
1,0018
1,0021
1,0015
1,0006
1,0002
1,0000
1,0000
1,0000
1,0000
1,0000
1,0001
1,0002
1,0005
1,0008
а»15
1,0017
1,0018
1,0020
1,0016
1,0008
1,0003
1,0001
1,0000
1,0000
1,0000
1,0000
1,0001
1,0003
1,0007
1,0010
2,0
2,5
3,0
3,5
4
5
6
7
8
9
10
12
14
16
18
а=12
1,0008
1,0017
1,0025
1,0032
1,0037
1,0045
1,0050
1,0052
1,0054
1,0055
1,0056
1,0057
1,0057
1,0057
1,0057
а=13
1,0011
1,0020
1,0029
1,0036
1,0042
1,0050
1,0055
1,0058
1,0060
1,0061
1,0062
1,0062
1,0063
1,0063
1,0064
а=14
1,0012
1,0022
1,0031
1,0039
1,0046
1,0055
1,0061
1,0066
1,0067
1,0068
1,0069
1,0069
1,0069
1,0069
1,0069
о = 15
1,0015
1,0026
1,0035
1,0043
1,0050
1,0058
1,0064
1,0067
1,0070
1,0071
1,0072
1,0073
1,0073
1,0073
1,0073
908
Продолжение табл. XXX
Функция /
г*
20
25
30
35
40
45
50
а = 12
1,0057
1,0057
1,0057
1,0057
1,0058
1,0058
1,0059
а=13
1,0064
1,0064
1,0065
1,0065
1,0066
1,0067
1,0068
а = 14
1,0068
1,0068
1,0068
1,0068
1,0068
1,0068
1,0068
а=15
1,0073
1,0073
1,0074
1,0074
1,0075
1,0075
1,0075
Т*
60
70
80
90
100
200
а=12
1,0060
1,0061
1,0062
1,0063
1,0064
1,0074
а=13
1,0069
1,0071
1,0072
1,0073
1,0074
1,0084
а = 14
1,0069
1,0070
1,0071
1,0072
1,0073
1,0081
а = 1б
1,0077
1,0078
1,0079
1,0080
1,0082
1,0091
Функция / *
т*
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
6
а=12
1,0027
1,0029
1,0027
1,0020
1,0006
1,0002
1,0002
1,0003
1,0003
1,0003
1,0002
1,0002
1,0002
1,0004
1,0008
1,0013
1,0026
1,0038
1,0049
1,0058
1,0070
1,0077
а=13
1,0027
1,0028
1,0032
1,0023
1,0009
1,0002
1,0001
1,0001
1,0001
1,0001
1,0001
1,0001
1,0003
1,0006
1,0011
1,0016
1,0031
1,0045
1,0056
1,0065
1,0078
1,0085
а =14
1,0027
1,0029
1,0033
1,0025
1,0011
1,0003
1,0001
1,0000
1,0000
1,0000
1,0000
1,0001
1,0004
1,0008
1,0013
1,0019
1,0034
1,0048
1,0060
1,0071
1,0085
1,0094
а » 15
1,0027
1,0028
1,0032
1,0026
1,0013
1,0005
1,0001
1,0000
1,0000
1,0000
1,0000
1,0002
1,0005
1,0010
1,0016
1,0022
1,0039
1,0054
1,0067
1,0077
1,0091
1,0099
г*
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
а=12
1,0081
1,0085
1,0087
1,0088
1,0089
1,0089
1,0089
1,0088
1,0089
1,0089
1,0089
1,0090
1,0090
1,0091
1,0092
1,0093
1,0096
1,0097
1,0099
1,0101
1,0115
а=13
1,0090
1,0093
1,0095
1,0097
1,0098
1,0099
1,0099
1,0100
1,0100
1,0101
1,0101
1,0102
1,0103
1,0104
1,0105
1,0108
1,0111
1,0112
1,0114
1,0116
1,0132
а =14
1,0105
1,0104
1,0106
1,0107
1,0109
1,0108
1,0108
1,0108
1,0107
1,0106
1,0106
1,0106
1,0106
1,0106
* 1,0107
1,0108
1,0109
1,0110
1,0113
1,0114
1,0127
а» 15
1,0105
1,0109
1,0111
1,0112
1,0114
1,0114
1,0115
1,0115
1,0114
1,0115
1,0115
1,0116
1,0116
1,0117
1,0118
1,0120
1,0122
1,0124
1,0125
1,0127
1,0141
909
Таблица XXXI
Функция Р для вычисления изотропной термодиффузии [55] для видоизменен-
ного потенциала F-ехр) Букингема
г*
0
0,1
0,2
0,3
0,4
0,5
•0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
6
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
[к
а=12
0,3133
0,2844
0,2047
0,0738
-0,0271
-0,0829
-0,1038
-0,1031
-0,0902
-0,0690
-0,0456
0,0071
0,0580
0,1050
0,1476
0,1841
0,2570
0,3103
0,3469
0,3744
0,4111
0,4312
0,4414
0,4494
0,4531
0,4536
0,4536
0,4512
0,4469
0,4441
0,4387
0,4321
0,4245
0,4188
0,4161
0,4118
0,4093
0,4051
0,4048
0,4034
0,4022
0,4024
0,4110
м, - м,
а=13
0,3133
0,2865
0,2264
0,1110
0,0157
-0,0389
-0,0616
-0,0623
-0,0509
-0,0317
-0,0088
0,0419
0,0923
0,1386
0,1797
0,2161
0,2883
0,3404
0,3786
0,4060
0,4437
0,4643
0,4778
0,4858
0,4889
0,4911
0,4917
0,4919
0,4887
0,4866
0,4826
0,4764
0,4713
0,4681
0,4648
0,4641
0,4616
0,4598
0,4590
0,4583
0,4584
0,4586
0,4703
а=14
0,3133
0,2887
0,2401
0,1369
0,0484
-0,0037
-0,0256
-0,0270
-0,0162
0,0023
0,0244
0,0734
0,1226
0,1680
0,2083
0,2438
0,3145
0,3658
0,4035
0,4313
0,4679
0,4911
0,5044
0,5131
0,5181
0,5208
0,5233
0,5225
• 0,5209
0,5187
0,5190
0,5140
0,5106
0,5066
0,5058
0,5038
0,5035
0,5032
0,5044
0,5071
0,5093
0,5095
0,5277
а = 15
0,3133
0,2887
0,2468
0,1545
. 0,0727
0,0229
0,0020
0,0005
0,0105
0,0287
0,0498
0,0981
0,1466
0,1911
0,2310
0,2665
0,3380
0,3890
0,4270
0,4544
0,4931
0,5134
0,5283
0,5358
0,5427
0,5446
0,5494
0,5495
0,5496
0,5485
0,5479
0,5452
0,5427
0,5429
0,5426
0,5426
0,5429
0,5443
0,5460
0,5474
0,5505
0,5534
0,5727
910
Таблица XXXII
Величины а*, в* и с* для вычисления коэффициентов переноса смесей [55]
для видоизмененного потенциала F-ехр) Букингема
Функция А*
т*
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
6
а=12
1,0065
1,0060
1,0374
1,0731
1,0976
1,1108
1,1170
1,1193
1,1194
1,1185
1,1173
1,1146
1,1126
1,1114
1,1111
1,1111
1,1130
1,1161
1,1193
1,1226
1,1284
1,1335
а = 13
1,0065
1,0146
1,0374
1,0693
1,0932
1,1067
1,1131
1,1154
1,1154
1,1143
1,1126
1,1096
1,1070
1,1052
1,1044
1,1044
1,1056
1,1081
1,1113
1,1141
1,1194
1,1238
а = 14
1,0065
1,0247
1,0445
1,0729
1,0946
1,1070
1,1128
1,1145
1,1142
1,1126
1,1106
1,1067
1,1037
1,1013
1,0999
1,0990
1,0987
1,1002
1,1026
1,1050
1,1101
1,1147
а=15
1,0065
1,0251
1,0435
1,0695
1,0905
1,1028
1,1088
1,1108
1,1104
1,1091
1,1071
1,1032
1,1000
1,0978
1,0964
1,0956
1,0958
1,0973
1,0995
1,1018
1,1064
1,1103
г*
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
а = 12
1,1377
1,1417
1,1449
1,1477
1,1532
1,1583
1,1625
1,1665
1,1702
1,1790
1,1865
1,1938
1,2005
1,2067
1,2123
1,2230
1,2326
1,2416
1,2492
1,2569
1,3103
а=13
1,1277
1,1309
1,1336
1,1361
1,1406
1,1447
1,1485
1,1517
1,1548
1,1623
1,1691
1,1752
1,1813
1,1867
1,1917
1,2008
1,2092
1,2168
1,2240
1,2300
1,2754
а = 14
1,1189
1,1224
1,1255
1,1281
1,1326
1,1366
1,1397
1,1423
1,1449
1,1502
1,1542
1,1578
1,1612
1,1643
1,1668
1,1715
1,1759
1,1796
1,1831
1,1858
1,2068
а = 15
1,1136
1,1163
1,1189
1,1210
1,1246
1,1276
1,1300
1,1322
1,1343
1,1385
1,1421
1,1454
1,1480
1,1508
1,1531
1,1573
1,1610
1,1642
1,1674
1,1700
1,1885
Функция В*
г*
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
а=12
1,1851
1,1988
1,2528
1,3028
1,3127
1,3003
1,2808
1,2601
1,2403
1,2239
1,2079
1,1837
1,1662
1,1536
1,1458
а=13
1,1851
1,1955
1,2407
1,2895
1,3019
1,2922
1,2737
1,2539
1,2346
1,2172
1,2025
1,1780
1,1612
1,1490
1,1394
а=14
1,1851
1,1926
1,2328
1,2788
1,2922
1,2844
1,2668
1,2476
1,2289
1,2125
1,1973
1,1733
1,1561
1,1440
1,1347
а-15
1,1851
1,1928
1,2268
1,2698
1,2840
1,2768
1,2609
1,2424
1,2238
1,2074
1,1930
1,1689
1,1523
1,1392
1,1298
г*
2,0
2,5
3,0
3,5
4
5
6
7
8
9
10
12
14
16
18
а = 12
1,1387
1,1288
1
1
]
]
1,1250
1,1217
1,1211
1,1225
1,1242
1,1252
1,1278
1,1298
1,1317
1,1351
1,1376
1,1384
1
,1405
а=13
1,1324
1,1222
1,1167
1,1136
1,1128
1,1126
1,1131
1,1155
1,1176
1,1185
1,1203
1,1225
1,1247
1,1259
1,1271
а = 14
1,1279
1,1174
1,1113
1,1084
1,1067
1,1048
1,1059
1,1067
1,1079
1,1094
1,1103
1,1127
1,1135
1,1149
1,1153
а=15
1,1226
1,1119
1,1057
1,1024
1,1002
1,1001
1,0989
1,1006
1,1009
1,1025
1,1026
1,1050
1,1056
1,1066
1,1070
911
Продолжение табл. XXXIГ
Функция В*
г*
20
25
30
35
40
45
50
а=12
1,1404
1,1426
1,1426
1,1427
1,1427
1,1418
1,1407
а = 13
1,1274
1,1282
1,1281
1,1279
1,1268
1,1270
1,1255
а = 14
1,1178
1,1176
1,1178
1,1162
1,1159
1,1148
1,1142
а=15
1,1077
1,1070
1,1058
1,1055
1,1050
1,1035
1,1023
Т*
60
70
80
90
100
200
а = 12
1,1388
1,1378
1,1365
1,1350
1,1331
1,1206
а = 13
1,1236
1,1218
1,1198
1,1180
1,1154
1,1021
а=14
1,1119
1,1106
1,1096
1,1077
1,1049
1,0911
а = 15
1,1003
1,0980
1,0950
1,0940
1,0922
1,0779
Функция С*
т*
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,2
1,4
1,6
1,8
2,0
2,5
3,0
3,5
4
5
6
а-12
0,8889
0,8835
0,8697
0,8463
0,8279
0,8172
0,8128
0,8127
0,8151
0,8193
0,8240
0,8348
0,8453
0,8549
0,8636
0,8709
0,8855
0,8959
0,9031
0,9084
0,9155
0,9195
а=13
0,8889
0,8844
0,8736
0,8531
0,8360
0,8257
0,8212
0,8209
0,8231
0,8269
0,8315
0,8419
0,8521
0,8615
0,8698
0,8771
0,8913
0,9014
0,9088
0,9141
0,9213
0,9254
а = 14
0,8889
0,8853
0,8765
0,8581
0,8421
0,8325
0,8283
0,8280
0,8301
0,8338
0,8383
0,8482
0,8582
0,8673
0,8753
0,8823
0,8961
0,9059
0,9131
0,9183
0,9253
0,9298
а-15
0,8889
0,8853
0,8779
0,8613
0,8467
0,8376
0,8337
0,8334
0,8354
0,8391
0,8434
0,8532
0,8629
0,8718
0,8797
0,8867
0,9005
0,9102
0,9174
0,9226
0,9298
0,9337
г*
7
8
9
10
12
14
16
18
20
25
30
35
40
45
50
60
70
80
90
100
200
а-12
0,9216
0,9232
0,9241
0,9242
0,9245
0,9243
0,9237
0,9234
0,9226
0,9218
0,9209
0,9203
0,9202
0,9198
0,9197
0,9196
0,9202
0,9205
0,9208
0,9214
0,9271
а-13
0,9280
0,9295
0,9302
0,9307
0,9311
0,9313
0,9310
0,9307
0,9302
0,9295
0,9291
0,9289
0,9288
0,9290
0,9290
0,9293
0,9298
0,9303
0,9309
0,9316
0,9377
а=14
0,9324
0,9342
0,9352
0,9359
0,9366
0,9367
0,9366
0,9364
0,9366
0,9361
0,9358
0,9354
0,9355
0,9355
0,9356
0,9360
0,9367
0,9375
0,9383
0,9387
0,9445
а-15
0,9365
0,9381
0,9394
0,9399
0,9409
0,9412
0,9414
0,9413
0,9414
0,9413
0,9412
0,9415
0,9417
0,9420
0,9423
0,9430
0,943?
0,9444
0,9453
0,9461
0,9520
912
Таблица XXXIII
Функции, необходимые для вычисления второго вириального коэффициента для
обобщенных сфероцилиндрических и эллипсоидальных молекул Кихара [43]
кТ
—IgX
-0,4
-0,3'
-0,2
-0,1
0,0
0,1
Q.2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,1
1>2
1,3
1,4
1,5
1,6
1,7
1,8
1,9
2,0
F3(X)
-9,859
-6,138
-4,003
-2,673
-1,795
-1,189
-0,7587
-0,4465
-0,2170
-0,0469
0,0794
0,1729
0,2415
0,2911
0,3259
0,3493
0,3638
0,3715
0,3737
0,3718
0,3668
0,3594
0,3501
0,3396
0,3281
F(X)
-5,211
-3,008
-1,776
-1,027
-0,5424
-0,2151
0,0132
0,1758
0,2930
0,3779
0,4392
0,4829
0,5134
0,5336
0,5459
0,5521
0,5534
0,5510
0,5457
0,5381
0,5287
0,5179
0,5062
0,4937
0,4806
Fx(X)
-1,784
-0,7761
-0,2221
0,1091
0,3198
0,4600
0,5562
0,6234
0,6710
0,7045
0,7279
0,7436
0,7536
0,7591
0,7611
0,7603
0,7575
0,7528
0,7468
0,7396
0,7320
0,7228
0,7135
0,7038
0,6937
F_x(X)
1,591
1,478
1,403
1,351
1,319
1,297
1,284
1,276
1,274
1,277
1,282
1,291
1,302
1,315
1,330
1,346
1,364
1,383
1,403
1,424
1,446
F_S(X)
2,683
2,392
2,204
2,084
2,009
1,968
1,953
1,957
1,977
2,012
2,058
2,116
2,184
2,262
2,349
2,446
2,553
2,669
2,794
2,930
3,075
F_6(X)
3,82
3,38
3,11
2,96
2,87
2,85
2,87
2,92
3,01
3,14
3,29
3,47
3,68
3,92
4,20
4,51
4,86
5,25
5,68
6,16
6,69
F_7(X)
5,12
4,55
4,24
4,07
4,02
4,06
4,17
4,35
4,6a
4,91
5,2?
5,74
6,27
6,91
7,63.
8,46
9,42
10,5a
11,79.
13,24
14,90.
913
Таблица XXXIV
Приведенные усредненные эффективные сечения &B>2)* для полярных молекул
кТ/е
1,0
1,2
1,4
1,6
1,8
2,0
2,2
2,4
2,6
2,8
3,0
3,2
3,4
3,6
3,8
4,0
5
6
7
8
9
10
11
12
13
14
15
16
32
64
128
256
512
0,00
1,5938
1,4568
-1,3557
1,2800
1,2216
1,1751
1,1377
1,1066
1,0803
1,0579
1,0385
1,0214
1,0063
0,9928
0,9807
0,9696
0,9265
0,8960
0,8725
0,8536
0,8378
0,8242
0,8123
0,8017
0,7922
0,7836
0,7756
0,7883
0,6939
0,6262
0,5634
0,5056
0,4528
0,25
1,7578
1,5872
1,4622
1,3688
1,2965
1,2390
1,1923
1,1537
1,1212
1,0935
1,0696
1,0488
1,0304
1,0141
0,9996
0,9864
0,9359
0,9010
0,8750
0,8545
0,8376
0,8232
0,8108
0,7999
0,7902
0,7814
0,7734
0,7660
0,6924
0,6259
0,5638
0,5061
0,4533
0,50
1,9312
1,7302
1,5812
1,4689
1,3813
1,3112
1,2539
1,2063
1,1662
1,1321
1,1026
1,0769
1,0544
1,0345
1,0168
1,0009
0,9411
0,9014
0,8727
0,8507
0,8330
0,8183
0,8058
0,7948
0,7851
0,7765
0,7686
0,7613
0,6897
0,6251
0,5640
0,5065
0,4539
0,75
2,0806
1,8661
1,7026
1,5756
1,4739
1,3907
1,3220
1,2643
1,2155
1,1737
1,1377
1,1065
1,0792
1,0552
1,0339
1,0149
0,9446
0,8994
0,8677
0,8440
0,8254
0,8102
0,7975
0,7865
0,7769
0,7684
0,7606
0,7536
0,6846
0,6213
0,5612
0,5047
0,4527
д*
| 1,00
2,1963
1,9881
1,8224
1,6878
1,5759
1,4817
1,4026
1,3349
1,2769
1,2270
1,1837
1,1460
1,1129
1,0838
1,0581
1,0351
0,9510
0,8980
0,8618
0,8356
0,8154
0,7994
0,7863
0,7752
0,7656
0,7572
0,7497
0,7429
0,6782
0,6181
0,5593
0,5035
0,4518
1,25
2,3025
2,1035
1,9395
1,8019
1,6843
1,5832
1,4957
1,4198
1,3537
1,2961
1,2456
1,2012
1,1621
1,1274
1,0965
1,0690
0,9674
0,9037
0,8609
0,8304
0,8077
0,7901
0,7760
0,7644
0,7546
0,7462
0,7388
0,7322
0,6717
0,6151
0,5579
0,5028
0,4515
1,50
2,4920
2,2749
2,0945
1,9430
1,8128
1,6999
1,6015
1,5155
1,4401
1,3738
1,3153
1,2636
1,2178
1,1770
1,1406
1,1С81
0,9874
0,9118
0,8614
0,8260
0,8000
0,7802
0,7648
0,7523
0,7421
0,7334
0,7260
0,7195
0,6637
0,6114
0,5563
0,5021
0,4512
1,75
2,5916
2,3782
2,2051
2,0599
1,9339
1,8224
1,7232
1,6343
1,5544
1,4827
1,4182
1,3601
1,3078
1,2606
1,2179
1,1794
1,0334
0,9401
0,8777
0,8342
0,8028
0,7793
0,7613
0,7471
0,7356
0,7262
0,7182
0,7114
0,6569
0,6078
0,5547
0,5014
0,4508
2,00
2,6199
2,4257
2,2713
2,1413
2,0263
1,9217
1,8258
1,7373
1,6559
1,5812
1,5126
1,4499
1,3926
1,3401
1,2922
1,2485
1,0797
0,9694
0,8946
0,8422
0,8043
0,7760
0,7544
0,7376
0,7243
0,7134
0,7045
0,6971
0,6462
0,6033
0,5528
0,5006
0,4505
Таблица взята из работы Кригера [561. Параметры для двенадцати полярных газов, которые необхо-
димы при использовании этой таблицы, даны в табл. 92, стр. 468.
914
Литература 915
ЛИТЕРАТУРА
1. Johnston H. L., G r i 11 у Е. R., Journ. Phys. Chem., 46, 938 A942).
2. Landolt-Bo*rnstein, Physikalisch-Chemische Tabellen, Berlin (см. перевод:
Ландольт-Бернштейн, Физико-химические таблицы, М.—Л., 1939.
3. J о h n s t о n H. L., М с С 1 о s k е у К. Е., Journ. Phys. Chem., 44, 1038 A940).
4. Trautz M., Melster A., Zink R., Ann. d. Phys. E), 7, 409 A930).
,5. Trautz M., Binkele H. E., Ann. d. Phys. E), 5, 561 A930).
6. T r a u t z M., Zimmerman H., Ann. d. Phys. E), 22, 189 A935).
7. T r a u t z* M., В a u m a n n P. В., Ann. d. Phys. E), 2, 733 A929).
8. Trautz M., Kurz F., Ann. d. Phys. E), 9, 981 A931).
9. van Itterbeek A., Claes A., Physica, 5, 938 A938).
10. Trautz M., Heberling R., Husseini I., Ruf F., Freytag A., Ann.
d. Phys. E), 20, 118 A934).
11. Trautz M., Weisel W., Ann. d. Phys. D), 78, 305 A925).
12. Trautz M., Sorg K. G., Ann. d. Phys. E), 10, 81 A931).
13. К a n d a E., Bull. Chem. Soc. Japan, 12, 465 A937).
14. Braune H., Linke R., Zs. phys. Chem., A148, 195 A930).
15. Rankine A. O., Proc. Roy. Soc, 88, 582 A913).
16. Braune H., Basch R., Wentzel W., Zs. phys. Chem., A137, 176, 447 A928).
17. Trautz M., Narath A., Ann. d. Phys. E), 7, 427 A930).
18. Epstein L. F., Powers M. D., Journ. Phys. Chem. (в печати).
19. T i t a n i Т., Bull. Chem. Soc. Japan, 5, 98 A930).
20. T i t a n i Т., Bull. Chem. Soc. Japan, 8, 255 A933).
21. M i с h e 1 s A., W о u t e r s H., Physica, 8, 923 A941).
22. M i с h e 1 s A., G о u d e к е t M., Physica, 8, 347 A941).
23. M i с h e 1 s A., G о u d e к е t M., Physica, 8, 353 A941).
24. В u с к i n g h a m R. A., Proc. Roy. Soc, A168, 264 A938).
25. Hoi born L., Otto J., Zs. Phys., 33, 1 A925).
26. M i с h e 1 s A., W i j к e r Hub., W i j к е г Hk., Physica, 15, 627 A949).
27. В e a 11 i e J. А., В a r r i a u 11 R. J., В r i e r 1 e у J. S., Journ. Chem. Phys., 20,
1613 A952).
28. N e w i 11 D. M., Design of High Pressure Plant and the Properties of Fluids at High
Pressures, Oxford, 1940.
29. Beattie J. A., Barriault R. J., Brierley J. S., Journ. Chem. Phys., 19,
1222 A951).
30. H о 1 b о r n L., S с h u 11 z e H., Ann. d. Phys., 47, 1089 A915).
31. H о 1 b о r n L., Otto J., Zs. Phys., 10, 367 A922).
32. Michel s A., Wouters H., de Boer J., Physica, 1, 587 A934).
33. С о r n e r J., Proc Roy. Soc, 58, 737 A946).
34. Michels A., Michels C, Proc Roy. Soc, A153, 201 A936).
35. MacCormack К. Е., Schneider W. G., Journ. Chem. Phys., 18, 1269A950).
36. Hirschfelder J. O., McClure F. Т., Curtiss С F., Os borne D. W.,
NDRC Rep., A116, November, 1946.
37. Michels A., Nederbragt G. W., Physica, 2, 1000A935).
38. MacCormack К. Е., Schneider W. G., Journ. Chem. Phys., 19, 849 A951).
39. M i с h e 1 s A., G e 1 d e r m a n s M., Physica, 9, 967 A942).
40. В i r d R. В., S p о t z E. L., University of Wisconsin, CM-599 A950).
41. Bird R. В., Spotz E. L., Hirschfelder J. O., Journ. Chem. Phys., 18, 395
A950).
42. d e Boer J., Диссертация, Amsterdam, 1940.
43. К i ha г а Т., Journ. Phys. Soc. Japan, 6, 184 A951).
44. d e В о e r J., M i с h e 1 s A., Physica, 6, 409 A939).
45. de Boer J., van Kranendonk J., Compaan K-, Physica, 16, 545 A950).
46. Wen tor f R. H., Buehler R. J., Hirschfelder J. O., Curtiss C. F.,
Journ. Chem. Phys., 18, 1484 A950).
916 Литература
47. Н i г s с h f e 1 d e r J. О., Bird R. В., S р о t z E. L., University of Wisconsin, CF-857,
April 8, 1948; Journ. Chem. Phys., 16, 968 A948).
48. Rowlinson J. S., Trans. Farad. Soc, 45, 974 A949).
49. В i r d R. В., Диссертация, University of Wisconsin, 1950.
50. Rowlinson J. S., Journ. Chem. Phys., 19, 827 A951).
51. H о 11 e r a n E. M., H u 1 b u r t H. M., Journ. Chem. Phys., 19, 232 A951).
52. Ко tan i M., Proc. Phys.-Math. Soc. Japan C), 24, 76 A942).
53. Buckingham R. A., Corner J., Proc. Roy. Soc, A189, 118A947).
54. Rice W. E., H i r sc h f e 1 d e r J. O., Journ. Chem. Phys., 22 A954).
55. Mason E. A., Journ. Chem. Phys., 22 A954).
56. К r i e g e r F. J., The Viscosity of Polar Gases, Project Rand Report RM-646, July 1951.
ОГЛАВЛЕНИЕ
Стр.
Предисловие редактора перевода 5
Из предисловия авторов 7
Обозначения 11
Глава 1. Введение и основные понятия 19
§ 1. Уравнение состояния. Вириальные коэффициенты 19
A. Простейшая теория уравнения состояния разреженных газов 20
Б. Простейшая теория уравнения состояния плотных газов и жидкостей .. 22
B. Введение в строгую статистическую теорию уравнения состояния 23
§ 2. Кинетическая теория газов. Коэффициенты переноса 24
A. Элементарная кинетическая теория разреженных газов 25
Б. Введение в строгую кинетическую теорию газов 30
B. Уравнения переноса и их применения 33
§ 3. Межмолекулярные силы и потенциал межмолекулярного взаимодействия 34
A. Источники сведений о межмолекулярных силах 35
Б. Различные составляющие межмолекулярных сил 36
B. Эмпирические потенциальные функции межмолекулярного взаимодей-
ствия 40
§ 4. Классическая механика 44
A. Уравнения движения классической механики 45
Б. Теорема Лиувилля 47
B. Теорема вириала 48
§ 5. Столкновения молекул в классической механике (одноатомные молекулы) 50
A. Сумматорные инварианты столкновения 50
Б. Траектории частиц при столкновении 51
B. Угол отклонения при столкновении 55
§ 6. Квантовая механика 56
A. Экспериментальные данные; о «неклассических» свойствах систем .... 56
Б. Квантовомеханическое описание систем 57
B. Операторы в волновой механике 59
Г. Неразличимость тождественных частиц 61
Д. Приближенные методы решения уравнения Шредингера 63
Е. Теорема вириала в квантовой механике 67
§ 7. Столкновения молекул в квантовой механике 68
А. Взаимодействие двух частиц; фазовые сдвиги rj^x) 69
Б. Вероятность отклонения на угол % 71
Задачи 73
Литература 74
918 Оглавление
ЧАСТЬ I
РАВНОВЕСНЫЕ СВОЙСТВА
Глава 2. Статистическая механика 79
§ 1. Описание статистических ансамблей в классической механике 79
A. Конфигурационное пространство, пространство импульсов и фазовое
пространство 80
Б. Ансамбли и функции распределения 81
B. Изменение плотности вероятности во времени 83
Г. Ансамбли, представляющие замкнутые системы в состоянии равновесия 83
Д. Ансамбли, представляющие открытые системы в состоянии равновесия 85
§ 2. Описание статистических ансамблей в квантовой механике 86
A. Квантовомеханическое описание отдельных систем 86
Б. Определение матрицы плотности вероятности 87
B. Физический смысл матрицы плотности вероятности 88
Г. Другие матрицы плотности вероятности 89
Д. Зависимость матрицы плотности от времени; ансамбли для замкнутых
систем в состоянии равновесия 89
Е. Ансамбли для открытых систем (большой канонический ансамбль) в со-
стоянии равновесия 90
§ 3. Основы статистической, механики 91
A. Обоснование микроканонического ансамбля 91
Б. Использование микроканонического ансамбля для получения выражения
распределения энергии по макроскопическим подсистемам 91
B. Средние по ансамблю и флуктуации 93
Г. Распределение энергии межцу молекулами в газе 95
Д. Обоснование канонического ансамбля 96
Е. Вычисление средних по ансамблю 98
§ 4. Основы статистической термодинамики 98
A. Статистическая сумма 99
Б. Внутренняя энергия и первый закон термодинамики 100
B. Температура, энтропия и второй закон термодинамики 101
Г. Энтропия при абсолютном нуле и третий закон термодинамики 102
Д. Выражение термодинамических величин через статистическую сумму .. 103
§ 5. Вычисление термодинамических свойств идеальных газов 103
A. Статистическая сумма идеального газа 103
Б. Распределение энергии между молекулами идеального газа 104
B. Влияние поступательного и внутреннего движения молекулы на термо-
динамические свойства 106
Г. Смеси идеальных газов 110
§ б. Теория флуктуации 111
А. Флуктуации плотности, выраженные через термодинамические величины 112
Б. Флуктуации плотности, выраженные через радиальную функцию рас-
пределения 115
Задачи 116
Литература 118
Глава 3. Уравнение состояния для газов малой и умеренной плотности 120
§ 1. Формальный вывод уравнения состояния в статистической механике .... 121
A. Метод статистической суммы 121
Б. Метод, основанный на теореме вириала 122
B. Эквивалентность уравнения состояния, полученного методом статисти-
ческой суммы и с помощью теоремы вириала 124
Оглавление 919
§ 2. Вывод вириального уравнения состояния с помощью статистической суммы 124
A. «(/-функции» 125
Б. Групповые интегралы Ъх 126
B. Вычисление статистической суммы 128
Г. Уравнение состояния в вириальной форме 129
§ 3. Вывод вириального уравнения состояния с помощью теоремы вириала 130
A. «Модифицированные (/-функции» , 130
Б. «Модифицированные групповые интегралы» 131
B. Выражение бинарной функции распределения через плотность 131
Г. Уравнение состояния в вириальной форме 132
§ 4. Вириальные коэффициенты 132
A. Предположение об аддитивности межмолекулярных сил 132
Б. Вириальные коэффициенты для не зависящих от углов потенциалов 134
B. Вириальные коэффициенты для потенциалов, зависящих от углов 135
Г. Вириальные коэффициенты для смесей 136
Д. Определение вириальных коэффициентов из данных зависимости между р, V и Т 137
§ 5. Вириальные коэффициенты в случае простых потенциалов взаимодействия,
не зависящих от углов 139
A. Модель твердых сфер 139
Б. Модель точечных дентров отталкивания 140
B. Модель Сюзерленда 140
Г. Модель прямоугольной потенциальной ямы 141
§6. Вириальные коэффициенты для потенциала F-12) Леннарда-Джонса .... 143
A. Вычисление второго вириального коэффициента 144
Б. Определение межмолекулярных сил по значениям второго вириального
коэффициента 146
B. Вычисление третьего вириального коэффициента 149
Г. Коэффициент Джоуля—Томсона 152
§ 7. Второй вириальный коэффициент для более сложных сферически симметрич-
ных потенциалов 155
А. Потенциал Букингема—Корнера 155
Б. Модифицированный потенциал (б-ехр) Букингема 156
§ 8. Второй вириальный коэффициент для несферических молекул 157
A. Рассмотрение модели твердых выпуклых молекул методом Ишихары ... 158
Б. Обобщенная модель сфероцилиндрических молекул Кихары 160
B. Обобщенная модель эллипсоидальных молекул Кихары 163
Г. Четырехцентровая модель длинных молекул Корнера 164
§ 9. Обсуждение результатов, полученных для нескольких потенциальных функ-
ций межмолекулярного взаимодействия неполяриых молекул 166
A. Сферические молекулы 166
Б. Несферические молекулы 172
B. Сравнение различных потенциальных функций межмолекулярного взаи-
модействия 174
§ 10. Вириальные коэффициенты для газов, состоящих из полярных молекул .. 176
A. Модель твердых сфер, содержащих точечные диполи 177
Б. Второй вириальный коэффициент для потенциала Штокмайера 178
B. Определение параметров потенциала Штокмайера по значениям второго
вириального коэффициента 180
Г. Коэффициент Джоуля—Томсона для потенциала Штокмайера 182
Д. Третий вириальный коэффициент для потенциала Штокмайера 183
Е. Вычисление для смесей 184
Ж. Диполь-квадрупольное взаимодействие сложных молекул 186
Приложение А. Разложение Кихары третьего вириального коэффициента .... 189
Приложение Б. Выражение термодинамических свойств через уравнение состоя-
ния и вириальные коэффициенты 190
Задачи 192
Литература 193
920 Оглавление
Глава 4. Уравнение состояния плотных газов и жидкостей 197
§ 1. Закон соответственных состояний 197
A. Эмпирический закон соответственных состояний 198
Б. Обобщенные диаграммы сжимаемости и термодинамических свойств плот-
ных газов и жидкости (по Хогену и Ватсону) 201
B. Теоретический вывод закона соответственных состояний для газов, со-
стоящих из сферических неполярных молекул 205
Г. Закон соответственных состояний для многоатомных молекул 208
Д. Закон соответственных состояний для полярных молекул 209
§ 2. Эмпирические уравнения состояния 210
A. Уравнения состояния с двумя постоянными 210
Б. Уравнение состояния Битти—Бриджмена 212
B. Уравнение состояния Бенедикта—Вебба—Рубина 216
Г. Эмпирические зависимости для жидкостей 218
§ 3. Газы при очень высоком давлении 219
A. Уравнение состояния пороховых газов 219
Б. Уравнение состояния газа при детонации 219
B. Использование теоремы о вириале для изучения деформаций молекул в
условиях экстремально высоких давлений 220
Г. Квантовомеханическое рассмотрение деформации молекул при высоком
давлении 223
Д. Оптические и электрические методы изучения деформации молекул при
высоких давлениях 225
§ 4. Некоторые общие положения метода ячеек » 225
A. Кристаллическая структура как основа метода ячеек 226
Б. Понятие коллективной энтропии 227
B. Концепция свободного объема 229
§ 5. Упрощенная модель ячеек для жидкостей и плотных газов 232
A. Приближенные выражения для свободного объема и энергии решетки .. 232
Б. Уравнение состояния Эйринга 233
B. Давление насыщающих паров, правило Гильдебранда и правило Трутона 234
Г. Теплоемкость 235
Д. Изменение энтропии при плавлении 236
§ 6. Уравнение состояния для твердых сфер, не обладающих силами притяжения 237
А. Точный и «сглаженный» свободный объем 237
Б. Уравнение состояния при малой плотности; строго и нестрого центри-
рованные свободные объемы 240
§ 7. Уравнение состояния Леннарда-Джонса и Девоншайра 242
A. Вывод уравнения состояния 243
Б. Модификация уравнения состояния Леннарда-Джонса и Девоншайра,
учитывающая взаимодействие с тремя оболочками 245
B. Сравнение с экспериментальными результатами 249
Г. Модификация уравнения состояния Леннарда-Джонса и Девоншайра,
учитывающая двойное заполнение ячеек 253
§ 8. Теория дырок для жидкости и плотного газа 254
A. Общая теория дырок для жидкостей 254
Б. Линейное приближение логарифма величины свободного объема 256
B. Сравнение вычислений, проведенных на основе теории дырок, с экспериментом 258
§ 9. Выражение уравнения состояния через бинарную функцию распределения 261
A. Свойства бинарной функции распределения 261
Б. «Потенциал усредненной силы» 263
B. Вывод интегральных уравнений для бинарной функции распределения 264
Г. Решение интегрального уравнения для бинарной функции распределения.
Суперпозиционное приближение 266
Задачи 270
Литература 271
Оглавление 921
Глава 5. Равновесие жидкость—пар и критические явления 274
§ 1. Промежуточная область между жидкостью и паром 275
A. Определение поверхностного натяжения 275
Б. Расчет поверхностного натяжения методом свободного объема 278
B. Расчет поверхностного натяжения методом радиальной функции распре-
деления 282
Г. Влияние радиуса кривизны на поверхностное натяжение 283
Д. Расчет поверхностного натяжения в первом приближении 285
Е. Уравнение Мак-Леода и парахор 287
§ 2. Фазовые переходы в однокомпонентных системах 289
A. Методы определения критической точки 290
Б. Стабильные и метастабильные состояния 293
B. Термодинамические свойства критической области 296
Г. Поведение вещества в критической области 300
Д. Статистическая теория конденсации 303
§ 3. Фазовые переходы в двухкомпонентных системах 306
А. Критическая точка и обратное поведение 306
Б. Термодинамическое рассмотрение 311
Приложение А. Химический потенциал смеси газов, подчиняющейся урав-
нению Ван дер Ваальса 312
Задачи 313
Литература 313
Глава 6. Квантовая теория уравнения состояния 315
§ 1. Предварительные сведения из статистической механики 316
А. Матрица плотности вероятности для канонического ансамбля 316
Б. Матрица плотности вероятности в классическом пределе 317
§ 2. Вывод уравнения состояния из статистической механики 320
A. Метод статистической суммы 320
Б. Метод, основанный на квантовомеханической теореме вириала 320
B. Эквивалентность уравнений состояния, полученных из статистической
суммы и с помощью теоремы вириала 321
Г. Окончательное выражение квантовомеханического уравнения состояния 321
§ 3. Свойства идеального газа 322
A. Сумма Слетера идеального газа, состоящего из молекул со спином, рав-
ным нулю 322
Б. Сумма Слетера идеального газа, состоящего из молекул со спином, не рав-
ным нулю 323
B. Уравнение состояния идеального газа 324
Г. Конденсация Бозе—Эйнштейна 325
§ 4. Второй вириальный коэффициент реальных газов при очень низких темпера-
турах 326
A. Выражение второго вирйального коэффициента через фазовые сдвиги и
энергию стационарных состояний 327
Б. Вычисление фазовых сдвигов для некоторых простых потенциалов 329
B. Вычисления для потенциала Леннарда-Джонса 331
§ 5. Второй вириальный коэффициент реальных газов при «промежуточных
температурах» 334
A. Общие квантовые выражения второго вирйального коэффициента 335
Б. Представление квантовой поправки к В(Т) в виде ряда (потенциал Лен-
нарда-Джонса) 336
B. Вычисления для потенциала Леннарда-Джонса 337
Г. Вычисления для потенциала Букингема—Корнера 338
922 Оглавление
§ 6. Закон соответственных состояний в квантовой механике 339
A. Закон соответственных состояний в квантовой механике 339
Б. Квантовые эффекты в жидкой фазе 340
B. Свойства изотопов гелия и водорода 343
§ 7. Квантовые эффекты в двухатомных газах 346
А. Краткая сводка теоретических результатов 346
Б. Сравнение теоретических и экспериментальных данных для водорода .. 347
Задачи 348
Литература 349
ЧАСТЬ I I
НЕРАВНОВЕСНЫЕ СВОЙСТВА
Глава 7. Кинетическая теория разреженных газов 353
§ 1. Функция распределения кинетической теории 353
A. Физическое описание неравновесных систем 354
Б. Физический вывод уравнения Больцмана 355
B. Вывод уравнения Больцмана из теоремы Лиувилля 359
Г. Распределение по скоростям 361
§ 2. Уравнения переноса 361
A. Скорости молекул и скорости потока 362
Б. Векторы потоков 364
B. Общее уравнение переноса 366
Г. Исчезновение интегралов столкновений для сумматорных инвариантов 367
Д. Частный вид уравнений переноса 368
§ 3. Решение уравнения Больцмана методом Энскога 370
A. Н-теорема (решение для случая равновесного состояния системы) 371
Б. Разложение функций распределения в ряды по методу Энскога 372
B. Решение уравнения Больцмана в первом приближении 374
Г. Интегральные уравнения 374
Д. Некоторые важные интегральные теоремы 377
Е. Вариационный принцип 378
Ж. Приложение вариационного принципа (разложение по полиномам Сонина) 378
§ 4. Формулы для вычисления коэффициентов переноса 381
A. Выражение коэффициентов диффузии и термодиффузии через коэффициент
разложения по полиномам Сонина 381
Б. Выражение коэффициента вязкости через коэффициенты разложения по
полиномам Сонина 383
B. Выражение коэффициента теплопроводности через коэффициенты разложе-
ния по полиномам Сонина 384
Г. Интегралы ?(Zs) 386
Д. Точные формулы для коэффициентов переноса, выраженные через интег-
ралы &**> 386
§ 5. Решение уравнения Больцмана методом Грэда 391
А. Уравнения моментов 392
Б. «Тринадцатимоментное» приближение 393
§ 6. Влияние химических реакций и внутренних степеней свободы молекул ..» 394
A. Уравнения переноса для реагирующей газовой смеси 395
Б. Влияние внутренних степеней свободы молекул на явления переноса
(поправка Эйкена) 396
B. Формальная кинетическая теория многоатомных молекул 398
Г. Некоторые специальные модели (твердые овалоиды, шероховатые сферы,
нагруженные сферы) 402
Приложение А. Значения «скобочных» выражений, используемых при выводе
коэффициентов переноса, записанные через интегралы Q^1^ 406
Задачи 407
Литература 407
Оглавление 923
Глава 8. Явления переноса в разреженных газах 409
§ 1. Векторы плотностей потоков и коэффициенты переноса 409
A. Перенос массы и коэффициенты диффузии 411
Б. Перенос импульса и коэффициент вязкости 414
B. Перенос энергии и коэффициент теплопроводности 415
§ 2. Сводка формул кинетической теории для чистых газов и многокомпонентных
смесей 416
A. Величины ?<'>s>* 416
Б. Коэффициент вязкости 420
B. Коэффициент теплопроводности 423
Г. Коэффициент диффузии 427
Д. Коэффициент термодиффузии 429
§ 3. Вычисление коэффициентов переноса для простых потенциалов 431
A. Твердые упругие сферы 431
Б. Точечные центры отталкивания 432
B. Модель Сюзерленда 435
Г. Прямоугольная потенциальная яма 436
§ 4. Вычисление коэффициентов переноса для потенциала F-12) Леннарда-
Джонса 437
A. Динамика парных столкновений и вычисление эффективного сечения
столкновения 438
Б. Коэффициент вязкости чистых газов 441
B. Коэффициент вязкости смесей 447
Г. Коэффициент теплопроводности 451
Д. Коэффициент диффузии 454
Е. Термодиффузионное отношение 458
§ 5. Сравнение результатов вычисления коэффициентов переноса для некоторых
сферически симметричных потенциальных функций, описывающих взаи-
модействие между неполярными молекулами 462
§ 6. Вычисление коэффициентов переноса чистых газов и газовых смесей, содер-
жащих одну полярную компоненту (приближенное рассмотрение) 467
А. Вязкость чистых газов 467
Б. Коэффициенты вязкости и диффузии смесей, содержащих одну полярную
компоненту 469
Приложение А. Формулы для высших приближений коэффициентов переноса ... 473
Задачи 477
Литература 477
Глава 9. Явления переноса в плотных газах и жидкостях 480
§ 1. Закон соответственных состояний 481
A. Обзор экспериментальных данных о коэффициентах переноса при высоких
плотностях 481
Б. Закон соответственных состояний для сферических неполярных молекул 484
B. Приложения закона соответственных состояний 485
Г. Закон соответственных состояний для полярных молекул 488
§ 2. Теория Эйринга явлений переноса в газах и жидкостях 489
A. Коэффициент вязкости 490
Б. Коэффициент диффузии 494
B. Коэффициент теплопроводности 495
§ 3. Теория Энскога явлений переноса в плотных газах и жидкостях 496
А. Видоизменение уравнения Больцмана для плотных газов 497
Б. Векторы плотности потоков 499
924 Оглавление
В. Уравнения переноса 500
Г. Решение видоизмененного уравнения Больцмана 502
Д. Коэффициенты переноса 503
Е. Краткий обзор результатов теории плотных газов Энскога для твердых сфер 506
Ж. Приложение результатов к реальным газам 507
§ 4. Рассмотрение явлений переноса в плотных газах и жидкостях методами ста-
тистической механики неравновесных систем 509
A. Уравнение Лиувилля и общее уравнение переноса 510
Б. Макроскопические переменные 510
B. Макроскопические переменные, выраженные через функции распределе-
ния низших порядков 512
Г. Векторы плотности потоков и уравнения переноса 513
Д. Вычисление коэффициентов переноса 515
Приложение А. Теория скоростей реакций Эйринга 516
Задачи 521
Литература 521
Глава 10. Квантовая теория и явления переноса 523
§ 1. Квантовая статистическая механика неравновесных систем 523
А. Общие положения статистической механики 524
Б. Уравнение Больцмана для разреженной смеси газов 525
§ 2. Явления переноса при очень низких температурах 527
A. Диффракционные эффекты 528
Б. Эффекты симметрии : 529
B. Вычисления для случая очень низких температур 531
§ 3. Явления переноса при промежуточных температурах 534
A. Определение фазовых сдвигов методом ВКБ 534
Б. Выражение сечения в виде ряда по степеням постоянной Планка 537
B. Вычисления для потенциала отталкивания, обратно пропорционального
двенадцатой степени расстояния 539
§ 4. Закон соответственных состояний в квантовой механике 540
Задачи 541
Литература 542
Глава 11. Гидродинамические применения уравнений переноса 543
§ 1. Уравнения гидродинамики 543
A. Применимость уравнений переноса 544
Б. Сводка уравнений переноса 545
B. Уравнение энтропии 547
§ 2. Термодинамика необратимых процессов 550
A. Соотношение взаимности Онзагера 550
Б. Применение термодинамики необратимых процессов к явлениям переноса 553
B. Применение термодинамики необратимых процессов к переносу количества
движения 554
Г. Применение термодинамики необратимых процессов к явлениям переноса
массы и энергии 556
Д. Сводка результатов 559
§ 3. Передача энергии излучением 562
А. Поток излучения в двух частных случаях 563
Б. Стационарный фронт излучения 566
§ 4. Теория распространения звука 567
А. Распространение без поглощения 568
Б. Распространение с поглощением 569
Оглавление 925
§ 5. Распространение волн конечной амплитуды в одном измерении 573
A. Метод характеристик Римана 574
Б. Применение метода характеристик к случаю идеального газа 576
B. Образование ударных волн в идеальном газе 577
§ 6. Одномерные уравнения переноса в стационарном случае 581
А. Основные уравнения системы в общем случае 581
Б. Основные уравнения для системы вблизи равновесия 585
§ 7. Теория распространения пламени 588
A. Качественная теория пламени горелки Бунзена 588
Б. Одномерная стационарная теория распространения пламени 591
B. Простой пример пламени : мономолекулярная обратимая реакция 594
Г. Простой пример пламени (кинетической энергией и диффузией пренеб-
регается) 598
Д. Простой пример пламени (кинетической энергией пренебрегается, а диф-
фузия учитывается) 601
Е. Простой пример пламени (диффузией пренебрегается, кинетическая энергия
учитывается) 604
§ 8. Теория распространения ударных волн 606
A. Соотношения Гюгонио 607
Б. Применение соотношений Гюгонио к идеальному газу 608
B. Структура и толщина ударной волны в идеальном газе 611
§ 9. Теория детонации 615
A. Соотношения Гюгонио и условие Чепмена—Жуге 616
Б. Применение теории к случаю детонации идеального газа 620
B. Структура детонационной волны 622
§ 10. Поток топливных газов в ракетных двигателях 626
A. Уравнения гидродинамики 627
Б. Течение идеального газа в сопле 629
B. Термохимия смесей реальных газов 631
Г. Течение реального газа в сопле 634
Задачи 637
Литература 638
ЧАСТЬ III
МЕЖМОЛЕКУЛЯРНЫЕ СИЛЫ
Глава 12. Электромагнитные основы теории межмолекулярных сил 643
§ 1. Электростатика 644
A. Кулоновское взаимодействие зарядов и распределений заряда 644
Б. Электростатический потенциал и напряженность электрического поля .. 645
B. Мультипольные моменты 646
Г. Одноцентровое разложение электростатического потенциала распределения
заряда 648
Д. Двухцентровое разложение потенциальной энергии взаимодействия двух
распределений заряда 649
Е. Свойства электрического диполя 651
§ 2. Поляризация вещества и диэлектрическая восприимчивость 655
А. Поляризуемость и поляризация 655
Б. Векторы JZ) и g 656
926 Оглавление
В. Вектор напряженности локального поля ^лок-) 658
Г. Выражение диэлектрической восприимчивости через диэлектрическую
постоянную 659
Д. Выражение диэлектрической восприимчивости через молекулярные вели-
чины 660
§ 3. Электромагнитные уравнения Максвелла 662
A. Уравнения Максвелла в вакууме 663
Б. Скалярный и векторный потенциалы; магнитные мультиполи 665
B. Намагничивание вещества 667
Г. Уравнения Максвелла для материальной среды 667
§ 4. Намагничивание вещества и магнитная восприимчивость 669
A. Векторы J( и JS 669
Б. Локальный вектор магнитной индукции «$$(лок«) 671
B. Выражение магнитной восприимчивости через магнитную проницаемость 672
§ 5. Классическая теория поглощения света и коэффициента преломления 674
A. Осциллирующий диполь (вибратор Герца) 674
Б. Уравнение движения заряженных частиц 675
B. Коэффициент преломления (теория Друде) 677
§ 6. Квантовомеханическая теория поглощения света и коэффициента прелом-
ления 678
A. Вероятности переходов молекулы в электромагнитном поле 678
Б. Индуцированное поглощение и испускание света 680
B. Коэффициент преломления 682
§ 7. Рассеяние электромагнитных волн 684
А. Рассеяние видимого света 686
Б. Рассеяние рентгеновских лучей 689
Приложение А. Коэффициенты двухцентрового разложения Bl^b{rit rj ; rab)... 690
Приложение Б. Коэффициенты представления трехмерной группы вращения 695
Приложение В. Матричные компоненты дипольного момента для оптических
переходов 700
Задачи 702
Литература 702
Глава 13. Теория межмолекулярных сил 704
§ 1. Функции потенциальной энергии межмолекулярного взаимодействия 706
A. Концепция функции потенциальной энергии межмолекулярного взаимо-
действия 706
Б. Приближения, вводимые разделением электронного и ядерного движений
(разделение Борна—Оппенгеймера) 710
B. Сведения о межмолекулярных потенциалах, получаемые с помощью тео-
ремы вириала 714
Г. Эквивалентность межмолекулярных сил, вычисленных на основе класси-
ческой и квантовой механики 716
Д. Квантовомеханический расчет потенциальной энергии межмолекулярного
взаимодействия 720
§ 2. Поляризуемость молекул 722
A. Применение вариационного метода для расчета поляризуемостей 723
Б. Поляризуемость молекулярного водорода 726
B. Аддитивность поляризуемостей связей 727
Г. Оценка поляризуемости и других свойств атомов путем использования
экранировочных постоянных 730
§ 3. Дисперсионные силы Лондона, действующие между симметричными моле-
кулами 733
А. Упрощенная теория дисперсионных сил, основанная на модели Друде 733
Оглавление 927
Б. Дисперсионные силы как возмущения второго порядка 737
В. Члены высших приближений в выражении для дисперсионной энергии 740
Г. Влияние «запаздывания» на дисперсионные силы на больших расстояниях 742
§ 4. Дисперсионные силы, действующие между асимметричными молекулами .. 743
A. Дисперсионные силы, действующие между асимметричными молекулами
на больших расстояниях 744
Б. Дисперсионные силы, действующие между асимметричными молекулами на
средних расстояниях 745
B. Дисперсионная энергия взаимодействия длинных ароматических молекул
с сопряженными двойными связями 747
§ 5. Силы, действующие между молекулами, имеющими постоянный электриче-
ский момент 754
A. Энергия индукции 754
Б. Потенциальная энергия взаимодействия, усредненная по всем ориента-
циям молекул 755
B. Относительные значения вкладов в межмолекулярный потенциал 757
Г. Водородные связи как электростатические силы 758
§ 6. Квантовомеханическая трактовка резонансных и электростатических сил 759
A. Природа резонансных сил 760
Б. Квантовомеханическое взаимодействие двух идеальных диполей, вставлен-
ных в линейные молекулы 764
B. Квантовомеханическое взаимодействие двух идеальных диполей, встав-
ленных в симметричные волчки 769
Г. Далекие взаимодействия между протоном и атомом водорода или атомом
гелия 771
Д. Силы квадруполь-квадрупольного взаимодействия атомов, ни один из
которых не находится в S-состоянии 775
§ 7. Определение межмолекулярных сил из данных по уширению микроволнового
спектра вследствие давления 781
А. Уширение линий в микроволновом спектре 781
Б. Сведения о дальнодействующих силах из данных по уширению вследствие
давления 784
§ 8. Определение квадрупольного момента молекулы воды 787
А. Теоретическое определение 788
Б. Эмпирическое определение 790
§ 9. Определение межмолекулярных сил из свойств кристаллов при абсолютном
нуле 792
A. Потенциальная энергия кристаллической решетки при абсолютном нуле 793
Б. Нулевая энергия кристаллической решетки при абсолютном нуле 793
B. Определение сил взаимодействия между атомами благородного газа 797
Приложение А. Полный гамильтониан системы во внешнем электрическом и маг-
нитном полях 798
Приложение Б. Отношение кинетической энергии электронов к потенциальной
энергии в молекулярной системе 799
Задачи 801
Литература 802
Глава 14. Квантовомеханические вычисления межмолекулярных сил 805
§ 1. Взаимодействие двух атомов водорода 808
А. Состояние г2, соответствующее нормальной молекуле Н2 811
Б. Состояние 327, соответствующее отталкиванию двух атомов водорода, на-
ходящихся в ls-состоянии 813
928 Оглавление
§ 2. Энергия взаимодействия атомов благородного газа, находящихся в основных
состояниях 814
A. Взаимодействие двух атомов гелия 815
Б. Взаимодействие двух атомов неона 818
B. Взаимодействие двух атомов аргона 820
§ 3. Взаимодействие атома водорода с молекулой водорода 821
А. Полуэмпирический метод Эйринга 822
Б. Прямой расчет дисперсионной энергии в первом порядке теории возму-
щений 825
§ 4. Взаимодействие двух молекул водорода 826
A. Химическая, или валентная, энергия 827
Б. Энергия взаимодействия на больших расстояниях 830
B. Полная энергия взаимодействия и сравнение с экспериментом 832
§ 5. Взаимодействие атомов или молекул водорода с различными ионами водо-
рода 834
A. Взаимодействие Н + Н+ 834
Б. Взаимодействие Н + Н" 835
B. Взаимодействия Н + Н^, Н2 + Н+ и Ня + НJ 835
Г. Взаимодействие На + Н" 836
Д. Группа ионов 837
§ 6. Взаимодействие атома гелия с возбужденным атомом гелия или с протоном 838
А. Взаимодействие нормального и метастабильного атомов гелия 838
Б. Взаимодействие нормального атома гелия с протоном 841
Приложение А. Интегралы, применяемые при расчетах энергий межмолекуляр-
ного взаимодействия 842
Задачи 845
Литература 846
ПРИЛОЖЕНИЕ
Таблица I. Постоянные сил взаимодействия для потенциала F-12) Леннарда-Джонса 851
Таблица II. Второй вириальный коэффициент и коэффициент Джоуля—Томсона при
нулевом давлении для потенциала F-12) Леннарда-Джонса 854
Таблица III. Третий вириальный коэффициент и его производные для потенциала
F-12) Леннарда-Джонса 856
Таблица IV. Функция А(Т*) для оценочных значений третьего вириального коэф-
фициента для газовых смесей 857
Таблица V. Коэффициенты разложения для второго и третьего вириальных коэффи-
циентов (и квантовые поправки ко второму вириальному коэффициенту) для
потенциала F-12) Леннарда-Джонса 858
Таблица VI. Фазовые сдвиги rjfao) для Не4, рассчитанные с помощью потенциала
F-12) Леннарда-Джонса 859
Таблица VII. Фазовые сдвиги B/+ l)fJi(xa) для Не8, рассчитанные с помощью потен-
циала F-12) Леннарда-Джонса 860
Таблица VIII. Фактор сжимаемости pV/NkT, основанный на модели Леннарда-Джонса
и Девоншайра с учетом трех оболочек 861
Таблица IX. Значение поправки на неидеальность газа для приведенной внутренней
энергии O'/Ne, основанное на модели Леннарда-Джонса и Девоншайра с учетом
трех оболочек 863
Таблица X. Значение поправки на неидеальность газа для приведенной теплоемкости
C'vjNk, основанное на модели Леннарда-Джонса и Девоншайра с учетом трех
оболочек 865
Таблица XL Значение поправки на неидеальность газа для приведенной энтропии
S'/Nk, основанное на модели Леннарда-Джонса и Девоншайра с учетом трех
оболочек 867
Оглавление 929
Таблица XII. Значение интегралов &1'** для вычисления коэффициентов переноса
с помощью потенциала F-12) Леннарда-Джонса 868
Таблица XIII. Величины А*, в* и с* для вычисления коэффициентов переноса в
смесях с помощью потенциала F-12) Леннарда-Джонса 870
Таблица XIV. Функции для вычисления высших приближений для коэффициентов
переноса 871
Таблица XV. Функция [кт]* для вычисления изотопического коэффициента диф-
фузии 872
Таблица XVI. Расстояние наибольшего сближения и угол отклонения 873
Таблица XVII. Значения второго вириального коэффициента для газа полярных
молекул (потенциал Штокмайера) 880
Таблица XVIII. Коэффициент Джоуля—Томсона для полярных газов (потенциал
Штокмайера) 884
Таблица XIX. Функция (Т*//*)Б*, применяемая для определения параметров в по-
тенциале Штокмайера по экспериментальным значениям второго вириального
коэффициента 888
Таблица XX. Третий вириальный коэффициент для полярных газов (потенциал Шток-
майера) 890
Таблица XXI. Расстояние наибольшего сближения и угол отражения для отталки-
вающего потенциала, обратно пропорционального двенадцатой степени рас-
стояния 891
Таблица XXII. Интегралы <Qa'e)*, применяемые при расчете коэффициентов переноса
и вычисленные для модели потенциальной ямы 892
Таблица XXIII. Интегралы ?(lt#>*, применяемые при расчете коэффициентов пере-
носа и вычисленные для потенциала Сюзерленда с притягивающим членом,
обратно пропорциональным шестой степени расстояния 893
Таблица XXIV. Второй вириальный коэффициент, рассчитанный с помощью потен-
циала Букингема—Корнера 894
Таблица XXV. Коэффициент Джоуля—Томсона при нулевом давлении, рассчитанный
с помощью потенциала Букингема—Корнера 896
Таблица XXVI. Первая квантовая поправка для второго вириального коэффициента,
рассчитанная с помощью потенциала Букингема—Корнера 898
Таблица XXVII. Первая квантовая поправка для коэффициента Джоуля—Томсона
при нулевом давлении, рассчитанная с помощью потенциала Букингема—Кор-
нера 900
Таблица XXVIII. Второй вириальный коэффициент, рассчитанный с помощью моди-
фицированного потенциала F-ехр) Букингема 902
Таблица XXIX. Интегральные функции столкновения, применяемые при расчетах
коэффициентов переноса и вычисленные для модифицированного потенциала
F-ехр) Букингема 904
Таблица XXX. Функции для вычисления высших приближений коэффициентов пере-
носа для видоизмененного потенциала F-ехр) Букингема 908
Таблица XXXI. Функция р для вычисления изотропной термодиффузии для видоиз-
мененного потенциала F-ехр) Букингема 910
Таблица XXXII. Величины А*, в* и с* для вычисления коэффициентов переноса
смесей для видоизмененного потенциала F-ехр) Букингема 911
Таблица XXXIII. Функции, необходимые для вычисления второго вириального
коэффициента для обобщенных сфероцилиндрических и эллипсоидальных мо-
лекул Кихара 913
Таблица XXXIV. Приведенные усредненные эффективные сечения ?B»2)* для по-
лярных молекул 914
Литература 915
Дж. Гиршфельдер, Ч. Кертисс, Р. Берд
МОЛЕКУЛЯРНАЯ ТЕОРИЯ ГАЗОВ
И ЖИДКОСТЕЙ
Редактор Л. В. ГЕССЕН
Художник А. П. Купцов
Технический редактор Е. С. Потапенкова
Корректор Е. В. Кочегарова
Сдано в производство 25/1X 1959 г.
Подписано к печати 28/XI 1960 г.
Бумага 7OxlO8Vie = 29,1 бум. л.
79,8 печ. л.
Уч.-изд. л. 74,7. Изд. № 2/4610
Цена 5 р. 43 к. Зак. 2017
* * *
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, 1-й Рижский пер., 2
¦ * *
Типография Академии, Будапешт
Стр.
45
48
50
321
399
410
468
483
507
544
820
Строка
Формула D.3)
Формула D.17)
4 св.
19 св.
Формула F.25)
8 сн.
20 св.
1 сн.
Подпись к фиг. 146
12 сн.
18 св.
Опечатки
Напечатано
т\
dF .
dpi Ч*
ЗФа - 6ФГ
kj/L
г\>
дабан
Хе - С2Н2
у = (pV/RT) -
крупного
. ВЗАИМОД ВИЕ
Следует читать
г)
dF .
dpi Pt
ЗФа - ЬФГ
kjL
rijr)
дебай
Хе - С2Нв
у = pv/RT - 1
круглого
В. ВЗАИМОДЕЙСТВИЕ