/













































































































































































































































































































Текст
МИНИСТЕРСТВО НЕФТЯНОЙ ПРОМЫШЛЕННОСТИ СССР ТРУДЫ ВСЕСОЮЗНОГО НАУЧНО-ИССЛЕДОВАТЕЛЬСКОГО ИНСТИТУТА ПО ПЕРЕРАБОТКЕ СЛАНЦЕВ (ВНИИПС)
ВЫПУСК II
химия И ТЕХНОЛОГИЯ ПРОДУКТОВ ПЕРЕРАБОТКИ СЛАНЦЕВ
j „ . -м ....................?
| И И И Ч (' Н < ,-А ; Г., |
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО НЕФТЯНОЙ И ГОРНО-ТОПЛИВНОЙ ЛИТЕРАТУРЫ
ЛЕНИНГРАДСКОЕ ОТДЕЛЕНИЕ Ленинград 1954
15—5—4
В книге изложены результаты изучения сланцевой смолы как сырья для производства различных видов жидкого топлива, смазочных масел и различных химических продуктов (растворителей, пластмасс и т. д.).
Книга рассчитана на научных и инженерно-технических работников, связанных с химией твердого и искусственного жидкого топлива.
Редакционная коллегия:
Коллеров Д. К., Зеленин Н. И., Гарновская Г. Н.
ПРЕДИСЛОВИЕ
Сборник трудов Всесоюзного научно-исследовательского института по переработке сланцев (ВНИИПС), выпуск IE так же как и выпуск I, изданный в 1948 г., посвящен, в основном, работам по исследованию состава и свойств смол и подсмольных вод прибалтийских сланцев.
Большие успехи в развитии сланцеперерабатывающей промышленности в Ленинградской области и ЭССР вызвали необходимость углубленного изучения состава продуктов переработки горючих сланцев, так как, наряду с получением бытового газа и смолы, используемой в качестве котельного топлива, наметились перспективы переработки смолы на моторное топливо и различные химические продукты. Это нашло отражение в Законе о пятом пятилетием плане развития СССР на 1951—1955 годы, которым только по Эстонской ССР предусматривается, наряду с увеличением производства газа из сланцев в 2—2,5 раза, рост производства искусственного жидкого топлива из того же сырья на 80%.
Исследование свойств сланцевой смолы как сырья для производства различных сортов жидкого топлива и получения различных химических продуктов играет важнейшую роль в исследованиях ученых, занятых проблемой переработки горючих сланцев.
Наиболее важной задачей научно-исследовательских работ в области химии сланцевой смолы является установление ее состава. Этот вопрос, несмотря на его большую историю, не получал должного освещения в работах исследователей главным образом потому, что принципиальные установки в этом деле были ошибочны. Сланцевая смола рассматривалась только как некий аналог нефти и подвергалась исследованию по аналогии с последней. Поэтому были известны только общие, валовые анализы сланцевой смолы, принципиально устанавливавшие сложность ее состава, но совершенно не вскрывавшие характера этой сложности.
Рациональное решение вопроса о химической переработке смолы было невозможно без значительного расширения имеющихся сведений о природе смолы. Это стало особенно очевидным после того как было установлено, что сланцевая смола
1 *
Предисловие
может служить исходным материалом для получения таких продуктов, которые не могут быть непосредственно получены из нефти, из этого следует, что химическая переработка смолы в методическом отношении должна существенно отличаться от химической переработки нефти.
За последние годы применение более совершенных и разнообразных методов исследования позволило значительно расширить имеющиеся сведения о составе и свойствах сланцевой смолы. Значительно обогатились сведения о групповом составе смолы и ее фракций. Принципиально важным оказалось установление характера большинства кислородных соединений сланцевых смол, которые составляют основную массу смолы (в данном случае речь идет о сравнительно лучше изученной смоле прибалтийских сланцев). Было установлено, что наряду с соединениями фенольного характера значительное место среди кислородных соединений занимают соединения с карбонильной группой. Так были выделены и описаны кетоны, принадлежащие к различным гомологическим рядам. Исследование свойств фенолов, содержащихся в смоле, также показало, что эти фенолы весьма разнообразны по своей природе. Установлено, в частности, что в смоле содержатся не только одноатомные и двухатомные фенолы, но и нафтолы. Найдены также омыляемые вещества и другие кислородные соединения. Кроме кислородных соединений, в смоле найдены углеводороды, принадлежащие к различным классам: ароматические с одним, двумя и тремя циклами, алифатические и др.
Публикуемые в настоящем сборнике работы посвящены, в основном, вопросам химии сланцевой смолы. Они являются продолжением работ, частично опубликованных в ранее выпущенном сборнике трудов института (выпуск I). Вместе с тем ш: следует рассматривать как дальнейший этап систематической исследовательской работы ВНИИПСа по химии сланцевой смолы.
В сборник включены также работы по некоторым методическим и другим вопросам, связанным с нуждами сланцевой промышленности.
В. Ф. Полозов
ОКИСЛЕНИЕ КЕРОГЕНА ПРИБАЛТИЙСКИХ СЛАНЦЕВ
Органическое вещество сланцев прибалтийского бассейна отличается постоянством состава. По нашим данным содержание углерода и водорода в сланцах не изменяется сколько-нибудь значительно даже по отдельным пачкам. Однако, как это было показано Н. Н. Жуковой, большое содержание минеральной части не дает возможности с достаточной точностью определить состав органического вещества и его свойства. Поэтому данные различных авторов о составе органического вещества несколько отличаются друг от друга по содержанию углерода, водорода и органической серы [1].
В последнее время, в связи с выяснившейся возможностью обогащения прибалтийских сланцев методом флотации, задача изучения их органического вещества значительно облегчилась.
Получение концентратов флотационным методом и последующее их обогащение при помощи соляной или фтористо-водородной кислот [2] позволяют выделять кероген прибалтийских сланцев почти в неизмененном виде.
Органическое вещество сланцев характеризуется весьма высокой стойкостью по отношению к реактивам. К прежним сведениям об устойчивости керогена по отношению к щелочи следует добавить, что действие на него кислот носит также весьма ограниченный характер.
Так, кипячение 90 %-кого концентрата с фосфорной кислотой при температуре 166—185° в течение 13,5 час. практически не изменило свойств керогена. Он не растворялся в бензоле и щелочи; его эфирное число даже после такой интенсивной обработки возросло только с 36 до 60, т. е. осталось величиной прежнего порядка.
Действие брома на органическое вещество сланца также выражается очень слабо. Так, бромное число 90 %-кого концентрата составляло всего 22,3% при числе замещения 9,0, и 17-часовая выдержка увеличила количество присоединенного брома только до 31,8% при числе замещения 26,6. Таким образом, реакции замещения получают большее развитие с увеличением продолжительности взаимодействия.
В. Ф. Полозов
Более легко кероген сланца поддается окислению. В этом отношении интересно было проследить поведение органического вещества и изменение его свойств в условиях сушки, т. е. при температурах в пределах 105—170°.
Для опытов был взят концентрат, содержащий 74% органического вещества. Часть этого концентрата была обработана сначала соляной кислотой, а затем, после промывки водой, плавиковой кислотой по методу Н. М. Караваева. В результате был выделен концентрат, содержащий 97% органического вещества. Характеристика его приводится ниже.
Химический состав 97%-ного концентрата керогена, %:
Зола (Ас).................................2,84
Сера общая (80бщ)........................2,0
„ пиритная (Snlipr).....................0,66
, сульфатная (8сульф)...................0,01
„ органическая (Sopr).................. 1,33
С....................'.................75,41
Н...................................... 9,33
Исходя из этих аналитических данных, рассчитываем элементарный состав органического вещества. Вводя в расчет поправку на пирит, находим минеральную часть концентрата АМиН:
Амин = 2,84 + 0,625 X 0,66 = 3,25%.
Отсюда, пренебрегая другими поправками на изменение веса минеральной части, получаем элементарный состав керогена, %:
С.......... 78,00 S ........... 1,40
Н.......... 9,64 O + N.................10,96
Большое количество органического материала в концентратах позволяет довольно точно определить удельный вес керогена из сопоставления двух образцов с примерно одинаковой по составу минеральной частью.
В табл. 1 помещены анализы 74%-ного и 85%-кого концентратов, мало отличающихся по величине отношения Ас:Асо2-
Таблица 1 Характеристика двух концентратов керогена
Наименование концентрата Уд. вес при 20°С Влага W, % Зола А % ’ Минеральная СО2, АСО2’ % Ае+ Асо2,%
74%-ный концентрат . 85%-ный концентрат . 1,33 1,23 1,0 2,0 21,06 12,18 4,94 2,26 26,00 14,44
Окисление керогена прибалтийских сланцев
7
Решая два уравнения с двумя неизвестными, можно рассчитать удельный вес керогена, который равен 1,11.
На рис. 1 показано изменение удельного веса концентратов в зависимости от содержания в них органического вещества.
Окисление концентрата проводилось в термостате, в который были поставлены противни с навеской и контрольными бюксами.
Органическая масса
Рис. 1. Изменение удельного веса концентратов в зависимости от содержания органического вещества.
Навеска концентрата распределялась слоем толщиной ~5 мм. Специальная дверца термостата с отверстиями для термометров, закрывая только среднюю часть камеры, позволяла осуществить конвекционный поток воздуха. Измерение температуры производилось в центре противней внутри слоя концентрата. Через каждые 7 часов нагрева производилось взвешивание противней и контрольных навесок в бюксах. Опыты проводились при температурах 140 и 170°. Продолжительность окисления достигала 44 часов.
После определенного времени окисления материал подвергался исследованию.
Результаты опытов по окислению и режим процесса приводятся в табл. 2 и графически показаны на рис. 2.
8
В. Ф. Полозов
Таблица 2
Опыты по окислению концентрата керогена прибалтийских сланцев
Начальное содержание органического вещества 74%
Показатели Характеристика концентрата
До окисления (опыт 1) После окисления, при температуре
120-110= С в течение 11 час. (опыт 2) 120 150=' С в течение 20 час. (опыт 3) 170° С в течение 20 час. (опыт 4) 170° С в течение 44 час. (опыт 5)
Привес на органическое вещество, % 1,03 2,2 1,9 2,2
Температура битуминизации,°C 390 — 410 не плав.
Элементарный состав концентрата, %: содержание С 57,80 56,80 57,20 55,50 53,30
н 7,13 6,90 6,93 6,31 5,51
отношение С : Н ... 8,13 8,28 8,80 8,80 9,70
Элементарный состав органического вещества, %: содержание С .... 78,2 77,0 77,3 75,0 72,0
. Н .... 9,44 9,33 9,35 8,53 7,45
„ S+O+N . 12,36 13,67 13,35 16,47 20,55
Продукты полукоксования по расчету на концентрат, %: смола 49,1 42,6 37,7 24,6 21,0
полукокс ....... 37,5 37,5 41,6 62,5 55,0
пирогенная вода . . . 5,0 6,0 6,0 10,5 9,0
По расчету на органическое вещество, %: смола . . 66,3 57,5 51,0 33,3 28,4
полукокс 15,1 15,1 21,1 40,3 39,2
пирогенная вода . . . 6,8 8,1 8,1 14,2 12,4
газ+потери 11,8 19,3 19,8 12,2 20,0
зола Ас 21,06 — 20,88 21,13
минеральная СО2 • . . 4,94 — 4,28 4,46
' Как видно из таблицы, при температуре 120—150° окисление идет весьма медленно, и по элементарному составу не наблюдается ни уменьшения содержания водорода, ни изменения отношения С : Н. Однако температура битуминизации повышается с 390 до 410е. Параллельно с этим снижается выход смолы при. полукоксовании за счет увеличения выхода полукокса и газа. Выход пирогенной воды также .заметно возрастает.
Окисление керогена прибалтийских сланцев
9
Представляется вероятным, что в интервале температур 120—150° происходит присоединение кислорода без заметного отщепления Н2О и СО2. Только при термическом разложе-
уводя с собой часть водорода, необходимого для образования смолы.
Интересно отметить, что при окислении получается весьма незначительный привес, составляющий ~ 2% на органическое вещество сланца.
При температуре 170° наблюдается более глубокое изменение свойств окисленного продукта. Цвет его становится бурым; окисленный концентрат совершенно не плавится. Выход смолы, рассчитанный на органическое вещество, за 20 часов уменьшился в два раза — с 66,3 до 33,3%. Увеличение продолжительности опыта до 44 часов привело к еще более низкому выходу смолы, равному всего 28,4%.
Это является, очевидно, следствием изменения элементарного состава керогена, а именно, понижения содержания углерода и водорода и увеличения содержания кислорода. Очевидно, что повышение температуры на 20° приводит уже к отщеплению воды и углекислоты.
Сравнивая элементарный состав керогена до и после окисления, можно представить процесс окисления в виде химических Уравнений.
о
В. Ф: Полозов
Таблица 3
Изменение элементарного состава керогена в процессе окисления1
Кероген Содержание, % 1 Изменение со- ! держания, %
С И O4-S С Н 1. 0
Исходный кероген до окисления . Кероген после окисления при 170° С в течение 20 часов (опыт 4) . . 78,0 75,0 9,5 8,5 12,5 16,5 —3,0 —1,0 4-4,0
Кероген после окисления при 170° С в течение 44 часов (опыт 5) . . . 72,0 7,5 20,5 -6,0 —2,0 4-8,0
Из табл. 3 видно, что удаление углерода и водорода компенсируется присоединением кислорода. Приводя изменение содержания элементов при окислении к весу атомов, легко заметить, что отщепление одного атома углерода и четырех атомов водорода связано с присоединением одного атома кислорода.
Исходя из этого и применяя условную формулу керогена СгзНззОз, получим уравнения:
1) С2вН38О34- 2,5О2 —>2Н2О4- СО2 4- С25Н34О4
2) С2вН38О3 4-5О2 ->4Н2О + 2СО2 + С21Н30О5
В этих уравнениях принято, что водород и углерод отщепляются в виде воды и углекислоты.
На основании сопоставления приведенных уравнений можно утверждать, что процесс окисления при температуре 170° в продолжение 44 часов происходил по одному направлению. Характерными признаками процесса являются отщепление кислорода в виде окислов и присоединение его в отношении 4:1. Кроме того, во время окисления выделяются вода и углекислота в молекулярном отношении 2:1.
Таким образом, происходит удаление группы СН2 и присоединение атома кислорода, вероятно, с образованием карбонильной группы.
При рассмотрении балансов приведенных выше реакций окисления легко заметить, что в результате отщепления от керогена четырех атомов водорода и присоединения одного атома кислорода не должно происходить изменения веса окисляемого вещества, поскольку одновременно отщепляется один атом углерода. Однако опыт показывает небольшое увеличение веса (2%), ко-
1 При окислении вес керогена почти не изменяется.
Окисление керогена прибалтийских сланцев
11
торое остается постоянным в течение всего времени окисления. Очевидно, что предложенная химическая схема процесса окисления керогена не учитывает образования промежуточных продуктов окисления, которые находятся в состоянии динамического равновесия, т. е. все время образуются и разрушаются, не накапливаясь в количестве более 2% от веса органического вещества сланца.
Принимая во внимание, что этими промежуточными продуктами окисления могут быть перекиси и карбонильные производные, можно предложить более детальную схему окисления. Эта схема учитывает наличие в керогене прибалтийских сланцев большого количества групп СН2.
Следует отметить, что скорость реакцйи определяется наиболее медленной реакцией образования гидроперекисей, которая при большом избытке воздуха и большом количестве групп СН2, реагирующих последовательно, не зависит до определенного предела от глубины реакции.
Действительно, если активация СН2-групп происходит лишь по соседству с карбонильной группой, то количество образующихся гидроперекисей будет все время одно и то же, независимо от числа СО-групп, находящихся в промежутке между группами СН2. Этим объясняется, вероятно, и нулевой кинетический порядок реакции.
Изменение свойств при окислении концентрата в условиях сушки наблюдалось также при измерении сопротивления току газа в момент расплавления органического вещества по методу Фоксвелла [3].
После калибровки прибора на коксующемся угле (рис. 3) на нем были испытаны исходный 74%-ный концентрат и продукты опытов 2 и 3.
При испытании прежде всего наблюдается резкое различие кривых давления для угля и концентрата (см. рис. 3 и 4).
Кривая давления в опыте с концентратом является более пологой и не имеет резкого максимума. Вероятно, разложение органического вещества сланца начинается задолго до наступления точки плавления.
После окисления концентрата в течение 20 часов на рис. 5, 6 нельзя заметить максимума давления, соответствующего точке плавления. Однако при плавлении в пробирке явление битуминизации наблюдалось при температуре 410°.
Очевидно, поток газа уносит смоляные пары и не допускает растворения твердых продуктов термического разложения в смоле.
Поэтому более правильным оказался метод определения температуры битуминизации путем нагревания вещества в стеклянной пробирке с мешалкой.
Рис. 3. Опыт 24. Калибровочное испытание на плавкость, по Фоксвеллу, коксующегося угля коксогазового завода.
Рис. 4. Опыт 12. Испытание на плавкость, по Фоксвеллу, исходного 74%-ного концентрата керогена.
Окисление керогена прибалтийских сланцев
13
Рис. 5. Опыт 20. Испытание на плавкость, по Фоксвеллу, продукта окисления керогена, полученного в опыте 2
(температура 120—150° С, время И часов).
Рис. 6. Опыт 21. Испытание на плавкость, по Фоксвеллу, продукта окисления керогена, полученного в опыте 3 (температура 120—150° С, время 20 часов).
14
В. Ф. Полозов
Сопоставляя кривые плавкости коксующегося угля и обогащенных сланцев, можно прийти к выводу, что органическое вещество прибалтийских сланцев, даже после окисления, не обнаруживает при термическом разложении характерных для коксующихся углей свойств.
ЛИТЕРАТУРА
1. А. Ф. Добрянский. Горючие сланцы. Ленгостоптехиздат, 1947.
2. Г. А. 3 и к е е в и А. Н. Корелин. Анализ энергетического топлива. Госэнергоиздат, 1948.
3. Г. Л. Стадников. Химия коксовых углей. Госхимтехиздат, 1934.
4. ГОСТ 2962—45.
Н. И. Зеленин и О. С. Куратова
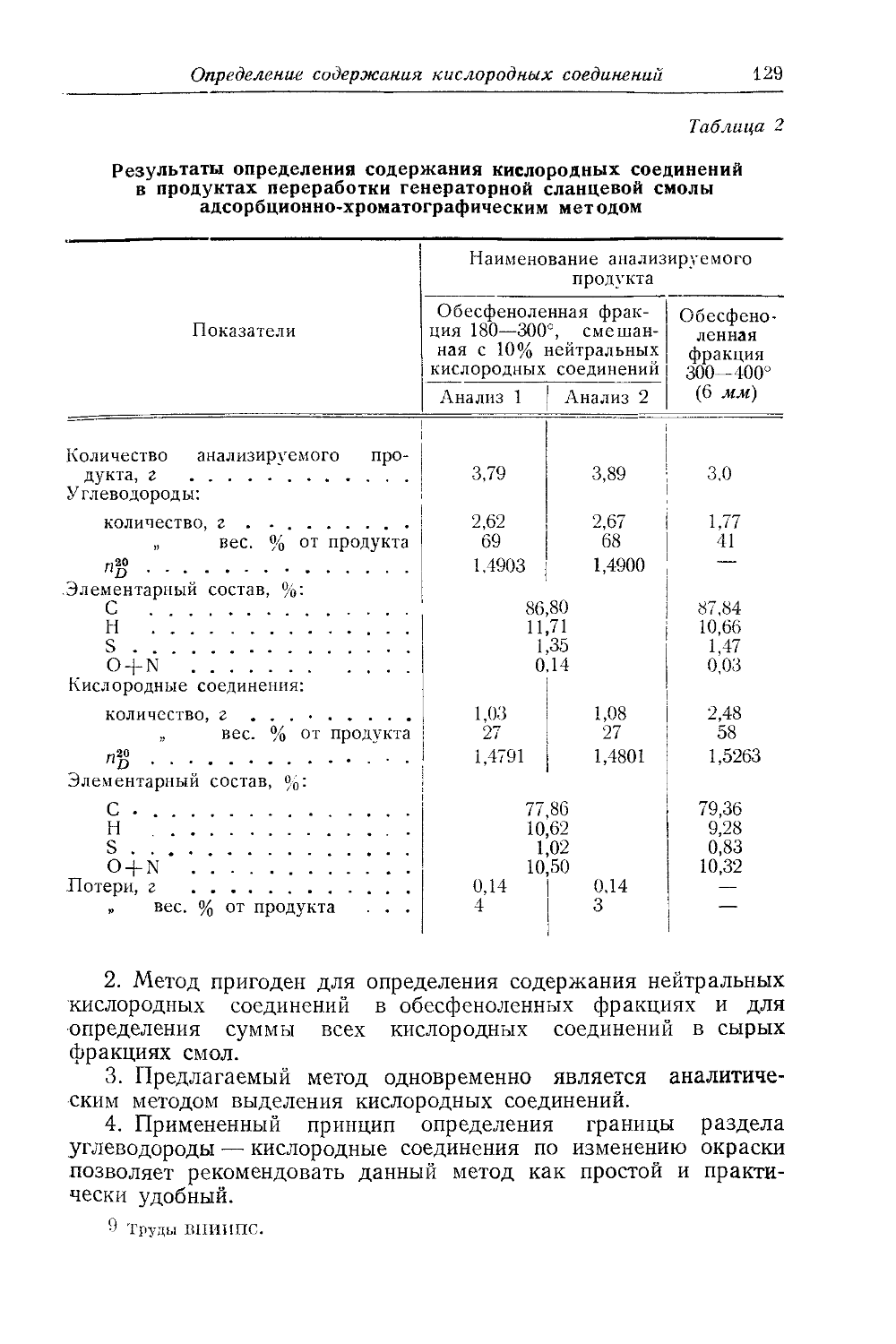
О СОСТАВЕ И СВОЙСТВАХ СЛАНЦЕВОЙ СМОЛЫ КИСЛОРОДНЫЕ СОЕДИНЕНИЯ
Основной особенностью смол прибалтийских сланцев является преобладание в них кислородных соединений.
По фракционному составу кислородные соединения распределяются следующим образом: фракция до 200° содержит 8—10% нейтральных и до 2% кислых кислородных соединений; фракция до 200—325° содержит до 30—35% нейтральных и до 25% кислых кислородных соединений; фракция выше 325° содержит около 50% нейтральных и до 20% кислых кислородных соединений.
В неразогнанной смоле содержание кислородных соединений составляет до 2/з всей массы смолы.
Кислородные соединения следует разделить на три группы: 1) кислоты, 2) фенолы и 3) нейтральные кислородные соединения.
Если первые две группы имеют более или менее определенную природу и основные их представители выделены и известны, то характер нейтральных кислородных соединений до самого последнего времени был совершенно неизвестен. Только теперь мы можем говорить более или менее определенно об их природе.
Кислоты. О наличии кислот в сланцевой смоле известно давно. П. К- Когерман [1] установил наличие жирных кислот, насыщенных и ненасыщенных, содержащих 5 и более атомов углерода, выделил и охарактеризовал некоторые из них. Ю. Ю. Хюссе [2] нашел карбоновые кислоты в высших фракциях сланцевой смолы, но не установил их природу. Содержание их в различных фракциях колебалось от 1,5 до 5,0%. Особенностью работы Ю. Ю. Хюссе было то, что исследование проводилось с вакуумными фракциями.
Мы установили наличие уксусной и масляных кислот в сланцевой смоле, а Б. И. Иванов и Н. Ф. Шаронова нашли их в подсмольной воде генераторной и туннельной смол. К. Вальдек [3], продолжая работу П. К- Когермана, доказал наличие в смоле, правда в очень небольших количествах, жирных кислот от С5
Iti
И. И. Зеленин и О. С. Куратова
до Си нормального строения. Следовательно, карбоновые кислоты содержатся во всех, без исключения, фракциях сланцевой смолы, и часть их безусловно принадлежит к жирному ряду и имеет нормальное строение. Точных систематизированных данных о количественном содержании отдельных кислот в смоле пока еще нет. Общее содержание уксусной, масляной, капроновой и других найденных и идентифицированных кислот значительно меньше суммы кислот, выделяемых бикарбонатом натрия.
Общее содержание кислот составляет до 2% от смолы. Установить, какую часть из этого количества составляют выделенные и изученные кислоты, пока нельзя из-за различия аналитических материалов, применявшихся различными авторами, и вследствие больших потерь при определении отдельных индивидуумов. Во всяком случае то, что нам известно, не покрывает, повидимому, и четверти суммарных «карбоновых кислот».
В. А. Ланин пытался выяснить природу кислых веществ сланцевой смолы посредством фракционной сорбции силикагелем. Он выделил несколько групп таких веществ и установил в них наличие карбоксильных, гидроксильных и карбонильных групп. Выделенные вещества высокомолекулярны (молекулярный вес около 500) и непредельны, легко уплотняются и изменяются. В. А. Ланин полагает, что в каждой молекуле таких веществ содержится не менее 3—4 ароматических или нафтеновых колец. Пока нельзя причислить эти вещества к какому-либо определенному классу соединений. В. А. Ланин называет их или асфальтенами, или «силикагелевыми» смолами. Во всяком случае анализы этих веществ показывают, что в них содержится кислород с практически всеми присущими ему функциями. Кроме собственно кислот, удалось установить наличие большого количества оксикислот. Соответствующие анализы показывают, что оксикислоты составляют, повидимому, главную массу веществ, содержащих карбоксил. Однако еще не выделено и не изучено ни одного индивидуального соединения из этого класса веществ.
Фенолы. Фенолы составляют значительную часть смолы. Общее содержание их в последней составляет 15—19%. Следует, однако, оговориться, что под термином «фенолы» в данном случае подразумевается щелочная вытяжка или из самой смолы, или из ее фракции. Собственно фенолов, т. е. гидроксильных производных ароматических углеводородов, в этой вытяжке немного. Основную массу составляют кислые вещества невыясненного строения и природы, повидимому, вещества того типа, с которыми имел дело В. А. Ланин.
Следует учесть, что метод выделения и определения фенолов несовершенен и носит явно условный характер. Так, концентрация щелочи оказывается небезразличной для выхода фенольной вытяжки: чем крепче щелочь, тем большее количество кислых
О составе и свойствах сланцевой смолы
17
компонентов увлекается в раствор. Особенно большое значение концентрация щелочи имеет при обработке высококипящих фракций. В этом случае при переходе от 5 %-ной к 20 %-ной концентрации щелочи количество фенольной вытяжки удваивается. Кроме того, раствор фенолятов и других кислых соединений оказывается растворителем некоторых компонентов смолы, в первую очередь нейтральных кислородных соединений. Нами были проанализированы фенолы, выделенные из средней фракции генераторной смолы при обработке последней 10%-ным раствором щелочи. Данные анализа (табл. 1) показывают, что среди фено-
Таблица 7
Характеристика фенолов, выделенных из средней фракции генераторной смолы 10%-ным раствором щелочи
Показатели Сырые фенолы Нейтральные масла из фенолов Фенолы
Удельный вес при 20° С . . . . Молекулярный вес Рефракция . ^Содержание группы ОН,% . . н 0,9949 ! 155 1,5248 7,65 0,9286 192 1,4990 0,95 1,02 148 1,5415 11,04
"‘Нейтральные НИЯ, % Нейтральные Фенолы, % ,, -CZ=O,% кислородные соед масла, % . . . . те- ।" ’у иядаь? %: Д' Ч- < •> 5,60 94,27
лов присутствует до 25% нейтральных масел, в основном соединений с карбонильной группой. Понятно, что в этом случае решающую роль играет концентрация полученных фенолятов и других растворенных веществ. С. Раудсепп, подробно изучивший методы выделения фенолов из смолы водными растворами щелочей, утверждает, что часть фенолов вообще неспособна растворяться в водных растворах щелочей, несмотря на наличие гидроксила, способного реагировать со щелочью. Эта особенность, по мнению Раудсеппа, зависит от способности алкилированных фенолов растворяться больше в смоле, чем в водном растворе щелочи. Ввиду большой склонности соответствующих фенолятов к гидролизу, появляется возможность почти полного сохранения алкилированных фенолов в исходной смоле или ее фракциях. Автор приходит к выводу, что водные растворы щелочей далеко не полностью увлекают фенолы из смолы, и данные, полученные этими методами анализа, совершенно не достоверны. Во всяком случае возможно, что алкилированных фенолов в смоле значи-
2 Груды ВНИИПС.
18
И. И. Зеленин и О. С. Куратова
тельно больше, чем это следует по известным в настоящее время анализам.
Значительную роль в процессе выделения фенолов из смолы или ее фракций играет не только физическое растворение каких-либо компонентов в фенолятах или смоле, но и химическое воздействие щелочи на кислые и нейтральные соединения смолы. Как мы установили, растворы кислых веществ в щелочи оказываются чрезвычайно активными к различным превращениям. Так, эти растворы легко поглощают кислород, превращаясь частью в смолы. Растворы щелочи сами по себе оказываются сильно действующим средством для полимеризации нейтральных кислородных соединений, вызывающим образование трудно растворимых смол. Кроме того, растворы щелочи, в зависимости от концентрации и времени обработки смолы, вызывают омыление эфирообразных веществ смолы, что, в свою очередь, ведет к совершенно искаженным результатам, поскольку нет возможности обеспечить, во всех случаях, абсолютно тождественные условия обработки растворами щелочи.
Наконец, по данным Б. И. Иванова, последующая нейтрализация щелочи и отгонка растворителя от выделенных фенолов связаны с активной конденсацией нейтральных кислородных соединений и фенолов по типу образования бакелитовых смол. Характерно, что неразогнанная смола, ее фракции и особенно фенольные вытяжки легко реагируют с формальдегидом в присутствии минеральных кислот, образуя при этом лакообразные вещества. Так называемый «эстолак» до сих пор получается именно этим способом и находит себе применение в строительном деле. Мы наблюдали образование подобного «эстолака» и без всякого добавления формальдегида, при простой обработке концентратов кислородных соединений в присутствии концентрированных щелочей. Это является доказательством того, что фенолы сланцевой смолы в определенных условиях конденсируются с другими кислородными соединениями, содержащимися в этой смоле. Следовательно, процесс выделения «фенолов» является сложной химической операцией, где образование фенолятов и солей карбоновых кислот является только частью процесса, а при наличии больших масс нейтральных кислородных соединений — даже неглавной частью общего процесса. С этой точки зрения трудно говорить о какой-либо идентичности выделенных кислых веществ с теми, которые первоначально находились в смоле.
По данным П. К. Когермана и Н. Вайдерпасса [5], фенолы распределяются по фракциям генераторной смолы следующим образом:
200—250°............... 8,2%
250—300°................12,4%
300—350°................21,6%
350-370“................22,8%
О составе и свойствах сланцевой смолы
19
Анализы технических фракций туннельной смолы комбината Кивиыли, выполненные Б. В. Валандером в 1951 г., дают следующее распределение фенолов:
в тяжелом масле..................25,4%
в среднем масле..................27,7%
в легком масле ...................9,4%
в печном бензине ................ 1,5%
Эти данные неоднократно получали подтверждение в исследованиях целого ряда других авторов, в которых учитывались возможные колебания в результатах анализа, обусловленные изложенными выше причинами.
Природа фенолов оставалась до последнего времени почти совсем не освещенной. По старым работам П. К. Когермана [1, 5], А. С. Броуна [6], Д. И. Андреевского [7] и других было установлено наличие только простейших представителей одноатомных фенолов. Были идентифицированы: карболовая кислота; орто-, мета- и паракрезолы; 1, 2, 4, -1, 3, 4, -1, 4, 2- ксиленолы и параэтилфенол.
За последние годы имеющиеся сведения о природе фенолов значительно расширились. Б. И. Иванов с сотрудниками детально изучили состав фенолов подсмольной воды и показали наличие в ней значительного количества двухатомных фенолов и карболовой кислоты, основная масса которых легко вымывалась из смолы водой, чем и был затруднен их анализ в смоле. Двухатомные фенолы оказались исключительно представителями гомологов резорцина (идентифицирован 4,6-диметилрезорцин). Пирокатехина и его гомологов не было найдено. Последующими работами по изучению состава фенолов смолы было установлено, что, наряду с представителями фенолов, в их составе находятся также и нафтолы. Трехатомные фенолы пока не обнаружены.
С. Раудсепп применил для выделения фенолов высокоэффективную ректификационную колонну, на которой были разогнаны выделенные из смолы суммарные фенолы. Ему удалось выделить ректификацией фенол, орто- мета- и паракрезолы. Исследование отдельных узких фракций дало возможность установить наличие в смоле туннельных печей следующих фенолов: фенола; орто-, мета- и паракрезолов; 2,3 -2,5, -3,4- и 3,5-диметилфенолов; 2,6-диметилгидрохинона; а- и ^-нафтолов. Интересно, что Раудсепп не обнаружил производных резорцина. Этот факт, расходящийся с данными Б. И. Иванова, требует объяснения.
Аналитические данные по содержанию различных фенолов в сланцевой смоле еще недостаточно обработаны. Однако не подлежит сомнению, что выделенные и изученные представители фенолов не составляют основной их массы, содержащейся в смоле, 2*
20
И. И. Зеленин и О. С. Куратова
особенно в высоко кипящих фракциях. Этот вопрос еще ждет своих исследователей.
Нейтральные кислородные соединения. Факту наличия в сланцевой смоле нейтральных кислородных соединений до самого последнего времени не придавалось никакого значения. Это обстоятельство не поддается объяснению. В самом деле, нейтральные кислородные соединения представляют собой количественно преобладающую и самую активную часть сланцевой смолы. Можно без преувеличения сказать, что они определяют главные особенности смолы, и их свойства ближе всего объясняют свойства сланцевой смолы в целом. В течение 20 лет исследователи проходили мимо этих веществ, отмечая иногда то или иное их влияние на применяемые методики анализа или способы обработки смолы. Это тем более удивительно, что содержание, повидимому, больших количеств таких веществ в сланцевой смоле было всем хорошо известно.
До войны была известна только одна работа, в которой исследователь попытался проникнуть в природу нейтральных кислородных соединений. Ю. Ю. Хюссе [21 подверг детальному обследованию нейтральные вакуумные фракции и установил в них наличие большого количества нейтральных кислородных соединений самых различных функций. В табл. 2 приведены данные Ю. Ю. Хюссе для 6 фракций, полученных при остаточном давлении 8 мм. Оставляя в стороне вопрос о правильности выбранных Хюссе методов определения отдельных функций кислорода, можно установить, что основную массу вакуумных фракций составляют соединения с гидроксильной и карбонильной группами,
Таблица 2
Анализ вакуумных фракций сланцевой кмолы, полученных при остаточном давлении 8 мм
Температура кипения, °C Молекулярный вес Кислоты, % Фенолы, % Нейтральные, % Эфиры в нейтральных, % Содержание в нейтральных фракциях соединений, %
гидроксильных карбонильных неизвестных
150-175 193 2,12 19,41 77,91 24.5 30,1 44,4 1,0
175—200 229 5,16 26,52 67,11 17,8 34,8 24,0 23,4
200—225 262 1,75 31,87 65,82 15,3 43,6 18,1 23,0
225—250 270 1,46 35,18 62,98 7,4 29,6 18,0 45,0
250—275 322 1,44 22,72 75,38 7,4 26,3 5,4 60,9
275—300 348 1,41 18,94 79,06 12,2 36,0 3,9 47,9
О составе и свойствах сланцевой смолы
21
а также кислородные соединения неизвестных функций. Если учесть, что вакуумных фракций можно извлечь из сланцевой смолы до 85% от ее массы, станет ясно, какую решающую роль играют нейтральные кислородные соединения в сланцевой смоле. Работа Ю. Ю. Хюссе прошла незамеченной и после нее фактически не было на эту тему каких-либо других публикаций. Ю. Ю. Хюссе вновь вернулся к своей работе в 1950 г. и опубликовал отдельную брошюру [8], в которой целиком перепечатал свою старую статью. Кроме того, он дополнил работу и новым компилятивным материалом.
В последние годы положение изменилось. Работами коллектива ВНИИПС было показано, что нейтральные кислородные соединения являются веществами, определяющими успех или неуспех всех способов переработки сланцевой смолы на моторное топливо и другие продукты. Углеводородный материал сланцевой смолы, представляющий собой основной потенциал сланцевого жидкого топлива, как бы окружен «кислородным барьером», который ограничивает возможность использования и переработки сланцевой смолы на моторное топливо. Процессы крекинга, ароматизации, обработки хлористым алюминием, серной кислотой и прочие оказались в случае сланцевой смолы совершенно бесперспективными именно из-за наличия в ней нейтральных кислородных соединений.
В настоящее время развернуты большие работы по установлению природы нейтральных кислородных соединений. Результаты этих работ еще далеки от завершения и окончательных выводов, но уже сейчас имеется достаточно данных, чтобы решить ряд принципиальных вопросов, касающихся основной массы нейтральных кислородных соединений. В число твердо установленных групп нейтральных кислородных соединений входят альдегиды, кетоны и эфиры.
В 1946 г. при объяснении механизма полимеризации кислородных соединений мы высказали предположение о наличии альдегидов в сланцевой смоле [9]. Детальные исследования подтвердили это предположение. Б. И. Иванов и Н. Ф. Шаронова нашли в подсмольной воде ацетальдегид, а затем и пропионовый альдегид. С. С. Семенов, исследуя нейтральные кислородные соединения фракции 200—325°, нашел по методу Пондорфа до 20% альдегидов в суммарной фракции нейтральных кислородных соединений и различные количества альдегидов во всех одноградусных фракциях, на которые была разделена исходная фракция.
Кетоны в настоящее время являются наиболее изученными представителями нейтральных кислородных соединений. Уже давно известен факт наличия ацетона и метилэтилкетона в подсмольной воде.
22
Н. И. Зеленин и О. С. Куратова
Исследование кислородных соединений средних фракций позволило С. С. Семенову не только определить содержание кетонов во фракции от 8 до 15%, но и выделить и идентифицировать следующие кетоны: ацетон, метилэтилкетон, метилбутилке-тон, метиламилкетон, метилгексилкетон, метилгептилкетон, ме-тилоктилкетон и метилнонилкетон. С. С. Семенов нашел также ароматический кетон метилгидриндон. В. А. Ланин выделил сорбцией некоторые карбонильные соединения и охарактеризовал их. Так, им был выделен кетон Ci0Hi6O, которому он приписывает строение 1-метил-4-изопропилциклогексанона. Выделены также кетоны СцН16О и С3Н22О, у которых также предполагается циклическое строение.
Природа эфиров, содержащихся в смоле, еще не установлена. Если присутствие жирных кислот в смоле является вполне доказанным, то, несмотря на длительные поиски, в ней до сих пор не удалось найти ни одного представителя спиртов жирного ряда. Присутствие в смоле эфиров фенолов и жирных кислот также не получило пока прямого подтверждения. Наличие этих эфиров может быть доказано косвенным путем: в случае их присутствия после омыления фракций, из которых предварительно были удалены фенолы, анализ должен снова обнаружить некоторое количество фенолов.
Содержание омыляемых соединений по фракциям смолы показано в табл. 3.
Таблица 3
Содержание омыляемых соединений в вакуумных и соответствующих им атмосферных фракциях генераторной и туннельной смол (в вес. %)
I до 100° II 100-150° III 150—200° IV 200—250°
Вакуумные фракции генераторной смолы . . 3,8 4,2 12,3 17,9
Вакуумные фракции туннельной смолы . . . 0,03 4,1 11,6 13,6
Атмосферные фракции генераторной смолы . 1,1 2,8 6,0 7,5
Атмосферные фракции туннельной смолы . . — 0,3 3,8 7,9
В. А. Ланин омылил отдельные фракции и выделил кислоты с молекулярным весом около 1000. Кислоты быстро твердели; на основании этого было сделано предположение, что они отличаются высокой непредельностью.
О составе и свойствах сланцевой смолы
23
Исследование природы кислородных соединений сланцевой смолы только начато. В ближайшем будущем мы рассчитываем установить наличие в этой смоле многочисленных и разнообразных групп соединений — оксикислот, альдолей, гликолей, возможно, кето-и альдоспиртов и многих других, — пока не обнаруженных, но присутствие которых можно предполагать.
УГЛЕВОДОРОДЫ
Углеводороды составляют главную массу легких фракций — до 90%; в средних фракциях 200—300° и 225—325° содержание их составляет около 45—50%. В высших фракциях, составляющих главную массу смолы, углеводороды пока не найдены. Может быть, они там и присутствуют, но все применяемые в настоящее время методы выделения и разделения нейтральных веществ высококипящих фракций сланцевой смолы не дают возможности получить материал, не содержащий кислорода. Поэтому пока не представляется возможным получить какие-либо сведения о наличии углеводородов в высших фракциях смолы и, тем более, об их количестве и природе.
Углеводородные компоненты легких фракций смолы изучены лучше всех других. Это понятно, так как легкие фракции смолы обычно рассматриваются как сырье для получения бензина и под этим углом зрения были детально исследованы. Углеводородная часть легких фракций сланцевой смолы по своему составу примерно соответствует углеводородам крекинг-бензинов с той лишь разницей, что в сланцевых легких фракциях преобладают, более чем в каких-либо других крекинг-бензинах, олефиновые углеводороды. Наиболее достоверные данные по групповому составу фракций, кипящих до 150° С, приводятся в работах Р. Цейд-лера [10] и Н. В. Ершова [4] (табл. 4).
Таблица 4
Состав углеводородов легких фракций (до 150°С) туннельной смолы, %
Группы углеводородов Эстонская смола (Цейдлер) Гдовская смола (Ершов)
Непредельные углеводороды .... 56,8 62,0
Ароматические „ .... 9,5 11,4
Парафиновые „ . 29,2 18,6
Нафтеновые « .... 4,5 8,0
По нашим и А. П. Сиверцева данным, углеводородная часть фракций туннельной смолы имеет состав, показанный в табл. 5.
24
Н. И. Зеленин и О. С. Куратова
Таблица 5
Состав углеводородов туннельной смолы
Группы углеводородов
С концом кипения । 225° С, % |
С концом кипения 200° С, %
Всего углеводородов во фракции Непредельные углеводороды Ароматические „ । . . .
Нафтеновые „ >
Парафиновые „I • •
90,0 (100)
63,7
12,5
23,8
87,0 (100)
60,0
17,2
16,0 / 22,8
А. П. Сиверцев установил следующий состав углеводородной части средних фракций генераторной смолы с концом кипения 320° С (табл. 6).
Таблица 6
Состав углеводородной части генераторной смолы в границах кипения 158—320° С
Группы углеводородов
Олефины ...................•...............
Ароматические углеводороды........... . . .
Парафиновые „
Нафтеновые „ ................
43,6
39,0
10,4
7,0
Наряду с количественными определениями группового состава сланцевой смолы имеются данные о присутствии в ней целого ряда индивидуальных представителей углеводородов и их строении. Эта работа проведена в основном А. П. Сиверцевым. В настоящее время из сланцевой смолы выделены и идентифицированы следующие углеводороды: алканы — н- и изобутаны, н-пентан, н-гексан, н-гептан, н-октан; алкены-—гексен-1, гептен-1, октен-1, нонен-1 и, как их спутники, в небольших количествах — гексен-2, гексен-3, гептен-2, октеп-3, октен-4; ароматические углеводороды — бензол, толуол, орто-, мета- и параксилолы; 1, 2, 3-триметилбензол; 1, 2, 3, 4-тетраметилбензол, нафталин и а-метилнафталин, а также антрацен и его производные. Представители нафтенов пока не найдены и не выделены. В. А. Ланин предполагает, что непредельные нафтены составляют главную массу углеводородов средних фракций смолы.
Следует обратить внимание на строение углеводородов. Наиболее легко выделяются углеводороды нормального строения,
О составе и свойствах сланцевой смо^ы
25
что объясняется прежде всего относительно большим количеством их в смоле, позволяющим выделять их фракционировкой. Олефины в основном имеют двойную связь в a-положении и нормальное строение. Указанные выше группы углеводородов — парафины нормального строения и олефины также нормального строения с двойной связью в а-положении — составляют основную массу углеводородов сланцевой смолы.
В табл. 7 и 8 приведены последние данные о групповом составе сланцевой смолы по фракциям.
Таблица 7
Состав легких фракций туннельной смолы, %
Группы соединений Туннельная смола
до 200° до 225°
Непредельные углеводороды . . . 52,0 56,2
Ароматические „ ... 15,0 11,6
Парафиновые » ... 14,0 | 20,5
Нафтеновые „ ... 6,0
Кислородные нейтральные соедине- НИЯ ... ........... 8,0 9,7
Кислородные кислые соединения (фенолы) 2,0 2,0
Сернистые соединения 3,0 —
Таблица 8
Состав средних фракций сланцевой смолы, %
Группы соединений Генераторная 160— 320° Туннельная 200-325°
Кислородные нейтральные соединения 26,2 25,0
Кислородные кислые соединения (фенолы) • 14,0
Сернистые соединения 7,0 —
Непредельные углеводороды .... 28,3 26,0
Ароматические „ .... 26,7 24,0
Парафиновые „ .... 7,0 | и,о
Нафтеновые , .... 4,8
Для высших фракций никаких сведений нет, кроме анализов, приведенных в табл. 2 и 3, и тех, которые разбираются далее (табл. 11 и 12). Установлено содержание фенолов в коли
26
II. И. Зеленин и О. С. Куратова
честве 20% и нейтральных кислородных соединений — около 50%, что в сумме составляет 70%. Состав остальных 30% неизвестен. Вероятно это — асфальтообразные вещества, продукты глубокого уплотнения кислородных соединений.
Производя соответствующий расчет, можно выразить состав сланцевой смолы (содержание в ней основных составляющих) следующими ориентировочными цифрами, %:
Туннельная смола
Углеводородов в неразогнанной смоле............................. около 30
Кислородных соединений кислых и нейтральных ........................ до 55
Сернистых соединений ............... до 5
Генераторная смола
около 20
до 60 до 5
Азотистые соединения в случае смолы прибалтийских сланцев можно исключить из рассмотрения, поскольку количество их ничтожно.
Из приведенных данных вовсе не следует, что уже известна природа соединений, принадлежащих к установленным выше группам. Более того, в смолах содержится 10—15% веществ, которые вообще еще никак нельзя классифицировать.
Таким образом, следует подчеркнуть, что в области изучения природы сланцевой смолы сделаны лишь первые шаги. Можно не сомневаться в том, что в результате дальнейшей систематической исследовательской работы в этом направлении откроются практически неограниченные возможности использования сланцевой смолы в качестве химического и энергетического сырья.
ОСНОВНЫЕ ТЕРМИЧЕСКИЕ ПРЕВРАЩЕНИЯ СМОЛЫ
Выше кратко рассмотрены современные данные о составе смолы прибалтийских сланцев. Из этих данных следует, что часть составляющих смолы принадлежит к химически активным веществам, это должно существенным образом отразиться на поведении смолы в условиях переработки и, прежде всего, при термическом воздействии.
К- Лутс [11] первый обратил внимание на то, что обыкновенная перегонка смолы представляет собой не простой процесс дестилляции, а более глубокий процесс — крекинг высокомолекулярных веществ, сопровождающийся выделением газов и изменением плотности фракций дестиллата, получающегося при атмосферной перегонке, по сравнению с фракциями вакуумной разгонки. Лутс не установил глубины происходящих превращений и не оценил большой важности сделанного им наблюдения, хотя оно, в сущности, в очень большой степени определяет выбор технологии переработки смолы и ставит ряд очень серьез-
О составе и свойствах сланцевой смола
27
них теоретических вопросов, касающихся образования отдельных компонентов смолы и их дальнейших превращений.
В самом деле, является фактом то, что сланцевая смола, являющаяся продуктом термического разложения сланца, была получена и находилась в парообразном состоянии при температурах 350—450°, затем была охлаждена и перешла в жидкое состояние в соответствующей аппаратуре, т. е. смола прошла весь путь термических превращений, какие были для нее возможны при температурах, превышающих температуру обычной перегонки. Тем не менее при обычной перегонке она не только вновь может испариться, но, наоборот, подвергается глубокому разложению с образованием уже неперегоняющегося остатка. Следовательно, в смоле произошли большие изменения.
Исследователи процесса полукоксования давно обратили на это внимание. Были высказаны соображения о туманообразном выносе тяжелых, асфальтообразных масс из зоны полукоксования и вторичном образовании тяжелых частей смолы во время крекинга в паровой фазе. Я- И. Хисин [12] видел объяснение образования тяжелых, высокомолекулярных масс в низкой температуре газосливных устройств полукоксовых аппаратов, что, по его мнению, ведет к конденсации тяжелых частей смолы и их последующему распаду с образованием тяжелых и легких продуктов и т. д. Этому процессу Я- И. Хисин дал наименование «реконденсация — редестиллация». Справедливость выводов Я. И. Хисина не вызывает сомнений, но эти выводы не исчерпывают вопроса и должны быть дополнены. Рассмотрим этот вопрос с точки зрения химического состава смолы на основе имеющегося опытного материала.
При перегонке смолы по мере подъема температуры начинается выделение газа, которое равномерно продолжается во все время разгонки; одновременно в дестиллате появляются, наряду с фракциями, соответствующими температуре гонки, фракции, кипящие значительно ниже. Это говорит об образовании легких дестиллатов, т. е. о том, что, наряду с образованием газа, происходит и распад перегоняемого сырья с образованием более сложных молекул.
В табл. 9 приведены сравнительные балансы вакуумной и атмосферной р азгонок смолы и состав газов, полученных при атмосферной разгонке. Перегонка велась до конца выделения дестиллатов и получения в остатке малоподвижного пека.
Наибольший интерес в данных этой таблицы представляют образование воды и состав газа. Последний соответствует примерно составу газа, который получается при полукоксовании. Особенно интересным является наличие в газе значительных количеств метана и водорода, которые, вообще говоря, обра-
28
Н. И. Зеленин и О. С. Куратова
Таблица 9'
Сравнительные балансы вакуумной и атмосферной разгонов генераторной и туннельной смол
Разогнано 1000 г смолы. Разгонке подвергнута смола, лишенная легких фракций (до 200°)
Контролируемая величина Вакуумная раз-гонка, ост. давл. 6 мм Атмосферная разгонка
генераторная туннельная генераторная туннельная
Максимальная температура паров
смолы,°C 251 240 359 345
Выход дестиллата, вес. % 64,5 63,1 46,1 54,5
Остаток в кубе, вес. % 35,5 34,2 47,1 39,3
Вода, вес. % нет нет 1,0 0,9
Газ, вес. % 3,0 2,2
Содержание в газе, %:
СО2 9,9 5,2
СО 12,1 8,3
спнт 20,3 22,0
н2 15,2 19,4
сн4 42,5 45,1
Потери, вес. % 3,3 2,7 2,8 3,1
зуются в процессах пирогенетического разложения при значительно более высоких температурах, чем та, которая может быть достигнута при обычной перегонке. Следовательно, образование указанных выше компонентов газа должно быть обусловлено какими-то реакциями, в принципе отличными от тех, которые отвечают нашим обычным представлениям о термическом крекинге с его простыми термодинамическими, обусловленными соответствующими равновесиями, возможностями образования тех или иных газовых компонентов. Из таблицы видно также, что атмосферная разгонка дает значительно меньше дестиллата,. чем вакуумная, т. е. в процессе атмосферной перегонки часть потенциального дестиллата распадается на газ и кокс.
Табл. 10 дает более обстоятельное представление об изменениях, происходящих в смоле во время перегонки. В ней приведены данные анализа газов и фракционный состав жидких де-стиллатов, которые отбирались параллельно с отбором газа, т. е. таким образом, что одновременно со сменой заполненного приемника газа производилась смена приемника жидкого дестиллата.
Из табл. 10 видно, что выделение воды происходит в определенном температурном промежутке (250—300° в парах и 320—400° в жидкости). Далее вода уже не выделяется, т. е.
О составе и свойствах сланцевой смолы
29
Таблица 10
Состав газа и фракционный состав дестиллатов атмосферной перегонки смолы
до 233 233—270 105 32,0 68,0 88,0 70,9 I 4,2 39,4 I 8,6 6,4 13,3 4,0 6,7 14,5 32,0 200 255 250 325 20,0 1,0
270—280 ПО 5,0 26,0 64,0 27,6 43,1 15,5 5,8 38,0 318 380 1,0 0,02
280—290 92 12,0 32,0 68,0 12,1 14,0 15,2 5,3 53,4 328 400 2,0 0,04
290—300 78 11,0 26,0 61,0 10,5 20,2 10,5 5,4 53,4 336 410 .— —
300-330 — — — 8,0 28,0 7,2 10,8 46,0 342 420 — —
330—350 90 10,0 17,0 62,0 5,9 18,6 6,8 12,7 56,0 350 425 —
весь материал, способный ее выделить, успевает разложиться к тому моменту, когда будет достигнут указанный выше верхний температурный предел. В дестиллате появляется больше 1/з легких компонентов, обязанных своим происхождением распаду тех веществ, которые выделили воду; при этом в высоко-кипящих фракциях, полученных разгонкой смолы при атмосферном давлении, значительно снижается содержание омыляемых и гидроксилсодержащих соединений (табл. 11 и 12).
Следовательно, в состав сланцевой смолы входят высокопо-лимерные, термически мало устойчивые гидроксилсодержащие соединения, способные к распаду, с выделением воды, при сравнительно низких температурах. К таким веществам могут быть отнесены альдолы, оксикислоты, оксикетоны и спирты.
Из той же табл. 10 видно, что выделение углекислоты происходит при самых низких температурах, без образования конденсата (фракция 1). Невидимому, в этих условиях разлагается главная масса органических кислот (судя по составу газа). В дальнейшем кислоты разлагаются во все уменьшающемся количестве. Появление водорода и метана в газе можно объяснить только распадом карбонильных соединений, так как только эти вещества могут образовать водород и метан при температуре 250—300°, когда все без исключения углеводороды еще совершенно устойчивы. Тот факт, что распад карбонильных соединений наблюдается в широком температурном диапазоне, говорит
30
И. И. Зеленин и О. С. Куратова
Таблица 11
Анализ вакуумных и атмосферных фракций генераторной смолы
Вакуумная разгонка Разгонка при атмосферном давлении
Контролируемая величина 1 2 3 4 1 2 3 4
до 100- 150- 200- 150— 190- 230- 250-
100° 150° 200° 250° 190° 230° 255° 350*
У д. вес при 20° С . . 0,8729 0,9076 1 0,997 Г 1,0246 0,7684 0,7917 0,8069 0,9496
Вязкость при 50° С
в “Е Растворяется в 1,00 1,95 3,02 13,74 1,00 1,07 1,16 1,16
10%-нон NaOH, % — 5,86 25,59 27,91 1,98 6,61 9,63 21,21
Молекулярный вес . Содержание, %: 157,7 171,5 213,4 228,8 136,8 154,5 163,7 169,3
углерода .... — 86,86 82,46 82,35 — 84,00 84,33 79,74
водорода . . . — 11,11 9,53, 9,35 — 11,03 10,75 9,48
серы ..... 1,30 1,03 0,85 0,69 1,27 0,90 0,81 0,78
кислорода и азота — 1,00 7,16 7,61 — 4,07 4,11 10,00
Кислотное число . . 5,6 4,2 2,2 3,3 5,6 4,1 2,0 3,1
Число омыления . . Содержание, %: 19,2 18,0 43,9 47,1 10,3 14,4 22,7 28,0
карбоновых кислот гидроксильных со- — 0,45 0,53 0,63 0,53 0,61 0,76 2,66
единений . . карбонильных со- — 1,50 3,44 5,62 — 1,50 1,20 2,44
единений . . . омыляемых соеди- — 37,2 27,7 67,2 — 19,9 31,7 45,2
нений .... 3,8 4,2 12,3 17,9 1,1 2,8 6,0 7,5
о том, что представители этих соединений, содержащиеся в смоле, принадлежат к различным классам и рядам, обладающим различной прочностью.
Из материала приведенных таблиц также следует, что атмосферная и вакуумная разгонки являются, с точки зрения их влияния на химический состав перегоняемого сырья, совершенно различными процессами и, следовательно, продукты этих разго-нок должны резко отличаться друг от друга. В табл. 11 и 12 дана сравнительная характеристика фракций вакуумной и атмосферной разгонок смол туннельной и генераторной.
В этой сравнительной характеристике атмосферных и вакуумных фракций сланцевой смолы наиболее резко выражены различия в величине четырех показателей: 1) плотности, 2) молекулярного веса, 3) содержания омыляемых и 4) содержания кислот. В нижеследующей сводке суммированы эти важные различия.
Плотность. Плотность атмосферных фракций не превышает 0,950 и достигает этого предела лишь в случае фракций, кипящих выше 250°, при содержании «фенолов» 21%, в то время как плотность вакуумных фракций доходит до 1,025.
О составе и свойствах сланцевой смолы
31
Таблица 12
Анализ вакуумных и атмосферных фракций туннельной смолы
Вакуумна? разгонка Разгонка при атмосферном давлении
Контролируемая величина 1 2 3 4 1 2 3 4
до 100 150— 200- 150- 195- 238- выше
50° 150° 200° 250° 195° 238° 275° 275'
Уд. вес при 20°С . 0,7302 0,8167 0,9920 1,0242 0,7886 0,8061 0,8397 0,868
Вязкость при 50°, °Е 1,0 1,24 9,53 14,43 1,00 1,07 1,52 1,52
Растворяется в 10%-ной NaOH, % 13,02 27,96 28,74 1,64 8,14 20,54 21,48
Молекулярный вес . 105,8 158,0 206,2 221,4 133,2 153,5 184,5 185,6
Содержание, % углерода _ . . — 86,04 82,67 84,30 - 81,36 79,35 79,83
водорода .... — 9,87 8,81 8,35 — 10,80 9,56 8,65
серы 1,32 1,10 0,84 0,68 1,20 0,95 0,62 0,52
кислорода и азота — 2,99 7,68 6,67 — 6,89 10,47 11,00
Кислотное число . . 7,0 5,0 3,7 4,2 7,4 4,8 4,1 4,6
Число омыления . . 7,2 19,7 35,5 38,9 6,4 5,9 15,8 28,3
Содержание, %: карбоновых кислот 0,29 0,61 0,89 0,57 1.33 4,20 4,55
гидроксильных соединений . . 2,10 4,05 5,59 1,47 2,18 1,69
карбонильных соединений . . . — 31,8 39,5 31,2 — 23,0 27,7 29,9
омыляемых соединений . . . 0,03 4,1 11,6 13,6 — 0,3 3,8 7,9
М о л е к у л я р I -I ы й вес. Мо. г1екул5 1рный вес ваку умных
фракций значительно выше молекулярного веса атмосферных фракций. Максимальный молекулярный вес имеют атмосферные фракции 170— 185 и вакуумные 220—228.
Содержание омыляемых. Содержание омыляемых соединений в вакуумных фракциях практически в два с половиной раза превышает содержание их в атмосферных фракциях (4—14% для вакуумных против 1—7% для атмосферных фракций) .
Содержание кислот. В атмосферных фракциях содержание кислот выше, чем в вакуумных.
Детальный анализ цифрового материала обеих таблиц мог бы выявить и другие различия в составе фракций (например, различия в содержании фенолов) и это, конечно, представляло бы значительный интерес. Однако, учитывая условный характер некоторых аналитических данных, связанный с небезупречной точностью соответствующих методов анализа, необходимо, во избежание поспешных и ложных выводов, воздер-
32
Н. И Зеленин и О. С. Куратова
жаться от более широкой интерпретации приведенного экспериментального материала.
Что же касается тех данных, которые мы приняли в рассмотрение, то они не вызывают сомнений. Плотность, молекулярный вес, числа кислотности и омыления достаточно хорошо и безошибочно характеризуют вещество. Эти показатели, вместе взятые, позволяют сделать достаточно обоснованные выводы.
Главное в различии дестиллатов вакуумной и атмосферной разгонки заключается в упрощении молекул, обусловленном в данном случае распадом омыляемых и гидроксилсодержащих веществ при более высокой температуре атмосферной разгонки. На это указывают основные для этого процесса показатели: плотность, молекулярный вес, содержание омыляемых соединений и кислот. Можно представить, что это упрощение молекул эфирообразных веществ происходит во вполне определенном направлении. Нет никаких сомнений, что связь, способная омылиться, явится и при термическом воздействии местом, с которого должны начаться все превращения, т. е. что мы имеем дело в данном случае с каким-то процессом разложения, но не крекинга, в обычном смысле этого слова.
Вакуумные разгонки показывают наличие в смоле большого количества омыляемых, высокополимерных нестойких веществ, которые могли образоваться в условиях полукоксования сланца как за счет распада высокополимерных кислородных молекул керогена сланца, так и в результате реакций уплотнения и конденсации образовавшихся реакционноспособных карбонильных соединений в последующих стадиях получения смолы. Таким образом, следует полагать, что часть смолы, полученной в виде простых реакционноспособных веществ, какими являются кислоты, спирты, альдегиды и кетоны, образовала полимерные вещества, среди которых немалую долю составляют омыляемые соединения. Таким образом, любая термическая переработка сланцевой смолы, начиная с первичного получения смолы, связана с глубокими изменениями, приводящими к образованию малолетучих веществ, газа и углеводородов.
Исходя из этих соображений и тех сведений, которыми мы располагаем о составе сланцевой смолы, можно построить рабочую гипотезу происхождения отдельных составляющих сланцевой смолы и наметить возможные пути подтверждения и дальнейшей разработки этой рабочей гипотезы.
ПРОИСХОЖДЕНИЕ СОСТАВЛЯЮЩИХ СЛАНЦЕВУЮ СМОЛУ
О механизме распада керогена ничего неизвестно, хотя этому вопросу посвящено очень много работ. Установлены общие закономерности разложения органической массы сланцев, количественные выходы смолы, воды, газа, отдельных компонентов
О составе и свойствах сланцевой смолы
33
таза и влияние на процесс различных факторов (температура, скорость прогрева, способ эвакуации паров и другие). О том, как разлагается кероген, каковы основные пути его химических превращений — пока ничего неизвестно.
Для тех представлений об изменениях смолы, которые мы себе составили, наиболее важным и единственно доступным при современном состоянии знаний опорным экспериментальным материалом является групповой функциональный состав смолы. Поэтому ниже рассматриваются пути превращения составляющих сланцевой смолы именно с точки зрения превращения функциональных кислородных групп, так как другие группы в керогене, по крайней мере в случае прибалтийских сланцев, не играют какой-либо существенной определяющей роли.
Согласно последним данным, кероген представляется не как смесь веществ, характеризующихся вполне определенными химическими свойствами и функциями и поддающихся разделению известными способами, а как высокополимерное многофункциональное вещество, в котором характерные особенности отдельных химических составляющих определенным образом замаскированы и обезличены большим размером молекулы и многообразием представленных в ней функций. Следует считать установленным [13], что полимерные молекулы керогена имеют, в основном, кислородную природу, характеризующуюся функциями карбоксила, карбонила, гидроксила и их производных. Кислоты, альдегиды и кетоны представляют, повидимому, главную массу кислородсодержащих компонентов керогена. Так, К- Лутс [11] считает, что в составе керогена имеется до 25% кислотных соединений, до 40% карбонильных и до 20% фенолов.
Хотя полной уверенности в точности количественных данных, приведенных К- Лутсом, нет, все же мы считаем не подлежащим сомнению, что карбонил, гидроксил и карбоксил являются главными функциональными группами в составе керогена, определяющими, в основном, химическую природу последнего, характер тех исходных веществ, метаморфизм которых привел к образованию керогена.
Трудно себе представить, что функциональные группы СООН, СО и ОН появились в результате каких-то вторичных процессов, например, за счет окисления первичного вещества. При таком предположении следовало бы допустить, что исходное вещество представляет собой углеводородный материал, превращающийся при окислении в то, что называют керогеном. Подобное допущение противоречит всем без исключения фактам и представлениям о метаморфизме каустобиолитов.
С другой стороны, не исключена возможность окисления керогена, имеющего структуру кислородных полимеров, но в этом
3 Труды ВНИИПС.
34
И. И. Зеленин и О. С. Куратова
случае богатство функциональных групп только увеличится, при сохранении основной структуры, так как в противном случае глубокое окисление полностью разрушило бы кероген. Следует думать, что при образовании полимерной формы керогена в значительной степени сохранились функции составляющих кислородных соединений, что, вообще говоря, вполне отвечает природе кислородных соединений, способных создавать сложнейшие комплексы веществ. В этих комплексах первоначальные химические и физические свойства исходного материала исчезают, но прекрасно сохраняются функциональные особенности групп, являющихся обычно «замками» в цепи строения больших молекул. Любой ключ, открывая эти «замки», как бы возвращает исходным веществам, в той или иной мере, их начальные свойства и, прежде всего, их основные химические функции, несмотря на происшедшее общее изменение молекул.
Следовательно, начальную форму образования смолы можно представить как образование высокополимерных кислородсодержащих молекул типа сложных эфиров, оксикислот, лактонов, карбоновых кислот и их ангидридов. Указанные соединения, будучи термически мало устойчивыми, способны в процессе полукоксования сланца при сравнительно низких температурах, под влиянием катализирующих веществ сланцевой золы, претерпевать превращения с образованием целой гаммы не только кислородсодержащих соединений, но и углеводородов различных классов.
Р. X. Оболенцев и Ю. Н. Усов [14] показали, что распад высокополимерных сложных эфиров одноатомных спиртов в присутствии алюмосиликатных катализаторов протекает при сравнительно низких температурах 195—200°С с образованием кислот, непредельных углеводородов и углекислоты без других газообразных продуктов.
При наличии в керогене сланца высокополимерных сложных эфиров последние будут претерпевать превращения на первой ступени термической обработки сланца, а именно, на стадии дезоксидации и термолиза, если принять, что превращение керогена сланца протекает по следующей схеме:
Кероген
У I. Дезоксидация
Пиробитум
II. Крекинг
4-
Смола
| III. Пиролиз
Легкие углеводороды
термолиз —> СО2, СО, Н2О, Н2
->СО2, СО, Н2О, н2, сн<, с
->Н2, CH*, спнт, с
->с, н2
О составе и свойствах сланцевой смолы
'-о
Продуктами распада эфиров жирного ряда явятся непредельные углеводороды и кислоты, которые при дальнейшем повышении температуры будут подвергаться дальнейшим превращениям.
Кислоты жирного ряда под влиянием углекислого кальция сланцевой золы способны дать альдегиды и кетоны. Интенсивность процесса образования альдегидов увеличивается по мере усложнения молекулы, с одновременным появлением в продуктах реакции непредельных углеводородов [15]. В ароматическом ряду связанный в форме эфиров кислород является источником образования фенолов и соответствующих кислот, в зависимости от строения радикала кислоты.
Наличие кислот в основной массе первичных продуктов разложения керогена является доказанным [16]. Карбонильные группы тоже найдены как в самом керогене, так и в любых формах его распада и, прежде всего, в первичных формах; фенолы также найдены в продуктах разложения, но нет никаких, даже косвенных, указаний на присутствие спиртов жирного ряда.
Наличие фенолов в смоле еще не является доказательством их наличия в керогене. Не исключена возможность образования ароматических соединений и их производных в смоле путем синтеза их из альдегидов, кетонов и спиртов, так как до сих пор пет убедительных данных о существовании ароматических колец в керогене и даже, наоборот, имеются некоторые основания предполагать обратное. А. Ф. Добрянский, с особой обстоятельностью изучавший вопрос о составе сланцевой смолы и, в частности, о составе и структуре керогена [13], не смог прийти к определенному выводу по вопросу о наличии ароматических колец в керогене и оставил его открытым. Мы также не видим пока экспериментальных данных, достаточных для обоснования категорических суждений по этому вопросу.
Совсем недавно Н. М. Караваев и И. М. Венер [17] еше раз подробно исследовали состав сланцевого битума на содержание в нем ароматических колец. Этим исследователям, как и многим другим, не удалось непосредственно обнаружить ароматические кольца в битуме, а следовательно, и в керогене. Применяемый для этой цели метод окисления керогена перманганатом в щелочной среде оказывается в данном случае непригодным. Тем не менее, применив остроумный метод косвенного доказательства путем сравнения результатов окисления битума и формальдегидрезорциновой смолы, названные выше авторы пришли к заключению о наличии ароматических структур в сланцевом битуме. Все же их выводы не являются решающими по двум причинам: во-первых, ими ие дано прямого доказательства иат линия ароматической структуры в битуме и, во-вторых, исследо-3*
36
Н. И. Зеленин и О. С. Куратова
ванне битума не может быть приравнено к исследованию самого керогена. Полагаем, что компоненты керогена и продукты его распада, по своей химической природе, представляют собой материал, вполне достаточный для синтеза разнообразных фенолов, а следовательно, и ароматических колец. Таким образом, нет никакой необходимости доказывать непосредственное происхождение фенолов из соответствующих структур керогена. Альдегиды и кетоны жирного ряда могут образовывать фенолы и другие ароматические кислородные соединения, а также углеводороды, в тех условиях, в которых образуется смола. Нет нужды отрицать наличие фенолов в составе керогена, важен тот факт, что их отсутствие в нем не является помехой их появлению в продуктах распада керогена. Таким образом, фенолы можно исключить из рассмотрения начальных форм превращения смолы.
Основными веществами, составляющими сланцевую смолу и определяющими ее дальнейшие превращения, являются:
1) кислоты, присутствие которых в самом керогене и в продуктах его распада является бесспорно доказанным;
2) альдегиды и кетоны, присутствие которых если не в самом керогене, то в продуктах его распада может считаться доказанным;
3) спирты, которые, несмотря на то, что пока нет доказательств присутствия их в керогене и продуктах его распада, должны так или иначе содержаться в сланцевой смоле, поскольку целый ряд превращений кислородсодержащих компонентов последней неизбежно связан с появлением спиртов.
Проследим пути превращения этих групп.
Кислоты. Наиболее важными термическими превращениями кислот являются:
I. CnHmCOOH ->С„Нт+1 + СО2
II. 2СпНтСООН -> (СпНт)2СО + СО2+ Н2О
Оба типа превращений играют большую роль в процессах распада органической массы сланца и сланцевой смолы.
Появление углекислоты в самых начальных стадиях разложения керогена, а впоследствии и при разложении смолы обусловлено, главным образом, распадом кислот, так как всякое разложение последних характеризуется прежде всего выделением углекислоты.
Реакция I является очень важной для превращений смолы, так как она, сохраняя свое значение для различных гомологических рядов, показывает источник образования углеводородов, не только метановых, но и всяких других, в зависимости от природы радикала кислоты. Следует, однако, иметь в виду, что значение указанной реакции в случае сланцевой смолы ограни
О составе и свойствах сланцевой смолы
37
чено тем, что найденные в составе смолы кислоты (насыщенные и ненасыщенные) принадлежат почти исключительно к жирному ряду. Это хорошо объясняет наличие в смоле парафинов и олефинов, но не дает решения вопроса о происхождении других углеводородов.
Превращения I и II протекают в различных температурных условиях, в зависимости от молекулярного веса и строения кислот. Для низших членов рядов кислот необходимы довольно жесткие условия (400—600°), тогда как высшие представители претерпевают превращения при сравнительно низких температурах (200° и выше). Во всяком случае оба типа превращений должны быть обычными для условий образования и переработки сланцевой смолы. Реакция II имеет для судьбы составляющих сланцевой смолы огромное значение, так как она открывает широкие перспективы для всевозможных превращений, поскольку карбоксильные соединения характеризуются, как известно, исключительно высокой реакционной способностью. Отметим, что реакция II протекает для высокомолекулярных кислот, таких как стеариновая, пальмитиновая, миристиновая, лауриновая, с почти количественным выходом соответствующих кетонов при температурах 270—300° [18]. Следовательно, условия полукоксования и дальнейшей переработки смолы обеспечивают течение подобных превращений. Это важно еще и потому, что, вне зависимости от наличия кетонов в первичной смоле, возможность их дальнейшего присутствия обеспечивается при наличии кислот в исходном материале.
Альдегиды и кетоны. Наиболее важными превращениями для этих веществ являются следующие.
III. Разложение с выделением окиси углерода СпНтСНО-+СпН т+1 + СО
IV. Разложение с выделением окиси углерода и водорода СпНтСНО -^СпНт_1 + со + н2
V. Альдольная конденсация
2СпНтСНО^СпНтСНОН—СпЩ-iCHO
VI. Конденсация по типу кротонового альдегида (дегидрата-ционная конденсация)
2CnHmCHO -э СпНтСН = С пНт_2СНО + Н2О
VII. Сложноэфирная конденсация
Н
2СпНтСНО ^СпНтСО—О—С—СпНт
Н
И. И. Зеленин и О. С. Куратова
38
VIII Гидратация
Н
СпНтСО-О-С—СпНт + Н20 -> СпНтСООН + СН2ОН—СпНт Н
Кетоны, которые также содержат карбонильную группу, но связанную с радикалом, являются менее активными. Для них наиболее характерными являются реакции конденсации с последующей дегидратацией, аналогичные реакциям V и VI.
Сведения, которыми мы располагаем (см. выше), говорят о том, что главная масса кетонов, содержащихся в сланцевой смоле, принадлежит к жирному ряду, откуда следует, что и основное количество углеводородов, содержащихся в смоле и продуктах ее распада, должно также принадлежать к жирному ряду. Следует добавить, что распад альдегидов и кетонов протекает обычно не по наиболее простой схеме III. Значительную роль начинают играть реакции IV, связанные с распадом до олефинов, с выделением водорода и окиси углерода, даже при низких температурах [18]. Хотя эти реакции и не являются определяющими, они легко могут объяснить факты образования водорода при сравнительно низких температурах.
Превращения V, VI и VII следует рассмотреть более подробно.
Альдольная конденсация V является одной из важнейших реакций карбонильных соединений. К превращениям этого типа склонны все карбонильные соединения, и границы их протекания практически не могут быть очерчены, поскольку рассматриваемая реакция совершается очень легко под влиянием различных катализаторов, кислот, щелочей, действия света, тепла, т. е. в таких условиях, которые вполне возможны в случае превращений сланцевой смолы как в процессе ее получения, так и в последующих процессах ее переработки.
Этим путем, скорее всего, происходит уплотнение кислородных соединений и образование высокомолекулярных соединений. Подобный процесс является тем более возможным, что в нем могут принимать участие не только собственно кетоны и альдегиды, но и всевозможные более сложные вещества, содержащие, наряду с карбонилом, и другие функциональные группы, в результате чего могут образовываться эфирообразные и другие вещества.
В случае альдегидов и кетонов реакции V и VI могут сопровождаться уплотнением не только двух, но и значительно большего числа молекул с выделением воды в последующих фазах
О составе и свойствах сланцевой смоли
39
реакции при сравнительно низких температурах (примером может служить образование из ацетона окиси мезитила и форона), чем может быть объяснено появление* воды при превращениях смолы.
Реакции конденсации по типу кротонового альдегида изучены в последнее время достаточно хорошо. При рассмотрении причин образования полимеров, в случае обработки сланцевых 'кислородных соединений глинами, эту реакцию следует считать одной из главных причин образования малолетучих, высокомолекулярных соединений, которые, в результате постепенного отщепления от них воды, превращаются во все более ненасыщенные вещества, переходящие с повышением температуры в труднорастворимые продукты.
Таким образом, конденсация по типу кротонового альдегида показывает нам путь образования пека и кокса в технологии сланцевой смолы, а возможно, и всякой другой смолы полукоксования.
Реакция VII имеет не меньшее значение в процессе превращения карбонильных соединений в условиях полукоксования сланца. Не отождествляя свойств альдегидов и кетонов и их поведения в реакциях сложноэфирной конденсации, которая еще почти не разработана для алифатических и других кетонов, можно полагать, что общий характер превращений кетонов дает основание рассматривать возможности сложноэфирной конденсации для кетонов наравне с альдегидами.
Остается решить вопрос, происходит ли сложноэфирная конденсация в условиях термических превращений альдегидов и кетонов? Ответ на этот вопрос, имеющий решающее значение для объяснения очень многих свойств сланцевой смолы, находим в последних работах А. Я. Ларина и А. В. Фроста [191, из которых следует, что сложноэфирная конденсация имеет место во всех превращениях карбонильной группы при термическом воздействии, по крайней мере в присутствии алюмосиликатов, что вполне отвечает условиям образования сланцевой смолы и ее переработки. По данным А. В. Фроста [23] кетоны жирного ряда претерпевают превращение с образованием непредельных углеводородов и соответствующих карбоновых кислот по двум направлениям:
О
СН3—С-СН3 о
II сн3-с-сн3
СНз
сн,
со-сн3
о н
; | ..... | ....
Й с—сн2
снзч ->сн3соон+ >с = сн2
сн/
(1)
40
Н. И. Зеленин и О. С. Куратова
2СН3—С—СН3 ——- 3\с = СН СО • СН3
I! * СН/
О
СНЗХ i СНЗХ
>С = СН-СО-СН3-> )С = СН2+СН3СООН сн/ i сн/
+ н—он
Конечными продуктами реакции в том и другом случаях являются карбоновые кислоты и непредельные углеводороды одинакового строения. В схеме (2) образованию последних должна предшествовать реакция конденсации двух молекул кетона с отщеплением воды, т. е. конденсация по типу кротонового альдегида с образованием непредельных кетонов, которые вновь присоединяют воду как углеводороды ряда дивинила. При дальнейшем изучении процесса превращения окиси мезитила на указанном катализаторе, а также на основании исследования промежуточных и побочных продуктов реакции авторы приходят к выводу, что превращение кетонов протекает через соответствующие непредельные кетоны до кислот и олефинов по схеме (2).
Судить о механизме превращения кетонов над алюмосиликатными катализаторами по образующимся продуктам реакции очень трудно. Основным продуктом реакции по схеме (1) должен быть сложный эфир третичного спирта — соединение, легко гидролизующееся водой даже на холоде [20]. Поэтому при повышенных температурах более вероятным является присутствие не самих сложных эфиров, а продуктов их гидролиза — спиртов и кислот. В свою очередь, образующиеся спирты, в зависимости от их строения, отщепляют воду, давая непредельные углеводороды или простые эфиры. Третичные спирты легко дегидратируются с образованием только непредельных углеводородов под влиянием даже таких мягких реагентов как щавелевая кислота. Наличие непредельных углеводородов и кислот, как продуктов конденсации альдегидов и кетонов жирного ряда, может быть объяснено реакцией (1). Наряду с этой реакцией протекает и параллельная ей реакция уплотнения по типу кротонового альдегида, но при более мягких условиях.
Для подтверждения указанного предположения были проведены опыты с бензойным и гексиловым альдегидами при тех же условиях и на том же катализаторе, что и в опытах А. В. Фроста.
Опыты с бензойным альдегидом были поставлены, главным образом, потому, что для этого альдегида исключаются реакции альдольного уплотнения.
О составе и свойствах сланцевой смолы
41
Опыты проводились над активными глинами при температуре 200—300° С. При 300° были достигнуты максимальные результаты превращения альдегида в бензойную кислоту — 22%-ный выход кислоты, считая на исходный альдегид, и 15%-ный выход высокополимерного смолистого остатка с молекулярным весом 300. Кислота имела молекулярный вес 122, температуру плавления 122°С и кислотное число 459,7, что полностью соответствует константам чистой кислоты и показывает возможность превращения ароматических альдегидов по типу сложноэфирной конденсации.
Известно, что альдегиды и кетоны легко вступают в реакцию конденсации с ароматическими углеводородами и их производными. Конденсация альдегидов с фенолами проходит с образованием высокополимерных продуктов. Глубина уплотнения может быть настолько значительной, что продукты конденсации будут обладать плохой растворимостью в органических растворителях.
Сланцевые дестиллаты в своем составе содержат не только карбонилсодержащие соединения, но и фенолы. В процессе полукоксования сланца образующиеся карбонильные соединения и фенолы будут участвовать в реакциях конденсации с образованием высокополимерных молекул.
Проведенные нами опыты со смесью фенола и бензальдегида над активными глинами при температуре 250 и 300° и объемной скорости 0,25 л/ч показали возможность образования высокополимерных кислых и нейтральных кислородных соединений. Среди кислот выделены кислые высокомолекулярные продукты, хорошо растворимые в бензоле и высаживающиеся из бензольного раствора нефтяным эфиром, по внешнему виду напоминающие асфальтены, выделяемые из сланцевых смол.
Опыты с гексиловым альдегидом должны были показать возможность превращений альдегида по обеим указанным выше схемам, а именно, по типу альдольного уплотнения с последующей дегидратацией и по типу сложноэфирной конденсации.
Для работы был взят гексиловый альдегид, имеющий следующую характеристику (в скобках указаны соответствующие теоретические константы):
Уд. вес при 15° С ................ 0,8255
Молекулярный вес ..................... 96
Содержание группы — С , .... 27,9
Хн
Температура кипения, °C ............ 118—130
(0,8340) (100,1)
(29,0)
(128)
Условия опытов: температура 150, скорость 0,25 л/ч; продолжительность
200, 250 и 300°; объемная
2 часа. Результаты опы-
42
' II. И. Зеленин и О. С. Куратова
тов, сведенные в табл. 13 и 14, показывают, что наибольший процент превращения альдегида наблюдается при температуре 300°, при которой уже имеет место незначительное газообразование. Основными продуктами реакции являются соединения, соответствующие альдольной и сложноэфирной конденса-
Таблица 13
Превращение гексилового альдегида над активной глиной
Продолжительность контакта 2 часа
Показатели
Выход катализата, %................
Содержание воды в катализате, % . Остаток на катализаторе-(-потери, %
О
Содержание — С^ в катализате, %
чн
О
Превращено — %............
Н
Температура опыта,°C
150 200 250 300
19,3 10,2
51,3 62,5
66,3
5,9
27,8
9,2
78,5
ции. Выделить муравьиную кислоту, отвечающую реакции гидратации непредельных альдегидов, не удалось. Идентификация кислот проводилась по их бариевым солям и кислотным числам. Для водорастворимых кислот содержание бария составляло 37,2% и кислотное число 492, что соответствует капроновой кислоте, для которой теоретическое содержание бария составляет 37,3% и кислотное число — 483. Для кислот, выделенных из остатка выше 130°, содержание бария равнялось 27,1, кислотное число — 325 и содержание ОН — 4,4%, что отвечает смеси капроновой и оксидодециловой кислот.
Опыты, проведенные с гексиловым альдегидом, наглядно показывают, что превращение альдегидов первой группы над активными глинами протекает по двум направлениям: 1) по типу альдольного уплотнения и 2) по типу сложноэфирной конденсации.
Характеристика продуктов превращения гексилового альдегида над активной глиной при температуре 150—ЗОО’С и продолжительности контакта 2 часа
Таблица 14
Показатели Исходное сырье С6Н12О Фракция1 60— 80° С, полученная при температуре, °C Остаток >150° С, полученный при температуре, °C Полимеры,2 полученные при температуре, °C
250 300 150 200 250 300 150 200 250 300
Уд. вес при 15° С 0,8255 0,6860 0,6954 0,9185 0,879 0,872 0,872 ___ —_ —
Молекулярный вес 96 79,5 82,3 185,0 200,0 205,1 209,0 350 380 394 392
Показатель преломления . — 1,392 1,394 1,4358 1,457 1,474 1,484 — — — —
Число омыления — 0 0 195,0 153,9 — 108,2 21,5 35,8 — 50,0
Кислотное число Содержание, % — 0 0 130,5 118,7 — 63,0 18,1 19,2 — 21,7
ОН о 0 0 2,2 1,3 1,1 0,63 0 0 0 0
-с^ \1 27,9 3,4 3,2 3,2 3,4 0 0 — — — —
Йодное число Содержание непредельных соедине- 0 154,8 142,2 8,0 52,0 74,2 — —
НИЙ, % — 47,5 45,4 6,6 40,9 60,1 — — —
Выход фракции, % — 30,0 31,5 29,2 26,9 39,0 33,0 - - — —*
1 Выход фракции в опытах при 150° = 2,0%, при 200° — 3,7%.
Экстрагированные (снятые с катализатора) растворители.
44
Н. И. Зеленин и О. С. Куратова
Ввиду наличия в продуктах полукоксования сланца значительных количеств альдегидов и кетонов, сложноэфирная конденсация и уплотнение по типу кротонового альдегида должны быть отнесены к числу важнейших превращений кислородных соединений смолы, конечными продуктами которых являются углеводороды, вода и карбоновые кислоты, в первую очередь уксусная, а также высокополимерные продукты — альдолы и соответствующие им кислоты.
Как видно из сказанного, основными продуктами сложно-эфирных превращений альдегидов и кетонов являются сложные эфиры, спирты, углеводороды и кислоты. Их судьба в основном уже известна. Появлению кислот, спиртов и углеводородов должен предшествовать гидролиз эфиров, который и происходит в действительности над алюмосиликатами в условиях повышенных температур и в присутствии воды.
Продукты гидролиза сложных эфиров — кислоты, — в зависимости от их строения, по мере повышения температуры будут претерпевать дальнейшие превращения. Известно, что наиболее термически устойчивой является уксусная кислота, которая не разлагается до температуры темнокрасного каления. Пропионовая, масляная и изовалериановая кислоты также устойчивы до температуры 460°. Кислоты от С!2 уже склонны к распаду при температуре 270—290°. Сланцевая зола, являющаяся грубым алюмосиликатным катализатором, будет каталитически воздействовать на образовавшиеся кислоты, в результате чего преобладающее значение приобретет реакция образования симметричных кетонов. А. В. Фрост и А. В. Очкин [21] показали, что образование дипропилкетона из масляной кислоты над алюмосиликатным катализатором протекает при температурах 250— 300°. С другой стороны, низкомолекулярные кислоты — уксусная, пропионовая и масляная, — будучи хорошо растворимыми в воде, будут частично удаляться из зоны швелевания с водяными парами и концентрироваться в подсмольной воде. Действительно, среди карбоновых кислот в подсмольных водах Н. Ф. Шароновой обнаружены уксусная и пропионовая кислоты.
Спирты. Основной реакцией для спиртов, получающихся в результате гидролиза сложных эфиров, является реакция отщепления воды. Рядом авторов показано, что спирты легко дегидратируются над алюмосиликатными катализаторами при сравнительно низкой температуре с последующей изомеризацией непредельных углеводородов. Не только спирты, но и другие гидроксилсодержащие соединения являются термически мало устойчивыми соединениями, которые в условиях полукоксования сланца будут подвергаться дегидратации при сравнительнонизких температурах.
О составе и свойствах сланцевой смолы 45
Нашими опытами показано, что в случае термокаталитического превращения гексилового альдегида содержание группы ОН в катализате при повышении температуры от 150 до 300° падает с 2,2 до 0,63%, а йодное число увеличивается с 8 до 74,2 (см. табл. 14).
Следовательно, сильно выраженная дегидратирующая способность алюмосиликатных катализаторов приводит к образованию не только непредельных углеводородов, но и непредельных кислот из оксикислот. Поэтому представляется мало вероятным присутствие в сланцевых дестиллатах спиртов и />-оксикислот, имеющих водородные атомы в а-положении.
Образующиеся непредельные углеводороды, карбоновые кислоты и карбонилсодержащие соединения, в зависимости от их строения и времени пребывания в зоне полукоксования, будут претерпевать дальнейшие превращения, среди которых возможны реакции уплотнения с образованием высокополимерных пековых остатков. Таким образом, источником образования тяжелых остатков в сланцевой смоле будут служить не только первичные продукты термического распада сланца, но также и термически менее стойкие кислородные и непредельные соединения, являющиеся вторичными продуктами распада органической массы сланца.
Процессы превращения спиртов и кислот, конечно, нельзя сводить только к реакциям дегидратации и декарбоксилирования. Превращения их представляют собой целую гамму реакций, продукты которых (альдегиды и др.) могут принимать активное участие в последующих процессах образования различных органических соединений.
В проведенной нами работе показано, что альдегиды, находящиеся в контакте с активными глинами, претерпевают процесс превращения уже при температуре 150°, давая высокополимер-ные продукты типа альдолов. С повышением температуры начинают преобладать легкие фракции, содержащие предельные и непредельные углеводороды (см. табл. 14). Степень превращения альдегида достигает 80%.
Таким образом, следует ожидать, что альдегиды, как соединения более реакционноспособные, чем кетоны, будут содержаться з сланцевых дестиллатах в меньших количествах и будут служить источником образования углеводородов (непредельных и предельных) и высокополимерных соединений. В самом деле, легкая и средняя фракции смолы прибалтийских сланцев содержат значительный процент непредельных углеводородов, среди которых преобладают олефины нормального строения с двойной связью, расположенной ближе к периферии. Образование таких непредельных углеводородов может протекать по следующей схеме:
46
Н. И. Зеленин и О. С. Куратова
Н
.° I
2R-C" —> R—СО—О—С—R
Н |
Н
н
R—CO-O-C-R + H2O -э R-COOH + R-CH2OH
Н
R-CH2OH —° - R-CH = СН2+ Н2О
Обобщая приведенные выше факты и соображения, можно прийти к предположению, что углеводороды в основной своей массе являются продуктами вторичного происхождения, образующимися в результате разнообразных превращений кислородных соединений. Эта мысль, достаточно доказанная фактами, развивается в последнее время авторами новейших гипотез происхождения нефти А. Ф. Добрянским [22] и А. В. Фростом [23, 24].
Можно думать, что и в случае сланцевой смолы указанное выше положение может занять определенное место в той гипотезе происхождения углеводородов, которая может быть построена в рамках общей схемы метаморфизма каустобиолитов.
Приведем некоторые соображения, относящиеся к рассматриваемому вопросу.
Как видно из уравнения, выражающего превращение (1), кислоты, распадаясь на углеводород и углекислоту, могут стать источником образования углеводородов любого класса, так как радикал кислоты определяет характер и строение полученного углеводорода. В соответствии с тем, что известно о природе кислот сланцевой смолы, можно считать, что основную массу углеводородов, получающихся при разложении этих кислот, будут составлять парафины и олефины нормального строения, так как все карбоновые кислоты, которые известны и выделены из сланцевой смолы, принадлежат исключительно к жирным кислотам нормального строения. Это подтверждается и строением выделенных и изученных углеводородов сланцевой смолы.
Действительно, наряду с нормальным строением кислот, в последнее время доказано, что и природа кетонов, содержащихся в сланцевой смоле, отвечает сказанному выше. С. С. Семенов и А. П. Сиверцев показали нормальное строение ряда кетонов. Большинство олефинов в сланцевой смоле также имеет нормальное строение с двойной связью на самом конце цепи.
О составе и свойствах сланцевой смолы
47
Образование фенолов в процессе термического разложения сланца может привести к конденсации альдегидов с фенолами и появлению высокополимерных кислых продуктов типа асфальтенов, что и было замечено в опытах с бензальдегидом.
Из всего сказанного выше видно, что скорость эвакуации смоляных паров и скорость нагрева сланца при коксовании будут существенным образом влиять на количественный выход и качество смолы, но практически всегда получаемые продукты коксования будут содержать соединения, способные к процессам окисления и смолообразования, а получаемые смолы — пековые остатки.
Для увеличения выхода смолы и улучшения ее качества необходимо проводить процесс коксования так, чтобы избежать образования термически нестойких кислородсодержащих соединений еще на первой ступени термического разложения керогена сланца, что возможно лишь при ведении процесса в присутствии таких веществ, которые будут способствовать образованию не кислот и спиртов, а их производных, т. е. соединений, являющихся термически более стойкими.
Таким образом, только при рассмотрении химии сланцевой смолы, как химии кислородных соединений, и при глубоком изучении природы керогена сланца, как высокомолекулярного кислородного соединения, можно правильно подойти к вопросам термического разложения сланца и к методам переработки получаемых сланцевых смол.
ЛИТЕРАТУРА
1. П. К. К о г е р м а н. Химия эстонских сланцев. ОНТИ, Госхимтех-издат, 1934.
2. Ю. Ю. Хюссе. Состав и свойства высших фракций эстонской сланцевой смолы. Химия эстонских сланцев, сборник ОНТИ, Госхимтсх-издат, 1934
3. К. В а л ь д е к. Доклад на сессии АН ЭССР. Таллин, 1947.
4. Н. В. Ершов. ЖПХ, т. XII, № 6, стр. 867, 869 (1939).
5. П. К- К о г е р м а н и Н. Вей дерн асе. Sitz, berichte v. Natur-forscher gcsell. Tartu, m. 33, 1926.
6. А. С. Броун, А. С. Цейтлин. XTT, II, стр. 367, 1936.
7. Д. И. Андреевский, А. Ф. Че г и с. XTT, V, стр. 469, 1936.
8. Ю. Ю. Хюссе. Химическая характеристика высших фракций гене-ратортГой смолы Прибалтийского сланца. Тарту. Научная литература, 1949.
9. Н. И. Зеленин, О. С. Куратова. Труды Bcecoiosrforo научно-исследовательского института по переработке сланцев, ВНИИПС, вып. 1, стр. 57, 1948.
10. Р. Цейдлер Новые пути использования горючего сланца, ОН ГИ, стр. 54, 1934.
И. К. Луге. Термический распад керогена. Химия эстонских сланцев, ОНТИ, Гостоптехиздат, 1934.
12. Я. И. Хисин. Термическое разложение горючих сланцев, Гостоптехиздат, 1948.
48
И. И. Зеленин и О. С. Куратова
13. А. Ф. Доб'рянский. Горючие сланцы СССР. Ленгостоптехиздат, 1947.
14. Р. X. Оболенцев и Ю. Н. Усов. ДАН СССР, т. XXXI, № 3, 489, 1950.
15. Сабатье. Катализ в органической химии. Стр. 176, Госхимиздат, 1932.
16. Н. А. Орлов, О. А. Радченко. ХТТ, V, № 6, 506, 1931.
17. Н. М. Караваев, И. М. Венер Труды ИГИ, изд. АН СССР, т. II, 1950.
18. Ч. Д. Херд. Пиролиз соединений углерода. ОНТИ, 1938.
19. А. Я. Ларин, А. В. Ф[рост. ДАН СССР, т. IX, № 7, 1927, 1948.
20. К. Элли с. Химия углеводородов, т. I, ОНТИ, 1936.
21. А. В. Фрост и А. В. Очкин. Вестник МГУ, 2, № 5, 73, 1947.
22. А. Ф. Добрянский. Геохимия нефти, Гостоптехиздат, 1948.
23. А. В. Фрост. Роль глин в образовании нефтей в земной коре. «Успехи химии», XIV, 6, 501, 1945.
24. А. В. Фрост. Ученые записки МГУ. Труды кафедры физической .химии, вып. 1, стр. 2, 1946.
С. С. Семенов и Б. Е. Гуревич
ВЫДЕЛЕНИЕ НЕЙТРАЛЬНЫХ КИСЛОРОДНЫХ СОЕДИНЕНИЙ СРЕДНИХ ФРАКЦИЙ СЛАНЦЕВОЙ СМОЛЫ
Исследованиями, начатыми в 1946 г. и продолжающимися до настоящего времени, установлено, что смолы полукоксования, получаемые из прибалтийских сланцев на существующих промышленных агрегатах, больше чем наполовину состоят из соединений неуглеводородного характера. В состав смолы, кроме углеводородов, входят фенолы, кислоты, кетоны и другие соединения, содержащие кислород и серу. Во фракциях смолы содержание гетероатомных соединений возрастает с повышением температуры кипения фракции.
Углеводороды содержатся только в легких и средних фракциях. Остатки смолы, кипящие выше 350° С, практически полностью состоят из кислородных соединений различных функций. Эго значит, что все физические и химические свойства сланцевых смол и их дестиллатов определяются кислородными соединениями.
Необходимость познания химической природы этих соединений совершенно очевидна. Исследовательская работа в этом направлении представляет большой теоретический и практический интерес. Знание химического состава и свойств основных составляющих сланцевую смолу позволит сознательно подходить к выбору методов ее переработки на товарные продукты.
То обстоятельство, что фракции сланцевой смолы представляют собой очень сложные смеси углеводородов и гетероатомных соединений, делает задачу изучения их состава более трудной, чем изучение фракций нефти. Обесфеноленная дизельная фракция, кипящая в пределах 200—350° С, содержит до 25% нейтральных кислородных соединений. Они оказывают ощутимое влияние на свойства фракций, снижая ее топливные качества.
Химический состав и свойства нейтральных кислородных соединений сланцевой смолы к моменту начала нашей работы еще не подвергались систематическому изучению. Лишь в 1950 г.
4 Труды вниипс.
50
С. С Семенов и Б. Е. Гуревич
была опубликована работа В. А. Ланина и М. В. Прониной [1], давшая общие сведения о нейтральных кислородных соединениях средней фракции сланцевой смолы, без указания метода их выделения.
Для того чтобы изучать нейтральные кислородные соединения сланцевой смолы, необходимо получить их в виде концентрата, свободного от соединений углеводородного и кислого характера.
Метод отделения нейтральных кислородных соединений должен обеспечить наиболее полное извлечение их, получение концентрата, достаточно богатого ими, и, главное, выделение этих соединений в неизмененном виде.
Метод селективного растворения с применением растворителя, не вступающего в химическое взаимодействие с нейтральными кислородными соединениями, может обеспечить выполнение этих требований.
ОПЫТНАЯ ЧАСТЬ
Для получения концентрата нейтральных кислородных соединений нами использовалась обесфеноленная и необесфеноленная средняя фракция сланцевой смолы (табл. 1).
Таблица Z Характеристика исходной фракции
Контролируемые величины Показатели
Удельный вес при 20°С сырой фракции , . 0,9291
„ » „ 20°С обесфеноленной фракции 0,8975
Содержание фенолов, вес. % 18,7
Содержание нейтральных кислородных соединений (по FeCl3), объемн. % 27,7
Начало кипения, °C 178
Выкипает до 325°С, % 81
Конец кипения, °C . . . 353
В качестве избирательного растворителя для извлечения нейтральных кислородных соединений использовался метиловый спирт. Последний не вступает в химическое взаимодействие с соединениями, входящими в состав фракции, и как соединение, имеющее температуру кипения значительно более низкую, чем начало кипения фракции, легко и полностью регенерируется отгонкой. Метиловый спирт, как и другие испытанные нами растворители, растворяя в себе гетероатомные соединения, образует смесь, растворяющую в себе и углеводороды фракции. Следовательно, успешное решение поставленной задачи зависит от
Выделение нейтральных кислородных соединений
51
соответствующего подбора подходящих условий, обеспечивающих наибольшее извлечение нейтральных кислородных соединений при наименьшем растворении углеводородной части.
На процесс извлечения нейтральных кислородных соединений влияют концентрация спирта, содержание фенолов в разделяемой фракции, температура и количественное соотношение фракции и растворителя. Отдельно было изучено влияние каждого из факторов.
Опытами было показано, что применение метилового спирта различных концентраций дает возможность разделить фракцию
Сырье
Сырье
Сырье
Сырье
Сырье
Р3~1цикл f^-П цикл Р3~Шцикл
Р.-Шцикл Рецикл Р3~Ицикл
Рис. 1. Схема трехступенчатого псевдопротивотока.
на две части, резко отличающиеся по составу: одна из них, рафинат, представляет собой смесь углеводородов, а вторая — экстракт — является концентратом гетероатомных соединений.
Для получения концентрата нейтральных кислородных соединений из необесфеноленной дизельной фракции последняя обрабатывалась метиловым спиртом 95%-ной концентрации методом трехступенчатого псевдопротивотока, по схеме, показанной на рис. 1.
Такая обработка обеспечивает извлечение ~ 91 % всех нейтральных кислородных соединений, содержащихся в исходной фракции. Полученный при этом экстракт Э3 содержит 27 — 28% фенолов, что соответствует содержанию фенолов в исходной фракции, 64—65% нейтральных кислородных соединений и 7—8% углеводородов. Экстракт представляет собой раствор всех этих соединений в метиловом спирте. Если понизить растворяющую способность растворителя (метанола), понизив его концентрацию введением в экстракт воды, образуются два слоя, один из которых будет в главной своей массе состоять из углеводо
4*
52
С. С. Семенов и Б. Е. Гуревич
родов, а другой — из спирта, фенолов и нейтральных кислородных соединений. Соотношение компонентов будет различным, в зависимости от количества добавляемой воды. В результате такой операции можно было рассчитывать на большие абсолютные выходы концентратов нейтральных кислородных соединений и фенолов во вторичных экстрактах. Количество добавляемой воды вводилось из расчета доведения спирта до 92,5—90 и 85 %-ной концентрации.
Результаты выделения нейтральных кислородных соединений (н. к. с.) приводятся в табл. 2.
Таблица 2
Результаты опытов по выделению нейтральных кислородных соединений (н. к. с.) из средней фракции сланцевой смолы путем экстракции метиловым спиртом
Показатели
Уд. вес необесфеноленной фракции при 20° С.......................
То же, обесфеноленной фракции . Содержание фенолов, вес. % . . . Содержание и. к. с., объемн. % от обесфеноленной фракции . . .
Выход, вес % от исходной фракции
То же, объемн. %..............'
Выход обесфеноленного продукта, % от обесфеноленной фракции .
Извлечено фенолов, % от содержания их в исходной фракции . . .
Извлечено н.к. с., % от содержания их в исходной обесфеноленной фракции.........................
Вторичные экстракты, полученные путем добавления к Э3 воды в количестве, %
Исходная фракция Экстракт 1,6 э13 3,3 4,3 6,7
Э14 Э15 э16
0,9291 0,9846 0,9957 1,0054 1,0084 1,0124
0,8975 0,9379 0,9453 0,9562 0,9625 0,9864
18,7 34,7 36,5 40,5 42,1 45,3
27,7 62,0 70,0 85,0 91,0 100,0
100 53,0 45,6 37,8 36,8 31,7
100 50,0 42,4 36,7 33,6 28,6
100 40,6 33,8 25,9 24,6 19,2
100 100 99,4 82,0 82,8 76,8
100 90,8 85,4 79,5 80,7 69,3
Схематически процесс извлечения нейтральных кислородных соединений из необесфенбленной дизельной фракции представлен на рис. 2.
Для извлечения нейтральных кислородных соединений из обесфеноленной дизельной фракции применялся метиловый спирт 100%-ной концентрации, так как отсутствие фенолов во фракции
Выделение нейтральных кислородных соединений
53
Необесфено-ленна. я дизельная фракция Метанол
Обесфеноленный концентрат н. к. с.
Рис. 2. Схема извлечения нейтральных кислородных соединений из необесфеноленной дизельной фракции.
54
С. С. Семенов и Б. Е. Гуревич
не обеспечивало достаточной степени извлечения нейтральных кислородных соединений слабым спиртом.
Последовательной трехкратной обработкой фракции спиртом извлекаются все нейтральные кислородные соединения. Три полученных экстракта смешивались и разбавлялись водой, что приво-
Сырье 100 %-иетанол
I Г
Рис. 3. Схема выделения нейтральных кислородных соединений из обесфеноленной дизельной фракции.
дило К выделению углеводородной части в виде вторичного рафината, а получаемый при этом вторичный экстракт представлял собой 100%-ный концентрат нейтральных кислородных соединений (по FeCl3).
Схема выделения нейтральных кислородных соединений из обесфеноленной дизельной фракции представлена на рис. 3.
Выделение нейтральных кислородных соединений
55
Характеристика продуктов разделения обесфеноленной дизельной фракции приведена в табл. 3.
Таблица 3
Характеристика продуктов, полученных при разделении обесфеноленной дизельной фракции путем экстракции метиловым спиртом
№ п/п. Продукт 1 Выход, объемы. % Степень извле- Концентрация ! ч®ния и'к-с-2 нкс 2 1 % от содеР- объем'н."% ; жания их в /и исходном продукте
1 Обесфеноленная фрак-
ция 27,7 100
2 Р1 63,9 15,8 36,0
3 Эх 33,6 57,0 69,0
4 р2 45,8 6,2 10,3
5 Э2 17,2 49,1 30,2
6 Рз 32,0 следы
7 Э3 . 13,0 30,3 14,2
8 р4 45,1 28,6 46,5
9 э4 13,2 100,0 47,9
10 р5 28,0 22,3 22,6
И э5 13,5 65,8 32,0
12 Ре 15,8 11,5 6,5
13 э6 — — —
14 р, 9,8 7,0 2,5
15 э7 6,8 29,3 7,2
16 р8 . . . .. . . 19,8 35,6 25,5
17 э8 3,7 100,0 13,4
18 Пробы для анализа . . 16,5
19 Потери 3,0
Вторичные экстракты, полученные по схеме 1 или 2, освобождаются от метанола отгонкой последнего, после чего экстракт, полученный из необесфеноленной фракции, обесфеноливается. Таким образом получаются концентраты неизмененных нейтральных кислородных соединений. Характеристика полученного концентрата приводится в табл. 4. Наличие такого концентрата делает реальной возможность изучения химического состава и строения нейтральных кислородных соединений средней фракции • сланцевой смолы.
1 Р — рафинат; Э—экстракт.
2 н, к. с. — нейтральные кислородные соединения.
56
С. С. Семенов и Б. Е. Гуревич
Таблица 4
Характеристика полученного концентрата нейтральных кислородных соединений
Контролируемая величина
Показатели
Удельный вес при 20° С.....................
Показатель преломления при 20° С . . . . . .
Молекулярный вес.................
Содержание нейтральных кислородных соединений (по ЕеС13), % ......................
Вязкость при 20° С ест.....................
Элементарный состав, Оо: С .................... .... .............
н ....................................
S.....................................
N + О ................................
Содержание, %:
гидроксилсодержащих соединений . . . . карбонилсодержащих , . . . .
альдегидов (по методу Пондорфа) .... афиров (омыляемых веществ)............
0,9646 1,5210
175
100,0
11,1
79,51
9,65 1,44
9,40
21,5
49,7
21,0
11,2
ВЫВОДЫ
1. Разработан метод получения концентрата нейтральных кислородных соединений из обесфеноленной и необесфеноленной дизельной фракции сланцевой смолы.
2. Метод обеспечивает возможность выделения нейтральных кислородных соединений в неизмененном виде и, следовательно, возможность изучения химической природы и свойств этих важных компонентов сланцевой смолы.
ЛИТЕРАТУРА
1. В. А. Ланин, и М. В. Пронина. ДАН СССР, т. XXIV, № 1, 115, 1950.
С. С. Семенов и Б. Е. Гуревич
ИЗУЧЕНИЕ НЕЙТРАЛЬНЫХ КИСЛОРОДНЫХ СОЕДИНЕНИЙ СРЕДНИХ ФРАКЦИЙ СЛАНЦЕВОЙ СМОЛЫ
Кислородные соединения нейтральных функций, выделяемые методом селективного растворения из средних фракций сланцевой смолы, 1 представляют собой сложную смесь соединений, содержащих карбонильную, гидроксильную и другие функциональные группировки. Для более глубокого изучения нейтральные кислородные соединения из дизельной фракции смолы полукоксования прибалтийских сланцев, содержащие 79,51 % углерода, 9,65% водорода, 1,44% серы, 9,40% кислорода и азота и имеющие молекулярный вес 175, были разогнаны в вакууме (при остаточном давлении 10 мм рт. ст.) на 5-градусные фракции. Разгонка производилась из стеклянной колбы с ректификационной колонкой, имеющей погоноразделительную способность около 23 теоретических тарелок. Некоторые свойства и групповой состав полученных фракций приведены в табл. 1.
Данные группового химического состава узких фракций показывают, что карбонилсодержащие соединения в них представлены в наибольших количествах, в сравнении с другими соединениями.
Обращают на себя внимание данные по содержанию альдегидов во всех фракциях, определяемых методом Пондорфа, причем количество альдегидов повышается с повышением температуры кипения фракций. Имеются суждения о том, что примененный нами метод определения альдегидов недостаточно специфичен, вследствие чего аналитические данные, свидетельствующие о высоком содержании альдегидов в составе нейтральных кислородных соединений сланцевой смолы, могли быть получены за счет других соединений, не содержащих альдегидной группы. Не имея других данных, полученных более точными методами, мы воздерживаемся от возражений против указанных суждений формального характера, но считаем необходимым указать, что
1 См. статью С. С. Семенова и Б. Е. Гуревич: «Выделение нейтральных кислородных соединений из средних фракций сланцевой смолы», стр. 49.
58
С. С. Семенов и Б. Е. Гуревич
Таблица 1
Свойства и групповой химический состав 5-градусных фракций нейтральных кислородных соединений (н. к. с.)
1 № фракции 1 Температура ки- 1 пения фракций при остаточном 1 давлении 10 мм.аС Удельный вес при 20° С Показатель преломления при 20° С Групповой состав, вес. %
Сумма альдегидов и кетонов Альдегиды Эфиры (омыляемые вещества) Гидроксилсодержащие соединения
1 До 65 0,8758 1,4628 64,0 11,0 3,9 0,0
2 65-70 0,8892 1,4710 104,5 16,0 8,3 0,0
3 70-75 0,8921 1,4755 62,3 9,0 4,7 0,0
4 75—80 0,9023 1,4879 78,1 13,0 9,5 1,4
5 80-85 0,9184 1,5070 63,5 6,0 4,5 1,2
6 85-90 0,9308 1,5141 61,5 6,0 3,1 3,0
7 90—95 0,9152 1,5013 52,2 10,0 4,2 1,7
8 95—100 0,9101 1,4988 53,2 15,0 4,5 3,0
9 100—105 0,9192 1,5052 58,9 15,0 3,8 1,3
10 105—110 0,9221 1,5056 63,1 16.0 3.3 9,9
11 110—115 0,9260 1,5076 60,4 18,0 3,1 14,2
12 115—120 0,9314 1,5108 42,2 19,0 3,5 16,5
13 120—125 0,9341 1,5127 44,9 16,0 2,2 8,1
14 125-130 0,9408 1,5151 62,5 20,0 2,8 15,7
15 130—135 0,9475 1,5180 66,0 22,0 2,8 21,5
16 135—140 0,9511 1,5200 47,0 26,0 3,7 25,3
17 140—145 0,9564 1,5222 39,0 28,0 3,5 25,7
18 145—150 0,9634 1,5268 37,8 30,0 4,1 25,2
19 150—155 0,9699 1,5300 37,2 34,0 5.8 27,9
20 Исходные
н. к. с. 0,9626 1,5210 49,0 21,0 11,5 21,5
Б. И. Ивановым и И. Ф. Шароновой’ реально выделены из подсмольной воды и идентифицированы уксусный и пропионовый альдегиды. Этот факт строго доказанного присутствия альдегидов в продуктах термического разложения тех же прибалтийских сланцев позволяет предполагать наличие альдегидов и в более высококипящих фракциях сланцевой смолы.
Общее содержание карбонильных соединений определялось фенилгидразиновым (по Штрахе) и гидроксиламиновым методами, причем оба метода давали достаточно хорошо сходящиеся результаты. Гидроксилсодержащие соединения определялись методом ацетилирования, а эфирообразные вещества рассчитывались по числам омыления.
Следует заметить, что при разгонке кислородных соединений, значительная часть их заполимеризовалась, о чем свидетель-
См. наст, сб., стр. 164.
Изучение нейтральных кислородных соединений
59
ствует остаток в колбе в количестве 37% от веса взятых на раз-гонку н. к. с. Остаток представлял собой черную массу с вязкостью при 20° С 106 сст, удельным весом 1,0449 и элементарным составом С — 80,26 %, Н — 8,73 %.
При разгонке рафинатной части той же дизельной фракции, из которой предварительно была удалена основная масса нейтральных кислородных соединений, в тех же условиях остаток в колбе составил 23,4%. Остаток представлял собой черную жидкость с вязкостью при 20° С 58,90 сст, удельным весом 0,9549 и элементарным составом С — 83,74%, Н — 10,60%.
Учитывая наличие больших количеств карбонилсодержащих соединений в н. к. с., целесообразно было подвергнуть их более детальному исследованию. С этой целью суммарные (исходные) кислородные соединения были разогнаны на одноградусные фракции в вакууме при остаточном давлении 5 мм рт. ст. Раз-гонка производилась из стеклянной колбы с ректификационной колонкой, имеющей эффективность около 50 теоретических тарелок. Полученные 102 фракции были проверены на выкипание при атмосферном давлении, причем оказалось, что они полностью выкипают в пределах 3—4°.
Во всех полученных фракциях были определены удельный вес, показатель преломления и молекулярный вес. Кроме того, в большинстве фракций определена сумма карбонильных соединений, элементарный состав и др. Содержание карбонильных соединений не падало ниже 43%, а в отдельных фракциях доходило до 84%.
Многие фракции при охлаждении до температуры минус 40— 70° С частично или в преобладающей своей массе кристаллизовались.
Содержание кислорода во фракциях колебалось от 4 до 14%.
В более низкокипящих фракциях оно было максимальным и постепенно падало с повышением температуры кипения фракции.
В табл. 2 приведены данные анализов для ряда фракций, полученных при разгонке н. к. с.
Наиболее трудным вопросом в работе было отделение карбонилсодержащих веществ от прочих соединений, находящихся в смеси, особенно при получении индивидуальных кетонов. Естественно, этому вопросу было уделено наибольшее внимание.
Для выделения карбонилсодержащих соединений из смеси были использованы два метода, краткое описание которых приводится ниже.
1. Метод кристаллизации. Выделение методом кристаллизации при низких температурах проводилось в специальном приборе (рис. 1), позволяющем производить кристаллизацию и отфильтровывание кристаллов от жидкости при неизменной низкой температуре Поскольку при охлаждении, как правило, резко
С. С. Семенов и Б. Е. Гуревич
Таблица 2
Характеристика узких фракций нейтральных кислородных соединений
1 № фракции | Удельный вес при 20° С Показатель преломления при 20° С Молекулярный вес Элементарный состав, вес. % Содержание карбонильных соединений, вес. % Температура кипения фракции, °C
С Н S O + N при"оста-точном давлении 10 и 5-и.ирт. ст. при атмосферном давлении
2 0,8643 1,4430 110,0 75,17 10,88 2,45 11,49 33-34710
7 0,8473 1,4310 114,0 72,85 11,17 2,02 13,96 82,8 38—39/10 147—151
12 0,8752 1,4530 115,0 76,86 10,88 3,77 8,49 67,8 43—44/10 151—156
18 0,8757 1,4600 121,0 77,69 10,85 2,52 8,94 61,8 49—50/10 159—163
22 0,8778 1,4595 124,0 79,03 10,94 1,06 8,97 84,3 53-54/10 165—169
32 0,8803 1,4690 134,0 81,00 10,89 1,91 6,20 64,9 63—64/10 179—183
37 0,8818 1,4686 — 77,90 10,81 1,79 9,50 53,1 51—52/5 181—186
42 0,8921 1,4686 — 80,33 10,89 1,40 7,38 — 56—57/5 188—193
48 0,8954 1,4740 142,0 78,65 10,30 1,26 9,79 — 62—63/5 192—199
55 0,9030 1,4910 146,0 81,67 10,75 1,77 5,81 42,8 69—70/5 203—207
57 0,9029 1,4848 145,0 80,73 10,44 1,98 6,85 55,0 71—72/5 207—209
62 0,9097 1,4884 — 82.21 10,52 2,02 5,25 — 76—77/5 211-215
69 0,9019 1,4908 —— 82,24 10,65 1,60 5,51 — 83-84/5 221—226
73 0,9117 1,5002 — 82,52 10,42 1,74 5,02 — 87—88/5 226—229
77 0,9233 1,5058 — 83,41 9,79 1,73 5,07 — 91—92/5 232—236
83 0,9224 1,5025 —. 82,96 10,40 1,63 5,01 97—98/5 241—245
87 0,9300 1,5090 — 82,60 10,31 1,82 5,27 101—102/5 246-250
97 0,9419 1,5135 186,0 i 83,40 10,81 1.58 4,21 — 111—112/5 256—263
повышалась вязкость фракций, вследствие чего затруднялось фильтрование, они растворялись предварительно в сухом этиловом или нефтяном эфире. В этом случае фильтрация проходила нормально, и достигалось наиболее полное отделение некристал-лизующейся части фракции от кристаллов. Двухкратная перекристаллизация обеспечивала получение чистых кристаллов, представляющих собой кетоны жирного ряда. Несмотря на простоту и удобство этого метода, следует отметить и большой его недостаток, состоящий в больших потерях карбонильных соединений.
2. Семикарбазидный метод. Для наиболее полного выделения карбонильных соединений из фракций применялся метод обработки всей фракции водным раствором солянокислого семикарбазида. При такой обработке карбонильные соединения вступали в реакцию с семикЯрбазидом с образованием семикарбазонов, частично выделяющихся в кристаллическом состоянии, частично растворяющихся в некарбонильной части кислородных соедине-
Изучение нейтральных кислородных соединений
61
ний. Кристаллические семикарбазоны после отфильтровывания и промывки охлажденным нефтяным эфиром уже не содержали маслянистой части, а семикарбазоны, находящиеся в растворе, выделялись путем отгонки маслянистых продуктов в вакууме при остаточном давлении 5 мм рт. ст.
Отгонка маслянистой части производилась до появления кристаллов семикарбазонов на горлышке отгонной колбы и в отводной трубке. В отгонной колбе оставались семикарбазоны с примесью маслянистой части, освобождение от которой производилось путем промывки холодным Hei Полученные семикарбазоны разлагались 5— 10%-ным раствором соляной кислоты; при этом карбонильные соединения выделялись в более чистом виде, а семикарбазид растворялся в воде.
Полученные карбонильные соединения подвергались исследованию.
Изучение карбонильных соединений из фракции № 7. Фракция № 7 была разделена на карбонильные соединения и остаточную часть методом кристаллизации; при этом были выделены кетоны общей формулы С7Н14О и промежуточные продукты. Произведенными анализами было установлено, что выделенное карбонилсодержащее соединение представляет собой индивидуальный кетон. Из 15 изомеров кетонов состава С7Н)4О только два кипят при температуре,
ния выделенного кетона: этил-н-бутилкетон, кипящий при 149— 150° С, и метил-н-амилкетон, кипящий при 151,45° С. Все остальные изомеры кипят значительно ниже. В табл. 3 приводится сравнение констант выделенного кетона с литературными и теоретическими данными для указанных двух изомеров.
На основании температур плавления чистого кетона и его производных (семикарбазона и 2,4-динитрофенилгидразона), а также других констант, можно утверждать, что выделенный кетон представляет собой метил-н-амилкетон.
Если сравнить теоретическую молекулярную рефракцию с подсчитанной по удельному весу и показателю преломления, оказывается, что выделенный кетон ближе отвечает теории, чем
Рис. 1. Прибор для кристаллизации и фильтрования при низких температурах: а — фильтрация,
б — кристаллизация.
1 — охлаждающий сосуд; 2 — внешняя пробирка; 3 — внутренняя пробирка; 4 — пробка; 5—фильтр с пористой пластинкой.
кипе-
близкой к
62
С. С. Семенов и Б. Е. Гуревич
кетон, описанный в литературе. Это дает основание предполагать, что имеющиеся в литературе данные по удельному весу и показателю преломления метил-н-амилкетона являются неточными.
Таблица 3
Характеристика кетона С7Н14О, выделенного из фракции № 7 н. к. с. сланцевой смолы и соответствующие литературные данные для метил-н-амилкетона и этил-н-бутилкетона
я я . а ч с<5 Я О А,
Я CU я с» Я-
5 я. о s « я Я СО £ сх з
Показатели а а *? я ес Я Он®
JTOH, 1Й из 7 н. 4^5? s я а н о я о н а, :ил-н-н (ли ie ДЭ1
А я о >> < а н Э1 то нь
Удельный вес d 20/4 0,8286 0,8196
Показатель преломления при 20° С . 1.4225 1,4143
Молекулярный вес 114 114
Элементарный состав, вес. %:
С . 73,86 73,68
Н 12,23 12,28
О 13,91 14,03
Молекулярная рефракция ..... 34,90 (37,1)1 34,54
Температура кипения при 760 мм, °C 149—151,5 151,45 149—150
Температура плавления, °C .... —34—35 —35,5 —
Температура плавления семикарба-
зона, °C . 120 120—123 99—100 (111)
Температура плавления динитрофе-
нилгидразона, °C 87 89 —
Изучение карбонилсодержащих соединений фракций н.к. с. № 14—22. Фракции н.к. с. от № 14 до № 22 были смешаны вместе и обработаны водным раствором солянокислого семикарбазида в присутствии уксуснокислого натрия для извлечения карбонилсодержащих соединений в виде семикарбазонов. После отделения бескарбонильной части путем промывки нефтяным эфиром и отгонки в вакууме полученные семикарбазоны были разложены соляной кислотой; при этом были получены кетоны, имеющие характеристику, приведенную в табл. 4.
Из приведенных данных видно, что выделенные кетоны представляют собой смесь кетонов С7 и Сз. Удельный вес и показа-
1 Молекулярная рефракция, подсчитанная по удельному весу и показателю преломления.
Изучение нейтральных кислородных соединений
63
Таблица 4
Характеристика кетонов, выделенных из фракций н. к. с. № 14—22
Показатели Кетоны из кристаллической части семикарбазонов Кетоны, растворенные в бескарбонильной части фракции
Удельный вес при 20° С 0,8639 0,8732
Показатель преломления при 20° С . 1,4326 1,4354
Молекулярный вес ' 121 122
Температура плавления семикарбазонов,°C 84—85 75—80
Температура плавления 2,4-динитро-фенилгидразонов, °C 50—52 —
тель преломления позволяют предположить наличие в них как жирных, так и циклических кетонов, вероятнее всего метилциклогексанов. Не исключена возможность присутствия и непредельных кетонов с открытой цепью.
О наличии непредельных кетонов можно судить и по тому, что последние при обработке солянокислым семикарбазидом присоединяют две молекулы семикарбазида с образованием семикарбазидсемикарбазона. Семикарбазидсемикарбазоны присоединяют молекулу кислоты и поэтому хорошо растворимы в воде. При подщелачивании водного раствора семикарбазидсемикарбазоны выпадают в осадок в виде мелких белых кристаллов. Такое явление наблюдалось почти во всех случаях обработки фракций кислородных соединений водным раствором семикарбазида.
При окислении кетонов йодом в щелочной среде, наряду с предельными кислотами, получались оксикислоты, что также подтверждает наличие непредельных кетонов в составе н. к. с. сланцевой смолы.
Изучение к арб они л с одерж ащих соединений фракций н. к. с. № 23—26, 30—31, 45—46, 57—60, 76, 97— 102 и других. Отделение карбонилсодержащих соединений от бескарбонильной части производилось вышеописанными методами; при этом получены кетоны, физико-химические свойства которых приведены в табл. 5, 6, 7, 8 и 9.
Данные табл. 5 позволяют утверждать, что выделенный из фракции 23—26 кетон по своим константам полностью отвечает метил-н-гексилкетону.
В свою очередь данные, помещенные в табл. 6, соответствуют циклическому кетону 2-, 4-диметилциклогексанону. В этой же таблице помещены данные для смеси кетонов, которые, вероятно,.
С. С. Семенов и Б. Е. Гуревич
Таблица 5
Характеристика кетона из фракций н. к. с. № 23—26
Кетон, С8Н16О, Метил-н-гексил-
Показатели выделенный из кетон (литера-
сланцевых н. к. с. турные данные
Удельный вес при 20° С ..... . 0,8213 0,8202
Показатель преломления при 20°С . 1,4112 1,4112
Молекулярный вес 128 128
Температура плавления, °C .... —18,5 —16
„ кипения, °C 70,1—70,4/20 мм 172,9/760 мм
56—60/11 мм
Молекулярная рефракция 39,05 38,96 (38,8)1
Поверхностное натяжение при 20° С 26,99 —
Парахор 356,6 355,2
Температура плавления семикарба-
зона, °C 118 122
Температура плавления 2,4-динитро-
фенилгидразона, °C 57—58 58
Элементарный состав, вес. %:
С 75,00 75.00
Н 12,63 12,50
О 12,37 12,50
Таблица 6
Характеристика кетонов, выделенных из фракции н. к. с. №30—31
Показатели Кетон, выделенный из сланцевых н. к. с. 2,4-диметилциклогексанон (литературные данные) Изомеры (литературные данные)
цис-изомер транс- изомер
Удельный вес при 20° С . . . 0,9006 0,9004‘в 0,8785
Показатель преломления при
20° С 1,4426 1,44291в 1,4398
Молекулярный вес .... 126 126 129
Температура кипения, °C . . 58/9 мм 177 171 59-61/8 мм
Температура плавления семи-
карбазона, °C 164—166 190 136 166—8
Температура плавления 2,4- 102-108
динитрофенилгидразона . . 136-8 — —-
Элементарный состав, вес. %: 76,57
С 76,27 76,19
н 11,40 11.11 12,05
о 12,33 12,70 11,38
1 Молекулярная рефракция, подсчитанная по показателю преломления и удельному весу.
Таблица ?
Труды ВШПШС.
Характеристика кетонов, выделенных из фракций н. к. с. №45—46
Показатели Кетон С9Н18О, вы-деленный из сланцевых и. к. с. Метил-н-геп-тилкетоп (литературные данные) Изомеры
Фракция 61,2—62°С/5 мм Фракция 62—65° С/5 мм
Удельный вес при 20° С 0,8272 0,8188 (22/4) 0,8540 0,8617
Показатель преломления при 20° С 1,4248 1,4175 при 22° 1,4396 1,4432
Молекулярный вес 142 142 140 140
Температура плавления, °C -11 —15 -25,-24 -26,-25
„ кипения, °C 62,2—62,5/5 мм 194—196/760 .ил 75-77/12 мм 61,2—62,0/5 лл 62,0 - 65,0/5 мм
Молекулярная рефракция . 44,11 43,96 (43,6) 1 —
Поверхностное натяжение 27,47 — — —
Парахор 393,0 394,2 — —
Температура плавления семикарбазопа, °C Элементарный состав, вес. %: 120 119-120 116—117 111—117
С .... . ... 76,55 76,06 76,74 76,43
и 12,60 12,67 11,72 11,52
о . 10,85 11,27 11,54 12,05
Температура плавления 2,4-дипитрофеиилгидразопа, °C . . — — 190
1 Молекулярная рефракция, подсчитанная по показателю преломления и удельному весу.
Таблица 8
Характеристика кетонов, выделенных из фракций н. к. с. № 57—60
Показатели Кетон с10н20о. выделенный из сланцевых II. к. с. Метил-н-ок-тилкетон (литературные данные) Изомеры
Фракция 74,5—15,5°/Ьмм Фракция 75,5—80°/5 мм
Удельный вес при 20° С 0,8268 0,8230 0,8693 0,8771
Показатель преломления при 20°С 1,4272 1,4263 при 22° 1,4478 1,4498
Молекулярный вес . 156 156 154 154
Температура плавления, °C +3 + 2,5 —14—13 — 16—14
„ кипения, °C 74,2/5 мм 209/750 мм\ 95—7/12 мм — —
Молекулярная рефракция 48,36 48,60 (48,90)1 — —
Поверхностное натяжение 27,85 — — —
Парахор . . ..... ... 432,65 433,2 — —
Температура плавления семикарбазона 125—126 (122) 126(121) 123—124 118—121
„ р-нитрофенил гидразона 98,0 — — —
„ 2,4-динитрофенилгидразона Элементарный состав, вес. %: — — — 72
С 76,87 76,92 76,67 76,15
Н 12,78 12,82 11,53 11,52
О 10,35 10,26 11,80 12,33
1 Молекулярная рефракция, подсчитанная по показателю преломления и удельному весу,
Изучение нейтральных кислородных соединений
67
Таблица 9
Характеристика кетона, выделенного из фракций н. к. с. № 97—102
Показатели
Показатель преломления прн 100° С Молекулярный вес.................
Температура плавления, °C . . . . Температура кипения, °C: ........
Температура плавления, °C:
семикарбазона ...............
2, 4-динитрофенилгидразона оксима ......................
фенилгидразона ..............
производного бензальдегида . . Элементарный состав, вес. %:
С ...........................
Н ...........................
О............................
Метилгидриндон, выделенный из сланцевых н. к. с. 4-метил-1 -гидрин-дон (литературные данные)
1,5357
146 146
98 95
255-7 —
164—8 1 —
274 1 —
158 —
132 1 132 1
137—8 —
82,27 82,19
7,04 6,85
10,69 10,96
представляют собой смесь циклических кетонов с жирными предельными и непредельными кетонами, имеющими от 8 до 9 атомов углерода в молекуле.
В табл. 7 помещены данные для индивидуализированного кетона, метил-н-гептилкетона и его изомеров.
В таб I. 8 приводятся данные для кетона, выделенного из н. к. с., метил-н-октилкетона и изомеров, состоящих, повидимому, из смеси жирных предельных и непредельных с циклическими ди-и триметилциклогексанонами.
В табл. 9 приводятся данные для кетона, выделенного из н. к. с. (фракций 97—102), соответствующие данным для 4-ме-тил-1-гидриндона, имеющего бициклическую структуру
СН3 СН
\Х \
С С-СН2
I И I
СН с сн2
X /\/
сн со
При выделении кетонов из фракций 97—102 был получен кетон с температурой плавления 27° С. Судя по температуре кипения фракции, характеру кетона и его температуре плавления можно предполагать, что это — метил-н-ундецилкетон.
1 Плавится с разложением.
5*
68
С. С. Семенов и Б. Е. Гуревич
При выделении кетонов из фракций 51—52 было получено ограниченное количество кетона, который по всем признакам и температуре плавления (20° С) отвечал ацетофенону. В этих же фракциях установлено наличие мета-метилацетофенона.1
При окислении кетонов, выделенных из фракций 75 и 76 йодом в щелочной среде, были обнаружены декановая и ундекановая кислоты, что говорит о присутствии в н. к. с. этих фракций метилнонил- и метилдецилкетонов.
ВЫВОДЫ
1. При исследовании карбонилсодержащих соединений сланцевой смолы выделены в чистом виде и охарактеризованы соответствующими данными: а) метил-н-амилкетон; б) метил-н-гексил-кетон; в) метил-н-гептилкетон; г) метил-н-октилкетон; д) 2,4-диметилциклогексанон; е) 4-метил-1-гидриндон.
2. При окислении йодом в щелочной среде смесей кетонов, выделенных из фракций нейтральных кислородных соединений сланцевой смолы, получены декановая, .ундекановая и метатолуиловая кислоты, что доказывает наличие в сланцевой смоле нонил- и децилкетонов и метаметилацетофенона. Не исключена также возможность присутствия пара- и ортоацетофенона.
3. Качественно установлено присутствие ацетофенона (температура плавления 20° С) и метилундецилкетона (температура плавления 27° С).
4. Выделены изомеры кетонов, состоящие из смеси предельных и непредельных, жирных и циклических кетонов. Непредельные кетоны почти всех фракций образуют семикарбазидсемикарбазоны, а при окислении йодом в щелочной среде дают оксикислоты.
1 См. статью С. С. Семенова, Ю. И. Корниловой и Б. Е. Гуревич: «Об окислении некоторых кетонов йодом в щелочной среде», стр. 69.
С. С. Семенов, Ю. И. Корнилова и Б. Е. Гуревич
ОБ ОКИСЛЕНИИ НЕКОТОРЫХ КЕТОНОВ ЙОДОМ В ЩЕЛОЧНОЙ СРЕДЕ
Одним из наиболее распространенных методов доказательства строения органических соединений является метод окисления.
Как было показано многими исследователями, механизм реакции окисления зависит не только от строения окисляемого вещества, но также и от окислителя. Кроме этого, следует отметить, что при употреблении одного и того же окислителя, путем создания различных условий, реакцию окисления можно остановить на различной стадии. Так, например, при окислении ацетофенона с помощью марганцовокислого калия, изменяя температуру, можно получить различные продукты: при пониженной температуре в разбавленных растворах образуется бензоилмуравьиная кислота, в то время как при повышенной температуре получается бензойная кислота.
Наиболее часто применяемыми окислителями для органических соединений являются марганцовокислый калий и хромовая смесь. По своему окисляющему действию они несколько отличаются друг от друга: марганцовокислый калий избирательно легче действует на карбонильные и карбинольные группы.
Кроме приведенных окислителей, в последнее время широкое применение находят и другие, как, например, селенистый ангидрид, тетраацетат свинца, перекись водорода, галогены и др.
В настоящем исследовании рассматривается окисление некоторых карбонильных соединений с помощью йода в щелочной среде.
Из литературных данных [1] известно, что при обработке метилкетонов раствором йода в щелочной среде наблюдается образование йодоформа (с количественным выходом) и соли кислоты, содержащей на единицу меньшее число углеродных атомов, чем исходный метилкетон.
Следует отметить, что данная реакция протекает как с метил-кетонами алифатического ряда, так и с метиларилкетонами.
70
С. С. Семенов, /О. И. Корнилов и и Б. Е. Гуревич
Химизм реакции иллюстрируется следующим итоговым уравнением:
R—CO-CHL + 6J + 4КОН -> CHJ3 + R-COOK + ЗК J + ЗН2О
В первой фазе реакции образуется йодюр следующего строения:
R—СО-СН3 + 6J 4- ЗКОН R—СО—CJ3 + 3KJ + ЗН2О
Во второй фазе йодюр распадается на йодоформ и соль кислоты :
R—СО—С J3 + КОН -> R—COOK + CHJ3
Для полного окисления метилкетона в соответствующую соль кислоты необходимо брать избыток йода, который в щелочной среде реагирует следующим образом:
2J + КОН -»КJO 4- КJ + Н2О
При подкислении выделяется свободный йод:
К JO 4- К J 4- H2SO4 K2SO4 + Н2О 4- 2J
За последнее время появились сообщения о том Г2|, что йод в щелочной среде может действовать окисляющим образом не только на метилкетоны, но также и на альдегиды непредельного строения.
Следует отметить, что авторы указанных сообщений рекомендуют окисление йодом в щелочной среде как количественный метод определения непредельных альдегидов. По их данным, чувствительность его превышает чувствительность бисульфит-ного и гидроксиламинового методов.
Кроме приведенных выше реакций, следует отметить, что йод действует также окисляющим образом на моносахариды [3], так, например, глюкоза под действием йода в щелочной среде окисляется до глюконовой кислоты.
Приведенные выше данные в достаточной степени убедительно показывают, что йод в щелочной среде с успехом может применяться в качестве окислителя для карбонильных соединений.
Данная реакция представляет интерес еще и в том смысле, что окисление с помощью йода в щелочной среде может служить методом разделения смеси кетонов, содержащих, наряду с ме-тилкетонами, кетоны другого строения.
Основываясь на приведенных выше данных, была сделана попытка осуществить разделение карбонильных соединений, выделенных из некоторых фракций сланцевой смолы.
Об окислении кетонов йодом в щелочной фазе
71
Прежде чем перейти к непосредственному исследованию карбонильных соединений, выделенных из сланцевой смолы, представлялось интересным произвести предварительные опыты по окислению карбонильных соединений известного строения. Для этого были взяты ранее полученные в . нашей лаборатории алифатические метилкетоны: метилгексилкетон, метилгептилкетон и метилоктилкетон.
В результате окисления метилгексилкетона была получена энантовая кислота:
СН3-СН2—СН2—СН2-СН2—СН2-СО-СН3-к^2н-
-> СН3-СН2 СН2 -СН2—СН2—СН2-СООК + CHJ3
Полученная энантовая кислота была идентифицирована путем титрования. Йодоформ идентифицировался во всех опытах по температуре плавления и на основании температуры плавления пробы смешения.
В результате окисления метилгептилкетона была получена каприловая кислота.
Реакция может быть представлена следующей схемой:
СН,-СН2—СН2—СН2-СН2-СН2—СН2-СО—СНз-jT^
-э СН3—СН2-СН2-СН2—СН, -СН, -СН,—COOK + СНJ3
Полученная каприловая кислота была идентифицирована аналогично энантовой кислоте.
При окислении фракции, в которой предполагалось наличие метилоктилкетона, были получены две кислоты: нониловая и декановая. Полученные кислоты, помимо титрования, были идентифицированы по температуре плавления. Наличие декановой кислоты может служить подтверждением того, что фракция метил-нонилкетона содержит примесь метилдецилкетона.
После изучения реакции окисления алифатических метилке-тонов с помощью йода в щелочной среде было интересно произвести окисление жирноароматического кетона в тех же условиях. Для этого был выбран простейший жирноароматический кетон — ацетофенон.
В результате окисления были получены бензойная кислота и йодоформ по реакции:
C6Hs-CO-CH31<4h C6H5COOK + CHJ3
Доказав в результате изложенного выше исследования, что в примененных нами условиях как алифатические, так и жирноароматические метилкетоны под влиянием йода в щелочной
72
С. С. Семенов, Ю. И. Корнилова и Б. Е. Гуревич
среде окисляются до карбоновых кислот, мы перешли к окислению фракций карбонильных соединений, представляющих собой смеси метилкетонов с кетонами другого строения.
В качестве исходных продуктов были использованы карбонильные соединения, полученные из одноградусных фракций нейтральных кислородных соединений (н. к. с.). Фракции н. к. с. были обработаны семикарбазидом с целью отделения карбонильных соединений от других нейтральных кислородных соединений, а из полученных семикарбазонов были затем обратно выделены свободные карбонильные соединения.
Полученные карбонильные соединения были охарактеризованы до обработки их йодом в щелочной среде. Были определены удельный вес, молекулярный вес, показатель преломления, содержание карбонильных соединений и, в некоторых случаях,, элементарный состав.
В результате описанной выше подготовительной работы было получено семнадцать образцов карбонильных соединений, характеристика которых приводится в табл. 1.
Таблица 1 Характеристика исходных карбонильных соединений, выделенных из одноградусных фракций нейтральных кислородных соединений сланцевой смолы
№ фракций Температура кипения фракции н.к.с, из которой выделены карбонильные соединения, °C Температура кипения выделенных карбонильных соединений, °C м 20 2» nD
1 73,7—75,5, р = 5 мм 149 0,8428 1,4365
2 — 101-103, р^-_ 3,5 „ 180 0,8900 1,4680
3 — 54,5—54,8, р= 9 „ 120 0,8658 1,4329
4 — 54,8-56,0, р= 9 „ 122 0,8615 1,4315
5 — 60,0—64,0, р= 6 , 137 0,8562 1,4346
6 197 — 140 0,9020 1,4642
7 197 — 143 0,9367 1,5006
8 199 — 137 0,9228 1,4986
9 199 140 0,9019 1,4646
10 200 — 143 0,9205 1.4982
11 200 147 0,9009 1,4642
12 200 .— 133 0,9432 1,5042
13 202 — 147 0,9108 1,4924
14 202 — 144 0,8933 1,4606
15 203 151 0,8324 1,4250
16 229 — 159 0,8954 1,4846
17 229 — 164 0,8862 1,4672
Все выделенные фракции представляли собой смесь карбонильных соединений, которые реагировали на холоде с марганцовокислым калием и бромной водой, что может служить под,-
Об окислении кетонов йодом в щелочной фазе
73
тверждением присутствия в них соединений с ненасыщенными связями. Кроме этого, можно было предполагать, что, наряду с метилкетонами, в них могли содержаться и неметилированные соединения.
Все описанные выше фракции с целью выделения из них ме-тилкетонов были подвергнуты обработке йодом в щелочной среде.
Опыты проводились в трехгорлой колбе с механическим размешиванием при комнатной температуре.
Небольшое количество (1—2 г) карбонильных соединений загружалось в трехгорлую колбу и растворялось в нескольких миллилитрах метилового спирта, после чего добавлялось небольшое количество щелочи. Далее к реакционной смеси из капельной воронки приливался по каплям раствор йода в йодистом калии, йод приливался всегда с избытком, примерно в количестве 10 г. По мере добавления йода в растворе происходило образование желтого осадка — йодоформа. На протяжении всего опыта в реакционной колбе поддерживалась щелочная реакция.
По окончании приливания йода реакционная смесь 'отфильтровывалась от осадка йодоформа. Далее щелочной раствор тщательно обрабатывался эфиром с. целью удаления оставшихся нейтральных продуктов. После отделения нейтральных продуктов к раствору добавлялась концентрированная соляная кислота до минерально-кислой реакции по конго; при этом соль органической кислоты разлагалась с образованием свободной кислоты, и одновременно происходило выделение свободного йода.
Затем выделившийся йод связывался тиосульфатом, а органическая кислота извлекалась эфиром. Эфирная вытяжка сушилась свежепрокаленным сульфатом натрия, после чего эфир отгонялся, а оставшиеся кислоты высушивались в вакуум-эксика-торе и анализировались.
Нейтральные продукты, оставшиеся после реакции окисления йодом, были охарактеризованы, причем были определены их удельный и молекулярный веса и показатель преломления.
Следует отметить, что молекулярный вес оставшихся нейтральных продуктов, как правило, был выше молекулярного веса соответствующих исходных карбонильных соединений, В дальнейшем было установлено, что повышение молекулярного веса нейтральных продуктов объясняется присутствием в последних йода. К настоящему времени еще не удалось установить, чго представляет собой эта примесь йода — йод йодоформа, частично растворенного в нейтральных продуктах, или йод, химически связанный с карбонильными соединениями.
В результате проведенного исследования установлено, что с повышением температуры кипения фракций карбонильных соединений содержание метилкетонов в последних падает.
74 С. С. Семенов, Ю. И. Корнилова и Б. Б. Гуревич
На основании результатов исследования можно предполагать, что метилкетоны, окисленные йодом, имеют ненасыщенные связи. Это предположение вытекает из следующих соображений.
Молекулярный вес кислот, вычисленный по результатам титрования, как правило, был выше молекулярного веса, полученного при омылении. Это может -быть объяснено тем, что непредельные метилкетоны в процессе окисления превращаются в оксикислоты, которые, в результате дальнейших превращений, в условиях титрования не могут быть обнаружены.
На основании данных титрования кислот, полученных при окислении различных фракций, было показано, что в результате окисления образуется смесь кислот. В дальнейшем мы сделали попытку разделить эти кислоты.
При окислении фракций 9—15 включительно было замечено, что, наряду с кислотами маслянистой консистенции, получалось небольшое количество кристаллической кислоты, которую в дальнейшем удалось выделить и проанализировать. Однако следует отметить, что кристаллическая кислота получалась в очень небольших количествах, что очень затрудняло ее изучение.
Выделение кристаллической кислоты было осуществлено двумя путями.
1. Смесь кислот отжималась фильтровальной бумагой, в результате чего кристаллическая кислота оставалась в виде осадка, а маслянистая впитывалась бумагой. Кристаллическая кислота далее перекристаллизовывалась из воды, а маслянистая извлекалась экстрагированием пропитанной ею бумаги.
2. Другой метод отделения кристаллической кислоты заключался в многократной обработке горячей водой, в которой кристаллическая кислота растворялась лучше.
В результате применения этих методов извлечения удалось получить около 0,5 г кислоты, которая после перекристаллизации из воды представляла собой кристаллы исключительно игольчатой формы, имеющие температуру плавления 90—98°; элементарный состав: С — 70,00%, Н = 5,99% и молекулярный вес М = 138.
Соответствующие литературные данные для толуиловых кислот следующие:
ортотолуиловая кислота: т-ра плавл. 10! ।
метатолуиловая , , „ 110° С = 70,60%; Н = 5,90%: М=136.
паратолуиловая „ „ „ 181° )
Для установления строения полученной кислоты последняя была подвергнута спектрографическому исследованию в ультрафиолетовой области спектра. Анализ проводился методом сравнения спектра анализируемой пробы со спектрами мета- и парато-луиловых кислот. Анализ показал, что полеченный спектр ана
Об окислении кетонов йодом в щелочной фазе
логичен спектру метатолуиловой кислоты: положение первого максимума в спектре последней 2873 А, а в спектре анализируемого образца — 2872 А. Однако здесь следует отметить, что присутствие пара-изомера в данном случае спектральным методом обнаружить нельзя.
Анализируя экспериментальные данные, можно предполагать, что не исключена возможность наличия в исследуемой кислоте орто- и мета-изомеров с преобладанием последнего.
Имея сравнительно небольшие количества кислоты, мы попытались все же разделить ее. С этой целью была осуществлена дробная возгонка кислоты, в результате которой было получено несколько образцов с температурой плавления 90—91,5, 91—93, 95—97,5, 96—98°.
На основании полученных результатов можно предполагать, что исследуемая кислота состоит из смеси изомеров, которые имеют один и тот же молекулярный вес и элементарный состав, но различную температуру плавления; естественным результатом совместного присутствия этих изомеров в различном количественном соотношении является непостоянная и пониженная температура плавления исследованных препаратов кислоты.
В дальнейшем полученная кислота была подвергнута окислению с помощью марганцовокислого калия в щелочной среде, которое производилось с механическим размешиванием при температуре 60°.
В результате окисления была получена изофталевая кислота, которая была идентифицирована по температуре плавления и данным спектрального анализа.
В процессе исследования, кроме метатолуиловой кислоты, удалось качественно доказать наличие бензойной кислоты в продуктах окисления одиннадцатой фракции.
Метатолуиловая кислота могла получиться при окислении в описанных выше условиях только за счет мета-метилацетофе-нона, а бензойная — за счет ацетофенона. Наличие ацетофенона в одиннадцатой фракции было качественно доказано ранее.
ВЫВОДЫ
1. На основании проведенного исследования установлено, что смесь карбонильных соединений может быть разделена на ме-тилкетоны и остаток путем обработки йодом в щелочной среде.
2. Количество метилкетонов с повышением температуры кипения фракций карбонильных соединений уменьшается.
3. Результаты проведенного исследования дают основание сделать вывод, что в сланцевой смоле содержатся не только алифатические метилкетоны, но и жирноароматические.
С, С. Семенов, Ю. И. Корнилова 'и Б. Е. Гуревич
4. В процессе исследования удалось доказать наличие в сланцевой смоле мета-метилацетофенона и, качественно, ацетофенона.
ЛИТЕРАТУРА
1. к. И. Ногин. Сухая перегонка дерева лиственных и хвойных пород, стр. 425, ГОНТИ, Ленхимсектор, 1931.
2. Р. И. Векслер. Труды комиссии по аналитической химии, т. III, стр. 363, 1951.
3. Губен. Методы органической химии, т. II, вып. 1, стр. 61, ГОНТИ, химии, литература, 1941.
Г. Н. Гарновская
ДЕЙСТВИЕ ЕДКОЙ ЩЕЛОЧИ НА КИСЛОРОДНЫЕ СОЕДИНЕНИЯ СЛАНЦЕВОЙ СМОЛЫ1
Растворы едких щелочей применяются в технологии переработки сланцевой смолы при очистке топливных фракций и для получения кислых соединений (фенолов).
Обычно всю щелочную вытяжку смолы или ее фракции квалифицируют как фенолы. Однако понятие это совершенно условно, так как действие щелочи на сланцевую смолу отнюдь не сводится только к образованию фенолятов и солей карбоновых кислот.
Еще П. К. Когерман [1] обратил внимание на то, что количество увлеченных в раствор щелочи веществ определяется концентрацией применяемой щелочи. П. Г. Когерман объяснял это явление разной растворимостью нейтральных масел в щелочах различной концентрации, а также конденсацией составляющих смолы, причем конденсация идет тем в большей степени, чем выше концентрация щелочи.
Большинство исследователей, имевших дело с действием щелочи на сланцевую смолу, отмечали условность метода определения фенолов в сланцевых продуктах и зависимость этого определения от концентрации применяемой щелочи, температуры, характера и времени обработки, а также фракционного состава исследуемого продукта.
В 1946—1947 гг. Н. И. Зелениным с сотрудниками, при исследовании вакуумных фракций туннельной и генераторной смол, было показано, что определить содержание фенолов, применяя растворы щелочей, в тяжелых фракциях практически невозможно, так как даже параллельные определения с одним и тем же раствором щелочи давали различные результаты.
При выделении фенолов из средних фракций генераторной смолы изучалось влияние концентрации щелочи на выходы кислых кислородных соединений и увлекаемых вместе с ними нейтральных масел.
1 В работе принимала участие техник Е. А. Калинина.
78
Г. Н. Гарновская
Результаты, полученные при обработке щелочными растворами различной концентрации фракции генераторной смолы, выкипающей в интервале температур 200—330° С, приведены в табл. 1.
Таблица 1
Результаты обработки щелочными растворами средней фракции генераторной смолы
) Ks опыта | Щелочной раствор Фенолы Нейтральные масла Общее количество щелочной вытяжки, % Выход гидроксильных соединений, % от сырья % извлечения гидроксильных соединений
Выход, вес. % Содержание гидроксильных соединений, % 1 Выход, вес. % Содержание гидроксильных соединений, % 1
1 3%-ная NaHCO3 . . . 1,48 88,0 0,13 1,61
2 5%-ный КОН .... 9,30 78,0 4,72 24,6 14,02 8,38 49,0
3 10%-ный КОН .... 11,30 75,7 12,58 13,8 23,88 10,28 68,0
4 20%-ный КОН .... 20,30 66.0 28,80 10,8 49,10 16,50 96,4
5 40%-ный КОН .... 24,80 59,3 24,10 6,4 48,90 16,30 95,4
Извлекаемые щелочью продукты, так же как ' и исходная фракция, характеризовались по содержанию гидроксильных соединений. Последнее рассчитывалось па основании определения содержания гидроксила и молекулярного веса (расчет везде велся на одноатомные фенолы). Определение содержания гидроксила проводилось методом ацетилирования раствором уксусного ангидрида в пиридине (по Верлею).
В исходной фракции содержание гидроксила оказалось равным 1,7%, что при молекулярном весе 171 соответствовало 17,1% гидроксильных соединений.
Разделение извлекаемых щелочью продуктов на фенолы и нейтральные масла проводилось путем извлечения щелочной вытяжки бензолом, причем обработка бензолом проводилась многократно до получения слабо окрашенного раствора, после чего бензольный раствор промывался дестиллированной водой, и промывные воды возвращались в щелочной раствор фенолятов. Естественно, что таким путем достигалось лишь относительное разделение.
Раствор щелочи, наряду с фенолами, извлекает много веществ, не содержащих гидроксила. Как видно из данных табл. 1, «фенолы», извлеченные щелочью и отмытые от нейтральных масел, представляют собой 80—60%-ные концентраты гидроксильных соединений; некоторое количество гидроксильных соединений содержится и в нейтральных маслах.
Действие едкой щелочи на кислородные соединения ”9“
Из приведенных данных видно, что повышение концентрации щелочи с 5 до 20% в 3,5 раза увеличивает общее количество веществ, извлекаемых щелочью. Десятипроцентный раствор щелочи’"извлекает примерно 70% гидроксильных соединений, содержащихся во фракции. Сорокапроцентный раствор щелочи извлекает 95% гидроксильных соединений, что составляет ’/з от всей щелочной вытяжки.
Сланцевая смола — сложная смесь органических веществ. Около 2/з составляющих смолу падает на долю кислородных соединений, среди которых найдены представители самых различных классов. Доказано присутствие в сланцевой смоле карбоновых кислот, фенолов, альдегидов, кетонов. Кроме того, есть основания предполагать .наличие в составе смолы соединений с несколькими функциональными группами (оксикислоты, оксиальдегиды, оксикетоны, альдокислоты и т. п.).
Обращают на себя внимание высокие числа омыления сланцевой смолы и особенно ее высших фракций. По всей вероятности, омыляемые соединения представляют собой вещества типа сложных эфиров; в то же время работами Таллинского Политехнического института установлено, что в сланцевой смоле содержится много так называемых «нейтральных» фенолов, не растворимых в 10%-ном растворе щелочи, но растворяющихся в концентрированных водных или спиртовых растворах последней. «Нейтральные» фенолы, так же как и сложные эфиры, могут давать высокие числа омыления.
Все перечисленные соединения небезразличны к действию щелочи. Вполне очевидно, что в присутствии щелочи кислоты будут образовывать соли, фенолы дадут феноляты, сложные эфиры могут омылиться; альдегиды и кетоны, в зависимости от условий обработки, будут испытывать различные реакции полимеризации и конденсации. Представляло интерес проследить действие щелочи на нейтральные кислородные соединения сланцевой смолы.
Излагаемая в настоящей статье работа посвящена, в частности, изучению процесса полимеризации нейтральных кислородных соединений средней фракции генераторной смолы в присутствии щелочи.
ОПЫТНАЯ ЧАСТЬ
Исходное сырье. Полимеризации подвергались концентраты нейтральных кислородных соединений, выделенные из-средней фракции генераторной смолы метиловым спиртом.
Характеристика исходных продуктов приведена в табл. 2.
Для изучения природы получающихся в результате полимеризации продуктов была проведена обработка щелочью фракций нейтральных кислородных соединений. Характеристика этих фракций приведена ниже.
80
Г. Н. Гарновская
В качестве катализатора для полимеризации применялась твердая химически чистая калиевая щелочь или ее водные растворы.
Табийща 2
Физико-химическая характеристика сырья
Показатели Обесфеноленный экстракт метанольной очистки Концентрат нейтральных кислородных соединений
Удельный вес d®® ....... 9,9236
Молекулярный вес 188 196
Число омыления, мг КОН на 1 г 6,6 22,5
Содержание нейтральных кислород-
ных соединений (по FeCl3),1 % . 60 100
Элементарный состав, %:
углерода .... 82,7 81,75
водорода .... 10,96 9,16
серы, азота и кислорода . , , 6,34 9,09
Фракционный состав, %:
выкипает до 200° С — 3,0
„ 250° С ..... 26,0 32.0
,, 300° С 66,0 68,5
Конец кипения, °C/% остатка . . . 360/8 350/6
Содержание эфиров, % 2,2 7,9
Методика исследова НИЯ. Опыты по полимеризации
проводились в стеклянной колбе, снабженной термометром, обратным холодильником и мешалкой. Для проведения опыта в колбу помещалось взвешенное количество сырья и щелочи. Полимеризация проходила при непрерывном перемешивании и определенной температуре, которая поддерживалась строго постоянной.
Продукты реакции разделялись в делительной воронке на масляный слой и щелочную вытяжку. Масляный слой, совместно с нейтральными маслами, извлеченными бензолом из щелочной вытяжки, отмывался водой от щелочи, сушился и анализировался.
Щелочной раствор, вместе с водой от промывки масляного слоя, после извлечения из него нейтральных масел и полимерных смол 2 подкислялся серной кислотой.
1 Как показали исследования, проведенные Л. И. Гуляевой. 100%-ные по FeCl3 концентраты н. к. с., выделенные из средних фракций смолы метиловым спиртом, содержат значительные количества ароматических углеводородов. Однако для наших целей достаточно показательны и эти относительные цифры.
2 Выделяются при добавлении к щелочной вытяжке бензола в виде третьего (промежуточного) слоя. После отделения хорошо растворяются в воде, при подкислении выпадают из раствора в виде хлопьев или очень вязкой смолистой массы.
Действие едкой щелочи на кислородные соединения
81
Выделившиеся кислые органические соединения извлекались бензолом и разделялись по их растворимости в трехпроцентном растворе бикарбоната натрия и десятипроцентном растворе едкого натра. Для выделения водорастворимых кислот последние •отгонялись с водяным паром из остатка после экстракции. В ряде опытов водный раствор, освобожденный от нейтральных масел и полимерных смол, подкислялся и перегонялся с водяным паром. Нелетучий остаток упаривался досуха и извлекался метиловым спиртом.
Водорастворимые кислоты идентифицировались в виде бариевых солей.
Условия полимеризации. Образование смолистых веществ заметно даже при обработке сланцевой смолы разбавленными растворами щелочей на холоде. Так, например, при количественном определении фенолов наблюдается образование промежуточного слоя во время извлечения бензолом нейтральных масел из фенолятов.
Естественно, что глубина полимеризации должна определяться условиями проведения реакции.
Нами исследовалось влияние на процесс полимеризации таких факторов, как температура, продолжительность, концентрация и количество щелочи, концентрация нейтральных кислородных соединений. Результаты исследования приведены в табл. 3, 4, 5, б, 7.
Таблица 3
Баланс продуктов, полученных при различной температуре полимеризации
Условия полимеризация: время полимеризации 30 минут; 60%-ный водный раствор КОН в количестве 50% от веса сырья (на безводный КОН)
Полученные продукты Выход, % при температуре полимеризации, °C
50 100 140
Нейтральные вещества 95,32 94,32 91,22
Кислые вещества 4,56 5,56 8,73
В том числе: растворимые в 10%-ном NaOH . 2,49 0,90 1,03
» в 3%-ной NaHCO3 0,74 0,76 1,03
» в воде 1,33 1,18 1,82
полимерная смола , , — 2,72 4,85
Потери 0,12 0,12 0,05
Всего 100,0 100,0 100,0
Н Труды ВШ1Ш1С.
82
Г. Н. Тарнавская
Таблица 4
Баланс продуктов, полученных при различной продолжительности полимеризации
Условия полимеризации: температура 50° С, остальные условия те же, что и в табл. 3
Полученные продукты Выход, % при продолжительности (минуты)
30 300 600
Нейтральные вещества Кислые вещества В том числе: растворимые в 10%-ном NaOH . , в 3%-ной NaHCO3 » в воде ...... полимерная смола отери 95,32 4,56 2,49 0,74 1,33 0,12 83,50 15,63 2,20 1,42 2,65 9,36 0,87 80,17 19,53 3,93 1,37 1,93 12,30 0,30
Всего 100,0 100,0 100,0
Таблица 5
Баланс продуктов, полученных при полимеризации с различным количеством щелочи
Условия полимеризации: температура 50° С; продолжительность полимеризации 5 часов; остальные условия те же, что и в табл. 3, 4
Полученные продукты Выход, % при количестве щелочи, % от веса сырья
100 50 25 5
Нейтральные вещества 81,92 83,50 88,05 92,19
Кислые вещества 16,67 15,63 11,25 7,13
В том числе:
растворимые в 10%-ном NaOH . 1,08 2,20 1,21 3,05
. в 3%-ной NaHCOs 1,34 1,42 2,02 1,95
„ в воде . . 1,60 2,65 1,30 2.13
полимерная смола . • 12,65 9,36 6,72 —
Потери 1,41 0,87 0,70 0,68
Всего 100,0 100,0 100,0 100,0
Из приведенных данных видно, что под действием щелочи нейтральные кислородные соединения превращаются частично в кислые продукты, причем выход последних во всех случаях
Действие едкой щелочи на кислородные соединения
83
Таблица 6
Баланс продуктов, полученных при полимеризации в присутствии щелочи различной концентрации
Условия полимеризации: температура 50° С; продолжительность полимеризации 5 часов; количество щелочи 50% от веса сырья (на твердую щелочь)
Полученные продукты Выход, % при концентрации щелочного раствора, %
100 60 20
Нейтральные вещества 88,85 83,50 95,19
Кислые вещества 9,89 15,63 3,44
В том числе: растворимые в 10%-ном NaOH . 1,06 2,20 0,65
„ в 3%-ной NaHCO3 1,08 1,42 0,50
» в воде 0,70 2,65 2,29
полимерная смола 7,05 9,36 —
Потери 1,26 0,87 1,37
Всего 100,0 100,0 100,0
Таблица 7
Баланс продуктов, полученных при полимеризации нейтральных кислородных соединений различной концентрации
Условия полимеризации: температура 50° С; продолжительность полимеризации 5 часов; щелочь — 60%-ный водный раствор в количестве 50% от веса сырья; растворитель — гептан
Полученные продукты Выход, % при концентрации нейтральных кислородных соединений, %
100 50 25 12,5
Нейтральные соединения 74,88 76,37 74,90 61,6
Кислые вещества 23,48 20,19 19,57 29,05
В том числе:
растворимые в 10%-ном NaOH . 2,69 3,47 13,38 14,80
, в 3%-ной NaHCO3 1,75 0,92 0,84 6,25
» в воде 0,14 0,30 0,58 1,00
полимерная смола 18,90 15,50 4.77 7,00
Потери1 1,64 3,44 5,53 9,35
Всего 100,0 100,0 100,0 100,0
1 Большие потери объясняются потерей нейтрального продукта с отгоняемым растворителем.
6*
84
Г. И. Гарновская
значительно превышает количество кислот, которые могут образоваться при омылении сложных эфиров, найденных анализом в исходном сырье. По всей вероятности, под действием щелочи на нейтральные кислородные соединения сланцевой смолы прежде всего идут реакции сложноэфирной конденсации альдегидов и кетонов:
0 0 О
// // //
R—С 4-Ri—С R—СН2—О—С—Rj
н чн
Образовавшийся сложный эфир тут же омыляется с образованием соли кислоты и спирта:
О О
// //
R—СНг—О-С +КОН -> R—СН2ОН 4-С—ОК
Кроме образования растворимых кислот, наблюдается образование высокополимерных продуктов. Последнее, очевидно, обусловлено протеканием реакций альдольной конденсации:
/Р
О О ОН Cf
// // II хн
R—СН2—С + Ri—СН2—С -»R—СН2—СН—СН—Rx
чн ^Н
Глубина реакций полимеризации увеличивается с повышением температуры и продолжительности процесса.
Наибольшие выходы продуктов полимеризации при постоянной температуре и при постоянном времени полимеризации наблюдаются под действием концентрированных водных растворов щелочи.
Действие безводной щелочи повышает выход высокополимер ных смол, но при этом мало образуется растворимых кислот.
Решающую роль для проведения процесса играет концентрация нейтральных кислородных соединений (см.‘ табл. 7). Меняя концентрацию этих соединений, можно направить процесс в сторону, получения тех или иных продуктов. Так, если из концентрированных нейтральных кислородных соединений получается до 20% полимерных смол и. только около 5% растворимых кислот, то, подвергая полимеризации нейтральные кислородные со
Действие едкой щелочи на кислородные соединения
85
единения, разбавленные н-гептаном, можно получить до 22% растворимых кислот, из которых 1,1%—летучие с водяным паром.
Исследование продуктов полимеризации. Для исследования были взяты продукты полимеризации двух фракций нейтральных кислородных соединений (фракция 220— 280°С и фракция 280—370°С). Полимеризация проводилась при следующих условиях: температура 100° С; продолжительность 5 часов; щелочь — КОН, 60 % -ный водный раствор в количестве 100% на кислородные соединения; концентрация нейтральных кислородных соединений 50%; растворитель н-гептан.
Баланс полученных продуктов (приведен в табл. 8) полностью подтвердил данные предыдущих опытов. Под действием щелочи прошли реакции уплотнения нейтральных кислородных соединений и превращения их в кислые вещества.
Таблица 8
Баланс продуктов, полученных при полимеризации фракций нейтральных кислородных соединений
Полученные продукты Выход, вес. % при полимеризации фракций нейтральных кислородных соединений, °C
220—280 280—370
Нейтральные вещества 80,84 80,16
В том числе: выкипающие в пределах кипения исходного сырья 53,20 80,16
кипящие выше конца кипения . исходного сырья 27,64 —
Кислые вещества 18,87 19,21
В том числе: полимерная смола 4,46 13,00
летучие с водяным паром, растворимые в воде ...... 0,17 0,63
летучие с водяным паром, растворимые в 10%-ном NaOH . 0,34 1,62
нелетучие с водяным паром, растворимые в 10%-ном NaOH . 13,90 2,25
Потери 0,29 0,63
Всего 100,0 100,0
После проведения полимеризации и разделения полимеризата на кислую часть, полимерную смолу и нейтральные соединения, кислая и нейтральная части полимеризата анализировались.
86
Г. Н. Гарновская
В табл. 9 приведены результаты анализа исходных нейтральных кислородных соединений и нейтральных продуктов полимеризации.
Таблица 9
Физико-химическая характеристика исходных нейтральных кислородных соединений и нейтральных продуктов полимеризации
Показатели
Уд. вес при 20° С Молекулярный вес Содержание ОН, % Содержание —
О
-С , % . . .
н
Число омыления .
Содержание эфиров, % . . . .
Растворимость в
FeCl3, объемн. %
0,9363 163 1,37
0,9580 188 1,28
0,9450
170
0,70
1,015 210 2,40
0,9870 203 2,40
0,9840 203 2,13
0,9531 181 1,62
0,9779 186 2,22
1,051 275 3,16
Предварительно нейтральная часть продуктов перегонялась в вакууме с отбором фракции, выкипающей в пределах температур кипения исходного сырья и вышекипящего остатка.
При полимеризации фракции 220—280° наблюдалось образование около 28% остатка, кипящего выше 280°, и около 5% высокополимерных смол. При полимеризации фракции 280—370° фракционный состав нейтральной части мало менялся по сравнению с фракционным составом исходной фракции, но получалось 13% полимерных смол.
В нейтральных продуктах полимеризации полностью отсутствуют омыляемые соединения, но наблюдается повышенное содержание гидроксильных и карбонильных групп. Последнее особенно заметно на остатке %>280°, полученном при полимеризации фракции 220—280°. Содержание карбонильных и гидроксильных групп в остатке, кипящем выше 280°, увеличивается больше, чем
Действие едкой щелочи на кислородные соединения 87
в два раза, и это указывает на то, что действие щелочи на нейтральные кислородные соединения прежде всего вызывает реакции альдольного уплотнения.
Наряду с образованием нейтральных продуктов уплотнения и высокополимерных смол (молекулярный вес 500), имеющих кислый характер, под действием щелочи идут реакции сложноэфир-ной конденсации и омыления, в результате которых образуются кислоты. При этом образование кислых веществ может быть только частично объяснено реакцией омыления сложных эфиров и нейтральных фенолов, содержавшихся в исходном сырье; основная же масса кислых веществ может быть получена лишь за счет сложноэфирной конденсации карбонильных соединений. Характеристика кислых продуктов приведена в табл. 10.
Таблица 10
Характеристика кислых продуктов реакции
Продукты Си К >> о И Кислотное число, Элементарный состав, %
Моле; НЫЙ 1 мг КОН С Н О Ва
Нелетучие с водяным паром, растворимые в NaOH: из фракции 220—280° . . „ » 280—370° . . 222 191 119,6 273,9 69,65 55,90 8,98 5,0 21,55 38,40 —
Летучие с водяным паром, растворимые в NaOH: из фракции 220—280° . . 280-370° . . 157 166 16,3 30,0 76,35 70,65 9,6 9,3 14,05 20,05
Летучие с водяным паром, растворимые в воде: Бариевая соль
из фракции 220—280° масляная кислота1 ... из фракции 280—370° Пропионовая кислота 1 ... 88 88 74 74 610 628 750 756 54,10 54,30 48,50 48,60 9,1 9,1 8,1 8,1 36,80 36,60 43,40 43,30 44,28 44,10 48,34 48,30
Из продуктов полимеризации обеих фракций нейтральных кислородных соединений были выделены летучие и нелетучие с водяным паром кислоты, а также летучие с водяным паром,
Литературные данные.
88 Г. Н. Гарновская
растворимые в 10 %-ном NaOH, вещества со слабо кислыми свойствами. Последние, очевидно, должны быть отнесены к классу фенолов.
Среди растворимых кислот были идентифицированы масляная и пропионовая кислоты.
Нелетучие с водяным паром кислоты подробно не анализировались.
ЗАКЛЮЧЕНИЕ
Обработка сланцевой смолы и ее фракций щелочью представляет собой сложный физико-химический процесс. Наряду с извлечением кислых веществ щелочные растворы увлекают значительное количество нейтральных соединений.
В зависимости от концентрации щелочного раствора, температуры и времени контакта, помимо реакций образования фенолятов и солей карбоновых кислот, под действием щелочи могут идти различные реакции полимеризации, конденсации и омыления нейтральных кислородных соединений, ведущие к образованию смолистых и кислых продуктов.
На основании рассмотренного экспериментального материала следует полагать, что действие концентрированных водных растворов едкой щелочи на нейтральные кислородные соединения средних фракций смолы, в первую очередь, вызывает реакции альдольной конденсации, сложноэфирной конденсации и омыления.
Действие щелочи на нейтральные кислородные соединения может быть использовано в целях получения органических кислот и гидроксильных соединений.
ЛИТЕРАТУРА
1. П. К. Когер май. Химия эстонских сланцев. ОНТИ, 1934
С. С. Семенов и Б. Е. Гуревич
ПОЛИМЕРИЗАЦИЯ ОЛЕФИНОВ СЛАНЦЕВОЙ СМОЛЫ НА МАСЛА В ПРИСУТСТВИИ ХЛОРИСТОГО АЛЮМИНИЯ 1
Бензиновая, лигроиновая и дизельная фракции смолы прибалтийских сланцев содержат от 50 до 35% олефиновых углеводородов.
Присутствие таких количеств олефинов в моторных топливах исключает возможность хранения последних в течение продолжительного времени. Олефиновые углеводороды легко окисляются, образуя смолы со всеми вытекающими отсюда последствиями.
Целесообразно использовать олефины в качестве исходного материала для синтезов. Одним из путей использования непредельных углеводородов является полимеризация их с хлористым алюминием в смазочные масла. Путь этот весьма заманчив, так как высокие требования и спрос на смазочные масла возрастают, а ресурсы природных масел ограничены.
Работы Ф. В. Сулливана [1], Л. Г. Жердевой [2], Л. А; Бухмана [3], М. Г. Мамедли [4] и других, посвященные изучению вопросов полимеризации непредельных углеводородов, достаточно полно выясняют значение факторов температуры, времени контакта с хлористым алюминием и количества последнего.
Известно, что хорошие масла получаются только из непредельных углеводородов нормального строения. Изоолефины и особенно циклические непредельные соединения не дают масел с высоким индексом вязкости.
Указанные авторы проводили работы с крекинг-дестиллатами парафинов, гача, отеков и сураханской пробки. Эти работы положили начало синтетическому получению высококачественных смазочных масел из технических продуктов.
Промышленное производство смазочных масел из бензина (до 225° С), получаемого крекингом парафина из смолы фушунь-ских сланцев (по данным В. И. Долгополова), показывает перспективность использования олефинов, кипящих в широких пределах температур.
1 В работе принимала участие лаборант Е М. Иванова.
9(1 С. С. Семенов и Б. Е. Гуревич
Получению смазочных масел из дестиллатов смолы прибалтийских сланцев не уделялось специального внимания, за исключением экспериментов Н. И. Зеленина ,[5], проведенных с различными фракциями смолы гдовских сланцев. Однако в этих опытах моторные масла не были получены.
Представлялось интересным провести работу с бензиновыми, лигроиновой и дизельной фракциями смолы прибалтийских сланцев.
Для работы были приготовлены сырые и очищенные фракции: бензиновые с концом кипения 160 и 225° С, лигроиновая— 160— 230° С и дизельная — 225—325° С.
Характеристика фракций, подвергавшихся полимеризации с А1С1з, приведена в табл. 1 и 2.
Опыты полимеризации производились в трехгорлой колбе, снабженной мешалкой с ртутным затвором, термометром и обратным холодильником.
Поскольку в условиях рабочих температур газообразования не наблюдалось, холодильник сообщался с атмосферой через хлоркальциевую трубку.
Обогрев осуществлялся с помощью водяной или глицериновой бани.
После полимеризации углеводородный слой сливался с комплексного соединения. Последнее разлагалось водой, и выделившееся комплексное масло исследовалось.
Углеводородный слой нейтрализовался и подвергался разгонке. При атмосферном давлении отгонялся дестиллат до 225° С, а в вакууме (при остаточном давлении 5—12 мм) —до 350— 390° С (при пересчете на атмосферное давление).
Остаток выше 350—390° С и представлял собой так называемое «остаточное масло», количество и качество которого характеризовали процесс.
Во всех опытах с бензиновыми дестиллатами хлористый алюминий применялся в количестве 5% от веса исходного сырья. Первая серия опытов была проведена с обесфеноленной фракцией, выкипающей при температуре до 225° С (см. табл. 1).
Из табл. 3 видно, что получаемое остаточное масло весьма низкого качества: индекс вязкости, по Дину и Девису, значительно ниже нуля, а стабильность масла настолько низка, что через 1 —1,5 месяца хранения на поверхности масла и на стенках сосуда образуется полувысыхающая, липкая пленка. Выход масла также очень, низок. Причиной низкого выхода масла и его нестабильности, повидимому, является наличие нейтральных кислородных и сернистых соединений в исходном сырье. Эти соединения, в первую очередь, подвергаются воздействию хлористого алюминия, что ведет к образованию высокомолекулярных смолистых продуктов и отравлению хлористого алюминия, после
Полимеризация олефинов сланцевой смолы
91
Таблица 7
Характеристика бензиновых фракций
Показатели Фракция до 160° С Фракция до 225° С
обесфе- ноленная очищенная 5% H2SO4 (d = 1,84) обесфе-ноленная очищенная 5% H2SO4 (d=l,84)
Уд. вес при 20° С 0,7575 0,7509 0,7753 —
Содержание S, вес. % . . . . 0,73 0,43 1,04 0,77
Групповой состав, объемн. %: нейтральные кислородные соединения 6,67 0,71 9,7 0,30
непредельные углеводе-роды 57,20 63,70 57,20 63,70
ароматические углеводороды 14,60 14,30 11,60 12,00
нафтеновые + парафиновые углеводороды . . . 21,53 21,29 21,50 24,00
Фракционный состав: н. к., °C 75 79 75 95
до 160° выкипает, % . . 93,0 86,0 — —
до 225° „ . . — — 97,5 95,0
к. к., °C, % 184/98 175/94 225/94,5 225/95
Таблица 2
Характеристика лигроиновой и дизельной фракций
Фракция Фракция
Показатели 160 230° С t 225 325° С
сырая | рафинат i сырая рафинат
Уд. вес при 20° С 0,8436 0,8232 0,9270 0,8639
Содержание фенолов, вес. % . 3,92 нет 14,99 нет
Элементарный состав, %:
С 83,19 85,38 82,51 84,14
н 12,92 13,24 10,47 12,03
S 1,22 0,92 0,95 0,97
N + О 2,67 0,46 6,07 2,87
Групповой состав, объемн. %:
нейтральные кислородные
соединения 12,3 7,7 24,5 6,8
без фенолов без фенолов
непредельные углеводо-
роды — — — 34,29
ароматические углеводо-
роды — — — 37,57
нафтены + парафины . . — — — 21,34
ракционный состав:
н. к., °C 155 185
выкипает, % до 160° С . . 5 —
„ „ 230° С . . 84,0 10,0
, „ 270° С . . — 50,0
„ „ 325° С . . — ! 85,0
к. к., °C, % .... . . 260/92 ! 340/90
92
С. С. Семенов и Б. Е. Гуревич
Таблица 3
Влияние температуры на процесс полимеризации
| № опыта I Исходное сырье Температура опыта, °C Время перемешивания, часы Выход полимеров, вес. % от исходного сырья Уд. вес масла при 20° С Выход остаточного масла, вес. % Вязкость, °Е Индекс вязкости, по Дину и Девису Содержание S в масле, % Содержание S в оставшемся бензине, %
при 50° С 1 при 100° С |
2 50 4 14,60 0,9658 6,36 32,8 2,63 <—40 0,53
3 60 4 14,64 0,9597 7,54 33,3 2,66 <-40 2,83 0,38
4 70 4 15,18 0,9499 7,38 22,3 2,22 <-40 2,44 0,43
5 Фракция 80 4 19,88 0,9548 9,68 32,5 2,68 <—40 2,25 0,49
6 до 225°С 90 4 24,65 0,9491 11,95 29,6 2,57 <-40 2,19 0,63
7 обесфе- 100 4 26,38 0,9440 13,28 24,5 2,39 <-40 1,77 0,72
8 коленная НО — 28,26 0,9417 13,56 34,6 2,64 <-40 1,63 0,82
13 120 4 26,97 0,9622 12,67 36,9 2,94 -35 1,69 0,69
14 125 4 28,26 0,9497 14,26 28,5 2,70 -10 0,71 0,71
16 100 50 — 0,9568 11,40 не течет — —
Таблица 4
Выход и качество масел, полученных
из очищенной фракции до 225° С
Количество А1С13 — 5%
№ опыта | Исходное сырье Температура опыта, °C Время контакта, часы Выход । полимеров Выход масла Уд. вес масла при 20° С Вязкость, °Е ! Индекс вязкости
при 50° С | ..... । при 100° С
% исхо,г СЫ] от 1НОГО эья
7 Фракция до 225° С, обес-феноленная 100 4 26,4 13,28 0,9440 24,6 2,39 <-40
10 Фракция до 225°, очищенная насыщенным раствором SnCl4 .... 100 4 25,6 15,6 0,9382 18,0 2,34 +25
11 Фракция до 225° С, очищенная насыщенным раствором FeCl3 . . . 100 4 33,3 19,0 0,9295 17,9 2,32 +25
19 Фракция 225° С, очищенная 5% H2SO4 (d=l,84) 97 16 29,1 11,40 0,9242 27,0 2,84 +25
24 Смесь фракций до 225° С от предыдущих опытов 97 12 31,4 13,10 0,9095 12,5 2,0 +20
Полимеризация олефинов сланцевой смолы
93
чего ни повышение температуры, ни увеличение времени контакта не приводят к улучшению результатов.
Для выяснения влияния нейтральных кислородных соединений на процесс полимеризации были проведены опыты с фракцией до 225° С, очищенной различными методами. Результаты опытов сведены в табл. 4.
Как и следовало ожидать, устранение нейтральных кислородных соединений хлорным железом и очистка фракции серной кислотой повысили качество получаемого масла. Такое масло не образовывало высыхающей пленки при длительном хранении. Индекс вязкости возрос до +25, однако повысить индекс вязкости выше +25 не удалось даже при полимеризации бензина, получаемого от предыдущих опытов, т. е. такого бензина, в котором нет ни кислородных, ни других смолообразующих соединений. Это заставляет предполагать, что основная масса непредельных углеводородов сланцевого бензина состоит из разветвленных или кольчатых молекул, полимеризацией которых вообще нельзя получить высокоиндексных масел.
Были проведены опыты с фракцией, кипящей до 160° С, сырой и очищенной серной кислотой (см. табл. 1). Результаты опытов приведены в табл. 5.
Таблица 5 Результаты полимеризации фракций до 160° в присутствии 5% А1С13
Вязкость, °Е
25
27
29
35
33
36
37
Фракция до 160° С, обес-феноленная ..........
Фракция до 160° С, очищенная 5% H2SO4 (# = 1,84)...........
Фракция до 160° С, очищенная насыщенным раствором FeCl3 . .
Фракция до 160° С, очищенная 5% H2SO4 (#=1,84) ............
То же ................
97
97
97
80
125
97
97
12 13,0 7,8
12 38,1 19,9
12 42,9 21,4
12
12
25
50
i
0,9333. 61,0
0,9171 12,9
1,85
2,07
1,97
2,09
+25
—5
—30 +20 +5
+ Ю
Результаты опытов с сырьем различной очистки при прочих равных условиях показали, что устранение нейтральных кисло-
родных соединений из сырья ведет к увеличению выхода масла вдвое, в сравнении с сырой фракцией.
94
С. С. Семенов и Б. Е. Гуревич
При полимеризации бензина, очищенного серной кислотой, повышение температуры от 100 до 125° С равносильно увеличению времени контакта от 12 до 50 часов.
Опыты эти подтвердили, что на качество получаемого масла оказывают влияние присутствующие в исходном сырье ароматические соединения, которые при полимеризации в значительной мере переходят в полимеры и остаточное масло.
Масла, полученные при полимеризации бензинов, обладают низким качеством. Для повышения качества масел некоторые из них очищались активированной глиной и нитробензолом. Результаты такой очистки представлены в табл. 6.
Таблица 6
Качество масел после очистки
36 Сырое..............
Отбеленное глиной .
Рафинат нитробен-зольной очистки
37 Сырое..............
Отбеленное глиной .
Рафинат нитробен-зольной очистки
Наименование
масла
0,9017
0,8979
0,8973 0,9058 0,9019
0,8995
12,4
12,0
12,4
14,6
14,3
16,0
1,97 4-5
1,98 4-25
2,00 4-25
2,09 4-10
2,09 4-20
216
217
223
— 0,04
1,1 отс.
0,57 0,026
0,55 0,100
0,82 отс.
0,25 отс.
0,044
—14
-16
0,006—16 отс. —12 0,015 —14
отс. —14
414
427
413
Из табл. 6 видно, что по качеству получаемые масла, даже после очистки глиной или селективным растворителем, не превышают автолов марок 10 и 18.
Опыты полимеризации бензиновых фракций позволили прийти к заключению, что целесообразно вести процесс только с сырьем, предварительно очищенным. Очистка должна обеспечить максимальное снижение содержания нейтральных кислородных и ароматических соединений.
Из бензиновых фракций нейтральные кислородные соединения могут быть удалены серной кислотой или полимеризацией их над активированными глинами. После этого бензин уже пригоден для полимеризации его на масла, но вряд ли это будет целесообразно, так как бензин представляет собой большую ценность, а выход масел из него невелик, не говоря уже о невысоком качестве этих масел.
Полимеризация олефинов сланцевой смолы
95
Для выяснения возможности получения смазочных масел из лигроиновых и дизельных дестиллатов были проведены опыты по полимеризации этих фракций как обесфеноленных, так и очищенных серной кислотой и другими реагентами.
Из этих фракций не удалось получить хороших масел и значительных их выходов. Й в этом случае кислородные, а возможно, и ароматические соединения, содержащиеся в сырых фракциях, сыграли вредную роль (см. табл. 2).
Сольвентный метод позволяет рафинировать фракции 160— 230° и 225—325° до полного удаления из них фенолов, основной массы нейтральных кислородных и некоторого количества ароматических соединений.
В качестве сольвента применялся метанол, очистка велась псевдопрстивотоком или противотоком при комнатной температуре.
Характеристика рафинатов дана в табл. 2.
При полимеризации рафинатов лигроиновой и дизельной фракций также выяснялось влияние факторов температуры, времени контакта и количества хлористого алюминия.
Хлористый алюминий вводился не сразу, а порциями в три приема. Углеводородный слой после выключения мешалки отстаивался в течение 12—24 часов, после чего сливался с комплексного соединения, нейтрализовался окисью кальция и одновременно отбеливался активированной глиной. Операция нейтрализации и отбеливания проводилась при 125° С в течение 2 часов. Окись кальция и глина применялись в количествах, составлявших по 5% от веса углеводородного слоя. Нейтральный и отбеленный углеводородный слой подвергался разгонке под вакуумом.
В случае полимеризации лигроинового рафината отгонялись фракции: 1) до 225° С (лигроин), 2) 225—370°, т. е. до начала кипения масла (дизельное топливо) и 3) остаток >370° С (остаточное масло). Сумма двух последних фракций составляла общий выход полимеров.
В случае дизельного рафината отгонялась фракция до 370— 380° С (дизельное топливо); остаток выше этой температуры: представлял собой остаточное масло;
Результаты опытов полимеризации рафинатов лигроиновой и дизельной фракций сведены в табл. 7.
Из данных, приведенных в табл. 7, видно, что повышение температуры полимеризации ведет к увеличению выхода масла, но лишь до известного предела. В дестиллатах, отогнанных до температуры конца кипения исходного сырья или начала кипения полимеров, что одно и то же, остается значительное количество сульфирующихся компонентов, которые представляют собой в основном ароматическую часть исходного рафината. В дизель-
Полимеризация рафинатов фракций 160—230 и 225—325° С
Количество А1С1В—10% 1
Таблица 1
Показатели Исходное сырье и условия полимеризации Рафинат дизельной фракции, очищенной H2SO4
Рафинат лигроиновой фракции 160—230° С Рафинат дизельной фракции 225-325° С
Опыт 53 80° С 24 часа Опыт 48 100° С 24 часа Опыт 65 125° С 24 часа Опыт 63 100° С 48 часов Опыт 45 1 100° С 12 часов Опыт 49 100° С 24 часа Опыт 64 125° С 24 часа Опыт 62 100° С 48 часов
Выход полимеров, вес. % от ис-
ходного рафината Лигроин: 37,9 45,6 43,6 48,5 — — — — —
выход, % 19,2 17,2 20,5 17,1 — —
уд. вес при 20° С содержание сульфирующихся, 0,7855 0,7795 0,7813 0,7717 — — — —
% —— 14,0 И,4 10,6 — — — ___ —
Дизельное топливо: *
выход, % ......... — — -- — 57,7 31,4 37,9 33,0 29,8
уд. вес при 20° С содержание сульфирующихся, —. — — — 0,8673 0,8535 0,8379 0,8422 0,8438
% —— — —. — — 26,6 — 18,0 24,6
Остаточное масло: i
выход, % 19,7 22,0 25,5 26,7 21,4 27,8 31,2 30,6 26,2
уд. вес при 20° С вязкость, °Е: 0,9100 0,9214 0,9230 0,9180 0,9536 0,9226 0,9310 0,9310 0,9238
при 50° С 16,80 19,80 1 16,90 17,37 9,6 16,9 17,7 18,35 —,
» 100° С —* — 2,32 2,38 -— — 2,43 2,45 —
Индекс вязкости — — ; 40 48 — 50 47 —
J В опыте 45 количество А1С13 равно 5%.
Полимеризация олефинов сланцевой смолы
97
шом топливе, например опыта 62, из 18,0% сульфирующихся — 14,1% ароматических и только 3,9% непредельных. Следовательно, дальнейшее повышение температуры и увеличение времени контакта вряд ли может привести к увеличению выхода масла. Как видно из табл. 7, увеличение времени контакта вдвое, с 24 до 48 часов, приводит лишь к незначительному увеличению выхода масла и не ведет практически к изменению его качества.
Опыты, проведенные с количеством хлористого алюминия, меньшим 10%, дали низкие выходы масла плохого качества. Получалось темное масло, нестабильное при хранении. При 5% хлористого алюминия уже через 12 часов перемешивания комплексное соединение оказывалось твердым, неактивным.
Эти наблюдения привели нас к выводу, что из расходуемых 10% хлористого алюминия (количество, которое применялось во всех последующих опытах) какая-то часть (4—5%) тратится не на прямые цели полимеризации, а на устранение наиболее реакционноспособных составных частей рафината, из которых не может получиться хорошего масла.
Таким образом, оптимальными параметрами полимеризации рафинатов лигроиновой и дизельной фракций являются: температура 100—125° С, время перемешивания 24 часа, количество хлористого алюминия 7,5—10%.
При оптимальных условиях опытов были приготовлены образцы масла и произведено специальное исследование их для получения полной качественной характеристики.
Масла, получаемые полимеризацией олефинов, имеют непредельный характер: они растворяются в серной кислоте удельного веса 1,82 на 75—80%.
Можно было ожидать, что такие масла будут мало стабильными, а следовательно, не удовлетворяющими существующим спецификациям. Логично было ожидать, что полимерные масла будут иметь высокие коксовые числа, большую кислотность после окисления, высокую способность к образованию осадков при окислении, низкую термостабильность и т. д.
Результаты исследования (табл. 8) опровергли все эти опасения. Коксовые числа у полимерных масел оказались несколько выше, нежели у нефтяных. Однако причина этого заключается не в непредельности полимерного масла, так как после 50-часового окисления коксовое число его возрастает не больше, чем у масла М3 и меньше, чем у моторного ‘масла Т. Завышенные коксовые числа полимерных масел являются следствием неполного отделения комплексного,, соединения от углеводородного слоя. Для полного удаления комплексного соединения нужно либо увеличить время отстаивания, либо производить эту? операцию в центрифуге. , :
l Труды вниипс.
Таблица 8
Основные физико-химические свойства полимерных масел
Наименование испытаний
Удельный вес при 20° С...........
Вязкость, °Е:
при 50° С ...................
прн 100°С ...................
Индекс вязкости по номограмме До-ксея ............................
Кокс, по Конрадсону, % ........
Кислотное число, мг КОН на 1 г масла ...........................
Термическая стабильность ........
Окисляемость (14 час., 140° С):
кислотное число после окисления содержание асфальтенов после окисления ...................
Коррозия свинцовистой бронзы, г/.ц2 Коэффициент ТреНИЯ, KZfCM1- . . . . Кокс, по Конрадсону, после окисления (50 час,, 140° С) ...... .
Молекулярный вес ................
Элементарный состав, %:
С............................
Н............................
N-J-O + S ...................
Температура застывания, °C ....
Нефтяные масла Масло из рафината фракции 225—325° С
МК М3 Моторное Т
0,902 0,894 0,913 0,931
23,32 11,92 8,49 17,69
3,10 2,26 1,83 2,43
83 85 52 50
0,47 0,51 0,40 1,42
0,064 0,27 1,17 0,011
31 30 22 23
0,43 0,34 1,5 0,70
0,05 0,152 0,47 0,03
4-23,0 4-5,23 + 145,0 +4,86
0,021 — 0,017 0,026
1,4 0,73 1,6 3,2
— — 405
— ___ 87,60
—— — — 11,67
— — — 0,81
1 1 __
Масло из рафината фракции 160-230° С Масло из рафината фракции 225—325° С, полученное в укрупненном масштабе в присутствии
10% А1С13 7,5 % А1С1,
0,918 0,939 0,941
17,37 14,8 20,7
2,38 2,28 2,’65
48 62 53
0,55 1,81 1,87
0,010 0,040 0,001
23 13 15
0,85 1,02 1,43
0,027 0,003 0,004
+4,0 — —
— — —
1,6 — —
— — —
— — .—
— —
— — —
при —20° С не застывает
Полимеризация олефинов сланцевой смолы
99
На основе полученных лабораторных данных были проведены опыты полимеризации рафината дизельной фракции в укрупненном масштабе.
Опыты проводились в мешалке открытого типа с обогревом глухим паром через змеевик. Поддерживалась температура 100° С, время контакта 30 часов. Хлористый алюминий вводился в 4 приема. Комплексное соединение, образуемое первой и второй порциями А1С13, уже через 4 часа работы становилось неактивным (твердым) и удалялось из мешалки вычерпыванием; третья и четвертая порции хлористого алюминия давали жидкое и, следовательно, активное комплексное соединение. Использование этого комплексного соединения вместо первой порции свежего А1С13 в последующем опыте позволило снизить расход хлористого алюминия до 7,5% без ущерба для выхода и качества остаточного масла.
Оборудование, в котором проводились опыты укрупненного масштаба, несколько не соответствовало технологическим требованиям: мешалки герметически не закрывались, разгонная аппаратура была загрязнена кубовыми остатками торфяной смолы. Неудивительно, поэтому, что полученные масла только после дополнительной обработки 82 %-ной серной кислотой и глиной достигали качества лабораторных образцов.
Все данные по процессу полимеризации, полученные в лаборатории, подтвердились и в опытах укрупненного масштаба.
Результаты исследования качества масел, выполненные Р. К- Платоновым, приведены в табл. 8.
Рассматривая остаточное масло, как основной продукт, следует отметить также количественные и качественные показатели, относящиеся к побочным продуктам изученных процессов полимеризации.
При полимеризации олефинов фракции, выкипающей до 160°, получается до 30% остаточного (предельного) бензина, имеющего следующую характеристику.
Уд. вес при 20° С .................................. 0,7328
Содержание S, вес. %...........................’ 0,49
Фракционный состав: н. к., °C......................................... 91
выкипает до 101°, % ............................... 10
„ до 122°, %.................................. 50
„ до 155°, % ................................. 90
К. к., °C, % 162/94
Содержание непредельных углеводородов, % 0,8
„ ароматических , 0,8
, парафиновых и нафтеновых углеводородов . 98,4
Октановое число......................................... 45
Промежуточная фракция от 225° до начала кипения масла, получаемая при полимеризации олефинов бензиновых, лигроино-7*
100
С. С. Семенов и Б. Е. Гуревич
вых и дизельных фракций в количестве до 32—37%, представ-'ляет собой превосходное дизельное топливо, имеющее следующую характеристику.
Уд. вес при 20° С.................................. 0,8400
Вязкость, °Е, при 20° С.............................. 1,95
, °Е, при 50° С.............................. 1,30
Содержание непредельных углеводородов, % .... 3,9
» ароматических , .... 14,1
„ нафтеновых и парафиновых углеводородов 82,0
Границы кипения, °C.................................. 225—325
Цетановое число........................................>54
Комплексное соединение разлагалось водой, в результате чего получалось 20—25% комплексного масла, считая на исходное сырье. Попытка разогнать это комплексное масло приводила к разложению его уже при 230—250° С. Дестиллат быстро темнеет, а остаток в колбе представляет собой весьма вязкую или твердую массу.
Комплексное масло имеет следующую характеристику.
Удельный вес при 20°С............................... 0,9782
Вязкость, °Е, при 75°С ............................ 1,81
Температура вспышки, по Бренкену, °C . ............. 94
Зола, % . • ;.....................................0,03
Содержание серы, вес. % ..................... 0,79
ВЫВОДЫ
Проведенная работа позволяет сделать следующие выводы.
1. Высокое содержание нейтральных кислородных соединений в сланцевом бензине обусловливает быстрое отравление хлористого алюминия и низкое качество масел, получаемых полимеризацией этого бензина.
Полимерные масла имеют отрицательные индексы вязкости и с течением времени высыхают с образованием пленки.
2. Из сланцевого бензина, очищенного серной кислотой, могут быть получены смазочные масла типа автолов марок 18 и 10 с выходом 28—30%.
3. Остаточный бензин имеет низкое октановое число, равное 40—45. Выход предельного бензина 30—32%.
4. Выходы масел при полимеризации олефиновых углеводородов, содержащихся в рафинатах сланцевых дестиллатов, составляют: в случае фракции, выкипающей при температурах 160—230° С от 25 до 27%, а в случае фракции, выкипающей при температурах 225—325° С от 31 до 33% на исходный рафинат.
5. Вследствие высокого содержания олефинов в рафинате фракции 225—325° С, выходы остаточных масел в 2—3 раза выше, чем выходы их, получаемые на сланцеперерабатывающих
R Ф’/НГУЧР HDH Qrrtrm* и тпм wo плгулпа yттлпигтлгг» япкъ V* и Л. U ТСГ У Д-XJ J ££ 11 1 х fLJy л * V/ /.X X 4Z xtA л л.ъ — * ч* л ЪЖ» л
Полимеризация олефинов сланцевой смолы
101
миния. Это обстоятельство благоприятно отражается на экономике процесса получения масел из рафинатов фракций прибалтийских сланцевых смол.
6. Подробным исследованием масел из рафинатов установлено, что, несмотря на их непредельный характер, эти масла по многим техническим показателям подходят к нефтяным смазочным маслам.
7. Опыты в укрупненном масштабе подтвердили результаты лабораторного исследования.
8. При полимеризации олефинов всех испытанных фракций получаются дестиллатные масла, представляющие собой дизельное топливо высокого качества. Цетановое -число его выше 54.
9. Наиболее подходящими рабочими условиями процесса полимеризации олефинов сланцевых дестиллатов в смазочные масла являются температура 100—125° С, время перемешивания 25—30 часов и количество хлористого алюминия 7,5—40% на полимеризуемое сырье.
ЛИТЕРАТУРА
1. F. W. Sul 1 iv ап и др. Ind. Eng. Chem. 23, № 6, 604, 1931.
2. Л. Г. Жердева. «Нефт. хоз.» № 10 (1932) и № 4, 1934.
3. Л. А. Гу хм ан и др. «Азерб. нефт. хоз.» № 7—8, 117, 1934.
4. М. Г. Мамед л и. ЖПХ № 3—4, т. XVI, 143, 1934.
5. Н. И. Зелени и. Статья в книге А. Ф. Добрянского — Крекинг с хлористым алюминием. ОНТИ, Химтеорет, 1938.
С. С. Семенов и Б. Е. Гуревич
О СТАБИЛЬНОСТИ ДИЗЕЛЬНОГО ТОПЛИВА ИЗ СМОЛЫ ПОЛУКОКСОВАНИЯ ПРИБАЛТИЙСКИХ СЛАНЦЕВ
Использование все возрастающих количеств смол (дегтей), получающихся в промышленных процессах полукоксования различных видов твердого топлива, до сих пор встречается с целым рядом трудностей, обусловленных специфическим химическим составом этих продуктов и недооценкой указанной специфичности исследователями и технологами.
Смолы полукоксования сланцев, углей, торфов отличаются от нефтей присутствием кислых соединений (фенолов, кислот и др.), непредельных углеводородов и кислородных соединений нейтральных функций (кетонов, альдегидов, спиртов, эфиров и др.). Часто кислые и нейтральные кислородные соединения представлены в смолах в количествах, доминирующих над углеводородной частью, и поэтому получение товарной топливной продукции из таких смол возможно только в случае применения новых методов очистки дестиллатов. Копирование широко применяемых в нефтяной промышленности методов очистки (щелочной и сернокислотной) приводит к совершенно неудовлетворительным результатам.
Одним из продуктов, получаемых из смол полукоксования, является дизельное топливо. Однако дизельное топливо, например, то, которое получается из смол прибалтийских сланцев, в случае применения только щелочной очистки совершенно не удовлетворяет требованиям, предъявленным ГОСТом. Это обстоятельство связано с химическим составом дизельной фракции, содержащей после дефеноляции до 25% (объемных) нейтральных кислородных соединений. Топливо такой очистки имеет очень резкий, неприятный запах, обладает низким цетановым числом, высоким коксообразованием и т. д. Кроме указанных нежелательных свойств, такое топливо нестабильно при хранении, в нем появляются кислые продукты с одновременным ростом коксообразующей способности. Вопросу стабильности дизельных топлив нефтяного происхождения не уделялось сколько-нибудь серьезного внимания по той простой причине, что такие топлива в основе своей состоят из углеводородов, сохраняю
О стабильности дизельного топлива
103
щихся длительное время без существенных изменений состава и свойств.
Сланцевое дизельное топливо, как, впрочем, и другие топлива аналогичного происхождения, имеет иной химический состав и поэтому вопросу стабильности их следует уделить должное внимание. Задача заключается в отыскании и разработке методов очистки дизельных фракций, которые позволяли бы производить топливо, стабильное при хранении.
Для выяснения вопроса о химической стабильности сланцевого дизельного топлива, получаемого из смол полукоксования прибалтийских сланцев, и влиянии метода очистки на стабильность этого топлива были приготовлены и поставлены на длительное хранение: I) сырая дизельная фракция, 2) та же фракция, предварительно обесфеноленная 10 %-ной щелочью, 3) рафинат, полученный селективной очисткой сырой дизельной фракции, и 4) обесфеноленный экстракт. Физико-химические и другие свойства этих образцов представлены в табл. 1.
Образцы были помещены в одинаковые склянки и поставлены на хранение (20.3. 1947) в темноте, при свободном Таблица 1
Сравнительная характеристика сырой дизельной фракции сланцевого происхождения и дизельных фракций, очищенных ______________________различными методами________________________
Показатели Сырая дизельная фракция Обесфеноленная щелочью дизельная фракция Рафинат дизельной фракции Обесфено* ленный экстракт дизельной фракции
Удельный вес при 20° С Содержание фенолов, 0,9270 0,8821 0,8639 0,9207
вес. % Содержание нейтральных 14,99 нет нет нет
кислородных соединений, объемн. % . . . 24,5 6,8 60,2
Коксовое число, по Кон-
радсону, % Фракционный состав: 0,23 0,18 0,08 0,44
н. к., °C выкипает до 225° С, 178 149 175 200
% выкипает до 325° С, 17,0 19,0 15,0 14,0
% 81,0 86,0 82,0 84,0
к. к., °C, % . . . . Элементарный состав 353/93 352/94 347/94 354/96
Содержание, % 82,14
С 83,22 85,97 85,91
Н 10,43 10,68 11,61 9,67
S + N-+-0 .... 6,35 3,35 2,48 8,19
Таблица 2
Изменения в характеристике испытуемых образцов сланцевых дизельных фракций при хранении
| № п/п | Дата отбора пробы Общая длительность хранения, месяцы Сырая дизельная фракция Дизельная фракция, обесфеноленная щелочью Метанольный рафинат дизельной фракции Обесфеноленный экстракт
Уд. вес при 20° С ... Фенолы, % Н. к. с.1, % Кокс, по Конрадсону, % Уд. вес при 20° С Фенолы, % I Н. к. с. \ % Кокс, по Конрадсону, % J Уд. вес при 20° С Фенолы, % | Н. К. с. *, % Кокс, по Конрадсону, % Уд. вес при 20° С ; Фенолы, % Н. к. с.1, % Кокс, по Конрадсону, %
1 20/111 1947 0 0,9270 14,99 25,0 0,23 0,8821 нет 24,50 0,18 0,8639 нет 6,80 0,08 0,9207 нет 60,18 0,44
2 22/IV 1947 1 0,9259 14,06 23,4 — 0,8876 0,35 22,0 ——• 0,8638 » 6,95 — 0,9197 нет 54,20 —
3 21/V 1947 2 0,9281 14,0 18,0 — 0,8907 0,42 20,0 — 0,8634 W 6,27 — 0,9219 нет 53,80 •' 1 '«
4 23/VI 1947 3 0,9284 13,82 23,0 0,8893 0,49 21,42 — 0,8696 5,74 — 0,9266 0,37 51,20 —
5 21/VII 1947 4 0,9330 13,75 20,0 — 0,8895 0,61 20,0 0,8692 — — 0,9235 0,46 50,0 —
6 20/VIII 1947 5 0,9353 14,20 24,0 — 0,8894 0,41 30,0 — 0,8695 » 8,0 — 0,9256 0,46 52,0 —
7 9/II 1948 11 0,9397 16,6 20,9 — 0,8892 2,60 24,6 — 0,8692 » 6,7 — 0,9248 2,80 50,0 —
8 14/IV 1948 13 0,9363 19,9 23,8 1,35 0,8937 1,81 22,34 0,85 0,8688 » 4,25 0,05 0,9284 1,63 52,08 1,40
9 10/1 1949 22 0,9420 19,4 26,3 2,12 0,8969 2,20 26,6 1,02 0,8696 5> 5,0 0,05 0,9321 2,18 55,9 2,18
10 10/VII 1950 40 0,9432 20,6 33,3 3,36 0,8975 1,90 25,0 1,09 0,8704 » 6,1 0,45 0,9374 5,10 48,8 4,2
11 10/VI 1952 63 0,9466 24,1 — 4,04 0,8979 1,90“ — 1,66 0,8702 1 ” — 0,52 0,9404 2,60» — 4,28
1 Нейтральные кислородные соединения.
2 Кроме того, 3,95% кислых смол (эмульсия).
3 Кроме того, 9,52% кислых смол (эмульсия),
О стабильности дизельного топлива 105-
доступе сухого воздуха. В период хранения отбирались пробы, причем определялись удельный вес, содержание кислых веществ, содержание нейтральных кислородных соединений и кокс по Конрадсону. Данные приведены в табл. 2.
Во время хранения подопытные образцы подвергались окислению кислородом воздуха с образованием смолистых веществ. Особенно заметные изменения произошли в образцах щелочной очистки и сырой фракции. Внешний вид этих образцов напоминает исходную смолу, из которой была получена дизельная фракция. Рафинат селективной очистки по внешнему виду не претерпел изменений, хотя удельный вес его за 5 лет и 3 месяца повысился с 0,8639 до 0,8702 и, соответственно, повысилось коксовое число с 0,08 до 0,52%.
Во всех остальных образцах удельный вес возрос значительно больше, чем в рафинате. Наряду с этим количество кокса, определяемого по Конрадсону, в этих образцах возросло в 10— 18 раз в сравнении со свежими. Обращает на себя внимание появление кислых продуктов, количество которых непрерывно возрастает с увеличением длительности хранения. После пятилетнего хранения в исходной, необесфеноленной дизельной фракции количество кислых веществ возросло с 15,0 до 24,1 %, в обесфеноленной, соответственно, с 0 до 1,9% и в обесфеноленной экстракте с 0 до 2,6%. Кроме того, в последних двух образцах при выделении кислых веществ щелочью появляется значительная эмульсия, особенно при отмывке от щелочи. Собранная отдельно эмульсия после высушивания имеет кислый характер, подобно кислым смолам. Количество эмульгируемых веществ составляет от 4 до 9% на навеску. Таким образом, количество кислых веществ, с учетом эмульсии, в обесфеноленной дизельной фракции и экстракте составляет около 6—12%.
Полученные данные свидетельствуют о том, что в процессе хранения отдельные классы соединений, входящие в состав дизельной фракции, ведут себя по-разному. Наиболее изменчивыми компонентами оказываются гетероатомные соединения. Углеводородная часть дизельной фракции, несмотря на наличие свыше 50% непредельных углеводородов, оказывается более устойчивой при хранении. Практически важным является основанный на этих наблюдениях вывод о том, что для получения химически стабильного дизельного топлива необходимо возможно полное удаление гетероатомных соединений кислого и. нейтрального характера. Селективная сольвентная очистка дизельной фракции полностью обеспечивает такое удаление нежелательных компонентов, в то время как щелочная очистка оказывается недостаточной для получения дизельного топлива, удовлетворяющего существующим требованиям по запаху, цетановой характеристике,, коксообразованию и химической стабильности..
Е. Е. Феофилов и Г. Н. Гарновская
ХИМИЧЕСКИЙ СОСТАВ И ПУТИ ПРАКТИЧЕСКОГО ИСПОЛЬЗОВАНИЯ ПОЛИМЕРОВ КАТАЛИТИЧЕСКОЙ СТАБИЛИЗАЦИИ ФРАКЦИЙ СЛАНЦЕВОЙ СМОЛЫ
При каталитической очистке бензиновой фракции сланцевой смолы на алюмосиликатных катализаторах образуется до 17% полимеров, кипящих выше температуры кипения бензина.
Нами изучались свойства смеси полимеров, полученных при разгонке катализата и продувке паром катализатора после стабилизации легкой фракции смеси генераторной и туннельной смол.
Стабилизация фракции была проведена на опытной установке ВНИИПС на катализаторе 2 (кембрийская голубая глина) при температуре 200—300° С.
Фракционный состав полимеров:
н. к. 202° С
10% выкипает до 214° С 60% выкипает до 304° С
20% .. 230° С 70% 333° С
3)% .. 243° С 80% ,, 365° С
40% „ 259° с 90% „ 380° С
50% „ 278° с
Полимеры были разогнаны на три фракции при остаточном давлении 10 мм рт. ст.:
1) фракция, выкипающая до 104° С; 2) фракция, выкипающая в интервале температур 104—125° С, и 3) фракция, выкипающая выше 125° С.
Характеристика фракций дана в табл. 1.
Первая и вторая фракции содержат незначительное количество кислородных соединений и являются, повидимому, продуктами уплотнения непредельных соединений.
Третья фракция, представляющая основную массу полимеров, содержит около 30% нейтральных кислородных соединений,
определяемых по хлорному железу.
В табл. 2 приведен элементарный состав третьей фракции и ее ориентировочный групповой состав, вычисленный на основании
Химический состав и использование полимеров
107
Общая характеристика фракций
Таблица 1
Показатели 1-я фракция 2-я фракция 3-я фракция
Выход фракции, % 20,0 20,0 60,0
Удельный вес при 20° С Нейтральные кислородные соедине- 0,8268 0,8620 0,9440
ния по FeCl3, объемн. % .... Кислые кислородные соединения, 4,6 5,1 30,0
вес. % отсутствие 1,31
Молекулярный вес 154 181 284
Йодное число 62,7 22,0 14,4
% гидроксила — 0,22 0,85
/° % группы — С\ — — 0,50
Число омыления —— 1,7 45,0
Кислотное число Фракционный состав: — — 1,1
н. к., °C 198 218 224
10% выкипает до °C 200 224 250
20% „ „°C 202 227 280
30% „ „°C 203,5 231 296
40% „ „°C 205 233 306
50% „ „°C 208 236 310
60% ,, „°C 210 239 330
70% „ „°C 215 243 338
80% , „°C 219 252 360
90% „ „°C 224 260 —
к. к., °C, % остатка 227/3 265/4 360/20
Таблица 2
Элементарный и групповой состав третьей фракции
Показатели %
Элементарный состав: 84,5
углерод
водород 10,5
сера 1,4
кислород 3,6
Групповой состав: 14,4
гидроксильные соединения
альдегиды 3,8
омыляемые соединения 22,8
непредельные соединения ... 16,5
прочие соединения 42,5
108
Е. Е. Феофилов и Г. Н. Гарнозская
расчета по молекулярному весу, содержанию функционально
ных групя ОН, —Сс , числу омыления и йодному числу.
ХН
Для дальнейшего исследования третья фракция полимеров была разогнана при остаточном давлении 4 мм рт. ст. на более узкие фракции. Выходы и физико-химическая характеристика полученных фракций приведены в табл. 3.
Таблица 3 Характеристика фракций
Показатели Границы кипения фракций при атмосферном давлении, °C
250-300 300—350 350—400 >400
Выход, % от 3-й фракции 31,60 30,00 16,60 21,80
Уд. вес d™ 0,9150 0,9246 0,9594 0,9936
Н. к. с. noFeCl31 объемн. % 7,2 22,50 25,00 70,00
Карбоновые кислоты, вес. % 0,54 0,24 0,29 0,19
Фенолы, вес. % 1,00 1,00 1,06 0,84
Молекулярный вес .... 208 253 279 463
Йодное число ...... 16,2 15,1 13,7 9,8
Кислотное число ..... 1,2 1,0 1,2 0,7
Число омыления 16,0 26,0 25,0 51,0
% гидроксила 0,32 0,47 0,63 0,40
% группы — С''7 .... \н — 0,23 0,90 1,40
Спирты, % 4 7 8 11
Альдегиды, % — 1,2 4,1 16,25
Омыляемые соединения, % 5,5 11,4 21,8 41,30
Цетеновое число 53 38 28 —
Как видно из приведенных в табл. 1, 2, 3 данных, полимеры каталитической стабилизации сланцевого бензина представляют собой концентраты каких-то кислородных соединений, возможно, непредельного характера. Не вызывает сомнений присутствие в составе полимеров веществ, имеющих спиртовую и альдегидную функциональные группы, а также каких-то легко омыляемых соединений, причем последние находятся в наибольшем количестве.
Детальное изучение подобных сложных по групповому составу высококипящих смесей очень затруднительно из-за отсутствия надежной методики исследования и нами не проводилось.
Химический состав и использование полимеров
109
До последнего времени предполагалось использовать полимеры каталитической стабилизации как компонент дизельного топлива.
Проведенные предварительные исследования полимеров, установившие наличие в них большого количества омыляемых соединений, позволили предложить новый путь практического использования полимеров в качестве сырья для производства консистентных смазок.
Образцы различных фракций полимеров были испытаны для приготовления смазок типа солидолов в ЦНИЛ треста нефте-маслозаводов. Лучшая по качеству смазка была получена из широкой фракции полимеров, характеристика которой приведена выше (третья фракция, табл. 1 и 2).
По заключению ЦНИЛ треста нефтемаслозаводов применение полимеров для целей мазеварения позволило сократить расход жиров и минерального масла.
Полученная смазка обладала повышенной пластичностью при высокой температуре плавления и высокой пластичностью при низких температурах. Смазка отличалась высокой коллоидной и химической стабильностью.
В 1951 г. была проведена работа по составлению различных композиций для варки смазок на основе полимеров каталитической стабилизации. Полимеры были получены при стабилизации бензиновой фракции туннельной смолы и имели характеристику, аналогичную приведенной выше.
Кроме полимеров, для приготовления смазки брались кислоты окисленного парафина (молекулярный вес 320, кислотное число 130 мг КОН на а) и минеральное масло «машинное Л» (вязкость Е°5о — 5,4).
Результаты работы приведены в табл. 4.
Таблица 4
Состав консистентных смазок
Состав смазки, вес. % Температура каплепадения, °C
парафиновые кислоты полимеры минеральное масло вода
25 25 50 1,8 68,5
25 50 25 1,8 70,5
25 12,5 62,5 1,8 68,0
12,5 37,5 50 1,7 37,0
50 —- 50 1,3 86,0
25 — 75 1,4 57,5
30 50 20 1,5 71,0
25 50 25 1,4 73,0
110
Е. Е. Феофилов и Г. Н. Гарновская
Смазка № 8, содержащая 50% полимеров, имела следующую хар актеристику. Содержание мыла, %..................................... 13
Температура каплепадения, °C .................... 73
Пенетрация при 25° С............................. 294
Содержание, %: воды ..................................... 1,4
свободных щелочей.......................... 0,13
механических примесей....................... 1,1
механических примесей, %, не растворимых в соляной кислоте ..................... отсутствие
Проба на коррозию............................выдерживает
Полученная смазка удовлетворяет требованиям ГОСТ 4366-50 на универсальные среднеплавкие консистентные смазки.
Н. И. Зеленин и Г. В. Татаркина:
СОСТАВ ЛЕГКОЙ ФРАКЦИИ СМОЛЫ КАШПИРСКОГО СЛАНЦА >
Химический состав смолы волжских, в частности кашпирских, сланцев до сих пор не расшифрован. Более того, не известен даже групповой состав смолы и ее фракций, в том числе и легкой фракции, обычно легче других поддающейся анализу.
Существует мнение, что основными представителями фракций смол волжских сланцев являются сернистые соединения. Это мнение основано на значительном содержании серы в смоле. Как известно, в смоле волжских сланцев содержится до 7 % серы, причем в легких (до 200°) фракциях сернистые соединения как бы концентрируются — содержание серы доходит до 11,5%.
Многочисленные, хорошо известные в литературе [1] попытки изолировать сернистые соединения оказались бесплодными. Несмотря на установившуюся традицию считать сернистые соединения смолы волжских сланцев представителями ряда тиофена, последние выделены не были и их индивидуализация ни в одной работе не может быть признана вполне достоверной.
Объяснение этому обстоятельству следует искать в той методике, которую исследователи применяли для целей выделения и индивидуализации сернистых соединений. Следует считать мало полезным применение реактивов, связывающих сернистые соединения ряда тиофена в комплексные соединения, так как, наряду с тиофенами, в смоле присутствуют кислородные соединения, не менее склонные к образованию комплексных соединений, с различными неорганическими веществами.
При последующих разделениях компонентов комплексов, как правило, получались не менее сложные смеси веществ, чем до образования этих комплексов.
Ректификация позволяла получать очень высокие концентраты сернистых соединений. Так, по данным А. П. Сиверцева, можно получить фракцию 130—135°, содержащую 15—18% S, фракцию 155—160°, содержащую 13,71 % S, и др.
1 В работе принимали участие Е. А. Калинина и Е. Б. Калашникова
112
Н. И. Зеленин и Г. В. Татаркина
Однако концентраты, полученные ректификацией, оказывались также смесями веществ различных классов. Ректификация показала также возможность выделения углеводородов, до сих пор никем еще не выделенных из состава волжских смол.
Наименьшее содержание серы в таких углеводородных фракциях, по1 тем же данным А. П. Сиверцева, было не менее 2,2% во фракции 70—75° и 2,3% во фракции 90—95°.
Таким образом, до сих пор неизвестны случаи выделения из смесей, составляющих волжские сланцы, чистых сернистых соединений и углеводородов.
При решении чисто практических задач переработки смолы волжских сланцев мы встретились с необходимостью принципиального решения вопроса о наличии углеводородов в составе смолы и проверки однородности состава последней по отдельным фракциям.
Единственно пригодным для таких целей в настоящее время является метод хроматографии, хорошо освещенный в литературе. Успехи этого метода в разделении чрезвычайно сложных смесей общеизвестны. Для разделения смол прибалтийских сланцев на составляющие хроматографический метод стал применяться в самое последнее время, и успех его применения виден из работы Л. И. Гуляевой [2].
В качестве объекта исследования в настоящей работе была использована фракция до 200° кашпирской смолы, очищенная от оснований и кислых соединений, следующего состава.
Удельный вес при 20° С................ 0,8859
Коэффициент преломления ................ 1,4760
Содержание серы, % ....................... 10
Молекулярный вес ......................... 120
Йодное число..............................22,5
Фракционный состав, объемн. %:
н. к............ 127° С
130°.............. 4 180°..........76
140°..............10 190°..........86
150°.............. 26 200° ..... 92
160°.............. 46 210°..........96
170°.............. 64 212° .... 98
Разделение фракции на отдельные классы соединений произведено хроматографическим адсорбционным методом на силикагеле при различной высоте и объеме, адсорбента. Применялся силикагель производства завода «Красный Химик», отвечающий временным техническим условиям для хроматографии.
Выбранный метод адсорбционного вытеснения заключался
R Г.ПРТТХГТЛТТТРЛЛ- кпгтгшия ТТПРГТГТДиТТаТЛТТТЯСТ ГТР1ГТГСТПТЛЛ7ВП
-------------------
Состав легкой фракции смолы кашпирского сланца
113
трубку диаметром 25 мм и высотой 150 см, наполнялась адсорбентом — силикагелем, фракция вводилась под давлением воздуха, подававшегося со скоростью 60—80 л!ч, до тех пор, пока уровень жидкости не достигал нижнего конца адсорбента; после этого добавлялся вытеснитель, метиловый спирт 90—95 %-ной концентрации.
Во время опыта давление поддерживалось постоянным, фильтрат собирался через каждые пять минут. В пробах определялись показатели преломления и содержание серы.
Выделенные фракции подвергались повторному .хроматографированию, что' позволило получить более четкое разделение.
Данные, приведенные в табл. 1, показывают результаты разделения фракции до 200° кашпирской смолы. При высоте слоя адсорбента до 120 см можно получить фракции почти чистых углеводородов, которые при повторном хроматографировании оказывались совершенно очищенными от сернистых соединений. С другой стороны, удалось выделить фракцию, содержащую 22 % серы.
Преобладающее количество фракций получено с содержанием серы 11%; эти фракции составляют по отношению к исходному материалу 72,4%. Результаты повторного хроматографирования этих фракций приведены в табл. 1—6.
Таблица 1
Начальное разделение фракции
Высота слоя 120 см
№ пробы (отбор через 5 минут) Количество отобранной фракции, % Характеристика отобранной фракции
20 nD Содержание S, %
1-4 1,17 1,4182—1,4189 0,01
5-8 1,53 1,4205—1,4280 0,03
9—14 2,14 1,4300—1,4497 0,96
15-20 2,24 1,4529—1,4680 6,1
21—46 11,30 1,4700—1,4780 10,7
47—149 61,10 1,4800—1,4815 11,8
150—155 3,63 1,4835—1,4980 13,6
156—164 4,53 1,5000—1,5100 17,0
165—168 2,22 1,5080—1,4900 22,1
169—170 1,06 1,4791—1,4698 16,3
171 0,42 1,4606 10,5
172 1,0 1,4525 6,9
173—175 1,33 1,4460—1,4410 6,4
176—180 4,15 1,4395—1,4390 5,9
181 Остаток — 4,5
Остаток 4-потери 2,18 — —
8 Труды вниипс.
114
И. И. Зеленин и Г. В. Татаркина
Таблица 2
Повторное разделение выделенной фракции
Проба 21-149. = 1,4800; S = ll,3%
№ пробы (отбор через 5 минут) Количество отобранной фракции, % Характеристика отобранных фракций
20 nD Содержание S, %
1—2 2,33 1,4190—1,4265 0,0
3—5 2,80 1,4350—1,4475 1,24
6—16 7,15 1,4522—1,4695 4,2
17—44 57,5 1,4702—1,4838 10,6
45—47 4,22 1,4858—1,4920 12,5
48—49 1,60 1,4962—1,4990 13,4
50—55 7,30 1,5010—1,5088 15,0
56-62 11,30 1,5100-1,5120 17,5
Остаток 5,8 3,7
Повторное разделение
Проба 150—155. = 1,4908; S=13,6%
Таблица 3
№ пробы (отбор через 5 минут) Количество отобранной фракции, % Характеристика отобранной фракции
20 nD Содержание S, %
1—2 5,65 1,4450—1,4631 5,9
3—4 11,55 1,4740—1,4852 10,4
5—8 67,60 1,4900—1,4996 14,0
9—10 9,70 1,5103—1,5118 17,4
Остаток + потери 5,50
Проба 156—164. S = 17,0%
№ пробы (отбор через 5 минут) Количество отобранной фракции, % Характеристика отобранной фракции
20 nD Содержание S, %
1 2,9 1,4962 11,4
2—3 78,7 1,5071-1,5080 17,1
4 12,2 1,5120 18,5
Остаток 4- потери 6,2
Состав легкой фракции смолы кашпирского сланца
115
Повторное разделение
Проба 15—20. Пр = 1,4600; S = 6,0%
Таблица 4
№ пробы (отбор через 5 минут) Количество отобранной фракции, % J Характеристика отобранной фракции
м20 nD Содержание S, %
1—2 3-4 5-6 7-8 9 10 Остаток + потери 3,9 i 1,4182—1,4215 12,2 1 1,4320—1,4360 14,7 i 1,4518—1,4590 46,5 1 1,4602—1,4623 15,6 : 1,4961 2,72 , 1,5098 4,38 i Повторное разделение Проба 165—170 0,07 0,65 4,7 6,3 12,1 15,6 Таблица 5
№ пробы (отбор через 5 минут) Количество отобранной Характеристика отобранной фракции
фракции, % „20 nD Содержание S. %
1-2 3 4-5 6—7 8—9 10 11 14,05 5,9 15,1 14,1 14,05 5,8 6,8 1,5112—1,5100 1,5040 1,5022—1,5015 1,5010 1,4959—1,4929 1,4908 1,4862 18,2 22,0 21,5 21,2 19,0 18,5 17,0
Таблица 6
Повторное разделение
Проба 1—14. Содержание серы 0,0—1,0%
№ пробы (отбор через 5 минут) Количество отобранной фракции, % Характеристика отобранной фракции
„20 nD Содержание S, %
1-2 6,1 1,4180—1,4190 0,0
3—4 11,9 1,4200—1,4266 0,4
5-7 45,4 1,4299-1,4362 0,6
8 29,5 1,4403 1,0
Остаток + 7,1
потери
8*
116
Н. И. Зеленин и Г. В. Татаркина
Из этих таблиц видно, что основная масса фракций, содержащая около 11 % серы, представляет собой не однородный
Рис. 1. Ректификация на колонке фракции, содержащей 17% S.
Рис 2 Ректификация на колонке Рис. 3. Ректификация на колонке фракции, содержащей 21,5% S. фракции, не содержащей серы.
умеренно сернистый материал, а смесь высокосернистых и бес-сернистых соединений; это видно из того, что при любом повторном хроматографировании, наряду с большим количеством умеренно-сернистых фракций, обязательно выделялись как бес-
Состав легкой фракции смолы кашпирского сланца
117
сернистые или малосернистые фракции, так и высокосернистые фракции, содержащие больше серы, чем исходная проба. Это говорит о том, что для такого рода смесей, как смолы волжских сланцев, общеизвестная методика хроматографирования нуждается в улучшении. Одновременно эти результаты говорят о том, что роль сернистых соединений в составе кашпирской смолы преувеличивается, так как, несмотря на их высокую концентрацию в определенных фракциях, удается выделить значительное количество бессернистых фракций, что вообще ранее считалось мало вероятным.
Очень интересным является то обстоятельство, что выделенные хроматографическим методом группы веществ (высоко сернистые, умеренно-сернистые и бессернистые) являются сложными смесями веществ и по температурам кипения, т. е. что и сернистые и углеводородные компоненты смолы находятся практически во всех возможных дестилляционных фракциях смолы (см. рис. 1, 2, 3).
ЛИТЕРАТУРА
1. А. С. Броун и А. П. Си верцев. Химия сернистых соединений Жидкого топлива, ОНТИ, 1936.
2. Л. И. Г у л я е в а и Н. И. П ы ш к и н а. Наст, сборник, стр. 124.
В. Н. Лапин
МОЮЩИЕ, СМАЧИВАЮЩИЕ И ЭМУЛЬГИРУЮЩИЕ СРЕДСТВА ИЗ СЛАНЦЕВОЙ СМОЛЫ
В 1933 г. профессором А. М. Беркенгеймом [1] было высказано мнение, что сульфопроизводные различных погонов кашпир-ской сернистой сланцевой смолы могут быть использованы в качестве эмульгаторов. Последним, по мнению автора, предстоит играть такую же роль, какую играют сульфированные животные и растительные масла в нашей промышленности.
В 1933 г. А. М. Беркенгеймом были проведены лабораторные опыты по использованию сульфопроизводных средней фракции смолы (200—300°) в качестве эмульгирующего средства в производстве красок и тиокреолина. В Московском отделении научно-исследовательского института по мыловарению Рождественский и Ребиндер исследовали физико-химические свойства сульфомыла из сланцевой смолы и сравнили их с соответствующими свойствами нафтенового мыла. Оказалось, что по своей эмульгирующей способности сланцевое сульфомыло в 13 раз превосходит нафтеновое мыло [1].
В 1934 г. производились опыты по применению сульфомыл сланцевой смолы в качестве эмульгаторов в производстве охлаждающих жидкостей для резания металла [2]. В 1942 г. сотрудниками ВНИИПС была проведена работа по изготовлению эмульсолов, эмульгаторов и эмульсий на основе сульфомыл сланцевой смолы. Однако все эти опыты не вышли за пределы лабораторных испытаний, и сульфомыла сланцевых смол применения не нашли.
Причина практической безуспешности упомянутых работ, несмотря на открытие целого ряда ценных свойств сланцевого сульфомыла, заключается в малой изученности природы сланцевой смолы и ее изменений под влиянием тех или иных реагентов. Этим же объясняется отсутствие успеха в применении технологических методов переработки кашпирской сернистой сланцевой смолы на химические продукты.
Долгое время существовавший среди специалистов в буржуазной Эстонии взгляд на сланцевую смолу как на смесь угле
Моющие, смачивающие и эмульгирующие средства
119
водородов, аналогичную нефти, не позволял подойти правильно к вопросу о ее переработке на топливные и химические продукты. Следует отметить, как это ни странно, что и до настоящего времени многие специалисты продолжают рассматривать сланцевую смолу как материал углеводородного характера, без учета ее специфического состава, чем в значительной степени объясняются мало удовлетворительные результаты переработки смолы б заводском масштабе. Дело в том, что еще в 1946— 1948 гг. в работах ВНИИПС Н. И. Зелениным, Е. Е. Феофило-вым, С. С. Семеновым и В. Н. Лапиным было показано, что смолы полукоксования прибалтийских и волжских сланцев на 2/з состоят из гетероатомных соединений. Содержание углеводородов в сланцевых смолах не превышает 30—35%, причем основное количество углеводородов находится в легкокипящих фракциях сланцевой смолы.
Таким образом, смолы полукоксования сланцев содержат 65— 70% гетероатомных соединений, в частности, смола прибалтийских сланцев содержит в указанном количестве кислородные соединения, а смола кашпирских сланцев — сернистые и кислородные соединения. Наличие последних, вследствие их высокой реакционной способности и термической нестойкости, приводит к целому ряду побочных реакций в технологических процессах, которые не учитывались исследователями и технологами. Вследствие этого затруднялось получение качественных товарных продуктов.
С учетом особенностей химического состава смолы сотрудниками ВНИИПС были разработаны методы переработки смол полукоксования на различные продукты и, в частности, на моющие, эмульгирующие и смачивающие средства.
Средняя фракция (200—300°) смолы кашпирских сланцев, как видно из табл. 1, кроме сернистых соединений, содержит значительное количество кислородных соединений.
Таблица 7
Характеристика фракции 200—300° кашпирской смолы без азотистых оснований и фенолов
Контролируемая величина Показатели
Удельный вес при 20° С ..... . Содержание нейтральных кислородных соединений, % . . Содержание, %: серы углерода . • водорода кислорода ... 0,9200 58,3 7,60 78,40 9,70 4,30
120
В. Н. Лапин
Последние при действии крепкой серной кислоты подвергаются полимеризации и конденсации, образуя тяжелые смолы, кипящие выше исходной фракции.
В результате сульфирования фракции 200—300° получаются сульфопродукты (сульфокислоты или сульфомыла) с большой примесью продуктов уплотнения нейтральных кислородных соединений, именуемых в дальнейшем смолистыми примесями. Последние придают сульфомылу неприятный специфический запах и черный цвет. Содержание смолистых примесей в сульфомыле достигает 30%, как это показано в табл. 2.
Таблица 2
Характеристика сульфомыла
Контролируемая величина Показатели
Содержание, %:
ВОДЫ 35,0
свободной щелочи 0,10
сульфата натрия 2,8
смолистых примесей 30,0
Такое большое количество примесей ухудшает свойства сульфомыла, в частности, эмульгирующие, смачивающие и моющие свойства и делает его непригодным для применения в промышленности.
Для получения сульфосолей, свободных от смолистых примесей, следовало бы сульфировать фракции смолы, свободные от нейтральных кислородных соединений.
Так, например, после удаления активных нейтральных кислородных, непредельных и, частично, сернистых соединений из легкой фракции над активными глинами от нее была отобрана фракция до 200—225°, которая содержала только 3% нейтральных кислородных соединений. Полученное из этой фракции сульфомыло не содержало смолистых примесей. Однако разделение кашпирской смолы на группы химических компонентов или хотя бы удаление кислородных соединений из средних фракций смолы является сложной и еще не разрешенной проблемой. Поэтому был избран путь последующего удаления из сульфопродуктов смолистых примесей, в частности растворителями.
Оказалось, что смолистые примеси из сульфопродуктов легко извлекаются легкой фракцией кашпирской сланцевой смолы, кипящей в пределах 100—200°.
Достаточно обработать сульфокислоты или сульфомыла этой фракцией, в которой хорошо растворяются продукты уплотнения нейтральных кислородных, непредельных и части сернистых со
Моющие, смачивающие и эмульгирующие средства
121
единений, чтобы отделить смолистые примеси от сульфопродуктов.
Для отделения смолистых примесей от сульфопродуктов последние разбавляются водой в отношении 1 :4 и троекратно обрабатываются в мешалке 10%-ным количеством легкой фракции, считая на обрабатываемый раствор.
После разделения верхний слой (легкая фракция с растворенными смолистыми примесями) сливается, а нижний слой (сульфопродукты) выпаривается до необходимой концентрации 60-70%.
В табл. 3 приведена характеристика сульфомыла, очищенного описанным выше способом; характеристика этого сульфомыла до очистки дана в табл. 2.
Таблица 3
Характеристика сульфомыла, обработанного растворителем для удаления смолистых примесей
Контролируемая величина
Показатели
Содержание, %:
воды........................
свободной щелочи.............
сульфата натрия ............
смолистых примесей..........
35,0
0,14
4,0 следы
После удаления продуктов уплотнения упомянутых выше нестойких соединений сульфомыло теряет свой неприятный запах и приобретает светложелтый цвет. Это сделало возможным применение сульфопроизводных сланцевой смолы в промышленности в качестве моющих, эмульгирующих и смачивающих средств.
В текстильной промышленности очищенное сульфомыло было испытано во всех операциях варки, отбеливания и крашения ткани, вместо жирового мыла, ализаринового масла и контакта. Испытания дали положительные результаты [3]. Прежде всего сульфомыло было испытано в процессе обработки тех сортов ткани, где наличие смачивателей, моющих средств и эмульгаторов является совершенно необходимым для получения кондиционного товара. С большим успехом сульфомыло было испытано в процессе крашения бумазеи-корд. Это — тяжелая в обработке техническая ткань и ее крашение должно укладываться в очень жесткую качественную и цветовую вилку. В рецепт крашения этого сорта ткани обязательно входил или контакт, или ализариновое масло.
При введении в рецепт сульфомыла взамен ализаринового масла в том же количестве были получены хорошие результаты,
122 В. Н. Лапин
которые позволяют считать сульфомыло равноценным по качеству ализариновому маслу.
Бумазеи-корд выпускается промышленностью миллионы метров в год, и потребность в дефицитном ализариновом масле только для выпуска этой ткани измеряется десятками тонн в год.
Исключительно положительные результаты в смысле повышения капиллярности ткани были получены при введении сульфомыла в варочные щелока при отварке партий вуали. Дело в том, что высокая капиллярность ткани ведет к значительному улучшению дальнейшей обработки последней и, в частности, к повышению восприимчивости ткани к красителям.
Обычно для повышения капиллярности ткани в состав варочных щелоков вводится мыло из растительных и животных жиров. При введении мыла из жиров в состав щелоков в количестве 0,5% норма капиллярности после отбелки равна 150 мм за 30 минут. Сульфомыло, введенное в варочные щелока в том же количестве 0,5%, взамен жирового мыла, не только заменяет, но и значительно превосходит последнее по своим техническим качествам: капиллярность ткани увеличивается до 175 мм за 30 минут.
Введение сульфомыла в ванну темносернистого крашения также дало положительные результаты.
Таким образом, в текстильной промышленности сульфопроизводные фракции кашпирской сланцевой смолы с успехом могут заменить дефицитные смачиватели, моющие средства и эмульгаторы.
Испытания сульфомыла в металлообрабатывающей промышленности на ряде заводов в качестве эмульгатора для приготовления охлаждающих эмульсий также дали положительные результаты. По данным одного из заводов, 10%-ная эмульсия с эмульгатором из кашпирской смолы в процессе нарезания резьб вполне заменяет ранее используемую для этих целей масло-олифу. Введение сульфомыла вместо контакта или мыл из жиров в режущие жидкости улучшает их свойства. Это объясняется тем, что во фракциях кашпирской смолы в значительном количестве содержатся сернистые соединения, которые будучи поверхностно-активными веществами, придают эмульсии лучшие режущие и охлаждающие свойства. Кроме того, замечено, что эмульсия со сланцевым эмульгатором обладает антисептическим действием, не загнивает и исключает возможность образования на коже рабочих масляных угрей.
В области приготовления медицинских препаратов, таких как ихтиол, ихтиоловая паста, в качестве эмульгатора тиофеновых фракций пользуются сульфопроизводными II фракции кашпирской смолы. Однако сульфирование этой фракции без предварительного удаления нейтральных кислородных соединений при
Моющие, смачивающие и эмульгирующие средства
123
водит к образованию сульфопродуктов с большим количеством (иногда до 403Ь) смолистых примесей. Последние также не удаляются из сульфопродуктов в процессе их производства. В результате получаются медицинские препараты, нестабильные, имеющие черный цвет и неприятный запах, обусловленный смолистыми примесями. Применение таких препаратов в качестве средств для дезинфекции одежды и помещений встречает противодействие со стороны потребительских организаций, потому что эти препараты имеют неприятный запах и оставляют неудаляе-мые смолистые пятна на тканях одежды и стенах помещений.
Медицинские препараты, приготовленные на эмульгаторах из сланцевой смолы, из которых удалены смолистые примеси, не имеют упомянутых недостатков. Медицинские препараты, изготовленные нами на сульфосолях, очищенных от продуктов уплотнения нейтральных кислородных соединений, стабильны и имеют светлокоричневый цвет. На дезинфицируемых предметах такие препараты совершенно не оставляют смолистых пятен и не сообщают им устойчивого неприятного запаха.
Таким образом и в области производства медицинских препаратов очищенные от смолистых примесей сульфопроизводные могут найти широкое применение.
ВЫВОДЫ
1. На основании изложенного можно сделать заключение, что сульфопроизводные погонов кашпирской сернистой сланцевой смолы могут быть использованы в промышленности в качестве эмульгаторов, смачивающих и моющих средств, вместо контакта, мыл из растительных и животных жиров, ализаринового масла и других применяемых в настоящее время средств.
2. Необходимым условием получения таких сульфопроизводных является удаление из фракции сланцевых смол или из сульфопродуктов, главным образом, нейтральных кислородных, непредельных и, частично, сернистых соединений и продуктов их уплотнения.
ЛИТЕРАТУРА
1. А. М. Бер к ен гейм. «Горючие сланцы» № 4, 25, 1933.
2. Н. К а р а с и к и В. Д об ад к ин. «Горючие сланцы» № 6, 34. 1934.
3. М. Г. К а ч у р и tf и В. Н. Л а и и н. «Текстильная промышленность» ,№ 11, 1948.
Л. И. Гуляева u Н. И. Пышкина
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ КИСЛОРОДНЫХ СОЕДИНЕНИЙ АДСОРБЦИОННО-
ХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ В СМОЛАХ СЛАНЦЕВ ПРИБАЛТИЙСКОГО МЕСТОРОЖДЕНИЯ
В настоящее время известно очень мало методов определения содержания нейтральных кислородных соединений в жидких продуктах полукоксования и коксования горючих ископаемых. Н. Г. Титовым [1] предложен метод, основанный на применении реактива Гриньяра. Сложность технического выполнения явилась причиной того, что этот метод не получил распространения. А'. С. Осокин [2] предложил определять кислородные соединения с помощью специально приготовленного хлористого магния. В. А. Ланин [3] показал ненадежность этого метода.
Широкое распространение получил предложенный Г. Л. Стадниковым [4], метод, основанный на открытой Р. Робинзоном [5] способности органических кислородных соединений давать комплексы с хлорным железом, растворенным в крепкой соляной кислоте. Однако во многих случаях результаты этого метода оказываются сомнительными [6].
Существующие в настоящее время методы определения содержания кислых кислородных соединений основаны на обработке анализируемого вещества раствором щелочи и определении количества увлеченного в щелочь продукта весовым [4] или рефрактометрическим [7] способами.
Однако известно, что при анализе сланцевых смол результаты зависят от концентрации применяемой щелочи, и метод, следовательно, не является абсолютным.
Современное состояние развития переработки сланцевых смол требует надежных сведений о содержании в них кислородных соединений. Поэтому разработка иных методов в настоящее время представляет не только бесспорный интерес, но и необходимость.
Настоящая статья посвящена описанию разработанного нами адсорбционно-хроматографического метода, пригодного для определения содержания нейтральных кислородных соединений в обесфеноленных фракциях, или суммарного содержания кис
Определение содержания кислородных соединений
125
лых и нейтральных кислородных соединений в сырых фракциях сланцевых смол и продуктах их переработки. Предлагаемый метод одновременно является аналитическим методом выделения кислородных соединений.
ОПЫТНАЯ ЧАСТЬ
Поскольку практически невозможно составить искусственные смеси индивидуальных компонентов, отвечающие составу изучаемой фракции смолы, разработка методики была проведена на метанольном экстракте обесфеноленной фракции 180—300° генераторной смолы, полученной с комбината Кохтла-Ярве, и проверена затем на ряде других продуктов. Содержание кислорода в продуктах разделения указывало на ту или иную степень разделения.
В качестве адсорбента применяли силикагель, выпускаемый заводом «Красный Химик» по временным техническим условиям для хроматографии.
Проявителями служили бензол и метанол.
Ниже кратко описывается примененная нами методика. В колонку диаметром 10 мм засыпали определенное количество силикагеля, утрамбовывали до тех пор, пока силикагель не переставал опускаться, и затем заливали взвешенное количество анализируемого продукта. После того как весь продукт входил в силикагель, в колонку сверху последовательно приливали определенное количество бензола, вымывающего углеводородную часть продукта, и метанол, вытесняющий кислородные соединения. Из колонки снизу раздельно отбирали бензольную и метанольную фракции в предварительно взвешенные колбочки. Обычно в практике хроматографического анализа о переходе от одного класса соединений к другому судят по изменению значения коэффициента преломления пъ' в последовательно отбираемых фракциях. В данном случае этот способ неприменим, так как значения коэффициента преломления По для смесей углеводородов и кислородных соединений могут быть очень близкими друг к другу. Поэтому мы применили иную методику: в анализируемый продукт, перед заливом в колонку, вводили незначительное количество азокрасителя «оранжевый». Адсорбируемость на силикагеле этого азокрасителя, так же как и кислородных соединений, значительно превышает адсорбируемость углеводородов любого строения. Поэтому при отборе фракций из низа колонки появление окрашенного фильтрата указывало на переход от бензольно-углеводородной фракции к метанольной, содержащей кислородные соединения. Подобный принцип был применен О’Коннор [8] при анализе синтола.
126
Л. И. Гуляева и Н. И. Пышкина
Растворитель из отобранных фракций отгоняли на водяной бане. Остатки растворителя удаляли высушиванием фракции до постоянного веса в вакуум-эксикаторе при комнатной температуре. Выход фракции, оставшейся после отгона метанола, соответствовал содержанию кислородных соединений в анализируемом продукте.
О точности разделения судили по содержанию кислорода в продуктах разделения, определяемому элементарным анализом (по разности).
Аналогично способу, предложенному Г. С. Ландсберг и Б. А. Казанским для анализа бензинов прямой гонки, количество вещества, которое может быть разделено находящимся в колонке силикагелем, рассчитывали, исходя из активности последнего, определение которой проводили следующим образом: 20 мл смеси, состоящей из 10% ацетона и 90% бензола (Пр смеси = 1,4855), пропускали через 10 г силикагеля, помещенного в бюретки, имеющие диаметр 10 мм. В последовательно отбираемых из низа колонки фракциях объемо?л в 0,5 мл, определяли коэффициент преломления п~и . по значению которого судили о появлении ацетона. Исходя из количества выделенного чистого бензола, рассчитывали активность, выраженную в количестве ацетона, адсорбирующегося ста граммами силикагеля. Так, для применяемого нами силикагеля получили 8,5 мл чистого бензола.
Следовательно, активность силикагеля aY равна: __ т-п-100 _ 8,5-10-100
С1^ (100-(г)й - (100—10) • 10 = У’4’
где: т — количество выделившегося чистого бензола, мл-,
П—процентное (по объему) содержание ацетона в исходной смеси;
£ —количество взятого силикагеля, г.
Приняв, что адсорбируемость кислородных соединений смолы на силикагеле не ниже адсорбируемости ацетона, рассчитывали количество продукта х, которое может быть разделено определенным количеством силикагеля (в данном случае вес силикагеля был равен 30 г). Расчет величины х производили по формуле:
„ щК 9,4 • 30 • 0,7 х = ----= 4,9 = о мл,
о 40 ’
где:£—количество силикагеля, г;
£ —предполагаемое примерное содержание кислородных соединений в анализируемом продукте, объемн. %;
К —коэффициент, учитывающий снижение эффекта разделения, вследствие наличия полициклических ароматических углеводородов и неполного использования силикагеля.
Определение содержания кислородных соединений 127
Далее, путем экспериментального подбора, нашли, что на 1 г углеводородов, содержащихся в анализируемом продукте, требуется при разделении не менее 10 мл бензола и не менее 10 мл метанола на 1 г взятого на анализ продукта. Установили влияние на эффективность разделения отношения высоты слоя адсорбента к диаметру колонки и времени фильтрования. Оказалось, что изменение значения -j- от 15 до 70 практически не сказывается на четкости разделения. Также не имеет значения изменение времени фильтрования в пределах от 14 до 3 часов.
Установив условия адсорбционного хроматографического разделения на примере метанольного экстракта дизельной фракции генераторной смолы, мы проверили применимость данного метода на различных продуктах переработки сланцевой смолы. Были проанализированы сырая и обесфеноленная фракции 180— 300° генераторной смолы, рафинат и экстракт метанольной обработки обесфеноленной фракции 180—300° и обесфеноленная фракция 300—400° той же смолы, отобранная при остаточном давлении 6 мм.*
Полученные результаты представлены в табл. 1 и 2.
Во всех случаях содержание кислорода в углеводородной части не превышает 0,3%, т. е. находится в пределах ошибки опыта.
Содержание кислорода во фракции кислородных соединений колеблется от 10 до 13%, в зависимости от ее состава. Следовательно, происходит четкое отделение кислородсодержащих соединений. Расхождения между параллельными определениями не превышают 1 %. Потери при разделении 3—4 г продукта составляют 2—5%.
Одинаково хорошие результаты были получены при анализе как обесфеноленных фракций, так и сырых фракций смолы. В первом случае отделяются нейтральные кислородные соединения, во втором — нейтральные и кислые. Четкое разделение было получено и при разделении фракции, выкипающей в пределах 300—400°.
В заключение следует отметить, что предлагаемый нами метод прост, требует применения сравнительно дешевых реактивов и позволяет анализировать малые количества продукта.
ВЫВОДЫ
1. Разработан адсорбционно-хроматографический метод, определения содержания кислородных соединений во фракциях смол сланцев прибалтийского месторождения, выкипающих в пределах 180—400°, и продуктах их переработки.
Таблица 7
Результаты определения содержания кислородных соединений в продуктах переработки генераторной сланцевой смолы адсорбцнонно-хроматографнческнм методом
Наименование анализируемого продукта
Сырая необесфенолен-ная фракция 180—300° Обесфеноленная фракция 180—300° Рафинат метанольной обработки обесфе-нолениой фракции 180—300° Экстракт метанольной обработки обесфеноленной фракции 180—300°
Анализ 1 | Анализ 2 Анализ 1 | Анализ 2 Анализ 1 | Анализ 2 Анализ! 1 | Анализ 2
Количество анализируемого продукта, г 3,95 4,40 3,64 4,04 3,84 3,85 4,16 15,20
Углеводороды:
количество, г 2,94 3,29 2,94 3,28 3,32 3,28 2,34 8,81
„ вес. % от продукта . . . 71 74 80 81 86 85 57 58
„20 nD 1,4908 1,4900 1,4880 1,4890 1,4780 1,4788 1,5491 1,5487
Элементарный состав, %:
С 86,87 87,0 87,45 88,50 88,00
Н 11,70 11,98 11,90 9,03 9,34
S 1,13 0,93 0,63 2,30 2,07
O + N 0,30 0,10 0,02 0,17 0,34 + 0,25!
Кислородные соединения: 0,53
количество, г . 0,94 1,00 0,53 0,47 0,47 1,60 6,17
„ вес. % от продукта . . . 24 22 15 13 12 12 39 40
„20 nD 1,5180 — 1,4789 1,4755 1,5007 1,5017 1,4789 1,4783
Элементарный состав, %:
С 76.86 78,75 77,75 78,20 77,81
Н 9,32 11,05 10,32 10,47 10,66
S 0,35 0,87 1,40 0,70 0,84
O+N 13,50 9,33 10,46 10,63 10,23 + 0,46
Потери, г 0,20 0,11 0,17 0,23 0,05 0,10 0,18 0,12
„ вес. % от продукта 5,0 4,0 5,0 5,7 2,0 3,0 4,0 1,6
1 Азот.
Определение содержания кислородных соединений
129
Таблица 2
Результаты определения содержания кислородных соединений в продуктах переработки генераторной сланцевой смолы адсорбционно-хроматографическим методом
Наименование анализируемого продукта
Показатели Обесфеноленная фракция 180—300°, смешанная с 10% нейтральных кислородных соединений Обесфеноленная фракция 300—400° (6 мм)
Анализ 1 Анализ 2
Количество анализируемого про-
дукта, г 3,79 3,89 3.0
Углеводороды:
количество, г . . 2,62 2,67 1,77
„ вес. % от продукта 69 68 41
п20 nD 1.4903 1,4900 —
Элементарный состав, %:
С 86,80 87,84
н 11,71 10,66
S 1,35 1,47
О-: \ 0,14 0,03
Кислородные соединения:
количество, г . . . 1,03 1,08 2,48
„ вес. % от продукта 27 27 58
п20 nD ' 1,4791 1,4801 1,5263
Элементарный состав, %:
С 77,86 79,36
н 10,62 9,28
S 1,02 0,83
O-j-N 10,50 10,32
Потери, г 0,14 0,14 —
» вес. % от продукта . . . 4 3 —
2. Метод пригоден для определения содержания нейтральных кислородных соединений в обесфеноленных фракциях и для определения суммы всех кислородных соединений в сырых фракциях смол.
3. Предлагаемый метод одновременно является аналитическим методом выделения кислородных соединений.
4. Примененный принцип определения границы раздела углеводороды — кислородные соединения по изменению окраски позволяет рекомендовать данный метод как простой и практически удобный.
9 Труды ВПИППС.
130
Л. И. Гуляева u Н. И. Пышкина
ЛИТЕРАТУРА
1. Н. Г. Тито в. ХТТ, № 7, 1931. стр. 7.
2. А. С. Осокин. ЖОХ, 1939, т. IX, стр. 13—14.
3. В. А. Ланин. Рефераты научно-исследовательских работ за 1945 г. АН СССР, отдел технических наук.
4. Г. Л. Стадников. Анализ и исследование углей, изд. АН СССР, 1936, стр. 169.
5. R. Robinson. J. Chem. Soc., 1925, т. L, стр. 127, 768.
6. Л. И. Гуляева и Н. И. Пышкина. Настоящий сборник, ст'р. 131.
7. А. М. Кунин, А. Н. М е л ь н и к о в а и Т. С Л агу тки на. Труды ВНИГИ, 1952, выл. IV, стр. 234.
8. О’Копп о г. Ind. and Eng. Ch., 1948, т. 40, № 11, стр. 2102.
9. Инструкция № 2. Хроматографический адсорбционный анализ, из книги — Определение индивидуального состава бензина прямой гонки. Проект инструкции АН СССР, 1950, стр. 28.
Л. И. Гуляева u Н. И. Пышкина
к ВОПРОСУ ОБ ОПРЕДЕЛЕНИИ СОДЕРЖАНИЯ НЕЙТРАЛЬНЫХ КИСЛОРОДНЫХ СОЕДИНЕНИЙ В СЛАНЦЕВЫХ СМОЛАХ ХЛОРНЫМ ЖЕЛЕЗОМ
Единственным способом определения содержания нейтральных кислородных соединений, получившим широкое распространение, является способ, предложенный Г. Л. Стадниковым [1]. Этот способ основан на открытой Р. Робинзоном [21 способности органических кислородных соединений давать комплексные соединения с хлорным железом, растворенным в крепкой соляной кислоте.
Предлагаемая указанными выше авторами методика заключается в следующем: замеренное количество исследуемого продукта взбалтывают в делительной воронке с равным объемом насыщенного раствора хлорного железа в крепкой соляной кислоте (уд. вес 1,19) в течение нескольких минут и затем оставляют в покое; вскоре происходит разделение смеси на три слоя: 1) верхний, содержащий углеводороды, 2) средний, представляющий густую, почти черного цвета массу комплексных соединений и 3) нижний, состоящий из солянокислого раствора хлорного железа; слои разделяют, комплексные соединения разлагают действием водного раствора едкой щелочи, а выделившиеся кислородные соединения отгоняют с водяным паром, сушат и взвешивают.
В другом варианте анализа, более распространенном в практике лабораторных работ, комплекс не выделяют, а ограничиваются замером объема невошедшей в реакцию части смолы и по разности определяют объем прореагировавших кислородных соединений. Анализ проводят в приборе типа сульфатора.
Этот вариант анализа широко применяется в настоящее время как при анализе сланцевых смол, так и полукоксовых смол других горючих ископаемых.
Однако результаты, получаемые по этому методу, часто оказываются сомнительными. В первые годы своего существования метод подвергался критике, причем, главным образом, с позиций изучения реакции с хлорным железом индивидуальных
9*
Л. И. Гуляева u Н. И. Пышкина
соединений. И. Б. Раппопорт [3] показал, что хлорное железо: 1) в крепкой кислоте частично реагирует с углеводородами, имеющими в молекуле третичный атом углерода, 2) хлорирует непредельные углеводороды, 3) образует комплекс с сернистыми соединениями. К- С. Курындин [4] наблюдал, что если кислородные соединения находятся в среде, богатой ароматическими углеводородами, комплексное соединение хлорного железа способно растворяться в этих углеводородах. Н. Н. Шуйкин и Е. А. Тимофеева [5], занимаясь изучением химической природы светлых фракций дегтя будаговских сапропелитов, отмечали, что концентрированный раствор хлорного железа в соляной кислоте, наряду с нейтральными кислородными соединениями, удаляет значительное количество непредельных соединений. Однако никаких конкретных поправок не предложено и не введено до настоящего времени, и описываемый метод продолжают применять в его первоначальном виде.
Занимаясь изучением состава дизельной фракции генераторной смолы и продуктов ее переработки адсорбционным хроматографическим методом, мы столкнулись с фактом очень большого расхождения результатов, получаемых по методам Г. Л. Стадникова (с хлорным железом) и адсорбционно-хроматографическому (табл. 1). Так, содержание нейтральных кислородных соединений в обесфеноленной фракции сланцевой смолы 180— 300° равно 22% по методу Г. Л. Стадникова и 14,5% по адсорбционно-хроматографическому, а в метанольном экстракте той же фракции — 92% по методу Г. Л. Стадникова и только 41,5% по адсорбционно-хроматографическому методу. Та же картина была получена при анализе специально составленных искусственных смесей с различным содержанием кислородных соединений. Компонентами для этих смесей послужили углеводородная фракция и фракция кислородных соединений, полученные хроматографическим разделением обесфеноленного метанольного экстракта фракции 180—300° сланцевой генераторной смолы. Представленный в табл. 2 элементарный состав этих компонентов свидетельствует о полном отсутствии кислородсодержащих соединений в углеводородной фракции.
Таким образом, судя по данным, представленным в табл. 1, во всех приведенных случаях содержание нейтральных кислородных соединений, найденное по методу Г. Л. Стадникова, значительно выше найденного хроматографическим методом, причем разница в некоторых случаях достигает 50% (абсолютных). Результаты же хроматографического метода отличаются от фактического содержания всего лишь на 1—1,5%.
В табл. 3 приведены результаты анализа 3 продуктов по методу Г. Л. Стадникова при условии разбавления анализируемых продуктов различным количеством гептана.
К определению содержания нейтральных кислородных соединений 133
Таблица 1
Сравнительные результаты определения нейтральных кислородных соединений в сланцевых продуктах по методам Г. Л. Стадникова и адсорбционно-хроматографическому
Наименование анализируемого Содержание нейтральных кислородных соединений, объе.мн. %
продукта по методу Г. Л. Стадникова по адсорбционнохроматографическому методу
Кислородные соединения метанольного экстракта обесфеноленной фракции 180300е Углеводородная фракция метанольного экстракта обесфеноленной фракции 180— 300° Рафинат метанольной обработки обесфеноленной фракции 180—300° Обесфеноленная фракция сланцевой генераторной смолы 180—300° ...... Смесь, содержащая 74% углеводородной фракции и 26% кислородных соединений метанольного экстракта Экстракт метанольной обработки обесфеноленной фракции 180—300° Смесь, содержащая 26% углеводородной фракции и 74% кислородных соединений метанольного экстракта 100 3 14,0 22,0 58,5 92,0 97,0 100 0 12,5 14,5 25,0 41,5 72,5
Таблица 2
Характеристика компонентов искусственных смесей
Компонент искусственной смеси 20 nD ^20 20 Элементарный состав, %
с Н S ’'O+N
Кислородные соединения . . 1,4685 0,9090 79,12 10,37 0,41 10,10
У глеводороды 1,5507 0,9581 87,10 10,78 2,12 0
Оказалось, что с увеличением степени разбавления содержание кислородных соединений, определяемое этим методом, снижается, приближаясь к данным хроматографического метода (кривые, рис. 1).
134
Л. И. Гуляева и Н. И. Пышкина
Таблица 3
Результаты анализа трех сланцевых продуктов, разбавленных гептаном, по методу Г. Л. Стадникова
Наименование анализируемого продукта Разбавление, объемы гептана на 1 объем анализируемого продукта Содержание н. к. с., объемн. % Разница в результатах определения двумя методами, абс. %
по метод}' Г. Л. Стадникова по хроматографическому методу
Смесь, содержащая 74%
углеводородной фракции и 26% кислород-
ных соединений . . . 0 58,5 25,0 33,5
То же 0,7 30,8 25,0 10,8
1,0 27,7 25,0 2,7
. 3,0 26,0 25,0 1,0
Экстракт метанольной
обработки обесфеноленной фракции 180—
300° 0 92,0 11,5 50,5
То же 0,3 76,0 41,5 34,5
1,0 59,0 41,5 17,5
1,8 48,5 41,5 7,0
3,0 . 44,0 41,5 2,5
5,7 43,3 41,5 1,8
Смесь, содержащая 26%
углеводородной фракции и 74% кислород-
ных соединений . . . 0 97,0 71,5 25,5
То же 3,0 82,0 71.5 10,5
5,2 72,0 71,5 0,5
” ” 9,0 72,0 71,5 0,5
Следовательно, в процессе анализа, при взбалтывании анализируемого продукта с раствором хлорного железа, происходит физическое растворение углеводородов в образующемся комплексном слое. Чем выше содержание кислородных соединений в продукте, тем, естественно, требуется и большее разбавление для извлечения растворенных в комплексном слое углеводородов.
Г. Л. Стадников [6] в 1934 г. отмечал наблюдаемое им растворение комплексным слоем декалина и производных тиофена. Данных о растворимости комплексным слоем других углеводородов в опубликованной к настоящему времени литературе нет. При исследовании фракций 180—300° сланцевой генераторной
К определению содержания нейтральных кислородных соединений 135
смолы адсорбционным хроматографическим методом нами были выделены продукты, представляющие собой концентраты углеводородов различного строения. Принадлежность этих продуктов к тому или иному классу углеводородов и их чистота были доказаны физико-химическими константами и подтверждены химическими реакциями. Из этих концентратов были приготовлены
Число частей гептана на 1 часть исследуемого продукта
Рис. 1. Содержание нейтральных кислородных соединений, определяемое солянокислым раствором РеС13 по методу Г. Л. Стадникова при различном разбавлении анализируемого продукта:
4—смесь, содержащая 74% углеводородной фракции и 26% кислородных соединений; 2 — экстракт метанольной обработки обесфеноленной фракции 180—300° С; 3 — смесь, содержащая 26% углеводородной фракции н 74% кислородных соединений.
смеси с концентратом кислородных соединений (характеристика последнего приведена в табл. 2) в соотношении 1:1. В полученных смесях определено содержание кислородных соединений по методу Г. Л. Стадникова (обычным способом, без разбавления). Оказалось (табл. 4), что наибольшей растворимостью в комплексном слое обладают ароматические углеводороды — производные нафталина (смесь 3). В данном случае они целиком растворились в комплексном слое, и найденное содержание кислородных соединений оказалось равным 100%, вместо 50%-ного фактического содержания.
Несколько слабее растворяются ароматические углеводороды — производные бензола (смесь 2). При анализе смеси этих
136
Л. И. Гуляева u Н. И. Пышкина
Таблица 4'
Результаты определения нейтральных кислородных соединений в искусственных смесях их с различными углеводородами по методу Г. Л. Стадникова
Наименование
углеводородного
концентрата-ком-
s понента анализи-g руемой смеси g
Смесь парафиновых, нафтеновых и олефиновых углеводородов ............
Ароматические углеводороды, производные бензола . . . .
Ароматические углеводороды, производные нафталина . . .
Характеристика углеводородного компонента смеси.
Элементарный состав, %
Содержание в смеси нейтральных кислородных соединений, объемн. %
d20
20
„20 nD
0,56
0
0,59
50
50
50
58,7
87,0
100,0
1
2
3
С
Н
S
O + N
углеводородов с кислородными соединениями найденное содержание последних оказалось равным 87 %, вместо 50%.
Заметной растворимостью обладают также и неароматические углеводороды (смесь 1), из 50% этих углеводородов, содержавшихся в анализируемой смеси, в комплексном слое растворилось 8,7%.
ВЫВОДЫ
1. При определении содержания нейтральных кислородных соединений во фракциях сланцевых смол и их продуктах переработки (кипящих в пределах 180—300° С) объемным методом Г. Л. Стадникова получаются завышенные результаты за счет, растворения комплексным слоем, главным образом, ароматических углеводородов различного строения.
2. Определение содержания нейтральных кислородных соединений по методу Г. Л. Стадникова следует производить при разбавлении исследуемого вещества гептаном или нефтяным эфиром (фракция 60—78°).
К. определению содержания нейтральных кислородных соединений 137
3. Определение следует производить несколько раз, постепенно повышая степень разбавления. Окончательным следует считать результат, не отличающийся более чем на 1—2% от предыдущего.
ЛИТЕРАТУРА
1. Г. Л. Стадников. Анализ и исследование углей. Изд. АН СССР, стр. 169, 1936.
2. R. R о b i n s о п. J. Chem. Soc, стр. 127, 768, 1925.
3. И. Б. Раппопорт. ХТТ, т. VI, вып. 5, стр. 444, 1935.
4. К. С. Курындин. ХТТ. т. VI, вып. 6, стр. 539, 1935.
5. Н. Н. Ш у й к и н и Е. А. Т и м о ф е е в а. Изв. АН СССР, отд,, химии, наук № 2, 1951.
6. Г. Л. Стадников. ХТТ, т. V, вып. 2, стр. 136, 1934.
Г. Н. Гарновская
РЕФРАКТОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ФЕНОЛОВ В СЛАНЦЕВЫХ ПРОДУКТАХ1
Неоднократно отмечалось, что весовой способ определения содержания фенолов в сланцевых продуктах является условным, не отличается большой точностью и требует длительного времени для выполнения анализа.
В 1950 г. сотрудниками ВНИГИ был предложен способ определения содержания фенолов по коэффициенту преломления фенолятного раствора.
Показатель преломления раствора какого-либо вещества в растворителе, не взаимодействующем с растворенным веществом, является аддитивной величиной и может быть вычислен по формуле:
_ X /7! +(100—х) п2
ПР !00
где: пр — показатель преломления раствора;
— показатель преломления растворенного вещества;
п2 — показатель преломления растворителя;
х —процентное содержание растворенного вещества в растворе.
При взаимодействии между растворенным веществом и растворителем аддитивность может нарушаться. Однако С. С. Стрелковым (ВНИГИ) было экспериментально показано, что п растворов фенолов в водной щелочи в первом приближении определяется суммой парциальных показателей преломления фенолов и водного раствора щелочи, причем, по данным автора, это правило остается справедливым как для растворов, содержащих избыточное количество щелочи, так и для растворов, содержащих свободные фенолы.
Для щелочных растворов фенолов могут считаться установленными следующие положения: 1) показатели преломления различных фенолов в щелочном растворе мало отличаются друг
1 В работе принимали участие В. В. Сизоненко и О. В. Постнова.
Рефрактометрический способ определения фенолов
139
от друга и 2) показатель преломления щелочного раствора фенолов увеличивается пропорционально повышению концентрации фенолов в растворе.
На основании этих двух положений рефрактометрический способ был рекомендован для определения суммарного содержания фенолов в различных полукоксовых смолах.
При определении фенолов по этому методу навеска анализируемого вещества, растворенная в бензоле, обрабатывалась раствором едкого кали с известным показателем преломления, после чего определялся показатель преломления полученного фенолятного раствора. Расчет производился по формуле:
где: х— содержание фенолов в исследуемой фракции;
а —количество фенолов в граммах, соответствующее показателю преломления раствора фенолятов;
Ь — навеска фракции в граммах.
Величина а определяется по табл. 1, составленной ВНИГИ для определения весового количества фенолов во фракции полукоксовых смол.
Простота определения и хорошая сходимость получаемых результатов (по данным ВНИГИ среднее относительное отклонение при определении не превышает 1 %) делают очень заманчивым применение метода для анализа сланцевых продуктов.
Нами была проведена работа по проверке применимости описанного выше рефрактометрического способа определения фенолов к сланцевым продуктам.
А. М. Кунин нашел, что показатель преломления Ид* различных фенолов, взятых для определения в количестве 1 г, колеблется в пределах 1,3762—1,3770 (см. табл. 2).
При определении показателя преломления фенолятных растворов отдельных узких фракций фенолов дизельной фракции генераторной сланцевой смолы были получены результаты, представленные в табл. 3. В той же таблице даны показатели преломления щелочных растворов а- и /3- нафтолов. Как видно из приведенных данных, сланцевые фенолы как будто подчиняются общей закономерности, хотя изменение значений По для щелочных растворов одно-двухградусных фракций этих фенолов несколько больше, чем то, которое наблюдал А. М. Кунин. Величина п2г> колеблется от 1,3750 до 1,3780.
Обращает на себя внимание резкое увеличение По для щелочных растворов чистых а- и нафтолов.
На фракции 272—282° С фенолов, выделенных из генераторной смолы, была проверена зависимость Ид щелочного рас-
140
Г. Н. Гарновская
Таблица 1
Определение весового количества фенолов во фракциях полукоксовых смол по показателю преломления раствора фенолятов
(стандартное количество раствора едкого кали — 10 мл) (данные ВНИГИ)
Весовое содержание фенолов во взятой навеске фракции, г Показатель преломления „20 nD Весовое содержание фенолов во взятой навеске фракции, г Показатель преломления „20 nD
0,0000 0,0493 1,3585 1,3595 1,0718 1,1008 1,3769 1,3773
0,0996 1,3602 1,1187 1,3777
0,1490 1,3611 1,1523 1,3780
0,1998 1,3620 1,1689 1,3783
0,2530 1,3630 1,1998 1,3787
0,3009 1,3640 1,2186 1,3791
0,3516 1,3647 1,2497 1,3795
0,3993 1,3655 1,2700 1,3799
0,4531 1,3662 1,2996 1,3804
0,5000 1,3672 1,3203 1,3806
0,5527 1,3680 1,3500 1,3809
0,6007 1,3691 1,3702 1,3813
0,6514 1,3700 1,4016 1,3818
0,7010 1,3708 1,4190 1,3822
0,7517 1,3717 1,4500 1,3828
0,8004 1,3722 1,4682 1,3831
0,8497 1,3730 1,4994 1,3837
0,8720 1,3735 1,5222 1,3841
0,9012 1,3740 1,5485 1,3846
0,9216 1,3746 1,5894 1,3850
0,9502 1,3752 1,6018 1,3855
0,9714 1,3754 1,6222 1,3857
1,0011 1,3758 1,6510 1,3863
1,0216 1,3761 1,6712 1,3865
1,0523 1,3765
Таблица 2
Показатель преломления щелочных растворов различных
фенолов (данные ВНИГИ)
Наименование продукта Навеска, Количество раствора КОН, „20 nD
МЛ
Карболовая кислота 1,0 10,0 1,3776
Орто-крезол . . . 1,0 10,0 1,3770
Крезольная фракция . . 1,0 10,0 1,3762
Резорцин .... 1,0 10,0 1,3770
Пирогаллол . . . 1,0 10,0 1,3770
Фенолы, выделенные из
керосиновой фракции
журинской полукоксо-
вой смолы • . 1,0 10,0 1,3767
Рефрактометрический способ определения фенолов
141
Таблица 3
Показатель преломления щелочных растворов узких фенольных фракций генераторной сланцевой смолы и индивидуальных нафтолов
Наименование продукта Навеска, г Количество раствора КОН, мл w20
•Фенолы дизельной фракции генераторной смолы . 1,о 10,0 1,3780
То же, фракция 77— 78° (р = 3 мм) . . . 1,0 10,0 1,3770
„ „ 88-- 89° (р = 3 мм) . . . 1,0 10,0 1,3750
100—ЮГ (р=3 мм) . . . 1,0 10,0 1,3760
„ „ 128—130° (р=3 мм) . . 1,0 10,0 1,3770
142—144° (р = 3 мм) . . 1.0 10,0 1,3770
„ „ 144—146° (р = 3 мм) . . . 1.0 10,0 1,3775
,, „ 148—149° (р = 3 мм) .'. . 1,0 10,0 1,3780
а - нафтол 1,0 10,0 1,3840
[1 - нафтол ... 1,0 10.0 1,3880
твора от концентрации в нем фенолов. Результаты определений даны в табл. 4.
Эти результаты полностью подтвердили данные ВНИГИ, и в дальнейшем исследовании мы пользовались приведенной выше подробной калибровочной таблицей, составленной ВНИГИ.
Таблица 4
Зависимость щелочного раствора от концентрации в нем фенолов (от навески анализируемого продукта)
Навеска, г п20 nD Навеска, г и20 "°
0,0000 1,3585 0,5999 1,3685
0,0500 1,3600 1,0003 1,3770
0,2002 1,3620 1,2000 1,3790
0,4005 1,3655 1,4001 1,3820
Как видно из результатов определения содержания фенолов во фракциях сланцевой смолы весовым и рефрактометрическим способами (табл. 5), рефрактометрический способ дает очень хорошую сходимость в параллельных пробах.
В то же время по абсолютной величине результаты определения содержания фенолов рефрактометрическим способом для всех фракций значительно превышают количества фенолов, определяемые весовым способом. Особенно это становится заметным при анализе высших фракций фенольных концентратов. В нашем
142
Г. Н. Тарнавская
Таблица 5
Определение фенолов в сланцевых продуктах
Наименование анализируемого продукта Содержание фенолов, вес. % Относительная ошибка определения, %
весовой метод рефрактометрический метод весовой метод рефрактометрический метод
Фракция туннельной смолы, от н. к. до 225° С Фракция генераторной смолы 200—325° С . . 0,9—1,34 1,96—1,97 38,6 0,508
14.2—15,8 15,2—15,2 10,7 0,00
Фракция генераторной смолы 325—400° С . . 26,4-31,3 37,4—37,35 17,0 0,134
распоряжении имелись две вакуумные фракции сланцевых фенолов, выкипающие в температурных пределах 300—380° и 380—420°, выделенные щелочью из генераторной смолы. При определении в этих фракциях нейтральных масел весовым способом было найдено: в 1-й фракции 18—20% и во 2-й фракции 18—17% указанных масел.
Учитывая условность метода определения нейтральных масел, было определено содержание фенолов в 1-й и 2-й фракциях рефрактометрическим способом. Результаты этих определений даны в табл. 6.
Таблица б Определение фенолов в тяжелых фракциях
Наименование анализируемого продукта Количество КОН, мл Взято продукта, г л20 nD Найдено фенолов, г Относительная ошибка, %
( 10,0 0,2500 1,3650 0,3700 48,0
Фракция 1 (300—380°) 1 10,0 10,0 0,5002 1,0000 1,3690 1,3780 0,6000 1,1523 20,0 15,2
1 10,0 2,0000 1,3940 2,1402 7,0
Фракция 2 (380—420°) | 10,0 10,0 0,5010 1,0050 1,3690 1,3780 0,6000 1,1523 20,0 15,2
Из данных табл. 6 видно, что даже если считать исследуемые продукты за чистые фенолы, не содержащие нейтральных масел, при определении получаются чрезвычайно завышенные цифры. При этом нарушается прямая пропорциональная зависимость между и концентрацией фенолов в щелочном растворе. Повидимому, причинами этого являются усложнение структуры фенолов и наличие среди них нафтолов, которые, как уже указывалось выше, не подчиняются общей закономерности.
Рефрактометрический способ определения фенолов
143
Ранее было установлено, что извлечение фенолов щелочью при весовом методе определения сопровождается растворением в щелочи большего или меньшего, в зависимости от концентрации применяемой щелочи, количества нейтральных кислородных соединений. При определении фенолов рефрактометрическим способом этот источник ошибки не исключается, часть нейтральных кислородных соединений переходит в фенолятный раствор, что определенным образом отражается на значении показателя преломления. При определении фенолов весовым способом мы обычно пользовались 10%-ным раствором щелочи, тогда как ВНИГИ рекомендует работать с 15%-ным раствором.
Проведенная Г. В. Татаркиной проверка влияния концентрации щелочи, применяемой для анализа, на количество определяемых фенолов показала, что изменение концентрации щелочи от 10 до 15% не сказывается на результатах анализа, при условии взятия достаточно большой навески (согласно методике ВНИГИ). Применение 5%-ного раствора КОН в количестве 10 мл не обеспечивает полноты извлечения фенолов. В случае малых навесок (1 г для смолы) в раствор переходит большое количество нейтральных масел.
Таблица 7
Определение фенолов в присутствии карбоновых кислот
Наименование анализи- руемого продукта Количество КОН, мл Навеска, 2 Л30 nD Найдено «фенолов» г
Кислоты генераторной смолы, фракция 200— 250° С 10,0 0,0885 1,3620 0,1998
То же 10,0 0,3071 1,3605 0,1000
10,0 0,6372 1,3639 0,3005
10,0 1,0304 1,3645 0,3510
Фенолы дизельной фракции + кислоты фракции 200—250° С 10,0 1,0076 0,5003 1,3731 0,8497
Фенолы дизельной фракции + 10,0 1,0002 1,3700 0,6514
кислоты фракции 200—250° С Фенолы дизельной фракции + 10,0 0,0712 1,0000 1,3750 0,9502
кислоты фракции 200—250° С . . . . Фенолы дизельной фракции 10,0 0,0250 1,0008 1,3759 1,0019
144
Г. Н. Гарновская
При определении фенолов в щелочной раствор, кроме нейтральных масел, переходят карбоновые кислоты; чтобы оценить их влияние на результаты определения были определены п~о щелочных растворов карбоновых кислот, выделенных из дизельной фракции генераторной смолы, а также По° щелочных растворов смесей фенолов и кислот различного состава.
Результаты этих определений приведены в табл. 7.
Из таблицы видно, что величина По щелочных растворов .карбоновых кислот, вне зависимости от концентрации, колеблется в пределах от 1,3620 до 1,3645; присутствие карбоновых кислот в смеси с фенолами мешает рефрактометрическому определению последних.
ВЫВОДЫ
1. Рефрактометрический способ определения содержания фенолов применим для анализа легких и средних фракций сланцевой смолы.
2. Метод неприменим для анализа тяжелых фракций и тяжелых смол, а также для анализа фенольных концентратов, в которых можно предполагать присутствие бициклических фенолов.
3. При определении содержания фенолов рефрактометрическим способом необходимо предварительное удаление карбоновых кислот.
ЛИТЕРАТУРА
1. А. М. Кунин, А. Н. Мельникова и Т. С. ЛагуткиИа. Рефрактометрический метод определения фенолов в продуктах термической переработки топлив. Труды ВНИГИ, вып. IV, 1952.
Е. И. Томина, К. Б- Чернышева и Е. М. Дементьева
СПЕКТРОГРАФИЧЕСКИЙ АНАЛИЗ НЕКОТОРЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ СЛАНЦЕВОЙ СМОЛЫ
Анализ органических соединений методом фотографической спектрофотометрии в ультрафиолетовой части спектра в последнее время нашел широкое распространение в различных областях исследования. В частности, этот метод с успехом используется при контроле производства в нефтяной промышленности, заменив менее эффективные химические методы анализа.
Применение спектрального анализа в сланцевой промышленности освещено лишь в работах Л. Уцу [1, 2, 3], который исследовал легкие фракции смол сланцев месторождения Пуэртол-лано (Испания). Работы, посвященные исследованию фракций, выкипающих выше 200° С, в литературе отсутствуют.
В настоящей статье показана возможность исследования тяжелых фракций сланцевых смол, подвергнутых предварительному адсорбционному разделению на силикагеле.
Работа велась в двух направлениях: 1) контроль полноты хроматографического отделения углеводородной части фракций от кислородсодержащих соединений; 2) идентификация ароматических углеводородов в узких хроматографических фракциях.
Хроматографические фракции для исследования, выделенные из более широких (180—300° С) фракций камерной и генераторной смол, были предоставлены нам Л. И. Гуляевой.
С целью дробного разделения некоторые хроматографические фракции, содержащие полициклические ароматические углеводороды, обрабатывались (Гуляевой Л. И.) пикриновой кислотой с последующим выделением свободных углеводородов из полученных пикратов.
Следует, однако, отметить, что приведенные ниже температуры плавления пикратов в большинстве случаев не отвечали тем углеводородам, которые были спектрально обнаружены в исследуемых фракциях. Последнее объясняется присутствием примесей.
10 Труды ВНИИПС.
146
Е. И. Томина, К. Б. Чернышева и Е. М. Дементьева
Для систематизации излагаемого материала ограничиваемся указанием, кроме температур плавления пикратов, интервала выкипания исследуемых фракций.
Все исследования проводились на отечественном кварцевом спектрографе ИСП-22. Источником излучения служила водородная лампа ГОИ.
Длины волн полос поглощения' определялись с помощью
Рис. 1. Нафталин в гексане.
анализируемого вещества. Идентификация углеводородов производилась путем сравнения спектров поглощения исследуемых фракций со спектрами индивидуальных продуктов.
В " тех случаях, когда отсутствовали чистые углеводороды,, которые могли служить для получения спектров сравнения, использовались литературные данные.
Полную спектральную характеристику нафталина и его производных в ультрафиолетовой области можно найти в работах Т..Де-Ласло [4], Р. Мортона [5] и Л. Ружички [6]. На рис. 1, 2, 3, 4 и 5 показаны некоторые кривые поглощения указанных соединений.
Из рассмотрения приведенных кривых следует, что характер распределения интенсивности в спектре нафталина сохраняется и для всех его производных. Введение метильной группы в нафталиновое ядро вызывает смещение кривой поглощения в красную часть спектра приблизительно на 80 А, в то время как замещение водорода ядра второй метильной группой связано с незначительным сдвигом кривой и возрастанием коэффициента
Спектрографический анализ ароматических углеводородов
147
200 250 300 200 250 300 X А
Тметил- 7-этимафталин Тметил-5-зтилна,фп:алин
Рис. 3. Кривые поглощения алкилпроизводных нафталина.
10*
148
Е. И. Томина, К. Б. Чернышева и Е. М. Дементьева
погашения более, чем в 10 раз. Абсорбционные полосы при этом имеют тенденцию сближаться, и количество их соответственно уменьшается.
Имеется очень мало сведений относительно действия алкенов в боковой цепи на спектр поглощения нафталиновых производных. Однако можно отметить следующее правило: двойная связь в боковой цепи, находящаяся в сопряженном положении к нафталиновому ядру, вызывает смещение абсорбционной кривой
Рис. 4. 2,3,5-триметилнафталин в гексане.
в красную часть спектра и возрастание коэффициентов погашения, причем спектры в этом случае обычно не имеют четкой структуры (см. рис. 5).
Рис. 5. 1—7-метилперинафтен; 2—аценафтилен в этаноле.
Двойная связь, не сопряженная с ядром, почти не влияет на форму и положение спектра. Закрытие алифатического кольца сопровождается деформацией связей и углов валентностей. Подобная деформация создает натяжение в молекуле, что ведет к повышению четкости структуры спектров поглощения.
Ниже приведен, в качестве примера, подробный анализ хроматографических фракций камерной смолы, содержащих в основном углеводороды нафталинового ряда, и дано сравнение их по качественному составу с соответствующими хроматографическими фракциями генераторной смолы.
АНАЛИЗ СЛАНЦЕВЫХ СМОЛ
Камерная смола
Исследовалась фракция 180—300° С камерной смолы, отобранной с промышленного блока комбината Кохтла-Ярве в мае 1951 г.
dffj фракции............ 0,9842
п?? фракции..............1,5712
Спектрографический анализ ароматических углеводородов
149
Содержание ароматических углеводородов (по данным хроматографии) составляет 75—80%.
1. На рис. 6 приведены спектрограммы исследуемой фракции 1 (/кип 54—69° С при р = 5 мм) и нафталина, принятого
Рис. 6. 7 — фракция 1; 2 — нафталин; 3 — фракция 2.
за эталон. Положение полос поглощения в спектрах дано в табл. 1.
Таблица 7
Положение абсорбционных максимумов в спектре фракции (7КИП 54—69° С при р = 5 мм) и нафталина
Наименование анализируемого вещества О Длины волн максимумов поглощения, А
I II III IV V VI VII VIII IX
Фракция 1 . . 3148 3110 3068 3040 ЗОЮ 2994 2975 2930 2881
Нафталин 2 . . 3148 3110 3067 3040 ЗОЮ 2994 2975 2930 2880
Из рис. 1 и 6 и табл. 1 очевидно, что положение полос поглощения и общий характер распределения интенсивности в спектре фракции совершенно точно совпадают с данными для чистого нафталина.
2. На рис. 6 показана спектрограмма фракции 2 (/кип 83—94° С при р = 5 мм), на рис. 7 — спектры поглощения чистых /?-метилнафталина 1, а-метилнафталина 3 и смеси а-и /?-метилнафталинов 2.
150
Е. И Томина, К- Б. Чернышева и Е. М. Дементьева
В табл. 2 помещены длины волн полос поглощения фракции чистых продуктов.
Таблица 2
Положение абсорбционных максимумов в спектрах a-и /3-метилнафталинов
Из сопоставления приведенных данных следует, что исследуемая фракция содержит смесь а- и /?-метилнафталинов, с преобладанием /3-изомера.
Рис. 7. 1 — /J-метнлнафталин; 2— а-метилнафталин; 3 — смесь а- и ^-метилнафталинов.
3. В спектре поглощения фракции 3 (/кип 94—105° С при р = 5 мм) наблюдаются полосы поглощения при следующих длинах волн: 3236, 3222, 3216, 3181, 3134, 3087 А.
Из сопоставления с литературными данными [5, 6, 7] можно сделать заключение, что фракция содержит смесь диалкилзаме-TTTC1TJ ТТТ-Т V ттпгК'по тттлигчп ncivp 1 CUiri.UWD.
Спектрографический анализ ароматических углеводородов
151
Из этой фракции, обработанной пикриновой кислотой, было выделено два пикрата (/„л 111 и 98° С).
Спектрограммы углеводородов, полученных после разложения пикратов, представлены на рис. 8 и 9, положение абсорбционных максимумов — в табл. 3.
Таблица 3
Положение абсорбционных максимумов в спектрах углеводородов, выделенных из пикратов (1ПЛ 111 и 98° С)
Наименование анализируемого вещества О Положение полос поглощения, А
I II 111 IV V VI VII
Фракция З1 /пл пикрата 111° С 3220 3215 3175 3150 3075 2920 2820
Фракция З2 /пл пикрата 98° С — 3215 3170 3151 3100 3075 3000
Рис. 8, Фракция З1.
Рис. 9. Фракция З2.
В табл. 4 помещены значения максимумов полос поглощения для чистых углеводородов, на рис. 3, 4 — соответствующие кривые поглощения.
152
Е. И. Томина, К. Б. Чернышева и Е. М. Дементьева
Таблица 4
Положение абсорбционных максимумов в спектрах некоторых алкилнафталинов
Исследованные соединения
О
Абсорбционные максимумы, А
1,7-диметилнафталин . . . 3220 3175 3150 3075 2915
1,3-диметилнафталин . . . 3218 — 3170 — 3080 2915 2830
1-метил-7-этнлнафталин . . — 3215 3172 3150 3070 2918 2818
2, 3, 5-триметилнафталин 3232 —— 3186 3130 3078 2960 2838
1,6-диметнлнафталин . . . 3220 — — 3150 3080 2910 2815
Из рассмотрения приведенных данных следует, что фракция З1 содержит смесь диметилнафталинов (возможно присутствие 1,7-, 1,3- и 1,6-изомеров). Фракция З2 содержит, главным
Рис. 10. Кристаллы, выделенные из фракции 3 Скип 94—105° С, р = 5 мм).
образом, 1-метил-7-этилнафталин с небольшой примесью три-метилнафталина, очевидно изомера 2, 3, 5.
Из остатка фракции, не дающего соединения с пикриновой кислотой, были выделены белые кристаллы с /пл = 72° С.
Сопоставление спектрограммы кристаллов (рис. 10) и спектра поглощения чистого дифенила (рис. 11) показывает, что исследуемый продукт представляет собой дифенил.
4. Из фракции 4 (/кип 105—120° С при р = 5 мм) было получено три пикрата с температурой плавления 132, 127 и 115° С.
Спектрограммы углеводородов, выделенных из пикратов, представлены на рис. 12, 13, 14. Положение полос поглощения дано в табл. 5.
Спектрографический анализ ароматических углеводородов
153
Рис. 12. Фракция 41.
Рис. 13. Фракция 42.
154
Е. И. Томина, К. Б. Чернышева и Е. М. Дементьева
Таблица 5
Положение абсорбционных максимумов в спектрах углеводородов, выделенных из пикратов (1ПЛ 132, 127 и 115° С)
Наименование анализируемого вещества
Положение полос поглощения, А
I II III IV V VI VII VIII IX
Фракция 41 /пл пикрата 132° С
Фракция 42 /пл пикрата 127° С
Фракция 43 7ПЛ пикрата 115° С
3395
3395 3227
3395 3335
3009 3167
3120 3079
3209 3167
3106 3065
ЗОЮ 2955
3150 3100
3020 —
3065 3015
Положение первого абсорбционного максимума во фракциях о
41 и 42 при 3395 А говорит о том, что рассматриваемый спектр
Рис. 14. Фракция 43.
не является характерным для алкилзамещенных нафталинов, так как последние поглощают в интервале длин волн 320—330 м д
Спектры производных нафталина с сопряженной двойной связью в боковой цепи, хотя и сдвинуты в красную часть спектра, но не имеют, как указывалось выше, четкой структуры. Из рассмотрения же приведенных спектрограмм (рис. 12 и 13) видно, что спектры поглощения фракций (4* и 42) имеют резко выраженные абсорбционные максимумы. Поэтому остается предположить, что исследуемые фракции содержат углеводород нафталинового ряда с замкнутым алифатическим циклом.
Действительно, при сравнении литературных данных с полученными спектрограммами, последние дают хорошее совпадение со спектром аценафтилена. Во фракциях присутствуют также,
Спектрографический анализ ароматических углеводородов 155
очевидно, аценафтен и алкилзамещенные нафталины, так как при Л = 2815 А— минимума поглощения для чистого аценафтилена — наблюдается широкая полоса поглощения.
На рис. 5 приведена кривая поглощения аценафтилена по данным Крэйга [7]. В табл. 6 помещены литературные и наши данные для чистого аценафтилена, спектрограмма которого приведена на рис. 15.
Таблица 6
Положение абсорбционных максимумов в спектрах аценафтилена
Данные ° • Длина волны, А Растворитель
I II III IV V
Крайг [7] ВНИИПС . 3395 3395 3335 3335 3280 3260 3260 3212 Этиловый спирт W 1»
Рис. 15. Аценафтилен.
С целью подтверждения присутствия во фракциях 41 и 42 аценафтилена последние были подвергнуты воздействию ультрафиолетовых лучей для получения димера [8].
Параллельно было поставлена проба со спиртовым раствором чистого аценафтилена; через некоторое время из всех облучаемых образцов выпали бледножелтые кристаллы димера аценафтилена, не растворимые в спирте и обычных растворителях (см. формулу):
^-СН-СН-^
СГ>-с1н-с'н-<~>
156 Е. И. Томина, К- Б. Чернышева и Е. М. Дементьева
Димер аценафтилена
Спектрограммы димера, выпавшего из фракций 41 и 42, показаны на рис. 16, из спиртового раствора чистого аценафти-
Рис. 16. Димер аценафтилена (из фракции).
лена •— на рис. 17. Положение абсорбционных максимумов на всех приведенных спектрограммах одно и то же: 3220, 3190,
Рис. 17. Димер аценафтилена (из спиртового раствора чистого аценафтилена).
3145, 3127, 3100, 3045, 2992, 2966 А. Начало широкой абсорбционной полосы — при 2766 А, максимум ее — при 2715 А.
Тождество спектрограмм с несомненностью подтверждает присутствие аценафтилена в исследуемых фракциях. Интересно отметить, что спектр поглощения аценафтилена напоминает спектр димера. Однако спектр димера сдвинут в коротковолновую часть на 170 А, т. е. расположен в области поглощения алкилзамещенных нафталинов. Это является вполне понятным, так как молекула димера не содержит двойной связи.
Спектрографический анализ ароматических углеводородов 157
Из рассмотрения данных табл. 5 и рис. 18 следует, что фракция 4s 6 содержит аценафтен. Спектр аценафтена весьма похож на спектр нафталина, но сдвинут по отношению к нему в красную часть на 100 А. Положение абсорбционных максимумов в спектре было определено для двух образцов чистого аценаф-
Рис. 18. Аценафтен.
тена: фирмы Кальбаум и образца, предоставленного нам ИОХ АН СССР; в качестве растворителя был использован этиловый спирт и гептан. Результаты определений приведены в табл. 7.
Таблица 7
Положение абсорбционных максимумов в спектре аценафтена
5. Из фракции 5 (/кип 120—130° С при р = 5 мм} были получены пикраты с /Пл 134 и 118° С. Спектрограммы углеводородов, выделенных из пикратов, даны на рис. 19, 20.
Длины волн соответствующих абсорбционных максимумов сведены в табл. 8.
Из данных табл. 8 и рис. 19 очевидно, что фракция 51 представляет собой аценафтилен; фракция 52 — триметилзамещенные нафталина с примесью аценафтена (см. табл. 7 и рис. 18).
6. В спектре фракции 6 (/кип 55—212° С при р = 5 мм) наблюдаются полосы поглощения при следующих длинах волн: 3750, 3690, 3555, 3505, 3458, 3380, 3336, 3306, 3230 А (рис. 21).
158
Е. И. Томина, К. Б. Чернышева и Е. М. Дементьева
Таблица 8
Положение абсорбционных максимумов в спектрах углеводородов, выделенных из пикратов (/пл 134 и 118° С)
Наименование анализируемого вещества О Положение максимумов, А
I II III IV V VI
Фракция 51 /пл пикрата 134° С 3395 3335 «' 3280 3260 3212
Фракция 52 /пл пикрата 118° С 3234 3209 3165 3105 3060 3018
Рис. 19. Фракция 51.
Рис. 20. Фракция 52.
При сопоставлении полученных результатов с данными табл. 9 следует сделать вывод, что фракция содержит смесь антрацена и фенантрена.
Спектрографический анализ ароматических углеводородов
159
7. Спектрограмма фракции 7(/КИ1212—280° С при р == 5 мм} приведена на рис. 22, где для сравнения показан также спектр
Рис. 21. Фракция 55—212° С.
Таблица 9
Положение абсорбционных максимумов в спектрах антрацена и фенантрена
Наименование анализируемого вещества
О
Положение полос поглощения, А
Растворитель
III IV
Антрацен
(ВНИИПС) . Антрацен [9] . Фенантрен
(ВНИИПС) . Фенантрен1 [10]
3750
3750
3690
3690
3555
3556
35053380.
3505
3378
3335
3225 3180
3080
3459 3382 3306 3227
345 338 330 324
3160
316
308
2933
Этиловый спирт,, гептан
Этиловый спирт
Этиловый спирт,, гептан
Нефтяной эфир
Рис. 22. 1 — фракция 55—112° С; 2 — фракция 212—280° С.
фракции 6 (/Кип 55—212° С, р = мм} . Характер распределения интенсивности в спектрах обеих фракций один и тот же,.
1 Длины волн даны в мм.
Сравнительная качественная характеристика камер
Камерная смола Г енератор
№ фракций t . °C КИП’ V при р = 5 мм 'пл пикрата, °C Качественная характеристика фракций Атмосферная разгонка
№ фракций t °C. ‘кип’ р = 5 мм 'пл. °C Качественная характеристика фракций
1 2 З1 З2 з3 41 42 4з 51 52 6 7 54—69 83—94 94—105 94—105 94—105 105—120 105-120 105—120 120—130 120-130 55—212 212-280 111 98 Не дает пикрата 132 127 115 134 118 Нафталин Смесь а- и jg-метилнафта-линов Диметилнафта-лины (возможно присутствие 1,7-1,3,1,6-изоме-ров) 1-метил-7-этил-нафталин, три-метилнафта-лин (2, 3, 5-изомер) Дифеиил Аценафтилен (с примесью алкилнафта-линов) Аценафтилен (с примесью аценафтена) Аценафтен Аценафтилен Триметилнафта-лины (с примесью аценафтена) Смесь антрацена и фенантрена Смесь моиоме-тилантраценов и 9, 10-дифе-иилаитрацена 1 2 3 4 5 6 73—83 83—93 93-109 109—120 120—134 134—150 119 116—117 144—146 108—109 110—111 Нафталин Нафталин, а-/?-метилнафта-лины Диметилнафта-лин (возможно присутствие 1-изомера), бутил нафталин Аценафтен Диметилнафта-лин (2,6 и 2,3-изомеры) Триметилиафта-лины
Таблица 10
ной и генераторной смол (фракция 180 — 300° С)
ная смола
1 Вакуумная разгонка Медленная разгонка с дефлегмацией
№ фракций 1 , °C, кип’ ’ р = 5 мм 'пл, °C Качественная характеристика фракций i \ °с, Качественная характеристика фракций
| В - р — Ьмм
1 2 3 4 5 66—82 82—95 95—108 108-120 120-134 i 114—115 118—119 108—109 109 1 Нафталин а- и /1-метил-нафталины Аценафтен Аценафтен Триметилнафта-лины (примесь аценафтена) 1 2 3 4 6 7 i i 66-79 i 79-94 94—108 108-120 120—134 134—150 134—150 Нафталин, а- и /1-метилнафта-лины а- и /1-метилна-фталины Диметилнафта-лины (возможно присутствие 1,2-изомера) примесь /1-метил-нафталина Аценафтен Триметилнафта-лины (1, 3, 5-изомер) и аценафтен Антрацен и три-метилнафта-лины Производные антрацена, триметилнаф-талины
.1
Труды ВНИИПС.
162 Е. И. Томина, К- Б. Чернышева и Е. М. Дементьева
однако спектр фракции сдвинут в длинноволновую часть (I max — 3920 А). Положение абсорбционных максимумов при 3920, 3865, 3720, 3668 А свидетельствует о присутствии во фракции алкилзамещенных антраценов, очевидно, смеси монометил-антраценов и 9, 10-дифенилантрацена.
Результаты качественного спектрального анализа камерной и генераторной смол сопоставлены в общей табл. 10.
Фракция 180—300° С выделялась из генераторной смолы, отобранной с промышленного блока Кохтла-Ярве в январе 1952 г. (^20 = 0,909). Содержание ароматических углеводородов (по данным хроматографии) равно 30—35%.
Разгонка смолы с целью выделения указанной фракции производилась при различных условиях: при атмосферном давлении, под вакуумом, путем медленной перегонки с дефлегматором.
ВЫВОДЫ
1. Показана возможность идентификации методом фотографической спектрофотометрии углеводородов нафталинового ряда, выделенных адсорбционным хроматографическим разделением на силикагеле из фракции 180—300° С камерной и генераторной сланцевых смол.
2. Доказано присутствие следующих полициклических углеводородов.
а) В камерной смоле: нафталина, а-метилнафталина, /Рметилнафталина, диметилнафталинов (очевидно изомеров 1, 7-, 1,6- и 1,3-), 1-метил-7-этилнафталина, триметилнафталина, дифенила, аценафтилена, аценафтена, фенантрена, антрацена, производных антрацена — монометилантраценов и 9, 10-дифенилантрацена.
б) В генераторной смоле:
в случае атмосферной разгонки — нафталина, а- и /3- метил-нафталинов, диметилнафталинов (очевидно изомеров 1,2- и 2,6-), бутилнафталинов, аценафтена и триметилнафталина;
в случае вакуумной разгонки—нафталина, а- и /3-метил-нафталинов, аценафтена, триметилнафталинов;
в случае разгонки с дефлегматором — нафталина, а- и /3-ме-тилнафталинов, диметилнафталина (изомера 1, 2-), аценафтена, триметилнафталинов, антрацена (в незначительном количестве).
3. Результаты проведенных исследований подтверждают имеющиеся данные о существенном различии качественного состава полициклических ароматических углеводородов камерной и генераторной сланцевых смол. Это различие выражается в более глубокой циклизации и дегидрогенизации ароматической части камерной смолы, о чем свидетельствует присутствие
Спектрографический анализ ароматических углеводородов
163
в последней значительного количества аценафтилена, фенантрена, антрацена и его производных, отсутствующих в генераторной смоле.
4. Из сравнения спектральных качественных характеристик фракции 180—300° С, выделенной из генераторной смолы при различных условиях, следует, что по своему качественному составу фракции, выделенные при атмосферном давлении и путем медленной разгонки с дефлегмацией, весьма близки друг к другу, тогда как фракция, выделенная путем вакуумной разгонки, значительно отличается от них.
ЛИТЕРАТУРА
1. L. U z u. Inst. nacl. te. aeronaut., Madrid, № 3, 34, 1944. Chem. Abstr., t. 43, № 14, 5577, 1946.
2. L. U z u, J. Doblas. Inst. nacl. tec. aeronaut., № 7, 1945.
3. L. Uzu, J. Doblas. Combustible (Zaragoza), 6, № 35/36, 113— 119, 1946.
4. De-Lazlo. Zeit. Phys. Chem., 117, № 369, 1925.
5. R. Morton. Journ. them. Soc., 916, 1934.
6. L. Ruzicka. Helv. Chem. Acta., 27, 1—3, 195, 1944.
7. L. Graig, W. Jacobs. Journ. Biolog. Chem., 139, 277, 1941.
8. K. Dzeiwonski. G. Rapalski. Ber. 45, 2491, 1912.
9. N. I ones. Chem. Rev, vol 41, № 1, 1947.
10. E. Helbronner, H. Doniker. Helv. Chem. Acta., XXVII.
1723, 1949.
11*
Б. И. Иванов и Н. Ф. Шаронова
ХИМИЧЕСКИЙ СОСТАВ ПОДСМОЛЬНОЙ воды
СЛАНЦЕВ ПРИБАЛТИЙСКОГО МЕСТОРОЖДЕНИЯ 1
Ущерб, наносимый спуском подсмольных вод в реки и озера, настолько значителен, что всем новостроящимся заводам вменено в обязанность производить очистку сточных вод в соответствии с нормами Госсанинспекции. Правильное решение задачи обезвреживания сланцевых подсмольных вод имеет большое народнохозяйственное значение.
Подсмольные воды, как правило, являются балластом для производства и требуют больших затрат на их обезвреживание. Главным препятствием к спуску подсмольных вод в водоемы является наличие в них фенолов и других органических веществ, которые оказывают пагубное действие на животный и растительный мир водоемов, а также делают воду непригодной для питья и коммунальных нужд. Обезвреживание этих вод может быть объединено с извлечением химических продуктов.
Знание химической природы указанных выше вредных веществ может благоприятно отразиться на эффективности и экономике процесса обезвреживания.
До настоящего времени органические вещества сланцевой подсмольной воды еще не подвергались подробному исследованию.
Присутствие в подсмольной воде разнообразных органических веществ обусловлено процессом вымывания их из смолы, происходящим при конденсации последней в холодильниках. Вследствие этого химический состав подсмольной воды во многом определяется растворимыми веществами смолы, а именно, фенолами, кислотами, альдегидами, кетонами и азотистыми основаниями. Для сопоставления состава подсмольных вод, полученных ъри термической переработке прибалтийских сланцев в туннельных, генераторных и камерных печах, в табл. 1 приводится их состав.
J В работе принимала участие Е. А. Косарева.
Химический состав подсмольной воды
165
Таблица 7
Состав подсмольных вод
Название (происхождение) подсмольной воды Фенолы летучие, г/л Фенолы нелетучие, г/л Кислоты летучие, г/л | Кетоны, г/л Аммиак общий, г/л
Туннельная . . . 1,2 8,7 1,0 2,4 0,3
Генераторная . . 0,6 6,0 0,4 0,1 0,4
Камерная .... 1,5 13,8 0,4 0,2 1,0
Объектом для данного исследования служила подсмольная вода, полученная при полукоксовании прибалтийского сланца в туннельных печах. Были исследованы альдегиды, кетоны, кислоты, фенолы и органические основания.
КЕТОНЫ И АЛЬДЕГИДЫ
Кетоны и альдегиды отгонялись из подсмольной воды на колонне, и в дальнейшем полученные концентраты укреплялись повторной перегонкой, сушились и окончательно разделялись ректификацией.
Для ректификации была использована стеклянная колонна с высотой ректифицирующей части 150 см и диаметром 11 мм. Колонна заполнялась насадкой из одиночных витков стеклянной спирали, имеющей внешний диаметр около 4 мм, изготовленных из стеклянной нити диаметром около 1 мм. Такая колонна, при флегмовом числе около 30, обладает погоноразделительной способностью в 24—20 теоретических тарелок.
В начале работы были определены условия получения кетон-ного концентрата. Для этого от подсмольной воды отгонялись пробы дестиллата, в которых определялись кетоны йодометрическим методом. Сущность этого метода заключается, как известно, в том, что йод в щелочной среде образует йодоформ, а избыточный йод, не вошедший в реакцию, оттитровывается гипосульфитом [1]. Результаты отгонки кетонов из подсмольной воды приведены в табл. 2.
Данные таблицы показывают, что кетоны концентрируются в первых погонах подсмольной воды, и практически для полного их извлечения достаточно отогнать незначительное количество дестиллата.
Отгонка альдегидо-кетонного концентрата проводилась из железного 15-литрового куба. *
Газообразные продукты, получающиеся при отгонке альдегидо-кетонного концентрата из подсмольной воды, пропускались через раствор димедона. При этом было обнаружено образова-
Таблица 2
Отгонка кетонов из подсмольной воды
Туннельная подсмольная вода Генераторная подсмольная вода Исходная концентрация 0,25 г/л
От газового бензина От печного бензина От легкого масла От среднего масла
Исходная концентрация 40,97 г/л Исходная концентрация 5,62 г/л Исходная концентрация 3,50 г/л Исходная концентрация 0,11 г/л
Концентрация <етоновв дестиллате, % Кол-во отогнанных КС-ТОНОВ, % от общего кол-ва кетонов воды Концентрация кетонов в дестиллате, % Кол-во отогнанных кетонов % от общего кол-ва кетонов воды Концентрация кетонов в дестиллате, % Кол-во отогнанных кетонов, % от общего кол-ва кетонов воды Концентрация кетонов в дестиллате, % Кол-во отогнанных кетонов, % от общего кол-ва кетонов воды Концентрация кетонов в дестиллате, % Кол-во отогнанных кетонов, % от общего кол-ва кетонов воды
67,8 71,0 62,0 67,8 62,0 52,3 20,0 0,4 16,57 17,35 15,14 16,57 15,14 12,78 4,88 1,57 S Гоо 34,7 12,7 4,8 2,0 1,1 0,6 0,1 0,1 61,72 22,58 8,61 3,55 2,07 1,03 0,22 0,22 S 100 25,8 5,2 1,9 1,2 0,9 73,66 14,76 5,52 3,31 2,75 S 100 09 0,2 0,06 78,03 16,76 5,21 27100 2,3 0,2 92,32 7,68 27100
Химический состав подсмольной воды
167
ние белого кристаллического осадка. После нескольких перекристаллизаций из 50 %-кого спирта было получено вещество с температурой плавления 175°, соответствующей температуре плавления продукта конденсации формальдегида с димедоном.
Кроме того, известно, что при действии аммиака на альдегиды последние присоединяют его по месту двойной кислородной связи и образуют твердые кристаллические соединения, называемые альдегидаммиаками. Поэтому для улавливания легко летучих альдегидов несконденсировавшиеся пары из холодильника пропускались через охлаждаемый льдом сухой эфир, куда одновременно вводился сухой аммиак. При этом выделился белый осадок, который имел специфический мыщиный запах и содержал 22,4% азота. Эти данные указывают на то, что полученный белый осадок состоит из ацетальдегидаммиака, для которого содержание азота равно 22,9%.
Вначале было собрано 40 л дестиллата, затем это количество путем ректификации было доведено до 3,5 л. Для индивидуализации отдельных представителей полученный концентрат тщательно ректифицировался на фракции, соответствующие ацетону и его гомологам. Были отобраны фракции до 55, 55—62, 62—72, 72—84, 84—96, 96—104°. Выход фракций, полученных при ректификации, приводится в табл. 3.
Таблица 3
Выход фракций при ректификации кетоиного концентрата
Температура кипения фракций, °C Выход, г Выход, %
до 55 120 4,9
55—62 1865 76,5
62—72 42 1,7
72—84 362 14,9
84—96 51 2,0
Как видно из этих данных, полученный концентрат на 76% состоит из ацетоновой фракции.
Фракция, выкипающая до 55°, исследовалась на альдегиды. После повторной ректификации этой фракции было получено 68 г вещества, которое нацело выкипало точно при 21° С. Последнее обстоятельство показывало, что вещество, соответствующее по температуре кипения ацетальдегиду, было получено в чистом виде.
С целью идентификации выделенный ацетальдегид был окислен [2] марганцовокислым калием в щелочной среде на холоде.
168
Б. И. Иванов и И. Ф. Шаронова
Полученная при этом безводная кислота, имевшая резкий запах уксусной кислоты, была затем идентифицирована по температуре плавления анилида и процентному содержанию бария в бариевой соли.
Характеристика продукта окисления фракции, кипящей при 21°, приводится в табл. 4.
Таблица 4
Характеристика кислоты, полученной окислением фракции, кипящей при 21° С
Температура кипения, °C Температура плавления анилида, °C % бария в солях
полученной уксусной полученной уксусной смеси полученной уксусной
кислоты 1 кислоты кислоты кислоты кислот кислоты кислоты
117 1 118 114 114 114 53,6 i ! 53,7
Приведенные результаты с достаточной убедительностью показывают, что выделенное соединение представляет собой уксусную кислоту, образовавшуюся в результате окисления ацетальдегида.
Следующая фракция, которая исследовалась, выкипала в пределах 49—53°.
Фракция была окислена марганцовокислым калием до соответствующей кислоты по методу, описанному ранее. Из кислоты была получена бариевая соль, которая и подвергалась дальнейшему исследованию.
Осаждение бариевой соли проводилось фракционно, в три приема. Для этого к общему количеству полученной кислоты прибавлялась третья часть общего количества барита, необходимого для полной нейтрализации. Затем кислота, не вступившая в реакцию, отгонялась с паром, а раствор бариевой соли выпаривался досуха. В сухом остатке подсчитывалось процентное содержание бария. После этого дестиллат, отогнанный с паром, вновь нейтрализовался следующей порцией барита и с ним производились те же операции, что и при первом осаждении. Вновь отогнанный дестиллат окончательно нейтрализовался по фенолфталеину и упаривался досуха.
Ниже приводятся экспериментальные данные по 3-ступенчатому осаждению бариевых солей.
I осаждение:
израсходовано 0,5900 г Ва(ОН)г или 0,4729 г Ва;
вес сухого остатка 0,9714 г;
количество бария в сухом остатке 48,68%.
Химический состав подсмольной воды
169
II осаждение:
израсходовано 0,4729 г Ва;
вес сухого остатка 1,0034 г;
количество бария в сухом остатке 47,12%.
III осаждение:
израсходовано 0,4587 г Ва;
вес сухого остатка 0,9958 г;
количество бария в сухом остатке 46,06%.
В сухом остатке, полученном при I осаждении, кроме определения количества бария, было установлено содержание углерода и водорода.
Результаты анализа этого остатка приведены в табл. 5.
Таблица 5
Характеристика пропионовокислого бария, полученного из фракции 49—53° С
% бария в пропионовокислом барии % углерода в пропионовокислом барии % водорода в пропионовокислом барии
найдено требуется для пропионовой кислоты найдено требуется для пропионовой кислоты найдено требуется для пропионовой кислоты
48,6 48,4 26,00 25,40 3,45 3,52
Таким образом можно считать установленным наличие пропионового альдегида в сланцевой подсмольной воде.
В одной из перегонок подсмольной воды, с целью накопления альдегид-кетонного концентрата, неконденсирующиеся газы и пары были пропущены через раствор воды для улавливания альдегидов.
Полученный водный раствор альдегидов окислялся марганцовокислым калием в щелочной среде. Полученные кислоты в дальнейшем были подвергнуты фракционной обработке гидратом окиси бария. Образовавшиеся соли были подвергнуты анализу, результаты которого соответствовали уксусной, пропионовой и масляной кислотам. Для последней были получены следующие данные: на титрование израсходовано 0,2098 г Ва(ОН)2 или 0.1618 г Ва; вес бариевой соли 0,3804 г; следовательно, процентное содержание бария в соли равно 44,1%, что точно соответствует маслянокислому барию. Таким образом, в подсмольной воде установлено присутствие масляного альдегида.
Из фракции, выкипающей при температуре 55—62°, была выделена узкая фракция 56—58°, соответствующая ацетону, и определены ее константы. Результаты элементарного анализа,
170
Б. И. Иванов и И. Ф. Шаронова
определения молекулярного веса и показателя преломления этой фракции хорошо совпадают с соответствующими константами ацетона.
Эти данные представлены в табл. 6.
Таблица 6
Характеристика фракции 55—58° С
Показатели Исследуемая фракция Ацетон (диметилкетон)
Температура кипения, °C . . 56—58 56,7
<14 0,8130 0,8125
"D 1,3596 1,3591
Молекулярный вес 58 58
Содержание углерода, % . . 61,44 62,06
„ водорода, % . . 10,28 10,34
Все попытки обнаружить метиловый спирт во фракции '62—72° были безуспешными. В результате нескольких разгонок была получена фракция, выкипающая в пределах 64—66°, в количестве 4,55 г. Однако произведенные с этой фракцией качественные реакции на метиловый спирт дали отрицательные результаты.
В дальнейшем исследовалась фракция 72—84°. В процессе ее ректификации было обнаружено, что она, в основном, выкипает в пределах 73—74°. Из литературы [3] известно, что метилэтил-кетон с водой образует азеотропную смесь с температурой кипения 74°. После тщательного высушивания поташем и нескольких перегонок на колонне из азеотропной смеси выделено вещество с температурой кипения 78,6° С, соответствующее метилэтилкетону. Элементарный анализ, показатель преломления и удельный вес этого вещества хорошо совпадают с данными для метилэтилкетона. Результаты анализа приведены в табл. 7.
Таблица 7
Результаты анализа фракции 78,6°
Показатели Исследуемая фракция Метилэтил-кетон
Температура кипения, °C . . 78,6 78,6
г/р 0,8093 0,8100
П1!О 1,3804 1,3807
-Содержание углерода, % . 66,71 66,66
„ водорода, % . . . 11,12 11,11
Химический состав подсмольной воды
171
Чтобы иметь более полное представление о составе кетонов подсмольной воды, для разгонки был взят кетоновый концентрат, полученный на сланцеперегонном комбинате в Кивиыли. При ректификации последнего, кроме выделенных ранее диметилке-тона и метилэтилкетона, удалось получить вещество с температурой кипения 102°, соответствующее метилпропилкетону. В качестве производных этого кетона были получены семикарбазон и 2, 4-динитрофенилгидразон.
Характеристика метилпропилкетона, выделенного из подсмольной воды, приводится в табл. 8.
Таблица 8
Результаты анализа метилпропилкетона, выделенного из подсмольной воды
Показатели Получено Требуется для метилпропилкетона
Температура кипения, °C 102 102
df 0,8091 0,8089
"d 1,3929 1,3894
Температура плавления семикарбазона, °C 110 ПО
Температура плавления 2,4-динитро-фенилгидразона, °C 140 141
При исследовании фракции, выкипающей в пределах 62—84°, одна из разгонок велась в присутствии твердой щелочи, для того чтобы связать, возможно, имеющиеся в ней альдегиды. При этом было обнаружено выделение значительных количеств аммиака. Это навело на мысль, что в исследуемой фракции находятся нитрилы, которые, как известно, при омылении щелочью выделяют аммиак.
Некоторым указанием на присутствие в подсмольной воде нитрилов служит наличие неприятного изонитрильного запаха, который появляется при перегонке подсмольной воды, причем изонитрилы в этих условиях довольно легко переходят в нитрилы.
Из натриевой щелочи после обработки серной кислотой и перегонки была выделена кислота. Ее бариевая соль содержала 53,3% бария, что соответствует соли уксусной кислоты. Последняя могла получиться в результате омыления ацетонитрила щелочью. Температура кипения ацетонитрила 81,5°.
При исследовании карбонильных соединений в подсмольной воде генераторных и камерных печей было установлено присутствие примерно одних и тех же альдегидов и кетонов.
172
Б. И. Иванов и И. Ф. Шаронова
кислоты
Для выделения кислот было взято 880 л подсмольной воды, полученной при полукоксовании сланца в туннельных печах.
Чтобы выяснить течение процесса перегонки кислот из подсмольной воды и установить предел отгонки летучих кислот, были проведены соответствующие опыты с отбором отдельных фракций дестиллата. Расчет производился на уксусную кислоту. Результаты этих опытов приведены в табл. 9.
Таблица 9
Распределение летучих кислот по фракциям подсмольной воды
Количество исходной п/воды................... 400 мл
Концентрация кислот в исходной п/воде......... 2,05 г/л
Количество отбираемой фракции дестиллата п/воды . . 40 мл
Порядковый №, 40 мл фракции дестиллата
1
2
3
4
5
6
7
8
9
Содержание кислот, % от содержания их в исходной п/воде
16,35 9,46 8,97 8,36 8,42 8,36 8,91 8,79 8,79
Весь дестиллат . . .
86,38
Как видно из приведенных данных, летучие кислоты равномерно распределяются по всем фракциям.
Для выделения кислот из подсмольной воды был выбран следующий путь.
Кислоты вначале концентрировались в виде натриевых солей, путем упаривания подщелоченной подсмольной воды, примерно на 90%. В остатке находился концентрат натриевых солей карбоновых кислот в смеси с фенолятами летучих и нелетучих фенолов. Чтобы отделить летучие вещества от нелетучих, концентрат солей подкислялся серной кислотой до слабокислой реакции. Выделившиеся свободные карбоновые кислоты и летучие фенолы отгонялись из 2-литровой колбы. Для полноты отгонки летучих веществ необходимо было отогнать не менее 90% дестиллата.
Для отделения летучих кислот от летучих фенолов, дестиллат, отогнанный с водяным паром, предварительно подщелачи
Химический состав подсмольной воды
173
вался едким натром до неполной нейтрализации кислот и затем окончательно нейтрализовался бикарбонатом натрия. Нейтрализованный раствор упаривался досуха, при этом в остатке были только соли кислот, а летучие фенолы отгонялись вместе с водой.
Перед тем как поставить нейтрализованный раствор упариваться, было проверено, не имеет ли место гидролиз солей при их кипячении. Для этого был проведен следующий опыт.
От 100 мл раствора солей отгонялось 10 мл пробы дестиллата, и каждая проба титровалась щелочью. Результаты титрования приводятся в табл. 10.
Таблица 10
Расход щелочи на титрование последовательных 10-миллилитровых проб дестиллата, отогнанных от раствора солей жирных кислот
Расход щелочи № пробы
1 2 3 4 1 5 6 | 7
Количество мл 0,1 N КОН 0,66 0,66 0,74 0.82 0,82 0,82
Как видно из приведенных данных, расход щелочи на титрование был очень незначительным, следовательно, и гидролиз солей при упаривании солевого раствора был очень мал.
По мере упаривания концентрата солей, из раствора выпа
дали кристаллы, которые периодически отделялись от маточного раствора. Натриевые соли имели белый цвет и прекрасно кристаллизовались. Таким путем было получено 2025 а сухих солей карбоновых кислот.
Разложение солей проводилось расчетным количеством крепкой серной кислоты при сильном охлаждении. Для отделения
выделившихся жирных кислот от сернокислого натрия применялась отгонка в вакууме при остаточном давлении 2 мм. Дестиллат при вакуумной отгонке собирался в приемник, охлаждаемый снегом с солью. Так были получены кислоты, имеющие 73,3%-ную концентрацию, считая на уксусную кислоту.
Для разделения смеси кислот была применена ректификация. Ниже приводятся температуры кипения гомологов уксусной
кислоты и рядом с ними пределы кипения соответствующих фракций, отобранных для разгонки и дальнейшего исследова-
ния (идентификации):
муравьиная кислота уксусная пропионовая изомасляная н-масляная изовалериановая
101 до 110
118 110—130
141 130-117
154 147—158
162 158—170
177 170—181
174
Б. И. Иванов и И. Ф. Шаронова
Ректификацией были выделены кислоты, представленные в табл. II.
Таблица 77
Кислоты, выделенные из подсмольной воды
Наименование кислот Выход
в граммах % от общего количества кислот
Уксусная 838,0 90,5
Пропионовая 68,5 7,3
н-Масляная 19,5 2,2
Как показали результаты разгонки, смесь кислот, выделенная из подсмольной воды, в основной массе представлена уксусной кислотой. Пропионовая и н-масляная кислота присутствуют в незначительных количествах. Более высококипящих гомологов нет, так как вся смесь кислот, взятая для разгонки, нацело переснялась до 163°.
Из фракции ПО—130° путем ректификации была выделена уксусная кислота 98,75 %-ной концентрации. Нагреванием безводной кислоты с анилином был получен ацетанилид, для которого была определена температура плавления.
Кроме анилида уксусной кислоты, была получена ее бариевая соль. По количеству гидрата окиси бария, пошедшего на титрование, и по весу осадка подсчитывалось процентное содержание бария. Кроме процентного содержания бария, был произведен элементарный анализ кислоты, который дал следующие результаты:
найденное вычисленное
Содержание С, %............... 39,27; 39,27 40,00
„ Н, %............... 6,13; 6,65 6,66
Кроме этого, для уксусной кислоты была определена температура кристаллизации, которая оказалась равной 14,33° С. Такая температура кристаллизации соответствует содержанию 1,21% воды или концентрации кислоты 98,79%.
Почти такая же концентрация кислоты (98,75%) была найдена на основании результатов определения удельного веса.
В табл. 12 приведены сравнительные данные для чистой уксусной кислоты и выделенной из подсмольной воды.
Химический состав подсмольной воды
175-
Таблица 12
Характеристика уксусной кислоты, выделенной из подсмольной воды
Показатели Выделенная кислота 100%-ная СН3СООН
Температура кипения, °C ..... . 118 118
dl° 1,0523 1,0517
Температура кристаллизации, °C . . 14,33 —
Температура плавления анилида, °C . Количество бария в уксусно-кислом 114 114
барии, % 53,7 53,7
Концентрация кислоты, % 98,75 100
Молекулярный вес 60 60
Как уже было отмечено выше, при ректификации смеси кислот, кроме уксусной кислоты, были получены пропионовая и н-масляная кислоты, но в значительно меньших количествах.
Для этих кислот были определены молекулярные веса методом титрования. Полученные значения молекулярных весов были весьма близки к теоретическим данным. Кроме молекулярного веса, для пропионовой и н-масляной кислот было определено процентное содержание бария в бариевых солях. Результаты этого определения приведены в табл. 13.
Таблица 13
Результаты определения бария в бариевых солях, выделенных пропионовой и н-масляной кислотами
Кислоты Количество Ва (ОН)а, пошедшее на титрование, г Количество Ва в Ва (ОН)2, пошедшем на титрование, г Количество образовавшейся соли, г Барий в соли, % Теоретическое количество Ва, %
Пропионовая . . . 0,0710 0,0569 0,1178 48,3 48,4
н-Масляная .... 01285 0,1030 0,2340 44,0 44,1
Эти результаты также подтверждают, что отобранные нами узкие фракции при ректификации смеси кислот соответствуют пропионовой и н-масляной кислотам. Их характеристика приводится в табл. 14.
Исследованием нелетучего остатка подсмольной воды показано отсутствие в ней оксикислот и многоосновных кислот.
17G
Б. И. Иванов и И. Ф. Шаронова
Таблица 14
Характеристика выделенных пропионовой и н-масляной кислот
Показатели Пропионовая кислота Н-масляная кислота
выделенная из п/воды 100%-ная выделенная из п/воды 100%-ная
Температура кипения, °C 139—140 141 159-162 162
1,014 1,013 0,9782 0,978
Содержание, %: углерода . - . . . 48,72 48,65 54.65 54,54
водорода 8,19 8,10 9,32 9,09
Молекулярный вес . . 74 74 87 88
подсмольных
камерной
При исследовании
вод, проведенном при участии Ю. А. ствие в них муравьиной, уксусной, кислот.
генераторной
Козак, установлено присут-пропионовой и масляной
ФЕНОЛЫ
Для исследования были взяты фенолы, извлеченные уксусно-амиловым эфиром из подсмольной воды, полученной при полукоксовании сланца в туннельных печах.
Эти фенолы представляли собой смесь одноатомных и многоатомных фенолов. Поэтому дальнейшая задача получения более однородных веществ заключалась в отделении одноатомных фенолов от многоатомных. Для предварительного разделения фенолы были разогнаны на две фракции. Первая фракция представляла собой смесь одноатомных фенолов, а вторая — многоатомных. В количественном отношении последняя составляла главную массу фенолов.
Из 360 л подсмольной воды было получено 2444 г фенолов. Разгонка их проводилась из железного 7-литрового куба при 3—4 мм остаточного давления.
Результаты разгонки фенолов приведены в табл. 15.
Таблица 15
Фракции Пределы кипения фракций при нормальном давлении, °C Выход фракций
г %
I до 240 135 5,5
11 240—330 1621 66,4
Остаток + по-тери — 688 28,1
Химический состав подсмольной воды 177
Во время разгонки отмечено, что фенолы, кипящие выше 240°, резко отличались по вязкости от нижекипящей фракции. В то время как последние представляли собой легкоподвижную жидкость, фенолы, кипящие выше 240°, были чрезвычайно вязкими. Для окончательного разделения фенолов была использована различная растворимость одноатомных и многоатомных фенолов в легком бензине: многоатомные фенолы почти не растворяются в бензине, а одноатомные смешиваются с ним в любых отношениях. Фракция 240—330° была растворена в 40 %-ном этиловом спирте и затем обработана легким бензином. При этом в водный раствор этилового спирта перешло 82% фенолов.
Соображения о том, что многоатомные фенолы должны находиться преимущественно во фракции, кипящей выше 240° и что при разделении бензином и спиртом они перейдут в раствор последнего, подтверждаются результатами определения гидроксильных групп. Определение последних проводилось по методу Чугаева-Церевитинова. Фенолы, растворимые в спирте, содержали 24% гидроксильных групп, что соответствовало диметиль-ному или (этильному) производному двухатомного фенола.
Для подробного изучения одноатомных фенолов фракция I была ректифицирована на колонне на фракции, представленные в табл. 16.
Таблица 16 Результаты разгонки фракции одноатомных фенолов
Наименование фракций
Фенольная ............
Ортокрезольная . . . Мета- и паракрезольная Ксиленольная ......... Ксиленольная ......... Остаток ..............
Температура кипения, ° С Выход фракций
г %
до 184 19,7 14.5
184—194 16,6 12,2
194—205 30,9 23,0
205—214 15,0 11,2
214—217 10,1 7,3
— 42,7 31,8
Рассматривая данные, полученные при разгонке, можно сказать, что одноатомные фенолы подсмольной воды в значительной части состоят из крезолов.
Фракция, кипящая до 184°, сразу же после разгонки закристаллизовалась, что доказывает чистоту полученного вещества. Проведенные с кристаллами качественные реакции на фенол дали положительные результаты. С хлорным железом в водном растворе получилось фиолетовое окрашивание; от добавления 12 Труды ВНИИПС.
178
Б. И. Иванов и Н. Ф. Шаронова
бромной воды выпадал желтовато-белый осадок. Кристаллический фенол представлял собой бесцветные кристаллы, обладающие характерным запахом.
Для характеристики фенола были определены температуры кипения, элементарный состав и получены некоторые производные.
В качестве одного из производных фенола была получена арилгликолевая кислота [4], для которой были определены температура плавления и молекулярный вес.
Все данные, характеризующие кристаллический фенол, выделенный из сланцевой подсмольной воды, приведены в табл. 17.
Таблица 77
Характеристика фенола, выделенного из сланцевой подсмольной воды
Показатели Выделенный фенол 100%-ный фенол
Температура кипения, °C 181 181
Содержание, %: углерода 76,55 76,60
водорода 6,51 6,40
Температура плавления арилгликолевой кислоты, °C 98—99 98—99
Молекулярный вес арилгликолевой кислоты 152 152
Температура плавления трибромфенола, °C 90 91,5
Из крезольных фракций были получены крезоксиуксусные кислоты, которые характеризовались по молекулярному весу и температуре плавления. Те же самые методы идентификации были применены и к ксиленольным фракциям. Полученные в результате конденсации вышеупомянутых фракций с монохлоруксусной кислотой арилгликолевые кислоты, после нескольких перекристаллизаций из воды, показали постоянные температуры плавления. Результаты этих определений приведены в табл. 18.
Рассматривая результаты, приведенные в таблице, можно сказать, что все три изомерных крезола присутствуют в сланцевой подсмольной воде. Кроме того, при помощи арилгликоле-вых кислот были изолированы два ксиленола, а именно 1,2,4- и 1,3,4-ксиленолы.
При исследовании летучих фенолов камерной подсмольной воды, проведенном Ю. А. Козак, установлено наличие в этой воде фенола, мета- и паракрезолов и 1,3,5-ксиленола.
В генераторной подсмольной воде доказано присутствие карболовой кислоты.
Химический состав подсмольной воды
17
Таблица 18
Характеристика арилгликолевых кислот, полученных из выделенных фенолов
Наименование арилгликолевых кислот Температура плавления, ° С Молекулярный вес
найденная по литературным данным найденный теоретический
о-Крезоксиуксусная .... 150—151 150-151 166 166
м-Крезоксиуксусная 102—103 102—103 166 166
р-Крезоксиуксусная 125 125—126 166 166
1,2,4-ксиленоксиуксусная . . 162—163 162,5 180 180
1,3,4-ксиленоксиуксусная . . 142—143 141,5 179 180
Фракция II, выкипающая в пределах 240—330°, представляла собой очень вязкую жидкость желтого цвета, которая постепенно закристаллизовалась в сплошную массу.
С целью выделения чистых фенолов указанная кристаллическая масса была растворена в бензоле. По охлаждении раствора до 15° из него выпали белые кристаллы, количество которых оказалось равным 24%. Всего кристаллических фенолов было получено 300 г. Они были вновь перегнаны в вакууме. В интервале температур 240—275° отогналось около 3 мл от всего количества фенолов, взятых для разгонки, основная же масса фенолов перегналась в довольно узком температурном интервале — в пределах 275—290°. Это обстоятельство указывает на то, что полученные кристаллические фенолы однородны по составу. Для этих фенолов были определены элементарный состав и количество гидроксильных групп. В табл. 19 приводятся результаты этих определений.
Таблица 19
Элементарный состав и содержание гидроксильных групп в кристаллических фенолах, выделенных из фракции 240—330° С
Показатели Найдено Требуется для фенола С8Н10О2
Содержание, %: 24,0 24,6
гидроксильных групп .
углерода 69,28 69,56
водорода 7,28 7,24
кислорода ...... 23,44 23,20
12*
180
Б. И. Иванов и И. Ф. Шаронова
Приведенные данные показывают, что исследуемый продукт представляет собой фенол, соответствующий формуле С9Н19О2-
Такой эмпирической формуле соответствуют двухатомные фенолы с двумя метильными или одной этильной группами. Последние представляют собой кристаллические вещества, температуры плавления которых значительно отличаются от температуры плавления диметильных двухатомных фенолов.
С кристаллическими фенолами были проведены качественные реакции на двухатомные фенолы. Реакции на гидрохинон и пирокатехин были отрицательными. Наоборот, реакции, характерные для резорцина [5], образование флуоресцеина с фталевым ангидридом и малиновокрасное окрашивание с едким кали в присутствии хлороформа были отчетливо положительными и не отличались от реакций, наблюдавшихся в контрольных опытах с чистым резорцином.
Кроме того, исследуемые фенолы были подвергнуты абсорбционному спектральному анализу в области ультрафиолетовой части спектра с целью уточнения их строения.
Были получены снимки спектров поглощения испытуемых фенолов и для сравнения — спектры поглощения резорцина, пирокатехина и гидрохинона.
Из сопоставления полученных спектрограмм можно было сделать вывод, что испытуемые фенолы являются производными резорцина:
полосы поглощения резорцина .......... 2825 А
I тх полосы поглощения испытуемого фенола . . 2825—27 А
Спектры поглощения пирокатехина и особенно гидрохинона сдвинуты, по отношению к спектру^ поглощения резорцина, в длинноволновую часть спектра.
Кристаллические фенолы путем фракционной кристаллизации из бензола и дихлорэтана удалось разделить на несколько фракций с различными температурами плавления.
Нами были получены фенолы с температурами плавления 137, 147 и 156,4°. Таким температурам плавления по литературным данным соответствуют диметильные производные резорцина. В табл. 20 помещены температуры плавления диметильных производных резорцина и характеристика нелетучих фенолов, выделенных из подсмольной воды. Определение температуры плавления, во избежание разложения, проводилось в капиллярах, запаянных под вакуумом.
Как видно из приведенных данных, температура плавления 2,5-диметилрезорцина несколько отличается от литературных данных, но температура кипения его вполне соответствует последним. Содержание гидроксильных групп и элементарный
Химический состав подсмольной воды
181
состав вполне согласуются с теоретическими и литературными данными для диметильного производного двухатомного фенола.
Таблица 20
Характеристика выделенных двухатомных фенолов — производных резорцина
Показатели 4,5-диметил-резорпин 2,4-диметил-резорцин 2,5-диметил-резорцин
теоретические данные найдено теоретические данные найдено теоретические данные най- дено
Температура плавления, 163 156
°C 136 137 147 147
Температура кипения, °C нет 284 нет 278 277 277
Удельный вес при 20° С Содержание, %: 1,1994 1,2114 нет 1,2177
группы ОН ... 24,6 23,7 24,6 23,9 24,6 69,56 25,0
углерода . . . 69,56 69,51 69,56 69,37 69,38
водорода .... 7,24 7,30 7,24 7,33 7,24 7,39
Были определены удельные веса при 20° и температуры кипения при атмосферном давлении.
Ранее одним из авторов было доказано присутствие в туннельной подсмольной воде 4,6-диметилрезорцина.
При исследовании многоатомных фенолов, выделенных из камерной надсмольной воды, установлено, что они тоже являются производными резорцина. Так, в камерной надсмольной воде доказано присутствие 4,5-2,4- и 2,5-диметилрезорцинов.
ОРГАНИЧЕСКИЕ ОСНОВАНИЯ
Для извлечения оснований была взята подсмольная вода, полученная при полукоксовании сланца в туннельных печах в количестве 3000 л.
Подсмольная вода для выделения оснований подщелачивалась и перегонялась. Выделяющиеся пиридиновые основания и аммиак улавливались серной кислотой. После этого раствор образовавшихся солей упаривался досуха. Для выделения оснований в свободном состоянии сухие соли разлагались щелочью.
Водный раствор оснований отделялся от сульфата натрия, высаливался поташом, и основания извлекались серным эфиром, который затем отгонялся.
Было установлено, что основания в главной своей массе состоят из аммиака, на долю органических оснований приходится 10%- Концентрация их в подсмольной воде равна 0,05 г/л.
182
Б. И. Иванов и И. Ф. Шаронова
В результате всех проведенных операций было получено 175 г пиридиновых оснований. Результаты разгонки оснований представлены в табл. 21. Начало кипения оснований 125°.
Таблица 2Z
Фракционный состав органических оснований, выделенных из подсмольной воды
№ фракций Пределы кипения фракций, ° С Количество фракции, объемн. %
I 125—150 18
II 150-175 51
III 175—200 15
IV 200—225 5
V 225—250 5
Остаток-(-потери 6
Фракции оснований, выкипающие до 175°, были бесцветны, а фракции, кипящие выше этой температуры, слегка окрашены в желтоватый цвет.
Все фракции оснований имели характерный запах пиридина.
Изучение химической природы оснований заключалось в разделении их на первичные, вторичные и третичные основания методом диазотирования.
При диазотировании первичные алифатические амины давали спирты, а вторичные — нитрозамины. Последние извлекались серным эфиром, а нерастворимые диазосоединения разлагались нагреванием на водяной бане при 50° С.
Выделившиеся при нагревании фенолы экстрагировались эфиром и в дальнейшем отделялись щелочью. Часть веществ, не извлекаемая из эфирного раствора щелочью, очевидно, является нейтральными азотистыми соединениями.
Третичные основания извлекались из подщелоченного серпокислого раствора эфиром. Результаты диазотирования представлены в табл. 22.
Таблица 22
Результаты диазотирования оснований
(групповой состав оснований)
Наименование оснований
% от общего количества оснований
Алифатические первичные и вторичные [ амины ..................................... 9,8
Анилины + гомологи..................I 1,0
Нейтральные азотистые соединения . ' ! 0,5
Третичные основания ........! 74,5
Остаток + потери..................11.2
Химический состав подсмольной воды
183
Таким образом, результаты диазотирования показывают, что основания сланцевой подсмольной воды состоят, главным образом, из третичных оснований.
В дальнейшем основания подвергались ректификации на 5-градусные фракции. Результаты разгонки приведены в табл. 23.
Таблица 23
Фракционный состав органических оснований, выделенных из подсмольной воды
Пределы кипения фракций, °C Количество фракций
г вес. %
80-105 2,0 1,1
105—120 2,5 1,4
120—125 1,7 0,9
125—130 6,4 3,6
130—135 5,0 2,8
135—140 14,3 8 2
140—145 9,9 5,6
145—150 18,1 10,3
150—155 15,9 9,0
155—160 13,4 7,6
160—165 17,5 10,0
165-170 8,9 5,1
170—175 6,6 3,8
175—180 6,9 3,9
180—185 5,8 3,1
185—190 4,8 2,7
190—195 4,0 2,3
195-200 3,7 2,1
200—205 3,1 1,8
205—210 3,4 1,9
210—215 2,6 1,5
215—220 2,9 1,6
220-225 1,3 0,7
225—230 1,2 0,7
Остаток 7,9 4,5
Потери 5.2 3,8
Все фракции, полученные при разгонке, окрашивали сосновую лучинку, смоченную соляной кислотой, в яркокрасный цвет, что указывало на присутствие пирроловых и индоловых оснований.
Наибольшее количество оснований, а именно 65,6% от их общего количества, было отобрано при температуре до 170°. До этой температуры выкипают метильные и диметильные производные пиридина. Если же взять температурный интервал 140—170°, то в нем выкипает 47,6%. Диметильные производные
184
Б. И. Иванов и Н. Ф. Шаронова
пиридина имеют температуры кипения от 142,5° (2,6-димегил-пиридин) до 170° (3,5-диметилпиридин).
На основании изложенного выше можно сделать вывод, что основания сланцевой подсмольной воды на 65,6% состоят из пиколинов и лутидинов, причем на долю последних приходится 47,6%. Выход остальных пятиградусных фракций был невысок и лежал в пределах 1—3%.
Учитывая то, что количество оснований, имевшихся в нашем распоряжении, было незначительным и то, что они представляют собой сложную смесь различных замещенных пиридина, мы не ограничились одной ректификацией, а для некоторых фракций провели вторичную разгонку.
Пятиградусные фракции, от 120 до 160°, были соединены вместе и разогнаны на фракции: до 122, 122—137, 137—146,5, 146,5—152 и 152—160°.
Фракции оснований, полученные после вторичной ректификации, были подвергнуты исследованию: прежде всего были определены их средние молекулярные веса методом титрования, удельные веса и содержание азота по методу сжигания.
Результаты этих определений приведены в табл. 24.
Таблица 24
Характеристика фракций, полученных при вторичной разгонке органических оснований сланцевой подсмольной воды
Пределы кипения фракций, °C Молекулярный вес "о азота
...... 122—137 । 97,2 0.9554 13,3
137—146,5 ! 106,2 0,9421 12,9
146,5-152 ! 108,7 0,9418 12,1
152—160 1 112,6 0,9383 11,6
160—165 I 117,5 0,9380 11,8
165—170 117,3 0,9380 11,8
170—175 122,4 0,9376 10,9
175-180 127,4 0,9372 10,2
180—1.85 137,7 — —
185—190 139,8 — —
190—195 147.6
195-200 155,4 —
Результаты определения молекулярного веса фракций позволяют заключить, что основания сланцевой подсмольной воды представлены производными пиридинового ряда.
Так, пиколины имеют молекулярный вес 93, лутидины—197 и коллидины— 121. Во фракции, выкипающей в пределах 180 — 185° и имеющей молекулярный вес 137,7, может находиться
Химический состав подсмольной воды
18а
2,6-диметил-4-этилпиридин, температура кипения которого равна 186°. Во фракции, выкипающей в пределах 185—190°, с молекулярным весом 139,8 могут быть 2,4-диэтилпиридин (температура кипения 187—188°) и 2-бутилпиридин с температурой кипения 189—192°. В вышекипящих фракциях, невидимому, находятся более высокомолекулярные гомологи пиридина.
Результаты определения азота также позволяют подтвердить высказанное выше положение, что основания сланцевой подсмольной воды относятся к гомологам пиридина. Действительно, если сопоставить температуры кипения азотистых оснований и процентное содержание в них азота, можно видеть, что исследуемые фракции по содержанию азота ближе всего подходят к производным пиридинового ряда.
В целях выделения отдельных индивидуумов из смеси оснований был применен пикратный метод. Осаждение производилось в спиртовом растворе. Как правило, легче всего удавалось выделение пикрата из первого осаждения; второе и, тем более, третье осаждение давали смесь трудно или совсем неразделимых пикратов.
Вся работа по идентификации оснований проводилась путем приготовления пикратов, достижения постоянной температуры плавления их, после ряда перекристаллизаций, затем давалась подробная их характеристика. В тех случаях, когда имелось достаточное количество пикрата, последний разлагался щелочью, и свободное основание после высаливания поташом извлекалось эфиром. Чистое основание после высушивания и отгонки растворителя подвергалось анализу.
Исследование фракции 105—120°. Эта фракция, имевшаяся в количестве 2,5 г, была разделена на две части. Одна часть была обработана насыщенным на холоде спиртовым раствором пикриновой кислоты, а другая — насыщенным раствором хлорной ртути. При действии последней на фракцию выпадал белый осадок, который, при перекристаллизации из сильно разбавленной соляной кислоты, образовал бесцветные кристаллы, с температурой плавления 175,5° С. По литературным данным, температура плавления двойной ртутной соли пиридина равна 177°.
Пикрат при кристаллизации из горячей воды выделил желтые кристаллы в виде коротких иголочек с температурой плавления 166° С. Пикрат пиридина плавится при 167°.
Исследование фракции 122—137°. Фракция подвергалась дробному осаждению спиртовым раствором пикриновой кислоты, взятой из расчета на пиколин. Первое осаждение дало пикрат, который после перекристаллизации из воды и ацетона плавился при 164,5°. Второе осаждение дало смесь пикратов. После их разделения получены пикраты с температурой
186
Б. И. Иванов и Н. Ф. Шаронова
плавления 149° (пикрат /3-пиколина имеет температуру плавления 149—150°) и 161° (температура плавления пикрата 2,6-лу-тидина 16Г°).
Пикрат с температурой плавления 164,5° был оттитрован 0,1 N КОН в присутствии фенолфталеина. Ниже приведены результаты титрометрического определения, показывающие, что этот пикрат представляет собой соль пиколина.
Навеска пикрата, г............................. 0,3068
Пошло на титрование 0,1 N КОН, мл................. 9,5
Найдено пикриновой кислоты, % 70,9
Найдено основания, % ..............................29,1
Найденный молекулярный вес основания ..............93,9
Молекулярный вес пиколина ......................... 93
Судя по температуре кипения фракции 122—137°, взятой для осаждения, можно предполагать, что полученный пикрат является пикратом «-пиколина, который имеет температуру кипения 129°. С целью проверки этого предположения пикрат с температурой плавления 164,5°, в количестве 6,6 г, был разложен щелочью, и свободное основание после перегонки извлекалось эфиром. После высушивания и отгонки растворителя получено 1,7 г основания с температурой кипения 129°. Молекулярный вес основания оказался равным 93.
Определение азота дало следующие результаты: найдено в двух навесках 14,65 и 14,69 или в среднем 14,67% N; требуется по теории для пиколина 15% N.
Исследование фракции 137—146,5°. Фракция осаждалась в три приема спиртовым раствором пикриновой кислоты, взятой из расчета на лутидин. Все три осаждения дали пикрат с температурой плавления 161°. Было получено 35 г этого пикрата. В результате титрования пикрата найден молекулярный вес, точно соответствующий диметилпиридину.
Навеска пикрата, г............................. 0,2335
Пошло на титрование 0,1 N КОН, мл................ 6,95
Найдено пикриновой кислоты, %......................68,2
Найдено основания, % ..............................31.8
Найденный молекулярный вес основания .............. 107
Молекулярный вес лутидина ......................... 107
При определении азота получены следующие результаты: найдено в двух навесках 16,3 и 16,6 или в среднем 16,45%N; требуется для пикрата лутидина 16,6% N.
В качестве производного этого основания была получена двойная соль с хлорной ртутью.
Для ее получения фракция растворялась в соляной кислоте и осаждалась насыщенным на холоде раствором хлорной ртути. При медленном охлаждении из горячего раствора выпала ртут
Химический состав подсмольной воды
187
ная соль в виде кристаллических иголочек, собранных в пучки. После перекристаллизации из воды, содержащей 2% концентрированной соляной кислоты, они плавились при температуре 187,5° и, следовательно, представляли собой соль 2,6-лутидина с 1 молекулой сулемы, для которой температура плавления равна 187—188°.
Все вышеприведенные данные позволяют сделать вывод, что из фракции, кипящей в пределах 137—146,5°, выделен 2,6-лутидин.
Затем пикрат с температурой плавления 161°, в количестве 29,4 г, был разложен едким натром с целью выделения свободного основания, которое после перегонки извлекалось эфиром. После высушивания и отгонки растворителя выделенное основание кипело при 142—143°, имело удельный вес с/Г’ 0,9285, молекулярный вес 106,5 (по методу титрования) и содержало 12,75% азота.
Исследование фракции 152—160°. Фракция в количестве 4,69 г была осаждена насыщенным спиртовым раствором пикриновой кислоты, взятой из расчета на лутидин. Первое осаждение сразу дало пикрат с температурой плавления 167°, которая не изменилась при последующей перекристаллизации. Такая температура плавления соответствует пикрату 2,5-диме-тилпиридина.
Анализ пикрата дал следующие результаты: навеска пикрата 0,1194 г, получено 0,2024 a СО2 и 0,0390 а И2О; навеска пикрата 0,0770 а, полученный объем N2 11,2 мл при давлении 757,9 мм и 20° С.
Найдено: 46,23% С, 3,63% Н и 16,51% N.
Вычислено: 46,42% С, 3,57% Н и 16,67% N.
При титровании пикрата получен молекулярный вес основания, соответствующий лутидину.
Навеска пикрата, г...........................0,1263
Пошло на титрование 0,1 N КОН, мл...............3,75
Найдено пикриновой кислоты, % ..................68,0
Найдено основания, % ...........................32,0
Найденный молекулярный вес основания..........107,7
Молекулярный вес лутидина .................... 107
Исследование фракции 165—170°. Фракция в количестве 5,7 а была растворена в спирте и затем осаждена спиртовым раствором пикриновой кислоты, взятой из расчета на триметилпиридин.
При перекристаллизации пикрата обнаружено, что по охлаждении из раствора одновременно выпадали различные по внешнему виду кристаллы — длинные иглы желтого цвета и более короткие. Эти кристаллы были отделены друг от друга. Таким путем были получены длинные кристаллы с постоянной
188
Б. И. Иванов и Н. Ф. Шаронова
температурой плавления, равной 156°, и короткие, температура плавления которых не была постоянной.
Температура плавления пикрата, равная 156°, соответствует пикрату 2, 4, 6-триметилпиридина.
Результаты определения молекулярного веса основания титрованием пикрата и элементарный состав подтверждают, что полученный пикрат является пикратом коллидина.
Навеска пикоата 0,1065 а; получено 0,1870 a СО2 и 0,0400 а Н2О.
Навеска пикрата 0,0857 а; полученный объем N2 12 мл при 777,2 мм и 25° С.
Найдено: 47,87% С, 4,16% Н и 15,90% N.
Вычислено: 48,0% С, 4,0% Н и 16,0% N.
Ниже приведены результаты титрования пикрата.
Навеска пикрата, г ...................... 0,2174 0,2330
Израсходовано 0,1 N КОН, мл .............. 6,20 6,65
Найдено пикриновой кислоты, % ..... 65,3 65,3
Найдено основания, % 34,7 34,7
Найденный молекулярный вес основания . . 122 122
Молекулярный вес коллидина.......... 121
ВЫВОДЫ
I. Проведено подробное исследование сланцевой подсмольной воды, выделены и идентифицированы следующие кислород-
и азотсодержащие соединения:
1) муравьиный альдегид
2) уксусный альдегид
3) пропионовый альдегид
4) масляный альдегид
5) диметилкетон
6) метилэтилкетон
7) метилпропилкетон
8) муравьиная кислота
9) уксусная кислота
10) пропионовая кислота
11) масляная кислота
12) ацетонитрил
13) фенол
14) ортокрезол
II. Оксикислоты, многоосновн в подсмольной воде отсутствуют.
15) метакрезол
16) паракрезол
17) 1,2, 4-ксиленол
18) 1, 3, 4-ксиленол
19) 1, 3, 5-ксиленол
20) 4, 5-диметилрезорцин
21) 2, 4-диметилрезорцин
22) 2, 5-диметилрезорцин
23) 4, 6-диметилрезорцин
24) пиридин
25) а-пиколип
26) 2, 6-лутидин
27) 2, 5-лутидин
28) 2, 4, 6-коллидин
е кислоты и метиловый спирт
ЛИТЕРАТУРА
1. К. И. Ногин. Сухая перегонка дерева, ГНТИ, стр, 425, 1931.
2. П. Д. Прянишников. Практикум по органической химии, 6,9. Госхимиздат, 1950.
3. Coulson and Holt, J. of the societu of Chemical industry № 8, стр. 267, 1937.
4. Holz in a n and P i 1 a t, Brennstoff Chemie, 20, 409, 1930.
5. Г. Мейе p. Анализ и определение строения органических веществ, стр. 308, 1935.
Б. И. Иванов, Ю. А. Козак и Н. Ф. Шаронова
ДЕФЕНОЛЯЦИЯ ПОДСМОЛЬНОЙ ВОДЫ ПРИБАЛТИЙСКИХ СЛАНЦЕВ БУТИЛАЦЕТ АТОМ 1
Вопрос о дефеноляции (обезвреживании) подсмольных вод, получающихся при термической переработке прибалтийских сланцев, изучался сравнительно мало.
А. П. Сиверцев и Д. А. Александрович Г1] получили хороший эффект дефеноляции сланцевой подсмольной воды при кипячении ее с формалином и серной кислотой. Этот метод основан на образовании фенолальдегидных смол, которые нерастворимы в воде и выпадают в осадок. С целью замены дорогостоящего формалина дальнейшие опыты велись с сосновыми опилками. При расходе последних 4,8%, серной кислоты 1,6% (на подсмольную воду) и 4-часовом кипячении подсмольной воды эффект извлечения фенолов составлял 95 %. Однако этот метод является очень дорогим, и аппаратурное оформление его представляет большие трудности, особенно на крупных заводах, где в сутки образуется более 1000 м3 подсмольной воды.
Примененные упомянутыми авторами для дефеноляции подсмольной воды бензол и различные фракции обесфеноленных сланцевых масел дали незначительный эффект извлечения фенолов.
Метод хлорирования сточных фенольных вод, предложенный Я- И. Чулковым [2] и проверенный ЛенВНИГИ на сланцевых подсмольных водах, оказался в аппаратурном отношении очень громоздким и связан с большим расходом хлора.
Обезвреживание сланцевых подсмольных вод активированным углем было проведено А. И. Козько, который имел дело со случайным образцом подсмольной воды, обладавшей низкой концентрацией летучих фенолов (0,27 г/л) и нелетучих фенолов (0,47 г/л). Можно заранее сказать, что регенерация активированного угля будет весьма сложной операцией, связанной с большими потерями.
Промышленный цех дефеноляции подсмольных вод в Кохтла-Ярве предусматривает экстракцию фенолов бутилацетатом,
1 В работе принимали участие М. В. Монахова и В. А. Даукшт.
190
Б. И, Иванов, Ю. А. Козак и И. Ф. Шаронова
с последующей доочисткой на биофильтрах. Решающим моментом в процессе дефеноляции подсмольной воды является экстракция фенолов растворителем, причем извлечение фенолов может идти псевдопротивотоком в смесителях, с последующим разделением жидкостей в отстойниках и в экстракционных колоннах. Аппаратурное оформление первого способа связано со значительными металловложениями, сам процесс более сложен в эксплуатации и требует большого расхода электроэнергии. Следует еще отметить, что в псевдопротивотоке длительность экстракции фенолов значительно больше и это, несомненно, создаст более благоприятные условия для окисления и полимеризации фенолов. Это обстоятельство не только ухудшает качество фенолов, но и затрудняет расслаивание жидкостей, вследствие образования эмульсии. Преимущество псевдопротивотока состоит в том, что благодаря применению насосных смесителей можно достигнуть большой дисперсности растворителя и тем самым обеспечить полноту извлечения фенолов из подсмольной воды.
Для получения сравнительных данных были проведены лабораторные опыты по экстракции фенолов из подсмольной воды псевдопротивотоком в делительной воронке и в экстракционной колонне.
ПСЕВДОПРОТИВОТОЧНАЯ МНОГОСТУПЕНЧАТАЯ ЭКСТРАКЦИЯ В ДЕЛИТЕЛЬНЫХ ВОРОНКАХ
Дефеноляция подсмольной воды псевдопротивотоком проводилась в 4 и 5 ступеней с применением 5% бутилацетата и в 3,
Рис. 1. Схема псевдопротивоточной экстракции.
4 и 5 ступеней с 10% растворителя.
Экстракция фенолов псевдопротивотоком производилась в делительных воронках, причем взбалтывание длилось 10 мин. и отстаивание 30 мин.
Схема псевдопротивоточ-ной 4-ступенчатой экстракции представлена на рис. 1.
По этой схеме определенный объем подсмольной воды (1) обрабатывался бу-
тилацетатом, раствор фенолов сливался, а оставшаяся под
смольная вода шла на вторую, третью и четвертую экстракции, каждый раз со свежим растворителем. Дефенолированная подсмольная вода сливалась. Вторая порция свежей подсмольной воды (II) обрабатывалась в 1-й, 2-й и 3-й ступенях раствори
Дефеноляции подсмольной воды бутилацетатом
191
телями 2, 3, и 4, соответственно прошедшими одну экстракцию, и в 4-й ступени — чистым растворителем. Третья порция свежей подсмольной воды (III) обрабатывалась в 1-й и 2-й ступенях растворителями, прошедшими две экстракции, в 3-й ступени — растворителем, прошедшим одну экстракцию, и в 4-й ступени — чистым растворителем. Четвертая порция свежей подсмольной воды (IV) обрабатывалась в 1-й, 2-й и 3-й ступенях растворителями, прошедшими соответственно три, две и одну экстракции, а в 4-й ступени — чистым растворителем.
Четвертая порция подсмольной воды анализировалась на содержание летучих, нелетучих и суммарных фенолов после 1-й, 2-й, 3-й и 4-й ступеней (IVi, IV2, IV3 и IV4).
Аналогично велись экстракции в 3 и 5 ступеней.
Исходная подсмольная вода была взята из общей ямы (смо-лоловушка) 4-й газогенераторной станции комбината в Кохтла-Ярве. Влажность поступающего сланца была 10%, температура газослива 180° и производительность печи 40 т в сутки сланца.
Результаты анализов представлены в табл. 1, 2, 3, 4 и 5, в которых указывается остаточное содержание фенолов после каждой ступени.
Таблица 1 4-ступенчатая экстракция с 5% бутилацетата
Фенолы Исходная концентрация фенолов, г/л Остаточная концентрация фенолов, г/л, после ступени
IV, IV2 iv3 IV4
Летучие 0,62 0,16 0,09 0,09 0,08
Нелетучие 6,26 3,57 3,22 3,00 1,88
Суммарные 6,88 3,73 3,31 3,09 1,96
Таблица 2
5-ступенчатая экстракция с 5% бутилацетата
Фенолы Исходная концентрация фенолов, г/л Остаточная концентрация фенолов, г/л, после ступени
V, v2 v3 v4 v5
Летучие .... 0,62 0,15 0,12 0,10 0,05 0,01
Нелетучие . . . 6,26 3,49 3,31 2,83 2,41 1,55
Суммарные . . . 6,88 3,64 3,43 .2,93 2,46 1,56
192
Б. И, Иванов, Ю. А. Козак и Н. Ф. Шаронова
Таблица 3
3-ступенчатая экстракция с 10% бутилацетата
Фенолы Исходная концентрация фенолов, г/л Остаточная концентрация фенолов, г/л, после ступени
Шх 1П2 П13
Летучие 0,62 0,16 0,08 0,06
Нелетучие 6,26 2,93 1,71 0,92
Суммарные 6,88 3,09 1,79 0,98
Таблица 7
4-ступенчатая экстракция с 10% бутилацетата
Фенолы Исходная концентрация фенолов, г/л Остаточная концентрация фенолов, г/л, после ступени
IVi IV2 iv3 iv4
Летучие 0,62 0,15 0,06 0,06 0,05
Нелетучие 6,26 2,91 1,94 1,25 0,65
Суммарные 6,88 3,06 2,00 1,31 0,70
Таблица 5
5-ступенчатая экстракция с 10% бутилацетата
Фенолы Исходная концентрация фенолов, г/л Остаточная концентрация фенолов, г/л, после ступени
Vi v2 v3 V, v5
Летучие .... 0,62 0,22 0,07 0,04 0,03 0,03
Нелетучие . . . 6,26 2,75 1,86 1,66 0,62 0,23
Суммарные . . . 6,88 2,97 1,93 1,70 0,65 0,26
Дефеноляция подсмольной воды бутилацетатом 193
Необходимо отметить, что при дефеноляции важным моментом является не только степень извлечения фенолов, выраженная в процентах, а конечное, остаточное содержание фенолов, так как для биофильтров, куда подается дефенолированная бутилацетатом подсмольная вода, существенное значение имеет концентрация фенолов в поступающих сточных водах. Эта величина, в условиях комбината Кохтла-Ярве, не должна превышать 0,35 г/л, что, с учетом разбавления фекально-хозяйственными водами в четыре раза, дает предельную допустимую концентрацию около 0,1 г/л фенолов.
Из приведенных в таблицах данных, с учетом допустимой концентрации по фенолам (0,35 г/л), следует, что 4- и 5-ступен-чатые экстракции с применением 5% бутилацетата, равно как и 3- и 4- ступенчатые экстракции с применением 10% растворителя, не дают удовлетворительных результатов. Лишь 5-ступенчатая экстракция с 10% бутилацетата дает хорошие результаты, причем остаточная концентрация фенолов равна 0,26 г/л.
ЭКСТРАКЦИЯ В КОЛОННАХ
Отрицательным моментом в работе экстракционных колонн является отсутствие хорошего контакта двух жидкостей, обусловленное незначительной раздробленностью последних. Это обстоятельство, естественно, снижает степень извлечения фенолов из подсмольной воды.
С целью изучения работы экстракционной колонны и выяснения вопроса об извлекаемости фенолов из подсмольной воды был поставлен ряд опытов, в которых устанавливалось влияние различных факторов.
Вначале были проведены опыты на колонне, имеющей следующие размеры: диаметр 67 мм, рабочая высота 4350 мм, полная высота 5000 мм, насадка из колец Рашита 14X14X1 мм, объем колонны 15,2 л, объем насадки 3,2 л, свободный объем колонны 12,0 л.
Опыты проводились по общей, описанной ниже методике. В качестве растворителя применялся бутилацетат, в количестве 10% весовых или 11,5% объемных от подсмольной воды. Сплошной фазой являлась подсмольная вода, дисперсной фазой — бутилацетат. Объемная скорость подсмольной воды была равна 6,3 м3/м2/час, а бутилацетата — 0,6 м5/м^/час. Линейная скорость подсмольной воды была равна 0,0017 м/сек. Бутилацетат подавался снизу, без специального устройства для раздробления, подсмольная вода поступала в верхнюю часть колонны. Отбор проб производился на расстоянии 1,75 и 4,35 м от верха колонны.
13 Труды ВНИИПС.
J 94
Б. И. Иванов, Ю. А. Козак и И. Ф. Шаронова
Подсмольная вода, прошедшая колонну, вновь возвращалась на верх колонны для дальнейшей экстракции, причем операции повторялись до тех пор, пока остаточная концентрация фенолов не становилась постоянной.
Для опыта была взята смешанная подсмольная вода, состоящая из 11 частей генераторной и 6 частей камерной подсмольной воды, т. е. представляющая собой среднюю подсмольную воду, получающуюся на комбинате Кохтла-Ярве.
Результаты опытов представлены в табл. 6.
Таблица 6
Извлечение фенолов из подсмольной воды на экстракционной колонне Рабочая высота колонны 4,35 м
Отбор проб на расстоянии 1,75 и 4,35 м от верха колонны
Фенолы
Остаточная концентрация фенолов, г/л, после экстракции
Летучие
Нелетучие
Суммарные
Полученные результаты показывают, что при 5-й экстракции извлечение из подсмольной воды фенолов прекращается. Однако в этом случае получается очень высокая остаточная концентрация суммарных фенолов, равная 1,3 г/л, что соответствует 90,8%-ной степени дефеноляции подсмольной воды. Причина такой плохой очистки подсмольной воды кроется либо в высокой исходной концентрации фенолов (14,57 г/л), более чем в два раза превышающей обычное содержание фенолов, либо в неудовлетворительной работе колонны. Однако поставленные дополнительные опыты по экстракции фенолов из той же подсмольной воды в делительной воронке в 8 ступеней с применением 10% растворителя показали, что остаточная концентрация суммарных фенолов может быть снижена более чем в два раза.
Результаты этих опытов представлены в табл. 7.
Дефеноляция подсмольной воды бутилацетатом
195
Таблица 7 Опыты по экстракции подсмольной воды
Фенолы Концентрация фенолов в исходной воде, г/л Остаточная концентрация фенолов, г/л, после ступени
6-й 7-й 8-й
Летучие Нелетучие 1,11 13,46 0,11 0,98 0,12 0,51 0,12 0,43
Суммарные 14,57 1,09 0,63 0,55
Полученные данные показывают, что после 8-й ступени экстракции, несмотря на то, что равновесие, отвечающее растворимости фенолов, еще не было достигнуто, остаточная концентрация фенолов была значительно ниже, чем при экстракции на колонне (1,3 г/л). Опыты на последней со скоростью сплошной фазы выше 6,3 М'1м'! час нельзя было провести из-за захлебывания колонны. Результаты опытов, проведенных со скоростью 4,3 м31м21час, дали еще менее удовлетворительные результаты, несмотря на то, что в одном случае было взято 20% бутилацетата. В первом случае с применением 10% бутилацетата остаточная концентрация фенолов была равна 1,90 г/л, а во втором случае с применением 20% бутилацетата — 2,00 г/л.
В. В. Кафаров и М. А. Плановская [3] исследовали гидродинамические факторы работы насадочных экстракционных колонн, определяющих эффективность процесса экстракции. Они различают три вида движения жидкостных потоков в колонне, а именно: пленочное, турбулентное и вихревое.
Пленочное движение характеризуется небольшой раздробленностью капель, растекающихся в виде пленки на насадке. Это движение потоков связано с небольшими скоростями жидкостей и дает наихудший эффект экстракции.
Турбулентное движение, отличающееся значительной раздробленностью капель, связано с большими скоростями потоков и дает значительно лучший эффект экстракции, чем пленочное движение.
Вихревое движение характеризуется еще большей дисперсностью растворителя, приближающейся к дисперсности веществ в эмульсиях, причем в этом случае благодаря сильно возросшей поверхности контакта получается максимальный эффект экстракции. Вихревое движение потоков связано с наибольшими скоростями, превышение которых приводит к затоплению колонны.
196 Б. И, Иванов, Ю. А. Козак и И, Ф. Шаронова
Создание указанных выше потоков может достигаться изменением скорости либо одной фазы, либо обеих, причем важное значение при этом приобретает отношение скоростей сплошной и дисперсной фаз. Большое значение имеет сопротивление колонны, которое складывается из сопротивлений насадки и опоры для насадки. Следует отметить, что при больших сопротивлениях затопление колонны может наступить без предварительного возникновения турбулентного или вихревого движения.
С целью изучения гидродинамики процесса экстракции была сделана стеклянная колонна следующих размеров: рабочая высота 1270 мм, полная высота 1500 мм, диаметр 46 мм, насадка 11 X 11 X 1,2 мм, объем пустой колонны 2,12 л, объем насадки 0,59 л, свободный объем колонны 1,53 л. Дисперсная фаза подавалась через четыре форсунки, диаметр которых был равен 0,5 мм.
Вначале были поставлены опыты с насадкой, имеющей размеры 6 X 6 X 0,8 мм; однако уже при скорости сплошной фазы 9 м31м21час наступило затопление колонны, и дальнейшая работа была проведена с насадкой, имеющей размеры 11 X И X 1,2 мм.
Для большей наглядности опыты велись с подкрашенной водой и бутилацетатом, причем последний был дисперсной фазой, а вода — сплошной. Были взяты следующие скорости: V* = 9; 18; 27 м31м21час и соответствующие скорости Уд** 1,35; 1,35 и 1,35 м3/м2[час.
В другой серии опытов изменялась скорость как сплошной, так и дисперсной фазы. В табл. 8 представлены результаты этих опытов с визуальным контролем.
Эти опыты наглядно, хотя и в качественной форме, показали, что при одинаковой насадке и постоянстве других факторов
1аблица 8
Влияние скорости потоков на дисперсность жидкостей
Ус = 9 м3/м Уд=1,35 Ус= 18 м3!м Уд=1,35 С С Г) II 11^ "|| ”|| Ус = 27щ3/ш Уд = 2,7 II II Q «=(
1 опыт 2 опыт 3 опыт 4 опыт 5 опыт 6 опыт
Пленочное движение, крупные капли То же, что и в опыте 1 Пленочное движение, более раздробленные капли Пленочное движение, еще более раздробленные капли Еще более раздробленные капли, чем в опыте 4 Движение, приближающееся к турбулентному
* Vе —объемная скорость сплошной фазы.
** X—объемная скорость дисперсной фазы.
Дефеноляция подсмольной воды бутилацетатом 197
увеличение скорости сплошной и дисперсной фазы приводит к повышению дисперсности последней, в данном случае бутилацетата.
Не менее важным обстоятельством, влияющим на извлечение экстрагируемого вещества, является выбор сплошной и дисперсной фаз. Например, если в качестве сплошной фазы выбрать бутилацетат, поверхность контакта подсмольной воды с растворителем значительно возрастет, в результате чего в этом случае становится возможным в широких пределах изменять по желанию количества и скорости бутилацетата.
Дальнейшие опыты были поставлены по извлечению фенолов из подсмольной воды бутилацетатом. В качестве сплошной фазы был выбран бутилацетат, который заполнял колонну на высоту 1170 мм при рабочей высоте колонны 1270 мм. Количество бутилацетата составляло 15% весовых или 17,5% объемных от подсмольной воды. Скорость дисперсной фазы (подсмольной воды) составляла 20 м3/м2/час, а сплошной фазы (бутилацетата) — 3,5 мл/м^/час. Соответствующие линейные скорости были равны 0,0055 и 0,001 м/сек. Количество бутилацетата и взятые скорости потоков соответствовали данным, запроектированным Ленгипрогазом для промышленной колонны в Кохтла-Ярве. Через колонну было пропущено 18 л подсмольной воды, причем это количество семь раз возвращалось на колонну. Это мероприятие позволило вести опыты со все возрастающей высотой колонны. Отбор проб для анализа фенолов производился после установления равновесия, что определялось отдельными анализами.
Результаты опытов представлены в табл. 9, где указывается остаточная концентрация фенолов и окисляемых веществ.
Полученные результаты показывают хорошую дефеноляцию подсмольной воды, достигающую 96,2%, при остаточной концентрации фенолов — 0,28 г/л. Высокий эффект извлечения фенолов Таблица 9 Экстракция на колонне
Скорость бутилацетата Ус = 3,5 м3/м2/час', скорость подсмольной воды Уд = = 20 м3/м2/час', рабочая высота колонны 1,27 м
Определяемые анализом вещества Концентрация в исходной п/воде, г/л Остаточная концентрация фенолов, г/л, при числе экстракции
1 2 3 4 5 6 7
Летучие фенолы . . . 0,71 0,16 0,09 0,11 0,09 0,08 0,08 0,04
Нелетучие , ... 6,60 1,49 1,07 0,61 0,37 0,38 0,26 0,24
Суммарные , ... 7,31 1,65 1,16 0,72 0,46 0,46 0,34 0,28
Окисляемые вещества 8,36 2,00 1,60 1,11 0,76 0,90 0,63 0,66
198
Б. И. Иванов, Ю. А. Козак и Н. Ф. Шаронова
подтверждается и результатами определения окисляемых веществ, количество которых снизилось на 92,2 %.
Для получения сравнительных данных была проведена экстракция фенолов из подсмольной воды в делительных воронках в 6 ступеней. Результаты этих опытов представлены в табл. 10. Они показывают остаточную концентрацию фенолов.
Из полученных данных видно, что 6-ступенчатая экстракция фенолов в делительных воронках соответствует 7 экстракциям на колонне, имеющей рабочую высоту 1,27 м, и что оба метода экстракции могут обеспечить практически одинаковую степень дефеноляции.
Таблица 10
6-ступенчатая экстракция в делительных воронках
Фенолы Концентрация фенолов в исходной п/воде, г/л Остаточная концентрация фенолов, г/л, после ступени
3-й 4-й 5-й 6-й
Летучие 0,71 0,08 0,08 0,07 0,03
Нелетучие 6,60 0,49 0,42 0,28 0,21
Суммарные 7,31 0,57 0,50 0,35 0,24
Следующие опыты были проведены при повышенных скоростях потоков, а именно, при скорости бутилацетата Ис — 4,4 лё/м^/час и скорости подсмольной воды Ид = 25 лР/лР/ 'ас.
Результаты опытов представлены в табл. 11.
Таблица 11
Опыты на экстракционной колонне
Скорость бутилацетата Ус = 4,4 л3/.«2/час; скорость подсмольной воды Vд = = 25 м3/м2/час', рабочая высота колонны 1,27 м
Фенолы Концентрация фенолов в исходной п/воде, г/л Остаточная концентрация фенолов, г/л, при числе экстракций
1 2 3 4 5 6 7
Летучие Нелетучие 0,62 5,10 0,25 2,29 0,05 0,62 0,04 0,55 0,04 0,47 0,04 0,44 0,04 0,39 0,04 0,31
Суммарные .... 5,72 2,54 0,67 0,59 0,51 0.48 0,43 0,35
Дефеноляция подсмольной воды бутилацетатом
189
Увеличение скорости потоков в данном случае не дало лучших результатов, чем те, которые были получены в предыдущих опытах. Полученная, несколько завышенная, концентрация фенолов, равная 0,35 г]л, объясняется, скорее, не гидродинамическим режимом колонны, а окислением фенолов подсмольной воды ввиду более длительного ее хранения.
ВЫВОДЫ
Проведенные опыты по удалению фенолов из подсмольной воды путем псевдопротивоточной экстракции в делительной воронке и экстракции на колонне позволяют сделать следующие выводы.
1. Псевдопротивоточная экстракция фенолов из подсмольной воды с начальной концентрацией фенолов 6,88 г/л дает хорошие результаты при 5 ступенях и применении 10% бутилацетата.
2. В случае экстракции на колонне на раздробленность дисперсной фазы, а следовательно, на достижение лучшего контакта между фазами, большое влияние оказывает гидродинамический режим процесса экстракции. Большие скорости движения потоков обеспечивают достаточно высокую степень раздробленности дисперсной фазы.
3. Для достижения достаточно высоких скоростей потоков необходимо, чтобы сопротивление колонны не было большим.
4. Применяя бутилацетаг в качестве сплошной фазы, можно получить хороший эффект дефеноляции подсмольной воды на колонне высотой в 4,35 м при экстракции в 2—3 ступени с применением 17,5 объемных процента от подсмольной воды растворителя.
ЛИТЕРАТУРА
1. А. П. Сиверцев и Д. А. Александрович. «Промышленность органической химии» № 17—18, 281, 1937.
2. Я. И. Чулков. Химическая промышленность № 12, 48, 1934.
3. Е'. В. К а ф а р о в и М. А. Плановская. Журнал прикладной химии № 6. 624, 1951.
Б. И. Иванов и Ю. А. Козак
ОБЕЗВРЕЖИВАНИЕ ПОДСМОЛЬНЫХ ВОД ПРИБАЛТИЙСКИХ СЛАНЦЕВ ЗОЛОЙ И КОКСОМ1
В отечественной и иностранной литературе имеются указания, что сточные воды, в частности фенольные воды, в некоторых случаях могут быть обезврежены золой и шлаком.
Л. В. Милованов и Р. В. Суворова [1] исследовали сорбционные свойства золы и шлака каменных углей, бурых углей и торфа по отношению к водным растворам фенолов, причем продолжительность контакта не превышала 30—45 мин.
Результаты этого исследования представлены в табл. 1.
Сорбционные свойства золы и шлаков
Наименование образцов Потери при прокаливании, % Концентрация фенольной воды, мг/л 1 кг образца снимает мг фенолов
ДО опыта после опыта
Шлак и зола донецкого тощего каменного угля .... 0,56 300,6 298,3 7,7
Шлак бурого подмосковного угля 0,70 333,7 320,9 42,7
Летучая зола бурого подмосковного угля . 1,11 300,6 253,0 158,7
Торфяной шлак из-под решеток 5,18 300,6 30,3 901,0
Летучая торфяная зола из-под экономайзера ....... 5,84 300,6 3,5 990,3
Летучая торфяная зола из циклона 8,90 300,6 1,95 997,8
Как видно из полученных результатов, те образцы золы, в которых имеется больше несгоревших частиц кокса, обладают наибольшей адсорбционной способностью. Особенно активно поглощает фенолы несгоревший кокс торфяной золы. Наоборот,
1 В работе принимали участие Е. А. Косарева, Е. А. Даукшт и М. В. Монахова.
Обезвреживание подсмольных вод золой и коксом
201
зола, не содержащая кокса, совершенно не обладает сорбционными свойствами. Наряду с этим, зашлакованность золы снижает ее адсорбционные свойства.
Особенно интересным представлялось изучение возможности обезвреживания подсмольных вод прибалтийских сланцев золой и коксом, так как последних образуется в 2—3 раза больше, чем подсмольных вод. Этот способ обезвреживания заманчив тем, что требует незначительных капитальных и эксплуатационных затрат ввиду возможности вести процесс обезвреживания непосредственно на коксозольных отвалах.
Предварительные опыты, поставленные в лаборатории, имели своей целью выяснить действие золы или кокса на фенолы подсмольной воды. Вначале было установлено, что зола и кокс впитывают в себя до 50% подсмольной воды, причем последняя моментально темнела, приобретая бурый цвет. Благодаря присутствию в коксе и золе свободной извести между фенолами подсмольной воды и известью протекают реакции образования фенолятов кальция, хорошо растворяющихся в воде. Действие на подсмольную воду золы, содержащей свободную известь, сопровождается незначительным выделением аммиака и пиридиновых оснований.
В силу гидратации извести происходит разогревание всей массы. В первых опытах четыре образца генераторной золы были смочены половинным количеством подсмольной воды и затем 1-й образец был сразу промыт двойным объемом чистой воды, 2-й, 3-й и 4-й были промыты, соответственно, через 15, 30 и 45 дней. Полученные водные вытяжки анализировались на летучие и нелетучие с водяным паром фенолы. Результаты представлены в табл. 2.
Таблица 2
Влияние золы на растворимость фенолов подсмольной воды
Фенолы Исходная подсмольная вода содержала, мг Вымыто
сразу после смачивания через 15 дней через через 45 дней
30 хней
мг % мг % мг % мг %
Летучие . . . 44 25 57 8 18 2 5 2 5
Нелетучие . . 148 93 63 63 42 39 26 26 17
Суммарные . . 192 118 61 71 37 41 21 28 15
202 Б. И. Иванов и Ю. А. Козак
Полученные результаты показывают, что адсорбция фенолов золой из подсмольной воды незначительна и равна 39% (61 % вымывается водой). Однако с течением времени феноляты кальция заметно теряют растворимость в воде и, спустя 45 дней, лишь 15% суммарных фенолов могут быть вымыты из золы. Заметное побурение подсмольной воды, при соприкосновении с золой, и потеря растворимости фенолятов кальция, при хранении их на воздухе, указывают на процессы окисления и полимеризации фенолов, ведущие к образованию нерастворимых в воде веществ.
В естественных условиях, на открытом воздухе, благодаря действию прямых солнечных лучей и ветра процессы окисления, и полимеризации должны идти быстрее.
В практических условиях обезвреживание подсмольной воды должно осуществляться следующим образом: зола или кокс, смоченные подсмольной водой, направляются по канатной дороге на отвалы, где они накапливаются в виде гор.
Однако на практике, ввиду большой высоты коксозольных гор, достигающих 70 м и более, встретились трудности в передвижке канатной дороги. Для устранения этого работники газосланцевого комбината в Кохтла-Ярве стали применять размыв поступающей золы и кокса водой. В качестве последней бралась смесь подсмольных и технологических вод с общей концентрацией фенолов около 1 г/л.
Исследования, проведенные с фенольными водами, непосредственно стекающими с зольной и коксовой гор, показали совершенно различные их свойства.
В то время как коксовая гора содержит крупные, довольно прочные куски, зольные горы в основном содержат мелочь, сце-ментировавшуюся в довольно плотную массу. Фенольные воды свободно проходят через всю толщу коксовой горы, как через своего рода фильтр, и вытекают значительно обесцвеченными и прозрачными. Следует отметить, что фенольные воды, вытекающие из подошвы коксовой горы, имеют температуру 60° и выше.
Совсем иное происходит на зольных горах. Фенольные воды не проходят внутрь горы, а стекают по откосу, без заметного осветления, одним потоком, который у основания разбивается на многочисленные ручейки, охватывающие всю гору полукругом, радиусом в 300—500 м. Эти ручейки пропитывают на своем пути размытую золу, создают большую поверхность испарения и лишь частично достигают канавы.
Ввиду того что при контакте подсмольной воды с золой образуются феноляты кальция, то, несмотря на значительное испарение стекающих вод, фенольный запах совершенно отсутствует. Наоборот, фенольные воды, стекающие с коксовой горы, благо
Обезвреживание подсмольных вод золой и коксом 203
даря наличию в последней сернистого кальция имеют сероводородный запах.
Исследование фенольных вод, вытекающих из коксовой и двух зольных гор, а также из общей канавы, куда они направляются, было проведено ВНИИПСом и Центральной лабораторией газосланцевого комбината в Кохтла-Ярве под руководством Э. Е. Сухотина.
Исходные фенольные воды подаются двумя насосными станциями, из которых одна находится у перекачки и подает воду на 1-ю зольную гору, а вторая находится у ГГС-4 и подает воду на 2-ю зольную гору и 3-ю коксовую гору.
Результаты анализов сведены в табл. 3.
Из полученных данных видно, что понижение концентрации фенолов и окисляемых веществ происходит больше у 3-й коксовой горы и меньше у 1-й и 2-й зольных гор, однако остаточная концентрация фенолов во всех случаях велика и является недопустимой для сброса в открытые водоемы.
В дальнейшем был произведен анализ стока, находящегося в 8 км от коксозольных отвалов, но и там концентрация фенолов была не ниже 0,10 г/л.
В 20 км от комбината канава, в которую сбрасываются все фенольные стоки, соединяется с небольшой речкой Пурца, впадающей в Финский залив.
Содержание фенолов в стоке у р. Пурца равно 0,02 г/л.
Так как средняя скорость сточных вод в канаве равна 0,2 м/сек и общая протяженность пути до залива не превышает 30 км, то время, в течение которого фенольные воды аэрируются до впадения в залив, определяется небольшой величиной, равной 40 часам.
По всей вероятности, этого времени недостаточно для того, чтобы фенолы могли полностью окислиться. Для подтверждения такого предположения были поставлены опыты, в которых подсмольная вода, с начальной концентрацией фенолов 100, 300 и 500 мг/л, смешивалась с золой в отношении 5:1, после чего пробы выставлялись на открытый воздух. По истечении определенного времени пробы анализировались на содержание фенолов и окисляемых веществ. Результаты исследования представлены в табл. 4.
Как видно из полученных данных, подсмольная вода, обработанная золой, окисляется медленно, и уменьшение фенолов идет хорошо лишь для многоатомных (нелетучих), которые более склонны к окислению, чем одноатомные (летучие).
Доказательством того, что уменьшение фенолов обусловливается окислением и полимеризацией, с последующим выпадением полимеров в осадок, служит прямое определение окисляемых веществ, количество которых также снижается.
29/VII 1/VIII 4/VIII 9/VIII 11/VIII 13/VIII 17/VIII 26/IX 29/IX 3/X 6/X 11/X 15/X 19/X 24/X 27/X 1/XI 4/XI 13/XI 17/XI 24/XI Среднее Дата Насосная ГТС-4
P ю — с>оррро —pp —pc> c>c>c>05 с> — с> 00 О — О 00 оо 'ф>- О Ю — оо OJ 05 Сл QO 4^ КЗ 00 Go 05 О Сл СО КЗ 0> —• 05 ОС Ф GO 05 Оз Сл КЗ 0п — — — -4 05 Сл Сс КЗ — Суммарные фенолы, г/л
КЗ КЗ — —‘„-рР О КЗ —ФО — — — О КЗ—-4 — ф СО co 1 4^00 1 1 4^ Сл 'ф СО Ф — ф 05 Сл 4- 4^ 1С - КЗ КЗ 1 КЗ О ’ ’ -4 со КЗ Сл 05 4-* 4^ о “« Окисляемые вещества, г/л
а 2 ~Х~Х х X XXXХХ7Р < < < < < • 1—11—1 . —Z ZZZ —-о Дата Перекачка
Ф РгРРРРРРР^Р.^ Фрррр о — о о 05 О Ф 05 4^ 05 05 КЭО1'^ Ф ОС СС О 00 GJ ф — 7—— 05 СЛ СО СО КЗ О СО СО КЗ со ю 05 О С — СС — СЛ ф ф го со Суммарные фенолы, г/л
р — КЗ —ФФ05О, •—* — . — — кзф —ф —фф — сл — — фРГф'^ } КЗ 05 1 1 СЛ4^ СО Оо — 05 со Со Со со со СО со СО КЗ СО 4-* со о 05 to СЛ 05 4-* -4 to 4-* 4^ Окисляемые вещества, г/л
Г) to —1— ко ко — — ко ко — — р “ , X 4^-4СО4^ — “«4^0>СЛ05СОф)05“<1 — Слрр^р^р^ га S 1В XX 2 ******X - - - - Дата У подножья 1-й горы (зольной)
СО ФФФ05ФООФФФФО. Ф О Ф Ф О О С to Со'-4 4- 0> —КЗ СО юР 4^ СО СО 1 СО — СО “О КО СО СО — to СО 05 — to СО Сл -4 со со to со Со — Сл 4-* Со СО 4-* Суммарные фенолы, г/л
СО — to со | со | 05 | со . .ффффф.фоф со С0 05'“<1 1 СО 1 4S*. 1 05 1 1 4^ Со'-4 ОС Со | КО КЗ Со СО Ю05-4 —‘ -4 to 4-* Сл -4 to СО 05 00 КЗ Окисляемые вещества, г/л
/-ч^з —— r<ito^-^-“- to to — — Д^4*<со4* -^<ДОСОС1>~С5СООО'.<Н- Сл <~"< <4 го g х'х'х'х х х'х'х'х х'х'х'хрВзр.-; < < < Р >— >— « >—« •—1 г* r^< r^< r*S кх ЗО I - —Z Дата У подножья 2-й горы (зольной)
СО р>- со СО СО СО со СО СО СО СО СОСО СО О СО СО СО СО рр 7и 7— 4^'4^ 05 4^ Со''<Г‘<1 р* 05 со'—* Сл'--Йо'— СО со”—1 о СО со со СО “<1 05 со —- GO со СО со со со — to со — СО 05 сл о Суммарные фенолы, г/л
—* to ’“*|——* СО СО I । СО । । СО С> । jO J-* '—* -4 со-4 4^co'-j to 1 1 '-«'со ! 1 со о* СО Сл СО 1 СлСо 05 — -“-О СО “< -о Сл КЗ со со—* 4- со — Окисляемые вещества, г/л
, to — — to to — — — to to — — QO C2-4 Х^^-ОСО^^^^СОСЛ — 05 CO CO 05^4 — СЛ<Рр.^-< га g 'х'х х'х'х'х XX XX XX^"^ /7^ £ < < < < Дата У подножья 3-й горы (коксовой)
co co co co co co co co co co co co co co co co co o co co о 05 co to 1-* CO CO CO CO CO CO — CO CO КЗ co — co co — to co'— 'p CO 05 CO’to CO’oc'GO СЛ CO’to CO’—-4 — "4 to СЛ-4 о-<1 — Сл Суммарные фенолы, г/л
CO ppp CO CO —‘pppp . p—ppp I p 1 p *05 4^'to'4^ Co 05'to CO CO 4^ Co 1 ! Co CO ^4 P 4^ 1 'ос 1 0: Co — 05 СЛ 05 СЛ tO tO 05 05 СЛ 00 05 4^ СЛ 05 05 СЛ Окисляемые вещества, г/л
П to — — to — — — toto — .^pp^j а Ду — -“-4 CO — — -ОСОСЛ — co co 05 — <P<P-P ro g / g/ p / z / z'/ - < < < < Дата Канава общих стоков 1
p co co cop CO CO CO COppppppppp — Io 'to 'to — '— CO CO '— CO CO 'o* o 'co Io 05 co co СЛ to CO Co — Сл GO 05 to — — Сл to co CO co 00 05 Суммарные фенолы, г/л
Ф o — — — — O|C0O. ppp | p | — 00 '-4 CO Сл olo'05 1 4*'-4 1 РГоо'сл i 'to i tO । 05 -4 — 05’05 00— GO— -4 CO GO — CO Окисляемые вещества, г/л
to — to to — X Л-J o. CO 4- — —4 4^ CO ro g X~X X X X X X Дата После терри-коника ГГС-5
05 pp coppp CO о co co co'о О 'o'- СО co о Сл О — Сл — Суммарные фенолы, г/л
0> О О О О 05 О | со 05 СО сл СО СО со 1 05 со — о — 4-* Окисляемые вещества, г/л
Анализ фенольных стоков
Обезвреживание подсмольных вод золой и коксом
205
Таблица 4
Окисляемость фенолов подсмольной воды обработанной золой, в зависимости от времени
Исходная Компоненты грация, Опыт 1 по истечении Опыт 2
ПО 14 истечении
10 21 пня 28 д ней
дней дней
п/воды, о — — — 'ч©
подсмольная вода определяемые я о id К рация рация обез-НИЯ, °/ к 5 Д’ R3 О- рация обез-ния, 9
анализом СТЗ н н
R я о о я £ 5 S
Д 35 а и *
ког M2t КО! сте вре S S' а Ч KOI M2k сте вре
П/вода, содержащая 100 мг/л фенолов ) Летучие фенолы : Нелетучие : Окисляемые веще- ; ства 12 88 136 9 о 5 0 25 58 100 82 8 7 21 6 0 14 50 100 90
П/вода, содержащая 300 мг/л Летучие фенолы Нелетучие Окисляемые веще- 264 24 9 23 0 36 100 21 29 14 4 61 99
фенолов ! ства 408 88 78 83 44 89
П/вода, содержащая 500 мг/л фенолов Летучие фенолы 60 44 28 53 27 1 19 68
Нелетучие „ Окисляемые веще- 440 131 60 86 67 ! 5 99
ства 680 — 236 66 150 । ! 81 88
Однако в этих опытах, как и в случае пропитки золы и кокса подсмольной водой, а также размывки коксозольных отвалов, обезвреживание идет очень медленно, и для полного завершения этого процесса требуется время, исчисляемое, по крайней мере, 1—2 месяцами.
ВЫВОДЫ
1. Практика размыва фенольными водами коксозольных отвалов, с целью обезвреживания этих вод, показала, что дефе-ноляция происходит, но недостаточно эффективно, и остаточная концентрация фенолов, равная 100 мг/л, не позволяет спускать сточные воды в открытые водоемы.
2. Коксовая гора благодаря наличию органических веществ, обладающих активной поверхностью, обезвреживает подсмольную воду лучше, чем зольная гора. С другой стороны, подсмольная
206
Б. И. Иванов и Ю. А. Козак
вода загрязняется сернистым кальцием, который она извлекает из кокса.
3. Процесс обработки золы или кокса подсмольной водой связан с образованием фенолятов кальция. Окисление последних идет значительно быстрее в случае многоатомных фенолов, чем в случае одноатомных, однако для полного окисления фенолятов кальция требуется значительное время, не менее 1—2 месяцев.
4. Обезвреживание подсмольных вод прибалтийских сланцев золой или коксом не обеспечивает достаточной полноты дефено-ляции, вследствие чего этот метод обезвреживания не может быть рекомендован для промышленности.
ЛИТЕРАТУРА
1. Л. В. Милованов и Р. В. Суворова. Производственные сточн'ые воды, вып. II, стр. 79, Медгиз, 1950.
A. E. Драбкин
К ВОПРОСУ ОБ ОЧИСТКЕ ГОРЮЧИХ ГАЗОВ
ОТ СЕРОВОДОРОДА РАСТВОРАМИ АМИНОСПИРТОВ
За последние годы в промышленности значительное распространение получил способ очистки газов от сероводорода и углекислоты водными растворами аминоспиртов.
Обладая высокой абсорбционной способностью по отношению к H2S и СО2, аминоспирты образуют с ними нестойкие соединения — соли, легко разлагающиеся при нагревании с выделением исходного абсорбента, что позволяет весьма просто осуществить процесс очистки газа с непрерывной регенерацией поглотительного раствора.
В зависимости от требований производства, подбором соответствующих аминоспиртов и технологического режима, при одновременном присутствии углекислоты и сероводорода в газе, можно организовать преимущественное извлечение последнего.
Исследования, посвященные способу очистки газов от H2S и СО2 аминами и их производными, весьма многочисленны. Они касаются, главным образом, кинетики, статики и технологии процесса. Однако в литературе нет почти никаких указаний о влиянии на процесс кислорода, содержащегося во многих технических газах. Между тем, как показали наши опыты, проводившиеся длительное время на промышленной установке, наличие в газе даже небольших количеств кислорода (1 % и менее) приводит к интенсивному образованию стойких соединений, практически неразлагаемых при температуре регенерации (102—103° С) и, следовательно, к понижению концентрации активных аминов в растворе, который быстро теряет свои поглотительные свойства.
МЕТОДИКА АНАЛИЗА РАБОЧЕГО РАСТВОРА
При взаимодействии кислых компонентов газа (СО2, H2S) с водными растворами аминоспиртов можно ожидать, что в присутствии кислорода, кроме углекислых солей и сульфидов, образуются также такие соли, как сульфат и тиосульфат соответствующих аминов.
208
A. E. Драбкин
Анализ такой сложной смеси представляет известные трудности, особенно при определении сернистых соединений. Поэтому считаем необходимым кратко изложить принятую нами методику анализа.
Систематический ход анализа сводится к тому, что определяются свободные амины, соли, разлагаемые при нагревании (сульфиды, карбонаты), и стойкие соли, к которым относятся сульфат, сульфит и тиосульфат аминов.
Титрованием раствора 0,1 N серной кислотой устанавливается общее количество титруемых аминов, т. е. свободных и связанных в виде сульфида и карбоната. Затем раздельно находят количество аминов, связанных в виде указанных солей. Вычитая это количество из общего количества титруемых аминов, определяют количество свободных аминов.
Сульфат определяют осаждением с хлористым барием весовым методом или титрованием в присутствии родизоновокислого натрия как индикатора.
Сульфит, сульфат и тиосульфат определяют йодометрически, а карбонат — по количеству углекислоты, выделяющейся при его разложении.
Определение S", SO3" и S2O3". Вначале берут навеску анализируемого амина и титрованием 0,1 N раствором йода находят количество последнего, расходующееся на титрование суммы анионов S", SO3" и S2O3", затем берут другую навеску и обоабатывают ее уксуснокислым цинком. Осадок ZnS отфильтровывают, промывают и титруют 0,1 N раствором йода. Таким образом прямо определяется содержание аниона S". Разность между результатами первого и второго титрования, отнесенная к единице навески, соответствует содержанию суммы SO3" и S2O3"
Далее, к фильтрату прибавляют в избытке формалин; при этом сульфит переходит в альдегидобисульфитное соединение, которое не реагирует с йодом. Подкислив затем раствор уксусной кислотой, титруют йодом тиосульфат.
Анализ многих рабочих растворов триэтаноламина показал, что сульфит и сульфат образуются в незначительных количествах (менее 1%), что позволяет ограничиваться определением сульфида и тиосульфата.
Прописи анализа последних и карбоната помещены ниже.
Определение сернистых соединений. В колбу емкостью 200 мл вводят 20 мл 0,1 N раствора йода, 10 мл 2N уксусной кислоты и 50 мл 5%-ного водного раствора глицерина. Затем туда же вносят навеску 5—10 г анализируемого раствора. После легкого взбалтывания дают смеси стоять 30 мин. в темном месте. По истечении этого времени оттитровывают избыток йода, не вступившего в реакцию, тиосульфатом натрия. Коли
Очистка горючих газов от сероводорода 209
чество вступившего в реакцию йода, отнесенное к единице навески, соответствует его расходу на титрование суммы анионов S2O3" и S".
Далее определяют расход йода на титрование S". Для этого в колбу емкостью 200 мл наливают 20 мл 3%-ного раствора уксуснокислого цинка, 7 мл 2 N уксусной кислоты и 50 мл 5%-ного водного раствора глицерина. В полученную смесь вводят навеску 10—15 г анализируемого раствора амина. После стояния колбы в темноте в течение 30—40 мин. отфильтровывают осадок ZnS. Промывают его подкисленной водой (10 мл Н2О + 1 мл СНзСООН) и затем вместе с фильтром переносят в широкогорлую колбу, в которую предварительно было введено 10 мл 0,1 N раствора йода, 5 мл 2 N уксусной кислоты и 50 мл дестиллированной воды. По истечении 10—15 мин. оттитровы-вают тиосульфатом натрия избыток непрореагировавшего йода.
Если обозначить количество йода, потраченное на титрование S" и1 S2O3" через а, а на титрование S" через Ь, то концентрации этих анионов в растворе определяются из уравнений:
Q„_ & 0,0112-1000 ,
о — 2
o ~ „ (a—b) -0,0016- 1000 ,
S2Os = —-----------------г/л,
где: g —навеска, г;
0,0112— количество S2O3", соответствующее 1 мл 0,1 N раствора йода;
0,0016 —количество S", соответствующее 1 мл 0,1 N раствора йода.
Определение амина, связанного в виде к ар бон а та. В колбу 1 (рис. 1), емкостью около 300 мл, после продувки всей системы азотом (или воздухом, очищенным от СО2) помещают навеску анализируемого амина и при непре-кращающейся подаче азота из капельной воронки 2 вначале на холоде, затем при нагревании вводят 20—30 мл 30 %-ной H2SO4. Выделяющуюся при разложении СО2 улавливают в змеевиковом адсорбере 3 титрованным раствором Ва(ОН)2, избыток которого по окончании реакции оттитровывают 0,lN НС1.
Количество СО2 определяют из уравнения
Vco • 1,113,
CO2 g
14 Труды ВНИИПС.
210
А. Ё. Драбкин
где: VCOa — количество СО2, мл]мл раствора;
а—количество мл O,1N раствора Ва(ОН)2, введенное в абсорбер;
b—количество мл 0,1 N раствора НС1, пошедшее на обратное титрование;
kt и /с2— коэффициенты нормальности, соответственно для Ва(ОН)2 и НС1;
1 113 — количество мл СО2, соответствующее 1 мл 0,1 N Ва (ОН)2.
Рис. 1. Схема установки для определения амина, связанного с углекислотой:
1 — колба; 2 —капельная воронка; 3 — змеевиковый абсорбер.
ОПЫТНАЯ ЧАСТЬ
Очистке подвергался газ следующего среднего состава (в объемных %):
СО2..................13,7 Спнш................5,1
H,S....................0,5 н2 ..................28,4
СО...................11,7 СН4..................19,8
О2 ....................1,0 N, ..................20,5
Концентрация сероводорода иногда повышалась до 0,7%, а содержание кислорода колебалось от 0,7 до 1,1%.
В качестве рабочего поглотительного раствора применялся 25%-ный водный раствор технического триэтаноламина, содер
Очистка горючих газов от сероводорода
211
жащего (в весовых %): триэтаноламина 82,0, диэтаноламина 14,6 и неперегоняющегося остатка 3,5.
Технологическая схема установки изображена на рис. 2.
Газ после эксгаустерного отделения, пройдя аммиачный скруббер, в котором он охлаждался водой до 25—40° С, и масляный скруббер, в котором улавливалась часть газового бензина, поступал в абсорбер 1. Последний представлял собой железный сварной цилиндр диаметром 3,5 м и высотой около 36 м, заполненный в нижней части хордовой деревянной насадкой и оборудованный в верхней части пятью колпачковыми тарелками. Подача раствора могла быть осуществлена либо на хордовую насадку, либо на тарелки. В наших опытах раствор подавался на тарелки. Над тарелками помещалась отбойная насадка из керамиковых колец 50X50 мм- Газ, частично освобожденный в абсорбере от H2S, направлялся далее на окончательную доочистку в башни, заполненные болотной рудой.
Регенерированный раствор подавался в противоток газу. Раствор из абсорбера, насыщенный сероводородом и углекислотой, забирался насосом 3 и подавался в жидкостные теплообменники 4, в которых, за счет тепла регенерированного раствора из десорбционной колонны, нагревался до 60—80° С. Далее он дополнительно подогревался до 90—95° С в паровых подогревателях 5 и поступал в десорбционную колонну 6 (диаметр 2,5 м и высота 22 >и), оборудованную 32 колпачковыми тарелками. Раствор подавался на 19-ю тарелку. Из десорбционной колонны раствор стекал в кипятильник 7, где он, с целью окончательной регенерации, доводился до температуры кипения (102—103°) глухим паром.
Образующиеся в кипятильнике пары поступали в нижнюю часть десорбционной колонны.
Регенерированный в кипятильнике раствор забирался насосом 8 и направлялся, в противоток насыщенному раствору, через теплообменники 4 и холодильники 9 на абсорбционную колонну 1. Температура поглотительного раствора колебалась в пределах 35—45° С.
Выхлопные газы из колонны, вместе с парами воды, этанол-амина и газового бензина, который конденсировался в системе, вследствие недостаточной очистки в масляном скруббере, поступали на охлаждение в конденсаторы 10 и затем на разделение в сепаратор 11. Сероводород и углекислота выбрасывались в атмосферу, водный слой возвращался наверх десорбционной колонны насосом 12, а газовый бензин собирался в специальный резервуар, не показанный на рис. 2.
Влияние интенсивности орошения на степень очистки газа от H2S. Была проведена серия опытов.
Рис. 2. Схема очистки газа раствором триэтаноламина:
1 — абсорбер; 2 —скруббер; 3 —насос; 4—жидкостные тешообменпики; 5—паровые подогреватели; 6 — десорбционная колонна; /—кипятильник; S— насос; 9 — холодильники; 70 — конденсатор; 11 — сепаратор; 12— насос.
Очистка горючих газов от сероводорода
213
с различной интенсивностью орошения от 1,5 л/м3 д,о 6,4 л/м3 газа. Данные этих опытов приведены в табл. 1.
Таблица 1
Очистка газа от H2S при различной интенсивности орошения (Производительность установки 10 000 м3!час)
Концентрация триэтаноламина в растворе, вес. % Температура раствора, °C Температура газа, °C Интенсивность орошения, л /м3 H2S в газе до очистки, г/м3 H2S в газе после очистки, г/м3 Степень очистки от H2S, г/м3
16,0 34 27 .,5 9,2 7,54 29,0
21,0 39 28 2,0 8,0 5,4 32,3
23,0 39 29 2,4 7.45 4,90 41,0
23,0 37 28 2,6 7,0 4,0 43,0
20,0 37 22 3,0 10,4 5,75 47,0
20,8 42 23 4,0 9,8 4,75 52,7
15,7 . 30 27 4,8 11,75 2,98 74,7
15,0 30 27 6,4 8,28 1,78 78,5
Дак видно из этой таблицы, при орошении 1,5 л/.w3 газ очищается только на 29 %. С увеличением плотности орошения степень очистки повышается, достигая 78,5% при 6,4 л/м3.
Следует учесть, что к тому времени, когда начались опыты с более плотным орошением, концентрация свободных аминов в растворе снизилась, вследствие образования тиосульфата, с 23 до 15%. Это не могло не отразиться на полученных результатах, однако в производственных условиях мы не смогли сохранить исходную концентрацию свободных аминов в растворе.
О влиянии концентрации аминов в растворе на глубину очистки можно судить по опытам, показанным в табл. 2, в которых при одинаковом технологическом режиме изменялась только
Таблица 2
Влияние концентрации свободных аминов в растворе на очистку от H2S
Концентрация аминов в растворе, % Интенсивность орошения, л/м3 Степень очистки от H2S, %
11,8 2,4 32,8
23,0 2,4 41,0
11,9 4,17 38,4
20,8 4,0 52,7
A. E. Драбкин
концентрация раствора. Нетрудно видеть из этой таблицы, что увеличение концентрации активных аминов в растворе с 11—12 до 20—23% повышает степень очистки газа от сероводорода на 9—12%.
Таким образом, можно ожидать, что при концентрации триэтаноламина 25% степень очистки газа от H2S должна достигнуть значения порядка 90%.
Интенсивности орошения, равной 6 л раствора на 1 м? газа при диаметре абсорбера 3,5 м. и производительности 10 000 м-Дчас, соответствует плотность орошения
10000 6-4 с о „ . ,
'3 14 352 ~6,3 мЛ на 1 лг сечения в час.
Такая плотность является обычной для скрубберных процессов.
Интересно отметить, что при максимальной очистке от сероводорода улавливалось только 24% углекислоты от общего количества ее, содержащегося в газе. Из сравнения степени очистки газа от H2S и СО2 можно видеть, что поглотительная способность раствора по отношению к H2S почти в четыре раза больше, чем по отношению к СО2.
Влияние кислорода, содержащегося в газе. Кислород, как это имеет место в содовом или мышьяково-содовом способах очистки газа, энергично воздействует на процесс в направлении образования тиосульфата:
2 (Rs) N + 4H2S + 502 = 2 (R3N) S2O3 + 4Н2О.
В систему очистки было загружено к началу опытов около 60 т 25 %-кого водного раствора триэтаноламина. Данные о накоплении тиосульфата триэтаноламина в растворе собраны в табл. 3 и графически показаны на рис. 3.
Таблица 3
Накопление тиосульфата аминов в растворе
Связалось аминов с
Время S2O3" тиосульфатом в % от
в сутках г!л раствора исходного количества
I аминов
4 8,0
8 13,55 14,4
12 21,30 21,2
16 21,9 21,8
20 24,8 26,4
24 28,4 30,6
28 29,1 31,0
32 32,2 34,8
36 34.8 37,0
40 35,5 37,7
Очистка горючих газов от сероводорода
215
Как видно из табл. 3, за 40 суток эксплуатации установки в связанное состояние перешло около 40% аминов, что составляет в среднем около 150 кг триэтаноламина в сутки или
Рис. 3. Изменение концентрации свободных и связанных аминов в растворе, в зависимости от продолжительности процесса очистки газа.
с образованием тиосульфата, по балансовым испытаниям составили всего 0,03 кг/1000 нмэ газа.
Таким образом, очистка газа от сероводорода сопровождается таким сильным образованием тиосульфата триэтаноламина, что промышленная реализация процесса без регенерации амина из его тиосульфата становится нецелесообразной.
ВЫВОДЫ
1. При промышленной очистке сланцевого газа, содержащего до 1% кислорода, 25%-ным водным раствором триэтаноламина степень очистки его от сероводорода достигает 78,5% и от углекислоты — 24 %.
2. В процессе очистки газа, содержащего кислород, происходит интенсивное образование тиосульфата амина, снижающего активность поглотительного раствора.
3. Промышленная реализация процесса без регенерации амина из его тиосульфата является нецелесообразной.
Д. К- Коллеров
О ЗАКОНЕ ВЯЗКОСТИ БАЧИНСКОГО
В 1912 г. А. И. Бачинским [1] было опубликовано первое сообщение об открытом им законе вязкости, выраженном уравнением
^-v~’ CD
где: pt—абсолютная вязкость в пуазах при температуре t\
С —константа, характерная для данного вещества;
Vt— удельный объем вещества при температуре t;
а>— некоторая постоянная, близкая к величине b уравнения Ван-дер-Ваальса.
На специальном заседании Московского отделения Всесоюзного химического общества им. Д. И. Менделеева (1938 г.) [2], посвященном 25-летию закона Бачинского, А. И. Бачинским был прочитан доклад, в котором он изложил основную сущность и значение найденной им формулы вязкости жидкостей.
Из выражения (1) видно, что в условиях применимости формулы (1) объем Vt жидкости не может быть меньше величины а>. Эту величину А. И. Бачинский и назвал «предельным объемом», а разность V — со, которая в процессах изменения состояния жидкости может принимать разнообразные значения, — «свободным объемом».
Предельный объем со представляет собой такое состояние жидкости, при котором значения термодинамических параметров, а следовательно, и удельного объема должны составлять некоторую определенную долю параметров в критической точке. Действительно, подсчет показывает, что величина со равна 4/1з критического объема. Таким образом на основании измерений вязкости формула (1) позволяет подсчитать величины критических объемов жидкости, важные для целого ряда технологических и термодинамических расчетов и часто трудно поддающиеся определению опытным путем.
В приложении к капельным жидкостям формула вязкости А. И. Бачинского является наиболее простой из всех существую-
О законе вязкости Бачинского
217
щих формул вязкости и правильно отражает связь между вязкостью и основными физико-химическими свойствами жидкости. Несмотря на эти очевидные преимущества формулы вязкости А. И. Бачинского перед всеми другими, предложенными до сих пор теоретическими и опытными уравнениями вязкости смесей или температурных зависимостей вязкости, она до сих пор не получила распространения в практике технологических расчетов. Дело в том, что выполнение расчетов по формуле Бачинского предполагает знание не только величин вязкостей при нескольких температурах, но и удельных весов при этих температурах для нахождения модуля вязкости С и предельного объема. Сочетание параллельных измерений этих двух величин в обычной практике почти никогда не встречается.
С другой стороны, определение вязкости смеси или зависимости вязкости от температуры, например, по уравнениям Вальтера, предусматривает знание только одной вязкости составных частей смеси или вязкости жидкости при двух температурах, а эти данные встречаются почти всегда. По этой причине в практике пока предпочитают пользоваться диаграммами Вальтера, хотя отсчеты по ним недостаточно точны и хотя эти диаграммы не дают возможности надежной экстраполяции вне пределов измеренных величин вязкости.
Положение могло существенно измениться, если бы были найдены зависимости между основными параметрами жидкости, например, плотностью и температурой кипения и модулем вязкости, предельным объемом и температурными коэффициентами плотности. При наличии этих зависимостей определение вязкости жидкостей по формуле Бачинского не представляет никакого труда и, конечно, имеет все преимущества перед существующими в настоящее время способами расчета. Рассмотрим некоторые обнаруженные нами новые зависимости.
УДЕЛЬНЫЙ ОБЪЕМ ЖИДКОСТИ
В недавно опубликованной работе автора [3] для капельных жидкостей, плотность которых подчиняется закону dt = d0 (1—at), была установлена зависимость температурного коэффициента
( у тк \
плотности a i(d)F\—tf— !, согласно которой величину а можно определить по значению плотности d?° и температуре кипения. Для всяких сложных смесей и технических продуктов, а также широких фракций за температуру кипения берется средняя объемная температура кипения. Вычисление dt по найденному значению а по уравнению dt = d0(l — at) дает среднюю точность 0,2% при изменении температуры на 100°. Удельный
218 Д. /С Коллеров
объем жидкости Vt при температуре t можно вычислить по формуле
V ,=____1_____
at)
В случае применения установленной зависимости
a — f(d)F ,
для вычисления потребуется знание только температуры кипения и стандартной величины плотности.
ПРЕДЕЛЬНЫЙ ОБЪЕМ
Предельный объем жидкости несомненно связан с ее химической природой. Возникает вопрос, возможно ли эту связь выразить в каких-либо соотношениях. Теоретические предпосылки позволяют искать решение вопроса разными способами.
Если рассматривать только капельные жидкости (неассоциированные) , то их химический характер находит объяснение в величине молекул, атомов и групп, в структуре молекулы и внутриатомных связях. Таким образом, химический характер жидкости может выражать группа таких свойств, как силы сцепления молекул, молекулярный объем или плотность, модуль расширения и др. В этом случае и следует искать взаимосвязи между величиной oj и этой группой свойств жидкости.
Если взять для характеристики какие-либо подобные системы, то должна обнаружиться связь между величиной со и тем критерием, который характеризует эти подобные системы. За
г 1/F-
такой критерий нами принят показатель К — ---- «приведенных»
Ф°
состояний и, следовательно, должна быть обнаружена зависимость w = jf(K). Зависимость этого вида является наиболее простой. Она интересна тем, что величину со определяют здесь наиболее простые и общедоступные данные: плотность и температура кипения. В табл. 1—4 помещена сводка величин т. взятых по вычислениям А. И. Бачинского для разных жидкостей и по данным наших работ для некоторых индивидуальных продуктов и разных технических фракций (смесей).
Вычисления производились по данным измерения вязкости при двух температурах и плотности при этих же температурах.
В табл. 4 сведены результаты определений величин предельных объемов жидкостей по данным работы А. И. Бачинского
О законе вязкости Бачинского
219
Таблица 1
Опытные данные по вязкостям и плотностям фракций сернистой смолы кашпирских сланцев
Фракция ГС П ’Zt CD C 10~6
115-125° 15 0,8614 1,162 0,00722 1 AAQ 71
/ср=П6 50 0,8301 1,200 0,00503 1 .UDO / ID
125—150° 15 0,8772 1,140 0,00975 1 ЛА2
'ср = 139 50 0,8474 1,181 0,00605 O/U
150—170° 15 0,8883 1,126 0,01253 1 Л77 Л1 1
/ср=165 50 0.8596 1.164 0,00707 1 ,U/ / Oil
175-205° 15 0,8969 1,115 0,01498 77П
/ср = 189 50 0,8689 1,153 0,00893 1 ,vOO i tv)
200—225° 15 0,9083 1,102 0,02968 1 Л7/1 7/1П
'ср =211 50 0,8824 1.134 0,01287 1 /4У
Таблица 2
Опытные данные по величинам плотности и вязкости фракций смолы полукоксования прибалтийских сланцев
Фракция t° C dt ’It “cp C • 10~6
до 150° 20 0,8204 1,219 0,010011 —
t —120 40 0,8049 1,244 0,007624 1,144 1,144 753
60 0,7896 1,266 0,006092 1,144
150—175° 20 0,8375 1,195 0,011300 -
t —163 40 0.8220 1,216 0,008557 1.134 1,128 705
70 0,7987 1,253 0,005973 1.122
175—200° 20 0,8459 1,182 0,013013 —
/ —188 40 0,8303 1,205 0,009461 1,119 1,120 784
70 0,8074 1,240 0,006517 1,121
200—225° 20 0,8982 1,166 0,015960 —
z =213 40 0,8430 1,186 0,011360 1,115 1,118 775
70 0,8212 1,218 0,007554 1,119
225—250° 20 0,8770 1,140 0,02263 —
t. — 230 40 0,8620 1,160 0,01506 1,103 1,107 870
70 0,8403 1,191 0,00943 1,11
250—275° lCp =263 20 0,8977 1,115 0,3799
40 70 0,8830 0,8620 1,133 1,160 0,02303 0,01320 1,087 1,092 1,09 898
220
Д. К. Коллеров
Таблица 3
Величины со и С продуктов перегонки сернистой смолы _________________кашпирских сланцев_________
Zk. ср. К ГС dt vt СО "ср с io—«
по 8,52 20 75 0,8524 0,8007 0,006843 0,004081 1,075 1,075 713
135 8,34 20 50 75 0,8846 0,8622 0,8373 0,008382 0,006286 0,004937 1,052 1,050 1,051 697
165 8,50 20 75 0,8942 0,8986 0,01149 0,00544 1,060 1,060 527
177 8,48 20 75 0,9037 0,9581 0,01191 0,007135 1,045 1,045 820
191 8,45 20 50 75 0,9158 0,8922 0,8708 0,01893 0,01207 0,008448 1,048 1,050 1,049 815
203 8,42 20 50 75 0,9276 0,9052 0,8844 0,02128 0,01287 0,009332 1,042 1,042 1,042 816
217 8,37 20 50 75 0,9423 0,9205 0,9001 0,02957 0,01652 0,01113 1,033 1,035 1,034 796
218 8,47 50 75 0,9396 0,8961 0,2928 0,01157 1,045 1,045 583
203 8,45 20 75 0,9181 0,8785 0,02363 0,009854 1,062 1,062
231 8,26 20 75 0,9635 0,9117 0,3734 0,01299 1,046 1,046 662
Таблица 4
_ со _
Величины со и уравнения Бачинского для разных веществ
1№ п/п| Вещество Формула 0 <Г° 4 К СО СО ~К Отклонение от среднего значения, %.
1 2-Метилбутан . . С5Н12 27,8 0.6195 10,82 1,3390 0,1238 0,72
2 н-Пентан . . . . С5н12 36,1 0,6263 10,8 1,3331 0,1235 0,96
3 Гексан с6н14 68,7 0,6595 10,6 1,3143 0,1239 0,64
4 Изогексан . . . с6н14 60,3 0,6581 10,54 1,3057 0,1239 0,64
5 Изогептан . . . С,н16 79,3 0,6797 10,5 1,3031 0,1240 0,56
6 н-Гептан .... С7Н16 98,4 0,6835 10,52 1,3028 0,1239 0,64
7 н-Октан .... С8н18 125,6 0,7022 10,48 1,3024 0,1243 0,32
8 н-Декан .... С1он22 174 0,747 10,47 1,323 0,1265 1.45
О законе вязкости Бачинского
221
Продолжение табл. 4
Вещество Формула 'к К О) ТС Отклонение от среднего значения, %
9 Диаллил .... C6Hi0 60 0,6887 10,06 1,2574 0,1248 0,08
10 Триметилэтилен С5н1о 38,4 0,6646 10,20 1,2369| 0,1236 0,88
11 Изопрен .... с5н8 32,6 0,6710 10,04 1,232210,1210 2,97
12 Этилизобутиловый
эфир С6Н14О 80 0,7346 9,62 1,2016 0,1245 0,16
13 Дипропиловый
эфир С6Н14О 89,5 0,7440 9,58 1,1993 0,1251 0,32
14 Этилпропиловый
эфир ..... С5Н12О 61,7 0,7343 9,46 1,1803 0,1246 0,08
15 Метилизобутило-
вый эфир . . . 0.7301 1,1793
16 Метилпропиловый
эфир C6Hi0O 39,1 0,7255 9,45 1,1624 0,1231 1,29
17 Этиловый эфир . . С4НХ0О 34,6 0,7135 9,46 1,1585 0,1225 1,77
18 Метилпроп илкетон С5н10о 103,3 0,8068 8,95 1,1175 0,1248 0,08
19 Диэтилкетон . . . с6н10о 102.7 0,8140 8,86 1,0983 0,1239 0,64
20 Метилэтилкетон . с4н8о 79,6 0,8094 8,73 1,0952 0,1253 0,48
21 Параксилол . . . С 8^10 138,3 0,8629 8,62 1,0744 0,1245 0,16
22 Ортоксилол . . . CgHjo 144,4 0,8766 8,52 1,0699 0,1257 0,81
23 Ацетон С3н6о 56,1 0,7961 8,67 1,0698 0,1234 1,04
24 Этилбензол . . . С8Н10 136,2 0,8673 8,56 1,0675 0,1247 0,0
25 Метаксилол . . . CgH10 139,1 0,8643 8,61 1,0655 0,1238 0,72
26 Толуол С7Н8 110,6 0,8656 8,39 1,0547 0,1256 0,72
27 Бензол с6н6 80,1 0,8790 8,04 1,0476 0,1300 4,27
28 Диизопропил . . С6Н14 58,0 0,6617 10,45 1,340 0,1282 2,81
29 а-Пинен 155 0,8628 8,73 1,1078 0,1268 1,68
30 Кумол ..... С6Н12 152.4 0,8723 8,62 1,0878 0,1262 1,2
31 Фракция смолы 120 0,8204 8,92 1,144 0,1282 2,81
32 полукоксова- — 163 0,8375 9,05 1,128 0,1247 0,0
33 НИЯ эстонских — 188 0,8459 9,12 1,120 0,1230 1,37
34 сланцев (гра- — 213 0,8582 9,16 1,118 0,1222 2,01
35 ницы кипения — 238 0,8770 9,1 1,107 0,1219 2,25
36 фракций 25°) — 263 0,8977 9,05 1,090 0,1205 3,37
37 Фракции смолы ( — — 7,6 0,953 0,1254 0,56
38 коксования •! — 116 0,8614 8,47 1,063 0,1253 0,48
39 углей 1 139 0,8772 8,5 1,063 0,1250 0,24
40 — 165 0,8883 8,55 1,077 0,1257 0,8
41 189 0,8969 8,73 1,065 0,1220 2,17
42 211 0,9083 8,67 1.074 0,1238 0,72
43 ПО 0,8524 8,52 1,075 0,1262 1,2
44 135 0,8846 8,39 1,051 0,1253 0,48
45 Фракции серии- 165 0,8992 8,50 1,060 0,1248 0,08
46 стой смолы 177 0,9040 8,48 1,045 0,1234 0,97
47 кашпирских 191 0,9158 8,45 1,049 0,1241 0,48
48 сланцев 203 0,9276 8,42 1,042 0,1237 0,8
49 217 0,9423 8,37 1,035 0,1237 0,8
50 218 0,9396 8,47 1,045 0,1235 0,96
51 203 0,9181 8,45 1,062 0,1256 0,72
52 — 230 0,9635 8,26 1,046 0,1267 1,6
Среднее . . 0,1247| 1,04
О законе вязкости Бачинского
223
и нашим исследованиям. Оказалось, что между величинами 3 Г---
/(= и w существует достаточно хорошо выраженная прямо-
линейная зависимость (рис. 1). Оказалось далее, что величина = 0,1247 4- является постоянной величиной. По взятым
К о
„СО
50 жидкостям среднее отклонение от средней величины составляет 1 %.
ПОСТОЯННАЯ С (МОДУЛЬ ВЯЗКОСТИ)
Природа постоянной С была вскрыта А. И. Бачинским на основании теоремы Камерлинг-Оннеса из теории подобия молекулярных систем. Согласно этой теореме в соответственных состояниях для различных жидкостей имеет одну и ту же величину выражение:
иТ,. 1/в
.. пр-,-,— = Const.
„х/вр
Так как п = ~гг-- и в соответственных состояниях V и
1 V —оу
(V—ю) для разных жидкостей составляют одну и ту же долю критического объема, то для всех жидкостей, следующих закону вязкости Бачинского, должна быть постоянной величина
СП<р1/6
== Const.
Так как для всех капельных жидкостей Z^Kp^Kp 1 —- = Const, •* кр т т
то, заменяя Укр через —~ или Ркр через -тД- , получаем Z^Kp Z4*' Кр
всеобщие постоянные:
С/?/2рД/з 5/Р— = Constx, 'Г 5/б if 1 кр Сд1/в Т х/2у х/з — Const2- 1 кр кр
Принимая во внимание, что величина со пропорциональна критическому объему Укр, получаем третью всеобщую по-
стоянную
с//б д>1/3
Constj.
Д. к. Коллеров
По вычислениям А. И. Бачинского:
Constj = 0,000907 Const., =0,0000358 Const" = 0,0000531
А. И. Бачинский замечает: «Постоянство каждой из рассмотренных величин является не особенно точным. Первая дает отступления в размере до 9% от средней арифметической, вторая до 7% с небольшим; самое лучшее согласие обнаруживают между собой значения Const3, где наибольшее отступление составляет 6,2% от арифметической средней. Присутствие таких отступлений является вполне понятным и законным потому, что в основе теоремы Оннеса лежит то заведомо неточное допущение, что молекулы различных веществ имеют одинаковую форму и что центры сил, которыми они действуют на расстоянии, лежат в геометрической точке их».
По поводу связи между объемом молекулы и величинами со и С А. И. Бачинский пишет, что если связь между V и со несомненна и внешне определенна, то связь между со и С, хотя и не является ясно выраженной, но все же существует. В общем по мере возрастания V и со величина С также возрастает. По нашим данным это заключение не совсем верно. Увеличение V и со достаточно ясно соответствует одновременному увеличению С лишь в том случае, когда рассматриваются жидкости сходного химического характера, например ряд парафинов. Отметив это обстоятельство, мы сделали попытку найти зависимость вида C = f(d)F(K) или С ~ f(tK}F(K). Оказалось, что такие зависимости действительно существуют, но соблюдаются также недостаточно точно и поэтому не могут быть рекомендованы для практического использования. По крайней мере, так они представляются на данной стадии разработки этого вопроса.
Если заменить вязкость обратной ей величиной — текучестью, — то уравнение Бачинского может быть написано в виде
?уС = Vt— со.
(2)
Для двух температур имеем:
<р±С = Vt—со
cpfi = V2—ш или
£1 _ У] —о>
<f>2 ^2-Ш ’
откуда
VzVl-CPiVz •
О законе вязкости Бачинского
225
Это выражение представляет собой уравнение прямой линии: (3
<Р* = 9’0-77
где текучесть жидкости у0 отнесена к тому случаю, когда удельный объем жидкости равен ее удвоенному предельному объему.
Сравнивая между собой уравнение (2) и уравнение (3), находим
<Рй = ~с
Следует обратить внимание на одну особенность величины у0.
0,980
0,960
0,940
0,920
0,900
0,880
\*0,860
3 0,840
0&20
0,800
0,780
0.760
10 11 12 18 14 75 76 7/ 18 Ю 20 21 22 %
Рис. 2. Зависимость между и tpQ.
Рассматривая вязкость или текучесть жидкости при одних и тех же давлениях и температурах, мы, хотя и относим данные к одним и тем же условиям, но фактически имеем разное состояние жидкостей, так как при этом отношение — для каждой данной жидкости имеет свое значение. Величина «0 является текучестью жидкости при одном и том же состоянии, т. е. когда удельный объем жидкости равен ее удвоенному предельному объему. С точки зрения природы явления вязкости здесь имеем соответственное состояние жидкостей. Поэтому можно думать, 15 Труды вниипс.
226
Д. К. Коллеров
что между величиной %0 и другими свойствами жидкостей могут быть найдены более точные и определенные зависимости.
Так как величина <р0 не зависит от температуры и давления, то, естественно, предположить связь %0 с величиной относительного предельного объема жидкости, т. е. с величиной -у- = wd. Если для характеристики относительного предельного объема принять плотность при с/40, то в первом приближении должна существовать зависимость <р0 = /(с/1°<«).Величина у>0, невидимому,
Таблица 5
Значение величин у0 и для ряда исследованных жидкостей
Вещество rf20 4 СО d20co 4 То
2-Метилбутан 0,6195 1,3390 0,829 2180
н-Пентан 0,6263 1,3331 0,834 2175
Гексан 0,6595 1,3143 0,865 2030
Изогексан . 0,6581 1,3057 0,86 2040
Изогептан 0,6796 1,3031 0,884 2040
н-Гептан 0,6835 1,3028 0,889 1992
н-Октан 0,7022 1,3024 0,914 1957
Триметилэтилен 0,6646 1,2369 0,823 2190
Изопрен 0,6710 1,2322 0,825 2215
Этилизобутиловый эфир ...... 0,7346 1,2016 0,882 1998
Дипропиловый эфир 0,7440 1,1993 0,890 1970
Этилпропиловый эфир ....... 0,7343 1,1803 0,865 2045
Метилизобутиловый эфир 1,7301 1,1793 0,863 2025
Метилпропиловый эфир 0,7255 1,1624 0,843 2140
Метиловый эфир 0,7135 1,1585 0,827 2229
Метилпропилкетон 0,8068 1,1175 0,9 1825
Этилкетон 0,8140 1,0983 0,895 1810
Метилэтилкетон 0,8094 1,0952 0,887 1890
Параксилол 0,8629 1,0744 0,927 1950
Ортоксилол 0,8766 1,0699 0,937 1840
Этилбензол 0,8673 1,0675 0,925 1860
Метаксилол 0,8643 1,0655 0,920 1897
Толуол - 0,8656 1,0547 0,910 1867
Бензол 0,8790 1,0476 0,920 1806
Диизопропил 0,6617 1,340 0,885 2040
а-Пинеп 0,8628 1,1078 0,954 1472
Кумол 0,8723 1,0878 0,950 1810
0,8204 1,144 0,940 1520
0,8375 1,128 0,944 1600
Фракции смолы полукоксования при- 0,8459 1,120 0,947 1430
балтийских сланцев . 0,8582 1,118 0,958 1445
0,8770 1,107 0,970 1370
0,8977 1,092 0,980 1218
Фракции ретортной и генераторной
смолы сернистых кашпирских сланцев • , . . ( 0,9423 1 0,9276 1,034 1,042 0,980 0,966 1310 1286
1 0,8772 1,063 0,932 1585
О законе вязкости Бачинского
227
также должна зависеть от конфигурации молекул, их взаимного притяжения и, возможно, их массы. Однако, если ограничиться рассмотрением каких-либо «подобных» систем, например, углеводородов или других соединений, включающих в молекулу атомы с близким углероду атомным весом (например, азот, кислород), то зависимость = f (dm) должна достаточно ясно обнаружиться. В табл. 5 дана сводка соответствующих величин для рассмотренных жидкостей, а на рис. 2 — зависимость <р0 = —/(^4°со)- Она действительно обнаруживается.
ЛИТЕРАТУРА
1. А. И. Бачинский. «Phys. Zeitsahr.» 24, 1157, 1912. «Временник» Общества содействия успехам опытных наук н нх практическому применению. Приложение № 3, 21, 1913.
2. Сборник «Современные проблемы физико-химии и химической технологии», II, стр. 113, 1938. Доклад А. И. Бачинского.
3. Д. К. Коллеров. Физико-химические свойства сланцевых и каменноугольных смол. Гостоптехиздат, 1951.
15*
Д. К. Коллеров
К ВОПРОСУ О ФИЗИЧЕСКОЙ СУЩНОСТИ ПОКАЗАТЕЛЯ
к = УГЛЕВОДОРОДНЫХ жидкостей
Недавно автором была опубликована работа [1], в которой все основные физико-химические свойства углеводородных жидкостей были определены ка'к функции температур кипения, плотности и некоторого показателя
К
3,_
утк
Эти зависимости были найдены как вытекающие из уравнения Бачинского a=CB(d — zl)4 и уравнения Этвеша
(ПР-П
а также на основании анализа зависимостей d = / (Тк) и д=/(Тк) для парафинов, непредельных углеводородов, нафтенов, ароматических углеводородов, кетонов, эфиров, альдегидов.
Выражение физико-химических свойств жидкостей в зависимости от показателя К было основано на простых предпосылках, однако математические соотношения, которые можно было вывести из этих предпосылок, были или недостаточно точны, или имели общий вид.
Решение этих зависимостей было сделано на основе большого количества опытных данных графическим путем, а также дано в виде соответствующих расчетных диаграмм.
Построенные расчетные диаграммы были проверены более чем на 2500 данных по различным свойствам различных жидкостей.
В настоящей статье автор вкратце излагает результаты даль-НсИШёГО ИССЛсДОВаНИЯ ЭТОГО ВОПрОСЯ.
К вопросу о физической сущности показателя К
229
Выбранный для целей обобщения физико-химических свойств жидкостей показатель
в данной формулировке не имеет физического смысла. Однако то обстоятельство, что при помощи этого показателя ТС все основные физико-химические свойства жидкостей удалось связать в стройную систему, позволяет предполагать наличие у показателя К определенного физического смысла. Вскрыть этот физический смысл оказалось возможным при анализе установленных зависимостей и диаграмм.
Для этой цели за исходное положение были взяты жидкости с температурами кипения в промежутке от 50 до 300° С и с показателем К =10,5; А =10; К = 9; К = 8; К = 7.
Для выбранных температур кипения и величин показателя А по диаграмме плотностей были отсчитаны плотности d%Q, а по диаграмме молекулярных весов — молекулярные веса. Для « и j20
найденных таким путем плотностей ад , по диаграмме темпера-турных коэффициентов плотности а были определены их величины и по уравнению плотностей
dt = d0(l — at)
по найденным величинам а рассчитаны плотность при температуре кипения и величина
/ К \®/в yW; •
(о
Результаты проведенных расчетов помещены в табл. 1. Они с очевидностью показали тот неожиданный факт, что жидкости с показателем
„ г .
К =------= Const
а™
являются такими жидкостями, которые имеют одинаковую плотность при температуре кипения.
Таким образом показатель А является критерием, характеризующим жидкости с одинаковыми плотностями при температуре кипения.
Анализ данных табл. 1 показывает, что между показателем К и плотностью при температуре кипения существует простая зависимость, показанная на рис. 1.
230
Д. К. Коллеров
Таблица 7
Исследование диаграмм плотности, молекулярных весов, модулей расширения и уравнения (1)
а™ по диаграмме al О- 6 по диаграмме /г по диаграмме Т-ра кипения ^КИП. \2Cj
°C °К
Жидкости с К=10,5
0,650 1400 83 50 323 0,620 0,00415 1836
0,685 1190 105 100 373 0,620 0,00421 2280
0,716 1050 133 150 423 0,619 0,00437 2755
0,742 950 164 200 473 0,616 0,00445 3255
0,767 870 198 250 523 0,615 0,00450 3790
Жидкости с К=10
0,713 1190 100 94 367 0,650 0,00540 2225
0,750 1030 130 150 423 j 0,650 0,00555 2755
0,780 930 160 200 473 1 0,650 0,00565 3255
0,812 830 200 256 529 j 0,650 i 0,00545 3850
Жидкости с К = 9
0,798 1065 95 100 373 | 0,730 0,00862 2280
0,834 955 118 150 423 0,730 0,00886 2755
0,876 850 145 200 473 0,730 0,00920 3255
0,894 810 175 250 523 I 0,728 i 0,00962 3790
Жидкости с К = 8
0,900 990 86 100 373 0,830 0,0142 2280
0,940 895 106 150 423 0,831 0,0152 2755
0,975 820 130 200 473 0.830 0,0157 3255
1,005 760 154 250 523 0,829 0,0160 3790
Жидкости с К = 7
1,115 770 120 200 473 0,960 0,0301 3255
1,152 725 140 250 523 0,960 0,0301 3790
1,190 660 162 300 573 0,965 0,0310 4360
К вопросу о физической сущности показателя К
231
Оказалось, далее, что показатель К является очень хорошо выраженной линейной функцией обратной величины плотности или удельного объема при температуре кипения
K = a+bVK. (2)
Таким образом линии постоянных величин /С на построенных диаграммах физико-химических свойств жидкостей являются по
Рис. 1. Зависимость между показателем К и удельным объемом жидкостей при температурах кипения.
существу линиями постоянных значений плотностей или удельных объемов жидкостей при их температурах кипения. Линия К = 10,5 соответствует плотности 0,619; линия К — 10 — плотности 0,650; линия К = 1 — плотности 0,960. При составлении диаграммы зависимостей свойств жидкостей, следовательно, можно было бы целиком отказаться от применения критерия Л', заменив его вполне определенной физической величиной — плотностью при температуре кипения, однако этой величине, как физической константе, до сих пор не придается никакого значения. Обычно она не приводится в справочниках и в сущности выпала из науки о свойствах жидкостей.
Принимая за основу стандартные плотности dl° при попытках вывести обобщающие зависимости, мы впадаем в ту же ошибку как и при попытках вывести закономерности при сравнении вязкости разных жидкостей при одной температуре, из-за сравнения жидкостей в несравнимых состояниях. Только вводя
232
Д. К. Коллеров
во взаимосвязи плотности жидкостей при их температурах кипения удалось установить стройную систему этих взаимосвязей. Таким образом плотность при температуре кипения оказалась очень важной константой жидкости.
Установление критерия К позволяет нам воспользоваться этими важными константами жидкостей на основе стандартных аналитических данных: температуры кипения и плотности d't". Так как плотность при температуре кипения не является величиной, характерной для данной жидкости и так как целые группы жидкостей, сходных по своему химическому характеру, могут иметь одни и те же плотности при температуре кипения, то, следовательно, в этой величине должны находить отражение не частные, а общие свойства жидкостей.
Сделанный на основании анализа найденных зависимостей вывод о том, что все жидкости с К = Const имеют dK = Const, представляло большой интерес проверить на фактическом материале. В нашем распоряжении не имелось непосредственных измерений плотности при температурах кипения. Приняв за основу уравнение плотностей di = d, (1—at), мы рассчитали величину dк по формуле
1 —at
(j —______!S. //so
к 1 — a 20 4 ’
пользуясь фактически измеренными значениями а.
Из имеющихся справочников нами были выбраны все углеводороды с Const si0,5, для которых и были сделаны упомянутые расчеты.
Результаты их сведены в табл. 2.
Из приведенных в табл. 2 данных видно, что, действительно, жидкости с К = Const имеют Const. По взятым 10 углеводородам с температурами кипения от 69 до 252° это постоянство
Таблица 2
Плотность при температурах кипения жидкостей, имеющих /С Const s 10,5 по фактическим данным
Химическая формула Наименование К tK а 10-6 1—а/к d20 4 Отклонение от среднего значения, %
1—-а20
С6Н14 н-Гексан . . . 10,58 69 1255 0,937 0,660 0,618 0.49
С7н1в 2, 2-Диметил-пентан , . . 10,48 79,2 1268 0,922 0,6736 0,623 1,3
С7Н16 2, 4-Диметил-пентан . . . 10,5 80 1284 0,921 0,6727 0,620 0,81
С7Н16 2-Метилгексан 10,5 90,0 1232 0,911 0,6786 0,617 0,33
К вопросу о физической сущности показателя К
233
Продолжение табл. 2
Химическая формула Наименование К 'к а 10—&> !-Щк J20 4 dK Отклонение от среднего значения, %
1—«20
с,н16 З-Метилгексан 10,4 91,8 1228 0,910 0,6871 0.623 1,3
С7Н16 С9Н20 н-Гептан . . 3, 3, 4, 4-Тетра- 10,5 98,4 1216 0,903 0,6838 0,618 0,49
метилгексан 10,46 150 1106 0,853 0,7176 0,612 0,49
С12Н2в н-Ундекан . . 10,48 197 985 0,822 0,741 0,609 0,98
С1зН28 н-Тридекан . . 10.52 234 913 0,802 0,757 0,607 1,3
С14Н30 н-Тетрадекан . 10,53 252 887 беднее 0,792 0,765 0,606 0,615 1,5 0,9
соблюдается со средними отклонениями от среднего значения в 0,9% при максимальном отклонении 1,5%.
Найденное среднее значение Ф- для взятых углеводородов с К = 10,5 равно 0,615. На основании сделанного анализа диаграмм физико-химических свойств жидкостей найдено для жидкостей с К = 10,5, dK = 0,619, т. е. почти та же самая величина.
Поскольку константа уравнения Бачинского
входит в формулу парахоров и поскольку парахоры обладают свойством сложи мости, а жидкости с К = Const являют собой химически подобные системы, при К— Const должно быть и СБ = Const.
В табл. 3 приведены значения молекулярных парахоров, плотностей, температур кипения, молекулярных весов и удельных р 1
парахоров Ру = —— = СБ/4. Все эти величины были получены для Л
некоторых жидкостей с К = 10,5; Л'=10; Д = 9 и Д = 8 следующим образом: по диаграмме парахоров брались плотности, по диаграмме плотностей — соответствующие этим плотностям температуры кипения, и по диаграмме молекулярных весов — соответствующие найденным температурам кипения молекулярные веса. На основе этих данных по формуле
рассчитывалась постоянная Бачинского Съ для жидкостей. с К = 10,5; Д = 10; Д = 9 и Д = 8.
234
Д. К. Коллеров
Таблица 3
Расчет константы Бачинского (СБ) по диаграммам парахоров, плотностей и молекулярных весов
/<=10,5 К=10
Пара- хоры Уд. вес d2O 4 Т-ры кипения по диаграмме [1] Молекулярный вес по диаграмме [1] Удельные парахоры Уд. вес rf20 4 Т-ры кипения по диаграмме [1] Молекулярный вес по диаграм-ме [1] Удельные парахоры
200 . . . . 0,663 25 70 2,86
250 0,653 55 84 2,98 0,694 65 86 2,91
300 0,677 87 100 3,00 0,720 100 104 2,88
350 0,698 120 116 2,92 0,741 140 124 2,83
400 0,715 150 133 3,01 0,761 170 143 2,86
450 0,730 175 149 3,02 0,778 198 159 2,83
500 0,745 208 168 2,98 0,792 222 177 2,82
Сред1 iee значение 2,98 2,86
удельных парахоров
К==9 К = 8
Пара- Уд. Т-РЫ ки-| удель- вес пения по л. „„ ные уд Т-ры ки- МолекГ v веС пения по ЛЯРНЫИ У*ель-
хоры d™ Диаграм-диаграм пара- 4 ме Ы ме [1] ор пнягпям вес П0 ные ме П| диаграм- парахоры L J ме {1]
200 0,760 52 75 2,67 0,886 81 78 2,56
250 0,796 92 92 2,72 0,926 135 100 2,50
300 0,825 136 111 2,70 0,960 180 120 2,50
350 0,850 175 132 2,65 0,990 221 140 2,50
400 0,872 203 147 2,72 1,015 264 163 2,45
450 0,890 241 170 2,65 1,040 305 185 2,44
500 0,910 270 195 2,63 1,060 335 205 2,44
Среднее значение
удельных парахоров 2,68 2,485
Как видно из приведенных в табл. 3 данных, постоянная Бачинского действительно выдерживается достаточно хорошо для всех жидкостей, имеющих постоянную величину К, или, иначе, имеющих равные плотности при температурах кипения. Этим подтверждается заложенное в основу составления зависимостей положение о том, что жидкости с равными значениями показателя К имеют сходный химический характер и что для всех этих жидкостей взаимосвязи физико-химических свойств
К вопросу о физической сущности показателя К
235
могут быть выражены определенными количественными соотношениями.
Таблица 4
Соотношение между СБ и плотностью при температуре кипения
К Плотность при температуре кипения ру сБ ig сБ 7К
10,5 0,616 2,98 79,0 1,897 1,624
10,0 0,650 2,28 66,7 1,824 1,539
9,0 0,730 2,68 51,5 1,712 1,370
8,0 0,830 2,485 38,0 1,580 1,205
Из приведенных в табл. 4 сводных данных и рис. 2 видно, что между величинами показателя К и логарифмом постоянной Бачинского также существует простая линейная зависимость
1g СБ = 0,565 + 0,127 К. (3)
Рис. 2. Зависимость между постоянной уравнения Бачинского СБ и удельным объемом жидкости при температуре кипения.
Из рис. 3 видно, что такая же зависимость имеется между удельным объемом жидкости при температуре кипения и логарифмом постоянной Бачинского
VK = 0,8+l,281gCB. (4)
* * *
При анализе физической сущности показателя К было установлено, что он является критерием, при помощи которого
236
Д. К. Коллеров
объединяются все жидкости, имеющие одинаковую плотность при температурах кипения. При = Const плотность при температурах кипения dK == Const.
Выведенная простая линейная зависимость между величинами К и удельным объемом жидкости при температурах кипе-
Рис. 3. Зависимость между показателем К. и постоянной уравнения Бачинского СБ.
ния относится к случаю нормального давления, равного 760 мм рт. ст.
Было сделано предположение, что зависимость вида tK=f(d)F(K)
должна сохраняться для любых давлений, что и было доказано построением диаграммы плотностей, .отнесенной к температурам кипения при 20 мм рт. ст. Так как показатель К выражает плотность при температуре кипения, то последняя должна сохраняться постоянной для любых давлений, меняясь лишь по своему абсолютному значению. В табл. 5 приведены результаты анализа диаграммы, составленной для температур кипения при 20 мм рт. ст., которые показывают, что это и есть в действительности.
* *
*
При выводе уравнения молекулярных весов лг» 1 ,5
к
/ Кэ
где„’ С = I j , был сделан ряд упрощений.
К вопросу о физической сущности показателя 1\
237
Таблица 5
Плотности жидкостей при температурах кипения при давлении
20 мм рт. ст.
d20 4 по диаграмме плотностей [1] а 10~6 по диаграмме коэффициента а [1] м по диаграмме молекулярных весов [1] Температура кипения по диаграмме при давлении 20 мм [1] ^КИП
°C °К
Жидкости с К = 10
0,713 1190 100 4 277 0,725
0,715 1030 130 54 327 0,723
0,780 930 160 95 368 0,724
0,812 830 200 152 425 0,725
Жидкости с К = 9
0,798 1064 95 6 283 0,809
0,834 955 118 51 324 0,810
0,876 850 145 106 379 0,813
0,894 810 175 134 407 0,811
Жидкости с
0,900 990 86 17 290 0,902
0,940 895 106 59 332 0,906
0,975 890 130 98 371 0,910
1,005 760 154 134 407 0,916
Из этого уравнения следовал важный вывод о том, что молекулярные веса жидкостей являются функциями не только температур кипения, но и плотностей жидкости. Выше было показано, что для жидкостей, имеющих равные плотности при температурах кипения (К = Const), постоянная Бачинского СБ также имеет одну и ту же величину. Принимая за постоянную величину постоянную Этвеша, мы приходим к выводу, что для всех ( А115
жидкостей с К = Const величина С tv- также должна \ сб 7
быть постоянной.
В табл. 1 дан подсчет этой величины. Он показывает, что для жидкостей с К — Const постоянство коэффициента С действи
238
Д. К. Коллеров
тельно приближенно соблюдается. Имеющиеся отступления законны, если принять во внимание упрощения, принятые при выводе уравнения (1).
* * *
На основании принятых предпосылок было выведено приближенное уравнение температурных коэффициентов плотности жидкостей:
_ <Ткр~273)0,3- (Пр-Л0’3 а~ (Т'~ 273)0’3 (Тк — 273)
или, принимая во внимание правило Гульдберга,
(1,5 Тк—273)0,3—(0,5 Т )°’3
(1,5 Гк —273)0’3 (Гк —273) v '
Согласно этому уравнению температурный коэффициент плотности жидкости или ее модуль расширения является в пер-
Рис. 4. о — углеводороды разного химического характера; х — кетоны и простые и сложные эфиры; • — амины и нитрилы.
вом приближении функцией температуры кипения жидкости. На рис. 4 дан график зависимости а = / (/к), вычисленной по уравнению (5). Там же помещены фактические величины коэффициентов а для разных жидкостей. В табл. 6 сведены результаты расчетов величин а по уравнению (5).
А вопросу о физической сущности показателя К
239
Таблица 6
Температура кипения Модуль расширения Температура кипения Модуль расширения
°C °К Рассчитано по уравнению (5) Среднее по опытным данным °C °К Рассчитано по уравнению (5) Среднее по опытным данным
47 320 0,00158 0,00142 247 500 0,000710 0,000800
107 380 0,00117 0,00114 307 560 0,000630 0,000730
167 440 0,000933 0,000940 367 670 0,000550 —
Из приведенного графика 4 видно, что расчетная кривая для величин а достаточно удовлетворительно согласуется с опытными данными, если принять во внимание упрощения, принятые при выводе уравнения (5).
* *
*
Анализом выведенных зависимостей установлено, что показатель К есть критерий «приведенного» состояния жидкостей и что между показателем К и удельным объемом жидкости при температуре кипения существует линейная зависимость.
А. И. Бачинским было доказано, что предельный объем жидкости составляет 0,307 или 4Лз критического объема, т. е.
— Const, следовательно, должно существовать постоянство Укр
и другого отношения = Const.
Действительно, оказалось (4), что для взятых 50 жидкостей величина Const = 0,1247. Среднее отклонение величин отношения от среднего значения для взятых 50 жидкостей оказалось около 1% при крайнем отклонении 4,2%.
Так как
ш = 0,307 Укр и
со = 0,1247 К,
то, следовательно, величина К составляет определенную долю критического объема и ему пропорциональна.
0,1247/< = 0,307 VKP, K = 2,46VKP. (6)
Совершенно определенную долю критического объема должен составить и удельный объем при температурах кипения, это следует из того, что величина К пропорциональна удельному объему при температурах кипения.
240
Д. К. Коллеров
Так как в уравнении (2) а —0,63 и в = 6,1
/< = 0,63 + 6,1 Ук, (7)
то из соотношений (6) и (7) VK = 0,1034(3,9 VKp—l).
Соотношение (6) с очевидностью объясняет, почему показатель К оказался характеристической величиной.
В табл. 7 сведены данные по критическим плотностям и критическим объемам жидкостей, взятые из различных литературных источников. В этой же таблице приведены для сравнения величины критических объемов этих жидкостей, рассчитанные по уравнению (6):
VKP = -2Д0- • (8)
Таблица 7
Сравнение критических объемов жидкостей, рассчитанных по уравнению (8), с табличными данными
Продукт Химическая формула к Критическая плотность по табличным данным Критический объем Расхождение с табличными данными, %
по табличным данным | Р. 3] по формуле (8)
2-Метилбутан . с5н12 10,7 0,232 4,31 4,35 0,92
н-Пентан . . . ^5^32 10,7 0,234 4,28 4,35 1,61
н-Гексан . . . СеН14 10,6 0,234 4,28 4,31 0,7
2,3-Диметилбутан с6н14 10,46 0,241 4,15 4,23 1,93
н-Гептан . . . с7н16 10,49 0,241 4,15 4,26 2,58
2,5-Диметилгексан С8н18 10,46 0,233 4,24 4,26 0,7
н-Октан .... C8Hi8 10,47 0,237 4,22 4,22 0
н-Нонан .... свн20 10,43 0,232 4,31 4,24 1,65
Циклогексан . . свн12 9,08 0,273 3,67 3,69 0,81
Бензол с„н6 8,04 0,305 3,27 3,27 0
Толуол . ... С7Н8 8,36 0,292 3,43 3,41 0,58
Диизопропил . . С6Н14 10,46 0,241 4,15 4,23 1,93
Этиловый эфир . С4Н10 9,47 0,262 3,82 3,85 0,79
Диэтиламин . . Этилпропиловый с4ни 9,67 0,246 4,06 3,93 2,95
эфир С5Н12О 9,49 0,258 3,86 3,87 0,26
Метилбутират С6Н10О2 8,04 0,300 3,33 3,27 1,8
Метилизобутират C5Hi0O2 8,04 0,301 3,32 3,27 1,51
Пропилацетат С5н10о2 8,14 0,296 3.31 3,38 2,11
Триэтиламин . . Метилэтиловый С6н15 9,79 0,251 3,98 3,98 0
эфир С3Н8О 9,88 0,270 3,71 3,81 2,7
Среднее . . . 1,3%
К вопросу о физической сущности показателя К
241
Для взятых 20 жидкостей среднее расхождение между критическими объемами, рассчитанными по этому простому соотношению, и табличными данными составило 1,3% при крайнем расхождении 2,45%.
ЛИТЕРАТУРА
1. Д. К. Кол леров. Физико-химические свойства сланцевых и каменноугольных смол, Гостоптехиздат, 1951.
2. Техн, энциклопедия. Справочник физико-химических величин, т. V.
3. М. Ф. Нагиев. Термодинамические расчеты процессов переработки нефти и данные по свойствам химических соединений. Гостоптехиздат, 1950.
4. Д. К. Коллеров. Настоящий сборник, стр. 216.
16 Труды ВНИИПС.
Г. И. Скрынникова
ЭКСПЕРИМЕНТАЛЬНОЕ ИССЛЕДОВАНИЕ КОЭФФИЦИЕНТА ТЕПЛОПРОВОДНОСТИ ЖИДКИХ СЛАНЦЕВЫХ ПРОДУКТОВ
Количественные данные, характеризующие физико-химические свойства и, в частности теплопроводность жидкостей, лежат в основе технологического расчета всевозможных процессов и установок.
Коэффициент теплопроводности входит в критерий Нусельта и Прандтля и является необходимой величиной для решения многих технологических вопросов производства.
Экспериментальных исследований по определению теплопроводности жидких сланцевых продуктов до настоящего времени не проводилось.
В литературе имеются многочисленные эмпирические и теоретические формулы для расчета теплопроводности, но в большинстве случаев они дают значения коэффициента теплопроводности, резко отличающиеся от надежных экспериментальных данных даже для чистых индивидуальных соединений.
Из опубликованных за последние годы исследовательских работ в области изучения явления теплопроводности наибольший интерес представляют работы советских ученых А. С. Пред-водителева [1, 4], Д. Л. Тимрот [2], Н. Б. Варгафтика [3] и их учеников.
А. С. Предводителев [1], исходя из теоретических представлений о том, что тепловое движение в жидкости носит в себе черты теплового движения в газах и твердых телах, и развивая теорию инвариантных количеств, вывел уравнение для теплопроводности
Л = ДСрр4/з м-1/з,
где: А — инвариантная, по данным Предводителева, величина, зависящая от температуры, но не зависящая от природы вещества; А = 3,5 10-3 при / = 30° С;
Ср—теплоемкость при постоянном давлении;
р— плотность;
М—молекулярный вес.
Исследование коэффициента теплопроводности
243
Уравнение дает удовлетворительное совпадение с опытными данными только для теплопроводности при комнатных температурах и для капельных жидкостей.
Н. Б. Варгафтик, на основании большого экспериментального материала по теплопроводности, видоизменил формулу Предво-дителева, применил ее к любым жидкостям (структурным и бесструктурным) и установил зависимость теплопроводности от температуры.
Формула Варгафтика имеет следующий вид:
Я = -1-Ве4/з; B = ACfM~lh
а — фактор, учитывающий степень упорядоченности молекул (степень ассоциации).
Ассоциация веществ может быть обнаружена и количественно оценена при отступлении от законов сложимости. Структурные жидкости дают отклонение от правила Кистяковского, Троутона, Этвиша, Льюиса и Траубе.
Варгафтик заменил коэффициент А = 3,5810~3 на А = = 4,2810~3; при этой замене получается лучшее совпадение с экспериментальными данными. Максимальное расхождение для 12 исследованных Варгафтиком веществ не превышает 5%; среднее расхождение равно 3%.
Последними работами А. С. Предводителева [4] для капельных жидкостей теоретически доказана зависимость коэффициента теплопроводности от температуры через плотность в первой степени
2 = ПРИ * = вв,х=1
где: А —постоянный множитель;
С„ — величина, сходная с теплоемкостью при постоянном объеме, но не зависящая от температуры;
Z— фактор, учитывающий ассоциацию;
Q — плотность;
М — молекулярный вес.
Все эти формулы проверялись в большинстве случаев на чистых индивидуальных продуктах и, за очень небольшим исключением, применялись для расчета теплопроводности сложных смесей.
Жидкие сланцевые продукты представляют собой сырье сложного химического состава с разнообразными физико-химическими свойствами, различное по происхождению, методам получения и обработки.
В 1951 г. Д. К. Коллеровым [5] был предложен очень простой графический метод расчета коэффициента В формулы 16*
244
Г. Н. Скрынникова
Предводителева— Варгафтика для сланцевых жидких продуктов. Для определения теплопроводности по этому методу необходимы данные только по плотности и среднеобъемной температуре кипения. Однако экспериментального подтверждения диаграмма Коллерова до настоящего времени не имела, так как опытных определений теплопроводности сланцевых продуктов в литературе не было.
Диаграмма Коллерова представлена на рис. 1.
Множитель В уравнения теплопоовадности X=-^8dt
Рис. 1. Диаграмма теплопроводности жидкостей.
Необходимая для расчета величина плотности q находится по другой диаграмме Д. К. Коллерова, представленной на рис. 2. Для определения значения плотности по этой диаграмме необходимо знать величины плотности р?" и средней температуры кипения.
Целью данного исследования и явилось экспериментальное определение коэффициента теплопроводности жидких продуктов переработки сланцев при различной температуре.
Исследование коэффициента теплопроводности
245
Продукты переработки сланцев очень разнообразны и насчитывают большое количество отдельных представителей. Экспериментальное исследование, охватывающее все эти продукты, потребовало бы очень много времени.
Нам казалось более целесообразным произвести прецизионные исследования теплопроводности нескольких наиболее харак-
tSOth
Б
g, ШЮ
<к>
O>
Sr
/зоа
ffOO\
100O\
Iiaaaaiak BaiiiiiBMaiiiiiiiiiiiiiiiiiiuiiiHiiiiuiimimi aaiaaiaaiiaiBiiiiaimiiiiiiiiiiiiiiiiiiiiiHHiiiiiifinif! aaaaiaio tiiaaiiaaaaiiiuiamiiuiiiiiiiiiiiaiiiuiiiiiiiHi SBaiBiakirMiiiaiaiiiiiiiiiiiiitiiiiiiiiiiiMHiiniiiiiii •!» iKehiaaiiiiaiitaiiiiiiiiiiiiiiiiiiiiiiiiiiiHiiii aaaiiaavi.
iiiifit iaiia tlSMaiMiiMMiii iiihiiiiitiiiiiHiiiHiiiiiif aaaaaiai aaiikk^jjrRaiiiiaiiiiimiiiiiiiiiiiiiiiiiiiiiiHi aaaaiaaiiit iaVKIb<?tiiiiaiiitiiiiiiiiii
aaeiBaajiaiaiiiAEflimiaiiiiiiiiiiiBiiiiiiiiutiiiuiiiHi aaaBiaaaivai авшк ar
aaaiiiMi 11.41 iinikiKPiiiituiiiiiiiiiHiiiiitiimiiiii waaaiaaaaamiikii/ia/mi ilia iiiiauiii iiihiiiii iiiiiiiiit aaaataaaaiiai.'ra.'iauauii'iniiiiiiaaiiiiiaiiiiiituiiiiiii laiiiiiBiiaui’ikaikiikiiUMiEtfiiiiiitiiiiiiiiiiiiiiiHti aaaaaaaaaaaa unn ааипшал mtiiiiiiiliiiiiiiiiiiiiiiii aaoaaiaaiaikaa nikian»i. in 'iiiiiiuimtiii iiiaiinii aaiaaiiaaf iiimi min 'ikiin 'in'iiiiiiiiiiitiiiiiniiiiii aaaiaaaaiiiaii m iitiania 41111111411111111111111111111111 ааааавваааамв'ав m Mi.iiHiiniiiHiiiiuiiiiiuiimiiiit aaaaBBaaBiaaii лак aiuia'iiui lit 1111411111(111111111111111
LI 111111 ft {ff|[|
aviiiiiaaimiiiciiiMiiiriiiMiiriikiiiKMIiiiiiiiiiii •BBaaaiaaaaiaaiai 'iiiiaa 'ii»4m4ia.nV0iiiiiiiiiiii aaaiiiiaiiBiiiiiikiikuaiiriiHiinMih'tHh'iiiiiiiiiii
11 44
700
600— 0,60
иluiuif si in mi Л1Ш1ЫНП 'iiiniiii riiiiiiit aaaaaaaaiiaiifliiiiiumiiriinuMiiiPiiiritiiii until aiiiaiaaiaiiiniiiii.’iiiiiMiiktiuuiiiiiiiJiiiihMini «iHiiiaiiiiiimmwtiih iiihiiim iiMiik4iin:iiii aiaaaaaaiaaaiiiaiiiiikUB.iiiLieiHiiriiii.uiiriiiii.Hi aaaiaaaii taiaiiaamiai'ariUkiiU'iH'iim'iiiriiiiiiir ------------------------ ш Hi Hl
«I ««iiia
0,70
0,80 0,90 1,00 fJO 120
Удельный Вес
I s
§
1
I
£
9ОЛ
Ж
Рис. 2. Диаграмма температурных коэффициентов плотности.
терных жидких продуктов переработки сланцев и, одновременно с этим, произвести изучение других физико-химических свойств этих продуктов (температурной зависимости теплоемкости, плотности, вязкости, среднеобъемных температур кипения, молекулярного веса и т. д.) для установления связи коэффициента теплопроводности с другими физико-химическими константами, а также для проверки применимости к жидким продуктам переработки сланцев предложенных формул и графических методов расчета коэффициента теплопроводности.
В качестве объектов исследования были взяты: фракции генераторной смолы, фракции камерной смолы (представляю
246
Г. Н. Скрынникова
щие собой ароматизованные жидкости), широкие фракции кислородных соединений 1 и фракция кашпирской смолы (высокосернистые соединения).
МЕТОД ИССЛЕДОВАНИЯ
При выборе метода определения теплопроводности всегда приходится считаться с тем, что передача тепла может происходить не только теплопроводностью, но и в результате конвекции и излучения.
Лучшим методом, с точки зрения устранения конвективного переноса тепла, является метод горизонтального плоскостного слоя при потоке тепла сверху вниз, но этот метод чрезвычайно сложен, в связи с необходимостью устранения боковых потерь, и требует сложной экспериментальной установки (создания специальных охранных нагревателей). Мы остановились на изучении теплопроводности методом нагретой нити. Этот метод наиболее полно разработан советскими учеными Д. Л. Тимротом и Н. Б. Варгафтиком. Метод нагретой нити заключается в том, что исследуемая жидкость помещается между двумя коаксиальными цилиндрами. Роль внутреннего цилиндра выполняет платиновая нить, которая является одновременно и источником тепла (нагревателем) и термометром сопротивления. Роль внешнего цилиндра выполняет стеклянный капилляр, на внешней стороне которого намотан платиновый термометр сопротивления.
Коэффициент теплопроводности определяется по формуле:
. _ Q In d2/rf! _ AQ.
217141 ~^At ’
где: А — постоянная прибора, равная А = ’
Q — количество тепла, выделяемое нагревателем и проходящее через жидкую прослойку;
rf2 и d1~диаметры внутреннего капилляра и проволоки;
I — длина проволоки внутреннего платинового термометра сопротивления;
At — перепад температур в слое жидкости.
Крауссольдом [6] на основании экспериментальных исследований было показано, что конвективная передача тепла в методе нагретой нити может отсутствовать при условии, когда произве
1 Фракции генераторной смолы, камерной смолы и нейтральных кислородных соединений были получены и предоставлены нам лабораторией смолы ВНИИПС (С. С. Семеновым, О. С. Куратовой и М. В. Кобыльской).
Исследование коэффициента теплопроводности
247
дение безразмерных критериев Грасгофа и Прандтля меньше 1000, т. е. когда
GrPr = -^?3 Л/<1000, va ’
где: Gr — критерий Грасгофа;
Рг — критерий Прандтля;
g ускорение силы тяжести;
/1 — коэффициент объемного расширения;
а — коэффициент температуропроводности;
v — кинематическая вязкость;
ст—линейный размер;
zit — разность температур на границах слоя.
В 1952 г. В. В. Шингареев установил, что зависимость, выведенная Крауссольдом, относилась к капиллярным трубкам с зазорами больше 7 мм и что для более узких трубок существуют несколько другие соотношения, а именно конвекция в узких зазорах (например, при S = 4 мм) практически отсутствует, если
GrPr< 2500.
При конструировании прибора это основное условие отсутствия конвекции было выполнено, и при самых неблагоприятных соотношениях параметров во всех случаях оно соблюдалось, т. е. конвекционная передача тепла отсутствовала.
ЭКСПЕРИМЕНТАЛЬНАЯ УСТАНОВКА
Измерительная трубка схематически представлена на рис. 3. Капилляр трубки изготовлялся из молибденового стекла. Капил-
Рис. 3. Измерительная трубка:
J—• платиновый термометр сопротивления — нагреватель; 2 —платиновая пружина; 3— платиновый термометр сопротивления; 4 — отверстия для заполнения; 5 — крючки для крепления проводов.
ляр отбирался строго однородного сечения по всей длине. Диаметр измерялся по весу столбика ртути, длина измерялась при помощи прецизионной измерительной линейки, снабженной оптическими линзами.
Для центрировки прибора был изготовлен специальный станок, на котором трубка закреплялась неподвижно, а платиновая нить, пропущенная внутрь капилляра, при помощи специальных
248
Г. Н. Скрынникова
винтов могла быть отцентрирована по оси капилляра. Положение платиновой нити просматривалось при помощи бинокулярной лупы в двух взаимно перпендикулярных направлениях по всей длине капилляра и, после того как удавалось добиться хорошей центрировки, производилось запаивание нити в оттянутые концы трубок.
Прибор для центрировки изображен на рис. 4.
Рис. 4. Прибор для центрировки.
Платиновая проволока «Победа» отвечала требованиям, предъявляемым к платиновым термометрам сопротивления:
«‘,ю = 3,91710' для проволоки d 0,1 мм;
aj00 = 3,91910-3 для проволоки d 0,05 мм.
Характеристика трубок представлена в табл. 1.
Характеристика измерительных трубок
Таблица 7
№ прибора Диаметр внутренний d2, см Диаметр внешний ds, см Диаметр платиновой проволоки di, см Длина нагревателя 1, см / 2ЛГ 1 — п d d_ U2 2П1/.С1
1 0,0778 ±0,001 0,177 ±0,001 0,010 ±0,0002 8,8 ±0,01 3,7083 1,4858 'чт
2 0,0842 ±0,001 0,0185 ±0,001 0,010 ± 0,0002 10,5 ±0,01 3.227 1,1921
Исследование коэффициента теплопроводности
249
В качестве изотермической ванны применялся двойной масляный калориметр, схема которого представлена на рис. 5.
Опыты велись при условии постоянства температуры в обоих
стаканах калориметра, контроль дифференциальной хромелькопе-левой термопары.
Измерительная электрическая схема представлена на рис. 6. Она состоит из потенциометрической установки УПН3/з завода «Эталон», в которую входят потенциометр ПН-4, зеркальный гальванометр ГПЗ-2 и нормальный элемент II класса, из образцовых катушек I класса, источников тока (система аккумуляторов большой емкости), инверсионного переключателя, штепсельных реостатов (на схеме показаны ползунковые реостаты), контрольного амперметра и миллиамперметра.
ПОПРАВКИ И АНАЛИЗ ПОГРЕШНОСТЕЙ ИЗМЕРЕНИИ
При обработке результатов, полученных методом нагретой нити, необходимо внести следующие поправки.
I. Поправки к количеству пе-
осуществлялся с помощью^
Рис. о. Масляный калориметр:
реданного тепла:
а) поправка на отвод с концов измерительной проволоки;
б) поправка на передачу тепла
наружный стакан калориметра; 2—внутренний стакан калориметра; 3— трубка-с исследуемой жидкостью; 4 — мешалка.
излучением.
II. Поправки к перепаду температур:
а) поправка на перепад в стенке измерительной трубки;
б) поправка к первоначальной калибровке.
III. Поправка на эксцентричность (поправка на неточность Центрировки).
1. Поправка на отвод с концов измерительной проволоки рассчитывалась по формуле, предложенной Варгафтиком,
zlQ _ r|/21n7?/r l/’T” Q ~ I У А,
Рис. 6. Принципиальная схема УПН-3/3.
Исследование коэффициента теплопроводности
251
Потенциометрические провода, при помощи которых измерялась разность потенциалов, на концах измерительного участка, изготовлялись из проволоки, имеющей диаметр 0,05 м; диаметр нагревателя был равен 0,1 мм. Поправка на отвод с концов для й 1 ал п/ е. Л®
первого прибора -q- =1,04%, для второго прибора ~q- = = 0,86%.
2. Поправка на излучение вводилась расчетным путем по формуле Стефана—Больцмана
„-с к т^~( мЪ
* р loo 7 к loo у J ’
где: Ср — коэффициент излучения платины;
7\ и Т2 — абсолютные температуры стенки и нагревателя в градусах Кельвина;
F — излучающая поверхность.
Зависимость коэффициента излучения платины от температуры была взята из работы Д. Л. Тимрот и Н. Б. Варгафтика. Поправка на излучение вводилась только в случае расчета теплопроводности воздуха, так как жидкости обладают малой теплопрозрачностью.
Поправки на излучение составляют:
для 1 прибора при t 50° 0,19, при t 115° 0,23%;
для 2 прибора при t 50° 0,23, при t 115° 0,38%.
3. Поправка на перепад температур в стенке измерительной трубки рассчитывалась по формуле
л Q In
где: Q — количество тепла прошедшего через слой стенки;
dg — внешний диаметр капилляра;
d2 — внутренний диаметр капилляра;
I — длина внутреннего термометра сопротивления;
Яст — теплопроводность молибденового стекла, рассчитанная по формуле Л = 0,77 (1+0,0013 О-
В наших приборах поправка на перепад температур в слое стенки составляла 5—6% общего перепада.
4. Поправка на калибровку. Вследствие неравномерного отжига проволоки и натяжения пружины, начальное сопротивление проволок несколько менялось, и для того чтобы устранить ошибки, вызванные этим явлением, перед каждым опытом при малых силах тока производилось определение температур проволок нагревателя и термометра сопротивления. Всегда, как правило, оба термометра показывали несколько различную тем
252
Г. Н. Скрынникова
пературу. Эта разность температур составляла от нескольких сотых до двух десятых градуса.
Поправка на калибровку рассчитывалась и вносилась с соответствующим знаком.
5. Поправка на центрировку. Из-за неточной центрировки фактор формы А может быть определен недостаточно точно.
Положение нити в капилляре фотографировалось и рассчитывалось отклонение нити от оси капилляра.
Расчет производился по формуле:
1 n V(R + ry- — a24- V(R^rY^
~ ]/(/? +г)2 —о2 — V ^R—-r)2 — а2
2П1
где: 7?— внутренний радиус трубки;
г — радиус проволоки;
а — расстояние положения центра проволоки от оси трубки.
Введение поправки на отклонение центра расположения платиновой нити от центра капилляра приводит к небольшим изменениям фактора формы А и составляет максимально 0,08%.
ОЦЕНКА ПОГРЕШНОСТИ ОПЫТОВ
Для вычисления погрешности измерений необходимо рассчитать относительную погрешность определений фактора формы А, количества тепла Q и перепада температур At, а также относительные погрешности для всех поправок, которые вносились в определяемые значения Q, At и А. Следует отметить, что относительные ошибки рассчитывались при самых неблагоприятных условиях измерения.
Относительная погрешность измерений:
для 1 прибора 1,63-% 1,41-% 0,082 = 3,12% ;
для 2 прибора 1,14-J- 1,24 + 0,071 = 2,45%.
ПРОВЕРКА УСТАНОВКИ
Для проверки установки были взяты химически чистые бензол и толуол, о чем свидетельствуют приведенные ниже физико-химические константы (температура кипения, показатель преломления и плотность) и результаты спектрального анализа (рис. 7).
Бензол .................. 80,2 1,502 0,8790
Толуол................... 110,6 1,495 0,8669
Теплопроводность этих жидкостей изучалась многими исследователями и поэтому литературные данные, использованные
Исследование коэффициента теплопроводности
253
нами в качестве контрольных при проверке установки, являются наиболее достоверными. Кроме того, установка проверялась по воздуху, теплопроводность которого также хорошо изучена.
Рис. 7.
Результаты опытов представлены в табл. 2.
Таблица 2
Проверка установки по бензолу, толуолу и воздуху
Вещество ГС нагревателя ГС стенки Л1° С Q1* /Ж°С
Бензол . . 34,64 31,18 3,46 —0,20 0,1079 33,0 0,123
Толуол . . 26,79 22,88 3,49 -0,22 0,1205 24,8 0,117
56,22 40,14 16,78 —0,17 0,1327 47,5 0,0239
Воздух . | 125,54 113,25 12,25 -0,16 0,1023 119,0 0,0271
Сравнение полученных экспериментальных данных с результатами определений других авторов представлено в табл. 3.
Из приведенной таблицы следует, что полученные на установке данные по теплопроводности бензола и толуола хорошо соответствуют наиболее надежным данным Варгафтика.
Q1 в таблице приводится со всеми поправками.
254
Г. Н. Скрынникова
Результаты определения теплопроводности воздуха находят хорошее подтверждение в данных Варгафтика, Керженцева и Чернеевой.
Таблица 3
Сравнение результатов определения коэффициента теплопроводности бензола, толуола и воздуха, полученных на установке ВНИИПС, с результатами других авторов
Результаты определения теплопроводности Авторы исследований
Бензол Толуол Воздух
Л / Л t 1
30 70 0,123 0,115 30 85 0,115 0,104 44,1 106,2 0,0230 0,0268 | Варгафтик
18 0,136 30 70 0,130 0,122 | Бриджмен [8]
10—20 20-40 0,121 0,111 0-20 20—40 0,140 0,126 — | Абас-Заде [7]
20 0,120 20 0,110 — — Вебер [2]
— — — — 60 102 0,0244 0,0272 | Чернеева
— — — — 35 69 0,0231 0,0241 | Керженцев
33 0,123 24,9 0,117 47,5 119,0 0,0239 0,0271 | Скрынникова
Для всех исследуемых образцов были рассчитаны произведения критериев Грасгофа и Прандтля, и во всех опытах эти произведения были меньше 2500; следовательно, конвекционная передача тепла в опытах отсутствовала.
Кроме того, проводились опыты с различным значением <1/, которые показали, что Я не зависит от At, что также является косвенным доказательством отсутствия конвекции.
На основании того, что полученные на установке данные по теплопроводности жидкостей на 2—3% отличаются от надежных экспериментальных данных других авторов и погрешность измерений может быть оценена также в 2—3%, мы считали установку пригодной для проведения на ней исследований.
—температура, °C; /. — теплопроводность, —-------.
М/Ч(1С
Исследование коэффициента теплопроводности
255
РЕЗУЛЬТАТЫ ЭКСПЕРИМЕНТАЛЬНЫХ ОПРЕДЕЛЕНИЙ ТЕПЛОПРОВОДНОСТИ ЖИДКИХ СЛАНЦЕВЫХ ПРОДУКТОВ
В табл. 4 представлена общая характеристика исследуемых фракций, а в табл. 5 — результаты экспериментального определения их теплопроводности.
‘ Таблица 4
Характеристика исследованных фракций
| № фракций I Название фракций Молекулярный вес, М Среднеобъемная температура кипения, I1 Плотность, Р20 4 Вязкость кинематическая, стоксы
*4 +20° С ’4 +50° С ’-’t 4-75° С
1 Узкие фракции ней- 145 222 0,9274 0,025 0,014 0,008
2 тральных кислород- 165 243 0,9305 0,039 0,017 0,009
3 ных соединений, вы- 179 261 0,9455 0,0568 0,0264 0,0127
4 деленные из генера- 214 310 1,0009 0,692 0,124 0,028
5 торной смолы Широкая фракция нейтральных кислородных соединений 176 273 0,9440 0,0709 0,0288 0,0136-
6 Фракция ретортной каш-пирской смолы . . . 138 184 0,9116 0,0163 0,0103 0,0079
7 Фракция камерной смолы 142 214 0,9585 0,0253 0,0135 0,009
8 Фракция камерной смолы 148 238 0,9902 0,0443 0,0201 0,0141
Из данных, приведенных в таблице 5 и кривых рис. 8, следует, что для всех исследованных фракций жидких сланцевых продуктов коэффициент теплопроводности уменьшается с повышением температуры.
Теплопроводность жидких сланцевых продуктов может быть представлена уравнением прямой вида ^t = A(l—fit).
Значения коэффициентов теплопроводности различных фракций близки между собой и лежат в пределах от 0,110 до 0,121 при 0° С. Рассчитанные по уравнениям значения хорошо воспроизводят опытные данные. Относительная погрешность не превышает 2%.
1 Среднеобъемная температура кипения t °C определялась по стандартным энглеровским разгонкам.
Рис. 8а—ж. Коэффициент
Исследование коэффициента теплопроводности
257
Таблица 5
Коэффициент теплопроводности жидких сланцевых продуктов
№ фракций Л/ж Q /ж *оп опытная рассчитанная Jz Л % Уравнения
1 3,88 0,116 26,3 0,110 0,109 0,4
4,14 0,119 60,6 0,105 0,106 0,9 zt = 0,114(1—0,001491)
4,0 0,104 105,5 0,097 0,096 0,6 J
2 3,30 0,101 28,3 0,114 0,114 0.4
4,15 0,124 56,6 0,109 0,110 0,9 zt = 0,118 (1-0,001251)
4,11 0,115 100,0 0,103 0,103 0,0
3 3,27 0,096 26,7 0,109 0,110 0,4
3,23 0,096 23,1 0,110 0,110 0.0
3,40 0,098 39,3 0,108 0,108 0,0 zt = 0,112 (1-0,0009551)
4,03 0,113 85,1 0,104 0.103 0,9
4,83 0,132 112,6 0,100 0,100 0,0
4 3,90 0,119 32,0 0,114 0,114 0,0
3,79 4,10 0,114 0,121 62,0 93,2 0,112 0,109 0,113 0,009 0,8 0,0 /.t = 0,116 (1—0,000621)
4,08 0,119 114,0 0,107 0,108 0,9
5 3,32 0,097 36,2 0,108 0,108 0,0
3,43 0,098 57,8 0,106 0,106 0,0 7-t = 0,lll (1-0,00075/)
3,55 0,099 95,7 0,103 0,102 0,6
6 3.45 0,107 29,6 0,115 0,115 0,0
3,65 0,113 48,1 0,114 0,114 0.0 zt = 0,117 (1—0,00059/)
3,69 0,113 52,2 0,113 0,113 0,0
7 3,08 0,110 29,8 0,115 0,115 0,0
3,24 3,22 0,113 0,113 54,2 54,1 0,111 0,112 0,111 0,110 0,0 1,8 = 0,120 (1—0,00145 f)
3,53 0,114 94,7 0,104 0,104 0,0
8 3,22 0,118 23,2 0,117 0,118 0,8
3,16 0,118 23,1 0,118 0,118 0,0
3,58 3,55 0,124 0,124 67,3 68,7 0,111 0,111 0,111 0,111 0,0 0,0 zt = 0,121 (1—0,00121 /)
3,53 0,116 107,7 0,107 0,105 1,7
3,53 0,116 109,5 0,106 0,106 0,0
® таблице 5:
Лж — перепад в слое жидкости с учетом всех поправок;
Q — количество тепла, переданное через слой жидкости с учетом всех поправок;
/ж — средняя температура жидкости при определении теплопроводности;
zon— теплопроводность по данным опыта;
Л — теплопроводность, рассчитанная по уравнению.
17 Труды ВНИИПС.
258
Г. Н. Скрынникова
ИССЛЕДОВАНИЕ ФИЗИКО-ХИМИЧЕСКИХ ХАРАКТЕРИСТИК ВЗЯТЫХ ОБРАЗЦОВ ЖИДКИХ ФРАКЦИИ СЛАНЦЕВЫХ ПРОДУКТОВ
Зависимость плотности от температуры
Плотность определялась методом пикнометра. Изотермическая ванна имела терморегулятор, который обеспечивал постоянство температуры с точностью до +0,05° С. При расчете плотности вводились поправки на приведение веса к пустоте. Определения производились при 4—5 температурах в интервале 0—125° С.
Опытные данные обрабатывались по способу наименьших квадратов. В табл. 6 представлены опытные данные по плотности [р0] и рассчитанные по уравнению [рр] при температурах опытов.
Для всех фракций получены уравнения вида Qt = 2о (1 — а0> которые хорошо описывают зависимости, отвечающие опытным данным: максимальное расхождение между экспериментальными-и расчетными данными равно +0,11 %.
Определение теплоемкости исследуемых образцов
Теплоемкость изучаемых фракций определялась калориметрическим способом по методу охлаждения навески.
Метод подробно описан в книге М. М. Попова [11] и применялся в работах ВНИИПСа.
Медные, с завинчивающимися крышками, ампулы заполнялись жидкостью настолько, чтобы обеспечить при нагревании свободное расширение жидкости. Поправка на радиацию определялась по упрощенной формуле А. Н. Щукарева [12].
Экспериментально полученные данные обрабатывались по способу наименьших квадратов.
Опытные данные по теплоемкости сопоставлялись с данными, рассчитанными по диаграмме для теплоемкостей, предложенной; Д. К- Коллеровым [5].
Ср = -^- (2,2881g 0,992),
где: п — среднее число атомов в молекуле; М — молекулярный вес;
и — некоторая функция, связанная с гок и с тем-
Gt
пературой кипения гкип в °К;
Т — температура, к которой отнесена величина истинной теплоемкости в °К.
В табл. 7 представлены результаты калориметрических определений теплоемкости восьми фракций, а в табл. 8 сопоставлены опытные данные и рассчитанные по диаграмме Д. К. Коллерова.
Исследование коэффициента теплопроводности
259
Таблица 6
Зависимость плотности от температуры
№ фракций 1°С Плотность Уравнения
опытная рассчитанная
1 0 0,9399 0,9412
20 0,9274 0,9256 gt = 0,9412 (1—0,00083
50 0,9016 0,9018
75 0,8826 0,8820 = 0,06%
80,8 0,8773 0,8770 е
107,3 0,8566 0,8566
2 0 0,9433 0,9440
20 0,9305 0,9288 et=0,9440 (1—0,000807 1)
50 0,9050 0,9080
75 0,8874 0,8869 — = 0,01%
80,8 0,8821 0,8825 Q
106,8 0,8631 0,8625
3 0 0,9574 0,9584
20 0,9455 0,9436 et=0,9584 (1—0,00077 t)
50 0,9205 0,9224
80,7 0,8985 0,8986 —— = 0,1%
107,3 0,9790 0,8789
4 0 1,0123 1,0142
20 1,0009 0,9994 et = 1,0142 (1—0,000662 /)
50 0,9771 0,9770 Л л
80,7 0,9551 0,9542 —= 0,09%
107,3 0,9385 0,9344
5 0 20 0,9585 0,9440 0,9584 0,9440 ) gt = 0,9584 (1—0,000753 t)
50,3 0,9218 0,9211 [ —= 0,05%
80,2 0,9007 0,9008 J e
6 0 0,9284 0,9285
20 0,9116 0,9119 gt = 0,9285 (1—0,000888 t)
50,3 0,8878 0,8873 Ao
75,5 0,8669 0,8662 —- = 0,05%
101,5 0,8442 0,8448 Q
7 20 0,9585 0,9585 > gt = 0,9745 (1—0,000821 1)
50 0,9342 0,9345 I Zip Л .
75,5 0,9145 0,9145 I —— = 0,01% 1 e
8 0 20 1,006 0,9902 1,0053 0,9902 1 et= 1,0053 (1—0,000761 t}
50 0,9663 0,9663 1 — — 0,04%
75,5 0,9476 0,9475 J e
17*
260
Г. Н. Скрынникова
Таблица 7
Калориметрические определения теплоемкости жидких сланцевых продуктов
№ фракций /°C Теплоемкость Уравнения
опытная рассчитанная
1 39,9 43,6 46,1 52,3 58,6 0,436 0,442 0,450 0,453 0,463 0,437 0,442 0,444 0,455 0,463 ct = 0,382+ 0,00139/
2 47,8 60,2 65,6 67,6 78,13 0,432 0,438 0,444 0,445 0,449 0,432 0,439 0,443 0,444 0,449 Ct = 0,407+ 0,000545/
3 53,6 64,9 67,9 70,7 73,9 76,1 76,5 0,444 0,458 0,463 0,466 0,472 0,472 0,474 0,446 0,454 0,463 0,467 0,470 0,474 0,473 Ct = 0,382+0,00118/
4 50,4 66,4 68,6 80,9 0,454 0,464 0,463 0,471 0,455 0,463 0,464 0,471 ct = 0,423 4- 0,000528/
5 38,2 46,4 57,7 66,9 76,7 185,5 0,452 0,457 0,465 0,476 0,488 0,492 0,452 0,459 0,466 0,476 0,484 0,491 Ct = 0,420 + 0,000888/
6 52,6 63,2 65,7 74,7 82,1 0,445 0,457 0,459 0,468 0,477 0,445 0,456 0,459 0,468 0,475 Ct = 0,393+0,00100/
7 57,18 66,38 0,434 0,436 ct = 0,405 + 0,000205/
8 43,7 67,7 0,414 0,418 Ct = 0,403 + 0,000324/
Исследование коэффициента теплопроводности
281
Таблица 8
Сравнение опытных и рассчитанных по диаграмме Д. К. Коллерова данных по теплоемкости при 0° С
Теплоемкость Фракции
1 2 3 4 5 6 7 8
Ср 20® с ’ опытная • > * Ср90о с ’ рассчитанная по 0,410 0,418 0,405 0,439 0,439 0,410 0,407 0,409
диаграмме Коллерова . 0,437 0,439 0,435 0,435 0,455 0,446 0,405 0,409
Расхождение, % .... 6,1 5,1 7,4 0,9 3,9 8,7 0,5 0,0
Расхождения между опытными и расчетными данными по теплоемкости составляют в среднем + 3,9 %
Молекулярный вес определялся криоскопическим методом.
В табл. 9 суммированы результаты экспериментального изучения физико-химических свойств рассматриваемых 8 фракций.
Таблица 9
Физико-химические свойства фракций жидких сланцевых продуктов
№ фракций 1 Зависимость коэффициента теплопроводности zt от температуры Зависимость плотности ot от температуры Зависимость теплоемкости Ct от температуры М О м 0х
1 0,114 (1—0,00145 Г) 0,9412(1-0,000838 t) 0,382 + 0,00139 / 145 000 0,410
2 0,118(1—0,00125/) 0,9440 (1—0,000806 /) 0,407 + 0,00051 / 165 243 0,439
3 0,112(1-0,000955/) 0,9584 (1—0,000778 /) 0,382 + 0,00118/ 179 261 0,447
4 0,116(1-0,000622 /) 1,0142(1—0,000622 /) 0,428 + 0,000528 / 214 310 0,439
5 0,111 (1—0,000801 /) 0,9588 (1—0,000753 t) 0,419 + 0,000838 / 176 273 0,437
6 0,117(1—0,000591 /) 0,9285(1-0,000888 /) 0,389 + 0,00105 / 138 184 0,410
7 0,120 (1—0,00145 /) 0,9745 (1—0,000821 t) 0,405 + 0,000205 / 142 214 0,407
8 0,121 (1—0,00121 /) 1,0053(1—0,000761 /) 0,420 + 0,000324 / 148 238 0,409
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ ИССЛЕДОВАНИЯ
Некоторые выводы из предложенных расчетных формул Теоретически выведенное уравнение Предводителева
* ZM Qt
и уравнение Варгафтика
Л СР _ J/3
м1/я Qt
(2)
262
Г. Н. Скрынникова
можно привести в соответствие с полученным нами эмпирическим уравнением теплопроводности, выражающим линейную зависимость
at=;0(i • /), (3)
где — температурный коэффициент теплопроводности.
Действительно, подставляя в уравнения (1) и (2) значение плотности
et=e0(i — аС
получаем из уравнения (1)
АС,.
^ = ~zm-QoO ~at), (4)
а из уравнения (2)
(5)
Из уравнения (4) следует, что
Разложив (1—а/)4/з в ряд и, отбрасывая члены высшего порядка, в первом приближении получим:
(1 — а/)4'3 (1----at; .
\ о z
Если считать в уравнении (5), что
то уравнение (5) может быть переписано в следующем виде 4
^ = 4(1 - f <*t>- (6)
Таким образом, оказывается, что температурный коэффициент теплопроводности уравнения (3) численно равен модулю расширения (из соотношения Предводителева) или 4/з модуля расширения (из соотношения Варгафтика), т. е.
= а или Р — i/3a. (7)
Указанные равенства представляют большой практический интерес.
Определение модуля расширения производится путем измерения плотности легко и с достаточно высокой точностью.
Имея это в виду, мы попытались проверить равенство (7) на имеющемся в литературе и полученном нами опытном материале.
Исследование коэффициента теплопроводности
263
В табл. 10 произведено сравнение модуля расширения и температурного коэффициента теплопроводности. Таблица составлена по результатам проведенных нами исследований для жидких сланцевых продуктов и по данным Варгафтика и Чернеевой для теплопроводности жидких индивидуальных соединений. Значения модуля расширения были рассчитаны по уравнениям для зависимости плотности от температуры, взятым из Технической энциклопедии [10] и частично дополнены нашими измерениями.
Таблица 10
Сравнение температурного коэффициента теплопроводности с модулем расширения
Вещество СО 1 © т—1 С СО 1 О СО 1 О С со % расхождения между а и р % расхождения между Р и 4/3 а
0,838 1,49 1,12 37,0 24,8
0,806 1,25 1,07 35,5 14,4
0,778 0,855 1,03 18,5 7,8
Фракции жидких сланцевых 0,736 0,620 0,881 18,7 42,0
продуктов 0,755 0,801 1,006 5,7 2,5
0,821 1,45 1,09 43,4 24,8
0,761 1,21 1,01 37,1 16,5
0,776 0,683 1,03 13,3 50,00
Бутиловый спирт 0,848 0,988 1,13 14,1 14,30
Толуол 1,02 1,56 1,36 34,6 12,8
Бензол 1,18 1,50 1,57 21,3 4,6
Ксилол 0,966 1,65 1,29 41,5 21,8
Нитробензол 0,807 1,19 1,08 32,1 9,2
Анилин 0,833 1,08 1,11 22,8 2,08
Касторовое масло 0,661 0,633 0,881 4,42 39,5
Вазелиновое масло 0,566 0,535 0,733 5,79 37,5
Уксусная кислота 0,836 1,00 1,11 16,4 11,0
Муравьиная кислота 0,981 1,33 1,31 26,2 1,5
Даутерм 0,761 1,23 1,02 38,2 17,0
Как видно из табл. 10, температурный коэффициент теплопроводности jS не равен модулю расширения и в отдельных случаях расхождение достигает 30—40%.
Анализ проведенных измерений и расчетов показал, что при существующих методах определения теплопроводности с точностью +3%, возможные колебания в точности величины температурного коэффициента теплопроводности достигают 40—50%.
Из табл. 10 следует, что установленные на основании опытных данных расхождения между величинами а и Д лежат в пределах этой точности.
264
Г. Н. Скрынникова
Следует отметить, что среднее значение температурного коэффициента теплопроводности ближе к 4/з модуля расширения, чем к самому модулю расширения, что видно из следующих данных.
Среднее значение а для 20 веществ ....... ... 0,832 • 10~3
для тех же жидкостей...................... 1,09 • 10—3
4/з« ,, , , ,, ................ 1,09 -ИГ3
Нами были подсчитаны значения коэффициента теплопроводности по трем уравнениям при температурах 20° и 100° С.
I Я* = Я0(1-#);
II 2t = Я0 (1 — а/);
ш at=20'/i — 4-<4,
\ 3 /
где: а — модуль расширения;
Р — температурный коэффициент теплопроводности.
Таблица 7 7
Подсчеты теплопроводности при помощи модуля расширения и температурного коэффициента теплопроводности
Вещество Теплопроводность при 20° С по уравнению Расхождение в % между Теплопроводность при 100° С по уравнению Расхождение в % между
I П III I и II I и III I II III I и II I и III
0,111 0,112 0,111 0,9 0,0 0,097 0,104 0,101 7,2 4,1
0,116 0,117 0,116 0,86 0,0 0,103 0,108 0,101 4,8 2,9
0,110 0,106 0,110 3,6 0,0 0,101 0,103 0,101 1,9 0,0
Фракции жидких слан- 0,114 0,114 0,114 0,0 0,0 0,109 0,107 0,105 1,8 3,67
цевых продуктов . 0,109 0,109 0,109 0,0 0,0 0,102 0,102 0,100 0,0 1,96
0,116 0,115 0,114 0,86 1,72 0,110 0,106 0,103 2,72 6,2
0,116 0,118 0,117 1,72 0,86 0,103 0,110 0,107 6,8 3,9
0,118 0,119 0,118 0,84 0,0 0,106 0,110 0,109 3,7 2,8
Бутиловый спирт . . 0,130 0,130 0,130 0,0 0,0 0,120 0,120 0,116 0,0 3,3
Толуол 0,117 0,118 0,118 0,85 0,85 0,102 0,108 0,104 5,88 1,9
Бензол 0,126 0,126 0,125 0,0 0,8 0,110 0,115 0,110 4,5 0,0
Нитробензол .... 0,133 0,133 0,132 0,0 0,75 0,120 0,122 0,118 1,66 1,6
Анилин 0,157 0,158 0,157 0,63 0,0 0,148 0,147 0,142 0,66 4,0
Ксилол 0,115 0,117 0,114 1,7 0,86 0,100 0,109 0,105 9,0 5,0
Вазелиновое масло 0,106 0,105 0,105 0,94 0,94 0,097 0,099 0,097 2,0 0,0
Уксусная кислота . . 0,157 0,158 0.157 0,68 0,0 0,148 0,147 0,142 0,68 4,1
Муравьиная кислота . 0,218 0,219 0,216 0,4 0,9 0,194 0,197 0,184 1,5 5,19
Среднее 0,81 0,45 Среднее 3,6 2,98
Исследование
коэффициента
теплопроводности
265
Результаты подсчетов представлены в табл. 11, которая показывает, что модуль расширения может быть использован для расчета коэффициента теплопроводности при различных температурах.
Замена температурного коэффициента теплопроводности на модуль расширения изменяет значение теплопроводности в среднем на +3,0% при 100° С, в то время как замена на 4/з модуля расширения при той же температуре дает среднее расхождение на +2,7%.
Применимость расчета теплопроводности жидких продуктов переработки сланцев по теоретическим и эмпирическим формулам
(формуле Н. Б. Варгафтика) и диаграмме Д. К. Коллерова
Расчет коэффициента А по опытным дан-н ы м. На основании опытного материала по физико-химическим свойствам изученных фракций был подсчитан коэффициент А в формуле Варгафтика
4, АСП<+3
я=в<?4/з=—р^~-
откуда:
Результаты подсчета представлены в табл. 12.
Таблица 12
Подсчет ко эффициента в формуле Варгафтика для расчета теплопроводности жидких сланцевых веществ_________________
Фракции /°C Др.10~3
№ \(М — 145; Ср20 — 0,410) | 0 100 0,9224 0,8208 4,71 3,30
№ 2(М -165; Ср20 -0,418) | 0 100 0,9264 0,8278 4,76 4,08
№ 3(М - 179; Ср20 — 0,405) | 0 100 0,9440 0,8440 4,86 3,77
№ 4 (А4 -215; Ср20 — 0,437) { 0 100 1,0190 0,9202 4,42 4,13
№ 5(М — 176; Ср20 — 0,436) | 0 100 0,9447 0,8512 4,36 3,36
№ 6(М - 138; Ср20 — 0,410) | 0 100 0,9058 0,8000 4,57 4,00
№ 7 (М — 142; Ср20 — 0,407) | 0 100 0,9757 0,8619 4,40 3,89
№ 8 (М — 148; Ср20 — 0,409) | 0 100 1,0070 0,9062 4,32 4,09
266 Г. Н. Скрынникова
На основании табл. 12 можно заключить, что формула Вар-гафтика применима для расчета жидких сланцевых продуктов.
Средний коэффициент А, подсчитанный на основании опытных данных для сланцевых продуктов при 30° С, равен 4-27 -10- , в то время как Варгафтик для той же температуры рекомендует 4 28 • 10~3 .
Проверка применимости диаграммы Д. К. К о л-леровадля расчета теплопроводности жидких сланцевых продуктов. В основу диаграммы Д. К. Кол-лерова для коэффициента теплопроводности была положена формула Н. Б. Варгафтика
= — BQt, где
В = А0СР0М-1/зр10/з.
Построение диаграммы теплопроводности было проведено только по данным стандартной плотности е|° и средним температурам кипения на основании установленных Д. К- Коллеро-вым взаимосвязей между этими величинами и другими основными свойствами жидкостей, представленных в виде расчетных диаграмм (плотности, молекулярного веса, теплоемкости и модуля расширения).
В табл. 13 собраны данные: 1) по стандартной плотности
j/ у7-
2) средней температуре кипения, 3) показателю К = —,
Bi
4) коэффициенту В, взятому по диаграмме, 5) теплопроводности, рассчитанной по диаграмме и 6) теплопроводности на основании опытных данных.
В качестве надежных опытных данных были использованы данные Варгафтика, Кей и Хигенса и Вебера.
Диаграмма проверялась на 29 жидких продуктах.
Результаты представлены в табл. 13, в 7-й графе которой данные автора, Варгафтика и Вебера отмечены, соответственно, цифрами 1, 2 и 3, заключенными в скобки.
Проверкой диаграммы на 29 жидкостях установлено, что по диаграмме Д. К. Коллерова можно рассчитывать теплопроводность жидкостей, так как средний процент отклонения от. надежных опытных данных не превышает 4,8.
Для жидких сланцевых продуктов теплопроводность, рассчитанная по диаграмме, дает хорошую сходимость с опытными данными и при высоких температурах. Среднее расхождение равно 2,7%'.
Исследование коэффициента теплопроводности
267
Таблица 13
Проверка диаграммы Д. К. Коллерова для теплопроводности жидких продуктов
Вещество io Vi о 20 к В t Расхождение
222 0,9274 8,4 0,120 0,113 0,114 1 0 0,9
0,120 0,103 0,097 1 100 6,2
243 0,9305 8,5 0,119 0,112 0,118 1 0 5,0
0,103 0,103 1 100 0,0
261 0,9455 8,7 0,114 0,109 0,112 1 0 2,6
0,101 0,101 100 0,0
305 1,0142 8,2 0,114 0,115 0,116 1 0 0,86
Фракции жидких слан- 0,103 0,107 1 100 3,7
цевых продуктов . 273 0,9584 8,5 0,125 0,109 0,111 1 0 1,8
0,101 0,102 1 100 0,9
184 0,9285 8,3 0,123 0,117 0,117 1 0 0,0
0,114 0,110 1 100 3,6
214 0,9795 8,0 0,123 0,119 0,120 1 0 0,83
0,110 0,103 1 100 6,8
239 1,0053 8,1 0,124 0,124 0,121 1 0 2,4
0,115 0,107 1 100 7,5
Бензол ........ 80 0,879 8,1 0,138 0,123 0,123 2 30 0,0
Толуол 110,8 0,860 8,4 0,132 0,113 0,115 2 30 1,7
Ксилол 139,3 0,880 8,5 0,128 0,112 0,113 2 30 0,9
Даутерм 258 1,060 7,6 0,132 0,139 0,116 2 30 19,8
Вазелиновое масло . . . 340 0,882 9,7 0,101 0,089 0,107 2 30 16,8
Этилбензол 136,2 0,867 8,6 0,128 0,11 0,109 3 30 0,9
Этилацетат 77,1 0,901 8,3 0,148 0,128 0,138 3 30 7,1
Пропилацетат .... - 101,7 0,886 8,3 0,134 0,118 0,117 3 30 0,8
Метилбутират 102,3 0,898 8,0 0,136 0,121 0,120 3 30 0,8
Этилбутират 121 0,894 8,4 0,135 0,120 0,120 3 30 0,0
Метилвалервт 127 0,910 8,1 0,132 0,119 0,124 3 30 4,0
Этилвалерат 145 0,877 8,5 0,127 0,109 0,110 3 30 0,9
Гексан 69 0,659 10,5 0,155 0,098 0,087 3 30 12,6
Октан 125 0,703 10,5 0,136 0,096 0,077 3 30 24,7
Нонан 150,7 0,718 10,5 0,134 0,096 0,083 3 30 15,6
Г ептан 98,4 0,684 10,5 0,146 0,092 0,085 [3 30 8,2
Додекан 214 0,749 10,4 0,128 0,096 0,083 [3 30 15,6
Масляная кислота . . . 162,5 0,959 7,9 0,132 0,127 0,129 3 30 1,5
Изомасляная кислота 154 0,949 7,95 0,131 0,124 0,122 3 30 1,61
Валериановая кислота 187 0,939 8,3 0,125 0,117 0,117 3 30 0,0
Изовалериановая кислота 176 0,995 8,2 0,127 0,119 0,123 [3 30 3,2
Простота расчета и использование в нем двух простых величин, легко определяемых в условиях заводских лабораторий — стандартной плотности (?|° и среднеобъемной температуры кипения — дают основание рекомендовать диаграмму Д. К- Коллерова для определения теплопроводности жидких сланцевых продуктов.
268
Г. И. Скрынникова
ЗАКЛЮЧЕНИЕ
Проведено экспериментальное исследование теплопроводности жидких сланцевых продуктов методом нагретой нити при одновременном определении других физико-химических свойств (плотности, вязкости, теплоемкости, в зависимости от температуры, среднеобъемных температур кипения и молекулярного веса).
На основании анализа полученных данных и их сопоставления с опытными данными для чистых веществ установлено, что:
1) для расчета теплопроводности жидких продуктов при различных температурах может быть использован модуль расширения;
2) для расчета теплопроводности жидких сланцевых продуктов может быть использована формула Н. Б. Варгафтика;
3) теплопроводность новых жидких сланцевых продуктов целесообразно рассчитывать по диаграмме Д. К. Коллерова, так как для расчета по этой диаграмме необходимо знание стандартной плотности q^° и среднеобъемной температуры кипения, определение которых не представляет труда и не требует большой затраты времени и применения сложных установок.
ЛИТЕРАТУРА
1. А. С. Предводите л ев. Журн. физ. химии, XXII, № 3, стр. 339, 1948.
2. Д. Л. Т и м р о т и Н. Б. В а р г а ф т и к. Журн. техн, физики, IX, 1940.
3. Н. Б. Варгафтик. Известия ВТИ, № 8, 1949.
4. А. С. Предводителе в. ДАН СССР. Новая серия, т. 72, № 2, 1950.
5. Д. К. К о л л е ‘р о в. Физико-химические свойства жидких сланцевых и каменноугольных продуктов. Гостоптехиздат, 1951.
6. Kraussold. Forschung, 4, 351, 1934.
7. А. К. Аба с-Заде. Журн. физ. химии, XXII, 6, 737, 1949.
8. Бриджмен. Физика высоких давлений. ОНТИ, 1935.
9. W eber. Ann. d Pays., о4, 325, 1917.
10. Техническая энциклопедия, т. V.
11. М. М. Попов. Термометрия и калориметрия. Гостоптехиздат, 1934.
12. А. Н. Щука ре в. Испытания теплотворной способности топлива. Техиздат, 1928.
И. Н. Каменская
ПРЕВРАЩЕНИЯ АЛИФАТИЧЕСКИХ КЕТОНОВ
НАД АЛЮМОСИЛИКАТНЫМИ КАТАЛИЗАТОРАМИ
Превращения алифатических кетонов над алюмосиликатными катализаторами представляют большой практический и теоретический интерес с точки зрения выяснения:
1) происхождения циклических и других соединений, содержащихся в сланцевой смоле;
2) механизма стабилизации фракций сланцевой смолы и
3) возможности образования нефти из «протонефти» согласно гипотезам А. Ф. Добрянского и А. В. Фроста.
Задачей настоящей работы являлось выяснение качественных изменений индивидуальных кетонов — ацетона, метилэтилкетона и ацетофенона — над алюмосиликатными катализаторами при прогрессирующей интенсивности процесса (при увеличении времени контакта).
Работы А. В. Фроста с сотрудниками [1, 2, 3] нельзя признать исчерпывающими в этом отношении, так как предложенный авторами механизм превращения кетонов с образованием кислот и алкенов может служить объяснением только одного из возможных направлений реакции и не охватывает всей совокупности процессов изменения кетонов над алюмосиликатными катализаторами. Кроме того, предложенная авторами схема образования продуктов реакции (кислоты и алкена) не подкреплена выделением предполагаемых промежуточных продуктов реакций — непредельных кетонов или сложных эфиров.
В отличие от опытов, проведенных А. В. Фростом с сотрудниками при атмосферном давлении в динамических условиях протекания кетона через слой катализатора, наши опыты проводились в статических условиях, в автоклаве под давлением паров реагентов при выбранной температуре (220—250° С).
В качестве катализаторов в работе были использованы кембрийская голубая глина, гумбрин и сланцевая зола из топки ТЭЦ. Предварительные опыты по превращению ацетона над кембрийской голубой глиной и гумбрином показали, что
270
И. Н. Каменская
в равных, выбранных нами условиях, кембрийская голубая глина является катализатором менее активным, чем гумбрин. Поэтому основная часть опытов по превращению ацетона, ме-тилэтилкетона и ацетофенона проводилась с кембрийской голубой глиной с целью облегчения задачи выделения первичных продуктов реакции. По ходу исследования интересно и необходимо было изучить продукты превращения промежуточных соединений— окиси мезитила (фр. 125—127° С) и гомофорона (фр. 206—210° С), а также продукты превращения ацетона (или другого кетона) над сланцевой золой.
Исследование продуктов превращения ацетона, метилэтил-кетона и ацетофенона над кембрийской голубой глиной при температуре 220—250° С и времени контакта 4 и 10 часов (6 часов для ацетофенона) показало, что основной реакцией является реакция дегидратации с образованием продуктов конденсации двух и трех молекул исходных кетонов.
Продуктам дегидратационной конденсации ацетона соответствовали: окись мезитила, мезитилен, изофорон и ксилитон; продуктам дегидратационной конденсации метилэтилкетона соответствовали гомомезитоны (З-метил-З-гептенон-5 и 3,4-диметил-3-гексенон-2), гомофорон и гомоизофорон; продукту дегидратационной конденсации ацетофенона соответствовал трифенил-бензол.
Наряду с продуктами конденсации в состав продуктов превращения кетонов неизменно также входили алкены, кислоты, газы, содержащие углекислоту, водород и т. д. и смолообразные кислородсодержащие продукты.
Из рассмотрения опытных данных было видно, что при увеличении продолжительности контакта, одновременно с уменьшением выходов продуктов конденсации, соответствующих двум молекулам исходных кетонов, увеличивались выходы продуктов конденсации, соответствующих трем молекулам исходных кетонов, а также выходы алкенов, кислот и смолообразных продуктов реакции.
Так, при переходе от четырехчасовой продолжительности контакта к десятичасовой, в опыте с ацетоном, одновременно с уменьшением выхода окиси мезитила, увеличивались выходы мезитилена, изобутилена в газе, уксусной кислоты и смолообразных продуктов реакции; в опыте с метилэтилкетоном, наряду с уменьшением выхода гомомезитонов, наблюдалось увеличение выходов гомофорона, гомоизофорона, алкенов (2-метилбутена- 1 и З-метилпентена-2), кислот (уксусной и пропионовой) и смолообразных продуктов реакции. Ароматический углеводород, аналог мезитилена для метилэтилкетона, отсутствовал в катализате метилэтилкетона, полученном над кембрийской голубой глиной.
Превращения алифатических кетонов
271
Во фракции гомофорона, кипящей в пределах 206—210° С? гомофорон может быть представлен пятью изомерами:
I. СН3—СН2-С = С— СО-СН=С—СН2-СН,
СН3 СН3 СН3
II. сн3- сн2—с=сн—с=сн—со—СН2 СН3
СНз сн2
СНз
III. СН3—СН2—С=СН—С = С—СО—СН3
СНз СН2 СНз
СНз
IV. СН3—СН2—С = С—С=СН—СО—СН2—СН3
Z Z I
СН3 СНз СНз
V. СНз—СН2—С=С—С = С-СО—СНз
Z Z I I
СН3 СН3 СНз СНз
Изомеры гомофорона можно рассматривать как промежуточные продукты при образовании ароматических углеводородов из метилэтилкетона. Тогда, дальнейшая дегидратация гомофорона должна была бы привести к образованию ароматических углеводородов.
Опыт, проведенный с гомофороном над кембрийской голубой глиной при температуре 300° С и времени контакта 10 часов, показал, что ароматических углеводородов в этих условиях не образовалось; продуктами реакции были гомоизофорон и кислоты — уксусная и пропионовая.
Образование кислот указывало на протекание реакций гидролитического расщепления изомеров гомофорона по схеме А. В. Фроста, аналогично гидролитическому расщеплению окиси мезитила:
СНз—СН2—С=СН—С=СН—СО—СН2—СНз+Н2О ->
I I
СНз СН2
СНз
II изомер гомофороиа
-> СНз—сн2—с=сн—с=сн2 +сн3-сн2—соон
I I пропионовая кислота
СН3 СН2
сн,
2-этил-4-метилгексадиен-1, 3
272
И. Н. Каменская
СН8—СН2—С=СН—С = С-СО—CHS+H2o ->
I I I
CHS СН3 сн3
сн3
III изомер гомофорона
->сн3— сн2—с=сн—с = сн—сн3 +сн3—соон
1 I уксусная кислота
сн3 сн2
сн3
3-этил-~-мстилгепта.тнен-2. 4
Таким образом условия превращения гомофорона над кембрийской голубой глиной были недостаточными для образования из него ароматических углеводородов, но оказались достаточными для гидролитического расщепления, приводящего к образованию кислот (уксусной и пропионовой).
Для выяснения возможности образования ароматических углеводородов, соответствующих продуктам конденсации трех молекул метилэтилкетона, был поставлен опыт по превращению метилэтилкетона над гумбрином при температуре 300° С и времени контакта 10 часов.
Катализат метилэтилкетона над гумбрином, в отличие от катализата метилэтилкетона над кембрийской голубой глиной, не содержал кислородных соединений — гомофорона, гомоизофорона, — а состоял из углеводородов. Применение более активного дегидратирующего катализатора — гумбрина — позволило осуществить синтез из метилэтилкетона ароматических углеводородов: 1, 3, 5-триэтилбензола; 1, 2-диметил-З, 5-диэтилбензола; 1, 2, 3, 4-тетраметил-5-этилбензола.
Образование ароматических углеводородов в ходе ступен-чато-протекающей реакции дегидратации метилэтилкетона можно представить схемой, приведенной на стр. 273.
Следовательно, на пути образования ароматических углеводородов из кетонов по мере их усложнения (переход от ацетона к метилэтилкетону) встает препятствие в виде гидролитического расщепления a,ft -непредельных кетонов, или диенонов, которые можно рассматривать как исходные вещества для построения ароматических углеводородов.
Течение реакции гидролитического расщепления а, /?-непре-дельных кетонов с образованием алкенов и кислот—схема А. В. Фроста [1, 2, 3] — подтверждается также произведенным расчетом количественных соотношений главных продуктов реакции в опытах по превращению ацетона и метилэтилкетона над кембрийской голубой глиной. При рассмотрении природы смолообразных продуктов реакции, снимаемых с кембрийской голубой глины растворителем, обращает на себя внимание факт
IS Труды ВНИИПС.
Схема образования ароматических сн, С,Н5 1 1 С»Н,—СН-СН-С=СН—СО—сан5 II изомер гомофорона сн, С2Н5 CHS 1 1 / с3н6—с=сн—с=с—со—сн, HI изомер гомофорона сн,сн,сн3 С3Н5— с = с - С=СН—СО—С,Н- IV изомер гомофорона СН, сн, СН, СН, 1111 сн,—сн2-с=с —с = с—со—сн, V изомер гомофорона углеводородов в ходе ступенчато-протекающей реакции дегидратаций метилэтилкетона ^аН5 —-С2Н5 \/ “ 1 с,н, *****.^в "** /У /> з-триэтилбензол сн, ~нго сн,-А 1, 2-диметил-З,5-днэтилбензол -н, 0 сн,-/\-сн, - ' ——* C2HS—сн3 1, 2» 3, 4-тетраметнл-5-этилбензол СНд -нп сн,-' 1-сн, £ X/ сн, гексаметилбензол
И. Н. Каменская
близости элементарного состава смолообразных продуктов к элементарному составу а, /3-непредельных кетонов.
Большое количество аналогичных по элементарному составу смолообразных продуктов реакции было получено в опытах по превращению окиси мезитила над кембрийской голубой глиной при времени контакта 6 часов.
Окисление смолообразных продуктов реакции, снятых с кембрийской голубой глины в опытах с ацетоном и окисью мезитила, привело к образованию щавелевой и уксусной кислот — продуктов окисления окиси мезитила.
Анализы смолообразных продуктов и соотношения главных продуктов реакции дали все основания рассматривать смолообразные продукты как состоящие, в основном, из полимеров. а, /3-непредельных кетонов (двух и трех молекул). Очевидно, полимеризация а, -непредельных кетонов, наряду с гидролизом их, служила препятствием образованию ароматических углеводородов из кетонов. Смолообразные продукты реакции претерпевали, в свою очередь, изменения над алюмосиликатными катализаторами, на что указывали изменения в составе газа и в составе смолообразных продуктов реакции при изменении условий превращения кетонов.
Исследование продуктов превращения ацетона над сланцевой золой при температуре 220—250° С и времени контакта 10 часов показало, что последняя также является дегидратирующим катализатором.
В продуктах реакции установлено наличие окиси мезитила (5,5%, считая на катализат, отдутый водяным паром), форона (2,7%), изофорона (32%) и ксилитона (8,3%).
Мезитилена в катализате не обнаружено.
Избирательное направление конденсации ацетона в сторону образования алкилированных циклогексенонов (40%, считая на конденсат) связано, очевидно, с наличием в составе сланцевой золы щелочных компонентов (СаО).
Необходимо также отметить, что основное направление изменения кетонов в условиях отсутствия катализаторов не менялось: реакция дегидратационной конденсации наблюдалась в холостых опытах с ацетоном и ацетофеноном, причем глубина изменения составляла 4—8%, считая на исходный кетон.
ОБЪЯСНЕНИЕ ДВУХ НАПРАВЛЕНИЙ РЕАКЦИИ ГИДРАТАЦИИ ОКИСИ МЕЗИТИЛА
Образование ацетона (13,5%), наряду с изобутиленом и уксусной кислотой (4,36%), в опыте по превращению окиси мезитила над кембрийской голубой глиной объяснялось распределением электрических зарядов у первичного продукта гидра-
Превращения алифатических кетонов
275
тации окиси мезитила-диацетонового спирта под влиянием нуклеофильных заместителей:
СН3. СН8.
I. }С=СН-СО-СН3+Н2О -> \С—СН,—СО—СН,
сн3/ сн3/ I
он
снзх+ + + _
>С—сн,-с—СН3+ н—он
СН/ I II
4- —
Н—ОН
СНЗЧ + _ + снзч
)С—СН2—С-СН3-> )С=СН2 + СН3СООН + Н2О
СН/ I II сн/
он °
он н
СН3 + - +
)С—СН„—С- СН3 -> 2СН3СОСН3+Н2О
Cl/ / II
он ~о
ОБЪЯСНЕНИЕ АКТИВНОСТИ АЛЮМОСИЛИКАТНЫХ КАТАЛИЗАТОРОВ
Меньшая активность кембрийской голубой глины, по сравнению с гумбрином, может найти объяснение в рассмотрении минералогического состава этих алюмосиликатов и природы каталитического действия алюмосиликата вообще [4].
Алюмосиликат — кислота, каталитическое действие которой обусловлено наличием иона Н (как известно, щелочные агенты являются ядами алюмосиликатов).
Минералы монтмориллонитовой группы — наиболее активные алюмосиликатные катализаторы — имеют состав, отвечающий следующей формуле:
А12О3 4SiO2 Н2О nRSiO3 mH2O. [5]
Отношение окиси алюминия к окиси кремния в монтмориллоните равно 1 : 4. Кристаллическая решетка монтмориллонита отвечает полному замещению атомов кремния атомами алюминия в кремнекислородных тетраэдрах. Результатом этого является увеличение отрицательного заряда тетраэдра на еди-18*
276
И. Н. Каменская
ницу и возможность насыщения его водородом. Как известно, гумбрин является представителем такого типа группы минералов.
В состав кембрийской голубой глины входят минералы хлоритовой группы и групп слюды и полевых шпатов [6].
Кристаллические решетки минералов групп хлоритовой, слюды и полевых шпатов характеризуются лишь частичным замещением кремния алюминием в кремнекислородных тетраэдрах [7]. Отсюда и меньшая возможность насыщения отрицательных зарядов тетраэдров водородом, а следовательно, и образования молекул алюмосиликатной кислоты.
ОПЫТНАЯ ЧАСТЬ
Методика исследования
Кетон и катализатор в весовом отношении 1 : 1 загружались в автоклав, в котором поддерживалось давление, равное давлению паров реагентов при температуре 220—250° Сив отдельных опытах при 300° С. Из охлажденного автоклава при комнатной температуре газ собирался и замерялся в газометре и анализировался на приборе ВТИ сернокислотным способом по А. Ф. Добрянскому. Жидкие продукты реакции отгонялись с водяным паром и фракционировались на лабораторной колонке. Узкие фракции, достаточно крупные по количеству, характеризовались (физическими и химическими константами. Неотгоняемая с водяным паром часть продуктов реакции снималась с катализатора растворителем в экстракционном аппарате, замерялась и анализировалась.
С целью накопления материала для каждого кетона в выбранных условиях проводилось по пять-шесть опытов. Емкость автоклава была равна 1 л, весовые количества кетона и катализатора — 250 г.
Катализаторы — глина месторождения Гумбри (Кутаиси) и кембрийская голубая глина (Ленинград) — активировались путем обработки 10%-ной серной кислотой при 100—105° С в течение 6 часов, с последующей отмывкой кислоты и просушкой при температуре 300° С.
Катализатор формовался в виде цилиндров, имеющих размеры: I = 4—5 мм, d — З мм.
Кислоты идентифицировались, где это было возможно, по температурам кипения, и во всех случаях—с помощью бариевых и серебряных солей.
Во избежание ошибки в количественном определении кислот за счет их связывания с железом автоклава, проводилось на
Превращения алифатических кетонов
277
гревание отдельных навесок кетона и катализатора в стеклянных запаянных ампулах в реакционной зоне. После охлаждения автоклава и вскрытия ампул количества уксусной и бензойной кислот определялись титрованием, по Фрезениусу [15], водного раствора, полученного при перегонке с паром содержимого ампул.
Раздельное количественное определение уксусной и пропионовой кислот производилось путем получения основных свинцовых солей по методике, описанной С. И. Сухановским и Е. В. Рогинской [8].
Характеристика исходных кетонов, катализаторов, а также условия и результаты опытов по превращению кетонов и характеристики продуктов превращения кетонов над различными алюмосиликатными катализаторами приводятся в помещаемых ниже табл. 1—20.
Таблица 1
Характеристика исходных кетонов1
Наименование кетона Температура кипения, °C ,20 d20 20 nD Литературная ссылка
Ацетон 56 (56,2) 0,7870 (0,787) 1,36152 (1,36157) 2 9
Окись мезитила 129—130 (129,5; 130,1) 0,863 (0,86532) 1,4446 (1,44478) 9
Метилэтилкетон 78—79 (79,6) 0,8060 (0,80591) 1,38140 (1,38140) 2 9
Гомофорон 206—210 (206—210) 0,8918 (0,8857) 1,4796 (1,4792) 16
Ацетофенон 200 (202,0) 1,025 3 (1,0236) 3 1,53416 (1,53418) 9
1 В графах 2—4 под экспериментальными данными помещены в скобках соответствующие литературные данные.
2 nD-
3 d25.
25
27S
И. Н. Каменская
Химический состав катализатора
Состав, вес. %
Наименование
катализатора
Гумбрин неактивированный Гумбрин активированный Кембрийская голубая глина неактивированная . . . .
Кембрийская голубая глина активированная ..........
Сланцевая зола с ТЭЦ . .
Таблица
5,62
6,75
6,96
6,83 10,9
Таблица 3
Условия и результаты опытов по превращению ацетона над кембрийской голубой глиной
Общие условия опытов: весовое отношение ацетона к катализатору 1 :1; температура 220—250° С
Контролируемая величина (показатель) % от исходного ацетона при времени контакта
4 часа 10 часов
Продукты превращения ацетона:
Газ:
общее количество 0,13 0,66
изобутилен . 0,046 0,130
Катализат, отдутый водяным паром:
общее количество 8,70 11,20 1 вл
окись мезитила (125—128° С) . . 4,60 4 9Q
мезитилен (160—162° С) 1,03 1 1 ВЛ
Уксусная кислота 1,105
Смолообразные продукты, снятые 7 98
с катализатора растворителем . . 2,52 9 65
Углеродистые соединения 4,04 ВО
Непрореагировавший ацетон 80 6
Потери 4
Таблица 4
Характеристика узких фракций Катализата, полученного в опытах по превращению ацетона над кембрийской голубой глиной
Общие условия опытов: весовое отношение ацетона к катализатору 1: Г, температура 220— 250э С
х® О'
СО Фракция катали- X
ЬЙ зата и сравнивав- X та со та Темпера-
X мые с ними СХ ,20
5 х индивидуальные ° г тура 'С
2: та соединения СО СО та * кипения, ’С
Он 5“ о и
X со bi о
4 I 52,8 125-128 0,8591
4 и 9,4 160—162 0,8657
10 I 13,4 125—128 0,8590
10 II 35,7 161-163 68—78 0,8628 0,9001
10 III 5,0
5 мм 128—138
6,0 0,9407
10 IV 5 мм
Окись мезитила . (130,1) (129,5) (0,86532)2
Мезитилен . . . —— (164,8) (0,8634)
Изофорон . . . . (205—210) (0,925)
Ксилитов . . . . — (252—253) (0,936)
Характеристика фракций катализата1
Элементарный состав, % Температура плавления, °C
и20 riD м С Н > семикарба-’ зона 1 4 s ! ’ § ч i 6 о £ s я i a, m V 5 § ! s £ о oi о- 1 та к я тримезино-вой кислоты
Литературная ссылк,
1,4430 98 74,0 10,2 156 201
1,4945 118 III - — — —„
1,4387 97 74,0 10,2 156 201 — —
1,4956 120 90,0 10,0 — 86 375
1,4864 134 80,0 10,32 188 — —
1,5271 170 82,2 10,67 — — — —
(1,448403 (98) (73,4) (Ю,1) 1(156) (201) — — 9
(1,4967) (120) (90,0) (10,0) — — (86) (375) 9,17
(1,4860) (138,1) (78,26) (10,14) (190) — — и
— (178) (80,90) (10,10) — — П
1 В графах 4—13 под экспериментальными данными помещены в скобках соответствующие литературные данные для индивидуальных соединений, указанных в графе 2.
2 d20
3 „13
nD
280
И. Н. Каменская
Таблица 5
Условия и результаты опыта по превращению ацетона над сланцевой золой
Условия опыта: температура 220—250° С; время контакта 10 часов
Контролируемая величина (показатель)
Продукты превращения ацетона:
газ....................................
катализат, отдутый водяным паром . . . . смолообразные продукты, снятые с катализатора растворителем .................
углеродистые соединения ...............
Непрореагировавший ацетон-{-реакционная вода
Потери + газ..................................
% от исходного ацетона
см. ..Потери"
12
6,8
14,4
58,8
8
Таблица 61
Характеристика узких фракций катализата, полученного в опытах по превращению ацетона над сланцевой золой
Условия опыта: весовое отношение ацетона к катализатору 1:1; температура 220—250° С; время контакта 10 часов
CC О , и S 5 2 К О 5 s Температура, °C Элементарный состав, % г- «О Г»,'-» я: «3 о-
Фракции лизата и сравнива । С НИМИ 1 видуальв соединен кипения плавления /720 “20 „20 nD М С Н Температу] плавления карбазона, Литерап ссылка
1 2 3 4 5 6 7 8 9 10
I Окись ( мезитила ( 127—130 (130,1; 129,5) —" 0,8625 (0,8630) 1,4435 (1,44397) 100 (98) 73,21 (73,40) 10,13 (10,10) 156 (156) 9
II Форон | 196—198 (197,2; 198,2) 28 (28) 0,880 (0,885) 1,4833 (1,4998) 134 (138) 79,00 (78,26) 10,23 (10,14) — 9
III Изофорон IV V Ксилитон 206-208 (205-210) 134—140 19 мм 165—170 19 мм (225; 252-253) 0,9256 (0,9255) 0,9358 0,9380 (0,9360) 1,4770 (1,4711) 1,5192 1,5300 138 (138) 175 177 (178) 78,30 (78,26) 81,29 81,30 (80,90) 10,17 (10,14) 10,20 10,32 (10,10) 190 (190) i 11 11
1 В графах 2—9 под экспериментальными данными помещены в скобках соответствующие литературные данные для индивидуальных соединений, указанных в графе 1.
Превращения алифатических кетонов
281
Таблица 7
Условия и результаты опытов по превращению метилэтилкетона (МЭК) над кембрийской голубой глиной
Общие условия опытов: весовое отношение МЭК к катализатору 1:1; температура 220—250° С
% от исходного метилэтилке-
Контролируемая величина тона при времени контакта
(показатель)
4 часа 10 часов
Продукты превращения МЭК:
газ 0,13 0,32
2-Метилбутен-1 0,79 0,275
З-Метилпентен-2 1,57 10,00
уксусная кислота . 1,18 9,40
пропионовая кислота ...... катализат, отдутый водяным паром: 1,00 3,20
общее количество 9,2 10,80
гомомезитоны (157—165°) 5,73 2,62
гомофорон и гомоизофорон .... смолообразные продукты, снятые с. 1,49 4,85
катализатора растворителем . . . 1,10 5,07
углеродистые соединения 3,70 4,00
Непрореагировавший МЭК 70,4 44,5
Потери 11 и Таблица S
Характеристика низкокипящих жидких продуктов превращения метилэтилкетона (МЭК) над кембрийской голубой глиной1
Общие условия опытов: весовое отношение МЭК к катализатору 1:1; температура 220—250° С
Время контакта, в часах Фракции катализата и сравниваемые с ними индивидуальные соединения Характеристика фракций катализата 1 2 Литературная ссылка
Температура кипения J20 “20 л20 nD М Элементарный состав, °/ /о Бромное число |
с Н
4 I II 30—40 65—68 0,6600 0,7061 1,3790 1,4213 70;75 82;86 85,53 85,43 14,19 14,20 222 186
10 I II 30—40 65—68 0,6593 0,7061 1,3792 1,4210 70;72 86;88 86,10 85,85 14,00 14,12 220 185
2--Метилбутен-1 З-Метилпентен-2 (29) (31) (0,6504) (65,7) (68,0) (0,6956) (1,3785) (1,4086) (70) 1(85,71) (84) (85,70) (14,29) (14,30) (228) (190) 10 10
1 Продуктов, кипящих до 100°С.
2 В графах 3—9 под экспериментальными данными помещены в скобках соответствующие литературные данные для индивидуальных соединений, указанных в графе 2.
Таблица 9
Характеристика узких фракций катализата, полученного в опытах по превращению метилэтилкетона (МЭК) над кембрийской голубой глиной
Общие условия опытов: весовое отношение МЭК к катализатору 1:1; температура 220—250° С
Время контакта, в часах Фракции катализата и сравниваемые с ними индивидуальные соединения Количество фракции, % от катализата Характеристика фракций катализата 1 Литературная ссылка
Температура кипения, °C d2° а20 п20 nD А1 Элементарный состав, °/ /о Температура плавления семикарбазона, °C 10 Температура плавления дисемикарбазона, °C
с Н 9
1 2 3 4 5 6 7 8 11 12
4 4 4 4 10 10 10 10 1 мещв! I (гомомезитоны) 3,4-диметил - 3-гексе-нон-5 20 157—161 (157 161; 65/20 мм) 0,8618 (0,8660; 0,8683)1 1,4410 (1,4410) 125 (126,11) 76,33 (76,15) 11,13 (11,10) 182 (180) — 9, 18, 19
11 (гомомезитоны) З-метил-З-гептенон-5 Окись гомомезитила 28,8 163—165 (165—167) (66/18 мм; 160) 0,863 (0,83516) (0,862); (0,863)4 1,4440 (1,4503) (1,4476; 1,4451) 126 (126,11) (126,11) 76,42 (76,15) (76,15) 11,07 (11,10) (11,10) 116 (П6) 240 (240) 9, 18, 19 12
III2 Гомофорон 5 206—210 (206—210) 0,8904 (0.8851)4 1,47,96 (1,4792) 180 (180) 79,95 (80,00) 11,02 (Н.1) — — 13, 16
IV2,3 Гомоизофорон и 242—252 (242—252) 0,93944 (0.9492)4 1,50410 (1,5092) 181 (180) 79,84 (80,00) 11,35 (П.1) — — 13
I II III IV В графах 4—11 под экс 1ы в скобках соответст 8 8 9 10 перимс вующи 157—161 163—165 206—210 242—252 штальным? е литерат> 0,8635 0,8597 0,8824 0,9200 данными фные дан 1,4410 1,4421 1,4788 1,5021 но- вые НИЯ 125 125 178 185 2 Опреде хлористс 76,5 76,55 80,44 80,73 ление чис го водоро 11,20 11,20 11,43 11,15 ла двойнь да дало с 181 115 lx связей ледующие путем пр результа исоедине-ты:
для индивидуальных соединении, указанных в графе 2 3 = 57,4; для гомоизофорона R2D = 57,52. Фракция, °C Навеска, Количество присоединивше- Число двойных гося НС1, г связей
206—210 242—252 0,1893 0,1931 0,7668 1 2 0,2835 | 1
Превращения алифатических кетонов
283
Таблица ТО
Условия и результаты опыта по превращению метилэтилкетона (МЭК) над гумбрином
Условия опыта: весовое отношение МЭК к катализатору 1 :1; температура 300° С; время контакта 10 часов
Контролируемая величина (показатель)
Продукты превращения МЭК:
газ ......................................
низкокипящие жидкие продукты (до 100° С) । катализат, отдутый водяным паром . . . . I
уксусная кислота . . ...............:
пропионовая кислота.....................|
смолообразные продукты, снятые с катализа- | тора растворителем .....................
углеродистые соединения ..............
Потери......................................
% от исходного метилэтилкетона
3,2
11,36
18,53
11,52
19,57
14,50
9,25
7,17
Таблица 11
Характеристика низкокипящих жидких продуктов превращения метилэтилкетона (МЭК) иад гумбрином1
Общие условия опыта: весовое отношение МЭК к катализатору 1:1; температура 300° С; время контакта 10 часов
Фракции катализата и сравниваемые с ними индивидуальные соединения Характеристика фракций катализата 2 Литературная ссылка
Температура кипения, °C «20 nD М Бромное число
I 2-метилбутен-1 26-35 (29; 31) 0,6500 (0,6504) 1,3731 (1,3785) 74; 76 (70) 220 (228) 10
II З-метилпентен-2 60—68 (65,7) (68,0) 0,6972 (0,6956) 1,4002 (1,4086) 90 (84) 181 (190) 10
1 Продуктов, кипящих до 100° С.
s В графах 2—6 под экспериментальными данными помещены в скобках соответствующие литературные данные для индивидуальных соединений, указанных в графе 1.
Характеристика продуктов превращения метилэтилкетона (МЭК) над гумбрином ,а0'л
Условия опыта: весовое отношение МЭК к катализатору 1:1; температура 300° С; время контакта 10 часов
Фракции катализата и сравниваемые с ними индивидуальные соединения Количество фракций, % от катализата Характеристика фракций катализата 1 Бромное число Литературная ссылка
Температура кипения, °C ail н20 nD М Элементарный состав, %
С Н
J 2 3 4 5 6 7 8 9 10
1 2,3,4,4-тетраметил гексен-2 2,5 150—160 /45—50 ч \ 15 мм' 0,7676 (0,7697) 1,4278 (1,4385) 144 (140) 85,75 (85,75) 14,0 (14,25) 79,8 (114,2) 10
II 3-этил-6-метил-гептен-2 3,0 160—165 /157—158 \ \ 750 мм ) 0,7774 (0,7520) 1,4331 (1,42708) 148 (140) 85,55 (85,75) 14,25 (14,25) 80,0 (114,2) 10
III С13Н24 из З-метилпентена-2 3,6 85-90 19 мм (196-199) 0,8011 (0,798; 0,809) 1,4540 157 (168) 84,0 (85,72) 13,0 (14,28) 73,4 (Ю8) 20
IV 2,4,4-триметил-2-этилгептен-2 2,2 90-93 19 мм (193-197) 0,8208 (0,786) 1,4599 (1,4510) 163 (168) (85,72) (14,28) 70,5 (Ю8) 10
V 4,0 115-117 19 мм 0,8613 1,4835 183 87,25 12,11 —- —
VI 3,3 128-130 19 мм 0,8680 1,4830 191 87,00 12,88 — —
VII 7,2 133—138 19 мм 0,8749 1,4862 203 88,98 11,00 — —
1 В графах 3—9 под »кспериментальными данными помещены в скобках соответствующие литературные данные для индидидуальных соединений, указанных в графе 1.
Таблица 13
Характеристика продуктов окисления фракций 115—117° (19 мм), 228 и 240—244° С катализата, полученного в опыте по превращению метилэтилкетона (МЭК) над гумбрином1 2
Условия опыта: весовое отношение МЭК к катализатору 1 :1; температура 220—250° С; время контакта 10 часов
Исходные фракции, продукты их окисления и сравниваемые с последними индивидуальные соединения 3 Температура кипения, °C Температура плавления, °C /72° и20 п20 nD м Элементарный состав, % Эмпирическая формула % гидроксильной группы Эквивалент нейтрализации
С Н
Исходная фракция .... 115—117 19 мм —, 0,8613 1,4835 183 87,25 12,11 — — — —
Продукт ее окисления . . 375 с возгонкой —— —. 210 51,85 3,00 — 24,05 —
Тримезиновая кислота . . — (350, 375, 380) — — (2Ю) (51,50) (2,80) — 24,3 — —
Исходная фракция3 . . . . 228 0,8662 1,4902 160 88,88 11,35 С1аН18 — — 53,5
Продукт ее окисления . — 238—240 — — — 47,57 2,38 — — 62,85 •—
Мелофановая кислота . . . — (238,53) — — (47,25) (2,36) — — (63,50) •—
Исходная фракция 4 . 240—244 — 0,8666 1,4967 165 89,23 11,28 Cis^is — — 55,7
Продукт ее окисления - . — 228 — — — 44,34 2,07 — — 59,2 —
Бензол-пентакарбоновая кислота . — (228—230) — -— (44,30) (2,03) — — (59,6) —
1 В скобках помещены литературные данные для соответствующих индивидуальных соединений.
2 Окисление исходных фракций производилось по Ульману.
3 Выделена из фракции 128—130° С (19 мм).
4 Выделена из фракции 133—138° С (19 мм).
286
И. Н. Каменская
Таблица 74
Условия и результаты опыта по превращению гомофорона над кембрийской голубой глиной
Условия опыта: весовое отношение гомофорона к катализатору 1:1; температура 300°; время контакта 6 часов
Фракции катализата и сравниваемые с ними индивидуальные соединения Характеристика фракций катализата 1 Литературная ссылка
Температура кипения, °C U20 п2° nD м Элементарный состав, %
С Н
1 Томом езитон 155—160 0,8625 (0,863) 1,4445 (1,4451) 38,29 (38,70) 124 (126) 76,5 (76,15) 11,29 (И,1) 19
II Гомоизофорон 248-253 0,9406 (0,9492) 1,5035 (1,5045) 56,35 (80,00) 178 (180) 79,39 (80,00) 11,15 (П.1) 13,14
III Гомоизофорон 280—285 0,9695 (0,9693) 1,5163 (1,5115) 58,40 (80,00) 187 (180) 79,69 (80,00) 10,92 (Н,1) 16
1 В графах 3—8 под экспериментальными данными помещены в скобках соответствующие литературные данные, указанные в графе 1.
Таблица Т5
Условия и результаты опыта по превращению окиси мезитила над кембрийской голубой глиной
Общие условия опыта: весовое отношение окиси мезитила к катализатору 1:1; температура опыта 220—250° С; время контакта 6 часов
Контролируемая величина (показатель)
Продт кты превращения окиси мезитила: газ ................... ......................
ацетон ..................................
катализат, отдутый водяным паром . . . . уксусная кислота .......................
смолообразные продукты, снятые с катализатора растворителем ...................
углеродистые соединения ................
Потери.........................................
% от исходной
окиси мезитила
2,75 13,5 28,1
4,36
22,6
6,25 22,5
Превращения алифатических кетонов
287
Таблица 16
Характеристика продуктов превращения окиси мезитила над кембрийской голубой глиной
Условия опыта: весовое отношение окиси мезитила к катализатору 1:1;
температура 220—250° С; время контакта 6 часов
Фракции катализата и сравниваемые с ними индивидуальные соединения Характеристика фракций катализата 1 Литературная । ссылка
Температура кипения, ° С d20 20 20 М Элементарный состав, % Температура i । плавления семикарбазона, °C 1 । Температура плавления динит- I I ропроизводно- 1 го,° С 1
С Н
I Ацетон II Мезитилен 56 (56,2) 164 (164,8) 0,789 (0,787) 0,863 (0,8634) 1,4933 (0,4967) 122 (120) 62,0 (62,15) 90,2 (90,0) 10,15 । 188 (10,34) (188) 10,15 | — (10,0)| - 86 (86) 9 9,17
1 В графах 2—8 под экспериментальными данными помещены в скобках соответствующие литературные данные для индивидуальных соединений, указанных в графе 1.
Таблица 17
Условия и результаты опыта по превращению ацетофенона над кембрийской голубой глиной
Условия опыта: весовое отношение ацетофенона к катализатору 1:1; температура 220—250° С; время контакта 6 часов
Фракции ката- ! “ лизата и 1 5 g сравниваемые с ними инди- § х s видуальные соединения § 2 ° С - У* GJ -1- -S- х Характеристика фракций катализата 1 Литературная ссылка
Температура кипения, °C Температура плавления, °C ,20 d 20 20 м Элементарный состав, % С I Н Бромное число |
1 а-Метилстирол 11 1,0 19,0 55—60 19 мм (161,2) 121 250 0,9095 (0,9134) 1,5300 (1,5384) 122 (И8) 90,55 1 8,45 141 (152,3) 9
(91,5) 69,0 (8,5) 5,0
Бензойная кислота (121) (122) (249,5) — — — (69,0) (4,9) — 9
III 10,0 300 52;53 — — 236 91,60 8,36 0
1, 1,3-триме-тил-3-фенил-гидринден (300) (51) (52) (53) — — (236) (91,52) (8,48) (0) 21
IV2 — — 171 — — 306 94,25 94,25 6,00 5,85 —
Симметричный трифенилбензол —— — (169) (172) — — (306) (94,25) (5,75) — 14
1 В графах 3-8 под экспериментальными данными помещены в скоб-
ках соответствующие литературные данные для индивидуальных соединений, указанных в графе 1.
2 Твердое вещество, выпавшее из катализата и перекристаллизованное из уксусной кислоты.
Таблица 18
Выход и характеристика газообразных продуктов превращения кетонов над алюмосиликатными катализаторами (сводная таблица)
Общие условия превращения: весовое отношение кетона к катализатору 1:1
Условия опыта Состав газа, объемы. % О о и
Исходный кетон Катализатор 1 II Температура, Gc Время контакта, в часах м о о х" о 00 X О 1 X + СО X п и X м о о о и X л + С л X е О Уд. вес газа при 20° и 760 мм Выход газа, % от ш ного кетона
Ацетон ..... I 220-250 4 20,0 33,30 3,33 6,70 3,30 26,70 6,70 1,065 0,13
,, • • • • • • I 220-250 10 49,70 19,70 2,50 0,86 1,14 1,64 21,49 3,60 1,180 0,66
Окись мезитила . . . Метилэтплкетон . . . I I 220-250 220-250 6 4 41,01 53,41 17,70 3,47 30,40 1,77 0,64 1,28 6,38 31,32 4,42 2,83 5,39 1,037 1,038 2,75 0,13
I 220—250 10 16,20 4,70 3,40 2,30 0,70 61,70 69,40 1,60 0,463 0,32
II 300 10 18,40 9,80 9,40 0,50 9,70 9,20 79,00 — 0,317 3,20
Гомофорон I 300 6 — — — — — — — — — 0
Ацетофенон I 220—250 б 2,05 0,90 31,27 52,10 13,68 0,368 0,59
1 I — кембрийская голубая глина.
II — гумбрин.
Таблица 19
19 Труды вниипс
Выход и характеристика смолообразных продуктов превращения кетонов, снятых с катализатора растворителем (сводная таблица)
Общяе условия опытов: весовое отношение кетона к катализатору 1:1; температура 220—250° С 1
Кетой Катализатор 1 Время контакта, в часах Выход „смол“, % от исходного кетона Свойства смолообразных продуктов Продукты окисления „смол“
<?° 20 М Кислотное число Число омыления Бромное число Элементарный состав, %
С Н
Ацетон I 4 2,52 1,005 260 5,2 21,3 82,5 83,37 10,0 Уксусная кислота
Ацетон I 10 7,96 0,976 290 5,6 23,0 73,5 84,97 10,60 Щавелевая кислота. Небольшое количество тримезино-вой кислоты
Окись мезитила I 6 22,50 0,9884 312 25,08 24,21 71,0 85,02 10,25
Метил этил кетон I 4 1,10 0,9820 244 21,00 24,40 84,0 77,33 10,36
I 10 6,07 0,9398 232 19,40 21,20 96,0 83,14 11,30
» II 10 14,50 0,9477 350 32,00 — 36,0 89,55 10,45
1 I — кембрийская голубая глина.
II — гумбрин (прн температуре 300° С).
290
И. Н. Каменская
Таблица 20
Характеристика кислот, отгоняющихся с водяным паром из продуктов превращения кетонов над алюмосвликатными катализаторами
Показатели1
Выделенные фракции кислот и соответствующие индивидуальные соединения
Элементарный состав, %
С Н
% бария в соли
% серебра в соли
Выделенная фракция
уксусной кислоты . . 113 — — —
Уксусная кислота . . (118,1) — — —
Выделенная фракция
пропионовой кислоты — — — —
Пропионовая кислота . — — — —
Выделенная фракция
бензойной кислоты . 249—250 121 69,0 5,0
Бензойная кислота . . (250) (121) (69,0) (4,9)
(122)
54 (53,75)
64,15
(64,15)
58,90
(59,70)
ВЫВОДЫ
1. Проведено сравнительное экспериментальное изучение превращений индивидуальных алифатических кетонов — ацетона, метилэтилкетона и ацетофенона — над типовыми алюмосиликатными катализаторами при температуре 220—250° С и продолжительности контакта 4 и 10 часов.
2. Сравнение полученных результатов показывает, что превращения кетонов в изученных условиях протекают в одних и тех же направлениях и представляют собой сложную совокупность реакций дегидратационной конденсации, гидратации, полимеризации, циклизации, декарбоксилирования и диспропорционирования водорода.
3. Основным направлением является реакция дегидратационной конденсации. Выделены продукты конденсации двух и трех молекул исходного кетона.
4. Предложено электронное истолкование реакций гидратации, происходящих в рассматриваемой системе. Выделены продукты гидролитического расщепления непредельных кетонов — алкены, кислоты — и, в случае расщепления окиси мезитила, — ацетон.
5. Сопоставление состава смолообразных продуктов полиме-
В скобках помещены литературные данные.
Превращения алифатических кетонов
291
ризации, полученных в различных условиях, приводит к заключению, что при прогрессирующей интенсивности процесса (при увеличении продолжительности контакта) происходит обеднение этих продуктов кислородом и обогащение их непредельными соединениями.
6. Сопоставление изменений в составе смолообразных и газообразных продуктов превращения приводит к заключению, что смолообразные продукты, адсорбирующиеся на катализаторе, претерпевают изменения, обусловленные процессами декарбоксилирования и диспропорционирования водорода.
7. Механизм образования ароматического кольца из карбонильного соединения в условиях превращения последнего над алюмосиликатным катализатором может быть представлен следующей схемой ступенчато-протекающей реакции дегидрата-ционной конденсации:
СН3х
2 СН.СОСНз ;с=снсосн3
СН3/
сн3
СНЗЧ „ п снзч I
>С=СНСОСН34-СН3-СО—СН3 >с=сн-с=снсосн3
сн/ сн/
сн3
СН,
/с=сн—с=сн—со—сн3 | I
сн/ СН3 — 1 /- СНз
На пути образования ароматического кольца встают препятствия в виде реакций гидролитического расщепления и полимеризации того диенкетона, который является исходным материалом для построения этого кольца.
СН3 СН3
СНЗХ | /СН3 f \
п >С=СН—С = СНСОСН3-> )С=СН—С=СН—СО—СНз
сн/ 'сн/ /п
где п от 2 до 3
СН3
СНЗЧ | СН3.
)С=СН—С=СН—СО—СН3 +Н2°. )С=СН-С=СН2+СН3СООН СН3/ СН3/ I
СНз
8. Предложено основанное на сопоставлении минералогических характеристик (особенностей кристаллической решетки) испытанных алюмосиликатных катализаторов объяснение того факта, что при прочих равных условиях гумбрин обладает более
19*
292
И. Н. Каменская
высокой дегидратирующей активностью, чем кембрийская голубая глина. Что касается образования ароматических углеводородов в результате дегидратационной конденсации трех молекул исходного кетона (отщепления трех молекул воды), то оно обус-
Рис. 1. Схема превращения алифатических кетонов над алюмосиликатными катализаторами.
ловливается не только «кислотной» активностью алюмосиликатного катализатора, но и положением алифатического кетона в гомологическом ряду.
9. Результаты исследования общей совокупности превращений алифатических кетонов над алюмосиликатными катализаторами при температуре 250° С, соответствующем этой температуре давлении паров реагентов и прогрессирующей продолжительности контакта (экспериментально исследовано влияние продолжительности контакта в пределах 10 часов) могут быть представлены в виде схемы, изображенной на рис. 1.
Превращения алифатических кетонов
293
10. Превращение ацетона над сланцевой золой может также рассматриваться и интерпретироваться на основе приведенной схемы (рис. 1), причем и в этом случае основным направлением является ступенчато-протекающая дегидратационная конденсация ацетона, приводящая к образованию циклогексенонов, изофорона и ксилитона (крайнее правое направление в схеме).
11. Обнаруженные закономерности в поведении алифатических кетонов над алюмосиликатными катализаторами могут найти применение в объяснении механизма стабилизации фракций сланцевой смолы и образования в промышленных условиях многих соединений, входящих в состав последней, поскольку общеизвестна та значительная роль, которую играют кетоны в сланцевой смоле и подсмольной воде.
Не лишено также интереса использование изложенного выше материала при рассмотрении возможных химических источников образования нефтей в свете гипотез А. В. Фроста и А. Ф. Добрянского.
ЛИТЕРАТУРА
1. А. В. Фрост, А. Я. Ларин. ДАН СССР, 59, № 7, 1927, 1948.
2. А. В. Ф р о с т, А. Я. Ларин. ДАН СССР, 54, № 5, 411, 1946.
3. А. В. Фрост. Проблемы кинетики и катализа, т. IV, изд, АН СССР, 1949.
4. А. П. Б а л л о д, К- В. Топчиева. «Успехи химии», 20, вып. 2, 161, 1951.
5. Неметаллические ископаемые СССР. Изд. АН СССР, 1941. Глина и каолин, т. IV. Глины отбеливающие, стр. 43.
6. П. А. Земятченский. Глины СССР. Общая часть. Изд. АН СССР, 1935.
7. А. Г Бетехтин. Курс минералогии. Госгеолиздат, стр. 408, 437, 422, 1951.
8. С. И. С у х а н о в с к и й, Е. В. Рогинская. «Лесохимии, пром.» № 5—6, 26, 1934.
9. Словарь органических соединений, т. I, стр. 15, 137, 224; т. II, стр. 194, 592, 593, 697; т. III, стр. 811, 847. Изд-во иностр, лит-ры, 1949.
10. Р. Д. Оболенцев. Физико-химические константы компонентов легких моторных топлив. Гостоптехиздат, 1943.
11. А. Д. Петров, В. Н. Ипатьев. ЖРФХО, 58, 1029, 1926.
12. Техническая энциклопедия, т. I, стр. 273.
13. А. Д. Петров. Вег. 60, 2548, 1927.
14. А. Д. Петров, В. Н. Ипатьев. ЖРФХО, 59, вып. 5—6, 1927.
15. F г е s е п i u s. Zeitschrift analyt. Chem. 598, 1908.
16. Ekele.y, Howe. J. Am. Chem. Soc. 45, 1917, 1923.
17. Fit tig. Lieb. Ann. 141, 133.
18. Bodroux, Faborv. Bull. Soc. Chim. 3, 831, 1908.
19. I. Collonge. Bull" Soc. Chim. [4], 49, 141, 1931.
20. J a w e i n. Lieb. Ann. 195, 261.
21. E. Bergmann, H. Taubadel, H. Weiss. Ber. 64, 1493, 1931
СОДЕРЖАНИЕ
Стр.
Предисловие........................................................ 3
В. Ф. Полозов. Окисление керогена прибалтийских сланцев . . 5
Н. И. Зеленин и О. С. Куратова. О составе и свойствах сланцевой смолы..................................................15
С. С. Семенов и Б. Е. Гуревич. Выделение нейтральных кислородных соединений средних фракций сланцевой смолы ... 49
С. С. С е и ейов и Б. Е. Г у р е в и ч. Изучение нейтральных кислородных соединений средних фракций сланцевой смолы ... 57
С. С. Семенов, Ю. И. Корнилова и Б. Е. Гуревич. Об окислении некоторых кетонов йодом в щелочной среде ... 69
Г. Н. Г а р н о в с к а я. Действие едкой щелочи на кислородные соединения сланцевой смолы...................................77
С. С. Семенов и Б. Е. Гуревич. Полимеризация олефиНов сланцевой смолы на масла в присутствии хлористого алюминия .......................................................89
С. С. Семенов и Б. Е. Гуревич. О стабильности дизельного топлива из смолы полукоксования прибалтийских сланцев . 102
Е. Е. Феофилов и Г. Н. Гарно век а я. Химический состав и пути практического использования полимеров каталитической стабилизации фракций сланцевой смолы . .....................106
Н. И. Зеленин и Г. В. Татаркина. Состав легкой фракции смолы кашпирског'о сланца...................................111
В. Н. Л а п и н. Моющие, смачивающие и эмульгирующие средства из сланцевой смолы.............................................118
Л. И. Гуляева и Н. И. И ы ш к и н а. Определение содержания кислородных соединений адсорбционно-хроматографическим методом в смолах сланцев Прибалтийского месторождения . . 124
Л. И Гуляева и Н. И. П'ышкина. К вопросу об определегСии содержания нейтральных кислородных соединений в сланцевых смолах хлорным железом......................................131
Г. Н. Гарновская. Рефрактометрический способ определения содержания фенолов в ^сланцевых продуктах.....................138
Е. И. Томина, К. Б. Чернышева и Е. М. Дементьева. Спектрографический анализ некоторых ароматических углеводородов сланцевой смолы.......................................145
Б. И. Иванов и Н. Ф. Шаронова. Химический состав подсмольной воды сланцев Прибалтийского -месторождения . . . 164
Б. И. Иванов, Ю. А. К о з а к и Н. Ф. Ш ар он о в а. Дефеноля-ция подсмольной воды прибалтийских сланцев бутилацетатом . 189
Б. И. И в а н о в и Ю. А. К о з а к. Обезвреживание подсмольных вод прибалтийских сланцев золой -и коксом...................200
А. Е. Драбкин. К вопросу об очистке горючих газов от сероводорода растворами аминоспиртов............................207
Содержание 295
Стр.
Д. К. К о л л е р о в. О законе вязкости Бачинского........216
Д. К. К о л л е р о в. К вопросу о физической сущности показателя 3 ,----
утк
К——углеводородных жидкостей................................228
Г. Н. Скрынникова. Экспериментальное исследование коэффициента теплопроводности жидких сланцевых продуктов . . . 242
И. Н. Каменская. Превращения алифатических кетонов Над алюмосиликатными катализаторами .... .... 269
Редактор В. В. Кедринский
Технич. редактор Е. В. Соколова Корректоры: Н. Е. Месман и В. В. Опарина
Сдано в набор 16/1-54 г. Поди, к печ. 30/П1-54 г. Формат бум. 60x921/16* Печ. л* 1 8,5 (усл. 18,5). Уч.-изд. л. 17,25. Тираж 1125 экз. М-23679.
Индекс. 15—5—4.
Гостоптехиздат (Ленинградское отделение), Невский проспект, 28. Издательский Кд 52. Зак. №55. Типография «Красный Печатник», Ленинград, проспект имени И. В. Сталина, 91.
Цена 13 р. 60 к.
ОПЕЧАТКИ
Страница Строка Напечатано Следует читать По чьей вине
27 11 сверху может испариться не может испариться изд.
37 16 сверху карбоксильные карбонильные авт.
53 рис. 2 дефеполят дефенолянт »
*33 27 снизу метилциклогексанов метилциклогексаио-пов корр.
70 13 сверху 2.7 6КОН-> 2J4-2KOH—> авт.
32 Табл. 1 отерп Потери тип.
175 Табл. 13 солях, выделенных пропионовой и н-мас-ляной кислотами солях выделенных пропионовой и п-мас-: ляной кислот вед. ред.
210 8 сверху 1113 1,113 тип.
26(5 4 сверху 4 • 27 10“ 4 • 27 10“3
Труты ВНИИПС