/
































































































































































































































































































































































































Текст
A. E. ЧИЧИБАБИН
ОСНОВНЫЕ НАЧАЛА ОРГАНИЧЕСКОЙ ХИМИИ
ИЗДАНИЕ ПЯТОЕ, ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Под редакцией |/7. Г. СЕРГЕЕВА]
Том II
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО ХИМИЧЕСКОЙ ЛИТЕРАТУРЫ
МОСКВА . 1957
547
Ч—72
Книга представляет собой второй том пособия по органической химии, посвященный алициклическим, ароматическим и гетероциклическим соединениям. При рассмотрении алициклических соединений большое внимание уделено основам современной стереохимии циклических соединений. Специальные разделы посвящены терпенам, стероидам, небензольным ароматическим соединениям, алкалоидам, витаминам и антибиотикам.
Кнша предназначена для углубленного изучения органической химии и может служить справочным руководством для химиков-органиков всех специальностей.
К ЧИТАТЕЛЮ
Издательство просит присылать Ваши с-амечанчя и отзывы об этой книге по адресу: Москва. К-121
Новая площадь, 10, Госхимиздат
СОДЕРЖАНИЕ ВТОРОГО ТОМА
ЧАСТЬ ВТОРАЯ
От издательства ................................................................. 8
Изоциклические, или карбоциклические, соединения
Алициклический ряд
Классификация и номенклатура алициклических соединений .... 12
Стереохимия алициклических соединений.............,............... 20
Напряженность циклов........................................... 20
Изомерия и стереоизомерия одноядерных алициклических соединений 27
Способы получения алициклических соединений из соединений других рядов . . . ....................................... 37
Превращения циклов друг в друга................................... 46
Свойства алициклических углеводородов............................. 52
Природные источники алициклических соединений..................... 60
Одноядерные алициклические соединения............................. 66
Циклопропан и его производные ................................. 66
Циклобутан и его производные................................... 68
Циклопентан и его производные.................................. 70
Циклогексан и рго производные................................... 75
Производные циклогептана и циклооктана.......................... 83
Производные углеводородов с высшими циклами..................... 85
Двухъялерные алициклические соединения............................. 88
Соединения с циклами, разделенными цепочками из атомов углерода 88
Каротиноиды................................................. 88
Соединения с циклами, разделенными простой углерод-углеродной связью......................................................... 90
Спирановые, или спироциклановые, двухъядерные соединения ... 95
Двухъялерные соединения с конденсированными ядрами........... 97
Сантонин и родственные рму вещества.......................... 107
Соединения с мостиковыми бициклическими системами............... 108
Алициклические соединения с многими циклами....................... 112
Терпены и их производные ............................................... 115
Алифатические терпены ..........................................
Циклические терпены ............................................
Моноциклические терпены .....................................
Ментан и его производные .................................
Ментадиены и их производные...............................
Бициклические и трициклические терпены .....................
Группа карана ............................................
Группа пинана..................................... . . . .
Группа камфана ...........................................
Политерпены ...................................................
Сесквитерпены ............................................
Яриродные смолы и бальзамы ........................................
Смоляные кислоты ............................................
117 118
119 119 124 131 131
133 139 154 156 161 162
Стероиды........................................................... 165
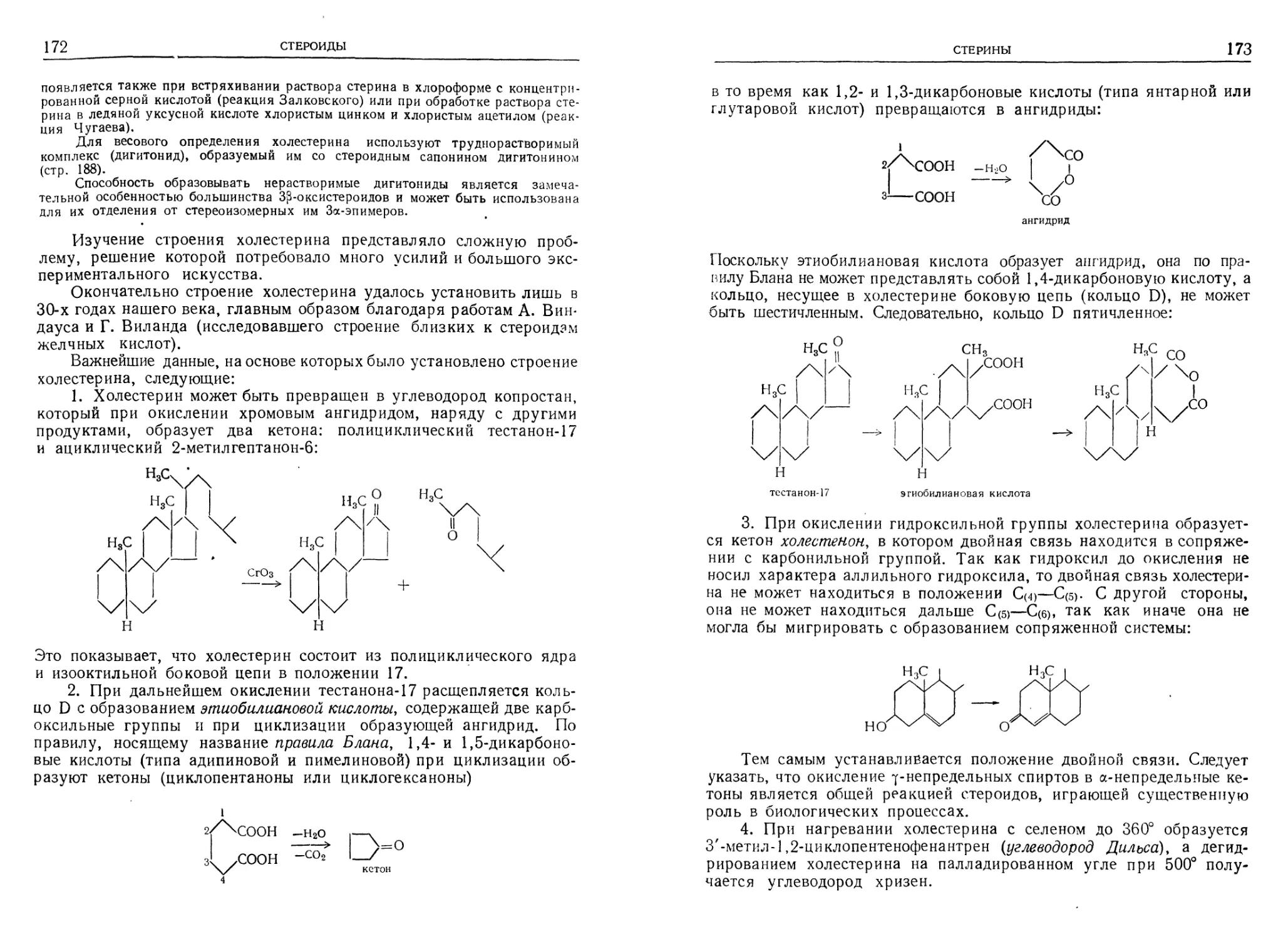
Стерины......................................................... 169
Желчные кислоты................................................. 179
Сердечные глюкозиды............................................. 182
Яды жаб и аглюконы морского лука................................ 184
Стероидные сапонины............................................. 186
Стероидные гормоны.............................................. 188
Ароматический ряд
Ароматические соединения.......................................... 198
Изомерия производных бензола...................................... 199
Синтезы ароматических соединений из соединений жирного ряда . . . 203
Реакции расщепления молекул ароматических соединений ............. 205
Взаимные переходы ароматических и гидроароматических соединений ......................................................... 205
У глеводороды ряда бензола и их одноатомные производные ............. 210
Углеводороды ряда бензола СЛН2Л_6 ................................ 210
Строение бензола............................................... 227
Дипольные моменты ароматических соединений..................... 236
Правила замещения в бензольном ядре............................ 238
Галоидные производные............................................. 243
Нитросоединения................................................... 247
Нитросоединения с нитрогруппой в бензольном ядре............... 247
Двойные соединения полинитропроизводных...................... 253
Нитросоединения с нитрогруппой в боковой цепи.................. 254
Таутомерия нипгросоединений с нитрогруппой в боковой цепи . . 254
Галоиднитросоединения ............................................ 255
Сульфокислоты..................................................... 257
Одноатомные фенолы и ароматические спирты......................... 261
Одноатомные фенолы ............................................ 261
Феноло-формалъдегидные счолы ................ 267
Галоидфенолы................................................... 269
Нитрофенолы.................................................... 271
Сульфокислоты фенола........................................... 274
Ароматические спирты .......................................... 274
Ароматические альдегиды .......................................... 275
Ароматические кетоны ............................................. 280
Стереохимия оксимов и гидразонов............................... 282
Ароматические одноосновные кислоты................................ 286
О так называемых пространственных затруднениях................. 296
Ароматические амины............................................... 297
Амины с группой NHj в бензольном ядре.......................... 298
Амины с группой NHj в боковой цепи ............................ 307
Анилины, замещенные в ядре на галоид, нитро- и сульфогруппы . . 309
Производные п-аминобензолсулъфамида ............ 311
Промежуточные продукты восстановления нитросоединений............. 312
Нитрозосоединения.............................................. 313
Р-Арилгидроксиламины........................................... 313
Азоксисоединения............................................... 314
Азосоединения.................................................. 315
Гидразосоединения.............................................. 317
Диазосоединения................................................ 318
Гидразины...................................................... 324
Производные бензола, содержащие фосфор, мышьяк и сурьму .... 326
Соединения фосфора........................................... 326
Соединения мышьяка......................................... 328
Соединения сурьмы ... 332
Металлорганические соединения ароматического ряда ............... 333
Многоатомные производные ряда бензола и соединения со смешанными функциями.......................................................... 337
Многоатомные фенолы............................................. 337
Фенолоспирты.................................................. 343
Лигнин........................................................ 344
Таутомерия фенолов............................................ 348
Хиноны ......................................................... 351
Хиноидные соединения и их таутомерия ......................... 357
Хиноли........................................................ 360
Многоатомные амины.............................................. 361
Аминофенолы.................................................. . 363
Азокрасители.................................................... 365
Таутомерия азокрасителей ..................................... 367
О связи между строением и окраской.............................. 368
Красители и крашение.......................................... 374
Многоосновные ароматические кислоты............................. 375
Ароматические оксикислоты ...................................... 381
Фенолокислоты................................................. 381
Дубильные вещества......................................... 385
Оксикислоты с гидроксильной группой в боковой цепи ........... 388
Ароматические аминокислоты...................................... 389
Аминобензойные кислоты........................................ 389
Аминокислоты с аминогруппой в боковой цепи.................... 391
Ароматические оксиальдегиды и оксикетоны........................ 392
Оксиальдегиды................................................. 393
О кси кетоны.................................................. 395
Ароматические аминоальдегиды.................................... 396
Соединения со смешанными функциями, содержащие мышьяк .... 396
О способах определения положения замещающих групп в бензольном ядре.......................................................... 398
ароматические соединения с ненасыщенными боковыми цепями ................. 401
Углеводороды.................................................... 401
Ионообменивающие смолы (иониты)............................... 404
Спирты и фенолы ................................................ 406
Альдегиды и кетоны.............................................. 407
Карбоновые кислоты.............................................. 407
Окси кислоты.................................................... 410
Многоядерные ароматические соединения ......................... 413
Простейшие многоядерные ароматические соединения. 413
Группа дифенила (бифенила).......................... 413
Стереохимия соединений бифенила ............. 416
Полифенилы.......................................... 418
Соединения с бензольными ядрами, связанными при помощи углеродных атомов, не входящих в кольцо............................... 420
Группа дифенилметана...................................... 421
Группа трифенилметана..................................... 424
Красители ряда трифенилметана ............. 426
Тетрафен и лметан . • • •................................. 432
Группа дибензила............................................... 432
Несимметрический дифенилэтан ............................... 434
Тетрафенилэтан.............................................. 435
Соединения трехвалентного углерода............................. 436
Свободные радикалы с аномальной валентностью неуглеродных атомов ..................................................... 439
Причины устойчивости свободных радикалов типа триарилме-тилов....................................................... 440
Магнитные свойства свободных радикалов...................... 444
Соединения с конденсированными бензольными ядрами.............. 445
Группа нафталина ........................................... 445
Стереохимия производных нафталина....................... 464
Инден...................................................... 466
Группа антрацена ........................................... 466
Таутомерия производных антрацена......................... 474
Амино- и оксипроизводные антрахинона..................... 475
Природные производные антрахинона........................ 478
Ализариновое крашение и протравные красители............. 479
Группа фенантрена.......................................... 480
Сложные конденсированные ароматические углеводороды............... 484 •
Многоядерные линеарные системы................................. 484
Многоядерные ангулярные системы................................ 486
Канцерогенные, или карциногенные, углеводороды................. 488
Небензольные ароматические соединения
Тропой........................................................... 494
Трополоны........................................................ 496
Азулены.......................................................... 502
ЧАСТЬ ТРЕТЬЯ
Гетероциклические соединения
Гетероциклический ряд............................................. 507
Ароматический характер непредельных гетероциклических соединений 514
Пятичленные гетероциклы ............................................ 519
Группа фурана ................................................... 519
Группа тиофена .................................................. 525
Группа пиррола .................................................. 528
Взаимные превращения пятичленных гетероциклов................. 533
Продукты присоединения водорода к пирролу и его производным . 534
Гемин, хлорофилл и родственные им вещества..................... 539
Группа индола, или бензопиррола ................................. 547
Производные индола и индолина, содержащие атом кислорода в пиррольной части ядра............................................ 553
Индиго и индигоидные красители................................... 558
Карбазол......................................................... 563
Группа пиразола.................................................. 563
Группа имидазола, или глиоксалина................................ 568
Другие пятичленные гетероциклы................................... 571
Шестичленные гетероциклы ........................................... 575
Группа пирана ................................................... 575
Хромоны и флавоны................................................ 583
Антоцианидины.................................................... 586
Ксантон .......................*................................. 587
Растительные инсектициды. Ротенон и родственные ему вещества . 588
Группа пиридина ............................................. 590
Продукты присоединения водорода к пиридиновому ядру ..... 605
Дипипилилы................................................. 609
Арсепидины................................................. 610
Соединения с конденсированными ядрами бензола и пиридина........ 611
Группа хинолина ............................................. 611
Изохинолин • ................................................ 620
Акридин...................................................... 621
Азины • • • • •................................................. 625
Группа пиразина .................. .......................... 625
Фенашн, феноксазин и фентиазин .............................. 628
Аналоги азинон, содержащие мышьяк............................ 631
Группа пиридязина .......................................... 632
Группа пиримидина ................................ 633
Гпуппа пупина . ........................................ .... 634
Группа птеридина ............................................ 640
Алкалоиды • •...••••••••••••••••................................ 642
Алкалоиды, содержащие экзоциклический азот................... 647
Алкалоиды, производные пиррола..................•............ 648
Алкал »и ты, производные 1-метилпирролизидина ............... 648
Алкалоиды, производные пиоитина и пиперидина ................ 648
Алкатоиды, сотержашие неконденсированные пяти- и шестичленный азотистые циклы............................................ 649
Алкалоиды, содержащие два неконденсированных шестичленных азотистых цикла................................................. 651
Алкалоиды, содержащие конденсированные пяти- и шестичленный азотистые циклы (группа трепана, ............................... 652
Алкалоиды, содержащие два конденсированных пиперидиновых цикла 657
Алкалоиды, производные хинолина ............................. 657
Алкалоиды, производные изохинолина ......................... 665
Алкалоиды, производные индопа ............................... 678
Алкалоиды, производные имидазола ............................ 686
Алка дои ты, содержа ние пуриновую группировку .............. 687
Стероидные алкалоиды (производные циклопентенофенантрена) , . . 687
'цтибиотики • •............................................... 689
Витамины .................................................... 703
Витамины алициклического ряда ............................... 704
Витамины ароматического ряда ................................ 707
Витамины гетероциклического ряда .......................... 709
Антагонисты витаминов (антивитамины) ........................ 715
Предметный указатель . . . • • ................................. 717
ОТ ИЗДАТЕЛЬСТВА
Второй том книги А. Е. Чичибабина «Основные начала органической химии» подвергся столь же существенной переработке, как и первый том. В связи с большим развитием химии алициклов и необходимостью изложения основ современной стереохимии алициклических соединений, в частности конформационного анализа, пришлось значительно увеличить объем этой части книги, а также ввести новый раздел, посвященный стероидам.
Несколько вырос также объем раздела, посвященного химии ароматических соединений, в котором даны современные представления о строении бензола. Специальный небольшой раздел посвящен химии открытых в последние годы небензольных ароматических соединений—трополонов, азуленов и др.
Раздел гетероциклических соединений ограничен в данной книге лишь основными понятиями и сведениями, так как даже для сжатого изложения химии гетероциклических соединений, ввиду весьма широкого ее развития, потребовалось бы издание самостоятельного тома. Здесь же кратко описаны только самые важные представители гетероциклических природных веществ, алкалоидов, витаминов и антибиотиков.
Чтобы дать представление о возможностях современного органического синтеза, в разделах, посвященных стероидам, алкалоидам и антибиотикам, приведены характерные схемы синтезов некоторых органических веществ очень сложного строения.
Общая редакция второго тома принадлежит ныне покойному П. Г. Сергееву, посвятившему много лет самоотверженного труда подготовке этой книги к печати. Раздел химии алициклических соединений обновлен и дополнен канд. хим. наук А. Л. Либерманом, материал по химии стероидов предоставлен канд. хим. наук Л. Д. Бергельсоном, материал по химии лигнина—докт. хим. наук Н. Н. Шоры-гиной, раздел, посвященный связи между строением и окраской, подготовлен действ, членом АН УССР А. И. Куприяновым, раздел химии тропонов и трополонов составлен при участии канд. хим. наук А. С. Хохлова, написавшего также раздел, об антибиотиках. Ценные советы и указания по общим и частным вопросам получены от акад. А. Н. Несмеянова, акад. Б. А. Казанского, члена-корреспондента АН СССР М. М. Шемякина, докт. хим. наук И. И. Бардыше-ва, проф. В. Н. Белова, проф. А. И. Королева, проф. А. Ф. Платэ, проф. Б. А. Порай-Кошица, проф. Н. А. Преображенского, проф. О. А. Реутова, проф. К). К- Юрьева, докт. хим. наук А. Н. Коста, канд. хим. наук И. К. Коробициной, а также от многих других химиков-органиков. Всем принявшим участие в этой работе издательство выражает свою глубокую признательность.
Часть вторая
ИЗОЦИКЛИЧЕСКИЕ, ИЛИ КАРБОЦИКЛИЧЕСКИЕ, СОЕДИНЕНИЯ
Алициклический ряд
Алициклические соединения, т. е. соединения, содержащиё в молекуле кольца или циклы из одних только атомов углерода (кроме бензола и его производных, выделяемых в особый ряд ароматических соединений), за немногими исключениями, сделались известными значительно позднее жирных и ароматических соединений.
Представление о неограниченной способности атомов углерода соединяться друг с другом цепеобразно* было в свое время воспринято быстро, без особых возражений, способность же углеродных цепей замыкаться в кольчатые, циклические системы вначале не казалась очевидной.
В правомерности таких представлений химиков убедило только успешное применение для бензола и его производных шестичленной циклической формулы Кекуле.
В то же время изучение широко распространенных в природе терпенов, часто представляющих собой производные гидрированного шестичленного бензольного кольца, и установление легкости замыкания, в ряде случаев, шестичленных кольчатых группировок наталкивали на мысль об исключительной устойчивости шестичленного цикла. При этом даже высказывалось сомнение в возможности существования циклов с числом углеродных атомов большим или меньшим шести.
Однако с начала 90-х годов прошлого столетия быстро стали накапливаться факты, опровергающие такие мнения. Около 1883 г. Перкин младший** синтезировал ряд производных циклобутана. Несколько ранее был получен циклопропан (Фрейнд, 1882 г.), хотя его строение не сразу было установлено. Затем были получены
* Недавно получен н-гектан С^Нз^, кристаллизующийся в виде пластинок темп, плавл. 115°). Углеродные цепи высокополимерных соединений могут включать еще большее число атомов углерода. Так, высшие фракции полистирола состоят из цепей, содержащих 10 000 связанных друг с другом углеродных атомов. Цепи еще большей длины (более 200 000 атомов углерода) получаются при полимеризации этилена по Циглеру при атмосферном давлении в присутствии триэтилалюминия *
** Вильям Перкин младший (1860—1929). Крупный английский химик-органик, сын основателя английской промышленности органических красителей (В. Перкина старшего). Работал в области алициклических соединений—производных циклобутана и циклопропана, в области алкалоидов и некоторых других природных органических соединений.
производные циклопентана (Перкин младший, 1885 г.). В 1893 г. был получен циклогептан, или суберан (В. В. Марковников).
Далее были получены соединения с кольцами атомов углерода до десятичленного, а начиная с 1926 г., благодаря систематическим работам Л. Ружички, сделались известными циклы от десятичленного до двадцатидвухчленного и, кроме того, кольца из 29 и 34 атомов углерода.
К настоящему времени открыто большое число методов получения углеродных циклов из соединений с открытыми цепями атомов углерода; изучен ряд реакций превращения одних циклов в другие; получено синтетически множество соединений этого класса; весьма значительное количество таких соединений найдено также в природе и для многих из них установлено строение, подтвержденное синтезом; изучены также химические и физические свойства различных рядов ‘этих соединений и установлены их взаимные отношения.
Как мы увидим далее, теперь многие алициклические соединения, помимо давно уже получивших практическое применение терпенов и их производных, приобрели важное значение в промышленности органического синтеза.
Исследование соединений с углеродными циклами сыграло большую роль в развитии теории органической химии.
Классификация и номенклатура алициклических соединений
Алициклические соединения* могут содержать в молекуле одно, два, три или более колец. Соответственно различают одноядерные, или моноциклические, двухъядерные, или бициклические, трехъядерные, или трициклические, и вообще многоядерные, или полициклические, соединения.
Соединениями, лежащими в основе классификации одноядерных алициклических соединений, являются углеводороды, состоящие из метиленовых групп, замкнутых в кольцо, так как это простейшие для каждого типа циклов углеводороды, содержащие только простые связи и имеющие состав СлН2/1.
Эти основные углеводороды в свое время было принято называть по числу метиленовых групп в кольце—триметилен, тетра-метилен, пентаметилен и т. д., а весь алициклический ряд— рядом полиметиленовых соединений. В настоящее время более принято называть их по женевской номенклатуре, которая дает этим основным циклическим углеводородам такие же названия, как и парафиновым углеводородам с тем же числом атомов углерода
Это назйание было предложено Бамбергером в 1889 г.
Н,С-----СН,
н,с СН, \н, пиклопентан (пентаметилен)
в молекуле, но с приставкой «цикло» впереди: циклопропан, цикло-бутан, циклопентан и т. д.
Н2С----СН, Н2С—СН2
\/ II
СН2 Н2С—СН,
циклопропан циклобутан
(триметилен) (тетра метилен)
си, н,с-сн,
Н2<6 \н, 11,6 \н,
II II
Н,С^ СН, Н,С^ /СН,
сн» чсн,
циклогексан циклогептан
(гексаметилен) (гептаметилен)
В соответствии с этим циклические углеводороды получили
,бщее название циклоалканов, цикланов, или циклопарафинов. Для
\тлеводородов с пяти- и шестичленными кольцами широко применяется предложенное В. В. Марковниковым и В. Н. Оглоблиным общее название нафтены (от слова нафта—нефть).
Циклогексан и его гомологи, не имеющие двух алкилов при зднОхМ и том же углеродном атоме, вследствие легкости их превращений в ароматические углеводороды и обратно, носят также название сагидробензолов или гидроароматических углеводородов.
В последние годы в литературе, наряду с приведенными выше изображениями формул алициклических соединений, часто встречаются и упрощенные ?чемы, например:
циклопропан метилциклобутан циклопентансл циклогексанон
Для наименования гомологов алициклических углеводородов очетают название цикла с обычным обозначением радикалов, завещающих атомы водорода в цикле, например:
Н2С-----СН-СН8
метилциклопропан
(метилтгиметилен)
Часто название производится также и от алифатической цепи: Н2С—СН8 н,с (1н,
сн3-сн,-сн-сн,-сн8
3-циклопентилпентан
Такие названия чаще всего употребляются для цикланов с длинными боковыми цепями, особенно если цикл находится не на конце цепи или если в цепи имеются другие заместители и ответвления:
Н2С----СН2
I I нгс сн,
I
сня—с—снгон
I
СН,
2-метил-2'Циклопентилпропанол
Если в цикле на радикалы замещенс более одного атома водорода, то относительное положение этих радикалов в кольце обозначается цифрами, например:
Н2С-СН,
1,1-диметилциклогексан^
н2с-сн2
СН3—н/ 'сН—СН, Н^Ь-СН,
1,4-Диметилниклогрксан
Положение двух боковых цепей при одном атоме углерода часто обозначают не цифрами 1,1, а приставкой гем (гемини— лат. близнецы).
Чтобы выбор цифр не был произвольным, соблюдаются определенные правила нумерации углеродных атомов кольца При наличии нескольких одинаковых боковых цепей номер 1 присваивается одному из углеродных атомов кольца, несущих боковую цепь, а все остальные атомы углерода в кольце нумеруются подряд. При этом выбор среди нескольких атомов с боковыми цепями и направление нумерации определяются требованием, чтобы все такие атомы получили наименьшие номера. Таким образом, из шести возможных способов нумерации изображенного ниже триметилциклогексана правильным является только первый:
1,2,4-гонметил;1иклогексан
Проше всего узнать, которая из возможных нумераций является правильной, сравнив суммы номеров атомов, несущих боко
вые цепи: правильной системе нумерации будет соответствовать наименьшая сумма, в рассматриваемом случае 1+24-4=7. Легко убедиться, что в остальных пяти случаях эта сумма будет больше.
Г ри наличии боковых цепей разной длины наименьший номер должен приходиться на самую короткую цепь. При нескольких боковых цепях разного строения, но с одним и тем же числом углеродных атомов, наименьший номер должен иметь радикал, произведенный из метила замещением меньшего числа водородных атомов. Таким образом, углеводороду
сн3
СН8—С—сн?
<!в
н2+ сн2
СНз—СН,—СН,—СН,— ucL СН—СН—СН,—СН,
сн3
должно быть дано название 1-н-бутил-3-втор-бутил-5-трвт-бутил*> циклогексан (радикалы можно назвать и по-другому, например Г-метопропил вместо я-бутил и Г, Г-диметоэтил вместо втор-бутил, или иначе; здесь следует обращать внимание только на цифры, указывающие местоположение радикалов разного строения). *
Менее строго регламентированы принципы номенклатурьГцик-ланов, имеющих заместители. Само название обычно составляется по общим правилам женевской номенклатуры, например:
Н2с----СН—С)
хлорииклопропан, или циклопропилхлорид
Н2С-СН2
I I
Н,С—СВг2
1,1-дибромцикло-бутан
н,с—сн, hJ СН—он
+н2
циклопентанол
СН2
\н2
hJ СН—соон \н,
циклогексанкарбоновая кислота
Однако порядок нумерации при наличии нескольких заместителей не вполне определен. По женевской номенклатуре номер 1 дается
атому кольца, связанному с атомом-заместителем, имеющим наименьший атомный вес. Поэтому соединение
СНЯ
СН н/7 'си, .H2i СН—он
\н"2
будет названо 1-метилциклогексанолом-3. Это правило соблюдается, однако, не всегда, и более принято (в соответствии с дополнениями к женевской номенклатуре,—согласно так называемой женевско-льежской номенклатуре) давать номер 1 главной функции молекулы. Таким образом, это соединение будет называться 3-метилциклогексанолом- 1 или просто 3-метилциклогексанолом.
Номенклатура двухъядерных цикланов. Двухъядерные углеводороды можно разделить на пять групп в зависимости от взаимного расположения циклов: а) циклы разделены цепочками из атомов углерода; б) циклы непосредственно соединены простой С—С связью между двумя атомами, принадлежащими этим циклам; в) циклы имеют один общий углеродный атом (спираны, или спироцикланы); г) циклы имеют два общих атома углерода (цикланы с конденсированными ядрами); д) циклы имеют три или более общих атомов углерода (мостиковые цикланы). Соединения последних двух групп иногда объединяют под общим названием собственно бициклических соединений. При замыкании каждого нового кольца число атомов водорода в молекуле уменьшается на два, а потому двухъядерные цикланы будут иметь состав СлН2п-2.
Для двухъядерных цикланов не имеется единой номенклатуры, и для наименования соединений каждой группы применяются свои системы наименований.
В основу номенклатуры соединений, в которых циклы разделены цепочками из атомов углерода, положены номенклатуры соединений алифатического ряда—рациональная и женевская—с использованием названий радикалов, производимых от названий цикланов: циклопропил, циклопентил и т. д.
Таким путем возникают названия дициклопропилметан, цикло-гексилциклопентилметан:
Как видно из приведенных схем, углеродные атомы одного из циклов нумеруются цифрами со штрихами. Если циклы неодина-
ковые, цифры со штрихами применяются для обозначения атомов углерода в меньшем кольце.
Если в алифатической цепочке имеется более одного атома углерода, приходится указывать положение циклов в цепочке, а также положение в ней других радикалов и групп. Для этого пользуются греческими буквами или нумеруют углеродные атомы цепочки цифрами без штрихов, обозначая углеродные атомы циклов цифрами с одним и с двумя штрихами.
Например, соединение
снгсн-сн-сн~снгад
II \б' 5/ >
ОН СНз 1
может быть назван о 3-метил-4-(3"-бромциклопентил)-6-(4/-хлорцик-логексил)-гекса нол ом-2 или а-(4-хлорциклогексил )-у-(3'-бромцик-лопен!нл)-о-ме'1 ил-е-оксигексаном или, наконец, менее последовательно, а,£-диметил-7-(3'-бромциклопентил)-е-(4-хлорциклогексил)-пен-танолом.
Для наименования соединений, у которых циклы связаны непосредственно ординарной связью, поступают следующим образом. Если циклы одинаковы, то соединение можно назвать по имени двух образующих его радикалов, например бициклогексил или дициклогексил:
Названия подобных соединений можно также строить подобно названиям моноцикланов с боковыми цепями, например циклогексил-циклотексан.
Если циклы разные, то пользуются только названиями второго типа, например циклобутилциклогептан. И в этом случае штрихами отмечаются номера углеродных атомов меньшего цикла.
Совершенно иначе строятся номенклатуры спиранов. Одна из них—менее удачная—основана на том, что каждый цикл называется отдельно и оба названия соединяются частицей «спиро». Таким образом, соединение
будет носить название циклобутанспироциклопентан. Нумерация атомов углерода производится, начиная с малого цикла и кончая общим атомом углерода. По другой системе номенклатуры в основе
названия лежит число углеродных атомов во всей циклической системе молекулы (без боковых цепей) с частицей «спиро», за которой в квадратных скобках указывается число углеродных атомов, связанных с общим атомом в каждом из циклов. Следовательно, приведенный выше спиран должен быть назван спиро-[3,4 ]-октаном. Так же строятся названия производных.
Например соединение
- О
2 |\1О/9 8\
„ X 7>-соон
з 4/ \5 6/
НО
будет называться: циклопентанол-3-он-1 -спироциклогексан-7-карбо-новая кислота (по первой системе номенклатуры) и спиро-[4,5]-деканол-З-он-1 -карбоновая-7 кислота, или спиро-[4,5]-декан-3-ол-1-он-7-карбоновая’ кислота (по второй номенклатуре). В настоящее время вторая номеклатура является наиболее распространенной .
Номенклатура бициклических соединений с конденсированными ядрами напоминает вторую номенклатуру спиранов, но только с заменой частицы «спиро» на частицу «бицикло». В квадратных скобках помещаются цифры, показывающие число атомов углерода, связанных в каждом цикле с узловыми (общими для обоих циклов) атомами углерода. Кроме того, ставится цифра 0, показывающая, что, кроме двух узловых атомов, больше общих атомов углерода не имеется.
Таким образом, соединение
будет называться бицикло-[0,2,6]-деканом или бицикл о-[6,2,0]-деканом*. Здесь нумерация также будет начинаться с малого цикла и кончаться общими атомами.
Применяются часто также ‘и тривиальные названия:
декалин
ш
гидриндан пенталан
Номенклатура мостиковых углеводородов очень близка к только что описанной, но в квадратных .скобках указывается число ато-
* Эта номенклатура была предложена Байером. Порядок расположения цифр в квадратных скобках не узаконен; например, в СССР более принят возрастающий, в Англии, наоборот, исходящий порядок.
мов (кроме узловых) уже в трех цепочках, связанных с узловыми атомами:
бицикло{1,1,2]-гексан бицикло-[2,2,2]-октан бицнкло[1,2,3|-октан
При графическом изображении мостиковых систем принято располагать в виде мостика наиболее короткую цепь.
Нумерацию углеродных атомов мостиковых систем начинают с одного из узловых атомов, продолжают вдоль самой длинной цепи до следующего узлового атома включительно, затем продолжают по следующей цепи до первого узлового атома, после чего уже нумеруют атомы мостика, например:
Как легко видеть, номенклатура бициклических соединений с конденсированными ядрами является таким частным случаем номенклатуры мостиковых углеводородов, когда мостик состоит лишь из простой связи между узловыми атомами.
Многие мостиковые соединения имеют тривиальные названия или названия, производимые от тривиальных^(см. терпены).
Необходимо подчеркнуть, что приведенные выше изображения бициклических углеводородов являются лишь плоскими проекциями действительных структур, тогда как все бициклические углеводороды с общими атомами углерода имеют совершенно определенное пространственное расположение. Так, три^нз приведенных углеводородов могут быть изображены следующим образом:
биигк 1О-[2,2,2уокта н
бипикло-[ ।. 2,3] октан
Номенклатура трехъядерных и многоядерных цикланов. Названия трехъядерных и многоядерных никла!.ов строятся по тем же принципам, что и наименования двухъядерных соединений. Здесь особенно часто применяются тривиальные названия.
Номенклатура ненасыщенных циклических соединений. При
рассмотрении номенклатуры ненасыщенных циклических соединений
приходился различать вещества с двойными связями в боковых цепях ^цикланы с ненасыщенными боковыми цепями) и вещества с двойными связями в циклах (циклены). Номенклатура первых ничем не отличается от номенклатуры моноцикланов, например:
СН=СН.
винилциклопропан, или этенилциклопропан
yj_CH,-CH=CH2 алл. jbi' клонен 1<1н, или прог.сн илциклопентан, или 3-циклопентилпропсн-1
Номенклатура цикленов основана на обычных принципах замены окончания ан на ен, ин, диен и т. п.:
циклобутен циклопента диен-1,3
Н2С—(СН2)1П—СН, I I
Н2С--CsC—сн2
циклогексадецин
Особые случаи представляют соединения с так называемой семициклической двойной связью, т. е. двойной связью ме/жду углеродным атомом кольца и первым атомом боковой цепи В названиях таких соединений используются наименования двухвалентных радикалов:
j—рсн-снэ _с/СНз
1Хсн3 этилиденциклобутан изопропилиденциклопентан
Кроме того, для указания положения двойной связи у ненасыщенных циклических соединений иногда пользуются специальной сим-вол и кой—греческой буквой А и цифрами при ней. Эта система, постепенно выходящая из употребления, все еще применяется в химии терпенов, стероидов, алкалоидов и др. Подробное описание ее дано в разделе «Терпены».
СТЕРЕОХИМИЯ АЛИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
НАПРЯЖЕННОСТЬ ЦИКЛОВ
После того как было доказано существование кольчатых систем с разным числом атомов углерода в кольце, возник вопрос об их относительной устойчивости, которая вначале исследовалась чисто эмпирически. Первоначально была установлена лишь непрочность четырехчленных и, особенно, трехчленных циклов, а шестичленному кольцу долгое время приписывалась исключительная, по сравнению со всеми другими циклами, устойчивость. Однако постепенно стало ясно, что все циклы, начиная с пятичленного, довольно близки по УСТ0ЙЧИВ0С1и.
Первые общие теоретические соображения, касающиеся относительной устойчивости различных циклов, были высказаны Адольфом Байером в 1885 г. на основе представления Вант-Гоффа о тетра-
эдрнческой модели углеродного атома. Байер исходил из двух предположений: во-первых, о том, что циклические структуры должны быть плоскими, а, во-вторых, что отклонение направлений валентных связей у атомов углерода от направлений к вершинам правильного тетраэдра, т. е. от угла 109°28', является мерой натяжения (напряженное/! и цикла), обусловливающего легкость или трудность образования цикла и его относительную устойчивость. Величины таких отклонений для каждого направления валентностей легко рассчитать- они должны составлять половину разности между 109°28' я величиной внутреннего угла соответствующего правильного многоугольника.
Для шести простейших циклов эти отклонения должны иметь с л еду ющие вел и чи ны:
Циклопропан Циклобутан Циклопентан
. . +24с44'
. . 4-9°44'
. . 4-0’44'
Циклогексан .... —5° 16'
Циклогептан .... —9 33'
Циклооктан .... —12~51'
Таким образом, по Байеру, наиболее устойчивым должен был быть пятичленный цикл, а следующим по устойчивости—шестичлен-ный. Четырехчленный и семичленный циклы должны обладать меньшей и почти одинаковой степенью устойчивости. Высшие же циклы должны быть все менее и менее устойчивыми. В дальнейшем оказалось, что этот вывод неверен.
Еше в 1890 г Саксе указывал, что шести- и семичленные циклы совсем не обязательно должны быть плоскими. Если же углеродные атомы циклических сисгем могут располагаться и не в одной плоскости, то тогда можно построить такие модели шестичленных и высших циклов, которые будут свободны от напряжения Однако соображения Саксе не привлекли к себе особого внимания вплоть до 1918—1922 гг., когда Э. Мор вновь поднял этот вопрос.
Дело к том, что долгое время применение плоских моделей шестичленных -карбоциклических и гетериникличрских структур (производные циклогексана, углеводы с пирановым кольцом' не приводило к серьезным противоречиям с опытом, <отя бы. например, в отношении числа предвидимых стереоизомеров С другой стороны, молекулярная ыория как раз в -;гот период :1890—-Щ20 гг.) подвергалась нападкам и критике со стороны философов и физиков, стоявших на идеалистических позициях Г.Цах Оствальд. Дю’ ем и др > Стереохимические модели многими расценивались как ненаучная игра фантазии тем более, что открытая около 1890г. вальдгнонская перегруппировка грозила совсем запутать вопрос о соответствии мест замещаемых и замещающих групп или атомов. В настоящее время, когда наука располагает методами исследования, позволяющими определять взаимные расстояния атомных ядер в молекуле и углы между направлениями валентностей, рассчитывать дипольные моменты молекул и т п , у химиков имеется возможность установления истинной пространственной конфигурации циклических структур. .
Противоречия с гипотезой Байера проявились особенно отчетливо, когда начали изучать соединения, содержащие циклы с числом атомов более шести. Например, если придерживаться представления
о плоской конфигурации, пятнадцатичленный цикл кет она мускона С15Н28О должен быть так же непрочен, как и трехчленный цикл циклопропана, поскольку у них величины отклонений от нормального угла между валентностями почти одинаковы (—23°16' и +24°44'). Между тем мускон, как оказалось, обладает примерно такой же прочностью, как и соответствующий парафиновый углеводород с нормальной цепью атомов—«-пентадекан.
Далее, предполагавшийся Байером рост напряжения по мере перехода к высшим циклам должен был бы сопровождаться значительным повышением в ряду циклопарафинов удельной теплоты горения, приходящейся на одну метиленовую группу, т. е. величины <?с„н9„ П 2П
П
Между тем оказалось, что и для углеводородов с высшими циклами эта величина остается равной 157—158 ккал и, следовательно, большого повышения запаса энергии в таких циклах не имеется. В настоящее время, благодаря работам Ружички, Циглера, Хюккеля, Штоля, Прелога и др., стало ясно, что никакого предела для числа атомов в цикле не существует.
Последующие исследования показали, что представления Саксе хй Мора все еще недостаточны для объяснения и предсказания всех особенностей геометрического строения циклов и их напряженности. Надо иметь в виду, что эти исследователи, как и Байер, учитывали только те напряжения, которые возникают в результате отклонения углерод-углеродных связей от их нормального тетраэдрического расположения.
Между тем, существуют и другие силы, создающие напряжение даже в молекулах простейших соединений с открытыми цепями, где нет никакого отклонения от тетраэдричности. Одной из таких сил является взаимное отталкивание нейтральных атомов (в частности атомов водорода), проявляющееся только тогда, . когда центры атомов сближаются на расстояние, близкое к сумме ван-дер-ваальсовских радиусов этих атомов. Существование таких сил отталкивания и их важная роль были установлены лишь около 20 лет тому назад, а их происхождение было разъяснено совсем недавно на основании квантово-химических соображений.
Открытие этого явления заставило пересмотреть прежние взгляды на свободное вращение вокруг простых связей. Даже в такой простой молекуле, как этан, различные положения одной метильной группы относительно другой оказываются энергетически неравноценными (см. том I, стр. 379). Самым невыгодным оказывается такое положение, при котором атомы водорода одной метильной группы находятся точно против' водородных атомов другой, как бы заслоняют их от наблюдателя, смотрящего на молекулу вдоль оси С—С-связи. Поэтому подобное состояние молекулы этана так и называют заслоненным, а также положением противостояния, или
оппозиции, атомов водорода. Наоборот, наиболее выгодным оказывается положение, при котором одна метильная группа повернута относительно другой на 60°. Разница в энергиях этих двух положений составляет хотя и небольшую, но вполне ощутимую величину— около 3 ккал (по последним данным 2,8 ккал). Благодаря этой энергетической разности свс«эдное вращение в молекуле этана вокруг оси, соединяющей метильные группы, становится неравномерным. Молекула стремится как можно быстрее перейти из наименее выгодного положения в наиболее выгодное, тогда как, попав в наиболее выгодное положение, молекула в нем задерживается, как бы тормозится. Поэтому такое положение молекулы называется заторможенным.
При небольшом запасе энергии молекула не может выйти из заторможенного положения, и полное свободное вращение метильных групп оказывается невозможным. Метильные группы лишь несколько отклоняются от заторможенного положения то в одну, то в другую сторону. Если же запас энергии молекулы превышает 2,8 ккал, молекула последовательно переходит из одного заторможенного состояния в другое, быстро проскакивая через промежуточные положения противостояния. Вращение делается полным.
Молекулы этана уже при обычной температуре сравнительно легко могут приобрести энергию, требуемую для полного вращения, а поэтому этан в этих условиях представляет собой динамическую смесь молекул, находящихся в указанных и в промежуточных между ними состояниях. Чем выше температура, тем больше вероятность приобретения энергии, необходимой для перехода, а потому тем больше концентрация молекул, находящихся в менее выгодном состоянии.
У гексахлорэтана разница между энергиями соответствующих состояний гораздо больше, чем у этана (10 — 15 ккал/моль против 2,8 ккал моль для этана), а потому свободное вращение в молекулах этого соединения при комнатной температуре почти полностью заторможено.
Молекулы в таких энергетически неодинаковых состояниях называют иногда поворотными изомерами. Реальность их существования подтверждается спектроскопически. Название «поворотные изомеры», однако, неудачно, так как при всех известных видах изомерии изомеризация осуществляется путем разрыва и возникновения связей между атомами. Так, при изомеризации к-бутана в изобутан, т. е. при переходе одного структурного изомера в другой, должны разорваться одна С—С-связь и одна С —Н-связь и соответствующие связи должны возникнуть в другом месте. При изомеризации цис-\ ,2-дихлорэтилена в траяс-изомер, т. е. при переходе одной пространственной конфигурации в другую, должна временно разорваться тс-связь между атомами углерода, а затем, после поворота одной части молекулы относительно другой, должна вновь
возникнуть та же связь между теми же атомами*. Для гнрехода же двух форм этана друг в друга не требуется никакого разрыва связей: переход осуществляется лишь путем свободного вращения. По этим соображениям выражение «поворотные изомеры», очень распространенное в спектроскопических работах, употребляется химиками довольно редко, и рассматриваемые энергетически неравноценные формы молекул, переходящие друг в друга только за счет свободного вращения, обычно называют конформациями (реже—констелляциями).
Итак, любая молекула с определенной пространственной конфигурацией может существовать в виде разных конформаций. Строго говоря, для каждой молекулы возможно бесконечное число разных конформаций, но большинство из них энергетически невыгодно, и любое реальное вещество представляет собой динамическую смесь молекул, большая часть которых находится в состояниях, отвечающих наиболее выгодным конформациям. Так, молекулам этана свойственна одна выгодная конформация; невыгодная конформация представлена в смеси весьма слабо. Промежуточные конформации встречаются в этане тем реже, чем больше их отклонение от наиболее выгодной конформации. При этом следует помнить, что любая конкретная конформация молекулы является лишь временным ее состоянием: молекулы постоянно переходят из одной конформации в другую. Конформационный состав вещества надо понимать только в статистическом смысле.
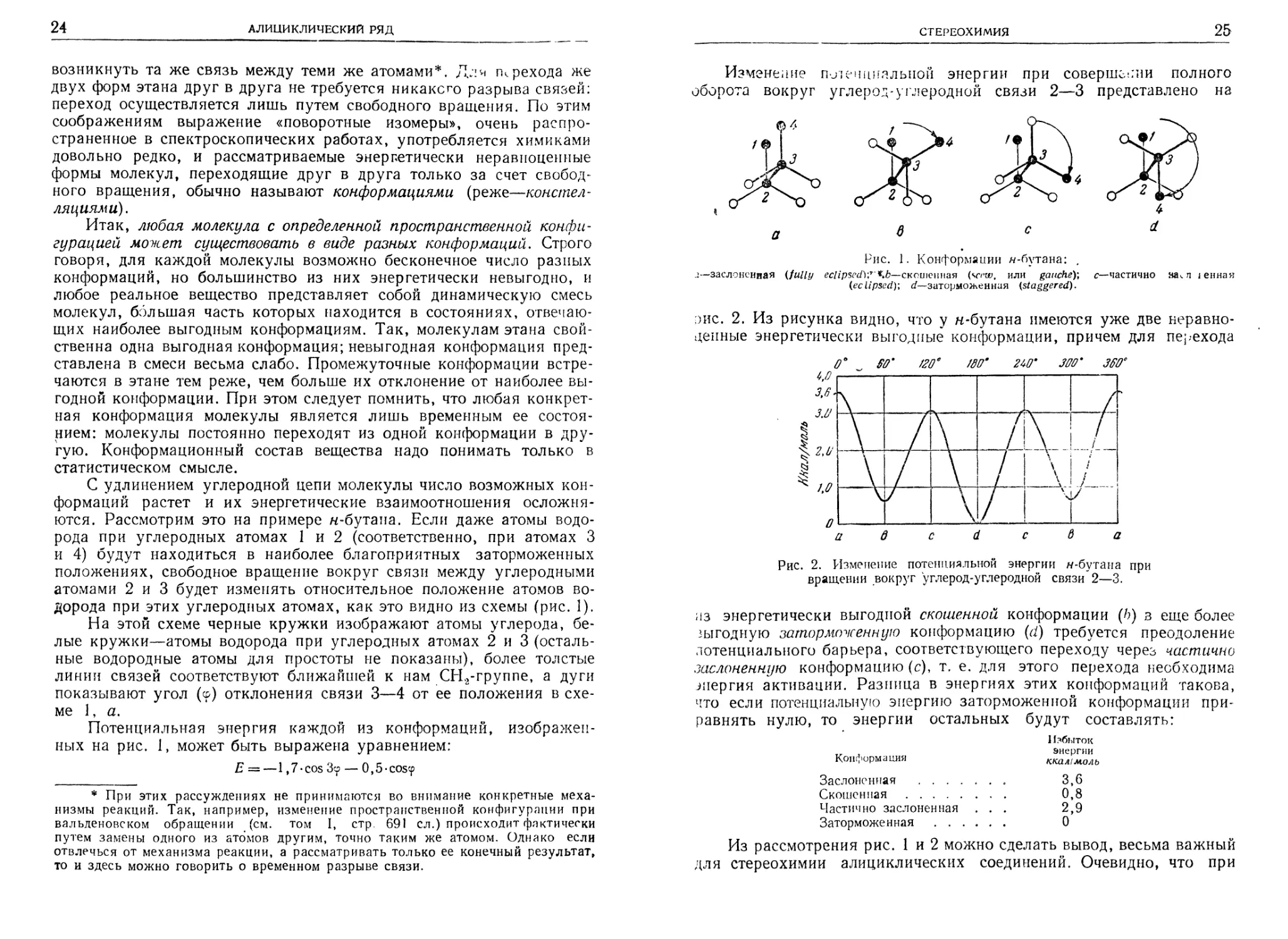
С удлинением углеродной цепи молекулы число возможных конформаций растет и их энергетические взаимоотношения осложняются. Рассмотрим это на примере н-бутана. Если даже атомы водорода при углеродных атомах 1 и 2 (соответственно, при атомах 3 и 4) будут находиться в наиболее благоприятных заторможенных положениях, свободное вращение вокруг связи между углеродными атомами 2 и 3 будет изменять относительное положение атомов водорода при этих углеродных атомах, как это видно из схемы (рис. 1).
На этой схеме черные кружки изображают атомы углерода, белые кружки—атомы водорода при углеродных атомах 2 и 3 (остальные водородные атомы для простоты не показаны), более толстые линии связей соответствуют ближайшей к нам СН2-группе, а дуги показывают угол (ф) отклонения связи 3—4 от ее положения в схеме 1, а.
Потенциальная энергия каждой из конформаций, изображенных на рис. 1, может быть выражена уравнением:
Е ~—1,7-cos Зср — 0,5-coscp
* При этих рассуждениях не принимаются во внимание конкретные механизмы реакций. Так, например, изменение пространственной конфигурации при вальденовском обращении (см. том I, стр. 691 сл.) происходит фактически путем замены одного из атомов другим, точно таким же атомом. Однако если отвлечься от механизма реакции, а рассматривать только ее конечный результат, то и здесь можно говорить о временном разрыве связи.
Изменение потенциальной энергии при совершении полного оборота вокруг углерод-углеродной связи 2—3 представлено на
в с
Рис. 1. Конформации w-бутана: ,
.1—заслоненная (fully eclipsed}?'скошенная (vr’w, или gauche)', с—частично ва^ л 1енная (eclipsed); «/-—заторможенная (staggered).
эис. 2. Из рисунка видно, что у н-бутана имеются уже две неравноценные энергетически выгодные конформации, причем для перехода
из энергетически выгодной скошенной конформации (6) в еще более выгодную заторможенную конформацию (d) требуется преодоление потенциального барьера, соответствующего переходу через частично заслоненную конформацию (с), т, е. для этого перехода необходима энергия активации. Разница в энергиях этих конформаций такова, что если потенциальную энергию заторможенной конформации приравнять нулю, то энергии остальных будут составлять:
11збыток тт , энергии
Конформация ккал! моль
Заслоненная ............. 3,6
Скошенная ...................... 0,8
Частично заслоненная ... 2,9
Заторможенная ............ 0
Из рассмотрения рис. 1 и 2 можно сделать вывод, весьма важный для стереохимии алициклических соединений. Очевидно, что при
построении колец циклопропана, циклобутана и циклопентана взаимное расположение атомов отвечает самому невыгодному энергетическому состоянию (заслоненная конформация). Это создает в таких циклах дополнительное, так называемое торсионное (поворачивающее) напряжение, помимо предусмотренного теорией Байера углового напряжения. В кольцах циклобутана и циклопентана, где имеется противостояние четырех или пяти пар атомов водорода, напряжение за счет взаимодействия этих атомов столь велико, что кольцо не может уже более оставаться плоским. Установлено, что один из углеродных атомов циклопентанового кольца находится вне плоскости цикла, причем выступающим из плоскости является поочередно каждый из пяти углеродных атомов. Это отклонение от плоскостности составляет около 0,2—0,3 А.
Естественно, что при выходе углеродного атома из плоскости кольца возрастает угловое напряжение, однако это увеличение с избытком компенсируется уменьшением напряжения благодаря частичному приближению одной из невыгодных заслоненных конформаций к более выгодной скошенной конформации, т. е. к уменьшению торсионного напряжения. В результате неплоская конформация циклопентана оказывается энергетически выгоднее плоской приблизительно на 4 ккал/моль.
По мере возрастания числа атомов в цикле увеличивается возможность осуществления в них более выгодных скошенных и заторможенных конформаций. Однако рассмотрение колец с числом атомов углерода от 7 до 12 показывает, что в них все же должны присутствовать и невыгодные группировки. В ряду циклов от трехчленного до двенадцатичленного исключение составляет лишь циклогексан, одна из конформаций которого (стр. 29 сл.) построена только из энергетически сравнительно выгодных скошенных конформаций. Это следует как из геометрических соображений, так и из термохимических данных.
Действительно, точные измерения теплот сгорания (в расчете на одну группу СН2) дают следующие величины (в ккал!моль)'.
Циклопропан . . Циклобутан .. . Циклопентан . . . Циклогексан . . и.иклогептан . . Циклооктан . . Циклодекан . . Жидкость . . . . 158,7 . . . 157,3 . . . . 156,1 . . . . 157,0 . . . . 157,3 . . . 157,6 Газ 166,3 158,7 157,4 158,3 158,6
Следовательно, наименьшей энергией обладает циклогексан, а на-
пряженность циклопентана и циклооктана одинаковая. Лишь в циклах с числом атомов углерода более 12 теплота сгорания уменьшается и становится равной величине, получаемой для открытых цепей, так как в этих циклах углеродные цепи постепенно приближаются к плоским заторможенным зигзагам, характерным для парафинов.
ИЗОМЕРИЯ и СТЕРЕОИЗОМЕРИЯ ОДНОЯДЕРНЫХ АЛИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
Для гомологов и производных циклопарафинов возможна изомерия четырех видов.
1. Изомерия соединений, содержащих различные циклы, или изомерия ядер. Например, изомерами являются метилциклопропан и циклобутан или диметилциклопентан и циклогептан.
2. Изомерия радикалов, замещающих водородные атомы цикла,—изомерия боковых цепей.
3. Изомерия, зависящая от относительного положения боковых цепей или функциональных групп в цикле,—изомерия положения. Так, для диметилциклогексанов возможны следующие изомеры: 1,1-, 1,2-, 1,3- и 1,4-.
4. Изомерия, зависящая от расположения боковых цепей в пространстве,—стереоизомерия.
Первые три вида изомерии ясны без дополнительных пояснений. Стереоизомерия алициклических соединений отличается рядом особенностей, которые невозможны для ациклических соединений. Так, у циклических соединений, имеющих боковые цепи или заместители, возникает новый вид изомерии, обусловленной тем, что эти заместители могут располагаться либо по одну, либо по разные стороны кольца.
Этот вид стереоизомерии (^wc-транс-изомерия) внешне сходен с геометрической изомерией непредельных соединений, но вызван совсем другой причиной: не отсутствием свободного вращения у связей, образованных к-электронами (см. том I, стр. 380 сл.), а невозможностью перенесения заместителя с одной стороны кольца на другую без разрыва одной из а-связей в кольце. Далее, неплоское строение высших циклов вносит (начиная с шести членного) дополнительное своеобразие во взаимное расположение боковых цепей (см. ниже).
В случае циклопропана все три углеродных атома, разумеется, могут лежать только в одной плоскости; при этом заместители могут располагаться над этой плоскостью или под ней.
При замещении одного атома водорода в молекуле циклопропана образуется только один продукт, так как все углеродные атомы циклопропана тождественны, а потому обе возможные формы (I) и (II) также тождественны:
Замещение двух атомов водорода в этом цикле одинаковыми группами X при разных атомах углерода приводит к двум стерео-
изомерам, называемым (по аналогии со стереоизомерами с двойной связью) цис- и транс-изомерами:
цис-изомер
транс- изомер
Однако из этих двух стереоизомеров транс-изомер может существо! ать в форме двух зеркально-подобных конфигураций, так как оруктура (II), в противоположность структуре (I), не обладает плоскостью симметрии, и зеркальное изображение ее (III) не может быть совмешено со структурой (II). Следовательно, транс-изомер может быть разделен на оптические антиподы.
К тому же выводу мы придем, исходя из того, что каждый из двух стереоизомеров (цис- и транс-) обладает двумя асимметрическими углеродными атомами, но у rf/c-изомера имеет место внутренняя компенсация, аналогично мезовинной кислоте, а у транс-изомера такая компенсация невозможна. Поэтому оба асимметрических углеродных атома транс-изомера будут или правовращающими, или левовращающими:
Н
X-------•
X-------
н цис-изомер
и
н
X
транс-изомс-
В случае двух различных заместителей при разных атомах углерода на оптические антиподы может быть разделен кт только транс-, но и цис-изомер, так как в молекуле цис-изомера плоскость симметрии также отсутствует (асимметрические атомы различны и внутренняя компенсация невозможна):
цис- изомер
транс-изомер
X
Углеродное кольцо циклобутана уже не является плоской системой, однако это не отражается ни на числе изомеров, ни на их стереохимических отношениях. Поэтому молекулу циклобутана можно условно рассматривать как плоский квадрат, в котором
два одинаковых заместителя при разных атомах углерода могут за. иимать положения 1,2- и 1,3-:
/^гс-1,2- транс-1,2 цис-1,3- транс~\,3
(рацем </)
Изомер ^ас-1,2 нельзя разделить на оптические антиподы, а изомер транс-1,2 может быть расщеплен на правую и левую формы. Изомеры же цис-1,3 и транс-1,3 будут существовать каждый только в одной, оптически недеятельной форме, поскольку их конфигурации обладают элементами симметрии: первая—двумя плоскостями симметрии, проходящими перпендикулярно к плоскости квадрата через его диагонали, а вторая—одной плоскостью симметрии, проходящей через атомы 1 и 3, и осью симметрии (через атомы 2 и 4).
Если заместители неодинаковы, то это скажется только на .{«с-1,2-изомере, теряющем единственный имевшийся у . него элемент симметрии—плоскость симметрии—и приобретающем в результате этого возможность существовать в оптически деятельных формах. Что касается 1,3-изомеров, то они теряют лишь часть элементов симметрии: wue-форма лишается одной из двух плоскостей симметрии (проходящей через атомы 2 и 4), а транс-форма теряет только ось симметрии. Поэтому, несмотря на разные заместители, эти изомеры нельзя разделить на оптически деятельные стереоизомерные формы.
Циклопентан, по Байеру, должен был бы образовываться с минимальной деформацией валентных углов углеродных атомов (всего лишь 0и44'). Однако, как было указано выше, его углеродный цикл ле является плоским (стр. 26). Отклонение циклопентанового кольца от плоского расположения, впрочем, невелико, и при рассмотрении пространственного размещения заместителей можно, как и в предыдущем случае, считать кольцо плоским. В отношении стереоизомерии боковых цепей и заместителей к циклопентану применим тот же метод рассуждения, который был использован для циклопропана и циклобутана.
Циклогексан и высшие циклы не могут быть плоскими без деформации валентных углов углеродных атомов. Для шестичленного цикла возможны две формы без углового напряжения: форма «ванны», или «лодки» (иногда ее называют С-формой), и форма «кресла» (или Z-форма):
Обе эти формы образованы без деформации валентных углов, но энергетически они не вполне равноценны. Рассмотрение про-
странственных моделей этих двух форм показывает, что «кресло» построено из шести скошенных конформаций, а «ванна» из четырех скошенных и двух энергетически невыгодных заслоненных конформаций. Поэтому форма «кресла» должна быть энергетически выгодней на 5,6 ккал, что легко подсчитать на основании таблицы на стр. 25.
Рентгенографическое изучение, кристаллов симметричных производных циклогексана, электронографическое исследование в паровой фазе самого циклогексана (шведский ученый Хассель), а также термодинамическое и спектроскопическое исследование циклогексана (Питцер) подтвердили, что в обычных условиях более устойчива форма «кресла» (т. е. Z-форма). Поэтому циклогексан.в обычных условиях почти целиком состоит из молекул в Z-форме. Для превращения Z-формы в С-форму надо, например, повернуть атомы 3 и 5 Z-формы вокруг осей 3—2 и 5—6. Такое превращение требует затраты энергии в 5,6 ккал. При повышении температуры равновесное содержание С-формы в циклогексане увеличивается и в связи с этим возрастает его теплоемкость.
Интересно отметить, что, согласно расчету, превращение неплоской Z-формы циклогексана в плоское кольцо (с теми же размерами межатомных расстояний С—С= 1,5 А и С—Н= 1, Ю А, но с изменением валентных углов С—С—С до 120°) требует затраты энергии около 31 ккал
Из того, что циклогексан существует главным образом в форме «кресла», вытекает целый ряд следствий. Для удобства рассмотрения изобразим эту форму следующим образом (см. рис. 3). Как видно из рисунка, углеродные атомы кольца ^Ось симметрии • циклогексана располагаются в двух па-
• -----раллельных плоскостях: в верхней—атомы
Zz/ X й\’ 1, 3 и 5, в нижней—атомы 2, 4 и 6. Рас-
\ I/ стояние между плоскостями составляет око-
/Х/ ~~ / ' ло 0*5 А. Форма «кресла» имеет ось сим-
k Ks Pv / ; метрии третьего порядка, т. е. обладает бо-4 ' лее высокой симметрией, чем форма «ванны».
“ - 2 Среди двенадцати связей углерода с
водородом (или с другими заместителями)’ Рис 3 в Z-форме циклогексана можно обнару-
жить два типа связей С—Н. Шесть связей направлены к периферии молекулы; они получили название экваториальных связей (^-связи). Шесть остальных связей, чередуясь, направляются три вверх, а другие три вниз, параллельно оси симметрии: их называют аксиальными (осевыми), или полярными* (a-связи, или р-связи).
* Не следует смешивать употребляемый в^этом случае (не совсем удачно) термин «полярная» (англ, polar) с представлением об ионной, полярной связи. Здесь «полярная» означает: «направленная к полюсу оси симметрии». Вследствие явного неудобства термина polar он в последние годы вытесняется из мировой литературы позднее предложенным термином axial (аксиальная—осевая). Термин «аксиальная связь» (a-связь) и будет нами применяться в дальнейшем.
Распределение этих связей ясно видно из приведенных ниже проекционных формул:
Для молекулы циклогексана оба кружка, обозначающие атомы водорода (заштрихованный и незаштрихованный), одинаковы, и обе изображенные конформации идентичны. В случае однозамещенных циклогексанов, например монохлорциклогексана или метилциклогексана, в зависимости от того, находится ли заместитель, отличный от водорода, в экваториальном (е) или в аксиальном (а) положении, одна из конформаций будет энергетически более устойчивой и при обычной температуре будет преобладать. Повышенная устойчивость экваториальной формы связана с тем, что заместитель в a-положении, всегда имеющий больший, чем у водорода, ван-дер-ваальсовский радиус, заметно приближается к атомам водорода в положениях 3 и 5 и вступает с ними в стерическое взаимодействие. В случае же ^-положения заместителя такого взаимодействия не будет.
Это наглядно видно из рис. 4, где показаны модели а- и е-кон-формаций метил циклогексана. В модели 1 (а-конформация) один
Рис. 4. Модели аксиально”! и экваториальной конформаций циклогексана: /—аксиальная; 2—экваториальная.
из водородных атомов метильной группы оказывается очень сближенным с аксиальными водородными атомами при углеродных атомах 3 и 5.
Более высокая устойчивость экваториальной конформации подтвердилась при изучении диффракции света молекулами однозамещенных циклогексанов. При этом было показано, что преобладающей является конформация с экваториальным расположением заместителя; энергия образования г-формы метилциклогексана оказалась на 1,8 ккал/моль меньше, чем a-формы. Это различие энергии образования, однако, столь мало, что разделить такие формы (е- и а-) нельзя; можно только установить физическими методами исследования преобладание одной из них.
У ближайших гомологов метилциклогексана разница в энергиях а- и г-форм несколько больше. При неразветвленной цепи (этил, н-пропил, н-бутил) она составляет постоянную величину 2,1 ккал, но разветвление, увеличивающее объем радикала, сказывается сильнее: 3,3 ккал для изопропил- и 5,4 ккал для гпргт-бутилциклогексана.
При наличии в молекуле нескольких заместителей одинакового характера обычно более устойчивым оказывается такое их расположение, при котором заместители, как несущие одинаковые электрические заряды, так и электронейтральные, стремясь отталкиваться друг от друга, займут максимально удаленные друг от друга места (если только не будут превалировать иные взаимодействия между этими заместител я ми *).
Для двузамещенных, циклогексанов из этого можно сделать следующие выводы. 1,1-Дихлорциклогексан может быть, разумеется, только (к,1а)-изомером. Дас-1,2-, транс-1,3- и цис-1 ,4-дихлор-циклогексаны могут существовать только в виде г,а-конформаций: (к,2а)-, (к,За)- и (к,4а)- или, что то же самое, (1а,2г)-, (1а,Зг)- и (1а,4г)-:
цис-1.2 траноХ^ цис-1,4
В то же время "транс-1,2-, цис-1,3- и транс-1,4-дихлорциклогексаны могут существовать либо в виде г,е-конформациш
* В случае таких заместителей, как гидроксильные или карбоксильные группы, где становится возможным образование водородных связей (см. том I, стр. П2 сл.), определение преобладающей конформации еще более усложняется.
либо в виде а,а-конформаций:
Конформация (1е,2е)- должна быть более устойчивой, чем конформация (1а,2а)-, поскольку расстояния от заместителей (атомов хлора) до ближайших атомов водорода в первом случае больше, чем во втором.
Подтверждением этих рассуждений служит тот факт, что вычисляемый для 'ц/с-1,2-дихлорниклогексана (для конформации г,а) дипольный моментр.==3,1 D совпадает с величинами, определяемыми как в растворах его в бензоле, так и в парах (при 236°).
В то же время для транс- 1,2-дихлорциклогексана наблюдаемая величина •1= 2.66D в бензольном растворе (при 40°) соответствует содержанию 74% конформации е,е- и 26% конформации а,а-.
Аналогично, конформация цис~\е,3е- значительно более устойчива, чем конформация цис-1а,За-.
Транс-1,4-дигалоидциклогексан в твердом состоянии существует исключительно в виде диэкваториальной конформации (1е,4е)-; в растворах же в зависимости от природы растворителя может преобладать либо та, либо другая форма (1е,4е- или 1«,40-конформация).
Из сравнения различных возможных конформаций двузамещенных циклогексанов вытекает, что цас-формы 1,2- и 1,4-производных по устойчивости приближаются к транс-1,3-производному. Точно так же транс-формы 1,2- и 1,4-двузамещенных близки по устойчивости к ^нс-форме 1,3-двузамещенного. Этот важный вывод не мог быть сделан на основании только классических стереохимических представлений, т. е. без учета отталкивания между нейтральными, не связанными друг с другом атомами. Поэтому долгое время цис-формам 1,3-двузамещенных циклогексанов приписывалось транс-строение, а транс-формам—£{ас-строение.
Эта ошибка была исправлена только в 1947 г. (Питцер) методами конформационного анализа* на основании термодинамических расчетов.
Стереохимия высших циклов еще мало изучена, но некоторые закономерности уже начинают выясняться. Так, пространственное строение цикланов с числом атомов углерода в цикле от 7 до И несколько
* Разбор и установление энергетических соотношений различных конформаций, а также вывод тех или иных следствий в отношении физических свойств, направления реакций, большей или меньшей реакционной способности различных геометрических конфигураций в связи с неодинаковой устойчивостью присущих им конформаций называется конформационным анализом.
напоминает форму кресла циклогексана. Однако связи, соответствующие в наиболее выгодных конформациях аксиальным связям циклогексана, не параллельны друг другу, а несколько наклонены к оси, проходящей через центр кольца (квазиаксиальные связи). Поэтому с каждой стороны кольца имеются атомы водорода, как бы смещенные к центру и тем сильнее, чем больше звеньев в кольцах. Это приводит к некоторому сближению таких квазиаксиальных атомов и взаимному их влиянию.
Взаимное влияние этих атомов, расположенных далеко друг от друга в цепи углеродных атомов, но сближенных в пространстве, приводит к весьма своеобразным реакциям (см. стр. 88). Оно получило название трансаннулярного эффекта {annularis—лат. кольцеобразный).
Установление ^геометрической конфигурации стереоизомеров. Установить, какому из полученных стереоизомеров следует приписать цис-, а какому транс-конфигурацию, часто очень трудно. Для решения этой задачи используют как химические, так и физические методы.
Химические методы установления геометрической конфигурации могут быть разделены на абсолютные и относительные.
Наиболее общими абсолютными методами являются расщепление на оптические изомеры, а также получение производных, которые возможны только для одной конфигурации. Так, например, геометрическую конфигурацию двух стереоизомерных 1,3-диметилолцикло-гексанов с темп. кип. 55° (при 0,1 мм) и 114° (при 0,1 мм) можно выяснить, превратив каждый из них в какой-либо оптически деятельный диуретан, а затем выделив оптические антиподы любым из известных способов (см. том I, стр. 506). При омылении уретанов получаются оптически деятельный диол с темп. кип. 114° (при 0,1 мм) и недеятельный диол, кипящий соответственно при более низкой температуре. Таким образом, вышекипящий диол является транс-формой.
Другое доказательство состоит в замыкании оксигрупп тех же диметилолциклогексанов в окисное кольцо.
Нижекипящий диол, имеющий ^ас-строение, дает циклическую окись, а для вышекипящего (транс-форма) замыкание окисного кольца невозможно:
Относительные методы доказательства состоят в синтезе вещества из какого-либо другого с ранее установленной конфигурацией. При этом следует использовать только такие реакции, которые не сопровождаются изомеризацией. Кроме того, нельзя затрагивать связи у атома, несущего пространственно ориентированные боковые цепи. Например» в циклопентаидиоле
ОН
он
нельзя производить замещение гидроксильных групп, так как это может повести к реакции, аналогичной вальденовскому обращению (см. том I, стр. 692), ио водородные атомы в гидроксильных группах можно заменять в мягких условиях, не опасаясь изомеризации. Примером является доказательство конфигурации двух стереоизомеров циклогексан-1,4-дикарбоновой кислоты (темп, плавл. 167 и 310°). Конфигурации стереоизомерных циклогексендикарбо-повых кислот
НС ОС—/^>—СООН
могут быть установлены расщеплением одной из них на оптически деятельные антиподы. Гидрирование одного из последних в присутствии платины (реакция, не вызывающая изомеризации) дает циклогексан- 1,4-дикарбоновую кислоту с темп, плавл. 310°. Таким образом, высокоплавкая форма кислоты обладает /ярдас-конфигурацией.
Из физических методов наибольшее значение имеет определение дипольных моментов, так как дипольный момент ф/с-формы всегда больше.
Особенные трудности представляет определение геометрической конфигурации углеводородов, так как перечисленные выше абсолютные методы для них обычно неосуществимы, а дипольные моменты их слишком малы для получения шдежных результатов. Относительные методы доказательства вполне возможны, но представляют обычно большие практические трудности. Поэтому Для определения конфигурации углеводородов часто пользуются эмпирическим правилом Уиесрса—Скита согласно которому /пршш-формы двузамещенных цикланов ки-• >ят ниже и имеют более низкие удельные веса и показатели преломления. Однако пользоваться этим правилом надо с осторожностью, так как недавно было показано. что 1,3-диметилпиклогексаны и 1,3-диметилниклопентаны не подчиняются правилу Ауверса—Скита и у- этих соединении более низкие константы имеют I : С-формы
Ненасыщенные алициклические соединения, их изомерия и стереоизомерия. Все циклопарафиновые углеводороды имеют общую формулу СпН2/;, т. е. они изомерны этиленовым углеводородам. При введении в молекулы циклопарафинов одной двойной связи получаются ненасыщенные
циклические углеводороды общей формулы СпНа ряды которых начинаются с членов:
нс=сн
I I
НС=СН НС=СН Н2с сн2 и т. д.
\ / II \ /
СН2 HjC-CH, сн2
циклопропен циклобутен циклопентен
Кроме четырех видов изомерии, перечисленных на стр. 27, здесь возможна изомерия, связанная с положением двойной связи. Так. для высших гомологов возможны изомеры, содержащие этиленовую связь в боковой цепи, например:
Н2С---СН-СН^СН2
этенолциклопропан (винплтриметилен, или винвлцпклопропан)
а также изомеры, содержащие двойную связь между циклом и боковой цепью (так называемая «семиниклическая» двойная связь), например для циклопен гена:
1РС-С- СН2
I !
Н2С-СН
ме Гил*, нцнклобутан (метилиден гстраметилеч)
Введение в цикл двух двойных или одной тройной связи приводит к рядам C,7H2/7-i. При еще большем числе ненасыщенных связей можно получить ряды СПН2,СпН2и-8 и т. д. Номенклатура этих соединений строится аналогично изложенной на стр. 20 номенклатуре циклических соединений с одной двойной связью.
Примерами углеводородов с двумя двойными связями могу! служить:
НС— СН
НС СН
ЙТц
циклопентадиен
СН п.сй Ан 'I I нс сн.
/
сн
циклогексадиеи-1,4
СН
циклогексадиен-1,3
Если не учитывать реакционной способности, свойственной двойным связям, то в отношении устойчивости и прочности такие углеводороды представляют собою полную аналогию соответствующим насыщенным циклическим углеводородам.
Введение тройной связи в циклы с четырьмя, пятью, шестью и семью членами оказалось невозможным (Вильштеттер, А. Е. Фаворский). Однако получение циклооктина и высших гомологов не представило особых затруднений (Н. А. Домнин, Прелог, Бломквист); в циклопентадекановое и в циклогептадекановое кольцо Ружичке удалосьфввести тройную связь еще в 1933 г. В боковые цепи трой ные связи легко вводятся обычными методами.
Все эти закономерности можно объяснить, исходя из стереохимических соображений. Как известно (см. том I, стр. 120), группировка С—С~С—С вся располагается по прямой линии. Слеювательно, образование циклобутинового кольца было бы сопряжено с необходимостью преодолеть колоссальное напряжение, значительно превышающее напряжение в кольце циклопропана. С возрастанием числа углеродных атомов напряжение уменьшается, но даже в циклооктане напряжение, хотя и преодолимое, все еще довольно велико:
/С-СЕС-С\ с с
......с/
В гомологических рядах ненасыщенных алициклических углеводородов могут быть получены производные обычных типов (галоидные соединения, спирты, кислоты и т. д.).
Особый вид стереоизомерии наблюдается в высших циклах с двойной связью. В то время как низшие циклы с двойной связью могут существовать только в одной ф/с-форме, например:
СН2 II
/ W
Н2с н
СН2 II
при достаточно большой длине замкнутой в кольцо цепи возможна и /пране-конфигурация. Наименьшим кольцом, где это может осуществиться, является восьмичленное:
II 11 н
н.^ Хсн2 с ILC-CZ ХСН2
1 1 Н2С сн2 । । ] Н2С н сн.
н^с—сн2 /(г/с-циклооктен н^с—сн2 znpawc-цпклооктен
Способы получения алициклических соединений из соединений других рядов
1. Д е й с т в и е металлов на д в у г а л о и д и ы е соединения с отдаленными друг- от д р у / а атомами галоида. Например:
/СН,—Вг /СН2
Н.,С +2Ха —> Н2с | +2\аВг
^СН,—Вг \СН2
Обыкновенно для этой цели применяется натрий (Фрейнд) или, еще лучше, цинковая пыль в спиртовом растворе (Г. Г. Густав-сон). Таким способом сравнительно легко замыкаются в цикл три и четыре атома углерода; соединения с пяти- и шестичлеииыми циклами получаются с ничтожными выходами.
2. Действие спиртовой щелочи на галоидопроизводные, содержащие карбонильную группу (или ци ан групп у). Под влиянием карбонильной группы атомы водорода, находящиеся при соседнем углеродном атоме, приобретают подвижность, благодаря чему легко происходит циклизация:
/СН2—Вг /СН2
Н2С 4- NaOIl —> Н2с I 4- \аВг + Н2О
хсн2-со—сн3 \сн—СО—СН3
Таким путем легче всего образуется трехчленное кольцо.
3. Действие жирных диазосоединений на соединения с этиленовой связью (Бухнер). Реакция приводит к образованию триметиленового цикла, причем промежуточно образуются пиразол ины:
С9Н5ООС СООС2Н5 С2Н5ООС СООС2Н5 СоН5ООС СООС2Н5
II II II
СН=СН НС--СН НС СН
, -* II \/ +N2
' Н2С N СН2
’ ch2n2 \ /
N
4. Действие гидразина на а, р-н е п р е д е л ь-ные карбонильные соединения (Н. М. Кижнер). В этом случае реакция также протекает с промежуточным образованием пиразолинового цикла, например:
СН=СН—СН(СН3)2 Н2С-СН-СН(СН3)2
I 4- —> I I >
сн3~ос nh2 СН3-С NH
h2n/
Н2сх
I СН-СН(СН3)2 + N, СНз-Нс/
5. Получение циклических кислот при помощи мало нового эфира. Эти синтезы представляют собой модификацию синтезов карбоновых кислот жирного ряда. Циклические кислоты могут быть получены двумя путями.
а) Действие двугалоидных соединений на натриймалоновый эфир (Перкин):
/СН2Вг /СН 2- СН(СООС2Н5)2
Н2С + NaCH(COOC2H5)2 Н2С 4- NaBr
\CH2Br \сН2Вг
,СН2—СН(СООС2Н5)2 /СН2—СХ’а(СООС2Н6)2
Н.2С + NaCH(COOC2H5)2 -> Н2С + СП2(СООС2Н5)2
\сН2Вг \сН2Вг
/CH2-CNa(COOC2H8)2 ^СООС2Н8
Н2С —» Н2С С + NaBr
XcH2Br Х'Н, ^COOQHg
СН2 СООС2Н8 СН2 СООН СН2
Н2(/ с -> н2с -> Н2С СН—СООН 4- со2
'YooQHg ^сн. 'соон \:н.
Метод Перкина был значительно усовершенствован Н. М. Киж-нером, предложившим применять в качестве дигалоидопроизводных хлоробромиды. Благодаря этому стадии образования хлоралкил-малонового эфира и замыкания кольца стало возможным проводить раздельно (при разных температурах).
При проведении реакции по Кижнеру значительно уменьшается выход побочных продуктов и увеличивается выход циклических кислот.
б) Циклизация эфиров четырехосновных кислот, получаемых из малонового эфира (см. том I, стр. 450). Так, например, в эфире
zCH2—СН(СООС2Нб\
Н2с \СН2-СН(СООС2Н6)2
который получается действием двух молекул натриймалонового эфира на триметилендибромид, атомы водорода групп СН можно заменить натрием и на полученное динатриевое производное действовать иодом:
/СН2—CNa(COOC2H5>2 уСН2—С(СООС2Н5)2
Н2С +2J —» Н2С | + 2NaJ
\CH2-CNa(COOC2Hs)2 \CH2-QCOOC2H8)2
Омылением и нагреванием такого эфира четырехосновной кислоты можно получить циклическую двухосновную кислоту:
уСН2—СН—СООН
Н2С |
ХСН2—СН—СООН
Легче всего этим способом получаются производные с пяти-и шестичленными циклами, но синтез соединений с меньшими кольцами также вполне возможен.
Аналогично синтезам при помощи малонового эфира могут быть осуществлены синтезы и с натрийацетоуксусным эфиром. Так, например, при действии
дибромэтана на ацетоуксусный эфир в присутствии алкоголята натрия получается эфир р-кетонокислоты циклопропанового ряда:
BrH2C CHNa—СОСН3 Н,С—СН-СОСН.,
|+| -> I I
BrH2C СООС2НВ BrH2C COOCjHg
н2с—сн—сосн3
I I + NaOC.H.
ВгН2С СООС2Н6
Н,С—CNa—СОСН3
I | + С2Н5ОН
ВгН2С СООС2Н8
Н2С— CNa—COCHj
ВгН,С COOCjM,
H,C< /СОСН8
-> I ;c^ +NaBr HtC/ ХСООС2НБ
Этот эфир после его омыления и отщепления углекислоты (см. кетонное расщепление ацетоуксусного эфира,—том I, стр. 526) дает метилциклопропилкетон:
COCH,
COOC2Hg
СОСН3
СООН
Н,С
Рсн-сосн3+со2 н2с/
6. П о л У 4 е н и е циклических .кетонов из двухосновных кислот. Подобно кальциевым солям одноосновных кислот, кальциевые соли двухосновных кислот при сухой перегонке отщепляют углекислый кальций и образуют кетоны. Однако в этом случае образование кетона происходит внутри одной молекулы, благодаря чему получается циклический кетон:
1+С-СН,—СН,—СОО\ н,с—сн,—сн,
j Са —> /СО + СаСО3
Н2С—СН2—сн,—соо/ Н,С—сн,-бн2
циклогептанон (суберон)
Реакция эта идет гладко при получении циклических кетонов с пяти-, шести- и семичленными циклами. Соединений с тремя и четырьмя атомами углерода в цикле этим путем получить не удалось. Образование кетонов с восьмичленным циклом (циклооктанона) и с девятичленным циклом (циклононанона) идет не гладко,— сопровождается значительной изомеризацией и разложением.
Кетоны с пятью-шестью атомами углерода в цепи легко могут быть получены также каталитически—нагреванием двухосновных кислот с некоторыми окислами металлов (MnO, ThO2, ВаО и др.). При этом отгоняются кетоны и выделяются вода и углекислота:
СН2—СН2—СООН Н2С-СН2
—> | СО ~р Н2О -р со2
СН2—СН2-СООН н2с—сн2
Благодаря высоким выходам и простоте условий проведения реакции, каталитический способ котонизации кислот применяется
в настоящее время для получения циклопентанонов почти во всех случаях, а разложением кальциевых солей пользуются лишь при кетонизации нестойких кислот, разлагающихся в нежелательном направлении при температуре катализа.
Ружичка нашел, что для получения более высокомолекулярных кетонов, начиная от циклооктанона до циклооктадеканона, лучше всего подвергать сухой перегонке под уменьшенным давлением ториевые соли двухосновных кислот. Хуже всего идет образование кетона с 10 атомами углерода в цикле (выход—не более 0,2% от теоретического). С дальнейшим увеличением числа атомов углерода в цикле выход кетона постепенно увеличивается и делается значительным для кетона С18Н34О.
Ружичка установил, что при сухой перегонке ториевых солей двухосновных кислот, наряду с монокетонами, образуются в небольшом количестве циклические дикетоны (из двух молекул двухосновной кислоты) согласно уравнению:
—О—ОС—(СН2Ц—СО—О—/СН,).
Th • т ОС CO + Th(CO3)2
О—ОС— (СН2)Х—СО—О-~ \сн2)д/
Th
Здесь —— эквивалент тория.
Этим путем из азелаиновой кислоты были получены дикетсны с 16 атомами углерода в цикле, из себациновой—с 18 атомами, из декандикарбоновой—с 22 атомами, из тетрадекандикарбоновой—с 30 атомами и др.
7. Конденсация эфиров и ди нитрилов двухосновных кислот. При действии натрия, алкоголята натрия или амида натрия на эфиры двухосновных кислот аналогично конденсации эфиров одноосновных кислот, приводящей к образованию (В-кетонокислот с открытой цепью, происходит конденсация с образованием циклов. В случае эфиров адипиновой и пимелиновой кислот и их гомологов цикл замыкается внутри одной молекулы (Дикман):
Н2С--СО—ОС2Н5 Н2С--со
I -> I I + С2н5он
Н2С СН2-СООС2Н5 Н2С СН-СООС2Н5
При конденсации эфира янтарной кислоты в образовании цикла участвуют две молекулы эфира:
СООС2Н6 СООС2Н6
I I
СН2—СН? СН—СН2
С2Н5О-ОС + СО—ОС2Н5 -» 0(4 \о + 2С,Н5ОН £, О 1 Z О . 1 U
СН2—СН2 СН2—СИ
I I
СООС2Нв СООС2Н5
Получающийся при этом так называемый сукцинилянтарный эфир представляет собой эфир р,р'-дикетоциклогександикарбоновой кислоты (циклогексан-2,5-дион-1,4-дикарбоновый эфир).
Подобные же конденсации, аналогичные образованию (З-дикетонов, возможны для эфиров у-кетонокислот:
Н2С----СН2
I I
ос со—ос2н5
"сНз
н2с—сн2
I I + С2Н5ОН
ОС со
Из динитрилов двухосновных кислот циклические кетоны могут образоваться таким путем (Торпе):
/CH2-C=N
(СН2). (СН2). C=NH
\сн2—GeeN \ /
+зн2о
1 СООН
о+ СО
2
Конденсация динитрилов может протекать не только внутримолекулярно с замыканием цикла, но и межмолекулярно, приводя к нежелательным линейным молекулам. Чтобы избежать этого, надо очень медленно прибавлять предельно малые количества динитрила к раствору конденсирующего агента (принцип Руггли—Циглера; тогда процесс внутримолекулярной конденсации успевает пройти до встречи одной молекулы динитрила с другой такой же молекулой. Этим путем Циглеру удалось получить, например, восемнадцатичленное кольцо циклооктадеканона с выходом до 70%.
8. Ацилоиновый метод. Как дальнейшее развитие описанного выше метода получения кетонов с большими циклами можно рассматривать разработанный в последние годы так называемый ацилоиновый метод (Штоль, Прелог). При кипячении эфиров дикарбоновых кислот с металлическим натрием происходит внутримолекулярная ацилоиновая конденсация'.
.-----СООС2Н5 !со
I I
(СН2). > (СН,).
I-----СООС2Н6 I-------------снон
Образующиеся оксикетоны могут быть легко превращены в циклические кетоны.
Для проведения ацилоиновой конденсации не требуется большого разбавления, и кетоны получаются с исключительно высокими выходами. Например, выход оксикетона с 21 атомом углерода достигает 96%, а циклы с 9—11 атомами углерода, для которых по методам Ружички и Циглера выход не превышает 0,5%, получаются с выходом около 40%.
9. Конденсация эфиров двухосновных кислот с гомологами ацетона. В эту реакцию легко вступают эфиры щавелевой кислоты, причем образуются трикетоны с пятичленным циклом:
R R
I i
,СН2 СО—ОС2Н5 /СН—со
ОС + | -> ОС | + 2С2Н5ОН
\сн, • СО—ОС.Н. \сн—со
I I
R R
С самим ацетоном реакция идет труднее. Вместо кетонов могут быть взяты и .ругне соединения, содержащие две активные метиленовые группы в р положении, щпример- эфир глутаровой кислоты:
СООС2Н5
/СН2
Н2С +
\сн2
I
СООС2Н5
СООС2Н5
I
СО—ОС2НЕ СН—СО
| —> Н2С | +2С.Н6ОН
СО—ОС2Н5 \сн—со
I СООС2Н8
10. Действие динитрила бром малоновой кислоты на кетон ы. Реакция протекает в присутствии йодистого калия по схеме:
>со +
R'z
CHBr(CN)2
CHBr(CN)2
R/ /CBr(CN)2 kj
R'//C\cBr(CN)2
R/ /CJ(CN)2
R'/C\cJ(CN)2
R\ zC(CN)2 >C< I + J2
Rz/ xC(CN)2
И. Циклизация замещенных дивинилкето-н о в. Пожалуй, простейшим методом получения многозамещенных соединений ряда циклопентана является путь через дивинил-ацетиленовые углеводороды, гидратацией которых могут быть получены непредельные кетоны. Под действием минеральных кислот эти кетоны легко циклизуются (И. Н. Назаров), например:
сн3—с Хсн
II II
СНз-СН СН
I сн3
СО СО
СН3—С ЧСНо
II I
сн3—с—сн I • СН3
12. Внутримолекулярная конденсация у- и о-д и к е-тонов. Аналогично конденсации уксусного альдегида в кротоновый альдегид и ацетона в окись мезитила протекает внутримолекулярная конденсация у- и о-ди кетонов:
си, сн3
' С
/С° / \
нос сн3 Н2С СН
I I -> I I 4- Н2о Н2С со Н2С со
13. Уплотнение с замыканием цикла ненасыщенных соединений, .содержащих этиленовые и ацетиленовые связи». Уплотнение простейших этиленовых соединений, содержащих одну кратную связь, приводит главным образом к образованию мало исследованных смесей циклических углеводородов с широкими пределами температур кипения. Такая смесь циклических углеводородов получается, например, при полимеризации этилена под давлением в присутствии окиси алюминия. Образование индивидуальных циклических соединений при полимеризации олефинов наблюдалось лишь в немногих случаях. Так, недавно димеризацией изобутилена под давлением был получен 1,1,3-триметилциклопентан (Мак1\инлей). Уплотнением углеводородов с этиленовыми связями, возможно, объясняется также образование многих циклических углеводородов при крекинге нефти.
Лучше изучено получение соединений с четырехчленным циклом из некоторых более сложных производных этилена. Такова димеризация аллена и его гомологов при нагревании (С. В. Лебедев):
Н2С=С-СН2 сн2=с=сн2 сн2—с=сн2
II I I
Н,С-С=СН, СН,=С=СН, сн,—с=сн2
Совершенно аналогично протекает димеризация кетенов (см. том I, стр. 399). Так, под действием триэтиламина хлорангидрид изомасляпой кислоты первоначально превращается в диметилкетен
СН3\
/СН-СОС1 —> >С=С=О 4- НС1
СН/ СИ/
далее димеризующийся в 1,1,3,3-тетраметилциклобутандион-2,4:
снзх >с=с=о сн3 >с-с= О
СН/ + /СН3 _ > CH3Z | | /СН3
0=с=с\сн3 о=с- с< "СН3
Хорошо изучена димеризация диеновых углеводородов с сопряженными двойными связями в соединения с шестичленными циклами (стр. 46, а также том 1, стр. 368).
Сравнительно недавно была открыта способность ацетилена полимеризоваться в особых условиях (см. том I, стр. 413) с образованием восьмичленного и десятичленного циклов. Поведение гомологов ацетилена е этом отношении еще не изучено.
14. Гидрогенизация производных бензо-л а. Алициклические соединания с шестью атомами углерода в цикле—производные циклогексана—проще всего получаются присоединением атомов водорода к бензолу и его производным, т. е. гидрогенизацией соединений ароматического ряда.
Присоединение водорода к различным ароматическим соединениям происходил с различной степенью легкости и в различных условиях. Двухатомные геиолы легко присоединяют водород in statu nascendi. Сравнительно легко гидрогенизируются ароматические двухосновные кислоты, труднее—одноосновные .ислоты (действием натрия на кислоту в кипящем растворе ее в изоамиловом спирте), еще тру л не с--углеводороды и особенно трудно—сам бензол. Правда, как установили еще Вертело и Ф. Р. Вреден*, ароматические углеводороды при нагревании с крепкой поди слово дородной кислотой присоединяют шесть атомов юдорода, но эта реакция сопровождается изомеризацией (Н. М. Кижнер).
Очень гладко присоединение водорода к ароматическим соединениям идет в присутствии мелкораздробленных металлов, действующих каталитически. Бензол и другие ароматические соединения легко присоединяют водород в присутствии мелкораздроб-leiiHoro никеля (Сабатье и Сандеран**), причем из бензола получает-я циклогексан
С6Н6 + ЗН2 —> С6Н12
з из других ароматических соединений—производные циклогексана. Еще лучше эта реакция протекает в присутствии мелкораздроблен-• юй платины при атмосферном давлении или в присутствии скелетного никелевого катализатора под давлением водорода.
15. Присоединение диеновых углеводородов ссо-пряженными двойными связями к ненасыщенным о е д и н е н и я м (диеновый синтез). Метод Дильса—Альдера (см. ;ом I, стр. 461) дает возможность получать различные алициклические соединения '• одним или несколькими циклами, например:
узн, /С\
НС сн. НС сн,
I + II -> II I
НС сн—сно НС сн-сно
' CIO сн>
бутадиен акролеин
Реакция протекает обычно чрезвычайно легко (иногда при простом смешении компонентов при комнатной температуре).
* Феликс Романович Вреден (1841 —1878). Профессор Петербургского горного института. Изучал действие иодистоводородной кислоты на бензол и нафталин; открыл камфарную кислоту.
** Поль Сабатье (1854—1941) и его долголетний сотрудник Жан Батист Сап-1еран (1856—1936)—французские химики. Поль Сабатье одним из первых применил парофазные контактно-каталитические реакции в органической химии.
В случае диеновых углеводородов с сопряженными двойными связями эта реакция при повышенной температуре может протекать и между двумя одинаковыми молекулами (С. В. Лебедев), например:
//~ / \
НС СН—СП=СН2 НС СН—сн=сп2
I 4- > -» I! I
нс сн2 НС сн.
Аналогично могут быть получены бициклические и трициклические соединения (стр. 97).
16. Прямая циклизация парафинов. Недавно была открыта реакция прямой циклизации парафинов в циклопентаны в присутствии платинового катализатора (Б. А. Казанский, А. Л. Либерман). Например:
9Н3 СИз Н2С—СН2
СН3-С—СН2—СН—С1’з —» | ^>СН—СН3-ь н2
СН3 СН3—с-сн2
сн3
Ряд случаев образования алициклов описан в разделе о терпенах (стр. 118 сл.).
Превращения циклов друг в друга
Одной из наиболее характерных особенностей химии алициклических соединений является то, что при некоторых химических реакциях может происходить превращение циклов друг в друга. Важно отметить, что в жирном и ароматическом рядах многие из этих реакций протекают без какой-либо изомеризации углеродного скелета.
Превращение циклов друг в друга состоит в том, что в кольцо углеродных атомов вступает один из атомов боковой цепи и таким образом происходит расширение цикла, или же, наоборот, один из углеродных атомов цикла переходит в боковую цепь и, следовательно, происходит сужение цикла. Подобные изменения углеродного скелета, относящиеся к атомным перегруппировкам, были впервые установлены для более сложных алициклов—бициклических соединений класса терпенов. При исследовании строения природных соединений этого класса превращения циклов часто вызывали большие затруднения. Позднее такие атомные перегруппировки были обнаружены и у соединений с одним углеродным циклом. Систематическое изучение алициклических соединений показало, что эти превращения не являются случайными, но составляют коренное свойство некоторых циклов.
' Закономерности, наблюдаемые при взаимных превращениях циклов, широко изучались Н. М. Кижнером и Н. Я. Демьяновым*.
1. Восстановление иодистоводородной кислотой. При достаточно высокой температуре восстановлением иодистоводородной кислотой галоидных соединений, в которых атом галоида связан с углеродным атомом, входящим в шести- или семичленный цикл, получается углеводород с циклом, содержащим на один атом углерода меньше {сужение цикла). Так, из иодциклогеп-тана при 250° получается не циклогептан, а метилииклогексан (В. В. Марковников):
Н2С-СН2 ^CIIJ
I ! -Н UJ н?с сн2
сн2
^СН-СНз
I I н,с сн.
+ J
а из иодциклогексана—метилциклопентан (Н. Д.^Зелинский). При восстановлении бензола крепкой иодистоводородной кислотой при 270° также получается не циклогексан, а метилциклопептан (Н. М. Кижнер).
2. Действие кислот на спирты. При действии кислот на спирты с трехчленными и четырехчленпыми циклами может происходить превращение низших циклов в высшие (Н. Я. Демьянов, Н. М. Кижнер). Например, при нагревании циклобутилкарби-нола со щавелевой кислотой главным продуктом отщепления воды является циклопентен:
Н2С-СН-СН2ОН
112С-СН2
Н2С—сч2
^СН + Н,0 н2с-сн
В ряду циклобутана при дегидратации изомеризуются даже третичные спирты:
НоС-СН-С—ОН
“I : I
н.,с-сн, сн3
Н2С—СН2
| Дс—си3
Н.,С—с
I СПз
* Николай Яковлевич Демьянов \ (1861—1938). Профессор Московской 'сельскохозяйственной академии, ныне Сельскохозяйственной академии имени К. А. Тимирязева. Впоследствии академик. Работал в области алициклических ^'единений; нашел и разъяснил многие реакции превращений одних циклов в ipy г не.
Во многих случаях с расширением цикла протекает дегидратация циклоалкилкарбинолов с пяти-, шести- и семичленными кольцами под действием щавелевой кислоты или хлористого цинка.
Действие бромистого водорода на циклопропилкарбинол даже при низких температурах приводит к образованию смеси нормального продукта реакции, содержащего трехчленный цикл, с цикло-бутилбромидом:
Н2С.
| ;сн—сн2он
Н2с/
Н2С
I ХСН—СН2Вг
Н2С—СНВг I I
Н2С-СН,
При действии же кислот на циклобутанол реакция может итти в сторону сужения цикла—циклобутанол отчасти превращается в циклопропилкарбинол:
н2с—сн—он h2q
|| -^ | ;сн-си2он
н2с—сн2 н2с/
Так же протекает реакция алкоголиза циклобутилхлорида и циклопропил-метилхлорида в разбавленном спирте:
Одинаковый состав продуктов реакции позволяет предположить, что в обоих случаях промежуточно образуется катион С4Н^“, легко изомеризующийся с изменением величины цикла в любом из двух направлений.
3. Действие азотистой кислоты на циклические амины. Из всех реакций превращения циклов эта реакция исследована лучше всего (Н. Я. Демьянов). Она изучена, начиная от трехчленного и кончая восьмичленным циклом. При действии азотистой кислоты на первичные амины, в которых аминогруппа находится при углеродном атоме, входящем в цикл, например на цнклогептиламин
Н2С—СН2
Н2С ^СНа
I I
н2с сн,
сн I nh2
должен был бы образоваться циклогептанол. В действительности же получается смесь циклогептанола с циклогексилкарбинолом, образующимся вследствие изомеризации с сужением цикла:
Н^С-СН*
Н3С
I 1
н2с Сна
\ / \ / сн
I он
сн* нХ Хсн, " н,с сн, снгон
Смесь тех же алкоголей получается и при действии азотистой
кислоты на аминометилциклогексан
СН*
i
ch2nh3
гак как в этом случае замена аминогруппы на гидроксил частично сопровождается расширением цикла. приводящим к образованию циклогептанола.
Вероятно, каки в предыдущем случае, изомеризация происходит на стадии карбоний-иона.
4. Пинаколи новые перегруппировки циклических пинаконов. Для алициклических пинаконов реакция вполне аналогична перегруппировке простейших алифатических пинаконов (см. том I, стр. 420).
Одним из возможных направлений этой реакции для пинаконов типа
С(ОН)—QOHZ
является превращение в пинаколин с расширением циклах
Наоборот,
пинакон типа
сн2
н2с
•Н-2
Н2С С(ОН)—сн3
С(ОН)
превращается
в
СН3 пинаколин с сужением цикла (С. С. Наметкин): Н2С----------СН2
I I /СН3 н,с с<
СН2
3
п и
лен нов
5. О тщ е лоидгидр реакция, аналогично
и е
цикли
предыдущей, протекает с сужением цикла:
СН3
о т гликолей.
галоидоводородов ч е с к и х
г а-Эта
СН
Н2<
сн2
СН3
Н2с---СН
Н2С СНВг
Н2(
СН2
С(ОН)
сн
со-сн3 на циклические а-г а-
СН3
6. Действие едких щел лоидкетоны. Реакция приводит к атомов углерода в цикле, причем возможно, что в качестве промежуточных продук-
тов образуются бициклические соединения СО
очей получению кислоты с меньшим числом
с пяти-
СО
и трехчленными кольцами:
Д'
-НС1
НС---СН
+н2о
Н2С-СН—СООН
Н2С сн.
2
Н2С СН2
СН.
7. Действие диазометана нон и его высшие гомологи.
н2<
СН2
СН2
на циклогекс а-При действии диазоме-
тана на циклогексанон получается с расширением кольца циклогеп-
танон:
I I
Н3С СН,
+ CH,Na
Н2С—СН2
Н2(С 'сН, I I + N.
н2с СН, 'со
Точно так же можно перейти от циклогептанона к циклооктанону, от циклооктанона к циклононанону и т. п.
8. Действие хлористого алюминия на у глеврдороды. При действии безводного хлористого алюминия углеводороды ряда циклобутана на холоду превращаются в углеводороды циклопентанового ряда (М. Б. Турова-Поляк), которые далее изомеризуются в циклогексановые углеводороды. Наоборот, при нагревании циклогексановые углеводороды дают в присутствии хлористого алюминия углеводороды с пятичленным циклом (Аскан, Н. Д. Зелинский, Неницеску). При превращении алкилциклопентанов в циклогексаны, как и при обратном переходе, всегда получается смесь тех и других, причем состав смеси зависит от температуры опыта. Углеводороды, содержащие более шести атомов углерода в кольце, в присутствии хлористого алюминия могут изомеризоваться только с сужением цикла, например циклогептан изомеризуется в метил циклогексан.
Эти закономерности (Н. Д. Зелинский, М. Б. Турова-Поляк) могут быть выражены схемой:
<-------------
при нагревании
на холоду
9. Нагревание циклогексан карбоновой кислоты с хлористым цинком. При этом отщепляется углекислота и получается смесь циклогексана и метилциклопентана (И. Д. Зелинский, И. Ф. Гутт).
10. Превращения тетрабромида пентаэритрита. Интересные превращения циклов происходят при реакциях, для которых исходным продуктом является четырехатомный спирт пентаэритрит (Г. Г. Густавсон). При действии цинковой пыли на тетрабромид пентаэритрита
Вг—Н2Ск ,СН2—Вг
XCZ
следовало бы ожидать получения углеводорода с двумя триметиленовыми циклами—так называемого спиропентана’.
Н2СЧ zCH2
|)<| H2CZ хсн2 Однако при этом как главный продукт получается метиленциклобутан (темп, кип, 41°, уд. вес 0,6950 при 10°)
Н2С—С—СН2
H2i-CH2
в смеси с метилциклобутеном (темп. кип. 37,5°; уд. вес 0,7052 при 18е) НС=С—СНЭ
Н,С-СН, в лишь небольшое количество спиропентаиа.
Продукт присоединения хлорноватистой кислоты к метиленциклобутан) в присутствии воды уже при обычной температуре, а еще легче—при нагревании, переходит в циклопентанон:
Н2С—С(ОН)—СН2СI Н2С—сн,
N - | уо + на н,с—-сн, н,с- сн,
Таким образом, здесь наблюдается последовательное превращение трехчленного цикла в четырехчленный, а последнего—в пятичлеиный, что в свое время чрезвычайно затруднило установление строения углеводородов, полученных Г. Г Гу ставсоном из пентаэритрита.
11. Фотохимический распад циклопентанона. Цик лопентанон может подвергаться фотохимическому распаду, давая с хорошим вы ходом циклобутан (Кистяковский, 1942 г.):
Н2С--СН, Н,С—СН,
1 J I I 4-СО
Н2С СН, щс—сн,
\о
Свойства алициклических углеводородов
Химические свойства. Естественно ожидать, что по химическим свойствам циклопарафины должны обнаруживать большое сходство с парафинами. Исследование этих углеводородов, однако, показало, что некоторые реакции низших цикланов близки к реакциям этиленовых углеводородов. Лишь начиная от углеводородов с пятичленным циклом, цикланы по многим свойствам оказываются сходными с парафиновыми углеводородами, оправдывая название циклопарафинов.
1. Действие галоидов. Действие галоидов на циклопропан и его производные чрезвычайно напоминает действие галоидов на этиленовые углеводороды.
При действии брома происходит размыкание кольца (т. е. разрыв связи между двумя углеродными атомами) и атомы галоида присоединяются по месту освобождающихся валентностей углерода:
Н2С--сн2
\ / 4- Вг? —> СН2Вг—СН,—СН,Вг
• ’ СН2
При действии хлора на циклопропан присоединение галоида сопровождается реакцией замещения:
Н2С—СН, Н2С----СНС1
' \ / 4-С1, —* \ / 4- HCI
СНа СН,
Гомологи циклобутана присоединяют бром гораздо менее энергич но, чем углеводороды с трехчленным циклом, а сам циклобутан присо
единяет его лишь с трудом при повышенных температурах. При действии галоидов на углеводороды ряда циклопентана и высшие циклические углеводороды цикл не размыкается, а происходит только замещение атомов водорода на галоид (металепсия):
н4С—сня н4с-сн2
I \сН, + С1, —► I \CHC14-HC1
н/-Сна Н2С—сн,
2. Действие галоидоводородных кислот. Действие этих кислот на соединения циклопропанового и циклобу-ганового рядов подобно действию галоидов, т. е. и здесь, особенно легко при действии иодистоводородной кислоты, происходит размыкание цикла и реакция присоединения.
Легче всего присоединяют иодистый и бромистый водород соединения триметиленового ряда, например:
Н4С--СН,
\ / 4- НВг СН3—СНа—СН2Вг
СН,
Для соединений ряда циклобутана реакция идет лишь, при нагревании, да и то иногда с трудом. Углеводороды с высшими циклами галоидоводородных кислот не присоединяют. Например, циклопен-гадекан и циклогептадекан, а также циклотриаконтан, даже при продолжительном нагревании при 250° с иодистоводородной кислотой уд. веса 1,96 не подвергаются никакому изменению, напоминая в этом отношении парафины.
.Однако при очень высоких температурах возможно размыкание и высших циклов. Так, действием крепкой иодистоводородной кис* лоты в этих условиях на продукты гидрирования бензола (состоящие, главным образом, из метилциклопентана) Н. М. Кижнер получил некоторое количество гексана. Впрочем, в таких жестких усло-зиях может происходить разрыв связей даже в парафинах (с образованием низших парафиновых углеводородов).
Для производных циклопарафинов размыкание циклов при действии галоидов и галоидоводородных кислот происходит с различной степенью легкости. Труднее всего размыкается кольцо циклических кислот, в которых карбоксильные группы связаны с углеродными атомами, входящими в цикл. Циклические кетоны, содержащие в цикле от 7 до 9 и от 12 до 18 углеродных атомов, при нагревании с концентрированной соляной кислотой при 180—190° лишь слегка обу гливаются.
3. Гидрогенолиз. Водород присоединяется к триметиленовым соединениям труднее, чем к этиленовым. Эта реакция может протекать в присутствии платины, палладия или никеля при несколько более высокой температуре.
Гидрогенолиз происходит с разрывом цикла и образованием предельных алифатических соединений:
Н2с---СН.,
\ / “ + Н, —» СН8—СН,—сн.
сн2
Температура, требуемая для присоединения водорода к тетраметиленовым соединениям, выше, чем для триметиленовых.
При еще более высокой температуре размыкаются с присоединением водорода не только низшие, но и пятичленные циклы. Эта реакция легко идет в присутствии платины (Н. Д. Зелинский, Б. А. Казанский, А. Ф. Платэ).
Подробное изучение этой реакции (Б. А. Казанский) показало, что разрыв пятичленного кольца легче всего происходит по углеродуглеродным связям, удаленным от атома углерода, несущего боковую цепь:
Если в молекуле углеводорода не имеется таких удаленных связей, например в 1,2,4-триметилциклопентане
цикл вообще почти не размыкается.
Аналогично гомологам циклопентана ведут себя гомологи циклопропана (см. также стр. 57); гидрогенолиз же гомологов циклобутана протекает по всем возможным направлениям:
Соединения, содержащие высшие циклы, в присутствии катализаторов водорода не присоединяют. Циклогептан, например, в присутствии платинового или никелевого катализатора не размыкается, а изомеризуется в метил циклогексан. Аналогичным образом из циклооктана получаются метилциклогептан и диметилциклогексан (Н. Д. Зелинский и др.).
4. Отношение к окислителям. В отношении к окислителям наблюдается резкая разница между этиленовыми соединениями и соединениями, содержащими циклопропановое кольцо.
При обычной температуре циклопропан и его гомологи лишь медленно окисляются перманганатом в щелочной или в нейтральной среде, а циклобутан и его производные, как и соединения с высшими циклами, ведут себя в этом отношении совершенно аналогично предельным углеводородам.
При более высоких температурах и при действии более энергичных окислителей, как и в случае парафинов, происходит окисление, сопровождающееся разрывом связей между углеродными атомами. Но в то время как для парафиновых углеводородов и их производных разрыв связей между углеродными атомами ведет к расщеплению молекулы, для циклических соединений характерно образование при окислении двухосновных кислот с тем же числом атомов углерода в молекуле.
Так, например, при окислении циклопентана получается глутаровая кислота:
/СН2-СН, /СН,—СООН
Н2С | +50 -> Н2С + н2о
\сн2—сн, \сн2-соон
при окислении циклогексана—соответственно адипиновая кислота и т. д.
Если при окислении какого-либо углеводорода получается двухосновная кислота с тем же числом атомов углерода в молекуле, это является несомненным доказательством циклического строения исходного вещества.
Необходимо, однако, помнить, что при окислении в кислой среде, например концентрированной азотной кислотой, могут происходить превращения циклов, особенно низших (см. выше), что может повести к неправильному выводу о величине цикла.
Алициклические углеводороды в жидком состоянии при повышенной температуре могут окисляться чистым кислородом или кислородом воздуха.
Первоначально образуются гидроперекиси, которые, распадаясь с отщеплением элементов воды, дают большей частью кетоны (Кри-гее; К. И. Иванов и В. К. Савинова):
^>СН2 + О2 —» ^>СН—ООН -» ^>С0 4- Н20
5. Нитрование. Нитрование углеводородов, содержащих пятичлеппый и высшие циклы, происходит в тех же условиях (см. том I, стр. 245), что и нитрование парафинов (М. И. Коновалов).
6. Отношение к высокой температуре. Соединения, содержащие триметиленовый цикл, при высоких температурах обыкновенно изомеризуются в этиленовые соединения, например:
Н2С---СН2
СНа-СН^СН*
Тетраметиленовые циклы иногда распадаются на две молекулы с этиленовыми связями:
н8с—сн^
| I —> 2СН2=СН,
Н2С-СН*
Имеются указания, что при высоких температурах соединения с восьмичленными и девятичленными циклами изомеризуются в со-единения с низшими циклами. О превращениях гекса гидробензо лов см. стр. 57. Невидимому, наиболее прочными циклами по отношению к нагреванию без катализаторов являются пятичленный и шестичленный циклы.
7, Особенности трехчленного цикла (сопряжение цикла с двойной связью в боковой цепи). Сходство циклопропанового цикла с двойной связью, уже отмеченное на стр. 52, проявляется особенно ярко в том, что при наличии в боковой цепи двойной связи возникают явления сопряжения, сходные с сопряжением двойных связей в алифатических соединениях. Сопряжение цикла с двойной связью в боковой цепи, проявляющееся как в химических, так и в физических свойствах подобных соединений, характерно только для трехчленного кольца и отсутствует у высших циклов.
Химическим проявлением эффекта сопряжения является характер реакций присоединения. Так, карбонильные производные циклопропана присоединяют галоидоводороды подобно а,р-ненасыщсн-ным алифатическим карбонильным соединениям; присоединение происходит в положении 1,4, в результате чего водород оказывается у менее гидрогенизированного углеродного атома и получаются д-за-мещенные соединения (Колер, Конант):
~СН2
|\сн=с-с6н5
СН5Вг
СН2Вг-СН,—сн,-с—с,н, I!
6
Аналогично протекают и некоторые другие реакции присоединения.
Особенно отчетливо можно проследить эффект сопряжения на простейших примерах—при гидроге юлизе углеводородов ряда циклопропана с двойной связью в боковой цепи (Б. А. Казанский, М. Ю. Лузина). Например, изопропенилциклопропан, в кольце которого в результате сопряжения оказываются ослабленными связи у атома углерода, несущего боковую цепь, дает при гидрогенолизе 2-метил-пентан:
СНг (рн3 СН3
I Н— С«=С Н2 С Н з~ С Нг” С Н 2“ С Н~ С Н 3
сн2
в то время как кольцо изопропилциклопропана разрывается нормально (стр. 54) с образованием 2,3-диметилбутана:
СН2 СН3 СН3 СНз
\ I +н2 I I
_ ^?СН—СН—СН,------> СН8—СН—сн—сн8
сн,
8. Особенности шестичленного цикла. Циклогексан, его гомологи и производные, не имеющие двух заместителей при одном атоме углерода, обладают способностью терять шесть атомов водорода (дегидрогенизация}. превращаясь в ароматические соединения.
Подобные углеводороды ряда циклогексана частично претерпевают дегидрогенизацию уже при пропускании их паров через раскаленные трубки. Легко и гладко эта реакция, обратная гидрогенизации бензола (стр. 45), протекает под действием катализаторов. Таким катализатором может являться мелкораздробленный никель (Сабатье), что свидетельствует об обратимости реакции Сабатье и Сандерана. Однако реакция здесь сопровождается сильным распадом бензола до метана.
Особенно активными дегидрогенизирующими катализаторами являются мелкораздробленные платина и палладий (Н. Д. Зелинский), в присутствии которых гексагидробензолы гладко и почти количественно превращаются в ароматические углеводороды
CeH18 > 4~ ЗН2
без какого-либо изменения углеродного скелета (см. также стр. 222).
Иначе ведут себя циклогексановые углеводороды, имеющие два заместителя при одном и том, зсе углеродном атоме. В присутствии платины они также превращаются в ароматические углеводороды, но либо с отщеплением, либо с перемещением одной из боковых цепей в орто- или в мета-положение (Б. А. Казанский, А. Л. Либерман);
Эта реакция протекает значительно медленнее, чем дегидрогед низация гидроароматических углеводородов.
Другой важной особенностью углеводородов с шестичленным кольцом является способность циклогексеновых и циклогексадиеновых углеводородов к диспропорционированию (перераспределению) водорода:
Диспропорционирование проходит очень легко при 80—150° в присутствии платинового или палладиевого катализатора.
Эта реакция, открытая Н. Д. Зелинским, была названа им необратимым катализом ввиду того, что она никогда не идет в обратную сторону.
При действии на циклогексан и его гомологи брома и бромистого алюминия получаются бромпроизводные ароматических углеводородов (М. И. Коновалов); например, из метилциклогексана в этих условиях получается пентабромтолуол:
СН8
СН сн8
\н, Br\J\/Br
| I + 8Вг2 —> II +11НВГ
н2с^ /Сн2 в/уХвг
СН2 Вг
Химические свойства производных алициклических углеводо-родов, за исключением мелких деталей, не отличаются от свойств аналогичных соединений жирного ряда, разумеется кроме особенностей, связанных с превращениями циклов.
Заслуживает внимания большая легкость, с которой циклические кетоны вступают в типичные для кетонов реакции—дают соединения с бисульфитом, конденсируются с альдегидами, особенно с ароматическими, и пр.
Например, обе метиленовые группы, находящиеся рядом с карбонилом, легко реагируют с бензальдегидом, выделяя воду:
СН2 СН2
\н2 н^ \н2
| I + 2СбН5—ОНО -> II + 2Н2О
Н.С СН2 CGH5-CH=C С=СН—сбнб
Л л ио и о
со со
дибензилиденциклогексанон
(темп, плавл. 118°)
Физические свойства. В пределах каждого гомологического ряда правильности в изменениях физических свойств (табл. 1) в общих чертах такие же, как в рядах жирных соединений.
Таблица 1
Алициклические углеводороды
Название Формула Температура плавления °C Температура кипения °C Удельный вес Показатель преломления 20 nD
Циклопропан . . . С3н6 —127,0 | —32,8 0,6807 (rf, при темп, кип.) 1,3723 (при темп кип.)
Метилциклопропан с4н8 +4,5 ' 0,6912 Ц-20) —
Циклобутан . . . Метил циклобутан . » i с5н)0 —90,2 + 12,6 ! 36,3 0,7038 0,6931 (d20) 1,3752 (при 0°) 1,3836
Циклопентан . . . » —93,2 49,3 0,7454 (df) 1,4065
Метил циклопентан C(jHi2 —142,5 71,8 0,7486 (d20) 1,4097
Циклогексан . . . +6,54 80,7 0,7786 (d20) 1,4262
Метилциклогексан с7ни — 126,4 1 100,8 0,7693 (d20) 1,4230
Циклогептан . . . » — 8,1 118,5 0,8109 (d20) 1,4449
Циклооктан . . . СвН1б 4-14,8 150,7 0,8362 (d20) 1,4585
Циклононан . . . СсД-Цз 4-10,8 178,4 0,8502 (df) 1,4666
Циклододекан . . ^12^24 4-61 118 (при 18 мм) 0,825 (d'5) —
Циклотридекан . . C13H06 4-17-18 112—113 (при 9 мм) — —
Циклотетрадекан . СцН28 +53 143 (при 16 мм) 0,825 (d80) —
Циклопентадекан . ^15^30 +60 110—115 (при 1 ММ) 0,824 (d78) —
Циклогексадекан . 016^32 +57 170—171 (при 20 мм) 0,819 (d79) —
Циклогептадекан С17Н34 4-65 — 0,820 (df) —
Циклооктадекан ОдНзв +72 — 0,820 (d76) —
Циклодокозан . . С20Н44 4-46 212 (при 1,6 мм) 0,817 (d75) —
Циклотетракозан . С24Н48 +47 222—228 (при 0,6 мм) — —
Циклогексакозан . ^26^5 2 4-44 218—219 (при 0,5 мм) 0,812 (d78) —
Циклооктакоза н ^28^56 4-48 213—214 (при 0,25 мм) 0,810 (df) —
Циклононакозан С29Н58 +47 215 (при 0,1 мм) 0,824 (d84) —
Циклотриаконтан . С3оНбо 4-58 — 0,822 (d73) —•
Из табл. 1 видно, что для углеводородов с небольшим числом углеродных атомов температуры кипения возрастают при переходе от низших циклов к высшим. То же относится к показателям преломления и к удельным весам: чем больше углеродных атомов входит в цикл, тем выше эти константы.
Исследования соединений с большим числом углеродных атомов в цикле показали, что в изменениях их физических свойств наблюдаются довольно сложные закономерности:
1. Удельные веса с увеличением числа углеродных атомов в цикле проходят через максимум. В ряду углеводородов этот максимум наблюдается, невидимому, для цикла Сн, а в ряду кетонов для цикла С10 (см. табл. 7 на стр. 87).
2. Температуры плавления до углеводорода С14 для циклов с нечетным числом углеродных атомов ниже, чем для двух соседних четных циклов, затем температура плавления повышается без колебаний до цикла С19, после чего следует крутой подъем до С22, а затем не вполне правильное постепенное повышение.
3. Удельные веса алифатических и алициклических соединений по достижении достаточно большого молекулярного веса делаются близкими.
Экзальтация молекулярной рефракции (см. том I, стр. 471) оказывается положительной для трех- и четырехчленных циклов (подобно экзальтации, вызываемой двойной связью), очень малой для пяти- и шестичленных и отрицательной для высших циклов (табл. 2).
Таблица 2
Экзальтация молекулярной рефракции циклов
Число членов цикла п Инкремент i рефракции । Число членов цикла । п Инкремент рефракции
3 4-0,62 И —0,96
4 4-0,32 12 — 1,71
5 4-0,10 13 —0,83
6 4-0,10 14 —1,05
7 '—0,11 15 —0,95
8 —0,29 16 —0,86
9 —0,39 17 -0,98
10 — 1,58 18 —0,54
Следует отметить, что повышение рефракции у циклических соединений вызывает семициклическая двойная связь. Инкремент семициклической связи в среднем для линии /7а равен 0,4, а для линии D натрия—соответственно 0,5.
ПРИРОДНЫЕ ИСТОЧНИКИ АЛИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
В настоящее время известны природные вещества, содержащие циклы с числом атомов углерода от 3 до 9, а также с 11 углеродными атомами. Соединения с пятичленным и шестичленным циклами чрезвычайно распространены в природе, а также могут быть получены из различных природных веществ. Низшие и высшие циклы найдены только в соединениях сложного строения: в терпенах и их производных (гумулен и др.), в алкалоидах (атропин, кокаин, псевдопель-тьерин, труксиллины) и в некоторых других веществах (пиретрины И др.).
ЦИКЛИЧЕСКИЕ УГЛЕВОДОРОДЫ НЕФТИ (НАФТЕНЫ)
Исследования Ф. Ф. Бейлыитейна* и А. А. Курбатова, и в особенности В. В. Марковиикова и его учеников, показали, что нефть Апшеронского полуострова на Кавказе (бакинская нефть) содержит очень много углеводородов ряда С„Н2/г, обладающих свойствами насыщенных углеводородов. В. В. Марковников и В. Н. Оглоблин дали этим углеводородам название нафтенов. Ими же были выделены из кавказской нефти циклопентан и узкие фракции состава от С6Н12 до С,5Н30. Позднее было доказано (Н. Д. Зелинский), что эти узкие фракции представляли собой смеси изомерных углеводородов с пятичленными и шестичленными циклами.
Работы Н. Д. Зелинского по дегидрогенизации углеводородов некоторых кавказских нефтей под влиянием палладия или платины показали, что гомологи циклопентана составляют иногда главную часть нафтенов.
В XIX и начале XX в. установление присутствия того или иного нафтена в нефти представляло огромные трудности, так как исследователи того времени пользовались почти исключительно чисто химическими методами и очень несовершенными приборами для перегонки. Только для доказательства присутствия в одной из кавказских нефтей простейшего нафтена—циклопентана—В. В. Марковников затратил много месяцев напряженного труда.
Применение развившихся в XX в. физических методов исследования (точная ректификация па очень эффективных колонках, азеотропная и экстрактивная перегонка, хроматография, кристаллизация при низких температурах из жидкого пропана или бутана, многоступенчатая экстракция и т. д.) позволило ускорить исследования и получать более надежные результаты, так как при физических методах исключаются ошибки, связанные с возможной изомеризацией исследуемых углеводородов. Подробные исследования физическими методами одной из американских нефтей проводились, начиная с 1928 г., Россини (в США). За 20 лет работы им было выделено более 90 индивидуальных углеводородов, причем большинство из них было получено в исключительно чистом виде. В число этих углеводородов входило 13 циклопентанов (циклопентан, метил-и этилциклопентаны, 1,1 -диметилциклопентан, трансЛ ,2-диметил-циклопентан, цис- и транс-\ ,3-диметилциклопентаны и др.) и 8 циклогексанов (циклогексан, метил- и этилциклогексаны, 1,1,2-и 1,1,3-триметилциклогексаны и др). Одновременно с подробным исследованием одной нефти Россини исследовал менее детальными, но зато более быстрыми методами бензины из семи различных
* Федор Федорович (Фридрих) Бейлыптейн (1838—1906). Родился в Петербурге. Учился в Германии, был доцентом Гейдельбергского университета, впоследствии профессор Петербурского технологического института, а затем академик. Особенно известен как автор основного справочного руководства по органической химии (Handbuch der organischen Chemie); последнее издание с дополнениями состоит из нескольких десятков томов
нефтей США. Во всех этих бензинах были найдены одни и те же углеводороды, но в разных количествах, причем из нафтенов меньше всего оказалось циклопентана и циклогексана. Россини пришел к выводу, что если распределить все содержащиеся в бензине индивидуальные углеводороды на 5 классов (нормальные парафины, изопарафины, циклопентаны, циклогексаны, бензол и его гомологи), то бензины разных нефтей будут различаться только относительным содержанием каждого из этих классов. Соотношение же отдельных индивидуальных углеводородов в пределах одного класса одинаково во всех исследованных бензинах.
В Советском Союзе в последние годы был разработан так называемый комбинированный метод исследования индивидуального состава бензинов (Б. А. Казанский, Г. С. Ландсберг и их сотрудники). По этому методу путем ректификации, хроматографии и дегидрогенизации циклогексанов* бензин разделяют на узкие фракции простого состава, которые затем исследуют с помощью спектров комбинационного рассеяния. Это дает возможность сравнительно быстро (за 2—3 месяца) определить качественный состав и количественное содержание индивидуальных углеводородов в бензине, причем расшифровать таким образом удается 85—90% состава бензина. Исследование комбинированным методом семи советских бензинов (в том числе пяти кавказских) показало, что они очень сильно отличаются по составу, даже если это бензины одного и того же месторождения, но с разных геологических горизонтов. Например, из двух таких бензинов в одном было найдено 6, а в другом 7 циклопентановых углеводородов состава QH14 и С8 H1G, но только два углеводорода присутствовали в обоих бензинах, а из остальных девяти—каждый был найден только в одном из бензинов. Отмеченное Россини для семи американских бензинов одинаковое соотношение количеств индивидуальных углеводородов в пределах каждого класса совершенно не соблюдалось для семи советских нефтей.
В советских нефтях были найдены все нафтены, обнаруженные Россини в американских нефтях, и, кроме того, некоторые диметилциклогексаны и триметилциклопентаны с различным пространственным расположением боковых цепей {цис-транс-изомеры).
Подавляющее большинство известных к настоящему времени природных нафтенов имеет пяти- или шестичленное кольцо. Сведения о наличии в нефти углеводородов с большим числом атомов углерода в цикле немногочисленны. Так, имеется указание В. В. Мар-ковникова, что в одной из кавказских нефтей содержится циклогептан; в другой кавказской нефти циклогептан недавно был обнаружен спектроскопически; немного циклогептана выделил Россини из исследуемой им нефти. Не исключено, что высшие фракции нефтей содержат нафтены и с иным числом углеродных атомов в цикле.
* На специальном железо-платиновом катализаторе, не вызывающем в уело виях опыта побочных реакций.
В нефти содержатся также в небольших количествах кислородсодержащие производные циклических углеводородов, в частности кислоты (нафтеновые кислоты). Имеется указание на содержание в нефти циклических кетонов (Тихвинский).
АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В ЭФИРНЫХ МАСЛАХ
В эфирных маслах содержатся циклические ненасыщенные углеводороды, а также кислородные производные насыщенных и ненасыщенных углеводородов: спирты, альдегиды, кетоны, кислоты, сложные эфиры и др.
Особенно часто встречаются соединения с 10 атомами углерода. Эти соединения по своим химическим свойствам весьма близки к бициклическим соединениям с тем же числом атомов углерода в молекуле и объединяются с ними в группу природных соединений, называемых производными терпенов. Поэтому некоторые из них будут рассмотрены в разделе о терпенах.
АЛИЦИКЛИЧЕСКИЕ ПРОДУКТЫ СУХОЙ ПЕРЕГОНКИ УГЛЯ И ДЕРЕВА
В продуктах сухой перегонки каменного угля всегда содержится интересный ненасыщенный углеводород с пятичленным циклом—циклопентадиен (темп, кип. 41°):
НС=СН | \сн, нс=сн
В этом углеводороде водородные атомы группы СН2 могут замещаться на ще лочные металлы. Углеводородный остаток в получающемся при этом пентадиенил-металле обладает особыми свойствами, сближающими его с углеводородами ряда бензола (стр. 492).
Под влиянием алкоголятов циклопента диен может вступать в реакцию конденсации с альдегидами и кетонами, давая окрашенные ненасыщенные углеводороды, называемые фульвенами, например:
нс=сн
| \СН2+СО(СН3)2 нс=ён
нс=сн
I 2с=с<сн3)2 + нао нс=сн
диметилфульвен
Фульвены и сам циклопентадиен легко окисляются кислородом воздуха. Циклопентадиен очень легко полимеризуется в дициклопентадиен. Процесс
синтезов за счет присоединения
димеризации протекает по типу диеновых молекул друг к другу в положениях 1,4 и 1,2:
/СН z/l
НС I
I СН2
НС I
хн
СН н<^|\н—сн
При нагревании, даже просто при перегонке» этот димер легко распадается* вновь образуя циклопентадиен. " - . .
В продуктах сухой перегонки каменного угля при низких температурах а вакууме А. Пикте обнаружил присутствие большого количества циклических алкоголей с шестичленным циклом. Продукты сухой перегонки дерева и бурого угли содержат циклопентанон и циклогексанон. Доказано также, что в продуктах сухой перегонки дерева содержится непредельный кетой метилциклопенте-(темп. кип. 157°):
Н2С—СО
| Сс-сн, н,с-сн
ПРИРОДНЫЕ АЛИЦИКЛИЧЕСКИЕ МНОГОАТОМНЫЕ СПИРТЫ (ЦИКЛИТЫ') И ОКСИКИСЛОТЫ
В соке дуба (Quercus), а также некоторых пальм, содержится циклический пятиатомный спирт ряда циклогексана—кверцит СвНпОь. Теоретически для него возможно шесть рацемических изомеров (способных разделяться на право- и левовращающие антиподы) и четыре лц?зо-изомера.
Природный кверцит (темп, плавл. 235°) вращает плоскость поляризации вправо; [а = + 25,6°. Азотной кислотой он окисляется в слизевую кислоту; обработка иодистым водородом приводит к образованию бензола, фенола и других ароматических соединений. Для кверцита наиболее’устойчивой является конформация еееаа:
Если условно рассматривать циклогексановое кольцо как плос-ю-е и отмечать положение гидроксильных групп над плоскостью к'^ьца номерами в числителе дроби, а гидроксильных групп под -угон плоскостью—номерами в знаменателе, то этот кверцит можно назвать 1,4/2,3,5-циклогексанпентолом.
В растительных и животных тканях широко распространены шестиатомные спирты ряда циклогексана—инозиты С6Н12О6. Теоретически возможно существование восьми стерических изомеров, из которых один может быть разделен на оптические антиподы, а ^стальные являются лсезо-формами. В настоящее время известны пять мезо-форм и оба оптически активных -антипода.
Это сциллоинозит (с конформацией ееееее), мезо- или миоинозит (еаееее), правый и левый инозиты (ааееее), эпиинозит (аеаеее), аллоинозит (аеааее) и мукой ноз и т (аааеее).
Важнейшим из изомерных инозитов является мезоинозит, впервые выделенный Ю. Либихом из мясного экстракта.
Мезоинозит, который может быть назван 1,2,3,5/4,6-циклогек-сангексолом, плавится при 225°; из воды кристаллизуется в виде дигидрата. Его можно синтезировать гидрированием гексаоксибен-зола (стр. 337).
Энергетически наиболее выгодная конформация мезоинозита может быть представлена следующим образом (еаееее):
При окислении щелочным перманганатом он дает с разрывом связи между пятым и шестым углеродными атомами рацемическую d,l-талослизевую кислотуг
СООН
но- —н
Н— —ОН
н— —он
Н— —он
с ;оон
Фосфорнокислый эфир мезоинозита в виде кальциевых и магниевых солей содержится в растениях, где служит резервным фосфорсодержащим материалом.
Свободный фосфорнокислый эфир мезоинозита—гексафосфат С6Н6(ОРО3Н2)6—может быть извлечен из жмыха различных семян; он находит применение в медицине под названием фитина как источник органически связанного фосфора.
Оказалось, что один из факторов, стимулирующих рост дрожжевых клеток (Биос I, см. раздел «Витамины»), является мезоинозитом (Когль).
Сциллоинозит (темп, плавл. 348,5°) встречается в тканях некоторых видов акул, а в растительном мире—в листьях кокосовых пальм.
Из природных растительных многоатомных циклических кислот наиболее важна циклогексантетраоксикарбоновая, или хинная, кислота С6Н7(ОН)4СООН (темп, плавл. 162°). Эта кислота оптически активна. Впервые она была выделена при переработке коры хинного дерева для извлечения из нее хинина и других алкалоидов. Хинная кислота довольно часто встречается и в других растениях. В зернах кофе и в млечном соке каучуконосов Ficus elastica она содержится в соединении с кофейной кислотой в форме депсида (стр. 382), носящего название хлорогеновой кислоты.
Хинной кислоте принадлежит строение 1,3,4/5-тетраоксицик-логексан-! -карбоновой кислоты:
но н
но н
При перегонке она разлагается на фенол, гидрохинон, бензойную кислоту и пр.; окисление ее перекисью марганца и серной кислотой дает хинон; при сплавлении со щелочами получается прото-катеховая кислота.
Нагреванием до 220° хинная кислота превращается в хинолак-тон—хинид (темп, плавл. 198°). Лактонизация происходит за счет ОН-группы при пятом атоме углерода.
Одноядерные алициклические соединения
Циклопропан и его производные
Циклопропан (триметилен) обладает наркотическим действием и имеет некоторое применение в хирургической практике как общий анестетик.
Циклопропан и его производные
Таблица 3
Название ; Формула Температура плавления °C Температура кипения °C Удельный вес .20 ^4 Показатель преломления 20 nD
Циклопропан (триметилен) QjHg — 127,6 —32,9 0,6886 К40) 1,3769 (При —40°)
Метилциклопропан . . . с3н5сн3 — +5 0,6912 (dr20) —
1,1 -Диметил циклопропа н. С3Н4(СН3)2 —109,0 20,7 \ о ) 0,6589 1,3668
Циклопропа нкарбоновая (триметиленкарбоновая) кислота С3Н5СООН 18 183 1,0885 1,4390
Циклопропан-1,2-дикарбоновая кислота: цис- ........ транс- ...... С3Н4(СООН), 139 : 175 । 1 — —
Технически циклопропан получают из 1,3-дихлорпропана действием цинка в спиртовом растворе с добавкой небольших количеств йодистого натрия:
/СН.,-а /:и,
H2(f -PZn —> Н2С I -FZ'iCp
\сн2—Ci \сн2
Другие способы его получения см. па стр. 37.
При пропускании через раскаленные трубки циклопропан превращается в пропилен. В присутствии силикагеля такая изомеризация происходит уже при комнатной температуре.
Циклопропан восстанавливается водородом в присутствии никеля при 120° с образованием пропапа.
Действием хлора из циклопропана получается хлорциклопропая (темп. кип. 43°):
Н2СХ ! "сие;
Н2с/
Метилциклопропан, как и циклопропан, представляет собой легко конденсирующийся газ.
1,1- Диметилциклопропан, кроме обычных способов синтеза алициклических соединений, может быть получен действием натрия на неопептилхлорнд:
IPG ZCH3 ПзС. zch2
-г Na -> | ,4- NaCl + Н
Нзс/ ЧСН2С1 Н3(У \сн2
Наряду с этой реакцией, в ограниченной степени идет также синтез Вюрца, приводящий! к образованию 2,2,5,5-тетраметилгексана-
сн8 сн3
I I
СН3—С—СН2—СН2—С—Ci 13
I I
сн3 сн, Циклопропен получен нагреванием гидрата окиси триметилниклопро'пилам-моиия:
Г н2сх -] Н2С.
j ;CH-N(CHS)3 он I Vh + N(CH9)s -г нго
H2CZ HCZ
циклопропен
Циклопропен—пахучий газ, сгущающийся в жидкость при —36°. Он жадно присоединяет бром (с образованием пламени), легко полимеризуется, поглощает кислород, образуя густую желтую жидкость.
При нагревании и под влиянием катализаторов циклопропен легко изомеризуется в аллен (см. том 1, стр. 347).
Циклопропанкарбоновая (тркметиленкарбоновая) кислота может быть приготовлена синтезом с натриймалоновым эфиром (стр. 38),
а также омылением нитрила, легко получаемого действием едкого кали на нитрил у-броммасляной кислоты:
СН2—СН2Вг _НЕг н2а Н2Сх
I ----> I ХСН—CN —> | СН-СООН
СН2—CN Н2с/ Н2с/
Она ведет себя, как типичная алифатическая кислота, но, кроме того, способна ко всем реакциям, характерным для трехчленного цикла.
Циклопропан-1,2-дикарбоновая кислота получена в цис- и mpawc-формах:
НООС СООН
{(пс-форма транс-форма
Дас-кислота плавится при 139°; с хлористым ацетилом дает ангидрид, плавящийся при 59°. Транс-кислота плавится при 175°. В соответствии с выводами стереохимии (стр. 28) эту кислоту удалось разделить на оптические антиподы, которые плавятся также при 175° и имеют удельное вращение [а]^ =84,8".
Циклобутан и его производные
Циклобутан (тетраметилен) получается восстановлением циклобутена водородом в присутствии никеля. Дальнейшее восстановление циклобутана до бутана происходит в этих условиях только при 180°.
Таблица -/
Циклобутан и его производные
Название Формула Температура плавления °C 1емпература кипения °C
Циклобутан с4н8 —90,2 4-12,6
Метилциклобутан с4н7сн3 — 36,3
Циклобутанол С4Н7ОН — 123
Циклобутанон С4нбо i _ (при 733 мм 99
Циклобутанкарбоновая (тетраметиленкарбоновая) кислота . С4Н7СООН 1 ! 191
Циклобутан-1,2-днкарбоновая кислота: цис- С4Н6(СООЫ)2 138 I
транс- 131 —
Циклобутан-1,3-дикарбоновая кислота: цис- С4Не(СООН\ 138 252
, транс- 171 Субл.
Циклобутен С4нб — +2,4
Другой путь получения циклобутана—фотохимический распад циклопентанона (стр. 52).
Диметилциклобутаны и некоторые другие гомологи циклобутана удалось разделить на цис- и транс-изомеры (Б. А. Казанский, М. Ю. Лукина).
Действием брома и щелочи на амид тетраметиленкарбоновой кислоты получается аминоциклобутан (темп. кип. 81°):
Н2С—СН—NIL
1 I
Н2С-СН2
С азотистой кислотой аминоциклобутан дает смесь циклопропилкарбинола и циклобутанола\
Н2С СН—СН2ОН
циклопропилкарбинол
Н2С-СН-ОН н2сДн2 циклобутанол
Циклобутен получен из аминоциклобутана путем исчерпывающего метилирования с последующим термическим разложением четвертичного аммониевого основания (аналогично синтезу циклопропена). Он представляет собой газ, сгущающийся в жидкость при +2,4°; уд. вес ^=0,733.
Циклобутанкарбоновые кислоты. Циклобутанкарбоновая (тетраметиленкарбоновая) кислота получается по Н. М. Кижнеру (стр. 39) из триметиленхлоробромида и натриймалонового эфира. Опа имеет запах жирных кислот; трудно растворима в воде.
ЦиклобутанЛ Д-дикарбоновая кислота
Н2С—СН—СООН
I
Н2С—СН—СООН
выделена в цис- и тракс-формах:
и СООН
СООН СООН
цнс-форма ттгралс-форма
Цпс-кислота плавится при 138°; образует ангидрид. Трпке-кислота рацемическая плавится при 131°, индивидуальные d- и /-формы— при 105°.
ЦиклобутанА ,Ъ-дикарбоновая кислота
Н2С-СН~СООН I I НООС—НС-СН,
также известна в цас-форме (темп, плавл. 138°) и в траке-форме (темп, плавл. 171°). Она получается нагреванием циклобутантетра
карбоновой кислоты с отщеплением двух молекул СО2 или отнятием элементов хлористого водорода от двух молекул этилового эфира а-хлорпропионовой кислоты:
СООС2Н5 COONa
Н—СН2—СН—Cl Н2С—СН
C2H5CNa 2 । .
+ НС—сн2
Cl—СН-СН.2—Н |
I NaOOC
СООСЛ
Эфир циклобутантетра карбоновой кислоты получается полимеризацией =»фира метиленмалоновой кислоты:
ХООС2Н
H2C=CZ \зоос2н
С2Н5ООС +
/С=СН2
C2H5OOCz
С2Н5ООС
с2н5ооо
,СООС2Н
н2с-с<
I I \СООС2Н
>с—сн,
5
5
5
Производными циклобутана являются также труксилловые и труксиновые кислоты—продукты димеризации коричной кислоты (стр. 409).
Циклопентан и его производные
Гомологи циклопентана удобнее всего получать из циклопента нона по схеме:
Н2С—СН2 Н2С—СН2 R Н2С—СН2
I \С0 + RMgBr^ I —Д I \CR
I / I / \пн '
н2с—сн2 н2с-сн2 Н2С-СН
Н2С—сн2 ‘ j ^CHR
Н2С-СН2
Важным способом получения углеводородов ряда циклопентана является также восстановление замещенных циклопентанонов. Эти кетоны могут быть получены непосредственно из адипиновой кислоты и ее гомологов (стр. 40), а также кетонным расщеплением эфиров кетонокислот, получаемых конденсацией сложных эфиров адипиновой кислоты и ее гомологов (стр. 43).
Кислоты ряда циклопентана получены главным образом синтезами с натриймалоновым эфиром (стр. 38) или из галоидопроизводных через магнийорганические соединения:
Н2С—СН2
/СН—AigBr -j-СО2
Н2С-СН2
Н2С—СНо н2о 1 ---> I /СН-СООН
Н2С—сн2
Физические свойства некоторых производных циклопентана приведены в табл. 5.
Производные циклопентана
Таблица 5
Название Формула Температура плавления °C Температура кипения °C Удельный вес Показатель преломления п20 nD
Циклопентан О>н10 —93,2 49,3 0,7454 1,4065
Циклопентен с5н8 — 134,6 44,2 0,7720 1,4225
Циклопентадиен-1,3 . . . QH, 35 42,5 0,7966 1,4404
(+ (nfи 25°)
Метилциклопентан . . . с5н„сн3 — 142,5 71,8 0,7486 1,4097
Этилциклопентан . . . . с6н9с2н5 — 138,4 103,6 0,7665 1,4198
1,1-Диметилциклопентан . 1,2-Диметилциклопентан: С6Н8(СН3), —69,8 87,8 0,7545 1,4136
цис- —53,9 99,5 0,7726 1,4222
транс- — 117,6 91,9 0,7514 1,4120
1,3-Диметилциклопентан: С5Н8(СН3),
цис- — 133,9 91,7 0,7488 1,4107
. транс- — 133,7 90,8 0,7448 1,4088
Хлорциклопентан . . . . С5Н9С1 — 115 1,0051 1,4510
Бромциклопентан . . . . С5Н9Вг — 138 1,400 (Л 1,4870
Иодциклопентан . . . . c5h9j — 78—79 (при 4Ь мм) Cl9j 1,7096 1,5447
Циклопентанол С3Н9ОН — 19,0 140,7 0,9488 1,4153
Циклопентилкарбинол . . С5Н9СН2ОН — 162 0,9313 1,4579
Циклопентанон С6Н8О —58,2 130,5 0,9480 1,4366
Циклопентилальдегид . . Циклопентанкарбоновая с5н9снр С5Н9СООН — 135 — —
кислота +8 216 1,0527 1,4532
Циклопентилуксусная кис-
лота С5Н9СН2СООН + 14 230 1,0216 1,4523
И) (при 18°)
Циклопентиламин . . . . c5h9nh2 —85,7 107 —
Циклопентандиол-1,2: С6Н8(ОН)2
цис- +48 130 (пои 20 мм) .— —
транс- +ю 130 (при 20 мм) — —
1 -Метилциклопента нкар-
боновая кислота . . . CSH8(CH3)COOH — 219,5 1,0270 1,4529
2-Метилциклопентанкар-
боновая кислота . . . . — 220 1,0159 1,4515
З-Метилциклопентанкар-
боновая кислота . . . . » — 220 1,0124 1,4480 (при 22°
Циклопентан и его низшие гомологи представляют собой легкоподвижные жидкости. В отличие от циклогексана и его гомологов, углеводороды ряда циклопентана при нагревании в присутствии никеля или платины не выделяют водорода. Бром в темноте не действует на циклопентан при обычной температуре, а на свету выделяется бромистый водород и получается бромциклопентан. Действием спиртовой щелочи на бромциклопентан получается циклопентен (темп. кип. 44,2°). Разбавленная азотная кислота под давлением нитрует циклопентан и метил циклопентан. Из метилциклопентана получаются при этом 1-метил-1-нитроциклопентан и 1-метил-2-нитро-цикл опентан (М. И. Коновалов):
Н2С—сн2
\зн—сн3 н2с—ён—no2
Таким, путем было доказано наличие этих углеводородов в кавказской нефти.
В присутствии платинового катализатора при 250—300° в атмосфере водорода циклопентан превращается в /i-пентан (Н. Д. Зелинский, Б. А. Казанский, А. Ф. Платэ). Благодаря высокому октановому числу (см. том I, стр. 173) и низкой температуре кипения циклопентан является ценной добавкой к моторному топливу, облегчающей запуск мотора («пусковая головка»).
Циклопентанон—жидкость с кетонным запахом. Он получается из адипиновой кислоты (стр. 40). При окислении азотной кислотой циклопентанон дает глутаровую кислоту, при восстановлении— спирт циклопентанол, представляющий собой жидкость с запахом плесени.
Циклопентанкарбоновая кислота имеет неприятный запах пота, почти нерастворима в воде. Эта кислота и ее гомологи составляют существеннейшую часть «нафтеновых кислот»—важнейших из кислородных соединений, входящих в состав нефтей.
Нафтеновые кислоты представляют собой трудно разделимую смесь различных циклических кислот. Они имеют промышленное значение, так как их соли применяются в качестве эмульгаторов, составных частей консистентных смазок, инсектицидов и др.
ПРИРОДНЫЕ ПРОДУКТЫ ЦИКЛОПЕНТАНОВОГО РЯДА
Из природных продуктов—производных циклопентана (точнее—циклопентена заслуживают внимания гиднокарповая кислота и особенно хаульмугровая кислота:
НС=СН
^СН—(СН2);0—СООИ
Н2С—СН2
гиднокарповая кислота
(темп, плавл. 60,5°)
нс=сн
| /СН—(СН2)12—СООН
н2с—сн2
хаульмугровая кислота
(темп, плавл. 68,5°)
Они содержатся в хаульмугровом масле, добываемом из семян тропических растений видов Hydnocarpus. Это масло и эфиры хаульмугровой кислоты применяются в терапии проказы.
К более сложным производным циклопентана относится жасмон, или 3-метил-2-(пентен-2'-ил)-циклопентен-2-он-1:
0= С---С—СН2—СН=СН—СН2—СН3
Н2С с—сн3
Это—ценное душистое вещество, найденное в эфирном масле жасмина {Jasminium grandiflorum)-, представляет собой маслянистую жидкость, перегоняющуюся при 108—110° (при 5 мм).
В близком родстве с жасмоном находятся высокоактивные инсектициды, выделяемые из цветочных головок (корзинок) пиретрума—далматской ромашки {Chrysanthemum cineraridefolium). Эти вещества представляют собой сложные эфиры:
О=С С—R"
। 3 । ц
\ Н2С с—сн
СН3 \ у
I сн—со—о—сн 3\с=сн-н£/
R'z
3
Индивидуальным веществам пиретрума отвечают в приведенной формуле следующие заместители R' и R":
R' R"
Пиретрин 1 . . . -СН3 —сн2—сн=сн—сн=сн2
Пиретрин 11 . . . —СООСН3 —сн2—сн=сн—сн=сн2
Цинерин 1 . . . . -СН3 —сн2—сн=сн—сн3
Цинерин 11 . . . —СООСН3 —сн2—сн=сн—сн3
Все эти вещества представляют собой густые желтые масла. При их гидролизе получаются кристаллические оптически активные кислоты ряда циклопропана:
I сн3—С
сн3
/СН—СООН
С=СН—СН
/npawc-хризантемовая кислота (темп, плавл. 17—21°)
сн3
сн3—с
Пен—СООН
СН3ч „|/
>С=СН—СН
HOOCZ хризантемовая дикарбоновая кислота (темп, плавл. 164°)
и кетоноалкоголи ряда циклопентана вакууме):
(жидкости, перегоняющиеся в глубоком
О=С----С—СН2—СН=СН—СН=СН2
Н2С С—СН3
^сн" । ОН (4-)-пи|.етрэлон
О=С С—CH.—СН=СН—СН3
1 Л
Н2С с— сн3
^С!^
I ОН (+)-цинеролон
Двойная связь в цикле этих кетоноалкоголей крайне трудно гидрируется. При восстановлении пиретролона амальгамой натрия получается дигидропире трон, повидимому индентичный жасмону.
Очень интересными природными производными циклопентана являются ауксины, играющие важнейшую роль в жизни растений. Ауксины образуются в окончаниях растущих побегов и вызывают их дальнейший рост (отсюда название— ростовые вещества). При освещении растения ауксины концентрируются на теневой его стороне, а потому затемненная часть побега начинает расти быстрее освещенной и побег изгибается кончиком к солнцу (явление фототропизма) При равномерном освещении ауксины концентрируются в горизонтальных побегах на нижней стороне, вследствие чего со стороны, обращенной к земле, побег начинает расти быстрее и кончик его поворачивается кверху.
Ростовые вещества, вызывающие эти явления, представляют собой циклические окси- и кетонокислоты:
НС=С—СН(ОН)—СН2—СН(ОН)—СН(ОН)—СООН I I
СН3—СН,—СН—НС СН—СН—СН,—сн3
I \ / I СН3 СН, сн3
ауксин А
НС=С—СН(ОН)—СН2—СО—СН2—СООН
СН3—СН,—СН—НС (Ь—СН—СН,—О13
СН3 СН3
ауксин В
Третьим ростовым веществом является ауксин-лактон, точное строение которого неизвестно (по-видимому, это лактон ауксина А). Еще одно важное ростовое вещество—гетероауксин—относится к гетероциклическим соединениям (см. стр. 552).
Производными циклопентана являются также кроконовая и лейконовая кислоты, получаемые из гексаоксибензола (стр. 337). Окислением гексаоксибензола получается циклический гексакетон—так называемый трихиноил. Под действием щелочи он перегруппировывается в соль кислоты ряда циклопентана. Эта кислота при подкислении и нагревании теряет СО2 и превращается \кроко~ новую кислоту (своим названием она обязана интенсивной желтой окраске ее щелочных солей, напоминающей цвет шафрана,-или крокуса):
СО
QC \о
I I
ОС со \о
Т.'ИХИНОВЛ
кон ОС СО H0SO4 °C СО _со2 ---* | | /СООК * I I /СООН *
ОС с< ОС с<
\ / \он \ / Х)Н
со со
ОС--СО ОС-С—он
I I /Н I ii
ОС с< ОС с—он
\ / Х)Н \ / со . со
кроконовая кислота
Окислением кроконовой кислоты получается лейконовая кислота*'.
ОС-----С—ОН 4-о ОС--СО
ОС С—ОН ОС СО
\о \о
кроконовая кислота лейконовая кислота
Строение лейконовой кислоты доказывается образованием пентаоксима при действии гидроксиламина и расщеплением при кипячении с карбонатами на глиоксаль и мезоксалевую кислоту:
ОС---СО ОНС---------СНО
I I -> +
ОС СО НООС СООН
\(Э \о
Циклогексан и его производные
В настоящее время простейшие производные циклогексана получают из ароматических соединений восстановлением их водородом в присутствии никеля или платины (стр. 45). Историческое значение имели синтезы циклогексана из пимелиновой кислоты через циклогексанон (стр. 40).
Представляющие теоретический интерес превращения сукцинилянтарного эфира (стр. 42) будут изложены в разделе о строении бензола. О получении других гидроароматических соединений будет указано при изложении свойств отдельных соединений ароматического ряда.
Физические свойства некоторых производных циклогексана приведены в табл. 6.
Циклогексан и метилциклогексан содержатся в значительных количествах в некоторых бензинах кавказских и дрогобычской нефтей, причем содержание в них метилциклогексана значительно больше.
При действии на циклогексан брома на свету получается бромциклогексан. Нитрование циклогексана и метилциклогексана разбавленной азотной кислотой приводит к образованию соответствующих мононитросоединений (аналогично циклопентану и метилциклопентану, стр. 72). Окислением циклогексана азотной кислотой или перманганатом получается адипиновая кислота. При пропускании циклогексана над платиновым катализатором при 300° он гладко превращается в бензол (Н. Д. Зелинский). В результате жидкофазного окисления кислородом образуется (через стадию гидроперекиси) смесь циклогексанола и циклогексанона.
* Способность лепконовой кислоты к солеобразованшо объясняется гидратацией:
\ \ /он
)С = О + Н2О )СХ
7 7 \он
Таблица Ь
Производные циклогексана
Название Формула Температура плавления °C Температура кипения °C Удельный вес </20 4 Показатель, преломле -ния
Циклогексан .... с.ни 4-6,5 80,7 0,7786 1,4262
Метил циклогексан . . 1,1 - Диметил цик логек- СвНцСН8 — 126,4 100,8 0,7693 1,4230
сан 1,2-Диметилциклогек-сан: Сб^ю(СН3)2 СбН10(СН3)2 —33,5 119,9 0,7810 1,4290
цис- —50,0 129,7 0,7963 1,4360
транс- 1,3-Диметилциклогек-сан: С«^1о(СН3)2 —88,3 123,4 0,7760 1,4270
цис- —75,6 120,1 0,7660 1,4229
транс- .... 1,4-Диметилциклогек-сан: С6Н1(1(СИ3)2 —90,1 i 124,5 0,7847 1,4309
цис- —87,4 ! 124,3 0,7829 1,4297
транс- 1,2,4-Триметилциклоге-ксан C5Hll(CHg)g —37,1 119,4 0,7525 1,4209
цис-цис-транс . . 1,3,5-Триметилцик-логексан: С5Н._,(СН3)3 — 142 0,786 1,439
цис- —43,2 138,4 0,7694 1,4263
транс- —107,5 140,5 0,7789 1,4310
Циклогексен . . . . СвН10 — 103,4 83,0 0,8110 1,4465
Циклогексадиен-1,3 свн8 — 104,8 80,5 0,8410 1,4755
Циклогексадиен-1,4- С6н8 — 86,5 0,8471 1,4679
Хлорциклогексан . . С«НИС1 —43,9 142,9 1,000 1,4626
Бромциклогексан . . СвНпВг — 165 1,3185 1,4932
Иодциклогексан . . . CeHnJ — 192 1,626 —
Циклогексанол . . . СвНцОН +25,4 161,1 0,9624 1,4648 (при 25°)
Циклогексилкарбинол Циклогександиол-1,4: свнпсн2он СсН10(ОН)2 — 182 0,9280 1,4649 I
цис- + 112 — — 1 ——
транс- + 143 — — —
Циклогексанон . . . СвН10О —31,2 155,4 0,9478 1,4507
Циклогексилальдегид . Циклогекса нкарбоновая свнпсно — 159,2 0,9253 1,4490
кислота свнпсоон 4-30,5-31 232,5 1,0344 1,4599 (при 22°) 1
Продолжение табл. 6
Название Формула Температура плавления ’С Температура кипения "С Удельный вес </20 4 Показатель преломления
Циклогексилуксусная кислота . 1 свнпсн,соон i 1 4-32 245 1,0007 1.4582
Циклогексан-1,2-дикарбо- новая кислота: Цис- СвН,0(СООН), +192
транс- (неактивная) । 221 | — — —
транс- (правовращающая) 179—183 — —
транс- (левовращающая) 179—183 — — —
Циклогексан-1,3-дикарбоновая кислота: цис- С6Н10(СООН), . 162
транс- . 148 — — —
Циклогексан-1,4-дикарбоновая (гексагидротере-фталевая) кислота: цис- . . i СбН10(СООН), 168—169
транс- ....... 310 — — —
Циклогексиламин . . . . сбнпхн? (сублимир.) 134 0,8191 1.4318
Нитроциклогексан . . . . C0H„NO, —34 205 ! 1,0668 1.4607
Циклогексанол (гексагидрофенол)
СН,
ЩС7 ^СН-ОН
I
Н9С СП,
получается в настоящее время в больших количествах каталитическим гидрированием фенола водородом в присутствии никеля под давлением. В чистом виде он образует гигроскопические кристаллы (темп, плавл. 4-25,4°), пахнущие камфорой и сивушным маслом и растворяющиеся в 28 объемах воды. При окислении он дает циклогексанон, а при более энергичном окислении—адипиновую кислоту. Циклогексанол обладает всеми характерными реакциями вторичных спиртов. Технический циклогексанол получается теперь в очень больших количествах. Он используется отчасти как хороший раство
ритель («гексалин»), но в основном является промежуточным продуктом в производстве синтетического полиамидного волокна—н: йло-па и капрона (см. том I, стр. 716).
Циклогексанон (подвижная жидкость, пахнущая ацетоном и мятой)
СН2 ^CHg । । Н2С сн2 \о
получается легче всего пропусканием циклогексанола над мелкораздробленной медью при 250—280° (см. том I, стр. 201). Он дает кристаллическое соединение с бисульфитом, легко вступает во все реакции, характерные для кетонов. При действии на циклогексанон бензойного альдегида и щелочи легко получается, с выделением двух молекул воды, дибензилиденциклогексанон (стр. 278 сл.).
Жидкий циклогексанон (как и его гомологи) представляет собой таутомерную смесь кетона с соответствующим енолом, содержащую при обычной температуре около 6% енола:
СН2
\н2
Н2С со
сн2 h2(/ \н2
I I
НоС С-ОН
сн
Этот енол, как и другие простейшие еноты, не дает окрашивания с хлорным железом.
Димедон. Конденсацией малонового эфира и окиси мезитила с помощью этилата натрия получается 5,5-диметил-1,3-дикетоцикло-гексан, или 5,5-диметилциклогексан-1,3-дион, названный димедоном:
Н3С СН3
^С^
НС^ сн2—СООС2Н5
I "И I
НО—С СО—ОС2Н5
^сн,
Н3С сн3
\н—СООСоН,
ос со
\н2
Он образует светложелтые или бесцветные кристаллы, плавящиеся при 145—147°. Цвет кристаллов зависит от условий кристаллпза^ ции и примененного растворителя.
Повидимому, окраска кристаллов димедона зависит от того, какая из таутомерных форм преобладает.
Димедон предложен как специфический реагент для открытия и идентификации альдегидов. При этом две молекулы димедона конденсируются с молекулой альдегида. Образующийся кристаллический аль-лимедон может терять воду, образуя циклический ангидрид. Для идентификации служат продукты обеих реакций:
СО I? СО
н/7 \—сн—с7 \н2
—* Н3С\ I : ! /CIL-4-ЦО
>с С—ОН но—с с<
Нзс/ \ / \ / ХС11.<
сн2 сн2
альдпмедон
I со сн со
ангидрид альдимедона
По первому пути реакция протекает в водно-спиртовой среде, второй продукт образуется в ледяной уксусной кислоте или при нагревании первичного продукта с 40%-ной H2SO4.
С формальдегидом димедон Образует продукты, плавящиеся соответственно при 189 и при 171°; с ацетальдегидом—продукты с темп, плавл. 139 и 173°.
Циклогексанкарбоновая (гексагидробензойная) кислота обыкновенно получается гидрогенизацией бензойной кислоты или из бромциклогексана через магнийорганическое соединение.
Циклогексанкарбоновая кислота имеет неприятный, сильно пристающий запах пота.
Циклоцитрали. Алифатические альдегиды с двумя двойными связями—цитрали (см. том I, стр. 395)
Н3С СН2 V
н3С СН3
V
i2C сн—сно
НС сн—сно
и
•СН3
Н2С С—СН3
н2с с
3,7-диметилоктадиен-2,6-аль-1 циклоцитрали, причем каждый
В
сн2
3,7-диметилокта диен-2,7-аль-1
легко циклизуются, превращаясь алифатический цитраль дает смесь а- и р-форм:
Н3с сн3 Н8С сн3
\z
h2Z сн—сно 1 Н^ чс—сно [1
1 Н2с с-сн3 1 н2с с—сн3
сн2 {3-циклоцитраль
сн
а-циклоцитраль
Иононы. Конденсацией а-циклоцитраля с ацетоном под действием щелочных агентов получается а-ионон, в присутствии концентрированной серной кислоты изомеризующийся в р-ионон:
Н3С сн3 Н3С СН3
Хс
СН—СНО + сн3—со—сн3 —> Н2С н2с
СН—СН=СН-СОСН3
н2с с-сн3 сн
с-сн.
а-ИОНОН
Н3С сн3
^С-СН-СН-СОСНз
! II
н2с с—сн3
\н2
3-ионон
Смесь у- и ^-иононов образуется также при циклизации под действием H2SO4 и Н3РО4 псевдоионона (см. том I, стр. 397). Процессу
собственно циклизации предшествует, повидимому, гидратация псевдоионона по месту двух двойных связей:
Н3С СН, V
Н(/ сн—сн=сн—сосн,
I II
Н2С с—сн3
| + 2Н2О
-н3с сн3 он
Н,С сн,—сн=сн—сосн3
а-Ионон—густая жидкость, перегоняющаяся при 123—124° (при 11 мм)', d2O=0,932, n~D=1,4980. В разбавленном состоянии а-ионон обладает интенсивным запахом фиалки (уtola odorata). (З-Ионон перегоняется при 127—128,5° (при 10 мм)', dXD—0y^, п о=1,521.
Иононы были впервые синтезированы Тиманом. Сравнительно недавно они были найдены в природе—в эфирном масле из Boronia megastigma.
Р-Иононная группировка обнаружена в витамине А (стр. 690) и в каротине (стр. 89), в котором содержится также и а-ионон-ная группировка.
Окислением а-ионона получается кетонокислота, превращающаяся далее в изогероновую кислоту (3,3-диметил-4-ацето-н-валериановую):
Н8с сн3 Н8с СН3 Н3с сп3 с
н2с \н—СН=СН—COCHg КМПО4 н2г ''сн-соон Н2С \н2
н,с 1 > с-сн. Н2с со—сн3 у 1 Н,с со—сн.
\х сн а-ионон СООН ^соон изогероновая кислота
кислота, а затем геро
Окислением р-ионона получается р-циклогераниевая новая кислота (1,1-диметил-4-ацето-к-валериановая): Н3С СН3 Н3С СН3
\:~СН=СН—СОСН3 кмпо4 Н2С \-COOH
I li -----> I h
Н2С с-сн3 Н2С с-сн3
Н3С СН3
V
НоС^ \оон
H2i со-сы.
р-ионон р-циклогераниевая
кислота
героновая кислота
Строение изогероновой кислоты доказывается превращением ее в р,р-диметил-адипиновую и, наконец, в а,а-диметилглутаровую; героновая кислота дает соответственно а,а-диметиладипиновую кислоту.
Ироны. Эфирное масло фиалкового корня (Iris florentina Ipallida) содержит смесь изомерных гомологов ионона—иронов. В природном ироне преобладают 7-форма (70—75%) и а-форма (около 25%); ^-изомер содержится лишь в небольшом количестве. Строение изомерных иронов и их взаимные переходы (Ружичка и др.) показаны на схеме:
Н3С сн8
V
Слабые кислоты
сн3—нс^ 'сн—сн=сн—сосн3 I I
н2с с=сн, V
Щелочи или сильные кислоты
^-ирон
н8с сн8
'с/
сн8—нс^ ^сн—сн=сн—сосн8
н2с с—сн3
Щелочи или сильные кислоты --------->
а-ирон
Н8с сн3
сн8—нс^ 'с—сн=сн—сосн3
н2с с—сн3 \н,
р-ирон
а-Ирон, обычно называемый просто ироном,—густое масло, перегоняющееся при 144° (при 16 мм). Он обнаруживает полноту своего аромата лишь при сильном разбавлении.
Производные циклогептана и циклооктана
Из производных циклогептана следует указать циклогептанол, или суберилоеый алкоголь (темп. кип. 185,2°), а также циклогептанон, или суберон (темп. кип. 179,5°, с?4°=0,951):
ОН
сн h2c^ ^сн2
Н2С /СН2
н2с—сн2
СО HjC^ ^сн2 н2 сн2
Н2С-СН2
циклогептанол
циклогептанон
Циклогептанон получен перегонкой кальциевой соли пробковой кислоты. При окислении он дает пимелиновую кислоту. В настоящее время его обычно получают действием диазометана на циклогексанон (стр. 50).
Особый интерес у химиков вызывает в последние годы система циклогептатриена (тропилидена)
производные которого—соединения, содержащие катион циклогеп-татриенилий (тропилий), кетоны—тропоны и, в особенности, кето-носпирты—трополоны обладают своеобразными «ароматическими» свойствами (подробнее о них см. на стр. 491 сл.).
Углеводород тропилиден (темп. кип. 116°) получен из алкалоида атропина.
Из алкалоида гранатовой коры псевдопельтьерина был впервые получен ряд производных циклооктана.
Наибольший интерес представляет полученный Вильштеттером из этого алкалоида циклооктатетраен (желтая жидкость; темп. кип. 42,3° при 17 мм; 4 = 0,943):
НС=СН
НС 'сн
II II
нс сн
нс=сн
В дальнейшем этот углеводород, наряду с циклодекапентаеном, был получен полимеризацией ацетилена (см. том I, стр. 413).
Несмотря на наличие замкнутой системы сопряженных двойных связей, он не обладает ароматическими свойствами (см. стр. 233 и 491): энергично присоединяет бром, быстро восстанавливает перманганат, водородом в присутствии катализаторов легко восстанавливается при комнатной температуре в циклооктан.
Гидрированием циклооктатетраена и циклодекапентаена в цик-лооктен и в циклодецен и последующим окислением могут быть получены синтетические субериновая (пробковая) и себациновая кислоты (см. том I, стр. 456).
Строение циклооктатетраена долго не удавалось установить из-за способности его давать три ряда продуктов реакции, как бы отвечающих формулам:
В большинстве случаев продукты реакции соответствуют формуле I. Так, при гидрировании циклооктатетраена получается циклооктзн, а при действии пер-бензойной кислоты образуется окись, гидрированием превращающаяся в циклооктанол:
Однако действием хлористого сульфурила цик ооктатетраен превращается в 5,6-дихлорбицикло-[0,2,4]-окта-1,3-диен, отвечающий формуле II:
Пиролиз продукта диеновой конденсации этого'соединения с нафтахиноном дает 3,4-дихлор циклобутен:
что являлось важным доводом в пользу принятия для циклооктатетраена формулы II. Наконец, с гипохлоритом в щелочной среде и с хромовым ангидридом в уксусной кислоте циклооктатетраен реагирует в соответствии с формулой 111, так как в первом случае получается терефталевый альдегид, а во втором—терефталевая кислота:
сно
СООН
Физические методы исследования также дали противоречивые результаты. Из электронографических данных следует, что молекула циклооктатетраена изогнута в пространстве, все углы С—С—С равны 120° и все связи С—С имеют одинаковую длину, тогда как из рентгенографических и некоторых других исследова-о ний вытекает, что длины простых и двойных связей неодинаковы (1,54 и 1,34 А).
В настоящее время считается, что циклооктатетраен имеет строение, отвечающее формуле I. Предположение, что он представляет собой смесь таутомеров, не подтверждается, и, повидимому, структуры II и III возникают лишь в момент реакции.
Производные углеводородов с высшими циклами
Соединения, содержащие кольца с большим числом атомов углерода, были синтезированы Ружичкой, исходя из азелаиновой и себа-циновой кислот. Восстановлением этих кислот натрием в спиртовом растворе он получил гликоли, которые затем превратил в дибромиды:
НООС—(СН2)7-СООН азе ла и новая кислота
I
НО—СН,—(СН,)7—СН,—ОН
ВгСН,—(СН,)7—СН,Вг
НООС—(СН,)8—СООН себациновая кислота
I
НО—СН2-(СН2)8—сн2—он
ВгСН2—(СН2)8—СН2Вг
Из этих дибромидов были получены двухосновные кислоты с большим числом атомов углерода в молекуле двумя путями:
1. Действием на дибпомиды цианистого калия и омылением образующихся двухатомных нитрилов. Таким образом получаются кислоты, содержащие на два атома углерода больше, чем исходные кислоты, например:
NC—СН2—(СН2)8—СН2—CN —» НООС—(СН2)10—СООН
2. Действием на дибромилы двух молекул натриймалонового эфира с последующим омылением получающихся эфиров четырехосновных кислот и нагреванием для отщепления СО2. Таким путем получаются двухосновные кислоты, содержащие на четыре атома углерода больше, чем исходные, например:
(ROOC)2CH-CH2-(CH2)8-CH2-CH(COOR)2 —> НООС-(СН2)12-СООН
Восстановлением этих высших двухосновных кислот Ружичка получал высшие гликоли, которые с помощью тех же реакций можно было превратить в двухосновные кислоты с еще большим числом атомов углерода. Таким образом он приготовил ряд двухосновных кислот от СпН20О4 до С35Н68О4 и сухой перегонкой ториевых солей в вакууме превратил их в циклические кетоны (табл. 7), из которых высший имел следующее строение:
СН2—(СН2)14—СН2
Н2(^ со
ХСН2-(СН2)14-СН2
Другие пути синтеза высших циклических кетонов описаны на стр. 41 сл.
При окислении хромовой кислотой циклические кетоны дают двухосновные кислоты с тем же числом атомов углерода. Восстановление кетонов приводит к циклическим углеводородам до циклотриаконтана С30Н60 включительно.
Низшие циклические кетоны пахнут камфорой, средние—имеют запах кедра, а начиная с С14Н26О—запах мускуса.
Циклогептадеканон CJ7H32O оказался тождественным продукту присоединения двух атомов водорода к пахнущему мускусом кетону—цибетону (кристаллы; темп, плавл. 32,5°; темп. кип. 158—160° при 2 мм):
НС-(СН2)7Ч II >со
НС-(СН2)/
Цибетон содержится в цибете—пахучем веществе, выделяемом железами виверры цибетовой (животное, водящееся в Индии, Иране, Абиссинии).
Пахучим началом настоящего мускуса, представляющего собой выделение желез самцов водящейся в Средней Азии и Тибете мускусной кабарги, является кетон, названный мусконом'.
сн3—СН----сн2
I I
. (СН^-СО
Мускон—густое масло; темп. кип. 142—143° (при 15 мм)\ df0— 0,920; вращает плоскость поляризации влево.
Цибет и мускус—ценные пахучие вещества, применяемые в парфюмерии. Мускус находит применение также в медицине как средство, усиливающее деятельность сердца.
Высшие циклические кетоны
Таблица 7
Название Формула Температура плавления °C Температура кипения °C Удельный вес
Циклооктанон Циклононанон Циклодеканон С8Н14О с9п160 Сю Н1ЬО 4-42 28 29 202 (при 720 мм) 74 (при 12 мм) 93—95 (при 12 мм) Ю6—107 (при 13 мм) 0,916 р743) 0,934 (^б) 0,953 р'9) Ж ИДК.
Циклоундеканон . . . . Циклододеканон . . . . Циклотридеканон . . . . Циклотетрадеканон . . . Циклопентадеканон (экза ЛЬТОН) Циклогексадеканон . . . Циклогептадеканон (ди-гидроцибетон) . . . . Циклооктадеканон . . . Циклононадеканон . . . Циклоэйкозанон . . . . Циклогенэйкозанон . . . Циклодокоза нон . . . . о о о о оо о о о о о о CJ ** «о 00 о СЧ СО 00 О OJ ^4 сч см сч с-1 со сосососо^ч) I I I I II IIIIII _1 СЧ СО Tf Ю СО I - 00 ®» О т~< сч и и и и о о о о о о о о 10 60 32 53 63 64 63 71 72 59 46 32 108 (при 10 мм) 126—128 (при 12 мм) 137—139 (при 12 мм) 155—156 (при 12 мм) 120 (при 0,3 мм) 138 (при 0,5 мм) 145 (при 0,3 мм) 157—159 (при 0,3 мм) 160 (при 0,3 мм) 170—171 (при 0,3 мм) 176—177 (при 0,3 ЛШ) 0,947 0,904 0,927 0,896 0,892 0,884 0,883 0,885 0,874 К) (<°) К) (45) К) «9
Циклотрикозанон . . . Циклотетракозанон . . . Циклогексакозанон . . . Циклооктакозанон . . . Циклононакозанон . . . Циклотриаконтанон . . . Циклотетратриаконтанон С2зН44О ^24^46^ Сге^боС С28^б4^ Сгв^ббО СзоНбвО С34НббО 39 36 42 45 47 56 66 175—177 (при 0,02 мм) 233—238 (при 0,4 мм) 208—218 (при 0,1 мм) 210 (при 0,25 мм) 220 (при 0,1 мм) 0,876 0,880 0,857 0,852 «з) (^°)
Под влиянием трансаннулярного эффекта (стр. 34) у некоторых соединений с высшими циклами наблюдаются очень своеобразные реакции. Так, циклононен и циклодецен при окислении надмуравьиной кислотой Н—СО—ООН дают не 1,2-ди-олы, как можно было ожидать, а соответственно 1,5- и 1,6-диолы:
ОН ОН
ОН ОН
При этом из цг/г-циклена получается транс-диол, а из тпрянс-циклена—цпс-диол. Циклооктен и циклоундецен также образуют диолы такого типа, но одновременно получаются и 1,2-диолы; циклогептен и циклододецен дают только 1,2-диолы.
Двухъядерные алициклические соединения
Химия двухъядерных алициклических соединений очень сложна. Исследование отдельных представителей таких алициклов часто осложнено многочисленными перегруппировками, протекающими при реакциях. Поэтому разные классы двухъядерных алициклических соединений изучены неодинаково подробно.
Номенклатура этих соединений дана на стр. 16 сл.
Соединения с циклами, разделенными цепочками из атомов углерода
По химическим свойствам соединения этого типа мало отличаются от моноциклических соединений с соответствующими кольцами. Для получения таких двухъядерных соединений могут быть использованы почти все способы синтеза моноцикланов и их производных при условии, что хотя бы один цикл уже имеется в исходном веществе, например:
D2Na г \ т—1
—СН2Вг---> |_/—Сн2“СН2—___|
СН2—CH2MgBr + O^Q —<3>—СН2-СН2_<02>
он
тСН2—СООН
/~\-сн2-ен -> O-CH’-GI=0
\:н2—СН2—СООН
Сравнительно легко доступны дициклогексилметаны, которые получаются гидрогенизацией диарил метанов, например:
сн,—^2/ < /—сн»~<^
КАРОТИНОИДЫ
В растительном мире широко распространены желтые и оранжево-желтые красящие вещества. Их цвета ярко проявляются после разрушения зеленых зерен хлорофилла в осенних окрасках увядающей листвы.
Вследствие растворимости в жирах эти вещества иногда называют липохромами. По предложению М. С. Цвета их чаще называют карот иноидам и.
К каротиноидам часто причисляют углеводороды нециклического строения типа ликопина, содержащего 13 сопряженных двойных связей в молекуле (см. том I, стр. 355). Здесь будут рассмотрены только соединения, содержащие, кроме сопряженных двойных связей* также циклические группировки.
К этим веществам относятся в первую очередь углеводороды С40Н56, содержащие две алициклические группировки, связанные между собой цепочкой из атомов углерода,—так называемые каротины.
а-Каротин (темнокрасные иглы; темп, плавл. 187°) легко растворяется в хлороформе и бензоле; нерастворим в метиловом и этиловом спиртах. При гидрировании он присоединяет 22 атома водорода, а при озонировании дает героновую и изогероновую кислоты. Это свидетельствует о его родстве с иононами. Кроме того, а-каротин является оптически деятельным веществом. Учитывая все эти обстоятельства, ему придают следующее строение:
СН3 СН3
С—(СН=СН—С=СН)„—CH=~CH-(CH=(L-CH=CH),—£н
героновая кислота изогероновая кислота
fi-Каротин плавится при 183°. При озонировании он дает две молекулы героновой кислоты; кроме того, 3-каротин оптически недеятелен. Ввиду этого ему придают строение:
СН3
С—(СН=СН—С=СН)2—СН
сн8
Н.С СН, V
СН—(СН=С—СН=СН)2—с
^-Каротин, плавящийся при 178°, содержит лишь одну циклическую группировку:
СН3 СН3
I ; I
С-(СН=СН-С=СН)2-СН=СН-(СН=С~СН=:СН)2-СН
/ /СНз \с С—сн3 н3с—с сн<
н3с/| | . I I 4(243
н,с сн, н2с сн2
4сн, \l42
Каротины являются провитаминами, так как они могут в животных организмах превращаться в витамин А (стр. 705) за счет левой половины (считая от пунктирных линий) приведенных выше структур а- и д-каротинов; [3-каротин может образовать две молекулы витамина А.
Ксантофилл С40Н56О2, второй желтый пигмент листьев, содержит две гидроксильные группы. Строение его еще не установлено окончательно; однако для аналогичного желтого пигмента маисовых зерен, зеаксантина С4оН5б02, установлено следующее строение:
СН3 СН3
С—(СН=СН—С=СН)2-—СН==СН—(СН=С—СН=СН)2-—С
Н,С СН2 Н2С СН2
• 4сн
он он
Красное красящее вещество венгерского красного перца (паприки) С35Н5вО3 обладает аналогичным каротинам углеводородным скелетом и, вместе с тем, имеет кетонный характер; строение его также окончательно не установлено.
Соединения с циклами, разделенными простой углерод-углеродной связью
Соединения этого класса почти не встречаются в природе; только в нефти обнаружены ди циклопентил, 3,3'-диметилдициклопентил, дициклогексил и, невидимому, имеются гомологи последнего.
Почти все способы получения соединений с циклами, разделенными цепочками из атомов углерода, пригодны (иногда с небольшими изменениями) также для синтеза соединений с непосредственно связанными циклами.
Применяются для этой цели и специальные методы, например восстановление циклических кетонов в пинаконы:
D=0+°-GI _ Dnci
но он
или кротоновая конденсация циклических кетонов в присутствии алкоголята натрия:
о=°+0 о=0 о-О
В В I
о о он
Реакции, аналогичные синтезу фульвенов (стр. 63), также приводят к образованию двухъядерных алициклических соединений:
ос - оа—о-ci
Наиболее доступны дициклогексил и его гомологи, которые могут быть получены из бифенила и его гомологов обычной гидрогенизацией.
Особенностью соединений с циклами, разделенными простой углерод-углеродной связью, является их способность превращаться в соединения ряда спиранов при реакциях, сопровождающихся расширением цикла. Например, дициклопентилдиол-1,Г при действии серной кислоты дегидратируется с перегруппировкой в спирокетон (Н. Д. Зелинский):
Стереоизомерия гомологов и производных двухъядерных соединений с непосредственно связанными кольцами часто более сложна, чем у моноциклических соединений. Так, например, для дицикло-гексилдиола-4,4' возможны уже три стереоизомера, которые (если условно рассматривать кольца как плоские) могут быть изображены
цыс-ц«с-дициклогексилдиол-4,4/ (темп, плаьл. 194—195°)
транс -транс-дициклогексил диол-4,4' (темп, плавл. 215—216°)
Еще более сложные комбинации стереоизомеров получаются, если заместители находятся в орто- или мета-положениях к связи между циклами, так как, например, все изображенные ниже вещества являются стереоизомерами:
VI
ш
Из этих шести изомеров четыре (II, IV, V и VI) могут быть разделены на оптически деятельные формы. Таким образом, для цикло-гексил-2,21-дикарбоновой (пергидродифеновой) кислоты возможно восемь различных оптически деятельных форм, две мезо-формы и четыре рацемата. Стереохимию этой кислоты подробно изучил Линстед, установивший пространственную конфигурацию двенадцати кислот (одну кислоту не удалось разделить на оптически деятельные антиподы). Для удобства условно принято рассматривать оба кольца как плоские и лежащие в одной плоскости и изображать молекулу так, чтобы обе карбоксильные группы были расположены рядом. При этом, если атомы водорода располагаются над плоскостью молекулы,
соответствующие углеродные атомы (например, атомы 1,1',2,2') обозначаются черными кружками
а если водородные атомы лежат под этой плоскостью, то обычным образом:
НООС CUOII
Если атомы водорода при атомах 1 и Г расположены по одну сторону плоскости
соединение получает название сия-формы, а если по разные стороны— ян/тш-формы. Тогда все представленные выше изомеры пергидроди-феновой кислоты можно изобразить (в том же порядке) следующим образом:
ноос соон
ноос соон
цис-син-цис-
НООС СООН
цис-анти-цис-
HOOC СООН
цис- син-транс-
III
НООС соон
транс-син-транс-
цис-анти-транс-
НООС СООН
транс-анти-транс-
Свойства изученных дициклогексил-2,2'-дикарбоновых кислот даны в табл. 8.
Таблица 8
Дициклогексил-2,2'-дикарбоновые кислоты
Название Формула Оптический энантиоморф Температура плавления °C Удельное вращение
Цис-син-цис- мезо- 289 —
Цис-сан-транс- d, /- 200 —
d- 170—174 +75
I- 171 — 174 —75
Т ранс-син-транс- мезо- 223 —
Цис-анти-цис- d, I- | 198 —
d- 238—240 +43
I- .238—240 —45
Цис-анти-транс- d, I- | 198 —
d- He выделены
I-
Т ране-анти-транс- d, I- | 247 —
d- | 257—259 +77,5
I- | 257—259 —79
Дициклопентил получен из циклопентил циклопентанона по методу Кижнера (см. том I, стр. 232—233):
h2n—nh2
и по Вюрцу из циклопентилбромида:
2 I3_ci
Этот углеводород кипит при 188—189,5° (при 744 мм).
Циклопентилиденциклопентанон (ушп. кип. 117—119° при 12лгл«) получается в результате конденсации циклопентанона в щелочной среде:
Он является важным промежуточным продуктом для синтеза 2-ал-килдициклопентилов.
Дициклогексил (темп. кип. 100° при 10мм\ темп, плавл. +4,5°) получается каталитическим гидрированием бифенила, дегидратацией с последующим гидрированием 1-окси- или 2-оксидициклогексилов, а также по реакции Вюрца из циклогексилбромида или циклогексил-иодида. Интересным способом его получения является электролиз натриевой соли циклогексанкарбоновой кислоты (реакция Кольбе).
Оксидициклогексилы. 1-Оксидиииклогексил легко может быть получен по Гриньяру:
а 2-оксидициклогексил получается нагреванием циклогексанола с твердым поташом:
Спирановые, или спироциклановые, двухъядерные соединения
Спирановые соединения характеризуются тем, что входящие в них циклы имеют один общий углеродный атом, например:
Простейшим спироцикланом является спиропентан:
Н2сч ,СН2
н2сх хсн2
Важнейшие способы получения спирановых соединений основаны на циклизации гелг-двузамещенных моноциклических соединений. При этом применяются обычные способы циклизации, например внутримолекулярная реакция Вюрца:
или реакция внутримолекулярной и межмолекулярной конденсации Дикмана:
,СН2—СНз—СООСзЩ
<н2—СООС2Не
C2H5ONa -------->
СООС2Н6
СООС2Н6
/Оед с=о
I с=о х0С2н6
COOCjH,
Остальные способы получения спиранов основаны на пинаколи-новых перегруппировках соединений с другими системами циклов. Один способ, уже рассмотренный выше (стр. 91), связан с расширением циклов.
Другой способ основывается на сужении циклов, например:
По химическим свойствам спироцикланы мало отличаются от моноцикланов с соответствующими циклами, однако спироциклановые соединения обычно менее устойчивы. Неожиданное исключение представляет спиро-[2,5\-октан
04
который, несмотря на наличие трехчленного кольца, выдерживает воздействие концентрированных кислот и щелочей, легко разрушающих сходные системы спиро- [2,4 ]-гептана и спиро- [2,61-нонана:
Спиро-[2,5]-октан и получается с необычайной легкостью. Например. из трех сходных между собой бромлактонов
гСНВг—С=0
СООН
/— \ /СНВг—с=о
। X ।
х— / \СН----О
соон
III
только бромлактон II при действии щелочи претерпевает перегруппировку, образуя почти исключительно соединение
—СООН
—СООН
ОН
содержащее систему спиро-[2,5|-октана, тогда как лактоны I и III дают главным образом оксилактоны, получающиеся без каких-либо перегруппировок—путем замены брома на гидроксил, т. е. в этих случаях спирановая группировка не возникает.
При дегидрогенизации многие спираны превращаются в ароматические углеводороды. Например, спиро- [4,51-декан под действием селена превращается в нафталин
DO СО
очевидно, с предварительным расширением кольца.
Некоторым спироциклановым соединениям свойственен особый вид стереоизомерии. Не имеющие асимметрических атомов спираны типа
могут быть расщеплены на оптически деятельные стереоизомеры, так как их молекулы не имеют ни одного элемента симметрии. Действительно, спиро-[3,3]-гептан-2,5-дикарбоновая кислота, а также многие- производные спиро-[5,51-ундекана были расщеплены на оптические антиподы*
Двухъядерные соединения с конденсированными ядрами
Алициклические соединения с конденсированными ядрами характеризуются тем, что их кольца имеют по два общих углеродных атома. Они могут получаться всеми методами синтеза моноцикли-ческих соединений, но в качестве исходных берутся соединения, уже имеющие в молекуле одно кольцо.
Примером могут служить реакции Дильса—Альдера, например:
СН2 // нс Hi ^CH2
со
нс" Хсн
п-бензохинон
сн2 со нс" \н 'сн
II I fl
нс сн сн
Хсн2 Хсо
Некоторые конденсированные бициклические системы лежат в основе очень важных природных соединений—терпенов. Поэтому, кроме рационального наименования (стр. 18), этим бициклическим соединениям придают названия и по соответствующему терпену с добавлением приставки нор. Эта приставка означает, что данное соединение является простейшим представителем соответствующего ряда терпенов, лишенным всех боковых цепей. Например, по аналогии с терпеновым углеводородом кареном и его дигидропроизводным караном. бицикло- [0,1,4J-гептан часто называют норкараном'.
Стереохимия алициклических соединений с конденсированными ядрами довольно сложна, так как молекулы этих веществ представляют собою трехмерные пространственные образования.
Даже простейший представитель этого класса соединений— бицикло- [0,1,1 ]-бутан
СН
Н2С^ '''CH, сн
изогнут в пространстве и имеет следующий вид:
Поэтому уже однозамещенные производные этого бицикла могут существовать в двух стереоизомерных формах:
С увеличением числа заместителей число стереоизомеров сильно возрастает.
Стереохимия бициклических соединений с конденсированными ядрами еще более осложняется, если один из циклов содержит пять и более атомов углерода, так как в этом случае сама бициклическая система может существовать в двух стереоизомерных формах даже при отсутствии заместителей. Действительно, при небольших цик-
л^х один цикл относительно другого может занимать только цис-положение (I); транс-положение (II) было бы слишком напряженным:
Расчет показывает, что наименьшее кольцо, которое может занять транс-положение относительно другого кольца, хотя бы и с напряжением, но не выходящим за рамки правдоподобного, должно иметь по крайней мере пять углеродных атомов. С энергетической стороны взаимоотношение различных возможных конформаций таких систем было подробно исследовано на примерах бицикло-[0,3,3]-октана (пенталана), бицикло-[0,3,41-нонана (гидриндана) и бицикло-[0,4,41-декана (декалина):
ш пенталан
гидриндан
декалин
Транс-форма пенталана является напряженной и жесткой системой; для нее возможна только одна конформация. Для цис-формы возможны две конформации (а и б), из которых а почти так же напряжена, как и /прайс-форма, тогда как конформация б почти свободна от напряжения и, очевидно, она и будет преобладать в цис-пентала-не при обычных условиях. Таким образом, цис-форма пенталана будет более устойчивой, что и подтверждается термохимическими данными (теплота горения цис-формы 1094,1 ккал/моль, а транс-формы 1100,9 ккал!моль).
В случае гидриндана число возможных конформаций гораздо больше:
конформации ц«с-гидриндана
Как и для пенталана, здесь также имеется только по одной энергетически выгодной транс- и ^e-конформации. В обоих случаях наиболее выгодны будут конформации с циклогексановым кольцом в форме кресла, т. е. конформации, обозначенные буквой а, в которых взаимное отталкивание водородных атомов будет наименьшим. Из этих же соображений следует, что тряяс-форма должна быть устойчивее ^wc-формы. Очень важно, что из классической теории напряжения вытекает противоположный вывод, так как только г{«с-формы в и г совершенно лишены углового (байеровского) напряжения. Од
нако термохимические данные ясно подтверждают, что тра«с-гидр-индан, действительно, более выгоден энергетически, так как его теплота горения (1345,7 ккал!моль) меньше, чем у гр/с-гидриндана (1347,5 ккал/моль). Разница в устойчивости гидринданов меньше, чем у пенталанов.
Еще больше различных конформаций может иметь декалин:
конформации qi/г-декаллна
Как расчет, так и электронографические исследования показывают, что преобладающими являются конформации, обозначенные буквами а, причем более устойчивым должен быть транс-декалин. Это подтверждается и термохимически, так как теплоты горения равны 1495 ккал/моль для транс-декалина и 1499,9 ккал!моль для цис- декалина.
Для изображения стереоизомерных бициклических соединений предложено два способа. Один из них был основан на том же принципе черных кружков, который был использован для изображения стереоизомерных дициклогексилдикарбоновых кислот (стр. 93). По другому способу связи атомов или групп, расположенных в плоскости бумаги или над ней, изображают простой черточкой, а расположенные под ней—пунктирной, например:
ц«с-декалин
/пракс-декалин
У декалина положения 1, 4, 5, 8 часто обозначают буквой а, а положения 2, 3, 6, 7—буквой р.
Пространственная конфигурация углеродного скелета стереоизомерных декалинов была доказана следующим путем.
Р-Декалол может существовать в четырех стереоизомерных формах:
Н Н Н Н
н н н н
Два из этих четырех веществ—р-декалол с темп, плавл. 105° и р-декалол с темп, плавл. 75°—при осторожном окислении дают два разных кетона.
Поскольку оба кетона, как установлено, представляют собой р-декалоны, они должны различаться только пространственным расположением водородных атомов при третичных атомах углерода, т. е. один из них является цис-, а другой траяс-Р-декалоном:
Н
Н
При более энергичном окислении тех же р-декалолов из каждого получалась смесь двух кислот—циклогексан-1-карбокси-2-пропионовой и циклогексан-1,2-диуксусной:
СН2 СН2
'cfT 'сН(ОН)
Н2С СН (!нг
СН» ,соон .
Н2С сн
I I
Н2С сн
'ХСН2—СН2—СООН
ZCH2—СООН
Н2С сн
I I
н2с сн
\н4 Хсн2—С°ОН
Однако полученные циклогександиуксусные кислоты оказались неодинаковыми.
Диуксусная кислота, полученная из высокоплавкого Р-декалола, плавилась при 159—161° и не поддавалась расщеплению на оптические изомеры, т. е. оказалась л^езо-формой. Диуксусная же кислота, полученная из низкоплавкого ₽-декало-ла, имела темп, плавл. 167° и путем кристаллизации цинхониновой соли была расщеплена на право- и левовращающую формы, т. е. оказалась рацемической смесью.
Следовательно, эти ^кислоты должны иметь строение:
Н
нУр
л«?зо-форма
Н
оптически деятельная форма
Но в таком случае декалолам, из которых эти кислоты получались, должны принадлежать формулы
Н Н
H2CJ CHs Н2С i/CH2
Н2С CZ 'сН(ОН) Had7 ^С СН(ОН)
III III
Н2С С СН2 Н2С с сн2
Н2С/|ХСН2 НгС^СН,
н н
цис-р-декалол
трсшс-З-декалол
в которых остается еще невыясненным вопрос о пространственном расположении гидроксильной группы.
Окислением этих декалолов в декалоны и восстановлением декалонов амальгамированным цинком и соляной кислотой были получены соответствующие декалины: цыс-декалин с темп. кип. 192° и транс-изомер, кипящий при 185®.
Бицикло-[0,1,3]-гексан (темп. кип. 80—81°) может быть получен действием цинка на 1-бромметил-2-бромциклопентан:
СН2Вг
Эта система лежит в основе некоторых терпенов, например са-бинена (см. стр. 154) и туйона (стр 153).
Бицикло-i 0,1,4 |-гептан-2,4-диен-1 -карбоновая, или норкарадиен-карбоновая, кислота (темп. кип. 108° при 13 мм) может быть получена из бензола и диазоуксусного эфира:
CHN2—СООС2Н6
—СООС2Н
5
При действии аммиака эфир норкарадиенкарбоновой кислоты превращается в амид (темп, плавл. 141°), гидролизом которого с помощью разбавленной серной кислоты получается кислота.
Бицикло-| 0,3,3 -октан, или пенталан, получен в виде цис-формы (темп. кип. 136° при 755 мм; di8=0,8719; 1,4629) и транс-
формы (темп. кип. 132° при 755 мм; dJ8=0,8626; п1* =1,4625). Первая получается при восстановлении цис-бицикло-[0,3,3 ]-окта-нона-1 по Кижнеру или по Клеменсену (стр. 211), а вторая—тем же путем из /пранс-бицикло- [0,3,3 1-октанона-2.
Об относительной устойчивости и конформационном анализе пенталанов см. стр. 99.
Бицикло-[0,3,ЗНоктаноны, или пенталоны. Бицикло-'0,3,3]-окта-нон-1, или пенталон-1 (темп. кип. 72° при 12 мм), существует только е ненапряженной цис-форме, так как сильно напряженная/прайс-форма легко изомеризуется в менее напряженную конфигурацию по еле» дующей схеме:
Наоборот, пенталоны-2
для которых такая изомеризация невозможна, существуют в двух стереоизомерных формах:
цис-пенталон-2 (темп. кип. 78° при 10 мм; темп, плавл. —28е)
/праяс-пенталон-г (темп. кип. 62° при 10 мм; темп, плавл. +18°)
Еицикло-[0,3,4]-нонан, или гидриндан. Бициклическая система этого углеводорода, о конформационном анализе которой уже шла речь на стр. 100, имеет большое значение в природе, так как она входит в состав скелета стероидов, где образует кольца С и D (стр. 165).
Индивидуальные стереоизомеры получаются восстановлением соответствующих p-гидринданонов. Наоборот, каталитическое гидрирование индена (стр. 466) дает смесь стереоизомеров:
цнг-гидриндан транс-гидриндан
(темп. кип. 166° при 760 мм; (темп. кип. 159° при 760 мм;
пр _ 1,4714) «р - 1,464з)
В присутствии платинированного угля при 300° гидриндан превращается в индан (стр. 466).
В отличие от quc-формы, тршус-гидриндан’может существовать в виде оптически деятельных энантиоморфов.
Гидринданоны (гидриндоны*). Подобно пенталонам, стереоизомерные гидринданоны-1 (а-гидринданоны) легко могут изомеризоваться друг в друга с промежуточным переходом в енольную форму. Поэтому при получении а-кетонов этого ряда из цис- и /пранс-ци-клогексан-1-карбокси-2-пропионовых кислот
Г’ \/\ СН2—СООН СООН
в обоих случаях получается смесь стереоизомерных а-гидриндано-нов одного и того же состава.
♦ Хотя название гидриндоны довольно употребительно, его следует избегать, чтобы предотвратить путаницу, так как в литературе оно часто употребляется и как синоним инданонов, т. е. кетоинданов:
0~1 - н-i
Гидринданоны-2 (p-гидринданоны) существуют в обеих стереоизомерных формах:
киогидринданон-2 (темп. кип. 225° при 754 мм; темп, плавл. 4-10°)
тра«с-гидринданон-2 (темп, кип 217,8° при 754 мм темп плавл. —12°)
Бицикло-Г0,4,4j-декан (декагидронафталин, или декалин). Система бицикло-[0,4,4 J-декана очень важна, так как она часто встречается в природе (нефть, сесквитерпены, сантонин). Кроме того, декалины, являясь гидроароматическими углеводородами, легко могут быть получены каталитическим гидрированием нафталинов. Благодаря этому стало возможным получение декалина в промышленном масштабе. Он применяется в технике в качестве растворителя.
При гидрировании нафталина получается смесь стереоизомерных декалинов (стр. 101), которая может быть разделена перегонкой на эффективной колонке:
цис-декалин
(темп. кип. 195,7° при 760 мм;
темп, плавл. —43.01°;
«£)°«1,4810; ^4°=0,8963)
транс-декалин
(темп. кип. 187,3° при 760 мм;
темп, плавл. —30,40°;
20 20
nD = 1,4692; dA = 0,8699)
Дегидрогенизация декалинов приводит к тетралину и нафталину. Так же ведут себя и гомологи декалина, не имеющие двух боковых цепей при одном атоме углерода (не являющиеся геж-замещенными) или боковых цепей в положении 9 и 10 (ангулярных групп). При наличии же г^-группировок или ангулярных метильных групп дегидрогенизация в присутствии платины или палладия (Линстед), а также селена или серы происходит с выделением метана или с перегруппировкой, например:
5Н2 +
+ СН4 + 4Н,
Декалоны. Дис-а-декалон (темп. кип. 126° при 20 мм; темп, плавл. +2°; п2с =1,4963) чрезвычайно легко изомеризуется в транс-а-декалон (темп, плавл. 33°) через промежуточный енол.
САНТОНИН И РОДСТВЕННЫЕ ЕМУ ВЕЩЕСТВА
Важными природными веществами, обладающими сильным физиологическим действием, являются некоторые а-лактоны кислот, содержащих систему бицикло-[0,4,4 ]-декана. К таким соединениям принадлежат извлекаемые из алантового корня токсичные аланто-лактон и изоалантолактон:
СН3 О—С=О с <!:н \=сн2
алантолактон (темп, плавл. 76°)
изоалантолактон (темп, плавл. 115°)
К этой же группе относится действующее начало цитварной полыни {Artemisia cina; Средняя Азия)—сантонин,—извлекаемый из молодых цветочных почек этого растения (так называемые Semen cinae), и сопровождающий его артемизин:
СН3 О—С=О СН3 О—С=О
сантонин артемизин
( темп, плавл. 270°) (темп, плавл. 203°)
Сантонин—средство против аскарид (глистов); в слишком больших дозах он вызывает нарушение зрительных ощущений (желтая окраска)» судороги, гиперемию мозга. Сантонин растворяется в щелочах с разрывом лактонного кольца, образуя соль сантониновой кислоты С15Н20О4. Нагреванием с соляной кислотой он изомеризуется в десмотропосантонин, являющийся уже фенолом:
СН3 О—С=О
I I \
ип С СН сн—сн3
V V V
I II I
НС с сн2 ^/\/ с сн2 I сн3
Соединения с мостиковыми бициклическими системами
Алициклические соединения с мостиковыми бициклическими системами характеризуются тем, что их кольца имеют по три и более общих атомов углерода.
Соединения этого класса широко распространены в природе; к ним принадлежат главным образом терпены. Так как терпенам посвящен большой раздел (стр. 115сл.)итам рассмотрены многие закономерности и характерные превращения, свойственные мостиковым соединениям, здесь будут даны только некоторые очень краткие общие сведения и отдельные данные главным образом о простейших представителях различных рядов мостиковых бициклических систем.
Синтез мостиковых систем может быть осуществлен путем замыкания второго кольца у производных моноциклических соединений. Сюда относятся способы внутримолекулярной циклизации с участием групп, находящихся в положениях 1,3 или 1,4, например:
СООН
ён
н2с Хсн2
Н2С сн, \н
СН2—СООН
сн
+ СО2 + Н2О
а также конденсация алифатических двугалоидных соединений с моноцикланами, имеющими две группы СН^ или СН с подвижными атомами водорода, например:
С!НвООС\/С^ нс с=о
I I о=с сн
Хсн,Хсоос,нЕ
СН,Вг
сн с,н6оосх//|\ нс сн, с=о I I I о=с сн, сн
Однако важнейшими методами синтеза мостиковых соединений являются синтезы Дильса—Альдера, например:
;СН
। I СН,
I +
НС I СН2
сн
НС I сн-со
сн, + || >0
нс I СН-СО
ХН
Эти реакции могут давать два типа продуктов присоединения в зависимости от взаимной ориентации реагентов.
Так, из циклопентадиена и малеинового ангидрида могут получиться следующие стереоизомерные ангидриды:
При гидролизе эти ангидриды дают соответствующие кислоты:
Конфигурацию, в которой связь углеродного атома одного из колец с заместителем направлена в сторону другого кольца, называют эядо-формой; если же связь с заместителем направлена прочь от другого кольца, то такой стереоизомер называют .экзо-формой. Таким образом, соединения I и 1а являются зкдо-формами ангидридов и кислог, а II и Па—их .экзо-стереоизомерами.
Оказывается, что при невысоких температурах реакция является стереоспецифичной (т. е. идет в сторону образования одного пространственного изомера) и приводит только к .эндо-конфигурациям. При более высоких температурах получаются смеси обеих форм. Отклонения от этой закономерности довольно немногочисленны.
Из особенностей мостиковых систем следует указать на закономерность, которая носит название правила Бредта.
Согласно этому правилу, при узловом атоме системы не может находиться двойная связь, т. е. соединения типа
невозможны.
Это правило, найденное сначала как эмпирическое, может быть выведено из стереохимических соображений, если учесть, что 6 атомов, входящих в группировку двойной связи
лежат в одной плоскости и все углы между связями в этой группировке равны 120°. Поэтому включение такой плоской группировки в бициклическую систему должно привести к огромному напряжению в молекуле. Однако из этих же соображений следует, что при очень большом цикле это напряжение уже будет проявляться не в полной мере, и в таких системах возможно нарушение правила Бредта. Расчет показал, что в тех мостиковых системах, где в одном из колец с узловым атомом связана цепочка из не менее чем 5 атомов углерода, правило Бредта теряет силу.
Многие мостиковые системы лежат в основе структуры терпенов. Поэтому их названия часто производят от наименований соответствующих терпенов.
Так, бицикло- 11,1,31-гептан часто называют норпинаном, а бицикло-11,2,2]-гептан—норкамфаном:
ло
бицикло-[1,1,3]-гептан, или норпинав
Оицикло-[ 1,2,2]-гептан, или норкамфан
Из мостиковых бициклических соединений, не входящих в чис-терпенов, следует упомянуть бицикло- И ,2,2]-гепгпадиен~2,5:
Это вещество в настоящее время производится в промышленном масштабе из циклопентадиена и ацетилена
так как оно является сырьем для очень эффективных инсектицидов— альдрина и дилъдрина*. Альдрин (вязкое масло) получается конденсацией бициклогептадиена с гексахлорциклопентадиеном;
С1
Дильдрин (темп, плавл, около 95°) является окисью альдрина:
♦ Дильдрин и альдрин названы так в честь Дильса (Diels) и Альдера (Alder). Поэтому надо признать совершенно неправильным встречающийся иногда в нашей литературе перевод названия первого из этих инсектицидов словом диальдрин.
Алициклические соединения с многими циклами
Химия алициклических соединений с многими циклами представляет собой большую и трудную область. При этом, однако, соединения с многими неконденсированными ядрами мало отличаются по свойствам от двухъядерных соединений тех же классов. При наличии же многих конденсированных ядер появляется большое число различных особенностей, в первую очередь стереохимических. Эти особенности будут затронуты в разделах, посвященных терпенам и, особенно, стероидам (стр. 165). Здесь будут приведены только два примера полициклических соединений—пергидрофенантрен и адамантан.
Пергидрофенантрен. Значение пергидрофенантрена
состоит в том, что его структура лежит в основе некоторых дитерпеноидов, например абиетиновой кислоты (стр. 162), а также, в сочетании с пятичленным кольцом, в основе ядра стероидов (стр. 165), играющих огромную роль в жизнедеятельности животных и растений. Он может существовать в шести стереоизомерных конфигурациях:
цис-анти-цис ^ккал/моль
транс-анти-цис (2,4 ккал/моль)
транс-син-транс (> 4,8 ккал/моль)
транс-син-цис (2,4 ккал/моль}
цис-син-цис (6,4 ккал/моль)
из которых первые четыре представляют собой смеси оптических антиподов, а остальные две не могут быть расщеплены, так как обладают элементами симметрии.
Цифры, указанные, в скобках под приведенными схемами, показывают избыток энергии по сравнению с транс-анти-транс-кон-фигурацией.
Во всех случаях здесь приведены наиболее устойчивые конформации. Любопытно отметить, что в /иранс-сш/-трянс-конфигурации кольцо В может существовать только в форме ванны.
Адамантан. Этот весьма интересный насыщенный полициклический углеводород состава С10Н16 был обнаружен в моравских нефтях. Затем он был синтезирован Прелогом из бицикло-! 1,3,3 ]-нонан-2,6-дион-3,7-дикарбоновой кислоты действием бромистого метилена на двунатриевое производное диметилового эфира этой кислоты с последующим превращением карбонильных групп в метиленовые через гидразоны (по Н. М. Кижнеру) и, наконец, отщеплением 2СО2 от свободной дикарбоновой кислоты:
ОС-
CHjOOC—СН
Н2С-
сн--СН2 ос--сн—сн2
I I СН2 |
СН2 НС—СООСН3 —>СН3ООС—CNa NaC-COOCHj^^
I I I
CH--CO H2C--CH—co
H2C-----CH----CH2
адамантан
Адамантан представляет собой кристаллическое вещество, образующее бесцветные октаэдры, плавящиеся при 268° и способные возгоняться; удельный вес его 1,07. Он обладает необычайно высокой (для насыщенных углеводородов) луче-преломляющей способностью (nD= 1,568).
Строение адамантана может быть представлено следующим образом:
Таким образом, адамантан представляет собой комбинацию из четырех сконденсированных циклогексановых колец в форме кресла. Расположение углеродных атомов в его молекуле почти точно повторяет расположение атомов углерода в кристаллической решетке алмаза. Это и по.лужило основанием назвать его адамантаном (от адамант—по-чешски алмаз).
Терпены и их производные
Терпенами издавна называют углеводороды состава С^Н^, встречающиеся в природе, особенно в смоле хвойных растений и во многих эфирных маслах. В тех же природных образованиях содержатся и многие кислородсодержащие вещества, по строению близкие к терпенам. Нейтральные продукты этого рода иногда объединяли (по имени важнейшего их представителя) под общим названием камфор.
Многие терпены путем различных химических реакций можно превратить в ароматический углеводород п-цимол, или и-метилизо-пропилбензол (стр. 119 и 226).
Скипидар, или терпентинное масло, получают из смолистых выделений хвойных растений, главным образом сосны: в Советском Союзе—из обыкновенной сосны (Pinus Silvestris), в Америке преимущественно из длиннохвойной сосны (Pinus palustr is), во Франции— из приморской сосны (Pinus mar it ima). В значительно меньших количествах используются для этих целей другие хвойные растения (ель, лиственница, пихта, кедр).
Если сделать надрез коры и наружных слоев древесины растущего хвойного дерева (эта операция называется «подсочкой»), то на месте ранения выделяется прозрачная жидкая смола, так называемая живица (ранее часто применялось также название терпентин). Смола, самопроизвольно вытекающая из трещин коры и подсыхающая на воздухе, в просторечьи называется «серой». При перегонке живицы (или серы) с парами воды отгоняется жидкий скипидар («живичный», или «серный», скипидар), и остается твердая смола, называемая канифолью (старое ее название «гарпиус» почти полностью вышло из употребления).
Смола, содержащая скипидар, добывается также экстракцией измельченных сосновых пней («осмол») различными растворителями, преимущественно высококипяшим бензином. При сухой перегонке осмола с выделением скипидарного погона получается скипидар худшего качества, чем из живицы. По типу аппаратов, в которых производится сухая перегонка осмола, различают печной, ретортный и котельный скипидар.
Скипидары перегоняются в пределах 155—180°, удельный вес их колеблется от 0,858 до 0,870. Они состоят преимущественно из терпенов. В русском и американском скипидарах преобладает правый
пинен, в французском—левый пинен; в русском скипидаре содержатся также значительные количества Д3-карена*.
Скипидар применяется как растворитель для смол, жиров, лаков, каучука, для приготовления красок для живописи, для получения синтетической камфоры, терпинеола, а также в медицине.
Эфирные масла—летучие маслянистые вещества, образующиеся в растениях и обладающие более или менее приятным запахом. Они могут быть выделены из различных частей растения; лучшие эфирные масла получаются из цветов. Обычно эфирные масла выделяют из растений отгонкой с водяным паром, реже—экстракцией растворителями или отжиманием (прессованием), а наиболее ценные сорта масел, испаряющихся из цветов,—также поглощением твердыми жирами (анфлераж).
Многие эфирные масла, кроме жидких компонентов—элеопте-нов, содержат вымораживаемые твердые вещества—стеаропгпены. Жидкие вещества состоят из углеводородов (часто терпенов), альдегидов, кетонов, спиртов, сложных эфиров; твердые—часто из спиртов или кетонов и т. п., являющихся производными терпенов и родственных им веществ.
Из сказанного можно заключить, что к терпенам и их производным относятся соединения, которые по рациональной систематике органических веществ принадлежат к различным классам и должны были бы рассматриваться при этих классах соединений. Так, среди терпенов состава С10Н16 встречаются вещества следующих классов:
1. Ненасыщенные жирные соединения с тремя двойными связями (алифатические терпены).
* Знак А и цифра вверху указывает, при каком атоме углерода находится двойная связь, причем подразумевается, что она лежит между обозначенным атомом и атомом с большим порядковым номером. Таким образом, А3 означает, что двойная связь расположена между атомами 3 и 4. Если атом углерода связан с несколькими атомами, имеющими больший порядковый номер, то номер второго из атомов, связанных двойной связью, указывается в скобках, например А4<8\ Положение двойных связей в соединении, скелет которого изображается схемой
будет обозначаться знаком д1<7), 2,4(5),8(9). этоТ способ обозначения, почти вышедший из употребления в большинстве разделов органической\химии, до сих пор широко применяется в химии терпенов, стероидов и в некоторых других.
2. Циклические соединения с двумя двойными связями, преимущественно производные циклогексана (моноциклическиетерпены).
3. Соединения с двумя конденсированными йеароматическими кольцами и одной этиленовой связью (бициклические терпены).
4. Соединения с тремя конденсированными циклами без этиленовых связей (трициклические терпены).
Ввиду общих генетических отношений и практической важности многих терпенов их обыкновенно выделяют в особую группу.
Наряду с терпенами С1о Н16 в смолах и эфирных маслах присутствуют родственные им вещества, состав которых, так же как и состав терпенов, является кратным от С5Н8. Так, различают сесквитерпены С16Н 24, или (С5Н8)3, дитерпены С20Н32, или (С5Н8)4, и вообще политерпены (СЬН8)Л. Все эти углеводороды, вместе с их кислородными производными, объединяют под общим названием терпеноиды.
Алифатические терпены
Алифатические терпены встречаются сравнительно редко. К ним относится мирцен (см. том I, стр. 354)
CPU /СН4
>С=СН—СН2—СН2—C<f
СН8/ \сн=сн2
а также близкий к нему по строению оцимен
Оцимен—жидкость (темп. кип. 73—74° при 15 мм; пя =1,4857; уд. вес d\b =0,8031). Он легко окисляется на воздухе; при восстановлении металлическим натрием в спиртовой среде переходит в дигидромирцен.
При нагревании оцимен легко изомеризуется в аллооцимен—углеводород с тремя сопряженными двойными связями (темп. кип. 192° при 760 мм; Пр=1,5448; уд. вес d45=0,8162)
сн8>
:=сн—сн=сн—с=сн—сн3
сн8>
сн8
В природе аллооцимен не встречается; Б. А. Арбузов получил его при термической изомеризации бициклического терпена—а-пинена.
В близких родственных отношениях с оцименом находятся спирты—цитронеллол, гераниол, родинол, нерол и линалоол (см. том I, стр. 392—393); альдегиды—цитраль, цитронеллаль и *родиналь (см. том I, стр. 395); кислоты—гераниевая (см. том I, стр. 395) и цитронелловая (см. том 1, стр. 406).
Для алифатических терпенов и их производных характерна легкость превращения в циклические терпены, а часто также в аромати
ческий углеводород цимол (стр. 226). В качестве примеров можно привести превращения цитраля в ионон (стр. 80), линалоола втерпинеол (см. том I, стр. 393), цитронеллаля в изопулегол (см. том I, стр. 395). Изопулегол образуется уже при хранении цитронеллаля, и особенно легко при действии на него уксусного ангидрида.
В аналогичных условиях родиналь превращается в ментон:
СН3 СН3
СН СН
Н^ 'сН, ЩС^ \н2
H2i <Lho н2с со
Il I
С(СН3)2 СН(СН8)г
родиналь ментон
Превращение цитраля в л-цимол под влиянием иодистоводородной кислоты или при кипячении в ледяной уксусной кислоте может быть изображено схемой: СН3 СН3 СН3
I I I
С С С(ОН)
/ Чч / Ч / \
Н2С СН Н2С СН +2Н2О ^2^ СН3 —зн2о
Н2С CHO H2i сн—он —* Н2С сн—он *
\н V ХхОН)
Il II I
С(СН3)2 С(СН3)2 СН(СНз),
цитраль сн3 I с нс^ \н
* НС\ <^Н ^с^ кцсн8), /2-ЦИМОЛ
Циклические терпены
Строение моноциклических и бициклических терпенов служило предметом многочисленных исследований в течение более чем полувека.
Только к началу нынешнего столетия, благодаря исследованиям Е. Е. Вагнера и Ад. Байера, использовавших обширный фактический материал, подготовленный трудами прежних исследовате
лей, особенно О. Валлаха* и его учеников, строение наиболее важных соединений этого класса стало выясняться. В настоящее время строение многих веществ из класса терпенов подтверждено синтезом, а некоторые из них, например камфора, являются уже предметом промышленного производства.
Моноциклические терпены
Большинство моноциклических терпенов представляет собой гидроароматические соединения, главным образом гидрированные производные n-цимола (реже—л<-цимола), содержащие две двойные связи. Эго означает, что они содержат шестичленный углеродный цикл с метильной и изопропильной группами в пара-положении и могут быть изображены схемой:
7
С
^02
sc Ь
I с—с-с 9 8 10
Цимол и продукты его окисления (n-толуиловая и терефталевая кислоты) часто образуются при различных превращениях моноциклических терпенов, особенно, если эти превращения сопровождаются окислением.
Ментан и его производные
Продуктом полного восстановления моноциклических терпенов и их производных является насыщенный циклический углеводород, по строению отвечающий п-цимолу,—п-метилизопропилциклогексан, называемый ментаном'.
СН3
СН
^СНг
Н2С СН£
\н
I
_________ СН(СН3)2
* Отто Валлах (1847—1931). Немецкий химик. Работал почти исключительно в области терпенов. Много сделал для отождествления природных терпенов различного происхождения, которые до его работ считались отличными друг от друга веществами и носили разные наименования.
Ментан существует в виде двух стереоизомеров: цис-ментан (темп. кип. 172,7°; темп, плавл.—89,84°) и/npauc-ментан (темп. кип. 170,5°; темп, плавл. —86,36°).
Ментол. Одним из важнейших кислородсодержащих производных моноциклических терпенов является ментол (ментанол, 1-ме-тил-4-изопропилциклогексанол-З)
СН8
*сн
СН(СН8)2
ментол
главная составная часть мятного масла (масло перечной мяты Mentha piperita), которая и придает ему запах мяты. Содержание ментола в мятном масле зависит от вида мяты и места ее произрастания. Особенно много ментола содержится в масле японской перечной мяты (свыше 80%).
Вследствие присутствия в молекуле ментола трех асимметрических углеродных атомов, для ментола и его производных следует ожидать существования 23—8 оптических изомеров, составляющих четыре пары антиподов.
Если бы кольцо циклогексана было плоским, заместители должны были бы располагаться по обе стороны этой плоскости следующим образом:
Это можно изобразить также условно по Фишеру:
н 11 н 2| Н С3Н7 3| 4| н L н 1 он с3н7 1 1
1 н3с li 1 1 1 ОН н 1 н3с 1'1 1 1 н н 11
Н3С 1 н 1 н С3Н7 1 1 Н3с 1 н он С3Н7 1 1
1 н 1 1 он н 4 н н
in IV
Если каждая из формул 1, II, III и IV будет изображать, например, правовращающий изомер, то зеркальные изображения этих формул будут отвечать левовращающим изомерам. Применив к изомерам ментола изложенные ранее (стр. 30) соображения об устойчивости креслообразной формы циклогексана и об аксиальных и экваториальных (а- и е-) связях, получим следующую картину стереохимических отношений изомеров:
Изомеры Взаимное расположение заместителей в пространстве Типы связей
СНз. ОН | ОН. С3Н7 СНз 1 он I С3Н7 | СНз | ОН I с3н7
Ментол (I) цис транс е е е а 1 а а
Неоментол (II) . транс цис е а е 4—^ । а I е а
Изоментол (III). транс транс а е е е а а
Неоизоме нтол (IV) цис цис а а е е 1 е а
Наиболее устойчивыми, а значит, и преобладающими конформациями будут приведенные слева от знака&обратимости превращений. Эти конформации можно изобразить следующими пространственными формулами:
(±)-изоментол
(±)-неоизоментол
Физико-химические характеристики изомерных ментолов приведены в табл. 9.
Таблица 9
Изомерные ментолы
Изомер Темпе ратура плавления °C Температура кипения °C Удельное вращение [“Ь Изомер Температура плавления °C Температура кипения °C Удельное вращение Мр
Ментол d- 42,5 216 +49 Изоментол d- . . . . 82,5 218 4-25,9
1- 42,5 216 —49 /- .... 82,5 — —25,9
d,l- . . . . 34,0 216 0 d.l- . . . 53,5 0
Неоментол d- —22,5 212 4-19,7 Нео изоментол d- .... —8 4-0,14
1- . . . — 212 —19,7 /- .... — — —
d.l- . . . . +51 212 0 d,l- . . . + 14 — 0
Обыкновенный ментол, выделяемый из масла перечной мяты, представляет собой /-ментол. Он применяется в медицине как антисептик и анестезирующее вещество, а также в парфюмерии и в пищевой промышленности для придания «мятного» запаха и вкуса. Его жидкие изомеры—неоментолы—обладают токсичностью, а потому ментол необходимо тщательно освобождать от их примеси. В масле перечной мяты содержится также в небольшом количестве ^-неоментол.
Помимо выделения из мятного масла, ментол получают гидрированием значительно более доступного фенола—тимола (стр. 267)
Ан(СН8)2
тимол
+ЗН2
сн8
сн
\н2
I I Н2С СН—он
I
СН(СН3)2 ментол
или гидрированием кетона—пулегона (стр. 123). Из получающихся смесей, содержащих жидкие изомеры, кристаллический ментол выделяют вымораживанием.
При гидрировании тимола с хорошим выходом получается d,/-ментол и образуется немного изоментола.
При окислении хромовой смесью ментол как вторичный спирт дает кетон ментон (темп. кип. 207°), а при дальнейшем окислении
хромовым ангидридом в уксусной кислоте образуется ₽-метил-8-изо« бутирил-нчралериановая кислота:
СН., I СН
н^ \н2 н2с со
I
СН(СН3)2 ментон
сн3 I сн
СН(СН3)2 р-метил-6-изобутирил-н-валериановая кислота
Ментон получается также при восстановлении пулегона:
СН3
СН н^ \н2 н2с со
(!(СН3)2
пулегон
СН3
н^ \н4
I I н2с со
СН(СН3)2 ментон
Пулегон (Д4(8)-ментенон-3) содержится в масле Mentha pule-Ц1ШП и представляет собой пахучую жидкость с темп. кип. 224°. Водными растворами кислот (особенно легко—муравьиной) пулеюн гидролизуется, образуя З-метилциклогексанон-1 и ацетон:
СН3
СН [1^ \н2
I I н2с со
II
С(СН3)2
+ (СН3)2СО
Отнятием воды от ментола или НС1 от ментилхлорида, образующегося при действии на ментол пятихлористого фосфора, получается
углеводород с одной двойной связью, называемый ментеном (Д3-ментен):
СН3
I
СН
\н2
I I
Н2С сн
Lh(CH3)2
Ментадие ны и их производные
Большое число моноциклических терпенов является п-мента-диенами, изомерия которых зависит от расположения двойных связей.
Некоторые ментадиены содержат асимметрический атом углерода и поэтому оптически активны.
Лимонен, или Д1>8(9>-n-ментадиен (темп. кип. 175,5—176°; б/Г= 0,8402; по =1,4743; [a]D=±125°)
СН3 d н^ %н
Н2С сн2 '*‘с^
сн2=с—сн8
может существовать в двух оптически деятельных формах (d- и /-лимонен) и в оптически недеятельной, рацемической форме, называемой обычно дипентеном (/ или г-лимонен).
d-Лимонен является главной составной частью масла померанцевой корки; в лимонном масле он содержится в смеси с пиненом. Большое количество d-лимонена содержится также в сельдерейном масле (до 60%) и в тминном масле (около 40%). l-Лимонен содержится в скипидаре из сосны обыкновенной, встречается в некоторых видах хвойных масел, например в Abies excelsa, в русском и американском мятном масле, а также в масле сосновых игл (в «лесной воде», получаемой перегонкой сосновой и пихтовой хвои с водяным паром). Пипентен встречается в некоторых скипидарах (в русском и во французском скипидаре), так как он получается при нагревании пинена, камфена и других терпенов до 250—270°, кроме того—в пихтовом масле и др. Он образуется также при сухой перегонке каучука и при димеризации изопрена.
Оптически активными терпенами являются также фелландрены'.
СН3
А
нс7 \н к I нс сн, '‘‘СН
Ан(СН3), а-фелландрен, или дЬ 5-п-мента диен (темп. кип. 58—59° при 16 мм)
I
СН(СН3), p-фелландрен, или д2.1(7)
-п-ментадиен (темп. кип. 57° при 11 мм)
которые при перегонке при нормальном давлении быстро полимеризуются.
В масле горького укропа, имбиря, в цейлонском коричном масле и в масле элеми преобладает d-a-фелландрен, в австралийском эвкалиптовом масле — Z-a-фел-Ландрен. d-$-Фелландрен содержится в масле водяного укропа (Phellandrium aqua. ticum). Z-P-Фелландрен найден в скипидаре пихты сибирской (Abies sibirica Ledeb).
Лимонены под действием кислот могут изомеризоваться в не содержащие асимметрических атомов углерода, а потому оптически недеятельные, терпинены
СН3
I
С h/Sh
I I
Н2С СН
Y
СН(СН3), a-терпинен, или д!’3-п-ментадиен (темп. кип. 174—175°)
и терпинолен:
сн2 (I с
Н2(/ \н2
Ан(СН3), 3-терпинен, или дЗ, Ц7)-п-ментадиен (темп кип. 173—174°)
СН3
I с нс сн,
I I н,с сн,
СН3
С /Ч
Н2С сн
I I
НС сн2 \ /
с
Ан(СН3), 7-терпинен, или Д Ь 4-п-мента диен (темп. кип. 183°)
А(СН3), терпинолен, или д 1»4^)-п-ментадиен (темп. кип. 183—185°)
Сырой терпинен обыкновенно состоит из a-терпинена (в преобладающем количестве) и ртерпинена. a-Терпинен содержится в кардамон
ном, кориандровом и других.маслах, ртерпинен— в тех же маслах, а кроме того, в лимонном. ^-Терпинен в природных продуктах не найден. Терпинолен найден в кориандровом масле и в масле из смолы элеми.
Из моноциклических терпенов с двумя двойными связями следует отметить также сильвестрен, который является производным лгцимола и имеет строение:
СН3 I с нс^
Сильвестрен не является собственно природным продуктом; его нахождение в скипидарах обязано изомеризации бициклического терпена Д3-карена (стр. 133), происходящей в процессе получения некоторых скипидаров.
Моноциклические терпены, содержащие две двойные связи, могут присоединять галоиды, галоидоводороды, воду и пр. в количестве одной или двух молекул.
Продукт присоединения брома к оптически деятельным лимоненам—лимонентетрабромид С10Н16Вг4—образует кристаллы с темп, плавл. 105°; бромированный рацемат—дипентентетрабромид—плавится при 125°.
При исследованиях терпенов часто используются продукты присоединения к ним хлористого нитрозила (обычно это кристаллические вещества, называемые нитрозохлор идами) и окислов азота (продукты присоединения N2O3 называются нитрозитами, продукты присоединения N2O4—нитрозатами).
Присоединением одной молекулы воды к лимонену получаются терпинеолы, или ментенолы, С10Н17ОН, а присоединением двух молекул воды—терпины, или ментандиолы, С10Н18(ОН)2. При осторожном окислении ментадиенов перманганатом в щелочной среде могут быть получены четырехатомные спирты, например из лимонена—лимонэритрит С10Н16(ОН)4 (Е. Е. Вагнер).
Из производных ментадиена, содержащих карбонильную группу, следует упомянуть карвон (Д1,8(9)-н-ментадиенон-6). Это жидкость (темп. кип. 230°), пахнущая тмином. Карвон содержит асимметрический атом углерода:
СН3
Н2С СН2
СНг=С—сн3
d-Карвон—главная часть тминного масла; /-карвон содержится в масле кудрявой мяты Mentha crispa.
Под действием щелочей карвон легко изомеризуется в фенол карвакрол (стр. 263):
СН3
С
У \
НС со
I I н,с сн2
СНз
СН2=С—СН3
карвон
НС с—он
I il
НС сн
\ / с I СН(СН3)2
карвакрол
Оксим карвона легко получается действием щелочи на нитрозохлорид лимонена:
сн3
/ ч
Н2С сн
н2с сн2
^сн
4- NOCi —>
сн2=с—сн3
Н3С CJ
сн3
с
Н2С C=NOH
Н2С сн2
^сн
I сн2=с—сн3
I I н2с сн2
V
I сн2=с-сн3
+ HC1
Терпин (я-ментандиол-1,8), являющийся продуктом присоединения двух молекул воды к лимонену, может существовать в двух, стереоизомерных цис- и тракс-формах:
Н3С ОН
2| !
Н2С С2Н
Н3С ОН \/
Н,(/ сн, I I н,с сн,
Н С(СН3)2ОН
мнс-тегпин
(темп плавл )05°: темп. кип. 258°)
ri' С(СНЯ),ОН
/пр-'шс-т'ерпив (темп плавл. 158°.
темп. кип. 265°)
Терпин был получен синтетически Леркиным следующим образом. При действии p-иодпропионового эфира на двунатриевое производное циан уксусного»
эфира протекает реакция, упрощенная схема которой дана ниже:
С,Н6ООС 1 СООС2Н6 С,Н6ООС COOCjHj 1 t
Н,с 4- сн2 - > н,с 1 CH, 4- 2NaJ
H2i 1 СН, н,с с'н,
\j NaCNa 1 С2Н6ОО^/ ''cN CjHjOCX^ \n
При омылении полученного соединения образуется четырехосновная кислота, которая теряет СО2 и дает трехосновную кислоту; последняя же при нагревании с уксусным ангидридом теряет Н2О и СО2 и превращается в циклическую кетонокислоту:
ноос соон ! I Н2С сн2
I
Н2С^/СН2 ноос/ \соон
Продукт присоединения 3CH3MgJ к сложному эфиру этой работке водой превращается в терпин:
кислоты при об-
Н3Ск zOMgJ
н2с сн2
Н2<2 СН,
I C(CH3)2OMgJ
Н2С сн,
I I н,с сн,
I C(CH3)20i i
Обыкновенный терпин представляет собой zji/c-форму, легко присоединяющую воду с образованием терпингидрата (транс-терпин не образует гидрата).
Терпингидрат, применяемый в медицине при заболеваниях верхних дыхательных путей, по мнению некоторых химиков является соединением с открытой цепью:
СН3
НОСН,—СН,—С(ОН)-СН2—сн2—сн2— ioH tl3 СН3
Однако большинство химиков считает терпингидрат обыкновенным кристаллогидратом терпина, так как при 100°, а над серной кислотой— даже при комнатной температуре, он теряет воду, образуя терпин. Обычно терпингидрат получают действием на скипидар спир
та и водной азотной или разбавленной (25%-ной) серной кислоты. При этом терпены скипидара, изомеризуясь в дипентен, присоединяют три молекулы воды с образованием терпингидрата.
Терпингидрат получается также в результате присоединения воды к гераниолу и циклизации образовавшегося триола:
HjC7 \н
i I + 2Н2О
Н2С СН2-ОН
\н
Н2С сн2 I I
Н8С сн2—он
^СНг i(CH3),0H
I I -н,0 и2с сн9
С(СН3)2 гераниол
С(СН3)2ОН
терпингидрат
(призмы; темп, плавл. 116—117°)
Действием на терпингидрат бромистоводородной кислоты получают стереоизомерные (цис- и транс-) дипентендигидробромиды С10Н18Вг2. Продуктом восстановления терпингидрата, например иодистоводородной кислотой, является ментан.
При действии водоотнимающих средств, например разбавленной серной кислоты при нагревании, терпин отщепляет одну или две молекулы воды. В результате отнятия двух молекул воды получаются дипентен и терпинолен; продуктами отщепления одной молекулы воды являются прежде всего ненасыщенные алкоголи— терпинеолы (ментенолы):
/ \ Z \
Н2С сн Н2С сн2
II II
Н2С сн2 Н2С сн8
4<jH
I ।
C(CH3)2OH сн2=с—снй
а-терпинеол р-терпинеол
(Д1-ментенол-8) (д8^9)-ментенол-1)
Терпинеолы обладают приятным запахом, напоминающим запах сирени и ландыша. Поэтому смесь терпинеолов, получаемая указанным способом (в ней преобладает оптически деятельный а-тер-пинеол), находит большое применение как душистое вещество.
а-Терпинеол имеет темп, плавл. 40° в оптически деятельной форме и 35° в недеятельной форме; кипит при 219°. (З-Терпинеол плавится при 32° и кипит при 210° а-Терпинеол как в оптически деятельной, так и в недеятельной форме содержится в ряде эфирных масел (кайе-путовое, кардамонное и пр.).
При действии на терпин серной кислоты кроме терпинеолов получается в небольшом количестве также цинеол—окись, отвечающая т^с-терпину как гликолю:
,-----С—СН3
I Н,<С \н.
о I |
н»с сн2 \н
------QCH3)s цинеол (темп кип 176°)
Цинеол—жидкость с камфарным запахом—содержится в большом количестве в эвкалиптовом и розмариновом масле, в масле сантонинной полыни и пр.
Единственным известным природным перекисным соединением является производное Д2-ментена—аскаридол'.
СН3
Н2С Y СН
СН(СН3)?
Он выделен из эфирного масла одного из видов полыни (Chenopodium antihelminticum) осторожной перегонкой в вакууме. Аскаридол— густое масло с удушливым запахом (d20=0,9985; Пв = 1,4743), перегоняющееся при 86° (при 8 мм); при нагревании до 130° он самопроизвольно разлагается со значительным повышением температуры. Он бурно разлагается также под действием концентрированных кислот.
Аскаридол получается окислением а-терпинена кислородом при аскаридола на палладиевом
облучении светом; при гидрировании катализаторе образуется гшс-терпин:
H2d сн3 А z Чн 1 + 02 сн3 с п,сТЬн 1 ° 1 -5- Н3Ск уОН н.Ч Чн2
н31 сн V 1 СН(СН3)2 Н2С 7 сн 1 СН(СН3)2 1 1 н2с сн2 ц/ \с(СН3)2ОН
Хеноподиевое масло, содержащее аскаридол, обладает глистогонным (антигельминтным) действием.
Бициклические и трициклические терпены
В бициклических терпенах и их производных чаще всего встречаются сочетания циклов, отвечающие норкарану (стр. 98), норпи-нану и норкамфану (стр. 111), а^ именно системы карана, пинана и камфана:
каран
(4,7,7-триметилбицикло-[0,1,4]-гептан)
пинан
(4,7,7-триметилбицикло-[1,1,3]-гептан)
камфан, или борнилан (4,7,7-триметилбицикло-[1,2,2]-гептан)
Из приведенных формул видно также, что строение всех трех основных углеводородов в известном смысле близко к строению /г-цимола.
Каран по строению близок, кроме того, к ти-цимолу^. а пинан— к о-цимолу.
Группа карана
Каран в природе не встречается. Он гладко получается из кетона карона разложением его гидразона щелочью, в присутствии платинированного фарфора (Н. М. Кижнер).
Можно получить каран также гидрированием Д3-карена. Каран имеет темп. кип. 169,5°, уд. вес d20=0,841 и показатель преломления 1,4567.
Наиболее доступным веществом группы карана являлся долгое время кетон карон, получаемый из моноциклического кетона карвона следующим путем:
С-СН3 Н(/ ХСО
I I Н2С сн2
Х'с'н
I сн2==с—сн, карвон
СН—сн3
н2с со + НВг I I-----------*
Н2С сн2
\н
сн2=<!;—сн3
сн—сн3 н2с^ \о н2с ^н2
С(СН3)2Вг
-НВг ---->
дигидрокарвон
карон
Карон при нагревании легко изомеризуется в моноциклический кетон кар-венон—производное п-ментана:
СН—СН3'
н/ Лео
н2с <Lh CH(CHS)2
карвенон
При нагревании карона с муравьинокислым аммонием получается карил-амин, под действием кислот изомеризующийся в моноциклический вестриламин, который при отнятии NH3 превращается в сильвестрен—производное лг-ментана:
СН3 I
к а рила мин
н2с
Н2С
сн3
сн
сн—nh2
I /СН3
сн—с< / СН2
вестриламин
сн3
с
/V
Н2С сн
I !
Н2С сн—
сильвестрен
При окислении карон дает кароновую кислоту (1,1-диметилциклопропан-2,3-ди карбоновую)
СНЗЧ ZCH—СООН
СН/ ^СН—СООН
В некоторых сортах скипидара (в индийском—из Pinas longt-folia, в шведском, русском и немецком—из Pinus Silvestris') обнаружены бициклические терпены, являющиеся производными карана (Симонсен, 1920 г.; в русском скипидаре—Б. А. Арбузов, 1928 г.) и названные каренами.
Твердо установлено существование А3- и Д4-каренов:
Д3-карен, „ли
4,7,7-триметилбицикло-[0,1,4]-гептен-3 (темп. кип. 170°; = 0,8645; = 1,4723)
Д4-карен, или
4,7,7-триметилбицикл о-[0,1,4]-гептен-4 (темп. кип. 165—167° при 707 мм)
При действии на карены хлористого водорода получается смесь сильвестрендигидрохлорида и дипентендигидрохлорида, т. е. происходит разрыв трехчленного кольца в двух направлениях:
сильвестрен- карен
дигидрохлорид
д ипе нтен дигидрохлорид
Группа пинана
Пинан также не встречается в природных продуктах. Гидрированием а- и ^-пиненов получаются препараты пинана с различными свойствами, зависящими от условий гидрирования. С. С. Наметкин
показал существование двух стереоизомерных форм этого углеводорода, названных им пинаном и пинокамфаном:
пинан (темп. кип. 169°) пинокамфан (темп. кип. 165°)
Нагляднее этот вид стереоизомерии можно представить так:
Из этих моделей ясно видно, что, несмотря на наличие в каждой из молекул трех асимметрических атомов углерода, возможно существование только двух оптически деятельных изомеров пинана и пинокамфана. Если бы не было группы СН3 при углеродном атоме 4, в молекулах имелась бы плоскость симметрии и происходила бы полная внутренняя компенсация, т. е. оптическая деятельность появляется только за счет одного центра асимметрии—углеродного атома 4.
Пинен. Главной составной частью скипидаров, получаемых из хвойных растений, является бициклический терпен ъ-пинен:
Часто его изображают для простоты схематической формулой
Подобные формулы вообще очень часто применяются для изображения терпенов.
а-Пинен—жидкость, обладающая запахом скипидара; темп. кип. 156°; 0,8585; п^=1,4653; 1а]^= ±51°. d-а-Пинен содержится
в русском, шведском и немецком скипидарах; /-а-пинен—во французском скипидаре.
В скипидарах вместе с а-пиненом почти всегда присутствует р-пинен, или нопинен, (темп. кип. 163—164°):
З-Пинен найден в русских скипидарах из сосны обыкновенной (Pinus stive-tris), ели (Picea excelsa) и других хвойных пород.
Приведенная выше формула а-пинена, правильно передающая его строение, была предложена и обоснована Е. Е. Вагнером в 1894 г. Осторожным окислением а-пинена перманганатом он получил пиненгликолъ, окислил его далее в кетоноспирт, дающий при окислении у.-пиноновую кислоту.
Строение а-пиноновой кислоты—производного циклобутана— было доказано окислением ее (через пиноилмуравьиную кислоту) в пиновую, а затем в очень прочную норпиновую кислоту
(СН3)2С—СН—сосн3
НООС—СН2— HtZ—СН2
а-пиноновая кислота
(СН3)2С—СН—СООН I I ноос—сн2—нс—сн, пиновая кислота
(СН3)2С—сн-соон
НООС—НС—сн2
норпиновая кислота
Норпиновая кислота оказалась идентичной синтетически полученной 1,1 -диметилциклобутан-2,4-дикарбоновой кислоте.
а-Пиноновая и пиновая кислоты были получены также непосредственно окислением пинена (А. Байер).
Установление строения пинена представило большие затруднения, так как циклобутановое кольцо пинена, сохраняющееся в продуктах окисления (пиноновой и пиновой кислотах), при большинстве других превращений пинена или разрывается, или изомеризуется в циклопентановое, а возникающая при этом группировка связей камфана в свою очередь способна к дальнейшим изомеризациям.
При действии кислот и при нагревании пинен может изомери-. зоваться в моноциклические терпены—дипентен, терпинен и терпинолен, благодаря чему из скипидара, содержащего пинен, можно получать терпингидрат, терпинеол и пр. Как показал Б. А. Арбузов, я-пинен при 350° изомеризуется в аллооцимен (стр. 117).
Продукт присоединения к пинену хлористого водорода, иногда неправильно называемый, вследствие своего запаха, «искусственной камфорой», представляет собой не пиненхлоргидрат, а производное камфена—борнилхлорид. Недавно было найдено, что при действии хлористого водорода на а-пинен при сильном охлаждении получается, действительно, пиненхлоргидрат, крайне легко отщепляющий НС1 (при действии слабых щелочей), а при обычной температуре изомеризующийся в борнилхлорид.
CI-HC
Н2С
борнилхлорид
пиненхлоргидрат
Пинен легко окисляется кислородом воздуха. Е. Е. Вагнер показал, что продукты окисления образуются с разрывом «мостика??, и установил их строение.
Одним из первых продуктов самопроизвольного окисления а-пинена влажным кислородом является собрерол, легко отщепляющий воду с образованием пинола:
а-пинен
собрерол (темп, плавл. 150°)
ппнол
(темп. кип. 184°)
При энергичном окислении а-пинена получаются последовательно кислоты пиноловая, терпениловая и теребиновая:
СН3 I
СО ноос \:н—
I I
Н2С СНо у (СН3)2С--------о
пиноловая кислота
ноос со—
H2i сн2
(СН3)2С----.0
со—
ноос сн2 ^сн .
I
(СН3)2С---о
терпениловая кислота (с 1 мол. НоО плавится при 57°; безводная—при 90°)
теребиновая кислота (темп, плавл. 175°)
Строение этих кислот находится в согласии с формулой строения пинена.
Пользуясь пространственными моделями, строение а-пинена и пинола можно изобразить следующим образом:
При взаимодействии а-пинена с сухим кислородом окисляется метиленовая группа, соседняя с двойной связью; при этом сначала образуются перекисные соединения, в дальнейшем дающие кетон вербеной и отвечающий ему спирт вербенол. Образование этих соединений является результатом следующих цепей реакций:
Вербеной—жидкость (темп. кип. 227°), обладающая своеобразным запахом, напоминающим одновременно запах камфоры и мяты.
При действии на пинен нитрозйлхлорида получается характерный продукт присоединения пиненнитрозохлорид (темп, плавл. 103°). Так как нитрозогруппа, если она находится при углеродном атоме, имеющем еще и атомы водорода, обычно перегруппировывается в оксимную группировку, пиненнитрозохлориду приписывается оксимное строение:
Однако молекулярный вес этого вещества отвечает удвоенной формуле.
Пиненнитрозохлорид при кипячении с анилином регенерирует пинен, а при кипячении со щелочами отщепляет НС1 и дает нитрозопинен (темп, плавл. 132ь).
При действии других аминов пиненнитрозохлорид образует нитроламины CiolIie(NO)(NHR). Во всех этих соединениях сохраняется «мостик» пинена.
При пропускании через раскаленные трубки пинен, как и дипентен, может распадаться на две молекулы изопрена: С10Н16 —> 2С5Н8.
Группа камфана
Камфора. Наиболее важным представителем группы камфана является обыкновенная камфора, или d-камфора, известная также под названием японской камфоры. d-Камфора добывается в больших количествах из дерева Laurtis camphora, растущего в Японии и в Китае (особенно на острове Тайвань). Из измельченной древесины перегонкой с водяным паром отгоняются камфора и камфарное масло. Камфору отжимают и очищают возгонкой. При этом она получается в виде прозрачной, бесцветной, вязкой кристаллической массы, обладающей своеобразным камфарным запахом. d-Камфора плавится при 175°, кипит при 209°; несколько летуча уже при обычной температуре.
Оптический антипод обыкновенной камфоры l-камфора встречается в природе в масле Matricaria parthenium, а также в некоторых видах полыни, например Artemisia leucoides (Средняя Азия), Artemisia maritima (район Астрахани). Смесь оптических антиподов— рацемическая г-камфора—плавится при 178,6°.
Камфора имеет весьма обширное применение как пластификатор в производстве целлулоида и кинопленки, в производстве взрывчатых веществ, а также в медицине. Ввиду этого, ее в значительных количествах получают и синтетически (стр. 145).
Камфора подвергалась огромному количеству исследований, в результате которых получено множество ее производных и продуктов расщепления. Установлено, что камфора представляет собой бициклический кетон, имеющий следующее строение:
камфора (формула Бредта)
Ввиду наличия двух асимметрических атомов углерода, камфора должна была бы существовать в четырех оптически деятельных формах, не считая рацемических смесей. Между тем, кроме одной недеятельной d,/-камфоры, известны только две оптически активные камфоры: правая (d-) и левая (/-). Это несоответствие требованиям теории находит себе объяснение в следующем.
Из рассмотрения модели молекулы камфоры видно, что введение в скелет циклогексана, производным которого является камфо-
ра, одночленной мостиковой связи в положение 1,4 не вызывает существенного искажения валентных углов и растяжения связей только при условии, что шестичленный цикл примет конфигурацию «ванны»:
При такой конфигурации молекулы камфоры оптическая изомерия может выражаться в наличии только двух зеркально-подобных форм всей молекулы в целом:
СН3 СН3
правая
левая
Углеродный скелет молекулы имеет плоскость симметрии, проходящую через атомы углерода 1,7,4. Замещение на кислород двух водородных атомов, при любом из атомов 2, 3, 5 или 6 лишает молекулу этой плоскости симметрии и создает асимметрию всей молекулы, что и обусловливает появление оптической деятельности. Обе формы можно изобразить и таким образом:
правая
левая
Здесь жирные линии мостиковой связи при углероде 7 обозначают, что этот мостик лежит над шестичленным циклом, а пунк-
тирные линии—что он располагается под этим циклом.
При окислении камфоры сначала образуется двухосновная камфарная кислота, содержащая пятичленный цикл, а при дальнейшем окислении—кислоты камфановая и камфороновая (триметилтри-карбаллиловая):
СН3
I
С—СООН и./!
-| С(СН3)2 н*с\сн
СООН камфарная кислота
сн3
I ^с------со
н9с |
| С(СН3)2
Н2С | \с---------о
I
СООН
камфановая кислота
сн3
/С—СООН Н2с !
I С(СНЙ)2 НООС |
СООН
камфороновая кислота
Строение камфороновой кислоты было доказано тем, что при перегонке она распадается на триметилянтдрную и изомасляную кислоты, уголь, водород и СО2:
(СН3)2С—СООН
I
СН3—С—СООН
I
СН2—СООН
(СН3)2С—СООН
СН3—(!н-СООН
4~ С 4~ П2 4~ СО о
(СН3)2СН—СООН + СН3—СН—СООН+ со2
СН3
Позднее строение этой кислоты было подтверждено синтезом.
Исследования строения камфарной кислоты также завершились синтезом (Комппа, 1909 г.). Конденсацией щавелевого эфира с эфиром диметилглутаровой кислоты был получен эфир диметилдикетоциклопентандикарбоновой кислоты. Последовательным действием Na и CH3J был введен в группу СН радикал СН3 и получен эфир дикетокамфарной кислоты, в котором рядом реакций восстановления обе группы СО были превращены в группы СН2 и получена камфарная кислота в виде ее эфира:
СН3
I
стг /СН—COOR X—COOR
ROOC | ОС I ОС I
I 4- С(СН3)5 -> | С(СН3), | С(СН3), ->
ROOC | ОС | ОС I
СНо—COOR \сн—COOR ^CH-COOR
СН3
уС—COOR Н2С |
> | С(СН3)2
Н,С I
xzh-coor
эфир камфарной кислоты
Синтез камфоры из камфарной кислоты был еще раньше произведен Бредтом по схеме:
СН3
уЬ-------со
Н2С I I восстановление
I С(СН3)2О----------->
Н2С | |
\сн—со
ангидрид камфарной кислоты •
X------со
Н2с | I
I С(СН3)2 О -
HgC | |
\сн------сн.
камфолид
сн3 I
/С—COOK
действие Н2С I омыление
— —— » С(СН3)2---------s
KCN Н2С |
СН—СН2—CN
СО С(СН3)2|
камфора
Таким образом был осуществлен полный синтез камфоры.
Камфора как кетон реагирует с гидроксиламином, фенилгидр-азином и пр., образуя оксим, гидразон и т. п. При действии иода камфора образует карвакрол, при нагревании с Р2О5 или P2S5—цимол. Обе реакции идут с разрывом мостика, образующего пятичленные кольца.
Водородные атомы группы СН2, находящейся рядом с СО, отличаются подвижностью, подобной подвижности их у других циклических кетонов. Так, благодаря наличию этой группы камфора вступает в реакцию с ароматическими альдегидами, давая продукты конденсации, например:
сн3 I *с
С(СН3)2|
*сн
с=снс6н5
С амилнитритом в присутствии алкоголята натрия камфора образует изо-нитрозокамфору, гидролизуемую кислотами в камферхинон'.
изонитрозокамфора
I С(СН3)2| нгс со
камферхинон
Борнеол и изоборнеол. При восстановлении камфоры получается смесь двух стереоизомерных вторичных спиртов—борнеола и изоборнеола:
Так как при восстановлении кетонной группы образуется в положении 3 асимметрический атом углерода
>сн—он
то и борнеол и изоборнеол, в свою очередь, существуют каждый в двух оптически деятельных формах.
d-Борнеол (борнейская камфора) содержится в выделениях дерева Dryobalanops camphora. растущего на островах Борнео и Суматра, а также в розмариновом и других маслах. Это кристаллы с темп, плавл. 203° и темп. кип. 212°.
l-Борнеол в виде уксусного эфира содержится в большом количестве в хвое пихты сибирской {Pinus sibirica), произрастающей в Сибири и на востоке Европейской части СССР. Он легко может быть выделен из пихтового масла омылением этого эфира. /-Борнеол и d,/-борнеол содержатся также в валериановом масле.
Очень интересен осуществленный Альдером диеновый синтез борнеола из 1,5,5-триметилциклопентадиена-1,3 и винилацетата:
СН3
НС I
I С(СН3)2
НС I
СН-СООСН.з
II
Сн2
омыление
гидрирование
I С(СН3)5| 112с сн
сн
борнеол
Побочно при этом образуется некоторое количество структурно-изомерного эпиборнеола за счет присоединения группы СН2 винилацетата к атому углерода, несущему метильную группу:
сн3 I
нс | сн2
I С(СН3)2 + ||
НС I СН—соосн,
хн
При действии на оптически деятельные борнеолы галоидоводородных кислот происходит рацемизация и получаются оптически недеятельные борнил галогениды, например борнилхлорид, который может быть получен также присоединением хлористого водорода к пинену (стр. 136).
При действии пятихлористого фосфора получается смесь бор-иилхлорида и стереоизомерного изоборнилхлорида (ср. выше борнеол и изоборнеол).
Изоборнил хлорид легко получается также присоединением хлористого водорода к камфену (стр. 146), причем реакция присоединения сопровождается глубокой изомеризацией углеродного скелета молекулы.
Борнилхлорид при действии уксуснокислых солей превращается в уксусный эфир борнеола, из которого омылением может быть получен d,/-борнеол. Последний может быть получен из борнилхло-рида превращением его в магнийорганическое соединение
С10Н17С1 + Mg C^H^MgCl
окисляемое затем кислородом воздуха:
C10H17MgCl + O C10H17-OMgCl
После гидролиза получается d,/-борнеол:
С10Н17—OMgCl 4- Н.О —> С10Н17ОН 4-MgCl(OH)
Из изоборнилхлорида аналогичными реакциями образуется стереоизомерный борнеолу изоборнеол (темп, плавл. 212°).
Изоборнеол может быть получен в виде уксусного эфира нагреванием камфена с ледяной уксусной кислотой в концентрированной серной кислоте.
При кипячении с натрием в ксилольном растворе изоборнеол изомеризуется в борнеол.
Как борнеол, так и изоборнеол при окислении (например, азотной кислотой, хлором и пр.) или при каталитическом дегидрировании превращаются в камфору.
На этом основано за рубежом производство недеятельной камфоры из скипидара (т. е. из пинена через борнилхлорид), а в СССР— производство /-камфоры из уксусного эфира /-борнеола пихтового масла.
Синтетическая камфора получается в СССР в основном по методу, предложенному В. Е. Тищенко.
Пинен скипидаров изомеризуют в парах в камфен над катализатором, содержащим двуокись титана. Присоединяя к камфену уксусную кислоту, получают борнил ацетат, который затем омыляют в борнеол.
Дегидрированием борнеола получается камфора.
Восстановлением борнеола может быть получен плавл. 156°; темп. кип. 160°):
камфан (темп.
СН3
С(СН3)2 |
камфан, или борнилан (4,7,7-триметилбицикло-[1,2,2]-гептан
Он не содержит асимметрических атомов углерода, а потому недеятелен. В природе камфан не встречается.
Камфен. При отнятии воды от борнеола и изоборнеола, а также при отщеплении галоидоводородов от борнил- и изоборнилгалоге-пидов, большей частью происходит глубокая изомеризация и получается камфен (темп, плавл. 53,5—54°; темп. кип. 160—16Г):
ОН
II
С сн
При действии концентрированных растворов КОН на борнилиодид, а также разложением изоборнилксантогената, получается, наряду с камфеном, чрезвычайно летучий терпен, отвечающий по строению борнеолу и называемый ином (темп, плавл. 113°; темп. кип. 146°): снз
С
I С(СН3)2|| Н2С сн
сн борнилен
Камфен представляет собой твердую массу, пахнущую скипидаром и камфорой. В природе он встречается в скипидарах и в различных эфирных маслах (цитронелловое, имбирное, камфарное, пихтовое и пр.). Камфен существует в трех модификациях: d-, /- nd,/-.
d,/-Камфен легче всего получается из б/,/-борнилхлорида отнятием молекулы НО. Чрезвычайно легко получить его также оь щёплением воды от изоборнеола. Изоборнеол, в свою очередь, легко получается присоединением воды к камфену.
Исследование камфена и его химических отношений представило большие затруднения вследствие глубоких изменений углеродных связей («атомных перегруппировок»), часто совершающихся при его образовании и превращениях. Камфен можно по справедливости назвать «органическим хамелеоном», и следует поражаться той проницательности, с которой Е. Е. Вагнер, один из талантливых учеников А. М. Бутлерова, еще в 1900 г. правил ьно разгадал его строение и объяснил многие его отношения.
Процесс окисления камфена щелочным перманганатом (Е. Е, Вагнер) можно представить так:
СН
Н2С С
I сн, |
Н2С
С(СН3)2
СН камфен
СН
СН
;=сн2
КМпО4
СН—СПО
С(СН»)2
Н2С
СП
Н,
СН камфенгликолъ
С(СН3)2
СН
камфеннлсвый альде; ид
CH-COOi 1
н?с с
I СН2 | н,с с
Nloch
Н2с
СООН
С'СН3).
СН камфен иловая кислота
сн
камфеновая кислеча
сн
СОО11
Н,С СО
I СИ’ I
Н,С
С(СНз)?
СН камфенилон
НООС
н2с
СООН
СН, |
С(СН3)2
СН изокамфанов и кислота
При взаимных превращениях камфена в изоборнеол (или в эфиры изобррнеола) происходят внутримолекулярные перегруппировки, аналогичные пинаколиновой (см. том I, стр. 420) и ретропинаколи-новой* перегруппировкам.
Первоначально предполагалось, что при превращении камфена в изоборнеол (или, например, в изоборнилацетат) продукт гидратации камфена сразу превращается из третичного спирта во вторичный [камфеновая перегруппировка первого рода, или перегруппировка Пагнера—Меервейна):
или
Однако последующие исследования показали, что промежуточно может происходить такая перегруппировка первичного продукта гидратации камфена, при которой одна из метильных групп обменивается местом с гидроксильной группой. Лишь после этого происходит окончательная перегруппировка молекулы, аналогичная приведенной выше.
Перегруппировка, идущая по второй схеме и называемая камфеновой перегруппировкой второго рода, или перегруппировкой Наметкина**, приводит к оптическому антиподу изоборнеола, полу-
* Ретропинаколиновой, т. е. обратной пинаколиновой, называется перегруппировка типа:
ХН3 СН3к /СН3
СН3—СН(ОН)—d--CH3 СН-С—ОН
\сн3 СН3/ \сн3
**Академик Сергей Семенович Наметкин (1876—1950). Выдающийся органик, ученик Н. Д. Зелинского. Работал в области алифатических нитросоединений, в особенности терпенов, впоследствии много занимался химией нефтяных углеводородов. Возглавлял Институт нефти АН СССР.
чаемого при перегруппировке первого рода. Действительно, конфигурация асимметрического атома углерода, отмеченного звездочкой, оказывается в результате перегруппировки второго рода зеркальным изображением конфигурации, полученной в первом случае:
Нагляднее это можно видеть, если рассмотреть обе перегруппировки на моделях или хотя бы воспользоваться графическими изображениями, учитывающими пространственное расположение углеродного скелета камфенгидрата (I) и продуктов его превращения:
При гидратации камфена обе перегруппировки идут одновременно, а потому из оптически деятельного камфена в определенных
|3 мстил камфен
условиях получается рацемическая смесь. Однако скорость этих двух перегруппировок далеко не всегда бывает одинаковой, а потому нередко происходит лишь частичная рацемизация или даже один из продуктов получается в преобладающем количестве.
В некоторых случаях камфеновые перегруппировки могут приводить уже не к оптическим антиподам, а к структурным изомерам. Так, при дегидратации 4-метилизоборнеола в результате перегруппировки Вагнера—Меервейна получается р-метилкамфен, а перегруппировка Наметкина приводит к а-метилкамфену (см. схему на стр. 150). Оба изомерных метилкамфена обнаружены в продуктах дегидратации.
Аналогичные перегруппировки происходят не только при реакциях гидратации—дигидратации, но и при некоторых других. Например, при действии хлористого водорода на камфен вместо нормального продукта присоединения получается равновесная смесь последнего с изоборнил-и борнилхлоридами (см. схему на стр 152).
При действии на дибромид пинена (тибромкамфан) цинковой пыли в спиртовом растворе образуется трициклен\
Трициклеи представляет собой кристаллическое вещество, плавящееся при 63—66° перегоняющиеся при 153—154s. Перманганатом он не окисляется. Трехчленное кольцо в трициклене легко разрывается: под действием НС1 из трицикле-на образуется камфенгидрохлорид и продукты его изомеризации. В природе трициклен не встречается.
Фенхан. Изомерный камфану бициклический углеводород с пя-тичлепным циклом—фен хан (темп. кип. 149,2—149,5°; б/4°=0,8317;
1,4459) сн3
Н2С
сн2
,сн3
сн
фенхан (4,6,6 триметилбицикло-[1,2,2]-гепган/ не встречается в природе.
Схема перегруппировок, происходящих при гидрохлорировании камфена
Фенхан был получен Н. М. Кижнером путем разложения гидразона фенхона—кетона, представляющего собой жидкость камфарного запаха, застывающую при температуре около 5—6° и кипящую яри 192°. Фенхон содержится в эфирном масле Phoeniculum pul pare. Восстановлением фенхона может быть получен фенхиловый с темп, плавл. 45° (оптически деятельный) и темп.
кип. 201—202°. Действием Р2О5 фенхиловый спирт может быть превращен в ,и-цимол:
Производные фенхана при различных реакциях часто претерпевают перегруппировки Вагнера—Меервейна и Наметкина.
Туйан—углеводород, содержащий трехчленное кольцо:
СН3
туйан (4*метил-1-изопропилбицикло-[0,1,3]-гексан)
Из производных туйана представляет интерес кетон ту ион или танацетон'.
СН3
СН(СН3)2
Туйон встречается в эфирных маслах из ТапасеЛит vulgare в некоторых видах полыни, в шалфейном масле. Это жидкость с приятным запахом, кипящая при 200—201°. Известны левовращающий
и правовращающий изомеры туйона; однако это не оптические антиподы, а структурные изомеры.
Отвечающие туйану ненасыщенные туйены, содержащие двойные связи в пятичленном цикле, получены только синтетически.
В эфирных маслах можжевельника встречается сабинен, содержащий экзо-циклическую (семициклическую) двойную связь:
СНо
II
С
Сабинен—жидкость, перегоняющаяся при 164—165°.
Политерпены
Кроме терпенов СюН16, в растительных эфирных маслах чрезвычайно распространены углеводороды того же состава, но более высокого молекулярного веса. Состав их может быть выражен формулой (CrJK)^ т. е. их можно считать полимерами изопрена QHs. Поэтому сам изопрен называют гемитерпеном (семитерпеном, полутерпеном) .
Политерпены делят на углеводороды Ci5H24—сесквитерпены (полуторатерпены), углеводороды С20Н32 дитерпены и т. д. Высшим членом этого ряда является природный каучук (СзН8)/2, для которого число п может быть очень велико. Таким образом, здесь наблюдаются отношения, подобные отношениям глюкоз и полиоз, с той лишь разницей, что в этом случае полимеризация происходит без отщепления каких-либо молекул.
Изопрен в природе не найден. Политерпены и их кислородные производные (пока выделены преимущественно алкоголи), особенно сесквитерпены, встречаются в эфирных маслах и распространены так же широко, как сами терпены и их кислородные производные. Вследствие сложности строения политерпенов и присущей им, как и терпенам, способности к всевозможным реакциям изомеризации в изучении сесквитерпенов достигнуты большие успечи благодаря трудам Земмлера, Шорма, и особенно Ружички. Теперь строение многих сесквитерпенов и родственных им алкоголей можно считать окончательно установленным.
Сескви- и политерпены, смоляные кислоты, полиеновые красители, стероиды, так же как и природные терпены, можно рассматривать как продукты полимеризации изопрена. Наличие в их молекулах углеродного скелета изопрена может быть проиллюстрировано следующими схемами:
кадинен
абиетиновая кислота
Однако происхождение всех этих веществ из изопрена или наличие в них «изопренового скелета» является чисто формальным представ-лением. Хотя алифатические терпены (с открытой цепью) могут образовываться из изопрена в довольно мягких условиях, тем не
менее происхождение самого изопрена в биологических условиях совершенно неясно. Поэтому было предложено много гипотез образования таких веществ в организмах растений. Г. Эйлер, например, считает, что терпены образуются из ацетона и ацетальдегида, уплотняющихся сначала в {3-метилкротоновый альдегид, обладающий углеродным скелетом изопрена:
СН3
I
сн3—с=сн—сно
Высказывались также взгляды, что источником образования изопренового скелета могут быть аминокислоты—продукты распада белковых веществ, например лейцин:
СН3 СН3
СН3—СН—СН2—СН— СООН —» СН3—СН—СН=СН2 + NH3 + со.,
NH2
Другие исследователи считают, что источником терпенов являются углеводы, подвергающиеся при окислительных процессах декарбоксилированию и дегидратации.
Сесквитерпены
Подобно терпенам, сесквитерпены могут относиться: 1) к алифатическим соединениям, т. е. содержать четыре двойные связи и не содержать колец из углеродных атомов; 2) к моноциклическим— с тремя двойными связями; 3) к бициклическим—с двумя двойными связями; 4) к трициклическим—с одной двойной связью; 5) к тетрациклическим—не содержащим двойных связей. Эти типы сесквитерпенов значительно отличаются друг от друга по молекулярной
рефракции:
Алифатические.................69,60
Моноциклические...............67,87
Бициклические ................66,13
Трициклические ................64,40
Тетрациклические ..............62,66
Благодаря этому можно сравнительно легко определить, к какому классу относится тот или другой представитель сесквитерпенов.
Все известные сесквитерпены—густые жидкости, кипящие между 250 и 280°; уд. вес их колеблется от 0,84 до 0,94.
Алифатические сесквитерпены. К производным алифатических сесквитерпенов относятся жидкие алкоголи С]5Н2бО: неролидол — из перуанского бальзама и цветов померанца, фарнезол—из мускатного ореха и цветов некоторых акаций (например, Acacia ^arnesiand), содержащийся также в цветах липы и придающим им их характерный запах.
.Неролидол был синтезирован Ружичкой из гераниола способом, не оставляющим никакого сомнения в его строении.
Третичный оптически деятельный неролидол при нагревании с уксусным ангидридом частью изомеризуется в недеятельный первичный спирт—фарнезол. частью отщепляет воду и превращается в сесквитерпен—фарнезен\
СН3
с сн2 'blj
I I
H.C CH2 C(OH)—CH3
‘\ \ /
CH CH
II
CH3—C—CH3
неролидол
CH3
I
C CH, \h?
H2C CH2OHC—CH.
\ \ //
CH CH
CH3—C—CH3 фарнезол
II CH3—C—CH3 фарнезен
Моноциклические сесквитерпены. При обработке фарнезола уксусной и серной кислотами получается уксусный эфир моноциклического третичного спирта i-бисаболода, при взаимодействии с НС1 дающего соединение С]5Н27С13 (темп, плавл. 80°), тождественное продукту присоединения трех молекул HCI к монони-клическому сесквитерпену бисаболену.
НО СН3 сн
Н2(^ с^ 'сн, н2с (*:н2 с—сн3 \ \ / сн сн II сн3—с—сн3 а-бисаболол
сн3
I
с сн2 /\/\
Н2С с сн2
H2i сн2 с-сн3
\н ^сн
II сн3—с—сн,
бисаболен
При нагревании с уксуснокислым натрием и уксусной кислотой продукт присоединения отщепляет ЗНС1, регенерируя бисаболен (жидкость; темп. кип. 268°; уд. вес 0,8725 при 20°). Бисаболен весьма распространен в природе. Он найден в масле сибирской пихты, в камфарном, лимонном, кардамонном и других маслах. Синтетический бисаболен, возможно, представляет собой смесь изомеров.
К моноциклическим сесквитерпенам относится содержащийся в имбирном масле левовращающий цингиберен (темп. кип. 137—139° при 17 мм рт. ст., уд. вес
0,8684 при 20°). Величина молекулярной рефракции цингиберена (68,37) указывает на присутствие в его молекуле сопряженных двойных связей.
Цингиберен присоединяет 3 молекулы водорода, но всего только 2 молекулы НС1, так как под действием соляной кислоты (как и под действием серной) он превращается в бициклический изоцингиберен.
СН3
СН СИ2
нс7" \н
н2с НС С—сн3
\ \/
сн сн
II сн3—с—сп3
сн8
I
сн сн2
I Ffc/ ^СН
I I II
НС НС с—сн„
V V
I
ci I.,—сн—сн3
цингиберен
изоцингиберен
Очень своеобразным моноциклнческим сесквитерпеном, содержащимся в хмеле, является гумулен (^.-кариофиллен).
Ранее ему приписывалось строение бициклического углеводорода, напоминающее структуру пинена. Однако недавно было показано (Шорм; Клемо и Гаррис), что на самом деле гумулен представляет собою моноциклический углеводород— 1,5,5,8-тетраметилниклоундекатриен-3,7,11:
СН3 з 11 СН с СНзк 5 ,// \г/ V
>с—сн сн2 сн СН3/| И|
н2с—с7н СН2—°сн2
I сн3
Невидимому, гумулен является первым известным природным веществом с один-надцатичленным циклом
Бициклические сесквитерпены. Из бициклических сесквитерпенов лучше всего изучены кадинен и селинен.
/-а-Кадинен (темп кип. 275е, уд. вес 0,918) содержится в скипидаре из сосны обыкновенной, кедра сибирского, а также в применяемом в медицине Oleum са-dinum (продукт сухой перегонки различных видов можжевельника). Кроме того, кадинен получается отщеплением молекулы воды от третичного спирта—-кодинола С1бН?5ОН, найденного в смоле персидских зонтичных растений, называемой гальбанумом.
d-Селинен (темп. кип. 128—132е при 11 мм, уд. вес 0,919 при 20°) получается из масла семян сельдерея.
При исследовании сесквитерпенов, особенно бициклических, очень полезной оказалась реакция их дегидрирования нагреванием с серой или селеном (Вестерберг; Ружичка).
В результате дегидрирования многие сесквитерпены превращаются в гомологи нафталина.
При нагревании с серой кадинен (также цингиберен и изоцингиберен) превращается в 1,6-диметил-4-изопропилнафталин, или кадалин:
СН3
С СН2
hq/ НС^ ^СН
I I !1
Н2С НС . С—СН3
СН3—СН—сн3
а-кадинен
сн . —сн—сн3
кадалин
а селинен— с отщеплением ангулярной метильной группы в виде CH4SH—в Ьме-тил-7-изопронилнафталин, или эудалин:
СН3
СН2 I СН2
3-сслинсн эудалин
Присутствие в кадинене и селииеие двух двойных связей устанавливается присоединением двух молекул НС1 и последующим осторожным их отщеплением от полученных хлоридов; при этом вновь получаются те же углеводороды. Расположение в них двойных связей, а также местоположение метильной группы в селинене установлено изучением продуктов окисления этих углеводородов.
К бициклическим сесквитерпенам относятся также интересные по своему строению углеводороды р- и у-кариофиллены, найденные в гвоздичном масле (из почек гвоздичного дерева Eugenia caryophyilaia)^ атакже в копайском бальзаме, цейлонском коричном масле и пр. Эти карниофиллены имеют следующее строение:
Н2С---СНо
СН3, / \ /СНз
х С—СН Z.C
сн/ I | Н—с/
Н..С-СН \.Сн2
•(-кариофиллен (нзокариофиллею
•'-кариофиллен (кариофиллен)
т. с. являются геометрическими изомерами. Строение 3* и у-кариофилленнов было установлено в 1952 г. Бартоном, Брууном и Линдси; ранее предлагавшиеся для них формулы с шестичленным или семичленным кольцом окзались ошибочными. Таким образом, 3 п у-кариофиллены являются природными продуктами с девяти членным циклом.
Легко видеть, что кариофиллены находятся в близком родстве с моноцикли-ческим сесквитерпеном гумуленом (стр. 158), углеродный скелет которого отличается лишь отсутствием мостика между четырех- и девятичленным циклами.
Под действием кислот кариофиллены гидратируются с одновременным возникновением нового мостика (бициклический сесквитерпен превращается в трициклический). Так, из p-кариофиллена получается трициклический ^-кариофилленовый спирт:
К производным бициклических сесквитерпенов относится алкоголь смолы гваякового дерева—гуайол Ci5H26O (темп, плавл. 93°; темп. кип. 288°):
СН3
СН
Трициклические сесквитерпены. Из производных трициклических сесквитерпенов лучше всего изучен санталол из масла сандала, получаемого перегонкой с паром измельченного сандалового дерева. Это масло применяется в медицине (Oleum santali) против воспаления слизистых оболочек.
Сырой санталол представляет собой смесь жидких алкоголей: правовращающего а-санталола (темп. кип. 159° при 10 мм рт. ст.; уд. вес 0,979 при 15°) и левовращающего р-санталола (темп. кип. 168° при 10 мм рт. ст.; уд. вес 0,973 при 15°). Санталол представляет собой первичный спирт, так как при окислении а-санталола хромовой кислотой получается санталовый альдегид с тем же числом атомов углерода. Из санталового альдегида может быть получена санталовая кислота С14Н2]СООН. При окислении озоном а-санталол отщепляет группу
\ /СН3
>С<
/ ХСН2ОН
и образует альдегид СПН17СНО, соответствующий экасанталовой кислоте Ci0H17COOH. При окислении уксусного эфира енольной формы этого альдегида С10Н1Б—СН=СН—О—СОСН3 получается норэкасанталовая кислота СюН15СООН, а из нее восстановлением—альдегид, который тем же путем превращается в тере-санталовую кислоту С9Н13СООН. Этим рядом превращений устанавливается наличие в санталовом альдегиде боковой цепи —СН2—СН2—СНО. Во всех продуктах окисления не имеется двойных связей, а следовательно, они содержат трициклическое ядро. Нагреванием тересанталовой кислоты с серной кислотой по-
ручается, благодаря изомеризации и отщеплению СО2, углеводород сантен (строение его доказано синтезом):
СН
сантен
На основании всех этих данных а-санталолу приписывается строение:
СН3 СН2 СН
I I снч
НС с--------сн
Природные смолы и бальзамы
Смолами называют твердые, при нагревании размягчающиеся, часто липкие выделения растений, нерастворимые в воде. Полужидкие выделения, содержащие, кроме собственно смол, отгоняемые с паром эфирные масла, называются бальзамами, Некоторые смолы, как, например, гуммигут, содержат примеси камедей, т. е. растворимых в воде смолоподобных веществ, и называются камедистыми смолами. На воздухе смолы обыкновенно окисляются; получающиеся при этом измененные смолы относятся к наиболее устойчивым растительным продуктам. Поэтому некоторые смолы находятся в осадочных породах и добываются как ископаемые. Таковы копалы африканские, южноамериканские, южноазиатские и новозеландские, янтарь, добываемый на восточных берегах Балтийского моря, и пр.
Смолы и бальзамы добываются чаще всего из хвойных, а также из ряда других растений экстракцией коры, древесины или других частей растения, а иногда из млечного сока. Для приготовления лаков применяются индийские смолы— даммар, драконова кровь и гуммилак (в очищенном виде называемый шеллаком), австралийская смола акароид, африканская—сандарак и др. Из бальзамов наиболее известны различные живицы, или терпентины, выделяемые из обыкновенных хвойных деревьев и дающие смолы, называемые канифолью (стр. 115 и 162), а также применяемые в медицине и парфюмерии перуанский и толутанский бальзамы, стиракс и др.
Смола бензойного бальзама (Индия), называемая росным ладаном, применяется для благовонных курений, смола бальзама элеми (Мексика и Филиппинские острова) —для приготовления лаков. Японский лак представляет собой млечный сок лакового дерева (Rhus vernicifera).
Составные части смол мало исследованы. По Чирху, они разделяются на следующие классы: резиноловые кислоты (смоляные кислоты), содержащиеся во многих смолах почти исключительно в свободном виде; резинолы—вещества алкогольного или фенольного характера, частью встречающиеся в виде сложных эфиров; резины—сложные эфиры; резены—углеводороды и другие неомыляемые нейтральные, бедные кислородом вещества.
Смоляные кислоты
Из составных частей смол лучше всего исследованы смоляные кислоты канифоли. В живице—клейкой массе, выделяющейся из пораненного места ствола хвойных деревьев,—содержится около 30% терпеновых углеводородов и около 70% смоляных кислот. Отгоняя углеводороды (скипидар) с паром, в остатке получают канифоль— твердую смолу, размягчающуюся при 50—70°.
Канифоль находит большое применение в промышленности. Свойство смоляных кислот образовывать с углекислыми щелочами соли—смоляные мыла, водные растворы которых обладают способностью пенообразования, используется при добавке их к жировым мылам. В бумажной промышленности канифоль применяется для проклейки бумаги. Ее соли с тяжелыми металлами (резинаты кобальта, марганца и свинца) используются как сиккативы для ускорения высыхания (окисления) олифы и масляных лаков. Канифоль применяется и как основа дешевых сортов лаков, заменяя частично или полностью дорогие смолы (шеллак, копал и т. п.). В состав лаков входит не сама канифоль, а главным образом эфиры ее смоляных кислот, например глицериновый, пентаэритритовый, этиленгликолевый и др. Канифоль вводят также в состав многочисленных композиций (например, так называемая менделеевская замазка—сплав канифоли с железным суриком Ёе2О3).
Канифоль представляет собою смесь кислот состава С19Н29СООН. Наиболее доступной из этих кислот является абиетиновая кислота, которая может быть выделена в чистом воде кипячением канифоли с ледяной уксусной кислотой с многократной перекристаллизацией выпадающего при охлаждении продукта. Наиболее чистые препараты абиетиновой кислоты плавятся при 172—175°; [a ]D= —106° (по другим данным —116°). Следующая по доступности, изомерная абиетиновой левопимаровая кислота имеет темп, плавл. 150—152°; [аЬ= —275°.
Н3С
Н2с I н2с
Н3С
СООН сн2
сн \н
I II
с с
сн \н
<4н, (?—СН(СН3)2 абиетиновая
кислота
Н3С СООН
СН,
Н.(^ \н \н2
I I I
Н.С с с
\ / \ / \
н,с сн сн
Н,С Lj. X СН(СН ) 3 СНо С—СН(СН3\> \ /
СН
левопимаровая кислота
Содержащаяся в канифоли в небольших количествах декспгро-пимаровая кислота (темп плавл. 218—219°; [a ]D= +73°) является не оптическим антиподом левопимаровой кислоты, а ее изомером:
декстропимаровая кислота
Дегидрированием абиетиновой кислоты действием серы (Вестер-берг) или селена получается углеводород ретен (1 -метил-7-изопро-пилфенантрен), а из декстропимаровой кислоты — углеводород пимантрен (1,7-диметилфена нтрен):
сн3
ретен
темп, плавл, 98—99°)
пимантрен (темп, плавл. 86°)
Из бурых углей и некоторых торфов был выделен углеводород фихтелит, обладающий структурным скелетом, лежащим в основе абиетиновой кислоты:
СН, СН сн2 н,^ \н2
I I I Н2С с сн щс^ ^сн" 'сн, н с । ।
сн2 сн \^\СН(СН,)4 фихтелит (темп, плавл. 46,5°
При дегидрировании с серой фихтелит, так же как и кислоты канифоли, отщепляя группу СН3 от четвертичного углеродного атома, образует ретен.
Из составных частей других природных смол почти окончательно доказано строение резинола из гваяковой смолы (гваяковая смоляная кислота) как производного дифенилбутена:
CHSO\ ' СН3 СН3 /ОСН3
НО—/ V-сн=с!---СН—сн8—/~"\—он
Весьма интересное строение имеет урушиол из японского и китайского лаков; он является производным пирокатехина и содержит неразветвленную боковую цепь из 15 атомов углерода. Природный урушиол представляет собой смесь гидроурушиола, содержащего насыщенную цепьС15Н31
НО/ /ОН <f~y-(CH2)14-CH3
и нескольких соединений, имеющих в боковой цепи от одной до трех двойных связей (Р. Манима).
Стероиды
Стероидами называют производные частично или полностью гидрированного 1,2-циклопентенофенантрена типа:
Кольца в этой системе принято для удобства обозначать прописными латинскими буквами. В большинстве стероидов R' и R"— метильные группы (иногда окисленные), которые в некоторых случаях могут и отсутствовать; R—различные алкилы, а также кислород-и азотсодержащие группы.
Стероиды широко распространены в природе и играют важную роль в жизнедеятельности животных и растений. Различные представители этого класса, весьма близкие по своему химическому строению, обладают сильно различающимся физиологическим действием. Это открывает большие возможности для применения их в качестве лекарственных препаратов.
В зависимости от их нахождения в природе и химического строения, стероиды подразделяются на следующие классы соединений: стерины, желчные кислоты, стероидные гормоны, стероидные сапонины, сердечные глюкозиды и стероидные алкалоиды.
Поскольку стероиды построены из конденсированных алициклических колец, для них возможна стереоизомерия, зависящая от цис- или транс-расположения колец относительно друг друга, аналогично стереоизомерии пергидрофенантрена (стр. 112). В природных стероидах кольцо С (см. схему) всегда занимает транс-положение относительно кольца В, а относительно кольца D—почти во всех случаях. Исключением являются лишь стероидные глюкозиды и яды жаб; в этих стероидах кольца С и D находятся друг с другом в цис-сочленении. Наоборот, кольцо А по отношению к кольцу В может быть как в цис-, так и в транс-конфигурации. В зависимости от этого различают два ряда стероидов: А/В-цнс-ряд (нормальный) и
А/В-транс-ряд (так называемый алло- ряд*), причем большинство природных веществ этого класса можно рассматривать как производные восьми углеводородов:
АВ-^мс-ряд
Теста н (этиохолан)
Прегнан Холан Холестан
А/В-тра«с-ряд Андростан Аллопрегнан Аллохолан Копростан
Радикал R,
н ................
с,н8................... ;
СН(СН8)СН2СН2СН3 . . . .
СН(СН3)СН2СН2СН'2СН( СН3)2
В приведенной схеме связи, соединяющие атомы и группы, не входящие в циклическое ядро молекулы, даны сплошными или пунктирными линиями, чтобы показать их расположение в пространстве; сплошными линиями даны связи, расположенные выше, а пунктирными—ниже плоскости циклической системы. Конечно, при этом нельзя забывать, что понятия «выше» и «ниже» здесь применяются лишь условно, так как абсолютная конфигурации асимметрических центров стероидной молекулы окончательно еще не выяснена; кроме того, молекулы стероидов на самом деле не плоские, а имеют сложную пространственную конфигурацию и могут существовать в разных конформациях. Так, например, наиболее устойчивые конформации тестана и андростана имеют следующий вид:
А По—лат. иной
При написании формул природных стероидов условно принято ангулярную метильную группу между кольцами А и В всегда помещать над плоскостью системы*. Из приведенных выше изображений тестана и андростана видно, что ангулярная метильная группа между кольцами С и D направлена в ту же сторону.
Известны также стероиды, в которых отсутствует ангулярная метильная группа между кольцами А и В. Эти стероиды являются производными углеводорода эстрана:
I *Z Н Lh^j ' * н* н
н
И в этом случае ангулярная метильная группа между кольцами С и D изображается лежащей над плоскостью системы.
Все перечисленные выше углеводороды оптически деятельны, так как отмеченные звездочками углеродные атомы в местах соединения колец асимметричны.
Это должно было бы приводить к громадному числу стереоизомеров среди стероидов. Так, например, только за счет стереоизомерии циклического скелета могло бы существовать 27—128 стереоизомеров прегнана, поскольку в этой системе имеется шесть асимметрических углеродных атомов. При введении в ядро заместителей, создающих дополнительные центры асимметрии, число стереоизомеров должно быстро расти. Действительно, при появлении двух дополнительных центров асимметрии число возможных стереоизомеров достигнет 512. Однако в природе количество встречающихся стереоизомеров весьма ограничено. Среди огромного числа природных стероидов найдено лишь четыре пространственные формы скелета (андростан, тестан и их изомеры с ^wc-сочленением колец С и D). Таким образом, синтез стероидов в природе осуществляется «с исключительно большой избирательностью. Такая избирательность не случайна. Следует учитывать, что структура андростана отвечает энергетически наиболее выгодной конфигурации пергидрофенантрена— транс-анти-транс-форме (стр. 112), а структура тестана соответствует конфигурации одного из двух следующих по энергетической выгодности стереоизомеров—цис-анти-транс-форме. Сочленение колец С и D этих углеводородов происходит за счет двух экваториальных связей кольца С, тогда как в zw-сочленении колец С и D должны участвовать одна экваториальная и одна аксиальная связь, что менее выгодно, а потому стероиды с такого рода строением встречаются реже.
* Синтетически были получены эпимеры природных стероидов, у которых ангулярная метильная группа находится по другую сторону плоскости молекулы. У таких соединений связь метильной группы с ядром изображается пунктиром; см., например, лумистерин (стр. 179).
Номенклатура. В основном номенклатура стероидов построена на тривиальных названиях одного из стероидов в каждом ряду. Названия производных выводятся из этих тривиальных названий по обычным правилам (например, холестанол из холестана). При этом нумерация углеродных атомов для определения положения заместителей несколько необычна. Для углеродных атомов ядра она показана ниже, а атомы боковых цепей нумеруются таким образом, что углеродные атомы радикалов R' и R" получают номера 18 и 19, а ближайшему к ядру углеродному атому радикала R дается номер 20. Остальные атомы этого радикала нумеруются один за другим по мере удаления от кольца и независимо от того, находится ли атом в основной цепи или в ответвлении, например:
Пространственную ориентацию заместителей обозначают с помощью букв аир. При этом ориентацию ангулярных метильных групп (С18 и С19) условно принято обозначать буквой р. Таким образом, все заместители, расположенные по ту же сторону плоскости молекулы, что и ангулярные группы, будут также иметь р-ориентацию. Все остальные атомы или группы будут иметь a-ориентацию, а следовательно, при графическом изображении молекул их связи с кольцом должны изображаться пунктиром (стр. 166). В названиях стероидов буквы аир ставятся после номера атома углерода, при котором находится данный заместитель, например: андростанол-За. Для обозначения пространственного положения заместителя при С<з> иногда пользуются приставкой эпи. Так, например, один из стероидных спиртов, в котором гидроксил при Со) занимает P-положение, называют холестанолом, а его эпимер—холестанол-За—эпихолестанолом:
холестанол
эпихолестанол
При этом следует иметь в виду, что приставка эпи сама по себе еще не дает указания на пространственную ориентацию 3-гидрок-сила по отношению к ангулярнь.м группам, а означает лишь противоположную ориентацию по сравнению с каким-либо определенным
соединением. Поэтому в ряде случаев элш-соеди нениями называют и стероиды с 3^-ориентацией гидроксила.
Особо надо отметить обозначение стереоизомеров при изомерии за счет асимметрических центров при C(i7> и С<20). Если при С<17)» кроме радикала R, находится еще какой-либо заместитель, то его конфигурация обозначается как обычно:
R И
17а-окси-
173-окси-
Если же заместитель имеется и при С<20), то здесь стереоизомерия носит уже другой характер: типа’ оптической изомерии винных кислот. Когда конфигурация оптического центра при С<2о> установлена, расположение заместителя при этом углеродном атоме относительно C(i7) и относительно всей системы также обозначается греческими буквами а и р, но при этом имеется в виду, что асимметрические центры C(i7) и С(20) изображены с помощью проекционной формулы Фишера:
R'
I Н—С—ОН
17а,20а-диОкси-
R' I Н—С—ОН
17р, 20а-диокси-
R'
I
НО—С— н
17а,2Ср-диокси-
R' I НО—С—н
173,203-диокси-
Таким образом, здесь буквы аир применяются в несколько ином смысле, чем это делалось при обозначении конфигурации других асимметрических центров.
Если же пространственная ориентация эпимеров при С(20> не установлена, то один из эпимеров условно обозначают буквой а, а другой буквой Ь, например: 20а-оксипрегнан или 20а,22Ь-диоксихолан.
Стерины
Стерины представляют собой вторичные предельные или ненасыщенные спирты, выделяемые из неомыляемой фракции жиров. Это бесцветные кристаллические вещества, почти нерастворимые в воде и хорошо растворимые в органических растворителях. Стерины содержат ^-гидроксильную группу при С(3) и встречаются в природе либо в виде свободных спиртов, либо в виде эфиров высших жирных кислот.
Все стер ины построены по общей схеме:
У многих стеринов в положении 5,6 имеется двойная связь.
Стерины, встречающиеся в животных организмах, называются зоостеринами\ стерины, выделенные из дрожжевых и плесневых грибков,—микостеринами, а стерины растений—фитостеринами. Характеристика важнейших стеринов дана в табл. 10.
Таблица 10
Стерины
Название R* Положение двойных связей Сочленение колец А/В Температура плавления, °C Оптическое вращение Ид Ист очники выделения
Холестерин Н 5 — 149 —39° I Мозг, нервы и другие ткани животного организма, кровь, яичный желток, желчные камни
Холестанол Н — транс- 142 +24° Желчь, кал
Копростанол (копростерин) Н — цис- 101 4-28" Кал
Эргостерин СН3 5, 7, 22 — 165 — 130- Спорынья, дрожжи
Сгигмастерин С2Н5 5, 22 — 170 —40° Соевое масло и масла семян других высших растений
* См. формулу выше.
Холестерин. Наиболее важным зоостерином является холестерин (холе—греч. желчь)
открытый Конради еще в 1775 г.
Холестерин содержится почти во всех клетках и жидкостях организма животных. Особенно много его в клетках центральной и периферической нервной системы, в кожном сале и в почках. Повидимому, он участвует в регуляции процессов диффузии и осмоса в клетке и служит источником образования других стероидных соединений в организме.
При атеросклерозе содержание холестерина в организме часто значительно повышается.
В небольших количествах холестерин выделяется с желчью. При некоторых заболеваниях в желчном пузыре и в желчных путях могут образоваться желчные камни, состоящие главным образом (иногда на 90%) из холестерина.
Холестерин содержится во многих пищевых продуктах, в частности—в яичном желтке и в молоке. Организм животных и человека снабжается холестерином не только за счет усвоения его из продуктов питания, но он и сам способен синтезировать холестерин из небольших органических молекул.
Это подтверждается, в частности, выделением радиоактивного холестерина из печени крыс после введения им уксусной кислоты, меченой радиоактивным углеродом.
Промежуточным продуктом этого синтеза, повидимому, является углеводород сквален.
Окислительным расщеплением продуктов биосинтеза доказано, что синтез холестерина в организме осуществляется по следующей схеме (жирными буквами С обозначен углерод метильной группы, а обыкновенным С—углерод карбоксильной группы ускусной кислоты):
Н2С
СН{СООН----- i
В промышленности холестерин получают, извлекая его органическими растворителями из костного мозга животных или из шерстяного жира (ланолина).
Для количественного и качественного определения холестерина и других стеринов имеется несколько чувствительных цветных реакций. При обработке холестерина уксусным ангидридом и концентрированной серной кислотой появляются быстро сменяющиеся характерные окраски (реакция Либермана). Окраска
появляется также при встряхивании раствора стерина в хлороформе с концентрированной серной кислотой (реакция Залковского) или при обработке раствора стерина в ледяной уксусной кислоте хлористым цинком и хлористым ацетилом (реакция Чугаева).
Для весового определения холестерина используют труднорастворимый комплекс (дигитонид), образуемый им со стероидным сапонином дигитонином (стр. 188).
Способность образовывать нерастворимые дигитониды является замечательной особенностью большинства 3^-оксистероидов и может быть использована для их отделения от стереоизомерных им За-эпимеров.
Изучение строения холестерина представляло сложную проблему, решение которой потребовало много усилий и большого экспериментального искусства.
Окончательно строение холестерина удалось установить лишь в 30-х годах нашего века, главным образом благодаря работам А. Вин-дауса и Г. Виланда (исследовавшего строение близких к стероидам желчных кислот).
Важнейшие данные, на основе которых было установлено строение холестерина, следующие:
1. Холестерин может быть превращен в углеводород копростан, который при окислении хромовым ангидридом, наряду с другими продуктами, образует два кетона: полициклический тестанон-17 и ациклический 2-метилгептанон-6:
Это показывает, что холестерин состоит из полициклического ядра и изооктильной боковой цепи в положении 17.
2. При дальнейшем окислении тестанона-17 расщепляется кольцо D с образованием этиобилиановой кислоты, содержащей две карбоксильные группы и при циклизации образующей ангидрид. По правилу, носящему название правила Блана, 1,4-и 1,5-дикарбоновые кислоты (типа адипиновой и пимелиновой) при циклизации образуют кетоны (циклопентаноны или циклогексаноны)
1
г/\сСЮН _ н2о
з1 ,СООН “с°2
О
кетон
в то время как 1,2- и 1,3-дикарбоновые кислоты (типа янтарной или глутаровой кислот) превращаются в ангидриды:
г/^СООН з'-СООН
-н2о
ангидрид
Поскольку этиобилиановая кислота образует ангидрид, она по правилу Блана не может представлять собой 1,4-дикарбоновую кислоту, а кольцо, несущее в холестерине боковую цепь (кольцо D), не может быть шестичленным. Следовательно, кольцо D пятичленное:
тестанон-17 этиобилиановая кислота
3. При окислении гидроксильной группы холестерина образуется кетон холестенон, в котором двойная связь находится в сопряжении с карбонильной группой. Так как гидроксил до окисления не носил характера аллильного гидроксила, то двойная связь холестерина не может находиться в положении С(4)—С<5). С другой стороны, она не может находиться дальше С(5)—С(б>, так как иначе она не могла бы мигрировать с образованием сопряженной системы:
Тем самым устанавливается положение двойной связи. Следует указать, что окисление ^-непредельных спиртов в а-непредельные кетоны является общей реакцией стероидов, играющей существенную роль в биологических процессах.
4. При нагревании холестерина с селеном до 360° образуется З'-метил-l ,2-циклопентенофенантрен (углеводород Дильса), а дегидрированием холестерина на палладированном угле при 500° получается углеводород хризен.
Эти превращения можно изобразить схемой:
Образование углеводорода Дильса объясняется обычным при дегидрировании отщеплением ангулярных метильных групп (стр. 106), а образование хризена—перегруппировкой с расширением пятичленного кольца, возможным в жестких условиях.
Эти данные показывают, что в основе скелета холестерина лежит ядро пергидроциклопентенофенантрена.
Результаты, полученные при дегидрировании холестерина, долгое время не признавались достоверным доказательством его строения, так как продукты дегидрирования удается выделить лишь с ничтожным выходом и трудно было решить, какой из них образовался без перегруппировки. Более -убедительные доказательства строения стероидного ядра были получены при окислительном расщеплении холестерина на многоосновные кислоты с более простым строением скелета. Окончательно этот вопрос был решен, когда Вудвордом (1951 г.) был осуществлен полный синтез холестанола по следующей схеме.
Продукт конденсации метокситолухинона (хиноны—см. стр. 351) с бутадиеном изомеризуется в щелочной среде из цис- в трсшоформу и восстанавливается литийалюминийгидридом в диол:
В результате омыления этого диола получается енольное соединение, которое изомеризуется обычным образом в диоксикетон, дегидратирующийся уже в процессе реакции с образованием непредельного оксикетона. Последний, при действии на него цинка в уксусной кислоте, восстанавливается в кетон. Этот кетон взаимодействием с муравьиным эфиром превращается в оксиметиленовое производное, дающее при конденсации с этилвинилкетоном дикетон, причем оксиметиленовая группа отщепляется:
Zn, сн3соон
СН3
СН3
Дикетон путем внутримолекулярной кротоновой конденсации превращается в трициклический кетон.
Окисление трициклического кетона четырехокисью осмия дает диоксикетон:
При этом окисляется .только одна двойная связь, так как остальные двойные связи, экранированные метильными группами, не подвергаются действию окислителя.
Диол представляет собой смесь цис- и /прсшс-форм и был разделен путем превращения ^wc-формы в ацетонид.
Ацетонид гидрировался водородом в присутствии палладия в неполярной-среде в положение 1,4:
Формилированием ацетонид превращается в оксиметиленовое производное, а для защиты оксиметиленовой группы гидроксил в ней замещается остатком метилани-лина:
Затем производится конденсация с акрилонитрилом, дающая после омыления антрильной группы и отщепления оксиметиленовой группы две изомерные кетокислоты. Одна из них переводом в лактон енола под влиянием уксусного ангидрида, действием йодистого метил магния и омылением превращается в производное ди кетон а:
которое путем внутримолекулярной кротоновой конденсации переводится в производное тетрациклического кетона:
При действии иодной кислоты оно окисляется с разрушением одного кольца
-и дает ди альдегид:
Внутримолекулярная конденсация диальдегида приводит к образованию пятичленного кольца D стероидного ядра
а в результате окисления оставшейся альдегидной группы и этерификации диазометаном получается метиловый эфир рацемической б/,/-3-кето-Д 4,9(11)1 -этио-холатриеновой кислоты. Восстановление эфира этой кислоты в смесь эфиров б/,/-3-а-и ^,/-33-оксикислот, выделение эфира d-Зр-оксикислоты с помощью дигитонинового комплекса (стр. 188) и окисление этого эфира привело к метиловому эфиру d-3-ке-то-Д4,9^1)’16 -этиохолатриеновой кислоты (темп, плавл. 188—190®; U]d= + 182+5°)
оказавшемуся идентичным соединению, получающемуся из природных стероидов.
Гидрирование этой кетокислоты дало смесь эфиров эпимерных кетокислот
из которой был выделен эфир 3-кетоэтиоаллохолановой кислоты. Затем эфир кетокислоты был восстановлен и омылен в окси кислоту, которая после ацетилирования оксигруппы была превращена в хлорангидрид.
Действие на хлорангидриз
диметилкадмия позволило превратить карбоксильную группу в ацетильную, а гриньяровская реакция с изогексилмагнийбромидом дала соответствующий спирт, дегидратация, гидриооначие и пиролиз которого привели к холестанолу*
Холестанол может быть получен гидрированием холестерина, а потому синтез Вудворда является окончательным доказательством строения последнего.
Из эфира 3-кетоэтиоаллохолановой кислоты могут быть получены также и некоторые стероидные гормоны, например андростерон, тестостерон и кортизон. Таким образом, рассмотренный синтез холестерина является одновременно и синтезом целого ряда других стероидов.
Эргостерин. Среди микостеринов наиболее хорошо изучен эргостерин (от ergot—франц, спорынья), найденный в дрожжах, плесневых грибках и в спорынье и являющийся отходом пенициллинового производства. Он применяется в производстве гормонов.
При облучении ультрафиолетовыми лучами эргостерин с размыканием кольца В переходит в витамин D2, недостаток которого в организме вызывает рахит и нарушение процессов образования костей и зубов.
Фотоизомеризация эргостерина в витамин D2 проходит через несколько промежуточных стадий:
витамин С2, или кальциферол
На схеме кальциферол изображен двумя способами, часто встречающимися в литературе. Формулы отвечают двум конформациям молекулы кальциферола, переходящим друг в друга за счет свободного вращения вокруг простой связи-между атомами С(б) и С(7) (здесь сохранена нумерация атомов углерода в ядре молекул стероидов). В настоящее время нет еще полной ясности, настолько ли велика энергетическая разница между этими конформациями, чтобы одна из них была преобладающей. Однако имеющиеся данные позволяют думать, что конформация А должна быть во всяком случае более напряженной вследствие стерического взаимодействия.
Близкие к витамину D2 соединения, обладающие таким же действием, образуются также при ультрафиолетовом облучении других стеринов, содержащихся в пищевых продуктах. Так, например, молоко под влиянием ультрафиолетовых лучей обогащается витамином D3 (витамином, находящимся в рыбьем жире).
Желчные кислоты
В близком родстве со стеринами находятся желчные кислоты, входящие в состав желчи. Важнейшая функция желчи состоит в ускорении усвоения жиров в кишечном тракте. В желчи содержатся не
свободные, а связанные с аминокислотами желчные кислоты (в виде натриевых солей). При омылении желчи выделяются свободные желчные кислоты, представляющие собой кристаллические бесцветные вещества*.
Почти все желчные кислоты являются производными не встречающейся в природе холановой кислоты:
Н
Желчные кислоты принадлежат к А/В-^ш?-ряду (стр. 166). Это—одноосновные кислоты, в большинстве случаев содержащие, кроме гидроксильной группы при С(3), еще одну-две гидроксильные группы в положениях 6, 7 или 12.
Интересная особенность желчных кислот состоит в их склонности к образованию комплексов с разнообразными органическими соединениями**. Благодаря этому в желчи растворяются многие нерастворимые в воде вещества, например каротин, жиры. Особенно важную роль это свойство желчных кислот играет в процессе усвоения жиров организмом. Предполагается, что жирные кислоты всасываются в виде комплексных соединений с желчными кислотами. Эти комплексы в стенках кишек распадаются, а освободившиеся желчные кислоты вновь вступают в реакцию с жирными кислотами, имеющимися в полости кишки.
Таким образом; относительно небольшого количества желчных кислот достаточно для всасывания больших количеств жирных кислот.
Холевая кислота. Важнейшая желчная кислота—холевая (За,7а, 12а-триоксихолановая кислота) находится в желчи ввиде амидов гликоколя (гликохолевая кислота} и таурина (таурохолевая кис* лота):
Радикал R
ОН Холевая
NHCH2COOH . . . Гликохолевая
NHCH2CH2SO3H . . Таурохолевая
* Золотисто-коричневая окраска желчи вызвана присутствием билирубина (стр. 514)
♦* Природа этих комплексных соединений еще не выяснена.
В желчи и в содержимом кишечника имеется также свободная холевая кислота, образующаяся омылением гликохолевой и тауро-холевой кислот под влиянием микроорганизмов.
Строение холевой кислоты установлено, в частности, путем превращения ее в те же продукты, какие получаются в результате химических превращений стеринов. Так, одним из продуктов окисления копростана (стр. 166) хромовым ангидридом является холановая кислота. Поскольку холановая кислота может быть получена дегидратацией и гидрированием холевой кислоты, холестерин и холевая кислота должны иметь один и тот же скелет. Строение холевой кислоты подтверждается также результатами дегидрирования. При нагревании с селеном при 360° холевая кислота образует углеводород Дильса и 5-метил-2',1'-нафта-П2-флуорен. В более жестких условиях (420°) происходит расширение пятичленного кольца D и образуются хризен и пицен:
углеводород Дильса
5-метил-2', l'-нафта-
1,2, флуорен
хризен
пицен
При дегидрировании 12-кетохолановой кислоты, синтезированной из холевой кислоты, образуется метилхолантрен (стр. 489), углеводород, оказывающий сильное канцерогенное действие:
12-кетохолановая кислота
дегидронорхолан
метилхолантрен
Предполагают, что аналогичные превращения желчных кислот и стеринов могут протекать в организме и, возможно, являются одной из причин раковых заболеваний.
Дезоксихолевая кислота. Вторая важнейшая желчная кислота— дезоксихолевая (За,12а-диоксихолановая) кислота
также содержится в желчи и в экскрементах животных.
Кроме холевой и дезоксихолевой кислот, в желчи человека, а также в желчи рогатого скота и других животных находятся еще За,7а-диоксихолановая (антроподезоксихолевая, хенодезоксихоле-вая*, или галлодезоксихолевая, кислота) и За-оксихолановая (лито-холевая** кислота).
В желчи многих животных содержатся, кроме того, особые, только им свойственные, желчные кислоты. Например, в желчи свиньи находятся а- и (3-гиодезоксихолевые (За,6(3- и 3[3,6а-диокси-холановые) кислоты, в желчи медведя найдена урсодезоксихолевая*** (3а,7(3-диоксихолановая) кислота.
Возможно, что желчные кислоты образуются в организме в результате окислительного расщепления холестерина. Так, при введении в организм животных холестерина, меченого дейтерием, была выделена холевая кислота, содержащая дейтерий.
Сердечные глюкозиды
Во многих растениях содержатся растворимые в воде весьма ядовитые глюкозиды, в очень малых дозах стимулирующие сердечную деятельность. При введении их в несколько больших дозах сердце останавливается.
Сердечные глюкозиды применялись народами Африки и Малайского полуострова для отравления стрел. В настоящее время они используются в'терапии сердечных, а также некоторых других заболеваний.
При гидролизе под действием кислот эти глюкозиды распадаются с выделением одного или нескольких сахаров и аглюкона— стероида.
Свободные аглюконы представляют собой сильные яды судорожного действия и в медицине не применяются.
* Антропос—греч. человек, хено—гусь.
** Эта кислота найдена также в желчных камнях {литое—-греч. камень).
*** Г ио—греч. свиноя, урсус—лат. медведь.
Аглюконы сердечных глюкозидов (генины) имеют рлактонное кольцо в положении 17 и (за исключением аглюконов морского лука) построены по общей формуле:
где Х^Н или ОН.
Все они содержат гидроксильные группы при и С(м). Во многих аглюконах имеются также гидроксил при С(5) и кислородсодержащий заместитель при С(ю).
Характерной особенностью этого класса стероидов является то, что у них кольца С и D сочленены в /^-положении, в отличие от большинства других стероидов. Так же сочленены и кольца А и В.
Поскольку стероидные аглюконы являются лактонами оксикислот типа
СООН
СН2ОН
ОН
их можно рассматривать как продукты окисления стеринов. Характеристика важнейших аглюконов дана в табл. 11.
Наиболее хорошо изученным аглюконом является строфантидин.
При действии щелочи строфантидин необратимо изомеризуется ь ивострофантидин'.
• грохавгидрн
изострофа нт идин
В пиридиновом растворе в присутствии нитропруссида натрия строфантидин дает со щелочью характерную темнокрасную окраску (реакция Легаля).
Таблица 11
Аглюконы сердечных глюкозидов (генины)
Название Положение ОН-групп (кроме 14?) Температура плавления °C Wp Источник выделения
Дигитоксигенин (нерийфолигенин, или теветигенин) сн3 33 250 + 18= Digitalis purpurea L. (красная наперстянка)
Дигоксигенин СН3 33,123 222 +18= Digitalis lanata L.
Гитоксигенин сн3 33,163 235 +34° Digitalis purpurea L.
Периплогенин сн3 3(3,53 185 +31,5° Periploca graeca B.
Сарментогенин СН3 33,11а 270 +21,5* Strophantus sarmentosus S.
Строфа нтидин (конваллатоксигенин) сно .33,53 235 +41° Strophantus kotnbe S., кендырь конопляный, Convallaria ma] al is (ландыш), Cleiranthu^ cheiri (желтофиоль)
Адонитоксигенин сно 33 178 Adonis vemails
* См. формулу на стр. 183.
При озонировании лактонное расщепляется, и в положении 17 цепь:
кольцо стероидных аглюконов остается оксикетонная боковая
Используя эту реакцию, можно превращать аглюконы в стероид»
ные гормоны коры надпочечников.
Яды жаб и аглюконы морского лука
Кожные железы жаб содержат сильные яды. Вытяжка из кожи китайской жабы, под названием чан-су, издавна применяется в китайской медицине при зубной боли, при воспалительных заболеваниях мозга и в качестве кровеостанавливающего средства. На сердце млекопитающих яды жаб действуют примерно так же, как сердечные глюкозиды. Это действие оказывают всегда присутствующие в них стероидные буфогенины*, содержащие в положении 17 непредельное 6-лактонное кольцо. Такого же типа соединения присутствуют (в ви-
Bufo—лат. жаба.
де глюкозидов) в морском луке (Scilla maritime?), применявшемся в качестве лекарства еще древними египтянами и римлянами, и в некоторых других растениях.
Буфогенины построены по общей формуле
причем, кроме гидроксилов в положениях 3 и 14, могут иметься и другие гидроксилы. Сочленения колец А—В и С—D у всех буфотенинов и близких к ним соединений осуществлены так же, как и у сердечных глюкозидов, рассмотренных выше.
Как видно из формулы, буфогенины представляют собой производные альдегидокислот типа
ОН
т. е., подобно аглюконам сердечных глюкозидов, могут рассматри-
ваться как продукты окисления стеринов.
Важнейшими из буфотенинов являются буфалин и буфоталин, Буфалин выделен из кожи китайской жабы (из чан-су). Он содержит два гидроксила в положениях 3^ и 14(3.
Буфоталин, содержащийся в выделениях кожных желез европейской жабы (в небольших количествах и у китайской жабы), имеет три гидроксильные группы—3[3, 14р, 16(3, причем ^-гидр-
оксил ацетилирован.
Из глюкозидов морского лука выделен сцилларен А, в котором генин соединен с остатком рамнозы с помощью За-гидроксила.
При омылении сцилларена А отщепляется вода и получается
аглюкон сцилларидин
Стероидные сапонины
Во многих растениях содержатся глюкозиды, которые образуют при взбалтывании с водой очень устойчивую пену,—так называемые сапонины*.
Сапонины очень ядовиты для рыб. Для теплокровных же, в том числе для человека, они ядовиты только при введении в кровь, а попадание их в желудочно-кишечный тракт практически безопасно. Поэтому рыба, убитая или парализованная сапонинами, остается съедобной для человека.
Это свойство сапонинов использовалось в тропических странах для ловли рыбы.
При кислотном гидролизе сапонины распадаются на одну или несколько молекул углеводов и на аглюконы—сапогенины, стероидное ядро которых соединено с циклической спирановой кетальной группировкой.
В зависимости от ее пространственной ориентации, сапогенины делят на соединения «нормального» ряда
и соединения «изо» ряда:
Сочленение колец у сапсгенинов в точности такое же, как и у стеринов.
При действии- хлористого водорода в спиртовом растворе сапогенины., нормального ряда переходят в изосапогенины.
* Sapo—лат. мыло.
При обработке уксусным ангидридом при 200° сапогенины как нормального, так и изо-ряда дают один и тот же псевдосапогенинг.
При окислении псевдосапсгенина хромовым ангидридом получаются сложные эфиры, которые омылением и последующей дегидратацией и гидрированием образующихся кетонов могут быть превращены в соединения ряда 20-кетопрегнана, родственные прогестерону (см. стр. 191):
дегидратация
Эту цепь превращений используют для промышленного получения стероидных гормонов из сапогенинов.
Большое значение для анализа и исследования стероидов имеет сапонин наперстянки—дигитонин, аглюконом которого является стероидный сапогенин изо-ряда—дигшпогенин'.
СИ,
н
Дигитонин обладает способностью образовывать избирательно нерастворимые комплексы с 3₽-оксистероидами (стр. 172), преимущественно с их d-формами.
Стероидные гормоны
Железами внутренней секреции вырабатываются специфические органические вещества, называемые гормонами (греческий корень слова «гормон» означает возбуждать, приводить в движение), которые являются регуляторами важнейших жизненных процессов и необходимы для нормальной деятельности организма.
По своей химической природе гормоны могут быть разделены на две группы: в одну группу входят аминокислоты и родственные им соединения (адреналин, тироксин), полипептиды и белки (инсулин, гормоны передней доли гипофиза). Вторую группу гормонов составляют стероиды, выделяемые половыми железами и корой надпочечников. Стероидные гормоны регулируют процессы обмена веществ, роста, созревания, старения и размножения, влияют на работоспособность и сопротивляемость организма. От их биологического действия зависят такие разнородные явления, как рост и выпадение волос, крепость мышц у мужчин, развитие молочных желез у женщин, половая окраска рыб и птиц и др.
В зависимости от биологического действия стероидные гормоны подразделяют на четыре группы:
1) андрогенные, или тестоидные, гормоны (по-гречески андрос-— мужчина, по латыни testis—семенники);
2) гестагенные, или лутоидные, гормоны (от gestatio—беременность, corpus luteum—желтое тело);
3) кортикостероиды (кортикула—кора надпочечников);
4) эстрогенные, или фолликулоидные, гормоны (эструс—течка, фолликулы—шаровидные скопления в яичниках).
Андрогены. Андрогенными гормонами называют вещества, способные восстанавливать вторичные половые признаки у кастрированных животных.
Гормон, образуемый семенниками, носит название тестостерон. Его выделение представляло значительные трудности, так как из 100 кг семенников удается выделить путем экстракции органическими растворителями всего 10 мг тестостерона. Это наиболее активный из всех известных андрогенов.
Тестостерон (темп, плавл. 155°; [а]г>= + Ю9°) представляет собой Д4-андростенол-17₽-он-3:
Синтетически тестостерон получают окислением холестерина хромовым ангидридом. Чтобы при окислении не разрушилось стероидное ядро, холестерин перед окислением ацетилируют, а двойную связь защищают путем присоединения брома.
После окисления бром отщепляют с помощью цинка, омы-ляют ацетоксильную группу и получают дегидроэпиандростерон (А5-андростенол-3^-он-17):
Несмотря “на низкий выход (5—10%), мировое производство дегидроэпиандростерона этим методом составило в 1950 г. около 1000 кг.
Дегидроэпиандростерон сначала восстанавливают в Д5-андростен-диол-3^,17р, а затем превращают этерификацией и неполным омылением в 17-моноацетат, который при окислении дает ацетат тестостерона.
Этот метод получения тестостерона из дегидроэпиандростерона основан на двух 'Стереохимических закономерностях.
Первая из них заключается в том, что восстановление 17 кетостероидов, вне зависимости от природы восстанавливающего агента, приводит главным образом к 17р-оксисоединениям.
Вторая закономерность состоит в меньшей реакционной способности 17Р-гидр-•эксила по сравнению с гидроксилом при С\з>, благодаря чему возможно частичное омыление диацетата А5-андростендиола-33 J73 в 17-моноацетат.
По другому, более совершенному методу дегидроэпиандросте* рон окисляют сначала в Д4-андростендион-3,17
дегидроэпиандростерон
Д4-андростенд1 он-3,17
который затем восстанавливают до диола с помощью литийалюминий-гидрида.
Образующуюся при этом смесь стереоизомерных андростендиолов окисляют двуокисью марганца, используя ее замечательную осо-
бенность—способность избирательно окислять аллиловые спирты, не затрагивая других гидроксильных групп:
но/М'Х/
тестостерон
При действии на* дегидроэпиандростерон магнгийиодметила с последующим окислением продукта реакции образуется V7-метил-тестостерон
Н3С |
о/х/х/ широко применяемый в медицине, так как, в отличие от тестостерона, активного только при подкожном введении, он высокоактивен также и при приеме внутрь.
Андрогенные вещества содержатся не только в семенниках, но и в моче и в надпочечниках. Так, из мочи выделены дегидроэпиандростерон и андростерон. Наряду с ними в моче содержатся также и продукты их восстановления—эпиандростерон и тестанол-За-он-\7 (не обладающий андрогенной активностью).
андростерон эпиандростерон тестанол-За-он-17
(темп, плавл. 183°; (темп, плавл. 174,5°; (темп, плавл. 131°;
[a]D = + 94.5°) [«]D = +88°) [«]D = -105°)
По международному соглашению андростерон служит стандартом для сравне-ния андрогенной активности различных препаратов: единицей андрогенной активности считают'такую активность, какую проявляют 100 \ьг андростерона.
Аналитическое определение андрогенов и близких к ним соединений (17-ке-тостероидов) в моче имеет значение в клинической практике (в диагностике), так как количество 17-кетосте^оидов в моче сильно возрастает или уменьшается при
различных заболеваниях. Количество 17-кетостероидов у здоровых людей также колеблется в зависимости от различных факторов, в частности—от усталости. Повышение содержания 17-кетостероидов в моче является настолько чувствительной реакцией организма на усталость, что может служить ее объективной мерой.
Андрогенное действие сильно зависит от химического строения вещества. Так, например, уже при переходе от тестостерона к его 17а-изомеру андрогенная активность снижается в 25 раз.
Лутоидные гормоны. В яичнике периодически увеличивается и атрофируется железа внутренней секреции, называемая желтым телом. В случае оплодотворения желтое тело не атрофируется, а разрастается и сохраняется на протяжении большей части беременности. Желтое тело вырабатывает гормон, необходимый для сохранения беременности. Этот гормон носит название прогестерон.
Если беременную крольчиху кастрировать, то ее организм больше не получает прогестерона и происходит выкидыш. Если же после кастрации ввести ей прогестерон, то плод развивается нормально.
Прогестерон (Л4-прегнендион-3.20)
сн3
прогестерон
(темп, плавл. 128°; [а]^ = +192°)
синтезируют из стеринов, имеющих в боковой цепи двойную связь в положении 22—23 (из ситостерина или эргостерина), или из стероидных сапогенинов.
В организме прогестерон восстанавливается в неактивный прег-нандиол-3а,20а, который выделяется с мочой.
В медицине применяется этинилтестостерон
Н3С |
который при приеме внутрь более активен, чем прогестерон. Для получения этинил-тестостерона дегидроэпиандростерон конденсируют с ацетиленом, а затем окисляют гидроксильную группу при С(3)«
Кортикостероиды. Кора надпочечников* выделяет большое число стероидных соединений, которые по биологическому действию можно разделить на минералокортикостероиды (регулирующие обмен электролитов и воды) и глюкокортикостероиды (регулирующие углеводный и белковый обмен и влияющие на работоспособность мышц).
При удалении надпочечников организм теряет способность удерживать ионы натрия и хлора; содержание хлористого натрия в организме резко уменьшается. Одновременно сильно увеличивается содержание калия в крови и через некоторое время наступает смерть. При введении минералокортикостероидов восстанавливается нормальное содержание ионов натрия и хлора в моче и ионов калия в крови, и животные не погибают.
Важнейшим минералокортпкостероидом является дезоксикортикостерон
СН,ОН
Н3С f°
Н3С | |
O/V'XZ
Дезоксикортикостерон может быть извлечен из надпочечников крупного рогатого скота. Практически, однако, приходится получать его синтетически, так как содержание его в надпочечниках исчезающе мало (25 рг в надпочечниках одного животного). Дезоксикортикостерон синтезируют, исходя из холестерина, который окисляется хромовым ангидридом в Зр-окси-Д5-этиеновую кислоту. Зр-Ацетат этой кислоты через хлорангидрид и диазокетон (см. том I, стр. 662) превращается в ацетат Д5-прегнендиола-Зр,21-она-20, окисляемый
затем в ацетат дезоксикортикостерона:
* Надпочечники представляют собой парные железы, состоящие из двух .различных тканей: коркового слоя и мозгового вещества. О гормоне мозгового .вещества надпочечников—адреналине—см. стр. 342.
Из глюкокортикостероидов наибольшее значение имеют кортикостерон, кортизон и гидрокортизон (17-оксикортикостерон):
кортикостерон
(темп, плавл. 182°;
[a]D= +223°)
кортизон (темп, плавл. 180;
= 4-299°)
гидрокортизон (темп, плавл. 220°;
= 4-167°)
Введение этих веществ животным с удаленными надпочечниками вызывает нормализацию углеводного обмена (т. е. повышает содержание гликогена в печени и сахаров в крови), в результате чего восстанавливается работоспособность мышц, резко понижающаяся при нарушении углеводного обмена.
Кортикостероиды применяются в медицине при заболеваниях надпочечников. Кортизон и гидрокортизон приобрели большое значение для лечения тяжелых ревматических заболеваний, воспалительных процессов, бронхиальной астмы и ряда других болезней.
Поэтому в последние годы было затрачено много усилий на осуществление синтеза кортизона из других стероидов, не имеющих кислородсодержащего заместителя в положении 11. Эту задачу удалось решить, однако все способы введения химическим путем кислорода в положение 11 трудоемки и с их помощью кортизон получается лишь с небольшим выходом. Практически для получения кортизона используют другой путь, основанный на окислении стероидов под влиянием микроорганизмов. Так, грибы Rhisopus nigricans избирательно окисляют прогестерон в положении 11 (с выходом 45%):
СН3
прогестерон
Поскольку lla-оксистероиды биологически мало активны, их приходится окислять хромовым ангидридом в 11-кетосоединения, восстановление которых (с помощью LiAlH4 или NaBH4) приводит к 11[3-оксистероидам.
Полный синтез кортизона был осуществлен, исходя из ацетонида тетрациклического кетона
полученного в качестве промежуточного продукта при полном синтезе холестерина (стр. 176).
Присоединением бромноватистой кислоты по двойной связи С(9)—окислением бромгидрина в бромкетон и заменой брома на водород был получен ацетонид ди кетона:
Окислением ацетонида иодной кислотой в диальдегид, подобно тому, как это делалось при синтезе холестерина (см. стр. 177), и циклизацией диальдегида действием уксуснокислого пиперидина получен непредельный ди кетоальдегид, имеющий уже стероидную циклическую систему:
Это соединение окисляли сначала перекисью водорода в окись по Двойной связи в пятичленном кольце, затем окисью серебра в щелочном растворе для превращения альдегидной группы в карбоксильную, после чего кислота
переводилась в хлорангидрид и, далее, при действии диазометана—в диазокетон» диазогруппа которого при кипячении с уксусной кислотой замещается на ацет-оксильную группу:
Расщепление окисного кольца под действием бромистого водорода и удаление брома восстановлением с никелем привели к ацетату кортизона:
СН2ООССН3
СО
СН2ООССН3
НВг
Кроме перечисленных соединений, кора надпочечников образует еще около двадцати других кортикостероидов, выделение и изучение которых осуществлено главным образом швейцарским химиком Рейхштейном.
Интересным кортикостероидом является альдостерон (электрокортин}
СН2ОН
отличающийся от кортикостерона наличием альдегидной группы вместо ангулярной метильной группы при С(13). Альдостерон активен не только как минерал окортико-стероид (в 100 раз активнее дезоксикортикостерона), но и как глюкокортикостероид (в 3 раза слабее кортизона).
Эстрогены. Яичники самок выделяют эстрогенные гормоны, вызывающие менструальные кровотечения или течку (эструс). Эстрогены образуются также и другими железами, в частности семенниками и надпочечниками, и выделяются организмом с мочой.
Введение таких эстрогенных веществ кастрированным самкам приводит к восстановлению функций, нарушаемых при кастрации, и может вызвать течку. На этом явлении основан метод определения эстрогенной активности.
Природные эстрогены отличаются от других стероидных гормонов наличием одного или двух ароматических ядер. Основными эстрогенами, образующимися в организме человека, являются эстрон, эстрадиол и эстриол.
В организме лошадей и других животных вида Equus, кроме того, образуется эквиленин—стероид, содержащий ядро нафталина.
Особенно много эстрогенов содержится в моче жеребых кобыл и в моче жеребцов (до 25 000 рг эстрона в 1 л), которая служит источником промышленного получения эстрона. Эстрон и эстриол встречаются также в растительном мире и были выделены из цветков ивы и из кокосовых орехов.
Строение эстрона (темп, плавл. 260°; ;а]о=+165°)
доказано восстановлением его карбонильной группы и дегидрированием продукта восстановления над селеном в 7-метокси-1,2-цикло-пентенофенантрен:
метиловый эфир эстрона
Строение эстрадиола (темп, плавл. 178°; ]<x]d= +81°)
доказано его получением путем восстановления эстрона. Образующийся при этом в качестве побочного продукта 17а-эстрадиол примерно в 40 раз менее активен, чем природный 17₽-эпимер.
Строение эстриола определяется, в частности/ образованием эстрона при его дегидратации:
Д)Н
эстриол (темп, плавл. 280е; [а]^ «= -ЬбГ)
Синтез эквиленина (темп, плавл. 259°; !a]z>= +87)
н,с ?
но
был осуществлен Бахманом в 1938 г. и явился первым полным синтезом стероидного гормона.
Большие трудности представил синтез эстрона, так как этот гормон содержит четыре асимметрических углеродных атома, и, следовательно, для него возможно существование шестнадцати стереоизомеров. Полный синтез этого гормона был впервые осуществлен только в 1948 г. (Мишер).
Андрогенное, лутоидное и кортикоидное действие проявляют только соединения со стероидным скелетом, причем даже незначительные изменения в структуре или конфигурации активных гормонов приводят большей частью к полному исчезновению или значительному снижению активности. Наоборот, в ряду эстрогенов обнаружено большое количество нестероидных веществ, обладающих гормональным действием. Наиболее важными из нестероидных эстрогенов являются стильбэстрол и гексэстрол, или синэстрол, стр. 433.
Ароматический ряд
Ароматические соединения
Под названием ароматических соединений объединяются все соединения, в молекуле которых содержится особая группировка из шести атомов углерода. Эта группировка в простейшем виде имеется в углеводороде С6Н6, носящем название бензола, а потому ароматический ряд называют также рядом производных бензола.
Название ряда «ароматический» произошло от того, что среди первых известных представителей этого ряда имелось большое число душистых (ароматических) веществ. В настоящее время это название потеряло прежний смысл, так как среди органических веществ других классов имеется не меньше душистых веществ, чем в ароматическом ряду, громадное же большинство производных бензола не обладает приятным запахом или даже вовсе не имеет запаха.
После того как было установлено, что многие вещества содержат «бензольное ядро», т. е. являются производными бензола, возникла необходимость выяснить его строение. Первую формулу строения бензола предложил Кекуле* в 1865 г. Принятие этой формулы имело громадное значение как для исследования многочисленных и важных ароматических соединений, так и для развития всей органической химии.
Согласно представлениям Кекуле, в бензоле имеется цикл из шести атомов углерода, соединенных между собой поочередно простыми и двойными связями; четвертая валентность каждого углеродного атома бензола затрачена на связь с атомом водорода:
Н
С
Н—с с—н
II I
Н—С С—н
\/z с I _________ н
* Август Кекуле (1829—1896); крупнейший немецкий теоретик-органик прошлого столетия. В 1857—1858 гг. сформулировал учение об атомности (валентности) элементов, в том числе о четырехвалентности углерода и способности его образовывать цепеобразные соединения. В 1865 г. предложил для бензола шестичлен-ную циклическую структуру и высказал гипотезу об осцилляции связей в бензольном цикле.
Приняв формулу бензола Кекуле, химики впервые признали возможность существования циклических соединений. Поэтому бензол можно было бы рассматривать как частный случай алициклического углеводорода.
По номенклатуре, принятой для алициклических углеводородов, бензол, если ему придавать формулу Кекуле, следовало бы называть циклогексатриеном-1,3,5 (стр. 20 и том I, стр. 123). Однако бензол и его производные обнаруживают много особых характерных химических свойств, отличных от свойств других соединений с двойными связями, как жирных, так и циклических. Эти особенности («ароматический характер»), а отчасти и важность ряда производных бензола, включающего гораздо больше изученных отдельных представителей, чем любой другой класс органических соединений, служат причиной выделения этих соединений в особый ряд органических веществ—ароматический ряд.
При дальнейшем рассмотрении свойств соединений ароматического ряда будут особо отмечены те их особенности, которые вызвали сомнения в правильности формулы Кекуле.
Изомерия производных бензола
Еще до установления для бензола формулы Кекуле было известно, что у однозамещенных производных бензола не существует изомеров*, а двузамещенные производные существуют в трех изомерных формах.
Из первого положения следует, что все шесть атомов водорода в молекуле бензола равноценны в том отношении, что при замещении любого из них получаются молекулы совершенно тождественного строения. Сочетание обоих положений возможно только в том случае, если атом водорода имеется при каждом атоме углерода, т. е. если в бензоле связаны между собой шесть групп СН и строение его можно выразить формулой (СН)6.
Из возможных формул циклического строения молекулы (СН)Ь формула Кекуле является одной из удовлетворяющих первому положению. Второму положению она может удовлетворять, лишь если допустить, что на изомерию влияет только относительное положение замещающих групп, а не расположение двойных связей. В самом деле, если отвлечься от расположения простых и двойных связей, то здесь будут возможны три различных относительных положения двух заместителей, а именно положения 1,2; 1,3 и 1,4:
X X X
I п X
in
Это положение правильно при отсутствии изомерии радикала-заместителя.
Очевидно, что положение 1,6 тождественно положению 1,2, положение 1,5 тождественно положению 1,3, а для положения 1,4 нет второго тождественного положения.
Относительное положение 1,2 называют орто-положением, 1,3—ж/тш-положением и 1,4—иоро-положением, а соответствующие изомеры—орто-, мета- и пара-изомерами. Очень часто применяется сокращенное обозначение изомеров русскими буквами о-, ж- и п- или латинскими буквами о-, т- и о-.
Согласно формуле Кекуле, следовало бы ожидать д: vx орто-изомеров (1а и 16), различающихся лишь положением двойной связи, а если два атома водорода в бензоле замещены двумя различными заместителями, то также и двух мета-изомеров (Па и Пб):
XX XX
1а 16 Па Пб
На самом же деле ни в одном случае подобные изомеры не могли быть получены. Для объяснения этого противоречия Кекуле предположил, что двойные связи в бензольном ядре не остаются неподвижными, но постоянно перемещаются таким образом, что формулы 1а и 16, а также Па и Пб представляют лишь две фазы состояния молекулы одного и того же вещества (осцилляционная гипотеза).
Если не принимать во внимание чередование простых и двойных связей согласно формуле Кекуле, то при замещении атома водорода тремя одинаковыми атомами или радикалами возможны три изомерные формы: 1,2,3—рядовой изомер (сокращенно v- или ряд.)} 1,2,4—несимметрический изомер (as- или нес.)} 1,3,5—симметрический изомер (s- или симм.)*'.
X X X
1,2,3 1,2,4 1,3,5
При замещении четырех атомов водорода также возможны три изомера: 1,2,3,4—рядовой} 1,2,3,5—несимметрический} 1,2,4,5—симметрический* **'.
— начальная буква латинского прилагательного vicinal—расположенный по соседству, рядом; as—asymmetricus; s—symmetricus
** Названия «симметрический» и «асимметрический» в сущности ус ловны, так как, например, молекула 1,2,3-изомера имеет ось симметрии, проходящую через углеродные атомы 2 и 5, а изомеры четырехзамещенного бензола все имеют оси симметрии: 1,2,3,4-изомер—ось, пересекающую средину отрезка 2—3; 1,2,3,5-изомер—ось, проходящую через углеродные атомы J2 и 5; 1,2,4,5-изомер— ось, пересекающую средины отрезков 1—2 и 4—5.
X
1,2,3,4
При замещении пяти или шести атомов водорода одинаковыми заместителями изомерия невозможна.
Число действительно наблюдаемых изомеров, а также их взаимные отношения вполне отвечают этим выводам*.
Равноценность водородных атомов в бензоле и существование двух пар атомов водорода, занимающих тождественные положения по отношению к любому углеродному атому молекулы бензола, были доказаны несколькими исследователями рядом прямых опытов. Приводим здесь некоторые из этих доказательств.
1. Наличие в молекуле бензола четырех равноценных атомов водорода было доказано Ладенбургом следующим образом.
Если в обыкновенном феноле С6НбОН гидроксил действием трехбромистого фосфора заместить на бром, то получается бромбензол СбН5Вг, который действием натрия и двуокиси углерода может быть превращен в соль бензойной кислоты СбНб—СООН. Ясно, что во всех этих соединениях заместители ОН, Вг и СООН занимают одно и то же положение, которое можно обозначить цифрой 1.
Та же бензойная кислота С6Н5—СООН может быть получена из любой из трех существующих оксибензойных кислот С6Н4(ОН)СООН заменой в них гидроксила на бром и восстановлением образовавшихся бромбензойных кислот CfiH4BrCOOH. Из этого следует, что во всех трех оксибензойных кислотах карбоксил занимает место, тождественное месту гидроксила в обыкновенном феноле (положение 1), а следовательно, гидроксилы в каждой из трех оксибензойных кислот занимают другие, различные между собой места (положения 2, 3 или 4). Но так как-все три оксибензой-ные кислоты при нагревании теряют СО2 и дают обыкновенный фенол, значит, положения 2, 3, 4 равноценны положению 1 и равноценны между собой.
2. Прямыми опытами доказано также, что по отношению к положению 1 имеются две пары симметрично расположенных атомов водорода.
О существовании одной такой пары свидетельствуют опыты Гюбнера и Петер-мана (см. схему 1). Эти исследователи подвергли нитрованию одну из упомянутых трех бромбензойных кислот, получаемых бромированием бензойной кис-
* Интересны подсчеты числа изомеров положения (сравн. с изомерией положения цикланов, стр. 27) для бензола (Эванс, Ле-Кен). Если для простоты опустить в формуле бензола атомы углерода, а водородные атомы, способные замещаться на группы В, С, D..., обозначить буквой А, то символ А6 будет равносилен формуле бензола С6Нб, символ А5В будет обозначать однозамещенный бензол С6НбХ (например, хлорбензол, нитробензол, толуол и т. д.). Число изомеров будет:
для
А4ВС........3
A3BCD .... 10
A2BCDE ... 30
ABCDEF ... 60
для
А4В2..........3
А3В2С.........6
A2B2CD .... 16
AB2CDE .... 30
ДЛЯ
А3В3..........3
А2В2С2 .... 11
AB2C2D .... 16
С помощью этой таблички легко определить, что, например, для тринитротолуола C6H2(CH3)(NO2)3, который можно обозначить символом А2ВС3, эквивалентным символу А3В2С, возможно существование шести изомеров.
лоты,—л-бромбензойную кислоту (I). Из полученных двух различных нитробромбензойных кислот (II и III) при восстановлении, когда бром заменяется на водород, а нитрогруппа переходит в аминогруппу, образовалась одна и та же о-амино-бензойная кислота.
Схема 1
СООН
II
СООН
о-аминобензойная кислота
СООН
СООН
СООН
III
о-аминобензойная кислота
Следовательно, положения 2 и 6 симметричны по отношению к положению 1.
Существование второй пары атомов водорода, расположенных симметрично по отношению к атому водорода в положении 1, доказано следующим образом (см. схему 2). Если n-толуидин (I) бромировать и затем из полученного ж-бром-/г-толуидина (II) диазореакцией (стр. 302 и 321) удалить аминогруппу, то получается /4-бромтолуол (Ш), при обмене брома на аминогруппу дающий ж-толуидин, в котором аминогруппа находится в ином положении, чем в о-аминобензойной кислоте (см. схему 1), а именно в положении 3.
Нитрованием того же лгбром-п-толуидина (II) получается соединение (IV). После замены в нем диазореакцией аминогруппы на водород (соединение V) и восстановления, при котором бром заменяется на водород, a NO2 переходит в NH2, также получается л/-толуидин.
Схема 2
сн8 I
сн3
'V'\nh2
льтолуидин
C^N/V^Br
2 I
nh2
IV
сн3
п
О2мС^/\Вг
сн3 I п
м-толуидин
Отсюда ясно, что положения 3 и 5 симметричны по отношению к положению 1.
Таким образом доказано, что в молекуле бензола четыре атома водорода (в положениях 1, 2, 3 и 4) идентичны друг другу и, кроме того, что две пары идентичных между собой атомов водорода расположены симметрично относительно положения 1. Водородные атомы 5 и 6, хотя и не входят в число идентичных атомов 1,
2, 3 и 4, но атом водорода в положении 5 равноценен водородному атому в положении 3, а водородный атом в положении 6—атому водорода в положении 2. Отвода с неизбежностью вытекает, что все шесть атомов водорода в молекуле бензола равноценны.
Синтезы ароматических соединений из соединений жирного ряда
В настоящее время известно чрезвычайно большое число синтезов ароматических соединений из соединений с незамкнутыми цепями углеродных атомов, причем иногда сразу получаются ароматические соединения, иногда же сначала образуются алициклические соединения, которые уже затем более или менее сложным путем превращаются в производные бензола. Все эти синтезы без исключения могут быть превосходно объяснены на основе формулы Кекуле. Ниже приведено несколько примеров простейших синтезов этого рода.
1. Полимеризация ацетиленовых углеводородов. Ацетилен легко полимеризуется в бензол, если его пропускать над активированным углем при 650° (Н. Д. Зелинский, 5. А. Казанский), а при более высоких температурах—и без катализатора (Вертело). Как на катализаторе, так и без него наряду с бензолом получаются и другие ароматические углеводороды (стирол, нафталин, антрацен и др.). Реакции образования бензола из ацетилена и мезитилепа из аллилена—см. том I, стр. 341.
2. Конденсация кетонов под действием серной кислоты. Например, из ацетона образуется мезити-лен (1,3,5-триметилбензол):
СН3
I
СО
Н3с/ сн3
+ I
СП3—ОС со—сн3
\снз
СН3 с
/ Ч
НС сн
II I + ЗНгО сн3—с с—сп3
\// сн
3. Конденсация малонового эфира. При нагревании до 150°
латриймалонового эфира с избытком малонового эфира происходит конденсация
малонового эфира, приводящая к получению флороглюцинтрикарбонового эфира:
СО—ОС2Н5
CJI5OOC— ЩС7 СН2—СООС2Н5
+ I
С2Н5О—ОС СО—ОС2Н5
^сн,
СООС2Н5
С2Н5ООС— НС7
-ЗС2Ы5ОН 2 3 * 5 I
со
\н—СООС2Н5 I
со
I СООС2Н5
Это соединение в своей другой таутомерной форме
ОН
у \
С2Н6ООС—С С—СООС2Н6
I ,1
I ю—с с—он
СООС3Н8
при омылении дает с отщеплением трех молекул СО2 флороглюцин:
ОН
НС сн но—(i с—он
\ / сн
4. Конденсация окиси углерода. Интересно образование фенолята гексаоксибензола из окиси углерода и металлического калия:
ОК I С \ КО—С С—ок 6СО4-6К —> |
ко—С С-ОК
ок
5. Конденсация некоторых альдегидо- и кетон окисло г. Конденсацией формилуксусного эфира (см. том I, стр. 516) получается эфир тримезиновой кислоты
СООН
ноос/ХХ\соон
а конденсацией пировиноградной кислоты (см. том I, стр. 517)—увшпинсвая кислота:
сн3
Ноос/Х/Хсоон
Реакции расщепления молекул ароматических соединений
Реакции расщепления бензольного ядра в ароматических соединениях также всегда приводят к продуктам, строение которых находите/, в согласии с формулой Кекуле:
1. Окисление гидрохинона. Гидрохинон, или л-диоксибензол (стр. 337 сл.), при окислении окисью серебра дает малеиновую кислоту и двуокись углерода:
ОН I нс Лен
II < I НС<^СН с4
I он
нс-соон
II
нс-соон
4- 2СО2 + Н?О
2. Окисление бензола кислородом возду-х а. Малеиновая кислота получается также окислением бензола в паровой фазе кислородом воздуха при высоких температурах в
присутствии окислов ванадия.
3. Озонирование бензола. При озонировании получается триозонид, распадающийся с образованием глиоксаля:
СН НС^СН И I нсх^сн сн
сно з I
сно
30
4. Восстановление салициловой кисло-т ы. Восстановлением салициловой, или о-оксибензойной,- кислоты (стр. 382) натрием в амиловоспиртовом растворе получается пиме-линовая кислота:
СН
\ СН2—СН2—СООН
НС С—СООН I
I --11. + 4Н + Н2О СН2
НС С—ОН I
\ / СН2-СН2-СОЭН
СН
Взаимные переходы ароматических и гидроароматических соединений
Ароматические соединения могут более или менее легко присоединять водород (стр. 206 и 218), причем получаются продукты присоединения двух, четырех или шести атомов водорода.
Обширные и тщательные исследования взаимных переходов от ароматических соединений к продуктам присоединения ео юрода, или к «гидроароматическим» соединениям, принадлежат немецкому
ученому Адольфу Байеру*. Особенно большое значение имели его исследования последовательного присоединения атомов водорода к простейшим двухосновным ароматическим кислотам (о-, jw- и п-фталевые кислоты), а также изучение производных сукцинил янтарного эфира (стр. 207), завершившиеся получением бензола из производных циклогексана.
Эти исследования привели к чрезвычайно важному для теории строения бензола выводу, что все превращения ароматических соединений в гидроароматические, равно как и обратные переходы от гидроароматических соединений к ароматическим, могут быть полностью объяснены на основе формулы Кекуле. Некоторые детали этих превращений нашли более полное объяснение на основе гипотезы Тиле (см. том I, стр. 378).
Ад. Байер показал, что при восстановлении фталевых кислот в результате присоединения к этим кислотам двух, четырех и, наконец, шести атомов водорода получаются производные циклогексана, образования которых следовало ожидать, если принимать для бензола формулу Кекуле.
При осторожном восстановлении, например амальгамой натрия, о-фталевой и n-фталевой (терефталевой) кислот вначале получаются продукты присоединения двух атомов водорода, или дигидрофталевые кислоты; при восстановлении же ле-фталевой (изофталевой) кислоты присоединяются сразу четыре атома водорода и получается тетрагидроизофталевая кислота. Образование гидроароматических кислот такого строения объясняется, согласно гипотезе Тиле, присоединением водорода к крайним атомам сопряженных систем.
В случае терефталевой кислоты конъюгация распространяется на систему из восьми атомов:
10-••
II 2С—ОН
ЗС
\
НС сн
5| || + Щ
НС сн
\ /
6С
I
7С—он
II 80-••
1,8-присоединение
----------------->
о—н
с-он
II с
I! II нс сн
та/томерное превращение
II с-он
I о—н
О—н
(Lo
/СН
нс \:н
И II
НС сн
\н
I
с=о
I
о—н
* Адольф Байер (1835—1917). Выдающийся немецкий органик. Главнейшие его работы—исследования в области индиго (начиная с 1860 г.), закончившиеся первым синтезом этого красителя в 1880 г.; открытие фталеинов (1871); исследования в области гидрированных производных бензола (1880—1895); теория «напряжения»; центрическая формула бензола; работы в области терпенов (1890—1900); исследования оксониевых соединений и явления галохромии (1900—1920). К числу его учеников принадлежали Эмиль и Отто Фишеры, Гребе, Карл Либерман, Виль-штеттер и многие другие.
а в случае изофталевой кислоты имеются две независимые конъюгированные системы (1,6 и к концам которых могут присоединиться четыре атома водорода:
10...
II 2С—ОН д н/ 1,6- и 1,4-
ПС. Сп • • • присоединение
5| ||2' 3' 4' + 2Н2 ---------—9
НС С—с=о- • •
'Ч / I
... сн он
о—н
с—он
li
с
" 1
НС С=С—он хс^2 он
таутомерное превращение
------------
II
С—ОН
сн
нс \н2 •
II I
НС СН—С^О
он
Присоединением галоидов к бензолу также можно перейти к производным циклогексана, например получить гексахлорциклогексан:
СН СНС1
НС СН CIHC7 ^CHCl
I II + ЗС12 -> I I
НС CH С1НС СНС1
\ / \ / CH CHC1
Примером обратного перехода—от производных циклогексана к производным бензола—может служить следующий ряд реакций.
Сукцинилянтарный эфир
СО
С2Н5ООС—НС^ ЧСН2
Н2С СН—соос2нв \о
получаемый конденсацией двух молекул янтарного эфира (стр. 41), превращается после омыления и отщепления СО2в 1,4-циклогександион: СО
Н2С
I
сн2 I сно
Этот дикетон при восстановлении дает двухатомный спирт, названный хинитом:
ОН с СП \н2
. Н2С сн2
\н
он
При действии на хинит бромистого водорода получается 1,4-5//-бромциклогексан, от которого действием спиртовой щелочи отщепляются две молекулы бромистого водорода и получается дигидробензол (1,3-циклогексадиен):
Вг ) СН сн
ЩС^ ^СНз н^ ^сн
H2i (in2 н2с
\н \н
Вг
В молекуле дигидробензола имеется сопряженная пара двойных связей, а потому при действии на него брома получается продукт присоединения* молекулы брома в положение 1,4
Вг
I
СН н^ \н
I II
Н2С СН
Вг
который при действии спиртовой щелочи превращается в бензол:
СН
нс^ 'сн
I II
НС сн ^сн"
Очень важным примером прямого перехода отсоединений ряда бензола к соединениям ряда циклогексана является реакция Сабатье и Сандерана (стр. 45), также вполне согласующаяся с формулой Кекуле для бензола.
Сравнение теплот горения бензола, продуктов его последовательного гидрирования и нормального гексана (табл. 12) показывает, что бензол является более устойчивым соединением, чем дигидробензол и тетрагидробензол. Различие теплот горения при переходе от дигидробензола к бензолу даже больше, чем при переходе от нормального гексана к циклогексану.
Таблица 12
Теплоты горения бензола и продуктов его гидрирования (при 25° и 760 мм рт. ст.)
Название Состав Формула Теплота горения (жидких) ккал/моль Разность ккал/моль
Бензол свн„ 781 66
1,2-Дигидробензол с„н8 0 847 45
Т етрагидробензол CeHio 892 45
Циклогексан СвН12 937 * 58
^-Гексан С,н14 СН3(СН2)4СН3 995
Углеводороды ряда бензола и их одноатомные производные
Углеводороды ряда бензола
СпНгп—б
Первый гомолог бензола—метилбензол, или толуол, С7Н8 не имеет изомеров, каки все другие однозамещенные производные*. Второй гомолог QH10 может существовать в четырех изомерных формах: этилбензол С6Н5—С?Н5 и три диметилбензола, или ксилола, СеН4(СН3)2 (орто-, мета- и пара-ксилолы, или 1,2-, 1,3- и 1,4-диметилбензолы). Из трех триметилбензолов рядовой изомер (1,2,3-) называется гемимеллитолом или гемеллитолом, несимметрический изомер (1,2,4-)—псевдокумолом, а симметрический (1,3,5-)—мезитиленом.
Остаток бензола носит название фенил, а названия остатков его гомологов производят от названий соответствующих углеводородов, добавляя к корню окончание ил (толил, ксилил, мезитил и т. п.) и обозначая буквами о-, л/-, и- или цифрами положение боковых цепей. Общее название для всех ароматических радикалов—арил, аналогично названию алкил для алифатических радикалов.
Названия высших гомологов бензола часто производят не от названия ароматического ядра, а от названия боковой цепи, т. е. называют их как производные парафиновых углеводородов, например:
СН3—СН—СН2—СН2—СН2—СН3
2-фенилгексан
СН3— СН2—СН—сн2—сн3
3-(л<-ксилил-2)-пентан
сн3
СН3— СН2—С—СН2—СН,—сн2— сн, о
СН3
3- метил-3-п-толил гепта н сн3—сн2—сн—сн2—сн3
W’ сн3 3-(л«-ксилил-4)-пентан
* Если нет изомерии за счет заместителя (изомерии радикалов).
При нумерации боковых цепей гомологов бензола, как и в номенклатуре алициклических соединений (стр 14), из возможных порядков нумерации следует избирать тот, при котором сумма цифр номеров заместителей будет наименьшей. Например, диметилэтилбензолу строения
С,н5 н3С\1
^^СНз
следует придать название 1,4-диметил-2-этилбензол (сумма цифр равна 7), а не 1,4-диметил-З-этилбензол (сумма цифр равна 8). Более короткие цепи всегда указывают в первую очередь.
Способы получения. Кроме способов получения ароматических углеводородов из соединений жирного ряда (стр. 203) и гидроаро-матических соединений (стр. 57 и 222), существуют способы их получения из различных веществ ароматического ряда. Многие из этих способов аналогичны способам получения парафиновых углеводородов из соединений жирного ряда, хотя здесь для проведения реакции часто требуются иные условия в отношении температуры, времени, применяемого реагента (например, восстановителя) и пр. Особенно большое значение имеют две синтетические реакции получения гомологов бензола—реакция Вюрца—Фиттига и реакция Фридел я—Крафтса.
1. Декарбоксилирование кислот. Для получения чистых ароматических углеводородов часто пользуются реакцией отщепления С0.2 от ароматических кислот при нагревании их солей с едкими щелочами или известью:
С6Н5—COONa + NaOH —> CeH6 + Na2CO3
2. Восстановление производных бензола. Из реакций восстановления производных бензола часто применяются следующие:
а) Восстановление диазосоединений кипячением со спиртом (стр. 321):
[C6H5N2]C1 + СН3-СН2ОН СвН6 + СН3СНО + N2 + НС1
б) Нагревание сульфокислот с водой в присутствии минеральных кислот (стр. 258):
C6H5-SO3H + Н2О C6H6 + H2SO4
в) Восстановление жирноароматических кетонов амальгамированным цинком и концентрированной соляной кислотой (восстановление по КлеменсенуУ
Zn + НС1 сбн5—со—сн3-------> сбнб—сн2—сн3
г) Перегонка фенолов (ароматических оксисоединений) с цинковой пылью:
C6H5OH + Zn —> СбНб + ZnO
д) Восстановление галоидных соединений действием натрия в спиртовом растворе:
С6Нб—Вг + 2Na + 2СН3ОН —> CeHe + NaBr + NaOCH3
3. Реакция В юр ц а—Ф и т т и г а*. Эта реакция является видоизменением способа получения парафиновых углеводородов по Вюрцу (см. том I, стр. 160). Она состоит в действии металлического натрия на смесь ароматического галоидного соединения и галоидного алкила:
СвН5—Вг + С2Н5-Вг + 2Na
CeH5—С2Н5 + 2NaBr этилбензол
/Вг ХСН3
С6Н4< + 2СН3—Вг + 4Na CeH,< + 4NaBr
ХВг ХСН3
ксилол
В качестве побочных продуктов образуются парафиновый углеводород (диалкил) и дифенил С6Н5—С6Н5 или его производные.
Реакция Вюрца—Фиттига протекает через промежуточное образование натрийорганических соединений (П. П. Шорыгин):
R— J 4- 2Na —> R—Na + NaJ
R—Na + R—J —» R—R + NaJ
Выдвигалась также точка зрения, что углеводород получается в результате взаимодействия двух свободных радикалов:
2RJ + 2Na —> 2R + 2NaJ
2R. R—R
В качестве доказательства образования свободных радикалов при' реакции Вюрца—Фиттига указывалось на получение ряда побочных продуктов этой реакции. Так, например, при действии натрия на хлорбензол в отсутствие растворителей, наряду с главным продуктом—дифенилом, образуются в некотором количестве бензол
k * Карл Адольф Вюрц (1817—1884); один из крупнейших французских химиков второй половины XIX века. Уроженец Эльзаса. Из наиболее важных его работ следует отметить: синтез первичных аминов при изучении гидролиза изоциановых эфиров (1849); открытие изобутилового спирта в сивушном масле (1853); получение алифатических углеводородов из галоидопроизводных и натрия или цинка— реакция Вюрца (1854); открытие гликоля (1856) и окиси этилена (1859).
Рудольф Фиттиг (1835—1910). Немецкий химик. Синтезировал в 1863— 1864 гг. толуол и этилбензол из бромбензола и иодистых алкилов с натрием— реакция Фиттига. Изучал окисление боковых цепей ароматических углеводородов. Начиная с 1877 г., много занимался исследованиями в области фумаровой и малеиновой кислот.
и трифенилен (стр. 486), очевидно, за счет диспропорционирования фенильных радикалов:
CeHs—Cl + Na -» CeHs* + NaCl
2СбН6* —> C6He 4* С6Н4 .
Еще несомненнее образование свободных радикалов при синтезе тетрадекана, где в качестве побочных продуктов получаются гептан и гептен:
2С7Н15—J + 2Na -» 2С7Н15* + 2NaJ
2С7Н1ьв —> С7Н1в С7НМ
Однако в дальнейшем было показано (Мортон), что свободные радикалы хотя и образуются одновременно с алкил- или арилнат-рием, но они, по крайней мере на 90%, диспропорционируются согласно приведенным выше схемам. Образование же углеводорода с удвоенным молекулярным весом происходит по схеме П. П. Шоры-гина.
Мортоном была открыта важная побочная реакция при синтезе углеводородов по Вюрцу—Фиттигу, которая в определенных условиях может стать и главной реакцией. При проведении синтеза в среде толуола может происходить взаимодействие натрийалкила с растворителем, например:
C6Hn-Na + CeHg—СН3 -> C6H6-CH2~Na + С5Н12
Получающийся бензилнатрий, реагируя с галоидным алкилом, дает со значительным выходом алкилбензол:
CeHs—СН3—Na + C5Hn—Cl -» С6Н5-С6Н13
В присутствии некоторых добавок (изопропилат калия, хлористый цезий и др.) выход н-гексилбензола из н-амилхлорида и толуола превышал 75%.
4. Реакция Фридел я—К р а ф т с а*. Эта реакция характерна для ароматического ряда. Она состоит в том, что под влиянием голоидных соединений алюминия, действующих каталитически, ароматические углеводороды вступают в реакцию с галоидными
* Шарль Фридель (1832—1899). Французский химик. Получил в 1862 г. изопропиловый спирт восстановлением ацетона. В 1872 г. осуществил первый синтез глицерина (совместно с Сильва). В 1863 г. совместно с Д. Крафтсом получил впервые тетраэтилкремний; дальнейшие работы в этой области проводил с Ладен-бургом. В 1876—1877 гг. совместно с Крафтсом синтезировал с помощью хлори стого алюминия амилбензол, этилбензол и т. д.
Джемс Крафтс (1839—1917). Американский химик, ученик А. Вюрца, в лаборатории которого вместе с Ш. Фриделем открыл и изучил реакцию, носящую их имя.
алкилами, выделяя галоидоводород и образуя соответствующие гомологи бензола, например:
С6Н5— Н + С1—СН3 -> С6Н5—СН3 + НС1
Обыкновенно реакция не заканчивается введением в бензольное ядро одного алкила, но идет и со второй, третьей и т. д. молекулами галоидного алкила, в результате чего получается смесь гомологов. При избытке хлористого метила конечным продуктом реакции является гексаметилбензол С6(СН3)6
Реакция Фриделя—Крафтса обычно осложняется побочными процессами, в результате которых из высших гомологов могут получаться низшие. Вследствие этого при действии хлористого алюминия, например, на толуол, наряду с ксилолами и высшими гомологами бензола может образоваться также и бензол. В некоторых случаях можно ясно установить перенос алкильных групп из одной молекулы в другую, например:
(СН3)3С-С6Н4-С(СН3)3 + С6Н6 2С6Н6—С(СН3)3
Вместо галоидных алкилов в реакции Фриделя—Крафтса можно применять и непредельные углеводороды, например:
СбН6 4-СН2=СН2 -> СбП5—СН2—СН3
/СН3 с6н6 + снз—сн=сн2 с6н5-сн<
Х2Н3
При этом фенильный остаток направляется всегда к наименее гидрогенизированному атому углерода (в соответствии с правилом В. В. Марковникова).
Реакцию Фриделя—Крафтса, повидимому, следует рассматривать как систему последовательных (консекутивных) обратимых реакций. Этим и объясняется образование, наряду с моноалкилбензолом, также ди-, три- и т. д. алкилбензолов:
С6Н6 + AlkX CGH5AIk + их
C6H5Alk + AlkX C6H4Alk2 + HX
C6H4Alk2 +AlkX C6H3Alk3 + HX и т. д.
Предлагались различные схемы протекания этих реакций. Широкое распространение приобрела следующая схема:
Aik—Cl + А1С13 Alk+ [A1C1J-
(образование иона карбония)
С6н6 —*
с«н5^-н
(поляризация ароматического ядра)
C^V4I + aik+[aiciJ —> С6Н5-А1К + н [A1C1J
H+[A1C1J" —> А1С134-НС1
Однако впоследствии было доказано, что кислота Н+[А1С14]’ в действительности не может существовать даже кратковременно Кроме того, приведенная схема не объясняет известного факта, что в отсутствие воды реакция Фриделя—Крафтса часто вообще не идет.
Недавно была высказана мысль (Неницеску), что катализатором является комплексное соединение А1С1з-Н2О, т. е. фактически кислота Н+[А1С13ОН]“, реаги
рующая с галоидными алкилами с образованием Aik+[AICI3OH]'. Последнее соединение, а не А1к+1А1С14]", и служит переносчиком алкила в цепной реакции по приведенной выше схеме.
Для алкилирования бензола олефинами предлагается следующая схема протекания реакции:
Чбнб с^н^-н
(поляризация ароматического ядра)
сн2=сн2 —► сн2—сн2
(поляризация олефина)
СбН5^-Н + сн2—сн2 —с6н5-сн2-сн3
Конденсирующим агентом и здесь являются комплексные соединения хлористого алюминия (Г. Г. Густавсон, Б. Н. Меншуткин, В. А. Плотников).
Объяснение протекания реакции по ионному механизму не является единственным. Возможно, что оно справедливо лишь для третичных и вторичных галоид-алкилов. Алкилирование же первичными алкилгалогенидами протекает, повидимому, по схеме нуклеофильного замещения через промежуточный комплекс.
5. Ароматизация парафинов. Ароматические углеводороды могут быть также получены из парафиновых углеводородов пропусканием их над катализаторами, путем так называемой дегидроциклизации (см. том I, стр. 363 и ниже, стр. 222).
Физические свойства (табл. 13). Ароматические углеводороды представляют собой частью жидкости, частью твердые тела с особым, нередко сильным, запахом. Изомеры с несколькими боковыми цепями, различающиеся положением алкилов в ядре (изомерия положения), имеют близкие температуры кипения. Значительно больше различаются их температуры плавления: при наиболее высокой температуре плавятся пара-изомеры. Температура плавления повышается также с накоплением метильных групп в молекуле ароматического углеводорода. При различии в строении, зависящем от изомерии радикалов, наблюдаются те же закономерности, что и в жирном ряду. Например, температура кипения ft-пропилбензола выше, чем у изопропилбензола.
Химические свойства. Химические свойства ароматических углеводородов, а также их производных, отличаются рядом особенностей, которые объединяются под названием «ароматического характера» этих веществ. Наиболее существенными чертами ароматического характера является особая склонность к реакциям замещения, а не присоединения, и большая устойчивость бензольного ядра. Особенно характерны для ароматических углеводородов, и вообще для ароматических соединений, легко протекающие реакции замещения под действием концентрированных серной и азотной кислот.
Однако работы, проведенные в последние десятилетия, показали, что грань, отделяющая ароматические соединения от других классов органических веществ, является не столь резкой, как это считали
ьо »—*
Oi
Ароматические углеводороды ряда бензола
Таблица 13
Название Формула Положение замещающих групп Температура плавления °C Температура кипения °C Удельный вес .20 d4 Показатель преломления 20 nD
Бензол с,нв — +5,53 80,10 0,8790 1,5011
Толуол с»н8сн3 1 —95,0 110,6 0,8669 1,4969
Ксилол орто- СвН4(СН3)2 1,2 —25,2 144,4 0,8802 1,5055
мета- 1,3 —47,9 139,1 0,8642 1,4972
пара- 1,4 + 13,3 138,4 0,8611 1,4958
Т риметилбензол рядовой (гемимеллитол, или гемеллитол) СвН3(СН8)3 1,2,3 —25,4 176,1 0,8944 1,5139
асимметрический (псевдокумол) 1,2,4 —43,8 169,4 0,8758 1,5048
симметрический (мезитилен) 1,3,5 —44,7 164,7 0,8652 1,4994
Тетраметилбензол рядовой (пренитол) 1,2,3,4 —6,3 205,0 0,9052 1,5203
дурол 1,2,4,5 +79,2 196,8 — —
изодурол 1,2,3,5 —23,7 198,0 0,8903 1,5130
Пентаметилбензол С6Н(СН3\5 — +54,3 231,8 —
Гексаметилбензол C6(CH3)g — + 165,5 263,5 — —
Этилбензол СвН8С2Н8 1 —95,0 136,2 0,8670 1,4959
н-Пропилбензол Свн5с3н, 1 —99,5. 159,2 0,8620 1,4920
Изопропилбензол (кумол) С,Н8СН(СН3)2 1 —96,0 152,4 0,8618 1,4915
н~ Бутилбензол свн8с4н, 1 —88,0 183,3 0,8601 1,4898
в тор-Бутилбензол /СН3 с6н.сн< ХС2Н8 1 —75,5 173,3 0,8621 1,4902
Изобутилбензол СвН8СН2СН2(СН3)2 1 —51,5 172,8 0,8532 1,4865
тр ет - Б у тил бе н зол СвНбС(СИ3)з 1 —57,9 169,1 0,8665 1,4927
^-Метилизопропилбензол (п-цимол) .сн, свН< ,, ХС3Н, 1 1,4 —67,2 177,1 0,8573 1,4909
УГЛЕВОДОРОДЫ РЯДА БЕНЗОЛА И ИХ ОДНОАТОМНЫЕ ПРОИЗВОДНЫЕ УГЛЕВОДОРОДЫ РЯДА БЕНЗОЛА
ранее, и там, где раньше усматривались резкие качественные различия, теперь признаются лишь различия количественного характера.
1. Восстановление. Восстановление бензола и его гомологов многими сильными восстановителями, легко превращающими обычные этиленовые связи в ординарные, например крепкой иодистоводородной кислотой, идет лишь с большим трудом (ср. стр. 47). Однако в присутствии мелкораздробленных металлов (никель, платина, палладий) ароматические углеводороды легко присоединяют шесть атомов водорода и переходят в соединения ряда циклогексана (см. стр. 45).
2. Д е й с т в и е галоидов. Хлор и бром в обычных условиях действуют замещающим образом:
СбНй 4- Вг2 С6Н5Вг + НВг
Наиболее энергично эта реакция идет в присутствии катализаторов. При пропускании же хлора или паров брома в кипящий бензол, особенно легко на прямом солнечном свету или при освещении ультрафиолетовыми лучами (кварцевая лампа), получаются продукты присоединения шести атомов галоида, например:
EgHg "Ь ЗС12 > С6Н6С16
Эти соединения, являющиеся шестигалоидными производными циклогексана, получаются в виде сложной смеси стереоизомеров.
Теоретически для 1,2,3,4,5,6-гексахлорциклогексана возможно существование восьми изомеров. Из смеси гексахлорциклогексанов, получаемой при хлорировании бензола, сначала были выделены четыре изомера (Менье, Ван-дер-Линден), в 1946 г.—пятый изомер, а в самое последнее время—остальные три:
Изомер Температура Изомер Температура
плавления плавления
°C °C
а...........157,5—158,5 в ...............218,5—219,3
3........... 309 т]........... 89,8—90,5
Y...........111,8—112,2 0............. 124—125
8 .......... 138—139 t(0.......... 88—89
Смесь изомеров может быть разделена систематической обработкой метанолом и хлороформом или путем избирательной адсорбции (хроматографически).
Оказалось, что у-изомер, образующийся при фотохимическом хлорировании бензола всего лишь в количестве 12—20%, обладает ценными свойствами как инсектицид, благодаря чему он получил теперь широкое применение.
Это бесцветный кристаллический порошок со своеобразным характерным запахом, нерастворимый в воде, но растворимый в органических растворителях, мало токсичный для высших теплокровных животных. Его называют гаммексаном (иногда линданом— по имени Линдена), а технический продукт—гексахлораном.
Наиболее устойчивым и наименее растворимым является [3-изомер, обладающий вместе с тем и наивысшей температурой плавления.
Все гексахлорциклогексаны при действии спиртовых щелочей отщепляют элементы хлористого водорода, образуя смесь изомерных трихлорбензолов.
Теоретически для гексахлорциклогексана можно построить (на основе наиболее устойчивой формы «кресла» для циклогексана, восемь устойчивых конформаций (стр 24) Шведскому ученому О Хасселю удалось показать, что выделенным изомерам гексахлорциклогексана принадлежат конформации с размещением атомов хлора по схемам:
а-изомер .... ааееее
[3-изомер .... ееееее
у-изомер .... аааеее
о-изомер .... аеееее
Е-изомер .... аееаее
уизомер .... ааеаее
0-изомер .... аеаеее
t-изомер (С-изомер) аеаеае
Конформацию у-хлорциклогексана можно изобразить схемой
в которой жирные точки соответствуют атомам хлора.
Интересно отметить, что молекулы а- и тризомеров не обладают элементами симметрии. Это можно видеть на примере а-изомера:
В данном случае имеется асимметрия молекулы без асимметрического атома углерода. Это было подтверждено выделением левовращающего изомера после отщепления хлористого водорода от оптически недеятельного а-изомера при помощи бруцина-основания. Повидимому, НС1 отщеплялся только от правовращающей формы:
4" 3/?С23Н2бNA >
г/г(—)*Сб14$С]б-f- 3/2C23H2eN2O4-HCl 4- 1/2CgH3Cl9
Левоврашаюший изомер имел темп, плавл. 128—132° (Кристоль).
3. Действие галоидных кислот. Хлорноватистая кислота присоединяется к бензолу в количестве трех молей
СбНб + ЗНС1О СвН6С13(ОН)3 присоединение же галоидоводородов к ароматическим соединениям не наблюдалось ни в каких условиях.
4. Действие эфира диазоуксусной кислоты. Эфир диазоуксусной кислоты присоединяется к бензолу с образованием эфира так назы*
ваемой псевдофенилуксусной кислоты, легко изомеризующейся в циклогеп татр иен-карбоновую кислоту:
НС
I
НС
СН
*хсн
I >сн-соон
X/CH сн
нс-сн
нс7 V*—СООН
НС<. ^CH2 сн
псевдофенилуксусная кислота
циклогептатриенкарбоновая кислота
5. Озонирование. Бензол легко присоединяет три молекулы озона, образуя триозонид СВНК(О3)3, строение которого соответствует формуле Кекуле. При действии воды триозонид распадается на три молекулы глиоксаля (см. стр. 205).
6. Действие окислителей. Одним из важнейших свойств бензола является его устойчивость по отношению к окислителям—к разбавленной азотной кислоте, хромовой кислоте и марганцовокислому калию. Этими окислителями он окисляется гораздо труднее, чем парафиновые углеводороды, и в настоящее время в этом видят наиболее резкое отличие ароматических углеводородов от обычных соединений с этиленовыми связями.
При энергичном действии сильных окислителей на гомологи бензола бензольное ядро оказывается более устойчивым, чем жирные радикалы (алкилы) боковых цепей. В результате такого окисления боковые цепи, какова бы ни была их длина, отщепляются от бензольного ядра; только ближайший к ядру атом углерода не отщепляется, а образует карбоксильную группу. Эта реакция может быть использована для определения числа боковых цепей и их относительного положения в ядре; при наличии одного замещающего радикала получается одноосновная бензойная кислота, при двух—соответствующие двухосновные кислоты (о-, м- и n-фталевая) и т. д.
При окислении орто-изомеров хромовой кислотой часто окисляется и бензольное ядро, а потому ароматические кислоты не получаются.
7. Нитрование. При действии концентрированной азотной кислоты на ароматические углеводороды происходит нитрование их, т. е. замена атомов водорода бензольного ядра на нитрогруппы:
С6Нв + НО—NOa —> CbH5— NOa4-H2O
8, Сульфирование. При действии концентрированной или дымящей серной кислоты на бензол и его гомологи происходит сульфирование их, т. е. замена атома водорода в ядре на сульфогруппу:
СН3—СвН5 + НО—SO3H СН3—С6Н4—SO3H + Н2О
При нагревании гомологов бензола с серной кислотой может происходить перенос алкильных групп, как и при действии хлористого алюминия (стр. 214).
Источники ароматических углеводородов. Основным источником бензола и прочих ароматических углеводородов на протяжении
XIX столетия и первых десятилетий XX века служила сухая перегонка (коксование) каменных углей. До середины XIX столетия коксование углей производили только для получения металлургического кокса или светильного газа. Получающаяся при коксовании каменноугольная смола являлась тогда обременительным отбросом. С развитием анилинокрасочной промышленности возник спрос на ароматические углеводороды, извлекаемые из этой смолы и в значительных количествах содержащиеся также и в коксовальном (коксовом) газе. Извлечение ароматических углеводородов из продуктов коксования и выделение их в виде индивидуальных продуктов стали производить попутно при переработке угля. Таким образом возникли коксохимические заводы.
На таких заводах коксующиеся (спекающиеся, богатые летучими продуктами) каменные угли после промывки, сортировки и дробления загружают в камеры коксовых печей. Выходящие из коксовых печей газы отделяют от каменноугольной смолы и подают в скрубберы, где газ отмывается сначала водой от аммиака, сероводорода, цианистых соединений, а затем—холодным поглотительным маслом, улавливающим пары бензола, толуола и других углеводородов. Поглощенные маслом углеводороды при нагревании отгоняются и далее конденсируются в виде так называемого сырого бензола, а масло после охлаждения вновь поступает в поглотительные скрубберы.
При коксовании угля получается (от веса угля) 75—80% кокса, 2,5—3,5% смолы, 0,2—0,4% аммиака, до 0,8—1,5% сырого бензола и выделяется около 300 м3 газа на 1 т угля. Коксовый газ содержит 30—50% метана (по объему) и 30—50% водорода; чем выше температура коксования, тем больше в газе содержится водорода и гем меньше метана.
Сырой бензол вместе с легкими погонами (кипящими ниже 160°), выделяемыми из смолы, подвергается разделению на бензол, толуол, смесь ксилолов и ряд других продуктов. Разделение ректификацией комбинируется с промывкой щелочами (для извлечения фенолов) и кислотами (для удаления пиридиновых и хинолиновых оснований).
Из средних (кипящих до 220—230°) и тяжелых (кипящих до 270°) масел, получаемых при перегонке смолы, путем кристаллизации выделяют нафталин. Антраценовое, или зеленое, масло, перегоняющееся до 360°, является источником получения антрацена, фенантрена, аценафтена, карбазола. Остаток от перегонки смолы (составляющий до 60% ее веса) представляет собой черную твердую, размягчающуюся около 60—75° массу—каменноугольный пек, применяемый в дорожном строительстве, строительном деле и для брикетирования коксовой мелочи.
В настоящее время, однако, основным источником ароматических углеводородов является, пожалуй, нефть.
Содержание ароматических углеводородов в нефтях обычно невелико, за исключением нефтей некоторых месторождений: нефть
с острова Борнео содержит до 20—40% ароматических углеводородов; довольно богата ими также нефть из Бирмы. Из нефтей Советского Союза сравнительно богаты ароматическими углеводородами нефти Западной Украины (до 6%), майкопская нефть, некоторые нефти «Второго Баку», сураханские нефти (Азербайджанская ССР), нефти Чусовских месторождений.
Однако ароматические углеводороды получают из нефти главным образом путем специальных методов ее термической переработки—крекинга, пиролиза, каталитической дегидрогенизации нафтеновых углеводородов и циклизации парафиновых углеводородов. Крекинг нефти и мазута ведут обычно при температурах порядка 450—650°, причем крекинг в жидкой фазе проводят при более низких температурах, а парофазный крекинг—при более высоких. Пиролиз ведут при еще более высоких температурах порядка 650—800°; в этом случае образуются большие количества ароматических углеводородов.
Наиболее перспективными являются сейчас процессы дегидрогенизации нафтеновых и каталитической циклизации (дегидроциклизации) парафиновых углеводородов.
Дегидрогенизация углеводородов ряда циклогексана в присутствии платины и палладия (позднее в присутствии никеля, отложенного на окиси алюминия) была открыта Н. Д. Зелинским еще в начале XX в. В дальнейшем было показано, что дегидрогенизация гомологов циклогексана может быть осуществлена и в присутствии окиси хрома, но при более высокой температуре (Адкинс, А. А. Баландин). В 1936—1937 гг. почти одновременно рядом советских исследователей было показано, что при 300—310° над платинированным углем (Б. А. Казанский, А. Ф. Платэ), при 450—470° над окисью хрома (Б. Л. Молдавский, Г. Д. Камушер) и при 500—550° над хромофосфорным катализатором (В. И. Каржев, М. Г. Северьяно-ва, А. Н. Снова) можно достичь довольно гладкого превращения парафиновых и олефиновых углеводородов в ароматические. Впоследствии список катализаторов для этой реакции был значительно расширен; оказалось, что для ароматизации парафинов могут быть использованы окислы ванадия, молибдена, тория, урана, церия и большое число смешанных катализаторов. В состав смешанных катализаторов в разных соотношениях могут входить почти все элементы периодической системы Менделеева (81 элемент).
Механизм реакции ароматизации в самом общем виде (для всех катализаторов) может быть выражен схемой:
т--------------------Парафины-------------—-----г
V I
Олефины I Циклог ексаны
I________________> Ароматические <--------------1
углеводороды
Многочисленные исследования механизма этой реакции показали, что на платине и на окисных катализаторах реакция протекает по-разному. На платине реакция идет через промежуточное образование углеводородов ряда циклогексана, например:
СН2
Н2С ^СНз
сн3
V
—3112
СН У \ НС CI1
I II
НС сн
сн
Вторая стадия протекает значительно (примерно в 100 раз) быстрее первой, вследствие чего и не удается выделить промежуточно образующийся циклогексан. В тех случаях когда строение получающегося при циклизации циклогексанового углеводорода затрудняет дегидрогенизацию (стр. 57)
н3с сн3 /с\ Н3с сн3
н2с сн2 Н2С сн2 1 2
Н2С 1 СН3 > 1 Н2С сн2
сн3
промежуточный продукт можно обнаружить.
При ароматизации парафинов на платине олефины не образуются, а если их добавить, ароматизация прекращается. Таким образом, на платине реализуется только правая ветвь приведенной выше схемы
На окисных катализаторах,, наоборот, частично всегда образуются олефины, которые могут затем превращаться в ароматические углеводороды (левая ветвь схемы) Часть парафинов превращается при этом в ароматические углеводороды, минуя стадию олефинов (центральная ветвь схемы) В зависимости от того, какой окисел служит катализатором, непосредственно в ароматические углеводороды превращается разная доля парафинов. Например, на окиси ванадия ароматические углеводороды получаются главным образом непосредственно из парафинов, а большая часть образовавшихся олефинов вообще не претерпевает дальнейших превращений и обнаруживается в продуктах реакции.
Происхождение ароматических углеводородов каменноугольного дегтя. До недавнего времени образование ароматических углеводородов приписывали только пирогенетическим процессам^ т. е. протекающей под влиянием высоких температур конденсации ацетилена и его гомологов, подобно синтезу бензола из ацетилена (Бер-тело). Более поздние исследования сухой перегонки каменных углей при сравнительно низких температурах и давлениях показали, что первоначальными продуктами этого процесса являются, невидимому, более сложные алициклические соединения. В ходе пиролиза они превращаются в сложные фенолы, при дальнейших пирогенетических процессах дающие простейшие фенолы, частью восстанавливающиеся в ароматические углеводороды.
А. Пикте* нашел, что в каменных углях содержатся некоторые сложные циклические углеводороды и их производные, которые могут быть экстрагированы из угля различными растворителями.
Бензол С6НР—легкоподвижная, сильно преломляющая свет, бесцветная жидкость со своеобразным нерезким запахом. При продолжительном вдыхании паров бензола появляется шум в ушах, слюнотечение и ощущение металлического вкуса, головокружение.
Бензол содержится в небольших количествах почти во всех нефтях. Технически он получается путем выделения из продуктов сухой перегонки каменных углей и при процессах ароматизации нефтей. Он имеет по сравнению со своими ближайшими гомологами довольно высокую температуру плавления (+5,53°).
Бензол был открыт М. Фарадеем (1825 г.) в жидкости, получающейся при компримировании светильного газа. Митчерлих** в 1833 г. получил бензол при сухой перегонке бензойной кислоты с избытком негашеной извести и доказал его тождество с углеводородом, полученным Фарадеем. Данное ему Митчерлихом название «бензин» Либих предложил заменить на «бензол».
Бензол служит основным исходным веществом для получения нитробензола, анилина, хлорбензола, фенола, этилбензола, изопропилбензола, стирола и т. д. Поэтому он является важным сырьем в анилинокрасочной и фармацевтической промышленности, в производстве синтетического каучука и пластических масс, в производстве высокооктановых добавок к авто- и авиамоторным топливам.
Бензол является прекрасным растворителем для многих органических соединений, он может смешиваться во всех отношениях со спиртами, эфирами, кетонами, с ледяной уксусной кислотой. Он весьма мало растворим в воде и, в свою очередь, очень мало растворяет воду. Соли минеральных кислот, едкие щелочи и вообще гетеропо-лярные соединения практически в нем совершенно не растворяются.
При высоких температурах (500—600°) в соприкосновении с металлами (свинец, железо) из бензола образуется дифенил (бифенил) и выделяется водород:
С6Н6 + С6Нб -» СвН5-СбН5 + Н2
При пропускании же паров бензола над силикатами (стекло, фарфор) образуется путем дальнейших конденсаций сложная смесь многоядерных ароматических углеводородов: 1,3-дифенилбензол, 1,4-ди-фенилбензол (терфенил), п.п'-дифенилбифенил (кватерфенил) и т. п.
При еще более высоких температурах образуется графит.
* Амё Пикте (1857—1937). Швейцарский химик. Известен своими исследованиями в области алкалоидов В последний период своей деятельности особенно интересовался процессами термического разложения углеводов и начал изучать вещества, извлекаемые органическими растворителями из каменных углей.
** Эйльгардт Митчерлих (1794—1863). Немецкий химик, ученик Берцелиуса. Открыл явление изоморфизма, в органической химии известен своими исследованиями каталитического превращения спирта в эфир. Открыл реакцию сульфирования бензола и реакцию образования его из бензойной кислоты.
Восстановление бензола фосфором и иодистоводородной кислотой ведет к образованию метилциклопентана, получающегося изомеризацией промежуточно образующегося циклогексана (Н. М. Кижнер). Однако при каталитическом гидрировании бензола в паровой фазе над платиной или палладием, а также над никелевым катализатором при 150—200°, получается нормально гексагидробензол— циклогексан (Сабатье, Сандеран). Эта реакция гидрирования обратима (Н. Д. Зелинский), и при повышении температуры до 300° циклогексан может дегидрироваться обратно в бензол.
По отношению к наиболее употребительным окислителям (КМпО4, СгО3 и т. д.) бензол очень стоек. С перекисью водорода он сам, как таковой, не реагирует, но при добавлении к смеси бензола с перекисью водорода небольших количеств солей закиси железа протекает бурная реакция, при которой образуются некоторые количества фенола, пирокатехина и продуктов их дальнейшего окисления.
В последние годы делались попытки высокотемпературным газофазным окислением бензола при помощи кислорода воздуха в присутствии бромистого водорода получать фенол. Схематически процесс может быть представлен таким образом:
2НВг + ЧА —> Вг2 4-Н2О
С6Нб + Вг2 -> С6Н5-Вг+НВг
С6НбВг + Н2О —> С6Н5—ОН + НВг
Бромистый водород должен, повидимому, играть роль как бы переносчика кислорода, все время возвращаясь в цикл реакций
Интересно отметить характерную для бензола реакцию, не обнаруживаемую его гомологами. При встряхивании растворов бензола в его гомологах или в бензине с аммиачным раствором цианистого никеля (К. Гофман) образуется почти бесцветный мелкокристаллический тяжелый осадок комплексного соединения Ni(CN)2(NH3)(C6H6).*
Толуол (метилбензол) С6Н5—СН3 был обнаружен впервые Сен-Клер-Девиллем в 1844 г. в продуктах перегонки толутанского бальзама, откуда и получил свое название. Толуол находит некоторое применение в анилинокрасочной промышленности, но основной областью его применения является производство одного из очень мощных взрывчатых веществ—тротила (тринитротолуола, или, сокращенно, тола).
Кроме извлечения из продуктов сухой перегонки каменного угля, толуол в значительных количествах получается при ряде пирогенетических процессов, которым подвергаются нефть и ее дестил-латы (ароматизация, дегидроциклизация, см. стр. 222).
* Возможно, что это соединение относится к соединениям включения, т. е. к соединениям, существующим только за счет включения одного вещества в кристаллическую решетку другого (включающееся вещество может быть и жидким). Хорошим примером таких соединений могут служить твердые, нерастворимые в спирте соединения мочевины с нормальными парафинами.
Ксилолы* (диметилбе изолы) С6Н4(СН3)2 были впервые получены в 1850 г. Кагуром из древесного спирта-сырца.
Технический ксилол представляет собой смесь всех трех изомеров (орто-, мета- и пара-) с примесью небольшого количества этилбензола. Из этой смеси сравнительно легко может быть выделен чистый л^-ксилол (Н. М. Кижнер и Г. Г. Вендельштейн) благодаря тому, что он сульфируется при более низкой температуре, чем орто-и пара-изомеры, а его сульфокислота гидролизуется при более низкой температуре, чем сульфокислоты других ксилолов. Сульфируя технический ксилол, а затем гидролизуя полученные сульфокислоты при строго определенных температурах, можно выделить ля-ксилол в очень чистом виде, а после его удаления выделить вымораживанием я-ксилол. о-Ксилол удается выделить из смеси изомеров перегонкой на очень эффективной ректификационной колонне.
Из всех трех изомеров только n-ксилол образует сравнительно высокоплавя-щийся комплекс с четыреххлористым углеродом состава n-C6H4(CH3)2-CCl4. Он плавится при температуре около —4° и при перегонке легко разделяется на компоненты. Этим методом недавно предложено пользоваться для выделения п-ксилола.
Этилбензол С6Н5—С2Н5 в настоящее время получается в громадных количествах из бензола и этилена в присутствии хлористого алюминия. Он служит исходным веществом для изготовления стирола, потребляемого в производстве синтетического каучука и пластических масс. Он может также служить отличной высокооктановой добавкой к моторному топливу.
Изопропилбензол (кумол) используется как высокооктановая добавка к моторному топливу и для получения фенола (стр. 266). Получается изопропилбензол алкилированием бензола пропиленом в присутствии хлористого алюминия в жидкой фазе или высокотемпературным парофазным алкилированием над катализаторами, содержащими фосфорную кислоту.
л-Метилизопропилбензол (n-цимол) содержится в значительных количествах в так называемом сульфитном скипидаре—углеводородном слое, образующемся при получении сульфитной целлюлозы из древесины хвойных пород. n-Цимол генетически связан с терпенами (см. стр. 119) и обладает углеродным скелетом, общим, например, с дипентеном:
СН3 СН3
С С\
НС \н Н2(/ хсн
II I II
нс сн н2с сн2
\/ с сн
СН3—СН-СНз сн2=с—сн3
n-цимол дипентен
* Цейлон—греч. дерево.
лг-Цимол может быть, кроме того, получен пиролизом а-пинена (стр. 134),главной составной части живичного скипидара (стр. 115).
СТРОЕНИЕ БЕНЗОЛА
Для теории ароматических соединений основной проблемой является строение самого бензола и прежде всего вопрос о том,, какая структурная формула наиболее правильно отражает строение бензольного кольца. Кроме того, теория должна объяснять следующие особенности ароматических соединений, наличие которых и составляет так называемый «ароматический характер» этих веществ:
1) своеобразную «насыщенность» бензольного ядра по сравнению с непредельными соединениями жирного ряда;
2) скачкообразное качественное и количественное изменение физических и химических свойств на последнем этапе перехода от циклогексана к бензолу через циклогексен и циклогексадиен (исчезновение непредельных свойств при переходе от циклогексадиена к формальному циклогексатриену);
3) особые физико-химические характеристики бензола (плоское расположение атомов кольца, равенство расстояний между центрами углеродных атомов, некоторые термохимические характеристики и т. п.);
4) повышенную кислотность оксипроизводных и пониженную основность аминопроизводных бензола по сравнению с аналогичными производными жирного ряда;
5) закономерности, наблюдаемые при реакциях замещения в ароматическом ядре
Для бензола, кроме формулы Кекуле (I), предлагались и другие формулы. Из них заслуживают упоминания: диагональная формула Клауса (II), призматическая формула Ладенбурга (III), хиноидная формула Дьюара (IV), центрическая формула Армстронга—Ьайера (V), формула Тиле с конъюгированными связями (VI):
I U Ш IV V VI
Каждая из приведенных формул имеет свои недостатки. Формула Кекуле не отражает равенства расстояний между всеми углеродными атомами в бензольном кольце (они были найдены равными 1,40 А, тогда как расстояния между атомами, связанными простыми связями, должны были бы составлять 1,54 А, а между связанными двойными связями—1,32 А). Теплота образования молекулы бензола больше, чем следовало бы ожидать для вещества, имеющего строение циклогексатриена (формула I).
Из химических свойств, не находящих объяснения на основе формулы Кекуле, можно указать следующие.
При озонировании бензола и последующем расщеплении триозонида образуются три молекулы глиоксаля. При озонировании же о-ксилола следовало ожидать, согласно нижеприводимым схемам, образования или одной молекулы глиоксаля и двух молекул метилглиоксаля
СН /СН3 нс^'сх
I II нс^ с сн сн3
сн=о
сн3—с=о
2 I
сн=о
или же двух молекул глиоксаля и одной молекулы диацетила:
СН ГНо нс^сх и ।
НСЧ /С сн гн3
< о
О/ I ХНо
>с J
X; сн3
iS >О 3
OV'
CH=O СНэ-С^О
2 I -ь | СН—О CH3-C=o
Фактически же образуется смесь всех трех продуктов, т. е. о-ксилолу как бы принадлежат и верхняя и нижняя формулы.
Формула Клауса (II) не дает возможности правильно предсказывать число дву- и трехзамещенных производных бензола. Для двузамещенных, по этой формуле, оказываются возможными лишь два, а не три изомера, например 1,2 и 1,3, так как три углеродных атома 2, 4 и 6 находятся в одинаковом отношении к углеродному атому 1, т. е. непосредственно с ним связаны, а следовательно, формулы
идентичны. Формула Ладенбурга (III) формально правильно передает число изомеров замещенных производных бензола. Однако эта формула была отвергнута еще Ад. Байером, поскольку она приводит к неверным выводам в отношении строения продуктов гидрирования фталевой кислоты и ее изомеров (стр. 206 сл.). Кроме того, эта формула, как и формула (II), несовместима с надежно установленным плоским расположением углеродных атомов в бензоле (стр. 229),
так как связи, которые на рисунках как бы пересекаются, не могут лежать в одной плоскости.
Формула (IV), предложенная Дьюаром, никак не согласуется со сравнительно насыщенным характером бензола и не позволяет объяснить образование глиоксаля при расщеплении продукта озонирования бензола. Кроме того, согласно этой формуле, возможно существование двух изомеров (1,2 и 2,3) для орто-замещенных производных бензола.
Ад. Байер и почти одновременно Армстронг предложили так называемую центрическую формулу (V). Эта формула предполагает отказ от объяснения строения бензола при помощи обычных связей и вводит представление о центрических связях, насыщенных не взаимно-попарно, а сразу всех шести. Такое представление логически не вытекает из теории химического строения, «привлечено произвольно и объясняет непонятное неизвестным» (А. Е. Чичибабин).
Формула Тиле (VI), пожалуй, с наименьшими натяжками могла бы служить для объяснения свойств бензольного ядра, исходя из теории парциальных валентностей и представлений о конъюгированных, сопряженных системах двойных связей.
Однако эта формула стала подвергаться сомнению после того, как был получен циклооктатетраен
с н - с и=с и сн - сн—сн
н/ /:н НС , 'к'"11
\н=сн-сн сн -сн-сн
и изучены его свойства. Казалось бы, и этот углеводород, подобно бензс>лу, должен обнаруживать «ароматическую» насыщенность, способность к реакциям замещения при сульфировании, нитровании, хлорировании и т. д., обусловливаемую взаимным насыщением парциальных валентностей. Однако, вопреки такому ожиданию, циклооктатетраен оказался веществом, обладающим отчетливо выраженным ненасыщенным характером (стр. 84). В дальнейшем это различие свойств бензола и циклооктатетраена получило объяснение (стр. 233).
Лучшее истолкование особенностей поведения бензола принесло с собой развитие современных электронных представлений о химической связи. Эти представления позволили дать удовлетворительные (по меньшей мере качественные) объяснения всех кратко перечисленных выше особенностей ароматических соединений.
В настоящее время можно считать твердо установленным, что молекула бензола является совершенно симметричным (дипольный момент |1=0) плоским образованием—шестичленным кольцом углеродных атомов, связанных друг с другом. В той же плоскости, в направлениях радиусов кольца, расположено шесть атомов водорода, каждый из которых связан со своим углеродным атомом. Расстояния между центрами углеродных атомов Саром.—Саром. составляют
1,40 А, расстояния С—Н равны 1,07 А, углы С—С—С и Н—С—С составляют 120°.
Остается, однако, вопрос—как, какими связями соединены друг с другом шесть углеродных атомов бензольного кольца?
Если рассчитать молекулярный объем или молекулярную рефракцию бензола, сделав предположение о существовании у него, в соответствии с формулой циклогексатриена, трех двойных и трех простых связей, то получится удовлетворительное совпадение с наблюдаемыми величинами. Например, наблюдаемая величина молекулярной рефракции бензола составляет:
М ("Ь°)2 — 1 78,11 1,50112—1
MRd “ d20 ’ (п20)2 + 2 - 0,8791’ 1,50112 4-2 -26’18
а вычисленная:
MRD = 2,418-64- 1,100-64- 1,733-3 = 26,31
Однако такое довольно близкое совпадение еще нельзя рассматривать как доказательство действительного присутствия трех двойных связей.
В самом деле, молекулярный объем и молекулярная рефракция являются мерой заполнения пространства веществом, как бы мерой объема, занимаемого молекулой. Но ведь расстояния между центрами углеродных атомов в бензоле (и в других ароматических соеди-о
нениях) представляют собой некоторое среднее (1,40 А) между расстояниями у простых связей С—С (1,54 А) и у двойных С=С(1,32'А). В таком случае в качестве меры объема, обусловленного наличием связей С—С в ароматическом ядре, надо брать некоторую среднюю величину инкремента рефракции между инкрементом для простой алифатической связи С—С (0,000) и для двойной связи С=С (1,733), т. е.
0,000 + 1,733 —----------- ^0,866
Если умножить эту величину на 6, по числу ароматических связей С—С, то получится, естественно, та же величина молекулярной рефракции для бензола, что и при предположении о наличии в его молекуле трех двойных связей.
Определение теплоты сгорания газообразного бензола дает величину (789 ккал) меньше вычисленной для гипотетического циклогексатриена, т. е. на основании формулы Кекуле (828,6 ккал). Это указывает, что бензол образуется с большим выделением энергии, чем можно было бы ожидать для соединения, отвечающего формуле циклогексатриена.
Величину этого превышения можно точнее экспериментально определить, используя тепловой эффект так называемой реакции
необратимого катализа Н. Д. Зелинского (стр. 58), когда циклогексен гладко превращается каталитически в смесь бензола и циклогексана. В самом деле, при этой реакции
сн2 сн2 сн Н(/ \н
Н2С сн н2с сн2
3 1 Н2С II сн -» 2 I Н2с 1 4~ сн2 1 || 4-36 ккал НС СН \ / сн
сн, чсн,
общее число всех связей С—С, С=С и С—Н остается неизменным, но три изолированные двойные связи переходят в систему трех сопряженных связей в молекуле бензола, в систему, приобретающую «ароматический» характер и большую устойчивость.
Таким образом, совокупность физико-химических характеристик бензола указывает на некоторый качественно особый характер имеющихся в нем углерод-углеродных связей. С точки зрения электронных представлений о природе химических связей этот особый характер, в частности насыщенность при формальной непределъ-ности, обусловлен специфическими свойствами сопряжения к-свя-зей в бензольном кольце.
Если в системе циклогексатриена учесть взаимное сопряжение всех к-электронов, выравнивающее С—С-связи, то при полном равенстве всех С—С расстояний не будет никаких оснований предпочитать у любогоС-атома сопряжение только с его правым или только с его левым соседом:
Необходимо допустить выравнивание состояния всей системы элект тронов, что может быть изображено схемой:
Такая схема будет выражать вероятность того, что к-электроны в данный момент находятся в некоторой области, представляющей
собой комбинацию двух соприкасающихся тел вращения, по форме приближающихся к торам* равной величины, с общей осью вращения. Такое геометрическое тело в перспективе и в проекциях разрезов имеет следующий вид:
Подобные представления в- известной степени объясняют особый, ароматический характер бензола и его производных. В отличие от линейной системы сопряженных связей, в которой повышенной способностью к присоединению обладают крайние атомы, в циклической системе сопряженных связей, какой является бензол, таких крайних атомов .нет, чем и объясняется замаскированность ненасыщенности ароматических соединений.
Активирование системы бензола в случае, например, присоединения (а не замещения) хлора достигается путем возбуждения молекулы квантами света. При каталитическом гидрировании активирование, вероятно, вызвано нарушением равномерности распределения электронной плотности в кольце (нарушением сопряжения связей) при адсорбции на катализаторах.
В молекулах, обладающих комбинациями а- и тг-связей, соответствующие пары углеродных атомов находятся во втором валентном состоянии (см. том I, стр. 119), характеризующемся наличием у такой пары атомов углерода пяти о-связей, которые, располагаясь в одной плоскости, образуют при каждом углеродном атоме три угла по 120°. При образовании циклогексатриена и при превращении его за счет сопряжения всех тг-связей в ароматическую систему бензола кольцо из шести углеродных атомов остается плоским. Из этих же представлений- вытекает и отличие шестичленного цикла бензола от гипотетического четырехчленного кольца циклобутадиена и от восьмичленного кольца циклооктатетраена. Для этих систем расположение всех тг-Связей в одной плоскости, что, согласно квантово-механическим представлениям, является обязательным
* Тор—тело вращения, образованное кругом, вращающимся около оси, лежащей в плоскости круга, но вне его.
условием полноты их сопряжения, было бы возможным только при значительной деформации валентных углов.
Так, система циклобутадиена должна была бы иметь плоские валентные углы, равные 90°. Однако это было бы возможно лишь при очень большой деформации связующих электронных облаков с изменением валентных углов на 30°. Между тем деформации валентных углов большей частью очень невелики и не превышают 5—10°. Поэтому система циклобутадиена существовать не может, и попытки получить такой углеводород приводили к получению вместо него двух молекул ацетилена:
НС—СН НС СН
II II III + III
НС—СН НС сн
Для полного выравнивания электронной плотности в кольце циклооктатетраена, при условии сохранения этого кольца плоским, валентные углы должны деформироваться примерно до 135°, т. е. почти на 15°. Такая деформация, очевидно, должна сопровождаться затратой энергии, т. е. возникновением некоторого напряжения, которое будет обусловливать меньшую прочность и устойчивость системы. Если же предположить, что система циклооктатетраена не является вполне плоской, то величины деформаций валентных углов могут быть и меньше, но тогда должна нарушиться равномерность распределения электронной плотности (иначе говоря, полнота сопряжения простых и двойных связей). Повидимому, у циклооктатетраена валентные углы несколько деформированы, но не настолько, чтобы все атомы находились в одной плоскости, и, следовательно, невозможно вполне равномерное распределение электронной плотности. Поэтому циклооктатетраен и не обнаруживает «ароматических» свойств, присущих системе бензола, хотя и отличается от циклоолефинов.
Для изображения ароматического кольца бензола продолжают пользоваться формулами Кекуле с двойными связями. Иногда указания на двойные связи вообще опускают, и бензол изображают просто шестиугольником.
Здесь мы будем пользоваться исключительно формулами с чередующимися двойными связями, причем при рассмотрении особенностей, обусловленных взаимным влиянием атомов, будем показывать смещение электронной плотности кривыми стрелками, указывающими направление этого смещения:
н
I
СН /ч С
нсх^сн (С% н—с^с-н
II I II7 ч
нсх<>сн • н-с^с-1-г
сн • с
I
и
В молекуле бензола электронная плотность совершенно равномерно распределена по отношению ко всем углеродным атомам кольца..
При вступлении в ароматическое ядро вместо атома водорода такого атома, который обладает свободными электронными парами (кислород, азот, галоиды), равномерность распределения электронной плотности нарушается
Так, в молекуле простейшего ароматического алкоголя—фенола—взаимодействие свободной электронной пары атома кислорода с системой сопряженных связей ароматического ядра вызывает дополнительное смещение электронной плотности, которое показано на схеме
Н
с
кривыми стрелками, изображенными вне кольца*.
Наличие взаимодействия именно в указанном направлении подтверждается гем, что дипольный момент фенола (ц= 1,40 D) значительно меньше дипольных моментов в ряду алифатических спиртов (для метилового спирта ц= 1,69 D), в молекулах которых такое смещение электронной плотности в направлении от атома кислорода к углеродным атомам ничем не может быть вызвано вследствие отсутствия к-связей.
В результате эффекта сопряжения электронная плотность в мо-• лекуле фенола оказывается повышенной в орто- и пара-положениях по отношению к гидроксильной группе.
Такое повышение электронной плотности мы будем обозначать знаком 8-.
Смещение электронной плотности в направлении от атома кислорода к углеродному атому бензольного ядра усиливает тенденцию
* В дальнейшем смещение электронной плотности в бензольном ядре, ведущее к ее выравниванию и изображенное выше кривыми стрелками внутри кольца, будет подразумеваться во всех случаях при изображении соединений ароматического ряда, хотя сами стрелки будут для простоты опускаться.
атома водорода гидроксильной группы фенола к отщеплению в виде протона и к образованию фенолят-иона:
Благодаря этому фенолы являются слабыми кислотами. Константа диссоциации фенола К25= 1,7 • 10-10, т. е. в 10 000 раз больше, чем у воды (/(= 1014); еще значительнее она превышает константы диссоциации алифатических спиртов.
В ароматических аминах взаимодействие свободной пары электронов атома азота с сопряженной системой ароматического ядра вызывает смещение электронной плотности в том же направлении, что и в случае фенола
h-nVh
вследствие чего электронная пара азота также входит в сопряженную систему и уже не является свободной.
Для того чтобы возникла возможность образования связи ме-?кду атомом азота и протоном какой-либо кислоты, электронная чара должна вновь перейти к атому азота, что требует затраты некоторого количества энергии.
Для аммиака и алифатических аминов такой затраты энергии не требуется, так как эти соединения не имеют двойных связей, с которыми электронная пара атома азота могла бы вступить в сопряжение. Действительно, в случае первичных алифатических аминов тепловой эффект солеобразования составляет 13 ккал, в то время хак при образовании хлоргидрата анилина тепловой эффект равен всего 7,4 ккал. Разность ^5,6 ккал, очевидно, и соответствует той затрате энергии, которая требуется для нарушения в анилине сопряжения ароматического ядра со свободной электронной парой азота.
Этим и объясняется ослабление основных свойств аминогруппы при вступлении ее в ароматическое ядро.
Эффект сопряжения в значительной мере обусловливает также повышенную прочность связи галоида в молекулах ароматических галоидопроизводных по сравнению с алифатическими галоидопроизводными, а также меньшую величину дипольного момента ароматических галоидопроизводных (например, р.= 1,83 D для СН3—С1 и ,J- — 1,70 D для С6Н5—С1).
ДИПОЛЬНЫЕ МОМЕНТЫ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ
Молекула бензола неполярна, что указывает на ее симметричность. Все однозамещенные производные бензола полярны, причем слабую, но ясно выраженную полярность обнаруживает уже толуол. Величины дипольных моментов производных бензола, содержащих разные заместители, в большинстве случаев отличаются от дипольных моментов соответствующих производных метана (см. табл. 14).
Таблица 14
Дипольные моменты производных метана и бензола
Производные метана Производные бензола
формула дипольный момент ц D формула дипольный момент ц D
СН8—СН, 0 С6Нв—сн. 0,37
СН3—С1 1,83 С6Н6—С1 1,70
сн3—он 1,69 С6Н5-ОН 1,40
C,H6-SH 1,56 CeH6-SH 1,33 (в бензоле)
ch's—NO, 3,54 СвН6— NO, 4,23
СН3—CN 3,94 CeH6—CN 4,39
СН3—СНО 2,69 СвН6—СНО 2,77
CHs-NH, 1,33 СвЩ—nh2 1,48
СН3—0—СНз 1,29 С-вЩ 0 Свнб 1,35
СН3—S—СН, 1,40 (в раств.) Св Н5—S С6Нб 1,50 (в бензоле)
Как видно из таблицы, у толуола имеется небольшой дипольный момент. Вопрос о направлении этого момента может быть решен следующим сопоставлением.
Дипольный момент n-хлортолуола (в бензольном растворе) равен 1,88 D, в то время как дипольный момент хлорбензола (также в бензольном растворе) составляет 1,570, т. е. он меньше, чем у п-хлор-толуола, на величину 0,31 D. Так как в диполе С—С1 отрицательный конец несомненно находится у атома галоида, направление дипольного момента n-хлортолуола соответствует схеме:
Сопоставление дипольных моментов нитробензола 4,01 D (в бензольном растворе) и п-нитрохлорбензола 2,55 D (также в бензольном растворе) показывает, что пониженный дипольный момент п-нитрохлорбензола объясняется противоположным направлением дипольных моментов отдельных полярных связей в соответствии со схемой
Меньшие величины дипольных моментов хлорбензола, фенола и тиофенола по сравнению с дипольными моментами соответствующих алифатических производных объясняются смещением свободных электронных пар атомов хлора, кислорода и серы в направлении к ароматическому ядру благодаря сопряжению с к-электронами ядра.
Это же обстоятельство вызывает сокращение расстояний С—С1, С—О и С—S и упрочнение таких связей в ароматических соединениях.
Повышенные дипольные моменты ароматических нитропроизводных по сравнению с дипольными моментами соответствующих алифатических соединений, наоборот, обусловлены смещением к-элек-тронов бензольного ядра по направлению к нитрогруппе, вызывающим некоторый добавочный момент, отсутствующий у алифатических соединений:
Аналогичные закономерности наблюдаются для ароматических нитрилов и альдегидов.
Надо, однако, заметить, что в более сложных случаях ограничиваться простым суммированием или вычитанием дипольных моментов отдельных связей нельзя,—приходится прибегать к векторному сложению, учитывая, например, углы между связями:
Q С
? А
Н НН
Дипольные моменты отдельных связей являются величинами направленными, а потому результаты их взаимодействия часто можно находить путем векторного сложения по формуле:
р- = у P-i + р-2 + 2p-rp-2*cos 9
где 6—угол между направлениями векторов.
Для двузамещенных ароматических соединений ряда бензола векторные суммы моментов будут выражаться приведенными ниже формулами*.
* Если дипольные моменты связей с обоими заместителями имеют одинаковое направление по отношению к углеродным атомам бензольного ядра, с которыми эти заместители связаны, то третий член подкоренного выражения входит со знаком (+) в случае орто-замещенных и со знаком (—) в случае мета- и пара-замещенных (соответственно значению cos 6). Если же дипольные моменты этих связей направлены в разные стороны, знаки меняются на обратные.
Если заместители различны:
для орто-двузамещенных р. = j/~ + р.| ± Р4-и2 для мета-двузамещенных pi = р^ + + р-гр-2 для пара-двузамещенных pt = + р.2
Если оба заместителя одинаковы:
для орто-двузамещенного pi = р.г -jZ-3
для мета-двузамещенного pi = р.х дл я пара-двузамещенного у = О
Формулы оказываются справедливыми в случае таких заместителей, для которых ось момента совпадает с линией, проходящей через углеродный атом в бензольном кольце и центральный атом замещающей группы. Такие заместители называются регулярными. К ним относятся: —СН3, —С1, —NO2, —CN и др. Для нерегулярных групп, у которых ось момента не совпадает с осью связи С—X (—ОН, —NH2, —NO, —ОСН3, —SH,—СООИ),надо вводить поправки на величины углов, образуемых осями их моментов и связей.
Приводим здесь (табл. 15) несколько примеров расчета дипольных моментов двузамещенных производных-бензола строения ХС6Н4СН3.
Таблица 15
Дипольные моменты двузамещенных производных бензола строения ХСбН4СН3
Заместитель X в орто-положении в мета-положении в пара-положении
, р. найд. [1 вычисл. р. найд. [X вычисл. р. найд. р. вычисл.
С1 . . . . 1,39 1,39 1,77 1,79 1,88 1,95
Вг ... 1,44 1,37 1,75 1,76 1,93 1,92
NO2 . . . 3,66 3,76 4,17 ‘ 4,16 4,44 4,35
NH2 . . . 1,57 1,74 1,43 1,46 1,27 1,25
он . . . 1,41 1,61 1,54 1,60 1,57 1,60
ПРАВИЛА ЗАМЕЩЕНИЯ В БЕНЗОЛЬНОМ ЯДРЕ
Если в бензольном ядре уже имеется замещающая группа, то положение, в которое вступает в ядро вторая замещающая группа, зависит от природы первого заместителя. Иначе говоря, замещающие радикалы направляют, или «ориентируют», вхождение второго заместителя. При этом имеется два типа заместителей. Заместители первого рода направляют последующее замещение в орто- и параположения, причем в большинстве случаев образуется смесь орто-и пара-изомеров, в которой иногда один из изомеров содержится в сильно преобладающем количестве. Мета-изомер в этих случаях
или не образуется вовсе, или образуется личестве.
Так, алкильные радикалы ориентируют положения. Поэтому, например, при действии получается смесь орто- и пара-изомеров:
в НИЧТОЖНОМ КО-
в орто- и параброма на толуол
сн3 п
I
Вг гьбромтолуол
сн3
о-бромтолуол
Атомы галоидов также относятся к заместителям первого рода. Так, при действии брома на хлорбензол или бромбензол получаются почти исключительно пара-изомеры:
С1 Вг
I I
или
I I
Вг Вг
Заместители второго рода направляют исключительно или почти исключительно в мета-положение. Имеются, впрочем, и такие заместители, при наличии которых могут получиться в значительных количествах все три изомера.
К заместителям, ориентирующим вновь вступающий заместитель преимущественно в орто- и в пара-положения, принадлежат:
С1, Вг, J, СН3, С„Н2п + 1, ОН, SH, ОСН3, SCH3, nh2
Это—заместители насыщенного типа, в большинстве случаев неспособные гидрироваться.
К заместителям, ориентирующим преимущественно в мета-положение, принадлежат:
NO2, СНО, СО—R, СООН, CN, SO3II
Это— заместители ненасыщенного типа, способные гидрироваться.
При наличии в ядре двух замещающих групп обе они могут оказывать ориентирующее влияние при вступлении следующих заместителей, причем одна из них может оказывать более сильное влияние, чем другая.
При наличии заместителей первого рода, ориентирующих в орто-пара-положение, реакции дальнейшего замещения идут значительно легче и быстрее, чем замещение атомов водорода в бензоле (исключение составляют галоиды). В случае же присутствия заместителей второго рода, ориентирующих в мета-положение, реакции Дальнейшего замещения идут труднее и медленнее, чем для самого бензола.
Необходимо также отметить, что, как показало детальное исследование реакционных смесей, фактически почти всегда образуются все три изомера, только в большинстве случаев или мета-изомер или орто- и пара-изомеры образуются в ничтожно малых количествах.
Голлеман, а позднее и многие другие исследователи изучали ориентирующее влияние заместителей в бензольном ядре с количественной стороны. Результаты этих исследований помещены в табл. 16, в которой изомеры расположены в порядке убывания их количеств. Цифрами, взятыми в скобки, обозначены изомеры, образующиеся лишь в очень небольших количествах.
Таблица 16
Ориентирующее действие заместите лей
Имеющийся заместитель в положении 1 Положение входящего заместителя
С1 Вг J so3H no2
С1 .... 4, 2, (3) 4 4 4 4,2
Вг .... 4, (2), (3) 4, (2), (3) — 4 4, 2 (3)
J 4, (2), (3) 4 4 4 4, 2
ОН ... . 4, 2 4, 2 4, 2 2, 4 (3) 2, 4
nh2 ... 4, (2) — 4 2, 4 4, (3), (2)
сн3 ... 4, 2 4, 2 4, 2 4, 2 (3) 2, 4 (3)
SO3H . . . — 3 — 3, (4) 3, (2), (4)
no2 ... 3 3 — 3, (2), (4) 3, (2), (4)
соон . . з, 2J4) 3 3 3J4) 3, 2, (4)
CN .... — —- 3
сно . . . 2, 3 — — — 3, 2
СН3СО . . — — — — 3, 2, 4
Иногда различают реакции прямого и непрямого замещения. При наличии таких орто-пара-ориентирующих заместителей, как Cl, Вг, J, СН3, или мета-ориен-тируюших заместителей второго рода (NO2, СО, СНО, COR, СООН, CN, SO3H) происходит прямое замещение атомов водорода бензольного ядра (при сульфировании, нитровании и др.).
При наличии же некоторых заместителей первого рода (ОН, NH2, NHBr) иногда сначала замещается атом водорода в ориентирующей группе, и лишь затем совершается перемещение нового заместителя в ядро, например:
HNO3
C6H6-NHa------> [CeH6-NH3]NO3 -> CeH5—NH—NO2 -> H2N-C6H4-NO2
(смесь орто- и пара-изомеров)
Объяснение преимущественной ориентации при введении второго заместителя в однозамещенный бензол следует искать в нарушении равномерности распределения электронной плотности, происходящем в результате вступления первого заместителя в молекулу бензола. Направление этого смещения электронной плотности в случаях наличия в бензольном ядре заместителей первого и второго рода различно.
При наличии в ядре таких заместителей первого рода, как ОН и NH2, эффект сопряжения приводит к повышению электронной
плотности в орто- и пара-положениях:
Избыточная электронная плотность в этих положениях отмечена знаками 8—.
В случае наличия в ядре атома галоида, также являющегося орто-пара-ориентантом, эффект сопряжения осложняется вследствие высокого сродства атома галоида к электронам.
Если в ядре имеется заместитель второго рода, эффект сопряжения сказывается в общем понижении электронной плотности в ядре, особенно значительном в орто- и пара-положениях (атомы углерода с наиболее низкой электронной плотностью отмечены знаками 8+):
Таким образом, при наличии в ядре заместителей второго рода электронная плотность в мета-положениях выше, чем в орто- и пара-положениях, хотя и не является избыточной.
Ориентация второго заместителя, вступающего в бензольное ядро, в котором равномерность распределения электронной плотности нарушена под влиянием уже имеющегося заместителя, зависит от характера атакующего реагента. Если атакующий реагент несет положительный заряд на атоме, вступающем в связь с углеродным атомом бензольного ядра, то новый заместитель, естественно, будет направляться к углеродному атому с наибольшей электронной плотностью. Отрицательно же заряженные атакующие группы будут направляться к тем углеродным атомам бензольного ядра, у которых электронная плотность понижена по сравнению с остальными.
Наиболее важными для практики реакциями замещения являются сульфирование, нитрование и хлорирование. При всех этих реакциях атакующие реагенты несут на атоме, вступающем в связь с углеродным атомом бензольного ядра, положительный заряд *
4- 4 4-
—SO3H —NO2 —Cl
и могут образовывать новую связь только за счет электронной пары этого углеродного атома. Подобные реагенты называют электрофильными, катионоидными или электроноакцепторными.
Естественно, что электрофильные реагенты, приближаясь к атакуемым молекулам, будут направляться к тем атомам (центрам), на которых сосредоточена наибольшая электронная плотность. Именно поэтому при атаке производных бензола, содержащих замес! ители первого рода, электрофильные реагенты будут вступать в орто- и пара-положения, а при наличии заместителей второго рода— в мета-положение (см. стр. 23б сл.).
Реакции замещения, в которых участвуют электрофильные атакующие реагенты, носят название реакций электрофильного замещения.
Гораздо реже встречаются и менее изучены реакции нуклеофильного замещения. Эти реакции протекают под действием несущих отрицательный заряд нуклеофильных, или анионоидных, реагентов, например гидроксил-иона. Вступающая в ароматическое ядро нуклеофильная гидроксильная группа, как и следовало ожидать, направляется заместителями первого рода к углеродным атомам, обладающим наименьшей электронной плотностью, т. е. в мета-положение, а заместителями второго рода—в орто- или пара-положения. Действительно, нитробензол реагирует с едким кали с образованием о-нитрофенолята (А. Воль); фенол образует мета-производное—резорцин и др.
Для проявления эффекта ориентации не требуется очень больших изменений электронной плотности, так как этот эффект сводится» повидимому» к конкуренции трех возможных реакций замещения (орто-, мета- и пара-замещения), скорости которых значительно отличаются. Поскольку для того, чтобы одна какая-либо реакция шла в 10 раз быстрее другой (отношение количества изомеров 10 : 1), достаточно разницы в энергии активации всего лишь на -^1,5 ккал (моль* *,—уже небольшие нарушения симметрии электронного облака в ароматическом ядре могут вызывать резкое изменение скоростей замещения в различных положениях, т. е. эффект ориентации. Так, например, постепенное накопление в боковой цепи атомов или групп с повышенным сродством к электронам может существенно изменить направление ориентации при нитровании толуола, благодаря относительному увеличению скорости мета-замещения:
а-Замещенные
толуолы ......... С6Н6—СН3 СвНв—СН,С1 СвН6—СНС19 С<НБ—СС13
Выход мета-нигропро-
изводного........ 4 12 33 48
При наличии в ядре различных заместителей для качественного сравнения их ориентирующего влияния пользуются следующим приемом: в двуза’мещенное производное бензола вводят третий заместитель и определяют относительные количества образующихся
___Е__
* Скорость реакции пропорциональна е » где Е—энергия активации»
обычно составляющая 20—25 ккал/моль.
продуктов. Например, при нитровании n-хлортолуола, получается больше 1 -метил-4-хлор-5-нитробензола, чем 1 -метил-4-хлор-6-ни-тробензола:
CH3-£j-C1 у СН3—С1
\\Оа Оам/
Это значит, что хлор ориентирует вступающую нитрогруппу сильнее, чем метильная группа.
Таким путем можно придти к рядам, характеризующим' (приближенно) направляющее действие различных заместителей.. Например, направляющее действие орто-пара-ориентирующих Заместителей убывает в следующем порядке:
ОН > NH2 > галоиды > СН8 а для мета-ориентирующих заместителей найден ряд:
N(CH3)3 > СООН > SO8H > NO,
Галоидные производные
Способы получения. 1. Действие галоидов на ароматические углеводороды, Галоидные производные ароматических углеводородов могут быть получены, при непосредственном действии галоидов (хлора и брома) на углеводороды (ср. том I, стр. 176). При этом характер продуктов реакции весьма сильно зависит от условий ее проведения. .
При низких температурах, особенно в присутствии катализаторов, галоид замещает водородные атомы бензольного ядра. Наиболее обычными катализаторами являются: галоидные соединения металлов, особенно алюминия и железа, иод, галоидная сера, галоидная сурьма. Подбирая условия реакции и катализатор, можно последовательно заменить все водородные атомы бензольного ядра на хлор и бром. Например, в присутствии бромного железа или иода можно легко получить монобромбензол и дибромбензол:
С6Н6 + Вга -» СвН5Вг + НВг
С6Н6 + 2Вг2 —> СвН4Вга + 2НВг
Самыми сильно действующими катализаторами’являются галоидные соли алюминия, в присутствии которых на галоид замещаются все водородные атомы бензольного ядра, например:
С6Нб + бС12 С6С18 + 6НС1
В гомологах бензола с одной боковой цепью в этих условиях замещаются пять атомов водорода бензольного ядра, например:
С6Н5-СН3 + 5Вг2 -> СбВг5-СН3 + 5НВг
В углеводородах с двумя боковыми цепями замещаются четыре атома водорода, например из ксилолов С6Н4(СН3)2 получаются те-трабромксилолы С6Вг4(СН8)2, и т. д.
При действии хлора и брома на гомологи бензола в отсутствие катализатора в газовой фазе при повышенной температуре замещение происходит почти исключительно в боковой цепи. Реакция хорошо протекает только на свету (прямые солнечные лучи или яркая лампа). Так, при пропускании хлора в пары кипящего толуола в этих условиях можно последовательно заменить на хлор все три атома водорода метильной группы и получить следующие три соединения: хлористый бензил С6Н5—СН2С1; хлористый бензилиден, или бензальхлорид, С6Н5—СНС12; хлористый бензенил, или бензотрихлорид, СвН6—СС13. Бензилом называется одноатомный радикал QH-—СН2—, бензилиден— двухатомный радикал С6Н5—СН<, бензенил—трехатомный радикал С6Н5—.
2. Введение галоида путем замены аминогруппы. Галоидные производные, содержащие галоид в ядре, могут быть получены путем обмена на галоид аминогруппы ароматических аминов через диазосоединения (стр. 321).
Этим способом можно синтезировать также и те изомеры, которые не удается получить путем непосредственного действия галоидов на углеводороды. Йодистые производные особенно легко получаются именно из ароматических аминов.
3. Введение галоида в боковую цепь. Галоидные соединения, содержащие галоид в боковой цепи, могут быть синтезированы всеми способами получения галоидных алкилов (см. том I, стр. 177 и сл.), например действием галоидоводородных кислот на соответствующие спирты:
CtHs—СН2ОН 4- HJ —» CeH5—CHtJ + Н,0
Йодистые соединения могут получаться из хлорпроизводных действием йодистого калия:
С6Н5—СН2С1 + KJ -> СвНб-СНв1 + КС1
4. Хлорметилирование. Ароматические хлорпроиз-водные, содержащие галоид в боковой цепи, могут быть получены методом так называемого хлорметилирования. При обработке смеси ароматических углеводородов и параформальдегида (или 40%-ного формалина) хлористым водородом в присутствии хлористого цинка или хлорпого олова атом водорода в ароматическом ядре замещается на группу —СН2С1 (Блан), например:
СвНв + crto + НС1 —> cf46— CH/1 + II2O
Физические свойства. Галоидные производные ароматических углеводородов представляют собой жидкости или твердые тела с удельным весом больше единицы, нерастворимые в воде, сравнительно легко растворяющиеся в спирте, эфире и бензоле. Моногалоидные и дигалоидные соединения обладают сильным запахом и несколько ядовиты; их низшие представители, содержащие атом галоида в боковой цепи, раздражающе действуют на слизистые оболочки.
Химические свойства. Соединения, в которых галоид находится в бензольном ядре, по химическим свойствам очень сильно отличаются от соединений, содержащих галоид в боковой цепи. Галоид, находящийся в боковой цепи, обладает не меньшей, а иногда и большей подвижностью, чем в галоидных алкилах, вступая во все свойственные им реакции, например:
СбН5-СП2С1 4- Н2О -» С6Н5—СН2ОН + НС1
С6Н5—СН2С1 4- KCN —» С6Н5—СН2—CN 4-КС1 и т. Д.
Галоидные производные бензола и другие соединения, содержащие галоид в ядре, с большим трудом вступают в реакции обмена. Лишь при высоких температурах и давлении эти реакции идут легко.
Большая прочность связи галоида с углеродом в ароматических соединениях объясняется взаимодействием свободных электронных пар атома галоида с ^-электронами ароматического ядра. По инертности галоида такие соединения аналогичны винилгалогенидам (ср. том I, стр. 384). Лишь в реакции с металлами, например с натрием или с магнием в эфирном растворе, эти соединения вступают почти так же легко, как галоидные алкилы, причем в первом случае получаются углеводороды (см. реакции Вюрца—Фиттига, стр. 212), а во втором—смешанные магнийоргапические соединения:
C6H5-Br + Mg -» c6Ha-MgBr
Однако Ульману* удалось найти катализатор, в присутствии которого при повышенных температурах обмен ароматически связанного галоида происходит легко. Это позволило осуществить ряд синтезов (аналогичных синтезам с галоидными алкилами), нашедших промышленное применение. Таким катализатором является мелкораздробленная металлическая медь—так называемая медная бронза, или бронзировальный порошок.
Благодаря реакциям Ульмана возникла возможность введения чисто ароматических радикалов в молекулы с подвижными атомами водорода или замещающими их атомами металла. Такие реакции носят название реакций арилирования.
Приведем несколько примеров реакций арилирования по Ульману:
С6Н5—Вг + NaOH -» СбН5—OH + NaBr
C6H5-J 4- KCN С6Н3—CN + KJ
C6H5—J 4-NaOC6H5 —> C6H5-O—C6H5 + NaJ
C6H5-J4-NH3 -> C6H3—NHa -f- HJ
И T. Д.
* Фриц Ульман (1875—1920). Немецкий химик. Применил для алкилирования диалкилсульфаты. Систематически исследовал реакции арилирэвання.
Таблица 17
Галоидные производные бензола и толуола
Название Формула Температура плавления Температура кипения Удельный вес Показатель преломления „20
°C °C 4 nD
Фторбензол CeH6F —39,2 85,2 1,0236 1,4677
Хлорбензол —45,2 132,0 1,1062 1,5246
Бромбензол С6Н5Вг —30,6 156,2 1,4952 1,5602
Иодбензол C„H5J —31,4 188,7 1,8307 1,6197
Дихлорбензол: с}нс412 —16,7 180,3 1,5476
орто-, или 1,2- 1,3064
(при 25°)
мета-, или 1,3- —24,4 171 1,2881 1,5457
пара-, или 1,4- +53,2 174,4 1,2264 (при 20,9°)
Дибромбензол: СбН4Вг2 223 \ 4 ) 1,6081
орто-, или 1,2- +6,7 1,956
«4)
мета-, или 1,3- —6,7 218 \ 4 J 1,9523 1,6087 (при 17°)
пара-, или 1,4- +87,3 220,2 1,8322 1,5743 (ПРИ 99,3°)
Гексахлорбензол ад 229,5 322,2 \ 4 J —
Г ексабромбензол 316 (возг.) — — —
Хлортолуол: СН3С6Н4С1 159,2 1,5248
орто-, или 1,2- —36,5 1,0824
мета-, или 1,3- —47,8 161,6 1,072 1,5214
пара-, или 1,4-Бромтолуол: СН3СвН4Вг +74,3 —28,1 162,0 181,8 1,070 1,4173 1,5199
ортог, или 1,2- —
мета-, или 1,3- —39,8 183,7 Г 4 ) 1,4099 —
пара-, или 1,4- +26,7 185,0 1,3898 (переохл. —
жидк.)
Пе нтабр омтол у ол CH3CeBrs 288 — — —
Хлористый бензил СвН6СН2С1 —39,0 179,4 \0994 1,5391
(бензилхлорид, а-хлортолуол) С„Н,СН,Вг ' — 3,9 ; 198 1,4380 (при 17,4°)
• омистый бензил —
(бензилбромид, а-бромтолуол) • : • • +24,1 ('?)
Йодистый бензил ! CeH6CH2J 93 1,7335
(бензилиодид, (при 10 мм) (rfo5)
а-иодтолуол) Хлористый бензилиден аденец —17,4 205,2 1,2557 1,5515
(бензальхлорид) Ю (При 19.4е)
БензотТрйхлорид адссц- •;-г—’4,8 220,6 1,3723 1,5573
Соединения многовалентных галоидов. Иодбензол и другие соединения с иодом в бензольном ядре обладают способностью присоединять два атома хлора с образованием кристаллических соединений, называемых иодидхлоридами, например хлористый иодбензол, или фенилиодидхлорид СвН5—JClg- При действии едких щелочей иодидхлориды обменивают хлор на кислород
CeH5—JC12 + 2KOH —> CeHe—J=O + 2КС1
При этом образуются твердые желтые иодозосоединения, например иодозобензол Се Н5—J—О.
Окислением иодозобензола хлорноватистой кислотой или окислением иод-бензола надсерной кислотой получается соединение СвН5—JO2, называемое иодо* бензолом. Иодобензол и иодозобензол являются сильными окислителями. Иодобензол при нагревании взрывает.
При действии влажной окиси серебра на смесь иодозобензола и иодобензола получается основание, называемое (по аналогии с аммониевыми соединениями) гидроокисью дифенилиодония-.
>0 Г СбН5Ч 1+
CeH5—J=O + C6H.—К + AgOH —> AgJOo + >j ОН*
L Свн/ J
Гидроокись дифенилиодония с иодистым калием дает трудно растворимый s воде и спирте иодистый дифенилиодоний
свн5х 1+
>J Г L Свн/
Это—солеобразное вещество, способное обменивать один атом иода по типу реакций обменного разложения. При нагревании он распадается на две молекулы иод-бензола. Соединения дифенилиодония напоминают по свойствам соединения закиси таллия.
Аналогичные соединения с многовалентным атомом галоида дают и бромзаме-щенные ароматические соединения, например бромидхлоридьк типа CeH6BrCL.
Нитросоединения
Нитросоединения с нитрогруппой в бензольном ядре
Способы получения. Одним из характернейших свойств арома* тических соединений является их способность при действии азотной кислоты легко выделять воду с замещением атомов водорода в бензольном ядре на нитрогруппу:
с,н5-н + но—no, —> C0He—NO, + Н,0
Для нитрования ароматических углеводородов обыкновенно требуется концентрированная азотная кислота, иногда дымящая азотная кислота (уд. вес 1,52). Часто пользуются также нитрующей смесью, т. е. смесью азотной и серной (иногда дымящей) кислот.
Действующим началом нитрующих смесей является катион нитрония, образующийся при действии серной кислоты на азотную:
HONO,j+ H,SO4 N02+ 4- HSOf 4- НО (Ганч)
или
HONO, 4-2H,SO4 NO^ 4-2HSO7 4-Н,0+ (А. И. Титов)
Собственно процесс нитрования ароматических углеводородов, например бензола, протекает в две фазы:
СбНб + NO+ —
no2
—> c6h5no2 + н+
Н+ + HSO7 H2SO4
При этом комплекс углеводорода с нитроний-катионом образуется сравнительно медленно, но перегруппировка его в нитросоединение происходит быстро. Понятно, что накопление воды в нитрующей смеси приводит к уменьшению концентрации ионов нитрония и постепенному ослаблению нитрующего действия смеси кислот.
Некоторые ароматические соединения, например фенолы, легко нитруются разбавленной азотной кислотой. Действующим началом в этих случаях являются окислы азота (А. И. Титов):
АгН + 2NO2 ArNO2 + HNO2 HNO2+HNO8 —» H2O + 2NO2
’ Установлено, что реакция не протекает, если в реакционной смеси отсутствует азотистая кислота, необходимая для регенерации NO2. Как видно из схемы реакции, азотистая кислота играет роль катализатора.
Нитрование протекает тем легче, чем больше боковых цепей содержит углеводород. Так как алкильные группы направляют в орто-и пара-положения, то, например, при нитровании толуола получается смесь о- и и-нитротолуолов. В зависимости от условий нитрования относительные количества орто- и пара-изомеров в этой смеси различны.
Нитрогруппа относится к числу заместителей, сильно ориентирующих в мета-положение. Поэтому, при дальнейшем нитровании мононитропродуктов, вторая нитрогруппа входит почти исключительно в мета-положение, и при нитровании, например, мононитробензола получается jw-динитробензол. Третья нитрогруппа вступает лишь с большим трудом и притом также в мета-положение, так что из бензола получается симметрический тринитробензол, а окончательным продуктом нитрования толуола является 1-метил-2,4,6-три-нитробензол, называемый обычно 2,4,6-тринитротолуолом или тротилом:
NO2 симм. тринитробензол
no2
1-метил-2,4,6-тринитробензол (тротил)
Больше трех нитрогрупп в молекулы бензола и толуола нитрованием ввести не удается.
Свойства. Мононитросоединения представляют собой жидкости или твердые тела очень слабо желтоватого оттенка, нерастворимые в воде, обладающие сильным запахом горьких миндалей. Они довольно ядовиты.
Полинитросоединения—желтые кристаллические вещества. Некоторые из них обладают также довольно сильным запахом. Полинитросоединения взрывчаты.
Таблица 18
Ароматические нитросоединения
Название Формула Темпера! ура плавления °C Темпера’ура кипения °C Удельный вес ,20 Показа гель преломлен и - п20
I1итробензол С6н6\о2 4-5,70 210,9 1,2033 1,5523
Динитробензол CeH,(NO2)2
орто-, или 1,2- + П7 319 <при 773 мм] — —
мета-, или 1,3- -r89,8 303 (при 770 мм) — —
пара-, или 1,4- 4-172 299 (при /77 мм) — —
Тр и нитробензол CeH3(NO.)3
симметрический + 122,5 — — —
(1,3,5-)
Нитротолуол CI13C6H4N('2
орто-, или 1,2- —3,2 с 1абильн.) —9,3 лаб льн. 221,7 1,1626 1,5467
мета-, или 1,3- + 16,1 232,6 1,1571 —
пара-, или 1,4- +51,6 238,4 1,1226 1,5382
(при 55°; (при 15 мм)
2,4 -Д и нитротолуол CH3CeH3(NO2)2 +70,1 — — —
2,4,6-Тринитрото- CH3CeH2(NO2)3 +80,1 — — —
луол
Фенилнитрометан C6HgCH2NO2 225—227 (с разл.) 135 (при 25 мм) 1,1596 1,5323
Нитросоединения с нитрогруппами в бензольном ядре представляют собой совершенно нейтральные вещества, нерастворимые ни в щелочах, ни в кислотах.
1. Реакции восстановления. Наиболее важными реакциями нитросоединений являются реакции восстановления нитрогруппы. Благодаря возможности восстановления нитрогруппы,
реакция нитрования имеет важное практическое значение и применяется в технике для получения многочисленных ароматических соединений разных классов из ароматических углеводородов.
Окончательными продуктами восстановления нитропроизводных бензола и его гомологов являются ароматические амины:
CeH6-NO2 + ЗН2 CeHs-NHa4- 2Н2О
Подбирая различные восстановители и соответствующие условия реакции, можно получить следующие продукты восстановления нитрогруппы (стрелки показывают последовательность, в которой могут происходить превращения):
р-мо2
н и т роп рои з водное
R-NO---------
нитрозо- ‘
производное
R—NHOH -------
р- арилгидроксиламин
----- -------------
I
О
азокси-производное w
R—N=N—R
азопроизводное
R-NH2 ---- R-HN-NH-R^
ариламин
гидразо-
производное
При восстановлении нитросоединений, кроме перечисленных выше веществ, могут получаться также продукты дальнейшего превращения промежуточных соединений, например n-аминофенол, образующийся из фенилгидроксиламина, бензидин—из гидразобензола и др.
Любые продукты восстановления нитросоединений могут быть получены также электролитическим путем при соответствующих условиях электролиза.
Изменяя потенциал катода путем подбора соответствующего катодного материала, плотности тока, температуры, pH электролита и т. п., можно в значительной мере влиять на процесс восстановления и получать различные продукты реакции. Например, при восстановлении нитробензола в кислой среде можно последовательно получать продукты различной степени восстановления: нитрозобензол -♦ фенилгидроксиламин -* анилин.
Более глубокое восстановление достигается при более отрицательных потенциалах катода, для чего электролиз ведут при больших плотностях тока на катодах с большим водородным перенапряжением. Например, на цинковых, свинцовых, ртутных катодах легко образуется амин. При менее отрицательных потенциалах, достигаемых при меньших плотностях тока на катодах с малым перенапряжением водорода (например, на платине, никеле, угле), получаются продукты менее глубокого восстановления—фенилгидроксиламин и бензидин.’
Существенное влияние оказывает также pH электролита. Например, при восстановлении нитробензола в щелочном электролите в качестве конечного продукта получается не анилин, а фенилгидрзксиламин. Кроме того, от pH зависит направление вторичных чисто химических реакций, протекающих между продуктами восстановления и исходным нитросоединением. Так, в щелочном растворе в результате взаимодействия фенилгидроксиламина с нитрозобензолом обра
зуется исключительно азоксибензол, в кислых же растворах фенилгидроксил-эмин легче переходит в п-аминофенол.
При восстановлении полинитросоединений могут быть созданы такие условия, что восстанавливаться будет только одна из нитрогрупп. Так, например, восстановлением л«-динитробензола сернистым аммонием, взятым в количестве, недостаточном для полного восстановления, можно получить л«-нитроанилин:
NO
Восстановление ароматических нитросоединений относится к числу хорошо разработанных процессов органической химии.
2. Реакции замещения. Водородные атомы бензольного ядра нитросоединений могут замещаться на галоиды или на сульфогруппу, но реакции замещения протекают гораздо труднее, чем для самих углеводородов. При этих реакциях заместитель вступает почти исключительно в мета-положение. Например, при бромировании нитробензола получается м-бромнитробензол, а при сульфировании—нитробензол-м-сульфокислота:
NO,
NO,
4/\so,h
3. Введение оксигруппы в нитросоединения. В л<-динитросоединениях, и в еще большей степени в симметрических тринитросоединениях, атом водорода, соединенный с углеродным атомом, находящимся между двумя нитрогруппами, приобретает способность окисляться в гидроксильную группу, например:
zno2
o2n—+ О \no2
/NO,
—> O,N—Ч—ОН 4no,
4. Замена нитрогруппы. В мононитросоединениях яитрогруппа связана очень прочно. В орто- и пара-динитросоединениях нитрогруппы, благодаря их взаимному влиянию, приобретают большую подвижность, чем в мононитросоединениях. Эта подвижность настолько велика, что у нитрогрупп возникает способ-
ность к сравнительно легкому нуклеофильному замещению, например:
NO, I
+ NaNO2
Как только одна из нитрогрупп будет замещена, вторая нитрогруппа теряет способность вступать в реакции обменного разложения, так как она больше не испытывает влияния заместителя второго рода.
Нитробензол С6Н5—NO2 производят в громадных количествах на химических заводах как полупродукт для получения анилина (стр. 305) и ряда других веществ. Раньше он применялся как дешевое парфюмерное средство под названием «мирбанового масла» или «мирбановой эссенции» главным образом для мыл (казанское мыло).
О- и n-Нитротолуолы, получаемые нитрованием толуола, разделяют вымораживанием я-нитротолуол а из смеси после предварительного ее фракционирования. Оба вещества имеют большое значение как исходные продукты для получения толуидинов (стр. 297) и как промежуточные продукты в производстве красителей и других ароматических соединений.
Нитропроизводные некоторых высших гомологов бензола получают в технике (в виде смеси изомеров) для восстановления в соответствующие амины.
2,4,6-Тринитротолуол (тротил, тол) является одним из важнейших бризантных взрывчатых веществ. При окислении тротила получается тринитробензойная кислота.
1,2,3,5-Тетранитробензол. Нитрованием не удается ввести больше трех нитрогрупп в ароматическое ядро, а потому тетранитробензол можно получить только косвенным путем.
Действуя на 1,3,5-тринитрофенетол гидроксиламином, получают пикрилгидроксиламин, который окисляют дымящей азотной кислотой:
Вещество кристаллизуется в желтых иглах, темп, плавл. 125—126°. Нитрогруппа, расположенная между двумя другими группами NO2, крайне подвижна; поэтому под действием разбавленных щелочей тетранитробензол превращается в пикраты, а действием аммиака—в пикрамид.
Тринитро-шреш-бутилтолуол (желтоватые иглы, темп, плавл. 97°) сн3
°2N\A/N°2 ^АС(снз)2
no2
благодаря своему сильному мускусному запаху находит значительное применение в парфюмерии под названием искусственного мускуса. Еще более ценный искусственный крезольный, или амбровый, мускус (темп, плавл. 85°)
СН3
°2N\X/N°2
СН3оА^
С(СН3)2
готовится из метилового эфира м-крезол а действием на него третичного хлористого бутила в присутствии хлористого алюминия и нитрованием полученного метило-гого эфира трет-бутил-л-крезола.
Так называемый кетонный мускус представляет собой 1,3-диметил-5-трет-бутил-2-ацетил-4,6-динитробензол; он получается ацетилированием 1,3-диметил-5-трс/ц-бутилбензола хлористым ацетилом в присутствии хлористого алюминия и последующим нитрованием получающегося кетона. Выяснению многих вопросов строения искусственных «мускусов» особенно способствовали работы А. Е. Чи-чибабина и Б. М. Дубинина.
ДВОЙНЫЕ СОЕДИНЕНИЯ ПОЛИНИТРО! 1РОИЗВОДПЫХ
Ди- и тринитропроизводные ароматических углеводородов (а также фенолов, хинонов и некоторых других ароматических соединений) обладают способностью образовывать со многими другими представителями ароматического ряда хорошо кристаллизующиеся двойные соединения, более интенсивно окрашенные и более трудно растворимые, чем исходные вещества. Такие соединения (их состав обычно отвечает мольным соотношениям 1 : 1 или 1 : 2) часто используют для идентификации, а иногда и для выделения индивидуальных веществ из смесей.
Примерами могут служить динитробензолаты—соединения м-ди нитробензол а
с анилином ............ красное (кристаллы, темп, плавл. 41°)
с диметил-п-*голуидином . черное (кристаллы, темп, плавл. 43")
н тринитробензолаты—соединения 1,3,5-тринитробензола
с нафталином ..........
с антраценом ..........
с анилином ............
с диметиланилином . . .
желтоватое (темп, плавл. 152,5s) яркокрасное (темп, плавл. 164°) оранжевое (темп, плавл. 123—124 ) темнофиолетовое (темп, плавл. 106—108)
Аналогичные соединения образует тринитротолуол (так называемые троти-латы), 2,4,6-трипитрофенол (пикраты, стр. 273), 2,4,6-трииитрорезоршш (стифнаты, стр. 341).
Можно предполагать, что такие продукты образуются за счет электроста-1ИЧССКОГО взаимодействия сильно полярных питрогрупп одного компонента с легко поляризующимися ароматическими или другими системами сопряженных связей второго компонента.
Нитросоединения с нитрогруппой в боковой цепи
Такие нитросоединения получаются обычными способами синтеза алифатических нитросоединений, например действием нитрита серебра на соответствующие галоидные соединения:
С6Нб—-СНаС1 + AgNOa -> СоНв-CH2NO2-f- AgCl
При действии на гомологи бензола разбавленной азотной кислоты под давлением при повышенной температуре может происходить нитрование в боковой цепи, а не в ядре (М. И. Коновалов), например:
СвНв—СН2—СН3 + НО—NO2 —> CeH6-CH(NO2)—сн3 + Н2О
Нитросоединения, у которых нитрогруппа находится в боковой цепи, обладают всеми свойствами алифатических нитросоединений. При их восстановлении получаются амины, например из фенилнитрометана С6Н5—CH2NO2 образуется бензиламин С6Н5—CH2NHa.
таутомерия нитросоединений с нигрогрупгюй^в боковой цени
'Первичные и вторичные нитросоединения, растворяясь в щелочах, дают соли изонитросоединений (см. том I, стр. 246—247). Явления таутомерии нитро- и изонитросоединений удалось особенно хорошо изучить на примере фенилнитрометана (Ганч), так как фенилизонитрометан обладает особенной устойчивостью и его удалось выделить в чистом состоянии в виде кристаллического вещества (темп, плавл. 84° при быстром нагревании), обладающего свойствами сильной кислоты.
Таутомерные формы веществ, которые могут существовать в двух состояних—нейтрального соединения и кислоты, часто называют псевдокислотами и аци-формами псевдокислот. Таким образом, сам фенил нитрометан является псевдокислотой, а фенилизонитрометан—его аци-формой:
фенилнитрометан
фенилизонитрометан
В растворах происходит превращение аци-формы в псевдокислоту, которое можно весьма удобно проследить на примере д^-нитрофенил нитрометана:
СН2—NO2
^\no.
лг-Нитрофенилнитрометан бесцветен, тогда как его аци-форма и ее соли интенсивно окрашены в желтый цвет. Если к соли аци-формы прибавить эквивалентное количество соляной кислоты, то желтая окраска постепенно исчезает (по мере превращения аци-формы в истинное нитросоединение).
Превращение псевдокислот и псевдооснований в соли изомерных им истинных кислот и оснований в большинстве случаев можно обнаружить по явлению медленной нейтрализации, объясняющейся тем. что такие превращения совершаются с измеримой скоростью. Обратные превращения можно проследить по уменьшению электропроводности. Кроме того, взаимные превращения истинных и изонитросоединений можно исследовать, измеряя показатели некоторых их физических свойств, например молекулярную рефракцию, которая у таутомерных форм различается на довольно значительную величину.
Г а лоиднитросое динения
При действии галоидов на нитросоединения получаются мета-галоиднитросоединения. Так как галоиды ориентируют в орто- и пара-полсжения, то при нитровании галоидбензолов получаются орто- и пара-галоиднитросоединения. Например, при нитровании хлорбензола сначала получаются о- и п-хлорнитробензолы:
С1
о-хлорнитробензол (темп, плавл. 32,5°)
NO, л-хлорнитробензол (темп, плавл. 83°)
а при дальнейшем нитровании —динитрохлорбензольг.
С1
NO,
2,6-динитрохлорбгнзол
2,4 динитрохлорбенз >л* (главный продукт)
2,4-Динитрохлорбензол применяется для получения сернистых красителей.
♦ Обыкновенная форма—ромбические кристаллы—плавится при 53,4’-Существует также в неустойчивой форме с темп, плавл. 43®.
Окончательным продуктом нитрования хлорбензола является 2,4,6-тринитрохлорбензол, или пикрилхлорид (почти бесцветные иглы, темп, плавл. 83°)
С1
°2N\A/N°2 и
no2
Присутствие нитрогрупп в орто- и пара-положениях (но не в мета-положении) к галоиду сообщает последнему подвижность, т. е. способность легко вступать в реакции нуклеофильного замещения (стр. 242), например:
С1 ОК
1 I
4-2КОН -> 0 +КС1 + Н2О
bJO2 NO,
Аналогично этому, хлор в zz-нитрохлорбензоле может замещаться на аминогруппу (стр. 298).
Особенно подвижен хлор в пикрилхлориде. Подобно хлорангид-ридам (см. том I, стр. 275), пикрилхлорид реагирует даже с водой, обменивая хлор на гидроксил с образованием тринитрофенола (пикриновой кислоты).
Легкость обмена галоида в ароматических галоиднитросоединениях под действием нуклеофильных реагентов объясняется влиянием нитрогруппы. Являясь заместителем второго рода (стр. 239), нитрогруппа вызывает смещение электронной плотности в ароматическом ядре, приводящее к наибольшему понижению ее в орто-и пара-положениях (по отношению к нитрогруппе), например:
ь
ОН' С1
При атаке нуклеофильным реагентом отрицательно заряженная группа, естественно, устремляется к углеродному атому с пониженной электронной плотностью. Поэтому нуклеофильное замещение галоида в галоиднитробензоле легко осуществляется только для орто- и пара-производных, а для мета-производных происходит значительно труднее.
Легкость нуклеофильного замещения галоида в галоиднитросоединениях может служить примером передачи реакционной спо-
собности по системе сопряженных связей—так называемого явления винилогии (см. том I, стр. 132). В самом деле, в хлорангидриде азотной кислоты хлор легко заменяется на гидроксильную группу; в соответствии с принципом винилогии следует ожидать, что подвижность галоида будет сохраняться, если группировки —NO2 и —С1 будут разделены . системой углеродных атомов, связанных двойной связью или сопряженными двойными связями:
о=\’—С1
I
О
0=N—С=С—С1 0=N—С= С—С=С—С1
В молекулах о- и п-нитрохлорбензолов заместители разделены как раз такими системами углеродных атомов. В случае же лг-нитро-хлорбензола винилогии не имеется:
Сульфокислоты
Легкость сульфирования при действии серной кислоты представляет собой такое же характерное свойство ароматических соединений, как и способность к нитрованию.
Сульфирующими агентами являются не ионы HSO^“ и SO* , а, вероятнее всего, нейтральные молекулы H2SO4, S03 или H2S2O7; возможно также, что роль сульфирующего агента играет катион H3SO^.
На основе работ Е. А. Шилова в настоящее время предложена схема реакции сульфирования, согласно которой молекула органического соединения промежуточно образует с электрофильным сульфирующим агентом комплекс, далее переходящий с отщеплением протона и сульфокислоту:
o-s4>
При сульфировании, например, толуола концентрированной серной кислотой получается смесь о- и м-толуолсульфокислот со значительным преобладанием пара-изомера. Кроме того, образуется немного мета-изомера. Дальнейшее сульфирование путем нагревания с дымящей серной кислотой приводит к образованию ?и-дисульфо кислоты (толуол-2,4-дисульфокислоты), так как сульфогруппа, как и нитрогруппа, сильно ориентирует в мета-положение. Трисульфокислоты получаются с большим трудом (сульфированием дымящей серной кислотой в присутствии сернокислого серебра).
Для выделения сульфокислот, образующихся при сульфировании ароматических углеводородов, их отделяют от избытка серной кислоты в виде растворимых в горячей воде бариевых или кальциевых солей, или же в виде натриевых солей, трудно растворимых в концентрированных растворах солей с одноименным катионом. Натриевые соли сульфокислот выделяются в кристаллическом виде при прибавлении к продукту сульфирования концентрированного раствора поваренной соли.
Свободные сульфокислоты представляют собой кристаллические вещества, обычно хорошо растворимые в воде, иногда расплывающиеся на воздухе. Это очень сильные кислоты, легко вытесняющие соляную кислоту из растворов хлористого натрия. Свободные сульфокислоты удобнее всего получать гидролизом сульфо хлоридов путем кипячения их с водой. Воду и образовавшуюся соляную кислоту удаляют азеотропной перегонкой с каким-либо парафиновым углеводородом, хлорбензолом и т. п. Твердые сульфокислоты можно также выделять из продуктов сульфирования кристаллизацией из концентрированной соляной кислоты.
Особая важность сульфокислот в химии ароматических соединений определяется тем, что сульфогруппа легко может быть заменена на некоторые другие функциональные группы, а так как многие сульфокислоты получаются сравнительно просто, они используются в лабораторной и промышленной практике в качестве легко доступных промежуточных соединений при синтезе различных производных ряда бензола.
Химические свойства. 1.Гидролиз. При действии на сульфокислоты водяного пара в присутствии минеральных кислот легко протекает реакция, обратная сульфированию (элиминирование сульфогруппы), причем регенерируются ароматические углеводороды:
CH8-CeH4-SOsH + Н,0 -> C6H6-CH3 + H2SO4
2. Сплавление со щелочами. При сплавлении солей сульфокислот с едкими щелочами происходит обмен сульфогруппы на гидроксил. При этом получаются фенол и сульфит натрия:
СвНБ—SO3Na + NaOH —» С6Н6—ОН + Na2SO3
Таким путем фенол и некоторые его гомологи и производные получают в промышленном масштабе.
. 3. Сплавление с цианидами. При сплавлении солей сульфокислот с цианистым калием происходит обмен сульфогруппы на^цианогруппу:
СвН8—SO8K + KCN -> CeH5—CN-|-K2SO8
4. Восстановление. Восстановлением сульфокислот можно получить ароматические меркаптаны, называемые пгиофено-лами:
ceH6-so3H + 6H -> C6H5-SH + 3H,0
Тиофенолы—вещества с кислотными свойствами, обладающие отвратительным запахом; окислением они снова могут быть превращены в сульфокислоты.
Нагреванием свинцовых солен тиофенолов можно получить ароматические тиозфиръг.
(C6H6S)2Pb -» (CeH6)2S + PbS
5. Образование х л оран гидр и до в. При действии на сульфокислоты пятихлористого фосфора получаются хлор-ангидриды сульфокислот, или сульфохлориды-.
С6Н5—SO3H + РС15 —» С6Н5—SO2C1 + РОС13 + НС1
Сульфохлориды могут быть получены непосредственным действием хлорсульфоновой кислоты на ароматические углеводороды:
СбНб + 2НО—SO2C1 —» С6Н5—SO2C1 + H2SO4 + НС1
Бензолсульфохлорид С6Н5—SO2C1—тяжелая жидкость (уд. вес dll =1,3766), застывающая при низких температурах в кристаллы с темп, плавл. +14,6°.
Чистый бензолсульфохлорид перегоняется при 252° под атмосферным давлением*.
о-Толуолсульфохлорид СН3—С6Н4—SO2C1—жидкость, перегоняющаяся в вакууме (21 мм) при 126°. п-Толуолсульфохлорид образует кристаллы с темп, плавл. 69°.
Сульфохлориды обладают характерным своеобразным запахом.
6. Образование сульфамидов. Действием на сульфохлориды аммиака (даже водного) получаются амиды сульфокислот, или сульфамиды.
СН3—СбН4—8О2С1 -4- NH3 CH3-QH4-SO2NH2 + НС!
Амиды сульфокислот легко кристаллизуются. Благодаря подвижности водородных атомов группы —SO2NH2 сульфамиды обладают кислотными свойствами и растворяются в едких щелочах.
Бензолсульфамид CGH5—SO2NH2 образует бесцветные листочки или иглы, плавящиеся при 153°. При действии на него щелочных гипохлоритов получаются: бензолмонохлорсульфамид, обычно выделяемый в виде натриевой соли [С6Н5—SO2NC1 ]Na-3H2O, и бензолди хлорсульфамид С6Н5—SO2NC12 (темп, плавл. 76°).
Дихлорсульфамид под названием хлорамина Б применяется как дезинфицирующее и дегазирующее средство. Его дезинфицирующее действие основано на окислительных свойствах препарата:
/С1
CeH6— SO2N< + 2Н3О СвНб—SO2NH2 + 2НС1 + 20
\ci
* Нечистые сульфохлориды даже при перегонке в неглубоком вакууме могут бурно разлагаться с выделением S02, НС! и обугливанием органического остатка.
о-Толуолсульфамид является промежуточным продуктом при производстве сахарина (ср. стр. 294). Из п-толуолсульфамида— отхода этого производства—получают натриевую соль п-толуол-монохлорсулъфамида
[CH3-C6H4-SO2NCl] Na-3H2O
и п-толуолдихлорсульфамид
СН3—СбН4—SO2NC12
применяемый под названием хлорамина Т для тех же целей, что и соответствующее производное бензола.
7. Образование сложных эфиров с у л ь ф о к и с л о т. При действии спиртов на хлорангидриды сульфокислот в присутствии щелочи получаются сложные эфиры сульфокислот:
CeH5-SO2Cl + NaOC2H6 -> CeH6-SO2-OC2H6 + NaCl
Эти эфиры имеют практическое применение, так как они, подобно эфирам серной’кислоты, являются алкилирующими средствами (см. том I, стр. 221), например-
СвНб—ONa + CaHsO-SO2-CeH6 -> С6^5-О-С2Н5 + NaOSO2-C6H5
Метиловый эфир n-толуолсульфокислоты (темп, плавл. +28°) и этиловый эфир с темп, плавл. +32° (эфиры n-толуолсульфокислоты называют иногда тозила-тами) широко применяются как алкилирующие агенты.
8. Образование сульфинов ых кислот и сульфонов. Осторожным*- восстановлением сульфохлоридов цинковой пылью можно получить соли ароматических сулъфиновых кислот'.
2С6Н6—SO2C1 + 2Zn —> (Cer i6SO2^2Zn 4- ZnCl2
Как побочные продукты при сульфировании ароматических углеводородов серной или хлорсульфоновой кислотой получаются сульфоны, например:
SOt'OH)2 + 2(у% -> СвНб-8О2-Сбнб + 2Н2О дифениле ульфон, или сульфобензид
Сульфоны можно получить также, действуя на углеводороды сульфокислотами в присутствии фосфорного ангидрида или хлорангидридами сульфокислот в присутствии хлористого алюминия:
CeHe-SO2-OH + Ce^6 -> C6H5-SO2-C6H5+H2O
Замещенные сульфокислоты. В процессе сульфирования галоидбензолов получается исключительно и-хлор(бром)-сульфокислота, а действие брома на бензол-сульфокислоту приводит к м бромбензолсульфокислоте.
При сплавлении галоидсульфокислот со щелочами и галоид и сульфогруппа замещаются на гидроксильные группы. Замечательно, что при перегревании плава все три изомера (орто-, мета- и пара-) могут дать один и тот же лс-диоксибензол, или резорцин («атомная» перегруппировка).
Нитрование бензолсульфокислоты и сульфирование нитробензола дают главным образом дрнитробензолсульфокислоту.
Одноатомные фенолы и ароматические спирты
Фенолами называются такие гидроксилсодержащие ароматические соединения, в которых гидроксильная группа замещает водородный атом бензольного ядра; если же гидроксил находится в боковой цепи, то такие соединения называются ароматическими спиртами.
Одноатомные фенолы
Первый представитель гомологического ряда фенолов—оксибензол С6Н.5ОН называется обычно престо фенолом или карболовой кислотой. Оксипроизводные толуола (метилфенолы, или метилоксибензолы) называются орто-, мета- и пара-крезолами. Соответствующие производные ксилолов называются ксиленолами. Изомерные ксиленолы именуются орто-, мета- или пара-ксиленолами в соответствии со строением ксилола, из которого они могут быть произведены заменой атома водорода в ядре на гидроксил; положение этого гидроксила указывают, прибавляя слова: рядовой, симметрический или несимметрический. Часто положения замещающих групп указывают, пользуясь цифрами, например: 2-окси-л«-ксилол, 2,4-ди-метилфенол, 1-окси-2,3-диметилбензол.
Фенол и его гомологи образуются при сухой перегонке дерева и в большем количестве—при сухой перегонке каменного угля. Их извлекают из каменноугольной смолы и кислых вод едкими щелочами, осаждают из щелочных растворов углекислотой или серной кислотой и разделяют дробной перегонкой.
Синтетические способы получения. 1. Сплавление солей сульфокислот со щелочами (см. стр. 258).
2. Действие азотистой кислоты на первичные ароматические амины. Эта реакция аналогична получению спиртов из аминов жирного ряда (см. том I, стр. 192), но здесь промежуточными продуктами являются диазосоединения, при кипячении- которых с водой и получаются фенолы (стр. 321).
3. Получение из ароматических галоидных соединений. Фенолы можно получать по Ульману действием едких щелочей на ароматические галоидные соединения в присутствии тонкого порошка меди (стр. 245), а также действием водяного пара при высоких температуре и давлении или в присутствии катализаторов (силикагель с добавками солей металлов).
4. Нагревание ароматических оксикислот. Например: /ОН с6н4< -> сбнбон + со2
\соон
5. Получение высших фенолов из низших. Высшие фенолы могут быть получены нагреванием фенолов со спиртами в присутствии* хлористого цинка
СбН5ОН 4- С2НбОН -> С2Н5-С6Н4ОН + Н2О
или изомеризацией жирно^-аромагических простых эфиров при нагревании с соляной кислотой (Н. И. Курсансв'ь например:
с2н6-о-с6нб -> С2Нб-С6Н4ОН
Можно также получать высшие фенолы алкилированием низших фенолов олефинами в присутствии кислотных катализаторов. Замещение идет в пара-положение:
СН3 .
I
ОН —> СН3— С— I
СН3
Физические свойства. Большая часть фенолов—кристаллические вещества, некоторые из них—жидкости, легко летучие с парами воды. Фенолы бесцветны, имеют интенсивный характерный запах и оказывают сильное антисептическое действие. В воде растворимы лишь низшие фенолы; в спирте, эфире и бензоле хорошо растворяются все фенолы.
Химические свойства. Химические свойства фенолов определяются, с одной стороны, наличием в них гидроксильной группы с весьма подвижным атомом водорода, а с другой—бензольным ядром с его характерными ароматическими свойствами, еще усиленными оксигруппой.
I. Реакции фенольного гидроксила. 1. Взаимодействие со щелочами. Фенолы являются третичными спиртами, но, вследствие взаимодействия электронной пары атома кислорода гидр оксильной группы с тг-электронами ароматического ядра (стр. 234), обладают повышенными кислотными свойствами. Они легко растворяются в едких щелочах, образуя аналогичные алкоголятам феноляты. Поэтому Рунге, открывший в 1834 г. простейший фенол (в каменноугольном дегте), назвал его «карболовой кислотой». В водных растворах феноляты, однако, сильно гидролизованы, и свободные фенолы могут быть выделены из щелочных растворов действием даже такой слабой кислоты, как угольная.
Образование интенсивно окрашенных в фиолетовый, синий или зеленый цвет фенолятов железа при взаимодействии фенолов с хлорным железом используется как характерная качественная реакция на фенолы.
2. Образование простых эфиров. При действии алкилирующих средств н*а феноляты получаются жирно-ароматические простые эфиры фенолов:
C6H5ONa 4- СН3С1 —> C6I I5-O—СН3 4-NaCl
Аналогично протекает реакция с солями монохлоруксусной кислоты:
СбНбО— Na 4- Cl—СН2—COONa —> СбН5—О—CH2COONa 4-NaCl
Получающиеся производные феноксиуксусной кислоты было предложено использовать для идентификации фенолов. Производные простейших фенолов
имеют следующие температуры плавления: Феноксиуксусная кислота ............................. 100—101э
о-Крезоксиуксусная » 151°
л<-Крезоксиуксусная » 103°
п-Крезоксиуксусная » 139°
Одноатомные фенолы
Таблица 19
Название Формула Температура плавле-ния, °C Температура кипения, °C Удельный вес Показатель преломления nD
Фенол Крезолы: орто- мета- пара- Свн6он СН3СвН4ОН +42,3 +30,8 + 11,9 +34,8 182,1 190,9 202,2 202,1 1,0596(d4°'6) 1,0415(4°) 1,0341(4°) 1,0340(47’7*) 1,5425 (при 40,6°) 1,5443 (при 20°) • 1,5432 (при 15°) 1,5361 (при 17,7°)*
о-Ксиленолы: рядовой, или 2,3-диметил-1 -оксибензол (СН3)аС,Н3ОН +75 218
несимметрический, или 3,4- диметил - 1-оксибензол +62,5 225
к-Ксиленолы: рядовой, или 2,6-диметил- 1 -оксибензол Го же +49 203
несимметрический, или 2,4-диметил-1-оксибензол +26 211,5 —
симметрический, или 3,5-диметил- 1-оксибензол +68 219,5 — —
п-Ксиленол, или 2,5-диметил-1-оксибензол То же +74,5 213,5 — —
2-Метил-5 - изопропил-1-оксибензол (карвакрол) сн3 >свн3он (СНз^СН/ +0,5 237,8 0,9760(4°) 1,5245 (при 20°)
5-Метил-2-изопропил-!-оксибензол (тимол) То же +49,6 232,9 0,9257(4°’') 1,4913 (при 80,1°)
Данные для переохлажденной жидкости.
Некоторые галоидфеноксиалкилкарбоновые кислоты (например, 2,4-ди-хлорфеноксиуксусная, 2-метил-4-хлорфеноксиуксусная, 2,4-дихлорфенокси-у-мас-ляная) в малых концентрациях способны стимулировать рост растений, напоминая в этом отношении природные ростовые вещества. В больших концентрациях они губительно действуют на растения. Можно подобрать такую концентрацию, при которой действие этих веществ носит гербицидный характер,—погибают одни виды растений (двудольные) и не затрагиваются другие (однодольные), что позволяет применять эти вещества для борьбы с сорняками, например с широколистными сорняками в посевах злаков.
Реакция образования простых эфиров фенолов особенно легко протекает со средними эфирами серной кислоты, например с диметилсульфатом. Чисто ароматические простые эфиры легко получаются по Ульману—действием галоидных арилов на феноляты в присутствии порошкообразной меди:
CeH5ONa + С6Н6С1 СбН5—О—СбНб + NaCl
3. Образование сложных эфиров. При действии кислот сложные эфиры фенолов получаются с неудовлетворительными выходами. Обычно их синтезируют действием на фенолы или феноляты хлорангидридов или ангидридов кислот, например:
C6H6ONa + СН3СОС1 —> СН8СО—OCeH5 + NaCl
Даже при действии на феноляты угольного ангидрида в отсутствие воды образуются соли кислых эфиров угольной кислоты:
CeH5ONa + COa NaOCO—ОСбН6
Кислые эфиры серной кислоты и фенолов содержатся в моче и образуются при гниении белков в кишечнике.
4. Взаимодействие с галоидирующими реагентами. Гидроксильная группа в фенолах лишь с трудом и с очень небольшим выходом замещается на галоид при действии галоидных соединений фосфора.
При действии трехгалоидных соединений фосфора образуются главным образом сложные эфиры фосфористой кислоты, например:
ЗСвН5ОН + РС13 Р(ОСвНб)8 + ЗНС1
Пятигалоидные соединения фосфора действуют главным образом как смесь галоида с трехгалоидным соединением, т. е. преимущественно получаются галоидозамещенные фенолы и эфиры фосфористой кислоты. Обработка фенолов хлорокисью фосфора РОС13 приводит к образованию эфиров фосфорной кислоты.
5. Восстановление. При нагревании фенолов с цинковой пылью происходит восстановление гидроксильной группы и получается углеводород.
II. Реакции бензольного ядра. Благодаря эффекту сопряжения (стр. 234) гидроксильная группа сообщает водородным атомам бензольного ядра способность необыкновенно легко замещаться на галоид, нитрогруппу или сульфогруппу; по этой же причине фенолы легко вступают в реакции конденсации. Кроме того, бензольное ядро в фенолах крайне легко подвергается окислению.
Гидроксил является одним из заместителей, наиболее сильно ориентирующих в орто- и пара-положения.
1. Действие галоидов. При действии галоидов (даже бромной воды) в молекуле фенола чрезвычайно легко замещаются на галоид последовательно три атома водорода. Конечным продуктом галоидирования является трибромфенол (мелкие иголочки, темп, плавл. 96°).
ОН
В\А/Вг у Вг
2. Нитрование. При действии на фенол очень разбавленной азотной кислоты получается смесь о- и и-нитрофенолов. Менее разбавленная кислота дает динитрофенолы и тринитрофенол, или пикриновую кислоту:
NO2
3. Сульфирование. При действии серной кислоты получается смесь о- и n-фенол сульфокислот.
4. Окисление. Фенолы медленно окисляются даже кислородом воздуха, причем они приобретают красный цвет или темнеют. Чем чище фенол, тем он устойчивее к действию воздуха, так что, быть может, примеси- играют роль катализаторов окисления.
При действии хромовой кислоты окисляется углеродный атом в пара-положении и образуются п-бензохинон (стр. 351) и продукты его превращения. Дальнейшее окисление приводит к разрушению бензольного кольца. При действии перекиси водорода, а также при сплавлении со щелочами образуются двухатомные и трехатомные фенолы.
5. Реакции конденсации. Фенолы легко вступают в реакции конденсации со спиртами (стр. 261), альдегидами, кислотами, ангидридами, хлорангидридами и пр. При этом всегда вступают в реакцию атомы водорода в орто- и пара-положениях.
6. Взаимодействие с азотистой кислотой (см. стр. 357).
7. Гидрирование. Водород при повышенной температуре и давлении в присутствии мелкораздробленного никеля присоединяется к фенолу. При этом образуется циклогексанол с примесью циклогексанона.
Фенол обыкновенный С6Н5ОН (карболовая кислота) представляет собой кристаллическое вещество (физические свойства— см. табл. 19) с характерным запахом. С водой он образует гидрат С6Н5ОН-Н2О (темп, плавл. 16°), а при дальнейшем прибавлении
воды растворяется в ней до содержания около 8% при комнатной температуре.
В растворах фенол ядовит, а в чистом виде может вызывать ожоги кожи.
Фенол получают из каменноугольного дегтя и, главным образом, синтетически—из бензола, через сульфокислоту или хлорбензол. В последнее время очень перспективным становится промышленный способ получения фенола из бензола следующим путем. Алкилированием бензола пропиленом (пропиленовая фракция газов 'крекинга нефти) получают изопропилбензол'.
<32/ + сна=сн—сна
Последний подвергают жидкофазному каталитическому окислению кислородом воздуха, получая гидроперекись изопропилбензола
которая под действием серной кислоты гладко расщепляется на фенол и ацетон, легко разделяемые ректификацией:
/СН3
ЧВз
В Советском Союзе этот метод осуществлен в промышленности с 1949 г. (П. Г. Сергеев, Р. Ю. Удрис, Б. Д. Кружалов).
Фенол обладает сильными антисептическими свойствами, но в настоящее время мало применяется как антисептик. Главное его применение—в качестве полупродукта для многочисленных промышленных органических синтезов. Фенол широко используется в производстве синтетических красителей, /?ля получения искусственных смол (конденсацией с формальдегидом), для гидрирования в циклогексанол (исходный материал для получения полиамидных воле кон), в производстве взрывчгтых веществ, в фармацевтической промышленности для приготовления салициловой кислоты и др.
Крезолы содержатся в каменноугольной смоле. Они находят широкое применение для получения искусственных смол (конденсацией с формальдегидом) и для синтеза красителей, а также для приготовления дезинфицирующих средств (лизол и пр.).
Крезолы содержатся также в продуктах гниения белковых веществ.
Тимол (2-изопропил-5-метилфенол) извлекают из некоторых эфирных масел (Thymus vulgaris, Psychotis Ajowan и пр.). Он применяется как малоядовитый антисептик (зубные пасты).
Простые эфиры фенолов (получение—см. стр. 262) щелочами не омыляются. При нагревании жирно-ароматических эфиров с бромистоводородной или иодистоводородной кислотой происходит отщепление алкильной группы в виде галоидного алкила и образуется свободный фенол:
СбН5—О—СН34-HJ C6H5OH4-CH3J
На этой реакции основано количественное определение «метоксильных» групп СН3О, содержащихся в сложных природных и синтетических веществах. На выделяющийся иодистый метил действуют спиртовым раствором азотнокислого серебра, а образующееся иодистое серебро определяют обычным путем (способ Цейзеля).
При многих реакциях замещения и конденсации простые эфиры фенолов реагируют так же, как и ароматические углеводороды, но реакция обычно протекает легче.
Метиловый эрир фенола СН3—О—С6Н5 называется анизолом и представляет собой жидкость с темп. кип. 153,8°; уд. вес a2Q = 0,9939, показатель преломления п2^ =1,5221, темп, плавл. —37,2°. Этиловый эфир, или фенетол, С2Н5—О—С6Н5 кипит при 172°, уд. вес его d20— 0,9666, показатель преломления п2£ = 1,5080, темп, плавл. —30,2°. Оба эфира обладают своеобразным запахом. При нитровании их получаются орто- и пара-нитросоединения, служащие исходными продуктами для получения соответствующих аминов и ряда синтетических красителей.
Дифениловый эфир (фениловый эфир) СбН5—О—С6Н5 плавится при 28°, кипит при 259,3°. Он имеет запах герани и в чистом виде применяется в парфюмерии. В значительных количествах дифениловый эфир используется в технике (обычно в смеси с дифенилом) в качестве теплоносителя, обладающего высокой температурой кипения, для нагревания автоклавов, аппаратов для контактно-каталитических реакций и т. п. взамен перегретого пара или пара очень высоких давлений.
ФЕНОЛО-ФОРМАЛЬДЕГИДНЫЕ СМОЛЫ
Большие количества фенола и смесей крезолов расходуются для производства феноло-формальдегидных смол, широко применяемых в промышленности пластических масс.
Изделия из пластических масс на основе феноло-формальдегидных смол (из так называемых фенопластов) изготовляют литьем или прессованием композиций, в большинстве случаев содержащих, кроме смолы, различные наполнители—древесную муку, асбест, хлопчатобумажную или стеклянную ткань, бумагу и т. д. Из композиций, содержащих в качестве наполнителя древесную муку, изготовляют карболит, на основе хлопчатобумажной ткани—текстолит, со стеклянной тканью—стеклотекстолит, с бумагой—гетинакс и пр.
Фенол о-формальдегидные смолы получают поликонденсацией (см. том I, стр. 494) фенола, крезолов, ксиленолов с формальдегидом. Невидимому, в начальных стадиях конденсации образуются метилольные производные фенолов—фенолоспирты, содержащие, в соответствии с правилами замещения в бензольном ядре (стр. 238). оксиметильные группы в орто- и пара-положениях:
ОН
I ZCH2OH и
он
НОН2С\/1чуСН,ОН и
СН2ОН
В результате ступенчатого процесса поликонденсации оксибензиловых спиртов, сопровождающегося на каждой стадии выделением воды
ОН ОН ОН ОН
I СН ОН I I I
(У + и " 0~снО +На°
СН2ОН СН2ОН
получаются высокомолекулярные продукты—смолы.
Если процесс поликонденсации проводить при избытке фенола, оксибензиловые спирты и начальные продукты их поликонденсации вступают в реакцию не только между собой, но и с фенолом, и на определенной стадии реакции укрупнение молекул прекращается ввиду отсутствия в концевых звеньях цепи реакционноспособных метилольных групп:
Такие смолы носят название новолачных. Они плавки, растворимы и неспособны переходить в стадию резита.
При избытке формальдегида получаются так называемые ре-зольные смолы, содержащие свободные метилольные группы:
В начальной стадии поликонденсации, когда молекулярный вес смолы составляет 700—1000, резольные смолы растворимы и легко плавятся. При дальнейшей поликонденсации (например, в процес
се прессования при повышенной температуре) резолы превращаются в неплавкие и нерастворимые резиты, имеющие трехмерное сетчатое строение.
При небольшом молекулярном весе (т. е. в стадии'резол а) ново-лачные и резольные смолы могут переходить друг в друга: новолач-ные—при добавлении формальдегида
а резольные—при добавлении фенола:
Поликонденсация с формальдегидом фенолов, незамещенных в орто- и пара-положениях, приводит к образованию смол трехмерного сетчатого строения:
Из производных фенола, содержащих заместители в орто- пли пара-положениях (например, из м-крезолов), могут получаться смолы только линейного строения.
Галоидфенолы
Галоидозамещенные фенолы могут получаться путем непосредственного замещения атомов водорода в фенолах (стр» 265), а также из ароматических соединений, содержащих, кроме галоида, другой заместитель, способный заменяться на гидроксил (например, из галоидозамещенных аминов действием азотистой кислоты через диазосоединения или из галоидозамещенных сульфокислот сплавлением со щелочами).
Галоидфенолы трудно растворимы в воде. Моногалоидфенолы, особенно орто-замещенные, обладают крайне навязчивым неприят: ным запахом.
Благодаря присутствию галоидов кислотные свойства фенолов усиливаются, и, например, трибромфенол является уже кислотой, вытесняющей угольную кислоту из углекислых солей.
Таблица 20
Галоидфенолы
Название
Температура плавления °C
Температура кипения °C
Хлорфенол орто- ...............
мета- ............
пара-............
Бромфенол орто- ................
мета- ............
пара.............
Иодфенол орто- ................
мета- ...........
пара- ...........
7*
31
43
Жидкость
32
66
43
40
94
172
214
219,7
195
236
238
186
* о-Хлорфенол существует в трех полиморфных модификациях. Из них вторая плавится при 0° и третья при —4°.
При сплавлении галоидозамещенных фенолов со щелочами при сравнительно более низких температурах получаются двухатомные фенолы соответствующего строения; при более высоких температурах происходит изомеризация и получается л«-диоксибензол, или резорцин.
2,4,6-Трибромфенол легко получается бромированием фенола; он представляет собой очень едкое вещество с темп, плавл. 96°. Его висмутовая соль, называемая ксероформом, находит применение как антисептическое средство.
Образование почти нерастворимого белого хлопьевидного осадка трибром-фенолброма при действии на водные растворы фенола избытка бромной воды является чувствительной реакцией на фенол.
Трибромфенолбром, по всей вероятности, является смесью двух таутомерных форм:
Нитрофенолы
Нитрофенолы получаются непосредственным нитрованием фенолов (стр. 265); при этом образуются о- и и-фенолы. Из нитроанилинов (стр. 309) через диазосоединения можно получить любые (орто-, мета-, пара-) изомеры нитрофенолов, в зависимости от строения исходного нитроанилина.
о- и г/-Нитрофенолы могут быть получены также обменом галоида на гидроксил в галоиднитробензолах (стр. 256). Из двух нитрофенолов, образующихся при нитровании, лишь 0-нитрофенол легко перегоняется с парами воды.
Уже мононитрофенолы разлагают углекислые соли. Динитрофенолы, и особенно тринитрофенолы, являются сильными кислотами (см том I, стр. 133).
Мононитрофенолы бесцветны или окрашены в лимонно-желтый цвет, полинитрофенолы имеют желтую окраску. Соли всех нитрофенолов имеют интенсивную оранжево-красную окраску.
Таблица 21
Нитрофенолы
Название
Температура плавления ох-
Температура кипения °C
Нитрофенол орто- ...............
мета- ...........
пара-........................ 114,1
Нитроанизол (метиловый эфир нитрофенола) орто- ................
мета- ...........
пара- ...........
217,2 194 (при 70 мм}
272
258
274
Повышение интенсивности окраски при образовании солей можно объяснить тем, что о- и n-нитрофенолы могут существовать в таутомерной (хиноидной) ацц-форме, способной к солеобразованию (Ганч).
Например, для солей n-нитрофенола возможно строение:
Na+
Действительно, метиловые эфиры о- и /2-нитрофенолов получены в двух формах: в виде обыкновенной—бесцветной и сильно окрашенной—хиноидной, отвечающей аци-форме.
Для солей ^-нитрофенола удовлетворительного объяснения интенсивной окраски (сам л<-нитрофенол почти бесцветен) не существует. Возможно, что полосы избирательного поглощения света у солей мета-изомера принадлежат к иному типу, чем полосы орто- и пара-изомеров.
Для динитрофенолов возможно шесть изомеров. Практическое значение из них имеет лишь ЪА'динитрофенол, получаемый нагреванием 2,4-динитрохлорбензола с растворами щелочей. Он образует кристаллы с темп, плавл. 115° и обладает отчетливым кислотным характером (/<25°= 1,0-10~4).
2,4-Динитрофенол служит промежуточным продуктом в производстве красителя «сернистого черного».
Пикриновая кислота (2,4,6-тринитрофенол) представляет собой окончательный продукт нитрования фенола. Она образуется также при кипячении с азотной кислотой различных животных и растительных веществ, например шелка, кожи, шерсти и пр. (впервые была получена в чистом виде в 1739 г. нитрованием шелка). Пикриновая кислота кристаллизуется из воды или спирта в виде желтых листочков или призм с темп, плавл. 122°.
Водные растворы пикриновой кислоты имеют горький вкус (отсюда и произошло ее название: ют греч. пикрос—горький) и весьма интенсивно окрашены, вероятно, вследствие того, что ионы пикриновой кислоты имеют хиноидное строение (см. стр. 357). В 100 частях воды при 20° растворяется 1,225 частей пикриновой кислоты, при ЧТ—3,89 частей.
Пикриновая кислота—довольно сильная кислота (/G5°=4,2- Ю-1). Такие высокие кислотные свойства пикриновой кислоты объясняются наличием в ее молекуле, кроме оксигруппы, трех нитрогрупп в орто- и пара-положениях.
Как было показано на стр. 234, уже фенол обладает кислотными свойствами вследствие сопряжения свободной электронной пары кислородного атома с z-электронными связями бензольного ядра: .
Н
Введение нитрогруппы в орто- или пара-положение усиливает оттягивание электронов кислородного атома, ослабляя его связь с атомом водорода:
При наличии в ароматическом ядре трех нитрогрупп отщепление водородного атома в виде протона еще более облегчается:
И здесь может итти речь о винилогии (см. стр. 257), а именно о винилогии о- и п-нитрофенолов с азотной кислотой:
Н—о—no2 н—о—с=с—no2 н—о—-с=сн—сн=с—no2
II II
Метиловый эфир пикриновой кислоты, получаемый действием метилирующих средств на ее соли (например, действием йодистого метила на серебряную соль), омыляется при кипячении со щелочами, а при действии аммиака образует пикрамид, или тринитроанилин. О свойствах хлорангидрида пикриновой кислоты—пикрилхлорида см. стр. 256.
Соли пикриновой кислоты—пикраты—хорошо кристаллизуются. Пикраты калия и аммония трудно растворимы в воде, что используется в качественном анализе для открытия этих катионов. Соли пикриновой кислоты, особенно пикраты тяжелых металлов, очень взрывчаты. В противоположность самой пикриновой кислоте, они взрывают не только при детонации, но при ударе или трении и даже при нагревании.
Пикриновая кислота образует также хорошо кристаллизующиеся, часто трудно растворимые соли со многими органическими основаниями. На этом основано широкое применение ее при исследовании синтетических аминов и природных оснований, например алкалоидов. Кроме того, пикриновая кислота образует хорошо кристаллизующиеся молекулярные соединения (также называемые пикратами) со многими высокомолекулярными ароматическими углеводородами (например, с нафталином) и их производными и применяется для выделения этих веществ в чистом виде. Щелочи, например аммиак, разлагают такие соединения. Пикраты углеводородов были впервые получены Ю. Ф. Фрицше* в 1858 г.
Пикриновая кислота раньше находила применение как желтая краска для шерсти и шелка. Применение ее как бризантного взрывчатого вещества для снарядов (под названием мелинита, или лиддита)
* Юлий Федорович Фрицше (1808—1871). Адъюнкт, затем экстраординарный и ординарный академик. Автор большого числа исследований в разных областях органической химии. Наиболее важные из них: получение .антраниловой кислоты и анилина из природного индиго и получение пикратов ароматических углеводородов.
также в настоящее время постепенно сокращается вследствие неполной безопасности в обращении с ней. Она является исходным материалом для получения хлорпикрина (см. том I, стр. 249), пикрами-новой кислоты и др.
При действии на пикриновую кислоту цианистого калия получается красный изопурпуровокислый калий, который, повидимому, имеет строение:
ОК
Nc/^XcONHs
no2
Эта реакция используется для качественного определения пикриновой кислоты.
Сульфокислоты фенола
о- и и-Сульфокислоты фенола получаются сульфированием фенола даже при обычной температуре. л^-Сульфокислота получается в результате обмена на гидроксил одной сульфогруппы в бензол-дисульфокислоте при сплавлении ее с едким кали. о-Сульфокислота при нагревании с минеральными кислотами легко переходит в n-сульфокислоту. При сплавлении сульфокислот фенола со щелочами получаются двухатомные фенолы (при высокой температуре из всех изомеров получается резорцин).
Продукты конденсации фенолсульфо кислот (также нафтолсульфокислот) с формальдегидом находят применение в качестве синтетических дубителег0. (нера-дол, ордоваль и др.).
Ароматические спирты
Продукты замещения на гидроксил ^водородных атомов в боковых цепях ароматических углеводородов, т. е. ароматические спирты, по способам получения и по свойствам первичных, вторичных и третичных соединений полностью аналогичны жирным спиртам.
Бензиловый спирт (фенилкарбинол) С6Н5—СН2ОН представляет собой простейший ароматический спирт. Он может быть получен из хлористого бензила С6Н5—СН2С1 путем обмена галоида на гидроксил различными способами, а также восстановлением бензойного альдегида С6Н5—СНО и бензойной кислоты С6Н5—СООН или ее эфиров и пр. Кроме того, его можно получить реакцией Кан-ницаро из бензойного альдегида (стр. 278).
Бензиловый спирт—трудно растворимая в воде жидкость (темп. кип. 205,5°, уд. вес dlQ— 1,0453), имеющая слабый запах. В виде сложных эфиров бензойной и коричной кислот бензиловый спирт содержится в перуанском и толутанском бальзамах и в других смолах, в виде уксусного эфира—в эфирном масле жасмина. Обменивая
гидроксил на алкоксил или кислотный остаток, бензиловый спирт образует простые и сложные эфиры (однако под действием серной кислоты легко происходит осмоление). Он восстанавливается иоди-стоводородной кислотой в толуол, окисляется в альдегид и кислоту и Т. д.
Фенилэтиловые спирты. э.-Фенилэтиловый спирт С6Н5-СН(ОН)-СН3 может быть получен, например, восстановлением кетона ацетофенона СС)Н5—СО—СН3 или взаимодействием бензойного альдегида CGH5—СНО с иодистым метилмагпием CH3MgJ. Его темп, плавл. -(-20,3°, темп. кип. 204°, уд. вес d2Q= 1,0142, показатель преломления /?£>—1,5230.
^-Фенилэтиловый спирт С6Н5—СН2—СН.2ОН представляет собой главную составную часть розового масла. Он имеет запах розы и получается в парфюмерной промышленности синтетически (искусственное розовое масло), например восстановлением сложных эфиров фенил уксусной кислоты С6Н,-—СН2—СООН, а также действием фенилмагнийхлорида на окись этилена (П. П. Шорыгин); его темп, кип. 219,8°, уд. вес d2,)~ 1,0235.
При нагревании а-фенилэтилового спирта с бисульфатом калия или натрия или при пропускании его паров над окисью алюминия гладко образуется стирол СбН5—СН = СН2 (стр. 401). Из 3-фепил-этилового спирта стирол получается нагреванием с едкими щелочами.
Фенилизопропиловый спирт (диметилфенилкарбинол)
сн.<
I
с6п5-с—ом
I
СНз
может быть получен действием метилмагнийбромида на ацетофенон (стр. 281) или фенил магнийбромида на ацетон. Он плавится при +37,5°; темп. кип. 103° (при 25 мм); уд. вес г/4°—0,9981; показатель преломления iid = 1,5240 (жидк.). При дегидратации фенил изопропилового спирта получается ^метилстирол.
Ароматические альдегиды
Способы получения. Ароматические альдегиды, независимо от того, связана ли альдегидная группа непосредственно с бензольным ядром или находится в боковой цепи, могут получаться всеми способами, применяемыми для получения жирных альдегидов, например окислением первичных спиртов, восстановлением кислот или их хлорангидридов, сухой перегонкой смеси кальциевой соли с солью муравьиной кислоты, через магнийорганические соединения и пр.
Для альдегидов с группой СНО, связанной непосредственно с бензольным ядром, существуют и другие способы получения.
1. Получение из галоидопроизводных. Большое практическое значение имеет способ получения таких альдегидов из двугалоидных соединений типа хлористого бензилидена нагреванием с водой и мелом (или известью) или с разбавленной серной кислотой:
СвН6—СНС1а + Н,0 —> CSHS~CHO + 2НС1
Можно получать альдегиды также омылением моногалоидных соединений с одновременным окислением, например разбавленной азотной кислотой или азотнокислым свинцом:
С6Н5—СШ 1 4- О —> СвНб—СНО + НС1
2, О к и с л е н и е углеводородов. Осторожное окисление ароматических углеводородов, содержащих метильные группы, приводит к альдегидам.
Например, из толуола получается бензальдегид:
СвН6-СН3 + 2О -> ОА-сно-ьн.о
Еще не очень давно эта реакция проводилась лишь в лабораториях с применением ^трудно доступных окислителей типа хлористого хромила СгО2С12 (реакция Этара). В настоящее время окисление производится в промышленном масштабе. Окислителем может служить перекись марганца с серной кислотой или хромовая смесь (двухромовокислые соли с серной кислотой), причем окислению способствует присутствие небольших количеств галоидоводородных кислот.
Тот же результат может быть достигнут при электролитическом окислении.
Жидкофазное окисление кислородом воздуха метильных групп, стоящих при ароматическом ядре, также может приводить к альдегидам через стадию^образования гидроперекисей:
СвНб-СН3 —> СвН&—СН2—О—ОН СвН5-СНО+Н2О
3. Синтез Г а тте р м а н а*—К о х а. Специальным способом получения^чисто ароматических альдегидов является прямое введение альдегидной группы при действии на ароматические углеводороды' смеси окиси углерода и газообразного хлористого водорода в присутствии хлористого или бромистого алюминия и небольшого количества однохлористой меди:
СН,-£> + со -» сн,-^у_сно
* Людвиг Гаттерман (1860—1920). Немецкий химик. Работал преимущественно в области ароматических соединений и в области тиоиндиго. Автор очень популярного, до сих пор переиздающегося руководства «Практические работы по органической химии».
Эту реакцию можно объяснить, допустив промежуточное образование хлорангндрида муравьиной кислоты НСОС1. В этом случае реакция будет аналогична реакции получения кетонов по Фриделю и Крафтсу.
4. Р е а к ц и я С о м м е л ё. Ароматические альдегиды непосредственно образуются при нагревании хлористого бензила или его гомологов с уротропином:
<УУ>-СП2С1 <уу> СНО
Механизм этой реакции не изучен.
Физические свойства. Ароматические альдегиды представляют собой большей частью жидкости, мало растворимые в воде. Альдегиды с группой СНО, связанной непосредственно с бензольным ядром, обладают приятным сильным запахом, альдегиды с группой СНО в боковой цепи имеют резкий запах, приятный лишь в сильном разбавлении.
Таблица 22
Ароматические альдегиды
Название । Формула i 1 Температура кипения Удельный вес Показатель преломления , 20 d
Бензойный (бензальдегид) Толуиловый С;Н5СНО i i 178.1 : 1,0498 1,5450 1
СН3С6Н4СНО
орто- ; 197 1,0375 1,5485
мета- ; 199 1,0206 1,5415
пара- i 204 ! 1,0194 (rf'u-7) ! 1,547
1 ( При 1 (-,6 »
Фе нил уксусный С6Н6СН2СНО 194 1,0272 (d‘9-6) j 1,5255 ' (при 19,6е -
Гидрокоричный СбН5СН2СН2СНО ; 223 1 — । —
Гидратроповый СсН5СН(СН3)СНО |203 1,0025 (d;SJ) 1,5176 I (при 17ДЮ
Кумиловый (куминол, (CHgljCHCeHjCliO 236,3 0,9775 1,5301
п -изопропилбонзой-ный) ! i
Химические свойства. Альдегиды с группой СНО в бсково. цепи по химическим свойствам совершенно аналогичны жирным альдегидам. Ароматические альдегиды, содержащие группу СНО. непосредственно связанную с бензольным ядром, обладают рядом особенностей, связанных с их «ароматическим характером». В тс-же время эти альдегиды вступают во все химические реакции, характерные для соединений с альдегидной группой (см. том ‘ стр. 227 ел.): аутоксидация кислородом воздуха, восстановленье
окиси серебра с образованием серебряного зеркала, присоединение синильной кислоты и бисульфита, образование оксимов, гидразонов и пр.
Аутоксидация кислородом воздуха была изучена наиболее подробно как раз на примере бензойного альдегида, для которого эта реакция особенно характерна. При этой реакции, как, повидимому, и во всех других случаях, вначале образуется продукт присоединения молекулы кислорода. В данном случае, по исследованиям Байера, таким продуктом присоединения является гидроперекись бензоила, или пербензойная кислота С6Н5—СО—О—ОН. Окисление ускоряется в присутствии следов тяжелых металлов, например меди.
При действии воды на пербензойную кислоту может образоваться перекись водорода:
СбН5—СО—О—ОН + Н2О С6Н6—СООН + ноон
Перекись водорода, являющаяся очень активным окислителем, может далее расходоваться на окисление еще одной молекулы бензальдегида в бензойную кислоту, которая и является здесь единственным окончательным продуктом окисления.
Из особенностей ароматических альдегидов с группой СНО, непосредственно связанной с ядром, наиболее важными являются следующие:
1. Действие аммиака. В отличие от продуктов взаимодействия аммиака с жирными альдегидами, при действии аммиака на ароматические альдегиды получаются так называемые гидрамиды: c6h5-ch=n4
ЗС6Н5—СНО + 2NH3 —> >СН—СбН5 + Н2О
С6Н5-СН=Ж гидробензамид
2. Реакция Канницаро*. При действии на ароматические альдегиды спиртовых, а иногда и водных растворов едких щелочей одна молекула альдегида восстанавливается в спирт, а другая окисляется в кислоту, например:
2С6Н5-СНО + КОН -> СбН5-СН2ОН + сбн5-соок
Реакция Канницаро, повидимому, является особым случаем реакции, впоследствии открытой В. Е. Тищенко и для жирных альдегидов (см. том I, стр. 235). В ароматическом ряду она, вероятно, лишь особенно легко протекает, и притом первоначально образующийся сложный эфир сразу же омыляется.
3. Реакции конденсации. Ароматические альдегиды особенно легко вступают во все реакции конденсации, например: с жирными альдегидами
СеН5— СНО + СН3—СНО С6Н5-СН=СН—СНО + Н2О
с кетонами
С6Н5— СНО + СН3—СО—СН3 —» СбН5-СН=СН—СО-СН3 + Н2О 2С6Нз-СНО + СН3—СО—СН3 —> СбН5—СН=СН—СО—СН=СН—С6Н5 + 2Н2О
* Станислао Канницаро (1826—1910). Итальянский химик. Активный участник борьбы за эсвобож щние Италии от азстрийского ига. Открыл в 1853 г. реакцию, названую его именем. Поборник и пропагандист молекулярно-атомисти ческой (унитарной) теории.
с ангидридами типа (R—СН2—СО)2О (реакция Перкина)
С6Н5—СНО + (СН3СО)2О -» С6Нб—СН=СН—СООН + СН3СООН
с ароматическими углеводородами и их производными
С6Н5-СНО + С6Н6 -> С6Н5—СН(ОН)—С6Н5
/С6Н4ОН
СбН5—СНО4-2С6Н5ОН с6н5—сь/ +н2о
\сС)Н4ОН
и Т. д.
Полимеров типа паральдегида ароматические альдегиды не образуют.
4. Бензоиновая реакция. Особым типом реакции конденсации является так называемая бензоиновая реакция, протекающая под каталитическим влиянием цианистого калия. Здесь образуется связь между углеродными атомами двух групп СНО
о он бензоин
Интересно, что бензоиновая конденсация может протекать и под влиянием энзима дрожжей, называемого карболигазой. Уксусный альдегид, образующийся при брожении, под действием этого энзима может конденсироваться с каким-либо добавленным в бродильную жидкость ароматическим альдегидом. Например, с бензойным альдегидом он дает фенилацетилкарбинол9.
СН
3_С—СН
5. Действие хлора. При действии хлора на альдегиды типа С6Н5—СНО водородный атом альдегидной группы замещается на хлор и получаются хлорангидриды ароматических кислот:
С6Н5—СНО + С12 —> С6Нб—СОС1 + НС1
6. Образование замещенных в ядре производных. При действии на ароматические альдегиды смеси азотной и серной кислот происходит нитрование. Группа СНО в этих условиях не направляет заместителей в какое-либо определенное положение, а является в отношении ориентации промежуточной. Действительно, главным продуктом нитрования оказывается уц-нитробензальдегид (темп, плавл. 58°), но в то же время получается довольно много о-нитробензальдегида (темп, плавл. 43,5°)* и немного и-нитробензальдегида (темп, плавл. 106,5°).
* Вторая полиморфная модификация имеет темп, плавл. 40,4°.
Альдегиды с другими заместителями получаются чаще всего общими методами введения альдегидной группы в ядро или в боковую цепь ароматических углеводородов, например:
С1—С6Н5—СЫС12 + Н2О —» С1—С6Н4—СНО + 2НС1
/СН3 /СНО
С6Н4< + 20 —> С6Н4< + Н90
Х)Н Х)Н
1! т. п.
Бензойный альдегид (бензальдегид) С6Н5—СНО распространен в растительном мире в виде глюкозида амигдалина (см. том 1, стр. 603). Особенно много бензальдегида в горьком миндале, откуда и произошло первоначальное название бензальдегида—горькоминдаль-ное масло.
Бензойный альдегид обладает сильным запахом горького миндаля и применяется в качестве душистого вещества. Кроме того, он является важным полупродуктом в синтезе красителей (например, малахитового зеленого), лекарственных и других сложных органических веществ.
Куминовый альдегид (куминаль, л-изопропилбензойный альдегид) содержится в масле римской ромашки и в других эфирных маслах (например, Cuminum cyminutri), имеет пряный запах и находит применение как душистое вещество.
Ароматические кетоны
Ароматические кетоны разделяются на две группы:
1. Чисто ароматические кетоны, содержащие два ароматических радикала (арила), связанных с карбонильной группой.
2. Жирно-ароматические (смешанные) кетоны, в молекулах которых с карбонильной группой связан лишь один арил и один жирный радикал (алкил).
Кетоны первой группы относятся к соединениям с несколькими бензольными ядрами (стр. 420).
Способы получения. Ароматические кетоны могут получаться обычными способами синтеза жирных кетонов и, кроме того, особыми способами, характерными для ароматических кетонов. Важнейший из них состоит в действии хлорангидридов кислот на ароматические углеводороды в присутствии хлористого или бромистого алюминия (реакция Фриделя—Крафтса, аналогичная реакции получения гомологов бензола):
С6Н6 + C1-CO-R —> С6Нб—СО—R + НС1
В отличие от реакций синтеза углеводородов, здесь установлено, что хлористый алюминий дает определенные двойные соединения как с хлорангидрида ми кислот (типа R—СОСЬА1С13), так и с образовавшимися кетонами. Комплексное соединение кетона с хлористым алюминием разлагается затем водой. Поэтому,
тогда как при синтезах углеводородов требуется лишь небольшое количество хлористого алюминия (действие каталитическое), при получении ароматических кетонов необходимо брать его в количестве, несколько превышающем эквимолекулярное по отношению к количеству хлорангидрида.
Комплекс кетона с хлористым алюминием, повидимому, не участвует в реакции , и большое количество хлористого алюминия приходится брать, чтобы получить некоторое количество свободного катализатора.
Ацетофенон CGH5—СО—СН3 получается также путем жидкофазного окисления этилбензола газообразным кислородом в присутствии солей тяжелых металлов с промежуточным образованием гидроперекиси этилбензола, распадающейся под действием ионов тяжелого металла:
С6Н5-СН2-СН3 + О2 ->
С6Н5-СН-СН3 о—он
-> С6Н5-С-СН3 4- НоО |.
6
Свойства. Физические свойства жирно-ароматических кетонов приведены в табл. 23.
Жирно-ароматические кетоны
Таблица 23
Название Формула Температура плавления °C Температура кипения °C Удельный вес 4° Показатель преломления п20 nD
Ацетофенон (метил-фенилкетон) с,.,и5сосн3 + 19,6 । 1 202, О' 1,0281 1,5342
I фопиофенон С6Н6СОС2Н5 +20 217,7 1,0120 1,5270
п -Метилацетофенон СН3СвН4С0СН3 — 226,3 1,0051 1,5335
Фенилацетон (ме-тилбензилкетон) с„п6сн2сосп3 л_ 27 i 216,5 ! i 1,0157 1,5168 (переохл. ж иди ость)
Жирно-ароматические кетоны вступают во все реакции, свойственные жирным кетонам, однако в реакциях конденсации может участвовать лишь алкил. Кроме того, ароматические кетоны могут нитроваться и сульфироваться.
Ацетофенон (метилфеяилкетон) СН3—СО—С6Н5 является простейшим представителем жирно-ароматических кетонов. Он нерастворим в воде, обладает запахом черемухи.
Водородные атомы метильной группы ацетофенона очень легко замещаются при действии галоидов; эта же группа ацетофенона участвует в реакциях конденсации, например:
СбН5-СО-СН3 + ОНС—сбн5 —> сбн6— СО—СН=СН—С6Ы5 + Н2О
При действии на ацетофенон хлора образуется хлорацетофенон С6Н5—СО—СН2С1. Это бесцветные кристаллы с темп, плавл. 60°. Он перегоняется без разложения при 247°. Пары хлорацетофенона, а также образующийся при его возгонке дым обладают чрезвычайно сильным слезоточивым действием. Общая токсичность его ничтожна. Хлорацетофенон омыляется спиртовыми растворами едких щелочей и т. п. с образованием бензоилкарбинола:
C6I-I5—СО—СН2С1 + NaOH -> СбН5—СО—СН2ОН + NaCl
При окислении ацетофенона карбонильная группа остается связанной с фенилом, так что количественно получается бензойная кислота.
СТЕРЕОХИМИЯ ОКСИМОВ И ГИДРАЗОНОВ
Органические соединения, содержащие группировку
R.
)C=N-R'z
могут существовать в двух стереоизомерных формах. Поэтому оксимы ароматических альдегидов часто получаются в виде двух стереоизомеров. Один из стереоизомеров, обычно имеющий более высокую температуру плавления, под действием уксусного ангидрида легко теряет воду, превращаясь в нитрил:
СбН5—CH=N—ОН -> С6Н5—CeN + H2O
Изомер с более низкой температурой плавления при.действии уксусного ангидрида образует ацетильное производное:
С6Н5—CH=N—ОН+ (СН3СО)2О -> С6Н5—CH=N—ООС—СН3 + СН3СООН
Из чисто ароматических кетонов изомерные оксимы дают лишь те, у которых радикалы, связанные с карбонильной группой, различны. Так, дифенилкетон (бензофенон) (С6Н5)2СО образует лишь один оксим (C6H5)2C=NOH. Если же ввести заместитель в одну из фенильных групп этого кетона (взять, например, СН3—С6Н4—СО—С6Н5 или Вг—С6Н4—СО—С6Н5), то всегда будут получаться два изомерных оксима.
В соединениях трехвалентного азота, в которых азот связан с другими атомами простыми связями, направления о-связей отвечают направлениям ребер трехгранной пирамиды с атомом азота в вершине. Плоские углы в такой пирамиде лежат в пределах от 90° для NH3 до 108° для N(CH3)3 (см. том I, стр. 309), т. е. молекула не имеет плоскостного строения. При наличии же в молекуле группировки X)C=N—, в которой атомы С и N связаны двойной (а-, к-) связью» все три заместителя X, Y, Z должны располагаться в одной плоскости под углами 120°.
Для таких плоских молекул становятся возможными две пространственные конфигурации
X Y X Y
II и ||
N N
которые условно могут быть изображены проекционными формулами: X—С—Y X—С—Y
II и II
N—Z Z—N
Как видно из этих формул, в одной конфигурации Z находится в большей пространственной близости к Y, а в другой—к X. Согласна предложению Вернера и Ганча, пространственную близость и отдаленность обозначают приставками сан (вместе) и анти (насупротив). В применении к номенклатуре оксимов альдегидов (альдоксимов) приставка син означает близость гидроксила к атому водорода. Например, для ош-бензальдоксима с темп, плавл. 128—130° предполагалась конфигурация:
СбН5—С—Н
N—ОН
пА//7ш-бензальдоксиму с темп, плавл. 34° придавалась конфигурация: С6Н5—С—Н
НО—N
Близостью гидроксила к атому водорода как раз и объясняли легкость отщепления воды от cz/н-бензальдоксима под действием уксусного ангидрида (стр. 282).
Конфигурации оксимов кетонов, или кетоксимов, обозначаются следующим образом, например:
СбН5-С-СбН4СН3
НО—N
шн-фенил-о-толилкетоксим (темп, плавл- 105°)
С6Н5—С—С6Н4СН3
N—ОН
пнтп-феш’л-о-толилкетоксим (темп, плавл. 69°)
Конфигурацию кетоксимов можно определять, пользуясь реакцией, называемой бекмановской перегруппировкой*, происходящей при действии на кетоксимы пятихлористого фосфора, бензолсульфохлорида, хлористого ацетила, серной кислоты и пр. Под влия-
гшем этих реагентов кетоксимы перегруппировываются в амиды кислот:
Rx
>C=N—ОН —> R—С—NHR
Rz
О
* Эрнст Бекман (1853—1923). Немецкий физикохимик. В начале своей деятельности работал в области органической химии. Перегруппировка, названная его именем, была им открыта в 1886 г.
Реакция протекает в две стадии: сначала гидроксильная группа обменивается местом с одним из радикалов
R—С—R R—С—ОН
Il II
NOH NR
а затем получившаяся енольная форма амида кислоты изомеризуется в конечный продукт реакции
R—С=О
NHR
Бекмановская перегруппировка служит удобным способом получения амидов кислот, часто трудно доступных другими путями.
В случае оксимов несимметричных кетонов в результате бек-мановской перегруппировки получаются амиды двух различных кислот, в зависимости от того, с каким из радикалов обменивается гидроксильная группа:
Rx > R—СО—NHR'
\C=N—ОН —
R'/ I—> R'—СО—NHR
Естественно, что из син- и анти-форм кетоксимов будут получаться разные продукты перегруппировки, вследствие неодинаковой пространственной близости гидроксила к каждому из двух радикалов. Первоначально предполагалось, что гидроксильная группа обменивается местом с ближайшим к ней радикалом (т. е. из цис-положения}. Однако Мейзенгеймер показал, что при бекмановской перегруппировке в подавляющем большинстве случаев обмен происходит не из цис-, а из транс-положения:
R—С—R' НО—С—R' О—С—R'
II —> г —> I
N—ОН N-R HN-R
Правильность утверждения Мейзенгеймера можно проиллюстрировать на примере бекмановской перегруппировки оксимов 2-бром-5-нитроацетофенона.
р-Оксим 2-бром-5-нитроацетофенона безусловно имеет строение сцн-кетоксима, так как он чрезвычайно легко при действии едких щелочей переходит в бициклическое соединение—метил-нитробензоксазол
O2N. O2N\
О----------^сн- Q“^-CH=
\вг НО—N X)—N
3-ОКСИМ
в противоположность неспособному циклизоваться а-оксиму:
а-оксим
При бекмановской перегруппировке р-оксим дает метил амид 2-бром-5-нитробензойной кислоты. Это означает, что обмениваются местами группы СН3 и ОН, находящиеся в транс-положении. а-Ок-сим образует 2-бром-5-нитроанилид уксусной кислоты, т. е. и здесь меняются местами группы, находящиеся в транс-положении. Таким образом, по продуктам бекмановской перегруппировки можно судить о пространственной конфигурации исходного кетоксима, однако делать это надо с известной осторожностью, так как все еще нельзя считать строго доказанным, что гидроксил во всех случаях без исключения обменивается с радикалом в транс-положении.
Жирные кетоксимы, повидимому, состоят из смеси стереоизомеров*, а для большей части жирно-ароматических кетоксимов известен лишь один стереоизомер, в которохм гидроксильная группа находится в 'пространственной близости к жирному, а не к ароматическому радикалу. Второй стереоизомер здесь, повидимому, является веществом неустойчивым.
Стереоизомерия этого рода наблюдается также для соединений других классов, содержащих в молекуле трехвалентный азот с двойной связью. Так, для гидразонов большого числа альдегидов, как жирных, так и ароматических, а также для гидразонов . кетонов с двумя разными радикалами, соединенными с карбонильной группой, было доказано существование двух стереоизомеров. Например, одна форма фенилгидразона уксусного альдегида плавится при 98— 101°, а другая—при 57°.
Однако пока не имеется возможности решить, которой форме принадлежит конфигурация спн-гидразона
СН3—СН
N—NHfCH5
и которой—бштн-гидразона
сн3—СН
II C6H5NH—N
Стереоизомерные формы гидразонов пока еще не были получены для многих простейших гидразонов, например для гидразона бензойного альдегида.
* Индивидуальные стероизомерные оксимы получены лишь в отдельных редких случаях, например для окиси мезитила.
Ароматические одноосновные кислоты
Простейшей ароматической одноосновной кислотой является бензойная кислота С6Н5—СООН. Ее ближайший гомолог может существовать в четырех изомерных формах: о-, м- и п-толуиловыс кислоты {о-, м-, n-метилбензойные кислоты) СН3—СсН4—СООН и кислота с карбоксильной группой в боковой цепи С6Н5—СН,2—СООН, называемая фенилуксусной или а-толуиловой кислотой. Диметилбензойные кислоты называются ксилиловыми кислотами.
Названия кислот с карбоксильной группой в боковой цепи обыкновенно производятся от названия алифатической кислоты с обозначением места замещения в ней атома водорода на ароматический радикал (арил). Например, кислота С6Н5—СН(СН3)—СООН называется а-фенилпропионовой. Отсюда происходит и общее название этих кислот—фенил-(арил}-жирные кислоты.
Способы получения. Ароматические одноосновные кислоты могут получаться всеми общими способами синтеза кислот жирного ряда.
Из специальных способов получения ароматических кислот с карбоксильной группой в бензольном ядре наиболее важными являются следующие:
1. Окисление гомологов бензола (стр. 220), а также различных ароматических соединений с заместителями в боковых цепях. Например, окислением толуола и других углеводородов с одной боковой цепью получается бензойная кислота. Она же может получаться и при энергичном окислении фенилуксусной кислоты и т. д. Частичным окислением ксилолов, проводимым так, чтобы окислилась лишь одна метильная группа, можно получить толуиловые кислоты и т. д.
Как окислители применяются азотная кислота, хромовый ангидрид или перманганат калия.
2. Г а л о и д и р о в а н и е гомологов бензола и гидролиз продуктов галоидирования. Из ароматических углеводородов с метильными группами кислоты могут получаться также путем предварительного галоидирования углеводорода и последующего взаимодействия галоидопроизводного с водой.
Например, при хлорировании толуола получается бензотрихлорид, при действии воды превращающийся в бензойную кислоту:
С,Н5—СС13 + 2Н2О —> С6Н5—СООН + ЗНС1
3. Омыление нитрилов. Цианогруппа может быть введена в бензольное ядро путем сплавления солей сульфокислот с цианистым калием (стр. 258). Кроме того, ароматические нитрилы могут получаться из аминов через диазосоединения (стр. 322).
4. Действие двуокиси углерода и натрия на галоидзамещенные бензолы (карбонизация арилнатрия). Реакция идет по схеме:
С6Н5С1 -г 2Na —» C6H5Na + NaCl
C6H5Na + CO2 —> C6H5—COONa
5. Действие двуокиси углерода на магний-органические соединения. Например:
С6Н5—MgBr + СО2 —> С6Н5—COOMgBr —> сбн5—СООН
6. Действие галоидангидридов угольной кислоты на ароматические углеводороды. В присутствии хлористого алюминия получаются производные ароматических кислот:
СбНб + Cl—СО—С1 —> CeH6—СОС1 + НС1 фосген
СбНв + С1—СООС2Н5 —> Свн5—СООС2Н5 + НС1
хлоругольный эфир
Ароматические одноосновные кислоты, например бензойная кислота, встречаются в природе—частью в свободном виде, частью в виде производных—в смолах, бальзамах, эфирных маслах, а также в животных организмах.
Физические свойства. Ароматические кислоты представляют собой кристаллические вещества. Их низшие гомологи несколько растворимы в воде, хорошо растворяются в спирте и эфире, летучи с парами воды. Они являются более сильными кислотами, чем жирные кислоты с тем же числом атомов углерода.
Таблица "24
Ароматические одноосновные кислоты
Название Формула Температура плавления °C Температура кипения °C Константа диссоциации /<25°0-'0
Бензойная Толуиловые: свн5соон СН3СвН4СООН 122,4 250,0 6,0
орто- 103,7 259 13,5
мета- 111,7 263 5,6
пара- 181 275 4,3
Фенилуксусная с6н8сн2соон 76,9 266,5 6,0
Гидрокоричная (3-фенилпро-пионовая) С6Н8СН2СН2СООН 48,6 280 2,3
Гидратроповая (а-фенилпро-пионовая) С8Н8СН(СН3)СООН Жидкость 265 4,25
Химические свойства. Карбоксильная группа ароматических кислот может подвергаться превращениям, при которых образуются всевозможные производные кислот. Многие из этих производных хорошо кристаллизуются. Наибольшее число производных (больше, чем для какой-либо другой органической кислоты) синтезировано для бензойной кислоты (табл. 25).
Водородные атомы бензольного ядра ароматических кислот могут замещаться на галоиды, нитрогруппу и сульфогруппу, причем карбоксильная группа направляет такие заместители в мета-поло-жение. Как и в других случаях мета-ориентации, реакции замещения водородных атомов бензольного ядра ароматических кислот протекают труднее, чем для ароматических углеводородов.
Ароматические кислоты гораздо легче, чем углеводороды, восстанавливаются в гидроароматические соединения. Так, уже действием амальгамы натрия бензойную кислоту можно превратить в тетрагидробензойную. При дальнейшей гидрогенизации (натрием в амиловом спирте или водородом в присутствии никеля) получается циклогексанкарбоновая, или гексагидробензойная, кислота.
Действием водорода в присутствии никеля при повышенной температуре под давлением можно и непосредственно превратить бензойную кислоту в гексагидробензойную.
Бензойная кислота и ее производные. Бензойная кислота содержится в некоторых природных смолах и в небольших количествах (для фармацевтических целей) получается перегонкой бензойной смолы. В больших масштабах ее получают из толуола непосредственным окислением или хлорированием с последующим омылением образующегося бензотрихлорида.
Бензойная кислота содержится в лошадиной моче в виде бензоил-гликоколя, или гиппуровой кислоты, С6Н5СО—NH—СН2—СООН, при омылении которой, например в процессе гниения, выделяется свободная бензойная кислота.
Бензойная кислота легко возгоняется, образуя белые пластинки, из растворителей же кристаллизуется в виде игл с темп, плавл. 122,4°. Растворимость бензойной кислоты сильно увеличивается при повышении температуры. В водной суспензии она расплавляется при 90°, образуя два слоя: нижний—раствор воды в бензойной кислоте (4,12% Н2О) и верхний—водный раствор, содержащий 11,2% бензойной кислоты; при 116° оба слоя смешиваются между собой. На холоду она почти не имеет запаха, пары же ее имеют интенсивный своеобразный запах, сильно раздражают слизистые оболочки и вызывают кашель.
Бензойная кислота и ее соли находят некоторое применение в медицине, а также в пищевой промышленности как консервирующее средство. В довольно значительных количествах она применяется в производстве красителей. Бензойная кислота служит также исходным материалом для получения перекиси бензоила—одного из инициаторов полимеризационных процессов.
Хлористый бензоил, или бензоилхлорид, С6Н5—СОС1 получается при действии хлористых соединений фосфора на бензойную кислоту или действием хлора на бензальдегид. Бензоилхлорид—жидкость с неприятным запахом хлорангидридов; пары его раздражают слизистые оболочки. Он чрезвычайно легко вступает в реакции, свойственные хлорангидридам, но реагирует не так бурно, как низшие хлорангидриды жирных кислот (см. том I, стр. 275). Так, например, с водой он реагирует медленно. При лабораторных синтезах, а отчасти и в технике, бензоилхлорид применяется как «бензоили-рующее» средство.
Для введения бензоильной группы на место гидроксильного водорода часто пользуются методом Шоттен-Бауманн а. Вещество, в которое надо ввести бензоильную группу, взбалтывают с хлористым бензоилом в присутствии раствора едкой щелочи или соды. Иногда реакцию проводят в неполярных органических растворителях (бензол, эфир и пр.) в присутствии сухой порошкообразной соды или органических третичных аминов, главным образом пиридина. Амины легко бензоили-руются уже при нагревании их солей с хлористым бензоилом.
Ангидрид бензойной кислоты, или бензойный ангидрид, (С6Н5СО)2О получается действием хлористого бензоила на сухую порошкообразную натриевую соль бензойной кислоты. Он является более слабым бензоилирующим средством, чем хлористый бензоил. С водой бензойный ангидрид при обычной температуре реагирует лишь чрезвычайно медленно, с кипящей водой—довольно быстро, но не бурно, образуя бензойную кислоту.
Перекись бензоила (С6Н5СО)2О2 легко получается бензоилированием по Шоттен-Бауманну (см. выше) перекиси водорода или перекиси натрия. Перекись бензоила применяется при лабораторных синтезах как потенциальный источник пербензойной кислоты.
Перекись бензоила находит промышленное применение как инициатор полимеризации многих винильных производных (см. том I, стр. 368 сл.). Повидимому, инициаторами полимеризации являются образующиеся при распаде перекиси бензоила свободные радикалы:
СбН5СО—О—О—ОССбН5 -> 2СбН5СО-О .
Гидроперекись бензоила (пербензойная, или надбензойная, кислота) С6Н5СО—О—ОН получается в виде натриевой соли действием алкоголята натрия на перекись бензоила:
С6Н5СО—О
| + NaOC2H5 —> СвНбСО—О—ONa + С6Н5СООС2Нб С6Н5СО—О
Из этой соли получают свободную гидроперекись бензоила, представляющую собой очень слабую кислоту. Обычно при этом ее тотчас же растворяют в СНС13 или СС14 и пользуются такими растворами. Гидроперекись бензоила является сильным окислителем, выделяет иод из йодистого калия, легко разлагается, особенно в растворах. Она легко летуча и обладает резким запахом, напоминающим запах озона или гипохлоритов.
Гидроперекись бензоила применяется также как реактив для определения строения соединений, содержащих этиленовую связь. При взаимодействии гидроперекиси бензоила с такими соединениями сначала получаются производные окиси этилена, или а-окиси (реакция Н. А. Прилежаева*):
^>С--0^-1- CeHs—СООН
Действием воды на а-окиси получаются гликоли
при дальнейшем окислении перманганатом превращающиеся в кетоны; таким путем надежно устанавливается положение двойной связи. В случае алициклических соединений с двойными связями гидроксильные группы всегда становятся в Чнс-положение. Таким образом, реакция Прилежаева является самым надежным способом получения циклических цис-гликолей.
Эфиры бензойной кислоты получаются обычными способами этерификации. Они* обладают сильным специфическим запахом и поэтому иногда используются для открытия этилового спирта.
Амид бензойной кислоты, или бензамид, С6Н5—CONH2 получается действием аммиака или углекислого аммония на хлористый бензоил. Он образуется в виде листочков, очень мало растворимых в холодной воде, лучше—в горячей. В бензамиде водородные атомы группы NH2 значительно легче, чем в жирных амидах, замещаются на металлы. Например, при действии натрия получается бензамид натрия C6H5CONHNa, из которого обменным разложением с солями серебра и ртути можно получить серебряную соль С6Н5—CONHAg и ртутную соль (CGH5CONH)2Hg.
Бензамид представляет собой один из простейших примеров соединения, способного к амидной таутомерии (см. том I, стр. 745). Так, при действии йодистого метила бензамид натрия дает метилбензамид (N-производное)
yNH—СН3 / J
при омылении распадающийся на бензойную кислоту и метиламин, чем доказывается связь метила с азотом. При действии же метилирующих средств на серебряную соль бензамида получается производное изобензамида, или имидоэфир (О-производное)
строение которого доказывается тем, что при его омылении получаются бензойная кислота, аммиак и метиловый спирт.
* Н. А. Прилежаев (1873—1944). Профессор Минского университета, действительный член Белорусской Академии наук. С 1920 г. много лет очень подробно разрабатывал реакцию, названную его именем.
Бензоилгликоколь CeH5CO—NH—СН2—СООН, называемый также гиппуровой кислотой, содержится в значительных количествах в моче травоядных (особенно лошадей) и в ничтожных количествах в моче прочих млекопитающих, Синтетически бензоилгликоколь легко получается из'хлористого бензоила и гликоколя по реакции Шоттен-Бауманна (стр. 290).
Гиппуровая кислота образует призмы, на холоду довольно трудно растворимые в воде и еще труднее—в спирте, но при повышенной температуре легко в них растворяющиеся.
Бензонитрил, или цианобензол, СвН5—CN, кроме указанных выше способов (стр. 286), может быть получен отщеплением воды от бензамида при помощи пятиокиси фосфора. Он вступает во все реакции, обычные для нитрилов.
При действии натрия бензонитрил легко полимеризуется, образуя циклическое соединение киафснин:
// \ N N
I И Сбнб-С с—свн5
'Ч / N
Бензонитрил в присутствии натрия легко присоединяет анилин, образуя фенилбензениламидин:
,NH
CeH5—CN + H2N-C,H6 -* CeH.-C^
\NH-c6H5
При действии хлористого бензоила на гидразин образуется бензоил-гидразин, который с азотистой кослотой дает бензоилазоимид:
С6Н5—СО—NH—NH2 + ON—ОН -> Q>HS— СО— N, + 2Н,0 бензоилгидразин бензоилазонмид
Бензоилазоимид—слабо взрывчатое вещество. При кипячении со щелочами он распадается с образованием солей бензойной и азэтисто-водородной кислот и выделением воды:
C6H6-CON3 + 2КОН -> СвН6-СООК:+KNS + Н2О
Для азоимидов (азидов) Курциус предположил циклическое строение:
ZN
-N< II
XN
Впоследствии Анджели и Тиле нашли, что свойства азидов лучше передаются линейной формулой:
- N=N=N
С точки зрения современных электронных представлений азидная группа должна иметь следующее строение
—N=N=N подтвержденное на примере фенилазида методом меченых атомов с помощью N15.
Для метилазида электронографическим методом найдены следующие расстояния между атомами линейной структуры:
Угол а здесь меньше 180°.
Замещенные бензойные кислоты. Галоидбензойные кислоты обыкновенно получают окислением галоидозамещенных гомологов бензола (например, окислением о-хлортолуола получается о-хлор-бензойная кислота C1CGH4—СООН) или из аминобензойных кислот диазореакциями.
Галоидозамещенные бензойные кислоты' являются более сильными кислотами, чем бензойная.
Таблица 26
Замещенные бензойные кислоты
Название Орто Мета Пара
температура плавления °C константа диссоциации температура плав-ления °C константа ! диссоциации температура плавления °C константа диссоциации
Фторбензойная 124 3,0-10“4 124 1,4-10—4 182,6 1,4-10—4
Хлорбензойная 140,7 1,28-Ю-3 154,9 1,56-10“4 239 7,8-10“5
Бромбензойная 149 1,6-10“3 156 1,5-10—1 254 —
Иодбензойная 162 1,4-10“ 3 187 1,63-io—4 267 —
Нитробензойная 147 6,2 • IO-3 141 3,4-10“'’ 240 4,2-10~4
Иодбензойные кислоты, присоединяя молекулу хлора, дают карбоксипроизводные фенилиодидхлорида:
сбн
zJCl2
‘\соон
которые под действием щелочей превращаются в иодозобе нзойные кислоты'.
с„н
zJO
Х200Н
Особенно легко иодозосоединение образуется при окислении о-иодбепзойпой кислоты. Окислением ее перманганатом легко получается соединение пятивалентного иода—о-иодобензойная кислота
СООН
Нитробензойные кислоты (орто- и пара-изомеры) получаются легче всего окислением о- и п-нитротолуолов. л-Нитробензойная кислота является главным продуктом нитрования бензойной кислоты. о-Нитробензойная кислота чрезвычайно легко получается также при самопроизвольном окислении о-нитробепзальдегида на прямом солнечном свету:
СНО
СООН
о-Нитробензойная кислота обладает сладким вкусом.
Вступление нитрогруппы, особенно в орто-положение к карбоксильной, сильно повышает константу диссоциации кислоты.
З,б-Л.инитробензойная кислота (темп, плавл. 205°) получается при энергичном ндароваиии бензойной кислоты. Ее хлорангидрид (темп, плавл. 67°), подобно хлорангидриду самой бензойной кислоты, применяется для идентификации окси-и аминосоединений.
2,^$-Тринитробензойная кислота легко получается окислением тротила. Она плавится при 210°, теряя СО2 и образуя симметрический тринитробензол.
Окончательными продуктами восстановления нитробензойных кислот являются аминобензойные кислоты. Были получены также промежуточные продукты восстановления нитрогруппы (стр. 250).
Сульфобензойные кислоты (орто- и пара-) получаются окислением о- и n-толуолсульфокислот. Сульфированием бензойной кислоты получается главным образом л/-сульфобензойная кислота.
Из сульфобензойных кислот наибольшее значение получила о-сульфобензойная кислота, вследствие того, что имид этой кислоты, или сульфинид бензойной кислоты, называемый сахарином
нашел большое применение благодаря чрезвычайно сладкому вкусу при, повидимому, полной безвредности в малых дозах. Сахарин имеет исключительно вкусовое значение, но не питательное, так как организмом он выделяется с мочой в неизмененном виде.
Производство сахарина обычно состоит из следующих процессов. •
1. Толуол сульфируют хлорсульфоновой кислотой (стр. 259), причем получается смесь п- и о-толуолсульфохлоридов:
СН3 СН3
A A/SO-CI
U U
I
SO2C1
Большая часть твердого п-сульфохлорида отделяется от жидкого орто-соединения вымораживанием.
2. Действием аммиака на жидкий о-сульфохлорид получают сульфамид
СН3
Jx/S0.2-NH, и
в котором преобладает орто-соединение*. Для отделения примеси пара-соединения (и небольшого количества мета-соединения) используют то обстоятельство, что о-сульфамид легче растворяется в щелочах, чем его изомеры.
3. Окислением о-толуолсульфамида перманганатом или хромовой смесью получают о-сульфамид бензойной кислоты
СООН
J'A-NH, и
который легко отщепляет воду, превращаясь в сахарин.
Сахарин представляет собою белый кристаллический порошок (темп, плавл. 224°), трудно растворимый в холодной воде и обладающий сильными кислотными свойствами. При нагревании с водой он медленно, а в присутствии кислот быстро, омыляется, образуя аммиак и о-сульфобензойную кислоту.
Натриевая соль сахарина, называемая кристаллозой
в 400 раз слаще тростникового сахара, чрезвычайно легко растворима в воде и отлично кристаллизуется. Она обыкновенно и поступает в продажу под названием сахарина. Аммонийная соль сахарина, или сукрамин, обладает еще более сладким вкусом, чем натриевая.
* о-Толуолсульфамид—короткие призмы, темп, плавл. 158,2°; п-пголуол* сульфамид—длинные иглы, темп, плавл. 137,5°. Их эвтектическая смесь плавится при 114°.
О ТАК НАЗЫВАЕМЫХ ПРОСТРАНСТВЕННЫХ ЗАТРУДНЕНИЯХ
Для производных ароматических кислот было замечено явление, названное пространственными затруднениями. Явление это состоит в том, что некоторые вещества лишь с трудом вступают в реакции, обычные для этого класса соединений, а иногда и вовсе не вступают. В большинстве случаев такие затруднения возникают тогда, когда функциональная группа, которая должна вступать в реакцию, окружена другими радикалами. Поэтому В. Мейер предположил, что соседние радикалы пространственно затрудняют доступ молекул реагента к находящейся между ними функциональной группе, осложняя таким образом протекание реакций.
Действительно, уже однозамещенные бензойные кислоты, содержащие заместитель в орто-положении, этерифицируются труднее, чем остальные изомеры. При двух же заместителях, находящихся в орто-положении к карбоксильной группе, т. е. для кислот типа
/Х
СООН
\х непосредственная этерификация* чрезвычайно затрудняется, а иногда и вовсе не происходит. Если же получить сложный эфир такой кислоты другим способом (например, действием галоидных алкилов на серебряную соль), полученный сложный эфир омыляется с трудом или вовсе не омыляется. Аналогичные затруднения наблюдаются также для двузамещенных в орто-положении соединений других классов. Например, кетоны типа
/СНз
со— R
Хсн3
не реагируют с гидроксиламином; ксилидин строения ХИ,
\сн8
нб дает с иодистым метилом солей четырехзамещенного аммония; нитрилы строения
/Х
O-CN
♦ Под непосредственной этерификацией подразумевается этерификация пу-ТВм нагревания кислоты с избытком спирта, например метилового, в присутствии небольших количеств серной кислоты или при пропускании в реакционную смесь хлористого водорода.
не омыляются в в динитротолуоле
обычных условиях в соответствующие кислоты; строения
zNO2
4no2
водородные атомы метильной группы не замещаются на хлор и т. д.
Подобные явления обнаружены не только для ароматических, но и для других классов соединений; так, с гидроксиламином, семикарбазидом и пр. не реагирует не только димезитил кетон, но и гексаметил ацетон:
/СН3 Н3С\ нзс-О-С0-О-СНз \сн3 Н3с/ димезитилкетон
сн3 сн3
I I
СН3—С—СО—С—сн3
I I
сн3 сн3 гексаметилацетон
Представления о пространственных затруднениях подтверждаются новейшими стереохимическими исследованиями.
Ароматические амины
Простейшим ароматическим амином является фениламин (также амино- или амидобензол) С6Н5—NH2, обыкновенно называемый анилином.
Впервые фениламин был получен в 1826 г. из индиго (по-испански индиго называется anil), а в 1834 г. выделен Рунге из каменноугольного дегтя. Метод получения анилина из индиго был улучшен Ю. Ф. Фрицше (1840 г.), давшим анилину это название. Синтетически анилин был впервые получен Н. Н. Зининым* в 1842 г. восстановлением нитробензола сернистым аммонием.
Следующий гомолог анилина—производное толуола—может существовать в четырех изомерных формах. Три изомера содержат группу NH2 в бензольном ядре и называются о-, м- и п-толу ид инами, а четвертый изомер, в котором группа NH2 замещает водородный атом метильной группы, называется бензиламином:
СН8—CtH4— NH толуидины
СвН5—СН,—NH, бензиламин
* Николай Николаевич Зинин (1812—1880). Блестяще окончив Казанский университет в 1833 г. как математик, с 1835 г. посвятил себя химии. В 1837 г. уехал в заграничную командировку; работал долгое время в лаборатории Ю. Либиха в Гиссене, затем у Пелуза в Париже. Вернулся в 1840 г. В 1842 г. получил анилин, названный им бензидамом, и а-нафтиламин; в 1844 г.—лс-фенилен-диамин и нафтилендиамин; в 1845 г.—льаминобензойную кислоту. В 1846—1847 гг. получил азоксибензол и открыл бензидиновую перегруппировку гидразобензола. С 1847 г. профессор Медико-хирургической академии в Петербурге, с 1858 г.— экстраординарный, а с 1865 г. ординарный академик. С 1868 г. первый президент Русского химического общества (до 1878 г.).
Аминопроизводные ксилолов, содержащие аминогруппу в ядре, называются ксилидинами, аминопроизводное мезитилена—мезидином, псевдокумол^—псевдокумидинами и т. д.
Изомеры более сложного состава различают, обозначая цифрами положения в ядре замещающих групп, например 1,2,4-триметил-5-аминобензол, 2,4,5-три-метиланилин и т. п.
Амины с группой NH2 в бензольном ядре
Амины, содержащие аминогруппу в бензольном ядре, иногда называют просто анилинами.
Способы получения. 1. Восстановление нитросоединений. Амины этого рода получаются почти исключительно восстановлением ароматических нитросоединений (стр. 246). Для этой цели применяют самые разнообразные восстановители. При лабораторных синтезах применяют чаще всего олово и соляную кислоту, а в промышленности используют более дешевые восстановители, например железо и серную кислоту.
2. Замещение гидроксила в фенолах. Способ получения аминов, состоящий в нагревании фенолов под давлением с аммиаком и хлористым цинком или хлористым кальцием
C6H5OH + NH3 —> C6H5-NH2 + H2O
а также с сернистокислым аммонием, применяется главным образом для синтеза более сложных аминосоединений, особенно производных нафталина.
3. Аминирование углеводородов. Пока только теоретический интерес представляет образование анилина при пропускании паров бензола с аммиаком через слабо накаленную трубку:
С6Н6 + NH3 -> C6H5-NH2 + H2
Аналогичная реакция непосредственного аминирования углеводорода происходит при нагревании нафталина с амидом натрия
С10Н8 + NH2Na —> С10Н7—NHNa ф- Н2 с последующим разложением натриевого производного водой.
4. Обмен галоида па аминогруппу. В последнее время этот способ (стр. 256) приобретает промышленное распространение, особенно в применении к замещенным аминам; например, /7-нитроанилин получается действием аммиака на п-нитро-хлорбензол при нагревании под высоким давлением.
Физические свойства. Ароматические амины (анилины)—бесцветные высококипящие жидкости или твердые вещества с своеобразным запахом.
Низшие ароматические амины несколько растворимы в воде и сами ее растворяют. Удельный вес их близок к единице.
Химические • свойства. Большая часть реакций анилинов, как первичных аминов, аналогична реакциям жирных первичных аминов. Лишь в некоторых реакциях (особенно в реакции с азотистой кислотой) проявляются отличия, характерные для ароматических соединений.
1 Основные свойства. Амины, содержащие аминогруппу в ядре, являются значительно более слабыми основаниями, чем жирные амины, вследствие включения свободной электронной пары атома азота аминогруппы в систему сопряжения с к-электро-нами бензольного ядра (стр. 241):
NII3 .............
CH3-NIL ..........
c6h5-nii2.........
2-10~5
5- 10"4
5,3- 1О"10
При замене атомов водорода в аминогруппе анилина.на алкильные группы константы диссоциации несколько возрастают, но не до уровня алифатических аминов. Это видно из следующего сопоставления:
С6Н5— NH2 ........ 5,3-10—10
С6Н5—NHCH3 ... 5,0-10"9
СбН5—N(CH3)2 . . . 1,0-10"9
При замене атомов водорода в анилине на фенильные остатки основные свойства получающихся дифениламина и трифениламина еще более ослабляются, и у трифениламина способность к образованию солей исчезает.
Растворы анилина не обнаруживают по лакмусу щелочной реакции и почти не проводят электрический ток. Со слабыми кислотами, например с угольной, они не образуют солей, а соли сильных кислот имеют кислую реакцию, и их можно титровать щелочами, как и свободные кислоты.
2. Образование жирно-ароматических вторичных и третичных аминов и четвертичных аммониевых оснований. При действии галоидных алкилов получаются смешанные жирно-ароматические амины, причем реакции их образования аналогичны реакциям получения жирных аминов (см. том I, стр. 251).
Например, анилин с иодистым метилом дает соль аммониевого основания, из которой при обработке щелочью получается метил-анилин:
С6Н5— NH2 + CH3J —> [С6Н5—NH2CH3]J —> С6Ы5—NH—сн3
Аналогично из метиланилина получается диметиланилин
с6нб—nh—сн3 + CH3J (СбН5—NH(CH3)2]J -> СбН5—N(CH3)2
а при дальнейшем действии йодистого метила—четвертичный иодисгпый триметилфениламмоний:
С6Нб—N(CH3)2 + CH3J -> [C6H6-N(CH3)3]J
Свободное четвертичное основание, являющееся сильной щелочью, можно выделить при помощи обмена с AgOH, а также действием растворов метилата или этилата натрия на растворы четвертичных хлоридов в соответствующих спиртах:
[СбН5— N(CH3)3]C1 + NaOC2H5 -> [C6H5-N(CH3)3]OC2H5 4- NaCl
[C6H5-N(CH8)3]OC2H5+H2o [C6H5-N(CH3)3]OH 4-C2H5O11
Вместо галоидных алкилов можно действовать спиртами на галоидиводород-ные соли аминов при нагревании в автоклаве под давлением. При этом галоидный алкил образуется из спирта и кислоты и действует в момент образования. Для получения жирно-ароматических‘аминов применяются также и другие алкилирующие средства. Так, алкиланилины можно получать пропусканием паров анилина и спирта через нагретые квасцы или через окись алюминия (также окись тория) при высоких температурах.
3. Образование чисто ароматических вторичных и третичных аминов. Чисто ароматические вторичные амины обыкновенно получают нагреванием анилинов с их хлористоводородными солями, например:
[СбН5—NH3]C1 + C6H6-NH2 -> С6Н5—NH—CGH5 Д-N114C1 дифениламин
Третичные амины этого рода легко получаются по реакции Ульмана (стр. 245) действием иодбензола или бромбензола на дифенид-амины в присутствии сухого поташа и небольшого количества порошкообразной меди, например:
(C6H5)2NH 4- C6H5J -> (C6H6>3N 4- HJ
трифенил-амин
Чисто ароматические четвертичные аммониевые основания неизвестны.
4. Образование анилидов. Анилидами называются амиды кислот, в которых водородный атом амидогруппы замещен на ароматический радикал (арил). Например, СН3СО—NHC6H5 есть анилид уксусной кислоты, или ацетанилид; С6Н5СО—NHC6H5— анилид бензойной кислоты, или бензанилид.
Анилиды образуются при нагревании кислот с анилином:
СбН5— NH2 4- но—СОСН3 —> C6H5-NH—СОСН3 4-Н2о
Еще легче они получаются при действии на анилин более сильных ацилирующих средств (сложных эфиров, ангидридов или галоид-ангидридов).
Таблица 27
Ароматические амины
Название Формула Температура плавления i °C Температура кипения Удельный вес Показатель преломления п20
Анилин (аминобензол) c„h5nh2 -6,1 184,40 1,0217 1,5863
о-Толуидин (1 -метил-2-амино-бензол) ch3c6h4nh2 —24,4(a) —16,4(b) 200,3 0,9988 1,5728
л^Толуидин (1 -метил-3-амшю- —31,2 * 203,4 0,9888 1,5686
бензол) 0,9615 (4°) -
п-Толуидин (1-метил-4-аминобензол) о-Ксилидины: » (CH3)2C»H3NH2 +43,7 200,5 1,5532 (при 59,1°)
рядовой (1,2-диметил-3-аминобензол) > — 223 0,9965 (при 15°) 1,5706 (при 15,3°)
несимметрический (1,2-ди-метил-4-аминобензол) > +51 226 1,0755 (при 17,5е) —
м-Ксилидины:
рядовой (1,3-диметил- » — 216 0,9796 1,5612
2-аминобензол)
несимметрический (1,3-диметил-4-аминобензол) > — 212 0,9783 ( 1,5547 (при 19,6°
симметрический (1,3-ди-метил-5-аминобензол) — 222 0.9791 (при 12. Г) 1,5618 (при 12,1°
п-Ксилидин (1,4-диметил-2-аминобензол) » + 15,5 213,4 0,9790 ( 1,5591 (при 21,3°)
Мезидин (1,3,5-триметил- (CH3)3C6H2NH2 —4,9 233 0,9633 —
2-аминобензол)
Псевдокумидин (1,2,4-три- » +68 234 — —'
метил-5-аминобензол) 1,5702 (при 21,2°)
Метиланилин (N-метиламино-- бензол) C.HBNH(CH3) —57 196,2 0,9895
Диметила нилин (М,Кт-диме-т ил аминобензол) CeH6N(CH3)2 +2,4 194,1 0,9561 1,5587
Диэтиланилин (М,Ы-ди этиламинобензол) C6H6N(C2H6)2 —21,3 (стаб. форма) —34,4 (нестаб.) 217,1 0,9348 1,5421
Дифениламин (CeH5)2NH +53,5 301,9 1,160 (тв. при 20°) —
•Трифени ламин (C.Hg)3N 4-127 365 -—
Бензиламин CeH6CH2NH2 — 184,5 0,9827 ( 1,5441 (при 19,5°^
а-Фенилэтиламин CeHE >CHNH, сн/ — 185,1 0,952 —
р-Фенилэтипамин CeH3CH2CH2NH, । 195 0,9640 И) —
Двухосновные кислоты дают два ряда анилидов, например щавелевая кислота образует следующие анилиды:
C6H5NH—СО—СООН C6H5NH—СО—СО—NHC6H5
оксаниловая кислота оксанилид
Анилиды являются замещенными амидами и реагируют подобно амидам. Например, при кипячении с водой в присутствии кислот или щелочей анилиды омыляются, присоединяя воду и образуя кислоту и анилин.
Ацетанилид C6H5NH—СОСН3 образует бесцветные листочки, трудно растворимые в холодной воде, легко—в горячей. Он находит некоторое применение в медицине под названием антифебрина. Антифебрин—первый синтетический жаропонижающий препарат (1886 г.). В промышленности ацетанилид применяется в качеств' промежуточного продукта при синтезе важных лекарственных веществ (например, сульфамидных препаратов) и красителей.
5. Образование шиффовых оснований. При действии ароматических альдегидов на анилины выделяется молекула воды и получаются анилы, или так называемые шиффовы основания:
С6Н5—NH2 + О=НС—СбН5 -> СбН5—N—СН—СбНб + Н2О бензилиденанилин, или бензальанил
В присутствии разбавленных кислот легко происходит обратная реакция.
С жирными альдегидами анилины образуют нестойкие анилы, легко переходящие в кислой среде в циклические соединения хинолинового ряда (хинальдиновый синтез Дебнера—Миллера, стр. 612).
6. Действие сероуглерода. Действием сероуглерода на анилин /S
получается не производное дитиокарбаминовой кислоты C6H5NH—С<; , а произ-
XSH
во ;ное тиомочевины—дифенилтиомочевина'.
2СбН5— NH2 + CS2 —> (C6H5NH)2CS + H2S
Э о соединение при действии кислот даст фениловое горчичное масло: (C6h5nh;2cs —> с6н5— n=c=s + c6ii5-nh2
7. Действие хлороформа и спиртовой щелочи. В этом случае реакция протекает так же, как и в жирном ряду (см. том I, стр. 286), и приводит к образованию фенилизонитрила (фенилкарбиламина) С6Н5—N = C.
8. Диазотирование. Особенно важной реакцией является действие на анилины азотистой кислоты. В противоположность жирным первичным аминам, сразу выделяющим азот и обменивающим аминогруппу на гидроксил, здесь получаются весьма важные промежуточные соединения, называемые диазосоединениями (см. стр. 318):
[C6H6NH3]C1 4- ON—ОН -> [CeH5N2]Cl 4-2Н2О •
Вторичные жирно-ароматические амины образуют с азотистой кислотой нитрозамины, например:
Под действием концентрированной серной кислоты эти соединения изомеризуются таким образом, что нитрозогруппа обменивается местом с водородным атомом ядра в пара-положении:
0N“<C^”’NH—СНз
нитрозометиланилин
Третичные жирно-ароматические амины, в противоположность жирным аминам, легко реагируют с азотистой кислотой, причем выделяется вода, а нитрозогруппа вступает сразу вместо атома водорода в пара-положение:
ON—ОН +<Q>-N(CH3)2 -> ON-<Q-N(CH3)2 + Н2О н итрозоди метила нилин
При нагревании со щелочами нитрозодиметиланилин обменивает группу N(CH3)2 на гидроксил, в результате чего получаются и-ни-трозофенол и вторичный амин (диметиламин):
ON—N(CH3)2 + КОН —» ON—<\^—ОН + NH(CH3)2
Так же можно получать и высшие диалкиламины.
При нагревании ароматических нитрозаминов с фенолом и серной кислотой с последующим растворением смеси в воде и насыщением едкой щелочью возникает интенсивное синее окрашивание (реакция Либермана на нитрозо соединения).
9. Восстановление. Анилины восстанавливаются с трудом. Однако при действии водорода под давлением в присутствии никеля анилин при нагревании переходит в циклогексиламин:
Наряду с циклогекснламином, в результате замены группы NH? на водород, может получиться также циклогексан.
10. Окисление. Подобно гидроксилу в фенолах, группа NH2 в анилинах сообщает бензольному ядру способность легко окисляться. При этом получаются киноны (стр. 351) или их производные.
Анилин медленно окисляется уже кислородом воздуха. Его соли легко окисляются в присутствии ничтожных количеств пятиокиси ванадия как катализатора. Окисление анилина и его солей происходит также при действии белильной извести или хромовой кислоты.
При этом образуется ряд окрашенных веществ, сначала зеленого или фиолетового цвета, по мере окисления все темнеющих, и, наконец, может получиться весьма важный в технике крашения тканей очень прочный черный краситель, называемый черным анилином или анилиновым черным*.Эту реакцию можно осуществить прямо на волокне. Промежуточно образующийся при окислении анилина темнозеленый краситель называется эмеральдином. Появление фиолетового окрашивания с хлорной известью и темнозеленого с хромовой кислотой является качественной реакцией на присутствие анилина.
С другими окислителями и в иных условиях можно получить из анилина все промежуточные продукты восстановления нитробензола (стр. 250) и самый нитробензол.
Так, например, действием перманганата на анилин можно получить азобензол СбН5—N=N—С6НБ и нитробензол.
При действии мононадсерной кислоты НО—SO2—О—ОН из анилина получается нитрозобензол СбН5—NO.
11. Действие галоидов, азотной и серной кислот. Подобно гидроксилу, группа NH, чрезвычайно облегчает реакции замещения в бензольном ядре, направляя замещающие группы исключительно в орто- и пара-положения.
Будучи растворен в концентрированной серной кислоте, анилин не реагирует с галоидами, а при его нитровании, наряду с п-питроанилином,' образуется также много л<-нитроанилина. Это объясняется, вероятно, тем, что в таком растворе отсутствует анилин со свободной аминогруппой, а содержится лишь'соль четвертичного аммониевого основания, вследствие чего изменяется характер ориентирующего действия заместителя.
При действии на анилины хлорной или бромной воды обычно происходит замещение сразу трех атомов водорода на галоид, причем образуется, например, триброманилин строения:
/Вг
Вг—NH,
^Вг
Чтобы получить однозамещенные анилины, галоидируют или нитруют обычно не сам анилин, а его производные, например анилиды, и затем уже, после омыления, получают замещенные анилины.
При сульфировании анилина легко получается zz-сульфокислота, так называемая сульфаниловая кислота.
Весьма вероятно, что действие заместителей первоначально направляется на аминогруппу и что, например, при действии галоидов сначала получается соединение типа CeH5NH—Вг, а при действии азотной кислоты—нитрамин строения C6H5NH—NO2. Действительно, такие соединения, полученные в других условиях, при действии кислот изомеризуются: Вг или NO2 обмениваются местами с водо
* О строении этого красителя см. на стр. 356.
родными атомами, находящимися в орто- или пара-положениях, с образованием замещенных анилинов.
12. Реакции к о*н денсации. Анилины, как и фенолы, очень легко вступают в реакции конденсации, причем особенно легко образуются производные дифенилметана и трифенилметана. Новые углерод-углеродные связи в орто-положении образуются несколько труднее, чем в пара-положении.
Легче, чем сами анилины, вступают в реакции конденсации моноалкил- и особенно диалкиланилины, например:
(CH3)2N-<^J + сн2° + O-N(CH3)2 -+
(CH3)2N-<^^-CH,-^2^>-N(CH3)2 + Н2О
о
К реакциям конденсации следует отнести и образование гомологов анилина при нагревании хлористоводородных солей диалкил-анилина.
Например, хлористоводородный диметиланилин дает при нагревании под давлением или в токе хлористого водорода смесь первичных аминов: /г-толуидина, несимметрического ju-ксилидина и мезидина.
Реакция, повидимому, происходит между ароматическим амином и хлористым метилом, который образуется при нагревании хлористоводородного диметиланилина по схеме:
C6H5N(CHs\.HC1 ±5 Cgm.-NH-CH3 + CH8C1
При взаимодействии хлористого метила с анилином атомы водорода в пара- и орто-положениях последовательно замещаются на метильные группы.
Анилин С6Н5—NH2 является одним из важнейших продуктов химической промышленности: служит исходным веществом для получения красителей (анилиновые красители), взрывчатых веществ и пр.
Ежегодное производство анилина измеряется многими десятками тысяч тонн.
Анилин не смешивается с водой В насыщенном водном раствора при 16е содержится 3,11% анилина, а в насыщенном водой при 25 анилине содержится 5% воды. Он смешивается со спиртом, эфиром и бензолом, легко летуч с парами воды.
Анилин весьма токсичное вещество, причем он может проникать в организм не только через слизистые оболочки дыхательных путей, но и через кожу.
Монометиланилин С6Н5—NH—СН3 (бесцветная жидкость, темп. кип. 196,2°) не дает характерной окраски с хлорной известью. С фосгеном он образует симметрическую дим?пшлдифенилмочевину
2CfiH5— NH—СН3 4- СОС12
-Лх /Сбн5
>N—С — N< 4-2HCI
СН/ \сн3
применяемую для стабилизации пироксилинового пороха.
Диметиланилин С6Н5—N(CH5)2 представляет собой жидкость своеобразного сильного дегтярного запаха, замерзающую при +2,4е; темп, кип 194,1°. Он применяется в больших количествах для производства красителей (малахитовый зеленый, метиловый фиолетовый и пр.), взрывчатых веществ (тетрил) и ряда других синтетических продуктов.
Диэтиланилин С6Н5—N(C2H5)2 (темп. кип. 217,1°; уд. вес. /=0,9348) применяется для получения диэтиламина через нитрозосоединение (стр. 303).
Дифениламин CGH,—NH—СВН5-кристаллическое вещество, имеющее слабый запах; темп, плавл. 53,5°, темп. кип. 302°. Он почти нерастворим в воде, легко растворяется в спирте, эфире и бензоле. Вследствие наличия в молекуле двух фенильных групп дифениламин—еще более слабое основание, чем анилин; соли его разлагаются водой. Как вторичный амин, он образует с азотистой кислотой нитрозосоединение:
х=/ । \=/
NO
При нитровании дифениламина получается гексанитродифенил-амин:
Серная кислота растворяет дифениламин; в присутствии даже следов азотной или азотистой кислоты такой раствор приобретает интенсивное синее окрашивание (чувствительная реакция на азотную
и азотистую кислоты). Однако подобное окрашивание вызывается также и некоторыми другими сильными окислителями.
Дифениламин производится в больших количествах как промежуточное вещество при изготовлении красителей (анилиновый синий, дифениламиновый оранжевый и др.), а также для стабилизации пироксилиновых порохов.
Трифениламин (C6H5)3N плавится при 127°. Он совершенно лишен основных свойств. В концентрированной серной кислоте при нагревании трифениламин растворяется с синей окраской.
Амины с группой NH, в боковой цепи
Амины, содержащие аминогруппу в боковой цепи, получаются обычными способами, применяемыми для синтеза жирных аминов. Так, при действии на аммиак хлористого бензила, как и при действии йодистого метила, получается ряд аминов:
первичный—бензиламин ............... С6Н5—СН,—NH,
вторичный—дибензиламин ............. (CfiH5—CH2)2NH
третичный—трибензиламин ............ (С6Н5—CH2)3N
четвертичный—соль тетрабензиламмония [(С6Нб—CH2)4NjCi
Для получения аминов этого рода особенно часто пользуются реакциями восстановления соединений с другими азотсодержащими радикалами. Так, например, бензиламин С6Н5—СН2—NH2 получается восстановлением фенилнитрометана С6Н5—СН2—NO2, бензонитрила С6Н5—CN, бензальдоксима С6Н5—CH^NOH и т. д.
Бензиламин обладает сильными основными свойствами и поглощает из воздуха углекислый газ, образуя твердый карбонат.
Для получения более сложных соединений с алифатически связанной аминогруппой часто берут в качестве исходных веществ альдегиды, которые конденсируют с различными азотсодержащими соединениями и восстанавливают продукт конденсации. Например, при восстановлении бензальанила СбНБ—CH=N—СбНБ, являющегося продуктом конденсации бензальдегида с анилином (стр. 302), получается вторичный амин СбНб—СН2—NH—СбНб, называемый бензиланилином (фенилбензиламин). Продукт конденсации бензальдегида с нитрометаном—ни-тростирол
СвНБ—СНО -F сн3— no2 -» С6Н5—сн=сн— no2 + Н,0 нитростирол
при восстановлении этиленовой связи и нитрогруппы дает первичный амин— $-фенилэтиламин СбНб—СН2—СН2—NH2.
Реакции аминов, содержащих аминогруппу в боковой цепи, почти не отличаются от реакций алифатических аминов. Бензольное ядро, не связанное непосредственно с атомом азота, не влияет или очень мало влияет на поведение аминогруппы.
Бензиламин СбНБСН2 -NH2 представляет собой жидкость с аммиачным запахом (темп кип. 187°), легко растворимую в воде и жадно притягивающую из воздуха углекислоту.
Дибензиламин (C6H5CH2)2NH кипит при 300е. При действии азотистой кислоты образует N-нитрозосоединение (CeH6CH2)2N—NO.
Таблица 28
Производные анилина, замещенные при азоте
Название Формула Температура плавления °C Температура кипения С Удельный вес
Метиланилин CelI6-NH-CH3 —57 196,2 0,9ь9б( df)
Диметила нил ин C«Hg-N(CH3)s 4-2,4 194,1 0,9361( d20)
Этиланилин CeH5—NH—С2Н5 —63,5 205,5 0.963 ( d20j
Диэтиланилин С,Н5—N(C2HB)2 -21,3 217,1 0.9348( d20)
Форманилид C6H5NH—СНО /стаб форма) -34,4 (нестаб.) 4-47,9 284 1,114 ( d33)
Ацетанилид (анти- CeH5NH—COCH3 115 303,8 1,207 ( d°)
фебрин)
Метилацетанилид zCH3 c0H5N< 102—104 253 0,977 ( d‘20)
(экзальгин) Анилидоуксусная XCOCH3 c,h5nh—сн2—соон 127
кислота (фенил-гликоколь, или фенилглицин)
Бензанилид CeH,NH—COCeHs 163 — 1,305 (тв.)
Оксаниловая кис- C,HSNH—CO—COOH 224 — —
лота
Окса нил ит CeHsNH— CO—CO—NHCeHj 254 >360 —
Фенилуретан CeH5NH-COOC2HR 53 237 —
Фенилмочевина CeHjNH—CO—NH, 147 ;с разлд 1.302
Дяфенилмочевина CeHsNH— CO—NHC»H6 239,5 260 (при 20°!
(карбанилид)
Фенилтиомочевина CeH6NH—CS—NH, 154 — —
Д ифе н и лтиомоче- C,HeNH-CS-NHC„H5 155 — —
вина
Фенилкарбиламин c8h5n=c Жидкость 166 0,977
(фенилизонитрил) Фенил изоцианат c9h6n=c=o Жидкость (с разл.) 166 (при 15°) 1,0961( d20)
(карбанил) X 4 /
Фениловое горчич- CeH6N=C=S —21 221 1,1304
ное масло Трифенилгуанидин /NHCeH, C«H8N=C< XNHCeHs 144 — »при 20°)
Трибензиламин (CeH6CH2‘3N (кристаллы с темп, плавл. 92°), присоединяя галоидные алкилы, образует соли четвертичных аммониевых оснований, но с галоидными арилами таких солей не дает.
Р-Фенилэтиламин CeHgCH,CHt—NHt (жидкость, темп. кип. 195е); имеет силь но основные свойства Он образуется при гниении белков и при нагревании фенилаланина C6HSCH2CH(NH2)COOH (стр. 391).
Производные р-фенилэтиламина, обладающие сильным физиологические действием, встречаются в природе и применяются как кровеостанавливающие средства (вызывают сужение периферических кровеносных сосудов). Таковы алкалоид эфедрин (стр. 647), адреналин (стр. 342) и пр. Сам р-фенилэтиламив также оказывает аналогичное действие, хотя и в более слабой степени.
Анилины, замещенные в ядре на галоид, нитро- и сульфогруппы
При галоидировании, нитровании и сульфировании анилинов заместители вступают в орто- и пара-положения в соответствии с правилами замещения атомов водорода бензольного ядра (стр. 238).
Орго- и пара-галоидные анилины получаются главным образом восстановлением галоиднитробензолов. лс-Нитроанилин получается неполным восстановлением д<-динитробензола.
В замещенных анилинах аминогруппа может подвергаться всем превращениям, типичным для анилинов; например могут получаться анилиды, диазосоединения и пр.
Таблица 29
Замещенные анилины
Название Формул^ Орг- j Мета Пар»
темпе гатура плав ления ICMCC 1 рлура । кипе I ния | темпе t pa-ivpa 1 плие | ления । емпе-атура кипения °C темпе ,«тура плав ления °C темп? ратуем княе- НИЯ °C
Фторанилин F-C6H4-NH2 —29 —. — 186,3 +0,8 187,4
(лоранилин ci-c6h4-~nh? —3,5 (стаб.) -14 <лаб.) 207,0 — 10,2 230,5 +72,5 232,3
5романилин Вг—С6Н4—NH, +32 229 + 17,5 251 +66,4 —
Иода нилин J—CeH4—NH, +60 — +33 —— +62,8 —
Нитроанилин NO2-CeH4-NH2 +71,5 — + 114,6 — + 147,8 —
В результате введения галоидов, и особенно нитрогрупп, основные свойства анилинов ослабляются, что иллюстрируется приводимыми ниже значениями констант диссоциации некоторых замещенных ароматических аминов:
Анилин ...........5,3-10“10 о-Нитроанилин . . 3,510”“н
я-Хлоранилин . . . 5,7-10“н л/-Нитроанилин . . 4 10’”12
п-Броманилин . . . 5,2-10"1! п-Нитроанилин . . 1,24-10“12
и-Броманилин . . 2,1 10~11
Нитроанилины представляют собой желтые кристаллические вещества. о-Нитроанилин легко перегоняется с парами воды и га-ким образом может быть отделен от нелетучего п-нитроанилина.
Летучесть о-нитрофенола и о-нитроанилина объясняют иногда образованием внутрикомплексной связи между обоими заместителями в ароматических ядрах, возможной благодаря пространственной близости заместителей:
Вследствие наличия такой связи молекулы вывать водородных связей с молекулами воды,
орто-изомеров не могут образо как пара- и мета-изомеры:
Н—N—Н---О—II
Н
H-L.. н—о
и потому более летучи.
При нагревании о- и n-нитроанилинов с водными щелочами аминогруппа легко замещается на гидроксильную группу.
Еще легче вступает в такую реакцию обмена 2,4,6-тринитро-анилин, называемый пикрамидом, который переходит при этом в пикриновую кислоту:
При действии азотной кислоты на пикрамид, растворенный в концентрированной серной кислоте, получается нитропикрамид
Со-
относящийся к классу нитрамидов. Его метильное производное, называемое те-трилом
легко получается нитрованием диметиланилина (при этом три нитрогруппы входят в ядро и, кроме того, четвертая нитрогруппа входит вместо метила к азоту).
Тетрил производится в больших количествах и является важным взрывчатым веществом; его темп, плавл. 129°; при нагревании до 190° он взрывает.
Нитроанилины имеют большое применение в производстве азокрасителей и других синтетических красителей как промежуточные продукты.
Аминобензолсульфокислоты. n-Аминобензолсульфокислота, или сульфаниловая кислота, получаемая сульфированием анилина, образует выветривающиеся кристаллы с темп, плавл. 280е (содержащие две молекулы воды), довольно трудно растворимые в холодной воде. Она имеет большое значение в производстве азокрасителей. Меньше применяется .м-аминобензолсульфокислота, называемая метаниловой кислотой'.
HO3S\
NI i.z
HO3S—nh2
сульфаниловая кислота
метаниловая кислота
Метаниловую кислоту получают восстановлением л!-нитробен-золсульфокислоты; она кристаллизуется с 1% молекулами воды в виде игл.
Сульфокислоты анилина растворяются в щелочах, но не дают прочных соединений с кислотами и, вероятно, представляют собой «внутренние» соли:
/SOI
ПРОИЗВОДНЫЕ п АМИНОБЕНЗОЛ СУЛЬФАМИДА
Очень большое значение в качестве лекарственных веществ имеют производные n-аминобензолсульфамида (амида сульфаниловой кислоты, или сульфаниламида), применяемые при лечении различных стрептококковых и пневмококковых заболеваний.
Простейшим лекарственным веществом группы сульфаниламидных препаратов является сам n-аминобензол сульфамид (темп, плавл. 164,5°)
H2N-<Q-SO2—nh2
известный под названием белого стрептоцида.
Более сложным продуктом является, например, так называемый дисульфан
H2N—SO2—NH—^^>—SO2~NH2
Если амидировать n-аминобензолсульфохлорид (вернее—его ацетильное производное) не аммиаком, а аминами гетероциклического ряда (производными тиазола, 1,2,4-тиодиазола, пиридина, пирими-
дина и др.), то получаются препараты, обладающие высокой активностью против кокковых бактерий и кишечных инфекций:
S---СН
H2N
норсульфазол (темп, плавл- 200—202°;
СН
НС7 \н
H2N-<Q>_SO2—NH—С Jit] сульфидин N
(темп, плавл 190—193°)
h9n
сульфазол 'темп плавл. 235—:
С—СН;
С—СН;
NZ ^СН
! 1,N—SO2—NH— С—СН,
сульфадимезин
(темп плавл 198—.а ! '
n-Аминобензолсульфамидную группировку содержит также красный стрептоцид, представляющий собой 4х-сульфамидо-2,4-диаминоазобензол и являющийся азокрасителем (стр. 365); его получают азосочетанием диазотированного я-аминобензолсульфамида с л/-фенилендиамином. Специфическим антибактериальным действием обладает 4-сульфамидобензиламин
H,N—CHS—SO,—NH,
известный под названием марфанияа, применяемый для лечения открытых ран, ожогов и т. п. Его хлористоводородная соль плавится при 256°. Активными антимикробными препаратами, применяемыми при лечении проказы, являются также п.п'-диаминодифениясуяьфон и его производные, например:.
СН3—CO—NH—so2—NH—СО—СН3
Прсайжуточные продукты восстановления нитросоединений
При восстановлении ароматических нитросоединений можно получить ряд продуктов неполного восстановления как с одним, так и с двумя атомами азота в молекуле. Эти же продукты могут быть получены и при окислении аминов (стр. 250). Такими путями можно получить соединения следующих классов (здесь Аг—арил):
Соединения с одним атомом азота:
Нитрозосоединения Аг—NO
Производные гидроксиламина Аг—NH—ОН
Соединения с двумя атомами азота:
Азоксисоединения Аг—N=N—Аг
Азосоединения Аг—N=N—Аг
Гидразосоединения Аг—NH—NH— Аг
Нитрозосоединения
Ароматические нитрозосоединения Аг—NO получаются: 1) электролитическим восстановлением нитросоединений; 2) окислением анилинов мононадсерной кислотой (стр. 304); 3) окислением Р-арилгидроксиламинов хромовой смесью, хлорным железом или кислородом воздуха, например:
СвН5—NHOH 4-О СРН<—NO 4-Н?О
Нитрозобензолы представляют собой бесцветные кристаллические вещества, которые в расплавленном состоянии и в растворах принимают интенсивную зеленую окраску. Это происходит потому, что в кристаллическом состоянии нигрозобензолы обладают удвоенным молекулярным весом, а при растворении и плавлении бесцветные димерные молекулы диссоциируют на простые окрашенные молекулы нитрозобензолов.
Нитрозосоединения можно окислить в нитросоединения я восстановить в амины. С аминами они дают азосоединения:
С6НБ—NO + NH2—С6Н6 —> CGH5—N==N—CeHg 4-Н?о
При действии азотистой кислоты на третичные ароматические амины, например на диметиланилин, образуются соответствующие п-нитрозосоедипения:
(CH3)2N—+ НО—NO —> (CH3)3N—NO + H./J
При действии азотистой кислоты на фенолы образуются /нш-грозофенолы, которые таутомерны /г-хинонмоноксимам (сгр. 357).
но—<\Zy—N==0 °=C>=N-0H
Нитрозобензол СбНб—NO образует прозрачные бесцветные кристаллы (темп, плавл. 68°), плавящиеся в изумрудно-зеленую жидкость и дающие растворы такого же цвета.
n-Нитрозодиметиланилин (CH3)SN—СвН4—NO образует зеленые таблички (темп, плавл. 85°). Его хлористоводородная соль—желтые иглы, плавящиеся при 177°.
п-Нитрозофенол НО—С6Н4—NO кристаллизуется в виде желтоватых игл, разлагающихся при температуре около 126е. Ею водные и спиртовые растворы окрашены в зеленый цвет. Растворы его фенолятов коричнево-красные.
Р-Арилгидроксиламины
Ароматические производные гидроксиламина называются р-замещенными, если арил связан с атомом азота, например Аг—NH—ОН, в отличие от а-замещен-ных гидроксиламинов, в молекулах которых арил связан с атомом кислорода, например Аг—О—NH2.
p-Арилгидроксиламины Аг—NH—ОН чувствительны к щелочам и кислотам, а потому получаются при восстановлении нитробензолов только в строго нейтральной среде, например действием цинковой пыли и раствора хлористого аммония или действием амальгамы алюминия на эфирные растворы нитробензола. Они
являются сильными восстановителями—восстанавливают аммиачное серебро и фелингову жидкость. Кислородом воздуха, особенно в присутствии щелочи, они окисляются в нитрозобензолы.
Важнейшей реакцией p-арилгидроксиламииов является легко протекающая в присутствии кислот изомеризация в п-аминофенолы:
H-<Z>-NH-OH -^HO-<^)-NH2
С альдегидами, особенно легко с ароматическими, р-арилгидроксиламины реагируют, образуя так называемые ншпролыл
ОН
Аг—N—Н + О=СН—R
НО он
Аг—N—СН—R
О
t
Аг—N=CH—R
fl-Фенилгидроксиламин образует кристаллы с темп, плавл. 82°. При действии азотистой кислоты p-фенилгидроксиламин дает N-нитрозофенилгидроксил-амин (бесцветные кристаллы, плавящиеся при 51°), который существует в двух таутомерных формах:
✓ОН /ОН ЛО
CeH6-N< + НО-NO -> С6Н5—N< С6Н5—N<C
\н XN=O ^N-ОН
О "существовании последней формы свидетельствует образование N-нитрозофенил-гидроксиламина из нитробензола и гидроксиламина:
CeHe-N<*° + H2N—О—Н CeH6—nZ° +Н2О
—О—Н
N-Нитрозофенилгидроксиламин—сильная кислота. Он образует с солями меди и железа характерные трудно растворимые осадки внутрикомплексных солей, например:
С6Н6— N—ох /O-N
II >CuZ II N—Си \o-n-c0h5
Благодар-я этому он применяется как специфический реактив на медь и железо под названием купферрона.
Интересным веществом является дифенилгидроксиламин (C6H5)2N—ОН, получаемый действием фенилмагнийбромида на нитрозобензол:
СвНб—N—О + СвНб—MgBr -> (C6H6)2N—OMgBr
с последующим разложением водой образовавшегося магниевого соединения. При окислении в эфирном растворе окисью серебра дифенилгидроксиламин теряет атом водорода и превращается в соединение четырехвалентного азота (C6H6)2N=O, которое можно рассматривать как органический аналог двуокиси азота O=N=O. Это соединение (кристаллы интенсивно красного цвета) является сильным окислителем, но в то же время способно к реакциям присоединения, в результате которых образуются производные высших степеней окисления азота.
Азоксисоединенйя
Восстановлением нитробензолов получаются азоксисоединения.
Азоксибензолы—желтые кристаллические вещества нейтрального характера. Электролитически или нагреванием с железными •опилками они восстанавливаются в азосоединения; действием сернистого аммония можно получить из них гидразосоединения и затем анилины.
В течение длительного времени азоксисоединениям приписывали формулу
Аг—N--N—Аг
Однако этой формуле противоречит существование изомерии в ряду несимметрических азоксисоединений. Такая изомерия легко объясняется на основании формулы с семиполярцой связью:
Аг—Аг—N=N—Аг'
I и I
О О
Восстановление нитробензолов в азоксисоединения легче всего протекает при кипячении с алкоголятами, например:
4СбН6—NO2 + 3CH3ONa —> 2С6Нб—N=N—СвНб + 3HCOONa + ЗН2О
При слабом нагревании с концентрированной серной кислотой азоксибён-юлы дают оксиазосоединения (перегруппировка Валлаха):
Аг—N=N—/ —> Ar—N=N—ОН
| \=/ О
Азоксибензол С6Н5—N2O—С6Н5 был открыт Н. Н. Зининым в 1845 г. Он образует блестящие светложелтые кристаллы с темп, плавл. 36°, нерастворимые в воде, легко растворимые в спирте и эфире.
Как и в случае оксимов (стр. 282), для азоксисоединений характерна геоме-*рическая изомерия, например:
О /СвН$
N=N c6Zh5
О, ........... N с6н5 с6н5
пгранс-форма ч^г-форма
При этом для цш>азоксибензола угол а составляет 106±15°.
Z/ис-формы азоксисоединений мало устойчивы и уже при кратковременном нагревании переходят в обычные транс-азоксибензолы, которые имеют более низкие температуры плавления и меньшие дипольные моменты.
Азосоединения
Азосоединениями называются вещества, в молекуле которых содержится группа —N = N— (азогруппа), связанная с двумя углеводородными радикалами. Соединения же, в которых один атом азота азогруппы связан с углеродным атомом, а другой—с атомом какого-либо другого элемента, относятся к классу диазосоединений Ar— N = N—X (стр. 318).
Название азосоединения, содержащего два одинаковых радикала, обыкновенно производят от названия углеводорода или его производного, остатком которого является радикал, например: азобензол С6Н5—N=N—СвНб, азотолуол СН,—СРН4—N = N—С6Н4—СН2. В названия же несимметрических азосоединений входят наименования обоих веществ, от которых произошли радикалы, причем частица «азо» ставится посредине, например: бензолазотолуол С6Н5—N—N—СеН4—СН3 (это соединение называют также монометил азобензолом).
В наименованиях всех азосоединений, кроме азобензола, приходится указывать положение замещающих групп в радикалах, например:
м,п' -азотолуол
2-метил-З' -этилазобензол
Азобензолы получают восстановлением нитросоединений или азоксисоединений, а также окислением аминов или гидразосоеди-нений. Это—яркокрасные или оранжевые вещества. С концентрированными кислотами они реагируют, образуя непрочные соединения. В разбавленных кислотах, щелочах и воде азобензолы нерастворимы; легко растворяются в спирте, эфире и бензоле.
Азобензол С6Н5—N—N—С6Н5 открыт Митчерлихом в 1834 г. Он образует блестящие оранжево-красные кристаллы, плавящиеся при 68°; кипит при 297,4°.
При интенсивном освещении обычный азобензол частично превращается в более растворимую и более интенсивно окрашенную форму, обладающую дипольным моментом. В этой форме он имеет температуру плавления на 2—3е выше, чем в обычной форме, в которую он переходит после плавления. Это обусловлено цис-транс-изомерией.
Обычный азобензол является /пранс-формой, так как не имеет дипольного момента и образует изоморфные кристаллы с транс-стильбеном:
с6н6Х С8Н5\ /И
N=N С=С
хс,н, н/ \свн6
азобензол
ргранс-стильбен
Г идразосоединения
При осторожном восстановлении азосоединений получаются гидразосоединения
Лг—N=N—Аг + 2Н —» Аг—NH—NH—Аг
представляющие собой диарилгидразины с симметрическим расположением заместителей. Восстановление чаще всего производят сернистым аммонием или цинковой пылью в щелочном растворе.
Гидразосоединения—кристаллические бесцветные вещества, нерастворимые в воде. Гидразобензолы почти нейтральны. Уже кислородом воздуха они окисляются в азосоединения. При перегонке они разлагаются, образуя азосоединения и анилин:
2C6H5-NH-NH-CeH5 -> CeH5—N=N—СбНБ -Ь 2CeH6—NHa
Чрезвычайно важной реакцией гидразосоединений является их превращение в диамины под действием минеральных кислот:
Легче перемещакися атомы водорода, находящиеся в пара-положении, со значительно меньшей легкостью—из орто-положения. Так как в случае гидразобензола главный продукт реакции называется бензидином (и^'-диаминодифенил), такое превращение было названо бензидиновой перегруппировкой. Наряду с бензидином получается немного дифенилина (о,п'-диаминодифенила)
H2N
Перегруппировку в орто-пара-положение иногда называют дифен плановой перегруппировкой.
В некоторых случаях, особенно если атом водорода в пара-положении замешен каким-нибудь радикалом, происходит преимущественно не бензидиновая, а так называемая семидиновая перегруппировка (т. е. полу бензидиновая), состоящая в том, что перемещение атомов водорода и связей происходит лишь в одной половине^ молекулы:
При семидиновой перегруппировке перемещение также может происходить ворто-и пара-положения. Семидины являются аминопроизводными дифениламина.
Бензидиновая и семидиновая перегруппировки являются частными случаями совершающихся под действием кислот характерных перемещений в производных ароматических аминов:
H-C6H4-NH-X -> X—С6Н4—NH2 t
при которых X вступает всегда в пара- или орто-положение, но не в мета-поло жение. Таковы следующие реакции:
СбНБ—NH—Hal (Cl,Br, J) —> Hal—СбН4—NH2
С6НБ— NH— NO2 -> O2N-C6H4—nh2
С6Нб—NH—СН3 -> сн3—с6н4—nh2 сбнБ—nh—он —> HO-C6H4-NH2
Гидразобензол СбН5—NH—NH—С6Н5 открыт Н. Н. Зининым в 1845 г. Он образует бесцетные листочки с темп, плавл. 131°; чрезвычайно легко окисляется до азобензола.
Бензидин H2N—С6Н4—С6Н4—NH2 также был впервые получен Н. Н. Зининым в 1845 г. Этот продукт имеет большое значение в производстве азокрасителей. В промышленном масштабе бензидин готовят восстановлением нитробензола цинковой пылью и водной щелочью с последующей обработкой полученного гидразобензола кислотами.
Диазосоединения
Диазосоединениями называются вещества общей формулы R—N2—X. Двухатомная группа из двух атомов азота в диазосоединениях, в отличие от азосоединений, связана только с одним углеводородным радикалом; второй остаток X (органический или неорганический) связан с группой R—N2— через посредство неуглеродного атома.
Под это определение не подходят жирные диазосоединения >C=N=N (см. том I, стр. 660). Однако вследствие аналогии их реакций с реакциями ароматических диазосоединений неправильное название—жирные диазосоединения—является все еще наиболее обычным.
Первые ароматические диазосоединения—непрочные продукты реакции азотистой кислоты с солями анилинов—были открыты Гриссом в 1858—1862 гг.
Ароматические диазосоединения могут иметь двоякое строение:
R—n=n—X и R—N X"
В течение нескольких десятков лет для всех ароматических диазосоединений принималась первая формула строения. Вторая же формула совершенно отвергалась. Последующие исследования, осо
бен но Бамбергера и Гаича, показали возможность существования как диазосоединений строения Аг—N = N—X (истинные диазосоединения), так и соединений строения
Аг— N
названных солями (замещенного) диазония, по аналогии с солями аммония, с которыми они очень сходны. Например, соединение
c6h5-n
называется хлористым фенилдиазонием или часто—хлористым бен-золдиазонием.
При осторожном действии азотистой кислоты на соли анилинов, в противоположность реакции ее с жирными первичными аминами, не происходит выделения азога и замены аминогруппы на гидроксил (см. гом I, стр. 255), а получается соль диазониевого основания. Чаще всего эту реакцию осуществляют, приливая раствор азотистокислого натрия (нитрита) к сильно кислому раствору амина
[C6H5NI I3]C1 + NaNO2 ф HCI —» C6H5N2C1 ф NaC] ф 2Н2О
при охлаждении снегом или льдом, так как многие соли диазония разлагаются уже при комнатной температуре.
Вместо солей азотистой кислоты иногда берут амилнитрит:
|C6H5NH3]NQ3 ф NO—ОС5Нп -> [C6H5N2]NO3 ф Н2О ф носбнп
Эта реакция носит название диазотирования и является одной из важнейших реакций в ароматическом ряду.
Соли диазониевых оснований в громадном большинстве случаев легко растворимы в воде, причем растворы их нейтральны. В растворах эти соли нацело диссоциированы на ионы, т. е. ведут себя, как соли сильных оснований.
В твердом виде соли диазониевых оснований можно получить, непосредственно пропуская окислы азота в твердые соли анилина, а также действуя азотистой кислотой на растворы солей первичных ароматических аминов в спирте или в ледяной уксусной кислоте. Соли многих диазониевых оснований трудно растворимы в спирте и уксусной кислоте и выделяются в кристаллическом виде.
При прибавлении к раствору соли диазония эквимолекулярного количества щелочи или влажной окиси серебра образуется гидрат диазониевого основания*.
Аг—N СГ ф AgOH
Аг—N ОН'фА§С1
Такие растворы имеют сильно щелочную реакцию и проводят электрический ток. Однако обычно щелочность и электропроводность их быстро уменьшаются.
В присутствии избытка щелочи происходит быстрая изомеризация первоначально образующегося гидрата диазониевого основания и получаются так называемые щелочные диазотаты, являющиеся уже истинными диазосоединениями:
Ar—N ОН" + NaOH —» Аг—N—N—ONa + Н2О
Согласно представлениям Ганча, щелочные диазотаты, подобно оксимам (стр. 282), могут существовать в двух стереоизомерных формах. Первоначально из гидратов диазониевых оснований образуются менее устойчивые син-диазотаты, которые под действием концентрированных щелочей превращаются в более устойчивые анггш-диазо-таты.
Аг—N Аг—N
и ।
NaO—N N—ONa
шя-диазотат анти- диазотат
Из цианистых и сернистокислых солей диазония также легко получаются -соединения, аналогичные диазотатам:
С6Н5— N CN’ —»
С6Н5— N=N—-CN диазоцианид
SO3Na- -» С6Н5—N=N—SO3Na диазосульфонат
которые могут существовать в син- и антп-формах.
Установление конфигурации основано на том, что сан-диазосоединения значительно легче, чем анти-диазосоединения, выделяют азот, причем радикал арил соединяется с остатком кислоты:
~ c6h5-n
II N2 + СбН5—CN
NC— N
что объясняется пространственной близостью соединяющихся остатков.
Против этого обычного выбора конфигурации имеются, однако, серьезные возражения (см., например, стр. 284).
При осторожном действии минеральных кислот можно получить из диазотатов кислоты, солями которых они являются:
CeH5—N—N—ONaНС1 —> C6HS—N=N—ОН-f-NaCl
Эти кислоты называются диазогидратами. В присутствии минеральной кислоты диазогидраты превращаются в соли диазониевых оснований:
СбН5—N=N—ОН + НС1 —>
С6Н5—N
Cl- + Н2О
Из сказанного видно, что гидраты диазониевых оснований являются настоящими сильными основаниями и в то же время псевдокислотами, а диазогидраты—настоящими кислотами (средней силы) и в то же время псевдооснованиями*.
Как соли диазониевых оснований, так и истинные диазосоединения являются веществами чрезвычайно непрочными. При нагревании все они с большей или меньшей легкостью и силой взрывают; некоторые из них чрезвычайно взрывчатые вещества. В растворах диазосоединения также легко разлагаются, частью уже при обычной температуре.
Диазосоединения относятся к числу наиболее реакционноспособных органических веществ. С их помощью можно получить все главные классы ароматических соединений, за исключением альдегидов и кетонов. Поэтому реакции диазосоединений в ароматическом ряду имеют чрезвычайно большое значение.
Реакции диазосоединений разделяются на два класса: I) реакции, протекающие с выделением азота, называемые диазо ре акциями', 2) реакции, происходящие без выделения азота.
Диазореакции. 1. Обмен диазогруппы на гидроксил. В кислых водных растворах солей диазониевых оснований происходит (быстрее при нагревании) замена диазо-остатка на гидроксил, и получаются фенолы:
[C6H5N2]HSO4 + H3O С6Н5ОН + N2 4-H2SO4
2. Элиминирование диазогруппы и обмен ее на алкокси л. При кипячении солей диазония со спиртом протекают две реакции, причем в каждом отдельном случае преобладает одна из них:
а) Превращение диазосоединений в ароматические углеводороды (реакция элиминирования диазогруппы), сопровождающееся окислением спирта в альдегид:
[C6H6N2]C1 + СН3СН2ОН -> СбН6 4-СН3СНО 4-N2 4-НС1
б) Обмен диазогруппы на алкоксил с образованием простых эфиров фенола:
[C6H5N2]C1 4- С2НБОН -> С6Н5-О-С2Н5 4-N2 4-НС1
3. Обмен диазогруппы на галоид. С различными галоидами реакция
[ArN2]X АгХ 4- N2
протекает в различных условиях:
* Вопросы строения диазосоединений, солей диазония, диазогидратов и т. д. выяснены в основном в результате долголетних работ двух крупных немецких органиков—Еугена Бамбергера (1857—1932) и Артура Ганча (1857—1935); истолкование результатов экспериментов часто вызывало острые дискуссионные споры между ними.
а) Обмен диазогруппы на иод с образованием иодистых арилов происходит при простом нагревании растворов солей диазония с йодистым калием.
б) Хлористые и бромистые арилы в отсутствие катализаторов образуются лишь с незначительными выходами, но эта реакция идет очень хорошо в присутствии специальных катализаторов.
В качестве катализатора Гаттерманом была предложена мелко раздробленная, так называемая молекулярная, металлическая медь. Особенно подробно эту реакцию разработал Зандмейер, показавший, что катализатором в действительности являются соли одновалентной меди (CuCl, CuBr и т. д.). Реакцию Зандмейера обычно проводят, прибавляя либо готовую медную соль, либо предварительно нагревая медную стружку с соответствующей кислотой, а затем постепенно вводя диазораствор.
Для достижения хороших выходов необходимо присутствие большого избытка хлористых или бромистых солей, или соответствующих галоидоводородных кислот.
в) Замена диазогруппы на фтор достигается путем осторожного термического разложения диазониевых солей борофтористоводородной кислоты:
[C6H5N2]BF4 C6H5F 4- N2 + BF3
Эти соли легко выделяются в кристаллическом состоянии при добавлении к раствору соли диазония борофтористоводородной кислоты, получающейся при внесении борной кислоты или борного ангидрида в растворы фтористоводородной кислоты. Борофториды, в отличие от других солей диазониевых оснований, не взрывчаты.
4. Обмен диазогруппы на циан. Обмен на циан легко происходит под действием цианистых солей меди, например:
2[C6H5N2]C1 4- KCu(CN)2 —> 2C6H6CN 4- 2N2 4- CuCl + KC1
5. Обмен диазогруппы на сульфгидрил. При действии на соли диазониевых оснований раствора ксантогеновокислого калия (см. том I, стр. 737) сначала получается ксантогеновокислая соль диазония:
[ArN2]Cl 4-KS-CS-OC2H5 [ArN2]S-—CS—ОС2Н5 4-КС1
которая при нагревании образует, с выделением азота, эфир ксантогеновой кислоты ArS—CS—ОС2Н5. При омылении этого эфира получается тиофенол Аг—SH (ароматический меркаптан).
6. Обмен диазогруппы на ароматический радикал. Иногда (особенно легко с сухими диазосоединениями) происходит обмен диазогруппы на ароматические остатки, например с образованием производных углеводорода дифенила:
[C6H5N2]C1 + С6Нв C6H5-C6H5 + N2 + HC1
Реакция обычно протекает с низким выходом.
СбНб- N
7. Обмен диазогруппы на остатки галоидных солей металлов (меркурирование и др.). В присутствии мелко раздробленной меди двойная хлористая соль фенилдиазония с хлорной ртутью дает смешанное металлорга-ническое соединение ртути (А. Н. Несмеянов):
[C6H5N2]CbHgCl2 + 2Cu C6H5-IIgCl + N2 + 2CuCl
Аналогично протекает реакция и с некоторыми другими солями (см. также стр. 336), например с SbCl3, BiCl3, SnCl4 (А. Н. Несмеянов, К. А. Кочешков).
Реакции диазосоединений без выделения азота 1. Образование диазоаминосоединений. При действии на соли диазониевых оснований первичных ароматических аминов в слабокислой среде обыкновенно получаются диазо аминосоединения, или анилиды диазосоединений, причем группа ArN2 вступает на место водородного атома аминогруппы:
СГ + NH2-—С6Н5 С6Н5—N—N—NHC6H5 Ч-НС1
диазоаминобензол (темп, плавл. 98°)
Под действием избытка минеральной кислоты протекает обратная реакция с образованием соли диазония и анилина:
С6Нб—N—N—NHC6H5 + 2НС1 —> C6H5N2C1 4- [C6H5NH3]C1
Диазоаминосоединения—желтые кристаллические вещества, при нагревании разлагающиеся со слабой вспышкой. При нагревании с солями анилинов они превращаются в аминоазосоединения, относящиеся к азокрасителям:
С6Н6— N=N4-NH--* C6H6-N==N-<^^.-NH3 аминоазобензол
В отличие от обычных реакций изомеризации производных анилина (стр. 318), здесь происходит межмолекулярное превращение: молекула анилина сначала отщепляется, а затем присоединяется в другом положении.
При действии солей фенилдиазония на толуидины и солей то-лилдиазония на анилин должны были бы получиться изомеры:
С6Нб—N=N—NH—С6Н4СН3 и СбН5—NH—N=N—СбН4СН3
Однако в обоих случаях получается вещество, которое, вступая в реакции, ведет себя как смесь двух изомеров, т. е. здесь наблюдается явление таутомерии.
2. Образование аминоазо- и оксиазосоединений. При действии солей диазония на третичные ароматические амины, а также на jw-диамины и на фенолы, остаток ArN2 вступает в бензольное ядро. В результате этой реакции получаются
аминоазосоединения (соответственно оксиазосоединения), относящиеся к азокрасителям:
[C6H5N2]C1 + C6H5-N(CH3)2 -> CeH8—N=N—C6H6-N(CH3)2 + НС1
[C6H5N2]C1 + C6H5OH —> CeH5—N=N—C6H5OH 4-HC1
Группа ArN2 вступает к имеющимся в ядре заместителям в пара-положение, а если последнее занято, то в орто-положение. Эти реакции называют реакциями сочетания.
3. Восстановление. При восстановлении диазосоединений получаются ароматические гидразины'.
[C6H5N2]C1 + 4Н —> C6H5NH—NH2 + НС1 •
4. Взаимодействие с подвижным водородом. При действии солей диазония на малоновый эфир, ацетоуксусный эфир и другие соединения. содержащие при углеродном атоме подвижные атомы водорода, получаются гидразоны, например:
[C6H5N2]C1 + СН2(СООС2Н5)3 -> C6H5-NH-N=C(COOC2H5)2 + HC1
гидразон эфира
мезоксалевой кислоты
Вероятно, промежуточным продуктом является смешанное (жирно-ароматическое) азосоединение С6Н5—N —СН(СООС2Н5)2.
5. Окисление. Окислением щелочных диазотатов можно получить соли фенилнитрамина С6Н5—NH—NO2, называемого также фенилдиазокислотой (стр. 304). Минеральными кислотами это соединение изомеризуется с образованием смеси о- и n-нитроанилинов (стр. 318).
Гидразины
При замещении атомов водорода в гидразине на ароматические радикалы получаются ароматические гидразины:
Ar—NH—NH2
Ai\
>N—NH2 Ar
и т. д.
К ароматическим гидразинам относятся также гидразосоеди-нения (стр. 317).
Первичные ароматические гидразины получаются восстановлением диазосоединений (Э. Фишер). Обычно соли диазония восстанавливают оловом и соляной кислотой. Фенилгидразин часто получают также восстановлением (цинком и уксусной кислотой) соли диазобензолсульфокислоты, полученной действием сернистокислого натрия на соль диазония (стр. 320):
CeH5—N=N—SO3Na + 2Н CtH5—NH—NH—SO8Na
CeH5—NH—NH—SO3Na + H2O —> CeH5—NH—NH2 + NaHSO4
фенилгидразин
Вторичные гидразины могут получаться восстановлением вторичных нитроз-аминов:
с.н6Ч свн5.
\bj—NO + 4H —> >N—NH2 + H2o
CH3Z СН/
При действии на первичные гидразины иодистых алкилов получается смесь изомерных веществ—вторичного гидразина и жирно-ароматического гидразосоеди-нения, например:
. СЛНЙ—NH—NH—СНЧ СеНб—NH—NH2 + CH3J —
сн/.......2
Продуктами исчерпывающего алкилирования являются четвертичные соли типа:
с6н5.
J~ и
С6Н5—N(CH3)2 I nh2 1
C6H5-N(CH3)2
NHCH3
J-
Ароматические гидразины представляют собой очень ядовитые жидкости или твердые вещества, трудно растворимые в воде, легко— в спирте и эфире. При обычном давлении они перегоняются с некоторым разложением. Являясь сильными основаниями, ароматические гидразины образуют прочные соли с одним эквивалентом кислот.
Первичные гидразины—очень сильные восстановители. Они медленно окисляются кислородом воздуха, принимая темную окраску, и восстанавливают фелингову жидкость уже при обычной температуре. Многие слабые окислители, такие, как соли окиси меди, хлорное железо и пр., окисляют первичные гидразины до ароматических углеводородов:
c6h5-nh-nh2 + о —> С6Н6 + n2 + Н2О
При окислении первичных гидразинов сначала, вероятно, образуются водородистые диаЗосоединения, или диазогидриды
С6Н5—NH—NH2 + О —> CeH5—N=N—Н + Н2О
При дальнейшем окислении могут получаться диазониевые основания или их соли
СбНб—NH—NH2 + 20 ГС6Н5—N1 ОН + Н20
СбН5—NH—NH2 + 2Вг2 —> ГС6Н5—N‘
Вг + ЗНВг
которые часто, особенно в присутствии катализаторов, уже в момент образования подвергаются дальнейшим превращениям, сопровождающимся выделением азота например:
c6h5-n-|
Вг СбН5Вг + N2
Под действием сильных восстановителей происходит разрыв связи между двумя атомами азота в гидразинах, например:
СвНб—NH—NH2 + 2Н -» C6H6-NH2 + NH3
Атом водорода группы NH в первичных гидразинах может замещаться на атом натрия. При действии ацилирующих средств на эти натриевые соединения получаются гидразиды кислот (а-гидр-азиды):
С6Н5—NNa—NH2 + СН3СОС1 —> СбНб—N(COCH3)—NH2 + NaCl
Действием кислот, их ангидридов или галоидангидридов на гидразины получаются изомерные а-гидразидам р-гидразиды:
С6Н5— NH—NH2 + СН3СООН —> СбНб—NH—NH—СОСН3 + Н2О
Важнейшей реакцией гидразинов, содержащих группу NH2, является их взаимодействие с карбонильной группой с выделением воды и образованием гидразонов (том. I, стр. 232), используемых для идентификации карбонильных соединений. Для этой цели часто применяются замещенные в ядре гидразины, например п-хлорфенил-гидразин (кристаллы с темп, плавл. 90°), п-бромфенилгидразин (темп, плавл. 106°), n-нитрофенилгидразин (темп, плавл. 157°). Такие замещенные гидразины получаются из соответствующих замещенных диазосоединений.
В настоящее время для идентификации карбонильных соединений, а также для их качественного и количественного определения, широко применяется 2,4-dw-нитрофенилгидразин (красные иглы с темп, плавл. 198°), получаемый действием гидразина на 2,4-динитрохлорбензол.
Фенилгидразин С6Н5—NH—NH2 образует кристаллы с темп, плавл. 19,6°; темп. кип. 243,5°. Он производится в больших количествах в промышленном масштабе, так как является важным полупродуктом в производстве красителей и лекарственных веществ.
. Дифенилгидразин (C6H5)2N—NH2 плавится при 34,5°. Он получается восстановлением дифенилнитрозамина. Дифенилгидразин образует гидразоны только с альдозами, но не с кетозами.
Тетрафенилгидразин (C6H5)2N—N(C6H5)2, получаемый окислением дифениламина, способен диссоциировать на две молекулы дифенилазота (C6H6)2N, являющегося органическим веществом типа окиси азота NO (см. стр. 439), т. е. содержащим двухвалентный атом азота (Ъиланд).
Производные бензола, содержащие фосфор, мышьяк и сурьму
Соединения ароматического ряда, содержащие фосфор, мышьяк и сурьму, можно разделить на три типа: 1) соединения, в которых элемент трехвалентен; 2) соединения с пятивалентным элементом; 3) соединения типа ониевых, в которых элемент образует катион четвертичного основания.
Соединения фосфора
При действии на бензол треххлористого фосфора в присутствии хлористого алюминия или, еще проще, при пропускании смеси паров бензола и треххлористого фосфора через накаленную (~^700°) трубку с осколками необожженного фарфора образуется фос-фенилхлорид (А. Е. Арбузов):
Фосфенилхлорид (фенилфосфиндихлорид, фенилдихлорфосфин) С6Н5—РС12 представляет собой бесцветную, дымящую на воздухе жидкость; темп. кип. 224,6°; уд. вес = 1,319. Он служит исходным веществом для получения большинства других ароматических фосфорорганических соединений.
Фосфенилхлорид энергично присоединяет хлор с образованием С6Н5—РС14, а также кислород, образуя С6Н5—РОС12. С элементарной серой он реагирует очень бурно, превращаясь в С6Н5—PSC12. Водой фосфенилхлорид гидролизуется:
СвНб-РС12 + 2Н2О -> СбНб—Р(ОН)2 + 2НС1
Образующаяся фенилфосфинистая кислота, или гидрат окиси фенилфосфина, представляет собой кристаллическое вещество, бесцветные листочки, плавящееся около 70°. При сухой перегонке она диспропорционируется:
ЗСбН5—Р(ОН)а -> 2СбН5-РО(ОН)2 + СбН5-РН,
фенилфосфинистая фенилфосфиновая фенилфосфин
(фен илфосфонистая) (фен илфосфоновая)
кислота кислота
Фенилфосфин—жидкость с отвратительным запахом, кипящая при 160—161°; уд. вес. d\5—1,001. На воздухе он быстро окисляется в фенилфосфинистую кислоту, с безводным иодистым водородом образует бесцветные кристаллы фенилфосфонийиодида [С6Н5—PH3]J, расщепляемого водой на HJ и С6Н6—РН2.
В противоположность анилину, аналогом которого он является, фенилфосфин не образует с фосгеном продукта, аналогичного ди-фенилмочевине, а претерпевает превращение:
С6Нб-РНа + 2СОС12 -» СбН5—РС1а + 2СО + 2НС1
Точно так же при нагревании фенилфосфина с хлороформом и едкими щелочами не образуется аналог фенилизонитрила.
Взаимодействием фенилфосфина с фенилфосфиндихлоридом можно получить фосфобензол С6Н5—Р=Р—С6Н5, аналог азобензола—желтоватый порошок, плавящийся при 149—150°.
Фенилфосфиновая (фенилфосфоновая) кислота С6Н5—РО(ОН)2 образует бесцветные кристаллы с темп, плавл. 158°, легко растворимые в воде. Она получается гидролизом С6Н5—РС14 или.С6Нь—РОС12, чаще гидролизом С6Н5—РС1? с окислением образующейся С6Н5Р(ОН)2.
При сплавлении натриевой соли фенилфосфиновой кислоты с едким натром не образуется фенол, а происходит отщепление бензола. Нагреванием этой кислоты с ее хлорангидридом получается фосфинобензол:
СбН5—РО(ОН)2 + С6Н5—РОС12 -» 2CgH5— РО2 + 2НС1
Фосфинобензол—аналог нитробензола, белый микрокристаллический порошок, плавящийся при 100°. Он довольно легко раство-
рим в бензоле, жадно поглощает влагу, переходя при этом в фенил-фосфиновую кислоту, ангидридом которой он является.
При длительном нагревании фенилфосфиндихлорида при 320° в запаянных трубках образуются треххлористый фосфор и дифенил-фосфинхлорид:
2СбН5—-РС12 (СбН5)2РС1 + РС13
Дифенилфосфинхлорид (дифенилхлорфосфин)—бесцветное масло, перегоняющееся при обычном давлении при 320°; уд. вес d]Q5 =1,2295. При его гидролизе образуются дифенилфосфиновая кислота и дифенилфосфин:
2(СбН5)2РС1 + 2Н2О -> (СбН5)2РО(ОН) + (СбН5)2РН + 2НС1
Дифенилфосфиновая кислота получается также гидролизом (С6Н5)2РС13 и окислением дифенилфосфина; это—бесцветные тонкие иглы, плавящиеся при 190°. Дифенилфосфин—густое масло с очень неприятным запахом; темп. кип. 280°; легко окисляется на воздухе.
При действии фенилмагнийбромида на треххлористый фосфор или при взаимодействии хлорбензола и треххлористого фосфора с натрием получается трифенилфосфин:
ЗСбН5С1 + РС13 + 6‘Na —> (СбНб)3Р + 6NaCl
Трифенилфосфин образует кристаллы, плавящиеся при 79° и перегоняющиеся при 188° (1 мм рт. ст.). Он легко окисляется воздухом, переходя в окись трифенилфосфина (бесцветные кристаллы с темп, плавл. 156°). В то время как трифениламин (C6H5)3N не способен к присоединению галоидных алкилов и к солеобразованию, трифенилфосфин присоединяет галоидные алкилы, образуя соли ал ки л тр ифе н ил фосфо н и я.
Виттигу удалось получить пентафенилфосфор (С6Н5)бР в виде нерастворимых в воде кристаллов, плавящихся при 124° (с разложением), действием фениллития на иодистый тетрафенилфосфоний.
Действием магнийорганических соединений на фенилфосфин-дихлорид и на дифенилфосфинхлорид можно получать смешанные • алкиларилфосфины. Интересно отметить, что окись метилэтилфенил-фосфина (С6Н5)(С2Н5)(СН3)РО удалось разделить на оптически активные стереоизомеры (Мейзенгеймер).
Соединения мышьяка
Интерес к ароматическим мышьяковистым соединениям за последние 40—50 лет проявился в связи с двумя обстоятельствами. Во-первых, работами Эрлиха (1910—1911) было показано химиотерапевтическое действие мышьяковоорганических соединений против сифилиса и некоторых заболеваний, вызываемых паразитами крови. После этого был синтезирован целый ряд многообразных мышьяковоорганических соединений (как известно, препарат Эрлиха—сальварсан—получил порядковый номер 606), для получения которых
химикам пришлось искать и разрабатывать новые синтетические приемы. С другой стороны, во время первой мировой войны некоторые мышьяковистые производные были применены в качестве боевых отравляющих веществ, и исследовательские работы в области мышьяковоорганических соединений продолжались в связи с поисками более эффективных средств такого рода.
Одним из простейших способов получения мышьяковоорганических соединений является действие натрия на смесь хлорбензола и треххлористого мышьяка, приводящее к образованию трифенил-арсина*.
ЗС6Н5С1 + AsCl3 + 6Na -» (C^H5)3As + 6NaCl
При нагревании трифенил арсина с треххлористым мышьяком образуются, в зависимости от количества AsCls, дифениларсинхлорид или фениларсиндихлорид (или их смесь):
2(C6H6)3As + AsCl3 -> 3(CeH5)2AsCl
(C6H5)3As + 2AsCl3 -> ЗСбНб—AsClj
Арсинхлориды могут быть получены также с помощью металл-органических соединений, например из ртутноорганических соединений:
(CcH5)2Hg + AsCl3 -> (C6H5)2AsC1 + HgCl2
C6H5—HgCl + AsCl3 -> C6H5-AsCl2 + HgCl2
Дифениларсинхлорид (дифенил хлорарсин) (C6H5)2AsC1-—кристаллическое вещество; стабильная форма плавится при 44,0°, лабильная—около 18,3°. При атмосферном давлении он перегоняется с некоторым разложением при 333°, а при 16 мм при 193°. Фениларсиндихлорид (фенил дихл орарсин) С6Н5—AsC12—подвижная бесцветная тяжелая жидкость, кипящая при 257°; уд. вес <4° =1,625.
Ароматические галоидарсины сравнительно легко гидролизуются водой по схемам обратимых реакций с образованием окисей арсинов:
2(C6H5)2AsC1 + Н-О-Н (C6H5)2As-O-As(C6H6)2 + 2НС1
C6H5-AsC12 ч- Н-О-Н С6Н5—AsO + 2НС1
Окись дифениларсина [(CGH5)2Asl2O образует кристаллы, почти нерастворимые в воде, плавящиеся при 92,5—93,5°. Окись фенилар-сина СсНб—AsO плавится при 144—146°. В виде димера
О
C6H5-As/ \as—С6Н5 ох
она плавится в интервале 210—220°, Окиси арсинов легко окисляются в соответствующие кислоты.
Фениларсиновая кислота (фениларсоновая, фенилмышьяковая) СБНБ—AsO(OH)2 кристаллизуется из воды в виде призм (темп, плавл. 158°). Дифениларсиновая кислота (дифенилмышьяковая) (C6H5)2AsO(OH) плавится при 178°. Галоидангидридами этих кислот могут считаться продукты присоединения хлора (или брома) к фе-ниларсиндихлориду и дифениларсинхлориду:
СвНБ—AsC12 + С12 -> CeH6—AsC14
(C,H6)2AsC1 + Cl2 -+ (CeH5)2AsCl3
так как при действии воды эти хлориды превращаются в фенилар-синовые кислоты. •
Фениларсинтетрахлорид (фенилтетрахлорарсин, четыреххлористый фенилмышьяк) C6H5AsC14 представляет собой масло, легко застывающее в кристаллы (темп, плавл. 45°).
• Дифениларсинтрихлорид (дифенилтрихлорарсин, треххлористый дифенилмышьяк) (C6H6)2AsC13—кристаллическая масса, плавящаяся при 191°.
Трифениларсиндихлорид (трифенилдихлорарсин, двухлористый трифенилмышьяк) (C6H6)3AsCl2—пластинки, плавящиеся при 174°. Он получается присоединением хлора к трифениларсину. При гидролизе трифениларсиндихлорида получается гидрат окиси трифенилар-сина (C6H5)3As(OH)2, при нагревании до 105—110° теряющий воду и переходящий в окись трифениларсина (C6H6)3AsO (темп, плавл. 189°).
Энергичным восстановлением арсиновых кислот, окисей арсинов и галоидарсинов могут быть получены и сами ароматические арсины. Например, восстановление фенил арсиновой кислоты или окиси фениларсина цинком и соляной кислотой приводит к образованию фениларсина:
CeH5—AsO(OH), 4-6Н —> CeH8-AsH2 + ЗН2О.
Фениларсин СвНБ—AsH2 представляет собой легко окисляющееся па воздухе масло с отвратительным запахом, напоминающим запах изонитр'илов; кипит при 148°. Этот мышьяковый аналог анилина не обнаруживает основных свойств.
Восстановлением дифенил арсиновой кислоты может быть получен еще более легко окисляющийся дифениларсин (CeH5)2AsH (масло, темп. кип. 163° при 20 мм).
Трифениларсин (C6H6)3As образует бесцветные кристаллы, плавящиеся при 58—60°; он кипит при атмосферном давлении выше 360° и перегоняется без разложения. Трифениларсин нерастворим в воде, но растворяется в органических растворителях. В противоположность трифениламину, он легко присоединяет галоидные алкилы, образуя ониевые соединения:
(CeH6)3As + CH8J -> KCH3)(CeH6)3As]J
Йодистый метилтрифениларсоний кристаллизуется из воды в виде бесцветных игл с темп, плавл. 121°. Хлорид, плавящийся так
же при 121°, легко растворим в воде. С окисью серебра хлорид дает гидроокись метилтрифениларсония—бесцветные кристаллы с темп, плавл. 125°.
Восстановлением окиси фениларсина или фениларсиновой кислоты фосфористой кислотой или гидросульфитами получается арсенобензол С6Н5—As=As—СбН5. Он образуется также при взаимодействии фениларсина с окисью фениларсина:
С6Н5—AsH2 4-O=As—С6Н5 СбН5—-As=As—-СбН5-f-Н2О
Арсенобензол—бесцветные иглы с темп, плавл. 242-^213°; почти нерастворим в воде, мало растворим в спирте, но растворяется в хлороформе и бензоле.
При восстановлении окиси дифениларсина фосфористой кислотой получается тетрафенилдиарсин, какодил ароматического ряда:
2Н (C6H5)2As-O-As(CeH5)2--> (CeH6)2As—As(C6H5)2 4- Н2О
Он образует кристаллы, плавящиеся при 130—130,5°. Вещество это очень легко окисляется на воздухе и при повышении температуры может самовоспламеняться.
Замещенные в бензольных ядрах мышьяковистые производные можно получать в некоторых случаях путем непосредственного арсенирования ароматических аминов и фенолов мышьяковой кислотой (реакция Вешана), например:
+ H3AsO4 -> H2N—AsO(OH)2 4- Н2О
Наиболее распространенным методом получения мышьяковоароматических соединений является реакция Барта:
CeH5—N2C1 4- Na2HAsO3 4- NaOH —> C6H6-AsO(ONa)2 4- NaCl 4~ N2 4- H2O
Этим путем получаются монофениларсиновые кислоты. Введением растворов солей окиси фениларсина в диазорастворы получают дифениларсиновые кислоты'.
СбНб—As—О 4- 2NaOH C6H5-As(ONa)2 4- Н2О
C6H5-N2C1 4- СбНб—As(ONa)2 -> (C6H5)2AsO(ONa) 4-NaCl 4-N2
Восстановлением арсиновых кислот можно получить мышьяковистые производные остальных типов.
Интересный метод получения одно- и двузамещенных мышьяковистых производных был открыт О. А. Зейде, С. М. [Перлиным и Г. И. Бразом, а именно—взаимодействие фенил гидразинов с мышьяковой кислотой в присутствии солей меди. Повидимому, здесь также промежуточно образуются соли диазония и диазосоединения.
Дифенилхлорарсин (C6H5)2AsC1 и получаемый из него обменным разложением с цианистым калием дифенилцианарсин (C6H5)2AsCN (бесцветные кристаллы, плавящиеся при 35,0°) в виде тонко дисперсных дымов вызывают резкое раздра
жение верхних дыхательных путей, делающее почти невозможным нормальный акт дыхания. В начальных стадиях действие это выражается в чихании и болезненном откашливании, а потому вещества этого типа во время первой мировой войны получили название чихательных отравляющих веществ. Для защиты от таких дымов в противогазе должен находиться противодымный фильтр, так как дымы не адсорбируются ни активированным углем, ни щелочным химическим поглотителем.
Фенилдихлорарсин, подобно льюиситу (см. том I, стр. 388), способен вызывать глубокие поражения при попадании на кожу и обладает высокой общей токсичностью; в смесях с дифенилхлорарсином он применялся в химических снарядах (немецкий «синий крест»).
Соединения сурьмы
При действии трех молекул фенилмагнийбромида на молекулу треххлористой сурьмы образуется трифенилстибин (C6H5)3Sb. Он кристаллизуется в виде пластинчатых кристаллов с темп, плавл. 53°. При избытке SbCl3 образуются переменные количества фенил-стибиндихлорида, или фенилдихлорстибина, С6Н5—SbCl2 (темп, плавл. 62°) и дифенилстибинхлорида, или дифенилхлорстибина, (CeH5)2SbCl (темп, плавл. 68°).
Фенилстибиндихлорид дает с хлором соответствующий тетрахлорид С6Н5—SbCl4, при гидролизе которого образуется фенилсти-биновая кислота С6Н5—SbO(OH)2—бесцветный кристаллический порошок, не имеющий определенной температуры плавления. Хлорированием дифенилстибинхлорида получается дифенилстибин-трихлорид (C6H5)2SbCl3, при обработке горячим разбавленным раствором едкого натра дающий натриевую соль дифенилстибиновой кислоты.
Дифенилстибиновая кислота (C6H5)2SbO(OH)—бесцветный, почти аморфный порошок. Подобно неорганическим кислородным соединениям сурьмы, эта кислота амфотерна—реагирует с соляной кислотой с образованием дифенилстибинтрихлорида или солеобразных продуктов его неполного гидролиза, например (C6H5)2Sb(OH)Cl2.
Из трифенилстибина можно получить трифенилстибиндихло-рид, или трифенилдихлорстибин, (CGH5)3SbCl2—иглы, плавящиеся при 143°. Гидролизом этого хлорида (или непосредственно окислением трифенилстибина перманганатом в щелочной среде) можно получить гидрат окиси трифенилстибина (C6H5)3Sb(OH)2—листочки с темп, плавл. 212°.
Стибинобензол С6Н5—Sb=Sb—CGH - получается восстановлением натриевой соли фенилстибиновой кислоты гидросульфитом.
Замещенные в бензольных ядрах сурьмянистые производные могут быть получены действием растворов солей диазония на щелочные растворы сурьмянистой кислоты, аналогично реакции Барта для мышьяковистых соединений. Некоторые ароматические сурьмянистые производные применяются как лекарственные препараты (для лечения заболеваний, вызываемых трипанозомами и паразитами—лейшманиями).
Метод получения сурьмяноароматических соединений окислением кислородом воздуха смесей арилгидразинов с треххлористой сурьмой в присутствии хлорной меди был разработан П. Г. Сергеевым и А. Б. Брукером:
С8НБ—NH—NH2.HCl + SbCl3 + O2 [С6Н5—N2]C1 - SbCI3 + Н2О
2[СбН5—N2]Cl-SbCl3 + CeH5—NH—NHj-HC! —>
[C6H5-N2]C1 + 2CeH5—SbCla + 4HC1 + N2
[C6H5—N2]C1-f-CeH5—SbCl2 -> (CeH5)2SbCl2 4-n2
Этим путем легко получаются первичные и вторичные сурьмяноорганические соединения. Можно исходить также и из пятихлористой сурьмы
Металлорганические соединения ароматического ряда
Соединения металлов первой группы. Среди органических соединений щелочных металлов первой группы в ароматическом ряду встречаются вещества трех типов:
а) бесцветные, крайне реакционноспособные, большей частью нерастворимые в органических растворителях;
б) интенсивно окрашенные, легче растворяющиеся;
в) интенсивно окрашенные продукты присоединения двух атомов по двойной или сопряженным двойным связям боковой цепи жирно-ароматических соединений.
В эфирных растворах окрашенные вещества обоих типов обладают электропроводностью, так как связь между металлом и углеродом носит в этих условиях ионный характер.
Примерами соединений первого типа являются фенилнатрий и фениллитий. Их можно легко получить действием щелочных металлов на дифенил ртуть (П. П. Шорыгин):
2Na + Hg(C6H5)2 2Na—CeH5 + Hg
Фенилнатрий (натрийфенил) можно получать также действием металлического натрия на хлорбензол в бензольном растворе. Оказывается, что хлорбензол взаимодействует с образующимся фенил-натрием гораздо медленнее, чем с натрием, вследствие чего реакция Вюрца—Фиттига
СсН5-С1 4- Na—С6Н5 СвН5-СвНб + NaCl
при этом практически не протекает. Из бромбензола или иодбензола, наоборот, легко образуется дифенил.
Фениллитий (литийфенил) можно получать, действуя металлическим литием на бромбензол в эфирном растворе. Получаемые таким путем растворы часто применяются взамен растворов магний-органических соединений, так как фениллитий реагирует с карбонильной группой, со сложноэфирной группировкой и т. п. гораздо энергичнее, чем фенилмагнийбромид.
Ароматические соединения щелочных металлов реагируют с углекислотой, образуя почти количественно ароматические карбоновые кислоты (реакция карбонилирования)',
С6Нб-Na + СО2 С6Н5-COONa
Примером интенсивно окрашенных металлорганических соединений второго типа является бензилнатрий, или натрийбензил, С6Н5—СН2—Na. Он получается в виде темнокрасных кристаллов при осаждении петролейным эфиром растворенного в этиловом эфире продукта взаимодействия (в бензольном растворе) дибензилртути с натрием.
Эфирные растворы бензилнатрия интенсивно окрашены в темный кирпично-красный цвет; они проводят электрический ток и, следовательно, вероятнее всего имеют строение [С6Н5—CH2]~Na+. Здесь отрицательный заряд не фиксирован на каком-либо определенном атоме, а рассосредоточен по всей системе аниона бензила.
Т рифенилметилнатрий (натрийтрифенилметил) (С6Н5)3С—Na получается при действии амальгамы натрия на эфирный раствор гексафенил этана:
(СвН6)8С С(СбН5'3 (СбН5)3С* + •С(С6Н5)3 (С6Н5)3С* + Na (CeH5)3C—Na
Можно также действовать избытком амальгамы натрия на эфирный раствор трифенилхлорметана.
Трифенил метил натрий—желтовато-красные кристаллы, растворяющиеся в эфире с образованием оранжево-красного раствора. Он реагирует с водой, аммиаком и с соединениями, способными к таутомерному превращению в енольную форму (например, СН3—СНО, СЬН5—СО—СН3 и т. п.). При этом, после разложения реакционной смеси водой, образуются трифенилметан и едкий натр. При взаимодействии трифенилмегилнатрия с углекислотой образуется натриевая соль трифенилуксусной кислоты. Кислород окисляет трифенил метил натрий с образованием перекиси натрия и перекиси три-фенилметила.
При действии на трифенилметилнатрий хлористого тетраметил аммония образуется тетраметиламмонийтрифенилметил—интенсивно окрашенный в красный цвет аналог натриевого соединения:
Na+[(C6H5'3C]“ + [N(CH3)4]+Cr [N(CH3)4]+[(C6H5)3C]'+ NaCl
Аналоги бензилнатрия могут быть получены также путем расщепления эфиров первичных или третичных жирно-ароматических спиртов действием натрия (иногда его амальгамы) и особенно калия (Циглер):
СбН5—СН2—О—СН3 + 2Na —> С6Н5—СН2—Na + CH3ONa
СН3 ' СН3
СбН5—С—О—СН3 + 2К -> сбн5-с-к + сн3ок
сн3 сн3
Углеводороды, содержащие по меньшей мере один ароматический радикал и сопряженную с ароматическим ядром двойную связь (или систему сопряженных связей), могут присоединять металл ал килы или однократно арилированные металлалкилы:
Сб^5\ СбНбк
>С—СН2 4- C2H5Li —> >С----СН2
с6н/ сбн/ I I
Li С2Н5
СвНб—СИ—СН—СбН5 4-СбН5—С(СН3)2К -> С6Н5-СН-СН-С6Н5 I !
к С(СН3)2С6Н5
Примерами металлорганических соединений третьего типа, образующихся присоединением двух атомов металла к непредельным жирно-ароматическим соединениям, могут служить фиолетовый динатрийдифенилэтан'.
СвН8-СН=СН-СвНБ 4-2Na —> С6Нб-СН-СН—С6Н5
Na Na темнокрасный динатрийтетрафенилэтан'.
СбНБ\ ZC6H5 С6Н5. /С6Н5
>С=С< + 2Na >С----С<
СвН/ ХСбН5 С6Н/ | | Ч6Н5
Na Na и кирпично-красный динатр ийдифенилбутен'.
СбН5—СН=СН—СН=СН—С6Н5 4- 2Na -» С6Н5—СН—СН=СН—СН-С61
Na Na
Очень легко реагирует с натрием антрацен (стр. 467).
Соединения металлов второй группы. Магнийорганические соединения ароматического ряда не представляют каких-либо особенностей по сравнению с алифатическими. Следует отметить, что в то время как хлор, бром и иод в ароматических соединениях обмениваются гораздо труднее, чем в алифатических, арилмагнийгало-гениды образуются лишь немногим труднее, чем алкилмагнийгало-ген иды.
Ртутноорганические соединения ароматического ряда могут быть получены действием амальгамы натрия на ароматические галоидопроизводные, например:
2CeH6Br4-(2Na-HHg) -> (C6H5)2Hg 4- 2NaBr
Легче, однако, получать их, действуя, например, бромной ртутью на арилмагнийгалогениды:
HgBr2 4- CeH5-MgBr СбН5—HgBr 4~ MgBr2
CeH5—HgBr 4- СбНб—MgBr (CGH5)2Hg4-MgBr2
дифенилртуть
Дифенилртуть (C6H5)2Hg—блестящие иглы с темп, плавл. 125°, сравнительно легко растворимые в бензоле, нерастворимые в воде.
>СбН4
Хлористая фенилртуть С6Нб—HgCl кристаллизуется из бензола в виде табличек, плавящихся при 251°, трудно растворимых в бензоле, нерастворимых в воде.
Ртутноорганические соединения могут быть также получены прямым меркурированием бензола и его производных. Например, нагревая при 110° бензол с уксуснокислой ртутью, можно получить сначала уксуснокислую фенилртуть, а затем и двузамещенное производное бензола:
(CH3COO)2Hg + С6Нб —> СН3СОО—Hg—сбн5
СН3СОО—Hg
CH3COO-Hg-C6H5 + (CH3COO)2Hg ->
CH3COO-Hg
Особенно легко меркурируются производные бензола, содержащие орто- или пара-ориентирующие группы. Производные, имеющие группы, ориентирующие в мета-положение, реагируют медленнее бензола. Независимо от характера имеющегося в бензольном кольце заместителя, при меркурировании получаются главным образом орто- и пара-соединения.
Универсальным методом получения ртутноорганических соединений является предложенный Л. Н. Несмеяновым метод разложения восстановителями двойных соединений галогенидов диазония с хлорной ртутью, так называемый диазометод, например:
[C6H6N2]CbHgCl2 + Zn -> C6H5-HgCl + N2 + ZnCl2
Модифицируя этот метод, А. Н. Несмеянов и К. А. Кочешков получили также оловоорганические, свинцовоорганические, висмут-органические и некоторые другие элементоорганические соединения ароматического ряда (стр. 323).
Многоатомные производные ряда бензола и соединения со смешанными функциями
Многоатомные фенолы
Способы получения. Наиболее важным способом получения многоатомных фенолов является сплавление с едкими щелочами («щелочной плав») многоатомных сульфокислот, а также галоидозамещенных и сульфокислот простейших фенолов. Так, например, из «м-бензол ди сульфо к и слоты получается резорцин (л<-диоксибензол)< Пирокатехин (о-диоксибензол) и гидрохинон (я-диоксибензол) могут быть получены из о- и и-сульфо к и слот фенола, а также из о- и n-хлорфенолов (с применением в качестве катализаторов солей • меди или йодистого калия), например:
Cl ONa
J\z°H 1 /ONa
+ 3NaOH -> 4-NaC14-2H,O
Если сплавление co щелочами вести при слишком высокой температуре, то происходит изомеризация, и вместо орто- и пара-изомеров получаются мета-изомеры.
Пирокатехин и гидрохинон могут получаться также восстановлением. соответствующих хинонов (стр. 353).
Большой интерес представляет окисление одноатомных фенолов или ароматических углеводородов в многоатомные фенолы перекисью водорода или солями надсерной кислоты.
Фенолы высшей атомности образуются из одноатомных фенолов также при непосредственном сплавлении фенолятов с едким натром (но не с едким кали), причем реакция протекает с выделением водорода. Из фенола таким образом получаются пирокатехин, резорцин и флороглюцин (1,3,5-гриоксибензол).
Весьма интересно синтетическое образование гексаоксибензола из простейших веществ. А именно, окись углерода и металлический калий, соединяясь, образуют фенолят гексаоксибензола Cfi(OK)e (стр. 204)—побочный продукт при получении калия из поташа. Гексаоксибензол может быть получен из этого фенолята действием разбавленной соляной кислоты в атмосфере СО8.
Некоторые многоатомные фенолы можно получить сухой перегонкой или нагреванием ароматических оксикислот, выделяемых
Таблица 30
Многоатомные фенолы
Название Формула Температура плавления °C Температура кипения °C Удельный вес | Пока ;атель преломления ПР
Пирокатехин (о-диоксибензол) СвН4(ОН)2 105 245 | 1,371 (тв.) 1,615 (при 20°)
Гваякол (1 -окси-2-метоксибензол) /ОН с6н4( xOCHt 28,4 205 1,1287(rfT’4) 1,5383 (при 21,4°)
Еератрол (1,2-диметоксибензол) СвН4(ОСН3), 22,5 207 1,0311 (d21-2) 1,5287 (при 21,2°)
Резорцин (м-диоксибензол) С„Н4(ОН), ПО 276,5 1,285 (d’/.тв.)
Монометиловый эфир резорцина (1 -окси-3-метоксибензол) /ОН С6н / хосн3 — 214,3 1,181 (<.'f) —
Диметиловый эфир резорцина (1,3-диметоксибензол) С.Н4(ОСН8'8 -52 217,7 0,552 (d2?) —
Гидрохинон (п-диоксибензол) СвН4(ОН), 170,5 286,2 1,358 (d20, тв.) —
Монометил овый эфир гидрохинона (1-окси-4-метоксибензол) /ОН с«н / хосн8 53 243 — —
Диметиловый эфир гидрохинона (1,4-диметоксибензол) С6Н4(ОСН8)2 56 212,6 l,0526(d5555) —
Орсин (3,5-диокситолуол) СН3С3Н8(ОН)2 107,5 (безводн.) 290 —
Гомопирокатехин (3,4-диокси-толуол) СН,СвН3(ОН), 65 252 1,1287( d-ч 1,5425 (при 73,6°)
Креозол (З-метокси-4-окситолуол) ось снлн<он ^3 5,5 222 1,0919 05) 1,5353 (при 25°)
Эвгенол (З-метокси-4-оксиаллил* бензол) /ОСН3 сн3=снсн2сбн3< хон 10,3 252,7 1,0664 05) 1,5420 (при 20°)
Изоэвгенол (З-метокси-4-окси-пропенилбензол) СН3СН=СНСвН3< /ОСН3 ЧОН — 10 267,5 1,0852 05) 1,5739 (при 19°)
О
Сафрол (3,4-метилендиоксиаллил-бензол) СН2=СНСН2С6Н3< \ /сн? О Н,2 234,5 1,100 05) 1,5383 (при 20°)
О
Изосафрол (3.4-метилендиокси-пропе нил бензол) сн3сн=снсвн8< ч /СН? oz
цис- 6,8 242—243 1,107 05) 1,5632 (при 15°)
транс- — 248—252 1,123 05) 1,5736 (при 15°)
Пирогаллол (1,2,3-триоксибензол) СвН3(ОН)3 133 309 1,453 (d 4. ТВ‘) —
Оксигидрохинон (1,2,4-триокси-бензол) » 140,5 — — —
Флороглюцин (1,3,5-триоксибен-зол) » 217-219 (безводн.) «— —
Л'НОГОАТОМНЫЕ ПРОИЗВОДНЫЕ РЯДА БЕНЗОЛА МНОГОАТОМНЫЕ ФЕНОЛЫ
из природных продуктов. Так, трехатомный фенол пирогаллол (1,2,3-триоксибензол) обыкновенно получается отщеплением СО? от галловой кислоты при ее перегонке:
ОН
Н ОН
ОН
СООН
галловая кислота
пирогаллол
Пирокатехин кюжет быть также получен сухой перегонкой ряда дубильных и смолистых веществ, например китеху—экстракта из древесины Acacia caiechu. Орсин (3,5-диокситолуол) получается сухой перегонкой орселлиноной (диокси-о-толуиловой) кислоты—продукта гидролиза некоторых веществ, выделяемых из лишайников (стр. 384). В состав продуктов cvxow перегонки дерева входят метиловые эфиры пирокатехина (например, его г’онометиловый эфир гваякол) и пирогалолла.
В некоторых эфирных маслах содержатся метиловые эфиры фенолов с ненасыщенной боковой цепью. Например, эвгенол (3-метокси-4-оксиалл илбензол) является главной составной частью гвоздичного масла. Едкими щелочами он изомеризуется в изоэвгенол (3-метокси-4-оксипропенилбензол):
СН2=СН—СН2—ОН \осн, эвгенол
СН3—СН=СН—С_2>—ОН %сн8 изоэвгенол
Окисление изоэвгенола является одним из способов получения ванилина (стр. 395).
В сассафрасовом масле содержится сафрол (3,4-метилендиокси-аллилбензол), изомеризующийся при нагревании со щелочами в изосафрол (3,4-метилендиоксипропенилбензол):
СН2=СН-СН
СН,
сн8—сн=сн
сн,
сафрол изосафрол
Окислением изосафрола получается пиперональ (стр. 395).
Свойства. Многоатомные фенолы чрезвычайно легко окисляются, особенно в щелочном растворе. На этом основано применение щелочных растворов пирогаллола для определения содержания кислорода в воздухе и в других газовых смесях. Легче всего окисляются фенолы, содержащие две или более оксигрупп в орто- или пара-положениях, несколько более стойки фенолы, содержащие оксигруппы в мета* положении друг к другу.
При осторожном окислении многоатомных фенолов часто от молекулы отрывается лишь один атом водорода (обыкновенно из
пара-положения к одной из оксигрупп) и возникает связь между двумя бензольными ядрами; это приводит к образованию оксипроизводных углеводорода дифенила (бифенила), например:
СвН4(ОН)а + О -> (НО)2СвН8—СвН3(ОН)а + Н2О
Орто- и, особенно, пара-соединения легко окисляются в соответствующие хиноны (стр. 351).
Многоатомные фенолы могут действовать как отрицательные катализаторы (ингибиторы) по отношению к реакциям окисления ненасыщенных органических соединений кислородом воздуха. Такие вещества называются антиоксидантами.
Так, достаточно 1 части гидрохинона на 20 000 частей акролеина, чтобы предотвратить его окисление и связанную с окислением полимеризацию. В качестве одного из очень эффективных ингибиторов полимеризации винильных соединений в технике применяется трет-бути л пирокатехин
СН8— С—СН8 I
получаемый алкилированием пирокатехина изобутиленом в присутствии ZnCla
Многоатомные фенолы весьма склонны к реакциям конденсации (см., например, стр. 421).
Чрезвычайно легко и энергично протекают реакции многоатомных фенолов с галоидами, серной и азотной кислотами. Например, при действии разбавленной азотной кислоты на резорцин получается тринитрорезорцин, или стифниновая кислота (темп, плавл. 176,7°)
ОН OaNxJx/NO,
NO,
Подобно пикриновой кислоте она является взрывчатым веществом. В лабораторной практике стифниновая кислота, как и пикриновая, применяется для выделения и идентификации органических оснований и ароматических углеводородов, с которыми она дает молекулярные соединения—стифнаты.
Пирокатехин. Пирокатехин, подобно другим орто-диоксисоединениям, дает с хлорным железом зеленое окрашивание, переходящее в красное в присутствии едких щелочей, карбонатов и ацетатов щелочных металлов.
Монометиловый эфир пирокатехина (гваякол) добывается из креозота—вязкого масла, получающегося при перегонке букового
дегтя. Гваякол получается также синтетически—из о-хлоранизола. Он находит значительное применение для изготовления душистых веществ (например, ванилина) и лекарственных веществ.
К лекарственным веществам принадлежат угол ьный эфир гваякола (дуотал) СН3О—С6Н4—OCOONa и смесь сульфокислот гваякола (тиокол).
Важным лекарственным препаратом является адреналин—гормон, выделяемый надпочечными железами (см. стр. 192). Он играет большую роль в передаче нервных импульсов в организме. Адреналин представляет собой производное пирокатехина:
НО.
H0~<\Z3~ СН( ОН)—СН2—NHCH,
Это бесцветный кристаллический порошок, быстро окисляющийся и темнеющий на воздухе; темп, плавл 215—216°. Адреналин весьма ядовит. Он обладает способностью суживать периферические кровеносные сосуды, т. е. является кровеостанавливающим средством.
Для синтетического получения адреналина пирокатехин конденсируют с хлоруксусной кислотой, причем получается хлорацетопирокатехин
НО.
но—\~У~ СО—СН,С1 который с метиламином дает адреналон'.
НО.
НО—СО—СН,—NHCH,
Восстановлением адреналона получается с?,Z-алреналин, разделяемый в виде солей винной кислоты, на природный /-адреналин и его d-стереои юмер. ^-Адреналин при нагревании рацемизуется и таким образом через dt/-адреналин может быть превращен в /-адреналин.
Раствор /-адреналина в разбавленной серной кислоте обладает удельным вращением |а|^ = —53,3°.
Резорцин находит значительное применение для изготовления красителей, резорцино.-формальдегидных смол, как антисептик, а также в производстве лекарственных веществ, в частности—для изготовления важного антигельминтного препарата—н-гекси лрезорцина (1,3-диокси-4-н-гексилбензол).
Гидрохинон и пирогаллол применяются как проявители в фотографии, пирогаллол—также в медицине и в газовом анализе для определения кислорода.
В промышленном масштабе гидрохинон получают восстановлением хинона, а пирогаллол из галловой кислоты (стр. 340). Гидрохинон содержится в некоторых растениях в виде глюкозида арбутина.
Флороглюцин кристаллизуется из водных растворов с двумя молекулами воды. Он содержится в растениях в виде глюкозидов; получается из некоторых природных смол сплавлением их со щелочами.
Синтетически флороглюцин получают из симметрического триаминобензола кипячением с кислотами:
НС1
CfiH3(NH2)3 -h ЗН2О-> CRH3(OH)3 + 3NH4C1
Флороглюцин применяется как реактив для определения фурфурола, для обнаружения лигнина в бумаге и для других аналитических целей.
Фенолоспирты
Фенолоспирты могут получаться синтетически конденсацией фенолов с альдегидами, например:
/ОН
СбНбОН -|- СН2О —> С6Н4<
\сн2он
Остаток молекулы альдегида вступает при этом в орто- и в пара-положение к фенольному гидроксилу. Эта реакция является начальной стадией процесса получения феноло-альдегидных смол (стр. 267).
Многие фенолоспирты получаются из природных веществ. Так, салигенин (о-оксибензиловый спирт) выделен из салицина, глюкозида ивовой коры, конифериловый спирт (3-метокси-4,13-диоксипропенил-бензол)—из кониферина, глюкозида коры хвойных растений. Конифериловый спирт представляет интерес вследствие того, что его производные являются составной частью лигнина (стр. 344), содержащегося в древесине.
Таблица 31
Фенолоспирты
Название Формула Температура плавле- НИЯ °C Температура кипения °C Удельный вес
Салигенин (о-оксибензиловый спирт) /ОН С6н / ХСН2ОН 86 возг. 1,16 (тв.)
м-Оксибензиловый спирт У 67 300 (с разл.) —
п-Оксибензиловый спирт » 125 252 —
Анисовый (п-метокси-бензиловый) спирт /ОСН3 Сбн4< чсн2он 25 258,8 М076 (^5)
Конифериловый спирт (13-оксипропенил-3-ме-токс и-4 -оксибе нзол) /СН=СНСН2ОН с6н3£осн8 хон 74 — —
ЛИГНИН
Более ста лет назад французский химик Пайен, отмечая неоднородность древесины, указывал, что более устойчивая к действию щелочей клетчатка механически соединена в древесине с инкрустирующими веществами; одну из фракций инкрустирующих веществ Пайен назвал лигнином (lignum—лат. дерево). Впоследствии было установлено, что стенки одревесневших растительных клеток содержат, во-первых, сравнительно легко гидролизуемые разбавленными минеральными кислотами полисахариды—пентозаны и гексозаны, получившие общее название гемицеллюлоз (24—30% от веса сухой древесины); второй составной частью является целлюлоза (30—50?6 по весу). Кроме того, в состав стенок клеток входят полиурониды. Часть древесины, не гидролизующаяся при действии минеральных кислот, называется лигнином. Его содержание составляет от 20 до 35% от веса древесины (в зависимости от породы дерева).
Известно большое число способов выделения лигнина из древесины. Одни из этих способов основаны на переводе в растворимое состояние углеводов; тогда лигнин остается в виде нерастворимого, окрашенного в желто-коричневый цвет аморфного продукта. При других способах в раствор переводится лигнин. К первым относятся кислотные способы, например гидролиз древесины холодной 72%-ной серной кислотой (способ Класона), гидролиз* холодной 41%-ной (дымящей) соляной кислотой (способ Вильштеттера), многократная попеременная обработка кипящей 1%-ной серной кислотой и холодным швейцеровым реактивом* (способ Фрейденберга) и др. Ко второй группе способов относятся алкоголиз (нагревание с абсолютными спиртами в присутствии НО), варка древесины с раствором бисульфита кальция или натрия, варка с раствором щелочи при 160—180°. Последние два способа применяются в промышленном масштабе для получения целлюлозы из древесины.
Лигнин не является индивидуальным веществом с вполне определенными свойствами и составом. Так, лигнины, выделенные из древесины различных растений, отличаются по содержанию углерода и водорода. Лигнины из древесины хвойных пород содержат 60,5—65% углерода, из лиственных—несколько меньше. Имеются различия и в элементарном составе лигнинов одного и того же растения, но выделенных разными методами.
Высокое содержание углерода в лигнине и многократно подтвержденное наличие ароматических соединений в продуктах его превращения заставляют предполагать, что лигнин имеет ароматический характер. Легкость меркурирования лигнина по Димроту ацетйтом ртути, сравнительная трудность отщепления метоксильных групп (аналогично поведению метиловых эфиров фенолов), способность растворяться в щелочах, подобно фенолам, а также данные исследования
* Водный раствор аммиаката окисной меди.
спектров поглощения лигнинов и продуктов их превращений в настоящее время привели к почти общепринятому представлению об ароматическом характере лигнина.
Лигнин содержит значительное количество функциональных групп, в первую очередь метоксильных и гидроксильных. Установлено, что все метоксильные группы лигнина связаны с ароматическими ядрами, в то время как гидроксильные группы являются либо фенольными, либо спиртовыми (имеются первичные, вторичные и, возможно, третичные ОН-группы). Вполне вероятно наличие в лигнинах также карбонильных групп и двойных связей.
К наиболее характерным превращениям лигнина, при которых получаются сравнительно простые соединения ароматического ряда, относятся следующие.
При сплавлении лигнина с едким кали получаются пирокатехин и протокатеховая кислота. Из плава, полученного в мягких условиях исчерпывающим метилированием диметилсульфатом, а затем окислением перманганатом, были выделены вератровая и изоге-мипиновая кислоты. При окислении лигнина древесины хвойных пород нитробензолом в присутствии щелочи образуется в значительных количествах (до 25%) ванилин (стр. 395), а при окислении в тех же условиях лигнина лиственной древесины, кроме ванилина, получается также сиреневый альдегид:
СООН
СООН
ОСН,
вератровая кислота
ноо
изогемипиновая кислота
сиреневый альдегид
Лигнины однолетних древесных пород наряду с ванилином дают при окислении п-оксибензальдегид. Недавно было найдено, что лубяные волокна коры дугласовой пихты при окислении нитробензолом и щелочью образуют, кроме ванилина, протокатеховый альдегид (стр. 394).
Следовательно, степень метоксилирования бензольных колец лигнина является особенностью, характерной для вида растения.
В некоторых случаях из лигнинов получаются низкомолекулярные соединения, являющиеся кислородсодержащими производными фенилпропана; например, из древесины ели был получен ряд соединений, родственных гваяцилпропану'.
НО—С—СН—СН3 II I
СН8о/ О ОС2Нб
1-гваяцил-2-этокси-пропанон-1
НО-^-Д-С-С-СНз
Il II
СН3О/ о о
1-гваяцилпропандион-1,2
НО—СНгСН,СН3 сн3о/
1-гваяцилпропаи
Из древесины лиственных пород получены, кроме того, соединения с еще одной метоксильной группой в положении 5, г. е. родственные сиреневому альдегиду и сириншлпропану:
СНзО^
н°—СН1СН2СНз сн3о/
При действии на лигнин щелочных металлов в жидком аммиаке получается дигидроэвгенол и 1-(4'-окси-3'-метоксифенил)-пропанол, образующиеся, повидимому. вследствие расщепления простых эфирных связей между структурными элементами лигнина. При каталитическом гидрировании лигнина под давлением в определенных условиях получаются производные пропилциклогексана, например 4-н-пропил циклогексанол.
Несмотря на то, что за последние 20—25 лет в области химии и биохимии лигнина велось большое число исследований, вопрос о строении этого важного природного продукта еще далеко не ясен. Предложены различные схемы строения лигнина, из которых некоторые уже опровергнуты новыми фактами. Повидимому, бесспорно, что лигнины представляют собой сравнительно высокомолекулярные продукты, образованные конденсацией структурных элементов, являющихся производными фенилпропана. Это фенолы, свободные или частично метоксилированные (в зависимости от происхождения лигнина). Для лигнина древесины хвойных пород характерными структурными звеньями являются производные гваяцил-пропана; в лигнинах лиственной древесины, наряду с производными гваяцилпропана, содержатся производные сирингилпропана. Для лигнина однолетних древесных пород характерно наличие производных п-оксифенилпропана и гваяциловых производных.
Структурные элементы, повидимому, связаны между собой простыми эфирными связями за счет фенольных гидроксилов и гидроксильных групп боковых цепей.
Фрейденберг предполагает, что в лигнинах хвойной древесины, кроме простых эфирных связей, имеются и связи С—С, образующие фурановые циклы в системах типа дегидродиконифери-лового спирта:
НО—сн—сн~ СН2ОН
СН3о/ СН=СН— СН2ОН
сн36
Такие системы образуются, по его мнению, при действии окислительных ферментов на конифериловый спирт, получающийся при ферментативном гидролизе глюкозида кониферина, содержащегося в камбиальном соке дерева.
Недавно Фрейденбергу удалось, обрабатывая кони Цериловый спирт окислительно-восстановительными энзимами (фенилредоксазой сока шампиньонов), получить аморфный светлокоричневый порошок, воспроизводящий почти все свойства лигнинов. Однако этого продукта получалось не более 30% Остальное количество кониферилово! о спирта полностью расходовалось на образование следующих остававшихся в растворе веществ:
н0—сн==сн-сн0
СН8о/
конифериловый альдегид
н0—<\=/—сн—сн—снг°н
СН3о/ (*)Н О—сн=сн-сн2он
конифериловый эфир гваяцилглицерина
но
сн
сн—СН-СН2ОН
О СН=СН— СН2ОН
СН8о/
дегидродиконифериловый спирт
пинорезинол
В зависимости от условий смешения реагентов в смеси преобладал либо дегидродиконифериловый спирт, либо конифериловый эфир гваяцилглицерина. Все эти вещества Фрейденберг считает строительными элементами лигнина, так как при дальнейшем действии того же энзима все они дают «синтетический лигнин». Таким образом, по мнению Фрейденберга, лигнин является полимером дегидрированного кониферилового спирта. Образование полимера и димерных продуктов он объясняет . сочетанием изомерных радикалов, образующихся из кониферилового спирта:
СН2ОН снаон СН2ОН 1 снаон 1
сн 1 сн •сн сн
у II 1 II
сн сн 1 сн II сн
э II П ; -А
у\эсн3 ^\эсн, у\)сн8 *\г'\осн8
он о» О он
Иногда в реакциях сочетания участвуют и молекулы воды.
По теории Гибберта, структурным элементом лигнина хвойной древесины является (3-оксиконифериловый спирт
НО—СН=С(ОН)— СН2°Н или НО—СН2~СО—СНаОН
сн8о/ сн3о/
молекулы которого связаны между собой преимущественно простыми эфирными связями. Подтверждением наличия главным образом эфирных связей является получение при расщеплении лигнина ели действием на1рия в жидком аммиаке значительного количества производных гваяцилпропана (Н Н Шорыгина).
Молекулярный вес получаемых лигнинов зависит от способа их выделения. Лигнины, выделенные из древесины кислотными методами, нерастворимы, неплавки. Повидимому, они имеют большой молекулярный вес и сильно разветвленное сетчатое строение. Лигнины, полученные в более мягких условиях, растворимы в некоторых органических растворителях и щелочах.
В настоящее время, в связи с развитием гидролизно-спиртовой промышленности, приобретает большое народнохозяйственное значение проблема использования лигнина, так как количество лигнина, являющегося отходом этой промышленности, почти вдвое превышает количество продуктов гидролиза целлюлозы.
ТАУТОМЕРИЯ ФЕНОЛОВ
Согласно формуле Кекуле для бензола, фенолы являются спиртами с гидроксильной группой при углеродном атоме, связанном двойной связью. Алифатические соединения этого рода—енолы— способны настолько легко изомеризоваться в соединения с карбонильной группой (см. том 1, стр. 389 и 520 сл ), что у алифатических соединений енольная группировка —С=С—ОН вообще неустойчива.
I I
Как и при перегруппировке алифатических енолов в кетоны, превращение фенола из енольной формы в кето-форму
должно сопровождаться выделением энергии (~13 ккал}. Отличие заключается лишь в том, что в случае фенола проигрыш энергии, вызванный нарушением сопряжения двойных связей в бензольном ядре, значительно превышает эту величину и составляет ~50 ккал. Поэтому для фенолов устойчивой формой является енольная, и переход в кето-форму не будет осуществляться.
При изомеризации резорцина образование двух кето-группировок должно происходить с выделением ^26 ккал. Хотя при этом
проигрыш энергии за счет разрушения системы ароматического кольца полностью и не компенсируется, но все же резорцин может уже в некоторой степени проявлять таутомерию.
Действительно, резорцин часто реагирует так. как если бы он имел одно из двух таутомерных строений с кето-группировками:
ОН ОН О
I I II
С с с
%н нс^ нс"/ \н2
II I ^ii^ll
нс с—он НС с=о НС с=о
\н Чг
При действии амальгамы натрия он легко присоединяет два атома водорода, образуя так называемый дигидрорезорцин
О
н// ^сн
I II
Н2С С—ОН
ХСН2
содержащий в молекуле по крайней мере одну карбонильную группу»
Еще чаще реагирует в кето-форме флороглюцин. В этом случае образование трех кето-группировок может почти полностью компенсировать проигрыш в энергии, вызванный нарушением ароматической системы. Поэтому флороглюцин отчетливо проявляет в ряде реакций характер 1,3,5-трикетогексаметилена:
ОН о
' II
с с
/ \ /\
нс сн н2с сн2
II I I I
НО—С С—ОН О=С С=О
\Н ^СН,
Например, при восстановлении амальгамой натрия он переходит в трехатомный спирт—флороглюцшп
О ОН
0=С 0=0 НО—НС СН—ОН
а при действии йодистого метила и алкоголята натрия реагирует аналогично р-дикеюнам и дает полностью метилированный циклогексан-трион:
О
(CHg)// \(CHJ2
I I о=с с=о \(СН3)2
С гидроксиламином флороглюцин как кетон дает триоксим, но с хлористым ацетилом образует триацетат отвечающий енольному строению, а с хлорным железом, подобно фенолам, дает сине-фиолетовое окрашивание.
Крайне интересен синтез флороглюцина из хлорангидрида малоновой кислоты и ацетона в присутствии мрамора, так как при этом флороглюцин должен был бы получаться в форме циклогексантриона (трикетогексаметилена):
' СН2 СН2
СЮ(^ \oci о<^ ''со
+ -* 1 I +2НС)
Н3С сн, нас сн2
\о
Синтез флороглюциндикарбонового эфира из малонового эфира нагревание:и с алкоголятом натрия также отвечает кетонному строению флороглюцина (Байер). В этом случае при действии алкоголята натрия часть малонового эфира превращается в смесь эфиров уксусной и угольной кислот:
Na
СН2(СООС2Нб)2 + С2Н5ОН-----> Ня-СООС2Нб + СО(ОС2Н5)2
Уксусный эфир с малоновым эфире j конденсируется в эфир ацетилмалоновой кислоты
СН3СО—ОС2Н6 + СН2(СООС2Нб)2 -> СН3СО-СН(СООС2Н5)2 + С2Н6ОН который с новой молекулой малонового эфира дает эфир флороглюциндикарбоновой кислоты:
О
''с(СООС?Н8)2
....н"""А..........
С,Н6О ОС2Н6
....I----1.........
О=С С=О \н,
флороглюциндикарбеновый эфир
+ 2CtHsOH
Кислота, полученная омылением этого эфира, при нагревании теряет 2СО, и дает обыкновенный флороглюцин.
Исследование спектра поглощения ультрафиолетовых лучей, однако, показывает, что в растворах флороглюцин содержится не в кето-форме, а только в енольной. Поэтому весьма вероятно, что продукты реакции, получающиеся как бы из кетоформы, на самом деле образуются в результате переноса реакционного центра в енольной форме в момент атаки электрофильным реагентом, например
Хиноны
Хинонами называются соединения, имеющие строение дикетонов, являющихся производными, собственно, не самого бензола, а дигидробензола. Такие вещества могут содержать кето-группы в пара-и орто-положениях, но не в мета-положении. Таким образом, хиноны разделяются на два класса: пара-хиноны и орто-хиноны. В ряду соединений с одним бензольным ядром легче получаются и лучше изучены /7-хиноны. Формулы простейших хинонов, или бензохинонов^ следовательно, будут таковы (формулы Фиттига):
О
Н(^ \н
о-бензохинон
!1
О п-бензохинон (обыкновенный хинон)
Рентгеноскопическое исследование n-хинона показало, что в его кольце расстояния между углеродными атомами неодинаковы, а именно: для ординарных связей С—С они составляют! ,50 А, а для двойных о
связей С=С лишь 1,32 А (в бензольном кольце расстояния между углеродными атомами для всех связей одинаковы и составляют 1,39 А).
Таким образом, в молекулах хинонов уже нет обычного бензольного кольца.
Наиболее простым способом получения хинонов является окисление двухатомных фенолов. Окислением гидрохинона получается обыкновенный бензохинон, или n-бензохинон, окислением пирокатехина—о-бензохинон. n-Бензохинон получается, кроме того, окислением многих замещенных в пара-положении производных бензола, например из n-аминофенола, сульфаниловой кислоты (стр. 304) и пр.
Обычно n-бензохинон получают окислением анилина хромовой кислотой.
Синтез п-хинонрв из а-дикетонов (см. том I, стр. 438) находится в полном соответствии с дикетонной формулой строения хинонов:
СО сн8—do \н3 + сн8 со—сн8
п-ксилохинон
Кетонное строение хинонов подтверждается также* тем, что при действии на них гидроксиламина получаются хиноноксимы, например:
NOH
NOH
п-хинондиоксим
Кроме того, в соответствии с кетонным строением, хинон присоединяет четыре атома брома, две молекулы диазоуксусного эфира; при
окислении хинона надсерной кислотой в присутствии солей серебра получается малеиновая кислота:
О
НС СН СН—СООН
II II + 30, -> II + 2СО2
НС СН СН—СООН
о
Хиноны являются сильными окислителями. При их восстановлении образуются гидрохиноны, например:
О ОН
о он
При этом путем присоединения двух атомов водорода в положения 1,6 сопряженной системы двойных связей хиноидная структура превращается в бензоидную.
Хлористый водород присоединяется к хинону в положения 1,4 с образованием хлоргидрохинона, в молекуле которого галоид связан с атомом углерода:
О О
ОН
X /С1
+ HCI
О ОН ОН
Возможно также присоединение двух молекул хлористого водорода, причем образующееся соединение
ОН
он
окисляется затем избытком хинона в 2,5-дихлоргидрохинон
он
С1
ОН
уксусного ангидрида получается три-
Действием на хинон ацетат оксигидрохинона:
ОСОСН3
\эсосн3
он
ОСОСНд
ососн, ососн.
При осторожном действии хлора сначала происходит присоединение по двойным связям и получаются дихлорид и тетрахлорид хинона
О
С1У\/\С1 II
тетрахлорид хинона
при нагревании отщепляющие НС1 или, соответственно, 2НС1 с образованием монохлорхинона и 2,6-дихлорхинона (наряду с 2,5-дихлор-хиноном):
ООО
монохлорхинон
2,6-дихлорхинон
2,5-Дихлорхинон
Исчерпывающее хлорирование n-бензохинона приводит к образованию тетрахлорхинона, называемого хлоранилом'.
о
о
Хинон может являться филодиеновой компонентой при диеновых синтезах Дильса—Альдера (см. том I, стр. 462), например:
К хинону могут присоединяться молекулы анилина, тиофенола, альдегидов и пр., причем получающиеся производные гидрохинона, например
ОН
(i
CeHsNH—(/ \н
НС С— NHCeHfi
I
ОН
обычно окисляются избытком хинона, превращаясь снова в производные хинона:
О
C6H6NH—Z \н
нс <L-nhc«h6
II о
дианилинохинон
n-Хиноны окрашены в золотисто-желтый цвет, обладают своеобразным резким запахом, перегоняются с водяным паром. С фенолами они способны давать чрезвычайно интенсивно окрашенные соединения, называемые фенохинонами\ к молекуле хинона присоединяются две молекулы одноатомного фенола. Соединения хинона с одной молекулой соответствующего гидрохинона называются хингидронами, Хингидрон С6Н4О2 С6Н4(ОН)2 кристаллизуется в виде черно-зеленых блестящих призм; фенохинон С6Н4О9 • 2С6Н5ОН образует красные кристаллы.
Хингидроны и фенохиноны представляют интерес для химии окрашенных веществ, а потому привлекли большое внимание химиков. Их относят к комплексным или молекулярным соединениям.
п-Хинон—золотисто-желтые кристаллы с темп, плавл. 115,7°. Он получен впервые А. А. Воскресенским* в 1838 г. окислением хинной кислоты, относящейся к производным циклогексана (стр. 65). В промышленном масштабе гг-хинон получается окислением анилина хромовой смесью.
Хлоранил (тетрахлорхинон С6С14О2 образует золотистые листочки, при нагревании возгоняющиеся; в запаянном капилляре плавится при 290°. Хлоранил
* Александр Абрамович Воскресенский (1809—1880 г.). С 1838 г. адъюнкт-профессор, а затем ординарный профессор Петербургского университета. Учитель Д. И Менделеева и многих других крупных русских химиков Один из основателей Русского физико-химического общества. Наряду с многочисленными чисто химическими работами впервые исследовал свойства донецких каменных углей.
может быть получен хлорированием хинона; получается действием белильной извести на анилин (отсюда его название), атакже на фенол и многие другие ароматические соединения. Он применяется в производстве красителей как окислитель.
О-Хинон получен в 1908 г. Вилльштеттером окислением пирокатехина окисью серебра в отсутствие воды. При этом сначала образуется бесцветная модификация в виде призм, которые вскоре превращаются в яркокрасные таблички, разлагающиеся при 60—70°.
Бесцветной модификации приписывают строение перекиси
ярко окрашенной модификации—-форму дикетона. При восстановлении о-бензохинон легко превращается в пирокатехин.
Галоидные производные о-бензохинона удалось получить действием хлора на пирокатехин еще до того, как был получен сам о-бензохинон.
Хинонимины. Ближайшими производными хинонов являются хинонимины, или хинонимиды. Так, осторожным окислением п-ами-нофенола и n-фенилендиамина получаются хинонмоноимин и хинон-диимин*.
хинонмоноимин
хинондиимин
Сложным производным хинондиимина является краситель анилиновый черный, или «черный анилин» (стр. 304), получающийся окислением солей анилина. Анилиновому черному, по Грину, принадлежит строение:
Энергичное окисление анилинового черного хромовым ангидридом при соответствующих условиях приводит к почти полному превращению его в п-хинон.
Осторожным действием хлорноватистых солей на аминофенолы и диамины могут быть получены хинонхлоримиды*.
Cl—N=<^^>=0 и C1-N=(2>=N-C1
Особенно важными производными хинониминов являются индамины и индофенолы
HN===<^J>=N—C6H4NH2 индамин
hn=<^22>=n~c»h«oh
индофенол
получаемые окислением смеси n-диаминов с аминами или, соответственно, с фенолами. Эти соединения являются красителями, пре-
имущественно синего цвета, которые сами мало применяются, но служат исходными продуктами для получения более сложные красителей.
ХИНОИДНЫЕ СОЕДИНЕНИЯ И ИХ ТАУТОМЕРИЯ
Соединения, содержащие характерные для хинонов сочетания простых и двойных связей, получили название хиноидных соединений, и их строение называют хиноидным в противоположность обычному бензоидному строению бензольного ядра.
Различают пар а-хиноидное и орто-хиноидное строение:
пара-хиноидная система связей
орто-хиноидная система связей
Пара-хиноидное строение часто называют просто хиноидным.
Многие соединения ароматического ряда обладают способностью в определенных условиях изменять свое строение—из хиноидного в бензоидное или обратно.
Иногда направление такого превращения является односторонним:
Хиноидная Бензоидная
форма форма
В других случаях наблюдаются взаимные превращения обеих форм:
Хиноидная _> Бензоидная
форма * форма
Часто отношения обеих форм приобретают характер настоящей таутомерии, носящей название хиноидной, или хинонной, таутомерии. Наиболее определенно и резко хиноидная таутомерия проявляется на ряде более сложных пара- и орто-замещенных ароматических соединений.
ТАУТОМЕРИЯ НИТРОЗОФЕНОЛОВ И ХИНОНОКСИМОВ
Действием азотистой кислоты на фенол, а также гидролизом ни-трозодиметиланилина под действием щелочей, получается соединение, которому было дано название я-нитрозофенол и приписано соответствующее строение:
НО—+ HONO
NO + НгО
(CH3)2N—NO + НОН НО—№ + (CH3)2NH
Однако то же самое соединение получается и при действии молекулы гидроксиламина на хинон, хотя в этом случае продукт реакции должен был бы иметь строение хинонмоноксима
O=</=\==NOH
Таким образом, здесь имеются характерные для хиноидной таутомерии отношения двух форм:
Н—О—NO и 0=^~^>=N—ОН
В аналогичных отношениях находятся между собой метиловый эфир нитрозофенола, получаемый окислением и-анизидина
СН3О—NH, + О, —» СН3О—NO + НаО
и метилхиноноксим
0=<^>=N-0CH3
который может быть получен действием йодистого метила на нитрозофенол в щелочном растворе.
Не вполне ясно, какая из двух форм—бензоидная или хиноидная—отвечает в каждом из этих случаев единственному существующему соединению. В первом случае предполагается, что само соединение есть нитрозофенол (бензоидная форма), а щелочные соли его являются солями хинонмоноксима (хиноидная форма).
ТАУТОМЕРИЯ О- и п-НИТРОФЕНОЛОВ
По Ганчу, окрашенность обычных препаратов о- и п-нитрофе-нолов объясняется присутствием в них небольших количеств окрашенных хиноидных таутомерных форм
названных им од//-нитрофенолами.
Нитрофенолы, не содержащие примеси а/4г/-форм, не должны быть окрашены. Действительно, Ганчу и Горке удалось выделить в виде бесцветных кристаллов не только n-нитрофенол, легко получаемый в таком виде, но и о-нитрофенол. Кроме того, метилированием л-нитрофенола в щелочном растворе Ганч получил наряду с обыкновенными бесцветными кристаллами нитроанизола
СН3О—NO,
также яркокрасные кристаллы, которым он приписывает строение эфира -формы нитрофенола:
=NO—ОСН
8
ТАУТОМЕРИЯ УГЛЕВОДОРОДОВ
Некоторые авторы допускают возможность таутомерии уже для толуола и других гомологов бензола:
^\^СН2 на<М
Присутствием орто-хиноидной формы объясняют, например, вступление галоидов в боковую цепь при повышенных температурах (П. П. Шорыгин)*.
Хиноидная форма возможна для углеводородов, которые могут быть произведены от хинона заменой атомов кислорода на метиленовые группы. Простейшим из них является п-хинодиметан
СН2 II
Сам хинодиметан неизвестен; известны лишь более сложные углеводороды, являющиеся его производными.
Углеводороды, содержащие хинондиметидную группировку, были получены из тетрафенил-/?-ксилола, трифенил-п-ксилола и их производных отнятием брома от их дибромидов (Тиле)
+ Zn
тетрафенилхинодиметан
а также отнятием галоидоводорода (нагреванием с третичными органическими основаниями) от их моногалоидных производных (А. Е. Чичибабин):
>с~< >С=< >=с< + НВг
свн/| I Хн5 с6н/ \=/
Вг Н
Тетрафенилхинодиметан и другие соединения этого класса представляют собою яркоокрашенные (красные) вещества, в растворах легко окисляющиеся кислородом воздуха, присоединяющие молекулу брома и галоидоводородов с образованием исходных галоидных соединений.
* А. И. Бродский и Г. П. Миклухин методом меченых атомов показали маловероятность таутомерии этого типа для толуола и мезитилена, что, однако, не исключает, по нашему мнению, ее наличия у некоторых других гомологов бензола [П. С J
Хиноли
Соединения, родственные хинонам, но имеющие лишь одну вне-кольцевую двойную связь, называются хинолями. В частности, хинолями называют соединения, содержащие гидроксильные группы в пара-или орто-положении к карбонилу, как, например:
Н8С ОН
О толухиноль, или 4-метилхиноль (темп, плавл. 75—76°)
Хиноли могут получаться путем перегруппировки п-алкилфенил-гидроксиламинов при осторожном действии кислот:
H2SO4 ----->
Окисление n-алкилфенолов надсерной кислотой (в присутствии MgCO3) приводит, хотя и с небольшим выходом, непосредственно к хинолям:
Повидимому, к хинолям относится горькое вещество хмеля (Humulus lupulus)— гумулон. Ему придают строение (Виланд):
(СН3)2СН—СН^СН ОН НО. Vх /ОН
СН8)2СН—CH2-CZ у ЧСН=СН-СН(СН3)2
Гумулон—кристаллическое вещество (темп, плавл. 66.5°) интенсивно горького вкуса, довольно трудно растворяющееся в воде. Водородный атом одной из гидроксильных групп гумулона настолько легко отщепляется в виде протона, что при титровании щелочью гумулон ведет себя как одноосновная кислота; поэтому гумулон часто называют а-лупулиновой кислотой.
Разбавленная серная кислота, а также разбавленные щелочи вызывают при нагревании перегруппировку, сходную с пинаколиновой.
Например, 4-метилхиноль превращается в толугидрохинон:
протекает с участием воды
Повидимому, перегруппировка
а может быть, и через карбоний-ион:
Этот переход хиноидной системы в бензоидную необратим.
Многоатомные амины
Наибольшее значение имеют двухатомные амины, или диамины. Диаминопроизводные бензола называются о-, м- и п-фенилендиами-нами, производные толуола—толуилендиаминами.
Положение аминогрупп в фенилендиаминах очень сильно сказывается на температуре плавления и менее резко—на температуре кипения:
Орто-Мета-Пара-
Температура плавления °C
102 62,8 139,7
Температура кипения °C
257 284 267
о-Фенилендиамин может быть получен восстановлением о-ни-троанилина, л!-фенилендиамин—восстановлением л1-динитробензола. n-Фенилендиамин получается восстановлением n-нитроанилина или аминоазобензола, дающего при этом смесь анилина и п-фенилёнди-амина:
C6H5-N=N-C6H4-NHs + 4H -> CeH6—NH, + NH,—С6Н4—NH,
Аналогичным образом из соответствующих аминоазокрасителей можно получать и другие двухатомные, а также трехатомные амины (см. стр. 366).
Ароматические диамины могут быть получены нагреванием под давлением дихлорбензолов или хлоранилинов с водным аммиаком в присутствии солей меди.
Многоатомные амины, особенно содержащие аминогруппы в орто-и пара-положениях, являются сильными восстановителями, благодаря чему они применяются в фотографии как проявители. Такие амины легко растворимы в горячей воде.
Для о-диаминов характерна легкость превращения их при различных реакциях в гетероциклические пяти- и шестичленные соединения (так называемые ангидрооснования). Так, они своеобразно реагируют с азотистой кислотой, образуя азимиды\
нох -----------NH
; 4- N -> I + 2HSG
v\NH, V\/N
С одноосновными карбоновыми кислотами о-диамины дают соединения с имидазольным, или глиоксалиновым, кольцом (стр. 568):
y\zNH<2
I ii
НО
Взаимодействие с а-дикетонами приводит к образованию соединений, содержащих азиновое ,кольцо (стр. 625),—так называемых хиноксалинов (реакция на а-дикетоны О. Гинзберга):
Характерным свойством >и-диаминов является легкость, с какой они вступают в реакцию с диазосоединениями, образуя азокрасители. При действии на и-фенилендиамин азотистой кислоты часть диамина обычно диазотируется и вступает в реакцию с другой молекулой диамина, образуя азокраситель основной коричневый (см. стр. 367).
п-Диамины особенно легко (легче, чем о-диамины) образуют при окислении хиноны и их производные (см. 351). Цветной реакцией на n-диамины является возникновение фиолетовой или синей окраски в присутствии сероводорода и хлорного железа. Окраска вызвана образованием красителей, содержащих гетероциклы с атомами серы (фиолетовый Лаута, метиленовый голубой).
При действии на м- и n-диамины ацилирующих средств получаются соединения, аналогичные анилидам (стр. 300).
Действием иодистых алкилов и других алкилирующих средств на диамины можно получить вторичные и третичные амины, а также соли четвертичных аммониевых оснований.
и-Аминодиметиланилин (диметил-/г-фенилендиамин) получают восстановлением n-нитрозодиметиланилина (стр. 313); его можно получить также восстановлением азокрасителя гелиантина:
HO3S—CeH4—N=N—CeH4-~N(CH8)2 + 2HS
-> HO3S-CeH4-NH2 + H2N—CfH4—N(CH8)2
n-Аминодиметиланилин— кристаллы с темп, плавл. 53°; темп, кип. 262°. С сероводородом и хлорным железом он дает великолепное синее окрашивание (метиленовый голубой). Эта реакция может служить для открытия сероводорода.
Аминофенолы
Аминофенолы можно получать восстановлением соответствующих нитрофенолов, лучше всего кипячением с водой и цинковой пылью. Они могут быть получены также замещением на аминогруппу одной гидроксильной группы в двухатомных фенолах путем нагревания их с аммиаком в присутствии хлористого цинка или хлористого кальция (стр. 298). Этим способом легче получаются ^-амино-фенолы.
n-Аминофенолы могут получаться восстановлением нитро- или нитрозофенолов, а также азокрасителей класса оксиазосоединений:
C6Hft—N=N— СвН4— ОН + 4Н —> H2N—С6Н4—ОН + СйНб—NH2
Технически п-аминофенол получают восстановлением нитрофенола железными опилками в кислой среде, а также электрохимическим восстановлением нитробензола в соответствующих условиях (стр. 250). В последнем случае гг-аминофенол получается в результате перегруппировки промежуточно образующегося фенилгидроксиламина (стр. 314).
Аналогичными способами можно получать и аминофенолы с несколькими аминогруппами.
Аминофенолы являются амфотерными соединениями. Они образуют соли с кислотами, но могут растворяться и в едких щелочах. В воде они также растворимы. Растворы аминофенолов, особенно щелочные, легко окисляются кислородом воздуха. Вследствие сильных восстановительных свойств аминофенолы применяются в фотографии как проявители.
В качестве фотографических проявителей применяют соли /?-аминофенола (под названием параамидофенол, или родиналь), соли метил-п-аминофенола (проявитель метол, применяемый обычно в смеси с гидрохиноном), соли 2,4-диаминофе-мола (амидол), а также «глицин* n-аминофенола НО—СаЩ—NH—СН2—СООН.
о-Аминофенолы, подобно о-диаминам, легко образуют циклические ангидрооснования, например с уксусным ангидридом—соединения группы бензоксазола:
/К ZNH2 0
fY А—N
|| + С— СН, -> II + сн3соон + Н2О
k А I к А /С-СН3
W>H О—COCHg /
° о
л-Аминофенол и его алкилированные при азоте производные находят применение в производстве азокрасителей как «вторая компонента» для сочетания с диазотированными аминами, а также для получения родаминов (стр. 432)—красителей ряда ксантона (стр. 588).
Как промежуточные продукты для получения азокрасителей имеют значение о- и я-аминофенолы, а также эфиры аминофенолов, которые можно получать, например, восстановлением эфиров нитрофенолов. Таковы анизидины C6H4(OCH3)NH? и фенетидины C6H4(OC2H5)NH2.
Ацетилированный при азоте /г-фенетидин (N-ацето-/г-фенетидин), так называемый фенацетин
ОС2НБ
6
I
NH—СОСН3 является одним из важнейших жаропонижающих и антиневралги-ческих средств.
Таблица 32
Аминофенолы
Название Формула Температура плавления °C Температура кипения °C
Аминофенолы* C6H4(OH)NH2 172
орто- —
мета- 122 —
пара- 186 —
Анизидины: C3H4(OCH3)NH2
орто- +5,2 225
мета- жидк. 251
пара- 59 240
п- Фенетидин C6H4(OC2H5)NH2 +2,4 254,6
Фенацетин (N-ацето- C3H4(OC3H5)NHCOCH:1 135 —
п-фенетидин) ,
Производное и-фенетидина и мочевины—дульцин (иглы; темп, плавл. 173°)
ОС2Н6
I
и
I
NH—СО—NH2
обладает очень сладким вкусом (в 200 раз слаще сахара)
АЗОКРАСИТЕЛИ
К азокрасителям относятся соединения, содержащие, кроме азогруппы —N = N—, также гидроксильные группы или первичные, вторичные или третичные аминогруппы, т. е. аминоазосоединения и оксиазосоединения.
Аминоазосоединения и оксиазосоединения обычно получают взаимодействием ароматических диазосоединений с аминами и фенолами (стр. 323). Эта реакция называется реакцией сочетания или азосочетания.
Диазотируемое аминосоединение, входящее затем в состав молекулы азокрасителя, носит название диазосоставляющей красителя. Амин или фенол, сочетаемый с диазосоединением, называется азо-составляющей красителя. Сочетание с аминами обычно’ производится в кислой среде, с фенолами—в щелочной среде.
Значительно реже азокрасители готовят непосредственно из азосоединений путем их нитрования или сульфирования, а затем восстановления нитрогруппы в аминогруппу или замены сульфогруппы оксигруппой.
Аминоазосоединения, содержащие первичную аминогруппу, могут быть снова диазотированы, а сочетанием полученного диазосоединения можно приготовить красители, содержащие в молекуле две, три и более азогрупп. Такие красители называются бис-азокрасителями, трис-азокрасителями, полиазокрасителями.
Для рационального наименования азокрасителя обычно поступают следующим образом. Сначала называют одну из составляющих, затем вставляют слово азо и, наконец, называют другую составляющую. Например, краситель СН3—С6Н4—N = N—С6Н4—NH2 называется толуол аминоазобензол ом. Если в основе обеих составляющих лежит один и тот же углеводород, то его указывают только один раз, например С6Н5—N = N—С6Н4—ОН называется оксиазобензолом.
В зависимости от того, вступает ли азогруппа в пара- или ортоположение к гидроксилу или аминогруппе, различают пара- и ортоаминоазосоединения (или пара- и орто-оксиазосоединения). Орто-соединения более интенсивно окрашены.
Аминоазосоединения являются основаниями, дающими с кислотами прочные соли; оксиазосоединения—слабые основания, обладающие одновременно свойствами фенолов.
Простейшие азокрасители—желтого, оранжевого и красного цвета, но получены и азокрасители всевозможных цветов, имеющие более сложное строение.
Чтобы придать нерастворимым азокрасителям растворимость, их часто сульфируют. Соли этих сульфокислот, а в некоторых случаях и сами сульфокислоты, хорошо растворимы в воде.
Аминоазосоединения относятся к классу основных красителей, а их сульфокислоты и оксиазосоединения—к классу кислых красителей.
При осторожном восстановлении азокрасителей получаются бесцветные гидразосоединения (лейкооснования азокрасителей}. легко окисляющиеся кислородом воздуха с образованием вновь исходного азокрасителя.
При энергичном восстановлении азокрасителей получается смесь соответствующих аминов или аминов с аминофенолами, таким способом определяют, из каких составных частей построены молекулы азокрасителей.
При обработке концентрированной азотной кислотой также происходит расщепление молекулы азокрасителя, причем обычно одна из составляющих дает соль диазония, а вторая составляющая выделяется в виде нитропроизводного, например:
NO,
o2n-
•N NOT4-O2N
III
N
8
Аминоазобензол СЯН5—N2—C6H4NH2 образует коричнево-желтые иглы (темп плавл. 126°); перегоняется без разложения при температуре выше 360°. Его соли применялись раньше в качестве красителей под названием анилинового желтого Теперь он служит только полупродуктом для синтеза более сложных азокрасителей. Смесь сульфокислот, получаемая сульфированием аминоазобензола, применяется как краситель для шерсти (кислотный желтый, или прочный желтый}.
Гелиантин HO3S—СвН4—N=N—CfiH4—N(CH8)2 (п-сульфокислота бензол азодиметил анилина) приготовляется из диазотированной сульфаниловой кислоты (стр. 304) сочетанием ее с диметиланилином. Гелиантин применяется как кислотно-щелочной индикатор под названием метиловый оранжевый (со щелочами дает желтое, с кислотами—розовое окрашивание); интервал перехода окраски при рН=3,1—4,4.
Метиловый красный НООС—СРН4— N=N—CeH4- N(CH.J? получается сочетанием диазотированной о-аминобензойной (антраниловой) кислоты с диметиланилином; применяется как индикатор (со щелочами бледная лимонно-желтая, с кислотами—розово-красная окраска) Переход окраски при рН = 4,2—6,3.
Хризоидин получается из хлористого фенилдиазония и л<-фе-нилендиамина.
Основной коричневый (везувин, бисмарк коричневый) получается действием азотистой кислоты в водном растворе на ти-фени-лендиамин. Это—смесь нескольких красителей, в состав которой входит триаминоазобензол (аминобензолазо-м-диаминобензол), образующийся диазотированием одной аминогруппы в лгфениленди-амине и сочетанием соли диазония со второй молекулой основания. Главными составными частями основного коричневого являются бисазокрасители, получаемые дальнейшим диазотированием аминогрупп и сочетанием с новыми молекулами фенилендиамина:
Красители, образующиеся путем сочетания диазосоединений с фенолами, иногда называют тропеолинами (хотя гелиантин также носит название тропеолин D).
Резорциновый желтый получается сочетанием диазотированной сульфаниловой кислоты с резорцином.
ТАУТОМЕРИЯ АЗОКРАСИТЕЛЕЙ
Оксиазосоединения и аминоазосоединения являются бензоидными формами, таутомерными с гидразонами хинонов и хинонимидов. Так, при действии фенил-гидразина на а-нафтохинон вместо гидразона хинона
образуется бензолазонафтол
получаемый сочетанием солей фенилдиазония с а-нафтолом. При действии же уксусного ангидрида на о-оксиазосоединения получаются ацетильные производные гидразонов хинонов, например
С6Н5—N(COCH3)—
СН3
О СВЯЗИ МЕЖДУ СТРОЕНИЕМ И ОКРАСКОЙ
Окраска вещества зависит от избирательного поглощения части лучей видимого спектра. Цвет вещества в проходящем свете всегда является дополнительным к цвету лучей, поглощаемых этим веществом.
Ниже приведены длины волн частей видимого спектра, соответствующие им спектральные и дополнительные к ним цвета:
Длина волны о А Цвет поглощаемых лучей Видимый цвет
4000—4350 Фиолетовый Желто-зеленый л 4 w
4350—4800 Синий Желтый g Ё
4800—4900 Зеленовато-синий Оранжевый § §
4900—5000 Сине-зеленый Красный <ь
5000-5600 Зеленый Пурпурный х X
5600—5800 Желто-зеленый Фиолетовый §
5800—5950 Желтый Синий 3 >>
5950—6050 Оранжевый Зеленовато-синий о £
6050—7500 Красный Сине-зеленый
Смещение области поглощения
к более длинным волнам, т. е.
переход от желтого цвета через красный и синий к зеленому, назы-
Ч) I I I I
I $
вается углублением цвета или бато-хромным смещением. Обратный переход из более длинноволновой области в более коротковолновую называется повышением цвета или гипсохромным смещением по гл още н и я.
Для характеристики окраски вещества недостаточно указать только его цвет, зависящий от длины волны (или частоты колебаний) поглощаемого света. Необходимо, кроме того, оценить интенсивность цвета окрашенного вещества. Мерой интенсивности цвета окрашенных растворов служит молярный коэффициент поглощения (молярная экстинкция), рассчитывае-
Рис. 5. Кривая поглощения спир-
мый по формуле:
тового раствора азокрасителя.
е==4" IgA а 1
где d—толщина слоя одномолярного раствора окрашенного вещества, см-,
—начальная интенсивность света;
/—интенсивность света, прошедшего через раствор.
Точной физической характеристикой цветовых свойств окрашенного соединения в растворе служит его кривая поглощения. Пример такой кривой дан на рис. 5.
Для построения кривой поглощения измеряют на спектрофотометре интенсивность поглощения раствором света различных длин волн, и полученные значения молярных экстинкций наносят на график против соответствующих длин волн или частот, отложенных по оси абсцисс.
Упрощенной характеристикой окраски вещества может служить длина волны kmax, при которой наблюдается максимальное поглощение.
Исследование спектров поглощения света является важным методом выяснения строения и идентификации органических соединений, особенно красителей, так как окраска органического соединения тесно связана с его строением. Поэтому делалось много попыток построить теорию цветности, связывающую избирательное поглощение света в видимой части спектра с особенностями строения вещества.
После создания А. М. Бутлеровым теории химического строения Гребе и Либерман в 1868 г. первыми высказали мысль, что окраска связана с ненасыщенностью. В 1876 г. О. Витт и, независимо от него, П. П. Алексеев развили эту мысль далее. Основываясь на данных о свойствах большого числа уже известных в то время синтетических красителей, они сформулировали так называемую хромофорную теорию окраски органических веществ. Цветность соединения, по этой теории, обусловлена присутствием в молекуле группировок, содержащих двойные связи (>С=С<, —N=N—, >С=О, —NO2 и др.), получивших название хромофоров*. Они же обратили внимание на усиление интенсивности окраски и углубление цвета окрашенного вещества при наличии в молекуле, содержащей хромофоры, таких групп, как —NH2, —NR2, —ОН, —OR, получивших название ауксохромов**. Эти же ауксохромные группы обычно обусловливают и сродство окрашенного вещества к растительным или животным волокнам. Без такого сродства окрашенное вещество еще не является красителем.
Несколько позже, в середине 80-х годов прошлого столетия, Армстронг и, независимо от него, Нецкий предложили хиноидную теорию цветности, согласно которой в молекулах большинства окрашенных соединений имеется хиноидная структура:
Хиноидная теория долго пользовалась популярностью; ее схемы часто применяются и до сих пор для изображения строения красителей. е
К 20-м годам текущего столетия были сформулированы и приобрели широкое распространение взгляды так называемой боннской школы (Дильтей, Вицингер и др.). Согласно этим представлениям, цвет
* Хромое—греч. окраска, цвет; форос—носитель.
♦♦ Ауксо—увеличиваю.
ность обусловлена наличием координационно ненасыщенных атомов в системе и ионоидным состоянием молекулы окрашенного вещества. При изучении связи между цветностью и строением окрашенных веществ давно уже ощущалась недостаточность обычных структурных формул для изображения окрашенных органических соединений и для описания их свойств. Это обстоятельство повело к ряду попыток применить представления об «осцилляционных» структурах (А. Байер в Германии, А. Е. Порай-Кошиц в СССР) и даже к представлениям о «делению? валентности (М. А. Ильинский) или «дроблению связей (Кауфман). В этом проявлялось своего рода интуитивное предвидение представлений о различиях в легкости возбуждения а- и к-связей и смещения в них электронной плотности.
Попытки развивать представления о так называемом мезо-состоянии окрашенных молекул делали уже давно А. Ганч, Кёниг и В. А. Измаильский. Позже Арндт в Германии, Льюис и Кельвин в США и другие зарубежные исследователи пытались использовать для объяснения явлений цветности так называемую теорию мезомерии (см. том I, стр. 140—143).
Чтобы подойти к правильным современным представлениям о природе возникновения цветности, надо ясно представлять себе механизм процесса поглощения света веществом и связь этого поглощения со строением молекулы.
Между поглощением в видимой и в ультрафиолетовой областях спектра принципиального различия нет. В обоих случаях кванты лучистой энергии поглощаются электронами, переходящими при этом на более высокие энергетические уровни. Кванты же инфракрасного излучения поглощаются не электронами, а атомами и радикалами. Здесь будет рассмотрено только поглощение, связанное с электронными переходами.
Согласно теории Бора, квант падающего на вещество света переводит электрон из его основного состояния, характеризующегося энергией £0, в возбужденное состояние с энергией Ех. Частота поглощенного излучения v связана с энергией кванта света соотношением:
- Eq h
Однако, так как не вся энергия кванта расходуется на возбуждение электронов, а некоторая часть ее (Es ) затрачивается также и на возбуждение колебаний атомов, более точным будет выражение:
E^Eq^Es
Частичным расходованием в энергии на возбуждение колебательных движений атомов и объясняется то обстоятельство, что полосы поглощения света в видимой и ультрафиолетовой областях спектра всегда несколько размыты.
Для возбуждения электронов, образующих простые связи (а-электронов), например в насыщенных углеводородах, требуются
весьма большие кванты энергии. Поэтому насыщенные соединения способны к поглощению только в далекой ультрафиолетовой области спектра—при длинах волн не более 2000 А. Электроны, участвующие в образовании двойных связей (^-электроны), возбуждаются гораздо легче. Так, этилен при 1900 А поглощает свет с такой же ин-о
тенсивностью, как этан при 1550 А. Но особенно легко возбуждаются ^-электроны сопряженных двойных связей, и притом тем легче, чем длиннее цепь конъюгации. С удлинением цепи конъюгации полоса поглощения вещества смещается в область более длинных волн, что можно видеть на примере кротоновой кислоты и трех ее винилен-гомологов (т. е. соединений, находящихся с ней в отношениях винилогии):
^maxj А
СН3—СН=СН—СООН ...... 2080
СН3—(СН—СН)2—СООН .... 2610
СН3—(СН=СН;3—СООН .... 3020
СН3—(СН=СН)4—СООН .... 3300
Когда в соединении имеется цепь конъюгации, происходит возбуждение не отдельных электронов, а всей системы подвижных электронов, функционирующей как одно целое (сопряженный резонатор). Для возбуждения такой системы требуются еще меньшие кванты энергии, и молекулы, содержащие сопряженные кратные связи, поглощают в видимой или на границе видимой области спектра.
В молекулах всех органических красителей имеются цепи конъюгации. В состав таких цепей обычно входят бензоидные и хиноидные ядра. Здесь уже нет двойных и простых связей в обычном смысле слова. При образовании общего к-электронного облака электронные плотности этих связей выравниваются частично или полностью (подобно тому, как это происходит в бензольном кольце). Точно так же ионный заряд, имеющийся у солеобразных красителей, нельзя считать фиксированным на одном определенном атоме,—он распределяется между несколькими атомами окрашенного иона.
В силу указанных причин одной классической структурной формулой со строго фиксированным распределением двойных связей и ионных зарядов нельзя изобразить истинное электронное строение молекулы красителя. Поэтому ни одна из приведенных ниже формул
372
многоатомные производные ряда бензола
с фиксированным положением положительного заряда на одном из трех атомов азота не может правильно изобразить действительное строение иона красителя парафуксина (стр. 426). Более правильно его строение передает формула
показывающая направление смещения электронной плотности по всей системе, приводящее к распределению заряда между всеми тремя атомами азота.
Аналогично можно написать формулу для иона оксиазобензола:
N = N
из которой видно, что смещение электронной плотности приводит к распределению отрицательного заряда между атомом кислорода и более удаленным от него атомом азота.
С современной точки зрения, основной хромофорной группой красителя является цепь конъюгации. Но вещества, содержащие в молекуле только хромофор, т. е. цепь конъюгации, еще не являются красителями. Даже если они поглощают свет в видимой области, их поглощение недостаточно интенсивно. Окрашенное соединение будет иметь большой коэффициент поглощения лишь в том случае, если в поглощении света примут участие ионные заряды. Именно поэтому большинство органических красителей представляет собой солеобразные вещества, содержащие окрашенный катион (основные красители) или окрашенный анион (кислотные красители). Однако не всякая солеобразующая группа имеет существенное значение для поглощения света. Как известно, мало влияет на окраску сульфогруппа, не усиливает поглощения группа —N(CH3)3 и другие группы, в которых электронные заряды фиксированы и не способны перемещаться по цепи конъюгации. Напротив, сильнейшее влияние на цвет—углубление цвета и повышение его интенсивности—оказывают атомы и группы, содержащие неподеленные электронные пары (доноры электронов). При непосредственном примыкании такой группы к цепи конъюгации ее электроны смещаются и вступают в цепь конъюгации; в результате эта группа частично теряет свой отрицательный заряд или даже приобретает некоторый положительный заряд.
К таким группам принадлежат ауксохромы старой теории цветности (NH2, NR2, ОН, OR), а также группы, способные принимать электроны (акцепторы электронов), приобретая таким образом отрицательный заряд (NO2, С=О).
Следовательно, красителями можно называть ионы, содержащие цепь сопряжения (хромофор) и по крайней мере два ауксохрома, между которыми распределен ионный заряд. В приведенных ниже формулах n-диметиламиноазобензола (в кислом растворе) и фенолфталеина (в щелочном растворе) цепи конъюгации—хромофоры—показаны жирными линиями, а ауксохромы обозначены пунктиром:
п- диметиламиноазобензол
фенолфталеин
Однако, кроме солеобразных красителей, существуют и несолеоб-разные—индигоидные, кубовые, антрахиноновые и др. Данное выше определение красителя можно применить и к ним, так как все несо-леобразные красители в большей или меньшей степени поляризованы и могут быть представлены в виде биполярных ионов. Так, например, электронное строение индиго может быть представлено следующим образом:
индиго
В результате поляризации на атоме кислорода появляется избыток. а на атоме азота—недостаток электронной плотности. Следовательно, и здесь имеются необходимые элементы строения молекулы красителя: хромофор (цепь конъюгации), ауксохромы и ионные заряды.
В настоящее время известно много полуэмпирических закономерностей, связывающих строение красителя с глубиной и интенсивностью его окраски. Так, например, установлено, что цвет углубляется по мере удлинения цепи сопряжения, что способность ауксохромов влиять на цвет зависит от степени их сродства к электронам.
Существенное значение имеет электронная симметрия молекулы или иона красителя:’ наибольшая глубина окраски наблюдается, когда ион вполне симметричен и ионный заряд равномерно распределен между двумя (или более) ауксохромами; нарушение же симметрии, связанное с частичной фиксацией заряда на одном из ауксохромов, ведет к ослаблению цвета.
В соответствии с требованиями квантово-механической теории поглощения света молекула красителя поглощает свет наиболее интенсивно, если ауксохромы и цепь конъюгации лежат в одной плоскости. Нарушение копланарности двух частей сопряженной системы связей, вызванное пространственными препятствиями, всегда ведет к уменьшению интенсивности поглощения.
Установленные закономерности позволяют химику-синтетику в известной степени предугадать оптические свойства синтезируемого красителя.
Для простейших случаев—комбинаций нескольких сопряженных связей в молекуле—положение kmax может быть определено (с не всегда одинаковой степенью приближения) квантово-механическими расчетами, независимо от каких-либо спектроскопических измерений.
Для бутадиена, например, расчет дает максимум поглощения о о
при 1900 А вместо наблюдаемого 2100 А, т. е. ошибка составляет 10%. Для бензола, нафталина и других циклических конденсированных систем получено несколько лучшее совпадение расчетных величин с наблюдаемыми:
X max вычисленное X max найденное Ошибка %
Бензол 2470 2550 3
Нафталин . . . 2950 2750 7
Антрацен . . . 3650 3700 1
Нафтацен . . . 4500 4600 2
Хотя результаты квантово-механических расчетов не всегда хорошо согласуются с наблюдениями, их тем не менее следует рассматривать как большой шаг вперед в деле установления связи между строением молекулы и поглощением света.
КРАСИТЕЛИ И КРАШЕНИЕ
Удовлетворительной общей научной номенклатуры красителей до сих пор не имеется, а производственники и изобретатели присваивают обычно красителям самые произвольные названия. Некоторые из этих названий укоренились довольно прочно и являются общепринятыми, например: фуксин, нильский голубой, метиленовый синий и многие другие. Однако иногда одно и то же вещество имеет несколько таких произвольных названий. Например, основной коричневый, бисмарк коричневый, везувин, манчестерский коричневый— это названия одного и того же продукта.
Довольно полный перечень существующих красителей имеется в специальном справочнике, так называемом Colour Index (сокращенно С. I.), в котором каждому красителю присвоен определенный порядковый номер. В этом справочнике по номеру красителя можно найти его строение, характеристику, названия, под которыми он выпускается, и важнейшую литературу.
Процесс крашения, т. е. такой фиксации красителя волокном, при которой окраска не смывается водой и мылом, весьма сложен и недостаточно выяснен. Во многих случаях, вероятно, образуются адсорбционные соединения, иногда твердые растворы (механическая теория крашения). Действительно, большое число красителей дает не истинные, а коллоидные водные растворы и, следовательно, может только адсорбироваться волокном. В других случаях, быть может, образуется химическое соединение волокна с красителем (химическая теория крашения).
Волокнистыми веществами могут фиксироваться также вещества, воспринимаемые глазом как бесцветные, но содержащие такие же группы, как и у красителей Подобные «бесцветные красители» пытались применять для защиты шерстяных тканей от моли, так как некоторые вещества, родственные соединениям, обладающим видимой окраской, «неприемлемы» для моли.
Процесс крашения может заключаться в непосредственном окрашивании волокна раствором красителя. Такие красители носят название субстантивных. По отношению к животным волокнам (шелку и шерсти) субстантивными обыкновенно являются красители кислотного характера (содержащие группы SO3H, ОН, СООН и пр.).
В других случаях для прочной фиксации красителя волокном требуется добавка так называемых протрав, т. е. таких веществ, которые прочно связываются одновременно и с красителем и с волокном {протравные красители). Протравами чаще всего служат соли и гидраты окисей тяжелых металлов, например: уксуснокислый алюминий, соли окисного железа, хрома, олова и пр. Осаждающиеся на волокне нерастворимые соединения красителя, например с гидроокисью алюминия, оловянной кислотой и т. д., носят название лаков.
Иногда, как, например, в случае красителя индиго, на волокно наносится растворимое в воде вещество, вследствие окисления кислородом воздуха легко образующее нерастворимый краситель на самом волокне. В этом случае процесс крашения называется кубовым.
Многоосновные ароматические кислоты
Простейшими двухосновными ароматическими кислотами являются три изомерные о-, м- и п-фталевые кислоты, содержащие оба карбоксила в бензольном ядре. о-Фталевая кислота называется обычно просто фталевой кислотой, ти-кислота—изофталевой, п-кис-лота—терефталевой кислотой. Они могут быть получены окислением изомерных ксилолов и других гомологов бензола, содержащих две боковые цепи.
Интересно, что при окислении о-ксилола хромовой смесью фталевая кислота практически не образуется, а происходит разрушение бензольного ядра. Такой процесс часто протекает и при окислении других орто-замещенных производных бензола.
О-Фталевая (обыкновенная фталевая) кислота получается в промышленности окислением нафталина концентрированной серной кислотой в присутствии солей ртути или кислородом воздуха в присутствии окислов ванадия как катализатора. Это более сильная кислота, чем бензойная: константа диссоциации фталевой кислоты (при 0°) для первого карбоксила равна 1,22-10-3, а для бензойной кислоты 6,05-10-5.
Фталевая кислота кристаллизуется в блестящих листочках, хорошо растворимых в горячей воде, в спирте и в эфире.
При нагревании фталевая кислота, не плавясь, теряет воду и переходит в циклический «внутренний» ангидрид:
О
фталевый ангидрид
Фталевый ангидрид—блестящие иглы или призмы (темп, плавл. 130,8°; темп. кип. 285°). Этот ангидрид очень легко вступает в реакции конденсации, в которых участвует преимущественно одна из групп СО.
При конденсации с бензолом и его окси- и аминопроизводными получаются производные дифенилметана и трифенил метана, например:
ОН
со со
о-бензоилбензойнагл кислота
О фталофснон
Особенно легко получаются продукты конденсации с двумя молекулами фенолов (фенолфталеины).
При восстановлении фталевого ангидрида кислородный атом одной из групп СО легко замещается на водород и получаеicn фта-лид (плавится при 74°, перегоняется при 290°)
представляющий собой лактон 7-оксикислоты
^\соон
Фталимид, или имид фталевой кислоты (темп, плавл. 238°>
получается пропусканием аммиака через нагретый фталевый ангидрид или нагреванием последнего с углекислым аммонием.
. Фталимид служит промежуточным продуктом для получения антраниловой кислоты (стр. 389), в производстве индиго (стр. 558> и др.
Действием на фталимид спиртового едкого кали легко получается фталимид калия
о II С
Он имеет большое значение для синтезов первичных аминов и, особенно, соединений со смешанными функциями, содержащих аминогруппу (синтезы Габриэля).
Реакции, протекающие при таких синтезах, можно проиллюстрировать на следующем примере: О
О
О
NK + СН2Вг—СН2Вг ->
N—СН2—СН2Вг + КВг
о о
—СН2—СН2Вг + Н2О
N—СН2-СН2ОН + НВг сХ
о
При ' омылении последнего
О
соединения протекает реакция:
N-CH2-CH2OH + 2Н2О
V'Xcooh
:оон
+ h2n—сн2—сн2он
этаноламин
(коламин)
О
Получаемые соединения О
N— СН2—СН2Вг
N—СН2—СН3—СН2Вг
могут вступать в реакции с цианистым калием, с натриймалоновым эфиром, с натрийацетоуксусным эфиром и т. д. Этим путем можно синтезировать аминокислоты, а также диаминокислоты (Э. Фишер) и пр.
Фталилхлорид (хлорангидрид фталевой кислоты) получается действием на фталевую кислоту пяти хлористого фосфора. Здесь наблюдается замечательный случай таутомерии, связанной с перемещением атомов не водорода, а хлора. А именно—фталилхлорид существует в двух таутомерных формах:
О
(темп, плавл. 16°)
О
и
С
о
О
Форма II получается, если реакция с пятихлористым фосфором протекает в присутствии А1С13, форма I—в отсутствие А1С13. Вторая форма легко превращается в первую. Первая форма реагирует быстрее с аммиаком и анилином, причем замечательно, что продукты реакции обеих форм тождественны.
Изофталевая кислота—трудно растворимые в воде тонкие иглы с темп, плавл. 348,5°. Эта кислота не образует ангидрида.
Терефталевая кислота—аморфный порошок, возгоняющийся, но не плавящийся. Она получается окислением тминного масла, содержащего цимол и куминовый альдегид, а также окислением гг-ци-мола, выделяемого из сульфитного скипидара. При конденсации терефталевой кислоты с этиленгликолем образуется высокомолекулярный эфир линейной структуры (полиэтилентерефталат):
----о—ОС—\Z=/~СО—О—СН2—СН2—О—ОС—^J^-CQ-G-СН2-СН2-------------
Практически сначала получают диметиловый эфир терефталевой кислоты (темп, плавл. 140,6°), который при нагревании с этиленгликолем отщепляет метиловый спирт и превращается в полиэтилентере-фталат. Этот смолообразный продукт, напоминающий глифталевые смолы, может в расплавленном состоянии выпрядаться в нити. Такой вид искусственного волокна получил название терилена.
Бензолполикарбоновые кислоты. Некоторые сведения о наиболее важных поликарбоновых кислотах приведены в табл. 33.
Таблица 33
Бензолполикарбоновые кислоты
Название Формула Температура плавления °C Форма кристаллов
Фталевые: С6Н4(СООН)2
обыкновенная (о-фталевая) ангидриди- Блестящие лис-
зуется точки
изофталевая (л«-фталевая) 348,5 Нежные иглы
терефталевая (п-фталевая) возгоняется Аморфный по-пптппк
Бензолтрикарбоновые: С6Н3(СООН)8 UUJJ-1U1Y
гемимеллитовая (1,2,3-бен- 194 (безв.) Иглы
золтрикарбоновая) тримеллитовая (1,2,4-бен- 224—225 »
золтрикарбоновая) тримезиновая (1,3,5-бен- 375—380 Иглы или приз-
золтрикарбоновая) мы
Бензолтетра ка рбоновые: СвН2(СООН)4 240
меллофановая (1,2,3,4-бен- Призмы
золтетракарбоновая) пренитовая (1,2,3,5-бензол- 253 >
тетракарбоновая) пиромеллитовая (1,2,4,5-бен- 275 (бесцв.) Таблички
золтетра карбоновая)
Бензолпентакарбоновая С6Н(СООН)6 238 Ромбы
Меллитовая (бензолгексакарбо- Св(СООН)в 288 Шелковистые
новая) (в запаянном капилляре) иголочки
Все бензол пол и карбоновые кислоты растворимы в воде и притом тем легче, чем большее число карбоксильных групп они содержат.
Растворимость их в воде уменьшается прй добавлении минеральных кислот (соляной или азотной). Калиевые, натриевые и аммониевые соли этих кислот также легко растворимы в воде. Соли прочих металлов вовсе нерастворимы или трудно растворимы.
Поликарбоновые кислоты с рядовым расположением карбоксильных групп легко образуют ангидриды.
Ангидрид гемимеллитовой кислоты (темп, плавл. 194°), образующийся при ее нагревании до 200°, теряет СО2 при 300°:
СООН
^l^COOH ^^соон
гемимеллитовая кислота
Меллитовая кислота при действии СН3СОС1 дает триангидрид, а при обработке SOC12—диангидрид:
ноос
ноос
соон
меллитовая кислота
SOC12
При сухой перегонке меллитовая кислота отщепляет 2Н2О и 2СОв, образуя диангидрид пиро меллитовой кислоты, а при перегонке с натронной известью теряет 6СО, и превращается в бензол.
Меллитовая кислота получается окислением графита, сажи, каменных и бурых углей, а также древесного угля азотной кислотой, перманганатом в щелочной среде, гипохлоритами. При окислении древесного угля, активированного с помощью хлористого цинка, азотной кислотой в присутствии небольших количеств пятиокиси ванадия меллитовая кислота получается с выходом до 40% от теоретического.
Алюминиевая соль меллитовой кислоты в виде так называемого • медового камня состава [C6(COO)JA12* 18Н2О встречается в залежах бурых углей.
Ароматические оксикислоты
Ароматические оксикислоты разделяются на два класса: 1) содержащие гидроксильные группы в ядре (фенолокислоты) и 2) содержащие гидроксильные группы в боковых цепях,
Фенолокислоты
Способы получения. 1. Действие азотистой кислоты и воды на аминокислоты. В качестве промежуточных продуктов образуются диазосоединения.
2. Сплавление со щелочами солей сульфированных ароматических кислот.
3. Окисление гомологов фенола.
4. Действие четыреххлористого углерода на фенолы в щелочных растворах:
/ОН
СбН5ОК + СС)4 + 4КОН -> С6Ч4< +4КС1 + 2Н2О
^СООК
5. Действие углекислоты на феноляты. Особенно важным способом получения фенолокислот является действие углекислоты на феноляты (Кольбе, 1859 г.)
/ОН
CeH6ONa + CO2 -> СвЧ4<
XLOONa
или на самые фенолы в присутствии сухого поташа при повышенной температуре. Обыкновенно при более низких температурах получаются о-фенолокислоты, при. более высоких—я-фенолокислоты. Многоатомные фенолы присоединяют углекислоту с образованием оксикислот уже при кипячении водных растворов с углекислым аммонием.
При действии углекислоты на феноляты при низких температурах образуются соли кислых эфиров угольной кислоты, или фе-нилуглёкислые соли, например CRH5O—СО—ONa. Поэтому раньше эти соли считали обязательными промежуточными продуктами реакции Кольбе. Теперь, однако, можно считать доказанным, что реакция эта состоит в непосредственном присоединении углекислоты к бензольному ядру.
Многие фенолокислоты содержатся в растениях в свободном состоянии или в виде солей, а также в виде сложных эфиров и других производных. Некоторые простые (метиловые) и сложные эфиры фенолокислот находят применение в медицине.
Свойства. Фенолокислоты могут вступать во всевозможные реакции, свойственные как кислотам, так и фенолам. Кроме того, они способны нитроваться, сульфироваться, галоидироваться и вступать в другие реакции замещения водородных атомов бензольного ядра, характерные для соединений ароматического ряда.
Метиловые эфиры фенолокислот (простые и сложные) легко получаются при действии диметил сульфата на .их щелочные растворы (стр. 264) или взаимодействием фенолокислот с диазометаном. При этом можно получить и продукты неполного метилирования гидроксилов/
Карбоксильные группы одной молекулы фенолокислоты могут вступать в реакцию с гидроксилами других молекул, образуя соединения типа сложных эфиров, например при действии хлорангид-ридов оксикислот на сами оксикислоты:
НО—С6Н4—СОС1 + НО—СбН4—СООН ->
—-> НО—СбН4-СО—О—СбН4—СООН + НС1
Аналогичным образом со второй молекулой хлорангидрида фенолокислоты может получиться соединение
НО—С6Н4—СО—О—СвН4—СО—О—СвН4—СООН и т. д.
Соединения этого типа, называемые депсидами или полидепсидами (по числу остатков фенолокислот различают дидепсиды, тридепсиды и т. д.), близки по строению к некоторым лишайниковым веществам и дубильным веществам типа таннина (стр. 387).
С фенолокислотами находятся в тесном родстве и другие важные природные продукты, например многие смолы, красители из желтых растительных экстрактов (см. ксантон, стр. 588, и флавоны, стр. 583), а также красящие вещества цветов и плодов—антоцианы (стр. 586).
Оксибензойные кислоты. Салициловая (о-оксибензойная) кислота— важнейшая из ароматических оксикислот. В промышленном масштабе она получается по способу Кольбе, усовершенствованному Шмидтом, нагреванием фенолята натрия с углекислотой в автоклаве при 130°.
Салициловая кислота образует кристаллы (темп, плавл. 159°), трудно растворимые в холодной воде. Она является значительно более сильной кислотой, чем бензойная; ее константа диссоциации К ./= 1,06-10““3. При осторожном нагревании она возгоняется, при более сильном нагревании расщепляется на фенол и углекислоту, причем фенол вступает в реакцию с салициловой кислотой, образуя сложный эфир НО—С6Н5—СО—ОС6Н5. Этот эфир, называемый салолом, обычно получают действием хлорокиси фосфора на смесь салициловой кислоты с фенолом.
С хлорным железом салициловая кислота дает фиолетовое окрашивание не только в водном,но и в спиртовом растворе (в отличие от фенола). Окрашивание, повидимому, обусловлено образованием внутрикомплексных солей; оно исчезает при добавлении к раствору минеральных кислот.
С гидратом окиси кальция о-оксибензойная кислота, в отличие от ее изомеров, легко образует нерастворимую соль циклического строения:
Салициловая кислота — сильное дезинфицирующее средство. Многие ее производные являются лекарственными препаратами.
Салициловокислый натрий применяется как жаропонижающее и антиревматическое средство. Метиловый эфир салициловой кислоты—пахучее масло с темп. кип. 224°—главная составная часть эфирного масла Gaultheria', применяется для втираний и мазей при ревматизме.
Фениловый эфир салициловой кислоты, называемый салолом (кристаллы; темп, плавл. 43°), применяется при кишечных заболеваниях и суставном ревматизме.
Наибольшее значение как лекарственное вещество имеет продукт ацетилирования фенольного гидроксила салициловой кислоты действием хлористого ацетила или уксусного ангидрида—ацетилсалициловая кислота, или аспирин'.
^\^^/ОСОСН3
I II
V'Xcooh
Аспирин (кристаллы кислого вкуса; темп, плавл. 135°) является одним из важнейших жаропонижающих, антиревматических и анти-невралгических средств.
Употребляющееся против головной боли средство салофен представляет собой сложный эфир салициловой кислоты и п-ацетаминофенола:
НО—С6Н4—СОО—С6Н4—NHCOCH3
Кроме медицинских целей, салициловая кислота употребляется в больших количествах для получения азокрасителей как вторая составляющая, а также для приготовления аминосалициловой кислоты, применяемой в качестве первой составляющей.
м-Оксибензойная кислота (темп, плавл. 201,3°) и п-оксибензой-ная (темп, плавл. 214,5°) по своей силе лишь немного превосходят бензойную кислоту. п-Метоксибензойная, или анисовая, кислота СН3О—С6Н4—СООН (темп, плавл. 184,9°) получается окислением анетола (стр. 406) и других метоксисоединений.
Протокатеховая (3,4-диоксибензойная) кислота получается из многих природных смол сплавлением их со щелочами. Синтетически ее получают нагреванием пирокатехина с углекислым аммонием. Протокатеховая кислота растворима в воде, кристаллизуется с одной
молекулой воды; безводная плавится при 199°, отщепляя СО2 и переходя в пирокатехин. С хлорным железом она дает зеленое окрашивание, при прибавлении соды переходящее сначала в синее, а затем в красное.
Простой монометиловый эфир протокатеховой кислоты называется ванилиновой кислотой, диметиловый эфир—вератровой кислотой, метиленовый эфир, или 3,4-метилендиоксибензойная кислота,— пиперониловой кислотой*.
СООН
СООН I
соон
'Vz'4och,
он
ванилиновая кислота (темп, плавл- 207°)
осн3
вератровая кислота (темп, плавл. 181°)
I I о—сн2
пиперониловая кислота (темп, плавл. 228°)
Орселлиновая (4,6-диокси-о-толуиловая) кислота получается гидролизом некоторых лишайниковых веществ, например эритрина (из лишайников Rocella Lecanora и др.), распадающегося на эритрит и орселлиновую кислоту (темп, плавл. 176°). Сухой перегонкой орселлиновой кислоты получается орсин (стр. 340). Простой метиловый эфир орселлиновой кислоты, или эверниновая кислота,
СООН
СООН
эверниновая кислота
орселлиновая кислота
является продуктом гидролиза лишайниковых кислот—эверновой и рамалевой.
Галловая (3,4,5-триоксибензойная) кислота—важнейшая из три-оксибензойных кислот. Она содержится в чае, в дубильных экстрактах (сумаховом и др.). Обыкновенно ее получают из таннина кипячением с разбавленными кислотами. Галловая кислота кристаллизуется в иглах с одной молекулой воды, хорошо растворима в кипящей воде, спирте и эфире, трудно—в холодной воде. При 210° она отщепляет СО2, образуя пирогаллол. Щелочные соли галловой кислоты окисляются кислородом воздуха, приобретая бурую окраску. Подобно пирогаллолу, галловая кислота восстанавливает серебро и золото из их солей, а потому применяется в фотографии. С солями окиси железа она дает черный осадок с синим отливом.
Основная висмутовая соль галловой кислоты применяется под названием дерматола как антисептик (для ран и при болезнях кожи).
СОО—В1(ОН)2 I
СООН
галловая кислота
дерматол
3,5-Диметиловый эфир галловой кислоты, называемый сиреневой кислотой СООН
/О
СН3О/ у \осн3
он
содержится в виде глюкозида в коре белой акации (Robinia pseudoacacia) и получается в результате расщепления ряда природных продуктов.
ДУБИЛЬНЫЕ ВЕЩЕСТВА
Под названием дубильных веществ объединяют различные вещества растительного происхождения, способные поглощаться кожей с образованием трудно проницаемой для воды и газов массы, т. е. превращать недубленую шкуру в дубленую кожу. Дубильные вещества содержатся в различных растительных экстрактах, особенно из коры древесных растений, в патологических образованиях дуба и других растений (в так называемых чернильных или дубильных орешках), а также в чае, кофе и пр. Дубильные вещества легко растворимы в воде (некоторые образуют коллоидные растворы), имеют сильно вяжущий вкус, дают с солями окиси железа черное или зеленое окрашивание, с солями свинца—нерастворимые осадки, вызывают коагуляцию белковых веществ.
Дубильные вещества разделяют на две большие группы:
1) дубильные вещества, способные гидролизоваться, т. е. присоединять воду под действием кислот или особых энзимов (например, танназы) и распадаться на соединения с меньшим молекулярным весом;
2) конденсированные дубильные вещества, неспособные подвергаться гидролитическому расщеплению.
Оба класса дубильных веществ находятся в тесном родстве с фе-нолокислотами.
Класс конденсированных дубильных веществ насчитывает гораздо большее число представителей, чем класс гидролизуемых дубильных веществ, но изучено лишь очень небольшое число таких продуктов. К классу конденсированных относится дубильное вещество дубовой коры, а также дубильное вещество южноамериканского дерева квебрахо. К наиболее простым веществам этого класса относится маклурин (желтые пластинки, темп, плавл. 220°), содержащийся в экстракте желтого дерева (Morus tinctoria). Он представляет собой пентаоксибензофенон,
строение которого установлено синтезом из нитрила протокатеховой кислота и флороглюцина:
Главной составной частью экстракта желтого дерева является мерин (стр. 584).
Из более сложных конденсированных дубильных веществ наиболее известны кристаллические вещества состава С]бН14О6, называемые катехинами. В различных дубильных экстрактах найдено несколько катехинов, повидимому.являющихся стереоизомерами, так как при нагревании с водой они могут превращаться друг в друга. В экстракте растения гамбир (Lincaria gambir} с острова Суматры содержится собственно катехин (темп, плавл. 174—175е), представляющий собою смесь правовращающего и недеятельного катехинов (J- и d,/-катехины). Экстракт катеху (Acacia catechu) содержит /-эпикатехин (темп, плавл. 245д; d-эпикатехин был выделен в небольшом количестве из гамбира. Для катехинов можно считать установленной формулу Фрейденберга
ОН
I сн2 ЛЭН
Х2Н Дао-» но/ХА/ ^<он который путем восстановления и метилирования превратил катехины в пентаметоксидифен илпропан
ОСН8
1 ^CHs-CHj-Cf I,—^2/*“ос"г
I I Хосн-
СН,(У/^/'Х0СН,
а также получил /-эпикатехин восстановлением цианидина (стр. 587).
Гораздо лучше исследован класс гидролизуемых дубильных веществ. Простейшие из них, особенно вещества, выделяемые из лишайников, принадлежат к классу полидепсидов (стр. 382). Такова, например, леканоровая кислота (темп, плавл. 175°)
уСН8 zch8
НО-^-СО-О-^-СООН
являющаяся дидепсидом орселлиновой кислоты (п-диорселлиновая кислота). Леканоровая кислота с хлорным железом дает пурпурово красное окрашивание.
м-Дигалловая, или м-галлоилгалловая. кислота (темп, плавл. 290°> НО^ zCOOH
но/ но/ \он
с хлорным железом дает черно-синюю окраску.
Другие дубильные вещества представляют собой сочетания глюкоз с фенолокислотами или с их депсидами. Они построены по типу глюкозидов или по типу сложных эфиров (иногда в них сочетаются оба типа связей).
Большая группа гидролизуемых дубильных веществ принадлежит к глюкозидам эллаговой кислоты, имеющей строение циклического внутреннего дидепсида:
/С“°\ /ОН
ч , НО—</ ^>----</ Ч—он
но/ /о-с/
/о
Наиболее важными веществами этого^класса являются таннины^ извлекаемые из дубильных орешков. Таннины в значительных ко-личествах применяются в качестве протрав при крашении хлоп» чатобумажных тканей азокрасителями. Кроме того, они находят применение в медицине благодаря вяжущим свойствам их растворов»
В течение многих десятков лет считалось, что таннин, выделенный из разных дубильных орешков, представляет собой одно и тс же вещество. Долгое время ему придавали строение дигалловой кислоты (см. выше), хотя многие исследователи указывали на то, что таннин оптически деятелен. В дальнейшем было окончательно доказано (Э. Фишер), что в состав таннина входит глюкоза, получаемая в свободном виде при его гидролизе. Более подробные исследования показали, что таннин, добываемый из китайских орешков (из растения Rhus semialata}, при гидролизе распадается на глюкозу и ж-дигалловую кислоту с выделением некоторого количества галловой кислоты.
Природные таннины не представляют собой индивидуальных продуктов. Таннин из китайский орешков, повидимому, содержит четыре остатка дигалловой кислоты и один остаток галловой, которыми этерифицированы пять гидроксилов глюкозы. У вещества турецких орешков из пяти гидроксилов глюкозы один не этерифици* рован, один этерефицирован дигалловой кислотой, а три—галловой кислотой.
Э. Фишер, заместив водородные атомы во всех гидроксилах глюкозы (точнее— f-глюкозы) на остатки дигалловой кислоты, получил пентадигаллоилглюкозу
[СвН2(ОН)8—СО—О—С,Н,(ОН),—СО]5С6Н7О6
Для приема внутрь при болезнях кишечника часто применяется осажденный таннином белок, или таннальбин. В желудке таннальбин остается неизмененным благодаря кислой среде, а в кишечнике под влиянием щелочи распадается ня белок и таннин.
Черные чернила (так называемые «ализариновые») получают из таннина или из дубильных экстрактов и солей закиси железа, к которым прибавляют какой-нибудь краситель, иногда клей или какой-нибудь другой загуститель и фенол (для предохранения от плесени).
Соединения дубильных веществ с закисью железа почти бесцветны, а потому сначала чернила обладают цветом прибавленного красителя. При медленном же окислении чернил на бумаге образуется соединение окиси железа черного цвета.
Оксикислоты с гидроксильной группой в боковой цепи
Миндальная (оксифенилуксусная) кислота С(5Н5—СН(ОН)—СООН является наиболее распространенной кислотой этого класса. Левовращающая миндальная кислота (темп, плавл. 133,3°) полу-чается при нагревании амигдалина с минеральными кислотами. Недеятельная кислота (темп, плавл. 120.5°), называемая параминдаяъ-ной кислотой, легко получается из бензойного альдегида присоединением к нему цианистого водорода и омылением образовавшегося оксинитрила С6Н5—СН(ОН)—CN. Кристаллизацией цинхониновых солей недеятельная кислота разделяется на d- и /-миндальные кислоты. Если при синтезе оксинитрила из бензойного альдегида прибавить к реакционной смеси хинин, то получается оптически деятельный оксинитрил (см. асимметрический синтез, том I, стр. 508 сл.), образующийся также расщеплением амигдалина под действием оптически деятельного энзима эмульсина.
Троповая и атролактиновая кислоты. Омылением алкалоида атропина получается троповая кислота (темп, плавл. 117—118°)
С,ЧВ—СН—СООН
СН2ОН
при дегидратации дающая ненасыщенную атроповую кислоту (стр. 410)
СвНб— С—СООН
сн2
Присоединяя хлористый водород, атроповая кислота образует кислоту С,НВ— СН-СООН
СН2С1
которая с водой обменивает хлор на гидроксил и дает троповую кислоту.
Присоединением синильной кислоты к ацетофенону и омылением образующегося оксинитрила на холоду получается изомер троповой кислоты—атролак-типовая кислота (темп, плавл. 93,5°)
ОН
I свнб—С—соон сн3
Если же омыл ять нитрил кипячением с соляной кислотой (с последующей обработ-кой содой), то образуется троповая кислота.
Атролактиновая кислота, теряя воду, так же как и троповая кислота, превращается в атроповую кислоту.
Ароматические аминокислоты
Аминогруппа в ароматических аминокислотах может находиться в ядре или в боковой цепи. В первом случае аминогруппа имеет такой же характер, как в анилине, во втором—как в жирных аминокислотах.
Аминобензойные кислоты
Все три простейшие аминобензойные кислоты могут быть получены восстановлением нитробонзийных кислот; они имеют следующие температуры плавления:
о-Аминобензойная . . . 145°
,и-Аминобензойная . . . 174°
я-Аминобензойная • • . 187°
Важнейшей из изомерных кислот является о-аминобензойная, гак называемая антраниловая кислота, В промышленном масштабе она получается действием хлорноватистокислых солей в щелочной среде на фталимид. Промежуточно образующаяся под действием щелочи фталаминовая кислота по реакции Гофмана (см. том I, стр. 282) превращается в антраниловую кислоту:
/С\ /COONa nh ^°д fY А\,/ 'V'V.ONIK фталаминовая кислота Реакция Гсф:.;а::а протекает, попидимому, :'о hoi СОО" СОО" _ л г ¥ •• 1 м А А/А/X'Hci \ ^/ ^cox’H^ с<о ос. - с a/OONj 1 II 'V'XnHj iному механизму* к ,соо~ Y ^сАг СОО" NH,
Вероятность такого механизма подтверждается тем, что промежуточные продукты выделены и идентифицированы.
Антраниловая кислота может быть получена также изомеризацией о-нитротолуола под действием концентрированной спиртовой или водной едкой щелочи:
СН3 СООН
Антраниловая кислота имеет сладкий вкус, легко растворяется в воде и спирте. При действии азотистой кислоты она дает соли диазония; является важным промежуточным продуктом в синтезе различных красителей (азокрасителей, синтетического индиго и др.). Метиловый эфир антраниловой кислоты (темп, плавл. 25,5°) применяется как душистое вещество. Он встречается в природе в эфирных маслах цветов померанца и туберозы.
п-Аминобензоиная кислота обладает высокой физиологической активностью. С несомненностью установлено, что л-аминобензойная кислота (сокращенноПАБ) является фактором роста микроорганизмов Так, бактерицидное действие сульфамидных препаратов (стр. 311) объясняют тем, что п-аминобензолсульфокислота и ее производные вытесняют необходимую для жизни бактерий л-аминобензойную кислоту. По некоторым данным, л-аминобензойную кислоту можно также рассматривать как витамин, способствующий росту и пигментации шерстяного покрова у животных.
Ценным противотуберкулезным препаратом является 4-амино-2-оксибензойная, или п-аминосалицилевая, кислота (сокращенно ПАСК). Она получается действием углекислоты (или бикарбонатов) на .м-аминофенол:
СООН
у\/он Л/он
Q —0
nh2 nh,
n-аминосалиииловая
кислота
Важно иметь в виду, что изомеры этой кислоты не оказывают бактерицидного действия, а один, из них — 5-амино-2-оксибензойная, или м-аминосалициловая, кислота
СООН
Hjn/'V'
обладает довольно высокой токсичностью.
Эфиры всех ароматических аминокислот в той или иной степени обладают физиологической активностью—списочноегыо вызывать местную анестезию. Эта способность особенно сильно выражена у эфиров ароматических аминокислот, преимущественно у содержащих аминогруппу в пара-положении к карбоксиль-30 1 П )эг<)му cii 1ьным анестезирующим действием обладают производные п-ами-яоЗэнзойной кислоты, применяемые для местной анестезии взамен кокаина. Так» хл johctoboiэродная соль этилового э|)ира n-аминобензойной кислоты (темп, плавл. 96— 9Г) применяется в медицине под названием анестезина. Особенно большое гииюнение имеет ц(И')киин—хлористоводородная соль р-диэтилами-яоэтилового эфира п-аминобензойной кислоты
HSN_\^_coo_CH2-CH’“N(c’Ht)s
Новокаин—бе?цветные, легко растворимые в воде иглы; темп, плавл. 156°. Свободное основание новокаина представляет собою маслянистую жидкость.
Аминокислоты с аминогруппой в боковой цепи
Из веществ этого класса наибольшее значение имеют производные |3-фенилпропионовой кислоты СЦНО—СНг—CHt—СООН, особенно фенилаланин и тирозин (/г-оксифенилаланин):
СН,—СН—СООН НО-^~Л—СИ»—С^—СООН
NH, NH,
фенилаланин тирозин
Фенилаланин и тирозин содержатся в продуктах гидролиза почти всех белковых веществ. Их присутствием обусловлены некоторые реакции белков, например ксантопротеиновая реакция /Миллона (см. том I, стр. 700).
Тирозин является одной из важнейших составных частей белков; его присутствие в белках безусловно необходимо для питания животных и человека Особенно много тирозина получаете при гидролизе казеина* (более 3,5%) и фиброина шелка (ди 10%). 1\ак продукт превращения белков тирозин был найден в самых различных животных и расти тельных веществах.
Тирозин был синтезирован по следующей схеме Конденсацией д-оксибенз-альдегида с гиппуровой кислотой щтр. 292)
СН2—СООН НО—СбН4—CI ь=с—соон
НО-С6Н4-СНО +1 | +Н2о
NH—COC6I Ie NH—СОСвНб
а присоединешем к продукту конденсации двух атомов водорода получается бензоил, пиразин
НО—С6Н4—СН?—СН—СООН I .
NH—СОСвН6 который при омылении дает недеятельный тирозин.
* Название тирозин как раз и произошло от греческого названия сыра, в котором он содержится.
Природный тирозин вращает плоскость поляризации влево. Бензоилтирозин был разделен с помощью оптически деятельных оснований на антиподы; один из антиподов при омылении дает природный /-тирозин, представляющий собой иглы, трудно растворимые в холодной, легче—в горячей воде.
Окислением тирозина при участии энзима тирозиназы получаются очень прочные коричневые красители разных оттенков, называемые меланинами. Цвет волос человека и высших животных и цвет кожи негров определяется присутствием аналогичного продукта окисления диоксифенилаланина («допа»), который может быть получен синтетически, а также из продуктов гидролиза белков Vici а faba.
Тироксин C15HnO4NJ4 содержится в щитовидной железе (около 0,027%); это гормон, регулирующий обмен веществ. Строение тироксин:: установлено в 1926 г. Харрингтоном, изучившим продукты расщепления тирозина и подтвердившим его строение синтезом. Взаимодействием монометилового эфира гидрохинона с трииоднитробензолом было получено иодированное производное дифенилового эфира:
\ \
СНЯО-<3-ОН + J—~N0* ch3o-<3-o-<3-no2 + HJ
j/ j/
монометиловый эфир трииоднитробензол
гидрохинона
Восстановление группы NO2 в NH2, замена группы NH2 на CN путем диазореакции и затем восстановление получаемого нитрила хлористым оловом дает альдегид. Этот альдегид был подвергнут конденсации с бензоилгликоколем:
ОН
Из полученного продукта конденсации действием иодистоводородной кислоты лолучен оксифенилдииодтирозин, который при действии иода дает тироксин:
\ \
но—о—сн,—сн—соон \=/ =/ I
У У nh2
тироксин
Ароматические оксиальдегиды и оксикетоны
Ароматические оксиальдегиды и оксикетоны могут содержать гидроксильную группу либо в ароматическом ядре, либо в одной из боковых цепей. Наибольшее значение имеют соединения первого типа.
Ароматические оксиальдегиды, содержащие альдегидную группу в орто-положении к фенольному гидроксилу, могут быть получены
действием на фенолы хлороформа и щелочи (реакция Тимана—Реймера):
ОН + СНС13 —> ОН + HCI
ЧСНС12
^>-OH + 2NaOH —> \;i 1С12
Наряду с орто-производным при этом образуется немного в пара-производного.
Пара-замещенные оксиальдегиды получаются при взаимодей» ствии фенолов с безводной синильной кислотой в присутствии соляной кислоты, причем сначала образуются соли иминов оксч льде-гидов:
н°—<^3^ + HCN-f-HCl —> H0-<3-CH^NH.HCl
НО— CH=NH-HC1 + Н2О —» НО-<^2^—СНО + NH4C)
Оксикетоны легко получить действием на фенолы хлорангид-ридов кислот (или кислот в присутствии галоидных соединений фосфора):
,ОН
С6Н5ОН + С1-СОСНЧ —> С6н/ +НС1
хсосн3
Особенно легко в эту реакцию вступают многоатомные фенолы.
Оксиальдегиды
Салициловый альдегид (о-оксибензальлегид) находится в эфирном масле Spiraea ultnaria. Он представляет собой жидкость с запахом миндалей, кипящую при 197°, замерзающую при —10°; уд. вес 1,1669 (при 20°).
Интересно, что этот альдегид обладает довольно сильными кислотными свойствами, и поэтому он был вначале даже назван сали-пилистой кислотой. Это является результатом смешения электронной плотности по направлению к кислородному атому карбонильной группы
сО-н
'с==о I н
облегчающего ионизацию протона в фенольном гидроксиле.
Щелочные растворы салицилового альдегида окрашены в желтый цвет; окраска объясняется образованием внутрикомплексных солей, например;
'%;
СН
Салициловый альдегид является промежуточным продуктом при синтезе кумарина (сгр. 411).
Оксим салицилового альдегида служит реактивом на ионы меди, с которыми образует светлозеленый осадок внутрикомплексной соли:
I
ч ZCU О
Анисовый альдегид (мет иловый эфир п-оксибензальдегида)
СНО
получается в промышленном масштабе окислением анетола (стр. 406). Он представляем собой жидкость пряного запаха с темп. кип. 248*; уд. вес 1.123 (при 20°). Анисовый альдегид применяется в технике как душистое вещество под названием обепин.
Нротокатеховый (3,4-диоксибензойный) альдегид (иглы с темп, плавл. 153—154°)
сно
I он он получается из пирокатехина по реакции Тимана—Реймера (стр. 393). Вго простые эфиры являются важными душистыми веществами.
Неполный метиловый эфир протокатехового альдегида—ванилин (белые иглы, темп, плавл. 81—82°, перегоняем при 170° и 15 мм) является действующим началом ванили; в боб^х ванили содержится 1,5—2,5% ванилина. Незначи1ельные количества ванилина обнаружены в очень многих растительных веществах (в смолах, бальзамах, в древесине растений) и даже в почве. В промышленности он готовится несколькими способами из гваякола (стр. 341) или окисле
нием изоэвгенола (стр. 340). Весьма перспективным является получение ванилина окислением лигнина (стр. 345). Полный метило-рый эфир протокатехового альдегида, или вератровый альдегид 'темп, плавл. 58°), по запаху напоминает ванилин.
СНО
СНО
он
ванилин
'у/\ссн8 ссн3
вератровый альдегид
Метиленовый эфир протокатехового альдегида, или пиперональ (темп, плавл. 37°, темп. кип. 263°), имеет запах гелиотропа и применяется как душистое вещество под названием гелиотропина. При действии НС1 пиперональ распадается да протокатеховый альдегид и уголь:
СНО
СНО
I I о—сн,
пиперональ
Оксикетоны
Из соединений этого класса заслуживают быть отмеченными следующие:
^-Оксиацетофенон, или бензоилкарбинол, СКН5—СО—СН2ОН (темп, плавл. 95°) получается при гидролизе ш-хлор- и ш-бром-ацетофенонов (непосредственно или с промежуточным образованием ацетильных производных).
п-Оксиацетофенон НО—С6Н4—СОСН8 (темп, плавл. 110°) содержится в иглах пихты в виде глюкозида пицеина. Из анизола и хлористого ацетила по реакции Фриделя—Крафтса можно получить метиловый эфир n-оксиацетофенона, при омылении дающий сам и-оксиацетофенон.
Цингерон (темп, плавл. 41°)—действующее начало имбиря— может быть синтезирован таким путем:
НО—сн=° + CHj-CO—СН, —> СН3о/
но—<\2у—сн=сн—со—cfj, — СН,о/
но—сн2—сн,-со-сн, СН8о/
цингерон
А роматическ ие амш юа ль дегиды
Лминобензальдегиды (о-, м- и п-) получаются восстановлением соответствующих нитробен *альдегидов. о-Аминобензальде^ид плавится при 39й, пара-соединение—при 7Г, .и-аминобензальдегид—• аморфное вещество.
о-Аминобензальдегид применяется для получения хинолиновых оснований.
п-Диметиламинобензальдегид (CHy)2N—СсН4—СНО получается действием едкого кали на продукт конденсации хлораля с диметил-анилином (CH3)2N—С(.Н4—Ci UOH)—СС1а. При этом, кроме аминоальдегида, получается хлороформ. Этот аминоальдегид применяется в производстве красителей, а также как реактив, способный давать характерные цветные реакции с рядом алкалоидов и с некоторыми другими гетероциклическими и ароматическими соединениями.
Соединения со смешанными функциями, содержащие мышьяк
Ароматические амины и фенолы могут вступать с мышьяковой кислотой в реакцию, подобную реакциям нитрования и сульфирования, При этом получаются производные ароматических арсино-вых кислот. Так, например, анилин с мышьяковой кислотой дает п-аминофениларсиновую, или арсаниловую, кислоту:
' + Н0~”As°(°h)2 —> H2NA^OiOHy 4- H?0
Арсаниловая кислота разлагается, не плавясь, при температуре около 280°. Натриевая соль этой кислоты, называемая итокси-лом, и натриевая соль ацетил арсиновой кислоты—арсацетин
СН3СО—NH—^-A.CXOH) (0Na}
являются тонизирующими средствами и обладают лечебным действием против сонной болезни.
При действии азотистой кислоты на арсаниловую кислоту получается п-оксифениларсиновая кислота
НО—< Аъ(Х°Н\
которая может быть также получена действием мышьяковой кислоты на фенол или из n-аминофенола по реакции Барта (стр. 331). Нитрованием n-оксифениларсиновой кислоты получается нитросоединение
—As°(°H)
НО
при восстановлении дающее диаминодиокснарсенобензол
As= As—*
H2n/ ^NHj
производное арсенобензола (стр. 331), названное Эрлихом* сальварсаном Сальварсан является сильнодействук шим средством против болезней, вызываемых спирохетами и трипанозсмами. К таким болезням относятся возвратный тиф, сифилис и сонная болезнь.
В настоящее время можно считать доказанным, что молекулы сальварсана не являются мономерными и он представляет собой смесь полимеров состава
причем п может варьировать от 7 до 15, от чего зависит растворимость препаратов (М. Я. Крафт).
В свое время сальварсан сыграл огромную роль в лечении сифилиса, долгое время считавшегося неизлечимой болезнью. Сальварсан-основание—желтый порошок, легко окисляющийся на воздухе с образованием ядовитых продуктов окисления. Вследствие вредного действия продуктов окисления он был в дальнейшем вытеснен неосальварсаном (новарсенол), получаемым путем присоединения к сальварсану формальдегидсульфоксилата (см. том I, сгр. 241).
Невидимому, неосальварсан представляет собой смесь двух соединений—собственно неосальварсана
—As=As—ОН
NaOSO—СН2—НЬк"\NH2
и миарсенола
НО—As=-As—^==^>—ОН NaOSO—CI 12-I IN'/ \nH-CH2—OSONa
Миарсенол выпускается как индивидуальный препарат.
Средством против сифилиса является также 3-ацетамино-4-оксифениларсиновая кислота, называемая стоварсолом.
* Пауль Эрлих (1854—1915). Крупный немецкий химик и биолог. Начав свою работу с поисков красителей, избирательно окрашивающих микроорганизмы, он создал рабочую гипотезу о причинах паразитотропного действия не только красителей, но и лекарственных веществ и вообще химических препаратов. Целеустремленно добиваясь модификации свойств группы мышьяковистых препаратов и впервые широко применив систему биологического контроля, он достиг крупных практических успехов в терапии сифилиса и некоторых тропических болезней.
О СПОСОБАХ ОПРЕДЕЛЕНИЯ ПОЛОЖЕНИЯ ЗАМЕЩАЮЩИХ ГРУПП В БЕНЗОЛЬНОМ ЯДРЕ
Положение замещающих групп в бензольном ядре в настоящее время обычно определяют путем такой замены этих групп другими радикалами, чтобы исследуемое вещество превратилось в соединение, для которого положение заместителей уже известно. Так, для определения положения боковых цепей в двузамещенных гомологах бензола устанавливают, какая из фталевых кислот получается при окислении боковых цепей в карбоксильные группы. Положение нитрогрупп в jw-динитробензоле подтверждается образованием при его восстановлении лг-фенилендиамина и т. д.
Разумеется, такой способ определения дает правильные результаты лишь при условии, что превращения происходят без перемещения замещающих групп в бензольном ядре. При громадном большинстве реакций замещающие группы, действительно, сохраняют свое положение. Перемещение заместителей в ядре происходит лишь в редких случаях, которые в настоящее время достаточно хорошо изучены. Так, известно, что при сплавлении сульфокислот фенолов и галоидсульфокислот со щелочами при высокой температуре оксигруппы из орто- и пара-положений могут перемещаться в мета-положение (стр. 260 и 337).
Таким образом, в большинстве случаев, заменяя одни заместители в бензольном ядре другими, можно установить строение любого производного бензола. Однако, если в настоящее время для установления положения заместителей в бензольном ядре достаточно превратить изучаемое соединение в какое-либо другое, уже известное, с таким же расположением заместителей, то прежде, когда еще не было известно таких соединений, этот путь был неприменим. Для первоначального определения расположения замещающих групп требовались не относительные, но абсолютные доказательства положения заместителей хотя бы в одном из веществ каждого ряда взаимно превращающихся производных. Одним из методов такого абсолютного доказательства является правило Кернера.
Кернер еще в 1875 г предложил определять принадлежность двузамещенных производных бензола к орто- и пара-ряду по числу изомерных веществ, получающихся при замещении третьего атома водорода в бензольном ядре на какой-нибудь атом или радикал. Действительно, двузамещенному орто-соединению с двумя одинаковыми заместителями X отвечают два трехзамещенных изомера
мета-соединению—три изомера
а при замещении третьего атома водорода в пара-соединении, в котором все четыре незамещенных водородных атома одинаково расположены по отношению к заместителям, может получиться лишь одно вещество:
X
о
X
Кернер показал, что из трех дибромбензолов получается отвечающее этому правилу число мононитролибромбензолов Почти одновременно Грисс установил строение трех фенилендиамнноь на том основании, что один из них может сыть получен нагреванием двух из шести известных изомерных диаминобензейных кислот СбИН^СООН. второй—из трех других, а третий—лишь из одной, последней Таким образом, первый является о-фениленлиамином, второй имеет метастроение, а третий—пара-строение. Позднее Нельтинг (1885 г., установил, что трем ксилолам отвечает число нитроксилолов, соответствующее правилу Кернера.
Кроме правила Кернера, возможны и другие способы абсолютного установления положения заместителей. Строение обыкновенной фталевой! кислоты как орто-кислоты устанавливается синтезом ее путем окисления нафталина.
Доказательством строения лг-ксилола (и изофталевой кислоты, получающейся при его окислении) является его синтез из мезитилена
СН, I
J
НзС/^ХсНз
Так, окислением мезитилена можно получить одноосновную кислоту
сн8 5
HgC/^/^COOH
при нагревании теряющую СО2 с образованием л<-ксилола. Симметрическое же строение мезитилена доказывается тем, что этот углеводород может быть синтезирован из ацетона.
Положение заместителей трех- и четырехзамещенных производных бензола устанавливают в большинстве случаев путем исследования их отношений к двузамещенным производным. Так, например, строение хлорнитроанилина C6H3CI(NO.?)NH.? доказывается тем, что он получается нитрованием л<-хлоранилина, а при замене аминогруппы на водород (диазореакцией со спиртом) дает п-хлорнитро-бензол:
С1-Q —» Cl—N02 —> ci—NO,
\nh2 \nh2
Строение пикриновой кислоты
/NOa
0,N-^^>—ОН
\мо2
доказывается тем, что она может быть получена нитрованием любого из двух динитрофенолов:
Строение первого из этих линитрофенолов доказано образованием <его при нитровании как о-, так и «-нитрофенола, строение второго— тем, что он получается при нитровании ^-нитрофенола, а при замене ОН на NH2 (нагреванием с ZnCl2 и NH3) и последующем обмене NH, на Н (диазореакцией со спиртом) дает лг-динитробензол.
Ароматические соединения с ненасыщенными боковыми цепями
Углеводороды
Стирол. Простейшим арома 1ическим углеводородом с ненасыщенной боковой цепью является фенилэтилен С6Нб—СН=-СН2, называемый обычно стиролом.
Стирол—приятно пахнущая жидкость, застывающая при низких температурах в кристаллы, плавящиеся при—30,6\ темп. кип. 145,2°; уд. вес ^4°—0,9060; показатель преломления п2э-- 1,5468.
Впервые стирол был получен перегонкой жидкой смолы родственного платану растения (от названия этой смолы—стиракс—и происходит слово стирол). Стирол содержится также в каменноугольном дегте.
Синтетически стирол может быть получен медленной перегонкой коричной кислоты
СвНб—СН=СН—СООН —» СбНб—СН=СН2 + СО, дегидратацией фенилэтиловых спиртов
СвНь—СН2—СН2ОН С6н5—СН=СН2+Н,0
отнятием галоидоводорода от этилбензолов, содержащих галоид в боковой цепи
СвНб—СНВг—СН, -» С6Нб-СН=СН2 + НВг и другими способами, аналогичными способам получения простейших этиленовых'углеводородов.
В промышленных масштабах стирол получают в основном дегидрированием этилбензола в паровой фазе при 450—500° над катализаторами (окись хрома или цинка; носитель—окись алюминия):
СвНб—СН2—СН3 СвНб-СН=СН2+Н2^
Процесс ведут или в вакууме, или при разбавлении паров этилбензола водяным паром.
Небольшие количества стирола получают следующим способом. Этилбензол каталитически окисляют в жидкий фазе:
СвНб-СН2-СН3 + О2 -> СвНб-СО-СН3 + Н2О
Полученный ацетофенон каталитически же гидрируют в метилфеьгил-карбинол
С6Н5-СО-СН3 + Н2 -> СвНб-СН(ОН)-СН3 который дегидратируют в паровой фазе над окисью алюминия:
СбНб—СН(ОН)—СН3 —> ( 6нб—сн=сн9 + Н2О
Этим путем получается значительно более чистый стирол.
Стирол чрезвычайно склонен к полимеризации Уже при обычной температуре медленно, а при нагревании, особенно на свету, быстро, он превращается в стекловидный полимер метостирол. при перегонке вновь дающий стирол.
Благодаря способности полимеризоваться стирол в настоящее время приобрел очень большое практическое значение. Его высокомолекулярные полимеры—полистиролы
СН,—СН—
Е6Нб п с молекулярным весом от 20 000 до 300 000 и выше—отличные диэлектрики и применяются в электро- и радиотехнике как изоляционные материалы.
Наибольшие количества стирола используют для совместной полимеризации с дивинилом (см. том I, стр. 374) при производстве синтет ических каучуков.
Для предохранения стирола от преждевременной полимеризации в нем растворяют, при перегонке, элементарную серу, в большинстве случаев в качестве ингибиторов полимеризации добавляют такие вещества, как гидрохинон пирокатехин и т п.
Стирол легко присоединяет галоиды, галоидоводороды и пр. и образует стиролдихлорид С6Н5—CHCI—СН,С1 (темп. кип. 233—234°) или, соответственно, дибромид CfiH5—СНВг—СН.,Вг (иглы; темп, плавл. 74°). Такие дигалогениды при нагревании особенно гладко в присутствии спиртовой щелочи, отщепляют элементы хлористого или бромистого водорода с образованием а-галоидстиролов, обладающих резким запахом.
Стирол очень легко, практически мгновенно, окисляется перманганатом, причем первым продуктом окисления является фенилэтилен-гликоль
СвН5-СН(ОН>-СН?ОН
Азотной кислотой стирол нитруется, легко образуя соединение с нитрогруппой в боковой цепи—^-нитростирол
СвН5—СН=СН—NO,
То же соединение получается конденсацией бензальдегида с нитрометаном*
С6Нб—СНО + СН,—NO, —> С,Н,—СН=СН—NO, + Н,0
а-Метилстирол С6Н5—С(СН3) = СН2 представляет собою жидкость, кипящую при 162°; уд. вес. d4°=0,9080; показатель преломления п2п= 1,5350. При —22° он застывает, образуя кристаллическую массу. а-Метилстирол может быть получен дегидратацией диметилфен ил карбинол а С6Н5—С(СН3)2(ОН), образующегося, например, при взаимодействии феиилмагнийбромида с ацетоном. Он способен к полимеризации лишь при очень низких температурах под действием хлористого алюминия, хлорного олова и т. п (ср. полимеризацию изобутилена—том I, стр. 370), сополимеризация же его с дивинилом легко протекает в водных эмульсиях под действием перекисных соединений.
а-Метилстирол легко окисляется кислородом воздуха, образуя ацетофенон и формальдегид
СвНб—С=СН2 + О, CeH6—С—О + О—СН,
Ан3 сн3
Стирол в этих условиях окисляется до бензальдегида значительно труднее.
B-Метилстирол С6Н5—СН=СН—СН3 получается отщеплением воды от этилфенилкарбинола С6НЬ—СН(ОН)—СН9—‘СН3, синтезируемого действием этилмагнийбромида на бензальдегид. Он образуется также при перегруппировке аллилбензола под влиянием щелочей. p-Метилстирол представляет собой жидкость, кипящую при 177°; уд. вес d?°=0.9141, показатель преломления 1,5492. Этот продукт является смесью цпс-щраноизомеров.
Аллилбензол С6Н5—СН2—СН = СН2 получается действием бромистого аллила на фенилмагнийбромид; это—жидкость, кипящая при 156,3°; уд вес бЙ°—0,8930; показатель преломления ггя= 1,5126.
Превращение аллилбензола в р-метилстирол
СвНб—СН,—СН=СН2 С6Нб—СН=СН—СН8
является обратимым процессом; однако в обычных условиях равновесие почти целиком смещено вправо.
Фенилацетилен С6Н5—С=СН может быть легко получен из стиролдибромида С6Н5—СНВг—СН2Вг путем отщепления от вето двух молекул бромистого водорода. В качестве промежуточного продукта при этом образуется бромстирол С6Н5—СН=СНВг. Фенил ацетилен—жидкость; темп. кип. 141,6°; уд. вес df =0,9295; показатель преломления п2о= 1,5485. Как однозамещенный ацетилен он легко дает металлические производные СПН5—C=CNa, С6Н5—C=CAg, С6Н5—С=ССи (желтого цвета) Под действием серной кислоты фенилацетилен присоединяет воду, образуя ацетофенон С6Н5—СО—СН3.
ИОНООБМЕНИВАЮЩИЕ СМОЛЫ (ИОНИТЫ)
Высокомолекулярные полимеры стирола, феноло-алъдегидные и другие смолы, содержащие кислотные группы (сульфогруппы, карбоксильные группы), представляют собой нерастворимые кислоты, способные обменивать водородный атом кислотной группы на ионы металлов из растворов электролитов. Образовавшиеся соли, в свою очередь, могут обмениваться катионами с растворами других солей или вновь замещать катион на протон из растворов кислот. Такие смолы называются катионообменивающими, или катионитами.
Процесс катионного обмена можно схематически изобразить так:
[Катионит]Н 4- Na+ 4- СГ [Катионит]Ма 4- Н+ 4- СГ
В случае сульфированных полимеров стирола этот процесс протекает следующим образом:
4- nNaCl
СН—СИ,—’
4-пНС1
В зависимости от характера кислотной группы различают сильные катиониты (например, содержащие сульфогруппы), способные вступать в обменные реакции с нейтральными солями сильных минеральных кислот, и слабые катиониты, содержащие карбоксильные группы или фенольные гидроксилы.
Нитрованием и последующим восстановлением сополимеров стирола с дивинилбензолом* можно получать нерастворимые высокомолекулярные смолы, содержащие аминогруппы:
* Роль небольшой добавки дивинилбензола при сополимеризации заключается в создании поперечных мостиков, связывающих цепи линейных полимеров стирола в сетчатые трехмерные образования, способные лишь набухать, но не растворяться в обычных растворителях.
Такие смолы, называемые анионообменивающими, или анионитами, в водной среде способны связывать кислоты
СН-СН2—" СН—СН2—~
образуя соли типа аммонийных, которые под действием растворов щелочей вновь регенерируют аминосоединения.
Слабыми анионитами являются также смолы, содержащие иминогруппы, например продукты конденсации ж-фенилендиамина с альдегидами, имеющие, повидимому, следующее строение: ------------NH—СН2
NH—NH—СН2-------
^\сн2 I NH
Иминогруппы в таких смолах сохраняют основной характер, благодаря чему эти смолы могут поглощать из растворов кислоты и образовывать соли по типу аммонийных солей.
Более сильными анионитами являются смолы, содержащие четвертичные аммониевые группы. Они могут быть получены, например, путем замены галоида на аминогруппу в полимерах хлористого винила с последующим превращением аминогрупп действием диметил сульфата в четвертичные аммониевые группы:
----СН—СН2—СН—СНа-----
N(CH3)3 N(CH3)S
он он
Подобные аниониты способны вступать в анионный обмен со слабыми кислотами и с растворами солей.
Последовательной фильтрацией раствора, содержащего электролиты, через катионообменный и анионообменный фильтры можно извлечь из раствора все катионы и анионы и получить таким образом деионизированную воду, практически идентичную дестиллиро-ванной. Этот метод нашел широкое применение при обессоливании и умягчении воды, используемой для питания паровых котлов и в ряде отраслей химической промышленности. Ионообменивающие смолы с успехом применяются в сахарной промышленности для удаления из растворов солей, затрудняющих кристаллизацию сахара; для
извлечения ценных металлов из разбавленных растворов их солей; в производстве лекарственных веществ; в аналитической практике и т. п.
Сульфополимеры стирола могут быть использованы не только в качестве сорбционных фильтров для поглощения катионов из растворов, но и для разделения сложных смесей катионов. Этот процесс основан на различии в величине константы ионного обмена различных катионов смеси. При помощи сульфированного сополимера стирола и дивинилбензола удалось разделить сложные смеси катионов редкоземельных элементов, выделяя каждый из них в спектрально чистом виде.
Изменяя соотношение стирола и дивинилбензола в составе сополимера, можно регулировать степень набухания продуктов сульфирования, а следовательно, и проницаемость смолы для катионов различного размера. Эти свойства катионообменивающих сульфополимеров используют для разделения смесей органических катионов. отличающихся по величине молекулярного веса (молекулярные смоляные сита).
Разделение сложных смесей катионов в некоторых случаях облегчается при применении в качестве катионообменных фильтров продуктов совместной полимеризации дивинилбензола с ненасыщенными кислотами (акриловая, метакриловая). В таких сополимерах содержатся кислотные группы с меньшей степенью диссоциации, чем сульфогруппы, а различие в константе обмена между отдельными катионами раствора и нерастворимой кислотой возрастает с уменьшением степени диссоциации последней.
Важной областью применения ионообменивающих смол является использование их в качестве катализаторов процессов, ускоряемых протонами или гидроксильными группами (этерификация, дегидратация спиртов, синтез ацеталей, простых эфиров и т. д.). При применении ионообменивающих смол отделение катализатора от продуктов реакции становится совсем простой операцией.
За рубежом ионообменные смолы выпускаются под названием вофатитов различных марок, амберлитов и под другими фирменными названиями.
Спирты и фенолы
Простейшим из ароматических спиртов с ненасыщенной боковой цепью является коричный алкоголь С6Н5—СН=СН—СН2ОН (иглы; темп, плавл. 33°. темп, кип 257,5°) Он имеет запах гиацинта; содержится в стираксе (стр. 401) в виде эфира коричной кислоты
Фенолы с ненасыщенными боковыми цепями и их метиловые эфиры содержатся в некоторых эфирных маслах Так в масле из листьев бетеля содержится хавикол НО—СвН4—СН2—СН=СН2, а главной составной частью эстраголового масла является метиловый эфир хавикола—эстрагол СН3О—СвН,—СН2—СН=СН2. Изомерный эстраголу анетол СН3О—СвЩ—СН=СН—СН3 (темп плавл 22,5°; темп, кип 235,3°)—главная часть анисового масла, содержится также в укропном (фенхе-левом) и других эфирных маслах
Соединения эти получены и синтетически.
Альдегиды и кетоны
Коричный альдегид СбН5—СН=СН—СНО—пахучее вещество корицы—представляет собой житкость, замерзающую при —7,5°; при 20 мм его темп. кип. 130° а при атмосферном давлении 251е (с небольшим разложением); уд. вес ^4°=1,0497; показатель преломления 1,61949.
С аммиаком коричный альдегид образует гидрамид
С6Нб—СН=СН—CH=N>
СбНб—СН=СН—СН=№
СН-СН=СН—С6Н5
а действием водных щелочей превращается в коричную кислоту и коричный алкоголь.
Коричный альдегид может быть получен конденсацией бензойного и уксусного альдегидов:
С6Нб—СНО + СН3—СНО —> СвНб—СН=СН—СНО + Н2О
Аналогичной конденсацией бензальдегида с ацетоном (в присутствии разбавленных щелочей) могут быть получены ненасыщенные кетоны—бензальацетон и дибензальацетон.
Бензальацетон СбН5—СН=СН—СО—СН3 образует бесцветные таблички с темп, плавл. 41—42 ; запах его напоминает землянику.
Бензальацетон окисляется гипохлоритами в коричную кислоту:
NaOCl
СбНб—СН=СН—СО—СН8-------> СбНб—СН=СН—СООН + СНС13
Дибензальацетон С6Н5—СН=СН—СО—СН=СН—С6Н5 образует желтова-
тые таблички, плавящиеся при 112°.
В концентрированной серной кислоте бензальацетон растворяется с образованием оранжево-красных растворов; растворы дибензальацетона—вишневокрасные.
Карбоновые кислоты
Кислоты этого класса хорошо изучены. Некоторые из них встречаются в природе.
Наиболее важный синтетический способ получения таких кислот состоит в нагревании ароматических альдегидов с солями жирных кислот в присутствии их ангидридов {реакция Перкина)'.
Аг—СНО + СН2—COONa Ar-CH= С—COONa
| -> I +Н2О
Aik (или Н) Aik (или Н)
г По современным представлениям, механизм реакции Перкина состоит в том, что альдегид реагирует с натриевым производным енольной формы ангидрида кислоты—с анионом енолята (енолизация происходит в результате взаимодействия ангидрида с натриевой солью или каким-либо основанием).
Продукт присоединения далее разлагается с отщеплением, например, коричной кислоты:
СН3—С=О
H2Cj=C-fQ:
Na* + С6Н5—СН=О —>
СН,-С=О Ъ
Н2С-С=О
С6Н5—СН-О:
СН3-С=О
чо сн3-соон
—► н2с-с=о —* +
I сн—соон
с6н5—сн—он ||
СсН5-СН
Кроме того, кислоты с ненасыщенными боковыми цепями могут быть синтезированы всеми способами получения жирных кислот этиленового ряда, например отнятием воды от соответствующих оксикислот:
С6Н5—СН—СООН С6Н3—С—СООН
I _* II + н20
СН2ОН сна
Коричная кислота С6Н5—СН^СН—СООН и все ее гомологи могут существовать в стереоизомерных цис- и транс-формах (см. том I, стр. 379 сл.) Их реакции в основном аналогичны реакциям цис- и транс-форм акриловой кислоты и ее гомологов Как ароматические кислоты, они могут нитроваться, причем нитрогруппа может вступать также и в боковую цепь.
Обыкновенная коричная кислота (темп, плавл. 134°) является составной частью перуанского и толутанского бальзамов, содержащих, кроме смолистых веществ, главным образом сложные эфиры бензойной и коричной кислот с бензиловым и коричным алкоголями. Она же получается в большинстве случаев при синтезах коричной кислоты.
В отбросах производства алкалоида кокаина содержится второй стереоизомер коричной кислоты (темп, плавл. 68°), называемый аллокоричной кислотой. Эта кислота может быть получена каталитическим восстановлением фенилпропиоловой кислоты (стр. 410) на холоду в присутствии палладия, а также действием ультрафиолетовых лучей на обыкновенную коричную кислоту. Так как гидрирование тройной связи в указанных условиях приводит к преимущественному образованию цис-форм, а под действием ультрафиолетовых лучей транс-формы превращаются в цис-формы. аллокоричной кислоте приписывают цис-конфигурацию, а обыкновенной коричной—транс-конфигурацию.
Кроме этих двух коричных кислот, известны еще две изоко-ричные кислоты с темп, плавл. 57° и 43,5—46°. По всей вероятности,
это—неустойчивые полиморфные разности аллокоричной кислоты, так как при внесении в их растворы кристаллика аллокоричной кислоты (в качестве затравки) изокоричные кислоты превращаются в аллокиричную.
Труксилловые и труксиновые кислоты. Коричная кислота не образует высокомолекулярных полимеров, подобно стиролу, но при длительном освещении она димеризуется с образованием кольца циклобутана. В результате димеризации получаются димерные двухосновные кислоты, разделяемые на две группы изомеров:
С6Нб—СН=СН—СООН С6Н5—СН-СН—СООН
_|_ —> I I
НООС—СН=СН—С6Нб ноос—сн—сн—сбн6
труксилловые кислоты
сбнб—сн=сн—соон с6н5—сн-сн—соон
+ —> I I
сбнб—сн=сн-соон сбн5—сн—сн-соон
труксиновые кислоты
Теория позволяет предвидеть для труксилловых кислот пять цш?-гиранс-изомеров в отношении плоскости циклобутанового кольца; все эти изомеры были получены. Для труксиновых кислот возможно шесть стереоизомеров, из которых пока известно четыре.
Для одной из труксилловых кислот, так называемой а-труксилловой кислоты, строение, изображаемое формулой I
I
доказывается следующим образом (Штермер). При обработке а-труксилловой кис-лоты дымящей серной кислотой происходит внутримолекулярная конденсация с образованием связей между карбоксильными группами и находящимися к ним в цис-положении фенильными группами. В результате образуется трициклический ди кето н—труксон:
С6Н4—СН—СН—С—О
I III
О=С----СН—СН—СвН4
Правда, это было бы возможно и для изомера а-труксилловой кислоты, имеющего формулу II (пера-труксилловая кислота, темп, плавл. 266°
НООС \ ,С
Н:56//'
Ст—
НООС/^П 5
----/-С
// \
I/ н
—С
\ н
Однако а-труксилловая кислота может образовать моноанилид, который удается разделить на оптически деятельные антиподы. Значит такой моноанилид не обладает ни центром, ни плоскостью симметрии, что возможно только для формулы /. так как моноанилид, соответствующий формуле II, имел бы плоскость симметрии, проходящую через карбоксильные группы и, следовательно, не мог бы существовать в виде оптических антиподов.
Чтобы определить, какие из кислот, получающихся при димеризации коричной кислоты являются труксилловыми, а какие труксиновыми, прибегают к их осторожному окислению: при окислении труксилловых кислот получается бензойная кислота, а труксиновые образуют дикетон бензил С6Н5—СО—СО—С6Н5.
Димеры коричной кислоты при перегонке довольно гладко переходя! в мономерную коричную кислоту. Димерные коричные кислоты обнаружены и в природе; они входят в состав молекул побочных алкалоидов листьев кока
Атроповая (а-фенилакриловая) кислота получается дегидратацией гроповой кислоты (продукт расщепления алкалоидов атропина и гиосциамина):
С6Н6—СН—СООН
СН2ОН
троповая кислота (темп, плавл. 117—118°-
С6Н5—С—СООН
СН2 атроповая кислота (темп, плавл. 106°)
Атроповая кислота может быть получена и синтетически (стр. 388 сл.)
фенилпропиоловая кислота СвН5—С=С—СООН (темп, плавл. 137°) может быть получена действием СО2 на металлорганические соединения фенилацетилена С=С—Na и СвН5—С~С—MgHal, а также отнятием двух молекул бромистого водорода от продуктов присоединения брома к коричным кислотам. о-Нитро-фенилпропиоловая кислота легко превращается в индиго.
Оксикислоты
Кумариновая и кумаровая кислоты. Из ненасыщенных оксикислот наиболее важной является о-оксикоричная кислота
/ОН
'Ч/Хсн^сн—соон
которая может существовать в двух формах:
н—с—СвН4ОН
н—с-соон
стереоизомерных цис- и транс-
н—с-свн4он
II
НООС—с—н
кумариновая кислота
о-кумаровая кислота
tyUr-Кислота, называемая кумариновой, известна только в виде производных и солей. Попытки выделить ее в свободном состоянии,
например из солей действием кислот, приводят к отщеплению молекулы воды и превращению кислоты в лактон кумарин
лп°
который при нагревании со щелочами снова дает соли кумариновой кислоты.
Кумарин плавится при 68°. Он является душистым веществом (запах свежего сена), распространенным в растительном мире {Anthoxantum odoratum, Asperula odorata и пр.).
Наиболее удобным способом получения кумарина является синтез его из салициловой кислоты. Впервые кумарин был получен Перкиным конденсацией натриевой соли енольной формы салицилового альдегида с уксусным ангидридом, что явилось первым примером открытой им реакции конденсации ароматических альдегидов с ангидридами кислот (стр. 407). Обычно для получения кумарина нагревают смесь салицилового альдегида с уксусным ангидридом в присутствии ацетата натрия. Вероятным промежуточным продуктом реакции является ацетильное производное кумариновой кислоты {ацетилкумариновая кислота):
Qu О—СОСН3 О
/\/ п CfVOONa /~\/ соон
| II + (СН3СО)2О-------> I || \иин I || СО
Ч/\сно । J II I
сно \/\ Z
Кроме того, кумарин и соли кумариновой кислоты можно получать из салицилового альдегида и малоновой кислоты. Малоновая кислота теряет одну молекулу СО2, а гидроксил салицилового альдегида ацетилируется уксусным ангидридом, в результате чего получается ацетилкумариновая кислота.
При нагревании с алкоголятами кумариновая кислота изомеризуется в транс-кислоту, называемую о-кумаровой (темп, плавл. 208°), устойчивую в свободном виде. о-Кумаровая кислота лишь с трудом при действии водоотнимающих средств теряет воду, превращаясь в кумарин.
В растительном мире очень распространены различные оксипроизводные кумарина, которые встречаются в виде глюкозидов, а также образуются при сухой перегонке некоторых смол. Так, в коре благородного каштана содержится в виде глюкозида эскулетин (темп, плавл. 270°)
а при сухой перегонке смол гальбанум и асафетида образуется умбеллиферон (темп плавл. 225—227°):
Умбеллиферон является лактоном п-оксики мариновой, или умбелловой, кислоты, эскулетин—лактон триоксикоричной кислоты.
Эти окси кумарины получены также синтетически по реакции Перкина из соответствующих альдегидов.
Кофейная (3.4-диоксикоричная) кислота получается из дубильного вещества кофе кипячением с едким кали.
Пипериновая кислота содержится в перце как составная часть алкалоида пиперина (стр. 608, 649). Пипериновая кислота (темп, плавл. 217°)
Н2С— О
О—^2^—СН=СН—сн=сн—соон
может быть получена из альдегида пипероналя (стр. 395). Конденсацией пипероналя с уксусным альдегидом получается пиперонил, акролеин
Н,С—О
6—СН=СН—СНО
который по реакции Перкина дает пипериновую кислоту.
Многоядерные ароматические соединения
Кроме простейших ароматических соединений, содержащих одно бензольное ядро, существуют ряды соединений с двумя и более бензольными ядрами в молекуле. Эти ядра могут быть либо связаны простыми связями непосредственно или через промежуточные цепи атомов углерода
ГЛ Г^А-СН,—
\=/ \=/ \==/ \=/ либо могут иметь общие углеродные атомы:
и
00 СО и
Соединения последнего типа носят название ароматических соединений с конденсированными бензольными норами.
Простейшие многоядерные ароматические соединения
Группа дифенила 'бифенила)
В эту группу входят соединения, содержащие два бензольных остатка (два радикала фенила), непосредственно связанных между собой. Простейший из таких углеводородов—дифенил, называемый также бифенилом, имеет строение
или СвН5-С„Н5
Дифенил (бифенил) образует бесцветные, растворимые в спирте и эфире кристаллы (темп, плавл 70° темп, кип 254°) В небольших количествах он содержится в каменноугольном дегте, образуется при пропускании бензола через раскаленные трубки В промышленности дифенил получают пропусканием паров бензола через расплавленный свинец.
Дифенил, обычно в смеси с простым дифениловым эфиром С6Нб—О—С^НЬ (чтобы понизить температуру застывания), применяется как высокотемпературный теплоноситель. Благодаря высокой
температуре кипения таких смесей, с их помощью удается нагревать аппараты до температуры, для достижения которой потребовался бы водяной пар очень высокого давления.
При замещении в дифениле одного атома водорода метилом или какой-нибудь другой группой может быть получено три изомера, замещенных в орто-, мета- и пара-положениях по отношению к атом}! углерода, связанному со вторым фенилом.
Углеводороды группы дифенила получаются при действии металлов, чаще всего натрия (синтез Фиттига) или меди (синтез Ульмана), на галоидные производные бензола и его гомологов, например:
2СбНбВг + 2Си —> СЙН5—CeH5 + 2CuBr
Они образуются также из диазосоединений (стр. 322).
Производные дифенила легко получаются из одноядерных ароматических соединений, главным образом при помощи следующих двух реакций.
1. Действие кислот на гидразосоединения (стр. 317). Из гидразобензола, в результате бензидиновой перегруппировки, получается бензидин (ntn -диаминодифенил)
NH’ и немного дифенилина (о,п'-диаминодифенила).
Бензидин кристаллизуется из горячей воды в листочках (темп, плавл. 122°). В бензидине, а также в его сульфокислотах, обе аминогруппы легко диазотируются. Сочетанием образующегося бис-диазосоединения с сульфокислотами аминов и фенолов получаются многие важные азокрасители. Бензидин применяется также в аналитической практике для определения серной кислоты, так как сульфат бензидина обладает ничтожной растворимостью.
Подобно тому, как бензидин получается из нитробензола с промежуточным образованием гидразобензола, так из о-нитротолуола получается о-толидин (темп, плавл. 129°)
H2N—NH’
н3с/ ^СН8
а из о-нитроанизола—о-дианизидин (темп, плавл. 131,5°)
сн3о/ \осн8
Эти вещества, как и бензидин, являются важными исходными продуктами (диазосоставляющими) для получения азокрасителей.
2. Окисление различных ароматических соединений. Реакции протекают по одной общей схеме; примером может служить простейший случай—окисление бензола:
УЗУ-н + Ы~<Э + ° “* +112°
Такое окисление ароматических углеводородов может проге* катьпод действием некоторых перекисных кислот, например надсерной кислоты H2S?OR и особенно мононадсерной кислоты H9SO6.
Легче, чем углеводороды, окисляются фенолы. Так, диоксибифенилы, или так называемые дифенолы, могут быть получены окислением кислородом воздуха или другими окислителями расплавленных щелочных фенолятов. Например, феноля! калия легко образует п,п'-диоксибифенил
Многоатомные фенолы в щелочном растворе окисляются по этой схеме уже кислородом воздуха. Например, из пирогаллола таким путем легко получается гексаоксибифенил (НО)3СЯН9 — CFH.,(OH)3
Для п,п -диоксибис}енила и его производных характерна способность окисляться в хинон с двумя хиноидными ядрами:
°-оо=°
бифенохинон
Бифенохинон образует красные иглы. Он разлагается при температуре около 165е, не возгоняется и не летуч с водяным паром. Его производным являе1ся темносинее вещество, получаемое при очистке сырого древесного уксуса хромовой смесью и называемое церулиньоном или цедриретом'.
Это соединение образуется в результате окисления находящегося в древесном дегте диметилового эфира пирогаллола
Окислением фенантрена (стр 481) или фенантренхинона (стр. 483) получается бифенил-о,о'-дикарбоновая, или дифеновая, кислота (темп, плавл. 229°)
НООС СООН
При действии уксусного ангидрида дифеновая кислота легко дает ангидрид
ос со
а при нагревании аммониинои соли—имид.
СТЕРЕОХИМИЯ СОЕДИНЕНИЙ БИФЕНИЛА
Неожиданное открытие было сделано Кеннером и Кристи (1922 г.), показавшими, что некоторые производные дифеновой кислоты, например нитродифеновые кислоты изображенного ниже строения
OgN NO2
°3n—N0*
НООС СООН
o2n
ноос соон
могут быть расщеплены на оптические антиподы. Это осуществляется кристаллизацией солей, образованных ими с оптически деятельными основаниями (алкалоидами). Аналогичным образом Мейзенгеймер (1927 г.) разделил на оптические антиподы о,о'-диамино-о-дитолил
H2NCH3
н^: nh,2
Это означает, что такие вещества, несмотря на отсутствие в молекуле асимметрического атома, ведут себя подобно соединениям, содержащим асимметрический атом углерода.
Необходимо отметить, что дифеновую кислоту и кислоту строения
НООС dOOH
разделить на антиподы не удается.
Причиной возникновения оптической изомерии в рассматриваемом случае является то, что при наличии в орто-положениях в молекуле дифенила заместителей достаточно большого размера свободное вращение бензольных колец вокруг общей оси становится невозможным (или, по меньшей мере, затрудненным). В результате этого два бензольных кольца дифенила оказываются расположенными в разных плоскостях (Кеннер):
При определенном характере и размещении заместителей расположенная в двух плоскостях молекула не будет иметь плоскости симметрии, и для такого производного должны быть возможны две пространственные структуры, относящиеся друг к другу, как предмет к своему зеркальному изображению. В таком случае вещество можно разделить на два оптических антипода. Этот вид оптической изомерии, связанной с отсутствием свободного вращения из-за пространственных затруднений, получил название атропизомерии
(тропо—греч. вращаю, а—отрицание). Таким образом, условиями, необходимыми для появления атропизомерии производных дифенила, являются: 1) невозможность свободного вращения бензольных колец вокруг общей оси; 2) несимметричное замещение каждого кольца.
Для оценки возможности или невозможности свободного вращения можно воспользоваться следующей моделью (Миллс):
1.42
Если положить расстояния между атомами углерода в бензольных кольцах равными 1,42 А, а расстояние между связанными друг с другом углеродными о
атомами разных колец (С и С')—равным 1,48 А, то расстояние между атомами
С" и С'" будет равно 0,71+1,48+0,71 =2,90 А. Когда сумма межатомных расстоя-о
ний С—X и С' — X' больше 2,90 А, свободное вращение невозможно, когда же она о
меньше 2,90 А, вращения можно ожидать. В случае четырех различных замести-о
телей свободное вращение невозможно, когда обе суммы радиусов больше 2,90 А.
Согласие расчетных результатов с опытными иллюстрируется данными, приведенными в табл. 34.
Таблица 34
Условия для появления атропизомерии
Заместители в орто-положениях Сумма радиусов заместителей Найдена ли опытным путем возможность расщепления на оптические изомеры
X, Х2
Н Вг J СООН no2 F F Cl Cl no2 сн3 Нримеча нi 1 е. Н н н н н F NH„ СООН С1 СООН СН3 Атомные ра 1,88 2,82 2,94 2,50 2,86 2,78 2,95 3,25 3,38 3,48 3,00 1ССТОЯНИЯ Сар —• нет (да?) да нет » » да » » » о -X составляют (в А):
с—н . . . 0,94—1,04 C-J . . . 2,00 с-он . . . 1,45
C-F . . 1,39 С-СНз . . 1,50 с-соон . . 1,56
С—С1 С—Вг . . . 1,69 . . . 1,88 С—NH2 . . 1,56 NO2 . . 1,92 С—ОСНз . . 1,45
При достаточно больших размерах заместителя можно ожидать расщепления на оптические изомеры, если даже замещены только три, два или даже всего одно орто-положение. Примерами соединений, расщепляющихся на антиподы.
могут
Явления атропизомерии наблюдаются и в других системах, например у тер» фенила' (стр. 419), нафталина (стр. 464) и т. п. .
Полифенилы
При пропускании паров бензола через раскаленные трубки, креме дифенила, получаются также два дифенилбензола:
о-дифемилбензол
Симметрический трифенилбензол (иглы; темп, плавл. 17Г)
легко получается конденсацией ацетофенона, аналогично конденсации ацетона в мезитилен.
В настоящее время получены также такие углеводороды, как пентафенилбензол и гексафенилбензол—бесцветные кристаллические вещества:
пентафенилбензол гексафенилбензол
(темп, плавл. 252—253е) (темп, плавл. 449—450°)
Из продуктов разложения муравьиной кислотой сернокислого фенилдиазония в уксуснокислых растворах можно выделить углеводороды с линейным сочетанием бензольных ядер
квинквифенил (темп, плавл 395е)
Действием меди на 4-иодтерфенил получен углеводород, содержащий шесте бензольных колец:
сексифвиил
(темп, плавл. 475е)
Все полифенилы представляют собой бесцветные кристаллические вещества.
В природе встречаются окрашенные производные терфенила, содержащие хиноидное ядро. Так, красящие вещества, выделяемые из некоторых грибов, являются производными 2,5-дифенил-п-хинона (красные кристаллы: темп, плавл. 214°):
В некоторых видах паразитирующего на дубе грибка Polyporw> содержится полипоровая кислота (бронзовые таблички; темп, плавл. 305*), а в верхней кожице шляпок мухомора (Amanita muse aria} найдено 0,1% мускаруфина (оранжево-красные иглы, темп плавл. 275,5°).
полипоровая кислота
° п
H0\zkzv
ноос Y |
| " || соон
]=сн-сн=сн—cooii
I ''
'V'
мускаруфин
Соединения с бензольными ядрами, связанными углеродных атомов, не входящих в
при помощи
кольцо
Простейшим ме тан:
углеводородом этого типа является
дифенил-
следующих атомов водорода в остатке
Замещением пильными группами можно получить углеводороды трифенилметан и тетрафенилметан:
метана фе-
гетрафенилметан
в молекуле которого остатки разделены двумя углеродными атомами, является ческий дифенилэтан, называемый обычно дибензилом'.
трифенилметан
Простейшим углеводородом,
фенильные симметри-
Дальнейшее замещение атомов водорода в цепи на фенилы приводит к следующим углеводородам:
ceiu
СвН/
трифенилэтан
сл
CfHf-
£,Н,
•С.Н,
свн5
б
симметрический тетра фенилэтан
I zC.H5
С6Н5 аесиммегрический теграфенилэган с9н5 с,н5
-с------с—с9н4
С»н5 с9н4 гексафенил^тан
I ХН, с„н, пентафен илэган Замещение на фенилы атомов водорода в пропане, бутане и т. д.
дает бесконечный ряд многоядерных ароматических углеводородов. Известны также и фенилированные циклические углеводороды, например дифенилциклопропан
СвНб-СН-СН-СвНв
Группа дифенилметана
Способы получения. Наиболее важными являются следующие синтетические методы получения дифенилметана, его гомологов и производных из одноядерных ароматических соединений.
1. Реакция Фридел я—К р а ф т с а (стр. 213). Исходными галоидными соединениями могут служить алифатические двугалоидные соединения, в которых оба атома галоида находятся при одном и том же атоме углерода (начиная с хлористого или бромистого метилена)
СН,С1, + 2С6Н6 -> СбН6—СН2—CeH5 + 2НС1
или ароматические моногалоидные соединения типа хлористого бензила:
СвНб—СН2—С1 4- СеНб СбНб—СН2—С6НБ 4- НО
Действием галоидангидридов кислот на бензол или его гомологи
СбН5—СО—С1 + СвНб -> СвНБ—СО—СвН6 4-НС1
легко получаются ароматические кетоны, которые затем одним из обычных способов могут быть превращены в соответствующие дифенилметаны.
2 Конденсация жирных альдегидов или жирно-ароматических спиртов с ароматическими углеводородами. Конденсация жирных альдегидов или бензилового спирта, его гомологов и производных с ароматическими углеводородами и их производными
СН2О + 2С6Н6 -> СбНБ-СН2-СвН5 4- Н2О
СвНб-СН2ОН 4- Свнв сен5-сн2-с6н64-н2о
протекает под действием серной кислоты, хлористого цинка и других конденсирующих средств.
Чрезвычайно легко такая конденсация идет с фенолами, особенно с многоатомными, и с ароматическими аминами, легче всего с третичными, например:
/СбН4ОН
СН8-СНО 4- 2С6НбОН СН8-СН< 4-Н2О
ХС6Н4ОН
СбНб-СН2ОН 4- СвН4(ОН), -> СвНб-СНз-СвН^ОН), 4- Н2О
yC6H4N( СН8)2
СН2О + 2C6H6N(CH8)2 -> сн2< + н2о
VeH4N(CH8)2
3. Действие ароматических металлорга-нических соединений на ароматические альдегиды. Например:
С6Н5-СНО4-С6Нб~М-Гг -> СбНб-СН-СвН5
OMgBr
Действием воды на продук! присоединения получается вторичный спирт С6Н5—СН(ОН)—С«Н5.
4. Действие фенолов на нитрилы ароматических кислот. Реакции фенолов с ароматическими нитрилами протекают аналогично синтезу оксиальдегидов из фенолов и синильной кислоты (стр. 393). Сначала под действием хлористого водорода образуется хлористоводородная соль кетимина, которая действием воды превращается в соответствующий кетон (К» Геш, 1927 г.): НО ОН НО ОН ' но он
С1-
Дифенилметан (темп, плавл. 26°; темп. кип. 262°) представляет собой пахнущее апельсинной коркой кристаллическое вещество, легко растворимое в спирте и эфире.
Действием брома на дифенилметан при нагревании получается дифенилбромметан С6Н5—СНВг—С6Н5, из которого обменом Вг на ОН можно получить дифенилкарбинол С6Н5—СН(ОН)—-С6Н5, обыкновенно называемый бензгидролом (иглы; темп, плавл. 69*; перегоняется при 298,5°). Легче получить бензгидрол из бензойного альдегида и магнийбромфенила (см. выше).
Бензофенон. Окислением бензгидрола получается дифенилкетон, так называемый бензофенон С6НР CO~CGH5. Его проще всего получать реакцией Фриделя—Крафтса из хлористого бензоила и бензола в присутствии хлористого алюминия (стр. 280) или сухой перегонкой кальциевой соли бензойной кислоты. Бензофенон кипит при 306,1°. Обычно он кристаллизуется в ромбических призмах с темп, плавл. 48,4°, но легко образует также неустойчивую полиморфную разность с темп, плавл. 26°.
Бензофенон часто применяется как стандартное вещество для проверки шкалы термометров по его температуре кипения.
Производное бензофенона—ди-лг-димез ил аминобензофенон, называемый обычно кетоном Маклера (CH3)2N—CrH4—СО— С6Н4—N(CH3)2, получается действием фосгена на диметиланилин:
zC6H4N(CH8)2
СОС12 4- 2CeHe—N(CH8), -> ОС< 4- 2НС1
\CeH4N(CH8),
Кетон Михлера служит для получения красителей—производных трифенил-метана. Действуя на него, например, аммиаком в присутствии хлористого цинка, получают желтый краситель, называемый аурамином, основанию которого придается строение имина кетона Михлера
Солям аурамина придают хиноидное сгроеыие; как указывались выше (стр. 373), в соединениях такого типа смещение электронной плотности должно приводить к распределению положительного заряда между двумя атомами азота:
Многие оксибензофеноны встречаются в природе. Например, в коре кото (семейство Lauraceae) содержится котоин (темп, плавл. 131°) и ряд его метилированных производных; одной из составных частей экстракта желтого дерева Morus tinctoria является чаклурин (стр. 385):
юн
сн30-О-с°_<Э \он
\он
маклурин
котоин
Флуорен. При сочетании в одной и той же молекуле связей между ядрами, характерных для дифенила и для дифенилметана, получается соединение с тремя ядрами—двумя бензольными и циклопентадиеновым:
Это соединение называется флуореном. Флуорен бесцветен, плавится при 116°, кипит при 295".
Благодаря наличию пиклопентадиенового кольца флуорен легко вступает во многие реакции, характерные для этой циклической системы. Так, водородные атомы группы СН2 флуорена действием калия можно заместить на металл и получить, например, флуорен-калий:
СНК
С ароматическими альдегидами флуорен вступает в реакции конденсации, образуя производные дибензофульвена'.
Флуорен содержится в каменноугольном дегте. Он может быть также получен восстановлением кетона, называемого флуореноном'.
Флуоренон—кристаллы желтого цвета (темп, плавл. 84°). Он получается сухой перегонкой кальциевой соли дифеновой кислоты (стр. 415).
Группа трифенилметана
Способы получения. Соединения группы трифенилметана могут получаться из одноядерных и двуядерных (группы дифенилметана) соединений способами, аналогичными способам получения производных дифенилметана из жирных и одноядерных ароматических соединений.
1. Реакция Фриде л я—К р а ф т с а. Например:.
СНС13 + ЗСбНб -> СН(СбНб)8 + ЗНС1
СбНб—СНС12-j-2CeH6 C6H5-CH(CeH6)s + 2HCi
2. Конденсация ароматических альдегидов и кетонов с производными бензола. Производные трифенилметана получаются при конденсации с производными бензола одноядерных ароматических альдегидов
ХАЬДСН,).
CeH6-CHO + 2C6H5N(CH3)? СбНб-СН< Ч-Н3о
^C6H4N(CH8)?
или двуядерных алкоголей и кетонов (производных дифенилметана):
НОС6Н4-СН-СбН4ОН+С6Н5ОН -> СН(СбН4ОН)з + Н3О он
(CH3)2NC6H4-C-C6H4N(CH3)2 + C6H5N(CH3)2 [(CH3)2NC6H4]3C-OH
II о
R качестве конденсирующих средств применяются PCIs, РОС13 и др.
3. Реакции карбонилсодержащих соединений с металлорганическимисоединения-м и. Например:
C6H5-CO-C6H6 + C6H6MgBr (С6Нб)3С—OMgBr -> (С6Н5)3С-ОН
СбНб-СООС2Нб + 2C6H6MgBr (C6H6)3C-OMgBr + Mg(OC2H6)Br
Трифенилметан получается по реакции Фриделя—Крафтса действием хлороформа на бензол в присутствии хлористого алюминия или восстановлением трифенилхлорметана (см. ниже). Он образует бесцветные кристаллы (темп, плавл. 92,5°; темп. кип. 359,2°), легко растворимые в спирте, эфире и бензоле. Из бензола трифенилметан выделяется в виде кристаллического молекулярного соединения (С6Н5)3СН-С6Н6, плавящегося при 78°, на воздухе выветривающегося.
В трифен и л мета не водородный атом группы СН легко замещается бромом при нагревании, причем получается трифенилбромметан (С6Н5)3СВг. При действии на трифенилметан щелочных металлов этот атом водорода замещается на металл с образованием, например, (C6H6)3CNa.
Трифенилхлорметан (С6Н5)3СС1 (кристаллы; темп, плавл. 113°) получается из четыреххлористого углерода и бензола в присутствии хлористого алюминия:
СС14 + ЗСбН6 (С6Н5)3СС1 + ЗНС1
Он чрезвычайно легко обменивает атом хлора на гидроксил (с водой) и на остатки OR (со спиртами), например:
(С6Нб)3СС1 + нос2нб (С6Нб)3С—ОС2Нб + НС1
этиловый эфир триф ен илкарбинола
С магнием, слегка прогретым в парах иода («активированный» магний), трифенилхлорметан образует металлорганическое соединение (С6Нб)3С—MgCl, которое с СО2 дает слабую игрифенилуксусную кислоту (СбН5)3С—СООН, легко распадающуюся при 265° на трифенилметан и СО2.
Трифенилкарбинол (С6Н5)3С—ОН (кристаллы; темп, плавл. 162,5°) может быть получен реакциями с магнийорганическими соединениями (см. выше) или из трифенилхлорметана действием воды. Гидроксил трифенилкарбинола обладает большой подвижностью: с НС1 и с СН3СОС1 трифенилкарбинол дает трифенилхлорметан, а со спиртами в присутствии минеральных кислот необыкновенно легко образует простые эфиры.
С концентрированной серной кислотой, а также с хлорной кислотой, он образует яркоокрашенные солеобразные соединения, разлагаемые водой с выделением вновь трифенилкарбинола. С хлорной кислотой (70%-ной) образуется янтарножелтый трифенилме-тилперхлорат состава [(СбН5)3С]С1О4-Н2О, с серной кислотой—желтый кислый сульфат [(С6Н5)3С ]SO4H -H2SO4. Средний сульфат [(C6H5)3C]2SO4—темнокрасные кристаллы—образуется из трифенил-
хлорметана и сернокислого серебра при обменном разложении в жидком SO?. Цветность этих продуктов связана с их солеобразным характером как электролитов, причем носителем цветности является ион (С6Н5)3С+. Это явление получило название галохромии (гал—греч. соль, хромое—окраска).
Интенсивно окрашены (в большинстве случаев в яркооранжевый цвет) также кристаллические двойные соединения трифенилхлор-метана и его гомологов с^галоидными солями тяжелых металлов:
(СвН6)3СС1 • FeCl8 (СвН5)3СС1 • А1С13 (С6Н5)3СС1 • SnCl4
Этим соединениям можно придать строение [(С,Н8).,СС [FeClJ- [(CeH5)3Cl+[AlCUr
т. е. и в этом случае причиной возникновения цветности является образование катиона Аг3С+ (или Аг2НС+).
КРАСИТЕЛИ РЯДА ТРИФЕНИЛМЕТАНА
Наиболее важными производными трифенилметана являются его амино- и оксипроизводные, главным образом содержащие аминогруппы и оксигруппы в пара-положениях к «метанному» углеродному атому. Эти соединения лежат в основе многих важных органических красителей. Сами амино- и оксипроизводные трифенилметана бесцветны (лейкосоединения), но они очень легко окисляются, часто цаже кислородом воздуха, переходя в производные трифенил карбинола, дающие с кислотами (или соответственно со щелочами) красители. Например, в случае красителя парафуксина’.
тпи-п-аминотрифенил метан (лейкооснование парафуксина)
три-Д-жминотрифенилкарбинол (основание парафуксина)
хлористоводородная соль три-n аминотриФамилкарбимола (краситель парафуксии)
Естественно, что в катионе парафуксина, благодаря смещению электронной плотности, положительный заряд распределен между всеми тремя атомами азота:
Взаимные превращения трифенилметановых красителей в их основания были детально изучены Ганчем на примере красителя кристаллического фиолетового (стр. 429) путем измерения эле тро-проводности растворов. При приливании щелочи к раствору красителя Ганч наблюдал образование хорошо проводящего ток вещества, которое далее медленно превращалось в малоэлектропроводное соединение.
Это явление медленной нейтрализации заключается в медленной рекомбинации диссоциированной формы карбинола в недиссоцииро-ванную:
Ганч рассматривал этот процесс как таутомерное превращение хиноидной формы, которую он называл истинным основанием, в бензоидную форму, или псевдовснование, красителя.
Красители, содержащие первичную или вторичную аминогруппу, могут давать ангидридную форму. Например, ангидридная форма парафуксина (основание Гомолки) имеет строение:
(HaN-CeH4)aC=<^>=:NH
Такие ангидридные формы являются производными фуксонимина (C6H5)aC=<^>=NH
который был получен Байером в полимерной форме.
Из п-окситрифенплкарбииола аналогичным образом получается фуксон
(С6Н s),C=^^^>=0
Он образует оранжево-желтые призмы, плавящиеся при 168°. Производным фуксона является диоксифуксон—краситель аурин (стр. 429).
Трифенилметановые красители и их лейкооснования получаются главным образом путем реакций конденсации (стр. 305 и 424).
Действием серной кислоты на трифенилметановые красители получаются их сульфокислоты. Сульфирование красителей часто при
меняют в технике, чтобы получить из нерастворимых в воде красителей растворимые сульфокислоты или их соли («кислотные» красители).
Малахитовый зеленый. Лейкооснование этого красителя (тетраметил -zz-диаминотрифенилметан) получается конденсацией бензойного альдегида с диметиланилином в присутствии хлористого цинка или других водоотнимающих средств:
✓C6H4N(CH3)2
С6Нб-СНО 4- 2C6H6N(CH3)2 -> С6Н5-СН< + Н2О
xCeH4N(CH3)2
При окислении лейкооснования (например, перекисью свинца в сернокислом растворе) получается тетраметилдиаминотрифенил-карбинол (основание малахитового зеленого), образующий., как и лейкооснование, бесцветные кристаллы При растворении этого карбинола в кислотах получаются бесцветные растворы его солей, при нагревании переходящих в интенсивно зеленые соли хиноидного основания малахитового зеленого.
Парафуксин, или парарозанилин (строение см. на стр. 426), может быть получен окислением смеси анилина и zz-толуидина, например, нитробензолом. Метильная группа толуидина, вероятно, окисляется при этом в альдегидную группу, альдегид с анилином дает лейкооснование (так называемый парааейканилин), окисляющееся далее в основание (парарозанилин). При действии кислот получается самый краситель—парафуксин.
Строение лейкооснования—паралейканилина—доказывается таким образом. При диазотировании все три содержащиеся в нем аминогруппы диазотируются, а при последующем кипячении диазосоединения в спиртовом растворе диазогруппы заменяются на водородные атомы, в результате чего получается трифенилметан. При нитровании же трифенилметана получается три-п-нитротрифенилме-ган, который при восстановлении дает вновь паралейканилин (три-п-аминотрифенилметан).
Фуксин, или розанилин,—гомолог парафуксина, содержащий в одном из бензольных ядер метильную группу в орто-положении к аминогруппе. Основание фуксина (розанилин) можно получить окислением смеси анилина, zz-толуидина и о-толуидина:
Его получают также конденсацией и окислением смеси п,п'-ди-аминодифенилметана с о-толуидином:
H2N-C6H4-CH2-C6H4-NH2 + CH3-C6H4-NH3 + 20 ->
-> (H2N-C6H4)2C~C6H3(CH3)NH2 + H2O
он
Фуксин (и его сульфокислоты—«кислый» фуксин) является одним из старейших красителей этого класса; он получен польским химиком Я Натансоном в 1855 г. При его промышленном получении долгое время применялась в качестве окислителя мышьяковая кислота, замененная впоследствии нитробензолом (окисление в присутствии хлорного железа и железных опилок).
Алкилированные и арилированные фуксины. При введении метильных групп в аминогруппы фуксина получаются фиолетовые красители. Краситель основной фиолетовый К (метиловый фиолетовый, метилвиолет) содержит главным образом пентаметил розанилин наряду с более и менее метилированными соединениями. Он получается окислением диметиланилина, причем одна из метильных групп, окисляясь, дает формальдегид, который вступает в реакцию конденсации и образует «метанный» атом углерода. Другой путь—метилирование фуксина нагреванием его под давлением с метиловым спиртом и соляной кислотой.
Кристаллический фиолетовый (кристаллвиолет) может быть получен следующим образом Сначала конденсируют диметиланилин с фосгеном (стр. 422), получая тетраметилдиаминобензофенон (кетон Михлера), который затем снова конденсируют с диметиланилином, например при помощи хлорокиси фосфора:
(CH;»2NC6Hs + [(СН3).?С6Н4|.2СО -» [(CH8)2NC6HJ3C-OH
Основание кристаллического фиолетового представляет собой, таким образом, гекса.мегилтриаминотрифенилкарбинол; последний с кислотами образует соли, являющиеся красителями.
Введение в аминогруппу фенильных групп приводит к образованию синих красителей Анилиновый синий образуется при нагревании фуксина с анилином в присутствии катализатора—бензойной кислоты. При этом в реакцию вступают три молекулы анилина’на каждую молекулу фуксина и получается трифенил-розанилин:
(СбН6—NH—C6H4)2C-~С6Н3(СН3)—NH—СвНб
он
Красители, содержащие фенольные оксигруппы. Простейшим красителем этого рода является аурин',
110^СХ
Он образуется действием азотистой кислоты на парафуксин; при этом аминогруппы заменяются на оксигруппы. Аурин может быть получен нагреванием фенола с щавелевой и серной кислотами. Об
разующаяся из щавелевой кислоты муравьиная кислота, вступая в конденсацию с фенолом, дает «метанный» атом углерода. Получающееся лейкооснование (три-п-окситрифенилметан) серной кислотой окисляется в триокситрифенил карбинол, который, отщепляя воду, и образует аурин.
Гомолог аурина—розоловая кислота—находится к аурину в таком же отношении, как фуксин к парафуксину. Розоловая кислота в настоящее время в промышленном масштабе не получается. Она может быть синтезирована аналогично аурину из фуксина, а также окислением мышьяковой и серной кислотами смеси фенола с о- и /г-крезолами.
Фталеины. Фталеинами называются красители, получаемые из фталевого ангидрида (стр. 376) Они представляют собою производные трифенилметана, содержащие карбоксильную группу и фенольные оксигруппы (иногда и аминогруппы). Простейшим представителем их является фенолфталеин, получаемый конденсацией (при помощи серной кислоты) фталевого ангидрида с фенолом:
Бесцветный фенолфталеин—кристаллический порошок, плавящийся при 261°,—имеет бензоидное лактонное строение. При растворении его в щелочах получаются соли кислоты хиноидного строения:
Соли эти красного цвета. Возникновение красной окраски в щелочной среде используется в аналитической практике, где фенолфталеин применяется как индикатор на щелочь При добавлении избытка щелочи красное окрашивание исчезает, так как при этом образуется трехзамещенная соль бензоидного строения:
^N^ook
Флуоресцеины. При получении фенолфталеина конденсация части молекул фенола протекает таким образом, что гидроксильные группы оказываются в орто-положении к «метанному» атому углерода.
При отщеплении воды от такого продукта конденсации образуется соединеннее кислородсодержащим шестичленным кольцом, называемое флу ораном'.
НО ОН
флуоран
Флуоран нерастворим в щелочах.
Соединения, содержащие флуорановую группировку, особенно легко получаются при конденсации фталевого ангидрида с л^-диокси-соединениями и л-аминифенолами. Из л<-диоксисоединений при этом получаются красители, хорошо окрашивающие шелк и шерсть. В водных растворах они обладают необыкновенно интенсивной флуоресценцией.
Сплавлением фталевого ангидрида с резорцином получается флуоресцеин:
Это кристаллический порошок, окрашенный в темнокрасный цвет. Водно-щелочные растворы его имеют желто-красную окраску и обладают, особенно в разбавленном виде, великолепной зеленой флуоресценцией, исчезающей при подкислении.
Флуоресцеины, содержащие в остатках резорцина атомы галоида, называются эозинами.
Обыкновенный эозин—тетрабромфлуоресцеин—получается действием брома на флуоресцеин. При действии на флуоресцеин иода получается эритрозин.
Сплавлением фталевого ангидрида с л-аминофенолами получаются родами-ны, например:
(C2H6)2N
N(c2h6)2
о
О
родамин С
Родамин С получают из jw-диэтиламинофенола и фталевого ангидрида.
Родамины могут содержать первичные, вторичные или третичные аминогруппы.
Тетрафенилметан
Действием на трифенилхлорметан (стр. 425) фенилмагнийбро-мида получается тетрафенилметан'.
(С6Н5)3СС1 + C6H5MgBr (С6Нб)4С + MgClBr
Он образует бесцветные кристаллы (темп, плавл. 285°); кипит почти без разложения при 431° (760 мм рт. ст.).
Производные тетрафенилметана очень легко получаются конденсацией трифенилхлорметана или трифенилкарбинола с аминами или фенолами, например с анилином:
(C6H6)3CC1 + C6H6NH2 -> (C6H6)3C-CeH4NH2 + HCl
Группа дибензила
Дибензил (симметрический дифенилэтан) С6Н5—СН2—СН2—СбН5 (кристаллы; темп, плавл. 52,5°; темп. кип. 284°) может быть получен действием натрия или магния на хлористый бензил:
2С6НбСН2С1 4- 2Na -> С6НбСН2-СН2С6Н5 + 2NaCl а также взаимодействием дихлорэтана с бензолом в присутствии хлористого алюминия.
Стильбен (симметрический дифенилэтилен) С6Н5—СН = СН— содержащий этиленовую связь, при восстановлении переходит в дибензил. Он может присоединять бром, галоидоводороды и пр. Стильбен образует кристаллы с темп, плавл. 124°; темп. кип. 167° (при 12 мм рт. ст.). Известна и его неустойчивая стереоизомерная ^ис-форма—жидкость с темп. кип. 143° (при 12 мм рт. ст.).
Стильбен может быть получен различными способами, например отнятием воды от алкоголя С6Н5—СН2—СН(ОН)—С6Н5. Интересно, что он образуется при нагревании солей фенилизонитрометана:
2С6НбСН—N—ONa —> CeH5CH=CHCeH5 4-2NaNO2 !
О
Отнятием двух молекул бромистого водорода от стереоизомерных стильбендибромидов СбНб—СНВг—СНВг—С6Нб получается углеводород с ацетиленовой связью СвНб—С=С—СбНб, называемый толаном (темп, плавл. 62,5°).
Из производных стильбена представляют интерес синтетические эстрогенные гормоны (стр. 196). Простейшим препаратом такого рода оказался 4,4'-диоксистильбен, названный стильбэстролом'.
H0-^Z=/~CH=CH ~он
Однако его эстрогенное действие невелико (действующая доза 500 7).
Практическое значение получили а^-диэтилстильбэстрол и его дигидропроизводное, называемое гексэстролом или синэстролом. Диэтилстильбэстрол оказался близким по силе действия к натуральным эстрогенным гормонам (действующая доза 0,87); гексэстрол (действующая доза 0,2 7) даже превосходит их по физиологической активности.
Синтез диэтилстильбэстрола и гексэстрола осуществляется по схеме: О
II С2Н5ВГ
СН3О—свн4— с- СН2—С6Н4—осн3 ——
о с,нБ
II I C2H5MgBr
-> СН3О-С6Н4-С-СН-С6Н4-ОСН3----------S
С2не с2н5
-» СН3О-СвН4-С------с-с6н4—осн3
<!>н н
дегидратация
Деметилирование
С2Н6 С2Не
нос6н4—с =с-свн4он
др этилетильбэстрол
гидрирование
С,н6 С2Н6
НОСвН4—СН— СН—СвН4ОН
гексэстрол
Бензоин. Наиболее доступными веществами ряда дибензила являются продукты, получаемые из бензальдегида, прежде всего бензоин CfiH5—СН(ОН)—СО—С6Н5 (бесцветные призмы; темп, плавл. 137°).
Бензоин получается конденсацией двух молекул бензойного альдегида под влиянием KCN и является одним из наиболее типичных кетоно-спиртов a-ряда: сн реагирует с фенилгидразином, давая гидразон и озазон. При восстановлении бензоина получаются два оптически недеятельных стереоизомерных гликоля строения
СбН5—СН—СН—СбНБ
I I
ОН он
называемых гидробензоином (темп, плавл. 139°) и изогидробензоином (темп, плавл. 121°). Число стереоизомеров и их взаимные отношения здесь такие же, как для винных кислот: изогидробензоин, подобно виноградной кислоте, может быть разделен на два оптиче
ских антипода, а гидробензоин, подобно мезовинной кислоте, на оптические изомеры не расщепляется.
Бензил. Окислением бензоина получается а-дикетон—дибензоил С6Н5—СО—СО—СсН5, называемый чаще бензилом. Он образует желтые призмы, плавящиеся при 95°.
Действуя на бензил гидроксиламином, можно получить два стереоизомерных монооксима {син- и анти-) диоксима {син-, анти- и амфи-):
С6 14 5 С С С6 Н5
II И
N-OH НО—N сан- диоксим
При действии на бензил спиртовой щелочи
и
три стереоизомерных
свн4-с-с-с6н6
НО—N N— ОН
6Ш/7Ш-ДИОКСИМ
5
С6Н5-С------с
!| I!
НО—N НО—N имфг/-диоксим
происходит внутри-
молекулярная перегруппировка и получается с присоединением воды бензиловая, или дифенилгликолевая, кислота (С6Нд)2С(ОН)-~СООН (темп, плавл. 150°), относящаяся к производным дифенилметана. Эта так называемая бензиловая перегруппировка
НО он
СбН5—С—С—СвНБ
СвН,-С—С-СвН6
I I
НО он
+н+
бензил
И
Н—0+ он
он
—н?о
С6Нб—С—С—С6Нб I I но он
Свн5
свн6—с—с—свн5
НО
С6Н5 . . -н+ |
-» С.Н.-С—С—ОН--------> свн8—с-с—он
I I I
но он но о
бензиловая кислота
аналогична пинаколиновой перегруппировке (см. том I, стр. 420).
Несимметрический дифенилэтан
Несимметрический дифенилэтан (1,1-дифенилэтан, или а,а-дифе-нилэтан)
СвНбХ
>сн—сн3 свн/
жидкость, кипящая при 272°. Он получается конденсацией бензола с паральдегидом под влиянием серной кислоты.
В последнее время большое практическое применение в качестве инсектицида получил 2,2-{4,4'-дихлордифенил)-! Д Д-трихлорэтан, сокращенно обозначаемый ДДТ. Это—мелкие бесцветные кристаллы
или белый порошок (темп, плавл. 114°). ДДТ получается конденсацией хлораля, хлоральгидрата или хлоральалкоголята с хлорбензолом под действием концентрированной серной или хлорсульфоновой кислоты:
i_ О—-CH—CCL
CH—СС13 4-
Н2О
Вещество это в ничтожно1 малых концентрациях убивает при контакте взрослые формы ряда насекомых (мух, комаров, тараканов, клопов, вшей и т. п ь поражая постепенно их нервную систему.
При сильном нагревания и под действием щелочей дихлорди-фенилтрихлорэтан легко отщепляет элементы хлористого водорода, превращаясь в значиюльно менее токсичный дихлордифенилдихлор-этилен:
(С1С6Н4)2СЫ-СС13 (С1С6Н4)2С—CCi2-h НС1
Оба вещества сравнительно мало токсичны для высших живое ных и человека.
Тетрафенилэтан
Симметрический тетрафенилэтан (кристаллы; темп, плавя. 207э; темп. кип. 380е)
zcrh6
>СН--СН<
сбн/ ХС6Нб
был получен очень многими способами, например действием металлов на дифенилхлорметан. Он дает кристаллическое молекулярное соединение с бензолом.
Восстановлением бензофенона цинком и уксусной кислотой или действием фенилмагнийбромида на бензил может быть получен те' рафенилэтиленгликоль, или бензпинакон
0 0 НО ОН
С н2о с.н^ | | ?свн6
С6Нб—С—С-—С6Н5 + 2C6H6MgBr-------> Х-с<
с6н/ \свНб
образующий бесцветные призматические кристаллы, которые плавятся при 186°. При действии хлористого ацетила или минеральных кислот бензпинакон, теряя элементы воды, перегруппировывается в $-бензпинаколин (трифенилацетофенон):
= Н Н;
О :0:
Ce^5\ I i /Сбнб
>с-с<
С?н/
О Сбн5
CeHf-C-C-CeH5 + н2о
с6н5
Р-Бензпинаколин кристаллизуется в виде (темп, плавл. 182,5°).
а-Бензпинаколином называется соединение
свн с/СбНб Cetv \/Чн5
бесцветных иголочек
имеющее темп, плавл. 205°.
Тетрафенилэтилен (темп, плавл. 227°)
СвН5\с_с/СвН5 свн/ ~ \свн6
получается, например, при действии цинка на бензофенонхлорид (СвН5)2СС12. Он присоединяет хлор, но не присоединяет брома.
Дибифениленэтилен—красный, хорошо кристаллизующийся углеводород, плавящийся при 189^; он может быть легко получен нагреванием 9,9-дихлорфл уорена с порошком меди:
Это вещество долгое время было единственным известным интенсивно окрашенным углеводородом.
СОЕДИНЕНИЯ ТРЕХВАЛЕНТНОГО УГЛЕРОДА
Действием цинка, меди или молекулярного серебра на гри-фенилхлорметан в отсутствие воздуха Гомберг* получил в 1900 г. соединение, элементарный состав которого отвечает формуле гек~ сафенилэтана, т. е. С38Н30, или
Сбн5 сбнб
I I
С6Н5-С---С—С6Н5
I I Сбн6 сбнб
Однако свойства этого соединения, казалось бы, несовместимы со строением гексафенилэтана. Оно быстро обесцвечивает иод, образуя трифенилиодметан (C6H5)3CJ, присоединяет газообразный кислород, образуя перекись трифенилметила (СбН5)3С—О—О—С(С6Нб)3. В растворах под действием прямых солнечных лучей оно быстро изменяется; почти со всеми органическими веществами, даже со многими
* Моисей Гомберг (1866—1947); американский химик, известный своими многочисленными исследованиями в области свободных радикалов.
жирными углеводородами, дает кристаллизационные соединения, например с этиловым эфиром образует соединение состава [(С2Н5)2О]• 2[(С6Н5)3С], со сложными эфирами—соединения, в которых также на две молекулы [(С6Н5)3С ] приходится одна молекула сложного эфира.
Так как все эти свойства характерны для ненасыщенных соединений, Гомберг предположил, что это вещество содержит трехвалентный атом углерода, т. е. представляет собой свободный радикал трифе нилметил (С8Н5)3О. Такое предположение как бы подтверждалось способностью этого вещества под действием кислот образовывать, очень прочный углеводород, имеющий состав и молекулярный вес гексафенилэтана и плавящийся при 231°. Однако криоскопическое определение молекулярного веса предполагаемого радикала трифе-нилметила при комнатной температуре показало, что молекулярный вес его вдвое больше и довольно точно отвечает формуле гексафенилэтана.
В. В. Марковников, вопреки общепринятому мнению, что гекса-фенилэтан должен обязательно являться устойчивым веществом, высказал мысль, что связь между атомами углерода в остатке этана должна быть сильно ослаблена наличием в гексафенилэтане шести фенильных радикалов, а потому вещество, описанное Гомбергом под названием трифенилметила, вполне может быть и настоящим гексафенилэтаном, распадающимся при реакции с иодом, кислородом и пр. по месту связи двух остатков трифенилметила.
А. Е. Чнчибабиным эта мысль была подтверждена двумя экспериментами. Им было доказано, что прочный углеводород с темп, плавл. 23Г, образующийся из предполагаемого трифенилметила, представляет собой не гексафенилэтан, а п-бензгидрилтетрафенилметан (С6Н5)2СН—С6Н4—С(СбН5)3. Кроме того, он показал, что пентафенылэтан (С6Н5)3С—СН(СбН5)2, полученный Гомбергом из 1пифенилметилмагнийхлорида и дифенилметилбромида
(С6Н5){С— MgCl + Br—CH(CfiH5)2 —> (CeH5i3C— CHiCeH5)2 + MgClBr
и описанный как прочное вещество с обычными свойствами насыщенных аромати ческих соединений, при повышенной температуре распадается, как и гексафенил этан, по месту связи этанных атомов углерода. Распад происходит по следующему уравнению:
2(С6Н5)3С-СН(С6Н5\ (С6Н5)3С-С(СеН6)3 4- (С6Н5)2СН-СН(СбЫ5)2
т. е. молекула пентафенилэтана распадается на радикалы, которые затем снова соединяются, но уже в другой комбинации.
Бензгидрилтетрафен и лметан при этой температуре (150°) не изменяется, если же нагревание вести в присутствии хлористого водорода, то он реагирует с ним, образуя (СбН5)3СН и С1С(С6Нб)3
Позднейшие исследования (Шмидлин, Шленк) подтвердили, что кристаллический бесцветный «трифенилметил» есть гексафенилэтан, но что в растворах, особенно при повышенной температуре (например, при температуре плавления нафталина или фенола), гексафенилэтан диссоциирует, образуя желтый трифенилметил
(С6Н5)3С-С(СбН5)з 2(С6Н5)3О
(подобно тому, как бесцветный N2O4 диссоциирует с образованием бурого газа NO?). Такой легкий распад гексафенилэтана связан с тем, что теплота диссоциации молекулы гексафенилэтана на две молекулы трифенил мет ила составляет лишь 11—13 ккал (Циглер, 1928 г.), тогда как теплота разрыва простых С—С связей составляет 85 ккал, а теплота образования связей между углеродными атомами бензольного ядра—80 ккал.
Шленку удалось получить аналогичные углеводороды, уже при обычной температуре имеющие молекулярный вес, отвечающий свободному радикалу с трехвалентным углеродом, причем эти углеводороды, как, например, гри-п-бифениЛметил (СбНа—С6Н4)3С •, и в твердом виде ярко окрашены (в фиолетовый цвет/.
Из производных трифенилметила особенной устойчивостью отличается три-п-нитротрифенилметил (NO2—С6Н4)3С*, образующий зеленые кристаллы.
В настоящее время получено довольно много веществ, аналогичных трифенилметилу,- и возможность существования «свободных радикалов» с трехвалентным углеродом является общепризнанной.
Некоторые исследователи высказывали мысль, что трифен ил метил имеет иноидное строение, т. е. что трехвалентный атом углерода находится не в метанном остатке, а в ядре:
\=/
Это могло бы служить объяснением окрашенности простейших производных трифенилметана. Однако такое объяснение в настоящее время оставлено в связи с возникновением иных представлений о строении соединений ряда 1рифенилме-тила, лучше согласующихся с экспериментальными фактами (стр. 449 сл.).
Из углеводородов другого типа с трехвалентным углеродом особенно интересен по своей относительно большой устойчивости пентафен илэтил
Получены и такие соединения трехвалентного углерода, в ко-орых углеродный атом связан не только с ароматическими радикалами, но, кроме того, например, с третичным бутилом или циклогексилом (Марвел). Наиболее прочными соединениями этого типа являются те из них, у которых связанный с трехвалентным углеродом алифатический радикал содержит двойную связь, как, например, земнозеленый 1,1-дианизил-3,3-дифенилаллил Циглера:
Интересен также полученный Циглером (1925 г.) пентафенилцик-лопентадиенил (красно-фиолетовые кристаллы)
СвН5-С---С-С,Н8
II II
С.Н.-С С—CgHB
<1.Н.
как производное «пятичленного бензола» С5Н5.
Из соединений с аномальной валентностью углерода, принадлежащих к другим классам органических веществ, наиболее изучены так называемые металлкетилы, получающиеся при действии щелочных металлов на ароматические кетоны. Например, действием натрия на бензофенон получается соединение
свн5ч< с.н6>с-0№
Простой фениловый эфир кетила получается при кипячении раствора перекиси трифен ил метил а:
(СбН3).{С—О—О—С(СвН5)3 -> 2(СвН5)2С-ОС6Нб
Рассмотренные здесь соединения трехвалентного углерода в принципе ничем, кроме относительно большей прочности, не отличаются от простейших свободных радикалов (метила, этила), кратковременное существование которых многократно доказано (см. том I, стр. 360).
СВОБОДНЫЕ РАДИКАЛЫ С АНОМАЛЬНОЙ ВАЛЕНТНОСТЬЮ НЕУГЛЕРОДНЫХ АТОМОВ
По аналогии с трифенилметилом, содержащим трехвалентный углерод, Виландом была предположена возможность существования органических соединений, содержащих двухвалентный азот. Действительно, получающийся окислением дифениламина тетра-фенилгидразин (CeH5)2N—N(C6H5)2 при 90° в незначительной степени диссоциирует с образованием интенсивно окрашенного дифенил-азота (C6H5)2N« . Дифенилазот соединяется с NO, образуя нитрозодифениламин (C6H5)2N—NO, а также с трифенилметилом, образуя соединение (С6Н5)3С—N(C6H5)2.
Сравнительно сильно диссоциирует в растворах при повышенной температуре тетра-п-анизилгидразин (CH3OC6H4)2N—N(C6H4OCH3)2. Его растворы, при нагревании приобретающие окраску, при охлаждении обесцвечиваются (вновь получается тетраанизилгидразин). Еще сильнее диссоциирует тетра-(диметил аминофенил Ьгидразин [(CH3)2N — C6HJ2N— N[C6H4—N(CH3)2]2, который в нитробензольном растворе уже при обычной температуре содержит 21% соединения с двухвалентным азотом.
Большой стойкостью отличаются также соединения четырехвалентного азота. Так, например, при окислении дифенилгидроксил-амина (C6H5)2N—ОН действием Ag2O или РЬО2 получается окись дифенилазота (C6H5)2N=O (Виланд)—яркокрасное кристаллическое вещество, при восстановлении легко снова переходящее в дифенилгидроксиламин. Подобно двуокиси азота, окись дифенилазота является сильным окислителем.
Ряд аналогов окиси дифенилазота К. Мейер получил восстановлением продуктов реакции азотной кислоты с простыми эфирами фенолов. Таким образом получено, например, соединение
CHsO-CeH4-N-CeH4-OCH3
Не исключена возможность диссоциации соединений типа С6Н5—S—S—СеН5 с образованием радикалов с одновалентной серой, а также диссоциации органических перекисей с образованием соединений одновалентного кислорода, как, например, для перекиси трифенилметила:
(С6Н5)3С-О-О-С(СвН6)3 2(С6Н5)3С-О*
В настоящее время признано, что соединения с нормальными валентное 1ямп атомов углерода, азота и других элементов представляют собой лишь наиболее устойчивые типы соединений, а соединения с низшей валентностью атомов отличаются от них только кратковременностью своего существования. Поэтому возникновение свободных радикалов в качестве промежуточных продуктов постоянно учитывается при рассмотрении механизма различных реакций.
Раньше (см. том 1,стр. 359) уже было дано объяснение (Райс, 1931 г.) направлению крекинга углеводородов на основе представления о свободных радикалах', подвергающихся затем дальнейшим превращениям. Во многих реакциях промежуточным образованием радикалов быть может, объясняется появление мимолетных окрасок, исчезающих при окончании реакции.
ПРИЧИНЫ УСТОЙЧИВОСТИ СВОБОДНЫХ РАДИКАЛОВ ТИПА ТРИАРИЛМЕТИЛОВ
На протяжении 1900—1930 гг. в области свободных ароматических радикалов было проведено громадное число исследований. Эти исследования оказали большое влияние на развитие теоретических представлений в органической химии, хотя они и не привели к отчетливым, всесторонне обоснованным взглядам на причины сравнительной устойчивости, цветности этих радикалов и т. д.
Если сопоставить, например, трифенилметил с метильным или этильным радикалом, то причины различия в поведении этих радикалов проще всего было бы искать в том, что метильный атом углерода в трифенилметиле несет три тяжелых ароматических остатка, занимающих большое пространство около этого углеродного атома. Наоборот, в метильном радикале сродство центрального атома углерода затрачивается только на удержание трех атомов водорода, не требующих для своего размещения большого объема. Можно было
бы думать, что на образование связей с тяжелыми ароматическими остатками (фенильными группами) центральный углеродный атом затрачивает много сродства и четвертая связь в молекуле гексафе-нилэтана оказывается сильно ослабленной, что и влечет за собой легкость диссоциации такой молекулы на два радикала.
Однако такому объяснению противоречит ряд фактов. Например, тетрафенилметан оказывается прочным соединением, хотя, казалось бы, связь трифенилметила с четвертым фенилом должна быть ослаблена; декафенилбутан не отщепляет трифенилметила, а диссоциирует на два радикала пентафенилэтила:
С6НБ С6Н6 С6Н6 С6Н5 СбН6 С6Н6
Illi I <
С6Н6-С---С---С---С-СбН5 ^=± 2С6Н6—с—с«
I I ! I II
С6Нб С6Нб С6Нб сбнб С6Н6 С6Нв
(радикал пентафенилэтил может быть получен и отнятием гтлоида металлами от пентафенилэтилхлорида). Поэтому можно думать, что имеется какой-то фактор, под влиянием которого система из двух свободных триарилметильных радикалов оказывается более стабильной, чем система гексаарилэтана. Это фактор, очевидно, отсутствует у этана и его алифатических производных.
Естественнее всего искать объяснения стабилизации триарилметильных радикалов в том, что одинокий неспаренный электрон такого радикала оказывается способнььм вступать во взаимодействие (в сопряжение) с ^-электронными ароматическими секстетами арильных группировок. Поскольку, однако, ни монофенил метильный (бензильный*) радикал
сбн6-с<
ни дифенилметильный (бензгидрильный) радикал
свн6Х.
>с-н
С6н/
по устойчивости несравнимы с трифенилметилом, следует заключить, что устойчивость такого радикала в значительной мере определяется эффектом сопряжения неспаренного электрона «метанного» атома углерода с электронными системами всех трех ароматических ядер.
Сопряжение связей, как известно из предыдущего, возможно лишь при плоскостном расположении сопряженной системы. Очевидно, что и взаимодействие неспаренного электрона с обобщенными
* Бензильный радикал получается по методу Панета пиролизом тетрабен-зилсвинца. Свойства этого радикала аналогичны свойствам метила и этила (см. том I, стр. 359—360), хотя полупериод существования бензила немного больше (~0,006 сек., вместо 0,002 сек.).
облаками ^-электронов всех трех ароматических группировок возможно лишь при перестройке тетраэдрического расположения этих группировок вокруг «метанного» атома углерода, в результате которой вся система триарилметила примет плоскую конфигурацию с углами 120° между связями. При этом электронная плотность неспаренного электрона распределится между «метанным» атомом углерода и тремя бензольными ядрами.
Направления возникающих электронных смещений в системе можно передать следующим образом:
Здесь круговая стрелка вокруг «метанного» углерода означает, что плотность неспаренного электрона распределяется по всем трем связям.
Если система из двух свободных триарилметильных радикалов более устойчива, чем гексаарилэтан, то при диссоциации должно выделяться некоторое количество энергии
Аг3С—САг3 —> 2Аг3С*-4-Q
в то время как для аналогичной диссоциации этана приходится затратить значительную энергию (около 85 ккал). Это подтверждается для радикалов, существующих при обычной температуре практически в мономолекулярной форме. Другие гексаарилэтаны занимают промежуточное положение; при их диссоциации энергия не выделяется, а поглощается, но необходимая энергия диссоциации оказывается значительно ниже, чем для этана. Например, для диссоциации гексафенилэтана требуется только 19 ккал, для пентафе-нилэтана 27,6 ккал. Еще прочнее связь в тетрафенилэтане, однако имеются некоторые признаки ее ослабления: при нагревании симметрического тетрафенилэтана с калием образуется бензгидрил-калий:
(СвН5)8СН-НС(С6Н5)2 4- 2К -> 2(СбН5)2СНК
Таким образом, можно предполагать, что изменение расположения ароматических ядер в пространстве и возникновение взаимодействия неспаренных электронов с ароматическими системами приводит в случае гексафенилэтана к выигрышу энергии (сообщающему стабильность каждому трицЬенилметильному радикалу) в размере
85—19
С усложнением ароматических группировок и введением заместителей в молекулы гексаарилэтанов, во взаимодействие с неспаренными электронами вовлекаются более длинные цепи конъюгации. Это должно способствовать стабилизации соответствующих свободных радикалов и, действительно, в некоторых случаях приводит к почти полной диссоциации.
Для иллюстрации можно сопоставить содержание свободных радикалов в 0,08 М растворах гексаарилэтанов в бензоле при 5°:
Содержание
Го
Трифенилметил (СбН5)3О............... 3,0
Три-п-бифенилметил (СбНб—СвН4)3О ... 74,0 Три-о-анизилметил [СбН4—ОСН3]3С* .... 96—100 Три-п-нитрофенилметил (NO2—С6Н4)3О . . 100
Наличие арилированных винильных групп в качестве заместителей при «метанном» атоме углерода, как, например, в случае 1,1,3,3-тетрафенилаллила и 1,2,3,4,5-пентафенилциклопентадиенила
СбН5. ,cGH5
)с-сн--с( с6н5/ \cfiH6
1,1,3,3-гетрафенилаллил
1,2,3,4,5-пентафенилииклопента-диенил
также способствует стабилизации мономерных радикалов.
Если же по каким-либо стерическим причинам плоская конфигурация триарилметильного радикала оказывается невозможной, то становится невозможной и стабилизация таких радикалов.
Часто об образовании свободных радикалов судят по появлению (без доступа воздуха) окраски растворов. Однако непосредственной связи между глубиной окраски и способностью к диссоциации соответствующего замещенного этана установить нельзя, как это видно из приведенного ниже сопоставления их окраски:
Трифенилметил.............................
п-Хлортрифенйлметил.......................
Три-п-хлортрифенилметил...................
Тетраметилди-(дг-аминотрифенил)-метил . . . Гексаметилтри-(п-аминотрифенил)-метил . . . Дифенилбифен ил метил ....................
Фенил-ди-(бифенил)-метил.............. .
Трибифенилме! ил..........................
Три-£-нафтилметил.........................
Фенилксантил .............................
Фенилантранил ............................
9-Фенилфлуоренил .........................
ор а нже во- желт ый светлокрасный оранжево-красный желтый
желтый
оранжево-красный красный темнофиолетовый фиолетовый темнокрасный красный коричневый
Хотя дифенилдибифепилэтан очень мало способен к диссоциации (заметцая диссоциация этого соединения—появление коричневой
окраски—наблюдается только при нагревании в высококипящих растворителях, например в анизоле), 9-фенилфлуоренил оказывается глубоко окрашенным. Наличие в ароматических ядрах таких сильных ауксохромных групп, как две или три диметиламиногруппы, скорее даже повышает окраску соответствующих триарилметилов (действует гипсохромно, а не батохромно) по сравнению с незамещенным трифенилметилом.
МАГНИТНЫЕ СВОЙСТВА СВОБОДНЫХ РАДИКАЛОВ
Свободные радикалы отличаются от обычных органических соединений с четным числом электронов у всех атомов своими парамагнитными свойствами.
В обычных сложных органических молекулах с четным числом валентных электронов все элементарные магнитные моменты вращающихся и движущихся электронов взаимно компенсируются. В свободных же радикалах имеется неспаренный электрон, спин-момент и орбитальный момент которого оказываются нескомпенсирован-ными, что проявляется в наличии постоянного магнитного момента.
Экспериментально определяемая магнитная восприимчивость может быть представлена суммой двух слагаемых:
^набл. —
Здесь Td—диамагнитная восприимчивость, присущая каждому веществу; она не зависит от температуры и возникает под влиянием налагаемого магнитного поля, возмущающего движение электронов. Величина диамагнитной восприимчивости может быть выражена аддитивно, как сумма атомных восприимчивостей (Паскаль, 1920 г.).
Второе слагаемое—парамагнитная восприимчивость отличается от по знаку и сильно отличается по величине. Зависимость ее от температуры может быть представлена выражением (П. Кюри):
При наличии одного нескомпенсированного электрона (т. е. для свободного монорадикала) величина у., может быть найдена расчетным путем и при 20° составляет ~12/0-10~6.
Исследования показали, что парамагнитная "восприимчивость трибифенилметила и пентафенилциклопентадиенила, действительно, имеет величину, соответствующую полной диссоциации, т. е. мономерному радикалу. Парамагнитная восприимчивость а-нафтил-дифенилметила составляет лишь 570-10-6, т. е. ди-а-нафтилтетра-фенилэтан диссоциирован неполностью.
Полученный А. Е. Чичибабиным «бирадикал», которому можно было бы придать строение
о-о-С
должен иметь /р=2-1270-10-6=2540-10~6. Однако он оказался полностью диамагнитным и, следовательно, не является свободным радикалом, а имеет хиноидное (хинондиметидное) строение (Э. Мюллер):
С6^54 с /С6Н5
с6н6 ==/ \с6н5
Этим и объясняется его глубокая окраска (цвет растворов перманганата).
В то время как рассмотренный выше «бирадикал» может стабилизироваться, перегруппировываясь в молекулу хиноидного строения, аналогичное соединение
<э-с>
\ /СвН5
>о .С<
С.н/ \с6нб
должно существовать как бирадикал, так как мета-хиноидное строение невозможно. Это было подтверждено наличием у последнего соединения сильной парамагнитной восприимчивости.
Соединения с конденсированными бензольными ядрами
Группа нафталина
Нафталин С10Н8 был открыт в 1819 г. как составная часть каменноугольной смолы. Название «нафталин» возникло позднее и произошло от слова «нафта», обозначавшего летучую часть нефти. Элементарный состав нафталина был установлен А. А. Воскресенским в 1858 г.
Нафталин получают обычно из каменноугольного дегтя и из продуктов пиролиза керосина (стр. 222)—из фракций, кипящих при 170—240°. Для отделения твердого нафталина от жидких примесей его отжимают, очищают путем обработки небольшим количеством серной кислоты и затем отгоняют или перегоняют с водяным паром. Вместе с бензолом и другими ароматическими углеводородами нафталин образуется при пиролизе ацетилена, а также при сухой перегонке и пропускании через раскаленные трубки паров многих органических веществ.
Чистый нафталин кристаллизуется в блестящих листочках, плавится при 80,3° и кипит при 218°; он обладает характерным запахом и сравнительно большой летучестью. Нафталин нерастворим в воде, хорошо растворяется в горячем спирте, эфире и бензоле; в холодном спирте растворимость его невелика.
Нафталин, его гомологи и многие производные дают соединения с пикриновой кислотой (стр. 273). Этой способностью обладают также многие другие соединения с конденсированными бензольными ядрами. Превращая нафталин в пикрат состава C10H8-C6H2(NO2)3OH с темп, плавл. 149°, можно количественно определить содержание нафталина в какой-либо углеводородной смеси.
Для нафталина был предложен ряд формул. По аналогии с формулой Кекуле для бензола, Эрленмейером и Гребе*(1866—1868 гг.) было предложено выражать строение нафталина формулой оэ
Согласно гипотезе Тиле, для нафталина должна быть принята формула
Бамбергер предложил центрическую формулу нафталина
которая, однако, лишена физического смысла, как и соответствующая формула бензола (стр. 229). Кроме того, в формуле Бамбергера вообще отсутствует прямая связь между углеродными атомами 9 и 10, в то время как основой большинства других предложенных формул как раз является представление о системе нафталина, как о таком сочетании двух бензольных ядер, при котором два углеродных атома являются общими для обоих ядер. Эта мысль может быть выражена следующей общей схемой:
Правильность этой схемы была строго доказана исследованием окисления производных нафталина (Гребе). Действительно, при окислении одного из двух существующих нитронафталинов получается 3-нитрофталевая кислота. Таким образом доказывается наличие в нафталине бензольного кольца, обозначенного цифрой II:
ноос
ноос
♦ Карл Гребе (1841—1927). Крупный немецкий химйк-органик. Главнейшие его работы посвяшены исследованию многоядерных конденсированных ароматических углеводородов.
Одновременно положение нитрогруппы в 3-нитрофталевой кислоте указывает на положение ее в молекуле нитронафталина: нитрогруппа находится при углеродном атоме 1 (см. нумерацию атомов углерода в приведенной выше схеме).
Наличие бензольного кольца I может быть доказано восстановлением того же нитронафталина в аминонафталин, который при окислении образует с разрушением кольца II фталевую кислоту:
Эти и другие подобные исследования подтверждают правильность формулы с двумя конденсированными бензольными ядрами, предложенной Эрленмейером и Гребе; надо, конечно, помнить, что на нее распространяется сказанное об аналогичной формуле Кекуле для бензола (стр. 233).
- Число и строение изомерных производных нафталина строго соответствует этой формуле. Так, каждый из углеродных атомов 1, 4, 5 и 8, согласно формуле, находится в одинаковом отношении ко всем остальным атомам углерода, а потому замещение водородного атома при любом из них на радикал X должно приводить к одному и тому же продукту: ,
X
В таких же отношениях между собой и с другими атомами находятся углеродные атомы 2, 3, 6 и 7. В результате замещения при них атома водорода должен получиться второй изомер:
Положения 1, 4, 5 и 8 объединяют названием a-положение, а положения 2, 3, 6 и 7 называют ^-положением. Соответственно, одно-
замещенные изомеры различают,
мером, например:
называя их а-изомером и р-изо-
3-мегилнафталин
сн8
Другой способ обозначения производных нафталина основан на нумерации атомов углерода в молекуле нафталина (стр. 446).
Тогда я-метилнафталин будет называться 1-метилнафталином, а Р-метилнафталин—2-метилнафталином.
Если заместить в нафталине два атома водорода одинаковыми радикалами, можно получить 10 различных изомеров; при двух разных заместителях число возможных изомеров равно 14. С увеличением числа заместителей количество изомеров сильно возрастает. При 8 различных заместителях теоретически возможно ожидать получения 10 766 600 изомерных производных
Положение заместителей в многозамещенных производных нафталина обыкновенно обозначают цифрами, например:
2,6-диметилнафталин
Нахождение заместителей в- положении 2,6 иногда называют омфи-положением, а в 1,8—nepw-положением.
Синтезы нафталина и его производных из простейших ароматических веществ находятся в полном согласии с приведенной выше формулой Эрленмейера и Гребе. Например, я-оксинафталин (я-наф-тол) может быть получен из фенилизокротоновой (фенилвинилуксус-ной) кислоты:
При действии о-ксилилендибромида на динатрийэтантетракарбоновый эфир получается тетрагидронафталинтетракарбоновый эфир:
<СН2Вг
Na.
V(COOC2H6)2
ZC(COOC,H6),
СН2
\С(СООС2Н61,г
I + 2NaBr
✓QCOOCsHg)^
сн2
При омылении и нагревании этот эфир, отщепляя 2СО2, дает двухосновную кислоту, серебряная соль^которой при разложении образует нафталин:
сн2
\сн—COOAg
/CH-COOAg
СН2
_> | J | + 2Ag + Н2 + _2СО2 4/XZ
Синтез нафталина из ацетилена также хорошо согласуется с формулой углеводорода с двумя конденсированными бензольными ядрами. В качестве промежуточных продуктов^образуются бензол и стирол:
СН
Нафталин и его производные, подобно бензолу, обнаруживают «ароматический» характер, т. е., будучи формально непредельными, ненасыщенными соединениями, ведут себя все же скорее как насыщенные соединения и, вместо реакций присоединения, проявляют большую способность вступать в реакции замещения. Однако нафталин легче окисляется и восстанавливается, чем бензол.
Десять углеродных атомов молекулы нафталина образуют^две группы по шесть атомов, как бы два бензольных ядра, у которых два атома являются общими. Дипольный момент нафталина равен нулю. Поэтому строение нафталина можно изобразить следующим образом:
Подобно'бензолу,янафталин представляет собой плоскую систему. Однако, в отличие от бензола, углерод-углеродные связи имеют пазнуъ длину, зависящую от их положения в системе.^Это может быть выражено ^схемой (Робертсон):
Таким образом,^хотя химические свойства нафталина показывают, что электронные плотности в нем выравнены, однако это выравнивание менее полное, чем в бензоле. Такое распределение электронной плотности обусловливает значительное сходство и одновременно определенные различия между химическими свойствами бензола и нафталина. Вследствие выравнивания электронной плотности по всей системе нафталина эта система функционирует как
единое целое, а не как сочетание бензольного ядра и непредельного кольца с двумя двойными связями и не как система из двух независимых бензольных ядер. Это подтверждается хотя бы следующими химическими данными.
1. Ароматические аминосоединения являются более слабыми основаниями, чем аммиак и алифатические амины. Замещенные в разных ядрах {гетеронуклеарные) нафтилендиамины, такие, как, например, 1,5-диаминонафталин, вполне аналогичны фенилендиаминам.
2. Ароматические аминосоединения дают диазониевые соединения, способные к азосочетанию. Гетеронуклеарные диамины нафталина дают при диазотировании бис-диазосоединения, у которых обе диазониевые группы оказываются способными к азосочетаниям.
3. Для ароматических систем характерен метод введения гидроксильной группы путем сульфирования и последующего щелочного плавления. При сульфировании нафталина в условиях, при которых в молекулу вступают две сульфогруппы, всегда образуются смеси гетеронуклеарных дисульфокислот, из которых щелочным плавлением получаются соответствующие диоксинафталины.
Наряду с этими фактами, свидетельствующими о сходстве ароматической системы нафталина с бензолом, имеются данные и об их различии. Действительно, не вполне равномерное распределение электронной плотности в двухкольцевой системе, отражением которого является неравенство межатомных расстояний в молекуле нафталина, выражается в неодинаковой электронной плотности у а- и p-углеродных атомов, причем на а-атомах она выше. В соответствии с этим как реакции присоединения, так и реакции замещения направляются прежде всего на а-углеродные атомы, причем для нафталина все реакции протекают легче, чем для бензола.
Правила вступления дальнейших заместителей в нафталиновую систему значительно сложнее, чем для бензола. Осложнение обусловлено хотя бы тем, что у однозамещенного бензола оба орто-положения тождественны, тогда как в нафталине два соседних положения по отношению к заместителю, находящемуся, например, в положении 2, т. е. положения 1 и 3, неодинаковы.
Место вступления в нафталиновое ядро второго заместителя (электрофильного) зависит от природы и положения уже имеющегося заместителя. При этом возможны два случая:
1. Если первый заместитель принадлежит к числу ориентирующих в бензольном ядре в орто-пара-положения, то, находясь в положении а (1), он ориентирует вновь вступающий заместитель в положения 2, 4, 5, 7
X
а находясь в положении В (2),—в положения 1, 6 и 8: * * X
w
2. Если первый заместитель является ориентирующим в бензольном ядре в мета-положение, то, находясь в положении а (1), он ориентирует второй заместитель в положения 3, 6, 8
Y
а находясь в положении t3 (2),—в положения 4, 5 и 7:
Продукты восстановления нафталина. Восстановлением нафталина амальгамой натрия получается 1 А-дигидронафталин (темп, плавл. 25°; темп. кип. 212°)
CIU
строение которого подтверждается тем, что он легко окисляется в о-фенилендиуксусную кислоту:
^\зн2-соон
При нагревании с этилатом натрия 1,4-дигидронафталин превращается в изомерный \2-дигидронафтал11Н (темп, плавл. —7°):
Оба изомерных дигидронафталина легко присоединяют одну молекулу брома.
Процесс изомеризации 1,4-дигидронафталина совершенно аналогичен превращению аллилбензола под действием щелочей в пропенилбензол:
При восстановлении нафталина водородом в присутствии никеля под давленивхМ легко может быть получен тетрагидронафталин строения
СН2
А7
I I А
сн,
обладающий свойствами одноядерных ароматических углеводородов. Это вещество, обычно называемое тетралином, имеет большое техническое значение. Благодаря способности хорошо растворять смолы и относительно высокой летучести тетралин применяется взамен олифы при приготовлении лаков, как растворитель для жиров, как заменитель скипидара и, кроме того, в смеси со спиртом и бензолом— как топливо для двигателей внутреннего сгорания.
Хромовой смесью тетралин легко окисляется в кетон и-тетралон
СО
\зн,
Ч/\ /сн-’
сн,
Нитрованием, сульфированием и другими реакциями из тетралина и тетра-лона получено большое число производных. Некоторые из них являются красите лями, другие—лекарственными веществами.
При избытке водорода и при более высокой температуре в присутствии никеля происходит полное гидрирование нафталина. При этом получается декагидронафталин, называемый обычно декалином и представляющий собой бициклический углеводород с двумя шестичленными циклами (стр. 106).
Таблица 35
Нафталин и продукты его гидрирования
Температура плавления СС Температура кипения °C Удельный вес Показатель преломления „20 Г1о
Нафталин С10Н8 80,3 218 0,9752 1,5898
Тетралин C1(iH12 Декалин С10Н18 -35,8 207,6 (при 85°) 0,9702 (при 85°) 1,5411
цис- —43,01 195,7 0,8963 1,4810
транс- —30,40 187,3 0,8699 1,4692
Гомологи нафталина могут быть получены обычными способами синтеза ароматических углеводородов, например из нафталина• по способу Фриделя—Крафтса (стр. 213) или из бромнафталинов по
способу Вюрца—Фиттига (стр. 212). Они содержатся в каменноугольном дегте и в нефти.
а-Метил нафталин—жидкость с темп. кип. 244,6°—применяется как один из компонентов смесей с цетаном, составляемых для определения цетанового числа дизельного топлива.
^•Метилнафталин (твердое вещество, плавящееся при 34,6е; темп. кип. 241°) находит применение для получения р-метилнафто-хинона—синтетического продукта, обладающего свойствами витамина К (стр. 693).
Таблица 36
Гомологи нафталина
Название Строение Температура кипения °C Температура плавления °C Температура плавления пикрата °C
а-Метил нафталин
{3-Метил нафталин
1,6-Ди метил нафталин
2,6-Диметил нафталин
1 -Метил-7-изопропил-нафталин (эудалин)
1,6-Диметил-4-изопро-пилнафталин (кала-лин)
СН3
(СН3)2СН
СН3
СН(СН3)2
244,6
241
263
262
281
(при 7L0 мм)
291—292
(при 720 мм)
—30.6
34,6
— 14
112
141—142
115—116
114
142—143
95
115
Галоидопроизводные нафталина. а-Хлорнафталин и а-бром-нафталин получаются непосредственно действием хлора и брома на нафталин. При дальнейшем действии галоидов легко получается
смесь двугалоидных, а затем и полигалоидных соединений. В присутствии переносчиков хлора (SbCl3) получается октахлорнафта-лин С10С18. [З-Галоидзамещенные производные нафталина менее доступны; они получаются с умеренными выходами из р-дифенил-амина путем диазореакции.
Из продуктов присоединения к нафталину хлора известны жидкий нафта-линдихлорид С10Н8С12, уже при 50° распадающийся на а-хлорнафталин и НС1, и твердый (темп, плавл. 182°) нафталинтетрахлорид, под действием щелочи дающий дихлорнафталин.
Нитронафталины. При нитровании нафталина получается а-ни-тронафталин, без примеси [3-соединения. Более энергичное нитрование приводит к образованию 1,5- и 1,8-динитронафталинов.
(З-Нитронафталин получается диазотированием [3-нафтиламинов в азотнокислом растворе в присутствии закиси меди (обмен диазогруппы на NO2):
C1oH7-N—NO34-HNO2 —» Cl0H7NO2 н-N2 4-HNO3
N
Оба нитронафталина окрашены в желтый цвет.
При обработке щелочью в метиловом спирте [3-нитронафталин лишь отчасти восстанавливается в азоксинафталин и азонафталин, главным же образом превращается в растворимый в воде ацеталь (3-нафтохинонмоноксима; при последующем омылении ацеталя кислотами получается сам р-нафтохинонмоноксим:
О
/X Л /NOH
Сульфокислоты нафталина. Сульфирование представляет собой важнейшую реакцию в химии соединений нафталина, так как, изменяя условия, можно получать либо а-, либо ^-замещенные производные. Если на нафталин действовать концентрированной серной кислотой при 80°, получается почти исключительно а-нафталинсуль-фокислота. При 160° получается главным образом [3-нафталинсульфокислота, так как при этой температуре а-сульфокислота превращается в присутствии серной кислоты в [3-сульфокислоту. Превращение а-изомера в [3-изомер—обратимый процесс, а потому при промежуточных температурах получается равновесная смесь изомеров.
Дигидрат а-нафталинсульфокислоты плавится при 88°, тригидрат [3-нафталинсульфокислоты—при 83°; обезвоживаются они при 110°. Натриевые соли нафталинсульфокислот при обработке пятихлористым фосфором дают соответствующие сульфохлориды—бесцветные листочки, плавящиеся при 68е и при 77°.
Дальнейшее сульфирование а-нафталинсульфокислоты в мягких условиях приводит к 1,5- и 1,6-нафталинсульфокислотам. При дальнейшем сульфировании [3-нафталинсульфокислоты концентрированной серной кислотой при 140—160° получаются две изомерные
дисульфокислоты: 2,6- и 2,7-нафталиндисульфокислоты. Более глубокое сульфирование приводит к три- и тетрасульфокислотам. При этом соблюдается правило Армстронга и Винна: вступающая сульфогруппа никогда не занимает орто-, пара- или пери-положения по отношению к уже имеющимся сульфогруппам.
Схема сульфирования нафталина приведена ниже:
Как и сульфокислоты бензола (стр. 257), сульфокислоты нафталина и его производных являются важными промежуточными соединениями для получения более сложных производных этого ряда. При этом сульфогруппы в нафталинсульфокислотах еще подвижнее, чем в бензолсульфокислотах, и реакции обмена идут легче. Кроме реакций, описанных для сульфокислот бензола, в ряду нафталина возможна также и замена сульфогруппы на атом хлора.
Оксипроизводные нафталина получаются обычными способами синтеза фенолов. Практически оба монооксинафталина, называемые нафтолами, получаются сплавлением с едкими щелочами соответствующих изомерных нафталинсульфокислот. Нафтолы обладают всеми характерными для фенолов общими свойствами, в частности— растворяются в едких щелочах, однако гидроксильная группа в нафтолах легче вступает в реакции обмена, чем в простейших фенолах.
Таблица 37
Производные нафталина
Название Температура плавления °C Температура кипения °C Удельный вес Показатель преломления „20 nD
Фторнафталины
а- — 13 216,5 1,1340 1,5939
61 212,5 — —
Хлорнафталины
а- — 17 259,3 1,1938 1,6332
₽- 56,7 266 —
Бромнафталины
а- +6,2 281,1 1,4875 1,6582
₽- 59,0 281 1,605 —
Иоднафталины
а- — 305 1,7344 (при 15°) 1,7054 (при 14е)
3- 54,5 309 — 1,6617 (при 9Q,4°)
Нитронафталины
а- 61,5 304 1,2226 (при темп, плавл.) —
?- 79 165 (при 15 мм) — —
Ди нитронафталины
1,5- 217,5 возг. — —
1,8- 173,5 — — —
Нафталинсульфокислоты
а- ( 88—88(водн.) 1 124 (безводн.) — — —
83 (води.) — — —
Нафтолы
а- 96 288 1,224 (тверд, при 4°) —
₽- 122 294 1,217 (тверд, при 4°) —
Нафтиламины
а- 50 301 1,131 (при 51,2°) 1,6703 (при 51,2°)
3- 113 306,1 1,061 (при 98,4°) 1,6493 (при 98,4°)
С хлорным железом а-нафтол дает фиолетовый хлопьевидный осадок, р-нафтол— сначала зеленое окрашивание, а затем бесцветный осадок динафтола.
Нафтолы и их производные, особенно сульфокислоты нафтолов, находят большое применение, в частности в производстве органических красителей.
Метиловый эфир ^-нафтола С10Н7ОСН3 (темп, плавл. 75°) применяется в качестве душистого вещества под названием неролина или ярй-яра.
Динитро-а-нафтол (2,4-динитронафтол) C10H5(NO2)2OH получается действием азотной кислоты на нафтол-2,4-дисульфокислоту. Он является красителем
для шерсти и шелка. Сульфокислота 2,4-динитронафтола (нафтоловый желтый) применяется для окраски пищевых продуктов.
Диоксинафталины, содержащие оксигруппы в положениях 1,2 и 1,4, получаются восстановлением хинонов.
Сульфокислоты нафтолов могут получаться сульфированием нафтолов, сплавлением со щелочами нафталинполисульфокислот или заменой на гидроксил аминогруппы в сульфокислотах нафтил-аминов. Как и сами нафтолы, некоторые их сульфокислоты, особенно сульфокислоты ^-нафтола, находят значительное применение как составляющие азокрасителей.
При сульфировании а-нафтола получаются сульфокислоты, содержащие сульфогруппу в одном ядре с гидроксилом, а именно 1-нафтол-2-сульфокислота и 1-нафтол-4-сульфокислота (кислота Невиля—Винтера). ^-Нафтол в более мягких условиях и при непродолжительном действии серной кислоты дает 2-нафтол-1-сульфокислоту, тогда как при продолжительном действии концентрированной серной кислоты получается 2-нафтол-6-сульфокислота ф-кислота, или кислота Шеффера) и 2-нафтол-8-сульфокислота (а-кислота, или кроцеиновая кислота).
1-Нафтол-8-сульфокислота, в которой заместители находятся в пери-поло- ’ жении, легко выделяет воду и образует внутренний сложный эфир, подобный лактонам и называемый поэтому сулътоном'.
О— SO2
I I nn 4/\Z
Наибольшее значение имеют две дисульфокислоты ^-нафтола, получаемые прямым сульфированием ^-нафтола и разделяемые благодаря различной растворимости их натриевых солей. Эти кислоты имеют строение:
/YY0H HOgS/^^^SC^H 2-нафтол-3,6-дисульфокислота
SO3H
2*нафтол-6,8-дисульфокисло1 а
f Первая кислота была названа Р-кислотой, так как получавшиеся с ее уча стием красители имели чисто красные оттенки (rot, R-кислота); вторая кислота давала красители желто-красных оттенков и потому получила название Г-кисло-ты (gelb, G кислота). Натриевая соль Г-кислоты легко растворима в спирте.
Диазобензол с Р-кислотой образует краситель пунцовый Г; из а-диазонаф-талина и Р-кисЛоты получается краситель кислотный бордо; диазобензол с Г-кис-лотой дает краситель кислотный оранжевый светопрочный.
Значительное применение в производстве красителей имеет дисульфокислота 1,8-(или пери-) диоксинафталина, называемая хромотроповой кислотой'.
НО ОН
но3s/\/\Z\so3H
Получаемые с ее участием азокрасители обладают способностью резко изменять окраску при протравлении окрашенной ткани солями металлов; изменение окраски обусловлено образованием лаков (стр. 375). Например, азокраситель, приготовленный из фэнилдиазония и хромотроповой кислоты, окрашивает ткань в красный цвет; при протравлении ткани солями алюминия красный цвет переходит в фиолетовый, под действием хромовых солей—в сине-черный.
Для идентификации или выделения органических оснований (в том числе алкалоидов) довольно часто применяется 2,4-динитро-1-нафтол-7-сульфокислота, получившая название флавиановой кислоты,
Нафтиламины. а- и р-Нафтиламины могут быть получены восстановлением нитронафталинов. р-Нафтиламины обыкновенно получают из р-нафтола нагреванием его с аммиаком в присутствии хлористого цинка или хлористого кальция:
+ NH3
Особенно легко реакция протекает при нагревании р-нафтола с сернистокислым аммонием (аналогично получению аминофенолов из двухатомных фенолов).
а-Нафтиламин имеет неприятный фекальный запах, р-нафтил-амин* запаха не имеет.
Нафтиламины по свойствам сходны с анилинами. Они являются слабыми основаниями, способны нитроваться, сульфироваться, галоидироваться, диазотироваться и пр. Оба нафтиламина, а также их сульфокислоты находят обширное применение в производстве красителей.
Как и р-нафтол, р-нафтиламин и его производные дают при сочетании с ди-азосоединениями азокрасители типа
N=N—- Ai-
Из сульфокислот нафтиламинов большое применение имеет 1-аминонафталин-4-сульфокислота, так называемая нафтионовая кис-юта:
SO3H
* С р-нафтиламином необходимо обращаться осторожно, так как имеются указания, что он обладает канцерогенным действием и длительный контакт с ним может привести к появлению раковых опухолей.
Представляют интерес получаемые из нафтионовой кислоты красители конго красный и бензопурпурин. Краситель конго красный готовят из диазотированного бензидина и нафтионовой кислоты (взятой в виде соли):
нафтионовая кислота
нафтионовая кислота
' Сама кислота конго синего цвета, в то время как ее соли имеют красный цвет. Краситель конго красный применяется как индикатор на минеральные кислоты, так как органические кислоты в большинстве случаев слишком слабы, чтобы вытеснить кислоту конго из ее солей.
Бензопурпурин получают сочетанием диазотированного о-толидина с нафтионовой кислотой.
Бензопурпурин окрашивает растительные волокна в красивый цвет кумача, но выкраска не отличается прочностью.
Особую ценность для приготовления азокрасителей, окрашивающих растительные и животные волокна, представляет 1,8-ами-нооксинафгалин-3,6-дисульфокислота, так называемая аш-кислота (Н-кислота):
он nh2 ! I m [ io3$/'V''4^\so3i i
Для получения аш-кислоты сульфируют нафталин в 1,3,6-трисульфокислоту, которую затем нитруют, получая 1-нитро-3,6,8-трисульфокислоту нафталина. Нитросоединение путем восстановления железом превращают в 1-амино-3,6,8-трисульфокислоту. Сплавляя при 180° эту аминосульфокислоту со щелочью, заменяют 8-сульфогруппу на гидроксил и получают таким образом аш-кислоту, отделяемую в виде кислой натриевой соли.
Гидрированные нафтиламины и нафтолы. Нафтиламины и нафтолы при восстановлении натрием в амиловом спирте легко присоединяют четыре атома водорода, причем гидрируется только одно бензольное ядро. Гидрирование р-нафтиламина идет в том ядре, где
находится аминогруппа, и получается тетрагидронафтиламин строения:
СН,
-СИ,
Ввиду нахождения аминогруппы в гидрированном ядре, этот амин обладает свойствами не ароматических, но алициклических аминов, близкими к свойствам жирных аминов. Он является сильным основанием, с азотистой кислотой дает алкоголь, в отличие от ароматических аминов не образует диазосоединений.
При гидрировании а-нафтиламина атомы водорода, наоборот, присоединяются к углеродным атомам другого бензольного ядра, так что получается соединение
nh2
сн3 I
Н2С\ /\^ сн2
в котором аминогруппа находится в негидрированном бензольном ядре. Поэтому такой тетрагидронафтиламин обладает характером анилина. Бамбергер, исследовавший эти продукты, назвал первое вещество алициклическим тетрагидро^-нафтиламином (at-тетра-гидро-Р-нафтиламин), второе—ароматическим тетрагидро-я-нафтил-амином (а/'-тетрагидро-а-нафтиламин).
а-Нафтол при аналогичном восстановлении дает аг-тетрагидро-а-нафтол
он сн2 I
н2с
тогда как толов:
^п2
из р-нафтола получается смесь аг- и ас-тетрагидро-р-наф-
Ек 2 л zOf 1
CI12
' \:н,-он
2
н2сч У сна
ог-Тетрагидро-а-нафтол—типичный фенол. ас-Тетрагидро-р-наф-тол обладает свойствами гидроароматических алкоголей.
Нафтохиноны. Для нафталина возможны и известны три хинона:
а-нафтохллон (1,4-нафтохлнон)
^-нафгохинон (1,2-нафт охилон)
ал<ф/л-нафтохинон (2,6-нафгохинон)
а-Нафюхинон аналогичен n-бензохинону, а р-нафтохинон— о-бензохинону; третий «двухъядерный» хинон представляет собой особое характерное сочетание пара-хиноидных связей в различных бензольных ядрах.
%-Нафтохинон, как и бензохинон,—летучее желтое вещество с острым запахом (иглы; темп, плавл. 125°). Получается он при окислении хромовой кислотой а-нафтиламина, лучше 1,4-диоксинафталина (нафтогидрохинона) или 1,4-аминонафтола; при восстановлении легко превращается в нафтогидрохинон.
3-Нафтохинон получается при окислении 1,2-диаминонафтола и образует красные иглы, разлагающиеся при 120°. Он обладает свойствами а-дикетонов—сернистой кислотой восстанавливается в 1,2-диоксинафталин.
Для производных а- и (3-хинонов особенно отчетливо проявляется таутомерия бензоидных и хиноидных форм. Так, в тех случаях, когда должны получаться нафтохинонмоноксимы, в действительности образуются изомерные им нитрозонафголы, получаемые при действии азотистой кислоты на нафтолы:
N—O
NOH
^\А/он Ш
3-Нитрозо-х-нафтол (желтовато-зеленые иглы, темп, плавл. 152°) и а-к«-трозо-$-нафтол (темнооранжевые кристаллы, темп, плавл. 110°) образуют с некоторыми металлами характерные цветные осадки внутрикомплексных солей А. Ильинский):
O*-N—Me
Поэтому они находят широкое применение в аналитической химии как реактивы на ионы кобальта, железа, меди и других металлов.
Благодаря таутомерии, монофенилгидразоны а- и р-нафтохино-нов тождественны оксиазосоединениям, получаемым азосочетанием диазотированного анилина с а- и р-нафтолами, например:
ОН
N=N—СвИв
Амфи-нафтохинон получается окислением амфи-диоксинафта-лина (2,6-диоксинафталина) перекисью свинца. Это очень нестойкое вещество, кристаллизующееся в оранжево-красных иглах. По химическим свойствам он напоминает я-бензохинон (является сильным окислителем, выделяет иод из йодистого водорода и пр.), но нелетуч и не имеет запаха.
Оксинафтохиноны представляют интерес потому, что некоторые из них находят применение как красители, а некоторые являются природными красящими веществами.
5,8-Диокси-а-нафтохинон, или нафтазарин
ОН О
[ОН б
применяется как протравной краситель, образующий черно-фиолетовый лак с гидратом окиси хрома. Его получают, обрабатывая 1,5-динитронафталин (или его смесь с 1,8-динитронафталином) олеумом, в котором в качестве восстановителя растворена сера. По окончании реакции нафтазарин осаждают водой. Он образует коричневые, с металлическим блеском, иглы; растворим в щелочах с синей окраской.
В шелухе недозрелых лесных орехов содержится 5-окси-а-нафто-хинон, или юглон
ОН О
образующий коричнево-красные кристаллы (темп, плавл. 155°).
В настоящей хне (хенна, из индийского растения kawsonia alba} содержится 2-окси-а-нафтохинон, или лаусон\
ОН
Пигмент туберкулезных бацилл, фтиокол, представляет собой 2-метил-З-окси-я-нафтохинон (желтые кристаллы; темп, плавл. 173°)
О
Карбоновые кислоты нафталина. Одноосновные а- и g-кислоты, называемые нафтойными кислотами, могут быть получены окислением гомологов нафталина, из нитрилов (синтезируемых сплавлением солей сульфокислот нафталина с цианистым калием), а также другими обычными способами получения карбоновых кислот.
а-Нафтойная кислота плавится при 160°, при нагревании теряет СО2, образуя нафталин. Нафтойная кислота плавится при 185° и перегоняется без разложения выше 300°. Константа диссоциации (при 25°) а-кислоты К=2,04-10~4, g-кислоты /(=6,8-10~5.
Из двухосновных карбоновых кислот нафталина особенно интересна 1,8-кислота, или пери-нафталиндикарбоновая кислота (так называемая перикислота) \
НООС соон
66
Она легче всего может быть получена окислением содержащегося в каменноугольном дегте углеводорода аценафтена (темп, плавл. 95°; темп. кип. 277,5°):
Н2С—СН2
60
Аценафтен промежуточно окисляется в аценафтенхинон (желтые иглы; темп, плавл. 26Г):
О=С—С=О
. м
Это соединение, несмотря на свое название, не является хиноном, а проявляет свойства обычного дикетона (с бисульфитом, например, дает продукт присоединения). При дальнейшем окислении его получается перикислота.
Пери-нафталиндикарбоновая кислота, как и о-фталевая кислота, легко образует ангидрид
О
O=Q/ ^С=О
имид и пр.
Важное практическое значение имеет 2-окси-З-нафтойная кислота, получаемая синтетически из нафтолята натрия и С02 под давлением. Ее анилиды (так называемые азотолы) фиксируются из щелочного раствора на хлопчатобумажные ткани и при обработке растворами диазосоединений дают на самом волокне чрезвычайно прочные окраски. Например, с анилином получается азотол А (нафтол АС):
/ОН
\conhc0h5
Некоторые частично гидрированные производные*' нафталина, встречающиеся в природных эфирных маслах и бальзамах, относят обычно к бициклическим сесквитерпенам (стр. 158).
СТЕРЕОХИМИЯ ПРОИЗВОДНЫХ НАФТАЛИНА
У некоторых двузамещенных производных нафталина с заместителями при соседних атомах углерода или в пери-положении наблюдается своеобразная оптическая изомерия. Если такие заместители достаточно велики, свободное вращение их вокруг оси связи с ядром станет невозможным и молекула может лишиться плоскости симметрии, проходящей через плоскость ядра нафталина. Таким образом возникает возможность оптически антиподного расположения заместителей в пространстве, т. е. атропизомерия, сходная с атро-пизомерией бифенила (стр. 416).
Построенная Миллсом модель (рис. 6) для пери-производных нафталина типа
Х'ХХА\
показывает невозможность свободного вращения заместителя, находящегося в положении 1, если этот заместитель достаточно велик, например, если он имеет строение
HOOC-CH2-N--SO2—С6Нб
Следует иметь в виду, что при построении Миллсом модели расстояния между атомами углерода, кислорода и азота были приняты равными 1,5 А В действительности же расстояния между углеродными атомами о
(1,39 А), а следова1ельно, npoci ране i вен ное расположение заместителей еще более тесное.
Основываясь на этом выводе, Миллс и Эллиот (1928 г.) произвели увенчавшийся успехом опыт— при помощи солей бруцина они расщепили на оптические антиподы производное 1,8-нит сснаф-тиламина следующего строения:
/SO2 С6Нб
O2N
:н2—соон
в бензольном ядр^ меньше
Принцип атропизомерии был применен Мей-зенгеймером для абсолют него определения конфигурации некоторых оксимов нафталинового р-ла. Из двух оксимов 1-апето-2-оксинафтойной 3-кислоты на оптические изомеры (хотя и быстро рацемизуюшиеся) оказалось возможным разделить только ^-оксимы Следовательно, пространственные затруднения у Р-оксима больше, чем у а-оксима, а потому ему должно быть приписано cww-строение:
он
а-оксим
3-оксим
Однако, хотя а-оксим сам не разделяется на оптические изомеры, его N-метиловый эфир, имеющий конфигурацию, соответствующую а-оксиму, все же может быть расщеплен, так как вращение группы, находящейся в положении 1, оказывается стерически невозможным:
О t
H3C N
С
N-метиловый эфир а-оксима
сн8
ОН
соон
Инден
Инден—жидкий углеводород каменноугольного дегтя, кипящий при 182°. Подобно нафталину, это—соединение с двумя конденсированными ядрами, но второе ядро является не бензольным, а пятичленным ядром циклопентадиена:
-сн
СН2
Водородные атомы группы СН2, как и в циклопентадиене и флуорене (стр. 63 и 423), способны замещаться на щелочные металлы. Аналогично циклопентадиену, инден может конденсироваться с альдегидами и кетонами. Инден и многие его производные получены синтетически.
При гидрировании водородом в присутствии никеля (при 200°) или платины (при комнатной температуре) инден присоединяет два атома водорода, образуя гидр-инден, или индан (темп. кип. 177°; п2^= 1,5381; d24Q = 0,9645):
При более высокой температуре гидрируется уже и бензольное ядро и образуется гидриндан (стр. 105).
Группа антрацена
Антрацен С14Н10 содержится в количестве 0,25—0,45% в каменноугольном* дегте, а именно в антраценовом масле, кипящем выше 270° (стр. 221).
Выделенный из твердой смеси углеводородов «сырой» антрацен (30—50%-ный) очищают кристаллизацией из пиридина или сольвентнафты и перегонкой с водяным паром. Совершенно чистый антрацен кристаллизуется в бесцветных призмах с голубей флуоресценцией; темп, плавл. 217°, темп кип. 354°. Он легко растворяется в горячем бензоле, трудно—в спирте и эфире, нерастворим в воде. Как и нафталин, он образует пикрат (с одной молекулой пикриновой кислоты), имеющий темп, плавл. 138°.
Под действием прямых солнечных лучей антрацен полимеризуется в диантрацен; в темноте происходит обратное превращение в антрацен (обратимая фотохимическая реакция):
на свету 2С14Н10 ----С28Н20
в темноте
Строение антрацена вызывало много споров. Одно время антрацену придавали то строение, которое в действительности принадлежит фенантрену (стр. 480).
* Название антрацен произошло от греческого слова антракс—уголь.
Антрацену придавались следующие формулы:
иФи 033' формула Гребе и Либермана" формула Гинсберга
В качестве подтверждения правильности формулы Гребе и Либермана приводился синтез антрацена из бензола и тетрабромэтана в присутствии бромистого алюминия (синтез Аншютца):
СН
Л-ен-вг + П уч г .
Vх Вг-СН-Вг Vх %/\ zA> сн
Однако этот синтез не может служить доказательством, так как реакции конденсации под действием галоидных солей алюминия часто протекают с разрывом связей между атомами углерода. Помимо чисто химических соображений, эта формула должна быть отвергнута и потому, что связь между углеродными атомами, находящимися, как в этом случае, на расстоянии 2,85 А, невозможна.
Формула Гинсберга хорошо согласуется с такими физико-химическими данными, как, например, результаты исследований спектра поглощения и особенностей рефракции. Эту формулу иногда называют также «хиноидной», потому что наряду с бензольным кольцом в ней имеется система четырех сопряженных двойных связей, расположенных по тому же типу, как и в орто-хиноне:
Углеродные атомы 9 и 10 являются, таким образом, концами цепи сопряжения двойных связей, вследствие чего антрацен, как типичный диен, может присоединять по атомам 9, 10 малеиновый ангидрид, ацетилендикарбоновый эфир и даже л-бензохинон. Легко реагирует антрацен также с металлическим натрием в эфирной суспензии, образуя темносинее двунатриевое производное дигидроантрацена— 9,10-динатрийдигидроантрацен:
Na I сн сн
юю............-осю
СН сн
I
Na
* Карл Либерман (1842—1914). Немецкий органик. Совместно с К. Гребе в 1869 г. установил строение ализарина и осуществил его синтез.
Такие продукты присоединения при действии воды, спиртов и т. п. дают соответствующие производные дигидроантрацена, с углекислотой образуют антрацендигидродикарбонивые кислоты.
. С хиноидной формулой ан।ранена согласуется также го, что при синтезах антрацена и его производных чаще всего сначала получается дигидроантрацен, который уже за1ем превращается соответствующими реакциями в антрацен. Например, при действии натрия на о-бромбензилбромид получается дигидроантрацен
при окислении превращающийся в антрацен:
Особенно хорошо согласуется с приведенной формулой синтез антрацена через о-бензоилбензойную кислоту и антрахинон (Бер и Ван-Дорп, 1874 г.). Конденсацией фталевого ангидрида с бензолом получается о-бензоилбензойная кислота (стр. 376), с замыканием цикла дающая антрахинон
который при восстановлении образует антрацен:
О
Как и в случае нафталина, для изображения строения антрацена ’ асто пользуются упрощенной схемой:
Эта схема позволяет для однозамещенных производных предвидеть три изомера: замешенные в положениях 1, 4. 5 и 8 (а-положе-ние), в положениях 2, 3, 6 и 7 ((3-положение) и. наконец, в положениях 9 и 10 (у- или ^-положение, называемое также зжзо-положением и обозначаемое буквами ms) При наличии нескольких заместителей их положение обозначается цифрами.
Положение заместителей в системе антрацена часто может быть определено, кроме синтезов, также и следующим образом.
При окислении антрацена и его производных легко получается антрахинон и его производные, при сплавлении с едкими щелочами антрахинон и его замещенные производные могут распадаться по уравнению:
При этом получаются две молекулы производных бензойной кислоты, по строению которых и можно судить о строении производных антрацена.
Формально антрацен можно рассматривать как линейную комбинацию грех шестичленных циклов, как бы трех бензольных колец. Будучи плоским образованием, он представляет собою ароматическую систему, однако еще более, чем нафталин, отличающуюся от бензола. Так, если в бензоле все атомы углерода с одинаковой легкостью обменивают свои водородные атомы, а в нафталине а- и р-атомы уже различаются по реакционной способности, то в антрацене различие реакционной способности отдельных атомов выражено еще сильнее. Наиболее уязвимыми местами системы антрацена оказываются углеродные атомы среднею кольца—мезо-атомы. Как реакции присоединения, так и замещения легче всего идут в среднее ядро антрацена, т. е. в мезо-положение.
При осторожном восстановлении антрацена легко получается дигидроантрацен (кристаллы; темп, плавл. 108,5®; темп. кип. 313®):
При исчерпывающем восстановлении получается пергидроантрацен С14Н24 (темп, плавл. 89°, темп. кип. 270е).
Галоиды также сначала присоединяются к мезо-углеродным атомам (с образованием 9,10-дихлор- или 9,10-дибромдигидроантраце-на), а после отщепления галоидоводородной кислоты получаются 9-хлор- и Q-бромантрацен.
Концентрированная серная кислота сульфирует антрацен не в мезо-положениях, а в крайних ядрах, причем легко получается смесь дисульфокислот, содержащих сульфогруппы в а-положениях:
SO3H SO..H 8О.,Н
qco ссо
SO3H
1,5-дисульфокислста 1,8-дисульфокислота
антрацена антрацена
При низкой температуре образуется преимущественно 1,8-кислота, при высокой—главным образом 1,5-кислота.
Более разбавленная серная кислота при нагревании дает £-ан-траценсульфокислоту и две дисульфокислоты с сульфогруппами в ^-положениях:
При сплавлении сульфокислот со щелочами можно получить оксисиединения— -ант ролы, имеющие характер фенолов.
Азотная кислота окисляет антрацен в антрахинон. В более мягких условиях (в ледяной уксусной кислоте) получается 9-нитро-антрацен
no2
который может быть также получен последовательным дейсишем низших окислов азота (образуются продукты присоединения) и затем едкой щелочи:
н no2 NO,
ОСО - До =' ХС' н no2
Восстановлением 9-нитроантрацена может быть получен 9-антрамин, или мезоантрамин (темп, плавл. 145—150°). Нерезкая температура плавления и некоторые превращения позволяют предположить его таутомерию с антронимином'.
NH2 NH
ООО=600
сн,
мезоантрамин
антронимин
Под действием азотистой кислоты 9-антрамин отчасти превращается в антрахинон» отчасти в другие, более сложные продукты, но не образует диазониевых соединений.
1-Антрамин и 2-антрамин (1- и 2-аминоантрацены) получаются нагреванием с аммиаком под давлением соответствующих антролов или восстановлением аминоантрахинонов. Это—желтые кристаллические вещества, обладающие очень слабыми основными свойствами; их соли способны диазотироваться.
Антрахинон. Наиболее важным производным антрацена является антрахинон С14НЯО2, легко получаемый окислением антрацена. Раньше его получали окислением антрацена хромовой смесью (смесью двухромокислых солей с серной кислотой) и отделяли от примесей, растворяя в концентрированной серной кислоте, из которой он вновь выпадает при разбавлении водой. Теперь значительные количества антрахинона получают синтетически—из фталевого ангидрида и бензола.
Антрахинон кристаллизуется в светложелтых, легко возгоняющихся кристаллах (темп, плавл. 286°; темп. кип. 382°). Это очень устойчивое соединение, лишь с трудом поддающееся окислению, сульфированию, нитрованию и пр.
Строена? антрахинона выражается формулой:
О
II
Г'
Расположение карбонильных групп в молекуле антрахинона дока зывается следующим образом. Ангидрид бромфталевой кислоты с бензолом в присутствии хлористого алюминия дает бромантрахи-
о
Заменой брома на гидроксил (нагреванием с едким кали при 160°) получается оксиантрахинон, который при окислении дает фталевую кислоту:
Н00С\^
С1 va3J ~* Та
ноос
о
Следовательно, карбонильные группы антрахинона находятся в обоих бензольных ядрах в орто-пиложениях. Так как восстановлением антрахинона можно получить антрацен, то очевидно, что и в антрацене оба мезо-атома углерода связаны с углеродными атомами обоих крайних бензольных ядер в орто-положении, т. е. формально антрахинон представляет собою п-хинон. Однако, вследствие особенностей своего строения (оба крайних бензольных ядра являются настоящими бензоидными ядрами), он не имеет обычных свойств п-хинонов: нелетуч, не имеет запаха, не является окислителем. Свойства же, характерные для кетонов, проявляются в нем более отчетливо, чем в хинонах. Так, с гидроксиламином он дает оксим:
NOH
хо
II о
Этот же оксим получается изомеризацией 9-нитроантрацена под действием спиртового едкого кали (реакция Мейзенгеймера):
NOa NOH
(XX - ОСО
При сплавлении с едким кали антрахинон распадается на две молекулы бензойной кислоты:
Ф-*^соок\оосаО
о
Антрахинон можно бромировать, нитровать и сульфировать. При сульфировании антрахинона замечательную роль в качестве катализатора могут играть соли ртути (М. А. Ильинский*). Тогда как без ртути при сульфировании получается почти исключительно Р-сульфокислота, при прибавлении небольшого количества ртути образуется главным образом а-сульфокислота. Подобное влияние ртуть оказывает и на другие процессы замещения в антрахиноне. Возможно, что при этом сначала происходит меркурирование антрахинона сульфатом ртути в a-положении и лишь затем группа Hg+|HSO4|~“ вытесняется серной кислотой с регенерацией катализатора—сульфата ртути.
* Михаил Александрович Ильинский (1856—1941). Один из крупнейших специалистов в области синтеза антрахиноновых красителей. Почетный член Академии наук СССР.
Нитросоединения антрахинона очень легко обменивают нитрогруппу на сульфогруппу при нагревании со средними солями сернистой кислоты.
Сульфогруппы в сульфокислотах антрахинона отличаются сравнительной подвижностью. Они легко обмениваются на гидроксил, а при нагревании с аммиаком—и на аминогруппу (с образованием аминоантрахинонов).
Антрагидрохинон. При восстановлении антрахинона цинковой пылью с едким натром получается двунатриевый алкоголят антра-гидрохинона (оксантранола):
ОН
I ОН
В свободном виде антрагидрохинон образует коричневые кристаллы (темп, плавл. 180°). Его водные растворы обладают сильной зеленой флуоресценцией; при подщелачивании они приобретают темнокрасный цвет, а на воздухе обесцвечиваются, так как вновь образуется антрахинон. Эта реакция может быть использована как чувствительная качественная реакция на антрахинон.
При окислении антрагидрохинона кислородом воздуха образуется эквимолекулярное количество перекиси водорода:
Н~О
н-А
Образование перекиси водорода и других перекисных соединений часто наблюдается при процессах окисления органических соединений кислородом воздуха («активирование» кислорода). Эта реакция может быть использована для ^полу-чения высококонцентрированной, почти безводной перекиси водорода.
Антрагидрохинон таутомерен оксантрону\
В холодном спиртовом растворе в присутствии НО происходит взаимное превращение обоих веществ, причем равновесие наступает,
когда в растворе содержится 97% антрагидрохинона и 3% оксантро-на. Оксантрон бесцветен, не флуоресцирует в растворах; плавится при 167°.
При восстановлении антрахинона оловом и соляной кислотой получается антрон, находящийся в отношениях таутомерии с антра-нолом, или 9-оксиантраценом:
О ОН
а ।
антрон антранол
Антрон—бесцветные иглы, плавящиеся при 163°; антранол— коричнево-желтые листочки; при медленном нагревании плавится около 150\ Антранол образуется при кипячении антрона в щелочных растворах и, в меньшем количестве, при кипячении его в уксуснокислом растворе. В кислых растворах может происходить обратное превращение антранола в антрон.
При энергичном восстановлении антрахинона и продуктов частичного восстановления его карбонильных групп, например при перегонке их с цинковой пылью, получается антрацен.
ТАУТОМЕРИЯ ПРОИЗВОДНЫХ АНТРАЦЕНА
'Для мезо-гомологов антрацена наиболее ясно обнаруживаются явления углеводородной бензоидно-хиноидной гаутемсриз (cip.,357):
Однако таутомерия мезо-гомологов антрацена отличается от других ранее рассмотренных случаев бензоидно-хиноидной таутомерии одной особенностью—атом водорода мигрирует через цикл. Этот вид таутомерии иногда называют транс-аннулярной таутомерией^^. стр.34).
Особенно наглядные примеры транс-аннулярной таутомерии наблюдаются у некоторых мезо-алкилантраценов. Так, для 9-метил-1,5-ди-хлорантрацена удалось получить обе формы, легко превращающиеся друг в друга:
АМИНО- И ОКСИПРОИЗВОДНЫЕ АНТРАХИНОНА
Эти производные антрахинона имеют большое значение в технологии так называемых ализариновых красителей. Некоторые оксипроизводные (содержащие оксигруппы в мета-положении друг к другу) применяются в медицине как слабительные.
Оксипроизводные антрахинона легко получаются из его сульфокислот.
Кроме того, эти производные, особенно многоатомные оксисоединения, можно получать взаимодействием фталевого ангидрида или его замещенных производных с многоатомными фенолами, например:
ализарин
< 1,2-диоксиантрахинон)
Ализарин в свое время был важнейшим красителем после индиго. До 1869 г его приготовляли из корня растения, называемого краппом или мареной (Rubia hnctorui), которое разводили в большом количестве в Южной Европе,
В соке корней марены содержится несколько глюкозидов, из которых важ* с/ ’им является рубэритриновая кислота:
О
СН2- (СНОН)з— СН — О
I___—О------[
Этот глюкозид под влиянием энзимов или серной кислоты гидролизуется, 1авая ализарин и две молекулы сахаров (d-глюкозу и d-ксилозу).
Из других глюкозидов образуется 1,2,4-триоксиантрахинон—пурпурин.
Гребе и Либерман в 1865 г. установили, что ализарин является производным антрацена. Они сделали этот вывод на том основании, что антрацен образуется при перегонке ализарина с цинковой пылью, а затем подтвердили его синтезом ализарина (а также изомерного ему
гистазарина} из фталевого ангидрида и пирокатехина. Таким образом было установлено нахождение обеих гидроксильных групп в одном и том же бензольном ядре в орто-положении. Оставался лишь выбор между двумя формулами: О ОН
ОН
О
ОН
ОН
о
о
Окончательное заключение в пользу первой формулы для ализарина и второй—для гистазарина было сделано на том основании, что первое строение допускает образование двух мононитросоединений, в которых нитрогруппа находится в том же ядре, что и гидроксилы, а второе—лишь одного такого нитросоединения.
Вскоре Гребе и Либерманом был разработан метод получения ализарина из антрахинона.
Искусственное получение ализарина было первым техническим синтезом природного красителя, имевшим большое экономическое значение. Оно привело к прекращению культивирования марены.
Ализарин кристаллизуется в красных иглах (темп. плавл. 290°), легко возгоняется. В воде ализарин почти нерастворим, немного растворяется в спирте; как фенол он легко растворяется в едких щелочах, давая интенсивно окрашенные красно-фиолетовые растворы.
В технике ализарин получают по способу, предложенному еще Каро* и Перкиным**, т. е. сплавлением со щелочами ^-антрахинон-сульфокислоты. При этом происходит не только замена сульфогруппы на гидроксил, но и окисление второго углеродного атома того же бензольного зарина:
ядра и образование двунатриевого производного али-
О
SO3Na
+ 3NaOH + О
О
О
О ONa
ONa
+ Na2SO3 + 2HtO
* Генрих Каро (1834—1910). Немецкий химик-органик; впоследствии предприниматель и организатор немецкой химической промышленности.
** Виллиам Перкин старший (1839—1907). Английский химик и предприниматель
При нитровании ализарина получается главным образом 1,2-диокси-З-нитро-антрахинон
О ОН
О
долгое время применявшийся (с алюминиевой протравой) как краситель под названием ализарин оранжевый. При нагревании его с серной кислотой и глицерином получается ализарин синий БС, относящийся к производным хинолина (см. синтез Скрауна, стр. 612).
Син’.езированы и другие диокси- и полиоксиантрахиноны; многие из них применяются как весьма ценные красители. Например, действием фталевого ангидрида на гидрохинон получается 1,4-диоксиантрахинон—хинизарин
О ОН
В технике его получают окислением антрахинона серной кислотой при нагревании в присутствии борной кислоты.
Окисление этим способом называется реакцией Бона—Шмидта. Борная кислота образует сложные эфиры с получающимися при окислении фенольными гидроксилами, предохраняя их таким образом от дальнейшего окисления. Реакция Бона—Шмидта довольно широко применяется для введения гидроксильных групп в различные производные антрахинона.
Конденсацией бензойной и галловой кислот под действием серной кислоты получают 1,2,3-триоксиантрдхинюн, или антрагаллол:
Пурпурин (1,2,4-триоксиантрахинон) получается окислением ализарина перекисью марганца и серной кислотой.
Флавопурпурин (1,2,6-триоксиантрахинон) и антрапурпурин (изопурпурин, «ли 1,2,7-триоксиантрахинон) получаются из соответствующих антрахинондисульфокислот сплавлением с едким натром и бертолетовой солью. Первый краситель дает с алюминиевой протравой красный цвет с оранжевым оттенком, второй— алый цвет.
При сплавлении $-аминоантрахинона с едким кали и последующем окислении получается кубовый краситель индантрен, являющийся одним из самых красивых и прочных синих красителей.
Индантрену придают формулу с гетероциклическим (дигидроазиновым) кольцом:
При более высокой температуре в присутствии воды и воздуха получаете желтый краситель флавантрен:
О
II
V'X/XZ
Его получают в технике путем обработки (З-аминоантрахинона пятихлористой сурьмой в кипящем. нитробензольном растворе. Как кубовые красители применяются также галоидные и другие производные индантрена.
ПРИРОДНЫЕ ПРОИЗВОДНЫЕ АНТРАХИНОНА
Производные антрахинона широко распространены в природе. Например., действующим началом различных лекарственных растений, применяемых в качестве слабительного, являются производные 1,8-диоксиантрахинона. Из них наиболее важными являются эмодин алоэ, эмодин крушины и хризофановая кислота из ревеня:
хризофановая кислота
Получаемый из дерева Andira araroba (семейство бобовых) хризаробин применяется как средство против кожных болезней.
Хризаробин (метилдиоксиантранол) ОН ОН ОН оса.
L3 является продуктом восстановления хризофановой кислоты, которая образуется при окислении его кислородом воздуха.
Во многих грибах Boletus, в том числе в чрезвычайно ядовитом сатанинском грибе Boletus satanas, напоминающем по виду белый гриб, содержится пурпу-рин-а-карбоновая кислота—болетол:
ОН О СООН
НО
он о
Самки червецов Coccus ilicis, живущих в Южной Европе на некоторых видах дуба, содержат в своем организме кермесовцю кислоту'.
СН3О он
Кермесовая кислота является главной составной частькГ/о’рл/тз, или дубовой кошенили, применявшейся еще в древности в качестве красителя Кермес после открытия Америки был вытеснен в Европе мексиканской кошенилью—высушенными самками насекомого Coccus cacti, живущего на кактусах. Кошениль содержит карминовую кислоту
СН3 О ОН
11 Л /СО-(СНОН)4-СН,
НООС о он
алюминиевый лак которой и до сих пор применяется в живописи под названием кармина.
АЛИЗАРИНОВОЕ КРАШЕНИЕ И ПРОТРАВНЫЕ КРАСИТЕЛИ
Ализарин, как и другие полиокси- и аминооксисоединения антрахинона, не является непосредственным красителем, но с окислами тяжелых металлов он образует лаки—окрашенные соединения, прочно фиксируемые волокном. Такие красители называются протравными.
Ализарин дает с окисью алюминия и с окисью олова пунцовокрасные лаки, с окисью хрома—фиолетово-коричневый, с окисыс
железа—фиолетово-черный лак. Ализариновые красители являются одними из важнейших протравных красителей. Большая часть их отличается чрезвычайной прочностью по отношению к свету, к стирке и пр.
Ализариновое крашение обычно производится следующим образом. Ткань •обрабатывают раствором так называемого ализаринового масла*, высушивают и после высушивания пропитывают раствором соли (например, уксуснокислого алюминия). Затем ее обрабатывают щелочным раствором ализарина или другого ализаринового красителя, после чего пропускают пар в ьанну и промывают окрашенную ткань мылом и водой.
Способность к образованию красящих лаков часто связана с наличием в молекуле ализаринового красителя двух и более гидроксильных групп в орто-положении (К. Либерман и Костанецкий). В дальнейшем были обнаружены случаи участия в образовании лаков не только оксигрупп, но и аминогрупп в орто-положении, а затем этих же групп в пери-положении (для производных нафталина) и пр.
Образование лаков связано с возникновением комплексных соединений тяжелых металлов (Л. Чугаев), в которых атом металла входит в структуру пяти- или шестичленных циклов, например:
Me
'(Здесь Me обозначает один эквивалент металла: Sn, Си, Al, Fe и т. п.)
Группа фенантрена
Фенантрен С14Н1# содержится в каменноугольном дегте вместе с антраценом, изомером которого он является. Он кристаллизуется в блестящих табличках (темп, плавл. ЮГ; темп. кип. 340.2°), легко растворим в эфире и бензоле, труднее—в спирте. Растворы имею! голубую флуоресценцию.
Количество фенантрена, которое можно выделить из каменноугольной смолы, даже превосходит содержание в ней антрацена. Однако фенантрен до сих пор не нашел широкого технического применения.
* Касторовое масло, обработанное серной кислотой и содержащее сернокислый эфир оксиолеиновой кислоты
yOSO2OH
6*17
\соон
образующейся из ненасыщенной рицинолеиновой кислоты.
Строение фенантрена изображается формулой:
причем второй способ изображения является в настоящее время
более ynoi реби гельным.
Эта формула доказывается прежде всего окислением фенантре-
на в дифеновую кислоту
\/\ соон соон
Кроме того, она подтверждается рядом синтетических реакций. Так, фенантрен образуется при пропускании паров о-дитолила, дибензила и стильбена через раскаленные трубки:
дибензнл
Синтез фенантрена из дибензила над платиновым контактом при 300° представляет даже препаративный интерес.
Как и антрацен, фенантрен обладает более слабым ароматическим характером, чем нафталин, а тем более, чем бензол. Хотя молекулу фенантрена можно было бы представить как сочетание трех колец с тремя сопряженными двойными связями в каждом, но в действительности электронная плотность не распределяется столь равномерно. Ароматическая система среднего кольца здесь настолько нарушена, что связь между девятым и десятым атомами углерода приобретает в некоторой степени характер обычной двойной связи. Сюда в первую очередь направляется действие атакующих реагентов: окислителей, брома, азотной кислоты. Углеродные атомы 9 и 10 молекулы фенантрена наиболее легко вступают в реакции как присоединения, так и замещения.
Фенантрен можно рассматривать также как производное нафталина, несущее боковую цепь с системой двух сопэяженных двойных связей. Так, металлический литий присоединяется к фенантрену в положении 1,4:
Одним из наиболее важных синтезов производных фенантрена является синтез Пшорра, позволивший получить ряд соединений,
являющихся продуктами превращения некоторых природных алкалоидов (особенно группы морфина).
В простейшем случае Пшорр конденсировал по Перкину (стр. 407) о-нитробензальдегид с фенилуксусной кислотой и получил таким образом а-фенил-о-нигрокоричную кислоту:
СООН
Восстановлением нитропроизводного получается аминокислота, которая после диазотирования выделяет в присутствии порошкообразной меди N2 и образует 9-фенантренкарбоновую кислоту:
СООН
При нагревании эта кислота отщепляет СО2 и дает фенантрен.
Применяя замещенные производные о-нитробензальдегида и фенилуксусной кислоты, можно получить ряд производных фенантрена.
Из метилового эфира о-нитрованилина и фенилуксусной кислоты был получен метиловый эфир двухатомного фенола ряда фенантрена, являющегося продуктом превращения морфина и называемого морфолом:
ссн3
ch3ox/!x/no2
I II +1
^/\сно ^2
\зоон
СН3О СН О ' "
I II I 'Ч/\//\соон
метиловый эфир морфола
Фенантренхинон. Наиболее важным производным фенантрена является фенантренхинон
кристаллизующийся в оранжевых иглах (темп, плавл. 208°). Фенантренхинон не имеет запаха и нелетуч с парами воды. Он получается окислением фенантрена хромивой кислотой в ледяной уксусной кислоте. Фенантренхинон и его производные могут быть получены также сплавлением с хлористым алюминием бензила (стр. 434) и его производных:
+ Н2
Фенантренхинон является а-дикетоном; при действии гидроксиламина он дает моноксим и диоксим. Для моноксима обнаруживается
таутомерия:
Фенантренхинон часто применяется как реактив на о-диамины (стр. 362), с которыми он образует гетероциклические соединения, относящиеся к ряду хиноксалина (стр. 627), например:
Известны многочисленные производные фенантрена и фенан-тренхинона, а также ряд продуктов присоединения водорода к фенантрену.
Циклопентенофенантрен. Химия фенантрена, более сложными производными которого являются стероиды (стр. 165), привлекает
в настоящее время большое внимание органиков и усердно разрабатывается.
Синтез 1,2-циклопентенофенантрена (скелет его лежит в основе молекул всех стероидов) был осуществлен таким образом. Из 2-(а-нафтил)-этилхлорида и циклопентанона гриньяровской реакцией был получен карбинол (I), который дегидратировали действием бисульфата калия в а-нафтилэтил-Д1-циклопентен (II). В результате его внутримолекулярного алкилирования с образованием нового цикла получается 1,2-циклопентано-1,2,3,4-тетрагидрофенан-трен (III), при дегидрировании дающий 1,2-циклопентенофенан-трен (IV):
ОН
Сложные конденсированные ароматические углеводороды
Сложные системы с конденсированными бензольными кольцами представляют трудно обозримое многообразие. Здесь можно лишь бегло отметить некоторые из них.
Многоядерные линеарные системы
Если бензольные кольца комбинируются друг с другом так, что их центры оказываются лежащими на одной прямой, получаются так называемые линеарные системы (линеарное аннелирование колец). Простейшей из них является уже рассмотренный нами антрацен (стр. 466), который можно также назвать 2,3-бензнафталином. Следующим членом ряда таких систем будет 2,3-бензантрацен, называемый обычно нафтаценом, иногда также тетраценом'
Он может быть получен конденсацией 1,4-нафтогидрохинона с фталевым ангидридом под действием хлористого алюминия и последующим восстановлением (перегонкой с цинковой пылью) полу
ченного9,11-диоксинафтацен-10,12-хинона. Нафтацен окрашен в красно-желтый цвет, плавится при 343° и является значительно более реакционноспособным веществом, чем антрацен.
Частично гидрированная система тетрацена лежите основе строения важнейших антибиотиков—террамицина, биомицина, тетрациклина и др. (см. раздел об антибиотиках).
Конденсацией хлористого бензоила с At-ксилилфенилкетоном, последующей внутримолекулярной конденсацией полученного дикетона и дегидрогенизацией с образованием бензольных колец был получен 2,3,6,7-дибензантрацен, или пентацен (сине-фиолетовые иглы):
пентацен
Пентацен обладает такой высокой реакционной способностью, что для него даже было предложено строение бирадикала.
Весьма реакционноспособным соединением является также гексацен (темнозеленые со стальным блеском иглы), получаемый из 1,5-диоксинафталина по следующей схеме:
О О
II ОН II
С I с
о он о
12 13 14 15 h 1
гексацен
Рубрен—оранжево-красные кристаллы, плавящиеся при ЗЗГ,— представляет собою 9,10,11,12-тетрафенилнафтацен. Он может быть получен из 9,11-диоксинафтацен-10,12-хинона двукратным взаимо-
действием с фенилмагнийбромидом и последующим восстановлением диоксидигидрорубрена:
рубрен
Окрашенные, обладающие желтой флуоресценцией растворы рубрена в бензоле поглощают на свету кислород (один моль кислорода на моль рубрена), образуя бесцветный оксирубрен, хорошо кристаллизующийся с половиной моля бензола. Оксирубрен при нагревании (не выше 140°) отдает более 80% поглощенного кислорода, снова превращаясь в рубрен. Таким образом, он мог бы рассматриваться как модель гемоглобина.
Многоядерные ангулярные системы
Системы, в которых бензольные ядра комбинируются так, что на одной прямой лежат центры только части этих ядер, называются, в отличие от линеарных (см. выше), ангулярными (ангулярное анне-лирование колец). Простейшим представителем многоядерных ангулярных углеводородов является фенантрен (стр. 480). За ним следуют хризен (1,2-бензфенантрен) и трифенилен (9,10-бензфенантрен), а затем пицен (1,2,7,8-дибензфенантрен), представляющие собой сравнительно прочные, довольно трудно поддающиеся окислению
настоящие ароматические системы:
трифенилен
пицен
Хризен, бесцветные пластинки с темп, плавл. 255° (желтый цвет обычных препаратов, за который хризен и получил свое название, обусловлен окрашенной примесью), содержится в высших погонах каменноугольной смолы. Трифенилен плавится при 199°, а пицен— при 365°.
Другого типа углеводородами, повидимому не обладающими полностью ароматическими системами связей, являются также встречающиеся в каменноугольной смоле пирен (темп, плавл. 156°) и окрашенный в желтый цвет перилен (темп, плавл. 275°).
Пирен при окислении не дает аналогичного фенантренхинону 4,5- или 9,10-хинона, а образует 1,6-хинон:
О
пирен
пиренхинон
Для перилена характерна способность в нитробензольном растворе присоединять малеиновый ангидрид, что, как известно, типично для диенов с сопряженными связями; здесь такое присоединение идет в положениях 6,7 или 1,12:
перилен
При окислении перилена образуется 3,10-периленхинон
периленхинон
С теоретической стороны представляет большой интерес система из шести кольцеобразно соединенных друг с другом бензольных ядер:
12
7 б
Такой углеводород С24Н12, названный короненом, оказался веществом очень прочным. Коронен, синтез которого был осуществлен Шоллом, образует желтые иглы, плавящиеся при 437—440°. Растворы его обладают сине-фиолетовой флуоресценцией.
Из многоядерных ангулярных углеводородов следует также отметить бензантрен—бесцветные листочки, плавящиеся при 84°. Получается он восстановлением бензантрона (желтые иглы, плавящиеся при 170°), который, в свою очередь, проще всего получается спеканием фенил-а-нафтил кетона с хлористым алюминием:
фенил-а-нафтилкетон бензантрон бензантрен
Щелочным плавлением из бензантрона получаются очень ценные прочные кубовые красители (стр. 375): дибензантрон, или виолантрон (известный под маркой индантрен темносиний)
и изодибензантрон,
Канцерогенные, или карциногенные*, углеводороды
Раковые заболевания характеризуются внезапным нерегулируемым ростом и размножением клеток до тех пор нормально развивавшейся ткани. Существуют представления о том, что вещества, сти-, мулирующие беспорядочный рост клеток, принадлежат к аномальным гормонам, вырабатываемым самим заболевающим организмом.
В поисках веществ, вызывающих появление злокачественных опухолей, было обращено внимание на профессиональные заболевания раком кожи лиц, соприкасающихся с высококипящими фрак-
Канцер—лат. рак; карцинома—раковая опухоль.
циями каменноугольной смолы, с пеком и продуктами его переработки (производство угольных брикетов, кровельного толя и т. п.). Из каменноугольной смолы удалось выделить вещество, обладающее канцерогенной активностью (опыты проводятся на мышах) при нанесении его на кожу. Это вещество оказалось углеводородом 1 Д-бензпиреном
Канцерогенными свойствами обладают также полученные сив тетически 1 ,2-бензантрацен и 1,2,ЬЗ-дибензантрацен'
1,2-бензантрацен (темп. плав^. 160,5е)
1,2,5,6-дибензантрацен (темп, плавл 267,5°)
В дальнейшем была обнаружена высокая канцерогенная активность продукта расщепления желчных кислот (дезоксихолевой и холевой, стр. 180 и 182)—углеводорода, названного метил холан-треном (темп, плавл. 179,5—180,0°):
1 2
Н2С—сн2
8
На этом основании была выдвинута гипотеза, что некоторые виды злокачественных опухолей могут возникать при образовании метилхолантрена (или близких ему веществ) из желчных кислот или из половых гормонов заболевшего организма.
Простейшим из изученных до сих пор веществ, обладающим, хотя и слабым, но определенным канцерогенным действием, является ЪД-бензфенантрен:
Циклофаны. Интересным классом соединений являются циклофаны, содержащие ароматические ятра, объединенные с помощью алифатических цепей в алициклическую макросистему:
ортоаиклофаны
мегаиик.юфаны
ларациклоТаньт
Для обозначения отдельных представителей перед названием циклорана указывают в ква тратных скобках число атомов углерода [т и п) в соединяющих бензольные ядра цепях, например: [2,3|-метациклофан или |6,6|-парациклофан.
Особенно большое теоретическое значение имеют мета- и папани к лофаны, открытые и изученные в самые последние годы. Расчет показывает, что метациклофаны при т—\ и п — 2 и парапикло|эаны при т = п = 3 должны быть сильно напряжены. Была высказана мысль ^Робептсон), что в таких циклоранах бензольные кольца изгибаются тля уменьшения напряженности На примере [2,21-парациклофана это было пот тверже но рентгкюструктурным анализом (Браун, 1953 г.) Оказалось, что в этом соединении расстояния межту соответствующими углеродными атомами двух колец неодинаковы и кольца имеют форму ванны:
Пока это единственный доказанный случай некопланарности бензольного ядра.
Известны аналогичные соединения не только с бензольными, но и с нафталиновыми, антраценовыми и т. п. ядрами.
Небензольные ароматические соединения
При рассмотрении свойств бензола уже отмечалось, что его ароматичность зависит в первую очередь от равномерного распределения электронной плотности между всеми шестью углеродными атомами бензольного кольца (стр. 234). Равноценность всех атомов углерода и всех углерод-углеродных связей в молекуле бензола можно условно выразить схемой:
НС<->СН I ( ; I нс<~>сн сн
или
Если бензол рассматривать формально, просто как циклогекса-триен, то следовало бы ожидать, что ароматическим характером будут обладать и его аналоги—циклобутадиен и циклооктатетраен:
нс-сн
II II нс-сн
нс=сн нсх хсн II II НСХ ХСН нс=сн
В действительности, однако, это не оправдывается. Циклобутадиен до настоящего времени не получен, и можно думать, что даже если его удастся синтезировать, он окажется чрезвычайно неустойчивым вследствие огромного напряжения цикла
Хорошо изученный циклооктатетраен (стр. 83) имеет неплоскостное строение, а потому не обладает ароматическими свойствами и ведет себя, как сильно ненасыщенное соединение. Обе эти системы не содержат секстета ^-электронов, характерного для бензола.
Большой интерес с этой точки зрёния представляют производные двух циклических систем с нечетным числом углеродных атомов— циклопентадиена и циклогептатриена:
СИ,
НС СН
НС---СН
СН2
нс \:н
L 11
НС сн \ / нс=сн
Как таковые, эти два вещества проявляют свойства циклических ненасыщенных соединений. Однако они склонны образовывать ионы, имеющие ароматический характер.
Циклопентадиен, отдавая протон и сохраняя электронную пару о-связи, превращается в анион:
Н : С-Н ; С-Н
\н _н+ нс' ^сн
II II —* II II нс—сн НС—сн
Наоборот, система циклогептатриена будет легко образовывать катион, если один из водородных атомов группы СН2 будет предварительно замещен на какой-либо атом, способный отщепляться, унося с собой соответствующую электронную пару:
X : СН
НС^ \н —х~
II II--------
НС сн
+ сн
\ / нс-сн
нс сн
II II
НС сн \ / нс==сн
При этом система циклогептатриена приобретает положительный заряд.
В обоих случаях при переходе в ионы создается секстет к-элек-тронов, что обеспечивает равноценность всех атомов углерода и всех углерод-углеродных связей, т. е. придает этим ионам ароматический характер:
Благодаря способности давать анион, циклопентадиен может образовывать соли с металлами.
Например, он образует калиевое производное, в котором имеется катион калия и анион циклопентадиена:
НС
Ан
сн
К
Благодаря устойчивости этого аниона, устойчив и ряд его производных, например диазоциклопентадиен
/СН
hcVh
не разлагающийся при нагревании до 160° в присутствии меди.
Чрезвычайно устойчив и катион, соответствующий циклогепта-триену—циклогептатриенилий (тропилий)).
В 1954 г. были получены в чистом виде соли, содержащие катион тропилия, в частности тропилийбромид (циклогептатриенилийбромид):
НС—сн
НОС- ?СН
I \ ; |
НС^-^СН * сн
Вг*
Он получается при обработке углеводорода тропилидена бромом с последующим отщеплением бромистого водорода при нагревании:
Тропилийбромид кристаллизуется в виде желтых пргим (1емп. плавл. 203°, с разл.). Он нерастворим в эфире, четыпеххлористом углероде, хлороформе и других малополярных органических растворителях, но чрезвычайно хорошо растворяется в холодной воде. При прибавлении к его водному раствору азотнокислого серебра немедленно образуется осадок бромистого серебра. Это показывает, что тропилийбромид содержит ионогенный, а не ковалентно связанный бром.
Сходными свойствами обладают некоторые производные тро-пилийгалогенидов, в частности карбокситропилийбромид (темп, плавл. выше 300°)
СООН
вг“
также нерастворимый в неполярных органических растворителях, но хорошо растворяющийся в воде и содержащий ионогенный бром.
Эти соединения по многим свойствам напоминают бромиды одновалентных металлов или аминов.
В связи с указанными свойствами солей, содержащих такие катионы, не следует выражать их строение формулами с ковалентно связанным бромом:
ХН=СН
с \
II ' .СНВг И
С_ /
соон
„.С=СН
НС \
, II СНВг
НС^ /
сн=сн
Спектры поглощения тропилийбромида отличаются большой простотой, что свидетельствует о чрезвычайно высокой симметрии иона гропилия.
Спектры этого вещества имеют большое сходство со спектрами бензола. Все это указывает на ароматический характер иона цикло-гептатриенилия.
Устойчивость иона тропилия в значительной мере определяет устойчивость и ароматические свойства других циклических семичленных соединений с тремя двойными связями, в частности тропона, трополона, азулена и их производных.
Таким образом, среди карбоциклических соединений имеются три ряда ароматических веществ, в основе которых лежат: анион пентадиена, нейтральная молекула бензола и катион циклогептатриенил ия (тропилия)4.
Все они имеют плоскостное строение с обобщенными секстетами к-электронов, равноценными атомами углерода и одинаковыми углерод-углеродными связями.
Тропой
Из производных циклогептана обладает открытый в 1951 г. тропой
ароматическими свойствами (циклогептатриен-2,4,6-он-1):
с
НС^ ХСН
сн
HQ
нс=сн
или
О
Тропой представляет собой сильное основание, образующее с кислотами устойчивые кристаллические соли. В этих солях тропон
О
находится в виде катиона, строение которого можно выразить формулой:
Пунктир внутри цикла показывает, что все связи между атомами углерода одинаковы (стр. 492), а положительный заряд распределен равномерно между всеми атомами углерода.
В связи с указанным строением катиона сам тропой можно рассматривать как производное иона циклогептатриенилия, илитропилия (стр. 493):
Тропой является окисью тропился, и поэтому его строение можно изобразить следующим образом:
Вследствие выравненное™ электронной плотности тропой проявляет в некоторых отношениях ароматический характер. Так, при действии галоидов (например, брома) происходят в основном не реакции присоединения, а реакции замещения с образованием, в частности, 2^-дибромтропонах
О
II
О
II
Хотя тропой вступает в некоторые реакции, характерные для обычной кетогруппы,—например при действии гидроксиламина он образует оксим,—однако одновременно протекает и реакция замещения в положение 2, приводящая к 2-аминотропону\
h2noh
При взаимодействии тропона с гидразином образуется исключительно 2-аминотропон:
Гидразон тропона при этом не образуется.
Однако тропой не является вполне ароматическим соединением. Он легко присоединяет малеиновый ангидрид (т. е. ведет себя в этом отношении как диен, а не как ароматическое соединение), сравнительно легко гидрируется. Наиболее важными и подробно изученными производными тропона являются трополоны.
Трополоны
Другой интересной циклической системой с сопряженными двойными связями и семичленным кольцом, обладающей ароматическим характером, является система циклогептатриен-2,4,6-ол-2-она (2-оксициклогептатриен-2,4,6-он), для которого было предложено название трополон* (Дьюар, 1945 г.):
или
О ОН
Трополон был синтезирован несколькими путями. Одним из простейших и наглядных путей является синтез его, исходя из пробковой (субериновой) кислоты, по следующей схеме:
СООН
щс7 СООН
I I
Н2С сн2
I !
Н2С сна
SeO-2
Н2С "cHj I I н2с сна ^срС
звг2
ВгНС 'сВг,
I I
Н2С сн2
\ / с-с / ч ВгС СН
11 L
НС сн
о. он
* Название это напоминает о генетическом родстве такого цикла с семичленным циклом тропана (см. тропановые алкалоиды, стр. 652).
Трополон (бесцветные иглы; темп, плавл. 50—5Г) легко растворим в воде и в органических растворителях. Он обладает высокой упругостью пара и легко возгоняется при комнатной температуре.
Строение трополона доказывается каталитическим гидрированием в присутствии платинового катализатора (над палладием он не гидрируется). При этом сперва быстро поглощаются три молекулы водорода с образованием циклогептанолона, а затем медленно поглощается и четвертая молекула с образованием циклогептандиола-1,2. Последний окисляется перманганатом в пимелиновую кислоту.
О ОН О ОН \ / ч /
с—с C-CII
нс" сн и.С CIL,
! I 'I I
НС сн н.с сн.
Xf 1 Хн2
но
он
нс-сн
сн
сн2
сн2
К Мп О 4
ноос соон нас хсн2
I !
Н2С СН8
V Сг12
Благодаря наличию гидроксильной группы, аналогичной фенольному гидроксилу, трополон обладает амфотерными свойствами:
образование аниона трополона
О Н--О+
образование катиона трополона
Со щелочами он дает светложелтые растворы, вытесняет угольный ангидрид из растворов бикарбонатов. С катионами тяжелых металлов он образует растворимые в хлороформе комплексные соли: с Си44"—зеленую, с Fe+ + +—красную. Вместе с тем, сконцентрированной соляной кислотой трополон, реагируя как основание, образует хорошо кристаллизующуюся, но легко гидролизуемую соль. С пикриновой кислотой он, как и его производные, образует устойчивую соль.
Кислотные свойства трополона являются средними между свойствами фенола и алифатических кислот; рК лежит между 7,0 и 6,7.
Как и тропой, трополон обладает ароматическим характером. Рентгенографическое исследование натриевой и медной соли трополона показало, что кольцо трополона представляет плоский правильный семиугольник с выравненными связями С—С, длины которых составляют около 1,40 А, а углы между связями (128,6°) сравнительно мало отличаются от нормальных углов (120°) между направлениями a-связей при ^-гибридизации (см. том I, стр. 119). Дипольный момент трополона (для растворов в бензоле) составляет 3,71 D. Теплота образования трополона примерно на 30 ккал больше, чем можно было бы ожидать на основании расчета по аддитивной схеме, пользуясь соответствующими величинами для двойных и простых связей.
Ароматический характер трополона, как и у бензола, проявляется в том, что при формальной ненасыщенности он часто ее не обнаруживает в своих реакциях. Так, например, при действии брома присоединение по двойным связям не идет, а происходит замещение атомов водорода в положениях а и а' с образованием моно-и дибромтрополонов; труднее бром вступает в у-положение, а (3-по-ложения совсем не затрагиваются.
Азотная кислота не окисляет трополон, а нитрует его, преимущественно в положении у. Многие другие окислители также не разрушают системы трополона. Например, p-метилтрополон можно окислить двуокисью селена до соответствующего альдегида и этот альдегид действием окиси серебра перевести в кислоту:
О ОН О ОН О ОН
\ / Ч / \ /
/~~Ч SeO2 ' /“"% Ag2O /““Ч
11 А —> 11 ) —> 11 /
\^\:н3 \^х:но \^\зоон
Азотистой кислотой трополон нитрозируется с образованием рнитрозотрополона; если это положение занято, то нитрозирование, хотя и труднее, идет в положение а.
Трополоны способны вступать в реакцию азосочетания с солями диазониев; сочетание идет в положение у. Таким образом, своим поведением трополон во всех перечисленных случаях напоминает фенол.
При восстановлении нитро-, нитрозо- и азосоединений трополонов могут образовываться аминотрополоны. Они, подобно ароматическим аминам, способны в кислой среде диазотироваться, а соответствующие соли диазония трополонового ряда могут подвергаться превращениям, аналогичным превращениям солей бензолдиазония.
Трополон не вступает в реакции, присущие карбонильной группе (не дает гидразонов, семикарбазонов и т. п., трудно гидрируется в обычных условиях).
Можно было бы думать, что симметричный характер кольца трополона, легкость образования водородного мостика и чрезвычайная легкость таутомерных превращений типа
обусловлены тождеством обоих атомов кислорода и одинаковыми длинами связей С=^О и С—О. Однако, как показали рентгенографические исследования, во всяком случае в солях трополона, длины связей различны: длина связи С=О равна 1,25 А, а для связи С—О она составляет 1,34 А.
Повидимому, строение трополона будет правильнее представить следующим образом:
Однако, хотя ароматический характер трополона выражен сильнее, чем у тропона, он все же не является вполне ароматическим соединением,
Так, гидроксильная группа трополона не метилируется при действии диметилсульфата на щелочные растворы трополона, но зато метиловый эфир его может быть получен при помощи диазометана или путем обработки его метанольных растворов газообразным хлористым водородом. Таким образом, его поведение напоминает поведение органических кислот, а не фенолов. Да и сам образующийся метиловый эфир трополона ведет себя, как сложный эфир, а не как простой эфир фенола. В противоположность эфирам фенолов, эфир трополона легко гидролизуется водными щелочами и кислотами. С аммиаком его метоксильная группа замещается на аминную, и образующийся при этом 2-аминотрополон щелочами превращается в трополон.
Далее, грополоны, в противоположность бензолу и подобно i еко-горым гетероциклическим ароматическим системам, способны присоединять малеиновый ангидрид (реакция Дильса—Альдера, характерная для непредельных соединений*). Кроме того, некоторые реакции, типичные для бензола (реакция Фриделя—Крафтса, реакция сульфирования, хлорметилирования) не осуществляются с трополоном.
Неспособность трополонов сульфироваться серной кислотой, и даже олеумом, объясняется тем, что в условиях реакции они образуют
* Следует, однако, помнить, что некоторые ароматические соединения бензольного ряда тоже вступают в эту реакцию, например перилен (стр. 487).
соли, в которых трополоя имеет характер катиона. Эго препятствует его взаимодействию с таким электрофильным реагентом, как серная кислота*. По то) же причине грополоны не вступают и в некоторые другие характерные для бенюла реакции, перечисленные выше.
Для установления строения трополонов имеет большое значение их способность изомеризоваться (в некоторых условиях гладко) в производные бензола.
Так, например, а.д-динитротрополон очень легко, при простом нагревании в водных или спиртовых растворах, изомеризуется в 2,4-динитробензойную кислоту:
V.-NO,
С I
no2
О==С—Oil
с
НС^ %—NO,
II I
НС сн
V
NO,
Легко происходит также изомеризация эфиров трополона при нагревании со спиртовым раствором алкоголя га натрия; например:
О ОСН. О=С— ONa
НС Cl R0Na НС С—С1
нс (!:н * нс сн
Незамещенный трополон и алкилтрополоны изомеризуются труднее; например, у-изопропилтрополон (7-туяплицин) превращается в n-изопропилбензойную кислоту лишь при сплавлении с едким кали при 230°:
О ОН О=С—ONa
нс *сн кон НС сн
I I -------—> I
нс сн нс сн
V 9
сн,—сн—сн3 сн,-сн—сн,
* Сульфирование трополона можно осуществить действием сульфаминовой кис-
лоты.
В настоящее время известно довольно много производных трополона, частью полученных синтетически, частью являющихся продуктами жизнедеятельности растительных организмов.
Особый интерес к производным трополона и к химии его производных вызван, несомненно, и тем, что многие их представители обнаруживают фунгицидное, а в ряде случаев и бактериостатическое действие и являются, таким образом, антибиотиками (стр. 703).
Так, в древесине разных видов туи, например туи западной (Thuja plicaid}, содержатся три изомерных изопропилтрополона (а-, (В- и ^-туяплицины)\
CHg-CH—СН3
а ТУЯ1.ЛИЦИН ^-туяплчцин, или 7-ТУЯПЛИНИН
• темп, плзел. 34°) хиною тиол (теки, плавл. 82°)
(темг. плаьл 52°)
Стойкость древесины туи западной по отношению к грибкам, разрушающим древесину, обязана фунгицидному действию туяплицинов. Туяплицины получены и синтетически.
Из культуральной жидкости плесневого грибка Peniclllium stipi-tatum была выделена кислота С.НнО5, названная стипитатовой кислотой, а при культивировании Penicuhum puberutum были выделены сходные со стипитатовой кислоты пубсруловая С8Н6Ов и пуберулоновая С9Н4О7:
стипитэт<'вая кислота (темп плавл ?02—304°, с разл.) •
пуберул-'вая кислота (Tv.mii. плавл 31ь—318
пуберулоносая кислота (темп, пл а ел 298°. с разл.)
Стипитатовая и пуберелспая кислоты получены и синтетически. Пуберулоновая кислота легко превращается в пуберуловую кислоту при кипячении с разбавленной серной кислотой При этом происходит гидролиз ангидридной группировки с последующим отщеплением опчпй группы СО2
Строение пуберуловой кислоты доказано образованием аконитовой кислоты
при окислении щелочными растворами перекиси водорода
Я1 /СООН / ч. / НОСС с
I сн2 HOO^f
а в дальнейшем подтверждено и синтезом.
Давно известное красящее вещество, образующееся при окислении пирогаллола, так называемый пурпурогаллин, как выяснено в настоящее время, является триоксибензо-а,р-трополоном:
Н<?0 он
НО. 1 Ч_/
j) /)
пурпурогаллин
Синтетически получены и незамещенные а,(3-бензотрополон и Pj-бензотрополон, представляющие комбинацию бензольного и трополонового циклов.
Интересно отметить, что наличие трополонового цикла доказано в алкалоидах безвременника (Colchicum autumnale)—колхицине и родственных ему соединениях (стр. 647).
Азулены
В процессе дискуссии о строении бензола и об особенностях ароматических соединений представлялось весьма полезным выяснить возможность синтеза новых циклических непредельных углеводородов и их производных, которые, не заключая шестичленного бензольного кольца, все же обнаруживали бы ароматические свойства, подобные наблюдаемым у тропилийбромида, тропона и трополона.
Весьма интересны с этой точки зрения попытки ряда английских ученых синтезировать аналоги нафталина—вещества, названные ими пенталином и гепталином'.
НС=С-----СН
I I II
НС с сн
сн сн
нс=сн нс7
нЬ—сн
у которых, подобно бензолу, чередовались бы простые и двойные связи и можно было бы допускать наличие замкнутой системы сопряженных к-связей.
Однако эти попытки пока не увенчались успехом. Между тем было обнаружено, что соединение, промежуточное между ними, существует в природе и обладает до некоторой степени ароматическим характером.
Некоторые эфирные масла, например масло римской ромашки (Chamomtlla), эвкалиптовое масло (из Eucaliptus globulus), масла некоторых среднеазиатских видов полыни {Artemisia), содержат глубокоокрашенные в синий или сине-фиолетовый цвет углеводороды, получившие название азуленов.
Исследования Платнера и Пфау показали, что азулены являются гомологами бициклического углеводорода циклопентадиено-циклогептатриена, или бицикло-10,3,5Ьдекапентаена
.СН^СН НС=-(У \
I | СН
НС Схх f
\ / ^СН-СН
сн
или
который обычно называют просто азуленом.
Прочность этой системы частично может быть объяснена электростатическим взаимодействием между двумя поляризованными ядрами.
При обозначении положений заместителей чаще всего применяется следующая схема нумерации углеродных атомов в системе азулена:
Простейший азулен—циклопентадиеноциклогептатриен—легко
изомеризуется в нафталин:
На основе аналогичных превращений природных азуленов в нафталины известного строения удалось установить структуру этих веществ, например азулена ветиверового эфирного масла:
СН3 I
Сам циклопентадиеноциклогептатриен в природе не встречается. Он был получен синтетически из Д9-’,0-окталина и представляет собой кристаллическое вещество (темносиние листочки; темп, плавл. 98,5—99°).
Синтез азулена осуществлен следующим образом.
Озонированием Д^.Ю-окталина с последующим разложением озонида получается циклопентеноциклогептанон, который восстанавливают в соответствующий алкоголь.
Алкоголь подвергают дегидратации *и нагреваниегл получившегося непре
дельного бициклического углеводорода с элементарной серой (или дегидрирование i в парах над Ni) отщепляют еще шесть атомов водорода:
СН2 СН2 'сИ
i и I
Н2С с сн2 \ н2 \ н,
нсх
Хен--сн2
Н2С с
\н2
сн
СН=С1Т
ХСН
сн—сн
\зн2—сн2
Как и ароматические соединения с конденсированными ядрами, азулены дают характерные кристаллические комплексы с тринитробензолом, тринитротолуолом и т. п. Они легко адсорбируются на окиси алюминия. Ароматический характер азуленов проявляется также в том, что они, как и собственно ароматические соединения, легко получаются каталитической дегидрогенизацией гидроарома-тических алициклов в присутствии платинового -катализатора.
Однако азулены являются менее прочными веществами, чем нафта-.шн. Они легче подвергаются окислению. Будучи совершенно индифферентными по отношению к щелочам, они обладают способностью растворяться в сильных кислотах (некоторые, например, в 40%-ной серной кислоте) и при разбавлении таких растворов выделяться из них без изменения.
Сам азулен, в противоположность не обладающему дипольным моментом нафталину, имеет ц— 1,0 D.
Окраска азуленов обусловлена избирательным поглощением света в областях около 700, 640, 580
Часть третья
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Гетероциклический ряд
К гетероциклическому ряду относятся вещества, в молекуле которых содержатся циклы (кольца), состоящие не из одних только атомов углерода, но содержащие также один или несколько атомов других элементов. В настоящее время можно с уверенностью утверждать, что в образовании циклов могут принимать участие все многовалентные атомы. Наибольшее значение имеют гетероциклические соединения, в цикл которых входят атомы N, О и S. Соединения, содержащие в цикле эти элементы (особенно два первых), чрезвычайно распространены в природе, легко образуются из природных веществ, а также при различных синтетических реакциях; многие из них имеют важное теоретическое и практическое значение.
Известны гетероциклы как с малым числом атомов в цикле (начиная с трехчленных циклов), так и с очень большим числом атомов в цикле. Однако, как и в алициклическом ряду, легче всего образуются соединения с пятичленными и шестичленными циклами, и притом особенно легко—пяти- и шестичленные непредельные циклы, содержащие атомы углерода не в виде групп СН2, а в виде групп СН.
Как известно, первая циклическая формула в органической химии была предложена Кекуле в 1865 г. Уже в 1866 г. А. Байер предложил для таких известных к тому времени веществ, как фуран С4Н4О, пиррол C4H5N и индол C8H7N, циклические формулы, в которых атомы кислорода и, соответственно, азота входили в цикл. В 1869 г. циклическая формула была предложена Кернером для пиридина C5H5N. Эти формулы нашли в дальнейшем серьезное экспериментальное подтверждение.
После того как формула Кекуле для бензола получила общее признание, В. Мейером в 1882 г. был открыт тиофен C4H4S, оказавшийся поразительно сходным с бензолом. В течение последующих двух десятилетий было открыто и изучено большое число новых гетероциклов. К ним относятся, например, пятичленные гетероциклы—пиразолу (Кнорр, Бухнер), имидазолы (Ладенбург, Гинсберг, Джапп), оксазолы (Ладенбург, Габриэль), тиазолы (А. Гофман, Ганч), еще более многочисленные шестичленные циклы—пираны (Ганч, Пехман, Ост), пиримидины (Э. Мейер), пиразины (В. Мейер, Тредвелл), пиридазины (Тойбер), а также системы, содержащие гетероциклы, конденсированные с бензольными кольцами,— феназины, феноксазины, фентиазины и т. д. Примерно до 1900 г. синтезы и исследования в области гетероциклических систем были излюбленной областью работ химиков-органиков.
Рассматривая соединения с углеродными циклами, мы видели, что хотя циклическое строение придает алициклическим (полиметиленовым) соединениям несколько своеобразный химический
характер; но отличия их свойств от алифатических, или жирных, соединений, в общем, не слишком велики. Лишь при определенной непредельности, как, например, в случае бензола и соединений с конденсированными бензольными ядрами, циклические соединения приобретают резко своеобразный—«ароматический» характер. Подобными свойствами обладают и гетероциклы, если все атомы углерода входят в цикл не в виде групп СН,, а в виде групп СН.
Однако «ароматический» характер гетероциклов далеко не всегда означает полную аналогию их свойств со свойствами бензола и ароматических соединений. В этом отношении имеется ряд переходов от соединений, по свойствам весьма близких к соединениям ряда бензола, до веществ с почти алифатическим характером.
Кроме того, каждому циклу присущ ряд совершенно своеобразных химических свойств.
Все попытки объяснения «ароматических» свойств гетероциклов и их своеобразия тесно связаны с гипотезами и теориями, предложенными для ароматических соединений, но пока еще нет такой теории, которая могча бы дать исчерпывающее объяснение свойств гетероциклических соединений Можно, однако, думать, что именно разнообразие свойств непредельных гетероциклов позволит в будущем создать полную общую теорию непредельных циклов.
Систематическое выделение гетероциклических соединений в особый гетероциклический ряд вызвано все увеличивающимся значением соединений этого класса, составляющих в настоящее время самый обширный раздел органической химии.
Многие гетероциклы, особенно предельные, легко образующиеся из соединений других классов и снова легко в них переходящие, целесообразнее рассматривать в связи с их родоначальниками— соответствующими алифатическими соединениями. Так, уже рас-сматривалип в I томе циклические окиси (например, окись этилена, диоксан), циклические ангидриды двухосновных кислот (янтарный ангидрид, малеиновый ангидрид и т. д.), 7- и В-лактоны и лактамы, циклические формы углеводов, циклические тримеры альдегидов (паральдегид), циануровая кислота и т.д. Такие гетероциклические соединения, как фталевый ангидрид, фталимид, кумарин, были описаны в разделе, посвященном ароматическим соединениям.
В разделе «Гетероциклические соединения» будут рассмотрены главным образом те соединения, в которых гетероциклическая группировка сохраняется при переходе к ряду их призводных, т. е. где она является, так сказать, более прочной. Ввиду необычайного обилия известных гетероциклических соединений в данном руководстве могут быть освещены лишь немногие разделы химии гетероциклов, и притом в них могут быть сообщены только самые основные сведения. Больше внимания будет уделено важнейшим пяти- и шестичленным циклам, преимущественно непредельным, и отчасти соединениям с конденсированными бензольными и гетероциклическими ядрами.
Из трехчленных гетероциклов нужно отметить следующие:
н2с—сн2 н2с—сн.2 н2с—сн2
икись этилена \lH X's'/ этчленимив этвлевсульфкд
Кольцо окиси этилена (о ней см. также том I, стр. 422), как и всех а-окисей, образовано со значительным искажением валентных углов: угол С—О—С равен около 67°, а углы О—С—С составляют о
около 57\ Однако межатомные расстояния (1,54 А для С—G и 1,43 А для С—О) остаются такими же, как и в алифатическом ряду. Искажение валентных углов обусловливает напряженность цикла, т. е. наличие в нем некоторого избыточного запаса энергии, который может высвободиться при раскрытии * цикла. По этой причине а-окиси ведут себя почти как ненасыщенные соединения. Большинство их реакций сводится к присоединению элементов или группировок к концевым атомам С и О разомкнувшейся связи углерод—кислород. В отличие от присоединения по двойной связи здесь возможно присоединение только таких реагентов, которые способны расщепляться на отчетливо полярные группировки, главным образом реагентов, отщепляющих протон (галоидоводородных кислот, годы, спиртов, аммиака, первичных и вторичных аминов, синильной кислоты и т. п.). Например:
Н2С-.-СН2 + Н_С1 __ СН-СНгО
О он
н2с—сн2
О'
h-nh2 —
ch2-ch2nh2
он
Н2С—СН2 О'
СН?-СН2ОН
+ н-°н-*6н '
Интересно отметить, что а-окиси при повышенных температурах легко и гладко изомеризуются на некоторых катализаторах в карбонильные соединения. Изомеризация происходит непосредственно или с промежуточным образованием непредельных спиртов:
Н2С—сн2 гсн2-=сн СН3—СН
\ / -> I -> II
о он о
сн3—нс—сн ^о^
сна=сн—сн2 I
он
сн3—СН2—сн
II о
Окись этилена, особенно под влиянием примесей щелочного характера, бурно полимеризуется с выделением тепла, образуя высо-комол екул я р ные соединен и я:
---СН2—CHa-O-4CHa-CH2~O)w-CH2-CH2~O-----
Одна из а-окисей—эпихлоргидрин
О
находит очень широкое применение в производстве так называемых эпоксидных смол. Эпоксидные смолы, являющиеся высокомолекулярными продуктами конденсации эпихлоргидрина с фенолами, обладают рядом исключительно ценных свойств в качестве изоляционных материалов, материалов для защиты металлов от коррозии и т. п.
Этиленимин* (темп. кип. 55°; уд. вес 0,8321 при 24°)—токсичная жидкость со своеобразным резким аминным запахом; пары ее сильно раздражают органы дыхания. Кристаллический пикрат этиленимина плавится при 142°.
Этиленимин получается действием концентрированных щелочей на соли ^-бромэтиламина:
NaOH Н2С---СН?
ВгСН2—CH2NH2-НВг------> \ /
NH
Действуя на этиленимин галоидоводородами, можно вновь получить галоидоводородную соль р-галоидэтиламина:
Н2С--СН2 нс1
\ /---------> С1СН2—ch2nh2.hci
NH
В присутствии серной кислоты в водном растворе, а также в нейтральней водной среде, этиленимин присоединяет элементы воды, образуя этаноламин:
Н2С .СН2 н q \/ CH2OH-C1I2NH2
NH
С сернистой кислотой он реагирует с образованием таурина (см. том I, стр. 682):
^2^ СН2 Н)5Оз \/ Д12Х ch2nh2-ch2so3h NH
В присутствии малых количеств хлористого водорода этиленимин может быстро полимеризоваться даже при комнатной температуре.
Этиленоульфид представляет собой подвижную жидкость с резким запахом, кипящую при температуре около 55е; крайне быстро полимеризуется в бесцветный, неплавкий, порошкообразный полимер, не обладающий запахом.
* Этому веществу долго придавали строение виниламина СН2 = СН—NH2.
Соответствующие четырехчленные гетероциклы
Н2С-СН9 I I
H2C-S тримет иленсульфил
Н2С-СН2
I I h2c-nh триметиленимив
н2с—сн2
Н2С-0 окись триметилена почти совсем не изучены.
Окись триметилена—жидкость с приятным эфирным запахом, смешивающаяся с водой; перегоняется при 47,8е. Она получается из 1,3-триметиленхлоргидрина действием едких щелочей, однако реакция сопровождается значительным отщеплением галоидоводо-рода. Действием бромистого водорода окись триметилена превращается в 1,3-дибромпропан.
Триметиленимин (азетидин, азациклобутан) представляет собой жидкость с аминным запахом (уд. вес 0,843 при 20°), смешивающуюся с водой и кипящую при 63°. Он ведет себя, как вторичный амин. Пикрат его плавится при 166°. Триметиленимин получается отщеплением галоидоводорода от у-галоидаминов:
СН2---СН2 Н2С—СН2
I I +KOII | I +КВг+Н2О СН2Вг ch2nh2 h2c-nh
В последнее время стали привлекать внимание кетопроизводные триметилениминов—р-лактамы, которые можно назвать ацетидин онами. Наличие р-лактамного кольца обнаружено у важных антибиотиков—пенициллинов.
Триметиленсульфид—жидкость с неприятным запахом, малорастворимая в воде (темп. кип. около 94°; уд. вес 1,0284 при 23°). По свойствам он напоминает алифатические сульфиды. Окисление триметиленсульфида азотной кислотой приводит к образованию три-метиленсульфона (кристаллы, плавящиеся при 76°).
К пятичленным циклам с одним гетероатомом относятся следующие
важнейшие системы:
НС---СН
НС
НС—см
ПС сн
НС—сн
НС сы
S гиофсн
Nil
ПИррОЛ
О фуран системы являются иредставшелями нроиейших «ароманы гетероциклов. Их гидрированием получаются системы ди-дигидротиофена и, соответственно, (теграметиленнмина).
Эти ческих» гидрофурана и тетрагидрофурана (фуранидииа), и тетрагидротисфена (тиофана) и, соответственно, дигидг-опир-рола и тетрагидропиррола (теграметиленнмина). Гидрированные системы, утратившие все ароматические свойства, являются просто непредельными или предельными циклическими эфирами, сульфидами или иминами.
В ряду пятичленных циклов с двумя гетероатомами возможно еще большее разнообразие систем, так как оба гетероатома могут
быть одинаковыми или различными и могут отличаться также взаимным расположением. Здесь будут рассмотрены важнейшие системы ароматического характера:
нс=сн
НС-------NH
I! I нс сн
НС—о
НС сн
N пиразол
имидазол
оксазол
НС---S
тиазол
Им отвечают утратившие ароматический характер гидрированные системы: дигидро- и тетрагидропиразолы—пиразолины и пиразол-идины, дигидро- и тетрагидроимидазолы—имидазолины и имидазолидины, дигидро- и тетрагидрооксазолы и соответственно тиазолы—оксазолины и оксазолидины, тиазолины и тиазолидины.
Существуют пятичленные циклы с тремя, а для азота даже с четырьмя гетероатомами:
HC=N
I I НС NH
трпазол
НС------N
II !
N N
^NH гетразол
Из илестичленных циклов с одним гетероатомом ными являются:
наиболее важ-
СН
НС %Н
I' I нс сн
пиридин
СН2
НоС7 ^СН, ! i
Н2с СН2
NH пиперидин
сн2
пиран
ен, нс" \н
||
НС сн
тиопиран
К важнейшим
тестичленным циклам с двумя
гетероатомами
относятся:
NH
Н(^ ^сн
II i|
НС СН
оксазин
NH нс^ \н
II
FIC СН
\н
пиразин
СН НС^ \ г I НС сн \
N пиримидин
NII нс \н
НС сн
тиазин
Тетрагидрооксазин, называемый’ морфолином
NH
является по свойствам одновременно вторичным амином и циклическим эфиром.
Тетрагидропиразин носит название пиперазина
NH
Н^ \н2
I I н2с сн2 \н
Его дикетопроизводные—дикетопиперазины (см. том. I, стр. 688 сл.) являются циклическими ангидридными формами а-амино-кислот жирного ряда.
Из конденсированных гетероциклических систем наиболее изучены такие, где пяти- или шестичленные гетероциклы, носящие ароматический характер, конденсированы с одним или двумя бензольными ядрами. Здесь к числу важнейших систем принадлежат:
СН
бензофуран, или кумарон
СН
н</ \—сн
бензопиррол, или индол
СН н</ \—сн
I II II
НС с сн
\ /\ /
СН S
бензотиофен
бензопиран, или у-хромен
сн сн
СН СН
СН СН сн
а,3-бензопиридин, или хинолин
р,у-бензопиридин, или изохинолин
СН N СН
дибензопиридин, или акридин
I I II I
НС С С СН
дибензопиразин, или феназин
. СН NH СН
СН NH СН
дибензооксазин, или феноксазин
дибензотиазин, или фентиазин
Систем, образованных конденсацией двух гетероциклов, имеется громадное количество. Много хорошо изученных соединений такого типа принадлежит к производным пурина, ядро которого образовано сочетанием имидазольного и пиримидинового циклов. Производными пурина являются мочевая кислота, гуанин и алкалоиды: кофеин, теоброми н, теофилл и н.
В последнее время усиленно изучаются производные птеридина* скелет которого представляет собой сочетание пиразинового и пиримидинового циклов. Его производными являются витамины группы фолиевой кислоты (стр. 697), пигменты крыльев бабочек (стр. 640).
пурин, или имидазолопиримидин
птеридин, или пиразинопиримидин
Целый ряд своеобразных бициклических и трициклических гетеросистем встречается в природных алкалоидах, благодаря чему некоторые соединения с такими ядрами сравнительно подробно изучены. Вообще же за исключением немногих систем, по преимуществу содержащих гетероциклические ядра, конденсированные с бензольными ядрами, бициклические и полициклические гетеро* циклы изучены в настоящее время еще недостаточно хорошо.
АРОМАТИЧЕСКИЙ ХАРАКТЕР
НЕПРЕДЕЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
Гетероциклические предельные соединения по своему химическому характеру не представляют каких-либо исключительных особенностей! при сравнении их свойств со свойствами соответствующих соединений с открытыми цепями. Поведение гетероатома в таких гетероциклах также мало отличается от поведения его в соответствующих алифатических соединениях.
Примерами соединений, по свойствам сходных с алифатическими, могут служить: циклнческие эфиры
О
Н2С---СН, н/^ ''сн,
Н8(1 Jih, н2с сн,
тетрагидрофуран 1,4-диоксан
тиоэфиры (циклические сульфиды)
СН2
Н2С---СН, Н2(/ СН2
X сн2 н2с сн2
Xх7 Xх7
тетрагидротиофен, или тетрагидротиопиран, или
тиофан циклопентаметиленсулъфид
циклические сульфоны
СН2
н.с7 \н2 I I Н2с сн2
циклопентаметиленсульфон вторичные амины
СН2 NH
Н2Х Хн, Н2Х Хн„
If II
Н2С сн2 Н2С сн2
пиперидин, или пиперазин
циклопентаметиленимин
И т. д.
Совсем иначе ведут себя такие непредельные гетероциклические соединения, в молекулах которых имеется возможность сопряжения системы двойных связей с неподеленными электронными парами гетероатомов цикла. Эффект сопряжения делает такие гетероциклы в большей или меньшей степени сходными по химическим свойствам с бензольным циклом, сообщает им ароматический характер. Сопряжение неподеленных электронов гетероатома с двойной связью вместе с тем резко изменяет и химическое поведение гетероатома, входящего в цикл.
Все это можно очень отчетливо проследить на примере пятичленного гетероцикла с атомом серы.
Тиофан (тетрагидротиофен) обладает типичным неприятным запахом алифатических тиоэфиров, легко окисляется перманганатом, азотной кислотой и т. п. в сульфоксид и сульфон, может присоединять к атому серы два атома брома:
Н2С---СН, н,с—сн2 Н2С—сн,
I I I I —> I I
н2с сн3 н,с сн, н2с сн2
Х^ Xх7 х^
тиофан
о
О
Вг
Вг
Т иофен нс——СН
li II
НС сн
циклический сульфид, содержащий на четыре атома водорода меньше, чем тиофан (он должен был бы содержать две сопряженные двойные связи), ведет себя совсем иначе. Он не окисляется перманганатом, азотная кислота его нитрует (т. е., как в бензоле, атом водорода замещается на нитрогруппу), но он не образует ни сульфоксида, ни сульфона. Запах чистого тиофена напоминает запах бензола. Серной кислотой он сульфируется, и притом легче, чем бензол. Аминотиофен—слабое основание, подобное анилину.
Интересно отметить, что тиофен и ряд его производных обладают физико-химическими характеристиками, очень близкими к соответствующим характеристикам бензола и его производных. Так, например, температуры кипения бензола и тиофена равны 80,1° и 84,18°; толуол и а-метилтиофен кипят, соответственно, при 110,6 и 113° и т. д. Можно сказать, что при замене в бензоле группировки —СН=СН— на атом серы почти не изменяются физико-химические свойства вещества, т. е. обе эти группировки оказываются почти равноценными.
Особый ароматический характер бензола и его производных объясняется сопряжением шести к-электронов (по одному от каждого углеродного атома), образующих электронный ароматический секстет. В случае пятичленных непредельных гетероциклических систем четыре углеродных атома могут предоставить только четыре к-электрона, а соответствующий гетероатом, находящийся в цикле, может отдать еще два электрона своей неподеленной пары для образования устойчивого ароматического секстета. Этим и объясняется, с одной стороны, неспособность таких гетероциклов к реакциям присоединения, а с другой стороны, неспособность (или пониженная способность) гетероатома в цикле к таким реакциям, которые требуют участия его неподеленных электронных пар. Так обстоит дело с пятичленными циклами фурана, тиофена и пиррола, где ароматический секстет электронов образуется за счет четырех к-электронов (по одному от каждого углеродного атома цикла) и двух электронов неподеленной пары гетероатома:
НС-г-СН Пу <11 НС^уСН
нс—СИ I I нс ^сн
НС-т-СН нс.—.сн 1Г/1! I I
HC<A/CH HCJCH
НС-т-СН НС—сн
JI I I ноДсн нс< .сн
фуран
тиофен
пиррол
N I н
N I н
Для пятичленных систем с двумя гетероатомами (для имидазола» оксазола и тиазола) картина будет несколько иной. Здесь три угле’
родных атома располагают тремя к-элемронами, два электрона доставляются неподеленной парой атома серы и один—атомом азота, например в случае тиазола:
HC-T-.S
II 7 я НС, ,('Н
НС.—.S
Н'П /и
Как мы видим, углеродный атом, находящийся между обоими гетероатомами (так называемый М-атом), обладает несколько иными свойствами, чем два других углеродных атома. Неподеленная пара электронов атома азота обусловливает основные свойства тиазола, имидазола, оксазола.
В случае шестичленного цикла пиридина ароматический секстет электронов образуется за счет взаимодействия шести тс-элек-тронов (пять it-электронов от углеродных атомов и шестой электрон от атома азота). Неподеленная электронная пара азота остается в его обладании и обусловливает хотя и ослабленные, но все же отчетливые основные свойства пиридина:
с и
нс^ т 1
И л!
НЧ' >сн
НС<’<н
НС. <н
ПИРИДИН
Аналогичная система имеется в ядре пиразина:
N N
нс^сн нЛ -СН
нс' (сн НС- -СИ
\f пиразин
Шестичленные циклы -[-пирана или тиопирана приобретают в известной степени ароматический характер за счет взаимодействия четырех тг-электронов углеродных атомов и двух электронов неподеленной пары гетероатома:
СН2 СН2
нс7 \н НС- \н
нс сн НС- -сн
Х6Х
-[-пиран
Более резко отличный от непредельных соединений ароматический характер проявляется у тех производных пирана (сам он не получен), где атом 4 (’[-атом углерода) располагает к-электро-
нами, сопрягающимися с к-электронами других кольцевых атомов. Таковы, например, у-пирон и его производные
7-пирон у которых не обнаруживается способность присоединения по двойным связям углерод—углерод и маскировано формальное присутствие карбонильной группы.
Прочность ароматической системы бензола по сравнению с гипотетической системой циклогексатриена характеризуется энергией, могущей выделиться при превращении системы с тремя отдельными двойными связями в систему с шестью ароматическими связями. Эта величина составляет 40,6 ккал/молъ (ср. стр. 230). Для фуранового и пиррольного колец, в соответствии с их меньшей прочностью, аналогичные величины составляют 23 и 22,6 ккал, а для тиофена, более всего приближающегося по прочности к бензолу,— около 31 ккал.
Как известно, расстояния между атомами углерода в бензоле (1,40 А) близки к величине, средней между расстояниями углеродных атомов, соединенных простой связью (1,53—1,54 А) и двойными о
связями (1,34 А). Оказывается, что расстояния между углеродными атомами в пиридине, пиразине, пирроле и тиофене примерно такие же (1,39—1,44 А). Связи С—N в пирроле (1,42 А), пиридине(1,36А) и пиразине (1,36 А) также укорочены по сравнению со связью С—N в алифатических аминах (1,47 А). Расстояние С—S, в алифатиче-о
ских тиоэфирах равное 1,82 А, в гетероцикле тиофене сократилось до 1,74 А.
Все это находится в соответствии с более или менее отчетливо выраженным ароматическим характером всех этих непредельных гетероциклических систем.
Пятичленные гетероциклы
Группа фурана
Строение фурана выражается формулой ₽'НС—СН <3 II II а'НС5 2СНа ^б^ фуран
где положение заместителей в кольце обозначается цифрами. При наличии одного заместителя положения 2 и 5 (a-положение), а также 3 и 4 (^-положение) тождественны.
Способы получения фурана и его производных. Соединения группы фурана получают чаще всего двумя способами.
1. Отщепление воды от четырехатомных спиртов и их производных типа
X— СН(ОН)—СН(ОН)-СН(ОН)—СН(ОН)-Х'
Реакция протекает под действием кислот, а часто и при простом нагревании. Таким способом, например, из пентоз получается фурфурол (см. том I, стр. 558):
НО-НС----СН—ОН НС СН
II -> II II + ЗНЙО
Н2С СН(ОН)—СНО НС С—СНО
\н ^о^
пентоза фурфурол
2. Действие водоотнимающих средств на вещества, содержащие две карбонильные группы в положениях 1,4. Так, например, ацетонилацетон
СН3—СО—СН2—СНг—СО—СН3 СН3-С(0Н)=СН-СН=С(0Н)-СН3
при действии хлористого ацетила образует, с отщеплением воды, диметилфуран, причем ацетонилацетон, вероятно, реагирует в енольной форме:
НС—сн НС—сн
II II -> II II + н2о н3с—с с-сн3 н3с-с с-сн3
нА Ан ^о^
Химические свойства фурана и его производных. Фуран, его гомологи и производные обладают некоторыми «ароматическими» свойствами, хотя фурановое кольцо и не имеет такой прочности, как бензольное и некоторые гетероциклические кольца (например, фурановое кольцо легко разрушается при действии сильных кислот и окислителей). Ароматический характер фуранового кольца проявляется в способности к реакциям замещения,—правда, в особых условиях. Так, при действии на фуран ацетилнитрата (смесь уксусного ангидрида с азотной кислотой уд. веса 1,5) и пиридина получается 2-нитрофуран:
НС---СН ch3coonoq НС-----СН
Il II---------II II
НС СН ппридин НС с—ью2
Х/ Х^
Прямое нитрование азотной кислотой невозможно из-за разрушения фуранового кольца.
В результате бромирования фурана диоксандибромидом (продуктом присоединения брома к диоксану) получается 2-бромфуран, а с помощью продукта присоединения серного ангидрида к пиридину можно получить сульфокислоты фурана и его гомологов (А. П. Терентьев с сотр.).
Фурановый цикл может подвергаться ацилированию (реакция Фриделя—Крафтса)
НС---СН
|| II +(СН3СО)2О ->
НС СН
X7
НС—сн
II II
нс С-СОСНз
Х"7
+ СН3СООН
но в этом случае вместо действующего разрушающим образом хлористого алюминия предложено пользоваться как катализатором хлорным оловом (Я. Л. Гольдфарб, Л. М. Сморгонский).
Свойства окси- и аминопроизводных фурана аналогичны свойствам соответствующих производных бензола, но эти производные фурана значительно менее устойчивы, чем фенол или анилин, и сравнительно мало изучены. Моно- и поликарбоновые кислоты фурана являются, пожалуй, наиболее прочными соединениями этого ряда, хотя при высоких температурах они отщепляют двуокись углерода, превращаясь в соответствующие фураны.
Фуриловый альдегид (фурфурол) обнаруживает все свойства ароматического альдегида. Подобно бензальдегиду, он превращается по реакциям Канницарро в фуриловый спирт и фуранкарбоновую кислоту, дает с ацетатом натрия и уксусным ангидридом (по реакции Перкина) аналог коричной кислоты—фурилакриловую кислоту, с цианистым калием уплотняется в аналог бензоина—фуроин.
Сам фуран, а также фурфурол и пирослизевая кислота (фуран-а-карбоновая) легко окисляются перманганатом в щелочной среде
с образованием малеиновой кислоты. Азотная кислота может окислить фуран в малеиновый альдегид. Бромная вода окисляет фуран и фурфурол в броммуконовую кислоту, а в присутствии щелочей при этом образуется фумаровая кислота.
Фуран и его гомологи, в отличие от бензола, легко присоединяют малеиновый ангидрид, т. е. ведут себя в этом отношении, как диены с сопряженными двойными связями.
Присоединение идет в положении 2,5 и получаются трициклические группировки с кислородным мостиком:
НС---СН НС=-СН
НС—О-^СН НС—О—^СН
Hcici-i пс---СН
II II
ОС со ОС со
V
Фуран может быть получен нагреванием до 275° паров пирослизевой кислоты (стр. 523). Он представляет собой бесцветную жидкость своеобразного запаха, нерастворимую в воде (темп, кип. 32°; d24°=0,9366; лг?(;= 1,4216). Пары фурана окрашиваю! сосновую лучину, смоченную соляной кислотой, в зеленый цвет.
Гидрирование фурана идет с присоединением четырех атомов водорода и приводит к тетрагидрофурану, называемому также фуранидином (темп. кип. 65,5°; уд. вес ^0=0,8892; п^=1,40о0)
Н2С—сн2 I I н2с сн2
Тетрагидрофуран является отличным растворителем, но при его применении надо, еще более чем с диэтиловым эфиром, принимать меры предосторожности против накопления а-гидроперекиси—кристаллического вещества, способного при повышенных температурах к детонации.
Особенно гладко гидрирование фурана протекает в присутствии осмия или палладия при обычном давлении (Н. Д. Зелинский. Н. И. Шуйкин). Тетрагидрофуран получается также при дегидратации 1,4-бутандиола (см. том I, стр. 411). Из тетрагидрофурана путем ряда превращений можно синтезировать те же важные промышленные полупродукты, какие получаются из ацетилена при конденсации его с формальдегидом в бутипдиол и т. д. (см. том I, стр. 379).
Производными тетрагидрофурана являются янтарный ангидрид и его гомологи, бутиролактон, а также у-глюкозы, или фуранозы (см. том I, стр. 544). Производным дигидрофурана можно считать также аскорбиновую кислоту (см. том U стр. 574).
Гомологи фурана, в том числе а-метилфуран, или сильван (темп, кип. 65°), найдены в низших погонах дегтя сосны Pinus silvestris и букового дегтя.
Каталитическим гидрированием с одновременной гидратацией сильван может быть превращен в у-ацетопропиловый спирт (одно из промежуточных веществ в синтезе акрихина, стр. 624):
НС---СН
|| II +Н2 + Н2О
нс с-сн3
Х^
н2с—сн2
I I
Н2С С(ОН)—сн3
Х^
Н2С—сн2
I I
н2с со—сн3
Хн
К трициклическим производным тетрагидрофурана относится кантаридин— действующее начало шпанских мушек (Litta vesicatoria)
Н2С-СН2
нсХо—Хн
Кантаридин кристаллизуется в блестящих бесцветных листочках, плавящихся при 218°, почти нерастворим в воде, обладает нейтральной реакцией. Он ядовит и вызывает нарывы на коже. При нагревании со щелочами кантаридин присоединяет молекулу воды и образует соли двухосновной кантаридиновой кислоты СюН14О5.
Фурфурол (а-фурилальдегид)
нс---сн
II II
НС С—СНО
х^
важнейшее производное фурана. Он получается при кипячении с разбавленными кислотами пентоз (том I, стр. 558), а также соответствующих полисахаридов—пентозанов. Фурфурол представляет собой бесцветную жидкость, быстро темнеющую при доступе воздуха, с довольно приятным запахом, от которого отчасти зависит запах свежего ржаного хлеба. Он плавится при —36,5°, кипит при 161,7°; уд. вес а2^ =1,1598; показатель преломления п^°=1,5261.
При окислении фурфурола получается пирослизевая кислота:
НС---СН
II II
НС с—соон
Х^
Фурфурол обладает свойствами ароматических альдегидов, как, .например, способностью под действием цианистого калия конденси
роваться в фуроин С4Н3О—СНОН—СО—С4Н3О, с аммиаком образовывать гидрамид (С4Н3О—CH)3N2, со щелочами—пирослизевую кислоту и я-фуриловый алкоголь.
С флороглюцином фурфурол дает нерастворимый красный продукт
С6Н6О3 + С5Н4О2 CUHSO4 + H2O
применяемый для количественного определения пентоз и пентозанов
При получении бумажной массы из древесины получается много фурфурола, который является поэтому доступным продуктом. Фурфурол получают также из шелухи подсолнуха, коробочек хлопчатника, соломы злаков и из других подобных отходов, содержащих пентозаны, путем обработки разбавленной серной кислотой и отгонки с водяным паром.
Из производных фурана фурфурол представляет наибольший интерес как исходный продукт для промышленного органического синтеза. При пропускании паров фурфурола при повышенных температурах над некоторыми катализаторами довольно гладко получается фуран (реакция декарбонилирования):
НС----СН НС---СН
II II II II + СО
нс С—СНО НС СН
Фурфурол предложено применять вместо формалина для получения кон* сенсационных феноло-альдегидных смол (стр: 267).
Подочно тому, как из пентоз образуется фурфурол, из кетогексоз под действием кислот получается а-оксиметилфурфурол
НС----СН
HOH2C—С С—СНО
При дальнейшем кипячении с соляной или разбавленной серной кислотой он почти количественно превращается в левулиновую и муравьиную кислоты (см. том I, стр. 558).
Оксиметилфурфурол с резорцином и концентрированной соляной кислотой дает темнокрасный осадок. При помощи этой реакции можно отличить искусственный мед от естественного. Искусственный мед обыкновенно содержит инвертированный сахар, получаемый кипячением тростникового сахара с разбавленными кислотами, а потому в нем всегда имеется примесь оксиметилфурфурола.
Пирослизевая (я-фуранкарбоновая) кислота получается при сухой перегонке слизевой кислоты
НО—НС- но—НС —СН—ОН СН—соон 1 НС- 11 НС —сн || +со2 + зн2о с-соон
ноос или окислением 1 он фурфурола: НС СН II II НС С—СНО НС—сн II II нс с—соон
^о^ V
Это—кристаллы, легко растворимые в горячей воде, плавящиеся при 130°, легко возгоняющиеся.
При нагревании паров пирослизевой кислоты до 275е она превращается в фуран с отщеплением СО2, а при каталитическом восстановлении образует тетрагидропирослизевую кислоту (темп, плавл. 21°; темп. кип. 13Г при 14 мм рт. ст.):
Н2С--СН,
I I н2с сн—соон
Бензофуран, или кумарон:
содержится в каменноугольном дегте и представляет собой довольно прочное вещество. Это—жидкость с темп. кип. 174е. Кумарон присоединяет по двойной связи молекулу хлора или брома. Концентрированные кислоты полимеризуют его в смолообразный паракумарон. Отчасти в этом и заключается процесс образования «кумароновой смолы» при действии серной кислоты на некоторые погоны каменноугольного дегтя.
Кумарон был получен из а-галоидокумаринов действием щелочи:
СН ^С-Вг КОН ZS,--------СН ---сн
V\ /со V\ /с-соои V\ /сн
0 0 о
Строение кумарона наглядно доказывается его синтезом из о-оксихлорсти -рола:
----СН NaOH СН
II----------> II
V\ U\ /си
н
Дибензофуран, или окись дифенилена
также содержится в каменноугольном дегте, но может быть получен и синтетически из различных производных дифенила, например из о,о'-дифенола, нагреванием его с хлористым цинком. Дибензофуран образует бесцветные пластинки с темп, плавл. 81°; кипит при 288°.
Группа тиофена
Строение тиофена и изомерия его производных
р' HCs—5СН з
II II а' НС6 2СН а \1/
S полностью аналогичны фурану.
Способы получения тиофена и его производных. 1. Действие сернистых соединений фосфора на соединения, содержащие две карбонильные группы в положениях 1,4. Так, при действии P2S5 на ацетонилацетон и его гомологи получаются гомологи тиофена (реакция, аналогичная реакции образования производных фурана,— стр. 519):
Н2С---СН2 Мб НС--------------СН
I I ------------* II II
сн3—ос со—сн3 сн3—с с—сн3
Если в положении 1,4 хотя бы одна карбонильная группа находится в виде карбоксила, то вместо P2S5 применяют P2S3, обладающий восстановительными свойствами. Так, соли янтарной кислоты с P2S3 дают тиофен:
Н2С—СН2 p2s3 НС-СН
I I---------> II II
НООС СООН НС сн
Гомологи янтарной кислоты, а также левулиновая кислота и ее гомологи под действием P2S3 циклизуются с образованием гомологов тиофена.
2. Взаимодействие ацетилена с соединениями серы. При пропускании ацетилена через нагретый до 300° железный колчедан образуется тиофен:
GH СН НС---СН
сн сн
НС сн
S
Если ацетилен пропускать вместе с сероводородом через нагретую до 400—450° окись алюминия, действующую как катализатор (А. Е. Чичибабин), тиофен получается с хорошим выходом:
СН СН
III + III + HaS
СН СН
НС сн
+ Н2
Повидимому, для протекания этой реакции необходимо, чтобы в окиси алюминия содержалась примесь окиси железа (хотя бы следы).
3. Действие сероводорода на альдегиды. Тиофен и его гомологи могут получаться при пропускании через нагретую окись алюминия смеси паров альдегидов с сероводородом (А. Е. Чичибабин), например: 4
сн3 сн3 НС -сн
1 + 1 +H2s - -> II II 4 н2 + 2Н2О
сно сно НС СН
4. Действие сероводорода на производные фурана и пиролла (см. стр. 534).
5. Пиролиз органических веществ в пр и-сутствии серы. Тиофен и его гомологи легко образуются при пирогенетическом разложении органических веществ в присутствии серы или некоторых сернистых соединений. В каменноугольном дегте тиофен и его гомологи сопровождают бензол и ароматические углеводороды (в сыром каменноугольном бензоле содержится около 0,5% тиофена). Много тиофеновых соединений получается при сухой перегонке сланцев, содержащих серу. Небольшие количества тиофена и его гомологов содержатся в дегте бурого угля, а также в нефти.
Некоторые сорта нефти содержат много серы, повидимому, также в виде производных тиофена или его тетрагидропроизводного— тиофана C4H8S.
Химические свойства тиофена и его производных. Реакции тиофена поразительно сходны с реакциями бензола и ароматических соединений.
Из всех гетероциклических соединений тиофен в наибольшей мере обладает «ароматическим» характером, который проявляется в его устойчивости к реакциям окисления (несмотря на присутствие в молекуле тиофена атома двухвалентной серы), в трудности протекания реакций присоединения, в способности к реакциям замещения при действии галоидов, в способности легко нитроваться и сульфироваться и т. д. Галоид, нитрогруппа, сульфогруппа, ацильный остаток вступают при этом в молекулу тиофена в а-положение, где плотность электронного облака больше, чем в [В-положении.
Реакции замещения в большинстве случаев протекают для тиофена значительно легче, чем для бензола (например, при обработке серной кислотой тиофен сульфируется гораздо легче, чем бензол). Этим можно пользоваться для отделения тиофена и его гомологов от ароматических углеводородов.
Особенно характерна для тиофена легкость «меркурирования» солями ртути.
Так, при действии на тиофен уксуснокислой ртути получается смешанное ртутноорганическэе соединение:
НС--СН (CH3coohHg НС----СН
II II-----------> II II +СН3СООН
НС СН НС С—HgOCOCH3
При действии кислот на получающиеся ртутноорганические соединения вновь легко выделяется тиофен:
ЫС---СН 2НС1 НС----СН
il II ----> || II +HgCl2 + CH3COOH
НС С—HgOCOCHg НС сн
Ароматические углеводороды меркурируются труднее, чем тио» фен, что используется для количественного отделения тиофена от бензола (реакция Димрота). Однако многие из производных бензола также способны сравнительно легко меркурироваться (стр. 336).
При нагревании тиофена с изатином (стр. 55S) и серной кислотой получается интенсивное синее окрашивание (индофениновая реакция). С помощью именно этой реакции В. Майер открыл (1883 г.) присутствие гиофена в каменноугольном бензоле, так как оказалось, что бензол, полученный из бензойной кислоты, не дает индофениновой реакции Это открытие и обнаруженное затем поразительное сходство гиофена с бензолом привлекли в свое время большое внимание химиков.
Синее окрашивание с тиофеном дают также растворы элементарного селена в концентрированной серной кислоте; этой реакцией можно пользоваться для открытия селена (реакция В А. Ушкова и М. II. Прокунина).
Тиофен—бесцветная жидкость со слабым углеводородным нехарактерным запахом, кипящая при 84,18° (темп. кип. бензола 80,1°); уд. вес d'{,= 1,0648, т. е. значительно выше, чем бензола, и притом больше удельного веса воды; показатель преломления n^=l,5289; застывает тиофен при низких температурах и плавится при —38,2°.
В некоторых нефтях, повидимому, присутствуют производные тетрагидро-тиофена или тиофана Тиофан—жидкость с неприятным запахом, кипящая пру 121,12°; уд. вес d4°=0,9987, показатель преломления пРр—1,5048.
При нитровании тиофена смесью азотной кислоты с уксусным ангидридом получается а-нитротиофен. Это кристаллы с темп, плавл. 46,5^ и темп. кип. 225°, обладающие запахом нитробензола.
При восстановлении нитротиофена получается аминотиофен. или тиофенин (темп. кип. 6Р при 1 мм рт. ст.), который, однако, гораздо менее прочен, чем анилин. При хранении он превращается в густую смолу; хлористоводородная соль его более устойчива. Тиофенин не образует диазосоединений, но может служить второй компонентой азокрасителей.
При действии на тиофен брома и иода получаются а-бром- и а-иодтиофены, способные давать магнийорганические соединения.
Концентрированная серная кислота разрушает тиофен и его гомологи. При сильном разбавлении петролейным эфиром или ароматическими углеводородами происходит гладкое сульфирование. Сульфокислоты гомологов тиофена, получаемые при обработке серной кислотой продуктов сухой перегонки некоторых сланцев, применяются в медицине в виде аммонийных солей под названием ихтиола.
Гомологи тиофена могут получаться из тиофена способами, аналогичными способам получения гомологов бензола, т. е. по реакциям Фиттига и Фриделя—Крафтса, а-Метилтиофен (темп, кип. 113°) и fi-метилтиофен (темп. кип. 115,4°), называемые также тиотоленами, находятся в каменноугольном дегте вместе с толуолом (темп. кип. 110,6°). Д,иметилтиофены, шш тиоксены, сопровождают в каменноугольном дегте ксилолы.
Тетрафенилтиофен, или тионессаль (темп, плавл. 184°)
CeHs—С---С—С6Нб
СбН5~С С—СсНБ
S
наиболее давно известное производное тиофена. Он получен несколькими реак- • циями из ароматических соединений. Легче всего он получается нагреванием стильбена с серой:
с6нб-сн=сн-с6нб С8Н5—С—С—СбНб
+ 3S —> | +2H2S
С6Н6-СН=СН-С6Н6 CeH6-C=C-CeH6
Тетрафенилтиофен—очень прочное вещество.
а-Тиофеновый альдегид (темп. кип. 198°) обладает характером ароматических альдегидов. Кетоны ряда тиофена могут получаться реакцией Фриделя—Крафтса, т. е. действием на тиофен галоидангидридов кислот в присутствии хлористого алюминия. Ацильный остаток направляется к а-углеродному атому.
Кислоты ряда тиофена—к-тиофенкарбоновая (темп, плавл. 126,5°) и $-тио-фенкарбоновая (темп, плавл. 138,4°) получаются окислением метилтиофенов. Они образуют изоморфные смеси.
а-Кислота получается действием СО2 на магнийорганические соединения тиофена. Это значительно более сильная кислота, чем р-кислота и бензойная кислота. При восстановлении амальгамой натрия а-тиофенкарбоновая кислота присоединяет четыре атома водорода.
Группа пиррола
К этой группе принадлежат важнейшие из гетероциклических соединений с пятичленным кольцом. К производным пиррола относится ряд важных растительных алкалоидов, таких, как никотин, атропин, кокаин и пр. а-Пирролидинкарбоновая кислота является постоянной составной частью сложных белков. Ненцкий и его ученики показали, что пиррольное кольцо имеется в молекулах красящего вещества крови—гемоглобина и зеленого вещества высших растений—хлорофилла.
Формула пиррола установлена Байером в 1870 г. Для обозначения положения заместителей применяется следующая схема:
НС—3СН р
1 I а' НС5 2СН а ^NH
В ряду пиррола большое значение имеют продукты присоединения водорода (гидрированные пирролы). Продукты присоединения двух атомов водорода называются пирролинами, а продукты присоединения четырех атомов водорода, не содержащие двойных связей,—пирролидинами:
нс=сн
пирролин-3,4
Н2С—сн
I II
Н2С сн
пирролин-2,3
Н2С—сн2 н2с Ан, пирролидин
Производные таутомерной формы пиррола—пирроленина
НС=СН нА сн, V
и продукты его гидрирования мало изучены.
Способы получения пиррола и его производных. Большая часть способов получения соединений группы пиррола аналогична способам получения соединений групп фурана и тиофена.
1. Действие аммиака на производные четырехатомных спиртов типа X—(СНОН)4—X. Сам пиррол получается сухой перегонкой аммонийной соли слизевой кислоты (лучше всего при нагревании до 200° в глицерине, насыщенном аммиаком):
СН(ОН)—СН(ОН)—COONH4 СН—СН
| -» | \ын + 2СО, + NH3 + 4Н,0
СН(ОН)-СН(ОН)—COONH4 СН=СН
2. Действие аммиака на соединения, содержащие карбонильные группы в положении 1,4. Например, дикетоны-1,4 (реагирующие, как и при замыкании фуранового и тиофенового колец, вероятно, в таутомерной енольной форме) с аммиаком дают гомологи пиррола:
СН-С(ОН)—R СН—С—R
I +NHS —> | \nH + 2H2O
СН=С(ОН)—R СН=С—R
Из янтарного альдегида по этой реакции образуется пиррол.
Если вместо аммиака взять первичные амины, то получатся замещенные при азоте гомологи пиррола. Например, янтарный альдегид с метиламином дает N-метилпиррол
СН=СН
| ^>N—CH3
СН=СН
Этой реакцией было получено особенно много производных пиррола.
3. Действие аммиака на ацетилен. Пиррол образуется при пропускании ацетилена и аммиака через слабо раскаленные трубки:
НС СН НС--СН
III + III -> II II + н2
НС СН НС сн
NH3
4. Восстановление кислородных производных гидрированных пирролов. Пиррол и его гомологи могут образовываться при реакциях восстановления производных гидрированных пирролов. Производным тетрагидропиррола (пирролидина) может считаться, например, сукцинимид (2,5-дикетопир-ролидин)
Н2С---СН2
I I
ОС со
\н
При восстановлении сукцинимид может образовать пиррол.
Наиболее общей реакцией восстановления является нагревание производных гидрированного пиррола с цинковой пылью.
5. Конденсация производных a-а м и н о к е-тонов с соединениями, содержащими реакционноспособную метиленовую группу в а-п оложении по отношению к карбонильной группе. Например, из а-аминоацетоуксусного эфира* и самого ацетоуксусного эфира можно получить диэтиловый эфир 2,4-диметил-пиррол-3,5-дикарбоновой кислоты (Кнорр):
сн3—с=о н2с—соос2нб сн3—С---С—СООС2Н5
I + I тт II II + 2Н2О
с2н5оос—сн с—сн3 С2НбООС—с с—сн3
"\н2 Q
6. Пропускание паров производных фурана в струе аммиака надокисью алюминия при 4 5 0° (см. схему на стр. 534).
* а-Аминоацетоуксусный эфир легко получается из ацетоуксусного эфира нитрозированием с последующим восстановлением нитрозогруппы в аминогруппу.
Пиррол. Пиррол содержится в костяном масле*, а также в небольших количествах в каменноугольном дегте. Он представляет собой бесцветную, сильно преломляющую свет жидкость, запах которой напоминает запах хлороформа. Пиррол кипит при 131°; уд. вес его =0,9669; показатель преломления п2£ = 1,5035. В воде он почти нерастворим, смешивается со спиртом и эфиром.
Чувствительной качественной реакцией на пиррол и его гомологи является окрашивание сосновой лучины, смоченной соляной кислотой и высушенной (пиррол и его низшие гомологи окрашивают лучину в красный цвет). С изатином и серной кислотой пиррол, подобно тиофену, дает темносинюю окраску.
Ароматические свойства у пиррола выражены гораздо слабее, чем у тиофена.
Пиррол сравнительно легко окисляется; уже кислород воздуха медленно окисляет его, причем жидкость темнеет. При окислении пиррола щелочным раствором перманганата получается имид малеиновой кислоты (2,5-дикетопир ролин, или диоксипирролин}
НС=СН
I I
ОС со
\н
Пиррол непосредственно не нитруется и не сульфируется. Действием на пиррол продукта присоединения серного ангидрида к пиридину удается получить а-сульфокислоту пиррола (А. П. Терентьев). При действии галоидов получаются продукты замещения в пиррольном ядре. Например, действием иода и щелочи можно получить тетраиодпиррол, или иодол, C4J4NH, применяемый иногда как антисептик взамен йодоформа.
К кислотам пиррол весьма чувствителен. Он представляет собой очень слабое основание и растворяется только в концентрированных кислотах. Однако при этом быстро происходит полимеризация пиррола, совершающаяся более медленно под влиянием разбавленных кислот. При дальнейшем действии кислот получается (с выделением части азота в виде NH3) красное аморфное вещество, нерастворимое ни в щелочах, ни в кислотах, называемое красным пирролом.
Ароматический характер пиррола проявляется в том, что по ряду свойств он напоминает фенол (на эту аналогию впервые обратил внимание Чиамичиан). Так, атом водорода в группе NH пиррольного кольца может заменяться на металлы. Соединение C4H4NK, или пиррол-калий, может получаться при действии концентрированных растворов КОН на пиррол. Большим количеством воды металлические соединения разлагаются на пиррол и едкие щелочи.
* Так называемое костяное масло получается при сухой перегонке обезжиренных костей. Пирролы образуются в результате пиролиза коллагена (см. том I, стр. 711) и других белковых веществ.
Пиррол-калий (или пиррол-натрий) может служить для синтеза гомологов пиррола. Так, при действии иодистых алкилов на пиррол-калий получаются замещенные при азоте пирролы, например N-метилпиррол C4H4N—СН3, который при нагревании изомеризуется в а-метилпиррол.
При действии углекислоты на металлические производные пиррола сначала образуется а-пирролкарбоновая кислота, которая при высокой температуре (200—250°) превращается в (3-пирролкар-боновую кислоту. Эта реакция вполне аналогична синтезу салициловой и n-оксибензойной кислот из фенола по Кольбе—Шмидту (стр. 381 и 382).
Действием на пиррол-калий сложных эфиров или галоидангидри-дов кислот при нагревании получаются кетоны a-ряда, причем сначала образуется N-производное (здесь также проявляется аналогия с ацилированием фенолов):
НС—СН
I! II
НС СН \^сосн3
нс—сн
'I II
НС с-сосн3 \н
нс йн NK^
При действии на пиррол-калий муравьиного эфира образуется а-пирролальдегид'.
НС--СН • НС--СН
II II + НСООС2НБ —> II II +С2н5ок
НС сн НС с—сно
Кетоны получаются также при действии ангидридов кислот непосредственно на пирролы. Так, при нагревании N-метилпиррола с избытком уксусного ангидрида образуется сначала а-ацетил-N-метилпиррол, а в дальнейшем а,а'-диацетилметилпиррол:
НС---СН НС----СН НС---СН
II II -> II II • II II
НС СН НС С-СОСН3 СН3ОС-С С-СОСН3
\-СН3 \-СН3 \-СН3
Сходство пиррола с фенолами проявляется также в том, что при действии на него хлороформа и щелочи получается а-пирролаль-дегид—аналогично образованию салицилового альдегида из фенола (ср. реакцию Тимана—Реймера, стр. 393).
Все синтетические реакции, осуществляемые с помощью пиррол-калия, могут протекать и с соединением, получающимся при действии на пиррол магний-галоидалкилов:
НС---СН
II II
НС сн
^N—MgHal
Совершенно своеобразным является синтез гомологов пиррола при действии на него алкоголятов при нагревании (Г. Фишер):
НС---СН
II II +CH3ONa
НС сн
\н
нс—сн
II II + Н2О
НС с-сн3
NNa
Таким путем можно постепенно заменить на алкилы все четыре атома водорода пиррольного ядра, находящиеся при атомах углерода.
Чрезвычайно интересны реакции, при которых из пятичленного кольца пиррола получается шестичленное кольцо пиридина (Чиами-чиан). Так, а-метилпиррол при пропускании через слабо раскаленную трубку превращается в пиридин:
НС----СН
II II
НС с-сн3
NH
сн ’ /Ч нс сн
При действии на пиррол йодистого метилена и щелочи получается также пиридин, а при действии хлороформа и спиртовой щелочи, наряду с пирролальдегидом, образуется довольно много р-хлор-пиридина
N
Важно отметить, что гомологи пиррола являются продуктами превращения физиологически важнейших веществ животного и растительного мира—красящих веществ гемоглобина и хлорофилла (стр. 539), а также пигментов желчи (стр. 544).
ВЗАИМНЫЕ ПРЕВРАЩЕНИЯ ПЯТИЧЛЕННЫХ ГЕТЕРОЦИКЛОВ
Весьма интересный ряд взаимных превращений пятичленных циклов друг в друга описан Ю. К. Юрьевым (1936 г.). При пропускании паров многих производных фурана с избытком аммиака над окисью алюминия при 450° идет превращение фуранового цикла в пиррольный:
НС---СН
О
+ NH3
НС-----СН
II II
НС сн
+ Н2о
При действии сероводорода фуран превращается в аналогичных условиях в тиофен. Пропуская над катализатором пары тиофена с аммиаком, можно получить пиррол.
В зависимости от выбранных условий оказалось возможным превратить каждый из трех гетероциклов в любой из двух других, как это можно видеть на следующей схеме:
+н2о
+h2s
>НС---СН
+ NH3
4-Н2О
НС--СН 4-NH3 НС—СН
II II ...—II II
НС СН +H,s НС СН
Гомологи фурана, пиррола и тиофена могут подвергаться аналогичным превращениям, хотя и с худшими выходами.
Продукты присоединения водорода к пирролу и его производным
При восстановлении пиррола цинковой пылью и кислотами получается продукт присоединения двух атомов водорода (темп, кип. 90°; уд. вес d4°=0,9097; показатель преломления П£?= 1,4574), вероятно, имеющий строение пирролина-3,4, к производным которого относится имид малеиновой кислоты:
НС=СН I I
Н2С сн2 \н", пирролин-3,4
нс=сн
I ОС
I со
имид малеиновой кислоты
Гомологи изомерного пирролина-2,3 получаются при действии аммиака или первичных аминов на у-бромкетоны, например:
СН2—СН2 Н2С СН
| | + 2CH3NH2 —» | || + [CH3NH3]Br + Н2О
сн2 СО—сн3 Н2С с—сн3
\r . ^N- СН3
При энергичном восстановлении пиррола и его производных, например натрием в кипящем спирте или дымящей иодистоводород-ной кислотой при нагревании, получаются продукты присоединения четырех атомов водорода.
Из самого пиррола или тетраметиленимин
таким образом получается пирролидин,
Н2с-----СН2
I I
Н2С сн2 \н
Это бесцветная, сильно щелочная, дымящая на воздухе жидкость с острым аммиачным запахом, смешивающаяся с водой; темп. кип. 88,5°; уд. вес ^22=0,8520; показатель преломления 1,4426. Пирролидин обладает химическими свойствами жирных вторичных аминов. Небольшие количества его обнаружены в табачном дыме и в опии.
Имеется много способов образования пирролидина и его производных из алифатических соединений; важнейшие из этих способов приведены ниже.
1. Нагревание хлористоводородных солей 1,4-д и а м и н о в (см. том I, стр 445).
2. Действие аммиака или первичных аминов на 1,4-д вугалоидные соединения. Например:
Н2С---СН2
I I ноос—нс сн-соон
Вг/ + \вг nh2
I СНз
Н2с—сн2 ноос—НС СН—СООН + НВг
^NH Вг“
I
СНз
3. Нагревание о-б р ом а м и н о в. Например:
Н2С---СН2
I I
Вг—Н2С - сн2
nh2
н2с—сн2
I I
н2с сн2 \+/
NH2 Вг"
4. Циклизаци я N-б ро м а м и н о в; Весьма своеобразна реакция образования пирролидинов при нагревании N-бромаминов соответствующего строения с серной кислотой, протекающая с отщеплением бромистого водорода:
СН2—СН3 Вг
| ^N—СН3
СН2—СН2
сн2-сн2
I ^>N—СН3 + НВг
сн2—сн2
Ни четырехчленные, ни шестичленные кольца аналогичными способами не образуются, что свидетельствует об особой легкости образования пирролидинового кольца.
Из реакций пирролидиновых оснований важны следующие реакции, имеющие большое значение для установления строения не только пирролидиновых оснований,- но и вообще циклических предельных аминов.
1. Исчерпывающее метилирование (реакция Гофмана). Реакция протекает в несколько стадий (см. том I» стр. 257).
При действии йодистого метила на пирролидин сначала получается третичное основание
СН2—СНа
\n—сн3 сн2—сн2
а затем соль четвертичного аммониевого основания
СН2—СН2
I >N(CH3)2 J-_сн2-сн2
Эта соль под действием влажной окиси серебра переходит в основание, которое при нагревании отщепляет воду и дает ненасыщенный алифатический амин:
СН2-СН2
I >N(CH3)2
_сн2-сн2
ОН- —>
сн=сн2
I + н20
СН2—СН2—N(CH3)2
Ненасыщенный амин при действии CH3J и затем влажной окиси серебра дает гидрат аммониевого основания
СНа=СН—СН2—СН2—Й(СН3)3 ОН-
который при нагревании, отщепляя воду, распадается на дивинил СНа=СН—СН=СН2 и триметиламин N(CHS)3.
Прием «исчерпывающего» метилирования часто применялся для установления строения азотистых гетероциклов.
2. Действие бромистого циана на третичные циклические амины (Браун). Реакция идет в двух направлениях:
СН2—СН2
I ^>N-CH8
СН2—СН2
BrCN
СН2—СН2
| \n-cn + СН3Вг сн2—сн2
СН2—СН2—Вг
СН2—СН2— N(CN)—СН3
3. Действие пятихлористого фосфора на бензо и-лированные циклические амины. Эта ракция также может вести-к образованию двух конечных продуктов (Браун)
СН2—СН2 рсь СН2—СН2С1
I ^>N-COCeH5------| /N=CC1-C6H5 + PCC13
сн2—сн2 сн2—сн2
4-Н2О нагревание
СН2—СН2С1 СН2—СН2С1
I I 4-C6H6CN
сн2—сн2—NHCCC6H5 СН2—СН2С1
т. е. получается или бензоилированный галоидамин, или двугалоидное соединение с отдаленными атомами галоида и бензонитрил.
Производные пирролидина. К числу их относятся лактамы у-аминокислот (см. том 1, стр. 674), например бутиролактам, или пирролидон (а-пирролидон)
Н2С---СН2
Н2С со
\н
а-Пирролидон (темп, плавл. 25°; темп. кип. 250°) легче всего получается восстановлением сукцинимида. Он растворим в воде с образованием нейтральных растворов; кислоты и щелочи гидролизуют его в у-аминомасляную кислоту.
К производным пирролидина относится также сукцинимид
Н2С---СН2
ОС СО
Особенно важными производными пирролидина являются некоторые кислоты пирролидинового ряда, получаемые из природных продуктов. Сюда относится пирролидин-а-карбоновая кислота, называемая пролином
Н2С---СН2
I I* Н2С сн-соон
Пролин является постоянным продуктом гидролиза казеина (Э. Фишер, 1901 г.) и некоторых других природных белковых веществ. При гидролизе белков он получается частично в оптически деятельной /-форме. Синтетически пролин впервые был получен действием аммиака на а,В-дибромвалериановую кислоту (Вильштеттер). В настоящее время он синтезирован и другими способами, причем вы
делены обе его оптически деятельные формы (кристаллизацией цинхониновых солей ти-нитробензоилпролина и последующим омылением оптически деятельных Л1-нитробензоилпролинов). Рацемический пролин—иглы с темп, плавл. 205°, чрезвычайно легко растворимые в воде; d- и Z-пролины плавятся при 215—220°.
Кроме пролина, в продуктах гидролиза некоторых сложных белков, например желатины, содержится оксипролин (бесцветные таблички, темп, плавл. 274° с разл.), которому придается строение
НО—НС----СН2
I L Н2С сн—соон
NH
N-Метилпролин носит название гигриновой кислоты (безводная плавится при 164°), так как получается при окислении одного из алкалоидов листьев кока, называемого гигрином'.
Н2С--СН2
I I н2с сн—соон
сн3
N-метилпролин
Н2С—-сн2
I I
Н2С СН—СН2СОСН3
\—сн3
гигрин
Бетаин N-метилпролина,
называемый стахидрином
Н2С--СН2
Н,С СН—СОО-
\+/
СН8—N—CHS
чрезвычайно распространен в растительном мире. Он был найден впервые в клубнях Stachys tuberifera, затем в листьях сладких и горьких апельсинов, в лимонной корке, в далматском порошке (цветы растения Chrysanthemum cineranaefolium) и т. д. Стахидрин плавится при 235°.
В растениях найдены также бетаины оксипролина.
Из дцкарбоновых кислот пирролидина особенно важна тропиновая кислота
[12с—сн2
ноос—нс сн—сн2—соон
\—сн3
которая в оптически недеятельной форме (темп, плавл. 248° с разл.) получается окислением алкалоида атропина или продукта его гидролиза—тропина, а в оптически деятельной форме—окислением кокаина или полученного из него экгонина. Тропиновая кислота получена и синтетически.
ГЕМИН, ХЛОРОФИЛЛ И РОДСТВЕННЫЕ ИМ ВЕЩЕСТВА
Важнейшими в физиологическом отношении производными пиррола являются гемоглобин и хлорофилл—пигменты крови и зеленого листа.
В основе их строения лежит скелет порфина, в котором симметрично сочетаются четыре пиррольных кольца, поочередно связанных друг с другом четырьмя метинными группами:
Ъ
СН СН СН
N—
// \
YHC СН«
\ /
HN-C
СН сн сн р
Замещенные порфины носят название порфиринов.
В состав гемоглобина входит комплексно связанный атом железа, в состав хлорофилла—магний. Один из витаминов, витамин В12, обладает порфириновым скелетом, комплексно связанным с атомом кобальта (стр. 714).
Химическое родство и известное подобие строения этих веществ являются доказательством общности происхождения растительных и животных организмов и служат важным биохимическим аргументом в пользу эволюционной теории.
Гемин. Гемоглобин красных кровяных шариков представляет собой комбинацию глобина—растворимого в воде протеина, обладающего слабыми основными свойствами, и гемина—комплекса, образованного ионом железа и протопорфирином (стр. 543).
Гемоглобин непосредственно участвует в процессе кислородного обмена животных организмов с окружающей их атмосферой. Он обладает замечательным свойством обратимо присоединять кислород, превращаясь в оксигемоглобин (яркокрасные кристаллы, обусловливающие цвет артериальной крови). Синтез гемина был осуществлен в 1929 г. (Г. Фишер). Однако до сих пор не удалось найти какого-либо вещества основного характера, подобного глобину, которое сообщало бы гемину способность обратимо присоединять кислород.
Гемин (строение—см. стр. 543)—двухосновная кислота, дающая два ряда сложных эфиров, например монометиловый и диметиловый. Кроме того, он содержит две ненасыщенные боковые цепи -сн=сн2.
При действии на гемин HJ в уксусной кислоте получается мезопорфирин C34H36N4O4, содержащий два карбоксила, но уже не имеющий двойных связей в боковых цепях.
Обработка гемина HJ в жестких условиях (Ненцкий и Зибер) дает смесь пирролов и пирролкарбоновых кислот. Из этой смеси в конце концов было выделено восемь веществ: замещенные пирролы—гемопиррол, криптопиррол, опсопиррол, филлопиррол и их карбоновые кислоты:
СН3—С-------С—С2Н5
II II
СН3—с сн
гемопиррол
(2,3-диметил-4-этилпиррол; темп плавл 17°;
темп. кип. 88° при 12 мм)
сн3-с----С-С2Н5
II II
НС с-сн3
криптопиррол
(2,4-диметил-З-этилпиррол;
жидкость; темп. кип. 84° при 10 мм>
сн3—с—с—с2н4—соон
1 II
СНз—с сн
V
гемопирролкарбоновая
кислота
СН3—С-----С—С2Н4—СОО1)
II II
нс с—снэ.
криптопирролкарбоновая кислота
СНз—с-------С—с2н6
II II
нс сн
опсопиррол (З-метил-4-этилпиррол; жидкость; темп. кип. 74—75° при 13 мм)
СН3—С-------С—C2HS
II II
сн3—с с-сн,
\н филлопиррол;
(2,3,5-триметил-4-этилпиррол; темп, плавл. 67э; темп. кип. 88° при 10 мм)
сн3—с—с—с2н4—соон
II II
НС сн
\н
опсопирролкарбоновая
кислота
СН3—С-----С—С,Н4—СООН
II II
СНз—С С—СНз
филлопирролкарбоновая кислота
Синтез продуктов расщепления гемина был осуществлен следующим путем.
Конденсацией по Кнорру (см. стр. 530) а-аминоацетоуксусного эфира с ацетилацетоном получается эфир 2,4-диметил-З-ацето-пиррол-5-карбоновой кислоты:
сн3—с=о Н2С—сосн3
I I
С2Н6ООС-СН + с—сн3 \ nh2 о
сн3—--р сосн3
с2нбоос—у-сн3 NH
Последний по реакции Н. М. Кижнера через гидразон может быть превращен в криптопиррол (избыток щелочи, применяемый для разложения гидразона, одновременно омыляет и отщепляет карбоксильную группу в положении 5):
N—NHa
СН8-|——сосн3 с2н5оос—сн3
NH
CHg—И-T-C-CHg
C2H5OOCJ—CHg NH
CH3——гсн2-сн3
H—L—CH3
NH
Пользуясь тем, что нагреванием пирролов с алкоголятами натрия можно проалкилировачь их в a-положение (при более низкой температуре) или в ^-положение (при более высокой температуре), из криптопиррола можно получить филлопиррол
СН3—--—СН2—СН
н—у—сн3 NH
CH3ONa ------->
СН3—гг—й-сн2-сн8
сн3J J-CH3
NH
3
и аналогичными путями другие алкилированные пирролы.
При бромировании криптопиррола или гемопиррола, как показал Г. Фишер (1916 г.), образуются дипиррилметены, а впоследствии (1926 г.) он же обнаружил, что при нагревании последних с кислотами получаются порфирины. Так как давно уже известно, что эти соединения содержат четыре пиррольных ядра, то ясно, что порфирины должны образовываться из двух молекул* дипир-рилметенов.
Ближайшее исследование продуктов, получаемых бромированием криптопиррола, показало, что в этом случае получается смесь двух метенов I и II:
СН3—л----С2Нб СН3-^=—С2Нб
Вг—/-------СН=\+^!—СН4ВГ
NH NH Вг"
I
-с2нб сн3-=—с2нб
—сн=^-сн3 NH Вг" II
В г,
Оба вещества при обработке кислотами дают порфирин.
Образование порфирина из двух метенов I идет, вероятно, таким путем:
Механизм реакции с участием метена II менее ясен. Известно лишь, что если удалить пербромидный (внешний) бром, то образующийся при этом метен III
сн3—л--с2н5 СН3—=~С2Н5
ВгJ z----сн=х+^сн3
NH NH Br-
ill
уже не способен превращаться под действием кислот в порфирин.
С использованием указанных реакций был синтезирован дейтеропорфирин
дейтеропорфирин
Затем была приготовлена комплексная железная соль дейтеропорфирина и путем обработки ее уксусным ангидридом в присутствии четыреххлористого олова были введены две ацетильные группы, которые затем были восстановлены во вторичноспиртовые.
Таким образом был получен гематопорфирин:
гематопорфирин
Гематопорфирин теряет в вакууме при 105° две молекулы воды, превращаясь в протопорфирин, который с хлорным железом [дает гемин*.
протопорфирин
гемин
Гемоглобин содержит 0,338% Fe. Если бы в молекуле гемоглобина содержался 1 атом железа, молекулярный вес его должен был бы составлять 1(^° О5О5^8 ^16 700. Однако другими методами моле-U, doo
кулярный вес гемоглобина был найден равным ~68 ООО; следовательно, он содержит 4 группы указанного состава.
В близких генетических отношениях к гемину находятся красящие вещества желчи, например билирубин, мезобилирубин и др. Они, повидимому, являются продуктами превращения гемина с раскрытием цикла между двумя пиррольными кольцами:
билирубин мезобилирубин
В моче содержатся следы уропорфирина, представляющего собой многоосноз-ную кислоту:
сн2-соон уропорфирин
У больных особой тяжелой болезнью, порфирией, в моче появляются значительные количества таких веществ. Их присутствие сенсибилизирует организм по отношению к свету, что вызывает очень серьезные последствия.
Хлорофилл. Окраска листьев зеленых растений обусловлена присутствием в них пигмента, называемого хлорофиллом. Он содержится в листьях в так называемых хлорофилловых зернах в количестве от 0,6 до 1,2% веса сухого вещества. Хлорофилл представляет собой темнозеленое воскоподобное вещество, с водой дающее кол^ лоидные растворы, а с водным спиртом или водным ацетоном— истинные растворы. Растворы хлорофилла обладают интенсивной темнокрасной флуоресценцией. Хлорофилл применяют в пищевой промышленности как безвредную краску. В зеленых листьях хлорофиллу сопутствуют желтые пигменты—каротиноиды (стр. 88).
Отделение хлорофилла от сопровождающих его примесей впервые было успешно осуществлено М. С. Цветом* с помощью предложенного им адсорбционного хроматографического метода.
Метод заключается в медленной фильтрации раствора изучаемого вещества через высокий слой адсорбента (обычно каолин, углекислый кальций, окись алюминия и т. д.); при этом происходит адсорбция отдельных веществ последовательными слоями. Промывая столбик адсорбента другим подходящим растворителем, можно добиться четкого разграничения зон, в которых адсорбированы отдельные компоненты смеси. Этот процесс называют «проявлением» хроматограммы. Разрезая затем столбик на отдельные слои, извлекают (элюируют) из них разделенные компоненты смеси специально подобранными растворителями или реагентами (кислотами, щелочами и др.).
Для-выяснения строения хлорофилла решающее значение имели работы Вилльштеттера, проведенные во втором десятилетии текущего столетия. Хлорофилл зеленых листьев состоит на 3/4 из так называемого хлорофилла a (C55H72O5N4Mg) и на V4 из хлорофилла b (C55H70O6N4Mg). При действии щелочей на хлорофиллы магний остается в молекуле образующихся хлорофиллидов, обладающих характером двухосновных кислот, но при этом отщепляются молекула метилового спирта СН3ОН и молекула спирта С20Н39ОН, получившего название фитола (см. том I, стр. 393). Следовательно, хлорофиллы могут быть изображены таким образом:
( СООСН3 [ СООСН3
CagHgoON^Mg \ C32H28O2N4Mg <
( СООС20П39 I СООС20Н39
хлорофилл а хлорофилл Ъ
Обработкой щавелевой кислотой можно заменить в хлорофиллах атом магния на водород; при этом получаются феофитины, не обладающие кислотными свойствами. Это означает, что магний в хлорофиллах связан с азотом, а не с карбоксильными группами.
Гидролиз феофитинов под действием кислот приводит к удалению фитола и образованию монометиловых эфиров дикарбоновых кислот—феофорбидов:
f СООСН3 ( СООСН3
C32H32ON4 < c32h30o2n4 i
( соон ( соон
феофорбид а феофорбид b
Феофорбиды а и b гораздо легче разделить, чем воскообразные хлорофиллы и феофитины, поэтому разделение и начинают обычно на этой стадии.
Феофорбиды, феофитины и хлорофиллы могут быть, подобно гемину, превращены в смесь замещенных пирролов (гемопиррола, криптопиррола и филлопиррола), а в качестве промежуточных продуктов при жестком щелочном расщеплении образуются порфирины. Выделены и охарактеризованы родопорфирин, пирропор-
* Михаил Семенович Цвет (1872—1919). Ботаник и физико-химик, создавший адсорбционный хроматографический метод разделения смесей органических веществ.
фирин, филлопорфирин и не содержащий кислорода пирроэтио-порфирин. В результате пиролиза (с потерей СО2) родопорфирин превращается в пирропорфирин, который, в свою очередь, может, отдавая СО2, превращаться в пирроэтиопорфирин.
Эти порфирины были получены и синтетически (Г. Фишер).
В настоящее время для хлорофиллов наиболее обоснованными можно считать следующие формулы:
хлорофилл а
хлорофилл b
Полный синтез хлорофиллов пока еще не осуществлен.
Фталоцианины. Нагреванием динитрила фталевой кислоты с металлами (Си, Fe, Со и др.) и их солями были получены интенсивно окрашенные в яркие синие или зеленые цвета фталоцианины:
фталоцианин меди
Они являются металлическими производными азапорфина—циклической системы, подобно порфину составленной из четырех пиррольных колец, но связанных друг с другом в a-положениях не метанными группами, а атомами азота:
Атомы металла во фталоцианинах связаны весьма прочно; фталоцианин меди, например, возгоняется при 500—600°, растворяется в высококипящих органических растворителях и может быть таким способом перекристаллизован. Фталоцианин железа способен разлагать каталитически перекись водорода. Получен и свободный фталоцианин (C3?H16NR)H2. Фталоцианины являются очень прочными и красивыми красителями. Они находят применение в лакокрасочной промышленности и, особенно, для цветной печати.
Группа индола, или бензопиррола
Индол C8H7N содержит бензольное кольцо (Bz), конденсированное с пиррольным кольцом (Ру):
Индол и его производные вызывали большой интерес у химиков вследствие тесного родства индола с индиго—красящим веществом,
долгое время являвшимся одним из важнейших синих красителей. Индиго ранее выделяли из растений, а в настоящее время получают почти исключительно синтетически. Индол может быть получен при перегонке с цинковой пылью индиго, а также легко получаемых из индиго кислородных производных индола (индоксила, оксиндола, изатина и многих других). Изучение индола, начатое в 18Ь5 г., дало результаты, важные не только для химии индола, но и для всей органической химии.
Небольшие количества индола образуются при гниении белковых веществ. Вместе с ^-метилиндолом (скатолом) индол находится в испражнениях. Довольно значительные количества индола (2—2,5%) содержатся во фракции каменноугольного дегтя, кипящей в интервале 240—260°. Около 2,5% индола содержится в душистом масле жасмина; кроме того, он содержится в цветах померанца, белой акации (Robinia pseudoacacia) и в других цветах.
Способы получения индола и его производных. Существует ряд синтетических способов получения индола и его гомологов.
1. Конденсация ацетилена с анилином. Наиболее простым способом получения самого индола является открытая А. Е. Чичибабиным (1915 г.) реакция конденсации ацетилена с анилином:
СН ___
+ III -> I II II + н2
СН V'X/
2. Циклизация боковой цепи алкилани-линов. Индол и его гомологи образуются при пропускании через раскаленные трубки этиланилина или других алкиланилинов.
3. Конденсация ор т о-з а м е ще н н ы х анилинов. Например, из о-аминохлорстирола при действии этилата натрия образуется индол:
Л ?Н 2!^^ I I | + NaCi+C2HsOH
V\ CHC1 V\ )
NH2 NH
4. Конденсация ацилированных производных о-т о л у и д и н а при высоких температурах с выделением воды. Таким путем можно получить индол и его а-замещенные гомологи:
zs/CHg у\____'
I I I I II + н-°
VXhhZ0-01- ^V^ch,
а-метилиидол
5. Получение из фенилгидразонов. Наиболее старым (1886 г.) и общим способом является открытый Э. Фише-
ром способ получения гомологов и производных индола из фенилгидразонов алифатических альдегидов и кетонов (сам индол из фенилгидразона ацетальдегида получается? однако, с трудом) при нагревании их с хлористым цинком или однохлористой медью (А. Е. Арбузов). Таковы, например, реакции:
f \ СН2-СН3
I । <!н
' / ' Nf Е—
3-метилиндол
фенилгидразон пропионового альдегида
фенилгидразон ацетона
а-метилиндол
П |Нз
V\nh-n^c-cooh
^\<\СООН а-индолкарбоновая кислота
фенилгидразон пировиноградной кислоты
6. Конденсация ацетонилацето на с пирролом. Интересный способ синтеза бензольного кольца индола представляет конденсация аце-тонилацетона с пирролом:
СН3
Индол (бесцветные листочки; темп, плавл. 52,5°, темп. кип. 254—255°) обладает слабым неприятным фекальным запахом. Однако он является составной частью душистого масла жасмина и находит значительное применение в парфюмерии, так как в смеси с другими душистыми веществами он сообщает им освежающий цветочный запах. На воздухе индол темнеет и постепенно осмоляется. Сосновую лучину, смоченную соляной кислотой, он окрашивает в красный цвет.
С n-диметиламинобензальдегидом и соляной кислотой индол дает фиолетово-красную окраску; со следами азотистой кислоты в присутствии азотной кислоты в водном растворе дает красный осадок или красное окрашивание.
Индол по многим реакциям напоминает пиррол. Он очень чувствителен к кислотам (подвергается полимеризации)» но с пикриновой кислотой дает прочные соли.
С щелочными металлами, амидом натрия и концентрированными едкими щелочами индол образует металлические производные, реагирующие подобно пиррол-калию. С магнийорганическими соединениями индол также образует аналогичные производные:
При помощи этих соединений могут быть произведены все синтезы гомологов и производных, осуществляемые с металлическими производными пиррола (стр. 532).
С алкоголятами индол, как и пиррол, образует гомологи (стр. 533).
Реакции, которые ведут к получению из пиррола пиридина и его гомологов, для индола идут аналогично и приводят к образова нию хинолина и его гомологов.
Например, метилиндолы при пропускании через раскаленные трубки превращаются в хинолин:
При действии на индол хлороформа и спиртовой щелочи образуется (3-хлорхинолин
Как и для пиррола, для индола существуют производные таутомерной формы, называемой индоленином
Так, при действии на индол амилнитрита в присутствии алкоголята натрия получается изонитрозоиндо ленин
---C=N—ОН
Таутомерное строение имеет, вероятно, индол-^альдегид
получаемый действием хлороформа или муравьиного эфира на индол-натрий. Аналогия свойств индола и фенола в данном случае не выдерживается; фенолят натрия с хлороформом дает о-оксибензальдегид и /?-оксибензальдегид, здесь же альдегидная группа вступает не в а-, а в ^-положение.
Очень сложно протекающая реакция взаимодействия индола с иодистым метилом в присутствии спиртовой щелочи также может приводить к производным индоленина, например:
СИ.
ZK I
---С-С1)„
V\+^c-CHs
N
I
СН3
Индол легко окисляется воздухом и другими окислителями. Окисление индола озоном, перекисью водорода, иодом и бикарбонатом и т. д. приводит к обрасоганию индиго. При окислении перекисью водорода как промежуточный продукт получается индоксил
Присоединение водорода к индолу и его производным ведет прежде всего к образованию индолинов—продуктов присоединения двух атомов водорода. Получаемый электрохимическим восстановлением индола простейший индолин (жидкость, кипящая при 230°; уд. вес 1,069)
%z\
NH
по свойствам напоминает монометиланилин.
Метилиндолы. а-Метилиндол, или метилкетол (темп, плавл. 60°; темп. кип. 272°), имеет такой же запах, как индол. Ь-Метилин-дол, или скатол (темп, плавл. 95°; темп. кип. 266,2°), имеет отвратительный фекальный запах. Скатол образуется при гниении белков и находится в человеческих испражнениях.
Триптофан. р-Индолил-а-аминопропионовая кислота, называемая также триптофаном
С—СН3—CH(NH3)— СООН сн
является одним из важнейших продуктов гидролиза природных белковых веществ. Присутствие триптофана в продуктах переваривания белков под влиянием трипсина было обнаружено цветными реакциями еще в 1890 г., а в 1901 г. он был выделен в чистом виде.
Строение триптофана было установлено Эллингером, который синтезировал затем рацемический триптофан следующим путем.
Взаимодействием индола с хлороформом и щелочью был получен индол-•р-альдегид, который вступает в реакцию конденсации по Перкину (стр. 279 и 407) с гиппуровой кислотой:
Восстановлением продукта конденсации получается бензоилтриптофан
СН2—СН—NHCOCeH6
СООН
при омылении дающий рацемический триптофан.
При гидролизе белков образуется I-триптофан—безвкусные, малорастворимые в воде листочки с темп, плавл. 293°. В белках содержится лишь немного триптофана, но, повидимому, он имеет большое физиологическое значение и является одной из «незаменимых» аминокислот (см. том I, стр. 715).
При нагревании триптофана, а также под влиянием гнилостных бактерий происходит отщепление СО3 и получается триптамин
СН3—СН3—NH3
При гниении белков или продуктов их гидролиза, содержащих триптофан» наряду с индолом и скатолом получается $-индолилуксусная кислота, называемая также гетероауксином (об ауксинах см. на стр. 74)
----г-СН2—СООН
Р-Индолилуксусная кислота, будучи применена в ничтожных концентрациях (0,01—0,1%-ный раствор в воде), стимулирует рост и развитие растений.
Грибки спиртового брожения превращают триптофан в спирт триптефол
СНа—СН8ОН
В животном организме триптофан может частично превращаться в кинурено-вую кислоту, являющуюся производным хинолина:
ОН
Из триптофана, вероятно, образуется обнаруживаемый в моче так называемый индикан мочи (стр. 554).
Производные индола и индолина, содержащие атом кислорода в пиррольной части ядра
Эти производные могут реагировать в двух таутомерных формах. Отношения таутомерии наиболее важных из этих производных выражаются следующими схемами:
индоксил:
оксиндол:
диоксиндол:
изатин:
п 1°
L II ,,С—ОН
Индоксил находится в моче травоядных животных и человека в виде калийной соли индоксилсерной кислоты («индикан мочи»), представляющей собой кислый эфир серной кислоты:
----C-OSO3H
11
/СН
NH
индоксилсерная кислота
«Растительный индикан», содержащийся в растениях, дающих индиго, представляет собой глюкозид, под влиянием кислот или особых энзимов распадающийся на индоксил и глюкозу.
Синтетически индоксил был получен Байером (1881 г.). Восстановлением эфира о-нитрофенилпропиоловой кислоты сернистым аммонием он получил эфир индоксиловой кислоты:
а его омылением—индоксиловую кислоту, при нагревании теряющую СО2 с образованием индоксила:
Кроме того, индоксил образуется как промежуточный продукт при многих методах синтеза индиго, например при сплавлении фенил глицина со щелочами (стр. 560).
Индоксил образует светложелтые кристаллы с темп, плавл. 85°; в кислых растворах осмоляется. Он обладает свойствами фенолов—растворяется в едких щелочах. Щелочные растворы индоксила легко окисляются уже кислородом воздуха с образованием индиго.
В некоторых условиях индоксил реагирует в таутомерной кето-форме. Например, при взаимодействии его с амилнитритом и алкоголятами образуется а-изатиноксим
----С°
U\ /<!:=юн
NH
Подобно другим монооксимам а-дикетонов, изатиноксим находит применение в аналитической химии как реактив на некоторые тяжелые металлы.
При взаимодействии индоксила с соединениями, содержащими карбонильную группу, эта группа вступает в конденсацию с группой
СН2 таутомерного индоксила и получаются соединения, называемые индогенидами, например:
/\-----со ---со
I I
И ХН, + о=сн-свн6 -> LA /С=СН-С6Н6 + Н2О NH NH
К этому же типу относится реакция индоксила с изатином, ведущая к образованию красного индиго {индирубина)'.
Ч/\ / \ / \/\ / \ /
NH СО NH со
Оксиндол— внутренний ангидрид, или лактам, о-аминофенил-• уксусной кислоты:
Поэтому он может быть получен восстановлением о-нитрофенилук-сусной кислоты.
Оксиндол получается также восстановлением диоксиндола и изатина.
Это бесцветные кристаллы с темп, плавл. 126—127°, растворяющиеся как в щелочах, так и в кислотах. При нагревании щелочных растворов оксиндола получаются соли о-аминофенилуксусной кислоты. При окислении оксиндол легко превращается в диоксиндол.
Диоксиндол (темп, плавл. 180°) представляет собой лактам о-аминоминдальной кислоты и может быть получен восстановлением о-нитроминдальной кислоты. Он получается также восстановлением изатина. Окислением диоксиндола получается изатин, восстановлением—оксиндол.
Изатин был получен в 1841 г. окислением индиго. Он легко получается окислением индиго азотной или хромовой кислотой, а также смесью обеих кислот; может быть получен также окислением оксиндола и диоксиндола. Одним из синтетических способов образования изатина является обработка раствором КОН о-нитрофенил-пропиоловой кислоты:
о-нитрофенилпропиолсвая кислота
изатин
I
О"
изатогеновая кислота
В промышленном масштабе изатин получается по Зандмейеру. Сначала конденсацией анилина с хлоральгидратом и гидроксиламином получают изонитрозоацетанилид
СН(ОН)2
СС18
CH--NOH | + ЗНС1 4- НаО
\ /С0
NH
а затем действием концентрированной серной кислоты проводят замыкание пятичленного гетероцикла:
CH=NOH H2SO<
I + NH3 со
VxZ0 NH
Изатин образует оранжево-красные призмы с темп плавл. 20Г, трудно растворимые в холодной воде. Он легко растворяется в едких щелочах, давая сначала растворы темнофиолетового цвета. В этих растворах содержатся соли, несомненно образованные таутомерной формой изатина:
•СО
^-ONa
N
Натриевая соль, осажденная путем прибавления этилата натрия к раствору изатина в абсолютном спирте,—почти черного цвета.
При нагревании щелочные растворы изатина приобретают желтый цвет вследствие образования соли изатиновой (о-аминофенил-глиоксиловой) кислоты
Vx соон
NP, лактамом которой является изатин.
Изатин может давать производные, отвечающие обеим таутомерным формам. Так, при действии йодистого метила или диметилсульфата на натриевую соль изатина получается ^метилизатин
N— СН8
а при действии на серебряную соль—О-метилизатин /\---------------------------СО
yC—OCHg N
отвечающий лактимной форме изатина, названной так Байером (в отличие от первой—амидной, или лактамной, формы изатина). При действии на изатин PCU получается так называемый изатинхлорид, строение которого также отвечает лактимной форме:
С1
Байер считал лактимную форму изатина устойчивой, а лактамную—неустойчивой. Однако позднейшие спектроскопические исследования показали, что изатин в растворах обладает спектром поглощения, аналогичным спектру поглощения N-метилизатина, а не О-метилизатина, т. е что он имеет строение лактама.
Изатин реагирует как кетон, образуя оксим и гидразон, при* чем в реакцию вступает (В-карбонильная группа, т. е. образуется, например, р-оксим
а-Оксим получается действием азотистой кислоты на индоксил или действием гидроксиламина на метиловый эфир таутомерной формы изатина. Диоксим получается только из N-ацетилизатина действием гидроксиламина.
При действии анилина на изатин получается р-изатинанил
а а-изатинанил был получен при взаимодействии анилина с таутомерным метил -изатином или изатинхлоридом
+ NH2CeHf
+ НС1
а также действием нитрозобензола на индоксил:
+ Н2О
Кроме того, а-изатинанил может быть получен и рядом других синтетических методов. При действии сероводорода а-изатинанил превращается в индиго.
Восстановлением изатина в кислом растворе цинковой пылью, а также амальгамой натрия получается диоксиндол.
Хромовой кислотой изатин окисляется в изатовую кислоту СО
Изатин дает различные цветные реакции. С тиофеном он дает синее окрашивание, обусловленное образованием красителя индофенина из двух молекул тиофена и двух молекул изатина:
НС=СН НС-=СН
----с=с с=с с=с—-Z4
U\ /СО ОС/
NH NH
С пирролом изатин образует синий краситель пирролиндофенин аналогичного-строения.
ИНДИГО И ИНДИГОИДНЫЕ КРАСИТЕЛИ
Индиготин, или синее индиго, представляет собой краситель, применявшийся еще в самой глубокой древности и до введения новых кубовых (индантреновых) красителей сохранявший первенствующее значение, в технике крашения. В настоящее время индиго уже не является столь важным красителем, так как получаемые с его помощью выкраски не могут конкурировать в отношении красоты и тона окрасок и, что еще важнее, в отношении прочности на истирание и т. п. с более новыми красителями.
С древних времен индиго получалось в Индии из различных видов Indigo-fera (/ tinctona и /. pseudot incton а', В Южной Европе, особенно во Франции, культивировалось растение, называемое вайдой (/satis tinctorial дававшее гораздо меньше индиго (0.05% тогда как из Indigotera получалось 1,5—2%). В Китае выращивали также растение Polygonum tinctona (дс 0,75% индиго) В этих растениях содержится растительный индикан—глюкозид, распадающийся под действием энзимов (индэмупьсин) на глюкозу и индоксил. В щелочном растворе (вследствие образования аммиака из коровьей мочи, применявшейся при переработке растений) индоксил окисляется воздухом в индиготин. Кроме индиготина, при этом образуется немного изатина, конденсирующегося с индоксилом в индирубин, или красное индиго (стр. 555) Еще в конце прошлого столетия применялось почти исключительно природное индиго, получавшееся преимущественно из Бенгалии и с острова Явы.
Исследования индиго и продуктов его превращения были начаты Ад. Байером еще в 1865 г. и только через 20 лет привели к установлению строения этого красителя. Вначале индиго придавали ^с-конфигурацию. В настоящее время установлено, что его молекула имеет транс-строение:
NH СО
После успешного осуществления в 1870 г. промышленного синтеза ализарина (Гребе, Либерман, Каро) задачу получения синтетического индиго стали рассматривать как основную задачу химии ароматических соединений.
Еще задолго до установления строения индиго Байер (1870 г.) нашел способ его получения из производных индола, например из изатинхлорида (при действии цинковой пыли и уксусной кислоты), но изатин получали тогда только из готового, природного индиготина. Позднее (1881 г.) Байером был открыт первый метод синтеза индиготина из производных бензола, а именно—восстановление о-ни-трофенилпропиоловой кислоты, при котором как промежуточный продукт образуется индоксил (стр. 554). Второй метод синтеза—из о-нитробензальдегида и ацетона, нашедший некоторое промышленное применение, состоит в следующем. о-Нитробензальдегид действием щелочи на холоду конденсируется с ацетоном:
,^/СНО ^X/CI он>—сн2 -COCI ц
I Y + СНз-СОСН.з -> I Y
'V'KnO, 'V'XnO,
При нагревании полученного кетона со щелочью образуется индиготин:
4- 2СН3СООН + 2Н2О
Только в конце XIX столетия был разработан первый промышленный синтез индиго (синтез Геймана), который был осуществлен в 1897 г. сразу в крупноз.аводском масштабе на Баденской анилиновой фабрике. В синтезе Геймана исходным веществом служил нафталин. Окислением нафталина получали фталевую кислоту; имид фталевой кислоты действием хлора и щелочей превращали в антраниловую кислоту (стр. 389), которая с хлоруксусной кислотой образует фенилглицинкарбоновую кислоту. Действуя щелочью на фенилглицинкарбоновую кислоту, получали индоксиловую кис-
лоту . ,СООН
\ || С1СН2СООН
^/соон
сн2—соон
с—он
,1 + н20
с—соон
антраниловая кислота
о-фенил глицинкарбоновая кислота
индоксиловая кислота
и далее индоксил, дающий при окислении индиго:
Впоследствии большее техническое значение приобрел второй синтез Геймана—исходя из фенилглицина:
и т. д.
Чрезвычайно успешным оказалось применение для конденсации вместо едких щелочей амида натрия NaNH2 в присутствии цианистого калия как катализатора. Позднее для этой цели была с успехом применена негашеная известь.
Исходным продуктом для получения индиготина эти*м способом является, таким образом, не нафталин, а более доступный бензол, и вся схема получения индиготина такова:
бензол —’нитробензол—> анилин—* фен илглинин—-* индоксил—» индиго
Индиготин обычно представляет собой темносиний порошок. Он может быть получен в виде синих, с меднокрасным блеском, кристаллов путем кристаллизации из горячих анилина, нитробензола или ледяной уксусной кислоты. В других обычных растворителях он нерастворим. Индиготин может быть возогнан; пары его имеют темнокрасный цвет*.
Окислением индиготина легко получается изатин. Действие галоидов в присутствии воды ведет к образованию галоидозамещенных изатинов. Если же бромирование производить в отсутствие воды, то получаются продукты замещения на бром атомов водорода в молекуле индиго, т. е. различные броминдиго (моно-, ди-, тетра-). Некоторые из бромированных индиго приобрели большое значение как красители благодаря красоте оттенка и прочности выкрасок. Они получены также и синтетическими методами, например синтезами Геймана из моно- и дибромантраниловых кислот. Различные броминдиго обладают синей или красной окраской разных оттенков.
Интересно, что ^иброминдиго
оказалось тождественным со знаменитым в древности «античным пурпуром», применявшимся для окраски царских мантий и других драгоценных одежд. Этот пурпур добывался из пурпуровой ракушки Murex brandaris, выделяющей бесцветное вещество, на свету окисляющееся воздухом в краситель.
Серная кислота сначала растворяет индиго с возникновением зеленой окраски, а затем его сульфирует, причем получается главным образом 6,6' индиготиндисульфокислота. натриевая соль ко
* Остроумным приемом очистки индиготина является перегонка его с парами ртути в вакууме.
торой, называемая индигокармином, легко растворима в воде и находит значительное применение как синий краситель, особенно как субстантивный краситель для шерсти.
Осторожное восстановление синего индиго приводит к образованию растворимого в щелочах белого индиго, обладающего фенольным характером. Белое индиго имеет строение:
ОН
ОН
Щелочные растворы белого индиго окисляются кислородом воздуха в синее индиго. На этом основано крашение тканей посредством индиго. Синее индиго нерастворимо в воде и поэтому неспособно окрашивать ткани. Применяя различные щелочные восстановители, например цинковую пыль в присутствии щелочи, щелочные растворы глюкозы, растворы железного купороса и щелочи, гидросульфита и пр., приготовляют щелочные растворы белого индиго, получившие в технике название «куба». В такой «куб» опускают ткань, пропитывают ее раствором и затем, «завесив», оставляют на воздухе для «вызревания». При этом происходит образование синего индиго на волокнах ткани (повидимому, образуется какое-то адсорбционное соединение индиго с волокном ткани).
При нанесении рисунка «печатанием» ткань пропитывают раствором глюкозы и «печатают» смесью измельченного индиго, щелочи и «загустителя» (крахмальный клейстер, декстрин и пр.). Затем ткань «запаривают» (при этом происходит восстановление синего индиго в белое в местах печатания и поглощение последнего волокном), смачивают и оставляют на воздухе. Рисунок наносят также «вытравкой», т. е. окрашивают всю ткань в синий цвет, и печатают смесью загустителей с окислителями, превращающими индиго в изатин, или сульфоксилатом натрия и бензилированным аммониевым основанием (в этом случае образуется не изменяющееся на воздухе сульфированное бензилированное белое индиго).
Способ крашения, при котором растворимые в щелочах «лейко-красители» окисляются воздухом на самой ткани с образованием красителя, называется кубовым крашением, а красители этого рода— кубовыми красителями.
Причиной окраски индиго считается имеющаяся в его молекуле группировка сопряженных двойных связей (в сочетании с ароматическими ядрами) о
называемая индигоидной группировкой. Так как эта группировка связей сходна с группировкой связей в о-хинонах, подобные индигоидные красители можно отнести к хиноидным красителям с двухъядерной орто-хиноидной группировкой.
К индигоидным красителям, кроме самого индиго, его гомологов и производных, относятся индогениды (стр. 555), получаемые, подобно красному индиго, конденсацией индоксила с орто-дикарбо-нильными соединениями или конденсацией изатина с соединениями, содержащими группировку —СН2—СО—. К ним относится также получаемое синтетически тиоиндиго (производное бензотиофена)
Тиоиндиго (красное тиоиндиго) может быть, например, получено из тиоса->ициловой кислоты (приготовленной из антраниловой кислоты диазореакцией с сернистыми соединениями) действием хлоруксусной кислоты с последующим сплавлением со щелочами, т. е. способом, аналогичным получению индиго из антраниловой кислоты:
+ С1СН2—COONa —»
^^/COONa
И + NaCl
уСН2-COONa
S
Индигоидные красители все являются кубовыми. К кубовым Красителям относятся также другие двухъядерные хиноидные соединения, например церулиньон (стр. 415), являющийся двухъядер-Ным пара-хиноидным красителем, а также некоторые производные антрахинона, например продукт конденсации индоксила с антрахиноном, представляющий собой двухъядерный о,п-хинон:
О
Карбазол
Карбазол, дибензопир рол, или дифениленимид
содержится в каменноугольном дегте и выделяется из него вместе с сырым антраценом. При перегонке сырого антрацена над едким кали карбазол остается в виде карбазол-калия
из которого карбазол может быть выделен действием водных растворов кислот.
Синтетически карбазол получен пропусканием дифениламина через раскаленные трубки и другими методами.
Карбазол трудно растворим в обычных растворителях, кристаллизуется в табличках; темп, плавл. 246°; темп. кип. 355°. Он представляет собой, как и пиррол, лишь слабое основание, дающее прочную соль только с пикриновой кислотой; очень устойчив как к кислотам, так и к щелочам. Подобно пирролу, карбазол окрашивает смоченную соляной кислотой сосновую лучину в красный цвет и дает синее окрашивание с изатином и серной кислотой.
Производные карбазола мало исследованы. Сам карбазол применяется для получения ценных сенистых кубовых красителей (гидроновый синий и т. п.).
Группа пиразола
Пиразол—пятичленный гетероцикл, содержащий в ядре два связанных друг с другом атома азота,—имеет строение:
НС---СН
I* 3i|
НС5 2N ^NH
Замещение в положениях 3 и 5 ведет”к получению различных изомеров лишь в том случае, когда атом водорода группы NH замещен каким-либо радикалом. При различных заместителях у третьего и пятого углеродных атомов обнаруживается таутомерия, связанная с перемещением атома водорода от одного атома азота ядра пиразола к другому:
НС---С—R'
NH
НС--=С—R' I I R"—С NH
V
Продукты присоединения двух атомов водорода к пиразолу называются пиразолинами, а продукты присоединения четырех атомов водорода—пиразолидинами.
Производные пиразол ина и пиразол идина, содержащие в кольце группу СО, называются пиразолонами и пиразолидонами, например:
Н2С----СН
I II
Н2С N
\н
•пиразолин
Н2С—сн I I!
ОС N
\[н пиразолов-<5
Н2С—сн2
Н2С NH \гН
пиразолидин
Н2С—сн„
I I
ОС NH
пиразолидон 5
I
Возможны производные четырех таутомерных «ов (и пиразолидонов), например:
форм пиразоло-
Н2С---СН
NH '«мино-форма
н.с—сн
I |!
НО-С N
V
имидо-кислстпая форма
НС-=--СН
I
ОС
НС----СН
И II
НО—С N
NH диимино-форма
енольная форма
Фенилированные производные пиразола были получены Кнорром в 1883 г.; свободный пиразол Бухнер получил только в 1899 г
Способы получения пиразола и его производных. Наиболее важными являются следующие реакции получения пиразола, пиразолина и их производных.
1. Действие гидразина на непредельные альдегиды и кетоны. Из альдегидов и кетонов с этиленовой связью в положении 1,2 получаются пиразолины, а если в том же положении находится ацетиленовая связь, то соответственно— пиразолы. Так, например, при действии гидразина на акролеин получается пиразол ин
СН-----СНО СН---сн Н2С—сн
II + .11 I . I II
СН2 + NH2 СН2 N Н2с N
nh2 i4h2 ^nh
пиразолин
а при действии гидразина на ацетали пропаргилового альдегида— пиразол:
С----СН(ОС2Н5)2 с—сн нс—сн
III + 111 II II II
СН NH2 СН N НС N
nh2 nh2 Xih
пиразол
2. Действие алифатических диазосоед и--нений на соединения с двойной и тройной связью. Присоединением диазоуксусного эфира (см. том I,
стр. 662) к этиленовым соединениям получаются производные пиразолина
а из ацетиленовых соединений—производные пиразола:
СН СН—СООС2Н5 НС---C-COOQH6 НС—с—соон гп
ill + I. -> Il I и • —
СН N2 НС N НС N
\ н \н
НС-----СН
II II
НС N
3. Действие на г кислот. Таким путем (Кнорр):
и д р а з и н ы эфиров £-кетоно получаются производные пиразолона
Н.С---СО-СН3
I + с2н6о-ос nh2
NH—С6Н5
Н2С----С—СН3
-» I I +С2Н5ОН + Н2О
ОС N
n-c6h5 1-фенил-3-метилпиразолон-5
Пиразол—кристаллическое вещество с темп, плавл. 70° и темп-кип. 188°. Он гораздо устойчивее, чем пиррол, и обладает в значительно большей степени «ароматическим» характером. Так, пиразол поразительно устойчив по отношению к окислителям, нитруется, бромируется и сульфируется. Аминопиразолы обладают характером анилинов; они способны к образованию диазосоединений и к диазореакциям. С кислотами пиразол дает соли, но является слабым основанием.
При дейстгчи на пиразол йодистого метила получается N-метиллиразол
НС----СН
:1 '!
ПС
\—СНд
Водородный атом группы NH в пиразоле и его гомологах при действии' аммиачной окиси серебра легко замещается на атом серебра. При действии алии-
ли рующих средств на серебряные соединения получаются N-замещенные гомологи пиразола:
НС---СН
II ' II + AgJ
НС N
СН3
НС---СН
+ CH3J -»
Незамещенные при азоте гомологи пиразола имеют лишь слабый запах; N-замещенные пиразолы имеют сильный запах пиридиновых оснований.
Гомологи пиразола окисляются в пиразолкарбоновые кислоты, при нагревании отщепляющие СО2, что может быть использовано для получения пиразола и его замещенных. Особенно легко декарбоксилируются кислоты, содержащие карбоксил в положениях 3 и 5 (в этом проявляется сходство с производными пиридина, например с пиколиновой кислотой,—стр. 604).
Пиразолины значительно менее прочны, чем пиразолы. Они легко окисляются, а под действием кислот расщепляются. При нагревании пиразолины теряют N2 и превращаются в соединения с трехчленным циклом. Пиразолины и пиразолидины при окислении легко превращаются в пиразолы. Пиразолидины—сильные восстановители.
Пиразолин—жидкость, кипящая при 144°, с сильно основными свойствами.
1-Фенил-3-метилпиразолон-5 и его производные являются важнейшими соединениями группы пиразола. Хорошо изучен ряд его превращений, а также отношения трех его таутомерных форм:
Н2с--С—СН3 НС---С—СН8 НС=С—СН}
I il li II ^±||'
О=С N НО—С N О=С NH
\<^с8н6 Х^С6Н5 \^С6Н5
Фенилметилпиразолон выделен лишь в одной форме, кристаллизующейся в виде бесцветных призм с темп, плавл. 127°.
Нитрованием фен ил метил пиразол он а получается пикролоновая кислота (1-п-нитрофенил-3-метил-4-нитропиразолон), по свойствам напоминающая пикриновую кислоту. Кислотные свойства пикролоновой кислоты показывают, что она является производным енольной формы метилпиразолона:
O2N—С----С—СН3
Пикролоновая кислота плавится при 124° с обильным выделением газов. Сна применяется при исследовании органических оснований.
Антипирин. При действии йодистого метила и других метилирующих средств на фенилметилпиразолон, наряду с четвертичными основаниями, получается \-фенил-2,3-диметилпиразолон-б, называе
мый антипирином и являющийся производным таутомерной дииминоформы пиразолона:
С—СН3
I I
ОС N— СН3
\-С6Н5 антипирин
Антипирин образует листочки с темп, плавл. 114°, легко растворимые в воде и спирте. Он представляет собой сильное однокислотное основание. Водные растворы его окрашиваются с хлорным железом в красный, а с азотистой кислотой—в зеленый цвет. Антипирин применяется в медицине как жаропонижающее средство. Соль антипирина и салициловой кислоты называется салипирином.
При действии азотистой кислоты антипирин образует нитрозоантипирин, при восстановлении дающий первичный аминоантипирин (темп, плавл. 109°). Действием метилирующих средств аминоантипирин превращается в диметил-аминоантипирин, являющийся хорошим болеутоляющим и жаропонижающим средством и находящий широкое применение в медицине под названием пирамидона.
ON—С—С—СН3 (CH3)2N— С-=С—СН3
ОС N—СН3 ОС N—СН3
\-СвН5 С6Н6
нитрозоантипирин пирамидон
Пирамидон плавится при 108°, растворяется в 20 частях воды.
Введение в молекулу пирамидона группы Cn.2SO3Na (формальдегидсульфонатной группы) приводит к значительному усилению болеутоляющего действия. Такой препарат, называемый анальгином
CH2SO3Na
I
Н3С—N—С=С—СН3
I I
ОС N—СН3
XN-C6H6 приобретает все большее применение.
Бензпиразол, или индазол, представляет собой сочетание бензольного и пиразолового ядер:
Индазол и многие его производные получены рядом синтетических методов. Одним из наиболее общих методов синтеза является нагревание нейтральных растворов диазосоединений о-толуидина, его гомологов и производных:
Особенно легко индазолы получаются при нагревании N-нитрозобензоил-.о-толуидина и его производных:
+ С6Н5СООН
Индазол представляет собой слабое основание, плавится при 148°, кипи? при 270°.
Группа имидазола, или глиоксалина
Для имидазола принята формула
р НС---N
h4 311
а НС5 2СН р.
\тн
Как и в случае пиразола, если атом водорода при азоте не замещен, при введении двух разных заместителей в положения 4 и 5 обнаруживается таутомерия:
R'-C-----N
il II
R"—С СН
R'— С--NH
Наиболее общей реакцией получения имидазола и его производных является действие альдегидов и аммиака на соединения, содержащие рядом две карбонильные группы (Радзишевский, Любавин):
R—СО NH3
I +
R'—СО ОНС—R"
NH3
R-C---N
h + зн2о
R'-C С—R"
\lH
Имидазол получается при взаимодействии глиоксаля с аммиаком и формальдегидом х(таким способом он был впервые получен Дебу еще в 1858 г. и отсюда приобрел название глиоксалин).
Образование глиоксалина объясняется частичным расщеплением глиоксаля под действием аммиака на формальдегид и муравьиную кислоту.
Второй общей реакцией получения производных имидазола является действие роданистоводородной кислоты на а-аминоальде-гиды и аминокетоны.
При нагревании аминокетонов с роданистым калием и соляной или серной кислотой образуются сульфгидрильные производные
имидазолов, которые при окислении азотной кислотой переходят в имидазолы:
СН3—СО
сн2 +
NHj
N-H
II c=s
СН3—С--NH HNO, СН3-С NH
I! I —-X II I
HC C—SH HC CH
N N
Имидазол и его производные являются более сильными основаниями, чем изомерные им пиразолы. Константа диссоциации имидазола 1,2-10~7, а пиразола—всего лишь 3,0«10-i2. Температуры кипения имидазолов выше, чем пиразолов. Сам имидазол (призмы; темп, плавл. 90°) кипит при 256°.
При действии иодистых алкилов на имидазол сначала происходит замещение на алкил водородного атома имидной группы, а в дальнейшем получаются иодалкилаты:
НС----N пт НС---------N R/J НС---------N—R'
II || -М» в л у л j-
нс сн НС сн НС сн
\|Н \-R _ R
. N-алкилимидазол
При кипячении иодалкилатов с едкими щелочами происходит расщепление молекулы имидазола и образуются амины R—NH, и R'—NH2.
Характерным для имидазольного ядра является также расщепление его при действии галоидангидридов кислот в присутствии щелочей; N-замещенные соединения при этом не изменяются. При пропускании N-алкилимидазолов через раскаленные трубки происходит перемещение алкила от азота в положение 2.
4(5)-Метилимидазол, образуется из а-глюкозы и других гексоз при действии аммиачного раствора ZnO. Как промежуточный продукт, вероятно, образуется метил глиоксаль (см. том I, стр. 636).
Производные имидазола часто встречаются в природе. Они содержатся в продуктах распада белков. Имидазольное кольцо имеется в молекуле некоторых алкалоидов, например в пилокарпине (стр. 686), а в сочетании с пиримидиновым кольцом—в ксантине, кофеине, теобромине и пр. (стр.637—640); сочетание имидазольного кольца с пиримидиновым представляет собой и молекула мочевой кислоты (стр. 636). Наконец, имидазольное кольцо содержится в некоторых циклических уреидах, например в парабановой кислоте, гидантоине (см. том I, стр. 744—745). Сочетание имидазольного и тиофанового циклов имеется в молекуле биотина, или витамина И (стр. 712).
Постоянным продуктом гидролиза белковых веществ является гистидин, или имид азо лил аланин (выделяемый, например, из белка гемоглобина крови в количестве до 10%):
N---СН
II II
НС с—сн<
\н
\соон
Гистидин разлагается при 285°.
Z-Гистидин найден также в мясном экстракте, в ростках растений и в других животных и растительных продуктах. Он был син езирован из хлорметилимидазола и натрийхлормалонового эфира следующим рядо.м реакций:
N---СН N---СН
II II II ||
НС С—СН2С1 + Na—С1С(СООСгН5)2 -» НС С—СН2— СС1(СООС2Н5)2 -»
^NH
N---СИ
И II NH3
НС С—СН2—СНС1—соон------»
N--СН
II II /NH2
НС с—сн2—сн<
\ / хзоон
NH
Полученный синтетически рацемический гистидин был разделен на антиподы кристаллизацией его виннокислых солей.
При нагревании и под действием гнилостных бактерий гистидин теряет С0.2 и превращается в гистамин, или ^-имидазолил-этиламин
N---СН
!! '.I
нс с—ch2-ch2nh2
Гистамин плавится при 83—84° и может перегоняться в вакууме (18 мм рт. ст.) при 209—210°.
Гистамин—физиологически важное вещество. Он действует на гладкую мускулатуру, вызывает расширение кровеносных сосудов и падение кровяного давления, усиливает перистальтику кишечника, стимулирует секрецию желез. Накоплению гистамина в результате нарушения гисти динного обмена в организме приписывают ряд патологических явлений. В связи с этим в настоящее время много внимания уделяется синтезам и поискам его антагонистов—противогиста-минных лекарственных препаратов. Гистамин был синтезирован из р -имидазолилпропионовой кислоты методом Курциуса (см. том I, стр. 252) и восстановлением нитрила импдазолилуксусной кислоты.
Более сложным производным имидазола является один из продуктов окисления перманганатом мочевой кислоты, так называемый аллантоин (см. том I, стр. 745). Он содержится также в околоплодной жидкости у млекопитающих.
Нагреванием хлористоводородной соли этилендиамина с уксуснокислым натрием получается производное дигидроимидазола, или имидазолидина, которое применялось для лечения подагры под названием лизидина:
С112-\Н2 И2С---N
। [ ЮОС—С11ч —> | |1 4-21190
сн2 п2с с—СПз
\’Н£
ЛПЗИДЬН
Производным тетрагидроимидазола является легко получающаяся из этилендиамина так называемая этиленмочевина\
Н2С----NH
I I
Н2С со
\/н
Это—бесцветные иглы, плавящиеся при 131°, легко растворимые в воде, трудно растворимые в эфире.
О получении бензимидазола (темп, плавл. 170°) и его производных действием органических кислот на ароматические о-диамины см. стр. 362. Бензимидазольная группировка входит в состав молекулы витамина В12 (стр. 715).
Другие пятичленные гетероциклы
Оксазол и тиазол. Примером других пятичленных циклов с двумя гетероатомами являются оксазол и тиазол:
(ЗНС----N вне-----N
II4 3ll ‘ li4 31|
а НС5 2СНр. а НС5 2СНр.
оксазол тиазол
Оксазол, как таковой, пока еще не был получен. Алкилзамещен-ные оксазолы получаются конденсацией а-галоидкетонов с амидами кислот. Если принять, что оба реагента взаимодействуют в таутомерной форме, то реакцию, например, между монохлорацетоном и ацетамидом можно представить следующим образом:
СН8-С—ОН Н—N
II II
НС + с—сн3
I I
С1 н—о
СНз-С----N
II Я
НС С—СН3 + НС1 + Н2О
\)/
Оксазолы являются слабыми основаниями. Они легко разрушаются при нагревании с минеральными кислотами, а в определенных условиях также при действии восстановителей и окислителей.
Производные оксазола пока особого практического значения не имеют. Значительно большее значение имеют производные тиазола, поскольку тиазоловый цикл обнаружен в ряде важных природных продуктов. Так, производным тиазола (и пиримидина, или 1,3-диазина) является витамин В1? или тиамин (стр. 710). Тиазоловый цикл имеется также в молекуле важного антибиотика пенициллина (стр. 690), нашедшего широкое терапевтическое применение.
Тиазол и его производные легко получаются из тиоамидов (в том числе и из тиомочевины) действием а-галоидозамещенных альдегидов и кетонов (Ганч). Например, из тиоацетамида (реагирующего в таутомерной форме) получается соль 2,4-диметилтиазола по схеме, аналогичной приведенной выше схеме получения 2,4-диметилоксазола.
Из тиомочевины при этой реакции образуются 2-ам и но тиазолы
СН3—С----N
II II
нс с—nh2
СНз-С---NH
нс! (L=NH
которые обнаруживают отношения таутомерии и другие свойства, подобные свойствам а-аминопиридина (стр. 601).
Дигидротиазолы, или пшазолины, получаются действием на тиоамиды бромистого этилена и его гомологов:
CH2Br NH Н2С----N
I + II -> I 1| +2НВг
СН2Вг С—R Н2С С—R
ms'
Тиазол—легко растворимая в воде жидкость (темп. кип. 116,8°; уд. вес 1,198 при 17°)—является очень слабым основанием. Он находится в таком же отношении к пиридину, как тиофен к бензолу, обнаруживая с пиридином и по физическим и по химическим свойствам такое же поразительное сходство, как тиофен с бензолом.
2-Метилтиазол кипит при 130°, тиазолин—при 138°, 2-метил-тиазолин—при 144,5°; 2-аминотиазол плавится при 90°.
Производным тиазола является важный сульфамидный лекарственный препарат сульфатиазол, обычно называемый норсульфазолом (стр. 311).
Бензтиазол. Сочетание тиазолового кольца с бензольным носит название бензтиазола. Практическое значение получили производные этой системы, применяемые в качестве ускорителей вулканизации каучука в производстве резиновых изделий.
2-Меркаптобензтиазол, известный под названием каптакса, синтезируют нагреванием в автоклавах эквимолекулярной смеси анилина и сероуглерода с элементарной серой:
кап гаке
Это—желтое мелкокристаллическое вещество, плавящееся при 178—179°.
Каптакс—очень энергичный ускоритель вулканизации. Окислением его получается соответствующий дисульфид, применяемый в качестве более мягко действующего ускорителя под названием альтакс:
альтакс
Представляют практический интерес производные бензтиазола, относящиеся к тиоцианиновым красителям. Они не обладают светопрочностью и применяются для сенсибилизации бромосеребряных фотоэмульсий. Небольшие добавки таких красителей придают фотоматериалам чувствительность к зеленым, красным и даже инфракрасным лучам.
Тиоцианиновые красители могут быть получены конденсацией двух молекул иодалкилата 2-метилтиазола с ортоэфиром карбоновой кислоты, например:
Такие красители имеют фиолетовую окраску с оттенком от красного до синего. Полиметинная цепь, связывающая бензтиазольные группировки, может содержать и большее число углеродных атомов.
Триазолы. Из производных триазолов интересен дифенилэндоанилодигидро-триа<ол-[.'2А*, получивший название нитрт вследствие того, что он дает труднорастворимую в воде азотнокислую соль, применяемую для количественного весового определения азотной кислоты.
Нитрон C2UH16N4 (желтые кристаллы; темп, плавл. 189°) получается из карбодифенилдпимида СбН5И = С = МСбН5, образующегося при отщеплении сероводорода от дифсчилтиомочевины. Действием фенилгидразина карбодифенилдиимид превращается в трифениламиногуанидин, дающий при нагревании с муравьиной кислотой нитрон (Буш):
Н н И N— С6н6
N-С61’5 | |
|| + NH2-NH-CeH6 -> С6НБ—N N—С6Н6
С6Н5 N—С \|
C=N ; ар5оди(1снилдиимпд фенилгидразин трифениламиногуанидин
н
I
II Н N—С8Н5
I I
СвНб—N N—С6Н5
---- N
СН-----N—СвНЕ
/I6 1
Свн5-N4 N—С6Н5
нитрон
* В этом названии «эндоанило» указывает на наличие внутреннего (греч. эндо} мостика, сСразованного остатком анилина >N—СбНб.
Тетразол. Чрезвычайно интересным гетероциклом является тетразол, содержащий в цикле четыре атома азота:
НС----N
л л
Тетразол образует кристаллы с темп, плавл. 155е, легко сублимируется, легко растворим в воде и спирте. Он представляет собой одноосновную кислоту. При сильном нагревании тетразол и его соли взрывают.
Тетразол может быть получен нагреванием 1,5%-ного спиртового раствора азотистоводородной кислоты с безводной синильной кислотой:
С НС----N
t N \н
Из производных тетразола практическое значение приобрел пентаметилентетразол, или коразол
являющийся ценным лекарственным препаратом, возбуждающим сердечную деятельность (аналогично камфоре и кофеину).
Шестичленные гетероциклы
Группа пирана
Кольцо пирана состоит из пяти атомов углерода и одного атома кислорода. Большая часть известных и важных производных пирана содержит в ядре одну группу СО и две двойные связи, т. е. является производными пиронов.
В зависимости от положения карбонильной группы относительно атома кислорода различают производные а-пирона и 7-пирона:
О
7 II
С сн с
а-пирон -[-пирон
Пироны являются, собственно говоря, производными а-пирана и 7-пирана
СН СН2
/ Ч / \
НС СН НС СН
II I !!
НС СН2 НС сн
а-пиран -[-пиран
Однако ни а-, ни 7-пираны не известны; их дигидридами являются ангидриды енольных форм o-оксикетонов, а тетрагидридами— ангидриды ^-гликолей. Например, окись пентаметилена может быть названа тетрагидропираном'.
СН2 н2(/ 'сн, I I Н2с сна
Производными пирана являются шестичленные циклические формы ряда углеводов, так называемые пиранозы, которые можно рассматривать как пентаоксипираны (см. том. I, стр. 544).
Y-Пироны способны образовывать с кислотами довольно прочные соли, в которых роль катиона играет шестичленный гетероцикл, содержащий положительно заряженный атом кислорода. Этот катион получил название пироксония, или пирилия.
Способность 7-пиронов к солеобразованию, обнаруженная Колли и Тикле еще в 1899 г., привлекла в то время чрезвычайное внимание химиков. Согласно объяснению А. Байера, образование солей обусловлено основными свойствами кислорода, проявляющимися в данном случае сильнее, чем в соединениях других классов.
С точки зрения современных представлений в молекуле 7-пирона имеется следующее распределение электронной плотности:
нсх xcii
11 и-7
нс сн с
Поэтому в кислой среде протон кислоты присоединяется к отрицательному концу диполя, и вся система приобретает положительный заряд, т. е. становится катионом:
О—Н ~
нС \н
II I
НС сн
ион ппроксония, или пирилия
Этот катион весьма устойчив вследствие взаимного перекрывания всех “-электронных облаков в образовавшейся «ароматической» системе.
Производные обоих пиронов часто встречаются в природе как продукты жизнедеятельности организмов. Благодаря блестящим исследованиям Вильштеттера было найдено, что и соединения, относящиеся к классу солей пирилия, чрезвычайно распространены в растительном мире. Повидимому, соединения, содержащие в шестичленном цикле кислород, являются наиболее устойчивыми и легче всего образующимися веществами из класса оксониевых соединений.
Производные а-пирона. а-Пирон
СН нс^ ^сн I II о=с сн
представляет собой лактон ненасыщенной оксикислоты, для которой таутомерной формой является альдегидокислота:
При нагревании яблочной кислоты с концентрированной серной кислотой получается а-пиронкарбоновая кислота, называемая яу-малиновой кислотой. Образование ее может быть выражено следующим рядом реакций:
ОН
<!н ''соон
ноос
СН
Н2С
I ноос
соон
н
яблочная кислота
полуальдегид малоновой кислоты
сно
н^ сна—соон
I + I
ноос сн
сн
н/7 соон
I I
ноос сн
кумалиновая кислота
Кумалиновая кислота кристаллизуется в виде бесцветных игл (темп, плавл. 205—210°), трудно растворимых в холодной воде. Ее ртутная соль при нагревании распадается на ртуть, СО2 и а-пирон, называемый поэтому кумалином. Кумалин—пахучее масло; кипит, частично разлагаясь, при 217,7° (при 760 мм рт. ст.); в процессе хранения осмоляется.
При нагревании кумалиновой кислоты с аммиаком происходит характерное для пиронового кольца превращение в пиридоновое кольцо:
+ NH3
Под действием щелочей производные а-пирона разлагаются с размыканием кольца.
Из производных а-пирона наиболее известны бензо-а-пирон (1,2-бензопирон), называемый кумарином, и его аналоги—лактоны производных о-оксикоричной кислоты (стр. 411).
Кумарину изомерен 2,1-бензопирон, носящий название изокумарин*.
кумарин изокумарин
При нагревании с NH3 изокумарин превращается в производное изохинолина (кислородный атом кольца замещается на группу NH), а кумарин в этих условиях с аммиаком не реагирует.
Производные у-пирона и пироксониевые соединения. Производные у-пирона очень распространены в природе. Они приобрели большое теоретическое значение при исследованиях оксониевых оснований.
Наиболее общим способом получения производных у-пирона является отнятие воды от соединений, содержащих в молекуле три карбонильные группы, отделенные друг от друга группами СН2 или СН—Y.
Такие соединения общей формулы
Y Y
I I
X—ОС—СН—CO—CH—CO—X реагируют в таутомерной (диенольной) форме: ^СО СО
Y—С С—Y Y—С A—Y
II II v -> II II +Н2О
X—с С—X X—С с—х
НО OH АА
В этом направлении реакция обыкновенно протекает под действием кислот. Под влиянием же щелочей происходит обратная реакция, причем получаются исходные трикарбонильные соединения или продукты их распада.
При действии аммиака на соединения у-пирона, как и в случаэ а-пирона (стр. 577), происходит замена кислорода в цикле на группу NH и получаются производные пиридина (у-пиридон и его производные), например:
О О
11 А
О +NH3 -> f|| +Н2О
H3c/V^CH3 Н3с/\нХСН3
Обычная формула у-пирона
О
недостаточно точно отражает его свойства. Согласно этой формуле, у-пирон должен был бы быть аналогом непредельного кетона типа дибензальацетона
О
С
нс^ ^сн
II II
СбН5—НС СН—С6Н5
т. е. вступать в реакции, характерные для карбонильной группы (например, с фенилгидразином и другими подобными реагентами), присоединять Вг2 или НВг по двойным связям, образовывать cH2SO4, SnCl4 и т. п. окрашенные солевые комплексы (галохромия), типичные для системы
С=С-С—С=С
Однако подобными свойствами у-пирон не обладает.
Естественнее всего было бы предположить для у-пирона бетаиноподобное строение, допускающее выравнивание электронной плотности в цикле:
0“
I
О +
Подобно рпирону ведет себя его сернистый аналог Х-тио-упи-рон. Однако у-тиопирон можно окислить и, получив соответствующий сульфон, зафиксировать ранее неподеленные электроны атома серы вне шестичленного цикла:
о- О
S "0-5-0“ + + +
При этом сохранение бетаинного строения становится невозможным, молекула теряет ароматический характер и полученный суль-
фон оказывается способным реагировать как кетон, присоединять две молекулы брома, давать галохромное окрашивание с концентрированной серной кислотой.
Основным возражением против бетаинной формулы у-пирона и у-тиопирона является то, что они не обладают таким большим дипольным моментом, какого следовало бы ожидать при бетаинном строении. Величины дипольных моментов у-пирона и его производных больше отвечают формуле ненасыщенного циклического кетона.
Интересно отметить, что пироксониевые (пирилиевые) соединения образуются не только при действии на у-пирон галоидоводородных кислот, но и при действии галоидалкилов. Если подействовать на образующуюся при этом соль у-метоксипирона аммиаком, то происходит замена кислорода в катионе пирилия на имино-группу и получается соль у-метоксипиридина:
ОСН8-
нс сн
ЛНз
осн3
с нс^ ''сн
I II
НС сн
При действии на диметилпирон магнийиодметила получается производное пироксониевого основания
которое с хлорной кислотой образует прочную соль:
/\+/\ Н3С О сн3
OMgJ-
C107
Из этой соли при действии аммиака получается соль симметрического три-метилпиридина. Если же действовать едкой щелочью, то не выделяется свободное
пироксониевое основание, а происходит разрыв цикла и образуется непредельный ди кетон
СН3
НС=С—СН2
I I
сн3—С с-сн8
у-Пирон (темп, плавл. 32°, темп. кип. 217° при 742 мм), называемый также пирокоманом, может быть получен отщеплением СО2 от кислот у-пирона, например при нагревании хелидоновой кислоты (стр. 582).
Диметилпирон (темп, плавл. 132,1°, темп. кип. 249°, без разложения)
О
II
1Ы1
является одним из наиболее известных производных у-пирона. Он образует бесцветные кристаллы, легко возгоняется. Пары его чрезвычайно едки. Диметилпирон прекрасно растворяется в воде и спирте, хорошо растворим в эфире. С соляной кислотой он образует кристаллическую соль состава С7Н8О2-НС1-2Н2О.
Диметилпирон может быть получен действием НС1 на ди ацетил ацетон СН3СО—СН2—СО—СН8—СОСН3 или действием НС1 на дегидрацетовую кислоту 'стр. 582).
Кроме того, диметилпирон получается отщеплением двух молекул COt от кислоты строения
СО
НООС— \-COOH
JI 'I
Н3С—С С—сн3
^о^
эфир которой легко получается взаимодействием медного соединения ацетоуксусного эфира с фосгеном:
СОС1, СО
С2116ООС—НС—Си—СН—СООС.Н, -» С2Н4ООС-</ соос,н,
II II II
сн3—с с—сн3 сн3-с с-сн,
В соке хвойных растений очень распространен так называемый малыпол, или 2-метил-З-оксипирон, получающийся также при поджаривании солода; он образует бесцветные иглы (темп, плавл. 160,5°).
Хелидоновая и меконовая кислоты являются наиболее важными из при* родных соединений у-пирона:
О
НООО/'о/'\СООН
меконовая (3-оксихелидоновая) кислота
хелидоновая кислота
Хелидоновая кислота содержится в млечном соке чистотела (Chelidonium majus), меконовая—в опии в виде солей алкалоидов опия. Эги кислоты получаются также синтетически из ацетондищавелевой кислоты. Это очень сильные кислоты, трудно
растворимые в воде.
Хелидоновая кислота плавится выше ~260°; при дальнейшем нагревании она теряет СО2, превращаясь сначала в комановую кислоту, а затем в у-пирон.
Меконовая кислота теряет СО2уже при длительном кипячении с водой давая сначала коменовую кислоту, а затем [3-оксипирон, обладающий свойствами фенолов.
ООО
6 6/0Н fV0H
CJ^COOH о \соон
комановая кислота комеиовая кислота (3-оксипиров
Койевая кислота, или а-метилол-^'-окси-у-пирон (см. также том I, стр. 579)
представляет интерес вследствие того, что ее образование из глюкозы может служить примером возможности легкого превращения гексоз в простейшие производные пирона.
Дегидрацетовая кислота относится одновременно к группам а-и у-пирона. Она получается при нагревании паров ацетоуксусного эфира. Образование ее можно представить как конденсацию двух таутомерных форм ацетоуксусного эфира:
СО
СО—ОС2Н5
СН СН2—СОСН3
нс"7 ^Н—СОСН3
II I сн8-с со
+ 2С2Н6ОН
дегидрацетовая кислота
При действии концентрированных галоидоводородных кислот на дегидраце-товую кислоту получается диметилпирон.
Несомненно, что образованию диметилпирона предшествует размыкание пиронового кольца дегидрацетовой кислоты с присоединением воды и образованием промежуточного продукта, который с выделением СО2 и Н2О превращается затем в диметилпирон:
СО СО
/ \ юн / \
НС С=С< НС с—соон
II I 'СНд -> || ||
СНз-с^ соон СНз-с с-сн3
ОН но он
Дегидрацетовая кислота (кристаллы с темп, плавл. 109°, темп. кип. 270°) легко растворяется в воде, имеет кислую реакцию на лакмус.
ХРОМОНЫ И ФЛАВОНЫ
Соединение, в молекуле которого содержится бензольное ядро, сконденсированное с ядром у-пирона, называется бензо-^-пироному или хромоном'.
О II СН с /4\ НС6 С 3СН 3
I II II
НС7 С 2СН а
'Че/ \1/ сн о хромон
Хромон—бесцветное вещество, кристаллизующееся в иглах с темп, плавл. 59°.
Система, состоящая из дигидропиранового и бензольного циклов, получила название дигидробензопирана, или хромана:
СН СН2
НС6 С 3СН 2
I 11 I
НС7 С 2СН2
\8/ \i/ сн о хроман Эта система лежит в основе строения токоферолов (см. витамины группы Е, стр. 708).
При замещении атома водорода хромона в a-положении на фенильную группу получается а-фенилхромон, или флавон (темп, плавл. 99—100°); замещение водородного атома в p-положении дает (3-фенилхромон, или изофлавон:
О
п II СН с
^\/4\
НС* с зСН НС—сн
„I, II II 5'\
НС7 С 2С—Си ,4'СН
\8/ \1/ \* »'/
СН о нс=сн
о
II сн с
НС—сн
изофлавон
флавон
Флавон, как и хромон, бесцветен. Оксипроизводные флавона (и изофлавона) —желтые кристаллические вещества с высокими температурами плавления. Они растворимы в воде, в спирте, в разбавленных кислотах и щелочах. С солями тяжелых металлов они дают нерастворимые окрашенные осадки, ввиду чего издавна применяются как протравные красители для окраски тканей. Эти оксипроизводные часто именуются, не совсем правильно, вообще флавонами. Они широко распространены в растительном мире; обычно находятся в растениях в форме глюкозидов.
Строение природных флавонов установлено систематическими исследованиями Ст. Костаненкого* с учениками, разработавшего также пути синтеза этих веществ.
Из от (ельных представителей флавонов надо отметить следующие тетра-и пентаоксифлавоны.
3,7,3',4'-Тетраоксифлавон, или физетин (темп, плавл. 360°),—краситель, извлекаемый из сумахового дерева. 5,7,3',4'-Тетраоксифлавон, или лутеолин (темп, плавл. 328—329°),—краситель экстракта вау, извлекаемого из растения Reseda luteola\ содержится также в желтом пигменте почек тополя. Тетраоксифлавоны (в том числе и метилированные) входят в ‘ состав витаминов группы Р (см. стр. 707—708).
3,5,7,3',4'-Пентаоксифлавон, или кверцетин (темп, плавл. 313—314°),—краситель, обычно находящийся в виде глюкозида в коре Quercus tinctoria (в экстракте этой коры—кверцитроне) и во многих желтых цветах. Его 7-метиловый эфир— рамнетин—содержится в виде глюкозида в ягодах различных видов крушины (Rhamnus)\ эти ягоды употреблялись для крашения под названием «грушка» или «желтая ягода».
3,5,7,2',4'-Пентаоксифлавон, или морин (темп, плавл. 290°), содержится в экстрак >е желтого дерева Morus tinctoria наряду с 2,4,6,3',4'-пентаоксибензофе-ноном, или маклурином.
Строение флавонов устанавливается сплавлением их со щелочами, приводящим к отщеплению фенильной группы. По положению заместителей в бензольных
♦ Станислав Костанецкий (1860—1910). Польский химик. Работал в Швейцарии; профессор Бернского университета. Работы его были посвящены природным органическим красителям, их строению и синтезам.
ядрах продуктов, полученных при таком расщеплении, можно судить о строении исследуемого флавона, например:
ОН О
лутеолин
ОН
лутеолина, был осуществлен таким образов
Синтез флавонов, например (Костанецкий, Тамбор):
СН8О | со
fY \сн3 + с2н6оос-^Л-°сн3
CH3o/^\oCHs Х)СН3
флорацетофенон эфир вератровой кислоты
Производным изофлавона является содержащийся в виде глюкозида в соевых бобах 7,4'-диоксиизофлавон—дандзеин. Производными одновременно бензофурана и бензопирона являются действующие начала семян Ammi visnaga (растение из семейства зонтичных)—виснагин (темп, плавл. 139—140°) и келлин (темп, плавл. 153—154°), применяемые при лечении ряда сердечных заболеваний:
ОН О
виснагин
СН3О О
I II
келлин
АНТОЦИАНИДИНЫ
Как показали исследования Вильштеттера, к желтым красителям группы флавонов близки растительные пигменты, от которых зависит окраска цветов и плодов. Эти пигменты, названные антоцианами, являются глюкозидами и при нагревании с кислотами гидролитически расщепляются на сахара и красящие вещества— антоцианидины*.
По строению антоцианидины очень близки к флавонам, но в отличие от них относятся к классу пироксониевых оснований. Поэтому, хотя антоцианидины (антоцианы) не содержат атомов азота, •они тем не менее дают прочные ооли с кислотами. В то же время благодаря наличию фенольных гидроксилов они могут давать соли и с основаниями:
Исследование антоцианов из различных цветов и плодов привело . к поразительному выводу, что вся пестрая окраска цветочного ковра зависит в основном лишь от трех антоцианидинов: пеларгонидина, цианидина и дельфинидина'.
дельфинидин
♦ Некоторые антоцианы содержат кислоты (n-оксибензойную, п-оксикорич-ную и др.), этерифицирующие гидроксилы антоцианидина или углевода.
Эти антоцианидины и их метиловые эфиры в различных сочетаниях с кислотами и основаниями дают различную окраску. Так, например, синий цвет василька зависит от щелочной соли циани-дина, в то время как его оксониевая соль является причиной красной окраски розы и герани.
Содержание антоцианов в растениях колеблется в широких пределах—-вплоть до 7% от веса сухих цветов (в цветах пеларгонии и лесной мальвы).
Антоцианидины были получены Вильштеттером синтетически*, а также восстановлением желтых флавоновых красителей.
Так, например, кверцетин (стр. 584) при восстановлении магнием и соляной кислотой дает цианидин:
ОН О
ЮН
—ОН
осн3
ПГСНО сн3с/^Чэн
Многочисленные синтезы антоцианидинов были произведены Робинсоном и его сотрудниками.
Один из способов Робинсона можно проиллюстрировать на следующем примере:
сг
п-метокси-а>-метокси- тстраметиловый эфир
ацетофенон пеларгонидина
диметоксифлороглюциновый альдегид
К антоцианидинам оказались химически очень близкими дубильные вещества, относящиеся к группе катехинов (стр. 386).
Так, оптически недеятельный эпикатехин был получен Фрей-денбергом восстановлением цианидина водородом в присутствии платины:
цианидин-основание
эпикатехин
* Эти синтезы основаны на методах получения солей бензопирилия, разработанных уже раньше Деккером (1907 г.) и Веркиным (1903 г.).
КСАНТОН
Соединение, в молекуле которого с ядром у-пирона сконденсированы два бензольных ядра, называется дибензо-упироном, или ксантоном (бесцветные иглы, темп, плавл. 174°):
СН СО СН
У8\ /9\
НС7 С С 2 сн
I II II I
НС6 с с 3 сн
сн о сн
Ксантон получается синтетически отщеплением воды от фенилового эфира салициловой кислоты (салола):
О
НО—СО II
Ксантон не вступает в реакции, характерные для карбонильной группы. Восстановлением его получается вторичный спирт ксант-гидрол (мелкие бесцветные иглы, темп, плавл. 123°), который при действии кислот растворяется с желтой окраской, образуя соли оксониевого основания:
I I 1+н2о
СГ
Ксантгидрол дает с мочевиной очень трудно растворимое соединение— диксантилмочевину\ поэтому он применяется для определения мочевины, например, в крови, а также в других случаях, когда концентрация мочевины очень мала.
Оксипроизводные ксантона^ например эйксантон, или 4,6-ди-оксиксантон (темножелтый краситель, темп, плавл. 240°), являются составной частью некоторых природных красителей.
РАСТИТЕЛЬНЫЕ ИНСЕКТИЦИДЫ. РОТЕНОН И РОДСТВЕННЫЕ ЕМУ ВЕЩЕСТВА
Производными пиронов и, одновременно, кумаронов является большая группа растительных инсектицидов и ядов для рыб. Так, корка плодов Citrus bergamia (семейство Rutaceae) содержит бергаптен,. a Fagara xanthoxyloides, кроме того, еще и ксантотоксин'.
ОСН3 | СН нс-------/у ^сн
II I
нс\ /U\ /с=0 о о бергаптен
ксантотоксин
Более сложным веществом является пейцеданин, содержащийся в Рейсе danum officinale из семейства зонтичных (Шпет):
СН
СН3О-С-^сн
с"\н4 I I уС—о сн/с V^V
Тропические растения из семейства мотыльковых (Derric elliptica, Millettia towaniana Lonchocarpus, Thephrosia toxucaria), произрастающие главным образом на Филиппинах, на островах Зондского архипелага и в Малайе, но встречающиеся и в тропических областях Южной Америки и Африки, часто содержат ротенон и дегелин
ротенон
СН I (1 нс^ \о
СНз\с сн/ \ /V
О дегелин
а также некоторые другие, близкие к ним вещества. Из них наибольший интерес представляет ротенон (бесцветные таблички, темп, плавл 163°). Он оптически деятелен и является сильным инсектицидом. На рыб действует в разведении до 1 : 50 000 000, вызывая паралич центральной нервной системы. Для теплокровных животных ротенон сравнительно мало токсичен.
Ротенон и препараты измельченного корня дерриса получили широкое распространение для борьбы с вредителями сельскохозяйственных культур.
При действии очень мягких окислителей ротенон теряет два атома водорода и превращается в дегидроротенон, который при обработке щелочами дает дерри-совую кислоту (за счет размыкания у-пиронового кольца и кольца пирана после присоединения двух молекул воды):
деррисовая кислота
Группа пиридина
Пиридин (и его производные), наряду с тиофеном, является наиболее подробно изученным гетероциклическим соединением.
Обычная формула строения пиридина—с тремя двойными связями, предложенная Кернером в 1869 г., выражает глубокую аналогию его строения со строением молекулы бензола, от которой молекула пиридина отличается заменой одной группыСН на атом азота:
уСН
Р' НС5 3СН р
а' НС6 2СН а \1/ N
Формула пиридина Кернера имеет те же достоинства и недостатки, что и формула бензола Кекуле.
В соответствии с современными представлениями о строении бензола, для пиридина следует принять аналогичную формулу, отражающую взаимодействие it-электронов, приводящее к выравниванию электронной плотности в цикле:
пиридин бензол
• Соответственно длина связей С—С в пиридине, как и в бензоле, меньше длины простых алифатических связей С—С. В отличие от бензола, в пиридиновом-ядре имеется атом азота, сохраняющий неподеленную пару электронов. Этим обусловлены основные свойства пиридина и возможность образования иона пиридиния:
* Н
В реакциях электрофильного замещения (сульфирование, нитрование, галоидирование) пиридин ведет себя также несколько иначе, чём незамещенный бензол. Эти реакции протекают значительно труднее, чем в случае бензола, причем заместители вступают в ^-положение к кольцевому атому азота. Иначе говоря, пиридин напоминает в этом отношении нитробензол. При реакциях с нуклеофильными реагентами (едкое кали, амид натрия), наоборот, замещаются атомы водорода у а- и у-углеродных атомов. Например, с КОН пиридин дает калиевую соль а оксипиридина, подобно тому как нитробензол с порошкообразным едким кали дает о-нитрофенол. Интересно отметить, что с пиридином, как и с нитробензолом, но удается осуществить реакцию Фоиделя—Крафтса.
Формула Кернера предполагает возможность образования трех изомерных однозамещенных производных пиридина, обозначаемых а или а' (2 или 6), р или (3 или 5) и у (4). При двух одинаковых заместителях возможны шесть изомеров. Как и в формуле бензола Кекуле, при этом не принимается во внимание расположение двойных связей. При таком условии все выводы относительно числа изомеров и определения места вступления заместителей вполне совпадают
с опытом. В настоящее время имеется уже очень большое число производных пиридина, для которых положение замещающих групп известно. Практически при определении положения заместителей производные пиридина стараются превратить в соответствующие пиридинкарбоновые кислоты, свойства которых хорошо изучены.
Для гомологов пиридина широко приняты эмпирические наименования. Так, метилпиридины C6H7N называются пиколинами, гомологи состава C7HyN и, в частности, диметилпиридины называются лутидинами, основания состава C8HnN—коллидинами, С9 Н13N—парвол инами.
Пиридин и его гомологи содержатся в каменноугольном дегте, в продуктах сухой перегонки дерева, торфа и др. и особенно много— в продуктах сухой перегонки животных остатков (например, костей). Образование пиридиновых соединений, вероятно, объясняется реакциями конденсации с аммиаком альдегидов и ацетилена, могущих образоваться при сухой перегонке. Из каменноугольного дегтя пиридиновые основания извлекают кислотами. Пиридин и его производные' находят применение в качестве растворителей, для денатурирования спирта, для синтеза лекарственных препаратов. Производные пиридина чрезвычайно распространены в растительном мире. К их числу относятся многие растительные алкалоиды.
Способы получения пиридина и его гомологов. 1. Конденсация ацетилена с синильной кислотой. Аналогично получению бензола из ацетилена (стр. 203 и том I, стр. 341} пиридин получается при пропускании через раскаленные трубки смеси ацетилена и синильной кислоты (Рамзай, 1877 г.):
СН СН
\\\ / Ч
НС сн НС сн
III 4- -> II I
НС сн НС сн
/// \ Z
N N
2. Конденсация альдегидаммиаков. Одним из наиболее старых синтетических способов получения гомологов пиридина является нагревание альдегидаммиаков. Так, нагреванием акролеинаммиака получен ^-пиколин (Байер, 1870 г.), а нагреванием уксусного альдегида с аммиаком—коллидин, имеющий строение а-метил-f/-этилпиридина и называемый альдегидколлиди-ном (также альдегид ином). При нагревании пропионового альдегида с аммиаком получается парволин (Вааге, 1882 г.).
Строение этих гомологов пиридина, а также уравнения реакций их образования были установлены сравнительно давно. Однако лишь исследованиями А. Е. Чичибабина, М. П. Опариной, П. А. Мошкина и др была выяснена общность реакций образования пиридиновых оснований из альдегидов и аммиака, а также доказана возможность при этом различных путей их образования, ведущих к получению оснований, ранее не полученных таким способом. Кроме того, применение в качестве катализатора окиси алюминия чрезвычайно облегчило получение этим путем оснований и привело к повышению выходов.
Как показал А. Е. Чичибабин, реакции протекают следующим образом:
а) Для насыщенных альдегидов типа уксусного альдегида и его однозамещенных гомологов возможны два направления реакции:
R СНО R
Н// СН2 + I онс сно
R—Н2сГ NH3
+ ЗН20 + Н3
Из уксусного альдегида (R=H) получаются таким образом а- и у-пико-лины. из пропионового альдегида (R=CH3)—два парволина (а-этил-Р,Р'-диметил-и f-этил-р.Р'-диметилпиридины) и т. д.
б) Для ненасыщенных альдегидов (однозамещенных гомологов акролеина типа R—СН=СН—СНО) реакция образования пиридиновых оснований также может итти в двух главных направлениях:
СНО СН—R
/ //
НС сн
II + I R—НС СНО
NH3
R-CH CH-R R
Из акролеина (R=H) в обоих случаях получается Р-пиколин, но из кротонового альдегида (R=CH3), а также из уксусного альдегида, образующего при конденсации кротоновый альдегид, получаются два коллидина: 2-метил-5-этилпири-дин, или альдегидколлидин, и 4-метил-З-этилпиридин, или Р-коллидин. Из 2-ме-тил-5-этилпиридина может быть получен 2-метил-5-винилпиридин—важный мономер для синтеза каучука.
Применяя смесь двух альдегидов, а также смеси альдегидов с кетонами, можно синтезировать основания, которые нельзя получить из одного альдегида. Например, из смеси акролеина и уксусного альдегида получается пиридин:
СН, нс^ сн3 I + I онс сно
NH,
+ 2Н,О4-Н,
3. Каталитическая конденсация ацетилена с аммиаком. При пропускании ацетилена вместе с аммиаком над окисью алюминия при 400—450° получается смесь тех же пиридиновых оснований, что из уксусного альдегида с аммиаком (А. Е. Чичибабин). Вероятно, ацетилен с водой образует уксусный альдегид, который и реагирует с аммиаком. При пропускании же ацетилена с аммиаком через раскаленные трубки при температуре выше 600° без катализаторов получаются пиридин и смесь его гомологов, что возможно лишь при расщеплении молекулы ацетилена (Р. Мейер).
4. Получение производных пиридина из
производных пирона (стр. 577—578).
5. Получение производных пиридина из производных пиррола с расширением пятичленного цикла в шестичленный (стр. 533).
6. Конденсация ацетоуксусного эфира с альдегидаммиаками. Довольно широкое применение для получения производных пиридина имеет синтез Ганча. Он состоит в конденсации ацетоуксусного эфира с альдегидаммиаками, причем получаются дигидропиридиновые основания, легко окисляющиеся (иодом, азотистой кислотой и пр.) в пиридиновые основания. Так, конденсацией ацетоуксусного эфира с ацетальдегидом и аммиаком получается дигидроколлидиндикарбоновый эфир, а из него—кол-лидиндикарбоновый эфир. Омылением этого эфира получается двухосновная кислота, которая при нагревании с известью теряет 2СО2 и переходит в симметрический коллидин*.
СН3 СН8
СНО
ROOC-CH2 ch2-coor I + I СНз-СО со-сн3 NH3
сн3 сн3
ROCX^J^OOR ' X
-> I II I II
н3с/ VXCH3 н3с/\/хсн3
7. Перегруппировка галоидалкилатов пиридина. Гомологи пиридина могут получаться из самого пиридина путем нагревания его галоидалкилатов. При этом алкил вступает в ядро главным образом в а- и у-положения (Ладенбург):
- СН3~
Хлористая медь и хлористый алюминий являются катализаторами реакции (А. Е. Чичибабин).
этой
Таблица 38
Гомологи пиридина
Температура Температура Показатель
Название плавления °C кипения °C Удельный вес преломления Пй
Пиридин —42,0 115,3 0,9826 (4°) 1,5096 (при 20°)
Пиколины (метилпиридины):
а-, или 2-метилп иридии —64,2 128,8 0,954 1,5014 (при 20°)
р-, или 3-метилпиридии --20,8 143,8 0,9564 (d20) 1,5063 (при 20°)
у-, или 4-метилпиридин 4-1,6 145,4 0,9502 (при 25°) 1,5050 (при 20°)
Лутидины (диметилпиридины): 143,9
а,эс'-, или 2,6-диметил- — 0,9303 И5) 1,4953
пиридин 163,4 \ .4 / (при 25°)
а,^-, или 2,3-диметил- — —
пиридин а, у-, или 2,4-диметил- — 157 0,9271 1,4984
пиридин 163,5—164,5 (при 25°) (при 25°)
р,у-, или 3,4-диметил- — 0,9537 1,5099
пиридин 171,6 (при 25°) (при 25°)
₽,Р'-, или 3,5-диметил- — 0,9385 1,5032
пиридин 159—160 (при 25°) (при 25°)
а,Р'-, или 2,5-диметил- — 0,9261 1,4982
пиридин (при 25°) (при 25е)
Этилпиридины: а-, или 2-этилпиридин — 149 0,9411 (d<2'6) 0,9466 (42,8) 1,5016* (при 12,6° >
Р-, или 3-этилпиридин — 166 1,5069* (при 12,8°)
у-, или 4-этилпиридин — 165 0,9417 (d20) 1,5010
2-Метил-5-этилпиридин — 174—175 0,9184 (при 23°) —
P-Коллидин (4-метил-3-этилпи- — 196 0,9656 (при 0°) —
ридин) Симметрический коллидин 171 1,4977*
0,9166 (d22-1)
(2,4,6-триметилпиридин) (при 22,1°)
Конирин (2-, или а-пропил- 165,8 — —
пиридин)
• Помеченные звездочкой величины показателя преломления соответствуют желтой гелиевой линии (пт. ).
Не'
Физические свойства (табл. 38). Пиридин и его низшие гомологи смешиваются с водой, но высаливаются из нее солями, особенно содой г поташом. Они обладают резким, своеобразным запахом. По мере увеличения молекулярного веса растворимость в воде уменьшается, а запах делается болеё слабым и иногда приятным. Со спиртом, эфиром, бензолом и многими другими обычными растворителями низшие пиридиновые основания также смешиваются. С водой пиридин образует азеотропную смесь, приблизительно отвечающую составу C6H5N+3H2O и кипящую при 92—93°. Пиридин и его низшие гомологи обладают способностью легко растворять многие органические и минеральные вещества.
Химические свойства. 1. Основные свойства. Пиридин и его гомологи являются слабыми основаниями. С сильными минеральными и органическими кислотами они образуют кристаллические соли, в водных растворах сильно гидролизованные; не дают прочных солей ни с угольной кислотой, ни даже с кислотами такой силы, как уксусная кислота и ее гомологи. Константа диссоциации пиридина К20=1,8-10“9. Пиридин и его гомологи обладают большой склонностью к образованию двойных солей и других комплексных соединений.
Пиридин и его гомологи присоединяют галоидные алкилы, давая галоидалкилаты—соли четвертичных аммониевых оснований, называемых также пиридиниевыми основаниями. Например, с йодистым метилом получается иодметилат пиридина, или иодистый ме-тилпиридиний (темп, плавл. 118°):
Л
\+^ J _ N—СН3_
При действии на галоидалкилаты NaOH или AgOH получаются свободные пиридиниевые основания, способные реагировать внескольких таутомерных формах:
пиридиниевое основание
вторичный спирт (псевдооснование)
ненасыщенный жирный аминоальдегид
Существованием этих трех таутомерных форм объясняется чрезвычайное разнообразие реакций пиридиниевых оснований. При действии кислот на свободные основания снова получаются галоидалкилаты, т. е. соли основания. При окислении они реагируют, вероятно, в форме альдегида. Получающаяся кислота затем с выделением Н2О замыкается снова в пиридиновое кольцо и образуется кетон, соответствующий таутомерному вторичному спирту,—^-алкилпиридон
G-
В альдегидной форме пиридиниевые основания легко вступают в реакции конденсации с различными веществами, способными конденсироваться с альдегидами.
Установление ряда таутомерных превращений пиридиниевых оснований (Деккер, Ад. Кауфман и др.) имело большое значение для объяснения свойств многих природных алкалоидов.
При действии на пиридин веществ, имеющих характер галоидангидридов или ангидридов, например бромистого циана, сернистого ангидрида (или бисульфита), серного ангидрида и пр., получаются продукты присоединения к азоту.
Как и иодметилаты, такие N-производные легко превращаются, особенно под действием щелочей, в соединения алифатического ряда и являются, повидимому, производными одной из таутомерных форм глутаконового альдегида
СН
Из продукта присоединения к пиридину эфира хлорсульфоновой кислоты удалось получить таким путем и сам глутаконовый альдегид:
| j СНО —2^ II СНО + NHsSO8Na + C2HSOH bf Cl' 'nh V)Na
I |
SO,OC2Ht SO,OC,H8 c. i сн
/ 'Ч / 4.
НС CH 112C сн
Hi -> i I HC CHO OHC CHO
\oNa
Эта реакция является как бы подтверждением для пиридина формулы строения Кернера.
Некоторые из N-производных пиридина являются веществами сильно окрашенными («пиридиновые красители»). Например, продукт присоединения к пиридину динитрохлорбензола реагирует с анилином с образованием яркокрасного вещества
2. Реакции окисления. Кольцо пиридина является не менее стойким по отношению к окислителям (СгО8, КМпО4, HNO3 и т. д.), чем кольцо бензола.
Аналогично гомологам бензола, гомологи пиридина окисляются в пиридинкарбоновые кислоты.
3. Реакции замещения. Пиридин сульфируется, нитруется и бромируется значительно труднее, чем бензол. При этом всегда получаются (3-замещенные производные.
Таким образом, азот пиридинового кольца оказывает такое же ориентирующее влияние в реакциях замещения, как группа NO2 в нитробензоле, являющаяся, как правило, мета-ориентантом (стр. 241).
При действии галоидов на пиридин и его гомологи сначала получаются нестойкие продукты присоединения, вероятно N-галоидо-производные, а затем уже—3"галоидозамещенные.
4. Реакция с амидом натрия. При действии на пиридин амида натрия происходит выделение водорода и образуются аминопроизводные пиридина (А. Е. Чичибабин, О. А. Зейде):
4- NHoNa
А. + н, 'V^'NHNa
А + Н26
Xr/\NHNa
+ NaOH
'/\nh.2
При этом получаются главным образом а-аминопроизводные и небольшие количества у-аминопроизводных. у-Производные образуются в значительном количестве лишь в том случае, когда оба a-положения уже замещены.
При дальнейшем действии амида натрия получается а,а'-ди-аминопиридин (бесцветные пластинки, темп, плавл. 122°).
5. Действие безводных щелочей. При действии на пары пиридина сухого порошкообразного едкого кали (при температуре выше 400°) также выделяется водород и образуются калийные соли (феноляты) а-оксипиридинов (А. Е. Чичибабин):
(А + КОН -> А +Н,
6. Реакции восстановления. Чрезвычайно важными являются реакции восстановления пиридиновых оснований. При этом получаются продуиты присоединения двух атомов водорода (дигидропиридины), четырех атомов водорода (тетрагидропиридины,
или пиперидеины) и шести атомов водорода {гексагидропиридины, или пиперидины)'.
СН2 'сн, I ! Н2С сн2 \н
Пиперидины легче всего получаются при восстановлении пиридиновых оснований натрием в кипящем абсолютном спирте (реакция А. Н. Вышнеградского и Ладенбурга) или каталитическим восстановлением водородом в присутствии коллоидных платины или палладия (Н. Д. Зелинский). Восстановление пиридиновых оснований можно успешно вести и электрохимически.
7. Реакции конденсации, а- и у-Гомологи пиридина, особенно содержащие метильные группы, обладают характерной способностью вступать в реакции конденсации с альдегидами и фталевым ангидридом. Реакции легче протекают в присутствии катализаторов (ZnCl2) и в зависимости от условий могут иттис выделением или без выделения Н2О, например:
О + сн3—сно |р] • + н2о
V'4ch8 \^\зн=сн-сн3
Pl + СН2О -> f4]
V'\3H3 Хг/'Ч'СН2-СНгОН
В первом случае получаются производные пиридина с этиленовыми связями в боковой цепи, во втором—пиридиновые алкоголи.
Кроме алкоголей, получаемых конденсацией с одной молекулой СН2О, при конденсации с муравьиным альдегидом легко получаются также гликоли (с двумя молекулами СН?О) и трехатомные алкоголи (с тремя молекулами СН2О):
и п
х/\;н(сн3он)3 V^C(Ch2oh)3
Продукты конденсации пиколинов с ароматическими альдегидами называются гтильбазолами, а гидрированные стильбазолы (продукты присоединения к стильб-: золам восьми ^атомов водорода)—стилъбазолинами*.
стильбазол
сгильбазолин
Конденсацией пиридиновых оснований с фталевым ангидридом получаются пиридофталоны. Из них хорошо изучены лишь фталоны бензопиридина (хинофта-лоны, стр. 617).
Высокая реакционная способность метильных групп а- и у-пиколинов возможно связана с таутомерными превращениями двух форм:
и
\ ZVu
NH СН2
а-метилпиридин а-пиридонметид
Еще легче, чем а- и у-алкилпиридины, вступают в реакции конденсации га-лоидалкилаты а- и ^-пиколинов. Так, они легко реагируют с солями диазония, давая гидразоны, и с амилнитритом (в присутствии алкоголятов), ^образуя изонитрозосоединения (оксимы):
+ СбНп—ONO —>
Lch3—]
J- + C6HnOH
Галоидпроизводные пиридина. а-Галоидозамещенные пиридины могут быть получены действием на N-алкилпиридоны (стр. 595) пятихлористого фосфора в смеси с РОС13 (метод О. Фишера):
РС1з
СН3—
таутомерное ------------->
превращение
С Г нагревание
----LZ2-->
СНз—N С1
Однако а- и у-галоидпиридины проще получать из аминопиридинов (стр. 601).
p-Галоидпиридины получаются непосредственным галоидированием пиридина.
Галоидозамещенные пиридины, особенно а- и у-замещенные, легче, чем галоидопроизводные бензола, вступают в реакции обмена. В этом проявляется их сходство с о- и л-хлорнитробензолами.
С алкоголятами галоидпиридины образуют эфиры оксипиридинов, с аммиаком—аминопиридины\ особенно же легко они реагируют с гидразином и его производными и с KSH, образуя, например, a-пиридилгидразин H2N—NH—C5H4N и %-пиридилмеркаптан HS-C5H4N.
(З-Пиридинсульфокислота, сравнительно легко получаемая путем сульфирования пиридина, при сплавлении с едким кали дает $-окси-пиридин, а при сплавлении с цианистым калием—$-цианпиридин, или нитрил никотиновой кислоты (см. стр. 604).
Оксипиридины, а- и у-Оксипиридины впервые были получены путем ряда реакций из производных а- и у-пирона. В настоящее время их получают взаимодействием пиридина с едким кали (стр. 597), благодаря чему а-оксипиридин является одним из самых доступных производных пиридина.
Оксипиридины—кристаллические вещества, легко растворимые в воде; они дают соединения с кислотами, а с едкими щелочами образуют соли (подобные фенолятам), разлагаемые угольной кислотой.
а- и у-Оксипиридины таутомерны пиридонам'.
ОН о
а-оксипиридин а-пиридон 7-ОКСИПИрИДИН 7-ПИрИДОН
Это один из наиболее давно известных случаев таутомерии, на которых устанавливалось само это понятие. Однако до настоящего времени не выяснено, какое из двух таутомерных строений следует приписывать известным формам а- и у-пиридонов. Повидимому, в нейтральных растворах преобладает пиридонная форма.
При действии галоидных алкилов на пиридоны или их натриевые соли получаются N-производные пиридонов, например N-этил-а-пиридон (темп, плавл. +7°, темп. кип. 250°) и ^-метил-^-пиридон (темп, плавл. 89°)
О II С
N-этил-а-п при дон ЬГметил-^-пиридон
а при действии иодистых алкилов на серебряные соли* пиридинов— эфиры оксипиридинов, например а-этоксипиридин (темп. кип. 156°) и у-метоксипиридин (темп. кип. 191°):
ОСН3
\/чОС,Н8
а-этоксипиридин у-метоксипиридин
При высоких температурах эфиры оксипиридинов изомеризуются в алкилпиридоны.
О витамине Вв (адермин, или пиридоксин), являющемся оксипроизводным а-пиколина, содержащим две метилольные группы, см. стр. 709.
Аминопиридины были впервые получены из амидов пиридинкарбоновых кислот реакцией Гофмана (см. том I, стр. 252 и 282), т. е. действием брома в щелочном растворе. В настоящее время а-аминопиридин может быть чрезвычайно легко получен действием амида натрия на пиридин.
В то время как (3-аминопиридин по химическим свойствам подобен анилину, а- и 7-аминопиридины обнаруживают ряд особенностей, отличающих их как от анилинов, так и от жирных аминов.
В кислых растворах а- и раминопиридины существуют в виде катионов, строение которых ближе к строению амидинов (см. том I, стр. 283):
Поэтому они дают соли только с одним эквивалентом кислоты, в то время как ,8-аминопиридин
образует, например с соляной кислотой, дихлоргидрат.
При действии на соли а- и p-аминопиридинов азотистой кислоты не получаются диазониевые соли, а сразу выделяется азот и в присутствии кислородсодержащих кислот образуются пиридоны (оксипиридины). В присутствии концентрированной соляной кислоты происходит как бы диазореакция и из а-аминопиридина получается а-хлорпиридин.
Действием на а-аминопиридин амилнитрита в присутствии алкоголятов или'амида натрия получаются соли истинных диазосоединений, например:
О +C6Hn-ONO + C2H5ONa -» [Р) +С6НПОН + С2Н6ОН
4j/4NH, ''N^XN=N—ONa
Эти диазотаты способны сочетаться в спиртовом растворе с аминами и фенолами, образуя азокрасители. При действии на них кислот происходят диазореакции: например, с HJ они реагируют с выделением азота и образованием а-иодпиридина.
а- и у-Аминопиридины, как и оксипиридины, могут реагировать в двух таутомерных формах. Например, при взаимодействии с иоди-стыми алкилами в зависимости от условий реакции получаются производные либо аминопиридина, либо пиридонимина'.
и г\
• VXNHCH3 CHS-/XNH
метиламинопиридин N-метилпиридонимин
При наличии в пиридиновом ядре гидроксильных групп и аминогрупп чрезвычайно облегчаются реакции замещения. Тогда как
сам пиридин лишь с большим трудом нитруется, сульфируется, бромируется и пр., аминопиридины и оксипиридины вступают в реакции замещения почти с такой же легкостью, как анилин и фенол, ориентируя входящие радикалы, как и в бензольном ряду, в орто- и пара-положения.
Как показали исследования А. Е. Чичибабина, при действии нитрующей смеси на все моноаминопиридины на холоду нитруется прежде всего аминогруппа, например:
|Р] + HNO3 -> А + Н,0
V^NH, V\NH-NO2
Получаемый при этом а-пиридилнитрамин (белые иглы, темп, плавл. 184° с разл.) относится к наиболее прочным представителям нитраминов. Тогда как фенилнитрамин С6Н5—NH—NO2 уже в присутствии следов кислот изомеризуется в смесь о- и п-нитроанили-нов, а-пиридилнитрамин изомеризуется только под действием концентрированных кислот при нагревании, образуя смесь 2-амино-З-нитро-пиридина и 2-амино-5-нитропиридина:
и VXNH,
Восстановлением а-пиридилнитрамина можно получить а-пиридилгидразин (бесцветные иглы, темп, плавл. 46—48°)
п
nh,
который быстро окисляется на воздухе, превращаясь в темную смолу.
а-Аминопиридин легко реагирует (в таутомерной пиридиниминной форме) с образованием бициклических соединений. Например, с малоновым эфиром он дает соединение, содержащее кольцо пиримидина (стр. 633):
NH N
СООС2Н5 \со
1+1 -> I I +2С2Н5ОН
L/NH СН2 k/N\
(SgHjOOc/ со
С хлор ацетоном а-аминопиридин образует метилпиримидазол, в котором сочетаются пиридиновое и имидазольное кольца (стр. 568):
NH СО—СН,
II I
С 4- СН, if 'кн ci
+ НС1 + Н,0
Важнейшие характеристики галоид-, нитро-, амино- и оксипиридинов приведены в табл. 39.
Таблица 39
Производные пиридина
Название Температура плавления °C Температура кипения °C Удельный вес
Хлорпиридины
а- — 170 1,205 (при 15°)
р- — 148 (при 744 мм) —
т- — 147—148 —
Бромпиридины:
а- — 193 1,657 (при 15°)
₽- — 69 1,645 (d°)
Иодпиридины:
а- — 93 (при 13 мм) 2,004 (при 0°)
? +53,5 — —
V 100 (с patл.) — —
Нитропиридины:
а- 71 256 —
₽- 41 216 —
Аминопиридины:
а- 57,5 210 —
Р- 65 251 —
Y- 158 — —
Ацетаминопиридины:
а- 71 —— ——
₽- 133 — —
Y- 155 — —
Оксипиридины (пиридоны): 107 281
а- ——
₽- 129,5 300 (без разл.) —
Y- 148,5 (безв.) 92 (с 1 мол. Н2О) 350 (с небольш. разл.) —
а-Пиридилкарбинол 112 (при 16 мм) —
а-Пиколилкарбинол — 115 (при 9 мм) 1, ПН(^)
а-Пиридинальдегид — 181 —
а-П ир и ди л метил кетон — 192 —
Пиридинкарбоновые кислоты легче всего получаются окислением гомологов пиридина. а-Пиридинкарбоновая кислота, или пиколиновая кислота, хорошо растворима в воде; (3-кислота, называемая никотиновой (так как она была получена окислением алкалоида никотина), растворима лишь в горячей воде, а -[-кислота, или изоникотиновая, трудно растворима и в горячей воде и в спирте.
Многие одноосновные пиридинкарбоновые кислоту образуют характерные медные и серебряные соли. Пиколиновая кислота, а также все кислоты, содержащие хотя бы один карбоксил в а-положении, дают с солями закиси железа характерную желтую или краснооранжевую окраску.
Одноосновные пиридинкарбоновые кислоты являются очень слабыми кислотами; они способны образовывать непрочные соли также и с сильными минеральными кислотами.
Пиридинкарбоновые кислоты
Таблица 40
Название кислоты Положение карбоксильных групп Температура плавления °C Константа диссоциации Полные ме силовые эфиры
температура плавления °C температура кипения °C
Пиколиновая 2- (а-) 139 0,3 ю-5 + 14 232
Никотиновая 3- (Р-) 234—237 1,4-10-5 38 204
Изоникотиновая 4- (у-; 317 1,09-10~5 8,5 208
Хинолиновая 2,3- (а,?-) 190—195 3,2-10~3 54—55 —.
Лутидиновая 2,4- (а, у-) (ангидрид плавится при 134,5°) 248—250 7,010—3 60
Изоцинхомеро- 2,5- (а,3'-) 254 4,5-10~3 164 —
новая Дипиколиновая 2,6- (а,а'-) 252 6,2-10~3 121
Цинхомероновая 3.4- (3.Y-) 266—268 2,2-Ю-3 141 —
Диникотиновая 3.5- (3,3'-) (ангидрид плавится при 77—78°) 323 1,6-10—3 85
Бербероновая 2,4,5- (а,у,3'-> 243 — —— —
Пиридинкарбоновые кислоты образуют сложные эфиры, хлор-ангидриды, амиды и другие производные кислот. При нагревании с известью они отщепляют СО2; а-кислоты отщепляют СО2 уже при простом нагревании.
Важную физиологическую роль играет ^-пиридинкарбоновая, или никотиновая, кислота и ее производные.
Амид никотиновой кислоты (бесцветные .кристаллы, плавящиеся при 129— 131°), как и сама никотиновая кислота, является антипеллагрнческим* витамином (стр. 709). Он входит в состав ферментных систем, участвующих в процессах клеточного дыхания (Варбург, Г. Эйлер).
Диэтиламид никотиновой кислоты обладает физиологическим действием, подобным действию камфоры; он находит применение в медицине как средство, регулирующее деятельность сердца и кровообращение, под названием корамина.
Гидразиды изоникотиновой кислоты и продукты конденсации этих гидразидов с альдегидами и кетонами—гидразоны
гидра? ид
СО—NH—N- CH—R
гидразон
находят в последнее время широкое применение в терапии туберкулезных заболеваний (препарат фтивазид и др.).
Продукты присоединения водорода к пиридиновому ядру
Пиридин, его гомологи и производные могут восстанавливаться в различных условиях с присоединением двух, четырех и, наконец, шести атомов водорода, образуя гидропиридиновые основания. Наибольшее значение имеют продукты присоединения шести атомов водорода, или пиперидиновые основания.
Простейшим представителем этих оснований является пиперидин:
СН,
Н,^ \н, I I н,с сн,
Названия гомологов пиперидина производятся либо от пиперидина (например, 2-, или а-метилпиперидин), либо от названий соответствующих гомологов пиридина со вставкой второго слога «пе». Например, от а-, [3- и у-пиколинов производятся а-, р- и ^-пипе-колины; от лутидинов—лупетидины; от коллидинов—копеллидины и т. д. Соединения, замещенные на алкилы при азоте, называются N-алкилпиперидинами.
Начиная от пипеколинов, многие гомологи пиперидина содержат асимметрические атомы углерода, например
СН, н// \н, I I Н,С *сн—сн, \н
и потому могут существовать в оптически деятельных формах. Стереохимия пиперидиновых оснований аналогична стереохимии производных циклогексана (стр. 29 сл.).
Способы получения. Гидропиридиновые основания могут быть получены не только восстановлением пиридинов, но также и из алифатических соединений рядом синтетических реакций. В большинстве случаев способы их получения аналогичны способам получения пирролидина и его производных (стр. 535).
1. Нагревание солей пентаметилендиамина и его гомологов:
сн,2
НоС7 ^СНз
I I
Н2С сн2
I I
HC1-H2N nh2-hci
СН2
н2с сн2
I I + nh4ci
Н2С сн2
\h-hci
2. Действие бромид и его
аммиака на пентаметилендипроизводные. Например:
СН2 'сн,
ВгН2С СНВг—СООН
2NH,
СН2
Н2(^ \н2
I I 4-NH.Br
Н2С СН-СООН
\ii-HBr
3. Действие аммиака на некоторые бромкетоны. Из бромкетонов, содержащих группу СО и Вг в положениях 1 и 5, при действии NHg получаются аминокетоны, которые с выделением Н2О превращаются в пи-перидеины (тетрагидропиридины):
СН2 СН2 СН2
nh3 н2с сн2 Н2С СН
I I ----------> || -> I II + Н2О
н2с со—сн3 Н2С со—сн, Н2С С—СНз
\г \н2
4. Циклизация иминодипропионового альдегида. В результате циклизации иминодипропионового альдегида получается тетрагидропиридиновый альдегид:
( СНО СН
/ / \
Н2С СН.-СНО Н.С с—сно
II -> I I + Н2О
Н2С СН. Н2с СП,
\н
Свойства. Пиперидин, а также его гомологи и производные являются очень сильными вторичными основаниями, имеющими своеобразный запах. С азотистой кислотой пиперидин дает нитрозопиперидин , с уксусным ангидридом—ацетилпиперидин (темп, плавл.
109°; темп. кип. 227°), с бензоилхлоридом—бензоилпиперидин (темп, плавл. 48°):
\н, I I н2с сн2
NO
нитрозопиперидин
сн2 ''сн, I I н2с сн2 \-сосн3 ацетилпиперидин
сн2
H2(S 'сн, I I н2с сн2
\-сос6нБ
бензоилпиперидин
С метилирующими средствами получается сначала N-метил-пиперидин, а затем соль четвертичного аммониевого основания:
Исчерпывающее метилирование по Гофману протекает так же, как и с пирролидином (стр. 536), но вместо ожидаемого дивинилме-тана СН2=СН—СН2—СН=СН2, после отщепления триметиламина получается пипериленСН2=СН—СН—СН—СН3 (см. томД, стр. 346).
Реакции Брауна (стр. 536 и 537) для пиперидина протекают вполне аналогично реакциям с пирролидином.
Пиперидиновые основания весьма стойки по отношению к восстановителям. Однако восстановление в очень жестких условиях, например действием концентрированной иодистоводородной кислоты при нагревании, приводит к размыканию кольца с выделением аммиака и образованием нормального пентана:
СН2
\н2
2Н2
----> СН,—CIV-CH1-CH1-CH84- nh3
Окислители действуют с различной легкостью. Так, пиперидин довольно устойчив к хромовой кислоте, азотной кислоте и перманганату в кислом растворе, особенно при обычной температуре, но восстанавливает аммиачное серебро с образованием серебряного зеркала. При нагревании с солями серебра или окиси меди, а также с концентрированной серной кислотой он может терять шесть атомов водорода и превращаться в пиридин.
Характерно действие концентрированной перекиси водорода на пиперидин и N-метилпиперидин. Из N-метилпиперидина при этом получается N-окись
СН2
Нас/ ^СНа
I I
Н2С сн2
-o' ^CHj
а из пиперидина—аналогичное соединение, способное к таутомерным превращениям:
Пиперидин получается восстановлением пиридина, различными синтетическими методами, а также омылением пиперина (алкалоид черного перца, являющегося производным пиперидина и пипериновой кислоты (стр. 412):
Н2С—С\ сн2—СН
I / \2
О СН=СН—СН=СН—СО— N сн2
\н,—С"Н2
Пиперидин—щелочная жидкость резкого своеобразного аммиачного запаха, смешивающаяся с водой.
d-a-Пропилпиперидин—природный алкалоид болиголова (Coni-ит maculaturn), называемый кониином, отличается чрезвычайной ядовитостью, значительно превосходящей ядовитость самого пиперидина. Синтез кониина, исходя из a-пиколина, был первым синтезом природного алкалоида (Ладенбург*, 1886 г.).
Конденсацией a-пиколина с уксусным альдегидом был получен аллилпиридин C5H4N—СН=СН—СН3, который был восстановлен
* А. Ладенбург (1842—1911). Крупный немецкий химик-органик. Ему принадлежит много работ по выяснению положения заместителей в бензольном ядре, доказательство равноценности шести атомов углерода в бензоле (ср. призматическую формулу бензола). В содружестве с французским химиком Ш. Фриделем он положил основу химии кремнийорганических соединений. В 1885 г. установил строение шести карбоновых кислот пиридина.
натрием в абсолютном спирте. Полученный а-пропилпиперидин (недеятельный кониин)
СН2
н2с \н2
н2с in—сн2—сн2—сн3
кристаллизацией солей d-винной'кислоты был разделен на /-кониин и d-кониин (тождественные природным алкалоидам).
К числу производных гидрированных пиридинов относится ряд других алкалоидов (см. раздел об алкалоидах, стр. 648 сл.).
Важнейшие свойства некоторых пиперидиновых оснований приведены в табл. 41.
Таблица 41
Пиперидиновые основания
Название Температура плавления °C Температура кипения °C Удельный вес Показатель преломления nD
Пиперидин —9 106,3 0,8622 (df°) 1,4534 (при 20°)
а-Пипеколин-(а-, или 2-метилпиперидин)неак- 0,8436 (df>6)
тивный —5 119 1,4464 (при 23,6°)
Р-Пипеколин (р-, или 3-ме-тилпиперидин) неактив- 0,8446 (d*4’3)
ный — 126 1,4378 (при 21,6°)
у-Пипеколин (у-, или 4-метилпиперидин) — 126,5—129 0,8674 (при 0°) —
N-Метилпиперидин (1-ме-тилпиперидин) — 105,9 0,8207 (df) 1,4378 (при 20°)
d-Кониин (а-, или 2-я-про-пилпиперидин) . —2,5 166,5 0,8430 (d*1-9) 1,4512 (при 21,9°)
Дипиридилы
Известен ряд соединений, содержащих в молекуле два пиридиновых ядра. Таковы, например, дипиридим, для которых возможно 6 изомеров Из них наиболее доступны а,а'-дипиридил, получающийся при перегонке медной соли пиколиновой кислоты, и 7,у'-дипиридил, легко образующийся вместе с небольшим количеством а,а'-изомера при действии натрия на пиридин (при этом выделяется водород):
а.а'-дипиридил
7,7'-дипиридил
а,а'-Дипиридил образует бесцветные кристаллы, плавящиеся при 69,5% и перегоняется при 273°; 7,у'-дипиридил плавится при 73° и кипит при 304,8°.
Арсепидины
Аналог пиперидина, содержащий атом трехвалентного мышьяка вместо атома азота, называется арсепидином. Ряд производных арсепидина получен действием на алкилдихлорарсины магнийорганического соединения, получаемого из 1,5-дибромпентана:
СН2
/ \ сн2
Н2С сн2 / \
I I Н2С сн2
Н2С СН2 —> I I + 2MgClBr
/ \ Н2С сн2
BrMg + MgBr \ /
Cl—As-Cl
I сн
СН3
метиларсепиднн
Метиларсепидин представляет собой бесцветную ядовитую жидкость с запахом горчичного масла (уд. вес 1,218 при 18°, темп, кип 156е), нерастворимую в воде, легко растворимую в спирте и эфире Это—сильный восстановитель, окисляющийся даже кислородом воздуха с образованием окиси арсина (продукт присоединения атома кислорода к атому мышьяка).
Соединения с конденсированными ядрами бензола и пиридина
Группа хинолина
Хинолин, или а,р-бензопиридин, находится в таком же отношении к нафталину, как пиридин к бензолу, т. е. представляет собой как бы нафталин, в молекуле которого одна из групп СН в a-положении замещена на атом азота:
Согласно такому строению, каждый водородный атом хинолина занимает особое положение, и число изомерных однозамещенных производных равно семи. Положения заместителей обозначаются или цифрами, или буквами: а-, у- в пиридиновом ядре; о-, м-, п- и а- («ана»)—в бензольном ядре. Подобно пиридину хинолин обнаруживает свойства основания и с кислотами образует соли.
Хинолиновые основания содержатся в каменноугольном дегте*, а также в техасской нефти. Производными хинолина являются некоторые природные алкалоиды, из которых особенно важны алкалоиды хинной корки: хинин, цинхонин и др.
Синтетические методы получения хинолина и его производных. Методы получения хинолиновых оснований из соединений других классов чрезвычайно многочисленны. Из них укажем лишь несколько важнейших.
. 1. Циклизация аллил анилин а. Впервые хинолин был синтезирован (Кёниге, 1879 г.) пропусканием паров аллил-анилина через раскаленную окись свинца:
СН2
^сн
| || I + 20 -> | || 1 + 2Н2О
* Наряду с хинолином и его гомологами в каменноугольном дегте содержатся также изохинолин (стр. 620) и его гомологи.
2. Синтез С к р а у п а* Этот синтез состоит в нагревании ароматических аминов с глицерином и серной кислотой в присутствии окислителей, чаще всего нитробензола или мышьяковой кислоты (из глицерина в качестве промежуточного продукта образуется акролеин): 1
ОНО
СН
СН
Роль окислителя заключается в дегидрировании первоначально образующегося дигидрохинолина.
Синтез Скраупа является наиболее важным практическим методом получения хинолина и ряда его производных
3. Хинальдиновый синтез (синтез Деб-н е р а—М и л л е р а). Метод заключается в действии альдегидов на анилин в присутствии кислот. В простейшем случае, когда исходят из уксусного альдегида, вероятно, в качестве промежуточного продукта образуется кротоновый альдегид, далее реагирующий с анилином по следующей схеме:
Таким образом, окончательным продуктом реакции является а-метилхинолин, или хинальдин (поэтому реакцию и называют хинальдиновым синтезом) Из акролеина, по гой же схеме, должен получиться хинолин, что и происходит при синтезе Скраупа.
Вместо уксусного альдегида можно применять другие альдегиды, а также смесь двух альдегидов или смесь альдегидов с кетонами, кетонокислотами и др., т. е. соединения, содержащие метильную или метиленовую группу рядом с кар бон ильной группой и способные поэтому вступать в конденсацию типа кротоно
• Зденко Скрауп НЯ50—1910). Австрийский химик, по происхождению венгерец. Впервые дал правильные формулы грех монокарбоновых кислот пиридина. В 1881 г. открыл названную его именем реакцию. Провел много ценных исследований в поисках пути синтеза хинина.
вого альдегида. Например из смеси уксусного альдегида с ацетоном и анилина можно получить а.у-диметилхинолин:
СН3
^сн
II I +2Н2О+Н2
%/\ /С—СН«
N
из смеси формальдегида с ацетоном—у-метилхинолин или лепидин, и т. д.
Реакции Скраупа и Дебнера—Миллера весьма сходны с синтезами пиридиновых оснований из альдегидов и аммиака (стр 591). Действительно, реакция анилина с уксусным альдегидом напоминает конденсацию уксусного альдегида с аммиаком, причем здесь также возможны два направления кроме хинальдина, может получаться и лепидин, если реакцию проводить при высокой температуре над окисью алюминия (А. Е. Чичибабин):
4. Конденсация анилина с ацетоуксусным эфиром. Дальнейшим расширением синтезов Дебнера— Миллера и Скраупа является синтез оксихинолинов по Кнорру из ацетоуксусного эфира и анилина и их гомологов и производных. В этом случае реакция также может итти в двух направлениях с образованием производных хинальдина или лепидина:
ОС2Н5
СО
+ НаО + СаНбОН
СН3
1 +НаО + С2Н8ОН
V \ zc-0H
N
Синтез лепидина хорошо идете анилидом ацетоуксусной кислоты (Фирц-Да-вид), легко замыкающимся под действием H2SO4 в а-оксилепидин (лепидон):
Действием РОС13 на оксилепидин получается хлорлепидин, а замена в нем галоида на водород действием олова и несколько разбавленной соляной кислоты приводит к лепидину (Г. И. Михайлов).
5. Действие альдегидов и кетонов на о-аминобензальдегид. Особенно наглядным для уяснения строения хинолина является синтез Фридлендера, состоящий в действии альдегидов или кетонов на о-аминобензальдегид:
СНО СН
СН3 ^сн
I II +1 + II |Н+2Н-0
Ч/\ СНО %/\
NH2 N
6. Расширение кольца индола. Аналогично получению пиридина и его производных из пиррола (стр. 533) из индола можно получать хинолин и его производные (стр. 550).
Свойства хинолиновых оснований. Хинолин и его гомологи (табл. 42), подобно бензолу, могут нитроваться и сульфироваться, причем нитро- и сульфогруппы вступают в бензольное кольцо в положения 5 или 8. Одновременно они являются также основаниями, хотя и сла-
Таблща 42
Гомологи хинолина
Название Температура плавления °C Температура кипения °C Удельный вес Показатель преломления „20 п4
Хинолин —19,5 237,7 1,095 1,6268
Хинальдин (а-, или 2-метил-хинол’ин) —2 246,5 1,1013 1,6116
P-Метил хинолин (3-метилхи-нолин) 16 250 1,074 1,6039 (при 23,3°)
Лепидин Ст-, или 4-метилхинолин) Толухинолины: +9—10 260 1,0852 1,6206
яяя-,или5-метилхинолин + 19 262,7 1,0832 1,6220
п-, или 6-метилхинолин +ю 255 1,0654 1,6157
ж-, или 7-метилхинолин <—20 252,5 1,072 1,6150
о-, или 8-метилхинолин — 248 1,073 1,6161
быми. Они дают соли с кислотами, и в частности, применяемые для их характеристики прочные соли с пикриновой кислотой, а также хлорплатинагы. С алифатическими галоидалкилами они образуют соответствующие галоидалкилаты. При действии на хинолин амида натрия получается а-аминопроизводное, а с безводным порошком •едкого кали—а-оксипроизводное.
а- и у-Оксисоединения и аминосоединения хинолина обнаруживают те же явления таутомерии, что и соответствующие производные пиридина, а- и ^-Алкилхинолины могут (повидимому, в метидной форме) вступать в реакции конденсации (ср. стр. 59б).
Хинолин довольно прочен по отношению к окислителям, но при энергичном окислении перманганатом разрушается его бензольное кольцо и образуется 2,3-пиридиндикарбоновая, или хинолиновая, кислота:
ру'СООН
Чк/Хчсоон
При окислении гомологов хинолина боковые цепи окисляются до карбоксильных групп; более энергичное окисление приводит к разрушению или бензольного, или пиридинового ядра, с образованием карбоксильных групп или ациламидной группировки у атомов углерода, общих для обоих циклов. Например, хинальдин сначала окисляется в хинальдиновую кислоту, а затем пиридиновое кольцо разрушается и образуется главным образом ацетил антраниловая кислота:
ГТ1 ^соон
'^V'XcOOH 4/\nH—сосн3
хинальдиновая ацетилантраниловая
кислота кислота
Хинолин и его гомологи легко восстанавливаются, присоединяя четыре атома водорода и превращаясь в тетрагидрохинолины.
Синтезы Скраупа и другие доказывают присутствие в молекуле хинолина бензольного ядра; окисление в хинолиновую кислоту и ее производные доказывает наличие в его молекуле пиридинового ядра. Положение заместителей в хинолиновых основаниях устанавливается реакциями окисления или синтезами из соответствующих соединений ряда бензола.
Хинолин—жидкость, отчасти растворимая в воде, но не смешивающаяся с ней, обладающая своеобразным, сравнительно приятным запахом.
Хинолин получается или из каменноугольного дегтя, или синтезом Скраупа. Впервые он был получен перегонкой алкалоида цинхонина с едким кали (Жерар, 1842 г.).
При перегонке хинина с КОН получается n-метоксихинолин, или хинанизол
Это жидкость, кипящая при 193° (при 50 мм рт. ст.). Хинанизол получается также синтетически по Скраупу из n-анизидина с глицерином. Окислением хинина получается n-метоксицинхониновая, или хининовая, кислота
[СООН
Хинальдин и лепидин (а- и рметилхинолины) содержатся в каменноугольном дегте. При окислении этих оснований получаются соответствующие карбоновые одноосновные кислоты: а-хинолинкар-боновая, или хинальдиновая, кислота (темп, плавл. 156°) и у-хино-линкарбоновая, или цинхониновая, кислота (темп, плавл. 254°):
СООН
m
V^j/\cooh
хинальдиновая кислота
цинхониновая кислота
Хинальдиновая кислота, подобно а-пиридинкарбоновым кислотам, дает желтую окраску с FeSO4.
Цинхониновая кислота синтезирована Ад. Кауфманом следующим образом. Хинолин с CH3J дает иодметилат, образующий с KCN цианид таутомерного строения Этот цианид с иодом дает иодметилат ^-цианхинолина, при нагревании отщепляющий CH3J с образованием 7-цианхинолина, омыляемого далее в цинхониновую кислоту:
Из хинанизола Кауфман аналогичным путем синтезировал хининовую кислоту.
Хинальдин конденсируется с фталевым ангидридом, образуя хинофталон (ср. стр. 599), применяемый в качестве красителя под названием хинолинового желтого.
Хинофталон существует в двух
О
таутомерных формах:
О
Хинальдин и лепидин применяются также для получения красителей, называемых цианинами.
Простейшие цианины получаются при нагревании с водными щелочами галоидалкилатов хинолина и его гомологов, содержащих алкильные группы в а- или у-положениях. При действии щелочи, например, на иодэтилат хинолина сначала образуется хинолиниевое основание, которое перегруппировывается в N-алкил-а-оксихинолин, легко окисляющийся воздухом в N-алкил-а-хинолон:
Такой хинолон конденсируется затем, например, с иодэтилатом хинальдина, давая цианиновый краситель этиловый красный:
Конденсацией галоидалкилатов хинальдина с ортомуравьиным эфиром с помощью уксусного ангидрида можно получить карбоцианиновые красители, у которых хинолиновые циклы связаны цепью =СН—СН=СН—, например:
J- 4- ЗС2Н5ОН -i- HJ
Промежуточная цепь может содержать несколько винильных групп. Красители, в молекуле которых связаны ос- и а'- или а- и у-углеродные атомы хинолиновых циклов,—красного цвета; если же связаны у- и у'-углеродные атомы, получаются синие красители. Хинолиновые циклы могут быть заменены тиазоловыми (с । р 573).
Цианиновые красители применяются как сенсибилизаторы фотографических материалов.
Оксихинолины. а-Оксихинолин (а-хинолон, или карбостирил) получается с прекрасным выходом по способу А. Е. Чичибабина нагреванием сухого хинолина с твердым безводным едким кали:
а-Хинолон, как и а-пиридон, дает два ряда алкилпроизводных:
^V^OCHg
Последняя форма, называемая метилхинолоном, легко получается окислением иодметилата хинолина в щелочном растворе (Деккер, объяснение реакции см. стр. 595).
Карбостирил является ангидридом (лактамом) о-аминокорич-ной кислоты. Он иногда кристаллизуется с одной молекулой Н2О (темп, плавл. 200°, возгоняется без разложения).
у-Оксихинолин (кинурин) впервые был получен нагреванием кинуреновой кислоты (роксихинолин-р-карбоновая кислота), содержащейся в моче собак при обильном мясном питании. Кинурин кристаллизуется с тремя молекулами воды, которые теряет при нагревании, и плавится затем при 201°.
Из производных хинолина, содержащих гидроксильную группу в бензольном ядре, заслуживает упоминания 8-оксихинолин, или оксин (бесцветные кристаллы, темп, плавл. 75—76°), применяемый в аналитической химии как реактив для осаждения некоторых тяжелых металлов и алюминия. Пространственная близость третичного атома азота и гидроксильной группы в молекуле оксина облегчает •образование прочных, мало растворимых в воде внутрикомплексных солей (ср. том I, стр. 440).
Тетрагидрохинолин
сн2
/\4'а
обладает более сильными основными свойствами, чем хинолин.
Тетрагидрохинолин был получен впервые А. Вышнеградским* в 1880 г. восстановлением хинолина натрием в кипящем спирте. Он плавится при 20°; темп. кип. 25Г, уд. вес do — 1,0546, показатель преломления 1,5933; по запаху напоминает индол.
С иодистыми алкилами он дает N-алкилтетрагидрохинолины, производные которых обладают жаропонижающим действием, но не нашли применения вследствие повышенной токсичности.
Из производных хинолина приобрела большое значение как противопода-грическое средство (выделяющее из крови мочевую кислоту) а-фенилцинхониновая кислота (желтые микроскопические иголочки, темп, плавл. 212—213°), применяемая под названием атофан.
Атофан получают из ацетофенона и изатиновой кислоты (или изатина) в воднощелочной среде:
СООН соон
сн3
I II + I I II 1 + 2Н,0
'VXnh, со-с6н5 \/\/Х „
1N
Важным анестезирующим веществом является совкаин (перкаин), представляющий собой хлоргидрат сложного производного а-оксицинхониновой кислоты (темп, плавл. основания 65°):
СО—NH—СНа—CH2-N(C2H6)2
Совкаин получается синтетически, исходя из изатина.
Большой интерес как средства против малярии, применяемые самостоятельно и в сочетании с хинином, представляют вторичные замещенные амины, производные 6-метокси-8-аминохинолина. К ним относятся плазмохин и плазмоцид;
CILO
СН3О
NH
CH3-CH-CH2-CH2-CH2-N(C2H5)2
т N NH (bj-CH,—CH2—N(C,H5)2
плазмохин
плазмоцид
Плазмохин—желтое масло, перегоняющееся около 200° (при 2 мм); плазмоцид (темп, плавл. 24,6°, темп. кип. 191 —195° при 1 мм) применяется в виде соли с двумя молекулами метилен-бис-салициловой кислоты, получаемой конденсацией салициловой кислоты с формальдегидом. Неосторожное применение обоих этих препаратов может вызвать побочное действие—атрофию зрительного нерва.
* А. Вышнеградский (1851—1880). Ученик А. М. Бутлерова. Установил связь некоторых алкалоидов с пиридиновыми основаниями.
Из а- и р-нафтиламинов реакцией Скраупа получены а- и нафтолинолинък
а-нафтохинолин ₽-нафтохинолив
Изохинолин
Изохинолин, или Рд-бензопиридин, имеет строение
Он содержится в небольшом количестве вместе с хинолином в каменноугольном дегте. Так как изохинолин является более сильным Основанием, для отделения его от хинолина обрабатывают сырой Хинолин малым количеством кислоты.
Изохинолин образует кристаллы с темп, плавл. +24,6°, темп. Кип. 242,5°. Удельный вес жидкого изохинолина d4°=h0986, показатель преломления пр’4 = 1,6243. Запах его слегка миндальный, отчасти напоминает запах хинолина.
Окислением изохинолина получается смесь фталевой кислоты с цинхомероновой (Рд-пиридинкарбоновой):
^/.со°н И00С\Л
I I I i
^\соон ноос/х^
фталевая цинхомероновая
кислота кислота
Таким образом устанавливается строение изохинолина, подтверждаемое также синтезами его из производных бензола. Так, Например, изохинолин получается восстановлением имида гомофта-левой кислоты, имеющего строение
Производными изохинолина являются многие важные алкалоиды, например некоторые алкалоиды опия (морфин, папаверин, наркотин и пр.), кураре (курарин), а также эметин и сальсолин.
Важнейший путь получения изохинолина и его производных-метод Бишлера—Напиральского. Он заключается в действии фос
форного ангидрида или хлорангидридов фосфорной кислоты на р-фенилэтиламиды кислот. Возможно, что эти амиды сначала циклизуются (в таутомерной форме) в производные дигидроизохинолина. например:
Последующее дегидрирование их, например в парах над палладием, приводит к производным изохинолина, а гидрированием, наоборот, легко получаются производные тетрагидроизохинолина (алкалоиды как раз являются производными тетрагидроизохинолина).
Тетра гидроизохинол ин получается конденсацией р-фенилэтил-амина с формальдегидом: *
Конденсируя с фенилэтиламином другие альдегиды, получают замещенные в положении 1 тетрагидроизохинолины. Дегидрированием геграгидропроизводных можно получить изохинолин и его гомологи.-
Акридин
Акридин, или дибензопиридин
был открыт в каменноугольном дегте (Гребе и Каро, 1870 г.). Он содержится в антраценовом масле вместе с антраценом. Акридин оказывает на слизистые оболочки и на кожу раздражающее действие, откуда и произошло его название.
При окислении акридина получается акридиновая кислота (а,[3-хинолиндикарбоновая)
V' 'г/\зоон
чем, наряду с рядом синтезов производных акридина, доказывается его строение.
Для получения акридина и его гомологов применяются следующие способы:
1. Нагревание дифениламина с одноосновными карбоновыми кислотами в присутствии хлористого цинка. С муравьиной кислотой получается, сам акридин:
НСООН
+ сн
О О CV Y'l + 21 v>
2. Действие серной кислоты на фенилиро-ванные антраниловые кислоты. Таким путем получаются кетопроизводные дигидроакридина, называемые акри-донами:
N-Фенилантраниловые кислоты гладко получаются нагреванием о-хлор-или о-бромбензойных кислот с производными анилина в присутствии мелко раздробленной меди (Ульман), например:
СООН
СООН
NH
3. Действие альдегидов или кислот на ароматические амины. Этим путем получаются аминопроизводные акридина, особенно 3,Ь-диамины. Например, из л«-фенилендиамина с формалином сначала получается тетраминодифенилметан, который нагреванием с галоидными солями олова может быть конденсирован в диаминодигидроакридин:
Последний окисляется хлорным железом в диаминоакридин.
Хлорметилат этого диаминоакридина называется трипафлавином'.
СН
IT II I Cl-
I CHS
Трипафлавин убивает трипанозом и потому применяется в медицине как средство против сонной болезни.
В качестве антисептика очень широко применяется 2-этокси-6,9-диамино-акридин (желтые кристаллы, плавящиеся около 120°) в форме солянокислой или молочнокислой соли, известной под названием риванола Водные растворы этих солей окрашены в зеленовато-желтый цвет, флуоресцируют и на свету довольно быстро разрушаются.
Акридин образует бесцветные иглы с темп, плавл. 108° и темп, кип. 346°. Он летуч с парами воды, легко растворим в спирте и эфире. Растворы акридина и многих его производных сильно флуоресцируют.
Акридин—слабое основание, дающее с сильными кислотами соли, обладающие желтой окраской, в растворах сильно флуоресцирующие.
Восстановлением акридин легко превращается в 9,\0-дигидро-акридин
NH
не обладающий основными свойствами. При окислении его хромовой смесью и другими окислителями получается акридон
Акридон образует желтые иглы с темп, плавл. 354°; он возгоняется без разложения, дает растворы с интенсивной синей флуоресценцией.
Окси- и аминопроизволные акридина представляют собой настоящие красители (акридиновые красители).
Из производных акридина важное применение имеет так называемый акрихин,атебрин (мелкие желтые иголочки, темп, плавл. 248—250°):
СН3-СН—СН2—СН2—СН2—N-
ЮСН3
2СГ
Акрихин широко и успешно применяется один или в комбинациях с хинином для борьбы с малярией.
По одному из вариантов синтеза акрихина (И. Л. Кнуньянц, Г. В. Челинцев, 3. В. Беневоленская, Е. Д. Осетрова) отдельно синтезируют алифатическую цепь:
СООС2Н6^ сн3со—сн2—сн2—сн2он+ со2 + С2Н6ОН
СНоСО—СН2 Н2С-----СН2 т-ацетопропиловый
спирт
СН3СО—(СН2)2—СН2ОН ЛД. СН3СО—(СН2)2—СН2Вг NH(C2H5)?
-* CH3CO-(CH2)3-N(C2H6)2 NH2°h CH3-C-(CH2)3-N(C2H5)2
N—OH
— CH3 — CH — CH2—CH2 —сн2—N(C2H6)2
гш2
1-диэтиламино-4-аминопснтан
С другой стороны, из 2,4-дихлорбензойной кислоты и n-анизидина с последующим действием хлорокиси фосфора получают 2-метокси-6,9-дихлоракрилин:
Конденсацией его с 1-диэтиламино-4-аминопентаном получается акрихин.
В Советском Союзе акрихин получают по этому методу в производственном масштабе.
Азины
Азинами называются производные шестичленных циклов с двумя и более гетероатомами, из которых по крайней мере один является атомом азота. Соединения, содержащие в кольце два атома азота, называются диазинами, три—триазинами и т. д. Наиболее важными
являются азины, содержащие гетероатомы в пара-положении; из остальных наибольшее значение имеет jn-диазин, или пиримидин. Важнейшие азины:
N
НО Ч2Н
пиразин
СН
НС5 SN
I II
НС* 2СН
V
СН
НС6 8СН
НС’ 2N
N
аиридазин
пиримидин
I казна
Группа пиразина
Пиразин и его производные получаются конденсацией а-ами-ноальдегидов или а-аминокетонов (двух молекул) с* последующим окислением образующихся дигидропиразинов (см. том I, стр. 660).
Из аминоуксусного альдегида этим путем можно получить пиразин. Он образует кристаллы с темп, плавл. 53°; темп. кип. 118°. Пиразин обладает несильным приятным запахом, легко растворим в воде, спирте и эфире. Это—слабое основание, дающее соли с одним эквивалентом кислоты.
На холоду пиразин окисляется перманганатом; у гомологов пиразина при этом окисляются только боковые цепи, и образуются пиразинкарбоновые кислоты, легко отщепляющие СО2, превращаясь в пиразин. Несмотря на наличие в ядре двух атомов азота, пиразины могут присоединять только одну молекулу га-лоидалкила.
2,Ь-Диметилпиразин (темп, плавл. 15э; темп. кип. 155*; уд. вес d^°=0,990) получается, исходя из изонитрозоацетона, который восстанавливается в аминоацетон.
При действии кислот аминоацетон дает дигидродиметилпиразин, из которого окислением солями окиси ртути получается диметилпиразин:
H2N N N
сн3—со \н2 —2н2о сн3—^н2 +о сн3—с %н
I + I -------------> I ‘ I --------------> II I
сн2 СО-СН, Н2С с—сн3 НС с-снэ
\ \ Z \ Z
NH2 N N
Тот же диметилпиразин образуется вместе с р-пиколином при нагревании глицерина с солями аммония (особенно с NH4C1). Небольшие количества пиразина и его гомологов образуются при нагревании глюкозы с концентрирован ным раствором аммиака.
Наиболее важными соединениями группы пиразина являются продукты присоединения к пиразинам шести атомов водорода—так называемые пиперазины. Простейший пиперазин, или диэтилендиимин (кристаллы; темп, плавл. 104°, темп. кип. 145°)
NH \н2 I I Н2С сн2 \н легко растворим в воде, с которой образует шестиводный гидрат. Кроме восстановления пиразина, он был получен действием .аммиака на дибромэтан.
Если вместо аммиака взять его производные, например амины, то можно получить замещенные при азоте производные пиперазина. Например, из анилина и СН2Вг—СН2Вг получается дифенилпиперазин n-c6h5 щс/ \н2
I I н2с сн2 \-СвН6 Пиперазин и его гомологи—сильные основания, дающие соли с двумя молекулами НС1 и даже с угольной кислотой. Соли пиперазина и некоторых его гомологов применяются в медицине в качестве противоподагрических средств.
Производными пиперазина являются циклические ангидриды а-аминокислот, так называемые дикетопиперазины (см. том I, стр. 674 и 688 сл.), например:
Н2(/ \ э
I I
ОС сн2 \н
Восстановлением дикетопиперазинов можно также получить пиперазины.
Бензопиразин, или хиноксалин (темп, плавл. 30,5°, темп. кип. 226°), получается конденсацией о-фенилендиамина с глиоксалем:
Он легко растворим в воде на холоду (при нагревании растворы расслаиваются); является слабым однокислотным основанием. Его производные получаются из ароматических о-диаминов при действии а-дикетонов. При окислении перманганатом разрушается бензольное ядро хиноксалина и образуется 2,3-пиразиндикарбоновая кислота. Хиноксалины присоединяют одну молекулу алкилгалоге-нидов. Восстановление хиноксалина приводит к 1,2,3,4-тетрагидро-хиноксалину, образующему бесцветные таблички с темп, плавл. 97°, малорастворимые в воде. Феррицианид калия в щелочных растворах вновь окисляет его в хиноксалин.
Антразин. Комбинация пиразинового кольца с двумя антраценовыми дает антразин. Его можно получить щелочным плавлением р-аминоантрацена:
-зн2
Антразин—коричнево-красные иглы (темп, плавл. около 390°). Практически он нерастворим в обычных органических растворителях, кристаллизуется из горячего нитробензола.
Важное значение имеет дихинон дигидроантразина—синий ин-дантрен, получаемый щелочным плавлением [3-аминоантрахинона. Он образует синие иглы при кристаллизации из кипящего хинолина; не изменяется даже при температуре выше 400°. Обрабатывая его щелочным раствором гидросульфита натрия, получают темножелтый раствор двунатриевого производного дигидроиндантрена (индантреновый куб):
СО
Пропитывая ткани такими растворами и предоставляя им окисляться на воздухе, получают чрезвычайно прочные темносиние окраски.
Феназин, феноксазин и фентиазин
Большое значение имеют производные n-диазинов, п-оксазинов и n-тиазинов, содержащие в молекуле два конденсированных бензольных ядра, а именно:
феназин
феноксазин
фентиазин
Феназину обычно придается орто-хиноидное строение, его производным—орто-хиноидное или пара-хиноидное (см. ниже). Феноксазин и фентиазин образуют N-алкил производные, а также оксо-ниевые и тиониевые соединения, которые могут иметь как орто-хиноидное, так и пара-хиноидное строение.
Феназин (желтые иглы с темп, плавл. 17Г, легко сублимирующиеся) получается конденсацией о-фенилендиамина с пирокатехином при 220° в присутствии кислорода воздуха:
Он получается также при различных реакциях азотистых производных бензола, например действием нитробензола на анилин в присутствии твердого едкого натра при 140°:
Производные феназина могут получаться также действием о-диаминов на о-хиноны:
Вместо хинонов можно брать соответствующие двухатомные фенолы в присутствии окислителей.
Феназин, феноксазин и фентиазин — окрашенные соединения, так как пара-диазиновые группировки являются хромофорами.
Однако лишь с введением ауксохромных групп, особенно амино-и окси-групп, их производные приобретают характер настоящих красителей.
Таковы, например:
1) эуродины, или аминофеназины,
2) важные красители, являющиеся производными аммониевых оснований (например, сафранины, индулины),
3) азоксон иевые и
4) азтиониевые красители и пр.
Эти красители получаются из производных ароматических диаминов и моноаминов при реакциях конденсации, большей частью сопровождающихся реакциями окисления.
Окислением смеси нитрозодиметиланилина и ле-толуилендиамина получается сначала индамин (стр. 356), называемый толуиленовым голубым, а затем эуродин, называемый толуиленовым красным:
толуиленовый голубой
толуиленовый красный
Простейший сафранин, или феносафранин, впервые был получен окислением смеси анилина и rz-фенилендиамина (с промежуточным образованием индамина):
♦ О 4 но
СЛ
I с6н5
феносафранин
К сафранинам относится также первый промышленный синтетический краситель, а именно мовеин, полученный Перкиным в 1856 г. окислением неочищенного анилина.
Он состоит из смеси фен и лированного по одной из аминогрупп феносафранина и его гомологов, называемых толусафранинами.
Взаимодействием о-аминодифениламина с 2,4,6-трин итрохлорбензолом (пи-крилхлоридом) и последующим восстановлением о-(2,4,6-тринитрофен и ламино)-
дифениламина получается пинакриптол зеленый (N-фенил-! ,3-диаминофеназоний-хлорид):
I с6н5
НС1 »
пинакриптол зеленый
Этот изомер феносафранина—зеленый краситель—применяется как сильный десенсибилизатор в фотографии. Предварительная, до проявления, обработка пластинок и пленок разбавленными растворами пинакриптола позволяет вести проявление даже высокосветочувствительных материалов на рассеянном свету.
Производные нафтофеназина (N-фенилнафтофеназония), содержащие в нафталиновом кольце аминогруппы, называются розиндулинами.
индулиновый алый (розиндулиновый краситель)
нафтофеназин
Нагреванием нитрозодиметиланилина с р-нафтолом получается азоксон ие-вый краситель (производное оксазина), известный под названием нафтолового голубого Мелдолы, для которого возможны следующие две формулы:
N
_(СН3)2Й^'
СГ
Cl-
оксониевая соль
аммониевая сопь
о
Одним из наиболее известных красителей является метиленовый голуба), получаемый окислением диметил-п-фенилендиамина (приготовленного восстановлением нитрозодиметиланилина) в присутствии сероводорода. Он относится к аз-тиониевым красителям (производные тиазина), и для него возможно строение аммониевой или сульфониевой соли:
N
(СН3)2ы/0%р0
(CH3)2N
аммониевая соль
сульфониевая соль
Реакцию его образования можно
выразить, например, следующей схемой:
В промышленности этот краситель получают, обрабатывая /г-аминодиметил-анилин тиосульфатом натрия в присутствии бихромата натрия и хлористого цинка. При этом образуется диметил-n фенилендиаминтиосульфокислота. Эта кислота обрабатывается диметиланилином и бихроматом натрия в кислой среде. Тогда образуется тетраметилиндаминтиосульфокислота, которая в присутствии хлористого цинка и соляной кислоты при нагревании отщепляет элементы сернистой кислоты и превращается в краситель:
Аналоги азинов, содержащие мышьяк
При нагревании треххлористого мышьяка с дифениламином получается 9-хлордигидрофенарсазин (желтые кристаллы, темп, плавл. 193°):
AsCl
Хлордигидрофен арса зин был предложен в конце первой мировой войны американскими химиками в качестве боевого отравляющего вещества под названием адамсита. Адамсит легко возгоняется, давая тонкодисперсный, крайне сильно раздражающий органы дыхания дым, не задерживаемый в противогазах активированным углем или химическими поглотителями. Для защиты от таких дымов необходимо применять в противогазах противодымные фильтры. При нагревании хлордигидрофенарсазина или, лучше, продукта замещения в нем хлора на метоксигруппу получается фенарсазин. Известен также арсантрен, или дифенилендиарсин:
As As
В отличие от азинов соединения мышьяка легко окисляются иногда уже на воздухе, переходя в соединения пятивалентного мышьяка.
Фенарсазин присоединяет воду и переходит в окисные соединения трехвалентного мышьяка:
При действии на хлордигидрофен арсазин магнийорганических соединений хлор замещается на алкил или арил и получается замещенный при мышьяке дигидрофенарсазин
R
который при нагревании с соляной кислотой отщепляет алкилдихлорарсин е образованием дифениламина.
Группа пиридазина
Пиридазин, или 1,2-диазин
СН нс<41сн II I НС* .3N
был получен в 1895 г. Он представляет собой бесцветную, смешивающуюся с водой жидкость со слабым запахом белены. Водные растворы его имеют нейтральную реакцию, но с кислотами он дает соли. Пиридазин плавится при —8°, кипит при 208®.
Пиридазин может быть получен конденсацией гидразина с малеиновым диальдегидом
сн СН
Н(^ % H2N Н(^ \
II + I J 1+ 2Н»°
НС О H,N нс N
СН СН
Сочетания бензольного цикла с 1,2-диазином представляют собой 3,4-бензо-пиридазин, или циннолин, и 4,5-бензопиридазин, или фталазин:
циннолин
фталазин
Циннолин—легкорастворимые в воде желтоватые кристаллы плавящиеся при 38® и обладающие запахом, напоминающим запах герани. Фталазин также легко растворим в воде; он образует желтоватые иглы, плавящиеся при 90°. Растворы обоих этих веществ не обладают щелочной реакцией, но сами они даюг соли с минеральными кислотами.
Циннолин можно получить из о-изодиазотата коричной кислоты:
^^СН^СН-СООН
I II
^Z\n=n—он
Фталазин, аналогично пиридазину, может быть получен конденсацией гидразина с фталевым диальдегидом.
Группа пиримидина
Пиримидин является 1,3-диазином
Это наркотически пахнущие кристаллы с темп, плавл. 22° и темп, кип 124°, легко растворимые в воде Пиримидин в водных растворах имеет нейтральную реакцию, но в то же время дает прочный пикрат.
Производными пиримидина являются некоторые циклические уреиды, как, например, аллоксан, барбитуровая кислота и др. (см. том 1, стр. 744 сл.).
Из барбитуровой кислоты может быть получен и сам пиримидин:
СО
\н I СО
РС15 и РОС1з
СО СН
К производным пиримидина относятся также физиологически важные соединения, получаемые гидролизом нуклеиновых веществ клеточных ядер, например урацил (бесцветные иглы, темп, плавл. 338°), получаемый гидролизом нуклеопротеидов из дрожжей, и
тимин, или 5-метилурацил (бесцветные листочки, темп, плавл. 340°), получаемый гидролизом нуклеиновых веществ щитовидной железы.
Урацил и тимин таутомерны соответственно 2,6-диоксипирими-дину и 5-метил-2,6-диоксипиримидину (о таутомерии уреидов см. том 1, стр. 745—746):
СН СН
Н(/ \н Н(/ \
I I ;± I II
ОС со но—с с—он
\н
урацил
СН СН
СН3—\н СН3—с^ \
I i I II
ОС со но—с с—он
\|Н
тимин
Другой^гомолог урацила, 4-метилурацил, получен конденсацией ацетоуксусного эфира с мочевиной:
ОС—СН3 С—сн3
нД NHa НС^ \н
1+1 I I +С2Н5ОН + Н2О
С2Н6ООС со ОС со
Конденсацией о-фенилендиамина с аллоксаном может быть получен изо-аллоксазин, являющийся сочетанием хиноксалинового и пиримидинового циклов, т. е. 1,4- и 1,3-диазинов:
о-фенилендиамин аллоксан изоаллоксазин
Изоаллоксазиновая группировка входит в азотсодержащую часть рибо флавина, или витамина В2, имеющего важное физиологическое значение (стр. 712)
Группа пурина
В тесной связи с циклическими уреидами находится мочевая кислота CH4N4O3, являющаяся наряду с мочевиной важнейшим продуктом обмена азотистых веществ в животном организме. Веществами, родственными мочевой кислоте, являются нуклеиновые основания, присутствующие в крови и в некоторых выделениях органов животных, а также (в меньших количествах) в растениях. К ним от
носятся ксантин, гипоксантин, аденин и гуанин (см. ниже). К циклическим уреидам близки также очень важные растительные алкалоиды. метилированные при азоте ксантины—кофеин, теобромин, теофиллин. В основе всей этой группы соединений лежит сложный гетероцикл, являющийся сочетанием пиримидина и имидазола и названный Э. Фишером пурином
СН
N1 :>С------NH
I 7 I
НС2 4С 8СН
Благодаря классическим исследованиям Э. Фишера, объектом которых являлась мочевая кислота и ее производные, окончательно выяснены взаимные отношения всех этих веществ и предложен ряд новых синтетических методов, завершившихся синтезами самой мочевой кислоты и родственных ей алкалоидов (90-е годы XIX столетия) Эти исследования Э Фишера по справедливости считаются, наряду с его работами по углеводам и белковым веществам, одной из самых блестящих страниц синтетической органической химии прошлого века.
Пурин представляет собой бесцветное кристаллическое вещество (темп, плавл. 216—217°), легко растворимое в воде. Растворы пурина имеют нейтральную реакцию, но он образует соли как с кислотами, так и со щелочами. Сам пурин не встречается в природных продуктах, но его 2,6- и 8-оксипроизводные являются довольно распространенными природными веществами.
Для оксипроизводных пурина характерна так называемая амидная таутомерия (см. том 1, стр. 745). Например, Ъ-оксипурин, или гипоксантин, может реагировать в виде двух таутомерных форм:
С—ОН
У \
N С---NH
1 15 1
НС с сн
\ / \ // N N окси-форма
СО
оксо-форма (амидная)
Строение этих веществ чаще изображают в амидной форме.
Для алкилированных пуринов известны обе изомерные формы (N-алкил- из О-алкилпроизводные).
Мочевая кислота. Важнейшим из природных пуриновых веществ является мочевая кислота, или 2,6,8-триоксипурин
СО
/6 \
HN1 &с---NH
I I! ’I
ОС2 4С «со
\з/
. NH NH
В нормальной моче человека и млекопитающих мочевая кислота содержится лишь в незначительных количествах. При нарушении же обмена веществ ее количество в моче может увеличиваться, особенно при некоторых болезнях (подагра). У птиц и у пресмыкающихся мочевая кислота является главной составной частью экскрементов.
Технически мочевая кислота получается из гуано (скоплений помета морских птиц, особенно значительных на островах Южной Америки). В гуано содержится около 25% мочевой кислоты.
Мочевая кислота—бесцветный кристаллический порошок, крайне трудно растворимый в воде, нерастворимый в спирте и эфире. Она является очень слабой кислотой, однако может давать соли (ураты) с одним или двумя эквивалентами металла. Двуметаллические соли щелочных металлов довольно легко растворяются в воде, причем растворы имеют сильно щелочную реакцию. Однометаллические соли трудно растворимы в воде, за исключением литиевой соли.
Препараты солей лития и воду минеральных источников, содержащих эти соли, употребляют при подагре и уратных камнях в мочевом пузыре для вывода из организма солей мочевой кислоты.
Мочевая кислота восстанавливает окиси меди и серебра в щелочных растворах. Азотная кислота окисляет мочевую кислоту в аллоксан (см. том I, стр. 744), аллоксантин (см. том I, стр. 746) и парабановую кислоту, являющуюся продуктом дальнейшего окисления аллоксана; перманганат в нейтральной среде окисляет мочевую кислоту в аллантоин (см. том I, стр. 745):
СО СО
Hrf^C NH HNO3 Hbf'co HN CO
I II I ----------> I I —* I I
ОС C CO ОС co ОС co
\lH
мочевая кислота аллоксан парабановая
кислота
| КМпО,
ОС---NH
H,N—ОС—NH—Н(!: со
аллантоин
При действии йодистого метила и других алкилирующих средств на мочевую кислоту или ее соли получаются продукты последовательного замещения водородных атомов при азоте на метил или другие алкилы.
Моно-, ди- и триметилмочевые кислоты могут существовать в соответствующем числе изомерных форм. Окончательный продукт метилирования—1,3,7,9-тетраметилмочевая кислота, у которой все четыре атома водорода замещены на метилы, по существу уже не является кислотой.
С хлорокисью фосфора кислородные производные пурина реагируют в окси-форме. Мочевая кислота при этом превращается в 2,6,8-трихлорпурин, дальнейшее восстановление которого приводит к пурину.
Если подействовать на трихлорпурин этилатом натрия, то легче всего замещаются на этоксильные группы атомы хлора в положениях 2 и 6. При гидролизе полученного диэтоксихлорпурина кислотами получается 2,6-диокси-8-хлорпурин или 8-хлорксантин, который после восстановления дает 2,6-диоксипурин, или ксантин:
Из ксантина действием РОС13 можно получить 2,6-дихлорпурин и путем частичной замены хлора на гидроксил с последующим восстановлением притти к 6-оксипурину, или гипоксантину (стр. 635).
Из метилированных мочевых кислот можно аналогично получить метилированные хлорксантины, а из последних восстановлением синтезировать метилированные ксантины, в том числе и алкалоиды теобромин и кофеин.
Синтез мочевой кислоты (с ничтожными выходами) был впервые осуществлен чешским химиком И. Горбачевским в 1882 г. нагреванием мочевины с гликоколем или с амидом трихлормолочной кислоты СС13—СН(ОН)—CONH2. Механизм этих синтезов совершенно неясен.
Наиболее наглядным является синтез Э. Фишера, получившего мочевую кислоту из малоновой кислоты и мочевины через барбитуровую кислоту. Нитрозированием последней ее переводят в вио-луровую кислоту, которую восстанавливают в урамил (амино-барбитуровую кислоту).
Действием циановой кислоты на урамил получается псевдомоче-вая кислота. При нагревании ее с 20%-ной соляной кислотой идет замыкание имидазольного цикла с отщеплением воды и образуется мочевая кислота:
СО
Hbf \н—NH.
|| ~ + HNCO —>
ОС со
со
Hbf ^СН—NH
ОС СО /С0
NH>
со
llff \ NH ос с' со
урамил
псевдомочевая кислота
мочевая кислота
Ксанпин, или 2,&-диоксипурин. представляет собой мелкокристаллический порошок, практически почти нерастворимый в воде (1 : 14 400 частей воды при 16а), но растворимый в едких щелочах. Вместе с тем ксантин является очень слабым основанием. При нагревании он разлагается. Строение ксантина было подтверждено синтезом (М Траубе), исходя из мочевины и циануксусной кислоты.
Гипоксантин, или b-оксипурин. находится в мясном экстракте и часто встречается в растениях. Эго—бесцветные иголочки, разлагающиеся около 150°, очень трудно растворимые в воде, но легко растворимые в разбавленных щелочах и кислотах.
Аминопурины. В мясном соке, в печени, а также в соках некоторых растений находится аденин, или 6-аминопурин. Он может быть превращен в 6-оксипурин действием азотистой кислоты. Кристаллизуется из воды в виде игл, содержащих ЗН.?О. Обезвоженный при быстром нагревании он плавится с разложением при 360—365°.
Другой аминопурин, а именно гуанин (2-амино-б-оксипурин)
С—ОН
СО
т —nh
I II I
H2N—с с сн
N N
в значительном количестве содержится, наряду с мочевой кислотой, в гуано, а также в чешуе рыб. Он может быть получен синтетически, исходя из циануксусной кислоты и гуанидина.
Гуанин—бесцветный кристаллический порошок с перламутровым блеском. Он нерастворим в воде, но растворяется в водных щелочах и кислотах. Действием азотистой кислоты гуанин превращается в ксантин.
Важными метилированными при азоте производными ксантина являются растительные алкалоиды: кофеин (Рунге, 1819 г ), содержащийся в кофейных зернах (Coffea arabica), в чайном листе (Thea sinensis), в орехах кола (Cola acumnata), в парагвайском чае (Ilex paraguaiensis). теобромин (А. А. Воскресенский, 1842 г.), находящийся в бобах какао (Theobroma cicao), и теофиллин (Коссель, 1889 г), обычно в небольших количествах сопровождающий кофеин в чайном листе.
Кофеин является 1,3,7-триметил ксантином. Положение метильных групп в его копекуле доказывается окислением его в диметил-аллоксан и монометилмочевину:
СО
/ \ ; /СНЙ СН.—N С—N<
I II \ >сн
ОС С—fv
CHg-N^
СНЙ—N
СО
CHS—n" \о I !
ОС со
Он кристаллизуется из воды (в которой он на холоду мало растворим) в виде одноводного кристаллогидрата, имеющего форму длинных хрупких шелковистых игл. Безводный кофеин плавится при 236,5°, при осторожном нагревании может возгоняться. Он легко растворяется в хлороформе. Водные растворы имеют нейтральную реакцию, но с кислотами он образует соли. В чайных листьях (после ферментации) содержится 2—4% кофеина, в кофейных зернах (бобах Coffea arabica) 1,0%, в орехах кола—3%.
Кофеин возбуждает центральную нервную систему, сужает кровеносные сосуды, повышает кровяное давление и стимулирует сердечную деятельность; обладает также мочегонным действием. Он широко применяется в медицине.
Кофеин получают из отходов производства чая—чайной пыли. Его извлекают из чайной пыли водой (предварительно обработав ее известью для связывания кислотных веществ) или хлороформом. Экстракт упаривают и выпадающий кофеин повторно кристаллизуют из воды.
В промышленных масштабах кофеин получают также полусинте-гическим методом—из мочевой кислоты. При нагревании ее с уксусным ангидридом образуется 8-метилксантин, из которого метилированием диметилсульфатом получают 1,3,7,8-тетраметилксантин. При осторожном хлорировании последнего в метильную группу при углеродном атоме 8 вступают три атома хлора. Трихлорпроиз-водное омыляют кипячением с водой, а получаемая карбоновая кислота тотчас же отщепляет СО2 и превращается в кофеин:
СО СО
НьГ \----NH (снзСО)2О Hhf \----NH
I JI I-------------> । Il I
ос с с—он ос с с-сн,
\/\z \/\^
NH N NH N
(CH3>2SO4 + NaOH ---------------->
мочевая кислота
СО
СН3—N \-N—СН,
i II I
ОС С С—СН,
СН,-ьЛ\/^
со
п2 СН3—N \-------N—СН3 Н2о
---> I II I -----------------
ОС С С—СС13
СН3—
СО
СН8—N \------N—СН3
I II I
ОС с сн
Кофеин может быть получен также метилированием ксантина, который, в свою очередь, можно синтезировать из гуанина или содержащих его материалов.
Теобромин является 3,7-диметилксантином, так как он при окислении образует монометилаллоксан и монометилмочевину:
СО
СО т \о I I ос со
СНз—
HN—СН3 I
СО
н2г/
Это—бесцветный мелкокристаллический порошок, трудно растворимый в воде. Он плавится при 35Г, способен возгоняться. Легко растворяется в едких щелочах, давая, например, натриевую соль. Эквимолекулярная смесь этой соли с салицилатом натрия применяется в медицине под названием диуретина как мочегонное средство.
Теофиллин представляет собой 1,3-диметилксантин
Он образует бесцветные шелковистые иголочки, содержащие одну молекулу кристаллизационной воды. Теофиллин трудно растворим в холодной воде, плавится при 269—272°.
Группа птеридина
Птеридином была пиразина:
названа комбинация циклов пиримидина и
СН N
i II I
нс2 с 7сн
N N
Оксипроизводные птеридина получили название птеринов.
Еще в 1895 г. из крыльев бабочки-капустницы было выделено белое кристаллическое вещество, которое из-за большого сходства с мочевой кислотой было за нее и принято (Гопкинс). Значительно позже Виланд и Шепф показали отличие от мочевой кислоты этого вещества, названного лейкоптерином.
Лейкоптерин был синтезирован нагреванием 2,5,6-триамино-4-оксипиримидина со щавелевой кислотой (Пуррман):
ОН
С NH, /4\/ N3 5С СООН
I I! + I
С2 6С СООН
/ 'W \
H2N N NH2
он
| СО NH
с N /4\ /5\
/ \/ Ч HN» С GCO
N С С—ОН | ।
| |i | HN=C2 С 7СО
Н2\—С С С—ОН W\8/
ч /\ Ч NH NH
N N
лейкоптерин (2-амино-4,0,7-триоксиптеридин)
Из крыльев лимонницы—другого вида бабочек—был выделен краситель желтого цвета, названный ксантоптерином. При действии перекиси водорода ксантоптерин окисляется в лейкоптерин:
СО NH
HN7 СО +о
I II I —
HN=C С СН
СО NH
HN=C С СО \fH \ТН
ксантоптерин лейкоптерин
(2-амино-4,6-диокс1Штеридин)
Ксантоптерин содержится в очень малых количествах в моче человека и других млекопитающих.
Птерины—очень прочные вещества, выдерживающие нагревание до 400°. Для оксиптеридинов, как и для оксипуринов, очевидно, должна иметь место амидная таутомерия.
Ксантоптерин является витамином для рыб. Некоторые более сложные производные птеринов, как оказалось, являются витаминами для человека и высших животных, например фолиевая, или птероилглутаминовая, кислота (стр. 714). Поэтому химия производных птеридина привлекает к себе в последнее время большое внимание.
Алкалоиды
’ Изучение продуктов жизнедеятельности растительных и животных организмов было предметом органической химии с самого ее зарождения. В течение последней трети XVIII столетия были выделены и охарактеризованы многие природные органические соединения, носящие характер кислот. Особенно много сделал в этой области один из крупнейших химиков XVIII века шведский ученый Шееле. Наряду с веществами кислотного характера, для их нейтрализации, в организмах неизбежно должны присутствовать какие-то вещества основного характера (помимо минеральных оснований, обнаруживаемых в небольших количествах в золе органических продуктов). Поэтому с начала XIX столетия начали предприниматься попытки выделить и охарактеризовать органические основания. Первый успех в этом направлении был достигнут в 1806—1807 гг. немецким фармацевтом Сертюрнером, который выделил из опия морфин. В дальнейшем для выделения органических оснований много было сделано французскими аптекарями Пеллетье и Каванту, выделившими, в частности, хинин из хинной корки (1820 г.).
С развитием теоретической органической химии оказалось возможным классифицировать практически все органические кислоты в соответствии с рациональной классификацией органических соединений, построенной на основе структурной теории. В отношении природных органических оснований этого не удавалось сделать на протяжении всего XIX века и первой четверти XX века прежде всего потому, что строение большинства важнейших алкалоидов* было неизвестно или неясно и, во всяком случае, не было еще подтверждено синтезом. Кроме того, лишь сравнительно простым органическим основаниям можно было найти только одно определенное место в существовавших системах классификации. Более сложные основания могли быть отнесены с одинаковым успехом к нескольким классам органических соединений. Это обстоятельство и до сих пор затрудняет отнесение многих алкалоидов к определенному классу, ряду или группе органических соединений.
Под названием алкалоиды в настоящее время объединяют растительные (и животные) азотсодержащие вещества основного характера, не являющиеся непосредственно продуктами гидролитического расщепления белковых веществ, имеющие в большинстве случаев сложный состав и часто обладающие сильным физиологическим действием.
* Название «алкалоид» означает «подобный щелочи».
По определению А. П. Орехова*, наиболее характерным для алкалоидов является факт их нахождения в готовом виде в растительном организме, продуктом жизнедеятельности которого они являются.
Роль алкалоидов в жизни и развитии растений до сих пор остается неясной. Предположение, что они являются защитными веществами против вредителей, явно несостоятельно, так как алкалоиды содержатся далеко не во всех растениях и, кроме того, алкалоидоносные растения поражаются вредителями не меньше других. Другое предположение, что алкалоиды, подобно мочевине и мочевой кислоте (отбросным веществам животных организмов), являются той формой, в виде которой растение выводит из своего кругооборота избыточный азот, также весьма сомнительно.
Возможно, что алкалоиды это—своего рода растительные гормоны, участвующие в процессах развития растений на тех или иных стадиях.
Что касается вопроса о механизме образования алкалоидов в растении, то весьма вероятно, что они образуются из аминокислот, участвующих в построении белковых веществ и выделяющихся при их распаде (А. Пикте, Робинсон), а также из продуктов превращения углеводов. С этой точки зрения очень интересны успешные попытки Цепфа и Робинсона получить простейшие алкалоиды и соответствующие им гетероциклические соединения в условиях, близких к условиям существования растительных организмов, т. е. при обычной (не повышенной) температуре, в средах, приближающихся к нейтральной, в отсутствие грубо действующих химических реактивов.
Отдельные семейства растений очень богаты алкалоидоносными видами, в других же семействах вовсе не обнаружено таких видов. Особенно богаты алкалоидами семейства кутровых (Ароеупасеае), маковых (Papaveraceae), мотыльковых (Papilionaceae), бобовых (Le-guminosae), лютиковых (Ranunculaceae), мареновых {Rubiaceae}, пасленовых (Solanaceae), а также некоторые из родов сложноцветных (Compositae). Чрезвычайно редко содержат алкалоиды однодольные и хвойные растения. Замечательно, что в растениях больших семейств губоцветных (Labiatae), розовых (Rosaceae) и орхидных {Orchideae) до сих пор не найден ни один алкалоид.
' В природе алкалоиды встречаются в большинстве случаев в виде солей органических (реже минеральных) кислот. Особенно часто с алкалоидами связаны кислоты: яблочная, лимонная, щавелевая, янтарная и дубильные (в таннинах); менее распространены уксусная (опий), пропионовая, молочная, роданистоводородная
* Александр Павлович Орехов (1881 —1939). Получил высшее образование в Швейцарии, где некоторое время работал в высшей школе; с 1918 по 1928 гг. работал в Париже у проф. Тиффено, совместно с которым провел обширные исследования пинаколиновой перегруппировки Вернувшись в СССР в 1928 г., начал успешно работать в области химии алкалоидов; выделенные им алкалоиды анабазин, сальсолин и платифиллин нашли практическое применение.
(горчица), фосфорная, серная (опий). Такие кислоты, как фумаровая, аконитовая, хелидоновая и др., встречаются лишь в определенных семействах растений. Некоторые кислоты являются специфическими, как, например, меконовая—для опия, хинная—для хинной корки и пр.
Большей частью в растениях содержатся смеси нескольких алкалоидов. Так, из препаратов опия (млечный сок недозрелых головок мака Раpaver somniferum) выделено 22 алкалоида, из хинной корки— 24, из листьев кока—7, из листьев табака—не менее 10 и т. д.
Замечательно, что если в растении содержится несколько алкалоидов, то обычно они обладают весьма близким строением, образуя как бы группу родственных веществ. Иногда наблюдается родственная близость алкалоидов с имеющимися в том же растении безазо-тистыми веществами, например с кислотами, связанными с алкалоидами. Однако известны также случаи, когда одинаковые или близкие по строению алкалоиды содержатся в растениях, принадлежащих к далеким друг от друга семействам.
Алкалоиды чаще всего сосредоточиваются в каких-либо определенных частях растения. Например, алкалоиды хинного дерева находятся почти исключительно в его коре, алкалоиды кустарника Erythroxylon coca (группа кокаина) и чайного дерева—в листьях, алкалоид гидрастин содержится в корневище //ydrastis canadensis, стрихнин и бруцин—в семенах Strychnos nux-vomica (рвотные орешки) и т. д.
Содержание алкалоидов в данном растении обычно невелико, и растительное сырье считают вполне пригодным для переработки при наличии в нем 1—2% алкалоида (в пересчете на сухой вес). Известны, однако, случаи более высокого содержания алкалоидов. Например, в коре культурных экземпляров хинного дерева (Cinchona Ledgeriana) содержание главного алкалоида—хинина—достигает 7—10%, а в отдельных случаях и 15%.
Для выделения алкалоидов из растительного сырья, состоящего в основном из углеводов, белков, дубильных веществ, смол и т. д., применяются различные приемы. В некоторых случаях мелкоизмельченное растительное сырье предварительно обрабатывают щелочами, смачивая его растворами аммиака, едкого натра или соды или смешивая с гашеной известью. Выделенные таким образом свободные алкалоиды-основания затем извлекают какими-либо не-смешивающимися с водой растворителями, например хлороформом, дихлорэтаном, бензолом, керосином, петролейным эфиром, диэтиловым эфиром и т. п. Пропуская растворитель последовательно через ряд экстракторов, можно получить раствор, достаточно богатый алкалоидами, даже если содержание их в исходом сырье составляло только доли процента. Далее смесь алкалоидов извлекают из экстракта водными растворами соляной или серной кислот, очищают и выделяют индивидуальные алкалоиды дробной кристаллизацией их солей или оснований (из соответствующих растворителей).
Другой прием выделения алкалоидов из растительного сырья заключается в экстракции разбавленными кислотами или спиртами (метиловым, этиловым, изопропиловым), подкисленными соляной или иногда органическими кислотами. В этих случаях алкалоиды извлекаются в виде солей этих кислот. Трудно растворимые в воде алкалоиды можно осадить из полученных кислых растворов щелочами, а легко растворимые—извлечь (после добавления щелочи) органическим растворителем. Иногда кислые водные растворы предварительно обрабатывают растворителями для удаления ряда балластных веществ и лишь после этого обрабатывают щелочами. В некоторых случаях алкалоиды можно извлечь из водных растворов с помощью ионообменных смол.
Указать общие приемы окончательной очистки и разделения смесей алкалоидов невозможно. Для каждого вида растительного сырья приходится применять комбинации различных приемов, а при исследовательских работах пользоваться, например, дробной кристаллизацией пикратов, пикролонатов и т. п.
При исследовательских работах, целью которых является обкзпужение алкалоидов ь растительных эх?трактах, часто пользуются различными приемами для предварительного у аале:: и я белковых веществ (осуждение ацетатом свинца и т, п.) и я:иров (экстрагирование иотк! еденных экюракнч,в петро.тейным эфиреш, хлороформом и т. д.Т
Для обнаружения алкалоидов i pi> не. я ют гпрпкыльчые реактивы. Очень многие алкалоиды. даию при нсьюгил ьяюм -огю’жании ч.х в вотвем растворе, даю'! нерастворимые осадки с п?ак7??плм’’. । ю т \ чин lob ми названье ыущих реакти* все на алкалоиды. К числу чаких реактивов шнссюея: воглю-спнргэеый раствор гаянина, водные растворы кислот фосфорполюлибдеповой (реактив Зсянсншейна), фосфорновольфрамовой (реактив Шейблера), кремневольфрамовой, пикриновой, ликролоновой, стифяиновой, флажкановой, платипохлоркстоводородной, золото-хлористоводородной, раствор иода в иодислом калии (реактив Вагнера), растворы K2[CdJJ (реактив Мармё), K[BiJ4] (реактив Драгендорфа), K2lHg.JJ (реактив Майера).
Для идентификации уже выделенных алкалоидов очень часто (например, при судебно-химических исследованиях) пользуются цветными реакциями. Так, не-которые алкалоиды дают характерные окраски или последовательную смену окрасок с чистой концентрированной серной кислотой, а также с серной кислотой, содержащей ничтожное количество азотной кислоты (реактив Эрдмана), 1% молибдата аммония (реактив Фреде), 0,5% ванадата аммония (реактив Манделина), небольшое количество формалина (реактив Маркй) и т. п.
Определение строения алкалоидов. В настоящее время описанс свыше 800 алкалоидов, но число алкалоидов с установленным строением не превышает 200, а синтезом подтверждено строение лишь нескольких десятков из них.
Многие природные алкалоиды имеют весьма сложное строение, определение которого представляло очень трудную задачу. Так, например, строение морфия, или морфина, выделенного в чистом виде еще в 1806—1817 гг., было окончательно установлено и подтверждено синтезом лишь в 1952 г. Строение хинина было установлено тоже сравнительно давно, а полный его синтез был осуществлен в 1945 г. Только в 1954 г. был осуществлен синтез стрихнина.
Обычно при определении строения алкалоидов прежде всего стараются установить присутствие прочных циклических группировок. Часто это достигается перегонкой с цинковой пылью или с едкими щелочами, при которой во многих случаях образуются различные пиридиновые и хинолиновые основания. После этого различными типическими реакциями определяют наличие первичных, вторичных или третичных аминогрупп, присутствие гидроксилов, группировок простых или сложных эфиров, карбонильных и карбоксильных групп ит. д. Особенно часто в алкалоидах содержатся метильные группы, связанные с азотом, или метоксильные группы. В обоих случаях при нагревании с иодистоводородной кислотой происходит отщепление йодистого метила, который может быть определен количественно (способ Цейзеля). Наконец, очень часто, как и для других природных веществ, при окислении различными окислителями получаются осколки молекулы исходного алкалоида, по строению которых можно составить представление о его строении.
Исследования многих сложных алкалоидов показали, что в их молекулах может содержаться в различных сочетаниях несколько циклических ядер, конденсированные ядра (гетероциклические, конденсированные с бензольными или несколько гетероциклических ядер, конденсированных между собой), а также сочетания гетероциклических ядер с иными углеродными циклами, кроме шестичленных. Бициклические (и трициклические) ядра в молекулах алкалоидов во многих отношениях напоминают соответствующие углеродные ядра в молекулах терпенов и их производных. Аналогия в строении алкалоидов и терпенов влечет за собой то, что и здесь, как в классе терпенов, ряд реакций может сопровождаться превращениями циклов. Эти превращения изучены пока еще менее, чем соответствующие превращения бициклических терпенов.
Классификация алкалоидов. Ввиду сложности строения не удается строго и однозначно классифицировать алкалоиды по химическому строению. Наиболее приемлемой представляется химическая классификация, в основу которой положено строение основной углеродноазотистой группировки молекулы алкалоида. По этому признаку все алкалоиды, строение которых можно считать установленным, могут быть разделены на следующие группы:
1. Алкалоиды, содержащие экзоциклический (не входящий в цикл) азот.
2. Производные пиррола.
3. Производные 1 -метилпирролизидина.
4. Производные пиридина и пиперидина.
5. Содержащие неконденсированные пяти- и шестичленный азотистые циклы.
6. Содержащие два неконденсированных шестичленных азотистых цикла.
7. Содержащие конденсированные пяти- и шестичленный азотистые циклы (группа тропана).
8. Содержащие два конденсированных пиперидиновых цикла.
9. Производные хинолина.
10. Производные изохинолина.
11. Производные индола.
12. Производные имидазола.
13. Содержащие пуриновую группировку.
14. Стероидные алкалоиды (производные циклопентенофенантрена).
Помимо химических классификаций алкалоидов, предлагалось классифицировать их на основе ботанической классификации растений, в которых встречаются алкалоиды. Таким образом, например, различали группу алкалоидов опия, алкалоидов чилибухи, алкалоидов чемерицы и т. д. Для алкалоидов нерасшифрованного строения к такому принципу классификации приходится прибегать и теперь.
Алкалоиды, содержащие экзоциклический азот
К этой группе относятся алкалоиды l-эфедрин и d-псевдоэфедрин н и он н
II II
С6Н5—С—С—СН3 сбН5—с—с—сн3
II II
ОН NH—СН3 Н NH—СН3
эфедрин псевдоэфедрин
содержащиеся в различных видах эфедры {Ephedra sinica, Ephedra helvetica). Их строение и конфигурация подтверждены синтезом. Эфедрин обладает сильным физиологическим действием, в частности повышает кровяное давление и расслабляет спазмы бронхов.
Алкалоидами с экзониклическим азотом являются также содержащийся в кактусе мескалин и выделяемый из ростков солода горденин.
К этой же группе должен быть отнесен и колхицин из безвременника осеннего (Colchicum autumnale'). Колхицин был открыт Пеллетье и Каванту еще в 1819 г., но строение его установлено лишь недавно (Дьюар, 1945—1950 гг.). Он является очень слабым основанием; органические растворители извлекают его даже из кислых растворов. Слабые основные свойства колхицина объясняются тем, что он является замещенным ацетамидом. Оказалось, что он содержит трополоновое кольцо (стр. 496):
CH3O^;\/_^/NHCOCH3
IM В I сНзо/^/Х/Ч с4 '<2'=о । осн3
Колхицин представляет собой аморфные блестящие обломки, плавящиеся при 143—147°. Он очень токсичен, вызывает паралич дыхания и сердечной деятельности; терапевтически не применяется.
Алкалоиды, производные пиррола
К этой группе относятся замещенные тетрагидропироллы: пирролидин (в табаке и в листьях свеклы), N-метилпирролидин (в черном перце Piper nigrum), гигрин (в листьях Erythroxylon coca) и кускгигрин:
J—СН2—сосн3
N
сн3
гигрин
(темп, плавл. 33—34°)
N N
СН8 сн3
кускгигрин (масло; темп. кип. 185° при 32 мм)
Алкалоиды, производные 1-метилпирролизидина
Производным 1 -метилпирролизидина, или гелиотридаиа
исчерпывающе изученного Г. П. Меньшиковым (1932—1936 rrj. является, например, платифиллин C18H27O5N.
Платифиллин представляет собой циклический диэфир пли-тинецина и двухосновной цис-сенециониновой кислоты:
НО
сн2он
платинецин (темп, плавл. 148,5°)
ОН
ОС--С--сн2—си—с—со
I II III
он сн—сн3 сн3 СН3 он
севециониновая кислота (темп, плавл 151°''
Платифиллин выделен А. П. Ореховым и Р. А. Коноваловой из крестовника широколистного (Senecio platyphyllus). Он плавится при 125—126°.
Применяется как спазмолитическое средство, расслабляющее гладкую мускулатуру, а также в глазной практике взамен атропина.
Алкалоиды, производные пиридина и пиперидина
Представителями этой группы являются крайне токсичные алкалоиды болиголова (Conium maculatum)—кониин (d-a-пропилпи-неридин, см. стр. 608), коницеин и конгидрин; алкалоид корки граната (Punica granatum)—пелътъерин, алкалоид касторового семени (Ricinus communis)—рицинин, а также алкалоиды арековой
пальмы (Areca catechu)—арекаидин и ареколин (N-метил-!,2,5,6-тетра-гидройикотиновая кислота и ее метиловый эфир):
^\сн2-сн2—СНО
пелыьерин CgHjsON (темп плавл. 195°)
осн3
rS/,CN СНа- NZ^° рицинин CgHgOoNg ''темп, плавл. 201°)
арекаидин C7HhO2N
(темп плавл. 223° с разл.)
арсголин AH13O2N
(масло без запаха, кип 209е)
Кроме того, сюда же должны быть отнесены алкалоиды лобелии {Lobelia inf lata). Виланд выделил из лобелии 14 алкалоидов, из которых главным является лобелии C2?H,7O2N. применяемый в медицине как средство для возбуждения дыхания, например при отравлениях ядовитыми газами, при удхшьи и т. и. При окт./тении лобелина получается лобеланин, для которого Бпт.снд (ГАТ' г.) установил строение П-метил-2,5-ди- 1фек:шил ]-ниг;сри тика. 2в.^е\ин оказался соответствующим кето-спирт ом:
о
СвН5ОС— СН/ N Abl.-COCJIj I сн8
лобеланнн
А
СЙНГ)ОС-СН/ N- \CH2-CH-CsHf сн8 9й лобелии
Рацемический лобелия плавится при 110°, левовращающий—при 130—131°.
К той же группе алкалоидов может быть отнесен пиперин (темп, плавл. 128—129,5°)—алкалоид перца, являющийся пипери-дидом пипериновой кислоты (стр. 608).
Алкалоиды, содержащие неконденсированные пяти- и шестичленный азотистые циклы
Такие циклы содержатся в молекуле алкалоидов табака (A ico-tiana tabacum и Nicotiana rustica из семейства Solanaceae).
Никотин. В листьях и семенах табака найдено несколько родственных алкалоидов, из которых главным (2—12%) является никотин C10H14Nj.
Никотин имеет строение р-(К-метил-а-пирролидил)-пир/йдина (его можно также назвать К-метил-а-(р-пиридил)-пирроллидином):
N | СН
Это жидкость с темп. кип. 247°; уд. вес dl°= 1,0099, пя= 1,5239. Он имеет запах табака, растворим в воде, вращает плоскость поляризации влево.
Никотин весьма ядовитый алкалоид. В малых дозах он возбуждает центральную нервную систему и повышает кровяное давление; при больших дозах вслед за возбуждением наступает угнетение нервной системы, остановка дыхания и паралич сердечной деятельности. Широко применяется как инсектицид для борьбы с вредителями сельскохозяйственных культур.
Никотин выделен Поссельтом и Рейманом еще в 1828 г. Строение его доказано синтезом, проведенным А. Пикте. Перегонкой ф-аминопиридина со слизевой кислотой (том I, стр. 569) А. Пикте получил М-(£-пиридил)-пиррол, который при пропускании через слабо накаленную трубку изомеризовался в а-ф-пиридил)-пиррол: п-Cl п-У
N N
Ь1-(3-пиридил)-пиррол а-(3'ПИрИДИЛ)-ПИррОЛ
При действии CH3J на калиевое производное этого пиридил-пиррола получается никотирин, или ₽-(М-метил-ос-пиррил)-пиридин, который восстановлением может быть превращен в недеятельный никотин, или р-(М-метил-ос-пирролидил)-пиридин:
гуМ
N I
никотирин никотин
Разделением солей винных кислот были получены d-никотин и /-никотин, тождественный природному никотину.
Шпет осуществил более изящный и наглядный синтез никотина. Конденсацией этилового эфира никотиновой кислоты с N-метилпирролидоном и нагреванием продукта конденсации с соляной кислотой он получил у-метиламинопро-пил-р-пири дил кетон:
./S—СО-ОС,Н, Н.С—сн.
и + оё у N v <!н8
+н2с
(НС1)
-СО-СН2-СН2 ноос сн, 5^
—СО-CH.,—сн I сн
NH
<!:н3
сн3 _
Кетон восстанавливался в карбинол, в котором с помощью HJ группа ОН замещалась на атом иода. Образовавшийся иодид легко замыкался в никотин:
Никотин может давать соли с одним или двумя эквивалентами кислоты. Присоединением CH3J к пиридиновому или пирролидиновому кольцу можно получить два изомерных иодметилата.
При окислении 'никотина перманганатом получается никотиновая ($-пири-динкарбоновая) кислота.
При перегонке никотина с цинковой пылью отщепляются четыре атома водорода из пирролидинового кольца и получается никотирин C10H10N2, найденный также в небольшом количестве в табаке наряду с никотеином C10HJ2N2 и ни-котеллином C10H8N2. Эти алкалоиды отличаются от никотина лишь содержанием водорода.
Алкалоиды, содержащие два неконденсированных шестичленных азотистых цикла
Анабазин. К таким алкалоидам относится анабазин C10HuN2, открытый А. П. Ореховым (1929 г.) в среднеазиатском ежовнике (Anabasis aphylld).
Анабазин представляет собой (З-(а-пиперидил)-пиридин:
N
Строение его подтверждено синтезами (Г. П. Меньшиков и А. А. Григорович; Шпет, 1936 г.). При окислении анабазина получается никотиновая кислота, при дегидрировании—а,(3-дипи-ридил.
Анабазин (бесцветное масло, темп. кип. 276°) широко применяется как инсектицид; его инсектицидная активность выше, чем никотина.
Алкалоиды, содержащие конденсированные пяти- и шестичленный азотистые циклы (группа тропана)
Алкалоиды этой группы являются производными тропана— бициклического основания, ядро которого состоит из конденсированных пирролидинового и пиперидинового колец и образует в то же время циклогептановый углеродный цикл:
1з
Тропан—жидкость, кипящая при 167°; уд. вес di° = 0,931.
В растениях семейства пасленовых (Solanacea) содержатся алкалоиды, представляющие собой сложные эфиры спирта тропина (атропин, его оптический /-изомер гиосциамин и др.), а в листьях кока (семейство Erytliroxplacp(bA-~~?^ калоиды группы кокаина, являющиеся эфирами гропии-2 карбоновой кислоты, называемой экгонином:
Тро ’ин
Сложные эфиры тропина называются тропеинами.
Тропин (темп, плавл. 63°, темп. кип. 233'3’ оптически недеятелен.
□астворим в воде.
Как вторичный спирт тропин пинон (темп, плавл. 42*, темп, превращающийся в тропин. При основание тропидин (темп. кип.
может быть окислен в кетон тро-кип. 225°), при восстановлении дегидратации тропина полагается 162—163°):
тропинон
тропидин
При нагревании со щелочами тропин превращается в стереоизомерный псевдотропин, который может быть получен также присоединением воды к тропидину.
Восстановлением иодистым водородом тропина и псевдотропина получается тропан.
Установлено, что для системы тропана наиболее устойчивыми являются такие конфигурации, в которых циклогептановое кольцо имеет форму «кресла»,
а пиперидиновое—форму «ванны» (форма А) или циклогептановое форму «ванны>, л пиперидиновое—форму «кресла» (форма Б):
А Б
Преобладание той или иной конфигурации зависит от характера и положения заместителей в тропане. Повидимому, как для тропина, так и для псевдотропина преобладающей является форма А. Тропин и псевдотропин отличаются друг от друга расположением заместителей при третьем углеродном атоме:
Тропин и тропинов при окислении превращаются в тропиновую кислоту (полученную также и синтетически) и в N-метилсукцин-имид; этим доказывается их строение. Вильштеттер путем ряда превращений получил из тропинона суберон (стр. 83), а из него— тропилиден (темп. кип. 117е):
гропнновая кислота ?vf-sePOH тропилиден
(циклогепта гриен)
Тропилиден образуется также при исчерпывающем метилировании тропидипа; путем ряда реакций он превращается снова в тропидин.
Робинсон синтезировал тропинов (1917 г.) очень простым путем—действием метиламина на смесь янтарного альдегида и ацетона:
СН2—СНО
+NH2CH3
СН,—СНО
сн3
со
СИ
сн2—сн—сп2
I \
NCH3 СОд-2Н2О
I /
сн2—сн—сн2
Атропин и гиосциамин C17H23O3N. Эти алкалоиды содержатся в белладонне (Atropa belladonna), в белене (Hyoscyamus niger) и в других растениях семейства пасленовых. Содержание атропина и гиосциамина в растениях составляет 0,1—0,3% и редко превышает 0,5%. В тех же растениях содержатся в еще меньших количествах родственные атропину алкалоиды: атропамин (апоатропин) C17H21O2N, скополамин (гиосцин) C17H21O4N и др.
Атропин кристаллизуется из спирта в призмах (темп, плавл. 115—116°). Растворы его имеют горький вкус. Гиосциамин плавится при 108,5°. В присутствии небольших количеств щелочи он уже на холоду превращается в атропин (рацемизуется).
Как атропин, так и гиосциамин являются эфирами тропина и троповой кислоты (рацемической—в случае атропина и оптически активной /-кислоты—в случае гиосциамина):
/NCH3 ОСО—СН—СН2ОН НООС-СН—СН?О1 ]
~ 7 С6Н5 С6Н5
атропин (и гиосциамин) троповая кислота
(стр. 388)
При омылении атропина кислотами или щелочами получается тропин и рацемическая троповая кислота (темп, плавл. 117—118°). При нагревании троповой кислоты и тропина с соляной кислотой снова получается атропин. Осторожным омылением гиосциамина получается /-троповая кислота (темп, плавл. 129—130°).
Атропин и гиосциамин чрезвычайно ядовиты. Они сперва возбуждают центральную нервную систему, вплоть до галлюцинаций (отсюда выражение «взбеленился», «белены объелся»), после чего наступает прострация. В токсических дозах они вызывают остановку дыхания. Применяются эти алкалоиды главным образом в офтальмологической практике для расширения зрачка.
Кроме атропина и гиосциамина, тропеином является также природный тропакокаин (стр. 655). Из синтетических тропеинов в медицине находит применение гоматропин, получаемый из тропина и миндальной кислоты. Подобно атропину он расширяет зрачок, но действие его менее продолжительно и он менее ядовит, чем атропин.
Кокаин C17H2iO4N—главный алкалоид листьев кока (Erythro-xylon coca).' Это растение произрастает в Южной Америке (Перу, Чили), а в настоящее время культивируется на Яве и на некоторых других островах Индийского океана.
Природный кокаин, или Г кокаин, кристаллизуется из спирта в виде призм (темп, плавл. 98°); трудно растворим в воде, легко— в спирте и эфире. Он находит значительное применение в медицине (в виде хлоргидрата, плавящегося при 200—202°) как одно из наиболее сильных местноанестезирующих средств. В то же время кокаин является сильным наркотиком. Злоупотребление кокаином, который вызывает приятное опьянение, сопровождается тягостной психической реакцией и приводит к истощению организма (ко-каинизм).
Кокаин представляет собой метиловый эфир бензоилэкгонипаг
----у-----уСОО С Н3
NCH3 ^>-осо-С6Н5
При кипячении с водой кокаин омыляется с выделением метилового спирта и образованием бензоилэкгонина. При омылении кокаина щелочами получаются метиловый спирт, бензойная кислота и левовращающий экгонин.
Этерификацией экгонина метиловым спиртом можно получить метиловый эфир, под действием хлористого бензоила или бензойного ангидрида превращающийся снова в кокаин. Бензоилированием экгонина получается бензоилэкгонин, который этерификацией метиловым спиртом превращается в кокаин.
Этими реакциями пользуются в технике для частичного синтеза кокаина из экгонина, получаемого омылением соляной кислотой побочных алкалоидов листьев кока и остатков от очистки кокаина.
В сухих листьях кока содержится от 0,5 до 1% кокаина. Кроме того, в них имеется гигрин (стр. 648) и ряд алкалоидов, по строению родственных кокаину: циннамилкокаин C19H23O4N, а- и ^-труксиллины (С,9>2’ бензоилэкгонин CieH]9O4N и тропакокаин C15H19O2N. Листья кока с острова Ява почти не содержат кокаина, но содержат много циннамилкокаина.
Циннамилкокаин отличается от кокаина лишь тем, что вместо остатка бензойной кислоты в нем содержится остаток коричной кислоты; в труксиллипах бензоил заменен остатками двухосновных труксилловых кислот (стр. 469).
Экгонин представляет собой оксикислоту тропана—тропин-2-карбоновую кислоту:
*
Д-соон
NCH3 *У--ОН
В отличие от оптически недеятельного тропина, он содержит четыре асимметрических атома углерода, вследствие чего может существовать в 16 оптически деятельных и 8 рацемических стереоизомерных формах.
Обыкновенный экгонин, или /-экгонин, при нагревании с концентрированным раствором КОН изомеризуется во вращающий вправо изомер (псевдоэкгснин), • который не является оптическим антиподом /-экгонина, а отличается от него пространственным расположением гидроксильной группы при третьем углеродном атоме (цис-, трянс-изомерия, аналогично тропину и псевдотропину, стр. 653).
Конфигурация экгонина (Z-экгонина) соответствует цис-расположению карбоксила и гидроксила, а конфигурация псевдоэкгонина—/пранс-расположению iex же гр\пп при втором и третьем углеродных атомах системы тропана:
экгонин
псевдоэкгонин
При нагревании иодметилата экгонина с едкой щелочью получается (с выделением Н2О, HJ и диметиламина) циклогептатриенкарбоновая кислота:
СООН
При окислении экгонина хромовой кислотой отщепляется СО2 и получается тот же тропинон (стр. 652), который образуется при окислении тропина и псевдотропина. Это свидетельствует о связи кокаина с атропином.
Экгонин был получен Вильштеттером синтетически, исходя из тропинона В тропиноне один из водородных атомов группы СН2, находящейся рядом с группой СО, может быть замещен на натрий. При действии СО2 на это натриевое производное получается тропинонкарбоновая кислота, которая восстановлением превращается в экгонин:
----------CHNa ।-COONa .-----------------------СООН
^NCH, \CQ I Ч’СН, \СО — ' ЛСН8 \_он
гропинон-натрий i попинонкарбоновая кислота экгонин
Согласно новейшим данным при этом получается смесь большого количества r-псевдоэкгонина с небольшим количеством г-экгонина.
Из экгонина метилированием и бензоилированием был получен рацемический кокаин. Кристаллизацией кислых солей винной кислоты его удалось разделить на оптические изомеры, один из которых оказался тождественным природному кокаину.
В 1923 г. Вилыптеттер осуществил полный синтез кокаина. Исходным продуктом для этого синтеза служил ацетонди карбоновый эфир, из которого можно получить экгонин следующими двумя путями:
1. Электролизом кислого эфира ацетондикарбоновой кислоты (том I, стр. 446) получается гексадиондикарбоновый эфир:
СН2—СО—СН2—СООСН8
2НООС—СН2—СО—СН2—СООСНз —> 1 + 2СО2 + Н2
СН2—СО—СН2—СООСН8
При действии на него метиламина (стр. 530) с последующим восстановлением полученного соединения образуется эфир N-метилпирролидиндиуксусной кислоты:
-----х—СН2—СООСН3
/NCH3 _____/_СН2—СООСНз
Реакцией Дикмана, т. е. замыканием цикла при действии алкоголята натрия^ (стр. 41), далее получается тропинонкарбоновый эфир, восстановлением превращаемы й в метиловый эфир экгонина:
\ S—СООСНз ^NCH3 \СО —>- х—СООСН3 -> ^NCH3 \_он
2. Действием метиламина на ацетон ди карбоновый эфир в смеси с яшар-ным альдегидом Робинсон еще в 1922 г. получил тропинондикарбоновый эфир:
СН2—СНО СНа—СООСНз
I
+NH2CH8+ со
I
СНа—СНО сна—соосн8
-----х—СООСНз
;nch8 Vo
-----— СООСНз
+ 2Н2О
Этим же способом из кислого эфира ацетондикарбоновой кислоты получается с отщеплением СО2 тропинонкарбоновый эфир.
Условия конденсации диальдегидов с ацетондикарбоновой кислотой и ее эфиром, необходимые для получения хороших выходов тропинона и его карбоксильных производных, подробно изучены Шепфом.
Как было указано выше, возможно существование восьми стереоизомерных оптически недеятельных экгонинов. В описанных синтезах Вильштеттеру удалось выделить три оптически недеятельных метиловых эфира экгонина. Из них один, оказавшийся рацематом природного экгонина, послужил исходным продуктом для получения рацемического кокаина.
Второй рацемат, образующийся в небольшом количестве, оказался метиловым эфиром псевдоэкгонина. Метилированием и бензоилированием из него был синтезирован стереоизомер кокаина, который также был разделен на оптические изомеры. Правовращающая форма этого стереоизомера, или d-псевдококаин, оказалась значительно менее ядовитой, чем l-кокаин. В то же время по анестезирующему действию псевдококаин не уступает /-кокаину. Виннокислая соль такого синтетического d-псевдококаина в настоящее время применяется в медицине под названием псикаина.
Третий синтетический стереоизомер экгонина, который, в отличие от обыкновенного экгонина, при нагревании со щелочами не превращается в псевдоэкгонин, но отщепляет воду и дает ангидроэкгонин, также был превращен в стереоизомер кокаина.
Алкалоиды, содержащие два конденсированных пиперидиновых цикла
Сюда можно отнести псевдопельтъерин C9H]5ON (темп, плавл. 49°)—алкалоид гранатовой корки, иногда называемый N-метилгра-натаном. Ядро этого алкалоида состоит из двух конденсированных пиперидиновых колец и образует одновременно восьмичленное углеродное кольцо:
/ XNCH3
\ / 3 /
Из этой системы в свое время исходил Вильштеттер при исследованиях, приведших его к синтезу циклооктатетраена (стр. 83).
Алкалоиды, производные хинолина
Число более простых алкалоидов, содержащих хинолиновое кольцо, невелико, и среди них не найдено практически важных веществ. Однако надо отметить, что к простым алкалоидам относятся хинолин и а-хинальдин. В то же время в эту группу входят важнейшие алкалоиды, содержащиеся в коре хинного дерева,—хинин и цинхонин. Кроме хинина и цинхонина, в хинной корке в значительно меньших количествах содержатся стереоизомерные им
хинидин и цинхонидин, а также содержащие на два атома водорода больше (за счет гидрирования винильной группы) гидрохинин и гидроцинхонин и фенол, соответствующий хинину,—купреин. Остальные спутники хинина, обнаруженные в .хинной корке, содержатся в ней в ничтожных количествах.
Строение главных алкалоидов хинной корки выражается формулами:
хинин C20H24O2No
цинхонин C19H22ON9
Как видно из приведенных формул, все эти алкалоиды содержат систему связанных карбинольной группировкой ядер хинолина и бициклического хинуклидина, в свою очередь состоящего из двух конденсированных пиперидиновых циклов. Хинуклидин, названный так Кенигсом, является довольно сильным основанием. Это— жидкость с темп. кип. 158°.
Строение хинных алкалоидов установлено в результате продолжавшихся. более ста лет исследований сотен химиков (начиная с работ Пеллетье и Каванту, 1820 г.). Из более поздних работ большое значение имели исследования Скраупа и Кенигса, а затем работы Рабе. В результате таких длительных исследований было не только окончательно установлено строение этих чрезвычайно сложных веществ, но и произведены многочисленные частичные синтезы, а также полный синтез соединений, отличающихся от цинхонина лишь отсутствием винильной группы (Рабе). В 1945 г. был осуществлен и полный синтез хинина.
Хинин и цинхонин. Кинин (темп, плавл. безводного 177°) может кристаллизоваться с тремя молекулами воды. В воде он трудно растворяется, в спирте и эфире—легко. Растворы хинина и его солей вращают плоскость поляризации влево и имеют чрезвычайно горький вкус.
В больших дозах хинин довольно ядовит. Он нашел исключительное по важности применение в медицине как противомалярийное и жаропонижающее средство. По силе действия он значительно превосходит цинхонин.
Растворы хинина имеют щелочную реакцию. Хинин может образовывать соли с одним или двумя эквивалентами кислот. Растворы этих солей имеют соответственно нейтральную или кислую реакцию.
Средняя сернокислая соль хинина, кристаллизующаяся с восемью молекулами воды, трудно растворима в воде, на чем и основано отделение хинина от остальных алкалоидов хинной корки.
В медицине применяется как сернокислая соль, так и хорошо растворимая в воде хлористоводородная соль хинина C20H24O2N2-HCl-2H2O и некоторые другие соли.
Для открытия хинина применяется очень чувствительная так называемая пгаллейохинная реакция: при прибавлении к раствору хинина (или его солей) хлорной воды и затем избытка аммиака получается интенсивное изумрудно-зеленое окрашивание. Растворы многих солей хинина обладают сильной флуоресценцией.
Цинхонин трудно растворим в воде, спирте и эфире, плавится при 255°, возгоняется в токе водорода; растворы цинхонина и его солей вращают плоскость поляризации вправо.
При окислении цинхонина получается цинхониновая, или хинолинукарбоновая. кислота, а из хинина—хининовая, или п-метоксицин-хониновая. кислота (стр. 616). При сплавлении со щелочами алкалоидов хинной корки за счет хинолиновой части их молекул образуются хинолин (Скрауп) и соответственно хинанизол, а при перегонке с цинковой пылью—лепидин (рметилхинолин). Таким образом и доказывается строение хинолиновой части молекул цинхонина и хинина.
Гораздо труднее было определить строение хинуклидиновой части молекул цинхонина и хинина, которая оказалась тождественной для обоих алкалоидов. За счет хинуклидинового ядра при перегонке алкалоидов со щелочами образуются пиридиновые основания—p-этилпиридин и [3-коллидин (рметил-р-этилпиридин).
Присутствие винильной группы в хинуклидиновом ядре было доказано присоединением галоидов и восстановлением в этильную группу (при этом получаются алкалоиды гидроцинхонин и гидрохинин). Особенно веским доказательством наличия винильной группы, является то, что при осторожном окислении хинных алкалоидов на холоду марганцовокислым калием отщепляется муравьиная кислота и образуются карбоновые кислоты, содержащие в молекуле на один атом углерода меньше, чем цинхонин и хинин. Эти кислоты называются цинхотенином и хитенином.
Окислением хинуклидиновой части молекулы хинина получаются мерохинен и продукты дальнейшего его окисления: цинхолой-поновая и лойпоновая кислоты. Окислением хинуклидинового ядра гидрохинина получается цинхолойпон* \
мерохинен (темп, плавл. 223—224°, с разл.)
ноос
н.
ООН
НООС
сн2
-СООН
NH цинхолойпоновая кислота (две модификации: темп, плавл. 211° и 248°) Цинхолойпоновая кислота, а гексагидроцинхомероновых получены синтетически.
На основании строения продуктов окисления алкалоидов хинной корки Кениге предположил присутствие в их молекулах кольца хинуклидина; а в частности в хинине и цинхонине—наличие группы р-винилхинуклидина.
$-Этйлхинуклидин (жидкость, темп. кип. 190° при 720 мм) был синтезирован Кенигсом из р-коллидина следующим путем:
’ ' сн2он z/l\zC2H,
NH лойпоновая кислота (темп, плавл 259—-260°, с разл.)
также одна из стереоизомерных (пиперидин-3,4-дикарбоновых) кислот
- СН3 СН2
+СН2О РЕг5
к / к / N N
сн2
сн,
N+Br"
Положение гидроксильной группы в алкалоидах хинной корки .было установлено опытами Рабе, показавшего, что при окислении этих алкалоидов хромовой кислотой можно получить кетоны цин-
* Эта своеобразная терминология производится от греческих слов: нуклеос— яйцо, ядро (отсюда хинуклидин—вещество, содержащее ядро хинина); мерос—часть, доля (мерохинен—вещество, заключающее часть хинина); лойпос—остаточный, остальной (цинхолойпон—то, что осталось от хинуклидина, цинхонина, хинина).
хонинон и хининон и что, следовательно, хинин и цинхонин являются вторичными спиртами. При восстановлении указанных кетонов вновь образуются хинин и цинхонин в смеси с их стереоизомерами.
Рабе удалось также выяснить процессы изомеризации хинина и цинхонина, легко протекающие под действием кислот (при этом органические кислоты, например уксусная, действуют сильнее, чем минеральные). Изомеризацию впервые заметил Пастер, назвавший изомер хинина хиницином. Позднее этот изомер (найденный также в хинной корке) и соответствующий изомер цинхонина были названы хинотоксином и цинхотоксином вследствие их чрезвычайной ядовитости.
Изомеризация цинхонина протекает следующим образом (здесь Ch—остаток хинолина):
цинхонин цинхотоксин
Из цинхотоксина и хинотоксина можно снова получить^цинхо-нин и хинин. А именно, при действии бромноватистых солей на эти токсины получаются бромамины, которые действием щелочей превращаются в цинхонинон и хининон, при восстановлении дающие хинин и цинхонин, например:
цинхонин
Позднее Рабе осуществил полный синтез токсинов, отличающихся от цинхо токсина и хинотоксина лишь отсутствием винильной группы. Для этого он кондеи-
сировал в присутствии алкоголята натрия эфир 1Ч-бензоил-7-пиперидилпропионо вой кислоты с эфирами цинхониновой и хининовой кислот.
Омылением продукта конденсации и отщеплением СО2 получается токсин и, кроме того, бензойная кислота:
Завершением работ по изучению строения хинина явился полный синтез его через мерохинен, осуществленный в 1945 г. Вудвордом с сотрудниками. При этом синтезе исходили из ацеталя и .и-оксибензальдегида. Хлорированием ацеталя с последующим обменом хлора на аммиак получали аминоацеталь, который конденсировали с м-оксибензальдегидом, и продукт конденсации циклизовали серной кислотой в 7-оксиизохинолин:
(с2н5о)2сн-сн2
ОН
Конденсацией последнего с формальдегидом в присутствии пиперидина получали 7-окси-8-метиленпиперидиноизохинолин и от него отщепляли пиперидиновый остаток нагреванием до 220° с метилатом натрия (Робинсон, 1942 г.). Полученный таким путем 8-метил-7-оксиизохинолин гидрировали с платиновым катализатором в уксусной кислоте; при этом получался 1,2,3,4-тетрагидро-8-метил-7-окси-изохинолин. После ацетилирования иминогруппы этот продукт гидрировали при 160° и 200 ат с никелем Ренея и цис-изомер получившегося полностью гидрированного вторичного циклического спирта окисляли хромовым ангидридом в уксусной кислоте при 0°.- Образовавшийся (в виде гидрата) цш?-М-ацетил-7-кето-8-метил-..декагидроизохинолин обрабатывали этилнитритом в абсолютном этаноле в присутствии этилата натрия. Это приводило к расщеплению гидрированного бензольного кольца и образованию ццс-М-ацетилоксиминодигидромерохиненового эфира, который гидрированием с платиновым катализатором превращали в аминосоединение. Затем действием CH3J первичную аминогруппу превращали
в четырехзамещенную аммониевую, после чего расщепляли эту группу действием 60%-ного NaOH, одновременно омыляя ацетильный остаток у изохинолинового азота и эфирную группу. Этим путем получался гомомерохинен:
I \ .СН-СН f НгТ N(CH3)3
I
СО с н3
гомомерохинен
Обработав гомомерохинен сначала НС] и этиловым спиртом, а затем СбН5СОС1 с поташом, получали этиловый эфир N-бензоилгомомерохинена Нагреванием сплавленной смеси этого эфира и этилового эфира хининовой кислоты с сухим этилатом натрия была получена (после гидролиза действием соляной кислоты и обратного бензоилирования) рацемическая смесь d- и Z-бензоилхиноток инов:
бензоилхинотоксин
Из этой смеси был выделен трудно растворимый бЛбензоилхинотоксин-б/-тар-трат. Превращение же хинотоксина (получаемого омылением бензоильного производного) в хинин было осуществлено Рабе еще в 1911 г.
В молекуле хинина имеется четыре асимметрических атома углерода:
Расположение атомов водорода при 4-м углеродном атоме однозначно определяется для структуры хинуклидина как цис-положение. Водород при 8-м углеродном атоме должен находиться в транс-положении по отношении к водороду при 3-м атоме углерода. Иначе (при их цис-положении) группировка 9-го углеродного атома и винильная группировка оказались бы в цис-положении, что должно было бы вызвать образование эфирного кислородного мостика между углеродными атомами 9 и 10 вследствие присоединения элементов гидроксильной группы по двойной связи. В противоположность изомерам с цис-расположением при 3-м и 9-м углеродных атомах, у хинина этого не наблюдается.
Конфигурацию заместителей при 9-м углеродном атоме устанавливают путем сопоставления с известными конфигурациями эфедрина (слабое основание, лсезо-конфигурация) и псевдоэфедрина (сильное основание, рацемат, разделяемый на оптические изомеры). Поскольку хинин, в отличие от так называемого эпихинина (отличающегося только расположением заместителей при 9-м углеродном атоме), является слабым основанием, его конфигурация должна отвечать конфи-
, СН,
Is I |
Н—С—N— Н—С—NHCH.
I I
но—С—н но—с—н
Iе • I
Ch CeHg
эпихинин псевдоэфедрин
хинина можно изобразить так:
гурации эфедрина: СН3
H-C-N— Н—С—NHCH3
H—С9—ОН Н—С—ОН
I I
Ch С6Нб
хинин эфедрин
Пространственную конфигурацию Н Н
Н2С—с—с—сн=сн2
сн2
сн2 ил"
H—С—N—СН2
Н—С—он
Алкалоиды, производные изохинолина
Сравнительно простым алкалоидом является сальсолин, выделенный А. П. Ореховым и Н. Ф. Проскурниной (1933 г.) из Salsola Richteri. Он представляет собой 1 -метил-6-окси-7-метокситетрагид-роизохинолин:
СН3
Сальсолин был синтезирован одновременно А. П. Ореховым и Шпетом (1934 г.), исходя из изованилина по Бишлеру и Напираль-скому (стр. 621). Рацемический сальсолин плавится при 218—22Г, d-сальсолин—при 215—216°. Сальсолин понижает кровяное давление, расширяя периферические кровеносные сосуды; применяется в терапии гипертонической болезни.
Более сложными производными бензилтетрагидроизохинолина являются важные алкалоиды опия {папаверин, наркотин и др ), а также гидрастин, выделяемый из желтокорня {Hydrastis canadensis}.
Папаверин, в отличие от других опийных алкалоидов, обладает слабым анальгетическим действием; для него очень характерна способность расслаблять гладкую мускулатуру, действовать спазмолитически при кишечных и почечных коликах, при грудной жабе и гипертонии. Гидрастин обладает способностью вызывать сужение периферических кровеносных сосудов и применяется в медицине (в виде солей) как кровеостанавливающее средство. Имеют применение (для остановки кровотечений, особенно маточных) также продукты расщепления наркотина и гидрастина — котарнин и гидрастинин.
Хлористоводородный котарнин носит в медицине название стиптицина.
Особую группу производных гидрированного бензилизохинолина составляют важнейшие алкалоиды опия—морфин, кодеин и тебаин, содержащие скелет частично гидрированного фенантрена.
Сложными производными бензил изохинол ина, содержащими две частично гидрированные бензилизохинолиновые группировки, соединенные эфирными связями, являются алкалоиды группы кураре. Они входят в состав парализующих стрельных ядов, применявшихся южноамериканскими индейцами. Эти алкалоиды содержатся в растениях Chondrodendron tomentosum, Strychnos toxifera и в некоторых других.
Ядовитые алкалоиды группы кураре почти не действуют при приеме внутрь, но при введении под кожу вызывают полное расслабление поперечно-полосатых мускулов, парализуя все виды движения; в больших дозах вызывают смерть, наступающую вслед
ствие паралича дыхания. Выделенный из кураре впервые в 1935 г. кристаллический правовращающий тубокурариндихлорид находит применение как добавка к наркотизирующим препаратам при некоторых хирургических операциях, а также в терапии расстройств деятельности мускулатуры, например при хорее.
Тубокурарин является дихлоридом четвертичного аммониевого -основания, производного алкалоида хондродендрана, или бебирина:
хондродендрин СзвНзвОвЫа
т убокурариндихлорид
В главных алкалоидах ипекакуаны (корень южноамериканских растений Psychotria Ipecacuanha или Cephaelis Ipecacuanha)—цефаэлине C28H38O4N2 и его метиловом эфире эметине—изохинолиновый и хинолизиновый циклы связаны метиленовой группой.
Для эметина Робинсон (1948 г.) предложил и обосновал следующую формулу:
СНз°\/Ч^
II I NH
СН2
С М I
U\fY0C"’ I I! I \/^\0СН8
Строение эметина (1950 г.) и тубокурарина (1956 г.) подтверждено синтезом (Н. А. Преображенский и сотр.).
Цефаэлин содержит в изохинолиновом ядре в положении 4 вместо метоксильной свободную гидроксильную группу.
Эметин (темп, плавл. 74°) нерастворим в воде и в щелочах. Цефаэлин плавится при 115—116°; способен растворяться в едких щелочах.
Эти алкалоиды (а также экстракты корня ипекакуаны) в малых дозах применяются как отхаркивающее.
Эметин является специфическим средством при лечении амебной дизентерии.
Папаверин C20H21O4N (нерастворимые в воде призмы с темп, плавл. 147°) является одним из главных алкалоидов опия и представляет собой тетраметоксибензилизохинолин:
папаверин
При действии на него иодистоводородной кислоты отщепляются четыре молекулы CH3J и образуется тетраоксибензил изохинол ин, или папаверолин.
При сплавлении с едким кали папаверин окисляется и распадается на диметоксиизохинолин и вератровую кислоту:
СНз°\^\/Ч
I II i ch.cZ'V-^' диметоксиизохинолин
СООН I
''У^ОСНз осн3
вератровая кислота
При действии на папаверин CH3J получается иодметилат папаверина, который при восстановлении образует рацемический N-метилтетрагидропапаверин, разделенный при помощи солей хинной кислоты на оптические антиподы.
Правовращающий изомер оказался тождественным другому алкалоиду опия—лауданозину.
Папаверин был получен синтетически в 1910 г. А. Пикте и Гамсом.
Наркотин C23H23O7N и гидрастин C21H2IOeN являются, как и лауданозин, производными тетрагидроизохинолина:
наркотин (темп, плавл. 176°) гидрастин (темп, плавл. 132°)
При окислении наркотина и гидрастина получается опиановая кислота (темп, плавл. 150°)
ОСН3
^JyzOCHs
'^/'Х'СООН
СНО и котарнин (иглы, темп, плавл. 132—133°) или гидрастинин (иглы, темп, плавл. 116—117°), отличающийся от котарнина отсутствием метоксильной группы.
Котарнин и гидрастинин относятся к числу веществ, подвергающихся таутомерным превращениям, например для котарнина:
. СН3О СН3О ОН
СН, I п
СН3О
III
В твердом виде они, вероятно, представляют собой разомкнутые формы типа I, а в растворах происходит таутомерное превращение с образованием алкоголя, производного тетрагидроизохинолина (II). Этот алкоголь с кислотами дает желтые соли аммониевых оснований, представляющих собой соли третьей таутомерной формы (III) котарнина (аналогично и для гидрастинина).
При действии на наркотин или гидрастин слабых восстановителей, вместо опиановой кислоты получается лактон оксикислоты— меконин (темп, плавл. 102°)
ОСН3
1 J3CH8
у\со I I
Н2С—о
а вместо котарнина и гидрастинина — гидрокотарнин и гидрогидрастинин, содержащие взамен группы СН—ОН (форма II) группу СН2. Гидрогидрастинин можно получить также энергичным восстановлением гидрокотарнина. Окислением гидрогидрастинина можно получить гидрастинин. При кипячении в спиртовом растворе с меконином в присутствии поташа гидрастинин и котарнин вновь образуют исходные алкалоиды (Перкин, Робинсон).
Котарнин и гидрастинин были синтезированы Деккером, исходя из сафрола (стр. 340), по следующей схеме:
соль гидрастинина
Для синтеза котарнина исходным веществом является метоксипроизводное сафрола—миристицин.
Морфин, кодеин и тебаин. В опии, т. е. в высушенном млечнОхМ соке, вытекающем при повреждении недозрелых головок* мака (Papaver sommferum), помимо наркотина (стр. 668), содержащегося в количестве 5—6%, и папаверина (стр. 667), найдено еще около 20 алкалоидов, более или менее близких по строению. Главнейшим из них является морфин, или морфий, C17H19O3N, содержание которого в опии сильно колеблется, но в среднем составляет около 10%. В наиболее тесном родстве с морфином находятся алкалоиды кодеин
C18H21O3N (содержание в опии от 0,2 до 1,2%) и тебаин C19H2IO3N (0,2%). Эти три алкалоида, в отличие от слабого основания наркотина, являются сильными основаниями.
Морфин обычно кристаллизуется с одной молекулой воды, безводный плавится с разложением при 254°. В воде морфин мало растворим, еще менее— в эфире, этилацетате и бензоле. Хлористоводородная соль морфина кристаллизуется с тремя молекулами воды, хорошо растворима в горячей воде, трудно—в холодной, имеет нейтральную реакцию. Морфин оптически деятелен—вращает влево плоскость поляризации. Морфин и его соли чрезвычайно ядовиты; в малых количествах они действуют успокаивающим образом, в более значительных—вызывают состояние наркоза и затем сон. Хлористоводородная соль морфина находит значительное применение в медицине как болеутоляющее снотворное средство.
Кодеин кристаллизуется из воды и водного спирта с одной молекулой воды, из эфира и бензола—безводный (темп, плавл. 155°). Он мало растворим в воде, легкое эфире, спирте и бензоле, нерастворим в щелочах; обладает левым вращением. Кодеин и его соли применяются в медицине как средство против кашля, уменьшающее чувствительность нервов слизистых оболочек. Стой же целью применяется и получаемый алкилированием морфина его этиловый эфир, называемый дионином.
Тебаин—листочки с темп, плавл. 193°. Он чрезвычайно ядовит, подобно стрихнину вызывает судороги.
Строение алкалоидов группы морфина окончательно установлено Робинсоном (1925 г.). Все они содержат скелет частично гидрированного фенантрена:
тебаин
При таком написании структурных формул ясно видно, что эти алкалоиды являются производными частично гидрированного фенантрена.
Если же формулу морфина написать иначе, шестичленный азотистый цикл, образованный углеродными атомами 9, 14, 13 и цепью —СН2—СН2—N(CHS)—, и конденсированный с ним шестичленный углеродный цикл, образованный углеродными атомами 5, 6, 7, 8, 14. 13. принимают знакомую форму скелета изох и целина:
В такой форме видно родство алкалоидов группы морфина с папаверином и наркотином.
Родство этих алкалоидов с фенантреном проявляется в ряде их превращений в более простые производные фенантрена (стр. 674). Другой ряд превращений, ведущий к получению сложных производных изохинолина, сопряжен с глубокими атомными перегруппировками молекулы морфина. Например, при нагревании морфина с кислотами происходит разрыв окисного кольца, отщепление молекулы воды и перемещение цепочки —СН2—СН2—N—СН3отС13 к С8. В результате из морфина образуется апоморфин, содержащий уже не одно, как в морфине, а два ароматическихАкольца:
Легкость протекания этой перегруппировки объясняется образованием новой ароматической системы.
Строение апоморфина было окончательно подтверждено синтезом соответствующего ему диметилового эфира (Шпет, 1929 г.). Действием р-фенилэтиламина на хлорангидрид 3,4-диметокси-2-нитрофенилуксусной кислоты был получен соответствующий амид. Обработка этого амида пятиокисью фосфора привела к замыканию дигидроизохинолинового цикла, а действием бромистого метила дигидроизохинолиновое производное было превращено в бромметилат:
Восстановлением его азотистый цикл был полностью прогидрирован и одновременно нитрогруппа превращена в аминогруппу. Диазотированием соли амина и последующим нагреванием образовавшейся соли диазония с порошком меди была отщеплена молекула азота. При этом связи между бензольными ядрами замкнулись:
диметиловый эфир апоморфина
Апоморфин находит применение в медицине благодаря сильному рвотному, а в малых дозах—слизеотделяющему действию.
Из трех атомов кислорода в молекуле морфина один имеет фенольный характер, второй—спиртовый, а третий—эфирный.
Благодаря наличию фенольного гидроксила морфин растворяется в едких щелочах и с хлорным железом дает синее окрашивание. Эфирный атом кислорода образует непрочное окисное (гидрофурановое) кольцо.
Щелочные растворы морфина легко окисляются на воздухе. Молекула морфина при этом теряет один атом водорода, и морфин пе
реходит в диоксидиморфин, или псевдоморфин (найденный и в самом опии).
При действии уксусного ангидрида на морфин водородные атомы обоих гидроксилов замещаются на ацетильные группы и получается диацетилморфин, под названием героина применяемый в медицине подобно кодеину.
В щелочных растворах морфин при действии СН3Ли других метилирующих средств образует кодеин. Таким образом, кодеин это— метиловый эфир морфина, причем в метоксигруппу превращается фенольный гидроксил морфина. Эту реакцию используют в промышленности для получения кодеина из морфина.
В Советском Союзе для получения кодеина из морфина пользуются изящным методом (В. М. Родионов и сотр.) термического разложения морфината три-метилфениламмония. Соответствующее четвертичное основание получается действием CHjCl или метилового эфира л-толуолсульфокислоты на диметиланилин с последующей обработкой продукта присоединения алкоголятом натрия.
При окислении кодеина его спиртовая группа ОН окисляется в карбонильную и получается кетон—кодеинон. Этот же кетон получается с выделением метилового спирта из тебаина при действии разбавленных кислот.
Сравнение формул кодеина и тебаина (стр. 670) показывает, что тебаин представляет собой метиловый эфир енольной формы кетона, изомерного кодеину, и что получение из тебаина кодеинона связано с перемещением двойной связи.
Присутствие двойной связи в кодеине и кодеиноне доказывается тем, что оба они легко присоединяют два атома водорода, образуя дигидрокодеин и дигидрокодеинон.
При действии соляной кислоты, а также хлористых соединений фосфора на морфин и кодеин происходит замещение алкогольной гидроксильной группы на хлор.
Вследствие частичного вальденовского обращения (см. том I, стр. 691 сл.) при этом получается смесь двух стереоизомерных веществ, носящих название а- и $-хл*рморфидов и соответственно а- и $-хлоркодидов.
При обратном обмене (действием волы) атомов хлора на гидроксил из а- и Р-хлоркодидов получаются не только стереоизомерные кодеин и изокодеин, дающие при окислении один и тот же кодеинон, но, вследствие изомеризации, образуются также структурные изомеры кодеина: стереоизомерные друг другу псевдокодеин и аллопсевдокодеин, дающие при окислении структурный изомер кодеи-н она—псевдокоде и нон.
Кодеинон при восстановлении дает смесь кодеина и изокодеина, псевдоко-деинон—смесь псевдокодеина и аллопсевдокодеина.
Происходящая изомеризация является примером взаимных превращений типа
Х-СН(ОН)—СН=СН— Y X—СН—СН—СН(ОН)—Y
Действительно, тогда как кодеин и изокодеин содержат гидроксильную группу при 6-м атоме углерода в третьем бензольном кольце фенантрена, в псевдокодеине и аллопсевдокодеине гидроксил присоединен к 8-му атому углерода, что доказывается превращением кодеинона и псевдокодеинона в различные производные фенантрена.
Так, кодеинон при нагревании с уксусным ангидридом отщепляет молекулу метиламиноэтилового алкоголя и дает монометиловый эфир 3,4,6-триоксифенан-
псевдокодеинон монометиловый эфир 3,4,8-триоксифенантрена
Из морфина, кодеина, тебаина и многих их производных при перегонке с цинковой пылью Получается фенантрен. При действии щелочей на иодметилаты изомерных кодеинов (стр. 673) получаются с выделением HJ изомерные метил-морфиметины, которые при нагревании с уксусным ангидридом дают монометиловый эфир диоксифенантрена, названного морфолом и полученного синтетически по способу Пшорра (стр. 482)
При нагревании метилморфиметина со щелочами образуется метиловый эфир морфенола. Морфенол при сплавлении со щелочами дает 3,4,5-триоксифенантрен.
Тебаин, подобно кодеинону и метилморфиметинам, легко отщепляет боковую азотсодержащую цепь, образуя производные фенантрена. Так, при действии уксусного ангидрида он дает диметиловый эфир 3.4,6-триоксифенантрена, названный тебаолом. При нагревании тебаина с разбавленной соляной кислотой получается вторичное основание—пгебенин.'.
Получаемый из тебаина в результате изомеризации и гидрирования дигидро-кодеинон дает оксим, при действии РОС13 превращающийся в изооксим:
сксим дигидрокодеинона
изооксим дигидрокодеинона
Установлением строения изооксима как альдегидонитрила со связью цепочки —СН2—СН2—N(CHt)— с 13-м атомом углерода было окончательно доказано нахождение этой цепочки между 13-м и 9-м атомами углерода фенантренового ядра.
Превращение оксима дигидрокодеинона в изооксим подобно превращению в тех же условиях бензоиноксима (см. бекмановскую перегруппировку, стр. 283)
Окислением тебаина перекисью водорода или хромовой кислотой получается оксикодеинон C18H19O4N, который амальгамой натрия восстанавливается в дигидроокси,содеинон. C184alO4N, под названиями эукодала или текодина применяемый в медицине как наркотическое болеутоляющее средство Предполагается, что эти соединения отличаются от кодеинона и дигидрокодеинона наличием гидроксильной группы у 7-го атома углерода.
В 1952 г. был впервые осуществлен полный синтез кодеина, а из него и морфина (Гэтес и Чуди).
Сложный путь этого синтеза, включающего свыше 20 стадий, разумеется, непригоден для промышленного осуществления. Его значение заключается в окончательном подтверждении ранее установленного строения алкалоидов группы морфина и их стереоконфигураций.
Сравнительно недавно был получен синтетически ряд веществ, обладающих углеродноазотистым скелетом морфина.
Предложенный для их получения путь (Греве, 1946 г.) изображается следующей схемой:
C6H5CH2MgCl
Оксигруппа может быть введена на одной из первых стадий синтеза (при алкилировании изохинолина может быть взято соединение, уже содержащее оксигруппу) Переход от N-метилморфинана к 3-оксмпроизводному может быть также осуществлен нитрованием метилморфинана в ароматическом ялр<* с последующим восстановлением диазотированием и заменой диазогруппы на оксигруппу.
3-Ок.си-Ы-метилморфинан образует кристаллы с темп, плавл. 251—253*, его хлористоводородная соль плавится при 176—178*. Этс вещество, подобно морфину, вызывает интенсивную длительную анальгезию и является одним'из лучших его заменителей.
Для замены морфина в настоящее время предложен ряд синтетических препаратов, не вызывающих привыкания при постоянном уп о-треблении. К ним относятся 6-диметиламино-4,4-дифенилгептан-3 -он (I) и 6-морфолино-4,4-дифенилгептан-3-он (II):
Эти препараты обладают аналогичными морфину терапевтическими свойствами и выпускаются под названиями: амидон, долофин, метадон, фенадон.
В Советском Союзе в качестве заменителя морфина был предложен (И. Н. Назаров) промедол—сложный эфир пропионовой кислоты и 1,2,5-триметил-4-фенил-4-пиперидола:
о
X zoco—С2П5 нзС\/\
СН3—
Алкалоиды, производные индола
Гармин и гармалин. Из сравнительно простых алкалоидов к этой группе относятся алкалоиды степного могильника (Peganum harmala)—гармин C13H12ON2 и гармалин C13H14ON2:
гармин
(темп, плавл. 257—258°)
NH | сн.
гармалин
(темп, плавл. 250—251°)
Гармин был выделен Ю. Ф. Фрицше (1847 г.), гармалин—Гебелем (1841 г.). Синтез гармалина был осуществлен в 1927 г. (Перкин, Робинсон, Манске) довольно сложным путем. Проще гармалин синтезируется по реакции Бишлера—Напиральского (стр. 621) из метоксипроизводного р-индолилэтиламина (Шпет, 1930 г.):
Дегидрированием с порошкообразным палладием гармалин превращается в гармин.
Растворы солей гармина и гармалина обладают интенсивной флуоресценцией (соответственно синей и зеленовато-желтой), а растворы гармалина даже окрашены в желтый цвет. Оба эти алкалоида являются возбудителями центральной нервной системы; в больших дозах вызывают судороги. Гармин применяется как средство против некоторых нервных заболеваний (паркинсонизм).
Йохимбин и резерпин. В коре южноафриканского дерева семейства мареновых (Corynanthe Yohimbe) содержатся алкалоиды (выделено И оснований), из которых главным является выделенный. Шпигелем в 1896 г. иохимбин C21H26O3N2 (темп, плавл. 234,5°).
Йохимбин находится также в различных видах Rauwolfia—растения, содержащего до 25 алкалоидов, в частности очень важный алкалоид резерпин:
При перегонке с цинковой пылью иохимбин и резерпин дают гармин (вернее, скелет гармина без метоксильной группы), изохинолин и смесь крезолов. В иохимбине имеется пять асимметрических углеродных атомов, в резерпине—шесть. Строение иохимбина в самое последнее время подтверждено синтезом (Н. А. Преображенский, 1957 г.).
Иохимбин снижает кровяное давление; в малых дозах он возбуждает дыхание, в больших—вызывает паралич дыхательного центра. Он парализует сосудодвигательные центры продолговатого мозга, вызывает приливы крови к коже и слизистым оболочкам. Применяется в медицине и в ветеринарной практике. Резерпин применяется в медицине под названием серпазил как снижающее кровяное давление и успокаивающее средство.
Эзерин (физостигмин). В калабарских бобах—плодах южноафриканского растения Physostigma venenosum из семейства бобовых (эти бобы туземцы называли «эзере»)—содержится эзерин, или физостигмин, C15H21O2N3. Он известен в двух модификациях, с темп, плавл. 106° и 86—87°. При гидролизе щелочами эзерин распадается на СО2, метиламин и эзеролин—вещество, обладающее свойствами фенола. При перегонке же эзерина с цинковой пылью образуются а-метилиндол и N-метилиндол.
Для эзерина была предложена фоомула (бр. Полоновские, 1925 г.)
I Н | сн8 сн,
подтвержденная синтезом в 1935 г. (Джулиан и Пикл).
Эзерин очень токсичен. Он повышает кровяное давление, действует на центральную нервную систему и на мускулатуру, сперва возбуждая ее, а затем парализуя. Применяется в офтальмологической практике как антагонист атропина, сужающий зрачок.
Стрихнин и бруцин. Из семян, произрастающих на Филиппинах и в Индонезии видов Strychnos (чилибуха), в частности Strychnos nux-vomica (рвотные орешки) и Strychnos Ignatii, Пеллетье и Ка-ванту еще в 1818 г. выделили два алкалоида — стрихнин и бруцин.
Стрихнин образует мелкие призмы (темп, плавл. 286—288°); бруцин безводный плавится при 178°, образует кристаллогидрат с четырьмя молекулами воды, плавящийся при 105°.
Бруцин отличается от стрихнина наличием двух метоксильных групп взамен атомов водорода. При перегонке с цинковой пылью оба алкалоида дают индол и хинолин, при сплавлении с едкими щелочами—р-пиколин и р-коллидин, а при перегонке с известью — также и карбазол.
При окислении в определенных условиях (например, СгО,) из обоих алкалоидов получается одна и та же кислота, содержащая оба атома азота, но не содержащая метоксильных групп (кислота Ганнсена). Это доказывает, что при окислении разрушилось арома-^ижкое ядро молекулы, несущее метоксильные группы.
Изучением строения этих алкалоидов занимались многие исследователи (особенно следует отметить Леукса, Виланда, Робинсона, Прелога), и, в конце концов, в 1946 г. Робинсоном были предложены следующие формулы:
стрихнин C21H22O2N2
бруцин С23Н26^4^2
В 1954 г. эти формулы были подтверждены полным синтезом стрихнина (Вудворд с сотр.), включающим свыше 25 стадий.
Вудвард синтезировал стрихнин, исходя из 2-вератрилиндола, который можно получить, например, по методу Э. Фишера (стр. 548) конденсацией фенил-гидразина с ацетовератроном. Действуя на 2-вератрилиндол, уже содержащий кольца А и В скелета стрихнина, формальдегидом и диметиламином, он получил амин, превращенный сначала действием йодистого метила в четвертичный иодид, а затем действием цианистого натрия (в диметилформамиде)—в нитрил. Этот нитрил восстановлением с помощью литийалюминийгидрида или водорода каталитически был превращен в 2-вератрилтриптамин, конденсацией которого с этиловым эфиром глиоксиловой кислоты было получено шиффово основание:
CH2-N(CH3)2
СН,—CN
у\/осн
'V>N)ch
VNdch,
2-вератрилиндол амин
нитрил
сн,-сн, I NH,
А—,
/\Z\/0CH’
NH | !!
'V'^OCHs 2-вератрил триптамин
Н2С-
СН, !
НС—N
^/\осн3
шиффово основание
Действуя на шиффово основание п-толуолсульфохлоридом в присутствии пиридина, удалось замкнуть кельцо Е скелета молекулы стрихнина:
Н2С----СН2
HC=N
А I соос,н,
V\ Д^/0СНз
NH | ||
V'Xqch»
H,C------CH2
E I
СНз-СвЩ-ЮгС! ------------>
N I l!
^AcH3
7
Полученный продукт, содержащий уже не индольный, а индолениновый цикл, после частичного гидрирования с помощью натрийборгидрида и последующего ацетилирования был подвергнут озонированию
Озонирование привело к расщеплению метоксилированного бензольного цикла по связи между двумя углеродными атомами, несущими метоксильные
группы, и к образованию производного муконового эфира, для которого возможны две конфигурации:
При обработке последнего эфира раствором НС1 в метиловом спирте произошло отщепление ацетильной группы от индольного атома азота и метоксильной группы муконового эфира, сопровождавшееся замыканием кольца С скелета стрихнина*.
Н2С-----сн.
Н2С----сн2
СООСНз
сняо—ОС
---N—SO,—С7Н.
СООС2Н4 сн
А
В
Z—NH соон
м
<сн2—соон
сн
сн
Полученный продукт, после ацетилирования при иминном азоте и метилирования обоих карбоксилов, оказался способным к сложноэфирной конденсации с помощью метилата натрия. Таким образом удалось замкнуть шестичленное углеродистое кольцо D скелета стрихнина*.
Н2С-----С Н9
.H3ONa ------->
н2с-----сн2
Гидроксильная группа кольца D этерифицировалась п-толуолсульфохлори-пом в присутствии пиридина, а затем обменивалась на сульфидную группу взаимодействием с натриевой солью беч^илмер каптана. П\тем гидрирования полученного сульфида водородом на скелетном никелевом катализаторе было подокно соединение, не содержавшее серы, но имевшее двойную связь в кольце D. Дальнейшая обработка водородом (в присутствии палладия на угле', привела к селективному гидрированию этой двойной связи. Однако при этом был получен цис-эфир. Его оказалось возможным изомеризовать в рацемический щраис-эфир, из которого в форме соли с хинидином была получена левовращающая фоума, идентичная соответствующему продукту деградации природного стрихнина-
Ы2С-----СН2 ’ Н2С----------С112 НоС-----СН2
Далее задача заключалась в замыкании азотистого шестичленного кольца b скелета стрихнина. Для этого карбоксильную группу превращали последовательно в ацилхлоридную, диазокетонную, иодокстонную и, наконец, в метилкетон-ную Затем энергичным кислотным гидролизом была отщеплена ацетильная группа при азоте кольца Е, причем сначала была получена смесь цис- и /яранс-метилке-!онов. Выделенный из смеси mpawc-изомер при окислении метильной группы двуокисью селена в этиловом спирте дал дггидрострихнинон (замыкание кольца F':
н2с----сн н2с-----сн2 н2с-----сн2
Осторожным действием ацетиленида натрия (но не лития или калия) дегидро-стрихнинон удалось превратить в этинилкарбинол, который затем частичным гидрированием тройной связи с особым катализатором был превращен в соответствующий винилкарбинол. Действуя на этот винилкарбинол литийалюмипийгидридом, удалось восстановить кетонную группу в кольце F до метиленовой и частично гидрировать а-пиридоновое кольцо С. Полученное соединение при нагревании в запаянной трубке при 120° с раствором бромистого водорода в уксусной кислоте превратилось в изострихнин'.
этинилкарбинол
винилкарбинол
азострихнин
При переходе от винилкарбинола к изострихнину, кроме восстановления, происходит изомеризация вин ильной цепочки по той же схеме, что и при переходе от кодеина к пс^влокодеинам (стр. 673).
Превращение изострихнина в стрихнин, т. е. замыкание последнего семичлен* ного кольца G, 5ыло осуществлено еще в 1948 г. Прелогом с сотрудниками путем на» гревания изострихнина с безводным поташом в этиловом спирте:
Синтетический стрихнин оказался совершенно идентичным природному продукту.
Стрихнин и бруцин оптически деятельны. Бруцин обладает левым вращением la ]/j—120°. Он образует хорошо кристаллизующиеся соли и сравнительно дешев, поэтому его часто применяют в лабораториях для расщепления рацемических смесей кислот на оптические изомеры. Бруцин применяется также как чувствительный реактив на азотную кислоту, с которой дает кроваво-красное окрашивание.
Стрихнин—одно из самых ядовитых веществ (для человека смертельная доза его около 0,03 г) Он вызывает общее возбуждение центральной нервной системы, а при больших дозах—конвульсивные сокращения мускулатуры и смерть от удушья. В медицине стрихнин применяется как стимулирующее средство при общей слабости и слабости сердечной деятельности. Антагонистами стрихнина и бруцина и противоядием могут служить алкалоиды кураре (стр. 665).
Эргоалкалоиды. Главным действующим началом спорыньи (покоющейся формы грибка Clavtceps purpurea, паразитирующего на колосьях ржи и других злаков) является группа эргоалкалои* дов—производных лизергиновой или изолизергиновой кислот:
н СООН
ноос н
лизерг"новая кислота
изолизергиновая кислота
В основе строения этих кислот, отличающихся друг от друга лишь расположением заместителей у атома углерода, несущего карбоксильную группу, лежит система из четырех циклов (А, В, С и D), которые можно рассматривать как ядра индола (циклы А и В), гидрированных хинолина (циклы С и D) и нафталина (циклы А и С):
В настоящее время идентифицировано 12 алкалоидов этой группы, которые подразделяются на 6 пар; каждому левовращающему изомеру, физиологически активному, отвечает малоактивный правовращающий изомер.
Продуктом щелочного гидролиза активных изомеров является лизергиновая кислота (темп, плавл. 238е; la ]D= +40*), малоактивные изомеры даю1 изолизергиновую кислоту (темп, плавл. 218°; 1а+281°). Вторые продукты гидролиза эргоалкялоидов—аминокислоты, кетокислоты, ди кетопиперазины или аминоал коголи— связаны с группировкой лизергиновой кислоты при помощи амидного азота.
Изучение продуктов гидролиза эргоалкалоидов позволило предложить для этих алкалоидов достаточно обоснованные формулы. Например, эрготамин (темп, плавл. 213—214"; * ос j == 192°), дающий при расщеплении (см. пунктирные линии), кроме лизергиновой кислоты, пировиноградную кислоту, /-фенилаланин и с/шролин, повидимому, имеет следующее строение!
Эрготоксин, или эргокорнин (темп, плавл. 190—200® с разл.; (а]£>=226°), расщепляется на лизергиновую кислоту, диметилпиро-
виноградную, /-валин и d-пролин, и, следовательно, его пептидная группировка имеет строение:
Препараты спорыньи и эргоалкалоиды вызывают сокращение гладкой мускулатуры (особенно сокращение матки) и применяются в гинекологической практике. Гидрированные в кольце D дигидро-эрготамин и дигидроэрготоксин применяются в терапии сердечных болезней.
Алкалоиды, производные имидазола
,В природе найдены лишь немногие представители этой группы алкалоидов, в частности алкалоиды африканского растения Pilocarpus Jaborandi и некоторых других растений этого рода. Главный алкалоид Pilocarpus Jaborandi, называемый пилокарпином, возбуждает периферические окончания нервов, регулирующих деятельность слюнных желез и потоотделение; он понижает внутриглазное давление и сужает зрачок. Применяется пилокарпин главным образом в офтальмологической практике при глаукоме и других заболеваниях глаз.
Пилокарпин CnH1G09N9 (иглы с темп, плавл 34°) легко растворим в воде, спирте и эфире, обладает правым вращением. При нагревании со щелочами или при нагревании его солей пилокарпин превращается в стереоизомерный изопилокарпин (густое масло, темп, кип, 261® при 10 мм}.
Пилокарпин и изопилокарпин имеют следующее строение:
Соответственно лактонному строению они растворяются в щелочах, давая соли оксикислот пилокарповой и изопилокарповой.
Изопилокарпин при окислении отщепляет монометилмочевину и дает гомопилоповую кислоту; при более энергичном окислении по
лучается пилоповая кислота:
-СН,—СООН
СООН
пилоповая кислота
гомоп -лог овая кислота
Рацемическая пилоповая кислота плавится при 90—91е, право-и левовращающие—при 122°.
При окислении пилокарпина обычно получаются те же кислоты вследствие изомеризации его в изопилокарпин, но при окислении иодметилатов пилокарпина и изопилокарпина озоном были получены метиламиды двух различных кислот—го-мопилоповой и изогомопилоповой.
Строение пилоповой кислоты из изопилокарпина, а следовательно и лактонной части алкалоида, было доказано ее синтезом (А. Е. Чичибабин и Н. А. Преображенский, 1929 г.). Конденсацией этилянтарного эфира с муравьиным эфиром и восстановлением полученной смеси стереоизомерных формилэтилянтарных эфиров была получена смесь эфиров итамалевых кислот, которые при нагревании дали смесь эфиров двух стереоизомерных оптически недеятельных пилоповых кислот (в соответствии с наличием в молекуле двух асимметрических атомов углерода). Одна из них оказалась неустойчивой, и подобно пилокарпину превращалась при нагревании в более устойчивый изомер:
С2Нб—СН—СН2—СООС2Нб
С2НбООС
эфир этилянтарной кислоты
С2Нб—СН—СН—СООС2Н5 +Н2
С2Н6ООС сно —
эфир формилэтилянтарной кислоты
С2Нб-СН-СН-СООС2Нб нагревание с2н5—нс----СН—СООС2П5
С2НбООС СН2ОН ос сн,
V
эфир итамалевой кислоты эфир пилоповой кислоты
Обе стереоизомерные оптически недеятельные пилоповые кислоты кристаллизацией солей стрихнина и бруцина были разделены на оптические антиподы. Правовращающий антипод устойчивой кислоты оказался тождественным с пилоповой кислотой из изопилокарпина (а-изопилоповая кислота). ^/-Антипод менее устойчивой пилоповой кислоты, вероятно, по конфигурации соответствует пилокарпину (d-пилоповая кислота). Из пилоповой кислоты действием диазометана на ее хлорангидрид, а затем спирта в присутствии окиси серебра была получена гомопилоповая кислота.
Синтез пилокарпина из гомопилоповой и изогомопилоповой кислот был осуществлен Н. А. Преображенским (1936 г.).
Алкалоиды, содержащие пуриновую группировку
Сюда относятся ксантин, гуанин, теобромин, теофиллин и кофеин, рассмотренные на стр. 637—640.
Стероидные алкалоиды (производные циклопентенофенантрена)
К этой группе принадлежат алкалоиды, в основе структуры которых, так же как у стероидов (стр. 165 сл.), лежит скелет циклопентенофенантрена. Для этих алкалоидов, содержащих, как и стероиды, у третьего атома кольца А гидроксильную группу, характерно осаждение их в форме дигитонидов (стр. 188).
К стероидным алкалоидам принадлежит большая группа алкалоидов, выделенных из растений семейства пасленовых (Solanaceae). Эти алкалоиды являются вместе с тем сложными глюкозидами. Так, например, соланин C45H73OloN, будучи устойчивым по отношению
к щелочам, при нагревании с разбавленными минеральными кислотами расщепляется на ахотистый аглюкон—соланидин—и три молекулы углеводов (d-глюкова, d-галактоха и d-рамноза). Соланин содержится в картофеле (Solarium tuberosum). Он образует мелкие бесцветные иглы, плавящиеся с разложением при 285е и предварительно размягчающиеся при 235е.
Соланидин (иглы с темп, плавл. 219е) при дегидрировании с помощью селена дает З'-метил-l ,2-циклопентенофенантрен (углеводород Дильса), в то время как азотистый цикл отщепляется в форме 5-метил-2-этилпиридина:
З'-метил- i. 2-ц ик ло-пентеиофенантреи
На основании этого и многих других фактов для соланидина была предложена приведенная здесь формула. В молекуле соланина к гидроксильной группе при 3-ем углеродном атоме соланидина присоединена трисахаридная цепь. В аналогичных отношениях находится алкалоид соласонин (из Solarium sodomeum) C45H75OJ6N со своим аглюконом соласонидином C27H43O2N.
К стероидным алкалоидам относятся также алкалоиды чемерицы (Veratrum album). Долгое время считалось, что в чемерице, содержится индивидуальный алкалоид вератрин, представляющий собой в действительности аморфную смесь нескольких алкалоидов. В настоящее время изучено семь кристаллических алкалоидов чемерицы. Для одного из них—иервина и для легко получающегося из него изоиервина предложены следующие формулы:
СН3 nh СН3 мн
иервин изоиервин
В пылевидном состоянии алкалоиды чемерицы вызывают интенсивное чихание. Вератрин находит применение в ветеринарной практике.
Антибиотики
В последние годы очень большое внимание микробиологов, врачей и химиков привлекают природные соединения, называемые антибиотиками. Антибиотики (антибиотические вещества)—это органические соединения, образуемые живыми организмами (бактериями, грибами, высшими растениями и т. д.) и способные убивать микроорганизмы (бактерии, грибы, вирусы и т. д.) или подавлять их развитие. Подобно витаминами гормонам, антибиотики не составляют единой группы химических веществ; их представители относятся к самым различным классам органических соединений.
Систематическое изучение антибиотиков началось только в 40-х годах этого столетия, но исследования в этой области развивались настолько быстро, что к настоящему времени описано уже около 1000 антибиотических веществ, из которых примерно для 200 полностью (или в основных чертах) выяснено строение, а около 100 уже удалось синтезировать. Всего через несколько лет после начала изучения антибиотиков была создана антибиотическая промышленность, в которой очень своеобразно сочетаются биологические и химические процессы.
Такое быстрое развитие химии и технологии антибиотиков объясняется их чрезвычайно высокой практической ценностью. Введение антибиотиков в медицинскую практику произвело подлинный переворот в лечении многих очень опасных болезней. В настоящее время антибиотики—это один из важнейших видов лекарственных веществ, выпускаемых в очень больших количествах. В последние годы их начали применять также в сельском хозяйстве (для борьбы с заболеваниями растений, для стимулирования роста животных), в пищевой промышленности (для консервирования мяса, битой птицы и т. д.) и в других отраслях народного хозяйства.
Важнейшие антибиотики образуются бактериями, плесневыми грибами и актиномицетами, причем именно актиномицеты образуют наибольшее число практически важных или интересных по своему строению антибиотиков.
Пенициллины. Изучение пенициллинов было начато в 1928 г., когда английский микробиолог Флеминг, при случайном заражении культуры стафилококка плесенью Penicillium notatum, обратил внимание на ее способность угнетать развитие бактерий. Это действие Флеминг приписал образуемому плесенью веществу, которое было названо пенициллином.
Отдельные наблюдения о взаимоотношениях плесеней и бактерий делались и ранее. Еще в 1871 г. В. А. Манасеин отметил, что среда, на которой культивируется один из видов РетсИНшп, свободна от бактерий, а в 1872 г. Л. Г. Полотеб-нов наблюдал ускоряющее действие этой плесени на процесс заживления рай.
Начиная с 1939—1940 гг. в Англии, США и СССР была предпринята большая систематическая работа по изучению плесеней, образующих пенициллин, подбору условий его образования и выделения, изучению его химической природы. Эта работа оказалась чрезвычайно сложной и потребовала совместных усилий большого числа исследователей разных специальностей. Можно указать, например, что в изучении химии пенициллина, проводившемся в 1941 —1945 гг. совместно 39 научными учреждениями, принимало участие более 1000 высококвалифицированных исследователей во главе с Робинсоном, Чейном, Дю-Виньо, Вудвордом, Фолькерсом и др.
В 1942—1943 гг. было выяснено, что активным началом препаратов пенициллина является не одно, а несколько сходных веществ, которые получили условные названия: пенициллин 1, или F, пенициллин II, или G, и т. д. Из этих так называемых «природных» пенициллинов наиболее активным и стабильным оказался пенициллин G (пенициллин II), обычно называемый просто «пенициллином». В 1943 г. натриевая соль пенициллина G была получена в кристаллическом состоянии, что весьма облегчило его дальнейшее изучение.
Для производства пенициллинов большое значение имело открытие «предшественников», т. е. веществ, прибавление которых в питательную среду сильно стимулирует образование пенициллинов плесенью. Предшественниками пенициллина G оказались фенилуксусная кислота и ее производные. Применяя другие предшественники, удалось получить значительное число новых, так называемых «биосинтетических» пенициллинов.
В настоящее время известно около 100 различных пенициллинов, имеющих весьма сходное строение. В основе их лежит своеобразная бициклическая система, образованная двумя конденсированными кольцами: тиазолидиновым (А) и [В-лактамным (В). Пенициллины различаются только характером радикала R, в зависимости от которого строится их номенклатура:
S
R—СО—NH—СН—НС7 ЧС(СН3)2
в [ А ;
СО---N----СН—СООН
Радикал R
Условное
обозначение
2-Пентенилпенициллин
Бензилпенициллин п-Оксибензилпенициллин н-Гсптилпенициллин
Аллилмеркаптометилпенициллин Феноксиметилпенициллин Цефалоспорин N (синнематин В)
-сн2сн=СНСН2СН3
—сн2с6н5
-СН2С6Н4ОН
-СН2(СН2)5СН3
-Cbb-S-CH2CH =СЫо
-СНЮСбНб
-СН2СН2СН2СНСООН
nh2
F или 1
G- или 11 X или III К или IV
О
V
Пенициллины представляют собой сильные органические кислоты (рКа~2,7), в свободном состоянии очень неустойчивые (за исключением феноксиметилпенициллина), вследствие чего они получаются в виде солей с металлами (натрий, калий) или с органическими основаниями (новокаин, ЮХ'-дибензилэтилендиамин).
Пенициллины неустойчивы как в щелочной, так и в сильнокислой среде. Они быстро теряют активность также в присутствии энзима пенициллиназы, ионов тяжелых металлов (медь, свинец) и др. Во всех Э1их случаях в первхю очередь размыкается р-лак-тамное кольцо и образуются пенициллоиновые кислоты (или их производные):
S
R—СО—ХН—СН—НС7 С(О I л.
I ! (
НООС HN—сн—соон
Пенициллоиновые кислоты легко декарбоксилируются в пенил-лоиновые кислоты
8
R—CO—NH—СН.—НС С(СН3)2
i I н\---сн—соон
которые образуются также при нагревании растворов пенициллинов.
При кислотном гидролизе пенициллинов с последующим действием сулемой из всех пенициллинов получается одна и та же а-ами-нокислота, названная пеницилламином и представляющая собой оптически активный г/-р,р-диметилцистеин*:
HS—С(СН3).,
I.
h2n-ch—соон
Из маточного раствора, после выделения пеницилламина, можно с помощью 2,4-динитрофенилгидразина выделить пениллоальдегиды R—СО—NH—СН2—СНО. Радикал R в пениллиальдегиде тот же, что и в молекуле пенициллина, подвергшегося расщеплению. Поэтому, например^ случае бензилпенициллина получается фенилацетиламино-ацетальдегид С6Н5—СН2—СО—NH—СН2—СНО.
При взаимодействии пениллоальдегида с пеницилламином образуется пениллоиновая кислота, чем доказывается строение кольца А молекулы пенициллинов.
Для решения вопроса о характере кольца В в молекуле пенициллинов очень большое значение имело изучение реакции гидрогено-
* Интересно отметить, что пеницилламин обладает ^-конфигурацией, сравнительно редко встречающейся у природных аминокислот, имеющих обычно /-конфигурацию.
лиза бензилпенициллина с помощью скелетного никелевого катализатора. При этом наряду с фенилацетил-1-аланил-й-валином
С6Н6СН2—СО—NH—СН—СН3 СН(СН3)2
СО—HN—СН—СООН
был получен дестиобёнзилпенициллин
С6НбСН2—СО—NH—СН—СН2 СН(СН3)2
lB I I
СО—N—СН—СООН
в котором было доказано наличие ^-лактамного кольца. Это позволило обосновать приведенную выше формулу пенициллинов.
Установление строения пенициллинов чрезвычайно затруднялось их способностью даже в мягких условиях претерпевать разнообразные изомерные превращения. Поэтому для доказательства строения пенициллинов чрезвычайно большое значение имели различные физико-химические методы, в особенности рентгеноструктурный анализ солей бензилпенициллина, давший строгое доказательство (Кроуфут) указанной выше структурной формулы.
В настоящее время известно пять типов изомеров пенициллинов:
СООН
I
СН S
ьГ нс7 ЧС(СН3)2
II I I
R—С----N----СН-СООН
пенилловая кислота
S
R—CO-NH-CH—НС7 ХС(СН3)2
со—N-----сн—СООН
пенициллин
СГ S
R—C=NH—СН—НС^ ХС(СН3)2
i------СО HN—сн—соон
псевдспеницнллин
соон
С SH
bf НС ХС(СН8)?
II I I
R—С---N —СН—СООН
изопенилловая кислота
S
R—CO-N--------НС^ ''с(СН3)2-
I I I
Н2С—СО—N------СН—СООН
пениллоновая кислота
I
HS
R—C=N—С=НС ^CfCH,,), 1111 О-----СО HN сн—СООН
пеницилленовая кислота
Синтез бензилпенициллина был осуществлен в 1946 г. Дю-Виньо с сотрудниками, однако, ввиду крайне ничтожного выхода и неясности механизма реакции, он имел мало значения.
Практически бензилпенициллин получают путем биосинтеза. Процесс проводят в особых аппаратах (ферментерах), снабженных приспособлениями для продувания стерильного воздуха. Специально селекционированные плесени (обычно Penicillium chrysogenum) выращивают на жидкой среде, содержащей сахара, белки, жиры и производные фенилуксусной кислоты (предшественники). Затем саму плесень (мицелий) отфильтровывают, а из подкисленного фильтрата быстро извлекают пенициллины органическим растворителем (обычно бутилацетатом). Дальнейшую очистку пенициллина проводят путем экстракции водным раствором (рН~7), реэкстракции органическим растворителем и осаждением в виде солей.
Начиная с 1953 г., большое внимание привлекает феноксиметил-пенициллин, легко выделяемый в виде пластинок (темп, плавл. 118—120°). Благодаря устойчивости к кислотам он не разлагается в желудке и оказывает лечебное действие при приеме внутрь, тогда как менее устойчивый бензилпенициллин обычно приходится вводить внутримышечно. При биосинтезе феноксиметилпенициллина в качестве предшественника применяется феноксиуксусная кислота С6Н5ОСН2СООН. В 1957 г. осуществлен полный синтез феноксиметилпенициллина с удовлетворительным выходом (Шихан и сотр.).
В 1954—1955 гг. было выяснено, что пенициллином является также антибиотик цефалоспорин N (синнематин В), нерастворимый в органических растворителях и, в отличие от всех других пенициллинов, обладающий значительной активностью в отношении таких бактерий, как, например, возбудители брюшного тифа. Цефалоспорин N имеет в качестве боковой цепи остаток d-a-аминоадипино-вой кислоты.
Стрептомицин. Несмотря на очень успешное примение пенициллинов, вне сферы их действия оказывалась большая часть патогенных для человека микроорганизмов. Первым ценным дополнением к пенициллину оказался открытый в 1944 г. Ваксманом с сотр. стрептомицин, образуемый актиномицетом Streptomyces griseus*. Он обладает значительно более широким антимикробным действием, чем пенициллины, сильно подавляя многие микроорганизмы, и в Частности туберкулезную палочку. Однако он более токсичен, чем пенициллины, и нередко оказывает нежелательное побочное действие на организм.
Стрептомицин C21H39O12N7 представляет собой трехкислотное основание; его выделяют в виде солей (сульфата, хлоргидрата), которые обычно не удается получить в кристаллическом виде. Хорошо кристаллизуется двойная соль трихлоргидрата стрептомицина с хлористым кальцием («хлоркальциевый комплекс»).
Стрептомицин значительно устойчивее пенициллинов—не разрушается при кратковременном воздействии кислот и щелочей. При кислотном гидролизе он распадается с образованием двух веществ: сильного двукислотного основания C8H18O4N6, названного стрептидином, и своеобразного дисахарида—стрептобиозамина. Стрептидин—производное циклогексана, содержащее две гуанидиновые группировки и четыре гидроксила. Стрептобиозамин яв
* По другой классификации Actinomyces globisporus.
ляется М-метил-а-/-глюкозаминидо-^-/-стрептозой, причем обе мо-нозы, из которых составлена его молекула, имеют редкую для природных сахаров /-конфигурацию. Остаток стрептобиозамина присоединен через глюкозидный атом остатка стрептозы к четвертому атому стрептидина. 1аким образом, строение стрептомицина может быть выражено следующей формулой:
/-стрептоза
Стрептомицин, как и пенициллины, получается биосинтетическим путем. После отделения мицелия стрептомицин адсорбируют из фильтрата активированным углем или катионообменной смолой, десорбируют раствором кислоты и далее очищают путем вторичной адсорбции, превращения в двойную соль с СаС12 или другими приемами.
Хлоромицетин (левомицетин). В 1947 г. из культуральной жидкости актиномицета Streptomyces venezuelae был выделен новый антибиотик хлоромицетин, отличающийся широким спектром ан*-тимикробного действия и, в частности, активный в отношении возбудителей дизентерии и брюшного тифа, риккетсий (в том числе возбудителей сыпного тифа) и некоторых крупных вирусов. Он оказался d-(—)-трео-1 -(/?-нитрофенил)-2-дихлорацети ламино-1,3-пропандно-лом, т. е. сравнительно простым, хотя и довольно своеобразным соединением, по строению и пространственной конфигурации очень сходным с алкалоидом псевдоэфедрином (стр. 647):
3
н СН2ОН
o2n-^~4-c—с-н
\=/ I* I*
ОН NH—СО—СНС12
хлоромицетин (левомицетин)
В молекуле хлоромицетина имеется два центра асимметрии, а потому для него возможно четыре пространственных изомера.
Интересно, что только один из них—природный d-трео-изомер— обладает антимикробной активностью.
В молекулах с двумя центрами асимметрии, в которых не все заместители при одном из асимметрических атомов углерода повторяются при другом асимметрическом атоме, необходимо различать два ряда стереоизомеров: трео- иэритро-ряды. В /?грсо-ряду заместители при обоих центрах асимметрии расположены в одинаковом порядке; для зр//тро-ряда характерна разная («зеркальная») конфигурация оптических центров (аналогично мезовинной кислоте, см. том I, стр. 502). Чтобы убедиться в этом различии, отделим верхние половины проекционных формул
b b
I, |..=:
а—С—х а—С—х
I, к
у—С—а а—С—у
I I
b b
трео- эритро-
и повернем их в плоскости чертежа:
эршпро-
Теперь легко видеть одинаковую конфигурацию асимметрических центров в mpeo-ряду и противоположную—в зрш/гро-ряду—аналогично конфигурациям треозы и эритрозы (см. том I, стр. 538).
Если проекционная формула написана как-либо иначе, например:
а
I х—С—b
I а—С—у
b
надо в каждой половине проекционной формулы передвигать заместители по часовой стрелке вокруг центра асимметрии до тех пор, пока на вертикальной оси не окажутся одинаковые заместители. Если при этом «гетероатомы» х и у окажутся в проекционной формуле в транс-положении—вещество относится к трео-ряду. Цпс-расиоложение «гетероатомов» является признаком эрнтро-ряда.
При большом числе «гетероатомов» отнесение к трео- или эритро-рядам становится более сложным.
В молекулах хлоромицетина (левомицетина) и псевдоэфедрина оба центра асимметрии имеют одинаковую конфигурацию, вследствие чего эти соединения следует относить к mpeo-ряду, тогда как эфедрин, имеющий разную конфигурацию центров асимметрии, относится к эритро-ряду:
11 СН3
он сн3
ОН NH—СН3 псевдоэфедрин /тоео-ряД.)
эфедрин (эритро-ряд)
Несмотря на простоту строения, хлоромицетин (левомицетин) имеет две редкие для природных веществ особенности: наличие нитрогруппы и дихлорацетильного остатка.
Хлоромицетин представляет собой бесцветное кристаллическое вещество (темп, плавл. 150,5—151,5°). Он хорошо растворим в пропиленгликоле, в метиловом и этиловом спиртах, хуже—в эфире, практически не растворяется в бензоле. Растворимость в воде при 25° составляет около 0,25%.
Синтез хлоромицетина был осуществлен в 1949 г. американскими химиками и группой советских исследователей под руководством М. М. Шемякина. Последними проведены также большие работы по получению аналогов хлоромицетина и изучению связи между его строением и антибиотическим действием.
Синтез хлоромицетина (левомицетина) может быть осуществлен различными методами. Один из наиболее удобных путей заключается в следующем.
n-Нитроацетофенон действием брома превращают в ц-нитро-со-бромацетофенон, из которого взаимодействием с уротропином (гексаметилентетрамином) с последующим омылением соляной кислотой получают хлоргидрат п-нитро-а>-аминоаце-тофенона. Его ацетилируют уксусным ангидридом, а полученное ацетильное производное оксиметилируют формальдегидом в присутствии бикарбоната или ацетата натрия. В получившемся п-нитро-а-ацетиламино-3-оксипропиофеноне восстанавливают кетогруппу (не затрагивая нитрогруппы) по Меервейну—Понн-дорфу изопропилатом алюминия в среде изопропилового спирта. При этом образуется преимущественно с?,/-трео-изомер 1-(п-нитрофенил)-2-ацетиламино-1,3-пропанд иол а:
Вг2
(CH2)6n4
НС1
(СН3СО)2О г.—х.
-------» O2N-/ %—СО—СН2
сн2о
NH2.HC1
NH—СОСН3
СН2ОН
изопропилат алюминия
—С-Н
Н СН2ОН
NH—СОСН3
ОН NH—СОСН,
После омыления ацетильной группы рацемат амина разделяют на антиподы с помощью d-винной или d-камфорсульфоновой кислоты и выделяют необходимый d-изомер. При его дихлорацетилировании посредством метилового эфира дихлоруксусной кислоты получается хлоромицетин:
н СН2ОН
7=4 I | СНС12СООСН3
O2N—< С—С—Н---------------->
I | он nh2 £?-мзомер
н сн.он
/=\ I I
O.N—< >—С—С—Н
V-Z | |
ОН NH-СОСНСЦ хлоромицетин (левомицетин)
Хлоромицетин (левомицетин)—единственный антибиотик, про» изводимый в промышленных масштабах путем синтеза, а не биосинтеза. Кроме синтетического хлоромицетина, в СССР выпускается также его в два раза менее активный рацемат под названием синтомицин.
Сравнительная простота строения хлоромицетина и разработанность методов синтеза позволили получить большое число его аналогов (более 500), ни один из которых, однако, не превосходит по своим свойствам природный антибиотик.
Тетрациклины. Важную группу антибиотических веществ составляют тетрациклины, имеющие очень сходное строение и близкие свойства. К этой группе относятся хлортетрациклин (называемый также ауреомицином и биомицином), окситетрациклин (терра-мицин), тетрациклин (выпускаемый под названиями ахромицин, тетрацин и др.) и пока малоизученный бромтетрациклин.
Первым из антибиотиков этой группы был описан хлортетрациклин, или ауреомицин (1948 г.), образуемый актиномицетом Streptomyces aureofaciens. Строение его было установлено в 1952 г. Тетрациклин был описан в 1953 г. как продукт каталитического восстановления хлортетрациклина. Позднее были разработаны условия его биосинтеза с помощью того же актиномицета, который дает хлортетрациклин. В 1955 г. был получен бромтетрациклин, образуемый тем же актиномицетом на среде, совершенно не содержащей ионов хлора, но содержащей ионы брома. По строению он, по-видимому, аналогичен хлортетрациклину. Окситетрациклин, или террамицин, образуемый актиномицетом Streptomyces rimosus, отличается от тетрациклина наличием еще одной гидроксильной группы:
тетрациклин (темп, плавл. 170—175°)
С1СН3 он
но о но
N(CH3)2
он
Н С ИП ПН M/rHj
окситетрац ик лин, или террамицин (темп, плавл. 184,5—185,5°)
хлортетрациклин, биомицин, или ауреомицин (темп, плавл. 168—169°>
бромтетрациклин (темп, плавл. 170—172°)
о
По спектру антибиотического действия хлортетрациклин, окситетрациклин и тетрациклин очень сходны с хлоромицетином. Наиболее перспективен, по-видимому, тетрациклин, оказывающий наименьшее побочное действие. Хлортетрациклин, помимо медицины, применяется в последнее время в животноводстве (для стимулирования роста животных) и в пищевой промышленности (при хранении рыбы, мяса и других скоропортящихся продуктов).
Грамицидин С. Большую группу антибиотиков, образуемых бактериями и некоторыми актиномицетами, составляют полипептиды. В настоящее время описано более 60 таких антибиотиков. Типичным представителем этой группы является грамицидин С, открытый в 1942 г. Г. Ф. Гаузс и М. Г. Бражниковой.
Грамицидин С образуется бактерией Bacillus brevis var. G.—В; ввиду токсичности при подкожном и внутривенном введении, он применяется только для местного лечения ран, ожогов, ангин. Грамицидин С (бесцветные пластинки, темп, плавл. 256—258°; дихлоргидрат плавится при 268—270°) представляет собой циклический декапептид, в состав которого входят по два остатка /-пролина, Z-валина, /-орнитина, /-лейцина и J-фенилаланина. Его строение может быть выражено структурной формулой:
валин СН(СН3)2 орнитин ch2nh, 1 (СН3)2 ТТ \Т А I-JrVT пейцин СН(СН3)2 СН, 1 14 NT Г14 Г О : фенилаланин j С6Н5 1 1 : СН2 i 1 LT мгнггт пролин U-co \Т
HNCnCU N ОС— COCHNH COCHNH L : Г1. N С!1L. О COCHNH _ COCHNH
пролин СН2 1 СбН5 фенилаланин сн2 i i j СН(СН3)2 : лейцин :. (СН2)2 1 ch2nh2 орнитин СН(СН3)2 валин
Полный синтез грамицидина С осуществлен в 1956 г. Швице-ром и Зибером.
Полимиксины. Антибиотики-полипептиды образуются бактериями обычно не как индивидуальные соединения, а как большие группы очень сходных по строению и свойствам веществ, различающихся между собой лишь отдельными деталями строения. Поэтому выделение индивидуальных соединений из смесей представляет весьма сложную задачу, для решения которой приходится использовать различные новые приемы исследования (хроматографические методы, противоточное распределение).
Одну из таких групп антибиотиков-полипептидов составляют полимиксина, образуемые различными расами Bacillus polymyxa. Они обладают сильной избирательной активностью в отношении некоторых бактерий (например, синегнойной палочки), очень устойчивых к действию других антибиотиков. Полимиксины имеют 1лавным образом местное применение, так как они оказывают довольно сильное побочное действие на почки.
Уже установлено, хотя стро1 о не доказано, строение полимиксина Bt, в состав которого входит шесть остатков а,у-диаминомасляной кислоты (из них пять имеют а один d-конфигурацию), два остатка /-треонина, по одному остатку ^-фенилаланина и /-лейцина и один остаток жирной -6-метилоктановой кислоты:
дна ми но масляная кислота а,-('-диаминомасля- пая кислота : Iрсонин
ch2nh2 CH,NH.? ? СПр
1 сн2 СП, ; 1 снон
1 нхснсо —! • 1 : - HNCHCO \ 1 HNCHCO
а/'-диаминомасля-ная кислота
ch.,nh2
I сн2
I
—нхснсо
OCCHNH--------j----соснхн
I = I
СН., : Cil.
I : I
CH(CH3)2 ; CJI5
лейцин : фенила манн
СОСНУ H_________5----COCHNH а.рдиамино-
1 сн2 1 ch2nh2 I сн2 ch2nh масляная кислота
у-дпаминомлеля-
ная кислота
СН3СН2СН(СН,)4СО
СН3
С-мстилокта новая кислота
NHCHCO____=__NHCHCO
I i I
сн2 : снон
I \ I
CH2NH2 i сн3
а,-[-диаминомасля- : треонин
ная кислота •
Другие антибиотики этой группы содержат остатки других аминокислот (например, серина) или остаток другой жирной кислоты.
Для большинства антибиотиков-полипептпдов характерно наличие остатков «неприродных» d-аминокислот и циклическое строение.
Эритромицин. Большую группу антибиотиков составляют чакролиды—макроциклические лактоны, содержащие остатки своеобразных диметпламиносахаров. Типичным представителем этой группы антибиотиков является эритромицин, образуемый актиномицетом Streptomyces erythreus. По антибиотическому действию он близок к пенициллинам; применяется главным образом для борьбы с бактериями, приобретшими устойчивость к пенициллинам.
Эритромицин C37H67O13N (бесцветные кристаллы, темп, плавл. 135—140°) хорошо растворим в хлороформе, метиловом и этиловом спирте, умеренно—в холодной и очень плохо растворяется в горячей воде.
Строение эритромицина полностью установлено. Молекула его состоит из трен частей—диметиламиногексозы (дезозамина/ гептозы разветвленного строения (клади-нозы) и лактона с длинной цепью (эритронолида):
N(CH3)2
I
СН
H2GZ Хснон I I
н3с—нс сн—о
дезозамин
'сноп
О НС сн—сн3
XoZ кладиноза
СН3
•н он
СН3 СН3 сн3
•сн—с—сн—сн.,—<i—сн—сн—сн—сн—с==о
II I о он
СН3
СН3
---------о
эригронолпд
Выяснено строение еще нескольких антибиотиков—макролидов, имеющих сходное с эритромицином строение и похожим на него по многим физическим и биологическим свойствам. К ним относятся три изомерных антибиотика состава C?6H43O7N: пикромицин, образуемый Streptomyces telleus, метимицин и неометимицин (актино-минет точно не идентифицирован), а также антибиотик магнамицин ^карбомицин) состава C42H67O16N, образуемый актиномицетом Streptomyces halstedu.
Саркомицин. В последние годы среди антибиотиков обнаружены вещества, проявляющие в опытах на животных заметное противоопухолевое действие. Хотя два из них (саркомицин и актиномицин С) уже испытываются клинически для лечения опухолевых заболеваний человека, еще нельзя сделать окончательного заключения об их практической ценности. Наибольшее внимание привлекает открытый в 1953 г. антибиотик саркомицин, образуемый актиномицетом Streptomyces erythrochromogenes. Он представляет собой оптически активную 2-метилен-З-кетоциклопентанкарбоновую кислоту
Н3С=С—сн—соон
I I
о=с си,
СН2
Актиномицины. Значительно более сложное строение имеют актиномицины—большая группа родственных антибиотиков (насчитывающая более $0 представителей), образуемых различными актиномипетами. Наиболее изученный из этих антибиотиков—актиномицин С, образуемый актиномицетом Streptomyces chryso-malius, оказывает лечебное действие при некоторых болезнях крови и опухолевых заболеваниях лимфатической системы. Он представляет собой сложную смесь нескольких близких антибиотиков. Строение одного из них—актиномицина С3, соответствующего суммарной формуле C64H80O16N12, полностью выяснено (Брокман и сотр.).
Актиномицин С3 (ярко-красные бипирамиды, темп, плавл. 232—235°) состоит из гетероциклического хромофора, соединенного с двумя одинаковыми полипе-птидными цепями, содержащими по одному остатку/-треонина, саркозина, N-метил-Z-валина, Z-пролина и d-аллоизолейцина, причем карбоксильная группа N-метил-валина и оксшрзппа треонина соединены в лактонные кольца:
(СН3),нс СН(СН3)2
I I
О=С-----------НС N-метилвалин СН-----------------С=О
I I
СН3—N N—СН3
ос io
I I
Н,С саркозин СН2 I I
СН3—N N— СН3
.....I I.....
ос со
Н?С—НСЧ пролин ,СН—СН,
,1 >N N< |
Н2С—H2CZ | | XCH2—CH,
ос co
I I
C2H6—НС—НС изолейцин CH—CH—C,H6 II II
H3C . HN NH CH.,
о oc io i
Illi CH3—HC---------------НС треонин CH------------------CH—CH3
I I
HN NH
oc! ’ co I I
• C N C
НС С С C—NH,
I II I I
He c c c
in, in,
Другие актиномицины имеют сходное строение, отличаясь, по-видимому, лишь строением полипепгидных цепей.
Из рассмотренного материала видно, что среди антибиотиков имеются вещества очень своеобразного строения, содержащие редкие для природных соединений группировки (3-лактамное кольцо в пенициллинах, нитрогруппа в хлороми-цетине, ^-аминокислоты в пенициллинах, антибиотиках-полипептидах и актиномицинах и т. д.).
/Можно упомянуть еще несколько не имеющих практического применения антибиотиков, отличающихся необычными для природных соединений особенностями строения. Так, два актиномицета образуют антибиотики (обладающие противоопухолевым действием), являющиеся жирными диазосоединениями:
n2ch—соосн2— сн—соон n2ch—сосн2—сн2—сн—соон
I I
nh2 nh2
a?acq ин 6-диазо-5’Оксо-/-норлейцин
(О диазояцетлл-/-серии)
Актиномицетом Streplomine^ hepaticus образуется антибиотик элайомицин, обладающий in vitro сильным противотуберкулезным действием. Он является алифатическим азоксисоединением:
О СН2ОСН3
t ।
«-С6Н13СН—СН—N—СН !
СНОП I сн3
Чрезвычайно неустойчивым соединением (разлагающимся со взрывом при 75°) является антибиотик микомицин, образуемый актипомицетом Nocardia acidophilus. Он содержит тройные связи и алленовую группировку, придающую ему оптическую активность (см том 1, стр, 382):
НС=С—С=С—СН=С=СН—СН—СН—СН —СН—СН2—СООН
По нескольку тройных связей содержит еще ряд антибиотиков, образуемых высшими грибами, например агроцибин, диатретин-2 и др.
НОСН2—С^С—С=С—C=C-CONH2 НООС—СН=СН—СеееС—с—С—CeeN агроцибин Лиат ретин-2
(дгатрстиннитрил)
Противогрибковым действием обладают антибиотики трополоны (стр. 501), образуемые высшими растениями и плесенями.
Можно привести еще много примеров нахождения среди антибиотиков соединений, имеющих группировки, которые обычно считаются «неприродными». Это показывает многообразие путей биосинтеза, осуществл$ емого микроорганизмами.
Витамины
Долгое время среди физиологов господствовало мнение, что организм человека и животных для поддержания работоспособности и здоровья нуждается в качестве основных питательных веществ в белках, жирах и углеводах, а также в небольших количествах минеральных солей (преимущественно NaCl). Впоследствии было установлено, что организм животного нуждается также в некоторых дополнительных веществах, которые он сам не может синтезировать, причем отсутствие этих веществ в пище ведет к ряду заболеваний (цынга, рахит, ксерофтальмия, полиневриты, пеллагра и др.). Такие необходимые для нормального существования организмов вещества,—поступающие вместе с пищей, но не являющиеся источниками энергии, активные физиологически в малых дозах и вступающие в организме в состав сложных биокатализаторов, выполняющих различные функции в процессе обмена веществ,—получили название витаминов.
Отдельные витамины или их группы относятся к самым различным классам органических соединений.
1. Витамины алифатического ряда:
Витамин F—высшие ненасыщенные алифатические кислоты: линолевая С17Н31СООН, линоленовая С17Н29СООН и арахидоновая С]9Н31СООН.
Витамин С (аскорбиновая кислота)—производное лактона ненасыщенной полиоксикислоты (см. том. I, стр. 574).
Холин—производное (В-оксиэтиламина (см. том I, стр. 658).
Пантотеновая* кислота
СН3 Н I I но—СН2—С----С—СО—ХН—сн?—сн2—соон
I I сн3 он и пантетеин:
сн3 н I I он—СН2—С----С—СО—XH—CH2—СН2—СО—-NH—сн2—CH2—SH
I I
СН3 ОН
II. Витамины алициклического ряда: Мезоинозит—производное циклогексана (стр. 65).
Витамины группы А и каротины (провитамины А)—производные циклогексена с полиеновой изопреноидной цепью.
Витамины группы D (кальциферолы)—стероидные соединения.
III. Витамины ароматического ряда: n-Аминобензойная кислота (стр. 390).
Витамины группы К—производные нафтохинона.
* Название ее происходит от слова пан (греч. «все»), так как пантотеновая кислота выделена почти из всех растительных и животных продуктов.
Витамины группы Р (эриодиктин, гесперидии, рутин, катехины)—производные флавонов.
Витамины группы Е (токоферолы)—производные хромана.
IV. Витамины гетероциклического ряда:
Витамины РР (никотиновая кислота, никотинамид).
Витамины группы Вб (пиридоксин, пиридоксаль, пиридоксамин)—производные пиридина.
Витамин Вг (тиамин, или аневрин)—производное бициклической пиримидино-тиазольной системы.
Витамины группы Н (биотины)—производные конденсированной бициклической имидазолидо-тиофеновой системы.
Витамин В2 (рибофлавин)—производное изоаллоксазина.
Фолиевая кислота—производное птерина.
Витамины группы В12 (кобаламины)—кобальтовые комплексы нуклеотида диметилбензимидазола и порфириноподобной циклической системы.
По растворимости, а следовательно и по методам определения содержания витаминов в различных пищевых продуктах, витамины делятся на два класса: жирорастворимые и водорастворимые.
Несомненно, что перечисленными веществами не ограничиваются все разнообразные дополнительные факторы питания, и дальнейшие исследования принесут, вероятно, еще много нового. Тем не менее можно считать, что важнейшие вещества такого рода (встречающиеся в сравнительно больших количествах) уже известны. Многие из них не содержат азота, и само название «витамины»—от слов вита (лат. «жизнь») и амин—сохранилось от того времени, когда они еще не были выделены в качестве индивидуальных продуктов и когда считалось, что все витамины являются сложными азотистыми веществами, аминами.
Витамины алициклического ряда
Группа витаминов А (аксерофтолы). Недостаток этих витаминов вызывает в наиболее резких случаях нарушение нормального состояния оболочек глаза (ксерофтальмию), изменение эпителия носоглотки и бронхов, а также нарушение функций желез пищеварительного тракта (ахилия), надпочечников и т. д. Нормальная доза витамина А для человека составляет 3—5 мг в день (минимально 1 мг). Витамины группы А содержатся в рыбьем жире, яичном желтке, молоке и в других продуктах.
Витамин А образует желтые кристаллы, плавящиеся при 63—64°. может перегоняться в глубоком вакууме (0,00001 мм) при 137—138°, С растворами SbCl3 в хлороформе он дает характерное синее окрашивание. На воздухе легко окисляется в героновую кислоту:
Н3С СН3
Н^ ''СООН
Н2С СОСН3
Хсн2
После того как в 1928—1933 гг. было установлено строение каротиноидов (см. стр. 89) и было показано, что каротины являются провитаминами (т. е. исходным материалом, превращающимся в организме в витамины А), Карреру удалось установить строение витамина А.
Витамин А и его провитамин—3-каротин—потребляются в очень больших количествах в качестве добавки к пищевым продуктам. Извлечение из природных источников не может полностью удовлетворить потребности в этих витаминах. Поэтому в настоящее время в ряде стран (Швейцария, СССР, США, Англия и др.) витамин А производится и синтетически на основе метода Ислера.
Витамин А (аксерофтол) синтезируется по следующей схеме. Конденсацией 3-ионона (стр. 80) с хлоруксусным эфиром через промежуточные стадии образования хлоргидрина и глицидного эфира получается у-(2,2,6-тримстилциклогексен-i -п.!)-метилкротоновый альдегид:
СН3 V СН..С!
Х/СН=сн-со IO;R
\сн3
сн3
СИ
3 сн3
V/CH=CH—С—СНС1—COOR
А
ОН
Н3с СН:!
\/,СН=СН—С СН—COOR
I ¥
сн.
Н3С сн:
СН2—СН=С—СН(')
сн3
о
Этот альдегид конденсируется с магнийорганическим соединением 3-метилпентен-2-ин-4-ол а:
СН3
I с=с—с=сн—сн. I I
MgBr OMgBr
Н3с СН3
CH3
CH3 I сн2—сн=с—сн—с=с—с=сн—сн2
OMgBr
OMgBr
После омыления продукта конденсации, восстановления тройной связи полученного гликоля в двойную, последующей изомеризации и дегидратации гликоля
(при защищенной ацетилированием первичноспиртовой группе) получается витамин А:
СН3
СН3
Н3с СНз
СН2—СН=С—СН—СееС—с=сн—сн2
н
ОН Pd/СаСОз
н3с СНз
сн3
СНз
СН2—СН=С—СН—СН=СН—С--=СН—сн2
ОН
ОН
‘3
СН3
сн3
J2
Н3с СН3
СН2—СН-С—СН—СН=СН—С=-СН—сн2 I —н2о
он ’
Н3С
он
'з
СН3
сн3
сн3
СН=СН—С=СН—СН—СН—С=СН—СН2
витамин А (аксерофтол)
сн3
ОН
Для витамина А, содержащего в молекуле пять сопряженных связей, возможно 16 стереоизомерных (цис-, транс-) форм. Сам витамин А (аксерофтол) имеет полное 1ранс-строение.
Витамин А2 является 3,4-дегидроаксерофтолом, т. е. содержит еще одну двойную связь в иононовом цикле.
Группа витаминов D (кальциферолы). Недостаток этих витаминов вызывает рахит и остеомалляцию. Их происхождение и строение выяснены главным образом благодаря работам Виндауса.
По строению и свойствам витамины группы D близки к стери-нам; многие стерины являются провитаминами D. Препараты, богатые витаминами D2, получаются облучением дрожжей (эргостерина). При сравнительно коротком воздействии ультрафиолетовых лучей на эргостерин (стр. 178) получается витамин D2 (кальциферол):
СН3 СН3 СН3
R = сн—сн==сн—сн—сн—сн3
Витамин D2 образует кристаллы с темп, плавл. 116°; его спиртовые растворы обладают правым вращением [а^] = + ЮЗ°.
Потребность взрослого человека в витаминах группы D составляет около 2 7 в день, для растущего ребенка она достигает 15—25 у. Витамины группы D содержатся в рыбьем жире, молоке и др.
Кроме кальциферола, в состав природного комплекса витаминов D входят также витамины D3, D4 и D5, отличающиеся от D2 строением боковой цепи и происхождением. Так, витамин D3 получается из холестерина (стр. 170) через 7-дегидрохолестерин:
холестерин витамин D3
(темп, плавл. 83°)
Витамин D4 получается облучением 22-дигидроэргостерина.
Витамины ароматического ряда
Группа витаминов К. Недостаток этих витаминов в пищевом рационе вызывает кровоизлияния в подкожной клетчатке, мускулатуре и т. п. Они содержатся в хлебе, овощах, печени животных.
Строение витамина Кх выяснили Альмквист, Дойзи и Каррер. Этот витамин оказался производным 2-метил-1,4-нафтохинона и был синтезирован конденсацией его с фитолом (см. том I, стр. 393):
О
| || Y СН3 СНз СН3 СНз
^^|/\сН2-СН==С—(СН2)з-СН—(СН2)з—СН—(СН2)з—СН—СН3 Q витамин Ki
Витамин Кх—желтое масло, закристаллизовывающееся при температуре около —20°.
Витамин К2 содержит еще более длинную боковую цепь, состоящую из 30 углеродных атомов.
Интересно, что такие простые синтетические вещества, как 2-метил-1,4-нафт°-хинон (витамин К3, или метанон) и получаемая из него 2-метил-З-сульфокислота (вика со л) обладают активностью витамина К и используются в медицинской практике.
Группа витаминов Р. Из материалов, богатых аскорбиновой кислотой, была выделена флавонная фракция, содержащая витамины Р.
Недостаток этих витаминов обусловливает так называемую пурпурную болезнь—множественные подкожные точечные кровоизлияния. Активными составными частями флавонной фракции оказались витамины эриодиктин, гесперидии и рутин—глюкозиды, производные флавонов эриодиктола, гесперетина и кверцетина (стр. 584):
он
он
ОН
ОН
гесперечин (темп, плавл. 227—228°)
эрлодиктол (темп, плавл. 267°)
В эриодиктине и гесперидине остатки сахаров С12Н21О10 находятся при седьмом углеродном атоме, в рутине (производном кверцетина) остаток рутинозы связан с третьим углеродным атомом.
Аналогичной физиологической активностью обладают катехины (стр. 386), содержащиеся в листьях чая (А. Л. Курсанов и др.).
Группа витаминов Е (токоферолы). Витамины Е являются веществами, необходимыми для нормального размножения животных организмов. Их строение выяснено благодаря работам Эванса, Эмерсона, Тодда, Бергеля и Каррера. Из масла ростков пшеницы сначала были выделены два вещества: я-токоферол С29Н50О2 и р-токоферол С28Н48О2, в 1937 г.—третье вещество, оказавшееся изомером p-токоферола и названное у-токоферолом, а в последние годы— еще четыре изомерных витамина Е. Таким образом, в настоящее время выделены все 7 возможных изомерных форм.
Пиролизом я-токоферола был получен тетраметилгидрохинон (дурогидрохинон), из р- и ^-токоферолов—изомерные триметилгидро-хиноны. При окислении токоферолов хромовым ангидридом образуется кетон, получаемый также из фитола (см. том I, стр. 383). Следовательно, в молекулу токоферолов входит остаток фитола, который сконденсирован с гидрохиноном таким образом, что полу-чается система оксихромана. Это позволило предположить для токоферолов следующее строение:
тетраметил гидрохинон
а-токоферол
В этих формулах символом^ обозначена цепь:
—СН,—СН2—СН,—СН—СН2—СН,—СН,—СН—СН,—СН,—СН,—CM—CHS сн3 сн3 сн3
7-Токоферол отличается от р-токоферола расположением метильных групп не при С5 и С8, а при С7 и С8.
Каррер нагреванием триметилгидрохинона с фитилбромидом и хлористым цинком получил рацемический а-токоферол (природный, выделенный из зародышевой части пшеничных зерен, вращает плоскость поляризации вправо).
Токоферолы—бесцветные масла, обладающие свойствами фенолов. Для идентификации токоферолов применяют аллофановые эфиры (a-эфир имеет темп, плавл. 159°, 3-эфир—темп, плавл. 138°).
Витамины гетероциклического ряда
Витамины РР (никотиновая кислота и ее амид). Никотиновая кислота (ниацин) и амид никотиновой кислоты (см. стр. 603) оказались факторами, предупреждающими пеллагру человека. Пеллагра — тяжелое общее заболевание, затрагивающее функции щитовидной железы и надпочечников, а также нервной системы; особенно характеризуется огрубением и шелушением кожи. Потребные дозы никотиновой кислоты составляют 30—50 мг в день; лечебные—могут достигать 0,3 г.
Витамины В6 предохраняют некоторых животных от заболеваний, подобных пеллагре человека. К этой группе принадлежат пиридоксин и его производные.
Пиридоксин (адермин), как установили Стиллер и Р. Кун, оказался 2-метил-3-окси-4,5-ди-(оксиметил)-пиридином. Дегидрированием пиридоксина получается пиридоксаль, от которого восстановлением можно снова перейти к пиридоксину. Если на пиридоксаль действовать аммиаком в восстановительных условиях, то получается пиридоксамин:
СН2ОН СНО CH2NHo
I I I
с с с
/ Ч / Ч / Ч
НО-С С-СН2ОН -2Н НО-С С-СНоОН +NH3+2H но-с. с~сн2он
II I —> II I “-------------> II I
сн3-с сн сн3-с сн сн3-с сн
\ \ // \//
N N N
пиридоксин пиридоксаль пиридоксамин
Пиридоксин-основание плавится при 160°, пиридоксамин—при 193е. Хлоргидрат пиридоксина плавится при 207°.
Группа витаминов В6 входит в состав ферментов, принимающих участие в белковом обмене. Суточная потребность в этих витаминах для человека составляет 10—100 мг.
Витамин В2 (тиамин, или аневрин) является существенно важным добавочным фактором питания. Его отсутствие вызывает заболевание, известное в Японии под названием «бери-бери»—периферический неврит. Оно наблюдалось при питании «полированным»
рисом, т. е. рисом, лишенным оболочек и зародышевых тканей. Источником витамина Вх служат рисовые и другие отрубй, а также дрожжи. Присутствие в витамине Bt серы (почему он и был назван тиамином) доказал Винда ус, установивший и его суммарную фор-мулу C12H16ON4S.
При действии сульфита натрия тиамин расщепляется с образованием натриевой соли сульфокислоты (I), являющейся производным 2,5-диметил-4-аминопиримидина, и легко растворимого в воде основания (II), оказавшегося 4-метил-5-ф-оксиэтил)-тиазолом:
СН N--С—СН3
/V II II
N С—CH2SO3H НС . С
СН3—С С—NH2 X's// ХСН2—сн2он
\ // N 11
I
Путем осторожного окисления при помощи K3[Fe(CN)e] из тиамина C12HieON4S было получено вещество C12H14ON4S, оказавшееся тождественным тиохрому, ранее выделенному Р. Куном из дрожжей. Тиохром—ярко-желтые кристаллы, плавящиеся при 226— 227°; растворы его обладают красивой синей флуоресценцией.
В результате многочисленных исследований были предложены формулы .для тиамина и тиохрома (Вильямс), вскоре подтвержденные синтезом:
СН2
/\/ \+
N I N---ГСН3
II I II Р
н3с/ч/хйн3\/Чн2
6 I
СН2ОН
2СГ
тиохром
СН2ОН
тиамин
Хлоргидрат тиамина, витамин Вр плавится при 249—250°. Суточная потребность в нем для человека составляет от 1 до 4 мг.
В СССР витамин Вх (тиамин) получают синтетически по методу, предложенному Г. В. Челинцевым и 3. В. Беневоленской. Сначала раздельно синтезируют пиримидиновую и тиазольную части молекулы тиамина:
Пиримидиновая часть
СН2
|| С2Н5ОН
NaOH~
CN
СН2ОС2Н5
/ НСООС2Н5
^2 С2Н5О^
CN
NaO—СН СН2ОС2Н5
\ / (CH3)2SO4
С --------*
! CN
СН3О—СН СН2ОС2Нб
I CN
СН3О-СН СН2ОС2Н6
HN ЧС
II + I
С CN
н3с/ \nh2
сн
N С-СН2ОС2Н6
II I н3с—с c—nh2
на
ацетамидин
СН / ч
N С—СН2С1
II I +
СН3—С С—NH3
I
С1-
Тиазольная часть
Н3С О Н3С О Н3С О
\ / \ н2 \ / \ С12 \ / \
С СН---------> С СН2-----------> С1—С СН2
II II Pd II I II
НС---СН НС----СН2 Н—С-----СН2
сильван /
Н3С о
HN Cl—С СН2 N----------С—СН3
II + I I II II
НС Н—С-----СН2 НС С
\sH Clz хсн2—СН2ОН
тиоформ- II
амид
Конденсацией полученных продуктов (нагреванием соединения I с галоидной или азотнокислой солью тиазольного производного II) получается соль тиамина:
СН / \
N С—СН2С1
II I +
СН3—С С—NH3
\ //
N
N С—СН3
Il II
С1- + НС с
хсн2— сн„он
S 2
сн сн2
/ \/ \+
N с N--С—СН3
II I II II
СН3-С С НС С
\ ^7 \+ \ / Х™
N NH3 S । 2
СН2ОН _
2СГ
Витамин Н (биотины). Еще в 1901 г. было показано, что дрожжевые клетки выделяют в питательную среду какое-то вещество, стимулирующее рост самих дрожжей. Это вещество получило название биос (греч. «жизнь»). Значительно более поздние исследования голландского ученого Кегля (1935 г.) показали, что биос представляет собой смесь многих веществ. Одним из них оказался мёзо-инозит, или гексаоксициклогексан (стр. 65). Кроме того, Кегль выделил ранее неизвестное гетероциклическое соединение—^-биотин C10H16O3N2S, плавящийся при 220°. Он содержится в дрожжах и яичном желтке в очень малых количествах (из 250 кг сухого желтка было получено 1,1 же а-биотина). ,
В 1940 г. из печени быков и из молока был выделен особый витамин, названный витамином Н, похожий на а-биотин Кегля, того же состава C10H16O3N2S, но плавящийся при 232°. Его строение было установлено Де-Виньо с сотр. Это соединение получило название {^-биотина.
Строение а- и 3-биотинов может быть выражено формулами: СО
со
HN 1 NH । HN NH 1
* 1 нс- 1 •л 1 —сн НС- 1 * 1 СН
1 н2с 1 СН-СН-СН(СН3)2 Н2с СН-(СН2)4-СООН
S СООН а-биотин
S р-биотин
[З-Биотин является насыщенным соединением; при окислении он, присоединяя два атома кислорода, дает сульфон; при щелочном гидролизе отщепляет СО2 и дает диаминокислоту:
NH2—НС----CH—NHo
I 1
Н2С СН—(СН2)4—соон
При обработке фосгеном эта диаминокислота вновь образует 3-биотин, а при энергичном окислении дает адипиновую кислоту.
Как легко видеть, (3-биотин имеет три асимметрических атома углерода: продукт, идентичный природному ([а]р=+91°), до сих пор не был выделен.
Витамин В2 (рибофлавин, или лактофлавин) является главнейшей частью термостабильных факторов комплекса витаминов В. Он кристаллизуется в виде оранжево-желтых игл с темп, плавл. 292—293° (с разл.); водные растворы его обладают желто-зеленой окраской и зеленоватой флуоресценцией.
Витамин В2 первоначально был выделен из молочной сыворотки (Р. Кун), почему и был назван лактофлавином. Для получения 1 г препарата требовалось переработать 5,4 т сыворотки. Выделение витамина В2 из молочной сыворотки основано на адсорбции его из водных растворов так называемыми кислыми глинами—флоридином или фуллеровой землей.
При освещении рибофлавина в щелочных растворах идет отщепление C4HSO4 с образованием лумифлавина, оказавшегося 6,7,9-три-метилизоаллоксазином:
СН3
СН N \
СН3-С7 с с -со
I !
СН3—С6 С С 3NH
''Чб / Чд / СН N СО
Строение лумифлавина было доказано синтезом (Р. Кун), а в дальнейшем был осуществлен синтез и самого витамина В., (Р. Кун, Каррере
В настоящее время рибофлавин производится синтетически из (/-рибозы. л-4-ксилидина и барбитуровой кислоты. (/-Рибозу получают окислением глюкозы с последующим превращением образовавшейся d-арабоновой кислоты в </-рибоно-iivio и d-рибонолактон. Синтез рибофлавина проводится по схеме:
о-4-ксилидин
СНО I Н—С—ОН
I н—с—он
I н-с-он
сно
I н-с-он
4- Н-С-ОН —>
н-с—ОН
СН2ОН
(/-риоста
СНоОН
н-с-он
I н—с—он
Н—С—ОН
CgH5N2C1
СН2ОН I н-с-он
I н-с-он
СНоОН Н-с—он н—с— он н—с—он
I
н—с—он
сн2
NH—СН2
рибофлавин
Рибофлавин играет важную физиологическую роль, так как он входит в состав окислительных ферментов клеток в виде рибофлавинфосфорного эфира C17H21N4PO9, сочетающегося с протеинами.
Оптимальная суточная доза рибофлавина для человека составляет 2—4 мг.
Фолиевая кислота. В 1941 г. Митчелл с сотр. выделили из листьев шпината вещество состава C15H15O8N5, названное фолиевой кислотой (витамин Вс), и доказали наличие в нем птериновой группировки (стр. 640).
Фолиевая кислота—желтые иглы, обугливающиеся, не плавясь, при температуре выше 250°. Водные растворы фолиевой кислоты обладают сине-зеленой флуоресценцией. Повидимому она оказывает благоприятное терапевтическое действие при различных видах анемии.
Фолиевой кислоте принадлежит строение:
N N
НС7 С 2C~NH2
НООС—СН2—СН2-СН—NH—СО—NH—СН2—С6 С 3N
I Х=/
СООН N С
I
ОН глутаминовая n-аминобензойная птериновая группировка
кислота кислота (6-метил-2-амино-4-оксиптеридин)
В группу витаминов Вс входит ряд производных фолиевой кислоты, содержащих разное число остатков глутаминовой кислоты.
Кроме фолиевой кислоты, из экстрактов печени в 1948 г. было выделено содержащееся там в очень малых количествах вещество, названное витамином В12. Это вещество в крайне малых дозах (2,5—5,0 у) оказывает лечебное действие при злокачественной анемии.
Витамин В12 образует красные кристаллы, темнеющие при 190—215°, но не плавящиеся при температуре до 300°. Как оказалось, витамин В12 содержит около 4,5% кобальта; состав его отвечает формуле C63H90N14O14PCo.
Кристаллографические и рентгенографические исследования свидетельствуют о наличии в молекуле витамина В12 большой плоской группировки с атомом кобальта в центре. Высказывалось предположение, что эта группировка имеет порфириноподобную структуру (стр. 539), так как перегоняющаяся при сплавлении витамина В12 с едким натром жидкость дает положительную реакцию на пиррол (или его производные).
В 1954 г. Тоддом и Джонсоном для витамина В12 предложена структурная формула кобаламина'.
Эта формула учитывает все изученные к этому времени превращения кобаламина и данные рентгенографических исследований.
Антагонисты витаминов (антивитамины)
При изучении действия сульфонамидных препаратов (см. стр. 311) было обнаружено, что некоторые бактерии выделяют вещество, противодействующее активности этих препаратов. Как было установлено, таким веществом является м-аминобензойная кислота. Вместе с тем оказалось, что сульфонамидные препараты препятствуют нормальному действию м-аминобензойной кислоты (стр. 390) в организме.
Таким образом, производные м-аминосульфобензойной кислоты оказываются антагонистами /г-аминобензойной кислоты.
Пантоилтаурин является антивитамином пантотеновой кислоты:
сн2он СН2ОН
(СН3)2С—СН(ОН)—CONH—(СНз)2—SO3H (CH3)2C-CH(OH)-CONH-(CH2)2-COObi пантоилтаурин пантотеновая кислота
[6-Пиридинсульфокислота—антивитамин 0-пиридинкарбоновой (нико типовой) кислоты.
В ряде случаев аналогичные изменения в структуре витаминов, иногда незначительные, приводят к получению продуктов, способных парализовать или ослаблять полезное действие витаминов.
Антагонистическое действие во многих случаях объясняется тем, что антивитамины способны химически реагировать подобно витаминам, вступая, например, в систему того или другого фермента, однако получающиеся при этом продукты оказываются чуждыми организму и не могут выполнять нормально те функции, которые выполняют ферменты, образованные с участием витаминов.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абиетиновая кислота 112, 155, 162, 163
Аглюконы сердечных глюкозидов (Генины) 182—184, 688
Агроцибин 702
Адамантан 112—114
А д а мсит (9- X л ор ди г и д р офе н ар саз и н) 631
Аденин (6-Аминопурин) 635, 638
Адермин, см. Пиридоксин
Адипиновая кислота 40, 70, 72. 75, 172
диэтиловый эфир 41
Адкинс, дегидрогенизация гомологов циклогексана 222
Адонитоксигенин 184
Адреналин 188, 309, 342
Адреналон 342
Азапорфин 547
Азасерин (О-Диазоаиетил-/-серин) 701
Азациклобутан (Азетидин, Триметилен-имин) 511
Азелаиновая кислота 41, 85
Азиды 292, 293
Азимиды 362
Азиновое кольцо 362
Азины 625—630
аналоги, содержащие мышьяк. 631, 632
Азобензол 304, 316
Азоимиды (Азиды) 292, 293
Азокрасители 312, 323, 362, 364—367, 363, 387, 390, 414, 457, 601 лейкооснования 366
Азоксибензол 251, 297, 315
Азоксинафталин 454
Азоксисоединения 314 антибиотики 702
Азоксониевые красители 629, 630
Азонафталин 454
Азосоединения 315
Азосоставляющая 365
Азосочетания (сочетания) реакция 324, 365
Азотистые соединения с аномальной валентностью азота 439
Азотолуол 316
Азотолы (2-Окси-З-нафтойная кислота, анилиды) 464
Азтиониевые красители 629, 630
Азулен (Бицикло-[0,3,5]-декапентен, Циклопентадпепциклогептатриен) 502—504
Акароид 161
Аконитовая кислота 501, 644
Акридин (Дибензопиридин) 513, 621 Акридиновая (а,(3-хинолинди карбоновая) кислота 621
Акридоны (Дигидроакридин, кетопроизводные) 622, 623
Акриловая кислота 406
Акрихин (Атебрин) 522, 623—624 Акролеин 45, 341, 564, 592, 612 Акр олеин аммиак 591
Аксерофтолы (Витамины А) 81, 90, 703, 704
Аксиальная (осевая, полярная, полюсная) связь 13, 30, 31, 34, 121, 167 «Активирование» кислорода 473 Актиномицеты 689, 693, 697, 699, 702 Актиномицины 700, 701 Алантолактон 107
Ализарин 467, 475, 476, 479—480, 559 двунатриевос производное 476 оранжевый (1,2-Диокси-З-ннтро-антрахинон) 477 синий БС 477
Ализариновое масло 480
Ализариновые красители 475, 486
Алициклические соединения 11 —114 двухъядерные 16, 88—111 изомерия 27—37 классификация 12—20
ненасыщенные 35—37, 291 номенклатура 12—20 одноядерные 66—88 получение 37—52, 60—66 с конденсированными ядрами 16, 97—114, 504 стереохимия 20—37, 98—102, 109 физические свойства 58—60
Алициклические углеводороды (Циклоалканы, Циклопарафины, Цикланы) 12, 13, 59, 61 взаимные превращения 46—52 доказательства циклического строения 55 напряжение в цикле 20—22 природные источники 60 размыкание цикла 53, 54 свойства 52—60
Алкалоиды 569, 596, 608, 611, 615, 620, 621, 635, 637, 638, 642—688 выделение 644—645 классификация 646, 647J
Алкил арилфосфины 328
Алкилдихлорарсины 610
N-Алкилимидазол 569
N-Алкил-а-оксихинолин 617
N-Алкилпиперидины 605
Алкилпиридоны 595, 599, 600
N-Алкилтетрагидрохинолины 619
Алкилтрифенилфосфония соли 328 п-Алкилфенилгидроксиламины 360 л-Алкилфенолы 360 Алкилхинолины 615 Алкилхинолоны 617
Алкоголиз галоидных производных циклопропана и циклобутана 48
Аллантоин (4-Уреидо-2,5-диоксоимид-азол) 570, 636
Аллен 67, 702
Аллиланилин 611
•Дллилбензол 403, 451
Аллил бромистый 403
Аллилмеркаптометил пенициллин (Пенициллин О) 690
Аллилпиридин 608 d-Аллоизолейцин 700 Аллоинозит 64
Аллокоричная кислота 408, 409
Аллоксан 633, 634, 636
Аллоксантин 636
Аллооцимен 117
Аллопрегнан 166
Аллопсевдокодеин 673
Алло-ряд стероидов 166
Аллофановые эфиры токоферолов 709
Аллохолан 166
Альдегидаммиаки 591, 593
Альдегидколлидин (Альдегидин, а-Ме-тил-р'-этилпиридин, ’ 2-Метил-5-этиЛ'пиридин) 591, 594
Альдегиды ароматические 275—280 идентификация 79 |
оксимы, стереохимия 282 реакции 142, 277—279 с ненасыщенными цепями 407
Альдер 111
синтез борнеола 144
Альдимедон 79
Альдоза, гидразон 326
Альдоксимы, см. Оксимы альдегидов
Альдостерон (Электрокортин) 195
Альдрин 111
Альтакс 572
Амберлиты 406
Амбровый (крезольный) мускус 253
Амигдалин 280, 388
Амидины 601
Амидная таутомерия 291, 635, 641
Амидобензол, см. Анилин
Амидон 678
Амилнитрит 319, 550, 554, 601 л-Амилхлорид 213
Аминирование углеводородов 298 d-a-Аминоадипиновая кислота 693 Аминоазобензол 323, 361, 366 Аминоазосоединения 323, 364, 366 Аминоалкоголи 685
Аминоальдегиды 396, 568, 595, 614, 625
Аминоантипирин 567
Аминоантрахиноны 471, 473, 477, 627
З-Аминоантрацен 631 Аминоацетон 626 a-Аминоацетоуксусный эфир 530, 540
Аминобарбитуровая кислота (Урамил) 637
Аминобензальдегиды 396, 614
Аминобензойные кислоты (см. также
Антраниловая кислота и ПАБ) 294, 297, 389—391, 703, 714, 715
Аминобензол, см. Анилин п-Аминобензолсульфамид (Сульфаниламид, Белый стрептоцид, Сульфаниловая кислота, амид) 311 л/-Аминобензолсульфокислота (Мет-аниловая кислота) 311 л-Аминобензолсульфокислота, см.
Сульфаниловая кислота л-Аминобензолсульфохлорид 311 n-Аминодиметиланилин (Диметил-л-фе-нилендиамин) 363, 630 2-Амино-4,6-диоксиптеридин (Ксанто-птерин) 641 о-Аминодифениламин 629 Аминокетоны 568, 625
Аминокислоты 156, 179, 188, 378, 643, 685 ароматические 389—392
a-Аминокислоты, циклические ангидриды (Дикетопиперазины) 513, 626, 685
о-Аминокоричная кислота, ангидрид (Карбостирил, а-Оксихинолин, а-Хинолон) 618
у-Аминомасляная кислота 537
Аминометилциклогексан 49
о-Аминоминдальная кислота, лактам (Диоксиндол) 553, 555, 557
Аминонафталин 447
1-Аминонафталин-4-сульфокислота (Нафтионовая кислота) 458, 459
Аминонитропиридины 602
Аминооксибензойные (аминосалицило-вые) кислоты 383, 390
1,8- Аминооксинафталин-3,6-дисульфокислота, см. Аш-кислота
2-Амино-6-оксипурин, см. Гуанин
Аминопиразолы 565
Аминопиридины 597, 599, 601—603, 650
6-Аминопурин (Аденин) 635, 638 л/-Аминосалициловая (5-амино-2-окси-бензойная) кислота 383, 390
ц-Аминосалициловая кислота (ПАСК, 4-Амино-2-оксибензойная кислота) 390
гс-Аминосульфобензойная кислота 715 2-Аминотиазолы 572
Аминотиофен (Тиофенин) 516, 527 2-Амино-4,6,7-триоксиптеридин (Лей-коптерин) 641
1-Амино-3,6,8-трисульфокислота нафталина 459
Аминотрополон 498, 499
2-Аминотропон 495, 496
Аминоуксусный альдегид 625
Аминофеназины (Эуродины) 629 я-Аминофениларсиновая (арсанпловая) кислота 396
о-Аминофенилглиоксиловая (изатиновая) кислота 619
соль 556
о-Аминофенилуксусная кислота, лактам (Оксиндол) 548, 553, 555
Аминофенолы 250, 251, 314, 352, 356,
363—365, 390, 396, 431
Аминохинолины 615 о-Аминохлорстирол 548 Аминоциклобутан 69 Амины алициклические 48 ароматические 250, 297—312, 421, 622 — константы диссоциации 299 — многоатомные 361—363 — получение 298 — реакции 299, 302—305, 396 — соли 300 — физические свойства 299
Аммониевые основания 566
гидраты 405, 536
соли 299, 309, 325, 405, 536, 595
АлцЬи-положение 448, 462
Анабазин (р-(а-Пиперидил)-пиперидин]
643, 651
Анальгин 567
Ангидрооснования о-аминофенолов 364 о-диаминов 362
Ангулярные
группы 106, 167, 174
системы 486—489
Андрогенная активность 190
Андрогенные (тестоидные) гормоны 188.
Андростан 166, 167
Андростендионы 189
Андростендион 189
Д4-Андростенол-178-он-3 (Тестостерон) 178, 188
Андростерон 178, 190
Аневрин, см. Тиамин
Анестезин 391
Анетол 383, 394, 406
Анизидины 358, 364, 616
Анизол 267, 395
Анилидоуксусная кислота (фенил гл и-коколь, Фенилглицин) 308, 554, 560
Анилиды 300, 302, 304
диазосоедпнений 323
диаминов 362
Анилин (Аминобензол, 'Амидобензол, Бепзидам, Фениламин) 224, 235, 297—305, 361
идентификация 253
качественная реакция 304 окисление 313, 352, 355, 356 получение 250, 252, 298 производные 308, 309
реакции 302, 396, 432, 548, 556, 560,
572, 612, 622, 626, 628
сульфокислоты 304, 311 физические свойства 298, 301
Анилиновые красители 302, 305. 428
Анилиновый
желтый 366
синий 307, 429
черный («Черный анилин») 304, 356
Анилы (Шиффовы основания) 302, 681
Аниониты (Анионообменивающие смолы) 405
Анионоидные (нуклеофильные) реагенты 242
Анисовая (я-метоксибензойная) кислота 383
Анисовое масло 406
Анисовый альдегид (Обепин) 394
Анисовый (я-метоксибензиловый) спирт 343
Аннелирование колец ангулярное 486 линеарное 484
. Антибиотики (Антибиотические вещества) 689—702
Антивитамины 715
Антигельминтные препараты 131, 342
Антиоксиданты 341
Антипирин (1-Фенил-2,3-диметилпира-золон-5) 566, 567
Антифебрин (Ацетанилид) 300, 302, 308
Антц-форма 93, 94, 283—285
«Античный пурпур» (6,6'-Дпброминди-
Го) 560
Антоцианидины 586—588
Антоцианы 382, 586
Антрагаллол (1,2,3-Триоксиантрахинон) 477
Антрагидрохинон V (Оксантранол) 473, 474
Антразин 627
Антрамины 470, 471
Антраниловая (о-аминобензойная) кислота 202, 367, 377, 389, 559, 562 производные 622
Антрапурпурин (Изопурпурин,
1,2.7-Триоксиантрахинон) 477 Антрахинон 468—471, 476, 562 производные 471—477, 562 — природные 478
Антрацен (2,3-Бензнафталнн) 203, 335, 374, 466, 485, 621 идентификация 253 производные 469—479, 490 формулы Гинсберга, Гребе и Либермана 467
Антрацендигидрокарбоновые кислоты 468
Антраценовое (зеленое) масло 221, 466, 621
Антролы 470, 471
Антрон 474
Антронимин 470
Антроподезоксихолевая кислота, см. За,7я-Диоксихолановая кислота
Анииотца синтез 467
Апоатропин (Атропамин) 653
Апоморфин 671, 672
Арабоновая кислота 713
Арахидоновая кислота 703
Арбузов А. Е.
получение производных индола 549 синтез фосфенилхлорида 326
Арбутин 342
Арекаидин (N-Метил-!,2,5,6-тетрагид-роникотиновая кислота) 649
Ареколин (N-Метил-1,2,5,6-тетрагид-
роникотиновая кислота, метиловый эфир) 649
Арилгидразины 324 Арилгидроксиламины 313—314
Арилирования реакция, см. Реакция Ульмана
Армстронга—Винна правило 455 Ароматизация олефинов 222, 223 парафинов (Дегидроциклизация) 215, 222, 223
Ароматические соединения 198, 227 и гидроароматические, взаимные переходы 57, 75, 205—209, 222, 225 многоядерные 413—490 небензольные 491—504 ориентирующее влияние заместителей 240, 243, 450 расщепление 205 синтезы из соединений жирного ряда 203
Ароматические углеводороды ряда бензола 210—227 восстановление 218 действие галоидных кислот 219 — галоидов 218, 243, 244 — диазоуксусного эфира 219 — окислителей 220 источники 221, 223 нитрование 220, 246, 248 получение 57, 75, 203, 211—215. 221—223 сульфирование 220, 257 физические свойства 215—217
Ароматический характер 199, 215, 227 гетероциклических соединений 514— 518 иона циклопентадиена 492 — циклогептатриенилия 493, 495 нафталина 449 пиразола 565 пиррола 532 тиофена 526 трополона 498 тропона 495
Арсаниловая (n-аминофениларсиновая) кислота 396
Арсантрен 631
Арсацетин 396
Арсенирование (реакция Бешана) 331
Арсенобензол 331
производные 397
Арсепидины 610
Арсин, окись 610
Артемизин 107
Асимметрический атом углерода 28, 124, 126, 134, 139, 149, 167, 169
Асимметрический синтез 388 Асимметрия молекулы 140 без асимметрического атома 219
Аскаридол 130
Аскорбиновая кислота (Витамин С) 521, 703, 707
Аспирин (Ацетилсалициловая кислота) 383
Атебрин (Акрихин) 522, 623—624
Атоксил 396
Атофан (а-Фенилцинхониновая кислота) 619
Атролактиновая кислота 388, 389
Атропамин (Апоатропин) 653
Атропизомерия 417, 418, 464, 465
Атропин 83, 388, 410, 528, 538, 648,
653, 654, 656
Атроповая (а-фенилакриловая) кислота 388, 410
Ауверса—Скита правило 35
Ауксин-лактон 74
Ауксины 74
Ауксохромные группы 369, 373, 444, 628
Аурамин 423
Ауреомицин, см. Хлортетрациклин
Аурин (Диоксифуксон) 427, 429, 430
Аутоксидация бензальдегида 278
Ахромицин, см. Тетрациклин
Аценафтен 221, 463
Аценафтенхинон 463
Ацетальдегид, см. Уксусный альдегид
Ацетамид 571
Ацетамидин 711
З-Ацетамино-4-оксифенил арсиновая кис-
лота (Стоварсол) 398
Ацетаминопиридины 603 п-Ацетаминофенол 383
Ацетанилид (Антифебрин) 300, 302, 308
Ацетидиноны (Триметиленимин, р-лак-тамы) 511
Ацетилантраниловая кислота 615
Ацетиларсиновая кислота 396
Ацетилацетон 540
Ацетилен 111, 191, 203, 445, 449, 525,
530, 548, 565, 591
Ацетиленди карбоновый эфир 467
N-Ацетилизатин 557
N-Ацетил-7-кето-8-метилдекагидроизо-хинолин 662
Ацетилкумариновая кислота 411
Ацетилмалоновый эфир 350 а-Ацетил-М-метилпиррол 532
Ацетил нитрат 520
N-Ацетилоксиминодигидромерохине-
’новый эфир 662
Ацетилпиперидин 606, 607
Ацетилсалициловая, кислота (Аспирин) 383
Ацетовератрон 681
Ацетон 203, 266, 418, 559, 613, 653 фенилгидразон 549
Ацетондикарбоновый эфир 656, 657
Ацетондищавелевая кислота 582
Ацетонилацетон 519, 525, 549 1-Ацето-2-оксинафтойная кислота-3, оксимы 465
у-Ацетопропиловый спирт 522, 624
Ацетоуксусная кислота, анилид 614 Ацетоуксусный эфир 40, 324, 530, 581 ?
593, 613, 634
М-Ацето-/?-фенетедин (Фенацетин) 364 Ацетофенон (Метилфенилкетон) 281,282 418
получение 403
реакции 275, 389, 401, 619
Ацилоиновая конденсация 42
Дг{ц-формы
нитрофенолов 271, 358
псевдокислот 254
Аш-кислота (Н-кислота, 1,8-Амино-оксинафтал ин-3,6-дисульфокислота) 459
Байер 206
аутоксидация бензальдегида 278 гидрирование фталевых кислот 206 номенклатура бициклических соединений 18
окисление пинена 136
синтез индиго 559
— индоксила 554
— Р-пиколина 591
— флороглюцинкарбонового эфира 350
— фуксонимина 427 солеобразование у-пиронов 576 строение индиго 558 — терпенов 118
теория напряжения 20—22, 26, 29, 100, 206
— цветности 370 формула бензола 227, 229 — индола 507
— пиррола 507, 529
— фурана 507
Бальзамы 161 —164, 287
бензойный 161
копайский 159
перуанский 156, 161, 274, 408 толутанский 161, 225, 274, 408 элеми 161
Бамбергер гидрирование нафтиламинов 321 строение диазосоединений 319 формула нафталина 446
Барбитуровая кислота 633, 637, 713
Барта реакция 331, 332, 396
Батохромное смещение (углубление цвета) 368
Бахман, синтез эквиленина 197
Бебирин (Хондродендрин) 666
Бекмановская перегруппировка 283, 676
Белковые вещества гидролиз 569 гормоны 188 и алкалоиды 643
Бензазид (Бензоилазоимид) 289, 292
Бензальанил (Бензилиденанилин) 302, 307
Бензальацетон 407
Бензальдегид, см. Бензойный альдегид
Бензальдоксимы 283, 307
Бензальхлорид (Бензилиден хлористый) 244, 247, 276
Бензамид 289, 291
Бензанилид 289, 300, 308
1,2-Бензантрацен 489
2,3-Бензантрацен (Нафтацен, Тетрацен) 374, 484
Бензантрен 488
Бензантрон 488
Бензгидрилкалий 442 я-Бензгидрилтетрафенилметан 437 Бензгидрильный (дифенилметильный) радикал 441
Бензгидроксамовая кислота 289
Бензгидрол (Дифенилкарбинол) 422
Бензенил хлористый (Бензотрихлорид)
244, 247, 286, 288
Бензениламидин 289
Бензидам, см. Анилин
Бензидин (/?,п'-Диаминодифенил) 250, 317, 414, 459
Бензидиновая перегруппировка 297, 317, 318, 414
Бензил (Дибензоил) 410, 434, 435, 483
Бензил
анион 334
бромистый (Бензилбромид, а-Бром-толуол) 247
иодистый (Бензилиодид, а-Иодто-луол) 244, 247
радикал 441
хлористый (Бензилхлорид, а-Хлор-толуол) 244, 247, 274, 421, 432
Бензиламин 254, 297, 301, 307
Бензиланилиц (Фенилбензиламин) 307 Бензилбромид, см. Бензил бромистый Бензилиден хлористый (Бензальхлорид) 244, 247, 276
Бензилиденанилин (Бензальанил) 302
Бензилизохинолин 665
Бензилиодид, см. Бензил иодистый
Бензилмеркаптан, соль 683
Бензилнатрий (Натрийбензил) 213, 334
Бензиловая (дифенилгликолевая) кислота 434
Бензиловая перегруппировка 434
Бензиловый спирт (Фенилкарбинол) 274 408, 421
Бензилпенициллин (Пенициллин II, или G) 690, 691, 692, 693
Бензилхлорид, см. Бензил хлористый
Бензимидазол 571, 632
Бензины 62, 75
2,3-Бензнафталин, см. Антрацен Бензоидно-хиноидная таутомерия 271, 357, 361, 367, 427, 461, 462, 474
Бензол
гидроперекись (Надбензойная кислота, Пербензойная кислота) 278, 289, 291
хлористый (Бензоилхлорид) 289, 290, 422, 485, 607, 655
Бензоилазоимид (Бензазид) 289, 292 о-Бензоилбензойная кислота 376, 468 Бензоилгидразин 289, 292
Бензоилгликоколь (Гиппуровая кислота) 288, 289, 292, 391, 392, 552
N-Бензоил гомомерохинен, этиловый эфир 663
Бензоил карбинол (ю-Оксиацетофенон)
282, 395
N-Бензоил-1 -пиперидил пропионовая кислота 662
Бензоилпиперидин 607
Бензоилтирозин 391, 392
Бензоилтриптофан 552
d-Бензоилхинотоксин-^-тартрат 664 Бензоилхинотоксины 663
Бензоилхлорид, см. Бензоил хлористый Бензоилэкгонин 655
метиловый эфир, см. Кокаин
Бензоин 279, 433
Бензоиновая реакция 279
Бензоиноксим 676
Бензойная кислота 79, 220, 286—288
амид (Бензамид) 288, 289, 291
как катализатор 429
получение 282, 286, 410
производные 201, 288—295 реакции 274, 288, 422, 655, 662 сульфинид (Сахарин) 260, 294, 295 эфиры 289, 291
Бензойная смола 288
Бензойный альдегид (Бензальдегид, Горькоминдальное масло) 275—280 аутоксидация 278 восстановление 274
реакции 78, 278, 279, 388, 402, 407, 422
Бензойный ангидрид 289, 655
Бензоксазол 364
Бензол 224—226
ароматический характер 199 восстановление 218, 225 гомологи 211, 220, 225—227 действие окислителей 205, 220, 286, 414
максимум поглощения света 374
меркурирование 336
нитрование 220, 246, 248 озонирование 205, 220, 228 получение 57, 75, 206, 208, 221, 380, 591
Бензол
«пятичленный» 439 реакции 218—220, 243, 326, 413, 414, 418, 421, 422, 432, 449, 467, 468, 471, 527, 560
строение 201, 227—235, 590 сульфирование 220, 257 теплота образования 227 — сгорания 209
формулы 11, 206, 227—233
Бензолазодиметиланилин, /г-сульфокис-лота, см. Гелиантин
Бензол азонафтол 367
Бензол азотолуол (Монометил азобензол) 316
Бензолгексакарбоновая (меллитовая) кислота 379, 380
алюминиевая соль (Медовый камень) 380
Бензолдиазоний (Фенилдиазоний) 319, 367, 419, 458
л^-Бензолдисульфокислота 337 Бензолдихлорсульфамид (Хлорамин Б)
259
Бензолмонохлорсульфамид 259 Бензолполикарбоновые кислоты 379, 380
Бензолсульфамид 259 Бензолсульфокислота 266, 455 Бензолсульфохлорид 259, 283 Бензолтетракарбоновые кислоты 379, 380
Бензолтрикарбоновые кислоты 204, 379, 380
Бензонитрил (Цианобензол) 289, 292, 307, 537
Бензопиразин (Хиноксалин) 362, 483, 626, 627, 634
Бензопиран (у-Хромен) 513 3,4-Бензопиридазин (Циннолин) 633 4,5-Бензопиридазин (Фталазин) 633 а,р-Бензопиридин, см. Хинолин р,у-Бензопиридин, см. Изохинолин Бензопиридин, фталон, см. Хинофта-лон
Бензопирилий 587 Бензопиррол, см. Индол 1,2-Бензопирон, см. Кумарин 2,1-Бензопирон, см. Изокумарин Бензо-а-пирон, см. Кумарин Бензо-у-пирон (Хромон) 583, 584 Бензопурпурин 459
Бензотиазол 572, 573
Бензотиофен 513, 562
Бензотрихлорид, см. Бензенил хлористый
Бензотрополоны 502 Бензофенон (Дифенилкетон) 282, 422, 435, 439
Бензофенонхлорид 436
Бензофуран, см. Кумарон Бензохиноны, см. Хиноны а-Бензпинаколин 436
6-Бензпина кол ин (Трифенилацетофенон ) 435, 436
Бензпинакон 435
Бензпиразол (Индазол) 567, 568
1,2-Бензпирен 489
1,2-Бензфенантрен (Хризен) 173, 181 486, 487
3,4-Бензфенантрен 489
9,10-Бензфенантрен (Трифенилен) 213, 486
Бербероновая кислота 604
Бергаптен 588
Вертело
действие иодистоводородной кислоты на ароматические углеводороды 45
синтез бензола из ацетилена 203
Бетаиноподобное строение у-пирона и у-тиопирона 579, 580
Вешана реакция 331
Билирубин 180, 543, 544
Биокатализаторы 703
Биомицин, см. Хлортетрациклин
Биос 712
Биосинтез 171, 702 пенициллина 693 стрептомицина 694 тетрациклина 697
Биотины (Витамины Н) 569, 704, 712
«Бирадикал» Чичибабина 444, 445
Бисаболен 157
а-Бисаболол 157
Бис-азокрасители 365, 367
Бис-диазосоединения 450, 459
Бисмарк коричневый, см. Основной коричневый
Бифенил (Дифенил) 91, 95, 212, 224,, 322, 333, 413—414 стереохимия 416
Бициклические соединения, см. Двухъ* ядерные соединения
Бицикло-[0,1,1]-бутан 98
Бицикло-[0,1,3]-гексан 104
Бицикло-[ 1,2,2]-гептадиен-2,5 111
Бицикло-[1,1,3]-гептан (Норпинан) НО, 111
Бицикло-11,2,2]-гептан (Норкамфан)
НО, 111
Бицикло-[0,1,4]-гептан (Норкаран) 98
Бицикл о-[0,1,4]-гептан-2,4-диен-1-кар-боновая (норкарадиенкарбоновая) кислота 104
Бицикло-[0,4,4]-декан, см. Декалин
Бицикло- 0,3,5]-декапентен, см. Азулен
Бицикло-[0,3,4]-нонан (Гидриндан) 99, 105, 466
Бицикло-11,3,3]-нонан-2,6-дион-3,7-дикарбоновая кислота 113
Бицикло-[0,3,3]-октан (Пенталан) 99—101, 104
Бйцикло-[0,3,3]-октаноны (Пенталоны) 104
Бишлера—Напиралъского реакция 620, 665, 678
Блана правило 172, 173
Болетол (Пурпурин-а-карбоновая кислота) 479
•Бона—Шмидта реакция 477 Борнеолы 143—146 Борнилан, см. Камфан Борнилацетат 145
Борнилен 146
Борнилиодид 146
Борнилхлорид 136, 145, 147
Брауна реакции 536, 537, 607 Бредт камфоры синтез 142 — формула 139—141 правило 110
Броманилины 309
Бромантраниловая кислота 560
Бромантрахинон 471 9-Бромантрацен 469 Бромацетофенон 395 о-Бромбензилбромид 468 Бромбензойные кислоты 201, 293, 622 Бромбензол 201, 212, 239, 243, 333 Бромбензолсульфокислоты 260 Бромидхлориды ароматического ряда 246
Броминдиго 560
Бромлактоны 96
Броммалоновая кислота, динитрил 43
у-Броммасляная кислота, нитрил 68 1-Бромметил-2-бромциклопентан 104 Броммуконовая кислота 521 Бромнафталины 452—453, 456 2-Бром-5-нитроацетофенон, оксим 284, 285
2-Бром-5-нитробензойная кислота, ме-тиламид 285
л!-Бромнитробензол 251
Бромпиридины 603
Бромстирол 403
Бромтетрациклин 697
а-Бромтиофен 528
Бромтолуидин 202 а-Бромтолуол, см. Бензил бромистый Бромтолуолы 202, 247 п-Бромфенилгидразин 326 Бромфенолы 270
Бромфталевая кислота, ангидрид 471 2-Бромфуран 520 Бромциклогексан 75, 76, 79 Бромциклопентан 71, 72
Р-Бромэтиламин 510
Бруцин 680, 687
Бутадиен 45, 174, 374
«-Бутан 24, 25, 69
Бутандиол 521
трет-Бутил хлористый 253 Бутилбензолы 217
трет-Бутил-л«-крезол, эфир 253 трет-Бутилпирокатехин 341 трет-Бутилциклогексан 32 Бутиролактам (Пирролидон) 537 Бутиролактон 521
Буфа л ин 185
Буфогенины стероидные 184, 185
Буфотал ин 185
Вагнер
окисление камфена 147
— лимонена 126
— пинена 137
реактив 645
строение терпенов 118
формула а-пинена 135
Вагнера—Мейервейна перегруппировка 148—151, 153
Валериановое масло 143
/-Валин 686, 698
Валлаха перегруппировка 315
Вальденовское обращение а,р-хлормор-фидов и а,р-хлоркодидов 673
Ван-дер-ваальсовские радиусы 22, 31 Ванилин 340, 342, 345, 394, 395 Ванилиновая кислота (Протокатеховая кислота, метиловый эфир) 384
Везувин, см. Основной коричневый 2-Вератрилиндол 681 2-Вератрилтриптамин 681
Вератрин 688
Вератровая кислота (Протокатеховая кислота, диметиловый эфир) 345, 384, 585, 667
Вератровый альдегид (Протокатеховый альдегид, метиловый эфир) 395
Вератрол (1,2-Диметоксибензол) 338
Вербенол 138
Вербеной 138
Вестриламин 132
Викасол 707
Виланд
выделение алкалоидов из лобелии 649
получение соединений с азотом аномальной валентности 326, 439, 440
установление строения бруцина 680
---- гумулона 360
— — лейкоптерина 641
---- стрихнина 680 '
----холестерина 172
Вильштеттер выделение изомеров экгонина 657
исследование солей пирилия 576 получение антоцианидов 586, 587 — кокаина 656 — пролина 537 — о-хинона 356
— циклооктатетраена 83 — экгонина 656
способ выделения лигнина из древесины 344
установление строения тропина 653
--------хлорофилла 545 Винилацетат 144
Винилогия 257, 273, 371 0-Винилхинуклидин 660 Винилциклопропан (Винилтриметилен,
Этенилциклопропан) 36
Винные кислоты, соли 570, 609, 650, 656, 696
Виолантрон (Дибензантрон, Индан-трен темносиний) 488
Виолуровая кислота 637 Висмуторганические соединения 336 Виснагин 585
Витамины 604, 641, 703—716
А, см. Аксерофтолы алифатические 703 алициклические 703, 704 антагонисты (Антивитамины) 715 ароматические 703, 707 Вр см. Тиамин
• В2, см. Рибофлавин
В6 704, 709
В12 539, 714
Вс, см. Фолиевая кислота гетероциклические 704, 709 С, см. Аскорбиновая кислота D, см. Кальциферолы
Е, см. Токоферолы
F 703
Н, см. Биотины
К 703, 707
Классификация 703, 704
Р 584, 704, 707, 708
РР, см. Никотиновая кислота синтез 705, 710, 713
Витта хромофорная теория 369 Воскресенский получение теобромина 638 — п-хинона 355 установление состава нафталина 445
Восприимчивость диамагнитная, парамагнитная, магнитная 444
Восстановление нитросоединений 312—318 по Клеменсену 211
Восстановление
по Меервейну—Понндорфу 696 производных пиридина 605—С08 — пиррола 534—537 электролитическое 313
Вофатиты 406
Вудворд
синтез стрихнина 681
— хинина 662
— холестанола 174
Вышнеградского и Ладенбурга реакция 598
Вюрца реакция 67, 94, 95
Вюрца—Фиттига реакция 212, 245, 333, 414, 453, 528
Габриэля синтезы 377, 378 ^-Галактоза 688
Галловая (3,4,5-триоксибензойная) кислота 340, 384, 387
диметиловый эфир (Сиреневая кислота) 385
Галлодезоксихолевая кислота, см.
За,7а-Диоксихолановая кислота льГаллоилгалловая (л1-дигалловая) кислота 387
Галоидбензойные кислоты 293 а-Галоидкумарины 524 Галоиднитросоединения 255—257 Галоидные производные ароматического ряда 243—247, 453—454, 456 подвижность галоида 255—257
Р-Галоидпиридины 597 а-Галоидстирол 402 Галоидфенолы 269, 270 р-Галоидэтиламины 510 Галохромия 206, 426, 579, 580 Гальбанум 158
Гаммексан, см. Гексахлоран Ганнсена кислота 680 Ганч 283, 321, 370, 507 получение тиазолов 507, 571 превращения трифенилметановы х красителей 427 синтез 593 строение диазосоединений 319—321 таутомерия нитро- и изонитросоединений 254 — нитрофенолов 271, 358
Гармалин 678
Гармин 678
Гаттермана катализатор 322
Гаттермана—Коха синтез 276
Гваяковая смоляная кислота 164 Гваякол (1-Окси-2-метоксибензол) 338, 340—342, 395
сульфокислота (Тиокол) 342 угольный эфир (Дуотал) 342
Гваяцилглицерин, конифериловый эфир 347
Гваяцилпропан 345, 346, 348 1-Гваяцилпропандион-1,2 345 1 -Гваяцил-2-этоксипропанон-1 345 Гвоздичное масло 159, 340 Геймана синтезы индиго 559, 560 Гексабромбензол 247
Гексагидробензойная (циклогексанкарбоновая) кислота 51, 76, 79, 95, 288
Гексагидробензолы, см. Гидроаромати-ческие соединения, а также Циклогексан
Гексагидропиридин, см. Пиперидин
Гексагидротерефталевая (циклогексан-1,4-дикарбоновая) кислота 35, 77
Гексагидрофенол (Циклогексанол) 76 — 78, 95, 266
Гексагидроцинхомероновые (пиперидин-3,4-дикарбоновые) кислоты 660
Гексадионкарбоновый эфир 656
Гексалин 78
Гексаметилацетон 297
Гексаметилбензол 214, 217
Гексаметилен, см. Циклогексан Гексаметилентетрамин 696 Гексаметилтри аминотрифенил карбинол 429
Гексаметилтри-(п-аминотрифенил)-ме-тил 443
Гексанитродифениламин 306
Гексаоксибензол 74, 337 фенолят 204, 337
Гексаоксибифен и л 415
Гексаоксициклогексан, см. Мезоинозит
Гексафенил бензол 418
Гексафенилэтан 334, 420, 436—438, 441—443
теплота диссоциации 438
Гексахлоран (Гаммексан, Линдан) 218
Гексахлорбензол 243, 247
Гексахлорциклогексаны 207, 218, 219
Гексахлорциклопентадиен 111
Гексахлорэтан 23
Гексацен 485
н-Гексилбензол 213
н-Гексилрезорцин (1,3-Диокси-4-«-гек-силбензол) 342
Гексозаны 344
Гексэстрол (Синэстрол) 197, 433 н-Гектан 11
Гелиантин (Бензол азодиметиланил ин, сульфокислота, Метиловый оранжевый, Тропеолин D) 363, 366
Гелиотридан (1-Метилпирролизидин) 648
Гелиотропин, см. Пиперональ
Гематопорфирин 543
Геж-двузамещенные соединения 14, 95, 106
Гемимеллитовая (1,2,3-бензолтрикарбоновая) кислота 379 ангидрид 380
Гемимеллитол (1,2,3-Триметилбензол рядовой, Гемеллитол) 210, 216
Гемин 539, 540, 543, 544
Гемитерпен, см. Изопрен Гемицеллюлозы 344
Гемоглобин 486, 528, 533, 539, 543 Гемопиррол (2,3-Диметил-4-этилпиррол) 540, 541, 545
Гемопирролкарбоновая кислота 540 Генины (Аглюконы сердечных глюкозидов) 182—184, 688
Геометрическая конфигурация углеводородов, определение 34
Гептаметилен, см. Циклогептан Гепталин 502 Гептан 213 Гептен 213 «Тептилпенициллин (Пенициллин IV, или К) 690
Гераниол 129, 155
Героин (Диацетилморфин) 673
Героновая (1,1 -диметил-4-ацето-«-ва-лериановая) кислота 82, 89, 704
Гесперетин 708
Гесперидии 704, 708
Гетероауксин ф-Индолилуксусная кислота) 74, 552
Тетеронуклеарные соединения 450 Гетероциклический ряд 507—514 Гетероциклы
ароматический характер 514—518 конденсированные 513, 514, 611*— 624
пятичленные 511, 519—574 трехчленные 509, 510 четырехчленные 511
шестичленные 512—514, 575—610
Тетинакс 267
Геша синтез 422
Гигрин 538, 648, 655
Гигриновая кислота (N-Метилпролин) 538
Гидантоин 569
Гиднокарповая кислота 72
Гидразиды кислот 326
Гидразины 38, 324—326 Гидразобензол 250, 318, 414 Гидразоны 285, 324 326, 557 Гидразосоединения 317, 366 Гидрастин 665, 668, 669 Гидрастинин 665, 668
Г идратроповая (а-фенилпропионовая) кислота 286, 287
Гидратроповый альдегид 277
Гидриндан (Бицикло-10,3,4]-нонан) 99, 105, 466
Гидринданоны (Гидриндоны) 105
Гидринден (Индан) 466 Гидрирование ароматических кислот 79, 288 бензола и его производных 45 бифенила 91, 95 диарилметанов 88 дигидроизохинолина 621 нафталина 106 нафтиламинов 459, 460 пиррола и его производных 534—537 Сильвана 522 трополона 497 фенолов 265 фурана 521, 523
Гидроароматические алкоголи 460
Гидроароматические соединения 13, 106, 119, 206
Гидробензоин 433
Гидрогенизация, см. Гидрирование Гидрогенолиз 53, 72, 692 Гидрогидрастин 669
Г идрокоричная ф-фенилпропионовая) кислота 287 производные 391
Гидрокоричный альдегид 277
Гидрокортизон (17-Оксикортикостерон) 193
Гидрокотарнин 669 Гидролиз белков 391 древесины 344 Гидроновый синий 563 Гидроперекиси 55, 276 бензоила, см. Бензоил, гидроп ре-кись изопропилбензола 266 тетрагидрофурана 521 циклогексана 75 этилбензола 281
Гидропиридиновые основания 605—608 Гидроурушиол 164 .
Гидрохинин 658—660
Гидрохинон (п-Диоксибензол) 205, 337, 338, 341, 352 эфиры 338, 392
Гидроцинхонин 658, 659
Гинзберга реакция на а-ди кетоны 362 а- и ^-Гиодезоксихолевые (За,6^- и
Зр, 6а-диоксихолановые) кислоты 182 Гиосциамин 410, 652—654 Гиосцин (Скополамин) 653 Гипоксантин (6-Оксипурин) 635, 637, 638
Гиппуровая кислота, см. Бензоилглико-коль
Гипсохромное смещение (Повышение цвета) 368
Гистазарин 476
Гистамин (Р-Имидазолилэтиламин) 570
Гистидин (р-Имидазолилаланин) 569, 570
Гитоксигенин 184
Г-кислота (G-кислота, 2-Нафтол-6,8-дисульфокислота) 457
Гликоген 193
Гликоколь 637
Гликоли
ангидриды 575 высшие 85, 86 циклические 50, 291 Гликохолевая кислота 180 Глиоксалин, см. Имидазол Глиоксаль 75, 205, 220, 228, 568 Глиоксиловая кислота, этиловый эфир 681
Глифталевые смолы 379
Глицерин 612, 616, 626 трибензоат 289
Глобин 539
Глутаконовый альдегид 596
Глутаминовая кислота 714
Глутаровая кислота 173 диэтиловый эфир 43 получение 55, 72 производные 82, 141
Глюкоза 387, 554, 561, 582, 626
а-форма 569, 688
у-форма (Фураноза) 521
Глюкозиды 411, 584, 687, 688, 708 сердечные 182—184
Глюкокортикостероиды 192, 193
Гоматропин 654
Гомберг, исследования свободных радикалов 436, 437
Гомолки основание 427
Гомомерохинен 663
Гомопилоповая кислота 686, 687
Гомопирокатехин (3,4-Диокситолуол) 339
Гомофталевая кислота, имид 620
Горденин 647
Гормоны синтетические 439 стероидные 165, 178, 184, 187, 188— 197
Горчичное фениловое масло 302, 308
Горькоминдальное масло, см. Бензойный альдегид
Гофман А., реакция исчерпывающего метилирования 69, 389, 536, 601, 607, 653
Гофман К., реакция на бензол 225
Грамицидин С 698
Графит 224
Г ребе 206
окисление производных нафталина 446
открытие акридина 621
промышленный синтез ализарина 559
Г ребе связь окраски с ненасыщенностью вещества 369 строение ализарина 467 формула антрацена 467 — нафталина 446, 447
Гриньяра реакция 95, 484
Гуайол 160
Гуанидин 638
Гуанин (2-Амино-6-оксипурин) 514, 635 638, 640, 687
Гуммигут и Гуммилак 161
Гумулен (а-Кариофиллен, 1,5,5,8-Тет-раметилциклоундекатриен-3,7,11) 158
Гумулон (а-Лупулиновая кислота) 360 Г уставсон комплексы с хлористым алюминием 215 получение алициклических соединений 37 превращения пентаэритрита 51, 52
Даммар 161
Дандзеин (7,4'-Диоксиизофлавон) 585 Двухъядерные (бициклические) соединения алициклические 12, 16, 88—112 — номенклатура 16—20 — стереоизомерия 91—93 ароматические 413—418, 445—466 гетероциклические 524, 547—562, 567, 572, 574, 583, 609, 611 — 621, 627, 633, 634—641
ДДТ (2,2-[4,4'-Дихлордифенил]-1,1,1-трихлорэтан) 434, 435
Дебнера—Миллера хинальдиновый синтез 302, 612, 613
Дегелин 589
Дегидрацетовая кислота 581—583 Дегидрирование (Дегидрогенизация) 61, 163, 181, 222, 504 декалинов 106 дигидроизохинолина 621 сесквитерпенов 158 спиранов 97 углеводородов нефти 61 циклогексана и его гомологов 57, 62, 75, 222, 225 этилбензола 401
3,4-Дегидроаксерофтол (Витамин А2) 706 Дегидродиконифериловый спирт 346, 347
Дегидронорхолан 181 Дегидроротенон 589 Дегидрострихнинон 683 7-Дегидро холестерин 707 Дегидроциклизация, см. Ароматизация парафинов
Дегидроэпиандростерон 189—191
Дезозамин 700
Дезоксикортикостерон 192
Дезоксихолевая (За, 12а-диоксихолано-вая) кислота 182, 489
Деионизированная вода 405
Дейтерообмен в холевой кислоте 182
Дейтеропорфирин 542
Декалин (Декагидронафталин, Бицик-ло-[0,4,4]-декан) 99, 104, 106, 452 стереохимия 101, 102 физические свойства 106
Декалолы 102—104
Декалоны 102—104, 106
Декандикарбоновая кислота 41
Декапептид циклический 698
Декафенилбутан 441
Декстропимаровая кислота 163
Дельфинидин 586
Демьянов, превращения циклов 47, 48
Депсиды (Полидепсиды) 382, 386
Дерматол 385
Деррисовая кислота 589
Десмотропосантонин 107
Дестиобензилпенициллин 692
1,2- Диазин (Пиридазин) 625, 632, 633
1,3- Диазин, см. Пиримидин Диазоаминобензол 323
О-Диазоацетил-/-серин (Азасерин) 701
Диазобензол 457
Диазобензолсульфокислота 324
Диазогидраты 320
Диазогидриды 325 6-Диазо-5-оксо-/-норлейцин 701 Диазометан 50, 83, 499, 687 Диазометод . 336 а-Диазонафталин 457
Диазониевые основания 319—321, 325, 331
Диазореакции 321—323, 400
аминопиразолов 565 аминопиридинов 601 антраниловой кислоты 562 дифениламина 454
Диазосоединения антибиотики 701 ароматические 302, 318—324, 331, 414, 450, 458,. 459, 601 бисдиазосоединения 450, 459 жирного ряда 38, 318, 564 реакции без выделения азота 323, 324 — с выделением азота, см. Диазореакции
Диазосоставляющая 365
Диазосульфонат 320
Диазотаты 320
Диазотирование 302, 319
Диазоуксусный эфир 104, 219, 352.,
564
Диазоцианид 320
Диазоциклопентадиен 493
Диалкилсульфаты 245
Диамагнитные свойства 444 Диаминоакридин 622
хлорметилат, см. Трипафлавин Диаминобензойные кислоты 399 Диаминодигидроакридин 622 Диаминодиоксиарсенобензол (Сальварсан) 328, 397
о,о'-Диамино-о-дитолил 416
о,и'-Диаминодифенил (Дифенилин) 317, 414
м,и'-Диаминодифенил, см. Бензидин Диаминодифевилметан 429 п,и'-Диаминодифенилсульфон 312 Диаминокислоты 378, 712 а,у-Диаминомасляная кислота 699 1,5-Диаминонафталин 450 Диаминонафтолы 461 а,а'-Диаминопиридин 597 о-Дианизидин 414 Дианилинохинон 355 Диантрацен 466 Ди ар и л метаны 88
Диатретин-2 (Диатретиннитрил) 702 Диацетил 228
Ди ацетил ацетон 581 а,а'-Диацетилметилпиррол 532 Диацетилморфин (Героин) 673 Дибензальацетон 407, 579 1,2,5,6-Дибензантрацен 489 2,3,6,7-Дибензантрацен (Пентацен) 485 Дибензантрон, см. Виол антрон Дибензил (Дифенилэтан симметрический) 420, 432, 481
Дибензиламин 307 Дибензилиденциклогексанон 78 Дибензилртуть 334
Ы,П'-Дибензилэтилендиамин 691 Дибензоил, см. Бензил Дибензооксазин (Феноксазин) 513, 628, 629
Дибензопиразин (Феназин) 513, 628, 629 Дибензопиридин (Акридин ) 513, 621 — 624
Дибензо-у-пирон (Ксантон) 364, 382, 587, 588
Дибензопиррол, см. Карбазол Дибензотиазин (Фентиазин) 513, 628, 629
1,2,7,8-Дибензофенантрен (Пицен) 181, 486
Дибензофульвен 424
Дибензофуран (Дифенилен, окись) 524 Дибифениленэтилен 436 Дибромантраниловая кислота 560 Дибромбензолы 243, 399 а,&-Дибромвалериановая кислота 537 9,10-Дибромдигидроантрацен 469 6,6'-Диброминдиго («Античный пурпур») 560
Дибромкамфан (Пинен, дибромид) 15!
1,5-Дибромпентан 610
1,3-Дибромпропан 511
Дибромтрополон 498
2,7-Дибромтропон 495
1,4- Дибромциклогексан 208
Дибромэтан 626
Дивинил 402, 403, 536
Дивинилацетиленовые углеводороды 43
Дивинилбензол 404, 406
Дивинилметан 607
л/-Дигалловая (л/-галлоилгалловая) кислота 387
9,10-Дигидроакридин 623 кетопроизводные (Акридоны) 622, 623
Дигидроантразин, дихинон, см. Индан-трен
Дигидроантрацен 468, 469
Дигидробензол (Циклогексадиен-1,3) 36,
76, 208
Дигидробензопиран (Хроман) 583, 584
Дигидродиметилпиразин 625
Дигидроизохинолин, производные 62!
Дигидроимидазолы (Имидазолины) 512,
570
Дигидроиндантрен 627
Дигидрокарвон 132
Дигидрокодеин 673
Дигидрокодеинон 673, 675, 676
Дигйдроколлидиндикарбоновый эфир 593
Дигидромирцен 117
Дигидронафталины 451
Дигидрооксазолы (Оксазолины) 512
Ди гидроокси кодеинон (Эукодал, Текодин) 676
Дигидропиразины 625
Дигидропиразолы (Пиразолины) 512. 563—566
Дигидропиретрон 74
Дигидропиридины 593, 597
Дигидропирролы (Пирролины) 511, 529, 534
Дигидрорезорцин 349
Дигидротиазолы (Тиазолины) 512, 572
Дигидротиофены 511
Дигидрофенарсазин, производные 632
Дигидрофталевые кислоты 206
Дигидрофуран 511
производные 521
Дигидрохинолин 612
Дигидроцибетон (Циклогептадеканон)
86, 87
Дигидроэвгенол 346
22-Дигидроэргостерин 707
Дигидроэрготамин и Дигидроэрготоксин 686
Дигитогенин 187
Дигитоксигенин (Нерийфолигенин, Те-ветигенин) 184
Дигитониды 172, 187, 687
Дигитонин 187, 188
Дигитониновый комплекс 177
Дигоксигенин 184
Ди-п-диметиламинобензофенон (Кетон
Михлера) 422, 423, 429
Диеновый синтез 45, 97, 108, 354, 499
Дикетокамфарная кислота, эфир 141
Ди кетоны 41, 44, 529, 627
Дикетопиперазины (а-Аминокислоты, циклические ангидриды) 513, 626, 685
2,5-Дикетопирролидин (Сукцинимид) 530, 537
2,5-Дикстопирролин (Диоксипирролин, Малеиновая кислота, имид) 531 ’Р,(3'-Ди кетоциклогександи карбоновая
кислота, эфир, см. Сукцинилянтарный эфир
Дикмана реакция 41, 96, 656
Диксантилмочевина 588
Дильдрин 111
Дильса углеводород (3'-Метил-1,2-цик-лопентенофенантрен) 173, 174, 181, 688
Дильса—Альдера реакция 45, 97, 108, 109, 354, 499
Димедон (5,5-Диметил-1,3-дикетоцик-логексан, 5,5-Диметилциклогексан-1,3-дион) 78
Ди мезитил кетон 297
Димеризация 44, 46
Диметиладипиновые кислоты 82
Диметилаллоксан 638
Диметиламин 681
л-Диметиламиноазобензол 373
Диметиламиноантипирин (Пирамидон) 567
п-Диметиламинобензальдегид 396, 459
М,Й-Диметиламинобензол, см. Диме-тиланилин*
Диметиламиногексоза 700
6- Диметиламино-4,4-дифенилгептан-3-он 677
2,5-Диметил-4-аминопиримидин, производное 710
Диметиламиноэтиловый алкоголь 674
Диметиланилин (М,М-Диметиламино-бензол) 299, 301, 306, 308, 366 идентификация 253 реакции 310, 396, 422, 428, 631
1,1-Диметил-4-ацето-«-валериановая (героновая) кислота, 82, 89, 704
3,3-Диметил-4-ацето-«-валериановая (изогероновая) кислота 82, 89
2,4-Диметил-3-ацетопиррол-5-карбо-новая кислота, эфир 540
Диметилбензойные (ксилиловые) кислоты 286
Диметилбензолы, см. Ксилолы 2,3-Диметилбутан 57 1,3-Диметил-5-трет-бутил-2-ацетил-4,6-динитробензол (Кетонный мускус) 253
1,3-Диметил-5-трет-бутилбензол 253
Диметилглутаровая кислота 82 эфир 141
5,5-Диметил-1,3-дикетоциклогексан, см. Димедон
Диметилди кетоциклопентандикарбоновая кислота, эфир 141
Диметилдифенилмочевина 306 3,3'-Диметилдициклопентил 90
1,6-Диметил-4-изопропилнафталин (Кадалин) 159, 453
Диметилкадмий 178
Диметил кетен 44 1,3-Диметилксантин (Теофиллин) 514, 635, 638, 640, 687
3,7-Диметилксантин (Теобромин) 514,
569, 635, 638, 640, 687
Диметилмочевая кислота 636
Диметилнафталины 448, 453
2,5-Диметилпиразин 625, 626
Диметилпировиноградная кислота
686
Диметилпиридины (Лутидины) 591, 594, 605
Диметилпирон 580, 583
2,4-Диметилпиррол-3,5-ди карбоновая кислота, эфир 530
Диметилсульфат 264, 556, 639
2,4-Диметилтиазол 571
Диметилтиофены (Тиоксены) 528
Диметил-п-толуидин 253 1,7-Диметилфенантрен (Пимантрен) 163 Диметил-п-фенилендиамин (п-Амино-диметиланилин) 363, 630
Диметил-п-фенилендиаминтиосульфо-кислота 631
Диметилфенилкарбинол (Фенилизопропиловый спирт) 275, 403
Диметилфуран 519 а,у-Диметилхинолин *613 1,1-Диметилциклобутан-2,4-дикарбоновая (норпиновая) кислота 136
Диметилциклобутаны 69
5,5-Диметилциклогексан-1,3-дион, см.
Димедон
Диметилциклогексаны 34, 35, 54, 76
Диметилциклопентаны 35, 71
Диметилциклопропан 66, 67
1,1- Диметилциклопропан-2,3-дикарбоновая (кароновая) кислота 133
'^-р,[3-Диметилцистеин (Пеницилламин) 691
1,4-Диметил-З-этилбензол 211
2,3-Диметил-4-этилпиррол (Гемопиррол) 540, 541, 545
2,4-Диметил-З-этилпиррол (Криптопиррол) 540, 541, 545
1,2- Диметоксибензол (Вератрол) 338
1,3- Диметоксибензол (Резорцин, диметиловый эфир) 338
1,1-Диметоксибензол (Гидрохинон, диметиловый эфир) 338
Диметоксиизохинолин 667 3,4-Диметокси-2-нитрофени л уксусная кислота 671
Диметоксифлороглюциновый альдегид 587
Димрот, реакция меркурирования 344, 527
9,10-Динатрийдигидроантрацен 467 Динатрийдифенилбутен 335 Динатрийдифенилэтан 335 Динатрийтетрафенилэтан 335 Динатрийэтантетракарбоновый эфир 448
Ди-а-нафтилтетрафенилэтан 444 Диникотиновая кислота 604 Динитрилы, конденсация 42 Динитробензойные кислоты 294, 500 Динитробензолаты 253
Динитробензолы 248, 251, 253, 309, 398, 400
Динитронафталины 454, 456, 462
Динитро-а-нафтол (2,4-Динитронафтол) 456, 457
2,4-Динитро-1-нафтол-7-сульфокисло-та (Флавиановая кислота) 458, 645
Ди нитротолуол 249, 297 лу-Динитротрополон 500 2,4-Динитрофенилгидразин 326, 691 Динитрофенолы 265, 271, 400
Динитрохлорбензолы 255, 272, 326, 596
1,4- Диоксан 514
Диоксандибромид 520
1,4-Диоксиантрахинон (Хинизарин) 477 3,4-Диоксибензойная кислота, см. Протокатеховая кислота
3,4-Диоксибензойный (протокатеховый) альдегид 345, 394, 395
н-Диоксибензол, см. Гидрохинон о-Диоксибензол, см. Пирокатехин лг-Диоксибензол, см. Резорцин Диоксибензолы 260, см. также Гидрохинон, Пирокатехин и Резорцин
Диоксибифенилы (Дифенолы) 415, 524
1,3-Диокси-4-«-гексилбензол («-Гексил -резорцин) 342
Диоксидигидрорубрен 486
Диоксидиморфин (Псевдоморфин) 672 7,4'-Диоксиизофлавон (Дандзеин) 585
3,4-Диоксикоричная (кофейная) кислота 412
4,6-Диоксиксантон (Эйксантон) 588 1,4-Диоксинафталин (Нафтогидрохинон) 461
2,6-Диоксинафталин (амфи-Диокси нафталин) 462
Диоксинафталин(ы) 450, 457, 462, 485
1,8-Диоксинафталиндисульфокислота (Хромотроповая кислота) 457, 458
5,8-Диокси-а-нафтахинон (Нафтазарин) 462
9,11-Диоксинафтацен-10,12-хинон 485, 486
Диоксиндол (о-Аминоминдальная кислота, лактам) 553, 555, 557
1,2-Диокси-З-нитроантрахинон (Ализарин оранжевый) 477
2,6-Диоксипиримидин 634
Диоксипирролин, см. 2,5-Дикетопир-ролин
2,6-Диоксипурин (Ксантин) 569, 635, 637, 640, 687
4,4'-Диоксистильбен (Стильбэстрол) 197, 433
4,6-Диокси-о-толуиловая (орселлиновая) кислота 340
метиловый эфир (Эверниновая кислота) 384
3, 4-Диокситолуол (Гомопирокатехин) 339
3,5-Диокситолуол (Орсин) 338, 340, 384
Диоксифенантрен, монометиловый эфир (Морфол) 482, 674
Диоксифенилаланин (ДОП) 392
Диоксифуксон (Аурин) 427, 429, 430
За,7а-Диоксихолановая (антроподезок-сихолевая, галлодезоксихолевая, хенодезоксихолевая) кислота 182
За, 12а-Диоксихолановая (дезоксихо-левая) кислота 182, 489
Зос,73-Диоксихолановая (урсодезокси-холевая) кислота 182
За,6^- и Зр,6а-Диоксихолановые (а- и р-гиодезоксихолевые) кислоты 182
2,6-Диокси-8-хлорпурин (8-Хлор ксантин) 637
Дионин (Морфин, этиловый эфир) 665, 670
n-Диорселлиновая (леканоровая) кислота 386
Дипентен, см. Лимонен
Дипентендигидробромиды 129
Дипентендигидрохлорид 133
Дипентентетрабромид 126
Дипиколиновая кислота 604
Дипиридилы 609, 610, 651
Дипольные моменты азулена 504
Дипольные моменты
ароматических соединений 234—238 бензола 229
двузамещенных соединений алициклического ряда 35
1,2-дихлорциклогексана 33
производных метана 236
Диспропорционирование водорода 58
Дисульфан 311
Дитерпены 117, 154
о-Дитолил 481
Дитиокарбаминовая кислота 302
Диуретин 640
Дифенил, см. Бифенил
Дифенилазот 326, 439
окись 440
Дифениламин 299, 301, 306, 326, 563, 622, 631
Дифениламиновый оранжевый 307
Дифениларсин 330
окись 329, 331
Дифенил арсиновые (дифенилмышьяко-вые) кислоты 330, 331
Дифениларсинтрихлорид (Дифенилтри-хлорарсин, Дифенилмышьяк треххлористый) 330
Дифениларсинхлорид (Дифенилхлор-арсин) 329, 331
1,4-Дифенилбензол (Терфенил) 224, 418, 419
Дифенилбензолы 224, 418
п,п'-Дифенилбифенил (Кватерфенил) 224, 419
Дифенилбифенилметил 443
Дифенилбромметан 422
Дифенилбутен, производное 164
Дифенилгидразин 326
Дифенилгидроксиламин 314, 440
Дифенилгликолевая (бензиловая) кислота 434
Дифенилдибифенилэтан 443, 444
Дифенил-о,о'-дикарбоновая кислота, см.
Дифеновая кислота
Дифенилендиарсин 632
Дифенилен, окись (Дибензофуран) 524
Дифениленимид, см. Карбазол
Дифенилин (о,п'-Диаминодифенил) 317, 414
Дифенилиновая перегруппировка 317
Дифенилиодоний 246
Дифенилкарбинол (Бензгидрол) 422
Дифенилкетон (Бензофенон) 282, 422,
435, 439
Дифенилметан 305, 420—422
Дифенилметилбромид 437
Дифенилметильный (бензгидрильный) радикал 441
Дифенилмочевина (Карбанилид) 308
Дифенилмышьяк треххлористый, см.
Дифениларсинтрихлорид
Дифенил мышьяковые (дифениларси новые) кислоты 330, 331
Дифенилнитрозамин 326
Дифениловый (фениловый) эфир 267
Дифенилпиперазин 626
Дифенилртуть 333, 335
Дифенилстибиновая кислота 332
Дифенилстибинхлорид (Дифенил хлор-стибин) 332
Дифенилстибинтрихлорид 332
Дифенил сульфон (Сульфобензид) 260
Дифенилтиомочевина 302, 308, 573
Дифенилтрихлорарсин, см. Дифениларсинтрихлорид
Дифенилфосфин 328
Дифенилфосфиновая кислота 328
Дифенилфосфинхлорид (Дифенилхлор-фосфин) 328
Дифенилхлорарсин (Дифениларсин-хлорид) 329, 331
Дифенилхлорметан 435
Дифенил хлорстибин (Дифенилстибинхлорид) 332
Дифенилхлорфосфин (Дифенилфосфин-хлорид) 328
Дифенилцианарсин 331
Дифенилциклопропан 420
Дифенилэндоанилодигидротриазол-1,2,4 (Нитрон) 573
Дифенилэтан несимметрический (1,1-Ди-фенилэтан, а,а-Дифенилэтан) 434
Дифенилэтан симметрический (Дибеи-зил) 420, 432, 481
Дифенилэтилен симметрический (Стильбен) 316, 432, 481, 528
Дифеновая (дифенил-о,о'-дикарбоно-вая) кислота 415, 416, 481 стереохимия производных 416
Дифенолы (Диоксибифенилы) 415,
524
Дифенохинон (Бифенохинон) 415
Дихлорацетилирование 696
Дихлорбензолы 247, 362
2,5-Дихлоргидрохинон 353
9,10-Дихлордигидроантрацен 469
Дихлордифенилдихлорэтилен 435
2,2-(4,4'-Дихлордифенил)-1,1,1-трихлор-этан, см. ДДТ
Дихлорнафталин 454
1,3-Дихлорпропан 67
2,6-Дихлорпурин 637
2,4-Дихлорфенокси-f-масляная кислота 264
2,4-Дихлорфеноксиуксусная кислота 264
Дихлорфлуорен 436
Дихлорхиноны 354 3,4-Дихлорциклобутен 84 Дихлорциклогексаны 32 Дихлорэтан 432
Дициклогексил 90, 91, 95
Дициклогексил-2,2'-дикарбоновая (пер-гидродифеновая) кислота 92—94
Дициклогексилдиол-4,4' 91
Дициклогексилметаны 88
Дициклопентадиен (Циклопентадиен, димер) 63
Дициклопентил 90, 94
Дициклопентилдиол-1,1' 91
Диэтил амин 306
1-Диэтиламино-4-аминопентан 624
м-Диэтил аминофенол 432
Л и э т и л а н и л и и (N, N - Д и э т и л а м и н о б е н -зол) 302, 306, 308
Диэтилендиимип (Пиперазин) 513, 515,
626
а,(3-Диэтилстильбэстрол 433
Диэтоксихлорпурин 637
Долофин 678
ДОП, см. Диоксифенилаланин
Драгендорфа реактив 645
Драконова кровь 161
Дубильные вещества 385—388, 587, 643
синтетические 274
Дульцин 365
Дуотал (Гваякол, угольный эфир) 342
Дурогидрохинон (Тетраметил гидрохинон) 708
Дурол (1,2,4,5-Тетраметилбензол) 216
Дьюара формула бензола 227, 229
Енолы 284, 407, 519
Жасмина масло 73, 548
Жасмон (3-Метил-2-пент-2'-енилцикло-пентен-2-он-1) 73, 74
Желатина 538
Желчные кислоты 172, 179—182
Живицы (Терпентины) 115, 162
Жиры 169, 180, 645
Залковского реакция на холестерин 171 Замещение
в бензольном ядре 215, 218, 238 — 243
в ядре нафталина 450, 451
Зандмейера реакция 322
Зеаксантин 90
Зеленое (антраценовое) масло 221, 466,
621
Зелинский
гидрирование пиридина 598
— фурана 521
гидрогенолиз циклопентановых уг-
леводородов 54, 72
изомеризация углеводородов 47, 51, 54
исследования состава нефтей 61
каталитическое дегидрирование цик-
логексана 57, 75, 222, 225
Зелинский
необратимый катализ 58, 231
получение бензола из ацетилена 203
— спирановых углеводородов 91
Зинин
получение азоксибензола 315
— анилина 297
— бензидина 318
— гидразобензола 318
Зоненшейна реактив 645
Зоостерины 170
Иервин 688
Изатин 527, 531, 548, 553, 555—558, 619
в красителях 561—563
Изатинанилы 557
Изатиновая (о-аминофенил глиоксило-
вая) кислота 619
соль 556
Изатиноксим 554, 557
Изатинхлорид 557, 559
Изатовая кислота 558
Изатогеновая кислота 555
Изоалантолактон 107
Изоаллоксазин 634, 704
Изобензамид 291
Изобензо-а-пирон, см. Изокумарин
Изоборнеол 143—148
Изобор нил ацетат 148
Изоборнилксантогенат 146
Изоборнилхлорид 145
Изобутилбензол 217
Изобутилен 44, 341, 403
димеризация 44
Изованилин 665
Изовиолантрон (Изодибензантрон, Ин-
дантрен фиолетовый) 488
Изогексилмагнийбромид 178
Изогемипиновая кислота 345
Изогероновая (3,3-диметил-4-ацето-
н-валериановая) кислота 82, 89
Изогидробензоин 433
Изогомопилоповая кислота 687
Изодибензантрон (Изовиолантрон, Ин-
дантрен фиолетовый) 488
Изодурол (1,2,3,5-Тетраметилбензол)
216
Изоиервин 688
Изокамфановая кислота 147
Изокодеин 673
Изокоричные кислоты 408, 409
Изокумарин (Изобензо-а-пирон,
2,1-Бензопирон) 578
Изолизергиновая кислота 685
Изомасляная кислота 141
Изоментол 121, 122
Изомеризация
борнил- и изоборнилгалогенидов 146
двухатомных фенолов 270, 337
изострихнина 684
Изомеризация кодеина 673, 684 окиси этилена 509 пиррола 533 третичных спиртов циклобутанового ряда 47 хинина 661 циклических углеводородов 47, 51, 54
Изомерия азоксисоединений 315 азосоединений 316 алициклических соединений 27-—37 оптическая 416, 464 пенициллинов 692 производных бензола 199—203 пространственная, см. Стереоизомерия
Изомеры оптические 28, 34, 92, 97, 410, 416, 464 поворотные 23
Изоморфизм 224
Изоникотиновая (у-пиридинкарбоно-вая) кислота 603—605
Изонитрозоацетанилид 556
Изонитрозоацетон 625
Изонитрозоиндоленин 550
Изонитрозокамфора 143 Изонитросоединения 254 Изопенилловая кислота 692 Изопилокарпин 686, 687 а-Изопилоповая кислота 687 Изопрен (Гемитерпен, Семитерпен) 154—156
Изопреноидная цепь 703 Изопропенилциклопропан 56 Изопропилат калия 213 н-Изопропилбензойная кислота 500 n-Изопропилбензойный альдегид, см.
Куминовый альдегид
Изопропилбензол (Кумол) 217, 224, 226 а,р,у-Изопропилтрополон (а,р,у-Туя-плицин) 500, 501
Изопропилциклогексан 32 Изопропилциклопропан 57 Изопурпурин, см. Антрапурпурин Изопурпуровокислый калий 274 Изосапогенины 186
Изосафрол (3,4-Метилендиоксипропе-нилбензол) 339, 340
Изострихнин 684
Изострофантидин 183
Изсфлавон (р-Фенилхромон) 584 производные 585
Изофталевая (л1-фталевая) кислота 206, 220, 375, 379, 399
Изохинолин (р, у-Бензопиридин) -513, 578, 611, 620, 621, 677 в алкалоидах 621, 665—678
Изоцингиберен 158, 159
Изоцинхомероновая кислота 604
Изоэвгенол (З-Метокси-4-оксипропени, i -бензол) 339, 340, .395
Ильинский
«деление» валентности 370
комплексные соединения нитрозо-нафтолов с металлами 461
сульфирование антрахинона 472
Имбирное масло 125, 147, 157
Имидазол (Глиоксалин) 512, 517, 568^
569
кольцо 362, 635, 637
производные 570, 571, 602, 686, 687
Имидазолидины (Тетрагидроимйдазолы) 512
Р-Имидазолилаланин (Гистидин) 569,570
Р-Имидазолилпропионовая кислота 570
Имидазолилуксусная кислота 570
Р-Имидазолилэтиламин (Гистамин) 570
Имидазолины (Дигидроимидазолы) 512, 570
Имидазолпиримидин, см. Пурин
Иминодипропионовый альдегид 606
Индазол (Бензпиразол) 567, 568
Индамин (Толуиленовый голубой) 629
Индамины 356
Индан (Гидринден) 466
Индантрен (Дихинон дигидроантразп на) 477, 478, 627
темносиний, см. Виолантрон фиолетовый, см. Изовиолантрон 488
Инден 105, 466
Индиго 206, 297, 373, 377, 390, 410.
547, 548, 551, 558—562
белое 561
красное (Индирубин) 555, 558, 562
синее (Индиготин 558—561
Индигоидная группировка 561, 562
Индигоидные красители 373, 558—562
Индигокармин 561
Индиготин (Синее индиго) 558—561
6,6'-Индиготиндисульфокислота 560
Индикан мочи (Индоксилсерная кисло -та) 553, 554
Индирубин (Красное индиго) 555, 558, 562
Индогениды 555, 562
Индоксил 548, 551, 553—555, 559, 562
Индоксиловая кислота 559
эфир 554
Индоксилсерная кислота (Индикан мочи) 553, 554
Индол (Бензпиррол) 513, 547—553, 686 в алкалоидах 678—686 расширение пятичленного кольпа 550, 614
Индол-р-альдегид 551, 552
Индоленин 550, 551, 681 p-Индолил-а-аминопропионовая кисло-
та (Триптофан) 552, 553
?>-Индолилуксусная кислота (Гетероауксин) 74, 552
р-Индолилэтиламин 678
Индол ины 551
а-Индолкарбоновая кислота 549
Индофен ин 558
Индофениновая реакция 527
Индофенолы 356
Индулины 629
Индулиновый алый 630
Инозит 64
Инсектициды 72, 588, 650
Инсулин 188
Иоданилины 309
Иодбензойные кислоты 293, 294
Иодбензол 246, 333
хлористый 246, 293
Иодидхлориды ароматического ряда 246, 293
Иодистый бензил (Бензилиодид, а-Иод-толуол) 244, 247
Иодистый метил, см. Метил иодистый
Иодметилат
пиридина (Иодистый метил пиридиний) 595
у-цианхинолина 616
Иоднафталины 456
Иодобензол 246
Иодозобензойные кислоты 293
Иодозобензол 246
Иодозосоединения 246
Иодол (Тетраиодпиррол) 531
Иодпиридины 601, 603
Р-Иодпропионовый эфир 127
4-Иодтерфенил 419
а-Иодтиофен 528
а-Иодтолуол, см. Бензил иодистый
Иодфенолы 270
Иодциклогексан 47, 76
Иодциклогептан 47
Иодциклопентан 71
Иониты (Ионообменивающие смолы)
404—406, 645
Ионоидное состояние 370
Иононы 80—82, 705
Йохимбин (Иохимбиновая кислота, ме-
тиловый эфир) 679
Ироны 82, 83
Итамалевые кислоты 687
Ихтиолы 528
К адалин (1,6-Диметил-4-изопропил нафталин) 159, 453
Кадинен 155, 158, 159
Кадинол 158
Казанский 69, 72
Гидрогенолиз циклопарафинов 54. 56, 57
Исследование нафтонов 62
Циклизация парафинов 46
Казеин 391, 537
Кайепутовое масло 129
Какодил (Тетрафенилдиарсин) 331
Кальциферолы (Витамины D) 179 703.
706
Каменноугольная смола (Каменноугольный деготь) 221, 261,266,401,413, 424, 445, 453, 466, 480, 486, 489, 524, 526, 528, 548, 563, 591, 611, 615, 620
Каменный уголь, коксование 63, 221, 224, 261
Камфан (Борнилан, 4,7,7-Триметилби* цикло-[ 1,2,2]-гептан) 131, 146 производные 146, 147
Камфановая кислота 141
Камфарная кислота 141, 142
Камфарное масло 139, 147, 157
Камфен 124, 145—151
Камфенгидрат 149
Камфен гл и коль 147
Камфениловая кислота 147
Камфениловый альдегид 147
Камфенилон 147
Камфеновая кислота 147
Камфеновые перегруппировки 148— 150, 153
Камферхинон 143
Камфолид 142
Камфора 115, 139—143, 145, 155 синтез 142
Камфороновая (триметилтрикарбалли-ловая) кислота 141
d-Камфорсульфоновая кислота 696
Канифоль 115, 161, 162
Канницаро реакция 274, 278, 520
Кантаридин 522
Кантаридиновая кислота 522
Канцерогенные (карциногенные) угле водороды 181, 489
Каптакс (2-Меркаптобензтиазол) 572
Каран (4,7,7-Триметилбицикло-[0,1,4] гептан) 98, 131 —133
Карбазол (Дибензопиррол, Дифенилен* имид) 221, 563, 680
Карбанил (Фенилизоцианат) 308
Карбанилид (Дифенилмочевина) 308
Карбодифенилдиимин 573
Карбокситропилийбромид 493
Карболигаза 279
Карболит 267
Карболовая кислота, см. Фенол
Карбомицин (Магнамицин) 700
Карбоний-ион 49, 214, 361
Карбонилирование 334
Карбостирил, см. о-Аминокоричная кислота, ангидрид
Карбоцианиновые красители 617
Карвакрол (2;Метил-5-изопропил-1-оксибензол) ‘127, 142, 263
Карвенон 132
Карвон (Д1,8-п-Ментадиенон-6) 126, 127, 132
Кардамонное масло 126, 129, 157 Карен 98, 116, 126, 131, 133 Кариламин 132 а-Кариофиллен, см. Гумулен Кариофиллен(ы) 159, 600 p-Кариофилленовый спирт 160 Кармин 479
Карминовая кислота 479
Карон 131—133
Кароновая (1, 1-диметилциклопропан-2,3-дикарбоновая) кислсгга 133
Каротиноиды (Липохромы) 88—90, 544, 705
Каротины 81, 89, 180, 703, 705
Карциногенные (канцерогенные) углеводороды 181, 489
Касторовое масло 480
Катализаторы гидрирования и дегидрирования (Платина, Палладий, Никель), 45, 46, 53, 54, 57, 58, 61, 62, 68, 75, 77, 105, 106, 130, 222, 223, 225, 288, 408, 466, 481, 497, 504, 521, 523, 587, 598, 621, 662, 678, 683
Катехины 386, 587, 704, 708
Катеху 340
Катиониты (Катионообменивающие смолы) 404
Каучук
натуральный 154 1
синтетический 224, 226, 402
Квазиаксиальные связи 34
•Кватерфенил (п,п'-Дифенилбифенил) 224, 419
Кверцетин (3,5,7,3',4'-Пентаоксифла-вон) 584, 587, 708
Кверцит (1,4/2,3,5-Циклогексанпентол) 64
Квинквифенил 419
Кекуле, формула бензола 11, 198— . 200, 227—228, 446, 507
Келлин 585
Кермес 479
Кермесовая кислота 479
Кернера правило 398, 399
Кетен, димеризация 44
Кетил, фениловый эфир 439
Кетогексозы 523
Кетоксимы 283
Кетон Михлера (Ди-п-диметиламино-бензофенон) 422, 423, 429
Котонизация двухосновных карбоновых кислот 40
Кетонное расщепление 70 у-Кетонокислоты, эфиры 42 Кетоны
алициклические 40, 50, 53, 58, 63, 86, 87, 90
Кетоны ароматические 280—282, 407 жирные 55, 203 непредельные 43, 407, 564
20-Кетопрегнан 187
17-Кетостероиды 190
12-Кетохолановая кислота 181
3-Кетоэтиоаллохолановая кислота, эфир 177, 178
З-Кето-Д4’9^11)’16’ этиохолатриеновая кислота 177
Киафенин 292
Кижнер
взаимные превращения алициклических соединений 47
восстановление бензола иодистоводородной кислотой и фосфором 45, 225
выделение л«-ксилола 226 получение фенхона 152 применение хлоробромидов в синтезе алициклов 39
размыкание пятичленного кольца 53 реакция 38, 94, 131, 541
Кину реновая (у-оксихинолин-р-карбо-новая) кислота 553, 618
Кинурин (у-Оксихинолин) 618 а-Кислота, см. Кроцеиновая кислота р-Кислота, см. Кислота Шеффера Кислота Ганнсена 680
Кислота Невиля—Винтера (1-Нафтол-4-сульфокислота) 457
Кислота Шеффера (р-Кислота, 2-Наф-тол-6-сульфокислота) 457
Кислотные красители 372, 428 бордо 457
желтый (Прочный желтый) 366
оранжевый светопрочный 457
Кислоты алициклические 38—42, 70 ароматические многоосновные 375— 380
— ненасыщенные 407—410
— одноосновные 286—295
высшие двухосновные 86
Кладиноза 700
Класона способ гидролиза древесины 344
Клауса формула бензола 227, 228
Клеменсена реакция 211
Кобаламины 704, 714
Кодеин (Морфин, метиловый эфир) 665, 670, 673, 676, 684
Кодеинон 673, 674, 676
Койевая кислота (а-Метилол-р'-окси- .
у-пирон) 582
Кокаин (Бензоилэкгонин, метиловый эфир) 408, 528, 538, 652, 654—657
Коксование каменного угля 63, 221» 224, 261
Коламин (Этаноламин) 378, 510
0-Коллидин (4-Метил-З-этил пиридин) 592, 594, 659, 680
Коллидин симметрический (2,4,6-Три-метилпиридин) 580, 593, 594
Коллидиндикарбоновый эфир 593
Коллидины 591—594
Колхицин 502, 647,
Кольбе
способ получения оксикислот 381 электролиз циклогексанкарбоновой кислоты 95
Кольбе—Шмидта реакция 381, 532
Комановая кислота 582
Комбинационное рассеяние света 62
Коменовая кислота 582
Конваллатоксигенин (Строфантидин) 183, 184
Конгидрин 648
Конго красный 459
Конденсация 42—44, 108, 176, 278, 376, 421, 477, 591, 593 ацилоиновая 42 Дикмана 41, 96, 656 Кнорра 530, 540 кротоновая 91, 175—177, 612 Перкина 482, 552 сложноэфирная 682
Конденсированные многоядерные соединения 12, 16, 18, 97—114, 413, 652—688 .
стереоизомерия 98—101, 109, 112
Кониины (Прогшлпиперидины) 608, 609, 648
Конирин (а-Пропилпиридин) 594
Конифериловый альдегид 347
Конифериловый спирт (З-Метокси-4,13-диоксипропенилбензол) 343, 346, 347
Кониферин 343, 346
Коницеии 648
Коновалов бромирование циклогексана 58 нитрование алициклических углеводородов 55, 72
— алкилбензолов в боковой цепи 254
Константы диссоциации имидазола 569 нафтойных кислот 463 пикриновой кислоты 272 пиразола 569 фенола 235
Конформации гексахлорциклогексана 219 гидриндана 100 декалина 101 инозитов 64 кверцита 64 ментола 121
Конформации
пенталана 99
циклогексана 29—34
циклопентана 26, 29
Конформационный анализ 24—26 29—
34
Конъюгация 206, 371—373
Копалы 161
Копеллидины 605
Копростан 166, 181
Копростанол (Копростерин) 170
Коразол (Пентаметилентетразол) 574
Корамин (Никотиновая кислота, ди-
этил амид) 604
Кориандровое масло 126
Коричная кислота 70, 274, 401, 407,
408, 409, 655
о-изодиазотат 633
Коричное масло 159
цейлонское 125
Коричный алкоголь 406, 408
Коричный альдегид 406, 407
Коронен 487
Кортизон 178, 193, 194
Кортикоидное действие 197 Кортикостероиды 192—195 Кортикостерон 193 Костяное масло 531 Котарнин 665, 668, 669 Котоин 423
Кофеин (1,3,7-Триметилксантин) 514, 569, 635, 637—640, 687
Кофейная (3,4-диоксикорнчная) кислота 412
Кошениль 479
Красители 373—375
азокрасители 312, 323, 362, 365— 367, 383, 387, 390, 414, 457, 601
азоксониевые 629, 630
азтиониевые 629, 630
акридиновые 623
ачизариновые 475, 480
«бесцветные» 375
индигоидные 373, 558—562
карбоцианиновые 617
кислотные 372, 428
кислые 366
кубовые 373, 375, 488, 561—563
основные 366, 372
«пиридиновые» 596
протравные 375, 387, 579, 584
розиндулиновые 630
субстантивные 375, 531
тиоцианиновые 573
трифенилметановые 423, 426—432 хиноидная форма 427, 430, 562 фталоцианиновые 547 цианиновые 617, 618
электронное строение молекул 371
Крашение 374, 375, 480
ализариновое 479, 480
кубовое 373, 375, 561 протравное 375, 387, 579, 584
Крезоксиуксусные кислоты 262
Крезолы (Метилфенолы, Метилоксибензолы) 253, 261, 266, 679
Крезольный (амбровый) мускус 253
Креозол (З-Метокси-4-окситолуол) 339
Креозот 341
Криптопиррол (2,4-Диметил-З-этилпир-рол) 540, 541, 545
Криптопирролкарбоновая кислота 540 Кристаллвиолет (Кристаллический фиолетовый) 429
Кристаллоза 295
Кроконовая кислота 74, 75
Кротоновая кислота 371
Кротоновая конденсация 91, 175—177, 612
Кротоновый альдегид 592, 612
Кроцеиновая кислота (а-Кислота, 2-Нафтол-8-сульфокислота) 457
Ксантгидрол 588
Ксантин (2,6-Диоксипурин) 569, 635, 637, 640, 687
Ксантогенат диазония 322
Ксантогеновая кислота, эфир 322
Ксантон (Дибензо-у-пирон) 364, 382, 587, 588
Ксантопротеиновая реакция 391
Ксантоптерин (2-Амино-4,6-диоксипте-ридин) 641
Ксантотоксин 588
Ксантофилл 90
Ксероформ (Трибромфенол, висмутовая соль) 270
Ксиленолы 261, 263 поликонденсация 268, 269
Ксилидины 296, 298, 301, 305, 713
о-Ксилилендибромид 448
Ксилил 210
Ксилиловые (диметилбензойные) кислоты 286
.м-Ксилилпентаны 210
л/-Ксилилфенилкетон 485
Ксилолы (Диметилбензолы) 210, 212, 216, 221, 226 галоидирование 243 доказательство строения 399, 400 озонирование 228 окисление 286, 376
«-Ксилохинон 352
Кубовое крашение 373, 375, 561—563
Кумалин (а-Пирон) 575—578, 600
Кумалиновая (а-пиронкарбоновая) кислота 577
Кумарин (Бензо-а-пирон, 1,2-Бензо-пирон) 394, 410, 411, 578
Кумариновая (г{«с-о-оксикоричная) кислота 410 лактоны 578
о-Кумаровая (трсшс-о-оксикоричная) кислота 410, 411 лактоны 578
Кумарон (Бензофуран) 513, 524 производные 585, 588
Кумароновая смола 524
Куминовый альдегид («-Изопропилбензойный альдегид, Куминаль) 277, 280, 379
Кумол (Изопропилбензол) 217, 224, 226
Купреин 658
Купферрон (N-Нитрозофенилгидроксил-амин) 314
Кураре алкалоиды 620, 665, 684
Курциус метод 570 строение азидов 292
Кускгигрин 648
Ладенбург доказательство равноценности атомов водорода в бензоле 201 изучение имидазолов 507 — кремнийорганических соединений 213
—оксазолов 507
перегруппировка производных пиридина 593
реакция 598
синтез кониина 608
формула бензола 227, 228
Лаки 375
ализарина 479, 480
р-Лактамы 511, 557, 690, 692
Лактоны 96, 97, 107
кольцо 183, 184, 700
Лактофлавин, см. Рибофлавин Ланолин 171
Лауданозин 667
Лаусон (2-Окси-а-нафтохинон) 463
Лаута краситель 362
Лебедев
диенбвый синтез 46 димеризация аллена 44
Левомицетин, см. Хлоромицетин
Левопимаровая кислота 162
Левулиновая кислота 523, 525
Легаля реакция 183
Лейкокрасители 561
Лейконовая кислота 74, 75
Лейкооснования азокрасителей 366
Лейкоптерин (2-Амино-4,6,7-триокси-птеридин) 641
Лейкосоединения 426 Лейцин 156, 698, 699
Леканоровая (zz-диорселлиновая) кислота 386
Лепидин (4-, или у-Метилхинотин) 613, 614, 617, 618, 659
Лепидон (а-Оксилепидин) 614
Либерман К., 206, 467
исследование ализариновых красителей 480
промышлен. синтез ализарина 559 реакция на нитрозосоединения 303 — на холестерин 171
строение ализарина 476
формула антрацена 467
Лигнин 343—348, 395
Лиддит, см. Пикриновая кислота
Лизергиновая кислота 685, 686
Лизидин 570
Лизол 266
Ликопин 89
Лимонен (Дипентен, Д1,8^-п-Ментадиен) 124—126, 129, 136, 155, 226 нитрозохлорид 127
Лимонентетрабромид 126
Лимонная кислота, соль 643
Лимонное масло 124, 126, 157
Лимонэритрит 126
Линеарные системы 484
Линдан, см. Гексахлоран
Линолевая кислота 703
Линоленовая кислота 703
Липохромы (Каротиноиды) 88—90, 544, 690
Литийалюминийгидрид 174, 193
Литийфенил (Фениллитий) 328, 333
Литохолевая (За-оксихолановая) кислота 182
Лобеланин (Ы-Метил-2,5-ди-[фенацил]-пиперидин) 649
Лобелии 649
Лойпоновая кислота 660
Лумистерин 179
Лумифлавин (6,7,9-Триметилизоаллок-сазин) 713
Л у петидины 605
а-Лупулиновая кислота (Гумулон) 360
Лутеолин (5,7,3',4'-Тетраоксифлавон) 584, 585
Лутидиновая кислота 604
Лутидины (Диметилпиридины) 591, 594, 605
Лутоидное действие 197
Лутоидные гормоны 191
Льюисит 332
Магнамицин (Карбомицин) 700
Магнитные свойства радикалов 444, 445
Магнитный момент 444
Майера реактив 645
.Маклурин (2,4,6,3',4'-Пентаоксибензо-фенон) 385, 386, 423, 584
Макролиды’ 699
Малахитовый зеленый (Тетраметил-л-диаминотрифенилметан) 306, 428
Малеиновая кислота 205, 212, 353, 521 имид, см. 2,5-Дикетопирролин
Малеиновый ангидрид 109, 467, 487, 496, 499, 521
Малеиновый диальдегид 632
Малоновая кислота 411, 637 хлорангидрид 350
Малоновый эфир 38—40, 78, 203, 324, 350, 602
Мальтол (2-Метил-З-оксипирон) 581
Манделина реактив 645
Манчестерский коричневый, см. Основной коричневый
Маркиза реактив 645
Марковна ков 13
исследования нефти 61, 62 правило 214
превращения алициклических углеводородов 47
синтез циклопентана 12
трифенилметильный радикал 437
Марме реактив 645
Марфанил (4-Сульфамидобензиламин) 312
Масла
ализариновое 480 анисовое 406
антраценовое (зеленое) 221, 466, 621 валериановое 143 гвоздичное 159, 340
горькоминдальное, см. Бензойный альдегид
жасмина 73, 548
зеленое, см. Антраценовое масло
имбирное 125, 147, 157 кайепутовое 129 камфарное 139, 147, 157 кардамонное 126, 129, 157 касторовое 480 кориандровое 126 коричное 159 — цейлонское 125 костяное 531
лимонное 124, 126, 157 мирбановое, см. Нитробензол мятное 120, 122, 124, 127 пихтовое 124, 143, 145, 147 полыни 130
померанцевой корки 124
римской ромашки 280, 502
розмариновое 130, 143
розовое 275 сандаловое 160 сассафрасовое 340 сельдерейное 124, 158 сосновых игл 124
терпентинное, см. Скипидар
Масла
тминное 124, 127, 379
укропа 125
фениловое горчичное 302, 308
фиалкового корня 82
хаульмугровое 73
хеноподиевое 131
цейлонское коричное 125
цитронелловое 147
шалфейное 153
эвкалиптовое 125, 130
элеми 125, 126
эфирные'ВЗ, 73,81, 116, 152—154, 161, 274, 280, 287, 340, 393, 406, 503 Медовый камень (Меллитовая кислота, алюминиевая соль) 380
Меервейна—Понндэрфа метод восста-
новления 696
Мезидин 298, 301, 305
Мезитил 210
окись 78
Мезитилен (1,3,5-Триметилбензол симметрический) 203, 210, 216, 399, 400, 418
Мезоантрамин (9-Антрамин) 470, 471
Мезобилирубин 543, 544
Мезоинозит (Гексаоксициклогексан,
1,2,3,5/4,6-Циклогексангексол) 64, 703, 712
гексафосфат (Фитин) 65
Мезоксалевая кислота 75
эфир, гидразон 324
Мезо-(у- или р.-) положение антрацена 469
Мезопорфирин 539
Мезосостояние окрашен, молекул 370
Мейзенгеймер
определение конфигурации оксимов 465
развитие бекмановской перегруппировки 284
разделение на оптические изомеры диаминодитолила 416
—фосфинов 328
реакция 472
Меконин 669
Меконовая кислота 582, 644
Меланины 392
Мелинит, см. Пикриновая кислота
Меллитовая (бензолгексакарбоновая) кислота 379, 380
алюминиевая соль (Медовый камень) 380
Меллофановая (1,2,3,4-бензоптетракар-боновая) кислота 379
Менделеевская замазка 162 Д^^-и-Ментадиен, см. Лимонен Ментадиен(ы) 124—127
производные 128—131
Д^в-ц-Ментадиенон-б (Карвон) 126, 127, 132
Ментан (п-Метилнзопропилциклогек-сан) 119, 120, 129
Ментандиолы (Терпины) 126—130
Ментанол, см. Ментол
Ментен (Д3-Ментен) 124
Ментенолы (Терпинеолы) 126, 129, 130
д4(8)_ Ментенон-3 (Пулегон) 122, 123
Ментилхлорид 123
Ментол (Ментанол, 1-Метил-4-изолро-пил циклогексанол-3) 120—124
Ментон 118, 122, 123
Меркаптаны ароматические (Тиофенолы) 236, 237, 258, 259, 322
2-Меркаптобензтиазол (Каптакс) 572
Меркурирование
бензола и производных 323, 336
лигнина 344
по Димроту 344, 527
тиофена 526, 527
Мерохинен 660
Мескалин 647
Метадон 678
Метакриловая кислота 406
Мета ле пс ия 53
Металлкетилы 439
Металлорганические соединения ароматического ряда 333—336
Метаниловая кислота (ж-Аминобензол-сульфокислота) 311
«Метанный» атом углерода 426, 429.
430, 443
Мета- положение 200
Метастирол 402
Мета циклофаны 490
Метены 541, 542
Метил иодистый 299, 358, 380, 556,
565, 616, 636, 650
Метиламин 530, 653, 656, 657
N-Метил аминобензол (Метиланилин)
176, 299, 301, 306, 308
6-Метил-2-амино-4-оксиптеридин 714
Метиламинопиридин 601
у-Метиламинопропил-р-пиридилкетон
650, 651
Метил-п-аминофенол, соль (Метол) 363
Метиланилин (N-Метил аминобензол)
176, 299, 301, 306, 308
Метиларсепидин 610
Метилацетанилид (Экзальгин) 308
Метилбензамид 291
Метилбензил кетон (Фенил ацетон) 281
Метил бензоат (Бензойнометиловый эфир) 289, 291
Метилбензол, см. Толуол
Метилбензольные (толуиловые) кисло ты 119, 286, 287
М-Метил-/-валин 700, 701
2-Метил-5-винил пиридин 592
Метил виол ет (Основной фиолетовый К, Метиловый фиолетовый) 306, 429 2-Метилгептанон-6 172
Метилглиоксаль 569
Ы-Мети7-а-/-глюкозаминидо-р-/-стреп-тоза (Стрептобиозамин) 694
N-Метилгранатан (Псевдопельтьерин) 83, 657
Метилдиоксиантранол (Хризаробин) 478
5-Метил-2,6-диоксипир имидин 634
N-Метил-2,5-ди-[фенацил]-пиперидин (Лобеланин) 649
9-Метил-1,5-дихлорантрацен 474
Метилен
бромистый 113, 421
иодистый 533
хлористый 421
Метилен-бис-салициловая кислота, соль 619
3,4-Метилендиоксиаллилбензол (Сафрол) 339, 340
3,4-Метилендиоксибензойная кислота, см. Пиперониловая кислота
3,4-Метилендиоксипропенилбензол (Изосафрол) 339, 340
2-Метилен-З-кетоциклопентанкарбоно-вая кислота (Саркомицин) 700
Метиленмалоновая кислота, эфир 70
Метиленовый голубой 362, 374, 630
Метиленциклобутан (Метилентетраметилен) 36, 51, 52
Метилизатин 556, 557
4-Метилизоборнеол 150, 151
Р-Метил-б-изобутирил-н-валериановая кислота 123
,и-Метилизопропилбензол (л«-Цимол) 119, 153
я-Метилизопропилбензол (л-Цимол) 115,
118, 119, 142, 217, 226, 227, 379
4-Метил-1-изопропилбицикло-[0,1,3]-гексан (Туйан) 153, 154
5-Метил-2-изопропил-1 -оксибензол (2-Изопропил-5-метилфенол, Тимол) 122, 263, 267
2-Метил-5-изопропил-1-оксибензол (Карвакрол) 127, 142, 263
я-Метил изопропилциклогексан (Ментан) 119, 120, 129
1-Метил-4-изопропилциклогексанол-3, см. Ментол
1 -Метил-7-изопропилфенантрен (Ретен) 163
а-Метилиндол (Метил кетол) 548, 549, 551 ft-Метилиндол (Скатол) 548, 549, 551 N-Метилиндол 680
Метилирование исчерпывающее, см. реакция Гофмана
Метил камфены 150, 151
Метилкетол (а-Метилиндол) 548, 549, 551
P-Метил кротоновый альдегид 156 8-Метилксантин 639 Метилморфиметины 674, 675 N-Метилморфин 677 N-Метилморфинан 677
Метил нафталины 447, 448, 453
5-Метил- Г,2'-нафта-1,2-флуорен 181
2-Метил-1,4-нафтохинон 707
сульфокислота (Викассл) 707
Метил нитробензоксазол 284
Метилнитропикрамид (Тетрил) 306, 310 Метилнитроциклопентаны 72
Метиловый
красный 367
оранжевый (Бензолазодиметилани-1 лин, л-сульфокислота, Гелиан-тин, Тропеолин D) 368, 366
фиолетовый, см. Метил виолет Метилоксибензолы, см. Крезолы 2-Метил-3-окси-4,5-ди-(оксиметил)-пи-
ридин, см. Пиридоксин 1-Метил-6-окси-7-метокситетрагидро-
изохинолин (Сальсолин) 620, 643, 665
2-Метил-З-окси-а-нафтохинон (Фтиокол) 463
2-Метил-З-оксипирон (Мальтол) 581 4-Метил-5-(р-оксиэтил)-тиазол 710 а-Метилол-Р'-окси-у-пирон (Койевая кислота) 582
Z-6-Метилоктановая кислота 699
2-Метилпентан 57
3-Метил-2-пент-2'-енйлциклопентен-2-
он-1 (Жасмон) 73, 74 3-Метилпентен-2-ин-4-ол 705 Метилпиперидины (Пипеколины) 605, 607—609
N-Метилпиразол 565
М-Метил-а-(пи ридил)-пирролидин, см.
Никотин
Метилпиридины (Пиколины) 591,' 592, 594, 598, 599, 608, 626, 680
N-Метил-у-пир идон 600
N-Метилпиридонимин 601
Метилпиримидазол 602
р-(И-Метил-а-п иррил)-пиридин, см.
Никотирин
N-Метилпирр ол 530
р-(Ь1-Метил-а- п ирролидил)-пиридин,
см. Никотин
N-Метилпирро лидин 648
N-Метилпирро л идинуксусная кислота, эфир 656
N-Метилпирро л идон 650
1-Метилпиррол изидин (Гелиотридан)
648
N-Метилпролин (Гигриновая кислота) 538
Метилстиролы 275, 403 N-Метилсукцинимид 653 17-Метилтестостерон 190 N-Метил-! ,2,5,6-тетрагидроникотино-вая кислота (Арекаидин) 649 метиловый эфир (Ареколин) 649
N-Метилтетрагидропапаверин 667 2-Метилтиазол 572, 573 2-Метилтиазолин 572
Метилтиофены (Тиотолены) 516, 528
З-Метил-З-я-толилгептан 210 1-Метил-2,4,6-тринитробензол, см. Тринитротолуол
Метилтрифениларсоний 330 гидроокись 331
(З-Метилтрополон 498 4-Метилурацил 634 5-Метилурацил (Тимин) 634 Метилфенилкарбинол 402 Метилфенилкетон, см. Ацетофенон Метилфенолы, см. Крезолы а-Метилфуран (Сильван) 522, 711
Метилхиноноксим 358 а-Метилхинолин (2-Метилхинолин, Хинальдин) 612—618, 657
р-Метилхинолин (З-Метилхинолин) 614 у-Метилхинолин, см. Лепидин Метилхинолины (Толухинолины) 614 Метилхинолон 618
4-Метилхиноль (Толухиноль) 360, 361 Метил хлорнитробензолы 243 2-Метил-4-хлорфен окси уксусная кислота 264
Метилхолантрен 181, 489
Метилциклобутан 59
Метил циклобутен 51
Метилциклогексан 32, 47, 54, 59, 61, 75, 76
З-Метилциклогексанон-1 123
Метилциклогептан 54
Метилциклопентан 47, 59, 61, 71, 72, 225
Мет ил ци кл опентан ка р боновые к и с-лоты 71
3'-Метил-1,2-ци клопентенэфонантрен (Углеводород Дильса) 173, 174, 181, 688
Метнлциклопентенон 64 2-Метил-5-этилпиридин 591. 594 4-Метил-З-этил пиридин (р-Колл и дин) 592, 594, 659, 680
З-Мэтил-4-этилпиррол (Опсоппррол) 540
Метилэтилфенилфосфина окись 328 Метимицин 700
Метинэн (2-Метил-Г,4-нафтохинон, Витамин К3) 707
6-Метокси-8-аминохинолин 619 п-Метоксибензиловый (анисовый) спи рт 343
n-Метоксибензойная (анисовая) кислота 383
З-Метокси-4,13-диоксипропенилбензол (Конифериловый спирт) 343, 346, 347
2-Метокси-6,9-дихлоракридин 624
Метоксильные группы, количественное определение *267
п-Метокси-со-метоксиацетофенон 587
З-Метокси-4-оксиаллилбензол (Эвгенол) 339, 340
З-Метокси-4-оксипропенилбензол (Изоэвгенол) 339, 340, 395
З-Метокси-4-окситолуол (Креозол) 339 у-Метоксипиридин 580, 600 у-Метоксипирон 580
Метокситолухинон 174
л-Метоксихинолин (Хипанизол) 616, 659
7-Метокси-1,2-циклопентенофенантрен 196
л-Метоксицинхонпновая (хининовая) кислота 616, 659
этиловый эфир 662, 663
Метол (Метил-л-аминофснол, соль) 363
Меченые атомы, метод 293
Миарсенол 397
Микомицип 702
Микостерины 170, 178, 179
Миллона реакция 391
Миллс пространственная модель дифенила 417
— — псрп-производпых нафталина 464
расщепление производных 1,8-ни-тронафтиламина 465
Миндальная (оксифснилуксусная) кислота 388, 654
М и н е р а л о к о р ти к остер он д ы 191
Мирбановое масло, см. Нитробензол
Мир петиции 669
Мирцен 117, 155
Маклера кетон (Ди-и-диметиламино-бензофенон) 422, 423, 429
Многоядерные (полициклические) соединения
алициклические 12, 19, 112 -1И ароматические 413—490
Мовеин 629
Молекулярные смоляные сита 406
Молекулярный объем 230
Молочная кислота 643
Молярная экстинкция (Молярный коэффициент поглощения) 368
Моиобромбензол 201, 212, 239, 243, 333
Монобромтрополон 498
Монометил азобензол (Бензол азотолуол) 316
Монометил ал л оксан 640
Монометил анилин 304
Монометилмочевая кислота 636
Монометилмочевина 639, 640, 686
Мононитродибромбензол 399
Монофениларсиновые кислоты 331
Монофенилметильный (бензильный) радикал 441
Монохлорацетон 571
Морин (3,5,7,2',4'-Пентаоксифлавон) 386, 584
Морфенол, метиловый эфир 674, 675
Морфин (Морфий) 482, 620, 645, 655,
G69—678
метиловый эфир, см. Кодеин
этиловый эфир (Дионин) 665, 670
Морфинат триметилфениламмония 673
Морфол (Диоксифенантрен, монометиловый эфир) 482, 674
Морфолин (Тетрагидрооксазин) 512
6-Морфол ино-4,4-дифен ил гептаи-3-он 677
Мостиковые бициклические системы 18, 19, 108—111
/Мочевая кислота (2,6,8-Триоксипурин)
514, 569, 570, 619, 635—641, 643 соли (Ураты) 636
Мочевина 588, 634, 637, 643
Мукойнозит 64
Муконовый эфир 682
Муравьиная кислота 523, 573, 622
Муравьиный альдегид 598
Муравьиный эфир 687
Мускаруфин 419
Мускон 22, 86
Мус кус 86
кетонный 253
крезольный (амбровый) 253
Мышьяковоорганические соединения аналоги азинов 631, 632 аналог пиперидина 610 ароматические 328—332
/Аятное масло 120, 122, 124, 127
Надбензойная кислота, см. Бензоил, гидроперекись
Надмуравышая кислота 88
Назаров 30
синтез промедола 678 циклизация непредельных кетонов 43
Наметкин 148
исследования пинана 133 перегруппировка 148—150, 153
сужение шестичленного цикла в пятичленный 50
Напряженность циклов 20—26
Наркотин 620, 665, 668—671
Натансон, синтез фуксина 429
Натрийацетоуксусный эфир 40, 378
Натрийбензил (Бензилнатрий) 213, 334
Натриймалоновый эфир 38, 39, 67, 69, 86, 378
Натрийорганические соединения 212
Натрийфенил (Фенилнатрий) 333
Натрийхлормалоновый эфир 570
Нафтазарин (5,8-Диокси-а-нафтохинон) 462
Нафталин 97, 203, 221, 374, 445—466, 490, 559, 611
аминопроизводные 298, 458
восстановление 106, 451
галоидпроизводные 453—454, 456
гомологи 158, 452
изомеризация в азулен 503
нитрилы 463
нитропроизводные 454
окисление 376, 399
оксипроизводные 455—458
пикрат 273, 445
стереохимия 464, 465
сульфокислоты 454—456, 459
сульфохлориды 454
/гери-Нафтал и иди карбоновая кислота
(Перикислота) 463, 464
Нафталиновое ядро
в эргоалкалоидах 685
в эстрогенах 196
Нафталиндихлорпд 454
Нафталинсульфокислоты 454—456
Нафталпнтстрасульфокислоты 455
Н афта л 11 нтет р а хлор 11 д 454
Натфалиптрисульфокислоты 455, 459
Нафтацен, см. 2,3-Бензаптрацен
Нафтеновые кислоты 63, 72
Н афтен ы 13, 61 —63
Нафтиламины 297, 454, 456, 458, 4G1, ' 620
а-Нафтилдифепилметил 444
Нафтилендиампны 297, 450
2- (а- Н афт и л) -эти л х л о р ид 484
2-(а-Нафтилэтил)-Д1-циклопентсн 484
Нафтионовая кислота (1-Аминонафта-лин-4-сульфокислота) 458, 459
Нафтогидрохинон (1,4-Диоксинафта-лин) 4G1
1,4-Нафтогидрохинон 484
Нафтойные кислоты 463. 464
а-Нафтол (а-Оксинафталин) 367, 448,
455—457. 460
Нафтол АС (Азотол А) 464
B-Нафтол (,3-Оксинафталин) 455—458, 460, 630
метиловый эфир (Неролин, Яра-яра) 456
Н афто л - 2,4 - д и с у л ьф о к и с л от а 456
2-Нафтол-3,6-дисульфокислота (Р-кислота) 457
2-Нафтол-6,8-дисульфокислота (Г-кис-лота) 457
Нафтоловый голубой Мелдолы 630
1-Нафтол-4-сульфокислота, см. Кислота Невиля—Винтера
2-Нафтол-6-сульфокислота, см. Кислота Шеффера
2-Нафтол-8-сульфокислота, см. Кроцеи-новая кислота
Нафтофсназин 630
Нафтохинолины 620
Нафтохинон 462
Р-Нафтохинонмоноксим, ацеталь 454
Нафтохиноны 367, 461—463, 703
Невиля—Винтера кислота (1-Нафтол-4-сульфокислота) 457
Неницеску изомеризация"углеводородов 51 механизм реакции Фриделя—Крафтса 214
Необратимый катализ 58, 231
Неоизоментол 121, 122
Неоментол 121, 122
Неометимицин 700
Неопентилхлорид 67
Неосальварсан (Новарсенол) 397
Нерадол 274
Нерегулярные заместители 238
Нерийфолигенин, см. Дигитоксигенин
Неролидол 156, 157
Неролин, см. р-Нафтол
Несимметрический изомер 200
Несмеянов, диазометод 323, 336
Нефть 61—63, 90, ИЗ, 221, 225
Ниацин, см. Никотиновая кислота
Никотеин 651
Никотеллин 651
Никотин (Ы-Метил-а-[р-пиридил]-пир-ролидин, p-fN-Метил-а-пирроли-дил]-пиридин) 528
Никотиновая кислота (Витамин РР, Ниацин, p-Пиридинкарбоновая кислота) 603, 650, 651, 704, 709 амид 604, 704, 709, 715 диэтиламид (Корамин) 604
Никотирин (р-[Н-Метил-а-пиррил]-пи-ридин) 650, 651
Нильский голубой 374
Нитрилы ароматические 422 ц-Нитро-<о-аминоацетофенон 696 Нитроанизолы (Метиловые эфиры нитрофенола) 271, 358, 414
Нитроанилины 251, 271, 304, 309, 318,
324, 361, 602
9-Нитроантрацен 470, 472
п-Нитро-а-ацетиламино-р-оксипропио-фенон 696
ц-Нитроацетофенон 696
НиА'робензальдегиды 279, 294, 396, 482, 559
л-Нитробензоилпролин 538 Нитробензойные кислоты 293, 294, 389
Нитробензол (Мирбановое масло) 224 236, 246—252, 304, 345, 414, 429, 590, 612
восстановление 249—251, 297, 314, 363
получение 220, 246, 304
реакции 242, 249—251, 313, 560, 628
физические свойства 249
.w-Нитробензолсульфокислота 260, 31! ц-Нитро-(о-бромацетофенон 696 Нитробромбензойная кислота 202 Нитрование
алициклических углеводородов 55
нафталина 454
полимеров стирола 404
производных бензола 241, 242, 248
фенолов 265 хинолина 614
о-Нитрованилин, метиловый эфир 482
Нитродифеновые кислоты 416
Нитрозамины 303, 324
Нитрозоантипирин 567
N-Нитрозобензил-о-толуидин 568
Нитрозобензол 304, 313, 557
N-Нитрозодибензиламин 307
Нитрозодиметиланилин 303, 313, 357,
363, 629, 630
Нитрозодифениламин 439
Нитрозометиланилин 303
Нитрозонафтолы 461
Нитрозопинен 138
Нитрозопиперидин 606, 607
Нитрозосоединения ароматические 313
у-Нитрозотрополон 498
N-Нитрозофенилгидроксиламин (Купферрон) 314
и-Нитрозофенол 303, 313, 357, 358, 363
Нитроксилолы 399
Нитроламины 139
Нитролы 314
Нитрометан 402
о-Нитроминдальная кислота 555
Нитрон (Дифенилэндоанилодигидро-триазол-1,2,4) 573
Нитронафталин 446, 447, 454, 456, 458
Нитропиридины 603
Нитропруссид натрия 183
Нитросоединения ароматические 246— 257
восстановление нитрогруппы 249— 251, 298, 315, 363
получение 220, 246—249, 304
Нитростирол 307, 402
а-Нитротиофен 527
1-Нитро-3,6,8-трисульфокислота нафталина 459
Нитротолуолы 249, 252, 294, 390, 414
1-(и-Нитрофенил)-2-ацетиламино-1,3-пропандиол, см. Хлоромицетин
и-Нитрофенилгидразин 326
,и-Нитрофенилнптрометан 254, 255
1-п-Нитрофенил-З-метилнитропиразо-
лон (Пикролоновая кислота) 566, 645
о-Нитрофенилпропиоловая кислота 410, 555
эфир 554
о-Нитрофенилуксусная кислота 555
Нитрофенолы 265, 271—274, 309, 358, 363, 400, 590
о-Нитрофенолят 242
З-Нитрофталевая кислота 446, 447
2-Нитрофуран 520
п-Нитрохлорбензол 236, 298
Нитроциклогексан 77
Нитрующая смесь 246
Новарсенол (Неосальварсан) 397
Новокаин 391, 691
Новолачные смолы 268, 269
Нопинен (р-Пинен) 133—135^
Норкамфан (Бицикло-[ 1,2,2]-гептан)
110, 111
Норкарадиенкарбоновая (бицикло-
10, 1,4]-гептан-2,4-диен-1-карбоновая) кислота 104
Норкаран (Бицикло-[0,1,4]-гептан) 98 Нормальный ряд (А/В i^c-ряд) стероидов 165—166
Норпинан (Бицикло-11,1,3]-гептан) 110, 111
Норпиновая (1,1-диметилциклобутан-2,4-дикарбоновая) кислота 136
Норсульфазол (Сульфатиазол) 312, 572
Норэкасанталовая кислота 160
Нуклеиновые вещества 634, 635
Нуклеофильное замещение 252, 256 Нуклеофильные (анионоидные) реагенты 242
Обепин (Анисовый альдегид) 394
Одноядерные (моноциклические) алициклические соединения 12, 66—87
Озонирование бензола 205, 220, 228 Окись
арсина 610
дифенилазота 440
дифениларсина 329, 331
дифенилена (Дибензофуран) 524 мезитила 78 метилэтилфенилфосфина 328 пентаметилена (Тетрагидропиран) 575
триметилена 511
трифениларсина 330
трифенилфосфина 328 тропилия, см. Тропой фениларсина 329—331 этилена 275, 291, 509, 510
Оксазин 512, 625, 630
Оксазол 512, 517, 571
Оксазолидины (Тетрагидрооксазолы;
512
Оксазолины (Дигидрооксазолы) 512
Оксанилид 302, 308
Оксаниловая кислота 302, 308
Оксантранол (Антрагидрохинон) 473
474
Оксантрои 473, 474
Оксиазобензол 365
ион 372
Оксиазосоединения 315, 323, 363
Оксиальдегиды ароматические 392—395
Оксиантрахинон 471
9-Оксиантрацен (Антранол) 474 co-Оксиацетофенон (Бензоилкарбинол)
282, 395
о-Оксибензальдегид, см. Салициловый альдегид
Оксибензальдегиды, 345, 391, 393, 551, 662
Оксибензиловые спирты (Фенолоспир-ты) 268, 343, 349
о-Оксибензиловый спирт (Салигенин) 343
n-Оксибензилпенициллин (Пенициллин
III, или X) 690
о-Оксибензойная кислота, см. Салициловая кислота
Оксибензойные кислоты 201, 205, 266, 382—385, 532
Оксибензофенон 423
Оксигемоглобин 539
Оксигидрохинон (1,2,4-Триоксибензол) 339
триацетат 354 Оксидициклогексилы 95 7-Оксиизохинолин 662 Оксикетоны
алициклического ряда 42
ароматического ряда 393—395
S-Оксикетоны, ангидриды енольных форм 575
Оксикислоты
алициклического ряда 64—66
ароматического ряда 377, 381—385. 410—412, 578, 586
Окси кодеинов 676
Р-Оксиконифериловый спирт 348 Оксикоричные кислоты 410, 411, 578.
586
лактоны 578
17-Оксикортикостерон (Гидрокортизон)
193
/?-Оксикумариновая (умбелловая) кислота 412
Оксилактоны 97
а-Оксилепидин (Лепидон) 614
7-Окси-8-метиленпиперидиноизохи но-
лин 662
З-Окси-М-метилморфинан 677
а-Оксиметилфурфурол 523
1-Окси-2-метоксибензол, см. Гваякол 1-Окси-З-метоксибензол (Резорцин, ме-тиловый эфир) 338
1 -Окси-4-метоксибензол (Г идрохинон, метиловый эфир) 338, 392
1-(4'-Окси-3'-метоксифенил)-пропанол 346
Оксимы
ароматических альдегидов и кетонов 282—285
бензила 434
дигидрокодеинона 675
изатина 554, 557
нафтохинонов 461
салицилового альдегида 394
стереохимия 282—285
Оксин (8-Оксихиполин) 618
ос-0ксинафталин (а-Нафтол) 367, 448, 455—457, 460
Оксинафталины, см. а- и Нафтолы
2-Окси-З-нафтойная кислота, анилиды
(Азотолы) 464
2-Окси-а-нафтохинон (Лаусои) 463
Оксин (8-Оксихинолин) 618
Оксинафтохиноны 462. 463
Оксиндол (о-Аминофенилуксусная кис-
лота, лактам) 548, 553, 555
Оксипиоидины (Пиридоны) 578, 590,
597, 599—603, 618
р-Окснпирон 582
11а-Оксипрогсстерон 193
Оксипролин 538
Оксиптеридины 640—641
6-Оксипурпн (Гипоксантин) 635, 637, 638
Оксипуппны 635—640
Оксирубреи 486
Оксистероиды 172, 188, 193
Окситетрациклин (Тсррамицин) 697
я-Окспфениларсиновая кислота 396
О к с j j фе п и л д и и одт и р оз и н 392
/ ? - О к с и т р и ф е н и л к а р б 11 и о л 427
л - О ксифе 11 и л и р о п ан 346
') к с 11 фе н п л у к с у с и а я (миндальная) к и с -лога 388, 654
а-Окспхинолин, см. о-Аминокорнчная
кислтга, ангидрид
у-Оксихппэлин (К и пурин) 618
8-Окспхинолпн (Оксин) 618
Слскхпнолины 613—615, 618
у-Оксн хи нол и II-J3-карбоновая (кинуре-
нозая) кислота 553, 618
о - О к с и х л о р с т и р о л 524
3~-Оксихолановая (литохолсвая) кисло-
та 182
Оксихроман 768
2-Окси-2,4,6-ци кл огептатриен-1 -он,
см. Трополон
а-Оксинпнхониновая кислота 619
ЗгЗ-Окси-Д5-этисновая кислота 192
Оксониевые соединения 576, 588
Д9,10-Окталин 503
Октановое число 72
Октахлорнафталин 454
Олефины, ароматизация 222, 223
Оловоорганические соединения 336
Опиановая кислота 668
Опий 643, 644
Опсопиррол (З-Метил-4-этил пиррол)
540
Опсопирролкарбоновая кислота 540
Оптическая изомерия
производных дифенила 416
производных нафталина 464
Оптические антиподы (изомеры) 28, 29
34, 92, 97, 410, 416, 464
Ордоваль 274
Орехов 643
выделение анабазина 651
— и синтез сальсолина 665
получение платифиллина 648
Ориентация
в бензольном ядре 238—243
в замещенных нафталинах 450, 451
/-Орнитин 698
Орселлиновая (4,6-диокси-о-толуило-
вая) кислота 340
метиловый эЬир (Эверниновая кислота) 384
Орсин (3,5-Диокситолуол) 338, 340, 384
Ортомуравьиный эфир 617
Орто-положенпе 200
Ортоцпклофаны 490
Осевая связь, см. Аксиальная связь Основной
коричневый (Бисмарк коричневый, Везувии, Манчестерский коричневый) 362, 367. 374
фиолетовый К, см. Метилвиолет
Основание Гомолкп (Парафуксин, ангидридная форма) 427
Основные красители 366, 372
Осцплляпионная гипотеза 200
«Оспиллянионные» структуры 370
Оцимен 117
ПАБ (я-Аминобензойная кислота) 390, 703, 714, 715
Пантетеин 703
Пантоплтаурин 715
Пантотеновая кислота 703, 715
Папаверин (Тетраметоксибензилизохинолин) 620, 665, 667, 671
Папаверолин (Тетраоксибензилизохинол ин) 667
Параамидофенол (Родиналь) 118, 363
Парабановая кислота 569, 636
Паракумарон 524
Паралейканилин (Три-/г-аминотрифе'
в ил метан) 428
Парамагнитные свойства 444
Параминдальная (оксифенилуксусная недеятельная) кислота 388
Пара-положение 200
Парарозанилин (Парафуксин) 372,
426—429
ангидридная форма (Основание Го-молки) 427
Парациклофаны 490
Пар вол ины 591, 592
ПАСК, см. «-Аминосалициловая кислота
Пейцеданин 589
Пеларгонидин 586
тетраметиловый эфир 587
Пельтьерин 648, 649
Пениллоальдсгиды 691
Пенилловая кислота 692
Пепиллоиновые кислоты 691
Пениллонозая кислота 692
Пеницилламин (п'-ВД-Димети л цистеин) 691
Пспинилленовая кислота 692
Пенициллин 178, 511, 571, 689, 699
изомеры 692
Пенициллиназа 691
Пснициллоиновые кислоты 691
Пектабромтолуол 58, 247
«-Пентадскан 22
П ей т а ди г а л л ои л гл ю к оз а 338
Пенталам (Бицик ю-[0,3,3]-октан) 99 —
101, 101
Пенталин 502
Пентал сны (Бицикл о-! 0,3,3]-октаноны)
101
Пентамзтилбепзол 217
Псятамстплсч, см. Циклопентаи
окись (Тетрагидропиран) 575
Пентамстилеидиамип, соль 606
Пеитаметилеидпбромид 606
Пентаметилентетразол (Кор азо а) 574
Пента мсти лрозан клан 4 29
Пейта метокси дифен или рои а н 386
«-Пентан 607
2,4,6,3',4'-Пентаоксибепзофеноп (/4ак-
пурин) 385, 386, 423, 584
Пептаоксиппраны (Пиранозы) 575
3.5,7,2',4'-Пснтаоксифл -звон (Мори н)
5S1
З.э, / ,3',4'-Пентаокси(Ьлавоп (Кверцетин) 584. 587
7-метпловый эЬпр (Рамнетпн) 584
П-штафенилбензол 418
Пен тафе I > i т л фосфор 328
Пситафенилциклопеитадиен 439, 443
Пептафенилциклопснтадиепил 4 14
Пентафенилэтан 420, 437, 442
Пентафеиилэтил 438, 441
Пентафенилэтилхлорид 441
Пентацен (2,3,6,7-Дибензантрацсн) 485
Пентаэритрит, тетрабромид 51 2-Пентенилпенициллин (Пенициллин I, или F) 690
Пентоза 519, 522, 523
Пентозаны 344, 522, 523
Пербензойная кислота, см. Бензоил, гидроперекись
Пергидроантрацен 167, 469
Пергидродифеповая (Дииикшюгексил-
2,2'-дикарбоновая) кислота 92—94
Пергидрофенантрен 112, 165
Пергидроциклопентснофенантрен 174
Перегруппировки
Бекмана 283, 676
бензидиновая 297, 317, 318, 414
бензиловая 434
Вагнера—Мейервейна, см. Камфеновые перегруппировки
Валлаха 315
галоидалкилатов пиридина 593 дифенилиновые 317 камфеновые 148—150, 153
Наметкина, см. Камфеновые перегруппировки
пипаколиповая 49, 96, 360, 434, 643
ретроп и па кол и нова я 148
сем иди н с в а я (пол v бензидиновая) 317, 318
Перекись бензоила 288, 290 трифенклметпла 436, 439, 440
Пери кислота (пери-Нафтали иди карбо-
новая кислота) 463, 464
Перилец 487, 488. 499
Перилснхппон 487
Пернплогенин 184
Пеpi / - п о л о же ни е 4 4 8
Перкаин, см. Совкаин
Перкин В. младший 11 изучение алкалоидов 669 реакция 279, 407, 411, 412, 520, 552 с ] I нто з а л и н, и к л 11 ч с с к и х с ое д и н е н и й 11, 38
— гармалина 678
— кумарина 411
— солей бензопприлпя 587
— терпина 127
Перкин В. старший 11
синтез мосеипа 629 техническое получение ализарина 476
а- П п ко л и л к а р б и н о л (а-11 и к о л и л алкин) 603
Пиколиновая (^-пиридин карбоновая) кислота 603, 604 соль 609
Пиколины (Метилпирпдины) 591, 592, 594, 598, 599, 608, 626, 680
Пикрампд (2,4,6-тринитроаиплин) 252, 273, 310
Пикраминовая кислота 274
Пикраты 273
нафталина 273, 445
полинитропроизводных 252, 253 при разделении алкалоидов 645 триметиленимина 511
этиленимина 510
Пикрилгидроксиламин 252
Пикрил хлорид (2,4,6-Тринитрохлор-
бензол) 256, 273, 629
Пикриновая кислота (Лиддит, Мелинит, 2,4,6-Тринитрофенол) 272— 274, 341, 400, 645
получение 251, 256, 265, 272, 310 соли 497, 550, 563, 615
Пикролонаты 645
Пикролоновая кислота (1-п-Нитрофе-нил-3-метилнитропиразолон) 566, 645
Пикромицин 700
Пилокарпин 569, 686
Пилокарповая кислота 686
Пилоповая кислота 686, 687
Пимантрен (1,7-Диметилфенантрен) 163
Пимелиновая кислота 41, 83, 172, 205, 497
Пинаколиновая перегруппировка 49, 96, 360, 434, 643
Пинаколины 49, 50
Пинаконы 49, 90
Пинакриптол зеленый (М-фенил-1,3-ди-аминофеназонийхлорид) 629, 630
Пинан (4,7,7-Триметилбицикло-[ 1,1,3]-гептан) 131, 133 производные 134—138
(З-Пинен (Нопинен) 133—135
Пиненгликоль 135
Пиненнитрозохлорид 138
Пиненхлоргидрат 136
Пинены 116, 124, 133—136, 145, 155,227 дибромид (Дибромкамфан) 151
Пиновая кислота 135, 136
Пиноилмуравьиная кислота 136
Пинокамфан 134
Пинол 137
Пиноловая кислота 137
Пиноновая кислота 135, 136
Пинорезинол 347
Пипеколины (Метилпиперидины) 605, 607—609
Пиперазин (Диэтилендиимин) 513, 515, 626
Пиперазины (Тетрагидропиразины) 513, . 515, 626
Пиперидеины (Тетрагидропиридины) 598, 606
р-(а-Пиперидил)-пиридин, см. Анабазин
Пиперидин (Циклопентаметиленимин, Гексагидропиридин) 512, 515, 598, 605—609, 648, 649, 662
Пиперидин-3,4-дикарбоновые (гекса-
гидроцинхомероновые) кислоты 666
Пиперилен 607
Пиперин 412, 608, 649
Пипериновая кислота 412, 608, 649
Пиперональ (Гелиотропин, Протокате-ховый альдегид, эфир) 340, 395, 412
Пиперонилакролеин 412
Пиперониловая (3,4-метилендиоксибеп-зойная) кислота 384
Пиразин 512, 517, 625—627
производные 627—633, 640
Пиразинкарбоновые кислоты 625, 627 Пиразол 512, 563—569
Пиразолидины (Тетрагидропиразол ы) 512, 563, 564, 566
Пиразолидоны 564
Пиразолины (Дигидропиразолы) 512, 563, 566
Пиразолкарбоновые кислоты 566
Пиразолоны 564
Пирамидон (Диметиламиноантипирин) 567
Пиран 512, 517, 575—589 Пирана группа 575—589 Пиранозы (Пентаоксипираны) 575 Пиренхинон 487 Пиретрины 73
Пиретролон 74
Пиретрум 73
Пиридазин (1,2-Диазин) 625, 632, 033 ос-Пиридилгидразин 599, 602 я-Пиридилкарбинол 603 ос-Пиридилмеркаптан 599 ос-Пиридилметилкетон 603 а-Пиридилнитрамин 602 Пиридилпирролы 650
Пиридин 512, 517, 572, 611, 614, 681, 683
группа 590—609
получение 533, 591—593
производные 311, 578, 599—605, 648—651, 704
а-Пиридинальдегид 603
2,3-Пиридиндикарбоновая (хинолино-
вая) кислота 604, 615 Пиридиниевые основания 595 Пиридиний-ион 590 а-Пиридинкарбоповая (пиколиновая) кислота 603, 604
соль 609
[3-Пиридинкарбоновая кислота, см. Никотиновая кислота
7-Пиридинкарбоновая (изоникотиновая) кислота 603—605
3, 7-Пиридинкарбоновая (цинхомеро-новая) кислота 604, 620
Пиридинкарбоновые кислоты 591, 597, 601, 603—605, 609, 612, 615, 620
Пиридиновые алкоголи 598
Пиридиновые красители 596
Пиридиновые основания 221, 591, 593, 597, 598, 613, 646
р-Пиридинсульфокислота 599, 715
Пиридоксаль 704, 709*
Пиридоксамин 704, 709
Пиродоксин (2-Метил-3-окси-4,5-ди-
[ оксиметил]-пиридин, Адермин) 600, 704, 709
Пиридонимин 601 а-Пиридонметид 599 Пиридоны (Оксипиридины) 578, 590, 597, 599—603, 618 '
Пирпдофталоны 599
Пирилиевые (пироксониевые) соединения 578, 580, 581, 586
Пирилий-ион (пироксоний-ион) 576, 580
Пиримидин (1,3- или л-Диазин) 512, 625, 633, 634
производные 311, 569, 571, 602, 634, 635, 641, 710
Пировиноградная кислота 204, 549, 685
Пирогаллол (1,2,3-Триоксибензол) 339— 342, 384, 415, 502
Пирогенетнческие процессы 223
Пирокатехин (о-Диоксибензол) 164, 225, 337—342, 345, 356, 383, 394, 476
Нирокоман, см. у-Пирон
Пироксилиновый порох 306, 307
Пироксониевые (пирилиевые) соединения 578, 580, 581, 586
Пироксоний-ион (Пирилий-ион) 57), 580
Пиролиз ацетилена 445 керосина 445
11иромеллитовая (1,2,4,5-бензолтетра-карбоцовая) кислота 379, 380
а-Пирон (Кумалин) 575—578, 600 у-Пиров (Пирокомап) 518, 575, 579 — 581
производные 576, 581—583, 589, 593, 600
Пироны 518, 575
производные 576—589, 593, 600 а-Пиронкарбоновая (кумалиновая) кислота 577
Пирослизевая (ос-фуранкарбоновая) кислота 520, 522, 523, 524
Пиррол 511, 516, 528—534, 563, 614 группа 528—547 производные 532—547, 593, 648, 714
7-Ппрролальдегид 532, 533
Пирроленин 529
а-Пирролидинкарбоновая кислота (Пролин) 528, 537, 685, 686
Пирр ол иди ны (Т етр а метилен имины, Тетр а гидропирролы) 511, 529, 530. 535—537, 606, 607
производные 537, 539, 648, 651, 652
Пирролидон (Бутиролактам) 537
Пирролиндофенин 558
Пирролины (Дигидропирролы) 511, 529, 534
Пирролкарбоновые кислоты 532, 540
Пирропорфирин 546
Пирроэтиопорфирин 546
Пихтовое масло 124, 143, 145, 147
Пицеин 395
Пицен (1,2,7,8-Дибензофенантрен) 181, 486
Плазмохин 619
Плазмоцид 619
Платинецин 648
Платифиллин 643, 648
Поглощение света 368
Полиазокрасители 365
Полиамидные волокна 266
Полидспсиды (Депсиды) 382, 386
Полиеновые кислоты 155
Полимеризация ацетиленовых углеводородов 203 окиси этилена 509 стирола 404—406 этилена 11, 44
Полиметиленовые соединения, см. Алициклические соединения
Полимиксины 698
Полиоксиантрахиноны 477
Полипептиды 188 антибиотики 698
Полипоровая кислота 419
Полистиролы 11, 402
Политерпены 117, 154—161
Полифенилы 418—420
Полициклические (многоядерные) соединения
алициклические 12, 19, 112—114 ароматические 413—490
Полпэтилентерефталат (Терилен) 3/9
Полубензидиновая (семидиновая) перегруппировка 317
Полуторатерпены, см. Сесквитерпены
Полыни масло 130
Поляризация ароматического ядра 214
Полярная (полюсная) связь, см. Ак спальная связь
Померанцевой корки масло 124
Порфин 539
Порфирины 539, 541—543, 704, 714
Правило
Армстронга—Винна 455
Ауверса—Скита 35
Блана 172, 173
Правило
Бредта НО
замещения в бензольном ядре 238— 242
Кернера 398, 399
Марковникова 214
Прегнан 166
Прегнандиол-3а,20а 191
А 5-Прегнандиол-Зр,21-он-20 192
А4-Прегнендион-3,20 (Прогестерон)
191, 193
«Предшественники» антибиотиков 690, 693
Пренитовая (1,2,3,5-бензолтетракарбоновая) кислота 379
Пренитол (1,2,3,4-Тетраметилбензол рядовой) 216
Прилежаева реакция 291
Пробковая (субериновая) кислота 83,
84, 496
Провитамины 90, 703, 705, 706
Прогестерон (Д4-Прегнендион-3,20)
191, 193
Пролин (Пирролидин-а-карбоновая кислота) 528, 537, 685, 686, 698
бетаин (Стахидрин) 538
Промедол((1,2,5-Триметил-4-фенил-4-пи-
перидол, пропионовый эфир) 678
Пропаргиловый альдегид, ацеталь 564
Пропенилбензол 451
«-Пропилбензол 217
Пропилен 67, 226, 266
Пропплпиперидины (Кониины) 608,
609, 648
а-Пропилпиридин (Конирин) 594
4-«-Пропилциклогексанон 346
Пропионовая кислота, производные 643,
678
Пропионовый альдегид 549, 591
Пропиофенон 281
Пространственные затруднения 296,
297, 416, 417
Противостояния (оппозиции) положение 22
Протокатеховая (3,4-диоксибензойная) кислота 66, 345, 383
диметиловый эфир, см. Вератровая кислота
метиленовый эфир, см. Пиперониловая кислота
метиловый эфир (Ванилиновая кислота) 384
нитрил 385
Протокатеховый (3,4-диоксибензойный) альдегид 345, 394, 395
метиленовый эфир, см. Пиперональ метиловый эфир, см. Ванилин, Ве-ратровый альдегид
Псевдоионон 80
Псевдокислоты 254, 321
Псевдокодеин 673, 684
Псевдокодеинон 673, 674
Псевдококаин 657
виннокислая соль (Псикаин) 657
Псевдокумидин (1,2,4-Триметил-5-амп-нобензол) 298, 301
Псевдокумол (1,2,4-Триметилбензол асимметрический) 210, 216
Псевдоморфин (Диоксидиморфин) 672
Псевдомочевая кислота 637, 638
Псевдооснования 255, 321, 427, 595
Псевдопельтьерин (N-Метилграна-
тан) 83, 657
Псевдопенициллин 692
Псевдосапогенины 187
Псевдотропин 652, 655, 656
Псевдофенилуксусная кислота 220
Псевдоэкгонин 655, 657
Псевдоэфедрин 647, 664, 694, 695, 700,
701
Псикаин 657
Птеридин (Пиразинопиримидин) '514, 640, 641
Птерины (Птеридин, оксипроизводные) 640, 641
Птероилглутаминовая кислота, см. Фолиевая кислота
Пуберуловая кислота 501
Пулегон (Д4(8)-Ментенон-3) ’ 122, 12Я
Пунцовый Г 457
Пурин (Имидазолпиримидин) 514, 635, производные 635—640, 687
Пурпурин (1,2,4-Триоксиантрахинон)
475, 477
Пурпурин-а-карбоновая кислота (Бо-летол) 479
Пурпурогаллин (Триоксибензо-а,£-тро-полон) 502
Пшорра метод синтеза производных фе-
нантрена 482, 647
Радикалы
бензгидрпльный 441
бензильный 441
диспропорционирование 213
дифенилметил 441
магнитные свойства 444, 445
монофенилметил 441
свободные 212, 213, 290, 437, 439
трифенилметил 437, 443
Рамалевая кислота 384
Рамнетин (3,5,7,3',4'-Пентаоксифлавон,
7-метиловый эфир) 584
Рамноза 185, 688
Расширение цикла 46—50, 59, 91, 97
Реактивы на алкалоиды 645
Реакции
азосочетания (сочетания) 324, 365> арилирования 245, 261, 264, 300,-414, 622
Реакции ароматизации 215, 222, 223 арсенирования 331
Барта 331, 332, 396 бензоиновая 279 Вешана 331
Бишлера—Напиральского 620, 665, 678
Бона—Шмидта 477
Брауна 536, 537, 607
Вышнеградского и Ладенбурга 598
Вюрца 67, 94, 95
Вюрца—Фиттига 212, 245, 333, 414, 453, 528
Гинзберга 362
Гофмана (исчерпывающее метилиро-
вание) 69, 389, 536, 601, 607, 653
Гриньяра 95, 484
дегидроциклизации 215, 222, 223 диазотирования 302, 319
Дикмана 41, 96, 656
Дильса—Альдера (Диеновый син-
тез) 45, 97, 108, 109, 354, 499
Димрота 344, 527
Залковского 171
Зандмейера 322
индофен и нова я 527
исчерпывающего метилирования, см. реакция Гофмана
Канницаро 274, 278, 520
карбонилирования 334
Кижнера 38, 94, 131, 541
Кольбе—Шмидта 381, 532
Легаля 183
Либермана на нитрозосоедпнения 303
— на холестерин 171
Мейзенгеймёра 472
меркурирования, см. Меркуриро-вание
Миллона 391
Перкина 279, 407, 411, 412, 520, 552
Прилежаева 291
Соммеле 277 таллейохинная 659 Тимана—Реймера 393 Тищенко 278
Ульмана (арилирования) 245, 261, 264, 300, 414, 622
Ушкова и Прокунина 527
Фиттига, см. реакция Вюрца—Фиттига
Фриделя—Крафтса 213—215, 280,
395, 421—422, 424—425, 452, 520, 528, 590
хлорметилирования 244
Чугаева 171
Шоттен-Баумана 290, 292
Этара 276
^езены 162
Резерпин (Серпазил) 679
Резиноловые (смоляные) кислоты
155, 162—164
Резинолы 162, 164
Резины 162
Резиты 269
Резольные смолы 268, 269
Резолы 269
Резорцин (л/-Диоксибензол) 337, 338.
342, 523 диметиловый эфир (1,3-Диметокси-бензол) 338
монометиловый эфир (1-Окси-З-ме-токсибензол) 338
получение 242, 260, 270
реакции 367, 431
таутомерия 348, 349
Резорциновый желтый 367
Резорцино-формальдегидные смолы 342
Рентгенографические исследования 85.
490, 498, 499, 692
Ретен (1-Метил-7-изопропилфенантрен) 163
Ретропинаколиновая перегруппировка 148
Рефракция молекулярная бензола и его производных 230. 255 сесквитерпенов 156 экзальтация для циклов 60
d-Рибоза 713
d-Рибоновая кислота 713
d-Рибонолактон 713
Рибофлавин (Витамин В.ъ Лактофлавин) 634, 704, 712
фосфорный эфир 714
Риванол (2-Этокси-6,9-диаминоакридин. молочнокислая соль) 623
Римской ромашки масло 280, 502
Рицинин 648, 649
Рицинолеиновая кислота 480
Р-Кислота (R-кислота, 2-Нафтол-3,6-дисульфокислота) 457
Робинсон исследование котарнина и гидри-стинина 669
механизм образования алкалиидив 643
синтез антоцианидинов 587
— гармалина 678
— тропинона 653
— тропинондикарбонового эфира 657 установление строения морфина 670 эметина 666
формулы бруцина и стрихнина 680
Родамины 364, 432
Родиналь (Параамидофенол) 118, 363
Родионов, синтез кодеина 673
Родопорфирин 546
Розанилин (Фуксин) 374, 427—429
Розиндулины 629, 630
Розмариновое масло 130, 143
Розовое масло 275
Розоловая кислота 430
Россини, исследования нефтей 61, 62
Ростовые вещества 74, 264
Ротенон 589
Ртутноорганические соединения 329, 334, 335
Рубрен (9,10,11, 12-Тетрафенилнафта-цен) 485
Рубэритриновая кислота 475
Ружичка
изучение сесквитерпенов 154, 158
превращения иронов 82
синтез больших циклов 12, 22, 85 — высших циклических кетонов 41, 43
— неролидола 157
— циклов, содержащих тройную связь 36
Рутин 704, 708
Рутиноза 708
Сабатье и Сандеран каталитическое гидрирование бензола 45, 209, 225
применение никеля как катализатора дегидрогенизации 57
Сабиней 154
Салигенин (о-Оксибензиловый спирт) 343
Салипирин 567
Салицилистая кислота, см. Салициловый альдегид
Салициловая (о-оксибензойная) кислота 205, 266, 382, 411, 532, 619 метиловый э|)ир 333 соли 383, 567, 640
фениловый эфир (Салол) 382, 588
Салициловый альдегид (о-Оксибензаль-дегид, Салицилистая кислота) 393. 394, 411, 532
Салицин 343
Салол, см. Салициловая кислота, фениловый эфир
Салофен 383
С а л ь в а и с а и (Д и а м и н од и о кси а р се н обе н -зол) 328, 397
Сальсолин (1-Метил-6-окси-7-метокси-тетрагидрохинолия) 620, 643, 665
Сандаловое масло 160
Сандарак 161
Санталовая кислота 160
Санталовый альдегид 160
Санталол 160, 161
Сантен 161
Сантонин 107
Сантониновая кислота 107
Сапогенины 186, 191
Сапонины 165, 186
Саркозин 700, 701
Саркомицин (2-Метилен-З-кетоцикло-пентанкарбоновая кислота) 700
Сарментогенин 184
Сассафрасовое масло 340
Сафранины 629
Сафрол (3,4-А4етилендиоксиаллилбен-зол) 339, 340
Сахара 523, 586
Сахарин (Бензойная кислота, сульфи-нид) 260, 294, 295 соль (Сукрамин) 295
Свинцовоорганические соединения 336 Свободное вращение 22—24, 416, 464 Свободные радикалы 212, 213, 290, 437—439 магнитные свойства 444, 445 устойчивость 440—444
Связь аксиальная (осевая, полярная, полюсная) 13, 30, 31, 34, 121, 167 квазиаксиальная 34 сопряжение 206, 231—235, 299, 371—373, 441, 442 экваториальная 31, 121, 167
Себациновая кислота 41, 84, 85
Сексифен ил 419
Селинен 158, 159
Сельдерейное масло 124, 158
Семидиновая (полубензидиновая) перегруппировка 317
Семидины 317
Семитерпеи, см. Изопрен Сенециониновая кислота 648 Сергеев получение сурьмяноароматических соединений 333 — фенола 266
Серин 699
Серпазил (Резерпин) 679
Сесквитерпены (Полуторатерпены) 117, 154—161, 464
Сильван (а-Метилфуран) 522, 711
Сильвестрен 126, 132
Сильвестрендигидрохлорид 133 «Синий крест» 332 Синильная кислота 591
Синнематин В (Цефалоспорин N) 690, 693
Синтезы
Аншютца 467
Габриэля 377, 378
Ганча 593
Гаттермана—Коха 276
Геша 422 ,
Дебнера—Миллера (Хинальдиновый) 302, 612, 613 диеновый 45, 97, 108, 354, 499 Пшорра 482, 674
Синтезы
Скраупа 612, 613, 615, 616, 620
Фридлендера 614
Синтомицин (Левомицетин, рацемат) 697
Син-форма 93, 94, 283—285
Синэстрол (Гексэстрол) 197, 433
Сиреневая кислота (Галловая кислота, 3,5-диметиловый эфир) 385
Сиреневый альдегид 345
Сирингилпропан 346
Ситостерин 191
Скатол (Р-Метилиндол) 548, 549, 551
Сквален 171
Скипидар (Терпентинное масло) 115, И6, 124—126, 128, 129, 133—136, 145, 158, 162 живичный 227 сульфитный 226, 379
Скополамин (Гиосцин) 653
Скэауп
синтез 612, 613, 615, 616, 620, 659 строение хинных алкалоидов 658
Слизевая кислота 64, 529, 650
Смолы 161—164, 287
бензойная 288
глифталевые 379 ионообменивающие (Иониты) 404— 406, 645
каменноугольная,см. Каменноуголь-
ная смола
«кумароновая» 524
природные 161 —164
резольные 268, 269
резорцино-формальдегидные 342
феноло-альдегидные 267—269, 343, 404—406, 523
эпоксидные 510
Смоляные (резиноловые) кислоты 155, 162—164
Собрерол 137
Совкаин (Перкаин, а-Оксицинхонино-
вая кислота, производное) 619
Соланидин 688
Соланин 688
Соласонидин 688
Соласонин 688
Сочетания (азосочетания) реакции 324, 365
Спирановая кетальная группировка 186
Спираны (Спироцикланы) 16—18, 91,
95—97
Спиро-[2,4]-гептан 96
Спиро-[ 3,3]-гептан-2,5-ди карбоновая
кислота 97
Спиро- 4,5]-декан 97
Спиро-[2,6]-нонан 96
Спиро-[2,5]-октан 96
Спиропентан 51, 95
Спиро-[5,5]-ундекан 97
Спироцикланы, см. Спираны
Спирты
алициклические многоатомные (Циклиты) 64, 65
ароматические 274, 275
— ненасыщенные 406
Стахидрин (Пролин, бетаин) 538
Стеароптены 116
Стеклотекстолит 267
Стереоизомерия
алициклических соединений 27—37
ароматических нитрилов 282
двухъядерных соединений 91—93 В-декалолов 102
дициклогексилдиола-4,4' 91 иохимбина 679 кариофилленов 159 ментола 120—122
пергидрофенантрена 112
пинана 134
спироциклановых соединений 97
стероидов 165—169
трео—эритро- 695
Стереоспецифичные реакции НО
Стереохимия
алициклических соединений 20—37, 98—102, 109, НО
оксимов и гидразонов 282—285
пиперидиновых оснований 605 производных бифенила 416—418 — нафталина 464, 465
— циклогексана 91—94, 605
стероидов 165—169
терпенов 120—122, 134, 137, 139 — 140
тропановых алкалоидов 653, 655, 657
Стерины 169, 706
Стероидные гормоны 165, 178, 184.
187—197
Стероиды 20, 105, 165—197, 483, 687, 703
номенклатура 168—169
Стибинобензол 332
Стигмастерин 170
Стил ьбазол ины 598
Стильбазолы 598
Стильбен (Дифенилэтилен симметрический) 316, 432, 481, 528
Стильбендибромид 433
Стильбэстрол (4,4'-Диоксистильбен) 197, 433
Стипитатовая кислота 501
Стиракс 161, 406
Стирол (Фенилэтилен) 203, 224, 275, 401, 402, 404, 449
полимеры 11, 402
сульфополимеры 406
Стиролдибромид 402, 403
Стиролдихлорид 402
Стифнаты 341
Стифниновая кислота (Тринитрорезорцин) 341, 645
Стоварсол (З-Ацетамино-4-оксифенил-арсиновая кислота) 398
Стрептидин 693
Стрептобиозамин (Ы-Метил-а-/-глюко-заминидо-(3-/-стрептоза) 694 /-Стрептоза 694 Стрептомицин' 693 Стрептоцид
белый, см. п-Аминобензолсульфамид красный (4' -Сульфамидо-2,4-диами-ноазобензол) 312
Стрихнин 645, 670, 681, 682—684, 687
Строфантидин (Конваллатоксигенин) 183, 184
Суберан, см. Циклогептан
Субериловый алкоголь (Циклогептанол) 49, 83
Субериновая (пробковая) кислота 83, 84, 496
Суберон (Циклогептанон) 40, 50, 51, 83, 653
Субстантивные красители 375, 561 Сужение циклов 46—50, 96 Сукрамин (Сахарин, соль) 295 Сукцинилянтарный эфир 42, 207 Сукцинимид (2,5-Дикетопирролидин) 530, 537
Сультон 457
Сульфадимезин 312
Сульфазол 312
Сульфамидные (сульфаниламидные) препараты 302, 311, 312, 715 4-Сульфамидобензиламин (Марфанил) 312
4'-Сульфамидо-2,4-диаминоазобензол (Стрептоцид красный) 312
Сульфамиды ароматического ряда (Амиды сульфокислот) 259, 295
Сульфаниловая кислота (л-Аминобен-золсульфокислота) 304, 31 1, 352, 366, 390 амид, см. л-Аминобензолсульфамид
Сульфатиазол (Норсульфазол) 312, 572 Сульфидин 312 Сульфиновые кислоты 260 Сульфирование ароматических углеводородов 226, 241, 242, 257—260 нафталина 450, 454, 455 нафтолов 457 фенолов 265, 274 хинолина 614
Сульфобензид (Дифен илсульфон) 260 Сульфобензойные кислоты 294, 295 Сульфокислоты гетероциклических соединений 520, 528, 531
производных бензола 226, 251, 257— 260, 274, 294, 295
— нафталина 454—455, 457—458
Сульфоны 260, 712
Сульфохлориды 259, 454
Сурьмяноароматические соединения
332, 333
Сцилларен А 185
Сцилларидин А 185
Сциллоинозит 64, 65
Таллейохинная реакция 659
Талослизевая кислота 65
Танацетон (Туйон) 153
Таннальбин 388
Таннин 382, 384, 387, 388, 645
Таурин 510
Таурохолевая кислота 180
Таутомерия
азокрасителей 367
амидная 291, 635, 641
аминотиазолов 572
бензоидно-хиноидная 271, 357 — 359, 367, 427, 461, 462, 474
гидрастинина 668
глутаконового альдегида 596
диазосоединений 323
диоксиндола 553
изатина 553
имидазола 568
индоксила 553
индола 550
кето-енольная 203, 204, 284, 348—
351
котарнина 668
питрозофенолов и хиноноксимов 357—359
нитросоединений с нитрогруппой i боковой цепи 254
нитрофенолов 271, 358
оксиндола 553
пиколинов 599
у-пирона 578
производных антрацена 470—475
— индола 550, 551, 553
— пиразола 563
— пиридина 596, 600—603
— пиримидина 634
— фенантрена 483
— хинолина 615
псевдокислот и псевдооснований 255
резорцина 348, 349
трансаннулярная 474, 475
углеводородов 359
фенолов 348—351
хиноидная, см. бензоидно-хиноид-
ная
хинофталона 617
циклогексанона 78
Тахистерин 179
Тебаин 665, 669, 670, 673—676
Тебаол (3,4,6-Триоксифенантрен, диметиловый эфир) 675
Тебенин 675
Теветигенин, см. Дигитоксигенин
Текодин, см. Дигидрооксикодеинон
Текстолит 267
Теобромин (3,7-Диметилксантин) 514, 569, 635, 638, 640, 687
Теории цветности 367—374
Теофиллин (1,3-Диметилксантин) 514, 635, 638, 640, 687
Теребиновая кислота 137
Тересанталовая кислота 160
Терефталевая (л-фталевая) кислота 85,
119, 206, 220, 375 379
Терефталевый альдегид 85
Терилен (Полиэтилентерефталат) 379
Терпениловая кислота 137
Терпентинное масло, см. Скипидар
Терпентины (Живицы) 115, 162
Терпеноиды 117
Терпены 11, 19, 20, 63, 108, 115—164 алифатические 117, 118 бициклические и трициклические 131 — 154 моноциклические 118—131 нитрозопроизводные 126, 138 политерпены 154—161
Терпингидрат 128, 129, 136
Терпинепы 125, 130, 136, 155
Терпинеолы (Ментенолы) 126, 129, 130
Терпинолен 125, 126, 129, 136
Терпины (Ментандиолы) 126—130
Террамицин (Окситетрациклин) 485, 697
Терфенил (1,4-Дифенилбензол) 224, 418, 419
Тестан (Этиохолан) 166
Тестанол-За-он-17 190
Тестанон-17 172, 173
Тестоидные (андрогенные) гормоны 188, 190
Тестостерон (Д4-Андростенол-17[3-он-3) 178, 188
Тетра-л-анизилгидразин 439
Тетрабензилсвинец 441
Тетрабромксилолы 243
Тетрабромэтан 467
Тетрагидробензойная кислота 288
Тетрагидробензол 209
Тетрагидроизофталевая кислота 206
Тетрагидроизохинолин, производные 621, 668
Тетрагидроимидазолы (Имидазолидины) 512
Тетрагидрооксазин (Морфолин) 512
1,2,3,4-Тетрагидро-8-метил-7-оксиизо-хинолин 662
Тетрагидронафталин (Тетралин) 106, 452
Тетрагидронафталинтетракарбоновый эфир 448
Тетрагидро-р-нафтиламин алициклический (ас-) и ароматический (аг-) 460
Тетр а гидронафтолы 460
Тетрагидрооксазолы (Оксазол идины) 512
Тетрагидропиразины (Г1иперазины\ 513, 515, 626
Тетрагидропиразол ы (Пир азолидины)4 512, 563, 564, 566
Тетрагидропиран (Окись пентаметилена) 575
Тетрагидропиридиновый альдегид 606
Тетрагидропиридины (Пиперидеины) 598, 606
Тетрагидропирослизевая кислота 524 Тетрагидропирролы, см. Пирролидины Тетрагидротиазолы (Тиазолидины) 512 Тетрагидротиопиран (Циклопентаметил енсульфид) 515
Тетрагидротиофен (Тиофан) 511, 515, 526, 527, 569
Тетрагидрофуран (фуранидин) 511, 514, 521—524
1,2,3,4-Тетрагидрохиноксалин 627
Тетрагидрохинолины 615, 618, 619
Тетрадекан 213
Тетрадекандикарбоновая кислота 41
Тетра-(диметиламинофенил)-гндразпн
439
Тетразол 512, 574
Тетр а и одп и рр о л (Иодол) 531
Тетралин -(Тетрагидронафталин) 106, 452
а-Тетралон 452
Тетраметиламмоний хлористый 334
Тетраметил аммонийтрифенил метил 334 1,2,3,4-Тетраметилбензол рядовой (Пре нитол) 216
1,2,3,5-Тетраметилбензол (Изодурол) 216
1,2,4,5-Тетраметилбензол (Дурол) 216 2,2,5,5-Тетраметилгексан 67
Тетраметилгидрохинон (Дурогидрохи-нон) 708
Тетраметил-л-диаминотрифенил карбинол 428
Тетраметил-л-диаминотрифенилметан (Малахитовый зеленый) 306, 428
Тетраметилди-(л-аминотрифенил) -метил 443
Тетраметилен, см. Циклобутан Тетраметиленимины, см. Пирролидины Тетраметиленкарбоновая (циклобутан карбоновая) кислота 68, 69
Тетраметилиндаминотиосульфокислота 631
1,3,7,8-Тетраметилксантин 639
1,3,7,9-Тетраметилмочевая кислота 636
1, 1,3,3-Тетраметилциклобутандион-2,4
44
1,5,5,8-Тетраметилциклоундекатри-ен-3,7,11, см. Гумулен
Тетраметоксибензилизохинолин (Папаверин) 620, 655, 667, 671
Тетраминодифенилметан 622
1,2,3,5-Тетранитробензол 252
Тетраоксибензил изохинолин (Папаве-ролин) 667
3,7,3',4'-Тетраоксифлавон (Физетин) 584
5,7,3' ,4'-Тетраоксифлавон (Лутеолин) 584, 585
Тетрафенилаллил 443
Тетрафенилгидразин 326, Тетрафенилдиарсин (Какодил) 331
Тетрафенилксилол 359
Тетрафенил метан 420, 432, 441
439
9,10,11,12- Тетрафенилнафтацен (Рубрен) 485
Тетрафенилтиофен (Тионессаль) 528
Тетрафенилфосфоний иодистый 328
Тетрафенилхинодиметан 359
Тетрафенилэтан 420, 435
Тетрафенилэтилен 436
Тетрахлорхинон (Хлоранил) 355
Тетрацен, см. 2,3-Бензантрацен
Тетрациклин (Ахромицин, Тетрацин)
485, 697
Тетраэтил кремний 213
Тетрил (Метилнитропикрамид) 306, 310
Тиазин 512, 625, 630 >
Тиазол 311, 512, 517, 571, 572
производные 571, 618, 710, 711
Тиазолидины (Тетрагидротиазолы) 512
кольцо 690
Тиазолины (Дигидротиазолы) 512, 572
Тиамин (Аневрин, Витамин Вг) 571, 704, 709
Тиле
гипотеза 206
получение углеводородов с хинонди-метидной группировкой 359 формула бензола 227, 229 — нафталина 446
Тимана—Реймера реакция 393
Тимин (5-Мети л урацил) 634
Тимол (2-Изопропил-5-метилфенол, 5-Метил-2-изопропил-1-оксибензол) 122, 263, 267
Тиоамиды 572
Тиоацетамид 571
Тцобецзамид 289
Тиббензойная кислота 289
Тиодиазол 311
Тиоиндиго (Красное тиоиндиго) 562
Тиокол (Гваякол, сульфокислота) 342
Тиоксены (Диметилтиофены) 528
Тиомочевина 572
Тионессаль (Тетрафенилтиофен) 528
Тиопиран 512
Тиопирон 579
Тиосалициловая кислота 562
Тиотолены (Метилтиофены) 516, 528
Тиофан (Тетрагидротиофен) 511, 515, 526, 527, 569
Тиофен 511, 516, 534
группа 525—528
Тиофенин (Аминотиофен) 516, 527
Тиофенкарбоновые кислоты 528
Тиофеновый альдегид 528
Тиофенолы (Меркаптаны ароматические) 236, 237, 258, 259, 322
Тиоформамид 711
Тиохром 710
Тиоцианиновые красители 573
Тирозин 391, 392
Тирозиназа 392
Тироксин 188, 392
Тищенко реакция 278
Тминное масло 124, 127, 379
Тодд
строение витаминов Е 708
— витамина В12 714
Тозил аты (п-Толуолсульфокислота, эфиры) 260
Токоферолы (Витамины Е) 583, 704,
708, 709
Токсины 661, 662
Тол, см. Тринитротолуол
Толан 433
о-Толидин 414, 459
Толил 210
Толугидрохинон 361
Толуидины 202, 252, 297, 301, 428,
429, 548, 567
Толуилендиамины 629, 361
Толуиленовый
голубой (Индамин) 629
красный (Эуродин) 629
Толуиловые альдегиды 277
Толуиловые (метилбензойные) кислоты
119, 286, 287
Толуол (Метилбензол) 210, 221, 225,
236, 275, 516
реакции 213, 220, 225, 242, 248,
257, 276, 286, 288, 295
таутомерия 359
Толуоламиноазобензол 365 n-Толуолдихлорсульфамид (Хлорамин Т) 260
п-Толуолмонохлорсульфамид 260 а-Толуиловая (фенилуксусная) кислота 275, 286, 287, 482, 690, 693
Толуолсульфамиды 259, 260, 295
п-Толуолсульфокислота 257, 294
эфиры (Тозил аты) 260
Толуолсульфокислоты 220, 257, 294, 295
Толуолсульфохлориды 259, 295, 681
Толусафранины 629
Толухинолины (Метил хинолины) 614
Толухиноль (4-Метилхиноль) 360, 361
Трансаннулярная таутомерия 474, 475
Трансаннулярный эффект 34, 88
Z-Треонин . 699, 700, 701
Трео-ряд 695
Трехчленный цикл, см. Циклопропан
Триазины 625
Триазолы 512, 573
Триаминоазобензол 367
Триаминобензол симметрический 343
2,5,6-Триамино-4-оксипиримидин 641
Т ри-n- ами нотр ифен и л метан (П а р а л ей к -анилин) 428
Три-о-анизилметпл 443
Триарилметил 442
Трибензиламин 309
Трибензоат глицерина (Бензойный эфир
глицерина) 289
Трибифенилметил 438, 443, 444
Триброманилин 304
Трибромфенол 265, 270
висмутовая соль (Ксероформ) 270
Трибромфенолбром 270
Трииоднитробензол 392
1,3,5-Трикетогексаметилен (Циклогек-сантрион, Флороглюцин, кетоформа) 349, 350
Трикетоны 43
Тримезиновая (1,3,5-бензолтрикарбоновая) кислота 204, 379
Тримеллитовая (1,2,4-бензолтрикарбоновая) кислота 379
1,2,4-Триметил-5-аминобензол (Псевдокумидин) 298, 301
1,2,4-Триметилбензол асимметрический (Псевдокумол) 210, 216
1,2,3-Триметилбензол рядовой, см. Гемимел л итол
1,3,5-Триметилбензол симметрический (Мезитилен) 203, 210, 216, 399, 400, 418
4,6,6-Триметилбицикл о-[ 1,2,2]-гептан, см. Фенхан
4,7,7-Триметилбицикл о-[ 0,1,4]-геитан, см. Каран
4,7,7-Триметилбицикло-[ 1,2,2]-гептан, см. Камфан
4,7,7-Триметилбицикл о-[ 1,1,3]-гептан
(Пинан) 131, 133
производные 134—138
Триметил гидрохиноны 708, 709
Триметилен, см. Циклопропан
Триметилена окись 511
Триметиленамин 536
Триметиленимин (Азетидин, Азациклобутан) 511
р-лактам (Ацетидинов) 511
Триметиленкарбоновая (циклопропан-карбоновая) кислота 66, 67
Триметиленсульфид 511
Триметиленсульфон 511 Триметиленхлорбромид 69 Триметиленхлоргидрин 511 6,7,9-Триметилизоаллоксазин (Луми-флавин) 713
1,3,7-Триметилксантин (Кофеин) 514, 569, 635, 637—640, 687
Триметилмочевая кислота 636
2,4,6-Триметилпиридин (Коллидин симметрический) 593, 594 соль 580
Т риметилтрикарбаллиловая (камфороновая) кислота 141
Триметилфениламмоний иодистый 300 1,2,5-Триметил-4-фенил-4-пиперидол, _ пропионовый эфир (Промедол) 678
Триметилциклогексаны 61, 76
у-(2,2,6-Триметилциклогексен-1-ил)-метил-кротоновый альдегид 705
1,5,5-Триметилциклопентадиен-1,3 144
Триметилциклопентаны 44, 62
Триметилциклопропиламмоний, гидроокись 67
2,3,5-Триметил-4-этилпиррол (Филло пиррол) 540, 541, 545
Триметилянтарная кислота 141
Три-р-нафтилметил 443
2,4,6-Тринитроанилин (Пикрамид) 252, 273, 310
Тринитробензойная кислота 252, 294
Тринитробензол симметрический 248, 249, 253, 294, 504
Тринитробензолаты 253
Тринитрорезорцин (Стифниновая кислота) 341, 645
Тринитро-трет-бутилтолуол 253
2,4,6-Тринитротолуол (Тротил, Тол 1-Метил-2,4,6-тринитробензол) 201, 248, 249, 252, 253, 294, 504
1,3,5-Трини.трофенетол 252
о-(2,4,6-Тринитрофениламино) -дифениламин 629
Три-п-нитрофенилметил 438, 443
2,4,6-Тринитрофенол, см. Пикриновая кислота
2,4,6-Тринитрохлорбензол (Пикрилхлорид) 256, 273, 629
Триозонид бензола 205, 220
1,2,3- Триоксиантрахинон (Антрагаллол) 477
1,2,4- Триоксиантрахинон (Пурпурин 475, 477
1,2,6- Триоксиантрахинон (Флавопур-пурин) 477
1,2,7- Триоксиантрахинон, см. Антрапурпурин
3,4,5-Триоксибензойная кислота, см. Галловая кислота
1,2,3-Триоксибензол (Пирогаллол) 339— 342, 384, 415, 502
1,3,5-Триоксибензол, см. Флороглюцин
1,2,4-Триоксибензол (Оксигидрохинон) 339
триацетат 354
Триоксибензо-а,р-трополон (Пурпурогаллин) 502
Триоксикоричная кислота, лактон (Эскулетин) 412
2,6,8-Триоксипурин, см. Мочевая кислота
Триокситрифенилкарбинол 430
Три-/г-окситрифенилметан 430
3,4,6-Триоксифенантрен
метиловый эфир 673, 674
диметиловый эфир (Тебаол) 675
Триоксифенантрены 674, 675
За,7а, 12а-Триок(?ихолановая (холевая) кислота 180—182, 489
Трипафлавин (Диаминоакридин, хлор-метилат) 622
Триптамин 552
Триптофан (р-Индолил-а-аминопропио-новая кислота) 552, 553
Триптофол 553
Трис-азокрасители 365
Трифениламин 299, 300, 301, 307
Трифениламиногуанидин 573
Трифениларсин 329
Трифенидарсиндихлорид (Трифенилди-хлорарсин, Трифенилмышьяк двухлористый) 330
Трифенилацетофенон (р-Бензпинаколин) 435, 436
Трифенилбензол симметрический 418
Трифенилбромметан 425
Трифенилгуанидин 308
Трифенилдихлорарсин, см. Трифенил-арсиндихлорид
Трифен илдихлорстибин (Трифенилсти-биидихлорид) 332
Трифенилен (9,10-Бензфенантрен) 213, • 486
Трифенил подметан 436
Трифенилкарбинол 425, 426, 432
Трифенилксилол 359
Трифенилметан 305, 420, 423, 424— 425, 428
Трифенилметановые красители 426
Трифенилметила перекись 334, 436,
439, 440
Трифенилметилмагнийхлорид 437
Трифенилметилнатрий 334
Трифенилметилперхлорат 425 1
Трифенилметилсульфаты 425
Трифенилметильный радикал 437—443
Трифенилмышьяк двухлористый, см.
Трифениларсиндихлорид Трифенилрозанилин 429 Трифенилстибин 332 Трифенилстибиндихлорид (Трифенил-дихлорстибин) 332
Трифенилуксусная кислота 334, 425 Трифенилфосфин 328
Трифенилхлорметан 334, 425, 426, 432,
436
Трифенилэтан 420 Трихиноил 74
Трихлормолочная кислота, амид 637 2,6,8-Трихлорпурин 637 Три-п-хлортрифенилметил 443
Трициклен 151
Триэтилалюминий 11
Тропан 496, 652
Тропановые алкалоиды 652—657
Тропеины 652, 654
Тропеолин D, см. Гелиантин Тропеолины 367
Тропидин 652, 653
Тропилиден (Циклогептатриен) 83, 493, 653
Тропилий (Циклогептатриенилий) бромид 493 ион 83, 493—495 окись, см. Тропой
Тропин 538, 652—656
Тропин-2.-карбоновая кислота (Экго-
нин) 538, 652, 655, 656
Тропиновая кислота 538, 653 Тропинон 652, 653, 656, 657 Тропинондикарбоновый эфир 657 Тропинонкарбоновая кислота 656 Тропинонкарбоновый эфир 656, 657
Тропинон-натрий 656
Троповая кислота 388, 389, 410, 654
Тропококаин 654, 655
Трополон (Циклогептатриен-2,4,6-ол-
2-он, 2-Окси-2,4,6-цикл огептатри-ен I-oh) 496—502
производные 83, 501, 647, 702
Тропой (Циклогептатриен-2,4,6-он,
Окись тропилия) 83, 494—496 Тротил, см. Тринитротолуол
Тротилаты 253
Труксиллины 655
Труксилловые кислоты 70, 408—410,
655
Труксиновые кислоты 70, 409
Труксон 409
Тубокурариндихлорид 666
Туйан (4-Метил-1-изопропилбицикло-
[0,1,3]-гексан) 153, 154
Ту йены 154
Туйон (Танацетон) 153
Туяплицины (Изопропилтрополоны) 500, 501
Увитиновая кислота 204
Углеводород Дильса (З'-Метил-l ,2-цик-лопснтенофенантрен) 173, 174, 181, 688
Углеводы 156, 575, 643, 688
Углерод
«метанный» 426, 429, 443
радиоактивный 171
трсхвалентный 436—439
Укропа масло 125 •
Уксусная кислота
2-бром-5-нитроанилид 285
соли с алкалоидами 643
Уксусный альдегид (Ацетальдегид)
407, 591, 592. 608, 612, 613
Укусный ангидрид 411
Ульмана реакция 245, 261, 261, 300,
414, 622
Умбеллпферон 412
Умбелловая (n-оксикумариновая) кислота 412
Урамил (Аминобарбитуровая кислота) 637
Ураты (Мочевая кислота, соли) 63с
Урацил 633, 634
4-Уреидо-2,5-диоксоимидазол (Аллантоин) 570, 636
У рейды 569, 633
Уропорфирин 544
Уротропин 696
Урсодезокси холевая (За. 7р-диокси холановая) кислота 182
Урушиол 164
Ушкова и Прокунина реакция открытия селена 527
Фарнезен 157
Фарнезол 156, 157
Фелингова жидкость 314, 325
Фелландрены 125
Фенадон 678
Феназин (Дибензпиразин) 513, 628, 629
Фенантрен 221, 415, 480—483, 486, 674 производные 482, 670—672, 675 9-Фенантренкарбоновая кислота 482 Фенантренхинон 415, 483
Фенарсазин 632
Фенацетин (М-Ацето-/г-фенетедин) 364
Фенетедины 364, 365
Фенетюл (Фенол, этиловый эфир) 267
Фенил аз ид 293
а-Фенилакриловая (атроповая) кислота 388, 410
Фенилаланин 309, 391, 685, 698, 699
Фениламин, см. Анилин
Фенилантранил 443
N-Фенилантраниловые кислоты 622
Фениларсин 330, 331
окись 329—331
Фениларсиндихлорид 329, 330
Фениларсиновая (фениларсоновая, фе-
нилмышьяковая) кислота 330, 331
Фениларсинтетрахлорид (Фенилтетра-хлорарсин, Фенилмышьяк четы-реххлорпстый) 330
Феннлацетил-/-аланил-^-валин 692
Фенил ацетил аминоацетальдегид 691
Фенил ацетилен 403, 410
Фенилацетилкарбинол 279
Фенилацетон (Метилбензилкетон) 281
Фенилбензениламидин 292
Фенилбензиламин (Бензиланнлин) 307
Фенил бензоат (Бензой но-фениловый эфир) 289
Фенилвинилуксусная (фенилизокрото-новая) кислота 448
2-Фенилгексан 210
Фен ил гидр аз ин 324, 326, 331, 367, 573, 681
Фенплгидразоны 285, 462, 549
Фенилгидроксиламин 250, 314, 363
Фенилглинин (Фенилгликоколь) 308, 554 560
Фенилглицинкарбоновая кислота 559
Фенилдиазокислота (Фен ил нитрамин)
304, 324, 602
Фенилдиазоний (Бензолдиазоний) 319, 367, 419, 458
N-Фенил-1,3-диаминофеназонпйхлорпд (Пинакриптол зеленый) 630
Фенилди-(бифенил)-метил 443
1 -Фенил-2,3-диметилпиразолон-5 (Антипирин) 566, 567
Фенилдихлорарсин 332
Фенилдихлорстибин (Феннлстибиндн-хлорид) 332
Фенилдихлорфосфин, см. Фосфепил-хлорид
Фенилендиамины 297, 319, 356, 361 — 363, 366, 367, 398, 399, 405, 622, 626, 630, 634
о-Фениленуксусная кислота 451
Фенпл-(арил)-жирные кислоты 286
Фенил изокротоновая (фенил винил уксусная) кислота 448
Фенилизонитрил (Фенилкарбидамин)
302, 308, 327
Фенилизонитрометан 254, 432
Фенилизопропиловый спирт (Диметил-фенилкарбинол) 275, 403
Фенилизоцианат (Карбанил) 308
Фенилиодидхлорид (Иодбензол хлористый) 246, 293
Фенилкарбиламин (Фенилизонитрил)
302, 308, 327
Фенилкарбинол (Бензиловый спирт)
274, 408, 421
Фенилксантил 443
Фениллитий (Литийфенил) 328, 333
Фенилмагнийбромид 275, 314, 328, 332, 333, 422, 432, 435, 486
Фенилмагнийхлорид 275
1-Фенил-3-метилпиразолон-5 565, 566
Фенилмочевина 308
Фенилмышьяк четыреххлористый, см.
Фениларсинтетрахлорид
Фенилмышьяковая кислота, см. Фенил-арсиновая кислота
Фенилнатрий (Натрийфенил) 333
Фенил-ос-нафтилкетон 488
N-Фенилнафтофеназоний 630
Фенил нитрамин (Фенилдиазокислота) 304, 324, 609
Фенилнитрометан 249, 254, 307
Фениловое горчичное масло 302, 308
Фениловый (дифениловый) эфир 267
Фенилпропан, производные 345, 346
Фенилпропиоловая кислота 408, 410 а-Фенилпропионовая (гидратроповая) кислота 286, 287
р-Фенилпропионовая (гидрокоричная) кислота 287 производные 391
Фенилртуть, производные 336
Фенилстибиндихлорид (Фенилдихлор-стибин) 332
Фенилстибйновая кислота 332
Фенилстибинтетрахлорид 332
Фенилтетрахлорарсин, см. Фениларсинтетрахлорид
Фенилтетрахлорфосфин 327
Фенилтиомочевина 308
Фенил-о-толилкетоксим (син-, анти-) 283
Фенилуглекислые соли 381
Фенилуксусная (а-толуиловая) кислота 275, 286, 287, 482, 690, 693
Фенилуксусный альдегид 277
Фенилуретан 308
N-Фенилфеносафранин 629
9-Фенилфлуоренил 443, 444
Фенилфосфин 327
Фенилфосфиндихлорид, см. Фосфенилхлорид
Фенилфосфинистая кислота (Фенилфосфин, гидрат окиси) 327
Фенилфосфиновая (фенилфосфоновая) кислота 327
а-Фенилхромон (Флавон) 382, 583— ‘ 586, 704, 707, 708
p-Фенилхромон (Изофлавон) 584 производные 585
а-Фенилцинхониновая кислота (Атофан) 619
р-Фенилэтиламин 307, 309, 621, 671
Фенилфосфонийиодид 327 Фенилэтилен, см. Стирол Фенилэтиленгликоль 402
Фенилэтиловые спирты 275
Феноксазин (Дибензооксазин) 513, 628, 629
Феноксиметил пенициллин (Пенициллин V) 690, 691, 693
Феноксиуксусная кислота 262, 693
Фенол (Карболовая кислота) 201, 224, 234—236, 260—269
метиловый эфир (Анизол) 267, 395 реакции 77, 21 f, 248, 262—265.
268, 269, 272—274, 298, 331,
337, 356, 357, 381, 393, 396.
397, 421, 429, 430
способы получения 221, 223, 225,
258, 261, 262, 321
таутомерия 348
этиловый эфир (Фенетол) 267
Фенолы многоатомные 270, 337—343, 422, 628
одноатомные 260—274
таутомерия 348—351
Феноло-альдегидные смолы 267—269, 343, 403—406; 523
Фенолокислоты 381—385, 387
Фенолоспирты (Оксибензиловые спирты) 268, 343, 349
Феноло-сЬормальдегидные смолы 267--269 ‘
Фенолсульфокислоты 265
Фенолфталеин 373, 377, 430
Феноляты 381
Фенопласты 267
Феносафранин 629. 630
Фенохиноны 355
Фентиазин (Дибензотиазин) 513, 628,
629
Фенхан (4,6,6-Триметилбицикле-[ 1,2,2,]-гептан) 151 —153
Фенхиловый спирт 152, 153
Фенхон 152, 153
Фенэтиламины 301
Феофитин 545
Феофорбиды 545
Фиалкового корня масло 82
Физетин (3,7,3',4'-Тетраоксифлавои)
584
Физостигмин (Эзерин) 679, 680
Филлопиррол (2,3,5-Триметил-4-этил-пиррол) 540, 541, 545
Филлопирролкарбоновая кислота 540
Филлопорфирин 546
Фиолетовый Лаута 362
Фитилбромид 709
Фитин (Гексафосфат мезоинозита) 65
Фитол 545, 707, 708
Фитостерины 170
Фипипиг
реакция 212, 245,333,414, 453, 528
формула бензохинона 351
Фихтелит 163
Фишер Г.
синтез гемина 539
синтез гомологов пиррола 533
синтез дипирролкетонов 541
синтез порфиринов 546
Фишер О. 206
метод галоидирования 599
Фишер Э. 206 восстановление диазосоединений в первичные гидразины 324 гидролиз казеина 537 исследования мочевой кислоты и ее производных 635, 637 получение производных индола 549, * 681 проекционные формулы 169, 664 синтез аминокислот 378 строение таннина 387
Флавантрен 478
Флавиановая кислота (2,4-Динитро-1 -нафтол-7-сульфокислота) 458, 645
Флавон (а-фенилхромон) 382, 583 — 586, 704, 707, 708
Флавопурпурин (1,2,6-Триоксиантра-хинон) 477
Флорацетофенон 585
Флороглюцин (1,3,5-Триоксибензол) 204, 337, 339, 342, 343, 349—351, 386, 523
Флороглюциндикарбоновый эфир 350 Флороглюцинтрикарбоновый эфир 203 Флороглюцит 349
Флуор ан 431
Флуорен 423
Флуоренкалий 424
Флуоренон 424
Флуоресцеин 431
Фолиевая кислота (Витамин Вс, Птероилглутаминовая кислота) 641, 704, 714
Формальдегид (Формалин) 268, 269, 403, 568, 613, 619, 621, 622, 662, 681
Формальдегидсульфокси л ат 397
Форманилид 308
Формилуксусный эфир 204
Формилэтилянтарный эфир 687
Фосген 287, 306, 327, 423, 429, 581, 712 Фосфенилхлорид (Фенилфосфиндихло-рид, Фенилдихлорфосфин) 326— 328
Фосфинобензол 327
Фосфобензол 327
Фосфора ароматические соединения 326—328
Фотоизомеризация 179
Фототропизм 74
Фотохимический распад 52, 69
Фреде реактив 645
Фрейденберг
получение эпикатехина 587 предполагаемый состав лигнина 346, 347
способ выделения лигнина из древесины 344
формула катехина 386
Фриделя—Крафтса реакция 213—213, 280, 395, 421—422, 424—425, 452, 520, 528, 590
Фридлендера синтез хинолина 614
Фрицше
выделение анилина 273, 297
— гармпна 678
получение антраниловой кисли!ы *273
— пикратов углеводородов 2/3
Фталазин (4,5-Бензопиридазин) 633
Фталаминовая кислота 389
Фталевые кислоты 85, 119, 206. 220, 375, 379, 399, 447, 471, 620 динитрил 547 имид, см. Фталимид
Фталевый ангидрид 376, 377, 43и—
432, 468, 471, 475, 476, 485. 598, 599, 616
Фталевый диальдегпд 633
Фталеины 206, 430
Фтал ид 377
Фталилхлорид 378
Фталимид 377, 389
Фталимид калия 377
Фталофенон 376
Фталоцианины 547
Фтивазид 605
Фтиокол (2-Метил-З-окси-а-нафтохинлч) 463
Фторанилин 309
Фторбепзойная кислота 293
Фторбензол 247
Фтор нафталины 456
Фуксин (Розанилин) 374, 427 — 429
Фуксин «кислый» 429
Фуксон 427
Фуксонимин 427
Фульвены 63
Фумаровая кислота 212, 521, 644
Фуран 511, 516, 519—524, 534 производные 346, 521
Фуранидин (Тетрагидрофуран) 511. 514, 521—524
а-Фуранкарбоновая (пирослизевая) кислота 520, 522, 523, 524
Фуранозы (у-Глюкозы) 521 а-Фурилальдегид, см. Фурфурол Фурилакриловая кислота 520 Фуриловый спирт 520, 523
Фуроин 520, 523
Фурфурол (а-Фурилальдегид) 519, 520, 521—523
Хавикол 406
Хассель, конформационный анализ циклогексана и его производных 30, 219
Хаульмугровая кислота 72, 73
Хаульмугровое масло 73
Хелидоновая кислота 581, 582, 644
Хенодезоксихолевая кислота, см.
За,7а-Диоксихолановая кислота
Хеноподиевое масло 131
Хиноксалины 362
Хинальдин (2- или а-Метил хинолин) 612—618, 657
Хинальдиновая (а-хинолинкарбоновая) кислота 615, 616
Хинальдиновый синтез Дебнера—Миллера 302, 612, 613
Хинанизол (п-Метоксихинолин) 616, 659
Хингидроны 355
Хинид (Хинолактон) 66
Хинидин 658, 683
Хинизарин (1,4-Диоксиантрахинон) 477
Хинин 388, 611, 612, 616, 619, 623, 642—645, 657—662, 664
Хининовая (п-метоксицинхониновая) кислота 616, 659
этиловый эфир 662, 663
Хининон 661
Хи нит 208
Хиницин 661
Хинная (циклогексантетраоксикарбоновая) кислота 65, 66, 355, 644, 667
Хинные алкалоиды 657—663 п-Хинодиметан 359
Хиноидная таутомерия, см. Бензоиднохиноидная таутомерия
Хиноидная теория цветности 369
Хиноидные красители 271, 357, 361, 369, 371, 562
Хиноксалин (Бензопиразин) 362, 483, 626, 627, 634
Хинолактон (Хинид) 66
Хиноли 360, 361
Хинолин (а,(3-Бензопиридин) 513, 550, 611—616, 618, 620, 659, 680 производные 617—620, 657—665, 685
Хинолина группа 611—624 а,р-Хинолиндикарбоновая (акридиновая) кислота 621
Хинолиниевые основания 617
а-Хинолин карбоновая (хинальдиновая) кислота 615, 616
у-Хинолинкарбоновая (цинхониновая) * кислота 616, 659, 662
Хинолиновая (2,3-пиридиндикарбоновая) кислота 604, 615
Хинолиновые основания 221, 396, 611, 614, 646
Хинолиновый желтый 616
а-Хинолон, см. о-Аминокоричная кислота, ангидрид
о-Хинон (о-Бензохинон) 303, 351, 356, 562, 628
л-Хинон (л-Бензохинон) 97, *265, 303, 351—357, 362, 461, 467, 562 гидразон 367 хлориды 354
Хинонди имин 356
л-Хинондиоксим 352
Хинонимины (Хинонимиды) 356, 367
л-Хинонмоноксим 313, 357
Хинонхлоримиды 356
Хинотоксин 661
Хинофталон (Бензопиридин, фталон) 599, 616, 617
Xинуклидины 658—660
Хитенин 659
Хлораль 396, 435
Хлоральгидрат 556
Хлорамин Б (Бензолдихлорсульфамид) 259
Хлорамин Т (л-Толуолдихлорсульф-
амид) 260
Хлоранизол 342
Хлоранил (Тетрахлорхинон) 355
Хлоранилины 309, 362, 400
*Хлорантрацен 469
Хлор ацетон 602
Хлорацетопирокатехин 342
Хлорацетофенон 282, 395
Хлорбензойная кислота 293, 622
Хлорбензол 224, 236, 239, 247, 255,
256, 266, 328, 329, 333, 435
Хлоргидрохинон 353
9-Хлордигидрофенарсазин (Адамсит) 631
Хлоркодиды 673
8-Х лор ксантин (2,6-Диокси-8-хлорпу-рин) 637
Хлорлепидин 614
Хлор метил имидазол 570
Хлорметилирование 244
Хлорморфиды 673
Хлорнафталины 453,. 454, 456
Хлорнитроанилин 400
Хлорнитробензолы 255, 257, 400,
599
Хлоробромиды 39
Хлорогеновая кислота 65
Хлоромицетин (Левомицетин) 694, 698
Хлорофилл 528, 533, 539, 544—546
Хлорофиллиды 545
Хлороформ 533, 550, 551
Хлорпикрин 274
ХлорпиридиньГ 533, 601, 603
а-Хлорпропионовая кислота, эфир 70 Хлортетрацикл ин (Ауреомицин, Биомицин) 485, 697
а-Хлортолуол, см. Бензил хлористый
Хлортолуолы 236, 243, 247, 293 я-Хлортрифенилметил 443 Хлоругольный эфир 287 Хлоруксусная кислота 559, 562 эфир 705
н-Хлорфенилгидразин 326
Хлорфенолы 270, 337 {В-Хлорхинолин 550 Хлорхинон 354 Хлорциклогексан 76 Хлорциклопентан 71 Хлорциклопропан 67 Холан 166
Холановая кислота 180
Холевая (За,7а, 12а-триоксихолановая) кислота 180—182, 489
Холестан 166
Холестанол 168, 170, 174, 178
Холестаноны 173
Холестерин 170—178, 181, 182, 189, 192, 194, 707
Холин 703
Хондродендрин (Бебирин) 666
Хризантемовая дикарбоновая кислота 73
Хризантемовая кислота 73
X ризаробин (Метилдиоксиантранол) 478
Хризен (1,2-Бензфенантрен) 174, 181, 486, 487
Хризоидин 367
Хризофановая кислота 478, 4?9
Xроман (Дигидробензопиран) 583. 704 производные 708
Хроматографическая адсорбция 61, 545 у-Хромен (Бензопиран) 513
Хромой (Бензо-у-пирон) 583, 584
Хромотроповая кислота (1,8-Диоксинафталиндисульфокислота) 457, 458
Хромофорная группа 372
Хромофорная теория окраски 369
Хромофоры 369, 373, 374, 628
Цвета адсорбционный хроматографический метод 545
Цветности теории 367—374
Цедрирет (Церулиньон) 415, 562
Цейзеля способ определения метоксильных групп 267, 646
Цейлонское коричное масло 125
Церулиньон (Цедрирет) 415, 562
Цетан 453
Цетановое число 453
Цефалоспорин N (Синнематин В) 690, 693
Цефаэлин 666, 667
метиловый эфир (Эметин) 620, 666
Цианидин 386, 586, 587
Цианиновые красители 617, 618
Цианины 617
Цианобензол, см. Бензонитрил (З-Цианопиридин (Никотиновая кислота, нитрил) 599
Циануксусная кислота 638
эфир 127
7-Цианохинолин 616
Цибет 86
Цибетон 86
Циглер полимеризация этилена 11 получение калийорганических соединений 334
работы в области алициклических углеводородов 22, 43
синтез соединений с трехвалентным углеродом 438, 439
Цикланы, см. также Алициклические углеводороды высшие 12, 56, 85—87, 158, 159 мостиковые 16, 18, 19 с конденсированными ядрами 16, 97—114, 504
Циклены 20, 88
Циклиты 64, 65
Циклоалканы, см. Алициклические углеводороды
Циклоалкилкарбинолы 48 Циклобутадиен 233, 491
Циклобутан (Тетраметилен) 11, 13, 21, 26, 28, 29, 52, 59, 68—70 производные 11, 20, 44, 51, 68—70, 135, 136, 409 реакции 52—56, 69
Циклобутандикарбоновые кислоты 68— 70
Циклобутанкарбоновая (тетраметилен -карбоновая) кислота 68, 69
Циклобутанол 48, 68, 69
Циклобутанон 68
Циклобутантетракарбоновый эфир 70
Циклобутен 36, 68, 69 Циклобутилбромид 48
Циклобутилкарбинол 47
Циклобутилхлорид 48 Циклогексадекан 59 Циклогексадеканон 87 Циклогексадиен-1,3 (Дигидробензол) 36, 76, 208
Циклогексадиен-1,4 36, 76
Циклогексакозан 59 Циклогексакозанон 87 Циклогексан (Гексагидробензол, Гексаметилен) 13, 21, 26, 34, 45, 59, 61, 75—75, 209, 225, 303 дегидрогенизация 57, 62, 75, 222, 225
Циклогексан производные 39, 45, 75—83, 114, ‘ 139—143, 222, 223, 303 реакции 51, 55, 57, 58, 75, 231 стереоизомерия 29—35
1,2,3,5/4,6-Циклогексангексол, см. Мезоинозит
Циклогександикарбоновые кислоты 77
Циклогексан-1,4-дикарбоновая (гекса-гидротерефталевая) кислота 35, 77
Циклогександиол-1,4 76 Циклогександион-1,4 207 Ци клогексан-2,5-дион-1,4-дикарбоно-вая кислота, эфир, см. Сукцинилянтарный эфир
Циклогександиуксусные кислоты 103 Циклогексан-1-карбокси-2-пропионо-вая кислота 103
Циклогексанкарбоновая (гексагидро-бензойная) кислота 51, 76, 79, 95, 288
Циклогексанол (Гексагидрофенол) 75— 78, 95, 266
Циклогексанон 50, 75—78, 83
1,4/2,3,5-Циклогексанпентол (Кверцит) 64
Циклогексантетраокси карбоновая (хинная) кислота 65, 66, 355, 644, 667
Циклогексантрион, см. 1,3,5-Трикето-гексаметилен
Циклогексен 76, 231
Циклогексендикарбоновая кислота 35 Циклогексилальдегид 76 Циклогексиламин 77, 303 Циклогексилбромид 95
Циклогексилиодид 95
Циклогексилкарбинол 49, 76
Циклогексилуксусная кислота 77
Циклогенэйкозанон 87 Циклогептадекан 59 Циклогептадеканон (Дигидроцибетон) 86, 87
Циклогептан (Суберан, Гептаметилен) 12, 13, 21, 26, 51, 54, 59, 62 производные 83, 652
Циклогептандиол 497
Циклогептанол (Субериловый алкоголь) 49, 83
Циклогептанолон 497
Циклогептанон (Суберон) 40, 50, 51, 83, 653 *
Циклогептатриен (Тропилиден) 83, 493, 653
Циклогептатриенилий, см. Тропилий Циклогептатриенкарбоновая кислота 220, 656
Циклогептатриен-2,4,6-ол-2-он, см. Трополон
Циклогептатриен-2,4,6-он, см. Тропой
Циклогептен 88 Циклогептиламин 48 Циклодекан 26 Циклодеканон 87 Циклодекапентаен 84 Циклодецен 84, 88 Циклододекан 59 Циклододеканон 87 Циклододецен 88 Циклодокозан 59 Циклодокозанон 87 Циклононадеканон 87 Циклононакозан 59 Циклононакозанон 87 Циклононан 59 Циклононанон 51, 87 Циклононен 88 Циклооктадекан 59 Циклооктадеканон 87 Циклооктакозан 59 Циклооктакозанон 87 Циклооктан 21, 26, 54, 59 производные 83—85, 657
Циклооктанол 84
Циклооктанон 51, 87
Циклооктатетраен 83, 84, 229, 233, 491 , 657
Циклооктен 37, 84. 88
Циклооктин 36, 37
Циклопарафины, см. Алициклические углеводороды
Циклопентадекан 59
Циклопентадеканон (Экзальтон) 87
Циклопентадиен 36, 63, 109, 111, 491
димер 63
производные 423, 466, 502—504 Циклопентадиенилкалий 492 Циклопентадиеноциклогептатриен, см.
Азулен
Циклопентаметиленсульфон 515 Циклопентаметиленимин, см. Пиперидин Циклопентаметиленсульфид (Тетрагидротиопиран) 515
Циклопентан (Пентаметилен) 12, 13, 21, 26, 29, 51, 59, 61, 70—73 производные 12, 39, 70—75, 136, 141, 151, 181 реакции 53—55
Циклопентандиол 35, 71
Циклопентанкарбоновая кислота 71, 72-
Циклопентанол 71, 72
Циклопентанон 40, 41, 52, 70—72, 484 1,2-Циклопентано-1,2,3,4-тетрагидро-фенантрен 484
Циклопентен 36, 47, 71, 72
1,2-Циклопентенофенантрен 165, 484 производные 173, 174, 181, 687
Циклопентецоциклогептанон 503 Циклопентилальдегид 71 Циклопентиламин 71
Циклопентилбромид 94
Циклопентилиденциклопентанон 95
Циклопентилкарбинол 71
Циклопентаметиленимин, см. Пиперидин
Циклопентилуксусная кислота 71
Циклопентилциклопентанон 94
Циклопропан (Триметилен) 11, 21, 26— 28, 52—55, 59, 66—68 производные 20, 36—38, 40, 56, 59, 66—68, 96
Ци клопропан-1,2-дикарбоновая кислота 66, 68
Циклопропанкарбоновая (триметилен-карбоновая) кислота 66, 67
Циклопропан 36, 67
Циклопропилкарбинол 48, 69
Циклопропилметилхлорид 48
Циклотетрадекан 59
Циклотетрадеканон 87
Циклотетракозан 59
Циклотетракозанон 87
Циклотетратриакоптапон 87
Циклотриаконтан 59, 86
Циклотриаконтанон 87
Циклотридекан 59
Циклотридеканон 87
Циклотрикозанон 87
Циклоундеканон 87
Циклоундецен 88
Циклофаны 490
Циклоцитрали 80
Циклоэйкозанон 87
Циклы высшие 33, 88
м - Ц и мол (ж-Мети л изоп р’оп и л бе 11 зол)
119, 153
п-Цимол (n-Метил изопропил бензол)
115, 118, 119, 142, 217, 226, 227, 379
Цингерои 395
Цингиберен 157—159
Цинеол 130
Цинерины 73
Цинеролон 73
Циннамилкокаин 655
Циннолин (3,4-Бензопиридаз1Гн) 633
Цинхолойпон 660
Цинхолойпоновая кислота 660
Цинхомероновая (р,у-пиридинка'рбоно-вая) кислота 604, 620
Цинхонидин 658
Цинхонин 611, 615, 657—661
Цинхониновая (7-хинолинкарбоновая) кислота 616, 659, 662
Цинхонинон 661
Цинхотенин 659
Цинхотоксин 661
Цитрали 80
Цитронелловое масло 147
Чан-су 184, 185
Чернила ализариновые 388
«Черный анилин» (Анилиновый черный) 304, 356
Чичибабин
«бирадикал» 444, 445
исследование пиридина и его производных 591—593, 597, 602
получение индола 548
— карбостирила 618
— лепидина 613
— пилоповой кислоты 687
— тиофена 525, 526
распад пентафенил- и гексафенил -этана 437
строение мускуса 253
углеводороды с хинондиметидной группировкой 359
Ч у гаев комплексы тяжелых металлов с ализарином 480
реакция на холестерин 171
Шалфейное масло 153
Швейцера реактив 344
Шейблера реактив 645
Шеллак 161
Шеффера кислота (2-Нафтол-6-сульфо-кислота) 457
Шиффовы основания (Анилы) 302, 681
Шорыгин галоидирование гомологов бензола 359
механизм реакции Вюрца—Фитти-га 212
получение р-фенилэтилового спирта 275
превращения натрийорганических соединений 212, 213, 333
Шоттен.—Бауманна метод 290, 292
Щавелевая кислота 47, 48, 429, 545, 641 анилиды 302 соли 643 эфиры 43, 141
Эвгенол (З-Метокси-4-оксиалл и л бензол) 339, 340
Эверниновая кислота (4,6-Диокси-о-то-луиловая кислота, метиловый эфир) 384
Эверновая кислота 384
Эвкалиптовое масло 125, 130
Эзерин (Физостигмин) 679, 680
Эзеролин 679
Эйксантон (4,6-Диокси ксантон) 588
Экасанталовая кислота 160
Экваториальная связь 31, 121, 167
Эквиленин 196, 197
Экгонин (Тропин-2-карбоновая кислота) 538, 652, 655, 656
метиловый эфир 656
Экзальгин (Метилацетанилид) 308
Экзальтон (Циклопентадеканон) 87
Экзо-формы НО
Экстинкция молярная (Молярный коэффициент поглощения) 368
Элайомицин 702
Электрокортин (Альдостерон) 195
Электронная плотность
ароматических соединений 233—237, 240—242
выравнивание в пиридине 590
— в трополоне 498
— в циклобутадиене 491
— в циклооктатетраене 491
нафталина 449, 450
7-пирона 576
смещение 372
Электрофильные (катионопдные, электроноакцепторные) реагенты 241, 242
Электрохимическое восстановление индола 551 —нитробензола 250, 363 окисление 276
Элеми масло 125, 126
Элеоптены 116
Элиминирование диазогруппы 321 сульфогруппы 258
Эллиговая кислота 387
Эмеральдин 304
Эметин (Цефаэлин, метиловый эфир) 620, 666, 667
Эмодины 478
Эндо-формы НО
Эозин (Тетрабромфлуоресцеин) 431
Эпиандростерон 190
Эпиборнеол 144
Эпиинозит 64
Эпи катехин 386, 587
Эпимеры 168, 169, 172
Эписоединения 169
Эпихинин 664
Эпихлоргидрин 510
Эпихолестанол 168
Эпоксидные смолы 510
Эргоалкалоиды 684—686
Эргокорнин (Эрготоксин) 685
Эргостерин 170, 178, 191, 706
Эрготамин 685
Эрготоксин (Эргокорнин) 685
Эрдмана реактив 645
Эр иод иктин 704, 708
Эриодиктиол 708
Эритрин 384
Эритрозин 431
Эритромицин 699
Эритронолид 700
Эритро-ряд 695
Эрленмейера формула нафталина 446, 447
Эскулетин (Триоксикоричная кислота, лактон) 412
Эстрагол 406
Эстрадиол 196
Эстран 167
Эстриол 197
Эстрогенные гормоны 196, 197, 433
Эстрон 196, 197
Этана конформации 22, 23
Этаноламин (Коламин) 378, 510
Этара реакция 276
Этспилцпклопропан, см. Винилцикло-пропан
Этиланилин 308
Этилбензоат (Бензой но-этиловый эфир) 289
Этилбензол 217, 224, 226, 281, 401
дегидрирование 401 гидроперекись 281
Этилен 226
бромистый 572
окись 275, 291, 509, 510
полимеризация 11, 44
Этиленгликоль 379
Этилендиамин 571
Этилен имин 509, 510
Этиленмочевина 571
Этиленовые соединения, реакции 565
Этиленовые углеводороды 401
Этиленсульфид 509, 510
Этилнитрит 662
Этиловый красный 617
Этилпиридины 594, 659
N-Этил-а-пиридон 600
Этилфенилкарбинол, дегидратация 403
р-Этилхинуклидин 660
Этилциклогексан 61
Этилциклопентан 61, 71
Этинилтестостерон 191
Этиобилиановая кислота 173
Этиохолан (Тестан) 166
2-Этокси-6,9-диаминоакридин, молочнокислая соль (Риванол) 623
а-Этоксипиридин 600
Эудалин (1 -Метил-7-изопропил нафталин) 159, 453
Эукодал, см. Дигидрооксикодеинон
Эуродин (Толуиленовый красный) 629
Эуродины (Аминофеназины) 629
Эфедрин 309, 647, 664, 695
Эфирные масла 63, 73, 81, 116, 152—154, 161, 274, 280, 287, 340, 393, 406, 503
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 767
Эффект сопряжения 234, 264 трансаннулярный 34, Яды жаб 184 Янтарная кислота 41, 173, 525, 643 88 Янтарный альдегид 529, 530, 653. 65/ Янтарный ангидрид 521
Юглон (5-Окси-а-нафтохинон) 462 Янтарный эфир 41, 207
Яблочная кислота 577 соль 643 Янтарь 161 Японский лак 161 Яра-яра, см. р-Нафтол
АЛЕКСЕИ ЕВГЕНЬЕВИЧ ЧИЧИБАБИН
ОСНОВНЫЕ НАЧАЛА ОРГАНИЧЕСКОЙ ХИМИИ
Copyleft ® SanTcy incorporation
derevyaha + Q
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стр. Строка Напечатано Должно быть
15 13 сверху «-бутил и 1',1'-диметоэтил вместо втор-бутил, втор-бутнл вместо и 1',1'-димет оэтил /пре/п-бутил,
18 1 снизу исходящий нисходящий
1о4 ' 4 снизу в изучении сесквитерпенов достигнуты большие успечи они долгое время оставались мало изученными. В последние 40 лет в изучении сесквитерпенов достигнуты большие успехи
166 Нижняя строка таблички Холестан Копростан Копростан Холестан
167 18 сверху имеется шесть имеется семь
194 4 снизу (левая формула) NZ\cho ^ДуСНО ( 2Н3 /^сно \ /СН0
Н
219 Формула а-изомера перевернута на 180’
350 18 сверху Na > Н2-СООС2Н5 Na СН2—СООС2Н5
411 2-я формула сверху соон 1 С \ сн соон сн сн
О О он
475 1-я формула снизу II о— fYYY XZY/\Z о Il 1 о-fYYY Y/Y/Z li О
493 1 снизу (в части тиража) ром. бром.
495 3-я формула сверху О 0~
634 5 снизу (в части тиража) С H4N4O3 c8h4n4o3
670 1 сверху тебаин C19H21O3N тебаин С19Н2зО3Кт
710 В формуле тиохрома II 1 1
Зак. 1454. А. Е. Чичибабин, том II.