/



























































































































































































































































































































































Автор: Жигалова К.А. Розенфельд И.Л.
Теги: испытания материалов товароведение силовые станции общая энергетика металлургия металловедение
Год: 1966




Текст
И. Л. РОЗЕНФЕЛЬД, К. А. ЖИГАЛОВА
хш
238415
УСКОРЕННЫЕ МЕТОДЫ
КОРРОЗИОННЫХ
ИСПЫТАНИИ
МЕТАЛЛОВ'
(ТЕОРИЯ И ПРАКТИКА)
ИЗДАТЕЛЬСТВО «МЕТАЛЛУРГИЯ»
Москва 19 6 6
УДК 620. 1.99
АННОТАЦИЯ
В монографии излагаются научные принципы
ускоренных коррозионных испытаний и практиче-
ское их применение. Описываются -различные ме-
тоды испытаний металлов и сплавов и определе-
ния .защитной способности покрытий, смазок, ин-
гибиторов. Освещаются теория и практика опреде-
ления склонности металлов к межкристаллит-
ной коррозии, коррозионному растрескиванию, то-
чечной коррозии, кавитации. Рассматриваются так-
же методы испытаний реакторных материалов.
Книга представляет научный и практический ин-
терес для большого круга научных и ннженерно-
гехнических работников, занимающихся вопроса-
ми коррозии и защиты металлов в металлургичес-
кой, машиностроительной, химической и атомной
п ромышлеиности.
3-12-5
ОГЛАВЛЕНИЕ
Предисловие.......................................... 5
I
ОСНОВНЫЕ ПРИНЦИПЫ УСКОРЕННЫХ МЕТОДОВ
КОРРОЗИОННЫХ ИСПЫТАНИЙ
и
МЕТОДЫ УСКОРЕННЫХ КОРРОЗИОННЫХ ИСПЫТАНИЙ
Ц. ’Испытания при полном погружении в электролиты . .. !3-
2' Испытания при периодическом погружении в -электролиты 38
3. Испытания при периодическом обрызгивании электролитами 50‘
-)4. Испытания в атмосфере с постоянной влажностью , . 52‘
5. Испытания в атмосфере с постоянной влажностью в при-
сутствии коррозиоиио активных агентов...............60-
'6. Испытания, воспроизводящие условия конденсации , 74
Литература..................................... .88
ш
ВЫБОР ПОКАЗАТЕЛЯ КОРРОЗИИ И МЕТОДА ОЦЕНКИ
КОРРОЗИОННОЙ СТОЙКОСТИ '.
ч. L Изменение массы образцов (весовой метод). . . '90
2. Объемные методы '................................. . 99-
3. Определение глубины проникновения коррозии ... . . 104 -
'.4. Определение коррозии по изменению механических свойств 109-
5. Определение коррозии по изменению отражательной способ-
иости поверхности металла...............................110* ~
6. Определение -коррозии по изменению электрического сопро-
тивления ...................................... ... Illi
7. Оценка стойкости по времени до появления первого .коррози-
онного очага илн определенной площади коррозии . . . 114 -
8. Определение количества металла, перешедшего в‘ раствор
в процессе коррозии . .........................117
•9. Количественная обработка экспериментальных результатов 117'"
Литература . . ............... .... 118
— 3 —
IV
ЭЛЕКТРОХИМИЧЕСКИЕ И ЭЛЕКТРИЧЕСКИЕ МЕТОДЫ
КОРРОЗИОННЫХ ИСПЫТАНИЙ МЕТАЛЛОВ
^1. Измерение электродных потенциалов . ............119
v=2. Снятие поляризационных кривых . .... 136
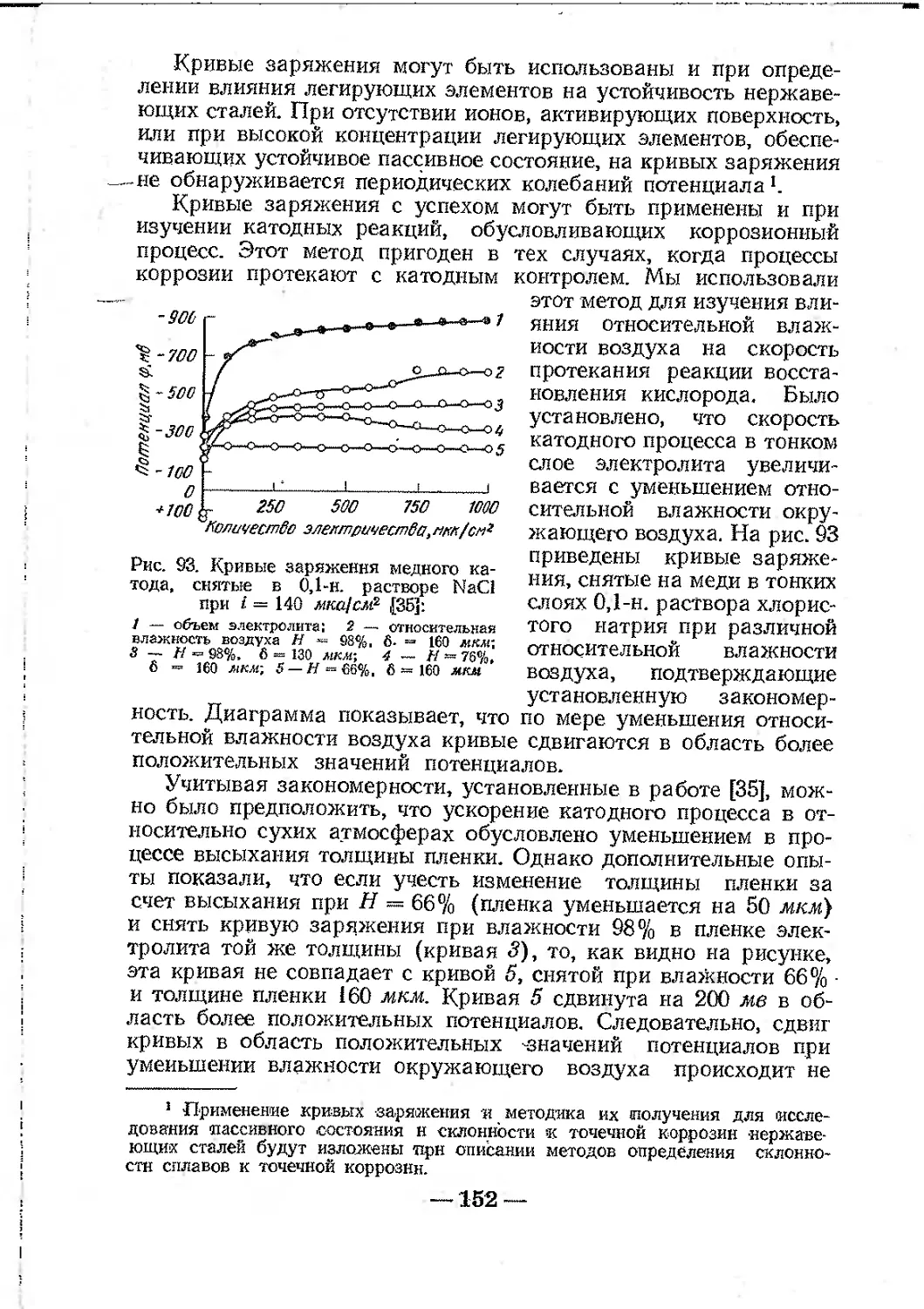
3. Получение кривых заряжения.....................150
4. Измерение силы тока, возникающего между двумя электро-
дами .......................................... ... 153
5. Электрические методы . . . . 158
Литература . ... 167
V
ОПРЕДЕЛЕНИЕ ЭФФЕКТИВНОСТИ ЗАЩИТНЫХ СРЕДСТВ
1. Металлические покрытия..................... .... 169
2. Неорганические пленки - . ... 180
-.. 3. Лакокрасочные покрытия . . . 184
4. Смазки.................................. ....... 211
5. Ингибиторы ... ..................220
Литература .... ... 237
VI
НЕКОТОРЫЕ СПЕЦИФИЧЕСКИЕ МЕТОДЫ
УСКОРЕННЫХ КОРРОЗИОННЫХ ИСПЫТАНИЙ
1. Определение склонности сплавов к межкристаллитной кор-
розии ........................................... 240
2. Определение склонности сплавов к коррозионному «растрес-
киванию ..........................................267
3. Определение склонности нержавеющих сталей к точечной
коррозии ........................................298
4. Определение устойчивости металлов в условиях кавитации 316
5. Определение устойчивости реакторных материалов при вы-
соких температурах и давлениях..................322
Литература...................................... 344
ПРЕДИСЛОВИЕ
Цель ускоренных методов коррозионных испытаний — полу-
чить в лабораторных условиях в возможно более короткий срок
данные, позволяющие оценивать коррозионное поведение метал-
лов, изделий и конструкций в естественных условиях в течение
длительного времени.
Этими методами должен быть обеспечен также контроль ка-
чества средств противокоррозионной защиты заводской продук-
ции в соответствии с требованиями государственных стандартов.
За последнее время в связи с расширением объема морских
перевозок, экспортом изделий и машин в страны с тропическим
климатом, консервацией дорогостоящей техники сильно возрос
интерес к ускоренным методам коррозионных испытаний. В Со-
ветском Союзе и за границей усиленно ведутся работы по усовер-
шенствованию и стандартизации существующих методов испы-
таний и по разработке новых. Между тем до сих пор нет руко-
водства по ускоренным методам „испытаний, доступного для
широкого круга исследователей и инженерно-технических ра-
ботников.
Учитывая крайнюю заинтересованность многих специалистов
и организаций в таком руководстве, авторы взяли на себя труд
изложить теоретические основы ускоренных методов коррозион-
ных испытаний металлов и практическое их применение.
Успехи, достигнутые за последнее время в раскрытии меха-
низма коррозионных процессов, дали возможность при решении
проблемы ускоренных испытаний в большей мере основываться
на научных предпосылках, чем это делалось раньше.
Разнообразие коррозионных процессов не позволяет в рам-
ках настоящей монографии рассмотреть все виды испытаний..
Поэтому мы рассмотрим лишь наиболее распространенные' виды
испытаний, опустив преследующие чисто специальные цели.
В связи с переходом в технической и справочной литературе
на новую Международную систему единиц СИ в настоящей кни-
ге материал излагается в основном в единицах системы СИ!
— 5 —
Для удобства читателей в таблицах, а также в расчетных
формулах стоят вначале наименования единиц в системе СИ, а
затем в скобках приведены единицы прежних систем.
Следует обратить внимание на то, что часто встречающийся
в литературе по коррозии термин «вес» неправильно исполь-
зуется для характеристики количества вещества, поэтому, когда
речь идет о потерях металла, употребляются выражения «изме-
нение массы металла», «метод определения изменения массы
металла» и т. п.
Для упрощения пересчета концентрации вещества выражены
в е/сЫ3 или жа/сЫ3, давление — в барах.
Настоящая монография, вероятно, не лишена недостатков,
поэтому все замечания читателей авторы примут с большой бла-
годарностью.
I
ОСНОВНЫЕ ПРИНЦИПЫ УСКОРЕННЫХ
МЕТОДОВ КОРРОЗИОННЫХ ИСПЫТАНИЙ
При разработке и выборе ускоренных методов коррозионных
испытаний следует руководствоваться одним весьма важным
принципом, который заключается в следующем. Коррозионная
стойкость — это ие абсолютное свойство самого металла; она
определяется не в меньшей степени и характером коррозионной
среды. Разноообразие условий внешней среды требует использо-
вания различных методов для определения коррозионной стой-
кости металлов. Поэтому невозможно создать универсальные
методы ускоренных коррозионных испытаний.
Методы испытаний должны учитывать характер коррозион-
ного процесса и основываться на современных представлениях
о механизме коррозии. Чисто формальные методы, ие учитываю-
щие требований теории, легко приводят, как отметил Г. В. Аки-'
мов, к ошибочным заключениям (1]. Для правильного выбора ме-
тода испытаний, необходимо, во-первых, учитывать условия экс-
плуатации изделия и, во-вторых, иметь ясное представление о
механизме процесса, протекающего в данных условиях.
Можно сформулировать ряд требований, которым должны
удовлетворять ускоренные методы испытаний, и наметить пути
их осуществления. Теоретические положения, обосновывающие^
эти требования, будут более подробно изложены при рассмот-
рении отдельных видов испытаний.
1. Ускорение коррозионного процесса не должно быть ре-
зультатом изменения его механизма. Для процессов коррозии,
протекающих .преимущественно с кислородной деполяризацией,
нельзя получить надежные результаты путем применения при ус-
коренных испытаниях кислых электролитов, в которых процесс
протекает с водородной деполяризацией. Так, например, по-
пытки разработать ускоренные методы испытаний алюминиевых
сплавов для оценки их стойкости в атмосферных условиях,
основанные на принципе разбрызгивания в камерах серной кис-
лоты, оказались безуспешными.
— 7 —
2. При выборе ускоренного метода испытания нужно учиты-
вать состав и свойства коррозионной среды, в которой будут эк-
сплуатироваться изделия. Нельзя применять один и тот же элек-
тролит для имитации всех коррозионных сред. Так, например,
промышленная атмосфера, характеризующаяся наличием в воз-
духе сернистого ангидрида, который впоследствии растворяется
в тонких слоях электролитов и изменяет характер течения ка-
тодного процесса, отлична от морской атмосферы, характеризу-
ющейся наличием в воздухе частичек хлористого натрия, влия-
ющих в основном, на анодный процесс. Изделия, эксплуатируе-
мые в морской и речной воде, нельзя испытывать в одном и том
же электролите.
Известно, что многие лакокрасочные покрытия, обладающие
высокими защитными свойствами в морской воде, оказываются
неустойчивыми в речной.
Если речь идет об атмосферной коррозии, то все атмосферы
следует разделить на три основные группы: промышленные, мор-
ские и сельские и применительно к ним выбирать электролиты
для ускоренных испытаний; при этом необходимо иметь в виду,
что в каждой группе имеются различные климатические и ат-
мосферные условия. Изделия, которые эксплуатируются или
транспортируются в тропическом морском климате, необходимо
испытывать при относительно высоких температурах и влажно-
сти в присутствии частичек хлористого натрия. Для изделий, эк-
сплуатируемых в северных приморских районах нашей страны,
температура испытания должна быть снижена. Если изделия,
имеющие защитные покрытия, подвергаются действию ультра-
фиолетовых лучей, то необходимо во время ускоренных испыта-
ний предусмотреть возможность имитации этих условий; особен-
но это относится к лакокрасочным покрытиям.
3. Ускоренный метод испытания должен учитывать условия
работы изделия. Процессы коррозии, протекающие в атмосфе-
рах, характеризующихся сильными перепадами температура
приводящими к периодической конденсации влаги, по своей при-
роде отличаются от тех, которые развиваются, например, на
конструкциях, подвергающихся периодическому смачиванию
брызгами морской или речной воды.
Ясно, что для этих различных по своему характеру коррози-
онных процессов должны быть выбраны и различные методы ис-
пытаний. Так, например, подбирая стали для свай оснований
морских нефтепромысловых сооружений, необходимо исходить
из того, что наибольшей коррозии сваи подвергаются в зоне пе-
риодического смачивания (части -свай, полностью погруженные
в морскую воду, корродируют медленнее) и ускоренные испыта-
ния проводить по методу переменного погружения. Сплавы для
конструкций и приборов, подвергающихся периодическому наг-
реву и охлаждению во влажной воздушной атмосфере, необхо-
димо испытывать, конденсационными методами.
4. Ускоренные методы испытаний необходимо разрабатывать
и выбирать для каждой группы сплавов в отдельности. Не мо-
жет быть единого метода ускоренных испытаний для всех спла-
вов и тем более единых коэффициентов пересчета' результатов
ускоренных испытаний иа длительную эксплуатацию, потому что
даннаццоррбзйонная среда или данный вид испытаний не в оди-
наковой степени ускоряет процесс коррозии различных метал-
лов. Так, периодическая конденсация влаги увеличивает корро-
зию стали и цинка, а коррозию никеля ускоряет незначительно,
если атмосфера не содержит промышленных загрязнений.
Железо и его сплавы, как и сплавы алюминия с медью, весь-
ма чувствительны к периодическому смачиванию электролита-
ми. Коррозия же кадмия и чистого алюминия при этом виде
испытания ускоряется в меньшей степени.
5. При оценке коррозионной стойкости сплавов и средств
} противокоррозионной защиты важно правильно выбирать пока-
V затель коррозии. Весовой показатель, удовлетворительно отра-
жающий поведение стали, меди и цинка, не совсем применим к
алюминиевым сплавам и нержавеющим сталям. Поведение пос-
ледних металлов оценивается наиболее правильно по глубине
проникновения коррозии и коэффициенту ее неравномерности.
При испытании низколегированных сталей определения потери
вещества должны "быть обязательно дополнены измерениями,
характеризующими глубину проникновения коррозии.
Определение качества защитных покрытий по изменению
массы образцов почти никогда не давало удовлетворительных
результатов. Эффективность защитных покрытий следует опре-
делять по изменению их толщины, времени появления первых
коррозионных очагов и площади, пораженной коррозией. Обыч-
но определяют'время, которое необходимо для того, чтобы 50%
поверхности подверглись коррозии. Иногда полезной оказыва-
ется оценка по 10-балльной шкале, устанавливающей балл в
зависимости от площади, занятой коррозией за данный проме-
жуток времени.
Разноречивые оценки свойств сплавов и средств защиты, по-
являющиеся периодически в литературе, в значительной степе-
ни обусловлены применением различных показателей коррозии,
а иногда и неправильным их выбором.
6. При выборе метода ускорения коррозионного процесса
необходимо учитывать контролирующий фактор. Скорость лю-
бого коррозионного процесса, протекающего по электрохимичес-
кому механизму, зависит от скорости течения двух сопряженных
электрохимических реакций — анодной, заключающейся в пере-
ходе ион-атомов металла из металлической решетки "В электро-
лит и сопровождающейся освобождением электронов, и
катодной, заключающейся в ассимиляции освобождающихся при
анодной реакции электронов. Иногда (чаще всего в макросисте-
мах) скорость процесса может определяться омическим сопро-
— 9 —
тивлением электролита. Не всегда обе реакции-в одинаковой сте-
пени определяют скорость коррозионного процесса, чаще всего
одна из них наиболее замедленная. Она и определяет скорость
коррозионного процесса или, как часто говорят, является кон-
тролирующей.
Общее правило, которым следует руководствоваться при вы-
боре метода ускорения процесса, состоит в следующем. В пер-
вую очередь необходимо влиять на тот фактор, который являет-
ся контролирующим. Если скорость коррозионного процесса оп-
ределяется скоростью электрохимической реакции, то ускорять
необходимо в первую очередь ту реакцию, которая наиболее за-
медленна и определяет общую скорость коррозии.
.Процессы, протекающие с катодным контролем, можно резко
усилить, лишь изменив кинетику катодного процесса. Добиться
заметного ускорения коррозии, протекающей с анодным ограни-
чением, можно, лишь увеличив скорость анодной реакции.
Коррозионные процессы, протекающие со смешанным конт-
ролем, можно ускорять, изменяя кинетику обеих электродных
реакций. В тех случаях, когда скорость коррозионного процесса
определяется и сопротивлением электролита, т. е. омическим па-
дением потенциала, можно ускорить испытания, увеличивая
электропроводность среды. Это применимо чаще всего к макро-
системам, поскольку омическое падение потенциала в микроэле-
ментах даже в сильно разбавленных электролитах, а также в
тонких слоях электролитов, ничтожно мало [2].
7. Метод и режимы испытаний должны обеспечивать усло-
вия протекания коррозионного процесса в течение значительной
части времени с большой скоростью. Существует несколько ме-
тодов повышения скорости коррозии. Применительно к атмос-
ферной коррозии илн случаям периодического воздействия элек-
тролита на металл наиболее простым является увеличение
времени воздействия электролита на металл. Известно, что в ат-
мосферных условиях время воздействия электролита на металл
ограничено и поэтому увеличение времени контакта металличес-
кой поверхности с электролитом может уже само по себе увели-
чить суммарный эффект и тем самым сократить время испыта-
ния. Поэтому простой путь ускорения испытаний, имитирующих
атмосферные условия, заключается в том, чтобы электролит
возможно дольше действовал на металлическую поверхность.
Считали поэтому, что лучше всего проводить испытания мето-
дом погружения образцов или изделий в объем электролита.
Однако при изучении кйнетики реакций в тонких слоях электро-
литов была установлена количественная зависимость между
скоростью катодного процесса и толщиной пленки электролита
и показано, что скорость кислородной деполяризации в тонких
слоях электролитов намного выше, чем в объеме [3].
Учитывая сформулированное выше положение о том# что не-
обходимым условием ускоренных испытаний является ускорение
— 10 —
контролирующей ступени процесса, а также то, что в нейтраль-,
пых электролитах процесс коррозии, как правило,-лимитируется
скоростью кислородной деполяризации, результаты можно полу-
чить быстро, увеличив скорость катодного процесса. Отсюда
следует, что испытания изделий, предназначенных для эксплуа-
тации в атмосферных условиях, необходимо проводить таким
образом, чтобы металл подвергался возможно дольше дейст-
вию тонких слоев электролитов. Подобные испытания к тому же
более близки к естественным условиям эксплуатации изделий в
атмосфере. Однако толщину пленки электролита на испытуемом
металле нельзя бесконечно уменьшать, так как в очень тонкие
пленках наряду с облегчением катодной реакции может насту-
пить вследствие концентрационных явлений резкое торможение
анодной реакции, что замедлит коррозионный процесс. Выбор
оптимальной толщины пленки, методы ее сохранения на поверх-
ности и регулирование скорости реакций рассматриваются ниже
при обсуждении отдельных методов испытаний.
8. При ускоренных методах коррозионных испытаний целе-
сообразно использовать возможность ускорения электрбхими-
ческих реакций, обусловливающих коррозионный процесс,
агрессивными компонентами или деполяризаторами. При испы-
тании металлов при полном погружении с целью увеличения ско-
рости катодного процесса можно вводить перекись водорода или
иные деполяризаторы. При атмосферных ускоренных испыта-
ниях можно ускорить процесс введением в атмосферу агрессив-
ных компонентов. При выборе одного из них необходимо учиты-
вать, содержится ли тот или иной компонент в атмосфере.
Поэтому при ускоренных испытаниях изделий, предназначенных
для эксплуатации в атмосфере морского воздуха, желательно в
камеру ввести частички хлористого иатрия, распределив их в
атмосфере в виде сухого аэрозоля или тумана. Для имитации
условий промышленной атмосферы желательно в конденсацион-
ную камеру или аппарат переменного погружения ввести сер-
нистый газ. Скорость коррозионного процесса можно при этом
увеличить в десятки, а иногда и в сотни раз.
9. При использовании в ускоренных коррозионных испытани-
ях температурного фактора как средства повышения скорости
коррозии необходимо учитывать характер протекающего про-
цесса. Повышение температуры — важный фактор, определяю-
щий скорость коррозии. Скорость электродных реакций с
повышением температуры увеличивается, но одновременно тем-
пература влияет на ряд других факторов — растворимость кис-
порода, свойства защитных пленок на металлах и т. п. Необхо- 5
димо иметь в виду, что температура увеличивает скорость кис- .«
дородной деполяризации лишь до определенного предела ;
(~60°С). Дальнейшее повышение температуры резко уменьша- j
ет растворимость кислорода, что приводит к обратным результа- •
там, т. е. к уменьшению скорости коррозии. Для процессов кор-
— 11 —
розии, протекающих с водородной деполяризацией (кислые элек-
тролиты), этих ограничений не существует и температуру можно
повышать вплоть до температуры кипения.
При этом необходимо учитывать изменение температурного
коэффициента процесса с температурой.
Если атмосферные условия имитируются путем создания на •
поверхности металла тонких пленок электролитов методом кон-
денсации или переменного погружения, последующее сильное
повышение температуры будет способствовать лишь быстрому
испарению электролита и уменьшению времени пребывания его
на металлической поверхности. Если не предусмотреть частое об-
новление электролита, повышение температуры не будет способ-
ствовать ускорению коррозии. Наиболее рациональные режимы
подобных испытаний рассматриваются ниже.
10. При ускоренных испытаниях не следует чрезмерно уско-
рять процессы коррозии. Скорость коррозии большинства ме-
таллов в сильной степени зависит от свойств образующихся за-
щитных пленок или. скорости их разрушения, если они имелись
на поверхности металла до. испытания. В естественных условиях
для возникновения защитных;.слоев или их разрушения требует-
ся длительное время. Сильноеа&окращение времени испытания
за счет увеличения агрессйвноси^реды приводит часто к из-
менению характера процесса, свойств родуктов коррозии и их
распределения по поверхности; меняются также защитные свой-
ства пленок и интенсивность коррозии. Для материалов, мало
отличающихся по коррозионной стойкости, продолжительность
испытания должна быть выбрана с таким расчетом, чтобы это
различие можно было оценить.
11. Ускоренные лабораторные испытания следует по возмож-
1 мости использовать для получения сравнительных данных. Учи-
тывая, что мы еще не располагаем надежными коэффициента-
ми пересчета результатов ускоренных испытаний на условия
длительной эксплуатации, целесообразно при ускоренных испы-
таниях новых сплавов или средств защиты параллельно испыты-
’ вать родственные сплавы или покрытия, по которым уже имеют-
ся надежные данные об их коррозионном поведении.
Сопоставляя эти результаты, удается, как правило, сделать
вполне обоснованные заключения о поведении материалов., и
средств защиты в условиях длительной эксплуатации.
ЛИТЕРАТУРА .
1. Акимов Г. В. Теория и методы исследования коррозии металлов. Изд-
во АН СССР, 1945.
2. Розенфельд И. Л., Павлуцкая Т. И. *ЖФХ, 1956, т. 30, вып. 6,
1427.
3. Розенфельд И. Л., Павлуцкая Т. И. ДАН СССР, 1953, т. 91,
№ 2. 315.
II
МЕТОДЫ
УСКОРЕННЫХ КОРРОЗИОННЫХ ИСПЫТАНИЙ
Известные до настоящего времени методы ускоренных испыта-
ний наиболее целесообразно, на наш взгляд, классифициро-
вать по характеру создаваемых условий:
1. Испытания при полном погружении в электролиты.
2. Испытания при периодическом погружении в электролиты.
3. Испытания при периодическом обрызгивании электроли-
тами.
4. Испытания в атмосферах с постоянной влажностью.
5. 'Испытания в атмосферах с постоянной влажностью в при-
сутствии коррозионных агентов.
6. Испытания, воспроизводящие условия конденсации.
Ниже рассматривается каждый метод в отдельности с изло-
жением научных принципов, методики проведения испытания и
аппаратурного оформления его.
1. ИСПЫТАНИЯ ПРИ ПОЛНОМ ПОГРУЖЕНИИ
В ЭЛЕКТРОЛИТЫ
Ускоренные испытания металлов при полном погружении в
электролиты следует проводить только в том случае, когда ме-
таллы предназначаются для работы в подобных условиях.
Скорость любого коррозионного процесса, протекающего по
электрохимическому механизму, зависит От скорости двух со-
пряженных реакций: катодной и анодной. О скорости этих реак-
ций лучше всего судить по изменению потенциала электрода при
пропускании через него электрического тока. Реакции, идущие с
большой скоростью, не приводят к сколько-нибудь заметным из-
Д«> л
менениям потенциала электрода —0.
AZ
Реакции, протекающие со значительным торможением какой-
либо из стадий суммарного процесса, сопровождаются’ замет-
— 13 —
ним изменением потенциала электрода —
Дг
оо. Поэтому ос-
новным методом исследования кинетики электродных реакций
считается метод построения поляризационных кривых, т. е. кри-
вых зависимости потенциала электрода от плотности тока,
О характере коррозионного процесса и факторах, определяю-
щих его скорость, обычно делают заключение на основании кор-
розионной диаграммы, которую строят по поляризационным
Рис. 1, Диаграмма коррозионного
процесса:
А Б — кривая аиодаой поляризации:
ВГ — кривая катодной поляризации;
АД — падение потенциала вслед-
ствие анодной поляризации; ДЕ —
омическое падение потенциала; ЕВ —
падение потенциала вследствие ка-
тодной поляризации; ОД' — кривая
омического падения потенциала;
ВИ — суммарная катодная поляри-
зационная кривая, учитывающая
омическое падение потенциала и по-
ляризацию
кривым (рис. 1). По -наклону по-
ляризационных кривых на корро-
зионных диаграммах можно всег-
да судить о том, какая из элек-
тродных реакций определяет сум-
марную скорость коррозионного’
процесса, и рассчитать относи-
тельную долю начальной разно-
сти потенциалов, которая теряет-
ся иа данном виде сопротивле-
ния. Эта величина называется
мерой контроля коррозионного
процесса данной электродной ре-
акцией или ’контролирующим
фактором. Мерой катодного и
анодного контроля могут слу-
жить величины тангенсов углов
наклона поляризационных кри-
вых аир.
Из диаграммы следует, что
уравнениями для катодной и
анодной поляризационных кри-
вых при наличии линейной зависимости скорости реакции от -по-'
тенциала будут:
<pKe<p£ — Ztg а (О
Pa = fa + /tg₽ = ^ + ^a/,
(2)
где RK и R& — катодное и анодное поляризационные сопротивле-
НИЯ.
В точке пересечения кривых, соответствующей максимально-
му току, потенциалы катодов и анодов равны: <рк == ?а, т-е-
<рк — RvJmax (^)
Из уравнения (4) следует, что в полностью заполяризован-
ных системах начальная разность потенциалов расходуется на
— 14 —
выво-
отве-
(5)
(6)
(7)
(8)
(9)
(Ю)
преодоление катодного и анодного поляризационного сопротив-
лений.
Системы, обладающие значительным омическим сопротивле-
нием, не могут считаться полностью заполяризованными - й при
определении тока коррозии в них приходится учитывать также
омическое падение потенциала в электролите 77?. Естествен но
что в этих условиях ток в системе не будет уже соответствовать
максимальному /, а выразится меньшей величиной
Уравнение коррозионного процесса в данном случае
дится следующим образом. Из рнс. 1 следует, что ток Г,
чающий омическому сопротивлению 7?, равен
R
Значения <р' и у/ определяются уравнениями:
<ра =я 7?а/1
<рк = — 7?кг.
Следовательно,
R
I'R -р RkJ' -р 7?а7/ = <рк- «ра,
Я+Як-РЯа’.‘
Выражение (10) является основным уравнением коррозион-
ного процесса, где учитывается поляризационное и омическое
сопротивления [1].
Из этого уравнения становится ясно, что скорость коррозион-
ного процесса можно увеличить изменением начальных потен-
циалов катодного и анодного процессов <ра и фк илн, если речь
идет о макроэлементе, увеличением начальной разности потен-
циалов. Таких же результатов можно добиться, если уменьшить
катодную и анодную поляризацию 7?к и 7?а или омическое сопро-
тивление, если оно определяет коррозионный ток. В связи с этим
необходимо иметь точное представление о механизме процесса,
который воспроизводится в ускоренных испытаниях, й знать,,
какая электрохимическая реакция его контролирует.
Как былр показано выше, коррозионный ток в общем опреде-
ляется скоростью течения двух сопряженных реакций н омиче-
ским сопротивлением. Поэтому, когда сила тока определяется
‘лишь скоростью протекания катодной реакции, говорят, что кор-
розионный. процесс протекает с катодным контролем. При этом
следует иметь в виду, что падение . потенциала приходится
в этом случае в основном иа катодное поляризационное сопро-
— 15 —
тивление, которое в свою очередь обусловлено медленностью
течения катодной реакции. Когда коррозионный ток в элементе
определяется скоростью течения анодной реакции, основное па-
дение потенциала в элементе приходится на анодное поляриза-
ционное сопротивление и тогда говорят, что процесс протекает
с анодным контролем.
Относительная доля контроля коррозионного процесса поля-
ризацией и сопротивлением определятся по поляризационным
кривым и вычисляется по формулам:
для анодного контроля
__ АД . АВ ' (11)
для катодного контроля
1' tga ЕВ . “ АВ ’ (12)
для омического контроля
ГЬ —Й = АВ (13)
В зависимости от того, какой электродной реакцией контро-
лируется коррозионный процесс, возможны три вида коррозион-
ных диаграмм (рис. 2). При катодном контроле (случай а) ско-
Рис. 2. Основные виды контроля коррозионных
процессов:
а — катодный контроль; б — «иодный контроль;
в — смешанный контроль
рость коррозионного процесса Zi контролируется преимущест-
венно скоростью катодной реакции; при анодном контроле (слу-
чай б) сила тока Z2, а следовательно, и коррозий определяются
преимущественно скоростью анодного процесса. Наиболее часто
наблюдаются коррозионные процессы, скорость которых опре-
деляется кинетикой обеих электродных реакций, т. е. и катод-
ной, и анодной. В таких случаях говорят, что контроль является
смешанным (случай в) .
— 16 —
Рассмотрим вначале случай, когда коррозионный процесс
контролируется катодной реакцией. При этом следует различать
процессы, протекающие с водородной деполяризацией:/
Н+.Цр + е^Н + НгО, (14)
и процессы, протекающие с кислородной деполяризацией:
О2 + 2Н2О + 4е - 4ОН“. (15)
Первые процессы реализуются при коррозии металлов в кис-
лотах, а также при коррозии металлов с амфотерными свойст-
вами в щелочах и частично магния в нейтральных электролитах;
вторые—при коррозии большинства металлов в нейтральных
электролитах.
Скорость катодного процесса разряда ионов водорода выра-
жается уравнением
о.Р ,
= 'е ", (16)
где [Н‘Ь] — концентрация ионов водорода;
<р — потенциал электрода;
Ф1 —потенциал на расстоянии ионного радиуса от по-
верхности электрода:
а— постоянная.
Анализируя уравнение (16), можно предполагать, какие пути
должны оказаться эффективными в увеличении скорости кор-
розии металлов, процесс коррозии которых контролируется ско-
ростью водородной деполяризации:
во-первых, — это увеличение концентрации водородных ио-
нов, смещающих потенциал водородного Электрода в положи-
тельную сторону и уменьшающих, кроме того, перенапряжение
водорода в концентрированных растворах сильных кислот. Этим
приемом следует пользоваться только в пределах концентра-
ций, не приводящих к появлению пассивности. Для железа,
например, концентрацию НС1 можно увеличивать беспредельно,
концентрацию H2SO4 —лишь до 50—55%, HNO3— до 30—35%;
во-вторых, — это введение анионов, которые, внедряясь в
двойной электрический слой, сдвигают потенциал в отрица-
тельную сторону и этим самым увеличивают скорость катодного
процесса. Не менее эффективными должны оказаться методы,
снижающие перенапряжение водорода (2], а также метод по-
вышения температуры электролита. Для металлов, обладаю-
щих высоким перенапряжением, повышение температуры на.
один градус приводит к снижению перенапряжения в среднем
на 2—4 ме.
Наконец, следует указать еще на одну возможность, связан-
ную с размешиванием электролита. Хотя подвижность ионов во-
дорода очень велика и их концентрация в кислых электролитах
достаточна для того, чтобы не наступала концен^р^^^ная по-
2 Зак. 1338 -- 17--
ляризация, в неразмешиваемых электролитах со временем мо-
жет начаться торможение процесса вследствие затруднения от-
вода продуктов растворения металла. Поэтому размешивание
кислых электролитов способно также сильно увеличить скорость
коррозионного процесса.
Итак, при ускоренных испытаниях металлов в кислых, элект-
ролитах рекомендуется ускорять процесс за счет увеличения
концентрации кислоты, снижения перенапряжения водорода, по-
вышения температуры и усиления размешивания. Для того что-
бы иметь представление, в какой области pH целесообразно из-
менять кислотность и насколько изменится скорость коррозии
того или иного металла при изменении концентрации водород-
ных ионов, приводим для некоторых наиболее технически важ-
Рис. 3. Зависимость скорости коррозии железа (а), алюминия (б)
и магния (в) от pH [2]
ных металлов диаграммы зависимости скорости коррозии от pH
j (рис. 3).
/ Для процессов коррозии, протекающих с кислородной депо-
; ляриза цией, положение более сложно и при выборе методов ус-
/ корения коррозионного процесса следует учитывать целый ряд
обстоятельств, осложняющих явление.
j Растворимость кислорода в нейтральных .электролитах весь*
j ма ограничена (в чистой воде ~8—9 мг/дм3 при 20° С), поэто-
• му, если вести испытания в неразмешиваемых электролитах,
• довольно быстро наступает концентрационная поляризация по
кислороду, и процесс сильно замедляется. Для того чтобы этого
не происходило, ускоренные испытания в нейтральных электро*
' литах необходимо проводить в движущихся жидкостях.
Для образца, обтекаемого электролитом, скорость катодного
процесса в зависимости от скорости движения жидкости выра-
жается, согласно теории В. Г. Левича [3], следующей формулой:
i^nFD'l’>l~'l‘vl'x~'k (С„ —Q, (17}
г де i — плотность тока;
п — число электронов, -ассимилирующихся в реакции;
F—число Фарадея;
р — коэффициент диффузии;
— 18 — . :
v — кинематическая вязкость раствора;
v — скорость движения жидкости;
х— расстояние от точки начала обтекания образца;
Со и Cs — концентрации кислорода в объеме электролита и
у поверхности электрода соответственно.
Из этого уравнения видно, что скорость катодного процесса,
лимитирующего обычно скорость коррозии в нейтральных элек-
тролитах, можно повысить при ускоренных испытаниях многи-
ми путями — увеличением коэффициента диффузии, скорости
движения электролита, кинематической вязкости электролита,
концентрации кислорода. Однако
следует пользоваться лишь теми Д
приемами, которые не изменяют ха- ,
рактера коррозионной среды. По
этой причине изменение кинемати- g 0.6 -
ческой вязкости среды нежелатель-
но. Наиболее эффективный путь по- ' .г
вышения скорости катодного про- |
цесса — изменение скорости враще- |
ния образцов относительно электро- § L , , , Л i
лита или электролита относительно
образцов. Оба приема увеличивают
доставку кислорода -к поверхности
металла и этим ускоряют катодный
процесс.
Для ускорения испытаний мож-
но также -пользоваться (приборами,
в которых образцы вращаются. Для
О 12 3 6 5 6 7
Ут, 2ярад/сек (об/сек)
Рис. 4. Зависимость пределу
кого диффузионного тока вос-
становления кислорода от ско-
рости вращения дискового
электрода [4]
дискового электрода, вращающегося в электролите, зависимость
плотности тока от угловой скорости вращения электрода вы-
ражается уравнением
i = 0,62nFP V0-‘/e {Со _
(18)
где со — угловая скорость вращения, рад]сек. Эта формула под-
тверждается опытом, о чем можно судить по кривой на рис. 4,
характеризующей зависимость предельного диффузионного тока
восстановления кислорода от скорости вращения дискового
электрода т [рад!сек (об] сек}], полученной Б. Н. Кабановым [4].
Как видно, зависимость между предельным диффузионным то-
ком и корнем квадратным из числа оборотов имеет прямолиней-
ный характер; это позволяет резко увеличить скорость кисло-
родной деполяризации, а стало быть, и коррозионного процесса,
поскольку он ею контролируется. Использование теории В; Г:
Левича [3] для моделирования коррозионных процессов рас-
сматривается ниже (стр. 38).
При выборе скорости вращения образцов или скорости дви-
жения электролита следует учесть, что увеличение доставки кис-
2* 19 —•
лорода не только повышает скорость катодного процесса, но и
в определенных условиях может затормозить анодный процесс,
что приведет к обратным результатам, т. е. не к ускорению про-
цесса коррозии, а к его замедлению. При этом сталь в речной и
морской воде корродирует по-разному (рис. 5 и 6). В речной
воде, где содержание хлор- и сульфат-ионов невелико (50—
150 лег/дм8), уже при скорости движения воды, равной 1 ж/сек,
скорость коррозии стали падает до нуля. В морской воде, где
концентрация ионов хлора велика, скорость коррозии непрерыв-
но увеличивается вплоть до 6—7 м!сек, поскольку даже увели-
чение доставки кислорода не может привести металл в пассив-
ное состояние. Дальнейшее увеличение скорости движения элек-
тролита приводит к кавитационным явлениям, способствующим
резкому возрастанию скорости коррозии.
0,100
О 12 3 9 5 6 7
Скорость 06и.ме t/ия боды, н/сек
Ряс. 5. Влияние скорости движения
морской воды иа коррозию стали :[5]
§ 0,020
't-0,0/0
S £
| 0,005
о 0,3 0.0 0,9 1,2
Скорость движения 6оды,я/сел
Рис. 6. Влияние скорости движе-
ния водопроводной воды
иа коррозию стали [6]
/ Поэтому при выборе скорости движения электролита при ус-
коренных испытаниях следует учесть эти особенности и для уве-
личения доставки кислорода, являющегося основным деполяри-
затором, применять скорости, движения жидкости в пределах
: первого восходящего участка кривых до максимума, когда еще
не сказываются ни пассивирующее действие кислорода, ни воз-
никновение кавитационных явлений.
I Усилить доставку кислорода к металлу можно не только раз-
/мешиванием, но и введением дополнительных деполяризаторов,
/ например перекиси водорода, которая сама по себе может вос-
; станавливаться от, кроме того, разлагаясь, увеличивать концент-
; рацию кислорода на поверхности металла. Поэтому при уско-.
1 ренных испытаниях к морской воде или 3 %-ному раствору хло-
ристого натрия часто добавляют 0,1%-ный раствор перекиси во-
дорода, особенно при испытании некоторых алюминиевых спла-
вов, предназначенных для использования и морских условиях.
Кроме перекиси водорода, для ускорения катодного процес-
са можно применять и другие катодные деполяризаторы, напри-
мер сернистый ангдрид, атомарный хлор, металлические катио-
ны, существующие в нескольких степенях окисления (Fe3* Сг6+),
20 —
а также кислородсодержащие анионы. Их применение особенно
оправдано, когда они могут хотя бы в небольших количествах
содержаться в электролитах, в которых предполагается экс-
плуатировать испытываемые изделия. Будет ли тот или иной
деполяризатор восстанавливаться на испытуемом сплаве, мож-
но заключить на основании значений потенциалов катодных
реакций, которые приведены в табл. 1 (7]. При использовании
этих данных следует учесть, что они указывают лишь иа тер-
модинамическую возможность течения реакции.
Таблица 1
ЗНАЧЕНИЯ ПОТЕНЦИАЛОВ КАТОДНЫХ РЕАКЦИЙ,
В РЕЗУЛЬТАТЕ КОТОРЫХ ВОЗМОЖЕН ПРОЦЕСС САМОРАСТВОРЕНИЯ
{КОРРОЗИИ) МЕТАЛЛОВ [7]
Деполяризатор Реакция Значение по- тенциала <₽° при pH «= 0, в
Водород (Н+) Н2О+ + е Н + Н2О 0,000
0, + 4Н+ + 4г -> 2НгО 4-1,299 ’
О2 + 2Н+ + 4г 2ОН“ 4-1,078
Кислород (О2) О2 + 2НаО + 4е -> 4ОН~~ 4-1,213
О8 4- 2НаО + 2е -* НйОа 4- 2ОН" 4-0,774 .
О2+2Н+ + 2е .НД 4-0,682
Перекись водорода (Н./Л) Н2О2 + 2Н+ + 2г - 2H./J 4-1,770
НЙОЙ + 2е - 2ОЬГ 4-1,766
Н2О2 + н+ + е - ОН“ + Н2О 4-0,720
Озон (О3) О8 + Н2О + 2е -> Ог4-2ОН~ 4-2,052
4H..SO3 + 4Н+ + 6г -> SsO,,2 - + 6Н2О 4-0,510
. H2SO8 + 4Н+ + 4г - > S + ЗН2О 4-0,450
Сернистый ан- гидрид (SOa) H2SOs + 2Н+ + 2г -> H2SO.j + Н2О 4-0,400
2H2SOs + Н+ + 2г -> HSjO, + 2Н2О 2HSO7 + 2Н+ + 2г - S2pJ + 2Н2О •—0,080 —0,103
HS2O7 + 7Н+ + Юг - 2S2— + 2Н++ —0,184
-HHgO
Fa 4~2е 2HF (води.) 4-3,080
Галогены Fa + 2е -> 2F- 4-2,650
(Fs, Вг2> Cl2, J,) С12 + 2е 2С1 4-1,359
Вг2 4~ 2е -» 2Вг“ 4-1,065
J2 -р 2е 2J- 4-0,535
Другой важный фактор, позволяющий ускорить коррозион-
ный процесс,— температура; этот фактор широко используется
при ускоренных испытаниях. Подсчет температурных коэффици-
— 21 —
ентов Вант-Гоффа, выполненный И. Л. Розенфельдом и
Г. В. Акимовым [2] по формуле
= (19)
Kt
где К/+!о»— скорость коррозии при t + 10°;
Kt — скорость коррозии при t,
показал, что коэффициенты меняются в широких пределах. Ча-
ще всего коэффициент Вант-Гоффа укладывается в пределах от
1 до 2, однако в некоторых случаях он достигает трех единиц
(А1 — Н3РО4). Температурные коэффициенты, определенные
А. И. Глуховой i[8] для железа и нержавеющих сталей в раство-
рах соляной и азотной кислот, в большинстве случаев изменя-
лись от 1 до 2.
Коэффициенты Вант-Гоффа позволяют судить не только о
том, во сколько раз можно ускорить коррозионный процесс, но
и получить данные о механизме процесса, так как одним из кри-
териев, отличающих диффузионный процесс от химического,
является температурный коэффициент. Для процессов, опреде-
ляемых скоростью химической реакции, он равен 7—10% на 1°,
а для процессов, определяемых диффузией, 1—3% на 1°. По ве-
личине этого коэффициента можно, таким образом, определить,
какая из реакций в суммарном процессе ускоряется. Коэффици-
енты, близкие к двум, могут свидетельствовать о том, что корро-
зионный процесс определяется скоростью протекания самой
электрохимической реакции, например реакции восстановления
кислорода или водорода. Коэффициенты, равные 1—1,5, указы-
вают на то, что скорость коррозионного процесса определяется
диффузией.
При выборе температуры для ускоренных испытаний следу-
ет иметь в виду, что ее не всегда можно увеличивать беспредель-
но. Иногда наблюдаются отклонения от общих закономерностей;
коррозионный процесс в определенном интервале температур
часто замедляется, а иногда и чрезмерно растет. Это происходит
благодаря тому, что на коррозионный процесс и его скорость
влияют многие факторы, которые нередко невозможно учесть.
Сильнее всего оказывают влияние продукты коррозии, свойства
которых изменяются с температурой; оказывает влияние и рас-
творимость кислорода или другого деполяризатора, которая
также зависит от температуры. Это хорошо иллюстрируется за-
висимостью скорости коррозии цинка в дистиллированной воде
от температуры, приведенной на рис. 7 [9]. Максимум коррозии
цинка наблюдается при 60—65° С. Это обусловлено свойствами
зернистых продуктов коррозии, которые образуются при 50—
95° С и плохо прилегают к поверхности металла. При темпера'
турах выше и ниже этого интервала образуются защитные слои,
хорошо сцепленные с поверхностью металла и защищающие его
от воздействия коррозионной среды. Аналогичную зависимость
— 22 —
от температуры обнаруживает железо в нейтральном электроли-
те при свободном доступе кислорода [10] (рис. 8, а). В этом слу-
чае зависимость обусловливается растворимостью кислорода в
электролите. Спад коррозии, наблюдающийся обычно в интерва-
ле 70—80 С, обусловлен резким
уменьшением концентрации кис-
лорода в электролите с твыше-
иием температуры. Зависимость
такого же характера отмечается
при коррозии дуралюмила ® рас-
творах хлористого натрия (рис.
8, б) [11]. Все эти закономерно-
сти надо учитывать при «выборе
температуры для ускоренных ис-
пытаний металлов в нейтраль-
ных электролитах. При желании
ускорить процесс рекомендуется
выбирать температуру испытаний
на «восходящей -ветви кривой рис. 7 зависимость скорости кор-
К— Т. розни цинка в дистиллированной
f В кислых электролитах на- и°Де от температуры [9]
? блюдается непрерывный рост
' коррозии с температурой обычно по. экспоненциальному закону,
’как это хорошо видно из приведенных нар рис. 9 кривых, харак-
Рис. 8. Зависимость скорости коррозии металлов от температуры
12. 10. И]:
а — железо в воде (система открытая); б — .дуралюмнн в 1«и. растворе
NaCl (pH “6); в — магний:
2 — в 0,1-н. на + 0,1-я. NaCl; 2 — в 0Д1-®. НС1 -ЮД-ш. NaCl;
3 — >в 1-н. iNaC; 4 — ® 0,2-и. NaCl (левая «шкала >для кривых 1 и 2}
теризующих наведение железа, стали и чугуна .в 5%-ном рас-
творе соляной кислоты [Г2]. При иопытаниях =в лабораторных
условиях для кислых электролитов ограничений в выборе тем-
пературы нет.
Каким же образом температура влияет на повышение скоро-
сти коррозионного процесса?
— 23 —
Когда процесс определяется скоростью электрохимической
реакции, например разряда ионов водорода или восстановления
кислорода, повышение температуры увеличивает энергию взаи-
модействующих частиц, благодаря чему уменьшается перена-
пряжение и увеличивается скорость реакции.
Однако, как уже указывалось, в нейтральных электролитах,
где концентрация основного деполяризатора (кислорода) мала,
процессом, лимитирующим скорость коррозии, становится диф-
фузия.
Исследование влияния температурного фактора на реакцию
восстановления кислорода, проведенное В. В. Герасимовым и
И. Л. Розенфельдом [13], показало,
что основная причина увеличения
предельного диффузионного тока с
температурой — изменение коэффи-
циента диффузии кислорода, а так-
же толщины диффузионного слоя.
Коэффициент диффузии <с темпера-
турой меняется по следующему за-
кону:
Рис. 9. Влияние температу-
ры на скорость коррозии
металлов в 5 %-ном растворе
соляной кислоты (12]:
1 —• железо; 2 — сталь 20; 3 —
сталь 45; 4 — высокопрочный
чугун
где К — постоянная Больцмана;
г — радиус диффундирующей
•частицы;
— вязкость раствора.
Из приведенного уравнения вид-
но, что коэффициент диффузии ки-
слорода должен расти с температу-
рой также вследствие изменения вязкости среды, которая с ро-
стом температуры падает.
Коэффициент диффузии большинства веществ в водных ра-
створах возрастает с температурой примерно на 2,5% на 1°, а
коэффициент вязкости уменьшается при этом на ту же величину.
Толщина диффузионного слоя при нагревании уменьшается
вследствие усиления саморазмешивания, обусловленного кон-
векцией; в первом приближении можно принять для неразмеши-
ваемых электролитов с естественной конвекцией следующую за-
висимость толщины диффузионного слоя (в мкм) от темпера-
туры:
lg6=l,ll+^. (21)
В тех случаях, когда коррозионный процесс определяется не
диффузией кислорода, а скоростью протекания самой электро-
химической реакции, что может наблюдаться при усиленном до-
ступе кислорода к поверхности металла, скорость коррозии бу-
— 24 —
дет, как уже указывалось, увеличиваться с температурой благо-
даря изменению перенапряжения для реакции восстановления
кислорода. Можно считать, что в интервале 20—100° С с повы-
шением температуры на Г перенапряжение для реакции’восста-
новления кислорода уменьшается в среднем на 2—3 мв. Зави-
симость скорости катодного процесса от перенапряжения имеет
логарифмический характер, поэтому даже небольшое изменение
перенапряжения приводит к значительному увеличению скоро-
сти коррозии.
Чтобы облегчить выбор температуры для тех или иных элект-г
ролитов и сплавов при ускоренных (испытаниях, были приведе-
ны некоторые зависимости скорости коррозии от температуры
(см. рис. 8 и 9).
Простая линейная зависимость скорости коррозии металла'
от температуры наблюдается сравнительно редко, например* для
магния в хлористом натрии и соляной кислоте (см. рис. 8,в).
Из изложенного выше следует, что при ускоренных испыта-
ниях использовать повышение температуры можно, только учи-
тывая зависимость скорости коррозии испытывающихся метал-
лов от температуры.
Зная эту зависимость, исходя из опытных данных или теоре-
тических предпосылок, температуру испытания выбирают с та-
ким расчетом, чтобы коррозия при ней шла с большей скоро-
стью, чем при нормальных условиях.
Рассмотрим теперь основные закономерности анодного про-
цесса, чтобы определить, какие имеются пути для ускорения
коррозии тех металлов, скорость коррозионного процесса кото-
рых контролируется анодной реакцией.
На рис. 10, а представлена схематическая потенциостатичес-
кая кривая для анодного процесса, характеризующая зависи-.
мость скорости растворения (или тока) металла от потенциала.
На кривой видны четыре характерных участка: область потен-
циалов от А до 5, при которых железо находится в активном
состояния; от Б до В, при которых железо находится на границе
активно-пассивного состояния; от В до Г, где железо находится
в устойчивом пассивном состоянии, и от Г до Д, где на электро-
де происходит разряд ионов гидроксила и выделение кислорода.
На кривой можно выделить несколько характерных точек,,
представляющих интерес в связи с рассматриваемой нами
проблемой ускоренных испытаний:
9® — потенциал, при котором скорость анодного растворения-
равна нулю;
?ст—стационарный потенциал металла, при котором скорость-
анодного растворения равна /кор;
<рп — потенциал пассивации, при котором наблюдается пере-
ход металла из активного состояния в пассивное;
<Рп.п — потенциал полной пассивации, при котором металл на-
ходится в устойчивом пассивном состоянии;
— 25 —
Zn —ток, необходимый для того, чтобы перевести металл в
пассивное состояние;
А1-п — скорость растворения металла из пассивного состояния;-
4ор — анодный ток при стационарном потенциале.
Указанные выше характерные точки имеются и на реальных
потенциостатических кривых (рис. 10,6). Положение характер-
ных точек на этих кривых в сильной сте-
пени зависит от состава электролита.
Как и следовало ожидать, анодное рас-
творение легче всего происходит в хло-
риде натрия (кривая 2). Хотя в этом
растворе потенциал пассивации и выяв-
ляется, однако скорость растворения из
пассивного состояния (область незави-
симости скорости растворения от потен-
циала) очень «велика (200 ма!см2). Это
указывает на то, что окисная плеика рас-
творяется в этом электролите с большой
скоростью.
Интересно наблюдать поведение же-
леза в азотнокислом натрии (кривая 3).
Рис. 10. Погенкиостатическне поляризационные кривые:
а — теоретическая кривая; б — кривые для железа, снятые в различ-
ных электролитах, I — 1-н. Na2SO4; 2 — 1-н. NaCl; 3 —• 1-н. NaNO3
В этом электролите железо из активной области растворяется
с гораздо большей скоростью, чем в сульфате натрия. Ток пас-
сивации в .нитрате равен 240 ма!см2, а в сульфате' 140 ма{см2.
В то же время нитраты, как известно, являются хорошими инги-
биторами для нержавеющей стали. Отсюда следует, что’дей-
ствие анионов .специфическое, что необходимо учитывать при
выборе электролитов для ускорения коррозионного -процесса.
— 26 —
Сложная зависимость скорости анодного процесса от потен-
циала обусловлена разнообразными явлениями, часто' проте-
кающими иа электроде одновременно. При смещении потенциа-
ла от точки в положительную сторону на электроде при от-
сутствии других усложняющих явлений (см. рис. 10, а) должен
идти процесс анодного растворения, скорость которого опреде-
ляется следующей зависимостью:
arup^F
' (22)
где К—константа скорости анодной реакции;
<ра — потенциал анода;
а — коэффициент, равный обычно 0,5 для случаев, когда
ионизация металла является замедленной стадией сум^
мерного процесса;
п—число электронов, освобождающихся при переходе
иои-атомов металла в раствор.
Из выражения (22) следует, что в активной области потен-
циалов скорость анодного процесса должна увеличиваться по
мере смещения потенциала в положительную сторону. Однако
это наблюдается лишь до определенного потенциала. В точке Б
(рис. 10, а) электрод внезапно, часто несмотря на отсутствие ка-
ких-либо видимых изменений поверхности, перестает раство-
ряться и переходит в пассивное состояние. Объясняется это сле-
дующим: уже при незначительном смещении потенциала от точ-
ки на электроде, как показывают термодинамические расче-
ты, может протекать не только реакция ионизации металла, но
и реакция электрохимического окисления железа, приводящаяк
возникновению тончайших адсорбционных и фазовых слоев.
Выполненный нами термодинамический расчет (14] показыва-
ет, что железо должно очень легко электрохимически окислять-
ся ионами гидроксила:
Fe 4- 2ОН~ FeO 4- Н2О 4- 2е; (23)
AF° == AFFe0 4- AFHe0 - 2AFoh- = — 58400 4-
-4- (— 56 690) + 2 (37 595) = — 168 кдж = - (39 900 кал).
о __ -39 900 •
__«л к — ' = — 0,665в.
•рн_14.5 и. 23060 2.23060
<?рН=7='Р?н=1<1,5+0,059(14,5—7,0) = -0,8654-0,44—3= —0,422в.
Окисление по другим схемам дает следующие результаты:
2Fe 4- 6ОН“ Fe2Os 4- ЗН2О 4- бе, 8рн«7 == — 0,431 в, (24)
3Fe 4- 8ОН“ Fe8O4 4- 4Н2О + 8е, 8рн=»7 = - 0,473 в. (25)
Если электрод находится при потенциалах, более положитель-
ных, чем указанные выше значения, должны протекать реакции
— 27 —
окисления; при потенциалах, более отрицательных (с учетом не-
которого перенапряжения) — реакции восстановления окисиых
слоев.
Если сравнить значения стационарных потенциалов железа.
в нейтральных электролитах (—0,3 ч 0,5 в) со значениями по-
тенциалов образования окисных слоев, то легко прийти к заклю-
чению, что уже при незначительном смещении потенциала от ста-
ционарного наряду с процессом ионизации происходит реакция
электрохимического окисления. По мере смещения потенциала
в положительную сторону процесс окисления все более усили-
вается, и при достижении потенциала пассивации электрод уже
в значительной степени покрывается окисным слоем, приводящим
к резкому уменьшению скорости ионизации металла; наступает
явление пассивности.
В некоторых случаях пассивность может наступить благодаря
концентрационным явлениям, обусловленным накоплением про-
дуктов анодной реакции вблизи поверхности электрода.
Пересыщение раствора приводит к выпадению продуктов кор-
розии на поверхности металла и уменьшению активной поверх-
ности электрода. Плотность тока на непокрытых участках резко
возрастает, что способствует появлению анодной пассивности.
Этой пассивности, называемой механической (в отличие от хими-
ческой), можно избежать, если усилить размешивание электро-
лита.
Начиная с потенциала полной пассивации на участке ВГ ско-
рость анодного процесса не зависит от потенциала; объясняется
это тем, что в этой области потенциалов происходит химическое
растворение окисной пленки и ток £п.п идет лишь на восстановле-
ние этой пленки.
В этой области потенциалов обычно оказываются металлы,
находящиеся в пассивном состоянии (нержавеющие стали, алю-
миниевые и титановые сплавы). Состояние пассивности легко на-
рушается ионами хлора, которые быстро адсорбируются при та-
ких положительных потенциалах. Поэтому для проверки устой-
чивости пассивного состояния этих металлов оправдано примене-
ние при ускоренных испытаниях ионов хлора.
На участке ГД может опять наступить усиленное анодное
растворение металла вследствие разрушения в каком-нибудь
месте окисной пленки или образования ионов высшей валент-
ности:
Fe6+ + 8ОН~ -+ FeO«- + 4Н2О.
Из анализа анодной потенциостатической кривой становится
ясно, какими путями можно ускорить коррозионный процесс.
Во-первых, испытания необходимо вести таким образом, что-
бы потенциал металла все время находился в активной области
(АБ). Важно, чтобы потенциал был по возможности смещен в по-
ложительную сторону от стационарного, но не выходил за пре-
— 28 —
деды потенциала пассивации. Этого можно достигнуть введением
s электролит окислителей в определенных концентрациях, а так-
же усилением подвода кислорода. Сместить потенциал можно,
также с помощью анодной поляризации, однако поляризация не
должна быть большой, а потенциал следует поддерживать на
уровне, более отрицательном, чем уровень потенциала пассива-
ции. Чтобы увеличить ток пассивации и избежать механической
пассивности, целесообразно применять размешивание, которое
будет отводить продукты анодной реакции от поверхности испы-
тываемых образцов.
Во-вторых, коррозионный процесс можно ускорить путем из-
менения состава коррозионной среды. При этом, как уже указы-
валось, следует иметь в виду, что’действие анионов является
-специфическим по отношению к каждому металлу. Например,
ионы SO?- действуют на железо почти так же, как ионы хлора.
В то же время сульфат-ирны не ускоряют коррозии алюминия и
нержавеющих сталей. Более того, как показано одним из авто-
ров работы [15], смесь ионов хлорида и сульфата играет пассиви-
рующую роль и при определенном соотношении способна полно-
стью подавить вредное влияние хлор-ионов. Поэтому при испыта-
нии нержавеющих сталей и алюминиевых сплавов увеличение
концентрации сульфат-иона не приводит к ускорению коррозион-
ного процесса. Такие сплавы надо испытывать в растворах, со-
держащих ионы хлора, и по возможности уменьшить концентра-
цию сульфат-ионов. Медные сплавы, наоборот, очень чувстви-
тельны к сульфат-ионам, поскольку растворимость сульфата
меди выше хлорида. При испытании низколегированных и мало-
углеродистых сталей применение смеси сульфата и хлорида так-
^ке допустимо.
При выборе электролита следует по возможности учитывать
состав коррозионной среды, в которой будет работать испытыва-
емое изделие, и увеличивать концентрацию того компонента, ко-
торый для данного металла наиболее агрессивен.
Поскольку большое количество металлов эксплуатируется
в морской воде, основная составляющая которой — хлористые
соли, в лабораторных испытаниях материалы, предназначенные
для эксплуатации в этих условиях, чаще всего испытываются в
растворах хлористого натрия, концентрацию которого выбирают,
исходя из общей концентрации хлористых солей, содержащихся
в морской воде. Обычно применяют 3%-ный раствор хлористого
натрия.
Если сравнить анализы воды в различных морях и океанах,
приведенные в табл. 2, то можно видеть, что они значительно от-
личаются по составу и, кроме хлористого натрия, содержат дру-
! гие составляющие. При коррозии металлов в морской воде на их
! поверхности образуются гидраты кальция и магния, которые, как
правило, снижают общую скорость коррозионного процесса. По-
этому 3%-ный раствор хлористого натрия более агрессивен, чем
— 29 —
Таблица 2
СОДЕРЖАНИЕ солей в ОКЕАНЕ и ЮЖНЫХ МОРЯХ [16]
Содержание от суммы солеи, %
Водоемы о о £ £
Океан - 3,94 6,40
Черное море . . . . 2,58 7,11
Каспийское море . . 6,92 23,58
Аральское море . 12,91 25,80
1,69 78,32 9,44 0,21 34,30
2,99 77,72 9,07s 1,59 18,60
1,21 62,15 4,54** 1,24 12,86
1,87 56,72 1,36 0,93 11,28
* 8,87MgCk -J- O.SOMeBrs-
** 4,47MgCre-}- 0.07A'.gBrs.
морская вода, что оправдывает применение первого при ускорен-
ных испытаниях.
Испытание материалов, предназначенных для эксплуатации
в речной воде, путем погружения в концентрированные растворы
хлористого натрия не может дать правильного представления
о поведении их в реальных условиях. Как видно из табл. 3, реч-
ная вода характеризуется относительно большим содержанием
SOf" по сравнению с С1". Концентрация же солей в 50—60 раз
ниже.
Для имитации речной воды в лаборатории ИФХ АН СССР
применяется раствор, содержащий 30 мг!дм* NaCl 4- 70 MzjdjvP
NasSO4. '
Таблица 3
СОСТАВЫ НЕКОТОРЫХ РЕЧНЫХ ВОД СССР [17]
Химический состав, мг'дм3
Река 1 X?
.и О 8 Z а
Аму-Дарья (г. Турт-Куль) . 45,4 78,9 140,4 11,4 89,5 3,2
Чусовая (д. Шелыги) . . . . 15,0 44,5 170,8 18,5 52,3 11,6
Волга (г. Вольск) . 19,9 112,3 210,4 12,5 80,4 22,3
Дон (с. Аксайская) .... 44,0 112,0 260,0 52,2 82,0 18,0
Днепр (с. Разумовка) .... 3,9 14,1 •231,8 2,3 55,7 11,9
Кама (с. Чистополь) .... 13,5 132,0 190,3 10,3 82,2 21,0
Енисей (г. Красноярск) . . 2,6 4,0 73,2 1.5 19,3 4,0
Для процессов, контролирующихся обеими электродными ре-
акциями, можно получить надежные результаты при ускоренных
испытаниях, применяя комбинированные методы, т. е. методы,
ускоряющие и катодный, и анодный процессы. К ним прежде все-
— 30 —
го можно отнести испытание в растворах хлористого натрия, со- ।
держащих ~ 0,1 % Н2О2- Это испытание, в частности, применяют \
для определения склонности алюминиевых сплавов к расслое-
нию. Результаты этих испытаний будут отражать поведение спла-
вов в морских условиях при длительной эксплуатации.
Введение в растворы кислот хлористого натрия в небольших
количествах при испытании металлов, находящихся в пассивном
состоянии, также будет способствовать увеличению скорости кор-
розии за счет влияния хлор-иона на анодный процесс.
В табл. 4 приведены некоторые составы электролитов, приме-
няющихся для ускоренных испытаний металлов в условиях пол-
ного погружения.
Таблица 4
ЭЛЕКТРОЛИТЫ ДЛЯ УСКОРЕННЫХ КОРРОЗИОННЫХ ИСПЫТАНИИ МЕТАЛЛОВ,
ЭКСПЛУАТИРУЮЩИХСЯ в условиях погружения
Условия ,, эксплуатации «Металлы Электролиты для ускоренных испытаний
Морская
вода
Растворы
НС1, H2SO4,
HNO3
Рудничные
воды, содер-
жащие H2S
Речная вода
Грунтовые
воды
Железо, малоугле-
родистые стали
Нержавеющие ста-
ли
Алюминиевые
сплавы
Медные сплавы
Цинк и цинковые
сплавы
Нержавеющие ста-
ли
Стали, алюминие-
вые сплавы
Стали, алюминие-
вые сплавы, цинк
Стали
Искусственная морская вода, 3%-ный
раствор NaCl
3%-ный раствор NaCl 4-0,1 % Н2О2
3%-ный раствор NaCl 4-0.1% Н2О2,
искусственная морская вода
Искусственная морская вода, 3%-ный
раствор NaCl
3%-ный раствор NaCl
Растворы соответствующих кислот сог-
ласно зависимости коррозии от pH
с добавками NaCl
3%-ный раствор NaCl 4" ОД—2%HaS
30 мг/дм* NaCl -J- 70 мг/дм9 Na2SO4
32,2 г/дм9 NaCl 4- 6 г/длг5 MgS04 4-
4-26 г/лг8 MgCl2; 0,78 e/cMt3
•Na2SO44-2,4 e/<WNaC14-
4-0,17 г/дж8 NaHCO34-0,84 г/дм9
MgSO44-0,66 e/d^MgClz
Испытания при полном погружении металла в электролит
наиболее распространены, так как они не требуют сложной
аппаратуры и могут быть проведены в любых сосудах из стекла
или другого инертного материала (плексигласа, эбонита, тефло-
— 31 —
на и т.п.). Однако следует иметь в виду, что это не лучший
метод быстрого определения коррозионной стойкости.
Рассмотрим способы аппаратурного оформления испытаний
при полном погружении металлических образцов в электролиты.
На рис. 11 представлены простейшие способы испытания об-
разцов в открытом сосуде. Испытания можно проводить в ком-
натных условиях и при термостатировании; в последнем случае
сосуды с образцами пометают *в термостат—чаще всего для
этой цели применяют термостаты, заполненные водой, темпера-
тура в которых поддерживается с помошью электрического на-
гревателя, включенного *в цепь реле контактного термометра
Рис. И. Способы испытания металлических
образцов при погружении в электролиты (1].
Спокойный электролит.'
,й — комнатные условия; б—при термостатировании;
размешиваемый электролит; в — мешалкой;
гад — пропусканием (воздуха
(см. рис. 11,6). При проведении испытания в условиях разме-
шивания в сосуд с образном помешают стеклянную мешалку,
как показано на рис. 11. в; мешалка вращается от электромо-
тора. Размешивание электролита можно осуществлять про-
пусканием через него потока воздуха под давлением 1—2 бар
(см. рис. 11, г) или присоединяя сосуд к водоструйному насосу;
в этом случае, как показано на рис. 11, <5, удобнее применять
сосуд несколько иной формы. Пропусканием потока воздуха че-
рез электролит, помимо механического размешивания, соз-
дается одновременно и дополнительная аэрация.
Размешивание электролита может осуществляться путем вра-
щения самих образцов. Установка для испытаний в таких усло-
виях, разработанная И. Л. Розенфельдом, И. С. Даниловым
и Т. И. Павлуцкой. показана на рис. 12. Испытания проводят
— 82 —
в термостатированных условиях, все сосуды для испытания по-
мещены в термостат 11. 'В специальные стеклянные сосуды 10,
имеющие (водяной затвор 9, 'помещают образцы 13. Образцы
крепятся на специальном держателе из плексигласа 12. Крышка
сосуда 8 выполняется из плексигласа или другого инертного
материала. Вращение образцов производится от мотора 1 через
Рис. 12. Установка для испытания в размешиваемом
электролите прн вращении образцов (ИФХ АН СССР):
1 — мотор, 2 и 5 — передаточные шкивы; 3 — рама; 4 — распреде-
лительный шкив; б — шкив, передающий вращение образцам; 7 —-
обратный холодильник; 8 — крышка сосуда; 9 — водяной затвор;
10 — сосуд для испытания; 11 — термостат; 12 - держатель образ-
цов; 13 — образцы
передаточные шкивы 2 и 5 и распределительный шкив 4\ послед-
ний имеет шесть направляющих для одновременного вращения
образцов в шести сосудах, на каждом из которых имеется от-
дельный шкив 6. Весь вращающий механизм укреплен на раме 3.
В установке предусмотрены две скорости вращения, которые по
желанию исследователя можно изменить соответствующим на-
бором диаметров передаточных шкивов.
При исследовании материалов, -предназначенных для разного
рода трубопроводов, работающих в условиях движения жид-
кости, в лабораториях обычно проводят испытания на спешь
3 Зак. 1338 ' — 33 —..
альных установках, в которых -предусмотрена циркуляция
жидкости. Циркуляция жидкости осуществляется различными
способами. Одна из наиболее простых установок — установка
Г’. В. Акимова [1] (рис. 13). В этой установке движение жидкости
создается за счет действия обычного -водоструйного насоса.
В качестве образцов можно применять трубчатые и обычные
цилиндрические или плоские образцы, помещенные в стеклян-
ные трубки, -как это схематично показано на рис. 13 справа.
Если необходимо создать большие скорости движения элек-
тролита, то вместо аэролифта с водоструйным насосом приме-
Рис. 13. Установка для коррозионных испытаний в условиях
циркуляции электролита (с аэрацией) i[ll
няют специальные насосы (водяные помпы). При применении
водяных помп обычно возникают осложнения в связи с загряз-
нением электролита посторонними примесями; особенно опасны
жировые вещества, вымывающиеся из сальников. За последнее
время эти трудности преодолевают, применяя бессальниковые
насосы [18].
При исследовании влияния скорости движущегося электро-
лита на коррозию алюминиевых труб И. Л. Розенфельд
и И. С. Данилов применяли установку, представленную на
рис. 14. В этой установке движение электролита из бака 1 объе-
мом 0,2 Л43 по трубопроводам 4 и 17 осуществлялось с помощью
авиационной -помпы, 'позволяющей получать скорости потока
3 м!сек при расходе электролита 0,04 м?[мин. Воздух подавался
в систему из газгольдера 11 через кран 14. Испытание проводили
на трубчатых образцах 2 и 13. Циркулирующий электролит подо-
гревался нагревателями 18, интенсивность нагрева регулирова-
— 34 —
лась реостатом 19. Скорость движения жидкости регулировалась
кранами 3 и 14, установленными на трубопроводе. Через кра-
ны 4 отбирали пробы электролита и измеряли скорость потока
при присоединении реометра.
Если движущимся электролитом служит кислота, установка
должна быть выполнена из инертных материалов. На рис. 15
приведена схема прибора [19}, состоящего из фаолитового насо-
са 1, двух винипластовых резервуаров 2 и 3 емкостью по 0,03 ж3
Рис. 14. Установка для испытания металлов в движущемся
электролите (ИФХ АН СССР);
1 — бак для электролита; 2 и /3 —образцы; 8 и 14 — краны; 4 — кран для
отбора проб электролита; б и 17 — трубопроводы: 6 и 7 — краны для пуска
воздуха ъ систему и газгольдер; 8 — трубка; 9 — «ран для спуска водь;
из газгольдера; 10 — кран; 11 — газгольдер; 12 — кожух газгольдера;
15 — -помп®; 16 — мотор (п — 3000 об/лигн, М «= 3 квг); 18 — нагреватели;
/9 „ реостат ♦
и соединяющих их труб с внутренним диаметром 32 мм, снаб-
женных фарфоровыми вентилями. В приборе испытываются
образцы диаметром 30 мм н толщиной 1 мм, которые закреп-
ляются неподвижно в отверстиях -вкладыша 4 (из винипласта).
Вкладыш вставляется на резиновой прокладке в паз трубы
и плотно прижимается к ней тремя скобами на болтах. Образцы
в отверстиях вкладыша закрепляются путем заливки..горячим
парафином,.. Скорость потока кнслоты регулируется при «помощи
вентилей и определяется -по времени заполнения верхнего резер-
вуара 3. Максимальная скорость потока (при полном открытии
вентилей) составляет 0,8 м[сек.
Условия, имитирующие обтекание образцов электролитом-
при отдаче ими тепла, воспроизводятся в установке, разрабо-
танной И. Л. Розенфельдом и И. С. Даниловым (рис. 16). Как
видно из рисунка, трубчатые образцы 3 нагреваются с помощью
з* — 35 —, ’
Нагревателя 1, помещенного внутри трубчатых образцов. На-
грев можно также осуществить, пропуская ток непосредственно
через образец, т. е. использовав его в качестве нагревательного
элемента. При этом необходимо принять меры к тому, чтобы не
было утечек тока через электролит; в противном случае могут
возникнуть анодные зоны, в которых металл будет электрохими-
чески активно растворяться. Образцы крепятся в корпусе испы-
тательного сосуда 6 с помощью тефлоновых уплотнителей 2 «и на-
кидных гаек 5. Циркуляция элек-
тролита осуществляется помпой ' \с^3 4 5
через соединительную трубку 9 и _ z
бак 10. Для контроля температу- /
Рис. 15.. Прибор для изучения кор-
розии металла в потоке кислоты (19]:
1 — насос; 2 1л 3 — резервуары для кис-
лоты; 4 — вкладыш для закрепления об-
разцов; 5 — образцы
Рис. 16. Установка для ускоренных
испытаний в циркулирующем элек-
тролите в условиях отвода тепла
от стенки образца:
1 — нагреватель; 2 — тефлоновый уплот-
нитель; 3 —- гермопара;' 4 —'трубчатый
образец-, 5 — накидная гайка уплотните-
ля; 6 — корпус сосуда, в котором раз-
мещаются образцы; 7 — термометр; 8 —
помпа; 9 — соединительная трубка;
10 — бак
ры в сосуд помещен термометр 7, температура самого образца
регулируется с помощью термопары.
Испытания при повышенных температурах даже в самом
простом случае требуют некоторого усложнения аппаратуры.
Образцы должны помещаться в специальные термостаты с за-
данной температурой; чаще всего испытания ведут в специаль-
ных установках. М. М. Куртеповым [20] для испытания метал-
лов в агрессивных средах при температурах до 140° С разрабо-
тана установка, состоящая из нескольких ванн, одна из которых
показана на рис, 17, с. Установленные на дне ванны трубчатые
нагреватели 1 обеспечивают равномерный нагрев теплоносителя
и сохранение 'постоянной температуры (±1°) в ванне, В качест-
ве теплоносителя могут быть использованы вода и водоглицери-
новые смеси, для более высоких температур — дифенил или
другие углеводороды с высоким молекулярным весом. Постоян-
ный уровень теплоносителя в ванне поддерживается при помощи
уравнительных бачков с -поплавковым регулятором. Управление
— 36 —
работой установки ведется со специального пульта, который
снабжен приборами для контроля силы и направления тока,
приборами для автоматической записи температуры в’в'аннах,
а также универсальными кнопочными переключателями
типа К-03 для включения и выключения пульта и отдельных
ванн. На пульте имеется световая и звуковая сигнализация.
В установке можно проводить массовые коррозионные испы-
тания, причем одновременно можно испытывать различные
Рио. 17. Установка для ускоренных коррозионных испытаний метал-
лов при повышенных температурах [20]:
а — ва-нна: I — трубчатый электронагреватель; 2 — -кожух ванны; 3 —
теплоизоляция; 4 — электропроводка к трубчатым нагревателям; 5 — при-
бор для коррозионных испытаний; б — реле; 7 — термометр; 8 — тер-
мометр сопротивления; 9 — трубопровод с охлаждающей водой; б —
прибор, в -который помещаются образцы
материалы в разных средах, так как образцы помещают в от-
дельные колбы (приборы) 5, устройство которых доказано на
рис. 17,6. Каждый такой 'прибор снабжен обратным холодиль-
ником для улавливания испаряющегося электролита и вмещает
тряпобразца.
"Кроме ускоренных испытаний, в лабораторной практике
часто возникает необходимость моделирования коррозионных
процессов, -в которых взаимодействие металла с агрессивной сре-
дой происходит в движении (трубопроводы, корабли и т. п.).
Эти 'процессы могут успешно моделироваться на вращающихся
дисковых образцах.
Исследования Новаковского В. М. [21] показали, что модели-
рование коррозионного процесса в трубе на вращающихся
— 37 —
дисковых образцах -возможно, если коррозионные процессы
в трубе и на вращающемся диске протекают при равной толщи-
не диффузионного слоя. Это условие, исходя из рассмотрения
гидродинамических условий течения в трубах, выполняется,
когда скорость потока жидкости в трубе v (м/сек) связывается
с числом оборотов образца п (рад/сек) следующим уравнением:
V = 0,26Р;Л' V"wr, (26)
где Рг— критерий Прандтля, величина которого принята рав-
ной 3,17;
v — кинематическая вязкость жидкости в стоксах.
Испытания -вращающихся образцов целесообразно проводить
в цилиндрических сосудах диаметром, в два раза большем диа-
метра армированного диска. Образец -погружается в жидкость
на глубину 10—15 мм. Диаметр образца, как следует из теории,
на диффузионные параметры влияния не оказывает. В указан-
ных выше исследованиях применяли образцы диаметром
50,5 мм.
И. Л. Розенфельд и О. И. Ваников [22] показали, что на вра-
щающемся электроде можно моделировать -процессы коррозии,
возникающие на обшивке корпуса движущегося судна. Так как
экспериментально определить зависимость толщины диффузион-
ного слоя на обшивке корпуса от скорости движения судна
непосредственно невозможно, то в данном случае соотношение
скорости вращающегося электрода и скорости движения ко-
рабля определяли, исходя из значений предельных диффузион-
ных токов. Диффузионные токи определяли экспериментально
на вращающемся электроде и на обшивке корпуса движущегося
судна. Проведенные расчеты показали, что число оборотов
электрода связывается со скоростью движения судна уравне-
нием
п = 66,5^,
где п выражается в рад/сек, а о — в узлах.
2. ИСПЫТАНИЯ ПРИ ПЕРИОДИЧЕСКОМ ПОГРУЖЕНИИ
В ЭЛЕКТРОЛИТЫ
Многие металлические конструкции подвергаются периоди-
ческому смачиванию электролитами. К таким конструкциям
относятся гидротехнические сооружения, -сваи оснований мор-
ских сооружений, корабли, плавающие доки и т. п.
В зонах, где металлы периодически смачиваются морской
или речной водой, «наблюдается самая большая коррозия. Она
значительно превосходит коррозию при полном погружении
в электролит [23].
— 38 —
Подобные условия эксплуатации изделий лучше всего ими-
тировать при ускоренных испытаниях методом переменного
погружения металла в электролит или методом обрызгивания.
Однако периодическое погружение в электролит широко ис-
пользуют при ускоренных испытаниях не только для изучения
коррозионной стойкости металлов и средств защиты, применяе-
мых <в судостроении и гидротехнических сооружениях, но и для
испытаний изделий, предназначенных для эксплуатации в атмо-
сферных условиях. При этом ви-
де испытания коррозионный про-
цесс большую часть времени
протекает в тонком слое электро-
лита, что, как было показано на-
ми {24], для целого ряда метал-
лов, процесс коррозии которых
определяется скоростью катод-
ной реакции, должно привести к
резкому сокращению сроков ис-
пытания. В цитируемой работе
изучалась зависимость скорости
кислородной деполяризации от
толщины пленки электролита и
было показано, что скорость ка-
тодного процесса в пленках на-
много выше, чем в объеме. Это
видно из поляризационных кри-
вых, представленных на рис. 18.
По мере уменьшения слоя элек-
тролита скорость кислородной
деполяризации все больше увели-
чивается. Необходимо при этом
Плотность тогго.па/сн2
Рис. 18. Зависимость катодной по-
ляризации железа от толщины
пленки 0,1-и. раствора NaCl:
1 — гв объеме; 2 — 330 Лклц-.
3 — 165 миг. 4 — 100 мкм
напомнить, что чрезмерное умень-
шение толщины пленки электролита может привести к сильной
анодной поляризации. Поэтому приходится выбирать оптималь-
ную толщину пленки. По данным наших работ, слой электролиз
та не должен быть меньше 30—50 мкм.
Как уже указывалось, для -правильного выбора метода уско-
рения процесса -необходимо иметь ясное представление -о том,
какая из реакций, обусловливающих коррозионный процесс,
является лимитирующей для данного металла или сплава. Если,
например, скорость коррозии определяется скоростью кисло-
родной деполяризации, то уменьшение толщины слоя электроли-
та ’приведет к ускорению испытаний. Если же процесс контро-
лируется скоростью анодной реакции, утоньшение слоя
электролита приведет к обратным результатам.
Рассмотрим эту сторону вопроса на примере атмосферной
коррозии. Расчет соотношения между омическим и поляриза-
ционным сопротивлениями, произведенный для различных- эле-
— 39 —
ментов —Fe— Си, Fe — Zn, покрытых тонкими слоями электро-
литов (NagSCU, NaCl), а также дистиллированной воды различ-
ной толщины (70—165 мкм), показал, что даже при расстоянии
между электродами, -равном 0,5 мм, омическое падение потен-
циала в электролите составляет всего 4—-6% от начальной раз-
ности потенциалов [25]. Ясно, что в микроэлементах^ где рас-
стояние между электродами на несколько -порядков ниже
(Z — 10“3—10~4 см), омическое падение потенциала остается
таким же, если даже толщину слоя уменьшить на несколько по-,,
рядков.
Анодное 'поляризационное сопротивление элементов при та-
ком же расстоянии между электродами составляет ® 0,1-®. рас-
творе NaCl 4,5%, в 0,1-н. растворе Na$SO4 10% и в дистиллиро-
ванной воде 26%. Доля катодного поляризационного сопротив-
ления во всех изученных электролитах была 'наибольшей и со-
ставляла 70—92%. Поскольку работа коррозионных микроэле-
ментов на стали под -видимыми тонкими слоями электролитов
почти не зависит от омического сопротивления, а определяется
лишь поляризацией электродов, притом в основном -катодной,
ускорение испытаний стало необходимо по возможности прово-
дить в условиях, обеспечивающих ускорение катодного процесса,
а именно при сохранении в течение длительного времени на
поверхности металла тонких пленок электролита.
Этот вид испытаний для металлов и изделий, предназначен-
ных для атмосферных условий, оправдан еше и тем, что он ими-
тирует условия эксплуатации. Основной материальный эффект
при атмосферной коррозии приходится на видимые пленки
электролитов, (возникающие на поверхности металлов либо
вследствие -прямого попадания атмосферных осадков в виде
дождя, снега, изморози или брызг морской или речной воды,
либо, что бывает чаще, -вследствие суточных колебаний темпера-
туры, приводящих к охлаждению атмосферного -воздуха и выпа-
дению росы. Толщина пленок в последнем случае составляет
100—200 мкм.
Для некоторых сплавов имеет значение и скорость анодной
реакции. В работе [25] было- показано, что коррозия алюминия
протекает (в этих же условиях уже с заметным анодным контро-
лем (23%). Еще -большее значение -приобретает анодный конт-
роль для алюминия при коррозии его в атмосферах, не содер-
жащих хлор-ионов. Поэтому для некоторых сплавов, условий
и сред ускорение испытаний не может быть достигнуто измене-
нием кинетики только катодного процесса.
Следовательно, для алюминия и некоторых его сплавов, а,
также нержавеющих сталей, корродирующих в значительной |
степени с анодным контролем, заметного ускорения испытаний (
можно добиться изменением кинетики анодного процесса, что 1
достигается введением в электролит активаторов (ионов!
хлора).
— 40 —
Поскольку процессы, которые контролировались бы на 100%
одной реакцией, (встречаются редко, на практике обычно исполь-
зуют оба пути, т. е. испытания 'проводят -в тонких слоях электро-
лита, снимающих катодное ограничение, а для ускорения анод-
ной реакции вводят в электролит активаторы
При разработке ускоренных испытаний металле®, предназна-
ченных для эксплуатации в атмосфере морского воздуха или
длительно транспортирующихся «по морю, необходимо, как уже
указывалось, предусмотреть введение в электролит хлористого
натрия. При этом, однако, уже бесполезно пытаться ускорить
испытания путем дополнительного облегчения анодной реакции,
поскольку она протекает в присутствии хлор-ионов легко и уже
не является контролирующей. Ускорить испытания в этих усло-
виях можно лишь за счет ускорения катодной'реакции восста-
новления кислорода (снижение толщины слоя электролита,
введение катодных деполяризаторов, снижение перенапряже-
ния).
Периодическое обновление на металле тонкого слоя электро-
лита создает условия для облегченного доступа кислорода
не только в связи с образованием тонкой пленки электроли-
та, но и в результате ее постоянного размешивания при смачи-
вании.
Исследование механизма переноса кислорода через тонкие
слои электролитов, проведенное нами [24], показало, что даже
в тонких слоях имеет место конвекционный -перенос кислорода;
толщина же слоя электролита, через который кислород будет
переноситься чисто диффузионным путем, значительно меньше
общей толщины слоя электролита.
Используя уравнение Эйнштейна, определяющее зависи-
мость коэффициента диффузии от вязкости среды, температуры
и размера диффундируюшей частицы
6ЯТ)Г
(27)
и уравнение для максимальной скорости кислородной деполяри-
зации «ю, наблюдающейся «при наибольшем значении градиента
концентрации кислорода в объеме и в диффузионном слое
(Ср — См) —> Ср
DnFCp
(28),
можно получить уравнение, которое выражает зависимость ско-
рости коррозии с кислородной деполяризацией от толщины
пленки электролита на поверхности Металла
RTnFC
6N блцг
(29)
где Лю—плотность тока;
— 41 —
Рис. 19. Зависимость скорости восста-
новления кислорода от толщины пленки
электролита:
а —• теоретическая кривая; б — эксперимен-
тальные кривые при давлении водяного па-
ра: 1 — 22 • 10—3 бар (17 мм рт. ст.)- 2 —
15- 10-3 бар (И,Б мм рт. ст.). Электролит —
0,1-н. раствор NaCl; материал катода —
мель
h— число электронов, ассимилирующихся при катодной
реакции;
N — число Авогадро;
R — газовая постоянная;
т) — коэффициент вязкости среды;
г — радиус молекулы кислорода;
F — константа Фарадея;
д — толщина диффузионного слоя;
С — концентрация кислорода в растворе.
Уравнение (29) применимо для случая, когда кислород пере-
носится к поверхности металла лишь посредством диффузии. »
Если подставить в это
уравнение все известные
величины, то получим вы-
ражение
(об = 0,190 • 10-5, (30)
которое представляет со-
бой уравнение гипербо-
лы. Расчет скорости вос-
становления кислорода
по данному уравнению,
выполненный авторами
[24], и сопоставление ре-
зультатов с эксперимен-
тально полученными дан-
ными показали, что экс-
периментальные резуль-
таты были во всех слу-
чаях, в том числе и для
толщин пленок, которые
ранее принимались как неразмешиваемые (0,05—0,07 см) [26,
27], выше теоретически рассчитанных (рис. 19) Это указывает,
вопреки существующим мнениям, на то, что и в тонких слоях
электролитов имеет место конвекционный перенос кислорода и,
•стало быть, эффективная толщина диффузионного слоя состав-
ляет всего часть слоя электролита. Таким образом, находящуюся
на металле пленку следует рассматривать как состоящую из
двух частей: диффузионной, в которой концентрация кислорода
меняется линейно, и конвекционной, простирающейся за пределы
эффективной толщины диффузионного слоя, в которой концент-
рация кислорода в любой точке поддерживается благодаря кон-
векции на постоянном уровне.
Авторами было обнаружено также, что скорость кислород-
ной деполяризации сильно изменяется, а стало быть, изменяется
и коррозия три испарении электролитов с металлической по-
верхности [24]. ’Прн этом было установлено, что испарение
электролита с поверхности электрода сопровождается ростом
— 42 —
тока, т. е. ускорением катодного процесса. Более эффективная
работа катода, как показали эксперименты, связана ие столько
с изменением толщины слоя электролита при испарении, сколько
с более сложными явлениями, происходящими на поверхности
электрода.
Было высказано -предположение, что само размешивание тон-
ких слоев обусловлено «изменением поверхностного натяжения
пленки от точки к точке. Это изменение может быть вызвано
температурными перепадами, которые возникают из-за неоди-
наковой скорости испарения, разной величиной электрического
заряда в различных точках поверхности, возникновением джоу-
лева тепла прн работе микроэлементов й т. л. Переменное по-
верхностное натяжение должно вызывать движение электроли-
та, под влиянием которого и усиливается доставка кислорода
к металлу.
Расчет В. Г. «Левина [3] подтвердил эту гипотезу. Оказалось,
что саморазмешивание тонких слоев возможно уже при незна-
чительных температурных -перепадах, а изменение ‘поверхност-
ного натяжения от точки к точке 'приводит к появлению танген-
циальных сил, действующих на поверхности жидкости и
приводящих ее в движение. Теория этого явления, названного
капиллярной конвекцией, разработана В. Г. Левичем [3J и имеет
важное значение для понимания .механизма коррозионных про-
цессов, развивающихся в тонких слоях.
При рассмотрении случая изменения -поверхностного -натя-
жения в результате незначительных температурных перепадов
необходимо иметь в виду, что они, естественно, вызовут обычное
конвективное движение. Однако «в тонких слоях, где поверхность
жидкости достаточно велика по отношению к объему, обычная
тепловая конвекция будет приводить к весьма малым скоростям
движения по сравнению с'теми, которые возникают из-за капил-
лярной конвекции. Для тонких слоев поверхностные эффекты
должны быть велики по сравнению с объемными, поскольку ве-
личина поверхности очень большая, а силы поверхностного натя-
жения весьма -значительны по сравнению с гравитационными.
Таким образом, представляется еще одна возможность уско-
рить испытания, создавая такие условия, при которых на по-
верхности металла пленка определенной толщины (~150 мкм)
сохраняется возможно дольше, а сам электролит постепенно
испаряется, способствуя этим увеличению скорости кислородной
деполяризации благодаря саморазмешиванию. Степень самог
размешивания зависит от толщины пленки ’электролита на ме-
талле. Пленки в термостатированных условиях практически
перестают размешиваться, как показали наши опыты, при тол-
'щине 0,035—0,040 см. Чем меньше влажность воздуха, тем
сильнее размешивается электролит и тем больше скорость кис-
лородной деполяризации при данной толщине пленки. На это
указывают кривые, характеризующие зависимость скорости
— 43 —
кислородной деполяризации от толщины пленки электролита
при различных относительных влажностях (ем. рис. 19).
Таким образом, особенно сильное ускорение коррозии при
периодическом смачивании можно получить тогда, когда основ-
ное время металл будет находиться в -воздухе ;и- кор.р_оди|ювать
в'тонком_слое электролита, который медленно испаряется. Пе-
риодическое смачивание исключает торможение анодного про-
цесса и обеспечивает благодаря усиленному подводу кислорода
эффективное протекание -катодного процесса.
Рис, 20. Влияние периодического смачивания 0,5-м. рас-
твором NaCl иа скорость коррозии металлов {28]:
1 — сплав Д16; 2 — алюминий; 3 — цинк; 4 — кадмий; 5 —
Ст. 3; 6 — чугун. Продолжительность испытаний 3 месяца;
В — погружение в электролит: □ — смачивание 4 раза в сутки
I Обычно хорошие результаты при периодическом увлажнении
получаются, когда около 15—20% всего (времени образцы нахо-
дятся в погруженном состоянии :в электролит и 80—85% време-
ни корродируют на воздухе в тонкой пленке электролита. Время,
в течение которого образцы погружены в электролит, во избе-
жание накопления продуктов коррозии -в тонких слоях целесо
образно разбить на несколько периодов, т. е. пленку возобнов-
лять несколько раз ® сутки или несколько раз »в час.
При периодическом "погружении в электролит процесс может
быть еще ускорен, если в промежутках между смачиваниями
создавать условия, при которых электролит высыхает.
Периодическое смачивание как способ увеличения скорости
коррозии может 'быть применено при ускоренных испытаниях
к тем металлам, коррозия которых контролируется в значитель-
ной степени скоростью доставки кислорода к металлу.
Действительно, опыты, проведенные нами (28], показывают,
что периодическое смачивание больше всего увеличивает ско-
рость коррозии таких металлов, как сталь и чугун; заметное
увеличение скорости коррозии наблюдается у цинка и дуралю-
мина [Д]-6]; чистый - алюминий и (кадмий мало чувствительны
к периодическому смачиванию, поскольку коррозия этих -метал-
— 44 —
Темпера туда* °с
Рис. 21. Зависимость скорости
коррозии стали в 0,5-и. рас-
твора NaCl от температуры (14]:
1 — в объеме электролита; 2 —
в невозобновляемой плеике элек-
тролита; 3 ~~ смачивание один
раз в сутки; 4 — смачивание
24 раза в сутки. В период между
смачиваниями образцы находятся
в атмосфере 100%-ной относитель-
ной влажности
лов контролируется в основном скоростью анодной реакции
1ионнзации металла (рис. 20).
Коррозию при периодическом смачивании электролитом
। можно усилить также (повышением температуры электролита.
[ Повышение температуры при периодическом смачивании увели-
чивает скорость электродных реакций, -в частности реакцию
! восстановления кислорода.
L- В отличие от процессов, протекающих в атмосфере, где по-
вышение температуры не всегда приводит (из-за -быстрого вы-
сыхания пленки) к увеличению скорости коррозии, повышение
температуры при периодическом
смачивании неизменно приведет
к увеличению скорости коррозии,
поскольку электролит постоянно
обновляется, что обеспечивает не-
прерывный контакт металла с аг-
рессивной средой.
Результаты лабораторных
опытов [14], приведенные на рис.
21, показывают, что повышение
температуры электролита неиз-
менно приводит к росту коррозии
стали, причем и при полном по-
гружении в электролит, и при
периодическом смачивании на-
блюдается примерно одна и та
же закономерность; наибольшее
увеличение скорости процесса
наблюдается в интервале от 20
до 40° С. При дальнейшем повы-
шении температуры коррозия
возрастает в меиьшей степени,
что обусловлено уменьшением
концентрации кислорода в элект-
ролите. Обращает на себя вни-
мание наклон кривых для перио-
дического смачивания, свиде-
тельствующий о том, что влияние температурного фактора в этих
условиях проявляется сильнее, чем в невозобновляемой пленке
или при полном погружении в электролит. Если иа поверхности
стали имеется пленка электролита, которая получена однократ-
ным смачиванием и не возобновляется, то коррозия, начиная с
определенной температуры, сильно замедляется. Это объясняет-
ся наряду с уменьшением растворимости кислорода более быст-
рым удалением электролита при повышении температуры с по-
верхности металла. При частых смачиваниях температура» начи-
ная с определенного значения, также перестает заметно влиять
на увеличение скорости коррозии, однако не в такой степени,
— 45 —
Рис. 22. Зависимость скорости коррозии
стали ^арки Ст. 3 от частоты смачивания
0,5-н. раствором NaCl. Продолжительность
испытания 7 суток. Пунктиром отмечен
уровень коррозии при погружении в объем
электролита
как в невозобновляемой пленке. В последнем случае фактором,
замедляющим коррозию, является лишь -уменьшение раствори-
мости кислорода. Повышение температуры при постоянном об-
новлении пленки способствует самор замешиванию пленки, ко-
торое, как указывалось выше, увеличивает скорость кислородной
деполяризации.
Учитывая, что в тонких слоях электролитов диффузионные
ограничения играют еще существенную роль, увеличение ско-
рости коррозии с «повышением температуры следует объяснить
облегчением диффузии кислорода к металлической поверхности.
Следовательно, при периодическом смачивании коррозия будет
заметно увеличиваться лишь до 50—60° С. Более высокие темпе-
ратуры при ускорен-
ных испытаниях мето-
дом переменного по-
гружения в нейтраль-
ных электролитах при-
менять нет смысла.
При ускоренных •
коррозионных испыта- !
ниях большую роль бу- i
дет играть также ча- j
стота смачивания; по;
мере увеличения коли-
чества смачиваний в i
единицу времени кор- |
розия будет возра- i
стать. Наибольшее
увеличение скорости •
коррозии при длитель-
ных испытаниях можно получить при смачивании приблизитель-
но один раз в час; дальнейшее увеличение частоты смачиваний
лишь слабо влияет на повышение скорости процесса. Это, оче- ~
видно, связано с тем, что при определенной частоте смачиваний
количество электролита, удерживающегося на поверхности, яв-
ляется таким, которое обеспечивает течение электрохимических
реакций с максимальной скоростью. Дальнейшее увеличение ча-
стоты смачиваний, которое способствует более длительному
пребыванию металла под воздействием более толстых слоев,
электролита, уменьшит скорость протекания катодной реакции
и поэтому уже не может привести к заметному росту коррозии.
Указанное выше хорошо иллюстрируется кривой, представлен-
ной на рис. 22. Кривая показывает зависимость скорости корро-
зии стали от частоты смачиваний 0,5-н. раствором хлористого
натрия при испытании в течение одной недели.
На рис. 23 «приведены «кривые, характеризующие зависимость
скорости коррозии стали от состава электролита при испыта-
ниях в условиях полного погружения в электролит, в невозоб*
— 46 —
новляемой пленке электролита -и при -периодическом смачивании
два раза в сутки. Кривые аюказывают, что и при погружении
в электролит и при: периодическом смачивании природа аниона
(С1~ >или SO|“) для -коррозии стали значения не имеет *; весь
эффект увеличения скорости коррозии возникает при смачи-
вании в результате облегчения катодной реакции восстановле-
ния кислорода, которая, как известно, мало зависит от харак-
тера аниона. Аналогичные зависимости наблюдались и для цин-
ка [14].
Повышение скорости коррозии при периодическом смачива-
нии наблюдается и в концентрированных, и «в разбавленных
растворах солей. Скорость коррозии при периодическом смачи-
вании, как видно из рис. 24, растет только до определенного
Рис. 23. Зависимость скорости кор-
розии стали от состава электро-
лита и режима испытания:
I — в объеме электролите; 2 — в нс-
возобновляемой пленке; 3 — при
смачивании 2 раза в сутки; а —
0,5-я раствор NaCl; -б. — 0,5-и. рас-
твор NagSOi
значения концентрации; для хло-
ристого натрия она соответствует
Рис. 24. Зависимость скорости корро-
зии стали марки Ст. 3 от логарифма
концентрации NaCl при смачивании
один раз в час. Продолжительность
испытаний 8 ч
Следовательно, максимального увеличения скорости корро-
зии при ускоренных испытаниях с периодическим смачиванием
в растворах хлоридов можно достигнуть, применяя концентра-
ции, равные 0,5-н. или близкие к ней; уменьшение концентра-
ции NaCl, а также ее увеличение приводят к снижению скорости
коррозионного процесса.
Характер коррозии при периодическом смачивании электро-
литами зависит ют условий испытания. Чем чаще производятся
смачивания и чем интенсивнее высушивается пленка, тем равно-
мернее распределяется коррозия по поверхности металла.
1 Возможно, что «при длительном -воздействии электролита (-годы), когда
начинают сказываться свойства продуктов коррозии и защитных пленок,
хлористые электролиты окажутся более агрессивными.
— 47 —
Периодическое смачивание металлов электролитами как ме-
тод, ускоряющий ^процесс коррозии, широко применяется в ла-
бораторных испытаниях. В (настоящее время многие исследова-
тели проводят испытания при периодическом смачивании
с разным соотношением времени погружения и высушивания.
Режимы испытаний черных и -цветных металлов методом пе-
риодического погружения, применяющиеся у нас и за грани-
цей [29], приведены -в табл. 5.
Таблица 5
РЕЖИМЫ ИСПЫТАНИЙ ПРИ ПЕРИОДИЧЕСКОМ ПОГРУЖЕНИИ ОБРАЗЦОВ
В ЭЛЕКТРОЛИТ (3%-НЫЙ РАСТВОР Nad).
Страны Время, мин Металлы
3 я ч ч и О S С S? 62 Е з- _ о S >. 1 время \ между по- i гружени- ями
СССР 60 10 50 Черные и цветные сплавы
США 15 9 I 3 14 6 ’Цветные сплавы » »
3—6 1—2 2—4 » »
60 10 50 » »
24 ч 16 ч 8 ч Окрашенные изделия
30 15 15 Черные сплавы
60 50 10 Алюминиевые сплавы
ГДР и ФРГ 30 10 20 » »
60 1—2 58 » »
Франция 60 30 30 Черные и цветные сплавы
60 15 15 Черные сплавы
Англия Ю I 9 Алюминиевые сплавы
Конструктивное оформление аппаратов для периодического
погружения образцов в электролиты различно. Применяются
рычажные или вращающиеся конструкции типа колес -или. бара-
банов, на которых укрепляются образцы; в последнем случае
исключается влияние электролита, скапливающегося на концах
•образцов.
Схема аппарата рычажной конструкции, разработанного
Гопиусом [30], приведена на рис. 25. Аппарат располагается на
крышке 6 деревянного стола 7. Образцы подвешиваются на
стеклянных палочках 1, укрепленных на деревянных штангах 2.
Образцы периодически погружают в электролит с помощью
кривошипа 8, часового механизма 12 и реле 11. Для уменьшения
влияния скопления электролита в нижией части образца целесо-
образно применять образцы, заостренные на конус.
Схема аппарата с вращающимся колесом приведена -на
рис. 26. Образцы 4, 'вращаясь -вместе с колесом 2, на котором
— 48 —
они укрепляются в радиально расположенных прорезях 3, -по-
падают на некоторое время в -ванну 6 с электролитом; остальное
время образцы корродируют в -высыхающей пленке электролита.
Высушивание пленки производится с помощью электроламп,
установленных в кожухе 1. В зависимости от скорости вращения
колеса образцы перед повторным погружением могут частично
Рис. 25. Схема прибора Гопиуса для уско
ренных испытаний металлов при перемен-
ном погружении в электролит (рычажная
конструкция) [30]:
/ — стеклянные палочки; 2 — деревянные штан-
ги, 3 — шток; 4 — направляющая; 5 — выклю-
чатель; 6 — крышка стола; 7 — деревянный
аол; 8 — кривошип; 9 — мотор; 10 — рубиль-
ник; 11 — реле; 12 — часовой механизм
или полностью высыхать. -Скорость высушивания образцов в пе-
риод между смачиваниями должна тщательно контролиро-
ваться и всегда -быть строго определенной, иначе результаты
испытаний не будут воспроизводиться.
Колесо, на котором укрепляются образцы, должно быть из-
готовлено из инертного материала (текстолит, эбонит, плекси-
глас); ванны для электролита, в которых производится смачи-
вание, обычно делаются из фаянса, керамики, металла, покры-
того специальной эмалью, или из любого (имеющегося в распо-
ряжении исследователя) инертного материала. При испытании
4 Зак. 1338 •— 49 —
алюминиевых сплавов -ванны можно изготавливать -из корро-
зиониостойкого алюминиевого сплава.
Скорость высушивания и частота смачиваний «при всех спо-
собах испытаний должны подбираться с таким расчетом, чтобы
в процессе высушивания пленка испарялась лишь частично
и к повторному погружению в
Рис. 26. Схема аппарата для уско-
ренных испытаний при периоди-
ческом погружении в электролит
(колесо) [29]:
I — кожух осушающей кодеры; 2 —
колесо: 3 — прорези для креплеиня
образцов; 4 — образец; 5 — стол;
С — ванна для электролита; 7
вращающийся вал
электролит величина ее на по-
верхности образца всегда состав-
ляла бы 30—50 мкм. Это необхо-
димо для того, чтобы коррозия
протекала в течение всего време-
ни испытания.
В качестве электролитов для
ускоренных испытаний при пе-
риодическом смачивании приме-
няют обычно 0,5-н. (3%-ный) рас-
твор хлористого натрия, искус-
ственную морскую воду или бо-
лее разбавленные растворы хло-
ристого натрия, например для
магниевых сплавов 0,001-н. рас-
твор.
При проведении испытаний
при повышенных температурах
предусматривается подогрев
электролита и- поддержание ок-
ружающего воздуха в заданном
режиме. Лучше всего аппараты помещать в воздушные термо-
статы или специально отведенные комнаты, где температура ре-
гулируется
3. ИСПЫТАНИЯ ПРИ ПЕРИОДИЧЕСКОМ
ОБРЫЗГИВАНИИ ЭЛЕКТРОЛИТАМИ
Путем периодического обрызгивания металлов электролита-
ми воспроизводят условия береговых районов, где конструкции
работают .в (непосредственной близости от моря ’или реки и «под-
вергаются систематическом'у воздействию электролита, а также
условия работы оборудования н приборов, расположенных на
палубах кораблей или в других местах, доступных для попада-
ния брызг. Периодическое обрызгивание различными электро-
литами способствует ускорению процессов коррозии, особенно
если электролит содержит ионы хлора. Ускорение в этом слу-
чае достигается, как и при периодическом смачивании, за счет
создания на металле тонкого слоя "электролита, в котором катод-
ные -процессы развиваются с большей скоростью, чем при
погружении *в электролит. Отличие этого испытания от испыта-
ния -при «периодическом погружении заключается главным обра-
— 50 —
РЕЖИМЫ ИСПЫТАНИЙ ПРИ ПЕРИОДИЧЕСКОМ ОБРЫЗГИВАНИИ МЕТАЛЛОВ ЭЛЕКТРОЛИТАМИ
Применение о Относительная влажность % Состав электролита Примечание
NaCl MgCb6H4O СаСЬ NasSO< КС! нго
Защитные по- крытия на стали и железе От до 19 21 100 (над во- дой) 23 е/Эле3 9,8 г/Эж3 1,2 г/Эж3 8,9 е/йж3 - Осталь- ное Образцы подвешива- ются вертикально; об- рызгивание один раз в сутки
Временные за- щитные покры- тия на стали От До 15 21 95 1,0% — — — — Осталь- ное Периодическое обрыз- гивание
Приборы связи 40±1 От 60 до 80 2,7% 0,6% 0,1% — 0,07% Осталь- ное Обрызгивание из руч- ного пульверизатора один раз в сутки; про- должительность испы- тания 7 суток
Временные за-, щитные покры- тия для слабоаг- рессивных усло- вий Комнат- ная 100 100% Образцы располагают- ся горизонтально; об- рызгивание один раз в сутки, высушиваются в течение одного-двух ча- сов, испытываются до по- явления ржавчины
Временные за- щитные покры- тия для слабоаг- рессивных усло- вий ’ 24 -27 100 100% Образцы располага- ются под углом 30° к горизонтали; высота падения воды 0,61 ж; обрызгивание непрерыв- ное
зом в том, что *при обрызгиваний смывающее действие электро-
лита отсутствует и (продукты коррозии будут более прочно
связаны с металлом. Кроме того, при обрызгивании в продуктах
коррозии будут (накапливаться соли, содержащиеся в электро-
лите.
В отличие от испытаний во влажных камерах, где при распы-
лении растворов электролиты непосредственно не попадают на
металл, при разбрызгивании струя мелких брызг направляется
на образцы. Непосредственное обрызгивание «применяется как
самостоятельный вид испытания и как дополнительное воздейст-
вие на металл при конденсационных испытаниях или при испы-
таниях в камерах искусственного климата.
При конденсационных испытаниях и в камерах искусствен-
I него климата обрызгивание производится периодически, один-
I два раза в течение одного цикла. В большинстве описанных
j в литературе методов ускоренных испытаний с периодическим
обрызгиванием применяют растворы хлористого натрия различ-
ной концентрации для создания условий береговой морской
атмосферы. В некоторых случаях для «воспроизведения климата
, с обилием дождей вместо обрызгивания образцов их периоди-
чески помещают под душ. Примером этого может служить ка-
мера заводов радиотехнической промышленности, имеющая
дождевальное устройство для орошения образцов. Схема этой
камеры рассматривается ниже (см. рис. 48).
• Обрызгивание электролитами применяется также при испы-
тании образцов с лакокрасочными покрытиями и с временными
защитными покрытиями, такими как смазки. Режимы таких
испытаний приведены в табл. 6.
Периодическое обрызгивание электролитами применяется
также иногда для увеличения скорости коррозии образцов, испы-
тываемых «в естественных условиях «а коррозионных станциях.
4. ИСПЫТАНИЯ В АТМОСФЕРЕ
С ПОСТОЯННОЙ ВЛАЖНОСТЬЮ
Наличие водяных паров в воздухе — необходимое условие
• появления коррозии. Вода, адсорбируясь или конденсируясь на
поверхности металла, создает тонкий слой электролита, в кото-
ром и происходят коррозионные процессы. Благодаря усилен-
ному доступу кислорода создаются благоприятные условия для
протекания катодного процесса. Однако в чистой пленке воды
больших коррозионных эффектов ожидать нельзя, «поскольку
анодная реакция «протекает медленно; образующиеся продукты
коррозии представляют собой гидраты, которые обладают низ-
кой растворимостью и благодаря этому со временем замедляют
коррозию.
Тем не менее влажность воздуха относится к числу важней-
ших факторов, определяющих скорость коррозии, поскольку при
— 52 —
определенных значениях ее коррозии- вообще может не-быть.
Для каждого металла характерна определенная величина крити-
ческой влажности, по достижении которой скорость коррозии
резко возрастает. Опытным путем для некоторых металлов были-
обнаружены две критические точки, получившие название пер-
вичной и вторичной критических влажностей. Первичная крити-
ческая влажность соответствует адсорбции на металле такого
количества влаги, которое достаточно, чтобы коррозия шла с за-
метной скоростью. Вторичная критическая влажность соответст-
вует моменту, когда за счет коррозионного процесса, протекаю-
щего хотя и с малой скоростью, накапливаются продукты корро-
зии, в присутствии которых конденсация 'влаги начинается уже
при малой относительной влажности (70—60%).
При наличии в атмосфере загрязнений (газы, частички солей)
критическая влажность заметно снижается и металлы начинают
корродировать с заметной скоростью при меньшей относитель-
ной влажности атмосферы.
Рассматривая влияние влажности воздуха на- коррозию ме-
таллов применительно к условиям ускоренных -испытаний, необ-
ходимо прежде всего заметить, что в атмосфере чистого воздуха
при отсутствии конденсации влаги нельзя ожидать значительно-
го увеличения скорости коррозии, как бы ии повышалась влаж-
ность. Наибольшего повышения скорости в этих условиях мож-
но достичь, если относительная влажность воздуха будет выше
критической. Лучше всего испытания проводить в-условиях, ко-
торые обеспечивают возможно большую адсорбцию водяных
паров и длительное сохранение их на поверхности металла. Эти
условия создаются, когда относительная влажность воздуха в
системе близка к состоянию полного насыщения пространства '
водяными парами («100%).
Однако, когда хотят воспроизвести условия сельской атмо-
сферы, в которой предполагают эксплуатировать изделия, иног-
да прибегают и к испытаниям при постоянной влажности в чи-
стой атмосфере. Срок испытания при этом должен быть удлинен
до 6—12 месяцев. Создание н обеспечение постоянной влажности
требуют определенных навыков и умения рассчитывать количе-
ство водяных паров, которое необходимо 'ввести в камеру или
реакционный сосуд.
Рассмотрим некоторые понятия (относительная влажность,
абсолютная влажность, влагосодержание), с которыми приходит-'
ся встречаться исследователю или инженеру, занимающемуся
испытаниями.
Количество -водяного пара в воздухе может быть вычислено
при помощи парциального давления (упругости) водяного пара
которое, как и давление воздуха, выражается в н/м2 (кг/м2).
Каждому значению температуры воздуха соответствует впол-
не определенное максимальное содержание водяного пара в нем.
Чем выше температура, тем больше значение этого максимума.
— 53 —
Водяной пар при полном насыщении пространства находится
в состоянии сухого насыщения.
Содержание водяных паров в воздухе при заданных темпе-
ратуре и давлении ограничено, таким образом, вполне опреде-
ленными пределами; оно не может превысить значение, при ко-
тором парциальное давление паров воды в смеси будет выше
давления насыщенного пара при данной температуре.
Упругость паров, насыщающих пространство над плоской по-
верхностью воды р, является в свою очередь функцией темпера-
туры и с повышением температуры быстро растет.
Теоретически эта зависимость выражается известным уравне-
нием Клапейрона [32]:
dp__ е
dT (у—У^Т '
(31)
где р — давление пара;
е — скрытая теплота парообразования -одного моля, дж(моль
(кал!моль);
Т — абсолютная температура пара;
V — объем одного моля пара;
Vo — объем -одного моля жидкости.
Допустив, что V — Vo = V (объем пара несравненно больше
объема жидкости) и учитывая, что для небольших темпер атур-
иых интервалов V = •— , получаем после интегрирования урав-
р
нения (31), что
Таким образом, зная давление паров при одной температуре,
можно легко вычислить давление пара при другой.
Имеются также эмпирические формулы, позволяющие рас--
считать упругость паров при любой температуре [33]:
л 1Л 7,63267
р — 6,1078 • 10 —---мбар. (33)
241,9+7
Кроме упругости паров, влажность может также характери-
зоваться такими показателями, как относительная и абсолютная
влажность воздуха, влагосодержание и удельная влажность
воздуха.
Относительной влажностью называется отношение фактиче
ского содержания водяных паров в воздухе к максимально воз-
можному в данных условиях. Обозначается эта величина Н:
(34)
где — плотность насыщенного водяного пара, находящегося
во влажном воздухе, кг[см3\
— плотность насыщенного водяного пара, кг[см\
— 54 —
Относительная влажность воздуха определяется часто и как
отношение парциального давления водяных паров, находящихся
в воздухе, pD к давлению насыщенного водяного пара при дан-
ной температуре ps .и выражается в процентах:
н = 100,
Ps
(35)
Таким образом, относительная влажность показывает, насколько
воздух близок к состоянию насыщения.
Абсолютная влажность а представляет собой количество во-
дяного пара, содержащегося в 1 м? воздуха, выраженное в грам-
мах. Эта величина соответствует плотности пара. Она приблизи-
тельно равна парциальному давлению пара (табл. 7).
Таблица 7
СОДЕРЖАНИЕ ВОДЯНЫХ ПАРОВ В НАСЫЩЕННОМ ИМИ ВОЗДУХЕ
ПРИ РАЗЛИЧНЫХ ТЕМПЕРАТУРАХ
{ПРИ ДАВЛЕНИИ 1,01 бая (760 ям рт. ап.))
Содержание водяных паров, г/м®
Темпера- туру 0 1 2 3 4 5 6 7 8 9
—10 2,10 1,98 1,81 1,66 1,52 1,40 1,28 1,18 1,08 0,98
— 0 4,84 4,47 4,13 3,81 3,52 3,24 2,99 2,75 2,'54 2,34
4- 0 4,84 5,18 5,84 5,92 6,33 6,76 7,22 7,70 8,22 8,76
+10 9,38 9,94 10,57 11,25 11,96 12.71 13,51 14,34 15,22 16,14
4-20 17,12 18,14 19,22 20,36 21.55 22,80 24,11 25,59 26,93 24,85
4-30 30,04 31,70 33,45 35,28 37,19 39,19 41,28 43.47 45,75 48.14
Связь между абсолютной влажностью воздуха а, упругостью'
пара Pd и температурой Т определяется следующим уравнением:
где а — абсолютная влажность воздуха, г/м3;
До —упругость водяного пара;
Т — абсолютная температура.
Влагосодержание. В технических расчетах широкое распро>
странеиие получила такая характеристика влажного воздуха,
как удельное влагосодержание, представляющее собой количе-
ство водяных паров, приходящихся на один килограмм сухого
воздуха в паровоздушной смеси. Влагосодержание обозначается
d (кг/кг)-.
d = 0,622 , (37)
p — HPs
где И — относительная влажность воздуха, %;
р—'давление (полисе) паровоздушной смеси, бар {кГ/м2)\
ps — давление насыщенного пара, бар (кГ/м2).
—55 —
Удельная влажность f представляет собой количество водя-
ного пара, содержащегося в одном килограмме влажного воз-
духа. Она по существу характеризует концентрацию водяного.-
пара в воздухе -и определяется как отношение массы пара, содер-
жащегося в 1 JH3 воздуха, к массе паровоздушной смеси:
/=0,622-----,
р—0,378pD
(38).
где рп — парциальное давление водяного пара, бар (кГ/м2);
р — полное давление паровоздушной смеси, бар (кГ]м2).
Р.ис, 27 Схема увлажняющей части установки
Вернона [34]
Величиной f удобно пользоваться при расчетах, так как по-
следняя не меняется при нагревании и охлаждении, а также
при сжатии и расширении воздуха, если только не происходит
конденсации водяного пара.
Плотность воздуха. Весьма важной характеристикой, кото-
рой приходится пользоваться при расчетах условий ускоренных
испытаний, является плотность влажного воздуха. Она зависит
от относительной влажности, температуры и давления и выра-
жается следующим уравнением:
ув = ус---0,013 кг/м3, (39)
где ув — плотность влажного воздуха;
ус — плотность сухого воздуха;
Т — абсолютная температура.
Из уравнения (39) видно, что при равном общем давлении
и температуре плотность влажного воздуха уъ ниже плотности
сухого воздуха ус.
Зная содержание водяных паров в воздухе, в котором пред-
полагается эксплуатировать изделие, или относительную влаж-
— 56 —
воздуха и водяного пара»
Рис. 28. Сосуд для испыта-
ния образцов во влажной
атмосфере при комнаткой
температуре {35]
ность воздуха, можно по формулам и таблицам, приведенным
выше, рассчитать количество водяных паров (воды), которое
надо ввести в камеру, где проводятся испытания.
Вернон и Унтби [34] достигали нужной влажности путем
смешения в предварительно эвакуированных сосудах опреде-
ленного количества сухого чистого
Схема увлажняющей части такой
установки приведена на рис. 27. На-
сыщенный «водяной пар из колбы 6,
в которой кипятилась вода, пропус-
кался через систему трубок 2, поме-
щенных в термостат 4, поддержи-
вающий температуру, равную тем-
пературе испытания. Требуемое ко-
личество водяного пара отбиралось
через отверстие /. Избыток влаги
конденсировался и собирался в ло-
вушку 3. При слишком обильном
образовании пара в колбе избыток
его выпускался через кран 5.
Другой широко распространен-
ный способ увлажнения воздуха,
подаваемого в камеру или сосуд, где
проводятся испытания, — пропу-
скание потока воздуха под не-
• большим давлением через столб
воды. На рис. 28 показан сосуд для
проведения коррозионных испыта-
ний при комнатной температуре при
100%-ной относительной влажно-
сти [ЗБ]. Увлажнение производится
пропусканием воздуха через колон-
ку 5, высота столба воды в которой
равна 1 At. Эта высота, по данным автора, обеспечивает нужную
степень увлажнения (100% относительной влажности). Увлаж-
ненный воздух поступает в сосуд /, в котором и а подставке 2
располагаются образцы.
Если по каким-либо причинам испытания необходимо прово-
дить в атмосферах, лишь частично насыщенных влагой, то это-
го можно достигнуть также просасыванием воздуха через на-
сыщенные растворы соответствующих солей. Прн этом необхо-
димо поставить дополнительные фильтры, предупреждающие
захват частичек соли.
Если в пространстве, где производятся коррозионные испы-
тания, находятся жидкости, создание атмосфер с заданным со-
держанием водяных паров производится помещением на дно-
соответствующей камеры или реакционного сосуда растворов
серной кислоты [36]:
— 57 —
Нормальность кислоты 2 3 4 6 7,6 9 10,6 12,4
Упругость водяного па-
ра 1,3-10~3 бар {мм рт.
ст.) .................. 23,0 22,2 21,7 19,6 17,8 15,7 13,4 10,9
Относительная влаж-
ность воздуха, % . . .96,0 93,5 91,5' 83.0 75,0 66,0 56,0 42,0
Таблица 8
ОТНОСИТЕЛЬНАЯ ВЛАЖНОСТЬ ВОЗДУХА НАД ВОДНЫМИ РАСТВОРАМИ
ГЛИЦЕРИНА 137]
Содержа- ние глицерина Относительная влажность, %, при температуре, °C Содержа- ние глицерина Относительная влажность, %, при температуре, °C
% 0 25 50 70 0 25 50 70
10,7 97,7 98,0 98,2 98,4 64,1 68,6 70,0 71,3 72,2
13,6 95,6 96,0 96,4 96,7 69,0 63,4 65,0 66,4 67,3
22,0 94,5 95,0 95,5 95,8 73,2 58,4 60,0 61,4 62,5
34,9 ' 89,2 90,0 90,7 91,2 77,0 53,3 55,0 56,5 57,6
44,1 84,0 85,0 85,9 85,6 80,3 48,3 50,0 51,5 52,6
52,0 78,8 80,0 81.1 81,8 83,4 43,3 45,0 46,6- 47,7
59,5 73,7 75,0 76,2 77,0 88,0 38,3 40,0 41,6 42,7
Не менее распространено также применение глицерина опреде-
ленной концентрации [36, 37] (табл. 8) илн насыщенных раство-
ров солей, находящихся в контакте с твердой фазой (табл. 9).
Таблица 9
СМЕСИ СОЛЕЙ С ИХ НАСЫЩЕННЫМИ ВОДНЫМИ РАСТВОРАМИ,
ПОДДЕРЖИВАЮЩИЕ ПОСТОЯННУЮ ВЛАЖНОСТЬ ПРИ 20° С [36]
Соль Относи* тельная влажность % Соль Относи- тельная влажность %
LiCblW 15,0 Na2S2O3-5H2O 78,0
ксгн3о2 20,0 NH4C1 79,5
СаС12-Н2О 32,0 (NH^SO. 81,0
СгО3 35,0 КВг 84,0
Zn(NC^)2-6H2O 42,0 khso4 86,0
kno2 45,0 КСгб4 88,0
KCNS 47,0 ZnSO4-7H2O 90,0
NaHSO4-H2O 52,0 NaBrOs 92,0
Na2Cr2O7-2H2O 52,0 К2НРО4 92,0
NaBr-2H2O 58,0 Na2SO4- ЮН2О 93.0
MglQjHjOA-WsO 65,0 nh4h2po4 93,1
NaNO2 66,0 Na2SO3-7H2O 95,0
№H4C1 и KNOs 72,6 Na 2HPO4- 12H2O 95,0
NaClO3 75,0 Pb(NOA 98,0
NaC2HsO2-3HsO 76,0 CuSO4-5H2O 98,0
H2C2O4-2H2O 76,0
— 58 —
При применении растворов, приведенных в табл. 9, следует
иметь в виду, что. поддерживаемая ими влажность изменяется
с изменением температуры, поэтому опыты следует вести в тер-
мостатированных. условиях, чтобы поддерживалась выбранная
влажность. Однако сущест-
вуют некоторые соли, с по-
мощью которых можно под-
держивать нужную посто-
янную влажность в широ-
ком интервале температур
[38] (рис. 29). В этом случае
термостатирование может
быть менее точным.
Аппаратура для прове-
дения испытаний в атмосфе-
рах с постоянной влаж-
ностью несложная; при ис-
пытании небольшого коли-
чества образцов пользуют-
I 100 Г-
| so -
во - * g
$ 70 ’ ут,
& -—... - < J
§ 60 - 4
|5Z?- 5
§ J0|---L__L__|-1-L..I..L_ I-1--1
*3 О 10 20 30 SO 50 60 70 80 SO 100
Температура °C
ся закрытыми сосудами, ча-
ще всего эксикатором или
гидростатом. Последний
представляет собой ванну
Рис. 29. Влияние температуры на от-
носительную влажность воздуха, соз-
даваемую насыщенными растворами
солей (38J:
I — KC1; 3 — NaCl; 3 — 'NaNOs;
4 — NaNOa; Б - CrO3
с гидравлическим затвором,
как показано на рис. 30, б. При испытании большого ко-
личества образцов применяют специальные камеры различной
конструкции. Если для ускоренных испытаний выбрана чистая
Рис. 30. Простейшие приборы для испытаний во влажной атмосфере:
а — эксикатор; б — гидростат; в — влажная камера простой конструкции
атмосфера, а влажность в течение опыта не изменяется, то конст-
рукция камеры может быть простой (рис. 30, в). Постоянная
влажность в такой камере поддерживается водой, налитой на
дно камеры.
Влажные камеры (гигростаты) типа Г-4 для испытаний за-
щитной способности лакокрасочных покрытий в атмосфере
— 59 —
100%-ной относительной влажности изготовляют на Хотьков-
ском заводе экспериментальной окрасочной технологии и аппа-
ратуры.
Испытания в такой камере должны проводиться в термоста-
тированных условиях, так как при 100%-ной относительной
влажности даже небольшие колебания температуры приводят
к конденсации влаги и изменению условий испытаний. С этим
в основном и связана плохая воспроизводимость результатов
испытаний: К аналогичному типу испытаний можно отнести ис-
пытания в камерах, где' распыляется дистиллированная вода.
Скорость коррозии -металлов в таких камерах будет несколько
выше, чем в замкнутых сосудах с постоянной влажностью
(100%), но и в этом случае процесс ускоряется-слабо.
При имитации условий промышленных или морских районов
для увёличения скорости коррозионных процессов в камеры
вводят такие агрессивные вещества, как хлористый натрий
и сернистый ангидрид. Их применение оправдано, поскольку
они в том или ином количестве всегда содержатся в подобных
атмосферах.
5. ИСПЫТАНИЯ В АТМОСФЕРЕ
С ПОСТОЯННОЙ ВЛАЖНОСТЬЮ
В ПРИСУТСТВИИ
КОРРОЗИОННО АКТИВНЫХ АГЕНТОВ
Как было показано в ряде работ4, значительное увеличение
коррозии металлов вызывает присутствие в атмосфере частиц
солей и газов (NaCl, MgCl2, SO2, Cl2, NH3).
Сернистыйангидрид. До сих пор предполагали, что механизм
действия этого газа -заключается в том, что он окисляется на
воздухе до SO8 и впоследствии, растворяясь в тонких пленках
влаги, конденсирующейся на металлической поверхности кон-
струкции, образует серную кислоту. Последняя, разрушая окис-
ные пленки, облегчает этим самым течение анодной реакции
ионизации металла. На этом основании Но многих ускоренных
испытаниях, имитирующих промышленные атмосферы, в каче-
стве электролита используют серную кнслоту. Ее применение,
на наш взгляд, лишено какого-либо научного обоснования.
Последними электрохимическими исследованиями [39] удалось
установить, что механизм действия сернистого газа является
принципиально иным. Оказалось, что этот газ при относительно
положительных потенциалах может восстанавливаться на боль-
шинстве Технически важных металлов, принимая, таким- обра-
зом, непосредственное участие в процессе катодной деполяриза-
ции. На рис. 31 показаны катодные поляризационные кривые,
снятые на железе в атмосфере, содержащей сернистый газ.
Скорость деполяризации в присутствии этого газа увеличивается
— 60 —
в десятки раз. Появление в системе нового катодного деполяри-
затора способствует тому, что катодная реакция, лимитирующая
чаще всего общую скорость коррозионного процесса, сильно
облегчается. Наиболее вероятно, что восстаиовление сернистого
ангидрида идет по следующей схеме:
2HSO3 2Н+ 2# -> S2O4 2HgO, <РрНа=2,8 = —0,268 в; (40)
85ОЦ + 2Н+ + 2г-^ЙгоГ4-Н2О, <ррн»2.8 = -0,973 с. (41)
В некоторых случаях возможно восстановление сернистого
ангидрида до серы по схеме
HESOa + 4Н+ 4- 4е - ЗН2О + S, <ррн™з = + 0,285 в. (42)
Кроме того, следует учитывать, что сернистый газ обладает
в 1300 раз большей растворимостью в воде, чем кислород. По-
этому даже при незначительном содержании сернистого газа
в воздухе концентрация его в электролите может стать соизмери-
мой с концентрацией кислорода, считающегося основным катод-
-JOOO
-900
\~воо
£-700
| -400
-300
~200[—1—1—1----1—।—।—।—।—1----1—।---1
О 0,5 1 1,5 2 2,5 3 3,5 4 4,5 5'5,5 6.0
Плотности motto t .на/снг
Рис. 31. Катодные поляризационные кривые для
железа, полученные в атмосфере, содержащей SO2
при 98% -ной относительной влажности в пленке
0,1-н. раствора Na2SO4 толщиной 160 jakav.
1 — -чистая атмосфера; 2 — 0,01% SO±; 3 — 0.1% SQal
4 — 1,0% SQj
ным деполяризатором. Если учесть, что концентрация кислорода
в воздухе составляет 21%, то при наличии в воздушной ^атмо-
сфере всего лишь 0,015% SO2 концентрация последнего в элек-
тролите становится равной концентрации, кислорода.
Влияние сернистого газа на скорость анодной реакции иони-
зации металла, по крайней мере, для видимых пленок электро-
литов значительно меньше, чем на катодную реакцию.
Учитывая рассмотренный выше механизм коррозии, при
ускоренных испытаниях .условия промышленной атмосферы сле-
дует воспроизводить не применением растворов серной кислоты,
а введением в атмосферу определенного количества сернистого
— 61 —
газа (0,01—0,1%). При этом происходит определенное подкисле-
ние коррозионной среды.
Хлор. Хлор в промышленных атмосферах встречается реже,
чем сернистый газ, и применять его при ускоренных испытаниях
следует лишь в тех случаях, когда нужно имитировать атмо-
сферу химических предприятий, занятых производством хлора,
Рис. 32. Катодные поляризационные кривые для алюминия,
полученные в атмосфере, содержащей хлор при 98%-ной отно-
сительной влажности в пленке 0,1-н. раствора NagSO^
толщиной 160 мкм:
f — чисгая атмосфера, 2 — 0,05% С1®; 3 — 0,1% С12; 4 — 1,0% Ci®
или атмосферу района, в котором расположено большое количе-
ство химических предприятий. Хлор —- сильный окислитель и так
же, как сернистый газ, может выступать в качестве мощного
катодного деполяризатора, усиливающего коррозионный про-
цесс (рис. 32). Действие хлора в процессе катодной деполяри-
зации выражается в восстановлении его и хлорноватистой
кислоты до ионов хлора.
Потенциал реакции восстановления молекул хлора до ионов
очень положителен:
72С12 + е -> СГ; <? = -р 1,3 в. (43)
Поэтому реакция (43) на большинстве технически важных
металлов может протекать даже при стационарном потенциале.
Кроме того, в воде возможен гидролиз хлора (диспропорциони-
рование) с образованием хлорноватистой кислоты, ионов хлора
и водорода по схеме
С1„ + Н8о Н+ + СГ + нею. (44)
Такнм образом, в электролите наряду с молекулами хлора
появляется значительное количество соляной и хлорноватистой
— 62 —
кислот; последняя является также сильным окислителем и спо-
собна восстанавливаться на катоде по двум схемам:
НСЮ 4- Н+ 4- е »/2С18 4- Н2О; <р « 4-1,63 г, (45)
НСЮ4-Н+4-2е СГ 4-Н2О; <j>==4- 1,49а. (46)
Хлористый натрий. При ускоренных испытаниях изделий,
предназначенных для работы в морской атмосфере, необходимо
применять растворы хлористого натрия. Дело в том, что
частички морских солей, уносимые с поверхности морей и океа-
нов, становятся впоследствии теми центрами, на которых идет
конденсация паров воды. Поэтому на металлическую поверх-
ность вместе с конденсирующейся влагой попадают хлористый
натрий н другие составляющие морской воды, вызывающие
сильную коррозию. Вблизи моря возможно и непосредственное
попадание частичек солей на поверхность металла. Ускорение
испытаний в этом случае происходит вследствие облегчения
анодной реакции ионизации металла (рис. 33).
Предполагают, что ион хлора обладает свойством легко про-
никать через защитные пленки, разрушая их [40]. Кроме того,
в его присутствии продукты коррозии содержат большое коли-
чество растворимых солей вместо гидроокисей, образующихся.
Рис. 33. Анодные поляризационные кривые для же-
леза и алюминия, полученные в объеме 0,5-н.
раствора NaCl и в дистиллированной воде:
1 - 0,5-н. раствор NaCl, алюминий; 2 0,5-н. раствор
NaCl, железо; 3 — дистиллированная вода, железо;
4 — дистиллированная вода, алюминий
например, в дистиллированной воде и тормозящих со временем
процесс. Существует, однако, и другая точка зрения, согласно
которой ускорение процесса коррозии в присутствии хлор-иона
происходит в результате адсорбционного вытеснения кислорода
с поверхности металла и нарушения благодаря этому пассивно-
го состояния.
Применение хлористых солей при ускоренных испытаниях
оправдывается в некоторых случаях и для тех сплавов, анодная
реакция на которых протекает беспрепятственно и, казалось бы,
— 63 —
не нуждается в ускорении. Дело в том, что образующиеся при
отсутствии активирующих ионов продукты коррозии способны
со временем (если испытания ведут в течение 30—60 суток)
сильно затормозить коррозионный процесс, поэтому для прове-
дения испытаний быстрыми темпами в течение всего времени
необходимо вводить в атмосферу частички хлористых солей.
При введении примесей в атмосферу применяющиеся для
ускоренных испытаний камеры усложняют, а растворы для рас-
пыления выбирают более агрессивные. Во влажных камерах
j в этом случае производят распыление не дистиллированной
[воды, а растворов хлористого натрия. Составы электролитов
* для распыления, рекомендуемые нами для воспроизведения раз-
личных атмосфер, приведены в табл. 10.
Таблица 10
СОСТАВЫ ЭЛЕКТРОЛИТОВ ДЛЯ РАСПЫЛЕНИЯ В КАМЕРАХ. РЕКОМЕНДУЕМЫЕ
ДЛЯ ВОСПРОИЗВЕДЕНИЯ РАЗЛИЧНЫХ АТМОСФЕРНЫХ УСЛОВИЙ
Состав электролита Воспроизводимая атмосфера
0,1—0,5-н. раствор NaCl Морская
0,01 -и. раствор NaCl 4- 0,01 ~ 2% SOg; 0,01-н. раствор NaySO^-'Н 0,01 — 2% SO2 Промышленная-
0,001-н. раствор NaCl; 0,001-н. раствор Na2SO4 ® SOs вводится в воздушное пространство камеры. Сельская
Современные камеры снабжены устройством для автомати-
ческой регулировки температуры и автоматического включения
пульверизаторов -через определенные промежутки времени.
Периоды включения пульверизаторов и время их действия раз-
личны. В одних лабораториях пульверизатор работает 16 ч
и выключается на остальные 8 ч, в других — включается
2—3 раза в суткн на непродолжительное время (15—20 мин),
а в третьих — включается на 30 сен каждые 15—20 мин. В не-
которых лабораториях пульверизатор работает непрерывно.
В табл. 11 приведены составы электролитов и режимы их рас-
пыления во влажных камерах, наиболее широко применяющие-
ся в различных странах.
Прн достаточной герметизации камеры частота распыления
не оказывает большого влияния на скорость коррозии. Важно,
чтобы все образцы в течение всего времени испытания находи-
лись в тумане. Разбрызгивание жидкости в камерах должно
производиться таким образом, чтобы струя распыляемой жидко-
сти не попадала непосредственно на образцы. Для этого пуль-
веризатор помещают в специальном закрытом отделении
камеры, имеющем прорези, или перед соплом пульверизатора
, _ 64 _
5 Зак. 1338
Таблица 11
СОСТАВЫ ЭЛЕКТРОЛИТОВ и РЕЖИМЫ РАСПЫЛЕНИЯ ИХ ВО ВЛАЖНЫХ КАМЕРАХ
Страна Состав электролита Температура °C pH Способ распыления Продолжительность распыления
СССР 3%-ный раствор NaCl Комнатная - Пульверизация Аэрозольный аппарат 0,5—1,5 мин через каж- дые 10 мин Через выбранные про- межутки времени
Гер мания D1N 50907 3%-ный раствор NaCl Комнатная - Пульверизация 5 мин в час
США ASTM В117-48Т, В117-57Т 5- или 20%-ный рас- твор NaCl 35 6,5-7,2 Пульверизация Непрерывно
США ASTM В 287'57 5%-ный растворNaCl 4- 4- сндсоон , 35 3,1—3,3 Пульверизация
Англия В5 1224, 1959 3,5 или 20 % -ный рас- твор NaCl 15-25 7-8 Пульверизация Непрерывно или через выбранные промежутки времени
Франция NF; A-91-10I; 1940 NaCl -j- -MgCla 4-CaCOg, суммарная концентрация 3% 15-25 8 Пульверизация Через выбранные про- межутки времени
Швейцария 0,05 %-ный раствор NaCl 4- СО2 - - Аэрозольный аппарат -
помещают стеклянную пластинку. Для более равномерного
распределения тумана в камерах устанавливают вентиляторы.
На рис. 34, а и б приведены внешний вид и схема одной из
таких камер. Объем камеры I м3, стенки двойные стеклянные.
Корпус 13 изготовляется из дуба. Внутренняя часть камеры по-
средством рамы 10 и поддона 11 соединена с воздухопрово-
дом 3. Корпус камеры установлен на металлический стол 12,
В патрубке поддона помещается осевой вентилятор /, с по-
мощью которого осуществляется беспрерывная циркуляция
воздуха в камере снизу вверх. Циркулирующий воздух обеспе-
чивает выравнивание температуры и влажности по всему объе-
му камеры. В воздухопроводе смонтирован калорифер 2 ц,пя
подогревания воздуха до нужной температуры. Обогрев кало-
рифера электрический. Система увлажнения воздуха состоит из
компрессора /4, двух распылителей форсунок 7 и бака 9 для
раствора. Сжатый воздух из компрессора поступает по резино-
вому шлангу к форсункам, засасывает из бака раствор и распы-
ляет его. Требуемые температура и влажность поддерживаются
регулятором 4. Вспомогательная аппаратура монтируется на
щите управления.
Электросхема камеры изображена на рис. 34,6. Температура
в камере регулируется автоматически с помощью дилатометри-
ческого регулятора с точностью ±0,5 1°. Постоянная влаж-
ность может поддерживаться автоматически с помощью волося-
ного гигрометра с точностью ±1%; может быть установлена
и циклически изменяющаяся влажность с длительностью цикла
10 мин с тремя периодами увлажнения (по выбору): 30 сек,
I мин, 1 мин 30 сек.
Камера предназначена для испытаний металлов и изделий
при повышенной влажности, которая создается распылением
пресной или соленой воды.
Результаты испытаний при применении простых пульвериза-
торов в значительной степени зависят от степени распыливания
электролита [41]. Поэтому в последние годы все чаще вместо-
пульверизаторов стали применять аэрозольные аппараты, обес-
печивающие более равномерное распыление электролита с вели-
чиной частиц от.0,1 до \б,мкм. При аэрозольном методе испы-
тания получаются более воспроизводимые результаты. Внешний
вид камеры с аэрозольным аппаратом и разрез аэрозольного-
аппарата показаны на рис. 35, а и б [42]. Камера выполнена
из плексигласа и по конструкции мало отличается от обычных
камер. Как видно на рис. 35, б, аэрозольный аппарат помещен
в защитный кожух [2], снабжен электромотором 1 и тарельча-
тым центробежным насосом 6. -Раствор из сосуда 7 засасывается
через патрубок 8, снабженный регулирующим клапаном 9. За-
тем, попадая на специальную решетку 12 и смешиваясь с воз-
духом, превращается в туман, который и подается в камеру.
Аппарат укреплен на специальной подставке 10, для защиты от
Рис. 34. Внешний вид и схема электрического управления влажной
камеры:
а — внешний вид камеры: 1 — вентилятор; 2 — калорифер; 3 — воздухопро-
вод; 4 — ре1улятор температуры; 5 — щнт управления; 6 — регулятор влаж-
ности; 7 — распылитель; 8 — трубка с воронкой; 9 — бак; 10 — рама;
И — поддон; 12 — стол; 13 — корпус камеры; 14 — компрессор; б — элек-
тросхема камеры: 1 — регулятор температуры; 2 — ртутные выключатели;
3 — электрическая лампа; 4 — нагреватель; 5 — предохранитель; 6—9— вы-
ключатели; 10 — электродвигатель вентилятора; 11 — электродвигатель ком-
прессора; 12 — магнитный пускатель; 13 — переключатель; 14 — телефонное
реле: 15 — выпрямитель; 16 — регулятор "влажности
5*=
67 —
попадания капель жидкости служит кожух /5, верхняя часть
аппарата имеет форму полусферы 14,
Аналогичные камеры с аэрозольными аппаратами приме-
няются в СССР; на рис. 36 представлена такая усовершенство-
дРазработанная в Институте физической химии
Ап СССР Ь. И. Палеолог. Внутри камеры находится вращаю-
щаяся подставка 6 для укрепления образцов, вверху — венти-
лятор 2; электролит распыляется аэрозольным аппаратом 10.
Рис. 35. Влажная камера и аэрозольный аппарат для распыления
электролита i[42 j:
а — внешний вид камеры; б — аэрозольный аппарат; 1 — электромотор; 2 — защитный
кожух; 3 — фланец; 4 — вертикальная ось; 5 — втулка; 6 — тарельчатый цеитрооеж-
ный насос; 7 — сосуд с распиливаемым раствором; 8 — всасывающий патрубок: 9 —
регулирующий клапан; 10 — нижняя часть аппарата;. П — воздушное пространство;
12 _ распиливающая решетка; 13 — кожух для 'защиты от попадания капель;
М —верхняя часть аппарата
Необходимая температура поддерживается с помощью нагрева-
телей 3 и контактного термометра /; влажность определяется
психрометром 5; на дне камеры расположена решетка 8 и испа-
рители 9.
В камере предусмотрена возможность проведения облучения
ультрафиолетовыми лучами через окно 7 и сжигание серы для
получения необходимой концентрации сернистого ангидрида
в пространстве камеры на специальном приспособлении 4. Для
обеспечения при необходимости конденсации влаги на образцах
в нижней части камеры располагают холодильники.
В камерах с неподвижными образцами результаты испыта-
ний в большей степени зависят от места расположения образ-
цов, поскольку концентрация солей, газов и водяных паров не-
— 68 —
одинакова по всему объему камеры. Поэтому в камере, изобра-
женной на рис. 36, образцы можно располагать и ча подставке,
вращающейся со скоростью 2,3 рад]сек (22 об{мин). Камера
изготовлена из плексигласа, аэрозольный аппарат обеспечивает
распыление раствора хлористого натрия частицами размером
0,1—10 мкм. Двуокись серы получают непосредственным сжнга-
иием серы в камере или вво-
дят через аэрозольный аппа-
рат. Рабочий объем камеры
0,85 м\
При отсутствии специаль-
ных камер ускоренные испыта-
ния можно проводить в упро-
щенных приборах, в которых
удается создать атмосферу за-
Рис. 36. Усовершенствованная влаж-
ная камера Института физической
химин АН СССР, разработанная
Е. Н. Палеолог:
1 — контактный термометр; 2 — лопасти
вентилятора; 3 — нагреватели; 4 — при-
способление для сжигания серы; 5 —
психрометр; 6 — вращающаяся подстав-
ка; 7 — окно для облучения; 8 — ре-
шетка; 9 — испарители; 10 — аэрозоль-
ный аппарат; 11 — гнезда для крепления
образцов
Рис. 37. Прибор для ис-
пытания во влажной
атмосфере, содержащей
сернистый ангидрид [44]
данного состава. Ускоренные испытания в упрощенных прибо-
рах применяли Престон и Санаул [43], 3. А. Иофа [44]. Прибор
3. А. Иофа изображен на рис. 37.
Указанный метод заключается в том, что испытания ведут
над растворами серной кислоты, к которым добавляется серни-
стокислый натрий. При этом в камере получается определенная
концентрация сернистой кислоты, над раствором которой
создается определенное парциальное давление сернистого анги-
дрида.
Концентрацию серной кислоты, наливаемой на дно сосуда,
выбирают в зависимости от влажности воздуха, которую хотят
создать (см. стр. 58).
— 69 —
Для создания в воздушной атмосфере заданной концентра-
ции сернистого ангидрида используют реакцию взаимодействия
серной кислоты с сернистокислым натрием:
H2SO. + Na2SO8 -> Na2SO4 HgSOs, (47)
HgSOg ? 4~ H2O. (48)
Поскольку парциальное давление сернистого газа над рас-
творами сернистой кислоты не- находится в строгой линейной
зависимости от любой концентрация кислоты, для создания
в реакционном сосуде соответствующей концентрации сернисто-
го газа рекомендуем пользоваться следующими данными:
Концентрация SO2 в воздухе, % (объ-
емн.) .....................
Давление SO2, 1,3-10“3 бар (мм рт.
•ст.) ........... ..............
Концентрация H2SO4, г/дм?.....
Концентрация SO2 в растворе, г/сЪи8 .
Отношение объема камеры к объему
раствора H2SO3 ... ............
0,001 0,010 0,100 0,500
0,0076 0,0760 0,7600 3,8000
0,0046 0,0460 0,4600 1,6800
0,003 0,030 0,320 1,120
5,0 5,0 5,0 4,2
Концентрация 8О2 в воздухе, % (объ-
емн.) ............................... 1,000 1,500
Давление SO*, 1,3* 10“z бар (мм рт.
ст.)................................. 7,6000 11,4000
Концентрация H2SO4, г/дм?........ 2,7500 3,7500
Концентрация SO2 в растворе, г/дм? . 1,830 2,500
Отношение обьема камеры к
объему раствора H2SO3................3,3 2,9
2,000
15,2000
4,7900
3,19.0
2,8
При проведении коррозионных испытаний на дно сосуда на-
ливают раствор серной кислоты, концентрацию которой выби-
рают по приведенным выше данным. Сернистокислого натрия
берут с небольшим избытком по сравнению с количеством, рас-
считанным по реакции (47). Сернистокислый натрий вводят
обычно в виде водного раствора после того, как поместят в ка-
меру образцы. В этом случае влажность в камере равна
« 100%.
При создании в камере или сосуде необходимой концентра-
ции сернистого газа количество серной кислоты, необходимое
для получения заданного парциального давления газа, часто
выбирают произвольно. Это приводит к ошибкам, обусловлен-
ным -значительным изменением концентрации сернистой кислоты,
поддерживающей данное парциальное давление сернистого
газа при больших объемах камеры. Разноречивые результаты
ускоренных испытаний, сообщаемые многими исследователями,
в" значительной степени обусловлены, по нашему мнению, тем,
что этот фактор обычно не учитывается.
Для получения воспроизводимых данных необходимо рас-
считывать количество раствора H2SO3, которое будет поддержи-
вать заданные парциальные давления SO2 и Н2О. Для этого
— 70 —
нужно задаться допустимой величиной отклонения концентра-
ции газа от исходной. Выше приведены отношения объемов
камеры и раствора H2SO3, обеспечивающие условия, при кото-
рых понижение концентрации SO2 не будет превышать 5%. Для
примера приведем расчет для сосуда объемом 10 дм3, в котором
создают концентрацию сернистого газа, равную 0,1% (объемн.).
Зная, что одна грамм-молекула газа при нормальных условиях
занимает объем 22,4 дм3, а молекулярная масса SO2 равна 64 г,
находим из отношения 22,4 : 64 = 0,01 :х количество SO2, со-
ответствующее концентрации 0,1% в замкнутом пространстве
указанного объема; оно равно 0,029 г. Концентрация SO2
в растворе, как видно из приведенных выше данных, составляет
0,32 г/дм3.
Если теперь задаться допустимым уменьшением концентра-
ции SO2 в растворе, равным, например, 5%, то количество SO2,
которое при этом поступит в воздушное пространство камеры
из 1 дм3 раствора, составит 0,32-0,05 — 0,016 а.
Следовательно, для поддержания в камере объемом 10 дм3
концентрации SO2, равной 0,01% (объемн.), необходимо взять
0,029: 0,016 « 2 дм3 раствора сернистой кислоты, содержащего
0,32 г/дм3 SO2. Отношение воздушного пространства камеры
к объему раствора сернистой кислоты при выбранных условиях
равно 5.
При проведении коррозионных испытаний в атмосфере сер-
нистого газа, особенно если скорость коррозии испытуемых
металлов велика, атмосферу необходимо обновлять. Обновление
можно производить один раз в сутки или один раз в несколько
суток в зависимости от количества образцов, объема камеры
и скорости коррозии, поскольку сернистый ангидрид восстана-
вливается в процессе коррозии. Количество расходуемого газа
мо>}<ет быть примерно рассчитано по скорости коррозии. Изме-
нение парциального давления SO2 в атмосфере не должно пре-
вышать 10%.
Сернистый ангидрид часто вводят в камеры при испытании
материалов для конструкций, эксплуатируемых в районах же-
лезных дорог. Сотрудники Британского ж.-д. департамента [45]
применяли комплексное испытание, в котором образцы сначала
испытывают в камере искусственного климата (рис. 38), где
они попеременно подвергаются обрызгиванию хлористым натри-
ем через форсунки 1 в течение 6 ч и облучению дуговыми лам-
пами 2 в течение 17 ч, а затем помещаются в аппарат объемом
0,06 м3. В аппарате образцы испытывают 6 ч, причем в течение
одного часа из сифона через трубку диаметром 6,3 мм, погру-
женную в воду на 38 мм, подается определенное количество
сернистого ангидрида. Испытания в аппарате проводят при
35—40° С в атмосфере, насыщенной водяными парами. Изменяя
соотношение времени нахождения образцов в обоих, агрегатах,
воспроизводят атмосферы различной агрессивности. Авторы
— 71 —
указывают, что поведение металлов при такого рода испытани-
ях соответствует поведению металлических конструкций вблизи
железных дорог.
Для изучения коррозии металлических и неметаллических
покрытий Кестер них [46] применил специальную камеру, воспро-
изводящую атмосферу индустриальных районов. Такого типа
камера применяется для проведения испытаний по DIN 50 018.
Схема камеры для испытания образцов, защищенных гальвани-
ческими и лакокрасочными покрытиями, приведена на рис. 39.
Влажность в камере поддерживается налитой на дно водой;
образцы укрепляются в держателях2 и вращаются при помощи
специального механизма^. Повы-
Рис 38. Камера искусственного
климата [45]:
образцов, защищенных гальвани-
ческими и лакокрасочными покры-
тиями [46]:
1 — форсунки для обрызгивания
электролитом; 2 —• дуговые лампы;
3 — образцы; 4 — кожух
/ — камера; 2 — держатели для об-
разцов; 3 — вращающий механизм;
4 — нагреватель; 5 — терморегулятор
•включении нагревателя 4 создают условия для конденсации вла-
ги на образцах. Заданную температуру поддерживают с по-
мощью терморегулятора 5. Температура воздушного простран-
ства контролируется термометром. Камера герметична и позво-
ляет вводить различные газы; автор применял ее для испыта-
ния различных покрытий, предназначенных для эксплуатации в
промышленной атмосфере.
Для изучения коррозий стали, покрытой различными смазка-
ми, Престон и Стронг [47] применили специальный аппарат,
в котором образцы подвергаются воздействию паров воды
и сернистого газа (см. гл. V).
Установка, позволяющая проводить ускоренные испытания
металлов при достаточно точном дозировании газов и водяных
паров, разработана нами совместно с Т. И. Лукониной [48]
— 72 —
и изображена на рис. 40. Она состоит из очистительной части А
и рабочей части Б. Воздух, просасываясь через серию поглоти-
телей 1—4, очищается от содержащихся в нем примесей: части-
чек масла, хлора, углекислого газа двуокиси серы. Затем воздух
просасывается через колонки увлажнителя 5, содержащие на-
сыщенный раствор соответствующей соли; перед входом в экси-
катор воздух пропускается через колонку с ватой 6 для задер-
жания частиц увлажняющего раствора, которые могут захваты-
ваться потоком воздуха. По достижении нужной влажности,
контролируемой гигрометром 8, водоструйным насосом создает-
ся небольшое разрежение в эксикаторе 7 и из газовой бюрет-
Рис. 40. Установка для исследования коррозии металлов при заданном
составе атмосферы ![48]:
1 — 4 — поглотители; 5 — увлажнитель; 6 — поглотительная колонка с ватой для
задержания частичек соли; 7 — эксикатор; 8 — гигрометр,- 9—13 — соединительные
краны; 14 — газовая бюретка
ки 14 вводится необходимое количество газа. Затем эксикатор
вновь подключается к системе и дополняется влажным возду-
хом, пока давление не станет равным одной атмосфере. Давле-
ние контролируется по вакуумметру. Последовательное подклю-
чение эксикатора в нужное положение производится открытием
соответствующих кранов 9—13.
Продолжительность испытаний определяется заданными
условиями (влажностью, содержанием газов). Относительную
влажность в реакционном сосуде следует поддерживать не ниже
критического значения: для железа и его сплавов 85—90%, для
алюминия и его сплавов ~80%. Наибольшего ускорения кор-
розии можно достигнуть при 100%-ной относительной влажно-
сти. Атмосферу необходимо обновлять через 2—3 суток.
Большое влияние на коррозию оказывают "находящиеся
в воздухе частички различных солей, а также пыль. В присут-
ствии таких частиц коррозия приобретает специфический харак-
тер, возникает так называемая нитевидная коррозия, при кото-
рой разрушение распространяется по каналам, размеры
— 73 —
которых изменяются в зависимости от агрессивности присут-
ствующих частиц. Надежность противокоррозионной защиты
с помощью лакокрасочных покрытий часто в значительной сте-
пени зависит от наличия таких частиц на поверхности, подлежа-
щей окраске. Поэтому существует особый вид испытаний, с по-
мощью которого контролируется чувствительность того или
иного защитного покрытия к наличию частиц, остающихся после
обработки на поверхности металлического образца (соль, наж-
дак, заводская пыль и т. п.) или попадающих из атмосферы
•в помещение. Этот вид испытаний заключается в том, что на
поверхность металла заранее наносится определенное количе-
ство мелких частиц солей, после чего образцы испытывают
в обычных камерах, рассмотренных выше. В тех случаях, когда
хотят проверить поведение лакокрасочных покрытий, образцы
после нанесения на их поверхность частичек покрывают по
•обычной схеме, после чего подвергают испытаниям. Этот вид
коррозии был изучен Верноиом [49] и рядом других исследова-
телей. Престон и Санаул [43, 50] исследовали образцы в атмо-
сферах с различной влажностью, на поверхность которых пред-
варительно наносились соли путем погружения в суспензию
мелкодисперсных солей в четыреххлористом углероде (1г соли
на 0,1 дм9 растворителя). После удаления растворителя на по-
верхности металла остаются равномерно распределенные части-
цы соли'
6. ИСПЫТАНИЯ, ВОСПРОИЗВОДЯЩИЕ УСЛОВИЯ
КОНДЕНСАЦИИ
Значительная часть коррозионных повреждений, наблюдае-
мых на конструкциях и приборах, эксплуатируемых в открытой
атмосфере, а также хранящихся на неотапливающихся складах
или^траиспортируемых по суше и морю, обусловлена конденса-
цией электролитов, возникающей вследствие периодического
•охлаждения атмосферного воздуха. Периодические колебания
температуры и влажности воздуха характерны для всех клима-
тических условий, но особенно большие перепады температуры
(до 20° С) ц относительной влажности (до 50% в сутки) наблю-
даются в некоторых тропических районах.
Коррозионные процессы вследствие конденсации возникают
и развиваются в специфических условиях, в большой степени за-
висящих от ее интенсивности и характера.
При охлаждении воздуха упругость водяных паров рп или
их абсолютное содержание не меняется до тех пор, пока ие
начнет конденсироваться вода. Упругость же пара, способного
насыщать пространство, т. е. насыщенного пара ps, как это
видно из приведенной табл. 12, с понижением температуры
уменьшается.
74—-
Таблица 12
ПЕРЕПАДЫ ТЕМПЕРАТУР. ВЫЗЫВАЮЩИЕ КАПЕЛЬНУЮ КОНДЕНСАЦИЮ
ПРИ РАЗЛИЧНОЙ ОТНОСИТЕЛЬНОЙ ВЛАЖНОСТИ ВОЗДУХА [151
100 4,5808 0 0
л 90 4,1227 —1 1
V 80 3,6646 —3 3
70 3,2065 —5 5
100 6,5394 5 0
90 5,8854 3 2
5 80 5,2315 2 3
70 4,5775 0 5
100 9,2040 10 0
90 8,2836 8 2
10 80 7,3632 6 4
70 6,4428 5 5
100 12,7840 15 0
90 11,5047 13 2
15 80 10,2264 11 4
70 8,9481 9 6
100 17,5297 20 0
90 15,7767 18 2
20 80 14,0237 16 4
70 12,2707 14 6
100 23,7530 • 25 0
25 90 21,3770 23 2
п- 80 19,0020 21 4
25 70 16,6270 19 6
100 31 8220 30 0
90 28,6330 28 2
80 26,4576 26 4
30 70 22,2754 24 6
100 42,1770 35 0
90 37,9593 33 2
35 80 33,7416 31 4
70 29,5239 29 6
100 55,3320 40 0
90 49,7988 38 2
40 80 44,2656 36 4
70 38,7324 34 6
100 71,8910 45 0
90 64,7020 43 2
45 80 57,5129 41 4
70 50,3238 38 7
100 92,5500 50 0
90 83,2900 48 2
50 80 74,0400 46 4
70 64,7800 43 7
Следовательно, относительная влажность воздуха
# = 2». 100. (49)
Ps
с понижением температуры повышается. Таким образом, пони-
жая температуру, можно довести относительную влажность
воздуха до 100%, не изменяя в нем содержание паров воды.
Значение температуры, при котором относительная влажность
воздуха с данным содержанием водяных паров становится
равной 100%, называется точкой росы г. При незначительном
охлаждении воздуха до температуры ниже точки росы пар на-
чинает немедленно конденсироваться, и появляются капельки
росы на конструкциях и приборах. Определенному содержанию
водяных паров в воздухе соответствует определенная темпера-
тура, при которой начинается капельная конденсация. Чем
меньше давление водяного пара, т. е. чем суше воздух, тем ниже
— 75 —
его точка росы и тем до более низкой температуры можно его
охладить без появления капельной конденсации. При высокой
относительной влажности воздуха незначительное понижение
температуры приводит его к состоянию насыщения. Разность
между температурой воздуха и точкой росы t—х для таких
атмосфер незначительна.
Из данных, приведенных в табл. 12, видно, что температур-
ный перепад, вызывающий капельную конденсацию, растет с по-
нижением влажности и падает с понижением температуры воз-
духа (при заданной влажности). Так, например, при Н = 80%
температурный перепад, вызывающий конденсацию в интерва-
ле от 5 до 50° С, равен 3—4°. Иными словами, при понижении
температуры всего на 3—4° достигается точка росы; воздух при
этом насыщается водяными парами, избыток которых должен
выделиться, т. е. должна начаться капельная конденсация. Для
воздуха, насыщенного влагой всего на 70%, температурный
перепад, вызывающий конденсацию, в интервале 5—50° С равен
5—7°. Даже для относительно сухого воздуха (Н — 50%)
перепад, вызывающий конденсацию, не слишком велик и со-
ставляет 9—10°.
Таким образом, если в замкнутом пространстве с помощью
растворов насыщенных солей или любым другим способом под-
держивать постоянную влажность, понижая температуру на
испытываемых изделиях или образцах, можно добиться конден-
сации паров воды. Для определения точки росы и температур-
ного перепада, необходимого для того, чтобы вызвать конден-
сацию в атмосфере с любой влажностью, можно пользоваться
табл. 12. На практике конденсацию часто вызывают периодиче-
ским помещением сосудов, в которых ведется испытание,
в камеры (холодильники) с более низкой температурой. В более
усовершенствованных камерах конденсация производится ох-
лаждением образцов непосредственно внутри камеры.
Режимы конденсационных испытаний, очевидно, выбирают
произвольно, совершенно не учитывая механизм процесса. Если
необходимость понижения температуры для создания конденса-
ции очевидна (но и в этом случае допускается неоправданно
длительное пребывание металла при низкой температуре), все
же не каждый исследователь представляет себе, каков должен
быть режим ускоренного испытания при высушивании.
Учитывая рассмотренные выше закономерности, температу-
ру, при которой происходит конденсация, и продолжительность
конденсации нужно выбирать с таким расчетом, чтобы пленка
влаги толщиной 100—150 мкм образовывалась за 10—15 мин.
Затем необходимо обеспечить постепенное испарение электроли-
тов с металлической поверхности или быстрое испарение их при
более частом возобновлении конденсации.
Конденсация должна возобновляться до полного высыхания
пленки, когда на металле еще сохраняется слой влаги толщиной
— 76 —
30—50 мкм. Больших скоростей коррозии можно достигнуть
при относительно больших скоростях высушивания и частом
повторении цикла.
Рассмотрим, каков должен быть режим ускоренных испыта-
ний при конденсации. До настоящего времени предполагали,
что тонкие слои электролитов начиная с 300-г-500 мкм не раз-
мешиваются и кислород в них переносится только путем диф-
фузии.
Основываясь на изложенных выше представлениях о меха-
низме переноса кислорода к поверхности металла, покрытого
тонкой пленкой электролита, следует заключить, что любые
явления, усиливающие размешивание электролита, должны при-
водить к уменьшению эффективной толщины диффузионного
слоя и увеличению скорости процесса восстановления кислоро-
да. Как было показано, значительное повышение скорости кор-
розии наблюдается при испарении электролита. При ускорен-
ных испытаниях необходимо иметь в виду, что для суммарного
коррозионного эффекта важна не только скорость коррозии, но
и длительность ее протекания. Последняя определяется време-
нем пребывания электролита на поверхности металла. Поэтому
испытания, значительно ускоряющие процесс коррозии, должны
строиться из циклов, обеспечивающих периодическое испарение
тонкого слоя электролита с поверхности металла с обязатель-
ным немедленным его возобновлением. Это относится не только
к испытаниям в условиях конденсации, ио и к другим любым ус-
коренным испытаниям с периодическим увлажнением поверх-
ности.
Электролиты с поверхности металлов высушивают либо
путем изменения влажности в замкнутом пространстве камеры,
либо путем повышения температуры, либо пропусканием над
образцами потока сухого воздуха.
При ускоренных испытаниях в условиях конденсации необ-
ходимо иметь возможность воспроизводить и контролировать
количество скоидеисированной влаги и характер конденсации
(капельная, пленочная), поскольку от этих факторов в значи-
тельной степени зависит скорость и интенсивность коррозии.
С этой точки зрения конденсацию лучше всего производить на
плоских, горизонтально расположенных поверхностях, хотя эти
условия не всегда идентичны условиям эксплуатации металла
на практике. При испытании вертикальных образцов электролит
стекает и накапливается в иижних частях их.
От характера конденсации сильно зависит скорость коррозии
и ее интенсивность. Коррозионные структуры при пленочной
и капельной конденсации должны быть различные. Характер
-конденсации зависит от природы металла и от подготовки по-
верхности, от наличия окисных пленок, загрязнений и т- п.
В реальных условиях эксплуатации на технических металлах
всегда будет возникать капельная конденсация, поэтому и при
— 77 —
ускоренных испытаниях на образцах необходимо получать
именно этот вид конденсации.
В зависимости от количества сконденсированной воды мож-
но получить любой вид коррозии — от язвенной до равномерной.
Наибольшая коррозия наблюдается при конденсации среднего
содержания воды, а не слишком большого или слишком малого.
По нашим данным [51], зависимость глубины коррозии от сред-
ней толщины слоя воды выражается кривой с максимумом,
показанной на рис. 41. Максимум соответствует толщине плен-
ки 150—200 мкм. Наличие максимума объясняется тем, что
в толстых слоях скорость коррозии
снижается из-за малой скорости ка-
тодной реакции восстановления кис-
лорода, а в малых слоях — вследст-
вие торможения анодной реакции
ионизации металла. Таким образом,
при ускоренных испытаниях нельзя
стремиться к получению возможно
большего количества сконденсиро-
ванной влаги на поверхности ме-
талла. Неравномерное распределе-
ние влаги на поверхности металла в
виде капелек различных размеров,
Средняя П70лщинй С ЛОЯ, НИН
Рис. 41. Зависимость глуби-
ны коррозионных поражений
от средней толщины слоя
сконденсированной влаги.
Продолжительность испыта-
ния 7 суток
получающихся при конденсации,
приводит к исключительной нерав-
номерности коррозии, которая кон-
центрируется в местах появления
этих капель.
Возможно, что развитию язвен-
ной коррозии способствует также
работа коррозионных элементов, возникающих вследствие не-
равномерного покрытия поверхности соответствующего металла
электролитом.
Количество сконденсированной воды, определяющее скорость
и интенсивность коррозии, в первую очередь зависит от темпе-
ратурного перепада; эта зависимость имеет одинаковый харак-
тер для сосудов или камер различной конструкции и объема.
Абсолютная скорость конденсации зависит от объема камеры
и конструктивных ее особенностей. Поэтому необходимо экспе-
риментально определить скорость конденсации на контрольных
образцах, в каждой камере, чтобы выбрать режимы, обеспечи-
вающие капельную конденсацию. Зависимость скорости конден-
сации воды от перепада температуры, полученная при испыта-
нии в приборах при температуре воздуха 25°С (рис. 42, а),
представлена на рис. 43. Как видно из кривой, с увеличением
температурного перепада скорость конденсации возрастает.
Зависимость, как и следовало ожидать, не носит линейного
характера.
— 78 —
При конденсационных методах испытаний скорость коррозии
зависит не только от скорости конденсации, но и от скорости
высушивания электролита. В этом можно убедиться по резуль-
Рис. 42. Схемы приборов для испытаний в условиях конденсации:
а — для плоских образцов: 1 — образец; 2 — отверстия для ввода газа; 3 — крыш-
ка; 4 — стеклянный сосуд; 5 — стеклянная подставка; 6 и 7 — термостаты;
б — для цилиндрических образцов: I— образцы; S—стеклянная труба; 3 — пробка;
4 — сосуд из плексигласа; 5 — дистиллированная вода
татам опытов, полученным авторами при изучении влияния
скорости высушивания после конденсации иа величину кор-
розии [27].
Опыты проводили в приборах, схема которых приведена на
рис. 42, а. Кондесацию и высушивание в этих приборах обеспе-
чивали поочередным пропу-
сканием через подставку 5
холодной или теплой воды.
Схема прибора для испыта-
ния цилиндрических образ-
цов е условиях конденсации
дана на рис. 40, б. Режимы
высушивания, применявшие-
ся при испытаниях, графиче-
ски изображены на рис. 44.
Подъем кривой соответству-
ет конденсации; по оси абс-
цисс он характеризует вре-
мя конденсации, а по оси
ординат увеличение услов-
ной толщины пленки. Спад
Рис. 43. Зависимость скорости кон-
денсации воды от перепада темпера-
туры; относительная влажность 100%,
температура воздуха 25° С
иа кривой характеризует процесс высушивания и уменьшение
толщины пленки во времени. Изучали четыре режима высушива-
ния. При коррозии в условиях режима 1 время конденсации бы-
ло в три раза больше времени высушивания (45 й 15 мин)-, плен-
ка влаги возобновлялась шесть раз в сутки в течение первых се-
ми часов испытания, остальное время образец находился под
пленкой влаги средней толщиной 150 мкм. Этот цикл повторял-
— 79 —
•ся семь раз. При коррозии по режиму 2 пленка влаги конденси-
ровалась 2,5 ч, после чего медленно высушивалась в течение
двух часов один раз в сутки. Режим 3 обеспечивает протекание
коррозии в невозобновляющейся пленке сконденсированной вла-
ги. Прн нем пленка влаги конденсировалась один раз в сутки в
течение двух часов. Для изуче-
Рис. 44. Режимы ускоренных испыта-
ний, применявшиеся для стали и чу-
гуна в условиях конденсации. Кон-
денсация пленки влаги средней
толщиной 150 мкм;
I — конденсация 45 мин, высушидалие
15 мин.', 2 — конденсация 25 ч, высуши-
вание 2 ч\ 8 ~ невозобновляемая плёнка;
4 — конденсация пленки влаги средней
толщиной 100 мкм в течение 45 мин,
высушивание 15 мин. Продолжительность
испытаний 7 суток
кия влияния толщины пленки
на скорость коррозии был вы-
бран режим конденсации с
меньшим перепадом темпера-
туры, обеспечивающий созда-
ние пленки с условной толщи-
ной 100 мкм (режим 4). Ско-
рость высушивания при этом
была такая же как и при ре-
жиме 1.
Результаты опытов (рис.
45) свидетельствуют о том, что
для малоуглеродистой стали и
цинка быстрое высушивание,
чередующееся с конденсацией,
Рис. 45. Скорость коррозии ста-
ли (а) и цинка (б) при режимах
конденсации и высушивания,
соответствующих рис. 44
значительно увеличивает скорость коррозии. Наибольшая ско-
рость коррозии наблюдалась при испытании по первому режи-
му, состоящему из часовых циклов (15 мин высушивание н
45 мин конденсация) при толщине пленки ' конденсирующейся
влаги 150 мкм.
Таким образом, при ускоренных конденсационных испыта-
ниях для значительного увеличения скорости коррозии следует
предусмотреть возможно частое периодическое высушивание с
последующим возобновлением конденсационной пленки.
— 80 —
Обычно в лабораторных условиях конденсационные испыта-
ния, как и испытания в атмосферах с постоянной влажностью,
проводят в закрытых сосудах или камерах с той лишь разницей,
что создается возможность конденсации влаги на поверхно-
сти образцов. Рассмотрим некоторые способы ускоренных испы-
таний в условиях конденсации. Такого рода испытания можно
проводить при постоянной и при периодически изменяющейся
температурах.
Рис. 46. Камера для ускоренных
испытаний автомобильных дета-
лей при циклически изменяющейся
влажности [52]:
а — схема камеры: 1 — увлажняющая
колонка;' 2 — гигрометр; 3 — коррози-
онная "камера; 4 — нагревательная
камера: 5 — уравнительный сосуд для
смачивающего раствора; б график
изменения влажности в течение одного
цикла испытания
Американская автомобильная компания разработала метод
ускорения испытания при периодически меняющейся влажности
при постоянной температуре [52] применительно к условиям
эксплуатации стальных дета л ей" автомобил ей, защищенных от
прямого попадания влаги. Влажность при этом испытании по-
степенно повышается от 10 до 100%. Схема камеры для подоб-
ных испытаний и график изменения влажности за один полный
цикл испытания приведены на рис. 46, а и б.
Повышение относительной влажности воздуха от 50 до 100%
производится пропусканием его через колонку с водой /, темпе-
ратура которой изменяется от 37 до 57° С. Низкие влажности
в камере создаются пропусканием воздуха через осушающее
отделение, в котором помещаются осушители — H2SO4 или
6 Зак. 1338 — 81 ---
СаС12 (на схеме не показано). Камера 3 изготавливается из
нержавеющей стали; в пространстве, где расположены образцы,
поддерживается температура 4-52° С. Результаты испытаний
показывают, что этот метод способствует значительному уско-
рению коррозии и дает удовлетворительную характеристику
поведения низколегированных сталей при образовании иа иих
продуктов коррозии, не обладающих защитными свойствами.
Конденсацию в этом испытании заменили смачиванием; это
увеличивает коррозию, по сравнению с испытаниями при цикли-
чески изменяющейся влажности, но*без смачивания, в 10 раз.
Рекомендуется применять смачивающий раствор следующего
состава: 1% NaClg + 1% СаС12 + 0,1% H2SO4 (по массе).
Испытания начинают с операции смачивания, которая
длится 25 мин, т. е. смачивающий раствор выдерживается это
время в камере. Затем, как’' показано на графике (рис. 46, б),
влажность воздуха попеременно уменьшается и увеличивается
в коррозионной камере. Последующие смачивания производят
в моменты достижения 100%-ной относительной влажности, но
не чаще чем через 25 ч. Изменение влажности регулируют так,
чтобы в течение каждых восьми часов три часа образцы вы-
сушивались. Смачивание образцов, кроме произведенного в на-
чале опыта, происходит трехкратным введением электролита
в камеру на 5 мин непосредственно из сосуда 5 через отверстие
в дне камеры. Образцы в камере располагают вертикально или
под углом в 15°. Сравнение результатов лабораторных испыта-
ний с результатами, полученными при испытании аналогичных
образцов в естественных условиях, показало, что этот метод
позволяет сравнивать коррозионную стойкость низколегирован-
ных сталей различного состава, на которых в процессе эксплуа-
тации образуются продукты коррозии, не оказывающие защит-
ного действия. Коррозионные потери за 20 суток испытания
' в этой камере соответствуют потерям при испытании в есте-
ственных условиях промышленной атмосферы в течение трех
I лет, а в морской — в течение 2,5 лет.
Этот метод испытания — наглядный пример успешного ис-
пользования в качестве ускоряющего коррозионного фактора
периодического высушивания и смачивания.
Разработке ускоренных методов лабораторных испытаний
с циклически меняющейся влажностью и температурой посвя-
щено много исследований. Некоторые циклы во многих странах
стандартизованы. На рис. 47 дано графическое изображение
изменения температуры и влажности при некоторых стандарт-
ных и исследовательских методах ускоренных испытаний, при-
меняющихся за рубежом, {53].
Испытания проводят при циклически изменяющихся усло-
виях, продолжительность одного цикла составляет 24 ч. При
испытании 1 продолжительность одного цикла может быть так-
же равной 12 и 6 ч. В исследовательском цикле 6 с нагревом
.— 82 —
до 65° С и охлаждением до —60° С продолжительность цикла
и влажность при минусовых температурах не оговариваются.
При кратковременных испытаниях 9 влажность поддерживается
равной 90%, а температура изменяется от 37 до 53° С шесть раз
07»
20 ‘‘--------•---1—.
О б 12 18 26 ч
о б 12 1в гьч
°C
во I
60
60
20
О 6 12 18 26 ч
О б 12 1в 26 ч
Рис. 47. Графическое изо-
бражение изменения тем-
пературы t и влажно-
сти (Н) прн стандартных
н исследовательских
ускоренных испыта-
ниях |[53]:
I — испытание по DINE
SO 016; 2 -— испытание ио
DIN Е 50 017 с конденса-
цией; 3 — длительные ис-
пытания по американским
нормам A'DB 09 105; 4 —
испытания по варианту А
французской нормали A/DB
09 105; 5 — испытания по-
варианту В . французской
нормали A/DB 09 105; 6 —
исследовательский цикл по>
американским нормам
MIL-STD 19999; 7 — ис-
пытание при конденсации
и введении SO2 и СО2 по
DIN 50 018; 8 —- испытание
по американским нормам
SC-D-16-268, 9 — кратко-
временные испытания во
американским нормам.
CCTU-01-01; 10 — испыта-
ние по американским нор-
мам SC-D-15-914: 11 — ком-
бинированное испытание,
по американским нормам
CCTU-01-01; сплошной ли
нией обозначено изменение-
температуры, пунктиром —.
изменение влажности
в час. При испытании 10 один цикл длится 48 ч, сначала про-
изводится нагревание до 65° С, затем охлаждение до 30° С,.
через 7ч — кратковременное охлаждение до 20° С с последую-
щим продолжением испытания при 30° С. Относительная влаж-
ность при этом испытании поддерживается в пределах 90—98%..
6* — 83 —
При испытании 7, помимо конденсации, предусмотрено вве-
дение по 0,8% (объемн.) СО2 и SO2.
Для изделий, предназначенных для работы в тропическом
климате, Международная электротехническая комиссия (МЭК)
рекомендовала специальные методы, по которым должны про-
водиться испытания на влагоустойчивость, сухой нагрев, пыле-
защищенность, грибоустойчивость, устойчивость к воздействию
солевого тумана и солнечной радиации и холодноустойчи-
вость {54].
В Англии, ФРГ, Франции и Чехословакии разработаны мето-
ды, принятые как стандартные при испытании изделий, пред-
назначенных для работы в тропическом климате. Британский
стандарт [55] предусматривает испытание узлов до сборки
изделия:
а) при влажности 100% и температуре 70° С в течение 16 ч,
затем охлаждение до комнатной температуры в закрытой каме-
ре в течение 6 ч. При этом выключается подача тепла и влаги.
Число циклов — три;
б) испытание на образование грибковой плесеии в течение
28 суток при 30°С и 100%-ной относительной влажности после
разбрызгивания на поверхности испытуемого материала водной
суспензии грибковых спор. В собранном виде машины испыты-
вают 6 ч при 40° С и влажности 95—100% и 12—18 ч при
охлаждении во влажной атмосфере до комнатной температуры.
Нормами DIN 5001056 предусмотрено испытание электротех-
нических изделий в камерах при 40- и 90 %-ной относительной
влажности.
Разработанные во Франции методы испытаний предусмат-
ривают возможность изменения температур от 16 до 60° С при
влажности 20—50% й от 10 до 40° С при влажности от 40
до 90%.
Методика испытания, принятая в Чехословакии, в основном
соответствует проекту публикации МЭК № 68.
Климатические условия в тропических странах различны
[57], поэтому так же, как для умеренного климата, не может су-
ществовать единого метода испытаний материалов, предназна-
ченных для эксплуатации в тропических условиях. Режим ис-
пытания следует подбирать так, чтобы материалы испытыва-
лись при повышенных температурах в сухом воздухе и при
более низких температурах при высокой относительной влаж-
ности и конденсации. Дождливые периоды года имитируются
периодическим смачиванием образцов с помощью дождеваль-
ных аппаратов. Обязательно также испытание на склонность к
образованию грибковой плесени.
Для испытания материалов и изделий на влагоустойчи-
вость и проверки работоспособности изделий с заданными па-
раметрами в условиях тропиков в СССР разработаны камеры
различной конструкции [58].
— 84 —
На рис. 48 изображена схема тепловлагокамеры Всесоюз-
(ВЭИ). Температура и
ного электротехнического института
влажность в этой камере поддер-
живаются автоматически при по-
мощи контактных термометров 2
и 3. Испытательная камера 7
окружена водяной рубашкой 8.
Внутри камеры расположены ис-
паритель 11с открытой водяной
поверхностью, вентилятор 6, ра-
ботающий от мотора 5, и конт-
рольный психрометр 4. Вода в
испарителе подогревается трубча-
тым электроподогревателем 13,
равномерный нагрев обеспечива-
ется размешиванием воды мешал-
кой 10, Вода нужной температу-
ры из бака для подогревания во-
ды 14 с помощью насоса 1 пода-
ется в рубашку камеры. Регули-
рование температуры воды, по-
ступающей в рубашку, произво-
дится включением электроподо-
гревателя 16 или змеевиков охла-
ждения 15. В камере можно под-
держивать высокую относитель-
ную влажность при температуре
до 50° С. Недостаток камер такой
конструкции — относительно
большая неравномерность темпе-
ратуры, доходящая до ±4°.
На рис. 49 приведена схема
тепловлагокамеры другой кон-
струкции, отличающейся от рас-
смотренной выше тем, что в ней
нагреватель 6 н испаритель 7 от-
делены от рабочего пространства
камеры 4. Воздух перемешивает-
ся при помощи осевого 'вентиля-
тора 3. Если нужно получить тем-
пературу, на 10—20° ниже окру-
Рис. 48. Схема тепловлагокамеры
Всесоюзного электротехнического
института {58]:
1 — насос; 2, 3 — контактные термо-
метры с магнитной регулировкой;
4 — контрольный психрометр; 5 —
электродвигатель вентилятора; 6 —
осевой вентилятор; 7 — испытательная
камера; 3 — водяная рубашка; S —
змеевик с проточной водой для осуш-
ки воздуха в камере; 10 — мешалка
для перемешивания воды в испари-
теле; 1,1 — испаритель с открытой
водяной поверхностью; 12 — змеевик
с водопроводной водой для охлажде-
ния воды в испарителе; 13 — трубча-
тый электроподогреватель воды в ис-
парителе; 14 — бак для подогрева
воды; 15 — змеевики для охлаждения
воды; 16 — трубчатый электроподо-
греватель мощностью 4 кет
жающеи, через змеевик 5 пропус-
кается охлаждающая вода или
газ. Увлажнение воздуха проис-
ходит в испарителе 7, расположенном вне камеры; вода в испа-
рителе подогревается нагревателем 8. Вентилятор увлажните-
ля 9 производит принудительную циркуляцию воздуха через ис-
паритель, затем подогретый в камере воздух увлажняется в ис-
— 85 —
парителе. Необходимая температура и относительная влажность
воздуха регулируются при помощи контактных термометров 1 и
2, включенных в блок сигнализации и автоматики.
В радиотехнической промышленности применяется камера
дождевальным устройством (рис. 50). Она представляет со-
Рис. 49. Схема тепловлагокамеры,
в которой нагреватель и испаритель
отделены от рабочего простран-
ства [58]:
/ и 2 — контактные термометры; 3 —
вентилятор; 4 — рабочее пространство
камеры; 5 — змеевик для охлаждения;
€ — электронагреватель мощностью
2.2 кет; 7 — увлажнитель; 8 — нагрева-
тель для подогревания воды в увлажни-
теле; 9 — вентилятор увлажнителя;
19 — изоляторы
Рис. 50. Схема камеры с дождеваль-
ным устройством, применяющейся
в радиотехнической промышлен-
ности [58]:
I — испытательная камера; 2 —- «влаж-
ные» контактные термометры; 3 — кон-
тактный термометр; 4 — дождевальное
устройство; 5 — трубчатый электроподо-
греватель; С — общий водяной бак; 7 —
электромагнитные блок-клапаны; 8, 12.
13 — воздуховоды; 9 — контактный' тер-
мометр; Ю — электронагреватель; 11 —
увлажнительный бачок; 14 — вентилятор
высокого давления
бой металлический шкаф, внутри которого расположены рабО?
чая камера 1, блок автоматики и сигнализации, увлажнитель-
ный бачок 11 с вентилятором высокого давления 14 и воздухо-
водами 8, 12, 13. На задней стенке камеры расположен
трубчатый электроподогреватель 5. Дождевальное устройство 4
при необходимости позволяет производить орошение образцов
Полезный объем камеры ОД м8, температура может устанав-
ливаться в пределах от 20 до 80° С с точностью ±1,5° С. Отно-
сительная влажность поддерживается постоянной, равной
100%.
— 86™
Существуют и другие конструкции камер. Среди них следует
отметить, например, камеру завода АТЭ-2, описанную
А. Г. Маркиным [59]. Эта камера, названная КТН-3, позволяет
изменять температуру и создавать различную влажность, а
также вводить грибковую плесень и воспроизводить атмосфе-
ру высокогорных районов, т. е. разреженный воздух.
При ускоренных испытаниях с конденсацией в некоторых
случаях предусматривают в камерах возможность охлаждения
Рис. 51. Схема камеры для ускоренных коррозионных испытаний
при периодической конденсации влаги [60]:
а — вид спереди; б — план; 1 — электромотор; 2 — дверь; 3 — пульверизатор;
4 — экран; 5 — охлаждающая стенка; 6 — вентилятор; 7 — камера высушивания;
8 — тамбур; 9 — направляющие; 10 — вращающееся колесо с образцами; 11 — холо-
дильник; 12 — спифъиь холодильника; 13 — охлаждающая смесь; 14 — насос для цир-
куляции охлаждающей смеси; 15 — рефрежератор; 16 — образцы; 17 — отверстие
для подачи ®оды
не окружающей атмосферы, а самих образцов путем временно-
го перемещения их в пространство с низкой температурой. Схе-
ма установки для комбинированных испытаний, при которых
предусматривается ускорение процесса коррозии за счет перио-
дической конденсации и высушивания, представлена на
рис. 51, с и б [60]. Установка представляет собой сложную ка-
меру, состоящую из нескольких отделений. Образцы располо-
жены на специальном колесе 10, вращающемся от электромото-
ра 1 со скоростью 3 об/ч. За одни оборот образцы 15 мин
увлажняются, 2,5 мин высушиваются и 2,5 мин охлаждаются.
Высушиваются образцы в камере 7, где установлен вентиля-
тор 6, затем они попадают в пространство с холодными стен-
ками 5. Температура холодных стенок поддерживается рефре-
жераторной установкой, отдельные части которой 11—15 по-
-87 —
казаны на рис. 51, б. Конденсация влаги на образцах происхо-
дит в пространстве, где пульверизатором 3 распыляется рас-
твор соли; перед пульверизатором устанавливается экран 4.
Охлаждение образцов производится до температуры +13° С.
Температура, при которой производится увлажнение, и темпе-
ратура сухого воздуха равны 43° С. Осматриваются образцы
через дверь 2.
Конденсацию применяют не только для проведения уско-
ренных испытаний различных металлов, но и для определения
защитной способности смазок, лакокрасочных покрытий и дру-
гих видов защиты металлов от коррозии. Эти методы испыта-
ний рассматриваются ниже.
ЛИТЕРАТУРА
1. Акимов Г. В. Теория и методы исследования коррозии металлов Изд-во
АН СССР, 1945.
2. Розенфельд И. JI., Акимов Г. В. Исследования -в области электро-
химического и коррозионного поведения металлов н сплавов. Оборонгиз,
1950, 179, 201.
3. Левич В. Г. Физико-химическая гидродинамика. Изд-во АН СССР, 1952;
Физико-химическая гидродинамика. Фнзматгиз, изд. 2, 1959.
4. Кабанов Б. Н. ЖФХ, 1948, т. 22, вып. 1, 53; 1949, т. 23, вып. 4, 428.
5. Сор son Н. R. Ind and Eng. Chem., 1942, v. 44, № 8, 1745.
6. Сб. «Коррозия металлов». Пер. с англ, под ред. В. В. Скорчеллетт-и. Гос-
химиздат. 1952.
7. Латимер В. М. Окислительные состояния элементов и их потенциалы
в водных растворах. ИЛ, 1954.
8. Глухова А. И. Диссертация на соискание ученой степени канд. хим.
наук. Ин-т физической химии АН СССР, 1959.
9. Сох G. L. Ind und Eng. Chem., 1931, v. 23, 902.
10. Спеллер Ф. H. Коррозия железа. ОНТИ, 1936.
II. Романов В. В., Акимов Г. В. Тр. Ин-та физической химии АН
СССР. М.-Л. Изд-во АН СССР, 1955, 50.
12. Акулов Н. С., Хнмченко Н. В. Сб. «Материалы в химической про-
мышленности». Госхимнздат, 1953, 117.
13. Герасимов В. В., Розенфельд И. Л. Изв. АН СССР, ОХН, 7,
279, 1956.
14. Розенфельд И. Л. Атмосферная коррозия металлов. Изд-во АН
СССР, 1960.
15. Розенфельд И. Л., Максимчук В. П. ДАН СССР, 1958, т. 119,
№ 5, 986.
16. В у л ь ф с о и В. И. Проблемы морской коррозии. Изд-во АН СССР,
1951, 44.
17. А л екни О. А. Основы гидрохимии. Гидр ометеоиз дат, 1953.
18. Черноусов Н. П. Химическое машиностроение, 1959, № 1, 14.
19. Путилова И. Н., Б а л е з и и С. А., Баранник В. П. Ингибиторы
коррозии. Госхимнздат, 1958.
20. Кур теп о в М. М. Заводская лаборатория, 1955, т. XXI, № 11, 389.
21. Новаковский В. М. Зашита металлов, 1965, № 2.
22. Розенфельд И. Л., Вашков О. И. Защита металлов, 1965,
№ 1, 70.
23. Негр ее в В. Ф. Сб. «Проблемы морской коррозии». Изд-во АН СССР,
1956, 90.
24. Розенфельд И. Л., Жигалова К. А. ДАН СССР, 1954, т. 99, № I,
137; 1955, т. 104, № 6, 876.
—-88--
25. Розенфельд И. Л., Пав'луцкая Т. И. ДАН СССР, 1953, т. 91,
№ 2, 315.
26. A g а г Т., Bowden F. Proc. Roy. Soc., 1944, v. 44, 262.
27. Glastone S., H i c k 1 i n g A. Electrolitic Oxidation and Reduction,
London, 1935.
28. Розенфельд И. Л., Жигалова К. А. Сб. «Коррозия металлов и
сплавов». Металлургнздат, 1963, 347.
29. Champion Т. A. Corrosion Testing Procedures, London, 1952.
30. Я к у б о в и ч С. В. Испытание лакокрасочных материалов и покрытий.
М.-Л. Госхимиздат, 1952.
31. Kutzelmig A. Werkstoffe und Korrosion, 1956, 7, 42, 65.
32 Касаткин А. Г. Основные процессы и аппараты в химической техно-
логии. Госхимиздат, 1948, 395.
33. Хргнан А. X. Физика атмосферы. Гостехтеоретиздат, 1953, 252.
34. Vernon W. >Н. J., Whitby L. Trans. Faraday Soc., 1931, v. 27, 248.
35. Todt F. Korrosion und Korrosionschutz, Berlin, 1955.
36. Справочник химика, т. 1. Госхимиздат, 1951, 48.
37. Техническая энциклопедия. Справочник физико-химических величин, т. 1,
1956, 456.
38. Carr D. S., Harfies В. L. Ind. and Eng. Chem., 1949, 41, Ns 9, 2014.
39. Розенфельд И. Л., Луконина Т. И. ДАН СССР, 1956, т. Ill,
№ 1, 136.
40. Н u d s о n 1. С. J. Appl Chem., 1953, v. 3, 86.
41. Kutzelning A. Der Prufung metallischer Dberzeuge, Berlin, 1960.
42. Hess W. Werkstoffe und Korrosion, 1955, H. 7.
43. Preston J. R. St., Sanual J. J. Appl. Chem., 1956, v 6, № 1, 26.
44. Ио фа 3 А. ЖФХ, 1957, t. 31, № 10, 2236.
45. Hair W. I. Corrosion Technology, 1931, v. 3, Ns 1, 8-
46. Kesternish W. Stahl und Eisen, 1951, № II, 28/1, 587/8.
47. Preston A. S. I., Strong E. G. J. Inst, of Petroleum. 1950, v. 36»
№ 319, 457.
48. Л у кони на, T. И. Жигалова К. А., Розенфельд И. Л. Завод-
ская лаборатория, 1956, т. XXII, № 12, 1463.
49. Vernon W. Н. J. Trans. Faraday. Soc., 1933, v. LX1V, 31, 1933.
50. Preston R. St. J., Sanual J. Chem. Ind., 1953, Ns 38, 995.
51. Розенфельд И. Л., Жигалова К. А. Заводская лаборатория,
1959, т. XXV, № 2, 172. ~ ;
52. О р i n s к у А. I., То ms оп В. Е., В о 1 g е h о 1 d A. L. Amer. Soc. Test.
Mater, Bulletin, 1953. № 187, 47.
53. Ledeburg H. Blech, 'I960, v. 7, № 4, 184.
54. Проект .публикации МЭК, Ns 68, 1958.
55. В. S., London, II, 33—1953; 1, 1961.
56. Проект стандарта DIN 50 010, ДК6 27. 19, Берлин 1952.
57. Информационный материал по данным министерств. ЦБТНИ, 1956.
58. Маслов В. В. Передовой науч.-техн. и производственный опыт, тема
52. Ns М—59—283/3, 1959.
59. М а р к и и А. Г. ЦБТНИ. Министерство автомобильной промышленности»
СССР 1957.
60. Pray Н., Gregg J. Proc. Amer. Soc. Test. Mater., 1941, v. 41, 758.
Ill
ВЫБОР ПОКАЗАТЕЛЯ КОРРОЗИИ
И МЕТОДА ОЦЕНКИ КОРРОЗИОННОЙ
СТОЙКОСТИ
Как уже указывалось, при ускоренных испытаниях, а также
в лабораторных исследованиях важно правильно выбрать
не только метод испытания, но и показатель 'коррозии.
Наиболее широкое распространение при оценке коррозион-
ной стойкости материалов -получили следующие показатели
коррозии:
• 1. Изменение массы образцов (весовой метод).
, 2. Изменение количества выделившегося в процессе корро-
зии водорода или количества поглощенного кислорода (объем-
ный метод).
• 3. Определение глубины проникновения коррозии.
* 4. Изменение в результате коррозии механических свойств
образцов
₽ 5. Изменение в результате коррозии отражательной способ-
ности поверхности металла.
„ 6. Изменение ® 'результате коррозии электрического сопро-
тивления образцов.
, 7. Определение времени до появления первого очага кор-
розии.
8. Определение очагов коррозии, «появляющихся за данный
промежуток времени.
• 9. Определение времени, необходимого для появления кор-
розии на определенной части поверхности (10 или 50%).
10. Определение количества металла, перешедшего в- рас-
твор в процессе коррозии.
1. ИЗМЕНЕНИЕ МАССЫ ОБРАЗЦОВ
(ВЕСОВОЙ МЕТОД)
Метод определения коррозии по потерям массы является
наиболее -простым и надежным, поскольку он непосредственно
указывает количество металла, разрушенного коррозией. Этот
— 90 —
метод используют в тех случаях, когда коррозия носит более или t
менее равномерный характер, и обычно применяют при изуче- ’
нии коррозии малоуглеродистых сталей, цинка и меди.
. Если т0—-первоначальная масса образца, т\— масса об-
разца после коррозии, F — поверхность образца и t — время, то
скорость коррозии Л -можно определить из соотношения
[Л « . (50)
Скорость коррозии обычно выражают в г/(я? • год) или
мг/(см2'ч). При определении этого показателя важно тщательно
удалить продукты коррозии. Иногда они удаляются простым
механическим воздействием (щетками или деревянными шпате-
лями). Если -продукты коррозии механически удаляются плохо,
то используют специальные электролиты, растворяющие про-
дукты коррозии, но не реагирующие с металлом. Состав элек-
тролитов, применяемых для снятия продуктов коррозии, приве-
ден в табл. 13. При использовании того или иного реактива
необходимо убедиться в том, что он не растворяет основной ме-
талл. Для этого ставят контрольные опыты. Если потери массы
некорродировавшего образца не превышают 3—5% общих по-
терь от коррозии, реактив можно применять.
Определение потери массы металла дает средние показатели
коррозии, -поэтому необходимо учитывать и «площадь, занятую
коррозией. При точных экспериментах -эту площадь можно опре-
делить с помощью рисовального аппарата, -приспособленного
к обыкновенному микроскопу (4]. При массовых опытах можно
пользоваться упрощенным -методом, заключающимся в том, что
после снятия продуктов коррозии, высушивания и взвешивания
образца на него накладывают кусок кальки и делают зарисовку
всех мест, -подвергшихся коррозии. Зарисованные участий
кальки вырезают и взвешивают на аналитических весах. Зная
вес единицы площади кальки, можно легко рассчитать -площадь,
занятую коррозией. Иногда прибегают и к планиметрированию,
однако этот метод сложнее и к тому же не дает более точных
результатов.
Степень неравномерности коррозии характеризуется «индек-
сом неравномерности», ‘представляющим собой отношение всей
площади образца к площади, занятой коррозией. Индекс «10»
означает, что лишь 10% поверхности занято коррозией.
В некоторых случаях коррозию можно определять и по уве-
личению массы образцов. Однако этим методом можно пользо-
ваться лишь в тех случаях, -когда продукты -коррозии хорошо
сцеплены с поверхностью -металла и есть уверенность >в том, что
они в процессе испытания не будут осыпаться. Метод дает
косвенные результаты, ибо для определения истинной коррозии
необходимо знать состав продуктов коррозии, который, к сожа-
лению, не всегда известен.
-—91 —
Таблица I3‘
ЭЛЕКТРОЛИТЫ, РЕКОМЕНДУЕМЫЕ ДЛЯ СНЯТИЯ ПРОДУКТОВ КОРРОЗИЙ
Сплав
Электролит
Режим
Примечание
Электрохимическая очистка, катодная обработка
Железо, 10%-ный раствор ли- Плотность тока
сталь, чугун мониокислого аммония 1,1 а[дм?
10%-ный раствор циа- Плотность тока нистого натрия , 11ЛОХНО^ТЬ T0Kd 1 1,6 а/Ом?-, температу- ра 20° С, время 20 мин 8—10%-ный раствор едкого натра Плотность тока 1,6 r а/ом?, температура 20 °C, время 20 мин 5%-иый раствор серной Плотность тока кислоты (по массе), со- 20 а] дм?, температу- держащий 0,2 мл/дм? ро- ра 75° С, время дана или 0,5 г/дм* за- 3—5 мин медлителя (диортотолила, тиомочевины, хинолина, этиодида, бетта-нафтола) Насыщенный раствор Плотность тока 1,6 лимоииой кислоты а]дм\ температура 75 °C Химическая очистка 10%-ный раствор вии- Температура 25-—- иокислого аммония -4-6% 70° С nh4oh 10%-ный раствор ли- Температура 25— мониокислого аммония-4- 70° С 4-4% NH4OH 10%-ный раствор сер- Температура 25° С. ной кислоты 4- 0,1% Кипячение в колбе с мышьяковистого аигид- обратным холодильни- рида ком (для прочно свя- занных продуктов кор- розии) 10%-ный раствор сер- Температура 20° С ной кислоты 4-0,5% тио- мочевины или 1% фор- малина 5%-иый раствор едкого Температура 80—90° иатра-|-цйвк граиулиро- С, время 30—40 мин ванный иди стружка 10%-ный раствор пер- Температура 25— сульфата аммония 70° С
— 92 —
Продолжение табл. 13
Сплав Электролит Режим Примечание
Железо, 650—700 г/дм9 едкого Температура 135— Разрыхляет
сталь» чугун натра 4~ 50 г/дм? азотно- 140° С окалину
кислого натрия 4-20 а/дл*8
азотистокислого натрия
200 г/йл'Л хромового ангидрида 4- 50 г/дм? ор- тофосфорной кислоты Температура 90° С 80— Для деталей с жестки- ми допуска- ми
Химическая очистка дет алей, не ограниченных
Медь и мед- ные сплавы Никель и никелевые сплавы Алюминий и алюмини- евые сплавь точными допусками 150—200 а/д-м8 серной Температура 60- кислоты (плотностью 70° С 1,84) 4- 3—5 a/dzi8 при- садки ЧМ 10%-ный раствор сер- Температура 18— иой кислоты [99% (объ- 25° С еми.)| 4- 1% формалина (объемн.) 55% фосфорной кисло- Температура 18— ты (плотностью 1,4— 25° С 1,6) 4- 25% этилового спи- рта 4-20% воды 5%-ный раствор серной Температура 10— кислоты 20° С, промывка в воде 5%-ный раствор серной Температура 20° С кислоты 4- 0,2% инги- битора 18%-ный раствор сер- Погружение на 2—- иой кислоты 3 мин. Очистка под водой щеткой 100 е/дж8 бисульфата Температура 20° С натрия 10—18%-ный раствор Погружение на 2 — серной кислоты 3 мин. Очистка под водой щеткой 5%-ный раствор азот- Температура 10— ной кислоты 15° С 5%-иый раствор азот- Температура 15— ной кислоты (плотностью 20<х С. Время-—до двух 1,4) 4- 1% хромпика часов концентрированная Температура ТО— азотная кислота 20° С, время 2—3 мин. Очистка под водой щеткой
— 93 —
Продолжение табл. 13
Сплав Электролит Режим Примечание
Алюминий и алюмини- евые сплавы 20%-иый раствор фос- форной кислоты -}-8% хромового ангидрида Температура 20°С, время 15—30 мин
65 г/дл8 хромового ан- гидрида 4- 280 а/дж8 фос- форной кислоты (плотно- стью 1,4) Температура 20— 25° С
50—60 а/дл? едкого натра Т емператур а 60— 70° С, время 2 мин
30 а хромового ангид- рида 4- 25 см9 85 %-ной фосфорной кислоты иа 1 дм9 воды Температура 20°С
2% хромового ангид- рида 4- 5% фосфорной кислоты Температура’80— 85° С, время 10 мин Применяется для предва- рительной обработки прочных продуктов коррозии пе- ред удалени- ем в 65% -иой НПО3
Олово и оловянные 5% -ный раствор соля- ной кислоты Температура 20°С
сплавы 5 %-ный раствор соля- ной кислоты 4-0,2% ин- гибитора Температура 20°С
15%-ный раствор нор- мальной фосфориоиатр ле- вой соли Температура кипе- ния, время 10 мин. Очистка щеткой
Свинец и свинцовые Насыщенный раствор уксуснокислого аммония Температура 20°С
сплавы 1%-иый раствор уксус- ной кислоты Очистка щеткой Для удале- ния РЬО
25%-ный раствор ук- суснокислого аммония При подогреве очистка щеткой Для удале- ния РЬО или PbSO4
80 а/дж3 едкого натра, 50 г/дм9 маниита 4- 4-0,62 а/дж8 серибкислого гидразина Предварительная очистка щеткой, затем кипячение 30 мин Для удале- ния плотной РЬО2
5 %-ный раствор серной кислота 4-0,2% ингиби- тора Температура 20°С
— 94 —
Продолжение табл, 13
Сплав Электролит Режим Примечание
Цинк И цинковые сплавы 10%-ный раствор хло- Температура 70° С, ристого аммония время 2,5—5 мин 5% хромового ангидри- Кипячение 15— Азотнокис- да + 1% азотнокислого 20 сек, лое серебро серебра растворяется отдельно и добавляется в кипящий раствор Насыщенный раствор Температура 20° С уксуснокислого аммония 10%-ный раствор пер- Температура 20° С сульфата аммония
Электрохимическая очистка, катодная обработка
Оцинкован- ная сталь 30 е/<Ы8 трехзамещен- Плотность тока ного фосфорнокислого 1,1 а[дмл, температу- иатрия + 15 г/йл'3 угле- ра 70° С кислого натрия +10 г/йл!8 едкого иатра 5%-иый раствор двух- Плотность тока замещенного фосфорио. 1,1 а) дм?, температу- кислого натрия ра 70° С, время 5 мин Химическая обработка 10%-иый раствор трехза- Время 2—15 ч мещеиного фосфорнокис- лого натрия 20%-иый раствор ам- Время 2—5 мин миака 10%-иый раствор пер- Температура ком- су льфата аммония иатная. Погружение в раствор до удаления ржавчины
Кадмирован- иая сталь и кадмий Магний и магниевые сплавы 10 %-ный раствор хло- Температура 70° С, ристого аммония время 2,5 мин 20% хромового ангид- Кипячение I мин рида + 1% азотнокисло- го серебра 15% хромового ангид- Кипячение 15 мин рида + 1% хромовокис- лого серебра
Нержавею- щие стали 10%-иый раствор ли- Температура 60° С мойной кислоты, 5%-ный раствор серной кисло- ты + 0,2% ингибитора
— 95 —
Продолжение табл. 13
Сплав Электролит Режим Примечание
Железо, сталь, чугун Механическая очистка Паста из 200 г окиси Протирание магния и I дм3 веретен- ного масла Паста из 200 г венской Протирание извести и I дм3 рафини- рованного веретенного масла Паста нз 200 а окиси Протирание хрома и I дм3 веретен- ного масла
Для удобства коррозию лучше всего выражать в толщине
разрушенного металла, которую называют проницаемостью.
Обыкновенно ее выражают в мм/год или мкм(гсд.
Классификация металлов =no их коррозионной стойкости
в отечественной практике производится -по 10-балльной шкале
(ГОСТ 5272—50) в соответствии с табл. 14.
Таблипа 14
ДЕСЯТИБАЛЛЬНАЯ ШКАЛА КОРРОЗИОННОЙ СТОЙКОСТИ МЕТАЛЛОВ
(ГОСТ 5272—50)
Группа стойкости Скорость КОррОЗИ и, мм/еод Балл
1. Совершенно стойкие Менее 0,001 1
11. Весьма стойкие Свыше 0,001 до 0.005 2
» 0 005 » 0,01 3
II1. Стойкие » 0,01 » 0,05 4
» 0,05 » 0,1 5
IV. Пониженно стойкие » 0,1 » 0,5 6
» 0,5 » 1,0 7
V. Малостойкие » 1,0» 5,0 8
» 5,0 » 10.0 9
VI. Нестойкие » 10,0 10
Пересчет потерь массы металла на проницаемость произво-
дится по формуле
П = А.Ю“3, (51)
У
где П — проницаемость, мм/год-,
К — скорость коррозии, г/(ж2 • год);
•у — плотность металла, г/см3.
— 06 —
7 Зак.
Т аб л ица 15 •
СОСТАВ ТРАВИТЕЛЕЙ, РЕКОМЕНДУЕМЫХ ПРИ МЕТАЛЛОГРАФИЧЕСКОМ ИССЛЕДОВАНИИ КОРРОЗИИ [5]
Сплав Что выявляет Состав травителя Режим
, Углеродистые стали Границы зерен феррита Линии деформации и расположение выделений при старении стали Границы ферритных и аустенитных зерен Границы аустенитных зерен в зака- ленной и отпущенной стали Линии напряжения деформирован- ной стали при неоднородном их рас- пределении 4 сл13 азотной кислоты -f- 100 см3 амилового спирта 2 см3 азотной кислоты + 50 см3 эти- лового спирта+«50 см3 воды 4 г пикриновой кислоты+100 см3 этилового спирта 3—5 см3 соляной кислоты 4- 4 г пикриновой кислоты+95—100 сл3 эти- лового или метилового спирта . 5 г хлорной меди + 40 см3 соляной кислоты + 25 см3 этилового спирта + + 30 см3 воды 60 сек, 20-30° С Травление и полиров- ка 2—3 мин Травление нагретого до 200’ С образца 10— 20 сек
Чугун серый, ковкий и др, Границы зерен между графитом и металлом 25 см3 соляной кислоты + 10 г хро- мового ангидрида+ 5 г хлористого никеля + 100 см3 воды t Травление 3—5 сек, промывка спиртом или аммиачной водой
Нержавеющие стали Границы зерен Границы зерен, межкристаллитная коррозия 25—35 см3 серной кислоты + 2—3 г хромового ангидрида+.65—75 см3 воды . 5 г хлорного железа + 15 см3 со- ляной кислоты + 50 см3 воды Травление при подо- греве
Медные сплавы Границы зерен, линии сдвигов и двойников в меди и однофазных спла- вах Границы зерен меди, латуни, брон- зы н сложных сплавов 10 ч. аммиака + 4 ч. 3%-ной пере- киси водорода + 1 ч. 20%-кого едкого кали + 5 ч. воды 5 ч. насыщенного водного раствора хлорного железа + 30 ч, 4%-ного вод- ного раствора соляной кислоты + 30 ч. амилового или этилового спирта Травление до 1 мин Травление 0,5—2 мин
Продолжение табл, 15
-86
Сплав Что выявляет Состав травителя Режим
Алюминиевые сплавы - Границы зерен алюминия и сплавов 5—20%-ный водный раствор едкого кали -
'. " с*' - ‘ • - Линии скольжения в алюминиевых 0,1т-1,0%-ный водный раствор Травление до потуск-
литых и отожженных сплавах ’• Лин'ии скольжения в деформирован- ных алюминиевых сплавах фтористоводородной кислоты 1 ч. азотной кислоты + 2 я. плави- ковой кислоты + 2—3 ч. глицерина нения Травление 10 сек, ос- ветление в 3—5%-ном растворе азотной кисло- ты
’Магниевые сплавы Границы зерен и структуру 1 см3 азотной кислоты + 75 си8 этиленгликоля — 24 см3 воды —
Границы зерен в деформированных сплавах • Границы зерен в деформированных сплавах Границы зерен в сплавах с марган- цем после ковки и- штамповки 10%-ный раствор винной кислоты 5 г пикриновой кислоты + 5 см3 35%-ной уксусной кислоты +10 мл воды + 100 см3 этилового спирта 5—10 а лимонной кислоты + 100 см3 воды . Травление 10—20 сек Травление 5—15 сек, промывка спиртом Травление 5—15 сек, промывка горячей водой
Титановые спла- вы Границы зерен a-фазы в сплавах с медью, серебром, беррилием, бором, кремнием, ванадием, хромом, молиб- деном, марганцем, железом, никелем, кобальтом и ниобием 2—3 см3 азотной кислоты + 3— 1,5 см3 плавиковой кислоты+ 95 см3 воды
Йодидный (чис- тый) цирконий —• 8—10 см3 плавиковой кислоты + + 60 см3 9%-ной перекиси водоро- да + 30 см8 воды Втирать 30—60 сек
Сплавы цирко- ния и гафния — 1—6 ч. (объемн.) азотной кисло- ты+3—5 я. (объемн.) плавиковой кислоты ’ ' Втирать несколько се- кунд, промывка в струе воды
При исследовании коррозии, а также «при массовых испыта-
ниях необходимо интересоваться не только коррозионными по-
терями, «но -и характером разрушений. Иногда при незначитель-
ных -коррозионных «потерях- может наблюдаться коррозионное
расслаивание, межкристаллитная коррозия, а также коррозион-
ное растрескивание. Последнее возможно -в «конструкциях, нахо-
дящихся -в -напряженном состоянии. Поэтому гравиметрические
измерения должны быть дополнены микроскопическими иссле-
дованиями. Необходимо изучать с помощью микроскопа состоя-
ние поверхности с неснятыми продуктами коррозии, а также
после их удаления.
Наличие межкристаллитной коррозии, «коррозионных тре-
щин, а также незначительное «расслоение лучше всего выяв-
ляются на поперечных шлифах. В качестве травителей для при-
готовления шлифов применяют реактивы, приведенные
в табл. 15 [5].
2. ОБЪЕМНЫЕ МЕТОДЫ
Одним из способов оценки коррозии является так называе-
мый объемный метод, определяющий «количество выделившегося
в процессе коррозии водорода или -поглощенного кислорода.
Объемный метод в 10—100 раз более точен, чем «весовой,_и позво-
ляет определить зависимость скорости коррозии от времени, не
удаляя продукты коррозии и -не прерывая, испытания. Примене-
ние объемных методов наиболее целесообразно, если процесс
коррозии идет преимущественно с выделением водорода или
с поглощением кислорода.
Для измерения объема «выделившегося -водорода, например
при коррозии магниевых сплавов «в растворах хлористого натрия,
алюминия и других сплавов «в «кислых растворах, применяют
приборы — «водородные коррозиметры. Коррознметры бывают
простые и усовершенствованные. Простейший коррозиметр,
который может быть применен в любой лаборатории, показан на
рис. 52. Такой прибор используют при испытании магниевых
сплавов в растворах хлористого натрия. В наполненный раство-
ром прибор устанавливают образец и измеряют объем -выделяю-
щегося «водородах помощью бюретки, установленной над образ-,
цом, каю показано на -рис. 52. Вначале отсчеты -производят часто
(через 0,5; I; 2; 5 мин, затем 10, 30 мин, 1, 2, 5 ч и т. д.). При
измерениях устанавливают условный нуль — время, необходи-
мое для помещения образца в раствор.
При массовых коррозионных испытаниях.более удобно поль-
зоваться коррозиметром, предложенным Г. В. Акимовым»
Л. И.. Стоклицким и Е. Н. Палеолст {6J, внешний в,ид которого
представлен на рис. 53. Прибор сострит, из комплекх^.Мёрных.
градуированных1 бюреток 3, «каждая1 из которых имеет $зйук>’
и широкую части и соответственно две ступени" отсчёта. Внизу
7* ^99 —
каждая бюретка заканчивается «небольшим «колоколом со стек-
лянным «крючком для ‘подвешивания образцов. Все бюретки, три
помощи разборного^ держателя 2, обеспечивающего их быстрый
подъем и опускание, укреплены в штативе 4. .Бюретки соединены
резиновым шлангом с водоструйным насосом и заполняются
раствором через кран /. Результаты испытаний -представляюг
простейшей Рис. 53. Усовершенствованный водородный
конструкции [1] коррозимегр [6]
где Крю массе) — количество металла, перешедшее в раствор
за время испытания, а;
А— атомная масса металла, а;
«—-валентность иона металла;
Ун = 23804 ои8 — объем 1 моля водорода при 17° С и давле-
нии 1 бар;
‘ К(обьемн)—объем водорода, выделившегося за'время
’ испытания, см*.
' Для случая растворения магния и его сплавов формула при-
обретает вид
. * К (по массе) == 1 ’ Ю ^С(объемн)‘ .
В описанном «коррозиметре количество выделившегося газа
определяется sjo'величине вытесненного объема раствора в гра-
дуированной бюретке. ' -
— 100 —
В. П. Задорожный (7] разработал 'конструкцию коррозиметра,
в котором предусмотрена автоматическая запись количества
выделившегося ‘водорода. Принципиальная схема этого прибора
приведена на рис. 54. Преимущество этого' прибора -в том, что
Рис. 54. Схема водородного коррозиметра с автоматической регистрацией
количества выделившегося газа (7J:
1 — реакционный сосуд; 2 — мотор Уорена; 3 — редуктор; 4 — движущийся бара-
бан; 5 — фонарик; 6 — шкив; 7 — проволоки; 8 — блок; Р — нить; 10 — храповое ко-
лесо; J1 — рычаг; /2 — электромагнит; 13 —-пружина; И—клапан; 15 — отверстие;
16 — манометр; 17 и 18 — контакты; 19 — регулятор контакта: 20 — электронное реле;
— 1 ком; /?->*= 2 ком; — 15 ком; — 500 ком
измерения в нем производятся всегда при одном и том же дав-
лении, а диапазоны измерений расширяются и объем бюретки
уже не лимитирует проведение испытаний с большим количест-
вом выделяющегося газа. Прибор'работает «следующим образом.
В сосуде / в результате (коррозионного процесса выделяется во-
дород. При закрытом клапане 14 отверстия 15 в результате не-
которого повышения давления в системе, создаваемого выделяю-
щимся водородом, происходит замыкание контактов 17,18 мано-
— 101 —
метра. 16. В момент замыкания «контактов манометра происходит
замыкание контактов электронного реле 20, что -приводит
к включению электромагнита 12. Последний притягивает кла-
пан, в результате чего открывается отверстие. Давлений в систе-
ме выравнивается до первоначального, контакты манометра
размыкаются, электромагнит включается и -клапан «под воздей-
ствием пружины 13 снова закрывает отверстие. Движение
клапана «передается на храповое «колесо 10 с «помощью рычага 11,
который жестко крепится с осью клапана. Вращение храпового
колеса «передается шкиву 6, сидящему на одной оси с колесом.
Шкив соединен иерастяжимой нитью 9, переброшенной через
блок 8, с -фонариком 5. Фонарик «проектирует иа фотобумагу
движущегося «барабана 4 узкий «пучок света»
Движение фонарика осуществляется «параллельно натянутой
проволоке 7, которая одновременно является «проводником пи-
тания. Равномерное движение -барабана осуществляется кото-
ром Уорена 2 через «редуктор 3. Для фотозаписи -приспособлена
фоторегистрирующая камера пирометра Курнакова. Манометр
снабжен регулятором высоты контакта уровня ртути 19, что дает
возможность проводить запись реакций различной интенсив-
ности. Этой же цели удовлетворяют манометры с различным
сечением
Скорость движения фонарика можно регулировать заменой
шкива 6 шкивом другого диаметра. Перед началом работ произ-
водится калибровка оси объемов «водорода по измерению ско-
рости «процесса с точно известным- количеством выделяющегося
водорода.
Для исследования процессов, протекающих с кислородной
деполяризацией, разработаны различные конструкции прибо-
ров (8—10]; .в таких приборах должна быть предусмотрена воз-
можность постоянного обновления атмосферы, чтобы испытания
не «проводились при недостатке кислорода. Рассмотрим конст-
рукцию одного из -приборов этого типа, применявшегося авто-
рами для исследования коррозии стали, цинка и алюминия
в нейтральных электролитах [10]. Доля водородной деполяриза-
ции для этих металлов незначительна и «поэтому количеством
выделяющегося при испытаниях водорода «можно 'пренебречь.
Как видно из рис. 55, «прибор, кроме реакционного сосуда 1,
имеет вспомогательный сосуд 2, назначение которого состоит
в том, чтобы измерения можно было -производить при определен-
ном давлении. Для того чтобы давление было- постоянным, необ-
ходимы вспомогательные сосуды очень большого объема, что
экспериментально представляет большие трудности. Чтобы из-
менения давления, «которые возникают вследствие поглощения
кислорода в -процессе коррозии, не сказывались на результатах
измерения, сосуды должны быть достаточно- большими. При’ма-
лых объемах сосудов и необходимости точных измерений строят
поправочную кривую, учитывающую изменение давления в вспо-
— 102 —
могательном сосуде «при перемещении капли раствора >в соеди-
нительной трубке.
Во всех случаях при определении коррозии по -количеству
поглощенного кислорода -необходимо- следить за тем, чтобы
испытания не проводились при недостатке кислорода. Для этой
цели необходимо периодически обновлять атмосферу в реакци-
онном сосуде. Обычно обновление •производят с таким расчетом,
чтобы количество поглощенного
кислорода ие превышало 10%
общего запаса кислорода в сосу-
де. Это легко определить, зная
объем сосуда и объем поглощен-
ного кислорода в процессе кор-
розии за данный промежуток
времени.
Прибор, приведенный на рис.
55, позволяет проводить испыта-
ния не только при погружении в
электролит, но и при периодиче-
ском смачивании образцов элек-
тролитом и просто во влажной
атмосфере. Специальное устрой-
ство в виде стеклянного поплав-
ка 9, на котором подвешиваются
образцы 8, периодически при за-
мыкании цепи электромагнита
// погружается в электролит, а
при выключении тока вновь
всплывает. Измерения произво-
дятся по передвижению капли
раствора 4 в манометрической
трубке 3. Пробка 5 и кран 7 слу-
жат для периодического освеже-
ния атмоферы. Одновременно в таком приборе может испыты-
ваться до восьми одинаковых образцов размером 20 X 30 мм.
При измерении коррозии объемными методами могут возни-
кать ошибки вследствие изменения температуры, наличия сме-
шанных деполяризационных процессов, выкрашивания мелких
частиц металла (например, при коррозии магниевых сплавов)
и изменения давления. При применении объемных методов необ-
ходимо тщательное термостатирование, а влияние изменения
давления (при точных измерениях) учитывать, приводя измерен-
ные количества газа к нормальным условиям.
Если коррозия сопровождается одновременным поглощением
кислорода и выделением водорода, конструкция прибора услож-
няется, в нем должна быть предусмотрена возможность выжига-
ния водорода, что достигается следующим образом. В крышку
сосуда впаивают ‘платиновые контакты, соединенные с платино-
— 103 —
Рис. 55. Прибор для определения
скорости коррозии, по количеству
поглощенного кислорода в усло-
виях периодического смачивания
электролитом н во влажной атмо-
сфере [10]:
I — реакционный сосуд: 2 — вспомо-
гательный сосуд; 3 — манометриче-
ская трубка; 4 — капля раствора,
в' котором производится испытание;
5 — пробка; 6 — термостат; 7 —•
кран; 8 — образцы; 9 — стеклянный
поплавок; 10 — железный сердечник,
впаянный в нижнюю часть поплавка;
II — электромагнит
вой спиралью, находящейся внутри прибора. Через спираль про-
пускается ток. Соприкасаясь с накаленной спиралью, находя-
щийся *в сосуде водород сгорает.
Если металл образует окислы или гидроокиси одной валент-
ности, количество поглощенного кислорода легко пересчитать на
количество разрушившегося -металла; если же в «процессе корро-
зии образуются соединения с различной -степенью окисления,
например у железа, то необходимо определить долю этих соеди-
нений в продуктах коррозии.
При ускоренных испытаниях оценку коррозии объемными
методами при смешанном характере деполяризации, очевидно,
применять нецелесообразно, поэтому описание таких приборов
не приводится.
3. ОПРЕДЕЛЕНИЕ ГЛУБИНЫ ПРОНИКНОВЕНИЯ
КОРРОЗИИ
Когда коррозия иосит исключительно неравномерный харак-
тер, что наблюдается, например, на алюминиевых сплавах, низ-
колегированных и нержавеющих сталях, показатель изменения
массы металла должен быть дополнен глубинным показателем,
который характеризует истинную глубину проникновения кор-
розии.
Существует ряд приборов для измерения глубины коррозии.
Наиболее точные — оптические приборы [4]. При достаточном
навыке глубина коррозионного поражения может быть опреде-
лена с помощью обычного микроскопа методом фокусирования
оптической схемы сначала на плоскость, совпадающую с верх-
ним краем язвы, а потом на плоскость дна язвы. По разности
отсчетов на микроскопическом винте судят о -глубине питтинга.
В «последнее время для определения глубины проникновения
коррозии применяют двойной микроскоп В. П. Линника, прин-
цип действия которого основан на создании оптического сечения
поверхности, -подлежащей исследованию [1I]. Оптическая схема
этого’ прибора приведена на рис. 56. Микроскоп имеет два ту-
буса А и В, смонтированных под углом <р к нормали Са^ Тубус В
представляет собой обыкновенный микроскоп с объективом Oj
и окуляром Л. Последний устанавливается таким образом, чтобы
точка изображения объектива Oi лежала ® его фокальной
плоскости.
Тубус А служит для направления узкого -пучка света на ис-
следуемую -поверхность /Ж Для этого в плоскости изображения
объектива О2 имеется светящаяся щель а. Если узкий пучок
света направить иа зеркально отполированную поверхность, то
изображение ах щели а спроектируется ib точке а2. В окуляре К
мы увидим изображение щели а, совпадающее с оптической
осью.
•—104-
Если плоскость передвинуть в'положение H^Hi на расстоя-
ние d, то легко показать, что изображение щели а -передвинется
в точку a'i на расстояние 2d от ai. В окуляре К изображение а [
спроектируется ® точке а2 вне фокальной плоскости окуляра, на
плоскости же получится размытое изображение щели а, отстоя-
щей от оси «на некотором расстоянии. При небольшом перемеще-
нии и небольшой числовой аппертуре объектива Oi изображение
будет еще достаточно резким и перемещение плоскости НН на
некоторое расстояние d будет эквивалентно ’перемещению неко-
торой точки ® предметной 'плоскости объектива Ог. Последняя
величина измеряется.
Таким образом, имея в толе зрения коррозионную язву, одно-
временно с ией можно наблюдать извилистую линию, точно- вос-
производящую профиль язвы в перевернутом -виде (рис. 57).
С помощью окуляра с визирным крестом и микрометрического
винта глубина язвы определяется в любом месте.
На этом приборе можно измерять коррозионные поражения
в .пределах от 10 до 100 мкм. При наличии специальной оптики,
пределы измерений могут быть расширены от 1—2 мм до 0,01—
3 мкм.
Для аналогичных целей -применяют и оптикомеханические
профилографы, относящиеся зю принципу действия к щуповым
приборам, например «профилограф ИЗП-17 Б. М. Левина [12].
Схема этого прибора -показана на рис. 58. В профилографе от
жестко связанных с иглой 12 зеркал 7 и 8, на которые от лам-
пы 2 проектируется узкий пучок света, отраженный» свет «прохо-
дит через систему линз телеобъектива 6 н попадает на зеркало 3..
Отразившись от зеркала 5, луч проходит через цилиндрическую-’
—105 —
линзу 5, которая фиксирует изображение нити лампочки в виде
точки на фотобумаге. Измерения могут производиться и визу-
ально 'по шкале 4. Ощупывающая исследуемую «поверхность
Рис. 57. Вид профилограммы
(а), разрез коррозионной язвы (б) [4]
игла 12 укреплена на пружине 10 и своим вторым концом, имею-
щим вид лезвия 13, упирается в оправу зеркала 14. Оправа
зеркала 14 подвешена на двух пружинах.
. Увеличение, осевого перемещения иглы осуществляется за
счет угла поворота зеркал и расстояния проекционного объектива
— 106 —
от барабана, на «котором укреплена фотобумага. Это расстояние
называется оптическим рычагом и 'поэтому приборы такой конст-
рукции относят к приборам, работающим на принципе опти-
ческого рычага. В настоящее время разработана новая усовер-
шенствованная модель профилографа ИЗП-18 [13].
Основное преимущество описанного профилографа в том, что
он дает возможность ие только измерить язву, но и получить
в увеличенном масштабе фотографическую запись микрогеомет-
рии поверхности образца, на ко-
торой образовались коррозион-
ные поражения. По профило-
грамме можно судить не только
о глубине, но и о форме обра-
зующихся питтингов.
Удачную и простую конст-
рукцию прибора, пригодного для
измерения относительно глубо-
ких язв, разработал С. Е. Пав-
лов [14]. Отличительная черта
глубиномера — возможность про-
изводить замеры не только иа
образцах, но и иа конструкциях.
Общий вид глубиномера пред-
ставлен .на рис. 59. Глубина по-
ражения измеряется при помощи
полого латунного цилиндра,
внутри которого двигается штифт
с иглообразным наконечником.
Торцовые плоскости патрона
должны быть тщательно отшли-
фованы и строго параллельны.
Перед измерением штифт выдви- Рис. 59. Схема тлубино-
гается из патрона и игла упира- мера [11J
ется в дио язвы, предварительно
очищенной.от продуктов коррозии. После этого нажимом руки
патрон приводится-в соприкосновение с .поверхностью образца
или детали. Игла при этом выступает над торцовой поверхно-
стью патрона иа глубину язвы. Поставив такой патрон на при-
паянный к нижней опоре микрометра алюминиевый диск, кото-
рый с целью электроизоляции анодирован, н присоединив его к
электрической схеме, как это показано' на ..рис. 59, получаем
возможность измерить глубину поражения. Вращением готовки
микрометра подвижный стержень приводится в контакт с остри-
ем иглы, и цепь замыкается. В этот момент загорается лампоч-
ка. Дальнейшее вращение головки приводит в соприкосновение
подвижную часть микрометра с торцовой поверхностью* патро-
на. По разности отсчетов судят о глубине коррозионного пора-
жения.
— 107 —
При определении глубинного показателя часто встречаются
с трудностями такого (порядка: ‘при наличии большого количест-
ва питтингов, глубину которых .промерить практически невоз-
можно, произвольный выбор определенного количества их может
привести к ошибочным результатам. В таких случаях рекомен-
дуется следующий прием. Образец необходимо разделить иа
определенное число квадратов и измерять глубину тех питтингов,
которые находятся на ‘пересечении линий, образующих квадра-
ты (узлы). Из общего количества измерений выводят среднюю
глубину коррозии. Кроме того, необходимо зафиксировать глу-
бину наиболее глубоких питтингов, встречающихся на поверх-
ности металла.
Важный показатель для низколегированных и нержавеющих
сталей, алюминиевых и титановых сплавов и вообще сплавов,
склонных к питтингообразованию,— коэффициент питтингообра-
зования Р. Он представляет собой отношение средней глубины
всех питтингов к условной глубине, вычисленной по потере массы
при допущении, что коррозия носит равномерный характер.
Например, если коэффициент равен 100 или 50, то это значит,
что глубина проникновения коррозии ® отдельных местах
в 100—50 раз больше по сравнению со средними разрушениями,
вычисленными по потере в массе. Коэффициент питтингообразо-
вания зависит, с одной стороны, от общей коррозионной стой-
кости сплавов и, с другой,—от склонности сплавов к точечной
коррозии. Для сталей корродирующих сильно, но более равно-
мерно, он будет меньше, чем для сталей, корродирующих с мень-
шей скоростью, но не слишком равномерно.
С помощью коэффициента питтингообразования можно при-
ближенно определить величину поверхности металла, занятую
коррозией [15].
Если hi — уменьшение толщины металла, вычисленное из по-
терь (по массе), гари допущении, что коррозия распределена
равномерно; Fj — площадь образца; h2 и Ё2—истинная глубина
коррозионных поражений и их площадь соответственно, то, при-
няв условно, что питтинги или язвы имеют цилиндрическую
форму, можно суммарную коррозию К в том и другом случае
выразить следующими уравнениями:
К2 =
где у — плотность металла.
Учитывая, что Ki — Кг, получим отношение
Р1 __ ^й
Ей
Поскольку коэффициент 1питтингообразования
Р =
Й1 ’
— 108 —
(54)
(55)
(56)
(57)
можно -приближенно вычислить -процент -площади, занятой кор-
розией:
F2=Tl.I00. (58)
Таким образом, для характеристики коррозионного поведе-
ния металлов, склонных к ‘питтинговой коррозии, необходимо
использовать следующие показатели:'' среднюю глубину корро-
зионных поражений, вычисленную по потере массы (при допу-
щении, что коррозия носит равномерный характер),-? среднюю
и‘Максимальную глубину коррозионных .поражений, измеренные
экспериментально, а также - коэффициент питтиигообразоваиия.
Этими показателями наиболее полно характеризуются свойства
металлов, склонных к (местной коррозии.
4. ОПРЕДЕЛЕНИЕ КОРРОЗИИ
ПО ИЗМЕНЕНИЮ МЕХАНИЧЕСКИХ СВОЙСТВ
При изучении коррозии алюминиевых сплавов часто исполь-
зуют такой характерный показатель, как изменение механи-
ческих свойств металла. Однако необходимо помнить, что при
отсутствии межкристаллитной коррозии механические’ свойства i
металлов в результате коррозии обычно -не • меняются, и этим
показателем лишь косвенно определяют изменение сечения об- i
разца вследствие коррозии. На самом деле, если определить
нагрузку, требующуюся для разрыва образца, до коррозии и эту :
же нагрузку после того, как металл прокорродировал, то -можно
определить фиктивный предел прочности, характеризующий
изменение сечения образца. Обычно определяют предел проч-
ности (5ъ [н/мм2 (кГ/мм2)] и удлинение 6 (%) при растяжении.
Если Ёо и Ei — начальное и конечное после коррозии сечение
образца, а иьс — предел прочности материала, то разрушающие
нагрузки
до коррозии
й = (59)
после коррозии
А = = (60)
фиктивный предел прочности о^, характеризующий изменение
сечения,
Pi . %(fo —
(61)
(62)
Obt = °ье I------.
\ ^0 /
— 109 —
Изменение предела прочности как меру коррозии удобнее
выразить в -процентном отношении:
Кь => . 100. (63>
ч
Аиалопичиым, образом выражают и изменение остаточного
удлинения:
Къ = А-6! . 100. (64>
Ofi
Предел прочности и -особенно остаточное удлинение—важ-
ные показатели при определении неравномерности коррозии»
а та-кже наличия межкристаллитной коррозии. Значение —г ха-
*0
растеризует неравномерность коррозии, поскольку разрыв воз-
никает в наиболее слабом сечении, а при возникновении меж-
кристаллитной коррозии предел прочности и удлинение резко
падают.
Описываемый метод наиболее применим для образцов тон-
кого сечения. Чем выше Fo, тем при одном и том же изменении
ЛЕ- &F
сечения за счет коррозии Лг отношение •— становится меньшим.
Го
Обычно этот метод дает .положительные результаты иа листовом
материале толщиной 1—3 мм и проволоке примерно такого же
диаметра. При использовании круглых образцов их размеры
следует пропорционально уменьшать исходя из диаметра образ-
ца 3—5 мм.
5. ОПРЕДЕЛЕНИЕ КОРРОЗИИ
ПО ИЗМЕНЕНИЮ ОТРАЖАТЕЛЬНОЙ СПОСОБНОСТИ
•ПОВЕРХНОСТИ МЕТАЛЛА
При изучении начальных стадий коррозии или процессов,
протекающих с малой скоростью, используют метод определе-
ния отражательной способности до коррозии и -после нее. По
изменению отражательной способности можно косвенно судить
о стойкости металла в данной атмосфере. Ясно, что этим мето-
дом абсолютные скорости процесса определить нельзя. Однако
он «позволяет сравнивать -между собой различные -металлы и по-
крытия. Он применим для изучения декоративных «покрытий,
нержавеющих сталей, а также эффективности летучих ингиби-
торов. Для «приборов и конструкций, отражательная способность
которых важна сама «по себе (прожекторы, отражатели фона-
рей, рефлекторы, архитектурные сооружения), метод позволяет
получить абсолютные 'показатели, В этом методе о начальных
стадиях коррозии (потускнении) судят по относительному коэф-
— 110 —
фициенту отражения К, вычисленному из величины -фототоков,,
измеряемых на 'специальной установке (рис. 60):
К = -£- • ЮО, (65>
/о
где /1—«величина, фототока для исследуемого образца, мка;
/0—то же, для стандартного зеркала.
В качестве стандартного образца можно применять алюми-
ниевое зеркало, полученное путем конденсации алюминия в ва-
кууме.
Чтобы-получаемые результаты были воспроизводимы, источ-
ник света должен всегда иметь постоянный накал; этого можно
Рис. 60. Схема установки для определения
относительного коэффициента отражения (16]:
У — вогнутое зеркало; 2 — лампа напряжением
12—16 в'. 3 — диафрагма; 4 — линза; 5 — испытуе-
мый образец; 6 — фотоэлемент
достигнуть, создавая одинаковое напряжение, но удобнее про-
изводить измерения при одном и том же значении фототока,,
например при 100 мка, контролируемом микроамперметром
(см. рис. 60) (16]. В этом случае отсчет по микроамперметру для
исследуемого образца, отнесенный к постоянной величине, и бу-
дет определять величину относительного коэффициента отра-
жения.
Изменение отражательной способности можно измерять на
фотометре типа ФМ-5-9 конструкции ГОИ.
6. ОПРЕДЕЛЕНИЕ КОРРОЗИИ
ПО ИЗМЕНЕНИЮ ЭЛЕКТРИЧЕСКОГО СОПРОТИВЛЕНИЯ
/
Коррозионные процессы, приводящие -обычно к изменению
сечения образцов, а иногда и к ’более глубоким изменениям
в самом материале ((межкристаллитная коррозия, образование
трещин, расслоение и т. и.), естественно, должны привести и
к изменению омического сопротивления. Поэтому методом изме-
рения омического сопротивления образков до коррозии и после
— 111 —
нее можно -получить ценные результаты, которые будут .харак-
теризовать поведение металлов в данных условиях.
Особенно сильное изменение сопротивления наблюдается
при появлении межкристаллитной коррозии (см. гл. VI).
Если р— удельное сопротивление образца; t0 — длина;
Do — исходный диаметр; Di — конечный диаметр после корро-
зии, то начальное электрическое сопротивление образца
/?„== . (66)
Вследствие коррозии изменится диаметр образца и сопро-
тивление возрастет до
(67)
я!>|
Абсолютное и относительное изменение сопротивлений опре-
деляется следующими уравнениями:
---LA; (68)
"И D* )
Учитывая, что
(Rt-RJn С2-В2_ (69)
Wo д?д2
/~)2 п2 .(70)
271 At * 4poio
(71)
Д^ Wo
jiDj j (72)
4Po4)
получаем
, £>?
1“^ = —’
Таким образом, измерив сопротивление до и после коррозии,
можно определить новый диаметр образца, а стало быть, и глу- •
бину проникновения коррозии. Учитывая, что £>f = (£>0— Д£>)2,
уравнение (73) можно преобразовать следующим образом:
j _ (Д„ —ДР)а = И, К<, . „4,
— /й> = 2ЛД _ ДД ~ 2ДД
«1 О. Д2 ~ До ’ ' '
— 112 —
’ Из уравнения (.75) следует, что лрв одинаковой глубине проник-
новения коррозии относительное изменение сопротивления бу-
дет тем выше., чем меньше начальный диаметр^юбразца.
Поэтому наибольшую чувствительность мётод дает при изу-
чении коррозии тонких листовых материалов’5и.1Проволоки. Для
выбора размеров образцов из различных металлов при оценке
коррозии по изменению электросопротивления можно пользо-
ваться кривыми, представленными на рис. 61 [17]. Размеры об-
Рис. 61. Значения парамет-
ров проволочных образцов,
величина коррозионных по-
терь которых может быть
определена по изменению
омического сопротивле-
ния [17]:
I — алюминий; 2 — сталь,
платина, фосфористая бронза;
3 — константан; 4 — нихром
Рис. 62. Схема установки для
измерения электросопротивле-
ния [18]:
1 и 2 — стабилизаторы СНЭ
120-Q1; S — выпрямитель ВСА-1:
Пх' — сопротивление исследуемого
образца; — эталонное сопро-
тивление; ППТВ и П4 — потен-
циометры
разцов (длина I и радиус г0), применяющихся для определения
коррозии по изменению электросопротивления, находятся на
кривой или в области, лежащей слева от нее. При выполнении
этого условия, по данным автора [17], измерения могут произ-
водиться на приборе, позволяющем определять сопротивление
с точностью 0,01 ом,. Чем левее от кривой лежат значения па-
раметров образцов, тем выше чувствительность метода.
Метод удобен тем, что позволяет проводить непрерывные
наблюдения за 'коррозией и изучать на одном и том же образце
развитие процесса во времени. Для измерения сопротивления
пользуются обычными двойными мостами, выпускаемыми про-
мышленностью (ММВ, РВ-16), а также установками, собранны-
ми в лабораториях. Одна из таких экспериментальных схем, при-
менявшаяся Ф..Ф.. Ажогииым ]18], показана на рис...62,; ..
8 Зак. 1333 __ррз
При изменении сопротивления важно обеспечить надеж-
ность контакта, ибо нарушение его -может вызвать такое увели-
чение сопротивления, которое превысит сопротивление,
возникающее за счет коррозии. Контакт создают обычно пайкой
или плотным клеммовым соединением, места контакта изолиру-
ют от коррозионной среды.
Чаще всего сопротивление измеряют отдельно до и после
коррозии, однако в литературе описаны установки, в которых
коррозионные испытания сочетали с непрерывными измерения-
ми сопротивления без извлечения образца из коррозионной
среды. Метод был с успехом применен Хадсоном при исследова-
иин атмосферной коррозии стальной проволоки [19], Н, И. Исае-
вым при исследовании атмосферной коррозии стальной и оцин-
кованной проволоки [17], а также для исследования коррозии
в присутствии ингибиторов [20].
Значения удельного сопротивления [(ом *мм2)/м] наиболее
употребительных материалов (при 20°С), которые необходи-
мо знать при использовании данного метода, приводятся ниже
[21]:
Алюминий . . . 0,028 Ртуть............ 0,958
Вольфрам .... 0,055 Свинец.......... 0,221
Латунь. 0,025—0,06 Серебро......... 0,016
Медь...... 0,0175 Сталь ........... 0,098
Молибден .... 0,057 Тантал .......... 0,155
Никель........... 0,100 Хром............. 0,027
Олово 0,115 Цинк............ 0,059
Иногда, не прибегая к вычислению глубины проникновения
коррозии, стойкость материала характеризуют косвенно относи-
тельным изменением сопротивления:
7^= (76)
7. ОЦЕНКА СТОЙКОСТИ ПО ВРЕМЕНИ
ДО ПОЯВЛЕНИЯ ПЕРВОГО КОРРОЗИОННОГО ОЧАГА
ИЛИ ОПРЕДЕЛЕННОЙ ПЛОЩАДИ КОРРОЗИИ
При изучении поведения металлов с защитными покрытиями
ни показатель изменения массы, ни глубинный показатель не
дают надежных результатов. Поэтому часто определяют время
появления первого очага коррозии. Этот метод, как отмечает
Г. В. Акимов [1], применим в тех случаях, когда очаг ясно выде-
ляется на фоне неизменившейся поверхности, например при
коррозии стальных изделий, покрытых защитными пленками
(металлическими, лакокрасочными, фосфатными, оксидными),
а также нержавеющих сталей н алюминневых сплавов.
Метод не дает указаний относительно скорости процесса,
а характеризует скорее всего вероятность возникновения кор-
— 114 —
розин. Он прост и поэтому широко применяется При контрольны#
испытаниях на заводах и вносится в том или ином варианте
в технические условия. Иногда оговаривают количество очагов
коррозии, которое может появиться за определенное время испы-
таний, иногда же задаются временем, необходимым для появле-
ния (первого очага коррозии.
Необходимо иметь в виду, что время появления первого оча-
га коррозии не может однозначно характеризовать коррозион-
ную стойкость изделия -в атмосферных условиях.
Кап показал Г. В. Акимов, из двух изделий А и Б, подверга-
ющихся коррозии в последовательности, указанной -в табл. 16,
наиболее стойким окажется не изделие А, на котором первый
очаг коррозии возник позже, а изделие Б, первый очаг коррозии
на котором появился раньше.
Таблица 16
ПОСЛЕДОВАТЕЛЬНОСТЬ ПОЯВЛЕНИЯ ОЧАГОВ КОРРОЗИИ
НА ИЗДЕЛИЯХ А и Б (КОЛИЧЕСТВО ОЧАГОВ)
Изделие Время, ч
1 2 5 10 15 20 25 30 35 40 45 50
А 0 0 0 0 0 1 5 22 100 — — —
Б 0 0 0 1 I 3 5 7 10 15 20 30
Исходя из этих соображений, правильнее оговаривать число
центров коррозии, «которое может появиться через определенные
промежутки времени, а не время появления первого очага кор-
розии. При оценке коррозионной стойкости материала необходи-
мо обязательно учитывать развитие -процесса во времени, ибо
появление очага коррозии может быть связано с каким-нибудь
случайным дефектом в металле или сплаве. Поэтому -наиболее
объективную оценку материала можно получить методом •по-
строения кривых зависимости скорости коррозии стальных об-
разцов (до 50% ржавления) от времени (рис. 63, а).
Точная оценка срока службы покрытия, как отмечают Блам
и Бреннер ;[:22], затруднительна *и по этим кривым. Одндко пло-
щадь влево от кривой является верным -показателем ^защиты.
Численно она равна средней абсциссе АБ, так как ордината ВГ
равна единице. Абсцисса АБ характеризует средний срок служ-
бы покрытия, ибо она равна числу лет, в течение которых по-
крытие защищало бы основной металл от коррозии, если бы она
корродировало равномерно.
При оценке устойчивости металлов по площади, занятой
коррозией, часто ‘прибегают к фотоэталояам (французский стан-
дарт FDA 91-020) или схематическим ’изображениям коррозион-
ных разрушений, подобным1 приведенным на рис. 63, б (DIN
53-210). • ' "
8=* —115 —
— 116 —
8. ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА МЕТАЛЛА,
ПЕРЕШЕДШЕГО В РАСТВОР В ПРОЦЕССЕ КОРРОЗИИ
Определение скорости коррозионного ’процесса по количеству
металла, •перешедшего в раствор за данный -промежуток, време-
ни, производится путем анализа раствора фотокалориметриче-
ским, аналитическим или полярографическим методом. Этот
метод может применяться лишь в тех случаях, когда продукты
коррозии полностью .растворяются в электролите. Основным
преимуществом метода является возможность непрерывного на-
блюдения за ходом коррозионного процесса.
9. КОЛИЧЕСТВЕННАЯ ОБРАБОТКА
ЭКСПЕРИМЕНТАЛЬНЫХ РЕЗУЛЬТАТОВ
При проведении ускоренных испытаний, а также лаборатор-
ных исследований необходимо учитывать, что (коррозионный
процесс зависит от большого числа факторов, нередко не под-
дающихся точному учету. Материал образцов обычно -бывает
неоднородным и по своей реакционной’ способности они слиш-
ком отличаются друг от друга. Состояние защитных , пленок
также зависит от структуры металла и качества обработки,„по?
верхности, которую трудно в каждом отдельном случае воспро-
извести. 'Частб'не удается воспроизвести и условия, зависящие
от коррозионной среды (конвекционные токи, изменения в кон-
центрации электролита, температурные перепады -в камерах по
высоте и т. «п.). Поэтому при испытаниях необходимо по возмож-
ности повышать точность эксперимента и увеличивать количе-
ство образцов, испытываемых параллельно. Для оценки точности
эксперимента необходимо определять вероятную ошибку опыта.
Вопросы статистической обработки результатов коррозион-
ных испытаний подробно разобраны в работах Г. В. Акимова,
Миерса и Эванса |1, 23—-25]. Мы рассмотрим лишь метод опре-
деления вероятной ошибки опыта и показателя точности.
Вероятная ошибка s данной серии опытов определяется с по-
мощью уравнения _______л
s = e./_____, (77)
|/ V(V—1)
где S — отклонение индивидуальных значений измеренных ве-
личин ait <z2, Оз, 04 от среднего значения:
Хй2 — сумма квадратов всех отклонений от среднего;-
V—число измерений;
Q — коэффициент, зависящий от числа измерений.
Ниже приводится зависимость коэффициента 6 от числа из-
мерений V (1]:
V................. 2 3 4* 5 6 7 10 со
в ...............0,816 0,766 0,740 0,728 0,718 0,703 0,674
— 117 —
Показатель точности Р выражает отношение “вероятной
ошибки к среднему значению коррозии (® %):
Р = — • 100. (78)
т
Увеличение количества параллельных образцов резко умень-
шает вероятную ошибку и показатель точности. Так, -например,
Акимов (1] июказал, что лишь одним увеличением количества
образцов вдвое (шесть вместо трех) вероятная ошибка s была
снижена с 0,00328 до 0,00053, а показатель точности Р— с 2,5
до 0,4%.
ЛИТЕРАТУРА
1. Акимов Г. В. Теория и методы исследования коррозии -металлов. Изд-во
АН СССР, 1945.
2. Бахвалов Г. Т., Турковская А. В. Коррозия и защита металлов.
Металлургиздат, 1947.
3. Сб. «Коррозия металлов». Пер. с англ, под ред. В В. Скорчеллетти. Гос-
/ химиздат, 1952.
4. Розенфельд И Л. Замедлители коррозии в нейтральных средах.
Изд-во АН СССР, 1953.
5. Панченко Е. В., Соколов Ю. Л., Попов К. В., Кремер Б. И.,
Арсентьев П. П., Хорни Я. Д. Лаборатория металлографии, Ме-
таллургиздат, 1957.
6. Акимов Г. В., Стоили цк ий Л. Па лес лог Е. П. Тр. Ин-та
физической химки АН СССР, Изд-во АН СССР, вып. 3, 1951, 33.
7. Задорожный В. П. Изв. Воронежского гос. пед. ин-та, 1964, 47, 105.
8. В о п g о и g h G. D., S t u a r t 1. M., Lee A. R. Proc. Roy. Soc., 1927,
A-116, 438, 89,
9. Ромашов H. Д., Матвеева T. В. Тр. Ин-та физической химик АН
СССР. Изд-во АН СССР, вып. 3, 1951, 39.
10. Розенфельд И. Л., Жигалова К- А. Заводская лаборатория, 1959,
т. XXV, № 2, 172.
II. Л ииник В П. ДАН СССР, 1933, т. I, 18; 1945, т. 46, № 9, 42.
12. Леви н Б. М. Современные приборы для измерения неровностей по-
верхности деталей машин. Изд-во АН СССР, 1950, 93.
13. Егоров В. А. Оптические и щуповые приборы для определения шеро-
ховатости поверхности. Мащгиз, 1961.
14. Павлов С. Е. Коррозия дуралюмина. Оборонгиз, 1949.
15. Р о з е н ф е л ь д И. Л., Данилов И. С. Тр. -межвузовской конференции
по коррозии металлов. Гостоптехиздат, 1962, 18.
16. Щиголев П. В. Диссертация на соискание ученой степени канд. хим
наук. Ин-т физической химии АН СССР, 1953.
17. Исаев Н. И. Тр. Ии-та физической химии АН СССР. Изд-во АН СССР,
вып. 8, I960, 144.
18. Ажогин Ф. Ф. Сб. «Коррозия и зашита металлов». Обороигиз, 1957,98.
19. Hudson J. С. Trans. Farad. Soc-., 1929. v. 25.-177.
20. Denman W. L. Corrosion, 1957, v. 13, № I, 59.
21. Те рещук P. M_, Дом-брузо-в P. M., Босый H. Д. Справочник ра-
диолюбителя. Киев. Изд-во АН УССР, 1962. 37.
22. Блам У., Бреи-иер А. Коррозия металлов. Пер. с англ, под ред. В. В
Скорчеллетти. Госхимиздат, т. 2, 1952, 857.
23. М е а г s R. В., D a n i е 1 s Н. Е. Trans. Electroch. Soc., 1935, v. 68, 375.
24. Э в а и с Ю. Р. Коррозия и окисление металлов. Пер. с англ, под ред.
И. Л. Розенфельда, Машгиз, 1962, 822.
25. Evans U. R. Trans. Electroch Soc., 1930, v. 57, 407..........
— 118 —
IV
ЭЛЕКТРОХИМИЧЕСКИЕ И ЭЛЕКТРИЧЕСКИЕ
МЕТОДЫ КОРРОЗИОННЫХ ИСПЫТАНИЙ
МЕТАЛЛОВ
Большинство коррозионных процессов, протекающих в усло-
виях эксплуатации «металлов -в различных средах (морская
вода, атмосфера, химические производства), носит электрохими-
ческий характер. Поэтому по различным электрохимическим
характеристикам можно судить о коррозионной устойчивости
металлов и сплавов (1]. В ряде случаев они могут заменить дли-
тельные испытания.
Электрохимические свойства металлов изучают в лаборатор-
ных условиях за сравнительно короткие промежутки времени,
поэтому они могут быть отнесены к ускоренным методам оп-
ределения коррозионной стойкости материалов в различных
средах.
1. ИЗМЕРЕНИЕ ЭЛЕКТРОДНЫХ ПОТЕНЦИАЛОВ
Общие положения. Несмотря на то что электродный потен-
циал не является абсолютным показателем устойчивости метал-
ла >в данной среде, его установившееся значение и характер
зависимости -потенциала от времени могут дать ряд важных
сведений о характере коррозионного (процесса и поведении -ме-
талла в естественных условиях. Метод измерения электродных
потенциалов, кроме того, широко используется в более сложных
электрохимических исследованиях, позволяющих получить дан-
ные о стойкости сплавов, покрытий. Поэтому любому исследо-
вателю, занимающемуся ускоренными испытаниями, знание этих
методов необходимо.
Подготовка электродов. Размеры образцов для измерения
потенциалов могут быть различными, поверхности их необходи-
мо выбирать в пределах 0,2—10 ся?\ Крепить образцы следует
с помощью металлических зажимов непосредственно или после
— 119 —
предварительного армирования образцов в изоляционные мате-
риалы (стекло, (плексиглас, полистирол, тефлон и т. п.). Способы
крепления и армирования образцов для измерения электродных
потенциалов показаны на рис. 64. Армирование позволяет пол-
ностью избежать или значительно сократить применение раз-
личного рода замазок и устранить 'влияние торцов образца. Ра-
бочей поверхностью в этом случае является лишь свободная от
армировки часть образца. Готовые образцы шлифуются сначала
Рис. 64. Способы крепления и армирования различных образцов
для измерения, электродных потенциалов [I—3]>
ВЛ — ватерлиния; 1 — провод; 2 — стеклянная трубка; 3 — бакелит; 4 — образец;
5 —спай с образцом; 6 — текстолитовый упор: 7 — изолирующая прокладка (поли-
этилен, тефлон)
на грубой, «потом на тонкой наждачной бумаге. Образцы цвет-
ных металлов следует шлифовать на стеклянной шкурке. После
шлифовки, образцы обезжиривают с помощью органических рас-
творителей или спирта. Для устранения возможного влияния
органических растворителей, адсорбирующихся поверхностью
металла, образцы обрабатывают на чистом, сукне окисью хрома,
алюминия или магния, промывают дистиллированной водой
и высушивают. Промытые и высушенные образцы перед испыта-
нием выдерживают в эксикаторе определенное время—обычно
10—24,5.
. Электролитические ячейки. Измерять электродный потенци-
аД.йрж'НО при погружении в электролиты и в тонких слоях элек-
тролитов, Существует способ измерения электродных потенциа-
лов в адсорбционных слоях электролитов, однако он сложен
— 120 —
методически и пригоден только при специальных исследованиях^
когда интересуются поведением металлов в сухих атмосферах
[4, 5].
Для измерения 'потенциалов применяют простые стаканы
или более сложные сосуды [1] (рис. 65, а — в), а иногда и спе-
Рис. 65. Приборы для измерения потенциалов (а, б, в) и установка
для массовых измерений потенциалов (а) [1, 61
циальные установки (рис. 65, г). Последние применяют в то»
случае, если необходимо произвести большое количество изме-
рений одновременно (6]. На рис. 65, а показан сосуд, в котором
измерениежпроизводят шри непосредственном погружении образ-
ца в электролит. На образце, вмонтированном в специальный
тубус прибора, измерение проводят, как показано иа рис. 65, б.
. Носик измерительного '.ключа подводится к поверхности образ-
ца. При измерениях электродных потенциалов может быть пре-
дусмотрено размешивание электролита.
— 121 —
В некоторых случаях удобно» чтобы образец располагался
горизонтально; тогда применяют сосуд, показанный на рис. 65, в.
Аппаратура для измерения потенциалов усложняется, если
необходимо воспроизвести специальные условия, -например про-
извести измерения в присутствии различных газов или ® усло-
виях, имитирующих коррозию в естественной атмосфере, когда
на поверхности металлов вследствие перепада температур или
при выпадении дождя образуются тонкие, видимые слои элек-
тролитов.
Вис. 66. Схема установки для измерения элек-
тродных потенциалов металлов в тонких слоях
электролитов [7];
1 — электролитический ключ; 2 — гайка; 3 — штуцер;
4 — крышка; 5 — каломельный э-чектрод; € — водяной
термостат; 7 — камера-, 8 — насыщенный раствор
CaSO< для поддержания определенной влажности;
9 — электрод
Схема установки для измерения электродных потен пи а лов
металлов в тонких слоях электролитов приведена- на рис. 66;
в установке предусмотрена возможность -снятия поляризацион-
ных кривых [7]. Электрохимическая ячейка, состоящая из двух
металлических электродов' 9 размером 4—5 мм, вмонтирован-
ных в стеклянную обойму, помещается ® плексигласовую каме-
ру 7, -в которой с помощью насыщенного раствора CaSO4 8,
содержащего избыток соли, создается влажность, близкая
к 100%. Электроды укрепляются на крышке 4 камеры с 'Помо-
щью штуцеров 3 и гаек 2. На поверхность стеклянного держа-
теля диаметром 35 мм наносится тонкий слой эл&сгролита;
толщина его рассчитывается исходя из веса и объема капель,
наносимых из микробюретки, и составляет обычно 30—300 мкм.
Если на установке измеряют только потенциалы, тр образцы не
соединяются электролитическим ключом. Для большинства
— 122 —
электрохимических измерений следует предусмотреть термоста-
тирование, так как перепады температуры.могут вызвать ‘воз-
никновение диффузионных потенциалов. Для измерения в тон-
ких слоях это ©ажно еще и потому» что резкие перепады темпе-
ратуры могут вызвать конденсацию. В установке, приведенной
на рис. 66, плексигласовая камера и электрод сравнения 5 по-
мещаются в водяной термостат 6.
2
1/50
Рис. 67. Принципиальная схема вращающегося электрода
(настольного):
I — мотор; 2 — передаточные. шкивы; S ~ .-натяжной ролик;
4 — кронштейн; 5 — даращающийся электрод; 6 — подставка;
7 — плита; 8 — штанга
Электродные потенциалы измеряют не только на стационар-
ных электродах, ио и на вращающихся. В последнее -время вра-
щающиеся электроды применяют очень широко, так как они
дают возможность изучать ряд теоретических вопросов, а также
определить практически (важные характеристики металлов (см.
гл. II).
Конструкция вращающегося электрода (настольного), приме
няемого в- Институте электрохимии АН СССР, представлена
на рнс. 67. Электроду сообщается вращательное движение от
мотора 1 типа ДО-50. В качестве передаточного механизма
используется система шкивов 2, соединенных капроновой ни-
тью. Конструкция обеспечивает семь скоростей вращения от 30
— 123 — '
до 550 padjceK (300—5500 об]мин). При сборке такой установки
необходимо сохранять вертикальное расположение электрода 5,
давление натяжного ролика 3 на капроновую нить должно быть
равно 350—500 а.
Изменить скорость вращения электрода можно при помощи
более совершенного способа — путем применения регулятора
оборотов электродвигателя. Схема программного регулятора
оборотов электродвигателя разработана Е. С. Богачевым [8].
Она позволяет регулировать скорости двигателя от 2 рад!сек
v (20 об]мин) до номинальных оборотов. Модулированный свето-
Рис. 66. Схема измерения электродного потенциала вращаю-
щегося электрода при наложении поляризующего тока
вой поток подается на фотосопротивление, с которого снимают-
ся импульсы напряжения» частота которых зависит только от
скорости двигателя.
Для измерения потенциала на вращающемся электроде при
наложении внешнего тока поляризации пользуются схемой, при-
веденной на рис. 68. На рисунке видно, что электрический кон-
такт с исследуемым электродом осуществляется через скользя-
щий контакт 3, стержень 4 из графитированной меди и втулку 5
из изоляционного материала, в которую вкладывается образец,6.
В сосуд с электролитом 9 (помещается электролитический ключ 2
каломельного электрода 1. Для проведения поляризации поль-
зуются вспомогательным электродом 8, расположенным в от-
дельном сосуде и соединенным с рабочим пространством П-об-
разной трубкой 7, заполненной электролитом. Носик измери-
тельного ключа подводится непосредственно к нижней —
рабочей части вращающегося электрода.
Электроды сравнения. Для электрохимических измерений
необходимы электроды сравнения; наиболее распространен-
ный — каломельный электрод. Для сильнокислых и щелочных
— 124 —
растворов этот электрод непригоден из-за появления диффузи-
онных потенциалов, величину которых не всегда удается точно
определить. Для измерения электродных потенциалов в ней-
тральных растворах хлоридов удобен также хлорсеребряный
электрод (Ag, AgCl). Основной его недостаток — светочувстви-
тельность, поэтому шри работе с ним надо принять меры к за-
щите его от гарямого воздействия солнечных лучей. В кислых
растворах можно применять .водородный и хингидронный элек-
троды, в щелочных — окиснортутный (Hg, HgO), обратимый по
отношению к ОН*-ионам электрод.
Ниже приведены потенциалы (в вольтах) наиболее распро-
страненных стандартных электродов при 25°С (по водородной
шкале) (1):
Каломельный:
Hg, Hg2Cl2 (твердый). 0,1-й. КС1 . . 0,3337
Hg, Hg2Cl2 (твердый), 1,0-н. КС1 . . . 0,2800
Hg, Hg2Cl2 (твердый), КС1 (иасыщ.) . . 0,2415
Хлорсеребряный Ag, AgCl (твердый), 0,1-и. КС1 0,2900
Окиснортутный Hg, HgO, 0.1-и. NaOH . . . 0,1650
Хингидронный Pt, Н2, 0,1-н. НС1 .... 0,6990
Водородный Pt, Н2, 1,0-н. НС1 ...... 0.0000
Потенциал металла по отношению к водородному электроду
определяют путем алгебраического сложения по уравнению
9 н. = 'Ы + 9 *• <79)
где <рНв — потенциал металла по отношению к недородному
электроду;
<рСр — потенциал электрода сравнения;
<рх—потенциал' металла, измеренный по отношению к элек-
троду сравнения. Потенциал металла имеет положительный
знак, когда он присоединён к положительному полюсу измеря-
ющего прибора.
Потенциалы ’металлов обычно изучают ® течение суток, про-
водя измерения сначала через 1, 3, 5, 10, 15, 30 мин, потом через
1, 2, 3, 4, 5, 6 ч и, наконец, через 24 ч, В некоторых случаях опыт
длится 10 суток, тогда измерения производят один раз в сутки.
'Интерпретация результатов измерений. Предположим, что
в результате введения «в сплав легирующего компонента или
изменения состава электролита, или нанесения защитного сред-
ства потенциал металла в данной среде сдвинулся в положи-
тельную сторону (фг > <pi). Как правило, большое смещение I
потенциала в положительную сторону указывает на то, что сЮй£^
но уменьшилась скорость анодной реакции, и сплав запассйви: |
ровался; на рис. 69 видно, что кривая анодной поляризации »
ф’23 переместилась в положение Можно заранее предска-
зать, что такая система -в естествейных условиях эксплуатации
окажется более стойкой (ток коррозии уменьшится с 4 до И).
Необходимо, однако, -иметь в виду, что иногда (таше случаи
—125 — '
встречаются редко) смещение потенциала в положительную
сторону может быть обусловлено ускорением катодного процес-
са (катодная «кривая сместилась из положения в положение
<Р°В). В этих условиях скорость коррозии должна возрасти
(?-з > G), что можно довольно быстро установить кратковремен-
ными испытаниями на коррозию.
Смещение потенциала в отрицательную сторону чаще всего
обусловлено облегчением анодной реакции ионизации металла.
На рис. 69, б видно, что смещение потенциала в отрицательную
сторону явилось результатом смещения анодной поляризацион-
ной кривой из положения в положение Это Должно
насторожить исследователя, ибо может привести к увеличению
коррозии (t*3>4).
Иногда потенциал может сместиться в отрицательную сторо-
ну благодаря уменьшению скорости катодной реакции, напри-
мер лрн удалении окислителя из электролита. В этом случае
скорость коррозии, как это видно из рис. 69, б, должна умень-
шаться из-за смещения катодной поляризационной кривой из
положения <р°Л -в положение <р®В (is > *i). Отличить эти случаи
можно постановкой кратковременных коррозионных опытов
с целью определения величины коррозии.
Рис. 69. Схема, поясняющая причины измене-
ния электродного потенциала металла:
а — потенциал смещается в положительную сто-
рону благодаря торможению анодной реакции или
облегчению катодной; б — потенциал смещается в
отрицательную сторону благодаря облегчению
анодной реакции или торможению катодной
По величине измеряемого электродного потенциала можно
судить и о характере коррозионного процесса и установить, та-
ким образом, какая из электрохимических реакций определяет,
скорость 'коррозии. Это очень важно три выборе (обязательно
с учетом контролирующего фактора) ускоренного метода кор-
розионных испытаний/
| Если электродный потенциал близок к потенциалу предпола-
*, гаемого анодного процесса, который обычно «можно установить
< —126 —
с некоторым приближением, то это означает, что коррозионный
процесс контролируется скоростью катодной реакции. Если из-
। меренный электродный потенциал близок к 'потенциалу катодной
реакции (как правило, это реакция восстановления кислорода
или разряд ионов водорода), то это значит, что коррозионный
‘процесс контролируется скоростью анодной реакции.
Рис. 70. Зависимость электрод-
ного потенциала нержавеющей
стали 18-8 в напряженном (кри-
вая /) и ненапряженном (кри-
вая 2) состояниях от времени при
погружений в 42%-ный раствор
хлористого магния JW]
О 0.5 1 1,5 2 25 3 3.5 О
1д военени (пип)
Рис. 71. Зависимость электрод-
ного потенциала стали от вре-
мени в присутствии нитрита
натрия [1Ц:
I — 0; 2 — 0.1 г]дм\ 8 — 1,0 г(дл<?
Метод изучения электродных потенциалов очень полезен
для быстрой оценки способности сплавов восстанавливать пас-
сивное состояние при его нарушении. Для этой цели измеряют
потенциалы металлов при зачистке их поверхности с целью
удаления окисных слоев под раствором. Этот способ измерения
был разработан Г. Б. Кларк и Г. В. Акимовым [9].
Легкопассивирующиеся металлы быстро приобретают пер-
воначальное значение потенциала после прекращения зачистки»
а труднопассивирующиеся металлы — медленно. Закономер-
ности изменения потенциала во времени при зачистке, а также
величина сдвига потенциала могут в некотором отношении
характеризовать поведение металла в той или иной среде и,,
таким образом, позволяют избежать длительных испытаний.
Другим удачным примером использования метода измере-
ния потенциалов может служить работа Хора и Хайнера [10].
Авторы по кривым изменения потенциала во времени судили о
развитии процесса коррозионного растрескивания нержавею-
щих сталей. Для напряженных и ненапряженных образцов за-
кономерности изменения потенциала различны, а начало появ-
ления трещины четко фиксируется по появлению характерного
пика иа кривой 1 (рис. 70).
И. Л. Розенфельд и сотрудники использовали метод изме-
рения потенциалов для быстрой оценки защитных свойств инги-
биторов. Эффективные ингибиторы, как правило, смещают
—127 —
электродный потенциал в положительную сторону и при дости-
жений защитной концентрации это смещение довольно устойчи-
во. И во времени (рис. 71). Методом измерения потенциалов
можно, таким образом, довольно быстро из большого числа
соединений отобрать те, которые обладают высокими защитны-
ми свойствами.
Наглядный пример использования данных измерений потен-
циалов в зависимости от pH — диаграммы коррозионной устой-
чивости металлов, полученные Пурбе [12].
Можно привести еще ряд примеров использования метода
измерения потенциалов металлов, однако и так ясно, что при
Рис. 72. Схема компенсационной установки с реохордом
для измерения электродного потенциала металла [131:
ЛБ — реохорд; С — подвижный контакт; ЭС — электрод срав-
нения; Me — металлический образец; П — переключатель;
НЭ — нормальный элемент
разумном применении его и правильном истолковании резуль-
татов измерений в ряде случаев можно и ие проводить длитель-
ные коррозионные- испытания^— J,
Измерительная аппаратура. Для измерения электродных по-
тенциалов при отсутствии специальных приборов (потенциомет-
ров, электродных вольтметров) можно пользоваться простыми
установками, основанными на принципе компенсации. Такие
компенсационные схемы собирают при помощи реохорда или
двух сопротивлений и нормального элемента Вестона [13].
В установке с реохордом (рис. 72) исследуемый элемент при-
соединяется к компенсационной цепи так, чтобы положитель-
ный полюс ячейки был подключен навстречу положительному
полюсу батареи. Переключатель П включает в компенсацион-
ную цепь нормальный элемент НЭ\ передвижением подвижного
контакта вдоль проволоки А Б находят положение, при котором
стрелка гальванометра при замыкании цепи не отклоняется.
Затем подключают в измерительную схему' исследуемый эле-
мент и снова находят место компенсации на реохорде. По со-
отношению плечей реохорда в первом и во втором случа.е ком-
128 —
пенсации определяют потенциал измеряемого электрода фж
по уравнению
<ех = 1,018^ , (80)
°-н
где ах и ан— длина печей реохорда.
С помощью такой установки электродный потенциал может
быть измерен с точностью до 0,002 в. Точность измерений будет
зависеть от постоянства сопротивления проволоки по всей дли-
не реохорда и постоянства напряжения питающей батареи.
Схема компенсационной установки с двумя сопротивлениями
показана на рис. 73; в качестве сопротивлений могут быть взя-
ты магазины сопротивления
по 10000 ом каждый. Изме-
рения производят аналогич-
но описанным для схемы, с
реохордом, только вместо
длины печей определяют
величину сопротивлений Ri
и при компенсации. Ве-
личину потенциала опреде-
ляют по уравнению
^=1,018^.. (81)
Рис. 73. Схема компенсационной уста-
новки с двумя сопротивлениями для из-
мерения электродного потенциала
металла [13]:
Mt и Mz — магазины сопротивления; НЭ —
нормальный элемент; П — переключатель
Проще и удобнее при
измерении потенциалов
пользоваться специальны-
ми приборами — потеицио-
метрами и электронными
вольтметрами, которые выпускаются промышленностью. Для
этих же целей можно использовать и pH-метры.,
При измерений пбтевдол^в ‘Необждймо руководствоваться
следующим правилом: входное сопротивление прибора должно
превышать сопротивление ячейки, по крайней мере, на два по-
рядка.
Выбор того или иного потенциометра для измерений зависит
от сопротивления ячейки и скорости изменения измеряемой вели-
чины. При сравнительно небольших сопротивлениях в измеря-
емой ячейке (до 1 ком) можно применять компенсационные
потенциометры, которые позволяют получать значение потен-
циала непосредственным отсчетом по шкале. К приборам этого
типа относятся потенциометры ППТВ-1 со ступенями измерения
0,00001 в и Р-305 со ступенями измерения 0,000001 в.
Схемы приборов обычно приводятся в описаниях или инст-
рукциях, прилагаемых к ним. На рис. 74 приведена схема од-
ного из этих приборов, предназначенного для измерений по ме-
тоду компенсации,— потенциометра ППТВ-1. Для регулировки
9 Зак. 1338 — 129 —
рабочего тока используются три декадных сопротивления
Къ'Кз и реостат сопротивлением 17 ом. В цепи гальванометра
имеется кнопочный переключатель, позволяющий подключать
последовательно с гальванометром сопротивление 50 ком или за-
корачивать гальванометр для успокоения стрелки (КЗ). Компен-
Рис. 74. Схема высокоомного потенциометра ППТВ-1:
НЭ — нормальный элемент; X — исследуемая электрохимическая ячейка
сирующее напряжение в приборе можно изменять ступенями с
помощью декадных сопротивлений, по которым и производится
отсчет показаний.
Цепь рабочего тока этого потенциометра питается от вспо-
могательной батареи. Рабочий ток прибора равен 0,0001 а.
С помощью входящего в комплект делителя напряжения ДН-1
верхний предел измерения прибора (1,2 в) может быть расши-
рен. Точность измерения потенциометром ППТВ-1 для измеряе-
мых напряжений порядка 1 в составляет 0,03%.
При сопротивлениях системы большой величины, когда из-
мерения ведутся в сильно разбавленных растворах или исполь-
зуются длинные капилляры в соединительных ключах, а также
при измерениях на электродах очень малой величины или при
значительной поляризации электродов следует применять лам-
повые потенциометры или катодные вольтметры. В настоящее
время промышленностью выпущены потенциометры типа П-4.
П-5, П-6 с ламповыми усилителями ЛУ-2, а также ламповые
потенциометры ЛП-3, ЛП-5 и ЛП-58. Наилучший из них — пос-
ледняя модель ЛП-58 с входным сопротивлением порядка
1 Мом. Для измерения электродных потенциалов может быть
использован переносный автоматический компенсатор ЭСК-1 с
входным сопротивлением также 1 Мом. Этот прибор выпускает
завод геофизической аппаратуры в Барнауле.
— 130 —
Если при измерении потенциалов необходимо производить
непрерывные отсчеты в течение всего опыта, используют катод-
ные вольтметры; некоторые из них имеют большое входное со-
противление и могут употребляться в системах с большим со-
противлением (>1 Мом).
Мастерскими Центракадемснаба (Москва) выпущен опыт-
ный катодный вольтметр, который представляет собой лабора-
торный переносной прибор с непосредственным отсчетом из-
меряемых напряжений постоянного тока в пределах от 0 до
3 в при точности измерения 4% и входном сопротивлении не ме-
нее 10 Мом. Схема прибора приведена на рнс. 75. Питание при-
бора осуществляется от сети переменного тока. Схема прибора
состоит из двух основных частей: измерительного блока и блока
питания. Измерительный блок состоит из микроамперметра
М-292 на 50 мка с четырьмя переключаемыми сопротивлениями
и балансового усилителя постоянного тока с катодной нагруз-
кой. Усилитель собран на лампе 6Н8С, между .катодами кото-
рой включается микроамперметр с добавочным сопротивлением,
соответствующим выбранной шкале. Установка нуля произво-
дится при закороченной сетке входной лампы. В блоке питания
имеется двойная стабилизация напряжения.
Рис. 75. Схема катодного вольтметра, разработанного Центральным
конструкторским бюро Центракадемснаба АН СССР
Удобен в работе ламповый вольтметр типа ВК7-3 (А4-М2),
который при измерении напряжений постоянного тока в пре-
делах от 0,1 до 100 в имеет входное сопротивление 11 Мом\ точь
ность измерения при этом составляет 5% от верхнего предела
измерения. Питается прибор от сети переменного тока.
Для автоматической записи потенциалов используют само-
пишущие приборы. В последние годы промышленностью выпу-
щены, например, саморегистрирующие потенциометры типа
ЭПП-09, СПР н др. Так как непосредственное назначение этих
s*
-—-131 —
Рис. 76. Схема включе-
ния усилителя в одно-
точечный саморегистри-
рующий потенциометр
[15|. Пунктиром указано
место включения лампо-
вого усилителя:
Bxt — вход 1; Вх2 — вход
2; Вых — выход усилителя
приборов—автоматическая запись температуры при помощи
Термопар, то для электрохимических исследований их можно
применять лишь при включении на вход прибора усилителя.
Дело в том, что такие приборы имеют очень низкое сопротив-
ление на входе. В зависимости от сопротивления электрохими-
ческой ячейки конструкции усилителей могут быть различны.
Практические схемы подобных усилите-
лей можно найти в литературе [14]. По
данным работы [15], для автоматической
записи электрохимических потенциалов
в системах с небольшим внутренним со-
противлением может быть использован
усилитель, собранный на сдвоенном
триоде 6Н8; входное сопротивление та-
кого усилителя равно 5—6 Мом.
Для исследования систем с разбав
ленными электролитами и образцами
малой площади усилитель собирают на
лампах пальчиковой серии 2Э2П; вход-
ное сопротивление его 12—15 Мом. Для
исследования работы микроэлементов
при коррозии под тонкими слоями элек-
тролитов необходим усилитель с вход-
ным сопротивлением 1011 ом, собранный
на электрометрическом тетроде 1Э1П.
На рис. 76 приведена схема включения
усилителя в одноточечный саморегистри-
рующий потенциометр, и а рис. 77 — схе-
ма усилителя на электрометрическом
тетроде.
Существует и другой способ увеличе-
ния входного сопротивления самопишу-
щего потенциометра ЭПП-09, заключаю-
щийся в небольшом изменении схемы са-
мого прибора. В Институте физической химии АН СССР Ан-
дреевым и Руслановым предложено простое изменение схемы
этого прибора, которое обеспечивает входное сопротивление е
1 Мом. Состоит оно в том, что в усилителе ЭПП удаляется
входной трансформатор, вход усилителя переделывается (рис
78). Выпрямитель на правом триоде лампы Л2(6Н9С) заменяет-
ся двумя диодами ДГ-Ц27. Накал лампы Лj (6Н9С) питается от
отдельной обмотки накала IV. Усилитель подключается к изме-
рительной схеме экранированным двухжильным кабелем. От
входа прибора отключается конденсатор фильтра С$. Выводы
обмотки возбуждения вибропреобразователя отпаиваются от
цоколя и выводятся наружу через отверстия в верхней части
экрана преобразователя. У самописца заземляется • входная
клемма — (минус). Если двигатель РД-09 будет вращаться в
—132 —
обратном направлении, необходимо поменять местами концы,
идущие от двигателя к выходному трансформатору усилителя.
Для того чтобы верхний предел самописца был равен 100 мв,
.сопротивление измерительного моста заменяется на сопро-
тивление, наматываемое из манганиновой проволоки, равное
приблизительно 50 ом-, точная величина сопротивления подби-
рается экспериментально. Класс точности прибора после пере-
делки не изменяется (0,5). Для расширения пределов измерения
по току и напряжению к самопишущему потенциометру ЭПП-09
подключают выносной блок, в котором смонтированы переклю-
чатель и сопротивления. Схема этого блока показана на рис. 79.
Рис. 77. Схема усилителя на электрометрическом
тетроде [15]:
П — переключатель ’усилителя; ПР — саморегистрирую-
щий прибор
Для автоматической записи электродных потенциалов в
электрохимических ячейках может быть применен потенциометр
ЭППВ-51, в котором также произведено изменение схемы, пред-
ложенное Щепихиным. Минусовый провод датчика отсоединя-
ется от контакта 6 электромагнита и присоединяется параллель-
но высокоомному сопротивлению. Для расширения пределов
измерения потенциалов применен делитель, смонтированный в
выносном блоке. Схема входной цепи потенциометра ЭППВ-51,.
приспособленного на четыре поддиапазона (0—0.5; 0—1,0; 0—
1,5; 0—2,0 в), приведена на рис. 80.
Кроме описанных самопишущих приборов, можно применять
и многопредельный переносной микроампермилливольтметр
Н-373, который выпускает завод электроизмерительных прибо-
ров Краснодарского совнархоза. Внутри этого самописца име-
ется усилитель Ф-17/1. Действие прибора основано на фотоком-
пенсационном принципе измерения. Для измерения малых
токов (до 10“8 а) и малых напряжений (до 10~6 в) может приме-
няться самописец с фотокомпенсационным усилителем марки
Ф-18; этот прибор выпускает Ленинградский совнархоз. Фото-
компенсационные усилители типа Ф-117 выпускает завод «Ви-
братор »; онн дают усиление от 104 до 106.
~ 133 — <
Рис. 78. Схема, поясняющая способ изменения входа самопишущего
потенциометра ЭПП-09
Измеряемое Нили I
К ЭПП-09
Выносной Book
Рис. 80. Схема входной цепи потенцио-
метра ЭППВ-51, приспособленного на
четыре поддиапазона для измерения
потенциалов
Рис, 79, Схема выносного блока для расширения пределов
измерения потенциометра ЭПП-09
по току и напряжению
В некоторых лабораториях нашел применение электрометри-
ческий усилитель ЭМУ-3, выпускаемый заводом радиотехниче-
ской промышленности. Усилитель ЭМУ-3 имеет входное сопро-
тивление 10й ом, снабжен специальным выводом для подклю-
чения самопишущего прибора с малым сопротивлением. К элек-
трохимической ячейке подключается вход электрометрической
лампы (выносной каскад ЭМУ-3), второй электрод заземляет-
ся, При измерениях в ячейке с большим сопротивлением кон-
такт электрода с выносным каскадом ЭМУ-3 должен во избе-
жание различного рода наводок тщательно экранироваться, а
сама ячейка — экранироваться и заземляться. Усилитель ЭМУ-3
можно использовать и как катодный вольтметр; для этой цели
его подключают к любому чувствительному вольтметру.
При работе с приборами, имеющими большое входное со-
противление (108 ом и выше), следует избегать включения раз-
личных моторов, электромагнитов и генераторов в непосредст-
венной близости от установки.
2. СНЯТИЕ ПОЛЯРИЗАЦИОННЫХ КРИВЫХ
Метод снятия поляризационных кривых применительно к
коррозии впервые предложен Ю. Р. Эвансом и получил широ-
кое распространение в работах Брауна и Миерса. Г В. Аки-
мова и его учеников [1, 16—20].
i Этот метод позволяет количественно судить о том, с какой
скоростью протекают в среде, в которой предполагают эксплуа-
I тировать металл, электрохимические реакции, обусловливаю-
] щие коррозионный процесс. В настоящее время почти все тео-
| ретические и практические вопросы решают, применяя этот
j метод.
Метод заключается в изучении зависимости скорости элек-
трохимической реакции (ионизации металла или восстановле-
i ния водорода или кислорода) от потенциала электрода.
Снятие поляризационных кривых можно производить галь-
ваностатическим или потенциостатическим методом. В первом
случае через ячейку пропускают определенную постоянную
силу тока и изучают изменение потенциала, во втором — иссле-
дуемому электроду задают определенный потенциал и измеря-
ют силу тока.
/ Потенциостатический метод рекомендуется применять лишь
• в тех случаях, когда металл склонен переходить в пассивное
1 состояние.
Гальваностатический метод. При таком методе образец по-
ляризуется катодным или анодным током постоянной величины.
Образец выдерживают при данной плотности тока некоторое
время, после чего измеряют потенциал электрода. Поскольку
процесс сдвинут от равновесного состояния, требуется очень
— 136 —
длительное время, чтобы потенциал приобрел постояннее зна-
чение (сутки). Поэтому исследователь обычно ограничивается д
одной выдержкой (от 1 до 15 мин) при всех значениях плотно- к
сти тока. Наиболее распространенная выдержка 5 и 10 мин. ;
В некоторых случаях потенциалы измеряют по достижении
Рис. 81. Схема установки для снятия поляризационных кривых
в объемах электролитов [20]:
/< — электрод, поляризуемый катодно; А —• электрод, поляризуемый аиодно;
/7 9 НЭ — каломельные полуэлементы; КВ — клеммы для присоединения катод-
ного вольтметра; НЭ (нормальный элемент Вестона; М — мешалка с гид-
равлическим затвором; Г — шЛифы с кранами для ввода газов; П — отбор
проб электролита; ГЛ — переключатель для включения катодного вольтмет
ра в цепь катода или анода; ГЛ и П3 — переключатели для включения в из-
мерительную схему элемента Вестона (включается при э. д. с. >18)
электродом постоянного значения потенциала при данной плот- ]
ности тока.. Принятое в исследованиях время выдержки следует
оговаривать в статьях и отчетах, чтобы можно было сопоста-
влять результаты различных исследователей.
В зависимости от поставленных задач установки для снятия
поляризационных кривых могут быть различными. Принципи-
альная схема установки для снятия поляризационных кривых в
объемах электролитов приведена на рис. 8L
— 137 —
Поляризационные кривые, по которым хотят определить ха-
рактер и скорость коррозионного процесса, протекающего в ат-
мосферных условиях, снимают в тонких слоях электролитов
Методы снятия поляризационных кривых в тонких слоях элек-
тролитов разработаны и описаны И. Л. Розенфельдом и
Т. И. Павлуцкой (7]. На рис. 82 приведена схема прибора для
снятия поляризационных кривых в тонких слоях электролитов
Рис. 82. Прибор для снятия анодных и катод-
ных поляризационных кривых в тонких слоях
электролитов (21J:
1 — стеклянный держатель; 2 — металлический
электрод; 3 — электролитический ключ для изме-
рения потенциала; 4 ~~ сосуд; 5 — электролитиче-
ский ключ для поляризации; 6 — пленка электролита
в чистой атмосфере; для снятия поляризационных кривых в
атмосферах любого заданного состава б крышке прибора преду-.
сматриваются отверстия для ввода и вывода газов.
Поляризация металлического электрода 2 (см. рис. 82) про-
изводится от вспомогательного платинового электрода через
четырехрожковый электролитический ключ 5, устанавливаю-
щийся иа стеклянный держатель /, в который вмонтирован об-
разец. Электролитический ключ 3 для измерения потенциала
^подводится в центр образца с нижней его стороны.
Как показали опыты [22], омическим падением потенциала
в пленках толщиной 150—300 мкм можно пренебречь, если в ка-
138-
честве электролита применять растворы концентрацией не ниже
0,1-н., а диаметр стеклянного держателя чтобы не превышал
35 мм. Для таких электролитов можно пользоваться Электро-
дом, изображенным на рис. 66. Измерения в более разбавлен-
ных электролитах следует проводить с помощью электрода,
показанного на рис. 82. В этом случае капилляр измерительного
ключа подводится непосредственно в центр металлического об-
разца н таким образом исключается возможное омическое па-
дение потенциала в тонком слое электролита.
При снятии поляризационных кривых в объеме электролита
при сравнительно малом сопротивлении его поляризация посто-
янной величиной тока, регулирование тока, а также измерение
величины электродного потенциала не представляют трудно-
стей. В тонких слоях электролитов, имитирующих атмосферные
условия, электролитическая ячейка обладает значительным со-
противлением, поэтому для регулирования налагаемой плотно-
сти тока необходимо пользоваться высокоомными сопротивле-
ниями и источниками питания с большим напряжением. Для из-
мерения потенциала металлов под тонкими слоями следует
применять потенциометры с большим входным сопротивлением
(ЛП-58, ВК7-3, ЭМУ-3).
- При выборе плотностей тока для поляризации следует учесть
характер коррозионной среды н металла.. Поляризующий ток
должен быть всегда больше тока саморастворения^ иначе нель-
зя заметно сдвинуть потенциал от стационарного .значения.
Для процессов коррозии, протекающих с водородной^поляриза-
цией (кислые среды), плотность поляризующего тока определя-
ется миллиамперами (от 0,25 до 5—10 ма/см2)-, для процессов,
протекающих с кислородной деполяризацией, плотность тока
составляет микроамперы (от 1 до 1000 мка!см2). Предельный
диффузионный ток по кислороду в неразмешиваемых электро-
литах невелик (15—30 жкй/см2), поэтому в таких случаях сле-
дует получать возможно большее число точек на катодной
поляризационной кривой в интервале малых плотностей тока
(от 1 до 25 мка!см2). Вообще, снимая кривые в нейтральных
электролитах, необходимо по возможности получать все три
участка кривой, характерных для реакции восстановления кис-
лорода, процессов диффузии н реакции разряда ионов водоро-
да. Для этой цели обычно достаточно довести величину потен-
циала металла при катодной поляризации до (—1,0) -5- (—1,2) в
по водородному электроду.»
При снятии анодной поляризационной кривой необходимо
сдвинуть потенциал от стационарного до потенциала + 1,2 —
+ 1,5 в, что даст возможность изучить поведений металла в ак-
тивном и пассивном состоянии и в состоянии перепассивации
или пробоя.
В последнее время получил широкое распространение по-
тенциостатический метод.
— 139 —
Потенциостатический метод изучения кинетики электрохими-
ческих реакций отличается от гальваностатического тем, что при
нем поддерживается постоянным не ток, а потенциал, в зависи-
мости от которого и изучается изменение тока. Потенциоста-
• тический метод имеет преимущество перед гальваностатиче-
ским, поскольку позволяет изучить зависимость скорости раст-
ворения от потенциала в широкой области потенциалов, в том
числе и в области, соответствующей переходу металла из ак-
. тивного состояния в пассивное, и наоборотА При гальваноста-
» тическом же методе, в котором постоянная плотность тока под-
Рис. 83. Блок-схема потенцио-
статического регулирования по-
тенциала электрода»® электро-
химической ячейке [23]
П — переключатель; К — иссле-
дуемый электрод; А — вспомога-
тельный электрод; ЭС — электрод
сравнения
держивается с помощью высоких
сопротивлений, наиболее инте-
ресная область потенциалов, в
которой металл переходит из ак-
тивного состояния в пассивное»
не выявляется.
Постоянное значение потен-
циала устанавливают с помощью
специального прибора — потен-
циостата. Конструкции потенцио-
статов различны. В Институте
физической химии АН СССР
М. Н. Фокин и А. Ф. Виноградов
[23] разработали несколько мо-
делей электронного потенциоста -
та. Блок-схема потенциостатиче-
ского регулирования потенциала
рабочего электрода в электрохи-
мической ячейке и принципиальная схема регулирующего блока
потенциостата третьей модели Института физической химии АН
СССР приведены на рис. 83 и 84 [23]. Регулирование системы
(см. рис. 83) заключается в поддержании постоянного перепада
потенциалов между исследуемым электродом К и электродом
сравнения ЭС, носик которого помещается в электролит в непо-
средственной близости от рабочего электрода. Постоянное зна-
чение потенциала на клеммах электрохимической ячейки обыч-
но не создается, так как в регулируемый объект в этом случае
входят две переменные величины -— поляризация вспомогатель-
ного электрода А и омическое падение напряжения в электро-
лите. Разность потенциалов электродов К и ЭС электролитиче-
ской ванны 1 сравнивают с заданным напряжением блока ком-
пенсации напряжения 3. Разность А<р — U — Е подается на вход
регулирующего блока 4, который регулирует ток в цепи элек-
тродов А и К электролитической ванны. Блок 5 — блок питания
регулирующего блока и источник автоматически регулируемой
составляющей тока, проходящего через ванну. Для измерения
тока в цепи электролитической ванны служит многопредельный
миллиамперметр с нулем посередине.
— 140 —
Для расширения диапазона изменения тока через ванну в
сторону больших значений применяется блок 6, создающий до-
полнительную, автоматически регулируемую составляющую
тока. Направление этого тока меняется переключателем П. Это
дает возможность использовать регулирующее устройство при
малых токах без захода в область малой крутизны характери-
стики выходной лампы регулирующего блока, а также прохо-
дить через нуль без снижения крутизны характеристики авто-
матической регулировки.
Рис. 84. Принципиальная схема регулирующего блока потенциостата [23]
Напряжение Е контролируется катодным вольтметром, на-
пример ЭКВ-3. Малые изменения на входе регулирующего бло-
ка приводят к соответствующему изменению тока через ваину
так, чтобы E—>U. Каждому заданному напряжению U от блока
3 соответствует значение тока / через ванну.
Регулирующий блок потенциостата (см. рис. 84) состоит из
усилителя постоянного напряжения с коэффициентом усиления
К и выходной лампы для регулировки тока с крутизной S. Та-
ким образом, ток, текущий через ванну,
I = A<pKS. (82)
Диапазон изменения тока, проходящего через ванну, опре-
деляется выходной лампой регулирующего блока. При этом
имеется в виду, что усилитель постоянного напряжения линеен
до напряжения на выходе, соответствующего максимальному
сеточному напряжению выходной лампы. При постоянных зна-
— 141 —
чениях К и S каждому значению I соответствует определенное
значение Д<р:
(83)
До
Абсолютное значение Дф тем меньше, чем выше коэффици-
ент усиления и крутизна выходной лампы. При достаточно
больших значениях К и S с определенной точностью можно
считать Е == U. В этом случае введенный в блок-схему катод-
ный вольтметр 2 может быть исключен, а отсчет напряжения
может производиться по шкале обычного вольтметра, введен-
ного в блок компенсации напряжения (БКН).
Регулирующий блок питается от отдельного блока пита-
ния— электронно-стабилизированного выпрямителя. БКН слу-
жит для установления определенного потенциала на одном из
электродов ванны. Диапазон потенциалов от БКН выбран рав-
ным ±2 в.
Потенциостат обеспечивает длительное устойчивое регули-
рование. Время срабатывания регулирующего устройства (ско-
рость регулирования) составляет 0,04 сек. Точность регулиро-
вания, соизмеримая с точностью измерительного катодного
вольтметра, составляет ±5 мв во всем диапазоне потенциалов.
Входное сопротивление потенциостата равно 20 Мом, благода-
ря чему ток поляризации электрода сравнения не превышает
10“7, а. При работе потенциостата замеряются токи от 1 мка
до 60 ма.
j В экспериментальной мастерской электронных приборов
j Научно-исследовательского физико-химического института
?им. Л. Я. Карпова разработан потенциостат, в котором для под-
держания постоянства потенциала рабочего электрода приме-
нен усилитель постоянного тока с кондуктивными связями меж-
ду каскадами {24]. Принципиальные схемы усилителя и преоб-
разователя катодного вольтметра приведены на рис. 85, а и б.
Первый каскад усилителя (см. рис. 85, а) собран по схеме па-
раллельного баланса, второй — по схеме вычитателя, третий
каскад является однотактным усилителем напряжения и чет-
вертый— усилителем мощности. В первом каскаде (лампа Л1)
используется двойной триод 6Н2П, отличающийся сравнительно
небольшими сеточными токами. На первый вход усилителя по-
дается напряжение от электрода сравнения, а на второй—на-
пряжение от источников эталонного напряжения.
Напряжения на входах усилителя могут принимать значения
от —3 до 4-3 в, отличаясь одно от другого на несколько милли-
вольт. В катодах двух первых каскадов усилителя имеются
довольно большие сопротивления Пу, #2 и создающие отри-
цательную обратную связь только по среднеарифметическому
напряжению на входах усилителя. Переключатель сопротивле-
ний Пу выполняет роль регулятора усиления.. Переменное со-
— 142 —
противление служит для грубой балансировки усилителя, а
/?5 — для плавной. Анодная цепь лампы третьего каскада пи-
тается от стабилизированного .источника мощностью +.700 в,
а катод подключен через газовый стабилизатор Л5 к; источнику
а
Рис. 85. Принципиальные схемы усилителя (а) и преобразователя (б)
потенциостата Научно-исследовательского физико-химического института
им. Л. Я- Карпова [24]
мощностью —700 в. Такое подключение обеспечивает необхо-
димое изменение напряжения на аноде этой лампы в пределах
—350 ч- +550 в.
В выходном каскаде усилителя лампа Л6 работает в режиме
катодного повторителя с отдельным источником питания эк-
ранной сетки. Через лампу Л7 на выход усилителя поступает ток
--143 —
30—40 ма отрицательного направления от источника с напря-
жением —750 в.
Газовый стабилизатор Л& поддерживает постоянным напря-
жение на экранной сетке^этой лампы, благодаря чему ее ток не-
значительно изменяется при изменении напряжения на ее ано-
де. Выходной ток усилителя, равный разности токов ламп Л6 и
Л7, изменяется за счет изменения тока лампы Л6. При равенст-
ве токов этих ламп напряжение на выходе усилителя равно
нулю.
Преобразователь' (см. рис. 85, б) катодного вольтметра,
контролирующего потенциал рабочего электрода, является
усилителем постоянного тока с двойным преобразованием изме-
ряемого напряжения и 100%-ной отрицательной обратной
связью по постоянному току. Он состоит из механического ви-
бропреобразователя, усилителя переменного тока, фазового
детектора и блока смешения шкал. Усилитель имеет два кас-
када усиления напряжения (лампа и каскад усиления мощ-
ности (лампа Л2). В качестве фазового детектора используется
кольцевой балансный модулятор (лампы и Л4). Синхрон-
ное напряжение на модулятор подается от обмотки силового
трансформатора, помещенного в силовом блоке. Переключатель
изменяет количество последовательно соединенных элемен-
тов в цепи обратной связи и полярность включения этих эле-
ментов.
Время установления заданного потенциала в указанном
потенциостате 2-10“4 сек при диапазонах тока +50-5-—30 ма
и напряжения +500 ч-----300 в при точности поддержания по-
тенциала ±10 ме.
В настоящее время Центральная лаборатория автоматики
Госкомитета по черной и цветной металлургии выпускает для
исследовательских целей потенциостат ППЭ-1, поддерживаю-
щий потенциалы от —8 до +8 в с точностью ±2 мв при времени
установления заданного потенциала 10“4 сек и диапазоне токов
поляризации от ±0.01 мка до ±200 ма.
В зарубежной практике для электрохимических и металло-
графических исследований разработано большое число различ-
ных схем потенциостатов [25, 26].
Преимущества потенциостатического метода снятия поляри-
зационных кривых хорошо видны на примере исследования за-
висимости скорости анодного растворения различно термообра-
ботанной стали 1Х13Н4Г9 в 10%-ной H2SO4 при 20°С [23]. На
рнс. 86 пунктиром показаны две области неустойчивости элек-
тродного потенциала, в которых не удается обнаружить раз-
личия в поведении двух одинаковых по составу сплавов, но ко-
торые отличаются по структуре (кривые 1 и 2, снятые гальва-
ностатическим методом). На поляризационных кривых 3 и 4 для
тех же сплавов, снятых потенциостатическим методом, можно
видеть отчетливое влияние структуры, проявляющееся в обла-
—144 —
сти неустойчивости потенциала. Преимущества потенциостати-
ческого метода при исследовании явлений пассивности очевид-
ны, особенно в интервале потенциалов, соответствующих состо-
янию пассивации (<р “ 4-0,154-1,3 в) и состоянию
перепассивации стали (<р == 4-1,35-*- 4-1,8 в).
Использование поляризационных кривых для определения
скорости коррозии. Поляризационные кривые дают не только
ценные сведения о характере коррозионного процесса, но в ряде
случаев позволяют количественно рассчитать его абсолютную
скорость.
Рис. 86. Зависимость скорости анод-
ного растворения стали 1Х13Н4Г9
после различной термообработки от
электродного потенциала в 10 %-ном
растворе H2SO4 при 20° С {23]:
1, 3 ~ закалка + отпуск при 650° С 24 ч
Рис. 87. Схема, поясняющая спо-
соб расчета скорости коррозии по
экстраполяции поляризационных
кривых
Было предложено много способов использования поляризаци-
онных кривых для определения скорости коррозии. Наиболее про-
стой из них — метод экстраполяции поляризационной кривой до
стационарного потенциала. Если скорость электродного процесса
контролируется скоростью электрохимической реакций, напри-
мер скоростью разряда ионов водорода или ионизации металла,
то в полулогарифмических координатах зависимость потенциала
от логарифма плотности тока выражается прямой линией. Экст-
раполируя эти прямые до значения стационарного потенциала,
т. е. потенциала металла при отсутствии внешнего тока, получа-
ют значение тока коррозии. Так, например, получив поляризаци-
онные кривые для одного и того же металла в двух- разных
электролитах (/ и 2), можно с достаточным основанием утвер-
10 Зак. 1338 — 145---
ждать, что во втором электролите скорость коррозии будет на не-
сколько порядков ниже, чем в первом (рис. 87).
Этот метод применим в тех случаях, когда не наблюдаются
явления пассивности; кроме того, он предполагает равномерное
растворение металла по всей поверхности. В некоторых средах
(H2SO4, НС1), являющихся активными электролитами, это усло-
вие выполняется.
На этом же принципе основан метод расчета скорости корро-
зии по уравнению, описанному Штерном [27]:
Рис. 88. Схема, поясняю-
щая, что скорость кор-
розии в широком интер-
вале потенциалов опре-
деляется величиной диф-
фузионного тока
1<3
(84)
где р — наклон поляризационной кривой
при соблюдении зависимости
Тафеля;
i0 — ток обмена для катодного про-
цесса.
В тех случаях, когда скорость корро-
зии контролируется кинетикой катодной
реакции, которая в свою очередь опреде-
ляется скоростью диффузии реагирующих
на катоде частиц (например, кислорода),
она, как это видно из рис. 88, в широком
интервале потенциалов определяется ве-
личиной предельного диффузионного
тока.
Поэтому для расчета скорости корро-
зии достаточно определить для данных условий эксплуатации
изделия величину предельного диффузионного тока.
Другой метод измерения скорости коррозии по поляризацион-
ным кривым основан на соотношении, выведенном в работе Пир-
сона [28]:
*КОр —'
гагк
4» + 4с
(85)
где /кор величина коррозионного тока;
tK — величина катодного поляризационного тока, при кото-
рой скорость анодного процесса оказывается равной
нулю;
«а — величина анодного поляризационного тока, при кото-
рой скорость катодного процесса оказывается равной
нулю.
Анализ поляризационных диаграмм для двуэлектродной си-
стемы показывает, что если то ZK0P « гк, и что если
то гКОр~*а (рис. 89).
Иными словами, для металлов, процесс коррозий которых
контролируется преимущественно катодной реакцией, коррози-
-—146—- <
онный ток близок к току1, необходимому для того, чтобы .сме-
стить стационарный потенциал металла до равновесного'потен-'
циала анодной реакции в данной среде (is). Для металлов, про-
цесс коррозии которых контролируется аиодной реакцией,
коррозионный ток близок к току, необходимому для смещения
стационарного потенциала до равновесного потенциала катодной
реакции в данной среде (iK). Для процессов коррозии, протекаю-
щих со смешанным контролем, коррозионный ток в зависимости
от доли контроля той или иной реакции будет находиться ближе
к га или гк.
Использование соотношения Пирсона для расчета скорости
коррозии основано на утверждении, что при достижении вслед-
Рис. 89. Схема, поясняющая метод расчета скоро-
сти коррозии по перегибам иа поляризационных
кривых (28]:
а ~ *кор”*а‘ б *корта/"к
ствие поляризации потенциалов равновесных реакций для анод-
ного и катодного процессов в данной среде <р° и ф® на поляри-
зационных кривых наблюдается излом, что позволяет опреде-
лить необходимые для расчета значения t'a и iK.
В ряде случаев эти изломы фиксировались довольно четко
и по ним удалось рассчитать скорость коррозии. Так, например,
Швердфегер и Мак Дорман [29] снимали поляризационные ха-
рактеристики со стальных образцов в различных почвах и по
изломам на кривых рассчитывали скорость коррозии. Получас-
лось удовлетворительное совпадение расчетных и эксперимен-
тальных данных (4%). При достижении потенциала, соответст-
вующего току i&, работа -микроэлементов прекращалась и сталь
полностью защищалась от коррозии.
Шашел и Марш [30] показали, что излом на катодной поля-
ризационной кривой, полученной в 3%-ном растворе хлористого
J Правильнее было бы в данном случае говорить о токе, при котором
скорость анодного растворения оказывается равной току обмена, ибо при
этом поля-риза1ционном токе достигается равновесный потенциал -анодного
процесса в данных условиях или, как иногда говорят, потенциал анодных
участков в разомкнутом состоянии.
10* — 147 —
натрия, близок к току коррозии, хотя и не совпадает с ним пол-
ностью.
Поскольку в уравнении (85) имеется в виду, что при токах
Поляризации 1а и «к работа микроэлементов прекращается, необ-
ходимо быть уверенным в том, что это условие выполняется.
Осциллографические поляризационные измерения на вращаю-
щемся электроде показали, что при достижении потенциала,
соответствующего току ia и гк, работа локальных элементов на
поверхности металла подавляется.
Не всегда изломы на кривых, необходимые для определения
4 и /к, выявляются четко, однако несомненно, что в тех случаях,
когда они хорошо видны, этот метод может быть .использован
для расчета скорости коррозии.
Скорость коррозии может быть также рассчитана по поля-
ризационному сопротивлению . Штерн и^Джиэри [31], ана-
лизируя поведение металлов в кислых средах, пришли к выводу,
что между скоростью коррозии и поляризационными характери-
стиками должно существовать следующее соотношение:
__РаРк zgg\
Д/ 2, Згкор (Ра -р Рк)
Лад
где —--------поляризационное сопротивление;
Д«
ра и — наклон анодной и катодной-поляризационных кри-
вых на участках, где наблюдается тафелевская
зависимость.
Из этого уравнения следует, что графическое изображение
в лографимических координатах зависимости коррозионного то-
ка от поляризационного сопротивления должно давать прямые
линии с наклоном, равным —L Поскольку это уравнение опре-
деляет соотношение между коррозией и поляризационным сопро-
тивлением вблизи стационарного потенциала, поляризация не
должна превышать 10 мв.
„ -Дф
Расчет, выполненный для различных значении — , позволил
получить прямые, которые удовлетворительно соответствовали’
экспериментальным результатам, полученным Эваисом и Келер
[32] при изучении коррозии алюминия в различных кислотах,
в также Швердфегером и Монуэл [33] для стали в 3%-ном раст-
воре хлористого натрия (рис. 90).
Хотя для других, систем и наблюдались отклонения, однако
•несомненно,, что этот метод расчета, скорости коррозии перспек-
тивный. Метод определения поляризационного сопротивления
предложен для случая, когда скорость коррозионного процесса
определяется скоростью электрохимической реакции; по мнению
Штерна, прямые линии на диаграммах должны получаться и
¥дгда‘,г когда- процесс коррозии контролируется диффузией.
—148 -т
Поляризационные кривые для количественных расчетов ско-
рости коррозии могут быть использованы также для качествен-
ной оценки влияния того или иного фактора.
В качестве примеров, иллюстрирующих возможности изуче-
ния метода электрохимических реакций, рассмотрим некоторые
кривые катодной и анодной поляризации. На рис. 31 (см. гл. II)
приведены кривые катодной поляризации железа в атмосферах,
содержащих сернистый ангидрид.
Из рисунка видно, что введение
SO2 сдвигает катодные кривые в
область более положительных по-
тенциалов, иными словами, увели-
чивается скорость катодной реак-
Плотносто том I, hm/chz
Рис. 91. Влияние характера
аииоиа на анодное растворе- ,
иие железа (20]:
1 — 1.0-н. раствор Na2SO« + 0,1-н.
раствор NaCl; 2а — 0,1-н. раствор
Na2SO4; 26 — 0,1-н. раствор NaCl +
0,1-н. раствор Na2SO5; 3 — За —
0,1-н. раствор NaCl; 36 — 0,1-нг
раствор NaCl 4- ОЛ-н. раствор
NaNOs; 4 — 0,1-н. раствор NaNO3;
5 — 0,1-н. раствор NaCl + 0,1-н.
раствор NaOH; 6 — 0,1-н. раствор
’ а/спг
о-a л.-5
Рис. 90. Зависимость скорости
коррозии от поляризационного
сопротивления £32, 33]:
1 _ ₽к-со, ра~0,12; 2-ра -
= О-.Эб; ₽к г= 0,06; с — алюминий
в различных кислотах; б — сталь
в 3%-ном растворе NaCl NaOH
ции. Зная, что в атмосферных условиях процесс контролируется
в основном катодной реакцией, можно предвидеть, что скорость
коррозии железа в присутствии SO2 резко возрастет.
Анодные поляризационные кривые для железа, иллюстрирую-
щие агрессивность различных ионов по отношению к железу,
приведены на рис. 91. Чем ниже- расположена кривая анодной
поляризации, тем соответственно меньше агрессивность введен-
ного иона. Кривая 6 (рис. 91) для 0,1-и. раствора NaOH соот-
ветствует устойчивому пассивному состоянию железа, кривая 5
характерна для неустойчивого состояния, кривые /—4 соответ-
ствуют растворам, в которых будет происходить растворение
металла. Из приведенных кривых можно заключить, например,
что по отношению к железу сульфаты действуют так же, как
— 149 —
и растворы хлорида, а также предположить, какие добавки ока-
жутся наиболее полезными для подавления вредного влияния
иоиов хлора.
3. ПОЛУЧЕНИЕ КРИВЫХ ЗАРЯЖЕНИЯ
Метод снятия кривых заряжения впервые был разработан
и применен советскими учеными А. Н. Фрумкиным, А. И. Шлы-
гиным.Д34]-для-изучения электрохимической кинетики. Кривые
заряжения, как показали наши работы [35, 36], могут быть с ус-
пехо^жпоХ'Б^бвйны'-к^при коррозионных исследованиях. Метод
заключается в наложении тока постоянной величины на электрод
и из'Мерении-потенциал'а во времени. Измерение потенциала при
медленных его измен^иях может производиться обычным пу-
тем, при быстрых же-^зменениях потенциал должен регистриро-
ваться автоматически. |
По характеру кр^в^х заряжения можно судить о процессах,
протекающих на электроде; изменение потенциала с увеличени-
ем количества пропущенного электричества в процессе опыта
свидетельствует о том, .кто, помимо простого заряжения электро-
да, возникает какбй-то|новый процесс, например адсорбция раз-в
личны-х-ионов,-разрушение или образование окисных пленок и т. п.*
Кривые заряжения могут сниматься при наложении анодно-
го и катодного тока. Этот метод исключает длительное протека-
ние электрохимического процесса на исследуемом электроде при
постоянном потенциале, так как последний непрерывно меняется
в зависимости от количества пропущенного электричества.
Метод снятия кривых заряжения применили И. Л. Розен-
фельд и В. П. Максимчук [36] для исследования устойчивости
пассивного состояния нержавеющих сталей в электролитах, со-
держащих различные анионы (Cl~, SO|~, NO7 и т. д.), а так-
же при изучении влияния легирующих добавок. Устойчивость
пассивного состояния определяется по кривым заряжения. На
рис. 92;“ й—в" приведены кривые заряжения нержавеющей стали
Х18Н9Т, полученные в электролитах различного состава при
плотности тока 2 mkoJcm\ Если в чистом хлориде (рис. 92, а)
потенциал стали претерпевает через 1,5 ч в среднем 12 колеба-
ний в минуту в пределах 0,55—0,95 в, а в смеси хлорида с суль-
фатом (1:1) 2—3 колебания в минуту в пределах 0,65—1,25 &
(рис. 92, б), то при 10-кратном содержании сульфата частота
колебаний равнялась нулю; сталь переходит в пассивное состоя-
ние и ее потенциал устойчив (рис. 92, в).
Таким образом, наблюдающиеся колебания потенциала сви-
детельствуют о неустойчивости пассивного состояния; чем боль-
ше частота колебаний в единицу времени, тем меиее устойчив
металл в данных условиях. На этом принципе авторы [36] раз-
работали быстрый метод определения склонности Нержавеющих
сталей к питтинговой коррозии.
— 150 —
Been я. мин
а
б
Рис. 92. Кривые заряжения нержа-
веющей стали Х18Н9Т, полученные в
электролитах различного состава [36]
при г'а = 2 ллка{сл^-.
а — 0,1-н. раствор NaCl; б — 0,1-й. рас-
твор NajzSO^ + 0,1-я. раствор NaCl;. в ~
0,1-н. раствор NaCl + 1,0-н. раствор
NasSO4
— 151 —
Кривые заряжения могут быть использованы и при опреде-
лении влияния легирующих элементов на устойчивость нержаве-
ющих сталей. При отсутствии ионов, активирующих поверхность,
или при высокой концентрации легирующих элементов, обеспе-
чивающих устойчивое пассивное состояние, на кривых заряжения
—не обнаруживается периодических колебаний потенциала*.
Кривые заряжения с успехом могут быть применены и при
изучении катодных реакций, обусловливающих коррозионный
процесс. Этот метод пригоден в тех случаях, когда процессы
коррозии протекают с катодным контролем. Мы использовали
Рис. 93. Кривые заряжения медного ка-
тода, снятые в 0,1-н. растворе NaCl
при 1 = 140 мка/см2 ,[35]:
1 — объем электролита: 2 —• относительная
влажность воздуха И 98%, б- = 160 мкм;
5 — # -= 98%. б = 130 мкм; 4 — Н 76%,
б = 160 мкм; 5 — Н = 66%, б = 160 мкм
этот метод для изучения вли-
яния относительной влаж-
ности воздуха на скорость
протекания реакции восста-
новления кислорода. Было
установлено, что скорость
катодного процесса в тонком
слое электролита увеличи-
вается с уменьшением отно-
сительной влажности окру-
жающего воздуха. На рис. 93
приведены кривые заряже-
ния, снятые на меди в тонких
слоях 0,1-н. раствора хлорис-
того натрия при различной
относительной влажности
воздуха, подтверждающие
установленную закономер-
ность. Диаграмма показывает, что по мере уменьшения относи-
тельной влажности воздуха кривые сдвигаются в область более
положительных значений потенциалов.
Учитывая закономерности, установленные в работе [35], мож-
но было предположить, что ускорение катодного процесса в от-
носительно сухих атмосферах обусловлено уменьшением в про-
цессе высыхания толщины пленки. Однако дополнительные опы-
ты показали, что если учесть изменение толщины пленки за
счет высыхания при Я = 66% (пленка уменьшается на 50 мкм}
и снять кривую заряжения при влажности 98% в пленке элек-
тролита той же толщины (кривая <?), то, как видно на рисунке,
эта кривая не совпадает с кривой 5, снятой при влажности 66%
и толщине пленки 160 мкм. Кривая 5 сдвинута на 200 мв в об-
ласть более положительных потенциалов. Следовательно, сдвиг
кривых в область положительных -значений потенциалов при
уменьшении влажности окружающего воздуха происходит не
1 Применение кривых -заряжения и методика их получения для иссле-
дования пассивного состояния н -склонности и точечной коррозии нержаве-
ющих сталей будут изложены при описании методов определения склонно-
сти сплавов к точечной коррознн.
— 152 —
столько за счет уменьшения толщины слоя электролита, сколько
за счет обнаруженного авторами эффекта саморазмешивания
вследствие изменения поверхностного натяжения при испарении,
приводящего к увеличению скорости кислородной деполяриза-
ции. Таким образом, используя этот метод, можно за относитель-
но короткий промежуток времени предсказать поведение металла
в той или иной среде.
Кривые заряжения можно строить в координатах потенци-
ал — время или потенциал — количество пропущенного электри-
чества.
4. ИЗМЕРЕНИЕ СИЛЫ ТОКА,
ВОЗНИКАЮЩЕГО МЕЖДУ ДВУМЯ ЭЛЕКТРОДАМИ
Метод измерения тока, возникающего между двумя электро-
дами, применяется часто для моделирования коррозионных эле-
ментов, изучения контактной и щелевой коррозии, влияния аэра-
ции, определения эффективности электрохимической защиты, за-
щитных свойств покрытий и т. д.
При массовых испытаниях желательно проводить опыты с
большим количеством коррозионных пар (необходимости в боль-
шом количестве измерительных приборов иет). В то же время
важно, чтобы ток одной пары в процессе измерения не оказывал
—153 —
влияния на работу другой пары электродов. Необходимо также
исключить влияние на результаты измерений внутреннего сопро •
тнвления прибора, которое меняется при переходе от одной шка-
лы прибора к другой. Чтобы исключить все погрешности, при-
меняют разнообразные схемы.
На рис. 94 представлена установка, разработанная И. Л. Ро-
зенфельдом и И. С. Даниловым для измерения силы тока корро
знойных пар. Один электрод от каждой пары постоянно присо-
Рис. 95. Конструкция штекера (а) и способ соедине-
ния штекера с клеммой (б):
I — изолирующий иакокечник; 2 — наконечник;
3 ~ штифт; 4 — контакт; 5 — пружина; 6 — изолятор;
7 — контактная втулка; 8 — контакт, к которому присоеди-
няется сопротивление; 9 — пружина
единен к миллиамперметру, второй электрод пары присоеди-
няется к прибору только в момент измерения тока с помощью
специальных штеккеров и клемм 5—8' В период между измере-
ниями пары с помощью переключателей 9—12 замыкаются на
сопротивления, равные внутреннему сопротивлению прибора. На
переключатели присоединены сопротивления, соответствующие
по величине сопротивлению прибора при всех пределах измере-
ния. Для переключения пары с прибора на сопротивление без
размыкания цепи применяют специальный штеккер, конструк-
ция которого показана на рис. 95, а. В положении Ь (рис. 95, б)
ячейка замкнута на сопротивление, в положении Ь’ ячейка
замкнута иа прибор.
Электроды электрохимических пар должны быть укреплены
на заданном расстоянии друг от друга. Образцы должны иметь
— 154 —
определенную поверхность. Соотношение поверхности анода
к поверхности катода выбирается исходя из поставленных задач
и учета возможного соотношения в -условиях эксплуатации рав-
ным 1:1, 1 : 10, 1 : 100, 1 : 1000.
Один электрод пары присоединяют к общему проводу, а вто-
рой— к одной из клемм переключателя; таким образом, в мо-
мент измерения пара замыкается на прибор с сопротивлени-
ем 7?, а в остальное время замкнута на сопротивление, равное
сопротивлению прибора. Это обеспечивает отсутствие хотя бы
временного размыкания цепи. Для измерения силы тока пары
пользуются чувствительными
гальванометрами или микроам-
перметрами марок М95, М-194,
М-82 и т. п.
После заполнения сосудов
электролитом току дают устано-
виться в течение приблизительно
30 мин, а затем ведут измерения
и строят кривую / — t. Величина
площади, очерченной кривой I—
t и осями координат, дает значе-
ние количества регенерируемого
парой электричества Q. Величина
Q/F, где F — площадь электрода,
характеризует влияние контакта.
При сравнении материалов отно-
сительное расположение кривых
/—t в данной среде укажет на
менее опасный контакт.
При измерении небольших то-
ков, например при изучении ще-
левой коррозии, когда исследует-
ся работа пары металл в объеме—
металл в зазоре, необходимы
очень чувствительные приборы,
большое сопротивление, которое
противлением» для измерения
силы тока пары {37]
Эти приборы обычно имеют
нежелательно вводить в цепь
создаваемого элемента. Поэтому такие токи лучше всего изме-
рить одним из следующих способов.
Первый способ состоит в том, что электроды замыкаются на
небольшое сопротивление и падение напряжения на этом сопро-
тивлении записывается чувствительным автоматическим потен-
циометром ЭПП-09. Шкала такого потенциометра должна быть
проградуирована в микроамперах.
Вторым способом ток измеряют на приборе с «нулевым со-
противлением» (рис. 96). Падение напряжения от исследуемой
гальванической пары на сопротивлении прибора и дополнитель-
ном сопротивлении R уничтожается наложением на сопротивле-
ние равного, но противоположного по знаку напряжения от внеш-
— 155 —
ней батареи. Таким образом, в измерительной схеме не происхо-
дит потери напряжения от исследуемой пары (сопротивление
схемы как бы равно нулю). Контроль за регулировкой схемы ве-
дут по гальванометру [37, 38].
Измерение силы тока пар всегда правильнее производить при
помощи схемы с «нулевым сопротивлением», так как в этом
случае моделируются условия работы короткозамкнутых пар,
возникающих при эксплуатации изделий или конструкций. В тех
случаях, когда внутреннее сопротивление пары значительно боль-
ше сопротивления измерительного прибора, а величина тока
значительная, измерения можно производить непосредственно
при подключении прибора в цепь, так как падение напряжения
от исследуемой пары на сопротивлении прибора будет незначи-
тельно и им можно без большой погрешности пренебречь.
Разделение катодного и анодного пространства при измере-
нии тока в паре с помощью электролитического ключа приводит
к созданию условий, чаще всего отсутствующих в практике, по-
этому падение напряжения на ключе необходимо также компен-
сировать по принципу «нулевой схемы», иначе результаты будут
сильно занижены. Уменьшить сопротивление между электродами
пары можно также, разделяя их электролитическим мостиком,
не имеющим шлифа; концы такого ключа заполняются агар-
агаром.
Более подробно методы измерения токов коррозионных пар
описаны в работе И. Л. Розенфельда и О. И. Башкова [39].
? Измерение силы тока пар можно производить в условиях дви-
: жущейся жидкости или в любых других условиях, воспроизво-
' дящих условия эксплуатации, например при различных темпе-
ратурах н соотношениях площадей электродов в различных
* электролитах и т. п.
На рис. 97 приведена схема установки, разработанной
И. Л. Розенфельдом [40] для изучения контактной коррозии
трубных материалов и определения дальности действия контак-
та. Установка позволяет производить измерения при движении
жидкости различной температуры (от 20 до 80° С). Исследуемый
14 электрод представляет собой составную трубку. Разные отрезки
{ трубы (электроды) изолированы друг от друга. Одна половина
; трубы собирается из электродов одного металла, вторая поло-
I вина — из электродов другого. Общая длина трубы зависит от
, ее диаметра и электропроводности электролита. В морской воде,
• например, труба общей длиной один метр была собрана из об-
’ разцов длиной 100—125 мм при диаметре 20—30 мм. Циркуля-
ция электролита в установке осуществляется за счет разрежения
в колбе, создаваемого водоструйным насосом. От каждого элек-
трода выводится электрический контакт на панель, служащую
для измерения тока. Во время испытаний каждая пара электро-
дов соединяется друг с другом поочередно. Схема позволяет из-
мерять ток при любой комбинации электродов, а также приво-
— 156 —
лить их в контакт на длительное время, когда измерение не про-
изводится. На рис. 98 показаны результаты измерений, проведен-
ных на этой установке при исследовании контактной коррозии
Рис. 97. Схема установки для
измерения силы тока пар [40]:
1 —- контактный термометр; 2 -~
колба с электролитом; 3 — элек-
тронагреватель; 4 — реле; 5 —
•стеклянные трубки; 6 — трубчатые
образцы; 7 — -переключатель; а.
Ь, с, d — клеммы для подключе-
ния пар; а’, Ь', tf, d' — рубиль-
ники для поочередного подключе-
ния миллиамперметра. Сплошны-
ми стрелками обозначен ток ®оды,
пунктирными — ток воздуха
пары медь — алюминий в цирку-
лирующем 3%-ном растворе хло-
ристого натрия.
При измерении тока моделей
коррозионных элементов следует
воспроизводить по возможности
условия эксплуатации контакти-
руемых металлов. Желательно
также ускорить процесс. Для про-
цессов коррозии, протекающих с
катодным контролем (нейтраль-
Рис. 98. Зависимость корро-
зионного тока пары Си —
А1 от температуры и рас-
стояния от места контакта.
Электролит — 3% -ный раст-
вор (циркулирующий) хло-
ристого натрия, ток изме-
рен через одну минуту пос-
ле замыкания цепи [40]
ные среды), наиболее эффективным способом оказывается уве-
личение площади катода (1 : 50, 1 : 100).
С помощью метода контактных пар можно получить корро- >
зионную диаграмму для определенной пары, не прибегая к слож- \
ному методу снятия поляризационных кривых. J
Если замкнуть два металлических электрода и ввести боль-
шое сопротивление так, чтобы в системе ие было тока, и потом
постепенно выводить сопротивление и изменять величину тока
— 157 —
пары и потенциал каждого электрода, то можно получить кор-
розионную диаграмму, по которой можно определить не только
величину тока контактной пары, но ® получить данные о том,
какие процессы (катодные или анодные) определяют величину
тока. Имея эти данные, можно наметить правильные пути умень-
шения коррозии.
5. ЭЛЕКТРИЧЕСКИЕ МЕТОДЫ
Электрические свойства защитных пленок, такие как элек-
тросопротивление, емкость, потенциал пробивания, представляют
определенный интерес для исследователей коррозии, и их изме-
рение находит все большее применение в практике для оценки
защищенных свойств пленок.
Измерение импеданса RuC.A. Н. Фрумкин с сотрудниками
применили метод измерения емкости электрода для исследова-
ния электрохимических процессов, протекающих на металлах.
Принцип этого метода заключается в том, что поверхности ме-
талла и электролиту, в который он погружен, сообщаются неко-
торые малые количества электричества прямого и обратного
направления и измеряется изменение потенциала электрода.
В дальнейшем этот метод получил развитие в работах М. А. Вор-
синой и А. И. Фрумкина, М. П. Борисовой, Б. В. Эршлера,
Б. Н. Кабанова и других [41—43]. Наряду с емкостью при изу-
чении сильноокнсляющихся металлов стали измерять омическую
составляющую. Г. В. Акимов, Г. Б. Кларк и Н. И. Исаев [44]
применили метод совместного измерения емкости и сопротивле-
s ния для изучения электрохимического поведения алюминия, по-
крытого защитными окисными слоями, и установили, что между
характером изменения этих величин во времени и коррозионной,
стойкостью материала существует соответствие.
Электрические методы исследования коррозионных процес-
сов начинают применять все шире, особенно в связи с развитием
исследований в области защитных полимерных покрытий.
На поверхности металлов, покрытых защитными пленками,
имеются участки, полностью покрытые пленкой (электрохимиче-
ски инертные, практически не пропускающие электронов), и
участки, покрытые пленкой малой толщины, способной пропус-
кать электроны. Последние участки могут выполнять роль като-
дов, а поры в тонкой пленке играют роль анодов. Как мы ука-
зывали выше, судить о коррозионном поведении металла можно
по поляризационным кривым, но при этом фактор омического
сопротивления непосредственно учесть нельзя. Поэтому в тех
случаях, когда на поверхности металлов есть защитные пленки,
окисные или лакокрасочные, их защитная способность в значи-
тельной степени может быть охарактеризована их сопротивле-
нием. Для измерения сопротивления пленок применяют различ-
ные методы.
— 158-
Граница раздела электрод — электролит обладает определен-
ной величиной импеданса. Емкость и сопротивление такойРси-
стемы будут изменяться по мере проникновения электролита
в поры и разрушения самой плёнки. Метод измерения сопротив-
ления и емкости защитных пленок применяется для ’ изучения
влияния различных свойств пленки на ее защитную'способность.
Применяя этот метод, через исследуемую ячейку пропускают
переменный ток.
Г. Б. Кларк и М. И. Михайловская [45] показали принципи-
альную возможность применения емкостного метода для изуче-
ния защитных свойств битумных покрытий. Метод определения
сопротивления и емкости для изучения свойств лакокрасочных
покрытий применяли различные авторы [46—48].
Сопротивление и емкость защитных пленок измеряют с по-
мощью обычной мостовой схемы (рис. 99). Плечи моста обра-
зуются сопротивлениями Ri и переменным сопротивлением
и переменной емкостью С3; последнее плечо — емкостью и
омическим сопротивлением исследуемого образца. В качестве
сопротивлений Rz и используются безреактивные 'магази-
ны сопротивлений типа Р-58 с пределами измерений 0,1—
111111 ом или типа Р-517 с пределами измерений 0,1—10000 ом.
Переменная емкость измеряется параллельно соединенными
магазинами емкости; один из них типа Р-513 с пределами'изме-
рений 0,0001—10 мкф, другой в зависимости от ожидаемой ем-
кости 10—100 мкф. Сопротивление и емкость на установке-, при-
веденной на рис. 99, измеряются на переменном токе; источником
его могут быть генераторы звуковой частоты типа ЗГ-З, ЗГ-10»
ЗГ-11, дающие синусоидальные колебания в интервале частот
20—20000 гц и выше (ЗГ-11). В качестве нулевого прибора мо-
жет служить телефон или, еще лучше, катодный осциллограф
типа ЭО-7, ЭО-4. Для увеличения точности измерения перед
осциллографом ставится низкочастотный усилитель с коэффи-
циентом усиления от 10 до 100. Конденсатор, включенный после-
довательно с генератором, обеспечивает стабильность работы
установки; емкость конденсатора 10 мкф.
Экспериментальные ячейки для определения емкости и со-
противления могут быть различной конструкции [45, 48]. Наи-
лучшей следует считать такую ячейку, где площадь исследуемого-
электрода во много раз меньше площади вспомогательного элек-
трода. Конструкция такой ячейки для определения емкости и со-
противления окрашенных проволочных образцов приведена на
рис. 100. Во вспомогательный электрод цилиндрической формы
помещается исследуемый электрод. Вспомогательный электрод
изготовляется из материала, не разрушающегося в данных усло-
виях. Обычно это платиновые электроды, поверхность которых
сильно развита благодаря специальной обработке (платиниро-
вание). Следует отметить, что измерения, проводимые некото-
рыми исследователями на двух одинаковых электродах с защит,-
— 159 —
ными пленками, с этой точки зрения неудачны. Если поверхность
вспомогательного электрода велика по сравнению с исследуемой,
то сопротивление такого электрода мало и им можно пренебречь
при измерениях больших величин сопротивлений, возникающих
на границе раздела^ исследуемый электрод — электролит.
При измерении импеданса электрохимической ячейки нема-
лую роль играют частота переменного тока и порядок включения
в плечо моста переменных емкости и сопротивления.
В начале опыта, когда изоляционный слой еще достаточно
сплошной и электролит не проник к металлу, измеряемая систе-
ма представляет собой параллельное вклю-
чение емкости и сопротивления; если в из-
мерительном мосте эти величины располо-
жены последовательно, то измеряемые и
реально существующие величины не экви-
Рис. 99. Принципиальная схема уста-
новки для измерения емкости и со-
противления защитных пленок при
наложении переменного тока:
и Р.1 — постоянные сопротивления;
/?8 — переменное сопротивление; С» ~
«агазин емкости; У — усилитель; О —
осциллограф; Г — генератор переменного
тока; X — исследуемая ячейка
Рис. 100. Экспериментальная
ячейка для определения емко-
сти и сопротивления окрашен-
ных проволочных образцов [45]з
I — проволочный образец; 2 —
стеклянный сосуд; 8 — платиновый
цилиндр
валентны и поэтому получаемые начальные значения заниже-
ны и зависят от частоты переменного тока [45]. По мере умень-
шения сопротивления покрытия все большее значение приобрета-
ют сопротивление электролита в порах и емкость электрохимиче-
ской системы. Измеряемую ячейку в этом случае можно рас-
сматривать как последовательное соединение R и С. В этом слу-
чае измеряемая величина R не зависит от частоты переменного
тока. Обратное явление наблюдается при изменении емкости.
В первый период емкость практически ие зависит от частоты, так
как относительная доля электрохимической емкости в общей
емкости мала. С увеличением электрохимической емкости в свя-
зи с нарушением сплошности покрытия зависимость емкости от
частоты увеличивается. Поэтому если интересуются механизмом
процесса, то изучают зависимость емкости от частоты перемен-
ного тока; изменяя частоту тока, можно также судить о том, ка-
кой эквивалентной электрической схеме соответствует строение
защитной пленки. Если же необходимо определить величину оми-
ческого сопротивления пленки и его изменение во времени, то
измерять сопротивление следует при больших частотах — поряд-
ка 10—20 кгц.
Во всех случаях наложения переменного тока на электрохи-
мическую ячейку его напряжение должно быть возможно мень-
шим (10—-50 мв) во избежание
поляризации исследуемого элек-
трода.
Используя зависимость сопро-
тивления и емкости полимерного
покрытия от времени при погру
жении окрашенного материала в
электролит, можно судить о за-
щитной способности покрытия в
этих условиях. Как видно из кри-
вых, полученных при наложении
переменного тока (рис. 101), рез-
кое измененне сопротивления и
емкости покрытия наступает по
истечении 9—12 суток. Оно соот-
ветствует моменту интенсивного
разрушения покрытия и появле-
нию сплошных разрушений плен-
ки и, следовательно, может харак-
теризовать устойчивость покры-
тия. Однако такая зависимость
наблюдается не для всех видов
покрытий.
Сопротивление покрытий мож-
но определять и при наложении
постоянного тока, например при
применении схем, приведенных на
рис. 102 и 103. При измерениях по
О 3 6 9 12 15 18 21
вреня. сутки
Рис 101. Зависимость сопротивле-
ния и емкости трехслойного по-
крытия (ФЛ-02), нанесенного 'на
сталь при погружении образцов
в 0,5-н. раствор NaCl. Измерения
проведены при наложении пере-
менного тока при f — 20000 гц
и U — 10 мв\ R — сопротивление;
С — емкость; Д1(ор — площадь, за-
нятая коррозией
ление рассчитывают по уравнению
схеме а (рис. 102) сопротив-
— _£.
Г*~ I
(87)
где гх— сопротивление исследуемого образца;
га — сопротивление амперметра;
U — напряжение, измеренное катодным вольтметром;
I — ток, измеренный прибором.
При измерениях по схеме б
считывают по уравнению
(рис. 102) сопротивление рас-
-V. (88)
1
где >1, — сопротивление вольтметра.
И Зак. 133S — 161 —
Измерения, проведенные с помощью двух указанных схем,
не отличаются высокой точностью, однако применение их при
ускоренных испытаниях вполне оправдано. Измерения должны
производиться при постоянных значениях напряжения V и то-
ка /.
а
Рис. 102. Схема для измерения сопротивления
методами амперметра (а) и вольтметра (6):
1 — электрохимическая ячейка с неизвестным
сопротивлением гх
6
Для определения больших сопротивлений, например, много-
слойных лакокрасочных покрытий, используют метод сравнения
измеряемого сопротивления с сопротивлением эталона. В каче-
стве последнего берут измерительную катушку или точный ма-
Z- const
Рис. 103. Схема для измерения сопротивлеиия методом
сравнения:
а — последовательное соединение; б — параллельное соедине
иие: / — электрохимическая ячейка с неизвестным сопротивле-
нием гх; 2 — образцовое сопротивление rs. 3 — измерительный
прибор
газин сопротивления. Так как величины сопротивления велики,
в цепи нетрудно поддерживать постоянное напряжение-при про-
ведении измерений. Измеряемое сопротивление и сопротивление
эталона могут включаться последовательно и параллельно. При
— 162 —
последовательном соединении (рис. 103, а) искомое сопротивле-
ние определяют по уравнению при I = const
(89)
с* о
где г0 — эталонное сопротивление;
Ux и Uo— падение напряжения, измеряемое на искомом
и эталонном сопротивлении соответственно.
Падение напряжения измеряется катодным вольтметром, со-
противление которого Должно быть во много раз больше сопро-
тивлений гх и Го. Метод сравнения можно применять и при па-
раллельном соединении измеряемого и эталонного сопротивления,
как показано на рис. 103, б. Искомое сопротивление рассчитыва-
ют по уравнению
гЛ = Го-^-, (90)
где /о и 1Х — сила тока в цепи при включении поочередно в од-
ну и ту же цепь искомого и эталонного сопротив-
лений.
В качестве измерительного прибора в этом случае можно при-
менять любой прибор, сопротивление которого мало по сравне-
нию с измеряемым сопротивлением. При измерении гальваномет-
ром сопротивление рассчитывают по уравнению (90), в котором
отношение токов заменяется отношением показаний гальвано-
метра. При проведении измерений необходимо следить, чтобы на-
пряжение было постоянным.
Метод сравнения хотя и точен, но требует применения катод-
ных вольтметров с очень большим входным сопротивлением.
Кроме того, измерение сопротивлений при наложении по-
стоянного тока всегда приводит к значительной поляризации
электродов, а следовательно, и к искажению измеряемых вели-
чин. Долю же поляризационного сопротивлення в общем сопро-
тивлении при таких измерениях определить невозможно. Поэто-
му применение методов с наложением постоянного тока целесо-
образно лишь для приблизительной оценки защитных свойств
пленок.
И. Л. Розенфельд, Е. К. Оше, А. Г. Акимов [49] разработали
осциллографический метод измерения сопротивления пленок;
позволяющий определить сопротивление в очень короткий проме-
жуток времени (10“4 сек) от начала опыта, когда поляризаци-
онное сопротивление еще не успевает развиться. Схема установи
ки для измерения сопротивления защитных пленок на металлах
осциллографическим методом приведена на рис. 104. В схеме
применяется осциллограф ЭНО-1 или 0-4, подключающийся сна-
чала в положение 1, а потом в положение 2. Таким образом, на:
экране осциллографа сначала измеряют падение напряжения;
возникающее от источника постоянного тока на известном сопро-
тивлении Rq, величина которого выбирается соизмеримой величи-
ц* —163 —
Л” осиитюгвафд
Рис. 104. Схема установ-
ки осциллографического
определения сопротивле-
ния защитных пленок на
металлах ,[491:
I и 2 — клеммы для при-
соединения осциллографа;
Ro — известное сопротив-
ление; Rx — измеряемое
сопротивление
не сопротивления ячейки Rx. Сопротивление ячейки, состоящей
из исследуемого и вспомогательного электродов, погруженных
в электролит, рассчитывается из полученных данных по уравне-
нию (89). Так как сопротивление вспомогательного электрода
мало, им обычно пренебрегают, а сопротивление электролита из-
вестно и всегда может быть учтено.
С помощью осциллографического метода можно легко опре-
делить долю омического и поляризационного сопротивлений в
общем сопротивлении пленки. Для этого
на осциллограммах, снятых в положении
1 и 2, кроме мгновенного значения напря-
жения, определяют и установившееся зна-
чение напряжения. При этом сила тока в
цепи должна быть постоянной; это кон-
тролируют с помощью включенного мик-
роамперметра.
Из полученных данных рассчитывают
величины омического и поляризационно-
го сопротивлений пленок. Накладывая
различные плотности тока анодного н ка-
тодного направления, можно получить
соотношение поляризационного и омичес-
кого сопротивлений в любых условиях»
которые возможны при эксплуатации из-
делия.
Определение пробивного напряжения.
В электротехнике для определения изоля-
ционных свойств различных материалов
применяют метод так называемого про-
бивного напряжения. В коррозионной
практике этот метод, заключающийся в
измерении величины напряжения, кото-
рое необходимо приложить, чтобы насту-
пил пробой пленки, используют при исследовании естественных
и искусственно получаемых окисных пленок.
Исследователь, как правило, интересуется не исходным зна-
чением потенциала пробивания, а его изменением во времени
под воздействием окружающей среды. В ряде случаев, напри-
мер, необходимо знать, изменяется ли контактная проводимость
металлов, имеющих на поверхности окисные слои или гальвани*
ческие покрытия. Г. Б. Кларк и Г. В. Акимов £60] для определения
••сопротивления окисных слоев на металлах и пробивного напря-
жения разработали специальный лабораторный прибор. Для
этой же цели удобно применять универсальную пробойную уста-
•новку УПМ-1М, выпускаемую промышленностью. На установке
’можно -испытывать электрическую прочность изоляции и окисиых
пленок шря наложении постоянного и переменного тока, а также
уценивать порядок сопротивления изоляции испытываемых дета-
— 164 —
лей. Максимальное напряжение, создаваемое на установке, со-
ставляет 10 кв. Испытания могут быть кратковременные (в те-
чение одной минуты), длительные и при наложении импульсов.
Схема установки приведена на рис. 105, откуда видно, что уста-
новка работает от сети переменного тока напряжением 220 в
и имеет три диапазона напряжения: от 0 до 10 кв, от 0 до 3 кв
и от 0 до 1 кв. Для получения высоких напряжений в установке
используется повышающий высоковольтный трансформатор ТР4.
Переменное напряжение снимается со всей обмотки трансформа-
Рис. 105. Принципиальная электрическая схема установки
УПУ-1М для определения пробивного напряжения
тора, а постоянное — с однополупериодного выпрямителя на
лампе Л2. Выпрямитель включен на часть обмотки так, чтобы
значение постоянного тока было равно действующему значению
переменного тока; для сглаживания пульсаций на выходе вы-
прямителя установлен фильтр. Регулировка высокого напря-
жения производится в первичной цепи высоковольтного транс-
форматора автотрансформатором ТрЗ, обмотка которого вклю-
чена параллельно трансформатору Тр4.
Определение толщины и сплошности изолирующих покрытий.
К числу электрических методов определения защитных свойств,
например лакокрасочных покрытий, могут быть отнесены и
методы измерения их толщины с помощью приборов, действие ко-
торых основано на изменении силы притяжения магнита к ферро-
магнитной подложке в зависимости от толщины магнитной плен-
ки. Такой прибор ИТП-1 выпускает в настоящее время Хотьков-
ский завод экспериментальной окрасочной технологии и аппа-
ратуры. Измеритель ИТП-1 имеет форму карандаша и представ-
ляет собой пружинный динамометр, снабженный магнитом, шка-
лой и номограммой (индивидуальной для каждого прибора).
— 165 —
С помощью номограммы показания шкалы измерителя перево-’
дятся в микроны, соответствующие толщине измеряемой пленки.
Этот прибор позволяет измерить пленки на магнитных подлож-
ках (стальных, чугунных) толщиной от 10 до 500 мкм с точ-
ностью ±10%.
Для измерения толщины и сплошности лакокрасочных пле-
нок на немагнитных материалах И. Л. Розенфельд и Ю. П. Оль-
ховников [51] разработали метод, основанный на определении
изменения емкости конденсатора в зависимости от толщины слоя
диэлектрика (измеряемой пленки) между ее обкладками. Изме-
ряется емкость конденсатора, образованного металлом подлож-
о б
Рис. 106. Конструкция датчи-
ка (с) и схема измерительной
установки (б)[ 51]
Рис. 107. Зависимость емкости
от толщины слоя покрытий [51]:
I — ПХВ-26 на стали; 2 — НФХ-2402
с ТЮа на магнии
ки, диэлектриком в виде покрытия и прикладываемым к поверх-
ности окрашенного металла электродом с постоянной площадью.
Для плоского-конденсатора емкость выражается уравнением
С=Г7- (91>
где 5 — площадь электрода, равная поперечному сечению поля
в любом месте;
е — диэлектрическая постоянная;
d — расстояние между электродами (толщина диэлект-
рика) .
Если площадь вспомогательного электрода постоянна, то ем-
кость зависит только от толщины слоя диэлектрика.
Разрабатывая этот метод, авторы использовали специальный
датчик, заменяющий вторую обкладку конденсатор а.
Конструкция датчика приведена на рис. 106, а. Он состоит
из плексигласового корпуса 3 с крышкой 2. Внутри корпуса
помещается войлочный электрод 4, смоченный электролитом,
к войлоку прижимается латунный контакт /, выведенный на
крышку датчика. В нижией части датчика войлок образует по-
верхность, которую приводят в соприкосновение с покрытием
—166 —
при измерениях. Эта поверхность имеет постоянную площадь;
равную площади отверстия в корпусе датчика. Преимущество,
жидкостного датчика — отсутствие возникновения каких-либо
воздушных зазоров за счет неровности измеряемой поверхно-
сти вследствие заполнения их электролитом.
На рис. 106, б изображена схема измерительной установки.
Измерения производят, присоединяя к металлической подлож-
ке 7 и датчику 6, установленному иа поверхности диэлектрика 8,
измеритель емкости 5 типа НИЕ-1, питающийся от сети пере-
менного тока. Метод может применяться не только к немагнит-
ным, но и к магнитным материалам. Толщину покрытия опреде-
ляют с помощью предварительно построенной градуировочной
кривой для данного покрытия. Градуировочные кривые строят
по данным емкости и толщины пленок, измеренных двойным мик-
роскопом В. П. Линника.
На рис. 107 приведены кривые для покрытия ПХВ-26 на ста-
ли и покрытия ИФХ-2402 с TiOg на магний, построенные таким
методом. С помощью этого метода можно контролировать не
только толщину покрытия, но и имеющиеся несплощности. При
наличии непокрытых участков или при появлении дефектов в по-
крытии в процессе его эксплуатации емкость резко возрастает.
ЛИТЕРАТУРА
1. Акимов Г. В. Теория и методы исследования коррозии металлов. Изц-
во АН СССР, 1945.
2. То м а ш о в Н. Д., Модестова В. Н. Заводская лаборатория, 1943,
т. XVI, № 3.
3. Миро любо в Е. Н., Курте пов М. М., Томатов Н. Д. Тр. Ин-та
. физической химии АН СССР, вып. 7, Изд-во АН СССР, i960, 51.
V 4’ а ш о в Н. Д., Михайловский Ю. Н. Заводская лаборатория,
1957, т. XXIII, № 12, 1462.
5. Скорчеллетти В. В.. Тукачинский С. Е. ЖПХ. 1953, т. XXVI,
№ 1, 30.
6. Куртепов М. М., Кольцова А. С. Тр. Ин-та физической химии АН
I СССР, вып. 1, Изд-во АН СССР, 1951, 83.
\/ 7. Розенфельд И. Л., П а в л у ц к а я Т. И. Заводская лаборатория, 1955, I
т. XXI, № 4, 473. ।
8. Богачев Е. С. Программный регулятор оборотов электродвигателя. Пе- 1
редовой иаучн.-техн. опыт. № 26—63—217/5, 1963.
9. Кларк Г. Б., Акимов Г. В. ДАН СССР, 1941, т. 30. № 9, 798.
10. Ноог Т. Р., Chainer R. Н. J. Iron and Steel Inst., 1962. 182. 124.
11. Розенфельд И. Л. Изв. АН СССР, № 6, 674, 1951.
12. Pottrbaix М. Werkstoffe und Korrosion, 1964, № 10, 821.
«и» 13. Бахвалов Г. Т., Тур ков ска я А. В. Руководство к лабораторным
работам по коррозии и гальваностегии, Металлургиздат, 1952.
14. Алексеев Н. Г., Прохоров В. А., Чмутов К. В. Электронные
приборы и схемы в физико-химических исследованиях. Госхимиздат, 1961.
15. Томатов Н. Д., Исаев Н. И. Тр. Ин-та физической химии АН СССР,
< вып. 7, Изд-во АН СССР, 1959, 96.
^16 . Эванс Ю. Р. Коррозия, пассивность и защита металлов. М.-Л. Метал-
лургиздат, 1941.
17. Т о м а ш о в Н. Д. Коррозия металлов с кислородной деполяризацией.
Изд-во АН СССР. 1947. |
—167 —
18. Розенфельд И. Л. Замедлители коррозии в нейтральных средах.
Изд-во АН СССР, 1953.
19. Голубев А. И. Коррозионные процессы на реальных микроэлементах.
Оборонгиз, 1953.
^20 . Розенфельд И. Л. Атмосферная коррозия металлов. Изд-во АН
СССР, I960.
21. Rosenfeld I. L. 1. Intern. Congress on Metallic corrosion, aprel 1961.
London, Butterworths, 1962, p. 243.
22. Розенфельд И. Л., Павлуцкая Т. И. ДАН СССР, 1955, т. 91,
№ 2, 315.
23. Фокин М. Н., Виноградов А. Ф. Электронный потенциостат для
электрохимических исследований. Изд-во филиала ВИНИТИ, тема 35,
№ П-59, 1959; № П-6057/5, I960.
24. Колотыркин Я. М., Брыскин И. Е., Флорианович Г. М., Че-
моданов А. Н. Проблемы физической химии, вып. 3. Госхимиздат,
1963, стр. 15.
25. Р raz a k М. Werkstoffe und Korrosion, 1958, Н. 9, 517.
26. Hickling A. Electrochemica acts, 1961, v. 5, 161.
27. Stern M. J. Electrochem. Soc., 1955, v. 102, 609.
28. Pearson I. M. Trans. Electrochem. Soc., .1942, v. 81, 485.
29. SchwerdtfegerW. 1, McDorman O. N. J. Electrochem. Soc-.,
1952, v. 99, 407
30. S c h a s c h e E.. Marsh G. A. Corrosion, 1952, v. 99, 407.
31. Stern M., Geary A. L. J. Electrochem. Soc., 1957, v. 104, 56.
32. Evans S., Koehler. J. Electrochem. Soc., 1961, v. 108, 509.
33. Schwerdtfeger W. I., Manuel R. I. J.,Res. Nat. Bur. Standarts,
1961, 65 C, 171.
34. Фрумкин A. H., Шлыгин А. И. Изв. АН СССР, серия хим., 773,
1956.
35. Розенфельд И. Л., Жигалова К- А. ДАН СССР, 1954, т. 99,
№ 1, 136.
36. Розенфельд^И. Л., Максимчук В. П. Заводская лаборатория,
I960, т. XXVI, № 3, 283. ‘
37. Sensetive Research Instrument Corp., Catalog 40, New York.
38. Розенфельд И. Л., Маршаков И. К- Щелевая коррозия и борьба
с ней. Изд-во филиала ВИНИТИ, № М-59-20/2, 1959.
39. Розенфельд И. Л., Башков О. И. Заводская лаборатория, 1964.
т. XXX, № 7, 813.
40. Розенфельд И. Л. Заводская лаборатория, 1955, т. XXI, № 1, 60.
41. Ворсина М. А., Фрумкин А. Н. ДАН СССР, 1939, т. 24, № 9, 918.
42. Борисова М. П., Э рад л е р Б. В;, Фрум к-и и А. Н. ЖФХ, 1948, т. 22,
8, 925.
43. Кабанов Б. Н. Тр. совещания по электрохимии. Изд-во АН СССРЛ-
1953, 134.
44. Акимов Г. В., Кларк Г. Б., Исаев Н. И. Тр. V Всесоюзного сове-
щания по коррозии и защите металлов. Изд-во АН СССР, 1956, 209.
45. Кларк Г. Б., Михайловская М. И. Тр. Ии-та физической химии
АН СССР, Изд-во АН СССР, 1959, 145.
46. Вассерман П. И., Колотыркии Я- М., Чеботаревский В. В.,
Феоктистова А. А. Коллоидный журнал, 1959, т. 21, №4, 392.
47. Worm well W., Brasher D. M. J. Iron and -Steel Inst., i960, v. 164,
№1, 141.
.48 . Ромашов H. Д., Лунев А. Ф., Игнатова 3. И. Тр. Ин-та физичес-
кой химии АН СССР, вып. 7. Изд-во АН СССР, 1960, 254.
49. Розенфельф И. Л., Оше Е. К., Акимов А. Г. Лакокрасочные ма-
териалы и их применение, 1964, № 6, 45.
50. Кл а р к Г. Б, Акимов Г. В. Тр. Ин-та физической химии АН СССР,
вып. 3. Изд-во АН СССР, 1951, 9.
51. Розенфельд И. Л., Ольховников Ю. П. Тр. Ин-та физической
химии АН СССР, вып. 7. Изд-во АН СССР, 1959, 155.
—168 ~
V
ОПРЕДЕЛЕНИЕ
ЭФФЕКТИВНОСТИ ЗАЩИТНЫХ СРЕДСТВ
Металлы с различными защитными покрытиями, будь то метал-
лические или лакокрасочные, различные временные защитные
слои (смазки и др.), представляют собой сложные системы и
требуют в зависимости от вида покрытия особого подхода к вы-
бору способов оценки их защитной способности.
Поэтому, несмотря на то, что разработка и усовершенствова-
ние ускоренных методов испытаний для таких систем в основном
базируются на уже изложенных принципах, необходимо учиты-
вать и некоторые специфические особенности того или иного спо-
соба защиты.
Способы ускоренных испытаний для оценки защитных свойств-
покрытий и способы защиты изделий путем обработки коррози-
онной среды будут в связи с изложенным рассматриваться каж-
дый в отдельности.
1. МЕТАЛЛИЧЕСКИЕ ПОКРЫТИЯ
Металлические покрытия широко применяются для защиты
металлов от коррозии во многих отраслях промышленности. Их
различают по способу нанесения: 1) погружение в расплавлен-
ный металл; 2) диффузионный; 3) распыление; 4) механотерми-
ческий (плакирование); 5) гальванический, а также по механиз-
му электрохимической защиты [1]. В последнем случае различа-
ют анодные и катодные покрытия.
По отношению к железу и его сплавам анодными являются
цинковые и кадмиевые покрытия, которые защищают основной
металл электрохимически. Все остальные покрытия относятся к
катодным, защищающим основной металл только благодаря изо-
ляции его от внешней среды. Они эффективны до тех пор, пока
не нарушена их сплошность. К катодным покрытиям относится
— 169 —
большинство защитно-декоративных покрытий, среди которых
наиболее распространены хромовые и никелевые.
В зависимости от назначения металлические покрытия долж-
ны удовлетворять различным требованиям, которые обычно ого-
вариваются техническими условиями. Выбор того или иного по-
крытия для защиты изделий определяется требованиями к его
внешнему виду и коррозионной устойчивости.
Коррозионная устойчивость покрытия зависит не только от
устойчивости самого металла покрытия, но и от работы много-
электродной системы, состоящей из основного металла, металла,
непосредственно прилегающего к защищаемому металлу, н на-
- ружного (защитного).
Несмотря на проводившиеся исследования и разработку раз-
I личных способов испытаний в СССР и за рубежом [2—7], все еще
\ нет надежного метода ускоренных испытаний металлических по-
крытий.
1 Существующие ускоренные методы испытаний металлических
покрьттйй следует разделить на методы, с помощью которых ус-
танавливают качество самих покрытий; и методы, определяющие
защитную 'мощность’покрытий'.
В первом случае представляет интерес, соответствует ли по-.
крытие требованиям, которые предъявляются техническими ус-
ловиями или ГОСТами. Такими испытаниями должно устанавли-
ваться наличие различного рода дефектов, определяться
пористость покрытия или его толщина. Косвенно по результатам
этих испытаний можно судить и о защитной способности покры-
тия; так, например, для анодных покрытий толщина нанесенного
слоя будет определять продолжительность его защитного дей-
ствия, для катодных покрытий большее значение будет иметь
пористость покрытия. Для такого рода испытаний точное вос-
произведение характера разрушения, наблюдающегося в услови-
•-ях эксплуатации, не обязательно, важно только, чтобы в возмож-
но короткий срок можно было установить, имеет ли покрытие
дефекты, которые могут вызвать разрушение в процессе эксплуа-
тации.
При ускоренных испытаниях, цель которых — определение за-
щитной способности различных покрытий, должны создаваться
такие условия, которые вызвали бы коррозионные разрушения,
подобные наблюдающимся на Практике, но за более короткий
-срок. В этом случае метод можно считать надежным лишь после .
сопоставления полученных результатов с результатами испыта-
ний в естественных условиях или с данными наблюдений за по-
ведением изделий в эксплуатации.
На этой основе за последние годы были разработаны новые
виды ускоренных испытаний гальванических покрытий, а также
усовершенствованы старые, результаты которых удовлетвори-
тельно характеризуют поведение покрытий в различных условиях
эксплуатации.
— 170 —
Ускорить коррозионный процесс, как было описано выше (см.
гл. I), можно, влияя на скорость электродных реакций. Так как
большинство металлических покрытий эксплуатируется в атмо-
сферных условиях при периодическом увлажнении, то следует
рассмотреть способы испытаний, имитирующие эти условия. Все
известные в литературе ускоренные испытания гальванических
покрытий, предназначенных для эксплуатации в атмосферных ус-
ловиях, проводятся в аппаратуре, описанной ранее. В качестве
ускоряющих процесс факторов во влажную атмосферу камер
вводят катодные деполяризаторы (SO2, CuCl2, FeCls) и ионы хло-
ра, нарушающие пассивное состояние металлов. Используют и
такие факторы, как повышение температуры окружающей среды,
а также периодическая конденсация влаги.
Широко распространенным испытанием защитной способно-
сти покрытий в лабораторных условиях долгое время было ис-
пытание в камере при распылении растворов хлористого натрия.
Этот вид испытаний постепенно был введен в стандарты многих
стран (США, ФРГ, Англии и др.) и начал применяться как уни-
версальный метод независимо от того, для каких условий пред-
назначается данное покрытие. Необходимо отметить, что приме-'
нение этого метода целесообразно только для определения
сравнительной коррозионной устойчивости тех покрытий, кото-
рые предназначаются для защиты изделий в морской атмосфере.
Кроме того, в процессе испытания вследствие образования про-;
дуктов коррозии изменяется pH раствора, и результаты испыта-
ний плохо воспроизводятся.
Концентрация распыляемых растворов хлористого натрия не г
должна быть больше 3—5%, так как с повышением ее умень- ;
шается растворимость кислорода и снижается скорость корро- ;
зии, а также создается опасность закупорки сопла пульверизато-
ра кристаллами хлористого натрия.
В настоящее время в США, согласно ASTM В-117-57Т, при-
меняют только 5%-ный раствор хлористого натрия. В ФРГ испы-
тания при распылении 3% -него раствора хлористого натрия как
универсальные исключены из стандартов и применяются, соглас-
но ДШ 50-907, только при исследовании легких сплавов, пред- i
назначенных для использования в морских условиях, причем
оговаривается, что заключение о поведении в других условиях
недопустимо [3].
Исследования Е. В. Ганушкиной [2] показали, что ускоренные
испытания цинковых покрытий во влажной атмосфере и в камере
при распылении раствора хлористого натрия не отражают пове-
дения металлических покрытий в естественных условиях главным
образом потому, что в последнем случае образуются раствори-
мые продукты коррозии, а при ускоренных испытаниях — защит-
ные слои продуктов коррозии.
Сравнение результатов различных методов ускоренных испы-
таний с данными, полученными при испытании в естественных
— 171 —
условиях [8], проведенное Е. В. Ганушкиной [2], показало, что
применительно к промышленной атмосфере лучшее совпадение
по характеру коррозионных разрушений по сравнению с испыта-
ниями во влажной камере и при распылении раствора хлористого
натрия дает испытание при введении во влажную камеру SO2.
Автор [2] рекомендует также испытание цинковых покрытий в ве-
зерометре при орошении образцов 0,025-н. раствором серной кис-
лоты при температуре испытания 50° С и скорости вращения ба-
рабана 0,1 рад!сек (1 об!мин). Установлено, что коррозия цинка
в везерометре в течение 12 ч соответствует коррозии цинка в про-
мышленной атмосфере в течение двух лет.
Рекомендация указанного метода определения устойчивости
цинкового покрытия основывается на выборе такой концентра-
ции кислоты для обрызгивания, при которой скорость коррозии
соответствовала бы определенному сроку испытания в естествен-
ных условиях. На наш взгляд, этот метод не совсем оправдан,
ибо серная кислота должна сильно изменять характер коррози-
онного процесса. Цинк в атмосферных условиях корродирует,
как правило, с кислородной деполяризацией. Изменение харак-
тера деполяризации катодного процесса может исказить резуль-
таты. Применение кислых электролитов при ускоренных испыта-
ниях оправдано в тех случаях, когда изделие работает в сильно-
загрязненной промышленной атмосфере, где конденсирующийся
на поверхности покрытия электролит приобретает вследствие аб-
сорбции сернистого газа слабокислую реакцию.
В последнее время в зарубежной литературе публикуются
многочисленные работы, посвященные созданию новых и усовер-
шенствованию существующих способов ускоренных коррозион-
ных испытаний металлических покрытий. Рассмотрим наиболее
тщательно разработанные методы. В первую очередь к ним от-
носится испытание с применением уксусной кислоты [4]. Предпо-
лагается, что с помощью кислоты можно имитировать кислые
электролиты, возникающие на изделиях с гальваническими по-
крытиями в промышленных городах.‘‘Образцы с металлическими
покрытиями помещают во влажную'камеру, где в качестве рас-
пыляемого раствора применяют 5%-ный раствор хлористого нат-
рия с добавкой уксусной кислоты (~1%), поддерживающей pH
в пределах 3,3—3,5 и тем самым создающей постоянство условий
на протяжении всего испытания. Этот вид испытания предлага-
ется для сравнения коррозионной стойкости металлических по-
крытий, предназначенных для морских и для промышленных ат-
мосфер, однако ускорение коррозионного процесса, достигаемое-
при этом испытании, невелико.' Поэтому Сьюкс [5] предложил еще
один вид испытаний, включающий применение ионов двухва-
лентной меди. Этот метод известен под названием СА SS-испыта-
ние. Сущность его заключается в том, что в распыляемый 5%-
ный раствор хлористого натрия, содержащий уксусную кислоту,
вводят дополнительный катодный деполяризатор СиС12 • 2Н2О
—’172 —
в количестве 0,3 г/сЬи3. Одновременно повышается температура;
испытание проводят при 49° С.
По данным [5], испытание с СиС12 дает хорошие результаты 5
для покрытий Си — Ni — Сг и ;Ni — Сг по стали и цинку, а также j
для анодированного алюминия. Срок испытания при введении |
CuCl2 сокращается по сравнению с испытанием при добавке i
только уксусной кислоты от двух до четырех раз. *
Для имитации условий промышленной атмосферы в камеру
вводят небольшие количества SO2. Эдвардс [6] предложил прово-
дить испытание во влажной атмосфере в присутствии сернистого
.ангидрида для контроля качества двухслойных и однослойных
покрытий, таких как -Ni — Сг по стали, меди и бронзе, хромовых
по бронзе и Sn — Ni по меди. Испытание проводят во влажной
камере при Н ~ 95% с содержанием SO2 1% и температуре 17—
25° С при отсутствии конденсации.
Степень ускорения коррозионного процесса в присутствии SO2
различна и зависит от характера покрытия металла и от кон-
центрации SO2. Концентрацию SO2 в камере выбирают в преде-
лах 0,01—2,0 % (объемн.). Выбор той или иной концентрации оп-
ределяется поставленной задачей; слишком малые количества
SO2 дают незначительное увеличение скорости коррозии, слиш-
ком большие не позволяют выяснить разницу в коррозионной
стойкости покрытий. Для определения сравнительной устойчиво-
сти покрытий в промышленной атмосфере во влажную камеру
обычно вводят 0,1% SO2. При проведении испытаний, основное
назначение которых — выявить качество покрытия и наличие от-
клонения от технологического процесса их нанесения, концентра-
цию сернистого газа в камере увеличивают. При стандартных ис-
пытаниях по немецким нормам DIN 50 0 18 предусматривается
введение 0,8% SO2 и 0,8% СО2. Дополнительное введение СО2
основывается на том, что некоторые металлы, например цинк и
свинец, очень чувствительны к наличию в воздухе этого газа, по-
скольку в присутствии СО2 образуются продукты коррозии за-
щитного характера.
Поэтому для постоянства условий проводимых испытаний
в камере всегда поддерживают постоянную концентрацию не
только SO2, но и СО2.
Испытание в присутствии SO2 можно значительно ускорить,
если на поверхности образцов периодически производить конден-
сацию влаги. Если испытание в присутствии SO2 проводится при
влажности, близкой к 100%, а конденсация не предусмотрена, то
необходимо тщательно контролировать температуру, так как да-
же незначительные колебания ее приведут к конденсации и резко
изменят скорость и характер процесса.
При повышении температуры испытания и введении сернис-
того газа растворимость SO2 уменьшается. По данным работы
[7], повышение температуры сверх 40° С поэтому нецелесооб-
разно.
— 173 —
Для имитации условий эксплуатации изделий с металличес-
кими покрытиями в автомобилях Пиннер [9] предложил метод,
состоящий в том, что образцы покрывают искусственной пастой,
а затем испытывают во влажной камере при относительной
влажности, близкой к 100%. Этот метод получил название кор-
родкот.
Состав пасты имитирует уличную грязь промышленного райо-
на и способствует значительному ускорению коррозионного про-
цесса вследствие введения катодного деполяризатора FeClg.
В результате большого количества анализов грязи, возника-
ющей на эксплуатируемом автотранспорте, и испытаний паст
различных составов был отобран и рекомендован следующий
состав пасты: 0,035 г Си (NOS)2-3H2O, 0,165 г FeCls-6H2O, 1,0 г
NH4CI, 30 г каолина и 50 см3 воды.
После нанесения пасты на образцы их помещают во влажную
камеру и выдерживают некоторое время.
Пиннер [10] сопоставил результаты ускоренных испытаний по
методу корродкот с результатами испытаний в различных естест-
венных атмосферах-и установил, что если испытания в естествен-
ных условиях продолжаются один год, а ускоренные 20 ч, то хоро-
шее совпадение результатов по характеру и скорости коррозии
наблюдается для промышленной атмосферы, коэффициент пере-
счета для которой 0,930 (Детройт и Мичиган). Коэффициенты
(’ пересчета для морской (Флорида) и сельской (Питтсбург и Пен-
сильвания) атмосфер составляют 0,833 и 0,697 соответственно.
Этот метод автор рекомендует применять в основном для сильно-
загрязненных промышленных атмосфер.
Оценивая различные ускоренные методы по тому, как они от-
ражают поведение никеля в естественных условиях, Ла-Кэ‘полу-
чил результаты, приведенные в табл. 17 [11]. Из таблицы видно,
что скорость коррозии никеля при ускоренных испытаниях значи-
тельно выше скорости коррозии его в естественных условиях;
наибольшая скорость коррозии никеля наблюдалась при испыта-
нии в атмосфере, содержащей SO2. Увеличение достигало при
этом 2000 раз. Поскольку при таком сильном ускорении процес-
са коррозии возможно искажение характера коррозионных раз-
рушений, ускоренное испытание в присутствии больших концент-
раций SO2 следует рассматривать как испытание, определяющее
качество покрытия, а не его защитную способность. Более уме-
ренное увеличение скорости коррозии наблюдалось, когда в ка-
мере распылялся 5%-ный раствор хлористого натрия, содержа-
щий уксусную кислоту и хлорную медь (СА SS-испытание), а
также при испытании по методу корродкот^ При СА SS-испыта-
нии скорость коррозии повышалась в 20 раз, при испытании ме=*
тодом корродкот — в 50.
При испытании многослойных покрытий, например таких, как
Си — Ni — Сг, на цинковом литье для воспроизведения промыш-
ленной атмосферы также лучше пользоваться методом коррод-
— 174 —
Таблица 17
СОПОСТАВЛЕНИЕ СКОРОСТИ КОРРОЗИИ ПРОКАТАННОГО НИКЕЛЯ
В ЕСТЕСТВЕННЫХ УСЛОВИЯХ И ПРИ УСКОРЕННЫХ ИСПЫТАНИЯХ [11]
Характер атмосферы, вид испытания
Скорость коррозии
Промышленная атмосфера, Нью-Йорк............. 0,890
Морская атмосфера, Кюр-Бич, 240 м от океана . 0,060
Сельская атмосфера, штат Колледж............... 0,052
Распыление:
5%-ного раствора NaCl...................... 1,600
20%-ного раствора NaCl..................... 0,280
5%-ного раствора NaCl, содержащего ~1%
СН3С00Н (pH ==--3,3) и 0,3 г/дм8 СиС12
(СА SS-испытание)......................17,500
Испытание:
при введении в камеру SO8................2031,000
при нанесении пасты, содержащей 0,035 г/дм8
Cu(NO3)a-3H2O, 0,165 г FeCls-6H2O, 1,0 г
NHaCl, 30 г каолина. 50 см8 воды (коррод-
кот-испытание).......................... 44,000
8,75 20 лет
0,25 15 »
0,25 20 »
6,27 7 суток
1,25 62 »
70,00 16 ч
825,00 20 ч
185,00 20 ч
кот, а для воспроизведения морской атмосферы, как это видно
из табл. 18, лучшие результаты дает испытание при распылении
5%-ного раствора хлористого натрия, содержащего уксусную
кислоту и хлористую медь. Испытание при распылении 5 %-кого <
хлористого натрия, содержащего только уксусную кислоту, в те- !
чение 192 ч создает коррозионные разрушения, меньшие, чем на- *
бл юдавшиеся в промышленной и морской атмосферах за один
год испытания.
Таким образом, в зависимости от вида покрытия и условий,
в которых предполагается эксплуатировать изделия, выбирают
одно из рассмотренных выше испытаний.
Оценку скорости коррозионных разрушений металлических <
покрытий чаще всего производят по условной шкале визуально. i
Определяют количество очагов коррозии или площадь, занятую 1
коррозией. В последнем случае вводят показатель коррозии, со-
ответствующий той или иной площади образца, занятой корро-
зией (оценка коррозионной стойкости по 5-балльной шкале):
Площадь, занятая коррозией, % . . 0—5 5—10 10—20 20—50® 50—100
Индекс коррозии............ 1 2 3 4’""'т g
В некоторых случаях определяют глубину коррозионных по-
ражений или потерю массы. Последнее возможно лишь в том
—175 —
«случае, если коррозия не выходит за пределы верхнего слоя по-
крытия.
Рассмотрев методы ускоренных испытаний покрытий для вы-
/ явления их защитной способности, необходимо отметить, что су-
ществует ряд свойств покрытий, которые в значительной степени
определяют их качество. К таким свойствам относятся внутрен-
ние напряжения, сцепляемость покрытия с основным металлом,
пористость и толщина покрытия.
Таблица 18
СРАВНЕНИЕ РЕЗУЛЬТАТОВ ИСПЫТАНИИ В ЕСТЕСТВЕННЫХ УСЛОВИЯХ
И УСКОРЕННЫХ ИСПЫТАНИИ МНОГОСЛОЙНЫХ ГАЛЬВАНИЧЕСКИХ ПОКРЫТИИ
(Си —Ni —Сг) НА ЦИНКОВОМ ЛИТЬЕ* [11]
Толщина покрытая мкм Испытания в естественных условиях (1 год) Ускоренные испытания
Си Ni Сг S'G’" Sew юмышлен- 1я атмосфе- t (Колум- w) я „ о “ Ф о S Я о о ание /слой той и •2Н»О 5-испы- ) 36 ч
о и— О Й-SCT о о а* § >5^
к = о, к и ало S УСЧ й К о и— я о МОУ £
10,0 20,0 4 0 5,8 4,7 8,6 5,2 3,0 4,7
17,5 32,5 4 0 7,0 5,8 9,1 5,6 3,6 6,2
10,0 40,0 4,0 7,7 7,0 9,6 7,4 4,4 - 6.8
10,0 20,0 4,0 1,5 3,9 6,0 5,0 3,4 3,2
10,0 20,0 4,0 6,7 6,2 9,7 6,8 4,4 6,0
10,0 20,0 8,0 9.9 8,9 10,0 9,2 9,7 9.4
* Оценка коррозионной стойкости производилась по 10-балльной шкале.
Отсутствие коррозии оценивается баллом 10.
Способы изучения свойств покрытий подробно описаны в кни-
ге А. Т. Баграмяна и 3. А. Соловьевой [12].
Коррозионная стойкость катодных покрытий в основном опре-
деляется их пористостью, поэтому исследователь должен уметь
быстро провести такое определение. Существуют различные ме-
тоды определения пористости покрытий; наиболее простые и рас-
пространенные — коррозионные и электрохимические.
Пористость коррозионными методами определяется без мало-
жения и при наложении тока. Образец с покрытием подвергается
действию коррозионного агента, который, разрушая подкладку,
не действует на металл покрытия. В местах пор появляются про-
дукты коррозии, которые и характеризуют пористость. Исходя из
сущности метода видно, что пористость может определяться и
при проведении уже рассмотренных коррозионных испытаний.
Однако удобнее определять пористость, применяя реактивы, да-
ющие с основным металлом окрашенные соединения. В табл. 19
приведены составы растворов для ряда покрытий и способы ис-
пытания пористости последних [12].
12 Зак. хззг —-177
Таблица 19
РЕКОМЕНДУЕМЫЕ МЕТОДЫ ИСПЫТАНИЯ ПОРИСТОСТИ ПОКРЫТИЙ [12]
Покрытие Основной металл Состав раствора Продол- житель- ность испытаний мин Способ испытания ГОСТ
Золото Медь и медные сплавы 10 z/дм3 красной кровяной соли 4- 4-60 z/дм3 хлористого натрия 4-. 4- 30 г/дм3 хлористого аммония 10-60 Наложение бумаги и после- — дующая обработка ее раство- ром желтой кровяной соли
Никель 10 г/Эл3 красной кровяной соли + 4- 20 г/дм3 хлористого натрия Ю—60 Наложение бумаги и после- — дующая обработка ее аммиач- ным раствором диметилглиок- сима
Медь Сталь 10 г/дм3 красной кровяной соли 4- 4- 20 г/дм3 хлористого натрия 20 Наложение бумаги 3247—46
Никель Сталь 10 г/дм3 красной кровяной соли 4- 4- 20 г/дм3 хлористого натрия 5 Наложение бумаги 3247—46
Медь и латунь 10 г/дм3 красной кровяной соли 10 Наложение бумаги и после- 3247—46 дующая обработка ее раство- ром желтой кровяной соли
Олово Сталь 2 г красной кровяной соли 4- Ю см3 0,5-н. серной кислоты 4-20 или 40 г пищевой желатины 4-200 см3 этило- вого спирта 4-790 см3 дистиллиро- ванной воды 10 Смазывание раствором 3265—46
» 10 г/дм3 красной кровяной соли 4- 4- 5 z/дм3 хлористого натрия 60 Наложение бумаги 3264—46
Медь и латунь 10 z/дм3 красной кровяной соли 4~ 4- 10 г/дм3 сернокислого натрия 5 Анодная обработка при плот- 3264—46 ности тока 0,5—0,6 а/дм3
•ш-
Пр одолжен и-е табл. 19
Покрытие Основной металл Состав раствора Продол- житель- ность испытаний мин Способ испытания ГОСТ
1 Свинец Сталь 40 а/дм3 желтой кровяной соли-f- 4-2 г/дм9 хлористого натрия 5 Анодная обработка при нап- — ряжении 6 в
Медь 10 г/дм3 красной кровяной соли -|- 4- 10 г/дм9 желтой кровяной соли 4- -j- 100 а/дм9 сернокислого натрия 5 Анодная обработка при плот- — ности тока 0,35—0,2 а/дм*
i Хром Сталь 10 а/дм9 красной кровяной соли 4- 4~ 60 а/сЫ3 хлористого натрия -J- 4- 30 г/дм9 хлористого аммония 10 Наложение бумаги 3247—46
Никель 10 а/дм3 красной кровяной соли 4- 4- 60 г/дм9 хлористого натрия 4- 4- 30 а/дм9 хлористого аммония 10 Наложение бумаги и после- 3247—46 дующая обработка ее раствором желтой кровяной соли
» 10 г/дм9 красной кровяной соли-|~ 4-60 а/дм9 хлористого натрия 4- 4- 30 г/дм9 хлористого аммония 10 Наложение бумаги и после- 3247—46 дующая обработка ее раство- ром желтой кровяной соли
» То же 10 Наложение бумаги и после- 3247—46 дующая обработка ее аммиач- ным раствором диметил глиок- сима
Цинк С га ль 40 а/дм3 желтой кровяной соли 4- 4- 2 г/дм9 сернокислого натрия или сернокислого магния 5 Анодная обработка при нап- 3265—46 ряжении 4 ± 0,4 <?
Медь, ни- кель, хром Сталь 10 г/дм3 красной кровяной соли 4- 4-15 а/дм9 хлористого натрия 4- 4- 20 а/дм3 пищевой желатины 5 Заливка раствором 3247—46
Для ускорения процесса растворения металла в порах-приме-
няют анодную поляризацию испытуемого образца; в некоторых
случаях применяют и катодную поляризацию. Электрографиче-
ский метод является некоторой разновидностью коррозионного
испытания с наложением тока. Определение пористости произво-
дят при определенном потенциале металла подкладки. Продукты
растворения, проникая через поры осадка, взаимодействуют с
проявителем и дают окрашенный отпечаток, характеризующий
распределение пор. При применении этого метода фотографиче-
скую бумагу, предварительно обработанную соответствующим
реактивом, накладывают на исследуемое покрытие и плотно за-
жимают между двумя электродами в прессе. Через 15 сек после
включения тока подложка отделяется и проявляется специаль-
ным реактивом. В местах пор образуются окрашенные пятна.
Метод рекомендуется только для определения пористости нике-
ля; наилучшим реактивом является раствор сернокислого калия.
Для определения стойкости анодных покрытий необходимо
прежде всего знать толщину покрытия. Толщину покрытий опре-
деляют различными методами. Наиболее распространенные из
них: метод снятия покрытия, метод непосредственного измерения
толщины покрытия, капельные и струйные методы, а также маг-
нитный метод. При определении толщины покрытия первым ме-'
тодом изделие взвешивают перед удалением покрытия и после
снятия покрытия в реактиве, не реагирующем с основным метал-
лом. Этот метод применим для мелких изделий и даёт возмож-
ность определить лишь среднюю толщину покрытия; Опрёделё---
ние толщины непосредственным измерением размеров изделий до
и после снятия покрытия производят микрометром.
Применение капельного метода позволяет определять толщи-
ну покрытия в данной точке. Сущность метода заключается в
растворении заданного участка поверхности покрытия под капе-
лей наносимого раствора за определенное время. Капли наносят
на изделие на 1 мин последовательно одна за другой с-предвари-
тельным высушиванием каждой предыдущей капли'- фильтро-
вальной бумагой. Нанесение капель продолжается до обнажения-
основного металла при однослойном покрытии или промежуток
кого слоя при многослойном покрытии. Расчёт толщины покры-
тия в данном месте производят по формуле . -
Т ~ (N —!)/<, (92)
где N—число капель, потребовавшихся для обнажения метал-з
ла подложки:’
К — толщина покрытия, снимаемая одной- каплей, при дан^
ной температуре в течение одной минуты; ' * t
Т т~ местная толщина, испытуемого покрытия,-* мкм. .
Если профиль детали не препятствует стеканию1 струи, то при-
меняют струйный метод, -который ио .существу является усовер- *
шенствованным капельным методом. .Точностьгстр-уй’нодо методзР
12* ^179.—.
выше капельного и составляет 10—15%. Участок,-выбранный для
определения толщины покрытия, подвергается действию струи
специального раствора, вытекающего с определенной скоростью
(10 см3 за 30 сек). Раствор выпускается из капилляра бюретки
с краном. Капилляр располагается на расстоянии 4—5 мм от по-
верхности детали и под углом 45°. Толщину покрытия рассчиты-
вают по формуле
Т = -у, (93)
где N — продолжительность действия струи до появления харак-
терного розового пятна контактно выделившейся меди
или оголения основного металла;
К—продолжительность растворения покрытия толщиной
в 1 мкм, сек. Значения коэффициента К определяются
для данной температуры и вида покрытия.
Магнитный метод позволяет определить толщину покрытия
в любой точке и без нарушения его. Сущность метода состоит в
измерении магнитного поля, изменяющегося в зависимости от
толщины слоя нанесенного металла. Для проведения измерений
по этому методу необходимо предварительно получить градуи-
ровочные кривые. Измерение толщины покрытия магнитным ме-
тодом производят с помощью специальных магнитных толще-
меров.
Описание других методов определения толщины покрытий
можно найти в работах [1, 3, 13, 14].
2. НЕОРГАНИЧЕСКИЕ ПЛЕНКИ
Неорганические защитные пленки получаются при обработке
металлов соответствующими электролитами, превращающими
поверхностный слой металла в химическое соединение.
Такого превращения достигают при химическом или электро-
химическом воздействии какого-либо реагента на металл. Состав
пленок^ получаемых при такой обработке, различный; чаще всего
этВУ&кйсные, фосфатные или хроматные пленки. Большинство
плф^бк, получаемых на стали, алюминии и магнии химическим
путем” самостоятельного значения для защиты металлов от кор-
розии не имеет. Но применение их в качестве подслоя под лако-
крйёочные покрытия значительно увеличивает защитную способ-
ность последних. В некоторых случаях, например при воронении,
окисные пленки при наличии на них слоя смазки могут оказывать
защитное действие в слабоагрессивных средах. Окисные пленки
на магнии используют для защиты изделий в межоперационный
период, при кратковременном хранении и транспортировке.
Испытания по. установлению защитной способности неоргани-
ческих пленок целесообразно проводить только в том случае,
если они предназначаются для защиты от коррозии.
-1180 —
К такого рода пленкам можно отнести: анодные пленки на
алюминии, окисные пленки иа стали с дополнительной смазкой,
фосфатные пленки на различных металлах, анодные пленки'на
магнии.
Определять защитную способность неорганических, пленок
при ускоренных, испытаниях следует, учитывая среду, в которой
предполагается эксплуатация изделий с этим видом защиты.
Методы ускоренных испытаний были подробно рассмотрены
выше. Отметим лишь некоторые особенности ускоренных ис-
пытаний для выявления защитной способности неорганических
пленок.
При испытании алюминиевых сплавов с анодной пленкой по
методу погружения в электролит необходимо предусмотреть час-
тую смену раствора, если анодная пленка наполнена хромпиком.
В первый период испытания раствор меняют каждые сутки, за-
тем один раз в пять суток. Смена раствора необходима потому,
что из наполненной пленки вымывается хромпик, который сни-
жает коррозионную активность -раствора. Алюминиевые сплавы
с анодной пленкой, предназначенные для применения в промыш-
ленной атмосфере, по данным работы [11], можно испытывать,
применяя ускоренный метод корродкот. Хорошие результаты по-
лучаются при испытании во влажных камерах, с распылением
•растворов хлористого натрия, содержащих, уксусную кислоту и
CuClg (СА SS-испытание),.
При ускоренных, испытаниях черных металлов с фосфатной
пленкой их погружают в раствор хлористого натрия или испыты-
вают во влажных камерах при распылении раствора хлористого
натрия или дистиллированной воды, насыщенной кислородом.
Защитная способность неорганических покрытий при ускорен-
ных. испытаниях определяется по потере массы образцов, количе-
ству основного металла, перешедшего в раствор, времени до по-
явления первого коррозионного поражения или по величине по-
верхности, занятой коррозией. Защитная способность неоргани-
ческих пленок в большей степени зависит от их пористости и тол-
щины. Поэтому, даже не проводя коррозионные испытания, неко-
торые данные о пленках можно быстро получить, применяя так
называемый капельный метод. Сущность метода заключается в
том, что после воздействия на пленку капли агрессивного ре-
актива пленка частично разрушается, и начинается коррозия
основного металла, о появлении которой судят по резкому изме-
нению цвета капли. Время до изменения цвета определяется по
секундомеру и служит характеристикой защитных свойств
пленки.
Капельный метод испытания качества анодной пленки на
алюминиевых сплавах состоит в том, что на испытываемую де-
таль наносят 1—2 капли раствора, содержащего 25 см3 соляной
кислоты плотностью 1,19, 3 а двухромовокислого калия и 75 см3
дистиллированной воды. При нанесении капель раствора на ано-
— 181 —
дированную поверхность алюминиевого сплава соляная кислота
проникает через покрытие до основного металла и реагирует с
ним, выделяя водород. При этом шестивалентный хром восста-
навливается до трехвалентного, в* результате чего оранжевая
окраска раствора переходит в зеленую. Качество пленки опреде-
ляют по времени, прошедшему с момента нанесения капли до на-
чала ее позеленения.
Обычно устанавливают определенные нормы, которые долж-
ны выдержать покрытия при капельной пробе. Эти нормы, как
видно из табл. 20, зависят от способа получения пленки и темпе-
ратуры испытания.
Таблица 20
ВРЕМЯ ВЫДЕРЖКИ ПРИ КАПЕЛЬНЫХ ИСПЫТАНИЯХ АНОДНЫХ ПЛЕНОК
ИА АЛЮМИНИЕВЫХ СПЛАВАХ В ЗАВИСИМОСТИ ОТ ТЕМПЕРАТУРЫ
И СПОСОБА АНОДИРОВАНИЯ [15]'
Температура испытания °C Нормы выдержки капли, мин. для пленок, полученных „ _ „ _А, в 20%-ном растворе ELSOe с по- в 8, 5 и 9,5%-ном растворе следующей обработкой в K2Cr2O, crtJs или горячей воде для плакирован- Для «еплакиро- для плакиро- для неплакиро- ного материала ванного мате- ванного мате- ванного мате- * риала риала риала
18—21 22—26 27—32 6,0 4,0 12,0 5,0 4,5 3,0 8,0 4,0 3,0 2,0 7,0 3,0
Окисные пленки на стали, полученные в щелочных растворах
с последующим промасливанием, применяют для защиты от ат-
мосферной коррозии и для декоративной отделки неответствен-
ных деталей при эксплуатации их в мягких условиях.
Качество оксидного слоя можно контролировать ускоренным
методом, погружая деталь или образец в 3%-ный раствор серно-
кислой меди или путем нанесения этого раствора на поверхность
изделия. Если в течение 30 сек на поверхности изделия не по-
явится участков, покрытых красной контактно высадившейся
медью, значит пленка обладает удовлетворительными свойства-
ми Допускается наличие 2—3 точек выделившейся меди на 1 см2
поверхности; размер точек в диаметре не должен превышать
1 мм.
Для оценки пористости пленки можно применять раствор, со-
держащий 10 г!дм3 K3[Fe(CN)6], 15 г!дм3 NaCl и 3—5 г!дм3 жела-
тины. Этим раствором смачивают фильтровальную бумагу и
плотно накладывают на поверхность -изделия, через 60 сек под-
считывают число синих точек. Пленка считается хорошего каче-
ства, если число синих точек на 1 см2 не превышает трех.
Для определения свойств фосфатных пленок Г. В., Акимовым
предложен метод, заключающийся в нанесении на поверхность
— 182 —
металла с фосфатной пленкой капли раствора, содержащего
40 см3 0,5-м. раствора CuSO4 • 5Н2О + 20 см3 10 %-кого раствора
NaCl + 0,8 см3 0,1-м. раствора НС1. Защитные свойства фосфат-
ных пленок на черных металлах определяют по времени до изме-
нения цвета капли раствора от сине-голубого до красновато-
желтого. В. С. Лопатухин [16] указывает, что время до изменения
цвета капли на поверхности черных металлов с фосфатными
пленками, полученными горячим способом, характеризует их
коррозионную устойчивость в соответствии со следующими дан-
ными:
Время до изменения цвета
капли, сек............ >300' 120-—295 60—115 <60
Характеристика защитных
свойств покрытия .... Высокая Средняя Пониженная Низкая
Как показали исследования В. С. Лопатухина, капельный ме-
тод Г. В. Акимова, предложенный для черных металлов, может
быть применен и для определения качества фосфатных пленок,
получаемых на цветных металлах. Время выдержки, характери-
зующее удовлетворительные свойства пленок, должно быть при
этом снижено. Так, например, пленки, полученные на стали при
холодном фосфатировании, обладают удовлетворительными за-
щитными свойствами, если они выдерживают капельную пробу в
течение 1,5 мин. Для фосфатных пленок на цинке, алюминии и
магнии время, в течение которого они выдерживают капельную
пробу, снижается до 40, 20 и 8—10 сек соответственно..
Из приведенных данных видно, что по результатам капельной
пробы фосфатные пленки, полученные различными методами
даже для одного и того же металла, сопоставлять нельзя. Для
одного и того же метода получения пленок капельная проба мо-
жет характеризовать изменение защитных свойств в зависимости
от режима или состава ванны.
Пленки, получаемые на магнии при электрохимическом окси-
дировании в щелочных или хромовокислых электролитах, бес-
цветны, характеризуются высокой твердостью и износостойко-
стью; обладают большей защитной способностью, чем пленки,
полученные химическим оксидированием.
Чтобы повысить защитные свойства пленок, их обрабатывают
в 5 %-ном растворе бихромата калия при 90—100° С в течение
20—30 мин.
Быстрое определение качества анодных пленок на магнии и
его сплавах производится визуально; не должно быть участков,
не покрытых пленкой, царапин, рыхлого налета. На оксидирован-
ную поверхность наносят каплю раствора, содержащего 1 % хло-
ристого натрия и 0,1% фенолфталеина (в виде спиртового рас-
твора). О качестве пленки судят по времени с момента нанесе-
ния капли до окрашивания ее в розовый цвет (табл. 21).
— 183 —
Таблица 21
ВРЕМЯ ВЫДЕРЖКИ ЗАЩИТНОЙ ПЛЕНКИ НА МАГНИЕВЫХ СПЛАВАХ
ПРИ капельных испытаниях
Марка сплава Время до изменения цвета капли (мин) при различных температурах испытания, °C
15 20 25 30 35 40
МАЗ (до 1,5 лш) 6,0 10,0 15,0 19,0 23,0 30,0
МАЗ (до 3 мм} 1.2 2,0 3,0 3,8 4,6 6,0
МА1 1,2 2,0 3,0 3,8 4,6 6,0
МА5 0,6 1,0 1,5 1,9 2,3 3,0
МЛ4 0,6 1,0 1,5 1,9 2,3 3,0
МЛ6 0,6 1,0 1,5 1,9 2,3 3,0
В тех случаях, когда неорганические пленки применяют толь-
ко в качестве подслоя под лакокрасочные покрытия или в соче-
тании с другими видами коррозионной защиты, ускоренным ис-
пытаниям должны подвергаться образцы с комплексной защи-
той. Метод ускоренного испытания, как и во всех других случаях,
должен выбираться исходя из условий эксплуатации.
3. ЛАКОКРАСОЧНЫЕ ПОКРЫТИЯ
Защита металлов полимерными пленками может осуществ-
ляться двумя путями: полной изоляцией металла от воздействия
окружающей среды или введением в состав пленки веществ, тор-
мозящих электродные реакции,— пассивирующих грунтов. В пер-
вом случае пленки должны быть непроницаемы для воды, кисло-
рода и ионов электролитов. Тогда защитные свойства полимер-
ного покрытия будут определяться лишь свойствами самого по-
крытия. Абсолютной непроницаемости покрытия, как правило,
достичь не удается, тем более что покрытие во времени стареет,
растрескивается и свойства его ухудшаются.
Действие большинства применяемых грунтов основано на
принципе торможения электродных реакций. Свойства таких ла-
кокрасочных покрытий определяются не только пассивирующими
.свойствами пигментов, входящих в состав покрытия, но и физи-
ко-химическими свойствами пленок, а также их адгезионной спо-
собностью.
Основа многих лакокрасочных материалов — высокомолеку-
лярные синтетические соединения. Ввиду недостаточно плотной
структуры и содержания гидрофильных групп в большинстве
плёнкообразующих они обладают способностью поглощать из
окружающей среды и пропускать через себя различные жидкие и
газообразные вещества.
— 184 —
Диффузия паров и газов через пленку представляется как
продвижение мигрирующей частицы пара или газа через времен-
ные поры, которые образуются и вновь исчезают вследствие гиб-
кости полимерных цепей. Поэтому скорость диффузии зависит от
многих факторов: величины временных пор, размеров мигрирую-
щей частицы, степени ее растворимости в полимере, а также от
подвижности цепей полимера и плотности их упаковки. В неко-
торых случаях сами полимерные пленки могут оказаться пере-
носчиками ионов среды к металлу.
Проникновение влаги в покрытия обусловливается также
осмотическими процессами в лакокрасочных пленках, представ-
ляющих собой полупроницаемые мембраны.
Поглощение пленкой влаги зависит от осмотического давле-
ния воздействующего раствора: при увеличении концентрации
раствора, а следовательно, при повышении осмотического давле-
ния адсорбция влаги пленкой уменьшается. Поглощение влаги
сопровождается увеличением объема пленки; при усиленном при-
токе влаги может нарушиться адгезия, что приведет к вспучива-
нию покрытия и его отслаиванию. Чем выше адгезия, тем мень-
ше склонность к вспучиванию.
Водонабухаемость не всегда характеризует степень проник-
новения воды через пленку. Абсорбция воды не зависит от про-
ницаемости покрытия.
Защитные свойства покрытий определяются поэтому рядом
физико-химических свойств (пассивирующая способность грунта,
диффузия электролитов, водонабухаемость, паро- и водопрони-
цаемость, адгезия, внутренние напряжения, механические свой-
ства, старение и т. д.). Весь комплекс свойств покрытий может
быть изучен путем раздельного определения физико-химических
и механических характеристик покрытия. Однако при ускорен-
ных методах испытаний часто достаточно определить лишь за-
щитную способность пленки при воздействии на нее окружающей
среды.
Методика определения отдельных свойств покрытий подробно
описана в работе С. В. Якубовича [17], поэтому мы ограничимся
лишь разбором способов ускоренного определения защитной
способности покрытий в целом.
Принцип классификации ускоренных методов по характеру
создаваемой коррозионной среды, принятый при рассмотрений
ускоренных методов испытаний металлов, сохраняется и для ла-
кокрасочных покрытий. Основные закономерности разработки
и выбора ускоренных испытаний для лакокрасочных покрытий в
основном остаются те же, добавляются лишь некоторые специ-
фические особенности, определяющиеся свойствами системы ме-
талл — лакокрасочная пленка.
Ускоренные испытания металлов, защищенных лакокрасоч-
ными пленками, проводятся при непрерывном и периодическом
погружении в различные электролиты в аппаратах, где воспро-
— 185 —
изводятся атмосферные условия или условия воздействия газо-
вых сред, электролитов и т. д.
Испытания при погружении в электролиты. При испытании-
защитных свойств покрытий необходимо прежде всего правильно
подготовить образцы. Они не должны иметь оголенных мест,
углы и края должны быть тщательно защищены, например за-
мазкой, состоящей из 60 ч. битума, 60 ч. канифоли и 30 ч. цере-
зина. Покрытие должно быть одинаковой толщины и наноситься
•на обе стороны пластины; нанесение различных покрытий на од-
ну пластину не допускается.
При испытании в условиях погружения образцы не должны
касаться друг друга и какого-либо другого металла. Лучше все-
го каждый образец испытывать в отдельном сосуде. Для этой
цели можно применять стеклянные и керамические ванны с пе-
регородками. Образцы помещают в отдельные ячейки и укрепля-
ют на стеклянных подставках или крючках.
Известно, что свойства лакокрасочных покрытий сильно за-
висят от воздействия света, поэтому даже при испытаниях в ус-
ловиях погружения должны соблюдаться одинаковые условия
освещенности. Электролитом для такого рода испытаний (в зави-
симости от условий эксплуатации, для которых предназначается
испытуемое покрытие) могут служить растворы хлористого нат-
рия, морская или речная вода и др.
Чтобы ускорить процесс коррозии под пленкой полимерного
покрытия, применяют те же методы, что и при испытании метал-
лов, например испытание в перемешиваемых растворах, когда
движется раствор или сами образцы, испытание при периодиче-
ском смачивании или обрызгивании образцов электролитами.
Аппаратура для таких испытаний не отличается от уже опи-
санной для ускоренных испытаний металлов без покрытий. До-
полнительно вводится лишь ультрафиолетовое облучение.
Образцы могут быть различной величины; чаще всего это
пластинки размерами 30 X 50, 50X100, 90X120, 70X150 мм. Вме-
сто пластинок можно применять металлические стержни диамет-
ром 12—15 мм, длиной 70—80 мм. Стержни закругляются снизу,
чтобы устранить влияние острого края.
Испытания, воспроизводящие атмосферные условия. Испыта^
ния лакокрасочных покрытий, предназначенных для эксплуата-
ции в атмосферных условиях, проводят во влажной чистой атмо
сфере, а также при введении различных коррозионных агентов,
содержащихся в атмосфере и ускоряющих коррозионный про-
цесс. В некоторых аппаратах предусматривается возможность
создания конденсационной пленки на поверхности окрашенных
образцов.
Широко распространено испытание окрашенных образцов на
устойчивость против коррозии в атмосферных условиях в аппа-
рате искусственной погоды. Для этой цели в СССР и за границей
применяют везерометры.
— 186 —
Аппараты искусственной погоды (везерометры) бывают раз-
личной конструкции. В настоящее время применяют аппараты
искусственной погоды, изготовленные в Государственном иссле-
довательском и проектном ин-
ституте (ГИПИ-4), а также
•аппараты марки ИП-1-3 (рис.
108), опытную партию которых
выпустил Хотьковский завод
экспериментальной и окрасоч-
ной технологии. Аппарат
ИП-1-3 состоит из изолиро-
ванной камеры, внутри кото-
рой установлен цилиндр, вра-
щающийся со скоростью
0,1 рад!сек (1 обIмин). В цент-
ре барабана помещены две
дуговые лампы и две ртут-
но-кварцевые лампы ПРК-2
для ультрафиолетового освеще-
ния. В аппарате находятся две
системы орошения: для созда-
ния влажности в камере и для
•орошения самих образцов. Об-
разцы в виде пластин распо-
Рис. 108. Внешний вид аппарата
искусственной погоды ИП-1-3
лагаются в аппарате верти-
кально. Температуру можно
поддерживать постоянной в
пределах 30—90° С.
Ниже приведены режимы испытаний в везерометре, рекомен-
дуемые С. В. Якубовичем [17]:
Режим I II
Продолжительность облу-
чения, мин ..... 17 50
Продолжительность оро-
шения, мин............ 3 10
Температура испытания,
°C .................... 60 60
III IV V VI VII
40 30 10 Непрерывно Непрерывно
20 30 50 — »
60 60 60 50 45-50
Режимы отличаются длительностью попеременного воздейст-
вия света и обрызгивания водой.
Осмотр образцов производят ежедневно, для чего аппарат
останавливают на один час. Режим выбирают в зависимости от
цели испытаний. Ускорение процесса увеличивается при перехо-
де от режима I к последующим. При испытаний по режиму I
выявляются все виды разрушений покрытий, кроме коррозии;
яри испытании по режиму V ускоряется главным образом про-
цесс меления; режим VI выявляет потерю глянца и цветоустой-
чивость покрытия при почти полном отсутствии меления. Наи-
— 187 —
большее ускорение разрушения покрытия наблюдается при ме-
тоде испытании по режиму VII (имитирует тропический влажный
климат).
Для проведения испытаний при низких температурах, кроме
везерометра, используют холодильник, в котором через опреде-
ленные промежутки времени образцы охлаждаются до —40° С.
При таких переменах температуры сильнее всего проявляется
склонность покрытий к растрескиванию, вследствие чего корро-
зионные процессы ускоряются.
Коррозионные разрушения покрытий при испытаниях уско-
ренными методами в аппаратах искусственной погоды и в естест-
венных атмосферных условиях иосят одинаковый характер.
Изменение состояния покрытия, происходящее во времени,
оценивают качественно по визуальному осмотру (ГОСТ 6992—
60). Полученные данные выражают отдельно по каждому виду
Таблица 22
ШКАЛА ОЦЕНКИ ДЕКОРАТИВНОГО ВИДА'ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
Баллы Виды разрушений
изменение глянца (блес- ка) (Г) изменение цвета и от- тенка (Ц) бронзировка (Бр) белесоватость (Б), пятна (П) грязеудержа- ние (Гр)
5 Едва замет- ное Едва замет- ное Отсутствует Отсутствует Отсутствует
4 (ДО 20%) Незначи- тельное Незначи- тельная Незначительная Незначи- тельное
3 (ДО 50%) Значитель- ное Значитель- ная Значительная, пятна местами Значитель- ное
2 (до 80%) Сильное Сильная Сильная, пятна по всей поверх- ности Сильное
1 Сильное (свыше 80%) Полная по- теря основ- ного цвета » Сильная, пятна по всей поверх- ности »
разрушения. Различают изменения декоративного вида лакокра-
сочного материала, которые оцениваются по пятибалльной шка-
ле (табл. 22), и изменения защитных свойств, оцениваемые по
десятибалльной шкале (табл. 23).
Окончательный вывод об устойчивости того или иного по-
крытия делают по двум показателям, найденным по указанным
таблицам. Декоративный вид покрытия определяют по измене-
нию глянца (блеска), цвета, бронзировки, белесоватости и гря-
зеудержания.
Изменение глянца покрытия — следствие начальной стадии
разрушения поверхностного слоя покрытия в результате фотохи-
мических процессов. Потеря глянца определяется фотоэлектриче-
ским методом при помощи блескомера марки ФБ-la и другими
— 188 —
681
Таблица 23
ШКАЛА ОЦЕНКИ ЗАЩИТНЫХ СВОЙСТВ ЛАКОКРАСОЧНЫХ МАТЕРИАЛОВ
Балл Виды разрушений
ы меление (М) выветривание (В) растрескивание (Р) отслаивание (О) пузыри (ГТ) коррозия (К)
10 Отсутствует Отсутствует Отсутствует Отсутствует Отсутствует Отсутствует
9 Едва заметное » » с» » »
8 Незначительное » Трещины или сетка поверхност- ные, видимые при 12-кратном увели- чении » » »
7 То же » Трещины или сетка поверхност- ные, местами види- мые при 4-кратном увеличении » » »
6 Значительное » То же, до 25% поверхности » Сыпь до 25% поверхности Отдельные точки коррозии только для стали и железа
5 То же Выветривание верхнего слоя ме- стами То же, до 50% поверхности Верхнего слоя местами Сыпь до 50 % поверхности Коррозия до 5% поверхности
4 Сильное. Про- Просвечивание То же, свыше От грунта или • Сыпь свыше 50% То же, до 10%
свечиванне грунта или подложки грунта или под- ложки 50% поверхности. Глубокие трещины местами от подложки до 100% поверхности поверхности. Ме- стами пузыри поверхности
3 Просвечивание грунта или подлож- ки до 25% поверх- ности Просвечивание грунта или подлож- ки до 25% поверх- ности Глубокие трещи- ны до 25% поверх- ности То же, до 25% поверхности Пузыря до 25% поверхности То же, до-25% поверхности
2 То же, до 50% поверхности То же, до 50% поверхности То же, до 50% поверхности .То же, до 50% поверхности То же, до 50% поверхности То же, до 50% поверхности
1 То же, свыше То же, свыше То же, свыше То же, свыше То же, свыше То же, свыше
50% поверхности 50% поверхности 50% поверхности 50 % поверхности 50% поверхности 50% поверхности
приборами или визуально — сравнением с эталоном. Остальные
виды изменения внешнего вида определяют только визуально и
применяя различные эталоны.
Изменение защитных свойств покрытия определяется сте-
пенью меления, выветривания, растрескивания, отслаивания, а
также наличием пузырей и коррозии.
Мелением называется поверхностное разрушение пигментиро-
ванной лакокрасочной пленки, приводящее к образованию сво-
бодных частиц пигмента, легко удаляющихся с покрытия. У пиг-
ментированных покрытий, содержащих различные белые
пигменты (окись цинка, двуокись титана и др.), на поверхности
образуется белый порошок, при удалении которого поверхность-
приобретает свой первоначальный вид. Меление возникает под
воздействием ультрафиолетовых лучей и активизируется под воз-
действием влаги и резких колебаний температуры. Меление, со-
гласно ГОСТ 6992—60, определяют по характеру и степени сво-
бодно отделяющегося от поверхности покрытия пигмента, путем
трения тканью (фланелью) — черной для светлых покрытий и
белой для темных. Существует и другой метод определения меле-
ния, описанный в работе [17]. Полоску фотобумапддириной 30 мм
накладывают на испытываемое изделие на 10 сек и надавлива-
ют на_ нее резиновым штемпелем. Предварительно полоски фото-
бумаги погружают на 2 мин в воду, излишек воды затем удаляют
фильтровальной бумагой, в результате чего фотобумага остает-
ся слегка влажной. Отпечаток на фотобумаге сравнивают со
шкалой меления, подбирая соответствующий ему индекс.
Остальные показатели коррозии определяют сравнением'с
фотоэталонами, приложенными к ГОСТ 6992—60.
Появление коррозии на окрашенной поверхности свидетель-
ствует о явном разрушении окрашенного металла. Коррозию оп-
ределяют по проценту поверхности, занятой ею. Для более точ-
ного определения на испытываемый образец накладывают сетку
и подсчитывают число клеток, в которых наблюдается тот или
иной вид разрушения. Это количество клеток соответствует про-
центу разрушения поверхности лакокрасочной пленки.
Все упомянутые методы оценки устойчивости лакокрасочных
пленок носят качественный характер и, конечно, не могут доста-
точно точно дифференцировать покрытия. В исследовательских
работах поэтому составляют графики для каждого вида разру-
шения в координатах длительность испытания — изменение со-
стояния покрытия в процентах или баллах.
Количественно коррозионную стойкость окрашенных образ-
цов определяют путем взвешивания пластинок др и после корро-
зии. Лакокрасочная пленка и продукты коррозии- перед взвеши-
ванием обязательно должны быть удалены.
Б. В. Бак [18] предложил метод оценки противокоррозионных
свойств лакокрасочных покрытий на железе по количеству ионов»
железа, перешедших' в раствор. Образцы испытывают в раство-
— 1-Q0—_
ре, содержащем 13,5 г надсернокислого калия и 15 а роданистого
аммония в I л воды. Концентрацию роданистого железа измеря-
ют через определенные промежутки времени с помощью фото-
колориметра.
Противокоррозионные свойства покрытий на легких сплавах
определяют методом электролиза по количеству меди, вытесняе-
мой из 0,02-н. раствора мед-
ного купороса основными
компонентами сплавов —
алюминием и магнием [19].
Испытания при периоди-
ческом погружении е элек-
тролит. Для испытания кор-
розионной устойчивости по-
крытий при периодическом
смачивании можно исполь-
зовать аппараты любой кон-
струкции, в которых преду-
смотрена возможность ук-
репления окрашенных образ-
цов с таким расчетом, чтобы
они периодически погружа-
лись в электролит, а в про-
межутки между погружени-
ями высушивались и подвер-
гались воздействию ультра-
фиолетового облучения.
Распространенным аппа-
ратом для испытания в усло-
виях периодического погру-
пытаний лакокрасочных покрытий под
воздействием воды, ультрафиолето-
вых лучей и тепла [17]:
1 — бутыль; 2 — кристаллизатор; 3 —
стеклянные сифоны; 4 — стеклянная во-
ронка; 5 — двугорлая склянка; 6 — ван-
на; 7 — кожух для кварцевой лампы
жения в электролит являет-
ся аппарат, разработанный Гопиусом, конструкция которого*
описана ранее (см. рис. 25).
Для испытания полимерных покрытий при периодическом
смачивании в настоящее время применяются различные прибо-
ры. Так, С. В. Якубович предложил прибор для испытания по-
крытий под воздействием воды, ультрафиолетовых лучей и теп-
ла (рис. 109). Он состоит из стеклянной бутыли емкостью 10 л^.
закрепленной в перевернутом виде. Горло бутылки соприкасается
с поверхностью воды, налитой в кристаллизатор, помещенный
под штативом. Кристаллизатор соединен через сифон и воронку
с двугорлой склянкой. Склянка соединена сифоном с эмалиро-
ванной ванной, помещенной под ртутно-кварцевой лампой, за-
ключенной в кожух. От ванны отходит металлическая трубка»,
которая отводит воду в сборник, помещенный на полу. Ток воды
можно регулировать зажимом на резиновой трубке, отходящей
0Т\ ваниы. Емкость склянки должна быть в два раза меньше ем-
кости-ванны; Образцы помещают на дно ванны и иа них попере-
—191 —
менно воздействуют вода и ультрафиолетовые лучи, а также
тепло, выделяемое лампой.
Цикл состоит из 15-мин воздействия ультрафиолетовых лу-
чей и воды и 30-лшн высыхания.
С. В. Якубович’и А. М. Грозовская предложили другой ре-
жим испытания на этом аппарате; суточный цикл при этом ре-
жиме состоит из попеременного облучения образцов кварцевой
лампой типа ПРК-2 (напряжением 375 в) при 60° С в течение
часа и облучения при погружении в воду (высота слоя воды
15 мм) в течение 30 мин. Затем через 15 ч на образцы наклады-
вается влажный компресс под небольшим грузом (1 /сг) и вы-
держивается 9 ч. Продолжительность испытания составляет
720 ч. Пластинки осматривают ежесуточно; разрушение оценива-
ют соответствующим баллом .по принятой системе.
Этот вид ускоренных испытаний позволяет сопоставить
полученные результаты с результатами испытаний в естествен-
ных условиях; коэффициент пересчета устанавливается по фор-
муле
К = , (94)
ь
где К — коэффициент пересчета; а — длительность нахождения
испытуемого образцз в атмосфере (на крыше здания), сутки:
b — длительность ускоренных испытаний, сутки.
Ускоренные и длительные испытания продолжают до одина-
ковой степени разрушения покрытия, оцененного по десяти-
балльной шкале ГИПИ-4. Коэффициент пересчета зависит от
типа покрытия и климатических условий. Коэффициент пересче-
та в условиях Москвы, по данным С. В. Якубовича, в среднем
равен 8.
Для ускоренных испытаний лакокрасочных покрытий при пе-
риодическом орошении, нагреве, охлаждении и освещении раз-
работан аппарат специальной конструкции [20]. Он представля-
ет собой колесо, на котором по окружности неподвижно укреп-
лено семь камер: холодильная, влажная, две световых, две.для
обогрева и одна дождевая камера. Схема этого аппарата приве-
дена на рис. НО. В холодильной камере обеспечивается темпе-
ратура —8° С пропусканием рассола с температурой около
—20° С по змеевикам, расположенным в камере. Во влажных
камерах поддерживается 100%-иая влажность пропусканием
мятого пара, конденсирующегося на стенках камеры. В свето-
вых камерах устанавливаются дуговые лампы. Тепловые камеры
снабжены лампами накаливания; температура в первой камере
40—50° С, во второй 25—30° С. В дождевой камере находятся
две форсунки, через которые подается водопроводная вода.
Одновременно на колесе можно испытывать 28 пластин. Ба-
рабан вращается от электромотора и делает один оборот в сут-
ки. Образцы находятся ,11 ч в холодильной камере, 1 ч вовлаж-
— 192 —
иой камере, в первой световой и первой тепловой камере по 3 ч
во второй световой и второй тепловой по 15 ч и в дождевой ка
мере 3 ч. Этот аппарат воспроизводит условия эксплуатации по
крытий.
Недостаток такого при- g
бора — его громоздкость; / | |
кроме того, время нахожде-
ния образцов в холодильной /
камере слишком завышено. /
Для подобных ускорен- 4 / 1г W
ных испытаний был также
разработан другой аппарат
[21] (рис. 111). Основное от-
личие этого аппарата от
Рис. 110. Колесо для ускоренных ис-
пытаний лакокрасочных покрытий
при периодическом орошении, нагре-
ве, охлаждении и освещении ,[20]:
1 — холодильная камера; 2 — камера по-
вышенной влажности; 3 — камеры света
и тепла; 4 — прорези для укрепления
образцов; б — камеры тепла; б — испы-
туемые образцы; 7 — каркас аппарата;
В — камера искусственного дождя; 9 —
сборник для воды; 10 — вращающийся
барабан
Рис. 111. Аппарат для ускоренных ис-
пытаний лакокрасочных покрытий при
периодическом орошении, нагреве,
освещении и высушивании искусст-
венным ветром [21]:
1 — колесо; 2 — кварцевая лампа; 3 —
электролампа (1000 ет); 4 — нагреватель-
ный элемент (инфракрасные лучи); 5 —
образец; б — подача воды; 7 — привод
от электромотора; В — сток воды; 9 —
приспособление для подачи воздуха от
вентилятора (искусственный ветер)
представленного на рис. 110 состоит в том, что образцы 5 распо-
лагаются на .колесе, .вращающемся в горизонтальной плоскости
со скоростью 11 об [сутки. Образцы располагаются иа горизон-
тальной поверхности колеса 1 и подвергаются воздействию воды
в виде искусственного дождя в течение 15 мин и тепла в виде ин-
фракрасных лучей от нагревательного элемента 4 в течение
25 мин, воздействию искусственного ветра со скоростью 10—
15 м[сек, света от электролампы мощностью 500—1000 ет в тече-
13 Зак. 1338 --193 —
ние 66 мин и ультрафиолетовых лучей от ртутной кварцевой лам-
.пы 2 в течение 20 мин при 20—30° С,
Продолжительность одного цикла 2 ч 6 мин. Двести циклов
такого испытания, по данным С. В. Якубовича [17],’ соответст-
вуют испытанию в течение
на коррозионной станции
Рис. 112. Экспресс-аппарат для
ускорения испытания лакокра-
сочных- масляных пленок при
увеличенном подводе кислоро-
да [22]:
1 — баллон с кислородом; 2 —
чашка с силикагелем; 8 — тер-
мометр; 4 — пластинка с испы-
туемым покрытием; 5 — крышка;
6 — кран; 7 — свечи; 8 — трубка
к вакуум-насосу; 9 — манометр;
10 — электрообмотка с асбестовой
изоляцией; 11 — корпус аппарата;
12 — электролампа (40 вт. 6 в);
13 — каркас для образцов; 14 —
трансформатор
6 месяцев в атмосферных условиях
в промышленном районе.
При изучении защитных свойств
покрытий важно также зиать, как
меняются их свойства под влиянием
старения. А. А. Дринберг и К. В. Ря-
шенцев [22] разработали метод уско-
ренного старения пленок масляных
покрытий. Метод основан на увели-
чении подвода кислорода за счет
создания повышенного давления в
камере, чередующегося с вакуумом.
Повышенное давление кислорода)
увеличивает скорость реакции меж-
ду молекулами масла и кислорода..
Испытания покрытий в процессе-
старения проводят при введении в
аппарат воды. Конструктивное офор-
мление этого метода испытаний, по-
лучившего название экспресс-аппа-
рат, дано на рис. 112. Аппарат пред-
ставляет собой камеру типа авто-
клава. Внутренний объем аппарата!
4 дм?. В крышке 5 вмонтирован ма-
нометр 9, патрубок для подвода и
вывода кислорода, термометр 3 и
осветительная лампа 12. Давление-
кислорода доводится до 4,5 бар, во>
время испытания температура под-
держивается равной 80—90° С; раз-
режение создается до 39-10—3 бар (30 мм рт. ст.). Чередо-
вание давления и вакуума в первые 4 ч происходит так: 1 ч ва-
куум, 1 ч давление, в последующие часы 2 ч давление, 1 ч ваку-
ум. Пар в аппарат пропускают каждый раз перед пуском кис-
лорода в количестве 15—20 г. Образцы осматривают каждые 4'
или 8 ч.
Авторы аппарата указывают, что результаты ускоренных ис-
пытаний покрытий на основе растительных масел отличаются от
полученных в атмосферных-условиях, но порядок расположения
различных покрытий по скорости их старения сохраняется. Что-
же касается защитных свойств, то коррозия-под масляной плен-
кой, появляющаяся в атмосферных условиях через каждые шесть
месяцев испытания, выявляется в экспреес-аппарате через;
48 часов.
— 194 —
В настоящее время на основании опыта научно-исследова-
тельских институтов лакокрасочной промышленности ускорен-
ные испытания стандартизированы и проводятся в соответствии
с руководящим техническим материалом (РТМ) 35—61 Всесо-
юзного научно-исследовательского института по нормализации в
машиностроении (ВНИИНМАШ). РТМ предусмотрены испыта-
ния при комбинированном воздействии ультрафиолетовых лучей,
тепла и влаги в аппаратах искусственной погоды или в более
простых установках, а также испытания во влажных камерах и
комбинированные испытания, воспроизводящие воздействие
тропического климата по режимам «тропики I» и «тропики 11».
При выборе метода ускоренных испытаний РТМ рекоменду-
ют пользоваться данными, приведенными в табл. 24.
При проведении ускоренных испытаний в аппаратах искусст-
венной погоды применяют стандартные режимы (табл. 25).
Испытания лакокрасочных материалов, предназначенных
для эксплуатации в районах с тропическим климатом, рекомен-
дуется в соответствии с РТМ 35—61 проводить как комбиниро-
ванные при последовательном воздействии различных климати-
ческих факторов. Разработано два режима испытаний «тропи-
ки I» и «тропики II» (табл. 26).
Если лакокрасочные материалы должны выдерживать рез-
кие температурные колебания и минусовые температуры, то при
испытаниях в аппаратах искусственной погоды по режиму I
и V вводится дополнительное охлаждение образцов в холодиль-
ной камере до —50° С. Продолжительность охлаждения 1 ч (см.
табл. 25).
Электрические методы определения защитной способности
лакокрасочных покрытий. Защитная способность покрытий ха-
рактеризуется некоторыми электрическими свойствами и их из-
менением во времени под воздействием коррозионной среды. К
таким свойствам относятся пробивное напряжение, сопротивле-
ние и емкость.
Пробивное напряжение характеризует одно из важнейших
свойств лаков и эмалей — пригодность их для изоляции. Связь
пробивного напряжения (электрической прочности) лакокрасоч-
ных материалов с их защитной способностью изучена еще мало.
Для оценки защитных свойств можно определять изменение
пробивного напряжения в процессе воздействия коррозионной
среды. Для такого рода испытаний обычно применяют свобод-
ную лакокрасочную пленку (ее наносят на папиросную бумагу
или алюминиевую фольгу, которую предварительно обрабаты-
вают тальком). Для определения изменения изолирующей спо-
собности материала величину пробивного напряжения измеряют
при воздействии воды иа медные окрашенные пластинки. Для
коррозионных исследований пленки следует наносить на тот
металл, для которого предназначено покрытие, и помещать его в•
коррозионную среду, имитирующую условия эксплуатации. Если
13s — 195—•
в лароютрютика те иды ускоренных
имитируемый климат атмосферных условий климатических испытаний
МЕТОДЫ УСКОРЕННЫХ ИСПЫТАНИЙ ЛАКОКРАСОЧНЫХ ПОКРЫТИЙ, ИМИТИ
Умеренно континен- Солнечная радиация, Испытания в аппарате
тальный осадки, промышленные и искусственной погоды
дымовые газы, морская марки ИП-1-2 (режим I)
а тмосфера. Температура
в пределах ±50° С. От-
носительная влажность Испытания в модерн и-
до 95% зированиом аппарате ис-
кусственной погоды мар-
ки ИП-1-2М (режим I)
Метод комбинирован-
ного воздействия ультра-
фиолетовых лучей, тепла
н влаги (режим II
ГИПИ-4)
Влажный субтропи- Солнечная радиация, Испытания в аппарате
ческий осадки, морская атмосфе- искусственной погоды
ра, сочетание высокой от- марки ИП-1-2 (режим V)
носительной влажности с
повышенной температу-
рой
Температура от 45 до Испытание в модерни-
10° С, относительная зированном аппарате ис-
влажность др 95%; за В ч кусственной погоды мар-
средиее изменение темпе- ки ИП-1-2М (режим V)
ратуры на 10° С; средняя
относительная влажность
при 35° С составляет 75%
Влажный тропичес- Высокая относительная Испытания в гидроста-
кий влажность в сочетании с тах (влажные камеры)
повышенной температу-
, рой, воздействие биоло-
гических факторов (гриб-
ковая плесень и др.), ро-
са. Температура от 45 до
3° С, относительная
влажность до 95%. Ис-
ключается воздействие
осадков и солнечной ра-
диации
— 196 —
Таблица 24
РУЮШИЕ РАЗЛИЧНЫЕ КЛИМАТИЧЕСКИЕ УСЛОВИЯ (РТМ 35—61 ВНИИИМАШ)
Факторы, воздействующие
на покрытие при
ускоренных испытаниях
Характер разрушений
(возможных)
Примечание
Свет дуговых ламп, по-
вышенная температура и
относительная влажность,
орошение водой (перио-
дическое)
Свет дуговых н ртутно-
кварцевых ламп, повы-
шенная температура и
относительная влажность,
орошение водой (перио-
дическое), действие низ-
кой температуры
Свет ртутно-кварцевых
ламп, повышенная темпе-
ратура, воздействие воды
(периодическое)
Свет дуговых ламп, по-
вышенная температура и
относительная влажность,
орошение водой (непре-
рывное)
Свет дуговых н ртут-
но-кварцевых ламп, по-
вышенная температура и
относительная влажность,
орошение водой (непре-
рывное), периодическое
действие низкой темпе-
ратуры только на спе-
циальные покрытия
Высокая относительная
влажность (постоянно) и
повышенная температура
(периодически), роса
Изменение глянца и
цвета, появление бронзи-
ровки и белесоватости,
меление (незначитель-
ное), появление трещин
Изменение глянца и
цвета, появление бронзи-
ровки и белесоватости,
меление (значительное),
появление трещин
Потеря глянца, выцве-
тание (сильное), появле-
ние белесоватости, меле-
ние (сильное), появление
пузырей или сыпи, кор-
розия (под пленкой)
Изменение глянца и
цвета, появление бронзи-
ровки и белесоватости,
меление (значительное),
появление трещин
Изменение глянца и
цвета, появление бронзи-
ровки и белесоватости,
меление (сильное), появ-
ление трещин
Изменение глянца и
цвета, появление белесо-
ватости, пузырей или сы-
пи, меление, коррозия
(открытая или под плен-
кой)
Рекомендуется для
испытания покрытий,
эксплуатируемых под
навесами
— 197 —
„ Характеристика Методы ускоренных
Имитируемый климат атмосферных условий климатических испытаний
Влажный тропиче- Солнечная радиация. Испытания по режиму
ский осадки, морская атмосфе- «тропики I» проводят
ра, сочетание высокой последовательно: в гидро-
относительиой влажности стате, камере солевого
с повышенной темпера- тумана, аппарате искус-
турой, воздействие био- огненной погоды и под
логических факторов воздействием ртутно-
(грибковая плесень, тер- кварцевых ламп ПРК-2
миты), роса
Температура от 45 до Испытания по режиму
3°С, относительная «тропики П» проводят
влажность до 95%; за последовательно: в гидро-
8 ч среднее изменение стате, камере сернистого
температуры на 10° С газа, аппарате искусст-
венной погоды и под воз-
действием ртутио-квар-
цевых ламп ПРК-2
испытания ускоренные, то соответственно выбирается и способ
ускорения разрушения покрытия.
Воздействию жидкой коррозионной среды подвергаются от-
дельные участки пленки, ограниченные восковыми кольцами, в
Рис. 113. Схема установки для определения про-
бивного напряжения лакокрасочной пленки [17]:
/ — блокировка; 2 — испытуемый образец;
3 — сигнальная лампа
которые наливают электролит. По истечении некоторого време-
ни кольца снимают, влага с поверхности образца удаляется вы-
сушиванием сначала фильтровальной бумагой, а потом сухим
воздухом в течение 5 мин при 20™ 25° С. Величину пробивного
напряжения определяют в местах, подвергавшихся и не подвер-
гавшихся воздействию среды. Полученные величины сравнива-
ют. При этом испытании очень важно, чтобы пленка была оди-
наковой толщины на всей поверхности образца.
— 198 —
Продолжение табл. 24
Факторы, воздействующие
на покрытие при
ускоренных испытаниях
Характер разрушений
(возможных)
Примечание
Высокая относительная
влажность, повышенная
температура, солевой ту-
ман, свет дуговых и ртут-
но-кварцевых ламп, роса
Изменение глянца и
цвета, появление белесо-
ватости, трещин, пузырей
или сыпи, Меление, кор-
розия (открытая или под
пленкой)
Рекомендуется для
испытания покрытий,
эксплуатируемых в
прибрежных районах
Высокая относительная
влажность, повышенная
температура, сернистый
газ, свет дуговых и ртут-
но-кварцевых ламп, роса
Рекомендуется для
испытания покрытий,
эксплуатируемых в
промышленных райо-
нах
Пробивное напряжение определяют на установке, описанной
в работе [17]; схема установки приведена на рис. .113. В установ-
ке находится трансформатор мощностью не менее 2 ква, снаб-
женный регулирующим приспособлением для плавного измене-
ния напряжения на обмотке высокого напряжения ступенями ие
«более 100 в.
Пробивное напряжение определяют при кратковременном
наложении напряжения, которое должно повышаться равномер-
но начиная от нуля до наступления пробоя со скоростью при-
мерно 500 в — 1 /се в секунду. Напряжение измеряют с помощью
(вольтметра, включенного на стороне низшего напряжения.
Электрическую прочность самой пленки определяют между
цилиндрическими электродами диаметром 2,5 мм каждый. Ниж-
ний электрод заземляется, верхний электрод должен весить
0,5 кг. Пробой производится в 8—40 местах пленки. После’испы-
тания измеряют толщину пленки с точностью до 5 мкм.
Пробивная напряженность пленки вычисляется как среднее
арифметическое из всех замеров по формуле
Д = —, (95)
d
где Ег — средняя пробивная напряженность, кв}мм\
]/а — величина пробивного напряжения, кв\
d — толщина пленки, мм.
Изменение пробивного напряжения лакокрасочных покрытий
под воздействием коррозионной среды можно определить с помо-
щью установки УПУ-1М, описанной выше (см. гл. IV, рис. 105).
— 199 —
200
Таблица 25
РЕЖИМЫ УСКОРЕННЫХ ИСПЫТАНИЙ ЛАКОКРАСОЧНЫХ ПОКРЫТИЙ
В АППАРАТАХ ИСКУССТВЕННОЙ ПОГОДЫ
Вид испытания Температура, °C Влажность. % Орошение Облучение Продолжительность испытания
Испытание в аппа- рате искусственной погоды 55—60 ("режим I) 50 (режим V) 60 (режим Oj 100 100 100 Дистиллированной водой по 3 мин каж- дые 17 мин Отсутствует » Непрерывное То же » » До появления изме- нений глянца, цвета, меления, растрески- вания и др., но не более 30 суток
Испытание во влажной камере Г4 50 (7 ч) от 50 до 20 при выключении обо- гревателей (15,5 ч) 95-100 95—100 - Общая продолжи- тельность испытания не менее 30 суток, но не более 60 суток
Испытание при комбинированном воз- действии ультрафио- летовых лучей, тепла н влаги1 50 (режим II, ГИПИ-4) 50 (режим II, ГИПИ-4 Без увлажне- ния Отсутствует Под слоем дистил- лированной воды 15 мм 1 ч 30 мин Не более 30 суток То же
18—23 (режим II, ГИПИ-4 40 (режим ВИАМ) Без увлажне- ния Влажный компресс Отсутствует 9 ч 8 ч » » » »
18—23 (режим ВИАМ) — Влажный компресс 16 ч » »
1 Применяется при отсутствии везерометра и проводится на установке, изображенной на рис, 109.
ЮГ
В последнее время для оценки защитных свойств лакокра-
сочных покрытий начинают применять методы, основанные на
определении электросопротивления и емкости и их изменении во-
времени под воздействием коррозионной среды. Установки для
измерения сопротивления и емкости описаны в гл. IV (стр. 159).
Результаты измерений емкости и сопротивления лакокрасочных
пленок на основе алкидной смолы, проведенные нами, показы-
вают, что изменение этих величин соответствует изменению их
защитных свойств. Последнее хорошо иллюстрируется данными,
приведенными на рис. 101. Как видно из кривых, резкое измене-
ние сопротивления н емкости наступает в момент резкого увели-
чения площади коррозионных разрушений.
Следовательно, этими измерениями можно пользоваться для
количественной оценки коррозионной устойчивости лакокрасоч-
ных покрытий. При этом необходимо, однако, иметь в виду, что
для некоторых систем лакокрасочных покрытий резкое падение
сопротивления и увеличение емкости не совпадают точно с мо-
ментом начала коррозии.
Электрохимические методы определения защитной способно-
сти лакокрасочных пленок. К ускоренным методам испытания
защитных свойств лакокрасочных покрытий относится и ряд
электрохимических методов.
Методы электрохимической оценки свойств покрытий в прин-
ципе ничем не отличаются от применяющихся для металлов без
покрытий.
Для лакокрасочных покрытий Н. Д. Тэмашов, В. С. Киселев и
М. М. Гольберг [23] предложили метод, заключающийся в испы-
тании окрашенного железного электрода в паре с цинковым в-
3%-ном растворе хлористого натрия. В этом случае окрашенный
электрод подвергается катодной поляризации. Расстояние меж-
ду электродами равно 3 см. Ток пары измеряется чувствитель-
ным микроамперметром, на который замкнута пара. Замеры
производят через определенные промежутки времени и строят
кривую плотность тока — время. О защитных свойствах покры-
тий судят по времени появления тока в системе. А. Я. Дринберг
и Е. С. Гуревич [24] предложили измерять силу тока пары и с
окрашенным катодом, и с окрашенным анодом. В этом случае
при испытании покрытий на железных образцах в качестве вто-
рого электрода сначала применяют цинковый, а затем плати-
новый.
Авторы [25] предложили метод определения защитных
свойств покрытий по току, возникающему в паре из окрашенно-
го и неокрашенного образца одного и того же металла. Этот ме-
тод, как показали исследования, лучше воспроизводит условия
работы покрытия, поскольку он моделирует коррозионные пары,
возникающие на поверхности металла при повреждении покры-
тия; кроме того, он не вызывает значительной поляризации элек-
тродов.
— 201 —
Таблица 26
РЕЖИМЫ ИСПЫТАНИЯ. ВОСПРОИЗВОДЯЩИЕ УСЛОВИЯ ТРОПИЧЕСКОГО
КЛИМАТА (РТМ 35—61 ВНИИНМАЩ)
Режим испытания Условия испытания Относитель- ная влаж- ность, % Температура °C Продолжи- тельность испытания ч Примечание
Гидростат 50—55 7
50—20 10 С включен- ным обо- гревателем
Солевая 95—100 35—40 2
«Тропики I» камера Аппарат 60±2 3 Без оро-
искусствен- ной погоды тения
Ртутно- кварцевые — S0±2 1
лампы
Комнатные 60—70 18—23 I Выдержка
условия образцов на воздухе
и осмотр
Гидростат 50—55 7
50—20 10 С выклю-
«Тропи- ки II» 95—100 ченным обогрева-
Камера с сернистым 50-55 2 телем
газом
(60 мг[длР)
Схемы установок для определения силы тока пар остаются
такими же, как и для испытаний металлов без покрытий (см.
рис. 94, 96).
Благодаря развитию теории электрохимической коррозии ме-
таллов стало возможно для быстрой оценки в лабораторных ус-
ловиях защитных свойств покрытий применять методы снятия
поляризационных кривых на образцах, покрытых лакокрасочной
пленкой и помещенных в различные коррозионные среды. С по-
мощью этого метода было установлено, например, пассивирую-
щее действие различных пигментов в лакокрасочных покрыти-
ях [25]. Установка для снятия поляризационных кривых иа окра-
шенных образцах приведена на рис. 114. Применяли образцы
малого диаметра (4 мм), армированные в стеклянные держате-
ли диаметром 35 мм. Лакокрасочная пленка наносилась иа всю
поверхность стеклянного держателя; предварительно поверх-
— 202 —
ность стекла и металла шлифовалась на наждачной бумаге и
промывалась растворителем и спиртом; все это обеспечивало
хорошую адгезию покрытия.
Ионная проницаемость. Важная электрохимическая характе-
ристика защитных свойств полимерных пленок,— ионная прово-
димость покрытия. Если предположить, что коррозионный про-
цесс может начаться лишь тогда, когда ионы из окружающей
среды достигнут поверхности металла, свойства пленки будут
Рис. 114. Установка для снятия поляризационных кривых
иа окрашенных образцах [25J
характеризоваться временем до начала появления ионной про-
водимости, а также величиной тока при прочих равных усло-
виях.
Ионная проводимость пленок будет определяться наличием
пор в пленке, способностью тех или иных ионов проникать через
них, величиной осмотического давления раствора, а в некоторых
случаях и участием материала покрытия в переносе ионов.
Для определения ионной проницаемости получают свободную
пленку, которую затем помещают в ячейку в качестве разделяю-
щей мембраны. О величине ионной проводимости судят по вре-
мени до появления тока между двумя электродами, на которые
подается постоянное напряжение, равное ~ 1 е. Электроды по-
мещаются по обе стороны от мембраны [26]. Херри и Майн [27]
изучали ионную проницаемость по изменению сопротивления
— 203 —
На
ГА
Рис. 115. Зависимость сопротив-
ления лакокрасочной пленки на
основе фенолформальдегидной
смолы от rk (pH раствора) {27]:
1 — 3-и. раствор КС1: 2 — 0.001-н.
раствор КС1
пленки при различных значениях pH раствора и постоянном со-
держании ионов калия и хлора в электролите. Полученная ими
зависимость сопротивления от rk представлена на рис. 115, где
rk = |&стК+ . (96)
Igaier^
Как видно на рисунке, кривые имеют резкий перелом. В об-
ласти перелома пленка, по мнению авторов, неустойчива. Таким
образом, по полученным данным можно судить об устойчивости
плеики в данной среде. На
рис. 115 видно, что резкое паде-
ние сопротивления наблюдается
при rk — 9.
По нашим данным, ионная
проницаемость пленок в сильной
степени зависит от характера
аниона раствора. Так, например,
как видно на рис. 116, через плеи-
ки на основе алкидной смолы лег-
че проникают такие ионы, как
NO г и СО?“, чем ионы С1~ и ио-
ны SO J-.
Поэтому сравнение лакокра-
сочных пленок по их ионной про-
водимости должно проводиться в
тех растворах, которые будут
имитировать условия работы.
Нельзя применять растворы хло-
ристого натрия как стандартные,
если изделие не будет работать
при погружении в морскую воду.
Ионная проницаемость лакокрасочных пленок может быть
также определена аналитическими методами, .в частности мето-
дом, разработанным И. Л. Розенфельдом, Ф. И. Рубинштейн и
С. В. Якубовичем [2&J. Метод заключается в определении скоро-
сти диффузии ионов хлора через исследуемую пленку, помещен-
ную в качестве разделительной мембраны в стеклянную ячейку,
состоящую из двух половинок (рис. 117). В одну половину ячей-
ки наливают 15 мл хлористого натрия (3%-ный раствор), в дру- •
гую 15 мл дистиллированной воды. Скорость проникновения
ионов хлора определяют после пяти суток выдержки приборов
в термостате при постоянной температуре титрованием 0,1-н.
раствором AgNO3 в присутствии 10%-ного раствора KgCrC^ по
уравнению
г aTk50 • 1000 .
С ~ ц Мг1^СМ * сУтки)> (97)
где а—количество AgNO3, израсходованное на титрование, мг;
'/'—титр 0,1-н. раствора AgNOs, по хлору;
— 204 —
k — поправка на титр;
S — площадь пленки, через которую происходила диффузия,
щи2;
/—.время, сутки.
Перед титрованием отобранную пробу (10 см3) разбавляют
до 50 см3.
Физико-химические методы оценки защитных свойств лако-
красочных пленок. При изучении защитной способности пленок
Рис. 116. Зависимость времени до появления тока
от толщины пленки на основе алкидной смолы
(лак ФЛ-021 при испытании ее в различных 0,5-н.
растворах:
1 — NaCl: 2 — Na2SO4; 3 — NasCOa: 4 — NaNO3
различных полимерных материалов нельзя, как уже указыва-
лось, ограничиться оценкой устойчивости по одной какой-либо
характеристике. Важен, очевидно, весь комплекс свойств, по-
этому при проведении ускорен-
ных испытаний лакокрасочных
пленок полезные сведения об их
защитных свойствах можно по-
черпнуть !и из данных о водопро-
ницаемости, паропроницаемости,
солепроиицаемости, водоиабу-
хаемости, а также об адгезион-
ных свойствах. Все перечислен-
ные свойства косвенно характе-
ризуют качество покрытия с точ-
ки зрения эффективности его за-
щитного действия.
Паропроницаемость лакокра-
сочных пленок характеризует спо-
собность паров воды проникать
через пленку. Ее определяют
Рис. 117. Прибор для опре-
деления ионной проницае-
мости лакокрасочных пле-
нок (28J.
методом, основанным на поглощении водоотнимающими веще-
ствами (например, фосфорным ангидридом) .паров воды, диф-
фундирующих через пленку. В соответствии с ОСТ 10086—39
М. И. 36 для испытаний принята специальная форма сосудов
— 205 —
(рис. 118). Сосуд составной, состоит из чашки Л на верхнюю
часть которой (диаметром 50 мм) воском прикрепляют испыты-
ваемую пленку толщиной 40—50 мкм, и стакана 3, в который
помещают фосфорный ангидрид (4—5 г). Чашку надевают на
стакан и помещают в эксикатор, в котором поддерживаются по-
стоянная влажность (95%) и температура (20° С). Сбоку чашки
находится небольшое отверстие, закрываемое пришлифованной
пробкой 2; оно служит для уравнивания давления в приборе в
момент сборки. Перед опытом и затем каждые 24 ч стакан вы-
нимают нз эксикатора, закрывают крышкой 4 и -взвешивают на
аналитических весах. Паропроиицаемость выражается количест-
Рис. 118. Прибор для опре-
деления паропроницаемости
вом водяного пара, прошедшего
в сутки через 1 см2 свободной
нленки; расчет ведут по уравне-
нию
D = 78^2 м&1(см2 ‘ сутки), (98)
где D — паропроиицаемость;
А — средний привес фосфор-
ного ангидрида за одни
сутки при испытании в.
течение 7—8 суток, мг*,.
b — толщина пленки, мкм;
cl — диаметр свободной плен-
лакокрасочных пленок: ки
? - томшкГ Водопроницаемость — это спо-
собность лакокрасочного покры-
тия пропускать воду; определяется качественно по ОСТ 10086—
39 М. И. 30. Мерой водонепроницаемости служит время, прошед-
шее с момента контакта пленки лакокрасочного покрытия, нане-
сенного на слой латекса, с водой. О проникновении воды судят
по появлению белых пятен на латексе в месте проникновения
влаги сквозь пленку. Осмотр производят с иижней части стек-
лянной пластинки.
Количественно водопроницаемость определяют в стаканах,,
на верхней части которых натянута пленка [17]. Стаканы запол-
няют водой и в перевернутом виде помещают в закрытую ка-
меру, на дно которой насыпают прокаленный хлористый каль-
ций. Водопроницаемость определяется по убыли воды в сосуде и
выражается в мг[ (см2 • сутки).
Водонабухаемостъ '(или водопоглощение) — это количество-
воды, поглощенной лакокрасочным покрытием при нахождении
его -в воде в течение установленного промежутка времени. Если
паро-, водо- и солепроницаемость определяют на специально по-
лученных свободных пленках, то водопоглощение — на окрашен-
ных для этой цели металлических пластинках; лучше всего»
— 206 —
применять коррозионностойкне металлы. В .соответствии -со стан-
дартным методом используют пластинки из черной или декапи-
рованной жести. Окрашенные пластинки помещают в ванну с
дистиллированной водой, через определенные промежутки вре-
мени вынимают, осторожно высушивают между листами филь-
тровальной бумаги до исчезновения серых пятен и тотчас взве-
шивают. Водопоглощение рассчитывают по уравнению
X = (г - с)100 , (99>
б — а
где х— прирост массы лакокрасочной пленки (за счет количе-
ства поглощенной влаги) к первоначальной массе плен-
ки, %;
а — масса металлической пластинки;
б-—масса пластинки с покрытием;
в — масса пластинки с краями, изолированными парафи-
ном;
г — масса пластинки после .испытания в течение определен-
ного промежутка времени.
Водонабухаемость можно определять не только весовым ме-
тодом. Брашер й Кингсбари [29] разработали метод определения
водопоглощення на основании измерения емкости с помощыо-
моста переменного тока. Лакокрасочное покрытие в этом случае
наносят на никелевые пластинки. Сопоставление полученных
данных с данными взвешивания показало, что по емкости мож-
но судить о водонабухаемости только тогда, когда распределе-
ние «пор в пленке не имеет определенной ориентации и вода про-
никает в нее равномерно по всей поверхности.
Применение метода определения водонабухаемости по изме-
рениям емкости основано на предположении, что изменение ем-
кости пленки в первый период испытания происходит благодаря
проникновению в пленку воды; диэлектрическая постоянная аб-
сорбированной воды не изменяется и равна 80; толщина измеря-
емой пленки и измеряемая емкость связаны линейной зависи-
мостью. Используя формулу Хартшорн, Мегсон и Растон [301
Брашер получила уравнение, по которому по данным .измерений
емкости рассчитывается объем поглощенной воды:
где Ст/Сто — отношение емкости, измеренной в некоторый
момент времени испытания н тотчас после погру-
жения;
80 — диэлектрическая постоянная воды.
На рис. 119 графически .представлены результаты, получен-
ные при определении водопоглощення по изменению массы плен-
ки и емкостным методом для лакокрасочного покрытия на осно-
— 207 —
ве фенолформальдегидной смолы. Как видно из приведенных
кривых, эти результаты хорошо совпадают, следовательно, ме-
тод емкости пригоден для определения водонабухаемости таких
лакокрасочных пленок. Преимущество емкостного метода за-
ключается в том, что в этом случае изменение водопоглощения
можно проследить на одном образце, не удаляя его из коррози-
онной среды. Метод, предложенный [29], рекомендуется только
для покрытий на основе фенолформальдегидных и каменноуголь-
ных смол при испытании в морской воде.
Адгезионные свойства лакокрасочной пленки в значительной
степени определяют ее защитные свойства, причем не столько
важно знать, какова адгезия покрытия к металлу вообще,
О 7 2 3 6 5 6 7 В 9 10 11 12 13 74
Время испытания,сутки
Рис. И9. Зависимость водопоглощения лакокрасочным
покрытием на основе фенолформальдегидной смолы
от времени при погружении в морскую воду (29J:
/ — весовой метод; 2 — метод емкости
сколько как оиа изменяется под воздействием коррозионной сре-
ды. К сожалению, связь адгезионных свойств с защитными изу-
чена недостаточно и в литературе имеется очень мало данных по
этому вопросу.
Существуют качественные и количественные методы опреде-
ления адгезии. В практике наибольшее распространение получи-
ли качественные методы. К ним относятся:
1. Метод решетчатого надреза с помощью бритвенного лез-
вия или иглы (ТУМХП 4202—54; СМИ-8). Надрезы наносятся
в двух -взаимно перпендикулярных направлениях; число надре-
зов должно быть не менее пяти. В результате надрезов образу-
ется .сетка -прорезов с четырехугольными ячейками. О степени
прилипаемости пленки к подложке судят по количеству ячеек,
отставших от поверхности во время прорезания покрытия. Если-
адгезия хорошая, отставания не наблюдается. Для более равно-
мерного надрезания применяют специальные приспособления, с
помощью которых надрез производится одновременно с помо-
щью жестко укрепленных лезвий бритв или патефонных >игл £17].
2. Метод звездчатого надреза с дальнейшим испытанием на
прессе Эриксена. На металлическую пластину размером 7 X
X 7 мм наносят испытываемое покрытие; делают четыре кресто-
образных надреза до подложки, после чего пластину зажимают
— 208 —
в приборе — прессе так, чтобы центр надреза совпал с центром
пуансона .прибора. Затем вращают штурвал прибора со скоро-
стью, при которой за 30 сек пуансон переместился бы на 8—
9 мм, и .следят за изменением пленки. За меру адгезии принима-
ют глубину продавливания, при которой начинается отслаива-
ние пленки от подложки. Испытания на прессе дают относитель-
ное представление об адгезии и пригодны главным образом для
покрытий с плохой адгезией, так как в противном случае не уда-
ется уловить разницу между отдельными покрытиями. При про-
ведении испытания по этому методу важно сохранять постоянны-
ми скорость вращения штурвала, толщину испытываемых пок-
рытий и подложки, влажность и температуру воздуха.
Некоторое понятие об адгезии можно получить, применяя .ме-
тод изгиба, удара, а также измеряя сопротивление истиранию и
твердость пленок при царапании.
Все качественные методы необъективны, так как они являют-
ся визуальными; полученные результаты зависят не только от
адгезии, ио и от целого комплекса других физико-механических
свойств.
Количественные методы оценки адгезии по способу приложе-
ния усилия делятся на методы отрыва, отслаивания, среза, сдви-
га и косвенные методы (по количеству попеременных сжатий я
растяжений, которые выдерживает покрытие, или по критичес-
кой (предельной) величине внутренних напряжений, измеряемой
оптическим методом].
Количественные методы определения адгезии часто дают
противоречивые результаты. Нельзя применять один и тот же
метод для любого покрытия. Исследование адгезии полимерных
пленок различными методами, проведенное П. И. Зубовым,
А Т. Санжаровским и М. С. Дыльковым [31], показало, что не
все количественные методы дают возможность определять номи-
нальную адгезию. С помощью таких методов, как отслаивание
покрытия от подложки и подложки от покрытия, срезания но-
жом, можно установить лишь практическую адгезию зависящую
от многих факторов.
Номинальную величину адгезии, обусловенную характером
взаимодействия пленки с подложкой, можно определить количе-
ственно, методами штифтов и нормального отрыва склеенных
подложек. Для прозрачных объектов величину номинальной ад-
гезии можно измерить оптически.
Метод штифтов заключается в определении силы отрыва или
силы скручивания штифта, находящегося в отверстии металли-
ческой пластины и заполированного заподлицо с ее поверхно-
стью, на которую нанесено покрытие. Для уменьшения трения
цилиндрическую часть штифтов натирают графитом. Относя си-
лу отрыва или скручивания к единице поверхности штифта, по-
лучаем значение прочности адгезии соответственно на сдвиг или
отрыв в кг)см2.
14 Зак. 1338 — 209 —
Метод нормального отрыва двух склеенных плоских поверх-
ностей часто применяют при исследовании адгезии полимеров.
Если получается адгезионный отрыв, то для определения адгези-
онной прочности необходимо зависимость прочности склеивания
от толщины покрытия экстраполировать на нуль. На принципе
измерения работы отрыва пленок от подложки работают пред-
ложенные Б. В. Дерягиным |[32] адгезнометры, определяющие
адгезию как при статическом, так .и при динамическом методе
отрыва. Эти приборы пригодны только для тех покрытий, у ко-
торых адгезия сравнительно невелика.
Н. А. Кротова и сотрудники (331 разработали несколько ва-
риантов конструкций роликовых адгезиометров типа АЗС; пос-
ледняя модель адгезиометра АЗС-2 позволяет определять рабо-
ту отрыва при отслаивании пленки, .наносимой на ролики;- н уси-
лие отрыва при разрыве двух цилиндрических металлических
образцов, торцы которых соединены .прослойкой полимера.
Пленка, нанесенная на вращающийся на шарикоподшипнике ро-
лик, может отрываться с различной скоростью—от l»10~4 до
НО5 см}сек. Усилие отрыва фиксируется с помощью динамо-
метрического устройства.
Хорошие результаты получаются также при определении
адгезии методом отслаивания подложки от покрытия, если его
наносить на алюминиевую фольгу толщиной 50 мкм [34]. После
нанесения первого слоя покрытия оно армируется, например
стеклотканью или стеклосеткой. Последующие слои покрытия
наносят уже на армирующий материал. Затем покрытие высу-
шивают, .после чего алюминиевую фольгу разрезают на отдель-
ные прямоугольные полоски шириной 10 мм, длиной 80—90 мм
и вручную частично отслаивают и отгибают на 180° по отноше-
нию к пленке.
Величину адгезии определяют по величине усилия, которое
необходимо приложить для отрыва пленки от подложки. Отрыв
производят на разрывной машине, причем в верхний зажим ма-
шины закрепляется отогнутая часть фольги, а в нижний — плен-
ка. Нерасслоенная часть образца удерживается в вертикальном
положении с помощью специального приспособления. При та-
ких испытаниях общая толщина пленки должна быть постоян-
ной. В исследованиях [34] она составляла 70—80 мкм.
Так как внутренние напряжения всегда оказывают существен-
ное влияние на адгезию покрытий, то, как указывается в работе
[31], необходимо или исключить их влияние, как это делалось в
уже описанных методах, .или, приняв их за основу, определять
по ним адгезию покрытия. На последнем принципе основан ме-
тод, предложенный С. А. Шрейнером и П. И. Зубовым (35],—
так называемый оптический метод нсследова-ния адгезии поли-
мерных покрытий по критическим напряжениям.
Метод заключается в нанесении покрытия на одну из граней
четырехгранной стеклянной прямоугольной призмы н определе-
— 210 —
нии величии двупреломления луча за пределами зоны формиро-
вания покрытия.
Двойное лучепреломление измеряют с помощью полярин
циониого микроскопа и поворотного кальцитового компенсато-
ра КПК.
Величину внутренних напряжений определяют по шка-
ле перехода, составленной на основе предварительно проведен-
ных’ измерений разности хода в призмах в зависимости от наг-
рузки ® пределах 98—1176 н}см2 (10—120 кГ[см2).
4. СМАЗКИ
Смазки широко применяют как средство временной защиты
от атмосферной коррозии металлов и изделий при транспорти-
ровке и хранении.
Смазки должны не только защищать от коррозии металл, на
который они нанесены, но и не оказывать на него вредного воз-
действия. Применяются они главным образом для защиты от
коррозии в атмосферных условиях, когда основными коррозион-
ными агентами являются влага, кислород и другие составляю^
щие воздуха. Возникновение коррозии можно предупредить с
помощью смазок, если они полностью или частично препятству-
ют доступу коррозионно активных составляющих атмосферного
воздуха к поверхности металла. Практически полностью изоли-
ровать металл с помощью смазок не удается, поэтому их отно-
сят к временным защитным мероприятиям, оказывающим за-
щитное действие на сравнительно короткий срок.
В последнее время наметилась тенденция к увеличению сро-
ка защитного действия смазок при помощи -введения в них спе-
циальных веществ (ингибиторов), которые обеспечивают защи-
ту металла при проникновении к нему влаги и других коррози-
онно активных агентов из воздуха.
Ускоренные испытания смазок делятся на испытания по
оценке коррозионной активности самих смазок и испытания по
определению их защитных свойств.
Оценку коррозионной активности смазок в СССР производят
в соответствии с ГОСТ 1037—41 и 5757—51 путем погружения в
смазки шлифованных пластинок при комнатной температуре и
при 100° С на определенное время, по окончании которого не
должно наблюдаться потускнения или изменения цвета пласти-
нок. В США по стандарту ASTMD 1261-55 проводят аналогич-
ные испытания. В. С. Лунева [36) в качестве показателя степени
коррозионной активности кальциевой смазки использовала из-
менение pH (А pH) раствора смазки по сравнению с чистым
растворителем. pH измеряли потенциометрически. В качестве
растворителя был выбран раствор следующего состава: 50 %-
С4Н9ОН, содержащий 15% Н2О + 25% СвН6 ф 25%СН2(ОН)2;
14* — 211 —
pH = 5,5. Для измерения изменения pH смазок использовали их
2 %-ный раствор.
Один из факторов, характеризующих коррозионную агрес-
сивность смазки, — содержание в ней активных кислот и осно-
ваний. Изменение pH благодаря уменьшению концентрации
кислых соединений принимается за положительное изменение,
а благодаря увеличению — за отрицательное.
Испытание защитных свойств смазок в лабораториях произ-
водится в условиях, когда действие того или иного фактора ат-
мосферной коррозии усиливается. Образцы металлов, для за-
щиты которых предназначается смазка, покрывают тонким сло-
ем ее и подвергают воздействию повышенной влажности, .паров
морской воды, воздуха, содержащего повышенные концентра-
ции двуокиси серы, периодической конденсации влаги или непо-
средственного полива струей морской воды, раствором хлори-
стого натрия или просто .водой [37—44].
Испытание при 100%-ной относительной влажности являет-
ся стандартным испытанием по ГрСТ 4699—53. Стальные шли-
фованные пластинки, покрытые слоем смазки (3—4 мм), поме-
щают в эксикатор над водой при температуре 50~1-60оС на
24—48 ч. По окончании срока испытания пластинки осматрива-
ют визуально; если коррозии не обнаруживается, смазка счи-
тается годной.
Испытания в атмосфере с высокой влажностью не позволяют
определить защитную способность смазок, обладающих отно-
сительно высокими защитными свойствами; сроки испытаний
таких смазок становятся слишком большими и испытания пере-
стают быть ускоренными. Были разработаны методы, позволяю-
щие сократить сроки испытания. К ним относится применявший-
ся ранее как стандартный ускоренный метод определения за-
щитных свойств смазок в условиях длительной конденсации.
Смазанные шлифованные или полированные пластинки кладут
поочередно сначала на один час в эксикатор, помещенный в со-
суд со льдом, а затем на то же или большее время в эксикатор,
помещенный в термостат при температуре 34—36° С. Образцы
покрываются равномерным тонким слоем смазки толщиной 0,1;
0,2; 0,5 или 1 мм с помощью специальных шаблонов. В эксика-
торах пластинки располагаются на подставках горизонтально
или подвешиваются вертикально.
Количество циклов в испытании назначается заранее или ис-
пытание ведут до появления видимой ржавчины. Защитную спо-
собность сравниваемых смазок оценивают визуально. О защит-
ных свойствах смазок можно судить также по характеру кон-
денсации влаги на смазанной поверхности. Если образуются
капли, которые легко удаляются фильтровальной бумагой и
быстро высыхают, значит смазка хорошего качества. Если смаз-
ка эмульгирует с водой или растворяется в конденсирующейся
воде, То защитные свойства такой смазки будут определяться
— 212 —
свойствами образующихся эмульсий и наличием в них ингиби-
торов; такая смазка защитного действия оказывать не будет.
Различные методы ускоренных испытаний защитных
свойств смазок в условиях конденсации влаги применяют и в за-
рубежной практике. Хор и Смит [45] предложили проводить
испытания, имитируя конденсационную влагу нанесением на по-
верхность смазанных образцов определенного числа капель
дистиллированной воды. На стальные образцы размером 4 X
X 5 см наносили 10 капель воды, затем образцы помещали во
влажную атмосферу при 25° С. Через 24 ч (или несколько боль-
ший срок) определяли число капель, под которыми началось
ржавление; отмечали также растекание капель по поверхности,
что указывает на способность смазки образовывать водную
эмульсию. Испытания проводили в сравнении с успешно приме-
нявшейся ранее смазкой.
Основной недостаток метода — произвольное расположение
капель в местах, которые могут ие соответствовать участкам об-
разования капелек влаги при конденсации.
При более совершенных конденсационных методах ускорен-
ных испытаний качества смазок конденсацию строго регламен-
тируют.
Необходимым условием -всех ускоренных испытаний, в кото-
рых создают конденсацию на поверхности смазанного металла,
является обеспечение постоянной скорости конденсации, так как,
помимо влияния количества сконденсированной влаги, на харак-
тер коррозионного воздействия большое влияние будет оказы-
вать смазывающее действие влаги, особенно если образцы рас-
положены вертикально. Роден [46] предложил испытание при по-
стоянной конденсации (рис. 120).
Испытания проводили на полых стальных образцах 4; кон-
денсация создавалась пропусканием через них холодной воды.
Постоянная температура воздушного пространства в сосудах 6
создавалась помещением его в специальный водяной термостат
(на рис. 120 не показан). Испытания можно производить одно-
временно в нескольких сосудах, количество которых опреде-
ляется размерами термостата. Количество сконденсированной
воды регулируется изменением разницы температур пропускае-
мой воды и воды термостата. Автор указывает, что на результа-
ты испытаний сильно влияет подготовка поверхности образцов,
их расположение и, как уже указывалось, скорость конденса-
ции. Если постоянные условия испытания выполняются, то ре-
зультаты хорошо воспроизводятся. При проведении испытаний
в тех или иных аппаратах, отличающихся по конструкции или
размерам, даже при одном и том же перепаде температуры ско-
рость конденсации будет различная и результаты испытания
оказываются ие сопоставимыми.
Сравнение результатов испытаний, проведенных в соответ-
ствии со спецификацией военного ведомства США 2-126 во
— 213 —
влажной камере .и в закрытых сосудах Родена [46], показало, что
в последнем случае разброс данных гораздо меньше; результа-
ты испытаний хорошо воспроизводятся, уменьшается срок ис-
пытания. Так, например, средняя продолжительность испытаний
смазок во -влажной камере составляла 552 ч, а в сосудах Родена
Рис. 120. Сосуд для изучения
защитных свойств смазок [46]:
1 — трубки для ввода и вывода воды;
2 — резиновые пробки; 3 — ниппель
для крепления образца; 4 — полый
образец; 5 — термометры; 6 — стек-
лянный сосуд; 7 — дистиллированная
вода
она снизилась до 158—94 ч (по-
верхность образцов опескоструи-
•валась). Защитные свойства сма-
зок в обоих случаях оценивались
по времени до появления первых
трех коррозионных поражений.
Скорость конденсации при прове-
денных испытаниях составляла
0,31 см3/(дм2-ч).
Регламентировать условия
конденсации можно, применяя
специальные устройства внутри
обычных влажных камер. На
рис. 121 представлено такое уст-
ройство, примененное Думай н
Чайкор [47] для испытания пане-
лей большого размера, Панель 1
с нанесенной на нее смазкой яв-
Рис. 121. Схема устройства, позво-
ляющего регулировать скорость кон-
денсации при ускоренных испытаниях
во влажной камере [47]
ляется стенкой коробки, изготовленной, из нержавеющей стали.
Внутрь таких коробок, соединенных с резервуаром 2 помпой 3,
подается холодная вода, которая затем под действием собствен-
ного веса собирается в бак 4.
Этот метод испытания защитной -способности смазок прин-
ципиально ничем не - отличается от описанного выше; ускорения
— 214 —
коррозии в обоих случаях достигают за счет непреры-виой кон-
денсации, влаги на поверхности смазанного образца. Оценка
скорости коррозии под смазкой производится визуально по вре-
мени до появления первых коррозионных поражений, а также
по потере веса.
Более жестким испытанием является испытание во влажной
атмосфере в присутствии двуокиси серы. При испытаниях, где
создаются условия для конденсации >в чистой атмосфере, корро-
зионный процесс под смазкой значительно ускоряется, но эти
Рис. 122. Внешний вид ап-
парата для испытания сма-
зок в присутствии SO2 при
одновременной конденсации
влаги (48]
Рис. 123. Кривые зависимости
смываемости различных смазок
дождем от температуры |]50].
Смазки:
1 - № 21; 2 — ЦИДТИМ-201; 3 -
пушечная; 4 — КВ; 5 — ружейная
испытания не могут отразить полностью условий .промышленной
атмосферы. Поэтому для получения результатов, которые мож-
но было бы отнести и к промышленной атмосфере, во влажную
камеру или в камеру, где создаются условия для конденсации,
вводят сернистый газ.
Престон и Стронг [48] проводили испытания в присутствии
SO2 в довольно простом аппарате, внешний вид которого пока-
зан на рис. 122. Образцы подвешивали под крышкой аппарата
над нагретым до 48° С раствором, содержащим 0,005 или 0,01%
SO2. Испытание проводили для определения защитной способ-
ности различных смазок, нанесенных на стальные пластинки
размером 150 X 50 мм. Коррозию оценивали по появлению кор-
розии на образцах и по потере веса, которую определяли после
снятия смазки. Несмотря на явное несовершенство аппарата —
неопределенность условий конденсации, изменяющуюся кон-
— 215 —
центрацию SO2 (его вводили два раза в сутки) — по данным ав-
торов [48], результаты испытаний хорошо воспроизводили ре-
зультаты, получаемые при испытании в естественных условиях.
Сопоставление результатов ускоренных испытаний с результа-
тами .испытаний в естественных условиях показало, что потери
в весе при ускоренных испытаниях в течение одной недели (кон-
центрация SO2 = 0,01%) составляют 0,92 потерь при испытании
в промышленной атмосфере в течение трех месяцев.
Помимо изложенных выше способов испытаний, в различ-
ных отраслях промышленностип рименяют специальные методы
определения защитной способности смазок, отражающие спе-
цифику их применения для защиты изделий данной отрасли
промышленности.
В работе [49] описывается, например, испытание смазок,
предназначенных для подшипников. Испытание проводят при
вращении смазанного подшипника в стеклянной подставке, по-
мещенного® сосуд, в котором создается 100%-ная относительная
влажность с помощью воды, наливаемой на дно сосуда. .Исследо-
вания проводят в течение двух недель при 25° С. Состояние под-
шипника после испытания определяют визуально по трехбалль-
ной шкале.
Характерно также испытание смазок, предназначенных для
защиты от коррозии вооружения на смываемость смазок водой
(дождем) [50].
Испытание на смываемость проводят на специальном прибо-
ре, представляющем собой сосуд емкостью 5 дм8 и миниатюрный
душ, из которого на смазанную пластинку, укрепленную с накло-
ном в 45°, подается вода под напором в 1 м. Смываемость смазок
определяют по разности в массе пластинок со смазкой (слой
смазки —0,5 мм) до и после испытания. Взвешивают образцы,
предварительно высушив их после испытания. Смываемость вы-
ражают в процентах по отношению к массе смазки, нанесенной
на пластинку. Испытания проводят при различных температурах.
По данным [50], смываемость некоторых смазок в зависимости от
температуры выражается кривыми, представленными на рис. 123.
Как видно на рисунке, сравнительно хорошо сопротивляются
смыванию смазки № 21 и ЦИАТИМ->201.
Важной характеристикой для смазок, предназначенных для
временной защиты в районах с жарким климатом, является кап-
лепадение, которое определяют в соответствии с ГОСТ 6793—53
в специальном приборе, представляющем собой стеклянную ча-
шечку с отверстием в дне, вставленную в пробирку. Нижний
край чашечки должен находиться на .расстоянии 25 мм от дна
пробирки. Если определяемая температура каплепаденйя не пре-
вышает 80° С, пробирку погружают в стакан с водой; при более
высоких температурах в стакан наливают вазелиновое масло или
глицерин. Стакан с жидкостью нагревают со скоростью 1° в ми-
нуту; по термометру, установленному в пробирке, отмечают тем-
. — 216 —
пературу, при которой падает первая капля или столбик нефте-
продукта, выступающий из отверстия чашечки, касается дна про-
бирки.
В большинстве испытаний защитную способность смазок оце-
нивают визуально. Это основной недостаток ускоренных методов.
Критерием оценки защитной способности смазок при массо-
вых испытаниях является время до появления первого коррози-
онного поражения. Различные исследователи определяют его как
время, необходимое для появления одного или нескольких пора-
жений на определенной площади, чаще всего площади .испыты-
ваемого образца.
В. В. Скорчеллетти, А. А. Бухбиндер и В. Е. Пискорский [51}
оценивали защитную способность тонкослойных смазок при испы-
таниях их в атмосфере 100%-ной относительной влажности при
содержании сернистого ангидрида 0,00015% (объемн.) по разра-
ботанной ими шкале баллов (табл. 27) [52]. Оценка производи-
лась путем сравнения образца с эталонами.
Таблица 27
ШКАЛА ОЦЕНКИ ЗАЩИТНОЙ СПОСОБНОСТИ СМАЗОК [521
Балл Состояние поверхности образца
1 Ржавчина покрывает всю поверхность; иногда наблюдаются
отдельные чистые участей
2 Многочисленные очагн ржавления и мелкие точки, покры-
вающие всю поверхность, но разделенные участками чис-
той, незаржавевшей поверхности
3 Редкие, разбросанные по всей поверхности очаги ржавления
и отдельные точки
4 Единичные очаги ржавления, точки нли потускнение пятнами
5 Один или два мелких очага ржавления или одна-две точки
на всей поверхности образца
6 Следов ржавления нет
Наиболее распространенный количественный метод оценки
защитных свойств смазок—определение потери массы металла
за время испытания, однако он возможен только при значитель-
ной коррозии или весьма чувствительных методах анализа, кото-
рые сейчас начинают интенсивно вводить в практику, но которые*
еще не получили широкого применения.
В настоящее время разработан ряд методов, позволяющих ко-
личественно оценивать защитные свойства смазок. В. С. Луне-
ва [53, 54] .предложила методы определения количества металла,,
перешедшего во время испытания в смазку, основанные на поля-
рографическом нли радиографическом методах. Существует
также ряд электрохимических методов для определения защит-
ных свойств смазо,к. Р. Г. Гаджиева [55] оценивала защитную-
— 217 —
способность смазок по изменению электропроводности слоя
смазки.
В работе [56] описан метод оценки защитной способности сма-
зок, основанный на определении момента нарушения сплошности
слоя смазки электрохимическим методом. Смазанный обра-
зец в паре со вспомогательным электродом погружают в раствор
хлористого натрия. На чувствительном миллиамперметре фикси-
руется момент появления тока в цепи при наложении на образ-
цы небольшого постоянного напряжения. Момент появления то-
ка соответствует моменту нарушения сплошности слоя смазки.
В. М. Мартынов и М. В. Морозова [57] разработали электро-
химический метод испытания смазок, в котором о защитных свой-
ствах судят по величине тока, воз-
никающего в паре железо — медь,
разделенной слоем диэлектрика,
Рис. 124. Схема прибора для элек-
трохимического определения за-
щитных свойств смазок |[57]:
1 — стальной полый цилиндр; 2 — ко-
жух. заполненный парафином: S —
металлический кожух; 4 — текстолито-
вая пластина: 5 — пропарафинирован-
ная бумага: 6 — медный стержень:
7 — слой смазки
Рис. 125. Схема измерения контакт-
ного угла, образуемого каплей воды
на слое смазки [58]:
а — контактный угол; 1 — вода;
2 — масло; 3 — сталь
при помещении пары в различные коррозионные среды. Для про-
ведения таких испытаний авторы разработали специальный при-
бор, схема которого показана иа рис. 124. Прибор представляет
собой -стальной полый цилиндр 1, помещенный в металлический
кожух 3, заполненный парафином с таким расчетом, чтобы при
помещении стального цилиндра на текстолитовую пластину 4
цилиндр выступал на 5 мм. Внутрь стального цилиндра
помещают медный стержень 6, изолированный от него пропара-
финированной бумагой 5. На торцовую часть изготовленной та-
ким образом пары наносят слой смазки. От стального цилиндра
и медного стержня выводят контакты на чувствительный ампер-
метр. Весь прибор помещают во влажную атмосферу или в рас-
твор хлористого натрия; по величине возникающего тока судят о
защитных свойствах смазки; чем меньше ток и чем позднее он
•появляется, тем выше защитные свойства смазки.
Сравнивая полученные этим методом результаты с результа-
тами испытаний смазок во влажной камере и с поведением сма-
зок в естественных условиях консервации изделий, авторы нрнхо-
— 218 —
дят к выводу, что метод дает правильное представление о за-
шитных свойствах.
Пир л и Фелей {58] предложили оценивать свойства смазок
косвенным методом по величине контактного угла, образующего-
ся при нанесении на поверхность образцов с масляной пленкой
капли воды (рис. 125). Этот метод был предложен для случая,
когда смазанная поверхность будет контактировать с каплями
•воды, т. е. в условиях конденсации. Метод измерения контактно-
го угла дает наилучшие результаты, если величина угла нахо-
дится в пределах 10—80°, а количество доба-вок к маслу не пре-
вышает 10%. Контактный угол измеряют с помощью микроскопа
н гониометрического окуляра через три минуты после нанесения
капли. Измерение контактных углов, как показали опыты авто-
ров, может характеризовать, защитную способность смазки. Кон-
тактный угол жидкого минерального масла равен 70—85°; оно
защищает сталь во влажной атмосфере (камера) в течение не-
скольких часов; ингибированное масло, контактный угол которо-
го равен 55°, защищает сталь уже 100 ч, а при контактном угле,
равном 30—35°, — в течение 200 ч.
Для проведения ускоренных коррозионных испытаний смазан-
ных металлических образцов может применяться аппаратура,
в которой воспроизводятся атмосферные условия и предусматри-
вается усиленное воздействие одного или нескольких факторов,
определяющих коррозионный процесс в естественных условиях,
которые были описаны выше.
Следует отметить, что при ускоренных испытаниях смазок
особенно важно соблюдать постоянные условия конденсации.
Необходимо проводить сравнительные испытания, т. е. одновре-
менно с новыми смазками испытывать смазку с уже известными
свойствами.
Ценные результаты при определении защитных свойств сма-
зок можно получить -методом определения паропроницаемости,
а также проницаемости агрессивных газов, присутствующих
в воздухе (SiO2, H2S). В. В. Скорчеллетти и С. Д. Васильев [59]
определяли паропроницаемость масла, помещая стакан с водой,
на поверхности которой находился слой смазки, в эксикатор,
в котором находился сосуд с определенным количеством погло-
тителя влаги. О паропроницаемости судили по привесу сосуда
с поглотителем. В качестве поглотителя ларов воды использова-
ли безводный медный купорос. При определении проницаемости
SO2 .или H2S через пленки масла в эксикатор вводили небольшое
количество газа [0,4% (объемн.)], а в качестве поглотителя ис-
пользовали 0,1-н. раствор едкого иатра. Исследования [59] пока-
зали, что надежность защиты металла слоем смазки в значитель-
ной степени определяется степенью его проницаемости для паров
воды н агрессивных в коррозионном отношении газов. Так как
кислород в жидких углеводородах растворим и поэтому предот-
вратить его доступ к металлу невозможно, то процесс коррозии
— 219 —
под смазкой можно устранить или сильно затормозить, если
смазка окажется препятствием для второго, необходимого для
возникновения коррозионного процесса вещества — воды. Про-
цесс коррозии сильно ускоряется, если сквозь смазку, кроме па-
ров воды, проникают и другие агрессивные составляющие воз-
духа.
Поэтому одним из'важных показателей эффективности за-
щитного действия смазок может служить их проницаемость для
к'оррозионно активных веществ.
5. ИНГИБИТОРЫ
Защита металлов от коррозии с помощью ингибиторов осно-
вана на свойстве некоторых веществ, добавленных в незначитель-
ных количествах в коррозионную среду, тормозить или полностью
подавлять коррозионный процесс.
По влиянию ингибиторов на скорость электродных реакций,
обусловливающих коррозионный процесс, их можно разделить на
три группы: а) ингибиторы, тормозящие только анодную реак-
цию; б) ингибиторы, тормозящие только катодную реакцию;
в) ингибиторы, тормозящие обе реакции [60].
Анодные замедлители при введении их в коррозионную среду
полностью подавляют коррозионный процесс, однако при недо-
статочном количестве ингибитора в среде возникает опасность
увеличения интенсивности коррозии на отдельных участках по-
верхности металла. Поэтому анодные замедлители, добавленные
в недостаточных количествах, весьма опасны, и надежной защи-
ты можно достигнуть только при добавлении такого количества
ингибитора, которое обеспечит полное прекращение коррозии.
Катодные ингибиторы уменьшают коррозию вследствие тор-
можения отдельных стадий катодной реакции; некоторые ингиби-
торы этой группы уменьшают площадь катодных участков. Ка-
тодные замедлители совершенно безопасны и никогда не приво-
дят к концентрации коррозии на ограниченной площади и к уве-
личению ее интенсивности, даже если они добавлены в корро-
зионную среду в недостаточных количествах. Однако они, как
правило, менее эффективны.
Смешанные ингибиторы тормозят обе электродные реакции.
Оии менее опасны, чем чисто анодные замедлители, и в ряде слу-
чаев могут не приводить к росту интенсивности коррозии при не-
достаточной их концентрации. При преимущественном торможе-
нии катодного процесса их свойства приближаются к свойствам
катодных ингибиторов, т. е. они становятся безопасными. Поэто-
му главное, что должен выявить ускоренный метод — это меха-
низм действия ингибитора с тем, чтобы избежать опасных по-
следствий, которые может вызвать анодный ингибитор. Это мож-
но установить электрохимическим способом, или определением
— 220™
коррозии при различных концентрациях ингибиторов (как ма-
лых, так и больших).
Методы испытания, устанавливающие защитную мощность ин-
гибиторов, целесообразно рассмотреть, классифицируя их по ха-
рактеру коррозионных сред и условиям эксплуатации.
Ингибиторы применяют для защиты от коррозии в жидких
средах (нейтральных, кислых и щелочных), а в последнее время
и в газовых. Для защиты от атмосферной коррозии находят все
большее применение так называемые летучие ингибиторы. Изве-
стно также применение ингибиторов для повышения защитных
свойств смазок и лакокрасочных покрытий.
При определении защитных свойств ингибиторов в жидких
средах прежде всего необходимо определить защитную способ-
ность выбранного вещества, его защитный эффект.
Защитный эффект Z, получаемый от введения ингибитора
в коррозионную среду, выражается в процентах и вычисляется
по формуле
7.= . ЮО, (101)
Ко
где Ко — скорость коррозии без ингибитора;
Хи — скорость коррозии в присутствии ингибитора.
Для вычисления защитного эффекта необходимо прежде все-
го получить кривую, выражающую зависимость -скорости корро-
зии металла, который хотят защитить, от концентрации ингиби-
тора в данной среде. Скорость коррозии можно определять по
изменению массы или объемным методом. Если испытывают ин-
гибиторы для нейтральных сред, то коррозию можно определить
по количеству поглощенного кислорода. Если испытывают инги-
биторы для кислых сред, коррозию определяют по количеству вы-
делившегося водорода (см. гл. III, стр. 99).
На рис. 126 приведена такая зависимость, полученная для же-
леза в растворе, имитирующем речную воду; в качестве ингиби-
тора вводили нитрит натрия [60]. Кривая имеет характерный мак-
симум в области малых концентраций ингибитора, следователь-
но, неполная защита железа от коррозии с помощью нитрита нат-
рия опасна. Полной защиты можно достигнуть, применяя кон-
центрации, которым соответствуют ничтожно малые скорости
коррозии.
При оценке защитного эффекта ингибитора необходимо так-
же определять зависимость глубины и площади, занятой корро-
зией, от его концентрации (рис. 127). Это особенно важно для
анодных ингибиторов. Площадь, занятая коррозией, и глубина
коррозионных поражений могут быть определены методами,
описанными в гл. III.
Второй важный показатель, определяющий защитное дейст-
вие ингибитора, — температурный коэффициент Т}( процесса кор-
розии при различных концентрациях ингибитора, поскольку не-
— 221— *
(102)
которые ингибиторы теряют при повышенных температурах свои
защитные свойства.
Температурный коэффициент выражается формулой
Гк = Л^_.100>
тде К,. — скорость коррозии при температуре /2;
Kt, — скорость коррозии при температуре Л;
?2 —- 4 — разность температур.
Температурный коэффициент определяют из кривых зависи-
мости коррозии -------~ -----
0,032
0,030
0,028
0.028
0,02Ь
металла от концентрации ингибитора, полу-
ченной при различных температурах.
Чтобы правильно выбрать ингибитор и
его оптимальную концентрацию, нередко
применяют электрохимические методы, кото-
/о.о
90
so Z
О 20‘/О 8080100
Концентрация NoNOg.яг/дм*
Рис. 127. Зависимость площа-
ди коррозии и средней глубины
ее проникновения в металл от
концентрации нитрита натрия
[60]. Состав электролита:
0,03 г/дл3 NaCl + 0,07 г/dzt3
NaaSCb,
j. о, ого
~%ЛО1В
^-0.010
§ c.im
^О.огг
=§ О.О7О
0,008
0,008
0,0ОЬ
0,002
О
О 0,7 0,2 0,3 01/ 05 06 07 OjB 0,9 1,0
Концентрация №Мг,е/дн3
Рис. 126. Влияние концентрации нитрита
натрия иа коррозию железа {60]. Состав
электролита: 0,03 е/сЬи3 NaCl + 0,07 а/Йл«3
NagSOfl
рые подробно описаны в гл. IV. Поляризационные кривые, сня-
тые в растворах в присутствии ингибитора и без него, указывают
на характер исследуемого соединения, т. е. к какому типу инги-
биторов он относится по влиянию на скорость электродных реак-
ций. На рис. 128 приведены поляризационные кривые, снятые на
железе в присутствии нитрита натрия и без него. Из кривых
видно, что нитрит натрия оказывает большое влияние на анод-
ный процесс (см. рис. 128, б) и поэтому должен быть отнесен
к группе анодных ингибиторов. Из этих же кривых можно заклю-
чить, что для данного электролита концентрация, равная 0,1 г/л,
не вызывает заметной анодной поляризации и, следовательно, не
— 222 —
обеспечит защиты. Сильная анодная поляризация наблюдается
при концентрации электролита 1,5-—5,0 г/л, которая обеспечит на-
дежную защиту металла от коррозии.
Все способы коррозионных испытаний металлов при погруже-
нии в нейтральные электролиты могут быть использованы и при
испытании ингибиторов, предназначенных для кислых сред. По-
скольку -в этих условиях коррозия протекает -в основном с водо-
родной деполяризацией, ее можно определять по количеству вы-
деляющегося водорода. Напри-
мер, на установке И. П. Ано-
Рис, 128. Влияние добавок нитрита натрия к электролиту на катодную (с)
и анодную (б) поляризацию железа {60]. Состав электролита: 0,1-н, раствор
NaCl + 0,53 г/дм3 NagCO3 + 0,42 г/дм3 МаНСО8; pH = 9. Добавка NaNO3.
г/дм3-.
На и б)—0,0; 2(й и б)—0.1; 3(a)— 2,0; 3 (б) — 1,5, 4-б£
щенко [61] можно оценивать коррозию не только объемным мето-
дом, но и одновременно определять потери массы; испытания
можно проводить при комнатной и при повышенных температу-
рах (рис. 129). В этой установке электролитический водород очи-
щается и высушивается, последовательно проходя три сосуда 1
с перманганатом калия, плюмбитом натрия и концентрированной
серной кислотой. Затем через шлиф 2, кран 3 (левый), соедини-
тельную трубку со шлифами и кран 10 (левый) водород посту-
пает по трубке в нижнюю часть сосуда, в которой на стеклянной
подставке помещается исследуемый образец в виде цилиндрика
диаметром 10 мм и высотой 10 мм. Из прибора водород в про-
цессе продувки выходит в атмосферу через трехходовой кран 16,
бюретку 18 и стакан с водой. При работе в кислородной атмосфе-
ре кислород проходит такой же путь, как и водород. Для иссле-
дования в водородной атмосфере раствор кислоты перед опытом
— 223 —
освобождают от кислорода, для чего электролитический водород
пропускают через кран 3 (левый) и шлиф 4 (левый) в колбу
с раствором, откуда его через трубку <в стакане с водой выбрасы-
вают в атмосферу. Кислота после продувки ее газообразным во-
дородом подается из колбы через кран 8, шлиф 9 (правый) .и
кран 10 (правый) в сосуд, где уже находится исследуемый обра-
зец. После заливки кислоты подача водорода прекращается, из-
быток водорода выпускается в атмосферу, показание водяного
Рис. 129. Схема установки для исследования защитного действия ингибиторов
в кислых средах объемным методом (61]:
1 — сосуды для очистки и осушки; 2 — шлиф; 3 — краны; 4 — шлифы; 5 — трубка;
б кран; 7 — крышка колбы; 8 — кран; 9 — шлифы; 10 — краны; II — подставка;
12 — образец; 13 — сосуд, в который помещается образец; 14 — змеевик; 15 — мано-
метр: 16 — трехходовой кран; 17 — сосуд с водой; 18 — бюретка для сбора газа;
19 — кран
манометра становится равным нулю и при помощи трехходового
крана и шлифа система соединяется с колбой 17, заполненной
водой. После открытия крана 10 (правый) колбы из насоса -вы-
деляется некоторое количество воды, которое исключается из об-
щего количества воды, собираемой в мерный цилиндр.
Сосуд с образцами находится в водяном термостате. Водород,
выделяющийся в процессе коррозии, поступает в измерительную
бюретку, заполненную водой и погруженную в стакан.
Применение ингибиторов для защиты стального оборудова-
ния нефтяных скважин требует изучения их защитной способно-
сти в условиях одновременного действия морской воды, сероводо-
рода и кислорода. В лабораторных исследованиях такие условия
— 224 —
моделируются с помощью, .например, установки (приведена на
рис. 130), разработанной В. Ф. Негреевым и Г. X. Монаховой [62].
Из сосуда 1 пластовая вода, содержащая сероводород, поступает
в сосуд 5 с испытываемыми образцами 4. В сосуд 5 подается
воздух через отверстие 6 и ингибитор из бюретки 3. Скорость по-
тока регулируется краном 2. Коррозию определяют по потере
массы образцов при испытании с ингибиторами и без них.
Для аналогичных усло-
вий Баррет и Клейтор [63]
применили более сложную
установку (рис. 131). Испы-
тания на такой установке
проводят в динамических ус-
ловиях при движении жид-
кости. В качестве коррози-
онной среды используют
нейтральный и подкислен-
ный сероводородом (до pH =
= 5,7) деаэрированный рас-
твор хлористого натрия.
Как видно на рисунке, рас-
твор, приготовленный в ос-
новном сосуде 1 объемом
16 дм&, подается помпой 2
Рис. 130. Схема установки для опреде-
ления защитной способности ингиби-
торов коррозии стального оборудова-
ния нефтяных скважин 162]:
1 сосуд с пластовой водой, содержа-
щей сероводород; 2 — реТулятор потока
воды; 3 — бюретка с ингибитором; 4 —•
стальные образцы; 5 — сосуд с испы-
тываемыми образцами; б — отверстие
для -ввода воздуха; 7 — выпуск воды
и воздуха
в соединенные между со-
бой сосуды, в каждый из
которых помещают по од-
ному образцу 3. Скорость
движения и температура
электролита контролирует-
ся реометром 4 и термомет-
ром 5. Ингибитор вводится
через воронку 6 при присое-
динении ее к трехходовому
крану 7. Ингибитор вводится перед началом испытания на
10 сек. и затем удаляется из системы, оставляя на образцах
защитную пленку. Защитное действие ингибиторов авторы опре-
деляли по изменению электросопротивления образцов. Показа-
телем служило время сохранения защитной пленки ингибиторов
на образцах. Эта установка позволяет изучать также влияние
скорости движения электролита и содержания кислорода на
защитное действие ингибиторов'.
Определить защитную способность ингибиторов, предназна-
ченных для защиты от коррозии -в атмосферных условиях, не-
сколько сложнее, чем ингибиторов, вводимых в жидкие среды.
При выборе или разработке метода испытания ингибиторов
для газовых сред в лаборатории, так же как и для всех других
ускоренных испытаний, прежде всего необходимо, чтобы резуль-
15 Зак. 1338
— 225 —
тэты испытаний были известны -в возможно более короткий срок,
т. е. были ускоренными и не искажали механизм коррозионного
процесса. Кроме того, результаты ускоренных испытаний долж-
ны быть сопоставимы с поведением ингибиторов в естественных
условиях.
Наиболее распространенными ингибиторами для газовых сред
являются ингибиторы, применяющиеся для защиты от атмосфер-
ной коррозии. Это могут быть ингибиторы, действующие при на-
несении на поверхность металла, а также летучие ингибиторы,
Рис. 131. Схема установки для определения защитной способности ингиби-
торов в условиях движущихся электролитов, содержащих хлористый натрий,,
сероводород и кислород [63]
которые различаются по способу попадания из носителя на по-
верхность металла: непосредственный контакт металла с носите-
лем ингибитора (ингибитированная бумага) и отсутствие непо-
средственного контакта с носителем, когда ингибитор попадает
на металл, адсорбируясь из атмосферы, насыщенной парами ин-
гибитора.
В соответствии с указанными условиями применения летучих
ингибиторов существуют два способа, ускоренных испытаний за-
щитного действия.. При испытаниях, когда металл находится в не-
посредственном контакте с носителем ингибитора, образцы иссле-
дуемых металлов упаковывают в .бумагу, пропитанную водным
или водно-спиртовым раствором летучего ингибитора и высушен-
ную. Поверх ингибитированной бумаги образцы упаковывают,
в какой-либо .барьерный материал; обычно -это те -материалы,
— 226’—_
которые предполагают использовать в практике (бумага, поли-
мерная пленка, картонная упаковка и т. д.).
Упакованные образцы помещают во влажную камеру, где под-
держивается 100%-ная относительная влажность или пропу-
скается поток увлажненного воздуха. Для испытаний во влажной
атмосфере могут быть использованы влажные камеры, описан-
ные в гл. II.
Схема установки Стронга и Вернона [64] для испытаний за-
щитных свойств летучих ингибиторов в потоке увлажненного воз-
духа представлена на рис. 132. Через трубку 2 воздух подается
Рис. 132. Схема установки для определения защитных свойств летучих ин-
гибиторов динамическим методом при контакте носителя летучего ингибитора
с металлической поверхностью {641:
а — общий вид установки: 1 — сосуд, наполненный водой; 2 — трубка для ввода воз-
духа; 3 — воронка; 4 — каплеуловители; 5 — сборник конденсата; 6 — реакционная
камера; 7 — термометр; 8 — рама с комплектом образцов;
б — рама: !— образцы; 2 — стеклянная перегородка; 3 — подставка из нихромовой
проволоки; 4 — скрепляющая проволока; 5 — картон и стеклянная пластина; 6 — гори-
зонтальная подставка для комплекта; 7 — держатели для крепления образцов
в сосуд /, наполненный водой, нагретой до 90—95° С. Освобо-
дившись от избытка влаги в каплеуловителях 4, насыщенный во-
дяными парами воздух попадает в камеру 6, в которой находится
рама с комплектом упакованных образцов 8. Скорость потока
воздуха регулируется так, чтобы полное обновление атмосферы
в камере 6 происходило за 1,5 мин.
Влажная камера 6 представляет собой стеклянный колоколо-
образный сосуд емкостью 6 дм3, термостатированный войлоком
толщиной 12 мм. В крышке сосуда монтируется резиновая проб-
ка с термометром и трубкой, подводящей воздух. Образцы для
испытания прокладывают с обеих сторон ингибитированным кар-
тоном и стеклянными пластинками; размер картона и стеклян-
ных' пластинок больше образцов. Для лучшего контакта упако-
ванные образцы перевязывают проволокой и затем монтируют
15* — 227 —
на раме из стеклянных трубок. Для предотвращения попадания
конденсата на образцы между рядами прокладывают наклонные
•стеклянные перегородки.
В тех случаях, когда в промышленных условиях адсорбция
летучих ингибиторов металлом происходит из газовой фазы, ла-
бораторные исследования защитных свойств ингибиторов прово-
дят в замкнутых стеклянных сосудах, в которых создается на-
сыщенная водяными парами атмосфера. По методу 164] образцы
подвешивают в колбах или стеклянных сосудах, на дно которых
наливают 50 см3 водного раствора ингибитора определенной кон-
центрации (рис. 133). Испытания
проводят в условиях конденсации
влаги на поверхности образцов, для
чего сосуды 8 ч выдерживают в, тер-
мостате при 35° С, а остальное вре-
мя суток при комнатной температу-
ре. Продолжительность испытаний
установлена авторами и равна шес-
ти неделям.
Конденсации при другом вари-
анте испытаний достигали, помещая
сосуды в термостат только наполо-
вину. Верхняя часть сосудов, где
располагались образцы, находилась,
вне термостата; конденсация в этом
случае происходила непрерывно.
Срок испытаний сокращался до
двух недель.
В реальных условиях примене-
ния ингибиторов они могут посту -
из жидкой, но и из твердой фазы —
ингибитированной бумаги; в этом случае в сосуды для испыта-
ний следует помещать не растворы ингибиторов, а ингибитиро-
ванную бумагу. При этом нужно иметь в виду, что прежде, чем
создать условия 100%-ной влажности или конденсации, нужно
обеспечить предварительную выдержку образцов в атмосфере
летучего ингибитора, в результате которой должна произойти
адсорбция ингибитора поверхностью металла.
Такого рода испытания также можно проводить в камерах
или специальных сосудах, в которых создаются описанные выше
условия. Стронг и Вернон [64] проводили испытания, подвешивая
образцы в стеклянных трубках, открытых с нижнего конца, во
влажной камере. Внутреннюю часть стеклянных трубок выкла-
дывали ингибитированной бумагой.
Более жесткий метод испытания защитных свойств летучих
ингибиторов — динамический [65], при котором испытания прово-
дят в движущемся, предварительно увлажненном и насыщенном
парами летучего ингибитора воздухе. Схема установки для ди-
— 228 —
Рис. 133 Схема установки для
определения защитных свойств
летучих ингибиторов [64]-
/ — резиновая пробка; 2 — ис-
пытуемый образец; 3 — стеклян-
ный сосуд; 4 — термостат
пать к металлу не только
намических испытаний приведена на рис. 134. В реакционный со-
суд помещают испытываемый образец. Образец монтируют
в стеклянной обойме с помощью бакелитового лапа так, что ис-
пытанию подвергается только одна его поверхность. При скоро-
сти просасывания воздуха 120 смешан и температуре воздуха
69° С время испытаний, по данным авторов, составляет 44 ре-
зультаты испытаний хорошо согласуются с поведением ингиби-
торов при испытаниях в естественных условиях.
Вход
Рис. 134. Схема установки для динамических ис-
пытаний защитных свойств летучих ингибиторов
при отсутствии контакта между носителем лету-
чего ингибитора и металлической поверхностью
(65J:
1 — увлажняющий сосуд; 2 — каплеуловители; 3 —
сосуд для насыщения воздуха парами ингибитора (вы-
ложен ингибитированной бумагой); 4 — реакционный
сосуд (выложен ингибитированной бумагой); 5 — испы-
тываемый образец; 6 — стеклянная обойма; 7 — диск
из ингибитированной бумаги
Динамические испытания отражают лишь условия, когда ин-
гибиторы применяют для защиты, изделий или конструкций, под-
вергающихся обдуванию воздухом; в промышленных же усло-
виях нередки случаи, когда испарение летучих ингибиторов из
твердой фазы в защищаемое пространство и их удаление во
внешнюю атмосферу затруднены, а конденсация и испарение с по-
верхности металла происходят в замкнутом пространстве.
Более правильны поэтому испытания, основанные на ускоре-
нии процесса за счет увеличения интенсивности воздействия
реально существующих в эксплуатации факторов, таких как кон-
денсация, чередующаяся с испарением, и агрессивные составляю-
щие атмосферы, например сернистый ангидрид.
С. А. Гинцберг и А. В. Шрейдер [66] определяли защитное дей-
ствие летучих ингибиторов во влажной камере, в которой цикли-
чески изменялась температура (1 ч—40° С, 2 ч—комнатная тем-
пература). Относительная влажность, равная 94—98%, создава-
— 229 —
Рис. 135. Прибор для определения
защитных свойств летучих ингибито-
ров, адсорбирующихся ина поверхно-
сти металла из атмосферы, насыщен
ной ингибитором [67]:
1 — стеклянная трубка с плоской
поверхностью; 2 — образцы; 3 — сосуд
с ингибитором
лась в камере при помощи водных растворов глицерина (см.
гл. П, табл. 9).
Каждый ингибитор помещали в отдельный герметизирован-
ный отсек камеры; в отсеках поддерживали концентрацию серни-
стого газа, равную 0,01 мг[дл&. ккюры отмечают, что испытание
ингибиторов в течение 35 суток в камере эквивалентно 6 месяцам
нахождения их в тропическом влажном климате индустриально- .
го района.
В. П. Персианцева и И. Л. Розенфельд [67] разработали ме-
тод испытания защитного действия летучих ингибиторов корро-
зии, адсорбирующихся на
поверхности металла из ат-
мосферы, насыщенной инги-
битором. Прибор, предло-
женный ими, позволяет осу-
ществлять периодически че-
редующиеся конденсацию
влаги на образцах и ее вы-
сушивание. Скорость корро-
зии в зависимости от коли-
чества циклов в сутки мо-
жет варьироваться в широ-
ких пределах.
На рис. 135 представлена
схема, этого прибора, состо-
ящего из вакуумного экси-
катора, по периферии кото-
рого расположена стеклян-
ная трубка с плоской верх-
ней стенкой. На стеклянной
трубке располагают испы-
тываемые образцы, нижнюю
сторону которых покрывают лаком, устойчивым в данной атмо-
сфере. Авторы, в частности, использовали грунт (ВХГМ). Кон-
денсации и высыхания влаги на образцах достигают периодиче-
ским пропусканием по трубке горячей или холодной воды. В со-
судах заданную влажность создают, пропуская через прибор
предварительно увлажненный воздух. Испытания проводили по
следующему режиму: 45-лш« пропуск через трубку холодной
воды при температуре 10—15° С, остальное время суток вслед-
ствие повышения температуры до комнатной происходило испа-
рение влаги. Для увеличения скорости испарения через стеклян-
ную трубку можно пропускать воду, нагретую на 5° выше темпе-
ратуры окружающего воздуха.
Насыщение парами исследуемых соединений пространства до-
стигалось путем естественного испарения ингибитора (твердого
или жидкого), помещенного на дно камеры в открытом б.юксе.
Образцы предварительно выдерживали в атмосфере ингибитора
— 230 —
и только после этого в сосуде создавалась влажная атмосфера.
Продолжительность испытания составляла 10 суток. Ингибитор
считали годным, если после 10 суток испытания при визуальном
осмотре на образцах не обнаруживалось коррозионных пора-
жений. Кроме того, защитные свойства ингибиторов можно оце-
нивать по изменению массы металла, а также по времени появле-
ния первых коррозионных очагов или по площади, занятой 'кор-
розией. В этих случаях испытания ведут в приборах, описанных
выше (стр. 79), в которых можно наблюдать за образцами, не
раскрывая их.
Сопоставление результатов коррозионных испытаний, прове-
денных этим ускоренным методом, с результатами испытаний
в условиях 100 %-ной влажности в течение 9 месяцев показало,
что ускоренный метод позволяет удовлетворительно оценивать
защитные свойства летучих соединений [67].
Метод, предложенный В. П. Персианцевой и И. Л. Розенфель- .
дом, в отличие от других описанных методов позволяет получать,
конденсацию влаги на металле благодаря охлаждению самих об-
разцов, а также вести испытания в присутствии -SOs-
Ускоренные испытания защитных свойств летучих ингибито-
ров можно проводить в аппаратуре, описанной в гл. II, но при вы-
боре способа испытания необходимо учитывать условия приме-
нения ингибитора и характер защищаемого изделия. Например,
для ингибиторов, предназначенных для защиты замкнутых си-
стем или емкостей, не сообщающихся с атмосферой при хране-
нии, испытание в условиях обильной конденсации не будет отра-
жать поведение ингибитора в реальных условиях. Смывающее
действие конденсирующейся влаги на практике не будет наблю-
даться и поэтому не должно иметь места при ускоренных испыта-
ниях. Такие системы надо испытывать в атмосфере водяных па-
ров (100%-ная относительная влажность).
Рассмотрим-испытания-ингибиторов, предназначенных для за-,
щиты от атмосферной коррозии и наносимых непосредственно на.
металл в виде загущенных или спиртовых растворов. Ингибито-
ры этого типа, такие как нитрит -натрия, загущенный крахмалом
или другими загустителями, а также некоторые соединения, на-
носимые на металл путем омывки спиртовыми растворами, могут
испытываться во влажной чистой атмосфере и в атмосфере, со-
держащей агрессивные компоненты.
Характерной особенностью испытания ингибиторов, предназ-
наченных для защиты от атмосферной коррозии, является необ-
ходимость в каждом отдельном случае при выборе того или ино-
го метода ускоренных испытаний исходить из реальных условий
применения ингибитора. Если предполагается, что ингибитор на-
носится на бумагу, то испытывать надо изделия, завернутые
в ингибитированную бумагу. Если предполагается омывать изде-
лие спиртовым или водным раствором ннгибитора, то и при уско-
ренных испытаниях <»на образцах следует создавать методом
— 231 —
Омывки тонкую пленку ингибитора, после чего помещать образцы
в камеру для испытания. Если летучий ингибитор предполагают
использовать в замкнутом пространстве, подвешивая его в ме-
шочках, следует эти условия воспроизводить и при ускоренных
испытаниях, чтобы определить дальность действия ингибитора и’
скорость насыщения им пространства.
При ускоренных испытаниях для определения защитного дей-
ствия летучих ингибиторов в атмосферных условиях часто опре-
деляют потери массы металла. Хотя эти данные полезны для
оценки свойств ингибиторов, но необходимо помнить, что для ле-
тучего ингибитора важна полная защита. Поэтому, если даже и
будет наблюдаться заметное снижение коррозии* например на
50—70%, такой ингибитор все равно нельзя рекомендовать для
защиты сложных изделий и приборов, поскольку уже незначи-
тельная коррозия может полностью вывести их из строя.
Наиболее правильно в этом случае пользоваться оценкой кор-
розии по времени до появления первого коррозионного пораже-
ния. Этот метод оценки будет отражать условия и требования,
предъявляемые к способу защиты изделий или конструкций при
транспортировке и хранении.
Ускоренные коррозионные испытания для выявления защит-
ной способности того или иного соединения необходимо прово-
дить в сравнении с известными ингибиторами, сведения об успеш-
ном применении которых уже существуют.
Для оценки защитных свойств летучих ингибиторов можно
с успехом применять и электрохимические методы, позволяющие
получить результаты в довольно короткий срок.
Несмотря на то что механизм действия летучих ингибиторов
еще нуждается в исследовании, в настоящее время получены
экспериментальные данные, указывающие на то, что их тормозя-
щее действие связано с воздействием на кинетику электродных
реакций, обусловливающих коррозионный процесс. И. Л. Розен-
фельд, В. П. Персианцева и др. [68] показали, что, используя ме-
тод снятия поляризационных кривых под тонкими слоями элек-
тролитов в атмосфере летучего ингибитора и метод измерения
электродных потенциалов металла, выдержанного в атмосфере
ингибитора различное время, можно судить об эффективности за-
щитного действия ингибиторов.
При электрохимических методах исследования коррозионных
процессов, протекающих на металлах под тонкими слоями элек-
тролитов в присутствии летучих ингибиторов, должны обеспечи-
ваться условия самопроизвольного попадания ингибитора на
металлическую поверхность до нанесения на электрод пленки'
электролита; для этой цели'в крышку прибора, используемого
для исследований электродных процессов под тонкими, слоями
электролитов, протекающих в присутствии агрессивных газов,
дополнительно впаивают воронку, которая позволяет наносить
слой электролита на электрод в замкнутой «атмосфере, насыщен-
— 232 —
нои парами летучего соединения (рис. 136). На дно прибора по-
мещают исследуемое соединение и дистиллированную воду для
поддержания 100%-ной относительной влажности.
В тех случаях, когда исследователь не располагает специаль-
ными приборами для проведения электрохимических исследова-
Рис. 136. Прибор для исследования
электрохимических процессов, проте-
кающих на металлах под тонкими
пленками электролита в атмосфере
заданного состава J67J:
I — краны для подключения системы,
создающей атмосферу заданной влажно-
сти; 2 — проводник от исследуемого
электрода; 3 — капельная воронка; 4 —
платиновая проволока (вспомогательный
электрод для поляризации): 5 — измери-
тельный ключ (соединяется через проме-
жуточный сосуд с каломельным полуэле-
ментом); 6 — поляризационный ключ.
7 — сосуд с ингибитором; 8 — электррд
ний в тонких слоях электроли-
тов, можно довольно быстро
из имеющихся соединений ото-
брать наиболее эффективные.
Рис. 137. Влияние времени предва-
рительного контакта стального
электрода с парами летучих ин-
гибиторов на значение начального
потенциала [68]. Ингибиторы:
I _ морфолин; 2 — морфолин угле-
кислый; 3 — морфолин ’коричнокис-
лый; 4 — дициклогексиламин; 5 —
дициклогексиламин азотистокислый;
6 — бензиламин; 7 — бензиламин уг-
лекислый; 8 — циклогексиламин;
$ — циклогексиламин углекислый
применяя обычные методы изучения электрохимической кинети-
ки в объеме электролита. Однако в дальнейшем необходимо
определить, обладает ли данное соединение достаточным давле-
нием паров для того, чтобы оно могло быть использовано в ка-
честве летучего ингибитора > 1,3 • 10~2 н/м2 (> 10~4 мм рт. ст.).
Рассмотрим некоторые данные электрохимических методов
испытаний ингибиторов, полученные Розенфельдом с сотрудни-
ками [68, 69].
На рис. 137 представлены кривые изменения начального по-
тенциала стального электрода после выдержки его в атмосфере,
— 233 —
насыщенной различными ингибиторами. Из кривых видно, что
ингибиторы, адсорбированные предварительно на поверхности,
заметно смещают потенциал в положительную сторону. По на-
правлению смещения потенциала и величине его сдвига можно
-судить об эффективности ингибиторов. Эти электрохимические
характеристики подтверждаются и коррозионными опытами,
-woo
-800
-600
-ООО
Рис. 138. Влияние ингибиторов на
ход потенциостатической кривой
для железа в I-н. растворе NazSO.,
[69]:
а — 2%-ный гексаметилеиимин; б —
2%-<ный нитрит дициклогексиламинз.
в — 2%-ный 3.5-динитробензоат гек-
саметиленимина
§ 200
I *00
$ 600
| 800
'/ООО
1200
/ООО
/бОО^^-1-^ ' ...... ' 1 ' I
50003020/0 О 3.0 2.0/.0 0 /,02.0 0 0080720
Плотность тока,на/смг
указывающими на то, что амины по отношению к стали более
•эффективны, чем соли аминов.-По значениям электродных по-
тенциалов, можно судить и о механизме действия ингибиторов,
т. е. определить, к какой группе они относятся, и тём самым
установить степень их опасности при малых концентрациях.
Потенциостатические кривые, характеризующие зависимость
скорости электродных реакций от потенциала, представлены на
рис. 138, а — в. Гексаметиленнмин (см. рнс. 138, а) резко тормо-
— 234 —
зит анодную реакцию ионизации железа, что характеризуется
сильной анодной поляризацией. Судя по этой кривой, которую
можно в лабораторных условиях получить довольно быстро, этот
ингибитор должен хорошо защищать от коррозии железо. Дли-
тельные и всесторонние испытания подтверждают данные элек-
трохимических исследований.
Нитрит дициклогексиламина и 3,5-динитробензоат гексаме-
тиленимина (см. рис. 138, б и в), судя по ходу кривых, должны
оказаться эффективными. Однако механизм их действия принци-
пиально различен. Нитрит дициклогексиламина замедляет
коррозию главным образом благодаря торможению анодной
реакции ионизации железа, в то время как 3,5-динитробензоат
гексаметил еним ина влияет благодаря ускорению катодного
процесса, приводящем)' к смещению стационарного потенциала
до значения, при котором железо пассивируется.
Такая высокая окислительная способность 3,5-динитробен-
зоата гексаметиленимина, связанная с восстановлением нитрит-
ной группы, достигнута благодаря введению в бензольное колы
цо второй нитрогруппы и карбоксильной группы, обладающих
высокими электрофильными свойствами. Это сочетание групп в
бензольном кольце оттягивает электроны от азота, что создает
на нем положительный заряд и обусловливает высокую скорость
окисления.
Наличие двух принципиально различных механизмов дейст-
вия этих ингибиторов отчетливо видно из схем гидролиза этих
соединений. Нитрит дициклогексиламина:
NHHNOg Ч- Н2О NHSOH ф HNO8
£ N~bH«+ ОН” + н+ + NO?; (ЮЗ)
3,5-динитробензоат гекса метиленим ина:
NO8
___ ________/ _______________
>NHHOOC—> NH*OH+
^NOs
NO8 ;
__/ _____
+ НООС—>N~H4a + OH“ + H+-|~
XNO2
no8
+ РОС—’ (104)
no2
-235 —
В результате гидролиза нитрита дициклогексиламина в элек-.
тролите появляется органический катион и ион иитрита, вы-
зывающие сильную анодную поляризацию. Наличие отрица-
тельного заряда затрудняет подход к катоду нитрит-иона, что
не позволяет получить при этом высокую скорость восстанов-
ления'.
При гидролизе 3,5-динитробензоата гексаметиленимина в
электролите возникают органические катионы и сложные ионы.
Поскольку заряд на группе NO2 стал положительным вследст-
вие оттягивания электронов, его восстановление сильно облег-
чается.
Эти ингибиторы относятся к разным типам и тем не менее
оба они могут в определенных неблагоприятных условиях вызы-
вать коррозию. Нитрит дициклогексил амина может приводить к
образованию коррозии на малой поверхности, т. е. увеличивать
интенсивность при малых концентрациях, а 3,5-диннтробензоат
гексаметиленимина — при попадании в среду больших концент-
раций активаторов, например хлор-ионов.
Из изложенного видно, что электрохимические методы позво-
ляют относительно быстро, не проводя длительных испытаний,
предсказать поведение ингибиторов в реальных условиях экс-
плуатации.
Для оценки эффективности защитного действия ингибиторов,
предназначенных для атмосферных условий,- помимо описанных
ускоренных испытаний, необходимо определить некоторые физи-
ческие или химические свойства этих веществ.
Важнейшей характеристикой летучих ингибиторов является
давление насыщенного пара соединения (летучесть). Соедине-'
ния, обладающие слишком большим давлением паров, очень
быстро насыщают пространство, в котором находится защищае-
мое изделие, но также быстро улетучиваются из него, если упа-
ковка недостаточно герметична. Соединения со слишком малым
давлением пара вследствие медленной диффузии не сразу до-
стигают поверхности защищаемой конструкции или изделия; в
этот начальный период неполного насыщения пространства ин-
гибитором возможно возникновение коррозии. Поэтому для
каждого соединения, которое предполагают использовать в ка-
честве летучего ингибитора, необходимо знать упругость его па-
ров н ее изменение с температурой.
Существует несколько способов измерения давления паров
[<1,3-10"3 бар {<1 мм рт. ст.)]! слаболетучих соединений.
Однако достаточно прецизионен лишь метод Кнудсена, сущ-
ность которого заключается в измерении скорости эффузии
газа через малое отверстие. Если в замкнутое пространство по-
местить сосудик-испаритель с ингибитором и сделать в нем
калиброванное отверстие площадью, намного меньшей поверх-
ности испарения, то довольно быстро установится равновесие
между конденсированной фазой и паровой, и давление пара в
— 236 —
сосуде можно считать истинным давлением пара данного ве-
щества:
где р — давление пара ингибитора, 1,3• 10~3 бар (мм рт. ст.);
Т — абсолютная температура, °C;
М — молекулярная масса ингибитора;
т— время истечения;
ДС— масса испарившегося вещества;
S -— поверхность отверстия;
А — коэффициент пропорциональности.
Поэтому, если определить количество испарившегося вещества,
можно по уравнению рассчитать давление пара.
Количество испаряющегося вещества определяют с помощью
весов Мак-Бена в вакуумной установке, описанной в работе
И. Л. Розенфельда с сотрудниками [70].
Иногда для определения летучести ингибиторов их помеща-
ют в сосуд с отверстием и периодически взвешивают его. По
у бы пи вещества судят о летучести соединения.
Применяют и такой метод, как пропускание воздуха над по-
верхностью ингибитора с определением количества уносимого
вещества при различных скоростях движения воздуха. Экстра-
полируя иа нулевую скорость, определяют давление. Эти мето-
ды неточны и могут дать лишь качественные характеристики
ингибитора.
Важной характеристикой ингибиторов, наносимых на защи-
'чщаёмое изделие путем омывки спиртовыми или водными рас-
творами, является также растворимость их в воде. Соединения,
хорошо растворяющиеся в воде,, при возникновении на металле
конденсационной влаги могут быстро смываться с отдельных
участков поверхности; при слишком малой растворимости .этих
соединений в воде или других растворителях трудно обеспечить
на металле защитную концентрацию ингибитора.
ЛИТЕРАТУРА
1. Лайнер В. И., Кудрявцев А Т. Основы гальванотехники, т. I и II
Мета ллургиз дат, 1953.
2, Ганушкина Е. В. Новые методы ускоренных испытаний металлопо-
крытий. ЦБТИ тракторе- и с.-х. машиностроения, 1959.
3. Kutzelnigg A. *Der priifung metllischer Uberzeugen, 4, Galvanotech-
nik, 1960.
4. ASTM, B—287—54.
5. Silkes G. L. Metall Finish, 1959, v. 57, № 12, 59.
6. E d w a r d s J. 46-th. Annual Techn. Amer. Electroplaters Soc., Newark,
1959, p. 154.
7. Sa nual В., В had war D. M. J. Scient and Industr., Res., 1959, A 18,
№ 2, 69.
-237 —
8. Берукштис Г. К-, С т а л ъ с к а я 3. С. Атмосферная- стойкость гальва-
нических покрытий, тема 13, № M-57-253/I3. ВИНИТИ, 1957.
9. Pinner W. L. Plating, 1955, № 42, 1043.
10. Pinner W. L. 48-th. Annual- Techn. Amer Electroplaters. Soc., Newark,
№ 1, 1961, p. 27.
IL La QueF. L. 46-th. Annual Techn. Amer. Electroplaters Soc., Newark, 1959.
12. Баграмян A. T., Соловьева 3. А. Методы исследования электро-
осаждения металлов. Изд-во АН СССР. 1960.
13. О р л о в а С. И., Абрамсон Д. С. Контроль электролитов и качества
гальванических покрытий. Машгиз, 1950.
14. Вайнер Я. В., Д а с о я н М. А., Д р и н б ер г А. Я., Тарасенко А. А.,
Хайн И. И. Справочник по защитно-декоративным покрытиям. М.— Л.
Машгиз. 1951.
15. Сцнборовская Н. Б. Оксидные и цинко-фосфатные покрытия метал-
лов. Обороигиз, 1961.
16. Лопатухин В.. С. Фосфатирование металлов. Машгнз, 1958.
17. Якубович С. В. Исследования лакокрасочных материалов и покрытий.
М.— Л. Госхимиздат, 1952.
18. Бак Б. В. Сб. «Техника окраски», т. II. Госхимиздат, 1941; Бюллетень
z обмена опытом малярной- техники, 1938, № 4—5, 21.
19. Бак Б. В., Гершанок И. М. ЖПХ, 1940, т. 13, № 7, 961.
20. Я к у б о в и ч С. В. Физико-химические методы испытания лакокрасочных
материалов и покрытий. ГОНТИ, 1938.
21. Др и и б ер г А. Я., Рохлин М. Г. ЖПХ, 1951, т. 24, № 2, 213.
22. Дринберг А. Я., Ряшенцев К. В. ЖПХ, 1942, т. 15, № 34, 346.
23. Т о м а ш о в И. Д, Киселев В. С., Г о л ь б е р г М. М. Йзв. АЙ СССР,
серия хим., 1949, 2, 152.
24. Дринберг А. Я-, Гуревич Е. С. Тр. ЛХТИ, вып. 24, 1947.
25. Розенфельд И. Л., Рубинштейн Ф. И., Якубович С. В, Лако-
красочные материалы и их применение, 1961, № 3, 50.
26. Т о м а ш о в Н. Д., Михайловский Ю. Н. Тр. Ин-та физической химии
АН СССР, вып. 7. Изд-во АН СССР, I960, 249.
27. Charry В. W., Mayne J. Е..О. 1-th. International congress on metallic
corrosion, London, 1961, p. 44.
28. Розенфельд И. Л.» Рубинштейн Ф И., Якубович С. В. Лако-
красочные материалы и их применение, 1962, № 2, 58.
29. Brasher D. М., Kingsbury А. Н. Appl. Chem., 1954, №4, 62.
30. Hartshorn L., Meg son N. J. L.± Rush ton E. J. Soc. chem., Ind.,
London, 1937. 56, .266 T.
31. Зубов П. И., Санжаровскнй A. T„ Дыльков M. С. Лакокрасоч-
ные материалы и их применение, 1963, № 2, 48.
32. Дерягин Б. В., Сорокин С. М. Тр. НИИОГИЗ, ч. 2, 1937, 207.
33. Кротова Н. А., Морозова Л. П.. С-околнна Г. А. ДАН СССР,
1959, т. 127, № 2, 302; Проспект ВДНХ. Адгезнометр АЗС-2, 1962.
34. Якубович С. В„ Масле и н и к о в а Л. Н. Лакокрасочные материалы
и их применение, 1962, № 4, 20.
35. Шрейн ер С. А.. Зубов П. И. ДАН СССР, 1959, т. 124, № 5, 1102.
36. Лунева В. С. Передовой научн.-техи. и произв. опыт, тема 16, КМ-60,
265/27, I960.
37. Беликовский Д. С. Консистентные смазки. Гостоптехиздат,. 1945.
38. Кузнецов В. Г., Решетников Е. Ф. Тезисы докладов Всесоюзного;
совещания по коррозии и защите металлов. Профйздат, 1958.
39. Inst, of Petroleum., XLI1, № 386, 50, 1956.
40. Fisch К R a oth. NLGZ, Spok., XXIII, № 3. 102, 1959. =
41. Бон ер К. Дж. Производство и применение консистентных смазок. Пер.
с англ. Гостоптехиздат, 1958.’ ? - ' ,
42. Ellis С. G. Sclent. Lubr., IX, № 2, 25, 1957; IX, № 3, 27, 1957.
43. Кажд.ан П, И. Защитные смазки. Гостоптехиздат, 19.49. :
44. Синицын В. В'. -Заводская лаборатория, т- XXVII, № I, 1961. ’
45. Hour Т. Р._ Smith G. С. J. Inst, of Petrol, 36, № 319, 457, '19&К’•
— 238 —
46. Rod,en H. Bulletin, ASTM, № 223, 55, 1957.
47. Die man E. A., Gaywer I. W. Corrosion, № 6, 60, 1958.
48. Preston R. St. J., S t г о n g E. G. J. Inst, of Petroleum, 36, № 319, 457,
1950.
49. Ad dm s F. W. NLGI, Spok, XXI, № 7, 21, 1957.
50. Поддубный В. Н.» Иконников С. Н., Колом ыцевП Т. Защита
авиационного вооружения и боеприпасов от коррозии. Воеинздат, 1955.
51. Скор чел л етт-и В. В., Бухбнидер А. А., Пискорский В. Е..
ЖПХ, 1954, т. 27, вып. 4, 454.
52. Скор чел летти В. В., Пискорский В. Е. ЖПХ, 1953, т. 26, вып. 7,
730.
53. Лунева В. С. Тр. ВНИИНП, вып. 7, 449, 1957.
54. Лунева В. С. Тезисы докладов иа Всесоюзном совещании по коррозии
и защите металлов. Машгиз, 1959.
55. Гаджиева Р. Г., Трифель М. С. Тр. Гипроморнефть, № I, 178, 1954.
56. Лунева В. С., К о в а л е в В. А. Тр. ВНИИНП, вып. 6, 210, 1957.
57. М а р т ы и о в В. М., Морозова М. В. Химия и технология топлив
и масел, I960, № 3, 22.
58. Pi I G. Р., Farley F. F. Ind. Eng. Chem., 1946, v. 38, № 5, 601.
59. С к о p ч e л л e т т и В. В., Васильев С. Д. ЖПХ, 1953, т. 26, № 10, 1033.
60. Розенфельд И. Л. Замедлители коррозии в нейтральных средах.
Изд-во АН СССР, 1953.
61. Аиощенко И. П. Сб. «Методы исследования ингибиторов коррозии
металлов», № 7. Профиздат, 1958, 89.
62. Н е г р е е в В. Ф., Монахова Г. X. Тр. межвузовской научной конфе-
ренции по борьбе с коррозией. Гостоптехиздат, 1962, 375.
63. Barrett I Р С I a u t о г Е. Е. Corrosion, 1962, v. 18, № 7, 2774.
64. Strong Е. G.,’ Vernon W. H. I. J. Appl. Chem., 1952, v. 2, № 3, 166.
66. Wax ter A., S с у T., Stillman N. Corrosion, 1951, v. 7, № 9, 284.
66. Г и и ц б e p г С. А., Шрейдер А. В. Передовой научно-техн, и производ-
ственный опыт. ЦТЭИН, № М-59-425/5, 1959.
67. Персиянцева В. П., Розенфельд И. Л. Тр. Ин-та физической хи-
мии АН СССР, вып. 7. Изд-во АН СССР, 1959. 41.
66 Розенфельд И. Л., Персианцева В. П., Кузнецова М. М.,
Полтера М Н., Терентьев Б. П. ЖПХ, 1961, т. 34, вып. 10. 2234.
69. Розенфельд И. Л., Пер’сианнева В. П. Вестник АН СССР, 1962,
№ 8, 57.
70. Розенфельд И. Л., Персианцева В. П., П о л т е в а М. Н. ЖПХ,
1961, т. 34, вып. 10, 2056.
VI
НЕКОТОРЫЕ СПЕЦИФИЧЕСКИЕ МЕТОДЫ
УСКОРЕННЫХ КОРРОЗИОННЫХ ИСПЫТАНИЙ
До сих пор мы рассматривали обычные, приемы проведения
ускоренных испытаний. Однако существует ряд систем> где
важны особые свойства материалов, которые необходимо вы-
явить при испытании. К таким видам испытаний относятся:
1. Определение склонности сплавов к межкристаллитной
коррозии.
2. Определение склонности сплавов к коррозионному рас-
трескиванию.
3. Определение склонности нержавеющих сталей к точечной
коррозии.
4. Определение устойчивости металлов в условиях кави-
тации.
5. Определение устойчивости реакторных материалов при
высоких температурах и давлениях.
1. ОПРЕДЕЛЕНИЕ СКЛОННОСТИ СПЛАВОВ
К МЕЖКРИСТАЛЛИТНОЙ КОРРОЗИИ
Механизм межкристаллитной коррозии нержавеющих сталей
и основные принципы выбора электролита
для ускоренных испытаний
Межкристаллитная коррозия — наиболее опасный вид элек-
трохимического разрушения сплавов, поскольку она приводит к
потере металлом прочностных свойств без заметного изменения
внешнего вида.
Вследствие неправильной термической обработки исходных
материалов, а также при определенном термическом воздейст-
вии в процессе сварки или при других видах технологической
— 240 —
обработки этому виду коррозии подвержены: аустенитные хро-
моникелевые стали; высокохромистые стали, содержащие более
13% хрома; аустенитные стали, легированные молибденом; не-,
ржавеющие стали, легированные молибденом и медью.
По современным воззрениям преимущественное разрушение
границ зерен обусловлено электрохимической неоднородностью
поверхности, возникающей в определенном для данного сплава
интервале температур в результате структурных превращений.
Так, например, при нагреве хромоникелевых сталей типа 18-8
в интервале 600—800° С происходит выделение из твердого рас-
твора сложных карбидов, содержащих хром, железо и никель:
(Cr, Fe, .Ni)4C, (Cr, Fe, NOrCs- Эти карбиды выпадают преимуще-
ственно по границам зерен, поскольку в этих местах скаплива-
ются примеси, которые становятся центрами кристаллизации.
Кроме того, особые энергетические свойства границ зерен дела-
ют возможным перераспределение атомов вблизи границ при
таких относительно невысоких температурах, при которых по-
движность атомов недостаточна для перераспределения внутри
зерен. Поскольку хром более сильный карбидообразующий эле-
мент по сравнению с никелем и железом, выпадающие по гра-
ницам карбиды содержат преимущественно хром и их можно
отождествлять с соединениями типа СГ4С или Сг7Сз-
Экспериментально было установлено, что в карбидах хрома
на каждую весовую часть углерода приходится 10—12 ч. хрома.
Поэтому выпадение карбидов хрома по границам зерен приво-
дит к сильному обеднению отдельных зон сплава хромом. Наи-
более сильное обеднение хромом наблюдается в зоне, непосред-
ственно прилегающей к границе зерна. Здесь концентрация хро-
ма может упасть до 5—8%. В зонах, удаленных от границы
зерна, концентрация хрома снижается в меньшей степени: пола-
гают, что всего на 2—3%. Такое неравномерное обеднение гра-
ниц и зерна хромом объясняется очень большим коэффициентом
диффузии углерода по сравнению с хромом в твердом растворе
ре — Сг — Ni. Схематически изменение концентрации хрома
около выпавших карбидов изображено на рис. 139, откуда вид-
но, что в результате нагрева нержавеющей стали в опасной зоне
температур (600—800° С) на поверхности стали появляются три
зоны, сильно отличающиеся по своему химическому составу. Эти
три структурные составляющие будут и в электрохимическом
отношении неоднородны, так как электродный потенциал твер-
дого раствора железо — хром сильно зависит от концентрации
хрома (рис. 140).
Поверхность такой стали уже следует рассматривать не как
однородную, а как систему, состоящую из трех электродов:
I —карбид (Fe, Сг, Ni^C; II — граница зерна Fe/Cr(Cr < 8%);
Ш — зерно Fe/Cr(Cr > 8%). В такой электрохимической систе-
ме наиболее отрицательным потенциалом будут обладать участ-
ки, непосредственно прилегающие к границам зерен, поскольку
16 Зак. 1338 — 241 —•
концентрация хрома в них наименьшая. При соприкосновении с
агрессивной коррозионной средой начинают растворяться преи-
мущественно границы зерен.
Они находятся под воздействием большого катода (зерно с
повышенйым содержанием хрома), коррозия концентрируется
преимущественно в узкой зоне и приобретает опасный-характер.
Теория обеднения границ хромом, по которой межкристал-
литная коррозия нержавеющих сталей происходит вследствие
выпадения карбидов, наиболее правильно трактует существо яв-
лений [1], однако необходимо учитывать также другие процессы.
Рис. 139. Изменение кон-
центрации хрома около
выпавшего карбида [1]:
/ — активное состояние;
II — пассивное состояние
Рис, 140, Зависимость потенциала твер-
дых растворов Fe—Сг в слабоокислитель-
иом растворе от концентрации хрома [1]
В частности, для нержавеющих сталей, содержащих молибден,
большое значение приобретает выпадение сигма-фазы. Эта фаза
содержит значительно больше хрома и молибдена, чем твердый
раствор, и при ее выпадении участки, окружающие ее, тоже-
сильно обедняются хромом. Поэтому появление в структуре
сплава сигма-фазы делает его в определенных средах склонным
к межкристаллитной коррозии. По мнению Варена [2], сетка вы-
делений сигма-фазы приводит к межкристаллитной коррозии
только в азотной кислоте, в то время как сетка карбидов вызы-
вает сильную межкристаллитную- коррозию и в других кис-
лотах.
Перераспределение хрома в нержавеющих сталях возможно
и в результате выпадения высокохромистого феррита — продук-
та распада аустенита. Б. И. Медовар [3] выдвигает предположе-
ние о том, что межкристаллитная коррозия сварных швов в ста-
лях типа 18-8 вызывается обеднением границ зерен аустенита в
-*242 —
результате выпадения не карбидов хрома, а вторичного хроми-
стого феррита.
Существует мнение, что в появлении склонности к межкри-
сталлитной коррозии определенную роль играют и внутренние
напряжения, возникающие вследствие фазовых превращений [4].
Таким образом, следует, что особо чувствительными к межкри-
сталлитной коррозии должны быть нержавеющие стали с по-
вышенным содержанием углерода, и такие стали необходимо
тщательно проверять на склонность к этому виду разру-
шения.
На основании теории обеднения границ зерен в результате
выпадения карбидов считали, что нержавеющие стали с инзким
содержанием углерода не могут быть подвержены межкристал-
литной коррозии. Однако за последнее время были получены
данные, указывающие на то, что стали с низким содержанием
углерода (С <0,05%) в определенных условиях подвергаются
межкристаллитной коррозии. Поэтому целесообразность борьбы
с межкристаллитной коррозией нержавеющих сталей лишь по-
средством выплавки сталей с весьма низким содержанием угле-
рода (0,02%) в настоящее время еще не ясна, тем более что
получать такие стали в производственных условиях весьма
трудно.
Следовательно, стали с очень низким содержанием углерода
также подлежат испытаниям на склонность к межкристаллитной
коррозии. Эффективным методом борьбы с межкристаллитной,
коррозией оказалось легирование стали элементами, обладаю-
щими гораздо большим’ сродством к углероду, чем хром (Ti,
Nb, Та). Дело в том, что эти элементы как сильные карбидооб-
разователи дают устойчивые карбиды при более высоких темпе-
ратурах (1100—1200°С), чем хром. Поэтому хром практически
не участвует в процессе карбидообразования и не происходит
обеднения прилежащих зон в местах выделения карбидов. Кар-
биды титана или ниобия, по. мнению Г. В. Акимова [1], кроме
того, устойчивы и не переходят в твердый раствор при закалке
стали даже.с очень высоких температур (1100—1200° С).
Все вместе это обеспечивает возможность получения струк-
туры, которая в электрохимическом отношении более благо-
приятна. Вместо трехэлектродной системы, в которой единст-
венным анодом является узкая зона металла, обедненного хро-
мом вблизи выпавшего карбида, возникает двухэлектродная
система: I — Fe/Cr (>10% Сг) и II—карбид (Ti, Nb, Та).
В такой системе карбиды уже не могут быть эффективными ка-
тодами, поскольку в них нет хрома или его содержание очень,
мало. Кроме того, если бы потенциал карбидов титана или хро-
ма н оказался более положительным, чем потенциал твердого
раствора Fe/Cr, эти катоды, по мнению Г. В. Акимова, не могли
бы представить опасности, так как они распределены равномер-.
но среди зерен твердого раствора, =
16* — 243 —
Концентрация стабилизирующего элемента зависит от содер-
жания углерода; обычно считают, что она должна в 4—10 раз
превысить концентрацию углерода; предельная концентрация
углерода в этих условиях значения уже не имеет.
По мнению Ю. Р. Эванса {5j, это справедливо для большин-
ства коррозионных сред. Однако наблюдались случаи (при
10 %-ной концентрации H2SO4), когда повышенное содержание
углерода (0,1%) приводило к усилению коррозии; отсюда сле-
дует вывод, что даже при введении титана и ниобия концентра-
ция углерода имеет значение. Поэтому повышение содержания
углерода может быть одной из причин появления склонности к
межкристаллитной коррозии у стабилизированных сталей, что
необходимо учитывать при ускоренных испытаниях.
Теоретически отношение содержания титана в сплаве к со-
держанию углерода должно равняться 4, что соответствует об-
разованию из всего титана карбида TiC с учетом некоторого
количества титана, израсходованного на соединение с азотом.
В СССР содержание титана в нержавеющих сталях регла-
ментируется следующим отношением: % Т1 (% С — 0,03%) X
X 5. При стабилизации ниобием %(Nb=%C*8. Концентрация
ииобия в стали не должна быть более 1,0%. При более высоких
концентрациях ниобия пластичность стали также снижается и
она становится склонной к трещинообразованию. Поэтому кон-
центрация углерода в сталях не должна превышать
0,08—0,1%.
Учитывая изложенное выше, необходимо, обнаружив при
ускоренных испытаниях склонность стали к межкристаллитной
коррозии, в первую очередь проверить, соблюдается ли необхо-
димое соотношение компонентов.
Наличие температурного интервала, в котором нержавею-
щие стали становятся склонными к межкристаллитной корро-
зии, делает особенно чувствительными к этому виду коррозии
сварные конструкции. Дело в том, что в процессе сварки различ-
ные зоны претерпевают неодинаковый нагрев, и на некотором
удалении от сварного шва металл может подвергаться непреду-
смотренной термической обработке как раз в интервале опасных
температур.
Обычно склонность к межкристаллитной коррозии возникает
у нержавеющих сталей типа 18-8 в результате кратковременно;
го нагрева до 700° С или при длительном нагреве при более вы-
соких температурах. Судить о появлении склонности нержавею-
щей стали к межкристаллитной коррозии можно на основании
кривых, характеризующих зависимость межкристаллитной кор-
розии от времени и температуры нагрева (рис. 141).
Изучая кривые распределения температуры в зоне сварного
шва, можно выбрать режим сварки, который обеспечит такое
время воздействия температуры на металл, которое не будет
вызывать появления склонности к межкристаллитной коррозии.
— 244 —
Подробно вопросы, относящиеся к межкристаллитной коррозии
сварных соединений, рассматриваются в монографии Б. И. Ме-
довара [3].
При не соблюдении нужного режима сварки, а также вслед|
ствие выгорания стабилизирующих элементов (Ti, Nb) из свар<
ной проволоки иногда наблюдаются случаи, когда сварные кон|
струкции из стабилизированной стали приобретают склонности
к межкристаллитной коррозии.
Поэтому ускоренные испытания должны включать проверку
не только исходной стали, но и сварных соединений и самой ап-
паратуры. Иногда достаточно
ограничиться испытаниями
сварных образцов-свидетелей,
которые должны быть сварены
одним и тем же сварщиком и в
том же режиме. Учитывая, что
в реальной конструкции усло-
вия для возникновения внут-
ренних напряжений иные, чем
на образцах-свидетелях, реко-
мендуется для особо ответст-
венной аппаратуры испытывать
специально изготовленные об-
разцы в виде коробочек или
миниатюрных аппаратов, ими-
тирующих расположение свар-
ных соединений в реальной
конструкции. Такие аппараты
помещают в электролит, при-
Рис. 141. Зависимость между време-
нем и температурой, необходимой для
появления склонности нержавеющей
стали к межкристаллитной корро-
зии {6]
меняемый для ускоренных испытаний, и после соответствующей
выдержки при заданной температуре проверяют на наличие меж-
кристаллитной коррозии.
Учитывая температурную зависимость склонности нержаве-
ющих сталей к межкристаллитной коррозии, считают возмож-
ным избежать этого опасного вида коррозионного разрушения,
не только применяя стали с низким содержанием углерода или
посредством введения стабилизаторов (Ti, bib, Та), но и путем
повторного нагрева сварной конструкции выше 1100° С с после-
дующим быстрым охлаждением для перевода всех карбидов в
твердый раствор.
Этот метод наиболее эффективен для малогабаритной аппа-
ратуры, в этом случае можно применять стали без стабилизи-
рующих элементов. Кроме закалки, возможен стабилизи-
рующий отжиг, приводящий к выравниванию концентрации
хрома и коагуляции выпадающих карбидов непосредственно
внутри зерна. Стабилизирующий отжиг при 800—900° С в
течение длительного времени ликвидирует склонность к межкри-
сталлитной коррозии, возникшую в результате нагрева при
— 245 —
1
600—700° С. Однако по мнению Ю. Р. Эванса [5], такая обра-
ботка может увеличить склонность к межкристаллитной корро-
зии, которая обусловлена нагревом при 1300° С, если только со-
отношение между титаном и углеродом не повысить до семи.
Несмотря на то что закалка и стабилизирующий отжиг явля-
ются эффективными средствами борьбы с межкристаллитной
коррозией, их применение для крупногабаритных изделий за-
труднено в связи с отсутствием больших термических печей.
Исходя из электрохимической теории межкристаллитной
коррозии нержавеющих сталей, представляется возможным
обосновать ускоренные методы коррозионных испытаний. Если
коррозия обусловлена электрохимической неоднородностью по-
верхности, то любой реактив, пригодный для быстрого опреде-
ления коррозии, должен действовать на границы зерен, обед-
ненные хромом, ответственные за межкристаллитную коррозию,
оставляя в пассивном состоянии сами зерна. Если это условие
не будет соблюдаться, то начнут корродировать зерна и меж-
кристаллитная коррозия перейдет в общую.
В начальный период применения нержавеющих сталей элек-
трохимическая теория коррозии вообще и межкристаллитной в
частности еще не была так хорошо разработана, как в настоя-
щее время, тем не менее эмпирическим путем был подобран ряд
электролитов, хорошо выявляющих склонность нержавеющих
сталей к межкристаллитной коррозии. Наиболее старый из
них — смесь серной кислоты и сернокислой меди, предложенная
Гатфильдом. Этот реактив действует на обедненную хромом
зону вдоль границ и не действует на зерно.
Металл, подвергшийся воздействию этого электролита при
наличии межкристаллитной коррозии, теряет в результате раз-
общения зерен металлический звук и электрическую проводи-
мость. За последнее время этот метод был усовершенствован
введением в раствор медиой стружки, что позволило сократить
срок испытания.
В США нашел широкое применение метод испытания в горя-
чей азотной кислоте с добавками фторидов и без них. Этим ме-
тодом выявляется в основном склонность к межкристаллитной
коррозии, обусловленная выделением сигма-фазы. Использова-
ние его для определения'склонности нержавеющих сталей к
межкристаллитной коррозии в любых средах химической про-
мышленности часто приводило к неправильному выбору мате-
риала. Это объясняется тем, что продукты взаимодействия.азот-
ной кислоты с нержавеющими сталями, например хромовая кис-
лота, ускоряют процесс коррозии. Наблюдались случаи, когда
стали, не выдержавшие испытания в горячей азотной кислоте,
успешно эксплуатировались в химических производствах. Ис-
пытания в серной кислоте с сернокислой медью давали в этих
случаях более согласующиеся результаты. Испытания в горячей
азотной кислоте, очевидно, более приемлемы для сталей, пред-
— 246 —
назначенных для эксплуатации в этой же кислоте, чем в других
средах. Более подробно этот вид испытания рассматривается в
работе Шерли и Трумена [6].
За последнее время для определения склонности, нержавею-
щих сталей к межкристаллитной коррозии был предложен кис-
лый раствор сульфата трехвалентног-о железа Fe2 (864)3. Этот
метод обнаруживает склонность к межкристаллитной коррозии,
возникающую в результате выпадения карбидов хрома. Он
•очень чувствителен, ибо процесс коррозии довольно быстро раз-
вивается по границам зерен, вызывая сильное изменение элект-
росопротивления и механических свойств. Метод в отличие 01
азотнокислого не приводит к устранению коррозии благодаря
появлению продуктов реакции, поэтому в одном сосуде можно
испытывать несколько образцов.
Ввиду того что методы испытания в серной кислоте с серно-
кислой медью, а также в азотной кислоте требуют длительного
времени, были предприняты меры к разработке электрохимиче-
ских методов, которые позволяли бы быстро определить склон-
ность нержавеющих сталей к межкристаллитной коррозии.
В связи с этим представляет интерес предложенный
Штрейхером [7]i метод анодного травления стали в 0,1-н. раство-
ре щавелевой кислоты, позволяющий выявить склонность к
межкристаллитной коррозии за 10—20 мин. Этот метод, по
мнению автора, пригоден для предварительной оценки склонно-
сти к межкристаллитной коррозии, в большей степени обуслов-
ленной выделением карбидов, чем сигма-фазы.
Рассмотрим сначала основные научные принципы проведе-
ния этих ускоренных испытаний, позволяющие выявить роль от-
дельных компонентов электролитов и более определенно выби-
рать условия испытания тех или иных сталей.
Интересные работы в этом направлении были выполнены в
последнее время И. А. Левиным совместно с И. Г. Великовой
18]. Исходя из электрохимического механизма межкристаллит-
ной коррозии, изложенного выше, авторы полагают, что одно-
фазную нержавеющую сталь, в которой появилась склонность
к межкристаллитной коррозии, можно рассматривать как двух-
электродную систему, в которой основная часть зерен практиче-
ски не корродирует, а пограничная часть зерна принимает на
себя подавляющую часть анодного процесса.
Если построить анодны'е потенциостатические кривые для
таких двух электродов, то получится диаграмма, изображенная
на рис. 142. Кривая. / относится к телу зерна. Ее, по мнению
авторов, можно отождествлять с анодной поляризационной кри-
вой, полученной на стали, ие склонной к межкристаллитной кор-
розии. Кривая II представляет собой анодную кривую для гра-
ниц зерен.
Из этой диаграммы видно, что существует определенная об-
ласть потенциалов ф4—<р-е, в которой тело зерна находится в
— 247™
пассивном состоянии, а границы (пограничные зоны) — в ак-
тивном. В этой области межкристаллитный характер коррозии
проявляется особенно отчетливо, так как само тело зерна анод-
но растворяется очень слабо.
Если стационарный потенциал нержавеющей стали находит-
ся между ф1 и ф2, то границы зерен поляризуются катодно, а те-
ло зерна находится в активном состоянии и корродирует.
В интервале потенциалов фз— ф4 и тело зерна и его границы
находятся в активном состоянии, следовательно, коррозия не
носит межкристаллитного харак-
l2 h h
Рис. 142. Схематическое изображе-
ние анодных кривых для основной
части зерна (/) и границ зерен
(Я) .[8] однофазной нержавеющей
стали
тера. В области потенциалов
Фз — Ф4 плотность тока на грани-
це зерна больше, чем на теле зер-
на. В этих условиях будет наблю-
даться межкристаллитная корро-
зия, однако, поскольку тело зерна
не находится в пассивном состоя-
нии, может иметь место и интен-
сивная общая коррозия зерен.
Наибольшая скорость межкрис-
таллитной коррозии должна на-
блюдаться при потенциале В
интервале потенциалов фв—фу
характер коррозии также должен
быть межкристаллитный. В обла-
сти перепассивации ф > фу, по
мнению авторов, также есть усло-
вия, при которых скорость корро-
зии границ зерна больше скорости коррозии самих зерен, но в
этих условиях коррозия самого тела зерна также значительна.
Отсюда следует, что, задавая нержавеющей стали опреде-
ленный потенциал, можно вызвать общую и межкристаллитную
коррозию.
Поскольку величина стационарного потенциала для двух-
электродной системы определяется пересечением суммарных по-
ляризационных кривых для анодной и катодной реакции, мож-
но соответствующим подбором концентраций активаторов к
окислителей (деполяризаторов) сместить стационарный потен-
циал до таких значений, при которых будет наблюдаться пре-
имущественное растворение границ зерен. Условия, обеспечи-
вающие выявление межкристаллитной коррозии, авторы опреде-
ляют из диаграммы, представленной на рис. 143, где схематиче-
ски изображена суммарная анодная кривая / для стали, склон-
ной к межкристаллитной коррозии, и анодная кривая I/ для
зереи, аналогичная анодной кривой для стали, не склонной к
межкристаллитной коррозии.
При малой концентрации деполяризатора (катодная кри-
вая /) суммарная анодная кривая пересекается с катодной при
— 248 —
потенциале, соответствующем преимущественной коррозии зе-
рен. Поэтому, в рассматриваемых условиях должна наблюдаться
значительная общая коррозия стали. Межкристаллитная кор-
розия отсутствует. При большой концентрации деполяризатора
(катодная кривая 2) пересечение происходит в пассивной обла-
сти и для большого числа случаев практически никакой меж-
кристаллитной коррозии наблюдаться не будет; при концентра-
ции деполяризатора, которой соответствует катодная кривая 3„
сталь, не склонная к межкристаллитной коррозии, будет нахо-
диться в пассивном состоянии, а склонная к межкристаллитной
коррозии будет интенсивно растворяться. Если окислительно-
Рис. 143. Поляризационная диаграмма,
характеризующая склонность нержавею-
щей стали к межкристаллитной корро-
зии {8]
восстановительный потен-
циал раствора сдвинуть в
область более отрица-
тельных потенциалов, на-
пример к потенциалу <р3,
то у стали, склонной к
межкристаллитной корро-
зии, с увеличением кон-
центрации катодного де-
поляризатора будет на-
блюдаться переход от об-
щей коррозии (кривая 4)
к межкристаллитной
(кривая 5).
Так как наибольшая
скорость коррозии будет
наблюдаться при пересе-
чении суммарных анодных ж .
тенциалов ф4 — <ре, то растворы для ускоренных испытаний сле-
дует выбирать так, чтобы катодная поляризационная кривая пе-
ресекала анодную кривую в области пассивного зерна и актив-
ных границ. Выдвинутые выше положения проверяли на нержа-
веющей стали типа 18-8 в растворах серной кислоты с добавка-
ми таких деполяризаторов, как сернокислое окисное железо и
медный купорос.
катодных кривых в интервале по-
и
Анодные кривые снимали на закаленной с температуры
1100° С (выдержка 60 мин) и отпущенной (650° С, 4 ч) стали.
Путем сравнения этих кривых была определена область потен-
циалов, при которых в отпущенной стали должно наблюдаться
преимущественное растворение границ верен. Было установле-
но, что на отпущенной стали преимущественное растворение
границ зерен происходит при потенциалах от —0,2 до 4-0,2 в.
Наиболее интенсивно протекает процесс при потенциалах от
—0,1 до 4-0,1 в. При более положительных потенциалах даже
длительная выдержка не приводит к повышенной травимости
границ. На закаленной стали во всем диапазоне потенциалов
наблюдается равномерное растворение.
— 249 —
Область потенциалов, в которой наблюдается межкристал-
литная коррозия (если сталь склонна к ней), -может быть до-
стигнута не только посредством анодной поляризации, но и вве-
дением в серную кислоту окислителей, которые играют роль ка-
тодных де1юляризаторов. Концентрация деполяризатора при
этом зависит от скорости его восстановления, т. е. от хода поля-
ризационной кривой. Поэтому необходимо изучить катодное
восстановление деполяризаторов.
Рис. 144. Анодные я катодные поляризационные кривые,
снятые на стали 1Х18Н9 в 15 %-ной серной кислоте [8].
Анодные кривые:
1 — для закаленной стали, 2 —- для отпущенной стали.
Катодные кривые:
3 — 15%-ная HaSO* + 8 г/дм3 FeHSOOs -9HSO + 1,0 г/дм3
FeSO*; 4 — 15%-ная HaSO4 F 23 г/дм3 Fea(SO«)s • 9НгО +
F 1.0 г/дм3 FeSO4; б — 15%-ная H?SO4 + 35 г/дм3 Fe2(SO4)3'
• 9HjO + ,1,0 г/дм3 FeSO<; 6 — 15%-яая ILSO, + 62 г/дм3
Fe2(SO4)s • 9Н2О + .1,0 г/дм3 FeSO4
Катодные кривые снимают на закаленной стали, т. е. на
сплаве, не склонном к межкристаллитной коррозии. По пересе-
чению катодных поляризационных кривых с анодными кривыми
можно судить о характере и скорости коррозии нержавеющей
стали (рис. 144).
Полученная поляризационная диаграмма дает основание ут-
верждать, что при малом содержании сернокислого железа
(8 г/дм3) и закаленная, и отпущенная стали должны находиться
в активном состоянии. При среднем содержании сернокислого
железа (23—25 г/дм3) закаленная сталь должна находиться в
пассивном состоянии, а отпущенная — в активном. При высоком
содержании деполяризатора {62 г/дм3 Ре(8О4)з-9Н2О] обе стали
должны быть пассивными. Для того чтобы в растворах серной
кислоты и сернокислого железа скорость растворения границ бы-
— 250 —
ла на много больше скорости растворения сердцевины зерна,
анодные кривые должны пересекаться с катодными в интервале
потенциалов (—0,1) 4- (+0,1 в).
Непосредственные коррозионные испытания активированных
образцов закаленной и отпущенной стали при различном содер-
жании окислителей, проводившиеся в течение 20 ч, подтвердили
предположения, вытекающие из электрохимических измерений.
При изучении влияния сернокислой меди было установлено,
что для достижения потенциалов, при которых наблюдается
преимущественное растворение границ зерен на отпущенной ста-
ли [(+0,1) +- (—0,1 в], концентрация сернокислой меди в при-
сутствии медной стружки не должна быть столь высокой, как это
предусмотрено ГОСТ 6032—58 по методу AM (160 а/л).
При этой концентрации деполяризатора анодные и катодные
кривые пересекаются при потенциале +0,2 в и отпущенная сталь
корродирует с небольшой скоростью. Требуется значительное
время, пока в растворе накопятся продукты коррозии, которые
сместят потенциал в отрицательную сторону до +0,1 в, при ко-
тором уже наблюдается усиленная межкристаллитная коррозия.
При введении в сернокислый раствор медного купороса мед-
ной стружки окислительно-восстановительный потенциал рас-
твора сразу принимает значение, равное +0,1 в, и межкристал-
литная коррозия с самого начала протекает с большой ско-
ростью, что позволяет сократить сроки испытаний. Разрушения,
наблюдавшиеся в кислом растворе сернокислой меди за 96 ч, в
присутствии медной стружки были достигнуты за 12 ч.
Роль медной стружки, по мнению И. А. Левина [8], не огра-
ничивается лишь сдвигом окислительно-восстановительного по-
тенциала. Медь в контакте с нержавеющей сталью обладает
более положительным потенциалом и, следовательно, в рассмат-
риваемой системе является катодом. Поскольку ее поверхность
намного больше поверхности испытываемого образца, катодный
ток резко возрастает, и концентрация деполяризатора может
быть заметно снижена.
Кроме того, следует учесть, что если сталь в малой степени
склонна к межкристаллитной коррозии, то без медной стружки
ее можно не обнаружить даже за длительное время, поскольку
пересечение анодной и катодной поляризационных кривых проис-
ходит при потенциалах, более положительных (+0,3 в), чем те
значения, при которых наблюдается интенсивная межкристал-
литная коррозия.
Поэтому испытания сталей типа 18-8 при отсутствии медной
стружки малоэффективны, ибо они требуют длительного времени
и в ряде случаев и не выявляют межкристаллитной коррозии.
На основании электрохимических исследований можно ре-
шить вопрос о пригодности сернокислых растворов сульфата
меди с медной стружкой для ускоренных испытаний однофазных
сталей, отличных от стали типа 18-8. Для этого, по мнению ав-
— 251 —
И
тора, достаточно снять анодные поляризационные кривые в
15 %-ной H2SO4 при температурах, близких к температуре кипе-
ния, на стали, склонной и не склонной к межкристаллитной
коррозии.
Если фладе-потенциал (потенциал полной пассивации) для
стали, не склонной к межкристаллитной коррозии, лежит отри-
цательнее +0,1 в, а для склонной —положительнее этой величи-
ны и предельный ток диффузии сернокислого раствора медного
купороса выше критического тока пассивации для стали,
склонной к межкристаллитной коррозии, то этот раствор можно
применять для ускоренных испытаний на межкристаллитную
коррозию.
Если же образец находится в контакте с поверхностью мед-
ной стружки, то межкристаллитная коррозия будет протекать
при таких концентрациях окислителя, предельный ток диффу-
зии которых меньше критического тока пассивации стали.
Руководствуясь этим принципом, авторы {8] установили, что
растворы, применяемые для выявления межкристаллитной кор-
розии на сталях 18-8, могут быть рекомендованы и для феррит-
ных хромистых сталей XI7T и Х25Т.
На основе электрохимических исследований были сформули-
рованы общие принципы подбора растворов для ускоренных ис-
пытаний нержавеющих сталей на склонность к межкристаллит-
ной коррозии, которые сводятся к следующему: основа раствора
(его состав, концентрация и температура), определяющая анод-
ную поляризацию стали, должна обеспечить определенную об-
ласть потенциалов, при которой сердцевина зерна находится в
пассивном, а границы — в активном состоянии, причем скорость
коррозии границ в этой области потенциалов высока. Эта об-
ласть потенциалов легко определяется сравнением анодных поля-
ризационных кривых для сталей в состоянии склонности к меж-
кристаллитной коррозии и в состоянии стойкости против нее.
Катодный деполяризатор должен обеспечивать -силу тока,
превышающую критический ток пассивации стали, не склонной
к межкристаллитной коррозии. Ход катодной кривой должен
быть таким, чтобы ее пересечение с анодной кривой происходило
в указанной выше области потенциалов. Целесообразно, чтобы
катодное восстановление деполяризатора протекало с малой
поляризацией.
Методы испытания нержавеющих сталей
для выявления склонности к межкристаллитной коррозии
Стандартный метод испытания А нержавеющих сталей по
ГОСТ 6032—58—кипячение в растворе, содержащем 110 г!дм3
CuSOvSHsO + 55 см3 H2SO4 (плотностью 1,84), в колбах с об-
ратными холодильниками в течение 24 ч. Этот метод применяют
для сталей Х13Н4Г9, Х14Г14НЗТ, Х22Н6Т, 2Х18Н9, Х20Н14С2,
-252--
ЭИ694, ЭИ695 и их сварных соединений. Образцы укладывают в
колбы на стеклянные палочки, бусы или стеклянную вату; раз-
решается укладка в несколько рядов с промежуточным слоем
бус. На один образец размером 20 X 80 мм требуется прибли-
зительно 100 см3 раствора.
Для сталей типа 1Х18Н9Т, Х22Н6Т, ЭИ694 и ЭИ695 время
испытания увеличивают не менее чем до 78 ч и оговаривают спе-
циальными техническими условиями.
Чтобы ускорить испытание, в кипящий раствор медного ку-
пороса и серной кислоты вводят медную стружку, механизм
действия которой был рассмотрен выше.
Исходя из этого, метод AM по ГОСТ 6032—58 предусматри-
вает для сталей марок 0Х18Н9, 1Х18Н9, Х23Н18, 0Х18Н9Т, 1X18,
1Х18Н11Б, Х18Н12М2Т, Х18Н12МЗТ и их сварных соединений
кипячение в растворе 160 г]дм3 CuSO^SHgO + 100 см3 H2SO4
(плотностью 1,84) с медной стружкой, которую помещают на
дно сосуда. Продолжительность испытания 24 ч.
Для сталей марок 0Х23Н28МЗДЗТ, Х23Н28МЗДЗТ,
Х23Н27М2Т и их сварных соединений медную стружку заменяют
5 г!дм3 цинковой пыли (метод В, ГОСТ 6032—58). Длительность
испытания 144 ч.
В США [2] стандартным методом испытания для выявления
склонности нержавеющих сталей к межкристаллитной коррозии
является кипячение в 65 %-ной азотной кислоте. Потери массы
за 240 ч являются показателем коррозии. У сталей, склонных к
межкристаллитной коррозии, потери массы выше, чем у сталей,
не склонных к этому виду разрушения. Слабую склонность к
межкристаллитной коррозии этим методом выявить не удается.
В СССР на предприятиях металлургических и машинострои-
тельных заводов для контроля правильности технологии сварки
и других технологических операций изделий из кислотоупорных,
жаропрочных и нержавеющих сталей аустенитного и аустенит-
но-ферритного класса марок Х23Н27М2Т, 0Х23Н28МЗДЗТ так-
же проводят испытание в 10%-ном растворе (по массе) HNOs +
+ 2% HF при 80°С (метод Г, ГОСТ 6032—58). Фтористый нат-
рий вводят в нагретый раствор, в котором уже находятся образ-
цы. Испытание проводят в два цикла по одному часу каждый, а
для сталей марок Х23Н27М2Т, Х23Н23МЗДЗТ, 0Х23Н28МЗДЗТ
и их сварных соединений — в три цикла со сменой раствора по-
сле каждого цикла.
Для проверки готовых изделий и полуфабрикатов из хро-
моникелевых сталей марок 0Х18Н9, 0Х18Н9Т, 1Х18Н9, 2X18Н9
и IX18H9T применяют ускоренный метод испытания, называю-
щийся методом анодного травления (метод Б, ГОСТ 6032—58).
Метод заключается в анодном травлении контролируемых участ'
ков поверхности детали, включенной в цепь постоянного тока
при напряжении 5—9 в и плотности тока 0,65 а!см2. Схема уста-
новки для определения межкристаллитной коррозии сталей ме~
— 253 —
1
Рис. 145. Схема установки
для определения межкрис-
таллитной коррозии сталей
методом анодного травле-
ния (ГОСТ 6032—58):
Z — свинцовый сосуд; 2 — ре-
зиновая манжета; 3 — обра-
зец; 4 —источник постоянного
тока; 5 — амперметр с ценой
деления не более ОД а- 6 —
реостат или магазин сопротив-
ления; 7 — переключатель
или ключ
тодом анодного травления приведена на рис. 145. Катодом слу-
жит свинцовый сосуд, укрепленный на поверхности испытывае-
мого изделия с помощью резиновой манжеты. Температура
испытания 20 ± 10° С. Продолжительность испытания после
включения тока 5 мин-, при повышенной травимости продолжи-
тельность испытания сокращают до 1—2 мин. В свинцовый
сосуд наливают 3—5 смг 60%-ного раствора серной кислоты и
0,5% уротропина или другого ингибитора для серной кислоты.
Детали, предназначенные для работы в азотной кислоте кон-
центрацией до 65% при температурах от 60° С до кипения, ис-
6 7 пытывают при кипячении в 65%-ном
—v9wv—________ растворе азотной кислоты (метод Д).
Склонность к межкристаллитной кор-
розии определяют по потере массы за
48-ч цикл; продолжительность испыта-
ния составляет 3 цикла. Считают, что
образцы не выдержали испытания, ес-
ли скорость коррозии стали после лю-
бого цикла превышает 1,5 г/(м2-ч)
или если после металлографического-
исследования отдельных изогнутых
образцов наблюдается разрушение
границ зерен металла даже на один
сЯой кристаллов.
Кроме описанных стандартных ис-
пытаний, существуют и другие качест-
венные методы испытаний, -позволяю-
щие установить наличие склонности
материала к межкристаллитной корро-
зии. Эти методы отличаются от стан-
дартных лишь составом применяющихся электролитов и режи-
мом испытания.
Анодное травление, как уже указывалось, можно произво-
дить в растворах 10%-ной щавелевой кислоты {10] или 65%-ной
фосфорной кислоты ,{11]. Плотность тока при травлении в этих
растворах следует подбирать таким образом, чтобы тело зерна
находилось в пассивном состоянии или растворялось с малой
скоростью, а границы зерен — в активной. Для щавелевой кис-
лоты, по данным работы (10], плотность тока должна быть рав-
ной 1 а!см2-, при этом необходимо следить, чтобы образец не
перегревался.
Методы оценки склонности к межкристаллитной коррозии
нержавеющих сталей после испыгпаний
В описанных выше стандартных методах наличие межкри-
сталлитной коррозии в испытанных образцах после воздействия
электролита определяют качественно ло потере образцами ме-
— 254 —
таллического звука при ударе о металлический предмет, по по-
явлению трещин на поверхности образцов при загибе их на 90°.
Если загиб невозможен, то проводят металлографическое ис-
следование на шлифах при увеличении в 300—400 раз.
Сталь можно считать склонной к межкристаллитной корро-
зии, если под микроскопом будут видны разрушенные границы
зерен.
И. Л. Розенфельд с сотрудниками [12] предложил металло-
графический метод оценки наличия или отсутствия межкристал-
литной коррозии на готовой аппаратуре. Для этой цели на сом-
нительных участках аппаратуры сошлифовывают лыску длиной
10—25 и глубиной 0,5—1,0 мм, которая затем просматривается
в лупу при увеличении в 75—150 раз. При наличии межкристал-
литной коррозии, помимо продольных борозд от шлифовки, вид-
ны поперечные, пересекающие эти борозды, извилистые линии,
являющиеся разрушенными границами зерен.
Существует и ряд других, принципиально отличающихся друг
от друга методов оценки наличия склонности сплавов к межкри-
сталлитной коррозии, основанных на определении изменения
физических свойств вследствие коррозии.
’ К таким методам прежде всего относится оценка степени
склонности к межкристаллитной коррозии по изменению элект-
росопротивления сплава [13]. Измерять сопротивление сплава
можно на установках, описанных в гл. III.
Изменение электросопротивления пересчитывают на показа-
тель степени поражения поперечного сечеиия образца т по
уравнению
т = = -^=.^....., (106)
5 Р(Р2 — Р1)
где 5— общая площадь поперечного сечения образца;
S2 — площадь поперечного сечения образца, пораженная
коррозией:
р —удельное электрическое сопротивление образца после
коррозионного испытания;
pi — удельное электрическое сопротивление стального об-
разца до коррозии;
р2 — удельное электрическое сопротивление прокорроди-
ровавшей части стального образца.
Обычно принимают с некоторым допущением:
т = - р~рг — JLzA. t /j07)
Р R 4 1
где 7? — электрическое сопротивление образца после коррозии;
jRi — электрическое сопротивление образца до испытания.
— 255 —
Зная размеры образца, можно определить глубину проник-
новения коррозии. Для проволочного образца ее определяют по
уравнению
б = |(1-УТ^, (Ю8)
где D — диаметр проволоки.
Для образца -прямоугольного сечения уравнение принимает
вид
_ (а + 6) — Vfc + Ъ^ — ^аЬт
где а и b — размеры поперечного сечения образца.
К числу методов определения межкристаллитной коррозии по
изменению электрических характеристик сплава относится так
называемый метод высокочастотного электрического резонанса
[14]. Межкристаллитную коррозию определяют путем соприкос-
новения испытываемого образца с катушкой самоиндукции из-
мерительного контура таким образом, чтобы он пронизывался
высокочастотным магнитным полем, которое -вызывает в метал-
ле вихревые токи. Эти токи создают свое магнитное поле с об-
ратным знаком, уменьшающее самоиндукцию контура и нару-
шающее электрический резонанс. Электрические колебания
в контуре, срываясь с резонансного контура вниз, уменьшаются
по амплитуде. Изменения в амплитуде колебаний характеризу-
ют склонность материала к межкристаллитной коррозии; изме-
нения тем больше, чем больше склонность к этому виду разру-
шения.
М. А. Веденеева и Н. Д. Томашов [15] применили -новый
чувствительный метод определения межкристаллитного разру-
шения путем измерения внутреннего трения -и частоты собствен-
ных колебаний образца. Проведенные ими исследования пока-
зали, что изменение частоты собственных колебаний близко
к изменению электросопротивления, а увеличение внутреннего
трения в несколько раз превышает изменение этих характери-
стик под влиянием -межкристаллитной коррозии. Поэтому метод
измерения внутреннего трения можно отиести к чувствительным
методам оценки межкристаллитного разрушения.
Измерение внутреннего трения образца можно производить
двумя методами: снятием -резонансной кривой при возбуждении
в образцах поперечных колебаний и методом затухания свобод-
ных крутильных колебаний. Первым_методом измеряют внутрен-
нее трение на установке сконструированной в специальной лабо-
ратории Московского института стали и сплавов J16] путем оп-
ределения амплитуды колебаний при резонансной и близкой к
ней частотах
— 256 —
Из значений резонансных частот до и после коррозии рассчи-
тывают изменение модуля упругости Е:
гдеА^2=т2— т2 ;
v — резонансная частота колебаний образца до коррозии;
vi — резонансная частота колебаний прокорродировавшего
образца.
При определении внутреннего трения по методу затухания
свободных колебаний используют проволочные образцы длиной
310 мм. Измерения производят на приборе РКФ-МИС [17] при
частоте около 1 гц. Внутреннее трение характеризуется величи-
ной логарифмического декремента затухания. Расчет ведут по
формуле
(Г1 = — , (111)
nN k 7
где п — —;
Ci — начальная амплитуда колебаний:
G.V — амплитуда через 7V колебаний;
N — число колебаний, требуемое для изменения амплиту-
ды в п раз. Отношение— выбирается равным 1,5 н 2,0.
aN
Критерием межкристаллитного разрушения служит измене-
ние измеряемой величины у образцов в результате коррозии,
выраженное 'в процентах по отношению к значениям, измерен-
ным до коррозии. Внутреннее трение, по данным авторов, явля-
ется более чувствительной характеристикой при оценке склонно-
сти стали к межкристаллитной коррозии, чем оценка по измене-
нию электросопротивления.
Для исследовательских работ качественные стандартные и
нестандартные методы испытания сталей на межкристаллитную
коррозию недостаточны. И. А. Левин и С. А. Гинцберг [18] раз-
работали микро-электрохимический ускоренный метод испытания
сплавов, подверженных межкристаллитной коррозии. Он заклю-
чается в снятии поляризационных кривых для зерен и их границ
при различных способах термической обработки. Применение
этого метода требует изоляции границ зерен при снятии кривых
для поверхности зерен.
Как было установлено, для межкристаллитно корродирующей
стали наблюдается очень слабая анодная поляризуемость границ
зерен.
Для исследования, межкристаллитной коррозии применяют и
микроэлектрохимические -методы, с помощью которых можно
измерить потенциалы структурных составляющих и их измене-
17 Зак. 1338 — 257 —
ние, например в различных зонах сварного шва [19]. Микроэлек-
трох-имические методы безусловно должны сопровождаться
металлографическими исследованиями.
Механизм межкристаллитной коррозии алюминиевых сплавов
и принципы выбора электролитов
г для ускоренных испытаний
Алюминиевые сплавы, содержащие медь, «при неправильной
термической обработке становятся склонными к межкристал-
литной коррозии. Этому виду коррозии подвержены иногда и
алюминиевые сплавы, содержащие магний и кремний, однако
скорость процесса коррозии намного меньше, чем алюминиево-
мед ных сплавов.
Межкристаллитная коррозия алюминиевых сплавов появля-
ется в результате неправильной термической обработки, а ино-
гда и при длительном воздействии солнечных лучей во многих
средах — морской воде, морской и промышленной атмосферах.
Т а б л ид а 28
ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ АЛЮМИНИЕВЫХ СПЛАВОВ
И ИХ СТРУКТУРНЫХ СОСТАВЛЯЮЩИХ, в [221
По данным Акимова и Олешке По данным Босхардта По данным Брауна По данным Голубева
Электрод 3%-ный 3%-ный 0.5%-ный раствор 3%-ный
раствор раствор +0,05 %-ный раствор
NaCl NaCl раствор н2о8 NaCl
Алюминий ЧИСТЫЙ . . -0,55 —0,50 —0,59 —0,57
Алюминий с 0,3% Си в твердом растворе . . -0,48
Алюминий с 4% Си в твердом растворе . . . -0,44-0,41 , —0,44-^0,28
СиА12 -0,37 — — —0,36
Алюминий с 4% Си, отожженный - —0,48 -
Теория межкристаллитной коррозии алюминиевых сплавов
была разработана на основе теории многоэлектродных систем
Акимовым и его учениками (1, 20, 23]. Согласно этой теории, при
искусственном старении алюминиевых сплавов или других ви-
дах термического воздействия в процессе технологической обра-
ботки в интервале температур 90—270° С происходит распад
твердого раствора А1 — Си с выделением фазы, по составу близ-
кой к интерметаллическому соединению СиА12. преимуществен-
— 258 —
но по границам зерен. В результате Этого вблизи границ воз-
никает зона, сильно обедненная медью. Концентрация меди
в этой зоне может упасть с 4 до 0,3—0,5%. Внутри зерна выпа-
дение интерметаллических соединений происходит в меньшей
степени, поэтому -в зонах, удаленных от границ, твердый раствор
обедняется медью меньше (рис. 146, 147).
После термического воздействия в указанном .выше интервале
температур -поверхность алюминиевого сплава в электрохимиче-
ском отношении уже !не может рассматриваться как однород-
ная. По измерениям Миерса и Брауна (21], разница в потенци-
алах границ зерен и тела зерна на алюминиевом сплаве, содер-
жащем медь (4%), после искусственного старения -при 150°С
в течение 16 « составляла ~ 100 мв.
По измерениям Г. В. Акимова,
А. И. Голубева, Босхардта и Брау-
на [1, 22], электродные потенциалы
алюминиевых сплавов, твердых рас-
творов на основе алюминия и ин-
Рис. 146. Схематическое изо-
бражение граиииы между зер-
нами сплава AI + 4% Си после
искусственного старения
при большом увеличении
Рис. 147. Схематическое изо-
бражение сплава А1 + 4% Си
после искусственного старения
в виде трехэлектродиой
системы [1]
тер металлических соединений характеризуются данными, приве-
денными в табл. 28.
Из этих данных следует, что между электродными потенци-
алами структурных составляющих алюминиевых сплавов суще-
ствует значительная разница. Поэтому алюминиевомедный
сплав после искусственного старения следует рассматривать,
как трехэлектродную систему [1]:
I п ш
CuA12 — Al/Cu (4 %) — Al/Cu (0,3 %).
Простой анализ показывает, что в такой системе наиболее
отрицательным электродом является зона вблизи границы зер-
на обеднения медью (111). Находясь под воздействием двух
катодов, обладающих более положительным потенциалом [одийо
из них (И) занимает большую часть -поверхности], зоны у гра-
ниц зерен подвергаются усиленному разрушению.
17* — 259 —
Достоверность подобного электрохимического механизма
межкристаллитной коррозии алюминиевых сплавов, содержа-
щих медь, 'подтверждается тем, что на основе этой теории уда-
ется предсказать методы борьбы с этим опасным видом разру-
шения. Если бы удалось создать в системе электрод с более
отрицательным потенциалом, зоны у границ зерен, вероятно,
перестали бы разрушаться. Это можно, например, осуществить,
понизив потенциал тела зерна. Опыты подтвердили, что, если
в такой сплав ввести небольшое количество магния, -склонность
сплава к межкристаллитной коррозии резко снижается. В этом
случае коррозия концентрируется в основном на теле зерен, за-
нимающих основную часть поверхности, и плотность тока у гра-
ниц ничтожна. На аналогичном принципе и основана электро-
химическая защита протекторами или плакирующими слоями,
обладающими более отрицательным потенциалом.
В последнее время была выдвинута иная точка, зрения на
механизм межкристаллитной коррозии алюминиевых сплавов,
содержащих медь [22]. По мнению автора, выпадающая на гра-
нице зерен б'-фаза, близкая по своему составу к интерметалли-
ческому соединению CuAlg, растворяется избирательно, и раз-
рушение границ в основном обусловлено преимущественным
растворением из этой фазы алюминия.
Вопрос о межкристаллитной коррозии алюминиевых спла-
вов до сих пор не имел такого большого значения, как вопрос
о коррозии нержавеющих сталей, поскольку алюминиевомедные
сплавы применяли в основном для клепаных конструкций.
С разработкой свариваемых сплавов этой системы проблема
межкристаллитной коррозии может стать более острой, ибо
в процессе сварки отдельные зоны могут быть подвергнуты
термическому воздействию в интервале температур, вызываю-
щих склонность к межкристаллитной коррозии..
Межкристаллитная коррозия алюминиевомедных сплавов —
чаще всего результат неправильной термической обработки,
в особенности крупногабаритных профилей, которые не удается
равномерно прогреть. Применением соответствующей термиче-
ской обработки, обеспечивающей благоприятное распределение
потенциалов по поверхности, удается, как правило, исключить
межкристаллитную коррозию алюминиевых сплавов.
Правильная термическая обработка должна заключаться
в том, чтобы сделать сплав более гомогенным и перевести воз- •
можно большее количество меди в твердый раствор, который
фиксируется быстрой закалкой. Поскольку полностью исклю-
чить выпадение интерметаллических соединений по границам
зерен нельзя, то необходимо принять меры к тому, чтобы они не
располагались по границам зерен сплошной цепочкой.
Оптимальная коррозионная стойкость дуралюмииа обеспечи-
вается при резкой закалке с 480—500° С в холодную воду
(40° С) и дальнейшем естественном старении. После такой тер-
— 260 —
за-
Рис. 148. Зависимость предела
прочности Сь и удлинения 6
неплакироваиных алюм иниевых
сплавов Д1 и Д16 толщиной
0,6 мм от продолжительности
задержки закалки [20]. Корро-
зия при переменном погруже-
нии в 3%-ный раствор хлори-
стого натрия; длительность ис-
пытания 15 суток
мической обработки сплав не -подвергается в атмосферных усло-
виях межкристаллитной коррозии. Нарушения в термическом
режиме, которые могут возникнуть в процессе закалки и -при
дальнейшей технологической обработке’сплава, могут сообщить
последнему склонность к межкристаллитной коррозии.
Задержка сплава перед закалкой на 'воздухе увеличивает
склонность к межкристаллитной коррозии (рис. 148).
Нормально закаленный сплав Д1, который 'испытывали в ус-
ловиях переменного погружения в 3%-ный раствор NaCl, не
обнаруживал признаков межкристаллитной коррозии, а подвер-
гался лишь 'местной коррозии. По мере увеличения времени
держки перед закалкой (до 10—
20 сек) сплав становился все более
склонным к межкристаллитной кор-
розии.
Указать точно, при какой за-
держке сплав начинает приобретать
склонность к межкристаллитной
коррозии, невозможно, поскольку
этот параметр зависит от содержа-
ния в сплаве меди, толщины листа
или профиля и т. п. Ясно лишь, что
закалку следует производить воз-
можно быстрее. Считают, что если
температура сплава вследствие вы-
держки на воздухе снизилась до
400° С, сплав будет подвержен меж-
кристаллитной коррозии уже неза-
висимо от того, какую бы скорость
охлаждения не применяли.
Другой важный фактор — состав
и температура закалочной ваины.
На рис. 149 приведено влияние за-
калочной среды на сопротивление
коррозии неплакировэнного дур-
алюмина Д16, который закаливался
тественно состаренном состоянии. Наибольшая потеря механиче-
ских свойств, обусловленная межкристаллитной коррозией,
наблюдается у сплава, закаленного на воздухе и в масло. Наи-
меньшая потеря механических свойств была обнаружена у спла-
ва, закаленного в холодную воду, наибольшая — при закалке
на воздухе.
Температура воды играет существенную роль; с ее повыше-
нием 'склонность сплава к межкристаллитной коррозии растет
(рис. 150). Оптимальной считается температура 40° С, посколь-
ку она обеспечивает получение таких же свойств, как при 20° С,
но зато уменьшает коробление тонких профилей вследствие не-
которого уменьшения температурного перепада.
490° С и испытывался в ес-
Весьма чувствительны к появлению межкристаллитной кор-
розии изделия сложной конфигурации, а также профили тол-
стых сечений, -поскольку скорость охлаждения различных
участков при закалке различна.
В этом отношении интересные результаты были получены
С. Е. Павловым [23], который определил изменение механических
свойств после коррозии образцов, взятых из различных мест,
по сечению тяжелого лонжеронного профиля из сплава Д16.
Профиль
70
предварительно подвергали гомогенизирующему на-
греву при 490° С, закаливали в холод-
ную воду и подвергали естественному
старению.
Минимальная потеря прочности по-
коррозии была обнаружена у об-
60
50
20
Рис. 149. Влияние закалочной
среды на сопротивление корро-
зии неплакироваиного дуралю-
мина Д16 толщиной 0,6 мм [20].
Потери Сь после 15 суток пере-
менного погружения в 3%-иый
раствор NaCl. Закалка:
/ — в холодную воду; 2 — в ки-
пящую воду; 3 — в масло; 4 —
на воздухе
£ 30
еле
Рис. 150. Влияние темпера-
туры закалочной воды на
сопротивление коррозии ие-
плакированного дуралюмииа
Д16Т толщиной 1,2 jam [20].
Потери Сь и 5 после 10 су-
ток коррозии в 3%-ном
растворе NaCl + 0,1 % Н2О2
разцов, вырезанных из наиболее тонкой части сечения (полки).
Образцы, взятые из толстой части сечения профиля, обнаружили
значительно большую потерю прочности. Выявилась также раз-
ница в поведении внутренних и внешних слоев металла профиля,
имевшего толстое сечение. Образцы, вырезанные из внутренней
части сечения, потеряли прочность в большей мере, чем образцы,
вырезанные из материала, выходившего на поверхность.
Микроскопическое исследование показало, что образцы из
внутренних слоев металла оказались в сильной степени подвер-
жены межкристаллитной коррозии, а образцы, вырезанные из
поверхностных слоев, коррозии не подверглись.
Такое поведение сложных профилей и толстых сечений обу-
словлено неодинаковой скоростью охлаждения при- за-калке,
приводящей к неравномерному распаду твердого раствора и по-
— 262 —
явлению в местах сечеиияг подвергающихся медленному охлаж-
дению, склонности к межкристаллитной -коррозии.
Последнее необходимо учитывать при выборе режима терми-
ческой обработки- изделий сложных конфигураций.
Следует иметь в виду, что правильная термическая обработ-
ка еще полностью не исключает возможности появления меж-
кристаллитной коррозии, если сплав в дальнейшем подвергается
термическим воздействиям (сварка, горячая штамповка, горя-
чая сушка лакокрасочных покрытий и т. п.) в неблагоприятном
интервале температур.
С. Е. Павлов и С. М. Амбарцумян [23] установили минималь-
ное время, необходимое для появления у сплава Д16 склонности
к межкристаллитной коррозии вследствие нагрева сплава при
различных температурах:
Температура, °C Время, ч s Температура, °C Время
90 300 150 30 мин
95 50 160 20 '»
100 15 170 15 »
110 8 180 10 »
120 2 200 2 »
130 1 240 10 сек
140 1 270 5 »
Как видно, повышение температуры от 90 до 270° С сокра-
щает продолжительность нагрева, достаточную, чтобы вызвать^
чувствительность к межкри-
сталлитной коррозии более
чем в 200 тыс. раз. Такое по-
ведение сплава связано с
ускорением распада твердо-
го раствора по границам
кристаллитов благодаря по-
вышению температуры.
Целесообразным выбо-
ром режима искусственного
старения в сочетании с пред-
варительной деформацией
удается, как было показано
в работе [23], получить ма-
териал, практически не
склонный к межкристаллит-
ной коррозии. На рис. 151 и
152 приведены данные о по-
Рис. 151. Влияние искусственного ста-
рения иа сопротивление коррозии
дур алюмина Д16, не подвергавшегося
предварительной деформации [23]. Ус-
ловия коррозии: 5 суток в 3%-ном
растворе NaCl + 0,1% НаО2
тере механической прочности сплава Д16 в процессе коррозии
в недеформированном состоянии и после предварительной де-
формации на 1%. Оптимальный режим старения, (на рисунках-
отмечен стрелками), обеспечивающий лучшие механические
свойства сплава при наличии деформации, близок к оптималь-
— 2'63 —
1
ному режиму по коррозионной стойкости, если старение произво-
дится при 190° С в течение 12—16 ч.
Оптимальным режимом -по коррозионной стойкости счйтаег-
ся естественное старение закаленного сплава (на рисунках от-
мечено пунктирной линией). В естественно состаренном состоя-
нии сплав Д16 обычно подвергается местной коррозии.
Исходя из электрохимического механизма межкристаллит-
ной коррозии алюминиевых сплавов, выбор электролита для
ускоренных испытании должен
О U в 12 16 20 2Ь 28
Яроволжшпельмость старения//
Рис. 152. Влияние искусственного
старения на сопротивление корро-
зии дуралюмина Д16, предвари-
тельно деформированного на 1%
[23]. Условия коррозии: 5 суток в
3%-ном растворе NaCl+0,1 % Н2Оо
основываться на принципе соз-
дания таких условий, при ко-
торых тело зерна находилось
бы в пассивном состоянии или
растворялось бы с малой ско-
ростью, а границы зерна — в
активном состоянии.
Очевидно, следует приме-
нять электролиты, которые со-
держат активатор и пассива-
тор в такой концентрации, что-
бы катодный ток, возникаю-
щий за счет восстановления
окислителя, был достаточен
для пассивации тела зерна и
недостаточен для пассивации
границ зерен. В качестве акти-
ватора применяют хлористый
натрий, а в качестве деполяри-
затора— перекись водорода и
ионы водорода, являющиеся
в известном смысле также
сильными окислителями. Сме-
щения потенциала сплава в желаемом направлении (пассивация
зерна и активация состояния границ) можно также достигнуть,
посредством анодной поляризации, поэтому некоторые ускорен-
ные методы испытаний основаны на этом принципе. Иногда для
выявления склонности алюминиевых сплавов к межкристаллит-
ной коррозии применяют и периодическое погружение в элек-
тролиты. При этом испытании ускорение катодного процесса
достигается за счет того, что процесс длительно протекает в тон-
ком слое электролита.
Методика проведения испытании
Для ускоренных испытаний алюминиевых сплавов типа дур-’
алюмина на склонность к межкристаллитной коррозии предло-:
жены следующие электролиты [20, 24, 25):
А— 3%-ный раствор ‘NaCl + 0,1 % Н2О2 (полное погруже-
ние), 10 суток;
— 264 т—
0
Б — 3%-ный раствор NaCl + 1,0% HCI (плотностью 1,14),
полное погружение, 24—48 ч\
В — 3%-ный раствор NaCl, переменное погружение, 15—
20 суток (10 мин в электролите, 50 мин на воздухе);
Г— 1-н. раствор NaCl, анодная поляризация, i = 10 ма!см\
20 мин.
Испытания ведут в таких же приборах и аппаратах, которые
списаны выше (см. гл. И).
После испытаний по методу А наличие межкристаллитной
коррозии определяют по-потере прочности и металлографически,
путем просмотра полированных шлифов под микроскопом при
увеличении в 100—150 раз. Травления шлифов при этом не тре-
буется.
При использовании стандартного метода выявления склон-
ности к межкристаллитной коррозии алюминиевых сплавов
в реактиве, содержащем 3% NaCl и 1% НС1 (метод Е), при на-
личии межкристаллитной коррозии поверхность сплава покры-
вается осадком меди, придающим ей красноватый оттенок.
При слабой склонности к межкристаллитному разрушению оса-
док меди может быть плохо заметен, тогда необходим дополни-
тельный металлографический контроль.
Переменное погружение (метод В) также часто применяется
для определения стойкости в межкристаллитной коррозии. Ре-
жим испытаний такой же, как и для общей коррозии. Однако-
эти испытания длительны и требуют 15—20 суток. После испы-
таний изготовляют шлифы, на которых металлографически оп-
ределяется наличие межкристаллитной коррозии. Для испыта-
ний на межкристаллитную коррозию можно также применять,
стандартные образцы для механических испытаний. При нали-
чии межкристаллитной коррозии резко снижаются механиче-
ские свойства материала, в особенности такое свойство как
удлинение.
Чтобы установить наличие межкристаллитного разрушения-
изделий из дур алюмина непосредственно на конструкции,.
С. Е. Павло® {26] предложил метод, заключающийся в приго-
товлении шлифа -непосредственно на поверхности детали. Про-
корродировавшая деталь в выбранном месте полируется пастой»
ГОИ после предварительной обработки шлифовальной бумагой.
При наличии межкристаллитной коррозии иа полированной по-
верхности детали при осмотре под микроскопом выявляется*
сетка разрушенных границ.
Если нужно определить склонность к межкристаллитной-
коррозии деталей или полуфабрикатов, еще не бывших в упо-
треблении, рекомендуется и а поверхность изделия наклеивать,
стаканчик из пластелина. и наливать в него 3%-ный раствор хло-
ристого натрия и 1 % соляной кислоты. После воздействия этого-
электролита -в течение 24 ч раствор удаляется и поверхность ис-
следуется металлографически.
— 265 —
Чтобы обнаружить межкристаллитную коррозию алюминие-
вых сплаве® при глубине поражения не менее 0,1 мм, А. Боро-
виков и С. Попов [27] предложили метод, заключающийся в на-
несении и а поверхность сплава сначала слоя красной -краски,
состоящей из 10—30 г анилинового красителя Судан, 20—30%
бензина и 70—80% осветительного керосина, и затем слоя белой
краски, представляющей собой смеси из 70% белой нитроэмали
ДВ, 20% разжижителя РДВ и 10% цинковых белил. О наличии
очагов межкристаллитного разрушения судят -по появлению
следов красной краски на белом фоне.
За последнее время в связи с разработкой высокопрочных
алюминиевых сплавов сильно возрос интерес к испытаниям по
определению склонности алюминиевых сплавов к расслаиваю-
щей коррозии.
К этому виду коррозии склонны прокатанные листы и прес-
сованные изделия после термической обработки, если она про-
водилась при относительно низкой температуре или при незна-
чительных выдержках. Расслаивающая коррозия, по мнению-
Эванса, представляет собой особый вид межкристаллитной кор-
розии, которая благодаря резко выраженной (направленности
зерен в материале развивается вдоль направления деформации.
Одиако при расслаивании межкристаллитная коррозия наблю-
дается не всегда. Причиной коррозионного расслаивания неко-
торых алюминиевых сплавов могут служить также дефекты,
возникающие в процессе прокатки (плохая приварка частей
расслоившегося на ранних стадиях прокатки слитка вследствие
окисления, ликвация меди). '
При воздействии коррозионной среды на алюминиевые спла-
вы, образующиеся .вдоль плоскостей, параллельных поверхности
сплавов, продукты коррозии, как правило, занимают больший
объем, чем сам металл. Поэтому в месте такой коррозии мате-
риал вспучивается, а продукты коррозии будут стремиться
раздвинуть промежуточные слои, не подвергшиеся коррозии.
Разделение материала иа слои открывает новые пути для дви-
жения электролита и дальнейшего развития коррозионного -про-
цесса.
Коррозионное расслаивание может быть выявлено при про-
ведении обычных усокрениых испытаний в 3%-ном растворе
.NaCl-I-0,1 %-ном растворе Н2О2 при полном погружении и,
дальнейшем исследовании микрошлифов из испытанных об-
разцов.
Время выявления склонности к расслоению зависит от
свойств сплава. Испытания поэтому следует проводить до появ-
ления расслаивания, на что иногда требуется 3 месяца. Элек-
тролит меняют через каждые 2 недели, перекись водорода —
через каждые пять суток. Образцы осматривают вначале че-
рез несколько суток, в дальнейшем производят осмотр через
неделю.
— 266 —
Специальный электрохимический метод быстрого определе-
ния склонности сплава к коррозионному расслаиванию описан
в работе [28]. Метод заключается в .выдерживании исследуемых
образцов в 3%-ном растворе хлористото натрия при строго ^оп-
ределенном потенциале. Таким образом создаются условия
избирательного травления определенных фаз сплава при анод-
ной поляризации.
Авторы рекомендуют следующие режимы ускоренных испы-
таний:
Сплав . .
Потенциал
?Н-К. э-> в
.А1— Си, Al — Mg —Си
. —0,6-г—0,65
Продолжитель-
ность поляри-
зации, ч
Al —Mg ALMg-Zn, Al-Cu-Mg-Zn
-0,60-5- — 0,60-*—0,75
—0,75
•16
8-16
4—16
Травление -при этих режимах позволяет выявить склонность
сплавов к расслаивающей коррозии при наличии межкристал-
литной коррозии и ее отсутствии.
Испытания на межкристаллитную коррозию цииковых спла-
вов с алюминием (от 3 до 5% AI) Бюро стандартов США реко-
мендует проводить в атмосфере водяных паров при 95=ё2оС
в течение 240 ч. Если после этого испытания не наблюдается
изменения удлинения и предела прочности, сплавы считаются
пригодными для эксплуатации. Рекомендуется также опреде-
лять изменение размеров образцов, так как эти сплавы склонны
к росту.
2. ОПРЕДЕЛЕНИЕ СКЛОННОСТИ СПЛАВОВ
К КОРРОЗИОННОМУ РАСТРЕСКИВАНИЮ
Общие положения
Коррозионное растрескивание сплавов возникает при одно-
временном воздействии коррозионной среды и статических ра-
стягивающих напряжений. Напряжения могут быть внешние и
внутренние. Коррозионному растрескиванию под напряжением
подвержены некоторые алюминиевые сплавы, матниевые и мед-
ные сплавы, а также высокопрочные сплавы и нержавеющие
стали.
Склонность сплавов к этому опасному виду коррозионного
разрушения определяется структурой металла, величиной и ха-
рактером напряжений, а также составом коррозионной среды.
Коррозионные среды, вызывающие избирательное разрушение
сплава, способствуют коррозионному растрескиванию его. •
Механизм коррозионного растрескивания изучался многими
исследователями, но до сих пор еще окончательно не выяснен.
Однако большинство исследователей считает, что .в возникнове-
— 267 —
нии и развитии процесса коррозионного растрескивания огром-
ную роль играют электрохимические факторы; возникновение
на поверхности сплава гальванических элементов под влиянием
различных причин приводит к образованию концентраторов на-
пряжения. Концентраторы напряжений в свою очередь способ-
ствуют коррозионному растрескиванию сплавов. Растрескивание
напряженного металла можно условно рассматривать как про-
цесс, состоящий из нескольких стадий: начальной, когда разру-
шение идет только в одной микрообласти, и последующих ста-
дий, во время которых происходит углубление начального микро-
разрушения, -приводящего к мгновенному разрушению металла.
Некоторые исследователи считают, что на отдельных стади-
ях разрушения часто играет роль расклинивающее действие
коррозионной среды в микрощелях (29].
Трещины .в материале при коррозии под напряжением разви-
ваются в плоскости, перпендикулярной направлению макси-
мальных растягивающих напряжений, и распространяются в
зависимости -от структуры сплава и его электрохимической не-
однородности по границам зерен или по телу зерна.
Многие исследователи отмечают, что процесс развития тре-
щин не является непрерывным (30, 31].
Для коррозионного растрескивания, как уже указывалось,
необходимы растягивающие напряжения, с увеличением кото-
рых сопротивление материала этому виду разрушения умень-
шается. Для многих сплавов обнаружено так называемое «кри-
тическое напряжение», ниже которого в данной среде материал
практически не склонен к коррозионному растрескиванию (23, 32,
33]. Величина «критического напряжения» складывается из ве-
личины извне приложенных напряжений и (Величины внутренних
напряжений. В большинстве случаев величину внутренних растя-
гивающих напряжений рассчитать не удается, поэтому «крити-
ческое напряжение» отождествляется с напряжением, создавае-
мым извне приложенным усилием. Это создает известную не-
определенность и поэтому многие исследователи оспаривают
положение о -наличии «критического напряжения». Полагают,
что если сплав склонен к коррозионному растрескиванию, то
рано или поздно разрушение произойдет. Вопрос решается -вре-
менем. При высоких напряжениях коррозионное растрескивание
выявляется ’быстро, при низких медленно.
Большинство исследователей считает, что коррозионное рас-
трескивание происходит при напряжениях, близких к пределу
текучести. Однако на некоторых сплавах наблюдалось рас-
трескивание и при значительно более низких напряжениях. Оче-
видно, что такая неопределенность в основном связана с тем,
что неизвестна величина внутренних местных напряжений.
Сплавы, (подверженные коррозионному растрескиванию -по
границам зерен и упрочняющиеся благодаря выделению второй
фазы, имеют три участка с различными электрохимическими
“—268 —
характеристиками: зерна твердого раствора, выделившаяся по
границам зерен фаза или переходное состояние этой фазы
и участки, прилегающие к границам, обедненные -каким-либо
компонентом.
Наличие электрохимической неоднородности приводит к пре-
имущественному растворению отдельных структурных состав-
ляющих или разрушению защитных пленок, что способствует
появлению концентраторов напряжения.
Влияние структуры материалов
Алюминиевые сплавы. В практике известны случаи корро-
зионного растрескивания сплавов типа дуралюмина, сплавов
систем А1— Mg, Al — Mg—Zn и Al-—Mg — Zn — Cu (34 j.
Сплавы алюминия с медью подвергаются -коррозионному рас-
трескиванию под напряжением при наличии на их поверхности
анодной пленки, а также если в изделиях возникала склонность
к межкристаллитной коррозии, например вследствие замедлен-
ного охлаждения с температуры закалки или применения искус-
ственного старения, случайного -нагрева при различных техно-
логических операциях или в процессе эксплуатации в интервале
опасных температур. Коррозионное растрескивание этих сплавов
происходит -по границам зерен благодаря возникновению галь-
ванического элемента, состоящего из 'большого по площади
катода (тело зерна) и малого анода (граница зерна) (1,34—36].
Согласно другой точки зрения (22], склонность к коррозионному
растрескиванию под напряжением объясняется способностью
самого интерметаллического соединения разрушаться избира-
тельно.
Сплавы алюминия с высоким содержанием магния склонны
к межкристаллитной коррозии и к коррозии под напряжением;
наиболее склонны к коррозионному растрескиванию сплавы, со-
держащие 7—9% Mg, причем с увеличением содержания магния
эта склонность увеличивается. Появление склонности к корро-
зионному растрескиванию алюминиймагниевых сплавов связано
с выпадением из пересыщенного твердого раствора интерметал-
лического соединения Mg2Al8 -по границам зерен.
При термической обработке, -приводящей к выделению интер-
металлического соединения в .виде сплошной сетки по границам
зерен, сплав склонен к межкристаллитной коррозии и к корро-
зии под напряжением. К такой обработке в первую очередь от-
носится нагрев после закалки при температурах от 60* до
250° С. Стойки к коррозии под напряжением те сплавы, у кото-
рых в результате стабилизирующей обработки по границам
* Для появления склонности -к коррозионному растрескиванию п-ри этой
температуре требуется длительный нагрев, исчисляемый тысячами часов.
\ — 269 —
зерен располагаются -крупные частицы второй фазы, а в теле
зерна имеются мелкие выделения этой фазы.
По данным Бренера [37], склонность к межкристаллитному
разрушению вызывается не только наличием непрерывной сетки
выделений второй фазы -по границам зерен, но и влиянием гра-
ниц между зернами с -различно ориентированными плоскостями.
Наличие более быстрого выделения второй фазы на границе раз-
лично ориентированных зерен приводит к образованию анодных
участков, с которыми и связано появление местных трещин,
являющихся концентраторами напряжений в процессе эксплуа-
тации.
На склонность к коррозионному растрескиванию алюминий-
магниевых сплавов оказывает влияние и степень холодной
деформации. По данным работы [37], после деформации при хо-
лодной прокатке сопротивление коррозионному растрескиванию
сплавов с 5% Mg повышается более чем на 30%. Автор объяс-
няет это дополнительным распадом твердого раствора не только-
по границам зерен, но и по плоскостям скольжения, что созда-
ет условия к переходу коррозии из Местной в равномерную кор-
розию.
Склонность к коррозионному растрескиванию -высокопроч-
ных сплавов системы А1— Mg—Zn долгое время была пре-
пятствием к их применению.
Сплавы с суммарным содержанием циика и магния не более
6% не склонны к коррозии под напряжением после любых видов,
термической обработки. При увеличении содержания этих эле-
ментов в сплавах склонность их к растрескиванию под напряже-
нием резко .возрастает. Как и другие алюминиевые сплавы,
сплавы системы А1— Mg—-Zn приобретают склонность к кор-
розионному растрескиванию под напряжением при определен-
ных .видах термической обработки. Так как распад твердого рас-
твора в этих сплавах •наступает уже при комнатной температуре,
то -в отличие от дур алюмина они ® естественно состаренном
состоянии • обладают наибольшей склонностью к коррозии под
напряжением. Повышение температуры старения приводит
к улучшению коррозионной стойкости сплавов А1— Mg — Zn
и Al —Mg — Zn — Си под напряжением.
Зависимость ‘Коррозионной стойкости под напряжением про-
мышленного сплава В95 этой системы от температуры старения
и времени приведена на рис. 153. Сплав В95 склоней к межкри-
сталлитной коррозии, ио .в ненапряженном состоянии она не
приводит к резкому снижению прочности сплава. Наличие на-
пряжений растяжения 'резко усиливает разрушение по границам
зерен. При этом происходит направленное разрушение, приво-
дящее к коррозионному растрескиванию.
Дикс [36], чтобы Объяснить коррозионное растрескивание
таких ’сплавов, высказывает предположение, что растягивающее
напряжение разрывает материал вдоль зон обедненного твер-
— 270 —
дого раствора -по линиям, соединяющим очаги коррозии,- где
ранее -находились частицы диспепсией фазы.
Исследования С. Е. Павлова [23] -показали, что, -несмотря на
высокую -степень деформации слитка, >в процессе изготовления
из -него полуфабрикатов в материале остаются межкристаллит-
ные пустоты или микротрещины, которые способствуют корро-
зионному растрескиванию под напряжением, создавая локали-
зованный характер коррозионно-
го разрушения по границам зерен
даже при отсутствии цепочки вы-
делений новой фазы.
Склонность алюминиевых
сплавов к коррозионному рас-
800
^700
| 6О0
у о о
§ ЬОО
|л»
200
| 100
о
О 610161826 303662 6856
Продолжительность
старения, и
Рис. 153. Зависимость коррози-
онной стойкости под напряже-
нием сплава В95 в морской во-
де от температуры и времени
старения [23]
Рис. 154. Зависимость длительно-
сти жизни состаренных при 120° С
за 24 ч сплавов алюминия с 4,5%
Zn и 1,8% Mg {38]:
1 — на чистейшем алюминии; 2 — на
техническом алюминии; 3 — на тех-
ническом алюминии + 0,8% Мп; 4 —
иа техническом алюминии + 0,75%
Мп + 0.17% Ti; 5 — на техническом
алюминии + 0,8% Мп + 0,12% Zr
•грескиванию может быть резко снижена выбором оптимального
содержания цинка и магния, введением в сплав различных леги-
рующих элементов, а также применением оптимального режима
термической обработки.
В. А. Ливанов и сотрудники {38], исследуя коррозионную
стойкость.под напряжением сплавов А1 — Mg — Zn, показали,
что сопротивление этих сплавов коррозионному растрескиванию
можно значительно повысить введением в них 0,6% Мп и не-
больших количеств (0,15%) Ti и Zr.
Кривые, приведенные на рис. 154, показывают, что наилуч-
шие результаты получаются при одновременном введении
в сплав алюминия, содержащий 4,5% Zn и 1,8% Mg, -марганца,
титана и циркония. Если сплав легирован только марганцем или
— 271 —
марганцем и титаном, его -сопротивление коррозии под напря-
жением сильно зависит от температуры закалки (рис. 154, кри-
вые 3 и 4). Поэтому при изучении склонности сплавов к корро-
зионному -растрескиванию необходимо сравнивать сплавы при
различных режимах закалки.
Авторы полагают, что введение марганца, титана и цирко-
ния в сплав А1 — Mg — Zn сильно увеличивает его стойкость при
коррозии под напряжением благодаря гетерогенизации струк-
Рис. 155. Зависимость дли-
тельности жизни сплавов,
закаленных с 450° С от тем-
пературы старения (38]. Обо-
значения те же, что и на
рис. 154
Рис. 156. Зависимость удельно-
го электросопротивления от
длительности жизни сплавов
алюминия с 4,5% Zn и 1,8%
Сплавы изготовлены:
1 — на чистейшем алюминии;
2 — на техническом алюминии [38]
туры; возможно также, что происходит дополнительное тонкое
изменение структуры.
Температура старения также оказывает большое -влияние на
-сопротивление коррозионному растрескиванию сплавов
А1— Mg — Zn (рис. 155). При режиме, обеспечивающем наилуч-
шие прочностные характеристики, коррозионная стойкость спла-
ва оказывается, -как правило, наименьшей. Поэтому необходимо
выбрать оптимальные режимы, обеспечивающие удовлетвори-
тельную коррозионную стойкость без существенного понижения
механических свойств.
Данные, приведенные на рис. 155, показывают, что все кри-
вые зависимости длительности жизни сплава от режима старе-
ния, за исключением кривой 5 для сплава с 0,8% Мп и 0,12% Zr,
имеют минимумы. Такой ход кривых объясняется, очевидно, тем,
— 272 —
что наименьшую стойкость -при коррозии под напряжением
•сплавы имеют после старения по режиму, который обусловли-
вает некоторую среднюю стадию распада пересыщенного твер-
дого раствора. Эта стадия, вероятно, 'близка к стадии, обуслов-
ливающей максимальное упрочнение. Такой вывод хорошо
подтверждается данными по изменению электросопротивления
образцов после различных режимов старения, приведенными на
рис. 156.
Минимальная длительность жизни образцов, наблюдаемая
у сплавов с 4,5% Zn и 1,8% Mg (кривые 1 и 2) после старения
при 100 и 120° С, а у сплава, дополнительно содержащего
0,8% Мп, при 140° С, соответствует некоторым средним значе-
ниям удельного электросопротивления, которые в свою очередь
соответствуют, по мнению авторов, некоторой средней степени
распада твердого раствора. Когда происходит сильный распад
твердого раствора или когда он очень мал, длительность жизни
образцов увеличивается.
Магниевые сплавы. Магниевые сплавы можно разделить на
практически не склонные к коррозионному растрескиванию
(сплавы магния с марганцем) и сплавы, обладающие в той или
иной степени склонностью к этому виду коррозионного разруше-
ния [23, 39]. К последней группе относятся сплавы, содержащие
алюминий и цинк; с увеличением содержания легирующих эле-
ментов сопротивление ‘коррозии под напряжением понижается.
Магниевые сплавы подвержены коррозионному растрескива-
нию в деформированном состоянии, литые сплавы практически
слабо склонны к коррозионному растрескиванию под напряже-
нием.
Термическая обработка влияет на склонность магниевых
сплавов к коррозии под напряжением по-разному.
Возникновение склонности к коррозии под напряжением
у магниевых сплавов исследователи объясняют по-разному. Не-
которые [40] высказывают предположение, что образование
железо-алюминиевой составляющей, выделяющейся преиму-
щественно параллельно определенным кристаллографическим
плоскостям, способствуют созданию электрохимической неодно-
родности, так как эта фаза может служить катодом по отноше-
нию к твердому раствору.
Предполагают также, что склонность к растрескиванию маг-
ниевых сплавов может возникнуть благодаря образованию
«тонкой структуры», которая создает предпосылки для появле-
ния склонности к питтинговой коррозии, а следовательно,
создаются условия избирательного разрушения сплава [41].
Е. М. Зарецкий [23] считает, что коррозионное растрескива-
ние магниевых сплавов связано с избирательным разрушением
твердого раствора Mg — Al, в результате чего изменяется пара-
метр решетки, что в свою очередь вызывает возникновение
в поверхностных слоях дополнительных напряжений. М. А. Ти-
18 Зак- 1338 — 273 —
монова (39] установила, что наиболее вероятными участками, на
которых происходит локализация коррозионного разрушения,
являются интерметаллические -соединения и пересыщенный твер-
дый раствор алюминия >в магнии. Исходя из этого, она -предпо-
лагает, что вследствие избирательного растворения легирую-
щего компонента на этих участках образуется зародыш микро-
надреза, который пои отсутствии напряжений не развивается.
Напряжения растяжения приводят к быстрому развитию микро-
надреза в глубину и образованию затем трещин.
Таким образом большинство исследователей [33, 39—42J
приходит к выводу, что склонность магниевых сплавов к корро-
зионному растрескиванию также обусловливается электрохими-
ческой неоднородностью различных участков сплава. Разруше-
ние сплавов под напряжением происходит при этом и по телу
зерна, и по границам зерен благодаря избирательной -коррозии.
Снизить склонность магниевых сплавов к коррозионному
растрескиванию можно -путем отжига, снимающего внутренние
напряжения. С увеличением температуры отжига сопротивление
магниевых сплавов коррозии -под напряжением повышается..
Старение после закалки снижает устойчивость магниевых спла-
вов против коррозии под напряжением.
Медные сплавы. Широко известно, что коррозионному рас-
трескиванию подвержены сплавы меди с цинком — латуни.
Иногда упоминаются случаи коррозионного растрескивания
меди и бронз.
Коррозионное растрескивание латуней чаще всего наблю-
дается в средах, содержащих аммиак; значительную роль -при
этом играют различные примеси и добавки к сплаву. Наиболее
склонны к растрескиванию латуни с высоким содержанием цин-
ка, особенно латунь с 40% цинка (рис. 157).
Растрескивание латуни, по мнению А. В. Бобылева [43],
в первую очередь обусловлено наличием ® ней цинка. Взаимо-
действие цинка с окружающей средой опасно лишь при наличии
в поверхностных слоях изделий или полуфабрикатов растяги-
вающих напряжений.
Уменьшение напряжений >в самом сплаве может быть достиг-
нуто путем рационального выбора технологического процесса.
Нежелательны с этой точки зрения следующие операции: свертка
и вытяжка путем диаметрального обжатия, давильные операции,
волочение труб без оправки, обжим. Если по условиям произ-
водства последней операцией является процесс, создающий
напряжения растяжения, то для устранения склонности к корро-
зионному растрескиванию необходимо производить отжиг при
230—300° С.
Зависимость механических свойств и скорости коррозионного
растрескивания латуни марки Л68 над 25%-ным раствором
аммиака от температуры отжига по данным А. Е. Гопиуса и
Ю. А. Смирновой [23] приведена на рис. 158.
— 274 —
На склонность латуней к коррозионному растрескиванию
большое влияние оказывает величина зерна. Менее стойка ла-
тунь с крупными зернами.
Различные исследователи по-разному судят о влиянии окру-
жающей среды на коррозионное растрескивание латуни.
В. В. Скорчеллетти и В. А. Титова придают 'Первоочередное
значение адсорбционному .понижению прочности металла за счет
проникновения жидкости
Рнс. 157. Зависимость скорости
растрескивания от состава латуни
(образцы поперечные) при испы-
тании иад растворами аммиа-
ка {23]:
I — 5%-иый NH3; 2 — 25%-ныЙ NH3;
3 — медноаммиачный раствор
© зародышевые клиновидные ще-
ли [44].
Очевидно, правильным будет по-
ложение, объясняющее коррозион-
ное растрескивание этих сплавов
совместным влиянием растягиваю-
Температура отжига, °C
Рис. 158. Зависимость механических
свойств и скорости коррозионного
растрескивания латуни марки «П68
над 25%-ным раствором NHS от тем-
пературы отжига [23]
щих напряжений и коррозионной среды при наличии в сплаве
электрохимической неоднородности. Коррозионное растрескива-
ние латуней носит межкристаллитный характер.
Стали. К числу сплавов, склонных к растрескиванию под
напряжением, следует отнести высокопрочные сплавы, мало-
углеродистые и нержавеющие стали.
На склонность малоуглеродистых сталей к коррозии под -на-
пряжением -большое влияние оказывают содержание в них угле-
рода и степень раскнсления стали. Растрескивание спокойных
сталей, содержащих 0,2% углерода, наблюдается только при
пластической деформации, вызванной какими-либо технологи-
ческими операциями. Мягкие нераскисленные стали, содержа-
щие 0,03—0,1% углерода и незначительные количества азота
и кислорода, обладают сильной склонностью к коррозионному
растрескиванию, что связано с -выделением карбидов и нитри-
18* — 275 —
дов по границам зерен в результате медленного разложения
пересыщенного феррита [32].
Искаженные границы зерен вследствие выделения на них
карбидов анодны по отношению к зернам феррита и разруша-
ются в -процессе коррозии избирательно. Действие растягиваю-
щих напряжений в этом случае еще больше -способствует иска-
жению границ зерен и может сделать их еще более анодными по
отношнию к телу зерна [45].
Наиболее склонны к коррозионному растрескиванию круп-
нозернистые стали и стали, имеющие структуру мартенсита.
Предполагают, что снизить склонность к коррозионному
растрескиванию стали -после сварки и холодной обработки мож-
но путем отпуска при температуре вблизи точки Act (600—
700°С). Однако длительные выдержки при этих температурах
приведут к увеличению склонности стали -к коррозии под напря-
жением.
Наиболее часты случаи коррозионного растрескивания
-аппаратуры с остаточными напряжениями после сварки. По-
этому при изучении склонности той или иной стали к коррозии
под напряжением надо проверять и сварные соединения.
Коррозионное растрескивание нержавеющих сталей наблю-
дается главным образом в сталях мартенситного класса (12%
хрома). Аустенитные стали типа 18-8 более склонны к коррозий
под напряжением, чем полуферритные хромистые стали. Значи-
тельное влияние на склонность к коррозии под ‘Напряжением
оказывает стабильность аустенита. Характер -коррозионного рас-
трескивания в большинстве сред транскристалл итный, если
сталь не склонна к межкристаллитной коррозии. Если сталь
склонна к межкристаллитной коррозии, то растрескивание про-
исходит по границам зерен.
При увеличении в нержавеющей стали содержания углеро-
да и никеля до определенного предела склонность к коррозион-
ному растрескиванию снижается. Хром при добавке его к сталям
с высоким содержанием никеля улучшает стойкость стали про-
тив коррозии под напряжением [32].
По данным некоторых авторов, склонность к коррозионному
растрескиванию хромоникелевых сталей устраняется отпуском
при температуре -выше 840—870° С.
Аналогичный эффект у высокопрочных сталей вызываем по.
данным Фелпса и Логинова [46], высокотемпературный отжиг.
Данные, приведенные на рис. 159, показывают, что для ехали
с 12% хрома наибольшая склонность к коррозионному рас-
трескиванию наблюдается при температуре отжига, равной при-
мерно 400° С. Дальнейшее повышение температуры резко сни-
жает склонность сплава к коррозионному растрескиванию.
Склонность к коррозионному растрескиванию низколегирован-
ной стали с 2% Сг и 1,5% Si также заметно снижается при от-
жиге с температур выше 570° С.
— 276 —
Эти результаты получаются при испытании в промышленной
атмосфере (один год) и при ускоренных испытаниях методом
погружения в 3%-ный раствор NaCl (40 суток), хотя в ко-
личественном отношении не всегда наблюдается полная корре-
ляция.
Следует иметь в виду, что область опасных температур, вы-
зывающих появление склонности -к межкристаллитной коррозии,
не всегда соответствует области, где сталь разрушается под на-
пряжением с развитием трещин по границам зерен.
Рис. 159. Влияние температуры отжига иа коррозионную стойкость
сталей при напряжениях, равных 75% ае [46]:
а — нержавеющая сталь с 12% Сг; б — низколегированная сталь с 2% Сг и
l.s% Si: 1 — при погружении в аэрируемый 3%-ный раствор NaCl (40 суток);
2 — в морской атмосфере Ходин год). Стрелки означают, что образцы не раз-
рушились по истечении выбранного срока испытания
Склонность к коррозионному растрескиванию под напряже-
нием высокопрочной конструкционной стали ЗОХГСА, ото данным
Ф. Ф. Ажогина [47], снижается, если сталь после закалки -в мас-
ло отпускается при 200 и 300° С. Если же отпуск производится
при 250° С, то склонность к растрескиванию этой стали увеличи-
вается; это может 'быть связано с тем, что при 250° С происходит
выпадение карбидов с максимальной интенсивностью.
Изложенные «выше данные о механизме коррозионного рас-
трескивания сплавов определяют и принципы проведения уско-
ренных коррозионных испытаний, в процессе которых необходи-
мо выяснить следующие вопросы:
1. Зависимость склонности сплава к коррозионному рас-
трескиванию от -величины напряжений. Для этой цели строят
кривые зависимости склонности к коррозионному растрескива-
нию от величины напряжения. По форме кривой и ее изгибу
можно судить о наличии «критического напряжения». Если оно
обнаруживается, то в ряде случаев можно допустить примене-
— 277 —
ние сплава, склонного к коррозионному растрескиванию, огово-
рив верхний предел допустимых -напряжений.
2. Влияние температуры и -скорости охлаждения при закалке
на склонность сплава к коррозионному растрескиванию. Не
всегда режимы термической обработки, обеспечивающие наи-
меньшую склонность к коррозионному растрескиванию, совпа-
дают с режимами, сообщающими сплаву наилучшие механи-
ческие свойства.
Однако в ряде случаев. удается -найти такой режим терми-
ческой -обработки, который 'Обеспечивает сплаву два необходи-
мых свойства-—высокую прочность и сравнительно низкую
склонность к коррозионному растрескиванию.
3. Влияние температуры и продолжительности старения или
отпуска. Изменением температуры старения и его продолжи-
тельности часто удается привести сплав ® состояние, при кото-
ром он практически не склонен к коррозии под напряжением.
Это хорошо иллюстрируется результатами, достигнутыми за
последние годы в области разработки высокопрочных алюми-
ниевых сплавов, применение которых долгое время ограничи-
валось из-за их высокой склонности к коррозионному растрески-
ванию.
4. Влияние предполагаемых технологических операций (на-
грев, деформация). В процессе технологической обработки (раз-
вальцовки, штамповки, гиба и т. п.) склонность материала
к коррозионному растрескиванию может появиться -вновь благо-
даря структурным изменениям, происходящим под «влиянием
кратковременного нагрева и деформации. Чтобы выяснить влия-
ние технологических операций, необходимо построение кривых
зависимости склонности к коррозионному растрескиванию от
кратковременных нагревов, возможных при технологической об-
работке, а также от величины 'пластической деформации.
5. Склонность к коррозионному растрескиванию сварных
соединений. Проведение этих испытаний диктуется тем обстоя-
тельством, что на некоторых сплавах после сварки появляется
склонность к коррозионному растрескиванию или непосредствен-
но в зоне сплавления, или на некотором расстоянии (10—15 мм)
от сварного шва, где происходит термическое воздействие в не-
благоприятном режиме.
Выбор коррозионной среды
Кратко рассмотрев влияние структуры сплавов на их склон-
ность к коррозионному растрескиванию и необходимые условия
проведения ускоренных испытаний, перейдем к обсуждению кор-
розионных сред, в которых должны проводиться коррозионные
испытания.
Коррозионные среды надо выбирать с таким расчетом, чтобы
сплавы в них подвергались избирательной коррозии, скорость
— 278 —
которой значительно превышала бы -скорость общей коррозии
в данной среде.
•Склонность сплавов к коррозии под напряжением следует
проверять в тех коррозионных -средах, для которых предназначе-
ны сплавы. Ускоряющим коррозионный процесс фактором в этом
случае должно быть усиление действия главного .фактора; опре-
деляющего скорость коррозии материала в естественных усло-
виях. Так, например, для условий морской атмосферы — испыта-
ние -в камере с распылением раствора хлористого натрия; для
сельской атмосферы — испытание при повышенной влажности;
для условий, когда «возможно периодическое увлажнение,—
испытание при периодическом погружении в электролиты. Ре-
зультаты таких испытаний надо сопоставлять с результатами
испытаний в естественных условиях или с данными о поведений
сплавов в условиях эксплуатации.
Если необходимо определить только, склонен ли сплав к это-
му -виду коррозии, проводят испытания в специально подобран-
ных для этой цели средах, которые не обязательно должны
воспроизводить условия эксплуатации, а должны лишь сохранять
характер коррозионного разрушения (избирательное разруше-
ние).
Так как процесс коррозии большинства металлов в нейтраль-
ных электролитах и атмосфере определяется скоростью катод-
ного процесса, то при ускоренных испытаниях применяют раз-
личные электролиты, увеличивающие скорость катодного про-
цесса.
К растворам хлористого натрия добавляют перекись водоро-
да, соляную кислоту, хромовокислый калий. Результаты от
введения этих добавок не всегда одинаковы. Добавка соляной
кислоты ведет к изменению механизма процесса коррозии; -вме-
сто кислородной деполяризации происходит водородная. Поэто-
му такой метод применим лишь тогда, когда, например, испыты-
вают один и тот же сплав в двух состояниях—склонном и не
склонном к коррозии под напряжением.
Введение перекиси водорода часто не дает положительных
результатов из-за каталитического разложения ее, особенно
в присутствии меди. По данным Ю. Р. Эванса [5], применение
хромовокислого калия -дает хорошие результаты; испытания
материалов с различной термообработкой в растворе хлористого
натрия, содержащем хромовокислый натрий, дают такие же ре-
зультаты, как при испытании -в атмосферных условиях.
При испытаниях алюминиевых сплавов Ферми и Эванс [48]
применяли растворы 0,5-м. хлористого натрия + 0,005-;м. бикар-
боната натрия; ускорение процесса при -этом испытании дости-
гается за счет .влияния бикарбоната натрия как вещества,
препятствующего нейтрализации накапливающейся в трещинах
кислоты в результате протекания преимущественно анодной ре-
акции.
— 279 —
Таких же результатов, как и при введении добавок, уско-
ряющих коррозионный процесс, можно достигнуть, применяя
при ускоренных испытаниях сплавов анодную поляризацию от
внешнего источника тока, .приводящую к смещению стационар-
ного потенциала в положительную сторону. Если образец скло-
нен к коррозионному растрескиванию, то растворение должно
концентрироваться в трещинах, а если склонности к коррозион-
ному растрескиванию нет, то коррозия будет протекать по .всей
поверхности образца.
Чтобы выявить склонность алюминиевых сплавов к корро-
зионному растрескиванию, можно рекомендовать ускоренные
испытания при полном погружении и при периодическом увлаж-
нении 3%-ным раствором хлористого натрия (10 мин в растворе,
50 мин на воздухе).
При полном погружении, когда испытания ведутся в 3%-ном
NaCl, алюминиевый сплав считается выдержавшим испытания,
если образцы в виде петель в течение шести месяцев не рас-
трескались. Электролит следует менять через одну-две. недели.
При этом на 1 см2 поверхности образца берется ~5—10 см3
электролита.
Испытания при переменном погружении также проводят
в 3%-ном растворе NaCl по режиму, указанному выше. Если
в течение трех месяцев образцы в виде петель не растреска-
лись, можно считать этот сплав пригодным для эксплуа-
тации. Электролит также целесообразно менять через одну-две
недели.
Для определения склонности к коррозионному растрескива-
нию магниевых сплавов испытания чаще всего проводят во
влажных камерах при распылении 0,001-и. раствора хлористого
натрия или при погружении в слабые ..растворы хлористого нат-
рия, содержащие бихромат калия (0,5-м. NaCl+0,05-м. KgCrgO?).
Склонность латуни к коррозионному растрескиванию опреде-
ляют >в средах, содержащих аммиак, сернистый ангидрид, угле-
кислоту и пары воды. Применяют и среды, содержащие азотно-
кислую ртуть. Считают, что действие ртути как поверхностно
активного вещества сводится к быстрому проникновению ее
в тонкие трещины и зазоры.
Склонность к коррозионному растрескиванию низколегирован-
ных сталей, высокопрочных сплавов и нержавеющих сталей
определяют чаще всего при погружении напряженных образцов
в 42 %-ный кипящий раствор MgCl2. Однако наличие склонно-
сти к коррозионному растрескиванию хорошо выявляется и при
увлажнении 3%-ным раствором NaCl, а также в камере с соле-
вым туманом.
Составы электролитов, применяющиеся для оценки склонно-
сти сплавов к коррозионному растрескиванию, приведены в
табл. 29.
— 280™
Таблица 29
СОСТАВЫ ЭЛЕКТРОЛИТОВ, РЕКОМЕНДУЕМЫХ ДЛЯ УСКОРЕННЫХ ИСПЫТАНИИ
НА СКЛОННОСТЬ к КОРРОЗИОННОМУ РАСТРЕСКИВАНИЮ
Сплавы Состав электролита Режим
Магниевые 3,5 % -ный раствор NaCi-|-2$6 КвСгО4 0,001-н. раствор NaCl 0,5-м. раствор NaCl4-0,05-м. KsCr2O3 0.001-н. раствор NaCl 0,001-н. раствор NaCl Погружение Переменное по- гружение Распыление в камере
Алюминие- вые 3%-ный раствор NaCl; 3%-ный раствор NaCl4-0,1 % Н2О2; мерекая вода 3%-ный раствор NaCl То же Погружение Переменное по- гружение Распыление в камере
Медные 10 % -ный раствор Сп(ОН)2 в 20 %-ном растворе NHS 20 %-ный раствор NaOH, насыщенный Сп(ОН)2 5 %-ный раствор NaNQj-l-4% CuS04-5HsO 1%-ный раствор HgCl24-l% НС1 1%-ный раствор Hg(NO8)4-l% HNOS 25%-ный раствор NH8* 5 %-ный раствор NH3* 0,1 см*! дм* H2SO44-0,l г! дм* NasSOs 0,3%-ный раствор NH84-2,3% (НН4)аСО3 Погружение Над'растворамн
Железные (низколеги- рованные стали) 3 4-6%-ный раствор NH4NO84~504-57% Ca(NO8)2 42%-ный раствор MgCl2 130—860 а/дл8 CafNOA-f-SO а/дл8 NH4NO8 25 4- 50 % -ный раствор NaOH 50%-ный раствор NH4NOS 3%-ный раствор NaCl То же Погружение при кипячении Погружение при кипячении Переменное по- гружение Распыление в камере
— 281 —
Продолжение табл. 29
Сплавы
Состав электролита
Режим
Железиые
(нержавею-
щие стали)
10 %-ный раствор HNOg-f-3% HF, 70—80°
60 %-ный раствор CaCl.2+0,l-?I,0%HgCl2
42%-ный раствор MgCls
65 %-ный раствор HNO3
ПО г/дл13 СивОН-бб слР/дл? HgS04
50—57 % -ный раствор Ca(NO3)2-|-3'^-6%
NH4NOs
20 %-ный раствор HNO^+I%HF
Погружение
Погружение прн
кипячении
3%-ный раствор NaCl
То же
Электролитиче-
ское травление
при 20°С
Переменное по-
гружение
Распыление в
камере
На 1 дм3 объема камеры берется 15 ли3 раствора.
Способы создания напряжения при ускоренных испытаниях
Если при ускоренных испытаниях, применяемых для выявле-
ния склонности сплавов к коррозионному растрескиванию, чрез-
мерное увеличение агрессивности коррозионной среды, по мне-
нию многих исследователей, не желательно в связи с возможным
искажением результатов, то при выборе величины напряжений
обычно исходят из того, что в реальных условиях всегда воз-
можны местные высокие напряжения, значительно превосходя-
щие расчетные. Поэтому проведение испытаний для определения
склонности к коррозионному растрескиванию при напряжениях,
близких к пределу текучести материала (~90% os), считается
оправданным.
Аппаратурное оформление ускоренных испытаний для выяв-
ления склонности сплавов к коррозии под напряжением ослож-
няется необходимостью создания в испытываемых образцах на-
пряжений.
Напряжения можно создавать двумя путями: созданием
постоянной деформации и приложением постоянной нагрузки.
Наиболее прост первый метод; он широко применяется в массо-
вых лабораторных и ускоренных испытаниях. Образцы деформи-
руются растяжением или изгибом и укрепляются в специальных
приспособлениях, иа которых затем и проводят испытание. Эти
приспособления относительно просты и легко могут быть изготов-
лены в любой лаборатории. Испытания при постоянной нагрузке
имеют ряд преимуществ, но требуют сложной аппаратуры, кото-
рая есть далеко не во всех лабораториях.
При всех способах нагружения испытания должны проводить-
ся как сравнительные, т. е. одновременно должны испытываться
— 282 —
ненапряженные образцы, а склонность к коррозионному растрес-
киванию должна устанавливаться по сравнению с уже известным
материалом, применявшимся в данных условиях, для которых
предназначаются испытываемые материалы.
Испытания при постоянной реформации
Наиболее простая и распространенная форма образца, при-
меняющегося для определения склонности сплава к коррозион-
ному растрескиванию под напряжением при массовых испытани-
Рис. 160. Форма и размеры образца «петли» для
определения склонности сплавов к коррозионному
растрескиванию по$ напряжением
ях, — «петля», предложенная Маттесом [49]. Форма и размеры
образца показаны на рис. 160. Образцы готовят из полос листо-
вого материала шириной 15 мм. Длина полосы должна равнять-
ся 80-кратной толщине +60 мм. Концы полоски на расстоянии
30 мм отгибаются на 30°. После этого полоска изгибается на
оправе в виде петли, как это показано на рис. 160. Диаметр
оправы равняется 16-кратной толщине испытываемого материала.
После пластической деформации образец подвергают упругой
деформации путем сближения концов петли и закрепления их в
специальной рамке из инертного материала (рис. 161). Изготов-
лять петлю надо после окончательной термической обработки
материала. Образцы для изготовления петель следует вырезать
поперек волокон.
Величина растягивающих напряжений, возникающих на греб-
не петли, может быть, по данным Е. М. Зарецкого [50], прибли-
зительно оценена, если рассматривать петлю как нагруженную
консоль. Величину напряжений рассчитывают по формуле
O==2£==JZL (112)
ww
где М — изгибающий момент;
W — момент сопротивления образца;
— 283 —
р — устанавливаемое опытным путем усилие, необходимое
для сближения концов петли;
/ — плечо момента.
Петли, по мнению С. Е. Павлова (51], имеют существенный
нельзя точно рассчитать упругие
недостаток: во-первых, в иих
Рнс. 161. Способ крепления образ-
цов, в ваде «петли» при испытании
напряжения, и, во-вторых, эти
напряжения в процессе опыта
снижаются вследствие удлине-
ния образца и возникновения
небольших коррозионных тре-
щин. Тем не менее эта форма
образцов благодаря своей про-
стоте находит широкое приме-
нение и позволяет получать на-
дежные данные, необходимые
для сравнительной оценки раз-
личных материалов.
Другой вид образцов, позволяющий создавать в материале
различные напряжения, — скоба (рис. 162). Изменяя стрелу
прогиба, можно создавать различные значения напряжения. Рас-
Рпс. 162. Образец в виде скобы:
а — подготовленный для испытания; б — в приспособлении с опорой
в середине [51, 52]
чет упругих напряжений в этом случае, при допущении, что
образец изгибается по дуге окружности, ведется по формуле
4Ehf
1*-р4р *
(113)
где а — растягивающее напряжение, н/мл? (кГ/мм2);
Е — модуль упругости;
— 284 —
h — толщина образца, мм;
f — стрела прогиба, мм;
I — расстояние между опорами.
С целью варьирования приложенных напряжений использо-
вали приспособление {52], позволяющее изменять стрелу прогиба
образца с помощью винта, расположенного в центре приспособ-
ления (рис. 162, б). Напряжение при таком способе нагружения
вычисляют по формуле
О = _6ВД 114
/2 ' 7
Применение вкладыша
(рис. 163, а) позволяет соз-
дать более равномерное
распределение напряжений
по рабочей длине образца.
Нагрузка прикладывается
в этом случае к образцу в
двух точках. Стрела проги-
ба рассчитывается по фор-
муле
,(115)
1 3Eh 4
Рис. 163. Образец для коррозионных
испытаний сварных соединений:
а — образец в приспособлении с вклады-
шем, имеющим две точки опоры; б об-
разец для коррозионйых испытаний; в —
образец для механических испытаний
где f — стрела прогиба в
точках опоры;
а — задаваемое напря-
жение;
Е — модуль упругости;
1,а,Ь — расстояния между
опорами (см. рис.
163);
Л —толщина образца.
Испытания сварных соединений ведут в приспособлениях,
изображенных на'рис. 163, а. При этом воспроизводимые резуль-
таты получаются при удалении усиления (наплавленного вали-
ка). Поскольку трудно удалить наплавленный металл шва
заподлицо с основным материалом, рекомендуется применять •
образец с выфрезерованным углублением, изображенный на
рис. 163, б.
Напряжения в этом случае рассчитывают по формуле
E^k
(Н6)
где f — стрела прогиба в месте опор, мм;
— 285 —
O' — задаваемое напряжение; для сварных соединений берет-
ся обычно 0,5—0,8 crs, к/лш2 (кГ/жлг2);
а — ширина выфрезерованной площадки (обычно 30 мм),
мм;
Ь — расстояние между опорами вкладыша;
di — толщина образца в месте, где снято усилие (61 дол-
жно быть не менее 0,86g);
k — коэффициент, равный /
\6i /
62— толщина образца до снятия усиления, мм.
Если образец за время, принятое для испытаний (обычно
3 месяца), не разрушился, то после ускоренных испытаний из
него изготавливают по чертежу, указанному на рис. 163, в, обра-
зец для разрывных испытаний. По изменению механических
свойств (оъ, 6) судят о поведении металла в напряженном со-
стоянии. Если изменение механических свойств не превышает
5—10%, то можно считать, что сварное соединение может в на-
пряженном состоянии работать.
Образцы закрепляют в согнутом состоянии в рамке из инерт-
ного материала или из сплава того же состава, что и испытывае-
мые образцы. Иногда пользуются обычными образцами, приме-
няемыми для механических испытаний на растяжение. Одни
образцы испытывают в согнутом состоянии, другие в свободном.
Сравнивая изменение механических свойств после испытания у
тех и других образцов, выявляют роль напряжений в -коррозион-
ном процессе. Влияние напряжений можно также определить по
времени, необходимому до появления первой трещины или полно-
го разрушения образца. Однако более надежные результаты
получаются при испытании в напряженном состоянии образцов
в виде полосок, из которых изготавливают образцы для механи-
ческих испытаний после коррозионных опытов. Это следует де-
лать, чтобы исключить влияние торцов, по которым обычно раз-
вивается местная коррозия, очаги которой могут стать концент-
раторами напряжения.
Напряжения изгиба в образцах при ускоренных испытаниях
создают равными 80—90% от предела текучести материала при
изгибе о' [53]. Величину о' можно найти по уравнениям:
о;= (Ц7>
os-j-6S£
где 6S принимается равной 0,002.
Для некоторых сплавов, как это видно из приведенных ниже
данных, предел текучести os значительно выше предела пропор-
циональности опл, Поэтому при выборе величины напряжения;
— 286 —
создаваемого в образцах, необходимо следить, чтобы это на-
пряжение не превосходило предела пропорциональности:
опл1 9,8 Мн]м* (кГ/мм2)
gs, 9,8 Мн!я? (кГ/мя?) .
gs ,9,8 Мн/м* (кГ/мму*
АМг-6 АМг-6
отожжен- нагарто- АЦМ А К 8 ЭЙ-257
мый ванный
14,0 23,0 34,0 34,8 154
15,8 29,4 40,0 37,7 176
21,6 37,2 49,2 46,5 206
Образцы в виде петель и скоб применяются обычно для ли-
стовых материалов, толщина которых не превышает 3 мм. При
использовании образцов в виде скоб возникают серьезные труд-
Рис. 164. Образцы, отлитые в виде вилки для испытания
на коррозию под напряжением прессованных н литых материа-
лов больших сечений
ности в связи с появлением щелевой коррозии в местах -упора-;
особенно это относится к алюминиевым сплавам и нержавею-
щим сталям.
Однако, как показывает опыт, этого можно избежать, приме-
няя тонкие прокладки из полиэтилена, фторопласта и других
инертных материалов. Применение для этих целей лаков и кра-
сок дает худшие результаты.
При испытании прессованных литых изделий больших сечений
применяют образцы в виде вилки, изображенные на рис. 1'64,
а также образцы, отлитые специально для испытания в виде
кольца (рис. 165). Для сечений, равных 6 мм и больше, образ-
цы изготавливают толщиной 6 мм. для меньших сечений —
толщиной, равной толщине материала.
Напряжения создаются сближением концов вилки. Для этой
цели образцы перед испытанием помещают в специальную рамку
из инертного материала (плексиглас, текстолит). Размер выреза
рамки рассчитывают таким образом, чтобы после установки
образцов расстояние между концами вилки уменьшилось на
50—75%. Для изучения склонности материалов к коррозионному
растрескиванию можно воспользоваться также плоским образ-
цом упрощенной формы, предложенным Бреннером [54]. Внеш-
— 287 —
ний вид образца и приспособление для создания напряжений
приведены на рис. 166, а и б. Изгибающий момент по длине об-
разца получается при этом постоянным. Сближением концов
приспособления-колодки с помощью пресса можно задать вполне
определенную нагрузку. Расстояние между концами колодки,
Рнс. 165. Образец, отлитый
в виде кольца для испыта-
ний литых материалов на
коррозию под напряжением
Рис. 166. Упрощенный образец, применяе-
мый для выявления склонности материала
к коррозии под напряжением (а), и при-
способление для создания напряжений в
образце (б) (54]
б
зависящее от величины нагрузки, фиксируется с помощью бол-
та. Напряжения в образце вычисляют по формуле
Ра
о = ----- ,
Ь&
где Р — нагрузка;
а — плечо рычага;
b — ширина образца;
б— толщина образца
При изучении склонности сплавов к коррозионному растрес-
киванию в зависимости от величины напряжения можно исполь-
зовать образцы переменного сечения (рис. 167). Напряжения
в металле создаются при помощи болта с кольцевой головкой 2,
свободно вращающегося на оси. Образец 3 укрепляется в осо-
бом приспособлении Г-образной формы с помощью двух сталь-
ных винтов 6.
Нагружение образцов производится на машине Шопера по-
средством пуансона, устанавливаемого на опоре особой приз-
мы 4, которая надевается на болт и упирается в специальный
вырез на образце. »
— 288 —
Усилие, необходимое для создания напряжений (обычно от
40 до 90% 0's), рассчитывают по формуле
р cb№
6х ’
где Р — необходимое усилие;
а— напряжение;
b — ширина образца;
А — толщина образца;
х — расстояние от точки приложения усилия до данного
сечения.
Толщину и ширину образца измеряют обычно в нескольких
местах, для каждого из них рассчитывают величину нагрузки и
выводят среднее значение. После приложения нагрузки начина-
ют завинчивать гайку 5 до полного падения нагрузки на маши-
не. Все металлические части (держатель, болты, гайки и приз-
мы) необходимо тщательно изолировать водостойким лаком.
Рис. 167. Приспособление для испы-
тания образцов переменного сечения
на коррозию под напряжением:
1 — корпус; 2 — болт с кольцевой голов-
кой; 3 — образец; 4 — призма; 5 —- гай-
ка; 6 —• стальные винты
Рис. 168. Образец в
виде кольца для ис-
пытания -труб на кор-
розию под напряже-
нием
Для испытания на коррозию под напряжением труб применя-
ют кольца шириной 15 мм, вырезанные из трубы (рис. 168).
Напряжения в образце создаются при помощи болта, проходя-
щего сквозь отверстия в кольце, просверленные по диаметру.
Изгибающий момент в наиболее напряженных сечениях МА при
незначительном сдавливании образца рассчитывают по формуле
^ = -£d„(l-Aj = O,O9i™m. (121)
Напряжение растяжения при этом сг — - ~ ,
где Mi — изгибающий момент;
W — момент сопротивления.
19 Зак. 1338 — 289 —
4
Нагружение образцов путем создания постоянной деформа-
ции применяют в массовых и сравнительно кратковременных
испытаниях. Деформация образцов достигается главным обра-
зом изгибом. ' ' ' ' *
Рассмотренные способы напряжения образцов путем изгиба
создают неравномерное по рабочей длине образца напряжение;
исключение составляют лишь образцы переменного сечения,,
в которых благодаря плавному уменьшению сечеиия с удалени-
ем от места жесткого крепления образца напряжение остается
одинаковым по всей длине. К недостаткам испытаний при по-
стоянной деформации следует также отнести и изменение на-
пряжений по времени, достигающее 30 и даже 50% от исходной
величины (46, 54].
Испытания при постоянной нагрузке
Недостатки испытаний с постоянной деформацией преодоле-
ваются при проведении испытаний с постоянной нагрузкой; на-
пряжение в этом случае осуществляется с помощью приборов
Рис. 159. Прибор Г. В, Акимова {!]
для испытания на коррозию под на-
пряжением при растяжении-:
I — зажим; 2 — коррозионный раствор,
3 — образец; 4 — сосуд; 5 — резиновая
или корковая пробка; б — нагружающий
рычаг
рычажной и пружинной конст-
рукций.
На рис. 169 приведено при-
способление Г. В. Акимова для
испытания на коррозию под
Рис. 170. Приспособление для ис-
пытания образцов на коррозию
-под напряжением при постоянной
нагрузке [39]
напряжением при постоянной нагрузке (1]. Нагружение образцов
производится с помощью рычага и грузов. Образцы для испыта-
ний на этом приспособлении изготавливают из проволоки или
^.290— •
листового материала. Образец 3 подвергается действию просто-
го растяжения; коррозионнный раствор А наливают в сосуд 4;
пробка 5 в нижней части сосуда должна плотно прижимать об-
разец; в некоторых случаях необходимо применять замазку.
Рис. 171. Общий вид ячейки установки ИНК-1
ЦНИИТМАШа для испытаний на коррозию под
напряжением при постоянной нагрузке (56]:
1 — нагружающее устройство; 2 — резервуар с нихро-
мовой обмоткой; 5 — станина; 4 —• металлический стол
В приспособлении, предложенном Е. М. Зарецким [23]
(рис. 170), образцам, укрепленным между двух траверс, сооб-
щается растягивающая нагрузка на машине. Приложенная на-
грузка фиксируется затем с помощью пружии, дисков и гаек,
расположенных на нижней траверсе. О передаче усилий гайкам
судят по началу снижения нагрузки, показываемой динамомет-
ром. Испытания ведут до разрыва напряженного образца в кор-
розионном растворе. Соскакивание верхней траверсы в момент
разрыва образца предупреждается головками шпилек.
19* — 291 —
Специальная установка ИНК-1 для испытаний при постоян-
ной нагрузке разработана в ЦНИИТМАШе [55]. Общий вид од-
ной ячейки этой установки приведен на рис. 171. Установка со-
стоит из четырех самостоятельных ячеек, что дает возможность
Испытывать сразу четыре образца. Нагружающее устройство /,
создающее растягивающие напряжения на образце, представля-
ет собой горизонтальный рычаг первого рода с соотношением
плечей 1 :50 и червячно-винтовой ручной привод. Станина 3
предназначается для монтажа силоизмерительного рычага и на-
74
Рис. 172. Схема машины Института физической химии АН СССР
для коррозионных испытаний под напряжением (56]:
I — подшипники; 2 — трехгранная призма; 3 — серьга; 4 — болты; 5 — стакан;
6 — образец; 7 — зажим; 8 — маховик; 9 — червячный винт; 10 — направляющие
рычаги; // — установочный -винт; 12—-каретка; 13 — груз; 14 — блок; 15 — стрелка;
16 — амортизатор
гружающего устройства. Приспособление для установки образ-
цов представляет собой герметически закрывающийся сосуд для
агрессивной среды, который с помощью специальных штанг вме-
сте с помещенным в иего образцом укрепляется в захваты уста-
новки. Нихромовая обмотка внутри резервуара 2 дает возмож-
ность испытывать образцы при повышенных температурах.
Установка монтируется на столе 4. При разрыве образца или по
истечении установленного срока испытания образец разгружает
ся и приспособление освобождается из захватов.
В Институте физической химии АН СССР для испытания
образцов при постоянной нагрузке применяют специальную ма-
шину [56]; схема этой машины показана на рис. 172. Преимуще-
ство этой конструкции — в возможности приложения нагрузки
единовременно, т. е. мгновенно и плавно с постоянной скоростью,
путем наращивания нагрузки перемещением груза по рычагу
с соответствующей шкалой.
— 292 —
Машина состоит из двенадцати одинаковых ячеек, каждая
из которых изготовлена по принципу рычага первого рода (см.
рис. 172). На одном плече рычага закреплен испытываемый об-
разец 6, на другом груз 13. Осью вращения рычага служит /пер-
пендикулярный ему стержень, свободно вращающийся в Шари
Рис. 173. Схема машины системы ЦНИИ МПС для испытаний на изгиб
и растяжение (57):
1 — плита; 2 — колонка; 8 — поперечная планка; 4 — указатель напряжения; 5 —
натяжная гайка; б — пружина; 7 — штанга; 8 — упорная скоба; 9 —• соединитель-
ная проушина; 10 — шатун; 1.1 — стакан; 12 — валик; 18 — рукоятка; 14 — корпус
подшипника; ,16 — фланец; 16 — ограничитель; 17 — станина передаточного меха-
низма; 18 — динамометр; 19 — валик; 20 — фланец; 21 —- головка центрального
вала; 22 — установочный винт; 23 — шестерни; 24 — редуктор; 26—электромотор;
26 — муфта; 27 — образец для испытания; 28 — аажнмной патрон; 29 — отверстия
для закрепления различных деталей во время испытания
новых подшипниках 1. Нагрузка на образец устанавливается
перемещением груза, подвешенного на шариковой опоре к ка-
ретке 12 по направляющим рычагам 10 при помощи установоч-
ного винта 11. Образец, зажатый в зажимы вместе с укреплен-
ным на нем стаканом, подвешивается серьгой 3 на грань тре-
угольника призмы 2. Чтобы устранить перекос образца, его кре-
пят с помощью двух взаимно перпендикулярных болтов 4.
Напряжение образцов плавно или мгновенно (в течение 2—
— 293 —
3 сек) может осуществляться с помощью червячной передачи
13, 14, состоящей из маховика 8 и винта 9. Для плавного
нагружения рукоятку переносного ключа заменяют шкивом, свя-
занным с. мотором. В этом случае нагрузку, составляющую 15—
20% от предела текучести, можно плавно с постоянной скоростью
наращивать вплоть до разрыва образца.
Для исследования устойчивости материалов при повторно
переменном плоском изгибе и статическом напряжении в Цент-
ральном научно-исследовательском институте Министерства пу-
тей сообщения применяют специальную машину, схема которой
приведена на рис. 173. Машина позволяет моделировать усло-
вия работы паровых котлов. Основная растягивающая нагрузка
создается с помощью пружины 6. Изгиб образца производится
при помощи шатунно-эксцентрикового механизма 10. На обра-
зец 27, имеющий концентратор напряжения в виде канавки, на-
девают специальный стакан, в который наливают коррозионный
раствор. Коррозионным раствором служит 41%-ный раствор
щелочи при температуре кипения и смесь растворов азотнокис-
лых солей аммония и кальция. Раствор подогревается электро-
нагревателем в виде кольца, надетого на стакан.
Проводя испытания малоуглеродистой стали на описанной
машине, Л. С. Лебедева и С. Г. Ведении» {23] установили, чго
трещины в кипящих щелочных растворах вызываются в основ-
ном статическими растягивающими напряжениями, лежащими
вблизи предела текучести. Трещины не образуются, если в рас-
твор щелочи вводить нитрит натрия или иатриевую селитру.
Методы оценки склонности сплавов
к коррозионному растрескиванию
Мегер®, оценки склонности сплавов к коррозионному растре-
скиванию обсуждаются в работах (58, 59]. Обычно склонность
сплавов к коррозионному растрескиванию определяют по вре-
мени, необходимому для появления первой трещины или полно-
го разрушения образца, а также путем сравнения изменения ме-
ханических свойств материала в напряженном и ненапряженном
состоянии за время испытания.
При массовых коррозионных испытаниях показателем корро-
зии обычно служит время до разрушения образца или, как иног-
да говорят, средняя продолжительность жизни образца. Резуль-
таты испытания выражают при этом обычно в виде кривых
зависимости средней продолжительности жизни образцов от
температуры закалки, отпуска, напряжений и т. д. Кривые мож-
но изобразить и в полулогарифмических координатах. Если об-
разцы за данный промежуток времени, принятый в испытаниях
за основу, не растрескались, полезно проверить, произошло ли
в результате воздействия коррозионной среды изменение меха-
нических свойств (оь и 6).
— 294 —
В результате такой обработки необходимо получить относи-
тельно простые показатели, которые должны характеризовать,
о одной стороны, склонность сплава к растрескиванию, а с дру-
гой— скорость развития процесса во времени. Легко показать,
что по результатам испытаний не всегда можно безошибочно
сделать заключение о склонности того или иного сплава к кор-
розионному растрескиванию. Особенно это относится к случаям,
когда одна часть образцов разрушилась, а другая нет. Безоши-
бочная оценка долговечности образцов может быть произведена
лишь тогда, когда материалы сильно отличаются друг от друга
по своей склонности к коррозионному растрескиванию.
Рассмотрим это на примере испытания сплава МА-2. При
изучении влияния термической обработки на склонность сплава
МА-2 к коррозионному растрескиванию были получены результа-
ты, приведенные в табл. 30.
Таблица 30
СКЛОННОСТЬ СПЛАВА МА-2 К КОРРОЗИОННОМУ РАСТРЕСКИВАНИЮ
В ЗАВИСИМОСТИ ОТ ТЕРМИЧЕСКОЙ ОБРАБОТКИ
Темпера- Время _ . Средняя иродолжи-
TVna °C ч Время до разрушения образцов, сутки* * тельность жизни
' ’ образцов, сутки
300 1
350 1
450 1
500 1
35 (—1)** 37 (—2) 43 (— 1) 44 (—1)
37 (—3) 72 (—2)
37 (—2) 40 (—2) 42 (—1)
25 (—2) 42 (—1) 90 (4-2)
* Испытания проводили при переменном погружении в 0.001 %-ный раствор
NaCl, 50 мин на воздухе, 10 мин в электролите.
** Цифра в скобках со знаком минус означает число образцов, разрушившихся
за данный промежуток времени; цифра в скобках со знаком плюс означает число
образцов, не разрушившихся за данный промежуток времени.
Исходя из данных табл. 27, можно утверждать, что, напри-
мер, из двух температур 350 и 500° С наиболее благоприятна
последняя. Что же касается температур 300 и 450° С, то без до-
полнительного анализа отдать предпочтение какой-либо из них
трудно. Обйчно определяют среднюю продолжительность жизни
•образцов и на основании этой цифры судят о склонности сплава
к коррозионному растрескиванию.
Однако необходимо иметь в виду, что сам по себе средний
показатель долговечности не может полностью характеризовать
склонность сплава к коррозионному растрескиванию. Следует
также учитывать развитие процесса во времени, а также про-
цент треснувших образцов.
Предположим, что мы испытали шесть композиций сплавов
с целью выбора сплава, менее склонного к коррозионному рас-
трескиванию, и получили результаты, приведенные в табл. 31.
-г-295 —
Таблица 31
ПРОДОЛЖИТЕЛЬНОСТЬ ЖИЗНИ ОБРАЗЦОВ, ИСПЫТАННЫХ НА СКЛОННОСТЬ
К КОРРОЗИОННОМУ РАСТРЕСКИВАНИЮ, СУТКИ
Д'» пи. Количество образцов, разрушив- шихся за данный промежуток времени Средняя про- должитель- кость жизни образцов и сутки Треснувшие образцы %
10 20 30 40 5
1 1 1 — 1 W 34 60
2 —— 1 4 — 34 100
3 — 2—2 1(+) 34 80
4 — 1 2 1 1 34 100
5 I — 1 2 1(+) 34 80
6 2 — — — 3(+) 34 40
* Плюс в скобках означает, что образец не разрушился.
Из данных табл. 28-. следует, что средняя долговечность об-
разцов для всех шести композиций одинакова (34 дня), но из
этого вовсе не следует, что они все в одинаковой степени склон-
ны к коррозионному растрескиванию. Очевидно, что одним из
основных показателей склонности сплава к коррозионному рас-
трескиванию является процент треснувших образцов. В данном'
случае этот показатель для всех шести сплавов неодинаков и
меняется в широких пределах — от 40 до 100%.
Можно указать на недостаточную обоснованность таких рас-
четов, поскольку не все образцы разрушились, однако, если
ждать полного разрушения всех образцов, мы сталкиваемся с
необходимостью проводить длительные испытания. На практике
это неосуществимо, в особенности, когда мы имеем дело со спла-
вами/ слабосклонными к коррозионному растрескиванию. Какой
же выход существует из этого положения? Очевидно, что, рас-
смотрев показатели, приведенные в табл. 2</ целесообразно про-
следить также за развитием процесса во времени, построив
соответствующие кинетические кривые, например зависимости
количества разрушившихся образцов от времени (рис. 174).
Подобные кривые наглядно показывают развитие процесса во
времени и процент разрушившихся к данному времени образцов.
Площадь, заключенная между кривой и осью абсцисс, которую
можно выразить в процентах от общей площади диаграммы, мо-
жет служить дополнительным показателем, характеризующим
склонность сплава к коррозионному растрескиванию; чем мень-
ше эта площадь (на рис. 174 она заштрихована), тем сплав бо-
лее стоек. На основании этой диаграммы удается дополнительно
дифференцировать сплавы, характеризующиеся одинаковыми
показателями по долговечности и проценту треснувших образ-
цов. Так, например, из диаграммы видно, что сплавы 2 и 4
характеризуются одинаковым процентом треснувших образцов,
но отличаются между собой по развитию процесса во времени.
— 296 —
Поскольку полное разрушение образцов наступает у сплава 2
уже через 30 суток, для него получен и наибольший процент
заштрихованной на диаграмме площади.
То же самое можно сказать и о сплавах 5 и 5. Хотя они
и характеризуются одинаковыми показателями по количеству
разрушившихся до конца испытания образцов (80%) й одина-
Рнс. 174. Зависимость количест-
ва’ разрушившихся образцов от
времени:
1, 2, 3, 4, 5, 6 — сплавы в соот-
ветствии с табл. 34 (с) и склон-
кость сплава АМГ6 к коррозион-
ному растрескиванию (Кр) в за-
висимости от степени деформации
(отпуск 200° С). Коррозия во влаж-
ной камере при распылении
3%-ного раствора NaCl (б)
ковой средней долговечностью (34 дня), тем не менее они долж-
ны несколько отличаться по склонности к коррозионному рас-
трескиванию. Последнее вполне определенно отражается ходом
кинетических кривых и по площади, заключенной между кри-
выми и осью абсцисс. Наибольшая площадь у сплава 3. Если
поделить площадь Fь заключенную между кинетической кривой
и осью абсцисс, на площадь F2, лежащую выше этой кривой,
то можно получить коэффициент, характеризующий склонность
сплава к коррозионному растрескиванию:
Кр = 4-. (122)
— 297 —
В качестве примера приведем данные для алюминиймагние-
вого сплава АМГ6, полученные одним из авторов совместно с
М. А. Новицкой, А. И. Сафроновой и Л. Н. Поляковой
(рис. 174, а). Точность определения склонности к коррозионно-
му растрескиванию по предлагаемой методике может быть резко
повышена, если увеличить количество испытываемых образцов,
например, с 5 до 10. '
Итак, о склонности сплавов к коррозионному растрескиванию
следует судить по нескольким показателям — средней долговеч-
ности образцов, проценту треснувших образцов, наклону кривой
развития процесса во времени и коэффициенту склонности спла-
ва к коррозионному растрескиванию.
При проведении испытаний на машинах появляется возмож-
ность оценить влияние величины приложенных напряжений на
склонность материала к растрескиванию, проследить за разви-
тием деформации образца во времени, а также снимать электро-
химические характеристики металла (потенциал, поляризуе-
мость) под напряжением в процессе испытания и в момент раз-
рыва образца.
Во всех случаях для выяснения характера коррозионных
трещин рекомендуется проводить микроисследование.
3. ОПРЕДЕЛЕНИЕ СКЛОННОСТИ НЕРЖАВЕЮЩИХ
СТАЛЕЙ К ТОЧЕЧНОЙ КОРРОЗИИ
Механизм точечной коррозии нержавеющих сталей
и принципы проведения ускоренных испытаний
Нержавеющие стали, обладающие высокой коррозионной
стойкостью в окислительных средах, легко активируются хлор-
ионами [59—61J. Характерные особенности коррозионного про-
цесса в этих условиях — концентрация коррозионных поражений
на отдельных участках поверхности и появление питтингов с
проникновением коррозии в глубь металла.
Возникновение питтинговой коррозии в процессе эксплуата-
ции изделия представляет большую опасность и поэтому при
определении устойчивости нержавеющей стали- в тех или иных
условиях важным показателем является ее склонность к питтин-
гообразованию.
Так как образование точечной коррозии связаио с наруше-
нием пассивного состояния стали, рассмотрим, от чего зависит
устойчивость пассивного состояния и какие факторы способству-
ют его нарушению.
Устойчивость пассивного состояния нержавеющих сталей
в растворах хлоридов зависит от состава сплава, его структуры,
напряжений, а также от состава электролита. Этот вопрос -под-
робно изучен в работах [62—68].
— 298 —
Свойства того или иного сплава, а также активирующее
влияние среды особенно четко выявляются при анодной поля-
ризации. Это хорошо иллюстрируется кривыми анодной поля-
ризации нержавеющих сталей различного состава, представлен-
ными на рис. 175. Для всех сталей выявляется характерная осо-
бенность, заключающаяся в том, что устойчивая поляризация
наблюдается лишь до определенных значений потенциалов, по
достижении которых сплав переходит в активное состояние. Зна-
чение потенциала, при котором сплав ак-
тивируется в отдельных участках, не оди-
наково для различных сплавов. Этот по-
тенциал может быть назван потенциалом
активирования и характеризует сопро-
тивление сплава активирующему влия-
нию хлор-ионов. Чем положительнее по-
тенциал активирования, тем более устой-
чиво пассивное состояние данного спла-
ва в растворах хлоридов.
Отсюда следует, что склонность нер-
жавеющих сталей к питтинговой корро-
зии может быть довольно быстро выяв-
лена анодной поляризацией.
Основной компонент, препятствую-
щий активированию поверхности ионами
хлора — хром: чем выше его концеитра-
ция, тем более устойчив сплав. Никель
сам по себе довольно легко активируется
ионами хлора, однако введение его в
сплав Fe — Сг резко повышает сопротив-
ление сплава активирующему действию
хлоридов. Никель влияет главным обра-
1.6 -
I—।—1-------1----
О 10 20 ЬО
Плотность тола i
нла/сн2
Рис. 175. Анодная по-
ляризация различных
нержавеющих сталей в
0,1-н. растворе NaCl[67]:
1 — 1X13; 2 - 1Х13М2; 3 —
Х17; 4 - dXlSHST; 5 — Х28;
6 — Х18Н12М2Т; 7 —
Х18Н12МЗТ
зом благодаря изменению структуры сплава. В его присутствии
сталь приобретает аустенитную структуру, обладающую повы-
шенной устойчивостью в растворах хлоридов, что уменьшает
склонность к питтингообразованию. Ферритная сталь Х17 намно-
го слабее анодно поляризуется в растворах хлоридов, чем сталь,
содержащая никель и такое же количество хрома, являющегося
основным элементом, препятствующим активированию.
Повышенная склонность нержавеющих сталей к питтиигооб-
разованию часто является результатом структурных изменений.
Так, например, в работе >[64] было установлено, что различные
партии сталей Х18Н12М2Т при одинаковом химическом составе
активировались в растворах хлоридов при анодной поляризации
по-разному. Наиболее устойчиво сохраняли пассивное состояние
в растворах хлоридов стали с полностью аустенитной струк-
турой. Анодная поляризуемость уменьшалась по мере увеличе-
ния ферритной составляющей. Повторная закалка сталей на
аустенитную структуру снова приводила к устойчивой анодной
— 299 —
поляризации. Поэтому для обнаружения в ускоренных испыта-
ниях повышенной склонности стали к питтингообразованию не-
обходимо проверить, ие произошли ли в сплаве какие-либо струк-
турные изменения в результате неправильной термической об-
работки.
Другие полезные элементы, препятствующие активированию
нержавеющей стали ионами хлора, — молибден и кремний. Это
наглядно видно из поляризационных кривых (рис. 175) для ста-
лей Х18Н12М2Т и Х18Н12МЗТ, содержащих различные концен-
трации молибдена, а также из данных, характеризующих склон-
ность сталей к питтингообразованию (I = 1 мка{см2, 0,1-н рас-
твор NaCl, время 30 ждя).
Марка стали.............1Х18Н9Т XL8H12M2T X18HL2M3T
Количество питтингов на
1 см*.................. 50—60 18—22 12
Легирование молибдеисодержащих сталей кремнием при од-
новременном сохранении аустенитной структуры за счет введе-
ния небольших добавок азота позволяет, как показали И. Л. Ро-
зенфельд и В. П. Максимчук [64J,
получить сплавы, обладающие бо-
лее высоким сопротивлением пит-
тингообразов анию.
Данные, полученные электро-
химическими исследованиями,
подтверждаются и непосредст-
венными опытами по определению
склонности нержавеющей стали
к питтиигообразованию. Как вид-
но из рис. 176, наибольшая веро-
ятность возникновения питтингов
из изученных сталей оказалась
устали 1X13 и .наименьшая — у
стали Х18Н12МЗТ. По средней
глубине питтинга, т. е. по скоро-
сти проникновения коррозии,
стали располагаются в обратном
порядке. Следовательно, чем вы-"
ше вероятность возникновения
точечной коррозии, тем меньше
скорость ее проникновения вглубь. На некоторых сталях (XI7,
1Х18Н9Т) наряду с большим количеством питтингов в отдельных
точках наблюдается относительно сильное проникновение в глубь
металла.
Что касается свойств электролита, то в этом направлении цо-
лучены результаты, указывающие на то, что соответствующим
регулированием его состава, скорости движения и температуры
— 300 —
Рис. 176. Склонность различных
нержавеющих сталей к точечной
коррозии [2% NH^FeCSO^Jg-
. 12Н2О+3% NH«C1] (68]:
1 — XI8H12M3T;
X18H12M2T;
Х18Н15М2Б;
2 — Х28; 3 —
4 ~ .1X18HST; 5 —
6 — Х18Н11Б; 7 — Х17;
8 — JXI3
можно заметно снизить склонность нержавеющих сталей к то-
чечной коррозии.
В растворе хлористого натрия, содержащего кислород, не-
ржавеющая сталь 1Х18Н9Т при анодной поляризации находится
на границе активно-пассивного состояния, что выражается в пе-
риодических колебаниях потенциала, обусловленных активиро-
ванием и пассивированием поверхности (рис. 177, кривая /).
При введении в электролит определенных количеств сульфат-,
хромат-, иитрат-, хлорат- и перхлорат-ионов активация поверх-
Рис. 177. Кривая заряжения стали 1Х18Н9Т
(i = 2 мкаЗслР) [64}:
/ — в 0,1-31. растворе NaCi; Q — ® 0,1-н. растворе
NaCl 4- 0,04-<к. раствор НаНОз
ности хлор-ионами прекращается, и на кривых заряжения исче-
зают периодические колебания потенциала. Получаются типич-
ные кривые, характерные для металла, находящегося в пассив-
ном состоянии (рис. 177, кривая 2).
Предельные отношения концентраций пассивирующего анио
на и активатора, при которых прекращается активация сплава
1Х18Н9Т хлор-ионами (Смах •' Скаа; pH = 7), таковы: SO|-10;
CrOf- 7; NOj- 0,4; CIO7 2; ClOf 0,5.
По пассивирующей способности, определяемой отношением
концентрации пассиватора и активатора, при котором сплав пе-
рестает активироваться, анионы могут быть расположены
в следующий ряд:
МОГ > СЮ? > С1ОГ > СгОГ > SO|~
Из полученного ряда вытекает важный вывод, заключающий-
ся в том, что пассивирующие свойства анионов не определяются
однозначно их окислительной способностью. Самым эффектив-
ным пассиватором против точечной коррозии для сплава
— 301 —
1Х18Н9Т оказался анион NO-f, обладающий значительно мень-
шей окислительной способностью в нейтральных средах, чем
ионы CrOf- СЮ7 и C1OJ-. Ион SO|~, не обладающий окисли-
тельными свойствами и не образующий труднорастворимых
соединений ни с одним из компонентов стали, дает примерно та-
кой же эффект, как СгО|", являющийся сильным окислителем.
Такое поведение анионов (см. ниже) обусловлено тем, что
их пассивирующие свойства определяются ие только окисли-
тельными свойствами, но и способностью адсорбироваться на по-
верхности стали и вытеснять с нее хлор-ионы. Важное значение
при этом имеет строение самого аниона.
Пассивирующие свойства анионов сильно зависят от pH сре-
ды, на что указывают кривые заряжения сплава 1Х18Н9Т, сня-
тые в щелочных и кислых растворах при постоянной концентра-
ции хлор-ионов.
Пассивное состояние стали нарушается хлор-ионами легче
в кислом электролите, чем в нейтральном, потому что периоди-
ческие колебания потенциала начинаются в кислом электролите
при более отрицательных значениях потенциала (на 0,2 в). Ам-
плитуда колебания потенциала в кислом электролите меньше,
чем в нейтральном (0,35—0,60 против 0,55—0,90).
Следует отметить, что, несмотря на сильное активирующее
действие хлор-ионов в кислых электролитах, пассивирующие
свойства такого аннона, как SO|~, проявляются сильнее в кис-
лом электролите, чем в нейтральном.
Ниже приведены данные о влиянии pH на предельное отно-
шение концентраций NasSO4: NaCl, при котором прекращается
активирующее действие ионов хлора:
pH ............... 2 7 12
Отношение концентрации 2 10 2
В кислом и щелочном электролитах требуется в пять раз
меньше сульфата, чтобы подавить активирующее действие хлор-
ионов.
Важной характеристикой сплава является потенциал активи-
рования. Учитывая, что активирующее действие хлор-ионов про-
является начиная лишь с определенного значения потенциала,
важно определить это предельное значение, чтобы, например,
катодной поляризацией или введением веществ, смещающих по-
тенциал в отрицательную сторону, перейти эту опасную границу.
Судя по кривой, представленной на рис. 178, а также на рис. 177
(кривая /), потенциал активирования стали 1Х18Н9Т равен
в обескислороженном электролите +0,2 в, а в электролите,
содержащем кислород, +0,3 в. Таким образом, если поддержи-
вать потенциал стали на уровне, несколько более отрицательном,
чем указанные значения, можно полностью исключить активи-
рующее действие хлор-ионов. Этот вывод подтвержден экспери-
— 302 —
ментально; электрохимическая защита предотвращает возник-
новение питтинговой коррозии.
Смещение потенциала в положительную сторону (за крити-
ческое значение) должно при отсутствии пассивирующих анио-
нов привести к активирова-
нию поверхности. При этом
безразлично, из-за чего сме-
щается потенциал стали —
вследствие анодной поляри-
зации или изменения состава
электролита. Подтверждени-
ем изложенного могут слу-
жить результаты, характери-
зующие изменение потенциа-
ла стали 1Х18Н9Т во време-
ни (рис. 179). При отсутст-
вии анодной поляризации
потенциал стали 1Х18Н9Т в
0,1-н. растворе NaCl относи-
тельно устойчив, поскольку
он более отрицателен, чем
+0,3 в, и поверхность, стало
быть, не может активиро-
ваться хлор-ионами. Однако
стоит сместить потенциал
стали в положительную сто-
рону, что достигается введе-
нием перекиси водорода
(0,3%), как он становится
неустойчивым; начинает про-
являться активирующее дей-
ствие хлор-ионов. После вве-
дения в электролит Na2SO4
в таком количестве, чтобы
получить 1-н. раствор, коле-
бания потенциала прекра-
щаются. Он устанавливает-
ся на уровне 0,62 в, совпа-
дающем со значением, при-
' Рис. 178. Кривая заряжения ста-
ли 1Х18Н9Т в обескислороженном
0,1-н. растворе NaCl (i —
~ 2 мка/см2} [64]
0.0
>________1 I L
0 30 i 60 90 120
время,huh
Рис. 179. Изменение стационарно-
го потенциала-стали 1Х18Н9Т в
растворе 0'5-н. NaCl+0,3 % Н2О2
во времени [64]. Стрелкой указан
момент введения 1-н. Na2SO4 [64]
обретаемым электродом в 1-н. растворе Na2SO4 + Н2О2 (0,3%N
не содержащем хлор-ионов.
Процессы возникновения и развития питтинговой коррозии
на нержавеющей стали были подробно изучены И. Л. Розен
фельдом и И. С. Даниловым [68, 69]. Некоторые результаты
этих работ, имеющие отношение к проблеме ускоренных испыта-
ний, приводятся ниже. На рис. 180 представлены кривые, харак-
теризующие зависимость питтинговой коррозии (числа питтин-
гов, их средней и максимальной глубины) от концентрации
— 303 —
окислителя и активатора. На рисунке видно (кривая /), что вна-
чале по мере увеличения концентрации окислителя число пит-
тингов резко возрастает, достигает максимума, а затем начи-
нает падать. Таким образом, окислитель играет двойственную
роль: при малых концентрациях он способствует активированию
процесса во многих точках поверхности, а при больших — чис-
ло активных центров уменьшается. Средняя глубина питтинга
также растет лишь до определенной концентрации окислителя.
Аналогичная зависимость наблюдается и при изменении кон-
центрации активатора, однако в количественном отношении мак-
Рис. 180. Развитие питтинговой коррозии на стали IXI8H9T:
а — под влиянием пассиватора NHsFeCSOJa • 12H/D в 0,5%-ном рас-
творе NHiCl; б — под влиянием активатора NH«CI в 0,5%-ном рас-
творе NH4Fe(SO4)s • I2H«O. Продолжительность 25 ч:
1 — число питтингов; 2 — средняя глубина питтингов; 3.— макси-
мальная глубина питтингов [68]
симум (по количеству возникающих питтингов) выявляется не-
четко. Если количество возникающих питтингов и средняя их
’глубина начиная с некоторых концентраций окислителя и акти-
ватора начинают падать, то в отдельных точках, где процесс
развивается с небольшой скоростью, этого не наблюдается. Харак-
терно то, что вероятность появления точечной коррозии (число
питтингов) в электролитах с высокими концентрациями пасси-
ваторов намного выше, чем в электролитах с высокими концент-
рациями активаторов.
Исходя из изложенных выше закономерностей, соотношение
между концентрациями окислителя и активатора в электроли-
тах, применяемых для ускоренных испытаний, должно быть
вполне определенным. При чрезмерно большом содержании окис-
лителя и активатора вероятность выяНления склонности нержа-
веющих сталей к точечной коррозии уменьшается.
При использовании смесей железо-аммиачных квасцов и хло-
ристого аммония наиболее полно склонность сталей к точечной
коррозии выявляется при содержании 2% NH4Fe(SO4)2-2H2O
и 3% NH4C1 [69].
— 304 —
При изучении склонности нержавеющих сталей.к питтинговой
коррозии немалое значение имеет метод построения кривых рас-
пределения коррозионных поражений. Если разделить все возни-
кающие на поверхности питтинги по размерам, то получится
типичная кривая распределения, указывающая на то, что про-
цесс в отдельных точках развивается исключительно неравно-
мерно (скорости могут отличаться в 50—100 раз). На поверх-
ности нержавеющей стали, как правило, возникает большое
Рис. 181, Кривые распределения
питтингов по глубине для стали
1Х18Н9Т [68]:
О 0.25 0.50 0,75 1,0 1,25
Глубина питтингов, им
Рис. 182. Кривые распреде-
ления питтингов по глубине
для стали 1Х18Н9Т при раз-
личном времени воздейст-
вия электролита, содержа-
щего 2% пассиватора и
3,0% активатора [68]
1 — 0.5% NH4Ci + 0,5% NH4Fe(SO<)s • 12ЩО;
2 - 0.5% NH.C1 + 2,0% NH4Fe(SO,)2 12HSO;
3 — 0,5% NH.Ci + 8,0% NH4Fe(SO4)s - 12НгО
число питтингов средней глубины и небольшое число малой и
средней глубины (рис. 181 и 182). С увеличением концентрации
окислителя кривые распределения в целом сдвигаются влево, за
исключением первых ветвей, сдвигающихся вправо. Это указы-
вает на то, что при увеличении концентрации окислителя про-
цесс коррозии в большинстве точек замедляется и на поверхности
остается лишь небольшое число активных центров (~2—3%),
в которых процесс развивается интенсивно.
В этих активных центрах наблюдается непрерывное увели-
чение глубины питтинга с ростом концентрации окислителя и ак-
тиватора (рис. 180, кривая 5).
Для ускоренных испытаний представляет определенный ин-
терес и развитие процесса во времени. Опыты показывают, что
питтинги зарождаются лишь в начальный период испытания
(в первые 15—30 мин), после чего количество их не увеличи-
вается. Это объясняется тем, что каждый возникший питтинг
резко уменьшает вероятность появления питтингов в других слач
20 Зак. 1338 305 —
бых местах, поскольку он играет роль точечного протектора.
защищающего остальную поверхность от коррозии.
Кривые распределения питтингов по глубине (см. рис. 182)
показывают, что не все питтинги, зародившиеся на поверхности,
развиваются во времени равномерно. В большинстве точек про-
цесс во времени замедляется, часть мелких питтингов вовсе
перестает расти и лишь в небольшом числе активных центров
о 5 ю 15 го
время, v
Рис. 183. Зависимость коэффициента
неравномерности коррозии нержавею-
щих сталей от времени воздействия
электролита [2,0% NH4Fe( SO4)2.
. 12Н2О + 3,0% NH4C1)1 [68]:
/ - 1Х18Н12МЗТ; 2 — X18H12M2T; 3 -
Х28; 4 — IX18H9T; 5 — Х18Ш5М2Б; б -
Х18НПБ; 7 — Х17; 3 — 1X13; la, 4а. За-г
по средней глубине питтингов
коррозия непрерывно развива-
ется в глубь металла.
Поэтому при ускоренных
испытаниях необходимо обяза-
тельно определить скорость
проникновения коррозии в на-
иболее активных центрах, ибо
в конечном счете именно они
определяют поведение аппара-
туры в эксплуатации.
Другая важная характери-
стика при ускоренных испыта-
ниях, выявляющая склонность
нержавеющих сталей к питтин-
говой коррозии, — определение
коэффициента неравномерно-
сти. Он характеризует отноше-
ние глубины двух-трех наибо-
лее глубоких питтингов к сред-
ней глубине питтингов, изме-
ренных непосредственно или
определенных на основе по-
терь массы. Как было показа-
но в гл. Ш, он косвенно харак-
теризует площадь, занятую
коррозией. С одной стороны,
этот показатель свидетельству-
ет о степени опасности этого вида коррозии, а с другой, если
изучать его изменение во времени, можно получить представле-
ние о том, в каком направлении преимущественно развивается
процесс: в глубь металла нли коррозия распространяется и по
поверности.
Исследования нержавеющих сталей в смеси железо-аммиач-
ных квасцов и хлористого аммония показали, что коэффициент
питтингообразбвания во времени падает (рис. 183). Число пит-
тингов уже после 15—30 мин остается практически одинаковым.
Это указывает на то, что со .временем питтинги начинают расти
вширь.
Было также установлено [68], что склонность нержавеющих
сталей к питтинговой коррозии сильно зависит от температуры
отпуска стали и состояния ее поверхности. Температурные воз-
— 306 —
действия, способствующие возникновению межкристаллитной
коррозии, вызывают и сильную питтинговую коррозию. Поэтому
изучение этих факторов при ускоренных испытаниях сталей в
различных средах должно также находиться в поле зрения ис-
следователя.
Рассмотрим, в чем заключается механизм питтинговой кор-
розии. При наличии в коррозионной среде хлор-ионов становит-
ся возможным активирование поверхности в отдельных ее точ-
ках, где пассивное состояние по каким-либо причинам менее
устойчиво, чем на остальной части поверхности. Такими участ-
ками могут быть: неметаллические включения, структурные де-
фекты или участки с менее совершенной фазовой или адсорбци-
онной пленкой, границы зерен и т. д. На этих участках хлор-ноны
относительно легко вытесняют кислород с поверхности и спо-
собствуют началу развития коррозии.
Коррозионный процесс, начавшись в отдельной точке, резко
уменьшает, как будет показано ниже, вероятность своего появ-
ления в других точках. Поскольку значительная часть поверх-
ности находится в пассивном состоянии, возникает мощный кор-
розионный элемент, в котором анодом является питтинг (один
или несколько), а катодом — остальная часть поверхности. Раз-
ность потенциалов в таком коррозионном элементе достигает
0,5—0,6 в.
Для того чтобы такой элемент мог эффективно работать, сле-
дует соблюдать ряд условий. Во-первых, необходимо, чтобы в
системе был деполяризатор, который восстанавливался бы с
большой скоростью н мог поддерживать течение анодной реакции
в питтинге. Обычно таким деполяризатором является кислород
или другой окислитель. Некоторые исследователи полагают, что
чем больше концентрация окислителя, тем выше будет интен-
сивность точечной коррозии. На самом деле, как было показано,
существует какая-то оптимальная концентрация окислителя,
зависящая от концентрации активатора, при которой наблюдает-
ся максимальная питтинговая коррозия. Это объясняется тем,
что увеличение концентрации окислителя не только усиливав?
скорость катодного процесса, но одновременно уменьшает веро-
ятность нарушения пассивного состояния в слабых местах. Кроме
того, даже в большинстве точек, где началась питтинговая корро-
зия, при наличии высокой концентрации окислителя процесс ср
временем приостанавливается, очевидно, в результате частичной
анодной пассивации. Лишь в тех точках, где возникающая анод-
ная плотность тока недостаточна для того, чтобы сильно замед-
лить анодный процесс, коррозия с увеличением концентрации
окислителя продолжает непрерывно расти вследствие большой
скорости катодного процесса.
Второе необходимое условие развития питтинговой кор-
розии— постоянное нахождение питтингов в активном состоя-
нии. Это условие соблюдается в связи с .тем, что преимущесж-
20* — 307 —
"е(0н)„ ne(0Hin
Рис. 184. Схематическое
изображение питтинга:
а — открытого;
б — закрытого |69. 72]
венное развитие в питтингах анодных реакций, протекающих
с большой скоростью, приводит к заметному подкислению кор-
розионной среды вследствие выпадения гидратов. Непосредствен-
ные измерения pH в питтингах показывают, что эта величина
часто достигает 3—4 единиц.
И, наконец, третье важное условие стабильного протекания
интенсивной коррозии в питтинге — это, по мнению Франка [70],
«охранение заметной разности потенциалов между питтингами
и остальной частью поверхности.
И. Л. Розенфельд и И. С. Данилов [69] в последнее время
по-новому трактуют механизм питтинговой коррозии. Они вы-
сказали идею о том, что питтинго-
вую коррозию следует рассматри-
вать как особый вид щелевой кор-
розии и интерпретировать ее, исходя
из теоретических представлений о
щелевой коррозии, развитых в рабо-
тах [71]. В обычно принятых пред-
ставлениях [72] питтинг рассматри-
вается как относительно глубокое
открытое точечное разрушение спла-
ва (рис. 184). На самом же деле,
как показывают исследования, пит-
тинги, возникающие на нержавею-
щих сталях в самых разнообраз-
ных электролитах [NHjFe'CSO^s *
• 12Н2О 4- NH4C1; FeCl3 • 6Н2О;
Fe2(SO4)3; NaCl], представляют со-
бой, как правило, относительно за-
крытые очаги коррозии. Оказывает-
ся, что хлор-ион проникает под пленку или поверхностный слой
металла, и процесс коррозии развивается в закрытой области
под пленкой, как это изображено иа рис. 184, б. При таком ха-
рактере развития процесса затрудняется доступ в питтинг ки-
слорода или другого пассиватора, необходимого для поддержа-
ния пассивного состояния.
Уменьшение концентрации пассиватора в питтинге выводит
его стационарный потенциал за пределы потенциала полной пас-
сивации, что сопровождается потерей металлом, находящимся
в питтинге, пассивного состояния и ускорением анодной реакции
ионизации металла.
Таким образом, уже с самого начала зарождения питтинг
представляет собой своеобразную щель, в которой из-за недо-
статочного доступа пассиватора создаются благоприятные ус-
ловия для развития с большой скоростью преимущественно анод-
ных процессов.
Очевидно, что если питтинговая коррозия, представляет осо-
бый вид щелевой коррозии, в которой особенности электрохими-
— 308 —
ческого поведения металла обусловлены, как известно, лишь
различием в доступе коррозионной среды и отводе продуктов
коррозии, то при обеспечении одинакового доступа электролита
к питтингу и к остальной части поверхности питтинговая кор-
розия должна прекратиться. Опыты, поставленные с этой целью,
подтвердили предположения. Оказалось, что если вскрыть воз-
никшие на поверхности металла
питтинги и этим самым обеспечить
к ним свободный доступ электроли-
та, то они перестают расти, в то вре-
мя как невскрытые питтинги, куда
доступ электролита затруднен, про-
должают расти непрерывно
(рис. 185).
Интересные результаты были по-
лучены и при изучении числа цент-
ров, в которых могло происходить
активирование сплава и могли воз-
никать питтинги. Обычно считают,
что на поверхности нержавеющих
сталей имеется какое-то ограничен-
ное число точек, обусловленное
структурными особенностями спла-
f| 0.50
Рис. 186. Зависимость количества
питтингов, возникающих на стали
1Х18Н9Т, от времени воздействия
электролита [69]:
I —• питтинги ие вскрывали; 2 — воз-
никавшие после каждого цикла (0.5;
1.0; 2,5; 5.0; 7.5; 10 ч) питтинги вскры
вали; 3 — возникавшие через каждые
15 мин питтинги вскрывали (электро-
лит: 20 г/дм* NH,Fe(SO4)2 - 12HSO +
+ 30 г/дм* 4 — в том же
растворе, содержащем 11.5 г/дм?
FeCh • 6HaO + 25,5 г/дм* NaCl
ореня.ч
Рис. 185. Зависимость глубины пит-
тингов на стали 1Х18Н9Т от времени
воздействия (20 г/длг3 NH4Fe(SO4)2 •
•12Н2О + 30 г/дм? NH4Cl):
/ — вскрытых питтингов (после 30
2 — невскрытых питтингов
ва, в которых может возникнуть питтинг (73]. Опыты показали,
что это не совсем так; число питтингов может непрерывно воз:
растать по мере вскрытия ранее образовавшихся питтингов
(рис. 186). Обычно основная часть питтингов возникает в пер-
вые 15—30 мин и, если их не вскрывать, новые питтйнги, как
правило, не появляются (рис. 186, кривая /). Объясняется это
тем, что каждый возникающий вначале питтииг можно рассмат-
ривать как точечный протектор, сильно уменьшающий вероят-
ность возникновения питтингов в других точках поверхности.
Если, однако, возникающие после каждого цикла питтинги
— 309 —
вместо ~400, возникающих, когда
0,3 Г За
О 2 Ь 6 6 10
всем я. ч
вскрывать и этим самым прекращать их рост, на поверхности
нержавеющих сталей появляются все новые и новые питтинги
(рис. 186, кривая 2).
Если сильно сократить время опыта и- каждый раз вскрывать
вновь возникающие питтинги, на поверхности металла образует-
ся все большее число питтингов. Оно достигает 5700 на 1 дм2
питтинги не вскрывали. На-
блюдается йрямолинейная
зависимость между количе-
ством возникающих на по-
верхности питтингов и вре-
менем (рис. 187, кривые 5,
4). Таким образом, должно
быть поставлено под сомне-
ние положение о наличии на
поверхности металла какого-
то ограниченного числа
центров, чувствительных к
питтинговой коррозии. На
самом деле их очень много,
и лишь благодаря протек-
торному эффекту от ранее
возникших питтингов новые
питтинги не возникают.
Изучение кинетики про-
никновения коррозий в
глубь металла показало, что
при вскрытии ранее образо-
вавшихся питтингов сильно
уменьшается их глубина.
При выполнении этой опе-
рации в начальной стадии
процесса можно прекратить
рост большинства питтингов и сильно ограничить глубину про-
никновения коррозии..
Исходя из рассмотренного выше механизма питтинговой кор-
розии и установленных закономерностей, представляется воз-
можным наметить наиболее рациональные пути борьбы с
питтинговой коррозией и проведения ускоренных методов испы
таний.
Эффективным методом борьбы с питтинговой коррозией дол-
жно оказаться (там, где это возможно) применение сталей с по-
вышенным содержанием хрома, являющимся основным элемен-
том, препятствующим активированию хлор-ионами поверхности
нержавеющих сталей.
По результатам наших исследований, молибден- также резко
уменьшает вероятность появления питтинговой коррозии и чис-
ла питтингов на поверхности, но не уменьшает глубину тех
— 310 —
Рис. 187. Влияние времени на глуби-
ну проникновения питтинговой корро-
зии на стали 1XI8H9T [69]:
1. 1а — средняя и максимальная глуби-
на невскрывавшихся питтингов; 2, 2а —
средняя и максимальная глубина вскры-
вавшихся питтингов при неравномерных
циклах; 3, За — то же, при равномерных
циклах (электролит: 20 г/дм3 NH«Fe(SO«)s *
• 12Н9О 4- 30 г/дм3 NH,Cl); 4, 4а — сред-
няя и максимальная глубина вскрывав-
шихся питтингов при равномерных циклах
(электролит: 11.5 г/ом3 FeCle-6H2O 4-
+ 25,5 г/дм3 NaCl)
единичных точек, где процесс начал развиваться. По другим
данным, молибден уменьшает и глубину питтингов.
Хорошие результаты также должны дать технологические
процессы, приводящие к получению листа без дефектов, посто-
ронних включений, и правильная термическая обработка, исклю-
чающая появление склонности к питтинговой коррозии (закал-
ка с 1050—1100° С).
По данным Улига [74], обработка нержавеющих сталей пос-
ле окончательной отделки в 10—20 %-ной азотной кислоте при
55—60° С (в течение 15—30 -мин) способствует ’ устранению
скрытых дефектов. Такой обработке может предшествовать
предварительное травление стали в смеси, состоящей из
10 %-ной концентрированной азотной кислоты и 1—2 %-ной пла-
виковой кислоты.
Резко уменьшить питтинговую коррозию можно также, воз-
действуя на коррозионную среду. Там, где это возможно, необ-
ходимо исключить попадание в нее хлор-ионов или довести их
концентрацию до минимальной. Повышение окислительной спо-
собности растворов, способствующее устойчивости пассивного
состояния, а также увеличение скорости движения электролита
и равномерного его доступа к различным частям конструкции
также должны вести к уменьшению питтинговой коррозии. Ис-
ходя из этих же позиций, следует избегать застойных плохо
аэрируемых зон.
Благоприятные результаты дает повышение pH растворов и
введение в коррозионную среду пассивирующих анионов, кон-
центрация которых зависит от концентрации активатора.
Ниже приведено отношение концентраций ингибиторов и
хлористого натрия, при которых активирующее действие хлор-
ионов на сталь 1Х18Н9 не проявляется [67].
Ингибитор
. . Хромат Нитрат
Перхлорат Хлорат
pH .......... 8,5 7,0
Отношение кон-
центраций . 6—7 0,4
7,0 7,5
2,0 0,5
Сульфат
7,'0~2Д) 12,"о
9—10 2,0 2,0
Снижение температуры электролита уменьшает склонность
сталей к питтинговой коррозии и глубину ее проникновения.
Учитывая, что нарушение пассивного состояния нержавеющих
сталей хлор-ионами зависит от потенциала, весьма эффектив-
ной оказывается электрохимическая защита, удерживающая
потенциал стали на более отрицательном уровне, чем значение
критического потенциала (+0,3 в для электролита, содержаще-
го кислород, и +0,2 в для обескислороженного электролита).
Опыты показывают, что эксплуатация нержавеющих сталей в
море в контакте с обычными малоуглеродистыми сталями,
имеющими потенциал ~ —0,3 в, полностью исключает питтин-
говую коррозию нержавеющих сталей. .' t
—811
Ускоренные методы испытаний для выявления склонности
сплавов к точечной коррозии должны основываться на смеще-
нии потенциала в положительную сторону от критических зна-
чений. Это может быть-достигнуто посредством анодной поля-
ризации или введением в электролит окислителей.
На первом принципе основаны метод Бреннерта [75] и метод
кривых заряжения, предложенный И. Л. Розенфельдом и
В. П. Максимчуком [76], на втором — испытания в хлорном же-
лезе и в смеси железо-аммонийных квасцов с хлористым аммо-
нием [69—76], рассматриваемые ниже.
Методы определения склонности нержавеющих сталей
к точечной коррозии
Метод Бреннерта [75] заключается в поляризации исследу-
емого образца‘анодным током при одновременном измерении
Рис. 188. Установка для определения склонности
нержавеющих сталей к точечной коррозии [75]:
/ — исследуемый образец; 2 — кольцо из нержавеющей
стели; 3 — носик каломельного электрода специальной
конструкции; 4 — стеклянный цилиндр; 5 — 1%-ный
раствор NaaSO.» для' предотвращения выделения хлора;
6 — коррозионный раствор; 7 — платиновая проволока;
S — пористые сосуды; $ — насыщенный каломельный
электрод; 10 — потенциометр
электродного • потенциала образца. При достижении определен-
ного для данного состава стали значения потенциала наступает
разрушение защитной пленки на образце, в одной или несколь-
— 312 —
ких точках, вследствие чего наблюдается падение потенциала.
Максимальное значение потенциала, при котором начинается
резкое изменение его, получило название «потенциала пробоя».
Схема установки Бреннерта приведена на рис. 188. Как видно
на рисунке, поляризация образца осуществляется в электриче-
ском поле с помощью двух электродов, между которыми поме-
щается исследуемый образец. Ток пропускается в направлении^
при котором образец поляризуется анодно. С помощью такой
установки получают кривые зависимости потенциала и тока от
времени (рис. 189) или строят кривую потенциал — приложен-
ное напряжение; в последнем
пробоя». По полученным та-
ким образом кривым судят о
склонности сталей к точечной
коррозии. На крнвых потен-
циал—-'время и ток — время в
момент образования питтин-
га появляется резкий перегиб,
потенциал становится сильно
отрицательным, а величина то-
ка возрастает. Этот метод поз-
воляет просто и быстро вос-
произвести точечную корро-
зию. Недостаток метода —
сложность монтирования об-
разца в горизонтальном поло-
жении на определенном рас-
стоянии от тубуса сосуда.
Г. В. Акимов и X. Б. Кларк
[1] предложили более простую
установку, в которой преду-
случае определяют «потенциал
кого потенциала и тока от вре-
мени при испытании нержавею-
щей стали на склонность к то-
чечной коррозии [75]:
— потенциал, соответствующий
моменту образования питтинга
смотрено вертикальное расположение образцов в строго фикси-
рованном положении; кроме того, величина поляризующего об-
разец тока изменяется реостатом не от руки, а с помощью мотор-
чика Уорена, соединенного с подвижным контактом на реохорде
(рис. 190). После погружения исследуемого образца в электро-
лит (обычно 0,1-н. раствор NaCl) и выдержки его в течение 20—
30 мин. до установления постоянного значения потенциала про-
изводят анодную поляризацию образца возрастающим током.
Авторы считают, что этим методом можно выявить не только
влияние изменения состава стали на склонность к точечной кор-
розии, но и влияние термической обработки.
И. Л. Розенфельд и В. П. Максимчук (76] предложили элек-
трохимический метод определения склонности нержавеющих
сталей к точечной коррозии, заключающийся в снятии кривых
заряжения на установке, приведенной на рис. 191. Установка
позволяет измерять и автоматически записывать электродный
потенциал стали при погружении в электролит.
— 313 —
В качестве регистрирующего прибора 1 использовали потен-
циометр ЭПП-09, на вход которого для увеличения входного со-
противления включали усилитель 2. Для увеличения пределов
Рис. 190. Установка для определения склонности стали к точечной
коррозии (вертикальное расположение образца) [1];
1 — пористые стаканы; 2 — вспомогательные платиновые электроды; 3 — образец;
4— внутренний сосуд с тубусом; 5 — каломельный электрод
измеряемого напряжения на вход усилителя включали высоко-
омный делитель напряжения. В качестве электрода сравнения
применяли каломельный электрод 4. Для того чтобы получать
значения испытуемого элек-
Рис. 191. Схема установки для
снятия кривых заряжения при
автоматической записи электрод-
ного потенциала -стали [76]
трода непосредственно по во-
дородной шкале, в цепь кало-
мельного электрода включали
компенсирующую э. д. с., рав-
ную потенциалу каломельного
электрода. Поверхность элек-
тродов, армированных в поли-
стирол, составляла 0,5 см2,
Для стабилизации плотности
тока последовательно с ячей-
кой 5 включалось большое со-
противление. Каломельный
электрод соединялся с ячей-
кой соединительным ключом 3.
Полученные иа этой установке
кривые заряжения (см. рис. 177) показывают, что при неболь-
шой скорости подачи ленты (120 мм/ч) можно судить о пределах
колебаний потенциала, при большой скорости подачи ленты
(4800 мм}ч) — о частоте и форме колебаний потенциала. Часто-
та и границы колебаний потенциала -характеризуют устойчи-
вость стали против точечной коррозии. Наибольшей устойчиво-
— 314 —
сгью будет обладать сталь с наименьшим числом колебаний по-
тенциала в единицу времени и наибольшими пределами измене-
ния колебаний. С помощью этого метода можно оценивать и
эффективность защитного действия ингибиторов, применяемых
для борьбы с точечной коррозией. Если сталь не склонна к то-
чечной коррозии, получается кривая заряжения, характерная
для абсолютно стойкого металла без каких-либо видимых коле-
баний потенциала (см. рис. 177, кривая 2).
Данные, полученные этим методом для нержавеющих сталей
в 0,1-н. растворе NaCl при анодной поляризации (Z = 2 мка/см2),
приведены ниже:
Марка стали............. 1Х18Н9Т
Число колебаний потенциалов в 1 мин 11—14
Пределы изменений потенциалов, е . 0,55—0,95
Х18Н12М2Т Х18Н12МЗТ
6—7 4—5
0,65-1,05 0,70—1,25
Из этих данных видно, что число колебаний потенциала
уменьшается при переходе от стали 1Х18Н9Т и Х18Н12МЗТ.
В таком же порядке сдвигаются в положительную сторону верх-
няя и нижняя границы значений потенциалов, а также пределы
его изменения.
Склонность указанных выше сталей к точечной коррозии в
реальных условиях также уменьшается по мере перехода от
стали 1Х18Н9Т к стали 1Х18Н12МЗТ.
Другая группа методов Ускоренных испытаний склонности
сталей к точечной коррозии основана, как уже указывалось,
на принципе повышения окислительной способности растворов.
Для этой цели в растворы активаторов вводят различные
окислители.
В методе, предложенном Смитом [73], образцы нержавеющей
стали помещают в 0,05%-ный раствор НС1, содержащий
10,8% FeCl3. Так как Fe3+—более активный деполяризатор»
чем кислород, то потенциал смещается выше критических значе-
ний, при которых усиливается адсорбция ионов хлора, активи-
рующих поверхность. Обычно бывает достаточно 4-ч выдержки
в этом растворе, чтобы сравнить поведение различных сталей.
По окончании испытания определяют потерю массы, количест-
во и глубину коррозионных поражений.
И. Л. Розенфельд и И. С. Данилов {69] при изучении склон-
ности сталей к точечной коррозии использовали раствор, состоя-
щий из хлористого аммония и железо-алюминиевых квасцов.
Хлористый аммоний создавал необходимую концентрацию ио-
нов хлора, активирующих поверхность сталей, а железо-алю-
миниевые квасцы, содержащие ионы Fe3* обладая окислитель-
ными свойствами, сообщали среде относительно высокий окис-
лительно-восстановительный потенциал. Опыты проводили на
установке, схема которой приведена на рис. 12 (см. гл. II). Ис-
пытания вели в течение 1—25 Наиболее благоприятное соот-
ношение концентрации окислителя и активатора при ускорен-
— 315 —
ных испытаниях создается при содержании в растворе
2-% NH4Fe(SO4)2- 12Н2О и 3% NH4C1.
Штоффельс и Швенк [77] при изучении склонности нержа-
веющих сталей к питтингообразованию применили раствор, со-
держащий около 1 г KsFe(CN)6 в 100 см* воды, и хлористый
натрий, концентрация которого менялась от 5,0 до 0,001 -н. При
взаимодействии ионов Fe(CN)|~~c ионами Fe2+ образуется сое-
динение ярко-голубой окраски. Поэтому места возникновения
питтингов в таком электролите четко выявляются.
4. ОПРЕДЕЛЕНИЕ УСТОЙЧИВОСТИ МЕТАЛЛОВ
В УСЛОВИЯХ КАВИТАЦИИ '
При больших скоростях движения электролитов наблюдает-
ся особое разрушение металла, возникающее благодаря сов-
местному воздействию электрохимических и механических фак-
торов.
Разрушающая сила потока жидкости зависит от скорости ее
движения, угла наклона струи к поверхности металла и наличия
в ней пузырьков воздуха. Этот вид разрушения встречается на
гребных винтах, в насосах, гидротурбинах, трубопроводах и на
многих других изделиях. Жидкой средой является обычно мор-
ская или речная вода, действие которой усиливается, если в ней
содержатся различные механические примеси.
Некоторые исследователи различают два вида разрушения
металла под влиянием движущейся жидкой среды: коррозион-
но-эрозионное, когда механическое воздействие среды сводится
к разрушению защитных пассивных пленок или продуктов кор-
розии, и разрушение механическое, которое сводится уже к раз-
рушению структуры самого металла. Одни исследователи ос-
новную роль отводят механическому фактору [78—80], другие
коррозионному [81—83].
Очевидно, в зависимости от условий работы конструкции со-
отношение механического и коррозионного факторов может из-
меняться. До некоторой величины скорости потока жидкости
влияет главным образом коррозионная устойчивость металла,
выше этого предела основным является механический фактор.
Если сила удара не создает напряжений, превосходящих предел
текучести металла, эрозия способствует развитию усталости в
металле, образованию линий сдвигов и усилению коррозии по
ним, а также возникновению усталостных трещин. Если сила
ударов создает напряжения, превосходящие предел текучести,
эрозия разрушает металл непосредственно и роль коррозии сни-
жается, но не устраняется.
Как отмечают И. Н. Богачев и Р. И. Минц [84], в представ-
лении о механизме кавитационного разрушения, следует исхо-
дить из двух основных воздействий на металл — эрозии и кор-
— 316 —
розни. Механизм кавитационного разрушения можно представить
следующим образом. В результате развития кавитации под воз-
действием гидравлических ударов поверхность металла в от-
дельных участках деформируется и подвергается наклепу. По-
являются линнн сдвига н скольжения. Коррозия, развивающая-
ся вначале слабо, постепенно ^усиливается, и скорость ее стано-
вится значительной. Наличие деформации и коррозии приводит
к образованию трещин, которые снижают прочность металла,
вызывая местные концентрации напряжений. Если наступает
усталость металла, то могут появиться н усталостные трещины.
Благодаря наличию на поверхности металла участков, находя-
Рис. 192. Схема диффузора для изучения кавита-
ции [85]:
1 — выступающий винт, -вызывающий кавитацию;
2 — испытываемый образец (60X30X8 мм]- 3 — дроссель;
4 — зона кавитации
щихся в напряженном и ненапряженном состоянии, возникают
коррозионные элементы, также усиливающие коррозионный
процесс. Поверхность металла покрывается микротрещинами,
частички металла выкрашиваются, разрушение его быстро раз-
вивается в ширину и глубину.
Необходимость подбора материалов, устойчивых в условиях
кавитации, привела к созданию ряда лабораторных методов
испытаний металлов. В этих ускоренных методах стремились
по возможности создать условия которые воспроизводили ка-
витационный характер разрушения за короткий промежуток вре-
мени.
Первые коррозионные испытания в условиях кавитации про-
водили в так называемых кавитационных трубах или диффузо-
рах (рис. 192), в которые жидкость подавалась под большим
напором (высота столба 350 м) [85]. Выдвижением винта 1 ме-
няется сечение потока в диффузоре, что дает возможность соз-
давать многочисленные кавитационные пузырьки, которые за-
тем уносятся потоком жидкости над образцом 2, укрепленным
в трубе. Позади трубы находится дроссель 3, который регули-
рует поток воды на выходе из трубы, создавая противодавление.
Результаты при таком методе испытания получают за несколь-
ко часов, определяя потерю массы образцом в зависимости от
— 317 —
времени. Диффузор чаще всего используют при изучении гид?
родинамики процесса.
В последнее время наибольшее распространение получили
два метода испытания: ударно-эрозионный (метод удара стру-
ей) и магнитострикционный.
Ударно-эрозионный метод заключается в направлении
струи воды на вращающиеся образцы. Образцы укрепляют иа
вращающемся диске радиально. Диск вращается от электромо-
Рис. 193. Схема ударно-эрозионной установки ЦНИИТ-
МАШа [86]:
1 — насос: 2 — электродвигатель; 3 — шкив; 4 — сливные трубы;
ч — вал диска; 6 — камера диска; 7 — крышка смотрового окна;
8 — образец; 9 — диск; 10 — фильтрующая сетка; 11 — сопло;
12 — манометр; 13 — трубопровод; 14 — бак; 15 — змеевик; 16 —
термометр; 17 — край для воды; 18 — сливной вентиль
тора. Число оборотов мотора в разных исследованиях было раз-
личное, в опытах [84] оно равнялось, например, 290 рад/сек
(2870 об!мин) при силе удара струи 2,7 н/см*- (0,28 кГ!см2),
диаметре выходного отверстия сопла от 5 до 8 мм и окружной
скорости 78 MjceK.
На установке ЦНИИТМАШа [86], схема которой приведена
на рис. 193, режим испытания был более жесткий. На диске
диаметром 800 мм, вращающемся от мотора с постоянным чис-
лом оборотов 220 рад!сек (2100 об/мин), укрепляют два образ-
ца. За каждый оборот образцы пересекают три струи с напором
воды 15 н!см2 (1,5 кГ]см2). Сопла диаметром 10 мм расположе-
ны перпендикулярно к плоскости диска. Окружная скорость об-
разцов 89,3 м!сек.
— 318 —
На результаты испытаний оказывает влияние расстояние
сопла от образцов; оптимальное расстояние. для данного сопла
находится эмпирически. Значение этого расстояния при прове-
денни опыта всегда должно учитываться; только в этом случае
можно получить сравнимые результаты в различных экспери-
ментах.
'Магнитострикционный метод впервые предложен Гейнсом
[87]. В СССР этот метод применяли многие исследователи, при-
чем в опытах использовались вибраторы различных систем [88—
91]. В основу метода положено
явление магнитострикции —
изменения линейных размеров
ферромагнитных материалов в
переменном магнитном поле.
Преимущество магнито-
стрикционного метода — воз-
можность проследить последо-
вательность разрушения и ис-
следовать продукты разруше-
ния па различных этапах, а
также изучить влияние раз-
личных жидкостей на кавита-
ционное разрушение. Принци-
пиальная схема установки
ЦНИИТМАШа [92] для опре-
деления кавитационных
свойств магнитострикционным
Вода
Рис. 194. Принципиальная схема
магнитострикционного вибратора
[92]
методом представлена на
рис. 194. Она состоит из усилителя мощности 7, в котором ис-
пользуются два предварительных каскада усиления, никелевой
трубки 2 с соответствующей магнитной цепью и катушками: об-
ратной связи 7, возбуждения II и подмагничивания III.
Колебания, поступающие от усилителя, подаются на катуш-
ку возбуждения II магнитострикционного вибратора. В магнит-
ной цепи вибратора отдельный источник (селеновый выпрями-
тель) создает постоянный магнитный поток, на который накла-
дывается поток, вызванный катушкой возбуждения. Результи-
рующий магнитный поток пульсирует от минимального значе-
ния, когда поля катушек II и III направлены навстречу друг
ДРУГУ> До максимального значения, когда эти поля складывают-
ся. Благодаря явлению магнитострикции пульсация магнитного
потока вызывает периодическое изменение длины стержня. Ко-
лебания его резко усиливаются по амплитуде, если частота
пульсации магнитного потока совпадает с частотой колебания
стержня. Прн колебаниях стержня в катушке обратной связи /
наводится э. д. с„ поступающая на выход усилителя. Колебания
стержня всегда происходят в резонансных условиях, так как
частота переменного тока задается частотой собственных коле-
— 319 —
баний стержня через катушку обратной связи. Чтобы изменить
частоту автоколебаний, переключателем изменяют емкость
-входного контура. Для регулирования амплитуды колебаний
стержня предназначен электрический амплитудомер IV, изме-
ряющий напряжение, возникающее в специальной катушке об-
ратной связи /
В описываемой установке магнитострикционным вибратором
является никелевая трубка 2 длиной 300 мм и диаметром 18 мм.
В трубку снизу ввертывается образец, который своей рабочей
частью погружается в ванну с жидкостью 3. Частота колебаний
трубки, определяемая ее размерами и массой образцов, состав-
ляет 7000—8000 гц. При такой частоте переменного магнитного
поля велики потери на вихревые токи. Для уменьшения этих
потерь и нагрева трубки в ней делается узкая прорезь по обра-
зующей (шириной 0,5 мм), которая затем заклеивается. Трубка
охлаждается проточной водой, подающейся в ванну, затем че-
рез распылитель вода поступает в никелевый стержень, стекает
по стенкам вниз и оттуда откачивается водоструйным насосом.
При проведении испытаний особое внимание необходимо обра-
щать на поддержание постоянной амплитуды колебаний трубки,
так как установлена прямая связь между амплитудой колеба-
ний и потерями массы образца.
Слишком большие амплитуды колебаний затрудняют изуче-
ние начальных стадий разрушения, кроме того, • происходит ис-
кусственное подавление процесса коррозии. Необходимо выби-
рать такие условия, чтобы развивались и ударно-мехаиические, и
электрохимические процессы. По данным работ [89, 93], сравне-
ние результатов опытов, проведенных ударно-эрозионным мето-
дом и с помощью магнитострикционного вибратора, показало,
что между ними существует хорошее качественное совпадение:
испытываемые материалы по стойкости располагаются в одной
и той же последовательности.
Критерием стойкости при испытаниях служит определение
потери массы образцов за определенные промежутки времени,
продолжительность которых определяется условиями опыта и
качеством исследуемых материалов.
Сопоставление результатов, полученных на ударно-эрозион-
ной установке и в магнитострикционном вибраторе, сделанное
Л. А. Гликманом [94], показало, что качественно онн совпадают.
Количественно же результаты отличаются, что, по мнению авто-
ра, связано с различием в механических условиях испытаний.
Однако можно предположить, что оказало влияние и различие
в коррозионных средах, так как в ударно-эрозионной установке
использовалась водопроводная вода, а в магнитострикционном
вибраторе — искусственная морская вода.
Данные, приведенные на рис. 195, хорошо иллюстрируют ка-
чественное совпадение результатов вибрационных и ударно-
эрозионных испытаний чугунов. В обоих случаях наибольшую
— 320 —
устойчивость обнаруживает высокопрочный чугун, содержащий
3,49% кремния, и наименьшую — обычный серый чугун.
Сопротивляемость стали кавитационному разрушению под-
робно изучали И. Р. Крянин и М. Г. Тимербулатов [86]. Иссле-
дования показали, что результаты испытаний на ударно-эрози-
онной установке ЦНИИТМАШа, где были созданы более жест-
кие условия, чем в работе ,[94], не совпадают с данными* полу-
магиитострикционной установке.
ченными при испытаниях на
На ударно-эрозиоиной уста-
новке для стали 1Х18Н9Т по-
лучается низкое значение ха-
рактеристики кавитационной
стойкости, а для закаленной
стали У8 — очень высокое.
Данные, получаемые при-
магнитострикционном методе
испытаний сталей продолжи-
тельностью 3 ч, согласуются
по количественной последова-
тельности со стойкостью ЭТИХ'
сталей в эксплуатационных
условиях. Более точных коли-
чественных данных, характе-
ризующих поведение сталей в
эксплуатационных условиях,
даже этим методом получить
не удается. Попытки усовер-
шенствовать метод вибрации
путем увеличения сроков испы-
тания результатов не дали.
Предполагают, что это связа-
но с тем, что для сталей, под-
Рис. 195. Результаты сравнитель-
ных вибрационных н ударно-эро-
зионных испытаний чугунов [94]:
/ — серый чугун типа 18-36; 2 — мо-
дифицированный чугун; 8 — высоко-
прочный чугун. Пунктирными линиями
обозначены ударно-эрозионные испы-
тания, сплошными — вибрационные
испытания
верженных коррозии, большую роль при кавитационном воздей-
ствии играет продолжительность коррозионного воздействия.
Исходя из представлений, что при слабом и умеренном кави-
тационном воздействии металлы разрушаются преимуществен-
но путем предварительного окисления и последующего отрыва
поверхностного слоя в виде окисных пленок и продуктов корро-
зии, И. Р. Крянин и М. Г. Тимербулатов [86] показали, что при
магнитострикционном методе следует чередовать продолжитель-
ное коррозионное воздействие в спокойной воде с кратковремен-
ным воздействием кавитации на вибраторе. Режим испытания
рекомендуется следующий: 24 ч коррозия в воде и 5 мин кави-
тационного воздействия иа вибраторе при общей продолжитель-
ности испытания, составляющей 36 циклов'
Результаты испытаний на вибраторе, полученные без чере-
дования и при чередовании коррозии и кавитации, приведены
на рис. 196. Применение .чередования, как видно из рисунка,
21 Зак. 1238
— 321 —
шение сталей при испытании
на магнитострикционном вибра-
торе [95]:
1 — непрерывная вибрация 3 ч;
« __ чередование коррозии (24 ч)
и вибрации (5 пин), продолжи-
тельность испытания 36 суток
позволяет более четко выявить разницу, в поведении низколеги-
рованных и нержавеющих сталей; так, например, -при обычных
.испытаниях на вибраторе сталь 18ДГСЛ близка к стали
20X1ЗНЛ, а при чередующихся воздействиях интенсивность
разрушения стали 18ДГСЛ больше, чем нержавеющих сталей^
в 3,8—5,6 раза, а стали 20ГСЛ— в 4,9—7,3 раза.
Надежность получающихся данных при чередующихся воз;-
действиях коррозии и кавитации подтверждается анализом дан-
ных, полученных при обследовании 28 турбин на различных
электростанциях.. .Было установ-
лено, что наиболее стойкой про-
тив кавитации в условиях эксг
плуатации поворотно-лопастных
гидротурбин является нержавею-
щая сталь 1Х18Н9Т; приемлема
также сталь 20Х13НД. Низколе?-
тированная сталь 18ДГСЛ усту-
пает .до -кавитационной стойкости
указанным нержавеющим ста-
лям.- В аналогичной, последова-
тельности располагаются ;метал-
лы по результатам испытаний
по рекомендуемой методике..
Из изложенного выше следу-
ет, что правильную оценку кави-
тационного воздействия на сталь
можно получить магнитострик-
ционным методом при непрерыв-
ном воздействии кавитации.,. ;
Если же необходимо Определить степень влияния коррозион-
ного фактора, то следует применять чередующееся воздействие
кратковременной кавитации с длительным влиянием спокойной
коррозионной среды. Очевидно, этот метод получит в дальней-
шем более широкое применение.
5. ОПРЕДЕЛЕНИЕ УСТОЙЧИВОСТИ
РЕАКТОРНЫХ МАТЕРИАЛОВ ПРИ ВЫСОКИХ
ТЕМПЕРАТУРАХ И ДАВЛЕНИЯХ1
В связи с развитием атомной промышленности в последние
годы возникла необходимость в разработке и испытании новых
материалов, к которым предъявляются особые требования. Это
прежде всего материалы, применяемые в атомной энергетике.
1 Эта часть книги написана И. JI. Розенфельдом совместно с Ю. П. Оль-
ХОВ'НИКОВЫМ.
— 322 —
Наиболее важные конструкционные узлы-атомных.электростан-
ций работают в условиях высоких температур и давлений, кото-
рые в лабораторных условиях лучше всего воспроизводятся в
автоклавах из нержавеющей стали 1Х18Н9Т. Измерение, регу-
лировка и поддержание нужной-температуры в рабочем объеме
автоклава производятся с помощью регулирующих потенцио-
метров.
ТенлершщюаХ
Рис. 197. Зависимость давления р и удельного
объема воды V от температуры на линии насы-
щения:
«кр= 374,15е С; ркр= 225,65 бар багл); Икр= 3,26 см?(г
правку температуры можно осуществить по давлению, так как
в равновесных условиях каждой температуре жидкости (воды)
соответствует совершенно определенное давление, которое лег-
ко находится из таблиц в работах [95, 96] или по графику
(рис. 197). Длительность коррозионных испытаний должна
быть разумно ограничена. На практике часто применяют крат-
ковременные испытания, которые позволяют получить надеж-
ные результаты в сравнительно сжатые сроки. Обычно продол-
жительность таких испытаний, составляет несколько тысяч чаг
сов [97] *. Полученные результаты экстраполируются до более
длительных периодов в предположении, что скорость коррозии
остается примерно постоянной. В некоторых случаях, когда ме-
ханизм коррозии остается неизменным при изменении темпера-
туры, как это наблюдается у сплава циркалой-2 (98], для сокра-
* Срок службы деталей обычно составляет -от нескольких тысяч до пе-
сятков тысяч часов. • • -
21* Зак. 1338 — 323 —
щения сроков испытаний целесообразно повысить температуру;
что может дать значительный выигрыш во- времени.
За последние годы получили более широкое применение
электрохимические исследования в условиях высокой темпера-
туры и давления. Разработаны автоклавы для электрохимиче-,
ских измерений и надежная изоляция электровводов в них. Все
это дало возможность выяснить электрохимическое поведение
металлов в высокотемпературной воде.
Классификация коррозионных испытаний в воде
при высокой температуре
В зависимости от выбранных методов коррозионного испы-
тания сначала находят конструтивное оформление установки,
иа которой оно будет проводиться.
Испытания, проводимые в автоклавах, где коррозионная
среда в течение длительных отрезков времени находится в отно-
сительном покое, называются статическими.
Испытания в статических условиях имеют целый ряд пре-
имуществ. Это прежде всего простота и компактность оборудо-
вания. Например, испытания в ампулах (см. ниже) можно про-
водить в обычных лабораторных условиях.
При коррозионных испытаниях обычно стремятся воспроиз-
вести если ие полностью, то хотя бы частично те условия, в ко-
торых исследуемый материал будет работать иа производстве.
С этой точки зрения статические испытания имеют существен-
ный недостаток, так как в реальных условиях коррозионная сре-
да (теплоноситель в атомных энергетических реакторах) обычно
движется с довольно большой скоростью. Тем не менее резуль-
таты статических испытаний можно рассматривать как предва-
рительную оценку коррозионной стойкости материала. "На осно-
вании этих результатов производят отбор наиболее коррозиоЩ
иостойких сплавов, которые в дальнейшем проверяют на стен-
дах и в действующей аппаратуре иепосредствеино на произ-
водстве.
Наиболее полно воспроизводятся производственные условия
при так называемых- динамических- испытаниях, где
коррозионная среда движется и омывает образцы. Динамиче-
ские испытания проводят чаще всего в петле. Петля представ-
ляет собой в большинстве случаев замкнутый контур, внутри
которого под действием насосов циркулирует коррозионная
среда. Перед камерой, в которую помещают образцы, стоит на-
греватель для подогрева жидкости до необходимой темпера-
туры.
Необходимость применения насосов, работающих при высо-
ких температурах и давлениях, сильно усложняет конструкцию
петли, делает ее громоздкой и дорогостоящей. Поэтому в неко-
— 324 —
торых случаях приходится ограничиваться испытаниями в
статических условиях, а в динамических условиях проверять
лишь те материалы, которые прошли предварительные испы-
тания.
Некоторые авторы {99] выделяют еще -одну форму коррози-
онных испытаний — так называемые полустатические ме-
тоды. По л у статические испытания характеризуются тем* что
скорость движения воды ограничивается той минимальной ве-
личиной, которая обеспечивает обновление воды у испытывае-
мых образцов и поддержание ее состава. Конструкция контура
для таких испытаний менее сложна, чем для динамических ис-
пытаний.
Аппаратура
Рис. 198. Ампула для
коррозионных испытаний
металла - при высоких
тёмпературах:
1 —• накидная гайка из пт-
лёрод'йсТой стали; 2 — проб-
ка из стали 1X18H9T; 3
корпус из Стали 1Х18Н9Т;
’ -4 — образец;
Для статических методов коррозионных (испытаний, как уже
указывалось, обычно применяют установки, -где в качестве со-
суда для испытаний используется автоклав.
Образцом простейшего автоклава мо-
жет служить ампула, схема которой
представлена на рис. 198. Это толсто
стенный сосуд с небольшим рабочим
объемом (~10 см*), на внешней сторо-
не которого имеется резьба для накид- .
ной гайки. Сосуд закрывается пробкой с
шаровой поверхностью, входящей в ко-
ническое расширение верхней части со-
суда, и затягивается накидной гайкой.
В данном случае применено уплотнение
«шар по .конусу».
Сосуд и крышку изготавливают из
стали 1Х18Н9Т. Накидная гайка может
быть изготовлена из любой другой ле-
гированной стали или из обычной угле-
родистой стали. Разнородность сталей,
из которых изготовлены корпус и гайка,
совершенно необходима, так как в про- .,
тивном случае при открывании возмож-
ны задиры на резьбе.
В каждую такую ампулу помещают,
один образец испытываемого металла и
заливают необходимое количество'воды.
Объем заливаемой/ воды рассчитывают по таблицам в работах
[95, 96] или графикам-состояния воды (см. рис. 197). При этом
необходимо учитывать общий объём ампулы и Температуру .ис-
пытания. Залйвать воды надо столько, чтобы в процессе расши-
рения при нужной температуре она полностью’ закрывала обра-
зец. Однако не следует заливать такое количёСТвЬ^оды; которое,
21е* — 325 —
расширяясь при нагреве, заняло бы при заданной температуре
объем, больший или равный внутреннему объему ампулы».
В этом случае может нарушиться герметичность, и- вода испа-
рится. Набор таких ампул помещают в обычную муфельную
печь, которая нагревается до заданной температуры-с учетом ее
перепада в реакционном сосуде.
Испытания в ампулах имеют-ряд недостатков:
а) давление и температуру внутри ампул практически не
контролируют, а определяют лишь расчетным путем. При пару-:
шении герметичности изменение режима испытаний сразу может
быть и не обнаружено;
б) объем коррозионной среды ограничен, что приводит к весь-
ма малым значениям отношения количества воды к поверхности
образца;
в) ограниченный объем коррозионной среды позволяет испы-
тывать в одной ампуле не более одного образца;
г) ненадежность уплотнения приводит иногда к утечке кор-
розионной среды в виде пара.
Несмотря на эти недостатки, ампулы имеют и;целый-ряд пре-
имуществ. Прежде всего — это простота конструкции и изготов-
ления их. Небольшие размеры ампул позволяют применять для
их нагрева любую лабораторную печь,-в которой можно поме-
стить 10—-15 ампул одновременно. Кроме того, в ампулах можно
исследовать все продукты коррозии, возникшие в результате
коррозии одного и того же образца, (и проводить анализ электро-1
лита после испытаний.
‘Для длительных массовых испытаний ампулы вряд ли при-?
годны, но для быстрой предварительной оценки коррозионных
свойств материалов они вполне себя оправдали:
"К наиболее распространенным аппаратам .для ускоренных
испытаний относятся автоклавы, в которых * можно контролиро-
вать температуру и давление. Существует большое количество
конструкций’ автоклавов/ которые отличаются друг от друга раз-
мерами или способом введения термопар и штуцеров для мано-
метрических трубок. Все они представляют собой-толстостенные
цилиндрические сосуды,» изготовленные преимущественно из спе-
циальной нержавеющей-стали, типа 1Х18Н9Т, ’ е массивными
крышками. . • • .
Толщина стенок зависит от условий работы, т. е-, от темпера-
туры и давления в рабочем объеме. Эти параметры конструктор
учитывает при расчете.- . - . •
Для удобства в работе'при конструировании автоклавной ус-
тановки рекомендуется также учитывать следующее:
а) корпус автоклава и нагревательную -печь ^желательно кре-
пить на жесткой арматуре; б) штуцера для ввода трубок от ма-
нометров следует устанавливать на корпусе; в) термопарный
чехол надо вводить либо через» дно, либо (что делается-гораздо
чаще) через крышку.. • •' = •
—326 —
Конструкция современной автоклавной усатновки включает
два основных, узла:
1) рабочий узел, представляющей собой жесткую арматуру,
на которой крепятся сам автоклав, нагревательная печь и мано-
метры (контрольный и электроконтактный);
2) щит управления, на котором устанавливаются приборы^
регистрирующие ток и температуру, а также приборы автома-
тической регулировки режима, регулировки тока и выключа-
тели. .
Рис. 199. Схема автоклавной установки с авто-
матической регулировкой рабочего режима:'
/ — перепускной и сбросной клапан; 2 — крышка; 3 —
корпус; 4 ~ электрическая нагревательная -печь; 5 —
термопарный чехол; 6 — автотрансформатор; 7 — маг-
нитный пускатель; 8 — автоматический"' регулирующий
потенциометр; 9 — рабочий электроконтактный мано.-
метр ЭМ; 10 — термопара; 11 — коиуррльный манометр
M.KD
Схема автоклавной.установки.с автоматической регулировкой
рабочего режима, принятой в исследованиях, проводимых в Ин-
ституте физической химии АН СССР , [100], представлена на
рис. 199. В установке, собранной по этой схеме, контроль рабо-
чего режима осуществляется по давлению и температуре. Тер-
мопара 10 включена в автоматический регулирующий дотенц'ио*
метр 8 (например, ЭПВ2-1А, или ПСР-1), который с помощью
магнитного пускателя 7 включает пли выключает нагреватель-
ную печь 4. В ту же цепь магнитного пускателя включены кон-
такты электроконтактного манометра 9 типа ЭМ. Таким образом,
осуществляется надежный двойной контроль режима работы
установки, который, почти полностью исключает возможность пе-
регревов при выходе из строя или появлении .погрешностей в ра-
— 327 —
боте одного 'из регулирующих приборов. Ток электропечи регу-
лируется с помощью автотрансформатора 6. Недостаток этой
установки в том, что регулировка температуры производится от
термопары, помещенной внутри автоклава, что может привести
к возникновению тепловых циклов. Этот недостаток можно уст-
ранить введением еще одной термопары на внешней стороне ав-
токлава.
' Сравнительно недавно разработанный метод коррозионных
испытаний в автоклавах специальной конструкции [101] позволя-
ет вводить образцы непосредственно в среду с нужными пара-
Рис. 200. Схема простейшей
установки для полустатиче-
ских коррозионных испы-
таний [99]
метрами. Сущность этого метода
заключается в следующем. Два ав-
токлава расположены один над
другим и могут нагреваться одно-
временно. Они соединены трубкой
с перепускным клапаном. Воду за-
ливают в верхний автоклав при за-
крытом клапане. В нижний авто-
клав помещают образцы, и он за-
полняется инертным газом (аргон).
Затем оба автоклава нагревают.
Когда температура в них достигнет
нужной, степени, открывается кла-
пан и вода перетекает в нижний ав-
токлав с образцами. Таким обра-
зом, образцы сразу подвергаются
воздействию воды при заданной
температуре, т. ё. отсутствует ее
воздействие в течение .периода на-
* грева.
Автоклавы других конструкций описываются в работах [99,
102, 103].
Выбор того или иного типа автоклава зависит' от многих
факторов, в том числе от задаваемой величины отношения объе-
ма коррозионной среды к поверхности образцов (V/S), от типа
й количества испытываемых образцов, .желаемой точности под-
держания заданного режима и т. д.
б установках для полустатических испытаний, как и. в пре-
дыдущем случае, основным элементом является автоклав, кото-
рый включен в замкнутый, контур. С помощью специальных уст-
ройств вода в контуре- циркулирует с небольшой скоростью. •
' Схема простейшей установки для коррозионных испытаний
алюминиевых сплавов [99] приведена на рис. 200. В этой схеме
контур не замкнут. Смена среды обеспечивается за счет ее не-
прерывной подачи из компенсационного объема 1 с помощью
насоса 2 в автоклав 4, откуда она поступает в холодильник 5
и сбрасывается посредством спускного клапана-#. Таким обра-
зом, в рабочем объеме происходит постоянное восполнение-по-
— 328 —
терянной при сбросе воды. Скорость обмена воды в этой уста-
новке невелика ’(10—15 см?!мин). В нижнюю часть автоклава во-
да подается предварительно подогретой в подогревателе 3. Дав-
ление в автоклаве регулируется спускным клапаном, Автоклавы
могут быть нагреты до температур, не превышающих температу-
ры насыщения пара при заданном давлении.
Рис. 201. Схема контура общего назначения для пояустйтиче-
ских коррозионных испытаний [99]:
7 — подвод воды; 2 — деионизатор; 3 — автоматический запорный кла-
пан; 4 — ячейка для измерения электропроводности воды;. 5 — резерву-
ар очивденной воды; 6 — расходный бак; 7 ~ термометр; 8 — ячейка-
для контроля электропроводности, воды; 9 —- теплообменник-подогрева-
тель; 10 — абсорбер; 11 — баллон с кислородом; 12 — фильтр низкого
давления; 13 — насос; 14 — фильтр высокого давления; 15 — буферная
емкость с баллоном водорода; 16 к 20 — манометры; 17 — расходомер;
18 — пробоотборники с холодильником; 19 —- нагреватель; 21 — автоклав
с образцами; 22 — другие автоклавы; 23 —, клапан для регулировки рас-
хода воды; 24 — магистраль обратной воды' 25 — теплообменник-холо-
дильник; 26 — регулятор давления; 27 — капиллярная трубка; 28 —
фильтры; 29 — предохранительные клапаны; 30 — отстойник и грязевой
насос; 31 — отвод в дренаж
Более сложная схема установки, применяемой для полуста-
тических испытаний с деминерализованной, насыщенной кисло-
родом водой, дана на рис. 201. В этом контуре легко осуществ-
ляется переход от одного водного режима к другому. Для рабо-
ты с деаэрируемой водой достаточно, направить воду из резер/
вуара с очищенной водой в деаэрирующую башню, а из нее
к насосу 13. ' ;
.— 329—7?
Работа контура осуществляется следующим1 образом. Вода 3
из водопровода проходит через смешанный катион-анионный
деионизатор 2 и поступает в резервуар 5, откуда подается в теп-
лообменник-подогреватель 9. Подогретая вода насыщается ки-
слородом в абсорбере 10 и перекачивается насосом 13 через на-
греватели 19 в автоклавы 21, 22. Каждый автоклав имеет на-
греватель, который только в нем поддерживает нужную темпе-
ратуру. Выходящая из автоклавов вода пропускается через
фильтры 28 и возвращается в деионизатор. При работе вода из
водопровода лишь компенсирует утечки и отбор проб..
При необходимости каждый автоклав может быть изолиро-
ван от системы и использован для статических испытаний.
Статические и полустатические методы испытаний не могут
'воспроизвести полностью условий работы реакторов, где вода
движется со значительной скоростью. Для этого требуются
динамические методы испытаний, осуществляемые в контуре
(петле).
Различают контуры с естественной (конвективной) и прину-
дительной циркуляцией.
Чтобы избежать кипения в контуре, работа ведется при дав-
лении выше давления насыщения для данной ’температуры [99].
В конвективном контуре циркуляция воды осуществляется
посредством расположения нагревателя в нижней, а холодиль-
ника в верхней части системы. Возникает разность плотностей
воды между восходящей и нисходящей ветвями петли. Схема
испытательного контура такого типа приведена на рис. 202 [99].
На рис. 203 представлен коррозионный стенд с естественной
циркуляцией воды, применяемый для исследования коррозии ,
металла паровых котлов {104]. Он состоит из барабана-сборни-
ка 2, опускной 7 и подъемной И циркуляционных труб, между
которыми в нижней части помещен грязевик 10. Подъемная тру-
ба контура снабжена электрической печью 12 из четырех само-
стоятельных секций. В верхней части спускной трубы размещен
водяной холодильник 3. При форсированной работе контура
включается еще один холодильник 1, расположенный в паровой
части контура. Трубчатые образцы 4 располагаются в специаль-
ных испытательных участках и электроизолируются от труб кон-
тура, что позволяет вести электрохимические измерения. Таким
образом, трубчатые образцы из испытуемого металла являются
составной частью контура, и вода циркулирует через них. Они
имеют собственную печь для подогрева и собственную регули-
ровку температуры. Запорные приспособления 9 предназначены
для пуска и заливки стенда. Вода в контур подается из бачка-
деаэратора 8, снабженного электропечью для кипячения воды
и создания необходимого для перепуска воды в контур давле-
ния. Скорость циркулирующей воды измеряется при помощи
диафрагмы 5 и дифманометра 6. Недостатком конвективного
контура является незначительная разность плотностей при ра-
— 330 —
боте в области высоких температур, так как скорость движения
жидкости в этом случае низка.
Более высокие скорости (до 18 м/сек) могут быть получены
в контуре с принудительной циркуляцией. На рис. 204 представ-
лена схема контура, где циркуля-
ция воды осуществляется при по-
мощи насоса. Желательно, как
отмечают авторы [99], во избежа-
ние загрязнения воды применять
насос с герметизированным рото-
ром, в котором подшипники ра-
ботают в циркулирующей воде,
Рис. 203. Испытательный стеид с
естественной циркуляцией воды
для исследования коррозии метал-
ла паровых котлов [104]
Рис. 202. Схема контура с естест-
венной циркуляцией воды [99]:
1 — клапан; 2 — манометрический
указатель уровня; 3 — задающий дав-
ление котел; 4 — предохранительная
мембрана; 5 — нагреватель котла; 6 —
нагреваемый участок: 7 — расходо-
мер; 8 — испытательный участок; 9 —
манометр; 10 — ребристый охлаждае-
мый участок; 11 — вентилятор; 12 —
отбор проб и соединение с иоиооб-
мекником; 13 — пишущий регулятор
температуры. Высота контура Л око-
ло 5 м; диаметры восходящей и ни-
сходящей ветвей около 25 мм каждый
т. е. в таких насосах отсутствует сальниковая набивка. Вода на-
гревается до нужной температуры нагревателем 5, а котел 10 с
нагревателем 9 поддерживает давление в контуре выше уровня
насыщения. Чтобы компенсировать утечки, в контур с помощью
пневматических насосов, работающих автономно, периодически
при необходимости добавляется вода. Образцы для испытания
располагают в специально предназначенных для этого участках
3 и 8. Расход, а следовательно, скорость циркуляции воды регу-
лируются перепускным клапаном 2.
— 331 —
Тот же автор [99] приводит схему многоканальной петли, ко-
торая позволяет одновременно исследовать влияние скорости
протекания теплового цикла и частично температуры в воде од-
ного и того же химического состава. На рис. 205 видно, что
к коллектору присоединены двенадцать отдельных - каналов,
в которых можно создавать различные рабочие условия. Темпе-
ратура и расход воды измеряются и регулируются отдельно
в каждом канале. Циркуляция- воды обеспечивается насосом,
ротор которого герметизирован. В контур подается деминерали-
зованная в ибнообменниках и фильтрах вода. Большим преиму-
Рис. 204. Схема контура с принудительной циркуляцией
воды [90]:
1 — насос; 2 — клапан для регулировки расхода воды; 3 и
в— участки с образцами; 4 — диафрагма; 5 и 9 — нагревате-
ли; 6 — пробоотборник; 7 — иоиообменник с- холодильником;
10—задающий давление котел
ществом этой петли является то, что каналы по мере надобности
могут включаться и отключаться. Это позволяет, во-первых, из-
влекать и загружать образцы, не останавливая остальную часть
установки, и, во-вторых, производить немедленный пуск в канал
воды рабочих параметров и немедленно его отключать.
Существует еще целый ряд конструкций петель, но мы оста-
новимся еще на контуре, позволяющем вести испытания в усло-
виях пароводяной смеси высоких параметров, т. е. в потоке
влажного пара [103]. В этой установке (рис. 206) деминерализо-
ванная и дегазированная вода перекачивается поршневым на-
сосом, Сухой перегретый пар, получаемый в -аккумуляторе 4,
амортизирует давление в петле и уменьшает колебания’скоро-
сти воды. После подогрева в теплообменнике 5 вода попадает
в электрический нагреватель 6, где образуется пароводяная
смесь желаемого состава,, которая направляется в испытатель-
ную секцию 8..Затем пар попадает в теплообменник для подогре-
— 332 —
ва притекающей воды. Из холодильника 10 вода проходит через
систему понижения давления 1L Охлажденная вода проходит
частично через очистительное устройство 12 и вновь попадает
в резервуар 1б, затем процесс
повторяется.
В заключение следует от-
метить, что конструкции уста-
новок для статических и осо-
бенно полустатических и дина-
мических коррозионных испы-
таний в воде высоких пара-
метров в большинстве случаев
сложны и громоздки. В перио-
дической литературе приводят-
ся обычно схемы их устройст-
ва, объясняющие основные
принципы работы, поэтому при
проектировании установки в
каждом отдельном случае не-
обходимо учитывать характер
и режим предполагаемых ис-
пытаний, а также наличие не-
обходимого оборудования и
площадей.
За последнее время был
разработан ряд установок, по-
Рис. 205. Схема многоканальной пет-
ли для испытаний на коррозию (99]:
1 — маслоохладитель; 2 — масляный
бак и насос; 3 — насос марки 100-А; 4 —
нагреватели; 5 — котел, задающий дав-
ление; 6 — спускной клапан; 7 — предо-
хранительная мембрана; 8 — указатель
уровня воды; 9 — насос; 10 — сосуды с
образцами; 11 — высоконапорный холо-
дильник; 12 — низконапорный холодиль-
ник; 13 — ионсобменник; 14 — подвод
охлаждающей воды; 15 ~ отвод охла-
ждающей воды; 16 — плоские образцы;
17 — образцы при высокой скорости про-
текания горячей воды; 18 — образцы при
высокой скорости протекания холодной
воды; 19 — ветвь ионообменннка высо-
кого давления; 20 — образцы при низкой
скорости протекания холодной воды; 21 —
образцы при низкой скорости протекания
горячей воды; 22 — образцы в сосуде для
визуального наблюдения; 23 — пробоот-
борник; 24 — камера лля введения доба-
вок газа; 25 — перепускная линия
зволяющнх косвенно оценивать стойкость материалов электро-
химическими методами, а также производить электрохимические
измерения при высоких температурах и давлениях. Конструкци-
онная особенность этих установок — устройство электровводов в
22 Зак. 1338 — '333 —
измерительную ячейку, представляющую собой обычный авто-1
клав.
Несмотря на кажущуюся простоту идеи, конструирование
и эксплуатирование автоклавных установок для электрохимиче-
ских измерений при высоких температурах и давлениях сопря-
жены со значительными трудностями. Ввиду отсутствия надеж-
ных изолирующих материалов, сохраняющих свои свойства
в электролитах при высоких температурах, с одной стороны,
Рис. 206. Схема контура для испытаний на коррозию в пароводяной
смеси [103]:
1 — насос; 2 — выпускной клапан; 3 — мембрана; 4 — аккумулятор; 5 — тепло-
обменник; £ — нагреватель; 7 — трансформатор; 8 — испытательная секция; 9 —•
газовый сепаратор; /0 — холодильник; 11 — система понижения давления; 12 —
фильтр и смешанный слой смолы: 13 — резервуар; 14 — индикатор; /5 — приборы
для измерения скорости потока (самописцы); 16 — контрольные термопары;
/7 — приборы для завися температуры; 18 — манометры
и необходимости работы реакционного сосуда при высоком дав-
лении, с другой, ввод электродов в металлический автоклав пре-
вращается в сложную проблему. Другая трудность связана
с электродом сравнения. Обычно применяемые электроды срав-
нения здесь непригодны в связи с разрушением в высокотемпе?
ратурной воде стекла и других инертных материалов, а также
недопустимости помещения электрода сравнения, например
Hg/HggCls, непосредственно в автоклав. Не ясно также;, как ме-
— 334 —
няется потенциал' электрода сравнения с температурой и давле-
нием в такой нетипичной для электрода сравнения среде, как
дистиллированная вода. При помещении электрода сравнения
снаружи автоклава возникают другие трудности,- связанные
с включением в измеряемую величину омических падений потен-
циала, изменениями потенциала благодаря температурным пе-
репадам и т. д.
Как указывалось выше, большие затруднения возникают при
конструировании электровводов в автоклав. На риС/207 изобра-
жена схема крышки с
электрическими вводами
для электрохимических
измерений в
Института
химии АН СССР. Эта
схема, как показал мно-
голетний опыт, весьма
надежна и позволяет ис-
пользовать в качестве
изоляции обычные мате-
автоклавах
физической
20
19
з
ю
Рис. 207. Крышка
с злёктровводами
для электрохимических
измерений в автоклаве:
1 — проводник: 2, 10. 14,
20 — гайки; 3 — карман
для термопары; 4 — труб-
ка-изолятор; 5 — крышка:
б — штуцер; 7 —= кольцо
(резина); 8 — кожух; 9 —-
оливка; 11 — кольцо (гети-
накс, эбонит); 12 — залив-
ка (бакелит, сургуч); 13, 15,
13 — втулки; 16 — стержень
контакта; 17 — -ниппель:
. 19 — контакт
риалы — гетинакс, эбонит, фибру и т. п. Электрический контакт
представляет собой латунный цилиндр с глухим резьбовым от-
верстием внизу, в который ввинчивается'проводник 1 от образца.
Проводник от измерительного прибора подключается к оерхией
части стержня контакта 16 при помощи гайки. Контакт изоли-
руется-от ниппеля 17 и штуцера 6 посредство^ втулок 13, 15 и
22* — 335 —
18, изготовленных из эбонита или другого изоляционного мате-
риала. Проводник / помещается в фарфоровую трубку-изоля-
тор 4. Трубка вставляется в отверстие втулки 13, а зазор между
трубкой и втулкой заливается бакелитом, пицеином и т. п. Таким
образом, проводник, идущий от образца к контакту, полностью
изолирован от крышки 5 и штуцера 6. Герметичность иа вводе
достигается посредством 'втулки 13, которая при затяжке гай-
ки 14 служит одновременно уплотнительной прокладкой. Для
удобства резьба гайки 14 мм ввинчивания ниппеля 17 сделана
обратной.
Средняя часть штуцера имеет меньший диаметр. На штуцер
надевается кожух 8. В пространство между корпусом штуцера
и кожухом пропускается вода от водопровода. Созданная таким
Рис. 208. Автоклав для электрохимических измерений [105]:
1 — образец; 2 — изоляция; 8 — электроввод; 4 — крышка; 5 — гайка; 6 — пробка;
7 — наконечник; 8 — трубка; 9 — ниппель; 10 — корпус
образом водяная охлаждающая рубашка предотвращает под-
вод тепла к контактному узлу, и последний в процессе работы
автоклава находится при комнатной температуре. Именно это
обстоятельство позволяет использовать в контактном узле обыч-
ные изоляционные материалы. Применяя крышку автоклава
с описанными вводами, можно измерять коррозионные токи при
изучении контактной коррозии, а также коррозионное поведение
металлов в условиях анодной и катодной поляризации при нало-
жении тока от внешнего источника. В качестве электродов
сравнения в наших исследованиях используются платинирован-
ная платина, золото, а также нержавеющая сталь.
Многие упрощения были сделаны в конструкци автокла-
вов для электрохимических измерений (рис. 208). В такой кон-
струкции электрод сравнения находится вне автоклава и соеди-
— 336 —
нен с рабочим пространством электролитическим ключом.
Конструкция ключа позволяет без нарушения герметичности
осуществлять электролитический контакт со средой, в которой
находится исследуемый образец. Как видно на рис. 208, роль
электролитического ключа выполняет пропитанная водой пробка
Рис. 209. Автоклав для электрохимических
измерений при высокой температуре [106]:
1 — тефлоновые прокладки; 2 — слюда; 3 — уплотняю-
щая гайка; 4 — платиновый стержень; 5 — шайба,
припаянная к платиновому стержню: 6 — проходная
прокладка
из дерева (береза), помещенная в металлический кожух, от ко-
торого она изолирована фторопластом. Кожух вварен в стенку
автоклава н охлаждается проточной водой. Наконечник 7 слу-
жит для подвода электрода сравнения, который находится при
комнатной температуре. Ток для поляризации подводится к об-
разцу 1 через электровводы в крышке 4 автоклава. Вводы от
— 337 —
крышки изолированы с помощью слюдяной прокладки 2, кото-
рая уплотняется гайками. В этом автоклаве катодное и анодное,
пространства разделены путем отделения поляризующего элек-
трода от коррозионной среда электролитическим мостом почти
такой же конструкции, как и для электрода сравнения.
Недостаток описанной конструкции — возникновение термо-
диффузионного потенциала вследствие наличия градиента тем-
ператур в электролитическом ключе, точная оценка которого за-
труднена. Кроме того, учитывая, что процессы происходят в та-
кой плохой проводящей среде, как дистиллированная вода,
в величину измерения потенциала при поляризации неизбежно
включается омическое падение потенциала.
Еще одна конструкция автоклава для электрохимических из-
мерений и электроввода к нему представлена на рис. 209 [106].
Этот автоклав применяли для изучения поведения сплавов алю-
миния в условиях катодной и анодной поляризации при темпе-
ратурах до 325° С. Конструкция электроввода в данном случае
проста, но имеет существенный недостаток: уплотняющая гайка
ввинчивается с внутренней стороны крышки автоклава, что
уменьшает надежность уплотнения, так как в случае появления
течи устранить ее в ходе работы невозможно. Другой недоста-
ток-использование в качестве уплотнителя тефлоновой про-
кладки. При температурах выше 250° С уплотнение размягчается
и становится ненадежным. Наконец, контакт образца с вводом
находится в рабочим объеме автоклава, что в значительной сте-
пени уменьшает его надежность в процессе испытания из-за воз-
можного окисления.
Методы контроля температуры и давления
(схемы приборов, принципы работы их и т. д.)
Существенную роль при проведении коррозионных испытаний
в автоклавах играет поддержание принятого рабочего режима,
т. е. сохранение в течение опыта на выбранном уровне, темпера-
туры и давления. Контроль и регулировка температуры и давле-
ния осуществляются при помощи специальных приборов.
Наиболее распространенный и надежный инструмент для из-
мерения температуры — потенциометр с термопарой. Различают
два основных типа потенциометров: регистрирующие и автома-
тические регулирующие.
Регистрирующие потенциометры используют в тех случаях,
когда требуется лишь контроль температуры, а заданный режим
поддерживается при помощи специально предназначенного для
этого устройства или вручную. Однако для нас представляют
гораздо больший интерес автоматические регулирующие потен-
циометры, поскольку они являются составной частью большин-
ства используемых в настоящее время электрических схем, при
-338-
помощи которых поддерживается заданный рабочий режим в' ав-
токлавных установках.
В схемах автоматической регулировки рабочего режима мо-
гут быть использованы автоматические регулирующие потенцио-
метры отечественного производства следующих типов: ЭПВ-2,
ПСР-1, ЭПД. Последние два снабжены самопишущими приспо-
соблениями, осуществляющими непрерывную запись температу-
ры на диаграммную бумагу. Все три типа потенциометров элек-
тронные и позволяют регулировать температуру с точностью
±5° С.
Давление внутри установок для коррозионных испытаний
в высокотемпературной воде контролируется с помощью мано-
Рис. 210. Схема автоматической регулировки
рабочего режима в автоклаве:
/ — автоклав; 2 — электрическая печь; 3 — электромаг-
нитное реле; 4 — автотрансформатор; 5 — потенцио-
метр регулирующий ЭПВ-2; 6 — электроконтактный ма-
нометр; 7 — термопара; 8 — клапан для сброса пара
метров. Можно использовать манометры различных типов с пре-
делами измерения, достаточными для выбранной температуры
испытания. Как уже указывалось, между температурой и давле-
нием насыщенного пара в замкнутом объеме существует строгая
зависимость (см. рис. 197), поэтому манометры одновременно
могут служить и для регистрации температуры. Эта зависимость,
используется также для автоматической регулировки рабочего
режима. В этом случае обычно применяют электроконтактные
манометры типа ЭМ (или ЭКМ).
Электроконтактный манометр представляет собой обычный
стрелочный манометр, снабженный разрывными контактами, ко-
торые включаются в схему автоматической регулировки.
Использование в схеме автоматической регулировки одновре-
менно электроконтактного манометра и автоматического регули-
— 339 —
рующего потенциометра с* термопарой позволяет осуществлять
двойной контроль рабочего режима. На рис. 210 представлена
такая схема, успешно примененная нами в Институте физической
химии АН СССР. Автоклав 1 помещается в электрическую
печь 2. Величина тока, подаваемая на спираль печи, регулирует-'
ся трансформатором типа РНО-250-5 (если печь небольшой мощ-
ности, то можно применять обычный лабораторный автотранс-
форматор типа «Латр» РНО-260-2) и контролируется ампермет-
ром переменного тока. Чехол с термопарой 7, регистрирующей
температуру, введен в рабочий объем автоклава через крышку.
Термопара подключена к потенциометру 5. Потенциометр в за-
висимости от температуры может замыкать или размыкать
цепь, в которую включена обмотка электромагнитного реле 3.
Реле в свою очередь включает или выключает электрическую
печь. В качестве реле могут быть использованы магнитные пу-
скатели типа П-222МР, электромагнитные реле типа ЭП 41/21-Б
или любого другого типа, контакты которого рассчитаны на ток,
достаточный для питания выбранной печи. Установка может
включаться и выключаться при помощи ручного переключа-
теля.
Описанная схема надежна в работе, так как автоматический
регулирующий потенциометр и электроконтактный манометр,
включенные последовательно в цепь обмотки магнитного пуска-
теля, практически исключают возможность нарушения режима
вследствие неисправности одного из указанных приборов. Они
как бы подстраховывают друг друга в работе.
Недостаток этой схемы, как уже указывалось, заключается
в том, что термопара находится далеко от источника тепла.
Трубчатые электрические печи могут быть сплошными и разъ-
емными, с открытой или закрытой спиралью. * Их мощность
и конструкция зависят от величины и формы корпуса автоклава,
а также от рабочих параметров, принятых для данного исследо-
вания.
Методы оценки коррозии
и исследования защитных пленок
Оценка коррозии — существенная часть всяких коррозионных
испытаний. Выбор метода оценки зависит прежде всего от ха-
рактера коррозии.
При высоких температурах в воде можно наблюдать практи-
чески все основные виды коррозии: общую, местную, межкри-
сталлитную и хрупкое разрушение. Соответствующие методы
оценки этих видов коррозии описаны в гл. Ш и VI.
Принципиально возможны четыре метода количественной
оценки скорости коррозии: 1) по потере металла; 2) по расходу
на реакцию воды (кислорода); 3) по количеству образовавше-
гося окисла; 4) по количеству образовавшегося водорода.
— 340 —
Трудности в учете количества воды, расходуемой на реакцию
в процессе коррозии, настолько велики, что второй метод прак-
тически не применяется.
При общей равномерной коррозии чаще всего применяют
методы, .указанные в п. 1 и. 3, которые представляют, собой два
варианта известного метода определения изменения массы. Этот
метод, состоящий в определении разности массы образцов до
и после испытания, в зависимости от качества пленки продук-
тов коррозии, а иногда от цели исследования, применяется в со-
ответствующем варианте. Если пленка достаточно прочная, не
осыпается й плотно прилегает к поверхности металла, причем
нет достаточно надежных способов ее снятия с этой поверхности,
интенсивность коррозии характеризуется увеличением массы об-
разца на единицу поверхности в единицу времени или просто
увеличением Массы на единицу поверхности в течение всего пе-
риода испытания. Если продукты коррозии могут быть удалены
с поверхности испытываемых образцов, определяется потеря
металла, отнесенная к площади образцов.
Первый вариант используется, например, при исследований
коррозионного поведения таких металлов, как цирконий, титан,
ниобий и сплавов на их основе. В частности, на цирконии и его'
сплавах при коррозии в воде высоких параметров образуете^
плотная пленка окисла ZrOg. Надежных методов снятия пленок
без растворения самого металла пока не существует, так как
химические реактивы, растворяющие этот окисел, не менее ин-
тенсивно растворяют и основной металл. Вплоть до того момен-
та, когда пленка начинает разрушаться (т. е. до «перелома» на'
кинетической кривой), количественные результаты, полученные
этим методом, с достаточной точностью отражают кинетические
особенности процесса. Однако после «перелома», когда часть
продуктов коррозии осыпается и учесть эти потери трудно, ме-
тод уже не позволяет точно оценить коррозию.
Этот метод как наиболее простой часто применяется и для
других металлов и сплавов в тех случаях, когда есть уверен-
ность, что продукты коррозии достаточно прочно удерживаются
на поверхности образцов. Это прежде всего относится к спла-
вам на основе алюминия.
Наиболее правильные результаты должны получаться только
тогда, когда есть возможность снять продукты коррозии с по-
верхности образцов и применить вариант оценки коррозии по
потере металла. Преимущество этого способа очевидно, так как
он позволяет с одинаковой высокой точностью изучать кинетику
процесса коррозии как до, так и после «перелома», наблюдаю-
щегося на кинетической кривой, независимо От того, теряются
ли продукты коррозии (за счет осыпания или растворения)
в процессе испытания.
Методы снятия продуктов коррозии с Таких распространен-
ных металлов, как железо, известны (см. гл. Ш), но для Многих.
-341 —
вновь созданных сплавов, используемых в реакторостроении, они
еще недостаточно разработаны. Сравнительно недавно был
предложен метод снятия продуктов коррозии со сплавов алюми-
ния с никелем 1106]. Снятие пленки этим методом осуществляется
в два этапа. Сначала образцы с пл.енкой электрохимически об-
рабатывают в насыщенном растворе борной кислоты путем
пропускания между образцом и платиновым электродом пере-
менного тока плотностью 20 ми!см2 (при напряжении 500—
600 ме) в течение 5—10 мин при комнатной температуре. Раз-
рыхленный и несвязанный с поверхностью окисел удаляется
щеткой. Затем пара образец — платина промывается в дистил-
лированной воде и переносится в раствор, содержащий 4%
СгОз + 10% Н3РО4, где образец подвергается катодной обработ-
ке в течение двух часов при температуре 80° С. Для сплавов
.алюминия с никелем метод дает хорошие результаты.
Пленка, образующаяся на сплавах алюминия, содержащих
кремний, может быть удалена обработкой в течение 5—15 мин
при 80° С в растворе следующего состава: 3% Н3РО44-7%'НС1О3,
причем контрольные образцы теряют при этом 0,02 мг!см2 [107].
Алюминиевые сплавы более сложного состава можно обраба-
тывать в растворе 20 а СгОз + 35 см? Н3РО4 на 1 дм? в те-
чение 30 мин при температуре 100° С с последующим погруже-
нием в 65%-ный раствор HNO3 на 5 мин. Обработка повторяет-
ся дважды [109].
Сплавы, испытываемые при высоких температурах и давле-
ниях, подлежат проверке на наличие межкристаллитной корро-
зий. Методы выявления межкристаллитной коррозии на алюми-
ниевых сплавах и нержавеющих сталях описаны в гл. VI, п. 2.
Для определения скорости коррозии по количеству выделив-
шегося водорода может быть использован метод, описанный
в работе [108]. Сущность его заключается в-том, что определяет-
ся количество водорода, выделяющегося в процессе высокотем-
пературной коррозии. Для этой цели применяется особый обра-
зец в виде капсулы, в которых заливается коррозионная среда.
Капсулы изготавливают тонкостенными (0,5—0,7 мм) длиной
~30 мм с внутренней поверхностью —5 см2. Внутренняя по-
верхность тщательно обрабатывается, чтобы добиться мини-
мальной шероховатости. После заполнения средой капсула зава-
ривается. Предварительно капсулы сплющивались с таким рас-
четом, чтобы при нагреве возникающее после расширения воды
гидравлическое давление компенсировалось за счет возвращения
капсулы к прежней форме. Таким образом, коррозионная среда
(вода) заполняет весь внутренний объем капсулы, а давление
внутри нее несколько превышает давление пара жидкости при
температуре испытания. Авторы применяли этот метод для оп*
ределения скорости коррозии стали.
В процессе коррозии выделяющийся на границе раздела меж-
ду жидкостью и стенкой капсулы атомарный водород может,
— 342 —
<с одной стороны, диффундировать через стенку капсулы наружу
и здесь рекомбинировать в На; с другой стороны, он может ре-
комбинировать и внутри капсулы, создавая определенное давле-
ние. Повышение давления водорода внутри капсулы приводит
к тому, что его выделение в процессе коррозии внутрь капсулы
уменьшается и в то же время какая-то часть молекулярного во-
дорода внутри капсулы при повышенной температуре диссоции-
рует на атомы, которые могут диффундировать через стенку.
В конце концов наступает такой момент, когда количество вы-
делившегося внутрь капсулы водорода и количество диссоции-
рованного водорода, продиффун-
дировавшего через стенку капсу-
лы, будут равны. Другими ело*
вами, количество водорода, диф-
фундирующего через стенку кап-
сулы в вакуумную систему, и ко-
личество водорода, образующе-
гося ® результате коррозионного
процесса, будут равны.
Количество выделившегося
на внешней стенке капсулы водо-
рода определяется в вакуумном
приборе. Схема установки для
испытаний представлена на
рис. 211. Образец в виде капсу-
лы 5 висит на нити с противове-
сами 3, которые перемещаются
при помощи внешнего магнита.
Стеклянный отросток системы,
в котором подвешена на молиб-
деновой гибкой проволоке капсу-
ла, помещается в электрической
печи 6 с термоизоляцией 7. Си-
стема герметически закрывается
и эвакуируется путем открытия
вакуумного крана 9 в трубопро-
вод 8, ведущий к вакуумному насосу. После эвакуации систе-
мы кран закрывается и капсулу опускают в печь. Капсула очень
быстро нагревается. Давление в вакуумной системе резко воз-
растает, что фиксируется манометром Пирани 12, связанным с
самописцем проводниками /. Измерительная система предвари-
тельно калибруется чистым сухим водородом. После достиже-
ния давления, равного 65-10~3 бар (50 мм рт. ст.), которое еще
обеспечивает достаточную разность между давлением внутри
капсулы и снаружи, система вновь откачивается до давления
<1,3-10~4 бар (0,1 мм рт. ст.). Кран опять закрывается до тех
пор, пока давление не достигнет 65 ♦ 10“3- бар (50 мм рт. ст.).
Этот процесс повторяется так часто, как это необходимо..Перво-
— 343 —
Рис. 211. Схема установки для
измерения выделившегося в про-
цессе коррозии водорода [108]:
/ — провода к самописцу; 2 — крыш-
ка; 3 — противовесы; 4 — боковое
плечо; Б — капсула; 6 — электриче-
ская печь; 7 — изоляция; S — трубо-
провод; 9 — вакуумный кран; 10 —
охлаждаемый отросток; И — место
спайки стекла с металлом; 12 — ма-
нометр Пирани; 13 — известный объем
начально процесс повышения давления протекает очень быстро;
но по прошествии некоторого времени замедляется.
Течь в капсуле и появление в системе воды легко обнаружи-
ваются с помощью охлаждаемого жидким азотом стеклянного
отростка Р, в котором появляется лед (снег).
Расчет скорости коррозий ведут по количеству водорода
и уравнению реакции металла с водой.
При тщательной подготовке и выполнении опыта, а также
при известном навыке метод дает хорошо воспроизводимые ре-
зультаты.
Наряду с описанным выше методом определения коррозии по
изменению массы исключительно ценные ’сведения о свойствах
образующихся на металле в процессе высокотемпературной кор-
розии защитных пленок можно получить, применяя электроно-
графические и электрохимические методы их исследования.
К ним относится определение электрического сопротивления
и емкости, скорости роста пленки при электрохимической поля-
ризации, а также химический и рентгеноструктурный анализ са-
мих пленок.
Методы исследования пленок, описанные в гл. V, могут быть
применены и к пленкам, образующимся в процессе коррозий
в высокотемпературной воде.
Дополнительные сведения по этим вопросам можно почерп-
нуть из работ {99, 102—104, 110, 111].
ЛИТЕРАТУРА
I. Акимов Г. В. Теория и методы исследования коррозии металлов..
Изд-во АН СССР, 1945.
2. Warren D. Corrosion, v. 15, 221, Д959.
3. Медовар Б. И. Сварка хромоникелевых аустенитных сталей. Киев.-
Машгиз, 1954.
4. Левин И. А. Тр. Ин-та физической химии АН СССР, вып. 2. Изд-во
АН СССР, 1951, 188.
5. Эванс Ю. Р. Коррозия и окисление металлов. Пер. с аигл. под ред.
И. Л. Розенфельда. Машгиз. 1962.
6. Shirly Н. Т., Truman J. Е. J. Iron and Steel Inst., 1952, № 171, 354;
J. Appl. Chem., 1954. № 4, 273.
7. Streicher H. R. Werkstoffe und Korrosion, 1954, H 5, 363.
•8. Л e в и и И. А., В о л и к о в а И. Г. Заводская лаборатория, 1964, т. XXIX,
№ 2, 180.
9. Standarts, Л S. Т. М., I, 1949.
10. Roach I. D., HoIIeubeck Н. Н. Iron Age, 1954, 174, № 25, 100.
1)1 >. Кунина А. И., Пелен Т. Сб. НИИХИММАШ, № 117, Конструкцион-
ные неметаллические материалы и коррозия металлов. Машгиз, 1954.
12. Розенфельд И. Л., Вруцевнч 3. А., Веганов М. В. Заводская
лаборатория, 1955, т. XXI, № 5, 557.
13. Левин И А., Бабаков А. А., Л юта ев а 3. В. Вестник инженеров,
и техников, .1950, № 2.
14. Еремин Н. И., Евг.р.афов А. В. Тезисы докл. иаучно-технич. сове-
щания по магнитным методам производственного контроля и новым
методам магнитных измерений. Уральский филиал АН СССР. Свердловск,.
1956.
— 344 —
К 5. Веденеева М. А., Томашов Н. Д. Сб. «Межкристаллитная корро-
зия и коррозия металлов под напряжением» Машгиз, 1960, 46.
16. Веденеева М. А., Панов А. В., Томашов Н. Д. Заводская лабо-
ратория, 1957, т. XX1II, К® 1, 64.
17. Постников В. С.. Пигузов Ю. В. Прибор для определения моду-
ля сдвига и внутреннего трения проволочных образцов. ИТЭИН,
№ ПС-55-448, 1955.
18 Л евнн И. А., Г инцбер г С. А. Тр. Ин-та физической химии, вып. Ш.
Изд-во АН СССР, 1951, 69.
19. Т о м а ш о в Н. Д., Дерягина О. Г. Заводская лаборатория, 1957,
т XXIII, № 6, 679.
20. Павлов С. Е. Коррозия дуралюмина. Оборонгиз, 1949.
21. Mea-rs К. В., Brown R. Н. Industr. Eng. Chem., 1941, 33, 1001.
22. Голубев А. И. Коррозионные процессы на реальных микроэлементах.
Оборонгиз, 1963.
23. Сб. «Коррозия металлов и методы борьбы с ней». Оборонгиз, 1955.
24. Logan Н. L. Metall. Progress, 1950, 58, 211.
25. Ketch'om S„ J., Beck W. Corrosion, 1960, 16, Ke I, 125.
26. Павлов С. E. Коррозия плакированного дуралюмина. Оборонгиз, 1940.
27. Борови-ков А., Попов С. Гражданская авиация, 1956, К® 3, 29.
28. Budd М. К., Booth F. F. Corrosion, 1962, v. 18, № 5, 197t.
29. Ребиндер П. А., Лихтман В. И. ДАН СССР, 1947, т. 56, № 7.
30. ‘ G i 1 b е г f О. Т., Н u d d е п S. Е. J. Inst, of Metals, 1950, v. 77, 237.
31. E d e 1 e а п у С. E. J. Inst, of Metals, 1952. v. 80, 187.
32. Шварц Г. Л., Кристалль М. М. Коррозия химической аппаратуры.
Машгиз," 1958.
-33. Зарецкий Е. М. ЖПХ, 1951, т. 24, К® 6, 614.
.-34. Павлов С. Е. Сб. «Коррозия и защита металлов». Оборонгиз, 1957, 184.
35. Mears R. В., Brown R. Н., D i х Е. Н. Symposium on Stress—Corro-
sion Cracking of Metals, ASTM—AIME, 19'44, p. 323.
36. D i x E. H. Amer. Inst, of Metals, Symposium on Stress—Corrosion Crac-
king of Metals, ASTM—AIME, 1944, p. 2.
37. В г e e n e r P. T. fflr Metallkunde, 1953, 44, K® 3, 85.
38. Ливанов В. А., Елагин В. И., Е л и и а Г. В., Ш т е й н и и г е р В. Р.,
Коряковская Л. А. Тр. МАТИ, вып. 55. Оборонгиз, 1962, 49.
39. Тимонова М. А. Сб. «Коррозия и защита металлов». Оборонгиз,
1957, 260.
40. Р г е i s t D. К., В е с k Е. Н., F о п t а и а М. G. Trans. ASTM, 47, 473, 1955.
41. Hiedenreich A. D., Me Nuli'y R.. Gerould R. C. Trans. AIME,
166, 15, 1946.
42. Романов В. В. Коррозионное растрескивание металлов. Машгиз, 1960.
-43. Бобылев А. В. Коррозионное растрескивание латуни. Металлургиздат,
1955.
44. С ко рч ел летти В. В., Титова В. А. ЖПХ, 1953, т. 26, К® 1, 41.
45. Parkins Р. W. J. Iron Steel Inst, 1962, К® 172, 149.
46. Phelps Е. Н., Login о w A. W. Corrosion, 1960, № 7.
47. А ж о г и н Ф. Ф. Сб. «Коррозия и защита металлов». Оборонгиз, 1957, 98.
48. Farmery Н. К., Evans U. R. J. Inst. Met, 84, 413, 1955—1956.
49. Matthaes К. Jahrb. Ljlientahl Geseltsch. Luftforsch., 1936, p. 404.
«$50. Зарецкий E. M. ЖПХ, 1947, t. 20, № 9, 830.
51. Павлов С. E. Заводская лаборатория, 1947, т. XIII, К® 10, 1234.
52. Fontana М. G. Industrial and Engineering Chem.. 1955, 47, K® 6, 91A.
53. Тимонова M. А., Ершова T. И. Заводская лаббоатория, 1961.
т. XXVII, № 4, 447. '
54. Brenner P. Z. fur Metallkunde, 1938, 30, 23.
55. Тр. ЦНИИТМАШа, K° 77, 1955.
56. Томашов H. Д„ Модестова В. H„ Блиичевский Г. X Тр.
Ин-та физической химии АН СССР. Изд-во АН СССР, 1959, 64.
57. Р о м а н о в В. В. Заводская лаборатория, 1961, т. XXVII, К® 11, 1377.
58. Зарецкий Е. М. Заводская лаборатория, 1961, т. XXVII, К® 11, 1378.
— 345 —
59. А к и м о в Г. В. Основы учения о коррозии и защите металлов. Метал*
лургиздат, 1946.
60. Томатов Н. Д., Керни ч Н. П., Ж У к Н. П. Сб. «Производство
и обработка стали и сплавов», № 38, 1958, 584.
61. Бабаков А. А. Сталь, 1945, № 5, 189.
62. Максимчук В. П., Розенфельд И. Л ДАН СССР, I960, т. 131г
№ 2, 354.
63. Rosenfeld I. L. und Maximtschuk W. P. Zeitschr. fur Physika-
lische chemie, I960, 215, 25.
64. Розенфельд И. Л., Максимчук В. П. ЖФХ, 1961, г. 35, № 8,
1882; № 11, 2561.
65. Колотыркии Я. М. Успехи химии, 1982, т. 31, вып. 3, 322.
66. Т о м а ш о в Н. Д., Чернова Г. П., Маркова О. Н. Сб. «Коррозия
металлов и сплавов». Металлургнздат, 1963, 73..
67. Розенфельд И. Л.. Максимчук В. П. Тр. Всесоюзной межвузов-
ской конференции по борьбе с коррозией. Гостоптехиздат, 1962, 6.
66. Розенфельд И. Л., Данилов И. С. Тр. Всесоюзной межвузовской
научной конференции по борьбе с коррозией. Гостоптехиздат,' 1962, 17;
ДАН СССР, 1961, т. 139, № 2, 414.
69. Розенфельд И. Л., Данилов И. С. ДАН СССР, 1962, т; 147, № 6,
1417.
70. Frank U. F. I-st International Congress on Metallic Corrosion, London,.
1961, p. 197.
71. Розенфельд И. Л., Маршаков И. К. ЖФХ, 1956, т. 30, вып. 12;
1274; 1957, т. 31, вып. 10, 2328; 1959, т. 33, вып. 1, 260.
72. Robinson Е. F. Corrosion Technology, I960, № 8, 237.
73. Streicher М. A. J. Electrochem. Soc., 1956, 103, № 7, - 375.
74. Сб. «Коррозия металлов», т. I, И. Пер. с англ, под ред. В. В. Скорчел-
летти. Металлургнздат, 1952.
75. Brennert S. J. Iron, and Steel Inst., 1937, v. 135, № 1, 101P.
76. Розенфельд И. Л., Максимчук В. П. Заводская лаборатория,
1960, т. XXVI, № 3. 288.
77. Stoffels A., Schwenk W. Werkstoffe und Korrosion, 1961, 12, H. 8,'
493.
78. Sielberrad D. Engineering, 1912, № 28.
79. P я ж с к а я T. К. Металлург, 1939, № 12.
80. Speller F., La Gue F. Corrosion, 1954, v. 20, № 5.
81. В r i m e I о w I. Engineering, 1956, № 6.
82. Гликман Л. A., 3 о б а ч e в Ю. E. Металловедение и обработка метал-
лов, 1956, № 11.
83. Том а шов Н. Д. Прикладная химия, 1953, т. 26, № 12.
84. Богачев И. Н., Минц Р. И. Кавитационное разрушение железоугле-
родистых сплавов. М.— Свердловск. Машгиз, 1959.
65. Меттер И. Успехи физических иаук, 1948, т. 35, вып. 1, 52.
86. К р я и и и И. Р., Т и м е р б у л а т о в М. Г. Тр. ЦНИИТМАШа, № 27,
1962. 4. *
87. G a i h е s N. Physics, 1932, № 3, 209.
88. Гликман Л. А.. Зобачев Ю. Е. Заводская лаборатория, 1954, т. XX,
№ 5, .593. 9,
89. П и с а р е в с к и й М. М. Котлотурбостроение, 1953, № 6.
90. Иванов Н. М. Сб. статей ВИГМ, вып. И, 1940.
91. Гавранек В. В. Тр. Харьковского политехнического ии-та, г. 9. серия
металлургий., вып. I. 1957.
92. Б о г о с л о в с к и й Ю. В., К о м а р ь Г. Д.,' Рыкова А. В. Заводская
лаборатория, 1954, т. XX, № 6.
93. Воскресенский И. Н. Коррозия и эрозия судовых гребиых винтов.
Судпромгиз, 1949.
94. Гликмаи Л. А. Коррозионно-механическая прочность металлов.
М.— Л., Машгиз, 1955.
— 346 —
95. ВукаловичМ. П. Термодинамические свойства воды и водяного пара.
Машгиз. 1951.
96. Теплофизические свойства веществ. Под ред. Н. Б. Варгафтика. Госэнер-
гоиздат, 1956.
97. Green N. D. J. Chem. Engin., 1956, № 4.
98. Tomas D. E., Kass S- J. Electrochem. Soc., 1957, 33, 2, 95.
99. Коррозия и изиос в водоохлаждаемых реакторах. Под ред. Де Поля.
Гос. союзное изд-во судостроительной промышленности, 1959.
100. Розенфельд И. Л., Ольховников Ю.*П., Сударикова А. А.
Влияние состава воды иа коррозию циркониевых сплавов при высоких
температурах и давлениях. Corrosion of Reactor Materials Intern. Atomic
Energy Agency, Vienna, 1962.
101. Mac Lennen D. F. Corrosion, 1961, 17, 4, 181—184.
102. Фр ей май Л. И. Сб. «Коррозия реакторных материалов», под ред.
В. В. Герасимова. Атомиздат, 1960.
ЮЗ. Bas si A., Brutto Е. et al. Zircaloy—2 and Stainless Steel Corrosion
in Flowing Wet Steam. Corrosion of Reactor Materials Intern. Atomic
Energy Agency. Vienna, 1962.
104. Ако л ьз ин П. А. Коррозия металла паровых котлов. Госэяергоиздат,
1957.
105. Герасимов В. В., Громова А. И. и др. Сб. «Коррозия реакторных
материалов», под ред. В. В. Герасимова. Атомиздат, 1960.
106. Dr ale у 1 Е., Ruther W. Е. Corrosion, 1956, 12, 480—490.
107. V i d е m К- Pitting Corrosion of Aluminium in Contact with Stainless
Steel, Corrosion of Reactor Materials Intern. Atomic. Energy Agency, Vien-
na, 1962.
108. Bloom M. C„ Krufeld M. J.'Electroch. Soc., 1957, 104, 264—269.
109. Герасимов В. В., Громова А. И., Шаповалов Э. Т. Сб. «Кор-
розия реакторных материалов», под ред. В. В. Герасимова. Атомиздат,
1960.
ПО. Кубашенекий О., Гопкинс Б. Окисление металлов и сплавов. ИЛ,
1955.
111. Том а шов Н. Д Теория коррозии и защиты металлов. Изд-во АН
СССР,- I960.
Иосиф Львович РОЗЕНФЕЛЬД
Кира Александровна ЖИГАЛОВА
УСКОРЕННЫЕ МЕТОДЫ
КОРРОЗИОННЫХ ИСПЫТАНИЙ
МЕТАЛЛОВ
в
Редактор издательства М. И. Заславская
Технический редактор Н. А. Коровина
Корректоры С. Н. Степанова и Л. И. Тубина
Переплет художника Ю. А, Давыдова
@
Сдано в производство 1/VI 1965 г.
Подписано в печать 22/XI 1965 г.
Бумага 60 X 9O’/16 — 10,68 бум. л.
21,75 печ. л.
Уч.-иад. л. 22, 64
Заказ 1338 Изд. № 4486
Т-15421
Тираж 2810 экз. Цена 1 р. 40 к.
Сводный темппан по металлургии
1965 г. п. 143
®
Издательство «Металлургия»
Москва Г-34, 2-й Обыденский пер., 14
Экспериментальная типография
ВНИИПП Государственного комитета
Совета Министров СССР по печати
Москва, Цветной бульвар, 30
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Стр. Строк Напечатано Должно быть
27 6 СИ. 0,44—3 0,443
43 4 си. 0,040 см 0,040 мм
82 12 св. I % NaClg 1% NaCl
128 14 сн. электродных электронных
164 4 сн. УПМ-1М УПУ-1М
265 23 св. стойкости в стойкости к
.315 16 св. 1Х18Н9Т и 1Х18Н9Т к
Зак. 1338.