/















































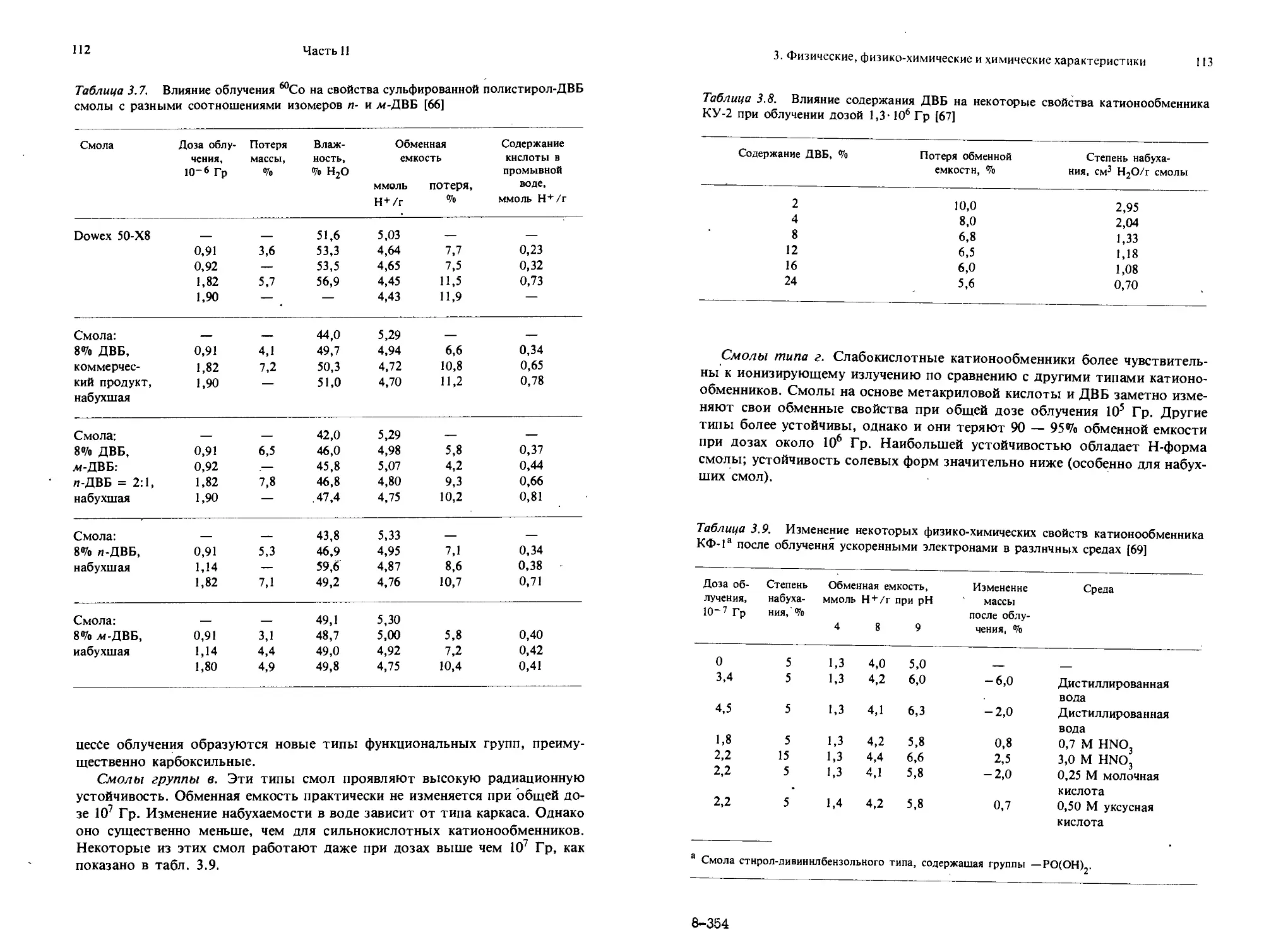
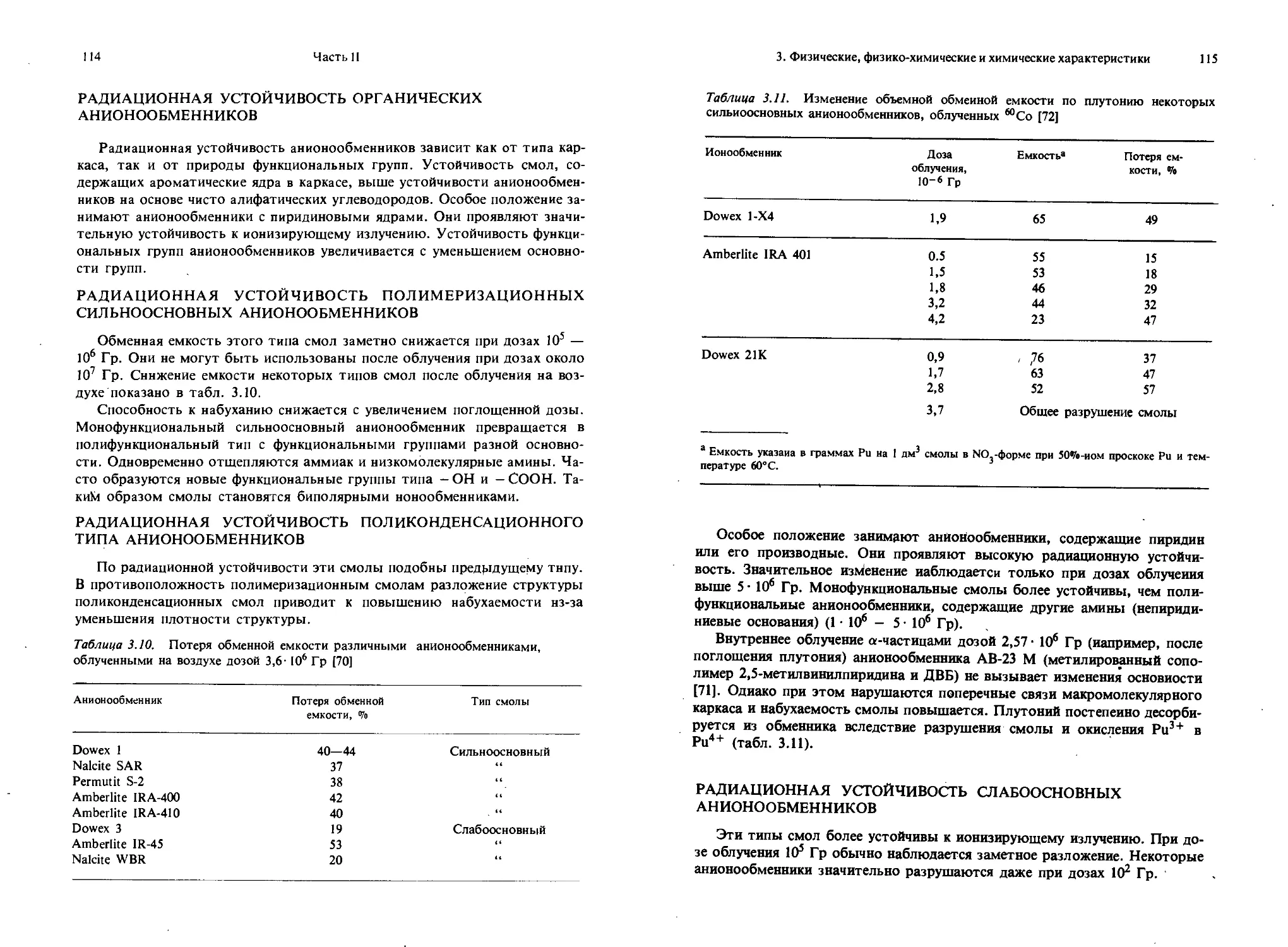



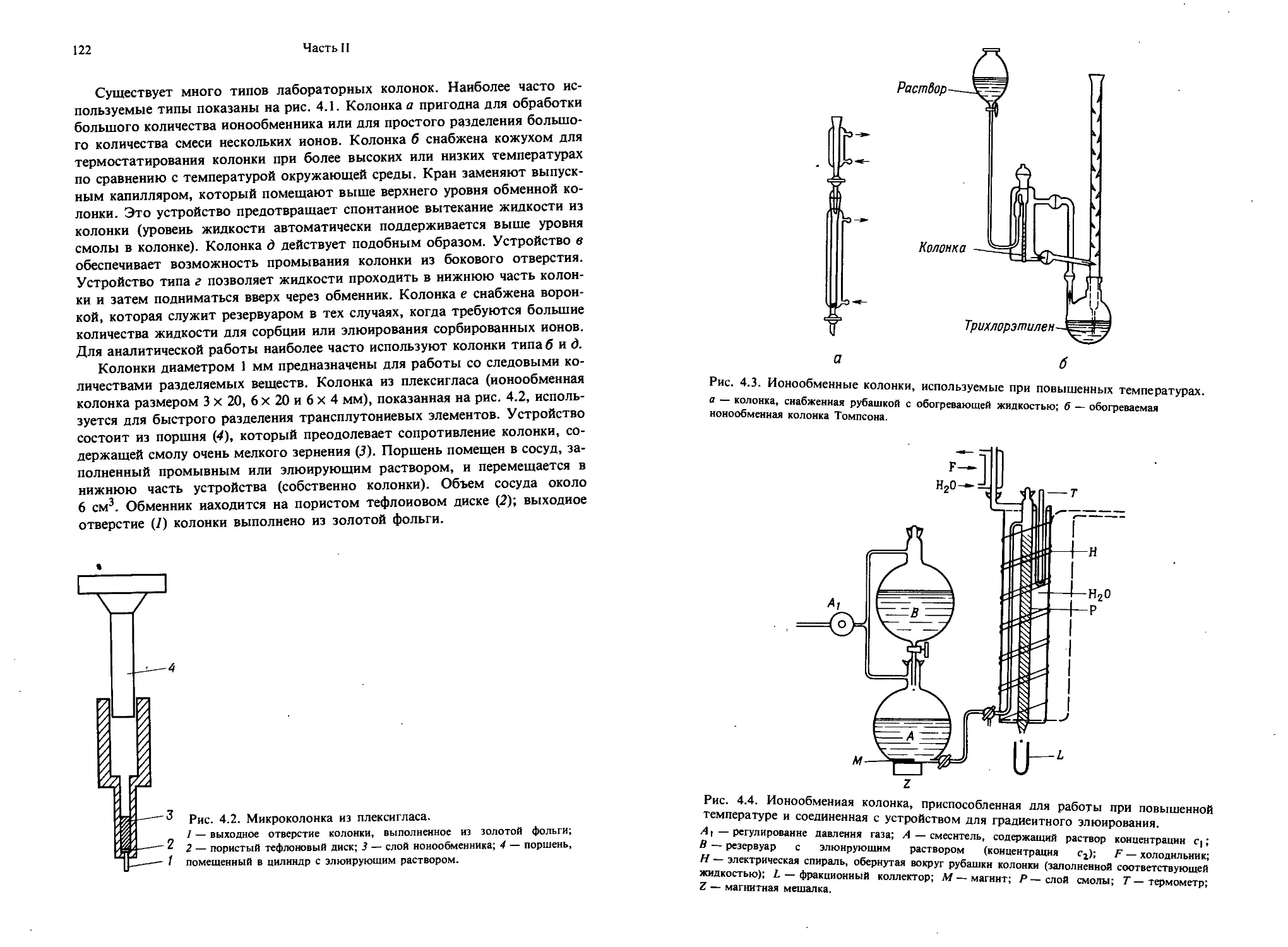








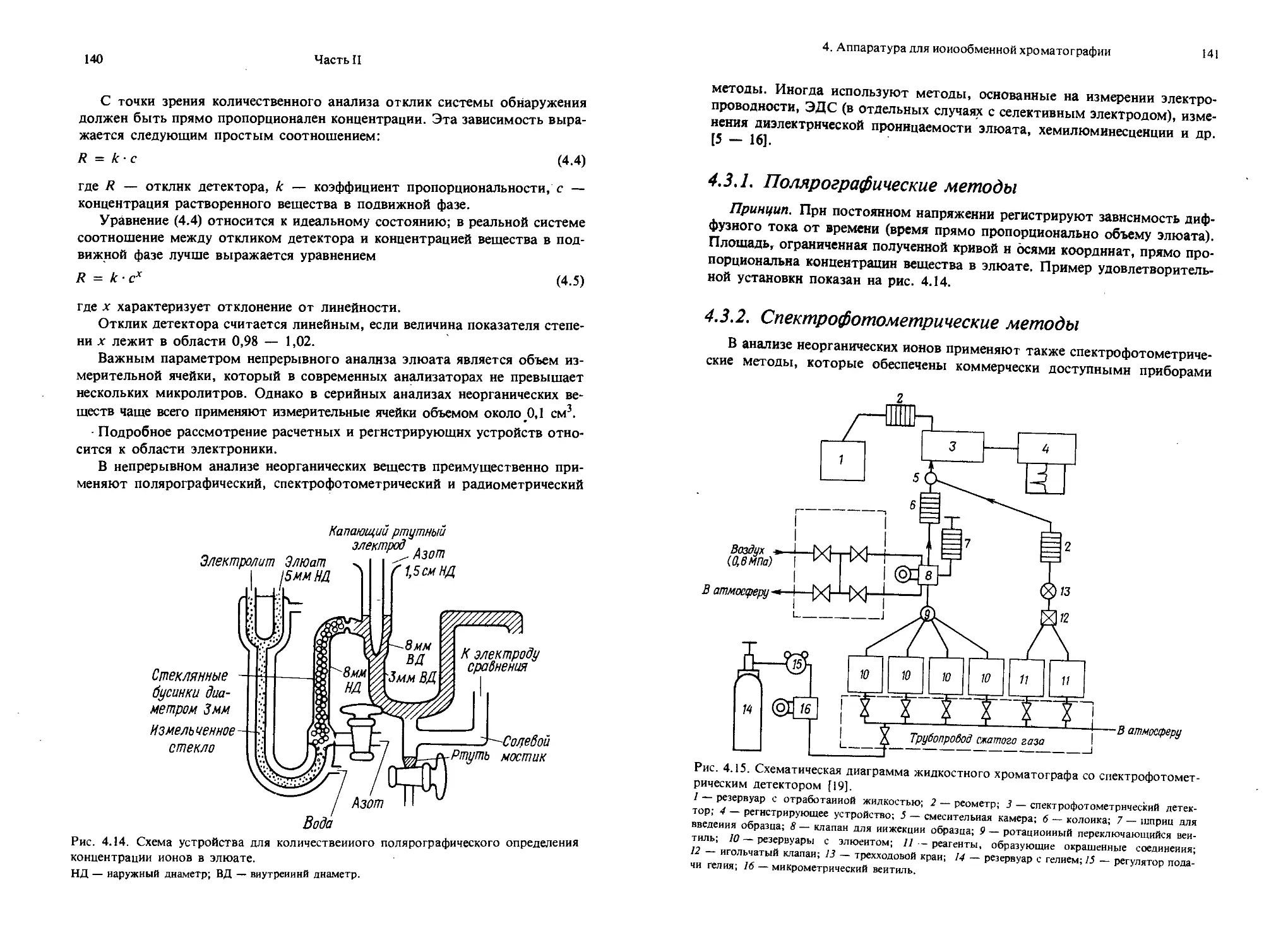














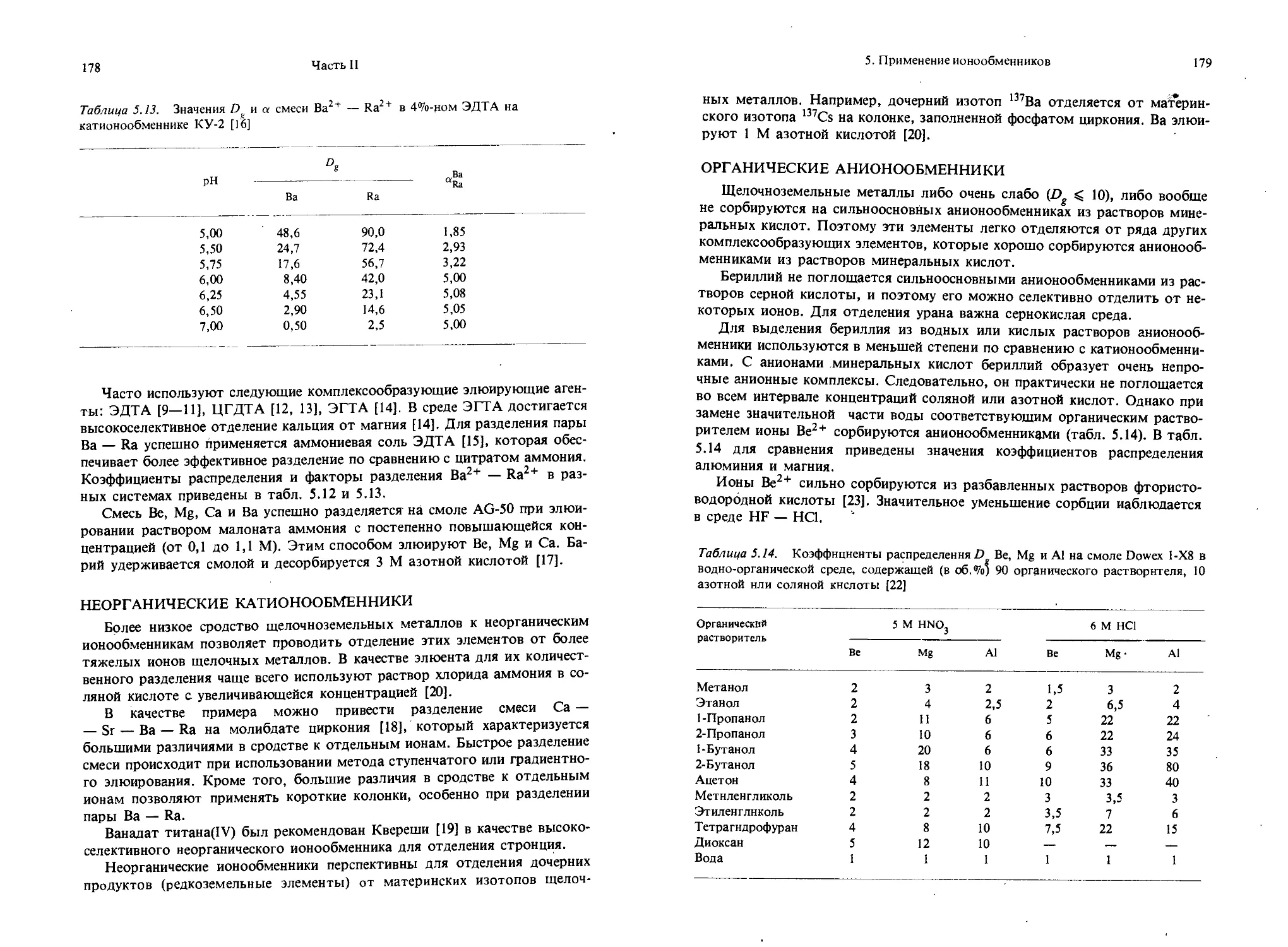










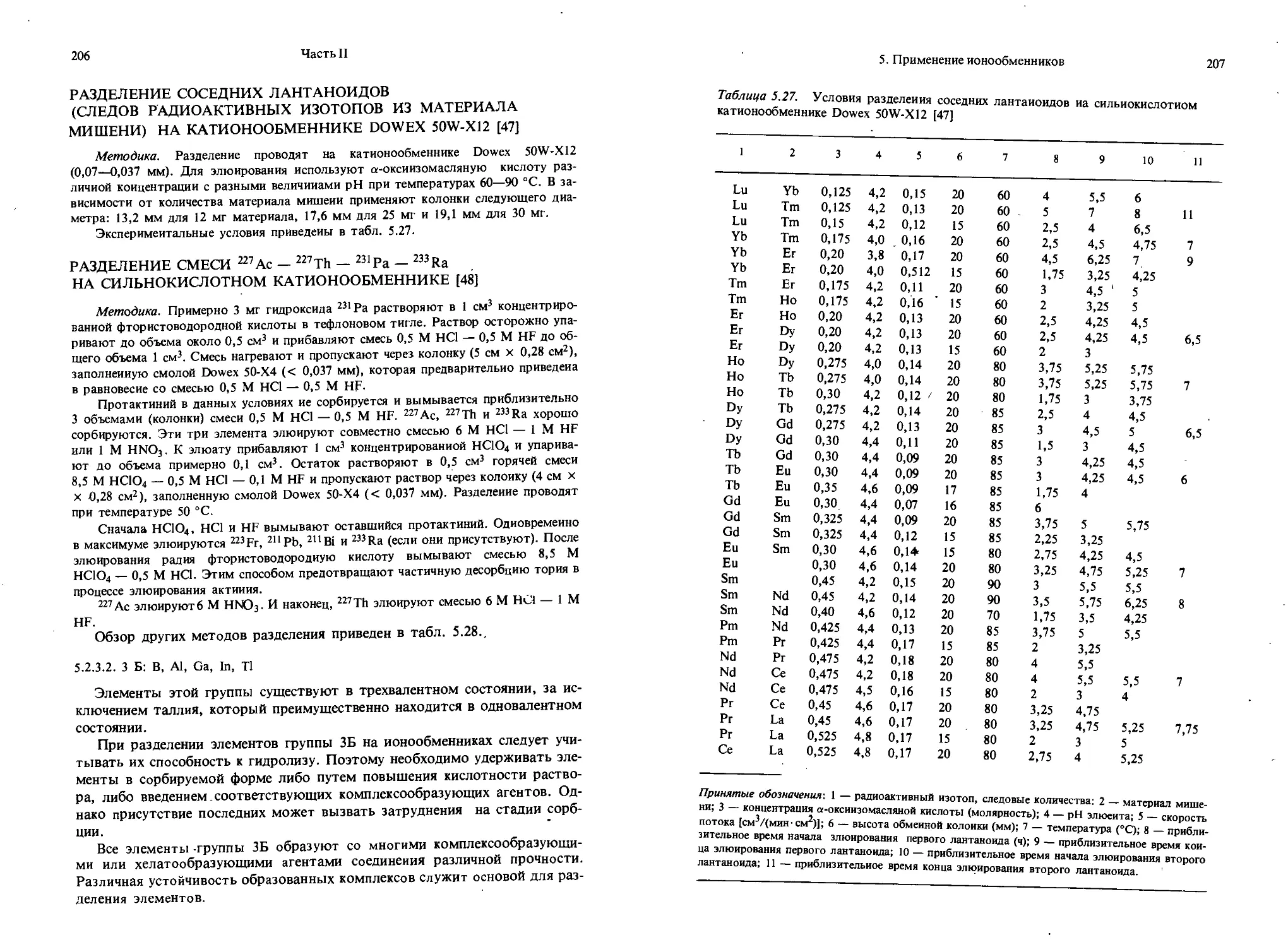
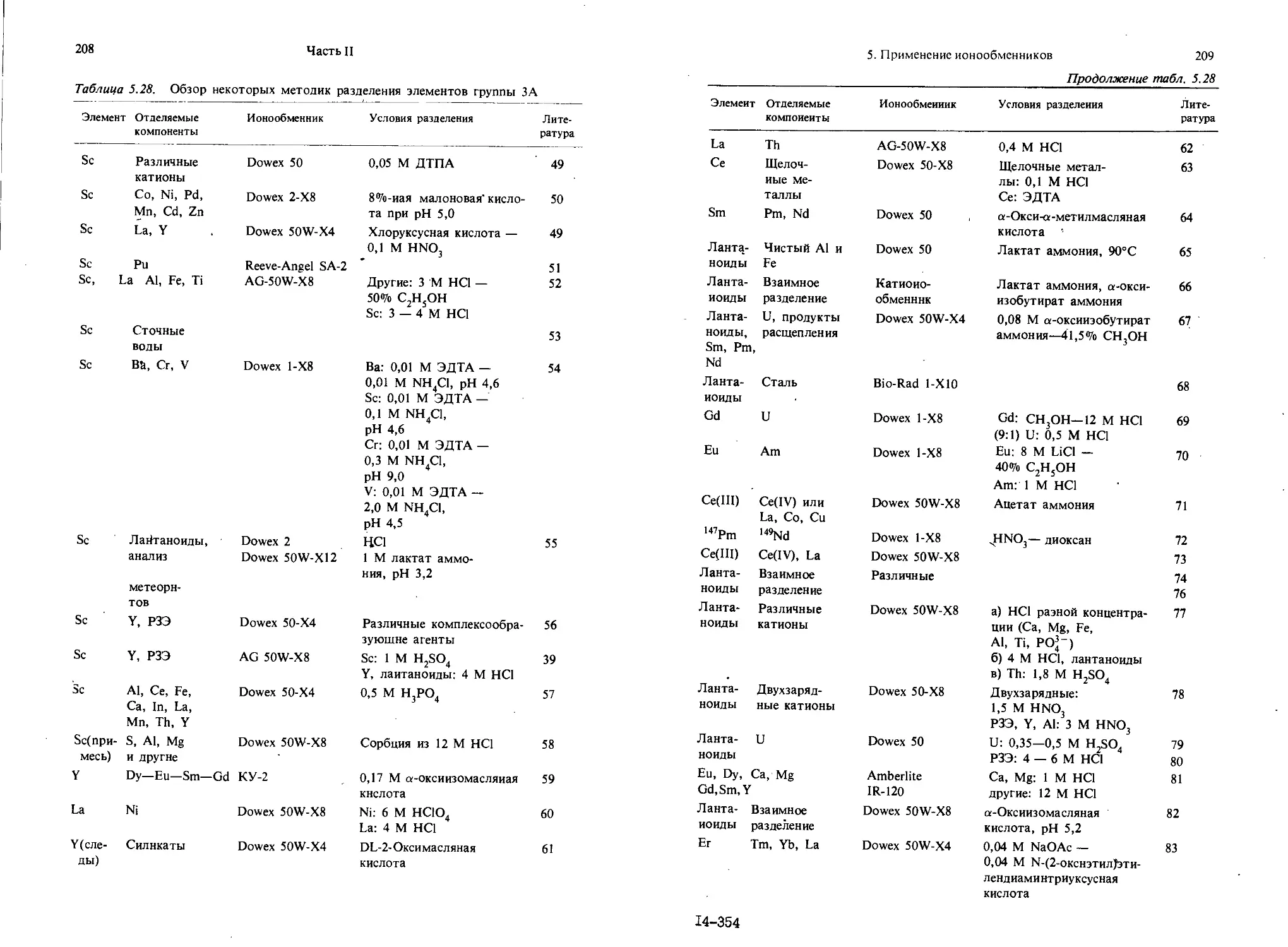








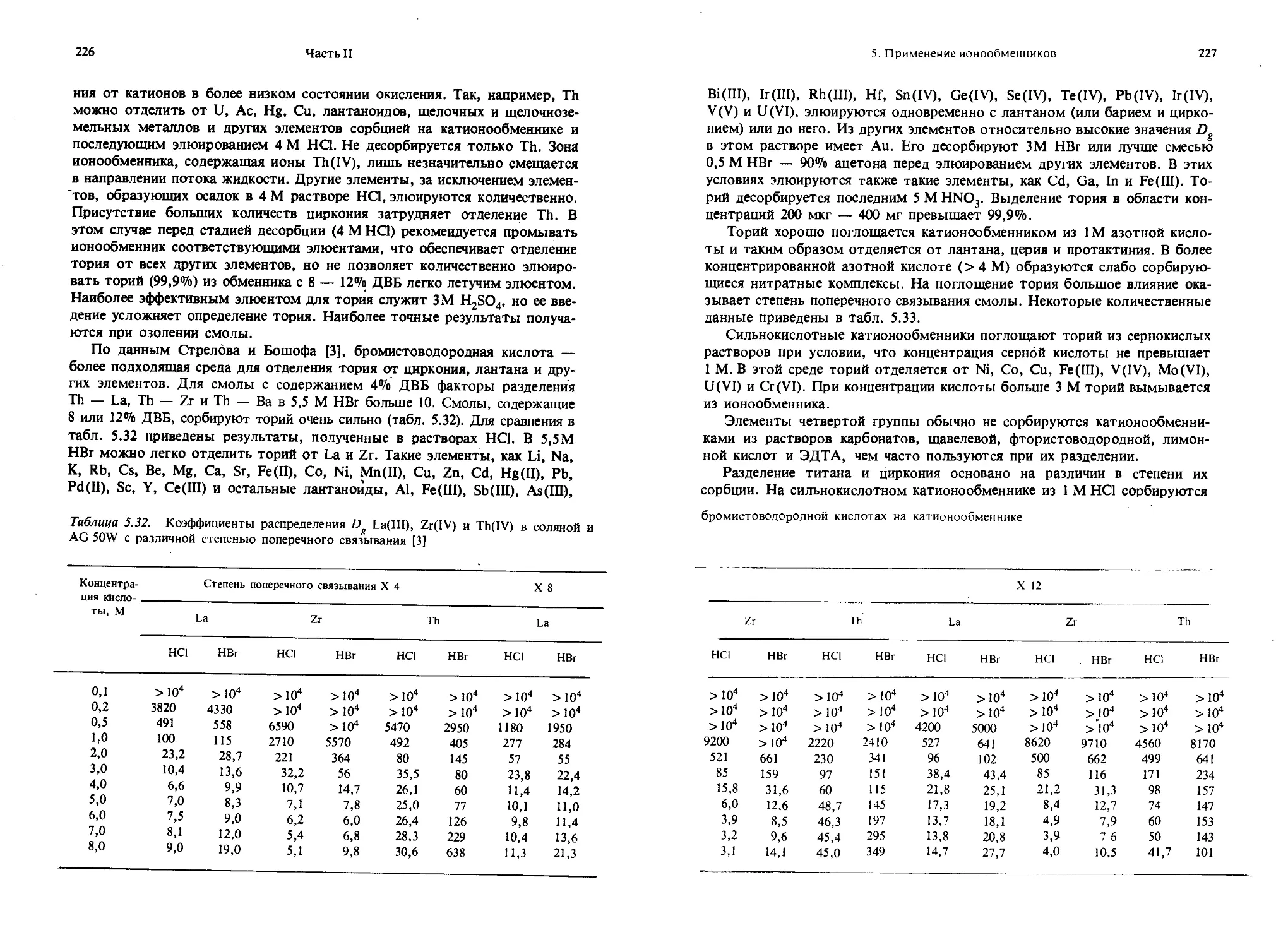


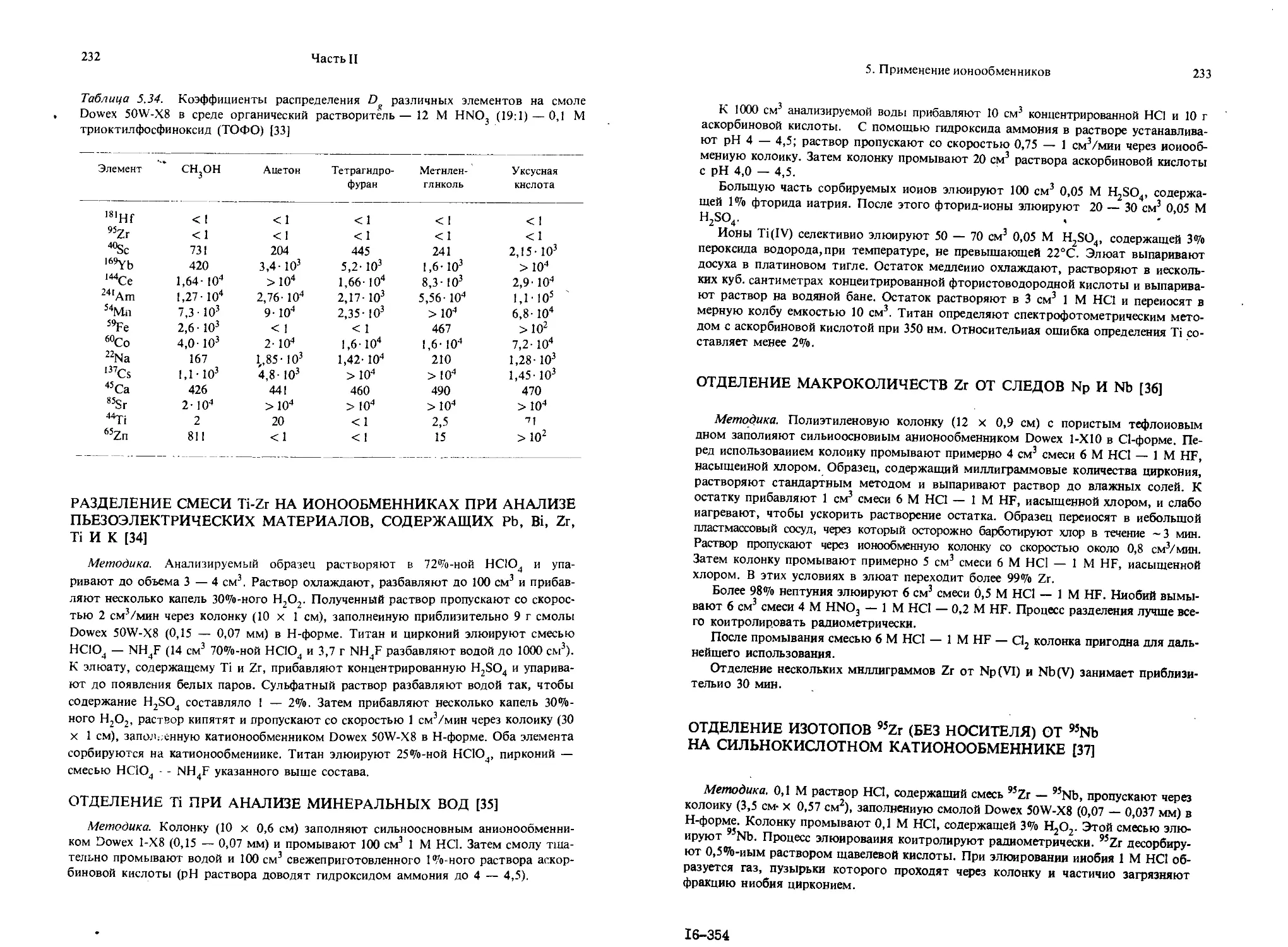













Текст
Milan Marhol
Ion Exchangers in Analytical Chemistry
Their Properties and Use in Inorganic Chemistry
Academia / Prague 1982
М. Мархол
Ионообменники в аналитической химии
Свойства и применение в неорганической химии
В 2-х частях
1
Перевод с английского канд. хим. наук О. П. Швоевой
Москва «Мир» 1985
ББК 24.4
М 29
УДК 543
Мархол М.
М 29 Ионообменники в аналитической химии: В 2-х частях. Ч. 1.
Пер. с англ. — М.: Мир, 1985. — 264 с., ил.
В книге, написанной автором из ЧССР, собраны обширные данные по ионообменникам различных классов, рассмотрено применение ионообменников для решения конкретных задач, приведены обобщающие таблицы н графики по свойствам и сорбционным характеристикам многих систем, представляющих интерес для аналитиков. В русском издании книга выходит в двух частях.
В ч. 1 описаны основные свойства ионообменников и ионообменных процессов, рассмотрены некоторые теоретические аспекты, связанные с использованием ионообменников в аналитической химии, обобщены физические свойства ионообменников и методы их определения, уделено внимание лабораторному оборудованию. Здесь помещена первая часть главы по применению ионообменников для разделения смесей неорганических ионов.
Книга представляет интерес не только для химиков-аналитиков, но может служить в качестве хорошего справочника для специалистов, работающих в других областях химии: радиохимии, биохимии и пр.
1804000000 — 113
М---------------------93 — 85, ч. 1
041(01) — 85
ББК 24.4
543
Редакция литературы по химии
© Milan Marhol, 1976, 1982
Translation © Vdclav Vesely, 1982
© Перевод на русский язык, «Мир», 1985
Предисловие
Обмен ионов, обнаруженный в почвах в 1862 г., вначале казался совершенно необычным, научно непознаваемым явлением. Однако за 125 лет, прошедших с момента этого открытия, он прочно занял свое место в ряду физико-химических сорбционных процессов. Создание теории ионообменных процессов, исследование и синтез разнообразных минеральных и органических, природных н синтетических сорбентов, разработка современной аппаратуры и приборов — все это, дополняя друг друга, определило применение ионообменных процессов в научных и технологических целях.
Во многих работах ионообменные процессы были предложены в качестве способа решения химико-аналнтических задач. В самом общем виде в гетерофазной системе «ионообменный сорбент — раствор» можно осуществить абсолютное и относительное концентрирование определяемого компонента. Конечно, эти процессы в ходе аналитического определения являются вспомогательными, но во многих случаях они необходимы, иначе их применение было бы неоправданным на фоне интенсивно развиваемых разнообразных прямых химических, физико-химических и физических методов современной аналитической химии. При недостаточном пределе обнаружения существующих или доступных в конкретной ситуации методов анализа прибегают к абсолютному концентрированию, например, путем упаривания, экстракции, осаждения. В ионообменном методе абсолютное концентрирование проводят поглощением определяемого элемента ионообменным сорбентом н регенерацией последнего малым объемом специально подобранного реагента (элюента). При недостаточной селективности существующих или доступных методов анализа прибегают к относительному концентрированию — отделению определяемого элемента от мешающих примесей. При ионообменном отделении мешающих элементов, далеких по ионообменным свойствам от определяемого компонента, относительное концентрирование выполняют простым пропусканием анализируемого раствора через слой (колонку) ионита в так называемых «динамических» проточных условиях (например, поглощение щелочноземельных металлов катионитом при титриметрическом определении сульфатов). Наконец, при отделении мешающих элементов, близких по свойствам к определяемому элементу (например, смеси щелочных, щелочноземельных, редкоземельных элементов, галогенов и пр.), относительное концентрирование осуществляют методом ионообменной хроматографии, т. е.. методом разделения сме
Предисловие
сей в колонке ноннта, основанным на различии скоростей перемешення определяемого и мешаюших компонентов по слою сорбента.
Аналитическому применению ионообменных процессов посвящено огромное число оригинальных работ и много специальных монографий. По существу, эти же вопросы рассмотрены и в книге М. Мархола. Однако своеобразие и достоинства книги вполне очевидны хотя бы из краткого рассмотрения ее основного содержания.
Несомненным достоинством книги М. Мархола является всестороннее освещение вопроса применения ионообменииков в аналитической химии. В ней дается общее представление о синтетических органических (иониты) и различных неорганических (оксиды и гидроксиды, гетерополикислоты, фос-форомолнбдаты и пр.) ионообменных сорбентах, подробно описаны основные свойства ионообменных сорбентов и методики их определения, а также кратко изложены вопросы теории: ионообменное равновесие и теория тарелок. Основное внимание автор уделяет изложению хроматографических методов разделения ионов по группам (подгруппам) периодической таблицы Д. И. Менделеева, включая редкоземельные и трансурановые элементы (материал этого раздела занимает почти половину книги). Кратко описано применение ионитов для определения общего солесодержания растворов и удаления мешающих ионов. Специальная глава посвящена технике выполнения ионообменных опытов.
Отличительная особенность монографии М. Мархола — высокая информативность. Наряду с описанием различных методик в ней приведен огромный материал справочного характера — таблицы характеристических свойств ионообменных материалов. Так, например, для 553 сильно- и слабокислотных катионитов, высоко- и низкоосновных анионитов, производимых 50 изготовителями в различных странах, в таблицах приведены сведения о способе получения матрицы ионита, функциональных группах, форме и размере зерен, числе поперечных связей, насыпной плотности, полной обменной емкости, термической устойчивости.
К достоинствам книги М. Мархола относится также и то, что она имеет характер руководства, облегчающего работу экспериментатора при проведении ионообменных процессов. Например, при описании ионообменных сорбентов формулируются рекомендации по их выбору для успешного решения конкретных аналитических задач, обосновывается выбор типа ионита (катионит или аннонит), степени его сшитости н зернения, приводится перечень основных свойств ионитов различных типов. Здесь очень полезна таблица, в которой сравниваются свойства однотипных ионитов, производимых в различных странах илн различными крупными фирмами, что облегчает пользование опубликованными в литературе методиками. В книге подробно изложена техника собственно хроматографических экспериментов: выбор и наполнение колонок, вспомогательные устройства (напорные емкости, коллекторы фракций) и методы непрерывного анализа хроматографических фильтратов (полярография, спектрофотометрия, радиометрия). В основной (пятой) главе книги, посвященной хроматографическому групповому разделению элементов, большое число методик описано на
Предисловие
столько подробно, что их можно воспроизвести, не прибегая к изучению оригинальных статей.
Конечно, как и всякая монография, книга М. Мархола не лишена некоторых недостатков. В этой связи представляется необходимым отметить два момента, которые надо иметь в виду при ее чтении.
Во-первых, при всестороннем, комплексном освешении вопроса применения ионного обмена в аналитической химии материал в книге излагается несколько фрагментарно. Так, полезные сведения по теории ионного обмена не используются при выборе условий проведения опытов и в лучшем случае выполняют чисто эвристическую, познавательную функцию. Равным образом выбор конкретной методики не базируется на свойствах ионообменных сорбентов, описанных в тексте и приведенных в справочном разделе. Не аргументируется и выбор технических средств эксперимента: формы колонки, метода наблюдения за ходом процесса. Было бы неправильным требовать от автора целостного изложения всего огромного аналитического материала, но наличие хотя бы отдельных примеров такого подхода было бы весьма желательным.
Во-вторых, в книге недостаточно отражены современные достижения в области аналитического применения ионообменных процессов. Формально это выражается в том, что в библиографии цитируется мало работ, появившихся в печати за последние годы (число работ, опубликованных с 1978 г., не превышает 15% от общего числа). Это приводит к ряду заметных пробелов. Так, явно недостаточное внимание уделено селективным (хелатообразующим) сорбентам, столь интенсивно и успешно разрабатываемым и используемым в настояшее время в аналитических и даже технологических целях. Практически не упоминаются волокнистые ионообменные сорбенты, весьма эффективные в анализе разнообразных неорганических объектов. Наконец, отсутствует описание и даже упоминание о сравнительно новом методе — ионной хроматографии, являющейся самым современным и высокоэффективным методом ионообменного анализа смесей близких по свойствам компонентов, который очень быстро развивается и имеет, несомненно, большое будущее.
С учетом всего сказанного при подготовке русского издания этот недостаток в некоторой степени компенсирован дополнением библиографии ссылками на более поздние работы, в которых подробно освещены указанные проблемы.
Несмотря на сделанные выше замечания, книга М. Мархола заслуживает в целом высокой положительной оценки, и ее с интересом и большой пользой прочтут специалисты, начинающие заниматься ионным обменом, инженерно-технические работники, перед которыми стоят сложные химико-аналнтические задачи, студенты и аспиранты химического и химико-технологического профиля. Книга поможет им ознакомиться с теорией и практикой интересной и перспективной области ионообменных процессов и послужит импульсом для более детального изучения оригинальной литературы последних лет.
М. Сенявин
Предисловие к чешскому изданию
В серии Comprehensive Analytical Chemistry публикуются работы, дающие всеобъемлющую информацию по изучаемой проблеме. В тех случаях, когда это невозможно, приведенный материал должен служить основой для последующих аналитических исследований.
Серия стремится к возможно более широкому охвату аналитических тем при детальном рассмотрении материала. Методы необходимо излагать настолько подробно, чтобы ими могли воспользоваться не только профессиональные аналитики, но и специалисты смежных областей. Если детальное изложение материала невозможно, указывается полная ссылка на соответствующую литературу.
Настоящий том посвящен только одной теме — аналитическому применению ионообменников. В т. II В Comprehensive Analytical Chemistry уже было дано краткое изложение этой проблемы. Но этот том появился 12 лет назад, и сейчас назрела "Острая необходимость в публикации нового более подробного обзора. Д-р Мархол любезно согласился адаптировать текст своей удачной чешской книги к нуждам серии, избегая по возможности перекрывания с материалом т. 11 В и акцентируя внимание на развитии науки об ионообменниках в последнее десятилетие.
Заслуживает благодарности вклад м-ра С. Джордана (Королевский университет, Белфаст) и д-ра С. Грэхема (Университет Бирмингема, Англия) в подготовку настоящего тома.
Г. Свехла
Моей жене Ольге
Предисловие автора
Ионообменники играют важную роль в современной аналитической химии. Они находят применение при разделении смесей ионов, концентрировании микроэлементов из чрезвычайно разбавленных растворов, в процессах получения и очистки растворов, реактивов, воды и во многих других случаях.
Из большого числа ионообменников в монографии отдано предпочтение обменникам на основе синтетических органических высокомолекулярных полимеров. Другим типам обменников, в частности производным целлюлозы, жидким ионообмеииикам, ионообменным бумагам и неорганическим ионообменникам, уделено относительно немного внимания.
Монография посвяшена ионообменникам, применяемым в аналитической химии неорганических систем. Ввиду ограниченного объема в книге не нашло отражения использование ионообменников в органическом и биохимическом анализе.
Существует некоторая связь отдельных разделов книги с главой «Ионный обмен» Ф. Севилле, опубликованной в т. II В Comprehensive Analytical Chemistry.
Монография состоит из трех частей (8 глав и 2 приложения). В первой части изложены общие понятия и основные характеристики ионообменников и ионообменных процессов. В нее включены также теоретические разделы, связанные с использованием ионообменников в аналитической химии, в частности для разделения смесей ионов.
Во второй части рассмотрены физические и физико-химические свойства ионообменников и методы их определения. Много внимания уделено лабораторному оборудованию и аппаратуре, применяемой при работе с ионооб-менниками. В самой большой главе этой части описано практическое применение ионообменников, приведены лабораторные методики.
Последняя часть книги представлена в форме таблиц и рисунков. В этих разделах приведены характеристики ионообменных материалов, выпускаемых различными фирмами, и большое количество данных по равновесным величинам коэффициентов распределения ионов в ряде систем. Наличие таких данных обусловливает возможность выбора соответствующего обменника, системы и лабораторной методики для решения конкретной аналитической задачи.
10 ’
Предисловие автора
Обзор литературы охватывает период до конца 1978 г.; работы, опубликованные до 1965 г., упоминаются относительно редко.
В книге использована система единиц СИ; перевод в эту систему иногда приводит к непривычным выражениям для ряда величин. Это, в частности, относится к выражению концентрации в миллимолях там, где раньше использовались миллиграмм-эквиваленты (например, обменная емкость обменника теперь выражается в миллимолях ионов Н1" или ОН-, относящихся к 1 г (или 1 см3) обменника, т. е. в общем виде: ммоль Мт±/т, где т — валентность рассматриваемого иона М.
В заключение автор благодарит д-ра В. Веселы (Институт ядерных исследований, ЧССР) за тщательный перевод книги с чешского языка на английский, инж. Дж. Штамберга (Институт макромолекулярной химии), инж. Дж. Алекса (Институт ядерных исследований) и инж. Ф. Дубски (факультет аналитической химии колледжа Химической технологии, Прага) за внимательное прочтение рукописи и критическое обсуждение, которое способствовало решению ряда проблем и улучшению рукописи. Автор выражает благодарность Н. Банковой за помощь в подготовке рукописи и сотрудникам библиотеки за помощь в обеспечении литературой.
Автор также признателен фирмам, производящим ионообменники, которые предоставили ему характеристики своих продуктов, а также обеспечили брошюрами и другой литературой.
Введение
Ионообменники можно рассматривать как гелеобразные дисперсные системы (за исключением так называемых «жидких ионообменников»). В качестве дисперсной среды служит низкомолекулярный растворитель, обычно вода; дисперсной фазой является трехмерный полимерный каркас ионо-обменника. По своему химическому составу полимерный каркас ионооб-менника может быть органического (ионообменники на основе органических полимеров целлюлозы, декстранов и т. п.) или неорганического (фосфат циркония, алюмосиликаты) происхождения. Полимерные цепочки соединяются между собой поперечными связями (метиленовые или дивинил-бензольные мостики, ионные связи) с образованием трехмерного каркаса, который препятствует перемещению полимерных цепей и их растворению. При контакте с растворителем наблюдается только набухание каркаса, которое зависит от характера, количества и длины поперечных связей.
Важным отличием ионообменников от гелей других типов является наличие ионогенных (функциональных, способных к обмену) групп. Ионогенные группы [ — SO3H, —СООН, —РО(ОН)2, —NH2, —N+R3 и др.] присоединяются к каркасу непосредственно или с помощью других групп (сложныё группы). Между ионами в растворе н ионогенными группами обменника происходил- обмен ионов, что является характерной особенностью гелей этого типа.
Ионообменный процесс характеризуется двумя особенностями: 1) обратимость (известно только немного исключений), 2) эквивалентность обмениваемых ионов в соответствии с принципом электронейтральности. Количество миллимолей иона, сорбированного обменником, соответствует количеству миллимолей равнозарядного иона, выделяющегося из ионообменника.
Способность ионообменников к обмену ионов в растворах обусловила их широкое применение в различных областях химии. В аналитической химии ионообменники успешно используют не только для разделения сложных смесей ионов, но также для концентрирования элементов из разбавленных растворов, выделения и удаления мешающих ионов, получения титрованных растворов, особо чистой воды и т. п.
Ионообменники применяют в широком интервале концентраций анализируемых веществ от радиоактивных индикаторов (без носителя) до макро-
12
Введение
количеств (граммы). Синтезированы специальные типы нонообмеиников, устойчивых при интенсивной радиации, повышенной температуре, в неводных н агрессивных средах.
Этот очень краткий обзор применения нонообмеиников в аналитической химии со всей очевидностью показывает, почему метод ионного обмена стал важным н необходимым в любой современной аналитической лаборатории.
1. Общие сведения о ионообменниках и ионообменных процессах
1.1. Основные понятия
1.1.1. Ионообменные матрицы
Любой нонообменник, органической или неорганической природы, представляет собой матрицу, содержащую способные к обмену ноногенные (функциональные) группы. Большинство современных синтетических органических нонообмеиников имеет каркас нз сополимера стирола с дивинил-бензолом (ДВБ). Эта эластичная трехмерная углеводородная сетка легко образуется н обладает достаточной физической н химической устойчивостью в различных условиях. Требуемые ноногенные группы могут быть относительно легко присоединены к этому каркасу при соответствующих химических реакциях.
Свойства каркаса определяются главным образом соотношением количеств индивидуальных мономеров, используемых в процессе синтеза. Ионо-обменники с низким содержанием ДВБ в каркасе сильно набухают в водных растворах. Большие ноны легко диффундируют через обменник, н скорость обмена высокая. Механическая прочность каркаса уменьшается с уменьшением доли ДВБ.
Ионообменннки с каркасами, доля поперечносвязываюшрго агента в которых велика (> 15% ДВБ), набухают в водных растворах в незначительной степени. Их механическая устойчивость выше. Для матриц с высокой степенью сшивания число ноногенных групп, которое может быть присоединено к каркасу, ограничено н уменьшается с увеличением процента ДВБ. Одновременно уменьшается скорость днффузнн обмениваемых ионов через трехмерный каркас. Обычно для синтеза нонообмеиников используют матрицы с 5 — 8% ДВБ.
Свойства матрицы в значительной степени зависят от чистоты сшивающего агента. Технический ДВБ является смесью м- н л-нзомеров. Помимо этих изомеров присутствует значительное количество днэтилбензола н других примесей (табл. 1.1). Степень поперечного сшивания, X, выпускаемых промышленностью продуктов (т. е. массовый процент ДВБ в исходной смеси для полимеризации) не реальная величина, а величина так называемого номинального поперечного связывания. Поскольку внедрение диви-иилбензола в растущие частицы сополимера происходит быстрее, чем внедрение стирола, полученная матрица имеет так называемую «островную» структуру. Кроме того, частицы сополимера, образованные в начале реак-
14
Часть 1
Таблица 1.1 Состав образцов технического дивинилбензола
Компонент
Содержание, масс.%
♦ образец I образец II образец
л«-Дивииилбензол 31,80 31,40 27,1
«-Дивинил бензол 23,40 13,46 25,2
л«-Диэтил бензол 2,00 1 1,6
л-Диэтилбеизол 1,90 6,67 2,1
о-Диэтилбензол 0,90 J 0,7
л«-Этилстирол 27,30 ] 48,22 24,5
л-Этилстирол 12,00 J 16,7
Нафталин 0,70 — 0,8
Остаток — 0,25 1,2
ции, сшиваются в большей степени по сравнению с частицами, сформированными позже.
Упомянутая выше неоднородность матрицы обусловлена колебаниями в содержании сшивающего агента в области ±0,5%, характерным для обычных коммерческих продуктов.
В настоящее время в качестве матриц находят также применение сополимеры дивинилбензола с акриловой кислотой. Другие винильные соединения (вииилпиридин, винилнафталин и т. п.) редко используют для синтеза ионообменных матриц.
Кроме матриц на основе винильных соединений в ограниченной степени применяют высокомолекулярные вещества, полученные поликонденсацией соответствующих мономеров. Эту группу полимеров составляют различные типы фенолформальдегидных смол и ионообменники, образованные поликонденсацией эпихлоргидринов или этилеихлорида с первичными и вторичными аминами. Главным недостатком поликонденсационных матриц следует считать меньшую воспроизводимость их свойств и более низкую химическую устойчивость по сравнению с матрицами полистирольного типа.
Другие матрицы — носители ионогенных групп, особенно обменники природного происхождения (целлюлоза и т. п.), редко применяются для анализа неорганических веществ.
Исследования в области синтеза органических высокомолекулярных матриц ионообменников продолжаются. Большое внимание уделяется степени дисперсности матрицы.
Наиболее хорошо изучены стирол-дивинилбеизольные матрицы. При синтезе такой матрицы без добавок других веществ (особенно обычных растворителей) в результате сополимеризации стирола и ДВБ образуется каркас гелевой структуры. Этот тип матрицы состоит из взаимопроникающих сеток, образованных индивидуальными цепями. Структура каркаса в
1. Ионообменники и ионообменные процессы
15
значительной степени неоднородна и включает низкопористые «острова», распределенные в более пористой среде. Размер пор (определяется расстоянием между индивидуальными полимерными цепями) очень низкий, и пористость обнаруживается только после набухания матрицы в соответствующем растворителе. Этот тип молекулярной пористости можно рассматривать как скрытый (микропористый или микросетчатый). Ионообменники, имеющие каркас с подобными свойствами, называются гелевыми (или микросетчатыми) смолами.
Каркас с достаточно большими порами, которые статистически распределены по всему объему, может быть получен дополнительным поперечным связыванием и модификацией пористой структуры (хлорметилиро-вание и последующее сшивание посредством этих групп) в предварительно образованной стйрол-ДВБ-матрице. Этот тип каркаса называется изопори-стым. у
Другой тип каркаса современных органических ионообменников представляет так называемая макропористая (макросетчатая) структура. Каркасы этого типа образуются при введении соответствующего растворителя (который легко растворяет мономер) в полимеризационную систему в процессе синтеза. Жидкая фаза затем легко отделяется от сополимера. Полученные гели имеют характерную губчатую структуру, состоящую из агрегатов сфер нормальной гелевой пористости, пронизанных порами негелевой структуры. Однако эти макропоры не являются частью гелевой структуры полимера. Размер пор можно регулировать в процессе получения каркаса. Могут быть получены структуры с размером пор порядка нескольких ангстрем в диаметре. Макросетчатые каркасы имеют большую внутреннюю поверхность (до 100 м2/г и более).
Чтобы предотвратить разрушение каркаса, необходимо вводить большее количество сшивающего агента. Обменники этого типа благодаря их структуре гораздо более устойчивы к осмотическим ударам. Они также меньше различаются по степени набухания в полярных и неполярных растворителях, имеют меньшую потерю объема в процессе высушивания и большую устойчивость к окислению. Кроме того, реакции, связанные с введением ионогенных групп в каркас, протекают легче и с большим выходом, чем для структур гелевого типа.
В настоящее время также интенсивно изучаются различные типы неорганических ионообменных кристаллов 'и осадков, образуемых множеством неорганических соединений.
1.1.2. Ионогенные группы
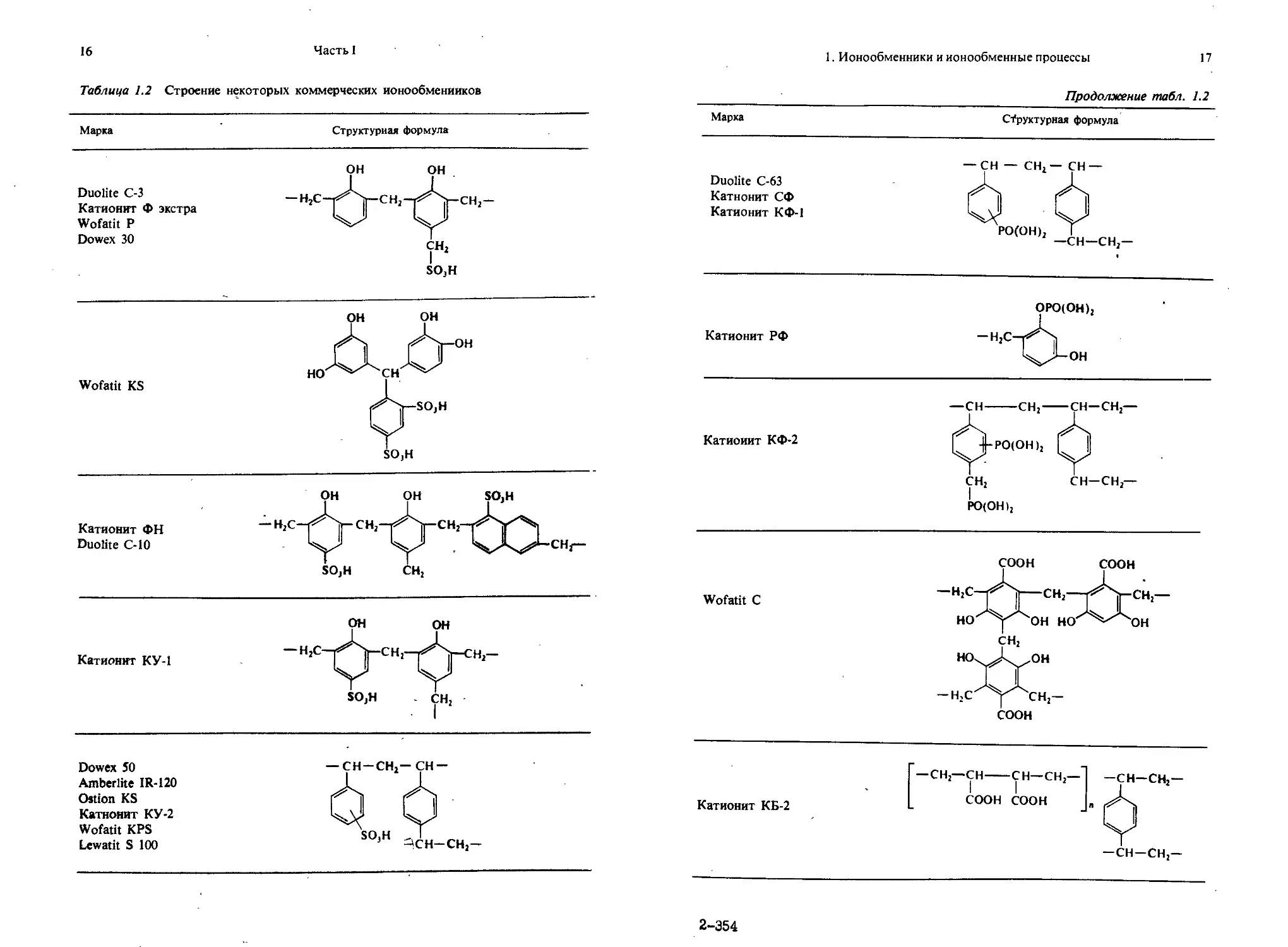
Полимер может быть использован в качестве ионообмеиника только после введения требуемого типа ионообменных групп в соответствующий каркас. Известно много ионообменников, различающихся как по типу каркаса, так и по типу присоединенных иоиогенных групп. В табл. 1.2 приведены структуры различных коммерческих ионообменников.
16
Часть 1
Таблица 1.2 Строение некоторых коммерческих ионообменииков
Марка Структурная формула
Duolite С-3 Катионит Ф экстра Wofatit Р Dowex 30 он он -h2c-^j-ch2-^j-ch2- сн2 SO3H
Wofatit KS он он Л Аон (Д^-SOjH SO)H
Катионит ФН Duolite С-10 OH OH SOjH SO2H CH2
Катионит КУ-1 OH OH —HzC~ГГСН1~ so,H CH2 1
Dowex 50 Amberlite IR-120 Ostion KS Катионит КУ-2 Wofatit KPS Lewatit S 100 — CH —CH2—CH — ij Ф SO.H 1 1 ACH—CH2—
1. Ионообменники и ионообменные процессы
17
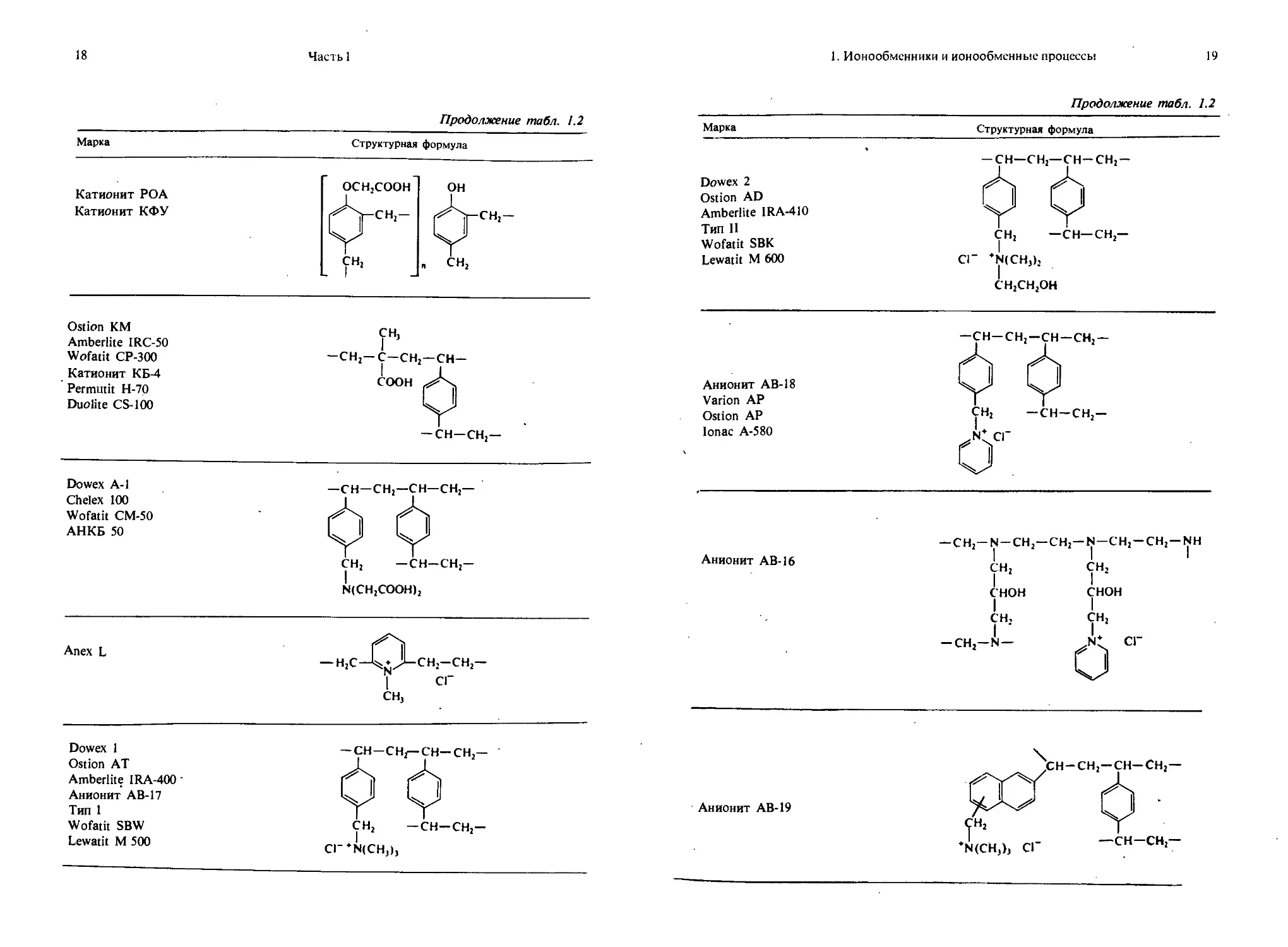
Продолжение табл. 1.2
Марка
Структурная формула
Duolite С-63 Катионит СФ Катионит КФ-1
Катионит РФ
ОРО(ОН)2
Катиоиит КФ-2
РО(ОН)2
Wofatit С
соон
Катионит КБ-2
— CH2—CH СН—СН,—
I I СООН соон
сн—сн2—
2-354
18
Часть 1
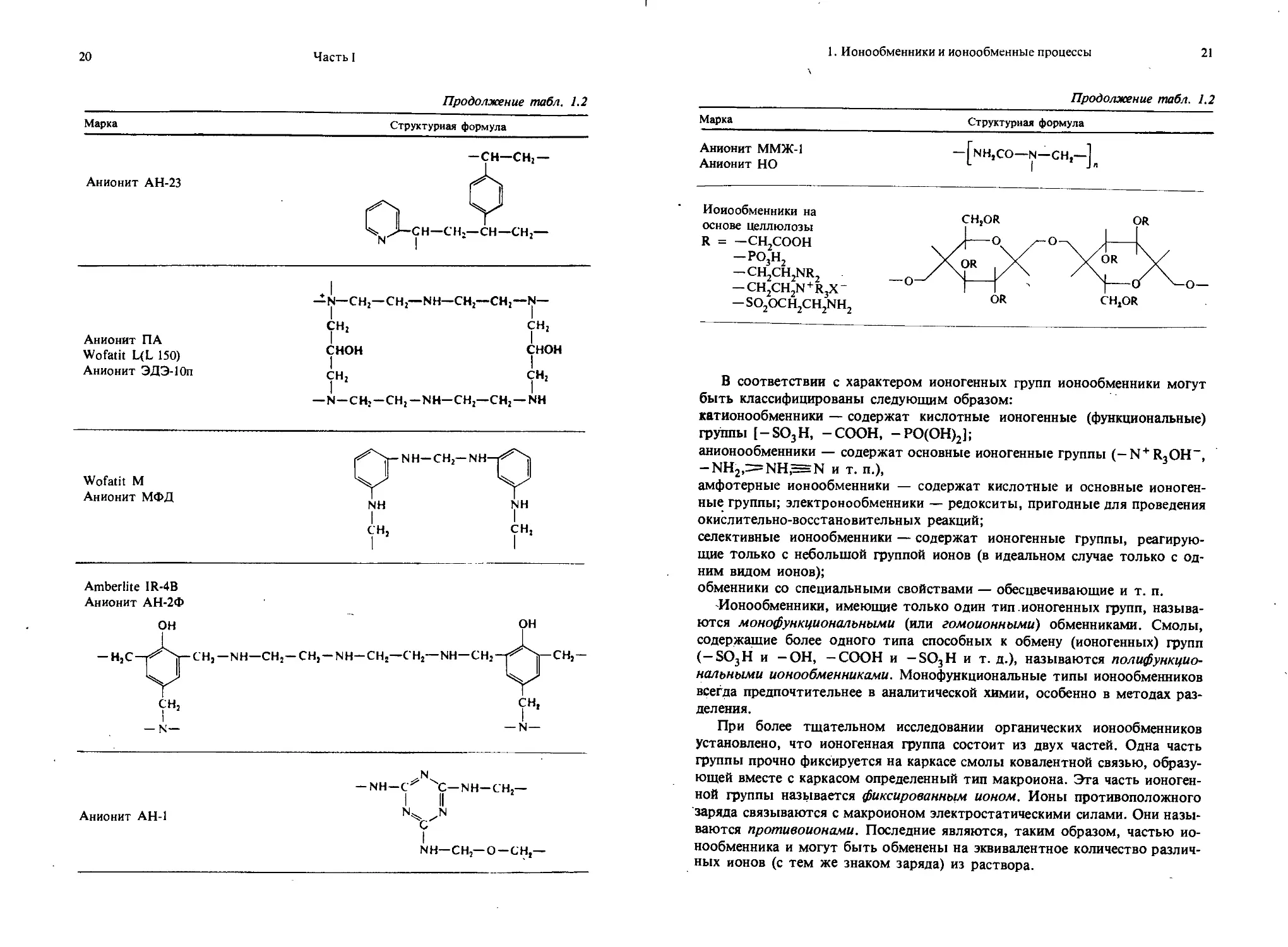
Продолжение табл. 1.2
Марка Структурная формула
Катионит РОА Катионит КФУ OCHjCOOH OH фрсн,- фрсн,- CH2 „ CH2 — 1 —
Ostion КМ Amberlite IRC-50 Wofatit CP-300 Катионит КБ-4 Permutit Н-70 Duolite CS-100 CH, -СНг-С-СНг —CH- COOH — CH—CH J—
Dowex A-l Chelex 100 Wofatit CM-50 АНКБ 50 —CH—CH2—CH—CH2— ф Ф CH; —CH-CH;- N(CH;COOH);
Anex L — H;C-C*jLcH;-CH;- 1 СГ CH,
Dowex 1 Ostion AT Amberlite IRA-400 ’ Анионит AB-17 Тип 1 Wofatit SBW Lewatit M 500 -CH-CH;— СН-СН,— ф Ф CH2 — CH—CH2— Cr*N(CH,),
1. Ионообменники и ионообменные процессы
19
Продолжение табл. 1.2
Марка
Структурная формула
Dowex 2
Ostion AD Amberlite IRA-410 Тип II Wofatit SBK Lewatit M 600
— CH—CH J— CH-CHj-
ф Ф
CH2 — CH— CH2—
СГ *N(CHj)2
CH2CH2OH
Анионит AB-18
Varion AP Ostion AP lonac A-580
—CH2-N-CHj—CH2—N—CH2—CH2—NH
Анионит AB-16
— CH2—
CH2 СИ:
1 CHOH CHOH 1
CH, CH2
N — X
СГ
Анионит AB-19
NICHj), СГ
—CH—CH2—
20
Часть I
Продолжение табл. 1.2
Марка
Структурная формула
Анионит АН-23
Анионит ПА Wofatit L(L 150) Анионит ЭДЭ-10п
—N—СН2—СН2—NH—СН—СН2—N—
| I
СН2 СН2
снон снон
СН2 СН2
— N-CH2-CH2-NH-CH2—СН2 —NH
Wofatit М
Анионит МФД
Amberlite IR-4B
Анионит АН-2Ф
Анионит АН-1
— NH—С' С— NH—СН,—
I II
N^ N
С I
NH—СН,—О—СН,—
1. Ионообменники и ионообменные процессы
21
Продолжение табл. 1.2
Марка Структурная формула
Анионит ММЖ-1 Анионит НО — Г NH,CO— N—СН,—1 L | J П
Ионообменники на основе целлюлозы R = —СН2СООН
-РО3Н2
-ch2ch2nr2
-ch2ch2n+r3x~
—so2och2ch2nh2
В соответствии с характером ионогенных групп ионообменники могут быть классифицированы следующим образом:
катионообменники — содержат кислотные ионогенные (функциональные) группы l-SO3H, -СООН, — РО(ОН)2);
анионообменники — содержат основные ионогенные группы (-N+R3OH~, — NH2p=NH;=N и т. п.),
амфотерные ионообменники — содержат кислотные и основные ионогенные группы; электронообменники — редокситы, пригодные для проведения окислительно-восстановительных реакций;
селективные ионообменники — содержат ионогенные группы, реагирующие только с небольшой группой ионов (в идеальном случае только с одним видом ионов);
обменники со специальными свойствами — обесцвечивающие и т. п.
Ионообменники, имеющие только один тип.ионогенных групп, называются монофункциональными (или гомоионными) обменниками. Смолы, содержащие более одного типа способных к обмену (ионогенных) групп (—SO3H и -ОН, -СООН и — SO3H и т. д.), называются полифункцио-нальными ионообменниками. Монофункциональные типы ионообменников всегда предпочтительнее в аналитической химии, особенно в методах разделения.
При более тщательном исследовании органических ионообменников установлено, что ионогенная группа состоит из двух частей. Одна часть группы прочно фиксируется на каркасе смолы ковалентной связью, образующей вместе с каркасом определенный тип макроиона. Эта часть ионогенной группы называется фиксированным ионом. Ионы противоположного заряда связываются с макроионом электростатическими силами. Они называются противоионами. Последние являются, таким образом, частью ио-нообменника и могут быть обменены на эквивалентное количество различных ионов (с тем же знаком заряда) из раствора.
22
Часть I
Вместе с противоионами противоположно заряженные ионы (по сравнению с противоионами) диффундируют из раствора внутрь смолы в процессе ионного обмена. Эти ионы носят название коионы и не рассматриваются как часть ионообменника.
Эта номенклатура иллюстрируется следующим примером. Если обозначить каркас смолы как R и присоединенную группу как — SO3H, то ионооб-менник можно символически записать как R — SO3H. При контакте смолы, например, с водным раствором хлорида натрия, с одной стороны, образуется макромолекулярный фиксированный ион RSO3- и, с другой стороны, первоначальный противоион Н+ обменивается на противоион Na+. В то же время ионы С1~ образуют коионы, которые диффундируют из раствора в смолу.
В ионогенной группе могут присутствовать различные способные к обмену ионы (противоионы). Для указания вида иона, присоединенного к смоле, используют название соответствующего иона, за которым следует термин «форма» или «цикл». Символ RSO3H, упомянутый выше, обозначает, что ионообменник находится в Н-форме. При обмене Н+ на ионы Na+, Ag+, Ва2+ или La3+ ионообменник получается в натриевой, серебряной, бариевой или лантановой форме. Анионообменники находятся в хло-ридной, сульфатной, нитратной и других формах.
В соответствии со способностью к диссоциации различных ионогенных групп ионообменники классифицируют на сильнокислотные, среднекислотные или слабокислотные (или основные). Эта классификация дана в табл. 1.3.
Кислотные ионогенные группы в Н-форме диссоциируют с выделением ионов Н+ :
-SO3H ** -SO3“ + Н+
-СООН** -СОО- + Н+ (1.1)
Таблица 1.3. Основная классификация ионообменников
Ионообменник Тнп Ионогенные группы
Катиоиообменник Сильнокислотный Среднекислотный Слабокислотный —SOjH (сульфоновые) — РО(ОН)2 (фосфоновые) —СООН (карбоксильные)
Анионообмениик Сильноосновный Среднеосновный Слабоосновный —N+(CHj)3OH- — N+(CH3)2C2H5OH он~ (смесь третичных аминов и четвертичных аммониевых групп) Амины, полиамины
1. Ионообменники и ионообменные процессы
23
Таблица 1.4. Кажущиеся константы диссоциации характерных ионогенных групп ионообменников (рК = -1g К’)
Ионогенная группа
— SO,H 1
—соон 4 — 5
— ОН(фенольный) 9 — 10
— N + RjOH_ 1
— NHR, —NR2 3 — 5
—nh2 6 — 9
Подобным образом основные ионогенные группы в ОН-форме выделяют ионы ОН _:
-NRjOH*4 — N+R3 + ОН-
-NH2 + НОН*4 -N+H3 + ОН" (1.2)
Процесс диссоциации ионогенных групп выражают количественно с помощью кажущихся констант диссоциации. Эти величины представлены в табл. 1.4 для некоторых основных типов ионогенных групп.
Каждый ионообменник содержит определенное число групп с подвижными обмениваемыми ионами. Число таких групп в единице массы (объема) ионообменника характеризует величину обменной емкости, которая теоретически может быть выражена в произвольно выбранных единицах. В области ионообменников строгой терминологии до сих пор не установлено. В данной монографии использована терминология, рекомендованная ИЮПАК в 1972 г. [1].
Qo — теоретическая удельная емкость. Она выражается общим числом миллимолей ионогенных групп в 1 г сухой смолы в Н- или Cl-формах.
Qv — теоретическая объемная обменная емкость. Она означает то же, что и Qo, но относится к 1 см3 набухшей смолы.
Qa — практическая удельная емкость. Она выражается общим количеством ионов (в миллимолях), поглощенных 1 г сухой смолы при данных специфических условиях (которые всегда должны быть указаны).
QB — емкость слоя ионообменника до проскока. Это практическая емкость слоя ионообменника, найденная экспериментально при заданных условиях пропусканием раствора, содержащего определенные ионы или молекулы, через колонку ионообменника до тех пор, пока первые следы вещества не появятся в фильтрате (элюате), или до достижения любой произвольно установленной проскоковой концентрации поглощаемого вещества. Емкость до проскока выражается в миллимолях, миллиграммах или других соответствующих единицах вещества, поглощенного 1 г сухой смолы или 1 см3 набухшей смолы.
24
Часть I
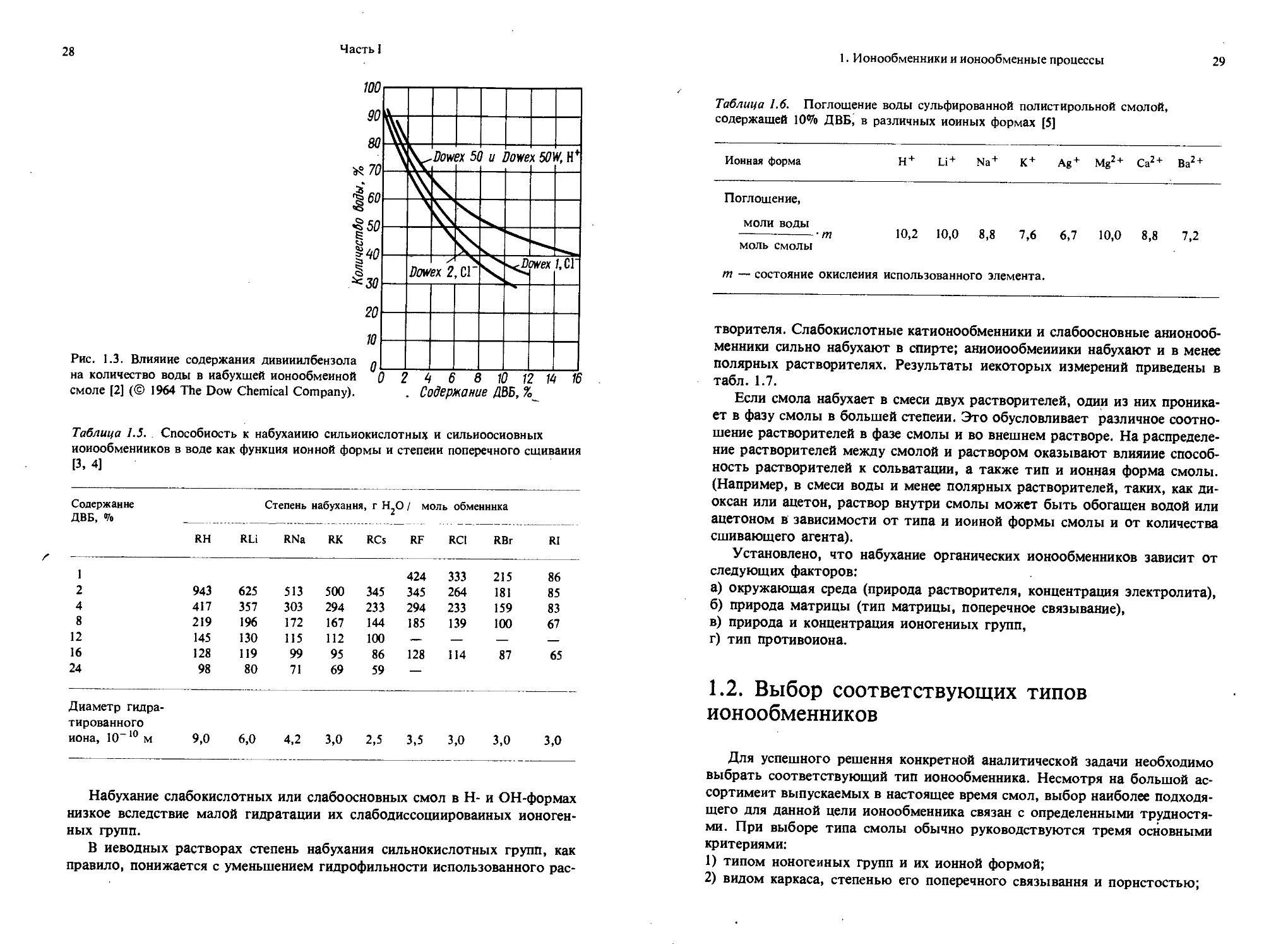
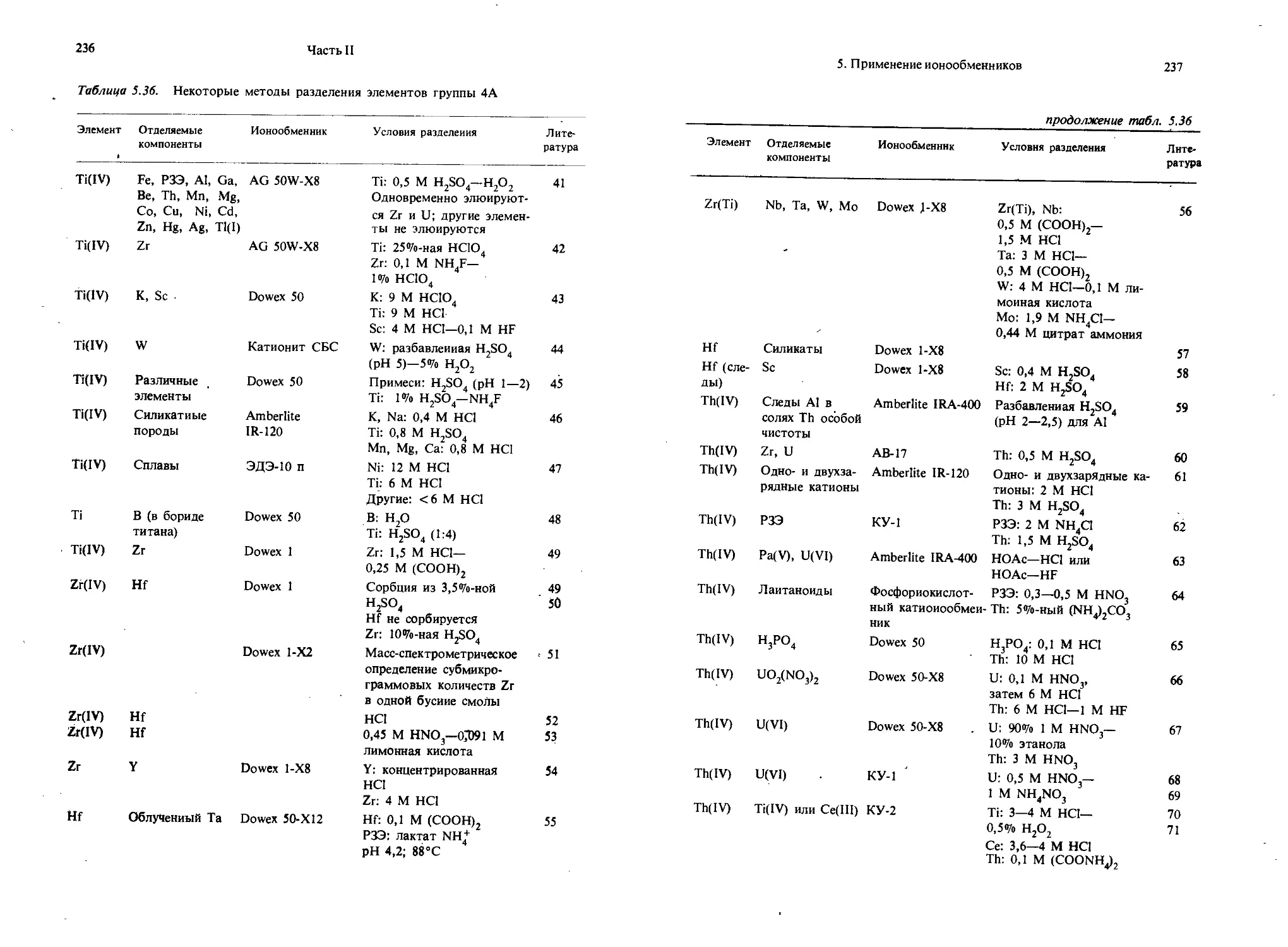
Рис. 1.1. Влияние содержания днвинил-бензола на обменную емкость сухого и влажного нонообмеиников [2] (© 1964 The Dow Chemical Company).
Величина обменной емкости в значительной степени зависит от структуры полимерной матрицы. Рис. 1.1 показывает влияние содержания сшивающего агента на величину обменной емкости различных коммерческих сорбентов.
1.1.3. Внешняя форма и размер частиц ионообменников
По внешнему виду ионообменники разделяются на зерненые и незерне-ные. К первому типу относятся сферические или гранульные частицы (гранулы неправильной формы). Вторую группу нонообмеиников образуют материалы в форме мембран, пленок, фильтров, волокон, тканей, нетканых полотен, трубок, пластин для тонкослойной хроматографии, а также жидкие ионообменники.
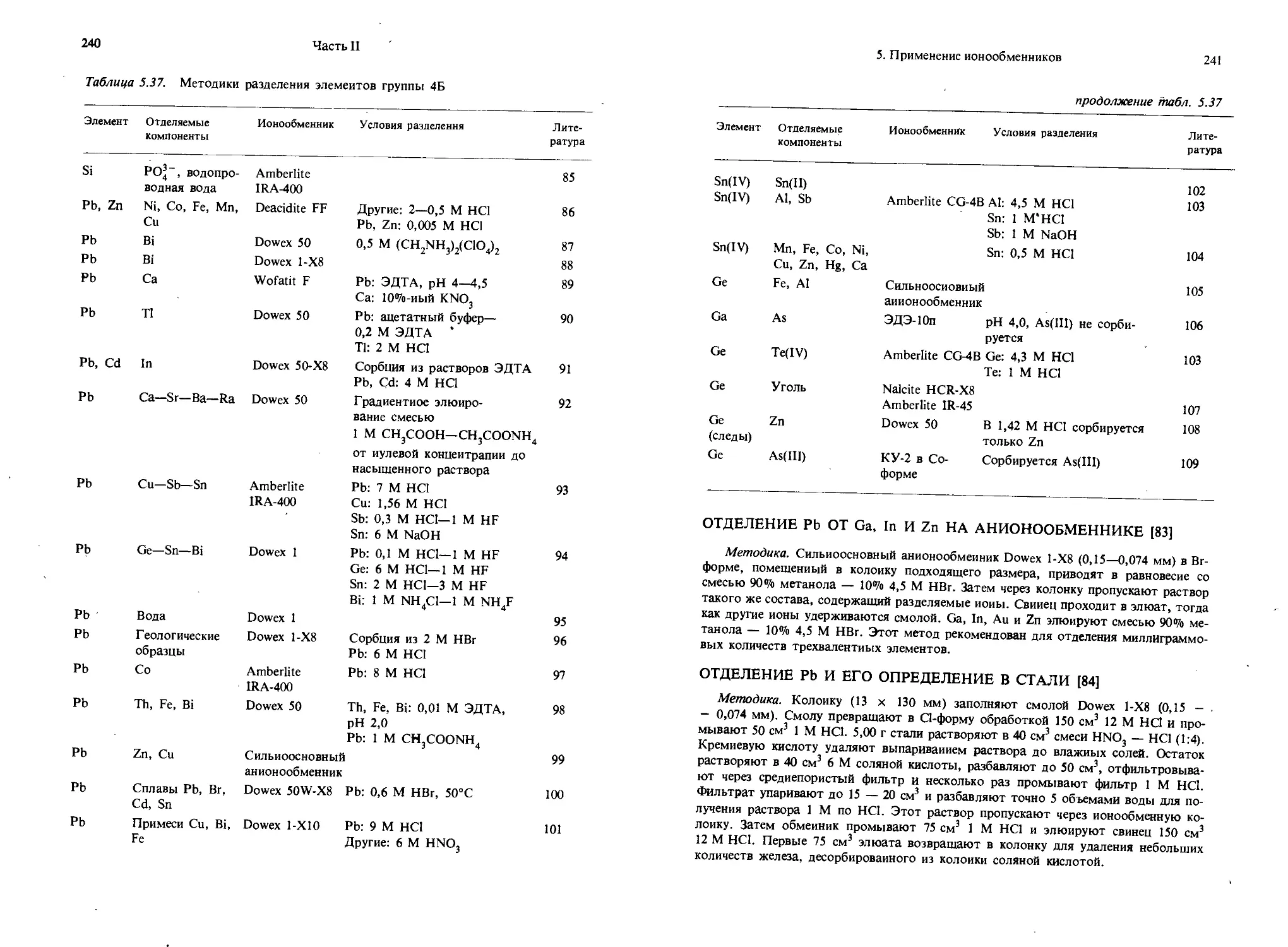
Кроме гелевых ионообменников различной пористости и ионообменников макросетчатого типа для решения специальных хроматографических задач разработаны пленочные ионообменники с поверхностными порами, характеризующиеся гораздо более высокой хроматографической эффективностью, чем обычные смолы. Эти ионообменные смолы в виде тонкой оболочки наносят на инертное твердое тело (обычно стекло в форме сфер или специально обработанный диоксид кремния).
Схематическое сравнение смол такой формы с классическими показано на рис. 1.2. Поскольку слой ионообменника очень тонкий, емкость пленочных смол низкая (< 10 мкмолей Н+/г). Однако этот недостаток компенсируется малым расходом анализируемых проб, хорошей эффективностью и быстрым установлением равновесия, что приводит к существенному сокращению времени, необходимого для разделения.
1. Ионообменники н ионообменные процессы
25
Рис. 1.2. Схематическое сравнение ионообменников разных типов.
Ионообменник: а — микросетчатый (гелевый): б — макросетчатый: в — пленочный; г — с поверхностными порами.
Неорганические ионообменники получают в форме микрокристаллов, кристаллов илн аморфных осадков. Учитывая трудности практического использования этих форм материалов, обменники часто гранулируют (с помощью соответствующего связывающего вещества) или смешивают с носителем.
Жидкие ионообменники, используемые в аналитической химии, обычно фиксируются на инертном пористом носителе, помещенном в колонку.
Размер зерненых частиц смолы указывают либо в миллиметрах, либо в мешах (приложение II). Он изменяется от —0,001 мм (коллоиды) до нескольких миллиметров.
Смолы с размером частиц 0,297 — 0,149 Мм (50 — 100 меш США) или бдльшим применяют в обычном макроанализе (простые операции, получение деионизованной воды и т. п.). Однако в более сложных методах (например, хроматографическое разделение) требуются смолы с размером частиц 0,149 — 0,074 мм (100 — 200 меш) или 0,074 — 0,038 мм (200 — 400 меш). Смолы с таким размером частиц выпускаются промышленностью. В хроматографии высокого разрешения используют смолы с размером частиц 0,001 — 0,010 мм.
26
Часть 1
1.1.4. * Сродство и селективность ионообменников
Если ионообменник вводится в водный раствор электролита, не содержащий комплексообразующих веществ, между ионообменником и ионами в растворе протекает обменная реакция (при условии, что противоионы смолы неидентичны ионам растворенного электролита).
Обменная реакция ионов одинакового заряда может быть записана как
RA + В+ 5=t RB + А+ (1.3)
Если эта реакция выполняется в замкнутой системе, то, поскольку ионный обмен является обратимым процессом, наступает равновесие. Равновесные концентрации ионов, принимающих участие в обмене, неидентичны. Они зависят как от величины относительного сродства реагирующих ионов к ионообменнику, так и от исходной концентрации. Экспериментально установлено, что сродство (или ионообменный потенциал) различных ионов к одной и той же смоле (в разбавленных растворах, <0,1 М) увеличивается с повышением ионного заряда исследуемого иона. Поливалентные ионы образуют с ионообменником более прочные связи (при одинаковых условиях), чем одновалентные ионы. Для ионов одинакового заряда сродство обратно пропорционально радиусу гидратиррванного иона. Однако это утверждение является только общим правилом; существуют исключения.
Более подробные сравнительные исследования показали, что сродство катионов к ряду ионообменников аналогично или идентично так называемому лиотропному ряду.
Для сродства анионов наблюдаются аналогичные закономерности. Кроме того, оно увеличивается с повышением поляризуемости анионов.
Для различных ионов найдены следующие ряды сродства: сильнокислотные ионообменники:
Li+ < Na+ < NH4+ < К+ < Rb+ < Cs+ < Tl+ < Ag+;
Mg2+ < Ca2+ < Sr2+ < Ba2+ < Ra2+;
Fe2+ < Co2+ < Ni2+ < Cu2+ < Zn2+ ;
Al3 + < Sc3 + < Lu3+ < Yb3+ < Tm3 + < Er3+ < Ho3+ < Y3+ < < Dy3+ < Tb3+ < Gd3+ < Eu3+ < Sm3+ < Pm3+ < Nd3+ < Pr3+ < < Ce3+ < La3+;
Th4+ > La3+ > Ca2+ > Na+ ;
сильноосновные ионообменники:
F" < СГ < Br" < I~;
SO^~ > AsO3" > MoO2~ > CrO2-;
I" > NO3~ > Br".
На порядок сродства ионов в каждом ряду слабо влияют индивидуальная природа обменника и условия. Различие в сродстве между индиви
I. Ионообменники и ионообменные процессы
27
дуальными ионами называется селективностью. Последняя определяется типом и концентрацией реагирующих ионов, а также природой растворителя и обменника.
Селективность ионообменников повышается с увеличением содержания сшивающего вещества. Катионы с меньшим эффективным радиусом гидратированного иона сорбируются предпочтительнее. Если ионогенная группа образует с реагирующим ионом ионную пару, селективность смолы к этому типу ионов увеличивается. При образовании в растворе труднодиссоци-ирующих соединений в первую очередь сорбируются ионы, которые образуют соединения с большей степенью диссоциации. С повышением температуры уменьшаются ионные гидратные оболочки, что приводит к уменьшению различия в радиусах гидратированных ионов и, следовательно, к снижению селективности смолы.
С аналитической точки зрения регулирование селективности смолы имеет важное значение. Наиболее удобным способом является введение комплексообразующих веществ в ионообменную систему (на стадии сорбции или элюирования). Поскольку ионогенные группы смол имеют различную селективность, подбором соответствующего агента можно легко разделить смеси ионов (либо селективной сорбцией, либо селективным элюированием).
1.1.5. Набухание ионообменников
При контакте смолы с водой (или другим соответствующим растворителем) ионогенные группы сольватируются и образуется «раствор» функциональных групп в фазе смолы. Смола проявляет тенденцию к переходу в раствор, но этот процесс предотвращается существованием эластичной трехмерной структуры смолы. В результате действия двух противоположных сил определенное количество растворителя поглощается смолой, и смола набухает.
Набухание можно рассматривать как результат различия в осмотическом давлении между внутренней частью ионообменной смолы и внешним, более разбавленным раствором. Концентрация ионов внутри смолы вследствие набухания уменьшается. (Для сульфированного полистирольно-го катионообменника с 8% ДВБ в Н-форме, содержащего при насыщении 12 молей воды на грамм-эквивалент, давление равно 16 МПа.)
Если органический воздушно-сухой ионообменник погрузить в воду, то, как можно видеть из рис. 1.3, количество присоединенной воды будет зависеть от структуры матрицы (т. е. степени поперечного связывания).
При погружении ионообменника в раствор электролита набухание опре-_ деляется концентрацией электролита. С увеличением концентрации электролита поглощение растворителя уменьшается, так как снижается различие в осмотическом давлении между внутренним и внешним раствором. На степень набухания оказывает влияние также природа электролита, что может быть иллюстрировано результатами измерения поглощения воды смолами с сильнокислотными и сильноосновными ионогенными группами (табл. 1.5 и 1.6).
28
Часть I
Таблица 1.5. Способность к набуханию сильнокислотных и сильиоосиовных иоиообменииков в воде как функция ионной формы и степени поперечного сшивания [3, 4]
Содержание Степень набухания, г Н^О / моль обменника
ДВБ, % ______________________________________ _____________
RH RLi RNa RK RCs RF RC1 RBr RI
1 424 333 215 86
2 943 625 513 500 345 345 264 181 85
4 417 357 303 294 233 294 233 159 83
8 219 196 172 167 144 185 139 100 67
12 145 130 115 112 100 — — — —
16 128 119 99 95 86 128 114 87 65
24 98 80 71 69 59 —
Диаметр гидра-
тированного иона, 10“10 м 9,0 6,0 4,2 3,0 2,5 3,5 3,0 3,0 3,0
Набухание слабокислотных или слабоосновных смол в Н- и ОН-формах низкое вследствие малой гидратации их слабодиссоциироваиных ионогенных групп.
В неводных растворах степень набухания сильнокислотных групп, как правило, понижается с уменьшением гидрофильности использованного рас-
1. Ионообменники и ионообменные процессы
29
Таблица 1.6. Поглощение воды сульфированной полистирольной смолой, содержащей 10% ДВБ, в различных ионных формах [5]
Ионная форма Н+ Li+ Na+ К+ Ag+ Mg2 + Са2 + Ba2
Поглощение, моли воды -------------~-т 10,2 10,0 8,8 7,6 6,7 10,0 8,8 7,2 моль смолы т — состояние окисления использованного элемента.
творителя. Слабокислотные катионообменники и слабоосновные анионооб-менники сильно набухают в спирте; аниоиообмеииики набухают и в менее полярных растворителях. Результаты некоторых измерений приведены в табл. 1.7.
Если смола набухает в смеси двух растворителей, одни из них проникает в фазу смолы в большей степени. Это обусловливает различное соотношение растворителей в фазе смолы и во внешнем растворе. На распределение растворителей между смолой и раствором оказывают влияние способность растворителей к сольватации, а также тип и ионная форма смолы. (Например, в смеси воды и менее полярных растворителей, таких, как диоксан или ацетон, раствор внутри смолы может быть обогащен водой или ацетоном в зависимости от типа и ионной формы смолы и от количества сшивающего агента).
Установлено, что набухание органических ионообменников зависит от следующих факторов:
а) окружающая среда (природа растворителя, концентрация электролита), б) природа матрицы (тип матрицы, поперечное связывание),
в) природа и концентрация ионогениых групп, г) тип противоиона.
1.2. Выбор соответствующих типов ионообменников
Для успешного решения конкретной аналитической задачи необходимо выбрать соответствующий тип ионообменника. Несмотря на большой ассортимент выпускаемых в настоящее время смол, выбор наиболее подходящего для данной цели ионообменника связан с определенными трудностями. При выборе типа смолы обычно руководствуются тремя основными критериями:
1) типом ноногеиных групп и их ионной формой;
2) видом каркаса, степенью его поперечного связывания и пористостью;
1
г? 1 ° | сп сп
сп оо 1 о | сн сп '
еЧ — 1 1
чп © 1 О I- чп ©
Tf сП 1 — 1 еч
— еч сп «л 1 § . I О чп 1 vn сп
Vй» сп 1 ° I 00 —
тГ \О 1 еч 1 еч —
Г- сп 1 I О оо
сп 1 еч 1 еч —
СЧ О \О — । । ГГ)
о | |
еч
00 00 1 ° 1 1 °
'cf OS 1 1 1
сп О ип О О | О
СП 00 00 00 I ©
сп еч —« 1
X к X
й О Л X EJ « Е. х S £ х о <2 л х _ ® й Й R £ О S о о cj а С
О й fifl Г) й < > ica
1. Ионообменники и ионообменные процессы
31
3) размером и однородностью зерен смолы (для негранулированиых смол — формой материала).
Дополнительными критериями при выборе смолы являются монофункциональность ионообменника, его чистота, механическая, химическая, термическая и радиационная устойчивость и т. п.
Выбор смолы для неорганического анализа в большинстве случаев ограничивается сильнокислотными или сильноосновными ионообменниками. Средне- и слабодиссоциированные типы ионообменников, а также неорганические, селективные, окислительно-восстановительные и другие типы смол используют главным образом для решения специальных задач.
Чаще всего катионообменники применяют для разделения смеси катионов металлов. Однако в ряде случаев разделение катионов легче выполнять с помощью анионообменников при условии предварительного перевода в соответствующие анионы. Например, для отделения ионов железа(Ш) от ионов никеля смесь катионов поглощают сильнокислотным катионообмен-ником и затем последовательно элюируют растворами соляной кислоты различной концентрации. На сильноосновном анионообменнике разделение этой смеси выполняют в одну стадию: при соответствующей концентрации соляной кислоты поглощаются только ионы железа(Ш), а ионы никеля остаются в растворе.
Для разделения катионов и анионов, например ионов РОд- и La3+ , могут быть использованы как катионообменники (ионы La3+ поглощаются, РО3- остаются в растворе), так и анионообменники (ионы РОд~ поглощаются, ионы La3+ остаются в растворе).
Степень сшивания смолы с азывает существенное влияние на степень набухания смолы, селективность и скорость установления равновесия. Низкая степень сшивания обусловливает высокую набухаемость при контакте с растворителем. Объем набухшей смолы в значительной степени изменяется с изменением свойств внешнего раствора. Это явление особенно нежелательно при работе в динамическом режиме. Низкая степень сшивания смолы способствует уменьшению различий в сродстве ионов к ионообменнику, что является одной из причин размывания зон при хроматографическом разделении.
В то же время высокая степень сшивания смолы способствует уменьшению набухаемости и повышению селективности. Вследствие малых изменений объема (даже при существенных изменениях свойств внешнего раствора) эти ионообменники имеют преимущества при работе в колонках.
К недостаткам смол с высокой степенью сшивания относятся меньшие величины удельной обменной емкости и низкая скорость обмена ионов. Кроме того, высокая плотность поперечных связей в ионообменнике ограничивает диффузию больших ионов.
На практике отдают предпочтение смолам на основе сополимера стирола с дивинилбензолом. Сильнокислотные катионообменники содержат 8% ДВЕ, а сильноосновные анионообменники — 4 — 6% ДВБ. Также выпускаются промышленностью продукты, содержащие 2 — 16% ДВБ.
32
Часть I
Наибольшее применение в аналитической практике находят смолы в виде зерен сферической формы. В настоящее время производятся ионообменники с различным размером зерен. Для решения простых аналитических задач пригодны смолы с диаметром зерен 0,1 — 0,3 мм. Хроматографические разделения выполняют на смолах с меньшим размером частиц (в отдельных случаях — до нескольких микрон) и, по возможности, с узким интервалом колебаний размеров. Некоторые ионообменники (особенно неорганические), получаемые в виде очень мелких частиц, не могут быть непосредственно использованы в колонках; в таких случаях необходимо вводить соответствующий носитель.
Ионообменные материалы в форме мембран, фильтров и пластин для тонкослойной хроматографии, а также жидкие ионообменники, нанесенные на соответствующий твердый носитель, находят ограниченное применение в аналитической практике.
В настоящее время большое внимание уделяется повышению селективности ионообменных процессов за счет использования селективных ионооб-менииков. Последние получают введением соответствующих функциональных групп (производных органических реагентов, известных в аналитической химии) в полимерную матрицу. Функциональные группы этих ионообменников обладают способностью образовывать комплексы или хелаты с некоторыми ионами и благодаря этому (при соответствующей обработке анализируемого раствора) селективно поглощают один вид или ограниченную группу ионов из сложных смесей ионов.
Например, смолы, содержащие группы — N(CH2COOH)2, селективно поглощают следовые количества тяжелых металлов в присутствии больших количеств ионов щелочных и щелочно-земельных металлов. Ионооб-мениик, содержащий группы — РО(ОН)2, в кислой среде селективно поглощает скандий из смеси скандий — редкоземельные элементы.
В настоящее время промышленный выпуск селективных ионообмении-ков ограничивается смолами, содержащими имииодиацетатные группы (Dowex А-1, Wofatit МС-50 и т. п.). Освоен также выпуск ионообменника Srafion NMRR, содержащего группы, селективные к некоторым металлам платиновой группы.
Многочисленные литературные данные свидетельствуют об интенсивном развитии селективных ионообменников. Описаны методы получения небольших лабораторных партий различных ионообменных смол.
Другую группу ионообменников, проявляющих повышенную селективность к некоторым ионам, образуют неорганические сорбенты. Недостатками этих ионообменников являются низкая устойчивость большинства из них. в щелочных растворах, склонность к пептизации, малая обменная емкость, а также трудности, связанные с получением их в форме, удобной для работы в динамическом режиме.
Жидкие ионообменники, нанесенные на соответствующий носитель, обладают преимуществами по сравнению с классическими высокомолекулярными органическими ионообменниками.
1. Ионообменники и ионообменные процессы
33
1.2.1. Основные характеристки ионообменников
СИЛЬНОКИСЛОТНЫЕ КАТИОНООБМЕННИКИ
Выпускают два типа ионообменников, содержащих группы — SO3H, иа стирол-дивинилбензольной и фенолформальдегидной матрицах. Группы — SO3H связаны с бензольным ядром непосредственно или через метиленовую группу. По степени ионизации иоиогеииых групп ионообменники сравнимы с сильными минеральными кислотами. Группы — SO3H, связанные непосредственно с бензольным кольцом, диссоциируют 'легче, чем группы — CH2SO3H.
При контакте ионообменников в Н-форме с растворами нейтральных солей иоиы Н+ переходят в раствор, а ионообменники превращаются в соответствующие солевые формы. Химические реакции нонообмеиников подобны реакциям серной кислоты. Обменная емкость смол практически не зависит от pH раствора, ионообменники могут быть использованы в кислых, нейтральных и щелочных растворах.
Селективность групп — SO3H повышается с увеличением атомного номера, валентности и степени ионизации обмениваемых иоиов и понижается с увеличением ионного радиуса гидратированных ионов. Как правило, селективность уменьшается в рядах (Dowex 50-Х8).
Ag+ > Cs+ > Rb+ > К+ > NH+ > Н+ > Li+;
Ва2+ > Sr2+ > Са2+ >‘ Mg2+ > Ве2+;
Ва2+ > Pb2+ > Sr2+ > Са2+ > Ni2+ = Cu2+ > Cd2+ > Со2+ > Zn2+ =
= Mg2+ > Mn2+ > Be2+ > UO^+ > Hg2+;
La3 + > Ce3+ > Cr3+;
Th4+ (NO3")4 > Fe3+ > Al3+ > Ba2+ > Tl+ (SO^-) = Pb2+ >
> Sr2* > Ca2+ > Co2+ > Ni2+ = Cu2+ > Zn2+ = Mg2+ > UO^+(NO3~)2 = = Mn2+ > Ag+ > Cs+ > Be2+(SO2-) = Rb+ > Cd2+ > NH+ = K+ > > Na+ > H+ > Li +
Эти ряды селективности справедливы для разбавленных растворов (приблизительно 0,1 моль/л хлоридов, если не указаны другие условия). В концентрированных растворах однозарядные ионы поглощаются лучше, чем многозарядные.
Для полного превращения ионообменников в Н-форму необходим большой избыток соляной кислоты высокой концентрации. Ионообменники стирол-дивинилбензольного типа устойчивы к действию окислителей и растворов кислот и щелочей.
СРЕДНЕКИСЛОТНЫЕ КАТИОНООБМЕННИКИ
В настоящее время выпускают ионообменники, содержащие группы — РО(ОН)2 или -ОРО(ОН)2, на различных полимерных матрицах. Химические свойства ионообменников подобны свойствам фосфористой или фосфорной кислот. По способности к диссоциации ионогенных групп смолы в
3-354
34
Часть I
Н-форме занимают промежуточное положение между сильнокислотными и слабокислотными катионообменниками.
Обменная емкость ионообменников зависит от pH внешнего раствора. Большая часть одно- и двухзарядных катионов наиболее эффективно поглощается при pH >5.
На селективность ионогенных групп большое влияние оказывает природа сорбируемого иона и pH раствора:
- РО(ОН)2 : Pb2+ > Cu2+ > Zn2+ > Cd2+ > Mn2+ > Со2+ > Ni2+; диаллилфосфат: Н+ > Ag+ > Cs+ > Rb+ > K+ > Na+ > Li+ (в кислой среде);
- РО(ОН)2 :Cs+>Rb+>K+>Na+> Li+(pH 6,7 — 8,5);
Cs+ > Rb+ > K+ > Li+ > Na+(pH 10,0);
Li+ > Na+ > Rb+ > Cs+ > K+(pH 12,6);
— PO(OH)2:Th4+ > U4+ > UO^+ > Fe3+ > РЗЭ > H+ > Cu2+ >
> Zn2+> Cd2+ > Mn2+ > Co2+ > Ni2+ > Ca2+ > Mg24- > Sr2+ > > Ba2+ > Na+.
Для полного превращения ионообменников в Н-форму из форм тех ионов, которые располагаются в ряду селективности после ионов Н+, необходим небольшой (по сравнению со стехиометрией) избыток сильной минеральной кислоты. Для других ионных форм требуется значительно больший избыток кислоты. Второй путь превращения смолы в Н-форму — предварительное элюирование катионов подходящим комплексообразующим веществом.
Стирол-дивинилбензольные ионообменники обладают высокой химической и термической устойчивостью (вплоть до 120 — 130° С). По механической прочности среднекислотные ионообменники уступают сильнокислотным ионообменникам аналогичной структуры.
СЛАБОКИСЛОТНЫЕ КАТИОНООБМЕННИКИ
Выпускают монофункциональные ионообменники, содержащие группы — СООН, на основе сополимеров акриловой и метакриловой кислот с диви-нилбензолом и ионообменники с группами —СООН и —ОН, полученные поликонденсацией фенолов с резорциловой кислотой. Ионообменники в Н-форме не выделяют ионы Н+ при контакте с растворами нейтральных солей; по степени ионизации соответствуют уксусной кислоте.
Обменная емкость ионообменников сильно зависит от pH раствора. Наиболее эффективная область pH находится в пределах pH 6 — 14.
Характерным свойством ионообменников являются высокая селективность к ионам Н+ и относительно высокое сродство к ионам щелочноземельных металлов. Ряды селективности для ионов металлов имеют обратный порядок по сравнению с сильнокислотными ионообменниками.
Ряд селективности при pH 7: Mg2+ < Са2+ < Ni2+ < Со2+ < Си2+;
1. Ионообменники и ионообменные процессы
35
Обычный ряд: Н+ > Са2+ > Mg2+ > Na+;
Aliasion CC:Ag+ > Li+ > Rb+ > Cs+ > Na+ > NH+ > K+;
Cu2+ ► Co2+ > Zn2+ > Ni2+.
Для превращения ионообменников в Н-форму достаточен небольшой избыток сильной минеральной кислоты. При изменении ионной формы смо--лы наблюдается существенное изменение объема. Ионообменники могут быть использованы в качестве буфера в системах с низкой активностью ионов Н+ и для выделения органических оснований.
Ионообменники на основе сополимера метакриловой кислоты — ДВЕ химически устойчивы; в растворах щелочей устойчивость сохраняется вплоть до 120° С.
СИЛЬНООСНОВНЫЕ АНИОНООБМЕННИКИ
Выпускают ионообменники, содержащие функциональные группы — N+(CH3)3C1~ (тип I), -N+(CH3)2C2H4OH • С1~ (тип II) или пиридиниевые группы, преимущественно на основе сополимеров стирол-ДВБ. Ионообменники в ОН-форме вытесняют из растворов нейтральных солей анионы; в результате этого обмена ионообменники превращаются в соответствующие солевые формы, а в растворе образуются гидроксиды. По степени ионизации ионогеиных групп ионообменники сравнимы с гидроксидами щелочных металлов. Ионообменники в ОН-форме поглощают даже слабодис-социированные кислоты (борную, кремневую). Основность смол типа I выше, чем смол типа II. Обменная емкость смол мало зависит от pH.
Селективность смол по отношению к анионам повышается с увеличением валентности ионов и снижается с увеличением радиуса гидратированного иона. Селективность уменьшается в рядах:
Dowex 1-Х8 (тип I): I- > HSO^ > NO^ > Br- > CN~ > HSO3~ > > NO2“ > СГ > HCO3“ > H2PO4“ > HCOO~ > CH3COO" > OH" > > F“;
SO2’ > CrO2- > цитрат > тартрат > NO3“ > AsO4~ > PO4" > > MoO2- > ацетат > I- > Br- > Cl- > F- .
Dowex 2-X8 (тип II): C1O4~ > I > HSO4“ > SCN“ > СС13СОО- > > CF3COO~ = NO3“ = Br- > NO£ = CN“ > СГ BrO3~ > > OH- > HCOf > H2PO4- > io3- > CH3COO- > F“.
Wofatit SBW: [Fe(CN)6]4- = [Fe(CN)6]3- = SCN“ = I" > NO3~ > Br“ > > hso- > CN* > no2- > Cl* > h2po- > so|- >
> C2O24~ > hpo^- > so2- > n2o^- > n2o|- >
> HCO^ > ацетат > PO|_ > F- > OH-.
Для полного превращения ионообменника в ОН-форму необходим большой избыток раствора гидроксида натрия (> 2%-ный раствор NaOH), осо
36
Часть I
бенно если ионообменник находится в Cl-форме. Ионообменники типа II легче превращаются в ОН-форму, чем ионообменники типа I.
Рабочая область сильноосновных анионообменников находится в пределах pH 1 — 13, но они могут быть использованы также в концентрированных растворах кислот (например, 12 М НО). Ионообменники проявляют достаточную устойчивость к действию кислот, щелочей и окислителей; смолы типа I более устойчивы, чем смолы типа II, особенно в ОН-форме. Смолы типа I в ОН-форме не рекомендуется применять при температуре выше 60°С, а смолы типа II — выше 50°С. Смолы в ОН-форме легко поглощают углекислый газ из воздуха.
СРЕДНЕОСНОВНЫЕ АНИОНООБМЕННИКИ
Ионообменники содержат как снльноосновные, так и слабоосновные ионогенные группы (преимущественно группы третичных аминов). Регенерированные раствором гидроксида натрия, ионообменники вытесняют анионы из растворов нейтральных солей и сорбируют слабые кислоты пропорционально содержанию сильноосновных групп. После регенерации растворами карбоната натрия или гидроксида аммония ионообменники ведут себя как слабоосновные.
СЛАБООСНОВНЫЕ АНИОНООБМЕННИКИ
Полимерной основой ионообменников, содержащих ионогеиные группы -NH2, -NHR, -NR]R2 (первичные или вторичные амины), являются стирол-дивинилбензольные, полиамин-эпихлоргидрииные и фенолформальдегидные матрицы. По степени ионизации иоиогеииых групп ионообмеини-кн сравнимы с гидроксидом аммония.
Ионообменники не реагируют с растворами нейтральных солей. При контакте с водой кислые формы ионообменников гидролизуются с выделением кислоты. В форме свободных оснований ионообменники сорбируют минеральные кислоты с образованием соответствующих солей. Обменная емкость смол сильно зависит от pH раствора и валентности сорбированного нона.
Некоторые смолы этого типа связывают вещества вандерваальсовыми силами. Аминогруппы ионообменников склонны к образованию прочных комплексов с Ag+, Cu^+ и другими катионами.
Для превращения ионообменников в свободные основания требуется небольшой (по сравнению со стехиометрией) избыток карбоната натрия, гидроксида натрия или аммония (или ароматических аминов).
Сродство анионов к иоиообмеиникам уменьшается в ряду ОН- > SO^“ > СгОд- > цитрат > тартрат > NO3~ > AsO4 > > РО’ > МоОд- > СН3СОО“ > I" = Вг“ > СГ > F".
Ионообменники достаточно устойчивы в разбавленных растворах кислот и щелочей.
1. Ионообменники и ионообменные процессы
37
1.2.2. Ионообменники для специальных целей
ОБЕСЦВЕЧИВАЮЩИЕ СМОЛЫ
Для обесцвечивания применяют различные типы смол. Ионообменники с фенолформальдегидными матрицами проявляют обменные свойства только в щелочной среде (pH > 8). Большая часть смол теряет способность к обесцвечиванию после высушивания. Смолы регенерируют 1 %-ным раствором NaOH. Некоторые типы смол после промывания водой активируют 0,1 М НС1 или 1,5%-ным раствором NH4C1.
В качестве обесцвечивающих смол применяют также слабоосновные анионообменники на основе полиамидов и снльноосновные (слабосшитые) стирол-дивииилбензольные аиноиообмениикн.
СЕЛЕКТИВНЫЕ ИОНООБМЕННИКИ
Иоиообменники этого типа при определенных условиях проявляют повышенную селективность к одному виду или небольшой группе ионов. Ио-ногеииые группы смол образуют с ионами, находящимися в растворе, хелатные комплексы, а также продукты взаимодействия свободных ионных пар и т. п. Типичный представитель селективных смол — коммерческий продукт Dowex А-1. Недостатком селективных ионообменников гелевой структуры является малая скорость обмена иоиов; на макропористых матрицах скорость обмена выше.
ЭЛЕКТРОНООБМЕННИКИ
Электроиообменники представляют собой высокомолекулярные смолы, содержащие окислительно-восстановительные группы, которые могут участвовать в окислительно-восстановительных процессах. Их сходство с иоиообменниками имеет формальный характер. Электроиообменники получают поликонденсацией или полимеризацией соответствующих мономеров (пирогаллола, гидрохинона, оксиантрахинона, ализарина, метиленового синего, вииилгидрохинона и т. п.).
Применение этих смол требует специальных приемов (отсутствие контакта с кислородом воздуха). Перед использованием смолы обрабатывают подходящим восстановителем [10%-иым раствором дитионита натрия в 1 М растворе гидроксида аммония, 10%-ным раствором хлорида титана(Ш) в 0,5 М серной кислоте или раствором сульфита натрия в серной кислоте]; воду для удаления растворенного кислорода кипятят.
В качестве электронообменников пригодны обычные катионо- или анио-иообменники в тех ионных формах, которые способны окисляться или восстанавливаться. Например, могут быть использованы следующие пары ионов: Fe2+ — Fe3+, Се3 + — Се4+, Ti3 + — Ti4+ и т. п.
38
Часть I
ЗАМЕДЛЯЮЩИЕ СМОЛЫ
Эти ионообменники содержат цепи с кислотными и основными ионогенными группами, которые имеют противоположный заряд и нейтрализуют друг друга. При контакте смолы с раствором электролита происходит ионный обмен, в результате которого оба типа ионов, присутствующих в растворе, поглощаются смолой. Последующее промывание ионообменника водой приводит к элюированию сорбированных ионов.
Вследствие различия в сродстве ионов к ионогенным группам и различной прочности связи с ними одни ионы при элюировании удерживаются на смоле в течение большего времени, чем другие. На этом принципе основано применение смол для быстрого разделения электролитов и неэлектролитов, единственным элюентом в этих системах является вода.
ЖИДКИЕ ИОНООБМЕННИКИ
Выпускаемые промышленностью среднекислотные катионообменники и слабоосновные анионообменники практически нерастворимы в воде, но растворяются в соответствующих углеводородах. Эти ионообменники можно использовать в виде жидкостей (подобно жидкостной экстракции) или в виде жидкой фазы, нанесенной на соответствующий инертный носитель (экстракционная хроматография). Жидкие ионообменники обычно применяют в виде приблизительно 5%-ных растворов в подходящих углеводородах; регенерация жидких ионообменников аналогична регенерации твердых смол.
ПРИРОДНЫЕ И МОДИФИЦИРОВАННЫЕ ОРГАНИЧЕСКИЕ ИОНООБМЕННИКИ
Природные органические ионообменники вследствие их малой обменной емкости и низкой химической и механической устойчивости не применяются в аналитической химии.
Сульфированные угли, принадлежащие к группе модифицированных ионообменных материалов, получают взаимодействием олеума или серной кислоты с подходящими видами углей. Эти ионообменники полифункцио-нальны; они обладают достаточной обменной емкостью, но используются только в специальных случаях.
Модифицированные целлюлозные материалы (хлопок, бумага и т. п.) и специально обработанная особо чистая целлюлоза с введенными ионогенными группами имеют важное значение в биохимии и фармацевтике. В неорганическом анализе этот тип ионообменников и производные полидекстранов (сефадексы) применяют редко.
1. Ионообменники и ионообменные процессы
39
НЕОРГАНИЧЕСКИЕ ИОНООБМЕННИКИ
Природные и модифицированные неорганические материалы вследствие их низкой химической стойкости, малой обменной емкости и неоднородности не нашли широкого применения в аналитической химии.
Синтетические неорганические ионообменники подразделяются на несколько групп. Слабокислотные катионообменники на основе алюмосиликатов известны давно; они используются только в нейтральных средах.
Вторую группу синтетических неорганических ионообменников образуют соли гетерополикислот, гидратированные оксиды (особенно четырехвалентных элементов) и нерастворимые соли поливалентных металлов. Эти ионообменники (главным образом катионообменники) начали применяться сравнительно недавно. Они характеризуются более высокой обменной емкостью, термической, радиационной и химической (в кислых средах) устойчивостью и в некоторых случаях высокой селективностью к определенным ионам. При pH > 8 большая часть ионообменников гидролитически разрушается.
1.3. Ионообменные процессы
Для того чтобы начался ионообменный процесс, необходимо ионообменную смолу привести в контакт с раствором, содержащим способные к обмену ионы. Существуют два метода осуществления контакта ионообменника с ионами в растворе: статический (встряхивание) и динамический (колоночный).
1.3.1. Статический метод
Ионообменник перемешивают или встряхивают с раствором в подходящем сосуде. После достижения равновесия между ионообменником и ионами в растворе фазы разделяют фильтрованием, декантацией или центрифугированием и анализируют на содержание в них ионов. Количественное поглощение ионов из раствора может быть достигнуто при большом избытке ионообменной смолы (одностадийный статический процесс) или последовательным прибавлением небольших количеств смолы в раствор. После установления равновесия каждая порция смолы отделяется от раствора. Этот метод, называемый многостадийным (каскадным) статическим процессом, является лабораторным, его применение ограничено вследствие большой затраты времени и возможности экспериментальных ошибок.
Многостадийный процесс целесообразно использовать при анализе систем, в которых в результате ионного обмена выделяются газы, а также в тех особых случаях, когда необходимо сдвинуть равновесие в сторону ионообменного процесса. Примерами таких процессов служат реакции нейтрализации, образования устойчивых комплексов и нерастворимых соединений, превращения нерастворимых веществ в растворимые формы. Напри
40
Часть I
мер, барий может быть переведен в раствор из нерастворимого сульфата бария
BaSO4 + 2RSO3Na = (RSO3)2Ba + Na2SO4 (1.4)
Смолу промывают водой и элюируют ионы бария 3 — 4 М соляной кислотой. Процесс проводят в присутствии большого избытка смолы при повышенной температуре. Аналогичную методику используют при анализе сульфатов свинца, стронция или кальция, хлорида свинца, нерастворимых фосфатов двухвалентных металлов и т. п.
Одностадийный статический процесс иногда применяют в качественном анализе: ионы концентрируют на белой или слабоокрашенной смоле и выполняют цветную реакцию на соответствующий ион непосредственно в фазе смолы.
Одно’й из наиболее важных областей применения статического метода является определение различных физико-химических параметров: структуры, устойчивости комплексов, коэффициентов селективности, равновесных характеристик для динамических опытов [уравнения (3.19) — (3.21)].
1.3.2. Динамический метод
Ионообменную смолу (набухшую в воде или подходящем растворителе) в виде гомогенной смеси с раствором помещают в вертикальную колонку (гл. 4) и пропускают анализируемый раствор. Другие методы пропускания раствора через колонку (противоточный) редко применяют в аналитической практике.
Проведение ионообменных процессов в колонках обеспечивает возможность количественного обмена ионов из раствора и разделение смесей с максимальной эффективностью.
Общая схема ионообменного процесса в колонках включает стадии сорбции, промывание соответствующим растворителем или раствором (чаще всего водой), регенерацию (элюирование сорбированных ионов). Эти операции подробно описаны в т. ПВ Comprehensive Analytical Chemistry (р. р. 230 — 236). Представляют интерес особые случаи применения этих операций в аналитической химии.
СЕЛЕКТИВНАЯ СОРБЦИЯ
Этот метод основан на выборе подходящих условий сорбции для одного элемента или для небольшой группы элементов, присутствующих в смеси. Для удержания нежелательных компонентов смеси в растворе часто применяют вещества, которые образуют с мешающими элементами достаточно прочные несорбируемые комплексы.
Например, для превращения кадмия (в смеси Zn — Cd) и железа в несорбируемые сильнокислотными катионообменниками комплексы вводят иодиды и цианиды соответственно; часто в качестве комплексообразующих
1. Ионообменники и ионообменные процессы
41
веществ используют такие комплексоны, как этилеидиаминтетрауксусная кислота (ЭДТА), этиленгликольтетрауксусная кислота (ЭГТА), циклогек-сандиаминтетрауксусная кислота (ЦГДТА) и другие.
Селективность сорбции зависит от pH раствора (разделение смеси Mg — Са с помощью ЭГТА, ч. 5.2.2. и др.). Количественное разделение смеси двух элементов методом селективной сорбции достигается, если коэффициенты распределения сорбируемых и несорбируемых ионов имеют следующий порядок:
DW1 > 100 - 300; DM2 < 3 - 10; D^/D^ >10 — 30
Метод селективной сорбции применяется также при разделении элементов на анионообменниках. Например, из растворов соляной кислоты, содержащей смесь элементов, при соответствующей концентрации кислоты на смоле может удерживаться только один ион, в то время как другие ионы проходят через колонку (отделение Fe от А1, Со от Ni и т. п.).
Селективность сорбции изменяют не только введением в раствор комплексообразующих веществ, но и использованием сильноосновных анионо-обменников в соответствующей анионной форме. Анионообменник в ЭДТА-форме не сорбирует щелочные металлы; другие элементы сорбируются избирательно в зависимости от pH раствора и констант устойчивости комплексов.
СЕЛЕКТИВНОЕ ЭЛЮИРОВАНИЕ
При селективном элюировании наблюдается картина, обратная селективной сорбции. Задача заключается в выборе условий, при которых один тип ионов десорбируется, а другие прочно удерживаются смолой. Процесс разделения ускоряется при использовании коротких колонок.
Процессы разделения в методе селективного элюирования основаны, как правило, на различиях в константах устойчивости разделяемых ионов. Типичным примером успешного применения метода селективного элюирования является разделение смеси Ni — Мп — Со — Си — Fe — Zn элюиро~ ванием соляной кислотой (с анионообменников). Индивидуальные элементы последовательно элюируются соляной кислотой соответствующей концентрации в указанном выше порядке.
Сочетание селективной сорбции и селективного элюирования значительно упрощает процесс разделения сложных смесей.
1.3.3. Хроматографические процессы
Разделение сложных смесей на индивидуальные компоненты возможно только при наличии высокоэффективных методов. Простое элюирование для этих целей непригодно.
Хроматографические колоночные методы подразделяются на три основных вида: фронтальный, вытеснительный и элюентный анализ. Фронтальный и вытеснительный виды хроматографического анализа (последний
42
Часть 1
может быть рассмотрен как частный случай элюентного анализа) имеют ограниченное значение при разделении смесей неорганических ионов, так как не позволяют количественно разделить смеси на индивидуальные компоненты.
Элюентная хроматография получила наибольшее распространение для разделения смесей неорганических ионов.вследствие высокой эффективности и возможности количественного разделения смесей на индивидуальные компоненты. Недостатком этого метода является ограниченное количество анализируемого вещества. При разделении элементов этим методом используется не более 5% общей обменной емкости колонки; при наличии больших количеств веществ разделение неэффективно вследствие перекрывания зон с разделенными компонентами.
Метод элюентной хроматографии основан на поглощении анализируемой смеси ионов в верхней части колонки в виде тонкого слоя и разделения с помощью соответствующего элюирующего раствора при продвижении его по колонке сверху вниз. В процессе перемещения раствора состав поглощенной пробы непрерывно изменяется: ионы, имеющие более низкое сродство к ионообменнику, двигаются вниз быстрее, а ионы с более высокой степенью сродства к ионообменнику — медленнее. После пропускания достаточного количества элюента индивидуальные компоненты анализируемой смеси распределяются вдоль ионообменной колонки в виде отдельных зон. В идеальном случае растворы, содержащие индивидуальные компоненты, вытекают раздельно и между индивидуальными зонами всегда есть некоторый объем элюирующего раствора.
Однако на практике этот метод разделения всегда сопряжен с определенными трудностями. Поскольку система неидеальная, внутри каждой зоны образуется разность градиентов концентраций. Концентрация компонентов с одной стороны зоны максимальная, с другой — уменьшается до нуля. Во время движения зон с разделенными ионами вниз по колонке максимальная концентрация постепенно уменьшается и первоначально узкая зона расширяется. В таких случаях хроматографическая кривая (т. е. кривая зависимости объема элюирующего раствора от концентрации элемента в элюате) имеет конусообразный вид и выражается математически кривой Гаусса (гл. 2).
1.3.4. Другие ионообменные процессы
В ионообменных процессах могут быть использованы не только гранульные ионообменники, но также материалы в форме бумаги, тонких пластин или мембран. Ионообменную бумагу получают введением тонкодисперсных частиц смолы в бумажную пульпу или проведением синтеза неорганического ионообменного материала непосредственно в слое бумаги. Практические методы работы с ионообменными материалами в форме бумаги, тонких пластин и мембран аналогичны приемам, используемым в бумажной и тонкослойной хроматографии и в электрохимических методах разделения.
1. Ионообменники и ионообменные процессы 43
Литература
1. ШРАС— Recomendation on Nomenclature for Chromatography (1972).
2. Dowex Ion Exchange, The Dow Chemical Company (1964).
3. Myers G.E., Boyd G. E., J. Phys. Chem., 60 (1956) 521.
4. Boyd G. E., Lindenbaum S., Myers G. E., J. Phys. Chem., 65 (1961) 577.
5. Gregor H. P., Sundheim B. R., Held К. M., Waxman M. X., J. Coll. Sci., 7 (1952) 511.
6. Bodamer G. W., Kunin R., Ind. Eng. Chem., 45 (1953) 2577.
2. Теория
В этой главе даны только основные принципы ионообменной теории, которые необходимы для понимания процессов разделения ионов на ионооб-менниках. Подробное изложение различных теоретических аспектов ионного обмена и ионообменных процессов можно найти в ряде монографий (приложение 2).
2.1. Ионообменные равновесия и коэффициенты селективности
Как указывалось ранее (разд. 1.1.4), если раствор электролита, не содержащий комплексообразующие или другие усложняющие процесс вещества, контактирует с ионообменником в замкнутой системе, то через некоторое время устанавливается равновесие между ионообменником и ионами в растворе. Эта равновесная система описывается уравнением w2rW|mj + wiM2'2 wlRm2M2 + W2M1"' (2.1)
где т1 и т2 — абсолютные заряды ионов М, и М2 соответственно, д?! Ф т2, R — ионообменная смола.
В случае обмена равнозарядных ионов уравнение (2.1) преобразуется:
RM, + Мр RM2 + M^i (2.2)
где От| = т2.
Состояние равновесия полностью обратимо и не зависит от того, с какой стороны оно достигнуто. На положение равновесия влияют не только относительные количества RM[ и ионов М2, но в значительной степени химическая природа ионов М| и М2, параметры смолы и очень часто другие вещества, присутствующие в растворе.
Как правило, в состоянии равновесия число обоих ионов в равновесном растворе и на смоле неодинаково. Смола предпочтительно сорбирует один тип ионов относительно другого (даже когда они присутствуют в эквивалентных количествах). Это явление в ионообменных процессах носит название селективности (или относительного сродства). Объяснение и предсказание селективности смол к различным ионам представляет одну из самых интересных проблем с теоретической и практической точек зрения.
При решении этой проблемы используют различные подходы. Строгая термодинамическая трактовка имеет ограниченное практическое значение вследствие необходимости большого количества точных измерений для выполнения расчетов. Поэтому, по-видимому, более легкий путь — непосредственное измерение селективности (коэффициентов селективности).
2. Теория
45
При формальном применении закона действия масс, например, к реакции (2.2) истинная термодинамическая константа равновесия Ка выразится уравнением
- . л"1!
Ка = -^1-----М*. (2.3)
аЪ/?г ' °RM1
где а — термодинамическая активность компонентов.
Для расчета Ка необходимо определить активности индивидуальных компонентов в обеих фазах. Одиако экспериментальное определение величин aRM) и aRMj очень сложно.
Уравнение (2.3) может быть преобразовано в выражение:
к _ IRM2]7rm2 • МЬм,
° ~ [М2)7м2 • IKMjbRM,
где квадратные скобки [ ] обозначают аналитические концентрации индивидуальных компонентов; у — коэффициент активности. Вводя соотношение
k = [RMjtMj] м2-м1 [M2HRMJ
(2.5)
получают уравнение для термодинамической константы Ка:
Ка — ^м2, М,
7rm27m, T'M27Rm1
(2.6)
Величина kM^ М) обозначает коэффициент селективности и выражает меру относительного сродства различных ионов к данному ионообменнику.
Соотношение 7м/7м2 (в растворе) во многих случаях может быть рассчитано достаточно точно. Однако определение величины 7rm2//7rm • т- е-соотношения коэффициентов активности в фазе смолы, вызывает серьезные трудности. Его величина зависит от выбранного стандартного состояния смолы. Если брать чистые смолы М| и М2 (обе в состоянии равновесия с чистой водой) в качестве стандартных состояний, то можно показать,
что
i
lg Ка = j 1g К • + поправочный коэффициент (2.7)
о
где К равно kM^ m^m/Tmj и wm2 — эквивалентная доля катиона М2 в смоле. Поправочный коэффициент выражает влияние концентрации раствора на удерживание воды смолой [1 — 7].
При обмене ионов разного заряда уравнение (2.5) принимает вид
= [RM2]m.[M,]m2
М2’ М1 [M2]'"1[RMi]'"2
где ш, и т2 — абсолютные заряды ионов М, и М2 соответственно. Уравнения (2.5) и (2.8) применимы для оценки коэффициентов селективности катионо- и анионообменников.
46
Часть 1
При обмене ионов равного заряда численное значение коэффициента ^м2 Mj не зависит от единиц, использованных для выражения концентраций ионов в обеих фазах. Напротив, при обмене разнозарядных ионов их концентрации входят в уравнение (2.8) с различными показателями степеней, и поэтому единицы концентраций оказывают влияние на численное значение коэффициента км^ м . Концентрация ионов во внешнем растворе обычно выражается нормальностью или молярностью, концентрация ионов в фазе смолы — моляльной (или эквивалентной) долей противоионов.
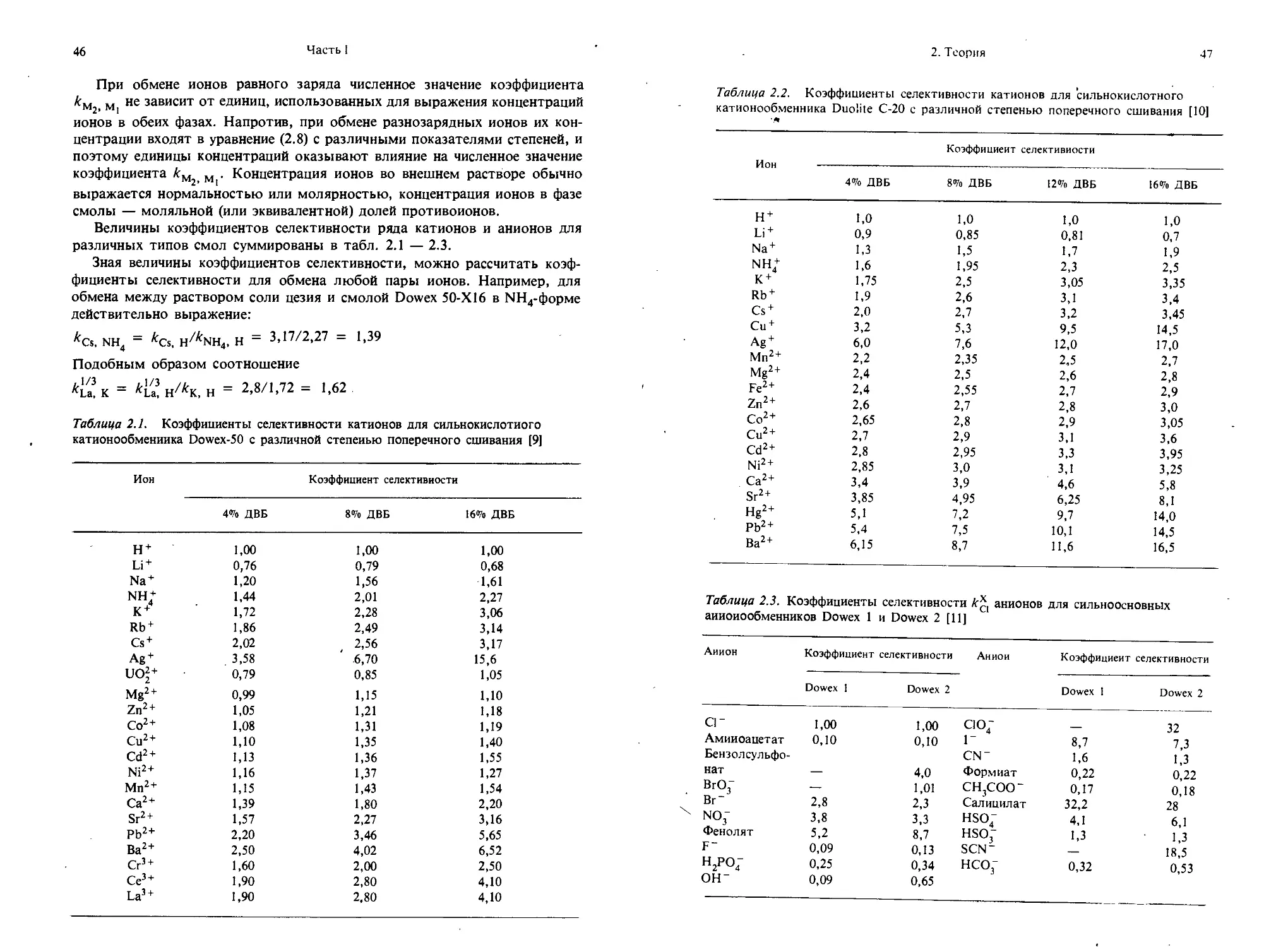
Величины коэффициентов селективности ряда катионов и анионов для различных типов смол суммированы в табл. 2.1 — 2.3.
Зная величины коэффициентов селективности, можно рассчитать коэффициенты селективности для обмена любой пары ионов. Например, для обмена между раствором соли цезия и смолой Dowex 50-Х 16 в МН4-форме действительно выражение:
kCs, nh4 = ^cs, h^nh,,, н = 3,17/2,27 = 1,39
Подобным образом соотношение
= 42н^к,н = 2,8/1,72 = 1,62
Таблица 2.1. Коэффициенты селективности катионов для сильнокислотного катионообмениика Dowex-50 с различной степенью поперечного сшивания [9]
Ион Коэффициент селективности
4% ДВБ 8% ДВБ 16% ДВБ
н+ 1,00 1,00 1,00
Li + 0,76 0,79 0,68
Na+ 1,20 1,56 1,61
NH + 1,44 2,01 2,27
К + 1,72 2,28 3,06
Rb + 1,86 2,49 3,14
Cs + 2,02 2,56 3,17
Ag + 3,58 6,70 15,6
uo2+ 0,79 0,85 1,05
Mg2 + 0,99 1,15 1,10
Zn2 + 1,05 1,21 1,18
Co2+ 1,08 1,31 1,19
Cu2+ 1,10 1,35 1,40
Cd2+ 1,13 1,36 1,55
Ni2+ 1,16 1,37 1,27
Mn2+ 1,15 1,43 1,54
Ca2+ 1,39 1,80 2,20
Sr2 + 1,57 2,27 3,16
Pb2+ 2,20 3,46 5,65
Ba2+ 2,50 4,02 6,52
Cr3 + 1,60 2,00 2,50
Ce3 + 1,90 2,80 4,10
La3 + 1,90 2,80 4,10
2. Теория
47
Таблица 2.2. Коэффициенты селективности катионов для сильнокислотного катионообменника Duolite С-20 с различной степенью поперечного сшивания [10]
Коэффициент селективности
Ион
4% ДВБ 8% ДВБ
12% ДВБ
16% ДВБ
н+ 1,0 1,0 1,0 1,0
Li + 0,9 0,85 0,81 0,7
Na + 1,3 1,5 1,7 1,9
NH + 1,6 1,95 2,3 2,5
К + 1,75 2,5 3,05 3,35
Rb + 1,9 2,6 3,1 3,4
Cs + 2,0 2,7 3,2 3,45
Cu + 3,2 5,3 9,5 14,5
Ag + 6,0 7,6 12,0 17,0
Mn2+ 2,2 2,35 2,5 2,7
Mg2+ 2,4 2,5 2,6 2,8
Fe2+ 2,4 2,55 2,7 2,9
Zn2+ 2,6 2,7 2,8 3,0
Co2+ 2,65 2,8 2,9 3,05
Cu2 + 2,7 2,9 3,1 3,6
Cd2+ 2,8 2,95 3,3 3,95
Ni2 + 2,85 3,0 3,1 3,25
Ca2+ 3,4 3,9 4,6 5,8
Sr2+ 3,85 4,95 6,25 8,1
Hg2+ 5,1 7,2 9,7 14,0
Pb2 + 5,4 7,5 10,1 14,5
Ba2+ 6,15 8,7 11,6 16,5
Таблица 2.3. Коэффициенты селективности /с* анионов для сильноосновных аииоиообменников Dowex 1 и Dowex 2 [11]
Аиион Коэффициент селективности Аниои Коэффициент селективности
Dowex 1 Dowex 2 Dowex 1 Dowex 2
cr 1,00 1,00 СЮ,- — 32
Амииоацетат 0,10 0,10 1- 8,7 7,3
Бензолсульфо- CN" 1,6 1,3
нат — 4,0 Формиат 0,22 0,22
ВгОГ — 1,01 сн3соо- 0,17 0,18
Br~ 2,8 2,3 Салицилат 32,2 28
NO” 3,8 3,3 HSO; 4,1 6,1
Фенолят 5,2 8,7 HSOj- 1,3 1,3
F~ 0,09 0,13 SCN- — 18,5
Н2РО4- 0,25 0,34 HCOf 0,32 0,53
ОН- 0,09 0,65
48
Часть I
(показатель степени указывает состояние окисления ионов) справедливо при обмене между раствором соли лантана и смолой Dowex 5О-Х8 в К-форме. «
Величина коэффициента селективности не постоянна и зависит от размера ионов и их общей концентрации в растворе, химической структуры смолы и в некоторой степени от ее обменной емкости. Влияние числа поперечных связей видно из данных, представленных в табл. 2.1 — 2.3.
Ионы, обладающие наибольшим сродством к обменнику, снижают на-бухаемость смолы до минимума.
При обмене ионов разной валентности влияние ряда факторов усложняется, однако общее правило, что селективность обмена выше для ионов с большей валентностью в более разбавленных растворах, выполняется.
2.1.1. Равновесие обмена микроэлементов
При хроматографических разделениях часто имеют дело с системами, в которых разделяемые ионы имеют очень низкую концентрацию на фоне большого избытка других ионов.
Томпкинс и Майер [8] вывели уравнения, характеризуюшие поглощение микроколичеств ионов. Заменяя выражение [RM2]mi/[M2]'”> в уравнении (2.8) на коэффициент распределения 2ЭМг> получаем n _ ^М2 - *м2, м, [Mij )
(2.9)
Ион М2 находится в растворе в следовых количествах и не связан в комплекс. М] — способный к обмену ион полностью диссоциированного электролита, присутствующий в растворе в большом избытке по отношению к иону М2. км^ м — коэффициент селективности, От] и /w2 — абсолютные величины зарядов ионов Mt и М2 соответственно.
Принимая во внимание большой избыток ионов М|, считаем их концентрацию в фазе смолы постоянной. Величина км м не изменяется при условии постоянства соотношения коэффициентов активности во внешнем растворе (ум/у&/т').
Тогда 1
DM = const -----^—7— (2.10)
Из уравнения (2.10) следует, что при определенной концентрации иона М, (макрокомпонента раствора) коэффициент распределения DM не зависит от фактической концентрации иона М2, т. е. поглощение иона М2 (микрокомпонента) прямо пропорционально его концентрации во внешнем растворе.
Уравнение (2.10) может быть использовано для приблизительной оценки влияния концентрации элюирующего агента, содержащего ионы М,, на величину коэффициента распределения микрокомпонента.
2. Теория
49
Преобразование уравнения (2.10) в логарифмическую форму дает возможность определять заряд микрокомпонента. Построением диаграмм типа 1g DM^ — lg[Mj] для ряда ионов с различным зарядом получают серию прямых линий, пересекающихся в определенных точках. Точки пересечения ’ прямых указывают концентрации ионов Mt, при которых разделения ионов М] и М2 в равновесных условиях не происходит.
2.1.2. Ионообменные равновесия в присутствии комплексообразующих веществ
КАТИОНООБМЕННИКИ
В присутствии комплексообразующих веществ в растворе катионы образуют комплексы, ионообменные свойства которых в значительной степени зависят от их заряда. Наибольшие изменения ионообменных свойств наблюдаются в тех случаях, когда катионы превращаются в комплексные анионы. Тогда на катионообменнике сорбируется лишь незначительное количество металла, и ионообменное равновесие определяется концентрацией положительно заряженного иона металла, не связанного в комплекс.
Если комплексообразующее вещество H^L, которое дает растворимые нейтральные или отрицательно заряженные комплексы типа М21/”* _ (не поглощаемые катнонообменником), вводят в водную фазу, содержащую ионы М^4", величина коэффициента распределения D’M выражается соотношением
= --------------------. _—— (2 Л1)
[\ф ]+ £ [M2L](n* - 1+ £
п = 1 п = 1
где — константа устойчивости комплексов M2L("* и [L* ] — равновесная концентрация аниона комплексообразующего вещества.
Присутствие комплексообразующего вещества приводит к уменьшению поглощения катиона М^г4" на катионообменнике. Это уменьшение пропорционально устойчивости образованного комплекса и показателю степени (л) равновесной концентрации комплексообразующего вещества, [L*-]”.
При условии образования в водном растворе только одного вида комплексов M2L„ для D^2 действительно выражение
D, _^2^_ РМ2 _^М2[Н + Г
м> [M2L„] ^'[HLf
где Кд — константа диссоциации одноосновной комплексообразующей кислоты HL (b = 1).
Фактор разделения двух ионов равного заряда в присутствии комплексообразующих веществ выражается следующим образом:
4-354
50
Часть I
Из уравнения (2.13) следует, что фактор разделения а' определяется только соотношением £>'/3"/£>"/3^ и не зависит ни от концентрации комплексообразующего вещества, ни от величины pH раствора.
Во многих случаях на соотношение /3" //3„ оказывает влияние изменение относительной диэлектрической проницаемости внешнего раствора. Эффективность разделения может быть улучшена путем введения смешивающихся с водой органических растворителей. Влияние добавок органических растворителей на эффективность разделения видно из соотношения
/в' \ в' X (2.14)
lg ( " =lg— + ------------F
v; /орг RT
где X — мольная доля органического растворителя и F — коэффициент, характеризующий перенос свободной энергии из водной в водно-органическую систему.
При положительном знаке величины F введение органического растворителя улучшает разделение компонентов. Второй член правой части уравнения (2.14) показывает, что эффективность разделения повышается с увеличением содержания органического растворителя в системе. Однако высокие концентрации органического растворителя способствуют уменьшению эффективности разделения вследствие неблагоприятного влияния изменения ионных сольватных оболочек, кинетики обмена и т. п.
АНИОНООБМЕННИКИ
Основное уравнение, описывающее обмен анионов, например, на анио-нообменниках в Cl-форме, имеет вид
£>RC1 + В*- RdB + дСГ
(2.15)
Коэффициент селективности выражается соотношением
. _ [RdB][Cl-f _ [СГ]*
B,cl [RC1]*[B*“] в [RC1]*
(2.16)
где DB — коэффициент распределения аниона В6- .
В присутствии катиона М, который может образовывать отрицательно заряженные комплексы, поглощаемые анионообменниками (например, МС1„ + р согласно уравнению
RC1- + МС1„ + ] * RMC1„ + ] + СГ
(2.17)
выражение для коэффициента селективности ^мс1„ + ь CI принимает вид
ЛМС1„ + ,, cl
[RMC1„ + ,][С1-] = [RMC1„ + ,] [Ma; + jKRCii [RC!]/3n + дм^нст]"
(2.18)
2. Теория
51
Поглощение комплекса МС1^ । увеличивается с повышением его устойчивости. Селективность разделения двух комплексных анионов улучшается с увеличением разности в их константах устойчивости.
2.1.3. Обмен анионов слабых кислот на анионообменнике
Коэффициент селективности fcAB обмена слабой кислоты НВ, фиксированной анионообменником, на кислоту НА при условии равенства зарядов анионов обеих кислот выражается соотношением
[RBHA-] [B"J[RA]
(2.19)
Если в обмене участвует только диссоциированная часть кислоты НВ, доля сорбированных ионов составляет
О - [в-1
Уэфф - ------
®обш
(2.20)
где сВо6щ= [В ] + [НВ], т. е. сВо6щ — общая (или аналитическая) концентрация В в растворе. Коэффициент распределения равен
D - [RB1 В с ®общ
(2.21)
Преобразуя уравнения (2.19) — (2.21), получаем
D - к О
~ *а,в уэФФ]^]
Принимая во внимание константу диссоциации НВ, находим
(2.22)
D = к *нв
в А'%в + [Н + ]
[RA]
[А-]
(2.23)
где /Гнв — константа диссоциации кислоты НВ.
Уравнение (2.23) позволяет оценить возможность разделения кислот или их анионов при различных концентрациях, а также выбрать оптимальную величину pH буферного раствора, используемого для разделения.
Следует отметить, что анионы буферных растворов, применяемых для установления pH внешнего раствора, не должны ни вытеснять анионы, сорбированные ионообменниками, ни влиять иа их коэффициенты распределения. Этим эффектом можно пренебречь при очень низкой концентрации буферного раствора и малой степени сродства анионов этого раствора к ионообменнику.
52
Часть I
2.2. Таредочная теория
При разделении веществ методами экстракции, дистилляции и вытеснительной ионообменной хроматографии процессы установления равновесия многократно повторяются. Исходя из аналогии этих процессов, для описания метода вытеснительной ионообменной хроматографии использована тарелочная теория. Первоначально эта теория была введена Мартином и Сингом [12] в распределительную жидкостную хроматографию, а затем адаптирована к ионообменной хроматографии. Разработана периодическая [13] и непрерывная [14, 15] модели процесса.
Под термином «тарелка» понимают теоретический поперечный слой ио-нообмениика, по площади сечеиия равный площади колонки. Выражение «высота, эквивалентная теоретической тарелке» (ВЭТТ) дается уравнением
ВЭТТ = L/N (2.24)
где L — длина колонки и Д' — общее число тарелок в колонке.
Для успешного применения тарелочной теории необходимы следующие условия:
а) равновесие между фазами ионообменника и раствора на каждой тарелке,
б) незначительные концентрации обмениваемых ионов в обеих фазах по сравнению с концентрациями элюирующих ионов в растворе и иоиообмеи-нике,
в) одинаковое распределение ионов между смолой и раствором на любой тарелке,
г) линейность изотермы сорбции.
Более ранняя периодическая теория Майера — Томпкинса осиоваиа на предположении, что движение жидкости в колонке с Д’ теоретическими тарелками осуществляется скачками «от тарелки к тарелке». Каждая тарелка имеет одинаковую массу смолы, т, и одинаковый объем раствора, Af. При пропускании жидкости через колонку объем раствора, Дг?, перемещается вниз по колонке от тарелки к тарелке. Компонент М также переносится вниз и распределяется между обеими фазами в соответствии с уравнением
D = 15^ (2.25)
[М$]
где D — коэффициент распределения, [MJ — количество компонента М в равновесном растворе, [RM] — количество компонента М в смоле в состоянии равновесия.
Доля компонента М, который вначале сорбировался в тарелке О верхней части колонки, а затем перешел в раствор Р-й тарелки, выражается уравнением
_ (N + Р - 1)! Dn - 1
Мм» — -------------- -------v -l р (2.26)
NP Р!(Д - 1)! (D + 1)" + р
2. Теория
53
где N — число кратных объема раствора Ду, уравновешенных с Р-й тарелкой.
Показано, что Мд,^ достигает максимального значения, когда через колонку пропущено число объемов тарелки, равное коэффициенту распределения, умноженному на число тарелок в колонке:
^max = PD (2.27)
Доля компонента в растворе в одном объеме тарелки, когда
MN, Р = max
и Ms max = (2xPZ?(l + D)J-1/2 (2.28)
выражается уравнением (2.26).
Если принять, что Vo( = PAv) — объем колонки (объем раствора в колонке) и В — число объемов Vo, пропущенных через колонку, т. е.
(общий объем, пропущенный через колонку \
' К Г
то ^тах = ^тт/Р (2.29)
и из уравнения (2.27) находим *тах = D (2-30)
Уравнение (2.27) — одно из основных уравнений тарелочной теории. Оно представляет зависимость между коэффициентом распределения и числом объемов колонки, пропущенных до того момента, когда концентрация компонента М в элюате достигает максимального значения.
Используя уравнение (2.26), можно теоретически оценить всю хроматографическую кривую для любой колонки при условии, что известны число тарелок и объем в пиковой точке для данной колонки.
Уравнение (2.26) может быть нормализовано в форме гауссова распределения (если хроматографическая кривая имеет форму конуса и приближается к нормальной кривой ошибок).
Глюкауф [14] в своей улучшенной модели предположил, что поток жидкости через колонку является непрерывным. Эта модель лучше соответствует действительности и дает возможность применить более удобную математическую обработку.
Исходная предпосылка модели Глюкауфа состоит в том, что колонка разделяется на единицы (тарелки) одинаковой длины. Обменник и раствор находятся в равновесии друг с другом, и концентрация компонента в каждой фазе одинакова.
Глюкауф выражает длину тарелки и расстояние от верха колонки в единицах геометрического объема колонки, Х\
X = L • А (2.31)
54
Часть 1
L — длина колонки, А — площадь поперечного сечения колонки, / — доля величины L и х — доля величины X.
Выделим тонкий слой ионообменной колонки высотой Д/. В случае частиц смолы сферической формы такую минимальную величину Д/ можно выбрать при условии постоянства основных механических и геометрических свойств частиц смолы.
В таком случае объем этого слоя Дх (высоты Д/) равен Д/ • А, где А — поперечное сечение колонки. Если объем колонки равен И, порции раствора объемом ДИ будут проходить через слой Дх. Концентрации раствора до прохождения через слой обменника и после него равны с(Л. _ и сх соответственно. Концентрация в слое Дх изменяется на ДИ(с^ _ дг) — сх).
Слой Дх до пропускания объема ДИ содержит определенное количество сорбированного компонента &xqy. После прохождения объема ДИ через слой Дх последний теряет некоторое количество компонента Дх<^и + (q — общая концентрация компонента в единице объема колонки). Истинное количество компонента, внесенное в тарелку Дх, равно
Д^(сСе - Аг) - сх) = Д*(?(Г + ДИ) _ Уу) (2.32)
qy—# общая концентрация рассматриваемого компонента, сх — концентрация раствора в тарелке.
Учитывая, что поток раствора через колонку непрерывный, ДИ выбирают как бесконечно малую величину (в противоположность величине Дх) и, вводя производные, получают
(2.33)
(2.34)
Подставляя выражения (2.33) и (2.34) в уравнение (2.32) и деля на
Дх • ДИ, находят
Для бесконечно малой величины ДИ действительно следующее уравнение
/дс \ /dq \ Дх / Э2с \
( — I + ( — ) - — ( —т ) = 0 (2.36)
\дх / у \dV/x 2 \дх2/у
Если смола и раствор находятся в равновесии и q = а сх (где а — постоянная и сх — концентрация раствора в тарелке), то
2. Теория
55
Дх /д2с\
(2.37)
Это общее уравнение после интегрирования при соответствующих граничных условиях применимо к любым типам колонок. Его можно использовать также при элюировании зон определенной ширины. В начале хроматографического разделения зона компонента занимает No верхних теоретических тарелок. Раствор в этих тарелках имеет одинаковую концентрацию с0.
При пропускании F теоретических тарелочных объемов растворителя (F = Р7аДх) концентрация раствора в ЛГ-й тарелке от верха колонки будет равна с.
В этом случае решение уравнения (2.37) записывают как
(2.38)
где At(t) — ошибка функции
1 С ( \
А>т = w J “р у т г <23”
о
Для успешного хроматографического разделения должны быть выполнены определенные условия, а именно N' > No, и хроматографическая кривая должна быть четкой, т. е. максимум величины с/с0 не должен быть слишком мал.
С учетом перечисленных выше приближений уравнение (2.38) принимает вид
т
exp
V(eA" V)
N'(aX' - И)2
2aX’ V
(2.40)
где N' = N — (N0/2); X' = TV' Дх (объем колонки от центра первоначальной зоны до низа); т = а с0 • х0 (общая масса компонента в зоне).
Таким образом действительно соотношение
Плах = « • Х' - (2‘41>
которое входит в экспоненциальный коэффициент уравнения (2.40). Уравнение (2.41) эквивалентно выражению Майера—Томпкинса в уравнении (2.8).
При пропускании раствора через колонку, содержащую более 25 теоретических тарелок, форму хроматографической кривой оценивают из уравнения
\ z у max у
(2.42)
56
Часть I
Рис. 2.1. Графическое представление обозначений, используемых в формулах, на хроматографической кривой.
Соотношение между числом тарелок и шириной зоны хроматографической кривой (/?) записывают в виде
(2.43)
где /3 — объем элюата, концентрация которого равна стах/е (рис. 2.1).
Концентрацию раствора в максимуме хроматографической кривой рассчитывают по уравнению
с max
/хг>\ 1/2 т I N \
V \ 2тг /
’ max X /
(2.44)
В соответствии с уравнениями (2.43) и (2.44) с помощью экспериментальной хроматографической кривой можно определить число теоретических тарелок в колонке. Уменьшение высоты теоретической тарелки приводит к образованию более четких пиков в максимумах хроматографических кривых, снижению перекрывания соседних зон и повышению эффективности разделения.
На основании рассмотренных выше теоретических представлений выведено два важных правила:
1) максимальная концентрация, стах, на вершине кривой обратно пропорциональна корню квадратному из длины колонки;
2) ширина хроматографической кривой прямо пропорциональна корню квадратному из длины колонки.
Число теоретических тарелок может быть оценено также по форме выходной кривой вещества, введенного в элюирующий раствор [14]. Действительно следующее уравнение:
2. Теория
57
И),5 ' И),159 (Н),5 - Н),159)2
(2.45)
где И0)5 — объем раствора, пропущенного через колонку при с/с0 = 0,5, И),159 ~ объем раствора, пропущенного через колонку при с/с0 = 0,159.
РАСЧЕТ ОБЪЕМОВ ЭЛЮИРОВАНИЯ
С точки зрения практического применения ионообменного хроматографического разделения необходимо знать величину Итах, т. е. объем элюирующего раствора, который нужно пропустить для получения максимума на хроматографической кривой. Эту величину легко рассчитать из равновесных данных с использованием простого уравнения
= f/c (2.46)
Л
где
АГ'=Х-|х0, (2.47)
X — объем загрузки колонки (см3). Второй член уравнения (2.47) обычно очень мал, и им можно пренебречь. Х$ — объем исходной заряженной зоны, f — равновесное количество компонента в 1 см3 загрузки колонки, с — концентрация компонента (ммоль Мт + /т в 1 см3).
Если массовый коэффициент распределения рассматриваемого вещества Dg известен, то с помощью уравнения (2.46) можно вывести следующее соотношение:
%®* = Dgpr + £,. = Dy + £,. (2.48)
X
Pr — масса сухой смолы в 1 см3 колонки, £; — относительный промежуточный объем колонки (£;- * 0,4).
Объем элюирующего раствора, необходимый для достижения стах, рассчитывают по уравнению
V = D • т ' (2.49)
где D& — массовый коэффициент распределения, т — масса смолы в колонке. Селективность разделения двух компонентов Mj и М2 определяется фактором разделения а:
Итах М2 DVM2 + Ei _ DgM$ + 1
а = -------=----------------~ (Z.jU)
V ч Duu + 1
ктах М| 1
Dy — объемный коэффициент распределения.
58
Часть 1
РАСЧЕТ ШИРИНЫ ЗОНЫ
Приближенное уравнение для расчета соотношения между числом теоретических тарелок и полушириной зоны введено Матесоном [16]
(2.51)
Более точное уравнение:
2Г (V 4- К)
— *maxV max tf
w2
(2.52)
W — полуширина зоны, И; — промежуточный объем колонки.
ВЫСОТА ТЕОРЕТИЧЕСКОЙ ТАРЕЛКИ
Высота, эквивалентная теоретической тарелке, Н, как функция параметров колонки определена Глюкауфом [15] согласно следующему уравнению:
0,26бА Л V2
X-------------+ — (2.53)
Ds(l + 70ги) и
г—радиус частицы, Dv—объемный коэффициент распределения, и — линейная скорость потока в миллилитрах на единицу площади в единицу времени, — относительный промежуточный объем, Dr и Ds — коэффициенты диффузии компонента в смоле и в растворе соответственно.
Величины Н, рассчитанные по уравнению (2.53), всегда ниже величин, полученных экспериментально для мелких частиц. Это несоответствие обусловлено такими факторами, как неравномерное заполнение колонки, неправильная форма частиц и образование каналов.
Уравнение (2.53) показывает влияние температуры на процесс разделения. Поскольку величина Н на стадии разделения должна быть по возможности малой, коэффициенты диффузии Dr и Ds во втором и третьем членах будут влиять на разделение. Коэффициенты диффузии увеличиваются с повышением температуры; чем выше их величины, тем меньше должна быть высота Н и тем больше число теоретических тарелок в колонке. Эффективность разделения при этом повышается.
Результаты экспериментальных исследований показали, что величина 1,64г должна быть в - 10 раз выше. При расчете оптимальных условий
2. Теория
59
работы по уравнению (2.53) последним членом уравнения пренебрегают, а величину первого члена умножают на два. Второй и третий члены в уравнении f2.53) приблизительно равны.
РАЗРЕШЕНИЕ
На основании формы пика хроматографической кривой, которая приближается к гауссовой кривой распределения ошибок, можно сделать вывод, что идеального физического разделения двух соседних компонентов не может быть достигнуто. В работах по хроматографии делаются попытки снизить загрязнение соседних компонентов или уменьшить перекрывание зон [17, 18]. Степень разделения двух соседних зон определяют как разрешение
у _ у
R = 2max 1 max (7 54)
0,5( И' + W2)
и W2 — средняя ширина хроматографических зон. Для достижения четкого разделения компонентов 1 и 2 должно выполняться следующее неравенство:
у _ у
2 max 1 max > । (2 55)
0,5( + Ш2)
Минимальное число тарелок выражается формулой
(у /у ц- 1 \ 2 * * * *
2тах -‘-ДВ*--1 (2.56)
^2 max'7 ВИ max — /
Это уравнение имеет большое значение и наряду с уравнением (2.50) позволяет рассчитать необходимое минимальное количество тарелок, если известны коэффициенты распределения веществ 1 и 2.
Зная величину Н, определенную экспериментальным или расчетным путем, из уравнения L = Н N находят оптимальную длину колонки.
В процессе ионообменного хроматографического разделения часто происходит расширение зон, обусловленное медленной диффузией внутри частиц ионообменника. Это приводит к замедлению установления равновесия при распределении компонентов между стационарной ц подвижной фазами при элюировании.
В тех случаях, когда хроматографический процесс определяется внутренней диффузией, число тарелок (в первом приближении) выражается как
LD
N’ » const,—+ const, (2.57)
Ни
где L — длина колонки, Dr — коэффициент диффузии компонента,
60
Часть 1
и — скорость потока, г — радиус частиц, const] и const2 — константы, зависящие от геометрических параметров колонки. ,
С целью сокращения времени, необходимого для разделения компонентов, увеличивают число тарелок N (чаше всего путем повышения скорости потока, и), но в этом случае должен быть уменьшен размер частиц смолы. Для поддержания приемлемой скорости потока используют высокое давление (квадрат радиуса частиц обратно пропорционален снижению давления в колонке при условии, что скорость потока постоянна). На этом принципе основана хроматография высокого давления.
Для повышения эффективности разделения целесообразно применять ионообменники, проявляющие высокую селективность к одному из разделяемых ионов, или изменять состав элюируюшего раствора. На практике часто предпочитают последнее, так как приготовление различных элюентов проше и быстрее. Для изменения состава стационарной фазы необходимо иметь селективные ионообменники со специфическими функциональными группами. В последние годы эти возможности значительно расширились в связи с накоплением опыта в получении и исследовании свойств большого числа селективных ионообменников (гл. 6).
2.3. Другие методы количественного описания хроматографического процесса
Для количественного описания хроматографического процесса Крауз и Море [19] предложили теорию, основанную на использовании констант элюирования.
Константа элюирования описывается соотношением
/ • А
Е = ~ (2.58)
V
где Е — константа элюирования, А — поперечное сечение колонки (см2), / — миграционное расстояние зоны (см) после заполнения объема V (см3) элюируюшего раствора.
Зависимость между коэффициентом распределения Dv и константой элюирования Е выражается уравнением
Е = . (2.59)
z + Dv
где i — доля растворителя в слое обменника.
Дженч приводит следующее соотношение между константой элюирования первых, Ej-, и последних, Et, следов разделенных вешеств в элюате:
Ef = (2.60)
2. Теория
61
Е, = — (2.61)
' V,
I — длина колонки (см), V? и — объемы элюента (см3), которые используют для вымывания из колонки первых и последних следов ионов соответственно.
Два иона разделяются количественно, если
^м,-£/м2>0 <2-62)
£ум2 — константа элюирования первых следов элемента М2, £/М1 — константа элюирования последних следов элемента Мг
2.4. Градиентная элюентная хроматография
Термин градиентное элюирование применим для методов; в которых состав элюирующего раствора непрерывно изменяется в процессе элюирования (концентрация элюируюшего агента повышается). Вдоль колонки создается таким образом определенный градиент концентрации [21 — 23]. Повышение концентрации элюирующего раствора сопровождается улучшением ряда параметров.
Во-первых, на кривых элюирования предотвращается образование хвостов. Вследствие повышения концентрации элюируюшего агента вытянутые концы кривых смещаются к главной зоне, и кривые элюирования становятся более симметричными.
Во-вторых, ускоряется элюирование следующих друг за другом веществ. Компоненты удерживаются в ионообменной колонке в течение меньшего времени и приближаются друг к< другу.
В-третьих, при использовании градиентного элюирования время разделения значительно сокращается, особенно при низкой концентрации веществ.
Однако в некоторых случаях применение градиента неблагоприятно. Если зоны разделяемых веществ находятся-близко друг от друга, то с увеличением концентрации элюирующего агента зоны настолько сближаются, что возможно их перекрывание. Поэтому вещества, легко разделяемые обычным элюированием, не могут быть разделены градиентным элюированием.
В ионообменной хроматографии градиентное элюирование осуществляют с помощью градиента концентрации (концентрация элюирующего раствора непрерывно изменяется) или градиента pH (pH элюируюшего раствора непрерывно изменяется).
В некоторых методах одновременно изменяют pH и концентрацию элюируюшего агента.
Формы градиента могут быть различными. Если изменение состава элюируюшего раствора постоянно в каждый промежуток времени, то градиент — линейный. Выпуклая форма градиента наблюдается при быст
62
Часть 1
ром изменении состава элюирующего раствора в начале процесса и медленном — в конце. Обратный порядок изменения концентрации приводит к образованию вогнутого градиента.
Градиент концентрации получают следующим образом: элюирующий раствор (концентрации ср поступает из резервуара в смеситель, содержащий элюирующий раствор более низкой концентрации, с2(с1 > с2) и затем в колонку. Состав раствора постепенно изменяется от концентрации с2 к концентрации q. На образование градиента влияет не только состав обоих растворов, но и соотношение объемов растворов в резервуаре и смесителе, а также скорости поступления растворов из резервуара в смеситель и из смесителя в колонку.
Непрерывное изменение состава элюирующего раствора достигается тремя путями:
1) изменением объема резервуара при постоянном объеме смесителя;
2) соединением смесителя и резервуара в систему сообщающихся сосудов и одновременным уменьшением уровня жидкости в обоих сосудах;
3) дифференциацией скоростей прокачивания в смеситель двух элюирующих растворов различного состава.
2.4.1. Градиент концентрации
СИСТЕМЫ С ОДНИМ ЗАКРЫТЫМ СМЕСИТЕЛЕМ ПОСТОЯННОГО ОБЪЕМА
Этот градиент легко получают с помощью установки, показанной на рис. 4.9. Более концентрированный раствор из резервуара А поступает в смеситель В, который заполнен разбавителем (раствором более низкой концентрации, чем в резервуаре А). Объем раствора в смесителе В сохраняется постоянным в течение всего процесса разделения. В этой установке образуется выпуклый экспоненциальный градиент.
Алм и др. [24] вывели следующее уравнение для градиента элюирующего раствора, поступающего из смесителя в колонку:
— = 2,303 1g Х (2.63)
V X - х
х — концентрация элюирующего раствора, поступающего из резервуара в смеситель в момент переноса объема v из смесителя в колонку, X — концентрация элюирующего раствора в резервуаре, V — объем смесителя. Наклон образованного градиента зависит от размера смесителя. Чем меньше смеситель, тем градиент более крутой и более выпуклый.
Черкин и др. [25] вывели уравнение, показывающее влияние концентрации исходного элюирующего раствора и соотношения объем потока/объем смесителя на разбавление вытекающего раствора элюента:
2. Теория
63
с = с0(1 - 1/ек) (2.64)
с0 — концентрация элюента в резервуаре, с — концентрация элюента, вытекающего из смесителя, к — отношение общего объема элюата (у) к объему разбавителя в смесителе (И) (к = v/V). Градиент имеет форму, близкую к линейной, при условии, что величина к < 1.
Другие авторы [26, 27] для этого типа градиента приводят следующее уравнение:
ск = Cz - (cz - с0) • e~v/v' (2.65)
ск — концентрация элюирующего раствора, поступающего в колонку, cz — концентрация элюента в резервуаре, с0 — исходная концентрация элюента в смесителе, v — объем элюирующего раствора, пропущенного через колонку, И] — объем жидкости в смесителе.
СИСТЕМЫ С НЕСКОЛЬКИМИ ЗАКРЫТЫМИ СМЕСИТЕЛЯМИ ПОСТОЯННОГО ОБЪЕМА
Схема установки показана на рис. 4.10. Элюирующий раствор поступает из резервуара в два смесителя постоянного объема, заполненных разбавителем. При этом образуются градиенты, которые зависят от соотношения объемов смесителей и имеют, как правило, менее выпуклую форму, чем в> системах с одним смесителем [26].
При условии неравенства объемов обоих смесителей для концентрации элюирующего раствора, поступающего в колонку, действительно следующее уравнение:
ск = сг - (сг - с01) ' е-^о, -
К01 “ К02
- ~ с“,Кт - <J •
L к01 “ к02 J
(2.66)
И01 — объем первого смесителя, И02 — объем второго смесителя, с01 — исходная концентрация элюента в первом смесителе, с02 — исходная концентрация элюента во втором смесителе. Другие обозначения те же, что и в уравнении (2.65).
Если объемы обоих смесителей равны (И01 = И02), ск выражается следующим уравнением:
ск - - (cz - с01) • • е v/v02 - (cz - с02) • e-v/(/02 (2,67)
К02
Символы идентичны символам, использованным в уравнениях (2.65) и (2.66).
Градиенты этого типа зависят от соотношения объемов сосудов В] и В2 и от исходной концентрации элюента в них. По форме градиенты менее
64
Часть 1
выпуклы, чем в системах с одним смесителем; могут образовываться также линейные и S-образные градиенты.
СИСТЕМЫ С ОТКРЫТЫМ СМЕСИТЕЛЕМ
Это наиболее распространенный метод получения градиента. Экспериментальная установка состоит из двух цилиндров, расположенных в форме сообщающихся сосудов (см. рис. 4.11). Соотношение поступающего и вытекающего потоков варьируют изменением поперечного сечения обоих цилиндров. Дрейк [26] вывел следующее уравнение для ск:
/ V \И0|/И02
ск = CZ ” - с02) ( 1 - у---) <2-68)
\ ' 0, обш /
Символы те же, что и в предыдущих уравнениях, И01 — объем резервуара, И02 — объем смесителя, И0>общ = И01 + И02.
При условии равенства диаметров цилиндров (И, = И2) уравнение (2.68) принимает вид
ск = cz~ (cz - с02^ - и/и0,общ) = с02 + <cz - с02)и/и0,обш <2’69)
Полученный градиент имеет линейную форму; наклон хвостовой части градиента повышается с увеличением диаметра цилиндрического смесителя.
2.4.2. Градиент pH
Проблему градиента pH активно исследуют с середины 50-х гг. Пиз [28] вывел ряд уравнений для описания градиента pH, получаемого в аппаратуре, состоящей из резервуара и смесителя.
Основное уравнение имеет вид
- f2
(2.70)
и' — объем исходного раствора, остающегося в смесителе, и0 — исходный объем раствора в смесителе. Другие символы те же, что и в уравнениях (2.67) и (2.71).
Градиент pH может быть получен тремя способами:
а) последовательным смешиванием двух буферных растворов;
б) смешиванием слабой кислоты (или основания) с буферным раствором;
в) смешиванием сильной кислоты (или основания) с буферным раствором.
Градиент, образованный смешиванием двух буферных растворов. Резервуар и смеситель заполняют буферными растворами, содержащими ела-
2. Теория
65
бую одноосновную кислоту и сопряженное с ней основание в разных соотношениях. Изменение pH зависит от концентрации буферных растворов и скоростей потоков и выражается уравнением
Г / V \х + 1/х 1
1 + I (1 + *— 1 - 1 ЛА-
ДрН = pH - рН0 = 1g-------L3------—Й---------------- <2'71>
1+K1 + *J _,гнА
где Яд = A~/Aq и Яна = НАг/НА0, индекс z относится к концентрации в резервуаре, индекс нуль — к первоначальной концентрации в смесителе, X = (j\ - /2)//г (/1 ~~ скорость поступления раствора в смеситель, /2 — скорость раствора, вытекающего из смесителя), v0 — первоначальный объем раствора в смесителе, V — вытекающий объем.
Соотношение концентраций соли и кислоты в буферном растворе ограничено областью 0,1 — 10. Для получения максимального изменения pH отношение А“/НАг должно быть равно 10, а отношение Aq/HA0 —0,1. Для буферных растворов одной и той же молярности действительно уравнение
ДрН = pH - рН0 = 1g
< i/\х + \/х
1 + X — )
< ”о/
' i/\A- + \/Х
1 + А'—)
ч "о/
1 0,1
(2.72)
При условии что смешиваются буферные растворы разной молярности (и если А£/НА2 = 10, А0/НА0 = 0,10,/j//^ = 1/4), получаем
ДрН = pH - рН0 - 1g
(2.73)
где Ям = А“ + HAz/Aq + НА0.
Приведенные уравнения имеют силу, если pH буферного раствора в резервуаре выше, чем в смесителе. В противном случае изменение pH происходит в обратном направлении.
Градиент, образованный смешиванием слабой кислоты (основания) с буферным раствором. Если в резервуаре находится слабое основание (в форме соли слабой кислоты), а в смесителе — буферный раствор, то
С Г / V \х + 1/х 1
ДрН = pH - рН0 = 1g ^1 + Hl + X --) - 1 1яА-j (2.74)
При условии что скорости вытекающих растворов равны (/^ = /2)> т. е. величина X равна нулю, уравнение (2.74) принимает вид
।
ДрН = pH - рН0 = lg[l + ev,vo - 1)ЯА-] (2.75)
5-354
66
Часть I
Если член /?А- равен 1, то соотношение линейно; если величина ЛА- не равна 1, градиент линейный только на небольшом отрезке.
При /?А- = 1, X Ф 1 и различных скоростях потоков уравнение (2.75) преобразуется:
ДрН = pH — рН0 =
V
1 + X — vo Когда J\/f2 > 1> образуется выпуклый градиент, при j\/f2 < 1 градиент вогнутый.
Если резервуар содержит раствор слабой кислоты вместо слабого основания, то в процессе образования градиента происходит уменьшение величины pH, градиент имеет противоположную форму и определяющим фактором является соотношение концентраций кислоты.
(2.76)
Градиент, образованный смешиванием сильной кислоты или сильного основания с буферным раствором. Если в резервуаре находится раствор сильного основания с высокой концентрацией ОН- ионов, а в смесителе — буферный раствор, полученный из слабой одноосновной кислоты и его соли, то градиент увеличивается в соответствии с уравнением
ДрН = pH — рН0 =
(2.77)
При j\ = f2 уравнение упрощается и для Ао /НА0 = 0,1 действительно следующее соотношение:
1 + (ev/vo - 1)22-
ДрН = pH - рН0 = 1g---------------------(2.78)
1 _ {е^0 - 1)22_
1ОАо-
Если резервуар содержит раствор сильной кислоты, то величины pH уменьшаются. Определяющим фактором является концентрация кислоты в резервуаре.
Приведенные уравнения действительны только в области небольших изменений pH. Для одновременного получения линейного градиента pH и градиента концентрации (практически во всем интервале pH) применяют так называемый последовательный градиентный метод элюирования [29, 30]. При использовании градиентного элюирования важно знать положение пика на хроматографической кривой. Шваб и др.[31] вывели уравнения, применимые к ионообменникам. Математические выражения, описывающие положение и ширину зон веществ, разделяемых хроматографическим методом, предложены Фрайлигом [32].
2. Теория
67
Градиентное элюирование имеет важное значение в хроматографии высокого разрешения. В ранних исследованиях основное внимание уделялось расчету удерживаемых объемов индивидуальных компонентов и меньшее внимание — влиянию градиента на расширение пика. Однако в хроматографии высокого разрешения расширение пиков имеет первостепенное значение. С целью получения информации о влиянии различных факторов на градиентное элюирование (без проведения трудоемких экспериментов) разработаны математические модели с использованием компьютеров. Эти модели дают возможность изучать действие градиента в условиях, когда другие параметры остаются неизменными [33].
2.4.3. Градиент температуры
Градиент температуры гораздо менее важен, чем градиенты концентрации и pH. Тем не менее представляет интерес дать краткое описание этого типа градиента.
Соответствующая установка для получения градиента температуры вдоль колонки описана, например, Литеану и др. [34].
Результаты многочисленных экспериментов показали, что градиент температуры оказывает заметное влияние на величину высоты теоретической тарелки (ВЭТТ), коэффициент селективности и форму хроматографической кривой.
Хорошо известно [35], что разрешение при данной длине ионообменной колонки является функцией высоты тарелки (Н) и фактора разделения (а). ВЭТТ выражается формулой
„ Dv 0,142г2и Dy 0,266г2и
И = 1,64г + ---------v-—, ---------+-------------------------------- (2.79)
(Dv + £/)2 Dr (Dy + £,)2АТ>5(1 + 70ги)
где г — радиус зерен смолы, Dy — коэффициент распределения компонента в обменнике, и — скорость потока элюента, Dr и Ds — коэффициенты диффузии в фазе смолы и раствора соответственно, £, — свободное пространство колонки, X — часть объема колонки, заполненная обменником (X =1 - £i).
При наличии продольной диффузии в обеих фазах в уравнение (2.79) вводятся два дополнительных члена [36, 37].
Как следует из уравнения (2.79), влияние температуры на ВЭТТ осуществляется посредством Dy и изменением величин Dr и Ds.
Изменение коэффициента диффузии, согласно Гилдингу [38], описывается уравнением
D = Doexp(-E/ffT) (2.80)
где Dq — коэффициент, Е — энергия активности, R — универсальная газовая постоянная, Т — температура в кельвинах.
Зависимость коэффициента распределения от температуры по Майерсу и Бойду [39] выражается формулой
68
Часть I
In D = In + In ХАВ + Д (KAR - KBR) (2.81)
>A T^BR RT
где 7a, 7b — коэффициенты активности иоиов А и В соответствеиио, 7AR, 7br — коэффициенты активности соединений AR и BR (смола) соответствеиио, PAR, PBR — парциальные мольные объемы соединений AR и BR соответственно, R и Т имеют те же значения, что и в уравнении (2.80).
Сравнение уравнений (2.80) и (2.81) с уравнением (2.79) показывает, что экспоненциальное увеличение коэффициента диффузии с повышением температуры способствует уменьшению величины ВЭТТ. Коэффициент распределения Dg ие оказывает существенного влияния на величину ВЭТТ. В некоторых случаях высота теоретической тарелки увеличивается с повышением температуры.
На основании изложенного сделан вывод, что повышение температуры в процессе разделения положительно влияет ие только на форму хроматографической кривой, ио и на весь процесс разделения. Повышение температуры, как правило, приводит к улучшению разрешения. Аналогичная картина наблюдается для зависимости ВЭТТ и формы хроматографической кривой от градиента температуры (вдоль колонки). Показано более сильное влияние на форму хроматографической кривой градиента температуры по сравнению с одноступенчатым изменением температуры вследствие непрерывного изменения скорости ионного обмена вдоль колонки. Хроматографические кривые более симметричны, хвостовая часть значительно подавлена (при условии выбора подходящего градиента), разрешение улучшается и время, необходимое для разделения, уменьшается.
Для оптимизации методик разделения с использованием градиентного элюирования целесообразно сочетать градиент температуры с градиентом другого типа.
Литература
1. Gaines G. L., Thomas Н. С., J. Phys. Chem., 21 (1953) 714.
2. Argersinger W. J., Davidson A. W., Bonner О. D., Trans. Kansas Acad.
Sci., 53 (1950) 404.
3. Cruickhank E. H., Meares P., Trans. Faraday Soc., 49 (1953) 968.
4. Cruickhank E. H., Meares P., Trans. Faraday Soc., 53 (1957) 1289, 1299.
5. Cruickhank E. H., Meares P., Trans. Faraday Soc., 54 (1958) 174.
6. Baumann E. W., Argensinger W. J., J. Amer. Chem. Soc., 78 (1956) 1130.
7. Helff erich F., lonenaustauscher, Band 1. Verlag Chemie Gmbh, Weinheim (1959)..
8. Tompkins E. R., Mayer S.W., J. Amer. Chem. Soc., 69 (1947) 2859.
9. Bonner O. D., Smith L. L„ J. Phys. Chem., 61 (1957) 326; 62 (1958) 250;
57 (1955) 719.
10. Duolite Ion Exchange Manual., Diamond Alkali Company (1960).
11. Peterson S., Ann. N. Y. Acad. Sci., 57 (1954) 144.
12. Martin A. J. P., Synge R. L. M., Biochem. J., 35 (1941) 1358.
2. Теория
69
13. Mayer S. W., Tompkins E. R., J. Amer. Chem. Soc., 69 (1947) 2866.
14. Glueckauf E„ Trans. Faraday Soc., 51 (1955) 34.
15. Glueckauf E., Ion Exchange and its Applications, Soc. Chem. Ind., London, 1954, p. 34.
16. cm. Tompkins E. R„ J. Chem. Educ., 27 (1949) 32, 92.
17. Giddings J. G., Anal. Chem., 39 (1967) 1027.
18. Said A. S., Separ. Sci. Technol., 13 (1978) 647.
19. Kraus K. A., Moore G. E., J. Amer. Chem. Soc., 75 (1955) 1460.
20. Jentzsch D., Pawlik L, Z. anal. Chem., 146 (1955) 880.
21. Snyder L. R., Chromatographic Reviews, 7 (1965) 1.
22. Mikes O., Chem. listy, 54 (1960) 577.
, 23. Liteanu C„ Gocan S., Pure Appl. Chem., 31 (1972) 455.
24. Alm R. S., Williams R. J. P., Tiselius A., Acta Chem. Scand., 6 (1952) 826.
25. Cherkin A., Martinez F. E., Dunn M. S., J. Amer. Chem. Soc., 75 (1953) 1244.
26. Drake B., Arkiv Kemi, 8 (1955) 1.
27. Bock R. M., Nan-Sing-Ling, Anal. Chem., 26 (1954) 1543..
28. PiezK. A., Anal. Chem., 28 (1956) 1451.
29. Mikes O., Tomasek V., Holesovsky K, Chem. listy, 53 (1959) 609.
30. Liebl V., Mikes O., Coll. Czech. Chem. Commun., 24 (1959) 809.
31. Schwab H., Rieman W„ Vaughan P. A., Anal. Chem., 29 (1957) 1357.
32. Freilig C. F., J. Amer. Chem. Soc., 77 (1955) 2067.
33. Pitt W. W„ Jr., J. Chromatogr. Sci., 14 (1976) 396.
34. Liteanu C., Gocan S., Hodisan T., NascuH., Marutoiu C., Rev. Roumai-ne Chim., 17 (1972) 497.
' 35. Dybczynski R., J. Chromatogr., 14 (1964) 79; 31 (1967) 155.
36. Dybczynski R., J. Chromatogr., 50 (1970) 487.
37. Hamilton P. B., Bogue D. C., Anderson R. A., Anal. Chem., 32 (1960) 1782.
38. Giddings J., Dynamics of Chromatography, Parti, in : Chromatography Sciences Series, Vol. 1, Marcel Dekker, N. Y., 1965, p. 232.
39. Myers G. E., Boyd G. E., J. Phys. Chem., 60 (1956) 521.
3. Физические, физико-химические и химические характеристики ионообменных смол и их определение
В настоящее время освоен выпуск ионообменных смол, удовлетворяющих основным требованиям в отношении ионообменных свойств, гомогенности, размера зерен, чистоты, механической, химической и радиационной устойчивости. Тем не менее некоторые операции по модификации смол перед их использованием по-прежнему необходимы. Главная цель этих операций — превращение смолы в нужную форму и требуемое физическое состояние.
3.1. Регулирование размера зерен, сортировка и очистка ионообменных смол
Органические типы ионообменных смол получают главным образом в форме сферических зерен различного размера. В лабораторных условиях обычно применяют смолы с величиной зерен 0,04 — 1,2 мм. Изготовители сортируют смолы по размеру гранул и степени чистоты.
Ионообменные смолы на основе целлюлозы поставляют в форме порошков, волокон, хлопьев или шариков разного размера.
Неорганические ионообменники получают в виде аморфных, микрокристаллических или кристаллических осадков разной степени дисперсности.
В некоторых случаях (особенно при хроматографическом разделении смесей) требуются смолы с очень мелкими частицами, размер которых изменяется в узком диапазоне. При отсутствии смол необходимого зернения смолы предварительно измельчают. Если имеют дело с органическими ио-нообменниками в форме сфер, этой механической обработки, невозможности, избегают. Сферическая форма неповрежденных частиц по сравнению с неправильной формой зерен обеспечивает более низкое гидравлическое сопротивление колонки (даже при очень тонком зернении). Кроме того, во время размалывания всегда образуется большое количество очень мелких частиц, которые являются отходами.
3.I.I. Размалывание и дробление частиц смолы
Если смола достаточно хрупкая, то она легко размалывается. Небольшие количества смолы доводят до частиц требуемого размера в агатовой
3. Физические, фишко-химические и химические характеристики
71
ступке или в ручной механической мельнице. Большие количества смолы в сухом или набухшем состоянии измельчают в шаровой мельнице. Следует отметить, что в металлических мельницах можно размалывать катионооб-менники только в Na-форме, а анионообменники — в Cl-форме. Измельчение катионообменников в Н-форме приводит к повышенному износу металлической поверхности мельницы и загрязнению смолы металлами.
В процессе измельчения часто образуется большое количество очень мелких бесполезных отходов. Поэтому при повторном измельчении смолы необходимо предварительно отделять частицы требуемого размера на соответствующих ситах.
3.1.2. Сортировка частиц смолы
Частицы смолы сортируют в соответствии с их размером сухим или мокрым просеиванием или фракционированием гидравлическим методом.
Сухое просеивание. Воздушно-сухую смолу просеивают через ряд стандартных сит вручную или с помощью механического вибратора. Фракцию смолы с требуемым размером частиц перед дальнейшим использованием суспендируют и несколько раз декантируют водой. Таким путем из смолы удаляют прилипшие частицы порошка.
Мокрое просеивание. Набухшую в воде смолу просеивают струей дистиллированной воды через ряд стандартных сит.'Другой метод основан на погружении сита в достаточно большой сосуд, заполненный водой. Сито медленно покачивают и опускают так, чтобы более мелкие частицы смолы проходили через отверстия сита. Этот метод не рекомендуется использовать для просеивания катионообменников в Н-форме во избежание загрязнения смол металлами (в результате взаимодействия смолы с материалом сита). Анионообменники, содержащие аминные группы, также сильно разъедают бронзовые сита.
Для фракционирования частиц смолы вместо просеивания может быть использован также метод седиментации [1, 2].
Перечисленные выше методы фракционирования частиц смолы не обеспечивают их хорошей сортировки. Для этой цели наиболее пригоден метод гидравлической седиментации в проточной установке. Частицы смолы различного размера при седиментации в противотоке жидкости распределяются в соответствии с их гидравлическими свойствами. Диапазон размера частиц смолы в отдельной фракции [3 — 8] определяется величиной линейной скорости потока жидкости в установке.
Скорость частиц Vr в направлении потока жидкости через фракционную установку равна разности между линейной скоростью потока Vp и скоростью седиментации Для задерживания частиц в определенном фракционном сосуде необходимо выполнение условия, при котором Vr = 0. Если линейная скорость потока выражается отношением объема жидкости (Ио), протекающего через систему в единицу времени, к площади поперечного сечения соответствующей седиментационной трубки и если скорость седиментации подчиняется закону Стокса, то
72
Часть 11
Рис. 3.1 А. Схема седиментационной установки для непрерывного определения распределения частиц смолы по размеру [6].
а, а' — бутыли Мариотта; Ь — пробка; с — бюретка; d — стекловата; е — регулирующий кран; f — реометр; g — делительная воронка (2000 см3); й — слив в отстойник; , j2 — отстойники, содержащие смолу; — бутыль для перекачки воды из отстойника обратно в бутыли Мариотта сжатым воздухом (трубопровод /).
Vo 2 г •$($,.-5,)
Vr = -Д---------—-------— = 0, (3.1)
r TR2
где Уг — скорость перемещения частиц (см/с), Уо — объемная скорость потока жидкости (см3/с),Л — диаметр фракционной трубки (см), g — постоянная силы тяжести (см/с), г — диаметр седиментированной частицы (см), SI — удельная масса жидкости (г/см3), Sr — удельная масса смолы (г/см3), Ч — динамическая вязкость жидкости [г/(см • с)].
Изменение линейной скорости потока достигается использованием фракционных трубок (сосудов) различного поперечного сечения при постоянной скорости потока жидкости через систему или изменением объемной скорости потока жидкости при постоянном поперечном сечении фракционных трубок.
3. Физические, физико-химические и химические характеристики
73
Рис. 3.1 Б. Схема проточной гидравлической установки для фракционирования частиц иоиообменника [7].
Г, — T'g — фракционные трубки; В — колба Эрленмейера (200 см3); Е — электромагнитная мешалка; М(, М2 — бутыли Мариотта; Kt — Kg — двухходовые краны; Н\ — резиновая трубка (внутренний диаметр 5 мм); Hi — пластмассовая трубка (внутренний диаметр 2 мм); F — пористая стеклянная пластина; L — капилляр; Z — резервуар для регенерированной жидкости; D — подача сжатого воздуха; V — шланг к вакуумному насосу.
В качестве разделяющей среды чаще всего используют дистиллированную или деионизованную воду. Если во время фракционировании происходит агрегация частиц, целесообразно воду заменять разбавленной соляной кислотой (0,001 — 0,5 моль/л). Для обеспечения постоянной величины коэффициента динамической вязкости жидкости в системе установку термоста-тируют.
Размер частиц фракций определяют под микроскопом.
Этот метод наиболее приемлем при фракционировании сферических частиц смол. Для частиц любой другой формы эффективность фракционирования ниже.
Таблица 3.1. Фракционирование смолы Dowex 50-Х12 (< 0,037 мм) гидравлическим методом3 [8]
Номер фракционной трубки 2 3 4 5 6
Размер частиц, 10—25 25—35 мкм а Для скорости потока жидкости 30 см3/мин. 35—45 45—55 55—75
74
Часть II
Схемы двух типов установок с различными принципами изменения линейной скорости потока жидкости представлены на рис. 3.1. В табл. 3.1 приведены данные по распределению смолы Dowex 50 (частицы <0,038 мм) в шестиступенчатой установке; частицы указанного размера составляют 90% каждой фракции [8].
3.1.3. Очистка ионообменников
Органические высокомолекулярные ионообменники выпускаются промышленностью главным образом во -влажном состоянии в запаянных полиэтиленовых мешках или бутылях. Неорганические ионообменники и смолы на основе целлюлозы выпускают, как правило, в сухом виде в упаковке, которая предотвращает загрязнение ионообменников при транспортировке и хранении.
Если органические высокомолекулярные ионообменники не соответствуют требуемой степени чистоты, то они должны быть предварительно очищены. Наиболее распространенными примесями являются растворимые в воде низкомолекулярные вещества, железо (III) и другие ноны, вводимые при синтезе. Процесс очистки в некоторой степени облегчает доступ ионов к функциональным группам смолы, и в то же время ионообменник проходит необходимую «тренировку».
МЕТОДЫ ОЧИСТКИ ОРГАНИЧЕСКИХ ИОНООБМЕННИКОВ
Метод А. Смолу помещают в стакан'соответствующей емкости, заполненный приблизительно на 1/3 объема дистиллированной водой. Воду прибавляют в таком количестве, чтобы ее уровень был по крайней мере на 10 см выше уровня смолы. Смолу оставляют для набухания в течение нескольких часов, перемешивают и после седиментации сливают воду декантацией. Эту операцию повторяют несколько раз. Последняя промывная вода должна быть прозрачной и бесцветной. Затем к смоле прибавляют раствор 3 — 4 М соляной илн серной кислоты (смола не должна содержать ионов, образующих нерастворимые соединения в этих растворах). Общая концентрация ионов Н+ (катионообменникн) нли соответствующих анионов (анионооб-менннки) должна превышать полную обменную емкость смолы приблизительно на 25%. Суспензию смолы оставляют в кислом растворе на 1 — 2 ч при периодическом помешивании. Кислоту декантируют и промывают смолу дистиллированной водой до нейтральной реакции промывных вод.
Очищенную смолу превращают в №(ОН)-форму обработкой 1 — 2М раствором гидроксида натрия в течение 1 — 2 ч. Затем смолу тщательно промывают водой и превращают в Н(С1 или SO^)-форму описанным выше методом. Весь цикл очистки повторяют 2 — 3 раза.
Если в растворе гидроксида натрия наблюдается заметное разрушение смолы, то вместо него можно использовать раствор ацетата натрия, который имеет более низкое значение pH и не растворяет смолу.
Метод Б, Суспензию полностью набухшей в воде смолы помещают в широкую стеклянную колонку с пористым стеклянным дном и сливным краном. После седиментации смолы в колонке устанавливают уровень воды на 5 — 10 см выше уровня смолы и медленно промывают обменник дистиллированной водой до тех пор, пока
3. Физические, физико-химические и химические характеристики
75
вытекающая вода не станет прозрачной и бесцветной. На этой стадии очистки предпочтительнее промывать колонку снизу.
Для превращения обменника в Н-форму последний обрабатывают приблизительно трехкратным (по объему) количеством 3 М соляной кислоты. Азотную кислоту не рекомендуется использовать, так как большинство органических ионообменников разрушается азотной кислотой умеренной концентрации при повышенной температуре. Скорость потока выбирают таким способом, чтобы указанное выше количество жидкости протекало за 30—40 мин. Затем обменник промывают водой (15 — 20 объемов обменника) и превращают в Иа(ОН)-форму обработкой 1,5 М раствором гидроксида натрия.
Полученный таким образом ионообменннк в №(ОН)-форме тщательно промывают водой до нейтральной реакции промывных вод; для слабокислотных или слабоосновных ионообменников промывание прекращают прн pH промывной воды, равном 8. Весь цикл очистки повторяют 2 — 3 раза. У слабосшитых смол наблюдается значительное изменение объема смолы вследствие сильного набухания в щелочных растворах. В некоторых случаях происходит разрыв стеклянной колонки. Поэтому после первой стадии цикла рекомендуется переносить обменник в стакан и заливать раствором гидроксида натрия. По достижении равновесия обменник снова переносят в колонку и промывают 2 — 3 объемами раствора гидроксида натрия. Если происходит сильное разрушение обменника в щелочной среде, используют раствор ацетата натрия, как описано в методе А.
Приведенные выше методики очистки ионообменников, как правило, являются удовлетворительными. Более высокая степень очистки достигается промыванием обменника (после полного цикла) горячей водой в течение 30 мин (Н-формы катионообменников при 90 — 95°С, Cl-формы анионо-обменников (при 60 — 80°С). Затем обменник промывают небольшим количеством холодной деионизованной воды и метанолом, этанолом или ацетоном до получения прозрачной и бесцветной промывной жидкости. После этого ионообменннк превращают в необходимую ионную форму. Эта методика может быть использована при условии, что ионообменннк устойчив в указанных растворителях при заданной температуре. Большинство ионообменников на основе сополимеров стирола с ДВБ устойчиво в этих условиях.
В специальных случаях некоторые авторы рекомендуют выполнять заключительную стадию очистки ионообменников с использованием растворов динатриевой соли этилендиаминтетрауксусной кислоты, 0,1 М лимонной кислоты или комплексообразующих и хелатообразующих веществ.
ОЧИСТКА ИОНООБМЕННИКОВ РАСТВОРОМ ЭДТА [9]
Методика. Ионообменннк (сильнокислотный стирол-дивинилбензольный катио-нообменник) обрабатывают 1 М раствором гидроксида натрия в течение 60 мин на кипящей водяной бане. Щелочной раствор декантируют, прибавляют 2 — 3 объема (относительно ионообменника) 0,2 М раствора динатриевой соли ЭДТА, устанавливают pH раствора 8 — 10 и перемешивают суспензию на кипящей водяной бане в течение 15 мин. По мере того как обменник желтеет или синеет, раствор ЭДТА заменяют и процесс повторяют. Затем ионообменннк тщательно промывают деионизованной водой и кипятят с 5 М раствором соляной кислоты на водяной бане в тече-
76
Часть II
иие ~ 60 мии. На этой стадии соляную кислоту меняют 3 — 4 раза. Окончательное промывание ионообменника выполняют'деионизованной водой.
Ионы железа (III) целесообразно удалять из обмеиннка раствором тиоцианата. Следы кремневой кислоты удаляют обработкой обменника 1 М раствором фтористоводородной кислоты в течение 24 ч; перед промыванием водой ионообменник оставляют в насыщенном растворе борной кислоты.
Сильнокислотные стирол-дивннилбензольные катионообменники иногда имеют интенсивную коричневую окраску, которую согласно Инцеди, можно ослабить или устранить превращением ионообменника в Т1(Ш)-форму с помощью раствора соли титана (III). После такой обработки обменник смешивают с раствором гидроксида аммония при 40°С и затем промывают 4 М соляной кислотой н водой.
Однако обменная емкость катионообменников при таком способе очистки немного уменьшается.
3.1.4. Превращение ионообменников в стандартное состояние и другие ионные формы
Стеклянную колонку подходящего размера заполняют очищенным и полностью набухшим органическим ионообменником, который промывают водой и превращают в стандартное состояние обработкой соответствующими растворами пб следующим методикам:
Сильнокислотные катионообменники в Н-форме получают обработкой обменников 5 — 8 объемами (относительно обменника) 7%-иой соляной кислоты. Затем обменник промывают деиоиизоваииой водой до исчезновения кислой реакции (метиловый красный) промывной воды.
Для перевода слабокислотиых катионообменников в Н-форму последние обрабатывают 6 — 8 объемами 2%-ной соляной кислоты и промывают деионизованной водой до исчезновения кислой реакции.
Сильноосновные анионообменники в С1-форме получают обработкой 10 объемами 7%-ной НО и промыванием деионизованной водой до отрицательной реакции на ионы О-.
Для превращения слабоосиовных анионообменников в форму свободного основания используют 10 объемов 4%-ного раствора NaOH. Ионообменник промывают водой до отрицательной реакции, по фенолфталеину.
Скорость пропускания кислых или щелочных растворов выбирают с таким расчетом, чтобы удельная загрузка колонки соответствовала S = 5 (т.е. 0,10 — 0,15 объема раствора на 1 объем обменника в 1 мии). Для промывании водой выбирают удельную загрузку колонки S, равную 10.
При необходимости превращения ионообменника в другую ионную форму обычно исходят из стандартного состояния. Однако Na-форма катионо-обмеииика и ОН-форма анионообмеиника часто более предпочтительны. Для полного превращения ионообменников в требуемую ионную форму применяют 2,5 — 3 - кратный избыток соответствующего вещества (по сравнению с теоретическим); в отдельных случаях необходимо регулирование pH раствора.
В процессе промывания водой некоторые формы слабокислотных или слабоосиовных ионообменников гидролизуются. Для предотвращения гидролиза используют минимальное количество воды или этанол.
3. Физические, физико-химические и химические характеристики
77
3.2. Физические свойства ионообменников
3.2.1. Определение размера частиц и фракционного состава ионообменников методом ситового
анализа
Для определения размера частиц и фракционного состава ионообменников применяют метод ситового анализа. Образцы анализируемых обменников должны быть в стандартном состоянии (Na-форма катионообменников) в воздушно-сухом или набухшем виде. Удобнее использовать полностью набухшие в воде образцы.
Если анализируемые ионообменники находятся не в стандартном состоянии, а в других ионных формах, то возможны небольшие отклонения в распределении частиц по размеру.
Методика А. Приблизительно 100 г воздушно-сухого ионообменника в стандартном состоянии (сильнокислотные ионообменники в Na-форме — исключение!) просеивают механически или вручную через набор стандартных снт в течение 30 мни. Фракции ионообменников, задержанные каждым ситом, количественно переносят в соответствующий сосуд и взвешивают с точностью до третьего десятичного знака. На основании полученных данных строят гранулометрическую кривую (интегральную или дифференциальную). Примечание: к анализируемой пробе полезно прибавлять приблизительно 50 мг антистатика.
Методика'Б. 150 — 200 см3 полностью набухшей смолы в стандартном состоянии (сильнокислотные катионообменники в Na-форме — исключение!) последовательно просеивают через набор стандартных сит. Грубое рассеивание выполняют струей воды. Окончательного тонкого разделения достигают ручным просеиванием отдельных фракций в соответствующих сосудах нз стекла, фарфора или других нержавеющих материалов. Сита поочередно поднимают и погружают в сосуд на 5 мин. Фракции ионообменника, задержанные каждым ситом, количественно перено-
Таблица 3.2. Распределение дерен обменника по размеру3
Размер отверстия сита, -мм Масса сухого обменника, г Количество удерживаемых частиц, % Общее количество частиц, %
1,19 0,00 0,00 0,00
0,84 10,38 18,88 18,88
0,59 22,08 40,04 58,92
0,42 13,00 23,65 82,57
0,297 7,08 12,88 95,45
0,25 2,50 4,55 100,00
— — —
55,04 100,00 100,00
a D = 0,36 мм; К = 1,92. эфф одн ’
78
Часть 11
Рис. 3.2. График ситового анализа (суммарная кривая распределения).
1 — величина, определяющая эффективный размер частиц; 2 — величина, необходимая для расчета коэффициента однородности.
сят в калиброванные мерные цилиндры и измеряют объем обменника после стабилизации его легким постукиванием дном цилиндра по твердой резиновой поверхности.
Распределение частиц анализируемой смолы по массе можно установить высушиванием индивидуальных фракций до постоянной массы.
Графическое представление полученных результатов дает кривую распределения. Примеры приведены в табл. 3.2 и на рис. 3.2.
По кривой распределения рассчитывают эффективный размер зерна (£>эфф) и коэффициент однородности (Коди). Первый определяется размером отверстия сита, задерживающего 90% частиц анализируемого ионообменника:
^эфф = ^90% (3-2)
Коэффициент однородности рассчитывают как отношение размеров отверстий сита, удерживающих 40 и 90% смолы соответственно (из кривой распределения):
„ _ d4on
^ОДН . (3.3)
“90%
Для ионных смол в форме сфер определяют сферичность и количество дефектных сфер.
ОПРЕДЕЛЕНИЕ СФЕРИЧНОСТИ
Методика. 10 г воздущно-сухой смолы высыпают с постоянной скоростью на металлический лист, наклоненный к горизонтали под углом в 10° Сферы при этом свободно перемещаются по листу вниз, не мешая друг другу. Затем определяют массу зерен, скатившихся с металлического листа.
Оценка. Количество неповрежденных сфер в процентах получают умножением массы скатившихся сфер на 10.
3. Физические, физико-химические и химические характеристики
79
ОПРЕДЕЛЕНИЕ КОЛИЧЕСТВА ПОВРЕЖДЕННЫХ СФЕР (ГРАНУЛ)
Методика. С помощью микроскопа или (лучше) микрофотографии определяют следующие параметры:
а) количество неповрежденных сфер,
б) количество сфер с трещинами на поверхности,
в) количество частиц, образованных дроблением сфер.
В процессе измерения оценивается около 300 частиц. Полученный результат выражается соотношением «б»/«в», отнесенным к 100 неповрежденным сферам.
Часто используют другие методы определения размера частиц (седиментация, фотографический метод).
3.2.2. Определение удельной массы
Удельную массу сухой и набухшей смолы определяют пикнометрическим методом.
ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ МАССЫ СУХОЙ СМОЛЫ
Методика. 1 — 5 г обменника в соответствующей ионной форме сушат до постоянной массы, взвешивают на аналитических весах и переносят в пикнометр. Сушить ионообменники рекомендуется в эксикаторе (над Р2О5) при пониженном давлении и температуре 60 — 80°С. Пикнометр заполняют до половины объема соответствующей жидкостью (и-октан или и-гептан) и оставляют приблизительно на 3 ч при периодическом покачивании. Пузырьки воздуха, прилипшие к зернам, при необходимости удаляют в вакуумном эксикаторе.
Затем доводят уровень жидкости в пикнометре до метки, пикнометр термостати-руют при определенной температуре и взвешивают. Удельную массу обменника рассчитывают по формуле
где 5 — плотность использованной жидкости при данной температуре, т — масса обменника (г), М { — масса пикнометра с жидкостью (г), М г — масса пикнометра с обменником и жидкостью (г).
ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ МАССЫ НАБУХШЕЙ СМОЛЫ
Методика А. 20 — 50 см3 набухшей смолы в соответствующей ионной форме помещают в воронку Бюхнера, накрывают влажным фильтром (для предотвращения потери жидкости в процессе фильтрования) и отсасывают с помощью водоструйного насоса в течение 5 мин или центрифугируют при G = 900. (Оценку величины G см. на стр. 84.) Затем отбирают 5 — 10 см3 образца, быстро и тщательно сушат между листами фильтровальной бумаги под небольшим давлением, переносят образец в предварительно взвешенный градуированный пикнометр и определяют массу образца (/и). После этого пикнометр частично заполняют кипяченой деионизованной водой, удаляют пузырьки воздуха, прилипшие к зернам обменника, в вакуумном эксикаторе, доводят уровень жидкости в пикнометре до метки, термостатируют и взвешивают. Удельную массу набухшей смолы (р) рассчитывают по уравнению (3.4).
80
Часть II
Таблица 3.3. Удельная масса некоторых ионообменников типа Duolite в различных ионных формах [10]
Иоиообмеииик Иоииая форма Удельная масса, г/см3
Duolite С-3 н 1,24
Duolite С-3 Na 1,26
Duolite С-20 Н 1,30
Duolite С-20 Na 1,32
Duolite А-30 ОН 1,15
Duolite А-30 CI 1,17
Duolite А-30 so4 1,18
Duolite А-102 он 1,08
Duolite А-102 CI 1,10
Методика Б. Образец обменника обрабатывают как описано выше. I — 2 см3 набухшей смолы переносят в мерную колбу, которая содержит насыщенную водой - смесь (1 : 1) четыреххлористого углерода и бензола, прибавляют бензол (уд. масса 0,866) или четыреххлористый углерод (уд. масса 1,595) до тех пор, пока частицы обменника не начнут свободно перемещаться в жидкости. После введения каждой новой порции жидкости закрытую колбу несколько раз переворачивают для гомогенизации смеси. По достижении необходимого соотношения обеих жидкостей (частицы обменника свободно перемещаются в жидкости) измеряют плотность смеси ареометром. Полученная величина является удельной массой обменника.
Если небольшая часть образца обменника всплывает на поверхность или опускается на дно колбы, то это свидетельствует о негомогенности обменника, которая может быть таким способом оценена.
Результаты, полученные описанным методом, для ряда обменников Duolite представлены в табл. 3.3.
ОПРЕДЕЛЕНИЕ КАЖУЩЕЙСЯ УДЕЛЬНОЙ МАССЫ СУХОЙ СМОЛЫ
Этот метод наиболее приемлем для макросетчатых обменников.
Методика. 1 — 2 г высушенного до постоянной массы обменника переносят в пикнометр, предварительно откалиброванный по ртути. Пикнометр помешают в эксикатор, снабженный устройством для подачи ртути в пикнометр. Эксикатор откачивают масляным диффузионным вакуумным насосом в течение 2 ч. Затем подают ртуть в пикнометр. Операцию повторяют до тех пор, пока давление в эксикаторе не будет равным внутреннему давлению, и ртуть перестанет поступать в пикнометр. После заполнения ртутью и термостатирования пикнометра систему взвешивают. Кажущуюся удельную массу рассчитывают, пользуясь уравнениями (3.4).
3. Физические, физико-химические и химические характеристики 81
ОПРЕДЕЛЕНИЕ КАЖУЩЕЙСЯ УДЕЛЬНОЙ МАССЫ НАБУХШЕЙ СМОЛЫ
Методика. 150 г полностью набухшей смолы в определенной ионной форме помещают в градуированную ионообменную колонку длиной 1000 мм и внутренним диаметром 25 мм; к нижней части колонки припаяны пористая стеклянная пластина и кран. Слой обменника в течение небольшого времени промывают водой снизу, выдерживают 2 мин (уровень воды на 5 см выше поверхности смолы) и пропускают через колонку сверху вниз воду со скоростью 60 см3/мин в течение 15 мин. После этого измеряют объем смолы И рассчитывают кажущуюся удельную массу набухшей смолы по уравнению
Масса смолы (г)
Ркаж = ---1- (3*^)
Объем смолы (см3)
3.2.3. Определение насыпной плотности и удельного объема сухой смолы
Методика. Определенное количество доведенной до постоянной массы смолы помешают в мерный цилиндр емкостью 100 см3 и постукивают дном цилиндра по твердой поверхности до тех пор, пока объем смолы не перестанет изменяться. Затем измеряют объем смолы и записывают результат в граммах на кубический сантиметр (объемная плотность) или в кубических сантиметрах на грамм (удельный объем).
ОПРЕДЕЛЕНИЕ НАСЫПНОЙ ПЛОТНОСТИ И УДЕЛЬНОГО ОБЪЕМА НАБУХШЕЙ СМОЛЫ
Методика А. Набухшую смолу в соответствующей ионной форме отсасывают на воронке Бюхнера (см. определение удельной массы, стр. 79) и определяют ее массу. Затем смолу переносят в мерный цилиндр, наполненный водой, постукивают дном цилиндра до тех пор, пока объем не перестанет изменяться, и измеряют объем набухшей смолы.
Методика Б (стандартный метод). 150 — 200 см3 набухшей смолы в токе дистиллированной воды помещают в градуированную трубку внутреннего диаметра 24 ±1 мм с таким расчетом, чтобы смола заняла 15О°7о исходного объема. Подачу воды прекращают, дают возможность смоле опуститься и измеряют объем смолы через 20 мин после начала его уменьшения. Затем смолу центрифугируют в течение 20 мин при G = 1200 и взвешивают. Объемную плотность выражают отношением массы смолы после центрифугирования к ее постоянному объему (г/см3). Удельный объем выражают в кубических сантиметрах на грамм.
3.2.4. Определение содержания воды в смоле
Для определения этого важного параметра применяют ряд методов, которые несколько различаются по технике выполнения и по полученным результатам.
Чаще всего используют метод, основанный на высушивании смолы до постоянной массы при 105°С (катионообменники) или при 60 — 80°С (анионообменники). Сильноосновные анионообменники в ОН-форме не рекомендуется сушить вследствие их низкой термической устойчивости.
6-354
82
Часть 11
Сушить смолу удобнее всего в эксикаторе над пентаоксидом фосфора при пониженном давлении и температуре.
Метод Карла Фишера дает хорошие результаты при определении содержания воды в макросетчатых смолах на основе сополимера стирол-ДВБ; ошибки определения (±0,75%) меньше ошибок, полученных в методе высушивания смол при 100°С (±2%).
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВОДЫ В СМОЛАХ МЕТОДОМ ВЫСУШИВАНИЯ
Методика. 0,200 — 2,000 г смолы в соответствующей ионной форме при выбранных условиях сушат до постоянной массы. Рекомендуется сушить смолу в вакуумном эксикаторе при 70°С над пеитаоксидом фосфора. Для сокращения времени сушки можно применять вакуумный эксикатор с водоструйным насосом.
Содержание воды в смоле обычно выражают в процентах.
ОПРЕДЕЛЕНИЕ ВОДЫ В НАБУХШЕЙ СМОЛЕ
Методика. Приблизительно 1 — 5 г набухшей смолы в определенной ионной форме помещают в стеклянный фильтрующий тигель с пористым дном. Тигель накрывают для предотвращения испарения воды с поверхности смолы и центрифугируют в течение 30 мин при G = 900 — 1200 (см., например, определение степени набухания, стр. 84). Затем тигель со смолой взвешивают и сушат до постоянной массы при заранее выбранных условиях. Аналогичные операции выполняют с пустым тиглем.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ВОДЫ КОСВЕННЫМ МЕТОДОМ [12]
Методика. Точно взвешенное количество сухой ионообменной смолы помещают в раствор подходящего органического красителя с определенной концентрацией. Молекулы выбранного красителя должны быть достаточно большими во избежание их диффузии внутрь зерна смолы. Количество воды, содержащейся в смоле, оценивают по изменению концентрации красителя в растворе. Применение этого метода ограничено. В качестве красителя рекомендуется использовать хлоразол лазурный FF.
Содержание воды в ионообменииках может бьръ определено также другими методами: титрованием реагентом Карла Фищера, азеотропной дистилляцией (с использованием толуола, ксилола, тетрахлорэтилена, трихлорэтилена, четыреххлори-стого углерода), обработкой обменника водой, меченой тритием, ЯМР-спектроскопическим, калориметрическим, изопиестическим. Эти методы критически рассмотрены в обзоре Питржика [25].
3.2.5. Степень набухания ионообменных смол
Набухание органических ионообменников является одним из наиболее • важных свойств этих материалов, оно зависит главным образом от типа и свойств каркаса матрицы, величины обменной емкости, заряда и размера противоиона, типа связи между этим ионом и функциональными группами ионообменника, концентрации электролита в растворе и полярности растворителя.
3. Физические, физико-химические и химические характеристики
83
ОПРЕДЕЛЕНИЕ СТЕПЕНИ НАБУХАНИЯ СМОЛЫ (ПО МАССЕ) МЕТОДОМ ЦЕНТРИФУГИРОВАНИЯ
Методика. Приблизительно 1 г набухшей смолы помещают в сосуд (рис. 3.3), в основание которого впаяна стеклянная пластина определенной пористости, а в верхней части находятся два небольших отверстия. Сосуд погружают в деионизованную воду при определенной температуре (25°С) и выдерживают там приблизительно 60 мин. Затем сосуд переносят в толстостенную центрифужную пробирку (о), покры-
Рис. 3.3. Сосуд для определения степени набухания смолы (по массе) методом центрифугирования [12].
а — толстостенная центрифужная пробирка; b — толстостенная трубка с двумя отверстиями е в верхней части и с основанием из пористой стеклянной пластины с; f — резиновая пробка, предотвращающая испарение воды из смолы в процессе центрифугирования; d — образец смолы.
Таблица 3.4. Соотношение между степенью поперечного сшивания и набухаемос-тью полистирол-дивинилбензольных катионообменников3, содержащих группы -SO3H [12]
Степень поперечного сшивания X Способность к набуханию W.R. Степень поперечного сшивания X Способность к набуханию W.R.
2 3,45 14 0,63
3 2,50 15 0,59
4 1,92 16 0,57
5 1,57 17 0,55
6 1,36 18 0,52
7 1,18 19 0,50
8 1,04 20 0,48
9 0,93 21 0,46
10 0,83 22 0,44
11 0,76 23 0,42
12 0,71 24 0,40
13 0,67 25 0,38
а X — степень поперечного сшивания, т.е. содержание ДВБ в полимере (%); W.R.— удерживание воды — максимальное количество воды (г), отнесенное к 1 г сухой смолы.
84
Часть 11
тую крышкой (/), и центрифугируют смолу при G = 400 около 40 мин. Точное время центрифугирования необходимо определить в предварительном опыте. Величину G оценивают из уравнения
4ir2rN2
G = ----- (3.6)
g
где г — расстояние образца от оси центрифуги, N — число оборотов в секунду, g — ускорение, обусловленное силой тяжести.
После центрифугирования сосуд, содержащий смолу, взвешивают, погружают в воду и операцию повторяют. Затем сосуд со смолой сушат при’ определенных условиях до постоянной массы.
Для определения массы пустого сосуда выполняют все описанные выше операции, включая высушивание. На основании полученных данных оценивают массу сухой и набухшей смолы. Поправку на поглощение воды поверхностными частицами смолы (50 мг/г) вводят, вычитая эту величину из массы набухшей смолы. Исправленный таким способом показатель используют для оценки количества воды, связанной набухшей смолой.
Если это количество воды относят к I г сухой смолы, то получают параметр, обычно называемый «удерживанием воды» (W.R.) и характеризующий способность к набуханию. Этим'методом можно определить также количество сшивающего агента в сильнокислотных катионообменниках на основе сополимеров стирола и ДВБ. Данные для смол с зернением 0,1 — 0,4 мм представлены в табл. 3.4.
ОПРЕДЕЛЕНИЕ СТЕПЕНИ НАБУХАНИЯ (ПО ОБЪЕМУ)
Методика. Приблизительно 10 см3 набухшей смолы помещают в мерный цилиндр и постукивают дном цилиндра до тех пор, пока объем смолы не перестанет изменяться. Затем измеряют объем слоя смолы и сушат ее до постоянной массы. Степень набухания (по объему) выражают в кубических сантиметрах на грамм сухого обменника. Для монофункциональных стирол-дивинилбензольных сульфокатионообменников в Н-, Na- или смешанной форме ее рассчитывают по уравнению
Го6 = W.R. + 0,63 (3.7)
Ко6 — объем набухшей смолы, отнесенный к 1 г сухой смолы [13].
Помимо рассмотренных выше методик определения набухаемости смол могут быть использованы и другие методы. Чаще других применяют метод разбавления электролита и красителя, а также микроскопический метод. Последний дает достаточно точные результаты при наличии хорошего микроскопа.
Отличный обзор с соответствующими практическими методиками опубликован Фриманом [14].
3.3. Физико-химические и химические свойства ионообменников
3.3.1. Определение обменной емкости ионообменников
Для выражения обменной емкости ионообменников существует ряд терминов. Практическая удельная емкость (QA) является характерной констан
3. Физические, физико-химические и химические характеристики
85
той индивидуального ионообменника. Теоретически она не должна зависеть от метода определения, однако на практике наблюдаются небольшие различия в величинах обменной емкости, определенной разными методами.
Кроме удельной емкости часто используют термин «объемная емкость», которая выражается в миллимолях на 1 см3 набухшей смолы.
Перед определением емкости проводят цикл очистки обменника (см. стр. 74). При работе с небольшим количеством обменной смолы (5 — 10 см3) цикл очистки можно сократить: катионообменник декантируют деионизованной водой и превращают сначала в Na-форму обработкой 3%-ным раствором NaOH, а затем в Н-форму обработкой 5%-ной соляной кислотой. Для превращения анцонообменника последовательно в ОН- и Cl-формы используют 2%-ный раствор NaOH и 5%-ную соляную кислоту соответственно. Неорганические ионообменники обычно переводят в Н-форму азотной кислотой, выбор концентрации последней зависит от характера обменника. Как правило, применяют 3 — 7%-ную азотную кислоту. В Na-форму эти ионообменники обычно не превращают, так как большинство современных синтетических неорганических ионообменников неустойчиво при значениях pH больше 6 — 8.
Обменники, обработанные таким способом, могут быть рассмотрены как исходная форма при определении их обменной емкости.
Принимая во внимание неопределенности при оценке влажности смол, величину обменной емкости относят к ионообменнику, высушенному при определенных условиях; ошибка определения обменной емкости ±1%.
Выбор метода определения зависит от природы функциональной группы и скорости реакции ионного обмена.
ОПРЕДЕЛЕНИЕ ОБЩЕЙ УДЕЛЬНОЙ ОБМЕННОЙ ЕМКОСТИ КАТИОНООБМЕННИКОВ
Методика. 1,000 — 2,000 г воздушно-сухого обменника в Н-форме оставляют для набухания в деионизованной воде. Можно использовать 1 — 3 г набухшего обменника, который предварительно отсасывают на воронке Бюхнера. Одновременно взве-‘ шивают 0,300 — 1,000 г обменника для определения содержания сухого вещества.
Набухшую смолу переносят в ионообменную колонку (высотой 6 — 10 см, диаметром 1 см) и пропускают определенное количество (200 — 300 см3) 0,1 М раствора гидроксида натрия со скоростью 2 — 3 см3/мин. Элюат собирают в мерную колбу емкостью 500 см3. После пропускания всего объема раствора оставшуюся в колонке жидкость переводят в мерную колбу сжатым воздухом. Промывание обменника водой может привести к гидролизу последнего. В аликвотной части элюата определяют оставшееся количество гидроксида титрованием 0,1 М НС1 и рассчитывают общую удельную обменную емкость в ммоль Н+/г, сухого обменника.
Для быстрого предварительного определения емкости катионообменни-ков используют следующую методику.
Методика. Определенное количество (1,000 — 3,000 г) набухшего катионообменника в Н-форме вносят (при перемешиваннн магнитной мешалкой) в 100 см3 0,1 М раствора NaOH и тщательно перемешивают. Для сильнокислотных катионообменников
86
Часть II
типа стирол-ДВБ (до 10% ДВБ) достаточно перемешивания в течение 8 — 10 мин, другие катиоиообменники необходимо перемешивать 20 — 30 мин. В аликвотной части раствора определяют оставшееся количество гидроксида и рассчитывают емкость обменника; Количество сухого вещества в ионообменнике определяют обычным способом.
Сильнокислотные катиоиообменники могут содержать дополнительные катионообменные группы, которые являются слабокислотными. Емкость обменных групп обоих типов определяют последовательным пропусканием через ионообменную колонку растворов нейтральной соли и гидроксида натрия.
ОПРЕДЕЛЕНИЕ СИЛЬНОКИСЛОТНЫХ И СЛАБОКИСЛОТНЫХ ГРУПП КАТИОНООБМЕННИКА
Методика А. Определенное количество (1 — 2 г сухого или 1 — 3 г набухшего ионообмениика в Н-форме) переносят в колонку соответствующего размера, пропускают 200 см3 1 М раствора хлорида натрия со скоростью 3 см3/мин и промывают водой до нейтральной реакции (в большинстве случаев достаточно 30 — 50 см3 воды). Элюат и промывные воды собирают в мерную колбу емкостью 500 см3. Аликвотную часть полученного раствора титруют 0,2 М раствором NaOH в присутствии метилового оранжевого в качестве индикатора. Количество выделившейся кислоты соответствует емкости сильнокислотных групп обменника.
После этого ионообменную колонку промывают 30 см3 0,1 М раствора NaOH (скорость потока 3 см3/мин) и 5 см3 деионизованной воды. Жидкость, оставшуюся в колонке, удаляют сжатым воздухом. Весь элюат собирают в стакан и титруют 0,1 М HCI в присутствии фенолфталеина в качестве индикатора. Обменную емкость слабокислотных групп рассчитывают по разности между общим количеством гидроксида и величиной, найденной титрованием.
Если сухой остаток обменника не определяют из отдельной навески, то после определения емкости обменник в колонке можно превратить в Н-форму, промыть водой и высушить до постоянной массы в предварительно взвешенном фильтрующем тигле.
Методика Б. Обменник (приблизительно 20 см3), превращенный в Н-форму по методике А, переносят в колонку и пропускают 250 см3 5%-ного раствора NaCI в течение 30 — 40 мин. Для определения выделившейся кислоты элюат титруют 0,1 М раствором NaOH в присутствии метилового оранжевого в качестве индикатора. Затем через колонку пропускают дополнительные порции 5%-ного раствора NaCI и выделившуюся кислоту снова титруют в аликвотных порциях элюата (5 см3). Операции повторяют до тех пор, пока расход 0,1 М раствора NaOH будет меньше 0,5 см3. Выделившаяся кислота соответствует сильнокислотным функциональным группам.
Для определения слабокислотных групп используют 250 см3 1%-ного раствора Na2HPO4. Раствор пропускают через колонку в течение 30 мин и затем титруют элюат 0,1 М раствором NaOH в присутствии фенолфталеина в качестве индикатора. Операцию повторяют, пропуская дополнительные, порции раствора по 50 см3 Na2HPO4. Определение заканчивают, когда расход раствора NaOH на 5 см3 элюата будет меньше 0,5 см3.
Различие в методиках А и Б состоит в использовании раствора фосфата натрия вместо раствора гидроксида натрия. Некоторые типы обменников частично разрушаются последним [15].
3. Физические, физико-химические и химические характеристики
87
ОПРЕДЕЛЕНИЕ ОБЩЕЙ УДЕЛЬНОЙ ОБМЕННОЙ ЕМКОСТИ АНИОНООБМЕННИКОВ
Методика. 1,000 — 2,000 г анионообменника в CI-форме (воздушно-сухого или набухшего, из которого удалена жидкость отсасыванием) количественно переносят в ионообменную колонку. Сухой обменник предварительно оставляют для набухания в воде. Затем через колонку со скоростью 3 см3/мин пропускают 200 см3 1 М раствора NaOH, нагретого до 40°С. Колонку промывают деионизованной водой и через обменник в ОН-форме пропускают 200 см3 0,1 М НС1 со скоростью 3 см3/мин. После этого колонку промывают 30 см3 воды и удаляют остаток жидкости из колонки сжатым воздухом. Всю пропущенную через колонку жидкость собирают в мерную колбу емкостью 250 см3 и определяют концентрацию соляной кислоты титрованием аликвотной части элюата 0,1 М раствором NaOH в присутствии метилового оранжевого в качестве индикатора. Емкость обменника оценивают по разности исходной и конечной концентраций НС1. Сухой остаток обменника определяют из отдельной навески.
ОПРЕДЕЛЕНИЕ СИЛЬНООСНОВНЫХ И СЛАБООСНОВНЫХ ГРУПП СИЛЬНООСНОВНЫХ АНИОНООБМЕННИКОВ
Методика А. Определенное количество обменника превращают в ОН-форму. Через ионообменную колонку пропускают 200 см3 1 М раствора NaCI со скоростью 3 см3/мин и промывают обменник 30 см3 деионизованной воды. Весь раствор собирают в мерную колбу емкостью 250 см3. Концентрацию ОН~ ионов, соответствующую сильноосновным группам обменника, определяют титрованием аликвотной части элюата 0,2 М соляной кислотой в присутствии метилового красного в качестве индикатора. Для определения .слабоосновных групп обменник промывают 200 см3 0,1 М HCI и затем 10 см3 деионизованной воды (3 см3/мин). Элюат собирают в мерную колбу емкостью 250 см3, остаток жидкости из колонки вытесняют в колбу сжатым воздухом. Уменьшение концентрации НС1, соответствующее слабоосновным группам, определяют титрованием аликвотной части элюата 0,1 М раствором NaOH.
Методика Б. В колонку помешают приблизительно 5 см3 набухшего анионообменника и промывают 100 см3 3 — 5%-ной НС1 и этанолом до получения отрицательной реакции на ионы хлора. Затем через обменник пропускают 50 см3 1%-иого раствора NH4OH. Элюат собирают в мерную колбу емкостью 250 см3. Аликвотную часть элюата подкисляют азотной кислотой и титруют 0,1 М раствором нитрата серебра. Этим способом определяют количество слабоосновных групп. После этого анионообменник промывают 250 см3 4 — 5%-ного раствора Na2SO4. Аликвотную часть фильтрата, собранного в мерную колбу, титруют 0,05 М H2SO4 в присутствии метилового оранжевого в качестве индикатора. Раствор подкисляют несколькими каплями концентрированной HNO3 и титруют 0,1 М раствором AgNOj. Суммарная величина, полученная в двух титрованиях, соответствует содержанию сильноосновных обменных групп. Сухой остаток обменника определяют из отдельной навески или после превращения анионообменника в Cl-форму, промывания этанолом и высушивания до постоянной массы.
ОПРЕДЕЛЕНИЕ ОБМЕННОЙ ЕМКОСТИ СЛАБООСНОВНЫХ АНИОНООБМЕННИКОВ
Методика А. 1 — 2 г анионообменника превращают в ОН-форму обработкой приблизительно 30 см3 1 М NaOH и промывают, водой до нейтральной реакции
88
Часть II
по фенолфталеину. Через ионообменник пропускают 200 см3 0,1 М HCI (2 — 3 см3/мин), промывают 10 см3 воды и 10 см3 этанола. Элюат собирают в мерную колбу емкостью 250 см3 и удаляют остаток жидкости из колонки сжатым воздухом. Расход кислоты, соответствующий количеству слабоосновных обменных групп, определяют титрованием аликвотной части фильтрата 0,1 М раствором NaOH.
Методика Б. Анионообменник в ОН-форме, предварительно обработанный по методике А, после промывания водой количественно переносят в мерную колбу емкостью 200 см3, прибавляют 20 см3 1 М НС1, разбавляют до метки водой н оставляют стоять 12 — 16 ч при периодическом помешивании. Обменную емкость анионо-обменника рассчитывают по расходу НС1 титрованием аликвотной части раствора. Сухой остаток обменника определяют обычным методом.
ОПРЕДЕЛЕНИЕ ОСНОВНЫХ ГРУПП В ОБМЕННИКАХ
ТИТРОВАНИЕМ РАСТВОРОМ ХЛОРНОЙ КИСЛОТЫ В ЛЕДЯНОЙ УКСУСНОЙ КИСЛОТЕ [16, 17]
Этот метод применим для количественного определения основных групп в ионообменниках разных типов. Он используется также для контроля промежуточных и конечных продуктов реакций синтеза специальных азотсодержащих обменных смол.
Метод основан на титровании освобождающихся основных групп 0,1 М раствором хлорной кислоты в ледяной уксусной кислоте. В качестве индикатора используют 0,5%-ный раствор кристаллического фиолетового в ледяной уксусной кислоте. Индикатор изменяет окраску от синей через голубую до зеленой.
Методика. Образец обменника, содержащий 0,3 — 0,4 ммоля ОН-титруемого основания, кипятят в 20 см3 ледяной уксусной кислоты в течение 1 мнн. После охлаждения прибавляют каплю индикатора и титруют раствором НС1О4 в ледяной уксусной кислоте до изменения окраски индикатора. Колбу с раствором нагревают на плитке до 100°С. При быстром изменении окраски индикатора раствор должен быть снова оттитрован хлорной кислотой. Затем раствор оставляют на ночь при температуре 40°С и в случае изменения окраски индикатора снова оттитровывают.
ОПРЕДЕЛЕНИЕ ОБМЕННОЙ ЕМКОСТИ ПО ДАННЫМ ЭЛЕМЕНТНОГО АНАЛИЗА
Метод определения емкости основан на количественном определении элементов, которые явлиются основными компонентами функциональных групп ионообменников. Этот метод применим главным образом к монофункциональным ионообменникам. Наиболее часто определяют серу и азот (или фосфор).
Метод используют также для расчета обменной емкости современных неорганических ионообменников по данным о количестве индивидуальных элементов, образующих ионообменники. Анализ выполняют после полного разложения материала. Однако универсальной методики разложения и анализа, применимой к любому типу ионообменников, не существует.
3. Физические, физико-химические и химические характеристики
89
Величины обменной емкости, рассчитанные из результатов элементного анализа, отличаются от данных, полученных другими методами. Это различие вызвано тем обстоятельством, что не все группы, содержащие анализируемый элемент, ведут себя как ионообменные. Поэтому данный метод дает завышенные величины обменной емкости.
Минерализацию органических материалов осуществляют классическим или мокрым методом.
Методика А. 0,2 — 0,5 г воздушно-сухого обменника (Н- или CI-форма) переносят в колбу Кьельдаля емкостью 30 — 50 см5, прибавляют 2 — 3 см5 концентрированной серной кислоты н нагревают смесь газовой горелкой до появления белых паров. Затем медленно, по каплям вводят 70%-ную НС1О4. Каждую новую порцию НСЮ4 прибавляют после окончания взаимодействия с предыдущей. При необходимости смесь нагревают. Во избежание опасности взрыва хлорную кислоту прибавляют очень осторожно.
Методика Б. Вместо хлорной кислоты используют азотную кислоту. В некоторых случаях разложение выполняют прн комнатной температуре. При необходимости смесь осторожно нагревают (содержимое колбы может выкипеть). Азотную кислоту прибавляют со скоростью 0,5 см3/мин.
Методика В. Способ разложения азотсодержащих ионообменников зависит от химической формы присутствующего азота.
Методика Г. Эту методику используют в том случае, если азот находится в обменнике в виде амина. В колбу Кьельдаля помещают 3 см3 концентрированной серной кислоты, 0,50 г катализатора, 0,200 г обменника и нагревают смесь до кипения. После того как раствор становится прозрачным, смесь нагревают еще 120 мин. Затем раствор охлаждают и количественно переносят, используя приблизительно 30 см3 воды, в дистилляционный аппарат для определения аммиака. Прибавляют 12 см3 10 М NaOH, разбавляют водой до 50 — 60 см3 и отгоняют аммиак в течение 5 мин (чистое время дистилляции) в приемник, содержащий 5 см3 4%-ного раствора борной кислоты н 5 капель индикатора. Отогнанный аммиак титруют 0,05 М хлорной кислотой.
Методика Д. Эту методику применяют, если вещество содержит азот в ннтро-или нитрозоформе. В колбу помещают приблизительно 0,200 г образца, прибавляют 5 см3 этанола, 5 см3 ледяной уксусной кислоты, приблизительно 0,5 г цинкового порошка, переносят колбу в водяную баню, нагретую до 60°С, и нагревают раствор на слабокйпящей водяной бане в течение 20 мин. Жидкость при этом почти полностью испаряется. Колбу охлаждают, прибавляют 3 см3 концентрированной H2SO4, 0,20 г катализатора и поступают как в методике Г. Перед дистилляцией необходимо увеличить количество 10 М NaOH до - 5 — 7 см3.
Одновременно проводят холостой опыт через все стадии анализа и его результат вычитают из объема 0,05 М НС1О4, израсходованной иа титрование отогнанного аммиака. Содержание азота относят к массе сухого обменника.
Расчет:
E-a-f
Содержание компонента А = ------ (3 8)
90
Часть II
где а — расход 0,05 М НС1О4 (см3), f — поправочный коэффициент для 0,05 М НС1О4, п — масса образца (мг). Е находят из следующей таблицы:
А Е
NH4 90,21
NOj 310,05
N 70,04
NO2 230,01
NO 150,01
Приготовление катализатора: 5 г металлического селена, 10 г сульфата меди (II) и 150 г сульфата калия растирают до получения гомогенной смеси.
Приготовление индикатора: 75 мг бромкрезолового зеленого и 50 мг метилового красного (кислота) растворяют в 100 см3 этанола.
Приготовление 0,05 М хлорной кислоты и определение ее поправочного коэффициента:
4,3 см3 70%-ной НС1О4 разбавляют до 1000 см3.
Определение поправочного коэффициента: Определенное количество (около 40 мг) оксида ртути(П) и 1 г иодида калия растворяют при слабом нагревании в 30 см3 кипящей воды. Раствор охлаждают, прибавляют 5 капель смешанного индикатора и титруют раствор 0,05 М НСЮ4 до перехода окраски индикатора из зеленой в красную.
Методика Е. Величину обменной емкости рассчитывают, исходя из структурной формулы ионообменника. Эта методика может быть иллюстрирована следующими примерами:
Структурная формула звена сильнокислотного (полностью сульфированного) по-листирольного катионообменника -СН2 —CH-C6H4SO3H, т.е. CgHgO3S, эквивалентная масса 184,2. Теоретическая величина максимальной удельной емкости (без учета содержания сшивающего агента) составляет 1000/184,2 = 5,43 ммоля Н+/г.
Звено сильноосновного анионообменннка типа I имеет формулу -CH,-CH-CtH.CH,N+(CH1).CI-, т.е. C,,H._NCI. Эквивалентная масса равна 201,8, максимальная величина удельной емкости составляет 4,96 ммоля ОН /г (с учетом поправки на содержание 8% ДВБ величина емкости равна 4,91 ммоля ОН“/г).
Для сильнокислотных стирол-дивинилбензольных катионообменников получено хорошее соответствие между расчетными и экспериментальными величинами обменной емкости. Однако для сильноосновных анионообменников подобной полимерной структуры экспериментально найденные величины емкости ниже.
3.3.2. Кривые нейтрализации (титрования) ионообменников
Кривая нейтрализации ионообменника является одной из важных его характеристик. По кривым нейтрализации катионообменников в Н-форме и анионообменников в ОН-форме оценивают характер обмениваемых групп, обменную емкость и некоторые другие параметры.
Кривые титрования монофункциональных сильнокислотных (или сильноосновных) обменников аналогичны кривым, полученным потенциометрическим титрованием сильных минеральных кислот или гидроксида натрия..
3. Физические, физико-химические и химические характеристики
91
Рис. 3.4. Кривые титрования катионообменников.
1 — слабокислотиый катионооб-менник с функциональными —ОН-группами; 2 — сильнокислотный катионообменник с —SOjH-rpyn-пами; 3 — среднекислотный катионообменник с функциональными —РО(ОН)2-группами; 4—слабокислотный катионообменник с функциональными —СООН-группами.
Расход титранта, ммоль КОН/г сухого обменника
Форма кривых титрования, как видно из рис. 3.4 и 3.5, показывает зависимость обменной емкости от pH внешнего раствора. Емкость сильнокислотных или сильноосновных ионообменников практически не зависит от pH. Функциональные группы этих обменников способны обменивать ионы в широком интервале pH, т.е. они могут диссоциировать в кислых, нейтральных или щелочных растворах.
Диссоциации функциональных групп слабокислотных или слабоосновных ионообменников так же, как их ионообменная способность, проявляется только в определенной области pH.
1 — слабоосновный анионообменник; 2 — сильно-основный анионообменник.
Расход титранта, ммоль НС1/г сухого обменника
92
Часть 11
Кривые титрования ионообменников могут быть получены двумя способами. Метод прямого титрования используют для обменников с сильно-диссоциирующими функциональными группами и хорошими кинетическими свойствами. Титрование обменников, которые содержат слабодиссоцииру-ющие функциональные группы и характеризуются медленным установлением равновесия, выполняют методом отдельных навесок. Этот метод может быть использован для любого типа обменников.
МЕТОД ПРЯМОГО ТИТРОВАНИЯ
Методика, В стакан емкостью 150 см3 вносят 0,500 — 1,000 г набухшего, отфильтрованного обменника (катионообменник в Н-форме, аниоиообмеииик в ОН-форме) и 50 см3 0,5 — 1,0 М раствора NaCI и в полученную суспензию помещают стеклянный электрод и электрод сравнения. Освобождающуюся кислоту (или гидроксид) титруют при перемешивании магнитной мешалкой. После прибавления каждой порции титранта суспензию еше некоторое время перемешивают для установления равновесия, затем выключают мешалку и измеряют pH. Титрант (обычно 0,5 — 1,0 М раствор) прибавляют порциями по 0,2 — 0,5 см3. На основании полученных данных строят график зависимости pH от объема титранта (см3).
МЕТОД ОТДЕЛЬНЫХ НАВЕСОК (МЕТОД ВСТРЯХИВАНИЯ)
Методика. По 0,500 — 1,000 г набухшего и отфильтрованного или отцентрифугированного иоиообменника (катионообменник в Н-форме, анионообменник в ОН-форме) взвешивают в сухие стеклянные (лучше полиэтиленовые) плотно закрывающиеся склянки емкостью 100 см3. В каждую склянку вводят по 50 см3 1 М NaCI, разные количества (1 — 10 см3) 1 М НС1 или NaOH и разбавляют растворы деионизованной водой до определенного объема. Эту операцию можно модифицировать следующим образом. В каждую склянку помещают по 0,15 — 0,30 г воздушно-сухого обменника в соответствующей ионной форме. Общее число склянок составляет 20. В каждую из этих склянок вводят постепенно уменьшающиеся количества (на 1 см3) 1 М раствора NaCI и постепенно увеличивающиеся количества (иа 1 см3) 0,1 М NaOH (или 0,1 М НС1) в 1 М растворе NaCI.
Для приведения в равновесие растворов со смолой плотно закрытые скляики встряхивают механически. Время встряхивания зависит от типа обменника. Сильнокислотные (основные) обменники обычно встряхивают 2 — 3 ч. Для других типов обменников время встряхивания устанавливают в предварительном эксперименте. Ориентировочное время, необходимое для установления равновесия, равно 2 — 4 сут.
После достижения равновесия измеряют величины pH каждого раствора. Результаты обрабатывают как описано в предыдущей методике. Одновременно определяют сухой остаток обменника.
3.3.3. Определение степени поперечного сшивания стирол-дивинилбензольных ионообменников
Степень поперечного сшивания стирол-дивинилбензольных ионообменников чаше всего определяют ИК-спектроскопическими методами [18, 19]. Возможно использование пиролитической газовой хроматографии [20].
3. Физические, физико-химические и химические характеристики
93
Этим методом может быть выполнен также структурный анализ ионообменного материала.
Полезно исследование суспендированных в воде ионообменных смол методом ядерного магнитного резонанса (ЯМР), который пригоден для характеристики анионообменников и катионообменников, предпочтительно с — 5О3Н-группами на основе сополимеров стирол-ДВБ; с его помощью могут быть оценены такие свойства, как гомогенность, степень поперечного сшивания, скорость обмена воды, гидратация и концентрация противоионов в фазе смолы [21, 25].
Для изучения и определения пористости ионообменных смол пригоден метод электронной микроскопии.
Молекулярную структуру обменников, включая сольватацию обменных групп, исследуют с помощью ИК-спектроскопии. В некоторых случаях могут быть определены ионы, удерживаемые ионообменником [26 — 30].
3.3.4. Определение констант диссоциации ионообменников
Ионообменники в виде полимерных кислот или оснований диссоциируют подобно своим мономерным аналогам. В соответствии с величинами их констант диссоциации ионообменники могут быть разделены на три группы: сильно-, средне- и слабокислотиые или основные. Ионообменники, принадлежащие к разным группам, различаются по их сорбционным свойствам.
Знание констант диссоциации сильнокислотных или основных обменников необязательно. Их обменная емкость в незначительной степени зависит от pH внешнего раствора вследствие слабого влияния pH на степень диссоциации функциональных групп ионообменников.
Величины констант диссоциации средне- и слабокислотных или основных ионообменников являются показателем изменения обменной емкости в зависимости от pH внешнего раствора.
Определение констант диссоциации может быть иллюстрировано следующим примером-
Катионообменник в Н-форме (RH) при контакте с водой диссоциирует согласно уравнению
RH # V + Н+ (3.9)
Если активности индивидуальных ионов заменить их аналитическими концентрациями, то кажущаяся константа диссоциации Х’каж будет равна
к _ каж
[R"][H+] [RH]
(З.Ю)
Квадратные скобки [ ] обозначают аналитические концентрации индивидуальных компонентов. Выражение [R~]/[RH] показывает соотношение дис-
94
Часть II
социированных и недиссоциированных групп в обменнике. При введении степени диссоциации, а, это соотношение приобретает вид
[R-] а
-----= ------- (3.11)
[RH] 1 - а
Поскольку — lgH+ = pH и — IgA" = рК, получаем
1 — а
pH = рА" — 1g-------- (3.12)
а
Принимая а = 0,5, т.е. при условии, что нейтрализуется 5О*7о всех обмениваемых групп слабодиссоциирующих обменников в Н-форме, получаем
pH = рК (3.13)
Однако было найдено, что величина рК сильнокислотных катионообменников ниже величин pH при 50%-ной нейтрализации всех обмениваемых групп.
Для более точного расчета величин рК в уравнение вводят дополнительные члены. Для катионообменников при а = 0,5 уравнение имеет вид рК = pH + lg[Na+] -lgx/2 (3.14)
Для слабоосновных анионообменников, титруемых соляной кислотой (при а = 0,5)
рК = 14 - pH + lg[CI~] - lgx/2 (3.15)
Величину рК определяют из кривой титрования данного ионообменника, построенной в координатах степень насыщения ионами Na+ (катионо-обменник) или С1~ (анионообменник) — pH внешнего раствора. Если концентрация ионов Na+ или С1~ во внешнем растворе достаточно высока (0,1 — 1 М), общая концентрация этих ионов меняется незначительно и нет необходимости в ее определении. Концентрация х (при а = 0,5) может быть рассчитана, если известна максимальная обменная емкость, удельная масса и содержание воды:
100 - Н2О%
Qa
(3.16)
р — плотность сухого обменника, QA — практическая удельная емкость сухого обменника (ммоль Н+ или ОН-/г).
Степень насыщения обменника ионами Na+ (катионообменник) или С1~ (анионообменник) определяют специальным методом. Из обменника удаляют поглощенный раствор центрифугированием, освобождают ионы Na+ и С1~ и определяют их концентрацию. Эту же величину можно определить (менее точно) титрованием равновесного раствора кислотой или основанием до pH 7,0. Концентрацию ионов Н+ или ОН- рассчитывают по расходу титранта.
3. Физические, физико-химические и химические
95
3.3.5. Определение моляльности функциональных групп обменника и концентрации обмениваемых групп в 1 см3 набухшей смолы
Исходя из пористой структуры органического ионообменника, можно рассчитать концентрацию обмениваемых групп относительно содержания воды в набухшей смоле. Концентрация, выраженная таким способом, представляет моляльность cR функциональных групп, т.е. количество (ммоль Н+) обмениваемых ионов, относящееся к 1 г воды в набухшей смоле. Моляльность функциональных групп обменника не постоянная величина. Она зависит от состояния и состава обменника, а также от состава внешнего раствора.
В полностью диссоциированном ионообменнике моляльность функциональных групп описывается уравнением
100 - W М°
CR = --„----' (3.17)
Jr М
где cR— моляльность функциональных групп полностью диссоциированного обменника, W — содержание воды в набухшей смоле (масс. %), М° — масса сухого обменника в стандартном состоянии (катионообменник в Н-форМе, анионообменник в Cl-форме), М — масса сухого обменника в измеряемой ионной форме, Qa — обменная емкость (ммоль Н+ /г или С1~ /г).
Следует различать моляльность и концентрацию cv, выражающую количество обмениваемых групп в 1 см3 набухшей смолы. Эту величину рассчитывают из уравнения
100 - W М°
" = ’Ъ.„-8* <’,8)
где Су — концентрация (мэкв) обмениваемых групп в 1 см3 набухшей смолы, р — плотность обменника в данной ионной форме. Другие обозначения те же, что и в уравнении (3.17).
Пример. Необходимо определить моляльность функциональных групп при условии, что обменник в Н-форме находится в равновесии с чистой водой. Из предположениям0 = М следует, что QA = 5,0 ммоль Н+/г, W = 45,0%, р = 1,25. Тогда
- 100 - 45
cR =----------• 5,0 = 6,11 ммоль Н+/г.
45
Су рассчитывают из уравнения
„ 100 - 45 .
Су = 1,25-----——• 5 = 3,43 ммоль Н/см3.
96
Часть II
3.3.6. Определение равновесного коэффициента распределения
Состояние равновесия в системе раствор электролита — ионообменник описывают разными способами. Наиболее часто используют параметр «коэффициент распределения», который равен отношению равновесных концентраций ионов в фазе обменника и в растворе. Коэффициент распределения D выражается уравнением
Равновесное количество компонента в фазе обменника
D = -------------------------------------------------- (3.19)
Равновесное количество компонента в жидкой фазе
Если коэффициент распределения рассчитывают на единицу массы (1 г) обменника и единицу объема (1 см3) раствора, то его обозначают символом Dg (часто Kd или KD). Если это количество относят к единице объема (1 см3) обменника, то используют символ Dv. Однако до сих пор нет единообразия в обозначениях коэффициента распределения, и в литературе встречается множество различных символов.
Соотношение между «массовыми» и «объемными» коэффициентами распределения выражается уравнением
= " Р (3.20)
где р — удельная масса обменника (масса сухого остатка обменника в 1 см3 загрузки колонки).
Для определения коэффициента распределения используют статический (перемешивание) или динамический (колоночный) методы.
СТАТИЧЕСКИЙ МЕТОД
Методика. Определенное количество ионообменника в известной ионной форме вводят в определенный объем стандартного раствора. После достижения равновесия (аппарат для механического встряхивания) определяют изменение концентрации анализируемого катиона.
Для более точных измерений необходимо определять количество ионов, сорбируемых обменником. Поглощенный раствор удаляют из обменника центрифугированием (обменник может быть промыт небольшим количеством воды) и элюируют сорбированные ионы соответствующим раствором.-Одновременно определяют сухой остаток обменника.
При лабораторных исследованиях чаще всего имеют дело с количествами обменника до 1,000 г и объемами растворов до 250 см3. Область исследуемых концентраций варьируется от следовах количеств ионов (для радиоактивных элементов) до 10"3 — 10~2 М.
Результаты выражают в виде массовых коэффициентов распределения Dg рассчитанных в соответствии с уравнением
Количество компонента/грамм сухого обменника
D =-----------------------------3----------------- (3.21)
Количество компонента/см подвижной фазы
3. Физические, физико-химические и химические характеристики
97
КОЛОНОЧНЫЙ МЕТОД
Объемный коэффициент распределения Dv рассчитывают по уравнению
_ Количество компонента в стационарной фазе/см3 объема слоя
Dv = ----------------------------------=----------------------- (3.22)
Количество компонента/см подвижной фазы
Выбор соответствующего экспериментального метода определения зависит от ожидаемой величины коэффициента распределения.
Методика А. Эта методика применима к определению высоких значений Dv. Небольшое количество исследуемого иона сорбируется в верхней части колонки, содержащей ионообменник в соответствующей ионной форме. Для макроизмерений обычно используют колонку размером 10 х 100 мм. Через колонку медленно пропускают раствор определенного состава и анализируют элюат. Выходную кривую строят, откладывая объем элюата (см3) относительно концентрации исследуемого иона. Для определения коэффициента распределения необходимо знать объем элюата Итах, соответствующий максимальной концентрации Стах исследуемого иона в элюате. Коэффициент распределения рассчитывают по формуле
V г, тах - _
Dv = - ei 0-23)
где А(. — поперечное сечение ионообменной колонки (см2), L — длина колонки (см), е. — коэффициент свободного объема колонки (относительный промежуточный объем) (см. с. 128)
е. = 1 - — (3.24)
Р
Масса набухшего обменника в колонке где а = ------------------------------- ,
Объем набухшего обменника в колонке
Р — удельная масса набухшего ионообменника.
Методика Б. Эта методика применима для определения средних величин Dv (10* - 102). Объем Итах определяют следующим образом: элюирующий раствор определенного состава, содержащий небольшое количество исследуемого иона (М), медленно пропускают через ионообменную колонку. Объем элюата определяют, когда концентрация иона М уменьшается в 2 раза по сравнению с исходной. В идеальном случае эта величина равна Утгх. Расчет Dy производится так же, как в' предыдущем случае.
Методика В. Эта методика применима к определению низких величин Dy. Элюирующий раствор, содержащий небольшое количество исследуемого иона, пропускают через ионообменную колонку до тех пор, пока концентрации исходного раствора и элюата не станут равны. Затем обменник промывают водой и после элюирования определяют- количество сорбированных ионов.
3.3.7. Коэффициент селективности и его определение
Если реакция обмена двух ионов А и В описывается уравнением
5RA + аВь+ aRB + 5Ао+ (3.25)
7-354
98
Часть II
то коэффициент селективности къ А выражается соотношением
_ [ИВ]°[А]* ^В’А " [RA]*[B]°
(3.26)
Квадратные скобки обозначают аналитическую концентрацию (молярность, моляльность) ионов А и В; а и b — абсолютные заряды ионов А и В соответственно.
Методика. Навеску иоиообмеииика в требуемой ионной форме помещают в склянку с притертой пробкой, прибавляют определенный объем раствора, содержащего известное количество второго исследуемого иоиа, и встряхивают содержимое (механически) до установления равновесия. Затем в аликвотной части раствора, тщательно отделенной от частиц ионообмениика, определяют равновесную концентрацию исследуемого иоиа и из полученных данных рассчитывают величину кц А в соответствии с уравнением (3.26). При условии что комплексообразующие ионы ие принимают участия в реакции, температуру системы поддерживают постоянной в интервале ± 1 — 2°С.
Время, необходимое для установления равновесия, определяют в предварительных кинетических экспериментах. Для сильнокислотных или сильиоосиовиых иоио-обмеиииков, как правило, достаточно 1 — 2 ч. Однако для слабокислотиых (основных), селективных или неорганических ионообменников равновесие может устанавливаться в течение нескольких суток.
Если иоиообмениик содержит слабокислотные (основные) группы и используется в Н (ОН)-формах, то распределение ионов между обменником и раствором является функцией pH системы.
При анализе ионообмениика возникают некоторые трудности, связанные с отделением жидкой фазы, поглощенной обменником. Жидкая фаза ие может быть удалена промыванием обменника водой без смещения равновесия. Этим обстоятельством можно пренебречь только в случае очень разбавленных растворов.
Если исследуют достаточно низкие концентрации ионов, то определяют равновесную концентрацию одного из ионов, а концентрацию второго иоиа рассчитывают. При использовании высоких концентраций иоиов необходимо определять оба компонента раствора. Ошибки в этих определениях заметно влияют иа рассчитанную величину коэффициента селективности, особенно при обмене ионов разного заряда.
3.3.8. Определение относительной скорости обмена
Методика. Определенное количество (приблизительно 10 ммолей) набухшего ио-иообмеиника в Н-форме (катионообменник) или ОН-форме (аиионообмеиник) помещают в трехгорлую колбу емкостью 500 см3, которую заполняют 250 см3 0,1 М NaOH (обмен катионов) или 250 см3 0,1 М НС! (обмен анионов). Эксперимент выполняют в термостатируемой бане при постоянной температуре. Систему ионо-обмеиник — раствор перемешивают с постоянной скоростью (приблизительно 400 об/мин). Через определенные интервалы времени отбирают пробы раствора (5 см3), определяют в них количества ионов Н+ и ОН" и, зная общую обменную емкость иоиообмеииика, рассчитывают степень нейтрализации (в данный момент). Для расчета используют следующее упрошенное уравнение:
3. Физические, физико-химические и химические характеристики
99
Таблица 3.5. Коэффициенты диффузии некоторых иоиов в сильнокислотные
катиоиообменники и сильиоосиовиые аииоиообмениики при 25°С
Противоион Ионообменннк D > cm2/c r
н+ Dowex 50-X4 X8 X16 9,14-IO"6 5,40-IO-6 2,20-10~7
Na+ Dowex 50-X4 X8 ' X16 X24 14,1-IO'7 2,88- IO"7 1,10-IO'7 1,00-ю-7
К + Dowex 50-X8,6 13,4' IO"7
Rb + Dowex 50-X4 X8,6 2,28- IO-7 13,8-Ю-7
Ag + Dowex 50-X8.6 X16 X24 6,42-Ю-7 2,75-Ю-7 1,13- Ю-7
Sr2+ Dowex 5O-X8 X16 3,38- Ю-8 2,98-Ю-9
Zn2 + Dowex 50-X8 X24 2,89- Ю-8 2,63-Ю-9
Y3+ Dowex 50-X8 X24 3,18- Ю-9 2,18-Ю-10
Th4 + Dowex 5O-X8 2,15-Ю-10
cr Dowex Z-X6 3,54-10-7
Br~ Dowex 2-X6 3,87-Ю-7
I" Dowex 2-X6 1,33-Ю-7
PO8-4 Dowex 2-X6 0,57- Ю-7
100
Часть II
— =l,08<7), (3.27)
-в»
где Ь = ---
г2
В' Концентрация обмениваемых ионов в момент времени t
Ва Равновесная концентрация обмениваемых ионов
Dr — коэффициент диффузии исследуемого иоиа в ионообменник, г — диаметр зерна ионообменника, t — время.
Величины коэффициентов диффузии для некоторых систем представлены в табл. 3.5.
ОПРЕДЕЛЕНИЕ ВРЕМЕНИ, НЕОБХОДИМОГО ДЛЯ ПОЛУНЕЙТРАЛИЗАЦИИ ИОНООБМЕННИКА
Время, необходимое для нейтрализации 50% функциональных групп ионообменника, является еще одним параметром, выражающим скорость ионного обмена. Сначала определяют время нейтрализации 40% и 60% групп (Н-форма катионообменника, ОН-форма анионообменника), а затем интерполяцией получают время 50%-ной нейтрализации.
Методика. В стакан емкостью 250 см3 помещают точно 10 ммолей катионообменника (Н-форма) и 50 см3 деионизованной воды. Одновременно получают второй раствор смешиванием 4,00 см3 1 М раствора NaOH, 50 см3 насыщенного раствора NaCl и 76 см3 воды. Обе системы термостатируют при 20°С.
Затем в стакан помещают мешалку, стеклянный и каломельный электроды для измерения pH и после включения мешалки второй раствор быстро выливают в стакан, содержащий ионообменник. Параллельно измеряют время, необходимое для установления значения pH, равного 7. Затем к ионообменнику прибавляют дополнительную порцию (11 см3) 1 М NaOH и перемешивают систему около 30 мин. В это время происходит полная нейтрализация обмениваемых групп смолы. Избыток NaOH определяют обратным титрованием 1 М НС1. Аналогичный эксперимент выполняют при другом составе раствора, прибавляемого к ионообменнику: 6,0 см3 1 М NaOH, 50 см3 насыщенного раствора NaCI и 74 см3 воды.
Интерполяцией между этими двумя величинами получают время, необходимое для достижения 50%-ной нейтрализации функциональных групп ионообменника.
Для исследования анионообменников в ОН-форме используют раствор, содержащий эквивалентное количество HCI вместо NaOH.
3.3.9. Характеристика неизвестных ионообменников
Для характеристики неизвестных ионообменников необходимо выполнить серию опытов.
О типе каркаса матрицы можно судить по внешнему виду смолы. Поли-меризационные ионообменники обычно белые, бледно-желтые или коричневые; поликонденсаты имеют цвет от красно-коричневого до черного.
3. Физические, физико-химические и химические характеристики
101
Сильноосновные анионообменники, содержащие четвертичные аммониевые группы, часто обнаруживают по аммиачному запаху, который появляется при встряхивании анионообменников с теплым раствором гидроксида натрия.
Для получения более подробных характеристик образец неизвестного ионообменника делят на четыре равные части. Если имеется в наличии большое количество образца, отбирают порции приблизительно по 10 г.
Набухшие в воде порции ионообменника переносят в соответствующие колонки и промывают разными способами: колонки 1 и 3 промывают приблизительно 2 М NaOH, колонки 2 и 4 — 2 М НС1 и измеряют pH элюата. После появления в элюате кислоты или щелочи продолжают промывание ионообменника приблизительно 10-кратным количеством раствора по сравнению с объемом, израсходованным до проскока. Окончательное промывание водой выполняют до нейтральной (или близкой к нейтральной) реакции элюата.
При необходимости определения ионной формы обменника производят анализ элюата. Качественную оценку Н-формы катионообменника (ОН-формы анионообменника) можно сделать по расходу промывного раствора до проскока кислоты (щелочи) при промывании колонок описанным выше методом.
При промывании кислотой катионообменников в Н-форме элюат становится кислым после прохождения свободного объема колонки. Если эти катионообменники промывают раствором гидроксида, первая порция элюата представляет собой нейтральную воду. Обратная картина наблюдается для ОН-формы анионообменника.
Когда катионообменник находится в солевой форме, ОН~-ионы появляются в элюате после прохождения свободного объема колонки. При этом в колонке может происходить осаждение гидроксида соответствующего металла. При промывании этого обменника кислотой элюат проявляет кислую реакцию. (В колонке образуется осадок, если катионообменник находится в Ag- или РЬ-формах.)
После промывания катионообменника в МН4-форме раствором гидроксида в элюате обнаруживается запах аммиака.
Анионообменники в солевой форме удерживают ионы ОН-. В то же время ионы Н+ быстро проходят через колонку, повышая кислотность элюата. Одновременно может выделяться летучая кислота (при условии, что ее анион сорбировался на анионообменнике), которая обнаруживается по специфическому запаху.
После промывания колонок 1 и 2 водой через обменники пропускают 0,1 М нейтральный раствор ацетата аммония. При этом элюат имеет либо кислую (ионообменник характеризуется как катионообменник), либо щелочную (запах аммиака, ионообменник характеризуется как анионообменник) реакцию.
Колонки 3 и 4 промывают 0,1 М нейтральным раствором хлорида натрия. Кислая реакция элюата из колонки 4 является признаком сильнокислотного катионообменника. Щелочная реакция элюата из колонки 3 указы
102
Часть II
вает на сильноосновный анионообменник. Нейтральный элюат в обоих случаях служит признаком либо слабокислотного катионообменника, либо слабоосновного анионообменника. В случае среднеосновных анионообмен-ников элюат из колонки 3 имеет слабощелочную реакцию.
Для точного определения природы функциональных групп ионообменника необходимо снять кривую титрования. По этой кривой определяют характер функциональных групп и общую обменную емкость неизвестного ионообменника.
Проведенные испытания должны быть дополнены данными элементного анализа на серу и азот. Для специальных типов ионообменников необходимы определение других элементов (фосфор, мышьяк) и дополнительные испытания в зависимости от предполагаемой природы функциональных групп. Лучшим способом характеристики неорганических ионообменников являются промывание их азотной кислотой достаточной концентрации для растворения обменника и качественный и количественный анализ раствора.
3.4. Термическая, химическая и радиационная устойчивость ионообменников
Термическая, химическая и радиационная устойчивость органических ионообменников зависит от типа полимерной матрицы, степени поперечного сшивания, типа ионогенных групп и их противоионов.
Помимо этих главных факторов на устойчивость ионообменников оказывают влияние чистота исходного сырья и метод синтеза.
Изменение устойчивости смолы становится особенно явным при изменении степени набухания, образовании новых групп (особенно слабокислотных) или при различной степени деструкции полимерной матрицы.
3.4.1. Термическая устойчивость
УСТОЙЧИВОСТЬ ПРИ НАГРЕВАНИИ НА ВОЗДУХЕ
Сульфированные катионообменники фенолформальдегидного типа в ll-форме проявляют первые признаки десульфирования при 100°С [31, 32]. Обменная емкость смол не изменяется при нагревании их до 70°С в запаянных ампулах в течение 60 суток. Для сильнокислотных стирол-дивинилбензольных катионообменников наблюдается противоположная картина.
С повышением температуры обменная емкость смол уменьшается вследствие повышения степени десульфирования.
R - SO3H - RH + H2SO4 (3.28)
Повышение температуры ускоряет процесс десульфирования, и при температурах > 150°С начинается реакция между образовавшейся серной кислотой и окисляемыми компонентами смолы. При этом выделяется диоксид серы [33, 34].
Rred + H2SO4 - Rox + Н2О + SO2 (3.29)
3. Физические, физико-химические и химические характеристики
103
Десульфирование наряду с другими реакциями является следствием нагревания на воздухе катионообменников на основе стирол-дивинилбензоль-ных матриц. Дифференциальная термогравиметрия для этого типа обменных смол в Н-форме обнаруживает три эндотермических превращения [35 — 39]: дегидратацию при 100 — 210°С, десульфирование [см. уравнение (3.28)] при 270 — 310°С и окислительную деструкцию матрицы при температурах выше 430°С. Кроме этих трех эндотермических эффектов при 37О°С наблюдается экзотермический эффект, соответствующий деполимеризации матрицы.
При более высоких температурах группы — SO3H реагируют с образованием сульфонов и дополнительных поперечных связей. Дальнейшее повышение температуры вызывает разложение сульфонов до SO2, а также приводит к значительной деструкции полимерной матрицы [35].
На процесс десульфирования сильнокислотных катионообменников большое влияние оказывают природа матрицы, место расположения сульфогрупп и присутствие других заместителей в ароматическом ядре (если каркас образован ароматическими соединениями). Сульфированные фенолформальдегидные ионообменники, содержащие как — SO3H-, так н ОН-группы, менее стабильны при нагревании на воздухе по сравнению с сильнокислотными стирол-днвинилбензольными катионообменниками.
Превращение Н-формы обменников в их солевые формы повышает термическую устойчивость [39 — 41].
Относительно мало внимания уделяется реакциям, протекающим при нагревании на воздухе слабокислотных катионообменников. Смолы на основе метакриловой кислоты и ДВБ довольно устойчивы при продолжительном нагревании до 120° С (Н-форма). Дальнейшее повышение температуры приводит к потере гидратационной воды. При температуре приблизительно 200° С наблюдаются реакции конденсации между ионогенными группами
СН, СН, СН3 СН3
Г Г I I
— R-СН —СН2 —С —R] - Н,О + -R-C-CH2-C-R<
I 2 I I I
СООН соон о = с-о — с = о
(3.30)
Продолжительное нагревание при этой температуре приводит к полному декарбоксилированию смолы [42] и затем к деполимеризации.
Термическая устойчивость слабокислотных катионообменников также увеличивается при превращении их в солевые формы.
На основании сравнения данных термического анализа (нагревание на воздухе) для разнообразных сильно- и слабокислотных катионообменников сделано заключение [35, 42, 43] о наличии процессов дегидратации в широком интервале температуры, десульфирования (илн потери СО2 для слабокислотных катионообменников) и окислительной деструкции матрицы. Температурный интервал, соответствующий этим процессам, изменяется в зависимости от типа смол н степени поперечного сшивания. Реакции де
104
Часть 11
струкции всегда протекают сначала в ионогенных группах, так как связи в этих группах обычно слабее, чем связи в полимерных цепях.
Высушивание на воздухе при комнатной температуре сильноосновных анионообменников типа I в ОН-форме приводит к уменьшению объемной обменной емкости их функциональных групп. Повышение температуры ускоряет потерю емкости.
Изменение обменной емкости сильноосновных групп обусловлено процессами деаминирования и разложения этих групп:
R-CH2N+(CH3)3OH- + Н2О - R-CH2OH + N+H(CH3)3OH~ (3.31, а) R—CH2N+(CH3)3OH“ + Н2О - RCH2N+H(CH3)2OH" + СН3ОН(3.31, б)
Однако наиболее вероятно, что главной реакцией является следующая: r-ch2n+ (сн3)2он- + н2о - rch2n+h(Ch3)2oh- (3 32)
'N+(CH3)3OH“ + rch2o
Когда сильноосновные анионообменники типа П в ОН-форме (группа — СН2СН2ОН связана с атомом азота менее прочно, чем группа — СН3) нагревают на воздухе, протекает следующая реакция:
R - CH2N+ (СН3)2ОН~ + Н2О—• RCH2N+H(CH3)2OH“ + ОНСН2СН2ОН
I (3.33)
СН2СН2ОН v ’
Солевые формы сильноосновных анионообменников более устойчивы при нагревании на воздухе, чем ОН-формы обменников. Их устойчивость уменьшается в ряду RC1 > R2SO4 > R3BO3.
Опубликованы разнообразные данные, характеризующие устойчивость при нагревании на воздухе анионообменников типа I в Cl-форме: до 70°С [45, 46], до 100°С [47] и даже до 160°С (короткое время нагревания).
При продолжительном нагревании обменника в Cl-форме наблюдается разложение части четвертичных аммониевых групп при 130°С:
RCH2N+ (СН3)3СГ - RCH2C1 + N(CH3)3 (3.34)
Дальнейшее нагревание до температуры 180°С приводит к заметному уменьшению количества сильноосновных групп вследствие реакции RCH2N+ (СН3)3С1~ - RCH2N(CH3)2 + СН3С1 (3.35)
Если при обработке смолы кислотой-окислителем (HNO3 и другие) образуется солевая форма, то устойчивость этой нитратной формы в значительной степени будет зависеть от температуры [46]; нитратная форма более устойчива, чем ОН-форма, вплоть до 140°С. Пиролитическое разложение и самовозгорание смолы наблюдаются при 160 — 200°С.
Ряд устойчивости для разнообразных типов ионообменников в NO3-форме имеет следующий вид [46]: Dowex 2 > Permutit SK > Amberlite IRA-411. С увеличением содержания сшивающего агента устойчивость смол уменьшается. Ионообменники в ЬЮ3-форме не рекомендуется сушить при температуре выше 60°С.
3. Физические, физико-химические и химические характеристики
105
Анионообменники, содержащие фосфониевые или сульфониевые ионогенные группы, при нагревании на воздухе гораздо менее устойчивы, чем их аммониевые аналоги.
Термическая устойчивость слабоосновных анионообменников выше устойчивости сильноосновных смол.
На основании вышеизложенного выработаны следующие практические рекомендации:
а. При определении сухого остатка Н-формы катионообменннка рекомендуется сушить смолы при низких температурах (80°С, вакуум, Р2О5). Нагревание выше 100°С противопоказано.
б. Не рекомендуется сушить илн хранить сильноосновные анионообменники в ОН-форме, Даже при комнатной температуре наблюдается разложение четвертичных аммониевых групп.
в. Не рекомендуется получать безводные ионообменники высушиванием при повышенной температуре ввиду опасности нх разложения. Например, если стирол-дивинилбензольный сильнокислотный катионообменник КУ-2 высушить до постоянной массы при 100°С, то он все еще содержит 9% воды в форме моногидрата сульфогруппы [48].
ТЕРМИЧЕСКАЯ УСТОЙЧИВОСТЬ ИОНООБМЕННИКОВ ПРИ КОНТАКТЕ С ВОДОЙ, ВОДНЫМИ РАСТВОРАМИ И ОРГАНИЧЕСКИМИ РАСТВОРИТЕЛЯМИ
Хорошо известные типы поликонденсационных сильнокислотных катионообменников в Н-форме используют при температурах, не превышающих приблизительно 60°С (иногда до 40°С) вследствие гидролитического расщепления сульфогрупп, связанных непосредственно с ароматическим ядром. В случае когда группа - SO3H связана через — СН2-мостик, гидролитическое расщепление наступает при более высоких температурах (80 — 90°С).
Авторы работ, рассматривающих термическую устойчивость ионообменников, констатируют, что полимерные типы сильнокислотных катионообменников, особенно на основе стирол-дивинилбензольных матриц, обладают достаточной устойчивостью при нагревании в воде. Согласно Линдсею [49], смола Dowex-50 устойчива при нагревании в воде до 105 — 115°С. При нагревании Н-формы Amberlite IR-120 в воде до 150°С полная потеря обменной емкости происходит через 12 сут. При 180°С аналогичная картина наблюдается через 24 ч.
Установлено, что макропористые ионообменные смолы при нагревании в воде более устойчивы, чем классические микропористые [50, 51]. После термической‘обработки некоторых типов макропористых ионообменников наблюдается изменение объема пор. Объемная обменная емкость при этом сохраняет свою величину.
На термическую устойчивость ионообменников сильно влияет природа противоиона.
106
Часть 11
Механизм термического разложения солевых форм сильнокислотных катионообменников может быть иллюстрирован следующими уравнениями: R-SO3M + Н2О - R-H + MHSO4 (3.36, а)
R-SO3M + MHSO4 - R -SO,H + M2SO4 (3.36, 6)
Освобождающиеся ионы Н'г вступают в обменную реакцию с противоионами смолы, превращая последнюю в Н-форму, что косвенным путем уменьшает термическую устойчивость смол.
Многие противоионы действуют как катализаторы процесса гидролиза и других стадий разложения. Показано, что скорость десульфирования — SO3H-rpynn фенолформальдегидных катионообменников в ионных формах щелочных металлов повышается в ряду Cs < Rb < К < Na < Li, т.е. с увеличением эффективного радиуса гидратированного противоиона.
Катионообменники, содержащие фосфониевые группы в стирол-дивинилбензольных матрицах, термически более устойчивы по сравнению с сульфосмолами. На устойчивость смол оказывают меньшее влияние форма катионообменника и степень поперечного сшивания.
В присутствии растворов сильных кислот или оснований при повышенной температуре гидролиз ионогенных групп все же происходит. В некоторых случаях скорость гидролиза выше скорости гидролиза в воде [50, 52, 53]. Помимо гидролиза функциональных групп кислоты окислители вызывают окислительную деструкцию полимерной матрицы. Более подробно эти и другие явления рассмотрены в обзоре Полянского [54].
Нагревание катионообменников в неводных растворителях сопровождается уменьшением обменной емкости. При взаимодействии растворителей с функциональными группами смолы последние катализируют реакции между молекулами растворителя, приводя к образованию продуктов с более высокой молекулярной массой. Эти продукты блокируют ионогенные групцы или удерживаются пористой структурой смолы [54].
В качестве примера рассмотрим нагревание сильнокислотного стирол-дивинилбензольного катионообменника в ацетоне [55]. При 150°С наблюдается потеря обменной емкости через 24 ч на 2,8%, через 48 ч на 8% и через 120 ч на 11,2%. Одновременно даже при температуре ниже 100°С происходит конденсация молекул ацетона, катализируемая смолой. Продукты конденсации фиксируются в пористой структуре смолы.
При нагревании смолы в присутствии алифатических спиртов ее термическая устойчивость уменьшается с увеличением числа атомов углерода в спирте.
Термическая устойчивость анионообменников в воде или в водных растворах в значительной степени зависит от природы полимерной матрицы, основности функциональных групп и их ионной формы.
Сильноосновные анионообменники в ОН-форме подвергаются классическому разложению Гофмана с образованием третичного амина и спирта в качестве продуктов разложения.
При нагревании анионообменников типа I разложение протекает следующим образом:
3. Физические, физико-химические и химические характеристики
107
s’ R-CH2N(CH3)2 + СН3ОН (разложение) (3.37,а) RCH,N (СН-а)-зОН
RCH2OH + N(CH3)3 (деаминирование) (3.37,6)
В соответствии с реакцией (3.37, а) остаток смолы приобретает слабоосновные свойства. Реакция (3.37, 6) приводит к образованию высокомолекулярного спирта (без обменных свойств) и триметиламина.
Гофмановское расщепление смол типа II в ОН-форме протекает, по-видимому, согласно эмпирическому закону /3-элиминирования главным образом следующим путем:
RCH2N + (CH3)2C2H5OH“ - RCH2N(CH3)2 + СН2=СНОН + Н2О (3.38)
Остаток смолы обладает слабоосновными свойствами. Другой продукт перегруппировывается в ацетальдегид.
Сильноосновные анионообменники более устойчивы в солевой форме, чем в ОН-форме. Их устойчивость уменьшается с увеличением содержания ДВБ в матрице смолы.
Анионообменники в ОН-форме претерпевают деаминирование и разложение в водно-органических растворителях. Потеря обменной емкости возрастает с увеличением концентрации органического растворителя в системе (при равных условиях большая потеря обменной емкости наблюдается в абсолютном спирте или ацетоне, чем в воде).
Термическая устойчивость неорганических ионообменников в общем удовлетворительна. Однако это не общее правило. При высушивании некоторых типов ионообменников наблюдается уменьшение обменной емкости вследствие процессов перекристаллизации или конденсации между функциональными группами.
3.4.2. Химическая устойчивость ионообменников
В зависимости от природы процессов расщепления, которые происходят при химической коррозии ионообменников, наблюдаются следующие типы реакций:
а) расщепление макромолекулярной цепи между пространственными мостиками;
б) химические изменения в группах, косвенно связанных с макромолекулярной цепью (ионогенные группы остаются неизменными);
в) замещение или расщепление функциональных группу
г) образование новых (часто слабокислотных) функциональных групп.
С точки зрения химической устойчивости ионообменников наиболее важной характеристикой является их сопротивление окисляющим агентам [56].
Если расщепление смолы вследствие недостаточной устойчивости к окислению идет по ее полимерной матрице, то степень поперечного сшивания уменьшается. Набухаемость смолы повышается и в предельном случае смола растворяется. Фенолформальдегидные матрицы подвергаются расщеплению легче, чем стирол-дивинилбензольные каркасы. Степень разло
108
Часть II
жения стирол-дивииилбеизольных смол зависит от количества ДВБ, присутствующего в полимере, и от чистоты мономеров, использованных при синтезе. Наблюдается также влияние изомеров ДВБ (или взаимного соотношения индивидуальных изомеров в техническом дивинилбензоле).
Смолы с более низким содержанием ДВБ подвергаются окислительному разложению в большей степени. Процесс окисления ускоряется в присутствии элементов, которые способны действовать как катализаторы (особенно железо, медь).
Фенолформальдегидный сильнокислотный катионообменник КУ-1 при нагревании до 70°С в 3%-ной Н2О2 в присутствии Fe3 теряет 97% своей массы в течение 24 ч. В тех же условиях стирол-дивинилбензольные суль-фокатионообменники теряют 62% массы при содержании ДВБ 1 %, 46% — при 2% ДВБ, 26% — при 4% ДВБ и 11,6% — при 8% ДВБ.
Сильноосновные анионообменники особенно чувствительны к перекиси водорода. Смолы, содержащие пиридиниевые группы, более устойчивы по сравнению с другими сильноосновными обменниками.
При контакте смол с кислыми растворами хроматов, перманганатов и ванадатов последние большей частью восстанавливаются, окисляя некоторые компоненты смолы [57 — 59].
В среде неокисляющих кислот смолы фенолформальдегидного типа менее устойчивы (особенно при нагревании) по сравнению со стирол-дивинилбензольными смолами, которые проявляют хорошую устойчивость независимо от того, являются ли они катионо- или анионообменниками. В среде окисляющих кислот (в частности, при нагревании и при высокой концентрации кислоты) фенолформальдегидные смолы подвергаются значительному разложению. Стирол-дивинилбензольный анионообменник Dowex 1 и Permutit SK проявляют удовлетворительную устойчивость в потоке 7 М HNO3 вплоть до 60°С при условии, что нитрит-ионы отсутствуют. Эксперименты в статическом режиме в 7 М HNO3 обнаруживают следы серьезного разрушения в течение двух недель вследствие автокаталитического образования нитрита [60].
Поликонденсационные смолы медленно растворяются в растворах щелочей, особенно при высоких концентрациях гидроксид-ионов. Стирол-дивинилбензольные смолы более устойчивы. Однако при контакте сильноосновных анионообменников с щелочными растворами высокой концентрации (> 1,0 М ) сильноосновные группы расщепляются. Не рекомендуется хранить такие смолы в ОН-форме в сильнощелочных растворах в течение продолжительного времени.
Макропористые сильноосновные анионообменники проявляют более высокую устойчивость к щелочным растворам, чем смолы гелевой структуры.
Обычные органические растворители (при комнатной температуре) незначительно разрушают ионообменники. Однако в некоторых случаях происходит потеря небольшого количества органического материала, что (иногда) мешает аналитическому определению.
3. Физические, физико-химические и химические характеристики 109
3.4.3. Радиационная устойчивость ионообменников
Воздействие радиоактивного излучения на ионообменники приводит к радиационному разрушению последних. В присутствии растворителя (воды) это первичное разложение сопровождается вторичными явлениями вследствие взаимодействия продуктов радиолиза растворителя со смолой.
При радиационном разрушении смол наблюдаются следующие явления: а) потеря обменной емкости, обусловленная разложением функциональных групп;
б) образование новых функциональных групп;
в) разрушение макромолекулярного каркаса. Повышение растворимости смолы вследствие уменьшения молекулярной массы (обратные явления наблюдаются при повышении степени поперечного связывания полимерного каркаса, особенно для слабосшитых смол);
г) изменение набухаемости смолы;
д) образование газообразных продуктов в результате радиолитического разложения органических смол.
Набухшая смола дополнительно подвергается косвенному радиолитическому воздействию (радиолиз воды, присутствие Н2О2 и т.п.). Важным следствием облучения смолы является изменение механических физико-химических свойств, селективности, кинетики обмена и т.д. [61, 62].
Устойчивость разных типов ионообменников к радиоактивному излучению уменьшается в следующем ряду: неорганические ионообменники > сульфоугли > органические катионообменники > органические анионообменники.
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ НЕОРГАНИЧЕСКИХ ИОНООБМЕННИКОВ
Вследствие низкой набухаемости этих обменников нет существенного различия в процессах разложения сухого или набухшего сорбента. Никаких значительных изменений не обнаружено при облучении обменников дозой 107 Гр. Доза 2- 106 Гр приводит к уменьшению обменной емкости силикагеля приблизительно на 15%. Некоторые типы неорганических ионообменников могут работать даже при дозах облучения вплоть до 107 — 108 Гр, когда использование органических ионообменников исключается [63 — 65].
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ ИОНООБМЕННИКОВ
НА ОСНОВЕ УГЛЯ
Ионообменники на основе разных типов углей в общем устойчивы к ионизирующему излучению. Облучение обменников вплоть до 107 Гр не зывает существенных изменений в их обменных свойствах. Вследствие низкой способности к набуханию не наблюдается существенного различия между устойчивостью сухого и набухшего обменника.
2 X efl S X 4- Е ® О 00 «П 00
rt D. ость силы S 5 а £ й &§ 3 4,8 3,1 4,8 3,3
* ж S 3
о V
X 4> енная
& 3 8 « +
«1 S X О <ч •л m
* □ 00 оо «гч
s гв
1 8 5 ГО m
s 'g i
I
g i а о. V) Г4
ев X o Влас НОС1 %н. 50, 54, 44, 49,
u
О
X
и
О
5
s 3
О X ! ношение юв облу % и необ 5 2 « о 1 5< ,940
s ш S О О
1 0 >§ | £
к
в после Потеря массы, It 20,1 18,2
&
а д
§ S 3 _ *е •ш
8 в 10 I- Й О 506 865 208 538
! Удельиь см3 н< смолы 3 Q ri <ч <ч <ч
О
ж
X
efl
X
-©
0) А & *© ъ
X я s U.
X Я £ _ 1 .
0) X 2. е? я ю s 1 Г? 1 г-
v " Й о х о о
я ® »tt
чв 2
<п 3 S ьв
г 5 К аэ
§. I & 00 оо <ч <ч ^4
Табли содерз Сол нне
88
3. Физические, физико-химические и химические характеристики
111
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ ОРГАНИЧЕСКИХ КАТИОНООБ МЕННИКОВ
По устойчивости к ионизирующему излучению катионообменники делятся на следующие группы:
а) сульфированные монофункциональные сильнокислотные катионообменники полимеризационного типа;
б) сульфированные полифункциональные сильнокислотные катионообменники поликонденсационного типа;
в) фосфорилированные моно- и полифункциональные среднекислотные катионообменники полимеризационного и поликонденсационного типов;
г) слабокислотные моно- и полифункциональные катионообменники полимеризационного и поликонденсационного типов.
Смолы группы а. Наблюдаются значительные изменения при дозах > 106 Гр. При облучении Dowex 5OW-X8 дозой 3,9- 107 Гр сохраняется только 5% обменной емкости. Данные, характеризующие изменение свойств и структуры смолы после облучения, приведены в табл. 3.6.
На радиационную устойчивость смол влияет природа сшивающего агента: смолы, сшитые дивинилбензолом, менее устойчивы по сравнению со смолами, связанными бутадиеном. Если при синтезе смолы используют дивинилбензол (это наиболее общий случай), то радиационная устойчивость обменника зависит от соотношения присутствующих изомеров диви-нилбензола. Результаты, полученные Вилеем, представлены в табл. 3.7.
Радиационная устойчивость смол зависит от содержания сшивающего агента: смолы с большей степенью сшивания относительно более устойчивы. Данные для катионообменника КУ-2 приведены в табл. 3.8.
Ионная форма смолы оказывает влияние на радиационную устойчивость: смолы в Н-форме наиболее чувствительны к облучению. Устойчивость смол повышается превращением их в соответствующую ионную форму [68].
При облучении смолы на воздухе образуются новые функциональные группы (преимущественно карбоксильные). Радиолиз сухих смол приводит к выделению газов (SO2, SO3 и особенно Н2).
При облучении набухшей смолы в ней появляются новые функциональные группы, чаще всего гидроксильные.
Из многочисленных литературных данных можно сделать вывод, что наблюдаются значительные изменения этого типа смолы при общих дозах облучения > Ю6 Гр. Дозы выше 107 Гр настолько сильно разрушают смолы, что последние вообще не могут быть использованы. По устойчивости к ионизирующему излучению нет заметного различия между макропористым и гелевым типом смол.
Смолы группы б. Эти смолы характеризуются большей устойчивостью, чем предыдущие типы. Заметное разрушение наблюдается только при дозах, больших чем 5- 106 Гр. В противоположность предыдущему типу нет различия в радиационной устойчивости солевой и Н-формы смол. В про-
112
Часть 11
Таблица 3.7. Влияние облучения ^Со на свойства сульфированной полистирол-ДВБ смолы с разными соотношениями изомеров п- и м-ДВБ [66]
Смола Доза облучения, ю-e Гр Потеря массы, % Влажность, <7о Н2О Обменная емкость Содержание кислоты в промывной воде, ммоль Н+/г
ммоль Н+/г потеря,
Dowex 5О-Х8 — 51,6 5,03 — —
0,91 3,6 53,3 4,64 7,7 0,23
0,92 — 53,5 4,65 7,5 0,32
1,82 5,7 56,9 4,45 11,5 0,73
1,90 — — 4,43 11,9 —
Смола: — — 44,0 5,29 — —
8% ДВБ, 0,91 4,1 49,7 4,94 6,6 0,34
коммерчес- 1,82 7,2 50,3 4,72 10,8 0,65
кий продукт, 1,90 — 51,0 4,70 11,2 0,78
набухшая
Смола: — 42,0 5,29 — —
8% ДВБ, 0,91 6,5 46,0 4,98 5,8 0,37
м-ДВБ: 0,92 — 45,8 5,07 4,2 0,44
л-ДВБ = 2:1, 1,82 7,8 46,8 4,80 9,3 0,66
набухшая 1,90 — .47,4 4,75 10,2 0,81
Смола: — 43,8 5,33 — —
8% л-ДВБ, 0,91 5,3 46,9 4,95 7,1 0,34
набухшая 1,14 —— 59,6 4,87 8,6 0,38
1,82 7,1 49,2 4,76 10,7 0,71
Смола: — 49,1 5,30
8% m-ДВБ, 0,91 3,1 48,7 5,00 5,8 0,40
набухшая 1,14 4,4 49,0 4,92 7,2 0,42
1,80 4,9 49,8 4,75 10,4 0,41
цесСе облучения образуются новые типы функциональных групп, преимущественно карбоксильные.
Смолы группы в. Эти типы смол проявляют высокую радиационную устойчивость. Обменная емкость практически не изменяется при общей дозе 107 Гр. Изменение набухаемости в воде зависит от типа каркаса. Однако оно существенно меньше, чем для сильнокислотных катионообменников. Некоторые из этих смол работают даже при дозах выше чем 107 Гр, как показано в табл. 3.9.
3. Физические, физико-химические и химические характеристики 113
Таблица 3.8. Влияние содержания ДВБ на некоторые свойства катионообменника КУ-2 при облучении дозой 1,3-106 Гр [67]
Содержание ДВБ, % Потеря обменной Степень набуха-
емкости, % НИЯ, см3 Н2О/г смолы
2 10,0 2,95
4 8,0 2,04
8 6,8 1,33
12 6,5 1,18
16 6,0 1,08
24 5,6 0,70
Смолы типа г. Слабокислотные катионообменники более чувствительны к ионизирующему излучению по сравнению с другими типами катионообменников. Смолы на основе метакриловой кислоты и ДВБ заметно изменяют свои обменные свойства при общей дозе облучения 105 Гр. Другие типы более устойчивы, однако и они теряют 90 — 95% обменной емкости при дозах около 106 Гр. Наибольшей устойчивостью обладает Н-форма смолы; устойчивость солевых форм значительно ниже (особенно для набухших смол).
Таблица 3.9. Изменение некоторых физико-химических свойств катионообменника КФ-1а после облучения ускоренными электронами в различных средах [69]
Доза облучения, 10-7 Гр Степень набухания, % Обменная емкость, ммоль Н + /г при pH Изменение массы после облучения, % Среда
4 8 9
0 5 1,3 4,0 5,0 — —
3,4 5 1,3 4,2 6,0 -6,0 Дистиллированная вода
4,5 5 1,3 4,1 6,3 -2,0 Дистиллированная вода
1,8 5 1,3 4,2 5,8 0,8 0,7 М HNO3
2,2 15 1,3 4,4 6,6 2,5 3,0 М HNOj
2,2 5 1,3 4,1 5,8 -2,0 0,25 М молочная кислота
2,2 5 1,4 4,2 5,8 0,7 0,50 М уксусная
кислота
а Смола стнрол-дивиннлбензольного типа, содержащая группы —РО(ОН)2.
8-354
114
Часть II
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ ОРГАНИЧЕСКИХ АНИОНООБМЕННИКОВ
Радиационная устойчивость анионообменников зависит как от типа каркаса, так и от природы функциональных групп. Устойчивость смол, содержащих ароматические ядра в каркасе, выше устойчивости анионообменников на основе чисто алифатических углеводородов. Особое положение занимают анионообменники с пиридиновыми ядрами. Они проявляют значительную устойчивость к ионизирующему излучению. Устойчивость функциональных групп анионообменников увеличивается с уменьшением основности групп.
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ ПОЛИМЕРИЗАЦИОННЫХ СИЛЬНООСНОВНЫХ АНИОНООБМЕННИКОВ
Обменная емкость этого типа смол заметно снижается при дозах 105 — 106 Гр. Они не могут быть использованы после облучения при дозах около 107 Гр. Снижение емкости некоторых типов смол после облучения на воздухе показано в табл. 3.10.
Способность к набуханию снижается с увеличением поглощенной дозы. Монофункциональный сильноосновный анионообменник превращается в полифункциональный тип с функциональными группами разной основности. Одновременно отщепляются аммиак и низкомолекулярные амины. Часто образуются новые функциональные группы типа — ОН и — СООН. Таким образом смолы становятся биполярными нонообменниками.
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ ПОЛИКОНДЕНСАЦИОННОГО ТИПА АНИОНООБМЕННИКОВ
По радиационной устойчивости эти смолы подобны предыдущему типу. В противоположность полимеризационным смолам разложение структуры поликонденсационных смол приводит к повышению набухаемости нз-за уменьшения плотности структуры.
Таблица 3.10. Потеря обменной емкости различными анионообменниками, облученными на воздухе дозой 3,6-106 Гр [70]
Анионообменник Потеря обменной емкости, Тип смолы
Dowex 1 40—44 Сильноосновный
Nalcite SAR 37 ч
Permutit S-2 38 < с
Amberlite IRA-400 42 < <
Amberlite IRA-410 40 tl
Dowex 3 19 Слабоосновный
Amberlite IR-45 53 < ‘
Nalcite WBR 20 <<
3. Физические, физико-химические и химические характеристики
115
Таблица 3.11. Изменение объемной обменной емкости по плутонию некоторых сильиоосновных анионообменников, облученных 60Со [72]
Ионообменннк Доза облучения, io-е Гр Емкость8 Потеря емкости, %
Dowex 1-Х4 1,9 65 49
Amberlite IRA 401 0.5 55 15
1,5 53 18
1,8 46 29
3,2 44 32
4,2 23 47
Dowex 21K 0,9 , ,76 37
1,7 63 47
2,8 52 57
3,7 Общее разрушение смолы
а Емкость указана в граммах Ри на 1 дм3 смолы в NO^-форме при 50%-иом проскоке Ри и температуре 60° С.
Особое положение занимают анионообменники, содержащие пиридин или его производные. Они проявляют высокую радиационную устойчивость. Значительное изменение иаблюдаетси только при дозах облучения выше 5- 10® Гр. Монофункциональные смолы более устойчивы, чем поли-функциональные анионообменники, содержащие другие амины (непиридиниевые основания) (1 • 10® - 5- 10® Гр).
Внутреннее облучение а-частицами дозой 2,57- 10® Гр (например, после поглощения плутония) анионообменника АВ-23 М (метилированный сополимер 2,5-метилвинилпиридина и ДВЕ) не вызывает изменения основности [71]. Однако при этом нарушаются поперечные связи макромолекулярного каркаса и набухаемость смолы повышается. Плутоний постепенно десорбируется из обменника вследствие разрушения смолы и окисления Ри3+ в Ри4+ (табл. 3.11).
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ СЛАБООСНОВНЫХ АНИОНООБМЕННИКОВ
Эти типы смол более устойчивы к ионизирующему излучению. При дозе облучения 105 Гр обычно наблюдается заметное разложение. Некоторые анионообменники значительно разрушаются даже при дозах 102 Гр.
116
Часть 11
РАДИАЦИОННАЯ УСТОЙЧИВОСТЬ ИОНООБМЕННЫХ МЕМБРАН
Облучение ионообменных мембран приводит к радиационному разрушению как обменных центров, так и материала носителя мембраны. Сильнокислотные и основные типы гетерогенных мембран обычно устойчивы к дозам облучения около 5- Ю^р. При дозах, близких к 107 Гр, наблюдается полная потеря обменной емкости, а также значительное повышение электрического сопротивления мембраны. Одновременно серьезно ухудшается механическая устойчивость мембраны.
Литература
1. MooreS., Stein W. Н., J. Biol. Chem., 192, (1951) 663.
2. MooreS., Stein W. H., J. Biol. Chem., 211 (1954) 893.
3. Hamilton P. B., Anal. Chem., 30 (1958) 914.
4. MooreS., Spackman D. H., Stein W. H., Anal. Chem., 30 (1958) 1185.
5. Simonson R., Svensk. Kem. Tidskr. 73 (1961) 531.
6.. Zmrhal Z. неопубликованные данные.
7. Koprda И, Fojtik M., Chem. listy, 62 (1968) 679.
8. Ayboin G., Laverlocheve J., J. Radioan. Chem., 1 (1968) 123.
9. Edge R. A., Chemist-Analyst, 47 (1958) 72.
10. Pollio F. X., Anal. Chem., 35 (1963) 2164.
11. Duolite Ion Exchange Manual (1960).
12. Pepper K. W, Reichenberg D., Hale D. K., J. Chem. Soc. (London), 1952, 3129.
13. Boyd G. E., Soldano B. A., Z. Elektrochem., 57 (1953) 162.
14. Freeman D. H., in: Ion Exchange, Vol. 1, Marinsky J. A., Ed., Marcel Dekker, N. Y., 1966, p. 173.
15. Ungar J., Anal. Chem., 34 (1962) 413.
16. Ficken G. E., Lane E. S., Anal. Chim. Acta, 16 (1957) 207.
17. Копылова В. A., Салдадзе К. M., Асамбадзе Г. Д. Ж. аналитич. химии, 24 (1969) 1727.
18. Calmanovici В., Rev. Chim. (Bucharest), 19 (1968) 222.
19. KvederH., Kem. Ind., 19 (1970) 141.
20. BtasiusE., Hausler H.., Z. anal. Chem., 277 (1975) 9.
21. Gilmer T. C, Pietrzyk D. J., Anal. Chem., 43 (1971) 1585.
22. Wilks A., Pietrzyk D. J., Anal. Chem., 44 (1972) 676.
23. Casey W. J., Pietrzyk D. J., Anal. Chem., 45 (1973) 1404.
24. Creekmoore R. W., Reilly C. N., Anal. Chem., 42 (1970) 570, 725.
25. Pietrzyk D. J., Crit. Rev. Anal. Chem., 6 (1976) 131.
26. Whittington D., Millar J.R., J. Appl. Chem., 18 (1968) 122.
27. Ben-zwi N., Heitner-Wirguin G., Israel. J. Chem., 10 (1972) 885.
28. Словохотова H. А., Никашина В. А., Сенявин M. M. ЖФХ, 35 (1961) 2387.
29. Freeman D. H., Separation and Purific. Sect., Superintend, of Docum. U. S. Gov. Print Office, Washington D.C.NBS Techn. Note 459 (Dec. 1968).
30. Лантых Г. В., Кириленко О. Д. ДАН Укр. ССР, Сер. Б., 35, 1973, 632.
3. Физические, физико-химические и химические характеристики
117
31. Jacubovic А. О., J. Chem. Soc., 1960, 4820.
32. Полянский Н. Г., Вишневская В. И. Миж-Мишин Ю. М., Высокомолек. соедин., 8, (1959), 1249.
33. Dusek К., Chromecek R., Chem. prumysl., 10 (1960) 267.
34. Кончева А. С., Морозова М. М., Олыианова К. М. Сб. «Синтез и свойства ионообменных материалов». — М.: Наука, 1968, с. 115.
35. Yamabe Т, Suzuki Т., TakaiN., Kogyo Kugaku Zasshi, 70 (1967) 1839; С. A., 69 (1968) 22356.
36. Эйриш 3. И, Эйриш М. В., Bummux М. В. Изв. АН Казах. ССР, сер. хим., 20 (1970)64.
37. Карпов О.Н., Тулупов П. Е, Волков В. И. ЖФХ, 45 (1971) 1759.
38. Карпов О. Н., Тулупов П. Е., Волков В. И. ЖФХ, 42 (1968) 2295.
39. Лейкин Ю. А., Смирнов А. В., Даванков А. В., Коршак В. В. Высокомолек. соедин. сер. А. 10 (1968) 157.
40. Calmon. С., Kressman Т. R. Е., Ion Exchange in Organic Chemistry and Biochemistry, Interscience Publ. Inc., New York, 1957.
41. Bauman W. C., Ind. Eng. Chem., 40 (1948) 1350.
42. Chapiro A., Seidler P., European Polymer J., 1 (1965) 189.
43. Nan и J., Gross J., 3 rd Lucr. Conf. Natl. Chim. Anal., 4 (1971) 323; C.A., П (1972) 89226 p.
44. Maura G., Pochetti F., Ann. Chim. (Rome), 61 (1971) 337.
45. Dobrevski I. D., Khim. Ind. (Sofia), 44 (1972) 22.
46. Barghusen J. J., Jonke A. A., Power Technology and Reactor Fuel Processing, 10 (1967) 2, 170.
47. Шабуров M. А. Ж. прикл. химии, 38 (1965) 2666.
48. Словохотова H. А., Никашина В. А., Сенявин М. М. ЖФХ, 35 (1961) 2387.
49. Lindsay F. К., Wirth L. F., Durinski А. М., Ind. Eng. Chem., 43 (1951) 1062.
50. Petrarin I., BarbintaE., Carpov A., Poinescu I., Mater Plast. (Bucharest), 10 (1973) 175.
51. Полянский H. Г., Полянская H. Л., Мазин В. И. ЖФХ, 45 (1971) 2858.
52. Полянский Н. Г., Матвеев В. П., Горбунов Г. В. Изв. Высш, учебн. за-вед., Химия и хим. технол., 17 (1974) 1013.
53. Шарапова Н. П., Коршунова С. П., Назаров П. П., Чмутов К. В. ЖФХ, 48 (1974) 546.
54. Полянский Н. Г, Тулупов П. Е. Усп. хим., 40 (1971) 2250;
55. Тулупов П.Е., Белоногова Ю. И. Ж. прикл. химии, 39 (1966) 2535.
56. Kuehne G., Martinola F, VGB Kraftwerkstechnik, 57 (1977) 173.
57. Евсинова Л. П., Куролап Н. С., Углянская В. А., Крысина О. Н. Теория и практика сорбционных процессов, №7 (1972) НО.
58. Мелешко В. П., Евсинова Л. П., Крысина О. Н. Теория и практика сорбционных процессов, №7 (1972) 142.
59. Олыианова К. М. и др. Изв. высших учебн. завед., Химия и хим. технол., 15 (1972) 1463.
60. Moody G. J., Thomas J. D. P., Lab. Pract., 21 (1972) 632, 717.
118
Часть II
61. PesekM., Radi V., Chem. Zwesti, 18 (1964) 502.
62. Pesek V., Radioisotopy, 8 (1967) 151.
63. Mathev J., Tandon S. N., Gill J. S., Radiochem. Radioanal. Lett., 30 (1977) 381.
64. Zsinka L., Szirtes L., Stenger V., Radiochem. Radioanal. Lett., 4 (1970) 257.
65. BornE., Paul G., Atomkernenergie, 31 (1978) 57.
66. Wiley R. H., Devenuto G., J. Appl. Polymer. Sci., 9 (1965) 2001.
67. Кузин И.A ., Семушин A. M. Ж. прикл. химии, 34 (1961) 1710.
68. Ichikawa T.,Hagiwara Z., J. Nucl. Sci. Technol. Japan, 10 (1973 ) 746.
69. Киселева E. Д. и др. ЖФХ, 36, (1962) 2457.
70. Паркер Г. В. и др. Ионообменная технология. — М.: Металлургиздат, 1959.
71. Парамонова Н. В. и др. Радиохимия, 12 (1970) 127.
Tl.RyanJ.L., Wheelwright Е. J. Proc. 2nd Int. Conf. Peaceful Uses of Atomic Energy, Vol. 17, Geneva, 1958. p.137.
4. Аппаратура для ионообменной хроматографии
В настоящее время в неорганическом анализе с применением ионообменников все еще используется так называемое «классическое» лабораторное оборудование. Анализируемые растворы поступают в колонки (обычно стеклянные) либо под действием силы тяжести сверху вниз (при постоянном уровне жидкости в резервуаре), либо с помощью разного типа насосов низкого давления с регулируемым объемом. Элюат, вытекающий из колонки, затем разделяется (при необходимости) на ряд фракций, которые анализируют обычным способом.
Применение соответствующих детекторов, соединенных с вычислительным устройством, делает возможным непрерывный анализ элюата с регистрацией результатов.
Используемое оборудование работает при небольшом избыточном давлении (< 0,5 М Па); аппаратура проста и собирается либо из элементов кустарного производства, либо из коммерчески доступных блоков. Это оборудование относительно дешево и до сих пор вполне удовлетворяло потребности.
Однако новые методы, разработанные в настоящее время, обусловили необходимость применения лабораторного оборудования нового класса. Одним из наиболее важных новых методов является высокоскоростная жидкостная хроматография (ВСЖХ), которая требует сложного и дорогостоящего оборудования. Поэтому применение этого метода в неорганическом анализе ограничено (анализируют только сложные смеси, например включающие лантаноиды, актиноиды).
Следует отметить, что более широкому распространению метода ВСЖХ препятствует не только высокая стоимость оборудования, но также то обстоятельство, что конструкционные материалы используемой аппаратуры не достаточно устойчивы к действию растворов минеральных кислот и других веществ, которые обычно применяют в процессе разделения неорганических ионов. Кроме того, возникают некоторые трудности вследствие многократного изменения концентрации элюирующего агента в процессе разделения и отсутствия соответствующих детекторов. Однако можно надеяться, что эти проблемы будут решены в ближайшем будущем.
120
Часть II
4.1. Ионообменные колонки
В большинстве методик ионообменного разделения смесей неорганических ионов используют динамические (колоночные) устройства. Однако статический метод, основанный на перемешивании суспендированного обменника в растворе, содержащем разделяемые компоненты, предпочтительнее в тех случаях, когда разделяемые вещества находятся в виде труднорастворимых осадков или когда в процессе разделения выделяется большое количество газов.
Динамические ионообменные разделения выполняют в ионообменных колонках, которые представляют собой трубки соответствующей конструкции, заполненные ионообменником. В простейшем случае ионообменной колонкой служит стеклянная трубка подходящего размера, наполненная набухшей смолой.
Материалом для изготовления лабораторных ионообменных колонок, как правило, служит стекло или кварц. В случае коррозионной опасности (например, в присутствии фтористоводородной кислоты и т.п.) колонки делают из других материалов (тефлон, плексиглас и другие пластмассы, нержавеющая сталь и т.д.), которые должны быть устойчивы к растворам, протекающим через колонку.
Жидкость обычно поступает в верхнюю часть колонки выше уровня поверхности смолы. Снизу раствор подается редко.
4.1.1. Терминология, используемая в колоночных методах
Объем колонки VL (или X) — объем той части колонки, которая содержит ионообменник (насадку). Объем слоя — синоним объема колонки, заполненной ионообменником. Промежуточный объем И,- — объем, занимаемый подвижной фазой, в части колонки с насадкой. Промежуточная фракция Ej (относительный промежуточный объем) — промежуточный объем на единицу объема колонки с насадкой (е( = Kz /И£).
4.1.2. Форма колонки
Колонка должна иметь такую конструкцию, чтобы поток раствора был равномерным по всему поперечному сечению. По этой причине точные стеклянные колонки (особенно малого диаметра) часто высверливают из стеклянных палочек или изготавливают из точных капиллярных трубок.
Слой ионообменника находится в колонке на плоской подложке. Рекомендуется использовать фриттованные стеклянные диски соответствующей пористости, которые впаивают в нижнюю часть колонки. В тех случаях, когда фриттованные стеклянные диски не могут быть использованы, применяют сетки из нержавеющей стали или платины. При работе с маленькими и узкими колонками достаточно заполнить нижнюю часть колонки стеклянной ватой, пористым тефлоном, полиуретаном или другим подходящим материалом.
4. Аппаратура для ионообменной хроматографии
121
Поток жидкости обычно регулируют краном, помещенным в нижней части колонки. При использовании колонок небольшого поперечного сечения поток регулируют давлением, необходимым для преодоления гидродинамического сопротивления слоя ионообменника. В этом случае в кране нет необходимости. Если колонка снабжена краном, то необходимо обеспечить минимальный объем пространства между поддерживающим диском и отверстием колонки. Выше уровня смолы в колонке должно находиться определенное количество жидкой фазы. Рекомендуется, чтобы высота слоя жидкости составляла 10 — 25% высоты колонки. Однако в случае непрерывного градиента элюирования это пространство (представляющее часть мертвого объема) должно быть сведено к минимуму.
Ионообменные колонки чаще всего используют в виде вертикально закрепленных трубок. Если их длина слишком велика (принимая во внимание требуемую методику разделения, например разделение изотопов), можно сконструировать колонки в форме вертикальной спирали.
Рис. 4.1. Различные типы ионообменных лабораторных колонок.
122
Часть П
Существует много типов лабораторных колонок. Наиболее часто используемые типы показаны на рис. 4.1. Колонка а пригодна для обработки большого количества ионообменника или для простого разделения большого количества смеси нескольких ионов. Колонка б снабжена кожухом для термостатирования колонки при более высоких или низких температурах по сравнению с температурой окружающей среды. Кран заменяют выпускным капилляром, который помещают выше верхнего уровня обменной колонки. Это устройство предотвращает спонтанное вытекание жидкости из колонки (уровень жидкости автоматически поддерживается выше уровня смолы в колонке). Колонка д действует подобным образом. Устройство в обеспечивает возможность промывания колонки из бокового отверстия. Устройство типа г позволяет жидкости проходить в нижнюю часть колонки и затем подниматься вверх через обменник. Колонка е снабжена воронкой, которая служит резервуаром в тех случаях, когда требуются большие количества жидкости для сорбции или элюирования сорбированных ионов. Для аналитической работы наиболее часто используют колонки типа б и д.
Колонки диаметром 1 мм предназначены для работы со следовыми количествами разделяемых веществ. Колонка из плексигласа (ионообменная колонка размером 3x20, 6х20и6х4 мм), показанная на рис. 4.2, используется для быстрого разделения трансплутониевых элементов. Устройство состоит из поршня (4), который преодолевает сопротивление колонки, содержащей смолу очень мелкого зернения (3). Поршень помещен в сосуд, заполненный промывным или элюирующим раствором, и перемещается в нижнюю часть устройства (собственно колонки). Объем сосуда около 6 см3. Обменник находится на пористом тефлоновом диске (2); выходное отверстие (7) колонки выполнено из золотой фольги.
Рис. 4.2. Микроколонка из плексигласа.
1 — выходное отверстие колонки, выполненное из золотой фольги; 2 — пористый тефлоновый диск; 3 — слой ионообменника; 4 — поршень, помешенный в цилиндр с элюирующим раствором.
Рис. 4.3. Ионообменные колонки, используемые при повышенных температурах. а — колонка, снабженная рубашкой с обогревающей жидкостью; б — обогреваемая ионообменная колонка Томпсона.
Рис. 4.4. Ионообменная колонка, приспособленная для работы при повышенной температуре и соединенная с устройством для градиентного элюирования.
A t — регулирование давления газа; А — смеситель, содержащий раствор концентрации q ; В — резервуар с элюирующим раствором (концентрация сг); F — холодильник; Н — электрическая спираль, обернутая вокруг рубашки колонки (заполненной соответствующей жидкостью); L — фракционный коллектор; М — магнит; Р — слой смолы; Т — термометр; Z — магнитная мешалка.
124
Часть И
Как правило, повышение температуры улучшает ионообменное разделение разных ионов, и поэтому разделение сложных смесей выполняют при повышенных температурах. Существует много конструкций колонок для работы при повышенной температуре и давлении. Две простые конструкции колонок этого типа показаны на рис. 4.3. Колонка а снабжена водяной рубашкой для нагревания колонки до*требуемой температуры (нагретая вода поступает из термостатируемой бани). Аппарат б состоит из ионообменной колонки, которая помешена в закрытый сосуд, соединенный с резервуаром, содержащим промывной раствор, и с другим сосудом, заполненным жидкостью с подходящей температурой кипения. Жидкость нагревают до кипения внешним источником тепла. Колонка и поступающий промывной раствор нагреваются паром этой кипящей жидкости.
Другой тип аппаратуры представлен на рис. 4.4. Водяная рубашка колонки нагревается обернутой вокруг нее электрической спиралью. Темпера-
Рис. 4.5. Ионообменная колонка с поршневым адаптером и кожухом для регулирования температуры.
4. Аппаратура для ионообменной хроматографии
125
туру воды регулируют изменением напряжения, подаваемого на спираль. Вместо спирали или нагревающего элемента можно использовать слой токопроводящего материала с высоким сопротивлением, которым покрывают кожух- колонки.
В последнее время разработаны новые типы колонок единой конструкции, которые легко монтируются. Для устранения мертвых объемов в обоих концах колонки и для компенсации набухания обменника колонку оборудуют поршневыми адаптерами (рис. 4.5). Трубки снабжены кожухами для регулирования температуры. Отдельные части, изготовленные из механически и химически устойчивой пластмассы, соединяются свинчиванием. Для поддержки слоя обменника служит пластмассовая пластина (полиамид, полиуретан и т.п.).
Внутренние стенки колонки гидрофобизуют соответствующим веществом. Этим путем предотвращают адсорбцию радиоактивных изотопов (следовые концентрации) на стенках колонки, а также нежелательные явления, происходящие между жидкостью и поверхностью колонки (особенно в очень узких колонках).
4.1.3. Размер колонки
Высота ионообменной колонки определяется фактором разделения ионов при данных условиях. Увеличение высоты колонки сверх оптимальных условий улучшает разделение. Одновременно увеличивается количество элюата, что приводит к улучшению выходных кривых.
Оптимальную высоту колонки рассчитывают с использованием равновесных данных или результатов предварительного эксперимента.
Поперечное сечение колонки выбирают в соответствии с количеством разделяемых веществ. Для нехроматографических простых операций используют обменные колонки, имеющие соотношение диаметра и высоты в интервале 1:10 — 1 : 20. В случае трудного разделения более сложных смесей выбирают колонки с соотношением 1 : 100 — 1 : 200.
При аналитических определениях макроколичеств веществ чаще всего используют колонки с внутренним диаметром 5 — 10 мм. Узкие колонки (< 5 мм) наиболее пригодны для работы с радиоактивными изотопами без носителей. Найдено эмпирическое правило, согласно которому диаметр колонки должен быть более чем в 20 раз больше диаметра частиц обменника.
4.1.4. Заполнение ионообменной колонки
Колонку, как правило, заполняют полностью набухшей смолой. Если колонка содержит сухую смолу, набухающую после контакта с водой, то колонка может растрескаться вследствие значительного изменения объема обменника.
Перед заполнением колонки необходимо деаэрировать обменник, т.е. удалить воздух. Смолу в стакане смешивают с водой и помещают в эксикатор, из которого затем откачивают воздух. По другой методике смолу,
126
Часть II
смешанную с водой или соответствующим раствором, помещают в водяную баню, температуру которой устанавливают приблизительно на 15° выше рабочей температуры; после этого колбу эвакуируют. Деаэрацию считают законченной после прекращения выделения пузырьков из смолы.
Деаэрация особенно необходима при проведении тонких хроматографических разделений при повышенной температуре.
Перед заполнением смолой колонка должна быть частично залита деионизованной водой, из которой удалены растворенные газы. Пористая пластина, поддерживающая смолу, должна быть полностью деаэрирована пропусканием небольшого количества воды через колонку.
Суспензию смолы наливают по стенке колонки таким образом, чтобы частицы оседали и образовывали гомогенный слой. Применяют и другой способ: воронку с широким основанием помещают в верхнюю часть колонки, которую предварительно полностью наполняют водой или другим соответствующим раствором. При этом воронка частично заполняется водой. Смешанную с водой смолу наливают в воронку и открывают нижний кран колонки. Этим способом быстро получают гомогенную колонку обменника.
В процессе заполнения и при последующих операциях необходимо исключить проникание воздуха в колонку. Присутствие воздуха может привести к образованию каналов в слое обменника и к ухудшению его качества. В заполненной колонке вода или другой соответствующий раствор всегда должны находиться выше верхнего уровня обменной колонки. Для заполнения колонки очень мелкими частицами смолы в высокоскоростной жидкостной хроматографии рекомендуются специальные приемы.
Если работа с колонками производится при повышенной температуре, то во всех операциях, включая приготовление растворов, необходимо пользоваться кипяченой деионизованной водой.
Заполненную колонку промывают водой или соответствующим раствором. Если последующие операции выполняют под давлением, то заполненную колонку промывают при том же давлений.
Хорошие результаты получены при условии первоначального промывания колонки снизу.
При попадании в колонку воздуха рекомендуется разрядить колонку и снова тщательно заполнить ее.
Вследствие изменений в составе элюирующего раствора может наблюдаться значительное изменение объема, обменной колонки. В таком случае перед поглощением разделяемой смеси желательно промыть обменную колонку элюирующим раствором. Последующее промывание не рекомендуется, или, если необходимо, колонку промывают только очень небольшим количеством деионизованной воды.
При работе в неводной или водно-органических средах может происходить растрескивание слоя обменника вследствие изменения его объема. Компактность слоя обменника часто нарушается при замене раствора, содержащего большие количества органического растворителя, на воду.
4. Аппаратура для ионообменной хроматографии
127
4.1.5. Емкость колонки
Массовая емкость Qm зависит от массы сухого обменника, помещенного в колонку. Она выражается общим числом миллимолей обмениваемых ионов. Помимо массовой емкости используют термин объемной емкости, Qv- Соотношение между этими двумя величинами дается следующим уравнением:
Qv - U _ s№Qm
(4.1)
Qv — объемная емкость колонки, Qm — массовая емкость, е; — относительный промежуточный объем, р — плотность набухшей смолы, S — содержание воды в смоле (масс. %).
Эффективная емкость QB колонки (так называемая проскоковая емкость) определяется как емкость в момент появления компонента в элюате. Это один из важных параметров при работе в динамических условиях.
Эффективная емкость определяется из выходных кривых.
Методика. Раствор, содержащий анализируемый иои с концентрацией с0, вводят в ионообменную колонку соответствующего размера и в индивидуальных фракциях элюата определяют концентрацию иона (с). Полученные результаты выражают в форме диаграмм с/с0 — объем элюата; эти кривые называются проскоковыми или выходными.
Эффективная способность колонки зависит от заданных условий сорбции (размера зерен обменника, скорости потока, состава раствора, температуры и т.п.). Для сравнения величин эффективной емкости при разных условиях необходимо определять ионы при одном и том же значении с/с0. Эффективная емкость определяется как количество вещества, удерживаемое обменной колонкой при достижении выбранного сравнительного значения с/с0.
4.1.6. Промежуточный объем колонки
Промежуточный объем (также называемый «пустым» объемом) ионообменной колонки — это пространство между частицами набухшей смолы (объем, занимаемый подвижной фазой в заполненной секции колонки). Этот объем Vj представляет собой разность между общим объемом слоя VL и объемом набухшей смолы.
Параметр К, используют при расчете данных по удерживающей способности смолы и при интерпретации экспериментальных результатов, полученных в динамическом режиме (например, при хроматографическом разделении смеси ионов).
Промежуточный объем имеет значение для ионообменников гелевого типа. Согласно Самуэльсону и Ларсону [1] для макропористых смол вопрос о промежуточном объеме является дискуссионным.
128
Часть П
Разработано множество методов определения И, для смол гелевого типа с учетом особенностей поперечного связывания смолы. Чаще других применяют две методики. Величины К,- для смол с более высокой степенью сшивания (> 8% ДВБ) определяют с помощью водорастворимых веществ с достаточно большими молекулами (например, поливиниловый спирт, полифосфаты, растворимые полисахариды, полистиролсульфокислота и т.п.), которые неспособны вследствие их большого мольного объема или высокого заряда проникать в фазу смолы (так называемый метод исключения ионов).
Величины для смол с низкой степенью поперечного связывания, которые поглощают простые (маленькие) ионы в соответствии с доннановским равновесием, легко определяют пропусканием через колонку раствора, содержащего ионы, одноименные с ионной формой обменника.
Помимо промежуточного объема К, часто используют параметр £,-, выражающий относительный промежуточный объем (или промежуточную долю)
“'К
е( — относительный промежуточный объем, К, — абсолютный промежуточный объем (см3), — объем слоя смолы (см3).
Величины £; зависят от формы и размера зерен смолы, а также от положения слоя смолы в колонке. Для гранулированных смол эта величина составляет 0,3 — 0,4, при оптимальных условиях до 0,26 (табл. 4.1 и 4.2). Специально получены смолы с размером зерен, величины £; которых доведены до 0,15. В то же время в случае дробленых частиц ионообменников значения £, могут подниматься до 0,70.
Существует ряд методов определения промежуточного объема колонки: а) прямое измерение, б) титрование, в) расчет.
Перед определением И,- необходимо установить уровень жидкости на верхней границе обменника. Предварительно должен быть определен объем жидкости, удерживаемый пористой подложкой, и объем между этой подложкой и выходным отверстием колонки. Эти два значения нужно вы-
Таблица 4.1. Относительные промежуточные объемы £, смолы Dowex 50-Х4 в растворах НС1—LiCl
Состав 1 М НС1 1 М НС1 + 1 М НС1 + 1 М НС1 + 1 М НС1 +
раствора + 2,0 М LiCl + 4,0 М LiCl + 6,0 М LiCl + 8,0 М LiCl
£,. 0,325 0,335 0,346 0,355 0,380
Обменная емкость смолы: 4,03 ммоля Н+ /г. Размер зерен: 0,044 мм.
4. Аппаратура для ионообменной хроматографии
129
Таблица 4.2. Относительные промежуточные объемы е, смол Dowex разных степеней поперечного связывания
Обменник е< Обменник e.
Dowex 5О-Х2 0,304 Dowex 1-Х2 0,351
Dowex 50-Х4 0,327 Dowex 1-Х4 0,350
Dowex 50-Х8 0,379 Dowex 1-X8 0,390
Dowex 50-Х16 0,395 Dowex 1-X16 0,396
Размер зерен: 0,037 — 0,074 мм.
читать из экспериментально найденной величины. Полученный результат равен величине К(-.
Методика А. Эта очень простая методика пригодна как для катионообменников, так и для аииоиообменииков. Раствор, содержащий непоглощаемый в данных условиях ион, медленно пропускают через обменную колонку. Измеряют объем элюата и время, когда ион появляется в элюате.
Методика Б. Эта методика, применимая к сильнокислотным катионообменни-кам, состоит в вымывании небольшого количества несорбируемого вещества из колонки. В верхнюю часть обменной колонки вводят следовое количество иодидного аниона в форме 1311 и определяют его проскок. При расчете величины И;. вводят упомянутые выше поправки.
Методика В. Эту методику применяют для сильнокислотных катионообменников. Ионообменник в Na-форме (хорошо промытый водой) переносят в колонку и промывают достаточным количеством деионизованной воды. Снижают уровень жидкости до границы поверхности смолы и медленно пропускают через колонку 0,002 М раствор полиметафосфата натрия. Объем этого раствора должен превышать объем обменника приблизительно в 3 раза. Можно использовать полиметафосфат, меченный 32Р. Уровень раствора снова снижают до границы поверхности смолы, удаляют раствор полиметафосфата из колонки и определяют его концентрацию. Зная исходную концентрацию, рассчитывают объем раствора, удерживаемый в колонке. Для аннонообменников эта методика неприменима (2].
Методика Г. Обменник превращают в Н-форму. В качестве несорбируемого Вещества используют 0,001 М раствор линейной полистирблсульфокислоты. В остальном методика аналогична методике В.
Методика Д. Эта методика пригодна для сильнокислотных катнонообменииков. Ионообменник приводят в равновесие с раствором, содержащим линейную полистиролсульфокислоту (0,001 М). (Концентрации элюента и элюата равны). Полистиролсульфокислоту, удерживаемую в колонке, удаляют промыванием последней исследуемым раствором, не содержащим поликислоту. Отдельные порции элюата собирают и анализируют на содержание полистиролсульфокислоты. Конечной считают порцию, в которой"концентрация этой кислоты превышает 0,0005 М. Вводят упомянутые выше поправки и определяют свободный объем колонки [3].
Методика Е. Методика применима для сильнокислотных катионообменников. Обменную колонку медленно промывают 0,10 М раствором соляной кислоты (до
9-354
130
Часть И
1000 см3). Затем уровень соляной кислоты снижают до границы смолы, вымывают соляную кислоту водой н определяют ее концентрацию титрованием. Найденная величина (после введения соответствующих поправок) представляет параметр Kf. В этом случае измеренная величина немного выше истинной вследствие того, что не учитывается диффузное проникание кислоты в смолу. Поэтому рекомендуется использовать только 0,1 М HCI. Вместо НС1 можно применять другие хлориды в соответствии с исходной ионной формой обменника.
Методика Ж. Методика пригодна для сильноосновных анионообменников. Обменник превращают в бромидную форму. В остальном методика аналогична методике В. В качестве неадсорбируемого вещества используют бромид полн-4-винил-М-алкилпиридиния. После удаления этого вещества из свободного пространства колонки определяют концентрацию бромид-нонов титрованием по Фольгарду.
Методика 3. Эта методика применима как для катионообменников, так и для анионообменников. Уровень жидкости (воды) в колонке, заполненной смолой, устанавливают на границе поверхности последней. Колонку взвешивают. После этого удаляют жидкость центрифугированием и снова взвешивают колонку. Величину V рассчитывают после введения обычных поправок; дополнительно из найденной величины вычитают 3% объема обменника в колонке [4].
Методика И. Промежуточный объем
Масса набухшей смолы/Объем навески смолы в колонке
V - 1------------------------------------------------(4.3)
Плотность набухшей смолы
4.1.7. Поглощение смеси ионов перед стадией разделения
Перед выполнением хроматографического разделения необходимо сорбировать смесь разделяемых ионов в верхней части колонки. Наиболее эффективное разделение достигается, когда количество разделяемых ионов невелико. Объем обменной колонки, удерживающей разделяемые ионы, должен составлять 3 — 5°7о общего объема. При разделении сложных смесей этот объем не должен превышать 1% объема колонки.
Эффективность колонки при селективном разделении может быть увеличена на — 50% при условии элюирования только одного из разделяемых ионов.
Небольшое количество пробы маленькой пипеткой наносят непосредственно на ионообменннк. Предварительно уровень жидкости в колонке снижают до уровня поверхности обменника. Определенный объем раствора просачивается через обменник под действием силы тяжести или под давлением. Стенки колонки несколько раз смывают водой. Этот простой прием неприменим, если процесс проводят при высоком давлении. В этом случае для отбора проб применяют петлю.
Вследствие значительного локального изменения pH в адсорбционной зоне колонки (особенно для Н- или ОН-форм смол) может происходить элюирование сорбированных ионов. Это явление предотвращают введением в раствор соответствующей буферной смеси.
4. Аппаратура для ионообменной хроматографии
131
4.1.8. Скорость потока
Скорость протекания жидкости в процессе хроматографического разделения выбирают таким образом, чтобы рабочие условия были близки к равновесному состоянию. В случае иехроматографических разделений, когда требование равновесии не должно строго соблюдаться, могут быть использованы более высокие скорости потока. Действительно общее правило, что повышение температуры и уменьшение размера частиц ионообмен-ника повышает скорость протекания раствора через колонку при сохранении хорошей разделяющей способности ионообменной колонки. Приблизительное представление о скоростях потоков, используемых в процессе разделения неорганических ионов, дают следуюшие данные о размере зерна (мм) и скорости потока [см3/(см2-мин)]: 0,074 — 0,088 : 1,3 ; 0,08 — 0,09 : 1,2; 0,25 — 0,30 : 0,2 — 0,4; 0,112 — 0,115 : 2,2 (100°С).
4.2. Дополнительное оборудование для ионообменной колонки
4.2.1. Система подачи раствора
Одним из основных условий успешного хроматографического разделения смеси ионов на колонке является постоянная скорость протекания жидкости через все поперечное сечение колонки.
Существует ряд методов, обеспечивающих пропорциональную подачу раствора в колонку. В случае коротких колонок, заполненных смолой с достаточно большим размером частиц (>0,10 мм), для поддержания постоянного уровня жидкости над смолой и приблизительно постоянной скорости протекания раствора используют сифон. Однако этот метод применим только для простых разделений, промывания колонки водой и т.п.
При работе с колонками большего размера или с более мелкими фракциями смол (гидродинамическое сопротивление колонки выше) постоянная скорость потока обеспечивается либо применением более высокого давления, либо использованием разных типов насосов.
Для обеспечения необходимого избыточного давления среды часто применяют газы. Газ непосредственно поступает из толстостенной склянки со сжатым воздухом через точный цилиндровый регулятор давления в резервуар с жидкостью, протекающей в колонку. Другой прием заключается в применении газгольдера, который обеспечивает избыточное давление газа на жидкость. Наиболее подходящим газом для этой цели является гелий, характеризующийся низкой растворимостью в жидкостях. Однако сжатый воздух или азот также пригодны. К недостаткам этих методов следует отнести растворимость газов в растворах, которая вызывает нарушение уплотненности ионообменной колонки, особенно при работе в режиме повышенной температуры. Поэтому непосредственного контакта сжатого газа с используемыми растворами необходимо избегать, например, с помощью слоя парафинового масла.
132
Часть II
Рис. 4.6. Перистальтический многоканальный микронасос (фирма Serva GmbH).
Таблица 4.3. Химическая устойчивость пластических материалов, используемых в перистальтических насосах
Материал Силикон Тайгон S-HL-50 Вайтон D ПЭ ПВХ Кель-F Тефлон
Ацетон + 0 — + +
Пропанол-2 — + + + — + +
Метанол 0 — 0 + + + +
Пиридин 0 + + + — + +
NH4OH конц. + + + + 0 + +
СН3СООН конц. + + 0 + 0 + +
HF 5%-ная + + + + — + +
HNOj 10%-ная 0 + + + + + +
HNOj конц. — — + — — + +
НС1 10%-ная + + + + + + +
НО конц. 0 0 + — + + +
H2SO4 — 0 . + + + + +
NaOH 50%-ный — 0 + + + + +
NaOH 10%-ный + + + + + + +
Принятые обозначения: ПЭ — полиэтилен, ПВХ — поливинилхлорид; + материал, пригодный для применения в данной среде (устойчивый в течение длительного времени), 0 — материал частично разрушается в данной среде (ограниченно устойчивый материал), — материал не рекомендуется.
4. Аппаратура для ионообменной хроматографии
133
Проблема переноса жидкости через колонку значительно упростилась в результате разработки соответствующих типов микронасосов. Приводимые в движение электромоторами миниатюрные насосы с электронным . управлением, обеспечивающим простое изменение скорости потока жидкости через колонку, получили широкое распространение, так как сохраняют постоянство любой выбранной скорости потока.
Разработаны поршневые, вращающиеся (перистальтические) и мембранные насосы. Промышленностью выпускается большое число насосов самой разнообразной конструкции (рис. 4.6).
Насосы низкого давления сконструированы в виде простых поршневых или перистальтических, достигающих давления в несколько килопаскаль. Количество прокачиваемой жидкости может непрерывно изменяться в результате хода поршня или изменения числа оборотов мотора.
Перистальтические насосы обладают некоторыми преимуществами по сравнению с поршневыми: небольшое мертвое пространство, легкая смена растворов, прокачиваемых через насос, возможность одновременного прокачивания одним насосом нескольких типов растворов из разных мест или в разные места. Однако их применение ограничено коррозионной устойчивостью используемых трубок (табл. 4.3). При скорости потока 0 — 10 см3/мин точность коммерчески доступных насосов обычно равна 2 — 3%.
4.2.2. Вспомогательное оборудование
Для соединения отдельных приборов или их частей используют трубки, сделанные из соответствующих материалов. Гибкие трубки для низких давлений изготавливают обычно из политетрафторэтилена, полиэтилена, силиконовой резины, полиуретана и поливинилхлорида. Внутренний диаметр соединительных трубок должен быть равен 2 — 3 мм или меньше во избежание изменений концентраций протекающего раствора в результате смешивания.
Хорошо известные фирмы, производящие и продающие хроматографическое оборудование, поставляют также муфты, клапаны, трубки, опорные части и т.п. детали, изготовленные из различных материалов и предназначенные специально для хроматографических исследований.
4.2.3. Фракционные коллекторы
В процессе хроматографического разделения смесей ионов часто возникает необходимость отбирать большое количество равных частей элюата. Для этих целей разработаны разные типы автоматического коммерчески доступного оборудования.
Автоматические фракционные коллекторы состоят из двух основных частей: а) установки для точной дозировки разных объемов фракций, б) управляемого устройства для замены сосудов-сборщиков, расположенных ниже колонки.
Работа установки для точной дозировки фракций основана на одном из следующих четырех параметров: время,, объем, масса, капля.
134
Часть II
Чаще всего используют дозировку посредством установления определенного интервала времени. Этот простой способ делает возможным применение любого реле времени илн системы для фиксирования соответствующего интервала времени. Вместо реле времени пригодны специально оснащенные моторы, вращающиеся с постоянным числом оборотов в минуту. Диск нли ряд сменных дисков, изготовленных из изоляционного материала, укрепляют на валу мотора. По окружности дисков размещают электрические контакты. Интервалы времени, через которые происходит замена сосудов, регулируют изменением числа оборотов мотора нли числа контактов на вращающемся валу.
Дозирование по принципу объема основано на переполнении сифона соответствующей формы дозированным фракционным объемом. Однако этот метод не рекомендуется использовать для точной работы, особенно с малыми количествами фракций, поскольку каждая фракция загрязняется небольшим количеством предыдущей фракции, оставшейся в сифоне. Современные установки и материалы конструкции обеспечивают малый объем остающейся фракции (меньше 0,3 см3).
Дозирование по принципу массы основано на том, что коллектор связан с коромыслом весов, которые отклоняются, когда определенное количество элюата вытекает в сосуд. Одновременно подается импульс к замене сосуда.
Наиболее широко распространенным и гибким является дозирование по принципу капли. Последний метод используется как при разделениях на больших колонках, так и при работе с малыми количествами веществ, разделяемых на микроколонках. Установка действует таким образом, что, когда капля жидкости капает нз устья колонки, она пересекает луч света. Этот факт регистрируется фототрубкой, обрабатывается соответствующим электронным устройством н поступает в форме электрического импульса в счетчик. После регистрации определенного (предварительно выбранного) количества капель счетчик посылает сигнал для смены сосуда.
. Помимо упомянутых выше принципов передачи сигнала для смены сосуда могут быть использованы-разнообразные свойства элюата, зависящие от его состава. Измеряют такие физические параметры элюата, как удельная электропроводность, pH, показатель преломления, радиоактивность и т.п. Сигналы подаются после достижения максимальной, минимальной или предварительно выбранной величины измеренного параметра.
Сравнение различных принципов дозирования показывает, что коллекторы, основанные на регулировании массы, имеют много недостатков (чувствительность к теплу, трудности установки весов и др.) н редко применяются. Коллекторы с регулированием времени требуют поддержания постоянной скорости потока жидкости. Этот тип коллекторов применяют довольно часто. В установках, регулирующих объем, нет необходимости в строгом постоянстве скорости потока жидкости. Вследствие некоторого, хотя н слабого, загрязнения раствором, оставшимся от предыдущей фракции, этот принцип применим только для больших объемов фракций. Счетчики капель полностью надежны при условии, что в процессе опыта не
4. Аппаратура для ионообменной хроматографии
135
Рис. 4.7 Панельный фракционный коллектор (фирма Serva GmbH).
происходит значительного изменения поверхностной активности элюата, ибо изменение этой величины влияет на объем капли.
Применяют два типа установок для смены сосудов-приемников:
а. Установка с движущимся рычагом может быть использована с фракционными коллекторами без электродвигателя. Вращение рычага осуществляется с помощью проволоки или струны, натянутой на вращающийся вал. На конце рычага (по периметру вращения) укрепляется сифон, который дозирует элюат. Сифон уравновешивается (на другом конце рычага) силой тяжести. В процессе заполнения сифона коромысло весов отклоняется н останавливается на коллекторе. После заполнения сифона жидкостью уровень последней снижается н жидкость протекает в сосуд. Коромысло весов поднимается, и рычаг движется по кругу до следующей остановки. Таким образом сифон оказывается над следующим сосудом.
б. Наиболее широко применяемым типом коллектора является вращающийся круглый стол с соответствующими сосудамн-приемниками (пробирками, колбами и т.п.) Стол приводится в движение с помощью маленького электрического мотора, управляемого импульсом от регулирующего устройства.
136
Часть II
Кроме круглого стола с сосудами по периметру круга применяют также вертикальные коллекторы, в которых сосуды размещены по спирали, по прямой линии или по сегментам.
Существует множество разных типов коллекторов. Эти коллекторы представляют собой один блок с различными приспособлениями. Их действие основано на упомянутых выше принципах. Интересный тип, производимый фирмой Serva-Technik (Гейдельберг, ФРГ), показан на рис. 4.7. На этом приборе одновременно можно получить до десяти разных хроматограмм. Установка состоит из одного блока и очень хорошо оснащена; возможна связь с программирующим устройством.
Подобные коллекторы выпускаются рядом других известных фирм.
ОБОРУДОВАНИЕ ФРАКЦИОННЫХ КОЛЛЕКТОРОВ
Полная автоматизация хроматографических процессов требует дополнительного оборудования фракционных коллекторов. Так, например, необходимы многочисленные клапаны, которые автоматически (будучи заранее запрограммированы) изменяют тип раствора, питающего колонку. Кроме того, требуется устройство для переноса растворов из разных мест в один резервуар.
Имеется в продаже оборудование, обеспечивающее отбор только тех фракций элюата, которые содержат разделенные вещества. Установка работает на принципе изменения соответствующего свойства элюата. Обнаруженное изменение дает импулье для автоматической смены сосуда. Схема одного из коммерчески доступных устройств показана на рис. 4.8.
Фракционный отбор пикоВ б несколько сосудов - приемников
Фракционный отбор
Рис. 4.8. Схема установки, использующей детектор пиков, с фракционным коллектором (фирма Gilson France).
I — ионообменная колонка; 2 — двухстворчатый клапан; 3 — фракционный коллектор; 4 — детектор; 5 — детектор пиков.
4. Аппаратура для ионообменной хроматографии
137
4.2.4. Установки градиентного элюирования
Простая, вполне удовлетворительная аппаратура для градиентного элюирования может быть легко собрана из обычных лабораторных сосудов.
а. Метод экспоненциального концентрационного градиента, использующий смеситель постоянного объема: схема установки показана на рис, 4.9. Элюирующий раствор соответствующей концентрации протекает из резервуара А в смеситель В, который полностью заполнен «разбавителем». Элюирующий раствор смешивается с разбавителем и течет в колонку. Получают градиентную кривую выпуклой формы. Этот метод часто применяют даже в тех случаях, когда получаемый градиент неоптимален для методики разделения. Если два последовательных смесителя наполняют разбавителем (рис. 4.10), то получают градиентную кривую, близкую к линейной.
б. Метод пропорционального концентрационного градиента, использующий сообщающиеся сосуды: резервуар А и смеситель В (рис. 4.11) представляют собой открытые сообщающиеся сосуды. Резервуар А содержит Элюирующий раствор соответствующей концентрации, смеситель В — разбавитель. В процессе элюирования уровни жидкости в обоих сосудах уменьшаются с одной и той же скоростью. В то же время жидкости смешиваются в разных, но постоянных пропорциях в зависимости от отношения поперечных сечений используемых сосудов. Это отношение определяет форму градиентной кривой. В случае равенства поперечных сечений (А = В) градиент линейный. При А < В градиентная кривая вогнутая, при А > В — выпуклая.
Рис. 4.9. Схема устройства для получения градиентной кривой выпуклой формы с использованием смесителя постоянного объема.
А — подача элюирующего раствора из резервуара; В — смеситель постоянного объема Ро , заполненный разбавителем; М — магнитная мешалка; К — ионообменная колонка.
Рис. 4.10. Схема устройства для получения градиентных кривых разного типа с двумя смесителями постоянного объема.
А — подача элюирующего раствора из резервуара; В [ и Вг — смесители постоянного объема и М — магнитная мешалка; К — ионообменная колонка.
138
Часть 11
Рис. 4.11. Схема устройства для получения градиентных кривых разного типа.
А — резервуар с элюирующим раствором объема В — смеситель объема М—магнитная мешалка; К — ионообменная колонка.
в. Метод, использующий непропорциональное смешивание двух жидкостей. В этом методе получают градиентные кривые разного типа. Последние зависят от профиля сосудов А и В, которые функционируют в качестве резервуаров элюирующего агента и растворителя соответственно. Жидкости из этих сосудов протекают в смеситель.
Помимо этих простых устройств имеется ряд коммерчески доступных приборов, обеспечивающих получение любого типа градиента. Приборы полностью автоматические. На рис. 4.12 показан прибор фирмы ISCO типа Dialagrad Model 190 для получения разных типов градиентов, образуемых
Рис. 4.12. Установка с программным управлением для получения требуемого градиента Dialagrad Туре 382 (фирма ISCO Inc).
4. Аппаратура для ионообменной хроматографии
139
5
Рис. 4.13. Схема автоматического устройства для элюирования с использованием требуемого градиента (фирма Gilson—France).
1 — колонка; 2--насос; 3 — смесительная камера; 4 — трехходовой тефлоновый вентиль;
5 — элюирующие растворы А и В; 6 — фракционный коллектор; 7 — дифференциальный спектрофотометр с регистрирующим устройством; 8 — электронное профильное устройство;
9 — фотоэлектрический счетчик показаний максимума; 10 — профиль градиента.
при смешивании двух компонентов. Прибор снабжен программным управлением. Время образования градиента колеблется от 10 мин до 12 сут. Насос может прокачивать жидкость со скоростью 0,5 — 500 см3/ч. Программа также предусматривает регенерацию насадки колонки выбранным агентом тотчас после окончания элюирования.
Другая коммерчески доступная установка для получения градиента схематически показана на рис. 4.13. Электронное устройство контролирует вычерчиваемый профиль градиента и дает инструкцию относительно его изменения. Одновременно контролируют концентрацию вымываемого вещества в элюате. Если концентрация вымываемого вещества превышает предварительно выбранную величину, устройство сохраняет постоянный состав элюирующег раствора. Если концентрация вещества становится ниже установленного предела, устройство автоматически регулирует градиент элюируюшего раствора в соответствии с предварительно выбранной величиной.
4.3. Непрерывный анализ элюата
Элюат можно анализировать периодически, т.е. проводить анализ отдельных фракций различными методами. Полная автоматизация процесса разделения требует непрерывного анализа элюата. Чаще всего используют физические или физико-химические методы анализа.
Установка для непрерывного анализа состоит из трех частей: соответствующего детектора, расчетной части и регистрирующего устройства.
Система детектирования должна обладать высокой чувствительностью (отношение отклика детектора к количеству образца) и низким фоновым откликом. Вообще вещество обнаруживается, если сигнал отклика детектора по крайней мере в два раза превышает фоновый.
140
Часть II
С точки зрения количественного анализа отклик системы обнаружения должен быть прямо пропорционален концентрации. Эта зависимость выражается следующим простым соотношением:
R = к • с (4.4)
где R — отклик детектора, к — коэффициент пропорциональности, с — концентрация растворенного вещества в подвижной фазе.
Уравнение (4.4) относится к идеальному состоянию; в реальной системе соотношение между откликом детектора и концентрацией вещества в подвижной фазе лучше выражается уравнением
R = к • сх (4.5)
где х характеризует отклонение от линейности.
Отклик детектора считается линейным, если величина показателя степени х лежит в области 0,98 — 1,02.
Важным параметром непрерывного анализа элюата является объем измерительной ячейки, который в современных анализаторах не превышает нескольких микролитров. Однако в серийных анализах неорганических веществ чаше всего применяют измерительные ячейки объемом около 0,1 см3.
• Подробное рассмотрение расчетных и регистрирующих устройств относится к области электроники.
В непрерывном анализе неорганических веществ преимущественно применяют полярографический, спектрофотометрический и радиометрический
Электролит Элюат
| [5мм на
Капающий ртутный электро^ Азот
С 1,5 см НД
Стеклянные бусинки диаметром Змм Измельченное стекло
Рис. 4.14. Схема устройства для количествеииого полярографического определения концентрации ионов в элюате.
НД — наружный диаметр; ВД — внутренний диаметр.
4. Аппаратура для ионообменной хроматографии
141
методы. Иногда используют методы, основанные на измерении электропроводности, ЭДС (в отдельных случаях с селективным электродом), изменения диэлектрической проницаемости элюата, хемилюминесценции и др. [5 - 16].
4.3.1. Полярографические методы
Принцип. При постоянном напряжении регистрируют зависимость диффузного тока от времени (время прямо пропорционально объему элюата). Площадь, ограниченная полученной кривой и осями координат, прямо пропорциональна концентрации вещества в элюате. Пример удовлетворительной установки показан на рис. 4.14.
4.3.2. Спектрофотометрические методы
В анализе неорганических ионов применяют также спектрофотометрические методы, которые обеспечены коммерчески доступными приборами
Рис. 4.15. Схематическая диаграмма жидкостного хроматографа со спектрофотометрическим детектором [19].
1 — резервуар с отработанной жидкостью; 2 — реометр; 3 — спектрофотометрический детектор; 4 — регистрирующее устройство; 5 — смесительная камера; 6 — колонка; 7 — шприц для введения образца; 8 — клапан для инжекции образца; 9 — ротационный переключающийся вентиль; 10— резервуары с элюентом; 11 •- реагенты, образующие окрашенные соединения;
12 — игольчатый клапан; 13 — трехходовой кран; 14 — резервуар с гелием; 15 — регулятор подачи гелия; 16 — микрометрический вентиль.
142
Часть П
Рис. 4.16. Простое устройство для непрерывного измерения гамма-излучения элюата.
1 _ трубка Гейгера-Мюллера; 2 — стеклянный змеевик.
для измерения в видимой и УФ-области. Для проведении соответствующей реакции раствор подходящего окращенного реагента вводят в анализируе-- мый раствор, который контролируют [9, 17, 18, 19]. Схема жидкостного хроматографа с спектрофотометрическим детектором приведена на рис. 4.15.
4.3.3. Радиометрические методы
Для качественного и количественного определения выбранного компонента в элюате измеряют его радиоактивность. Тип детектора зависит от вида регистрируемого излучения. Для измерения бета- и гамма-излучения пригодны ионизационная камера и сцинтилляционный детектор. Наиболее простая установка представляет собой трубку Гейгера — Мюллера, которая находится внутри тонкостенного стеклянного змеевика (рис. 4.16). Подобный метод (трубка изготовлена из пластмассы вместо стекла) может быть использован для измерения достаточно жесткого бета-изг^чения. Материал трубки препятствует адсорбции радиоактивных изотопов на стенках трубки.
Другой тип проточного детектора, который пригоден для измерения слабого бета-излучения, показан на рис. 4.17. Учитывая низкую проникающую способность бета-излучения, канал проточной камеры покрывают тонкой пленкой пластика или тонким слоем слюды. Проточную камеру помещают ниже окна трубки Гейгера — Мюллера и весь аппарат экранируют
Рис. 4.17. Проточное устройство для непрерывного измерения бета-радиоактивности нонов в элюате.
а — плексигласовый блок с отверстием; б — плексигласовый блок с просверленным каналом.
4. Аппаратура для ионообменной хроматографии
143
Рис. 4.18. Расположение приборов для отделения радиоактивных ионов на ионообменной колонке и для непрерывного анализа элюата.
А — резервуар с элюируюшим раствором; Б — ионообменная колонка; В — реометр; Г — трубка Гейгера-Мюллера; Д — свинцовый экран.
Регистрирующее устройство
свинцовой оболочкой. Проточная камера должна быть сконструирована таким образом, чтобы ее выступающая часть была по возможности небольшого размера (для работы с микроколичествами меньше 0,5 см3).
Материал камеры не должен адсорбировать радиоизотопы из раствора. В некоторых случаях проточную камеру изготавливают из пластмассы.
Схема аппаратуры для непрерывных измерений показана на рис. 4.18.
Существует другой метод непрерывного измерения. Элюат (или его фракция) по каплям падает на медленно движущуюся полосу, сделанную из алюминия или специальной бумаги. После испарения жидкости измеряют индивидуальные фракции элюата.
Для измерения больших количеств элюата сконструированы различные типы коммерчески доступных трубок Гейгера — Мюллера.
ИЗМЕРЕНИЕ РАСПРЕДЕЛЕНИЯ РАДИОАКТИВНОСТИ ВДОЛЬ КОЛОНКИ
При изучении процесса разделения ионов непосредственно на колонке контролируют распределение ионов вдоль колонки с помощью соответствующего радиоактивного индикатора. Трубку Гейгера — Мюллера или сцинтилляционный детектор помещают в свинцовую оболочку. На передней стенке экрана вырезают узкую щель. Детектор перемещают вдоль колонки вручную или автоматически с помощью электромотора и регистрируют распределение радиоактивных изотопов вдоль колонки.
144
Часть И
Литература
1. Larsson К., Samuelson О., J. Chromatogr., 134 (1977) 191.
2. Manolo G.P., Turse R., Rieman V. M. Ill, Anal. Chim. Acta 21 (1959) 383.
3. Jakob F, Park К. C., CiricJ., Rieman V. M., Ill, Taianta, 8 (1961) 431.
4. Kraus K. A., Moore G. E., J. Amer. Chem. Soc., 75 (1953) 1457.
5. Neary M. P., Seitz R., Hercules D. M., Anal. Lett., 7 (1974) 583.
6. Van Urk-Schoen A. M., Huber J. P. K., Anal. Chim. Acta, 52 (1970) 519.
7. McGuinness E. T., Cullen M. C., Chem. Educ., 47 (1970) 19.
8. Gilbert T. W., Dobbs R. A., Anal. Chem., 45 (1973) 1390.
9. Seymour M. D., Fritz J- S., Anal. Chem., 45 (1973) 1394.
10. Small H., Stevens T. S., Bauman JK C., Anal. Chem., 47 (1975) 1801.
11. Nomura T., Nakagawa G., Bunseki Kagaku, 20 (1971) 1570.
12. ArakiS., SuzukiS., Yamada M., Taianta, 19 (1972) 577.
13. Tanaka Y., Fujita L., J. Chromatogr., 108 (1975) 255.
14. Takata Y., Muto G., Anal. Chem., 45 (1973) 1864.
15. Takata Y., Japan Analyst, 24 (1975) 531.
16. Takata Y., Arikawa Y., Japan Analyst, 24 (1975) 762.
17. KawazuK., Fritz J. S., J. Chromatogr., 77 (1973) 387.
18. Fritz J. S., Stoy J. N., Anal. Chem., 46 (1974) 825.
19. King J. N., Fritz J. S., J. Chromatogr., 153 (1978) 507.
5. Применение ионообменников
В настоящее время в продаже имеется большое количество ионообменников с различными функциональными группами и разными свойствами. Многие из них производятся в промышленном масштабе, другие получают только в лабораторных условиях.
Ионообменники нашли широкое применение в аналитической химии для выделения и концентрирования следов элементов, удаления мешающих компонентов из анализируемого раствора, полного удаления ионов деионизацией, определения общего солесодержания, очистки и получения многих реактивов или растворов, качественного определения элементов и для решения многих других задач.
Одна из наиболее важных и широко распространенных областей применения ионообменников связана с разделением смесей ионов. Даже очень сложные смеси ионов, присутствующих в анализируемых растворах, можно разделить на индивидуальные компоненты методом ионообменной хроматографии с использованием соответствующей среды (водной, водноорганической или неводной) и подходящих элюентов. Очень часто эти разделения выполняются быстрее и с большей точностью, чем другими методами.
5.1. Предварительное концентрирование и выделение следов
В настоящее время наблюдается непрерывный рост потребности в проведении количественного определения чрезвычайно низких концентраций элементов в анализируемых материалах, особенно в связи с необходимостью анализа объектов окружающей среды, биохимических проб, полупроводников, внеземных материалов, металлов высокой чистоты и других. Часто требуется проводить количественные определения разных компонентов, содержащихся в количестве порядка 10"9 г или меньше.
Количественное определение таких низких концентраций элементов современными инструментальными методами ограничено пределами чувствительности этих методов. Для определения элементов с достаточной чувст-
10-354
146
Часть П
вительностью и требуемой точностью необходимо предварительное концентрирование следов элементов, отделение их от макроколичеств мешающих элементов или разделение анализируемой смеси на группы элементов, которые не мешают определению друг друга.
Предварительное концентрирование следов элементов осуществляют разнообразными методами. Наряду с экстракцией, осаждением, дистилляцией, сублимацией и электролизом широко распространены методы, основанные на использовании ионообменников.
Ионообменное концентрирование имеет ряд преимуществ по сравнению с другими методами концентрирования. Оно отличается простотой исполнения и возможностью полного, группового или индивидуального элюирования элементов, сорбированных ионообменником, с помощью соответствующих элюентов. Однако во многих случаях нет необходимости элюировать сорбированные элементы — смолу озоляют и определяют исследуемые элементы в золе. Можно также определять элементы непосредственно в смоле соответствующим инструментальным методом.
Разработаны методы предварительного концентрирования элементов, основанные на сорбции микро- или макрокомпонентов анализируемого образца. Одновременно можно выполнить хроматографическое разделение и концентрирование элементов.
В методиках предварительного концентрирования используют органические и неорганические ионообменники.
Особое положение занимают так называемые селективные ионообменники. Их функциональные группы проявляют (при определенных условиях) высокое сродство только к некоторым элементам, что обусловливает возможность эффективного предварительного концентрирования выбранного элемента (или группы элементов) даже из весьма сложных смесей различных ионов.
Для предварительного концентрирования элементов предложены новые типы сорбентов, которые можно рассматривать как гибриды неорганических сорбентов и синтетических хелатных смол. Их получают из неорганических сорбентов, чаще всего силикагелей, «прививкой» соответствующих функциональных групп [1—3].
Используют также зерненые формы ионообменников, ионообменную бумагу или мембраны. Круги бумаги, на которую нанесены высокоселективные ионообменные смолы, или ионообменные мембраны служат для концентрирования и разделения следов элементов в форме, являющейся оптимальной для проведения рентгено-спектроскопического и нейтронноактивационного анализа [4—6].
Применение ионообменников для предварительного концентрирования облегчает отбор представительной пробы на месте. Отпадает необходимость транспортировать большие объемы жидких проб от места отбора до лаборатории [7, 8].
В ультраследовом анализе использование ионообменников ограничено по двум причинам: а) загрязнением элюата смолой (следы металлов или органического материала из смолы); б) необратимой адсорбцией небольших количеств материала.
5. Применение ионообменников
147
Загрязнение элюата устраняют тщательной очисткой сорбентов. При неполном выделении сорбированных ионов смолу озоляют (на воздухе или в кислородной плазме [9, 10]) и определяют следы элементов в золе. Другой путь — определение следов элементов непосредственно в смоле соответствующим инструментальным методом (рентгеновская флуоресценция, нейтронно-активационный анализ и т. п.).
Во избежание внесения дополнительных загрязнений в анализируемый материал большое внимание уделяют чистоте используемых реактивов [11].
Важно знать форму, в которой присутствуют концентрируемые элементы. При концентрировании ионов, склонных к полимеризации в разбавленных растворах [например, Fe(III), Zr(IV), Nb(V)], в анализируемом растворе устанавливают определенную кислотность. Нецелесообразно, например, использовать ионообменники для предварительного концентрирования на-нограммовых количеств железа из ультрачистой воды, в которой оно образует коллоиды [12].
Аналогично при концентрировании радиоактивных изотопов без носителя большое внимание необходимо уделять физическому и химическому состоянию микроэлементов, присутствующих в растворах [13].
Сорбцию следов элементов рекомендуется проводить при более низких значениях pH или после соответствующей обработки раствора, которая обеспечивает переведение ионов в хорошо известную и легко сорбируемую форму (например, путем добавления восстановителей, окислителей или комплексообразующих агентов).
5.1.1. Природные воды
Концентрации следов элементов в природных водах, как правило, слишком низки для их прямого определения. Стадия, соответствующего предварительного концентрирования, или обогащения, неизбежна даже при использовании чувствительного метода обнаружения. Для проведения предварительного концентрирования элементов из природных вод ионообменники являются наиболее перспективными, так как желаемой эффективности анализа достигают пропусканием через ионообменную колонку практически неограниченных объемов.
При анализе вод с низким солесодержанием используют сильнокислотные или сильноосновные ионообменники. В анализируемых пробах устанавливают pH, при котором концентрируемые элементы находятся в форме, сорбируемой на ионообменнике. Такой же эффект наблюдается при введении соответствующих комплексообразующих веществ.
В связи с изучением окружающей среды и контролем за распределением следов элементов в природных водах (особенно нанограммовых концентраций Pb, Си, Zn и Cd) Коркиш и Сорио [14] предложили использовать для предварительного концентрирования Cd, Си и РЬ сильноосновный анионообменник Dowex 1-Х8. Пробу воды подкисляют бромистоводородной кис
148
Часть II
лотой и вводят аскорбиновую кислоту. Бромидные комплексы указанных элементов сорбируются ионообменником; для их элюирования используют 1 М азотную кислоту.
Сорбция этих элементов из очень разбавленных водных растворов подробно изучена советскими авторами [15].
Описан метод флуориметрического и спектрофотометрического определения ураиа в пробах природных несоленых вод [16]. После подкисления соляной кислотой пробу отфильтровывают, вводят аскорбиновую кислоту и KSCN (10 г/дм3) и пропускают через колонку, заполненную смолой Dowex 1 в SCN-форме. Урай сорбируется в форме тиоцианатного комплекса. После удаления железа и других сопутствующих элементов промыванием колонки смесью органический растворитель — НС1 (50 об. % тетрагид-рофураиа, 40 об. % метиленгликоля и 10 об. % 6 М НО) и затем одним б«М раствором НС1; уран элюируют 1 М НО.
Определение ураиа в элюате выполняют флуориметрическим или спектрофотометрическим методом с реагентом арсеназо III. Эта методика пригодна для серийных определений ураиа в пробах воды.
Аналогичный способ применяют при определении наиограммовых количеств кобальта в природных водах. Кобальт элюируют из обменника 6 М соляной кислотой и определяют спектрофотометрически с нитрозо-R-солью [17].
Этот метод пригоден также для предварительного концентрирования кадмия [18].
Известно, что хром(У1) представляет серьезную опасность для здоровья людей, но до сих пор его роли в загрязнении природных вод уделяется мало внимания. Поскольку хром содержится в водах в очень низких концентрациях (общее содержание Сг в незагрязненных водах составляет 0—50 мкг/л, в морской воде — 0,04 мкг/л) [19], перед его определением необходима стадия предварительного концентрирования [20, 21]. Лучшим методом определения является нейтронно-активационный анализ.
Ионообменники успешно применяют для предварительного концентрирования радия в родниковых водах [22], 2301 232Th [23] и следов цинка [24] в природных водах. На ионообменниках можно также сконцентрировать следы серебра в пробах дождевой воды, полученной из облаков, засеянных с помощью иодида серебра [25].
Для концентрирования и отделения ртути [26—28] используют как катиоиообменники, так и анионообменники. Последующее количественное определение ртути выполняют нейтронно-активационным методом. На сильноосновных аиионообменниках ртуть обычно сорбируется в форме комплекса HgCl2-; элюирование, как правило, затруднено. В качестве наиболее подходящего элюирующего агента рекомендуется тиомочевина. После элюирования ртуть осаждают сульфидом [29].
Метод Эмаиа и Хьюзенга [30], усовершенствованный Бовеном и др. [31], основан на облучении пробы тепловыми нейтронами и последующем концентрировании ртути на аиионообменнике в виде комплекса HgCl2-. Количество радиоактивного изотопа измеряют непосредственно на обмен
5. Применение ионообменников
149
нике. В этом методе устранена недостоверность, связанная с элюированием HgCl^~ из смолы. Он пригоден для определения 200 мкг/мл — 10 мкг/л ртути.
При одновременном определении Си, Zn, Cd и Hg в биологическом материале анионообменники позволяют проводить предварительное концентрирование и отделение ультрамалых количеств ртути (порядка 0,5 нанограмма).
Таблица 5.1. Данные по сорбции и элюированию (20 см3 элюента) следов элементов из морской воды смолой Chelex 100 [34]
Элемент pH сорбции Сорбция, %' Элюент Обшее выделение, %
Алюминий 7,6 0 0
Барий 5,0а 25 2 М HNOj 25
Ванадий (VO,’) 6,0а 100 4 М NH4OH 100
Висмут 9,0а 100 2 М НС1О4 100
Вольфрам (WO2-) 6,0а 100 4 М NH4OH 100
Индий 9,0а 100 2 М HNO3 100
Иттрий 9,0а 100 2 М HNO3 100
Кадмий 7,6 100 2 М HNOj 100
Кобальт 7,6 100 2 М НС1 100
Марганец 9,0а 100 2 М HNO3 100
Медь 7,6 100 2 М HNO3 100
Молибден (МоО2~) 5,0а 100 4 М NH4OH 100
Мышьяк (AsO3-) 7,6 0 0
Никель 7,6 100 2 М HNO3 100
Олово (Sn4+) 7,6 85 2 М HNOj 60б
Рений (ReO~) 7,6 90 4 М NH4OH 90
Ртуть (Hg2+) 7,6 85 2 М HNOj 40®
Свинец 7,6 100 2 М HNOj 100
Селен (SeO2-) 7,6 0 — 0
Серебро 7,6 100 2 М HNOj 90б
Скандий 7,6 100 2 М HNOj 100
Тадлий (Т1+) 7,6а 50 2 М HNOj 50
Торий 7,6 100 2 М H2SO4 100
Уран (ЦО2+) 7,6 0 — 0
Фосфор (РО3-) 7,6 0 — 0
Хром (Сг3+) 5,0а 25 2 М HNOj 10б
Цезий 7,6 0 — 0
Церий (Се3+) 9,0а 100 2 М HNOj 100
Цинк 7,6 100 2 М HNOj 100
а Оптимальная величина pH.
6 Максимальное количество вещества (в %), удаляемого из смолы. Размер зерна обменника составляет 0,149 — 0,297 мм.
150
Часть II
Хелатную смолу, содержащую иминодиацетатные группы, успешно применяют для предварительного концентрирования 3—2000 мкг/л Си, Zn, Pb, Cd и Мп из солоноватых и прибрежных вод и вод горячих источников. В пробах воды устанавливают pH 6—8. Элементы, удерживаемые обменником, элюируют 1 М азотной кислотой и затем определяют атомноабсорбционным методом [32].
Для предварительного концентрирования следов металлов из природных вод (Zn, Cd, Си, Pb, Со, Мп, Ni, V, Мо) часто используют смолу Che-lex 100 [33—37]. Наиболее полное извлечение металлов наблюдается в области pH природных вод, т. е. при pH 6—8 (табл. 5.1), где коэффициенты распределения Dg изучаемых металлов имеют величину порядка 105 [38].
Смола Chelex 100 пригодна также для предварительного концентрирования редкоземельных элементов и тяжелых металлов из речной воды. По-
Таблица 5.2. Влияние поверхностно-активных веществ на степень выделения металлов смолой Chelex 100 [40]
Поверхностно-активное вещество Концентрация, мг7дм3 Выделение металлов, %а
Zn Cd Си Ni Мп Со РЬ
Не введено 100 100 100 100 100 100 100
Катионный детергент6 100 98 95 98 100 97 100 94
Анионный детергент’ 100 94 95 98 97 97 98 95
Анионный детергент 10 98 98 —, — 99 — 97
Неионный детергентг 100 95 96 98 97 99 96 97
Промышленный детер-
гентд (типа LAS) 100 95 96 98 97 99 96 97
Детергент' известного
состава 500 95 95 92 99 97 95 96
Стиральный порошок* 500 94 95 93 98 90 99 98
Пирофосфат натрия 100 98 95 97 99 97 97 97
Триполифосфат натрия 10 98 97 — — 97 — —
20 96 96 — — 97 — —
100 96 95 100 100 97 97 97
Мыло 20 — — — — 78 80 —
НТА 10 37 70 95 4 91 55 48
НТА 100 32 58 90 4 91 50 46
а Из 200 см3 пробы. Концентрация введенных металлов,мг/дм3: 0,5 для Zn и Cd; 1,0 для Си, Ni, Со и Мп; 3,0 для РЬ.
6 Цетилдиметилбензиламмонийхлорид (100% активного материала).
* Линейный алкилированный сульфонат (LAS) (100% активного материала).
г Нонилфенолзтоксилат (100% активного материала).
д 74% детергента типа LAS и 6% трнполифосфата натрия.
’ Сульфированный алкилбензильный, нейтрализованный этвноламином кокосового масла (13% активного материала).
ж Продукт, содержащий 15% детергента типа LAS и 25% трнполифосфата натрия.
5. Применение ионообменников
151
следующее определение элементов выполняют нейтронно-активационным методом [39].
Методика. Для предварительного концентрирования указанных элементов из проб воды объемом 500 см3 используют 1,00 г набухшей смолы Chelex 100 в МН4-форме (pH 5—7). После промывания деионизованной водой и 0,1 М раствором ацетата аммония (для удаления Na+) смолу помешают в полиэтиленовый пакет, сушат при 75 °C, затем пакет запаивают и облучают тепловыми иейтроиами. После облучения смолу переносят в колонку, заполиеииую смолой Dowex А-1 в МН4-форме (высота слоя несколько сантиметров), обмеииик промывают 50 см3 0,1 М раствора KNO3 (для удаления Na+ , Вг~ , С1“) и элюируют редкоземельные элементы 100 см3 горячего 1 М раствора Na2CO3; тяжелые металлы вымывают 100 см3 2 М азотной кислоты.
Во многих природных водах присутствуют разнообразные катионные, анионные и неионные детергенты, стиральные порошки и триполифосфаты. Эти вещества могут влиять на сорбцию металлов смолой Chelex 100. Пакалус и др. [40] изучили влияние подобных соединений на концентрирование металлов смолой Chelex 100. Они показали, что выделение металлов превышает 95% даже при адсорбции на этой смоле поверхностно-активных веществ. Лишь в присутствии мыла или нитрилотриуксусной кислоты (НТА) степень сорбции металлов существенно снижается (> 20%). Это видно из данных табл. 5.2.
5.1.2. Сильносоленые воды и другие материалы
Морская вода представляет собой типичный пример сильносоленой воды, содержащей разнообразные элементы в следовых количествах. Для концентрирования элементов из морской воды часто используют смолы Chelex 100 и Dowex А-1 [41—43]. Наиболее подробно изучено поведение Cd, Pb, Zn, Со, Си, Fe, Мп, Ni, U, V и Au. Эти элементы чаще всего определяют атомно-абсорбционным, рентгено-спектрометрическим и иейт-ронно-активационным методами [44—48].
Согласно исследованиям Лейдена и др. [49], смола Chelex 100, используемая для предварительного концентрирования следов элементов из морской воды, имеет ряд недостатков. Наиболее существенным из них является почти одинаковое сродство смолы к щелочноземельным металлам и цинку. Кроме того, смола содержит противоионы, главным образом натрий. После обработки смолы морской водой (если применять нейтронноактивационный анализ) натрий необходимо удалить обменом на’ катион типа иона аммония.
Поэтому были изучены другие смолы. Смола, полученная из тетраэтиленпентамина и толуолдиизоцианата, проявляет гораздо более высокое сродство к переходным элементам, чем к щелочноземельным металлам [49]. Для тяжелых металлов предложена дитиокарбаматная смола [50]. Описана смола, содержащая гидразидные группы [51].
Из неорганических ионообменников для предварительного концентриро
152
Часть II
вания урана из морской воды успешно использовали гидрат ТЮ2 [52]. Даши и Састри сообщают о возможности его применения также для выделения и концентрирования 95Zr, 106Ru, 65Zn и 59Fe из морской воды [53].
Для концентрирования микрограммовых количеств благородных металлов из кислых растворов использовали новую селективную обменную смолу на основе 3(5)-метилпиразолона [54-56]. Концентрированию не мешают 1—2 мг/см3 Си, > 10 мг/см3 Fe, >50 мг/см3 Со, Ni и А1 и другие элементы. Эта смола пригодна для анализа благородных металлов и различных руд.
Другие примеры использования ионообменников для предварительного концентрирования и выделения следов элементов приведены в табл. 5.3.
Таблица 5.3. Предварительное концентрирование следов элементов методом иовного обмена
Элемент Среда Метод определения Обменник Литература
и Морская вода AM-10 57
In Морская вода Н.а.а. Dowex А-1 58
Ag Морская вода Н.а.а. Deacidite FF-IP 59
Hg Морская вода Бумага Amberlite IRA-400 60
Zn, Ca, Mg, Sr Водопроводная вода X Aminex А-27 61
Различные элементы Вода X Обменники на основе целлюлозы 62
Си Дистиллированная вода Н.а.а. 63
и Природные воды Н.а.а. 64
Различные элементы Вода X Ионообменная бумага 65
и Вода Н.а.а. 66
и Грунтовые воды X Chelex 100 67
А1 Кости Н.а.а. AG 50 W-X8 68
Au Си и Cd Н.а.а. Dowex 1-Х8 69
Pb Лунные образцы Анионообменник 70
РЬ Цирконы Анионообменник 71
РЗЭ Чистый иттрий КУ-2 72
Мп Металлические тантал, ниобий Спектрофотометрический Amberlite CG-120 73
РЬ Образцы стали А.а.с. Dowex 1-Х8 74
Fe Фосфаты Amberlite IRA-400 75
Принятые обозначения', н.а.а. — нейтронно-активационный анализ; а.а.с. — атомно-абсорбционная спектрофотометрия; X — рентгеновская спектрометрия.
5. Применение ионообменников
153
Литература
1. Leyden D. Е., Nutrell G. Н„ Anal. Chem. 47 (1975) 1612.
2. Leyden D. E., Nutrell G. H., Nonidel W. K., Werho D. B., Anal. Chem., 48 (1976) 67.
3. Leyden D. E., Nutrell G. H., Sloan A. E., De Angelis N. J., Anal. Chim. Acta, 84 (1976) 97.
4. Grenn T. E., Law S. L., Campbell W. J., Anal. Chem., 42 (1970) 1749.
5. Eisner U„ Mark H. B., Jr., Taianta, 16 (1969) 27.
6. Mark H. B., Jr., Eisner U., Rottschafer J. M., Berlandini F. J., Mattson J. S.., Environ. Sci. Technol., 3 (1969) 165.
7. Parslow G. R., Dwari I., J. Geochem. Explor., 8 (1977) 541.
8. Shuman M. S., Dempsey J. H., J. Water. Pollut. Control. Fed., 49 (1977) 2000.
9. Brooks R. R., Analyst, 85 (1960) 745.
10. Vitkovd J., Jambor J., Vrchlabsky M., Chem. listy, 72 (1978) 417.
11. Chalmers R. A., Pure Appl. Chem., 31 (1972) 569.
12. Hoffmeister W., Z. anal. Chem., 290 (1978) 289.
13. Kepdk F, in: Radiotracer Techniques and Applications. Eds. E. E. Evans, M. Muramatsu, Vol. 1, Marcel Dekker, N. Y., 1977, p. 339.
14. Korkisch J., Sorio A., Anal. Chim. Acta, 76 (1975) 393.
15. Чупахин M. С., Разяпов A. 3., Макарова С. Б., Дорофеев В. С. Ж. аналит. химии, 30 (1975) 2437.
16. Korkisch J., GodlL., Anal. Chim. Acta, 71 (1974) 113.
17. Korkisch J., Dimitriadis D., Taianta, 20 (1973) 1287.
18. Korkisch J., Gbdl L., Taianta, 21 (1974) 1035.
19. Chnecas I., Riley J. P„ Anal. Chim. Acta, 35 (1970) 240.
20. Pankow J. F., Janauer G. E„ Anal. Chim. Acta, 69 (1974) 97.
21. Lima F. W., Silva С. M., J. Radioanal. Chem., 1 (1968) 147.
22. Padmanabhan P. K., Joshi L. U., Venkateswarlu Ch., Ganguly A. K., J. Radioanal. Chem., 44 (1978) 49.
23. Meyer H. G., Born H. J., Stark H., Turkowsky C., Radiochim. Acta, 10 (1968) 128.
24. Korkisch J., GodlL., Gross H., Taianta, 22 (1975) 280.
25. Warburton J. A., Young L. G., J. Appl. Meteorol., 7 (1968) 433.
26. Kato K., Kakihana FL, Nippon Kagaku Zasshi, 84 (1963) 804.
27. Kuroda R., Kiriyama T., Ishida K., Anal. Chim. Acta, 40 (1968) 305.
28. Hilgeman F., Shimomura K., Walton W., Separ. Sci., 4 (1969) 111.
29. Marowski G., Z. anal. Chem., 253 (1971) 267.
30. Ehmann W. D., Huizenga J. R., Geochim. Cosmochim. Acta, 17 (1959) 125.
31. Bowen H. M., Gibbons D. A., Radioactive Analysis, Blackwell, Oxford (1963).
32. Satake M., Asano T., Ito A., Nagoasa Y„ Fukui Daigaku Kogakuku Kenkyu Hokoku, 24 (1976) 65. C. A. 85 (1976) 166393.
33. Lee C., Kim N. B., Lee I. C., Chung K. S., Taianta, 24 (1977) 241.
34. Riley J. P„ Taylor D., Anal. Chim. Acta, 40 (1968) 479.
154
Часть II
35. Riley J. P., Taylor D., Deep Sea Res., 19 (1972) 307.
36. Riley J. P., Taylor D„ Anal. Chim. Acta, 41 (1968) 175.
37. Windom H. L., Smith R. G., Deep Sea Res., 19 (1972) 727.
38. Leyden D. E., Underwood A. L., J. Phys. Chem., 68 (1964) 2093.
39. Hirose A., kobori K., Ishi D., Anal. Chim. Acta, 97 (1978) 303.
40. Pakalus P., Batley G. E., Cameron A. J., Anal. Chim. Acta, 99 (1978) 333.
41. Abdullah M. J., El-Rayls O. A., Riley J. P., Anal. Chim. Acta, 84 (1974) 363.
42. Holynska B., Radiochem. Radioanal. Lett., 17 (1974) 313.
43.. Florence T. M., Batley G. E., Taianta, 22 (1975) 201.
44. Korkisch J., Steffan I., Anal. Chim. Acta, 77 (1975) 312.
45. Smith R. G„ Jr., Anal. Chem., 46 (1974) 607.
46. Green T. E, Law S. L., Campbell W. J., Anal. Chem., 42 (1970) 1749.
47. Kingston H. M., Barnes I. L., Brady T. J., Rains T. C, Champ M. A., Anal. Chem., 50 (1978) 2064.
48. Riley J. P., Skirrow G., Chemical Oceanography, Vol. Ill, Academic Press, N. Y., 1975.
49. Leyden D. E., Patterson T. A., Alberts J. J., Anal. Chem., 47 (1975) 733.
50. Dingman J. F., Jr., Gloss К. M., Milano E. A., Siggia S., Anal. Chem., 46 (1974) 774.
51. Egrawa H., Maeda H„ J. Chem. Soc. Japan (Chem. Ind. Sect.), (1978) № 7, 1043.
52. Yamashita H., Ozawa Y., Nakajima F, Murat a T„ Nippon Kagaku Kaishi (1978) № 8, 1057.
53. Doshi G. R., Sastry V. N., Indian J. Chem., 15A (1977) 904.
54. Myasoedova G. V., Antokol’skaya I. L, Shvoeva О. P., Bol’shakova L. L, Savvin S. B., Taianta, 23 (1976) 866.
55. Мясоедова Г. В., Малофеева Г. И., Швоева О. П., Илларионова Е. В., Саввин С. Б., Золотов Ю. А., Ж. аналит. химии, 32 (1977) 645.
56. Malofeeva G. L, Myasoedova G. V., Volynets M. P., Mikrochim. Acta, 1978, 1, 391.
57. Ласкорин Б. H., Метальников С. С, Смолина Г. И., Ат. энергия, 43 (1977) 472.
58. Matthews A. D., Riley J. Р., Anal. Chim. Acta, 51 (1970) 287.
59. Kawabuchi K., Riley J. P., Anal. Chim. Acta, 65 (1973) 271.
60. Clechet P., Eschalier G., Rampon C., Vallouy S., Analusis, 5 (1977) 366.
61. Tanaka Y., Miyagi H., HirotaK., Arikawa Y., Japan Analyst, 26 (1977) 752.
62. Lieser К. H., Rober H. M„ Burba P., Z. anal. Chem., 284 (1977) 361.
63. Desai H. B., Padmanabhan P. K., Venkateswarlu Ch., J. Radioanal. Chem., 31 (1976) 525.
64. Gladney E. S., Owens J. W., Starner J. W., Anal. Chem., 48 (1976) 973.
65. Cesareo R., Gigante G. E., Water, air, soil pollut., 9 (1978) 99.
66. Baucom E. I., Ferguson R. B„ Wallace R. M., Rept. DC-MS-77-11 (1971).
67. Hathaway L. R., James G. W„ Anal. Chem., 47 (1975) 2035.
68. Blotcky A. J., Rack E. P., Recker R. R., Leffler J. A., Teitelbaum S., J. Radioanal. Chem., 43 (1978) 381.
5. Применение ионообменников
155
69. Hirose A., Ishii D., J. Radioanal. Chem., 41 (1977) 37.
70. Tera F., Wasserburg G. J., Earth Planet Sci. Lett., 13 (1972) 457.
71. Krogh T. E., Carnegie Inst. Year Book, 70 (1971) 258.
72. Рябухин В. А., Гатинская H. Г., Ермаков А. Н. Ж. налит, химии, 32 (1977) 909.
73. Fukasawa Т., Yamane Т., Japan Analyst, 24 (1975) 120.
74. Sellers N. G., Anal. Chem., 44 (1972) 410.
75. Mitchell J. W., Gibbs V., Taianta, 24 (1977) 741.
5.2. Разделение неорганических ионов
Ценность и универсальность метода ионообменного разделения подтверждаются многочисленными примерами. Ионообменники проявляют свои преимущества во многих трудных разделениях. Возможности этого метода иллюстрирует разделение смесей Zr — Hf, лантаноидов и трансурановых элементов.
Применение ионообменников для разделения неорганических ионов составляет важную область использования этих веществ в аналитической химии. В последние годы опубликовано большое число разнообразных методик разделения, что свидетельствует об интенсивном развитии данного метода. Каждый год публикуются сотни статей.
Учитывая большое количество литературных данных, не представляется возможным подробно описать все опубликованные методы или критически сравнить все предложенные методики разделения.
Поэтому в дальнейшем будут приведены только основные сорбционные характеристики отдельных элементов. В разделе «Лабораторные методики» рассмотрены либо методики разделения типичных смесей элементов, либо методики более общего характера.
Дополнительными параметрами для сравнения различных процессов разделения служат известные значения коэффициентов распределения. Эти данные (для различных сред) приведены в третьей части книги.
Методики разделения с использованием селективных или специальных ионообменников даны в гл. 6.
5.2.1. Группа 1
5.2.1.1. 1 A: Li, Na, К, Rb, Cs, Fr
Катионы одновалентных щелочных металлов легко сорбируются сильнокислотными катионообменниками. Для разделения щелочных металлов преимущественно используют эти смолы.
Наиболее широко применяется метод элюентной хроматографии. Элюирование выполняют разбавленными водными или водно-органическими растворами минеральных кислот, главным образом соляной или азотной. Метод градиентного элюирования с повышающейся концентрацией элюента имеет ряд преимуществ. Разделение можно улучшить введением в элю
156
Часть II
ент низкомолекулярного спирта, ацетона или фенола. Использование водно-органической среды особенно полезно при разделении смесей Li—Na или Na—К.
Щелочные металлы не образуют достаточно прочных комплексов, в виде которых их можно разделить на анионообменниках. Однако эти металлы легко отделяются от других элементов, образующих прочные анионные комплексы, которые сорбируются анионообменниками.
ОРГАНИЧЕСКИЕ- КАТИОНООБМЕННИКИ
При разделении ионов щелочных металлов применяют сильнокислотные катионообменники. Сродство этих элементов к ионообменникам повышается с увеличением атомной массы элемента и с уменьшением радиуса его гидратированного иона, т. е. в ряду Li < Na < К < Rb < Cs < Fr. Принимая во внимание опубликованные данные по величинам коэффициентов распределения [1—3], можно заключить, что щелочные металлы довольно' четко разделяются на две группы: Li, Na и Rb, Cs, Fr. Калий служит как бы мостиком между двумя группами.
Разделение ионов щелочных металлов на сильнокислотных катионооб-менниках полистирольного типа зависит от степени поперечного связывания последних. Эта зависимость иллюстрируется хроматограммами, показанными на рис. 5.1.
Влияние степени поперечного связывания обменника и температуры на разделение смеси щелочных металлов (элюирование соляной кислотой) подробно изучено Дубчинским [5]. Оптимальное разделение было достигнуто на смоле Dowex 50-W с 16% ДВБ при 25 °C. На смоле, содержащей 2% ДВБ, получают отдельный пик для Na+, но очень плохое разрешение для К, Rb и Cs. Применение смолы с 8% ДВБ приводит к заметному улучшению разрешения. При 75 °C (вместо 25 °C) наблюдается плохое разрешение при всех изученных степенях поперечного связывания.
Рис. 5.1. Влияние степени поперечного связывания на разделение смесей щелочных металлов на смоле Wofatit KPS [4]. Элюент: 0,2 М соляная кислота. Скорость элюирования: 1 см3/мин.
Содержание ДВБ в смоле в «й>: а — 8; б — 16; в — 26.
5. Применение ионообменников
157
Таблица 5.4. Факторы разделения а щелочных металлов для смола разных типов в среде НС1 и НС1 — 80% СН3ОН (по объему) [6 — 8]
Dowex 50 Duolite С-3 КУ-1
Смесь ионов 1 M HC1 1 M HC1 + + CHjOH 1 M HC1 1 M HC1 + + CHjOH HC1, M
22,24Na—Li 1,5 4,2 1,8 4,1 1,5 (0,25)
42K_22,24Na 2,3 3,9 2,3 5,4 1,8(0,50)
86Rb—42K 1,2 2,0 1,9 2,2 1,6 (0,50)
134Cs_86Rb 1,2 1,0 4,2 2,8 4,2 (0,80)
223Fr_134Cs — — 2,1 2,0 —
а Dowex 50 — сополимер стирола с ДВБ: —SOjH;
Duolite С-3 — фенолформальдегидный поликонденсат: —-CH^SOjH, —ОН; КУ-1 — фенолформальдегидный поликонденсат: —SOjH, —ОН.
Замена монофункционального катионообменника полистирол-ДВБ типа на сильнокислотный фенолформальдегидный катионообменник значительно повышает сродство к ионам цезия. Последние, особенно в щелочной среде, образуют достаточно прочные комплексы с фенольными группами смолы, что видно из данных табл. 5.4. При разделении пары Cs—Rb фактор разделения а для поликонденсационных смол Duolite С-3 и КУ-1 выше, чем для смолы Dowex 50. Данные табл. 5.4 показывают также влияние добавки метанола на качество разделения пар щелочных металлов.
Проведение разделения при высоком давлении может привести к изменению степени гидратации ионов, что несколько повлияет на качество разделения. Показано, что с повышением давления от атмосферного до 35 МПа обменная емкость для ионов Rb+ и К+ (с поправкой на сжатие) изменяется почти на 25%, в то время как для Li+ и Na+ остается практически постоянной [9].
Как показано Малдахером и Рогерсом [10], повышение давления оказывает подобный же эффект и в случае разделения некоторых анионов на ани-онообменниках.
Слабокислотные катионообменники плохо сорбируют ионы щелочных металлов (а также щелочноземельных металлов, Zn, Cd, Pb, Ag), что было подтверждено путем изучения влияния концентрации ионов Н+ на коэффициенты селективности.
На карбоксильной смоле в 1ЧН4-форме в нейтральных или слабокислых растворах можно отделить однозарядные катионы от многозарядных. В качестве элюирующего агента обычно используют хлорид аммония. Описаны методы отделения следов щелочных металлов от реакторного урана [11]; натрия и калия от кальция и магния [12]; щелочных металлов от Mg, Са, Sr, Ba, Ni, Си, Со, Zn, Al, Fe, Th [13].
158
Часть И
НЕОРГАНИЧЕСКИЕ ИОНООБМЕННИКИ
Для отделения тяжелых щелочных металлов (особенно для селективного отделения цезия) перспективны разнообразные неорганические нонооб-менники (см. гл. 6): нерастворимые гетерополикислоты и их соли [14], комплексные цианиды некоторых элементов и соединения типа фосфатов (15], арсенаты, молибдаты и вольфраматы четырехвалентных элементов (цирконий, титан, олово). Для селективной сорбции ионов натрия был приготовлен ионообменник на основе гидратированного пентоксида сурьмы [16, 17]. Ионы натрия сорбируются из 6—12 М НО; никакие другие элементы (кроме тантала и фторидов) не сорбируются.
Селективность неорганических обменников, за исключением гидратированного пентоксида сурьмы, по отношению к однозарядным ионам в кислых растворах повышается с увеличением атомной массы элемента. Селективность к цезию очень высокая. В табл. 5.5 приведены сравнительные данные по селективности фосфоромолибдата аммония и смолы Dowex 50 к ионам щелочных металлов.
Комплексные ферроцианиды цинка, кобальта, никеля, молибдена, ванадия и вольфрама также проявляют высокую селективность к ионам цезня [19-24]. По аналогии с другими неорганическими ионообменниками их селективность повышается в ряду Li < Na < К < Rb « Cs. Так как сродство к ионам Cs+ у некоторых неорганических ионообменников чрезвычайно велико, Cs+ очень трудно элюировать из обменника. В этом случае в качестве элюентов используют концентрированные растворы нитратов аммония, серебра или ртути(П). Если количественное элюирование цезия этими растворами невозможно, рекомендуется проводить химическое или термическое разложение обменника. Цезий не поглощается Th[Fe(CN)6] и Zr[Fe(CN)e] и лишь слабо сорбируется на (ThO)2[Fe(CN)6].
Обменники в форме гидратированных оксидов для солей многозарядных элементов имеют важное значение при разделении пары Rb—Cs и при выделении цезия из смеси продуктов расщепления урана. С этой целью чаще всего используют фосфат циркония [25—28], а для элюирования — растворы нитратов различной концентрации, хлорид аммония или азотную кислоту. Коэффициенты распределения ионов щелочных металлов на фосфате циркония представлены в табл. 5.6.
Таблица 5.5. Сравнение коэффициентов распределения Dg ионов щелочных металлов для смолы Dowex 50 и фосфоромолибдата аммония [18]
Смола Dz
Na+ К+ Rb+ Cs+
Dowex 50, NH^-форма Фосфоромолибдат 26 46 52 62
аммония 1 3,4 230 6000
5. Применение ионообменников
159
Таблица 5.6. Коэффициенты распределения Dg ионов щелочных и щелочноземельных металлов на фосфате циркония (Н-форма) [29]
Ион Концентрация раствора, M D g
Li + 0,014 17
Na+ 0,014 36
К+ 0,014 84
Rb + 0,013 182
Cs + 0,013 200
Mg2 + 0,014 20
Ca2 + 0,014 56
Sr2+ 0,012 63
Ba2+ 0,014 91
С помощью фосфата циркония отделяют следы щелочных и щелочноземельных металлов. В качестве элюента используют 1 М NH4C1. Всю операцию можно полностью автоматизировать. Содержание отдельных компонентов в различных порциях элюата определяют методом пламенной фотометрии. Достигнуты следующие пределы определения индивидуальных компонентов: для LiCl — 5 • 10“12 моля; NaCl — 4 • 10“13 КС1 — 5 • 10-14 и RbCl — 3 • IO"13 моля [30].
Интересным типом неорганического ионообменника, пригодного для разделения щелочных металлов, является «фосфоросурьмяная(У) кислота». Как показано несколькими авторами [31—34], осадок, образующийся при взаимодействии пентоксида сурьмы и ортофосфорной кислоты, представляет собой сильную одноосновную кислоту, очень хорошо сорбирующую щелочные металлы. (Подробности получения соответствующего сорбента приведены в статье Абе [35]). Селективность этого сорбента к щелочным металлам уменьшается в ряду Cs > Rb > К> Na > Li. Смесь ионов щелочных металлов разделяется на индивидуальные элементы (за исключением пары Li+ — Na+) на относительно маленькой колонке при элюировании раствором нитрата аммония. Для пар Rb—К н Cs—Rb получены более благоприятные факторы разделения а по сравнению с факторами разделения на смоле Amberlite IR-120 или Dowex 50 в растворе нитрата аммония. Сравнительные данные приведены в табл. 5.7.
Смесь Li—К—Rb (0,1 М NH4NO3) и Cs (1 М NH4NO3) удовлетворительно разделяется на фосфоросурьмяной кислоте. Как можно предположить из величин Dg, приведенных в табл. 5.7, для пары Li—Na разделение менее эффективно даже при элюировании более разбавленным раствором nh4no3.
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Ионы щелочных металлов не сорбируются на анионообменниках за исключением указанных ниже случаев. Ионы лития сорбируются на анионо-
5. Применение ионообменников
161
обменнике в ЭДТА-форме особенно в присутствии этанола [36, 37]. Ионы Rb+ и Cs+ сорбируются частично при условии, что в системе присутствует большое количество метанола или диоксана [38, 39]. На смоле Dowex 1-Х8 в смеси 0,49 М НС1 — 78% диоксана получены следующие коэффициенты распределения: 2,4 для Li; 2,1 для Na; 2,8 для К; 2,8 для Rb; 2,9 для Cs. При изменении содержания диоксана от 93 до 73% Li+ можно количественно отделить от ионов других щелочных металлов. Согласно данным Шабана и Руфа [40], натрий и цезий удалось успешно отделить от бария на анионообменнике Dowex 1-Х8 при элюировании смесью 2 М HNO3 — 75% С2Н5ОН; Ва элюируют 2 М HNO3. В среде соляной кислоты отделение проходит гораздо хуже. Метод пригоден также для разделения микроколичеств элементов [40].
Так как ионы щелочных металлов (за исключением упомянутых выше) не сорбируются анионообменниками, последнее можно использовать для отделения щелочных металлов от других элЩгентов. После превращения сопутствующих металлов в анионные комплексы с помощью ЭДТА, цитрат- или оксалат-ионов возможно отделение, например, алюминия, кальция, магния и других металлов. Анионные комплексы этих металлов сорбируются анионообменниками. Для образования хлоридных комплексов используют растворы соляной кислоты разной концентрации. Подобным же образом различные кислородсодержащие анионы (хроматы, вольфраматы, молибдаты, ванадаты, перренаты и т. п.) сорбируются сильноосновными анионообменниками в ОН-форме и таким путем отделяются от группы щелочных металлов. Для отделения ионов, образующих нерастворимые карбонаты, используют анионообменник в СОуформе.
Лабораторные методики
ОТДЕЛЕНИЕ ЛИТИЯ ОТ НАТРИЯ И КАЛИЯ [41]
Методика. Нейтральный раствор, содержащий разделяемые ионы (лучше всего в форме хлоридов), пропускают через колонку, заполненную сильнокислотным кати-онообменннком Amberlite IR-120 в Н-форме. Затем колонку промывают водой и элюируют литий смесью 0,2 М НС1 (7 объемов) и метанола (3 объема), В этих условиях ионы Na+ и К+ не элюируются. Скорости сорбции и элюирования равны 1 см3/мин. Для разделения смеси, содержащей 5 ммолей рассматриваемых иоиов, необходимо приблизительно 6 г смолы. Натрий можно элюировать 0,2 М НС1 в 10%-ном метаноле, а калий — 0,5 М НС1.
ОТДЕЛЕНИЕ НЕБОЛЬШИХ КОЛИЧЕСТВ Li ОТ ИЗБЫТКА К, Са И Mg [42]
Методика. Нейтральный раствор, содержащий разделяемые элементы, пропускают через колонку (120 X 1,5 см), заполненную сильнокислотным катионообменни-ком Amberlite IR-120 (0,85—0,15 мм) в Н-форме. После промывания колонки водой сорбированные ионы элюируют смесью концентрированной НС1 (10%) и метанола (90%). Литий элюируется первым.
П-354
162
Часть II
ОПРЕДЕЛЕНИЕ 137Cs В МОЧЕ ПОСЛЕ ЕГО ОТДЕЛЕНИЯ НА ФОСФОРОМОЛИБДАТЕ АММОНИЯ (ФМА) [43]
Методика. 200 см3 мочи подкисляют 20 см3 концентрированной HNO3 и выпаривают досуха. К остатку прибавляют еще 20 см3 концентрированной HNO3 и снова выпаривают досуха. Затем приливают 100 см3 воды и с помощью HNO3 доводят pH раствора до 1,0. Полученный раствор со скоростью 3 см3/мин пропускают через колонку, подготовленную следующим образом. Смесь 10 г ФМА, 5 г асбеста и 50 см3 0,1 М NH4NO3 переносят в колонку, промывают 0,1 М NH4NO3 и дают ей постоять несколько часов. После пропускания анализируемого раствора обменник промывают двумя порциями по 100 сМ3 4 М NH4NO3 так, чтобы весь промывной раствор вытек из колонки. Затем порциями по 10 см3 прибавляют 6 М раствор NaOH до полного растворения ФМА. Порции элюата собирают в колбы емкостью 125 см3, доводят pH раствора до 12,5 и экстрагируют 0,5 М раствором 4-втор-бутил-2-(а-метилбензнл)фенола в керосине. Экстракцию повторяют пять раз. Раствор выпаривают и измеряют радиоактивность остатка. Анализ 1 дм3 мочи длится 4,5 ч (1 ч работы оператора). Нет необходимости во введении неактивного носителя.
РАЗДЕЛЕНИЕ СМЕСИ 22Na — 86Rb — 134Cs НА МОЛИБДАТЕ ЦИРКОНИЯ [44]
Методика. Нейтральный водный раствор разделяемых ионов пропускают через колонку (6 х 0,5 см), заполненную 0,75 г обменника в ИН4-форме (0,15—0,07 мм). Скорость потока при разделении 7,5 • 10~4 ммоля каждого элемента составляет 0,15 см3/мии. Порядок элюирования (при скорости потока 0,13 см3/мин) следующий:
0,10 М NH4C1: Na (6 см3 элюата),
0,50 М NH4C1: Rb (14 см3 элюата), 1,00 М NH4C1: Cs (30 см3 элюата).
ОПРЕДЕЛЕНИЕ Na И К В МЕТАЛЛИЧЕСКОМ ВОЛЬФРАМЕ ИЛИ МОЛИБДЕНЕ [45]
Необходимое оборудование. Колонка (100 х 2 мм), заполненная вольфраматом циркония в NH^-форме; колонка (100х 7 мм), заполненная анионообменником Wofatit SBW в Cl-форме (размер зерен 0,06—0,09 мм).
Растворы. 1. Смесь фтористоводородной и соляной кислот в объемном отношении 3:1.
2. Смесь соляной кислоты, щавелевой кислоты и пероксида водорода в следующих (конечных) концентрациях: 0,1 М НС1, 0,5 М (СООН)2, 1 М Н2О2.
3. 0,1 Ми 0,3 М растворы хлорида аммония.
Методика. Приблизительно 100 мг образца облучают в ядерном реакторе тепловыми нейтронами. Одновременно облучают 1 мг Na и 1 мг К в качестве радиометрических стандартов.
Облученный образец растворяют в смеси HF—НС1 (раствор 1); после введения 100 мг смеси неактивных Na—К раствор выпаривают досуха. Остаток растворяют при нагревании в 10 см3 раствора 2. Раствор охлаждают и фильтруют со скоростью 10 капель/мин через колонку, заполненную анионообменником SBW. После этого кйлонку промывают 10 см3 раствора 2. Весь натрий и калий переходит в фильтрат.
К фильтрату прибавляют 0,5 см3 концентрированной H2SO4 н выпаривают досуха. Остаток растворяют в нескольких каплях воды и раствор количественно пере
5. Применение ионообменников
163
носят на колонку, заполненную неорганическим ионообменником. В этих условиях ионы натрия и калия сорбируются количественно.
Натрий элюируют 0,1 М NH4C1, калий — 0,3 М NH4C1 и определяют их концентрацию радиометрически.
Чувствительность метода 5 мкг/л при определении натрия и 50 мкг/л для калия.
При элюировании более концентрированными растворами эта методика применима для определения в указанных материалах рубидия и цезия.
Полный анализ (за исключением облучения в реакторе) длится приблизительно три часа.
ОПРЕДЕЛЕНИЕ СУММЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ В ПРИСУТСТВИИ НЕКОТОРЫХ ДРУГИХ ЭЛЕМЕНТОВ [37, 46]
Методика. Порции-по 100 см3 сильноосновного анионообменника Dowex 2 превращают в этилендиаминтетраацетатную (500 см3 0,2 М ЭДТА), гидроксильную (1000 см3 IM NaOH), оксалатную, цитатную и ацетатную (500 см3 0,2 М раствора соответствующих аммониевых солей) формы. Затем отдельные порции обменника промывают 1500—2000 см3 деионизованной воды.
Для разделения щелочных металлов используют две колонки, соединенные в ряд (размер 17 х 0,9 см). Первую колонку заполняют смесью смолы Dowex 2 в эти-лендиаминтетраацетатной, оксалатной, цитратной и ацетатной формах. Элюат из этой колонки протекает во вторую колонку такого же размера, заполненную смолой Dowex 2 в ОН-форме. Для улучшения поглощения щелочноземельных металлов можно использовать смесь воды и этанола (1 : 1).
В процессе сорбции в присутствии этанола наблюдается изменение объема обменника. В некоторых случаях образуются осадки. Во избежание загрязнения колонки рекомендуется следующая методика: 50 см3 образца, содержащего смесь щелочных металлов, щелочноземельных металлов, Y, Се, Pm, Zr, Nb, V(IV), Fe(III), Al, Cu, Ni, Co, Mn(II) и Zn (лучше всего в форме хлоридов), смешивают с 60 см3 этанола и соответствующим количеством обменника в этилендиаминтетраацетатной и ацетатной формах (1 : 1). При образовании осадка вводят дополнительную порцию обменника. Затем суспензию помещают в первую колонку, заполненную смолой Dowex 2 в упомянутых выше формах, и пропускают через колонку со скоростью 1—2 см3/мин. Колонку промывают 60%-ным раствором этанола. Элюат поступает во вторую колонку, заполненную смолой Dowex 2 в ОН-форме.
Сумму щелочных металлов определяют в элюате второй колонки титрованием 0,1 М НС1. Продолжительность определения составляет приблизительно 2 ч, точность ±0,2%.
РАЗДЕЛЕНИЕ Cs—Fr (МИКРОКОЛИЧЕСТВА) [8]
Методика. Смесь разделяемых ионов помещают в колонку (4 см х 0,28 см2), заполненную сильнокислотным катионообменником Duolite С-3 (0,044 — 0,037 мм). Разделение выполняют элюированием 4,5 М НС1 при 25 °C и скорости потока 1 см3/мин. Цезий десорбируется первым.
Смолу Duolite С-3 можно заменить обменником КУ-1. При использовании обменника стирольного типа (Dowex 50, КУ-2) разделение гораздо хуже.
Обзор некоторых методов разделения других смесей, содержащих щелочные металлы, дан в табл. 5.8.
Таблица 5.8. Выделение щелочных металлов из различных смесей
Элемент Отделяемые Смола Условия разделения Литература
компоненты
Li Na Сильнокислотный полимерный катионо-обменник Li: 0,5 M HC1 + 80% CH3OH 47
Li Na КУ-2 0,25 M HC1 7
Li Na, NH + , К Dowex 50 0,13 M ацетат кальция 48
Li Na, K, Ca, Mg, Sr Amberlite IR-120 MeOH — HC1 (9:1) 42
Li —Na—К Dowex 5OW-X8 Li: 0,6 M HC1 — 60% C2H5OH Na: 0,4 M HC1 — 40% C2H5OH K: 0,6 M HC1 — 20% C2H5OH 49
Щелочные Mg Amberlite IR-120 Кислота — спирт — фенол 50
металлы, Dowex AG 5O-X8 Пиридин — C2HSOH (1:1) 51
22Na
Na Mg КУ-2 Na: 0,1 M HC1 52
Щелочные Многозарядные Dowex 50 ЭДТА, pH 8, катнонообменник в 53
металлы ИОНЫ тетраметиламмониевой форме
Li, Na, К, Mg — Ba, Mn, Zeokarb 226 В растворе устанавливают pH 54
Rb, Cs Cd, Co, Ni, 2,5 ±0,5, смолу промывают
Zn, Al, Fe, U, Th, РЗЭ 0,1 М NH.C1 * 4
Li, Na, К Mg, Ca, Sz, Ba Li, Na, К: 0,25 М НС1 Mg, Са: 0,80 М НС1 Sr: 2 М НС1 Ва: 4 М НС1 55
Щелочные Нитрованный 56
металлы Amberlite IR-120
Cs Ce, Sr из Ферроцианид 57
морской воды молибдена
Na Биологический Sb2O5xH2O 58
материал
Щелочные Ферроцианид меди 59
металлы
Щелочные Щелочноземель- Ферроцианид титана 60
металлы ные металлы, Y
Rb Cs, Fr Молибдат циркония Rb: 1 М NH4NO3
Cs, Fr: 4 M NH4NO3 61
Rb, К, Cs Wofatit KPS-X16 2 M HF 62
Cs Na, NH.+ 4 Zeokarb 315 Na, NH+: 0,25 M HNO3 63
Cs: 3 M HCl
Cs Радиоактивная Dowex 5OW-X12 64
пыль
Cs Молоко, биоло- Dowex 1-X8
гические об- Dowex 50 65
разцы
Щелочные Цемент 66
металлы
Cs Продукты Dowex AG-5OW-X8 Теноилтрнфторацетон, пиридин 51
расщепления
Na К Фосфоромолибдат Na: 0,1 M HNO3
аммония на асбесте K: 0,25 M NH.NO, 67
Щелочные Образцы 4 3 68
металлы силикатов
166
Часть II
5.2.1.2. 1 Б: Си, Ag, Аи
Способность ионов меди, золота и в некоторой степени серебра образовывать комплексы различной устойчивости с рядом комплексообразующих агентов используется для селективного отделения этих элементов.
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Ионы меди(П) очень сильно сорбируются органическими катионообмен-никами как сильнокислотного, так и слабокиелотного типов. Значения коэффициентов распределения Dg при cHNo3 = 0,1 М составляют 1 • 103 — 1,5 103. Сорбция меди уменьшается с повышением концентрации кислоты. Так, при cHNO = 4 М значение Dg составляет порядка 2—7. Подобное поведение наблюдается для сернокислых растворов [69]. При введении подходящих органических растворителей Dg увеличивается.
С другой стороны, сорбция на катионообменниках ионов серебра(Т) гораздо слабее, чем ионов меди(П). Коэффициент распределения Dg при chnoj = 0,1 М равен 150 и уменьшается примерно до 4 при cHNO3 = 4 М.
Сорбция ионов золота(Ш), главным образом в форме AuCl^, из солянокислых растворов на катионообменниках очень низка [2]. Степень сорбции увеличивается с повышением концентрации кислоты. Коэффициенты распределения Dg при снс1 > 8 М для катионообменника Dowex 50-Х4 имеют величину порядка 102. Сорбция постепенно уменьшается с повышением степени поперечного связывания обменника.
Из сильнокислотных катионообменников ионы Си2+ легко элюируются приблизительно 4 М HNO3, 1—4 М H2SO4, 3—4 М CH3COONH4, 2—5 М NH4NO3 или 4—5 М NaNO3. Для элюирования непригодны лимонная, уксусная или винная кислота. Ионы серебра не элюируются 0,1—1 М соляной кислотой. Это обстоятельство может служить основой для их разделения (см. табл. 8.33).
Для отделения золота используют ионообменные свойства целлюлозы [70], на которой ие сорбируются граммовые количества золота, но поглощаются следовые количества Ag, Fe, Cr, Со, Мп, Zn и Cd.
При отделении золота от платины применяют бромистоводородную кислоту [71]. Золото легко отделяется от ртути или никеля в 2 М НС1 [72]. Сообщается о высокой селективности по отношению к ионам меди карбоксильного катионообменника. Эту смолу можно использовать для селективного концентрирования и отделения меди. Нанограммовые количества меди концентрируют из одного литра дистиллированной воды в маленькой колонке, заполненной смолой Zeokarb 226 (0,4 х 2 см), и облучают тепловыми нейтронами. После элюирования натрия раствором хлорида аммония определяют концентрацию меди [73].
Фритц и Миллен [74] сообщили о высокоселективной сорбции Au(III) на пористой полиакрилатной смоле XAD-7 из 0,1 М кислых растворов.
НЕОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Медь(П), как и ионы серебра, хорошо сорбируется неорганическими ка-тионообменниками. Следует отметить, что золото не сорбируется фосфатом циркония из разбавленных растворов соляной кислоты [75].
5. Применение ионообменников
167
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Большое значение при разделении элементов и отделении их от мешающих компонентов имеют галогенсодержащие кислоты. Сорбция элементов на смоле Dowex 1-Х10 повышается в ряду Си < Ag < Au и уменьшается с повышением концентрации кислоты [76, 77].
В 4 М НС1 получены следующие коэффициенты распределения Dv: < 101 для Cu2+, < 1 10* для Ag+, > 105 для Аи3+. Эти элементы легко отделяются от щелочных и щелочноземельных металлов, лантаноидов, иттрия, марганца, алюминия, тория и других элементов, которые либо не сорбируются на анионообменнике из растворов НС1, либо сорбируются в незначительной степени. Сорбцию золота можно полностью подавить введением достаточного количества органического растворителя (ацетон, этанол, тетрагидрофуран).
Ионы Си2+, как и ионы Ag+ , не поглощаются анионообменниками из фтористоводородной кислоты (1—24 М), в то время как ионы Аи3 + сорбируются легко. Для того чтобы добиться хорошей сорбции этих элементов анионообменниками, их превращают в цианидные комплексы [78—82].
Сорбция из среды азотной кислоты уменьшается в ряду Au > Ag > Си [78, 83].
Лабораторные методики
РАЗДЕЛЕНИЕ СМЕСЕЙ Си—Ni, Си—Cd, Си—Ag
И Си—РЬ НА КАТИОНООБМЕННИКЕ
Методика для смесей Си—Ni или Си—Cd. Смесь обоих элементов, содержащую до 30—35 мг Ni и неограниченное количество Cd, уравновешивают с сильнокислотным катионообменником в Н-форме.
Оба элемента количественно сорбируются из нейтральных или слабокислых растворов.
Обменник промывают водой и элюируют ионы Си2+ примерно 250 см3 раствора следующего состава: 5 г NaOH и 5 см3 глицерина растворяют в воде и разбавляют до 1000 см3. Ионы Ni2+ или Cd2+ элюируют 5%-ным раствором соляной кислоты.
Методика для смеси Си—РЬ. Ионы Си2+ и РЬ2+ сорбируются на сильнокислотном катионообменнике в Н-форме. РЬ2+ количественно элюируют 250 см3 водного раствора, содержащего (в 100 см3) 10 см3 концентрированного NH4OH и 3 г винной кислоты. Ионы Си2+ элюируют 5%-ной НС1.
Методика для смеси Си—Ag. Раствор, содержащий смесь Си2+ — Ag+ (разное соотношение Си : Ag), вводят в колонку, заполненную сильнокислотным катионообменником в ИН4-форме. Колонку промывают водой и элюируют ионы Cu2+ -250 см3 насыщенного раствора оксалата аммония, а ионы Ag+ — 1 М HNO3 (-20 см3).
ОТДЕЛЕНИЕ lnAg (БЕЗ НОСИТЕЛЯ) ОТ 109Pd [84]
Методика. Анализируемый, образец облучают тепловыми нейтронами и растворяют. В растворе устанавливают концентрации: 2 М НВг и 0,003 М В?2 — и пропускают через ионообменную колонку (6 см х 0,065 см2), снабженную водяной ру-
168
Часть II
Таблица 5.9. Результаты анализа смесей Ag — Pb — Hg [85]
Взято металла, мг Найдено металла, °7о
Ag Pb Hg Ag Pb Hg
6,1 11,9 29,6 99,5 101 100,8
12,1 11,9 29,6 102,8 98,1 98,5
30,2 29,8 59,2 101,3 102 98,7
башкой и заполненную смолой Dowex 1-Х4 в Br-форме (размер зерен 0,02—0,05 мм). Несорбируемые примеси (главным образом 24Na) вымывают 2 см3 смеси 2 М НВг + 0,0035 М Вг2.
Ионы Ag+ элюируют смесью 6 М НВг + 0,0035 М Вг2 со скоростью 2,3 см3/(см2 мин). После элюирования всего Ag+ (приблизительно 2 см3 смеси НВг + Вг2) колонку нагревают до 75 °C и элюируют 109Pd 5 мл смеси 10 М НВг + 0,0035 М Вг2. Разделение элементов длится приблизительно 90 мин. В обоих случаях получают радиоизотопы высокой радиохимической чистоты.
РАЗДЕЛЕНИЕ СМЕСИ Pb—Ag— Hg [85]
Методика. Раствор , содержащий нитраты разделяемых элементов, пропускают со скоростью 1 см3/мин через колонку, заполненную сильнокислотным катионооб-менником Dowex 5OW-X8 (0,15—0,07 мм) в Н-форме. После этого колонку промывают двумя порциями воды по 10 см3 каждая (при скорости 1 см3/мин) и элюируют ионы РЬ2+ 0,25 М раствором CH3COONH4. Ионы Ag+ элюируют 0,50 М раствором CH3COONH4, а ионы Hg2+ — 4 М CH3COONH4. Фактор разделения а д^ равен 3,7 в 0,25 М и 3,6 в 0,5 М растворе ацетата аммония. Некоторые из полученных результатов приведены в табл. 5.9.
ОТДЕЛЕНИЕ lnAg ОТ Pd МИШЕНИ [86]
Методика. 500 мг металлического палладия облучают нейтронами и затем растворяют в царской водке. Раствор выпаривают досуха в присутствии соляной кислоты. Остаток растворяют в 4 см3 10 М НС1 и переносят раствор в колонку (120 х х 5 мм), заполненную смолой Dowex 1, уравновешенной с 10 М НС1. I]IAg элюируют 4 см3 10 М НС1. Палладий элюируют 10 см3 12 М НС1.
РАЗДЕЛЕНИЕ СМЕСИ Ag—Hg НА КАТИОНООБМЕННИКЕ [87]
Методика. Раствор, содержащий исследуемые элементы (лучше в форме нитратов), разбавляют до 30 см3 и устанавливают pH 4,6 прибавлением ацетатного буферного раствора (60 см3 ледяной уксусной кислоты и 13,70 г CH3COONa • ЗН2О в объеме 100 см3). Затем вводят известное количество динатриевой соли ЭДТА (небольшой избыток). Полученный раствор пропускают со скоростью 5 см3/мин через колонку (15x1 см), заполненную смолой Dowex 5OW-X8 в Na-форме с размером частиц 0,30—0,15 мм.
5. Применение ионообменников
169
Смолу промывают 300 см3 воды. В элюате, содержащем избыток ЭДТА и комплекс Hg — ЭДТА, определяют количество ЭДТА титрованием и рассчитывают содержание Hg.
Иоиы Ag+ элюируют 250 см3 16%-ной HNO3 со скоростью 4 см3/мин. Для искусственных смесей ошибка определения составляла ± 0,5%. Ионы Си2+, Со2+, Ni2+ и Zn2+ образуют с ЭДТА комплексы и, подобно ионам Hg2+, проходят через колонку.
ОПРЕДЕЛЕНИЕ СЛЕДОВ Au В МЕТАЛЛИЧЕСКОЙ МЕДИ ПОСЛЕ ОТДЕЛЕНИЯ НА ИОНООБМЕННИКЕ [88]
Этот метод пригоден для определения микрограммовых количеств золота в металлической меди.
Методика. Образец растворяют в смеси НС1—HNO3 и переносят раствор в колонку (20 х 4 мм), заполненную сильноосновным анионообменником Amberlite IRA-400 в Cl-форме. Ионы Си2+ не сорбируются смолой. Смолу промывают 6 М НС1 и озоляют. Остаток растворяют и определяют содержание Au спектрофотометрическим методом. Ошибка определения 0,5—100 мкг/л Au составляет 5—10%. Продолжительность анализа 2—3 ч.
ОПРЕДЕЛЕНИЕ НЕБОЛЬШИХ КОЛИЧЕСТВ Ag В МЕДНЫХ РУДАХ [89]
Методика. Разделение выполняют на колонке (10 х 0,5 см), заполненной ~ 2,5 г анионообменника Wofatit L-150 (0,09—0,10 мм) в Cl-форме. Перед употреблением смолу промывают разбавленной соляной кислотой (1:1).
Навеску руды массой 2 г, содержащей 0,02—1% серебра, растворяют в 15 см3 концентрированный HNO3. Раствор упаривают до объема — 5 см3 и разбавляют 20 см3 серной кислоты (1 : 1). После этого прибавляют твердый нитрат аммония до получения прозрачного раствора и затем выпаривают его. Остаток растворяют в 50 см3 воды, раствор кипятят, фильтруют и промывают фильтр горячей водой. Фильтрат, объединенный с промывными водами, порциями пропускают через ионообменную колонку. Затем смолу промывают небольшим количеством соляной кислоты (порциями по мере вытекания каждой порции раствора). Этим способом удаляют задержанные сульфат-ионы. По окончании сорбции ионообменную колонку промывают 20 см3 НС1 (1 : 2) и 20 см3 НС1 (I : 4). Остаток сорбированных ионов РЬ вымывают из колонки разбавленной азотной кислотой.
Серебро связывается ионообменником в форме хлорида серебра, который элюируют сначала 20 см3 1,2 М, затем 2Q см3 5 М и, наконец, 20 см3 10 М гидроксида аммония. Элюат собирают в мерную колбу емкостью 100 см3 и подкисляют азотной кислотой. Определение серебра выполняют в аликвотной части раствора в виде AgCl нефелометрическим методом.
Перед повторным использованием колонку регенерируют раствором НС1 (1 : 1). Продолжительность анализа около 3 ч. Относительная ошибка определения ± 5%.
Обзор избранных методов разделения других смесей, содержащих Си, Ag и Au, дан в табл. 5.10.
Таблица 5.10. Отделение Си, Ag, Аи
Элемент Отделяемые Смола
Условия разделения
компоненты
Литература
Ag Ba Dowex 5OW-X8 Ag: 2 M CH3COONH4 Ba: 4 M CH3COONH4 90
Ag Sb(lll) Dowex 5OW-X8 Sb: 5%-ная винная ' кислота, pH 1,0 90
Ag Bi, Fe, Th Dowex 5OW-X8 Сорбция из 0,01 М ЭДТА, pH 2 — 2,2 Ag: 4 М HNO3 90
Ag Cu, U, Al, Dowex 5OW-X8 U, Al, Cu, Zn: 0,5 М HNO3
Zn, V(V) V(V): 0,5 М H2SO4 Ag: 4 М HNO3 90
Ag Ce(lV), Zr Dowex 5OW-X8 Сорбция из 5 %-ной лимонной кислоты, pH 2,7 Ag: 4 М HNO3 90
Ag Ni Zerolit 225 Сорбция из 0,1 М ЭДТА Ni: Н2О Ag: 7%-ный NaNO2 91
Ag Cu Lewatit MN Си: 2 М НС1 Ag: раствор NH4OH 92
Ag . Pb, Hg Dowex 5OW-X8 РЬ: 0,25 М цитрат аммония Ag: 0,50 М цитрат аммония Hg: 4,0 М цитрат аммония 85
11’Ag Na, Pd Dowex 1-X4 Na: 2 М НВг—Вг2 Ag: 6 М НВг —Вг2 Pd: 10 М НВг —Вг2 84
Cu Сплавы меди Diaion SA-100 93
Продолжение табл. 5.10
Элемент Отделяемые компоненты Смола Условия разделения Литература
Си Mg, Sr Zerolit 225 Сорбция нз раствора ЭДТА pH 3,5 Си не сорбируется 94
Си Bi Amberlite CG 400 Си: 3 М HNO3 95
Аи Ag, Cd, Си, Fe, Pb, Pt, Zn Lewatit S-100 96
Аи Ag, Cu Фосфат циркония Си: 0,1 М HCI Ag: 4 М NH4()H — NH4C1 97
Аи Медь особой чистоты Amberlite IRA-400 85
Аи Ta, Mo, W As, Sb, Sn Анионообменник AB-17 Сорбция из ра юавленной HF Sn, Mo, W, As: 17 М HF Та: 1 М HF — 2 М NH4NO3 Sb: 3 М НС1О4 Аи: 1 М CS(NH2)2 82, 98
Си Ряд металлов AG 5OW-X8 0,2 М НС1 — 80% ацетон элюнрует Ga, Fe(HI), Zn, Cd, Sn(lV), In, Tl(Ill), Ce(IV), As(lll), Se(IV), Mo(Vl), W(VI), Au(IlI), Pd, Pb(IV), Rh, Hg(ll) 0,2 M HCI — 85% ацетон: Cu(ll), U(VI) (частично); не элюируются данным раствором: Со, Ni, Мп, Li, Na, К, Rb, Cs, Be, Mg, Ca, Sr, Ba, Ti, V(V), Hf, Zr, Th, Al, Sc, Y, РЗЭ 99
Аи Другие элементы Amberlite XAD-7 Au: 1 M HCI — ацетон 100
Продолжение табл. 5.10
3
О
О
са $ о 4Л
сч о Cs о 'g <
Z * я 5 Z § ffi 1 и и JD (Z)
« * ffi X я §
3 Q. х О X X s s E5
£ 2 (Ч л s fls - a о s о X
X 60 & oh .. 2 3 4 s
< Е < < m
60 5
< <
3 <
ш <
з О
х si о
О <
so
<
5. Применение ионообменников
173
Литература
1. Strelow F. W. Е., Anal.Chem., 32 (1960) 1185,
2. Strelow F. W. Е., Rethemeyer R„ Bothnia C. J. C., Anal. Chem., 37 (1965) 106.
3. Nelson F., J. Chromatorg., 16 (1964) 538.
4. Scheibe F., Sauer R., Z. Chem., 5 (1965) 208.
5. Dybczynski R., J, Chromatogr., 71 (1972) 507.
6. Tsubota H„ Bull, Chem. Soc. Jap., 36 (1963) 1038.
7. Лилова О. M., Преображенский Б. К. Радиохимия, 2, 1960, 728.
8. Nelson F., Michelson D. С., Phillips Н. О., Kraus К. A., J. Chromatogr., 20 (1965) 107.
9. Prukop G., Rogers L. В., Separ. Sci., 13 (1978) 117.
10. Maidacker T. A., Rogers L. B., Separ. Sci., 9 (1974) 27.
11. Alhavale V. T., Krishnan С. V., J. Sci. Ind. Res., 21B (1962) 339.
12. Солдатов H. С, Беспалко M. С, Новицкая Л. H. Вести. АН Бел. ССР, Сер. хим. наук, 1965, 100.
13. Iyer S. G., Krishnan С. V., Venkateswarlu Ch., Ind. J. Chem., 6 (1968) 277.
14. Nonaka N., Sato K., Higuch H., Hamaguchi H., Radioisotopes, 25 (1975) 599.
15. Inoue У., J. Inorg. Nucl. Chem., 26 (1964) 2241.
16. Girardi F., Sabbioni E., J. Radioanal. Chem., 1 (1968) 169.
17. Phillips H. O., Kraus К. O., J. Amer. Chem. Soc., 84 (1962) 2267.
18. van Smith J. R., Nature, 181 (1958) 1530.
19. Koufim V., Rais J., Stejskal J., J. Inorg, Nucl. Chem., 26 (1964) 1761.
20. KouHm V., Rais J., Million B., J. Inorg. Nucl. Chem., 26 (1964) 1111.
21. Huys D., Baetste J., J. Inorg. Nucl. Chem., 26 (1964) 1329.
22. Krtil J., J. Chromatogr., 20 (1965) 384.
23. Krtil J., J. Chromatogr., 21 (1966) 85.
24. Krtil J., J. Inorg. Nucl. Chem. 27 (1965) 233.
25. Amphlett С. B., McDonald L. A., Burgess J. S., Maynard J. C, J. Inorg. Chem., 10 (1959) 69.
26. Лаврухина А. К., Малышев В. В., Родин С. С., Ж. всесоюзн. хим. общ. им. Д. И. Менделеева, 8 (1963) 227.
27. Kraus К. A., Carlson Т. A., Johnson J. S., Nature, 177 (1956) 1128.
28. Baetsle J., van Deyck D., Huys D., Guery A., Report CEN, BLG-267 (1964).
29. Amphlett С. B., McDonald L. A., Redmann M. J., J. Inorg. Nucl. Chem., 6 (1958) 220.
30. ArakiS., SuzukiS., Yamada M„ Taianta, 19 (1972) 577.
31. Бригевич P. Ф., Кузнецов P. А. Радиохимия, 9 (1967) 693.
32. Ito T„ Abe M„ Bull. Chem. Soc. Japan, 34 (1961) 1736.
33. Ito T., 'Abe M., Denki Kagaku, 33 (1965) 175.
34. Caletka R., Koneiny C., Radiochem. Radioanal. Lett., 9 (1972) 285.
35. Abe M., J. Chromatogr., 134 (1977) 507.
174
Часть П
36. Nelson F., J. Amer. Chem. Soc., 77 (1955) 813.
37. Samuelson O., Sjostrom E., Anal. Chem., 26 (1954) 1908.
38. Yi-Liang, Lan-Chiang, Acta Chim. Sinica, 30 (1964) 117, 120.
39. Ruch R. R„ Tera F„ Morrison G. H„ Anal. Chem., 36 (1964) 2311.
40. Shabana R„ Ruf H, Report KFK-2460, 1977.
41. Okuno H., Honda M., Ishimoru T., Japan Analyst, 2 (1953) 428.
42. Ratner R., Ludmer Z., Israel J. Chem., 2 (1964) 21.
43. Everett R. J., Mottola H. A., Anal. Chim. Acta, 54 (1971) 309.
44. Botanero P. S., May S., Bull. Chim. France, 1971 , 1935.
45. Doge H. G., Anal. Chim. Acte, 38 (1967) 207.
46. Samuelson O., Schramm K., Z. Elektrochem, 57 (1953) 207.
47. Sulcek Z., Povondra P., Stangl R., Collect. Czech. Chem. Commun., 30 (1965) 380.
48. Cornet C„ Coursier J., Нигё J., Anal. Chim. Acta, 19 (1958) 259.
49. Nevoral Z„ Z. anal. Chem., 195 (1963) 332.
50. Mazzei I., Gualandini C., Burana G., Ann. Chim. (Rome), 53 (1963) 368.
51. Korkisch J., Orlandini K. A., Anal. Chem., 40 (1968) 1127.
52. Близнукова В. А., Кретенко А. П., Укр. хим. ж., 34 (1968) 156.
53. Olsen E. D„ Sobel H. R„ Taianta 12 (1965) 81.
54. Ganapathy S., et al., Indian J. Chem., 6 (1968) 277.
55. Mazza L., Sardo L., Frache R., Dadone A., Gazz. Chim. Ital., 95 (1965) 610.
56. Ratner R„ Itzchaki J., Kohn D. H., J. Appl. Chem., 18 (1968) 48.
57. Kawamura S., Kurotaki K., J. Chromatogr. 45 (1969) 331.
58. TorbkG., Diehl J. F., Radiochim. Acta, 15 (1971) 96.
59. Козаков E. В., Карпова И. E. Вести. Ленинград, унив., Хим., № 2 (1966) 139.
60. Lieser К. Н, Bastian J., Hecker А. В. Н, Z. anal. Chem., 228 (1967) 98.
61. Родин С. С., Лаврухина А. К., Радиохимия, 4 (1962) 623.
62. Holzapfel Н., Ehrhard Н., J. Prakt. Chem., 24 (1963) 92.
63. Pratchett A. G., Report AERE-R-3404, 1961. C. A. 56 (1962) 916b.
64. Johnson J. E., Wilson D. W., Ward G. M., Thompson R. D., Anal. Chim. Acta, 34 (1966) 59.
65. Boni A. L., Anal. Chem., 38 (1966) 89.
66. PucherS., Zement-Kalk-Gips, 14 (1961) 346.
67. Goetzee C. J., Anal. Chim. Acta, 44 (1969) 293.
68. Strelov/ F. W. E., Liebenberg C. J., von Toerien F. S., Anal. Chim. Acta, 47 (1969) 251.
69. Dyer F. E., Leddicotte G. W., The Radiochemistry of Copper, Washington USAEC, 1961.
70. Muzzarelli R. A. A., Talanta, 14 (1967) 85.
71. Maleszewska'H., Dybczynski R., Nukleonika, 12 (1967) 1181.
72. Wilkine D. H., Hibbs L. E., Anal. Chim. Acta, 20 (1959) 273.
73. Desai H. B., Padmanbhan P. K., Venkatesv/arlu Ch., J. Radioanal. Chem., 31 (1976) 525.
74. Fritz J. S., Millen W. G., Talanta, 18 (1971) 323.
5. Применение ионообменников
175
75. Nunes da Costa M. J., Jeronimo M. A. S., J. Chromatogr., 5 (1961) 456.
76. Kraus K. A., Nelson F., J. Amer. Chem. Soc., 75 (1954) 984.
77. Gibbons D., Intern. J. Appl. Radiation Isotopes, 4 (1958) 45.
78. Faris J. P, Anal. Chem., 32 (1960) 520.
79. Sherma J., Talanta, 9 (1962) 775.
80. Sherma J., J. Chromatogr., 19 (1965) 458.
81. Sunderman D. N., Townley C. W., The Radiochemistry of Silver, Washington USAEC, 1961.
82. Калинин А. И., Кузнецов P. А., Моисеев В. В., Мурин A. H. ДАН СССР, 141 (1961) 98.
83. Faris J. P., Buchanan R. F., Report ANL-6811, 1964.
84. Maleszewska H., Dybczynski R., Radiochem. Radioanal. Lett., 6 (1971) 7.
85. De A. K„ Majumdar S„ Talanta, 10 (1963) 201.
86. Mirza M. Y., Radiochim. Acta, 14 (1970) 61.
87. Shirmal R. L„ Talanta, 18 (1971) 1235.
88. Hirano S„ Mizuike A., lidy Y., Japan Analyst, 9 (1960) 423.
89. Kemula W., Brajtner K., Cieslik S., Lipinska-Kostrowicka H., Chem. Anal. (Warszaw), 5 (1960) 225.
90. RangenekarA. V., Khopkar S. M., Mikrochim. Acta, 1965, 642.
91. Burriel H., Alvarez H., Inform quim. analit., 18 (1964) 61.
92. Kielczewski W., Chem. Anal. (Warszaw), 8 (1963) 691.
93. Matsuo T„ Funada S.,Shinozaki A., J. Chem. Soc. Japan, 68 (1965) 1999.
94. Marti F. B., Herrero C. A., Inform, quim. anal., 16 (1962) 121.
95. Sakamoto T., Nakashima T„ Fukada K., Mizuike A., Japan Analyst, 19 (1970) 1218.
96. Kutil J., Cuta F., Collect., Czech. Chem. Commun., 32 (1967) 1390.
97. Hirano S., Mizuike A., Yamada K„ J. Chem. Soc. Japan Ind. Chem. Sect., 62 (1959) 1494.
98. Калинин А. И., Кузнецов P. А., Моисеев В. В. Радиохимия, 4 (1962) 575.
99. StrelowF. W. E„ Victor A. H„ Anal. Chim. Acta, 59 (1972) 389.
100. Fritz J. S„ Millen W. G„ Talanta, 18 (1971) 323.
101. Bhatnagar R. P., Shikla R. P., Anal. Chem., 32 (1960) 777.
102. Nelson F., Michelson D. C., J. Chromatogr., 25 (1966) 414.
103. Iyer R. K., Krishnamoorthy K. R., J. Radioanal. Chem., 33 (1976) 243.
104. Wodkiewicz L., Dybczynski R., J. Chromatogr., 102 (1974) 277.
105. StrelowF. W., Victor A. H., Stern J., Lachmann H. H., Talanta, 23(1976)
173.
5.2.2. Группа 2
5.2.2.1. 2 A: Be, Mg, Ca, Sr, Ba, Ra
Катионообменники имеют большое значение для разделения этой группы ионов. Образование комплексов различной устойчивости с большим числом комплексообразующих агентов обеспечивает возможность эффективного и быстрого отделения ионов этой группы от других элементов и разделения ионов внутри группы.
Часть П
176
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Для разделения элементов этой группы преимущественно используют сильнокислотные катионообменники полистирольного типа. Сродство к функциональной группе — SO3H возрастает в ряду Mg < Ga < < Sr < Ba < Ra (в отсутствие комплексообразующих веществ в растворе).
Бериллий занимает особое место в этом ряду. Он легко сорбируется на катионообменниках из разбавленных растворов минеральных кислот; поглощение Ве2+ уменьшается с увеличением концентрации кислоты. Кроме тогр, сорбция, особенно в области более низких концентраций кислоты, зависит от природы кислоты и возрастает в ряду HCI < HNO3 < H2SO4. Таким образом, в 1,5 М растворе соляной кислоты бериллий отделяется от ионов щелочноземельных металлов, которые сорбируются на смоле Dowex 50.
Поглощение Ве2+ катионообменниками увеличивается с повышением pH раствора вследствие образования основных полиядерных комплексов. Максимальная сорбция Be24 на смоле Dowex 50-Х8 наблюдается при pH 6,5. По-видимому, наиболее селективный метод отделения Be основан на том, что он не образует прочных комплексов с ЭДТА и удерживается катионообменной смолой. Такие элементы, как Cu, Pb, Ni, Со, Zn, Al, Th, десорбируются 0,5%-ным раствором ЭДТА при pH 3,7. Для элюирования А1, Fe(III) или Ti(IV) использовали 0,5%-ный раствор ЭДТА с pH 3,3. Вместе с бериллием сорбируются только щелочные металлы, U(VI), U(IV) и Ag(I).
Более прочные комплексы бериллия с оксалат-ионами и особенно с салицилат-, сульфосалицилат-, лактат- и карбонат-ионами не сорбируются на катионообменных смолах.
Магний, кальций, стронций, барий и радий имеют разное сродство к сильнокислотным катионообменникам в растворах минеральных кислот, которое повышается в ряду Mg < Ca < Sr < Ba < Ra. Этого различия достаточно для успешного отделения кальция от магния (1,0 М НС1) или стронция (2,5 М НО).
Качество разделения существенно зависит от степени поперечного связывания смолы и температуры. Для разделения смеси Са — Sr — Ва предпочтительнее использовать смолу с 16% ДВБ. Хорошее разделение наблюдается при 50 °C; при более низких температурах разделение ухудшается. Снижение содержания ДВБ в смоле до 8% приводит к улучшению разделения Sr — Ва, но при разделении пары Са — Sr было получено худшее разрешение. Смолы, содержащие менее 8% ДВБ, характеризуются очень плохим разрешением [1].
Соляная кислота является подходящей средой для отделения ионов щелочных металлов и других ионов, образующих стабильные хлоридные комплексы [Cu, Hg(II), Zn, Cd, U(VI), элементы платиновой группы].
Фтористоводородная кислота имеет важное значение для отделения магния от элементов, образующих устойчивые фторидные комплексы (особенно для разделения пары Mg — Zr).
5. Применение ионообменников
177
Таблица 5.11. Факторы разделения пар щелочноземельных металлов на катнонообменннке КУ-2 нли Dowex 50-Х12 в различных средах [8]
Элюент
Фактор разделения а смеси
Mg-Be Са—Mg Sr—Ga Ва—Sr Са-Ва
Ацетат NH + 20 — 2,2 2,5 4,5 2,2
90 — 2,0 2,8 2,4 1,55
1,2 М лак- 20 — — 1,5 2,5
тат NH* 90 3,7 2,8 3,1 3,2 2,1
5%-ный цнт- 80 — 1,7 1,8 3,4
рат NH +, pH 5,4 НС1а 80 2,3 (1,5 М) 4,0 (1,5 М) 1,8 (2 М)
3,3 (2,5 М) 2,3 (4 М) 3,0 (4 М) 1,1 (6 М)
а В скобках стоит концентрация.
Наряду с минеральными кислотами для разделения элементов группы 2А часто используют комплексообразующие вещества, которые значительно повышают селективность.
В качестве элюентов для разделения смесей щелочноземельных металлов на сильнокислотных катионообменниках успешно применяют растворы, содержащие формиат- [2], ацетат- [3, 4], цитрат- [5], лактат- [б] или а-оксиизобутират- [7] ионы. Эффективность разделения уменьшается в ряду а-оксиизобутират > лактат > цитрат (> соляная кислота). Сравнение эффективности разделения элементов различными элюентами представлено в табл. 5.11.
Таблица 5.12. Сравнение значений Dg и факторов разделения смесн Ва2+— Ra2+ на ионообменниках разных типов в среде НС1 и цитрата аммония [16]
Ионообменник НС1 54^-ный цитрат Фактор
аммония, pH 7—9 разделения а
Ва Ra Ва Ra HCI цитрат
Dowex 5О-Х8 16,2 22,9 6,15 13,5 1,41 2,20
Dowex 50-Х10 23,8 36,0 6,3 16,0 1,51 2,54
КУ-2-Х8 16,2 22,9 6,08 12,8 1,41 2,11
SBS-P 2,37 3,20 1,20 1,8 1,35 1,50
РФ 0,20 0,30 3,2 4,6 — 1,44
12-354
178
Часть II
Таблица 5.13. Значения D* и а смеси Ва2+ — Ra2 + в 4%-ном ЭДТА на катионообменнике КУ-2 [Гб]
Ва Ra
5,00 48,6 90,0 1,85
5,50 24,7 72,4 2,93
5,75 17,6 56,7 3,22
6,00 8,40 42,0 5,00
6,25 4,55 23,1 5,08
6,50 2,90 14,6 5,05
7,00 0,50 2,5 5,00
Часто используют следующие комплексообразующие элюирующие агенты: ЭДТА [9—11], ЦГДТА [12, 13], ЭГТА [14]. В среде ЭГТА достигается высокоселективное отделение кальция от магния [14]. Для разделения пары Ва — Ra успешно применяется аммониевая соль ЭДТА [15], которая обеспечивает более эффективное разделение по сравнению с цитратом аммония. Коэффициенты распределения и факторы разделения Ва2+ — Ra2+ в разных системах приведены в табл. 5.12 и 5.13.
Смесь Be, Mg, Са и Ва успешно разделяется на смоле AG-50 при элюировании раствором малоната аммония с постепенно повышающейся концентрацией (от 0,1 до 1,1 М). Этим способом элюируют Be, Mg и Са. Барий удерживается смолой и десорбируется 3 М азотной кислотой [17].
НЕОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Более низкое сродство щелочноземельных металлов к неорганическим ионообменникам позволяет проводить отделение этих элементов от более тяжелых ионов щелочных металлов. В качестве элюента для их количественного разделения чаще всего используют раствор хлорида аммония в соляной кислоте с увеличивающейся концентрацией [20].
В качестве примера можно привести разделение смеси Са — — Sr — Ва — Ra на молибдате циркония [18], который характеризуется большими различиями в сродстве к отдельным ионам. Быстрое разделение смеси происходит при использовании метода ступенчатого или градиентного элюирования. Кроме того, большие различия в сродстве к отдельным ионам позволяют применять короткие колонки, особенно при разделении пары Ва — Ra.
Ванадат титана(1У) был рекомендован Квереши [19] в качестве высокоселективного неорганического ионообменника для отделения стронция.
Неорганические ионообменники перспективны для отделения дочерних продуктов (редкоземельные элементы) от материнских изотопов щелоч
5. Применение ионообменников
179
ных металлов. Например, дочерний изотоп 137Ва отделяется от материнского изотопа 137Cs на колонке, заполненной фосфатом циркония. Ва элюируют 1 М азотной кислотой [20].
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Щелочноземельные металлы либо очень слабо (Dg < 10), либо вообще не сорбируются на сильноосновйых анионообменниках из растворов минеральных кислот. Поэтому эти элементы легко отделяются от ряда других комплексообразующих элементов, которые хорошо сорбируются анионооб-менниками из растворов минеральных кислот.
Бериллий не поглощается сильноосновными анионообменниками из растворов серной кислоты, и поэтому его можно селективно отделить от некоторых ионов. Для отделения урана важна сернокислая среда.
Для выделения бериллия из водных или кислых растворов анионообменники используются в меньшей степени по сравнению с катионообменни-ками. С анионами минеральных кислот бериллий образует очень непрочные анионные комплексы. Следовательно, он практически не поглощается во всем интервале концентраций соляной или азотной кислот. Однако при замене значительной части воды соответствующим органическим растворителем ионы Ве2+ сорбируются анионообменниками (табл. 5.14). В табл. 5.14 для сравнения приведены значения коэффициентов распределения алюминия и магния.
Ионы Ве2+ сильно сорбируются из разбавленных растворов фтористоводородной кислоты [23]. Значительное уменьшение сорбции наблюдается в среде HF — НО.
Таблица 5.14. Коэффициенты распределения D Be, Mg и Al на смоле Dowex 1-Х8 в водно-органической среде, содержащей (в об. %) 90 органического растворителя, 10 азотной нли соляной кислоты [22]
Органический растворитель 5 М HNO3 6 M HCI
Be Mg Al Be Mg- Al
Метанол 2 3 2 1,5 3 2
Этанол 2 4 2,5 2 6,5 4
1-Пропанол 2 11 6 5 22 22
2-Пропанол 3 10 6 6 22 24
1-Бутанол 4 20 6 6 33 35
2-Бутанол 5 18 10 9 36 80
Ацетон 4 8 11 10 33 40
Метнленгликоль 2 2 2 3 3,5 3
Этиленгликоль 2 2 2 3,5 7 6
Т етрагндрофуран 4 8 10 7,5 22 15
Диоксан 5 12 10 — — —
Вода 1 1 1 1 1 1
180
Часть II
Азотная и особенно фтористоводородная кислота имеют большое значение для группового отделения этих элементов от других многозарядных ионов, а также от элементов, образующих устойчивые хлоридные или нитратные комплексы. Эту среду можно успешно применять для отделения радия от полония, урана, висмута, свинца и протактиния [24, 25].
Важное практическое значение имеет отсутствие поглощения ионов Ва2+ и Sr2+ анионообменниками в ОН-форме. На этом основано быстрое разделение пар изотопов ^Sr — и 140La — 140Ва (сорбируются только трехзарядные ионы).
Упомянутые выше хелатообразующие вещества дают возможность проводить как анионообменное отделение группы щелочноземельных металлов от других элементов, так и анионообменное разделение элементов этой группы на индивидуальные компоненты. Последовательное элюирование индивидуальных щелочноземельных металлов можно осуществить соответствующим изменением pH элюирующего раствора [26]. Разделение элементов, как и в случае катионообменников, происходит за счет селективной сорбции или элюирования. Эффективность разделения и порядок элюирования индивидуальных элементов зависит от типа присутствующего хелатообразующего агента и pH раствора. Сродство щелочноземельных металлов к анионообменнику Dowex 1 в цитратной среде, например, уменьшается в ряду Mg > Са > Sr > Ba > Ra. Эту среду можно успешно использовать для разделения смеси Ba — Ra (радий элюируется первым).
Несмотря на то что радий образует анионные комплексы (поглощаемые анионообменниками) со многими органическими кислотами, все же анионообменники не находят широкого применения для отделения радия (особенно от бария). Чаще всего анионообменники используют для отделения радия от продуктов его радиоактивного распада, которые образуют анионные комплексы в растворах минеральных кислот. Полоний, висмут и свинец отделяют от радия на смоле Dowex 1 в 2 М НС1. В этих условиях радий, как и барий, не элюируется [27].
Сильноосновные анионообменники перспективны для отделения радия от тория в 0,5 — 1,0 М растворе HNO3, содержащем 80 об. % метанола. При этом наблюдается только незначительная сорбция радия [28].
Лабораторные методики
ОТДЕЛЕНИЕ Ве ОТ РЯДА ЭЛЕМЕНТОВ [22]
Методика. Колонку (30 х 1 см), заполненную смолой Dowex 1-Х8 в С1-форме (размер зерен 0,15—0,07 мм), промывают 100 см3 раствора, содержащего 90% изопропилового спирта и 10% б М НС1. В анализируемый раствор вводят изопропиловый спирт и соляную кислоту до указанных выше концентраций и пропускают весь полученный раствор через колонку со скоростью 0,2 см3/мин. После этого обменник промывают смесью изопропиловый спирт — б М НС1 (9 : 1). Порции по 5 см3 (или ббльшие) анализируют на содержание бериллия. Бериллий всегда элюируется первым. Другие элементы удерживаются обменником и только очень медленно перемещаются вдоль колонки. При анализе смеси, содержащей 5 мг Ве и по 10 мг каждого
5. Применение ионообменников
181
из следующих металлов: Mg, Са, Sr, Ba, Си, Zn, Hg, Al, Ga, In, Sc, Y, La, Sn, Pb, Ti, Zr, Th, Bi, V, Cr, Mo, U(VI), Fe, Co, Ni, найдено 5 ± 0,01 мг Be. Вместо 6 M HC1 можно использовать 5 M HNO3 (см. табл. 5.13). Эта методика, в частности, рекомендуется для отделения Ва2+ от больших количеств ионов UO^+ .
РАЗДЕЛЕНИЕ СМЕСИ Be — А1 [29]
Методика. Раствор, содержащий оба иона, доводят до pH 2,5 и пропускают через колонку, заполненную сильноосновным анионообменником АВ-17 в СО3-форме. Колонку промывают 15—20 см3 воды и десорбируют А13+ 200 см3 4 М NH4OH или 70 см3 1 М NaOH.
Ве2+ элюируют 450 см3 2,5 М (NH4)2CO3. Оба элемента определяют полярографически.
РАЗДЕЛЕНИЕ СМЕСИ Be — Mg [30]
Методика. Смесь, содержащую около 2 ммолей бериллия и 1 ммоль магния, сорбируют на колонке (внутренний диаметр 15 мм), заполненной 5 г сильнокислотного катионообменника AG 5OW-8X в ИН4-форме (размер зерен 0,074—0,037 мм). После промывания колонки водой бериллий элюируют 0,10 М раствором малеата аммония (pH 7,5) со скоростью 2 см3/мин. Бериллий элюируется первым.
РАЗДЕЛЕНИЕ СМЕСИ Са — Sr 0,10 М РАСТВОРОМ ЭДТА [30]
Методика. Смесь обоих элементов сорбируют на колонке, заполненной 20 г обменника AG 5OW-X8 в ИН4-форме (размер зерен 0,074—0,037 мм). После промывания колонки водой элементы элюируют 0,10 М раствором ЭДТА, содержащим 0,30 М CH3COONH4 (pH 5). Кальций элюируется первым.
СЕЛЕКТИВНОЕ ОТДЕЛЕНИЕ Са (И ДРУГИХ МЕТАЛЛОВ) ОТ Mg РАСТВОРОМ ЭГТА [14]
Методика. В анализируемый раствор вводят приблизительно 10%-ный избыток хелатообразующего агента (относительно ожидаемого количества разделяемых элементов), доводят его до pH 6,5—7,0 и пропускают со скоростью 2—4 см3/мин через колонку, заполненную смолой Dowex 50-Х8 в NH4^opMe. Колонку предварительно промывают 0,05 М раствором ЭГТА с pH 6,5—7,0. После сорбции обменник промывают 30—50 см3 0,03 М ЭГТА (pH 6,5—7,0) и 50—100 см3 воды. В этих условиях на обменнике сорбируются только ионы Mg2+, которые легко элюируются 2—3 М НС1. Этот метод пригоден для отделения магния от больших количеств кальция, стронция, железа, меди и других поливалентных металлов.
СЕЛЕКТИВНОЕ ОТДЕЛЕНИЕ Са (Mg, Sr, Ва) от Sc, Fe, Ga, In, U и Th [31]
Методика. Для разделения используют полистирол-ДВБ обменник с — РО(ОН)2 функциональными группами в Н-форме. В раствор, содержащий перечисленные выше металлы, вводят азотную или соляную кислоту примерно до 0,25 М концентрации и пропускают со скоростью 1 см3/мин через ионообменную колонку. После завершения сорбции обменник промывают 0,2 М раствором кислоты. Ионы кальция (щелочноземельные металлы) количественно переходят в фильтрат, тогда как другие ионы очень прочно удерживаются обменником.
182
Часть II
ОТДЕЛЕНИЕ Sr ОТ БОЛЬШИХ КОЛИЧЕСТВ КАЛЬЦИЯ [32]
Методика. Смесь Sr — Са осаждают в исходном растворе в виде карбонатов. Осадки промывают и растворяют в минимальном количестве соляной кислоты при нагревании. Полученный раствор охлаждают и разбавляют до определенного объема.
Аликвотную часть раствора доводят до pH 9,0 гидроксидом аммония и титруют стандартным раствором ЦГДТА. В другую аликвотную часть раствора вводят небольшой избыток ЦГДТА (по сравнению с найденным титрованием) и устанавливают 0,4 М концентрацию ионов NHJ и pH 5,10. Полученный раствор пропускают со скоростью 3 см3/мин через колонку, заполненную смолой Dowex 50-Х12 (0,30— 0,15 мм) в Ь1Н4-форме. Затем колонку промывают 30—50 см3 воды. В фильтрат переходит только кальций. Сорбированный на колонке стронций элюируют 4 М НС1 или раствором состава 0,03 М ЦГДТА — 0,4 М NH4NO3 с pH 6,7. Можно использовать другой элюент такого состава: 10 см3 0,05 М ЭДТА — 20 см3 буферного раствора Шварценбаха, разбавленного водой до 100 см3. Дня приготовления буферного раствора 54 г NH4C1 растворяют в 200 см3 воды, прибавляют 350 см3 25%-ного раствора NH4OH и разбавляют водой до 1000 см3.
Элюат упаривают до 30 см3, вводят 30 см3 концентрированной HNO3 и выпаривают досуха. В остатке определяют Sr. Ошибка определения составляет ±1% при соотношении Sr:Ca около 200. Вместо смолы Dowex 50 можно использовать Amberlite IR-120. При разделении необходим точный контроль концентрации ионов NH^ и pH как. на стадии сорбции, так и на стадии элюирования.
РАЗДЕЛЕНИЕ СМЕСИ Mg—Са—Sr НА АНИОНООБМЕННИКЕ [33]
Методика. Колонку, заполненную макропористым сильноосновным анионооб-менником Amberlyste XN-1002 в МО3-форме, промывают 40—50 см3 смеси 95% этанола и 0,25 М HNO3. В анализируемом растворе (20—30 см3) устанавливают такие же концентрации этанола и HNO3 и пропускают .через колонку со скоростью 0,5 см3/мин. Ионы Mg2+ элюируют смесью 0,25 М HNO3 — 95% этанола, Са2 + элюируют смесью 0,25 М HNO3 — 95% метанола. Ионы Sr2+ десорбируют водой или метанолом.
Для приготовления смеси 0,25 М HNO3 — 95% С2Н5ОН 1 объем 5 М HNO3 смешивают с 19 объемами спирта. Воду прибавляют в количестве, соответствующем уменьшению объема при смешивании кислоты и спирта.
РАЗДЕЛЕНИЕ СМЕСИ Ba — Ra
Методика. Раствор, содержащий смесь микроколнчеств нонов Ва2+ — Ra2+ , выпаривают в тефлоновом сосуде практически досуха. К остатку прибавляют примерно 0,4 см3 0,01 М (NH4)2H2 ЭДТА, доведенного до pH 8,8 концентрированным раствором NH4OH. Колонку (12 х 0,6 см), заполненную смолой Dowex 5О-Х8 в МН4-форме (0,037 мм, высота слоя 30 мм), промывают ~ 2 см3 0,01 М раствора (NH4)2H2 ЭДТА со скоростью 0,6 см3/мин. Через колонку пропускают анализируемый раствор, 0,4 см3 промывного раствора и элюируют ноны Ва2+ 2,5 см3 этого раствора. Ионы Ra2+ десорбируют 1,7—2,0 см3 0,01 М раствора ЭДТА с pH 10,5. Продолжительность разделения около 30 мин.
5. Применение ионообменников
183
РАЗДЕЛЕНИЕ СМЕСИ Ba — РЬ [34]
Методика. Оба элемента сорбируют на сильнокислотном катионообменнике в МНд-форме. Колонку промывают водой и элюируют ионы Pb2 + 1 М раствором ацетата аммония. Ионы Ва2 + элюируют 5%-ным раствором соляной кислоты.
БЫСТРОЕ ОТДЕЛЕНИЕ 24Na и 27Mg от А1 [35]
Методика. Кусок алюминиевой фольги (54 мг) после облучения в ядерном реакторе растворяют при нагревании в 1 см3 6 М НС1 и прибавляют эквивалентное количество горячего раствора ЭДТА. Раствор охлаждают, устанавливают pH 7,5 с помощью раствора NH4OH и пропускают через колонку, заполненную сильноосновным анионообменником Dowex 2-Х8 в смешанной (1 : 1) ОН — СН3 СОО-форме. Скорость пропускания раствора 2,5 см3/(мин • см2). 24Na проходит через колонку; 27Mg элюируют 1 М СН3СООН. Ионы алюминия удерживаются обменником.
РАЗДЕЛЕНИЕ СВОБОДНЫХ ОТ НОСИТЕЛЯ ИЗОТОПОВ
90Sr _ 9оу и 140Ва - ,4oLa [36]
Методика. Разделение выполняют на колонке (7 х 0,4 см), заполненной смолой Dowex 1-Х8 в СО3-форме. Через колонку пропускают 50 см3 анализируемого раствора со скоростью 1,5 см3/мин и затем промывают 50 см3 0,01 М (NH4)2CO3 или воды. Изотопы стронция или бария количественно переходят в элюат. Сорбированные трехвалентные элементы элюируют 1 М НС1. В процессе десорбции из обменника выделяется СО2, что приводит к образованию в ионообменной колонке пузырьков. Компактность колонки восстанавливается при последующем промывании кислотой.
Образование пузырьков можно предотвратить, превратив обменник (перед элюированием кислотой) в Cl-форму промыванием разбавленным раствором NH4C1. Получены следующие значения коэффициентов распределения: 144 для La, 4,2 для Ва в 0,02 М (NH4)2CO3; 327 для La, 2,6 для Ва в 0,04 М растворе карбоната; 199 для La, 2,0 для Ва в 0,08 М растворе карбоната. Смесь 90Sr — 90Y можно также разделить на смоле Dowex 1-Х8 в 5О4-форме элюированием стронция разбавленным метанолом [37] или на обменнике в НСО3-форме [38].
Для разделения смесн 140 Ва — 140 La на смоле Dowex 50-Х8 в качестве элюента используют смесь 5%-ная лимонная кислота — цитрат аммония (pH 5,5) [39].
РАЗДЕЛЕНИЕ СМЕСИ Са—Sr—Ва ЛАКТАТОМ АММОНИЯ [7]
Методика. Смесь элементов сорбируют на катионообменнике Dowex 50-Х8 в МН4-форме и элюируют раствором лактата аммония следующих концентраций:
Са2+ 0,8 М раствором с pH 4,15,
Sr2+ — 1,0 М раствором с pH 5,00,
Ва2+ — 2,0 М раствором с pH 6,20 или 6 М НС1.
Можно также элюировать 1,5 М раствором лактата аммония [6].
РАЗДЕЛЕНИЕ СМЕСИ ИОНОВ ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ а-ОКСИИЗОБУТИРАТОМ АММОНИЯ [40]
Методика. Смесь ионов щелочноземельных металлов сорбируют на сильнокислотном катионообменнике в МН4-форме. Колонку промывают водой и элюируют элементы растворами а-оксиизомасляной кислоты различной концентрации, нейтрализованными NH4OH до указанных выше значений pH: Mg — 0,8 М раствором с pH 4,15; Са — 1,0 М раствором с pH 5,00; Ва — 2,0 М раствором с pH 6,20.
184
Часть II
РАЗДЕЛЕНИЕ МИКРОКОЛИЧЕСТВ 223 Ra — 227 Ас (В ПРИСУТСТВИИ 223 Fr, 211Pb, 211Bi, 231Ра) [41]
Методика. Для разделения используют колонку (10 х 0,6 см), заполненную сильнокислотным катионообменником Dowex 50 в Н-форме (0,074—0,037 мм). Колонку, снабженную водяной рубашкой, нагревают до 60 °C. В верхнюю часть колонки помещают 5 см3 смеси в 2 М НС1 при 80 °C и промывают колонку 5 см3 горячей 2 М НС1. Этим способом элюируют Fr, Pb, Bi и Ра. Радий десорбируют следующей порцией объемом 20 см3 горячей 3 М HNO3, актиний — 25 см3 горячей 6 М HNO3. Первую порцию (5 см3) элюата не анализируют.
Разделение подобных смесей (включая 227 Th) выполняют также на ионообменни-ке Dowex 50-Х4. Ионы Fr, Pb, Bi, Ра, Ra элюируют смесью 8,5 М НС1О4 — 0,5 М НС1. Радий десорбируется последним. Актиний элюируют 6,0 М HNO3, торий — смесью 6 М НС1 — 1 М HF [42].
ОПРЕДЕЛЕНИЕ ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ В СТЕКЛЕ {6, 43]
Методика. 1 г образца разлагают смесью H2SO4 — HF. Раствор выпаривают до появления белых паров SO), охлаждают, разбавляют до 5 см3 и прибавляют 40 см3 80%-ного этанола. Образующийся осадок, который содержит все щелочноземельные и другие присутствующие металлы, отфильтровывают и промывают смесью 80% С2Н5ОН — 10% H2SO4. Осадок переносят в стакан, фильтр озоляют и золу объединяют с основным осадком. Затем прибавляют 7 г твердого иодида аммония, 40 см3 концентрированной НС1, разбавляют до 150 см3 и выпаривают полученную смесь досуха. Избыток солей удаляют последующим нагреванием.
Остаток растворяют в небольшом количестве разбавленной НС1 с добавлением нескольких капель бромида. Элементы, образующие нерастворимые гидроксиды, удаляют повторным осаждением гидроксидом аммония. Объединенные фильтраты упаривают до объема 40 см3 и подщелачивают до сильнощелочной реакции. Щелочноземельные металлы осаждают в виде карбонатов введением 3 г карбоната натрия. Осадок карбонатов отфильтровывают, растворяют в небольшом количестве НС1 и выпаривают досуха. Остаток растворяют в 5 см3 воды.
Раствор пропускают со скоростью 1,4 см3/мин через колонку (19 х 1,5 см), заполненную смолой Dowex 50-Х8 в 1ЧН4-форме (мелкого зернения), которую предварительно приводят в равновесие с 1,20 М раствором лактата аммония и затем промывают водой. Колонку, содержащую сорбированные элементы, промывают водой и элюируют отдельные ионы 1,20 М раствором лактата аммония в следующем порядке: Са — Sr — Ва. Скорость элюирования около 1 см3/мин [10].
ОПРЕДЕЛЕНИЕ Sr В КОСТЯХ [10]
Методика. Пробу костей прокаливают и 1,000 г золы растворяют в соляной кислоте. Раствор профильтровывают и выпаривают досуха. Остаток растворяют в 200 см3 2%-ного раствора ЭДТА (аммониевая соль), в растворе устанавливают pH 4,8 и пропускают его со скоростью 1—2 см3/мин через колонку, заполненную ионообменной смолой Dowex 50-Х8.
Подготовка колонки. Колонку (28 х 1,2 см) , заполненную смолой Dowex 50-Х8 (0,30—0,15 мм), промывают 250 см3 3 М НС1 и 50 см3 воды. Затем смолу переводят в Ь1Н4-форму обработкой 150 см3 10%-ного раствора NH4C1 и промывают 50 см3 2%-ного раствора аммониевой соли ЭДТА с pH 5,3 (20 г ЭДТА растворяют в нескольких куб. сантиметрах NH4OH и разбавляют водой до 1000 см3).
£
I
ч
S
5
5
3
Таблица 5.15. Разделение смесей, содержащих ноны щелочноземельных металлов
5
X 6
Элемент Отделяемые Ионообменник Условия разделения Лите-
компоненты ратура
X о
b о О
и
("
Я
2
X
о X
U
Д
S
ей
5. Применение ионообменников
187
Кальций элюируют 250 см3 смеси NH^ — ЭДТА с pH 5,30; скорость потока 1—2 см3/мин. После промывания колонки 50 см3 воды магний и щелочные металлы десорбируют 250 см3 0,75 М НС1. Стронций элюируют 250 см3 3 М НС1. Элюат выпаривают досуха и остаток растворяют в воде. Содержание стронция определяют пламенно-фотометрическим методом.
Для регенерации колонки используют 2%-ный раствор NH4+— ЭДТА (pH 5,30).
Относительная ошибка метода меньше 5%. Примеры разделения других смесей, содержащих щелочноземельные элементы, приведены в табл. 5.15.
5.2.2.2. 2 Б: Zn, Cd, Hg
Эти элементы образуют прочные комплексы со многими неорганическими и органическими веществами, которые используются при их разделении и отделении от других ионов. Особое значение имеют комплексы с галогенсодержащими кислотами.
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Сорбция всех трех элементов уменьшается с повышением концентрации
<9
минеральной кислоты в растворе, особенно в среде галогенсодержащих кислот. В случае ионов Hg2+ снижение сорбции весьма значительно вследствие образования несорбируемых комплексов Исключение составляет нитрат ртути (II)].
Ионы Zn2+ сорбируются сильнокислотными катионообменниками только из разбавленных растворов соляной кислоты. На смоле Dowex 50-Х8 в 0,1 М НС1 получены следующие значения коэффициентов распределения Z>s: 1850 для Zn2+ , 510 для Cd2+ , 1,6 для Hg2+ . Сорбция Zn заметно снижается в > 1 М растворах НС1.
Низкое сродство ионов Cd2+ и Hg2+ к катионообменникам в кислых растворах используется для их отделения от. Си, U, Ti, Fe, Al, Th, Zr и других металлов; ионы Cd2+ легко элюируются разбавленной минеральной кислотой.
НЕОРГАНИЧЕСКИЕ ИОНООБМЕННИКИ
Сорбция ионов цинка, кадмия и ртути на неорганических ионообменни-ках увеличивается с повышением pH раствора. Установлен следующий ряд сродства: Zn2+ > Cd2+ > Hg2+ . Поскольку металлы этой группы легко разделяются на органических анионообменниках, неорганические ионообменники не.нашли широкого применения'для их разделения.
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Наиболее широко распространенные методы разделения элементов этой группы основаны на сорбцин их анионообменниками из растворов галогенсодержащих кислот.
Ионы Zn2+ образуют с галогенид-ионами комплексные соединения, устойчивость которых уменьшается в ряду ZnCI4~ > ZnBr4~ > Znl4“.
188
Часть 11
Для ионов Cd2+ действителен обратный порядок: Cdl4“ > > CdBr4“ > CdCI^- . Сорбция ZnCI^- на сильноосновном анионообменни-ке Dowex 1 увеличивается с повышением концентрации НС1. Найдены следующие величины D : 1 в 0,1 М НС1, 1800 (максимум) в 2 М НО, 55 в 12 М НС1.
Подобное сорбционное поведение характерно также для ионов Cd2+ и Hg2+, но в отличие от ионов Zn2+ сорбция этих двух элементов из разбавленных растворов НС1 более сильная.
Сродство комплексов Cd и Hg к ионообменникам в растворах броми-стоводородной кислоты выше, чем в растворах соляной кислоты. Для комплексов Zn2+ наблюдается обратное поведение [61].
Лабораторные методики
ОТДЕЛЕНИЕ Cd ОТ БОЛЬШИХ КОЛИЧЕСТВ ЦИНКА [62]
Методика. Колонку (25 х 2,5 см), заполненную катионообменником Dowex 50-Х8 (размер зерен 0,07—0,14 мм) в Н-форме, промывают смесью 0,15 М H2SO4—0,30 М Н1. Объем промывного раствора должен быть равен объему смолы; водой колонку не промывают.
Около 2 г анализируемого образца растворяют в серной кислоте в присутствии ио дистовод ородной кислоты, разбавляют до 150 см3 и устанавливают 0,15 и 0,30 М концентрации H2SO4 и Н1 соответственно.
Полученный раствор пропускают через ионообменную колонку со скоростью 7 см3/мин. Колонку промывают 3-кратным количеством (по сравнению с объемом смолы) смеси 0,15 М H2SO4 — 0,30 М HI. Ионы Cd2+ при этом элюируются количественно. Цинк десорбируют 2 М НС1.
РАЗДЕЛЕНИЕ СМЕСИ Мп — Со — Zn — Си — Fe НА АНИОНО-ОБМЕННИКЕ [63]
Этот метод пригоден для разделения микрограммовых количеств указанных элементов.
Методика. К ~2 см3 пробы, содержащей 10—15 мкг каждого из этих металлов, прибавляют тартрат натрия до 2,12 10“3 М концентрации, устанавливают pH 4,6 и пропускают раствор со скоростью 3—4 капли/мин через колонку (100 х 8 мм), заполненную анионообменником Dowex 2-Х8 в тартратной форме. В качестве элюеита используют 2,12 • 10-3 М раствор тартрата натрия (pH 4,6). Для элюирования максимального количества Мп требуется 5 объемов колонки, Со — 20 объемов колонки, Zn — 40 объемов колонки, Си — 70 объемов колонки. Ионы Fe3 + элюируют 1 М НС1.
ОПРЕДЕЛЕНИЕ Zn В СПЛАВАХ МАГНИЯ [64]
Сплавы магния могут содержать цинк, железо, алюминий, марганец и медь.
Методика. Колонку соответствующего размера (в зависимости от количества аликвотной пробы), заполненную сильноосновным анионообменником, промывают
Св О.
се о.
зГ: s: «гу ц S С о” С
I
S Я
S ч
CQ
чО Г-
чО 40
О —
® о
Z
X о, X
S
о" о z.
д •X
д. Z 2 I Z- z х u?iz
X Z
о и
о
Q <
С .•—S
N 5 2
Z « | „ | s
„ - аа о н
£ б £ 5 | р | 5 g
Я <’ m
- л4 '•« • я D. '
° <U r-f _ 35 =! СЗ S 3 О с Ё
u£3du2uu 2 5 О -S ч
ею ею
X X
s о
з iB
5 3 - 3 •и К О <
= .5? N X
О
О
190
Часть II
100 см3 2,5 М НС1. Образец сплава растворяют при нагревании в разбавленной соляной кислоте (1 : 1), содержащей несколько капель концентрированной HNO3. Раствор выпаривают досуха. Остаток растворяют в горячей 2,5 М НС1 и пропускают раствор через колонку, которую затем промывают 100 см3 2,5 М НС1.
Только цинк сорбируется ионообменником. Все другие упомянутые выше металлы (включая магний) проходят в элюат. Цинк элюируют 100 см3 0,02 М НС1 и определяют подходящим методом. Относительная ошибка при определении 0,2% Zn составляет около ± 5%, а при определении 1,2% Zn — около ± 2%.
ОПРЕДЕЛЕНИЕ Zn, Pb и Bi В МЕТАЛЛИЧЕСКОМ ВАНАДИИ [64]
Методика. Образец металлического ванадия растворяют в смеси НС1 — HNO3 (3 : 1). Раствор упаривают примерно до 2 см3, вводят 15 см3 концентрированной НС1 и выпаривают досуха. Эту операцию повторяют трижды.
К сухому остатку прибавляют 25 см3 2,5 М НС1 и полученный раствор со скоростью 0,5 см3/мин пропускают через колонку, заполненную анионообменником ТМ или ЭДЭ-Юп, предварительно промытым 100 см3 2,5 М НС1.
После этого смолу промывают 25 см3 2,5 М НС1 . Весь ванадий проходит в элюат. Цинк, свинец и висмут сорбируются. Ионы Zn2+ и РЬ2+ элюируют 100 см3 0,02 М НС1. Висмут десорбируют последним 1 М H2SO4. Относительная ошибка определения 2 • 10~3 — 2 • 10-4% Zn, Pb и Bi составляет около ± 10%.
РАЗДЕЛЕНИЕ СМЕСИ Ni — Мп — Со — Си — Fe — Zn [65]
Эта методика представляет собой типичный пример разделения, основанного на образовании хлоридных комплексов различной прочности, которые характеризуются разной степенью сродства к сильноосновному ани-онообменнику.
Методика. Колонку соответствующей длины (в зависимости от количества разделяемых ионов), заполненную анионообменником Dowex 1-Х8 в С1-форме, промывают 12 М НС1.
Хлориды разделяемых элементов растворяют в минимальном объеме (1—2 см3) 12 М НС1 и переносят раствор в верхнюю часть ионообменной колонки. Колонку промывают 12 М НС1; ионы Ni2+ не сорбируются анионообменником и количественно переходят в элюат. Ионы Мп2+ элюируют 6 М НС1. После изменения концентрации НС1 до 4 М десорбируются ионы Со2+ . Ионы Си2+ элюируют 2,5 М НС1, ионы Fe3+ — 0,5 М НС1 . Последними десорбируют ионы Zn2+ с помощью 0,005 М НС1.
Обзор других методик разделения различных смесей приведен в табл. 5.16.
Литература
1. Dybczynski R., J. Chromatogr., 71 (1972) 507.
2. Tsubota Н., Kitano Y., Bull. Chem. Soc. Japan, 33 (1960) 765, 770.
3. Eulitz G., Nucleonik, 2 (1960) 85.
4. Honda M„ Japan Analyst, 3 (1954) 132.
5. Tompkins E. R., J. Amer. Chem. Soc., 70 (1948) 3520.
5. Применение ионообменников
191
6. Lerner М., Rieman W., Anal. Chem., 26 (1954) 610.
7. Wish L., Anal. Chem., 33 (1961) 53.
8. Лилова О. M., Преображенский Б. К. Радиохимия, 2 (1960) 731.
9. Duyckaerts G., Lejeune R., J. Chromatogr., 3 (1960) 58.
10. Wade M. A., Seim H. J., Anal. Chem., 33 (1961) 793.
11. Weiss H. V., Lai M. G„ Anal. Chem., 33 (1961) 39.
12. Povondra P., Pribil R., Chemist-Analyst, 49 (1960) 109.
13. Povondra P., Sulcek Z., Chemist-Analyst, 50 (1961) 79.
14. MarcholM., Cheng K. L„ Anal. Chem., 42 (1970) 652.
15. Nelson F., J. Chromatogr., 16 (1964) 403.
16. Ласкорин Б. H., Ульянов В. С., Свирцов Р. А., Аржаткин А. М., Южин А. И. Атом, энергия, 7 (1959) НО.
17. Strelow F. W„ van Zyl С. R„ Nolte C. R., Anal. Chim. Acta, 40 (1968) 145.
18. Kraus K. A., Phillips H. O., Carlson T. A., Johnson J. S., Proc. 2-nd Intern. Conf. Peaceful Uses of Atom. Energy, United Nations, Geneva, 28 (1958) 3.
19. Quereshi M., Varshey K. G., Kabiruddin S. K., Can. J. Chem., 50 (1972) 2071.
20. Kraus K. A., Phillips H. O., Carlson T. A., Johnson J. S., Proc. 2-nd Intern. Conf. Peaceful Uses of Atomic Energy, United Nations, Geneva, 28 (1958) 3.
21. Phillips H. O.. Nelson F., Kraus K. A., Rept. ORNL-2159, 1956, p. 37.
22. Korkisch J., Feik F„ Anal. Chem., 37 (1965) 757.
23. Faris J. P„ Anal. Chem., 32 (1960) 520.
24. Baratta E., Herrington A., USAEC Report WIN-118, 1960.
25. Petrow H. G„ Nietzel O. A„ DeSesa M. A., Anal. Chem., 32 (1960) 926.
26. Осипова T. И., Cepeda Г. А. Ж. аналит. химии, 32 (1978) 890.
27. Nelson F., J. Chromatogr., 16 (1964) 403.
28. Заборенко К. Б., Богатырев И. О., Нитзолд Д„ Радиохимия, 5 (1963) 638.
29. Эристави В. Д. и др., Сообщ. АН Груз. ССР, 64 (1971) 325.
30. StrelowF. W. Е., Weinert С. Н., Taianta, 17 (1970) 1.
31. Marhol М., Cheng К. L., неопубликованные данные.
32. Noshkin V. Е., Mott N. S., Taianta, 14 (1967) 45.
33. Fritz J. S„ WakiH., Garralda B„ Anal. Chem., 36 (1964) 900.
34. Khopkar S. M„ De A. K., Anal. Chim. Acta, 23 (1960) 441.
35. Cerrai E., Gadda F, J. Inorg. Nucl. Chem., 16 (1961) 356.
36. Misumi S., Taketatsu T„ J. Inorg. Nucl. Chem., 20 (1961) 127.
37. Ziegler M., Rittner W., Z. anal. Chem., 211 (1965) 170.
38. Ogura T., Tarutani T., Musimi S., Fac. Sci., Kyushu Univ., 5 (1964) 129.
39. Ceasarano G., Pugnetti G., Gazz. Chim. Ital., 93 (1963) 28.
40. Pollard F. H„ Nickless G., Spincer D., J. Chromatogr., 13 (1964) 224.
41. Cabell M. J., Can. J. Chem., 37 (1959) 1094.
42. Nelson F„ J. Chromatogr., 16 (1964) 538.
43. Lerner M. W., Rieman W., Ill, Anal. Chim. Acta, 13 (1955) 439.
192
Часть II
44. Merril J. R., Honda M., Arnold J. R„ Anal. Chem., 32 (1960) 1420.
45. Karve V. M., Indian J. Appl. Chem., 3 1965, 391.
46. StrelowF. W„ Weinert С. H„ Anal. Chim. Acta, 83 (1976) 179.
47. Abdullah M. J., Riley J. P., Anal. Chim. Acte, 33 (1965) 391.
48. Ray Sarkar В. C., Chankan U. S. P., Indian J. Appl. Chem., 3 (1965) 542.
49. Povondra P.,- Sulcek Z., Pribil R., stangl R., Talanta, 8 (1961) 705.
50. Povondra P., Pribil R., Talanta, 10 (1963) 713.
51. BouquiauxJ. J., Gillard J. H. C„ Anal. Chim. Acta, 30 (1964) 273.
52. Elfers L. H., Hallbach P.yF„ Welten R. J., Anal. Chem., 36 (1965) 540.
53. Sixta V., Miskovsky M., Sulcek Z.,Coll. Czech. Chem. Commun., 38 (1973) 3418.
54. Akki S. B., Khopkar S. M. Z., Z. anal. Chem., 249 (1970) 228.
55. Winowski Z., Chem. Anal. (Warszaw), 13 (1968) 583.
56. Frache R., Mazzucotelli A. Talanta, 23 (1976) 389.
57. Winchester J. W., Pinson W. H., Jr., Anal. Chim. Acta, 27 (1962) 93.
58. Macasek A. T., <tech R., Chem. Zvesti, 19 (1965) 107.
59. Range A. T., Bhatki K. S., Anal. Chem., 38 (1966) 1598.
60. Senegancik M., Daljik S., Kristian J., Z. anal. Chem., 249 (1970) 39.
61. Andersen T., Knutsen A. B., Acta Chem. Scand., 16 (1962) 849.
62. Kallmann S., Oberthin H., Liu R., Anal. Chem., 32 (1960) 58.
63. Pistick G. F., Sweet .T. R., Morie G. P., Anal. Chem., 35 (1963) 995.
64. Степин В. В., Поносов В. И., Мельников Е. Е. Ионообменная технология. — М.: Наука, 1965, с. 93.
65. Kraus К. A., Moore G. Е., J. Amer. Chem. Soc., 75 (1953) 1460.
66. Kawabuchi К., Ito T., Kuroda R., J. Chromatogr., 39 (1969) 61.
67. Janauer G. E„ Madrid E. 0., Mikrochimica Acta (Wien), 1974, 769.
68. Lee K. S„ Lee D. W., Kang S. W., Anal. Chem., 43 (1971) 876.
69. Эристави Ф. И., Эристави В. Д., Кутладзе Г. С., Ж. аналит. химии, 26 (1971) 2234.
70. Majumdar А. К., Mitra В. К., Z. anal. Chem., 208 (1965) 1.
71. De А. К., Majumdar S. К., Z. anal. Chem., 184 (1961) 356.
72. Ruf H, Z. anal. Chem., 247 (1969) 297.
73. De A. K., Asit Sen K., Z. anal. Chem., 211 (1965) 243.
74. Suzuki T., Morinaga H., Japan Analyst, 25 (1976) 712.
75. Marowsky G., Z. anal. Chem., 253 (1971) 267.
76. Khasnobis S. K„ Dey B. R; Indian J. Appl. Chem., 32 (1969) 305.
77. Strelow F. W„ Anal. Chim. Acta, 100 (1978) 577.
5.2.3. Группа 3
5.2.3.1. 3 A: Sc, Y, ЛАНТАНОИДЫ, Ac
Элементы этой группы присутствуют в анализируемых растворах преимущественно в форме трехзарядных катионов. Исключение составляет церий, который может находиться также в виде четырехзарядного катиона. Некоторые лантаноиды при определенных условиях образуют двухзарядные катионы.
5. Применение ионообменников
193
Таблица 5.17. Состояние окисления и ионные радиусы Y, РЗЭ и трансплутониевых элементов [1]
Элемент Состояние окисления Ионный радиус Элемент Состояние окисления Ионный радиус
М3+, 10“ 10 м
М3+, 10- 10 М
Y 3 0,880 — Ат 3, 4, 5, 6 0,985 0,99
La 3 1,040 1,061 Ст 3, 4 0,979 0,986
Се 3, 4 1,018 1,034 Вк 3, 4 0,954 0,981
Рг 3 1,008 1,013 Cf 3, 2, 4 0,949 (0,944) 0,976
Nd 3, 2 0,995 0,995 Es 3, 2 0,97 —
Pm 3 0,976 (0,979) Fm 3, 2 —
Sm 3, 2 0,964 0,964 Md 3, 2 —
Eu 3, 2 0,950 0,950 No 2, 3 — —
Gd 3 0,938 0,938 Lr 3 —
Tb 3, 4 0,920 0,923 Ku 4 — —
(RO
Dy 3, 4 0,907 0,908
Ho 3 0,894 0,894
Er 3 0,881 0,881
Trn 3, 2 0,869 0,869
Yb 3, 2 0,858 0,858
Lu 3 0,848 0,848
Группа лантаноидов занимает особое положение. Ввиду большого сходства химических и физических свойств элементов этой группы очень трудно провести ионообменное разделение их смесей, основываясь только на различии в величинах радиусов гидратированных ионов (табл. 5.17).
В присутствии иттрия и скандия степень сродства к сильнокислотным катионообменникам уменьшается в ряду La > Се > Pr > Nd > Pm > > Sm > Eu > Tb > Dy > Y > Но > Er > Tm > Yb > Lu > Sc. Несмотря на заметное уменьшение сродства между La и Lu, различие в сродстве между отдельными соседними лантаноидами очень мало. Поэтому для их разделения на сильнокислотных и сильноосновных ионообменни-ках необходимо использовать соответствующие комплексообразующие агенты.
Скандий и иттрий ведут себя по-иному, поэтому их можно отделить от лантаноидов из растворов минеральной кислоты соответствующей концентрации.
Все элементы этой группы образуют комплексы разной прочности с рядом веществ, особенно с органическими кислотами; последние можно использовать для хроматографического разделения смесей лантаноидов. Разница между константами устойчивости комплексов, образованных двумя соседними элементами, обычно очень мала. Для того чтобы разделение было успешным, необходим тщательный подбор условий.
13- 354
194
Часть II
Таблица 5.18. Влияние температуры на факторы разделения пар лантаноидов молочной кислотой [2]
Разделяемые элементы
Фактор разделения
при 20°С при 87°С
La — Се 2,1 1,83
Се —Рг 2,04 1,73
Pr-Nd 1,38 1,36
Nd — Pm 1,35 1,33
Pm — Sm 1,32 1,32
Sm — Eu 1,20 1,28
Eli—Gd 1,04 1,22
Gd —Tb 1,50 1,73
Tb —Dy 1,65 1,56
Dy —Ho 1,45 1,38
Ho —Er 1,60 1,52
Er — Tm 1,54 1,43
Yb — Lu 1,40 1,31
С этой целью применяют преимущественно сильнокислотные катионообменники полистирольного типа с размером зерен 0,074—0,015 мм. Разброс в размере зерен должен быть по возможности небольшим. Эффективность разделения уменьшается с увеличением диаметра зерен смолы. Кроме того, наблюдается значительное перекрывание отдельных фракций элюата, содержащих различные лантаноиды.
Важным параметром при разделении лантаноидов является температура. Ее положительное или отрицательное влияние зависит как от природы комплексообразующего агента, так и от типа образующихся комплексов. Например, при использовании гликолевой кислоты в качестве элюирующего агента повышение температуры приводит к увеличению факторов разделения. Обратный эффект наблюдается в случае винной кислоты. В табл. 5.18 показано влияние температуры на факторы разделения пар лантаноидов при их элюировании молочной кислотой.
Эффективность разделения пар лантаноидов зависит также от скорости протекания раствора через колонку. Оптимальная скорость потока определяется природой смолы, размером зерен и температурой элюента. Для каждой комбинации этих параметров существует оптимальная скорость потока, при которой разделение наиболее эффективно. Применение более низких скоростей потока не оказывает существенного влияния на эффективность разделения. Оптимальная скорость элюента при температуре 80—90 °C при разделении лантаноидов на смоле Dowex 50 с размером зерен — 0,04 мм составляет около 1 см3/(см2 • мин). В некоторых благоприятных случаях допустима скорость до 3 см3/(см2 • мин). Однако при температуре разделения 20—25 °C на смоле с размером частиц 0,015—0,07 мм скорость потока необходимо снижать в 2—3 раза.
5. Применение ионообменников
195
Важным фактором является также загрузка колонки. При увеличении количества лантаноидов, подлежащих разделению, наблюдается постепенное уширение пика, приводящее к ухудшению разделения. Уширение становится особенно заметным, когда загрузка колонки превышает 5—10 мг смеси элементов на 1 см2 сечения колонки. Рекомендуемая максимальная загрузка колонки составляет 30—40 мг смеси элементов на 1 см2 поперечного сечения колонки при условии, что отношенйе высоты ионообменной колонки к ее диаметру 50. В этом случае происходит удовлетворительное разделение двух соседних лантаноидов.
Эффективность разделения зависит также от ионной формы смолы.
ВЫБОР ЭЛЮЕНТА
При выборе элюента исходят из различия в константах устойчивости образуемых ими комплексов. Для большинства элюентов, используемых в настоящее время, фактор разделения двух соседних лантаноидов равен 1,3—1,4. Этому критерию удовлетворяют многие комплексообразующие вещества. Однако большую часть их следует исключить вследствие плохой растворимости в воде и низкой буферирующей способности. Например, ЭДТА можно использовать только в виде растворов с концентрацией 0,0125—0,0025 М и pH 4. При более низких pH происходит выделение свободной кислоты в ионообменной колонке.
ВЛИЯНИЕ pH И КОНЦЕНТРАЦИИ ЭЛЮЕНТА
Эти два фактора определяют концентрацию комплексообразующего аниона в элюенте. В табл. 5.19 (где собраны данные различных авторов) приведены оптимальные pH элюентов с 0,25 М концентрацией комплексообразующего компонента при разных температурах.
Таблица 5.19. Оптимальные значения pH при разделении легких лантаноидов с помощью 0,25 М кислоты при двух разных температурах
Кислота
рНа
при 20°С при 90°С
Лимонная 3,05 3,5
Гликолевая 4,0 (4,5)
Молочная 3,7 4,2
а-Оксиизомасляная (4,2) 4,7
Виииая 3,5 (4,0)
Эриоксиглутаровая (3,1) 3,6
0,025 М ЭДТА — 3,5
0,0125 М ЭДТА — 3,9
а В скобках приведено приближенное значение.
196
Часть II
Таблица 5.20. Влияние концентрации и pH элюента (лимонная кислота) на фактор разделения «г"1 [3]
Ей 1 J
Концентрация кислоты, М pH Pm “Eu
0,46 2,40 1,52
0,46 2,72 1,47
0,46 2,85 1,46
0,23 3,08 1,44
0,23 3,25 1,40
0,115 3,63 1,33
0,115 3,90 1,18
0,118 4,25 1,16
0,118 4,73 1,14
Интересные сведения, иллюстрирующие влияние концентрации комплексообразующего аниона (лимонная кислота) и pH элюента на эффективность разделения, опубликованы Томпкинсом н Майером [3] (табл. 5.20).
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Иттрий и лантаноиды хорошо сорбируются сильнокислотными катио-нообменниками из растворов сильных минеральных кислот (вплоть до - 0,2 М концентрации). Коэффициенты распределения достигают значений 104. Для скандия значения коэффициентов распределения примерно на один порядок ниже. Отделение скандия от иттрия и лантаноидов в сернокислой среде основано на образовании сульфатных комплексов Sc [4, 5]. В этой среде [0,025 М H2SO4 — 0,3 М (NH4)2SO4] скандий отделяют от Са, Cd, Со, Cu, Al, Ga, In, Y, лантаноидов, Mg, Zn и Ni.селективным элюированием со смолы Dowex 50-Х8. Все перечисленные выше элементы сорбируются на смоле, в то время как скандий проходит в элюат [6].
Для аналитического фракционирования скандия на катионообменниках одним из наиболее подходящих элюентов является аммониевая соль а-оксиизомасляной кислоты. Применение ее в качестве элюирующего агента приводит к более высоким факторам разделения по сравнению с другими часто используемыми оксикарбоновыми кислотами (например, лимонной нли молочной). Элюирование выполняют при комнатной температуре.
Для эффективного отделения скандия применяют растворимые соли ЭДТА, НТА, ГЭДТА и других полиаминокислот, но самым распространенным комплексообразующим агентом является ЭДТА.
Гидратированные ионы Ас3 + хорошо сорбируются сильнокислотными катионообменниками. В < 1 М НС1 коэффициенты распределения имеют величину порядка 102 — 103. Сорбция Ас3+ , как и других лантаноидов, значительно уменьшается с повышением концентрации кислоты. Однако в растворах хлорной кислоты наблюдается очень хорошая сорбция Ас3+ ; величина Dg в 9 М НС1О4 составляет приблизительно 104. При отделении
5. Применение ионообменников
197
Ас3 + от лантаноидов элюированием лимонной, молочной или а-окси изо масляной кислотой [7, 8] лантаноиды десорбируются первыми, после них элюируется актиний.
При элюировании соляной кислотой актиний удерживается обменником более прочно, чем трехвалентные лантаноиды.
Все элементы этой группы хорошо сорбируются сильнокислотными ка-тионообменникаМй из уксуснокислой среды.
Растворы неорганических веществ непригодны для элюирования при разделении элементов лантаноидной группы. В более ранних публикациях есть упоминания о том, что в 0,1 М НС1 или НС1О4 коэффициенты распределения Dg с увеличением атомной массы элемента непрерывно уменьшаются. Однако фактор разделения двух крайних лантаноидов составляет только около 2, что позволяет проводить разделение пар лантаноидов, расположенных далеко друг от друга, но не соседних.
Неорганические вещества часто используют в качестве элюирующих агентов для отделения элементов группы лантаноидов от других элементов. Полная сорбция лантаноидов из разбавленных растворов минеральных кислот обусловливает возможность отделения их от различных одно-и двухзарядных ионов.
В растворах фтористоводородной или серной кислоты лантаноиды отделяют от UO| + и других ионов, образующих в этой среде комплексные аннонМ. Эффективность отделения UC>2+ от лантаноидов уменьшается в ряду HF > H2SO4 > HNO3 > HCI > НС1О4. Для отделения лантаноидов от различных элементов использовались также растворы, содержащие триполифосфат или пирофосфат натрия [8—13].
Значительное повышение поглощения лантаноидов сильнокислотными катионообменниками в солянокислой, азотнокислой или сернокислой среде наблюдалось при введении соответствующего органического растворителя.
Безуспешный поиск неорганических веществ в качестве элюирующих агентов для разделения лантаноидов обусловил необходимость тщательного исследования органических веществ, образующих с разделяемыми ионами комплексные соединения разной прочности.
Сначала были изучены растворы лимонной кислоты, нейтрализованные до различных pH. Результаты показали, что эффективность разделения цитратными растворами повышается с уменьшением их pH. Однако при этом увеличивается время, необходимое для полного разделения элементов. Несмотря на то что в более поздних исследованиях были найдены лучшие элюенты, все же цитратная среда до сих пор считается пригодной для разделения следовых количеств лантаноидов. Обычно используют 5%-ные или 0,3—0,5 М растворы (pH около 3). Рекомендуется применять градиент pH.
Более эффективным элюирующим агентом, например для разделения смеси Y — ТЬ — Ей — Sm, является лактат. Элюирование обычно выполняется при повышенной температуре и градиенте концентрации или pH. Очень хорошие результаты получены при элюировании смесью 0,4 М молочная кислота — 0,002 М ЭДТА [14, 15].
198
Часть II
При использовании молочной кислоты в качестве элюирующего агента ее растворы поражаются микроорганизмами или плесенью, что изменяет их состав. Кроме того, вследствие частичной димеризации молочной кислоты в растворе наблюдается плохая воспроизводимость элюирующих объемов индивидуальных элементов.
Молочную кислоту удаляют из отдельных фракций элюата подкислением до pH 2 и последующей экстракцией диэтиловым эфиром. После отделения элюирующего агента фракцию пропускают через колонку, заполненную сильнокислотным катионообменником. Редкоземельные элементы сорбируются обменником и затем элюируются б М НС1. Молочная кислота особенно эффективна при разделении более тяжелых лантаноидов. Разделение можно улучшить введением в элюент небольших количеств фенола.
Таблица 5.21. Влияние природы элюента и его температуры на факторы разделения а редкоземельных и траисплутоииевых элементов при их разделении на смоле Dowex 50 [20, 21]
Элементы Элюент и его температура
H3Cit (87°С) HLact (90°С) HGIyc (20°С) HGIyc (87 ° С) HBut (20°С) NH4But (25°С) NH4Lact $25°С)
Lu — Yb 1,31 1,31 1,31 1,36 1,45 1,54 —
Yb—Tm 1,53 1,43 1,37 1,33 1,37 1,70 —
Tm — Er 1,60 1,47 1,40 1,30 — 1,71 —
Er—Ho 1,50 1,52 1,28 1,23 — 1,73 —
Ho —Dy 1,37 1,38 1,23 1,62 1,95 1,80 —
Dy—Tb 1,40 1,56 1,04 1,88 1,84 2,30 —
Tb—Gd 1,37 1,73 1,68 2,2 1,57 2,40 1,45
Gd—Eu 1,12 1,22 1,00 1,4 1,54 1,65 —
Eu — Sm 1,19 1,28 1,08 1,6 2,06 1,88 —
Sm — Pm 1,38 1,32 1,33 1,82 1,57 2,13 1,19
Pm — Nd 1,09 1,33 • 1,83 1,61 2,0 1,54 1,40
Nd—Pr 1,18 1,36 1,37 1,57 — 1,68 1,70
Pr—Ce 1,69 1,73 2,2 1,6 — 1,85 —
Ce—La — 1,83 1,64 2,2 — 1,97 —
Am — Cm 1,20 1,21 1,25 1,45 1,4 — 1,28
Cm — Bk 1,63 1,54 — 2,22 2,7 — —
Bk —Cf 1,42 1,58 — 2,25 2,0 — —
Cf—Es 1,2 1,24 1,20 1,54 1,46 — —
Es—Fm 1,2 1,77 — 1,84 — — —
Fm — Md 1,18 1,3 — 1,4 — — —
Принятые обозначения: H3Cit — лимонная кислота, HLact — молочная кислота, HGIyc — гликолевая кислота, HBut — масляная кислота, NH4But — бутират аммония, NH4Lact — лактат
аммония.
5. Применение ионообменников
199
Недостатки двух предыдущих элюирующих агентов существенно устраняются при использовании а-оксиизомасляной кислоты. Водные растворы этой кислоты чаще всего применяют при разделении лантаноидов. Это наиболее эффективный элюент, применимый даже при температуре 20°С [16—19]. Однако для разделения более сложных смесей редкоземельных элементов необходима повышенная температура (87 °C). Обычно используют 0,2—0,4 М растворы а-оксиизомасляной кислоты (pH 4,0—4,6) и градиентное элюирование (для уменьшения хвостов на кривых элюирования). В табл. 5.21 сравниваются факторы разделения редкоземельных и трансплутониевых элементов, полученных для указанных выше оксикислот. Представленные данные показывают более высокую разделяющую способность производных масляной кислоты. Многочисленные примеры применения оксикислот при разделении лантаноидов даны в табл. 5.28.
Некоторые а-оксикислоты, гликолевая кислота и ее производные (диэтил-, дипропил-, метил-, изобутил-, изопропилметил-) имеют меньшее значение. Преимущество их по сравнению с молочной кислотой состоит в том, что в водных растворах они не образуют димеров. Эти оксикислоты обеспечивают удовлетворительное разделение смесей лантаноидов, однако чистота разделяемых элементов хуже, чем в случае а-оксиизомасляной кислоты. Винную и яблочную кислоты редко используют при разделении лантаноидов.
Наряду с а-оксикарбоновыми кислотами применяют натриевые или аммониевые соли полиамин оуксу сных кислот. Наиболее важные из
Таблица 5.22. Факторы разделения а пар лантаноидов с помощью ЭДТА и ГЭДТА [22j
Элементы ЭДТАа ГЭДТА
Lu —Yb 1,9 1,3
Yb—Tm 1,8 1,6
Tm — Er [3,1] 2,0
Er—Ho 1,8 1,2
Ho —Dy 2,6 -1,0
Dy—Tb 2,3 - 1,0
Tb —Gd [4,2] -1,0
Gd—Eu 1,05 0,7
Eu—Sm 1,5 - 1,0
Sm—Nd 3,2 2,6
Nd—Pr 1,8 1,8
Pr—Ce • 2,5 2,8
Ce—La 3,7 5,0
Dy—Y 1,6 —
Y—Tb 1,5 — .
a Величины, взятые в квадратные скобки, явно завышены.
200
Часть II
них — ЭДТА, ГЭДТА, НТА, ДТПА, ЭГТА и ДКТА. Эти соединения характеризуются высокими факторами разделения лантаноидов. Наиболее селективной среди них является этилендиамин-N, N, N', N'-тетрауксусная кислота (ЭДТА). Недостатком этих комплексообразующих агентов является их ограниченная растворимость, особенно при низких pH элюентов, что может приводить к осаждению свободных кислот в ионообменной колонке. Другой неблагоприятный фактор — это медленное установление ионообменного равновесия, что приводит к увеличению высоты теоретической тарелки (а также длины колонки) и к уменьшению эффективности разделения.
ГЭДТА характеризуется большей растворимостью, чем ЭДТА, что позволяет использовать ионообменник в Н-форме. Осаждения свободной кислоты в колонке не наблюдается. ГЭДТА применяют при разделении смесей La — Се — Рг — Nd — Y — Sm, Но — Er — Tm — Tb — Lu и особенно при разделении Tm — Yb — Lu в концентратах. Низкая эффективность разделения наблюдается, например, для смеси Sm — Ей — Gd — Tb — Dy — Но.
В табл. 5.22 и 5.23 сравниваются факторы разделения лантаноидов при помощи различных хелатообразующих агентов.
Порядок элюирования, как видно из данных табл. 5.24, зависит от природы хелатообразующего агента.
Другое поведение наблюдается при элюировании иттрия органическими элюентами. Его положение в ряду лантаноидов в зависимости от элюента варьирует в широких пределах: ЭДТА элюирует иттрий между Dy и ТЬ, ДТПА — вблизи Nd, ГЭДТА — вблизи Рг, цитрат при 10—20 °C — около Ей, а при 87—100 °C — около Dy — Но, лактат или тиоцианат — вблизи Dy — Но.
Для разделения лантаноидов применяли также фосфорсодержащие аналоги хелатообразующих агентов. С помощью аминофосфоновых кислот удалось разделить следовые количества ряда редкоземельных элементов.
Таблица 5.23. Сравнение приблизительных значений факторов разделения прометия, других лантаноидов и иттрня при разделении их различными хелатообразующими агентами на катионообменнике Dowex 50-Х8
Лантаноиды ЭДТА ГЭДТА ДТПА НТА
Pm — La 38,9 51,3 380,0 56,2
Pm — Се 8,3 7,1 34,7 19,1
Pm — Рг 3,6 3,5 4,9 5,0
Pm —Nd 1,8 1,7 2,0 2,3
Pm — Sm 1,8 1,7 2,0 2,3
Pm — Eu 2,4 1,9 2,3 4,7
Pm — Gd 2,5 1,5 3,0 4,1
Pm —Y 11,8 2,8 1,4 3,2
5. Применение ионообменников
201
Таблица 5.24. Порядок элюирования некоторых элементов со смолы Dowex 50-Х8 некоторыми хелатообразующими агентами
Элюент Порядок элюирования
0,012 М ЭДТА, pH 8,5 0,030 М ГЭДТА, pH 5,0 0,050 М ДТПА, pH 6,5 Zn, Al, Y, Gd, Eu, Sm, Pm, Nd, Pr, Ce(lll), La Zn, Pb, Al, Gd, Eu, Sm, Y, Pm, Nd, Pr, Ce(lll), La Zn, Pb, (Cm, Ho, Am), Gd, Eu, Sm, Y, Pm, Nd, Pr, Ce(IIl), La
0,105 М НТА, pH 6,4 Zn, (Cm, Y), Gd, (Eu, Am), Sm, Pm, Nd, Pr, Ce(lll), La
Европий был отделен от Се(Ш) элюированием 0,05 М раствором этилен-диаминдиизопропилфосфоновой кислоты (А) при pH 6,68. Подобным же образом Се(Ш) и Pm отделяются от Ей 0,1%-ным раствором N-оксиэтилендиаминтриметилфосфоновой кислоты (Б) при pH 4,64. Иттрий отделяют от Се(Ш) и Pm элюированием 0,1%-ным раствором этилендйа-минтетраметилфосфоновой кислоты (В) при pH 3,42 [23]. Для соединения А фактор разделения пары Се — Ей равен 4, для соединения Б факторы разделения пар Ей — Се и Ей — Pm равны соответственно 4,2 и 1,46, а для В и пар Pm — Се, Y — Се -г соответственно 3,22 и 7,25. Обычно факторы разделения ниже, чем при элюировании ЭДТА и НТА.
Помимо названных выше веществ изучены следующие фосфорорганические хелатообразующие агенты: этиленди(иминоизопропилиден)дифосфо-новая кислота (Г), этиленди(нитрилодиметилен)тетрафосфоновая кислота (Д), 1-оксидиэтилидендифосфоновая кислота (Е). Получены следующие факторы разделения: пары Y — Се 130 (соединение Д); Y — Се 15, Y — ТЬ 14, Y — Ей 3,9 (соединение Г); Y — Се 3,5, Y — Ей 1,5 (соединение Е) [24].
Изучение возможности повышения факторов разделения и улучшения эффективности разделения соседних лантаноидов привело к использованию водно-органических элюентов [25—27].
Для быстрого разделения лантаноидов разработан метод жидкостной хроматографии высокого давления (ВЭЖХ), основанный на использовании очень мелких частиц смолы (< 30 мкм) с малой дисперсией. Часто разделение проводят при повышенной температуре (80—100 °C). В качестве элюирующего агента обычно применяют а-оксиизомасляную кислоту. Вследствие высокого сопротивления ионообменной колонки для прохождения через нее элюента необходимо высокое давление. Возможность работы при повышенных скоростях потока [ > 10 см3/(см2 • мин)] обусловлена ускоренной диффузией ионов через мелкие частицы смолы [28—31].
При использовании обычных хроматографических методов длительность разделения сложных смесей лантаноидов составляет 42—48 ч. Применение метода ВЭЖХ приводит к сокращению времени разделения до < 5 ч. Разделение одной пары лантаноидов занимает около 30 мии.
202
Часть П
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Иттрий и лантаноиды не сорбируются сильноосновными анионообменниками из солянокислых растворов [32]. Таким образом, в 6 М НС1 от лантаноидов можно легко отделить элементы, хлоридные комплексы которых хорошо сорбируются на этих смолах [U, Fe(III), Pd, Ir, Cu, Au, Ga, In, Tl(III), Mo, W, Ru и т. n.].
В азотнокислых растворах лантаноиды частично поглощаются анионообменниками, особенно нитратные комплексы Се(Ш), Рг и Nd. Максимальное поглощение происходит из 5—6 М HNO3. Скандий, иттрий и другие лантаноиды не сорбируются при любой концентрации кислоты [33].
Подобное поведение наблюдается в смеси растворов HF — НС1 и фосфорной кислоты.
Необычным образом ведут себя [34] редкоземельные элементы в смесях 7 М HNO3 — метанол. Поглощение лантаноидов на смоле Dowex 1-Х8 в среде 7 М HNO3 — 95% СН3ОН- уменьшается в ряду La > Се(Ш) > > Nd М» Eu, Sm > Gd > Tb > Dy > Но > Tm, Lu > Yb. При снижении содержания метанола поглощение La остается неизменным, в то время как сорбция других, особенно тяжелых, лантаноидов значительно уменьшается. Этим способом можно отделить тяжелые лантаноиды от легких. В указанной среде от лантана также отделяются ионы Na, Mg, Al, К, Sc, Mn(VII) и Fe(III), которые имеют величины Dg < 1.
При замене кислоты подкисленными растворами литиевых или аммониевых солей (LiNO3, NH4NO3, LiCl) поглощение лантаноидов значительно повышается. Растворы этих солей можно применять в качестве элюентов при разделении лантаноидов. Для группового отделения лантаноидов от трансплутониевых элементов успешно использовали смесь 10 М LiCl — 0,1 М НС1.
Из растворов, содержащих серную кислоту, иттрий и лантаноиды не поглощаются анионообменниками. Но в этой среде наблюдается высокоселективная сорбция скандия. Последний отделяют от многих других элементов в смесях 0,05 М H2SO4 — (NH4)2SO4 или K2SO4.
Высокие значения коэффициентов распределения скандия были получены для подкисленных растворов тиоцианата. Как видно из данных, приведенных в табл. 5.25, в этих средах скандий можно отделить от кальция, лантана, самария и других элементов.
В среде фтористоводородной кислоты скандий можно отделить от лантаноидов, образующих нерастворимые фториды. Поглощение скандия зависит от концентрации кислоты. В 0,5—2,5 М HF скандий отделяется от ванадия, не сорбирующегося в этой области концентраций HF.
Анионные фторидные комплексы скандия хорошо сорбируются на смоле Dowex 1 в присутствии разбавленной соляной кислоты. С повышением' концентрации кислоты поглощение Sc уменьшается.
Согласно данным авторов работы [36], скандий образует с малоновой или аскорбиновой кислотой легко сорбируемые анионные комплексы. В 8%-ном растворе малоновой кислоты (pH 5,0) или 5%-ном растворе аскор-
5. Применение ионообменников
203
Таблица 5.25. Коэффициенты распределения Dg некоторых элементов на анионообменнике Dowex 1 в среде 0,5 М НС1 и NH4SCN (различной концентрации) [35]
Элемент Концентрация NH4SCN, М
0,2 0,4 0,6 0,8 1,0
Sc 56 250 905 5040 2828
Al 0 0 0 0 0
Sm 1 1 1 1 1
Lu 1 1 1 1 1
La 1 1 1 1 1
In 579 646 734 580 706
Ca 0 0 0 0 0
Cd, Cu 1 Hg, Zn / 104 104 104 104 104
Th 3,3 19 43 74 106
биновой кислоты (pH 6,5) на анионообменнике Dowex 2-Х8 скандий отделяется от щелочных и щелочноземельных металлов, T1(I), Hg(II) и Fe(II).
Иттрий легко поглощается анионообменниками из разбавленных растворов карбонатов щелочных металлов (0,01 М). Величины Dg составляют около 104. Эта среда имеет практическое значение при разделений пар Sr — Y, Ва — La, а также при отделении лантаноидов от урана или тория [37].
В отличие от катионообменников при разделении лантаноидов на анио-нообменниках органические хелатообразующие или комплексообразующие агенты не находят широкого применения. В большинстве случаев разделение соседних лантаноидов менее эффективно. По сравнению с катионооб-менниками наблюдается обратный порядок элюирования [32, 38, 39].
Вследствие большого размера молекул образующихся комплексов неблагоприятное влияние на разделение оказывают стерические факторы [40, 41].
Сильноосновные анионообменники обычно используют в той же ионной форме, что и хелатообразующий агент, поскольку смола в С1- или ОН-форме характеризуется низким сродством к хелатам металлов. Как правило, коэффициенты распределения хелатов отдельных лантаноидов повышаются при переходе от лантана к европию и затем существенно уменьшаются в ряду европий — лютеций. Что касается других элементов группы 3 А, то коэффициенты распределения скандия находятся между значениями для Тт и Ег, коэффициенты распределения иттрия — между значениями для Ег и Но.
204
Часть II
Лабораторные методики
ОТДЕЛЕНИЕ Sc ОТ У И ЛАНТАНОИДОВ [42]
Методика. 20 г сильнокислотного катионообменника AG 50W-X8 (0,15— 0,07 мм) помещают в колонку соответствующего размера, переводят в Н-форму и тщательно промывают водой.
Раствор, содержащий до 1,6 ммоля Sc и до 1,6 ммоля Y, La, Се, Sm, Gd, Er и Yb, подкисляют 3 М раствором минеральной кислоты и пропускают через ионообменную колонку. Обменник промывают водой и элюируют Sc 1 М серной кислотой со скоростью 3 см3/мии. Из колонки удаляют серную кислоту промыванием 50 см3 0,1 М НС1. Затем колонку промывают примерно 500 см3 4 М НС1 со скоростью 3 см3/мин. В этих условиях лантаноиды не десорбируются.. При разделении меньших количеств лантаноидов используют колонки меньшего размера. До 0,3 ммоля Sc отделяют от равных (молярных) количеств редкоземельных элементов на колонке, заполненной 5 г смолы (0,07—0,035 мм) при скорости потока 0,6 см3/мин.
Для сравнения эффективности отделения Sc от некоторых ионов редкоземельных элементов в табл. 5.26 приведены коэффициенты распределения Dg в 1 М НС1 и 0,5 М H2SO4.
ОТДЕЛЕНИЕ Sc ОТ ЛАНТАНОИДОВ НА КАТИОНООБМЕННИКЕ [43]
Методика. Образец, содержащий до 100 мг скандия, растворяют в соляной кислоте и выпаривают почти досуха. Остаток растворяют в 180 см3 воды, прибавляют 20 см3 насыщенного раствора щавелевой кислоты и пропускают через колонку, заполненную 2 г сильнокислотного катионообменника Dowex AG 5OW-X8 (0,15— 0,07 мм)'в Н-форме. Затем колонку промывают 10—15 см3 (СООН)2 со скоростью 10 см3/мин. В этих условиях только часть скандия удерживается смолой. Более 90% Sc проходит в элюат. Сорбированный Sc злюируют ~80 см3 0,10 М раствора щавелевой Кислоты.
Таблица 5.26. Коэффициенты распределения Dg скандия, иттрия и некоторых редкоземельных элементов на катионообменнике AG 50W-X8 в среде 1 М НС1 и 0,5 М H,SO. [42]
Элемент D g
в 1 м HCl в 0,5 M H-SO„ 2 4
La 265 326
Се(Ш) 265 318
Sm 217 269
Gd 183 246
Yb 153 249
Y 145 253
Sc 120 35
5. Применение ионообменников
205
Раствор щавелевой кислоты удаляют из колонки промыванием водой и элюируют лантаноиды (вместе с иттрием) 5 М азотной или соляной кислотой.
РАЗДЕЛЕНИЕ СМЕСИ Y3+ — Sc3+ НА АНИОНООБМЕННИКЕ [44]
Методика. Колонку (47 см х б мм), заполненную сильноосновным анионообменником Dowex 1-Х8 (0,07—0,035 мм) в Cl-форме, промывают абсолютным этанолом. Подкисленный раствор смеси анализируемых ионов выпаривают досуха на водяной бане. К остатку прибавляют 50 см3 абсолютного этанола и пропускают раствор через обменную колонку. Скандий проходит в элюат. Затем колонку промывают 150 см3 абсолютного этанола и элюируют иттрий раствором, состоящим из 8 частей этанола, 2 частей воды и 2 частей соляной кислоты.
ОТДЕЛЕНИЕ МИКРОГРАММОВЫХ КОЛИЧЕСТВ ЛАНТАНОИДОВ ОТ БОЛЬШИХ КОЛИЧЕСТВ U И Th [45]
Методика. Образец, содержащий примерно 1 г нитрата тория или уранила и микрограммовые количества редкоземельных элементов, растворяют в воде и прибавляют 12—15 г сульфосалициловой кислоты. Смесь разбавляют водой до объема 500 см3, с помощью гидроксида аммония устанавливают в ней pH 6,5, кипятят в течение короткого времени и охлаждают. Измеряют pH раствора и, если необходимо, снова устанавливают pH 6,5. Полученный раствор разбавляют до объема 1500 см3 и пропускают через колонку, заполненную катионообменником Dowex 50-Х8 в NH4-форме. Затем колонку промывают 50 см3 0,02 М раствора сульфосалициловой кислоты с pH 6,5 и водой.
Лантаноиды десорбируют 25 см3 насыщенного раствора хлорида аммония.
При pH 6,5 лантаноиды отделяются на катионообменнике от А13+ , Th4+, Fe3 + , МоО?” и UO+. 4 2
Отделение титана, ванадия и вольфрама проводят при pH 4,0. К элюату прибавляют 9,00 мг нитрата бериллия, 10 см3 6%-ного раствора купферона и несколько капель метилового оранжевого. При нейтрализации раствора гидроксидом аммония осаждается купферонат бериллия, с которым соосаждаются гидроксиды лантаноидов, скандия и тория. Осадок купфероната озоляют и в золе определяют редкоземельные элементы спектрографическим методом.
ОТДЕЛЕНИЕ Се ОТ ДРУГИХ ЭЛЕМЕНТОВ ПРИ АНАЛИЗЕ СТАЛИ [46]
Методика. Колонку, заполненную сильноосновным анионообменником Dowex 1-Х8 (0,15—0,07 мм) в NOj-форме, приводят в равновесие со смесью 5% 5М HNO3 — 95% СН3ОН. 1,0000 г стали растворяют в азотной кислоте и выпаривают раствор досуха. Сухой остаток растворяют в определенном количестве 5 М HNO3.
Порцию полученного раствора объемом 1 см3 разбавляют метанолом до объема 20 см3 и пропускают через ионообменную колонку со скоростью 0,25—0,30 см3/мин. Ионы Се3+ , РЬ2 + и Bi3+ хорошо сорбируются на анионообменнике. Ионы Fe3+, которые частично сорбируются на колонке, полностью вымываются смесью 95% СН3ОН — 5% 5 М HNO3. Церий элюируют 50 см3 смеси 90% СН3ОН — 10% 6 М НО. Ионы РЬг+ и Bi3+ в этих условиях не десорбируются. Ионы РЬ2+ элюируют 50 см3 1 М НО, а ионы Bi3+ — 50 см3 1 М HNO3.
206
Часть П
РАЗДЕЛЕНИЕ СОСЕДНИХ ЛАНТАНОИДОВ
(СЛЕДОВ РАДИОАКТИВНЫХ ИЗОТОПОВ ИЗ МАТЕРИАЛА МИШЕНИ) НА КАТИОНООБМЕННИКЕ DOWEX 50W-X12 [47]
Методика. Разделение проводят на катионообменнике Dowex 50W-X12 (0,07—0,037 мм). Для элюирования используют а-оксиизомасляную кислоту различной концентрации с разными величинами pH при температурах 60—90 °C. В зависимости от количества материала мишеии применяют колонки следующего диаметра: 13,2 мм для 12 мг материала, 17,6 мм для 25 мг и 19,1 мм для 30 мг.
Экспериментальные условия приведены в табл. 5.27.
РАЗДЕЛЕНИЕ СМЕСИ 227 Ас — 227 Th — 231 Ра — 233 Ra
НА СИЛЬНОКИСЛОТНОМ КАТИОНООБМЕННИКЕ [48]
Методика. Примерно 3 мг гидроксида 231 Ра растворяют в 1 см3 концентрированной фтористоводородной кислоты в тефлоновом тигле. Раствор осторожно упаривают до объема около 0,5 см3 и прибавляют смесь 0,5 М НС1 — 0,5 М HF до общего объема 1 см3. Смесь нагревают и пропускают через колонку (5 см х 0,28 см2), заполненную смолой Dowex 50-Х4 (< 0,037 мм), которая предварительно приведена в равновесие со смесью 0,5 М НС1 — 0,5 М HF.
Протактиний в данных условиях ие сорбируется и вымывается приблизительно 3 объемами (колонки) смеси 0,5 М НС1 — 0,5 М HF. 227Ac, 227Th и 233Ra хорошо сорбируются. Эти три элемента элюируют совместно смесью 6 М НС1 — 1 М HF или 1 М HNO3. К элюату прибавляют 1 см3 концентрированной НС1О4 и упаривают до объема примерно 0,1 см3. Остаток растворяют в 0,5 см3 горячей смеси 8,5 М НС1О4 — 0,5 М НО — 0,1 М HF и пропускают раствор через колонку (4 см х х 0,28 см2), заполненную смолой Dowex 50-Х4 (< 0,037 мм). Разделение проводят при температуре 50 °C.
Сначала НС1О4, НС1 и HF вымывают оставшийся протактиний. Одновременно в максимуме элюируются 223Fr, 2,1 Pb, 2,1 Bi и 233Ra (если они присутствуют). После элюирования радия фтористоводородную кислоту вымывают смесью 8,5 М НС1О4 — 0,5 М НС1. Этим способом предотвращают частичную десорбцию тория в процессе элюирования актииия.
227Ас элюируютб М HNOj. И наконец, 227Th элюируют смесью 6 М НС1 — 1 М HF.
Обзор других методов разделения приведен в табл. 5.28.,
5.2.3.2. 3 Б: В, Al, Ga, In, Т1
Элементы этой группы существуют в трехвалентном состоянии, за исключением таллия, который преимущественно находится в одновалентном состоянии.
При разделении элементов группы ЗБ на ионообменниках следует учитывать их способность к гидролизу. Поэтому необходимо удерживать элементы в сорбируемой форме либо путем повышения кислотности раствора, либо введением.соответствующих комплексообразующих агентов. Однако присутствие последних может вызвать затруднения на стадии сорбции.
Все элементы группы ЗБ образуют со многими комплексообразующими или хелатообразующими агентами соединения различной прочности. Различная устойчивость образованных комплексов служит основой для разделения элементов.
5. Применение ионообменников
207
Таблица 5.27. Условия разделения соседних лантаноидов иа сильнокислотном катионообменнике Dowex 50W-X12 [47]
1 2 3 4 5 6 7 8 9 10 11
Lu Yb 0,125 4,2 0,15 20 60 4 5,5 6
Lu Tm 0,125 4,2 0,13 20 60 . 5 7 8 11
Lu Tm 0,15 4,2 0,12 15 60 2,5 4 6,5
Yb Tm 0,175 4,0 0,16 20 60 2,5 4,5 4,75 7
Yb Er 0,20 3,8 0,17 20 60 4,5 6,25 7 9
Yb Er 0,20 4,0 0,512 15 60 1,75 3,25 4,25
Tm Er 0,175 4,2 0,11 20 60 3 4,5 ' 5
Tm Ho 0,175 4,2 0,16 15 60 2 3,25 5
Er Ho 0,20 4,2 0,13 20 60 2,5 4,25 4,5
Er Dy 0,20 4,2 0,13 20 60 2,5 4,25 4,5 6,5
Er Dy 0,20 4,2 0,13 15 60 2 3
Ho Dy 0,275 4,0 0,14 20 80 3,75 5,25 5,75
Ho Tb 0,275 4,0 0,14 20 80 3,75 5,25 5,75 7
Ho Tb 0,30 4,2 0,12 / 20 80 1,75 3 3,75
Dy Tb 0,275 4,2 0,14 20 85 2,5 4 4,5
Dy Gd 0,275 4,2 0,13 20 85 3 4,5 5 6,5
Dy Gd 0,30 4,4 0,11 20 85 1,5 3 4,5
Tb Gd 0,30 4,4 0,09 20 85 3 4,25 4,5
Tb Eu 0,30 4,4 0,09 20 85 3 4,25 4,5 6
Tb Eu 0,35 4,6 0,09 17 85 1,75 4
Gd Eu 0,30 4,4 0,07 16 85 6
Gd Sm 0,325 4,4 0,09 20 85 3,75 5 5,75
Gd Sm 0,325 4,4 0,12 15 85 2,25 3,25
Eu Sm 0,30 4,6 0,1* 15 80 2,75 4,25 4,5
Eu 0,30 4,6 0,14 20 80 3,25 4,75 5,25 7
Sm 0,45 4,2 0,15 20 90 3 5,5 5,5
Sm Nd 0,45 4,2 0,14 20 90 3,5 5,75 6,25 8
Sm Nd 0,40 4,6 0,12 20 70 1,75 3,5 4,25
Pm Nd 0,425 4,4 0,13 20 85 3,75 5 5,5
Pm Pr 0,425 4,4 0,17 15 85 2 3,25
Nd Pr 0,475 4,2 0,18 20 80 4 5,5
Nd Ce 0,475 4,2 0,18 20 80 4 5,5 5,5 7
Nd Ce 0,475 4,5 0,16 15 80 2 3 4
Pr Ce 0,45 4,6 0,17 20 80 3,25 4,75
Pr La 0,45 4,6 0,17 20 80 3,25 4,75 5,25 7,75
Pr La 0,525 4,8 0,17 15 80 2 3 5
Ce La 0,525 4,8 0,17 20 80 2,75 4 5,25
Принятые обозначения-. 1 — радиоактивный изотоп, следовые количества: 2 — материал мишени; 3 — концентрация а-оксиизомасляной кислоты (молярность); 4 — pH элюеита; 5 — скорость потока [см3/(мин- см2)]; 6 — высота обменной колонки (мм); 7 — температура (°C); 8 — приблизительное время начала элюирования первого лантаноида (ч); 9 — приблизительное время конца элюирования первого лантаноида; 10 — приблизительное время начала элюирования второго лантаноида; 11 — приблизительное время конца элюирования второго лантаноида. '
208
Часть П
Таблица 5.28. Обзор некоторых методик разделения элементов группы ЗА
Элемент Отделяемые Ионообменник Условия разделения Литература
компоненты
Sc Различные катионы Dowex 50 0,05 M ДТПА 49
Sc Со, Ni, Pd, Мп, Cd, Zn Dowex 2-X8 8%-иая малоновая'кислота при pH 5,0 50
Sc La, Y Dowex 50W-X4 Хлоруксусная кислота — 0,1 М HNO3 49
Sc Pu Reeve-Angel SA-2 51
Sc, La Al, Fe, Ti Sc Сточные воды AG-50W-X8 Другие: 3 М НС1 — 50% С2Н5ОН Sc: 3 — 4 М НС1 52 53
Sc B&, Cr, V Dowex 1-X8 Ва: 0,01 М ЭДТА — 0,01 М NH4C1, pH 4,6 Sc: 0,01 М ЭДТА — 0,1 м nh4ci, pH 4,6 Сг: 0,01 М ЭДТА — 0,3 м nh4ci, pH 9,0 V: 0,01 М ЭДТА — 2,0 М NH4C1, pH 4,5 54
Sc Лантаноиды, анализ метеоритов Dowex 2 Dowex 50W-X12 НС1 1 М лактат аммония, pH 3,2 55
Sc Y, РЗЭ Dowex 50-X4 Различные комплексообразующие агенты 56
Sc Y, РЗЭ AG 50W-X8 Sc: 1 М H2SO4 Y, лантаноиды: 4 М НС1 39
Sc Al, Ce, Fe, Ca, In, La, Mn, Th, Y Dowex 50-X4 0,5 М Н3РО4 57
Sc(npn-месь) s, Al, Mg и другие Dowex 50W-X8 Сорбция из 12 М НС1 58
Y Dy—Eu—Sm—Gd КУ-2 0,17 М а-оксиизомасляиая кислота 59
La Ni Dowex 50W-X8 Ni: 6 М НС1О4 La: 4 М НС1 60
Y(cne-ды) Силикаты Dowex 50W-X4 DL-2-Оксимасляная кислота 61
5. Применение ионообменников 209
Продолжение табл, 5.28
Элемеип г Отделяемые компоненты Ионообменник Условия разделения Литература
La Th AG-5OW-X8 0,4 M HCI 62
Се Щелочные металлы Dowex 50-Х8 Щелочные металлы: 0,1 М НС1 Се: ЭДТА 63
Sm Pm, Nd Dowex 50 а-Окси-а-метилмасляная кислота 64
Лантаноиды Чистый Al и Fe Dowex 50 Лактат аммония, 90°С 65
Ланта- Взаимное Катиоио- Лактат аммония, а-окси- 66
иоиды разделение обменник изобутират аммония
Ланта- U, продукты ноиды, расщепления Sm, Pm, Nd Dowex 50W-X4 0,08 М а-оксииэобутират аммония—41,5% СН3ОН 6?
Лантаноиды Сталь Bio-Rad 1-Х10 68
Gd и Dowex 1-X8 Gd: СН3ОН—12 М НС1 (9:1) U: 0,5 М НС1 69
Eu Ат Dowex 1-X8 Ей: 8 М LiCl — 40% С2Н5ОН Am: 1 М НС1 70
Ce(III) Ce(IV) или La, Со, Си Dowex 50W-X8 Ацетат аммония 71
147Pm 149Nd Dowex 1-X8 XHNO3— диоксан 72
Ce(III) Ce(IV), La Dowex 50W-X8 73
Лантаноиды Взаимное разделение Различные 74 76
Лантаноиды Различные катионы Dowex 50W-X8 а) НС1 разной концентрации (Са, Mg, Fe, Al, Ti, PO’“) 6) 4 M HCI, лантаноиды в) Th: 1,8 M H,SO„ 77
Лантаноиды Двухзарядные катионы Dowex 50-X8 Двухзарядные: 1,5 M HNO3 РЗЭ, Y, Al: 3 M HNO3 78
Лантаноиды и Dowex 50 U: 0,35—0,5 M H2SO4 РЗЭ: 4 — 6 M HCI 79 80
Eu, Dy, Са, Mg Amberlite Ca, Mg: 1 M HCI 81
Gd.Sm.Y IR-120 другие: 12 M HCI
Лантаноиды Взаимное разделение Dowex 50W-X8 а-Оксиизомасляная кислота, pH 5,2 82
Er Tm, Yb, La Dowex 50W-X4 0,04 M NaOAc — 0,04 M М-(2-окснэтил?эти-лендиаминтриуксусная кислота 83
14-354
210
Часть II
Продолжение табл. 5.28
Элемент Отделяемые Ионообменник Условия разделения Литература
компоненты
Ланта- Взаимное Катионе- вэжх 84.
ноиды разделение обменник 159
Лантаноиды V (в ванадатах РЗЭ) ARA-8 в NO3-форме Лантаноиды: Н2О V: 5 М HNO3 85
Ег Tm Dowex 5OW-X8 Элюирование ГЭДТА, pH 7,5 86
17|>Гт 160 Tb, 153Sm, 141 Се Dowex 5OW-X8 1%-ный НТА, pH 6,5 87
Nb, Pr, Се, La La, Sm, Eu (при- Взаимное разделение U ядерной чистоты KPS-200 0,5 М лактат аммония, 86°С 88 89
меси)
Лантаноиды Продукты расщепления Dowex 50W-X4 а-Оксиизомасляная кислота 90
Лантаноиды Взаимное разделение Dowex 5OW-X8 а-Оксиизомасляная кислота 91
Lu Минералы Dowex 50W-X8 а-Оксиизомасляная кислота, градиент 92
Y — Eu — Взаимное разделение Dowex 1-X4 ЦГДТА 93
Pm Y— Sc— Sm Tm—
Dy—La—
Pr
Лантаноиды Взаимное разделение Aminex A-5 а-Оксиизомасляная кислота 94
Ac 227Ac Ра Дочерние продукты Dowex 50 Ра: 0,5 М HF—0,5 М НС1 или 8,5 М НС1О4—0,5 М НС1—0,1 М HF, 50°С Ас: 6 М НС1—1 М HF или 6 М HNO3 21‘Pb: 2 М НС1 2"Ва, 223Ra: 2 М HNO3 227Ас: 4 М HNO3 227Th: 6 М HNO3 95 96
Ac РЬ, Ва HNO3 97
Ac Дочерние продукты (Bi, Th, Pb) AB-17-X8 Сорбция из НС1— ацетон или НС1 — 1-пропанол 98
5. Применение ионообменников
211
Амфотерное поведение Al, Ga и In также используют для их выделения из разнообразных смесей.
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Первый элемент этой группы, бор, практически не сорбируется катио-нообменниками. Сорбция других элементов (в трехвалентном состоянии) на сильнокислотных катионообменниках весьма высока. С другой стороны, ионы Т1(1) очень слабо сорбируются из растворов минеральной кислоты.
Как уже упоминалось, борат-ионы практически не поглощаются сильнокислотными катионообменниками (Dy < 1) из солянокислых растворов. Сорбция ионов А13 + уменьшается с повышением концентрации НС1. Максимальное значение коэффициента распределения (Dv = 80) получено для 0,1 М НС1. При этой же концентрации НС1 для Ga и In Dv равен соответственно 104 и — 10. Сорбция Ga увеличивается с повышением концентрации НС1 и в 6—12 М НС1 D v превышает 103. При концентрации НО выше 4 М ионы 1п3+ поглощаются в незначительной степени. Сорбция ионов Т13+ из разбавленных растворов НС1 очень низка. Для 4 М НС1 Dv равен 5 и постепенно увеличивается с повышением концентрации НС1 (в 12 М НС1 Dv = МО).
Подобным же образом ведут себя А13+ и Ga3+ в растворах бромисто-. водородной кислоты. Поглощение 1п3+ и Т13+ сильнокислотными катионообменниками из растворов НВг, особенно из концентрированных, очень велико. По сравнению с солянокислыми растворами среда бромистоводородной кислоты более подходит для сорбции 1п3+ и Т13+.
Элементы группы ЗБ в трехвалентном состоянии сильно поглощаются катионообменниками из растворов серной кислоты. Степень сорбции уменьшается в ряду Ga > In > Tl(III).
Растворы азотной кислоты также можно использовать для поглощения элементов группы ЗБ. Сорбция этих элементов уменьшается с повышением концентрации кислоты в ряду In3+ > Ga3+ > Т1+ . В разбавленных растворах хлорной кислоты 0,1 М) значения Dv всех элементов порядка 104; с повышением концентрации кислоты поглощение уменьшается, и минимальные значения Дополучены в области концентраций 5—6 М. В среде хлорная кислота — ацетон на катионообменнике можно осуществить простое и быстрое разделение смеси Ga — In — Т1 [99, 100].
Для А13+ в растворах уксусной кислоты Dg < 100. Поглощение Ga3+ и 1п3+ зависит от концентрации кислоты; значения D находятся в интервале 103—104.
Очень низкая сорбция Ga3+ и 1п3+ наблюдается из растворов муравьиной кислоты [101].
Ионы Al3+, Ga3+, In3+ и Т13 + легко сорбируются фосфорилированной целлюлозой. Они разделяются хроматографически элюированием 0,01 М раствором НС1 в смеси вода — метанол (1 : 4 по объему) или 0,5 М раствором НС1 в смеси вода — этанол (2 : 3 по объему). Элементы элюируются в порядке Т1 — А1 — In — Ga [102].
212
Часть II
НЕОРГАНИЧЕСКИЕ ИОНООБМЕННИКИ
Ионы Al3+, Ga3 + и 1п3+ сорбируются гидратированным оксидом циркония. Их поглощение лишь в незначительной степени зависит от pH раствора, достигая максимальной величины при pH около 3. Ионы Т13+ слабо сорбируются в области pH 1—6 (Dg < 10).
Поглощение ионов А13+ , Ga3+ и Т1+ на фосфате циркония увеличивается с повышением pH раствора. Ионы 1п3+ сорбируются при pH 3. Высокие значения коэффициентов распределения Т13+ получены на молибдате и вольфрамате циркония (pH > 2).
Галлий отделяют от Fe3+, А13+ и 1п3+ с помощью вольфрамата титана [103].
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Борат-ионы легко отделяются от других ионов на сильноосновных ани-онообменниках (pH раствора < 5). Борная кислота, как слабая кислота, непрочно связывается функциональными группами и количественно вымывается из обменной колонки водой (например, разделение Н3ВО3 — Н3РО4). При отделении борат-ионов часто рекомендуют сначала удалять катионы металлов, обменивая их с ионами Н+ на сильнокислотном катионообменнике. Отсутствие катионов, особенно тяжелых металлов, значительно облегчает отделение борат-ионов раствором гидроксида натрия (разделение ВО^~ — SiO3~ [104]). Слабоосновные анионообменники в ОН-форме также пригодны для отделения борат-ионов (например, разделение смеси ионов борат — фосфат — сульфат [105]).
При отделении ионов А13+ от большинства катионов металлов применяют солянокислую среду. В этих условиях ионы А13+ не поглощаются сильноосновными анионообменниками. Метод имеет практическое значение при отделении алюминия от железа [106].
Подобно А13+ , ионы щелочных и щелочноземельных металлов, Be, Mg, У, лантаноидов, Ac, Th и Т1(1) не поглощаются анионообменниками во всем интервале концентраций НС1.
Ионы А13+ слабо сорбируются из растворов НС1 — HF. Их сорбцией практически можно пренебречь. Цирконий, ниобий и гафний, напротив, сильно поглощаются анионообменниками из среды НС1— HF. Таким способом ионы А13+ легко отделяются от этих элементов (после промывания колонки раствором НС1 — HF).
Обратная картина наблюдается для ионов А13+ в среде чистой HF. Алюминий хорошо поглощается анионообменниками в форме фторидных комплексов.
Ионы А13+ плохо поглощаются из растворов серной кислоты, поэтому они легко десорбируются из анионообменника даже 0,01 М H2SO4. Это используют при отделении алюминия от железа; последнее десорбируют 1 М H2SO4. При добавлении к раствору серной кислоты метанола, этанола, 2-пропанола или ацетона ионы А13+ поглощаются анионообменником. Сорбция Ga, In и Al увеличивается с повышением доли органического компонента в смеси [107].
5. Применение ионообменников
213
Ионы А13+ не поглощаются сильноосновными анионообменниками из растворов бромистоводородной кислоты (в противоположность растворам HF). Однако при сНВг > 3 М поглощаются ионы Ga3+; степень сродства ионов 1п3+ в этой среде также очень велика. Сорбция 1п3+ увеличивается с повышением концентрации НВт. Бромидный комплекс трехвалентного таллия связывается очень прочно (коэффициент распределения Dg около 105). Таким образом, смесь Al — Ga — In — Т1 разделяют на анионообменнике Dowex 1-Х8 элюированием 7 М (А1), 2 М (Ga) и 10,1 М (In) растворами НВг После промывания смолы азотной кислотой и водой трехвалентный таллий элюируют 5%-ным раствором тиосульфата натрия [108]. Разделение указанной выше смеси можно проводить и с помощью соляной кислоты. Однако элюирование бромистоводородной кислотой обеспечивает лучшее и более быстрое разделение этой смеси. Кроме того, в этом случае нужны более короткие колонки.
Галлий, индий и таллий поглощаются анионообменниками из растворов соляной кислоты. Таллий предварительно окисляют до трехвалентного состояния, так как одновалентный таллий не сорбируется. Сорбция Ga максимальна в 7 М НО. При дальнейшем повышении концентрации кислоты сорбция уменьшается. Аналогично с повышением концентрации кислоты уменьшается поглощение Т1(Ш) (на сорбционной кривой нет максимума). Значительно меньшее поглощение [по сравнению с Ga(III) и Т1(Ш)] в среде НС1 наблюдается для ионов 1п(Ш). Максимальная сорбция достигается в - 3 М на.
Как упоминалось выше, смесь А1 — Ga — In — Т1 разделяют на индивидуальные компоненты элюированием соляной кислотой [109].
Эффективность разделения улучшается при элюировании смесью НС1 — органический растворитель. Согласно данным Коркиша и Хазана [ПО], при использовании соляной кислоты и соответствующего органического растворителя можно получить высокие значения коэффициентов распределения (табл. 5.29).
Таблица 5.29. Значения Dg Ga3 + , In3+ и А13+ на ионообменнике Dowex 1 в 0,6 М НС1 в присутствии некоторых органических растворителей [НО]
Среда Концентрация, об.®7о Ga3 + In3 + Al3 +
Ацетон 90 < 1 2 38
Ацетон 80 <1 10 25
Ацетон 70 20 36 8
МетиленгликолЬ 90 20 70 <1
Метиленгликоль 80 10 30 <1
МетиленгликолЬ 70 8 28 <1
214
Часть П
Таблица 5.30. Влияние температуры и степени поперечного связывания смолы Dowex 1 на разделение следовых количеств In и Ga 0,1 М ортофосфорной кислотой [ИЗ]
Температура, °C а‘п Ga '
Dowex 1-Х2 Dowex 1-Х4 Dowex 1-X8
15 12,08 13,00 36,00
25 11,80 8,66 42,30
45 8,33 7,95 11,69
65 6,00 6,50 8,86
85 4,84 5,56 6,36
Ионы Ga(III) отделяют от Fe(III) в растворах соляной кислоты после восстановления Fe(III) в Fe(II) [111].
Другой метод разделения Ga — Fe основан на использовании комплексных соединений элементов с щавелевой кислотой. Комплексы, образованные с 1 • 10“3 М щавелевой кислотой, сорбируются анионообменниками. Галлий элюируют 1 М NaOH [112].
Галлий отделяют от других элементов в виде галлатных анионов, а индий — в виде карбонатных комплексов.
В работе [113] подробно описано разделение Ga — In на анионообменнике Dowex 1 из фосфорнокислой среды. Эффективность разделения зависит от степени поперечного связывания обменников и от температуры элюирования (табл. 5.30). Смолу превращают в Н2РО4-форму промыванием 0,1—1,0 М раствором ортофосфорной кислоты. Галлий сорбируется в виде комплекса [Ga(HPO4)2]“, а индий — в виде комплекса [1п(НРО4)3]3“. Более быстрое уменьшение степени сорбции Ga (по сравнению с In) наблюдается при изменении концентрации ортофосфорной кислоты [114].
Ионы одновалентного таллия отделяют от многих других металлов в 2 М НС1. В то время как Zn2+, Cd2+, Pb2+, Sb3+ и Hg2+ связываются смолой, ионы Т1+ не сорбируются ею [115]. Для этой цели пригоден анионообменник ЭДЭ-10п.
Лабораторные методики
ОТДЕЛЕНИЕ И ОПРЕДЕЛЕНИЕ БОРНОЙ КИСЛОТЫ
В РАСТВОРАХ НИТРАТА УРАНИЛА [116]
Методика. Образец металлического урана в кварцевом сосуде растворяют в концентрированной азотной кислоте. Раствор осторожно выпаривают до появления кристаллов нитрата уранила. Твердую фазу растворяют в таком количестве диэтилового эфира, чтобы в 100 г его содержалось 10 г UO2(NO3)2.
Колонку (10 х 1 см), заполненную сильноосновным анионообменником Dowex 1-Х8 (0,015—0,07 мм) в NOj-форме, промывают водой, метанолом и эфиром. Через
5. Применение ионообменников
215
колонку со скоростью 5 см3/мин пропускают 300 см3 раствора нитрата уранила в эфире. Колонку промывают 100 см3 эфира и элюируют борную кислоту 25 см3 5 М НС1.
Концентрацию борной кислоты определяют методом пламенной фотометрии.
ОТДЕЛЕНИЕ БОРА ОТ ДРУГИХ ЭЛЕМЕНТОВ
НА КАТИОНООБМЕННИКЕ [117]
Методика. К - 150 см3 раствора, содержащего бор в форме борат-ионов, прибавляют НС1 или NaOH до pH 3. Полученный раствор пропускают со скоростью 4—5 капель/с через колонку, заполненную сильнокислотным катионообменником Dowex 50-Х12 (0,15—0,07 мм) в Н-форме. Элюат собирают. Размер ионообменной колонки зависит от содержания катионов. Для 10—15 ммолей катионов (в форме М2 + -ионов) требуется 250 см3 набухшего ионообменника и колонка внутреннего диаметра 3 см и высотой около 45 см.
Обменную колонку промывают затем примерно 500 см3 воды. Элюат и промывные воды объединяют и определяют количество бора подходящим методом.
ОТДЕЛЕНИЕ А1 НА СИЛЬНООСНОВНОМ АНИОНООБМЕННИКЕ. ОПРЕДЕЛЕНИЕ А1 В СТАЛЯХ [118]
Методика. Колонку (13 х 0,8 см), заполненную сильноосновным анионообмен-ником Amberlite IRA-400 в Cl-форме, промывают 100 см3 0,2 М НС1 и 100 см3 9 М НС1. Примерно 0,2 г (0,1—2% А1) образца стали растворяют в смеси 15 см3 НО (1:1) — 15 см3 Н004. Раствор упаривают до появления белых паров Н004, прибавляют несколько капель (10—15) концентрированной НС1 и продолжают выпаривание. Эту операцию повторяют 2—3 раза.
Остаток растворяют в 15 см3 концентрированной НС1 и выпаривают раствор досуха. Остаток растворяют в 15 см3 9 М НС1, раствор переносят в мерную колбу емкостью 200 см3 и разбавляют до метки 9 М НС1. Аликвотную часть раствора (5 см3) пропускают через колонку со скоростью 1 см3/мин. Алюминий элюируют 15 см3 9 М НС1 и выпаривают элюат досуха. Остаток растворяют в 0,5 см3 HCI (1 : 1) и разбавляют водой до 10 см3. Раствор количественно переносят в мерную колбу емкостью 50 см3 и определяют содержание алюминия спектрофотометрическим методом.
Если в образце стали присутствует никель, то он проходит в элюат вместе с алюминием. При спектрофотометрическом определении алюминия никель следует маскировать.
РАЗДЕЛЕНИЕ СМЕСИ Ga — In — Al НА АНИОНООБМЕННИКЕ В СРЕДЕ АЦЕТОН — НС1 [ПО]
Методика. Колонку подходящего размера, заполненную анионообменником Dowex 1-Х8 (0,15—0,07 мм) в Cl-форме, промывают 100 см3 смеси 80 об. % ацетона — 20 об. % 3 М НС1.
К 4 см3 3 М НС1, содержащей смесь разделяемых ионов, прибавляют 16 см3 ацетона. Полученный раствор пропускают через колонку со скоростью 0,2 — 0,3 см3/мин.
Ионы Ga3+ элюируют 200 см3 смеси 80% ацетона — 20% 3 М HCI, 1п3+ — 140 см3 смеси 90% ацетона — 10% 6 М НО, А13+ — 100 см3 смеси 70% ацетона — 30% 2 М HCI.
216
Часть И
РАЗДЕЛЕНИЕ СМЕСИ Al — In — Ga ПРИ АНАЛИЗЕ МЕТАЛЛИЧЕСКОГО АЛЮМИНИЯ [ПО]
Методика. 1 г алюминия растворяют в соляной кислоте. В процессе растворения постепенно првбавляют несколько капель азотной кислоты. Раствор выпаривают досуха. Остаток растворяют в нескольких куб. сантиметрах б М НС1 и снова выпаривают досуха. Остаток растворяют в 20,0 см3 5 М НС1. Аликвотную часть раствора (2 см3) разбавляют 18 см3 2-метоксиэтанола-1 и пропускают через колонку, заполненную смолой Dowex 1-Х8, которую предварительно промывают 100 см3 смеси 10% 6 М НС1—90% 2-метоксиэтаиола-1. Скорость пропускания раствора 0,2—0,3 см3/мин. Ионы А13+ элюируют 100 см3 смеси 30% 2 М НС1 — 70% 2-метоксиэтаиола-1. Смесь Ga—In разделяют элюированием 1 М НС1. Галлий десорбируется первым.
РАЗДЕЛЕНИЕ СМЕСИ Ga — Zn НА АНИОНООБМЕННИКЕ ИЛИ КАТИОНООБМЕННИКЕ В КАРБОНАТНОЙ СРЕДЕ [119]
Методика. Галлий в карбонатных растворах образует анионные комплексы, которые используют для его отделения от других элементов. При отделении больших количеств Ga3+ от малых количеств Zn2+ рекомендуется применять сильнокислотный катионообменник в МН4-форме. Колонку (35 х 0,8 см) заполняют 8—10 г кати-оиообмеииика КУ-2 (с частицами соответствующего размера), переводят смолу в ЫНд-форму и промывают водой. Образец, содержащий галлий и циик в соотношении (1000—10000) : 1, растворяют так, чтобы получить 2 М концентрацию (NH4)2 СО3. При образовании осадка вводят дополнительную порцию 2 М раствора (NH4)2CO3. С помощью гидроксида аммония доводят pH раствора до 9—10 и пропускают его через ионообменную колонку.'Затем колонку промывают 2 М раствором (NH4)2CO3. Галлий количественно переходит в элюат. После промывания колонки водой ионы Zn2+ элюируют 3 М НС1.
При соотношении Ga : Zn порядка 1:1 — 1 : 10 лучше применять аниоиообмеи-иик. Карбонатные комплексы галлия сорбируются обменником, в то время как ионы Zn(NH3)J+ проходят в элюат.
Методика с использованием анионообменника ЭДЭ-10п аналогична методике с катиоиообмеиником. Галлий элюируют 2,5 М НС1 .
ОТДЕЛЕНИЕ 67Ga ОТ БОЛЬШОГО ИЗБЫТКА Zn И Fe [120]
Методика. 1,5 г металлического цинка облучают нейтронами и растворяют в 10 см3 соляной кислоты. Для восстановления Fe3+ в Fe2+ вводят 100 мг иодида натрия. Раствор пропускают через колонку, заполненную 8,5 см3 сильнокислотного , катиоиообмеииика AG 50W-X8 (0,074—0,037 мм) в Н-форме. Затем смолу промывают 20 см3 концентрированной НС1, содержащей 1% Nal, если Fe3+ предварительно не отделено. Иодид натрия должен присутствовать во всех растворах, содержащих железо, так как он предотвращает окисление Fe2+ в Fe3+ и загрязнение фракции галлия железом. Для удаления всего присутствующего иодида натрия ионообменную колонку промывают б см3 концентрированной НС1 и элюируют галлий 20 см3 3,5 М НС1 . По этой методике от изотопа 67Ga (без носителя) отделяется более 99,998% цинка и 99,5% железа.
ОТДЕЛЕНИЕ Ga ОТ Al И ДРУГИХ ЭЛЕМЕНТОВ [121]
Помимо ионов Ga3+ на сильноосновном анионообменнике в 6 М НС1 сорбируются хлоридные комплексы Fe(III), Zn, Cd, Hg(II), Tl(III), Bi,
5. Применение ионообменников
217
Sb(V). Ионы Ga3+, Fe3 + и Sb(V) элюируют 1 М НС1 .Для отделения галлия от Fe(III) и Sb(V) прибегают к предварительному восстановлению последних.
Методика. Соответствующее количество образца растворяют в соляной кислоте и делают раствор б М по НС1 .Смесь пропускают через восстановительную колонку (6 х 1 см), заполненную металлическим серебром. Затем колонку промывают 20 см3 б М НС1. Объединенные фильтраты пропускают, через колонку (4 х 1 см), заполненную сильиоосиовиым анионообменником Dowex 1-Х4 (0,15—0,07 мм) в Cl-форме. После этого колонку промывают 60 см3 6 М НС1. Галлий элюируют 20 см3 1 М НС1 и определяют подходящим методом.
ОТДЕЛЕНИЕ Ga ОТ Ри В Ga—Ри СПЛАВАХ [122]
Методика. Разделение выполняют в колонке, заполненной иоиообмеиииком Dowex 1-Х4 (0,30—0,15 мм), предварительно приведенным в равновесие с раствором аскорбиновой кислоты в НС1 (1 г аскорбиновой кислоты рг -творяют в 100 см3 5 М НС1). Около 1 г образца растворяют в мерной колбе емкостью 25 см3 в ~ 5 см3 смеси 5 М НС1 — аскорбиновая кислота указанного состава (раствор должен быть всегда свежеприготовленным). Смесь прибавляют порциями по 0,5 см3. Растворение ведут при осторожном нагревании (смесь не должна кипеть). Раствор охлаждают и пропускают через колонку. Плутоний элюируют примерно 20 см3 смеси 5 М НС1 — аскорбиновая кислота со скоростью 4 см3/мии. В процессе элюирования окраска элюата меняется. Галлий элюируют 50 см3 0,5 М НС1.
РАЗДЕЛЕНИЕ СМЕСИ Т13+ — In3+ — Ga3+ — А13+ — Yb3 + НА СИЛЬНОКИСЛОТНОМ КАТИОНООБМЕННИКЕ В ВОДНО - ОРГАНИЧЕСКОЙ СРЕДЕ [100]
Методика. Колонку (19 х 2 см) заполняют 60 см3 катионообменника AG 50W-Х8 (0,07—0,03 мм) в Н-форме и приводят в равновесие с 0,1 М раствором НС1, содержащим 50% ацетона,
25 см3 раствора 0,1 М НС1 — 50% ацетона, содержащего до 0,5 ммоля каждого из вышеупомянутых элементов, пропускают через колонку со скоростью 3 см3/мии.
Отдельные элементы элюируют со скоростью 3,5 см3/мии растворами следующего состава: Т13+ — 200 см3 0,1 М НС1 — 50% ацетона; Ga3+ — 250 см3 0,5 М НС1 — 90% ацетона; 1п3+ — 200 см3 0,5 М НС1 — 50% ацетона; А13+ — 400 см3 3,0 М НС1 — 40% ацетона; Yb3+ — 300 см3 4,0 М НС1. Такие количества элюентов необходимы для отделения приблизительно 0,5 ммоля каждого из элементов.
Ионы А13+ отделяют от Fe3+,UO$+, Ве2+, Сц2+, Со2+, Mn2+ , Ga3+, In3+, Zn2+ , Cd2+ и Hg2+ элюированием смесью 2 М НС1 в 70% ацетона. Иоиы 1п3+ отделяют от Cu2+ , Со2+ , Mn2+ , Ni2+ и Fe3+ элюированием 0,5 М НС1 — 40% ацетона. Cd2+ и Sn4+ элюируются вместе с 1п3 + .
ОТДЕЛЕНИЕ In ОТ Cd, Zn, Pb, Ga, Al И Fe
НА АНИОНООБМЕННИКЕ AB-17 В СО3-ФОРМЕ [123]
Методика. 25 см3 раствора (pH 2,5), содержащего разделяемые элементы, пропускают со скоростью 1 см3/мин через колонку (9 х 1,6 см), заполненную сильиоосиовиым аииоиообмеиииком АВ-17 в С03-форме. Затем колонку промывают 15 см3 воды (5 см3/мии) и элюируют отдельные элементы в следующем порядке: Zn, Al,
218
Часть II
Ga — 250 см3 3 М NH4OH; In — 70 см3 1,5 М (NH4)2CO3; Pb — 50 см3 1 М NaOH; Fe — 160 см3 1,5 М НС1.
Этим методом отделяют микрограммовые количества индия от 100-кратного избытка Ga, Zn, Cd, Al и Fe. Максимальная относительная ошибка определения сопутствующих элементов не превышает т 10%.
ОТДЕЛЕНИЕ 115mIn ОТ 115mCd НА АНИОНООБМЕННИКЕ [124, 125]
Методика. Кадмий, облученный нейтронами (смесь In и Cd), растворяют в 2 М НС1 с добавлением нескольких капель пероксида водорода. Раствор выпаривают досуха. Остаток растворяют в минимальном количестве 2 М соляной кислоты и пропускают раствор через колонку (7 х 1,3 см), заполненную анионообменником Dowex 1-Х8 в Cl-форме (размер зерен 30—40 мкм). I15mIn элюируют 20 см3 0,15 М НО со скоростью 1 см3/мин. Его радиохимическая чистота выше 99,99%.
ОТДЕЛЕНИЕ ИОНОВ Т1+ ОТ РЬ2+ И ДРУГИХ ЭЛЕМЕНТОВ [126]
Методика. Колонку, заполненную на 20% смолой Dowex 50 (1,2—0,4 мм) в Na-форме, промывают щелочным раствором глицерина (0,5 М NaOH, 1 М глицерин).
Анализируемый раствор делают 0,5 М по NaOH и 1 М по глицерину и пропускают через колонку. Ионы Т1+ поглощаются количественно, тогда как глицератные ,комплексы свинца в данных условиях не сорбируются. Затем колонку тщательно промывают водой и элюируют ионы Т1+ раствором нитрата кальция.
В среде 1 М глицерин — 1 М NaOH таллий отделяют от Pb, Sn, Си, Bi, Zn, Al, Fe, As, Sb, Se и Те.
Обзор других методов разделения смесей, содержащих элементы группы ЗБ, приведен в табл. 5.31.
Таблица 5.31. Обзор избранных методик разделения элементов группы 3 Б
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
в Боросиликаты Dowex 50W-X8 127
в U, Ри Dowex 50W-X10 В: 0,2 М HNO3 128
в Сульфат тория ядерной чистоты 129
А1 Be Dowex 1 Al: 1 М HF—0,01 И НС1 Be: 1 И НС1 130
А1 Ti Сильноосновный Al: 12 М НС1 131
анионообменник Ti: 3 М НС1
А1 Ga ЭДЭ-10п Al: 7 М НС1 Ga: Н2О 132
А1 Fe Dowex 1 Al: 0,01 М H2SO4 Fe: 0,10 М H,SO„ 133
А1 Be Селективная Al: 4 M CH3COONH4 134
смола Be: 2 M HC1
5. Применение ионообменников
219
Продолжение табл. 5.31
Эле- Отделяемые Ионообменннк Условия разделения Лнте-
мент компоненты ратура
А1 Mg, Fe Dowex 50-X8 135
А1 Be Be: 0,5 М H2SO4 Al: 0,7 М H,SO. 136
А1 30 элементов Al-сплава при нейтронно-актива ционном анализе и радиохимическом разделении Dowex 50 Dowex 1 Среда НС1 и HF 137
Al.Ga In, Fe Си AB-17 Ga, Al: раствор NaOH 138
Другие: лактат аммония, pH 4,0
Al Силикаты Ti, Fe 139
Al 82Sr AG 1-X8 (ОН') Al: сорбция A1(OH)~ Sr: Sr(OH)2 в элюате 140
Ga Al Wofatit L-150 Ga: 4 M KSCN—0,1 M HC1 Al: 2 M KSCN—0,05 M HC1 141
Ga In Dowex 1-X8 Ga: 0,5 M KI In: 1 M NH4C1 (pH 1) 142
Ga In, Al Dowex 1-X8 Ga: 0,5 M KI (pH 1) In: 0,5 M (NH4)2SO4 или Ga + In: 1 M HC1 Al: 6 M HC1 143
Ga • In, Al Dowex 1-X8 Ga: 80% ацетона; 20% 3 M HC1 In: 90% ацетона; 10% 6 M HC1 Al: 70% ацетона; 30% 2 M HC1 НО
68Ga Cu, Ag, Sb, Fe Dowex 1-X8 Другие: в 14 M НС1 не сорбируются Ga: 4 M HC1 144
Ga Fe, Al, In Вольфрамат 103
титана
Ga In КУ-2 In: элюируется 0,5 M HC1— 40% ацетона Ga: 2,5 M HC1 145
Ga Zn, Cd ЭДЭ-10п Ga: сорбируется из цитратной, малонатной или 4-аминосалицилатной среды 146
220
Часть П
Продолжение табл. 5.31
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
Ga Cu, Ni ЭДЭ-10 и или КУ-2 Ga: карбонатный комплекс в 2 M (NH4)2CO3— NH4OH 119 147
Ga In, Tl, Al и другие Dowex 21К Малонатная среда 148
Ga In, Al Amberlyst 15 In: 0,3 М НС1—20% диметилсульфоксида Ga: 0,3 М НС1—90% ди-метилсульфоксида А1: 6,0 М НС1 149
In Sb—Те—Sn Amberlite IRA-400 In—Sb: 3 М НС1 Те: 1 М НС1 Sn: 2 М НС1О4 150
In Двухвалентные элементы Обмениик, содержащий —РО(ОН)2 группы Сорбция из 0,2—0,3 М НС1 или HNO3; сорбируется только In 151
In Zn, Cd, Pb Сильиоосновный анионообменник Тиосульфат натрия 152
In Cd Анионообменник 2,5—4,4 М LiCl 153
113In 113Sn Chelex-100 Bio-Rex 63 154
T1(I) Ag, Mg, Hg, Cu Amberlite IR-120 Ag, Mg, Hg, Cu: 2%-ный NaNO2 Tl: 10%-ная H,SO 2 4 155
T1 Морская вода De Acidite FF Tl: вода, насыщенная SO2 154
и воды различного происхожде-
НИЯ
Tl Wofatit P 156
Tl Hg AG 50W-X8 Tl: 12,5%-ная HC1O4 Hg: 12,5%-ная HC1O4 (tl первый) 157
Tl Hg Hg: NaNO2 Tl: Na,SO. 2 4 158
Tl In, Zn Zeokarb 226 и Dowex A-l 160
Литература
1. Scherrer V., WeiglF., van Ghemen M„ Inorg. Nucl. Chem. Lett., 3 1967, 589.
2, Преображенский Б. К., и др. Радиохимия, 2 (1960) 239.
5. Применение ионообменников
221
3. Tompkins Е. R., MayerS. W., J. Amer. Chem. Soc., 69 (1947) 2859.
4. Брынкина Г. Д. и др. Радиохимия, 8 (1966) НО.
5. Sekine Т„ Hasegawa Y., Bull. Chem. Soc. Japan, 39 (1966) 240.
6. Kuroda R. et al., J. Chromatogr., 22 (1966) 143.
7. Petrow H. G., Allen R. J., Anal. Chem., 35 (1963) 747.
8. Брандштетр И., Зварова T. С., Крживанек М., Малы Я. Радиохимия, 5 (1963) 694.
9. Subharaman Р. R„ Rajan К. S„ Gupta J., Z. anorg. allgem. Chem., 304 (1960) 191.
10. Hamaguchi H„ Kuroda R., Опита N., Talanta, 10 (1963) 120.
11. Carter J. A., Dean J. A., Appl. Spectrosc., 14 (I960) 50.
12. PietrzykD. J., Kiser D. L„ Anal. Chem., 37 (1965) 233.
13. Ripan R., Stanislav C., Chim. Analyst., 49 (1967) 469.
14. BriichnerE., Szarvas P., Acta Chem. Hung., 52 (1967) 31.
15’. BriichnerE., Szarvas P., J. Inorg. Nucl. Chem., 28 (1966) 2361.
16. Wish L., Foti C. S., J. Chromatogr., 20 (1965) 585.
17. Zeligman M. M„ Anal. Chem., 37 (1965) 524.
18. Wolfsberg K, Anal. Chem., 34 (1962) 518.
19. Campbell D. O., Ind. Eng. Chem. Process Develop., 9 (1970) 95.
20. Star? J., Talanta, 13 (1966) 421.
21. Deelstra H„ Verbeek J., J. Chromatogr., 17 (1965) 558.
22. Le Roy Eyring (Editor), Progress in the Science and Technology of the Rare Earth, Vol. 1, Pergamon Press, 1964.
23. Чувелева E. А., Харитонов Д. В., Гелис В. M., Назаров П. П. Чму-тов К. В. ЖФХ, 48 (1974) 2846.
24. Кунбазаров А., Рудомино М. В., Сорочан А. М„ Дятлова Н. М., СенявинМ. М. Ж. прикл. химии, 47 (1974) 2452.
25. Alexa J., Collect. Czech. Chem. Comrnun., 30 (1965) 1799.
26. Alexa J., Collect. Czech. Chem. Comrnun., 33 (1968) 2731.
27. Alexa J., Collect. Czech. Chem. Comrnun., 36 (1971) 3370.
28. Sisson D. H., Mode V. A., Campbell D. O., J. Chromatogr., 66 (1972) 129.
29. Campbell D. O., Buxton S. R., Ind. Eng. Process Des. Develop. 9 (1970) 89.
30. Campbell D. O., Ketelle B. H„ Inorg. Nucl. Chem., Lett., 5 (1969) 533.
31. PfrepperG., J. Chromatogr., 116 (1976) 407.
32. Faris J. P., J. Chromatogr., 26 (1967) 232.
33. Faris J. P., Buchanan R. F., Anal. Chem., 36 (1964) 1157.
34. Roelandts J., Duyckaerts G., Brunfelt A. J., Anal. Chim. Acta, 73 (1974) 141.
35. Hamaguchi H., Опита N„ KishiM., Kuroda R., Talanta: 11 (1964) 495.
36. Chakravorty M., Khopkar S. M., Chromatographia, 10 (1977) 100.
37. Taketatsu T., Talanta, 10 (1963) 1077.
38. Arnold R„ Ritchie J. F„ J. Chromatogr., 8 (1963) 205.
39. Minczewski J., Dybczynski R., J. Chromatogr., 7 (1962) 98, 568.
40. Dybczynski. R., J. Chromatogr., 50 (1970) 487,
222
Часть И
41. Dybczynski R., Wddkievicz L., J. Inorg. Nucl. Chem., 31 (1969) 1495.
42. Strelov F. W., Bothma C. J., Anal. Chem., 36 (1964) 1217.
43. OrlandiniK. A., Inorg. Nucl. Chem. Lett., 5 (1969) 326.
44. Wilkins D. H., Smith G. E, Taianta, 8 (1961) 138.
45. Das A. K. et al., Anal. Chim. Acta, 47 (1969) 162.
46. Ahluvalia S. S., Korkisch J., Anal. Chim. Acta, 31 (1964) 552.
47. Martin G. C„ Jr., J. Inorg. Nucl. Chem., 26 (1964) 1621.
48. Nelson E, J. Chromatogr., 16 (1964) 538.
49. Sherma J., van Leuten F. J., Separ. Sci., 6 (1971) 199.
50. Chakravorty M., Khopkar S. M., Chromatographia, 10 (1977) 100.
51. Hakkila E. A., Hurley R. G., Watebury G. R., Anal. Chem., 41 (1969) 665.
52. Strelov F. W., Baxter C., Taianta, 16 (1969) 1145.
53. Csdvari M., Czegtedi B., 2-nd Symposium on Ion-Exchange, Vol. 1, Budapest, 1969, p. 73.
54. Nelson F., Day R. A., Kraus К. A., J. Inorg. Nucl. Chem., 15 (1960) 140.
55. Mosen A. W., Schmitt R. A., Vasilevskis J., Anal. Chim. Acta, 25 (1961) 10.
56. Petru F., PeSek J., Collect. Czech. Chem. Commun., 36 (1971) 3752.
57. Pesek J., Petri F., Collect. Czech. Chem. Commun., 37 (1972) 2528.
58. Albert P„ Cleyrergue C„ Deschamps N„ ЬеНёпсу J., J. Radioanal. Chem., 1 (1968) 297.
59. Alimarin I. P. et al., J. Radioanal. Chem., 1 (1968) 139.
60. Deschuyter M„ Massart D. L„ Speecke A., Hoste J., Radiochim. Acta, 10 (1968) 11.
61. Mazzucotelli A., Frache R., Dadone A., BaffiF., Cescon P„ Anal. Chim. Acta, 99 (1978) 365.
62. Strelov F. W., Gricins A. J., Anal. Chem., 44 (1972) 1898.
63. Seim H. J., Johnson J. L., Stever K. R., Heady H. H., U. S. Bur. Mines Rept. of Invest., 6097, 1962.
64. Massart D. L., Bossaert W.., J. Chromatogr., 32 (1968) 195.
65. Gaittet J., Ann. Chim., 5 (1960) 1219.
66. Gusmini S., Dubuquoy C., Report CAE-R-4102, 1971.
67. Krtil J., Moravec A., MenclJ., Alexa J., Radiochem. Radioanal. Lett., 3 (1970) 113.
68. Kashuba A. T., Hines C. R., Anal. Chem., 43 (1971) 1758.
69. Wodkievicz L., Dybczynski R., Chem. Anal. (Wars.), 19 (1974) 175.
70. Гусева Л. И., Тихомирова Г. С. Радиохимия, 12 (1970) 771.
71. Eusebius А. К., Mahan A., Ghose А. К., Dey А. К., Indian J. Chem. Sect. А, 15А, (1977) 438.
72. Абраров О. А., Аминова М. М„ Докл. АН Узб. ССР, 1 (1976) 24.
73. LalitC. Т., Mahan А. Е., Ghose А. К., Dey А, К., Indian J. Chem. Sect. А, 15А (1977) 438.
74. Bleyl Н. J., Muenzel H., Radiochim. Acta, 9 (1968) 149.
75. Dybczynski R., J. Chromatogr., 31 (1967) 155.
76. Shibata M., Tanabe I., Oda K., Kadota K., Bull. Chem. Soc. Japan, 41 (1968) 2627.
5. Применение ионообменников
223
77. Hugson М. R., Sen Gupta J. G., Amer. Mineralogist, 49 (1964) 937.
78. Fritz J. S., Garalda В. B„ Taianta, 10 (1963) 91.
79. Braier H. A., 2-nd U. N. Conf, on the Peaceful Uses of Atomic Energy, Geneva, 1958, Rept. A/CONF 15/P/1568.
80. StrelowF. W„ J. South African Chem. Inst., 16 (1963) 38.
81. Kallamann S., Oberthin H. K., Hibbits J. O., Anal. Chem., 32 (1960) 1278.
82. Zeligman M. M., Anal. Chem., 37 (1965) 524.
83. Brunisholz G., J. Chromatogr., 75 (1973) 101.
84. Dubuquoy C., Metzger G., Analysis, 5 (1977) 314.
85. Федоров П. И., Филипова H. В., Андреев В. К., Ж. аналит. химии, 31 (1976) 392.
86. Hagiwara Z., Oki Н., Bull. Chem. Soc. Japan, 42 (1969) 3177.
87. Das N. R., Bhattacharyya N. S. Int. J. Appl. Radiat. Isot., 28 (1977) 731.
88. Лаврухина А. К., и др. Радиохимия, 11 (1969) 330.
89. Turkstra J., Behrens G. B., deWet W. J., J. South African Chem. Inst., 22 (1969) 111. Anal. Abstr., 19 (1970) 1148.
90. Wolfsberg K„ Anal. Chem., 34 (1962) 519.
91. FalcoffR., MayS., Piccot D., Bull. Soc. Chim. France, 1967, 3257.
92. Massart D. L., Hoste J., Anal. Chim. Acta, 42 (1968) 7, 15, 21.
93. Dybczinski R„ J. Chromatogr., 32 (1968) 394.
94. Hbhlein G., Voller H., Wienlander W„ Radiochim. Acta, 11 (1969) 172.
95. CabellM. J., Canad. J.Chem., 37 (1959) 1094.
96. Muller W. A., Radiochim. Acta, 9 (1968) 181.
97. Blair A. J„ Ihle H., Michael H., Radiochim. Acta, 10 (1968) 119.
98. Каралова 3. К., Мясоедов Б. Ф., Родионова Л. M., Пыжова 3. И., Ж. аналит. химии, 29 (1974) 1332.
99. StrelowF. W., Victor А. Н., Taianta, 19 (1972) ”49.
100. StrelowF. W., Victor A. H., van Zyl C. R„ h,jff C„ Anal. Chem., 43 (1971) 870.
101. Korkisch J., Hazan J., Taianta, 11 (1964) 1157.
102. Bilba D„ Fisel S., Mikrochim. Acta Wien, 1974, 985.
103. Qureshi M., Gupta J. P., J. Chem. Soc., 1970, A, 2620.
104. Suzuki T., J. Chem. Soc. Japan, Pure Chem. Sect., 1961, 82.
105. Callicoat D. L„ Wolszon J. D., Hayes J. R., Anal. Chem., 31 (1959) 1437.
106. Moore G. E., Kraus K. A., J. Amer. Chem. Soc., 72 (1950) 5792.
107. Плюснин А. В., Казанцев E. И., Пятин H. П. Изв. высш, учебн. завел. Цвета. металлы, 4 (1975) 72.
108. Andersen Т., KnutsenA. В., Acta Chem. Scand., 16 (1962) 849.
109. Kraus К. A., Nelson F„ Smith G. W., J. Phys. Chem., 58 (1954) 11.
110. Korkisch J., Hazan J., Anal. Chem., 36 (1964) 2308.
111. Gregory G. R., Jeffrey P. G„ Taianta, 9 (1962) 800.
112. Blasius E., Negwer M., Z. anal. Chem., 143 (1954) 257.
113. Shabana S., J. Radioanal. Chem., 41 (1977) 53.
114. Dybczynski R., Polkowska-Motrenko H, Shabana R. M., J. Chromatogr., 134 (1977) 285.
115. Ефремов Г. В., Зверева A. M., Кодевзурем Т. Зав. лабор., 28 (1962) 159.
224
Часть II
116. Muzzarelli A. A., Anal. Chem., 39 (1967) 365.
117. Master Analytical Manual Section 1, Metod No, 100701, USAEC, 1961.
118. Foglino M. L., Spagliardi G. P., Metalurgia Ital., 50 (1958) 372. Anal. Abstr., 7 (1960) 2232.
119. Алимарин И. П., Цнцевич E. П., Горохова A. P. Зав. лабор., 26 (1960) 144.
120. Neirinckx R. D„ van der Merwe M. J., J. Radiochem. Radioanal. Lett., 7 (1971) 31.
121. Gregory G. R., Jeffery P. G., Talanta, 9 (1962) 800.
122. Miner F. J., DeGrazio R. P., Anal. Chem., 37 (1965) 1071.
123. ЭриставиД. И., Эристави В. Д., Куртладзе Г. С. Ж. аналйт. химии, 26 (1971) 2234.
124. Torko J., Atompraxis, 12 (1966) 97.
125. Torko J., Magy. Kern. Foly., 72 (1966) 17.
126. Carobene G„ Vicedomini M., J. Chromatogr., 33 (1968) 566.
127. Povondra P., Hejl V., Collect. Czech. Chem. Commun., 41 (1976) 1343.
128. Wenzel A. W„ Pietri CH. E, Anal. Chem., 36 (1964) 2083.
129. Federgrun L., Abrao A., Rept. IEA-420, 1976. C. A. 88 (1978) 163374.
130. Nelson F„ Rush R. M., Kraus K. A., J. Amer. Chem. Soc., 82 (1960) 339.
131. Hibbs L. E, Wilkins D. H., Talanta, 2 (1959) 16.
132. Денисова H. E„ Цветкова E. В. Зав. лабор., 27 (1961) 656.
133. Kraus К. A., Nelson F., J. Amer. Chem. Soc., 75 (1953) 3273.
134. Blasius E., Janzen К. P., Fallot-Burghardt W., Talanta, 18 (1970) 549.
135. Danielsson L., Ekstrom T., Acta Chem. Scand., 21 (1967) 1173.
136. Vetejska K, MazaHek J., Collect. Czech. Chem. Commun., 25 (1960) 2245.
137. Beurton G„ Beyssier B., Angella Y„ J. Radioanal. Chem., 38 (1977) 257.
138. Пятницкий И. В., Коломиец Л. Л., Ж. аналйт. химии, 25 (1970) 479.
139. Калинин А. И., Калинина Н. Л., Ж. аналйт. химии, 26 (1971) 881.
140. Kulprathipanja S., Hnatowich D. J., Brownell G. L., J. Radioanal. Chem., 43 (1978) 439.
141. Kpcheva L. L., Draganova R., God. Sof. Univ. Khim. Fak., 62, (1967/68) 129. Anal: Abstr. 20 (1970) 2350.
142. Kocheva L. L., C. R. Acad.Bulg. Sci., 22 (1969) 447. Anal Abstr. 19 (1970) 107.
143. Kocheva L. L„ Anal. Abstr., 20 (1971) 2351.
144. Aubronin G., Diebolt J., Junod E., Laverlochire J., Proc. 2-nd Conf. Modern Trends Activation Analysis, 1965, p. 344.
145. Цинцевич E. П. и др. Вест. МГУ, Сер. хим., 1969, 91.
146. Sura nova. Z. Р., Morozov A. A., Grabchuk О. Ya, Anal. Abstr., 18 1970, 3817.
147. Цинцевич E. П„ Горохова А. Н. Изв. выс. учебн. завед., Химия и хим. технология, 1960, 245.
148. Chakravorty М., Krdpkar S. М., Chromatographia, 9 (1976) 230.
149. Janauer G. Е., van Wart Н. E„ Carrano J. T., Anal. Chem., 1970, 42.
150. Stronski I., Rocz. Chem., 34 (1960) 709.
151. Marhol M., Cheng K. L., неопубликованные данные.
5. Применение ионообменников
225
152. Sugisata R., Kimata К., Japan Analyst, 15 (1966) 974. C. A. 68 (1968) 18266.
153. Irwing H., Woods G. T., J. Chem. Soc. 1963, 939.
154. Bhatnagar R. P., Trivedi R. G., J. Indian. Chem. Soc., 42 (1965) 53.
155. DenniR., Adloff J. P, J. Inorg. Nucl. Chem., 30 (1968) 1112.
156: Matthews A. D., Riley J. P., Anal. Chim. Acta, 48 (1969) 25.
157. Panchev B. N., Izv. Geol. Inst. Big. An., 1963, 237-
158. Dosch R. G., Anal. Chem., 38 (1966) 826.
159. CampbellD. O., J. Inorg. Nucl. Chem., 35 (1973) 3911.
160. Iyer S. G., Padmanabhan P. K., Nair L. D., Venkateswarlu Ch., Talanta, 23 (1976) 525.
5.2.4. Группа 4
5.2.4.1. 4A: Ti, Zr, Hf, Th
Элементы группы 4A периодической системы образуют четырехзарядные катионы, которые имеют большую склонность к гидролизу. Помимо типичного четырехвалентного состояния титан может находиться в форме трехзарядного катиона. В водных растворах четырехзарядные катионы существуют в виде М4+ (М = Ti, Zr, Hf, Th) или MO2+ (М = Ti, Zr, Н0.
Принимая во внимание большую склонность к гидролизу и полимеризации четырехзарядных катионов в водных растворах, все операции с такими растворами проводят в довольно кислых растворах. Однако при повышении концентрации кислоты поглощение этих элементов катиоиообменника-ми уменьшается.
Элементы этой группы образуют достаточно прочные комплексы с целым рядом веществ. Большое значение в процессах разделения имеют пероксо-комплексы (титан), комплексы с щавелевой и фтористоводородной кислотами (цирконий, гафний), карбонатные и сульфатные комплексы (торий).
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
В присутствии сильных минеральных кислот поглощение Ti(IV), Zr, Hf и Th сильно зависит от природы и концентрации кислоты. Поглощение катионов Ti4+ на смоле Dowex 50 из растворов различных минеральных кислот уменьшается в ряду НС1О4 > HNO3 > НС1 > H2SO4. Аналогичное поведение наблюдается также для Zr и Hf.
Коэффициенты распределения Dg на сильнокислотном катионообменнике в растворах соляной и серной кислот (вплоть до 2 М) для Hf выше, чем для Zr. Заметное поглощение наблюдается только в более кислых растворах (3—10 М) НС1О4. В растворах других минеральных кислот (за исключением серной кислоты) значения Dg составляют около 1. Более подробные сведения содержатся в обзорах Винарова [1] и Мукхерджи [2].
Высокое сродство четырехвалентных элементов к сильяокислотным ка-тионообменникам в растворах соляной кислоты используют для их отделе
с~354
226
Часть II
ния от катионов в более низком состоянии окисления. Так, например, Th можно отделить от U, Ac, Hg, Си, лантаноидов, щелочных и щелочноземельных металлов и других элементов сорбцией на катионообменнике и последующим элюированием 4 М НС1. Не десорбируется только Th. Зона ионообменника, содержащая ионы Th(IV), лишь незначительно смещается в направлении потока жидкости. Другие элементы, за исключением элементов, образующих осадок в 4 М растворе НС1, элюируются количественно. Присутствие больших количеств циркония затрудняет отделение Th. В этом случае перед стадией десорбции (4 М НС1) рекомендуется промывать ионообменник соответствующими элюентами, что обеспечивает отделение тория от всех других элементов, но не позволяет количественно элюировать торий (99,9%) из обменника с 8 — 12% ДВБ легко летучим элюентом. Наиболее эффективным элюентом для тория служит ЗМ H2SO4, но ее введение усложняет определение тория. Наиболее точные результаты получаются при озолении смолы.
По данным Стрелдва и Бошофа [3], бромистоводородная кислота — более подходящая среда для отделения тория от циркония, лантана и других элементов. Для смолы с содержанием 4% ДВБ факторы разделения Th — La, Th — Zr и Th — Ba в 5,5 M НВг больше 10. Смолы, содержащие 8 или 12% ДВБ, сорбируют торий очень сильно (табл. 5.32). Для сравнения в табл. 5.32 приведены результаты, полученные в растворах НС1. В 5,5М НВг можно легко отделить торий от La и Zr. Такие элементы, как Li, Na, К, Rb, Cs, Be, Mg, Ca, Sr, Fe(II), Co, Ni, Mn(II), Cu, Zn, Cd, Hg(H), Pb, Pd(II), Sc, Y, Ce(III) и остальные лантаноиды, Al, Fe(III), Sb(III), As(III),
Таблица 5.32. Коэффициенты распределения Dg La(III), Zr(IV) и Th(IV) в соляной и AG 50W с различной степенью поперечного связывания [3]
Концентрация кисло- . ты, М Степень поперечного связывания X 4 X 8
La Zr Th La
HCI НВг HCI НВг НС1 НВг НС1 НВг
0,1 >104 >104 >104 >104 >104 >104 >104 >104
0,2 3820 4330 >104 >104 >104 >104 >104 >104
0,5 491 558 6590 >104 5470 2950 1180 1950
1,0 100 115 2710 5570 492 405 277 284
2,0 23,2 28,7 221 364 80 145 57 55
3,0 10,4 13,6 32,2 56 35,5 80 23,8 22,4
4,0 6,6 9,9 10,7 14,7 26,1 60 11,4 14,2
5,0 7,0 8,3 7,1 7,8 25,0 77 10,1 11,0
6,0 7,5 9,0 6,2 6,0 26,4 126 9,8 11,4
7,0 8,1 12,0 5,4 6,8 28,3 229 10,4 13,6
8,0 9,0 19,0 5,1 9,8 30,6 638 11,3 21,3
5. Применение ионообменников
227
Bi(III), Ir(III), Rh(III), Hf, Sn(IV), Ge(IV), Se(IV), Te(IV), Pb(IV), Ir(IV), V(V) и U(VI), элюируются одновременно с лантаном (или барием и цирконием) или до него. Из других элементов относительно высокие значения Dg в этом растворе имеет Аи. Его десорбируют ЗМ НВг или лучше смесью 0,5 М НВг — 90% ацетона перед элюированием других элементов. В этих условиях элюируются также такие элементы, как Cd, Ga, In и Fe(III). Торий десорбируется последним 5 М HNO3. Выделение тория в области концентраций 200 мкг — 400 мг превышает 99,9%.
Торий хорошо поглощается катионообменником из 1М азотной кислоты и таким образом отделяется от лантана, церия и протактиния. В более концентрированной азотной кислоте (> 4 М) образуются слабо сорбирующиеся нитратные комплексы. На поглощение тория большое влияние оказывает степень поперечного связывания смолы. Некоторые количественные данные приведены в табл. 5.33.
Сильнокислотные катионообменники поглощают торий из сернокислых растворов при условии, что концентрация серной кислоты не превышает 1 М. В этой среде торий отделяется от Ni, Со, Cu, Fe(III), V(IV), Mo(VI), U(VI) и Cr(VI). При концентрации кислоты больше 3 М торий вымывается из ионообменника.
Элементы четвертой группы обычно не сорбируются катионообменни-ками из растворов карбонатов, щавелевой, фтористоводородной, лимонной кислот и ЭДТА, чем часто пользуются при их разделении.
Разделение титана и циркония основано на различии в степени их сорбции. На сильнокислотном катионообменнике из 1 М НС1 сорбируются
бромистоводородной кислотах на катионообменнике
X 12
Zr Th La Zr Th
HCI HBr HCI HBr HCI HBr HCI HBr HCI HBr
>104 >104 >104 >104 >104 >104 >104 >104 >104 >10‘
>104 >104 >104 > 104 >104 >104 >104 >104 >104 >10‘
>104 >104 > 104 > 104 4200 5000 >104 >104 >104 > 10‘
9200 >104 2220 2410 527 641 8620 9710 4560 8170
521 661 230 341 96 102 500 662 499 641
85 159 97 151 38,4 43,4 85 116 171 234
15,8 31,6 60 115 21,8 25,1 21,2 31,3 98 157
6,0 12,6 48,7 145 17,3 19,2 8,4 12,7 74 147
3,9 8,5 46,3 197 13,7 18,1 4,9 7,9 60 153
3,2 9,6 45,4 295 13,8 20,8 3,9 7 6 50 143
3,1 14,1 45,0 349 14,7 27,7 4,0 10,5 41,7 101
228
Часть II
Таблица 5.33. Коэффициенты распределения Dg Th в растворах азотной кислоты на смолах AG 50W различной степени поперечного связывания [3]
HNO3, М Х4 Х8 XI2 Х16
0,1 > 104 > 104 > 104 > ю4
0,2 > 104 > 104 >104 > ю4
0,5 2070 > ю4 > ю4 > ю4
1,0 327 1180 3380 3670
. 2,0 52 123 271 332
3,0 21,4 43,0 90 122
4,0 14,8 24,8 55 68
5,0 12,6 19,7 39,4 49,6
6,0 12,8 21,3 41,2 52
лишь небольшие количества Ti(IV), в то время как цирконий количественно удерживается обменником. Ионы Ti(IV) полностью элюируются IM НС1 [4]. Аналогичным способом цирконий отделяют от хрома и алюминия [5], а также от железа и тория [6]. Цирконий элюируют 5М НС1.
Максимальная эффективность отделения циркония от гафния достигается в смеси азотной и лимонной кислот [7]. Цирконий элюируется первым смесью 0,4 М HNO3 — 0,09М лимонная кислота. Такие же результаты получены при элюировании 0,4 — 0,6 М серной кислотой [1].
Катионообменники фенольного типа преимущественно применяют при элюировании смеси Zr — Hf — La — Nd [9].
Титан (IV) в растворах серной (0,1 М) или соляной (0,4 М) кислоты превращается в несорбируемые комплексы и таким образом количественно элюируется из катионообменника. Пероксо-комплексы титана очень прочно связываются катионообменником. Из 0,5 — 0,7 М растворов соляной кислоты, содержащих 0,5% Н2О2, титан сорбируется количественно. В этих условиях ниобий полностью переходит в элюат. Аналогичным способом титан отделяют от V(V), Mo(VI) и W(VI) из растворов с pH 5, содержащих Н2О2.
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Ионы Ti(IV) не поглощаются сильноосновными анионообменниками из разбавленных растворов соляной кислоты. Комплексные анионы TiClg-сорбируются из более концентрированных растворов кислоты [10]. Ионы Ti(III) не сорбируются анионообменниками во всем интервале концентраций кислоты.
Цирконий и гафний поглощаются только из более концентрированных (>7 М) растворов НС1 [И]. В концентрированной соляной кислоте коэффициенты распределения выше 103. Отсутствие поглощения тория в солянокислой среде позволяет отделить его от урана, протактиния и многих других металлов.
5. Применение ионообменников
229
Титан, цирконий и гафний легко сорбируются сильноосновными анионообменниками из растворов фтористоводородной кислоты. Сорбция уменьшается с повышением концентрации кислоты [12]. Коэффициенты распределения в 1MHF приблизительно в 100 раз выше, чем в концентрированной HF (24 М). Введение HF в 1М раствор НС1, содержащий Zr и Hf, устраняет трудности, связанные с полимеризацией и гидролизом этих катионов. Очень прочные фторидные анионные комплексы Zr и Hf использованы Краузом и Мором [13], а также Гуфманом и Лилли [14] в их ранних исследованиях.
Титан (IV) удерживается анионообменниками только из очень разбавленных растворов серной кислоты (около 0,05 М). Даниэльсон [15] показал, что в ~0,2М H2SO4 Dg 1. В присутствии пероксида водорода никакой сорбции Ti(lV) на сильноосновных анионообменниках не происходит [16]. При использовании слабоосновного анионообменника Amberlite CG-4B наблюдается обратная картина. На этом основано отделение Ti(IV) от многих других металлов [17]. При отделении Ti(IV) от Al, Cu(II), Cr(III), Ga, La, Sm, Y и Mg на смоле Amberlite CG-4B сопутствующие элементы элюируют смесью 0,1 М H2SO4 — 0,15% Н2О2. Затем десорбируют Ti(IV) 2М H2SO4.
Поглощение Ti(IV) из сернокислых растворов увеличивается при введении фтористоводородной кислоты. Подобный эффект наблюдается в присутствии органических растворителей [18}г
Цирконий и гафний ведут себя аналогичным образом. Поглощение достигает максимума в 0,15 М H2SO4 и'уменьшается с повышением концентрации кислоты (значение Dg в 5 М H2SO4 находится около 0). Гафний в тех же условиях образует менее прочные сульфатные комплексы, чем цирконий, и всегда элюируется перед ним.
Поглощение тория сильноосновными анионообменниками уменьшается с повышением концентрации серной кислоты. Оптимальный pH находится в области 2 — 2,5 [19]: при pH > 3 возможно образование основных сульфатов, а при pH < 1 поглощение Th незначительно. Последнее обстоятельство используют для отделения Th от Zr и Nb, которые хорошо сорбируются в этих условиях.
Сорбции тория благоприятствуют системы, содержащие большие количества (80 — 90%) метанола или этанола. В таких системах поглощение тория возрастает приблизительно на 2 порядка по сравнению с водными растворами H2SO4 той же концентрации [20]. Некоторым недостатком является низкая растворимость солей Th (IV) в этих растворах.
Для отделения тория от некоторых металлов применяют слабоосновные анионообменники. С помощью Amberlite CG-4B в 0,1 М H2SO4 торий отделяют, например, от Be, Y, La и Sm; торий элюируют 2MH2SO4. Для предотвращения хвостовых явлений при элюировании ионов вследствие низкой скорости обмена на слабоосновном анионообменнике рекомендуется работать при повышенной температуре (около 50°С).
Слабое поглощение Ti, Zr и Hf наблюдается в растворах азотной кислоты (1 — 14 М). В этой среде цирконий удерживается более прочно, чем
230
Часть II
гафний [21]. Нитратная среда играет важную роль в методах отделения тория, основанных на поглощении анионообменником устойчивых комплексов типа [ThCNO^g]2-. Коэффициенты распределения достигают максимальных значений в 5 — ЮМ HNO3 [22, 23]. Элюирующая способность растворов, применяемых для десорбции тория, уменьшается в ряду H2SO4> > НС1 > HNO3 > (NH4)2SO4 > NH4NO3 > Н2О > нао4 [24].
Введение ортофосфорной кислоты приводит к значительному уменьшению поглощения Th.
Нитратные комплексы тория легко поглощаются сильноосновными анионообменниками из водно-органических сред с высоким содержанием органического растворителя [25]. Система ацетон — HNO3 более эффективна по сравнению с системой СН3ОН—HNO3. В растворах, содержащих ацетон, торий отделяется от всех других элементов, за исключением Pd, Pb, Au и Bi [26].
Торий полностью отделяется от урана и редкоземельных элементов в растворах азотной кислоты [27].
Для разделения элементов четвертой группы представляют интерес растворы, содержащие аскорбиновую кислоту. В отсутствие пероксида водорода аскорбатные комплексы титана сорбируются на анионообменниках. В разбавленных растворах аскорбиновой кислоты в присутствии Н2О2 титан не сорбируется [28, 29]. Цирконий также образует комплексы с аскорбиновой кислотой, пригодные для его отделения. Из растворов, содержащих аскорбиновую кислоту (pH 4 — 4,5), торий сорбируется сильноосновными анионообменниками. Вместе с торием на ионообменнике удерживаются уран, титан, цирконий, ванадий, вольфрам и молибден, в то время как другие элементы не сорбируются на нем.
В тартратной среде титан образует отрицательно заряженные комплексы (TiOC4H3O6)_, которые легко поглощаются сильноосновными анионообменниками.
Ситарам [30] тщательно исследовал применимость малоновой кислоты для разделения ионов и показал возможность селективного поглощения циркония в форме малонатного комплекса на сильноосновном анионообменнике Dowex 21 К. Цирконий селективно поглощается из малонатных растворов с pH 4,0 в присутствии щелочных и щелочноземельных металлов, не образующих малонатных комплексов в этих условиях. Слабосвязанные комплексы Ni, Со, Zn, Мп, Pd, Cd и Sb элюируют из смолы водой. Цирконий десорбируют 1 М НО.
Аналогичное явление наблюдается при введении в анализируемый раствор ЭДТА. Цирконий отделяют от щелочных и щелочноземельных металлов селективным поглощением из растворов с pH 2,2 и последующим элюированием 1 М НС1. В присутствии Al, Zn, Fe, Cd, Ni и Co их ЭДТА-комплексы элюируют 0,5 М NaCl.
Малонатные комплексы тория [31] отделяют при pH 4,3 на сильноосновном анионообменнике Dowex 21 К от Ba, Sr, Cs, Rb и Се, которые не сорбируются на нем. Слабо связанные малонатные комплексы Sb, Pb, Cd и Zn элюируют водой. Торий десорбируют 0,25 М H2SO4. В присутствии циркония используют 1 М H2SO4.
5. Применение ионообменников
231
Лабораторные методики
РАЗДЕЛЕНИЕ СМЕСИ Zr — Hf НА КАТИОНООБМЕННИКЕ
В СРЕДЕ МУРАВЬИНОЙ КИСЛОТЫ [32]
Методика. 1 М раствор НСООН, содержащий Zr и Hf, пропускают через колонку подходящего размера, заполненную смолой Dowex 5OW-X8, предварительно приведенной в равновесие с.1 М муравьиной кислотой. Сразу после сорбции цирконий элюируют 1 М муравьиной кислотой. Колонку промывают водой и элюируют Hf 4 М HNOj. Ошибка определения обоих элементов не превышает 2,5%.
ОТДЕЛЕНИЕ Zr ОТ Ti, Fe, Al И ДРУГИХ ЭЛЕМЕНТОВ
НА КАТИОНООБМЕННИКЕ [5]
Методика. Около 2 г образца, содержащего до 200 мг ZrO2, сплавляют с карбонатом натрия и растворяют плав в соляной кислоте. Если в образце присутствует кремний, его удаляют обычным способом в виде SiO2- Раствор разбавляют водой до ~500 см3 и подкисляют НС1. Полученный раствор пропускают через колонку (1,15 х 22 см), заполненную сильнокислотным катионообменником AG 5OW-X8 в Н-форме. Ионы Al3 + , Ti4+ и Fe3+ элюируют из обменника 400 см3 2 М НС1. Цирконий десорбируют сначала 400 см3 5 М НС1 и затем 250 см3 1,5 М H2SO4. Объединенные элюаты упаривают и определяют Zr в виде фосфата циркония в 1,75 М H2SO4.
Такой метод применим для отделения Zr от Ti, Fe(III), Fe(II), Al, Cu, Ni, Pb, Co, Mn, Zn, Cd, Be, Mg, Sn(II), Sn(IV), NH4’, Li, Na, К н Rb. Эти элементы элюируют 2 М НС1. Разделение Сг3+ и Zr неполное. Ионы Са2+ также десорбируют 2 М НС1. Стронций и барий этим методом не отделяются.
Фториды, оксалаты, фосфаты и большие количества сульфатов мешают разделению. Допускается присутствие нитратов н перхлоратов.
ОТДЕЛЕНИЕ Zr И Hf ОТ РЯДА ЭЛЕМЕНТОВ
НА КАТИОНООБМЕННИКЕ [33]
Методика. Сильнокислотный катионообменник Dowex 5OW-X8 (0,15 — 0,07 мм) в Н-форме оставляют набухать в течение 5 мин в 0,1 М растворе триоктилфосфинок-сида (ТОФО) в смеси метанол — 12 М HNO3 (19:1). Колонку (250 х 5 мм) заполняют набухшей смолой и промывают 5 — 10 см3 смеси ТОФО — СН3ОН — HNO3.
К 0,5 см3 12 М HNO3 (до 5 мг циркония и гафния в смеси ионов) прибавляют 9,5 см3 CHjOH, содержащего 386 мг ТОФО. Смесь охлаждают до комнатной температуры и пропускают через ионообменную колонку со скоростью 0,3 см3/мин. Zr и Hf проходят в элюат. В конце разделения колонку промывают 10 см3 смеси ТОФО — СН3ОН — 12 М HNO3.
Судя по значениям коэффициентов распределения, приведенным в табл. 5.34, этот метод позволяет отделять от титана, лантаноидов, щелочных и щелочноземельных металлов, железа, кобальта, марганца, цинка и других элементов цирконий и гафний в количествах от следовых до миллиграммовых.
При использовании соляной кислоты вместо азотной и другого органического растворителя вместо метанола эффективность разделения Zr — Hf снижается.
232
Часть II
Таблица 5.34. Коэффициенты распределения Dg различных элементов на смоле Dowex 5OW-X8 в среде органический растворитель—12 М HNO3 (19:1) — 0,1 М триоктилфосфиноксид (ТОФО) [33]
Элемент CH3OH Ацетон Тетрагндро-фуран Метнлен-глнколь Уксусная кислота
!8lHf < 1 < 1 < 1 < 1 < 1
95Zr < 1 < 1 < 1 < 1 < 1
40Sc 731 204 445 241 2,15 - Ю3
169уЬ 420 3,4- 103 5,2-103 1,6- 103 > 104
144Се 1,64- 104 >104 1,66-104 8,3-103 2,9- 104
^Am 1,27-Ю4 2,76- 104 2,17- 103 5,56-104 1,110s
54Мп 7,3 103 9-104 2,35- 103 > 104 6,8-104
59Fe 2,6 - 103 < 1 < 1 467 >102
60Со 4,0- 103 2- 104 1.6-104 1,6- IO4 7,2- 104
22Na 167 1,85- 103 1,42-104 210 1,28- 103
137Cs 1,1- 103 4,8- 103 > 104 > 104 1,45- 103
45Са 426 441 460 490 470
85Sr 2- 104 >104 > 104 >104 > 104
44Ti 2 20 < 1 2,5 Tl
65Zn 811 < 1 < 1 15 > 102
РАЗДЕЛЕНИЕ СМЕСИ Ti-Zr НА ИОНООБМЕННИКАХ ПРИ АНАЛИЗЕ ПЬЕЗОЭЛЕКТРИЧЕСКИХ МАТЕРИАЛОВ, СОДЕРЖАЩИХ Pb, Bi, Zr, Ti И К [34]
Методика. Анализируемый образец растворяют в 72%-ной НС1О4 и упаривают до объема 3 — 4 см3. Раствор охлаждают, разбавляют до 100 см3 и прибавляют несколько капель 30%-ного Н2О2. Полученный раствор пропускают со скоростью 2 см3/мин через колонку (10 х 1 см), заполненную приблизительно 9 г смолы Dowex 5OW-X8 (0,15 — 0,07 мм) в Н-форме. Титан и цирконий элюируют смесью НС1О4 — NH4F (14 см3 70%-ной НС1О4 и 3,7 г NH4F разбавляют водой до 1000 см3). К элюату, содержащему Ti и Zr, прибавляют концентрированную H2SO4 и упаривают до появления белых паров. Сульфатный раствор разбавляют водой так, чтобы содержание H2SO4 составляло 1 — 2%. Затем прибавляют несколько капель 30%-ного Н2О2, раствор кипятят и пропускают со скоростью 1 см3/мин через колонку (30 х 1 см), заполненную катионообменником Dowex 5OW-X8 в Н-форме. Оба элемента сорбируются на катионообменнике. Титан элюируют 25%-ной НС1О4, пирконий — смесью HCiO4 - - NH4F указанного выше состава.
ОТДЕЛЕНИЕ Ti ПРИ АНАЛИЗЕ МИНЕРАЛЬНЫХ ВОД [35]
Методика. Колонку (10 х 0,6 см) заполняют сильноосновным анионообменни-ком Dowex 1-Х8 (0,15 — 0,07 мм) и промывают 100 см3 1 М НС1. Затем смолу тщательно промывают водой и 100 см3 свежеприготовленного 1%-ного раствора аскорбиновой кислоты (pH раствора доводят гидроксидом аммония до 4 — 4,5).
5. Применение ионообменников
233
К 1000 см3 анализируемой воды прибавляют 10 см3 концентрированной НС1 и 10 г аскорбиновой кислоты. С помощью гидроксида аммония в растворе устанавливают pH 4 — 4,5; раствор пропускают со скоростью 0,75 — 1 см3/мии через ионообменную колонку. Затем колонку промывают 20 см3 раствора аскорбиновой кислоты с pH 4,0 — 4,5.
Большую часть сорбируемых ионов элюируют 100 см3 0,05 М H2SO4, содержащей 1% фторида натрия. После этого фторид-ионы элюируют 20 — 30 см3 0,05 М H2so4.
Ионы Ti(IV) селективно элюируют 50 — 70 см3 0,05 М H2SO4, содержащей 3% пероксида водорода, при температуре, не превышающей 22°С. Элюат выпаривают досуха в платиновом тигле. Остаток медленно охлаждают, растворяют в нескольких куб. сантиметрах концентрированной фтористоводородной кислоты и выпаривают раствор на водяной бане. Остаток растворяют в 3 см3 1 М НС1 и переносят в мерную колбу емкостью 10 см3. Титан определяют спектрофотометрическим методом с аскорбиновой кислотой при 350 нм. Относительная ошибка определения Ti составляет менее 2%.
ОТДЕЛЕНИЕ МАКРОКОЛИЧЕСТВ Zr ОТ СЛЕДОВ Np И Nb [36]
Методика. Полиэтиленовую колонку (12 х 0,9 см) с пористым тефлоновым дном заполняют сильиоосновиым анионообменником Dowex 1-Х10 в Cl-форме, Перед использованием колонку промывают примерно 4 см3 смеси 6 М НС1 — 1 М HF, насыщенной хлором. Образец, содержащий миллиграммовые количества циркония, растворяют стандартным методом и выпаривают раствор до влажных солей. К остатку прибавляют 1 см3 смеси 6 М HCI — 1 М HF, насыщенной хлором, и слабо нагревают, чтобы ускорить растворение остатка. Образец переносят в небольшой пластмассовый сосуд, через который осторожно барботируют хлор в течение ~3 мин. Раствор пропускают через ионообменную колонку со скоростью около 0,8 см3/мин. Затем колонку промывают примерно 5 см3 смеси 6 М НС1 — 1 М HF, насыщенной хлором. В этих условиях в элюат переходит более 99% Zr.
Более 98% нептуния элюируют 6 см3 смеси 0,5 М HCI — 1 М HF. Ниобий вымывают 6 см3 смеси 4 М HNO3 — 1 М НС1 — 0,2 М HF. Процесс разделения лучше всего контролировать радиометрически.
После промывания смесью 6 М HCI — 1 М HF — С12 колонка пригодна для дальнейшего использования.
Отделение нескольких миллиграммов Zr от Np(VI) и Nb(V) занимает приблизительно 30 мин.
ОТДЕЛЕНИЕ ИЗОТОПОВ 95Zr (БЕЗ НОСИТЕЛЯ) ОТ 95Nb НА СИЛЬНОКИСЛОТНОМ КАТИОНООБМЕННИКЕ [37]
Методика. 0,1 М раствор НС1, содержащий смесь 95Zr — 95Nb, пропускают через колонку (3,5 см- X 0,57 см2), заполненную смолой Dowex 50W-X8 (0,07 — 0,037 мм) в Н-форме. Колонку промывают 0,1 М НС1, содержащей 3% Н2О2. Этой смесью элюируют 95Nb. Процесс элюирования контролируют радиометрически. 93Zr десорбируют 0,5%-иым раствором щавелевой кислоты. При элюировании ниобия 1 М НС1 образуется газ, пузырьки которого проходят через колонку и частично загрязняют фракцию ниобия цирконием.
16-354
234
Часть II
ОПРЕДЕЛЕНИЕ СЛЕДОВ Hf В МЕТАЛЛИЧЕСКОМ Zr С ИСПОЛЬЗОВАНИЕМ СИЛЬНООСНОВНОГО АНИОНООБМЕННИКА [38]
Методика. Колонку (25 х 2,5 см) заполняют смолой Dowex 1-Х8 (0,07—0,037 мм) в CI-форме. Смолу переводят в 5О4-форму 10%-ным раствором H2SO4, промывают водой и приводят в равновесие с 3%-ной серной кислотой при скорости потока 3 см3/мин. В платиновом тигле при нагревании осторожно растворяют 0,5 г металлического Zr в концентрированной HF. Затем прибавляют 5 см3 концентрированной H2SO4, смесь упаривают до появления белых паров, охлаждают и разбавляют водой до 50 см3. Аликвотную часть образца (0,50 — 5,0 см3 в зависимости от содержания Hf) переносят пипеткой в стакан емкостью 100 см3, прибавляют 3 или 4 см3 разбавленной H2SO4 (1 : 1) и упаривают до появления белых паров SO3. Остаток охлаждают, прибавляют 10 см3 воды, снова охлаждают и прибавляют еще 50 см3 воды. Раствор пропускают через ионообменную колонку со скоростью 3 см3/мин. Первые 60 см3 элюата не анализируют. Гафний элюируют 3%-ной H2SO4 порциями по 50 см3. Каждую фракцию (50 см3) собирают в стакан емкостью 150 см3 и сразу же осаждают купфероном, как описано ниже. Раствор пропускают до тех пор, пока купферо-натный осадок не станет белым, что указывает на проскок циркония в элюат. Проскок циркония обычно наблюдается приблизительно в пятой фракции.
Осаждение купфероном выполняют следующим образом: к 50 см3 элюата, содержащего Hf, прибавляют 1 см3 раствора Fe3+ (50 мкг) и 5 см3 свежеприготовленного 1%-иого раствора купферона. Осадок отфильтровывают и определяют содержание Hf методом рентгеновской спектроскопии.
Следы циркония отделяют от больших количеств гафния модифицированным методом, который до операции элюирования всего гафиия 3%-ной H2SO4 порциями по 50 см3 аналогичен описанному выше. Цирконий элюируют 10%-иой H2SO4 тремя порциями по 50 см3. Концентрацию Zr в элюате определяют после осаждения купфероном методом рентгеновской спектроскопии.
Точность определения примеси гафиия в цирконии иллюстрируется данными табл. 5.35.
Таблица 5.35. Определение Hf в металлическом Zr [38]
Взято Zr, мг Введено Hf, мкг ’ Найдено Hf, мкг
10 5,0 5,0
50 10,0 10,0
10 25,0 25,3
50 25,0 25,0
— 50,0 50,2
10 50,0 51,0
10 50,0 51,0
50 50,0 50,0
10 100,0 98,5
50 100,0 100,4
10 200,0 196,0
50 200,0 197,0
5. Применение ионообменников
235
ОТДЕЛЕНИЕ Th ОТ РЯДА ЭЛЕМЕНТОВ
НА СИЛЬНОКИСЛОТНОМ КАТИОНООБМЕННИКЕ КУ-2 [39]
Методика. 25 см3 смеси 5 М НС1О4 — 0,05 М H2SO4, содержащей до 250 мг Th и до 10 мг каждого из сопутствующих элементов (Be, Fe, Cr, Ti, Zr, Ge, Sn, V, Mo, W, U), пропускают co скоростью 2 — 3 см3/мин через колонку (20 х 1 см), заполненную катионообменником КУ-2 в Н-форме. Колонку промывают смесью 5 М НС1О4 — 0,05 М H2SO4 со скоростью 4 — 5 см3/мии. Все указанные элементы, за исключением Th (IV) и Се (III), переходят в 300 см3 элюата. Затем колонку промывают водой и элюируют Се(Ш) 300 см3 1 М раствора NH4C1 с pH 5. Обменник промывают 0,01 М НС1 и элюируют Th(IV) 200 см3 0,5 М NH4HSO4 с pH 2.
Анализируемый раствор не должен содержать комплексообразующих анионов (фторидов, тартратов, оксалатов и т.п.). Если в образце присутствует ниобий, то послб отделения Th его десорбируют смесью 1 М H2SO4 — 0,5 М Н2О2.
РАЗДЕЛЕНИЕ СМЕСИ Th — Zr — Hf - Мо В СЕРНОКИСЛОЙ СРЕДЕ НА АНИОНООБМЕННИКЕ [16]
Методика. Колонку диаметром 20 мм заполняют 30 г четвертичного аммониевого анионообменника AG 1X8 (0,074 — 0,037 мм) в 5О4-форме. Раствор, содержащий разделяемые элементы, нагревают с H2SO4 до появления белых паров и непосредственно перед сорбцией разбавляют водой до получения раствора H2SO4 требуемой концентрации.
100 см3 смеси 0,62 М H2SO4 — 0,1% Н2О2, содержащей примерно по 0,5 ммоля Zr, Hf, Th и Мо, пропускают через колонку со скоростью 1,2 ± 0,2 см3/мин. Торий элюируют 600 см3 смеси 0,62 М H2SO4 — 0,1%-ный Н2О2; торий появляется в растворе после гафния. Затем десорбируют цирконий 50 см3 1 М H2SO4 и, наконец, Mo(VI) — 250 см3 смеси 2 М NH4NO3 — 0,5 М NH4OH. Разделение на индивидуальные элементы очень хорошее.
Примечание- Принимая во внимание «историю» растворов, перед разделением на колонке их необходимо нагревать до появления паров серной кислоты. При этом происходит деполимеризация полиядерных комплексов. Пренебрежение этой операцией приводит к плохому отделению циркония.
ОТДЕЛЕНИЕ Th ОТ U НА СИЛЬНООСНОВНОМ АНИОНООБМЕННИКЕ [40]
Методика. Колонку (12 х 0,6 см) заполняют сильноосновным аниоиообменни-ком Dowex 1-Х8 (0,15 — 0,07 мм) в Cl-форме, который приводят в равновесие со смесью 90% и-бутанола — 10% 6 М НС1. Смесь Th — U сорбируют из раствора того же состава. Торий элюируют смесью 90% СН3ОН — 10% 6 М НС1, уран — 1 М НС1.
Этим методом можно разделить элементы в области от микрограммовых до миллиграммовых концентраций.
Другие методики разделения приведены в табл. 5.36.
5.2.4.2. 4 Б: С, Si, Ge, Sn, Pb
Для разделения элементов этой группы, особенно элементов, образующих типичные электроотрицательные ионы, используют анионообменники. Амфотерный характер последних двух элементов позволяет применять как катионо-, так и анионообменники. Поскольку в водных растворах эти эле-
236
Часть II
Таблица 5.36. Некоторые методы разделения элементов группы 4А
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
Ti(IV) Fe, РЗЭ, Al, Ga, AG 50W-X8 Ti: 0,5 M H2SO4—H2O2 41
Be, Th, Mn, Mg, Одновременно элюируют-
Co, Cu, Ni, Cd, ся Zr и U; другие элемен-
Zn, Hg, Ag, T1(I) ты не элюируются
Ti(IV) Zr AG 5OW-X8 Ti: 25%-ная НС1О4 Zr: 0,1 М NH4F— Ио НС1О4 42
Ti(IV) K, Sc Dowex 50 К: 9 М НС1О4 Ti: 9 М НС1 Sc: 4 М НС1—0,1 М HF 43
Ti(IV) W Катионит СБС W: разбавленная H2SO4 (pH 5)—5% Н2О2 44
Ti(IV) Различные Dowex 50 Примеси: H2SO4 (pH 1—2) 45
элементы Ti: Ио H,SO,—NH.F 2 4 4
Ti(IV) Силикатные Amberlite К, Na: 0,4 М НС1 46
породы IR-120 Ti: 0,8 М H2SO4 Mn, Mg, Са: 0,8 M НС1
Ti(IV) Сплавы ЭДЭ-10 n Ni: 12 M HC1 Ti: 6 M HC1 Другие: <6 M HC1 47
Ti В (в бориде Dowex 50 B: H2O 48
титана) Ti: H2SO4 (1:4)
Ti(IV) Zr Dowex 1 Zr: 1,5 M HC1— 0,25 M (COOH)2 49
Zr(IV) Hf Dowex 1 Сорбция из 3,5%-ной 49
HjSO, Hf не сорбируется Zr: 10%-ная H,SO. 50
Zr(IV) Dowex 1-X2 Масс-спектро метрическое определение субмикро-граммовых количеств Zr в одной бусине смолы 51
Zr(IV) Hf НС1 52
Zr(IV) Hf 0,45 М HNO3—0JD91 М 53
лимонная кислота
Zr Y Dowex 1-X8 Y: концентрированная НС1 Zr: 4 М НС1 54
Hf Облученный Та Dowex 50-X12 Hf: 0,1 М (СООН)2 55
РЗЭ: лактат NH+ pH 4,2; 88°С
5. Применение ионообменников
237
продолжение табл. 5.36
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
Zr(Ti) Nb, Ta, W, Mo Dowex J-X8 Zr(Ti), Nb: 0,5 M (COOH)2— 1,5 M HC1 Ta: 3 M HC1— 0,5 M (COOH)2 W: 4 M HC1—0,1 M лимонная кислота Mo: 1,9 M NH.C1— 0,44 M цитрат аммония 56
Hf Силикаты Dowex 1-X8 57
Hf (следы) Sc Dowex 1-X8 Sc: 0,4 M H2SO4 Hf: 2 M H,SO4 2 4 58
Th(IV) Следы Al в солях Th особой чистоты Amberlite IRA-400 Разбавленная H2SO4 (pH 2—2,5) для Al 59
Th(IV) Zr, U AB-17 Th: 0,5 M H,SO. 60
Th(IV) Одно- и двухзарядные катионы Amberlite IR-120 Одно- и двухзарядные катионы: 2 М НС1 Th: 3 М H,SO. 61
Th(IV) РЗЭ КУ-1 РЗЭ: 2 М NH4C1 Th: 1,5 М H,SO. 62
Th(IV) Pa(V), U(VI) Amberlite IRA-400 НОАс—НС1 или НОАс—HF 63
Th(IV) Лантаноиды Фосфориокислот-ный катиоиообмеи-ник РЗЭ: 0,3—0,5 М HNO3 Th: 5%-ный (NH^CO, 64
Th(IV) Н3РО4 Dowex 50 Н3РО4: 0,1 М НС1 Th: 10 М НС1 65
Th(IV) UO2(NO3)2 Dowex 5O-X8 U: 0,1 М HNOj, затем 6 М НС1 Th: 6 М НС1—1 М HF 66
Th(IV) U(VI) Dowex 5O-X8 U: 9О«7о 1 М HNO3— 10% этанола Th: 3 М HNOj 67
Th(IV) U(VI) КУ-1 " U: 0,5 М HNOj— 1 м nh4no3 68 69
Th(IV) Ti(IV) или Ce(HI) КУ-2 Ti: 3—4 М НС1— 0,5% Н2О2 Се: 3,6—4 М НС1 Th: 0,1 М (COONH^j 70 71
238
Часть П
продолжение табл. 5.36
Элемент Отделяемые компоненты Условия разделения Условия разделения Литература
Th(IV) Zr—лантаноиды КУ-2 Zr: 3—4 M HC1 Th: 0,1 M (COONH4)2 72
Th(IV) 224Ra Снльноосновные анионообменники Ra: 80% метанола 0,5—1 M HNO, 73
Th(IV) U(VI) Dowex 1-Х8 U: 90% ацетона— 10% 10 M HNO, Th: 1 M HNO, ‘ 74
Th(IV) U(VI) Deacidite FF U: 6 M HNO3 (77°C) Th: вода 75
Th(IV) Лантаноиды (следы) Dowex 1-X8 РЗЭ: 5—8 M HNO, Th: 2,4 M HC1 8
Th(IV) Лантаноиды (следы), Y, Sc Dowex 1 Следы: 8 M HNO, 76
Th(IV) 228Th Получение 234Th из UO2(NO3)2 228Ra Amberlite IRA-400 U: 7—7,5 M HNO, Th: вода или 0,1 M HNO, 77 78
Th Изотопы Ra, Pu, Si, Fe, С в частицах, супендиро-ванных в морской воде Dowex 1-X8 [ HNO3, HC1 различной концентрации 79
менты легко гидролизуются, разделение проводят в сильнокислых растворах или в присутствии соответствующих комплексообразующих агентов.
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Катионообменники редко используют для разделения элементов этой группы. В некоторых случаях они играют важную роль при удалении мешающих элементов. В процессе отделения германия такие элементы, как медь, свинец, цинк, железо и т.п., удаляют сорбцией на катионообменни-ках; германий в анионной форме переходит в элюат. Катионообменники непригодны для отделения ионов Sn2+ или Sn4+. Гидролиз ионов Sn4+ предотвращают введением комплексообразующего агента, который образует несорбируемые комплексы. Только ионы РЬ2+ легко поглощаются катионообменниками, содержащими сульфогруппы.
Ионы РЬ2+ лучше всего сорбируются из слабокислых растворов. Сорбция значительно уменьшается с повышением концентрации кислоты. Лучшим агентом для элюирования РЬ2+ из сильнокислотных катионообменни-ков является 1 М раствор ацетата аммония. С помощью этого элюента можно отделить РЬ2+ от Sr, Ва, А1 и других элементов.
5. Применение ионообменников
239
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Германий, олово и свинец в форме анионных хлоридных комплексов сорбируются сильноосновными анионообменниками из солянокислой среды. Для поглощения Ge подходит 8 — 10 М НС1, для поглощения Sn4+ — 12 М НС1; в случае сорбции ионов РЬ2+ концентрация НС1 не должна превышать 8 М. При более высоких концентрациях НС1 сорбции не наблюдается. При 0,15 — 5,0 М концентрациях сорбция германия незначительна. Это можно использовать для отделения ионов, обладающих в данной среде высоким сродством к сильноосновным анионообменникам.
Один из наиболее приемлемых методов отделения свинца от ряда элементов основан на элюировании РЬ(П) из сильноосновного анионообмен-ника довольно концентрированной соляной кислотой. Sn, Sb и многие другие элементы в этих условиях остаются на смоле.
Германий в растворах фтористоводородной кислоты не сорбируется на анионообменниках. Сорбция РЬ(П) также невысока. Поглощение Sn(IV) уменьшается с повышением концентрации кислоты.
Сорбция в растворах азотной кислоты уменьшается в ряду Ge > Sn > > Pb.
Егоров [80] установил, что сорбцию Ge на среднеосновном анионообменнике ЭДЭ-10п можно улучшить введением подходящей органической кислоты (хромотроповой, пирокатехин-3,5-дисульфокислоты, ализарии-сульфокислоты).
Лабораторные методики
ОТДЕЛЕНИЕ РЬ2 + ОТ Bi, TI, Cd, Hg, Au, Pt, Pd И ДРУГИХ ЭЛЕМЕНТОВ [81]
Методика. Разделение выполняют в среде НВг на колонке (2x7 см), заполненной 10 г сильноосновного анионообменника AG1-X8 (0,15 — 0,07 мм). U(VI), Th, Ti(IV), Zr, Hf, Sc, Y, La, Gd, Yb, Al, Mn(II), Cu, Be, Mg, Ca, Sr, Ba и щелочные металлы элюируют 0,1 М НВг. Эти элементы находятся в первых порциях элюата. Pb, Cd, Hg(II), Bi, Tl(III), Au(III), Pt(IV) и Pd удерживаются смолой. Ионы Pb2+ элюируют 1 М HNO3. После этого тем же раствором десорбируют Cd и Bi. Другие ионы в данных условиях не сорбируются. При анализе бинарных смесей, содержащих свинец и один из сопутствующих элементов [Cd, Bi, Т1(Ш), Hg(II), Pd, Pt(IV), Au(III)], сорбированные из 0,1 M НВг ионы РЬ2+ элюируют смесью 0,30 М HNO3 — 0,025 М НВг. Другие элементы вымывают в следующем порядке: Cd(l,0 М NH4OH — 0,1 М NH4NO3 — 0,02 М НВг); Bi (0,02 М ЭДТА — 0,1 М NH4NO3), Т1 (1,0 М NH4OH и затем 0,50 М HNO3 — 0,10 М тиомочевина), Pd (1,0 М NH4OH), Pt (1,0 М НС1 — 0,10 М тиомочевина, 0,02 М НС1 — 0,10 М тиомочевина). Для определения Au смолу озоляют.
ВЫДЕЛЕНИЕ СЛЕДОВ РЬ ИЗ РАСТВОРОВ СУЛЬФАТА ЦИНКА [82]
Методика. Колонку (27 х 3,1 см) заполняют анионообменником Dowex 50-Х4 (0,074 — 0,037 мм) в Н-форме, промывают водой и затем пропускают 10 см3 нейтрального анализируемого раствора (125 г Zn/дм3). После этого смолу промывают 75 см3 воды и элюируют свинец 1 М НС1. Цинк десорбируют 8 М НС1.
240
Часть П
Таблица 5.37. Методики разделения элементов группы 4Б
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
Si PO^’, водопроводная вода Amberlite IRA-400 85
Pb, Zn Ni, Со, Fe, Мп, Cu Deacidite FF Другие: 2—0,5 M HCI Pb, Zn: 0,005 M HCI 86
Pb Pb Bi Bi Dowex 50 Dowex 1-X8 0,5 M (CH2NH3)2(C1O4)2 87 88
Pb Ca Wofatit F Pb: ЭДТА, pH 4—4,5 Ca: 10%-иый KNO3 89
Pb Tl Dowex 50 Pb: ацетатный буфер— 0,2 M ЭДТА ’ Tl: 2 М HCI 90
Pb, Cd In Dowex 50-X8 Сорбция из растворов ЭДТА Pb, Cd: 4 М НС1 91
Pb Ca—Sr—Ba—Ra Dowex 50 Градиентное элюирование смесью 1 м сн3соон—ch3coonh4 от нулевой концеитрапии до насыщенного раствора 92
Pb Cu—Sb—Sn Amberlite IRA-400 РЬ: 7 М НС1 Си: 1,56 М НС1 Sb: 0,3 М HCI—1 М HF Sn: 6 М NaOH 93
Pb Pb Ge—Sn—Bi Вода Dowex 1 Dowex 1 Pb: 0,1 М HCI—1 М HF Ge: 6 М HCI—1 М HF Sn: 2 М НС1—3 М HF Bi: 1 М NH.C1—1 М NH.F 4 4 94 95
Pb Г еологические образцы Dowex 1-X8 Сорбция из 2 М НВг РЬ: 6 М НС1 96
Pb Со Amberlite IRA-400 РЬ: 8 М НС1 97
Pb Th, Fe, Bi Dowex 50 Th, Fe, Bi: 0,01 М ЭДТА, pH 2,0 Pb: 1 М CH.COONH. 98
Pb Zn, Cu Сильиоосновный анионообменник 99
Pb Сплавы Pb, Br, Cd, Sn Dowex 50W-X8 Pb: 0,6 М HBr, 50°С 100
Pb Примеси Cu, Bi, Fe Dowex 1-X10 Pb: 9 М HCI Другие: 6 М HNO3 101
5. Применение ионообменников
241
продолжение табл. 5.37
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
Sn(IV) Sn(H) 102
Sn(IV) Al, Sb Amberlite CG-4B Al: 4,5 M HCI Sn: 1 M'HCl Sb: 1 M NaOH 103
Sn(IV) Mn, Fe, Co, Ni, Cu, Zn, Hg, Ca Sn: 0,5 M HCI 104
Ge Fe, Al Сильноосиовиый анионообменник 105
Ga As ЭДЭ-10п pH 4,0, As(III) не сорби- руется 106
Ge Te(IV) Amberlite CG-4B Ge: 4,3 M HCI Те: 1 M HCI 103
Ge Уголь Nalcite HCR-X8 Amberlite IR-45 107
Ge (следы) Zn Dowex 50 В 1,42 M HCI сорбируется только Zn 108
Ge As(III) КУ-2 в Co- Сорбируется As(III) форме 109
ОТДЕЛЕНИЕ РЬ ОТ Ga, In И Zn НА АНИОНООБМЕННИКЕ [83]
Методика. Сильиоосновный анионообменник Dowex 1-Х8 (0,15—0,074 мм) в Вг-форме, помешенный в колонку подходящего размера, приводят в равновесие со смесью 90% метанола — 10% 4,5 М НВг. Затем через колонку пропускают раствор такого же состава, содержащий разделяемые ионы. Свинец проходит в элюат, тогда как другие ионы удерживаются смолой. Ga, In, Au и Zn элюируют смесью 90% метанола — 10% 4,5 М НВг. Этот метод рекомендован для отделения миллиграммовых количеств трехвалентиых элементов.
ОТДЕЛЕНИЕ РЬ И ЕГО ОПРЕДЕЛЕНИЕ В СТАЛИ [84]
Методика. Колонку (13 х 130 мм) заполняют смолой Dowex 1-Х8 (0,15 - . - 0,074 мм). Смолу превращают в Cl-форму обработкой 150 см3 12 М НС1 и промывают 50 см3 1 М НО. 5,00 г стали растворяют в 40 см3 смеси HNO3 — НС1 (1:4). Кремневую кислоту удаляют выпариванием раствора до влажных солей. Остаток растворяют в 40 см3 6 М соляной кислоты, разбавляют до 50 см3, отфильтровывают через средиепористый фильтр и несколько раз промывают фильтр 1 М НС1. Фильтрат упаривают до 15 — 20 см3 и разбавляют точно 5 объемами воды для получения раствора 1 М по НС1. Этот раствор пропускают через ионообменную колонку. Затем обменник промывают 75 см3 1 М НС1 и элюируют свинец 150 см3 12 М НС1. Первые 75 см3 элюата возвращают в колонку для удаления небольших количеств железа, десорбированного из колонки соляной кислотой.
242
Часть II
Элюат выпаривают до влажных солей, остаток растворяют в 10 каплях HNO3 и 5 см3 воды и разбавляют раствор водой в мерных колбах емкостью 10 или 25 см3.
Концентрацию свинца определяют методом атомно-абсорбционной спектроскопии при 217 нм в пламени ацетилена.
Обзор других методов разделения приведен в табл. 5.37.
Литература
1. Винаров И. В. Успехи химии, 36, 1967, 5?2.
2. Mukherji А. К., Analytical Chemistry of Zirconium and Hafnium. Pergamcn
Press Inc., Oxford, 1970.
3. Strelow F. W., Boshoff M. D., Anal. Chim. Acta, 62, 1972, 351.
4. Белявская T. А., Алимарин И. П., Колосова И. Ф., Ломоносов М. В. Ж.
аналйт. химии, 13, 1958, 668.
5. StrelowF. W., Anal. Chem., 31, 1959, 1974.
6. Samedy S. R., Diss. Abstr., 20, 1960, 4256.
7. Benedict J. T., Schumb W. C., Coryell C. D., J. Amer. Chem. Soc., 76,
1954, 2036.
8. Danon J. J. Inorg. Nucl. Chem., 5, 1957, 237,
9. Легенченко И. А., Стриженюк С. Л. ЖФХ, 42, 1968, 1224.
10. Kraus К. A., Nelson F., Smith G. W., J. Phys. Chem., 58, 1954, 11.
11. Kraus K. A., Nelson F., Proc. Intern. Conf. Peaceful Uses At. Energy, Geneva, 7, 1956, 113.
12. Faris J. P, Anal. Chem., 32, 1960, 520.
13. Kraus К. A., Moore D. G., J. Amer. Chem. Soc., 71, 1949, 3263.
14. Huffman E. H., Lilly R. C., J. Amer. Chem. Soc., 73, 1951, 2902.
15. Danielsson L., Acta Chem. Scand., 19, 1965, 570.
16. StrelowF. W., BothmaC. J., Anal. Chem., 39, 1967, 595.
17. Kuroda R., Oguma K., Kono N., Takahashi Y., Anal. Chim. Acta, 62,
1972, 343.
18. Antal P., Korkisch J., Hecht F., J. Inorg. Nucl. Chem., 14, 1960, 251.
19. Nagle R. A, Murthy T. K. S., Analyst, 84, 1959, 37.
20. Janauer G. E., Korkisch J., J. Chromatogr., 8, 1962, 510.
21. Buchanan R. F., Faris J. P., Radioisotopes in the Physical Sciences and
Industry, IAEA, Vienna, 1962, p. 361.
22. Казанцев E. И., Коробейников В. Л. Изв. высш, учебн. завед. Цветн. металлы; 4, 1966, 89.
23. Gerontopulos Р., Kigali L., Radiochim. Acta, 3, 1964, 122.
24. Казанцев Е. И., Кудусов В. А. Радиохимия, 5, 1963, 231.
25. Walter С. W., Korkisch J., Mikrochim. Acta, 1971, 81, 137, 158, 181, 194.
26. Korkisch J., Ahluwalia S. S., J. Inorg. Nucl. Chem., 28 (1966) 264.
27. Kraus K. A., Nelson F, ASTM Spec. Techn. Publ., 195 (1958) 27.
28. Korkisch J., FaragA., Mikrochim. Acta, 1958, 659.
29. 'Korkisch J., Mikrochim. Acta, 1961, 262.
30. SitaramR., Khopkar S. M., Chromatographia, 8 (1975) 645.
31. Chakravorthy M., Khopkar S. M., Sepn. Sci., 10 (1975) 623.
5. Применение ионообменников
243
32. Qureshi М., Husain К., Anal. Chem., 43 (1971) 447.
33. Korkisch J., Orlandini К. A., Talanta, 16 (1969) 45.
34. Dosch R. G., Conrad F. J., Anal. Chem., 36, (1964) 2306.
35. Korkisch J., Z. anal. Chem., 178 (1960) 39.
36. Holloway J. H., Nelson F., J. Chromatogr., 14 (1964) 255.
37. Sano H., Shiomi R., J. Inorg. Nucl. Chem., 5 (1958) 251.
38. Luke C. L., Anal. Chim. Acta, 41 (1968) 453.
39. Набиванец Б. И., Кудрицкая Л. Н. Ж. аналйт. химии, 21, (1966) 40.
40. Korkisch J., Тега F., J. Chromatogr., 6 (1961) 530.
41. StrelowF. W., Anal. Chem., 35 (1963) 1279.
42. Кеппа В. T., Conrad F. J., Anal. Chem., 35 (1963) 1255.
43. Nelson F., Murase T., Kraus К. A., J. Chromatogr., 13 (1964) 503.
44. Рябчиков Д. И., Бухтияров В. Е. Ж. аналйт. химии, 15 (1960) 242.
45. Oki Y., Oki S, ShibataH., Bull. Chem. Soc. Japan, 35 (1962) 273.
46. Цитович И. К. Изв. выс. учеб, завед., Химия и хим. технология, 5 (1962) 194.
47. Bandi W. R., Buyok Е. G., Lewis L. L., Melnick L. M., Anal. Chem., 33 (1961) 1275.
48. Dimitrova M., Z. anal. Chem., 274 (1975) 384.
49. Machlan L. A., Hague J. L., J. Res. Nat. Bur. Stand., 66 (1962) 517.
50. Hague J. L., Machalan L. A., J. Res. Nat. Bur. Stand., 62 (1959) 13.
51. Walker R. L., Bottis J. L., Carter J. A., Constanzo D. A., Anal. Lett., 10 (1977) 251.
52. Vobecky M., Mastalka A., Collect. Czech. Chem. Commun., 26 (1961) 1716.
53. Benedict J. T., Schumb W. C., Coryell C. D., J. Amer. Chem. Soc., 76 (1954) 2036.
54. ForsbergS. et. al., Ark. Kemi, 23 (1965) 269.
55. Зайцева H. Г., Хоу-Мо-Луан. Радиохимия, 4 (1962) 738.
56. Melnick С. M., Buyok E. G., Lewie L. L., Bandi W. R., Anal. Chem., 33 (1961) 6275.
57. Greenland L. P., Anal. Chim. Acta, 42 (1968) 365.
58. BalsencL., Haerdi W., Monnier D., Anal. Chim. Acta, 48 (1969) 213.
59. Athavale V. T., Subramanian A. R., J. Sci. Ind. Res. India, B19 (1960) 431.
60. Каралова 3. К., Шибаева H. П., Пыжова 3. И. Ж. аналйт. химии, 21 (1966) 1133.
61. Nietzel О. A., Wessling В. W., DeSesaM. A., Anal. Chem., 30 (1958) 1182.
62. Жуков А. И., Оносов В. Н., Шевков Н. А.. ЖНХ, 7 (1962) 926.
63. Maher М., El-Dessouky, Mikrochim. Acta, 2 (1976) 461.
64. MarholM., Z. anal. Chem., 231 (1967) 265.
65. Menis O.. Mannig D. L., Goldstein G., Anal. Chem., 29 (1957) 1426.
66. Murase T., Lind E. L., Nelson F., J. Chromatogr., 14 (1964) 478.
67. Korkisch J., Tera F., J. Chromatogr., 8 (1962) 516.
68. Жуков А. И., Казанцев E. И., Яковлев А. В. Ж. прикл. химии, 36 (1963) 743.
69. Жуков А. И., Казанцев E. И., Вакуленко В. А. Ж. прикл. химии, 38 (1965) 43.
244
Часть II
70. Алимарин И. П., Медведева А. М., Бурлова М. А. Ж. аналит. химии, 18 (1963) 468.
71. Алимарин И. П., Медведева А. М., Бурлова М. А. Ж. аналит. химии, 19 (1964) 1322.
72. Алимарин И. П., Медведева А. М. Ж. аналит. химии, 19 (1964) 436.
73. Заборенко К. Б., Богатырев И. О., Ниттсол Д. Д., Радиохимия, 5 (1963) 638.
74. Urubay S., Janauer G. Е., Korkisch J., Z. anal. Chem., 193 (1963) 165.
75. Carswell D. S., J. Inorg. Nucl. Chem., 3 (1957) 384.
76. Faris J. P., Appl. Spectrosc., 12 (1958) 157.
77. Bjornholm S., Nielsen О. B., Nucl. Phys., 42 (1963) 642.
78. Tomida A. K., Abrao A., Radiochem. Radioanal. Lett., 29 (1977) 131.
79. Krishnaswami S.\ Sarin M. M., Anal. Chim. Acta, 83 (1976) 143.
80. Егоров A. M., Кац T. С. ЖФХ, 51 (1977) 2315.
81. Strelow F. IV., Toerien F. S., Anal. Chem., 38 (1966) 545.
82. Leclerq M., Duyckaerts G., Anal. Chim. Acta, 29 (1963) 139.
83. Korkisch J., Hazan I., Anal. Chem., 37 (1965) 707.
84. Sellers N. G., Anal. Chem., 44 (1972) 410.
85. Munaro F., Boll. Lab. Chim. Prov., 19 (1968) 404. C. A., 70 (1969) 6480.
86. Carson R., Analyst, 86 (1961) 198.
87. Fritz J. S., Karakker S. K., Anal. Chem., 32 (1960) 957.
88. Ahluwalia S. S., Korkisch J., Z. anal. Chem., 208 (1965) 414.
89. Liteanu C., Demian M., Studia Univ. Babes-Balgai, ser. I, 1961, 85. C. A., 57 (1962) 6589.
90. Ziemba S., Prdce Inst. Hut., 16 (1964) 261. Anal. Abstr., 12 (1965) 4482.
91. Doleial J., Povondra P., Stulik K., Sulcek Z., Collect. Czech. Chem. Comrnun., 39 (1964) 1538.
92. Eulitz G., Nukleonika, 2 (1960) 85.
93. Ariel M., Kirova E., Talanta, 8 (1961) 214.
94. Nelson F., Rush R. M., Kraus К. A., J. Amer. Chem. Soc., 82 (1960) 339.
95. Korkisch J., SorioA., Talanta, 22 (1975) 273.
96. Korkisch J., Gross H., Talanta, 21 (1974) 1025.
97. KasiuraK., MensM., Chem. Anal. (Warsaw), 23 (1978) 305.
98. Khopkar S. M., De A. K., Talanta, 7 (1960) 7.
99. Kemula W., Brajtner K., Rubel S., Chem. Anal. (Warsaw), 14 (1969) 1339.
100'. Fritz J. S., Greene R. G., Anal. Chem., 35 (1963) 811.
101. Alexandrov S., Krasnobaeva N., Acta Chim. Hung., 64 (1970) 11.
102. Husain W., Gulab M., Separ. Sci., 6 (1971) 737.
103. Kuroda R., Ishida K., Kirigamu T., Anal. Chem., 40 (1968) 1502.
104. MachirouxR., Mousty F., Anal. Chim. Acta, 48 (1969) 219.
105. Burriel-Marti C., Alvarez-Herrero C., Chim. Anal., 48 (1966) 602.
106. Саликова Г. E., Севрюков H. H. Зав. лабор., 36 (1970) 25.
107. Драницкая P. M., Ду Хен-хуан. Ж. аналит. химии, 19 (1964) 769.
108. Cabbel Т. R., Orr A. A., Hayes J. R., Anal. Chem., 32 (1960) 1602.
109. Драницкая Р. М., Тетервак А. Г. Ж. аналит. химии, 27 (1972) 1214.
5. Применение ионообменников
245
5.2.5. Группа 5
5.2.5.1. 5 А: V, Nb, Та, Ра
Эти элементы присутствуют главным образом в анионной форме, поэтому при их разделении наиболее широко применяются анионообменники. Катионообменники полезны при отделении четырехвалентного ванадия, а также для удаления катионов, мешающих определению этих элементов.
Из разнообразных комплексообразующих агентов, применяемых для разделения элементов группы 5 А, часто используют пероксид водорода, который образует несорбируемые комплексы с ванадат-ионами. При разделении ниобия и тантала применяют фтористоводородную кислоту, смеси HF — НО, HF — HNO3 или HF — H2SO4, щавелевую или винную кислоты. Тиоцианат аммония пригоден для отделения протактиния. Введение в анализируемые растворы комплексообразующих кислот предотвращает образование коллоидных частиц.
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Органические катионообменники применяются в основном для поглощения ванадия и протактиния. Ионы V(IV) сорбируются катионообмеиника-ми из 0,1 — 0,2 М растворов минеральных кислот (HCI, HNO3, HjSO^. Они имеют достаточно высокие коэффициенты распределения, которые уменьшаются с повышением концентрации кислоты. Коэффициенты распределения в некоторых кислотах приведены в табл. 5.38. Из этих данных видно, что в хлорной кислоте поглощение ванадия(1У) и особенно ванадия! V) увеличивается с повышением концентрации кислоты. В других минеральных кислотах наблюдается противоположное поведение.
В 0,1 М или менее концентрированной соляной кислоте ионообменная смола восстанавливает V(V) в V(IV). Поэтому необходимо работать в присутствии пероксида водорода или при pH > 5. При более низких pH пероксид водорода образует с V(V) несорбируемые комплексы [4 — 6].
Различие в степени поглощения V(IV) и V(V) обеспечивает возможность легкого отделения его от анионов. После превращения V(V) в V(IV) соответствующим восстановителем (например, SO2) четырехвалентный ванадий сорбируется катионообменниками, в то время как анионы проходят в элюат.
Два других члена этой группы, ниобий и тантал, практически не сорбируются из растворов соляной кислоты. Коэффициенты распределения Dg Nb(V) иа смоле Dowex 50 в 1 М азотной или серной кислоте имеют величину порядка 40 и быстро уменьшаются с повышением концентрации кислоты. В 9 М НС1О4, содержащей следы фторидов, получены высокие значения Dg (более 103).
Протактиний в 1 — 3 М растворах минеральных кислот (соляной, азотной, бромистоводородной и хлорной) образует положительно заряженные комплексы, сорбируемые катионообменниками. На кривой, выражающей зависимость поглощения от концентрации кислоты, наблюдается минимум, который характерен для данной кислоты.
246
Часть II
Таблица 5.38. Коэффициенты распределения D V(IV) и V(V) на смоле Dowex 50 в растворах некоторых минеральных кислот [1—3]
Концентрация кислоты. М D g
V(IV) V(V)
НС1: 0,1 — 13,9
0,2 230 7,0
0,5 44 5,0
1,0 7,2 1,1
3,0 2,0 0,2
6,0 0,7 0,8
9,0 -0,3 -2,0
HNO,: 0,1 495 20
0,2 157 10,9
0,5 35,6 4,9
1,0 14 2
2,0 4,7 1,2
3,0 3,0 0,8
4,0 2,5 0,5
H2SO4: 0,05 1230 27,1
0,1 490 15,2
0,25 140 6,7
0,5 46,6 2,8
1,0 11,5 1,2
1,5 2,4 0,7
2,0 0,4 0,4
НС1О,: 1,0 -10 < 1
3,0 4 - 1
6,0 7 -6
9,0 50 >102
10,0 102 >102
Однако катионообменники редко применяют для отделения Ра, так как в этом случае необходимо работать в разбавленных кислотах, в которых протактиний проявляет сильную склонность к гидролизу, полимеризации и сорбции на различных веществах. Описано использование катионообменников для отделения протактиния от тория [6, 7] или тантала [3].
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
В растворах НС1 наблюдается только слабое поглощение V(IV) на сильноосновных анионообменниках. Введением подходящего комплексообразующего агента ионы V(IV) превращают в слабокислой среде в анионные комплексы, сорбируемые на анионообменниках. Чаще всего применяют ЭДТА [8], цитраты [9], тиоцианаты [10], оксалаты [9], ацетаты [11] з тартраты [12]. Слабое поглощение ванадия (IV) в солянокислой среде ис
5. Применение ионообменников
247
пользуется для его отделения от элементов, сильно сорбирующихся из 9 — 12 М НС1. В некоторых случаях, например при отделении рения от ванадия, предпочтительнее слабокислая среда [13].
Поглощение V(V) на смоле Dowex 1 начинается при pH 2, а полная сорбция наступает при pH 8 [14, 15]. С увеличением концентрации ОН- небольшое количество ванадия (V) десорбируется. Полное элюирование проводят 1 М раствором Na2CO3 или ЗМ NaOH.
Ниобий и тантал в солянокислой среде поглощаются анионообменниками. Минимальные коэффициенты распределения получены для 4 — 5 М НС1. С повышением концентрации кислоты сорбция возрастает. В 8 М НС1 значения Dg равны 100 для Nb и 10 для Та. Протактиний ведет себя аналогичным образом. Его сорбция в 1 — 4 М НС1 незначительна. Количественное поглощение наблюдается только в 8 М НС1 (Dg > 1000).
В табл. 5.39 сравнивается сорбция Nb, Та и Ра из солянокислых растворов на анионообменниках с разным содержанием ДВБ. При увеличении содержания ДВБ от 2 до 10% коэффициенты распределения возрастают в 3 — 5 раз.
На сильной сорбции Ра из растворов НС1 (>6М) основано его отделение от элементов, не образующих хлоридных комплексов (Th) или образующих непрочные комплексы (Zr). При промывании обменника большим объемом 6 — 8 М НС1 количество Ра в элюате не превышает 0,10%. По
Таблица 5.39. Влияние концентрации НС1 и содержания ДВБ на коэффициенты распределения (Dg) Nb, Та и Ра на смоле Dowex 1 [16]
Содержание ДВБ, % Концентрация НС1, М
8,2 10,2 12,5
2 220 Ниобий 250 180
4 410 530 380
8 630 890 660
10 760 1230 810
2 Тантал 150 230
4 — 180 340
8 — 400 1060
10 610 1060
2 950 Протактиний 1600 2000
4 ИЗО 2900 4200
8 1360 3700 4300
10 1640 5990 9600
248 '
Часть II
сравнению с Pa(V) четырехвалентный протактиний не образует устойчивых хлоридных комплексов даже в концентрированной НС1. Таким способом его отделяют от элементов, хлоридные комплексы которых полностью сорбируются анионообменниками (Fe, V, Zr, Nb и т.п.) [60]. Ра (IV) легко отделяется от Pa(V) на смоле Dowex 1-Х10 элюированием 6 — 9 М НС1; протактиний (V) элюируют смесью 6 — 7М НС1 — 0,1М HF [17].
Фтористоводородная кислота непригодна для поглощения V (V) на анионообменниках; максимальная величина коэффициента распределения равна 10.
С другой стороны, ниобий, как и тантал, хорошо сорбируется из растворов HF. Их коэффициенты распределения превышают 102 и при понижении концентрации HF продолжают увеличиваться. Для Та получены более высокие значения Dg, чем для Nb, особенно при низких концентрациях кислоты.
Такое же сорбционное поведение этих двух элементов наблюдается в среде HF — НС1 или HF — HNO3 [18, 19]. Данные по сорбции различных элементов из растворов HF — HNO3 или HF — H2SO4 представлены в табл. 5.40. При разделении смеси Nb — Та на анионообменниках Dowex 1 или Deacidite FF наиболее подходящей средой оказалась смесь 0,1 М HF — 3 М НС1. Ниобий быстро элюируется смесью HF — НС1, тантал — смесью 4 М NH4C1 — 1 М NH4F [21, 22]. При элюировании смесью 0,05 М HF — 9 М НС1 первым также десорбируется ниобий [23, 24].
Таблица 5.40. Сравнение сорбционных свойств некоторых ионов металлов в среде 1 М HF—0,1 М HNO3 (или 0,05 М H2SO4) [20]
Сорбция D g Анионный обмен
Слабая <10 Ag, Al, As(III), Bi, Cd, Co(II), Cr(III), Cu(II), Fe(II), Fe(III), Mg, Mn(II), Ni, Pb(II), Sb(III), V(IV), Zn
Умеренная 10—100 As(V), V(V)
Хорошая >100 Mo(VI), Nb(V), Sb(V), Sn(IV), Ta(V), Ti(IV), W(VI), Zr(IV)
Сорбция Катионный обмен
Слабая Умеренная <10 Al, As(III), As(V), Cr(III), Fe(III)a, Mo(VI), Nb(V), Sb(IlI), Sb(V), Sri(IV), Ta(V), Ti(IV), V(V), W(VI), Zr(IV) Bia, Fe(II)a, V(IV)a
Хорошая >100 Ag, Cd, Co(II), Cu(II), Mg, Mn(II), Ni, Pb(Il), Zn
а При с’Нр = 0,1 М эти элементы сорбируются хорошо.
5. Применение ионообменников
249
С повышением концентрации HF до 0,1 М, но при той же концентрации НС1 (9 М) возможно отделение ниобия от вольфрама, титана, ванадия, циркония, бериллия, хрома и никеля. Эту методику используют при анализе разнообразных материалов [25, 29]. Отделение ниобия от тантала, присутствующего в стали, выполняют на анионообменнике Wofatit SBW смесью 0,05 М HF — 9 М НС1 [30].
Низкое поглощение обоих элементов наблюдается в чистой азотной кислоте [31]. Поглощение ниобия в среде HF — НС1 или HF — HNO3 выше, чем в среде HF — H2SO4. При повышении концентрации H2SO4 сорбция значительно ухудшается. С другой стороны, влияние концентрации H2SO4 иа поглощение тантала менее заметно.
Протактиний (V) проявляет высокое сродство к сильноосновиым анио-нообменникам в растворах фтористоводородной кислоты. В этих растворах Pa(V) образует ряд комплексов (PaF2~, PaF^, PaF|~ и т.п.), которые не подвергаются гидролизу или полимеризации и легко поглощаются ацио-нообмеиниками. Подобным образом ведут себя сульфатные и оксалатные комплексы. Для протактиния(V) на смоле Dowex 1-Х10 получены следующие значения коэффициентов распределения: 2 - 104 в 0,20 М HF; 1 • 102 в 7 М HF [32]. Даже в 25 М HF величина Dg превышает 3.
Однако сорбция Pa(V) значительно ухудшается после введения,небольших количеств НС1 или НС1О4 вследствие понижения диссоциации HF. Таким путем подавляется образование сорбируемых анионных комплексов.
Протактиний (V) успешно отделяется в этой среде от щелочных и щелочноземельных металлов, ниобия и продуктов радиоактивного распада Ра.
До сих пор мало внимания уделялось поглощению Pa(V) анионообменниками из растворов азотной кислоты. Тем не менее существует возможность отделения Pa(V) от элементов, не образующих анионных комплексов в этих растворах и потому не сорбирующихся анионообменниками (Y, Ce(III), Am, Sr). Поглощение в 4,0 М HNO3 уменьшается в ряду Np(IV) > > Pu(IV) > Th > Ра > U(V1) > Np(VI) > Np(V).
При элюировании ниобия и тантала со смол Dowex 2 или Amberlite IRA-400 раствором 1 М НС1 — 0,5 М (СООН)2 ниобий десорбируется первым [33]. При повышении концентрации НС1 в элюенте до 2 М и снижении концентрации (СООН)2 до 0,01 М первым элюируется тантал [34].
Среда НС1 — NH4SCN пригодна для отделения Ра от лантаноидов, урана или тория. Наблюдается следующий ряд сродства: U(VI) > Pa(V) > > Th (IV) [35]. Растворы лимонной кислоты используют для разделения смеси Та — Ра на анионообменнике Amberlite IR-4B [36].
Лабораторные методики
ОТДЕЛЕНИЕ V (И Ni) ОТ Fe И Си [37]
Методика. Сильноосновный анионообменник Dowex 1-Х8 (0,15 — 0,07 мм) в Cl-форме, помешенный в колонку, приводят в равновесие со смесью 6 М НС1 — метанол (1:9) и промывают 50 см5 этой смеси. К 100 см3 смеси 6 М НС1 — метанол (1:9)
17-354
250
Часть II
прибавляют примерно по 10 мкг каждого из разделяемых элементов. Раствору дают постоять 6 ч и затем пропускают через обменник. Колонку дважды промывают 10 и 5 см3 смеси 9 М НС1 — метанол. Этим способом количественно элюируют ванадий (и никель). Fe и Си элюируют 1 М НО. Данный метод использовали при анализе золы различных масел.
ОТДЕЛЕНИЕ Та ОТ ПРИМЕСЕЙ (МИКРОГРАММОВЫЕ КОЛИЧЕСТВА Fe, Mo, Nb, Ti, U, V, W) [19]
Методика. Колонку, изготовленную из полиэтиленовой трубки диаметром 1,5 см и заполненную 6 — 10 г смолы Dowex 1-Х8, промывают смесью 1 М HNOj — 1 М HF. 0,2 — 0,6 г анализируемого образца растворяют в смеси HNOj — HF, упаривают до объема 1 см3, устанавливают 1 М концентрацию кислот и пропускают раствор через колонку. Затем колонку промывают 150 см3 смеси 1 М HNOj — 1 М HF. Этим способом элюируют все указанные элементы, за исключением тантала. Тантал десорбируют смесью 12 М HNOj — 5 М HF. В процессе элюирования наблюдается частичное разрушение смолы.
ОТДЕЛЕНИЕ И ОПРЕДЕЛЕНИЕ СЛЕДОВ Nb
(ДО 1 мкг/г) В ТАНТАЛЕ [38]
Методика. Около 1 г тантала (порошок, стружка) растворяют в 8 см3 HF (1:1) и 2 см3 HNOj (1 : 1) при нагревании. Раствор переносят в полиэтиленовую "колонку, заполненную 10 см3 сильноосновного анионообменника Diaion SA-400, используя смесь 1,0 М HNO3 — 5 М HF. Ниобий элюируют 100 см3 смеси 5 М HNOj — 0,2 М HF. К элюату прибавляют 1,5 см3 концентрированной НС1О4 и 2 см3 H2SO4 (1:19) и упаривают до появления белых паров. Раствор переносят в мерную колбу емкостью 25 см3, содержащую 5 см3 HCI (1 : 1) и небольшое количество воды, прибавляют 2 см3 0,05%-ного раствора сульфохлорфенола С, 2,5 см3 ацетона и разбавляют водой до 25 см3. Через 60 мин измеряют ОП раствора при 650 им относительно раствора сравнения.
ОТДЕЛЕНИЕ Ра ОТ Th, ОБЛУЧЕННОГО НЕЙТРОНАМИ [35] -
Методика. Облученный торий частично растворяют в небольшом количестве 4 М НО при осторожном нагревании. Нерастворившийся остаток обрабатывают 4 см3 смеси 0,5 М НС1 — 0,2 М NH4SCN. Объединенные растворы пропускают через колонку (3 х 1 см), заполненную смолой Dowex 1-Х8. Колонку промывают смесью 0,5 М НС1 — 0,2 М NH4SCN со скоростью 0,3 см3/мин. Этим способом элюируют весь торий. Протактиний десорбируют смесью 2 М H2SO4 — 2 М (NH4)2SO4 со скоростью 0,3 см3/мин.
ОТДЕЛЕНИЕ МАКРОКОЛИЧЕСТВ 231Ра ОТ ДОЧЕРНИХ ПРОДУКТОВ (227Ас, 227Th, 223Ra) И ПРИМЕСЕЙ Nb [32]
Методика. Ра2О5, содержащий около 4 г.231 Ра, растворяют в смеси 9 М НС1 — 0,2 М HF и смешивают с равным объемом 9 М НС1, насыщенной хлоридом алюминия. Раствор пропускают через полиэтиленовую колонку, заполненную 300 см3 сильноосновного анионообменника AG 1-Х8 в Cl-форме. Затем колонку промывают большим объемом 9 М НС1. Этим способом удаляют продукты распада 231 Ра и ио* ны алюминия. Протактиний элюируют смесью 9 М НС1 с 0,2 — 1 М HF. Однако ниобий удаляется не полностью, и операцию очистки рекомендуется повторить.. '
5. Применение ионообменников
251
Таблица 5.41. Методики разделения элементов группы 5А
Эле-меи! Отделяемые ' компоненты Ионообменник Условия разделения Литература
V Zr, Ti, Mo, W, Nb 40
V Cr Dowex 1-Х8 V сорбируется в смеси 0,1 М НС1 и 5—6% Н2О2 41
V Zr, Ti, Cu, Mo Dowex 1-Х8 Сорбция на анионообменнике 42
Dowex 5О-Х8 из 9 М НС1 Zr: 6 М НС1 Си: 6 М НС1 Mo: 1 М НС1 Ti—V—Mg: 9 М НС1 Сорбция Ti—V—Mg в 11 М НС1: Ti удерживается; сорбция V, Mg на катионообменнике: V: 1 М HCI, Mg: 4 М НС1
V Различные ионы Dowex 5OW-X8 Маскирование ЭДТА, V: 1%-ный Н2О2 5
V Анализ стали Deacidite FF Элюирование 0,6 М NaOH 8 М и 1 М НС1 43
V Бинарные смеси с Са, Ni, Си, Со, Fe, Zn, Mg,Мп 44
V Mg, Са, Zn, Cd, ARA-8 в Ме2+: Н2О 45
Co, Ni NOj-форме V: 5 М HNO3
V Mo, Tl (мкг/г) Dowex 1 Сорбция из 6 М НС1—Вг2 Мо: 2 М НС1О4— 1 М НС1 Т1: водный раствор SO2 V: 6 М НС1 46
V Mo (водопровод- Dowex 1-X8 V: 6 М НС1 47
ная вода) (цитратная Мо: 2 М НС1О4—
форма) 1 М НС1
Nb Pa, Zr СН3СООН—HNO,—HF 48
Nb Ta Amberlite Nb: 0,3 М Н2С2О4 49
IRA-400 Та:, 1—2 М НС1
Nb Та в стали 25
Nb Та в минералах 50
Nb Та в стали Wofatit SBW-X4 9 М НС1—0,05 М HF 51
Nb Та, W, Ti в рудах 52
Nb В стали Dowex 1-X8 Сорбция из 0,05 М HNO3— 53
1 М HF
Элюирование: 4 М HNO3— 1 М HF
252
Часть II
продолжение табл. 5.41
Элемент Отделяемые компоненты Ионообменных Условия разделения Литература
Nb Сплавы Dowex 1-Х8 54
Та Примеси Dowex 1-Х8 Примеси: 1 M HF—1 М HNO3 Та: 12 М HNO3—5 М HF 19
Ра Nb, щелочноземельные элементы, продукты распада Dowex 1-Х10 Pa Сорбция из 2,5 М HF Ра: 17 М HF 22
Ра Th, U, лантаноиды Dowex 1-X8 Сорбция из: 0,5 М НС1— 1,5 М NH4SCN La: 0,5 М НС1—1,5 М NH4SCN Th: 8 М НС1 Ра: 8 М НС1—0,001 М HF U: 1 М НСЮ, • 36
Ра Та КУ-2 Ра: 3% оксиизомасляная кислота Та: 10 М НС1—1 М HF 55 ,
Ра Np (горное породы) Deacidite FF Np: 5,9 М НС1 Ра: 3,8 М НС1 56
ВЫДЕЛЕНИЕ 231Ра ИЗ НИТРАТА ТОРИЯ [39]
Методика. 1 г Th(NO3)4 растворяют в 10 см3 8 М НС1 и пропускают раствор через колонку, заполненную анионообменником Dowex 1-Х8 (0,30 — 0,15 мм) в Cl-форме. Затем колонку промывают со скоростью 2,5 см3/мин 50 см3 8 М НС1. Весь торий проходит в элюат. Протактиний элюируют 25 см3 3,8 М НС1, уран — 25 см3 концентрированной НС1О4.
Обзор других методов разделения приведен в табл. 5.41.
5.2.5.2. 5 Б: N, Р, As, Sb, Bi
Группа 5Б периодической системы включает как элементы исключительно электроотрицательного характера, сорбируемые только анионообменниками, так и элементы, которые можно перевести в соответствующее состояние окисления и затем поглотить катионо- или анионообменниками. При использовании катионообменников в случае сурьмы и висмута могут возникнуть некоторые трудности вследствие гидролиза этих элементов в водных растворах. Чтобы предотвратить гидролиз, повышают содержание соответствующей минеральной кислоты или вводят подходящий комплексообразующий агент.
5. Применение ионообменников
253
ОРГАНИЧЕСКИЕ КАТИОНООБМЕННИКИ
Азот в форме катиона NH^ удерживается катионообменниками в растворах сильных минеральных кислот легче, чем ионы Na, Li, V(V) или Mo(VI). С повышением концентрации кислоты коэффициент распределения (в 0,1 М НС1О4 равный 73) уменьшается [3, 57, 58]. Ионы РО4“ и AsOj- в растворах НС1 и НС1О4 не сорбируются катионообменниками. В растворе НВг наблюдается сорбция As (III), которая повышается с увеличением концентрации кислоты (Dg в 12 М НВг больше 1). В среде НС1 сорбция Sb (III) и Bi (III) очень низкая. Висмут сорбируется только в разбавленных растворах НС1 (Dg < 1 в 0,5 М НС1). Повышение концентрации НС1 приводит к быстрому уменьшению сорбции Bi(III). С другой стороны, Sb(V) очень хорошо поглощается катионообменниками. Максимальная сорбция наблюдается в 8 М НС1 (Dg > 2000). Высокое сродство Sb(V) к катионообменникам, используется в разнообразных методиках. Трудности, связанные с десорбцией сурьмы(У), предотвращают восстановлением ее до сурьмы(Ш) раствором иодида аммония в 12М НС1 после элюирования других отделяемых элементов [58].
Ионы Bi3+ сильно сорбируются из разбавленных растворов серной кислоты. В 0,1 М H2SO4 Dg > 104, в 0,2М НС1О4 Dg 104. Поглощение Sb(III) в этой среде гораздо ниже.
При низких pH (около 1,5) висмут образует с ЭДТА прочные комплексы, не сорбируемые катионообменниками. При pH 1,3 — 1,5 цинк, кадмий и свинец полностью сорбируются сильнокислотными катионообменниками в КН4-форме, в то время как комплексы Bi с ЭДТА количественно проходят в элюат.
Висмут практически не сорбируется (Dg < 1) из растворов НС1 и НВг, если часть воды в них заменить соответствующим органическим растворителем, лучше всего низкомолекулярным спиртом [59, 60]. Таким способом висмут отделяют от U(VI), Th, Се(Ш), Al и Fe(III) (также в среде’НВг). При отделении сурьмы на катионообменниках ее часто связывают винной кислотой в несорбируемые комплексы. Этим способом ионы Sn2+, Fe3+, Cu2+ и Cd2+ при подходящем pH отделяют от сурьмы. Последнюю десорбируют с катионообменников винной кислотой и некоторыми другими оксикислотами [61].
Сильнокислотные катионообменники часто применяют для удаления мешающих катионов при анализе нитратов, фосфатов или арсенатов [62, 63]. ’
ОРГАНИЧЕСКИЕ АНИОНООБМЕННИКИ
Сильноосновные анионообменники успешно используют при разделении кислот, содержащих фосфор в различных степенях окисления, а также при разделении смесей конденсированных фосфатов.
Для разделения смеси конденсированных фосфатов в качестве элюентов чаще всего применяют растворы хлорида калия или натрия, буферированные при pH 5,0 ацетатами, формиатами или боратами.
254
Часть II
Коэффициенты распределения отдельных полимеров с повышением их молекулярной массы увеличиваются. Для ускорения элюирования его часто проводят при концентрационном градиенте. Градиентным.элюированием отделяют линейные полимеры фосфорной кислоты, содержащие до 14 атомов фосфора в молекуле.
Высокомолекулярные полимерные анионы (например, полиметафосфат) сорбируются сильноосновными анионообменниками. Однако их очень трудно элюировать солевым раствором. Полимеры гидролизуют в кислой среде при повышенной температуре и затем элюируют.
Разработана автоматическая установка для серийных анализов полимерных фосфатов на сильноосновных анионообменниках [64].
При разделении линейных фосфатов на отдельные вещества (от орто- до октафосфатов) на смоле AG 1-Х8 в CI-форме экспоненциальным градиентным элюированием раствором хлорида натрия (0,12 — 0,32 М) при pH 7,0 гидролиз связей Р—О—Р, обусловленный присутствием следов тяжелых металлов, можно предотвратить введением 5 мМ ЭДТА. Эффективность маскирования тяжелых металлов с помощью ЭДТА выше, чем, например, с помощью лимонной кислоты [65].
При разделении H3AsO3 и H3AsO4 на слабоосновном анионообменнике используют разницу в константах диссоциации; сорбируется только мышьяковая кислота.
Сурьма (V) в растворах НС1 хорошо поглощается анионообменниками. Поглощение увеличивается с повышением концентрации кислоты (в > 10 М НС1 величина £>? больше 105). Максимальный коэффициент 'распределения Sb (III) в ~1 М НС1 приблизительно на 2 порядка ниже, чем Sb(V). Сорбция Sb(III) уменьшается с ловышением концентрации кислоты.
Акки и Кхопкар [66] провели систематическое исследование анионообменного поведения малонатных комплексов сурьмы (III). В 5%-ном мало-натном растворе при pH 4,8 образуется отрицательно заряженный мало-натный комплекс Sb(III), который сорбируется анионообменниками (Dowex 21К и т.п.) в малонатной форме. Растворы НС1, HNO3, НС1О4, H2SO4 или их натриевые соли и ацетат аммония (0,1 — 1,0 М) полностью элюируют сурьму. Вода оказывает аналогичное действие.
В этой среде сурьма селективно отделяется от Li, Rb, Cs, Be, Mg, Ca, Sr, Zn, Mn, Co, Ni, Cd, Ti и Pd, которые не образуют малонатных комплексов при pH 4,8 и не сорбируются анионообменниками.
Малонатные комплексы Си, Fe, Al, Th, U и Pb проявляют более высокое сродство к анионообменнику, чем комплекс Sb. Слабосвязанный комплекс Sb первым легко элюируется водой. После этого десорбируются Си, Fe, Al (4М НС1), Th (6М НС1), U (0,5 М HjSO^ и Pb (6 М HNO3). Сильно мешающие ионы Hg(II) и Bi(III) маскируют введением ЭДТА.
Bi (III) сорбируется анионообменниками во всем интервале концентраций НС1.
Ионы As (III), Sb (III) и Bi (III) легко сорбируются анионообменниками из смеси НС1 — HF. Быстрое разделение смеси As — Sb — Bi достигается при использовании подходящей смеси НС1 с HF или NH4F. Первым 3 М НС1
5. Применение ионообменников
255
элюируется As(III), затем смесью 0,3 М НС1 — 1 М HF десорбируется Sb(III) и, наконец, смесью 1 М NH4C1 — 1 М NH4F элюируется Bi(III) [67].
В некоторых случаях для элюирования Bi (III) из анионообменников выгодно применять смесь НС1 — HF. Элюирование чистой НС1 требует много времени вследствие высокого сродства ионов Bi(III) к обменнику.
Из чистой фтористоводородной кислоты на анионообменниках сорбируются только As(V), Sb(V) и Sb(III); As(III) и Bi(III) не сорбируются.
Значительное поглощение висмута(Ш) (Dg ~ 103) наблюдается из растворов НВг, содержащих метанол или этанол, тогда как Fe(III), Al, Th, U(VI), Ce(III), Mg, Ca, Sr, Mn(II), Co(II), Ni(II) и V(IV) практически не сорбируются (Dg < I). Это большое различие в сродстве используют для высокоселективной сорбции Bi(III) [68].
В системах, содержащих азотную кислоту, элементы группы 5Б ведут себя аналогичным образом. Но разница в величинах D между Bi (III) и другими элементами меньше, чем в среде НВг [69].
Лабораторные методики
ОПРЕДЕЛЕНИЕ Р В СТАЛИ, ЧУГУНЕ ИЛИ ФЕРРОВАНАДИИ [70, 71]
Методика А. Эта методика применима для анализа стали и чугуна. 0,1000 г образца растворяют при нагревании в 4 см3 HNO3 (1 : 1). Затем прибавляют 2 см3 0,2%-иого раствора КМпО4 и кипятят 5 мин. Образующийся диоксид марганца растворяют введением нескольких капель 10%-ного раствора нитрита натрия, прибавляют 4 см3 концентрированной НС1 и выпаривают досуха на водяной бане. Остаток обрабатывают 1 см3 концентрированной НС1 и снова выпаривают. Полученный остаток растворяют в 0,5 см3 концентрированной НС1, прибавляют 40 см3 воды и 0,1 г NH2OH • НС1 и кипятят раствор 3 — 4 мин. Смесь охлаждают, вводят несколько кристаллов NH2OH • НС1 и пропускают охлажденный раствор через колонку (15x1 см), заполненную катионообменником Amberlite IR-120 в Н-форме. Колонку промывают 0,2 М НС1. Элюат собирают в мерную колбу и в его аликвотной части определяют фосфор одним из стандартных методов.
Методика Б. Эта методика применима для анализа феррованадия. 0,1000 г образца растворяют в смеси 0,8 см3 H2SO4(1 : 1) и 3 см3 HNO3 (1 : 1). Раствор упаривают до появления белых паров SO3. Смесь охлаждают, прибавляют 1 — 2 см3 воды и снова выпаривают. Сухой остаток растворяют в 40 cmj воды, прибавляют 0,1 г сульфата гидроксиламина и кипятят в течение нескольких минут. Охлажденный раствор пропускают через колонку (см. методику А) со скоростью 5 см3/мин и промывают колонку таким количеством воды, чтобы получить 250 см3 элюата. Элюат упаривают и переносят в мерную колбу емкостью 50 см3. Весь присутствующий фосфор содержится в элюате. Содержание фосфора определяют фотометрически. Ионообменную колонку регенерируют 4 М НС1. Данный метод непригоден для анализа сталей с высоким содержанием хрома.
ОТДЕЛЕНИЕ As ОТ Fe ПРИ АНАЛИЗЕ СПЛАВОВ ЖЕЛЕЗА [72]
Методика. Отделение выполняют на колонке (13 х 1 см), заполненной смолой Dowex 5О-Х8 (0,30 — 0,15 мм) в Н-форме.
Примерно 0,15 г образца растворяют в разбавленной (1 : 1) HNO3 при нагревании. Раствор выпаривают досуха на водяной бане. Остаток растворяют в 2 см кон
256
Часть 11 .
центрированной НС1 и снова выпаривают раствор. Остаток растворяют в 5 см3 3 М НС1 и разбавляют водой до 40 см3. Сосуд с раствором накрывают часовым стеклом и тотчас погружают в ледяную воду. Раствор насыщают газообразным диоксидом серы и дают ему постоять в течение 1 ч. Затем раствор кипятят для удаления избытка SO2, разбавляют водой и пропускают через колонку со скоростью 5 см3/мин. Колонку промывают 50 см3 0,3 М НС1. Все порции элюата собирают и объединяют. Содержание мышьяка определяют титрованием иодом.
Железо элюируют 150 см3 2 М НО.
РАЗДЕЛЕНИЕ СМЕСИ Bi — U — Th НА АНИОНООБМЕННИКЕ [73]
Методика. Раствор состава 96% и-пропилового спирта — 4% 5 М HNO3, содержащий разделяемые ионы, пропускают через колонку (12 х 0, 6 см), заполненную смолой Dowex 1-Х8 (0,15 — 0,075 мм) в NOj-форме, которую предварительно приводят в равновесие с раствором упомянутого выше состава. Колонку промывают раствором того же состава и элюируют U смесью 80% метанола — 20% 5 М HNOj, Th—смесью 80% метанола — 20% 6 М HCI, Bi 1 М HNO,.
ОТДЕЛЕНИЕ Bi ОТ РАЗЛИЧНЫХ ЭЛЕМЕНТОВ НА
АНИОНООБМЕННИКЕ [74]
Этот метод применим для отделения микро- и миллиграммовых количеств висмута от Mg, Са, Pb, Cu, Zn, Cd, Со, Ni, Mn(II), Al, Ga, In, Fe(III), Sc, Yb, Zr, Hf и U.
Методика. Смолу Dowex 1-X8 (0,15 — 0,074 мм) в NOj-форме, помещенную в колонку (10 х 0,6 см), приводят в равновесие со смесью состава 90% метиленгликоля — 10% 5 М HNOj.
Раствор смеси нитратов разделяемых ионов выпаривают до влажных солей. К остатку прибавляют достаточное количество смеси указанного выше состава и пропускают раствор через колонку. Вместе с висмутом на смоле сорбируются лантан и торий. Затем колонку промывают смесью указанного выше состава и элюируют висмут 1 М HNOj.
ОТДЕЛЕНИЕ 210Bi И 210РЬ ОТ 210Ро [75]
Методика. Сильнокислотный катионообменник Dowex 50-Х4 помещают в колонку (3 см х 0,28 см2), снабженную водяной рубашкой, и приводят в равновесие с 12 М НВг, 4 М раствор HNO3, содержащий разделяемые элементы, упаривают с 1 см3 9 М НВг примерно до 0,05 см3. Остаток растворяют в 0,5 см3 12 М НВг и раствор пропускают через колонку. Затем обменник промывают 12 М НВг при 60°С, в результате чего сразу же десорбируются висмут и свинец. Полоний элюируют 6 М НС1.
ОТДЕЛЕНИЕ СЛЕДОВ Bi ОТ БОЛЬШОГО ИЗБЫТКА Си [76]
Методика А. Катионообменник Dowex 50-Х8 (0,15 — 0,074 мм) в Н-форме помещают в колонку размером 25 х 3 см и приводят в равновесие с Г М НС1. В смеси НС1 — HNO3 (1 : 1) растворяют 1 г металлической меди и выпаривают раствор досуха. Остаток растворяют в 25 см3 1 М НС1 и пропускают через колонку. Висмут элюируют 1 М НС1, медь — 12 М НС1.
Методика Б. Раствор образца, полученный как описано выше, пропускают через колонку размером 10 х 1,5 см, заполненную анионообменником Dowex 1-Х8 (0,15 —
5. Применение ионообменников
257
Таблица 5.42. Методики разделения элементов группы 5Б
Элемент Отделяемые
Условия разделения
Ионообменник
компоненты
Литература
NH4+ Удобрения Wofatit Р, . KPS-200 77
nh4+, Удобрения Amberlite 78
NO“ no2~ Вода IRA-400 и сильнокислотный катионообменник Анионооб- NOj": 5%-ный NaCl 79
As As Мо, W Sb менник Dowex 21 К 1 М NaOH—винная кислота 80 81
As Ge As(III) сорбируется, ' Sb(III) проходит As: 1 М H,SO, 2 4 Сильноосновный Смесь НС1—СН3СООН 82
PO’~ soj- анионообменник Diaion SA-100 РО’": 0,1 М HCI- 83
PO’~ 4 Железные руды О.19 М КС1 SO4“: 2,0 М КС1 Amberlite IR-120 84
pi-p3 Взаимное Dowex 1-Х10 Раствор KC1 85
PI~PIO разделение Wofatit SBW P], P2: 0,25 M KC1 P,: 0,50 M KC1 P4: 0,75 M KC1 AB-17 KC1, NaCl 86
P,-P5 Dowex 1-X4 или КО при различных pH 87,
PI-PI4 Dowex 2-X8 Dowex 1-X4 KC1, pH 8 88 89
H2po2- нро*-, НРО4~ Dowex 1-X8 KCl, градиент 90
Оксикис- Dowex 1-X8 CH3COONH4 различной 91
ЛОТЫ фосфора PO4~ р2°Г концентрации Amberlite Орто: 0,05 M НО 92
Различ- IRA-400 Пиро: 0,50 М HCI Amberlite Н2РО2: 0,1 М КО— 93
ные P-КИСЛОТЫ IRA-410 0,025 М глицин- 26,5 мМ КОН РО’~ + РО’-; 0,05 М НС1 Р2О*~: 0,3 М НС1 Р3О’“: 0,5 М НС1
258
Часть II
продолжение табл. 5.42
Элемент Отделяемые компоненты Ионообменник Условия разделения Литература
РО3~ 4 Пиро- и монофторфосфорная кислота Dowex 1-X8 Орто: 0,085 М КС1 Монофтор: 0,194 М КС1 Пиро: 0,218 М КС1 94
Sb Ge, Sn Dowex 1-X8 Ge: 3 М НВг Sn: 0,5 М НВг Sb: 1,0 М HNO3 95
. Sb In, Cd Dowex 1-X8 In: 0,25 М НВг Sb: 0,10 М НВг Cd:-1,0 М HNO3 95
Sb 4 In, Zn ЭДЭ-Юп Сорбция из 0,20 М НС1 Сорбируется только Sb Sb: 1,5 М H2SO4 96
Sb Sn(IV) Dowex 1-X8 Sb: 0,50 М винная кислота— 0,30 М НС1 Sn: 2,0 М H2SO4 97
Sb Различные элементы Dowex 21 К Сорбция из раствора малоновой кислоты, pH 4,80 98
Sb Bi (Cu, Fe, Pb) Dowex 5OW-X8 Sb: 1 М НС1О4 Bi: 3 М НС1 99
Sb Te(IV) AB-17 Раствор LiCl 100
Bi Различные элементы Dowex 5OW-X8 H2SO4 или HNO3 различной концентрации 101
Bi Cu, Zn, Cd КУ-2 Сорбция из раствора тай-рона: Bi не сорбируется 102
Bi As, Те, Cd Amberlite CG-4B As(III): 4,5 M HC1 Te(IV): 1,0 M HC1 Cd: 0,10 M HC1 Bi: 3 M H SO, 103
Bi Pb, Zn Сильноосновный анионо-обменник НС1 различной концентрации 104
Bi Al, Fe, B, Si, следы AB-17-X8 в SO*--форме Комплексы тайрона с Bi, Al, Fe; фторидные комплексы В, Si 105
Bi Hg, Cd, Sb Amberlyst 15 Градиент 0,05—0,5 М НВг 106
Bi sb2o3 Dowex 50-X2 Сорбция: винная кислота-г-нсю4 Bi: 3 М НС1 107
0,074 мм) в Cl-форме. Медь элюируют ~ 175 см3 1 М НС1, висмут — 150 см3 2 М H2SO4.
Обзор других методов разделения дан в табл. 5.42.
5. Применение иоиообменников
259
Литература
1. Китига К., Yokoyma Y., Sano Н., Mabuchi Н., Japan Analyst, 6 (1957) 637.
2. StrelowF. W., Anal. Chem., 32 (1960) 1185.
3. StrelowF. W., Rethmeyer R., Bothma C. J., Anal. Chem., 37 (1965) 106.
4. Ariel N., Manka J., Anal. Chim. Acta, 25 (1961) 248.
5. Fritz J. S., Abbink J. E., Anal. Chem., 34 (1962) 1080.
6.7 i:atsu S., KumagaiN., J. Chem. Soc. Jap. Pure Chem. Sect., 78 (1957) 1558.
7. Kuroda R., Ishida K., J. Chromatogr., 18 (1965) 18.
8. Shiskov D., Khimiya Ind. (Bulg.), 32 (1966) 69.
9. Walter R. I., J. Inorg. Nucl. Chem., 6 (1958) 58.
:0. Bok L. D., Schuler V. C., J. South African Chem. Inst., 13 (1960) 82.
)i. Sliepcevic Z., Siroki M., Stefanec Z., Mikrochim. Acta (Wien), 1973 , 945.
12. Qureshi M., Rathore H. S., Kaushik R. Ch., Anal. Chem., 47 (1975) 1710.
13. Дарбинян M. В., Даниелян А. А. Изв. АН Арм. ССР, Сер. хим. наук, 18 (1965) 462.
14. Goto Н., Hakita Y., Japan Analyst, 10 (1961) 904.
15. Priyadarshini U., Tandon S. C., Anal. Chem., 33 (1961) 435.
16. Keller C., Radiochim. Acta, 1 (1963) 147.
17. Pluchet E., Muxart R., Bull. Soc. Chim. France, 1961, 372.
18. Headridge J. B., Dixon E. J., Analyst, 87 (1962) 32.
19. Huff E. A., Anal. Chem., 36 (1964) 1921.
20. Danielsson L., Arkiv for Kemi, 27, No. 38 (1967) 467.
21. Nishimura K., Irokawa H., Japan Analyst, 13 (1964) 304.
22. Bergstresser K. S., Anal. Chem., 31 (1969) 1812.
23. Kraus K. A., Moore G. E., J. Amer. Chem. Soc., 71 (1949) 3855.
24. Kraus K. A., Moore G. E., J. Amer. Chem. Soc., 73 (1951) 9, 2900.
25. Kaliman S., Oberthin H., Liu R., Anal. Chem., 34 (1962) 609.
26. Miinchow P., Chem. Zt., 88 (1964) 37.
27. Dugain F., Laverlochere J., Anal. Chem., 37 (1965) 998.
28. Hibbits J. O., Oberthin H., Liu R., Kallmann S., Talanta, 8 (1961) 209.
29. Sawada T., Kato S. Nippon Kizoku Cakkaishi, 28 (1964) 180.
30. Spauszus S., Heimer M., Chem. Tech. (Berlin), 13, (1961) 96.'
31. Faris J. P., Buchanan R. F., USAEC Report ANL (1964) 6811.
32. Physico-Chimie du Protactinium, Publ. No. 154, Centr. Nat. Rech. Sci. Paris (1966).
33. Dragulascu C., Kyri L, Oprescu M., Acad. R. P. R. Baza Cercet. Stiint Timisoara, Stud. Cercet. Stiln. Chim., 10 (1963) 55.
34. Specke A., Hoste J., Talanta, 2 (1959) 332.
35. Hamaguchi H., Ishida K, Kuroda R., Anal. Chim. Acta, 33 (1965) 91.
36. Yang J. T., C. R. Acad. Sci., 231 (1956) 1059.
37. JanauerG. E., Korkisch J., Z. anal. Chem., 179 (1961) 241.
38. Hashitani H., Adachi T., Japan Analyst, 24 (1975) 303.
39. Pillai T., J. Indian. Chem. Soc., 42 (1965) 32.
260
Часть II
40. Ferraro Т. A., Taianta, 16 (1969) 669.
41. Kuroda R., Kiriyama T., Japan Analyst, 19 (1970) 1287.
42. Michaelis C., EveslageS., Coulter P., Fortman J., Anal. Chem., 34 (1962) 1764.
43. Hall F. M., Bryson A., Anal. Acta, 24 (1966) 308.
44. Marti F. B., Alvarez C., Rev. Cienc. Apl., 20 (1966), C. A. (1967) 61464 p.
45. Федоров П. И., Андреев В. К., Лиде Т. В-, Слотвинский-Сидак Н. П. Ж. аналит. химии, 30 (1975) 379.
46. Korkisch J., Steffan J., Arrhenius G., Anal. Chim. Acta, 94 (1977) 237.
47. Korkisch J., Krivanec H, Anal. Chim. Acta, 83 (1976) 111.
48. Kim J. I., Legally H., Born H. J., J. Inorg. Nucl. Chem., 33 (1971) 3547.
49. Dragulescu C. et. al., Nucl. Sci. Abstr., 19 (1965) 20054.
50. Kallmann S., Oberthin H, Liu R., Anal. Chem., 34 (1962) 609.
51. SpauszusS., Heimer M., Chem. Tech. (Berlin), 13 (1961) 96.
52. Foldziriska A., Chem. Anal. (Warsaw), 16 (1971) 821.
53. Danielson L., Jen Kontorets Ann., 15 (1967) 325. C. A., 68 (1968) 45953.
54. Ferraro T. A., Taianta, 15 (1968) 923.
55. Stronski I., Zielinski A., Nucleonika, 9 (1964) 801.
56. Morgan J. W., Lowering J. F., Anal. Chim. Acta, 28 (1963) 405.
57. StrelowF. W. E, Sondorp H, Taianta, 19 (1972) 1113.
58. Nelson F., Murase T., Kraus K. A., J. Chromatogr., 13 (1964) 503.
59. Korkisch J., Klakl E., Taianta, 16 (1969) 377.
60. Korkisch J., Ahluwalia S. S., Taianta, 14 (1967) 155.
61. Khorasani S. S., Kundkar M. A., Anal. Chim. Acta, 25 (1961) 292.
62. Васильева 3. В. Труды Инет, геологии рудн. месторожд., петрогр., минералогии и геохимии АН СССР (1961) 91.
63. PuschelR., Lassner Е., Z. anal. Chem., 174 (I960) 1.
64. Lundgren D. P., Lueb N. P., Anal. Chem., 33 (1961) 366.
65. Nakamura T., Yano T., Fujita A., Ohasi S., J. Chromatogr., 130 (1977) 384.
66. AkkiS. B., KhopkarS. M., Z. anal. Chem., 255 (1971) 130.
67. Nelson F., Rush R. M., Kraus К. A., J. Amer. Chem. Soc., 82 (1960) 339.
68. Klakl E., Korkisch J., Taianta, 16 (1969) 1177.
69. Walter C. W., Korkisch J., Mikrochim. Acta, 1971, 81, 137, 158, 181, 194.
70. Wigh K., Inczedy J., Erdey L., Magy Kem. Foly., 69 (1963) 73.
71. Venturello G., Gualandini C., Bull. Sci. Fac. Chim. Bologna, 17 (1959) 5.
72. Duyal G. R., Ironside R., Russel D. S., Anal. Chim. Acta, 25 (1961) 51.
73. Korkisch J., Tera F., Z. anal. Chem., 186 (1962) 290.
74. Feik F., Korkisch J., Taianta, 11 (1964) 1585.
75. Nelson F., J. Chromatogr., 25 (1966) 414.
76. Van DyckG., Verbeek F., Z. anal. Chem., 249 (1970) 89.
77. Kozak M., Krajc’P., Chem. Prum., 13 (1963) 246.
78. Kowalska E., SollorzJ., Z. anal. Chem., 210 (1965) 271.
79. Takayuki T., Akira S., Toshiu M., YujijT-, Aichi-Ken Kogai Chosa, Sento Shoho, 1977, 5,130, C. A. 88 (1978) 197328.
80. Gartner K., Ebert L., TaubigE., Acta Chim. Hung., 27 (1961) 215.
81. Chakravarty T. N., Sci Cult. (Calcutta), 28 (1962) 392. C. A., 58 (1963) 1896.
82. Korkisch J. , FeikF., Separ. Sci., 2 (1967) 1. C. A. 67 (1967) 39826.
83. Naosuke Shiraisi et al., Bunseki Kagaku, 14 (1965) 450.
84. Povondra P., RoubalovdD., Collect. Czech. Chem. Commun., 25 1960, 1890.
85. Wieker W., Z. Chem., 1 (1961) 211.
86. Украинская Д. M., Кузнецов-Фетисов Л. И. Изв. высш, учебн. завед., Хим. технол., 13 (1970) 1694.
87. Nakamura Т., Kimura М., Waki Н., Ohasi S., Bull. Chem. Soc. Jap., 44 (1971) 1302.
88. Luiansky D., Chem. Prum., 26 (1976) 514.
89. Ohasi S., Tsuji N., Ueno Y., Takeshita M., Muto M., J. Chromatogr., 50 (1970) 349.
90. Pollard F. H., Rogers D. E., Rothwell M. T., Nickless G., J. Chromatogr., 9 (1962) 227.
91. KoguchiK., NakiH., Ohasi S., J. Chromatogr., 25 (1966) 398.
92. Winad L., J. Chromatogr., 7 (1962) 400.
93. Беремзанов Б. А., Буркитбаев M. M., Муратбеков M. Б., Матвеева Г. X. Ж. аналит. химии, 32 (1977) 1694.
94. Benz С.,Kelley R. М., Anal. Chim. Acta, 46 (1969) 83.
95. Andersen T., Knutsen A. B., Acta Chem. Scand., 16 (1962) 849.
96. Кахман Б. X. Ж. аналит. химии, 23 (1968) 1234.
97. Casal A. R., DeNamor A. F., An. Quint., 68 (1972) 229.
98. Akki S. B., Khopkar S. M., Z. anal. Chem., 255 (1971) 130.
99. Sulcek Z., BoseovdM., Dolezal J., Collect. Czech. Chem. Commun., 34 (1969) 787.
100. Бусев А. И. и др. Ж. аналит. химии, 25 (1970) 1374.
101. Akki S. B., Khopkar S. M., Separ. Sci., 5 (1970) 707.
102. Головин П. В. Укр. хим. журн., 27 (1961) 261.
103. Kuroda R., Ishida К., Kirigama T., Anal. Chem., 40 (1968) 1502.
104. Степин В. В., Поносов В. И., Зобнина Е. В., Новикова Е. В. Тр. ВНИИ станд. образцов, 5 (1969) 75.
105. Слежка Н. И., Дядик И. С., Демешева Е. П., Чащина О. В., Отмахо-ва 3. И. Ж. аналит. химии, 33 (1978) 102.
106. Willis R. В., Fritz J.S., Taianta, 21 (1974) 347.
107. PetdkP., Koubova V., Analyst, 103 (1978) 179.
Милан Мархол
ИОНООБМЕННИКИ В АНАЛИТИЧЕСКОЙ ХИМИИ
ч.1
Научные редакторы Б. М. Комарова, И. Н. Лаврова. Художник В. Я. Медников. Художественный редактор М. Н. Кузьмина.
Технические редакторы Л. А. Тихомирова, Л. С. Тимофеева.
Корректоры Т. Е. Луганова, И. Н. Холкина.
ИБ № 3929
Подписано к печати 14.05.65 г. Формат 60 х 90У16. Бумага офсетная № 2.
Гарнитура тайме. Печать офсетная. Объем 8,25 бум.л. Усл.печ.л. 16,50.
Усл.кр.-отт. 16,50. Уч.-изд.л. 17,39. Изд. №3/2903. Тираж 2900 экз.
Зак. ?54 Цена 2 р. 90 к.
Набрано в издательстве «Мир» иа фотонаборном комплексе «Компьюграфик»
129820, ГСП Москва, 1-й Рижский пер., 2.
Тульская типография Союзполиграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли.
300600, Тула, проспект им. В. И. Ленина, 109.
Уважаемый читатель!
Ваши замечания о содержании книги, ее оформлении, качестве перевода и другие просим присылать по адресу: 129820, Москва, И-110, ГСП, 1-Рижский пер., д'. 2, издательство «Мир».
ИЗДАТЕЛЬСТВО «МИР» выпускает в свет в 1986 году
Мазор Л. Методы органического анализа. Пер. с англ., 38 л., 5 р. 70 к.
Книга из серии монографий по общим вопросам аналитической химии; посвящена одному из актуальных вопросов современной химии — органическому анализу. Автор книги — крупный венгерский ученый в области аналитической органической химии. Впервые в практике изданий, касающихся анализа органических соединений, объединяются методы качественного и количественного элементного и функционального анализа.
Для научных работников, преподавателей и студентов, специализирующихся в области аналитической и органической химии, биохимии.
Из отзыва профессора В. М. Потапова: «... книга интересна химику-органику, в задачу которого входит анализ синтезированных соединений. Химик-аналитик, занятый органическим анализом, найдет в ней ссылки на ряд методик, которые можно использовать для рещения конкретной задачи.»
Из отзыва доктора хим. наук М. А. Володиной: «... Данную книгу можно считать уникальным научным трудом, объединившим все стороны области органического анализа.»
Заблаговременно оформляйте предварительные заказы на интересующие Вас книги. Заказы принимают магазины, распространяющие научно-техническую литературу. Своевременно сданный заказ гарантирует приобретение нужных Вам книг.