




































































































































Автор: Пасынский А.Г.
Теги: вещества химия биология коллоидная химия учебное пособие высоко-молекулярные соединения
Год: 1959




Текст
Л.Г.ПАСЫНСКШ
сл
/ !'
или л:
ВЫСША1ШКОЛА
МОСКВА-
А. Г. ПАСЫНСКИЙ
КОЛЛОИДНАЯ
химия
Под редакцией акад. В. А. КАРГИНА
Допущено
Министерством высшего образования СССР
в качестве учебного пособия
для университетов
ГОСУДАРСТВЕННОЕ ИЗДАТЕЛЬСТВО
«ВЫСШАЯ ШКОЛА»
М о с к в а—1 9 5 9
ПРЕДИСЛОВИЕ
За последние 15—20 лет коллоидная химия, и особенно
учение о высокомолекулярных соединениях, получили
настолько значительное развитие, что изменились многие
принципы и основные положения этой науки; кроме того,
расширилась сфера приложения ее в области биологии
и многих народнохозяйственных проблем. В связи с этим
возникла необходимость в создании нового краткого курса
коллоидной химии.
Предлагаемое учебное пособие предназначается для
студентов биологических специальностей университетов;
книга, однако, может быть использована студентами ряда
других специальностей, а также научными работниками
и инженерами, студентами заочной сети при изучении кол-
коллоидной химии в объеме лекционного курса 30—35 часов
или при самостоятельном ознакомлении с этой наукой.
В книге использована специальная литература, опублико-
опубликованная примерно до середины 1958 г.
В рамках курса коллоидной химии книга кратко охва-
охватывает физико-химию дисперсных систем и высокомолеку-
высокомолекулярных веществ. Сравнительное-изучение различий и сход-
сходства этих систем соответствует, с точки зрения автора, не
только указанным выше условиям прохождения курса
коллоидной химии, как единой учебной дисциплины, но
и научному существу рассматриваемых проблем. Материал
книги рассчитан на читателя, знакомого с основным курсом
физической химии. Для удобства проработки материала к
каждой главе приложены краткие выводы и контрольные
вопросы.
Автор приносит глубокую благодарность акад. В. А. Кар-
гину за совместное обсуждение ряда проблем, а также акад.
П. А. Ребиндеру и коллективу кафедры химии МГУ и
проф. С. И. Соколову за ряд ценных и важных замечаний
при рецензировании рукописи книги.
Автор
2* з
Глава первая
ОБЩАЯ ХАРАКТЕРИСТИКА КОЛЛОИДНЫХ
СИСТЕМ
Системы, изучаемые физической химией, — газы, жид-
жидкости, растворы, — состоят из сравнительно небольших
молекул, редко содержащих более одного-двух десятков
атомов. Между тем существует огромное количество сие-
тем, отдельные частицы которых включают много сотен
и тысяч атомов и достигают иногда микроскопически види-
видимых размеров. Во многих случаях эти частицы представля-
представляют собой зародыши кристалликов, маленькие обломки
различных кристаллических решеток или аморфных ве-
веществ, или капельки жидкостей. В случае кристаллических
решеток, они по природе связей, соединяющих их
структурные элементы, могут быть разделены на ионные
(подобные решетке NaCl), атомные (решетки алмаза, гра-
графита), молекулярные (решетки антрацена, ZnS) и металли-
металлические (решетки Аи, Ag); в структурном отношении частицы
относятся к трехмерным или слоистым решеткам. Так,
например, в алмазе (рис. 1) весь кристалл можно рассмат-
рассматривать как одну молекулу, в которой все атомы углерода
связаны в пространственную сетку одинаковыми, тетраэдри-
чески расположенными, ковалентными связями С—С
(длиной 1,54 А); с другой стороны, в слоистой решетке гра-
графита (рис. 2) атомы углерода образуют плоскую, двухмер-
о
ную шестиугольную сетку с расстояниями С—С 1,42 А,
тогда как плоскости отстоят на 3,42 А и соединены лишь
весьма слабыми связями. Как пример трехмерной сетки
можно указать кремнекислородные группировки в силикате
ультрамарина и двухмерной сетки — в слюде (рис. 3).
Для перечисленных структур характерно то обстоятель-
обстоятельство, что они могут быть бесконечно продолжены в трех или
двух измерениях и при достаточном числе атомов, естест-
естественно, образуют со средой поверхности раздела; в этом
заключается первый основной способ образования крупных
частиц.
Другим основным способом образования больших час-
частиц является последовательное сочетание молекул в виде
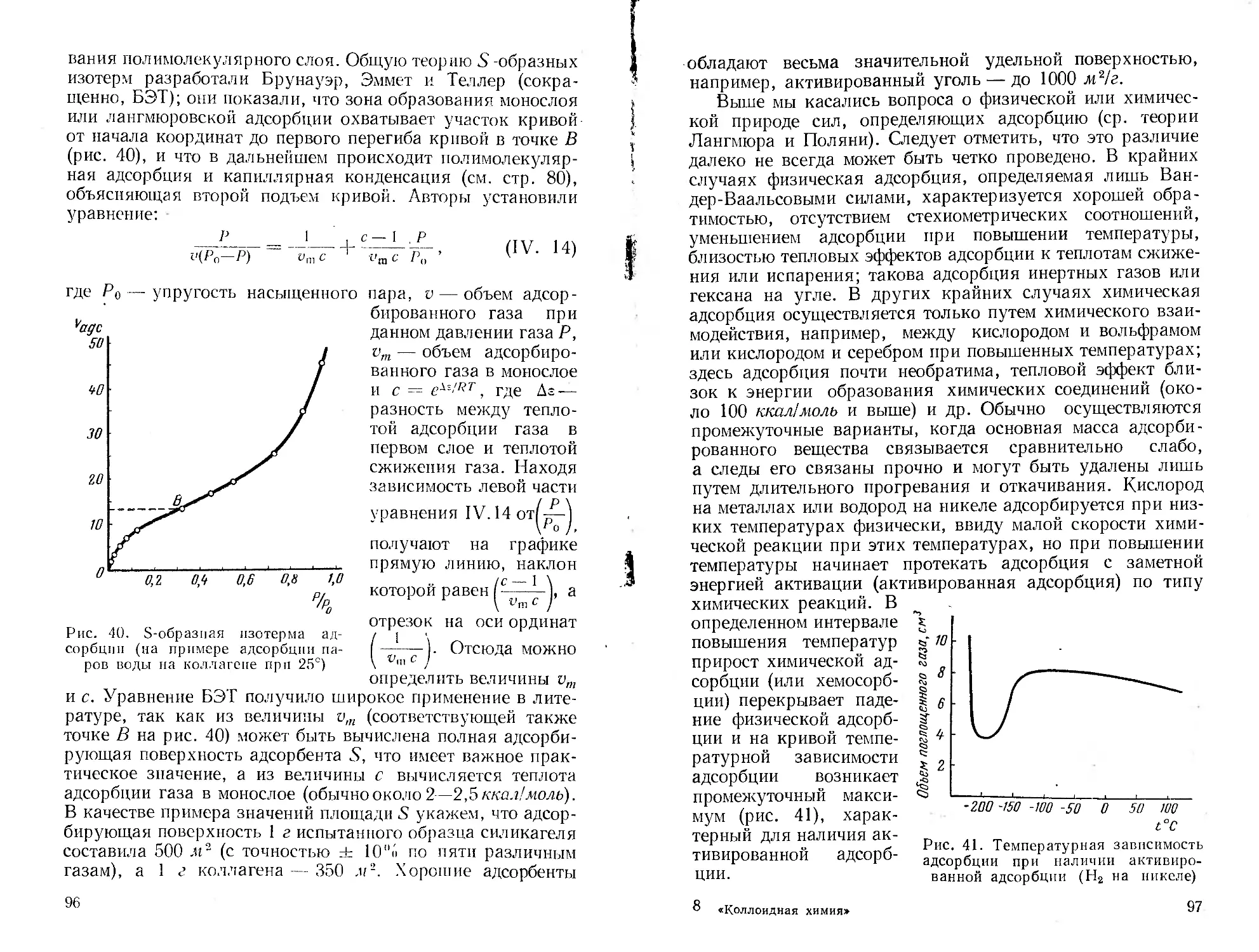
Рис. 1. Структура
алмаза
Рис. 2. Структура графита
линейных структур, которые могут быть бесконечно про-
продолжены лишь в одном измерении; например, цепные моле-
молекулы из нескольких сотен или даже тысяч звеньев дает
линейная последовательность тех же С—С атомов, сое-
соединенных между собой ковалентными связями:
Mill
—С—С—С—С—С— (в полиэтилене).
I I I I I
Если свободные валентности С-атомов насыщены
атомами водорода или одновалентными радикалами R,
то цепи могут развиваться лишь в одном измерении. В со-
состав цепей могут входить также атомы кислорода, азота,
кремния и других элементов (например, в молекулах бел-
белков, эфиров целлюлозы, кремнийорганических полимеров
и др.). Разумеется, если атомы цепи связаны с многовалент-
многовалентными атомами или радикалами R', то цепи также могут
развиваться в двух или трех измерениях, и тогда они пере-
переходят в структуры первого типа, обладающие поверхностями
раздела.
Однако условия соединения молекул между собою зави-
зависят не только от химических свойств этих молекул, но и
от свойств окружающей
среды. Молекулы хлор-
хлорного железа, мочевины
или сахара могут об-
образовывать кристалли-
кристаллические решетки, но в во-
воде эти решетки, вслед-
вследствие своей растворимо-
растворимости, быстро распадают-
распадаются. Это происходит по-
потому, что силы взаимо-
взаимодействия между моле-
молекулами в решетке сла-
слабее, чем взаимодействие
отдельных молекул с
водой. Если же эти
соединения поместить,
например, в бензол, то
размеры кристалликов
могут сохраняться до-
достаточно крупными, так
как растворения не про-
происходит. Напротив, ча-
частицы антрацена в бен-
бензоле распадаются до мо-
молекул, а в воде остают-
остаются неизменными. Длин-
Длинные цепные молекулы
также, могут образовать
в инертной среде кри-
;ypлcИc сталлические или амор-
тая структура (слюда), // — трех- фные частицы (кристал-
мерная сетка—каркас (ультрамарин) лы парафинов ИЛИ части-
цы смол в воде), тогда
как в растворителе, например в бензине, они распадаются
на отдельные молекулы, сохраняющие, однако, в отли-
отличие от предыдущего случая, свои значительные размеры.
Таким образом, крупные частицы, состоящие из сотен
или тысяч атомов или молекул, в зависимости от их хими-
химического строения и свойств среды, могут относиться лишь
к структурам двух типов: 1) трехмерные или двухмерные
структуры, для которых характерно наличие поверхно-
поверхностей раздела со средой и 2) длинные цепные молекулы или
макромолекулы.
Системы, образованные такими частицами, называются
коллоидными системами (более точное их определение дает-
дается ниже), а наука, изучающая физико-химические свойства
и закономерности этих систем — коллоидной химией.
ПРАКТИЧЕСКОЕ ЗНАЧЕНИЕ КОЛЛОИДНЫХ СИСТЕМ
В основе всех жизненных процессов, а также структур
живых организмов, тканей и клеток лежат такие вещества,
как белки, нуклеиновые кислоты, крахмал, гликоген, цел-
целлюлоза, построенные из гигантских цепных молекул. Про-
Продукты питания (хлеб, мясо, рыба, овощи), одежда и обувь
(текстильные ткани, искусственное волокно, кожа, рези-
резина, пластмассы) образованы различного рода коллоидными
системами. Изменение структуры и поглощающих свойств
почв, выветривание горных пород, вынос частиц ила и
глин реками, образование облаков и туманов — тесно свя-
связаны с коллоидными процессами. Производство строитель-
строительных материалов (цемент, гипс), добыча и переработка нефти
(бурение скважин, обезвоживание нефти), обогащение цен-
ценных руд методом флотации, производство лаков и красок,
кинофотоматериалов, бумаги, сажи, удобрений в значитель-
значительной степени основано на использовании свойств различных
суспензий и эмульсий. В фармацевтической промышлен-
промышленности многие лекарственные вещества производятся в
форме тонких суспензий или эмульсий, мазей, паст, кре-
кремов. Важное значение в промышленности, в сельском хозяй-
хозяйстве и в военном деле имеют различные дымы и туманы.
Развитие авиационной и автомобильной промышленности,
машиностроения и приборостроения было бы невозможно
без резины и различных пластмасс. Изделия из целлюлозы,
резины, пластмасс, искусственного волокна приобретают
все большее значение в технике и в быту. Можно сказать,
что материальная основа современной цивилизации и самого
существования человека и всего биологического мира свя-
связана с коллоидными системами.
ОЧЕРК РАЗВИТИЯ КОЛЛОИДНОЙ ХИМИИ
Большое практическое значение и своеобразные свой-
свойства коллоидных систем естественно привлекали внимание
исследователей в течение длительного времени, хотя осо-
особенно интенсивно коллоидная химия развивалась только
в последние десятилетия.
Первые практические сведения о коллоидах содержатся
в трудах Аристотеля и алхимиков, затем у Берцелиуса
A824—1834 гг., суспензии нерастворимых осадков), Сель-
ми A845 г. и позднее, истинные растворы и псевдорастворы),
Фарадея A857 г., изучение золей золота) и др.; в частности,
Сельми рассматривал псевдорастворы, как «распределе-
«распределение вещества в жидкой среде, не сопровождающееся раз-
разбавлением до молекул».
В трудах великого русского ученого М. В. Ломоносова
A751) имеются данные о ясном различии между явлениями
кристаллизации и свертывания, описывается получение
золей золота в воде и стекле (цветные стекла). Важное откры-
открытие адсорбции на угле было сделано Ловицем A789).
Рейсе A809) описал явления электрофореза и электроосмо-
электроосмоса. Давидов A851) разработал теорию капиллярных явле- ,
ний.
Впервые термин и понятие «коллоиды» были четко сфор-
сформулированы Грэмом A861), с появлением трудов которого
обычно связывается возникновение коллоидной химии.
Грэм разработал ряд методов приготовления и очистки
коллоидов, используя различия в диффузии и диализе,
и назвал коллоидами (что буквально означает клееобразные
вещества) такие вещества, как альбумин, желатину, гид-
гидрат окиси алюминия, не проходящие через мембраны
(неспособные к диализу), в отличие от обычных кристал-
кристаллических веществ.
В России основоположником коллоидной химии был
киевский профессор И. Борщов, в классической работе
которого «О свойствах и строении коллоидов, участвующих
в образовании растительных и животных организмов»
A869) четко сформулированы положения о сложности сос-
состава коллоидных частиц и значении связанной ими воды
для сцепления частиц. Позднее важные работы провели
И. Громека A879, развитие теории капиллярности),
Н. Любавин и А. Сабанеев A890, криоскопические опреде-
определения размеров коллоидных частиц), Ф. Шведов A889,
изучение упруго-пластичных свойств растворов желатины)
и др. Великий русский ученый Д. И. Менделеев уже в 1871 г.
указал, что «вопросы коллоидной химии должно считать
передовыми и могущими иметь большое значение во всей
физике и химии» и высказал идею о всеобщности коллоид-
коллоидного состояния. Эта идея была экспериментально широко
разработана и подтверждена Веймарном (в 1905—1916гг.),
который на сотнях объектов показал, что любое кристалли-
кристаллическое вещество при соответствующем подборе среды можно
получить в коллоидном состоянии.
К концу XIX в. в области физики и классической
физической химии был выполнен ряд фундаментальных
исследований, сыгравших позднее большую роль в разви-
развитии коллоидной химии. В их числе необходимо назвать
труды Лапласа A806, теория капиллярности), Гиббса
A878, правило фаз, теория поверхностных явлений),
Рэлея A871, теория рассеяния света), Эйнштейна A905,
теория броуновского движения, теория вязкости суспен-
суспензий), Смолуховского A906, теория броуновского движения)
и др. Работы Перрена A908) и Сведберга A912) эксперимен-
экспериментально обосновали молекулярное строение вещества и
теорию Эйнштейна—Смолуховского, а при помощи ультра-
ультрамикроскопа Зигмонди A903) удалось непосредственно наб-
наблюдать мельчайшие частицы в коллоидных раство-
растворах.
На основе этих исследований и работ Веймарна, Во. Ост-
Оствальда и др. было установлено, что в коллоидных раст-
растворах частицы находятся при высокой степени раздробления
или диспергирования, но что они гораздо больше молекул;
было найдено, что во многих случаях размер частиц приб-
приблизительно составляет от 1 до100ж[х. Степень диспергиро-
диспергирования частиц получила название дисперсности, дисперги-
диспергированные частицы — дисперсной фазы, среда, в которой
они находятся, — дисперсионной среды, а вся система
в совокупности — дисперсной системы; эти названия при-
применяются и в настоящее время. Веймарн, Во. Оствальд
и др. высказали мысль, что свойства дисперсных систем
определяются только размерами или степенью дисперсности
частиц; это представление получило выражение в предло-
предложении называть коллоидную химию дисперсоидологией.
Развитие этого представления было обусловлено тем, что
большинство известных в то время свойств коллоидных
систем — оседание под действием силы тяжести, мутность,
«Коллоидная химия» 9
способность к диализу и др., действительно в значительной
мере зависели от размеров частиц.
Однако дальнейшие исследования коллоидных систем,
особенно изучение зависимости их устойчивости от нали-
наличия и концентрации электролитов в растворе, детальное
изучение движения частиц в электрическом поле показали
недостаточность представлений дисперсоидологии для по-
понимания свойств коллоидных систем. Экспериментальные
данные по осаждению коллоидов электролитами (коагуля-
(коагуляция коллоидов) получили Шульце A882) и Гарди A900),
позднее обширные исследования произвели Фрейндлих
и Кроит; теорию кинетики коагуляции разработал Смолу-
ховский A916); большое значение имело также развитие
работ по теории адсорбции и строению поверхностных и
мономолекулярных слоев A917, Лангмюр; 1890, Рэлей и
др.). В.России в этот период важные работы провел Ду-
манский (с 1903 г., измерения электропроводности в кол-
коллоидных растворах, в 1913 г. применение центрифуги для
определения размеров частиц), который с 1912 г. начал
читать первый курс коллоидной химии. Весьма важным
явилось открытие хроматографии Цветом A903), иссле-
исследования поверхностного натяжения растворов Антоновым
A907) и Шишковским A908), исследования по адсорбции
Титова A910), Шилова A912) и Гурвича A912), создание
противогаза Зелинским A916) и т. д.
Наиболее четко критику дисперсоидологии дал Песков
A917), показавший, что свойства коллоидных систем зависят
не только от размеров частиц, но в гораздо большей мере—
от наличия поверхностей раздела со значительной свобод-
свободной поверхностной энергией. Песков отделил понятие кине-
кинетической устойчивости, обусловленной скоростью оседа-
оседания частиц (зависящей от их размера), от устойчивости
частиц к взаимному слипанию, которую он назвал агрега-
тивной устойчивостью; он указал, что коллоидным систе-
системам, вследствие их многофазности (гетерогенности) свойст-
свойственна агрегативная неустойчивость, преодолеваемая лишь
путем адсорбции ионов или других стабилизирующих
веществ на частицах дисперсной фазы. Таким образом,
агрегативно устойчивая коллоидная система в принципе
должка состоять из трех компонентов: диспергированных
частиц, среды и стабилизатора. Позднее аналогичная кон-
концепция была широко развита в работах Кройта, Фрейндли-
ха и их школ. Она давала объяснение основным свойствам
ю
коллоидных систем и отвечала установленному многими
методами строению коллоидных дисперсий или золей ме-
металлов, гидроокисей и сульфидов металлов и других не-
нерастворимых в воде веществ (гидрофобных золей). Было
показано, что частицы золей состоят из нерастворимого
ядра, на поверхности которого адсорбированы стабили-
стабилизирующие ионы, причем второй слой ионов противополож-
противоположного знака находится в растворе вокруг частицы; такая
сложная, в целом электронейтральная частица, называется
мицеллой. Эта картина строения частиц гидрофобных
золей остается правильной и в настоящее время (см. стр.
104). Следует отметить, что гидрофобные золи (Аи, Ag,
As2S3, Fe2O3) и др. в течение длительного времени были
основными объектами исследований в коллоидной химии.
Вторая группа коллоидных систем, отличавшаяся высо-
высокой устойчивостью к действию электролитов и сравнитель-
сравнительно хорошей растворимостью (белки, агар, желатина, крах-
крахмал и др.), называемых поэтому гидрофильными коллои-
коллоидами, изучалась по аналогии с гидрофобными золями.
Предполагалось, что частицы гидрофильных коллоидов
также состоят из нерастворимого ядра, на поверхности
которого, однако, адсорбированы не ионы, а электроней-
электронейтральные молекулы неэлектролитов (молекулярный стаби-
стабилизатор), чем обусловлена их сравнительно малая чувстви-
чувствительность к электролитам. Различия в свойствах гидрофиль-
гидрофильных и гидрофобных коллоидов (или, с включением систем
с органическими растворителями — лиофильных и лиофоб-
ных коллоидов) объяснялись различием в интенсивности
взаимодействия частиц этих коллоидов с растворителем,
сильным связыванием растворителя (сольватацией) в
лиофильных коллоидах.
Изложенные представления господствовали в коллоид-
коллоидной химии приблизительно до 30-х годов. Для развития
коллоидной химии большое значение имели работы класси-
классической физической химии этого периода: теория силь-
сильных электролитов Дебая и Гюккеля A923), рентгеногра-
рентгенографический метод Дебая и Шеррера A916), работы по строе-
строению двойного электрического слоя А. Н. Фрумкина (с1921г.).
Детальное изучение природы лиофильных коллоидов
стимулировалось все более интенсивным развитием промыш-
промышленности эфиров целлюлозы, натурального и синтетичес-
синтетического каучука, искусственного волокна, полиамидов, поли-
полиэфиров и других высокомолекулярных веществ или высоко-
1*
11
полимеров включавшихся в эту группу коллоидов, а также
разработкой ряда новых специальных методов исследова-
исследования. Успехи органической химии позволили синтезировать
большое количество новых важных видов высокополиме-
ров и неопровержимо доказали наличие в них длинных
цепных макромолекул с различной степенью гибкости
Многие физики (Кобеко, Александров, Кун, Флори и др)
выясняли природу механических и диэлектрических про-
процессов в высокополимерах и обосновали статистику гибких
макромолекул. Работами Каргина и его школы было
показано, что растворы высокомолекулярных веществ
подчиняются правилу фаз и являются термодинамически
ооратимыми молекулярными гомогенными (однофазными)
системами, способными сохранять агрегативную устойчи-
устойчивость без стабилизатора, в двухкомпонентном растворе-
в этом же направлении работали школы Мейера, Флори
и др. Проведенные исследования позволили отказаться от
прежних представлений Марка и Мейера о наличии в полиме-
полимерах постоянных кристаллических образований (тоже называ-
называемых мицеллами) с фазовой поверхностью раздела Свойст-
Свойства растворов полимеров оказались обусловленными не особо
высокой сольватацией, а в значительной мере теми же свой-
свойствами длинных цепных макромолекул, которые определяют
и поведение твердых полимерных материалов вне раствора
1аким образом, коллоидные системы оказались разделен- •
ными на дисперсные системы и высокомолекулярные ве-
вещества с их растворами: это деление существует и в настоя-
настоящее время. Не следует думать, что рассмотрение растворов
высокомолекулярных веществ, как лиофильных коллоидов
ооладающих высокой сольватацией, было простой ошиб-
ошибкой; как и во всяком значительном этапе развития науки
в этом представлении была определенная доля истины'
Ь настоящее время имеются довольно подробные данные о
количестве прочно связанного растворителя в растворах
простых электролитов, неэлектролитов, высокомолекуляр-
высокомолекулярных веществ и лиофобных золей; отношение молекуляр-
молекулярной массы на один моль связанного растворителя MIS
может служить обратной характеристикой относительной
сольватации растворенных веществ (она тем меньше, чем
больше MIS). Соответствующие данные, приведенные в
табл. 1, ясно показывают, что высокомолекулярные ве-
вещества в растворах действительно превосходят по сольвата-
сольватации лиофобные коллоиды на два порядка.
12
Таблица 1
Относительная сольватация растворенных
веществ и коллоидов
Вещество
Мол. вес, М
Сольватация
S, мол/мол
M/S
Na+
к+
Этиловый спирт . . .
Мочевина
Рафиноза
Яблочная кислота . .
Желатина •. • • • •
Нитроцеллюлоза
(в ацетоне) ....
Этилцеллюлоза (в аце-
ацетоне)
Золь As2S3
23
39,1
46
60
504
119
30000
252
(на звено)
218
(на звено)
г=15,8 Ж{л
заряд=86
электронов
6
6
2
4
19
11
500 (=0,3 г/г)
2
1,2
со 200
4
6,5
23
15
26
11
60
116
170
со 50 000
Таким образом, существо вопроса заключается не в том,
являются ли лиофильные коллоиды действительно «лио-
фильными», а в том, что наиболее характерные и своеоб-
своеобразные свойства растворов высокомолекулярных веществ
объясняются наличием длинных цепных молекул (см. вось-
восьмую главу), а не сольватацией, хотя для многих полярных
полимеров и белков сольватация остается главным фак-
фактором устойчивости их растворов; заметную роль играет
гидратация также в устойчивости таких коллоидов, как зо-
золи кремнекислоты, гидроокиси алюминия и др.
Важно отметить, что каждый этап развития коллоид-
коллоидной химии не был простым отрицанием предыдущего, а
преодолением исторической ограниченности предыдущего
этапа. Современное деление коллоидных систем на лиофоб-
лиофобные дисперсные системы и растворы высокомолекулярных
веществ не отрицает лиофильности последних, а подчерки-
подчеркивает невозможность сведения к лиофильности принципиаль-
принципиально иных свойств молекулярных растворов полимеров. Уста-
Установление природы агрегативной устойчивости коллоидных
систем не умаляло роли размеров коллоидных частиц, а
выяснило невозможность сведения всех свойств коллоид-
13
ных систем к количественным различиям в размерах частиц.
Как будет видно из последующих глав, современная кол-
коллоидная химия уделяет исключительное внимание изуче-
изучению размеров и формы частиц и учету роли этого фактора
в свойствах коллоидных систем, но эти исследования, ра-
разумеется, не имеют ничего общего с дисперсоидологией.
Наконец, дисперсоидология," установив всеобщность кол-
коллоидного состояния, не уничтожила его различий с кристал-
кристаллическим состоянием, а явилась преодолением прежнего
упрощенного противопоставления кристаллоидов и колло-
коллоидов. Таким образом, каждый этап развития знаний о кол-
коллоидах соответствовал все более широкому обобщению.
Несомненно, что и современный этап развития коллоид-
коллоидной химии не является окончательным и, возможно, что
развитие новых синтезов неорганических полимеров суще-
существенно изменит известную в настоящее время картину.
Не представляется возможным кратко охарактеризовать
работы последних десятилетий; этому в значительной мере
посвящена вся книга, насколько это допускается ее рамками.
Необходимо отметить, что в ряде основных проблем уче-
учения о дисперсных системах и высокомолекулярных соеди-
соединениях, благодаря работам Н. П. Пескова, И. И. Жукова,
А. И. Рабиновича, А. И. Рабинерсона, А. В. Думанского,
П. А. Ребиндера, В. А. Каргина, С. М. Липатова, Б. В, Де-
рягина, Д. Л. Талмуда, С. И. Соколова, Б. А. Догадкийа
и др. советская коллоидная химия занимает ведущее ме-
место в мировой науке. Коллоидная химия всегда развивалась
на базе достижений физики, физической химии и органиче-
органической химии. Это развитие в высокой мере стимулировалось
потребностями народного хозяйства и здравоохранения.
ПРИРОДА КОЛЛОИДНЫХ СИСТЕМ
Коллоидная химия, как указывалось выше, изучает
свойства систем с крупными частицами. С учетом исто-
исторического развития коллоидной химии можно дать следу-
следующее более точное определение предмета коллоидной химии.
Коллоидная химия—это физико-химия гетерогенных
высокодисперсных систем и высокомолекулярных систем*.
* «Коллоидная химия, будучи самостоятельным разделом фи-
физической химии, ставит своей задачей изучение, пользуясь ее ме-
методами, с одной стороны, свойств высокодисперсных, простираю-
простирающихся до молекулярных размеров гетерогенных систем, кладя в
14
Как видно из определения, к коллоидным системам
относятся два основных типа систем. Первому типу —
гетерогенным высокодисперсным системам — соответствует
первый указанный ранее тип укрупнения частиц путем
образования трехмерных и двухмерных структур в инерт-
инертной среде; он характеризуется наличием развитой поверх-
поверхности раздела. Условие высокодисперсности отделяет
коллоидные системы от грубых, быстро оседающих суспен-
суспензий и порошков с низкой кинетической устойчивостью.
Ввиду наличия частиц со свободной поверхностной энергией,
коллоидные дисперсные системы являются термодинами-
термодинамически неустойчивыми, потому что стремление этой энергии
к уменьшению приводит к агрегации частиц (см. четвертую
главу). Частицы не слипаются, т. е. оказываются агрега-
тивно устойчивыми лишь при условии, что на их поверх-
поверхности за счет свободной поверхностной энергии адсорби-
адсорбируются молекулы или ионы третьего компонента системы
или стабилизатора. Однако агрегативная устойчивость
этих частиц имеет индуцированный характер, и по истече-
истечении достаточного промежутка времени (путем рекристал-
рекристаллизации и др.) процесс слипания неизбежно наступает.
В этом смысле коллоидные дисперсные системы являются
необратимыми системами. Таковы основные черты первого
типа коллоидных систем, которые характеризуются, по
Пескову, как гетерогенные высоко дисперсные системы,
обладающие агрегативной устойчивостью только в присут-
присутствии стабилизатора.
Второй основной тип систем — высокомолекулярные си-
системы — соответствует второму структурному типу укрупне-
укрупнения частиц, ведущему к образованию цепных макромолекул.
Они дают при смешении с растворителями молекуляр-
молекулярные растворы, подобные обычным растворам низкомо-
низкомолекулярных веществ, но с очень длинными цепными моле-
молекулами. Такие растворы относятся к однофазным (гомоген-
(гомогенным) системам, как и растворы сахара или мочевины, они
образуются самопроизвольно, потому что сам процесс
растворения идет с уменьшением свободной энергии и не
требует наличия стабилизаторов. Растворы оказываются
вполне устойчивыми, независимо от длительности их сущест-
основу поверхностные явления на границе фаз; с другой стороны —
изучение физико-химических свойств высокомолекулярных и высо-
высокополимерных соединений как в твердом состоянии, так и в состоя-
состоянии раствора» (И. И. Жуков).
15
вования. Они являются молекулярными термодинамичес-
термодинамически равновесными и поэтому обратимыми системами в том
же смысле, в каком обратим, например, 10%-ный раствор
сахара при 20°, всегда обладающий определенными свой-
свойствами, независимо от пути его получения. Таким образом,
ко второму типу коллоидных систем относятся термодина-
термодинамически обратимые молекулярные гомогенные системы.
Вследствие большой длины молекулярных цепей,
коллоидные системы второго типа в ряде отношений отли-
отличаются от растворов низкомолекулярных веществ (глава
восьмая). Важно, что эти отличия характеризуют те же
свойства макромолекул, которые определяют их поведение
и в твердых полимерах (глава десятая). Поэтому твердые
полимеры изучаются в коллоидной химии наряду с их
растворами. Однако в инертной, нерастворяющей среде
полимеры образуют дисперсии со свободными поверхно-
поверхностями раздела, которые по своим- свойствам относятся к
первому типу систем.
В связи с тем, что устойчивость коллоидных систем в
значительной мере связана с их фазовой гетерогенностью
или гомогенностью, вопрос об определении фазы в колло-
коллоидных системах весьма интересен. Как известно, фаза пред-
представляет собой гомогенную часть системы, в пределах
которой температура, давление и удельный объем одинаковы
и которая отделена от другой фазы поверхностью раздела.
Как указывает Ван-дер-Ваальс, с молекулярно-теорети-
ческой точки зрения требуется, чтобы фаза содержала очень
большое число молекул, оправдывающее применение зако-
законов термического равновесия; гомогенная фаза должна
обладать достаточно большими размерами, чтобы можно
было экспериментально определить ее плотность, давление
и температуру, а находящееся вблизи поверхности раздела
количество вещества должно быть исчезающе мало по срав-
сравнению с той частью, которая находится внутри этой
фазы.
Для низкомолекулярных систем, изучаемых в курсе
физической химии, эти условия обычно удовлетворяются;
для отдельных макромолекул в растворе условия фазы
обычно отсутствуют, хотя Лангмюр считал возможным рас-
рассматривать энергию сил притяжения между большими мо-
молекулами как поверхностную энергию. В коллоидных дис-
дисперсных системах частицы, очевидно, должны быть доста-
достаточно большими, чтобы положение о наличии у них поверх-
16
ХностИ раздела имело определенный физический смысл.
* В этом отношении указание о приближенном интервале
размеров коллоидных частиц от 1 до 100 м\* (стр. 9),
хотя и не должно пониматься слишком строго, но все же
имеет то значение, что указывает приблизительные пределы
максимального развития удельной поверхности (см. стр.
74), выше которых частицы постепенно теряют кинети-
кинетическую устойчивость, а ниже — утрачивается физическая
поверхность раздела. Аналогично в цепных молеку-
молекулах должно содержаться достаточное число звеньев для того,
чтобы могли проявиться особые свойства макромолекул
(см. восьмую и десятую главы). Таким образом ясно, что
необходима определенная степень укрупнения частиц,
количественного увеличения их размеров для того, чтобы
в системах появились качественно новые свойства, в одном
типе систем связанные с доминирующей ролью поверхност-
поверхностной энергии, а в другом — с особыми свойствами цепных
макромолекул.
Вопрос осложняется в тех случаях, когда частицы в кол-
коллоидных системах находятся как бы на грани возникнове-
возникновения или исчезновения фазы, например, в случае кри-
критических и самопроизвольных эмульсий (стр. 155),
случайных статистических роев и ассоциаций больших
молекул, растворов мыл и детергентов (стр. 120), явле-
явлений кристаллизации в полимерах (стр. 232) и др.
Картин и Слонимский указывают на возможность расхож-
расхождения в этих случаях структурных и термодинамических
критериев фазового состояния, проявления гомогенности
по одним свойствам и гетерогенности — по другим. Однако
эти интересные вопросы еще являются предметом научного
обсуждения.
Важно отметить, что для многих свойств коллоидных
систем наличие или отсутствие поверхности раздела не
имеет существенного значения, хотя, как мы видели, оно
весьма важно в проявлении устойчивости. Такие важные
процессы, как установление равновесия при поглощении
ионов электролитов или нейтральных молекул (электро-
(электрохимическое, сольватационное равновесие и др.) зависят
лишь от природы и плотности расположения ионогенных
или полярных групп, но не от того, находятся ли эти группы
на дисперсной частице или макромолекуле, возникли ли
они путем адсорбции из раствора или диссоциации групп
на поверхности. Различными авторами было показано, что
17
при большом числе ионогенных групп диссоциация полиос-
полиосновных кислот приводит к тем же результатам, что и адсорб-
адсорбция ионов на поверхности раздела; доннановское равновесие
(пятая глава) также не зависит от природы фиксированных
коллоидных частиц, много общего наблюдается в обменной
адсорбции ионов и др. Таким образом, большинство проб-
проблем электрохимического равновесия можно рассматривать
одновременно для лиофобных коллоидов, белков и поли-
полиэлектролитов (см. пятую главу). Точно также динамичес-
динамическое равновесие при связывании сотен молекул воды на мо-
молекуле белка или сотен молекул ацетона на молекуле
нитроцеллюлозы не отличимо от равновесия сольватации
на дисперсных частицах. Образуется ли гидратный слой
на молекуле белка, поливинилового спирта или силикаге-
ле, — во всех случаях процесс идет с положительным теп-
тепловым эффектом, низкой начальной упругостью пара,
понижением энтропии растворителя и т. д. (восьмая и де-
девятая главы). Думанский нашел, что для самых различных
веществ теплота связывания 1 г воды составляет 80 кал
(восьмая глава). Особенности молекулярно-кинетических
свойств коллоидных систем — медленность диффузии, низ-
низкое осмотическое давление, неспособность к диализу, усло-
условия оседания и др. (вторая глава) — не зависят от наличия
поверхности раздела, а определяются лишь размерами и
формой частиц. При изучении механических свойств поли-
полимерных и структурированных коллоидных систем поль-
пользуются общими методами анализа объективных констант
деформации (десятая глава). В явлениях защитного эффек-
эффекта (шестая глава), действия наполнителей в полимерах
(десятая глава) поверхностные свойства диспер-
дисперсных частиц и свойства макромолекул непосредственно
связаны между собою. Много общего имеется в диэлектри-
диэлектрических свойствах (пятая глава), оптических свойствах
(например, в явлениях светорассеяния, в двойном луче-
лучепреломлении при течении, третья глава), гидродинамических
свойствах (восьмая глава).
С другой стороны, необходимо отметить значительные
различия в свойствах, вызываемые гибкостью макромоле-
макромолекул ряда полимеров (восьмая— десятая главы). Возмож-
Возможность изменения конфигурации и плотности расположе-
расположения активных центров в зависимости от внешних условий
является качественно новой особенностью, которая часто
резко отличает гибкие макромолекулы не только от дисперс-
IS
ных частиц, -но и от жестких макромолекул, обладающих
более фиксированной конфигурацией. Это различие прояв-
проявляется в термодинамических свойствах растворов полиме-
полимеров (восьмая глава), в свойствах эластичных гелей (девятая
глава), в электрохимических свойствах растворов полиэлек-
полиэлектролитов (пятая глава), в ряде молекулярно-кинетических
и оптических свойств растворов полимеров (восьмая глава),
в высокоэластических свойствах и кристаллизуемости по-
полимеров (десятая глава), в хроматографическом использо-
использовании ионообменных адсорбентов (девятая глава), в диэлект-
диэлектрических свойствах полимеров (десятая глава) — короче
говоря, оно проходит красной нитью через многие пробле-
проблемы коллоидной химии.
Таким образом, при изучении коллоидной химии
необходимо, с одной стороны, учитывать принципиальные
различия в природе устойчивости между основными типами
коллоидных систем, а, с другой стороны, сложную картину
влияния ряда факторов — гибкости или жесткости струк-
структурных элементов, размеров и формы частиц, условий ад-
адсорбционного равновесия и др., определяющих многообраз-
многообразные свойства коллоидных систем, их общность и различия,
но не сводящихся только к проблеме устойчивости. В этом
отношении сравнительное изучение коллоидных систем
способствует углубленному пониманию их характерных
особенностей подобно тому, как это достигается при изу-
изучении организмов методами сравнительной анатомии и
физиологии.
Итак, мы дали общую характеристику основных осо-
особенностей коллоидных систем в качестве введения к изу-
изучению последующих глав; однако этот раздел в известной
мере рассчитан на повторный просмотр после окон-
окончания курса. В заключение отметим, что коллоид-
коллоидная химия изучает закономерности систем с круп-
крупными комплексами молекул, связанных между собой раз-
разнообразными химическими связями или силами межмоле-
межмолекулярного взаимодействия; эти закономерности еще лежат
в пределах химической формы движения. Следующей сту-
ступенью являются системы сложных молекул, связанных
между собой уже не только различными силами взаимодей'
ствия, но и упорядоченной последовательностью хими-
химических превращений, — системы, изучаемые биохимией.
19
В предмете биохимии появляются уже качественно новые,
более высокие закономерности биологического порядка.
Таким образом, коллоидная химия является важным зве-
звеном, непосредственно связывающим химические науки
с биохимией и биологией.
МЕТОДЫ ПОЛУЧЕНИЯ КОЛЛОИДНЫХ СИСТЕМ
Получение коллоидных систем в инертной, нерастворяю-
щей среде, как уже указывалось, требует доведения вещест-
вещества до определенной степени дисперсности и наличия ста-
стабилизатора. Первая задача может быть решена двумя
путями — диспергированием более крупных частиц или
конденсацией более мелких частиц.
Методы диспергирования технически осуществляются
путем дробления, измельчения, истирания на дробилках,
жерновах, шаровых и вибрационных мельницах и др.
Очень тонкое раздробление (до 0,1—1 ц.) достигается на спе-
специальных коллоидных мельницах с узким зазором между
быстро вращающимся ротором A0—20 тыс. об/мин) и
неподвижным корпусом, причем частицы разрываются
или истираются в зазоре. Диспергирование обычно ведут,
добавляя стабилизирующие вещества, препятствующие сли-
слипанию раздробленных частиц.
В природе коллоидные системы образуются преимущест-
преимущественно путем механического диспергирования — при обва-
обвалах, выветривании, эрозии почв, замерзании воды в трещи-
трещинах и др.
Дисперсии металлов получают путем распыления под
водой или в органической жидкости в вольтовой дуге
(Бредиг), или в высокочастотном разряде (Сведберг),
хотя в этом случае большое значение имеет конденсация
паров металлов. Эмульсии получают путем диспергирова-
диспергирования действием ультразвука. При этом всегда образуются
различные окисленные продукты, стабилизирующие сус-
суспензии.
Методы конденсации-агрегации основаны на переходе
от молекулярных растворов к коллоидным системам путем
перевода веществ в нерастворимое состояние при помощи
различных химических реакций (восстановления, гидро-
гидролиза, двойного обмена и др.) с последующей агрегацией
и рекристаллизацией нерастворимых частиц; например,
для получения коллоидных растворов AgJ или Fe(OH)s
используются соответственно реакции
20
AgNOs + KJ= AgJ + KNOs;
FeCls + 3H2O-*Fe(OHK + 3HC1.
Стабилизаторами являются химические вещества, при-
присутствующие в среде. Эти методы непосредственно связаны
с возникновением новой фазы. Рост частиц происходит
"на существующих или вносимых в систему центрах или
зародышах кристаллизации, например, пылинках, неболь-
небольших добавках готового золя и др. При большом числе цент-
центров роста частиц получаются более высокодисперсные
системы, чем при малом числе зародышей. Рогинский и
Шальников разработали способ получения золей из так
называемых молекулярных пучков путем совместного испа-
испарения в вакууме вещества А и инертной
жидкости В (рис. 4); пары смешиваются в
объеме и конденсируются на поверхности
С, охлаждаемой жидким воздухом. После
удаления последнего золь оттаивает и сте-
стекает в сосуд D. Этим путем были получе-
получены многие труднодоступные золи.
При конденсации слипание частиц
происходит случайным, неупорядоченным
образом, вследствие чего частицы вна-
вначале получаются аморфными, но затем
они переходят в кристаллическое состоя-с"
ние, как это показано Каргиным и Бере-
стневой при помощи прямых измерений
электронной дифракции (стр. 68). Так,
например, золь золота, полученный
восстановлением хлорного золота белым
фосфором, оставался в аморфном состоя- ,,
нии лишь 5—10 минут, золь серебра —
около часа, золь ТЮг—15—20 минут, Рис. 4. Схема
а золь АЦОНЬ начинал кристаллизоваться прибора Ропш-
лишь через сутки. """ISJZ"*
В фармацевтической промышленности
некоторые препараты получают восста-
восстановлением металлов в присутствии защитных веществ (см.
стр. 147), например, препараты коллоидного серебра, защи-
защищенного солями лизальбиновой и протальбиновой кислот
(коларгол), или коллоидной окиси серебра, защищенной аль-
альбумином (протаргол) и др. В природе туманы и облака обра-
21
зуются при конденсации водных паров в атмосфере.
Растворы высокомолекулярных веществ, как уже указы-
указывалось, возникают самопроизвольно, без добавок стабили-
стабилизирующих веществ. В зависимости от условий растворения—
полного или ограниченного, образуются растворы, набух-
набухшие студни или продукты их расслоения (см. восьмую и
девятую главы).
Высокомолекулярные вещества получают из низкомоле-
низкомолекулярных веществ двумя основными методами: полимери-
полимеризации и поликонденсации, которые отличаются между со-
собой как методы присоединения или замещения в химии.
Молекулы исходных низкомолекулярных веществ, обра-
образующих полимер, называются мономерами, а в составе длин-
длинной цепной молекулы или макромолекулы полимера они
часто называются звеньями цепи (эти названия уже упот-
употреблялись выше); их число в макромолекуле, которое может
достигать нескольких тысяч, называется степенью поли-
полимеризации.
Полимеризацией называется соединение молекул низко-
низкомолекулярного вещества (мономеров) без изменения эле-
элементарного состава полимера по сравнению с исходными
веществами; например, поливинил хлор ид—(СШ—СНС1)Л—
образуется путем полимеризации п молекул винилхло-
рида п (СН2 = СНС1) за счет раскрытия двойных связей.
Продукты полимеризации разнородных мономеров назы-
называются сополимерами, например, сополимер винилхлорида
и винил ацетата:
х СН2 = СНС1 + х СН2 = СНОСОСНд >
(—сна—сна — сн2 — сн —)х.
О—СНдСО
Поликонденсацией называется образование полимера из
мономеров с отщеплением воды, спирта и других низкомо-
низкомолекулярных веществ, вследствие чего по элементарному
составу продукт поликонденсации отличается от исходных
веществ. Например, белки образуются путем поликонден-
поликонденсации аминокислот 2n(NH2CHRCOOH) -»¦ Н — (— NH—
—CHR — СО—NH—CHR—СО—)„—ОН + Bя—1)Н2О.
Таким образом, одновременно с продуктом поликонден-
поликонденсации всегда появляется низкомолекулярное вещество,
что характеризует эти реакции как реакции обмена.
22
Некоторые высокомолекулярные вещества могут быть
получены обоими методами, например, полиэтиленоксид
можно получить полимеризацией окиси этилена или поли-
поликонденсацией этиленгликоля (по Коршаку):
— СН9
(_СН2-СН2-О-);
(_СН2-СН2-О-Ь+ Н2О.
или
*HOCH2 — СН2ОН
ОСНОВНЫЕ ВИДЫ КОЛЛОИДНЫХ СИСТЕМ
Коллоидные системы, как указывалось, различаются
по химическому составу и структуре частиц или макромо-
макромолекул и по составу среды.
Гетерогенные высокодисперсные коллоидные системы в
большинстве .случаев существуют в виде дисперсий трехмер-
трехмерных и двухмерных структур как аморфных (включая жид-
жидкости), так и относящихся к различным кристаллическим
решеткам — ионной, атомной, молекулярной или металли-
металлической. Дисперсионной средой обычно является нераство-
ряющая жидкость, реже газы. Наличие высокоразвитой
поверхности раздела — наиболее характерная черта, общая
для всех коллоидных систем этого рода.
. Коллоидные дисперсные системы твердых частичек в
воде называются гидрозолями, в органических жидкостях —
органозолями; при низкой дисперсности они называются
суспензиями.
Приводим некоторые примеры гидрозолей:
Гидрозоль
Аи, Ag. Pt, Си, Hg
S, Р
Fe (ОНK; А1(ОНK; V2O5; Cr2O3; WO3;
МоО3; SiO2
As2 S3; Sb2 S3; CuS; HgS
AgJ; AgBr; берлинская лазурь
Натуральные и синтетические латексы,
дисперсии нитроцеллюлозы в воде,
желатины в спирте и др.
Свойства гидрозолей подробно рассмотрены ниже — в
главах третьей — шестой.
23
Вещество
Металлы
Неметаллы
Окислы и гидроокиси
Сульфиды
Соли
Полимеры
Дисперсии жидкостей в жидкостях называются эмуль-
эмульсиями, а газов в жидкостях — пенами; дисперсии твердых
и жидких частиц в газах называются аэрозолями. Свойства
этих видов дисперсных систем описаны в главе седьмой.
Высокомолекулярные системы образуются преимущест-
преимущественно на основе цепных линейных структур, звенья ко-
которых связаны между собой прочными химическими связя-
связями, вследствие чего молекулярные цепи сохраняются как
в твердых полимерах, так и в растворах. К высокомолеку-
высокомолекулярным системам относятся различные полимеры с линей-
линейными гибкими макромолекулами (каучуки, эластомеры),
линейными жесткими макромолекулами (целлюлоза и ее
эфиры), спиральными макромолекулами (белки, нуклеи-
нуклеиновые кислоты), разветвленными макромолекулами (крах-
(крахмал, гликоген) и др. Свойства этих систем подробно рассмот-
рассмотрены в ряде последующих глав (см. главы восьмую —
десятую). Приводим некоторые примеры линейных поли-
полимерных веществ, которые удобно записать следующей ти-
типовой формулой:
—СН2—CHR—СН2—CHR — СН2 — CHR—
Название полимера R
Полиэтилен (политен) —Н
Полихлорвинил —С1
Полистирол .... —С6Н3
Полиакрилонитрил —CN
Полиакриловая кислота —СООН
Поливиниловый спирт —ОН
Поливинилацетат —ОСОСН3
Полиметакрилат —СООСН3
Большое значение имеют различные сополимеры (см.
стр. 22); в последнее время их получают также в форме блок-
полимеров, в которых группа звеньев одного мономера А
чередуется с группой звеньев другого мономера В:
— Ап Вт Ап Вт кп Вт..., и графтполимеров или привитых
полимеров, построенных по типу
—А—А—А—А—А—А—
В,
В,
При помощи соответствующего чередования блоков и от-
ответвлений и подбора гидрофобных и гидрофильных цепей
24
удается широко изменять свойства полимеров. Оба эти
вида полимеров обладают более высокой упорядоченностью
строения, чем обычные сополимеры. Крупным достижением
в получении упорядоченных полимеров является синтез
стереоспецифических (или изотактических) полимеров. Так
например, в присутствии катализатора Циглера
АЦСгНбЬ + TiCU был получен чистый цш>1,4-полиизо-
прен, идентичный с натуральным каучуком, кристалли-
кристаллический полипропилен, а также кристаллический полисти-
полистирол, в котором вместо случайного расположения СбНб-
групп в обычном полимере (стр. 24) существует строго
упорядоченное чередование ряда D-конфигураций с рядом
L-конфигураций (Натта). Благодаря высокой упорядочен-
упорядоченности строения, стереоспецифические полимеры легко кри-
кристаллизуются и обладают рядом новых ценных качеств
(более высокой прочностью, теплостойкостью и др.) по
сравнению с обычными полимерами. Эти результаты пока-
показывают значение для свойств полимера не только химичес-
химического состава, но и пространственного строения цепи, и
приближают синтетические полимеры к высокоупорядочен-
ным структурам биологического происхождения.
Наконец, большой интерес представляют различные
элементоорганическке полимеры, содержащие неоргани-
неорганические элементы в цепях молекул. Среди них важное зна-
значение имеют кремнийорганические полимеры или полиор-
ганосилоксаны (Андрианов), в которых неорганическая
цепь из атомов кремния и кислорода (как в кварце) окру-
окружена углеводородными радикалами R,
R R R
I I I
—Si —О—Si —О—Si —О— ,
R
R
R
где R = СНз, С2Н5, С6Н5идр.
Эти полимеры обладают высокой термостойкостью
и ценными изоляционными свойствами и производятся в
виде жидкостей, эластомеров и смол. Развитием синтеза
этой группы полимеров является получение полиоргано-
металлосилоксанов, в состав неорганической цепи которых,
кроме атомов кремния и кислорода, входят атомы различных
металлов (как в природных силикатах) и других элементов.
Эти полимеры имеют общую формулу:
25
R R
_O_Si—O—E—O—Si—O—,
R
R
E-
OR R
— Al— ; — Ti —; —Sn—; — В
OR
OR
R
R
Sb—;
О
— P— и др.
OR
OC2H5
Они дают материалы с высокой термостойкостью, хорошо
растворимые в органических растворителях. Кроме того,
получены также полимеры с неорганической цепью, не
содержащей кремния; он входит лишь в боковые радикалы.
Такие полимеры называются полиорганосилил (элемент)
океаны и имеют общую формулу:
_Е_О—Е—О—Е—О,
OSiRg
I
где Е = —Ti —;
А1 —
и др.
OSiR3 OSiR3
Получены полимеры, вообще не содержащие кремния,
например
С6Н5
который, однако, при п = 3 выдерживает нагревание до
500 °. Таким образом, указывает Андрианов, не только
углерод и кремний могут использоваться для образова-
образования цепей полимерных молекул, как считалось еще недав-
недавно, но и алюминий, титан, бор, фосфор, магний и многие
другие элементы второй, третьей, четвертой и пятой групп
26
периодической системы элементов Д. И. Менделеева могут
участвовать в синтезе полимеров. Боковые органические
радикалы связывают эти полимеры с органическими
высокомолекулярными соединениями, а неорганические
цепи молекул сближают их с такими неорганиче-
неорганическими веществами, как кварц, силикаты, корунд, поли-
титанаты и др. При синтезе этих полимеров их легко полу-
получить не только с линейными, но также с неорганическими
разветвленными и пространственными цепями, что еще бо-
более сближает их со структурами неорганических веществ.
Несомненно, что при неограниченных перспективах полу-
получения различных веществ, которые открываются в этой об-
области синтеза полимеров, будет создан непрерывный пере-
переход между известными в настоящее время основными типа-
типами коллоидных систем.
Выводы
Коллоидная химия — это физико-химия гетерогенных
высокодисперсных систем и-высокомолекулярных систем.
Коллоидные системы имеют чрезвычайно большое биоло-
биологическое и народнохозяйственное значение. Гетерогенные
высокодисперсные системы обладают агрегативной устой-
устойчивостью только в присутствии стабилизатора (ионного
или молекулярного); растворы высокомолекулярных ве-
веществ являются термодинамически устойчивыми молекуляр-
молекулярными гомогенными системами. По структуре частиц системы
первого рода состоят из осколков трехмерных и двухмерных
кристаллических и аморфных тел, образующих в инертной
среде поверхности раздела фаз; они получаются методами
диспергации и конденсации-агрегации; к ним относятся,
например, гидрозоли металлов, металлоидов, гидроокисей
и сульфидов металлов, дисперсии высоко полимеров.
Высокомолекулярные системы образуются методами
полимеризации и поликонденсации из длинных цепных
молекул или макромолекул; к ним относятся многочислен-
многочисленные органические полимеры с линейными гибкими и
жесткими, спиральными и разветвленными макромолеку-
макромолекулами, различные сополимеры, стереоспецифические поли-
полимеры и различные элементоорганические полимеры, являю-
являющиеся переходом к неорганическим полимерам.
Резко различаясь по природе устойчивости, оба основ-
основных типа коллоидных систем имеют много общего в свойствах
27
обусловленных размерами и формой частиц, и в разнообраз-
разнообразных свойствах, не зависящих от наличия поверхности раз-
раздела. Изменения химического строения частиц и состава
среды создают многочисленные переходы между коллоид-
коллоидными системами.
Вопросы
1. Каковы основные структурные типы полимолекулярных
частиц, как они зависят от состава среды?
2. Охарактеризуйте основные этапы развития коллоидной химии.
3. Какова природа устойчивости дисперсных коллоидных
систем? Чем определяется устойчивость растворов высокомолеку-
высокомолекулярных веществ?
4. Каково значение размеров частиц, наличия поверхности
раздела, гибкости частиц для свойств коллоидных систем?
5. Охарактеризуйте предмет и задачи коллоидной химии.
6. Каковы основные методы получения дисперсных и поли-
полимерных коллоидных систем?
7. Назовите основные виды дисперсных коллоидных систем.
8. Назовите основные виды высокомолекулярных коллоидных
систем.
9. В каком отношении находится коллоидная химия к хими-
химическим и биологическим наукам?
10.- Охарактеризуйте биологическое и народнохозяйственное
значение коллоидных систем.
Глава вторая
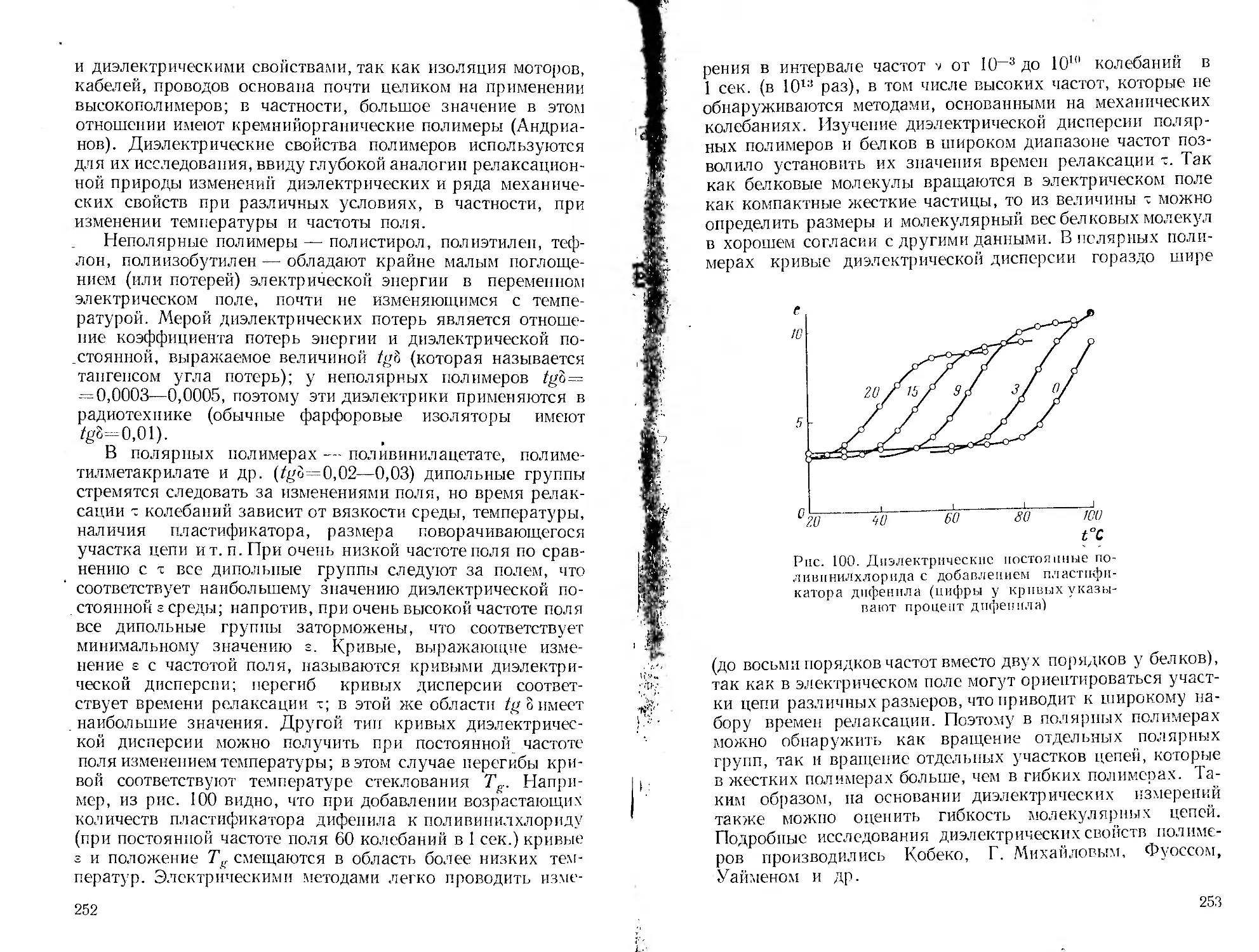
МОЛЕКУЛ Я РНО-КИНЕТИЧЕСКИЕ СВОЙСТВА
КОЛЛОИДОВ
БРОУНОВСКОЕ ДВИЖЕНИЕ
Коллоидные системы по молекулярно-кинетическим свой-
свойствам принципиально не отличаются от обычных раство-
растворов. Все молекулы и взвешенные в растворе частицы нахо-
находятся в постоянном беспорядочном тепловом движении.
Сталкиваясь между собою, частицы и молекулы обменива-
обмениваются своим количеством движения и в результате средняя
то» 3 R Т
кинетическая энергия каждой из частиц —~—— ~<f~jj-yc~
танавливается одинаковой, независимо от их размера. Одна-
Однако, если для молекул газов средние скорости движения изме-
измеряются сотнями метров в секунду, то для частиц размером
в 3—5 [А они весьма незначительны. Еще в 1827 г. англий-
английский ботаник Броун заметил, что микроскопические частицы
пыльцы растений находятся в воде в беспрерывном хао-
хаотическом движении, и по его имени это движение называется
броуновским. Оно хорошо заметно и в протоплазме клеток.
Теория броуновского движения была развита Эйнштейном
A905) и Смолуховским A905). Броуновское движение пред-
представляет большой интерес как непосредственное проявле-
проявление молекулярного движения. Наблюдаемое в микроскоп
смещение частицы (рис. 5) за определенный промежуток
времени является лишь статистическим результатом мно-
множества смещений частицы по различным направлениям в
пространстве (в их проекции в поле зрения микроскопа).
Любое смещение частицы в данном направлении может с
одинаковой вероятностью происходить в противоположном
29
направлении; поэтому среднее значение всех смещений
частицы за большой промежуток времени равно нулю,
и для того, чтобы определить среднюю величину смещений
частицы (без учета направлений смещения), необходимо
рассчитать величины квадратов
этих смещений. Как показал
Эйнштейн, среднее значение
квадрата смещения частицы ~Р ,
определенное из большого числа
измерений смещения ~~х за про-
промежутки времени t, равно:
RT
(II.1)
V
n — газовая постоянная,
температура,
Рис. 45. Броуновское^Гдви-
v. ¦_ жение частицы
где
Т — абсолютная температура,
N — число Авогадро, т] — вяз-
вязкость среды и г — радиус
взвешенных частиц. Помимо
поступательного движения, ча-
частицы обладают также враща-
вращательным броуновским движе-
движением, которое характеризуется
величиной "а2 — средним квад-
квадратом угла вращения за время t.
По Эйнштейну:
^а ' RT (II. 2)
t 4 к N y] г3 "
Измеряя на золях мастики с известными Т, у] и г величи-
ны поступательного смещения частиц -г- и вращения ча-
частиц — (вращение измерялось на частицах с неболь-
небольшими наблюдаемыми дефектами на поверхности), Перрен
вычислил по формулам (II. 1) и (II. 2) число Авогадро N =
= 6,5 • 1023, в близком согласии с другими известными
данными. Таким образом было показано, что закономер-
закономерности молекулярно-кинетического движения коллоидных
частиц и движения простых молекул в растворе одинаковы.
диффузия
Вследствие молекулярно-кинетического движения час-
частицы испытывают случайные смещения, при которых они
перемещаются вверх и вниз.
30
Однако если в растворе распределение частиц нерав-
неравномерно, например, содержание их у дна сосуда больше,
чем в верхнем слое жидкости, то число смещений снизу
вверх будет больше, чем в обратном направлении, и поэ-
поэтому частицы будут передвигаться снизу вверх. Такой
процесс выравнивания концентрации частиц по всему
объему раствора под влиянием теплового или броуновско-
броуновского движения называется диффузией; он также является об-
общим как для молекулярных, так и для коллоидных раст-
растворов. Поскольку равномерное распределение вещества в
растворе наиболее вероятно, в термодинамическом отноше-
отношении процесс диффузии идет с увеличением энтропии и
является самопроизвольным. Согласно закону Фика, ко-
количество вещества dm, продиффундировавшего за время dt
через площадь S, пропорционально изменению концентра-
концентрате-
ции на единицу расстояния или градиенту концентрации^- •
dm
(II. 3)
Коэффициент пропорциональности D называется коэф-
коэффициентом диффузии вещества. По Эйнштейну:
EL
TV
6 7Г Yj Г
(II. 4)
если рассматриваются сферические частицы, размеры ко-
которых гораздо больше, чем молекул растворителя. Это
уравнение — одна из основных формул коллоидной химии;
оно позволяет на основании измерений D определить ра-
радиус растворенных или взвешенных частиц, начиная от
малых молекул и кончая сферическими коллоидными час-
частицами и макромолекулами. Для частиц несферической
формы в уравнении (II. 4) выражение (бщг) заменяется
более сложными выражениями, причем для несферических
частиц величина D меньше, чем для сферических частиц
равной массы. :
Для измерения величины D устанавливают различными
способами скорость изменения концентрации в определен-
определенном слое раствора, в котором происходит диффузия; чаще
всего концентрацию определяют по оптическим свойствам
раствора — по изменению показателя преломления, пог-
31
лощения света и др. Особой чувствительностью обладает
метод поляризационного интерферометра (Цветков), поз-
позволяющий определить D при концентрации растворов всего
0,01% с точностью до 0,5%. При работе с биологически
активными веществами (ферментами, антибиотиками) иног-
иногда определяют D по скорости проникновения биологичес-
биологической активности из раствора в растворитель через пористый
диск, назначение которого заключается в предотвращении
конвекционного переноса (Нортроп). Метод пористого дис-
диска значительно менее точен, чем оптические методы измере-
измерений величины D, но он не требует выделения исследуемого
вещества в чистом виде, что иногда удобно при работе с
биологически активными веществами. Из уравнения (II. 4)
видно, что диффузия зависит от температуры, и поэтому
при измерениях диффузии необходимо поддерживать стро-
строгое постоянство температуры. Величина D имеет размер-
размерность см и обычно вычисляется для 20°С и вязкости
L сек \ м
воды (Ь2о', w). В табл. 2 приведены значения D20, w для
некоторых веществ ( о значении DJD см. ниже).
Таблица 2
Коэффициенты диффузии некоторых веществ
Вещество
Сахар
Конго красный
Яичный альбумин
Сывороточный альбумин . .
Препарат целлюлозы (в мед-
но-аммиачном растворе) .
Полистирол (в бензоле) . .
Вирус табачной мозаики . .
?> см* 1 сек
20, w '
4,6-10—6
5,4-Ю-6
7,8-10—7
6,1-Ю-7
2,4-Ю-7
8,3-10—8
5,0-10—8
D0/D
1
1,16
1,28
2,0
В некоторых случаях измерениями диффузии поль-
пользуются для исследования процессов ассоциации или дезаг-
грегации частиц в растворе.
Кроме коэффициента поступательной диффузии, может
быть также измерен коэффициент вращательной диффузии,
обусловленной вращательным действием броуновского дви-
32
жения. Для этого вызывают искусственную ориентацию
частиц в растворе, например, действием электрического
поля, которое затем в определенный момент внезапно
выключается, и оптическими методами измеряется скорость
самопроизвольной дезориентации частиц. Для ориентации
частиц может быть использовано и течение раствора (см.
стр. 65). В случае сферических частиц коэффициент
вращательной диффузии ©0 связан с радиусом частиц г
следующим уравнением:
Сравнивая уравнение (II. 5) и (II. 2), устанавливаем, что
во=2* ;
0 для асимметрических частиц см. уравнение (III. 9).
ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
Положим, что коллоидный раствор и чистая жидкость
разделены мембраной, проницаемой только для молекул
жидкости, но непроницаемой для коллоидных частиц. Этот
случай имеет важное значение не только в коллоидной хи-
химии, но и в физиологии, так как в любом организме большую
роль играют различные полупроницаемые мембраны. Ввиду
того, что наличие мембраны ограничивает свободное пере-
передвижение коллоидных частиц в сторону чистой жидкости,
различие в концентрации раствора по обе стороны мембраны
не может выравняться путем свободной диффузии.
В соответствии с законом Рауля упругость пара Ро
над чистым растворителем будет больше, чем упругость
пара Р над раствором:
1 +
«2
(II.6)
где /2Х — число молей растворителя и л2 — число молей
растворенного вещества. Тем же соотношением определяется
различие в активности растворителя в чистом виде и в
растворе. Поэтому растворитель переходит в раствор до
тех пор, пока этот переход не компенсируется встречно
направленным и возросшим, вследствие перехода части
растворителя, гидростатическим давлением со стороны
раствора, которое и называется осмотическим давлением
раствора.
«Коллоидная химия»
33
В состоянии равновесия осмотическое давление ~ раст-
раствора равно, по закону ВантТоффа:
RTc • (II. 7)
тс =
100М
где с — количество граммов растворенного вещества в
100 мл растворителя и М — молекулярный вес растворен-
растворенного вещества; если - выражено в атмосферах, то постоян-
г> ,тт _ г,г. ~„ см3!атм ЛГ
ная Н в уравнении (II. 7) составляет 82,07—j—^—. Урав-
Уравнение ВантТоффа позволяет вычислить молекулярный вес
М растворенного вещества по осмотическому давлению его
раствора. Интересно отметить, что осмотическое давление тс
равно тому давлению, которое оказывало бы на стенки со-
сосуда данное число молей растворенного вещества в том же
объеме, если бы оно находилось в виде газа, как это следует
из сравнения уравнения II. 7 и уравнения состояния идеаль-
идеальных газов р — nRT. Однако следует иметь в виду, что
осмотическое давление физически обусловлено не ударами
молекул о стенки сосуда.
Уравнение (II. 7) Вант-Гоффа, как и закон Рауля (П.6)
применимы лишь к разбавленным растворам, в которых
отношение rJc — по-
постоянная величина.
Обычно измеряют ве-
величину тс при не-
нескольких концентра-
концентрациях с и затем,
экстраполируя, опре-
определяют предельное
значение (тг/с)с->0,
которое и под-
подставляют в уравнение
П.7 для определения
М; примеры подобных
кривых для растворов
каучука (с М —
= 200000) в различ-
различных растворителях
приведены на рис. 6.
Вместо уравнения П. 7
О 0,2 ОМ 0,6 0,8 1,0 1,1 1М
с.%
Рис. 6. Осмотическое давление раство-
растворов бутадиенового каучука (М =200000):
)в бензоле; 2—в бензоле+15% ме-
спирта
бензоле; 2—в
тнлооого
применяют более общее уравнение
1 +Вс,
К Т с
м
(И.Ь)
где величина В характеризует отклонения от уравнения
Вант-Гоффа и измеряется по наклону кривых зависимости
(тс/с) от с (см. рис. 6) при малых концентрациях; при В = О
уравнение (II. 8) переходит в уравнение Вант-
Гоффа.
Отклонения от уравнения Вант-Гоффа при повышении
концентрации вызываются взаимодействием коллоидных
частиц между собою (это особенно заметно для вытянутых
частиц), изменениями ассоциации или диссоциации частиц
при изменении концентра-
концентрации, наличием электрических
зарядов на коллоидных ча-
частицах (доннановский эффект;
см. пятую главу). В раство-
растворах линейных полимеров зна-
значительные отклонения вызы-
вызываются гибкостью молекуляр-
молекулярных цепей (см. стр. 190).
В свою очередь, измерения
осмотического давления мо-
могут служить методом иссле-
исследования указанных свойств
в коллоидных растворах.
Экспериментально осмо-
осмотическое давление измеряют
в приборах — осмометрах.
Один из современных типов ос-
осмометров изображен на рис. 7.
Мембрана М из целлофана
или коллодия плотно закрепляется на пористом диске;
коллоидный раствор А B—3 мл) находится над мембраной,
а внешний раствор (растворитель) В — под мембраной,
заполняя часть резервуара С; верхнюю часть С и капилляр
D заполняют толуолом. Манометр Е позволяет задать
между А и В желаемую разность давлений. Определяют
разность давлений в Е, при котором уровень толуола в D
остается постоянным (вследствие компенсации внешнего
и осмотического давлений) в течение достаточно продолжи-
продолжительного промежутка времени. Измерения производятся
при постоянной температуре и, благодаря большой величине
отношения поверхности мембраны М к объему раствора А,
не требуют очень длительного времени.
Рис. 7. Осмометр Геппа-Скат-
чарда
34
35
При измерении давления в Е в сантиметрах водного стол-
столба и при 25°С
2,53•105 с
М
Например, если 1 г белка растворен в 100 мл воды (с =
= 1) при 25°, и молекулярный вес белка М ~ 10 000,
то осмотическое давление тс раствора будет равно:
2,53- Ю5-1,0 ог о и п
гс = -„,„ = 25,3 см Н„О.
Различие в упругости пара между растворителем и раст-
раствором можно измерять также непосредственно по пониже-
понижению упругости пара методом изотермической перегонки и по
понижению точки замерзания или повышению точки кипе-
кипения раствора. Однако эти методы, пригодные для обычных
растворов низкомолекулярных веществ, мало чувствитель-
чувствительны для измерений в коллоидных растворах. Так, в приве-
приведенном примере белковый раствор, дающий осмотическое
давление в 25,3 см НгО, вызывает понижение точки замер-
замерзания лишь на 0,00186°С, а упругость пара — лишь на
0,00 058 см НЮ, что явно затрудняет проведение измерений
(Булл). Однако, хотя метод осмотического давления наи-
наиболее чувствительный из перечисленных методов, он также
имеет определенные пределы применимости. Для низкомо-
низкомолекулярных растворов метод ограничен отсутствием под-
подходящих полупроницаемых мембран, а для очень крупных
частиц — низкой величиной осмотического давления при
возможных концентрациях этих частиц в растворе. Так,
в приведенном примере при молярной концентрации 10~3
осмотическое давление составляло хорошо измеримую
величину 25,3 см НгО, но уже при 10~5 мол/л измерение
тс = 0,25 см Н-2 О крайне затруднительно, а для 10~6 мол/л —
уже невозможно. Между тем, обычный золь золота с
концентрацией 0,1 % и радиусом частиц 2 м\ъ содержит около
1015 частиц в 1 мл, что соответствует около 2 • 10~6 мол;
поэтому в обычных лиофобных золях величины осмотичес-
осмотического давления слишком незначительны. Таким образом,
область применения осмотических измерений ограничена
веществами с молекулярным весом приблизительно от
10 тыс. до 200—300 тыс. и лишь при особых условиях —
до 1 млн., что включает, однако, ряд таких важных веществ,
как белки, каучуки, многие красители и др.
36
Величина осмотического давления пропорциональна
числу частиц растворенного вещества в растворе; поэтому
при наличии в растворе частиц различного размера (в раст-
растворах полидисперсных веществ, например, нефракциони-
рованиого каучука) осмотические измерения дают усреднен-
усредненный по числу частиц, или среднечисленный молекулярный
вес Мп.
Следует подчеркнуть, что источником осмотического
давления в наших примерах было ограничение возможности
равномерного заполнения коллоидными частицами всего
объема системы вследствие наличия полупроницаемой
мембраны. Однако ограничение диффузионной способ-
способности коллоидных частиц может быть осуществлено не
только при наличии мембраны, но также при соединении
коллоидных частиц между собой, например, в виде геля.
Поэтому осмотические явления имеют место также в гелях,
в ионообменных адсорбентах и в других системах (см. гла-
главы пятую и девятую).
В организмах осмотические явления играют важную
роль в жизнедеятельности всех живых клеток, но в этом
случае они осложняются сопряженными химическими реак-
реакциями в клетках, вследствие чего распределение веществ
может значительно отличаться от наблюдаемого в равно-
равновесных условиях.
ДИАЛИЗ И УЛЬТРАФИЛЬТРАЦИЯ
Полупроницаемые мембраны используются в коллоид-
коллоидной химии не только для измерений осмотического давления,
но и для очистки коллоидных растворов путем диализа
и ультрафильтрации. Диализ заключается в том, что кол-
коллоидный раствор А помещают внутри мешочка или гильзы М
из целлофана, коллодия, пергамента и других аналогич-
аналогичных материалов, которые снаружи омываются часто сме-
сменяемой или проточной водой В (рис. 8). Содержание кол-
коллоидных частиц при этом остается постоянным, так как
мембрана непроницаема для них, а низкомолекулярные
вещества (электролиты, органические вещества) постепенно
диффундируют в воду и удаляются; в результате происхо-
происходит очистка коллоидного раствора. Степень очистки огра-
ограничивается устойчивостью коллоидных частиц или процес-
процессами их гидролиза при удалении электролитов. В некото-
некоторых случаях внешнюю воду В не сменяют и процесс ведут
37
до равновесия (равновесный диализ) для изучения состава
жидкости, находящейся между коллоидными частицами,
или изменений в составе этой жидкости при различных
процессах. Обычно для ускорения диализа коллоидный
раствор перемешивают и диализа-
диализаторы устраивают с большим отноше-
отношением поверхности мембраны к объему
жидкости.
Значительного ускорения удале-
удаления электролитов из коллоидного
раствора добиваются, помещая его
(рис. 9) в постоянное электрическое
поле (при падении потенциала
20—50 е/см и выше) между двумя
электродами Е и Е1, отделенными
от коллоидного раствора А с обеих
сторон полупроницаемыми мембра-
мембранами. Через электродные пространства Е и Е1 непре-
непрерывно протекает дистиллированная вода. Этот процесс
Рис. 8. Схема ди-
диализа
Рис. 9. Электродиализатор Паули
называется электродиализом; он позволяет ускорить
очистку коллоидного раствора от электролитов в десят-
десятки раз.
Ультрафильтрацией называется фильтрование коллоид-
коллоидного раствора через полупроницаемые мембраны, которые
укрепляются на твердой пористой подкладке в специальных
ультрафильтрах (рис. 10). Обычно процесс ультрафильтра-
ультрафильтрации проводят под разрежением или под повышенным дав-
давлением. При ультрафильтрации низкомолекулярные элект-
электролиты и органические вещества, содержащиеся в коллоид-
коллоидном растворе, отделяются гораздо скорее, чем при диализе.
Периодически разбавляя коллоидный раствор водой, можно
38
Мембрана
при ультрафильтрации добиться значительной очистки
раствора и на конечной стадии путем отсасывания диспер-
дисперсионной среды достичь концентрирования коллоидного
раствора. Наконец, применяя мембраны для ультрафильт-
ультрафильтров с определенной степенью
пористости, можно в известной
мере разделить по размерам
коллоидные частицы и одно-
одновременно приближенно опреде-
определить их размеры. Этим путем
были определены размеры ча-
частиц ряда вирусов и бактерио-
бактериофагов.. Таким образом, ультра-
ультрафильтрация может служить так-
также одним из методов диспер-
соидного анализа и препаратив-
препаративного разделения коллоидов.
Наконец, методы ультра-
ультрафильтрации и электродиализа
применяются в сочетании в виде метода электроультрафильт-
рации коллоидов.
Гейман дает следующее сопоставление относительных
скоростей очистки по различным методам при сравнимых
условиях (табл. 3).
Таблица 3
Относительные скорости очистки раствора
Рис. 10. Ультрафильтр
Метод
Диализ
Электродиализ D0 в/см) . .
Ультрафильтрация
Электроультрафильтрация .
Удаляемое вещество
NaCl
1
163
14
182
сахар
0,3
2
14
14
СЕДИМЕНТАЦИЯ И УЛЬТРАЦЕНТРИФУГИРОВАНИЕ
Под действием силы тяжести все коллоидные частицы,
независимо от их природы, оседают в растворе; этот
процесс называется седиментацией. Скорость оседания
частиц зависит от размеров частиц, от разности плотностей
частиц d и среды о и от вязкости жидкости -ц. Для шарооб-
39
разных частиц с радиусом г, по закону Стокса, скорость
оседания:
^^йй, (П9)
Из уравнения (II. 9) видно, что скорость оседания особенно,
сильно зависит от размера частиц. Так, например, частицы
серебра при диаметре 200 \ь оседают в воде на 1 см за
0,05 сек., при диаметре 2[х — за 500сек., а при диаметре
20mjj.— лишь за 58 дней. Если частицы легче жидкости (на-
(например, в эмульсии масла, в воде), то (d—p) имеет обратный
знак, и вместо оседания наблюдается всплывание частиц, со-
согласно тому же закону. При отсутствии противодействую-
противодействующих сил седиментация коллоидных частиц за достаточно
продолжительный промежуток времени неизменно приводи-
приводила бы к осаждению всего коллоида на дне сосуда. Этого,
однако, обычно не происходит ввиду того, что оседанию
частиц (даже при полном покое раствора, при постоянстве
температуры, отсутствии конвекционных потоков и др.)
всегда противодействует броуновское движение, стремящее-
стремящееся равномерно распределить коллоидные частицы по всему
объему раствора. Чем меньше частицы, тем сильнее сказы-
сказывается влияние броуновского движения или диффузии
(табл. 4).
Таблица 4
Соотношение броуновского движения
и оседания частиц серебра (по Бертону)
Диаметр частиц
М[Х
100
1000
10 000
Расстояние, проходимое за 1 сек в му.
броуновское
движение
10 000
3162
1000
оседание
67,6
6760
676000
Из табл. 4 видно, что частицы серебра с диаметром менее
0,1 а не могут осесть на дне сосуда, а частицы с диаметром
10 ;х осядут почти полностью; частицы же промежуточных
диаметров распределяются тем ближе к дну сосуда, чем
больше их размер и вес. Для любых однородных частиц
относительное изменение их числа в единице объема —
в двух слоях жидкости, отстоящих на расстоянии h (слой 2
40
I
выше слоя 1), определяется уравнением:
щ
=е
A1.10)
где v — объем отдельной частицы. Это уравнение показы-
показывает, что с увеличением высоты при постоянной температуре
число частиц убывает, причем скорость убывания концент-
концентрации резко увеличивается с увеличением массы частиц
(следует учесть, что величины h и v находятся в показателе
степени), т. е. более тяжелые частицы сосредоточиваются
у дна сосуда. Перрен на однородной суспензии гуммигута
(смолы из высушенного млечного сока каучуконосов)
доказал количественную точность уравнения (II. 10) и
определил этим путем значение числа Авогадро N от6,5Х
ХЮ23 до 7,2-1023, а Вестгрен на золях золота получил
еще более точное значение 6,06 • 1023, что хорошо согла-
согласуется с данными других методов. Заметим, что тому же
закону подчиняется и распределение числа частиц или давле-
давления в столбе газа или в атмосфере, вследствие чего уравне-
уравнение (II. 10) называется барометрическим законом.
Если статистическое равновесие между действием сил
тяготения и броуновского движения таково, что основная
масса частиц за сравнительно короткое время оказывается
на дне сосуда, то система называется кинетически неустой-
неустойчивой, если же частицы в основном остаются распределен-
распределенными в объеме раствора, то система называется кинетически
устойчивой. Лиофобные золи и растворы высокомолеку-
высокомолекулярных веществ обычно имеют такие размеры и плотности
частиц, что 'обладают достаточно высокой кинетической
устойчивостью, если только в системе не происходит про-
процессов, резко понижающих их степень дисперсности (см.
шестую главу).
Однако в системе, обладающей при обычных условиях
высокой кинетической устойчивостью, можно все же выз-
вызвать заметное оседание частиц, если силу земного тяготения
заменить более значительной центробежной силой. Эта
идея была впервые высказана Думанским A913), применив-
применившим центрифугу для оседания коллоидных частиц, и затем
развита Сведбергом, разработавшим специальные центри-
центрифуги с большим числом оборотов, называемые ультрацентри-
ультрацентрифугами, дающими центробежную силу свыше 250 000 g.
Современная ультрацентрифуга представляет собой слож-
«Коллоидная химия»
41
ный аппарат, в котором вращение ротора Л (рис. 11) про-
происходит с частотой до 60 000 об/мин и выше при помощи
масляной турбины; имеются более новые конструкции с
использованием воздушной турбины или высокочастотных
электрических моторов. Ротор вращается в толстостенном
металлическом корпусе М в вакууме или в атмосфере водо-
водорода (для лучшей теплоотдачи); постоянство температуры
м
Рис. 11. Ультрацентрифуга Сведберга (ротор)
при вращении ротора поддерживается до 0,02°. В роторе,
имеются два сквозных отверстия В, в которых помещаются
кюветы с коллоидным раствором, емкостью всего на 0,5 мл.
По мере оседания частиц изменяется показатель прелом-
преломления или поглощения света по высоте кюветы, находя-
находящейся на пути луча S, что фиксируется оптическими ме-
методами на экране или фотопластинке.
В ультрацентрифуге оседают не только мельчайшие
частицы гидрофобных коллоидов, но и молекулы белков
и высокомолекулярных веществ. Скорость седиментации v
сферических частиц в ультрацентрифуге определяется также
42
уравнением (II. 9), в котором g заменяется ш2х, где «о —
угловая скорость вращения ротора их — расстояние от
частицы до оси вращения. Следовательно, в поле земного
тяготения ускорение, действующее на частицу, остается
постоянным, а в ультрацентрифуге оно увеличивается по
мере оседания частицы. Поэтому скорость оседания v надо
dx
выражать в виде -п-; величина этой скорости, отнесенная
к единице действующей силы, называется константой седи-
седиментации 5 частицы:
Величина 5 является характеристической константой раст-
растворенных молекул. За единицу измерения константы седи-
седиментации принята
5 = 1 сведберга 10-13 ™ ¦ .
сек-бич
Величины 5" колеблются от 2 (при молекулярном весе
около 104) до 200 (при молекулярном весе около 107).
Молекулярный вес М связан с величиной 5 уравнением
R T S
(П 12)
м=
D(l-vP)
где D — коэффициент диффузии, v — парциальный удель-
удельный объем и о — плотность растворителя.
Из уравнения (II. 12) видно, что для вычисления М надо
определить не только константу седиментации 5 вещества,
но и его коэффициент диффузии D. Уравнение A1.12) остает-
остается справедливым как для сферических частиц, так и для
частиц несферической формы, поскольку величины S и D
изменяются приблизительно одинаково с формой частиц.
Поэтому, определив по уравнению (II. 12) молекулярный
вес несферических частиц, можно затем рассчитать радиус г
сферической частицы, обладающей той же массой, и, под-
подставив эту величину г в (II. 4), рассчитать ожидаемый коэф-
коэффициент диффузии Do, которым обладало бы исследуемое
вещество, если бы его молекулы или частицы имели сфери-
сферическую форму. Сравнивая величину Do с фактически изме-
измеренной для данного вещества величиной коэффициента
диффузии D, можно найти отношение DOID, которое явля-
является мерой отклонения формы частиц от сферической.
Придавая уравнению (II. 4) более общий вид
3* 43
iV / '
где для шарообразных частиц D = Do и /0 = 6 тгт|г, можно
отношение (Do/D) заменить равным и чаще употребляемым
отношением (///0). Величина (///0) называется коэффициентом
дисимметрии и является характеристикой формы частиц.
Для шарообразных частиц (///0) = 1, для асимметричных ча-
частиц эта величина больше единицы (см. табл. 2). Если асим-
асимметричные частицы имеют форму эллипсоида, то величину
(///о) можно связать с отношением большой и малой осей
(-) эллипсоида, например, для (///0) = 1,2 (-J ~3—4,
для (///о) = 2,0 (|) ^10—11 и др. На величине (///0) отра-
отражаются как собственная асимметрия частиц, так и гидра-
гидратация частиц, влияние которой в необходимых случаях
должно особо учитываться.
Кроме «скоростных» ультрацентрифуг, применяемых при
измерении скорости седиментации (значительно преобла-
преобладающей над процессами диффузии), применяют также
ультрацентрифуги с меньшим числом оборотов (до
-20 000 об/мин), в которых скорости седиментации и диф-
диффузионного переноса близки и поэтому устанавливается
седиментационное равновесие. Молекулярный вес при опре-
определении по методу седиментационного равновесия,
2 RT In (С/О ш j.
таким образом, вычисляется лишь из разности концентра-
концентраций Съ и С г на расстояниях хг и хъ без использования
данных других методов.
Для определения размеров первичных частиц или
молекул необходимо производить измерения в достаточно
разбавленных растворах, так как при наличии ассоциации
измерения на ультрацентрифуге характеризуют размеры
ассоциированных частиц. При работе с растворами веществ
с сильно вытянутыми молекулами (целлюлозы, нуклеиновых
кислот, каучуков), которые легко взаимодействуют между
собой, часто приходится ограничиваться их 0,02—0,1 %-ной
концентрацией.
ПОЛИДИСПЕРСНОСТЬ И КРИВЫЕ РАСПРЕДЕЛЕНИЯ
Пользуясь методами седиментации и ультрацентрифуги-
ультрацентрифугирования, можно исследовать полидисперсность коллоидных
систем. Большинство коллоидных систем характеризуется
44
наличием частиц или молекул различных размеров, что
часто отражается на технических свойствах материалов.
Изучение полидисперсности в форме установления коли-
количественного распределения частиц или молекул по разме-
размерам (так называемых кривых распределения) производится
разными методами для различных дисперсных систем.
Системы с очень грубыми частицами (свыше 0,2 мм), напри-
например, порошки лекарств, исследуются простым ситовым
С
анализом при помощи на-
набора шелковых или ме-
металлических сит. Сус-
Суспензии же и эмульсии
с размером частиц в ин-
интервале 1—200 {х изуча-
изучаются простыми методами
седиментации, вслед-
вследствие чего анализ поли-
полидисперсности в этом
случае называется седи-
ментационным анализом.
В седимеитометре
Фигуровского (рис. 12)
к упругому стеклянному
или кварцевому стержню
С прикреплена на стек-
стеклянной нити К с крючком чашечка S, на которой на-
накапливается осадок суспензии. Прогиб плеча С измеряется
по шкале при помощи микроскопа. По мере оседания частиц
дисперсной фазы прогиб С увеличивается вначале быстро,
вследствие преимущественного выпадения более тяжелых
частиц, а затем все медленнее, почти до полного окончания
оседания. Этим путем получается кривая седиментации
(рис. 13), на абсциссе которой откладывается время оседа-
оседания, а на ординате — процент выпавшей суспензии Р.
Так как время оседания по (II. 9) связано с размерами осе-
оседающих частиц, а для каждого интервала размеров частиц
может быть определена доля выпавшей суспензии, то
анализ кривой седиментации на рис. 13 позволяет рассчи-
рассчитать кривую распределения для данной суспензии, которая
обычно имеет один максимум (рис. 14). Аналогичным обра-
образом исследуется по скорости всплывания капель распреде-
распределение частиц по размерам в эмульсиях, в которых плотность
частиц меньше плотности растворителя; при этом чашечка 5
Рис. 12.
Седиментометр Фпгуров-
ского
45
(рис. 12) заменяется небольшим колоколом. Наконец, для
медленно оседающих суспензий (например, с очень мелкими
частицами) пользуются седиментометрами в сочетании с
центрифугами, а для наиболее высоко дисперсных фракций
t, мин.
Рис. 13. Кривая седиментации
РЩ
иногда применяют определения размеров частиц при по-
помощи электронного микроскопа (стр. 67).
В высокодисперсных лиофобных золях (в золях золота)
и в растворах высокомолекулярных веществ полидисперс-
полидисперсность часто исследуют
при помощи ультра-
центрифугирования.В
«скоростной» ультра-
ультрацентрифуге полидис-
полидисперсность проявляет-
проявляется в размытости гра-
границы оседания; для
монодисперсных ве-
веществ граница оста-
остается весьма резкой;
в «равновесной» уль-
ультрацентрифуге поли-
полидисперсность проявляется в отклонении наблюдаемого
распределения концентрации по высоте столба от идеаль-
идеального логарифмического распределения, вычисленного по
формуле (II. 13). Для построения полной кривой рас-
распределения молекулярных весов полимеров измерения на
Рис. 14. Кривая распределения
ультрацентрифуге иногда сочетают с предварительным
фракционированием полимера методом осаждения (см.
стр. 186), суммируя кривые распределения для от-
отдельных фракций. В отличие от метода осмотического
давления измерения на ультра центрифуге дают усреднен-
усредненные по массе частиц значения молекулярного веса, или
средневесовой молекулярный вес Mw. Для монодиспер-
монодисперсных веществ значения Мп и Mw совпадают, для поли-
полидисперсных веществ расхождение этих значений (обычно
-~- > 1) может служить мерой полидисперсности.
Наконец, следует указать, что если количество оседаю-
оседающего вещества достаточно велико, седиментометры и спе-
специальные препаративные ультрацентрифуги могут быть
использованы для препаративного разделения и выделения
фракций с различными размерами частиц или молекул.
В последние годы препаративные ультрацентрифуги широко
применяются при выделении и очистке вирусов и входящих
в их состав нуклеиновых кислот и белков.
Значение ультрацентрифуг для определения размеров,
формы, ассоциации и полидисперсности частиц, а также
возможности их препаративного использования, делают
ультрацентрифуги одним из основных средств исследова-
исследования коллоидных систем.
вязкость
Вязкостью жидкости называется сопротивление перед-
передвижению одного слоя жидкости относительно другого
слоя. Единицей вязкости или пуазом (т; = 1) характери-
характеризуется вязкость жидкости, в которой слои в 1 см2, находя-
находящиеся на расстоянии 1 см, движутся друг относительно
друга со скоростью 1 см/сек под действием силы в 1 дн.
Для примера укажем, что при 20° вязкость воды равна
0,01 пуаза, вязкость сыворотки крови человека — 0,014—•
—0,019 пуаза, цельной крови—0,05 пуаза, а вязкость гли-
глицерина — около 8 пуаз. Размерность вязкости составляет
[ . При достаточно высоких скоростях течения сло-
[ см-сек J r r
истое или ламинарное течение жидкости сменяется вихре-
вихревым или турбулентным. Движение крови в тонких кровенос-
кровеносных сосудах и капиллярах имеет ламинарный характер но,
в большой аорте течение иногда приближается к турбулент-
47
м
ному (Булл). Вязкость обычно измеряют в пределах лами-
ламинарного течения. Если при изменении скорости течения в
пределах ламинарности вязкость жидкости остается посто-
постоянной, т. е. скорость течения прямо пропорциональна при-
приложенной силе, жидкость называется ньютоновой, если же
вязкость при этом изменяется и скорость течения жидкости
не пропорциональна приложенной силе, жидкость назы-
называется неньютоновой. Чистые жид-
жидкости и разбавленные растворы
коллоидов с сферическими части-
частицами характеризуются ньютоновой
вязкостью, тогда как растворы кол-
коллоидов с палочкообразными или
нитевидными частицами часто обла-
обладают неньютоновой вязкостью (по-
(подробнее см. стр. 244).
Вязкость обычно измеряют в
капиллярных вискозиметрах (рис.
15), в которых точно фиксируется
время истечения t объема жидкости
v между метками а я в под действи-
действием собственного веса- столба жид-
жидкости или приложенного к одному
из колен прибора определенного
давления Р; измерения произво-
производят при строго постоянной тем-
Рис. 15. Капиллярные пературе. Истечение в капилляр-
вискозиметры: / — по ных вискозиметрах подчиняется
Оствальду, //—по Убел- закону Пуазейля:
л оде
где / — длина капилляра иг — радиус капилляра. Иногда
исследуют движение жидкости в зазоре между двумя вло-
вложенными друг в друга цилиндрами (коаксиальные
вискозиметры). При высокой вязкости жидкости (касто-
(касторовое масло, глицерин) ее измеряют также по скорости
падения шарика выбранного размера на определенном
отрезке пути в жидкости (шариковые вискозиметры).
Оседание круглых пигментных включений в протоплазме
в центрифуге используется для определения вязкости про-
протоплазмы.
В коллоидных растворах наличие крупных (по сравне-
48
нию с молекулами растворителя) частиц нарушает равно-
равномерное течение жидкости, вследствие чего вязкость таких
растворов т] всегда выше, чем вязкость растворителя щ.
Если коллоидные частицы имеют сферическую форму и не
взаимодействуют между собою (разбавленные растворы),
то, как было установлено Эйнштейном,
1+0,5?
(II. 15)
• где ср — доля объема суспендированных частиц в единице
объема суспензии. Правая часть формулы (П. 15) может
быть выражена в виде ряда членов с возрастающими сте-
степенями ср. Для разбавленных растворов достаточно точным
является уравнение
tjAIo = 1 + 2,5 ср, (П. 15а)
если же учесть еще один член
rjrlo =\ + 2,5 ср+ 14,1 ср2, (Ц. 156)
то A1.15 6) может быть применено, например, для латекса
до концентрации 30%. Строго говоря, выражение A1.15)
применимо к растворам с твердыми частицами; однако
жидкие капельки при очень малых радиусах двигаются
подобно твердым шарикам. Если от коллоидных растворов
с сферическими частицами перейти к растворам, содержа-
содержащим палочкообразные частицы (золи V2O5, растворы по-
линуклеотидов), то в них вязкость относительно еще более
повысится благодаря тому, что при броуновском движении
и вращении палочкообразных частиц они занимают в
жидкости более значительный объем и сильнее нарушают
нормальное течение жидкости, чем сферические частицы.
В этом случае зависимость между вязкостью и объемной
концентрацией ср становится довольно сложной в соответ-
соответствии со степенью асимметрии частиц. Если представить
частицы в виде вытянутых эллипсоидов с отношением полу-
осей —, то в общем уравнении
7]/7j0 = 1 + А Ср + В
(II. 16)
в котором для сферических частиц Л= 2,5 и В = 14,1 (см.
II. 15, б), для вытянутых частиц коэффициенты Л и В будут
иметь различные значения, легко определяемые по рис. 16.
Для растворов высокомолекулярных веществ с очень
49
длинными жесткими или гибкими молекулами вязкость
связана с молекулярным весом частиц М обобщенным
уравнением Штаудингера
[гЛ = КМ\ (II. 17)
которое подробнее рассматривается на стр. 193—194.
Для перечисленных видов растворов предполагалось,
что частицы не взаимодействуют между собой и остаются в
растворе кинетически
индивидуальными. Ес-
Если же частицы каким-
либо образом перепле-
переплетаются между собой и
образуют пространст-
пространство венные структуры, то
их вязкостные свойства
сильно усложняются
(см. десятую главу).
Зависимость вязкости
коллоидных растворов
от температуры также
й
6,0
5.0
4.0
3.0
2.0
—
>
100
75
Z5
10
/5
Рис. 16. Зависимость коэффициентов
А я В от асимметрии суспендиро-
суспендированных частиц
имеет довольно слож-
сложный характер. В про-
простых случаях существу-
существует линейная зависимость lg-rj от \1Т, но обычно она на-
нарушается изменением состояния коллоида и степени аг-
агрегации частиц при колебании температуры.
Наконец, при наличии электрических зарядов на кол-
коллоидных частицах вязкость увеличивается (электровискоз-
(электровискозный эффект, по Смолуховскому), но в большинстве случаев
незначительно. При высоких концентрациях частиц и
низком содержании электролитов этот эффект следует
учитывать, но обычно его стремятся устранить повышением
концентрации электролитов.
Выводы
Коллоидные системы по своим молекулярно-кинети-
ческим свойствам (броуновскому движению, диффузии,
осмотическому давлению, седиментации) отличаются от
растворов низкомолекулярных веществ, главным образом,
лишь благодаря более значительным размерам своих час-
50
тиц. Поэтому многие методы установления размеров частиц
в коллоидных системах основаны на определении поступа-
поступательной и вращательной диффузии (II.4 и II. 5), осмотичес-
осмотического давления (II. 7), седиментации в поле тяготения (II.9)
и в ультрацентрифугах A1.12, 11.13), вязкости (II. 17).
Этими же методами могут быть изучены процессы ассоциа-
ассоциации частиц в коллоидных растворах.
Ряд молекул яр но-кинетических свойств (диффузия, се-
седиментация, вязкость) зависят от формы частиц, которая
может быть исследована этими методами.
Методы седиментации и ультрацентрифугирования поз-
позволяют также количественно исследовать полидисперсность
коллоидных систем и установить кривые распределения
по размерам частиц или по молекулярным весам. В поли-
полидисперсных системах методом осмотического давления мож-
можно определить среднечисленный молекулярный вес Мп,
а методом ультрацентрифугирования — средневесовой мо-
молекулярный вес Mw.
Важной характеристикой является поведение коллоид-
коллоидных частиц относительно полупроницаемых мембран.
Различие в концентрации раствора по обе стороны мембра-
мембраны приводит к различной активности растворителя, прояв-
проявляющейся в осмотическом давлении и изменении упругости
пара. Неспособность к прохождению через полупроницае-
полупроницаемые мембраны, являющаяся одним из характерных отли-
отличий коллоидных систем от растворов низкомолекулярных
веществ, используется в методах диализа и ультрафильтра-
ультрафильтрации для очистки и концентрирования растворов. Дополни-
Дополнительное наложение постоянного электрического поля —
в методах электродиализа и электроультрафильтрации —
позволяет значительно ускорить удаление электролитов
(см. табл. 3).
Изучение молекулярно-кинетических свойств коллоид-
коллоидных систем привело к установлению ряда основных зако-
законов (выражаемыхуравнениями II. 1; II.2; II. 4; II. 7; II. 10;
II. 15; расчет числа Авогадро и др.), общих для коллоидных
и низкомолекулярных систем.
Вопросы
1. Какие свойства коллоидных систем относятся к молекуляр-
но-кинетлческим? Почему они так называются? Какие основные
уравнения и законы выражают общность молекулярно-кииетичес-
ких свойств коллоидных и нпзкомолекуляриых систем?
51
2: Перечислите основные методы определения размеров кол-
коллоидных частиц, основанные на их молекулярио-кинетических
свойствах.
3. Какие из перечисленных методов характеризуют коллоид-
коллоидную систему в состоянии термодинамического равновесия?
4. По каким молекулярио-кинетическим свойствам можно су-
судить о форме коллоидных частич?
5. Какими методами можно исследовать полидисперсность
коллоидных систем? Укажите значение кривых распределения и
методы их построения.
6. Каково соотношение смещений коллоидных частиц, вызы-
вызываемых броуновским движением и седиментацией, для частиц раз-
различных размеров? Сопоставьте уравнения обоих процессов. Дайте
характеристику кинетической устойчивости коллоидных систем.
7. На чем основано наличие осмотического давления и каким
образом оно измеряется? Сопоставьте метод осмотического давления
с методами криоскопии и эбуллиоскопии.
8. Какими уравнениями характеризуется вязкость суспензий
сферических и вытянутых частиц?
9. Каковы назначение и сравнительная эффективность диали-
диализа, электродиализа и ультрафильтрации?
10. Объясните принцип устройства ультрацентрифуг, их наз-
назначение, способы определения молекулярного веса и полидисперс-
полидисперсности.
Глава третья
ОПТИЧЕСКИЕ СВОЙСТВА КОЛЛОИДОВ
Размеры частиц коллоидных систем соизмеримы с дли-
длиной световых волн, поэтому, кроме общих для всех раство-
растворов явлений преломления и поглощения света в различных
областях спектра, коллоидные растворы обладают также
рядом своеобразных оптических свойств. Благодаря тесной
связи оптических свойств с внутренним строением и формой
коллоидных частиц, а также вследствие удобства и точности
оптических методов измерений, они в настоящее время от-
относятся к числу основных методов исследования коллоид-
коллоидных систем.
СВЕТОРАССЕЯНИЕ
Допустим, что лучи падают на поверхность какой-либо
частицы, линейные размеры которой велики по сравнению
с длиной волны лучей; тогда эти лучи просто отражаются
по законам геометрической оптики. Если же линейные
размеры частиц составляют, например, лишь около 0,1
длины .волны падающих лучей, то наиболее характерным
процессом является диффракционное рассеяние света в
результате огибания частиц световой волной. Диффракцион-
Диффракционное рассеяние было впервые замечено Тиндалем A869),
который наблюдал образование светящегося конуса при
пропускании пучка сходящихся лучей через коллоидный
раствор. Внешне похожее явление можно наблюдать в за-
затемненной комнате, в кинотеатре, когда в луче света видно
сверкание частичек пыли в воздухе, незаметных простым
глазом в обычно освещенном пространстве.
Теория светорассеяния была разработана Рэлеем
A871). Согласно уравнению Рэлея, интенсивность рассеян-
53
иного света У:
24*2 Л2
, (III. 1)
где v — число частиц в единице объема, v — объем
частицы, X — длина волны падающего луча, А — его
амплитуда колебаний, m и йг — показатели преломления,
соответственно дисперсной фазы и дисперсионной среды.
Из (III. 1) видно, что светорассея-
светорассеяние пропорционально концентрации
частиц, квадрату объема частицы
(или, для сферических частиц, ше-
шестой степени их радиуса) и обратно
пропорционально четвертой степени
длины волны падающего света. Та-
Таким образом, рассеяние коротких
волн происходит относительно более
интенсивно; вследствие этого в про-
проходящем свете золь кажется красно-
красноватым, а в боковом, рассеянном све-
свете — голубым, что хорошо видно,
например, на высокодисперсных золях
серы. Голубой цвет неба и красно-
красноватый цвет заката объясняются по
существу теми же явлениями.
Рэлеевское рассеяние света или
опалесценцию коллоидных растворов
практически измеряют при помощи
нефелометров или специальных фото- Рис. 17. Прибор Доти
метров. В приборе Доти (рис. 17) Для визуального из-
свет от источника Л падает на рассей- меРения светорассея-
вающий раствор в термостатирован-
термостатированной кювете D и пластинкой В ча-
частично направляется на пластинку из молочного стекла
С, являющуюся «стандартом» мутности. Интенсивность
света, рассеянного под углом 90° в D, и стандартного пучка
из С сравниваются в фотометре Пульфриха Н и уравни-
уравниваются поворотом лимбов /, причем отсчеты на лимбах
Д и /2 характеризуют отношение интенсивностей рассеян-
рассеянного света.
Фотометр Н может быть заменен фотоэлементами с ум-
умножителями и гальванометром (фотоэлектрические нефело-
нефелометры, например, ФЭКН-54).
54
Ьлагодаря зависимости интенсивности рассеянного света
от числа и объема частиц, а также от разности показате-
показателей преломления частиц и среды (см. III. 1), нефелометри-
ческими измерениями часто пользуются для определения
концентрации коллоидного раствора или для изучения
происходящих в этом растворе процессов агрегации частиц
или изменения их состояния. Если два сравниваемых раст-
раствора отличаются только концентрацией частиц, то при урав-
уравнивании их мутности при помощи отсчетов на лимбах /i
и h (рис. 17) соблюдается соотношение
(III. 2)
по которому можно вычислить неизвестную концентрацию
(например, Ci).
Следует отметить, что рассеянный свет обычно частично
поляризован, если даже падающий свет является неполя-
ризованным, т. е. рэлеевское рассеяние света происходит
преимущественно в определенных плоскостях колебаний.
Этим, в частности оно отличается от флуоресценции многих
растворенных веществ, равно как и тем, что флуоресцен-
флуоресценция присуща лишь определенным веществам, тогда как
рэлеевское рассеяние проявляется во всех коллоидных
системах при соответствующей степени дисперсности и
разности показателей преломления частиц и среды.
Как указывалось, при увеличении размеров частиц
более 0,1 длины световых волн все возрастающее значение
получают явления геометрического отражения света. С дру-
другой стороны, по мере уменьшения размеров частиц, как
это следует из уравнения (III. 1), интенсивность рэлеевско-
го рассеяния света значительно ослабляется (пропорцио-
(пропорционально уменьшению v2). Поэтому максимальной опалесцен-
цией обладают именно коллоидные системы.
Однако в-молекулярных растворах также происходит
рассеяние света, хотя гораздо более слабое и обусловленное
другими факторами, чем в более грубодисперсных коллоид-
коллоидных системах. Благодаря тепловому движению молекул
в жидкостях и газах, возникают случайные сгущения частиц
или флуктуации плотности, а в растворах, кроме того,
флуктуации концентрации, которые также являются цент-
центрами рассеяния света (в рассмотренных выше системах
частицы далеко удалены друг от друга и «флуктуацией»
как бы являлась сама коллоидная частица).
55
Теория флуктуационного рассеяния света была развита
Эйнштейном A910) и особенно плодотворно применена для
растворов макромолекул Дебаем A947). В настоящее время
измерения светорассеяния являются одним из наиболее
важных методов исследования растворов белков и высоко-
высокополимерных веществ.
Для растворов макромолекул основное значение имеют
флуктуации концентрации раствора, а не плотности раст-
растворителя. При флуктуации концентрации раствора проис-
происходит изменение свободной энергии, которое можно рас-
рассматривать как работу осмотического давления, необхо-
необходимую для изменения концентрации в данном объеме v
на величину Ас (это обстоятельство объясняет внутреннюю
связь уравнений (III. 4) и (III. 5). Одновременно с изме-
изменением концентрации на Ас изменяется показатель прелом-
преломления раствора п на величину An.
Дебай показал, что мутность разбавленного раствора
32
3NX*
-сМ=НсМ,
(III. 3)
где п и п0 — показатели преломления раствора и раствори-
растворителя, с — концентрация раствора и М— молекулярный вес
растворенного вещества; константа Н включает члены, посто-
постоянные для данной серии измерений. Из A11,3) следует, что
т )
М
(Ш. 4)
По уравнению III. 4 можно рассчитать величину М,
исходя из экспериментальных данных Я, сит; III. 4 спра-
справедливо лишь для идеальных растворов и эквивалентно
уравнению ВантТоффа для осмотического давления тех
тс 1
же растворов -тгу— = ~тг (см. формулу II. 7). Для не-
неидеальных растворов светорассеяние и осмотическое давле-
давление также подчиняются эквивалентным уравнениям
1
RTc
Не
(III. 5)
56
отличающимся лишь коэффициентом в Вс, где множитель В
характеризует взаимодействие между растворенными мо-
молекулами; при В = О (II 1.5) переходит в (III. 4).
Экспериментально измеряется разность показателей пре-
преломления An — п—По, которая должна составлять около
0,001, но так как в (III. 3) она входит в квадрате, ее необ-
необходимо измерить с высокой точностью (до ±0,00 001). Кроме
того, необходимо установить мутность на точном фотометре
визуального (рис. 17) или фотоэлектрического типа.
Исследуемые растворы должны быть совершенно свободны
от источников посторонней мутности (частиц пыли и др.),
для чего их центрифу-
центрифугируют на супер- или | з
ультрацентрифуге; в ка-
качестве стандарта берут
многократно перегнан-
перегнанные чистые жидкости —
сероуглерод или толуол.
Концентрация исследуе-
исследуемых растворов состав-
составляет около 0,1 %. При
работе с растворами
макромолекул, размеры
которых больше Х/10,
необходимо , учитывать
рассеяние от различных
концов молекул, для че-
чего измеряют интенсив-
интенсивность рассеянного света
R под разными углами
от 20° до 150° (угловое
распределение интенсив-
ностей); асимметрию молекул часто характеризуют отноше-
отношением R^JRl3b. В растворах полидисперсных веществ
метод светорассеяния характеризует средневесовой моле-
молекулярный вес Mw.
Методом светорассеяния в настоящее время исследовано
большое количество высокомолекулярных веществ, бел-
белков, нуклеиновых кислот. В качестве примера в табл. 5 и на
рис. 18 приведены данные Доти, Цимма и Марка для раст-
растворов нескольких фракций полистирола в метилэтилкетрне.
Следует указать,что приведенные выше уравнениями. 1—
III. 5) относились к растворам веществ, не обладающих
57
0.6 0,8 1.0
С г но 100 мп
Рис. 18. Зависимость
от
концентрации для растворов
фракций полистирола в метилэтил-
кетоие
специфическим поглощением каких-либо лучей падающего
света. При наличии такого поглощения зависимость ин-
интенсивности рассеянного света от 1/Х4 и квадрата объема
частиц v2 в (III. 1) нарушается, изменяется степень поляри-
поляризации рассеянного света и др. Теорию оптических свойств
поглощающих дисперсных систем, к которым, например,
относятся золи металлов, разработал Ми.
Таблица 5
Сравнение молекулярных весов фракций
№ фракции
1
2
3
4
5
пэлистирола
Молекулярный
по осмотическому
давлению
172 000
198 000
91 000
по
вес
светорассе-
светорассеянию
178 000
182 000
107 000
190 000
445 000
ПОГЛОЩЕНИЕ СВЕТА
При прохождении света через молекулярные растворы,
пленки и другие однородные среды интенсивность прошед-
прошедшего света J всегда меньше интенсивности падающего света
/о, вследствие некоторого поглощения света в этой среде.
Согласно закону Ламберта—Бэра, в данном слое однородной
среды отношение J/Jo определяется только числом погло-
поглощающих частиц или молекул
j/jo==e~?C\ (HI. 6)
где /—толщина слоя, с—молярная концентрация раство-
растворенного вещества и г —молярный коэффициент поглоще-
поглощения вещества. Величина г не зависит от концентрации,
но изменяется с длиной волны, температурой и природой
растворителя. При выражении закона Ламберта—Бэра через
десятичные логарифмы величина е10 называется коэффи-
коэффициентом экстинции или погашения (s10 = 0,434s).
В окрашенных растворах поглощение измеряют,
пользуясь колориметрами с визуальным (рис. 19) или
фотоэлектрическим отсчетом. В колориметре Дюбоска
(рис. 19) свет от зеркала М проходит в осевом
58
направлении через стаканчики Л . и В, содержащие
испытуемый Сх и сравнительный Ci растворы, после чего
оба пучка света сравниваются при помощи окуляра Е.
Толщина поглощающего слоя изменяется при помощи от-
отсчетов на лимбах / или /'. Из (III. 6) видно, что в растворах
одного и того же вещества (когда г одинаково) при равной
освещенности полей в Е, т. е. при равной относительной
интенсивности проходя-
проходящего света J/Jo, CJX =
= Ci/i, откуда
Сх =
(III. 7)
ЕЛ
На основании фор-
формул (III. 6) и (III. 7)
производится колори-
колориметрическое определение
концентрации растворов
окрашенных веществ.
Измерения величины
J/Jo в монохроматиче-
монохроматическом свете при различ-
различных длинах волн X и
одинаковой концентра-
концентрации раствора с и тол-
толщине слоя / позволяют
выразить зависимость
коэффициента г от к в
виде спектральных кри-
кривых поглощения, харак-
характерных для каждого ве-
вещества. В качестве при-
примера на рис. 20 приве-
приведены спектры поглоще-
поглощения хлорофиллов а и в
в интервале длин волн от 400 до 700 мр, ясно указы-
указывающие на различия этих веществ. Спектры поглощения
измеряют на фотоэлектрическом спектрофотометре (нап-
(например, на отечественном приборе СФ-4), или на спектро-
спектрографе с последующим микрофотометрированием. В зави-
зависимости от примененных длин волн различают оптические,
ультрафиолетовые или инфракрасные спектры поглощения
(стр. 61).
59
Рис. 19. Колориметр Дюбоска
Справедливость закона Ламберта—Бэра, однако, ограни-
ограничена определенной областью концентраций в разбавленных
растворах; при более высоких концентрациях значения ?
могут изменяться. В концентрированных растворах откло-
отклонения от закона Ламберта — Бэра обусловливаются из-
изменением ассоциации или химического состояния вещества
(например, вследствие гидролиза) при изменении концент-
концентрации. В коллоидных растворах часть света теряется в
результате как истинного поглощения, так и рэлеевского
150
100
500
600
700
Рис. 20. Спектры поглощения хлорофил-
лов айв
рассеяния, что должно учитываться при измерениях. Ок-
Окраска коллоидных растворов вообще зависит от степени дис-
дисперсности. В высокодисперсных системах с непроводящими
частицами (золи SiCb и др.) агрегация не изменяет погло-
поглощения, но в золях с проводящими частицами, по мере
уменьшения степени дисперсности максимум поглощения
смещается в сторону длинных волн; поэтому при агрега-
агрегации частиц золота цвет золя изменяется от красного к си-
синему и, напротив, в золях серебра при повышении степени
дисперсности окраска золя переходит от синей к красной
и затем к желтой.
60
I
В растворах макромолекул и белков окраска зависит,
прежде всего, от истинного поглощения, максимумы кото-
которого, однако, часто расположены вне видимой области спект-
спектра. Поэтому растворы, кажущиеся бесцветными в видимых
лучах, могут обладать сильным поглощением в ультрафио-
' летовой или инфракрасной областях спектра.
Ультрафиолетовые спектры поглощения' определяются
возбуждением электронных уровней атомов и молекул и
обладают максимумами, положение которых характерно
для определенных атомных группировок, сопряженных
двойных связей и др. В белках ультрафиолетовые спектры
поглощения в основном определяются ароматическими ами-
аминокислотами—¦ фенилаланином (\макс= 260 мр), тирози-
тирозином и триптофаном (\)(вте= 280 мр), причем спектры пог-
поглощения могут быть даже использованы для аналитическо-
аналитического определения этих аминокислот. Нуклеиновые кислоты
и нуклеопротеиды обладают настолько резким максимумом
поглощения при 260—265 м\ь, что при помощи фотографи-
фотографирования в ультрафиолетовом микроскопе легко определить
их содержание в отдельных клетках (Брумберг). Зависи-
Зависимость ультрафиолетовых спектров поглощения от рН, сос-
состава среды, от образования комплексов с другими соеди-
соединениями позволяет исследовать изменения состояния раст-
растворенных веществ; так, по смещению максимума поглоще-
поглощения с 280 до 260—265 м \ь было обнаружено образование
комплекса между белками и полисахаридами (Розенфельд).
Линейные полимеры обычно не имеют интенсивных полос
поглощения в видимой и ближней ультрафиолетовой об-
областях спектра.
Инфракрасные спектры поглощения характеризуют
колебательно-вибрационные частоты связей в определенных
атомных группировках — карбонильной группе С = О
E,5 — 6,0 ji), в иминогруппе N—Н B,8—2,9 \у), в гидрок-
сильной группе ОН B,7—2,85 и), С—Н-связи в метильной
группе C,2—3,5" jx) и Др- Число возможных внутренних
колебаний в сложной молекуле полимеров чрезвычайно
велико, что затрудняет полную расшифровку инфракрасных
спектров; поэтому иногда приходится ограничиваться уста-
установлением характеристических частот, присущих опреде-
определенным атомным группам (рис. 21). Интенсивность поглоще-
поглощения, т. е. высота спектральных максимумов, характеризует
количественное содержание соответствующих групп в данном
веществе. Измерения инфракрасных спектров часто произ-
61
водят на пленках белков или полимеров, так как вода об-
обладает значительным максимумом поглощения при 3 jx
(волновое число 3300 смг1), что мешает определению групп,
поглощающих в той же области. Интересные результаты
дают измерения на ориентированных или вытянутых плен-
пленках полимеров с применением поляризованного инфра-
инфракрасного света, в котором электрический вектор колеба-
колебаний либо параллелен, либо перпендикулярен направлению
ориентации; в обоих случаях поглощение получается раз-
различным и по отноше-
Ъалентные нию величин поглоще-
Десрор.мационные У tacTmt>lj ния в обоих направле-
nh ниях (дихроическое от-
отношение) можно опре-
определить пространствен-
пространственное расположение свя-
связей в ориентированных
участках полимеров. Так
например, этим путем
Л было показано, что в
пептидных цепях связи
NH направлены периен-
зооо З^оо дикулярно пептидным
группам в вытянутых
JZOO
1Ш W00
Волновое число см *
Рис. 21. Инфракрасные спектры молекулах и параллель-
полипептида (/) и денатурированного ИО ИМ — В свернутых
кератина (//) молекулах полимеров
(Мицусима, Амброзе);
инфракрасный дихроизм исследовался также на пленках
полиэтилена и поливинил ацетата (Волькенштейн). Про-
Процессы окисления, термической деструкции, полимеризации,
денатурации и другие изменения полимеров, связанные с
появлением новых частот или изменением их интенсивно-
интенсивности, также могут быть исследованы с помощью инфра-
инфракрасных спектров поглощения.
ВРАЩЕНИЕ ПЛОСКОСТИ ПОЛЯРИЗАЦИИ
Полимеры, образованные из оптически-активных моно-
мономеров с преобладанием одного из антиподов (например,
белки построены исключительно из L-аминокислот,
полисахариды —¦ из D-углеводов и др.), изменяют
направление плоскости поляризации при прохождении че-
62
рез них поляризованного света. Вращение плоскости поля-
поляризации измеряется специальными приборами — поля-
поляриметрами. Если при прохождении через слой раствора /
при концентрации растворенного вещества С г на 1 г
раствора, поляризованный падающий луч поворачивается
на угол а, то удельное вращение [а] растворенного вещества
равно
где р — плотность раствора. Для белков удельное вращение
всегда отрицательно и колеблется для различных белков от
— 30° до — 60°. Величина оптического вращения в значи-
значительной степени зависит от рН, состава и конфигурации
полипептидной цепи и в настоящее время измерениями [а]
широко пользуются для изучения денатурационных из-
изменений в полипептидах и белках (Каузман, Доти и др.).
ОПТИЧЕСКАЯ АНИЗОТРОПИЯ
Характерным свойством коллоидных частиц является
их оптическая анизотропия, т. е. различие оптических
свойств по различным направлениям. В одних случаях
оптическая анизотропия обусловлена внутренним строением
частиц, в других -— их формой или искусственно вызванной
ориентацией частиц. Кроме того, исследование оптической
анизотропии при различных условиях — весьма важный
метод изучения структуры коллоидных частиц (с исполь-
использованием поляризованных лучей, т. е. лучей, имеющих
преимущественные плоскости колебаний, о которых уже
неоднократно упоминалось выше).
Экспериментально для исследования оптической ани-
анизотропии применяется поляризационная оптика, состоящая
из поляризаторов, создающих пучки плоско-поляризован-
плоско-поляризованного света при помощи призм Николя или поляроидов и
анализаторов с компенсаторами, пользуясь которыми можно
обнаружить и измерить поляризацию светового пучка.
Исследуемые растворы или пленки, в которых иногда соз-
создается, соответственно, течение или растяжение, помещают
между поляризатором и анализатором.
Плоско-поляризованный луч в анизотропной среде,
характеризующейся расположением анизотропных элемен-
элементов вдоль одной оптической оси, разделяется на два луча,
63
один из которых колеблется перпендикулярно оптической
оси среды (этот луч называется обыкновенным), а другой —
параллельно оптической оси среды (необыкновенный луч).
Скорость распространения обоих лучей в среде и их показа-
показатели преломления различны, вследствие чего возникает
двойное лучепреломление An, измеряемое разностью пока-
показателей преломления для необыкновенного (пе) и обыкно-
обыкновенного (п0) лучей. Двойное лучепреломление называется
положительным, если пе>по(Ап>0) и отрицательным,
если пе < по(Ап < 0). Если коллоид-
коллоидные частицы сами по себе оптически
анизотропны, что наблюдается во
многих лиофобных золях, то при-
присущая им величина An мало зависит
от показателя преломления диспер-
дисперсионной среды; в этом случае говорят
о собственном двойном лучепреломле-
лучепреломлении частиц. Однако оптическая ани-
j зотропия в коллоидной системе может
,/| быть также обусловлена просто ори-
ориентированным расположением асим-
асимметричных по своей форме частиц,
которые сами по себе могут и не об-
обладать оптической анизотропией; в
этом случае говорят о двойном луче-
лучепреломлении формы. Оно исчезает
при равенстве показателей преломле-
преломления коллоидных частиц и дисперсион-
дисперсионной среды. Таким образом, измеряя
величину An при различных пока-
показателях преломления среды, можно отличить оба вида
двойного лучепреломления.
Практически, однако, оптическая анизотропия колло-
коллоидных систем может определяться одновременно обоими
факторами; тогда при уравнивании показателей прелом-
преломления частиц и среды двойное лучепреломление полно-
полностью не исчезнет и лишь уменьшится до некоторой мини-
минимальной величины, которая и будет характеризовать соб-
собственную анизотропию частиц.
Например, из рис. 22 видно, что желатина (кривая //)
имеет лишь двойное лучепреломление формы, а миозин
(кривая /), кроме того, обладает заметным собственным
двойным лучепреломлением.
Рис. 22. Зависимость
двойного лучепрело-
лучепреломления миозина (/)
и желатины (//) от
показателя преломле-
преломления растворителя
64
Ориентацию коллоидных частиц или макромолекул в
растворах можно Еызвать различными способами и, соот-
соответственно, можно исследовать двойное лучепреломление
в электрическом поле (эффект Керра), в магнитном поле
(эффект.Коттона — Мутона) и при течении раствора (эффект
Максвелла). Коллоидный раствор с ориентированными
вытянутыми частицами приобретает описанные выше свой-
свойства одноосного оптически анизотропного тела, но полнота
ориентации частиц нарушается их вращательным броунов-
броуновским движением; в результате, в растворе устанавливается
определенное распределение ориентации, при котором угол
/ между направлением ориентации и оптической осью в
жидкости, в зависимости от силы ориентирующих воздей-
воздействий, изменяется от значения 45° при сла-
слабой ориентации до 0° при сильной ориента-
ориентации частиц.
Наибольшее значение при исследовании
коллоидных растворов получило изучение
двойного лучепреломления при течении (оно
называется также двойным лучепреломлением
в потоке). Для этого раствор помещают меж-
между двумя коаксиальными цилиндрами, из ко-
которых один вращается, а другой остается
неподвижным, и рассматривают поле между
цилиндрами в плоско-поляризованном моно-
монохроматическом свете при скрещенных нико-
лях или поляроидах. В неподвижном колло-
коллоидном растворе поле зрения кажется темным,
но при течении возникает ориентация вытя-
вытянутых частиц (например, V2O5 или вируса
-табачной мозаики), раствор приобретает
двойное лучепреломление и поле становится светлым.
При этом в поле зрения наблюдается характерная для
одноосного кристалла крестообразная фигура — крест
изоклин (рис. 23), поворот которой зависит от скорости
течения и может быть измерен при помощи компен-
компенсатора. Положение креста изоклин позволяет непо-
непосредственно определить угол /, характеризующий степень
ориентации частиц. Зная значение угла % при известной
скорости течения жидкости, можно вычислить коэффициент
вращательной диффузии В (см. стр. 33), который для
вытянутых эллипсоидных частиц с известным соотношением
большой и малой полуосей у—-I равен
» «Коллоидная химия» 6?
Рис. 23.
Крест изок-
изоклин в дву-
преломляю-
щей жидко-
жидкости, находя-
находящейся между
коаксиаль-
коаксиальными цилин-
цилиндрами
в =
ЗРТ
lb т. Nr.b3
(III. 9)
откуда может быть найдена длина коллоидных частиц
2 Ь. На основании измерений двойного лучепреломления при
течении были определены линейные размеры молекул мно-
многих высокополимерных веществ (В. Цветков, Садрон),
белков (Эдсалл), нуклеиновых кислот (Жоли), вирусов
(Бенуа) и др. (табл. 6).
Таблица 6
Длина некоторых частиц и макромолекул по данным
двойного лучепреломления при течении
Вещество
Зеин
Фибриноген человека . . .
Полистирол (М-700000) . .
Глобулин антител лошади .
Частицы золей V2O5 ....
Вирус табачной мозаики
Нуклеиновая кислота (ДНК)
Актомиозин
Отношение
(Ь/а)
16
18
—
20
—.
20
>20
—
Длина 1Ь,
о
А
350
700
800
1280
1600
3000
5000—8000
5000—8000
По мнению Эдсалл а, этот метод не следует применять
при установлении размеров молекул с длиной меньше
о
200 А, но в случае сильно вытянутых молекул он весьма
удобен как для определения линейных размеров молекул,
так и для изучения изменения их состояния при денатура-
денатурации, деструкции (в частности, этим методом был показан
распад нитей актомиозина при действии аденозинтрифос-
форной кислоты) и др.
Следует отметить, что при высоких скоростях течения
происходит не только ориентация асимметричных частиц,
но также их деформация или растяжение, а это приводит к
появлению нового вида двойного лучепреломления — дефор-
деформационного или эластического, которое устраняется при
снятии деформирующего усиления. Эластическое двойное
лучепреломление особенно удобно наблюдать в прозрачных
пленках и гелях полимеров (полистирол, полиметилметак-
рилат, желатино-глицериновые студни и др.), где это явле-
явление практически используется для изучения распределения
напряжений в образце (С. Соколов).
66
Интересный метод определения коэффициента враща-
вращательной диффузии (У и размеров молекул по поляриза-
поляризации флуоресценции разработан Вебером. Он получал хими-
химические соединения белков с флуоресцирующими красителя-
красителями (например, с /-диметиламинонафталин-5-сульфонил-
хлоридом) и измерял интенсивность флуоресценции по
различным направлениям. Было показано, что степень
поляризации флуоресценции наибольшая при малых 9,
тогда как при больших в флуоресценция полностью деполя-
деполяризована. Промежуточные значения степени поляризации
флуоресценции отвечают определенным значениям ©, отку-
откуда по формуле (III. 9), или по (II. 5.) вычисляются размеры
молекул. Вебер исследовал этим методом размеры ряда
белковых молекул и процессы их денатурации; Гейнц
применил этот метод к растворам полистиролов; Валь — к
растворам поливиниламинов и др.
Асимметрия формы коллоидных частиц проявляется и в
светорассеянии. Поляризация рассеянного света оказы-
оказывается наибольшей в направлении, перпендикулярном
направлению падающего луча. Кришнан показал, что если
поляризация рассеянного света не полная даже в том слу-
случае, когда падающий свет является вертикально поляри-
поляризованным, то это обстоятельство с несомненностью указы-
указывает на геометрическую или оптическую анизотропию
коллоидных^1 частиц. Интенсивность светорассеяния при
поляризованном падающем свете также зависит от формы
частиц, возрастая в том случае, если электрический вектор
падающего поляризованного луча параллелен длине палоч-
палочкообразной частицы или плоскости пластинчатой частицы.
Вызывая ориентацию асимметричных частиц течением жид-
жидкости, можно наблюдать, что относительное возрастание
интенсивности светорассеяния в поляризованном падаю-
падающем свете более значительно при течении растворов с
палочкообразными частицами, чем с пластинчатыми части-
частицами. Этим путем Диссельхорст и Фрейндлих показали
наличие палочкообразных частиц в золях V2O 5, пластин-
пластинчатых частиц — в золях РегОз, приблизительно сфери-
сферических частиц в золях Ag и AS2S3 и др.
ЭЛЕКТРОННАЯ МИКРОСКОПИЯ И РЕНТГЕНОГРАФИЯ
Пределом разрешающей способности обычного светового
микроскопа является диаметр частиц около 0,2 р., но при
6*
67
этом размере уже нельзя разобрать деталей формы. В ульт-
ультрафиолетовом микроскопе Брумберга нижний наблюдае-
наблюдаемый размер, который, как известно, тем ниже, чем короче
длина применяемых волн, может быть доведен до 0,1 ц.
Однако для коллоидных частиц эти пределы слишком грубы.
Используя явление тиндаллевского рассеяния света,
Зигмонди A903) сконструировал ультрамикроскоп, в ко-
котором при наблюдении в темном поле могут быть обнаруже-
обнаружены рассеивающие частицы размером всего до \7m\i, но при
этом изображение частиц представляется лишь в виде диф-
фракционных пятен; непосредственно определить форму
и истинные размеры частиц этим путем невозможно. В пос-
последние годы для наблюдения размеров и формы коллоид-
коллоидных частиц и некоторых макромолекул чаще всего поль-
пользуются электронным микроскопом, в котором применяются
пучки электронов .с длиной волны всего 0,02—0,05 А.
Ход электронного пучка в электронном микроскопе оди-
одинаков с ходом световых лучей в обычном микроскопе, но
фокусировка пучка производится не оптическими, а магнит-
магнитными или электростатическими линзами. Изображение
рассматривается на флуоресцирующем экране или фотогра-
фотографируется на пластинке, причем снимок может быть затем
увеличен. Разрешающая способность электронного микро-
микроскопа достигает 10—15 А, а полное увеличение превышает
300 тыс. раз. Этим путем были изучены размеры и форма
частиц многих лиофобных коллоидов, аэрозолей, молекул
различных полимеров и вирусов и др. Так, например,
Каргин и Берестнева изучили процессы образования зо-
золей золота, V2O5 и др. (см. стр. 21), Рогинский
исследовал строение ряда катализаторов, Уайков ¦=— моле-
молекулы вируса табачной мозаики (рис. 24), или частицы латек-
латекса полистирола (рис. 25). Значение электронного микроско-
микроскопа для исследования коллоидных структур может быть
сопоставлено в настоящее время лишь со значением свето-
светового микроскопа для исследования гистологических струк-
структур. Наблюдения в электронном микроскопе ограничива-
ограничиваются необходимостью тщательного высушивания объектов,
так как внутри электронного микроскопа поддерживается
высокий вакуум для прохождения электронного пучка;
кроме того, вследствие сильного поглощения электронов,
изучаемые образцы должны быть весьма тонкими A—10 р.);
наконец, следует учитывать изменения образца под влияни-
68
Рнс. 24. Молекулы вируса табачной мозаики; тол-
толщина палочкообразных частиц равна 150 А (уве-
лнч. 45000 раз)
Рис. 25. Суспензия латекса полистирола;
в части снимка видно два слоя частиц
(увелич. 7000 раз).
69
ем воздействия пучка электронов. В настоящеее время раз-
разработаны новые методы наблюдения в электронном микро-
микроскопе не самих образцов, а отпечатков с них, которые могут
быть получены и с влажных объектов.
Если в электронном микроскопе используется поглоще-
поглощение электронов для изучения внешней формы и размеров
коллоидных частиц и макромолекул, то методы рентгено-
рентгенографии и электронографии при исследовании внутренней
структуры коллоидных частиц и полимерных материалов
основаны на диффракции рентгеновых лучен, или, соот-
соответственно, электронов. При регулярном расположении
атомов, например в кристалле, интерференция рассеянных
волн приводит к определенной системе диффракционных
пятен. Положение пятен определяется законом Вульфа-
Брэгга:
«>. = 2flr-sina, (III. 10)
где а — длина волны падающих рентгеновых лучей, d —
расстояние между параллельными плоскостями, в которых
расположены рассеивающие атомные центры, а—угол,
характеризующий отклонение рентгеновского луча от атом-
атомной плоскости \\ п— целое число A, 2, 3...), соответствую-
соответствующее спектрам первого, второго и более высоких порядков.
Вполне упорядоченные трехмерные кристаллические ре-
решетки дают отражения в виде резких точечных пятен (рис.
26. I), волокнистые структуры — в виде небольших тонких
дуг (рис. 26. II), а для аморфных структур отражения сли-
сливаются в сплошные размытые кольца (рис. 26.III). Изу-
Изучение отражений и измерение их положения и интенсивнос-
интенсивности позволяет установить повторяющиеся расстояния в ве-
веществе и их взаимное пространственное расположение, а
также структуру рассеивающих центров, хотя эти исследо-
исследования в случае сложных молекул требуют чрезвычайно
большой вычислительной работы (часто с применением
электронных счетных машин.) В настоящее время методы
рентгеноструктурного анализа достигли настолько высокого
уровня, что с их помощью была полностью расшифрована
внутренняя структура, вплоть до определения простран-
пространственного положения каждого атома таких сложных моле-
молекул, как молекулы пенициллина или витамина Bis (Кроу-
фут), и достигнуты значительные успехи в расшифровке
строения молекул некоторых белков гемоглобина
(Перутц), инсулина (Лоу), рибонуклеазы (Карлайл) и
высокомолекулярных веществ, хотя эта проблема еще да-
70
лека от разрешения. -Помимо этого, изучение рассеяния
рентгеновых лучей под малыми углами в белковых раство-
растворах (Ритленд) позволило определить величину рассеиваю-
рассеивающего объема в растворах и этим путем получить новые
данные о размерах белковых молекул и величине их гидра-
гидратации. о
Так как длина рентгеновых лучей составляет около 1А
о
(от 0,1 до 10А), а пучка электронов— около 0.02А, то обэ-
Рис. 26. Рентгенограммы различных веществ: /—алмаза,
//—волокна рами, ///—аморфного каучука.
ими методами можно исследовать структуры различных раз-
размеров. Методы электронографии позволяют обнаружить
наличие кристаллической упорядоченности в расположении
отдельных атомов и наиболее удобно прилагаются к иссле-
исследованию частиц золей металлов и окислов металлов (Кар-
гин, Уайзер). Методы рентгенографии и электронографии
(особенно при их сочетании) позволяют глубоко исследовать
степень упорядоченности внутренней структуры коллоид-
коллоидных частиц и высокополимерных веществ, определить раз-
размеры и структуру упорядоченных участков и изучить их
изменения при различных процессах (вытяжке, набухании,
нагревании и др.).
Выводы
Оптические свойства коллоидов тесно связаны с разме-
размерами, формой и внутренней структурой коллоидных час-
частиц и поэтому имеют важное значение при изучении кол-
коллоидных систем. Характерными для коллоидных систем
свойствами являются: а ) дифракционное рассеяние света
на коллоидных частицах (уравнение III. 1), которое исполь-
71
зуется, в частности, при нефелометрических измерениях,
и б) флуктуационное светорассеяние на сгущениях концент:
рации молекул в растворах полимеров (уравнение III. 3),
применяемое, в частности, для определений молекулярно-
молекулярного веса и асимметрии формы макромолекул в растворах.
Изучение поглощения света в видимой, ультрафиолето-
ультрафиолетовой и инфракрасной областях спектра характеризует на-
наличие и природу специфических поглощающих группиро-
группировок в частицах, их концентрацию в растворах (III. 6 и
III. 7), а также позволяет исследовать влияние изменений
конфигурации макромолекул, их взаимодействия с различ-
различными веществами и другие изменения их состояния. В кол-
коллоидных растворах явления поглощения света осложняются
явлениями светорассеяния и зависимостью поглощения от
степени дисперсности частиц.
Оптическая и геометрическая анизотропия коллоидных
частиц исследуются методами поляризационной оптики,
среди которых основное значение имеет изучение двойного
лучепреломления, как собственного, обусловленного опти-
оптической анизотропией частиц, так и двойного лучепреломле-
лучепреломления формы, зависящего от ориентированного расположе-
расположения асимметричных частиц. Метод двойного лучепреломле-
лучепреломления при течении особенно широко используется для опре-
определения коэффициента вращательной диффузии (III. 9) и
линейных размеров вытянутых частиц; для той же цели
иногда изучают поляризацию флуоресценции.
Прямым методом определения размеров и формы кол-
коллоидных частиц, молекул вирусов и ряда макромолекул
является электронная микроскопия. Внутренняя структура
коллоидных частиц и ее изменение при различных, процес-
процессах изучаются методами рентгенографии и электроногра-
электронографии.
Вопросы
1. Чем отличаются яилення рэлеевского и флуктуашюшюго
рассеяния света? Сопоставьте уравнения обоих процессов.
2. Какими оптическими методами можно измерить размеры
коллоидных частиц?
3. В чем заключается закон Ламберта—Бэра и чем выливаются
отклонения от этого закона?
4. Сравните нефелометричеекпо и колориметрические методы
определения концентрации растворов.
5. Какими оптическими методами исследуется форма коллоид-
пых частиц?
6. Сравните спектры поглощения и пг.дпчои, ультрафполето-
72
V
вой и инфракрасной областях спектра. Укажите их значение и об-
области применения.
7. В чем заключаются основные различия в поглощении света
в истинных и коллоидных растворах?
8. В чем заключается различие двойного лучепреломления —
.собственного, зависящего от формы, и эластического? Каково
-значение каждого из них и каковы методы их экспериментального
изучения?
9. Укажите области применения поляризационной оптики в
коллоидной химии.
10. Сравните методы электронной микроскопии и электроно-
электронографии, их принципы и области применения. Какими методами
изучается внутренняя структура коллоидных частиц?
«Коллоидная химия»
Глава четвертая
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ И АДСОРБЦИЯ
Для всех коллоидных систем, частицы которых суспен-
суспендированы в инертной среде и образуют с ней физическую
поверхность раздела, процесс диспергации означает значи-
значительное увеличение поверхностной энергии системы. Легко
видеть, что если некоторое количество вещества в виде
кубика в 1 см3 имеет общую поверхность 6 см1, то это же
количество вещества в виде кубиков в 1 [Xs будет иметь
общую поверхность 60 000 см2. Таким образом, удельная
поверхность вещества (суммарная поверхность 1 см3
вещества) резко возрастает в диспергированном состоянии,
приблизительно пропорционально уменьшению линейных
размеров частиц. Значение этого фактора особенно велико
для лиофобных коллоидов, частицы которых инертны по
отношению к среде; в этом случае увеличение удельной
поверхности непосредственно связано с увеличением сво-
свободной поверхностной энергии, что лежит в основе наибо-
наиболее характерных свойств лиофобных коллоидов. Благодаря
большому значению поверхностных явлений для проблем
коллоидной химии, необходимо вначале рассмотреть на
простых примерах некоторые общие физико-химические
свойства поверхностей раздела.
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
Дисперсные частицы и дисперсионная среда в лиофоб-
лиофобных коллоидах всегда относятся к различным фазам. В пер-
первой главе уже указывалось, что для существования
фазы в ней должно содержаться достаточно большое ко-
количество атомов или молекул для того, чтобы имелась
74
возможность применения законов термического равновесия
и образования физической поверхности раздела. Поэтому,
когда мы говорим о наличии фаз в лиофобном золе,
предполагается, что указанные условия выполняются и что
между коллоидными частицами и средой действительно
существуют физические поверхности раздела, похожие по
своим свойствам на обычно изучаемые макроповерхности
раздела.
Молекулы вещества в данной фазе на границе раздела с
другой фазой всегда испытывают неодинаковое влияние
сил молекулярного взаимодействия, которые внутри каж-
каждой фазы полностью компенсируются. Например, силы
взаимодействия со стороны жидкости на молекулы В,
лежащие в поверхности раздела жидкость — газ, значи-
значительно больше, чем со стороны газа, тогда как в отношении
молекул Л они компенсируются (рис. 27). В результате из-
Гпз
Жидкость,
О О
Пограничный слой
О
Рис. 27. Поверхность раздела жид-
жидкость — газ
Рис. 28. Растяже-
Растяжение пленки жид-
жидкости
бытка сил притяжения со стороны жидкости молекулы В
стремятся втянуться внутрь жидкости, вследствие чего
поверхность раздела жидкости стремится к уменьшению.
В свободной капле жидкости стремление поверхности к
уменьшению, как известно, приводит к ее шарообразной
форме. Это изменение является частным проявлением из-
известного из термодинамики общего стремления свободной
энергии системы к уменьшению. В различных системах
эта общая закономерность проявляется по-разному.
В чистых жидкостях единственной возможностью умень-
уменьшения свободной энергии системы является уменьшение
величины поверхности. При образовании пленки жидкости
на проволочном каркасе А В (рис. 28) стремление пленки к
5*
75
уменьшению своей поверхности путем приближения про-
проволочки В к А в момент равновесия точно уравновешивает-
уравновешивается растягивающим усилием груза С, подвешенного к про-
проволочке В. Для увеличения поверхности пленки необхо-
необходимо, очевидно, увеличить груз С и затратить работу на
передвижение проволочки В вниз; одновременно возрастет
поверхностная энергия системы. Величина работы, ко-
которую необходимо затратить на увеличение поверхности
на 1 см2 (или на образование этой поверхности), выражен-
выраженная в эрг/см2, численно равна свободной поверхностной
энергии на 1 см2 поверхности; эта величина и называется
поверхностным натяжением. Таким образом, поверхност-
поверхностное натяжение а представляет собой избыток свободной
энергии, приходящийся на 1 см2 поверхности раздела.
Размерность поверхностного натяжения [эрг/см2] иден-
идентична размерности [дин/см], в которой эта величина также
часто выражается; она особенно удобна при рассмотрении
линейной границы между двумя поверхностями.
Среди многочисленных методов измерения поверхност-
поверхностного натяжения а известны методы, непосредственно осно-
основанные на схеме, приведенной на рис. 28; другие методы
связаны с определением среднего размера или
веса падающей капли жидкости или с измере-
измерением давления р , под которым выдавливается
из стеклянного капилляра пузырек газа в
жидкость, или капли одной жидкости в другой
жидкости (если нужно измерить ai2 на границе
двух жидкостей). В растворах для изучения
кинетики изменений а иногда измеряется дав-
давление, под которым размеры пузырька газа
остаются постоянными.
При измерениях по методу давления ши-
широко используется прибор Ребиндера, в кото-
котором газ или другая жидкость (рис. 29) про-
продавливаются под давлением через капилляр-
капиллярный кончик К (диаметром 0,1—0,3 мм), лишь слегка
погруженный в поверхность раздела. Величина давления р
устанавливается при помощи чувствительного манометра:
она пропорциональна величине поверхностного натяжения
на границе раздела
o = k-p, (IV. 1)
где к— константа капилляра — измеряется на жидкости с
известным -3.
Рис. 29.
Прибор
Ребинде-
Ребиндера (основ-
(основная
часть)
76
Приводим величины поверхностного натяжения различ-
различных жидкостей *.
Жидкость а, эрг/см2
Вода 72,7
.Ртуть ' 430
Спирт этиловый 22
Бензол 29
Эфир этиловый 16
Гексан ..... 21
Сыворотка крови человека (при 38°) . . . 46—47
Цитоплазма амеб (на границе с маслом) . . 0,5—1,5
Цитоплазма яиц морского ежа (на границе
с маслом) 1,0—2,0
Поверхностное натяжение с повышением температуры
уменьшается, и в критической точке, при которой поверх-
поверхность раздела жидкость — пар (мениск) исчезает, поверх-
поверхностное натяжение равно нулю. Вообще, чем меньше оХ2
на границе раздела двух жидкостей, тем выше взаимная
растворимость жидкостей и при критической температуре
полного смешения обеих жидкостей (например, для системы
вода—фенол при 66°) а12 = 0. По правилу Антонова, по-
поверхностное натяжение ai2 на границе двух взаимно огра-
ограниченно растворимых жидкостей равно разности поверх-
поверхностных натяжений этих жидкостей (однако, насыщенных
одна другой) на границе с воздухом:
ala=Ol —og. (IV. 2)
Правило Антонова справедливо при условиях, близких
к полному смешению жид-
жидкостей.
На границе раздела трех
фаз наблюдаются более слож-
сложные соотношения между меж-
фазными поверхностными на-
натяжениями, которые лежат в
основе важных явлений сма-
смачивания. Если на твердую
поверхность 3 (рис. 30) на-
нанесена капля воды / и обе
поверхности граничат с га-
газом 2, то капля образует с твердой поверхностью краевой
угол смачивания О (измеряемый в водной фазе). По
* Измерения а на границе с воздухом, при комнатной темпера-
температуре, за исключением оговоренных данных.
Рис. 30. Смачивание на
нице раздела трех фаз
Ребиндеру)
гра-
(по
77
уравнению Лапласа, величина cos В при равновесии связа-
связана с межфазными поверхностными натяжениями следую-
следующим соотношением:
cos 6=-^^-
°12
(IV.3)
где индексы а указывают границы раздела фаз. Из рис. 30
видно, что если равновесный угол в меньше 90°, то капля
имеет сравнительно плоскую форму и как бы растекается
по твердой поверхности: капля воды смачивает поверхность,
и поверхность является гидрофильной. В предельном слу-
случае, при 6 = 0, смачивание полное и капля полностью
растекается по поверхности. Напротив, если угол в больше
90°, то капля воды приближается по форме к сферической
(например, капля воды на парафине): поверхность в этом
случае гидрофобная с плохим смачиванием водой. Если
средой 2 (рис. 30) оказывается не газ, а углеводородная непо-
неполярная жидкость, то из рис. 30 видно, что при в<90°
поверхность 3 лучше смачивается водой (избирательное
смачивание), а при 9>90° — лучше смачивается угле-
углеводородной жидкостью. Таким образом, гидрофобная по-
поверхность является одновременно олеофильной, а гидро-
гидрофильная поверхность — олеофобной. По данным Ребин-
дера, полученным по методу избирательного смачивания,
гидрофильными поверхностями обладают гипс (©== 0°),
слюда A2°), кальцит B0°), малахит C0°); гидрофобные
поверхности имеют пирит F= 135°), графит A27°), тальк
A39°), сера A46°) и др. Лейкоциты обнаруживают значи-
значительную гидрофи ьность и иногда остаются окруженными
тонкой пленкой воды в масляной фазе (например, моноци-
моноциты кролика). Напротив, эритроциты млекопитающих и
птиц сравнительно гидрофобные и вода с их поверхности
легко вытесняется маслом. По Модду, смачивание поверх-
поверхности бактериальной клетки определяют под микроскопом
по той фазе, в которую втягивается клетка на границе раз-
раздела вода — масло.
Помимо краевого угла смачивания, другой мерой гидро-
фильности поверхности является теплота смачивания, так
как гидрофильные поверхности смачиваются водой с поло-
положительным тепловым эффектом, например, по данным Ду-
манского, при смачивании водой поверхности агара выде-
выделяется 44,8 кал/г, желатины — 32,5 кал!г, силикагеля —
22,7 кал!г и др. По Ребиндеру, гидрофильность поверхности
78
этим методом следует характеризовать по отношению теп-
лот ее смачивания водой qx и бензолом q%\
для гидрофильной поверхности
-^— > 1 (для агара 35);
для гидрофобной поверхности
Я*
(для угля 0,34).
Если поверхность раздела жидкости выпуклая, а не
плоская, то, как видно из рис. 31, ее молекулы (/) втяги-
ь
9-?4?-?-?
Рис. 31. Выпуклая (/), плоская (//) и вогнутая (///) поверхности
жидкости
ваются внутрь меньшим числом молекул жидкости или
слабее, чем из плоской поверхности (//), а из вогнутой
поверхности (///)—большим числом молекул жидкости или
сильнее, чем в остальных случаях. Между искривленной
и плоской поверхностью раздела существует, поэтому,
разность молекулярных давлений, которая называется
капиллярным давлением Р. Искривление поверхности
характеризуют радиусом кривизны г, направленным внутрь
жидкости при выпуклой поверхности (в этом случае г
считается положительным), и наружу от вогнутой поверх-
поверхности (в этом случае радиус г отрицателен). Для плоской
поверхности г — оо. По уравнению Лапласа
Р =
(IV. 4)
79
откуда видно, что для плоской поверхности Р = О, для
выпуклой поверхности Р> 0 и для вогнутой поверхности
Р < 0; эти давления Р суммируются с внешним давлением,
оказываемым на жидкость.
При опускании тонкого стеклянного капилляра в воду
(рис. 32, /) краевой угол смачивания близок к нулю,
вследствие чего мениск становится вогнутым, давление Р
по формуле (IV. 4) —¦ более низким, чем под плоской по-
поверхностью и, в резуль-
результате, мениск поднимает-
поднимается на высоту h, при
которой вес поднятого
столба жидкости урав-
уравновешивает разность
давлений между обеими
поверхностями. При
погружении капилляра
в несмачивающую жид-
жидкость (рис. 32) происхо-
происходит, напротив, опускание
уровня жидкости в ка-
капилляре. Измерение вы-
высоты капиллярного под-
поднятия также использует-
используется для измерения вели-
величины поверхностного на-
натяжения жидкости.
Ыз рис. 31 следует также, что на выпуклой поверхно-
поверхности молекулы легче могут перейти в пар, чем на плоской
или, тем более, на вогнутой; другими словами, давление
пара жидкости на выпуклой поверхности больше, а на вог-
вогнутой поверхности — меньше, чем нормальное давление
пара на плоской поверхности жидкости. Эта закономер-
закономерность выражается уравнением Томсона:
Рис. 32. Капиллярное поднятие (/) и
опускание (//) жидкости
Р
2аМ
RTpr
(IV. 5)
где (АР/Р) — относительное изменение давления насыщен-
насыщенного пара по сравнению с нормальным, М — молекуляр-
молекулярный вес, о — плотность жидкости иг — радиус капилляра
или капли жидкости. Из уравнения (IV. 5) следует, напри-
например, что для капель воды с г = 10~6 см упругость пара
на 10% выше, чем для воды с плоской поверхностью; поэто-
поэтому, если в замкнутой системе содержатся одновременно
крупные и мелкие капли, последние перегоняются к круп-
крупным каплям. Разумеется, коалесценция, или слияние ка-
капель при прямом их контакте, объясняется непосредствен-
непосредственно стремлением к уменьшению величины свободной поверх-
поверхности. Формула (IV. 5) объясняет также более высокую
растворимость мелких твердых частиц по сравнению с
крупными, явление капиллярной конденсации пара в уз-
узких капиллярах или порах, смачиваемых данной жидко-
жидкостью, и ряд других явлений.
АДСОРБЦИЯ НА ПОВЕРХНОСТИ РАСТВОРОВ
Свободная поверхностная энергия г равна произведению
величины поверхности 5 на поверхностное натяжение а:
г= 5а. Поэтому свободная поверхностная энергия может
быть уменьшена не только за счет уменьшения величины
поверхности S, но и за счет пони- „_^
жения величины поверхностного
натяжения, что достигается в ра-
створах изменением их состава.
Этот путь имеет важное значение
для многих проблем коллоидной
химии.
Поверхностное натяжение вод-
водных растворов всегда отличается
от поверхностного натяжения чи-
чистой воды. Растворение неорга-
неорганических солей несколько повы-
повышает поверхностное натяжение,
тогда как растворение органи-
органических веществ — мыла, детерген-
детергенты, спирты, жирные кислоты —
обычно вызывает значительное по-
понижение поверхностного натяжения. Вещества второй
группы называются поверхностноактивными. На рис. 33
для примера приведены кривые относительного понижения
поверхностного натяжения водных растворов ряда жир-
жирных кислот при различных концентрациях.
Понижение а в растворах поверхностноактивных ве-
веществ сопровождается повышением их концентрации в
поверхностном слое раствора, так как перенос молекул
этих веществ в поверхность, понижая поверхностное на-
П.ч м/л
Рис. 33. Понижение по-
поверхностного натяжения
в водных растворах жир-
жирных кислот A5°С).
81
тяжение, требует меньшей затраты энергии, чем перенос
молекул растворителя. Повышение концентрации раство-
растворенного вещества в поверхностном слое раствора (или
вообще на границе раздела фаз) называется адсорбцией.
Количественное соотношение между величиной адсорб-
адсорбции Г, т. е. избытком растворенного вещества в г-мол на
1 см2 поверхности, и изменением поверхностного натяжения
Id а\
с концентрацией раствора -т-1 , определяется уравне-
уравнением Гиббса ( 1878):
(IV. 6)
с do
~RT~d7~
Уравнение Гиббса может быть выведено по-разному.
Один из выводов, принадлежащий Оствальду, заключает-
заключается в следующем. Рассматривается раствор с поверхностью 5
(в см2), поверхностный слой которого содержит избыток
в 1 моль растворенного вещества; тогда-^- = Г. При пере-
о
ходе очень малого количества растворенного вещества из
раствора в поверхностный слой поверхностное натяжение da
уменьшается и, соответственно, уменьшается свободная
поверхностная энергия ds = Sda—~da. Это измене-
изменение поверхностной энергии в состоянии равновесия дол-
должно быть равно осмотической работе dF удаления из ра-
раствора того же количества растворенного вещества
dF = —vd-к, где v — объем раствора, содержащего 1 моль
растворенного вещества, a dtz — изменение осмотического
давления раствора. Приравнивая de = dF, получим
-jr da = — vd-к.
(IV. 6а)
Для разбавленных растворов, к которым применим закон
Вант-Гоффа, можно, на основании выражения (II. 7),
заменить в (IV. 6а)
откуда
и v=
RT
dc.
(IV. 65)
Из уравнения (IV. 66) непосредственно следует уравнение
(IV. 6).
82
Уравнение Гиббса неоднократно доказывалось опытным
путем прямым определением величин Г в поверхностном слое
и (-т-ч(Мак-Бэн, Фрумкин),с хорошим совпадением вычис-
\dc j
ленных и экспериментальных данных.
Из уравнения Гиббса видно, что, если растворенное ве-
da n
щество понижает поверхностное натяжение, то —г- < 0 и
Г>0, т. е. адсорбция растворенного вещества является
положительной (концентрация в поверхностном слое боль-
больше, чем в объеме раствора). Напротив, для поверхностно-
неактивных веществ — > 0 и Г<0 , т. е. адсорбция ока-
CLC
зывается отрицательной (концентрация в поверхностном
слое меньше, чем в объеме раствора).
При очень малых концентрациях, когда величина с
приближается к Ас, (IV. 6) переходит в уравнение
Да=а0—a~F=RTF, и так как величина Да аналогична
величине давления в поверхностном слое (см. ниже), а Г=-щ >
где 5 — площадь, занимаемая адсорбированным веществом
в поверхностном слое \——-\ то в результате из уравне-
уравнения (IV. 6) следует, что
F-S = RT. (IV. 7)
Уравнение (IV. 7) является двухмерным аналогом уравне-
уравнения состояния идеальных газов. Действительно, Лангмюр
A917) показал, что по формуле (IV. 7) для различных
поверхностноактивных веществ можно точно вычислить
значение газовой постоянной R. Таким образом, при очень
малых концентрациях молекулы адсорбированного вещест-
вещества находятся в поверхностном слое в «газовом» состоянии.
Фрумкин A92!э) ввел поправки, учитывающие собствен-
собственную площадь адсорбированных молекул 50 и силы взаимного
притяжения между ними а, и получил формулу:
Д ^ (IV. 8)
S-So ' S2
Легко видеть, что уравнение Фрумкина является двухмер-
двухмерным аналогом уравнения Ван-дер-Ваальса. Оно хорошо
передает концентрационную зависимость поверхност-
поверхностного натяжения растворов в широком интервале концентра-
83
ций. По аналогии с уравнением Ван-дер-Ваальса, урав-
уравнение (IV. 8) называется уравнением состояния адсорбцион-
адсорбционного слоя.
Исследуя поверхностное натяжение водных растворов
органических веществ, Траубе A891) установил правило,
что в любом гомологическом ряду при слабых концентра-
концентрациях удлинение углеродной цепи на СНг - группу увели-
увеличивает поверхностную активность в 3—3,5 раза. Это озна-
означает, что для последовательных гомологов концентрации,
вызывающие одинаковое понижение поверхностного натя-
натяжения, уменьшаются примерно втрое для каждого очеред-
очередного члена ряда (что видно, например, также из рис. 33).
Лангмюр выяснил, что правило Траубе справедливо лишь
при свободном расположении адсорбированных молекул
в поверхностном слое параллельно поверхности, что также
соответствует их «газовому» состоянию.
В общем виде зависимость поверхностного натяжения
от концентрации для водных растворов жирных кислот
выражается эмпирическим уравнением Шишковского A909)
Да = 61п (—-{- 1 ), (IV. 9)
где b и k — эмпирические постоянные, причем значение b
одинаково для всего гомологического ряда, a k изменяет-
изменяется в гомологическом ряду в 3—3,5 раза для каждого члена
ряда, в соответствии с правилом Траубе.
ОРИЕНТАЦИЯ МОЛЕКУЛ В ПОВЕРХНОСТНОМ СЛОЕ
Выше было показано, что при очень малых концентра-
концентрациях, когда величина адсорбции Г невелика, молекулы
в адсорбционном слое находятся в «газовом» состоянии (IV.
7); затем при увеличении концентрации раствора величина
адсорбции Г возрастает, и в уравнении состояния ад-
адсорбционного слоя (IV. 8) приходится учитывать собствен-
собственную площадь и силы взаимодействия адсорбированных
молекул. Наконец, при дальнейшем увеличении адсорбции
поверхностноактивного вещества Г достигает своего мак-
максимального значения Гтах, при котором площадь 5= 1/Гтах,
занимаемая молекулой в адсорбционном слое, также при-
приобретает свое предельное значение. Лангмюр сопоставляя
уравнения (IV. 6) и (IV. 9) при высоких концентрациях,
показал, что константа b из уравнения Шишковского равна
84
b = RTrmax. Поэтому постоянство значений b в пределах
гомологического ряда (стр. 84) означало постоянство пло-
площади 5 (например, для различных жирных кислот 5 =
о
=21 А2 на молекулу), занимаемой адсорбированными моле-
молекулами в поверхностном слое при Г=Гт&^. Между тем, оче-
очевидно, что длина молекул различных членов гомологичес-
гомологического ряда (например,уксусной, пропионовой и других кис-
кислот) должна быть различной. Это расхождение было объяс-
объяснено Лангмюром на основе представлений об ориентации
молекул в поверхностном слое.
МоЛГекулы многих поверхностноактивных веществ име-
имеют асимметричное строение. Алифатические спирты, жир-
жирные кислоты, мыла, многие детергенты и др. содержат по-
полярные группы (—ОН, —СООН, — СОН, — NH2 и др.),
локализованные лишь в одной части молекулы, тогда как
остаток молекулы имеет строение углеводородной цепи
(рис. 34). Расположение таких молекул в поверхностном слое
СН3 СН3
Рис. 34 Атомная модель молекулы мыла — стеарата
натрия: /—углеводородная цепь, //—полярная группа
энергетически наиболее выгодно при условии погружения
полярных групп в воду, а углеводородных цепей — в воздух
или в неполярную фазу, в соответствии с принципом неза-
независимости поверхностного действия функциональных групп
молекул, сформулированным Лангмюром. Было подсчита-
подсчитано, что вероятность ориентации гидроксильной группой
в воду уже для молекулы метилового спирта в сотни тысяч
раз больше, чем вероятность обратной ориентации. При
малой концентрации адсорбированных молекул в поверх-
поверхностном слое тепловое движение нарушает их ориентацию
и молекулы в основном лежат в поверхностном слое (о чем
уже говорилось в связи с правилом Траубе), но при
повышении концентрации усиливается взаимодействие
углеводородных цепей между собою, что благоприятству-
85
ет вертикальной ориентации молекул, и при насыщений
адсорбционного слоя (Гтах) создается возможность образо-
образования «молекулярного частокола» из вертикально распо-
расположенных молекул (рис. 35). В этом состоянии площадь
Рис. 35. Предельная ориентация молекул
в адсорбционном слое
So = ~— , занимаемая молекулой, определяется лишь
1 max
площадью полярной группы, общей для всех членов гомо-
гомологического ряда, что и объясняет постоянство константы &
в уравнении Щишковского. Напротив, длина углеводород-
углеводородных цепей и энергия их взаимодействия изменяются
при каждом удлинении цепи на СНг-группу —¦ это
объясняет изменение второй константы k в уравнении
Шишковского. Из величины этого изменения C—3,5) Ланг-
мюр вычислил, что энергия адсорбции на каждую СНг-
группу возрастает приблизительно на 640 кал/моль.
Естественно, что молекулы с несколькими симметрично
расположенными полярными группами (например, дикарбо-
новые кислоты и их эфиры) сохраняют горизонтальную
ориентацию в адсорбционных слоях вплоть до их насыще-
насыщения (Таубман). В этом случае коэффициент правила Траубе
уменьшается с 3,5 до 2,6, а площадь, занимаемая молекулой,
возрастает с увеличением длины цепи. Таубман также
показал необходимость учета гидратации полярных групп
в адсорбционном слое, что заметно увеличивает собственные
размеры молекул.
мономолекулярные слои
Помимо данных о насыщенных адсорбционных слоях,
представления об ориентации молекул в поверхностном
слое были в значительной мере обоснованы исследованиями
мономолекулярных слоев, образованных на поверхности
воды или водных растворов трудно растворимыми или не-
86
растворимыми веществами (Марселей, Лангмюр, Ребин-
дер).
Многие нерастворимые вещества при нанесении на по-
поверхность воды обладают способностью самопроизвольного
растекания на ней до очень тонкого слоя.' Условие расте-
растекания жидкости А на жидкости В, по Гаркинсу, заключается
в том, чтобы разность поверхностных натяжений
(ов~-оа-°ав)>0, (IV. 10)
где ов и ад — поверхностные натяжения, соответственно,
жидкостей Б и Л на границе с воздухом и аАВ — межфаз-
межфазное поверхностное натяжение между ними. Величину / =
= ав —аА —аЛВ Гаркинс назвал коэффициентом растекания;
при/> 0 жидкость растекается тонким слоем, а при / <0 —
остается в виде нерастекающейся капли. Значение коэф-
коэффициентов растекания на воде для некоторых жидкостей
приведены в табл. 7.
Таблица 7
Коэффициенты растекания на воде B0°) (по Гаркинсу)
Растекающиеся жидкости
н. Октиловый спирт . .
Гептиловая кислота . .
Олеиновая кислота . . .
Анилин
Бензол
Хлорбензол
Гексан
f.
эрг/см2
36,7
37,8
24,6
24.4
8.9
2,3
3,1
Нера стекающиеся
жидкости
Монобромбензол . . .
Сероуглерод .....
Моноиодбензол . . .
Вазелиновое масло . .
Йодистый метил . . .
эрг, см2
— 3,3
— 6,9
— 8,8
—13,5
—26,5
Физические причины растекания заключаются в том,
что силы молекулярного притяжения между верхней и
нижней жидкостями больше, чем силы молекулярного
притяжения в верхней жидкости; аналогичным образом
объясняется растекание некоторых жидкостей на твердых
телах.
Из табл. 7 видно, например, что олеиновая кислота
самопроизвольно растекается на поверхности воды; при
этом образуется тонкий слой, границы которого можно
сделать заметными, если посыпать поверхность воды таль-
тальком, так как олеиновая кислота оттесняет тальк. Если на
87
поверхность воды нанесено небольшое известное Коли-
Количество олеиновой кислоты, то после измерения величины
поверхности пленки легко вычислить толщину пленки,
которая оказывается всего около 10 А, т. е. состоит лишь
из одного слоя молекул. При нанесении других жидкостей
иногда монослой находится в равновесии с одной крупной
каплей, сидящей островком на поверхности. Некоторые
твердые вещества (глиадин и др.) при соприкосновении с
чистой водной поверхностью также приобретают способ-
способность самопроизвольно образовывать монослой.
Другим способом приготовления монослоев или очень
тонких слоев является нанесение на поверхность воды
небольшого количества раствора вещества в летучем раст-
растворителе, который затем испаряется.
Стремление вещества к растеканию не ограничивается
образованием насыщенного, сплошного монослоя, но при
наличии свободной поверхности может продолжаться до
образования все более разбавленных слоев, вплоть до
«газового» состояния пленки, подобного «газовому» состоя-
состоянию адсорбционного слоя при очень низких концентрациях
(стр. 83). Подобно тому как обычный трехмерный газ стре-
стремится к неограниченному расширению, оказывая давление
(в дин'см2) на ограничивающую поверхность, так и двухмер-
двухмерная пленка в своем стремлении к расширению оказывает
боковое давление на ограничивающую линию; однако это
давление, как и поверхностное натяжение, измеряется в
дин!см (поэтому и в уравнении IV. 7 величина понижения
поверхностного натяжения Аз играла роль бокового давле-
давления, с которым она по существу идентична).
Измерение зависимости площади пленок от величины
поверхностного давления относится к числу основных ме-
методов исследования монослоев. Эти измерения производятся
при помощи специальных приборов, называемых поверх-
поверхностными весами (Лангмюр, Вильгельмы, Адам и др.).
Поверхностные весы Лангмюр а (рис. 36) основаны на
прямом измерении силы, действующей на легкий поплавок
А, отделяющий пленку от чистой поверхности. Основная
кювета представляет собой низкий четырехугольный сосуд
(примерно, 60 X 14 X 1,5 см), до краев наполненный во-
водой. Пленка создается между планками X и А и может
сжиматься передвижением планки X. Давление на планку
А измеряется при помощи весового приспособления или
чувствительного динамометра (рис. 36) с двумя крутильными
88
нитями. Новые приборы позволяют измерять боковые дав-
давления до 0,01 дин/см и ниже.
С помощью метода измерения зависимости площади пле-
пленок от давления удалось изучить в монослоях все рассмот-
рассмотренные выше состояния молекул в адсорбционных слоях.
Было показано, что очень разбавленные пленки находятся
в газовом состоянии, при котором произведение бокового
Рис. 36. Поверхностные весы Лангмюра
давления на соответствующую поверхность пленки постоян-
постоянно, аналогично закону Бойля-Мариотта (см.уравнение IV.7).
По мере сжатия пленки (рис 37, /) , ее поверхностное
давление возрастает, ана-
аналогично увеличению давле-
давления при сжатии объемного
газа, а затем пленка при по-
постоянном поверхностном
давлении (рис. 37, 2) пере-
переходит в состояние конден-
конденсированной пленки, со зпа-
чительным уменьшением
поверхности, подобно тому
как сильно сжатый объем-
объемный газ переходит при по-
постоянном давлении в жид-
2
кость. В области постоям-
ного давления островки
сплошной пленки сущест-
Рис. '.if. Кривая сжатия пленки
(Г-.поперх}юсть, к_дзвлеНис рас-
ширен и я)
89
вугот среди участков газовой пленки. Наконец, при сжатии
конденсированных пленок с плотным расположением моле-
молекул (рис. 37,3) наступает резкое возрастание поверхностного
давления при малом изменении поверхности, аналогично
явлениям при сжатии жидкостей. Более детальное изуче-
изучение структурно-механических свойств монослоев — упру-
упругости, пластичности, прочности на сдвиг (Ребиндер и Тра-
Трапезников) и поверхностной вязкости при помощи двухмер-
двухмерного капилляра (Талмуд и Бреслер) позволило более
четко характеризовать изменения состояния пленок, раз-
различие жидких и твердых пленок, фазовые переходы в моно-
монослоях и др.
Помимо метода поверхностного давления, расположение
молекул в поверхностных слоях исследуется также методом
поверхностных потенциалов по Фрумкину, для чего изме-
измеряется разность потенциалов между водным раствором и
воздухом при наличии или в отсутствие пленки. Один
из электродов погружается в водный раствор, а на
кончике платинового электрода в воздухе помещается
небольшое количество полония, делающего пространство
между электродом и поверхностью воды проводящим.
Скачок потенциала измеряется при помощи потенциометра
и электрометра. Ориентация дипольных молекул в конден-
конденсированном поверхностном слое приводит к возникновению
двойного электрического слоя, величина и знак потенциала
в котором зависят от расположения диполей адсорбирован-
адсорбированных молекул.
Исследования конденсированных пленок в отношении
определения величины площади на молекулу дали обширный
материал, характеризующий ориентацию молекул на
поверхности. Для многих гомологических рядов с конце-
концевыми группами — СООН, — CH2NH2, — ОЬОН площадь
на молекулу оказалась постоянной B0,5—21,0а2), несмот-
несмотря на различную длину цепей — от Ci4 до С22, что с оче-
очевидностью указывало на перпендикулярную ориентацию
цепей к поверхности в конденсированных слоях. Величины
предельной площади на молекулу для некоторых соедине-
соединений приведены в табл. 8.
Ориентированное расположение молекул на границе
раздела, благодаря значительным энергиям взаимодей-
взаимодействия между ними, является одним из факторов понижения
свободной поверхностной энергии системы (см. выше),
оно наблюдается лишь в конденсированных слоях. В раз-
90
Таблица 8
Площади на молекулу S для некоторых соединений
(по Адаму)
t: ¦,
Гомологический ряд
Жирные кислоты
Амиды
Спирты . . . .
Фенолы . . . .
Нитрилы . . . .
Холлгтерол . . .
Хлорофилл . . .
Гемин
Концевая
группа
—СООН
—CON На
—СНоОН
— <~, он
-C=N
С о!
Ь, д
20,5
20,5
21.6
24,0
27,7
40,8
80—100
70
бавленных слоях ориентация нарушается, молекулы ста-
становятся наклонно или горизонтально к поверхности;
в газовых слоях преобладает горизонтальное расположение
молекул в поверхности раздела.
Большой интерес представляют мономолекулярные
слои белков и ферментов, которые получают, например,
на водных растворах сульфата аммония и других поверх-
поверхностях. При «газовом» состоянии белковых пленок в них
можно определить молекулярный вес белков по формуле
(IV. 7), которой, по аналогии с законом ВантТоффа для
осмотического давления (стр. 34), обычно придают следую-
следующий вид:
FA - nRT, (IV. 11)
где/" — поверхностное давление в дин/см, А — поверхность
о
пленки и я — число молей белка на поверхности А, при-
причем п — w/M, где w — вес белка и М — молекулярный
вес. Величину FA определяют при различных низких F
и находят предельное значение при F — 0. Этим путем
Булл, Дэвис и др. определили молекулярный вес яичного
альбумина, серумальбумина, гемоглобина, трипсина, инсу-
инсулина в хорошем согласии с другими известными данными.
Толщина белковых монослоев составляет около 10 А (Гор-
тер), что соответствует лишь толщине полипептидной цепи и
указывает на развертывание белковых молекул в поверх-
поверхностном слое, причем значительная часть гидрофильных
групп белка обращается к воде, а гидрофобных групп —
в воздух (Пчелин). Площадь на аминокислотный остаток
91
составляет около 15—20 А2. Белки в монослоях часто пере-
переходят в нерастворимое состояние (поверхностная денату-
денатурация) и приобретают упруговязкие свойства (Трапезников).
Площадь белков в монослоях заметно изменяется в зави-
зависимости от величины рН в результате изменения конфигу-
конфигурации и гидратации белковых молекул в поверхности.
Белки в монослоях часто сохраняют свои ферментативные
свойства (каталаза, трипсин) и способность к иммунным
реакциям и связыванию антител. На этих свойствах основано
исследование реакции специфических белков в ничтожно
малых количествах. Интересный метод последовательного
отложения многих белковых монослоев на твердых поверх-
поверхностях был разработан Лангмюром и Шефером.
В организме различные белковые, липопротеидные,
нуклеопротендные и другие молекулярные слои играют
большую роль в жизнедеятельности клеток. Взаимодейст-
Взаимодействие поверхностноактивных веществ с белками, проникнове-
проникновение гемолизирующих веществ (9-аминоакридина, фенотиа-
зона и др.) в белковые пленки, соединения холестерола с
глиадином и другими белками, реакция аденозинтрифос-
форной кислоты с миозином в монослое и др. — неоднократно
исследовались методами двухмерного давления и поверх-
поверхностных потенциалов (причем были разработаны также
специальные методы подобных измерений для межфазной
границы двух жидкостей).
АДСОРБЦИЯ НА ТВЕРДЫХ ПОВЕРХНОСТЯХ
В жидкостях и растворах свойства поверхности одно-
однородны в различных ее участках, тогда как на поверхности
твердых тел положение атомов фиксировано и строение
поверхности имеет сложный и неоднородный характер. Кро-
Кроме того, адсорбция на твердых телах имеет большое прак-
практическое значение, так как почти все адсорбенты и катали-
катализаторы, применяемые в промышленности и химической
защите, — твердые тела. Вследствие этего, процессы адсорб-
адсорбции на твердых телах явились предметом многочисленных
исследований.
При помощи разнообразных методов была установлена
адсорбционная неоднородность поверхности твердого тела.
Так, например, было показано, что теплота адсорбции
первых небольших порций газа обычно значительно больше,
чем последующих порций (при адсорбции кислорода на
9 '_>
угле теплота адсорбции падает от 220 ккал'моль до 40—
50 ккал/моль, при адсорбции кислорода на платине —• с 161
до 104 ккал/моль, при адсорбции водорода на вольфраме —
с 34,2 до 17,5 ккал/моль и т. д.), что объясняется адсорбцией
первых порций газа на наиболее активных участках поверх-
поверхности. Отравление катализаторов часто наблюдается при
адсорбции такого количества «ядов», которое покрывает
лишь доли процента поверхности твердого тела (Тэйлор)
и др. Таким образом, следует считать, что адсорбция проис-
происходит не на всей поверхности твердого тела, а лишь на ак-
активных центрах этой поверхности.
По теории Лангмюра, каждый активный центр поверх-
поверхности обладает лишь молекулярной сферой действия, так
как адсорбция определяется остаточными валентностями
на поверхности. С этой точки зрения адсорбция является,
по существу, химическим процессом и насыщенный адсорб-
адсорбционный слой имеет мономолекулярный характер. В про-
процессе адсорбции адсорбируемые молекулы остаются неко-
некоторое время связанными на активных центрах (это время
называется «продолжительностью жизни» в адсорбирован-
адсорбированном состоянии) и затем вновь отрываются. Таким образом,
состояние равновесия при адсорбции определяется ра-
равенством скоростей конденсации и испарения молекулы.
При этом, часть 9 адсорбирующей поверхности занята адсор-
адсорбированными атомами или молекулами, а часть A—9)
поверхности остается свободной. Величина 9 равна отно-
отношению адсорбированного количества вещества Г к макси-
максимальной адсорбции Г тах при полном заполнении поверх-
поверхности 0 = ——-. При постоянной температуре число столк-
I шах
новений молекул с поверхностью пропорционально давлению
р или объемной концентрации с газа. Так как, по Лангмю-
ру, молекулы адсорбируются лишь одним слоем, они не
могут адсорбироваться на уже занятой части поверхности
и поэтому скорость их адсорбции v (или конденсации)
на поверхности пропорциональна концентрации с и сво-
свободной части поверхности A —0):
v = kc{\~9), (IV. 12а)
где k — коэффициент пропорциональности. Напротив,
десорбция (или испарение) молекул происходит лишь с
занятой части поверхности, и поэтому скорость v' этого
процесса пропорциональна 0:
93
v' = k'& (IV. 12 6)
Приравнивая в состоянии равновесия скорости обоих прс-
цессов v и v', получим:
или
__
" ~^П^ - ~^^ ~ с~Т^ (IV. 12в)
Этим путем Лангмюр нашел для поверхности с равномерным
распределением однородных активных центров и при от-
отсутствии взаимодействий между адсорбированными моле-
молекулами, следующее уравнение:
max
с -f a '
(IV. 12)
где Г — величина адсорбции, с — объемная концентрация
газа на — отношение скоростей десорбции и адсорбции
(или испарения и конденсации). Уравнение (IK. 12) назы-
называется уравнением изотермы адсорбции Лангмюра. Оно по-
показывает, что при малых концентрациях (при с<^а) величи-
на адсорбции пропорциональна концентрации 1Г = -^^с =
= kc 1, а при высоких концентрациях (с ~^> а) величина
адсорбции стремится к предельному значению Гтах (рис. 38).
При адсорбции газов с может быть заменено пропорцио-
пропорциональной величиной давления газа р. При адсорбции из
раствора, для которой (IV.
12) в принципе также ос-
остается справедливым, с оз-
означает концентрацию рас-
растворенного вещества.
При неравномерном рас-
расположении активных цент-
центров, различной их природе
или при заметных силах
взаимодействия между ад-
адсорбированными атомами,
уравнение изотермы ад-
адсорбции усложняется. Ме-
Рис. 38. Изотерма адсорбции
Лангмюра
тоды расчета распределе-
94
ний активных центров по теплотам адсорбции на основа-
основании экспериментальных изотерм адсорбции разработа-
разработаны Рогинеким. Предполагая экспоненциальное распреде-
распределение активных центров по энергиям адсорбции, Зельдо-
Зельдович A934) теоретически получил уравнение, которое ранее
эмпирически было установлено Фрейндлихом A906):
4
х = р с п ,
где х — величина адсорбции на единицу веса адсорбента,
с — концентрация газа или растворенного вещества, р
и постоянные величины. Изотерма адсорбции Фрейнд-
лиха (рис. 39) отли-
отличается от изотермы
Лангмюра отсутстви-
отсутствием зоны насыщения.
Уравнение (IV. 13)
хорошо согласуется с
многими опытными
данными в области
слабых и средних кон-
концентраций, но форму-
формула (IV. 12) обладает
большой общностью
применения, охваты-
охватывая также области
концентраций насы-
насыщения при условии
образования мономо-
мономолекулярного адсорб-
адсорбС, моль/л
Рис. 39. Изотерма адсорбции Фрейнд-
лиха (на примере адсорбции пропи-
оновой кислоты активированным уг-
углем при 25е)
ционного слоя.
По теории Поляни, адсорбция определяется Ван-дер-
Ваальсовыми силами, радиус действия которых больше,
чем остаточных валентностей в теории Лангмюра, и поэтому
адсорбция не локализуется в первом молекулярном слое,
а приводит к образованию полимолекулярного слоя. Дей-
Действительно, при широком исследовании изотерм адсорбции
на различных адсорбентах, особенно при адсорбции паров,
было показано, что наиболее общим типом являются не
лангмюровские изотермы (рис. 38), а так называемые
5"-образные изотермы, в которых адсорбция не останавли-
останавливается на образовании монослоя, а продолжается до образо-
95
вания полимолекулярного слоя. Общую теорию 5-образных
изотерм разработали Брунауэр, Эммет и Теллер (сокра-
(сокращенно, БЭТ); они показали, что зона образования монослоя
или лангмюровской адсорбции охватывает участок кривой
от начала координат до первого перегиба кривой в точке В
(рис. 40), и что в дальнейшем происходит полимолекуляр-
полимолекулярная адсорбция и капиллярная конденсация (см. стр. 80),
объясняющая второй подъем кривой. Авторы установили
уравнение:
р \ с ] р
¦ -Ь* (IV. 14)
vm с '
где Ро—-упругость насыщенного пара, v — объем адсор-
адсорбированного газа при
данном давлении газа Р,
vm — объем адсорбиро-
адсорбированного газа в монослое
и с ~ eXz'/RT, где Дг —
разность между тепло-
теплотой адсорбции газа в
первом слое и теплотой
сжижения газа. Находя
зависимость левой части
/ Р
уравнения IV.Нот
получают на графике
прямую линию, наклон
которой равен
отрезок на оси ординат
——). Отсюда можно
Рис. 40. S-образпая изотерма ад-
адсорбции (на примере адсорбции па-
паров воды на коллагене при 25е)
определить величины vm
и с. Уравнение БЭТ получило широкое применение в лите-
литературе, так как из величины v,n (соответствующей также
точке В на рис. 40) может быть вычислена полная адсорби-
адсорбирующая поверхность адсорбента 5, что имеет важное прак-
практическое значение, а из величины с вычисляется теплота
адсорбции газа в монослое (обычнооколо2—2,5ккал!'моль).
В качестве примера значений площади 5 укажем, что адсор-
адсорбирующая поверхность 1 г испытанного образца силикагеля
составила 500 м~ (с точностью ± 10"('> по пяти различным
газам), а 1 г коллагена — 350 м-. Хорошие адсорбенты
96
обладают весьма значительной удельной поверхностью,
например, активированный уголь — до 1000 м У г.
Выше мы касались вопроса о физической или химичес-
химической природе сил, определяющих адсорбцию (ср. теории
Лангмюра и Поляни). Следует отметить, что это различие
далеко не всегда может быть четко проведено. В крайних
случаях физическая адсорбция, определяемая лишь Ван-
дер-Ваальсовыми силами, характеризуется хорошей обра-
обратимостью, отсутствием стехиометрических соотношений,
уменьшением адсорбции при повышении температуры,
близостью тепловых эффектов адсорбции к теплотам сжиже-
сжижения или испарения; такова адсорбция инертных газов или
гексана на угле. В других крайних случаях химическая
адсорбция осуществляется только путем химического взаи-
взаимодействия, например, между кислородом и вольфрамом
или кислородом и серебром при повышенных температурах;
здесь адсорбция почти необратима, тепловой эффект бли-
близок к энергии образования химических соединений (око-
(около 100 ккалЫоль и выше) и др. Обычно осуществляются
промежуточные варианты, когда основная масса адсорби-
адсорбированного вещества связывается сравнительно слабо,
а следы его связаны прочно и могут быть удалены лишь
путем длительного прогревания и откачивания. Кислород
на металлах или водород на никеле адсорбируется при низ-
низких температурах физически, ввиду малой скорости хими-
химической реакции при этих температурах, но при повышении
температуры начинает протекать адсорбция с заметной
энергией активации (активированная адсорбция) по типу
химических реакций. В
определенном интервале "V
повышения температур
прирост химической ад-
адсорбции (или хемосорб-
ции) перекрывает паде-
падение физической адсорб-
адсорбции и на кривой темпе-
температурной зависимости
адсорбции возникает
промежуточный макси-
максимум (рис. 41), харак-
характерный для наличия ак-
активированной адсорб-
10
s2
?
-ZOO -150 -WO -50 0 50
100
t°C
ванной адсорбции (Нг на никеле)
«Коллоидная химия>
97
Образующиеся при адсорбции кислорода на угле соеди-
соединения с переменным количеством кислорода, согласно Ши-
Шилову, имеют характер кислых или основных поверхностных
окислов, тогда как по теории Фрумкина адсорбированные
молекулы кислорода не входят в химическую связь с поверх-
поверхностными атомами углерода, а образуют слой ориентиро-
ориентированных диполей (изменяющий, в частности, электрохимичес-
электрохимическое поведение угля). На поверхности многих металлов,
кроме хемосорбированной кислородной пленки, существует
также физически адсорбированная кислородная пленка,
о
толщиною в 10—35А, способная полностью улетучиваться
в вакууме (Шишаков). Установлены случаи образования
при адсорбции поверхностных соединений с участием во-
водородных связей. Таким образом, явления физической и
химической адсорбции могут четко различаться лишь в
крайних случаях, между которыми возможны различные
переходы.
ЗНАЧЕНИЕ АДСОРБЦИОННЫХ СЛОЕВ В КОЛЛОИДНЫХ
СИСТЕМАХ
Ввиду наличия чрезвычайно развитой поверхности
раздела фаз в лиофобных коллоидных системах, значение
адсорбционных слоев обусловлено, прежде всего, тем, что
они резко изменяют природу поверхностей раздела, хотя
для их образования часто достаточно лишь очень неболь-
небольшого количества вещества. При ориентированном распо-
расположении молекул поверхностноактивных веществ в адсорб-
адсорбционных слоях полярными группами к гидрофильной
поверхности (или к гидрофильным участкам поверхности)
и углеводородными цепями наружу, поверхность становится
гидрофобной. Напротив, при расположении молекул угле-
углеводородными цепями к гидрофобной поверхности, а поляр-
полярными группами наружу, поверхность становится гидрофиль-
гидрофильной. Соответственно изменяется устойчивость дисперсных
систем в водной или углеводородной среде, например, гидро-
фобизированные частицы становятся менее устойчивыми в
водной среде, а гидрофилизированные частицы -— более
устойчивыми (см. главы шестую и седьмую). В ряде работ
Ребиндера и его школы было установлено важное явление
облегчения деформации и измельчения твердых тел при ад-
адсорбции на их микротрещинах поверхностноактивных ве-
веществ, что используется в процессах сверления, бурения,
истирания. В присутствии поверхностноактивных веществ
резко изменяются формы роста кристаллов — это может
иметь значение при образовании кристаллов лекарственных
препаратов. В фармацевтической практике применяются
препараты адсорбентов (например, медицинский активи-
активированный уголь) для понижения кислотности в желудке
и поглощения газов. За последние годы большое значение
получила адсорбционная хроматография, особенно моле-
молекулярная и ионообменная хроматография, однако, ввиду
преимущественного значения для технологии антибиоти-
антибиотических и лекарственных препаратов ионообменной хрома-
хроматографии и для цельности изложения, мы рассмотрим воп-
вопросы хроматографии в пятой главе (см. стр. 126).
Работами Ребиндера и его сотрудников была установлена
также способность поверхностноактивных веществ в адсорб-
адсорбционных слоях образовывать двухмерные структуры с
повышенными физико-механическими свойствами (упру-
(упругостью, прочностью и др.), что оказывает чрезвычайно боль-
большое влияние на устойчивость дисперсных систем (главы
шестая—седьмая). Вещества, обладающие способностью к
образованию структурно-прочных адсорбционных слоев
(желатина и др.), могут оказывать защитное действие; они
используются для получения высокоустойчивых золей
лекарственных препаратов и золей металлов (серебра,
радиоактивного золота и др.).
Выводы
Лиофобные дисперсные системы характеризуются зна-
значительной удельной поверхностью и свободной поверхност-
поверхностной энергией, которая, естественно, стремится к уменьше-
уменьшению. В чистых жидкостях это проявляется в стремлении к
уменьшению величины поверхности (образование капель,
коалесценция капель). Соотношение поверхностных натя-
натяжений двух жидкостей определяет условия растекания
(уравнение IV. 10), или равновесного контакта (IV. 2),
а соотношение поверхностных натяжений на границе раз-
раздела трех фаз, из которых одна является твердой, опреде-
определяет условия смачивания (IV. 3). Для искривленных жидких
поверхностей раздела характерно наличие капиллярного
давления (IV. 4) и изменение упругости пара (IV. 5).
В растворах стремление к уменьшению свободной поверх-
поверхностной энергии проявляется преимущественно в понижении
8* 99
поверхностного натяжения путем адсорбции поверхностно-
активных веществ из раствора на поверхности раздела.
Количественно связь между адсорбцией и понижением
поверхностного натяжения определяется уравнением Гиб-
бса (IV. 6). Изменение поверхностного натяжения с кон-
концентрацией растворов выражается уравнением Шишков-
ского (IV.9), а изменение поверхностной активности в го-
гомологическом ряду — правилом Траубе.
Поверхностноактивные вещества в адсорбционных и мо-
мономолекулярных слоях могут находиться в очень разбавлен-
разбавленном состоянии, аналогичном идеальным газам в объеме, —
в виде двухмерной газовой пленки (IV. 7). Газовое состоя-
состояние пленок позволяет определить молекулярные веса бел-
белков и других веществ в поверхностном слое (IV. 11).
Общее уравнение состояния адсорбционных слоев, по
Фрумкину, (IV. 8), является двухмерным аналогом урав-
уравнения Ван-дер-Ваальса.
При увеличении поверхностного давления пленки пере-
переходят в конденсированное состояние, а в насыщенных ад-
адсорбционных и мономолекулярных слоях происходит ориен-
ориентация асимметричных молекул поверхностноактивных ве-
веществ по типу «молекулярного частокола» (Лангмюр,
Ребиндер), что подтверждается измерениями предельной
площади на молекулу для различных гомологических ря-
рядов (табл. 8).
Адсорбция на твердых поверхностях определяется урав-
уравнениями Лангмюра (IV. 12) и Фрейндлиха (IV. 13), адсорб-
адсорбция по типу .S-образных изотерм — уравнением Брунауэ-
ра, Эммета и Теллера (IV. 14). Уравнение БЭТ позволяет
рассчитать по кривым низкотемпературной адсорбции газов
величину полной адсорбирующей поверхности адсор-
адсорбентов.
Образование адсорбционных слоев резко изменяет при-
природу поверхностей раздела между фазами и имеет основное
значение для устойчивости дисперсных систем (Ребиндер).
Вопросы
1. Какими способами измеряются поверхностное натяжение,
поверхностное давление, поверхностные потенциалы?
2. Дайте характеристику гидрофильных и гидрофобных по-
поверхностей. Каковы способы их изменения?
3. Какими отличительными свойствами обладают искривлен-
искривленные поверхности жидкостей?
100
4 Какие количественные зависимости связывают понижение
поверхностного натяжения раствора с величиной адсорбции и с
концентрацией растворов?
5 Каковы условия растекания жидкостей?
6 Почему гидрофобные вещества (уголь, графит) лучше адсор-
адсорбируют поверхностноактивные вещества из водных растворов,
а гидрофильные вещества (силикагель) — из углеводородных
растворов?
7. Как доказывается ориентация молекул поверхностноактив-
поверхностноактивных веществ в поверхностных слоях?
8 Что представляют собой газовые и конденсированные пленки?
Какие уравнения соответствуют уравнению состояния идеальных
газов и уравнению Ван-дер-Ваальса для двухмерного состояния?
9. Сопоставьте уравнение Лангмюра, Фрейндлиха и БЭТ,
укажите их физические основы и области применения.
10. Каково значение адсорбционных слоев в дисперсных си-
системах?
Глава пятая
электрохимия коллоидов
Электрохимические явления в коллоидных системах
имеют большое значение, ввиду распространенности взаи-
взаимодействий коллоидов с электролитами и роли этих взаимо-
взаимодействий для устойчивости коллоидов.
ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ
При взаимодействии коллоидов с электролитами обычно
на поверхности частиц адсорбируется преимущественно
один из ионов, сообщающий поверхности свой знак заряда.
Для простоты положим, что адсорбируются только ионы
одного знака (например, частицы йодистого серебра в 0,01 н.
AgNOs адсорбируют только Ag + -ионы) и что проти-
противоположные или компенсирующие ионы полностью оста-
остаются в растворе. Под действием сил электростатического
притяжения компенсирующие ионы стремятся располо-
расположиться возможно ближе к ионам, адсорбированным на
поверхности. В предельном случае образуется два слоя
ионов, из которых один расположен на поверхности, а дру-
другой — в растворе, на молекулярном расстоянии от первого
слоя (рис. 42 а). Такая система ионов, в целом нейтральная,
называется двойным электрическим слоем, по Гельмголь-
цу. Под действием теплового движения упорядоченное
расположение компенсирующих ионов в растворе нарушает-
нарушается, вследствие чего в другом предельном случае двойной
электрический слой приобретает структуру диффузного
двойного слоя, по Гуи (рис. 42 в); толщина этого слоя мо-
может составлять, например в 0,001 н. KQ — 10 ми,, а в
0,1 н. КС1 —¦ 1 м\1. Фактически следует представлять, что
часть ионов находится на молекулярном расстоянии от
102
поверхности, образуя гельмгольцевский двойной слой
(рис. 42 с, часть кривой АВ), а другая часть ионов образует
диффузный двойной слой (рис. 42 с, часть кривой ВС).
Таково строение двойного электрического слоя по Штерну
(рис. 42 с), теория которого является наиболее общей. При
разбавлении раствора структура двойного электрического
слоя приближается к структуре слоя Гуи, а при повышении
S
©
©
±)
©
©
©
©
©
+>
Рис. 42. Двойной электрический слой: /—по Гельмгольцу,
//—по Гуи,///—по Штерну; вверху—расположение ионов,
внизу—кривые падения потенциала
концентрации — слоя Гельмгольца. Необходимо указать,
что двойной электрический слой может образоваться на
непроводящих макроповерхностях и коллоидных частицах
лишь при наличии достаточно большого количества заря-
зарядов. В растворах низкомолекулярных электролитов вокруг
каждого иона, по теории Дебая-Гюккеля, образуется ионная
атмосфера, а не двойной электрический слой; напротив, в
растворах полиэлектролитов (полиакриловые кислоты,
нуклеиновые кислоты), на каждой молекуле которых со-
содержится большое количество ионогенных групп, создают-
создаются возможности образования двойного электрического слоя.
103
Структура двойного электрического слоя не зависит от
механизма возникновения зарядов на поверхности — воз-
возникают ли они путем преимущественной адсорбции ионов
из раствора, или путем диссоциации ионогенных групп
молекул, — а от плотности расположения зарядов на по-
поверхности. Если поверхность является непроводящей и
заряды расположены редко, то вокруг каждого заряда воз-
возникает по существу ионная атмосфера в растворе, если же
заряды расположены плотно и,
тем более, если поверхность яв-
является проводящей (электроды,
золи металлов), то обобществле-
обобществление ионных атмосфер дает струк-
структуру истинного двойного слоя.
Рассмотрим на нескольких
примерах строение двойного
электрического слоя различных
типов коллоидных частиц.
В золе AgJ в растворе KJ
(Кройт) преимущественно адсор-
бируютсяЛ — ионы (вообще легче
Рис. 43. Строение частицы адсорбируются ионы, общие с
золя AgJ в pacTBope'KJ (по ионами решетки частиц или изо-
Пескову и Прейс) морфные с ними),вследствие чего
частицы золя получают отрица-
отрицательный заряд, а компенсирующие ионы К+ располагаются в
растворе частично в гельмгольцевском слое, частично — в
диффузном слое (на рис. 43 условно поровну). В растворе
AgNCh частицы золя AgJ преимущественно адсорбируют
Ag+ -ионы и приобретают положительный заряд, а
компенсирующие ЫОз~-ионы располагаются в растворе,
подобно ионам К+ на рис. 43.
В золях AS2S3 на ядре частиц адсорбируются анионы
мышьяковистой или тиомышьяковистой кислот (Харин),
а в растворе располагаются компенсирующие ионы Н+,
Na+.
Частицы золей кремнекислоты в присутствии НС1 в
растворе прочно адсорбируют Н+-ионы (Каргин и Бере-
стнева) и приобретают положительный заряд, а компенси-
компенсирующие С1~ионы располагаются в растворе. Собственная
диссоциация кремнекислоты весьма незначительна. В зо-
золях АЬОз на поверхности частиц находится оксихлорид
алюминия (А12Оз)птА1(ОНJ+, а тС1~-ионов нахо-
104
дится в растворе. В золях V2O5 или WO3 источником заря-
зарядов является диссоциация ванадиевой или вольфрамовой
кислот на поверхности частиц. Интересно, что для всех пере-
перечисленных золей между ионами на поверхности частиц и
электролитами в растворе существует подвижное адсорб-
адсорбционное равновесие, вследствие чего наличие известной
концентрации электролитов в растворе — необходимое
условие самого существования коллоидов. В этом отношении
наиболее чистые лиофобные золи, по-видимому, были полу-
получены Н. Бах — золи платины в атмосфере водорода в вы-
сокоочищенной воде.
В частицах лиофобных золей всегда содержится ком-
компактное ядро, лишенное электрических зарядов. Напро-
Напротив, в частицах белков и полиэлектролитов подобное «яд-
«ядро» отсутствует, и они по всей своей массе несут способные
к диссоциации ионогенные группы. В белках ионогенные
группы имеют различную химическую природу — кислые
карбоксильные, основные амино-группы и др., вследствие
чего белки относятся к классу амфотерных электролитов.
При крайних рН резко преобладает диссоциация групп
одного знака и, например, при рН-2 белковая молекула
несет лишь положительные заряды. Однако тот же заряд
при кислых рН можно представить, по Бьерруму и Линдер-
штрем-Лангу, обусловленным не диссоциацией солеобразных
аминогрупп, а адсорбцией Н+-ионов из раствора и
подавлением диссоциации СОО~-групп на белковой мо-
молекуле. При любом предположении белковая молекула при
данном рН несет точно определенное число зарядов, а ком-
компенсирующие ионы располагаются в растворе с определен-
определенной плотностью распределения, что, соответственно,
измеряется при помощи кривых титрования и электрофоре-
тической подвижности (см. ниже).
Наконец, в молекуле линейных полиэлектролитов, на-
например полисульфостирола (рис. 44), вдоль цепи располо-
расположено большое количество одноименных ионогенных групп,
диссоциация которых сообщает отрицательный заряд мо-
молекулам полиэлектролитов, сохраняющийся до очень кис-
кислых рН.
Интересно отметить, что при большом числе ионогенных
групп или центров адсорбции на частицах уравнения за-
закона действующих масс и адсорбционной изотермы Ланг-
мюра (см. стр. 94) совпадают между собой, как было пока-
показано Линдерштрем-Лангом, Паули и Валько, Гимантом
«Коллоидная химия>
105
и др. В частности, Линдерштрем-Ланг A924) получил
идентичные формулы для ионного равновесия белков, ис-
исходя из изотермы Лангмюра и из теории диссоциации мно-
гоосновных кислот, по Бьерруму. Количественные критерии
в этом вопросе были установлены Фрумкиным A940); из
них легко рассчитать, что белки, исключая область изоточ-
ки (см. стр. 111), удовлетворяют условиям образования двои-
©so-
Рис. -14. Структура цепей полисульфостирола;
катионы
-[ обмениваемые
но го слоя, а для линейных полиэлектролитов, подобных
полисульфостиролу, это справедливо для всего интервала
рН, хотя эти же системы с полным основанием рассматри-
рассматриваются обычно в свете теории растворов электролитов.
Тикнм образом можно объяснить значительную общность
л:;офсбпых коллоидов и высокомолекулярных электроли-
электролитов (включая белки) в отношении таких электрохимических
свойств, как строение двойного слоя, точки нулевого заря-
заряда, закономерности обмена ионов, зависимость подвижности
частиц от заряда, наличие доннановского равновесия
и др. (см. ниже).
10G
ЭЛЕКТРОКИНЕТИЧЕСКИЕ ЯВЛЕНИЯ
Когда коллоидные частицы любого описанного выше типа
находятся в постоянном электрическом поле, в них, как
и в растворах простых электролитов, происходит движение
зарядов к противоположно заряженным электродам: кол-
коллоидная частица движется в одну сторону, компенсирую-
компенсирующие ионы — в другую. Если бы все компенсирующие ионы
были свободны в своем движении, то общая картина была
бы аналогичной простому переносу ионов. Однако коллоид-
коллоидная частица движется не только с адсорбированными на
ней зарядами (число которых, в отличие от простых ионов,
часто непостоянно), но и с той частью компенсирующих
ионов, которые непосредственно к ней прилегают, что при-
приближенно соответствует гельмгольцевской части двойного
электрического слоя (рис. 42). Таким образом, граница про-
противоположно направленного смещения ионных слоев колло-
коллоидных частиц в электрическом поле не совпадает с границей
поверхности частиц, а несколько смещена от нее в сторону
раствора (приближенно по пунктирной линии на рис. 42).
Это обстоятельство приводит к важным следствиям.
Скорость передвижения коллоидной частицы в электри-
электрическом поле пропорциональна ее потенциалу (см. ниже
уравнение V. 1). Если бы коллоидная частица передвига-
передвигалась без части компенсирующих ионов, то ее измеряемый
потенциал соответствовал бы полной разности потенциалов
между поверхностью частицы и глубиной раствора, т. е.
обычно определяемому в электрохимии полному или тер-
термодинамическому потенциалу ср (рис. 42). Однако в самом
деле часть компенсирующих ионов увлекается вместе с
коллоидной частицей, поэтому определяемый по передви-
передвижению в электрическом поле так называемый электроки-
электрокинетический потенциал частицы С (рис. 42) составляет
лишь часть потенциала ср.
Экспериментально определяется перенос в электрическом
поле коллоидных частиц (электрофорез) или среды (элект-
(электроосмос). Можно осуществить и обратные измерения раз-
разности потенциалов, возникающей при механическом дви-
движении коллоидных частиц (потенциал оседания) или сре-
среды (потенциал течения), но эти методы редко применяются.
Для измерения электрофореза наблюдают перемещение
границы между золем и наслоенной на него жидкостью в
электрическом поле. Совершенная техника электрофореза
7* 107
сложных смесей белков и других веществ была~разработана
Тизелиусом.
Электрофорез ведется при высоком падении потенциала
8—10 el см для более четкого разделения компонентов раст-
раствора по их электрофоретической подвижности. Для опти-
оптического наблюдения, которое обычно производится по от-
Рис. 45. Электрофоретическнй прибор Тизелиуса
клонениюлучана границе раздела,измеряемого при помощи
длиннофокусной оптики (методы Свенсона, Ламма, Троиц-
Троицкого) берут ^/-образную трубку (рис. 45) с плоскопараллель-
плоскопараллельными стенками. Во избежание нарушения четкости границы
при нагреве опыт ведут при охлаждении до 2—4Ъ, при
строгом постоянстве температуры. Электроды Е выносят
далеко от испытуемого раствора, чтобы предупредить за-
загрязнение раствора продуктами электролиза. Испытуемый
раствор (в нижней половине трубки U) и наслаиваемый
буферный раствор (заполняющий всю остальную часть
сосуда) строго выравниваются по рН, электропроводности
108
и ионной силе. Результаты измерений непосредственно
получаются в виде одного или нескольких пиков (рис. 46),
соответственно числу компонентов в растворе, передвигаю-
передвигающихся с различной скоростью. Относительная площадь
пиков характеризует со-
содержание компонентов
в растворе. Электрофо-
ретический анализ, по
Тизелиусу, получил ши-
широкое распространение
при исследовании нор-
нормальных и патологиче-
патологических сывороток (приме-
(примеры приведены на рис.46),
чистых белков и их сме-
смесей, нуклеопротеидов,
детергентов и др. Неко-
Некоторые модификации при-
прибора допускают возмож-
возможность препаративного
разделения наблюдае-
наблюдаемых компонентов.
Для исследования электрофореза " микроскопически
видимых частиц суспензий, бактериальных клеток, а также
белков, адсорбированных на частицах стекла или кварца,
используются методом микроэлектрофореза, наблюдая под
микроскопом перенос частиц в электрическом поле в спе-
специальной микрокювете.
Широкое распространение при исследовании и разделе-
разделении белков, нуклеиновых кислот, аминокислот, жирных
Рис. 46. Диаграммы электрофорети-
ческого анализа по Тизелиусу /—
нормальная сыворотка, //—плазма
крови при множественной миэломе;
///_СЬШоротка крови при нефрозе
Рис. 47. Электрофорез сыворотки на бумаге
кислот, стеринов и других биохимически важных соедине-
соединений получил метод электрофореза на бумаге (рис. 47).
Исследуемая смесь @,01—0,04 мл раствора в буфере)
наносится в виде узкой линии на середине полоски бумаги
(типа хроматографической бумаги, 40 X 6 см), края
109
которой соприкасаются с электродными растворами. Элект-
Электрофорез производится при падении потенциала около 15—
20 в/см и сил тока 3—5 ма. По окончании опыта бумагу
высушивают и прокрашивают красителем (бромфенолблау
и др.), хорошо связывающимся с белками; на бумаге прояв-
проявляется несколько полос, соответственно числу компонентов
смеси.
Электрофоретическая подвижность и измеряется по
величине перемещения, рассчитанной для времени 1 сек.,
и падения потенциала 1 в/см; и имеет размерность [см2 в
сек]. В качестве примера укажем, что для частиц различных
коллоидов (As2S3, берлинская лазурь, РегОз и др.) и =
=2—3-10~4, для белков (вблизи изоточки) #=0,4—0,8-10~4,
для эритроцитов (при рН крови) различных животных
и = 1,0—1,7 • 10~4, и т. д. Величина электрокинетичес-
электрокинетического потенциала С вычисляется при известной и по следую-
следующему уравнению:
1 = ^, (V. 1)
где k — постоянная, зависящая от формы частиц (для
малых сферических частиц k = 6, для цилиндрических
частиц k =4),т]— вязкость растворителя, D — диэлектри-
диэлектрическая постоянная, е — градиент напряжения поля. При
выражении С в милливольтах, е — в вольтах на 1 см,
и — в \х/сек и k=A, уравнение (V. 1) принимает вид:
С- 1,1295-10* ZL
D
(V. 1а)
Величина I, рассчитанная по (V. 1) или (V. 1 а), во многих
случаях составляет около 30—40 мв. По (V.1) отношение
(C/w) не зависит от размера частиц, однако, это справед-
справедливо, если размер частиц велик по сравнению с толщиной
двойного слоя. Для белко-
белковых молекул радиус частиц
сравним с толщиной двой-
двойного слоя и в этом случае
электрофоретическая под-
подвижность зависит как от
_l формы, так и от размеров
V частиц.
Измерения электроосмо-
Рис 48. Схема прибора для элек- са проводятся в «-образной
троосмоса трубке (рис. 48), снабжен-
ПО
ной в одном колене капилляром с делениями и краном.
В средней части трубки / (собирающейся на шчпфе) поме-
помещается диафрагма из испытуемого материала. В постоян-
постоянном электрическом поле происходит перемещение жидко-
жидкости Через диафрагму, которое наблюдается при помощи
капилляра 2. При электроосмотических измерениях
= 1>1295.106
X V Y]
(V.2)
где v — объем жидкости, протекающей через капилляр
(в см3/сек), х — удельная электропроводность (в обратных
омах), / — сила тока в миллиамперах и С выражено в мил-
милливольтах. При измерениях необходимо учитывать иска-
искажающее влияние поверхностной проводимости капилляров
диафрагмы.
Метод электроосмоса применяется преимущественно для
исследования материалов, которые трудно иметь в раство-
растворенном или высокодисперсном состоянии; например, Со-
Соколов исследовал этим методом электрокинетические свой-
свойства коллагена. Практическое применение электроосмос
находит при некоторых процессах обезвоживания пористых
материалов.
ЗАРЯД ПОВЕРХНОСТИ ЧАСТИЦ. ТОЧКА НУЛЕВОГО ЗАРЯДА
И ИЗОТОЧКИ
Электрокинетический потенциале, как указывалось, при-
приближенно характеризует диффузную часть двойного -элек-
-электрического слоя (рис. 42III, кривая ВС). При малых значе-
значениях С заряд tjc , сосредоточенный в этой части двойного
слоя, может быть связан с С по формуле
Dx
(V. 3)
где D — диэлектрическая постоянная и 1/V. — толщина
ионной атмосферы по Дебаю.
Для изучения структуры двойного слоя весьма н-тклое
значение имеет определение полного .заряда на no/.f;)\nocTii
частиц 7,0, соответствующего падению термодииг.^'-'.-ского
потенциала ср на рис. 42. Однако, ввиду ря/n тру;гч1,:тс,
измерения т@ пока представились возможшллп; лишь для
некоторых систем.
111
Фрейндлих и Эттиш сопоставили изменения термодина-
термодинамического потенциала ср и электрокинетического потенциа-
потенциала С для одной и той же поверхности стекла в растворах
различных электролитов. Измерения ср производились со
стеклянным электродом, измерения С — по методу потен-
потенциала течения. Полученные ими результаты приведены в
табл. 9.
Т а'б_л и ц а 9
Значения термодинамического и электрокинетического
потенциалов на стекле
Электролит
Концентрация электролита, мол.
ю-»
ю-»
ю—*
9-потенциал, мв
КС1 . .
ВаС12 .
La(NO3K
Th(NO3L
КС1 . ,
ВаС12
La(NO3K
Th(NO3L
— 92
— 120
— 89
— 134
— 84
— 108
— 64
— 120
— 63
— 103
— 56
— 88
- 35
— 29
— 11
— 3
(.-потенциал, мв
— 38
- 26
— 7
+ 21
— 22
— 17
— 1
+ 16
—54
—92
—38
—46
—4
—3
—0.5
+4
Из табл. 9 ясно видно различие ср и С — потенциалов
по величине и по влиянию на них изменений природы и
концентрации электролитов. Однако эти измерения все же
не дают прямого определения величины заряда поверх-
поверхности ТГ).
Одной из хорошо изученных в этом отношении систем
является макроповерхность ртути, так как изменение ее
потенциала ср (при помощи внешней разности потенциалов)
может быть непосредственно связано с легко измеримым
изменением поверхностного натяжения ртути а и вели-
величиной заряда поверхности т]. Кривые зависимости поверх-
поверхностного^ натяжения а от прилагаемого потенциала поля-
поляризации Е называются электрокапиллярными кривыми
(Липпман, Фрумкин) (рис. 49). По уравнению Липпмана
Те—** (V.4)
112
где у\ — заряд поверхности. Максимум электрокапиллярной
кривой соответствует скачку потенциала ср0 незаряженной
поверхности ртути или потенциала нулевого заряда ртути
относительно раствора, причем значение ср зависит от специ-
специфической (неэлектростатической) адсорбции на ртути ди-
польных молекул или ионов из раствора. Так, из рис. 49
видно, что адсорбция Вг~ или J~~-hohob на ртути сме-
смещает максимум электрокапиллярной кривой в сторону
\го
зго
А/ог SO,
0,5
-0,5
Е,в
Рис. 49. Электрокапиллярные кривые (по
Фрумкину)
/
более отрицательных потенциалов (например, при переходе
от КС1 к KJ — на 260 мв). Лишь при достаточно высокой
отрицательной заряженности поверхности ртути электро-
электростатическое отталкивание может преобладать над адсорб-
адсорбцией любых анионов, вследствие чего в правой ветви электро-
электрокапиллярные кривые сливаются. Для ртути в 1 н. NaCl
величина rl0, измеренная по электрокапиллярной кривой,
составляет 47—50 \ж/см2.
Другой хорошо изученной системой являются золи
Agj (Кройт), в которых заряд частиц определяется преиму-
преимущественной адсорбцией Ag+ или J--hohob: г10=е(ГАё—Гг).
В этом случае адсорбция обоих ионов могла измеряться
прямыми электрометрическими методами при помощи со-
соответствующих электродов.
Было установлено, что равенство адсорбции Ag+ и
J—-hohob, т. е. точка нулевого заряда, при которой
(rAg — /"j ) -= Q? соответствует концентрациям CAg+ —
113
^10 6 и С j^10~10, или, пользуясь обозначением pAg (=
= —\gaAg+), точка нулевого заряда лежит при pAg = 6.
В золях AgJ точка нулевого заряда мало чувствительна
к электролитам, но многовалентные и органические поверх-
ностноактивные ионы несколько смещают ее.
Наконец этот вопрос был детально изучен для белков,
так как в чистых водных растворах заряд белковых молекул
определяется только поглощением Н+ или ОН—-
ионов, что легко может быть измерено при помощи водо-
водородного или стеклянного электрода. Для белков погло-
поглощение Н+ - и ОН~ - ионов непосредственно сопоста-
сопоставимо с константами диссоциации ионогенных групп — кар-
карбоксильной Ка и аминогруппы Кв- Точка нулевого заряда,
т. е. точка равного поглощения Н+ - и ОН~ - ионов,
называется для белков изоионной (Зеренсен) или изопро-
тонной точкой. Положение изоточки (pJ) на шкале рН
определяется для /—/-валентного амфотерного электроли-
электролита уравнением Михаэлиса:
ИЛИ
J =
(V. 5)
где Kw—константа диссоциации воды (pKw= 13,9).
Правда, в белке имеются различные кислые и основные
группы, но формула (V. 5) остается приближенно правиль-
правильной, если отнести значения /Са и /Сб к наиболее сильным
группам. Для полного определения молярного содержания
различных ионогенных групп в белке и их констант диссо-
диссоциации измеряют ход поглощения Н+ - и ОН~ - ионов
в широком интервале рН (примерно, рН 2—12), прибавляя
к белковому раствору различные количества кислоты или
щелочи и измеряя равновесное рН водородным или стек-
стеклянным электродом. При помощи этих кривых титрования
белков определяют также максимальную емкость погло-
поглощения кислот и оснований, т. е. максимальное число заря-
зарядов на белковой молекуле. Например, для сывороточного
альбумина человека при рН = 2 эта величина составляет
vjo = 100 элементарных зарядов на молекулу белка.
В растворах электролитов при преимущественной
адсорбции анионов белками, например, J~ или CNS-,
114
изоионная точка (т. е. значение рН, при котором белковая
молекула является незаряженной по Н+ - и ОН~ - ионам)
смещается в сторону более щелочных рН или более
отрицательного заряда поверхности, подобно смещению
максимума электрокапиллярной кривой на рис. 49; однако,
если на ртути смещение точки нулевого заряда при переходе
от КС1 к KJ составило 260 мв в отрицательную сторону
(см. стр. 113), то на белке это отрицательное смещение сос-
составляет 0,3—0,6 рН, т. е. лишь 20—35 мв.
Нулевые точки заряда и изоточки для некоторых кол-
коллоидных систем приведены в табл. 10.
Таблица 10
Нулевые точки заряда и изоточки
Лиофобные
коллоиды
(в воде)
AgJ
AgBr
AgCI
Положение
нулевой точ-
точки заряда
pAg 6
pAg5,7
pAg 4,0
Белки
Яичный альбумин
Сывороточный альбумин
Желатина
Химотрипсин
Карбоксигемоглобин
Положение
изоточек
рН 4,71
рН 4,89
рН 5,05
рН 8,6
рН 6,87
Для белков между зарядом молекул и электрофорети-
ческой подвижностью существует прямая пропорциональ-
пропорциональная зависимость в широком интервале рН. Если при не-
некотором значении рН число всех положительных зарядов
на белковой молекуле равно общему числу отрицательных
зарядов, то при этом рН молекула не передвигается в элект-
электрическом поле. Значение рН, при котором электрофоре-
тическая подвижность белка равна нулю, называется изо-
электрической точкой. Для чистого водного раствора бел-
белка, в котором заряд молекул определяется только Н+ -
и ОН~ - ионами, изоэлектрическая и изоионная точки
белка, естественно, совпадают. Напротив, при преимущест-
преимущественной адсорбции анионов и щелочном смещении изоион-
изоионной точки (см.выше) на белковой молекуле остаются избыточ-
избыточные отрицательные заряды анионов, сообщающие молекуле
электрофоретическую подвижность в сторону анода. Для
ее компенсации следует сообщить молекуле эквивалентное
количество положительных зарядов, что может быть до-
достигнуто подкислением раствора, т. е. в этом случае изоэлек-
115
трическая точка сместится в кислую сторону. Таким образом,
в растворах солей изоионная и изоэлектрическая - точки
белков смещаются в разные стороны (Пасынский).
Помимо методов электрометрического титрования и
электрофореза, положение изоточки белков иногда опре-
определяется по ряду свойств, косвенно связанных с зарядом
частиц — по вязкости, набуханию, осмотическому давле-
давлению, оптическому вращению, кривые зависимости которых
от рН обладают минимумом в изоточке.
ВЗАИМОДЕЙСТВИЕ С НЕЙТРАЛЬНЫМИ СОЛЯМИ
При добавлении к коллоидному раствору нейтральной
соли имеют место два основных эффекта. Первый эффект
заключается в ионном обмене между ионами двойного слоя
и ионами нейтральной соли; второй эффект состоит в изме-
изменении толщины двойного слоя (или ионной атмосферы)
под влиянием изменения ионной силы раствора. Для про-
простоты положим, что ионы нейтральной соли не входят в
состав решетки или ионогенных групп частицы, вследствие
чего взаимодействие происходит при постоянстве заряда
на поверхности частицы.
Рассмотрим отрицательно заряженную коллоидную
частицу, например, золя AgJ (рис. 43), у которой компен-
компенсирующими ионами являются катионы К+. Добавляя к
золю раствор NaNCb, можно вызвать между ионами Na+
и К+ обмен, который в основном определяется просто
законом действующих масс, поскольку взаимодействие
J- - ионов, образующих внутреннюю обкладку двойного
слоя, с ионами К+ или Na+ приблизительно одинаково.
В этом случае распределение в растворе компенсирующих
ионов, т.е. теперь не только К+-ионов, но смеси К+- и Na+ -
ионов, мало отличается от исходной структуры двойного
слоя. Если же катионы добавленной соли (Си++, РЬ++
и др.) сильнее взаимодействуют с отрицательными зарядами
на поверхности, чем ионы К+ (что может быть результа-
результатом более высокой валентности добавленных катионов или
образования ими с J~- ионами на поверхности частицы
менее растворимого соединения), то обмен ионов и вытесне-
вытеснение некоторого количества К+ - ионов в раствор сопровож-
сопровождается переходом новых компенсирующих ионов Ме2+
из диффузной части в гельмгольцевскую часть двойного
слоя, т. е. перестройкой структуры двойного слоя. Внешним
116
проявлением этой перестройки двойного слоя, связанной
с уменьшением количества зарядов в диффузной части двой-
двойного слоя, является понижение электрокинетического
потенциала; при более полном переходе компенсирующих
ионов в гельмгольцевский слой значение С может упасть
до нуля. Аналогичные процессы происходят при взаимо-
взаимодействии Ва++-ионов с золем AS2S3 (вытеснение компен-
компенсирующих Н+-ионов), SO4—-ионов с золем РегОз или
АЬОз (вытеснение компенсирующих С1~-ионов), Са++-ио-
нов с белками (вытеснение Н+-ионов) и др. (Рабинович,
Каргин, Вейзер, Мукерджи). Большое значение имеет
обменное поглощение ионов почвами (Гедройц, Антипов—
Каратаев), бентонитовыми глинами, осадочными породами
и др. Основной процесс обмена ионов происходит в экви-
эквивалентных соотношениях, но он осложняется специфичес-
специфической адсорбцией одного из ионов соли, реакцией добавлен-
добавленных солей с электролитами, присутствующими в золе,
влиянием концентрации соли (при повышении которой
усиливается молекулярная адсорбция соли) и др. Сильно
адсорбируемые высоковалентные катионы или большие
органические катионы могут при отрицательном заряде
поверхности входить в гельмгольцевскую часть двойного
слоя в сверхэквивалентном количестве, вследствие чего в
диффузную часть двойного слоя входят также сопутствую-
сопутствующие им анионы, что приводит к изменению не только вели-
величины, но и знака (^-потенциала (явление перезарядки).
На рис. 50 подобные изменения С-потенциала показаны для
ионов Al3+,Th4+ и кристаллического фиолетового на отри-
отрицательно заряженной поверхности стекла (по данным Рут-
жерса).
Второй эффект действия солей, как указывалось, опре-
определяется изменением ионной силы раствора при добавлении
к нему электролита. В соответствии с теорией Дебая—Гюк-
келя, повышение- концентрации электролитов, особенно
содержащих многовалентные ионы, вызывает уменьшение
толщины A//.) ионной атмосферы или сжатие диффузной
части двойного электрического слоя. В результате сжатия
все более значительное количество компенсирующих ионов
оказывается в пределах гельмгольцевской части двойного
электрического слоя. Поэтому измеряемая величина С-
потенциала падает и при полном сжатии диффузной части
двойного слоя может дойти до нуля даже при неизменном
значении зарядов на поверхности частицы.
117
В растворах белков влияние нейтральных солей, поми-
помимо смещения изоточек (см. стр. 115), проявляется в повороте
кривых титрования и электрофоретической подвижности
вокруг изоточки в результате изменения ионной силы раст-
раствора.
-с
160,
/4/7
ПО
100
80
60
го
-20
ио
-60
-80
-100
-110
10s W W W 10 1 10 WO
ммоль/л
Рис. 50. Изменение знака С-потенциала стекла при добавле-
добавлении солей (по Рутжерсу)
При взаимодействии коллоидов с электролитами, ионы
которых могут входить в состав решетки или ионогенных
групп частиц, перечисленные эффекты осложняются воз-
возможными процессами изменения заряда поверхности, что
приводит к еще большему разнообразию электрохимических
изменений.
И
YV
\\
^»
\\\
\\\\
X N
\
\
\
\
ч
\
\
V
\
\
у
ч,
ч
\
д
с \
1
\
\
\
?
ч
»
•
\
V
\
к
ч
•.
>
\
ч
•
*•
\
•
ч
L
V
\
—У
ч.
w
с—
ч
\
*—-
у
с
у
?
DKCI
о Са (N03)z
x/)l(N03K
^. Кристалличес
кий сриолетовы
т.
ч
1
ч
с
>
/
J
¦,
N
>
ч,
й
118
РАСТВОРЫ ПОЛИЭЛЕКТРОЛИТОВ
Полиэлектролиты или высокомолекулярные электроли-
электролиты — полиакриловая кислота, полинуклеотиды, полиэтиле-
нимин, полилизин, полиглютаминовая кислота, полисуль-
фостиролы (рис. 44) и др. — обладают рядом важных осо-
особенностей. Они характеризуются высокой плотностью
расположения зарядов — по одному на каждый остаток
цепи (в то время как у белков приходится по 1 СООН-груп-
пе или NH2-rpynne на 6—8 аминокислотных остатков);
поэтому явления электростатического взаимодействия между
ионогенными группами, составляющего, например, в
полилизине более 3 ккал/моль на остаток, и взаимной элек-
электростатической инактивации этих групп выражены весьма
резко. Многие полиэлектролиты имеют гибкие молекулы,
вследствие чего электростатическое отталкивание между
одноименно заряженными группами может, в зависимости
от степени ионизации, приводить к значительной деформации
молекул. Нити полиакриловой кислоты при изменении рН
могут самопроизвольно растягиваться или сокращаться
в несколько раз (Качальский), тогда как в глобулярных
белках растяжение даже при крайних рН составляет ¦ не
более 1,5 раз (Пасынский). В последнее время получены
синтетические полиамфолиты в виде сополимеров акрило-
акриловой кислоты и 2-винилпиридина или сополимеров лизина
и глютаминовой кислоты (Доти); они обладают изоточкой
и имеют в растворе свернутую конфигурацию, которая
может заметно растягиваться при изменении рН. Многие
полиэлектролиты (полилизин и др.) характеризуются зна-
значительной биологической активностью. Белки также от-
относятся к амфотерным высокомолекулярным электро-
электролитам, но, благодаря относительно малой плотности рас-
расположения зарядов, более слабым электростатическим
взаимодействиям и наличию свернутых молекул с множест-
множеством внутримолекулярных водородных связей (см. стр.
237), они обладают рядом характерных особенностей
по сравнению с типичными линейными полиэлектролитами.
Трехмерные полиэлектролиты, т. е. сетки линейных молекул
полиэлектролитов, соединенных химическими поперечными
связями (рис. 44), широко применяются в хроматографии
в качестве ионообменных адсорбентов.
119
КОЛЛОИДНЫЕ ЭЛЕКТРОЛИТЫ
Высокомолекулярные электролиты (полиэлектролиты)
содержат сотни ионогенных групп на одной молекуле. В от-
отличие от них, коллоидные электролиты (как назвал эту
группу веществ Мак-Бэн) образуются путем ассоциации
группы молекул, каждая из которых имеет лишь 1—2 ио-
ногенные группы. Поэтому для существования коллоидных
электролитов необходимо наличие ассоциационного равно-
равновесия между продуктами ассоциации (мицеллами) и индиви-
индивидуальными молекулами, тогда как для существования
полиэлектролитов это условие не должно соблюдаться.
Длкипарипсулыронат -
Длкилсулырат-С
Жирная кислота
\
/
Рис. 51. Ассоциация в растворах детергентов (по Лундгрену)
К коллоидным электролитам относятся вещества, име-
имеющие большое практическое значение: синтетические де-
детергенты, мыла, дубители, красители.
Синтетические детергенты и мыла обладают высокой
поверхностной активностью. Они образуются длинными
углеводородными цепями (С8—С20), на одном конце кото-
которых содержатся ионогенные группы. Анионные детер-
детергенты включают отрицательно заряженные группы —
—SO;T, OSOJT, ОРОГ—GOO" и др. и в качестве компенси-
компенсирующих ионов — Ыа+-ионы; обычные мыла — соли выс-
высших жирных кислот — являются одним из классов анион-
анионных детергентов (рис. 51). Катионные детергенты (или ин-
вертные мыла) относятся преимущественно к алкилзаме-
щенным аммонийным солям (например, хлористый окта-
дециламмоний Ci8H37 NH3 С1) и высшим жирным аминам.
Наконец, имеются также неионогенные детергенты с по-
полярными группами. В водных растворах, благодаря силам
межмолекулярного притяжения, между углеводородными
цепями происходит ассоциация молекул с образованием
сферических или пластинчатых мицелл (рис. 51), содер-
содержащих несколько десятков молекул и имеющих общий
мицеллярный вес 12000—22000. В результате мицеллооб-
разования неполярные цепи образуют как бы углеводород-
углеводородную каплю, экранированную обращенными в воду по-
полярными группами; это состояние соответствует наи-
наибольшему уменьшению свободной энергии. Размеры мицелл
ограничиваются силами электростатического отталкивания
между сближенными ионогенными группами. Равновесие
ассоциации между мицеллами и отдельными молекулами
и степень их ионизации зависят от концентрации, темпе-
температуры, рН, наличия электролитов в растворе и др. Кри-
Критические (или минимальные) концентрации мицеллообразо-
вания лежат около 0,05% и ниже. Физико-химические
свойства растворов детергентов и мыл — эквивалентная
электропроводность, осмотическое давление, вязкость, чис-
числа переноса — проявляют ряд аномалий, обусловленных
наличием как простых ионов, так и мицелл в растворах.
Мицеллярные растворы мыл и детергентов обладают ха-
характерной способностью к солюбилизации или к повы-
повышенной растворимости углеводородов в водных растворах,
благодаря возможности поглощения углеводородов в не-
неполярных ядрах мицелл (Мак-Бэн, Ребиндер). На рис. 52
показана солюбилизация бензола в пластинчатой мицелле
детергента, с увеличением ее размера.
Номенклатура детергентов насчитывает в настоящее
время сотни наименований, в частности, применяются дю-
понол (додециллсульфат), игепон (алкилбензосульфонат),
аэросол ОТ (натриевая соль диоктилового эфира сульфоян-
тарной кислоты), цетилпиридиниумхлорид и др.
Во многих отраслях промышленности и в домашнем хозяй-
хозяйстве широко используется моющее действие детергентов
и мыл, основанное на сложном комплексе явлений смачи-
смачивания, отрыва загрязнений (твердых частиц, масляных
пленок) от ткани, образования механически устойчивых
адсорбционных слоев в пене и удержания частиц загрязне-
загрязнений в пене (Ребиндер). Благодаря высокой поверхностной
120
121
активности и способности к растворению белков, липоидов,
каротеноидов, диссоциации белковых комплексов, инак:
тивации вирусов и бактериальных токсинов, гемолизу и
мощному бактерицидному действию детергенты применя-
применяются для приготовления различных фармацевтических,
дезинфицирующих, бактерицидных и фунгицидных препа-
препаратов. В частности, в фармацевтической промышленности
72,7mju
Рис. 52. Солюбилизация бензола в мицелле детер-
детергента (по Уорду)
детергенты используются при приготовлении мазей против
грибковых и других кожных заболеваний, эмульсий рыб-
рыбного жира, профлавина, касторового масла; препараты
ал кил сульфатов натрия применяются для лечения язвы
желудка.
В растворах дубителей-таннидов — производных много-
многоатомных фенолов — ионогенными группами являются фе-
нольные и отчасти карбоксильные группы. Средний моле-
молекулярный вес первичных частиц таннидов, применяемых
в кожевенной промышленности, равен 1000—-2000, а сред-
средний мицеллярный вес продуктов ассоциации около 20000
(А. Михайлов). Ассоциация таннидов в значительной мере
определяется водородными связями.
122
В растворах красителей ионогенные группы также мо-
могут быть различной природы: сульфогруппы, карбоксиль-
карбоксильные, фенольные и аммонийные (вплоть до четвертичных
замещенных групп). Благодаря значительному молеку-
молекулярному весу большинства красителей, ассоциация в раст-
растворах красителей выражена сильнее, чем в растворах мыл,
причем равновесие ассоциации сильно зависит от концент-
концентрации и температуры растворов (Валько, Липатов).
Электропроводность и осмотическое давление в растворах
красителей (конгорот и др.) также проявляют аномалии,
связанные, как и в растворах детергентов, с образованием
мицелл.
ДОННАНОВСКОЕ РАВНОВЕСИЕ
Большой интерес представляют особенности взаимодей-
взаимодействия коллоидов с ионами электролитов при условии огра-
ограничения свободной диффузии коллоидных частиц, напри-
например, при помощи полупроницаемой мембраны. Общая тео-
теория этих явлений была разработана Доннаном A911).
Эта проблема, имеющая большое физиологическое значение,
представляет также специальный интерес для коллоидной
химии, ввиду отсутствия подобных условий в растворах
низкомолекулярных веществ.
Положим, что коллоид R в результате поглощения
некоторого количества НС1, образовал (с С1 - -ионами, как
компенсирующими ионами) недиффундируемые частицы
RH+, входящие в состав геля или отделенные мембраной
от внешнего раствора электролита:
Если всего поглощено Ci молей НС1, то концентрация RH+
и С]- также равна Ci. Добавим к раствору нейтральный
электролит с одним общим ионом, например, NaCl (при
концентрации Сг) и рассмотрим его распределение в объеме.
Казалось бы, что концентрация NaCl по обе стороны мембра-
мембраны должна быть одинаковой, но это не так. Положим, что
во внутреннюю жидкость, или в гель, перешло х молей
NaCl. Тогда концентрация каждого компонента по обе
стороны мембраны (или вовне и внутри геля) равна
+
R+ (CO
СГ (Ci-
Na+ (x)
+
Na^ (C8-jc)
СГ (C2 — x)
123
При равновесии, согласно термодинамике, произведение
концентрации диффундирующих ионов по обе стороны
мембраны должно быть одинаковым:
х
откуда
= (с2 ~
<v-6>
Из уравнения (V.6) следует, что при сх = 0 х — ^ > т. е. при
отсутствии недиффундирующих коллоидных ионов распре-
распределение NaCl действительно равномерное; если же Сг
значительно больше съ (с1>сг), то х очень мало, т. е. в
этом случае свободно диффундируемый электролит NaCl
все же не может проникнуть внутрь мембраны и распреде-
распределение NaCl между гелем и раствором, или по обе стороны
мембраны, оказывается неодинаковым (табл. 11).
Таблица 11
Мембранное равновесие
Концентрация
Cl
0,01
0,1
1,0
1,0
1,0
Концентрация
XT Г>1
JNaCl
1,0
1,0
1,0
0,1
0,01
0,01
0,1
1,0
10,0
100,0
% NaCl, прошедшего
через мембрану
49,7
47,6
33,0
8,3
1,0
Неравномерное распределение электролита по обе сто-
стороны мембраны вызывает отклонения в величине измеряе-
измеряемого осмотического давления, зависящие от концентрации
R+, заряда коллоидных частиц и от концентрации электро-
электролитов. Теоретически следует, что наблюдаемое осмотиче-
осмотическое давление может быть отнесено к коллоидным частицам
(стр. 33) лишь при очень низкой концентрации электролитов
во внешней жидкости (с2<^а), если же преобладает кон-
концентрация электролитов (c2^>ci), то измеряется лишь
124
половина собственного осмотического давления коллоида.
Фактически при наличии заряженных коллоидных частиц
измеряются промежуточные значения осмотического дав-
давления, в которые необходимо вводить поправку на вели-
величину доннановского эффекта.
Неравновесное распределение ионов по обе стороны
мембраны или между гелем и раствором приводит к возник-
возникновению разности потенциалов, которая может быть изме-
измерена в виде так называемого мембранного потенциала,
например в следующей цепи:
Мембрана
каломелевыи элек-
электрод с насыщен-
насыщенной КС1
внешний рас-
раствор
внутренний
раствор(или
гель)
каломелевыи
электрод с
насыщенным
КС1
Для гелей желатины мембранный потенциал точно от-
отвечает измеренным различиям в концентрации ионов (Леб),
но в структурах коллагена возможны расхождения (А. Ми-
Михайлов). Доннановский эффект наблюдается также при
набухании гелей (стр. 211). В организме доннанов-
доннановский эффект имеет большое значение для распределения
электролитов между кровью и лимфой, для возникновения
биопотенциалов, но следует учитывать, что условия в орга-
организме могут быть далеки от равновесия, ввиду зависимос-
зависимости распределения ионов от ряда сопряженных процессов
обмена (углеводно-фосфорного и др.). Например, в клетках
Valonia, живущих в морской воде, концентрации ионов
внутри и снаружи
Jk
= 41,6;
Na;
Nae
= 0,18;
I!*- = 158,5;
Cl
= 0,97.
Неравенство этих отношений настолько велико, что ясно
указывает на отсутствие осмотического равновесия живых
клеток со средой.
Интересно, что в лиофобных золях наблюдается изме-
изменение рН при оседании суспензии Fe2O3 или глин (суспен-
(суспензионный эффект по Пальману-Вигнеру), или изменение
PJ при оседании и центрифугировании золей AgJ (золь-
125
концентрационный эффект), которые также объясняются
доннановским эффектом. Хотя мембраны и гели в данном
случае отсутствуют, наличие недиффундирующих ионов,
«прикрепленных» к коллоидным частицам и оседающих
вместе с ними, приводит также к неравномерному распреде-
распределению электролита в растворе.
Наконец, при ультрафильтрации кол-
коллоидных растворов наличие доннановско-
го эффекта на мембране ультрафильтра
может привести к отклонениям состава
ультрафильтрата и межчастичной жид-
жидкости золя при значительном относи-
относительном объеме ультрафильтрата (Ра-
(Рабинович, Васильев, Гатовская).
ХРОМАТОГРАФИЯ
Явления ионного обмена и молеку-
молекулярной адсорбции, рассмотренные в чет-
четвертой и в настоящей главе, приобрели
особое значение в хроматографии, став-
ставшей за последние годы одним из важ-
важнейших методов препаративного разделе-
разделения и аналитического исследования
смесей различных соединений, в частно-
частности антибиотических и лекарственных
Рис. 53. Хромато-
грамма хлорофил-
рф
ла: 1—бесцветный
слой, //—ксанто- веществ. Адсорбционная хроматография
филл^р (желтый), была открыта М. С. Цветом A903), при-
р (желтоО-зеленый)Н менившим ее Для разделения компонен-
/К-хлорофиллпна тов хлорофилла (рис. 53).
(зелено-синий),1/- Экспериментально адсорбционная
^жел' хроматография проводится в колонке,
заполненной адсорбентом. Исследуемый
раствор смеси веществ непрерывно про-
пропускается через колонку, и изучается
концентрация вещества С в выходящей
жидкости. Ввиду неодинаковой адсорби-
руемости различных компонентов на
адсорбенте из колонки сначала выходит менее адсорбируе-
адсорбируемый компонент Си, затем — сильнее адсорбируемый — С22
(в смеси с первым компонентом С12) и т. д., вследствие
чего выходная кривая имеет вид, показанный на
рис. 54.
тый), VI—ксанто-
VI—ксантофилл а'(желтый),
VII — ксантофилл
а (желтый), VIII—
хлорофиллин (се-
ростальной)
126
С
Описанный метод анализа называется фронтальной
адсорбционной хроматографией (Тизелиус).
Чаще ограничиваются пропусканием через колонку срав-
сравнительно небольшого объема раствора исследуемой смеси
веществ. В этом случае растворенные вещества задержива-
задерживаются лишь в верхней части колонки. Затем через колонку
начинают пропускать чистый
растворитель и исследуют состав
выходящей жидкости, в которой
один компонент появляется за
другим. Такой метод анализа на-
называется элюционным; типичная
элюционная кривая для сложной
смеси аминокислот показана на
рис. 55, где каждый компонент
дает самостоятельный пик и мо-
может быть собран в виде самосто-
самостоятельной фракции. Вместо раст-
растворителя в тех же условиях мож-
можно применить раствор какого-ли-
какого-либо вещества, которое адсорбиру-
адсорбируется сильнее, чем любой из ком-
компонентов последующего раствора; тогда это вещество будет
вытеснять из адсорбента менее сильно связанное вещество,
последнее—еще слабее связанное и т. п. Выходная кривая
2.0
с»
С22
?,2
Рис. 54. Фронтальная ад-
адсорбционная хроматогра-
хроматография двухкомпонентного
раствора
"
вланин*
глмтаминодая
кислота
нргинин А
Л /V
Л
J.3 itiS si 59 75 8Z 100 WE 117 13b 1ЧУ WJ У/9
Ойьем дытнкшнй /KudxuHmu, мп '
Рис. 55. Элюционное разделение аминокислот на крахмальной ко-
колонке (по Штейну и Муру); растворители: н. бутанол, н. пропанол,
0,1 н. НС1 A:2: 1) и, после аспарагиновой кислоты, н. пропанол,
0,5 н. НС1 B:1)
в этом случае имеет ряд пиков в порядке возрастающей ад-
сорбируемости веществ на адсорбенте и заканчивается
127
выходом вытесняющего вещества. Такой метод анализа
называется вытеснительным.
Во всех перечисленных видах адсорбционной хрома-
хроматографии первичный акт адсорбции может иметь, в зави-
зависимости от природы адсорбента и разделяемых веществ,
характер молекулярной адсорбции или ионного обмена.
Первичные процессы молекулярной адсорбции при хро-
хроматографии подчиняются уравнениям адсорбционной изо-
изотермы Лангмюра или Фрейндлиха (см. стр. 94—95),
а первичные процессы при ионном обмене — уравнению
Никольского:
У тг _
У m2
2
У с2
(V. 6)
где mWi т%—количества поглощенных ионов (в миллиэкви-
валентах на 1 г адсорбента), С\ и С 2 — концентрации, или,
точнее, активности соответствующих ионов в растворе-
2i И22 — валентности ионов и К — константа обмена. Для
одновалентных ионов
?=Ф (V. 6а)
Величина константы ионного обмена К определяет соот-
соотношение ионов в поглощенном состоянии при заданной их
концентрации в растворе и должна заметно отличаться от
единицы.
Основной особенностью хроматографии является прин-
принцип многократного повторения элементарных актов адсорб-
адсорбции-десорбции или ионного обмена. Когда, например,
при элюционном анализе растворитель вымывает вещества
из адсорбированного слоя в верхней части колонки, то на
всем протяжении колонки эти вещества испытывают много-
многократную адсорбцию их на адсорбенте и десорбцию раствори-
растворителем. На высоте слоя адсорбента 1 еж при хроматографии
происходит несколько десятков или даже сотен элементар-
элементарных актов адсорбции-десорбции или ионного обмена
(Самсонов), причем общая высота колонки может составлять
десятки сантиметров и более. Понятно, что эффективность
разделения компонентов смеси при этом резко повышается
по сравнению с эффективностью единичного процесса.
128
Для полноты укажем, что процессы распределения ве-
веществ между двумя жидкими фазами при многократном
повторении лежат в основе еще одного важного метода
хроматографии— распределительной хроматографии. В рас-
распределительной колоночной хроматографии, внешне не
отличающейся от адсорбционной, один из растворителей
пропитывает материал (силикагель, крахмал, целлюлозу),
наполняющий колонку, причем этот материал является
лишь носителем одного растворителя. Исследуемая смесь
наносится вверху колонки. Второй растворитель протекает
через колонку и в процессе течения происходит многократ-
многократное распределение разделяемой смеси вещества между дву-
двумя растворителями и, в результате — полное разделение
компонентов. В качестве носителя неподвижной фазы может
быть взята фильтровальная бумага. Развитая на этой ос-
основе хроматография на бумаге (Мартин, Синг) получила
исключительное значение для целей анализа. Наконец,
многократное использование (до 250—1000 раз) распреде-
распределения между двумя жидкими фазами, без применения
носителя, также широко распространено в виде метода
противоточного распределения (Крэйг).
Существует еще много других методов хроматографии —
осадочная, газовая, газо-жидкостная и др., однако наиболь-
наибольшее значение при работе с веществами биохимического зна-
значения, антибиотиками, лекарственными препаратами и др.
имеют ионообменная и распределительная хроматографии.
Успехи ионообменной хроматографии в значительной мере
обусловлены развитием синтеза ряда специальных ионооб-
ионообменных полимеров или смол (ионитов). Различают два ос- ~7
новных вида ионитов: 1) катиониты, способные к обмену ка-
катионов, представляющие собой сетку высокомолекулярных
полиэлектролитов с многочисленными сульфогруппами (рис.
44) карбоксильными группами и др. (амберлит J/?-100,
дауэкс-50, отечественные КБ-4, СБС и др.) и 2) аниони-
ты, способные к обмену анионов (ОН-, С1~ и др.) и пред-
представляющие собой сетку высокомолекулярных катионов
(амберлит J/?A-400, дауэкс-2, вофатит-М, отечест-
отечественные ЭДЭ-10, ПЭК и др.). Поглотительные емкости ио-
ионитов доходят до 3—10 мэкв на 1 г ионита. Имеются также
окислительно-восстановительные иониты (получаемые пели-
конденсацией гидрохинона, пирогаллола и пирокатехина
с формальдегидом и фенолом), иониты с оптически-актив-
оптически-активными группировками (для разделения оптических изоме-
«Коллоидная химия»
129
ров), иониты с антигенными группировками (для отделения
антител) и др.
В настоящее время при помощи хроматографии произ-
производят полное удаление солей из воды (получение дистилли-
дистиллированной воды без перегонки), разделение сложных смесей
^ 3
I
1
Урация
Цитозин
Гуанин
100
Z00 300
500 600
Объем Zh.HU мл
Рис. 56. Разделение пурнновых и пирнмидиновых оснований на
колонке из катионнта дауэкс — 50, растворитель — 2н. НС1. Выход
98,5—101,5%; концентрация измерялась по оптической плотности
при 260 М[1
аминокислот и гидролизатов белков (см. рис. 55), разделе-
разделение сложных смесей фосфосахаридов, пуриновых и пири-
мидиновых оснований (рис. 56), фракционирование белков
(цитохрома, рибонуклеазы, инсулина и др.), очистку анти-
антител, выделение стрептомицина, хлортетрациклина, поли-
миксина и других антибиотиков, а также алкалоидов, гор-
гормонов, антигистаминных веществ. Большой интерес пред-
представляет также терапевтическое использование ионооб-
ионообменных смол для регулирования состава ионной среды в
желудочно-кишечном тракте и для диагностических целей.
130
ЭЛЕКТРОПРОВОДНОСТЬ КОЛЛОИДОВ. ДИЭЛЕКТРИЧЕСКИЕ
СВОЙСТВА КОЛЛОИДОВ
Электропроводность коллоидного раствора слагается
из электропроводности, обусловленной коллоидными час-
частицами, и электропроводности находящихся в растворе
электролитов. Если посторонних электролитов в растворе
очень мало (высокоочищенные растворы белков и полиэлект-
полиэлектролитов), измерениями электропроводности • можно вос-
воспользоваться для определения удельного заряда или
подвижности частиц, однако, в лиофобных золях определить
собственную электропроводность коллоидных частиц до-
довольно трудно. Существенное влияние на собственную
электропроводность частиц оказывает структура двойного
электрического слоя, так как подвижность компенсирую-
компенсирующих ионов ограничивается электрофоретическим торможе-
торможением со стороны коллоидных частиц (более медленно передви-
передвигающихся в поле, чем ионы) и скоростью перестройки ионной
атмосферы в переменном поле (эффект релаксации). В свою
очередь, измерениями электропроводности в широком диа-
диапазоне частот (дисперсия электропроводности) пользуются
при изучении структуры двойного слоя. В растворах поли-
полиэлектролитов (например, полиакриловой кислоты) измере-
измерения эквивалентной электропроводности X при различных
концентрациях представляют интерес для характеристики
формы молекул, так как значения X падают в той области
концентраций, в которой расстояния между молекулами
полимера становятся велики по сравнению с толщиной
двойного электрического слоя (Каргин). Измерения элек-
электропроводности коллоидных растворов при их взаимодей-
взаимодействии с нейтральными солями (метод кондуктометриче-
ского. титрования) широко применялись при исследовании
состава двойного слоя и процессов вытеснения из коллоид-
коллоидных частиц, например, подвижных Н+-ионов (Паули, Ра-
Рабинович).
С изучением поведения коллоидов в быстропеременном
электрическом поле связано исследование диэлектричес-
диэлектрических свойств коллоидных растворов. Многие золи лиофоб-
лиофобных коллоидов содержат вытянутые частицы со значитель-
значительной асимметрией в расположении электрических зарядов;
вследствие этого они имеют большие постоянные диполь-
ные моменты и легко ориентируются в электрическом поле,
кие Растворы характеризуются высокой диэлектричес-
10*
и 131
кой постоянной D, например, у 1 °о-ного золя V2O5 D = 400
(для сравнения укажем, что у воды D = 80, а у большинства
органических жидкостей — ниже 30). Высокий дипольный
момент был найден также у молекул аминокислот, белков,
нуклеиновых кислот; напротив, у большинства гидрофоб-
гидрофобных коллоидов он невелик. Изучение изменения диэлектри-
диэлектрической постоянной в широком интервале частот поля (дис-
(дисперсия диэлектрической постоянной) является одним из
важных методов исследования коллоидных систем. В гидро-
гидрофобных коллоидах (Fe-zCh, V2O5) этим путем исследовались
изменения систем во времени, в растворах белков — раз-
размеры и форма молекул, в полимерах — процессы их дефор-
деформации (стр. 252) и др. Диэлектрические измерения широ-
широко применялись также для изучения изменений в структуре
клеточных оболочек, эритроцитов крови и др.
Выводы
Электрические заряды на коллоидных частицах возни-
возникают в результате преимущественной адсорбции одного
из ионов электролитов из раствора или диссоциации соб-
собственных ионогенных групп. Независимо от механизма
возникновения зарядов на коллоидных частицах, при доста-
достаточной плотности расположения зарядов, образуется двой-
двойной электрический слой, состоящий из зарядов на поверх-
поверхности и из компенсирующих ионов в растворе; при этом,
по теории Штерна, компенсирующие ионы частично входят
в прилегающий к поверхности адсорбционный слой, а
частично — в диффузную часть двойного слоя. Изучение
заряда поверхности методом электрокапиллярных кривых
(на ртути, V. 4) и кривых титрования (золи AgJ, растворы
белков) позволили определить точки нулевого заряда (в
белках — изо ионную точку, V. 5) и установить их смеще-
смещение в растворах различных электролитов.
В постоянном электрическом поле коллоидные частицы
движутся вместе с частью компенсирующих ионов, причем
граница отрыва приближенно соответствует диффузной
части двойного слоя. Скорость перемещения в электричес-
электрическом поле определяется методами электрофореза и электро-
электроосмоса; по ней можно вычислить величину электрокине-
электрокинетического С-потенциала (V. 1 и V. 2). Величина С-потен-
С-потенциала сильно зависит от природы, и концентрации элек-
электролитов в растворе. С-потенциал отличается от термоди-
132
намического потенциала ср, определяемого зарядом поверхно-
поверхности, и может даже иметь различный знак. Для амфотерных
коллоидов измерения зависимости С от рН позволяют опре-
определить изоэлектрическую точку. При изучении состава
двойного слоя коллоидных частиц применяются также ме-
методы измерения электропроводности.
Взаимодействие коллоидов с нейтральными солями соп-
сопровождается двумя основными эффектами: 1) адсорбцией
ионов солей, обычно по типу ионообменной адсорбции с
изменением структуры двойного слоя, 2) сжатием двойного
слоя при повышении ионной силы раствора.
Характерные особенности растворов полиэлектролитов
состоят в высокой плотности расположения ионогенных
групп и возможности деформации молекул при изменении
величины заряда. Важную группу составляют коллоидные
электролиты — синтетические детергенты, мыла, дубители,
красители; их характерными особенностями являются
ассоциационное равновесие между мицеллами и простыми
молекулами, наличие явлений солюбилизации в растворах
детергентов и мыл и др.
При ограничении свободной диффузии коллоидных час-
частиц (мембраны, гели, суспензии и др.) имеет место харак-
характерное для коллоидных систем неравномерное распреде-
распределение электролитов (эффект Доннана, V. 6).
Процессы многократной ионообменной и молекулярной
адсорбции на трехмерных полиэлектролитах и адсорбен-
адсорбентах лежат в основе различных важных методов хромато-
хроматографии.
Вопросы
1. Как образуется двойной электрический слой и каково его
строение по теории Гельмгольца, Гуи, Штерна?
2. Чем различаются термодинамический и электрокинетичес-
электрокинетический потенциалы и как они зависят от состава среды?
3. Какими методами измеряется электрокинетический потен-
потенциал и как рассчитывается его величина?
4. Что такое нулевые точки заряда и изоточки, какими мето-
методами они исследуются в различных системах, как изменяются в
зависимости от состава среды?
5. Каковы основные эффекты взаимодействия коллоидных
систем с нейтральными солями? В чем выражаются специфические
явления при этом взаимодействии? Как изменяется структура
Двойного слоя в результате этого взаимодействия?
6. Каковы характерные особенности полиэлектролитов?
7. Какие вещества относятся к коллоидным электролитам,
каковы характерные особенности этой группы веществ, каковы
133
условия ассоциации в растворах коллоидных электролитов? Что
такое солюбилизация?
8. Укажите принципы и методы использования различных ви-
видов адсорбционной, ионообменной*и распределительной хромато-
хроматографии.
9. В чем заключается эффект Доннана и в каких формах он про-
проявляется в различных коллоидных системах?
10. Какое значение имеют хроматографические методы и ис-
использование детергентов в промышленности?
Глава шестая
УСТОЙЧИВОСТЬ И КОАГУЛЯЦИЯ КОЛЛОИДОВ
Лиофобные коллоиды являются термодинамически
неустойчивыми системами, стабильность которых обуслов-
обусловлена наличием адсорбционных ионных или молекулярных
слоев. Изменения состояния этих слоев, механизм обра-
образования и свойства которых были рассмотрены в главах
четвертой и пятой, сопровождаются изменением устойчи-
устойчивости лиофобных коллоидов и при определенных услови-
условиях могут приводить к потере устойчивости; внешне это
проявляется в агрегации и выпадении частиц из раствора
или в их коагуляции. Таким образом, теория коагуляции
тесно связана с выяснением природы устойчивости и само-
самого существования золей, что придает ей большое значение.
Условия коагуляции золей весьма различны и зависят от
природы стабилизующих слоев. Целесообразно, поэтому,
рассмотреть эту проблему отдельно для золей с ионными
и молекулярными адсорбционными слоями.
УСТОЙЧИВОСТЬ ЛИОФОБНЫХ КОЛЛОИДОВ С ИОННЫМИ
АДСОРБЦИОННЫМИ СОЛЯМИ. КОАГУЛЯЦИЯ ЛИОФОБНЫХ
КОЛЛОИДОВ ЭЛЕКТРОЛИТАМИ
Лиофобные золи, частицы которых несут двойные ион-
ионные слои, могут быть коагулированы любыми электроли-
электролитами при сравнительно невысокой концентрации, однако,
величины этих концентраций, в зависимости от природы
электролита, резко различаются между собой. Установ-
Установлено, что коагулирующее действие обычно оказывает ион
противоположного знака по отношению к знаку заряда
поверхности коллоидных частиц, причем это действие тем
сильнее, чем выше валентность иона (правило Шульце—
135
Гарди). Соотношение коагулирующих концентраций (или
порогов коагуляции) различных ионов оказывается обрат-
обратно пропорциональным высокой степени валентности z
(приблизительно пропорционально z6), как видно из
табл. 12 и 13.
Таблица 12
Средние значения порогов
отрицательно
Валентность
коагул ирующего
иона
Одновалентные
Двухвалентные
Трехвалентные
Четырехвалент-
Четырехвалентные ....
Золь
As2S3
55
0,69
0,091
0,090
заряженных
Отно-
Отношение
1
0,013
0,0017
0,0017
Золь
Аи
24
0,38
0.С06
0,0009
коагуляции (ммол/л)
золей (по Овербеку)
Отно-
Отношение
1
0,016
0,0003
0,00004
Золь
AgJ
142
2,43
0,068
0,013
Отно-
Отношение
1
0,017
0,0005
0,0001
Теоретиче-
Теоретическое от-
отношение
(Z»)
1
0,016
0,0013
0,00024
Таблица 13
Средние значения порогов коагуляции (ммол/л)
положительно заряженных ионов (по Овербеку)
Валентность коагулирую-
коагулирующего иона
Золь
Fe2O3
11,8
0,21
—
—
Отно-
Отношение
1,00
0,018
—
—
Золь
А12О3
52
0,63
0,080
0,053
Отноше-
Отношение
1,00
0,012
0,0015
0,0010
Теоретиче-
Теоретическое от-
отношение
<»¦)
Одновалентные
Двухвалентные .
Трехвалентные
Четырехвалентные
1,00
0,016
0,0013
0,00024
Интересно, что в тех же отношениях находятся агглю-
агглютинирующие концентрации различных солей при их дей-
действии на бактерии.
Правило Шульце—Гарди имеет лишь приближенный
характер, так как коагулирующее действие зависит не
только от валентности ионов; например, некоторые орга-
органические одновалентные основания (катионы морфина и др.)
обладают более сильным коагулирующим действием, чем
двухвалентные ионы. При одном и том же анионе пороги
коагуляции одновалентных катионов не одинаковы, и рас-
136
полагаются в ряд Lib у Na+ > K>>Rb' > Cs+", а при
одном и том же катионе одновалентные анноны располага-
располагаются в ряд J- > NO3- > Вг~ > C1-. Эти ряды ионов
называются лиотропными рядами (см. стр. 185). При
коагуляции золен смесями электролитов наблюдаются
дальнейшие усложнения, проявляющиеся в отклонениях
от аддитивности или взаимозаменяемости солей.
Ограничиваясь более простыми случаями, необходимо
подчеркнуть, что при коагуляции коллоидов обычно проис-
происходит адсорбция коагулирующих ионов коллоидными час-
частицами. Эта адсорбция не обязательно должна приводить к
потере зарядов на поверхности частицы; напротив, на при-
примере золей AgJ было показано, что пороги коагуляции
сравнительно слабо зависят от термодинамического потен-
потенциала поверхности. Гораздо важнее те изменения, которые
вызываются в структуре двойного электрического слоя в
растворе, и которые при коагуляции, в основном, всегда
направлены в сторону уменьшения диффузной части двой-
двойного слоя.
Как уже указывалось (см. стр. 116), при взаимо-
взаимодействии коллоидных частиц с электролитами обычно проис-
происходят реакции ионного обмена между коагулирующими
ионами и частью компенсирующих ионов, которые подроб-
подробно исследовались при помощи различных электрометриче-
электрометрических и аналитических методов (Рабинович, Каргин, Фрейнд-
Фрейндлих, Вейзер и др.). Например, при коагуляции золя AssS3
раствором ВаСЬ ионы ВаН- адсорбируются коллоидными
частицами, вытесняя приближенно эквивалентное количест-
количество Н+-ионов в раствор; при коагуляции золя ре*Оз раст-
раствором Na2SOa ионы SO i адсорбируются с вытеснением
С]---ионов из двойного слоя и т. д. Структура двойного
слоя при этом изменяется таким образом, что коагулирую-
коагулирующие ионы в большей мере, чем прежние компенсирующие
ионы, сосредоточиваются во внутренней, гельмгольцевской
части двойного слоя. Химически этот результат эквивален-
эквивалентен образованию на поверхности частицы менее раствори-
растворимого или менее диссоциированного поверхностного сое-
соединения (например, мышьяковисто-кислого бария вместо
мышьяковистой кислоты в золе AssSs; см. стр. 104).
Уменьшение количества компенсирующих ионов в диффуз-
диффузной части двойного слоя проявляется в понижении величи-
величины электрокинетического С-потенциала, обычно сопровож-
сопровождающем обменную адсорбцию (рис. 57). Во многих случаях
9 «Кил.ГС)НДП,1я \М\П!И>, ] -^-7
коагуляция наступает при понижении С-потенциала,
примерно, с 70 до 30 же, однако это изменение представляет
собой лишь внешнее проявление перестройки двойного
слоя, и само по себе не является причиной коагуляции,
как предполагалось ранее. В некоторых случаях коагуля-
коагуляция происходит при слабом уменьшении или даже при
Концентрация БаС12
Рис. 57. Кривые электрофоретической подвижно-
подвижности A) и обменной адсорбции B) при коагуляции зо-
золя As2S3 раствором ВаС12 (Рабинович и Фодиман).
Обменная адсорбция Ва++ с вытеснением Н+ из-
измерялась по росту электропроводности раствора
небольшом росте измеряемого С-потенциала (например,
при коагуляции золя HgS при помощи K4Fe(CNN).
Большое значение имеют также химические реакции
коагулирующего электролита с теми электролитами, ко-
которые содержатся в дисперсионной среде лиофобного золя,
ввиду того, что эти реакции смещают адсорбционное рав-
равновесие между коллоидными частицами и электролитами
дисперсионной среды и нарушают условия обменной адсорб-
адсорбции (Каргин). Необходимо также учитывать возможность
молекулярной адсорбции добавленного электролита (см.
стр. 117). Однако, несмотря на сложное взаимодействие
перечисленных факторов, основные изменения в структуре
двойного слоя при коагуляции, как указывалось, направ-
направлены в сторону его сжатия.
В пятой главе (стр. 117) отмечалось также, что второй
основной эффект действия солей на коллоидные частицы
138
обусловлен изменением ионной силы раствора при добав-
добавлении к нему электролитов. Весьма существенно, что это
электростатическое действие солей, как было показано,
также приводит к сжатию ионной атмосферы или электри-
электрического двойного слоя вокруг коллоидных частиц.
Существует ряд теорий коагуляции, пытающихся объяс-
объяснить ее только явлениями адсорбции (Фрейндлих) или
только электростатическим сжатием двойного слоя (Мюл-
(Мюллер), однако из сказанного следует, что теория не может
иметь такого одно-
одностороннего характе-
характера. Поскольку все
факторы коагуляции
различными путями
приводят к такой пе-
перестройке двойного
электрического слоя,
при которой происхо-
происходит его сжатие, воз-
возникает вопрос—поче-
вопрос—почему вообще сокраще-
сокращение толщины двойно-
двойного слоя должно при-
приводить к коагуляции?
Отвечая на этот вопрос, следует учесть, что для коагуля-
коагуляции коллоидных частиц они должны сблизиться на такое
расстояние, при котором энергия их взаимного молеку-
молекулярного притяжения, обусловленного Ван-дер-Ваальсо-
выми силами, была бы больше энергии теплового (броунов-
(броуновского) движения. Для этого при сближении сферических
частиц необходимо, чтобы наименьшее расстояние между
их поверхностями было мало по сравнению с радиусами
частиц. При достаточно малых расстояниях энергия
взаимодействия убывает обратно пропорционально первой
степени расстояния между поверхностями, но на больших
расстояниях, порядка сотен ангстремов, энергия взаимодей-
взаимодействия начинает очень быстро убывать (Лифшиц, Дерягин).
Однако сближению коллоидных частиц на достаточно ма-
малые расстояния препятствует электростатическое отталки-
отталкивание между их двойными электрическими слоями. Дерягин
показал, что эти силы электростатического отталкивания
возникают лишь при перекрытии ионных атмосфер кол-
коллоидных частиц Ai и Л 2 (рис. 58). Внешняя оболочка двои-
Рис. 58. Перекрытие ионных атмо-
атмосфер двух коллоидных частиц (по
Дерягину)
139
ного электрического слоя в значительной мере экранирует
заряд коллоидных частиц, и до тех пор, пока диффузные
слои коллоидных частиц практически не перекрываются,
электростатическое отталкивание не имеет существенного
значения. Лишь в зоне перекрытия ионных атмосфер
(рис. 58,А о) происходит перераспределение ионов с местным
изменением их концентрации, вследствие чего появляются
дополнительные электростатические и осмотические силы,
приводящие к результативному отталкиванию коллоид-
коллоидных частиц. Отсюда следует и ответ на поставленный выше
вопрос: при сокращении толщины двойного слоя коллоид-
коллоидные частицы имеют возможность более тесного сближения
без того, чтобы между ними возникли силы отталкивания,
и при достаточно малых расстояниях силы межчастичного
притяжения, наконец, преобладают над броуновским дви-
движением частиц; этот момент и соответствует слипанию
или коагуляции частиц.
Дерягин и Ландау показали, что величина порога коа-
коагуляции -у определяется следующим уравнением:
(VI. 1)
где С—константа, D — диэлектрическая постоянная,
А — константа Ван-дер-Ваальсового притяжения, е —
заряд электрона иг — валентность коагулирующего иона.
Из (VI. 1) видно, что величина порога коагуляции обратно
пропорциональна zu; это хорошо согласуется с данными,
приведенными в табл. 12 и 13.
Частным случаем коагуляции электролитами является
взаимная коагуляция двух гидрофобных коллоидов с раз-
различными знаками зарядов. Здесь структура двойных слоев
коллоидных частиц имеет обратный знак и перекрытие ион-
ионных атмосфер способствует не отталкиванию, а притяжению
коллоидных частиц. Таким образом, расстояние, на котором
преобладают силы притяжения (радиус взаимодействия
R) при взаимной коагуляции коллоидов, значительно боль-
больше фактических размеров частиц г; например, при взаим-
взаимной коагуляции положительно заряженного золя гидро-
гидроокиси меди и отрицательных частичек глины величина R
составляла до 65 г (Вигнер). Естественно, что наиболее
полная коагуляция происходит при взаимной нейтрализации
. зарядов частиц. При избытке одного из коллоидов наблю-
наблюдается перераспределение ионов и образование изменен-
140
ных двойных слоев вокруг агрегированных частиц, вслед-
вследствие чего система в целом может остаться устойчивой,
со знаком заряда избыточного коллоида.
При взаимодействии различных гидрозолей с одноимен-
одноименно заряженными частицами основное значение имеют хи-
химические реакции с электролитами межчастичной жидкости,
с последующим смещением адсорбционного равновесия
между этими электролитами и коллоидными частицами.
Например, реакция между отрицательно заряженными
золями серы и серебра в значительной мере объясняется
реакцией пентатионовых кислот, присутствующих в золе
серы, с частицами золя серебра (Скворцов).
При изменении концентрации золей или при коагуляции
смесями электролитов также происходит смещение адсорб-
адсорбционного равновесия между коллоидными частицами и
электролитами дисперсионной среды, вследствие чего ионы
двойных слоев перераспределяются, и пороги коагуляции
изменяются.
Для коагуляции золя смесями электролитов часто тре-
требуется большее количество поливалентного иона, чем
в отсутствии второго электролита. Это явление повышения
порога коагуляции в смеси электролитов называется анта-
антагонизмом ионов; оно обычно наблюдается для разновалент-
ных ионов,.например, для ионов А13+ и К+ при коагуляции
золя AgJ. Антагонизм ионов объясняется, с одной стороны,
смещением адсорбционного равновесия (конкуренцией ио-
ионов за адсорбционные центры на поверхности коллоидных
частиц, наличием специфической адсорбируемости ионов и
др.), а с другой стороны — электростатическим понижением
активности ионов в объеме раствора и в электрическом поле
коллоидных частиц (Кройт, Глазман, Лепинь). В некоторых
случаях при коагуляции смесями электролитов наблюдается
понижение порогов коагуляции (синергизм ионов).
Описанные явления не следует смешивать с явлениями
физиологического антагонизма ионов, под которыми пони-
понимают ослабление одним катионом токсического или иного
физиологического действия, вызываемого другим катио-
катионом. Например, соли Na+ и Zn++ в отдельности ядовиты
для зародышей морской рыбы Fundulus, но в смеси они
безвредны. Очевидно, что явления физиологического анта-
антагонизма ионов имеют гораздо более сложную природу.
Если в результате коагуляции возникает прямой контакт
поверхностей частиц (например, при коагуляции золей
Ml
благородных металлов) или происходит значительная пере-
перекристаллизация и срастание частиц, то коагуляция прак-
практически необратима. Однако если частицы в осадке сохра-
сохраняются обособленными и разделенными тонкими слоями
жидкости или сжатыми двойными слоями (например, в
свежих осадках гидратов окисей металлов), то, увеличивая
степень диффузности двойных слоев, можно вновь пере-
перевести коагулированные частицы из осадка в состояние
золя. Например, Васильев и Дешалыт прибавляли к золю
Fe(OHK, коагулированному SO" - ионами, раствор
ВаСЬ; вследствие выпадение осадка BaSO4, ионы SO7~~
удалялись из двойного слоя и вновь замещались С1~ -
ионами, с увеличением толщины двойного слоя и перехо-
переходом частиц в золь. Такой процесс обращения коагуляции
путем перехода коллоидных частиц из осадка в золь назы-
называется пептизацией (в отличие от образования золя путем
дробления и диспергации частиц). Пептизация может
происходить также при промывке осадков и удалении из
них избыточного электролита. Напротив, свободные от
электролитов осадки Fe(OHK можно пептизировать добав-
добавлением небольших количеств электролита (РеСЬ), один из
ионов которого (Fe3+) способен сильно адсорбироваться
на поверхности частиц, а другой (С1~) образует внешнюю
обкладку диффузного двойного слоя. Таким образом, при
известных условиях коагуляция лиофобных коллоидов
является обратимой, причем было показано, что пептизи-
рованные золи по своим электрохимическим свойствам
могут быть очень близкими к исходным золям.
Глазман исследовал для ряда отрицательно заряженных
золей (AS2S3, AgJ, V2O5, HgS) адсорбцию одноименно
заряженных ионов SO4 и POij~ при коагуляции,
пользуясь методом радиоактивных изотопов (Ss5 и Р32).
Оказалось, что адсорбция одноименных ионов крайне не-
незначительна A—2 pi моля на 1 г коллоида), в соответствии
с правилом Шульце—Гарди.
Большой интерес представляют исследования устойчи-
устойчивости золей в различных неводных средах (органозолей).
Было показано, что устойчивость органозолей металлов
и сульфидов металлов также объясняется образованием
двойного электрического слоя (Глазман, Прейс и Николае-
Николаева), причем устойчивость зависит от возможности образо-
образования внутрикомплексных соединений между частицами
металлов и растворителем, например, между W, Mo, Zr
112
и ортоксихинолином, фенилгидразином и др. (Натансон).
В целом, устойчивость лиофобных органозолей ниже, чем
золей в водной среде.
тиксотропия
Энергии взаимодействия W двух коллоидных частиц,
в зависимости от расстояния между их поверхностями, из-
изменяются по кривой, изображенной на рис. 59, где поло-
положительные значения W соответствуют отталкиванию, а
отрицательные W—притяжению частиц. Подобные кри-
кривые, получаемые суммирова-
суммированием сил межчастичного при-
притяжения и электростатическо-
электростатического отталкивания, называются
потенциальными кривыми.
На рис. 59 видно наличие
барьера отталкивания 5 на
расстоянии г0, препятствую-
препятствующего сближению частиц; при
достаточно высокой концент-
концентрации электролитов барьер 5
устраняется и преобладают
силы притяжения, сближаю-
сближающие частицы при коагуляции
до расстояния г2. Однако рас-,
четы показывают,что при боль-
больших расстояниях гх между час-
частицами (порядка нескольких
толщин двойного электрического слоя) на потенциальной
кривой существует второй, неглубокий минимум М. Для
обычных гидрофобных золей глубина М незначительна,
но для сравнительно крупных асимметричных частиц, имею-
имеющих форму палочек или пластинок, энергия взаимодействия
в точке М может быть в несколько раз больше энергии
теплового движения, что создает возможность доволь-
довольно устойчивого взаимного притяжения этих частиц на боль-
больших расстояниях. Из рис. 59 видно, что глубина минимума
М гораздо меньше, чем коагуляционного минимума F,
и поэтому связь между частицами на расстоянии гх гораздо
слабее, чем при истинной коагуляции. Слабые структуры,
образующиеся благодаря минимуму М, придают золю
гелеобразное состояние, но могут быть легко разрушены
143
Рис. 59. Энергия взаимодейст-
взаимодействия двух коллоидных частиц в
зависимости от расстояния
между их поверхностями
при простом встряхивании, причем получившийся золь
при стоянии снова превращается в гель и т. д.; такие пере-
переходы могут быть повторены десятки раз. Это интересное
явление изотермического обратимого золь ч^ гель пере-
перехода получило название тиксотропии (Фрейндлих).
Тиксотропные структуры возникают лишь при определен-
определенных концентрациях коллоидных частиц и электролитов и
по существу также относятся к коагуляционным структу-
структурам, но образующимся при своеобразных условиях. Период
застудневания при тиксотропии — величина постоянная для
каждой данной системы и часто используется в качестве
показателя ее устойчивости. Количественная характерис-
характеристика тиксотропии дается путем измерения кинетики само-
самопроизвольного восстановления различных механических
свойств (модуля упругости на сдвиг, прочности и др.;
см. стр. 243) в зависимости от времени стояния сис-
системы. Предельное развитие тиксотропной структуры харак-
характеризуется по кинетическим кривым. Явления тиксотропии
наблюдаются в золях V2O5, \УОз, РегОз, в различных сус-
суспензиях бентонита, в растворах вируса табачной мозаи-
мозаики, миозина и др., обладающих асимметричными частица-
частицами, а также в протоплазме клеток (особенно при их де-
делении).
Образование и разрушение тиксотропных структур
имеет большое значение в технике, при использовании
суспензий глин, типографских красок и др.
В растворах перечисленных выше золей с вытянутыми
частицами наличие сил межчастичного притяжения на
больших расстояниях приводит при определенных концент-
концентрациях золей и электролитов к ориентированной агрега-
агрегации частиц в виде оптически анизотропных образований
веретенообразной формы, называемых тактоидами. Расстоя-
Расстояния между частицами в тактоидах могут достигать несколь-
нескольких сотен ангстремов.
УСТОЙЧИВОСТЬ ЛИОФОБНЫХ КОЛЛОИДОВ
с молекулярными адсорбционными слоями.
ЗАЩИТНОЕ ДЕЙСТВИЕ
В лиоробных золях поверхность коллоидных частиц
является резкой границей раздела двух фаз и обладает
свободной поверхностной энергией, определяющей обра-
образование адсорбционных слоев на поверхности.
Ы4
При адсорбции ионов и создании двойного электричес-
электрического слоя на поверхности коллоидных частиц гидрофиль-
ность поверхности возрастает в результате собственной
гидратации ионов и влияния зарядов поверхности на ориен-
ориентированную адсорбцию дипольных молекул воды; так,
например, возрастает смачивание заряженной поверхности
ртути. Однако в этом случае гидратные слои полностью
зависят от ионных взаимодействий и при наступлении коа-
коагуляции коллоидов электролитами не препятствуют про-
процессам слипания частиц. Поэтому в типично лиофобных
золях (Аи, Ag, S, AS2S3 и др.) сразу после перехода порога
коагуляции наблюдается помутнение раствора, изменение
цвета, выпадение осадка и другие проявления коагуляции.
Совсем другой характер имеет устойчивость золей, если
поверхность коллоидных частиц сама является гидрофиль-
гидрофильной или способна к образованию молекулярных сольват-
ных слоев при участии Ваи-дер-Ваальсовых, водородных и
комплексных связей, сравнительно устойчивых к действию
электролитов небольших концентраций. В этом случае
даже при переходе порога коагуляции коллоидные частицы
могут сохраниться в состоянии золя, если они обладают
достаточной гидрофильностью. Так, например, высоко-
очищенные золи кремнекислоты и А1(ОН)з могут сохранить-
сохраниться в растворе даже, при падении электрофоретической под-
подвижности (С-потенциала) почти до нуля (Каргин и Берест-
нева).
Значение сольватных слоев для устойчивости коллоид-
коллоидных растворов было выяснено Дерягиным, который показал,
что для сближения частиц, разделенных сольватными слоя-
слоями, необходимо затратить работу на преодоление сопротив-
сопротивления («расклинивающего давления»), обусловленного сила-
силами молекулярного сцепления жидкости (воды) и поверхности
частиц. Это сопротивление ощущается приблизительно до
расстояния 10д или меньше, после чего силы взаимного
притяжения самих частиц уже преобладают над силами
сцепления в сольватном слое. На более значительных
расстояниях сольватные слои являются термодинамически
устойчивым стабилизующим фактором.
Таким же образом надо рассматривать значение адсорб-
адсорбционных слоев, образованных молекулами поверхностно-
активных веществ на поверхности коллоидных частиц, для
устойчивости коллоидных растворов, но в этом случае боль-
большое значение имеет и характер ориентации поверхностно-
145
активных молекул в адсорбционном слое (см. стр. 98),
так как повышение гидрофильное™ поверхности может
быть достигнуто лишь при определенной ориентации мо-
молекул (Ребиндер).
Устойчивость коллоидных систем в водной среде более
высокая, если полярные группы поверхностно-активных
молекул в адсорбционном слое обращены в воду, что повы-
повышает гидрофильность поверхности, и, напротив, более
низкая, если в воду обращены углеводородные цепи, ко-
которые в водной среде стремятся к взаимному соединению.
Разумеется, в неполярной углеводородной среде роль
ориентации молекул в адсорбционных слоях для устойчи-
устойчивости коллоидов оказывается обратной.
Наконец, роль ориентации поверхностно-активных мо-
молекул в адсорбционных слоях приобретает особое значение
в случае образования ими двухмерных гелеобразных струк-
структур, обладающих повышенными структурно-механическими
свойствами, которые подробно исследовались Трапезни-
Трапезниковым. Обладая довольно высокой упругостью и механи-
механической прочностью, подобные адсорбционные пленки мо-
могут эффективно защищать коллоидные частицы от возмож-
возможности слипания. Это явление лежит в основе защитного
действия желатины и некоторых мыл против коагуляции
лиофобных коллоидов. Так, например, при добавлении
всего 0,01 мг желатины на \0 мл золя золота можно защи-
защитить его от коагуляции 1 мл 10%-ного раствора NaQ.
Зигмонди назвал эту величину @,01 мг) «золотым числом»
желатины и определил подобные числа для ряда других
веществ. Аналогичным образом было определено защитное
действие в отношении золей серебра («серебряное число»),
конгорубинового («рубиновое число»), серы, берлинской
лазури, окиси железа (табл. 14), из которых методически
наиболее удобно определение «рубинового числа».
Золотые, рубиновые и другие числа являются обратной
мерой защитного действия, так как они тем меньше, чем
сильнее это действие. Измерения золотого числа спинномоз-
спинномозговой жидкости используются для диагностических целей
при некоторых заболеваниях.
Для желатины толщина защитного слоя на частицах
кварца или золота составляет всего около 10а, т. е. соот-
соответствует образованию одного мономолекулярного слоя
(стр. 91). Показано, что высокомолекулярная фракция
желатины обладает более сильным защитным дей-
146
Таблица 14
Защитное действие различных коллоидов
(по Овербеку)
Число
Защитные
коллоиды
Желатина
Казеинат натрия . .
Гемоглобин ....
Яичный альбумин
Гуммиарабик ....
Декстрин
Картофельный крах-
крахмал
Сапонин
Золотое
0,01
0,01
0,03—0,07
2,5
0,5
20
20
115 ,
Сереб-
Серебряное
0,035
1,5
1,25
100
35
Руби-
2,5
0,4
0,8
2,0
20
Серное
Для
берлин
ской
лазури
0,00012
0,025
0,024
0,125
0,015
0,05
25
5
250
2,5
Для
ОКИСИ
желе-
15
25
20
15
ствием в водных растворах (Липатов), тогда как органо-
золи вольфрама в толуоле лучше защищаются высокомоле-
высокомолекулярной фракцией каучука (Натансон), что, естественно,
объясняется более легким образованием гелеподобных струк-
структур этими фракциями. Электрофоретическая подвижность
частиц золей золота, кварца, РегОз, МпОг, защищенных
желатиной, совпадает с подвижностью чистых растворов
желатины (рис. 60), т. е. адсорбция не затрагивает
ионогенных групп желатины. Из определения золотого
числа видно, что защищенные лиофобные золи обладают
резко повышенной устойчивостью к коагуляции электроли-
электролитами; правило Шульце—Гарди к этим золям неприменимо.
В то же время они осаждаются теми веществами, которые
осаждают желатину, например, таннином.
Защитное действие представляет интерес для фарма-
фармацевтической промышленности при получении устойчивых
концентрированных золей серебра («колларгол»), ртути, зо-
золота и их радиоактивных изотопов.
При очень малых количествах желатины, недостаточных
даже для образования мономолекулярного слоя (т. е.
при неполном покрытии поверхности), может наблюдаться
не повышение, а, напротив, понижение устойчивости или
сенсибилизация гидрофобного золя. Возможно, что здесь
одна молекулярная цепь желатины приходит в соприкос-
соприкосновение с двумя или несколькими частицами лиофобного
147
золя и, благодаря своей гибкости, способствует прямому
контакту частиц; кроме того, молекулы желатины' могут
десорбировать часть стабилизирующих ионов с поверхности
коллоидных частиц (Песков и Прейс). В тех случаях, когда
желатина и лиофобный золь заряжены противоположно,
сенсибилизация может быть частным случаем взаимной
коагуляции коллоидов.
В целом, состояние и свойства ионных и молекуляр-
молекулярных адсорбционных слоев, рассмотренные в четвертой —
шестой главах, пол-
полностью опрсгяляют
устойчивость лиофоб-
ных коллоидов в от-
отношении агрегации
их частиц или агре-
гативную устойчи-
устойчивость коллоидов; на-
напротив, без этих слоев
лиофобные коллоиды
являются агрегатив-
но совершенно неу-
неустойчивыми система-
системами, ввиду наличия
на частицах значи-
значительной свободной
z 3
Л -зорь МпО,
Рис. 60. Электрофоретическая подвиж-
подвижность золей, защищенных желатиной.
Сплошная линия — чистый раствор
желатины
поверхностной энер-
энергии. Понятие агрега-
тивной устойчивости
было разработано
Н. Песковым (стр. 10). Агрегативная устойчивость, как
и кинетическая (стр. 41), является самостоятельной
характеристикой лиофобных коллоидов. Разумеется, обе
характеристики устойчивости внутренне связаны между
собой: агрегативная устойчивость имеет смысл только для
кинетически устойчивых систем, а кинетическая устойчи-
устойчивость быстро утрачивается в агрегативно неустойчивой
системе. Лишь сочетание кинетической устойчивости и
агрегативной устойчивости, обусловленной ионными и
молекулярными адсорбционными слоями, характеризует
природу стабильности лиофобных коллоидов *. Химичес-
* О природе устойчивости растворов высокомолекулярных ве-
веществ см. в главе восьмой.
118
кие и структурно-механические свойства адсорбционных
слоев в значительной мере определяют все поведение лио-
лиофобных коллоидных систем. Эти слои могут быть образова-
образованы или изменены небольшими количествами веществ, поэ-
поэтому создаются возможности регулирования ряда свойств
лиофобных коллоидов, что широко используется в различ-
различных практических приложениях.
КИНЕТИКА КОАГУЛЯЦИИ
Устойчивость лиофобных коллоидных систем имеет в
значительной мере кинетический характер. Система тем
более устойчива, чем медленнее она коагулирует. Скорость
коагуляции определяется бро-
броуновским движением частиц
и энергией их взаимодействия
при сближении; другими сло-
словами, она зависит от величины
радиуса сферы взаимного при-
притяжения частиц R и от ко-
коэффициента их диффузии D.
Теоретически наиболее про-
простым является вариант, когда
каждое столкновение частиц
приводит к их соединению,
т. е. когда частицы полностью лишены стабилизирующих
факторов. В этом случае происходит быстрая коагуляция,
скорость которой целиком определяется броуновским дви:
жением. Теория кинетики быстрой коагуляции однородных
сферических частиц была разработана Смолуховским A916).
По Смолуховскому, условием соединения двух частиц
является равенство расстояния между центрами частиц и
радиуса сферы их взаимного притяжения R. Если R = 2г,
где г — радиус одной частицы (рис. 61), то слипание насту-
наступает при прямом контакте двух сферических частиц; фак-
фактически, по Зигмонди, значение R лежит между 2 г и Зг.
В начальной стадии коагуляции, когда преобладают соу-
соударения индивидуальных частиц, процесс коагуляции
протекает как бимолекулярная реакция, т. е. общее число
частиц T.-J уменьшается по уравнению реакции второго
порядка
Рис. 61. Взаимное притяжение
частиц при R=2r
149
где k, по теории броуновского движения, связано с коэф-
коэффициентом диффузии частиц D и радиусом сферы притяже-
притяжения R уравнением
k =4nD/?.
Интегрирование (VI. 2) дает
1 -t
(VI. 3)
(VI. 4)
где v0 — общее исходное число частиц, at — время на-
наблюдения после начала коагуляции. Продолжительность
времени Т, в течение которого общее число частиц умень-
уменьшается вдвое, называют временем коагуляции. Из (VI. 4) ви-
0T — 1,
Дно, что при Ev = ^-должно быть
или
Т = — —
(VI. 5)
Через некоторое время после начала коагуляции образуют-
образуются двойные частицы и необходимо учитывать влияние столк-
столкновений их с индивидуальными частицами (с образованием
тройных частиц), а также между собой ( с образованием
учетверенных частиц) и т. д. Смолуховский показал, что
число частиц vn, содержащих п первичных частиц, изме-
изменяется во времени (в виде зависимости от отношения ЦТ)
по уравнению
'о[т) (VI. 6)
\«+1
т)
Таким образом, динамика изменения числа индивидуаль-
индивидуальных, двойных, тройных и т. д. частиц в процессе коагуляции
определяется уравнениями:
VI =
V+T)
150
ty
и т. д.
(VI. ба)
Графически ход изменения частиц vi, V2, V3... и общего
числа частиц Sv0 в зависимости от -_ показан на рис. 62.
В концентрированном золе AgJ, содержащем v0 = 4-1014
частиц в 1 мл при радиусе частиц г = 30 мр, время коагу-
коагуляции 7=^1/2000 сек.;
в золе AgJ нормальной
концентрации, обычно -^ 1 г
применяемом для оп-
определения коагуля-
коагуляции, значение Т изме-
изменяется от 1 сек. до 1
мин. Прейс использо-
использовала измерения Т при
коагуляции латекса
полистирола электро-
электролитами для количест-
количественной характеристи-
характеристики агрегативной ус-
устойчивости золя.
Мюллером было
показано, что в поли-
полидисперсном золе (при
неодинаковом размере
исходных частиц) ма-
маленькие частицы в
присутствии больших
исчезают относитель-
относительно быстрее; кроме того, частицы палочкообразной формы
коагулируют быстрее сферических или пластинчатых
частиц.
Теорию медленной коагуляции частиц, сохраняющих
некоторую стабильность, Смолуховский развил на осно-
основании аналогичных соображений, предполагая, что в этом
случае не каждое столкновение частиц приводит к их сли-
слипанию, и что эффективной является лишь часть столкно-
столкновений а; таким образом, предполагалось, что константа
скорости реакции уравнения VI. 3 равна kl = 4~DRa(Vl. 7)
Рис. 62. Зависимость числа частиц от
времени при быстрой коагуляции
151
с соответствующим изменением последующих уравне-
уравнений. По Фуксу, медленная коагуляция отличается от быст-
быстрой уменьшением числа столкновений вследствие наличия
остаточных энергетических барьеров между частицами
(по данным Харина, для золей серы при медленной коагу-
коагуляции величина этих барьеров составляет 2—3 ккал/моль).
Если обозначить через W отношение скорости быстрой коа-
коагуляции и скорости медленной коагуляции при наличии
потенциального барьера S, то для значительной области
медленной коагуляции золей с сферическими частицами, как
показал Рееринк, lg IF линейно зависит от логарифма кон-
концентрации коагулирующих электролитов, однако, деталь-
детальная теория кинетики медленной коагуляции еще не разра-
разработана.
Выводы
Устойчивость лиофобных коллоидов определяется ион-
ионными и молекулярными адсорбционными слоями на поверх-
поверхности частиц. Лиофобные золи, стабилизованные ионными
слоями, коагулируют при сравнительно низких концентра-
концентрациях электролитов, причем коагулирующее действие
преимущественно оказывают противоположно заряженные
ионы, особенно многовалентные ионы (правило Шульце—
Гарди). В результате обменной адсорбции и повышения
ионной силы раствора, при коагуляции коллоидов элект-
электролитами происходит сокращение толщины двойного
слоя, которое внешне обычно проявляется в снижении С-
потенциала. В теории Дерягина показано значение сжатия
двойного слоя для установления соотношения сил межчас-
межчастичного притяжения и электростатического отталкивания
частиц, приводящего к коагуляции, и дано теоретическое
обоснование правила Шульце—Гарди (VI. 1). Кривые
энергии взаимодействия имеют минимум на больших рас-
расстояниях, что создает для асимметрических частиц воз-
возможность возникновения тиксотропии и образования так-
тоидов. Коагуляция в ряде случаев может иметь обратимый
характер; образование золя из коагулированного осадка
путем повышения диффузности двойного слоя частиц
или сообщения заряда частицам называется пептнзацией.
При наличии молекулярных адсорбционных слоев
устойчивость коллоидов слабее зависит от концентрации
электролитов и не подчиняется правилу Шульце—Гарди;
основными стабилизирующими факторами являются ад-
сорбционно-сольватные слои и ориентированная адсорбция
молекул поверхностно-активных веществ, образующих
двухмерные структурированные слои (Ребиндер). Об-
Образование последних лежит в основе явлений защиты
лиофобных золей от коагуляции электролитами. Устой-
Устойчивость" коллоидных систем имеет кинетический характер,
т. е. является обратным выражением скорости коагуля-
коагуляции; кинетика процессов быстрой коагуляции количест-
количественно выражена в теории Смолу»ховского (VI. 6).
Вопросы
1. Чем определяется агрегативная устойчивость лиофобных
коллоидов, чем она отличается от кинетической устойчивости?
2. В чем заключается правило Шульце—Гарди? В каком отно-
отношении находятся пороги коагуляции для различных электролитов?
3. Каким образом происходит сокращение толщины двойного
слоя при коагуляции коллоидов электролитами и почему оно при-
приводит к коагуляции?
4. Что такое потенциальные кривые взаимодействия коллоид-
коллоидных частиц и какой ход они имеют? Каким образом на их основе
объясняются явления коагуляции и тиксотропии?
5. В чем заключается пептизация и какими методами она
осуществляется?
6. Почему при длительном отмывании многих осадков (суль-
(сульфидов тяжелых металлов, гидратов окислов) они начинают про-
проходить через фильтр?
7. Чем определяется значение сольватных слоев для устойчи-
устойчивости коллоидов?
8. Какое значение для устойчивости коллоидов имеют адсорб-
адсорбционные слои поверхностно-активных веществ и чем оно опреде-
определяется?
9. В чем заключаются защитное действие и сенсибилизация?
10. Каковы основные положения теории кинетики быстрой
и медленной коагуляции коллоидов?
Глава седьмая
ЭМУЛЬСИИ, ПЕНЫ, АЭРОЗОЛИ
Помимо коллоидных систем, рассмотренных в главах
четвертой-шестой, существуют другие классы дисперс-
дисперсных систем, свойства которых также определяются поверх-
поверхностными явлениями, и которые имеют важное практи-
практическое значение. К числу этих систем относятся, прежде
всего, эмульсии, пены и аэрозоли. Они обладают рядом
характерных особенностей; изучение этих особенностей
представляет большой практический и теоретический ин-
интерес и расширяет наши представления о коллоидных
системах.
ЭМУЛЬСИИ
Эмульсиями называют коллоидные системы, в которых
одна жидкость диспергирована в другой жидкости, т. е.
обе фазы являются жидкими. Разумеется, жидкости должны
быть нерастворимыми или, по крайней мере, слабо раство-
растворимыми друг в друге. Если одна из этих жидкостей вода,
то другая обычно обозначается «масло», хотя она мо-
может вовсе не быть маслом, а любой неполярной жид-
жидкостью. Эмульсии масла в воде называют прямыми [их
сокращенно обозначают (м/в)] а эмульсии воды в масле
(в/м) — обратными. Отличить оба типа эмульсий довольно
легко, так как в первом случае непрерывной (дисперсион-
(дисперсионной) средой является вода, а во втором случае — масло,
что резко сказывается на электропроводности, природе
растворяемых красителей и других свойствах системы.
Простейший пример эмульсий — критические смеси двух
ограниченно-растворимых жидкостей, например, воды и
фенола. При комнатной температуре они образуют два
154
расслаивающихся раствора — фенола в воде и воды в фе-
феноле, которые при взбалтывании дают очень нестойкую,
быстро разделяющуюся эмульсию. По мере повышения тем-
температуры составы обоих растворов сближаются, а межфазное
поверхностное натяжение на границе раздела уменьшает-
уменьшается, пока, наконец, при критической температуре смещения
F5,9°) оно не обращается в нуль, и обе жидкости смешивают-
смешиваются во всех отношениях. Если понизить температуру чуть
ниже критической, то самопроизвольно образуется эмуль-
эмульсия при крайне низком поверхностном натяжении, сос-
составляющем всего сотые доли дин/см. В этом состоянии
слабое стремление капель эмульсии к слиянию (коалес-
ценции) уравновешивается энтропийным фактором — стрем-
стремлением веществ к равномерному распределению в объеме.
Такая ^критическая эмульсия является термодинамически
устойчивой и строго равновесной системой. По мере охла-
охлаждения системы и возрастания различия состава капель
и среды поверхностное натяжение на границе их раздела
увеличивается и все более преобладает стремление к по-
понижению поверхностной энергии путем уменьшения по-
поверхности раздела, т. е. путем коалесценции капель и рас-
расслоения жидкостей. В этой возможности самопроизвольного
образования термодинамически устойчивых равновесных
систем, при условии очень низких значений поверхностно-
поверхностного натяжения, заключается одна из характерных особен-
особенностей эмульсий, отсутствующая у всех других коллоид-
коллоидных систем, не относящихся к растворам высокомолеку-
высокомолекулярных соединений.
Однако такие термодинамически устойчивые эмульсии
можно получить не только на основе критических смесей.
Можно осуществить сильное понижение поверхностного
натяжения з на границе раздела капель и среды добавле-
добавлением к системе поверхностно-активного вещества (стр.
81), если по условиям растворимости его можно
добавить в достаточно большом количестве. При понижении
а до десятых или сотых долей дин/см такая эмульсия —
на. этот раз с добавлением третьего вещества, стабилиза-
стабилизатора ¦— может стать самопроизвольно образующейся, тер-
термодинамически устойчивой эмульсией. Так, например,
получают самопроизвольное эмульгирование бензола в
воде при добавлении 0,1 н. NaCl и олеиновой кислоты до
а = 0,04 эрг/см2, или некоторых масел в воде при добавле-
добавлении в них до 10—40% натриевых мыл (эмульсолы). При
155
Поглощении жидкого жира организмом, он эмульгируется
в кишечнике и затем всасывается через стенку кишечника;
интересно, что система таурохолат (желчная соль)—моно-
глицеридолеиновая кислота при рН 6,0—8,5 действительно
обладает очень низким поверхностным натяжением (ниже
1 эрг/см2), при котором может происходить самопроизволь-
самопроизвольное эмульгирование.
Чаще при добавлении поверхностно-активного вещества
понижение а все же недостаточно для самопроизвольного
эмульгирования, и поэтому в дальнейшем мы вновь будем
рассматривать лишь системы с коллоидным механизмом
устойчивости. Однако и в этом случае устойчивость эмуль-
эмульсии выше при наличии стабилизаторов (называемых иначе
эмульгаторами), способных вызвать значительное пониже-
понижение поверхностного натяжения; например, в ряду жир-
жирных кислот лучшими эмульгаторами являются лауриновая
и миристиновая кислота, дающие, согласно правилу Траубе
(стр. 84), наибольшее понижение поверхностного натя-
натяжения по сравнению с предшествующими членами
ряда.
Другой основной путь получения устойчивых эмульсий
с применением эмульгаторов обусловлен способностью
эмульгаторов к образованию в адсорбционном слое двух-
двухмерных структур с повышенными механическими свойства-
свойствами (Ребиндер). Этот механизм устойчивости эмульсий анало-
аналогичен устойчивости других лиофобных коллоидов с моле-
молекулярными адсорбционными слоями (стр. 144); хорошими
эмульгаторами этой группы являются белки (включая
желатину), сапонин, камедь, агар, мыла и др.
Наконец, своеобразным и характерным лишь для
эмульсий типом стабилизаторов являются твердые порош-
порошкообразные вещества (гипс, графит и др.), способные скап-
скапливаться на границе раздела капель и среды благодаря
избирательной смачиваемости твердых тел (стр. 78).
Например, частицы гипса в эмульсии м/в (рис. 63),
благодаря своей гидрофильное™, почти полностью входят
в воду и лишь частично — в каплю масла, вследствие чего
они окружают каплю М сплошным слоем и препятствуют
ее слиянию с другой каплей масла. Легко видеть, однако,
что избирательное смачивание не должно быть полным,
так как в этом случае частицы стабилизатора сказались бы
целиком в водной фазе, и капли М остались бы незащищен-
незащищенными. Аналогично, при неполном избирательном смачива-
156
Рис. 63. Капля эмуль-
эмульсии м/в, стабилизо-
стабилизованная твердыми час-
частицами
нии гидрофобных частиц (графит, ZnS, CuS и др.) они могут
быть стабилизаторами эмульсии в/м. Таубман и Корецкий по-
показали, что большое значение при стабилизации эмуль-
эмульсий м/в твердыми частицами имеет образование на границе
раздела частица — масло адсорбционных слоев солей жир-
жирных кислот. При тщательной очистке масла стабилизация
достигается лишь при высокой концентрации твердых
частиц (до 4—5%). Стабилизация
твердыми частицами дает весьма устой-
устойчивые эмульсии.
Свойства эмульсий в высокой мере
зависят от применяемых эмульгато-
эмульгаторов. Во многих случаях не только
устойчивость, но и тип эмульсий
(м/в или в/м) обусловливается приро-
природой и концентрацией эмульгаторов.
Так, например, белки, камеди, дек-
декстрины, мыла щелочных металлов
дают эмульсии м/в, а стерины,
мыла многовалентных металлов —
эмульсии в/м. Для эмульсионных мазей типа в/м,применяе-
в/м,применяемых в фармацевтической промышленности, обычно в качест-
качестве эмульгаторов используют мыла Са, Mg, Zn, Al и соответ-
соответствующие соли смоляных кислот. При известных условиях
можно приготовить высококонцентрированные эмульсии,
содержащие свыше 90% в диспергированном состоянии.
Так, например, можно получить эмульсию м/в из 99%
бензола и 1% водного раствора олеата натрия, в ко-'
торой толщина промежуточных пленок воды (дисперсион-
(дисперсионной среды) составляет лишь 100а (Кремнев), или стойкую
эмульсию 95% бензола в 1%-ном растворе желатины —
ее можно резать ножом (Жуков и Бушмакин) и др. Устой-
Устойчивые концентрированные эмульсии, в которых раствор
эмульгатора растягивается в очень тонкие пленки, могут
быть получены лишь при условии достаточной механической
прочности и гелеобразной структуры этих пленок.
При добавлении поверхностно-активного вещества к
готовой эмульсии возможно разрушение эмульсии или
изменение типа эмульсии (обращение фаз).
Разрушение эмульсий является часто важной техничес-
технической задачей. При добавлении поверхностно-активного
вещества к эмульсии оно происходит в тех случаях, когда
это вещество обладает более высокой поверхностной актив-
157
ностью, чем прежний эмульгатор и может вытеснить его
с поверхности, но в то же время само не образует'прочных
защитных пленок, например, разрушение некоторых эмуль-
эмульсий при помощи амилового спирта. Для разрушения эмуль-
эмульсий применяют также различные методы электрического
переноса, суперцентрифугирования, фильтрования через
пористые, избирательно
смачиваемые материалы,
коагуляция электролита-
электролитами и др.
Изменение типа эмуль-
эмульсии (переход м/в в в/м,
или обратно) происходит в
том случае, если добавлен-
добавленное поверхностно-активное
вещество может быть ста-
стабилизатором обратного
типа эмульсии (обращение
фаз), например, при заме-
замещении натриевого мыла в
поверхностном слое мыла-
мылами двувалентных металлов.
Рис. 64.
кратной множественной эмульсии (по
Зейфрицу)
Микрофотография пяти- При обращении фаз внача-
ле образуются оба типа
эмульсин, но затем ос-
остается или преобладает
одна более устойчивая система; часто возникают слож-
сложные типы множественных эмульсий; капелька масла, вхо-
входящая в эмульсию м/в, может содержать в себе эмульсию
в/м и т. д. (рис. 64). Для обращения фаз известное значение
имеет также соотношение объемов фаз.
За исключением самопроизвольно образующихся эмуль-
эмульсий, в остальных случаях для получения эмульсий
необходимо применение сильного перемешивания, встря-
встряхивания, вибрации, действия ультразвука или специаль-
специальных гомогенизаторов. Например, в одном из типов гомоге-
гомогенизаторов (рис. 65) грубая эмульсия продавливается через
клапан Л с хорошо пригнанными трущимися поверхностями;
пружина В позволяет отжимать клапан А всего на сотые
доли миллиметра, причем клапан Л вибрирует во время
работы. При гомогенизации молока средний диаметр ка-
капель масла понижается с 3 до 0,2 а, и такое молоко уже не
отстаивается. При помощи ультразвука получают, напри-
158
мер, фармацевтические эмульсии рыбьего жира и его
смесей с сульфидином.
Устойчивость эмульсий измеряют по продолжитель-
ности ее существования - сек. в столбе высотой Н см: ~= —'
где v — скорость расслоения эмульсии в см/сек, либо по
продолжительности существования отдельных капель.
Таким образом, устойчивость эмульсий имеет кинетический
характер (Ребиндер). Устойчивость т возрастает с повыше-
повышением концентрации эмульгаторов до образования насы-
насыщенного адсорбционного слоя или поверхностной пленки
с оптимальными структурно-механическими свойствами
Рис. 65. Схема гомогенизатора (по Льюису)
В отличие от описанных концентрированных эмульсий,
высокоразбавленные эмульсии, содержащие всего 0,01—
0,1 % дисперсной фазы, могут быть стабилизованы только
ионными двойными слоями, подобно лиофобным золям
(главы пятая и шестая). Разбавленные эмульсии м/в не от-
отличаются от других лиофобных золей по природе своей
устойчивости, электрофоретической подвижности, условиям
коагуляции электролитами и др., за исключением способ-
способности к коалесценции капель после коагуляции. Именно
на разбавленных эмульсиях м/в была установлена связь
коагуляции с понижением С-потенциала (приблизительно
до 30 мв, по Повису), или с критическим отношением С2/7-»
где 1/-/. — толщина дебаевской ионной атмосферы (критерий
коагуляции Эйлерса и Корфа, теоретически обоснованный
Дерягиным) и др. Примерами ионностабилизованных эмуль-
эмульсий являются также разбавленные эмульсии ртути в воде.
Частицы эмульсий обычно имеют довольно большие раз-
размеры @,1—1,0а и выше) и поэтому заметно испытывают дей-
действие силы тяжести. Оседание или всплывание частиц
может быть вследствие этого исследовано методами седимен-
тационного анализа для определения размеров и степени
полидисперсности (кривых распределения) частиц (стр.
159
45—46). Кривые распределения размеров частиц могут
быть также определены по подсчетам на микрофотографиях
эмульсий; этим методом исследовалась кинетика медленной
коагуляции разбавленной эмульсии м/в (Лоуренс и Миллс)
и были определены энергетические барьеры v — коагуля-
коагуляции (стр. 152): v — 3,9 ккал/мол для чистой эмульсии
и v = 6,5 ккал/мол для эмульсии, стабилизованной олеа-
том натрия.
Эмульсии имеют большое практическое значение.
К эмульсиям относятся молоко, сливки, майонезы, марга-
маргарин, яичный желток, млечный сок каучуконосов, латексы,
битумные эмульсии в дорожном строительстве, препараты
для жирования кож, средства для опрыскивания растений,
эмульсии воды в нефти и мн. др. Эмульсионная полимери-
полимеризация применяется для получения синтетических латексов
(Догадкин). Водные дисперсии высокополимеров широко
применяются для изготовления пленок и различных покры-
покрытий (Воюцкий). В организме жиры и липоиды переносят-
переносятся кровью в виде эмульсий и комплексов с [3 -глобулином
(хиломикронные эмульсии), обеспечивая жировое питание.
В фармацевтической промышленности многие лекарствен-
лекарственные вещества применяются в виде эмульсий, причем обыч-
обычно эмульсии м/в используются в составе внутренних ле-
лекарств, а эмульсии в/м — наружных средств. В ряде слу-
случаев эмульгированием удается замаскировать или ослабить
неприятный вкус масел и смол, например, в эмульсиях
рыбьего жира, касторового масла и др. В качестве эмуль-
эмульгаторов жирных масел применяют крахмальный клейстер,
яичный желток, камедь, декстрин, желатину, казеинат
натрия и др. Можно указать также на эмульсии акрифла-
вина, этиламинобензоата (для местного анестезирования),
медицинского минерального масла, бактерицидные эмуль-
эмульсии в/м с 97% растительного масла (для лечения тепло-
тепловых ожогов), разнообразные эмульсионные мази, пасты
и др.
ПЕНЫ
Пены по своей природе близки к концентрированным
эмульсиям, но дисперсной фазой в них является газ, а
не жидкость. Как уже указывалось, в высококонцентриро-
высококонцентрированных эмульсиях пленки эмульгатора растягиваются до
очень тонких слоев, устойчивость которых определяется
160
их структурно-механическими свойствами; точно так же и в
пенах газовые пузырьки разделены тончайшими пленками
жидкости, образующими в своей совокупности пленочный
каркас, который и служит основой пен. В пенах нет ана-
аналогов разбавленным эмульсиям и в устойчивости пен
двойные ионные слои не имеют значения. Поскольку пены
являются концентрированными эмульсиями газа в жид-
жидкости (Ребиндер), устойчивость пен зависит от устойчивости
их пленочного каркаса. Если пленки образованы чистыми
низкомолекулярными жидкостями, то их прочность очень
невелика и, как известно, дисперсии газа в чистой воде или
в чистом спирте очень быстро разрушаются. Возможность
получения устойчивых пен полностью определяется свой-
свойствами молекулярных адсорбционных слоев, образующих
стенки газовых пузырьков. Хорошими стабилизаторами пен,
или пенообразователями, естественно, являются вещест-
вещества, которые служат эмульгаторами для эмульсий м/в (пос-
(поскольку в пенах дисперсной фазой является неполярная сре-
среда— газ): белки, мыла, сапонин, твердые частицы и др.;
напомним, что все эти вещества образуют прочные поверх-
поверхностные слои. В гомологическом ряду жирных кислот
устойчивость пен возрастает в соответствии с ростом поверх-
поверхностной активности этих веществ (Ребиндер, Талмуд).
На поверхности раствора существует разность между
капиллярным давлением р — 4- (см. стр. 79) в газовом
пузырьке и внешним атмосферным давлением /?0. Для
того чтобы пузырек оставался устойчивым, нужно, чтобы
прочность стенок газовых пузырьков была больше (р — р0),
в противном случае пузырек лопается.
Устойчивость пены, как и эмульсий, количественно
характеризуется продолжительностью существования от-
отдельного пузырька на границе раздела, или продолжитель-
продолжительностью разрушения столба пены, высотою Н см.(си. стр. 159).
Таким образом, устойчивость пен, подобно устойчивости
эмульсий и суспензий, определяется из кинетики их раз-
разрушения или коагуляции; пены также относятся к агрега-
тивно неустойчивым системам.
Одним из важных применений пен является их исполь-
использование для обогащения ценных руд. В измельченной руде
(до частиц 0,01—0,1 мм) частицы пустой породы имеют
гидрофильный характер и избирательно смачиваются во-
Дой, а гидрофобные частицы ценных минералов (сернистые
<Кол л о и л на я хи ми я >:
161
— _
Пустая
f
-4
-Jf Воздух
порода^
Ценная порода
%{ .Масло
W~~Boda ~~
серебро, свинец, медь и др.) преимущественно втягиваются
в масло (этот процесс называется флотацией). При вспени-
вспенивании суспензии измельченной руды и флотореагентов
масло окружает пузырек воздуха (рис. 66) и вместе с части-
частицами минералов уносится в верхний слой, который отде-
отделяется и после разрушения пены образует концентрат. При
помощи специальных добавок — коллекторов регулируют
избирательное смачивание частиц минералов, а при помощи
пенообразователей—ус-
пенообразователей—устойчивость пузырьков
пены.
Моющее действие
мыл и детергентов было
бы невозможно без об-
образования пен (см.
стр. 121).
Пены с твердыми
тонкими стенками (аэро-
гели) широко использу-
используются для изготовления
теплоизоляционных и
звукоизоляционных ма-
материалов, пенопластов,
спасательных средств и
др.; к твердым пенам
относятся также конди-
кондитерские пены, торты и др. Чрезвычайно велико значение
стойких пен в пожаротушении (Л- Розенфельд).
Однако может возникать необходимость и в разрушении
пен, образование которых часто нежелательно. Способы
пеногашения, естественно, основаны на замещении или
разрушении структурных адсорбционных слоев, стабили-
стабилизирующих пену. Некоторые пены разрушаются амиловым
спиртом, который, благодаря своей высокой поверхност-
поверхностной активности, как и при разрушении эмульсий, способен
вытеснять стабилизатор из поверхностного слоя, носам не
дает механически устойчивых слоев. В производстве анти-
антибиотиков применяют смесь лярда с 3% октадеканола и си-
силиконовые масла, образующие с двухмерными белковыми
пленками, стабилизирующими пены, более текучие и лег-
легче разрушаемые комплексы. Для разрушения стойких пен
используют также различные механические средства, теп-
тепловое «пережигание» пленок и др.
162
Рис. 66. Схема пенной флотации (по
Пескову и Прейс)
АЭРОЗОЛИ
Аэрозолями называют коллоидные системы, образован-
образованные жидкими или твердыми частицами в газах (обычно в
воздухе). Аэрозоли получают путем диспергирования
при различных взрывах, при истирании, измельчении и
др., и путем конденсации— из паров воды и углеводоро-
углеводородов, при испарении из распыленных растворов, при хими-
• ческих реакциях некоторых газов (реакции NHs и НС1
с выделением дыма NFhCl) и др. В природе аэрозоли обра-
• зуются путем диспергирования при обвалах, в водопадах,
при выветривании и эрозии почв, а путем конденсации —
при появлении облаков и туманов, при вулканических извер-
извержениях и др. Обычно методами диспергирования образуют-
образуются более грубодисперсные и неоднородные аэрозоли, чем
методами конденсации. Аэрозоли с жидкими частицами на-
называют туманами, аэрозоли с твердыми частицами, полу-
полученные путем диспергирования, — пылью, а конденсацион-
конденсационные аэрозоли с твердыми частицами — дымами.
Размеры частиц в аэрозолях весьма различны, от 1 м\у.
до 100 [х; в табачном дыме содержатся частицы от 0,2 \i до
1;а, в тумане H2SO4 — от 0,5 \i до 5 [х; в слоистых облаках
капли обычно от 4 до 10 [х и выше. Размеры частиц в аэро-
аэрозолях измеряют при помощи оптического микроскопа,
ультрамикроскопа и методами электронной микроскопии.'
Аэрозоли с размерами частиц ниже 0,1 ^ называют высоко-
высокодисперсными, а выше 1 ;х — грубодисперсными; изменения
свойств аэрозолей в зависимости от размеров частиц по-
показаны на рис. 67.
Частицы размером болег 1 и заметно оседают под дейст-
> вием силы тяжести, тогда как для частиц менее 1 \ь харак-
характерно преобладание броуновского движения (диффузия).
Благодаря низкой вязкости воздуха диффузия частиц
аэрозоля происходит с большой быстротой. Кроме того,
аэрозоли легко переносятся ветром и конвекционными
потоками в воздухе, что имеет большое значение для
распространения дымов и туманов.
Особенностью аэрозолей, отличающей их от всех осталь-
остальных коллоидных систем (помимо природы образующих фаз),
является наличие у них лишь кинетической устойчивости!
Аэрозоли полностью лишены агрегативной устойчивости
и каждое соприкосновение их частиц или частицы и стенки
приводит к слипанию (коагуляции). Так как число столкно-
12*
163
вений частиц быстро растет с увеличением объемной кон-
концентрации аэрозоля, то аэрозоли нельзя получить при
очень высокой концентрации. В результате броуновского
движения и быстрого слипания частиц с последующим tfx
оседанием, аэрозоль, независимо от начальной концентра-
концентрации частиц, уже через несколько минут содержит не более
Ю7 частиц в 1 см3 (напомним, что обычный золь золота со-
содержит около 1015 частиц в 1 мл). В полидисперсных аэро-
w-''
/
10'6
—
Диффузия
ч
*~~ III
10~5
.
^^
^^
ю-"
"Zr2
- 5
ш
'Оседание
10
-2
Радиус частиц t см
Сопротивление среды |
Скорость испарения v
Скорость охлаждения
Константа коагуляции
Рассеянии света
Область микроскопической
видимости
06пасть ультрамикроско-
пической Видимости
Преобладание диффузии
ипи оседания
Давление пара
{'водяньп капелек)
Рис. 67. За.шсимэст:> сгиГтстл аэрозолей от размеров
частиц (по Фуксу).
золях мелкие частицы сравнительно быстро перехватывают-
перехватываются более крупными частицами, поэтому монодисперсные ды-
дымы коагулируют сравнительно медленнее.
В атмосферных аэрозолях частицы либо совсем лишены
зарядов, либо несут всего 1—2 заряда при радиусе частиц
10—5СЫ;х (что несравненно меньше, чем на частицах лио-
фоэных золей). При увеличении числа зарядов на частицах
аэрозоля в результате ультрафиолетового облучения, элект-
электрических разрядов, поглощения газовых ионов и т. п.,
164
одноименно заряженные (униполярные) частицы разле-
разлетаются, но быстрее наталкиваются на стенки сосуда, а раз-
разноименно заряженные (биполярные) частицы быстрее сли-
слипаются между собой; вследствие этого увеличением числа
зарядов на частице при электрическом разряде пользуют-
пользуются для ускоренной коагуляции аэрозолей (см. стр. 166).
В парах поверхностно-активных веществ скорость коагу-
коагуляции'аэрозолей изменяется лишь незначительно.
Большое значение имеют оптические свойства аэрозолей,
ввиду широкого использования маскирующих свойств
дымов и туманов. В высокодисперсных аэрозолях рассея-
рассеяние света подчиняется уравнению Рэлея (стр. 54),
но при размере частиц 0,1 — 1,0;х, довольно обычном для
аэрозолей, они соизмеримы с длиной волны света, что при-
приводит к наложению явлений отражения и рассеяния света
и отклонениям от уравнения Рэлея. Поэтому нарушается
зависимость светорассеяния от 1/Х4 (показатель степени
приобретает более низкие значения). Аналогично, зави-
зависимость от радиуса частиц изменяется от пропорциональнос-
пропорциональности г6 (или v2) в уравнении Рэлея до г3 или г2 (см. рис. 67).
Вследствие сравнительно близких величин отражения и
рассеяния света различной длины волны, многие туманы
и дымы кажутся белыми. Наибольшей интенсивностью сум-
суммарного рассеяния света, или наибольшей маскирующей
способностью (принимаемой за единицу) обладает дым бе-
белого фосфора; широко используются также дымы, получае-
получаемые возгонкой антрацена и нашатыря, туманы H2SO4 и др.
Практическое значение аэрозолей чрезвычайно велико.
Общеизвестно значение облаков для формирования клима-
климата в метеорологии, биологическая роль переноса ветром
семян и пыльцы растений, спор бактерий и плесеней;
широко применяются в технике сжигание распыленного
топлива и распылительная сушка, в сельском хозяйстве—¦
распыление удобрений, средств борьбы с вредителями
растений, тепловая защита садов дымами и др. С другой
стороны, образование туманов является значительной поме-
помехой для авиации и других видов транспорта; большую опас-
опасность представляют радиоактивные аэрозоли, возникаю-
возникающие при взрыве атомных бомб; в промышленности тысячи
тонн ценных руд и различных химических веществ выносят-
выносятся дымами в атмосферу: борьба с этим явлением имеет боль-
большое санитарно-гигиеническое значение. Для народного
здравоохранения актуальное значение имеют различные
165
-ге-
-гепатогенные аэрозоли, так как этим путем передаются мно-
многие инфекции (при одном чихании в воздух вводится до
100 тыс. бактерий, частиц вируса гриппа и др.), или вызы-
вызываются профессиональные заболевания (силикоз и др.).
Таким образом, задачи борьбы с аэрозолями или защи-
защиты от них часто представляют собой не менее актуальные
проблемы, чем получение и использование аэрозолей.
Для осаждения частиц грубых аэрозолей с г = За и
ваше применяют центробежные пылеотделители — цикло-
циклоны, в которых газ движется по спирали внутри узких не-
неподвижных цилиндров (диаметром 5—15 см) и частицы
осаждаются на стенках приборов. Широко используются
тканевые и волокнистые фильтры, основанные на принципе
прилипания в процессе броуновского движения, инерцион-
инерционного осаждения, соответственно на нитях или волокнах
фильтровальной бумаги или фильтровального картона (ас-
бестоцеллюлозных фильтров) и др. Фильтры являются необ-
необходимой составной частью противогазов; различного рода
фильтры применяют также в промышленности для получе-
получения стерильного воздуха. Наконец, большое значение име-
имеют различные электрофильтры (аппарат Котреля и др.).
В этих приборах аэрозоль пропускают через коронный
электрический разряд, вызывающий усиленную отрицатель-
отрицательную зарядку частиц, которые осаждаются на положитель-
положительном электроде в постоянном поле высокого напряжения
G0 — 100 тыс. в).
При вдыхании аэрозолей человеком особенно много
частиц осаждается у разветвлений и поворотов дыхатель-
дыхательных путей, что указывает на значительную роль инерцион-
инерционного осаждения; при этом заряженные частицы осаждаются
сильнее, чем незаряженные. Грубодисперсные частицы
(более 1 [х) задерживаются в носоглотке и в верхних ды-
дыхательных путях. В медицинской практике часто применяют
аэрозоли для введения в организм лекарственных и антибио-
антибиотических веществ (ингаляции). Эти лечебные аэрозоли
должны проникать в легкие, для чего им сообщается дос-
достаточно высокая степень дисперсности.
Бы воды
Эмульсии, пены и аэрозоли образуют .группу коллоид-
коллоидных систем большого практического значения. Природа
устойчивости этих коллоидных систем различна. Устойчи-
166
вость разбавленных эмульсий обусловлена двойными ион-
ионными слоями, устойчивость концентрированных эмульсий
и пен — молекулярными адсорбционными слоями, особен-
особенно при образовании в них прочных двухмерных структур,
устойчивость аэрозолей основана на их кинетической устой-
устойчивости. Устойчивость всех перечисленных систем имеет
кинетический характер и измеряется по скорости их рас-
расслоения или коагуляции.
Тип и устойчивость концентрированных эмульсий в
значительной мере зависят от природы эмульгаторов, обра-
образующих молекулярные адсорбционные слои. При замещении
одного эмульгатора другим возможно разрушение эмульсии
или изменение типа эмульсии (обращение фаз). Эмульсии
могут также стабилизироваться твердыми частицами при
условии их неполной избирательной смачиваемости.
Пены стабилизируются пенообразователями (белки,
мыла, сапонин и др.), по своей природе близкими к эмуль-
эмульгаторам концентрированных эмульсий м/в. Пеногашение
происходит при замещении пенообразователя поверхност-
поверхностно-активным веществом или путем придания слою пено-
пенообразователя вязкотекучих свойств. Пенообразование и
избирательная смачиваемость твердых частиц используются
в явлениях флотации.
Аэрозоли — различные дымы, туманы и пыли — лише-
лишены агрегативной устойчивости и каждое соприкосновение
их частиц приводит к слипанию; концентрация аэрозолей
обычно не превышает 107 частиц в 1 см3, а частицы несут не
более 1—2 зарядов. Повышение зарядки частиц использует-'
ся для их ускоренного осаждения в электрофильтрах; инер-
инерционное осаждение и прилипание при броуновском движе-
движении применяется при фильтрации аэрозолей. По оптиче-
оптическим свойствам аэрозоли охватывают область рэлеевского
рассеяния света и отклонений от него, обусловленных явле-
явлениями отражения света; аэрозоли обладают высокой маски-
маскирующей способностью.
Практическое использование условий образования и раз-
разрушения эмульсий, пен и аэрозолей крайне разнообразно,
в частности, в медицинской практике и фармацевтической
промышленности.
Вопросы
1. Перечислите основные факторы устойчивости разбавленных
эмульсий, концентрированных эмульсий, пен и аэрозолей.
2. Каким образом измеряется устойчивость эмульсий и пен?
167
3. Укажите стабилизаторы эмульсий в'м или эмульсий м/в
среди следующих веществ, мел, сажа, гипс, сернистый свинец,
желатина, тальк.
4. В чем заключается обращение фаз и каким образом оно
осуществляется?
5. Какие свойства эмульсин и пен связаны с явлениями изби-
избирательного смаиивания?
6. Как ориентированы молекулы жирных кислот п пленках
мыльной пены?
7. Укажите способы образования аэрозолей в технике и в
природе и способы быстрого осаждения аэрозолей.
8. Каковы оптические свойства аэрозолей? Что такое мас-
маскирующая способность аэрозолей?
9. Коковы характерные размеры части \ дисперсной фазы
в эмульсиях, пенах и аэрозолях?
10. Охарактеризуйте практическое значение эмульсий, пен
и аэрозолей, в частности, в фармацевтической промышленности и
меди шнекой практике.
Глава восьмая
РАСТВОРЫ ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
В предыдущих главах была рассмотрена большая группа
коллоидных систем, обладающих развитой физической
поверхностью раздела и значительным избытком свободной
поверхностной энергии, стремление которой к уменьшению
делает эти системы термодинамически неустойчивыми. Бла-
Благодаря избытку поверхностной энергии, в таких системах
образуются ионные и молекулярные адсорбционные слои,
которые и обобщают агрегативную устойчивость коллоид-
коллоидным частицам. Легко видеть, что природа устойчивости
этих систем резко отличается от устойчивости обычных
истинных растворов низкомолекулярных веществ, напри-
например, сахара. Хотя каждая молекула сахара в растворе проч-
прочно связана, примерно, с 12—15 молекулами воды, нельзя
говорить, что молекула сахара окружена адсорбционно-
сольватным слоем воды, так как она не имеет поверхности
раздела и не образует фазы; водный раствор сахара являет-
является однофазной системой. Устойчивость раствора сахара
определяется тем, что связь молекул сахара с водой сильнее
их взаимной связи в решетке сахара (энергетический
фактор) и что растворенные молекулы сахара равномерно
распределены во всем объеме раствора (энтропийный фак-
фактор). Термодинамически это означает, что состояние раст-
раствора сахара, при постоянном давлении и температуре, мо-
может быть полностью описано изменением двух функций —
теплосодержания или энтальпии ДЯ и энтропии AS:
М - ДЯ — ГД5,
(VIII. 1)
где AZ — изменение свободной энергии при постоянном
давлении или изобарного термодинамического потенциала.
' «Коллоидная химия»
1G9
При равновесии раствора AZ = 0 или АН — T&S, лричем
эти условия характеризуют состояние системы независи-
независимо от того, каким путем система пришла к данному
равновесию; 15%-ный раствор сахара при 20° обладает
определенными свойствами, независимо от того, получен
ли он разбавлением 20%-ного раствора или концентрирова-
концентрированием 10%-ного раствора сахара, нагреванием или охлаж-
охлаждением до данной температуры и т. п.
ТЕРМОДИНАМИЧЕСКАЯ УСТОЙЧИВОСТЬ РАСТВОРОВ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Подобно истинным растворам низкомолекулярных ве-
веществ, растворы высокомолекулярных веществ обладают
термодинамической устойчивостью; в этом заключается их
основное отличие от ранее рассмотренных лиофобных кол-
коллоидных систем. Как было показано, наличие термодинами-
термодинамической устойчивости вместе с тем означает обратимость
состояния растворов высокомолекулярных веществ и
возможность достижения ими истинного равновесия,
определяемого уравнением VIII. 1.
Экспериментальное обоснование этих положений для
растворов высокомолекулярных веществ заключается, пре-
прежде всего, в доказательстве применимости правила фаз, ко-
которое справедливо лишь для термодинамически равновес-
равновесных систем. По отношению к белкам приложимость правила
фаз была показана Галеотти A904), Зеренсеном, Мак-Бэном
и др. Для типичных цепных макромолекул применимость
правила фаз и обратимость растворов высокополимеров
были впервые доказаны работами Каргина и сотрудников
A937—1941). Так, например, Каргин, Роговин и Папков
показали, что диаграмма состояния растворов ацетил цел-
целлюлозы (в дихлорэтане, хлороформе) вполне аналогична
диаграмме системы фенол-вода (см. стр. 155). Любой
заданной температуре ниже 55° соответствуют строго опре-
определенные концентрации ацетилцеллюлозы в верхнем и
нижнем слоях раствора (рис. 68), причем эти концентрации
могут быть достигнуты как путем нагревания, так и путем
охлаждения системы; при температурах выше 60е наблю-
наблюдается неограниченное смешение компонентов. Аналогич-
Аналогичные результаты получены авторами для растворов ацетил-
целлюлозы в метилэтилкетоне, этилацетгте, толуоле;
Каргиным и Тагер — для нитроцеллюлозы в моногцетине
170
и бутилкапроновом эфире; Роговиным и Цаплиной — для
растворов поливинилхлоридов в ацетоне, нитробензоле,
дихлорэтане; Флори — для полистирола в циклогексане
и др. Все эти данные показывают, что различные высоко-
высокомолекулярные вещества образуют при определенных ус-
условиях термодинамически устойчивые истинные равновес-
равновесные растворы. Разумеется, такие растворы нельзя получить
для лиофобных золей, где некоторые случаи обратимости
(например, при тиксотро-
пии) относятся, как было
показано в шестой главе,
не к равновесным, а к мета-
стабильным состояниям.
Однако принципиальное
отнесение растворов высо-
высокомолекулярных веществ
к термодинамически устой-
устойчивым равновесным систе-
системам не означает, что всегда,
"когда мы имеем дело с
раствором высокополиме-
ра, мы располагаем равно-
равновесной системой. Практи-
Практи70
60
50
ЬО
30
го
ю
о
1
1
J
i
s
\
1
\
\
\
.... .
1.0 2.0 3,0 <*,0 5,0 6.0 70
% ацешрцептопозь!
Рис. 68. Диаграмма состояния п:-
стемы ацетил цел л юл оз а-хлоро-
а-хлороформ
чески это условие далеко не
всегда осуществляется, в
виду того, что в растворах
полимеров достижение рав-
равновесия по ряду причин мо-
может быть сильно замедленным (в приведенных выше опытах
равновесие достигалось в течение ряда недель или месяцев).
В этом отношении растворы высокополимеров существенно
отличаются от истинных растворов низко молекулярных
веществ, которые (за исключением пересыщенных раст-
растворов) действительно всегда находятся в равновесном сос-
состоянии. Напротив, в растворах полимеров изменение взаим-
взаимного расположения длинных цепных, иногда перепутанных,
макромолекул не может происходить быстро; кроме того,
взаимодействие длинных цепей может сильно измениться
уже от образования нескольких связей между ними, для
чего достаточно крайне небольшого по весу количества со-
солей или других примесей в растворе. Наличие в полимере
молекул различных размеров (полидисперсности), разли-
различающихся по своей растворимости, диффузии и пр.,затруд-
11* ,7|
няет достижение равновесия возможностью взаимодейст-
взаимодействия молекул и т. п. При работе с разбавленными раство-
растворами высокоочищенных и фракционированных (монодиспер-
(монодисперсных) полимеров действие перечисленных факторов
ослабляется и состояние термодинамического равно-
равновесия достигается легче; поэтому в научной работе обычно
специально обеспечивают эти условия. Однако при работе
с-концентрированными растворами, особенно на производ-
производстве (резиновые клеи, прядильные растворы целлюлозы и ее
эфиров, концентрированные растворы желатины и др.)
следует учитывать, что такие растворы часто не находятся
в состоянии тер?*годинамического равновесия и могут дости-
достигать его лишь спустя длительное время.
Тем не менее, указанные особенности кинетики процес-
процессов в растворах высоко полимеров, несмотря на их прак-
практическое значение, не изменяют принципиальной харак-
характеристики природы стабильности этих растворов, как термо-
термодинамически устойчивых обратимых истинных растворов.
Эта характеристика, как указывалось, резко отличаетраст-
воры высокомолекулярных веществ от л иофобных коллоид-
коллоидных систем; она означает также, что растворы высокомо-
высокомолекулярных веществ подчиняются основному уравнению
VIII. 1. Рассмотрим теперь более подробно роль энергети-
энергетического и энтропийного членов уравнения VIII. 1 в термо-
термодинамическом состоянии растворов полимеров.
ИЗМЕНЕНИЕ ТЕПЛОСОДЕРЖАНИЯ И СОЛЬВАТАЦИЯ
В РАСТВОРАХ ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Изменение теплосодержания системы Д# при растворе-
растворении некоторого количества полимера в данном объеме
растворителя равно тепловому эффекту растворения Q,
взятому с обратным знаком:
Д#=—Q. (VIII. 2)
По данным Каргина и Тагер, значения фдля нитроцел-
нитроцеллюлозы в ацетоне равны 18—19 кал (в пересчете на 1 г
полимера в достаточно большом объеме растворителя),
а для ацетилцеллюлозы в ацетоне Q — 10—11 кал/г; по
данным Липатова и Путиловой, для желатины в воде
Q^=30 кал/г; по данным Думанского и Некряча, для крах-
крахмала в воде Q = 28 кал/г и др. Таким образом, для растворов
полимеров с полярными группами в полярных раствори-
растворителях (ацетон, вода и др.) значения тепловых эффектов
\71
раствзрения являются положительными и, следовательно,
ДЖО. Изменения теплосодержания Д// в этих растворах
обусловлены, главным образом, взаимодействием молекул
полимера и растворителя, т. е. сольватацией полимера.
Сольватацией называется такое взаимодействие раство-
растворенного вещества с растворителем, которое приводит к бо-
более низкой активности растворителя вблизи частиц раст-
растворенного вещества по сравнению с чистым растворителем.
В случае водных растворов сольватация называется гид-
гидратацией. Гидратация ионов обусловлена ориентацией
дипольных молекул воды в электрическом поле иона, а
гидратация полярных групп — в молекулах неэлектро-
неэлектролитов и полимеров — ориентацией молекул воды в резуль-
результате взаимодействия диполей и образования водородных
связей. В гидратном слое молекулы воды располагаются
более упорядоченным образом, но остаются химически не-
неизмененными, чем гидратация отличается от химического
соединения с водой окислов металлов и ангидридов кислот.
Благодаря постепенному падению энергии связи раство-
растворенного вещества с растворителем (по мере удаления от
молекулы растворенного вещества), сольватный слой имеет
несколько диффузный характер, но в основном энергия
взаимодействия и наибольшее падение активности раство-
растворителя сосредоточены в первом молекулярном слое. Раство-
Растворитель в сольватной оболочке обладает меньшей упру-
упругостью пара, меньшей растворяющей способностью, мень-
меньшей диэлектрической постоянной, меньшей сжимаемостью,
он труднее вымораживается, обладает большей плотностью
и т. д.; изменение любого из этих свойств раствора может
быть использовано для определения величины сольвата-
сольватации. Наиболее прямой метод измерения сольватации со-
состоит в установлении теплового эффекта поглощения на-
навеской полимера определенного количества растворителя
из смеси последнего с инертной к полимеру жидкостью;
например, Каргин и Папков определили, что сольватация
нитроцеллюлозы в ацетоне и пиридине составляет около
1 молекулы растворителя на одну полярную группу —
ONO2 полимера (табл. 15). Думанский и Некряч опреде-
определили гидратацию ряда полимеров по теплоте смачивания
(см. стр. 78), в частности, для крахмала найдено, что на
глюкозный остаток приходится 3 молекулы связанной во-
воды. Думанский установил также, что связывание воды
самыми различными веществами происходит с тепловым
173
Таблица 15
Сольватация некоторых полярных групп
и звеньев полимеров
Полярная группа или звено
полимера
-ОН
— соон
> СО; > NH; СОН
— NHo
— CONH —(в белке)
— ONO2 (в нитроцеллюлозе)
Глюкозный остаток (в крахмале)
Изопрен (в каучуке)
— ОС2Н5 (в этилцеллюлозе)
Дивинил (в каучуке)
Растворитель
Вода
Вода
Вода
Вода
Вода
Ацетон,
пиридин
Вода
Бензол
Ацетон
СС14
Сольватация
в молекулах
на группу
3
4
2
2—3
1
1
3
0,5
0,5
со0,1
эффектом около 80 кал на 1 г связанной воды; близость
этой величины к теплоте замерзания воды (около 85 кал/г)
указывает на упорядоченное состояние воды в гидратной
оболочке. Булл исследовал гидратацию ряда белков, опре-
определяя 5-образные изотермы упругости пара (см. стр. 96).
Пасынский показал, что сольватацию можно определить по
величине несжимаемой части растворителя, так как в
сольватной оболочке растворитель находится под большим
внутренним давлением; величина сжимаемости опреде-
определялась по скорости распространения ультразвуковых
волн в растворах. Этим путем была измерена сольватация
ряда электролитов, неэлектролитов и полимеров и опре-
определено число молекул растворителя, связанных с различ-
различными химическими группами. Известно много других мето-
методов определения сольватации. Хотя физический смысл выра-
выражения сольватации числом прочно связанных (инактивиро-
ванных) молекул растворителя следует считать условным,
все же весьма различными методами для ряда систем
были получены довольно близкие данные, которые хорошо
соответствуют возможному числу водородных связей или
геометрическим условиям координации молекул раствори-
растворителя около полярной группы полимера. Некоторые сводные
данные по характеристике сольватации приведены в табл.15.
174
В пересчете на полярную группу или звено полимера
сольватация (или гидратация) не отличается существенно
от сольватации низкомолекулярных веществ и не превышает
одного слоя молекул. В некоторых случаях пространствен-
пространственные ограничения препятствуют полной гидратации (поэтому
пептидные группы — CONH — в молекуле белка связы-
связывают лишь по одной молекуле воды), но свободные гидро-
гидрофильные группы в боковых цепях (например, ¦— СООН, —
¦— ОН и др.) связывают такое же количество воды, как и
в молекулах жирной кислоты или спирта. Расчеты сольва-
сольватации полимеров по числу полярных групп в молекуле, с
учетом данных табл. 15, хорошо сходятся с прямыми весо-
весовыми определениями сольватации (в граммах растворителя
на 1 г полимера), которые для ряда белков в воде состав-
составляют 0,25—0,35 г/г, для нитроцеллюлозы в ацетоне 0,47 г/г,
для крахмала—0,35 г/г, и др. Этот результат показывает,
что при сольватации молекулы растворителя располагают-
располагаются в виде одного слоя лишь вокруг полярных групп поли-
полимера, приблизительно пропорционально их содержанию в
цепи. Неполярные участки, лежащие между полярными
группами в полимерах, и составляющие, например, в бел-
белках или нитроцеллюлозе около половины веса полимера,
остаются свободными от растворителя, т. е. топография
гидратного слоя выражается рядом островков на моле-
молекуле полимера. На каждом из этих центров сольватации
связанные молекулы растворителя обладают определенной
продолжительностью жизни и статистическое равновесие
связывания и освобождения молекул растворителя на сот-
сотнях центров, существующих в молекуле полимера, анало-
аналогично электрохимическому равновесию при ионизации по-
поливалентных электролитов (стр. 105) или динамическому
равновесию адсорбции-десорбции, выражаемому изотермой
Лангмюра (стр. 93), хотя лишь в последнем слу-
случае процесс происходит на физической поверхности разде-
раздела. Этим объясняется, почему сольватационное равновесие
или взаимодействие полимерных молекул в растворе иног-
иногда выражают уравнением адсорбционной изотермы или го-
говорят об адсорбции молекул растворителя молекулами по-
полимера. В наличии внутренней связи этих различных про-
процессов заключается одна из характерных особенностей,
коллоидных систем, которая отсутствует в растворах низко-
низкомолекулярных веществ (см. главу первую).
В растворах полярных полимеров, как указывалось,
175
сольватация является основным фактором, определяющим
величину ДЯ (VIII. 2). При измерении полного теплового
эффекта Q при растворении твердого полимера в большом
объеме растворителя (см. стр. 172) величина Q называется
интегральной теплотой растворения; при измерении Q
в процессе смешения раствора полимера с растворителем
величина Q называется теплотой разбавления (обычно ве-
величины теплот разбавления невелики). Помимо прямых
измерений Q =¦ — АН при помощи калориметра, величину
ДЯ можно вычислить по формуле VIII. 12. При отнесении
ДЯ к изменению молярного содержания компонентов раст-
раствора измеряют парциальные молярные величины
ДЯ х =
адя.
адя
(VIII.3),
где индекс / обычно относят к растворителю, а индекс 2 —
к растворенному полимеру. Величину ДЯг называют пар-
парциальной или дифференциальной теплотой растворения.
В растворах неполярных полимеров взаимодействие с
растворителем очень невелико (ДЯ~О) или даже сопро-
сопровождается отрицательным тепловым эффектом (ДЯ>0);
кроме того, изменение теплосодержания вообще не является
единственным фактором в растворении. Большую роль
играют термодинамические изменения в растворах полиме-
полимеров, связанные с энтропийным членом в уравнении VIII. 1.
ИЗМЕНЕНИЕ ЭНТРОПИИ В РАСТВОРАХ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Изменением энтропии при смешении AS называется
разность между энтропией раствора и суммой энтропии чи-
чистых компонентов до смешения. Идеальная энтропия
смешения '
AS= — R(nilnni + n2lnn2), (VIII. 4)
откуда следуют парциальные значения идеальной молярной
энтропии смешения:
A-Sl ид =
\, ид —
A^2, ad = -fa-
— А 1П tl\,
= — R In Щ
С другой стороны, энтропию смешения можно эксперимен-
тально. определить по формуле VIII. 1:
AH-&Z
AS-
Д5, =
т
(VIII. 6)
Сопоставление ASt по (VIII. 6) и идеальной kSiud по
(VIII. 5) для ряда растворов полимеров показало, что эк-
экспериментальные значения ASi всюду гораздо больше
Siud, особенно для растворов неполярных полимеров.
Так, для системы каучук-толуол (или каучук-бензол) они
в сотни раз больше (Мейер и др.) Значения /\S2 для кау-
каучука также гораздо больше идеальной энтропии смешения
AS2UC по (VIII. 5), причем /\S-2 резко возрастает при раз-
разбавлении раствора (рис. 69).
Эти результаты объясняются характерными особенно-
особенностями химического строения цепных полимеров. Длинные
гибкие молекулы могут принимать в растворе множество
конфигураций, мало различающихся между собой по внут-
внутренней энергии. Известно, что состояние системы, которое
можно осуществить большим числом способов, обладает
большей термодинамической вероятностью w и, следо-
следовательно, характеризуется возросшей энтропией S = klnw.
Если цепная молекула состоит из х звеньев и каждое по-'
следующее звено может расположиться 7 способами, то
число конфигураций, которые может принять макромоле-
макромолекула, по сравнению с числом способов, которым могут рас-
расположиться х звеньев в виде отдельных молекул мономеров
G-1 Г
(рис. 70), возрастает с отношением
х\
(где х\ —
(VIII. 5)
= 1.2.3....х). Как легко можно убедиться на простых
численных примерах, это отношение при не очень малых
7 необычайно возрастает с увеличением х, что соответ-
соответствует росту AS>>. Увеличение Д5г особенно велико для
разбавленных растворов, в которых каждая цепь может
разместиться всеми возможными способами. При повыше-
повышении концентрации растворов цепи взаимно ограничивают
число способов размещения, вследствие чего значения
176
177
TAS2 падают (рис. 69, справа налево). Очевидно, что в
растворе число возможных конфигураций гибких макро-
макромолекул гораздо больше, чем в твердом полимере, вследствие
чего растворение полимера сопровождается большим воз-
возрастанием энтропии. Статистическая теория энтропии сме-
смешения была разработана Флори, Хэггинсом, Жуховицким
In оя =
8
7
. S
Ь
КО"
/
V
V
у
/
/
/
/
50 60 70 SO 90 100
30
Рис. 69. Изменение TAS2 каучука
при разбавлении раствора (Сх—
концентрация толуола в %) (по
Мейеру и др.)
оооооооооо
оооооооооо
о о о о о •—• о о о
о о о о ^-А •—* о о
О О О О о\ О О »-•
ооооовоооо
оооооооооо
оооооооооо
оооооооооо
оооооооооо
оооооооооо
оооооооооо
оооооввооо
оооо***#оо
ооооовоо**
ооооовоооо
оооооооооо
оооооооооо
оооооооооо
оооооооооо
л
Рис. 70. Модель размеще
ния макромолекулы (/) и
равного числа мономеров
(//) (по Флори)
и др.; они дали расчет этой величины при смешении компо-
компонентов с резко различающимися размерами молекул (что
имеет место в системе полимер — растворитель). По Хэг-
гинсу, активности растворителя ах и полимера аъ в растворе
определяются следующими уравнениями:
In ax = In
( 1 +
v2 -Ь
—^ )vx
(VIII. 7)
где vi и V2 — объемные доли обоих компонентов раствора,
vx и V2 — их парциальные молярные объемы и {х — кон-
константы, характеризующие взаимодействие с растворителем:
к этим уравнениям мы вернемся ниже.
Естественно, что энтропийные эффекты особенно зна-
значительны для неполярных полимеров с гибкими молекулами
(каучук, поливинил ацетат). Для полярных полимеров, обыч-
обычно обладающих жесткими цепями (поливиниловый спирт,
белки), число возможных конфигураций в растворе резко
уменьшается и, следовательно, величины энтропии смеше-
смешения приближаются к идеальным значениям; одновременно
для этих полимеров возрастает значение энергетического
члена АН.
ИЗМЕНЕНИЕ СВОБОДНОЙ ЭНЕРГИИ В РАСТВОРАХ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Изменение теплосодержания АН и энтропии AS при
растворении полимеров или разбавлении их растворов са-
сами по себе не определяют направления самопроизвольного
процесса при смешении полимера с растворителем. Для
этого необходимо определить изменение термодинамическо-
термодинамического потенциала системы AZ, который связан с изменениями
АН и AS уравнением VIII. 1; в системе могут самопроиз-
самопроизвольно происходить лишь те процессы, для которых
AZ<0. _
Экспериментально определение AZt производится при
помощи измерений упругости пара растворителя над раство-
раствором полимера рх и над чистым растворителем рх°
= /?Пп
А.
/V»
(VIII. 8)
Джи и Трелоар производили измерения упругости пара
в приборе, изображенном на рис. 71. После переливания
ртути из ED в В часть растворителя из калиброванной труб-
трубки С перегоняется к каучуку А, причем поглощенное ко-
количество жидкости измеряется по понижению уровня в
С. Затем ртуть из В вновь переливается в ED и при постоян-
178
179
ной температуре измеряется разность уровней в Е и D,
равная разности упругостей пара растворителя- и набух-
набухшего каучука при данной известной концентрации геля.
-ч Зная AZi, можно вычис-
^_ А лить AZ2 по термодина-
// ((?? ^==Щ§\ мическому
уравнению
J
1 —N*
N2 _„ (VIII. 9)
где TVi и 7V-2 -г- молярные
доли обоих компонентов
в растворе, и затем изме-
изменение свободной энер-
энергии всей системы
AZ =
(VIII. 10)
Рис. 71. Прибор для измерения
упругостей пара- набухшего
каучука
где /2i и пч—количество
молей обоих компонен-
_ тов.
Величину AZi можно также измерить методом осмоти-
осмотического давления - (стр. 33) на основании уравнения
AZX
(VIIJ. 11)
Следует указать, что измерения AZ при двух температу-
температурах Т\ и Тг позволяют рассчитать ДЯ (помимо прямого
калориметрического измерения по (VIII. 2) по следующему
уравнению:
TXLZT —ТгАЪтх
Д#= ^—= ¦ • (VIII. 12)
11
Наконец, как уже указывалось, определения AZ и
АН позволяют рассчитать величину AS по (VIII. 6).
Таким образом, измерения упругости пара растворителя
над раствором полимера при двух температурах и различных
концентрациях позволяют определить по - уравнениям
VIII. 6, VIII. 8—VIII. 10 и VIII. 12 термодинамические
параметры раствора.
180
L
Различные соотношения между термодинамическими
параметрами растворов полимеров по VIII. 1 приведены в
табл. 16.
Таблица 16
Соотношения между термодинамическими параметрами
растворов пэлимеров
дн
<о
<0
=0
>0
AS
>0
<о
>0
>0
А/
<о
(при ДН
>TAS)
<о
<0
если
дн<
<TAS)
Смешение
+
(экзотермич. елгеше-
ние)
+
(экзотерыич. смеше-
смешение)
+
(атермич. смешение)
+
(эпдотермич. сл;ен;е-
ние)
>
Примеры
Нитроцеллюлоза —
—циклогексаноя
Яичный альбумин —
— года
Полиизобутилен —
—изооктан
Каучук — толуол
Из табл. 16 видно, что самопроизвольное растворение
полимера (условием которого является AZ<O) может про-
происходить не только при положительном тепловом эффекте,
но также в его отсутствие или даже при поглощении тепла
за счет соответствующих изменений энтропии.
В табл. 16 одну из групп систем образуют такие растворы
неполярных полимеров с гибкими цепями (например, кау-
каучук-толуол), в которых основное значение при смешении
имеют большие положительные значения молярной эн-
энтропии смешения, в десятки и сотни раз превышающие
идеальную энтропию смешения, тогда как тепловые эффек-
эффекты являются слегка отрицательными (АЯ>О).
Далее идут атермические системы (АН— 0), например,
изученные Каргиным и Тагер растворы полимеров в своих
гидрированных мономерах, в частности, полиизобутилена в
изооктане. В этом случае энергии взаимодействия молекул
полимера между собою равны их энергии взаимодействия
с растворителем, поэтому при смешении ДЯ = 0 гибкость
цепей полимера не изменяется и растворение происходит
благодаря тому, что энтропия смешения больше идеальной
энтропии смешения (в приведенном примере — прибли-
приблизительно в 100 раз).
181
кал.
TaS
Следующей группой являются полярные полимеры с же-
жесткими цепями, для которых характерен положитель-
положительный тепловой эффект смешения (ДЯ<0) при уменьшении эн-
энтропии низкомолекулярного компонента в начальной ста-
стадии его поглощения. Так, например, Булл нашел, что при
гидратации яичного альбумина ДЯ = —1570 кал/моль,
a AZ=—892 кал/моль; для гидратации сывороточного
альбумина АН=—1450, а ДZ=—1034 кал/моль; для гид-
гидратации желатины
ДЯ=—3800, а ДZ=
—1537 кал/моль и др.
Сопоставление этих
цифр по VIII. 1 пока-
показывает, что AS про-
процесса гидратации от-
отрицательно, благода-
благодаря переходу молекул
воды в гидратной обо-
оболочке в более упоря-
упорядоченное состояние
(см. стр. 173). После-
Последующая стадия рас-
растворения гидратиро-
ванных молекул в из-
избытке воды происхо-
происходит уже с небольшим
положительным теп-
тепловым эффектом и воз-
возрастанием энтропии.
Аналогичное поведе-
поведение установлено для
системы ацетил целлю-
целлюлоза—ацетон (рис. 72) и полистирол — этилбензол.
Наконец, можно выделить системы, образуемые также
полярными полимерами с жесткими цепями в таких усло-
условиях, когда положительный тепловой эффект (ДЖО)
сочетается с увеличением энтропии (Д5>0). Так, например,
для нитроцеллюлозы в ацетоне—ДЯ =18—19 кал/г, а
молярная энтропия смешения больше идеальных значений,
правда, всего лишь в 2—3 раза. Другими примерами яв-
являются нитроцеллюлоза в циклогексаноне, влажный яич-
яичный альбумин в воде и др. Во всех этих случаях жидкости
оказываются хорошими растворителями полимеров.
П.5
1.0
Рис. 72. Термодинамические параметры
системы ацетилцеллюлоза-ацетон при
различных концентрациях
182
Таким образом, условия смешения зависят от свойств
полимера (гибкие или жесткие цепи, наличие полярных
групп и др.) и его взаимодействия с растворителем. В атер-
мических смесях изменение AS при растворении, по Кар-
гину и Тагер, может служить количественной характери-
характеристикой гиокссти цепей полимеров.
Приведенные в табл. 16 сочетания изменений ДЯ и
AS исчерпывают все возможные случаи смешения поли-
полимеров с растворителем; иные сочетания ДЯ и AS могут
приводить лишь к AZ>0, что исключает самопроизволь-
самопроизвольное смешение. Однако конкретные системы полимер —
растворитель, удовлетворяющие тому или иному соче-
сочетанию ДЯ и AS, могут существенно отклоняться от приме-
примеров, приведенных в табл. 16. Так, например, Тагер пока-
показала, что жесткие полимеры при плотной упаковке цепей
(поливиниловый спирт, полиакрилонитрил) обладают
ДЯ>0 и AS>0 (см.табл. 16), однако в этом случае AS лишь
очень мало отличаются от Д5„^, чего не наблюдается
для полимеров с гибкими цепями. Липатов и Меерсон при
изучении температурной зависимости растворения ряда
полимеров подчеркивают значение изменения энергии меж-
межмолекулярного взаимодействия (агрегации) для термоди-
термодинамических параметров раствора. Хэггинс выражает от-
отклонения в термодинамической активности компонентов
раствора величиной ^ (VIII. 7) и др. Эти исследования
имеют большое значение для выяснения природы термо-
термодинамических параметров растЕоров полимеров, но не из- .
меняют соотношений этих параметров в табл. 16, при кото-
которых может происходить смешение полимеров с раствори-
растворителями.
НАРУШЕНИЕ УСТОЙЧИВОСТИ РАСТВОРОВ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
(РАССЛОЕНИЕ, ВЫСАЛИВАНИЕ, КОАЦЕРВАЦИЯ)
Основные факторы, которые определяют растворение
высокомолекулярных веществ, при другом соотношении
параметров обусловливают явления их ограниченного ра-
стЕорения или нерастворимости. Переходы от полного
растворения к ограниченному растворению или нераство-
нерастворимости выражаются в различных процессах нарушены*
устойчивости растворов полимеров. Если условием сме-
смешения было AZ<C0 (табл. 16), то уже при небольших поло
183
жительных значениях AZ (Д2>0) наблюдаются явления
расслоения раствора полимера на две фазы. Многочислен-
Многочисленные примеры расслоения раствора на две фазы при пониже-
понижении температуры ниже критической температуры растворе-
растворения приведены на стр. 170—171. Другим фактором, вызы-
вызывающим расслоение, является изменение состава раствора
путем добавления жидкости, в которой полимер не раство-
. ряется. Ввиду резкой зависимости растворимости от зна-
знака AZ, большинство полимеров (при достаточно большом
молекулярном весе) либо
полностью растворяется,
Полистирол либо совсем не раство-
растворяется в жидкостях. По-
Поэтому при добавлении
нераствор яющей жидко-
жидкости, после определенного
предела, устойчивость
раствора резко изменяет-
изменяется и происходит выпаде-
выпадение полимера; например,
при добавлении воды к
раствору нитроцеллюло-
нитроцеллюлозы в ацетоне, раствори-
растворители м-крезол мость полимера в водном
ацетоне резко падает при
Рис. 73. Диаграмма состояния систе- определенном содержа-
мы полистирол-метакрезол-ацетон; нии вдды и нитроцеллю-
А — область неограниченного смеше- г
ния; В—область ограниченного сме- ЛОЗа выпадает ИЗ раСТВО-
шения ра. Если нерастворяю-
щая жидкость не от-
относится к прямым оса-
дителям полимера (как вода по отношению к нитроцеллю-
нитроцеллюлозе), но полимер способен хотя бы ограниченно набухать в
ней (см. стр. 201), то расслоение не доходит до полного
выпадения полимера, а обычно образуются две жид-
жидкие фазы, например, при добавлении ацетона к раствору
полистирола в метакрезоле (рис. 73).
Выделение белков из водных растворов может быть осу-
осуществлено повышением концентрации солей; этот процесс
называется высаливанием. Высаливание белков произво-
производится полунасыщенными или насыщенными растворами
солей (Na2SO4, (NH42 SO4 и др.) и совершенно отличается
от коагуляции лиофобных коллоидов слабыми концентра-
&
циями электролитов, в частности, к высаливанию непри-
неприменимо правило Шульце—Гарди (стр. 135—136). В явлениях
высаливания, по Дебаю, основное значение имеет вытесне-
вытеснение хмолекул растворенных веществ из электрического
поля ионов, которые сильно связываются с дипольными
молекулами растворителя. По своему влиянию на высали-
высаливание ионы располагаются в последовательности, называе-
называемой лмотропными рядами или рядами Гофмейстера, в част-
частности для натриевых солей; SO4 > цитрат > ацетат >
>Cr~>NOi~>Br~~>J~>CNS~, где левые члены ряда
способствуют высаливанию, а правые три члена препят-
препятствуют высаливанию; для хлористых солей Li+>Na+>
>K+>NH4". Лиотропные ряды проявляются также во
влиянии ионов на набухание, застудневание, поверхност-
поверхностное натяжение, повышение растворимости и др. Бернал
и Фаулер объясняют лиотропные ряды действием ионов
на структуру воды. Ионы, способствующие высалива-
высаливанию (SO-i~~, Liт) усиливают упорядоченность располо-
расположения молекул воды, что соответствует как бы понижению
температуры растворителя, а ионы, стимулирующие по-
повышение растворимости (J~, CNS~, К+), усиливают сте-
степень дезориентации молекул воды, что соответствует как
бы повышению температуры растворителя; таким образом,
они связывают лиотропные ряды с изменением «структурной
температуры» растворителя.
Высаливание белков концентрированными растворами
солей является одним из основных методов фракционирова-
фракционирования белковых смесей на альбумины и глобулины. Пониже-
Понижения растворимости белков можно достичь также добавле-
добавлением спирта и действием низкой температуры. На тонком
сочетании действия спирта, солей и охлаждения до —5°
основаны способы детального фракционирования белко-
белковых смесей по Копу; например, из сыворотки крови этим
путем выделено свыше 12 белков.
Растворимость белков также сильно зависит от рН, с
минимумом в изоточке (рис. 74); при смещении от изоточки
возрастают заряд и гидратация белковых молекул и повы-
повышается их растворимость, поэтому при высаливании бел-
белков всегда поддерживают рН близким к изоточке.
Растворимость полимеров и условия их выделения из
растворов при добавлении осадителя сильно зависят от их
молекулярного веса; для полимеров с меньшим молекуляр-
молекулярным .весом резкость явлений в критической области раство-
184
185
рения значительно ослабляется и для их осаждения требует-
требуется добавление большего количества нерастворяющей жид-
жидкости (соответственно, они легче растворяются). Поэтому в
растворах полидисперсных полимеров, представляющих
собой смесь гомологических полимеров с различной дли-
длиной цепи, постепенное добавление возрастающих количеств
нерастворяющих жидкостей приводит к выпадению ряда
фракций в порядке убывания молекулярного веса; для
woo
I 900
I i soo
t-a? ?oo
53 § 600
II чоо
ГС ЬОО
300,
\
\
s
i i
у
5,0
Рис. 1\. Растворимость яичного альбумина
в 26%-ном растворе (NH^SC^ в зависимо-
зависимости от рН
более четкого разделения фракций их осаждают из разбав-
разбавленных растворов полимеров. Для фракционирования
применяют также способ постепенного понижения температу-
температуры при постоянном составе жидкости. При наличии в раст-
растворе полимера компонентов, отличающихся по разветвлен-
ности цепей, по химическому составу (например, продуктов
окисления или посторонних полимеров) и др., разде-
разделение по фракциям может, разумеется, отклоняться от нор-
нормальной последовательности молекулярных весов. Пре-
Препаративное разделение ряда фракций полимеров, которые
затем можно отдельно исследовать, широко применяется
для научных и практических целей (в частности, для харак-
характеристики полидисперсности полимеров).
При растворении полидисперсных полимеров различие
критических условий растворения для различных фракций
186
i ¦$
может вызвать переход в раствор не всей навески полимера,
а некоторой непостоянной части фракций. Вследствие этого
возможна зависимость растворимости полимеров от коли-
количества навески (правило осадков, по Оствальду), тогда как
в истинных растворах чистых низкомолекулярных веществ
растворимость не зависит от количества твердой фазы на
дне сосуда. Это расхождение, которое ранее казалось ха-
характерной чертой коллоидных растЕоров, было устранено,
когда удалось показать, что для однородных, хорошо
фракционированных препаратов полимеров растворимость
также является постоянной величиной, не зависящей от
количества навески; более того, для белков, которые мо-
могут быть получены строго монодисперсными и очищенны-
очищенными, постоянство растворимости является одним из чув-
чувствительных критериев чистоты препаратов.
На понижении растворимости и переходе от полного
смешения к ограниченной растворимости основаны также
многочисленные случаи коацервации (Бунгенберг-де-Ионг).
Так, например, коацерваты с расслоением в капельно-
капельножидкой форме или в виде двух слоев могут быть получены
из водных растворов желатины добавлением спирта или
сернокислого натрия, из спиртовых растворов проламинов
при разбавлении их водой, из положительно заряженных
молекул желатины (при рН 1,2—4,8) и отрицательно заря-
заряженных частиц гуммиарабика или крахмалофосфорной ки-
кислоты, из растворов двух белков с сильно различными по-
положениями изоточек, из растворов белка и нуклеиновых
кислот и др. Во всех этих случаях коацерваты возникают
в условиях перехода к взаимно ограниченной растворимо-
растворимости компонентов раствора. Степень расслоения полимеров
при коацервации очень велика, например, при получении
коацервата из 1%-ного раствора желатины до 93% ее ко-
количества входит в состав коацерватного слоя, а при более
низких концентрациях — относительно еще больше; по-
поэтому оба слоя при коацервации резко различаются по
содержанию коллоидных веществ. Физико-химические свой-
свойства коацерватов в ряде отношений напоминают соответ-
соответствующие свойства протоплазмы, что привлекает к ним вни-
внимание биологов; согласно Опарину, коацервация имела
большое значение для пространственного отделения и орга-
организации коллоидных веществ в истории возникновения жиз-
жизни на Земле.
При полной потере растворимости полимеров в данной
187
среде могут быть получены дисперсии высокополимеров,
которые вполне уподобляются лиофобным суспензиям и
эмульсиям (главычетвертая — седьмая); таковы, например,
эмульсии каучука в воде — латексы (их свойства подробно
исследованы Догадкиным, Гликманом, Воюцким и др.).
Аналогично могут быть получены коллоидные дисперсии
нитроцеллюлозы в воде, желатины в спирте и др.
РАЗМЕРЫ И ФОРМА МАКРОМОЛЕКУЛ В РАСТВОРАХ
Термодинамические характеристики растворов полиме-
полимеров, как было показано, тесно связаны с цепным строением,
размерами и гибкостью макромолекул, а также с энергией
их взаимодействия с растворителем. Эти основные парамет-
параметры определяют также многие другие свойства растворов по-
полимеров, по которым, в свою очередь, можно судить о строе-
строении и свойствах макромолекул. Так, например, гибкость
цепей отражается не только на высоких значениях энтропии
смешения, но и на условиях передвижения молекул в раст-
растворах при диффузии, течении и др. В этом отношении изу-
изучение разбавленных растворов полимеров представляет
тем больший интерес, что оно выясняет строение и свой-
свойства индивидуальных макромолекул, лежащих в основе
всех полимерных материалов.
Влияние гибкости цепей в полной мере проявляется
лишь при достаточной длине цепей. Слишком короткие це-
цепи слегка изгибаются, но, вследствие ограничения изгибов
валентными углами, они все же не отклоняются сильно от
формы вытянутых палочек. По мере удлинения цепи (т. е.
увеличения молекулярного веса), изгибы цепей все более
приближают молекулы к свернутой форме, вначале, одна-
однако, довольно рыхлой, со многими просветами, через ко-
которые может протекать растворитель. Затем изгибы цепей
все более экранируют внутренние участки молекулы и,
в конце концов, образуется модель беспорядочно сверну-
свернутого клубка, через который растворитель уже не может про-
протекать; он лишь обтекает такую молекулу, подобно тому,
как он обтекает сплошную твердую частицу эквивалентной
формы (Дебай, Кирквуд). Размеры клубка обычно ^харак-
^характеризуются квадратом гидродинамического радиуса г2, ко-
торый приблизительно равен -g-Л2, где h2—средний квадрат
расстояния между концами цепи (см. главу десятую);
188
для вытянутых частиц /г1 и г2 больше, чем для свернутых
молекул. Очевидно, что гибкие цепи образуют непрони-
непроницаемые для растворителя клубки при меньшей длине цепи
(и меньшем молекулярном весе), чем жесткие цепи. Так,
например, дезоксирибонуклеиновая кислота при УИ = 4,5- 10е
имеет форму слегка изогнутой палочки (Роуэн), тогда
как многие гибкие макромолекулы образуют непро-
непроницаемые клубки уже при М ниже 500 000 (Цветков, Рос-
си). Кроме того, размеры и форма клубка могут изменяться
в зависимости от состава среды, так как взаимодействие с
растворителем (сольватация) изменяет гибкость цепи.
В главах второй и третьей был описан ряд методов, при
помощи которых исследовались размеры и фиксирован-
фиксированная форма жестких сферических и вытянутых частиц;
теперь необходимо рассмотреть особенности, вызываемые
наличием гибких макромолекул или изменением размеров
и формы молекулярного клубка в растворе.
Наиболее близко к термодинамическим характеристи-
характеристикам растворов полимеров находятся изменения их осмоти-
осмотического давления.
Изменения свободной энергии A Z растворов полимеров,
как уже указывалось, связаны с осмотическим давлением
растворов уравнением VIII. 11; с другой стороны__AZ
связаны с активностью компонентов раствора:
=RT\nal; отсюда следует, что осмотическое давление
RT .
— ¦=- In
(VIII. 13)
Из статистической теории растворов полимеров можно опре-
определить lnai по формуле (VIII. 7); подставляя ее в (VIII. 13)
и проведя некоторые преобразования, получаем уравнение
RT
RTdx
V-i
)c2, (VIII. 14)
где с — концентрации (в г! 100 см'л), d — плотности и
М — молекулярные веса, а индексы / и 2, как и в прежних
уравнениях, относятся к растворителю A) и полимеру B).
При небольших концентрациях (VIII. 14) имеет вид:
(VIII.14а)
189
Уравнение (VIII. 14) является основным уравнением, ха-
характеризующим осмотическое давление растворов полиме
ров. Графическое выражение зависимости леЕой части
(VIII. 14) от с дано на рис. 6 (стр. 34). Из (VIII. 14) или
(VIII. 14а) видно, что при экстраполяции до бесконечного
разбавления (с2->0) оно переходит в уравнение Вант—Гоф-
фа:— =
С2
"Ж
(глава вторая), откуда определяется моле-
молекулярный вес (по отрезку на оси ординат); однако, из
(VIII. 14) видно также, что по наклону кривой можно
определить тгл~т(у — V-i) .откуда легко найти констан-
константу (хг в (VIII. 7). Величина \it для ряда гомологичных поли-
полимеров или фракций мало зависит от молекулярного веса,
но заметно изменяется с природой растворителя, что видно
и на рис. 6. Величина отражает не только энергию, но и
энтропию взаимодействия с растворителем, т. е. включает
влияние гибкости цепей.
Приводим некоторые значения константы взаимодей-
взаимодействия полимера с растворителем (\xi):
Полимер
Натуральный каучук
Гуттаперча
Полистирол
Этилцеллюлоза
Бензилцеллюлоза
Растворитель
Бензол
Хлороформ
СС14
Толуол
Толуол
Этилацетат
Анилин
0,43
0,37
0,28
0,36
0,44
0.35
0,40
При известном значении ;xj, определенном по осмоти-
осмотическим данным, можно по (VIII. 7) рассчитать величину
In ci\ и отсюда AZi и относительную упругость пара раст-
растворов полимеров по (VIII. 7). Необходимо отметить, что
расслоение раствора или выпадение полимера при добав-
добавлении нерастворяющей жидкости (что изменяет jxi) проис-
происходит лишь при некотором критическом значении |-ч>икрит
!J-1, Крит
1
(VIII. 15)
190
где (vilv2) — отношение парциальных молярных объемов
компонентов (связанное с их молекулярными весами);
таким образом, (VIII. 15) позволяет в принципе рассчитать
условия расслоения растворов полимеров. По Флори и
Кригбауму, зависимость тг/с от концентрации выражается
следующим уравнением:
+/> + />2 + -'-] (VIII. 16)
где Г% и Г2. — коэффициенты разложения в ряд по с и
Г2. при определенных условиях равно E/8)А2; уравнение
(VIII. 16) ими сопоставляется с уравнением светорассея-
(VIII. 17)
ния (см. III. 4) и с уравнением концентрационной зависи-
зависимости диффузии по Манделькерну и Флори
= DQ[\
(VIII. 18)
Уравнения (VIII. 16—VIII. 18) интересны тем, что позво-
позволяют установить общие коэффициенты зависимости от кон-
концентрации для весьма различных свойств растворов поли-
полимеров; все они показывают отсутствие линейной зависимо-
зависимости от концентрации.
Величину гидродинамического радиуса клубка можно
определить путем сопоставления коэффициентов поступа-
тачьной Dt и вращательной Dr диффузии (см. главу вторую)
D~
(VIII. 19)
или из измерений светорассеяния под углами 45° и 135° к
падающему пучку света (с длиной волны X):
135°
1 Л- 37
(VIII. 20)
Для проницаемого клубка, через который растворитель
протекает, трение / пропорционально числу звеньев в
клубке, т. е. молекулярному весу; в этом случае коэффици-
коэффициент диффузии Dt должен быть обратно пропорционален
молекулярному весу М полимера. Напротив, для непрони-
непроницаемого клубка, обтекаемого растворителем, трение за-
191
висит от внешних размеров клубка в целом; в этом случае
величина Dt должна быть обратно пропорциональной)/М.
Для растворов фракций полиметилметакрилата (М
от 5-Ю5 до 6,5-106) в ацетоне Цветковым было показано,
что Ут2 растет пропорционально М0-56, а для растворов
дивинилстирола (М от 5,6-10* до 2,5-1G5) пропорционально
/И0-50; таким образом, при достаточной длине цепи гибкие
молекулы образуют свернутые клубки, непроницаемые для
растворителя. Росси и сотрудники для растворов поли-
полистирола в бензоле нашли приближенно применимость урав-
уравнения Эйнштейна (см. П. 3), что объясняется не очень
большой асимметрией формы молекулярных клубков. Так,
по данным Цветкова, асимметрия молекул полиметил-
полиметилметакрилата с М=4,2-106 в различных растворителях
изменялась от 2 до 5, тогда как при вытянутой форме мо-
молекул она должна была равняться сотням. Приводим дан-
данные о асимметрии молекул полиметилметакрилата в раз-
различных растворителях.
Растворитель
Хлорбензол
Бромбензол
Бромоформ
Ацетон . .
Этилацетат
Хлороформ
Асимметрия клубка
(в;а)
1,90
3,10
3,25
3,60
4,40
5,10
Из этих данных видно, что в растворителях с более вы-
высокой растворяющей способностью молекулярные клубки
обладают более вытянутой формой, чем в плохих раство-
растворителях. Липатов и Меерсон получили аналогичные дан-
данные для растворов этилцеллюлозы в различных раствори-
растворителях, а Доти, Цимм и Марк — для молекул полистирола.
Большое количество интересных исследований размеров
и формы молекул полимеров было произведено методом из-
измерений вязкости (см. стр. 47). Результаты этих
измерений обычно выражают рядом условно принятых
характеристик; так, например, отношение вязкости раство-
раствора Tj к вязкости растворителя г(„ называют относительной
вязкостью 7]птн = — ; величину '" — 1 — tJvi,
192
удельной вязкостью, а величину
— приведенной
(к единице концентрации) вязкостью; последнюю вели-
величину часто измеряют при нескольких концентрациях (все
измерения производят в разбавленных растворах) и затем
экстраполируют результаты к с-Ю; этим путем полу-
полу= [т(],
называемую предель-
предельчают величину Игл
но \ с
ной приведенной вязкостью или характеристической
вязкостью (в английской литературе она называется int-
intrinsic viscosity); со всеми этими величинами приходится
часто встречаться в литературе.
По Штаудингеру, удельная вязкость г1уд разбавленных
растворов полимеров пропорциональна молекулярному ве-
весу М и концентрации с (выраженной в молях мономера
цепи на 1 л раствора)
или
ч\уд = КМс
КМ
'(,уд
с
(VIII. 21)
(VIII. 21а)
где К—константа, характерная для данного гомологи-
гомологического ряда полимеров в данном растворителе, например,
для ряда парафинов в CQ 4 /Г= ] ,07 -10~4, для ряда псли-
стиролов в СС14/С=1,8-10~4, для ряда полиоксиметиленов
в хлороформе /С=2,4-10~4 и т. д. Уравнение Штау-
дингера позволяет замечательно простым способом
опредатять молекулярный вес полимеров, но оно справед-
справедливо только для сравнительно коротких цепей или более
длинных, но жестких цепей, которые еще сохраняют фор-
форму вытянутых или слегка изогнутых палочек (см. стр. 188).
В широком диапазоне молекулярных весов, а также при
изменении растворителя значения констант К изменяются.
При удлинении цепи свертывание молекулы в клубок
уменьшает ее сопротивление течению, вследствие чего за-
зависимость вязкости от молекулярного веса отклоняется
от прямой пропорциональности. По обобщенному урав-
уравнению Штаудингера
= КМ"
(VIII. 22)
где а— величина, характеризующая свертывание макро-
макромолекул в растворе (т. е. гибкость цепей). Для жестких
/а " «Коллоидная химия»
193
молекул, близких к палочкообразной форме, а^1 и
(VIII. 22) переходит в (VIII. 21а); для гибких молекул,
близких к сферической форме, а=0,5; у сильно заряжен-
заряженных молекул полиэлектролитов (стр. 119) ос=ё2. Зна-
Значения К и а обычно определяются для данного ряда по-
полимер гомологов в данном растворителе на основании точ-
точных измерений величины [т] ] и молекулярного веса (при
помощи методов осмотического давления, светорассеяния
и др.) нескольких высокоочищенных фракций данного по-
полимера; затем, при известных К и а, можно определять М
непосредственно из измерений вязкости по (VIII. 22).
Примеры для некоторых растворов полимеров приведены в
табл. 17.
Таблица 17
Зависимость [yj] от молекулярного веса
для некоторых растворов полимеров
Полимер
Полиизобутилел
Натуральный кау-
каучук
Полистирол
Поливиниловый
спирт
Поливияилаиетат
Поливинилацетат
Ацети лцеллюлоза
Целлюлоза
Растворитель
Диизобути-
лен
Толуол
Топуол
Вода
Ацетон
Буганон
Ацетон
Медноамм.
раствор
Тем-
пера-
пература
20
—
—
50
5Э
25
25
25
ы
3,6 -10-4 МО,64
5,02- 10-4 М0.67
1,28-10-4 мо. 70
5,9 -10-4 уио-67
2,8 -10-4 М0.67
4,2 -10-2 Af0.62
1,49-10-4 М0.82
0,85-10-4 уио.81
Область
молекулярных
весов 10Б
0,05—13,0
0,4 —15.0
5,5 —20,5
0,44— 1,1
0,77— 8,5
0,17—12,0
0,31— 3,9
Таким образом, для гибких каучуков а~0,64, а для
жестких молекул целлюлозы и ее производных а~0,81;
для растворов нитроцеллюлозы в ацетоне Мейергоф на-
нашел а=1,0. Для более низких молекулярных весов, чем
указано в таблице, значения а для всех полимеров по-
постепенно приближаются к а=1,0, в соответствии с урав-
уравнением (VIII. 21).
Флори и Фокс указали, что, если производить изме-
измерения вблизи критической температуры смешения 6, когда
молекулы полимера находятся на грани выпадения, в пре-
194
дельно «плохом» растворителе и поэтому наиболее свер-
свернуты, то
[т,Ь =КМ0'5 (VIII. 23)
где /С=2,Ы021 (нгг) '2, а ге—гидродинамический ра-
диус молекулярного клубка при температуре G; величина
/*е уже не зависит от природы растворителя, поскольку
растворы исследуются в соответственном состоянии. Под-
Подставляя значение К в (VIII. 23), получаем, что
2,1-1021
7з
(VIII. 24)
Уравнение (VIII. 23) было подтверждено Флори и со-
сотрудниками при измерениях в растворах полистирола, по-
поли изоб у тилена, полидиметилсилокеана и других полиме-
полимеров в широком интервале молекулярных весов.
Отношение г©, измеренного по (VIII. 24), к теоретиче-
теоретическому радиусу г0 сферической свернутой молекулы с данным
.молекулярным весом М, характеризует асимметрию клубка,
обусловленную неполной гибкостью молекулярной цепи и
природой растворителя. Вполне гибкая цепь в «плохом»
ГА 1 /-л 1
растворителе должна давать —— 1,0, фактически для по-
го
лиизобутилена в циклогексане (т. е. в хорошем раствори-
растворителе) было получено —= 1,5, а для жестких молекул
трибутиратцеллюлозы в метилэтилкетоне это отношение
возрастает до 3,5.
Уравнения (VIII. 23) и (VIII. 24) применяются также
для приближенной оценки разветвленности молекул поли-
полимеров (например, молекул декстрана). В этом случае ве-
величина гидродинамического радиуса больше, чем для ли-
линейных молекул с тем же молекулярным весом, причем
квадрат их отношения g равен
е « --?"±1 (VIII. 25)
где значение К приведено выше. Поддубный применил этот
метод, а также измерения коэффициента вращательной
диффузии (см. стр. 32), для характеристики степени раз-
разветвленности различных синтетических каучуков.
V,14* 195
Значения молекулярного веса, определенные по (Vlll.
22) и (VIII. 17), дают усредненный по весу частиц или сред-
невесовой молекулярный вес Mw, а значения М, опреде-
определенные по (VIII. 14), дают усредненный по числу частиц
или среднечисленный молекулярный вес Мп (см. главу
вторую). Для высокоочищенных фракционированных пре-
препаратов полимеров Mw= М„, тогда как для неоднородных
полидисперсных препаратов Mw>Mn. Чен Иен-юань и
Ши Лян-хо исследовали влияние полидисперсности на
зависимость осмотического давления и светорассеяния от
"концентрации в уравнениях (VIII. 16) и (VIII. 17) и наш-
нашли ее согласующейся с теорией Флор и и Кригбаума. Кор-
шак и Рафиков указали на возможность количественной ха-
характеристики полидисперсности по отношению MJMn и
подтвердили ее на смесях фракций полиэфиров.
Выводы
В отличие от лиофобных золей, растворы высокомоле-
высокомолекулярных веществ являются термодинамически устой-
устойчивыми обратимыми истинными растворами. Они подчи-
подчиняются правилу фаз и их устойчивость определяется соот-
соотношением энергетического (Д#) и энтропийного (TAS)
членов в уравнении (VIII. 1). Для растворов полярных
полимеров, обычно обладающих жесткими цепями, основное
значение имеют изменения АН, в значительной мере за-
зависящие от сольватации. Тепловые эффекты, изменения
упругости пара, сжимаемости и других свойств растворов
при сольватации указывают, что наиболее прочно связан-
связанная часть растворителя составляет около одного слоя мо-
молекул вокруг полярных групп полимера (табл. 15). Для
растворов неполярных полимеров с гибкими цепями основ-
основное значение имеют изменения энтропии смешения, во
много раз превышающие идеальные значения, и непо-
непосредственно связанные с гибкостью макромолекул в раст-
растворах. Различные соотношения АН и AS, приводящие к
возможности самопроизвольного растворения полимеров
(AZ<0) приведены в табл. 16. Нарушение устойчивости
растворов полимеров при понижении температуры, добав-
добавлении нерастворяющей жидкости или высоких концентра-
концентраций солей приводит к различным случаям расслоения на две
фазы, выпадения полимеров, высаливания белков и др.
Зависимость растворимости полимеров от молекулярного
196
веса создает возможность их фракционирования путем
дробного осаждения или растворения. Переход от полного
смешения к ограниченной растворимости определяет также
различные случаи коацервации.
Гибкость макромолекул при достаточной длине цепей
приводит к образованию свернутых форм молекул, причем
размеры и проницаемость для растворителя молекулярных
клубков влияют на ряд свойств растворов полимеров —
диффузию, светорассеяние, вязкость и др. Для сравнитель-
сравнительно коротких или жестких цепей наблюдается пропорцио-
пропорциональная зависимость от молекулярного веса М (VIII. 21),
для предельно свернутых форм молекул — зависимость от
М0-5 (VIII.22); в последнем случае может быть рассчитан
(по VIII. 24) гидродинамический радиус молекулярного
клубка, а также относительная степень разветвленности
молекул (VIII. 25). Измерения осмотического давления
(VIII. 14) позволяют определить константу (j-i взаимодей-
взаимодействия полимера с растворителем и среднечисленный моле-
молекулярный вес полимера Мп; измерения светорассеяния по
(VIII. 17) и вязкости — по (VIII. 21, 22) характеризуют
средневесовой молекулярный вес полимера Mw; отношение
Mw IMn характеризует полидисперсность полимера.
Вопросы
1. Какова природа устойчивости растворов полимеров, в чем
ее сходство и отличия от растворов низкомолекулярных веществ и
от лиофобных коллоидов?
2. Как доказывается применимость правила фаз к растворам
полимеров и какие выводы из этого следуют ?
3. Чем определяется энергетическое взаимодействие полимера
с растворителем и какими методами оно исследуется?
4. Как вычислить гидратацию желатины (в г на 1 г), если
в ней приходится по одной СООН- и NH2-rpynne на 10 аминокислот-
аминокислотных остатков?
5. Как изменяется энтропия смешения в растворах полиме-
полимеров и как она зависит от гибкости макромолекул?
6. При каком условии возможно самопроизвольное растворе-
растворение полимеров и при каких соотношениях ДН и AS оно выполняется?
7. Чем определяется расслоение растворов полимеров и ка-
каковы критические условия расслоения?
8. Каким образом производится высаливание белков, чем оно
определяется, как используется?
9. На каких свойствах растворов полимеров отражается гиб-
гибкость макромолекул, какие уравнения выражают эти зависимости?
10. Какими методами измеряется молекулярный вес, размеры
и форма свернутых молекул полимеров в растворах?
«Коллоидная химия»
197
Глава девятая
ГЕЛИ
Гелями называют структуры, образуемые коллоидными
частицами или молекулами полимеров в форме простран-
пространственных сеток, ячейки которых обычно заполнены раство-
растворителем. Гели отличаются как от компактных коагулятов
или твердых полимеров, так и от разбавленных растворов,
в которых каждая коллоидная частица или макромолекула
являются кинетически индивидуальными частицами. За-
Занимая в ряде отношений промежуточное положение между
растворами и твердыми полимерами, гели обладают также
многими своеобразными свойствами и имеют большое прак-
практическое значение. В частности, к гелям относятся колла-
коллаген, мясо скота н рыб, различные пористые и ионообмен-
ионообменные адсорбенты, ультрафильтры и искусственные мембра-
мембраны, а также волокна мышечных тканей, клеточные оболоч-
оболочки, хрящч, оболочки эритроцитов и различные мембраны
в организме.
В зависимости от природы образующих их веществ раз-
различают гели, построенные из жестких частиц, или хрупкие
гели, и гели, образованные гибкими макромолекулами,
или эластичные гели (студни).
ХРУПКИЕ ГЕЛИ
Хрупкие гели образуются коллоидными частицами БЮг,
ПО2, SnO2, Ре2Оз, УгО.-,, беизопурпурина и др.; типичным
примером является гель кремнекислоты. Благодаря жест-
жесткости частиц и всего каркаса хрупкого геля, его объем при
высушивании пли оводнении мало изменяется, поэтому хруп-
хрупкие гели называют также ненабухающими. Хрупкие гели име-
198
ют сильнопористую структуру с множеством узких жест-
жестких капилляров, диаметром до 20—40 А. При постепенном
оводнении высушенного геля первые порции воды или другой
жид-кости прочно адсорбируются на стенках капилляров в
виде тонких молекулярных пленок с очень низкой упру-
упругостью пара (рис. 75); при дальнейшем оводнении, т. е.
при повышении давления пара, начинается капиллярная
конденсация (см. главу четвертую). По уравнению Том-
сона (IV. 5), в узких капиллярах, в которых мениск более
вогнут, конденсация пара происходит раньше, чем в широ-
широких капиллярах. Ввиду полидисперсности размеров ка-
Поглощенное количество Виды
Рис. 75. Кривые упругости пара хрупких
гелей (SiO2) (по Ван-Беммелену): 1—при
обезвоживании, 2—при оводнении
пилляров, кривая относительной упругости пара в правой
части рис. 75 идет вверх по кривой 2 вплоть до значения
1,00, соответствующего упругости пара над плоской по-
поверхностью.
При высушивании оводненного геля (при температурах
ниже 150°) упругость пара соответственно падает по мере
удаления воды, вначале из широких, затем из все более
узких капилляров, но на стадии удаления адсорбционных
слоев кривая проходит ниже, чем при оводнении (рис. 75,
кривая /). Это расхождение кривых прямого и обратного
процессов называется гистерезисом; в данном случае Хок
объясняет наличие гистерезисной петли изменением ка-
капиллярной структуры при высушивании, а Песков и
Прейс — различием условий смачивания при наличии,
соответственно, воздуха или воды на стенках капилляров.
Внешне гель с пустыми или частично заполненными капил-
13*
199
лярами кажется мутным, но при заполнении их водой
он становится прозрачным.
Ввиду того, что капиллярная конденсация пара имеет
физическую природу и не является специфической, хрупкие
гели могут поглощать весьма различные жидкости, смачи-
смачивающие стенки капилляров (что необходимо для образования
вогнутых менисков). Так, например, Киселев исследовал
адсорбцию на силикагеле паров воды, метилового спирта,
пентана и бензола.
Измеряя объем поглощенной жидкости при различных
давлениях пара, которые по уравнению Томсона легко
пересчитать на соответствующие радиусы капилляров,
можно определить относительную емкость капилляров раз-
различных размеров или так называемые структурные кривые
пористых адсорбентов — гелей SiOs, активированных уг-
углей и др.
Образование хрупких гелей из лиофобных золей следует
рассматривать как один из видов коагуляционного струк-
турообразования (Ребиндер). При наличии жестких вытя-
вытянутых частиц, их сближение при понижении устойчивости,
в зависимости от концентрации электролитов и золя, может
вызвать появление тактоидов (см. стр. 144), простран-
пространственного каркаса в гелях или, наконец, беспорядочного
коагулята. Бентонитовые глины или частицы V2O5 могут
образовывать гели уже при концентрации ниже 0,1%, тогда
как золь гидроокиси железа дает гель при концентрации
5%, а при 0,5% он коагулирует в виде осадка. При слабых
силах взаимного сцепления, например, в 10%-ных суспен-
суспензиях бентонитовых глин или в гелях гидроокиси алюми-
алюминия, структура геля разрушается даже при небольшом ме-
механическом воздействии, но может легко восстановиться.
Такие обратимые коагуляционные структуры, связанные со
вторым минимумом на потенциальных кривых (см. стр.
143), называются тиксотропными. Напротив, при зна-
значительных силах сцепления, когда частицы в местах кон-
контакта, особенно на концах, разделены лишь очень тонкими
сольватными слоями, появляются прочные каркасы с тон-
тонкими хрупкими стенками, механическое разрушение ко-
которых происходит необратимо (например, в гелях крем-
некислоты). Добавление небольших количеств поверхно-
поверхностно-активных веществ, образующих молекулярные ад-
адсорбционные слои, может легко изменять условия контакта
частиц и прочность образующихся гелей (Ребиндер).
ЭЛАСТИЧНЫЕ ГЕЛИ
Эластичные гели или студни образуются цепными моле-
молекулами желатины, агара, каучука и других полимеров и
поэтому по своим свойствам во многом отличаются от хруп-
хрупких гелей. Благодаря гибкости цепей в пространственной
сетке, эластичные гели сравнительно легко могут изменять
свой объем при поглощении или отдаче растворителя, а при
высушивании сохраняют свою эластичность. Если в же-
жестких капиллярах хрупких гелей возможна капиллярная
конденсация паров различных жидкостей, то гели высоко-
высокополимерных веществ могут поглощать лишь те жидкости,
которые по отношению к ним служат растворителями, т. е.
эластичные гели обладают значительной избирательностью
поглощения. Поэтому в зависимости от природы поглощен-
поглощенной жидкости различают гидрогели, алкогели, глицерогели
и др., тогда как для хрупких гелей эта терминология обыч-
обычно не применяется.
Поглощение жидкостей эластичными гелями сопрово-
сопровождается значительным увеличением объема студня, или его
набуханием; поэтому эластичные гели иначе называют на-
набухающими гелями. Объем набухшего студня может в де-
десятки раз превосходить собственный объем полимера. Ес-
Если набухание непосредственно переходит в полное растворе-
растворение полимера (например, набухание каучука в бензине,
гуммиарабика в воде, нитроцеллюлозы в ацетоне), то оно
называется неограниченным. В этом случае набухание
является лишь первой стадией растворения полимера (гла-
(глава восьмая); оно имеет только кинетический характер и
обусловлено тем, что проникновение растворителя в поли-
полимер происходит быстрее, чем диффузия цепных молекул
полимера в растворитель.
Напротив, если студень поглощает определенное коли-
количество растворителя, но не образует раствора полимера, то
такое набухание называется ограниченным (например, же-
желатина в холодной воде). Ограниченное набухание может
переходить в неограниченное при повышении температуры
или изменении состава среды. Например, студень желати-
желатины растворяется в воде при нагревании выше 40—42°
или при комнатной температуре при добавлении 2М KCNS
или KI. Эти явления вполне аналогичны переходу ограни-
ограниченно-растворимых жидкостей, например, системы фенол-
вода, к полному смешению при нагревании выше 66° или
201
при добавлении салицилата натрия. В отличие от неогра-
неограниченного набухания, ограниченно-набухший студень, как
и система ограниченно-растворимых жидкостей, находится
в равновесном состоянии.
Ограниченное набухание может также иметь место при
химической модификации полимеров, которые сами по себе
способны к неограниченному набуханию. Например, на-
натуральный каучук может набухать в бензине до полного
растворения, однако, после вулканизации, когда его мо-
молекулы химически связаны некоторым количеством атомов
серы и образуют прочную пространственную сетку, набу-
набухание становится ограниченным; аналогично, задубленный
студень желатины даже при нагревании остается в ограни-
ограниченно-набухшем состоянии. В этом случае равновесие
при ограниченном набухании имеет вынужденный ха-
характер. Отрезки цепей между узлами пространственной
сетки выпрямляются при набухании, вследствие увеличе-
увеличения расстояний между этими узлами, но в то же время они
отходят от своего наиболее вероятного свернутого состоя-
состояния (см. стр. 188), поэтому при деформации энтропия
цепей уменьшается (А5(Э<?5б<0). С другой стороны, энтропия
смешения полимера и растворителя при набухании воз-
возрастает (см. стр. 176). Соотношение этих противо-
противоположных процессов изменения энтропии определяет на-
напряжение в полимерной сетке, ограничивающее степень на-
набухания (Флор и и Ренер).
При очень большом числе прочных связей между моле-
молекулами, например, в эбоните, короткие отрезки цепей меж-
между узлами пространственной сетки утрачивают гибкость,
гель перестает быть эластичным и теряет способность к на-
набуханию. Бахман показал, что студень желатины можно
превратить в хрупкий гель, подобный гелю кремнекислоты,
если при помощи ряда растворов с возрастающим содер-
содержанием спирта полностью удалить воду из студня и этим
путем придать гелю жесткость.
В полярных полимерах и белках процесс набухания, как
и растворение, начинается с сольватации полярных групп
растворителем. Ввиду сильного понижения активности раст-
растворителя в сольватных слоях, при поглощении жидкости
вначале наблюдается очень низкая упругость пара (рис.
76), как и при образовании первых адсорбционных сольват-
сольватных слоев в хрупких гелях (рис. 75). Удаление последних
следов жидкости из гелей обоих типов, вследствие низкой
\
упругости пара, дается с большим трудом, тогда как основ-
основное количество растворителя удаляется довольно легко.
В эластичных гелях кривые упругости пара имеют лишь
небольшой гистерезис, обусловленный, вероятно, неболь-
небольшими остаточными деформациями в структуре геля.
Как уже указывалось (см. стр. 173), растворитель в
сольватных слоях находится под большим внутренним дав-
давлением и обладает повышенной плотностью; кроме того,
его связывание сопровождается положительным тепловым
эффектом. Поэтому, хотя объем студня при набухании рез-
резко увеличивается, суммарный объем полимера и жидкости
W го зо
Поглощенное количество воды
Рис. 76. Кривая упругости пара эластично-
эластичного геля (желатины)
при набухании несколько уменьшается в результате не-
небольшого сжатия растворителя; при набухании также вы-
выделяется заметная теплота набухания О. Изменение сжатия
объема Аи и теплоты Q с увеличением количества погло-
поглощенной студнем жидкости i выражаются однотипными урав-
уравнениями Катца:
а а'1
Av =
Q
В -I-
A-
(IX. 1)
где а, р (или А, В)—-константы. Из (IX. 1) видно, что
при малых /эффекты пропорциональны /, а при больших i—
достигают постоянных величии. Отношение (lu/Q) при-
приблизительно постоянно для различных эластичных гелей.
Полная или интегральная теплота пчбгхячня, по дан-
данным Липатова исотрушиков. составляет дгл /мчатины в во-
203
де 30 кал/г, для ацетилцеллюлозы в метил ацетате 19 кал/г,
для каучука в толуоле 4,8 кал/г и для каучука в
бензоле 2,8 кал/г; по величине сольватации эти цифры
соответствуют для желатины 0,3 г/г, и для каучука около
0,17 г/г. Сопоставление приведенных данных с данными
по сольватации в растворах (стр. 172—175) указывает, что
тепловые эффекты набухания в основном определяются
образованием первого сольватного слоя. При этом, если
полная теплота набухания Q по мере поглощения жидко-
жидкости стремится по (IX. 1) к предельному наибольшему
значению (рис. 77, 1), тодиф-
??1 ференциальная теплота набу-
набухания q (в пересчете на 1 г
связанного растворителя) по-
постепенно падает (рис. 77, 2);
эти изменения величин Q и q
при набухании аналогичны их
изменениям при сольватации
в растворах.
Наконец, в начальной ста-
стадии набухания, при поглоще-
поглощении растворителя в количе-
количестве, примерно, 20—40% от
Рис. 77. Изменение интеграль- веса сухого вещества, также
ной (/) и дифференциальной наблюдается уменьшение энт-
B) теплоты набухания при уве- ропии в результате упорядо-
личенип количества поглощен- ченного расположения моле-
ного растворителя г
кул растворителя в сольват-
ном слое. Например, для кол-
коллагена при 25°, TAS = —2020 кал/моль, для агара при
5° TAS =—1350 кал/моль, для целлюлозы при 50°С TAS =
= —770 кал/моль и т. д.
Таким образом, целый ряд свойств, характеризующих
набухание полимеров: низкая начальная упругость пара,
сжатие объема, тепловые эффекты, уменьшение энтропии
растворителя — являются лишь непосредственным резуль-
результатом сольватации полимерных молекул. Сольватация весь-
весьма важна для всего процесса набухания, однако, на этой
стадии набухание еще не отличается существенно от обыч-
обычных процессов растворения.
Наиболее характерна следующая стадия набухания,
когда гель поглощает не 20—40%, а десятикратные коли-
количества растворителя, сильно увеличиваясь в объеме; на
204
этой стадии набухание происходит без заметного сжатия
общего объема и с крайне незначительным или нулевым теп-
тепловым эффектом. Эта вторая стадия набухания объясняет-
объясняется осмотическими явлениями. Благодаря более быстрому
проникновению молекул растворителя в полимер, чем мо-
молекул полимера в растворитель, и замедленной скорости
Рис. 78. Прибор для измерения давления набухания (по
Фрейндлиху и Позняку). Внизу—общий вид; вверху и
справа: разрез через трубку для набухания, т—нарезка
для соединения с манометром, к-—измерительный ка-
капилляр, п и пп—замазки, L—стеклянный цилиндр с
растворителем, Т—глиняный сосуд, R и g—стеклян-
g—стеклянные трубки, Q—набухающий гель
перегруппировки макромолекул, создается разность кон-
концентраций растворов внутри геля и в жидкости, приводя-
приводящая к соответствующим различиям в активности раство-
растворителя. При разделении раствора полимера и растворителя
полупроницаемой мембраной это различие в активности
растворителя проявляется в осмотическом давлении (см.
стр. 33), а при соприкосновении геля и растворителя —
в давлении набухания.
Фрейндлих и Позняк измеряли давление набухания в
приборе, изображенном на рис. 78. На дне пористого гли-
глиняного цилиндра Т находится гель (каучук, желатина);
растворитель из сосуда L проникает к гелю (или пленке)
и вызывает набухание. Измеряется давление (подаваемое
из баллона с сжатым воздухом через ртуть Hg), которое
необходимо для удержания геля при постоянном заданном
объеме. Было показано, что давления набухания достигают
очень значительных размеров; так, например, для 20%-но-
го геля каучука в бензоле—2,1 кг/см2, для 36%-ного студ-
у p.
?(/
35
30
•« 25
\ 20
QJ 14
1
10
V
/
/
/
/
f
j
f
ча во so wo по по то zoo гмгяозго
Количество желатины^ г на ЮОг воды
Рис. 79. Давление набухания желатины в воде
(при высоких концентрациях студия)
ня желатины —1,1 кг/см2, для 46%-ного студня желатины—
2,1 кг/см2 и т. п.
Таким образом, при поглощении растворителя в коли-
количестве, превосходящем сольватационное связывание, дав-
давление набухания еще составляет свыше 1 атм, постепенно
понижаясь до малых величин; при поглощении первых ко-
количеств растворителя давление набухания достигает де-
десятков атмосфер (рис. 79).
Неограниченное набухание сопровождается осмотиче-
осмотическим проникновением растворителя внутрь геля до пол-
полного растворения полимера; при наличии прочной простран-
206
ственной сетки (например, в вулканизованном каучуке)
проникновение растворителя продолжается, как указыва-
указывалось, до тех пор, пока эластическое напряжение набух-
набухшего геля не уравновесит давления набухания, после чего
гель остается в ограниченно набухшем состоянии. Скорость
достижения этого состояния вначале велика, потом она за-
замедляется приблизительно пропорционально приближению
системы к равновесному состоянию (Липатов).
В не очень концентрированных студнях (например, в
10%-ных студнях желатины) молекулярные цепи доста-
достаточно удалены друг от дру-
друга и слабо взаимодействуют
между собой. При неболь-
небольшом сжатии или растяже-
растяжении таких студней цепи,
оставаясь в том же взаим-
взаимном расположении в про-
пространственной сетке студ-
студня, легко изменяют свою
форму, благодаря своей
гибкости, и после снятия
деформирующего усилия
быстро восстанавливают
форму, что характеризует
упругие свойства студней.
Каргин, Зубов и Журкина
показали, что 10—20%-ные
w%
¦го%
0
Рис. 80. Деформация студней
желатины при различной ча-
частоте сжатия (при 20°)
студни желатины сохра-
сохраняют свои упругие свой-
свойства в широком интервале
частот деформации; из рис. 80 видно, что студни успевают
восстановить изменения формы и сохранить постоянство
размеров даже при частоте сжатия до 1000 раз в минуту
(/gv=3). При нагревании студня желатины внутренние
водородные связи в узлах сетки в определенном темпера-
температурном интервале разрываются и пространственная струк-
структура, ранее фиксировавшая взаимное расположение
молекул, распадается, а цепные молекулы получают воз-
возможность взаимного перемещения при действии дефор-
деформирующего усилия. Однако перемещение длинных цепных
молекул уже не может происходить слишком быстро, и при
высоких частотах деформации изменения формы не сразу
устраняются.- В результате появляются остаточные изме-
207
нения формы, характеризующие переход от упругих свойств
к пластическим деформациям, зависящим от скорости сжа-
сжатия (рис. 81; ср. рис. 80). Того же эффекта можно достиг-
достигнуть при деформации студня желатины в растворах 5 М
мочевины, 2 М KCNS, 2 М KJ, в которых разрыв межцеп-
межцепных водородных свя-
связей происходит уже
при комнатной темпе-
температуре.
Таким образом,
при определенных ус-
условиях (температуры,
состава среды) можно
вызвать разрушение
фиксированного вза-
взаимного расположения
цепных молекул в
пространственной сет-
сетке и их взаимное пе-
перемещение. Этот про-
цесс перехода студня
в состояние раствора
полимера называют
-Ю
о
10 20 30
Рис. 81. Температурная зависимость
деформации 30%-ного студня желати-
желатины при различной частоте сжатия: 1 —
1000; 2—100; 3—10, 4—1 колеб/ мин
плавлением студня.
Таким образом, раз-
различие между студнем
и раствором полимера
той же концентрации
заключается в наличии или отсутствии сетчатой структуры.
При наличии пространственной сетки (в студне) взаимное
перемещение молекул исключено и студень, если концен-
концентрация не очень высока, обладает лишь упругими свой-
свойствами, похожими на свойства твердого тела; при разруше-
разрушении пространственной сетки (в растворе) появляется воз-
возможность необратимого перемещения молекул или течения.
Потеря текучести и наличие упругих свойств являются
характерными свойствами гелей; при этом для устранения
текучести достаточно сравнительно небольшого количества
межцепных связей. Очень большое значение имеет природа
этих связей; если узлы сетки образованы водородными свя-
связями или взаимодействием диполей, то достичь плавления
студня легко нагреванием или изменением состава среды
(см. выше), если же цепи связаны между собой химически-
208
ми связями (вулканизация, дубление), то студень может
полностью утратить способность к плавлению.
Переход раствора полимера в состояние студня при той
же концентрации называется застудневанием, например,
при охлаждении 5%-ного раствора желатины он превра-
превращается в студень. Застудневание отчетливо проявляется в
прекращении броуновского движения в студне, оно не
сопровождается заметным тепловым эффектом или измене-
изменением объема, что объясняется малым числом образую-
образующихся межцепных связей. Влияние электролитов на ско-
скорость застудневания зависит от их положения в лиотроп-
ном ряду (см. стр. 185), начиная от сульфатов, ко-
которые наиболее сильно ускоряют застудневание. Напро-
Напротив, лиотропный ряд влияния электролитов на плавление
студней имеет обратную последовательность, так как наи-
наиболее сильное «расплавляющее» действие оказывают ро-
даниды и йодиды (см. стр. 208). Ввиду замедленной ско-
скорости установления равновесия в растворах полимеров
(см. стр. 171), их нагревание и охлаждение может со-
сопровождаться гистерезисом ряда свойств — вязкости, опти-
оптического вращения (мутаротация) и др., изменение которых
обычно отстает от скорости изменения температуры раство-
растворов. Интересно, что слишком сильное охлаждение не уско-
ускоряет, а тормозит процесс застудневания, благодаря за-
замедлению скорости образования межцепных связей. На-
Например, по Хоку, 1,5 %-ный раствор желатины в глицерине
застудневает при комнатной температуре в несколько дней,
а при 0° остается в течение нескольких недель в жидком
состоянии. В эластичных гелях при определенной концен-
концентрации полимера и электролитов застудневание раствора
может происходить в изотермических условиях, по типу
тиксотропных превращений. Разбавленный студень жела-
желатины можно получить тиксотропным, подобно гелю гидро-
гидроокиси железа; тиксотропными свойствами обладает также
протоплазма при некоторых клеточных процессах — во
время деления клеток, при возбуждении клетки, при дей-
действии наркотиков и др.
Процессы застудневания и возникновения тиксотропии
в растворах полимеров по существу аналогичны соответ-
соответствующим процессам при коагуляционном структурооб-
разовании в лиофобных золях, хотя в растворах полимеров
они являются результатом понижения растворимости. Суль-
Сульфаты, понижающие растворимость белков и способствую-
209
щие их высаливанию (см. стр. 184), ускоряют также
застудневание растворов желатины.
При значительном понижении растворимости полимеров
происходит расслоение раствора на две фазы (см. стр.
184). Однако если застудневание предшествует достиже-
достижению равновесия, то процесс расслоения продолжается в
геле, приводя к его разделению на более плотный осадок и
слой жидкости. Этот важный процесс самопроизвольного
расслоения геля называется синерезисом (Липатов, Фрейн-
Фрейндлих и др.). Синерезис имеет практическое значение в
-связи с явлениями расслоения студней пироксилина, ви-
вискозы, агара, разделения сгустка крови при ее свертыва-
свертывании и др. Аналогичные процессы уплотнения остатка геля
и отделения жидкости наблюдаются также в гелях кремне-
кислоты, гидроокиси железа, гидроокиси меди и др., в
результате коагуляционного структурообразования. Вслед-
Вследствие уплотнения гелей при синерезисе, их прочность повы-
повышается, и они теряют тиксотропные свойства.
ГЕЛИ ПОЛИЭЛЕКТРОЛИТОВ И БЕЛКОВ. МЕМБРАНЫ
Важную группу гелей составляют гели с большим коли-
количеством ионогенных групп, в том числе гели различных по--
лиэлектролитов, белков, в которых большую роль играют
электрохимические явления. Они приобретают особое зна-
значение в гелях полиэлектролитов, образованных гибкими
макромолекулами с высокой плотностью зарядов. В этом
случае изменение степени ионизации ионогенных групп
приводит к значительным изменениям объема геля, обус-
обусловленным электростатическим отталкивательным взаимо-
взаимодействием одноименно заряженных групп. Так, например,
Качальский показал, что в гелях или волокнах полиакри-
полиакриловой кислоты, содержащих по одной СОО~-группе в
каждом звене цепи, путем смещения рН или замены Na-
солей на менее диссоциированные Ва-соли, можно вызвать
обратимые удлинения в 8—10 раз; аналогичные опыты про-
производились на гелях полиальгиновой кислоты. По мнению
Кирквуда и Риземана, подобные явления могут иметь место
при мышечном сокращении, в результате процессов фермен-
ферментативного фосфор ил ирования идефосфорилирования. Заме-
Замечательно, что в описанных процессах происходит непосред-
непосредственный переход изменений химической энергии в меха-
механическую работу (хемомеханический процесс), который
210
несомненно лежит в основе мышечного сокращения, хотя
его конкретный механизм еще нельзя считать выясненным.
Большое значение имеют гели полиэлектролитов в
ионообменной хроматографии (стр. 126). В этом случае
обратимое набухание и сжатие ионита при обмене ионов
регулируют количеством межцепных химических связей;
например, вводя 6, 10, 17 или 23% дивинилбензола в поли-
сульфостирол (см. рис. 44), можно регулировать набуха-
набухание смолы и уменьшить объем геля, приходящийся на1 моль
сульфогрупп, соответственно от 300 до 100, 70 или 50 мл;
одновременно изменяются среднее расстояние между ионо-
генными группами, их электростатическое взаимодействие
и активность растворителя. Степень набухания определяет
для ряда органических ионов интенсивность ионного взаи-
взаимодействия и возможность проникновения в сетку геля и,
тем самым, избирательность поглощения, что имеет боль-
большое значение для хроматографии. Избирательность пог-
поглощения обычно характеризуют коэффициентом избиратель-
избирательности
Кп=^:^-, (IX. 2)
где п — молярное содержание катионов / и 2 в смоле /
и в растворе о. Теория обмена ионов на набухающих иони-
тах разрабатывалась Самсоновым, Грегором и др.; в част-
частности, Бреслер и Самсонов применили набухающие иони-
ты для выделения и очистки стрептомицина.
Доннан и Гугенгейм исследовали ионное равновесие
между набухающим ионитом и внешним электролитом
и установили следующую зависимость между отношением
Ка произведений активности ионов (см. стр. 123) и
давлением набухания в ионите тгг—тго:
RT\nKa = (*/ — *о) (Уа+ — Vb+Ь + const, (IX.3)
где (УлН—Vb +)i— разность парциальных моляльных объе-
объемов обменивающихся ионов в ионите.
Дополнительное давление набухания (^ — тго) в (IX. 3),
обусловленное доннановским распределением ионов, было
значительно ранее другим путем рассчитано Проктером и
Вильсоном для объяснения более сильного набухания бел-
белковых студней (желатины) в кислых и щелочных средах.
В студнях белков эти эффекты слабее, чем в невулканизо-
211
ванных гелях полиэлектролитов, ввиду меньшей плотности
расположения зарядов (в белках одна СОО-группа при-
приходится на 7—9 аминокислотных остатков), однако, в
них набухание, подобно осмотическому давлению, прохо-
проходит через четкий минимум в изоточке. Таким образом, к
начальному изоэлектрическому набуханию, механизм кото-
которого описан выше (стр. 202—205), в белковых студнях добав-
добавляется дополнительное набухание при изменении рН;
величина этого набухания, как указывалось, ограничи-
ограничивается упругим напряжением в набухшем студне. Теория
7000
6000
|S 5000
Is чооо
^5 2000
WOO
0
5 6
/7Н
8 9 10 11 1Z
Рис. 82. Набухание желатины при различных рН (/—
в отсутствие других электролитов, кроме НС1 и NaOH;
2—в присутствии 0,1 М NaCl)
Проктера и Вильсона прилагалась ими для объяснения
увеличения оводненности коллагена (нажор дермы в ко-
кожевенном производстве) в кислых и щелочных средах.
А. Михайлов показал, что в этом случае, кроме осмотиче-
осмотических явлений, необходимо также учесть дополнительную
гидратацию на заряженных группах белка и растяжение
цепей белка под влиянием одноименных зарядов. В при-
присутствии нейтральных солей, уменьшающих доннановский
эффект (см. стр. 124), явления дополнительного кислотно-
щелочного набухания значительно ослабляются (рис. 82).
Большое значение имеют эластичные гели в форме тон-
тонких мембран. В живом организме различные белковые,
белковолипоидные и другие мембраны способствуют избира-
избирательному поглощению и переносу различных веществ, про-
212
странственной организации процессов обмена веществ, ис-
использованию и превращению различных форм энергии.
Достаточно указать, что общая поверхность стенок крове-
кровеносных сосудов в организме человека достигает 6300 м2
(гго Крогу), не считая оболочек эритроцитов, клеточных
ядер, клеток, слизистых оболочек внутренних органов
и др. Некоторые свойства этих мембран напоминают просто
свойства тонкой неводной фазы, разделяющей две водные
фазы; например, проницаемость клеток для различных
неэлектролитов приблизительно пропорциональна их раст-
растворимости в масле; она возрастает, по Овертону, с удли-
удлинением углеводородной цепи или с увеличением числа ал-
кильных групп и падает при введении полярных групп
(гидроксильных, карбоксильных, аминогрупп), обращен-
обращенных к воде (см. главу четвертую); напротив, живые клетки
;адохо пропускают ионы электролитов. Однако двухмер-
двухмерный слой масла может быть лишь отдаленной моделью гомо-
ггенных мембран, действие которых основано на избиратель-
избирательной растворимости; строение этих мембран в организме
гораздо .сложнее, часто имеет многослойный или мозаич-
мозаичный характер и, кроме того, избирательный перенос ве-
веществ тесно связан с сопряженными процессами обмена
: веществ, поставляющими энергию для накопления веществ
в клетке при более высоких концентрациях, чем в окру-
окружающей среде. Фактически, мембраны в организме сле-
„дует рассматривать . как сочетание избирательно-раство-
избирательно-растворяющих (гомогенных) и пористых мембран (Булл); послед-
последние имеют также наибольшее техническое значение.
Пористые мембраны, к числу которых от-
относятся различные коллоидные, керамические, пергамент-
-иые и разработанные в последнее время ионообменные мем-
'браны, обладают в принципе ситовым механизмом дей-
¦ствия. Однако задержка ими различных растворенных
частиц объясняется не только величиной, но также ад-
сорбируемостью или знаком заряда частиц. Пористые мемб-
мембраны можно разделить на две основные группы — диали-
зующие мембраны и молекулярноионные сита.
Диализующие пористые мембраны применяются при диа-
диализе, ультрафильтрации, электродиализе и др. Они обла-
• дают пористостью, примерно, от 2 м\ь до 2\ь. Размер пор
коллодийных мембран регулируют подбором состава раст-
растворителей и условий испарения растворителя из пленки;
.добавление хороших растворителей к исходному раствору
213
коллодия понижает пористость образующейся мембраны (Ил-
форд). Для характеристики пористости мембран измеряют
зависимость от давления скорости протекания воды через
мембрану, смоченную изобутиловым спиртом или другой
несмешивающейся с водой жидкостью с небольшим меж-
межфазным поверхностным натяжением (Эрбе). Протекание
30
20
Ю
Л
А
л
f
i
¦
'¦4
¦-"
V/
\\\
II1
A
—
5 10 15 Z0 25 30 3S ?0
Р2
Рис. 83. Зависимость протекания воды через
мембрану от давления (по Жукову)
воды начинается при давлении Pi (рис. 83) через поры наи-
наибольшего размера, затем, по мере включения более уз-
узких пор, скорость течения (сплошная кривая) возрастает
быстрее, чем повышается давление, и, наконец, после дав-
давления Р%, при котором вода протекает через все поры мем-
мембраны, скорость течения пропорциональна приложенному
давлению. Отношение (AS/АР) в интервале давлений
Р1Р2 характеризует кривую распределения размеров пор
в мембране, а абсолютная величина пор рассчитывается из
величины давления Р по уравнению (II, 14 и IV.4).
214
Применяя ряд коллодийных мембран с градуированной
по Илфорду и все уменьшающейся пористостью, Грабар
измерял наименьший размер пор, при котором через мембра-
мембрану в процессе ультрафильтрации проходил раствор бел-
белков- или вирусов, и определял таким путем их размеры. Этот
метод применялся для выделения и изучения размеров ряда
вирусов. Использование диализа и ультрафильтрации
для очистки коллоидных систем описывалось во второй
главе.
Проникновение веществ через мембраны под действием
электрического тока называется ионофорезом и широко
используется в медицине для введения в организм через
кожу лекарственных препаратов. Этим путем вводят хинин,
новокаин, салицилат, ионы кальция, цинка, ртути, йода,
а также создают кожные ионные депо длительного действия.
Большое значение имеет применение пористых мембран
для электродиализа (см. стр. 38). При достаточно
высокой пористости мембран числа переноса ионов в мем-
мембранах мало изменяются по сравнению с таковыми в водных
растворах; так, например, Григоров показал, что число
переноса С1~-ионов в коллодийной мембране с размером
пор выше 65 м }х равно 0,504, как и в свободном растворе.
В этом случае мембрана является электрохимически не-
неактивной, хотя коллодийные мембраны, как и большинство
других мембран, приобретает в растворе электролитов
отрицательный заряд (положительный заряд приобретают
белковые мембраны). Электрохимически неактивные мем-
мембраны лишь отделяют среднюю камеру от электродных ка-
камер, препятствуя перемешиванию очищаемого раствора и
продуктов электролиза, уносимых с водой.
Однако при уменьшении размера пор коллодийной
мембраны до 9,7 и 0,9 м\ъ, числа переноса С1~-ионов в
мембране уменьшаются, соответственно, до 0,42 и 0,20
(Григоров). Такую мембрану выгодно поместить на катод-
катодной стороне электродиализатора, так как она увеличивает
число переноса катионов, т. е. является электрохимически
активной мембраной. Напротив, электрохимически-актив-
электрохимически-активную положительную мембрану выгодно поместить на анод-
анодной стороне электродиализатора, где она увеличивает чис-
число переноса анионов. В результате процесс электродиализа
может быть ускорен, и использование проходящего тока
значительно улучшено. Жуков и Маркевич выяснили ус-
условия регулирования рН и хода очистки в средней камере
215
путем правильного подбора электрохимически активных
мембран и состава жидкости в электродных камерах; при
неправильном расположении активных мембран в средней,
камере вместо очистки раствора от электролитов происхо-
происходит повышение их концентрации. Изменение числа перено-
переноса ионов А в мембране является мерой ее электрохимиче-
электрохимической активности. В предельном случае отрицательно за-
заряженная мембрана проницаема только для катионов, а
положительно заряженная мембрана проницаема только
для анионов. В таких идеально электрохимически актив-
активных мембранах (Жуков) разница между числами переноса
доходит до единицы (для каждой из мембран числа переноса,
соответственно, равны 1 и 0).
Мембраны с идеальной ионной избирательностью были
практически получены при достаточно малой величине пор;
они относятся к классу молекулярных или ионных сит и
обладают рядом особенностей. Зольнер получал электро-
электроотрицательные избирательные мембраны на основе окис-
окисленного коллодия или путем введения сульфированного
полистирола в раствор коллодия, из которого изготовляют-
изготовляются мембраны; эти мембраны имеют толщину всего около
20—40 }х. Для создания окисленных групп в мембранах
их подвергают действию ионизирующей радиации. Уилли
и Патнод готовили мембраны прессованием тонкой смеси
катионита и инертной смолы в виде дисков толщиной от
0,5 до 4 мм, но электрическое сопротивление таких мембран
было выше. Электроположительные избирательные мембра-
мембраны Зольнер готовил путем адсорбции основных белков
протаминов на коллодийных мембранах, а Синха — прес-
прессованием тонкой смеси анионита и полистирола при 120—
130° и давлении 280 атм. Ионообменные мембраны можно
также приготовить из каучуковых пленок путем их хлори-
хлорирования и последующего аминирования.
Одной из важных областей использования избиратель-
избирательных мембран является применение их в качестве мембран-
мембранных электродов. При соприкосновении двух растворов с
различной концентрацией одного и того же электролита на
их границе, как известно, возникает диффузионный потен-
потенциал; тот же потенциал образуется при разделении обоих
растворов мембраной с достаточно широкими порами,
полностью лишенной ионной избирательности (электро-
(электрохимически неактивной). При повышении избирательности
мембраны измеряемый концентрационный потенциал от-
216
клоняется от диффузионного, в соответствии с изменением
чисел переноса Д в мембране. Наконец, при идеальной
ионной избирательности мембраны концентрационный по-
потенциал точно соответствует разности потенциалов между
дбумя растворами электролита, измеряемой при помощи
обратимых электродов. Действительно, уравнение диффу-
диффузионного потенциала
и '
г-.
пг
(IX. 4)
где и+ и и~— подвижности катиона и аниона, при «+=
= 1,0 и иг—0 (в случае электроотрицательной избиратель-
избирательной мембраны), переходит в обычное уравнение концентра-
концентрационной цепи
? = S-ln — (IX. 4а)
Этим путем удобно измерять активность многих ионов в раст-
растворах (в том числе в биологически важных системах), для
которых не существует специальных обратимых электро-
электродов, например, анионов F~, NOi", С1ОГ. СНзССО~, JO^,
катионов Li+, K+, Na+, Rb+, Cs+, Mg++, Ca++, ЫН4+и др.;
кроме того, можно производить различные электрометри-
электрометрические титрования, измерения доннановского распреде-
распределения ионов, биоэлектрических потенциалов и др. Изме-
Измерения скорости переноса радиоактивных изотопов катио-
катионов Na, Zn, Cd в избирательных мембранах использова-
использовались для определения их коэффициентов диффузии.
Интерес представляют также опыты по применению
избирательных мембран для фильтрации солей из раство-
растворов. В избирательных мембранах в стенках пор содержится
большое количество фиксированных ионогенных групп с
соответствующими компенсирующими ионами; таким об-
образом, избирательные мембраны являются, по существу,
ионообменными мембранами. При достаточно узких порах
ионы электролитов, имеющие одинаковый знак с фикси-
фиксированными зарядами мембраны, вследствие электростати-
электростатического отталкивания, не смогут пройти через поры, по-
поэтому, ввиду необходимости сохранения электронейтраль-
электронейтральности, задержатся и сопутствующие противоположно заря-
заряженные ионы электролитов, если не считать небольшой
части их, которая сможет проникнуть в мембрану в про-
процессе ионного обмена. По окончании обмена компенсирую-
1. ^ 15 «Коллоидния химия»
217
щих ионов в мембране, основное количество электролита
задерживается мембраной: через нее проходит только
вода. Например, Уилли и сотрудники фильтровали мор-
морскую воду и 0,1 н. NaCl через катионитные избиратель-
избирательные мембраны «пермаплекс С-10», толщиной 0,6 мм, под
давлением около 100 кг/см2, и получили значительное
понижение солености воды.
Для химикофармацевтической промышленности пред-
представляет интерес то, что соли чувствительных к изменениям
рН органических кислот могут быть превращены в свобод-
свободные кислоты путем гидролиза на избирательных мембранах
"или диализа против кислоты без соприкосновения с кисло-
кислотой, которое неизбежно при реакциях осаждения (Зольнер).
Избирательные мембраны, по-видимому, являются также
важной составной частью природных мембран в организме.
ДИФФУЗИЯ В ГЕЛЯХ
Ввиду сетчатой структуры гелей, диффузия малых моле-
молекул и ионов в гелях мало отличается от их свободной диф-
диффузии; соответственно, электропроводность растворов мало
изменяется при их застудневании. Однако диффузия круп-
крупных молекул или диффузия в высококонцентрированные
студни, естественно, встречает затруднения. В производ-
производственных процессах крашения и дубления проникновение
красителей или таннидов внутрь геля в значительной мере
лимитирует скорость всего процесса. При дублении необ-
необходимо избегать образования пленки на поверхности кол-
коллагена (задуба), препятствующей дальнейшему проникно-
проникновению дубителя; с другой стороны, появление пленки при
действии таннидов на белковые вещества используется в
медицине при обработке ран (адстрингентные вещества).
Путем периодического сжатия хромированного коллагена
в растворе таннидов с частотой, соответствующей скорости
восстановления формы волокон коллагена (эффект «сжа-
«сжатия губки»), скорость диффузии дубителя в коллаген можно
ускорить в несколько раз (Пасынский и Тонгур).
Благодаря тому, что в гелях отсутствуют перемешиваю-
перемешивающие конвекционные потоки, реакции осаждения в гелях
имеют характерные особенности. Лизеганг показал, что
при диффузии вещества (например, AgNCb) в студень, со-
содержащий другое вещество (К2СЮ4), дающее с первым не-
нерастворимый осадок, наблюдаются явления периодического
218
f. осаждения (кольца Лизеганга), вместо обычного сплошного
осаждения. Подобные слоистые структуры появляются, в
Рис. 84. Срез желчного камня. Внутри видны кристал-
кристаллы холестерина, по периферии кольца Лизеганга
частности, при отложении холестерина (рис. 84) в печени и
других органах в виде камней.
Выводы
Гели — образуемые жесткими коллоидными частицами
(хрупкие гели) или гибкими макромолекулами (эластичные
гели или студни) пространственные структуры, обычно за-
заполненные растворителем. Они отличаются от растворов
упругостью формы и отсутствием текучести. Хрупкие гели
не набухают и способны к неспецифическому поглощению
паров жидкостей в результате образования адсорбционных
сольватных слоев и капиллярной конденсации. Эластичные
гели могут сильно набухать при избирательном поглощении
15 «Коллоидная .химия»
219
жидкостей. Набухание студней является результатом соль-
сольватации и осмотического поглощения растворителя; неогра-
неограниченное набухание — лишь первая стадия растворения;
ограниченное набухание происходит при ограниченном
смешении или наличии в полимере прочной пространствен-
пространственной сетки. Сольватационная стадия набухания сопровож-
сопровождается положительным тепловым эффектом, уменьшением
общего объема, понижением энтропии растворителя, низ-
низкой начальной упругостью пара и др. Осмотическое пог-
поглощение растворителя проявляется в давлении набухания.
Разрыв связей в узлах сетки при нагревании или изменении
"состава среды приводит к плавлению студня, образование
сетки связей в условиях понижения растворимости — к
застудневанию, иногда с последующим синерезисом. В ге-
гелях полиэлектролитов и белков значительные дополни-
дополнительные объемные изменения происходят при изменении
рН. Гели полиэлектролитов с регулируемой степенью на-
набухания имеют большое значение для хроматографии.
Важное биологическое и техническое значение имеют
эластичные гели в форме мембран. Мембраны разделяются
на гомогенные (с избирательной растворимостью) и по-
пористые (с ситовым механизмом действия). Изменение раз-
размеров пор изменяет электрохимическую активность мем-
мембран; избирательные мембраны значительно повышают
эффективность электродиализа; узкопористые мембраны с
высокой ионной избирательностью применяются в качестве
мембранных электродов и др.
Вопросы
1. Чем отличаются хрупкие гели и студни?
2. Какой вид имеют изотермы адсорбции-десорбции паров
для хрупких гелей и чем они объясняются?
3. Каковы характерные отличия неограниченного и ограничен-
ограниченного набухания студней?
4. В чем проявляется сольватационная стадия набухания?
5. Что такое давление набухания, чем оно объясняется, как
измеряется?
6. Как влияют электролиты на плавление и застудневание?
Какова природа синерезиса студией?
7. Какие объемные изменения происходят в гелях полиэлек-
полиэлектролитов и белков при изменении рН?
8. Каково значение электрохимической активности мембран
при электродиализе?
9. Опишите методы получения и применения мембран с высо-
высокой ионной избирательностью.
10. Каково биологическое значение гелей и мембран?
220
Глава десятая
ТВЕРДЫЕ ПОЛИМЕРЫ
Твердые полимеры—каучуки, целлюлоза и ее эфиры, бел-
белки, новые синтетические полимеры и пластмассы—составляют
обширный мир веществ, имеющих исключительно большое
значение в биологии и в народном хозяйстве. Все твердые
полимеры образованы цепными макромолекулами, с молеку-
молекулярным весом от нескольких тысяч до нескольких миллио-
миллионов; химический состав и структура макромолекул
лежат в основе замечательных свойств полимерных мате-
материалов.
ОСНОВНЫЕ СОСТОЯНИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ
ВЕЩЕСТВ
Линейные макромолекулы состоят из большого коли-
количества последовательно соединенных химическими связями
мономеров или звеньев цепи, положение ко-
которых в пространстве ограничено соседними
звеньями. При свободном вращении каждого
звена, например, около связи С—С, оно опи-
описывало бы полный конус, определяемый ва-
валентным углом (тг—а) (рис. 85). Однако из
рис. 85 видно, что расстояния между атома-
атомами С\ и Сз или Сх и С4 изменяются в раз-
различных положениях, и поэтому некоторые по-
положения могут быть энергетически более
выгодными. Для перехода из одного равно-
равновесного положения в другое требуется затрата
энергии Ео, характеризующая потенциальный
Рис. 85. Вра-
Вращение звень-
барьер вращения; чем выше Ео, тем более ус- ев в цепочке
15*
221
тойчивым является данное расположение звеньев в цепи.
Величины Ео для некоторых соединений приведены в табл. 18.
Потенциальные барьеры вращения
(по Волькенштейну)
Таблица 18
Молекула.
Н3С — СН3
Н8С - СН2 -СН3
НС - (СН3)з
С(СН3L
Н,С - С(СН3K
Связь, около которой
происходит вращение
НзС — СНд
Н3С — СН2
Н3С - СН
Н3С — С
Н3С - С =
1
Е0,к(м/моль
2750
3400
3870
4700
1800
В процессе теплового движения взаимное расположение
звеньев изменяется, причем эти изменения происходят
тем легче, чем выше энергия теплового движения. Поэ-
Поэтому высоту потенциального барьера Ео надо оценивать
относительно тех температурных условий, при которых
находится полимер. Вероятность изменения положения
звена при барьере вращения Ео пропорциональна е-Е°>кТ.
При обычных температурах величина kT близка к
600 кал/моль и значения Ео, приведенные в таблице,
соответствуют около 3—8 кТ\ при более высоких темпера-
температурах отношение EJtcT меньше и степень свободы вращения
звеньев возрастает. При значительной свободе вращения
(низкие Ео, высокие кТ) легко видеть из рис. 85, что поло-
положение атомов Сз, С4 и, тем более, последующих атомов
мало зависит от положения исходного звена С1С2, что
проявляется в сильной изогнутости и гибкости цепи; прак-
практически цепи с Ео~2>—4 кТ еще достаточно гибкие. Если
же Е0^кТ, звенья совершают преимущественно колеба-
колебательные движения около положения равновесия и взаим-
взаимная зависимость положения звеньев может охватывать
значительные участки цепей; в этом случае цепь является
более вытянутой и жесткой. При повышении температуры
отношение Ь0/кТ уменьшается и цепи становятся более
гибкими; так, например, молекулы полистирола, довольно
жесткие при комнатной температуре, становятся гибкими
при 80°.
222
Если звено при вращении может занимать не одно,
а несколько определенных положений равновесия с раз-
различными потенциальными барьерами вращения Ео, то
полимер в каждом из положений можно рассматривать как
поворотный изомер; в этом смысле Волькенштейн считает
полимеры равновесными смесями поворотных изомеров.
Они отличаются, однако, от цис- и транс-изомеров этиле-
этиленовых соединений в десятки раз меньшей высотой барьеров
и высокой скоростью взаимного превращения (до 1010
раз в 1 сек.); при этом, поворотные изомеры с наиболее
высокими Ео преобладают. Для простоты в дальнейшем мы
рассматриваем только свободное вращение звеньев (гиб-
(гибкие цепи), или ограничение вращения колебаниями около
одного положения равновесия (жесткие цепи).
В полиэтиленовых цепях величины Ео сравнительно
невелики. Наличие в молекулах полярных групп — ОН,
¦—СООН, —CN, —С1, сильно поляризующихся или боль-
больших по размеру групп заметно ограничивает свободу вра-
вращения и повышает жесткость цепей; так, например, поливи-
поливиниловый спирт или полихлорвинилы обладают сравнитель-
сравнительно жесткими цепями при комнатной температуре. Жесткие
цепи содержат также белки и нуклеиновые кислоты.
Не меньшее значение для величины Ео имеют межмоле-
межмолекулярные взаимодействия полимера с растворителем или
пластификатором (см. стр. 242), или полимерных цепей меж-
между собой. Таким образом, гибкость цепей полимеров зависит
от многих факторов —¦ от химического строения цепи,
количества и природы функциональных групп, темпера-
температуры, природы растворителя, межцепного взаимодействия
и др.
По мере понижения температуры вращение звеньев
все более ограничивается, вследствие увеличения отно-
отношения (Е01кТ) и усиления межмолекулярного взаимодей-
взаимодействия, пока, наконец, при некоторой температуре Tg вра-
вращательное движение звеньев не перейдет полностью в
колебания звеньев около фиксированных положений рав-
равновесия. Как известно, этот процесс происходит и при ох-
охлаждении низкомолекулярных веществ, но в этом случае
вместо звеньев цепи имеются отдельные молекулы, которые
при понижении температуры могут уложиться в кристал-
кристаллическую решетку; в ее узлах и происходят колебательные
движения; поэтому температура TgB низкомолекулярных
кристаллизующихся телах совпадает с температурой их
223
кристаллизации. Однако имеется много низкомолекуляр-
низкомолекулярных веществ, в которых при охлаждении настолько быстро
возрастает внутренняя вязкость, что передвижение и вра-
вращение молекул затормаживается еще до образования крис-
кристаллической решетки. Такие вещества называются аморф-
аморфными; к их числу принадлежат глицерин, некоторые
смолы, стекло, поэтому для аморфных тел температура
Tg называется температурой стеклования. В аморфных
телах Tg соответствует зафиксированной структуре жид-
жидкости. При температуре T<Tg аморфные тела не находят-
находятся в состоянии термодинамического равновесия; их устой-
устойчивость является вынужденной, обусловленной высокой
внутренней вязкостью (составляющей до 1011 пуаз и выше).
При нагревании аморфного низкомолекулярного вещества
до температур 7"> Tg межмолекулярные связи разрываются,
вязкость резко падает и аморфное тело переходит в жидкое
состояние; обычно эти переходы около Tg не происходят
очень резко и охватывают некоторый температурный интер-
интервал размягчения. Таким образом, в аморфных низкомо-
низкомолекулярных веществах существует два состояния — стек-
стеклообразное и жидкое, переход между которыми отмечен
температурой стеклования Tg.
В высокомолекулярных веществах охлаждение до тем-
температур, при которых сохраняются только колебания звень-
звеньев около положений равновесия, также соответствует обыч-
обычно состоянию их застеклования, а не кристаллизации.
В полимерах при охлаждении резко возрастает внутренняя
вязкость, а укладка длинных цепей в правильную решетку
встречает дополнительные затруднения (см. ниже); поэтому
кристаллизация полимеров при охлаждении наблюдается го-
гораздо реже, чем их переход в застеклованное состояние, в ко-
котором в полимере не только цепи, но и все звенья находятся в
фиксированном состоянии (сохраняются лишь колебательные
движения звеньев), деформация материала сильно затруд-
затруднена, он становится неэластичным и хрупким, как обычное
стекло; например, известно, что каучук при замораживании
теряет свою способность к растяжению и становится хруп-
хрупким. Так как морозостойкость полимерных материалов
заключается в сохранении ими эластичности при низких
температурах, то температура стеклования определяет мо-
морозостойкость эластичных материалов и имеет большое тех-
техническое значение. Переход полимеров в застеклованное
состояние также характеризуется температурами Tg; тех-
224
нические пределы морозостойкости обычно на несколько
градусов выше Tg. Некоторые значения температур стекло-
стеклования Tg высокомолекулярных веществ (по Бойеру и
Спенсеру) приведены ниже. Из этих данных видно, что
у'полимеров с жесткими цепями значения Tg выше, чем
у неполярных полимеров с гибкими цепями.
о
Полимер Т
Натуральный каучук —73
Стиролбутадиеновый каучук —61
Поливинилацетат ... . . . . 28
Этилцеллюлоза . . 43
Найлон 47
Нитроцеллюлоза 52,6
Пслиметилметакрилат 57—68
Полистирол 81
Поливиниловый спирт 85
Полиакриловая кислота ..... ... 80—95
При нагревании полимера до Ty-Tg звенья полимера
восстанавливают подвижность и свои вращательные дви-
движения, но благодаря химической связи многих звеньев
в макромолекуле проявляется существенное отличие от
низкомолекулярных веществ. В определенном интервале
температур сумма межмолекулярных связей по длине цепи
остается настолько значительной, что цепи не могут пере-
перемещаться друг относительно друга и их взаимное располо-
расположение остается неизменным. В то же время цепи, благодаря
подвижности звеньев или отдельных участков цепей,
приобретают гибкость. В этом состоянии полимерный ма-
материал обладает высокоэластическими свойствами, он лег-
легко деформируется и способен к значительным удлинениям.
Следует заметить, что различие в поведении макромолеку-
лярных цепей и в поведении их звеньев, впервые установ-
установленное в работах Каргина и сотрудников, вообще является
одной из характерных особенностей полимерных материалов,
обусловливающей многие их интересные свойства.
Лишь при дальнейшем повышении температуры поли-
полимера до некоторой температуры Tf межмолекулярное вза-
взаимодействие цепей настолько ослабляется, что они полу-
получают возможность взаимного смещения или изменения их
взаимного положения; это состояние соответствует течению
полимера и называется вязкотекучим, а величина Tf—
225
температурой текучести. Таким образом, аморфные высо-
высокомолекулярные материалы в отличие от низкомолекуляр-
низкомолекулярных веществ характеризуются не двумя, а тремя состоя-
состояниями: стеклообразным, высокоэластическим и вязкоте-
кучим, соответственно разделенными температурами Tg
и Tf (Каргин, Александров); эти состояния, конечно, не
-80
О
то
zoo °с
Рис. 86. Зависимости деформации S поли-
изобутилена с различным молекулярным
весом от температуры. Начало резкого
перегиба кривой соответствует температуре
Т/ (по Каргину и Соголовой) /—число
звеньев п в молекуле 102; 2—«=1200-
3—п= 10400; 4— я=28000; 5—«=62500
следует смешивать с агрегатными состояниями тел — твер-
твердым, жидким и газообразным.
Температура Tg определяет при нагревании восстанов-
восстановление подвижности и вращательного движения звеньев
или отдельных участков цепи и поэтому почти не зависит
от общей длины цепи; напротив,.температура Tf соответст-
соответствует началу движения молекулярных цепей и зависит от
их общей длины (рис. 86). На этом основании Каргин и
Слонимский показали, что величина интервала (Tf—Т )
должна быть связана с молекулярным весом полимера; на-
например, для полиизобутилена
где п — число звеньев в молекуле. Этот метод интересен
тем, что позволяет определить молекулярный вес полимера
226
без его растворения. Измерения температурной зависимос-
зависимости деформации в широком интервале температур (рис. 86),
называемых термомеханическими кривыми (Каргин), яв-
являются важным методом исследования полимеров.
ОСНОВНЫЕ ТИПЫ МАКРОМОЛЕКУЛ
Линейные гибкие макромолекулы. Способность моле-
молекулярных цепей изменять свою конфигурацию в зависимос-
зависимости от внешних условий, т. е. гибкость или жесткость этих
цепей, является кардинальной характеристикой макромо-
макромолекул, определяющей свойства полимерных систем. Раз-
Различие в поведении гибких и жестких частиц проявляется,
как указывалось, в электрохимических свойствах (глава пя-
пятая), в термодинамических свойствах растворов полимеров
(глава восьмая), в молекулярно-кинетических свойствах
коллоидных систем (главы вторая и восьмая), в свойствах
гелей (глава девятая) и др. Это различие связано и с основ-
основными характеристиками структуры и физикомеханичес-
кими свойствами полимерных материалов. Как уже указы-
указывалось, гибкость и жесткость макромолекул являются
относительными характеристиками, зависящими от ряда
внешних условий, прежде всего, от температуры; однако,
применительно к обычному интервалу средних температур,
полимеры с гибкими и жесткими макромолекулами доста-
достаточно отчетливо различаются между собой; влияние дру-
других факторов (пластификации, скорости деформации) опи-
описано ниже (стр. 242—251).
Линейные гибкие макромолекулы лежат в основе та-
таких важных полимерных материалов, как натуральный
каучук и каучукоподобные синтетические вещества (поли-
изобутилен, бутадиеновые каучуки и др.); все они
обладают высокой эластичностью и иногда называются
эластомерами. Из рис. 85 видно, что при свободном враще-
вращении звеньев достаточно небольшого числа звеньев для слу-
случайного расположения конца цепи относительно начала.
В вытянутой цепи между концом и началом возможно толь-
только одно расстояние rmax, тогда как в свернутой цепи они мо-
могут находиться на меньшем расстоянии /"<Утах, однако,
расстояние г оказывается более вероятным, так как оно
связано не с одной, а с различными конфигурациями це-
цепи (см. стр. 177). Вследствие теплового движения
величина г непрерывно изменяется. Кун показал, что
средний квадрат расстояния
227
7* = я/2
cos a.
(X. 2)
где /г — число звеньев в цепи, / — длина звена и (тс—а) ва-
валентный угол (рис. 85). При растяжении клубка цепь пере-
переходит в менее вероятное состояние и при снятии натяжения
самопроизвольно возвращается к более свернутой конфигу-
конфигурации; то же самое происходит при сжатии цепи. Обрати-
Обратимость этих изменений характеризует упругие деформации,
а большое различие между линейными размерами свернутого
клубка по уравнению (Х.2) и длиной вытянутой цепи, для
которой 72~-(nlJ (т. е. различие линейных размеров со-
составляет \гп раз), указывает на возможность больших
удлинений. Эта способность к большим обратимым дефор-
деформациям и определяет высокоэластические свойства.
Следует отметить, что высокая эластичность каучука
совершенно отлична от упругих деформаций кристалли-
кристаллических веществ или металлов, составляющих всего несколь-
несколько процентов от исходных размеров, тогда как каучук
можно растягивать в 10 раз. Резко различаются также не-
необходимые для деформации напряжения. Модуль упру-
упругости (или модуль Юнга) Е, характеризующий отношение
между приложенным напряжением-и относительным удли-
удлинением образца, составляет для стали около 20000 кг/мм2,
для стекла около 6000 кг!мм2, а для каучука лишь около
0,1 кг!мм2. Эти различия объясняются тем, что при упру-
упругой деформации кристаллов происходят небольшие изме-
изменения средних расстояний между молекулами и валентных
расстояний между атомами, связанные со значительными
изменениями внутренней энергии. Напротив, при чистой
высокоэластической деформации большие удлинения про-
происходят без изменения валентных расстояний, при постоян-
постоянстве внутренней энергии (во всяком случае, при удлине-
удлинениях до 3 раз). Лишь у идеальных газов можно также осу-
осуществить большие обратимые сжатия под действием неболь-
небольших напряжений без изменения внутренней энергии. Сжа-
Сжатый газ в замкнутом пространстве после снятия давления
вновь возвращается к первоначальному объему благодаря
тому, что этот процесс соответствует переходу в наиболее
вероятное состояние и происходит с увеличением энтропии.
Легко видеть, что механизм упругих деформаций газа,
несмотря на внешнее несходство, вполне аналогичен ме-
механизму эластической деформации каучука, причем модуль
228
упругости газов (около 0,01 кг!мм2) также близок к моду-
модулю упругости каучука и резко отличается от модулей упру-
упругости других материалов. Таким образом, высокоэласти-
высокоэластические свойства каучука, подобно упругости идеального
газа, обусловлены преимущественно энтропийным факто-
фактором и являются результатом теплового движения молекул.
Деформация каучука происходит при постоянстве объ-
объема и внутренней энергии системы Е, поэтому для каучука
дЕ\
= 0. Зависимость упругого напряжения каучука /
от энтропийных изменений при растяжении выражается
уравнением
\dlJT
(X. 3)
/
Соотношение энергетического и энтропийного членов при раз-
различной деформации реального каучука показано на рис. 87
(отрицательные значения ординаты соответствуют сжатию
каучука). Из рис. 87 видно, что опытные значения [-'j ) j,
не равны нулю, как
следовало ожидать
для идеальной моде-
модели, но они постоян-
постоянны до 3-кратного
удлинения и начина-
начинают возрастать лишь
при дальнейшем уд-
удлинении. Зависимость
. напряжения от де-
деформации в основном
определяется энтро-
энтропийным членом по
уравнению (Х.З).
Предполагая, что
энтропия макроскопи-
макроскопического образца рав-
равна сумме энтропии
отдельных цепей, Кун
получил следующее
уравнение для мо-
модуля упругости ка-
каучука (ср. с Х.2)
Ъ 20
I
S 1Z
S
О
-2
УУ
/уш
7/7/7
200 300
Удлинение, %
Рис. 87. Изменение напряжения,
внутренней энергии и энтропии ка-
каучука при растяжении
229
F = 3RTp (J- 1 + cos *)
M U/a ' l — cos a) '
(X. 4)
где р — плотность полимера. Из (Х.4) следует, что при
постоянной длине образца модуль упругости Е растет
пропорционально абсолютной температуре Т — это под-
подтверждается опытом; тот же вывод вытекает из (Х.З),
так как при постоянной длине / пропорционально Е (на-
(напомним, что Е = —). Но уравнение (Х.З) аналогично
уравнению для газов р = Т (^\ , где давление р=—/, а
/ заменено объемом; эта пропорциональность р и Т при
постоянном объеме газа соответствует закону Гей-Люссака.
Известно также, что при адиабатическом сжатии газа про-
происходит выделение тепла (эффект Джоуля); аналогично
при быстром растяжении каучука он нагревается (термо-
(термоэластический эффект); напротив, кристаллические тела
при растяжении охлаждаются.
Уравнение (Х.З) можно написать в следующем виде:
(X. 5)
так как из термодинамики известно, что /_) = —(—)
\dTJi \д1)т.
В связи с этим, Виганд и Снайдер предложили общее урав-
нение
dl Jt
[dTJi
(X. 6)
Значение (Х.б) заключается в том, что, измерив темпе-
температурный коэффициент напряжения (—) при закрепленной
длине образца в небольшом температурном интервале
Т\ Тг и величину напряжения / в середине этого интервала,
можно по (Х.б) рассчитать соотношение энергетического
и энтропийного члена в деформации полимера. Кинети-
Кинетическая теория упругости каучука была вначале развита
Куном и Марком с сотрудниками для модели изолирован-
изолированной макромолекулы со свободным вращением звеньев.
Бреслер и Френкель ввели в теорию учет ограниченного
вращения звеньев (см. стр. 223); Уолл, Гут и др. развили
теорию для пучка цепей; Догадкин и Гуль подробно ис-
230
т
I
\
I
следовали роль .межмолекулярных взаимодействии в Де-
Деформации каучука.
При больших удлинениях (выше 500%) условия дефор-
деформации каучука изменяются. В вытянутых цепях набор
возможных конфигураций уменьшается, что ограничивает
роль энтропийного фактора, роль же межмолекулярных
взаимодействий увеличивается. Появляется возможность
взаимного упорядочения расположения значительных участ-
участков цепей, вследствие чего в
каучуке возникает волокнистая
структура, которая хорошо вид-
видна, если каучук заморозить в
вытянутом состоянии (рис. 88, I);
после нагревания она исчезает
и каучук возвращается к исход-
исходной форме (рис. 88, II). Рентгено-
Рентгенограмма каучука в нерастянутом
состоянии дает лишь одно раз-
размытое кольцо, характерное для
аморфных веществ (рис. 89, I),
тогда как при растяжении на
500 °о на рентгенограмме виден
ряд отчетливых кристаллических
интерференции, хотя аморфное
кольцо не исчезает. Аналогичные
явления наблюдаются при дли-
длительном выдерживании каучука
при низкой температуре.
Явления кристаллизации ка-
каучука при сильном растяжении
и охлаждении представляют
большой интерес, так как они
наглядно характеризуют особен-
особенности высокополимеров по срав-
сравнению с низкомолекулярными ве-
веществами. Кристаллизация низ-
низкомолекулярных веществ проис-
происходит путем правильной укладки молекул в трехмерную ре-
решетку, имеющхю правильные периоды повторяемости по всем
трем направлениям. В высокомолекулярных веществах
укладываются лишь отдельные участки цепей. Для пол-
полной кристаллизации полимера потребовалось бы такое вы-
выпрямление цепей, которое статистически мало вероятно;
231
Рис. 88. Волокнистая
структура каучука, заморо-
замороженного в вытянутом со-
состоянии (/); тот же каучук
после нагревания (//) (по
Кобе ко).
кроме того, цепи полимеров не идентичны на всем своем про-
протяжении, что также должно вызывать отклонения в уклад-
укладке. В изогнутых отрезках цепей между закрепленными при
укладке участками создаются напряжения, цепи уже не
Рис. 89. Рентгенограммы нерастянутого каучука (/) и растянутого
на 500% (//) (по Воларовнчу)
могут вытянуться и остаются в неупорядоченном состоянии.
В результате одна и та же полимерная цепь проходит через
ряд упорядоченных участков или кристаллитов (по Кобеко)
и неупорядоченных или аморфных областей (рис. 90). Кар-
гин, Китайгородский и Слонимский считают, что этого
положения цепь достигает, заметно не перепутываясь с
соседними цепями.
Следует отметить, что в низкомолекулярных веществах
кристаллизация происходит до полного исчезнования амор-
аморфной фазы, благодаря независимой укладке молекул на
центрах кристаллизации; между тем, в полимерах до 40—
60°о вещества остаются в аморфном состоянии. В низко-
низкомолекулярных веществах кристаллы имеют поверхность
раздела и их можно полностью отделить от остальной массы,
тогда как в полимерах упорядоченные и неупорядоченные
участки меньше размера самих полимерных молекул и прин-
принципиально неразделимы. Наконец, в низкомолекулярных
веществах кристаллизация резко изменяет не только струк-
структуру вещества (что видно по изменению рентгенограмм),
но и термодинамические свойства (плотность, теплоемкость,
сорбцию паров и др.); в полимерах кристаллиты содержат
сравнительно небольшое число атомов, которого достаточ-
232
Но Для появления интереференции на рентгенограмме
(рис. 89), но не всегда достаточно для изменения термоди-
термодинамических свойств. На основании этих соображений Кар-
Картин и Слонимский критически оценивают применение поня-
понятий фазового превращения и гомогенности по отношению
к полимерам. Они указывают, что при решении этого вопроса
необходимо учитывать отмечавшееся выше различие в по-
поведении макромолекул и их звеньев. Кристаллический
полимер следует считать однофазной системой по отношению
к большим структурным единицам (группе макромолекул) и
двухфазной системой — по отношению к малым структур-
структурным единицам (группе звеньев). По взглядам других иссле-
Рис. 90. Схематическое изображение кристаллитов и по.шмерах
(по Кобеко)
дователен, кристаллические полимеры надо во всех случаях
считать двухфазными системами или дефектными по-
поликристаллами (Волькенштейн, Касаточкин).
Натуральный каучук при кристаллизации становится
мало эластичным, обладает измененной плотностью, тепло-
теплоемкостью, тепловым расширением и др.; теплота плавления
кристаллитов (при + 10°С) составляет около 1100 кал
на звено цепи. Таким образом, в натуральном каучуке
233
при охлаждении и растяжении происходит истинная кри-
кристаллизация упорядоченных участков цепи. В растянутом
каучуке кристаллиты ориентируются по направлению рас-
растяжения (рис. 88); после снятия напряжения они снова
быстро плавятся и каучук становится аморфным. Кристал-
Кристаллизуются при растяжении также полиизобутилен, терилен,
поливинилиденхлорид и др.
Необходимо отметить, что не все вещества с гибкими
макромолекулами могут кристаллизоваться; например, ди-
виниловые синтетические каучуки не кристаллизуются.
Это обьясняется тем, что молекулы дивинилового каучука
химически неоднородны; они содержат связи мономеров
в 1 : 2 и 1:4 — положении и, кроме того, частично раз-
разветвлены. В дивинилстирольном и дивинилнитрильном
каучуке чередование остатков в цепи может быть различ-
различным. Эти обстоятельства затрудняют укладку участков це-
цепей в правильную кристаллическую решетку. Не могут
также кристаллизоваться вещества, молекулы которых за-
закреплены в пространственной сетке — вулканизованный
каучук, эбонит и др.
Напротив, гибкие макромолекулы сравнительно про-
простого строения, с регулярной структурой, гораздо легче
укладываются в кристаллические решетки. К этой группе
относятся такие полимеры, как полиэтилен, тефлон, най-
найлон и другие полиамиды, в значительной мере образующие
кристаллиты уже при комнатной температуре без охлаж-
охлаждения или растяжения; например, полиэтилен при комнат-
комнатной температуре закристаллизован на 50—70%. Легко
кристаллизуются также полимеры стереоспецифического
регулярного строения (изотактические полимеры), моле-
молекулы которых обладают высокой химической однородностью;
они при комнатной температуре кристаллизуются почти
нацело. Такие полимеры называются кристаллическими,
тогда как все рассмотренные выше полимеры называются
аморфными. Они обладают значительной прочностью, но
гораздо менее эластичны, чем каучуки; у полиэтилена высо-
высокая эластичность проявляется лишь при температуре выше
115°. Температура плавления кристаллитов большинства
этих полимеров лежит выше 80°, причем ее положение сме-
смещается при растяжении полимера (Александров, Лазур-
кин). Поэтому при деформации кристаллических полимеров
происходит плавление одних кристаллитов и рекристал-
рекристаллизация других в направлении силы растяжения, что
234
I
i
объясняет так называемый процесс холодной вытяжки
этой группы полимеров (Каргин и Соголова). Величины
теплот растворения и сорбции паров ориентированными
•и неориентированными образцами кристаллических поли-
полимеров оказываются довольно близкими (Каргин, Ю. Ли-
Липатов, Гатовская), что свидетельствует об отсутствии за-
заметного перехода аморфных участков в кристаллиты при
растяжении.
Линейные жесткие макромолекулы. Важнейшим пред-
представителем полимеров с линейными жесткими макромоле-
макромолекулами является целлюлоза и ее эфиры; все полимеры
этой группы относятся к полярным полимерам. В то время,
как свойства каучукоподобных полимеров основываются на
наличии длинных цепных молекул со свободным вращением
звеньев и слабыми силами вза-
взаимодействия между цепями, свой-
свойства полярных полимеров с жест-
жесткими цепями определяются длин-
длинными цепными молекулами с
сильно ограниченным вращением
звеньев и значительными энер-
энергиями взаимодействия между
цеп ям и. Поэтому, в отличие от ка-
каучукоподобных полимеров, они
характеризуются небольшими об-
обратимыми удлинениями, преобла-
преобладанием энергетического меха-
механизма упругости, значительным
модулем упругости A00— Рис. 91. Рентгенограмма волок-
200 кг/мм2); в растворах жест- иа хлопка
кие макромолекулы обладают
сильно вытянутой формой и растворяются при Mi^TAS
(см. стр. 181—182).
В природной целлюлозе длинные цепи (с молекулярным
весом до нескольких миллионов) образуются в процессе
биосинтеза с высокой степенью взаимной ориентации,
вследствие чего рентгенограммы целлюлозы дают отчетли-
отчетливую картину волокнистой структуры (рис. 91). При по-
получении искусственного волокна (вискозы, ацетатного
шелка и др.) для обеспечения высокой ориентации це-
цепей и прядильные растворы (концентрированные ксанто-
генатные растворы целлюлозы и др.) продавливают через
тончайшие отверстия — фильеры и, после образования
V, 15- 235
волокон в осадительной ванне, подвергают значительной
вытяжке. Каргин и Н. Михайлов получили методами силь-
сильной вытяжки упрочненные ориентированные гидратцел-
люлозные волокна, с повышением прочности на разрыв,
примерно, с 20—30 до 40—50 кг/мм2. Рентгенографическое
изучение показало, что в ориентированном волокне частицы
в большей мере располагаются вдоль оси волокна и по по-
поперечному сечению волокна оно становится более однород-
однородным. Это объясняет повышение разрывной прочности.
Н. Михайлов и Файнберг при растворении нерастянутого
вискозного волокна и растянутого на 120% получили прак-
практически одинаковые теплоты растворения (около 35 кал/г),
следовательно, при ориентации цепей их энергии взаимо-
взаимодействия мало изменяются. Нагревание ориентированных
волокон приводит к дезориентации положения частиц от-
относительно оси волокна и вследствие этого к некоторому
сокращению его длины (усадке); значительное снижение
ориентации достигается также при обработке целлюлозы
жидким безводным этил амином.
Выше было показано, что гибкие макромолекулы про-
простого или регулярного строения при растяжении или ох-
охлаждении сравнительно легко укладываются отдельными
участками цепей в кристаллиты; напротив, для жестких мак-
макромолекул процессы точной взаимной укладки отрезков
цепей крайне затруднены. Отрезки цепи подобны пачке
карандашей, расположенных приблизительно параллельно,
но различным образом сдвинутых по длине и повернутых
вокруг своей оси. Здесь вновь можно подчеркнуть харак-
характерное для полимеров различие между поведением цепей
и звеньев молекул. При ориентированном расположении це-
цепей (измеряемом, например, по двойному лучепреломлению)
расположение звеньев остается все же неупорядоченным
(что видно по рентгенограммам); при охлаждении полярных
полимеров с жесткими цепями уже при высоких Tg (стр. 225)
происходит застывание как цепей, так и звеньев в стекло-
стеклообразном, а не в кристаллизованном состоянии. По Кар-
гину, процессы взаимной ориентации цепных молекул не
следует смешивать с кристаллизацией полимеров; в част-
частности, полимеры типа целлюлозы и ее эфиров, поливини-
поливинилового спирта и др., несмотря на высокую ориентацию
цепей, следует считать не кристаллическими, а аморфными.
Многие исследователи (Марк, Германе, Зайдес, Роговин,
и др.), однако, полагают, что целлюлоза обладает ми-
236
крокристаллическим строением.
Этот вопрос, как и вопрос
о кристалличности полимеров,
еще не выяснен полностью.
Спиральные макромолекулы.
Спиральной конфигурацией об-
обладают два важнейших вида по-
полимерных молекул, лежащих в
основе процессов жизнедеятель-
жизнедеятельности — белки и нуклеиновые
кислоты; интересно, что этой
своеобразной конфигурацией
они резко отличаются от всех
других типов высокополимеров.
В молекулах белков (альбу-
(альбумины, глобулины, ферменты и
др.) и полипептидов цепи постро-
построены из большого количества
разнообразных остатков L-ами-
нокислот. Помимо последова-
последовательно соединяющих их плоско-
плоскорасположенных пептидных свя-
зей- СО NH—, аминокислотные
остатки связаны большим коли-
количеством водородных связей с уда-
ленными остатками. Условия
максимального насыщения внут-
внутримолекулярных водородных
связей и максимальной плотно-
плотности упаковки аминокислотных
остатков в цепи, при соблюдении
обычных валентных углов и рас-
расстояний,—приводят к характер-
характерному свертыванию цепи в спира-
спирали. По теории Паулинга и Корея,
в глобулярных белках, а-кера-
тине и некоторых полипептидах
свертывание происходит по типу
а-спирали (рис. 92), где на
3 витка спирали приходится
по 11 остатков и через каждый
третий аминокислотный остаток
между пептидными группами об-
17
«¦Коллочдная ли.мця»
Рис. 92. Фрагмент а-спира-
а-спирали белковой молекулы (по
Паулингу и Корею). Ход
спирали отмечен жирной
линией; пунктирные ли-
линии—водородные связи.
237
разуется водородная связь, параллельная оси спирали.
Последовательность аминокислотных остатков различна
для каждого белка, что создает на поверхности спирали из
боковых цепей аминокислот специфичный рельеф, опре-
определяющий структуру центров ферментативной, антиген-
антигенной, гормональной активности белка. Взаимодействие боко-
боковых цепей вызывает также специфические для каждого белка
отклонения основного хода спирали. В фибриллярных бел-
белках—фиброин шелка, р-кератин, миозин и др., спирали вытя-
вытянуты и водородные связи соединяют цепи по перпендикуляр-
перпендикулярным к их осям направлениям. При денатурации происходит
перегруппировка части звеньев цепи с нарушением первона-
первоначальной специфической конфигурации и рельефа боковых
групп, вследствие чего изменяются илиутрачиваются различ-
различные свойства белковой молекулы. Спиральная конфигура-
конфигурация белковых молекул, с множеством внутримолекулярных
химических, водородных, солевых и других связей придает
всей молекуле значительную жесткость, что способствует
устойчивости структуры активных центров. Рассчитано
(Пасынский), что модуль упругости молекул альбуминов
составляет около 15—40 кг/мм2, почти на два порядка выше
чем у каучукоподобных полимеров. Благодаря высокой
однородности молекул глобулярного белка по форме, раз-
размерам и конфигурации, они образуют трехмерные крис-
кристаллы (размером до долей миллиметра) с хорошо развитыми
гранями. Влажные кристаллы, с включением 30—60%
воды, дают на рентгенограммах множество точечных интер-
интерференции, по которым изучаются размеры, молекулярный
вес и внутренняя укладка полипептидных цепей (Бернал,
Перутц, Кендрьюи др.). Однако эти кристаллы отличаются
тем, что у них регулярно расположены полипептидные цепи,
но вдоль полипептидной спирали нет повторяющихся групп
аминокислотных остатков; в этом смысле вся полипептид-
полипептидная цепь является уникальным элементом структуры.
В дезоксирибоиуклеиновой кислоте (ДНК) цепь построе-
построена из нескольких тысяч нуклеотидов, каждый из которых
содержит остаток фосфорной кислоты, углевод дезокси-
рибозу и одно из 4—5 пуриновых или пиримидиновых
оснований, причем плоскости оснований лежат перпенди-
перпендикулярно к длинной оси молекулы. Две цепи ДНК, по Уот-
сону и Крику, образуют совместную спираль (рис. 93),
в которой основания расположены внутри, а остатки фос-
фосфорной кислоты —снаружи. Расположение обеих цепей
238
в спирали взаимно строго обусловлено условиями упаковки
и образования водородных связей между аденином и тими-
ном и между гуанином и цитозином,— они всегда распола-
располагаются друг против друга, но внутри каждой цепи чередо-
' вание нуклеотидов своеобразно и специфично у каждого
вида организмов. Интересно, что шаг спирали на одно нукле-
отидное звено C, 4 д) практически совпадает с периодом
повторяемости на один аминокислотный остаток в вытяну-
вытянутой полипептидной цепи. Взаимоотношение между спираля-
спиралями ДНК и глобулярных белков (фер-
(ферментов) представляет исключитель-
исключительный биологический интерес и в насто-
настоящее время подробно исследуется.
Разветвленные макромолекулы.
Важнейшими представителями поли-
полимеров с разветвленными молекулами
являются крахмал, гликоген и некото-
некоторые другие полисахариды; частично
они образуются при техническом син-
синтезе линейных полимеров (например,
дивиниловых каучуков). Кроме того,
разветвленными молекулами обладают
различные граф'г полимеры (или при-
привитые полимеры, см. стр. 24).
В крахмале содержится 10—20%
амилозы, растворимой в воде и состо-
состоящей из линейных жестких молекул,
с молекулярным весом 10—60 тыс.;
основная часть крахмала состоит
из сильно разветвленных молекул
амилопектина (рис. 94. II, где струк-
структурные элементы разветвлений изоб-
изображены отдельно в 1 и II), с моле-
кулярным весом свыше 1 млн. Обра-
Обработкой крахмала а-амилазой можно
вызвать отсечение боковых ответ-
ответвлении и получить линейные от-
отрезки амилозы (рис. 94, 1). Некоторые исследователи
(Фрейденберг, Бир и др.) предполагают, что крахмал
также образует спирали из шести глюкозных остатков
в витке, расположенных гидроксильными группами наружу
и углеводородными группами внутрь спирали; с этой точки
зрения известную реакцию крахмала с иодом объясняют
Рис. 93. Молекулярная
модел ь дво ii ной сп нр ал и
дезоксирибоиуклеиновой
кислоты
239
поглощением иода по оси спирали, где он в углеводородной
среде дает синюю окраску (как и в чистых углеводородах),
тогда как в воде и полярных растворителях — красную.
Подробно коллоидные свойства крахмала исследованы
В. И. Назаровым, Самеком и др.
В гликогене молекулы построены по типу амилопектина,
но обладают более короткими и частыми боковыми ветвями.
Молекулярный вес гликогена составляет 1—4 млн. Строе-
Строение и свойства гликогена подробно изучали Степаненко,
Е. Розенфельд и др.
Пространственные полимеры. К пространственным поли-
полимерам относится большая группа разнообразных техниче-
технически чрезвычайно важных полимеров. Образование простран-
пространственных полимеров из линейных молекул охватывает
различные системы, начиная с гелей (глава девятая) и
Рис. 94. Схема строения крахмала
кончая продуктами вулканизации каучука, дубления бел-
белков и др. Каучуки и коллаген практически используются
преимущественно в виде трехмерных полимеров; шерсть
является природным пространственным полимером, в ко-
котором пептидные цепи соединены дисульфидными связями.
Пространственные структуры линейных полимеров обра-
образуются также при введении активных наполнителей (на-
(например, сажи в каучуки), где узлы сетки образованы дей-
действием поверхностных и химических сил на частицах
наполнителя. Истинные пространственные полимеры, с хп-
24U
мическими связями между линейными молекулами, обра-
образуются путем их реакции с бифункциональными молекулами
(например, дитиолами), с атомами серы или кислорода, при
действии излучений и др. Пространственные полимеры спо-
способны лишь к ограниченному набуханию и полностью лише-
лишены текучести; при малом числе поперечных связей (мягкие
резины) их эластические свойства соответствуют кинетичес-
кинетической теории упругости чистого каучука (см. стр. 229—230).
Увеличение числа связей между линейными молекулами
вызывает уменьшение длины свободных отрезков цепей и
их изгибаемости, возрастание жесткости полимера (эбо-
(эбонита) и, наконец, полный переход каучукоподобной элас-
эластичности в обычную упругость твердых тел. Теория прост-
пространственных сеток
линейных полимеров ГС
разрабатывалась Уол-
лом, Флори, Догад-
киным, Бартене-
киным, Бартеневым и др.
Другую основную
группу пространст-
пространственных полимеров об-
образуют фенол фор-
мальдегидиые, глице-
ринофталевые, мо-
чевино-формальдегид-
ные и другие смолы
(пластмассы). При
получении фенолфор-
мальдегидных смол
в зависимости от чис-
числа подвижных водо-
водородных атомов фено-
фенола, принимающих
участие в конденсации с альдегидами, и природы ка-
катализатора образуются продукты линейного или сет-
сетчатого строения. При трифункциональной реакции фе-
фенола пространственные полимеры появляются само-
самопроизвольно при нагревании или длительном хранении
(резольные смолы); при бифункциональной реакции фенола
вначале образуются линейные продукты, которые дают
. пространственные полимеры после нагревания с уротропи-
уротропином (новолачные смолы). На рис. 95 показано характерное
241
Нерастворимые
>? и ненпбу*аюш.ие
смолы У/,
50 i
%уротропинп
Рис. 95. Схема стадий затверде-
затвердевания феноло-формальдегидных смол
(по Слонимскому н Коварскон)
изменение свойств смол последней группы по мере увеличе-
увеличения густоты поперечных связей при повышении количества
уротропина и температуры нагрева. Для таких смол харак-
характерно только стеклообразное и эластическое состояние
вплоть до зоны температурной деструкции. В последнее
время применяется введение в пластмассы стеклянного во-
волокна (армированные пластики), что позволяет сильно
повысить их прочность.
ПЛАСТИФИКАЦИЯ
Помимо температуры важным фактором, определяющим
гибкость цепей полимеров, является взаимодействие поли-
полимера с веществами среды. Для практических целей приме-
применяют вещество, способное понизить потенциальные барьеры
вращения, так как при этом цепь становится более гибкой
и понижается порог температуры, при которой еще наблю-
наблюдается высокоэластическая деформация,т. е. понижается тем-
температура стеклования Те. Это позволяет расширить интер-
интервал температур, в котором полимер обладает высокоэластиче-
высокоэластическими свойствами. Кроме того, вещество должно хорошо
совмещаться с полимером для того, чтобы оно не расслаи-
расслаивалось или не «выпотевало» даже при низких температу-
температурах. Наконец, при совмещении происходит раздвижение
макромолекул и ослабление межмолекулярного взаимодей-
взаимодействия, что также увеличивает гибкость цепей и улучшает
эластические свойства1 полимеров. Вещества, удовлетворяю-
удовлетворяющие всем этим условиям, называются пластификаторами.
Таким образом, пластификаторами являются низкомоле-
низкомолекулярные органические вещества, хорошо совмещающиеся
с полимером и повышающие гибкость его цепей.
Введение пластификаторов в полимеры (пластификация)
широко используется в производстве для улучшения элас-
эластичности и морозостойкости полимеров. Практически в ка-
качестве пластификаторов для нитроцеллюлозы, ацетил цел-
целлюлозы и поливинилхлорида применяют дибутилфталат
или трикрезилфосфат, для поливинилового спирта — гли-
глицерин, и др. Например, при введении 40% трикрезилфос-
фата в нитроцеллюлозу величина Tg понижается с 40°
до —30°, а в ацетил целлюлозе — с 60° до—30°. Журков
показал, что в полярных полимерах смещение Tg пропор-
пропорционально лишь молярному количеству поглощенной
жидкости, а в неполярных полимерах величина смещения
242
оо
fg зависит не только от молярного количества, но также
0т размеров и формы молекул пластификатора. Для поляр-
полярного полимера полихлорвинила зависимость изменения
Tg и механических свойств от молярного содержания плас-
пластификатора установлена Соколовым и Фельдман. Эта за-
зависимость объясняется тем, что в полярных полимерах
межмолекулярное взаимодействие оказывается наиболь-
наибольшим в местах расположения полярных групп, а каждая
полярная группа полимера способна прочно связаться с
одной молекулой растворителя (см. стр. 175), откуда
следует пропорциональность эффекта действия пластифи-
пластификатора количеству поглощенных молекул. Напротив, в
неполярных полимерах для ослабления межмолекуляр-
межмолекулярного взаимодействия требуется раздвижение значительных
отрезков цепей, что лучше осуществляется более крупными
молекулами пластификатора. По данным Соколова, введе-
введение 40% пластификатора в полихлорвинил понижает
энергию активации перемещения звеньев цепи с 9500 до
6200 кал.
Другим способом повышения гибкости цепей и пониже-
понижения Tg является химическая модификация цепей путем сов-
совместной полимеризации; например, в сополимере стирола
и бутадиена B : 1) Tg лежит при —30°, тогда как в чистом
полистироле при +81° (стр 225,).
Совмещение пластификаторов с полимером является
проблемой, по существу близкой к проблеме совмещения
полимеров с растворителем (глава восьмая) и поэтому под-
подчиняется тем же термодинамическим условиям. В родствен-
родственной проблеме взаимного совмещения высокополимеров,
кроме условия AZ< 0, особое значение приобретает необхо-
необходимость установить возможно более близкую полярность
смешиваемых полимеров (Воюцкий, Слонимский). Это ус-
условие особенно строго должно соблюдаться при смешении
высокополимеров, более строго, чем при смешении полимера
и пластификатора, ввиду резкой зависимости критических
условий расслоения (глава восьмая) от молекуляр-
молекулярного веса.
ФИЗИКО-МЕХАНИЧЕСКИЕ СВОЙСТВА
ВЫСОКОПОЛИМЕРОВ И СТРУКТУРИРОВАННЫХ
СИСТЕМ
Гибкость цепей полимеров зависит от химического строе-
строения, температурных условий и состава среды (пластифика-
243
ция), однако, возможность проявления гибкости цепей в зна-
значительной мере обусловлена также условиями деформации.
Изменение конфигурации цепей происходит не мгновенно,
а требует известного времени; при слишком быстрой дефор-
деформации изменения конфигурации не успевают следовать за
полем, и цепь в этих условиях перестает быть гибкой; то
же самое относится к процессам перемещения цепей (те-
(течению). При быстром повторном действии деформирующих
усилий на величину деформации накладываются остаточные
влияния предыдущих деформаций и результирующее на-
напряжение в образце оказывается зависящим от его пред-
предыстории. Эти вопросы имеют важное значение для харак-
характеристики физико-механических свойств полимеров (растя-
(растяжения, сжатия, изгиба и др.), на которых главным образом
основано их техническое применение.
Остановимся вначале на некоторых исходных поня-
понятиях. В идеально-упругих твердых телах относительная
деформация г при сдвиге пропорциональна приложенному
напряжению о (закон Гука)
a^Ge, (X. 7)
где G — модуль сдвига. В отличие от этого в простых жид-
жидкостях с обычной истинной вязкостью, например, в воде
или глицерине, напряжение а пропорционально не г, а
скорости деформации (течению) ~ (закон Ньютона)
7tf>
(X. 8)
где т] — коэффициент вязкости (см. главу вторую). Течение
жидкостей, удовлетворяющих уравнению (Х.8), или нью-
ньютоновских жидкостей, изображено на рис. 96, кривая /.
Многие растворы полимеров отличаются тем, что у них
йг
скорость течения -~п- возрастает оыстрее, чем приложенное
напряжение, т. е. при возрастании напряжения сдвига из-
измеряемый коэффициент вязкости падает (рис. 96, кривая 2);
такие жидкости называются неныотоновскими. Характерно,
что в обоих видах жидкостей течение начинается при сколь
угодно малых значениях а, вследствие чего кривые прохо-
проходят через начало координат; жидкости не имеют начального
напряжения сдвига. Однако имеются системы, в которых
течение начинается лишь после приложения критического
244
напряжения сдвига или предела текучести af (рис. 96,5),
затем оно ускоряется до достижения напряжения сдвига
а , после чего становится прямолинейным (величина ав
• характеризует предельное напряжение сдвига, по Бинга-
му); такие системы называются пластичными; к их числу
относятся пасты из глины, фармацевтические пасты и
кремы и др. При а<af пластичная система ведет себя подоб-
подобно твердому телу; поэтому наличие су или ав в системе
показывает наличие в ней известной структурированнос-
структурированности, отличающей ее от жидкостей, хотя бы и обладающих
высокой вязкостью.
Деформации пластических систем или вязких жидкос-
жидкостей необратимы; напротив, упругие деформации твердых
тел и эластические де-
деформации каучукопо-
добных полимеров яеля-
ются обратимыми. Од-
Однако они не развивают-
развиваются мгновенно. В твердых
телах при наложении
напряжения мгновенно
развивается основная
часть деформации по
уравнению (Х.7) в ре-
результате изменений рас-
расстояний между соседни-
соседними молекулами и не-
небольших изменений ва-
валентных связей и уг-
углов, но затем развивает-
развивается в течение некоторого
времени меньшая часть деформации — также обратимой
(упругое последействие); то же происходит при снятии на-
напряжения. Мгновенная упругая деформация по (Х.7)
характеризуется модулем Gi, который имеет высокие зна-
значения, порядка 50—200 кг/мм2; деформация этого рода
существенна для полимеров в зоне стеклования. В элас-
эластической деформации полимеров доля упругой деформации
(по X. 7) невелика; собственно эластическая деформация
полимера, обусловленная изменением конфигурации цепей,
также пропорциональна приложенному напряжению, но
с другим модулем G?, величина которого составляет лишь
сотые доли кг/мм2 (величины модулей сдвига втрое меньше
Рис. 96. Зависимость скорости дефор-
деформации от напряжения сдвига; 1—
ньютоновская жидкость, 2—неньюто-
2—неньютоновская жидкость, 3—пластичные
системы
16 Коллоидная химия
245
модуля упругости, стр. 228). Однако в полимере деформи-
деформируются не изолированные цепи, а совокупность цепей,
поэтому изменения конфигурации цепи связаны с переме-
перемещениями отдельных звеньев в конденсированной фазе, что
учитывается в теории введением особого коэффициента внут-
внутренней вязкости т]2. Наконец, течение полимера характери-
характеризуется обыЧНЫМ Коэффициентом ВЯЗКОСТИ 7]з, ОТНОСЯЩИМСЯ
к перемещению значительных отрезков цепей (следует
заметить, что при течении полимеров цепи вообще не пере-
передвигаются целиком, а по частям, подобно движению гусе-
гусеницы); для низкомолекулярных веществ значения т]2 и
Tj3 совпадают.
Таким образом, три основных состояния полимера:
стеклообразное, высокоэластическое и вязкотекучее (см.
стр. 226) соответствуют трем видам деформации: мгновенной
упругой деформации, конфигурационной деформации и
течению, которые в целом характеризуются двумя моду-
модулями упругости Gi и G* и двумя коэффициентами вяз-
вязкости т] 2 и т]з. Общее выражение деформации е от времени t
при постоянном напряжении сдвига а, по Алфрею, равно
(X-9)
В зависимости от внешних условий, тот или иной член в
уравнении (X. 9) преобладает. При очень больших Gi
и т]з (как обычно в твердых полимерах) первый и третий
члены (Х.9) малы и деформация е определяется вторым чле-
членом, из которого видно, что деформация е при постоянном
о растет от нуля (при /=0) до предельного значения ?max=^r
(при /=оо). Напротив, при снятии напряжения падение
деформации происходит по уравнению
~-'/T (X. Ю)
Аналогично можно показать, что при постоянной деформа-
деформации (например, в растянутом до постоянной величины поли-
полимере) падение возникшего в образце напряжения проис-
происходит по уравнению
— Ч-
°/ = 3-е (X. 10а)
246
Из (X. 10) или (X. 10а) видно, что при /=- величина г/ = —^
или at — ^^- , т. е. деформация при постоянном на-
напряжении или напряжение при постоянной деформации
спадают до Me своего начального значения. Эта величи-
величина х называется временем или периодом релаксации.
При очень большом времени t воздействия постоян-
постоянного напряжения сдвига можно видеть, что в уравнении
(X. 9) преобладает третий член (течение), тогда как при
очень малом времени воздействия преобладает первый
член (упругие деформации); аналогично первый член преоб-
преобладает при низких температурах Т< Tg, а третий член —¦
при высоких температурах 7'>Т'й..При промежуточных
значениях температуры или времени воздействия силы
осуществляются промежуточные типы деформации, являю-
являющиеся сложным сочетанием основных элементов деформации.
Следует отметить, что в уравнении (Х.9) время релак-
релаксации входит только во второй член, ввиду того, что первый
и третий члены специально характеризуют мгновенную
и полностью необратимую часть деформации (соответствен-
(соответственно х=0, и т=оо). Суммарная же деформация полимера
г во всех случаях имеет релаксационный характер. По-
Поэтому учет зависимости деформации или напряжения от
времени воздействия совершенно необходим при работе
с полимерами.
Весьма существенно, что релаксационная природа де-
деформации свойственна не только полимерам, но и всем ре-
реальным телам, в зависимости от соотношения (th);n аморф-
аморфных твердых телах х велико (от секунд до многих часов)
и соответственно необходимо длительное действие силы для
заметного развития деформации; в жидкостях х малы (по-
(порядка 10~9—10"~6 сек.) и длительность воздействия должна
быть небольшой. Так, например, известно, что вар при ударе
ломается, как хрупкое тело, а при очень медленном действии
нагрузки ведет себя, как вязкая жидкость. Корнфельд и
Рыбкин показали, что при быстром поперечном ударе по
струе вязкой жидкости она также изгибается или ломается
в зависимости от быстроты удара (рис. 97). По Кобеко,
полиметилметакрилат ведет'себя, как хрупкая пластмасса
при частоте механического воздействия 1000 колебаний
в 1 мин., тогда как при той же температуре A40 ) и частоте
1 колебание в 1 мин. он обладает высокоэластическими
свойствами. Кобеко указывает также, что различие в тем-
16*
247
пературе морозостойкости при деформации резиновых
покрышек 100 раз в 1 сек. (соответствующей скорости авто-
автомобиля СО км!час) и 1 раз в 5 мин. составляет ATg=30°.
В полиизобутилене при частоте воздействия 10г> колебаний
в 1 сек. измеряется высокоэластический модуль сдвига
G2=0,4 кг/мм2, тогда как при частоте 107 колебаний в
1 сек., при которой может проявиться только упругая де-
деформация, модуль сдвига равен Gi=58 кг/мм2. Эти данные
ясно показывают общность релаксационного механизма
деформации и зависимость эластических и упругих свойств
полимеров от частоты деформации.
Разумеется, для различных реальных тел физический
смысл приведенных выше параметров (особенно G2 и rt2)
Рис. 97. Влияние удара по струе вязкой
жидкости; 1—вязкость 5000 пуаз, скорость
удара 19 м 'сек, //—вязкость 3000 пуаз, ско-
скорость удара 23 м'сек
неодинаков. Кроме того, для полимерных материалов
учет одного времени релаксации недостаточен, так как
в полимерах имеются большие периоды релаксации, свя-
связанные с перемещением значительных отрезков цепей, и
малые периоды релаксации (Ю-3—10~6сек.), связанные
с движениями отдельных звеньев. Полная теория дефор-
деформации полимеров с учетом наборов периодов релаксации,
развитая Слонимским и Каргиным, имеет очень сложный
характер. Практически удобный метод анализа кривых
248
деформации по предельным значениям констант был раз-
разработан Ребиндером.
Кривые деформации экспериментально определяются,
например,в приборе Вейлера—Ребиндера (рис.98), в котором
пластинка А в исследуемой системе при опускании столи-
столика С с постоянной скоростью растягивает пружину D, созда-
создавая в системе постоянное напряжение сдвига а; соответс-
соответствующая деформация измеряется по передвижению шкалы В
в поле микроскопа М. Прибор применяется для различ-
различных систем от слабоструктурированных золей до высоко-
высокопрочных структур. Измеряются кривые кинетики нарас-
нарастания деформации при постоянном напряжении сдвига
Рис. 98. Схема
прибора Вейле-
Вейлера—Ребпндера
80 120 160 200 г* 0 ?80 ПО 160
Г, /чин
Рис. 99. Кривые кинетики деформации
для четырех суспензий бентонита (по
Рсбппдеру)
а и кинетики спада деформации после снятия нагрузки
(а = 0); на рис. 99 изображены подобные кривые для четы-
четырех суспензий бентонитов. Анализ любой из этих кривых
позволяет полностью охарактеризовать механические свой-
свойства коллоидной структуры (сохраняя прежние обозначе-
обозначения) двумя модулями сдвига Gx и G2 (мгновенным и эласти-
эластическим) и двумя коэффициентами вязкести т,2 и tj3, соот-
соответствующими для структурированных систем вязкости при
полном сохранении и полном разрушении структуры. Кро-
Кроме того, определяется предельное напряжение сдвига if
(см. рис. 96); последняя величина, характеризующая меха-
механическую прочность структуры, может быть определена
в приборе Вейлера—Ребиндера, в коаксиальном вискози-
вискозиметре и др. Этим путем Ребиндеру и Ивановой-Чумако-
Ивановой-Чумаковой удалось охарактеризовать растворы полиизобутилена
в ксилоле и толуоле от чистого полимера до чистого раство-
растворителя, в интервале предельной вязкости т{з от 5-1010до
6-10~3 пуаз A3 порядков). Для характеристики возможного
диапазона изменений предельных величии вязкости в тик-
сотропных суспензиях можно указать, что в суспензиях
бентонита в интервале напряжений от 13 до 25 дин/см2
вязкость падает от rt2 = 107 пуазов до тK=10~2 пуаза (на
8—9 порядков). В неразрушенном состоянии тиксотропные
структуры обладают упругими свойствами в соответствии
с уравнением (Х.7). Таким образом, своеобразие тик-
сотропиых структур заключается в том, что предел упру-
упругости в них совпадает с пределом текучести при крайне
резком падении вязкости. В главах шестой и девятой ука-
указывалось, что тиксотропные структуры относятся к коагу-
ляционным структурам каркасного типа. Слабые силы сцеп-
сцепления до определенного предела сдерживают структуру
каркаса, а затем, при более значительных "усилиях сдвига,
структура сразу распадается на ряд независимых облом-
обломков, элементы которой сами по себе не деформируются. Если
коагуляционные структуры более прочны, например, в
гелях кремнекислоты (при срастании отдельных микрокрис-
микрокристалликов), то разрушение структуры происходит труднее
и имеет необратимый характер (по типу хрупкого разрыва).
Для этих систем характерно отсутствие высокоэласти-
высокоэластической деформации. В прочных структурах, подобных гелю
кремнекислоты, она полностью отсутствует; в тиксотропных
суспензиях бентонита или в разбавленных гелях гидрооки-
гидроокиси алюминия обнаруживается медленная эластическая де-
деформация, которая в 2—3 раза больше упругой, но она,
очевидно, является результатом относительной подвижности
структурных элементов каркаса, а не гибкости самих струк-
структурных элементов.
В студнях высоко полимеров также создаются простран-
пространственные сетки связей, которые обладают достаточной
прочностью для создания устойчивой формы; такая сетка
может быть разрушена слабыми механическими воздей-
воздействиями (в тиксотропных студнях желатины) и обладает
значительной эластической зоной. В растворах высокополи-
250
меров уже при небольших концентрациях (около 0,1-—0,2%)
создаются ассоциативные связи между цепями, разрываю-
разрывающиеся при течении, что приводит к явлениям аномальной
вязкости, иллюстрируемым кривой 2 на рис. 96. В концен-
концентрированных растворах полимеров и в твердых полимерах
проявляется весь комплекс параметров, охватываемых
уравнением (X. 9), включая высокоэластические деформа-
деформации, отсутствующие у жестких коагуляционных структур,
и упругость сдвига. Наконец, в пространственных поли-
полимерах и при сильных механических воздействиях на поли-
полимеры (см. стр. 254) создаются условия для необратимых на-
нарушений в структуре, вследствие чего вводятся параметры,
характеризующие прочность структуры.
Таким образом, физико-механические свойства всех
систем, начиная от высокомолекулярных веществ и их рас-
растворов и кончая структурированными дисперсными систе-
системами, могут в принципе исследоваться общими методами
реологии (реологией называется общее учение о деформации
и течении). Такие исследования имеют преимущество перед
простыми измерениями аномальной или структурной вяз-
вязкости неньютоновских жидкостей (рис. 96), потому что
структурная вязкость зависит от условий измерения, тогда
как реологические константы характеризуют материал
независимо от размеров прибора или режима течения.
Образование или разрушение различного рода структур или
пространственных сеток частиц или молекул с различной
прочностью связей и жесткостью структурных элементов
играет исключительную роль в дисперсных и полимерных
системах и во многих отношениях определяет их техни-
техническое использование. Поэтому изучение процессов дефор-
деформации, их кинетики, частотной зависимости, предельных
напряжений и др. имеет большое научное и техническое
значение. Установление релаксационного механизма де-
деформации и объективных методов характеристики процес-
процессов деформации является существенным успехом коллоид-
коллоидной химии, во многом обусловленном работами советских
ученых — Кобеко, Александрова, Каргина, Слонимского,
Ребиндера, Соколова, Догадкина и др.
ДИЭЛЕКТРИЧЕСКИЕ СВОЙСТВА ВЫСОКОМОЛЕКУЛЯРНЫХ
ВЕЩЕСТВ
Техническое значение полимеров определяется не толь-
только их физико-механическими или термическими, но также
251
и диэлектрическими свойствами, так как изоляция моторов,
кабелей, проводов основана почти целиком на применении
высоко полимеров; в частности, большое значение в этом
отношении имеют кремиийорганические полимеры (Андриа-
(Андрианов). Диэлектрические свойства полимеров используются
для их исследования, ввиду глубокой аналогии релаксацион-
релаксационной природы изменений диэлектрических и ряда механиче-
механических свойств при различных условиях, в частности, при
изменении температуры и частоты поля.
Неполярные полимеры — полистирол, полиэтилен, теф-
тефлон, полиизобутилен — обладают крайне малым поглоще-
поглощением (или потерей) электрической энергии в переменном
электрическом поле, почти не изменяющимся с темпе-
температурой. Мерой диэлектрических потерь является отноше-
отношение коэффициента потерь энергии и диэлектрической по-
постоянной, выражаемое величиной tgb (которая называется
тангенсом угла потерь); у неполярных полимеров tgb=
= 0,0003—0,0005, поэтому эти диэлектрики применяются в
радиотехнике (обычные фарфоровые изоляторы имеют
tgb=O,O\).
В полярных полимерах — поливинил ацетате, полиме-
тилметакрилате и др. (tgo=0,02—0,03) дипольные группы
стремятся следовать за изменениями поля, но время релак-
релаксации -z колебаний зависит от вязкости среды, температуры,
наличия пластификатора, размера поворачивающегося
участка цепи и т. п. При очень низкой частоте поля по срав-
сравнению с т все дипольные группы следуют за полем, что
соответствует наибольшему значению диэлектрической по-
постоянной г среды; напротив, при очень высокой частоте поля
все дипольные группы заторможены, что соответствует
минимальному значению г. Кривые, выражающие изме-
изменение в с частотой поля, называются кривыми диэлектри-
диэлектрической дисперсии; перегиб кривых дисперсии соответ-
соответствует времени релаксации т; в этой же области tg о имеет
наибольшие значения. Другой тип кривых диэлектричес-
диэлектрической дисперсии можно получить при постоянной частоте
поля изменением температуры; в этом случае перегибы кри-
кривой соответствуют температуре стеклования Tg. Напри-
Например, из рис. 100 видно, что при добавлении возрастающих
количеств пластификатора дифенила к поливинилхлориду
(при постоянной частоте поля 60 колебаний в 1 сек.) кривые
з и положение Tg смещаются в область более низких тем-
температур. Электрическими методами легко проводить изме-
252
рения в интервале частот v от 10~а до 1010 колебаний в
1 сек. (в 10г:! раз), в том числе высоких частот, которые не
обнаруживаются методами, основанными на механических
колебаниях. Изучение диэлектрической дисперсии поляр-
полярных полимеров и белков в широком диапазоне частот поз-
позволило установить их значения времен релаксации х. Так
как белковые молекулы вращаются в электрическом поле
как компактные жесткие частицы, то из величины z можно
определить размеры и молекулярный вес белковых молекул
в хорошем согласии с другими данными. В полярных поли-
полимерах кривые диэлектрической дисперсии гораздо шире
Рис. 100. Диэлектрические постоянные по-
ливннилхлорида с добавлением пластифи-
пластификатора дифенила (цифры у кривых указы-
указывают процент дифеимла)
(до восьми порядков частот вместо двух порядков у белков),
так как в электрическом поле могут ориентироваться участ-
участки цепи различных размеров, что приводит к широкому на-
набору времен релаксации. Поэтому в полярных полимерах
можно обнаружить как вращение отдельных полярных
групп, так и вращение отдельных участков цепей, которые
в жестких полимерах больше, чем в гибких полимерах. Та-
Таким образом, на основании диэлектрических измерений
также можно оценить гибкость молекулярных цепей.
Подробные исследования диэлектрических свойств полиме-
полимеров производились Кобеко, Г. Михайловым, Фуоссом,
Уайменом и др.
253
МЕХАНОХИМИЯ ПОЛИМЕРОВ
При действии сильных механических напряжений на
полимеры, например, при продавливании полимеров через
капилляры, очень быстром перемешивании или помоле,
в условиях, когда макромолекулы не успевают или не
могут перемещаться друг относительно друга, в них могут
возникать разрывы цепей по валентным связям с образова-
образованием свободных полимерных радикалов. Если формование
изделия проводится достаточно быстро, то воссоединение ра-
радикалов приводит к закреплению образованной формы изде-
изделия (Каргин, Слонимский, Согол.ова). Если подобным воздей-
воздействиям (механическому крекингу) подвергнуть смесь поли-
полимеров, можно после рекомбинации радикалов получить
новые химические сочетания полимеров. Берлин применил
для временного разрыва связей замораживание набухших
полимеров (крахмала, полистирола), используя для меха-
механических воздействий изменения объема при замерзании.
Подобные химические изменения при механическом воз-
воздействии на полимеры составляют область механохимии
полимеров. В* отличие от обычного течения высокополиме-
ров, при котором макромолекулы постепенно, отдельными
участками цепей, передвигаются друг относительно друга,
при механохимическом течении передвигаются обломки или
фрагменты сетчатой структуры полимера до момента их
рекомбинации, что уподобляет этот процесс обратимому
разрушению коагуляционных структур. Введение неболь-
небольших добавок защитных веществ, дезактивирующих сво-
свободные радикалы (бутилгидрохинона и др.), позволяет
регулировать процесс восстановления структуры, подобно
действию добавок поверхностноактивных веществ при коа-
гуляционном структурообразовании. Механохимия поли-
полимеров несомненно открывает новые пути в их технологи-
технологической переработке.
Выводы
Твердые полимеры образованы длинными цепными мак-
макромолекулами различного строения. Звенья молекулярной
цепи полимеров обладают способностью к взаимному вра-
вращению, что в макромолекуле приводит к гибкости цепи.
Величина гибкости цепей зависит от химического строения
цепи (потенциального барьера внутреннего вращения),
254
температуры, состава среды, условий межцепного взаимо-
взаимодействия и др. Кроме того, проявление гибкости цепей
зависит от условий деформации. Высокомолекулярные
вещества могут находиться в трех состояниях: стеклооб-
стеклообразном, высокоэластическом и вязкотекучем, соответ-
соответственно разделенных температурами Т„ и Tf, эти состояния
могут изменяться в зависимости от перечисленных выше
условий. Линейные гибкие макромолекулы (каучуки, эла-
эластомеры) способны к большим обратимым удлинениям с
малыми модулями, обусловленными преимущественно
энтропийными изменениями. Аморфные полимеры этой
¦ группы в большинстве способны частично кристаллизоваться
при охлаждении и растяжении; другие полимеры с гибкими
цепями простого или хорошо упорядоченного строения
могут оставаться частично кристаллическими при обычных
температурах (кристаллические полимеры). Кристаллиза-
Кристаллизация полимеров имеет ряд специфических отличий от крис-
кристаллизации простых веществ. Линейные жесткие макро-
макромолекулы (целлюлоза, ее эфиры) дают ориентированные
структуры; повышение степени ориентации является важным
методом изменения свойств волокон. Белки и нуклеиновые
кислоты характеризуются спиральной укладкой макромо-
макромолекул; крахмал, гликоген и некоторые синтетические поли-
полимеры — разветвленными молекулами; пластмассы и многие
технические полимеры имеют строение пространственной
сетки. Важным способом регулирования свойств полимеров
является введение пластификаторов — низкомолекуляр-
низкомолекулярных органических веществ, хорошо совместимых с полиме-
полимером и повышающих гибкость цепей. Механизм действия
пластификаторов различен для полярных и неполярных
полимеров. Физико-механические свойства полимеров (рас-
(растяжение, сжатие, изгиб и др.) имеют релаксационный
механизм; анализ кривых скорости деформации и течения
полимеров позволяет характеризовать материалы объектив-
объективными значениями модулей сдвига, вязкости и предельной
упругости сдвига, не зависящими от условий измерения;
этими же методами могут исследоваться другие струк-
структурированные системы. Диэлектрические свойства полиме-
полимеров также обладают релаксационным механизмом; в поляр-
полярных полимерах проявляется ориентация отдельных ди по ль-
льны х групп или участков цепей. При сильных механических
воздействиях на полимеры в них проявляются химические
изменения (образование свободных радикалов), которые
255
могут быть использованы при формировании изделий и
получении новых видов полимеров.
Вопросы
1. В каких состояниях могут находиться высокомолекуляр-
высокомолекулярные вещества? Чем характеризуются переходы между этими состоя-
состояниями?
2. Какие основные типы макромолекул образуют полимеры?
Назовите их основных представителей.
3. Чем определяются высокоэластические свойства каучуко-
подобных полимеров? Какие уравнения характеризуют их пове-
поведение?
4. Чем отличаются аморфные и кристаллические полимеры,
каковы характерные особенности кристаллизации полимеров?
5. Какими свойствами отличаются полимеры с линейными
жесткими макромолекулами, каковы особенности их ориентации?
6. Каковы особенности структуры белков и нуклеиновых
кислот?
7. В чем заключается пластификация, каковы различия в
механизме пластификации полярных и неполярных полимеров?
8. В чем заключается релаксационный механизм физико-ме-
физико-механических и диэлектрических свойств полимеров?
9. Какими константами характеризуется деформация поли-
полимеров и структурированных систем, чем отличаются условия их
деформации?
10. В чем заключаются механохнмические изменения поли-
полимеров и каковы возможные пути их использования0
Л ИД Е Р А Т У Р А
Адам Н. Физика и химия поверхностей. Гостехиздат, 1947.
А л ф р е й Т. Механические свойства высокополимеров ИЛ, 1952.
Б у л л Г. Физическая биохимия. ИЛ, 1949.
Д у м а н с к и й А. В. Учение о коллоидах. Госхпмиздат, 1948.
Д у м а и с к и й А. В. Библиографический очерк развития оте-
отечественной коллоидной химии. Вып. 1 и 2. Изд. АН УССР,
1949 и 1955.
Догадкин Б. А. Физика и химия каучука. Госхимиздат,
1951.
Жуков И. И. Коллоидная химия, ч. 1. Изд. ЛГУ, 1949.
Ионообменные смолы в медицине и биологии. ИЛ, 1956.
К р о й т Г. Р. Наука о коллоидах, т. 1. ИЛ, 1955.
Клейтон В. Эмульсии. ИЛ, 1959.
К о б е к о П. П. Аморфные вещества. Изд. АН СССР, 1952.
Ко р ш а к В. В. Химия высокомолекулярных веществ. Изд. АИ
СССР, 1950.
ЛипатовС. М. Физико-химия коллоидов. Госхимиздат, 1948.
Л ь ю и с У, С к у а й р с Л, Б р у т о н Дж. Химия коллоид-
коллоидных и аморфных веществ. ИЛ, 1948.
Михайлов А. Н. Физико-химические основы технологии ко-
кожи. Гизлегпром, 1949.
Н е й р а т Г. и Б э й л и К- Белки, т. I и II. ИЛ, 1956; т. Ill,
1958.
Песков Н, П. и Александров а-П р е й с Е. М. Курс
коллоидной химии. Госхимиздат, 1948.
Ребиндер П. А. Конспект общего курса коллоидной химии.
2-е изд. Изд. МГУ, 1958.
Самсонов Г. В. Хроматография. Медгиз, 1955.
Т а г е р А. А. Растворы высокомолекулярных соединений. Гос-
Госхимиздат, 1951.
Фукс Н. А. Механика аэрозолей. Изд. АН СССР, 1955.
Ш в а р ц А. н П е р р и Дж. Поверхностноактивные вещества.
ИЛ, 1953.
ПРЕДМЕТНЫЙ УКАЗА ГЕЛЬ
Агрегативная устойчивость 10,
15, 148
Агрегация при получении кол-
коллоидов 20
Адсорбционные слои 81—86,
98—99, 146
Адсорбция активированная 97
— в растворах 82—84
— на твердых поверхностях
92—98
— природа сил 97
— теория Поляни 95
— уравнение Лангмюра 94
Брунаэра (БЭТ 96
Фрейндлиха 95
Активные центры 93
Алфрея уравнение деформации
246
Аморфные вещества 224
Амфотерные электролиты 105
Анизотропия оптическая 63—
67
Аниониты 129
Антагонизм ионов 141
Антонова правило 77
Ассоциация измерения по ме-
методу диффузии 32
. — нефелометрии 55
Атермические растворы 181
Аэрогели 162
Аэрозоли 24, 163—166
Барометрический закон 41
Блокнолимеры 24
Броуновское движение 29—30,
40
Вант-Гоффа уравнение 34
Вейлера-Ребиндера прибор 249
258
Взаимная коагуляция 140—141,
148
Виганда-Снайдера уравнение
230
Вискозиметры 48
Вульфа-Брэгга закон 70
Высаливание 184—185
Высокоэластическая деформа-
деформация 228—230
Высокоэластическое состояние
полимеров 225—226
Вязкость 47—50, 192—195
Вязкость относительная 192
— приведенная 193
— структурная 251
— удельная 193
—• характеристическая 193
Вязкотекучее состояние поли-
полимеров 225—226.
Гели хрупкие 198—200
— эластичные 201—210
Гиббса уравнение 82
Гибкость микромолекул 18—
19, 188—189, 201, 222—223,
227 и ел., 242—243
Гидратация 173
Гидродинамический радиус 188,
191, 195
Гидрофильные поверхности
78—79
Гидрофобные золи, гидрозоли
11, 23
— поверхности 78
Гистерезис в гелях 199, 203,
209
Гомогенизаторы 158
Грофт-полимеры 24
Гука закон упругости 244
Давление набухания 205—206,
211—212
Двойное лучепреломление 64—
66
Двойной электрический слой
102—103
Дебая уравнение светорассея-
светорассеяния 56
Денатурация 238
Дерягина-Ландау уравнение
140
Детергенты 120—121
Деформация студней 207—208
— полимеров 246—248
Диализ 37
Диспергирование 20
Дисперсная система 9
— фаза 9
Дисперсионная среда 9
Дисперсоидология 9
Дифракция рентгеновских лу-
лучей 70
Диффузия вращательная 32—
33, 65, 191, 195
— в гелях 218—219
— поступательная 30—32, 191
Диэлектрические свойства кол-
коллоидов 131—132
полимеров 251—253
Доннановское равновесие 123
и ел.
Дубители как коллоидные элек-
электролиты 122
Заряд частиц 111—116
Застудневание 209
Защитное действие 99, 146—
147
Золотое число 146
Золо-концентрационпый эффект
126
Изотактические полимеры 25,
234
Изоионная точка 114
Изоэлектрическая точка 115
Инфракрасный дихроизм 62
— спектр поглощения 61 — 62
Ионообменные смолы 129
Капиллярное давление 79
Катиониты 129
Каучукоподобная эластичность
Кинетическая теория 229—230
Кинетика коагуляции 149—152
Кинетическая устойчивость 10,
41
Коагуляция коллоидов быстрая
149
взаимная 140—141, 148
— — медленная 151—152
порог 137
электролитами 135—143
Коацервация 187
Коллоидная химия, определе-
определение 7, 14, 19—20
Коллоидные системы, общие
свойства 17—18
определение 7
¦ практическое значение 7
основные типы 15—16
методы получения 20 и
ел.
Коллоидные электролиты 120—
123
Колориметрия 58—59
Константы деформации 249—¦
250
Коэффициент дисимметрии 44
— диффузии 31
— растекания 87
—¦ экстинкции (погашения) 58
Краевой угол 78
Красители как коллоидные
электролиты 123
Кривые распределения 45—46
Кристаллизация полимеров
231-234
Кристаллические полимеры 234
Критическая температура, раз-
разделение фаз 155, 170
Куна уравнение упругости 229
Ламберта-Бэра закон 58, 60
Лангмюра уравнение адсорб-
адсорбции 94
и закон действия масс
105
— поверхностные весы 88
Лапласа уравнение 78—79
Лпзеганга кольца 219
Линейные структуры 5
Лиотропные ряды 137, 185
Лиофильиые коллоиды 11—13
Лиофобмые коллоиды 11
259
«¦
Макромолекуты линейные гиб-
гибкие 227—235
жесткие 235—237
разветвленные 239—240
— спиральные 237-239
Мембранный потенциал 125
Мембранные электроды 216—
217
Мембраны гомогенные 213
— пористые 213—215
— диализующие 213
— электрохимически активные
215—216
Механохимия полимеров 254
Механохимическое течение 254
Мицеллы 11, 12
Модуль сдвига полимеров 245
— упругости полимеров 229.
238, 245
Молекулярный вес
определение по вязкости
50, 193—195
по механическим свой-
свойствам 226
¦— в монослоях 91
по осмотическому дав-
давлению 34, 189—190
по светорассеянию 56
ыа ультрацентрифуге
43
средневесовой 47, 57, 196
— — среднечислснный 37, 196
— «клубок» 188—192
Мономолекулярные слои спо-
способы получения 88
— — белков и ферментов 91—
92
Моющее действие 121
Мыла 120—121
Набухание 201—207
— неограниченное 201
— ограниченное 201
Наполнители активные 240
Напряжение сдвига 245
Нейтральные соли, взаимодей-
взаимодействие с коллоидами 116—118
I Гефелометрпя 54—55
Никольского уравнение обмена
ионов 128
Нулевые точки заряда 113—115
Ньютона закон вязкости 244
Ньютоновские жичкости 244—
245
Обмен ионов 117, 128, 137
Обращение фаз 157—158
Окраска коллоидных растворов
60
Опалесценция 54
Оптическая анизотропия 63—67
Оптическое вращение 62—63
Органозолн, устойчивость 142—
143
Ориентация молекул в поверх-
постном слое 85—86
в полимерах 236
Осмометр 35
Осмотическое давление 33—37,
189—191
Пеногашсние 162
Пены 24, 160—162
Пептнзания 142
Перенос ионов в мембранах 215
Период релаксации 247—248,
252
Плавление студней 208
Пластификация 242—243
Пластичные системы 244—245
Пластмассы 241—242
Поверхностноактивные вещест-
вещества 81, 98—99 (см. детерген-
детергенты, мыла)
Поверхностная энергия 81
Поверхностное давление 88
-- натяжение 74—81
Поверхностные потенциалы 90
Поверхность адсорбентов 96
Поворотная изомерия в поли-
полимерах 223
Поглощение света 58—62
Полидпсперсность 37, 44—47
Поликонденсация 22
Полимеризация 22
Полиэлектролиты 119, 210
Поляризация света 55, 63—-64
— флуоресценции 67
Поляриметрня 63
Полярные группы, ориентация
85—86
гольватация 174
Порог коагуляции 137
Потенциальные барьеры вра-
вращения 221—222
260
Правило осадков 187
Правило фаз в растворах по-
полимеров 170
Привитые полимеры 24
Пространственные полимеры
240—242
Противоточное распределение
129
Пуазейля закон 48
Равновесие садиментационнос
44
— термодинамическое в рас-
растворах полимеров 171-—172
Разветвленные макромолекулы
195—239
Размеры коллоидных частиц по
двойному лучепреломлению
66
поляризации флуо-
флуоресценции 67
электронной микро-
микроскопии 68
Разрушение эмульсий 157—158
L Расклинивающее давление 145
Расслоение 184, 190
Рауля закон 33
Релаксация в полимерах 247—
248, 252
Рентгеноструктурный анализ
70—71
Реология 251
Решетки слоистые 4
— трехмерные 4
Рубиновое число 146
Рэлея уравнение 54
Свечберг, единица измерения
43
Сведберга ультрацентрифуга
42, 44
Светорассеяние 53—58, 191
Сжатие двойного электрическо-
' го слоя 117, 139
f Седиментациониый анализ 45
Седиментация 39—40
Сенсибилизация золя 147
Синергизм ионов 141
Синерезис 210
... Смачивание 77—78
Г'* Смешение полимера с раство-
растворителем 181 — 183
Смешение полимера с пласти-
пластификатором 242
с полимером 243
Смолуховского теория коагу-
коагуляции 149—150
Смолы резольные 241
— новолачные 241
Солюбилизация 121 —122
Сольватация 12—13, 145, 173—
176, 202—204
Сополимеры 22, 24
Состояния высокомолекуляр-
высокомолекулярных веществ 224—226
Спектры поглощения 59—62
Спиральные макромолекулы
237—238
Стеклообразное состояние в
полимерах 223—224, 226, 236
Стокса закон 40
Структурные кривые адсорбен-
адсорбентов 200
Структурная вязкость 251
Суспензионный эффект 125
Тактоиды 144
Температура застеклования
224—225
— текучести 225—226
Теплота адсорбции 93
— набухания 203—204
— растворения 176
— сольватации 172
Термодинамический потенциал
двойного слоя 107, 112
Термомеханические кривые 227
Термоэластический эффект 230
Тизелпуса прибор для электро-
электрофореза 108
Тиксотропия 143—144
Томсока уравнение 80
Траубе правило 84
Удельное вращение 63
Ультрамикроскоп 68
Ультрафнльтрацня 38
Ультра фильтрация, роль дон
иаповского эффекта 12G
Ультрафиолетовый спектр по-
поглощения 61
Улмрацетрнфх гпрование 43—
44'
Упругие ц-формацин 24о
Упмугое последействие 245
261
Устойчивость высокодисперс-
высокодисперсных систем 15
. и электрскинетический
потенциал 137, 138
и обмен ионов 137
сжатие двойного слоя
139
— растворов полимеров 15—
16, 170—172
органозолей 142—143
— — пен 161
эмульсий 159
аэрозолей 163—164
Фаза в коллоидных системах
16—17
Флори и Фокса уравнение 195
Флотация 161 —162
Форма коллоидных частиц 43—
44, 49—50, 67, 68
Фракционирование полимеров
186—187
Фрумкина уравнение 83
Хроматография адсорбционная
126
—- вытеснительный анализ 128
¦— ионообменная 128, 211
Хроматография на бумаге 129
— распределительная 129
— элюционный анализ 127
Шишковского уравнение 84
Штаудингера уравнение 193—
194
Шульце-Гарди правило 135—
136
новского движения 30
Эйнштейна уравнение броу-
броуновского движения 30
вязкости 49
диффузии 31
Эластомеры 227
Электровискозный эффект 50
Электродиализ 38
Электрокапилляриые кривые
112—113
Электрокинетический потенци-
потенциал 107, ПО, 137—138
Электронная микроскопия 68
Электроосмос ПО—111
Электропроводность коллоидов
131
Электростатическое отталкива-
отталкивание частиц 139
Электроультрафильтрация 39
Электрофорез 107—108
— на бумаге 109—110
Элементоорганические полиме-
полимеры 25—26
Эмульгаторы 156—-157
Эмульгирование самопроиз-
самопроизвольное 155
Эмульсии 24, 154—160
Энергия притяжения частиц
139
Энтропия, изменение в раст-
растворах полимеров 176—178
— при набухании 204
ОГЛАВЛЕНИЕ
Предисловие
Глава первая. Общая характеристика коллоидных систем
Практическое значение коллоидных систем
Очерк развития коллоидной химии
Природа коллоидных систем
Методы получения коллоидных систем
Основные виды коллоидных систем
Выводы
Вопросы
Глава вторая. Молекулярно-кинетические свойства коллоидов
Броуновское движение
Диффузия
Осмотическое давление
Диализ и ультрафильтрация
Седиментация и ультрацентрифугирование .
Полидисперсность и кривые ^ распределения
Вязкость
Выводы
Вопросы
Глава третья. Оптические свойства коллоидов
Светорассеяние
Поглощение света
Вращение плоскости поляризации ....
Оптическая анизотропия ...
Электронная микроскопия и рентгенография
Выводы
Вопросы
Глава четвертая. Поверхностные явления и адсорбция
Поверхностное натяжение
Адсорбция на поверхности растворов
Ориентация молекул в поверхностном слое . . . .
Мономолекулярные слои
Адсорбчия на твердых поверхностях
Значение адсорбционных слоев в коллоидных системах
Выводы
Вопросы
7
8
14
20
23
27
28
29
29
30
33
37
39
44
47
50
51
53
53
58
62
63
67
71
72
74
74
81
84
86
92
98
99
100
263
Гласа пятая. Электрохимия коллоидов 102
Двойной электрический слой 102
Электрокинетические явления 107
Заряд поверхности частиц. Точка нулевого заряда и
изоточки 111
Взаимодействие с нейтральными солями ....... 116
Растворы полиэлектролитов 119
Коллоидные электролиты 120
Доннановское равновесие 123
Хроматография - - • 126
Электропроводность коллоидов. Диэлектрические свойства
коллоидов 131
Выводы !S2
Вопросы 133
Глава шестая. Устойчивость и коагуляция коллоидов J35
Устойчивость лиофобных коллоидов с ионными адсорб-
адсорбционными солями. Коагуляция лиофобных коллоидов
электролитами 135
Тиксотропия 143
Устойчивость лиофобных коллоидов с молекулярными
адсорбционными слоями. Защитное действие 144
Кинетика коагуляции 149
Выводы 152
Вопросы 153
Глава седьмая. Эмульсии, пены, аэрозоли • 154
Эмульсии 154
Пены 160
Аэрозоли 163
Выводы 166
Вопросы : 167
Глава восьмая. Растворы высокомолекулярных веществ 169
Термодинамическая устойчивость растворов высокомоле-
высокомолекулярных веществ 17о
Изменение теплосодержания и сольвата: ия в растворах
высокомолекулярных веществ 172
Изменение энтропии в растворах высокомолекулярных
веществ 176
Изменение свободной энергии в растворах высоко-
высокомолекулярных веществ . 179
Нарушение устойчивости растворов высокомолекуляр-
высокомолекулярных веществ (расслоение, высаливание, коацервацня) . 183
Размеры и форма макромолекул в растворах 188
Выводы 196
Вопросы 197
Глава девятая. Гели 198
Хрупкие гели 1д8
Эластичные гели 201
Гели полиэлектролктов и белков. Мембраны 210
Диффузия в гелях 218
264
Выводы - . 219
Вопросы 220
Глава десятая. Твердые полимеры 221
Основные состояния высокомолекулярных г.егцесги . .
Основные тины макромолекул 212
Линейные гибкие макромолекулы j*J
Линейные жесткиг макромолекулы &J
Спиральные макромолекулы ?>5
Разветвленные макромолекулы ~~'
Пространственные полимеры 9о9
Пластификация ? ?
Физико-механнческне свойства высоконолпмеров и ^*~
структурированных систем ,..
Диэлектрические свойства высокомолекулярных веществ ±vi
Механохимия полимеров ^.^
Выводы „
Вопросы ?92
Литература jfl
Предметный указатель zo°
Анатолий Германович Пасынский
коллоидная химия
. Редактор А. Б. Лукьянов
Редактор издательства Т. Г. Липкина
Технический редактор С. С. Горохова
Корректор Л. 3. Черникова
Сдано в набор 8/IV-59 г. Подписано к печати 23/IX-59 г. Бумага
84хЮ87з2- 16,75 печ. л., 13,73 усл. печ. л., 14,69 уч.-изд. л.
Тираж 12 000. T-06S99. Цена 5 р. 90 к. Зак. 33-1.
Издательство «Высшая школа»
Полиграфический комбинат Ярославского совнархоза,
г. Ярославль, ул. Свободы, 97.
т
К ЧИТАТЕЛЯМ
Издательство «Высшая школа» просит Вас
отзывы и замечания по данной книге
посылать по адресу:
Москва, Б-64, Подсосенский пер., 20.