






























































































































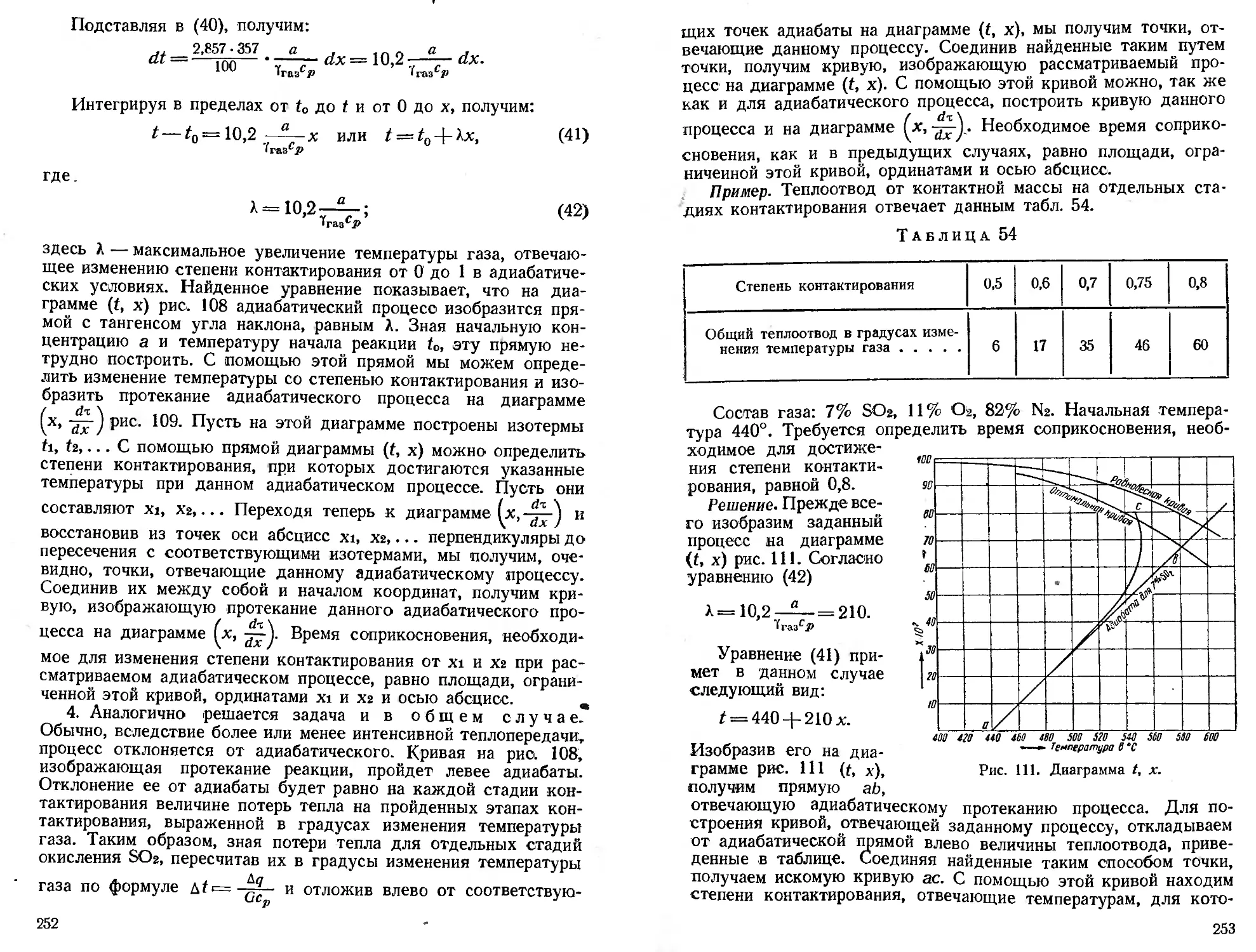









































































































































Текст
К. М. МАЛИН
Г. К. БОРЕСКОВ, И- Л. ПЕЙСАХОВ, М. Г. СЛИНЬКО,
н. и. СМЫСЛОВ, М. Н. ВТОРОВ, н. Л. АРКИН
ТЕХНОЛОГИЯ
СЕРНОЙ КИСЛОТЫ
И СЕРЫ
Утверждено Всесоюзным Комитетом по делам
высшей школы при СНК СССР в качестве учебника
для химико-технологических вузов
ГОСУДАРСТВЕННОЕ НАУЧНО-ТЕХНИЧЕСКОЕ ИЗДАТЕЛЬСТВО
ХИМИЧЕСКОЙ ЛИТЕРАТУРЫ
МОСКВА 1941 ЛЕНИНГРАД
К читателю
Прежде чем пользоваться книгой, исправьте следующие опечатки
Стр. Строка Напечатано Следует читать По вине
193 18-я снизу конденсации 2Н2О конденсации 2 г Н2О Ред.
229 15-я снизу составлению состоянию Тип.
254 уравнение (43) Й(хтх>0’8 Л(хт X)0’8 Корр.
в знаменателе
293 рис. 136 10 и 11 — кривые 10 н 11 — краны Корр.
297 15-я сверху (рис. 9) (рис. 97) Ред.
318 табл. 71, графа
4-я снизу 2NO2 2HNO2 Тип.
341 9-я снизу (Ре~ Pi*) Авт.
343 19-я снизу необходимо необратимо Ред.
402 18-я сверху (гл. 3) (стр. 301) Ред.
Зак. 5019. К М. Малин
22-5-2
661
М-18
Книга представляет учебник технологии серной
кислоты и серы для студентов химических втузов.
По каждому отдельному производству дается
описание схем производства, физико-химические
основы его, аппаратурное оформление процесса,
технологический режим, пути и ближайшие пер-
спективы развития. В книге показана связь серно-
кислотной промышленности с другими отраслями
народного хозяйства, как применяющими серную
кислоту, так и являющимися ее сырьевой базой.
В связи с этим в книгу введена специальная глава
об использовании н концентрировании отбросных
(металлургических и др.) сернистых газов. '
В книге освещены иаучио-исследовательские ра-
боты (советские и иностранные), связанные с про-
изводством серной кислоты и серы, а также описаны
последние технические усовершенствования.
Книга помимо учащихся представляет также
интерес для инженеров и работников научно-иссле-
довательских институтов химической промышлен-
ности.
Я ЧИТАТЕЛЮ
Издательство просит присылать Ваши замечания
и отзывы об этой книге по адресу: Москва, Новая
площадь, д. 10, подъезд 11, Госхимиздат
СОДЕРЖАНИЕ
Предисловие ..................................................... 9
Глава I. Введение............................................... 13
I. Элементарная сера......................................... 14
1. Физические и химические свойства серы.................. —
2. Применение серы...................................... 16
11. Сернистый ангидрид....................................... 18
1. Физические свойства сернистого ангидрида..........
2. Химические свойства сернистого ангидрида . ....... 21
111. Серный ангидрид........................................ 22
'•/ IV. Серная кислота......................................... 23
1. Химические свойства серной кислоты и ее применение . .
2. Физические свойства серной кислоты.................. 27
Плотность растворов серной кислоты .................... —
Теплоемкость......................................... 28
Температура замерзания . . . • •...................... —
Упругость паров и температура кипения растворов сер-
ной кислоты . . • • •.............................. 29
Теплота парообразования.............................. 31
Теплота растворения, разведения и смешения.........
Вязкость растворов серной кислоты.................... 32
3. Стандарты серной кислоты............................ 33
4. Примерные расчеты.................................... —
V Материалы для аппаратуры сернокислотного производства . • . . 35
1. Материалы природного происхождения................... —
2. Материалы искусственного происхождения.............. 36
Огнеупорный кирпич.................................... —
Каменный товар и фарфор........................... 37
Эмаль................................................. —
Кислотоупорные цементы и бетоны....................... —
3. Металлы....................•........................ 38
Черные металлы...................................... —
Кремнистые чугуны.................................... 39
Хромистые стали и чугуны ............................. —
Кислотоупорные бронзы . . 40
Свииец>‘....................•......................... —
Алюминий............................................. 41
VI. Хранение и перевозка серной кислоты ..................... —
1. Хранение серной кислоты ... —
2. Перевозка серной кислоты . 44
Литература......................................... 45
3
Стр.
Глава 2. Сернистое сырье и его подготовка....................... 48
I. Виды сернистого сырья ........................................ —
1. Самородные серные руды.................... . . . 49
2. Рядовой серный колчедан............................... 50
3, Флотационный колчедан.................................. 52
рОГазы металлургических печей............................. 53 \
5. Углистый колчедан .................................... 54
6. Отходы углеобогатительных фабрик...................... 55
7. Дымовые газы......................................
8. Сероводород и газоочистительная масса ................ 56
9. Доменные шлаки ....................................... 57
10. Кислые гудроны.......................................... —
11. Сульфаты.............................................. 58
12. Травильные растворы.................'.................. 60
13. Соотношение отдельных видов сырья....................... —
II. Дробление колчедана......................................... 62
1. Склад колчедана.......................................... —
2. Схема дробильной установки.........................• 64
3. Щековая дробилка...................................... 66
4. Вальцовая мельница ..................................... 68
5. Барабанный грохот..................................... 70
6. Подсобное оборудование дробильных отделений............. 71
7. Ситовой анализ.......................................... 72
III. Сушка флотационного колчедана.................• . . . . 73
Литература............................................... 78
Глава 3. Обжиг сернистого сырья и очистка обжигового газа . 80
I. Теоретические основы обжига..............................
1. Химизм горения колчедана и его примесей.................. —
2. Выход огарка, выгорающий процент серы, сера в огарке . 82
3. Состав обжиговых газов.................................. 84
4. Теплота горения колчедана............................... 88
5. Скорость горения колчедана.................• . . . . 90
6. Примерные расчеты....................................... 95
И. Печи для обжига сернистого сырья............................. 98
1. Механические печи для обжига колчедана................... —
Конструкции механических печей.....................’ —
Режим и работа механических печей...................... 106
Особенности сжигания флотационного и углистого колче-
дана ................................................ 111
Дальнейшие возможности интенсификации механических
печей............................................. —
2. Печи пылевидного обжига................................ 114
Конструкции печей пылевидного обжига................... —
Производительность и технологический режим печей пыле-
видного обжига....................................... 118
3. Горизонтальные вращающиеся печи........................ 120
4. Контроль за работой печного отделения.................. 124
5. Печи для сжигания серы................................. —
[ 6. Металлургические печи............................... 127
Печи медеплавильных заводов............................ —
/_ Печи для обжига цинковой обманки....................... 132
7. Получение сернистого газа из гипса..................... 134
III. Подача колчедана к печам и отвоз огарка................... 137
1. Подача колчедана к печам . . . •..........- ... —
- 2. Выгрузка и удаление огарка . ..................... 139
4
Стр.
IV. Газоходы печных отделений.............................. 142
V. Использование огарка................................... 143
1. Извлечение меди.......................................... —
2. Агломерация огарка..................................... 145
3. Другие возможности использования огарка................ 148
VI. Очистка печного газа от пыли........................... —
1. Механические способы отделения пылн.................... 149
Осаждение пыли под влиянием силы тяжести.............. —
Циклоны и мультиклоны................................... 152
2. Электрическая очистка газа............................. 155
Теоретические основы................................ —
Электрофильтры.......................................... 162
Подстанция.............................................. 168
Глава 4. Производство серной кислоты методом контактного
окисления................................................. 175
1. Основные схемы контактного процесса . . . . ............ —
11. Специальная очистка и осушка газа...................... 183
1. Очистка газа от мышьяка и тумана серной кислоты ... —
Способ Тентелевского завода............................. 185
Способ Герресгоф-Байера................................. 189
Система Кемико.......................................... 198
2. Осушка газов........................................... 199
Абсорбция паров воды серной кислотой ................... —
Сушильное отделение...............-................. 204
3. Фильтр-маслоотделитель................................. 207
4. Перемещение газов............................ ... 208
5. Контроль работы отделений очистки и осушки газа . . . 212
111. Теоретические основы контактного окисления SO2. ..... 213
1. Равновесие реакции 2SO2 -|- О2 — 2SOa............... —
Теоретически возможная степень контактирования .... 214
Влияние температуры .................................... 216
Влияние начального состава газа.......................... —
Влияние давления ............................. ..... 217
2. Скорость окисления SO2.................................. —
3. Катализаторы для окисления SO2 ........................ 221
Платиновые контактные массы............................. 223
Ванадиевые контактные массы............................. 225
Определение каталитической активности контактных масс . 227
4. Кинетика и механизм окисления SO2...................... 229
Платиновые контактные массы .... —
Ванадиевые контактные массы............................. 234
5. Отравление контактных масс............................. 238
6. Оптимальный температурный режим процесса окисле-
ния SO2................................, . . . . ... 244
7. Определение объема ванадиевой контактной массы для
заданной степени контактирования......................... 247
8. Оптимальная концентрация SO2........................... 254
9. Ведение контактного процесса при повышенном давле-
нии .................................................... 258
IV. Контактные аппараты и контактные узлы..................... 260
1. Классификация конструкций контактных аппаратов .... —
2. Контактные аппараты с промежуточным теплообменом . • 261
3. Контактные аппараты с внутренним теплообменом . . . . ' 268
Теоретически необходимый теплоотвод................... —*
5
Стр.
Трубчатые аппараты с загрузкой контактной массы вну-
три труб............................................ 571
Контактные аппараты с двойными теплообменными труб-
ками и расположением контактной массы в междутруб-
ном пространстве . . . . •.......................... 275
4. Сравнительная оценка различных конструкций. Схема кон-
тактного узла для работы на газе высокой концентрации 280
5. Контроль работы контактного узла...........• • . . . 282
V. Абсорбция серного ангидрида............................ 284
1. Теоретические основы................................ —
2. Непосредственная конденсация SO3 с парами воды . . . 291
3. Схемы и аппаратура абсорбционных отделений.......... 293
4. Режим и работа абсорбционного отделения............. 298
VI. Расходные коэфициенты.................................. ЗОЭ
VII. Производство высокопроцентного олеума................. 303
VIII. Особые формы контактных установок................• . . 305
1. Контактный завод, работающий на'сере................ —
2. Контактная система на принципе мокрого катализа • . . —
XI. Задачи в области интенсификации н дальнейшего усовершен-
ствования контактного процесса......................... 307
Литература................................................. 308
Глава 5. Производство серной кислоты при помощи окислов
азота..................................................... 311
I. Принципиальная с£ема башенного и камерного способов ... —
1. Камерный способ................................ . .' —
2. Башенный способ..................................... 312
3. Краткие исторические сведения....................... 313
II. Фнзико-хнмнческие основы нитрозного метода............. 314
1. Основные процессы н химические теории нитрозного ме-
тода ...................................................... —
2. Окислы азота в газах камерных и башенных систем . . . 318
Равновесие и скорость реакции 2NO -|- О2 = 2NO2- • . . 319
Реакция образования N2O4............................... 322
Реакция образования N2O3 . ........................... 324
3. Окислы азота в кислотах камерных и башенных систем . —
Растворимость окнслов азота в серной кислоте............. —
Скорость абсорбции окислов азота растворами серной
кислоты......................................... 329
Эксорбция окислов азота из нитрозы.................... 334
4. Зависимость скорости кислотообразования от различных
факторов .................................................. —
Скорость кислотообразования в камерах.................. 335
Скорость кислотообразования в условиях башенного про-
цесса ......................'................... 337
5. Влияние давления на процессы абсорбции и кислотообра-
зования .............................................. 340
6. Окисление аммиака................................... 343
III. Аппаратура башенных систем •.................... . . • 345
1. Замена свинца......................................... —
2. Башни.............................................. 346
3. Насадка башен....................................... 352
4. Приборы "Для разбрызгивания кислоты в башнях и воды
в камерах............................................... 356
6
Стр.
5. Хвостовые вентиляторы................................. 360
6. Брызгоуловители....................................... 362
7. Холодильники.......................................... 364
8. Баки и кислотохранилища............................... 366
9. Насосы.............................................. 367
Насосы Амаг-Гильперта................................... 369
Насосы Бейзе............................................ 370
10. Коммуникация.......................................... 373
11. Аппаратура для снабжения башен и камер окисламн
• азота.....................-........................... 375
12. Камеры................................•............... 377
IV'. Технологический режим башенных и камерных систем .... 379
1. Характер встречи газа п кислоты в башенных системах . —
2. Орошение башен............• . • - •................... 380
Схема орошения............................................ —
Количество орошения..................................... 381
Концентрация орошающих кислот........................... 382
3. Температура кислот и газа . . . •.............. . —
4. Концентрация SO2 при входе в систему.................. 383
5. Нитрозность кислот и денитрация нитроз................ 385
6. Переработка SO2, эксорбция и абсорбция окислов азота
по отдельным башням................................... 389
7. Тепловой баланс башенной системы и схема охлаждения
кислот.................................................... 391
8. Подготовка окислов азота к абсорбции.................. 394
9. Технологический режим камерной системы................ 397
10. Контроль производства башенных н камерных систем . . 399
11. Расходные коэфициенты в башенных н камерных систе-
мах ....................................................... 402
12. Задачи и пути дальнейшей интенсификации и усовершен-
ствования нитрозного метода ......................... 408
Литература...................................................... 412
Глава 6. Концентрирование серной кислоты.......................... 415
I. Теоретические основы......................................... —
И. Установки для концентрирования с непосредственным обогре-
вом кислоты............................................. 421
1. Схемы производства........................................ —
2. Аппаратура............................................. 424
Топки..................................................... —
Концентраторы........................................... 426
Аппараты для улавливания тумана и брызг серной кислоты . 429
3. Режим и работа концентрационных установок Кесслера и
Кемико................................................. 431
4. Контроль производства.................................. 434
5. Концентрирование башенной кислоты теплотой обжиговых
газов ... •................................................ 436
III. Установки с внешним подогревом............................. 438
1. Установки, работающие по принципу дефлегматорных
колонн ...................................................... —
2. Вакуум-концентратор..................................... 439
IV. Дальнейшие задачи в области концентрирования серной кис-
лоты ................................................... 445
Литература..................................................... Ж>
7
Глава 7. Производство элементарной серы .......................
I. Извлечение серы из самородных серных руд................
1. Термический метод ... •............................
Калькароны..................•.......................
Печи Жилля..........................................
Паровая выплавка серы...............................
2. Физико-тер.мнческий метод..................... - . .
Способ Фраша........................................
Выплавка серы в автоклавах.................... . .
3. Экстракция серы растворителями..................- .
4. Размол комовой серы................................
I] . Получение серы из сероводородсодержащих газов.......
1. Метод сухой очистки...............................
Очистка активированным углем.......................
2. Методы мокрой очистки............................
Нейтрализационный метод............................
Окислительный метод................................
Очистка железо-содовым и железо-аммиачным растворами
(способ Феррокс)...................................
Очистка мышьяково-содовым и мышьяково-аммиачными
растворами (способ Тайлоке)........................
Голландский способ Пти ............................
Электрический способ Фишера.........•..............
Способ Фельда......................................
3. Катасульфпроцесс ...............................
Ill. Получение серы из сероводорода......................
IV. Получение серы нз сульфидных руд и сернистого ангидрида .
1. Метод Шабалина....................................
2. Получение серы восстановлением SO2...............
3. Метод Оркла.................•....................
V. Очистка и рафинировка серы............ . . ..........
Литература...............................................
Глаза 8. Концентрирование сернистого газа....................
I. Метод вымораживания.........•.........................
II. Циклические методы обогащения SO2....................
1. Способ водного обогащения.........................
2. Обогащение посредством аммиачного раствора.......
3. Основной сульфат алюминия как абсорбционный раствор
при циклическом методе ..............................
4. Сульфидиновый способ..............•..............
111. Концентрирование SO2 методом адсорбции................
IV. Другие методы использования и концентрирования SO2 . . .
1. Аммиачный метод....................................
2. Кислотный и известково-кислотный методы использова-
ния SO2 топочных газов............................. . . .
V. Выбор метода использования и концентрирования SO2 . . .
Литература.................................................
Предметный указатель
Стр.
447
448
449
455
457
458
459
460
461
462
463
465
467
470
471
474
476
478
482
485
486
488
489
492
495
5.0
502
504
505
506
509
510
511
512
8
ПРЕДИСЛОВИЕ
В настоящем учебнике вопросы технологии серной кислоты
подвергнуты достаточно подробному рассмотрению, по техноло-
гии же серы дан лишь принципиальный обзор методов производ-
ства. Это объясняется тем, что в учебном плане курса специ-
альной технологии серной кислоты и серы вопросам технологии
серы из общего количества 80 час. отведено только 8 час. На
такое количество учебных часов не представляется возможно-
сти дать материал в большем объеме.
В отступление от действующей программы курса специаль-
ной технологии в связи с особой актуальностью проблемы ис-
пользования отбросных,' содержащих SO2, газов в книгу вве-
дена глава о концентрировании сернистого газа.
Вопросы контроля производства излагаются в связи с рас-
смотрением технологического режима каждой установки. Изло-
жение самих методов анализа, а также описание конструкций
контрольно-измерительной аппаратуры в книге не дается; эти
вопросы не входят в программу лекционного курса и изучаются
в связи с техническим анализом и специальной лабораторной
работой; этим вопросам посвящается подготовляемое НИУИФ
к изданию специальное пособие («Контроль производства сер-
ной кислоты» под редакцией К- М. Малина и Г. В. Рабов-
с к о го).
При подборе и расположении материала мы руководствова-
лись теми принципами, которые были положены в основу курса
«Технологии серной кислоты и серы» (Малин), изданного
в 1935 и 1937 гг. Против указанных принципов за истекшие
годы не было возражений ни со стороны втузовских, ни со сто-
роны заводских работников.
Основные главы курса изложены по следующей схеме:
принципиальная схема производства; физико-химические основы
производства; аппаратурное оформление процесса; технологиче-
ский режим; пути уменьшения расходных коэфициентов, воз-
можности интенсификации и дальнейшего развития данного
метода.
При описании каждого отдельного производства (на-
пример производство серной кислоты контактным методом)
материал расположен не по различным схемам производства
(Герресгоф-Байера, Тентелевская и др.), а по стадиям производ-
ственного процесса (очистка газа, осушка газа, контактное оки-
сление SO-. и др.). г
Наряду с общими основами каждого производственного про-
цесса в книге дается и практический материал технологиче-
ского и аппаратурного характера.
Однако более или менее детально мы описывали только са-
мые основные аппараты. Мы не давали также указаний практи-
ческого характера на все случаи производственной деятельно-
сти, полагая, что никакой курс не может разрешить те задачи,
которые разрешаются уже практическими навыками. Все необ-
ходимое для сознательного овладения основными процессами,
по нашему мнению, в книге дано.
Основы расчета производственных процессов и частично
аппаратуры в книге даются в связи с рассмотрением теорети-
ческих основ производства; отдельных расчетов в конце глав,
а также задач для самостоятельных расчетов не дано.
Авторы и подбором и характером изложения материала стре-
мились показать учащемуся:
1. Тесную связь сернокислотной и серной промышленности
с рядом других отраслей народного хозяйства как по линии
применения серной кислоты, сернистого газа и серы, так и по
линии использования отходов других -производств в качестве
сырья для сернокислотной и серной промышленности, а также
использования отходов сернокислотного производства.
2. Изменение производственных схем данной отрасли про-
мышленности, отмирание стареющих схем, появление новых бо-
лее совершенных схем в связи с развитием других отраслей
народного хозяйства (требующих продукта определенного сорта,
поставляющих новые виды сырья, строительные материалы и
оборудование) и достижениями науки.
3. Наличие противоречий в каждой производственной схеме,
в каждом принятом технологическом режиме, открывающих
возможности дальнейшего совершенствования.
Материал учебника показывает также, что изменение про-
цесса идет во взаимосвязи с изменением его аппаратурного
оформления. Так, интенсификация производственного процесса
выдвигает новые повышенные требования к материалу и кон-
струкции аппаратуры, а выполнение повышенных требований
к аппаратуре в свою очередь открывает новые возможности
более широкого использования технологических факторов для
дальнейшей интенсификации производственного процесса. Это
-положение должно воспитать в студенте одинаково серьезное
отношение и к вопросам технологии производства и к произ-
водственной аппаратуре, начиная от важнейших аппаратов и
кончая коммуникацией.
4. Зависимость общего результата производственной работы
от множества (технологических, аппаратурных, организацион-
ных) взаимосвязанных факторов.
В связи с этим курс специальной технологии должен пока-
зать студенту, что практически неисчерпаемые возможности
производства превращает в действительность лишь тот, кто учи-
(О
тывает, должным образом оценивает и направляет все стороны
производства вплоть до мелочей. Так, например, никакое знание
физико-химических основ процесса, никакое построение техно-
логического режима не обеспечат хорошей работы башенной
сернокислотной системы, если во-время не проверяются и не
исправляются оросительные приборы на башнях или если во-
время не очищаются от грязи кислотные холодильники. Ряд до-
стижений сернокислотного производства (интенсификация пе-
чей, повышение производительности башен и др.), нашедших
отражение в книге, показывает, что стахановцы-рабочие и ин-
женеры сернокислотной промышленности, сочетающие в себе
коммунистическое отношение к труду и овладение новой техни-
кой с правильной организацией труда, дали немало примеров
превращения возможностей в действительность.
В связи с тем, что серная кислота является полупродуктом
в громадном числе производств, поставленная XVIII Съездом
ВКП(б) задача — догнать и перегнать передовые капиталистиче-
ские страны также и в экономическом отношении — для серно-
кислотного производства имеет особую актуальность.
Материал технологии серной кислоты и серы показывает
также, что эти области производства особенно тесно связаны
с задачей ликвидации ряда народнохозяйственных диспропор-
ций, являющихся пережитками капитализма в экономике нашей
социалистической промышленности (выбрасывание огромных
масс сернистых газов в атмосферу, неиспользование кислых
гудронов, отходов углеобогащения и др.).
Для разрешения обеих этих основных проблем XVIII Съезд
ВКП(б) поставил перед сернокислотной промышленностью ряд
вполне конкретных задач: дальнейшее расширение производ-
ства; уменьшение расходных норм сырья и энергии; расширение
применения местного сырья и освоение новых видов местного
сырья; использование отходящих газов цветной металлургии и
электростанций и других отходов; развертывание производства
в восточных и дальневосточных районах. Данный в книге мате-
риал о громадных сырьевых ресурсах в • СССР об еще неис-
пользованных возможностях интенсификации действующих за-
водов, о возможностях разработки новых более совершенных
схем производства и др. показывает, что стоящие перед серно-
кислотной промышленностью задачи могут быть полностью раз-
решены.
Однако эти возможности могут быть превращены в действи-
тельность не в порядке самотека, а в процессе упорной работы и
борьбы. В связи с этим и при изучении курса специальной тех-
нологии нельзя ограничиваться только материалом имеющихся
учебников и учебных пособий, а необходимо использовать также
текущий практический опыт промышленности и ее отдельных
работников.
Разделы «Материалы для аппаратуры сернокислотного про-
изводства» и «Хранение и перевозка серной кислоты» (гл. I)
и
написаны Н. И. Смысловы м. Разделы «Сушка флотацион-
ного колчедана» (гл. 2) и «Подача колчедана к печам и отвоз
огарка» (гл. 3) написаны Н. Л. Аркины м. Разделы «Очистка
печного газа от пыли» (гл. 3) и «Очистка газа от мышьяка и
тумана серной кислоты» (гл. 4) изложены И. Л. П е й с а х о-
в ы м. Раздел «Теоретические основы 'контактного окисления
SO2» (гл. 4) написан Г. К. Боресковы м, а раздел «Кон-
тактные аппараты и контактные узлы» — Г. К. Боресковым
и М. Г. С л и н ь к о. Раздел «Аппаратура башенных систем»
(гл. 5) написан М. Н. Второвы м. Все остальное написано
К. М. Малины м, которым проведена также работа по редак-
тированию и увязке всего материала.
Кроме указанных авторов ряд товарищей оказал содействие
в работе по подготовке книги или просмотром отдельных ча-
стей рукописи (Г. П. Л у ч и н с к и й, А. Г. Амели н, 3. П. Р о-
зенкноп, И. Н. Шок ин, А. В. А в д е е в а, Г. Б. Б л ю м,
А. Г. С о к а л ь с к и й, Н. Е. Кириченко, Д. Ф. Терентьев,
М. А. К л о к а ч е в) или предоставлением необходимого
материала (М. Н. Степанов, М. А. К л о к а ч е в, 3. П. Р о-
зенкноп, Г. В. Рабовский, А. В. Авдеева). Всем этим
товарищам выражаем свою благодарность. Выражаем также
благодарность тт. В. Н. Ш у л ь ц у , Б. Д. М е л ь н и к у и
С. В. Б е н ь к о в с к о м у, просмотревшим всю рукопись в целом
и сделавшим по ней ряд ценных замечаний.
Все указания на недостатки этой книги будут приняты с бла-
годарностью.
К. Малин
20/Х 1940 г.
ГЛАВА 1
ВВЕДЕНИЕ
Сера была известна в самой глубокой древности. За 1600—
2000 лет до нашей эры в Египте применяли ее соединения для
отбелки тканей и для получения красок. В древнегреческой
поэме Гомера упоминается сера как дезинфекционное средство,
римляне применяли ее для фармацевтических целей. Во времена
Цезаря (I в. до н. э.) сера применялась для военных нужд. Осо-
бое значение она приобрела в XII в. с изобретением пороха.
Широкое применение серы в промышленности началось в конце
XVIII в. в связи с получением из нее серной кислоты.
По содержанию в земной коре (0,1%) сера среди химических
элементов занимает 14-е место. Полезная добыча (в составе
самородных руд и сульфидов) серы (для потребления в виде
элементарной серы и для переработки в сернистый газ и серную
кислоту, но не считая неиспользуемую серу, извлекаемую из
недр земли при добыче руд цветных металлов) составляет
7—7,5 млн. т в год.
Серная кислота впервые бурное развитие получила в связи
с производством соды по Леблану (большие количества, серной
кислоты требовались для получения сульфата натрия из пова-
ренной соли). Первые три четверти прошлого столетия произ-
водства соды и серной кислоты были ведущими и основными
отраслями химической промышленности. С вытеснением лебла-
новского метода производства соды более совершенным соль-
веевским методом, не требующим применения серной кислоты,
серная кислота, однако, не утратила своего значения как важ-
нейший полупродукт. Наоборот, в связи с возникновением новых
отраслей промышленности — искусственных удобрений, взрывча-
тых веществ, искусственных красителей — потребность в сер-
ной кислоте непрерывно увеличивалась и производство ее рас-
ширялось.
В XX в. круг потребителей серной кислоты продолжал рас-
ширяться (нефтяная промышленность, металлообрабатывающая
промышленность, промышленность искусственного шелка и др.),
и в настоящее время сернокислотная промышленность по тон-
нажу выпускаемой продукции остается в ряду самых мощных
отраслей химической промышленности: содовой, азотной и удо-
брений. По разнообразию же применения серная кислота до
сих пор несомненно продолжает удерживать первое место среди
всех химических продуктов.
J3
I. Элементарная сера
1. Физические и химические свойства серы
В обыкновенных температурных условиях сера находится
в твердом состоянии. Твердая сера существует в нескольких
молекулярных модификациях. Наиболее устойчивой молекуляр-
ной модификацией твердой серы является восьмиатомная сера,
, существующая в двух аллотропных формах. Ниже 95,5° устой-
чива обыкновенная желтая сера, кристаллизующаяся в ромбиче-
ской системе. Удельный вес ее 2,06, температура плавления
(при очень быстром нагревании) 112,0°. Выше 95,6 устойчи-
вой является форма, кристаллизующаяся в моноклинической
системе. Удельный вес моноклинической серы 1,96, температура
плавления 119,0°.
Другие молекулярные модификации твердой серы (шести-
атомная и четырехатомная) получаются в специальных усло-
виях и не имеют большого практического значения. Из разных
видов аморфной серы больше всего заслуживает внимания пла-
стическая, или каучукоподобная, сера, не растворимая в серо-
углероде. Такая модификация получается, если кипящую серу
вылить в холодную воду. Предполагают, что эта модификация
представляет собой смесь нескольких молекулярных модифи-
каций; она неустойчива и при стоянии быстро переходит в ром-
бическую серу.
Скрытая теплота плавления ромбической серы (при 112,8°)
9,4 ккал, а моноклинической (при 119°) 10,8 ккал на 1 кг.
При расплавлении сера переходит в желтую легкоподвиж-
ную жидкость. Выше 160° жидкость буреет и при 200° пре-
вращается в вязкую темнокоричневую массу. Выше 250'
вязкость опять уменьшается и при температуре 400 расплавлен-
ная сера, оставаясь темнокоричневой, опять становится легко-
подвижной. Эти изменения связаны с изменением соотношений
между различными молекулярными модификациями при повы-
шении температуры. Жидкая сера кипит при 444,6° (при нор-
мальном давлении).
Атомная теплоемкость твердой серы зависит от темпера-
туры; для ромбической серы эта зависимость будет:
C(J = 4,12 4-0,0047 7,
для моноклинической:
СР = 3,62 4- 0,0072 7.
Зависимость средней молекулярной теплоемкости газообраз-
ной двухатомной серы от температуры выражается так:
Ср = 4,49 4- 0,001 7.
Скрытая теплота испарения серы 362 ккал. Упругость насы-
щенных паров серы изменяется в зависимости от температуры
(табл. 1).
)4
Таблица 1
Температура в °C Упругость паров серы в мм рт. ст. Температура в °C Упругость паров серы в мм рт. ст.
49,7 0,00034 242 8,4
78 0,002 245 10,0
104 0,01 265 20,5
131,9 0,081 306,5 53,5
135 0,1 342 106
141 0,13 363 176
157 0,33 374 240
172 0,63 393 . 436
181 1,0 410 443
190 1.4 427 580
211,3 3,14 444,6 760
Газообразная сера в интервале температур 800—1400° со-
стоит, главным образом, из двухатомных молекул, при более
низких температурах двухатомные молекулы переходят в моле-
кулы Se и Ss; при температуре ~1700° газообразная сера со-
стоит из одноатомных молекул. В табл. 2 приводится состав
газообразной серы в зависимости от температуры выше точки
кипения при общем давлении паров серы, равном 1 ат.
Таблица 2
Темпера- тура в °C Давление паров в am Сера в виде $2 в % от об- щего коли- чества серы
s2 S6 S6
444,6 0,038 0,546 0,416 1.1
500 0,130 0,545 0,325 4.2
550 0,305 0,490 0,205 11,0
700 0,790 0,181 0,025 56,0
750 0,930 0,06.9 0,003 81,0
100 0,970 0,029 0,001 92,0
В связи с изменением атомности молекул парообразной серы
происходит изменение цвета паров: от оранжево-желтого
(вблизи точки плавления) до красного и затем до соломенно-
желтого (при 65(F).
В воде сера не растворима; она хорошо растворяется в не-
которых органических жидкостях, например в сероуглероде,
гидрированном нафталине и хлорированном бензоле, а также
в хлористой сере (табл. 3 и 4).
15
Таблица 3
Растворитель Температура и °C Растворяется серы на на 100 вес. ч. растворителя
Бензол .... Бензсл .... Толуол .... Эфир Фенол .... Хлорифсрм . . Анилин .... 26 71 23 23,5 174 22 130 0,965 4.377 1,479 0.972 16,''5 1,208 85,96
Сера плохо проводит тепло и электричество; под действием
трения заряжается отрицательным электричеством. Сера
является одним из самых активных химических элементов.
По своему химическому
характеру сера выступает
как окислитель (атом серы
присоединяет два электрона,
приобретая валентность — 2)
и как восстановитель (атом
серы отдает 4 или 6 элек-
тронов).
2. Применение серы
Сера и ее многочислен-
ные соединения имеют са-
мое разнообразное примене-
ние.
Таблица 4
Температура в СС Вес. ч. серы в 100 вес. ч. CS3
— 11 16,54
— 8 17,75
± 0 23,99
--I5 46,05
+ 38 94,57
--48 146,21
+ 55 181,34
Мировое производство элементарной серы составляет
~3 млн. т в год. Главные производители серы: США —
2092 тыс. т (1939 г.), Италия — 372 тыс. т (1937—1938 гг.) и
Япония— 175 тыс. т (1936 г.).
В дореволюционной России серной промышленности почти не
было: в 1901 г. было добыто 2589 т, а в 1911 г. добыча элемен-
тарной серы прекратилась совсем. Перед первой империалисти-
ческой войной в Россию импортировалось около 25 тыс. т серы
в год. Теперь создана советская серная промышленность, однако
производство серы и в настоящее время в СССР еще далеко не
достаточно.
Около половины всей мировой продукции элементарной серы
идет на производство серной кислоты (преимущественно
в США), около четверти (в виде SO2) потребляется бумажной
промышленностью. 10—15% мировой добычи серы идет на
борьбу с вредителями сельского хозяйства и остальные 10—
15% расходуются на все другие цели. Из этих других целей
радо прежде всего указать на производство сероуглерода, по-
16
лучаемого при взаимодействии паров серы с раскаленным углем.
Кроме того, элементарная сера применяется для приготовления
спичек, в пиротехнике, в резиновой промышленности, для про-
изводства ультрамарина и минеральных красок. Не так значи-
тельно по масштабу, но очень многообразно применение серы
в медицине. В последние годы сера начинает применяться в про-
мышленности строительных материалов (специальные цементы,
литые трубы, фильтры для очистки сильно корродирующих жид-
костей и др.), а также для пропитки дерева и бумаги.
Сера и ее соединения широко применяются и для военных
нужд.
В зависимости от требования потребителей, а также от усло-
вий производства сера выпускается в виде нескольких сортов-
стандартов. В СССР сера выпускается, главным образом, в двух
видах — комовая и дисперсная; последняя получается методами
размола, распыла, возгонки или осаждения. На комовую серу и
на дисперсную предусмотрено по четыре сорта. Требования к от-
дельным сортам указаны в табл. 5.
Таблица 5
Сорта серы Ингредиенты в %
сера не менее влага не более ЗОЛЬНОСТЬ не более кислотность не более селен и и мышьяк не более
1-й 2-й 3-й 4-й (концентрат) 99,5 97 95 75 0,2 0,5 1,25 5.0 0,3 2,3 3,0 19,5 0,02 0,1 0,15 Не норми- руется 0,001 0,1 0,1 Не норми- руется
(табл. 6).
Таблица 6
Сорт дис- персной серы Сито с чис- лом отвер- стий на 1 см2 Проходит через сито не менее в %
1-й 10000 98
2-й 6 400 96
3-й 5 000 95
4-й 5000 93
К дисперсной сере предъявляются также требования в отно-
шении степени дисперсности
Кроме того, дополнитель-
ные требования предъявля-
ются к сере, направляемой
в отдельные отрасли про-
мышленности. Так, напри-
мер, сера, идущая в резино-
вую промышленность, не
должна содержать углерода
и мышьяка (допускаются
только следы их).
Комовая сера в виде че-
шуек или кусков перево-
зится навалом. Некоторые предприятия пришютот для мкмпсюня,
комовой серы старые бочки. В этом случае 'жидкая выпу-
скаемая из автоклавов, разливается в бочки, где 4
2 Зак. 5019. Технология серной кислоты.
17
Дисперсная сера, молотая и осажденная, расфасовывается
в бумажные мешки по 50 или по 25 кг. Два бумажных мешка
далее зашиваются в один джутовый. К складу хранения серы
предъявляются требования огнебезопасности и наличия противо-
пожарных средств. Перевозить серу можно любым видом тран-
спорта. При железнодорожных перевозках сера грузится в кры-
тые вагоны навалом. Вагоны с серой должны иметь на дверях
наклейку с надписью «огнеопасно» и должны находиться
в конце состава, а при стоянке на станциях отводиться в тупик.
II. Сернистый ангидрид
1. Физические свойства сернистого ангидрида
Сернистый ангидрид SO2 применяется в разнообразных отра-
слях промышленности: в холодильной технике, для консервиро-
вания, для дезинфекции и т. д. Применяется он также для
синтеза отравляющих веществ: этилдихлорарсина и дифенил-
хлорарсина; получаемый из сернистого газа и хлора хлористый
сульфурил служит для получения фосгена и бромбензилциа-
нида.
Сернистый ангидрид получается при сжигании серы и ее не-
окисленных руд, при восстановлении шестивалентных соедине-
ний серы (например гипса углем) и при термическом разложении
серной кислоты и ее солей.
При окислении серы выделяются следующие количества
теплоты:
SPOM6 + °2 = SO2ra3 + 70,9 ккал;
' Sp0M6 + О2+aq = SO2aq + 79,1 кка..;
SPOM6 + l‘/2O2 = SO3ra3 + 91,99 ккал.
В нормальных условиях сернистый ангидрид — бесцветный
газ с удушливым запахом. При 0° и нормальном давлении плот-
ность его по отношению к воздуху 2,2638.
Сернистый ангидрид легко сжижается. Он первый из всех
газов был сжижен (в 1780 г. Г. Монжом и Л. Клуе). При нор-
мальном давлении сернистый газ конденсируется при —10,01° и
затвердевает при —72°.
Критическая температура его 157,3 ', критическое давление
77,7 ат.
В табл. 7 приведены основные свойства жидкого сернистого
ангидрида в зависимости от температуры.
Зависимость средней теплоемкости (от 0° до t°) газообраз-
ного SO2 от температуры на 1 норм, /и3 газа выражается фор-
мулой:
+ = 0,437 + 0,0000847 t.
18
Таблица 7
Сернистый ангидрид
Темпера- тура в °C Упругость паров в ат Теплота испарения в ккал Уд. вес в кг/л Уд. вес насыщенных паров в кг)м^ Теплосо- держание в ккал
+ 50 8,46 69,95 1,2957 25,000 17,70
+ 40 6,35 75,03 1,3264 18,282 13,82
+ 30 4,67 79,67 1,3556 13,210 10,19
--20 3,35 83,85 1,3831 9,374 6,68
+ ю 2,34 87,56 1,4095 6,592 3,28
=t 0 1,58 90,82 1,4350 4,490 rt 0
— 10 1,03 93,60 1,4601 3,024 — 3,16
— 20 0,65 95,92 1,4846 1,950 — 6,20
— 30 0,382 97,77 1,5090 1,217 — 9,31
- 40 0,221 99,17 1,5331 0,717 — 11,94
— 50 0,119 100,10 1,5572 0,402
Теплота растворения 1 моля газообразного SO2 в воде при
15—20° составляет 8200 кал, а жидкого 2300 кал. Следова-
тельно, теплота конденсации составляет ~ 5900 кал.
Сернистый газ значительно растворим в воде. В табл. 8 при-
ведены значения растворимости SO.2 в воде в зависимости от
температуры при давлении
SO2 над раствором 760 мм
рт. ст.
SO2 растворяется в спир-
те лучше, чем в воде; один
объем спирта при 0° погло-
щает 216,5 объема SO2.
В одном объеме камфоры
при 0° растворяется свыше
300 объемов SO2, в муравьи-
ной кислоте — 351, а в аце-
тоне — 589 объемов.
С хлористой серой SO2
смешивается во всех оро-
шениях.
Жидкий сернистый ангид-
рид сам растворяет очень
многие вещества; растворы
солей в жидком сернистом ангидриде хорошо проводят электри-
ческий ток.
Таблица 8
Темпера- тура в °C 1 л воды растворяет SO2 в г % SO2 в рас- творе Уд. вес рас- твора
0 228,5 18,56 1,0609
5 193,1 16,19 1,0590
10 161,9 15,93 1,0547
15 155,5 11,92 1,0420
20 112,7 10,12 1,0259
30 81,3 7,4
40 57,0 6,1
50 40,8 4,9
В табл. 9 приведены данные об упругости SO2 и НгО над
водными растворами сернистого ангидрида в зависимости от
температуры и концентрации SO2 в растворе. Из этих данных
видно, что растворимость SO2 в воде лишь немного отступает
от закона Генри.
2»
19
Таблица
Упругость паров SO3 и HSO (в мм рт. ст.) над водным раствором SO2 о001 о со 1 -ГКО ь- Щ • CM to со
о «ч X о со с* о о оо о
О о О’. о со 1 юю о CM tF CC Т* CM TF со о
О X о с\ 525 524 523 523
о О об о 1 см о со О ХКтГтГ —< со ю ь-
о X Ю tF rF СО СО Ю io ю ю ю ь- се со со со
о е* О ел TF Г-ICD ОО СМ со —< CM ^F Ю
о «ч X tF ’Ф СО СО СО со со со со со см см см см см
о О о G4 О сл 4-4 b- со ^F СМ — 4-4СМ ’TF со •— CM СО ^F CD
о см X to СМ О go_o — СП СП СП оо со со ^F ’~F ^F ^"4 4 4 ”* 4
о О to <м о со со г- со *—* со 00 CD ^F СО •— СП г-4 СМ СО тОО
о е* X Ю СО CNOOO’T см’см'смсм’"-'—*'—- 0 0 0 010 01(0
о rF с-1 о сл OOr-'tOCOCOr-rF ССМХтГ.^-ЩО Г-4 Г-4 СМ СО тО О ОС
о с-1 X со СМ О О С-О СО 10*10 I/O ^F lOtOiOtQtOtQUOiOtO
о СО сз о со СМЮСПОСООСМоОСОО TFC0CMb*b*C^C000CnO г-. г-. CM со vF LO о со
о X СО Г^Г- СО CD tOTf СО'CM 1-* о сосососооососососососо
о о см <м о со СПСПОсОг-4 tF СО г-< о CM GC*rF СП СМ10СПСМСПОС0г-00ОС0г-400 г-4г-4СМСО’Т’ТЮФ
о S'* to Ю СО со СМ СМ —« т-^0,0 СП ь? ь-Г г-Г с- о «-Ч Г“И «—< »—1 ₽-4 ’“4 ’“4 ’“*
о ст о СО г-СМ^ОООС0г-С>Ог-<С0ОО10ОС0О CM’=FOC0C0C'-CMF^CMb-CMt>CMG0C0G0’'^O Г-ir-CMCMCOCOTFTFLOOOOt^-t^-
о, X CM CM CM CM »-4~.— о О О, О. СП СП СП со со 00,00 с^спспсп'спспспспо^сп'сп'спсососбоосооооо
SO2 на 100 2 Н2О В 2 о ю о to о о о о о о О_ОО О О О 0,0 о, О о~ •—4^ СМ со ’Г 1О со* GO* СП О см со to о
20
2. Химические свойства сернистого ангидрида
При растворении SO2 в воде протекают следующие процессы:
Н2О + SO2 H2SO3 Н 4- HSO3' — 2Н’ + SO3".
Таким образом водный раствор сернистого ангидрида содер-
жит и растворенный SO2 и недиссоциированную H2SO3 и ионы
Н‘; HSO3' и SO3". Так как сернистая кислота двухосновная, то
она может давать и кислые (бисульфиты) и средние соли (суль-
фиты). В технике широко применяются и кислые и средние соли
сернистой кислоты — сульфит натрия в фотографии, для консер-
вирования пищевых продуктов и т. д.; бисульфит натрия — при
отбелке тканей, для консервирования, для получения органиче-
ских веществ (например нафтиламина), из которых производятся
краски и пр. При кипячении раствора ТЧагВОз с серой получается
гипосульфит (ГСагБгОз), который применяется в качестве анти-
хлора в красильном и ситценабивном деле, фотографии, гальва-
нопластике, в кожевенном деле, при обработке серебряных руд.
Бисульфит кальция применяется при переработке дерева в суль-
фитную целлюлозу.
При взаимодействии сернистого ангидрида с пятихлористым
фосфором получается хлористый тионил:
РС15 + SO2 = SOC12 + РОС!3.
Хлористый тионил применяется при приготовлении многих
органических веществ: красок, фармацевтических препаратов и др.
Известны многочисленные реакции присоединения SO2 (или
H2SO3) к органическим соединениям без изменения валентности
серы.
В качестве примеров реакций, идущих с уменьшением валент-
ности серы, прежде всего надо указать на взаимодействие SO2
с рядом восстановителей. SO2 в определенных температурных
условиях восстанавливается водородом, метаном, углем, окисью
углерода, сероводородом и др. до элементарной серы. Это поло-
жено в основу производства серы из сульфидных руд через по-
лучение сернистого газа с последующим восстановлением его
углем или генераторным газом.
Из реакций, в которые вступает SO2, где сера повышает свою
валентность, наиболее важна реакция окисления SO2 до серной
кислоты. Другой пример окисления SO2 — взаимодействие его
с хлором: SO2 + CI2 = SO2O2.
Сернистый газ уже при умеренных концентрациях при вды-
хании раздражающе действует на все наружные слизистые обо-
лочки. Вдыхание его в более высоких концентрациях вызывает
хрипоту, боли и чувство сдавленности в груди. Через некоторое
время становится невозможным разговор и глотание и часто
воспаляются соединительные оболочки глаз. При вдыхании сер-
нистого газа очень высоких концентраций быстро появляется
бронхит, одышка, цианоз и потеря сознания. Против отравления
сернистым газом совершенно необходимо проведение, всех
21
мероприятий, предусматриваемых на случай выделения SO2
в цех. В тех отделениях, где есть возможность при расстрой-
ствах процесса или авариях (печное отделение, контактное отде-
ление) появления SO2 в больших концентрациях, должна быть
предусмотрена хорошая вентиляция. Все работающие в этих
местах должны иметь противогазы.
III. Серный ангидрид
Свойства твердого серного ангидрида изучены еще недо-
статочно. По одним данным твердый серный ангидрид имеет три
модификации, по другим данным — четыре. Наиболее устойчи-
вой модификацией является льдообразная a-форма, имеющая со-
став SO3. Эта модификация плавится при 16,83е, при длительном
стоянии жидкости при температуре ниже 25° из нее выделяется
асбестообразная масса с шелковистым блеском. Это — другая
модификация серного ангидрида, p-форма, плавящаяся при 31°
и имеющая состав SaOe. Смесь различных модификаций серного
ангидрида устойчива даже в присутствии жидкой фазы, в связи
с чем при температурах ниже 25° можно наблюдать разные ве-
личины упругостей паров серного ангидрида.
Наиболее изучены физические свойства a-формы. Плотность
жидкого серного ангидрида в пределах температуры от 15 до
100° меняется от 1,929 до 1,529. Температура кипения 44,8°.
Скрытая теплота плавления при —30° равна 23,8 ккал, теплота
^испарения — 147,4 ккал, упругость пара над жидким SO3 равна:
Прн 24°........ 240 мм рт. ст. При 100°.......8,0 ат
При 44,8J . ... 760 „ „ „ При 200° .... 61
Зависимость упругости пара от температуры может быть
представлена уравнением:
lgJp= 10,022 — ^^.
Характерным свойством серного ангидрида является его
большая летучесть и чрезвычайная гигроскопичность; его пары,
вводимые в воздух, реагируя с водяным паром, образуют туман,
состоящий из взвешенных в воздухе частиц серной кислоты. На
этом основано применение серного ангидрида в качестве основ-
ного компонента дымообразующих смесей.
Газообразный серный ангидрид и пары серной кислоты при
их вдыхании вызывают воспаление верхних дыхательных путей,
которое может распространиться и на легкие. Концентрация
меньше 0,1 объемного процента уже делает дыхание невозмож-
ным. При попадании на кожу человека серная кислота произво-
дит сильный ожог с последующим образованием раны. Если
своевременно не промыть глаз, в который попала капля серной
кислоты то это может привести к потере глаза.
На сернокислотных заводах должна быть полностью исклю-
чена опасность упасть в какой-нибудь резервуар с серной кисло-
22
той: баки с кислотой должны быть закрыты, незакрытые резер-
вуары с кислотой должны быть ограждены; не должно до-
пускаться проведение ремонтных работ с кислотопроводами и
другими резервуарами, когда они заполнены кислотой, находя-
щейся под давлением; при обслуживании их всегда надо быть
в очках.
На тех рабочих местах и на тех работах, где есть опасность
попадания капель кислоты, необходимо быть в спецодежде (су-
конной). Ноги предохраняются резиновыми сапогами, руки —
резиновыми перчатками. Если на тело попала кислота, ее необ-
ходимо быстро смыть большим количеством воды, лучше слабо-
щелочной. Для этого в цехах на видных местах должны быть
установлены бачки с чистой, лучше с слабощелочной водой.
Обычно устанавливают бачки с раствором соды.
IV. Серная кислота
1. Химические свойства серной кислоты и ее применение
Серная кислота — одна из самых активных неорганических
кислот. Она применяется как сильная и нелетучая кислота, как
дегидратационное средство, как окислитель и для процессов
сульфирования.
По масштабам и разнообразию промышленного применения
из химических продуктов с серной кислотой конкурируют
только сода и аммиак.
Главнейшие типы химических реакций, являющиеся основой
промышленного применения серной кислоты, следующие:
1. Серная кислота дает ряд промышленных солей, реагируя
непосредственно с металлами. В качестве примера можно ука-
зать на производство сульфата цинка (Zn + H2SO4 = Z11SO4 +
+ Н2), потребляемого в производстве литопона.
2. Ряд промышленных солей получают при действии серной
кислоты на окислы металлов. Например производство сульфата
•грехокисного железа [Ре2Оз + 3H2SO4 = Fe2fSO4)s + ЗН2О],
применяемого как коагулянт; производство сульфата меди
(СиО H2SO4 = CuSC>4 + Н2О), применяемого в борьбе с вреди-
телями сельского хозяйства, а также в качестве минеральной
краски; приготовление квасцов [AI2O3 + 3H2SO4 = АЦЭС^з +
+ ЗН2О; A12(SO4)3 + K2SO4 + 24НгО = K2SO4 • A12(SO4)3 • 24НгО],
применяемых в красильном деле, при дублении кож, при получе-
нии лаков, в бумажном производстве, в водоочистке и др.
3. Реагируя с аммиаком или его водным раствором, серная
кислота дает сульфат аммония [(NHi)2SO4] — одно из важней-
ших минеральных удобрений.
4. Действуя на соли других кислот, серная кислота вытес-
няет более слабые или более летучие кислоты. Укажем следую-
щие примеры:
а) Вытеснение уксусной кислоты из ее кальциевой соли:
(СН3СОО)2Са 4- H2SO4 = 2СН3СООН -J- CaSO4. ’
23
По этой реакции из продукта сухой перегонки дерева полу-
чается уксусная кислота.
б) Из поваренной соли серная кислота вытесняет газообраз-
ный хлороводород:
2NaCl 4- H2SO4 = Na2SO4 Д 2НС1,
при абсорбции которого получается затем соляная кислота.
Раньше на этой реакции основывалось производство соды (по
Леблану).
в) При действии на чилийскую селитру серная кислота осво-
бождает азотную кислоту:
NaNO3 Д H2SO4 = HNO3 4- NaHSO4.
Раньше, когда не умели получать синтетическую азотную
кислоту, эта реакция была основой для производства азотной
кислоты.
г) При обработке серной кислотой фосфата кальция полу-
чают фосфорную кислоту:
Са3 (РОД 4- 3H2SO4 = 2Н3РО4 4- 3CaSO4
или суперфосфат:
Са8 (РОД 4- 2H2SO4 4- 5Н2О =
= Са (Н2РОД • Н2О4-2 (CaSO4 2Н2О).
д) При действии на плавиковый шпат серная кислота выде-
ляет фтористый водород:
CaF2 4- H2SO4 4- 2Н,0 = 3HF Д CaSO4 • 2Н2О.
Раствор фтористого водорода в воде — фтористоводородная
кислота — применяется для производства фторида натрия и
криолита.
5. Серная кислота (крепкая) жадно соединяется с водой. Это
ее свойство широко используется в производстве и в лаборато-
рии для осушки газов от влаги. На этом же свойстве основано
применение серной кислоты для концентрирования азотной кис-
лоты и при реакциях нитрования органических соединений, ле-
жащих в основе производства взрывчатых веществ (пикриновая
кислота, тротил, тетрил, нитроглицерин и др.), органических
красок, нитрошелка, некоторых пластических масс, в том числе
кинопленки и целлулоида. При нитровании водородный атом
заменяется нитрогруппой по реакции:
R • Н ДНО • NO2 = R • ЬЮ2Д Н2О.
Находящаяся в нитрующей смеси серная кислота связывает
выделяющуюся воду и тем самым сохраняет активность азотной
кислоты. Для нитрования применяются только высококонцен-
трированные сорта серной кислоты (купоросное масло и олеум).
Серная кислота жадно соединяется не только со свободной,
водой, но и отнимает ее от химических соединений. Так, при
действии серной кислоты на винный спирт получается этилен:
СН3СН2ОН — Н2О = С2Н4.
На этом же свойстве серной кислоты основано получение
эфиров из соответствующих спиртов или спиртов и кислот.
6. Как окислитель серная кислота применяется для очистки
нефтепродуктов.
7. Серная кислота (олеум) применяется для сульфирования
органических соединений.
Процесс сульфирования органических соединений состоит
в замене водорода ядра бензола и его гомологов на сульфо-
группу (т. е. SOgOH), например
/ОН /ОН
С6Не + SO2< = C6H5SO/ + Н2О.
хон
Этот процесс применяется в производстве сахарина и органи-
ческих красок.
Главным потребителем серной кислоты является химическая
промышленность. Производство суперфосфата и сульфата ам-
мония потребляет немного более половины всей мировой серно-
кислотной продукции. Остальные химические производства
берут 6—7% от мировой продукции серной кислоты, промышлен-
ность минеральных масел потребляет тоже 6—7%, на промежу-
точные продукты и краски из смол идет ~3%. В последние
годы сильно возросло потребление серной кислоты промышлен-
ностью искусственного волокна — до 6—7% от общей мировой
продукции. В связи с подготовляемыми и ведущимися уже вой-
нами в ряде стран возросло потребление серной кислоты на про-
изводство взрывчатых веществ.
Уже из вышеприведенного перечня, хотя и далеко не полного,
ясно, что серная кислота является одним из основных химиче-
ских полупродуктов, масштаб производства которого в значи-
тельной степени характеризует уровень развития химической
промышленности страны. В последние годы в число основных
химических полупродуктов встали также аммиак и азотная кис-
лота. Тенденции промышленного развития, вытесняющие приме-
нение серной кислоты, в основном состоят в следующем:
1) Развитие термической и электротермической возгонки фос-
фора, позволяющее получать фосфорную кислоту и фосфорные
удобрения без применения серной кислоты.
2) Внедрение новых методов очистки нефтепродуктов, не
требующих серной кислоты (обработка бензина хлоридом цинка,
применение методов экстракции в производстве смазочных масел
и др.).
3) Применение азотной кислоты для разложения фосфатов и
применение в качестве удобрения азота в нитратной форме. Ч
25
4) Развитие промышленности органического синтеза, даю-
щего возможность каталитического получения ряда продуктов
(уксусная кислота, муравьиная кислота, спирт и др.), производ-
ство которых раньше осуществлялось с применением серной
кислоты; получение синтетической соляной кислоты.
Однако роль серной кислоты в народном хозяйстве не умень-
шается, а абсолютная ее выработка продолжает возрастать. Рост
-мирового производства серной кислоты в тыс. т выражается
в следующих цифрах:
Годы тыс. m Годы тыс. m
1913 •....... 7 200 1935 ......... 13 800
1925 ......... 9050 1936 16000
1929 ........ 13100 1937 19000
1934 ........ 12500
Продукция сернокислотной промышленности по важнейшим
капиталистическим странам (в тыс. т) приведена в табл. 10.
Таблица 10
Страны Производство 100%-ной Н28С>4 в тыс. m
1913 г. 1929 г. 1934 г. 1935 г. 1936 г. 1937 г. 1938 г. 1939 г.
США 2100 4860 3500 3680 4350 5100 3820 4730
Япония — 710 1470 1720 2010 2620 —
Германия .... 1480 1704 1307 1567 1680 2050 2800
Великобритания . 1170 966 886 936 1043 1100 994
Франция 800 1440 810 875 1100 1180 1100
Италия 470 815 765 858 1021 1094 1150
Канада — НО 205 224 241 283 268
Рост производства серной кислоты связан с ее большим
значением для военных целей. Почти во всех странах, произво-
дящих серную кислоту, она потребляется на месте. В между-
народной торговле участвует только 1—2% от мировой серно-
кислотной продукции.
Б царской России сернокислотная промышленность вместе со
всей химической промышленностью была чрезвычайно отста-
лой. В 1913 г. серной кислоты было произведено около
165 тыс. т. Теперь один Воскресенский завод производит в год
гораздо большее количество кислоты. По производству серной
кислоты СССР сейчас стоит на четвертом месте в мире. Разре-
। шение поставленной XVIII съездом ВКП(б) задачи — догнать и
перегнать передовые капиталистические страны Европы и США
как в техническом, так и в экономическом отношении — должно
•вывести СССР по производству серной кислоты на первое место
в мире.
26
2. Физические свойства серной кислоты
В неорганической химии серной кислотой называют моногид-
рат серного ангидрида, т. е. соединение H2SO4. В технике под
серной кислотой подразумевают и смеси H2SO4 с водой, т. е.
H2SO4 + пНгО (водная серная кислота) и смеси H2SO4 с серным
ангидридом, т. е. H2SO4 + nSOs (олеум, или дымящая серная
кислота).
Серный ангидрид образует с водой-, несколько соединений.
Кроме Нг5О4(8Оз. НаО), называемым моногидратом, вполне
установлены при помощи термического анализа следующие сое-
динения SO3 о водой.
SO3-5H2O (57,6%-ная H2SO4),
SO3-3H2O (73%-ная H2SO4);
SO3-2H2O (84%-ная H2SO4);
2SO3-H2O (пиросерная кислота).
Исследования Бела (Bell) и Иепесена (leppesen) при помощи
спектра Рамана показали, что высококонцентрированная серная
кислота состоит из гомополярных молекул 50з(ОН)2, а разбав-
ленная— из гетерополярных молекул [Н2] [SO4].
Такие же исследования Шедена (Chedin) показали, что в сла-
бых растворах олеума имеются молекулы H2S2O7. По мере уве-
личения концентрации олеума в Раман-спектрё появляются ли-
нии, характерные для SOs и SaOe, а также свободной H2SO4.
С повышением температуры концентрация SaOe уменьшается за
счет увеличения концентрации SO3.
Плотность растворов серной кислоты. С увеличением кон-
центрации серной кислоты до 98,3% H2SO4 ее плотность возрас-
тает. С дальнейшим повышением концентрации H2SO4 плотность
ее падает. Плотность 100%-ной H2SO4 при 0° 1,853, а при 15°
(по отношению к воде при 4°) по Лунге и Нефу— 1,8384.
Плотность растворов серной кислоты измеряется ареометром. Для арео-
метров применяются три шкалы: по градусам Боме (° Вё), по удельному
весу и по градусам Тведделя.
Для перехода от градусов Вё к удельному весу справедливо
следующее приближенное соотношение:
где d —- удельный вес раствора, ап — число °Вё, показываемое
ареометром при погружении в раствор.
При повышении температуры объем серной кислоты увели-
чивается, а удельный вес понижается. В таблицах приводятся
удельные веса для водных растворов серной кислоты при темпе-
ратуре 15°. Если температура кислоты данной крепости (данного
процента H2SO4) не 15°, то удельный вес ее будет не тот, кото-
рый указан в таблице. К цифре удельного веса по таблице надо
сделать поправку на температуру. Величины этих поправок
в °Вё приведены в табл. И.
27
Таблица 11
Показания арео- метра в сВё Поправка на 1°С разности между 15° и температурой кислоты
0—30 ±0,07
30—45 0,06
45—65 0,05
65—66 0,04
Таблица 12
Удельный вес по замеру Поправка на ПС разности между 15° н температурой кислоты
1 —1,170 1,170—1,450 1,450—1,580 1,580—1,750 1,750—1,820 1,820—1,840 ± 0,0006 0,0007 0,0008 0,0009 0,0010 0,0008
Величина поправок на температуру в цифрах удельного веса
приведена в табл. 12.
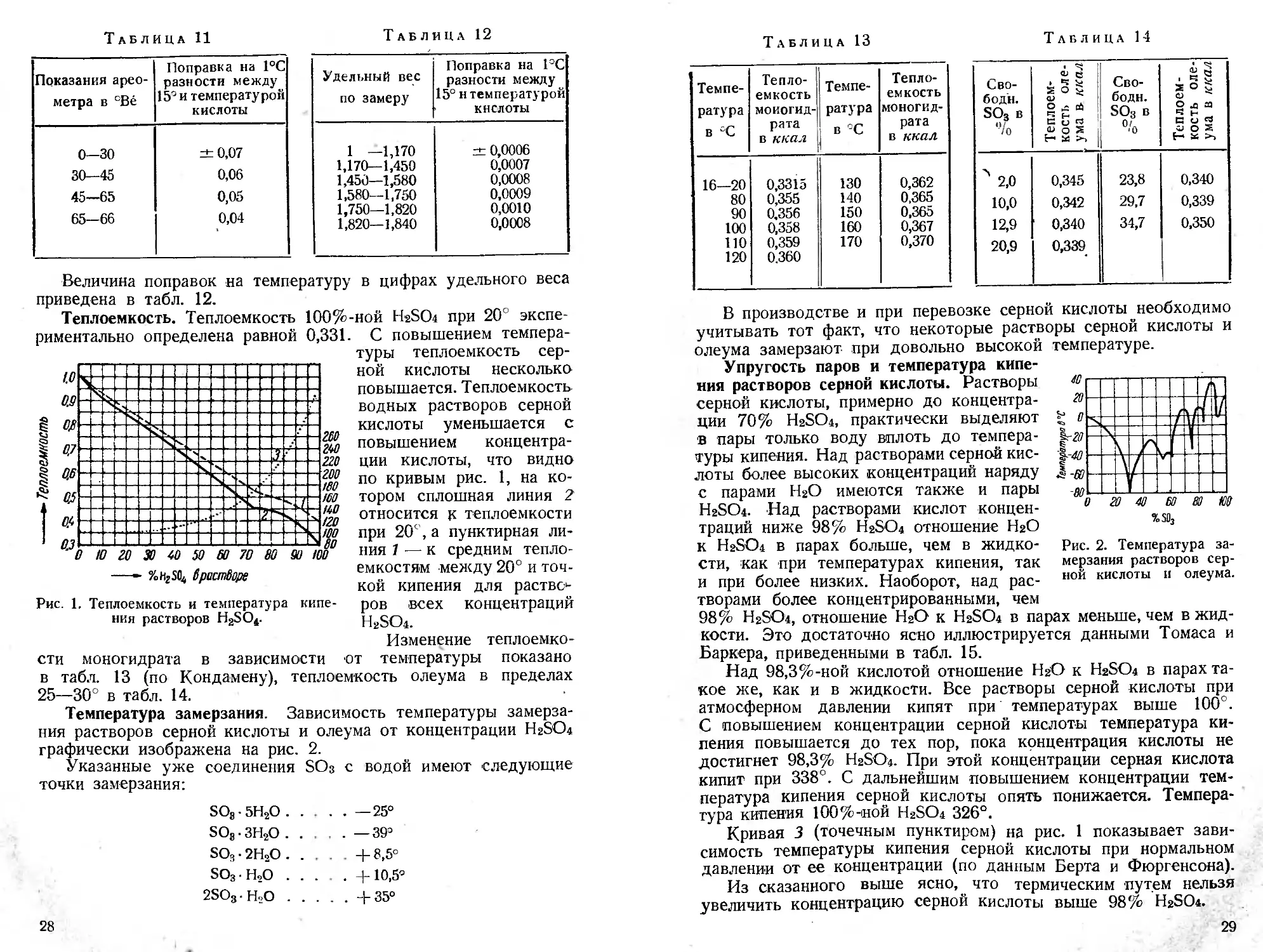
Теплоемкость. Теплоемкость 100%-ной H2SO4 при 20° экспе-
риментально определена равной 0,331. С повышением темпера-
Рис. 1. Теплоемкость и температура кипе-
ния растворов H2SO4.
туры теплоемкость сер-
ной кислоты несколько
повышается. Теплоемкость
водных растворов серной
кислоты уменьшается с
повышением концентра-
ции кислоты, что видно
по кривым рис. 1, на ко-
тором сплошная линия 2
относится к теплоемкости
при 20е, а пунктирная ли-
ния 7 — к средним тепло-
емкостям между 20° и точ-
кой кипения для раство-
ров всех концентраций
H2SO4.
Изменение теплоемко-
сти моногидрата в зависимости от температуры показано
в табл. 13 (по Кондамену), теплоемкость олеума в пределах
25—30° в табл. 14.
Температура замерзания. Зависимость температуры замерза-
ния растворов серной кислоты и олеума от концентрации Нг8О4
графически изображена на рис. 2.
Указанные уже соединения SO3 с водой имеют следующие
точки замерзания:
SOa • 5Н2О . .
SO8 • ЗН2О . .
SO3 • 2Н2О . .
SO3 H,O . .
2SO3 H<>O . .
. —25°
. —39=
+ 8,5°
. +10,5=
. +35°
28
Таблица 13
Темпе- ратура в °C Тепло- емкость моногид- рата в ккал Темпе- ратура в СС Тепло- емкость моногид- рата в ккал
16—20 0,3315 130 0,362
80 0,355 140 0,365
90 0,356 150 0,365
100 0,358 160 0,367
110 0,359 170 0,370
120 0.360
Таблица 14
Сво- боды. SO3 в О/ 70 Теплоем- кость оле- , ума в ккал Сво- боды. SO8 в 0/. 'U Теплоем- кость оле- ума в ккал
> 2,0 0,345 23,8 0,340
10,0 0,342 29,7 0,339
12,9 0,340 34,7 0,350
20,9 0,339
В производстве и при перевозке серной кислоты необходимо
учитывать тот факт, что некоторые растворы серной кислоты и
олеума замерзают при довольно высокой температуре.
Упругость паров и температура кипе-
ния растворов серной кислоты. Растворы
серной кислоты, примерно до концентра-
ции 70% H2SO4, практически выделяют
в пары только воду вплоть до темпера-
туры кипения. Над растворами серной кис-
лоты более высоких концентраций наряду
с парами НгО имеются также и пары
H2SO4. Над растворами кислот концен-
траций ниже 98% H2SO4 отношение НгО
к H2SO4 в парах больше, чем в жидко-
Рис. 2. Температура за-
сти, как при температурах кипения, так мерзания растворов сер-
и при более низких. Наоборот, над рас- ной КИСЛО1Ы 11 олеума,
творами более концентрированными, чем
98% H2SO4, отношение НгО к H2SO4 в парах меньше, чем в жид-
кости. Это достаточно ясно иллюстрируется данными Томаса и
Баркера, приведенными в табл. 15.
Над 98,3%-ной кислотой отношение НгО к H2SO4 в парах та-
кое же, как и в жидкости. Все растворы серной кислоты при
атмосферном давлении кипят при температурах выше 100°.
С повышением концентрации серной кислоты температура ки-
пения повышается до тех пор, пока концентрация кислоты не
достигнет 98,3% H2SO4. При этой концентрации серная кислота
кипит при 338°. С дальнейшим повышением концентрации тем-
пература кипения серной кислоты опять понижается. Темпера-
тура кипения 100%-ной H2SO4 326°.
Кривая 3 (точечным пунктиром) на рис. 1 показывает зави-
симость температуры кипения серной кислоты при нормальном
давлении от ее концентрации (по данным Берта и Фюргенсона).
Из сказанного выше ясно, что термическим путем нельзя
увеличить концентрацию серной кислоты выше 98% H2SO4.
29
Таблица 15
Темпера- тура в °C 99,23% H,SO4 98,0% H2SO4 95,06% H2SO4 91,26% H2SO4 89,25% H2SO4
Рнло, T’HjSO, T'lLSO, 1 Г. н..о T’HjSOj Рим T’HjSO, T’Hj.O
180 9,5 1.3 0,12 3,1 11,7 0,51 31,6 0,47 69,4
190 14,0 2,2 0,21 4,0 17,5 0,84 40,0 0,75 94,7
200 20,5 3,40 0,35 5,2 25,8 1.35 70,7 1,17 128,4
210 29,5 5,4 0,55 6,8 37,3 2,13 102,6 1,76 172,2
220 42,1 8,3 0,87 9,1 53,0 3,24 144,3 2,59 230,0
230 60,1 12,5 1,30 12,0 74,1 4,84 199,2 3,71 306,0
240 83,1 18,6 2,01 15,9 102,1 7,12 270,6 5,22 403,2
250 115,5 27,1 2,98 21,2 139,0 10,22 360,1 7,16 530,0
260 270 280 290 300 310 320 158,0 214,9 286,6 389,1 517,2 684,7 785,1 39,2 55,6 78,1 109,1 151,7 202,8 272,7 4,35 6,25 8,94 12,40 17,12 23,35 31,50 28,3 38,0 50,7 67,9 90,7 105,7 186,5 247,1 323,8 420,2 538,6 607,7 14,45 20,02 470,8 604,5 9,66 691,5
Полная упругость насыщенного пара над серной кислотой
любой концентрации при любой температуре может быть вы-
числена по уравнению:
Igp = 4— у,
где Т — абсолютная температура, р — давление в рт. ст.,
А и В — константы, зависящие от
Таблица 16
% H2SO4 A B % H2SO4 A В
95 9,790 3888 55 8,827 2400
90 9,255 3390 50 8,832 2357
85 9,239 3175 45 8,809 2322
80 9,293 3040 40 8,844 2299
75 9,034 2810 35 8,873 2286
70 9,032 2688 30 8,864 2271
65 8,853 2533 20 8',922 2268
60 8,841 2458 10 8,925 2259
концентрации кислоты. Значе-
ния А и В для различных
концентраций приведены в
табл. 16.
При 416° серная кислота
полностью разлагается. Сер-
ная кислота в парах диссо-
циирует по уравнению:
H2SO4 —SOs + H2O.
По Боденштейну и Ката-
яма связь константы равно-
весия этой реакции К,> =
pH п * PsG
= —------- с температурой
Ph2so,
выражается уравнением:
WP = —1,751g Т— 5,7- 10-4 Г+3.
В парах над олеумом присутствует только SOa. Упругость
30
SO3 над олеумом быстро растет с повышением концентрации
олеума и температуры. Данные об упругости SO3 над олеумом
приведены в гл. 4.
Теплота парообразования.
Теплота парообразования
(считая на 1 кг выпаренной
воды) повышается с увели-
чением концентрации серной
кислоты и с понижением
температуры (табл. 17).
Для определения тепло-
ты парообразования можно
пользоваться формулой:
£ = 83,9(365 — ty»>
где L — теплота испарения
в ккал на 1 кг Н2О, t —
Т АБЛИЦА 17
°/о SO3 в серной кислоте Скрытая теплота парооб- разования на 1 кг воды в ккал при температуре
15° 100’ 150’ 200’
20 595 545 510 467
50 659 612 579 538
60 724 681 650 611
70 844 810 784 750
80 1088 1077 1064 1044
температура кипения.
В некоторых случаях теплота парообразования самой H2SO4
представляет интерес. При 326° она равна 122,12 ккал на 1 кг
100%-ной H2SO4.
Теплота растворения, разведения и смешения. При соедине-
нии SOs с водой с образованием моногидрата выделяется те-
плота согласно реакции:
SO3 4- Н2О = H2SO4 Д-21,3 ккал.
твердый
Если к моногидрату прибавлять воды, то выделение теплоты
продолжается. На одну молекулу H2SO4 выделяется тем больше
теплоты, чем больше прибавляется воды. .Вначале выделение
теплоты идет очень интенсивно, а затем тепловой эффект пони-
жается.
Теплотой растворения называется то количество те-
плоты, которое выделяется при растворении граммолекулы ве-
щества в таком количестве растворителя, что дальнейшее при-
бавление растворителя не ведет к новому выделению теплоты.
Для серной кислоты теплота растворения равна 18',00 ккал. Ко-
личество тепла, выделяемое при растворении 1 моля H2SO4 в п
молях воды, называется теплотой разведения.
Если мы к кислоте, в которой на 1 моль H2SO4 приходится
щ молей НгО, прибавим воды в таком количестве, что на 1 моль
H2SO4 будет приходиться лг молей Н2О, то количество вновь
выделившейся теплоты называется диференциальной
теплотой разведения.
Для вычисления теплоты разведения с достаточной точно-
’ стью можно пользоваться формулой Томсена:
н_ и-17 860
П я+ 1,7983’
31
где п — число молей Н2О на моль H2SO4, а Н — число боль-
ших калорий на 1 моль H2SO4.
Диференциальная теплота разведения может быть опреде-
лена как разность:
ir ,, _______ /?2-17 860 гц-17 860
п.2 — Н1— Лг+ 1 798з — П1 _]_ 1>7983 •
При смешении двух растворов серной кислоты разной кон-
центрации получающийся тепловой эффект зависит от концен-
траций исходных растворов и от-
ношения их количеств. Этот теп-
ловой эффект называется тепло-
той смешения.
Вязкость растворов серной
кислоты. Вязкость растворов сер-
ной кислоты оказывает влияние
на ход процессов, поэтому вели-
чину вязкости необходимо в не-
которых случаях принимать во
внимание.
Так, при расчетах падения на-
пора в трубопроводах учитывает-
ся вязкость перекачиваемой жид- Й
кости. В сильной степени от вяз- |
кости жидкости зависит также §
коэфициент теплопередачи от |
жидкости к стенке и обратно. X
На рис. 3 построены кривые §
зависимости вязкости серной |
Рис. 3. Вязкость раство-
ров серной кислоты
Рис. 4. Вязкость растворов
серной кислоты и олеума.
кислоты (в сантипуазах) от ее концентрации (%H2SO4) при тем-
пературе 0, 20, 25, 40, 50 и 75° по данным Ландольта.
На рис. 4 показаны результаты работы В. Постникова и
Л. Кузьмина, охватывающие не только водные растворы серной
кислоты, но и некоторые концентрации олеума.
32
3. Стандарты серной кислоты
Серная кислота, выпускаемая из производства, обычно
, загрязнена различными примесями. Эти примеси попадают в сер-
ную кислоту из сырья, из которого она вырабатывается, или за-
носятся в нее в процессе производства в результате изнашива-
ния аппаратуры.
Серная кислота как товарный продукт выпускается в виде
нескольких сортов, отличающихся между собой различным со-
держанием SO3 и примесей. Качество каждого из выпускаемых
продуктов-стандартов зависит от условий производства и требо-
ваний, которые предъявляются потребителями.
Стандарты серной кислоты, выпускаемой в СССР, и их ос-
новные показатели (содержание H2SO4 или SO3 и окислов азота)
приведены в табл. 18.
кроме того, для напол- Таблица 18
нения аккумуляторов выпу-
скается особый сорт кон-
тактной кислоты под назва-
нием аккумуляторной.
Содержание H2SO4 в ней
должно быть 92—93%, же-
леза не более 0,015%, мышь-
як должен отсутствовать.
Сверх этого аккумуляторная
кислота должна удовлетво-
рять еще ряду требований:
она должна быть прозрачной
и бесцветной, не должна со-
держать примесей органиче-
ских веществ, металлов,
хлора и азотной кислоты.
Стандарты серной кислоты Содержание в %
H2SO4 NO + NC2
Камерная . Башенная н гловерная Купоросное масло . . Олеум . . . Не менее 65 Не менее 75-76,5 Не менее 92,5 18,5—20 свободного SO3 Не более 0,01 Не более 0,02
В зависимости от потребителя к товарной кислоте предъяв-
ляются в отдельных случаях и другие требования. Так, олеум,
предназначаемый для нитрующих смесей, должен содержать
свободного SO3 не менее 20%, железа не более 0,04% и твер-
дого остатка после прокаливания не более 0,15%. Купоросное
Масло, предназначенное для нужд пищевой промышленности и
промышленности искусственного волокна, не должно содержать
Мышьяка и окислов азота.
Стандарты на серную кислоту, как и на другие продукты
промышленности, систематически пересматриваются в связи
с изменяющимися условиями производства и требованиями по-
требителей.
4. Примерные расчеты
1. В специальных таблицах приводится содержание H2SO4 и SOa в про-
центах в водных растворах серной кислоты. Перевод процента H2SO4 в про-
цент SOs и обратно нетрудно сделать и без таблиц. Если процент H2SO4
3 Зак. 5019. Технология серной кислоты.
33
в водной серной кислоте обозначить через Рцзо,'8 процент 80s через PSOj>
то зависимость между ними будет следующая: на каждые 98 вес. ч. HsSO*
приходится 80 вес. ч. SOs, следовательно:
^so3 ~ с§ ^H2so«= 0,816 /3h2so4
или обратно
98
PHjS04 = 80 PSO3 = !'225 pso3-
2. Содержание свободного SOs и общей H2SO4 в весовых процентах
приводится в специальных таблицах. Зная 'какое-нибудь одно из этих дан-
ных, легко вычислить другое, не пользуясь таблицами; точно так же по-
любому из этих данных можно вычислить процент общего SO3.
Если процент свободного SOs в олеуме равен d, то в 100 вес. ч. олеума
80
будет d вес. ч. свободного SOs н (100 — d) В€С- ч- связанного SOs,.
а всего SOs:
d -I- (100 — d) Ц = 81,6 -J- 0,184 d.
Если процент общего SO3 в олеуме обозначить через С, то получим
соотношение:
С = 81,6 4- 0,184 d.
Если процент свободного SOs в олеуме равен d, то моногидрата H2SO4
в 100 ч. олеума будет:
100 — d.
Кроме того, из d частей свободного SOs может быть получено 98/80 d
моногидрата.
Следовательно, из 100 ч. данного олеума может быть получено моно-
гидрата:
98
^=100-d+g d
или
К= 100 + 0,225 d.
Эта величина и называется процентом моногидрата в олеуме.
3. Часто приходится получать кислоту заданной концентрации из
двух других растворов кислоты. Пусть А кг а%-ной H2SO4 надо разбавить
до б %-ной H2SO4. Пусть разбавление проводится с%-ной H2SO4. По
смыслу задачи ясно, что а > b > с. Требуется определить, сколько надо
взять килограммов с%-ной кислоты.
В более крепкой кислоте моногидрата кг; если более слабой кис-
X • С
лоты надо взять х $г, то моногидрата H2SO4 в ней будет —кг; в смеси
Лл I хс у
будет —— кг моногидрата HaSOs- Общий вес смеси будет равен
(Л + х) кг. По заданию:
(Лл + хс) • 100
~ (Л + х)-100 ’
откуда
Л(я —6)
Х-----Ь — с ’
или
х а — b
А Ь — с
34
4. Пусть 2 т 93%,-ной H2S0« смешиваются с 5 т 20%-ной H2SO«- Надо
определить величину теплового эффекта смешения.
Для 93%-ной H2SOa количество молей Н»О на моль H2SOt:
7 .93 п „
18 ’ 98 ~ 0,4 ’’
для 20%-ной:
SO 20 _
П-~ 18 ’98 —2 ,8‘
При смешении получим 7 т кислоты крепости:
(2000 • 0,93 + 5000 • 0,20) 100 лп оп, „
-----------7 -------------— 4U,8u/0H2ot>4.
Для этой кислоты:
«3 = 59^98 = 18-40,8 ’
Для первой кислоты: 0,41-17 860 0,41 +1,7983
—
Для второй:
я2 = 21,8-17 860 21,8+1,7983 165С0-
Для третьей:
/73 = 7,82- 17860 7,82 + 1,7983
Если бы 7 т 40,8%-ной H2SOj были получены разбавлением моногидрата
водой, то выделилось бы тепла:
Q3 = £90^0408 . jj500 = 423000 ккал
Но при получении 93%-иой H2SO4 из моногидрата и воды теплоты уже
выделилось:
~ 2000 - 0.93
Qi =-----по---• 3 320 = 63 000 ккал
Уо
и при получении 20% -ной HsSOi теплоты выделилось:
п 5000-0,20 ,ССЛПП
(?2 ------б«--* '9 500 = 168 000 ккал.
Следовательно, тепловой эффект смешения равен
т. е.
Q = 423 000 — (63 000 +168 000) = 192 000 ккал.
V. Материалы для аппаратуры
сернокислотного производства
1. Материалы природного происхождения
Из природных кислотостойких материалов на территории
СССР известны некоторые виды гранита, андезит, базальт, диа-
баз. Главной. составной частью этих пород является SiOs-
3*
25
Некоторые виды осадочных пород, как песчаник, кварцит,
также отличаются кислотостойкостью.
В СССР из природных материалов в строительстве сернокис-
лотных установок особенно широко применяется андезит,
добываемый на Кавказе. Он употребляется для футеровки ба-
шен и концентрационных аппаратов. Химический состав его при-
мерно следующий: SiOa — 62%, AI2O3 — 8,8%, БегОз— 5,9%,
СаО —5,4%, MgO —2,3%, R2O —5,3% и др.
Базальт и диабаз употребляются в естественном виде
и идут на изготовление кислотоупорного литья. При переплавке
их химический состав не меняется, но повышается однородность
строения и теряется пористость. Плавленый диабаз и базальт
обладают механической прочностью, термической стойкостью
(до 150°) и высокой кислотоупорностью. Из этих минералов от-
ливают плитки для футеровок аппаратов и газоходов, фасонные
части трубопроводов, краны, крыльчатки для насосов и пр.
Широко применяется в сернокислотной промышленности
кварц. В раздробленном виде до размера кусков 20—100 мм
он употребляется в качестве насадочного материала. Этот ми-
нерал обладает большой твердостью и высоким удельным весом
(2,6); химический состав его: 46,7% Si, 53,3% О2. В качестве на-
садочного материала может быть применен также кварцит —
естественно опрессованные мелкие кристаллы кварца.
Большое значение для сернокислотного производства имеет
асбест. В основном он представляет собой магниевый силикат
состава 3MgO • 2SiOa • 2Н2О. Асбест отличается волокнистым
строением, обладает значительной кислотостойкостью, повышаю-
щейся при увеличении содержания в нем кремнезема. Большое
количество асбеста идет на теплоизоляцию газоходов и аппара-
тов. Как кислотостойкий материал он применяется для набивки
сальников кислотных насосов, в качестве прокладочного мате-
риала — между фланцами кислотных коммуникаций.
Для прокладки он употребляется в виде картона и плетеного
асбестового шнура. Для плотности соединений во фланцах асбе-
стовые шнуры провариваются в льняном масле с добавкой гра-
фитового порошка. Можно проваривать шнуры также в техни-
ческом вазелине с небольшой добавкой канифоли и парафина.
Прорезиненный и спрессованный в1 листы асбест носит название
клингерита; последний применяется в качестве прокладоч-
ного материала.
2. Материалы искусственного происхождения
Огнеупорный кирпич. Наиболее широко применяется в серно-
кислотной промышленности огнеупорный кирпич — шамот для
кладки футеровки и сводов печи, для футеровки газоходов и
электрофильтров. Шамот обладает большой механической проч-
ностью, кислотоупорен и хорошо выдерживает высокие темпе-
ратуры. Состав его: SiO2 (60—67%), Бе20з, А12Оз, СаО, MgO.
36
В зависимости от природы примененной при изделии глины
температура начала размягчения шамота колеблется в пределах
1150—1400°.
Обычный строительный кирпич, отличаясь от шамота боль-
шим содержанием окиси алюминия, разрушается кислотами.
Каменный товар и фарфор. Под названиями каменный товар
и фарфор понимаются изделия, отформованные из смеси глины,
кварцевого песка, полевого шпата и обожженные до сплавления.
Так как полевой шпат плавится при более низкой темпера-
туре, чем глина, то после обжига черепок этих изделий полу-
чается не пористым, как у кирпича, а спекшимся, плотным и во-
донепроницаемым. Примерный химический состав таких изделий:
64—75% SiOa, 19—28% AI2O3 и небольшие примеси БегОз, СаО,
MgO и др.
Для изготовления фарфора идет чистая глина (каолин), не
содержащая железа. Для большей непроницаемости изделия ча-
сто покрываются глазурью.
Каменный товар и фарфор отличаются высокой стойкостью
к кислотам. Серная кислота начинает действовать на фарфор
лишь при температуре выше 400°. Температура начала разложе-
ния фарфора находится в пределах 1200—1400°. В сернокислот-
ном производстве каменный товар применяется для футеровки
башен и изготовления насадок (кольца Рашига); из фарфора из-
готовляются кольца Рашига и колпачки для аппаратов Кесслера.
Эмаль. Эмалью называется легкоплавкое стекло, нанесенное
на поверхность железного или чугунного предмета.
По механическим и химическим свойствам эмаль сходна со
стеклом и обладает высокой кислотостойкостью.
В сернокислотном производстве эмаль применяется для за-
щитного покрытия сборников, абсорберов и некоторых других
аппаратов.
Кислотоупорные цементы и бетоны. При производстве футе-
ровочных работ для связывания отдельных элементов кислото-
упорной футеровки между собой — футеровки с материалом
кожуха — и создания плотности в швах между отдельными эле-
ментами в сернокислотной промышленности употребляются сили-
катные цементы. Среди них различаются цементы, затвердеваю-
щие без добавки кремнефтористого натрия и с добавкой. К пер-
вым относятся цементы марки КЦ и КЦВ (Брянского завода),
ко вторым — андезитовый, диабазовый и др.
Основными материалами для цементов, затвердевающих без
добавки кремнефтористого натрия, являются кварцевый песок
(просушенный и измельченный в муку на шаровой мельнице),
растворимое стекло, сиштоф и церезит или льняное масло.
Цементы второй группы состоят из каменной муки, приготов-
ленной из андезита, диабаза или другого кислотостойкого, мине-,
рала, и добавки кремнефтористого натрия. При затвердевании
смеси этих двух компонентов на растворимом стекле получается
пластичная масса, способная затвердевать и превращаться
37
в прочный камень без искусственного нагревания. Цементирую-
щим веществом является кремнезем, получающийся в резуль-
тате взаимодействия растворимого стекла с кремнефтористым
натрием.
Химизм твердения цемента заключается в следующем.
В водном растворе кремнефтористый натрий и растворимое
стекло гидролитически расщепляются следующим образом:
Na2SiF6 + 4Н2О — 2NaF + Si (ОН)4 + 4HF;
Na2O • п SiO2 + 2Н2О 2NaOH -|- Н2О • п SiO2.
Продукты гидролиза реагируют между собой:
NaOH -|- HF = NaF -ф Н2О.
При достаточном количестве кремнефтористого натрия реакция
протекает так:
«Na2SiF6-|-iNa2OnSiO2 + cH2O = rfNaF-|-nH2O • «SiO2+ ^Н2О
и заканчивается полным разложением силиката с образованием
твердой кремнекислоты.
Кислотоупорный бетон приготовляется из заполнителя, в со-
став которого входит щебень кислотоупорных материалов и ки-
слотоупорная мука, ускорителя твердения (кремнефтористого
натрия) и растворимого натриевого стекла.
Соотношение количеств отдельных компонентов при приго-
товлении бетона несколько меняется в зависимости от качества
исходных материалов.
Для изготовления 1 м3 кислотоупорного бетона примерно
требуется:
Кислотоупорный щебень и песок............... 1300—1500 кг
Пылевидный заполнитель...................... 500— 600 ,
Растворимое натриевое стекло...........- • . . 200— 260 „
Кремнефтористый натрий...................... 30— 39 „
Перед приготовлением бетона составные части его тщательно
перемешиваются до получения однородной массы. Пылевидный
заполнитель и кремнефтористый натрий предварительно просу-
шивают, просеивают, удаляют комки и тщательно перемешивают
между собой. Кислотобетон затвердевает в 4—5 дней. Преиму-
щества кислотобетона в том, что он сравнительно дешев, нетру-
доемок в применении, монолитен и принимает любую форму. Он
применяется для постройки кислотохрапилищ, крышек башен,
полов и пр.
\] 3. Металлы
Черные металлы. Обычные железо и чугун являются основ-
ными материалами в строительстве аппаратов газовых и кислот-
ных коммуникаций сернокислотных заводов.
38
Высоконитрозные кислоты, легко пассивирующие поверх-
ность черных металлов, позволяют на башенных системах для
орошающих и вытекающих кислот ставить чугунные коммуни-
кации, строить железные (без футеровки) циркуляционные сбор-
ники, железные напорные бачки и железные змеевики для холо-
дильников. Высоконитрозные газы башенных систем также обла-
дают пассивирующим действием, что позволяет пользоваться
железом для изготовления газопроводов, крышек башен, хвосто-
вых вентиляторов и нефутерованных башен абсорбционной
зоны.
Разрушающе действуют на черные металлы слабые кислоты.
Возможность образования слабых кислот в железных сборниках
(вследствие поглощения зеркалом кислоты водяных паров из
воздуха) и угроза коррозии железа устраняются герметизацией
этих сборников. В тех точках системы, где условия не благо-
приятствуют образованию защитной пленки на металле, послед-
ний защищают от коррозии кислотоупорной изоляцией.
Железо и чугун применяются в строительстве многих аппара-
тов и коммуникаций контактных и концентрационных устано-
вок. В контактных системах для кислоты крепостью от 85% и
до моногидрата применяется чугун, для олеума — сталь.
Кремнистые чугуны. Добавка кремния сообщает чугуну чрез-
вычайно ценное качество — устойчивость в отношении корро-
зии. Наибольшую стойкость обнаруживают чугуны, содержащие
12—18% Si. Более высокое содержание кремния делает чугун
слишком твердым и хрупким, не увеличивая его химической
стойкости. Устойчивость кремнистого чугуна к кислоте
объясняется образованием на его поверхности тонкой пленки
SiO2.
Из иностранных марок особенно известны кремнистые чу-
гуны: термисилид, содержащий 14—16% Si, термисилид эк-
стра— более 16% Si, ацидур 17—18%> Si и др.
Для сернокислотных производств в СССР изготовляются из
высококремнистых чугунов краны, центробежные насосы и пр.
Основной недостаток кремнистых чугунов — их твердость и
хрупкость — они не куются и не обрабатываются режущими ин-
струментами. Аппараты и детали изготовляются исключительно
отливкой. Изделия из кремнистого чугуна вследствие его хруп-
кости требуют очень осторожного обращения с ними
Высококремнистые чугуны успешно противостоят действию
крепкой серной кислоты (начиная от 60% H2SO4 и выше) при
любой температуре вплоть до температуры кипения. С уменьше-
нием концентрации серной кислоты устойчивость кремнистых чу-
гунов понижается. Так, например, растворы 25%-ной H2SO4 (не
выше) уже разрушающе действуют на термисилид.
Хромистые стали и чугуны. Большой химической стойкостью
обладают хромистые стали. К ним относятся нержавеющие
стали, содержащие 13—18% Сг. Применяемые на практике
хромистые стали бывают: малохромистые с 4% Сг, среднехроми-
39
стые с 11—15% Сг и высокохромистые с 20—30% Сг.
Серная кислота на них действует слабо. Не разрушаются они
также азотной кислотой концентрации выше 11 %. Стойкость
к серной кислоте значительно повышается от введения в сплав
небольшого количества меди, а также кремния. Хромистые стали
пока мало применяются на сернокислотных заводах вследствие
высокой их стоимости.
Введение в хромистую сталь никеля еще более увеличивает
ее химическую стойкость. В СССР освоено производство хромо-
никелевых сталей, что дало возможность наладить производство
кислотостойких насосов и других аппаратов. Хромоникелевые
стали отлично противостоят серной кислоте, азотной и их сме-
сям. Несмотря на необыкновенно ценные свойства этих сталей,
масштаб их применения невелик по причине их дороговизны.
Прибавление хрома к чугуну также благотворно отражается
на его качествах — он приобретает кислотостойкость и жаростой-
кость. В СССР из хромистого чугуна изготовляется кислото-
стойкая аппаратура. Хромистый чугун применяется также для
отливки жаростойких зубьев для механических печей. Налажи-
вается-производство жаростойких гребков.
Кислотоупорные бронзы. В качестве сплавов, противостоя-
щих действию слабой серной кислоты, применяются кислото-
упорные бронзы. Так, например, сплав, состоящий из 77% меди,
15% свинца и 8% олова, применяется для изготовления деталей
вакуум-концентратора Симонсон-Мантиуса (гл. 6). К числу спла-
вов, устойчивых против действия слабой серной кислоты, отно-
сится также алюминиевая бронза, состоящая из 85,6—86% меди,
9,8—10,8% алюминия и 3,14—3,57% железа.
Свинец. Свинец на холоду хорошо противостоит корроди-
рующему действию безнитрозной серной кислоты крепостью до
купоросного масла. Слабые кислоты (ниже 80%) он выдержи-
вает даже при нагреве.
Устойчивость свинца объясняется образованием на его по-
верхности защитного слоя нерастворимого сульфата свинца
PbSOi. При действии же на свинец более крепких кислот
(концентрации выше купоросного масла) образуется раствори-
мый кислый сернокислый свинец Pb(HSO«)2. В этом случае за-
щитной пленки не образуется и кислоты таких концентраций
разрушают свинец. Стойкость свинца зависит от его чистоты.
Наличие в нем примесей посторонних элементов (цинк, сурьма,
медь и др.) делают его менее стойким.
Нитрозные кислоты действуют разрушающе на свинец, осо-
бенно при высоких температурах и высоких нитрозностях.
В условиях экстенсивной эксплоатации камерных и башенных
систем свинец оправдывал себя продолжительностью службы и
легкостью ремонта и монтажа аппаратуры и коммуникаций.
Условия интенсивного башенного процесса оказались весьма
неблагоприятными для свинца, а потому с переводом башенных
систем на интенсивную работу свинец быстро начал вытесняться
40
более устойчивыми в этих условиях черными металлами. В на-
стоящее время свинец как строительный 'материал сохранил
свое значение в полной мере только на камерных установках.
В контактных системах свинец употребляется для строитель-
ства некоторых аппаратов и коммуникаций промывного отделе-
ния, однако и здесь он может быть, в значительной мере, заме-
нен черными металлами.
Свинец применяется для монтажа сборников слабой кислоты
и конденсатов и коммуникаций для их перекачки. Он также упо-
требляется для защиты стенок стальных сборников и цистерн от
действия слабых кислот. Защитный слой свинца наносится на
сталь различными способами. Наиболее простой — это) погруже-
ние железа в расплавленный и нагретый до 350° свинец. Так как
свинец не пристает к стали, прибегают к помощи третьего ме-
талла — олова, которое сплавляется и со свинцом и со сталью.
Сталь сначала лудится оловом и уже на олово наносится сви-
нец. Покрытие свинцом производится также электролитически
или обрызгиванием поверхности стали расплавленным свинцом
с помощью аппарата Шоопа (шоопирование). При хорошем по-
крытии свинцом сталь свободно может заменить чистый свинец,,
если освинцованная сталь должна будет соприкасаться со сла-
быми кислотами.
Свинец в сплаве с сурьмой (4—12%, а иногда и олова 2—-
3%) под названием твердый свинец, или гартблей, употребляется
для изготовления кранов, вентилей и других мелких деталей,
однако эти детали с. успехом могут быть изготовлены из дру-
гого материала, например из соответствующего сорта чугуна.
Алюминий. Алюминий, если он достаточно чист, хорошо
противостоит корродирующему действию холодной азотной
кислоты-.
Стойкость обусловливается образованием на его поверхности
тонкой защитной пленки — окиси алюминия. При нагревании
пленка разрушается и металл быстро травится.
В сернокислотном производстве (башенном) алюминий приме-
няется для изготовления бачков под азотную кислоту и комму-
никаций для ее перекачки.
Алюминий употребляется также для изготовления деталей
аппаратов и коммуникаций для аммиачно-окислительных уста-
новок.
VI. Хранение и перевозка серной кислоты
1. Хранение серной кислоты
Башенная, гловерная кислота, купоросное масло и олеум
хранятся вне цеха на специальных складах, оборудованных же-
лезными баками. Наиболее распространенная емкость баков
335 ms.
При баках (обычно в отдельной пристройке) устанавливаются
насосы для перекачки кислоты в железнодорожные цистерны
4L
(иногда из цистерн в баки) или цехи-потребители. Для удобства
обслуживания баков над ними делается площадка. Хранилища
продукции для контактных цехов строятся в утепленных поме-
щениях.
На рис. 5 показана схема работы продукционного бака.
Готовая продукция из цеха подается в бак по трубопроводу 7.
Бак разгружается при помощи сифона 3. Зарядка сифона произ-
водится двумя способами. По первому способу сифон заливается
кислотой из бачка 4 по трубопроводу 5, снабженному вентилем
и присоединенному к колену сифона. При такой зарядке конец
Рис. 5. Схема работы склада готовой продукции.
сифона в баке предварительно закрывается пробкой 17. Второй
способ предусматривает использование сжатого воздуха. В бу-
рачок 6, предварительно залитый серной кислотой, подается че-
рез воздухопровод 7 сжатый воздух. Кислота из бурачка устрем-
ляется в бак по сифону 3 и по трубопроводу 8. Как только уро-
вень кислоты в бураке опустится ниже конца трубопровода 8,
воздух получит свободный выход в бак по трубопроводу 8 и
таким образом сифон 3 останется заряженным. В этот момент
сжатый воздух отключается и открывается вентиль на трубо-
проводе 9 к насосу. Кислота из бака по сифону через бурак пой-
дет в насос 12 и через кислотопровод 14 к цеху-потребителю.
Бачок 4 должен быть постоянно наполненным кислотой для
зарядки сифона. Для наполнения его служит трубопровод 10,
отведенный от общей кислотной магистрали 13.
Иногда завод получает кислоту извне в ж.-д. цистернах.
Кислота из цистерн сливается в приемный бачок 11, откуда
забирается насосом 12 и перекачивается по трубопроводу 2
в бак. Низ бака соединен с бураком при помощи патрубка 15,
снабженного запорным вентилем. Этот патрубок служит для
спуска остатка кислоты при чистке бака от грязи. Конец па-
42
трубка со стороны бака приспособлен под пробку 16, которая
используется лишь в случае порчи вентиля. Конец штока пробки
выведен под крышку бака.
На складах готовой продукции контактных цехов часто
устанавливаются подогреватели олеума для подогрева его
в морозное время года во избежание замерзания при хранении
или в цистернах в пути и смесители для приготовления кислоты
требуемой крепости.
Подогреватель (рис. 6) представляет собой вертикальный ряд
горизонтально поставленных стальных труб, соединенных между
Рис. 6. Подогреватель олеума.
собой коленами. Прямые участки труб заключены в паровые
рубашки — трубы большего диаметра, покрытые теплоизоляцией.
Паровые трубы также соединены между собой и образуют об-
щий канал для насыщенного при 4 ат пара. Олеум поступает
снизу и выходит подогретым сверху, а пар, наоборот, поступает
сверху и конденсат его стекает снизу.
Смеситель для кислот состоит из нескольких горизонтальных
рядов труб, расположенных вертикально один над другим
(рис. 7). Трубы каждого горизонтального ряда соединены между
собой коленами. Каждый горизонтальный ряд соединен с ниже-
лежащим рядом труб. Кислоты для смешения поступают в две
чаши, установленные на верху холодильника и соединенные
с трубами смесителя.
Процесс смешения производится постепенно и начинается
уже в чашах, причем во избежание бурной реакции в чаши по-
ступают кислоты с возможно небольшой разницей по концен-
трации. Получаемые в чашах две новые кислоты также не
сильно разнятся по концентрации между собой. Смешение этих
кислот происходит уже в самом смесителе — по выходе из
третьего сверху горизонтального ряда труб.
Два верхних ряда труб смесителя разделены соответственно
количеству чаш на две секции, что дает возможность кислотам
43
проходить через них, не смешиваясь и охлаждаясь. Поверхность
труб смесителя орошается водой. Пройдя всю систему орошае-
мых труб, продукт смешения выходит из аппарата в охлажден-
ном состоянии.
Рис. 7. Смеситель для серной кислоты.
2. Перевозка серной кислоты
Основная масса серной кислоты, не потребляемой на месте
производства, перевозится в ж.-д. цистернах.
При отправке небольших партий серной кислоты местным по-
требителям пользуются автоцистернами, контейнерами, сталь-
ными барабанами, керамиковыми турилами и, наконец, стеклян-
ными бутылями. Последними пользуются и для транспортировки
камерной кислоты на большие расстояния. В железнодорожных
цистернах перевозят башенную, гловерную кислоты, купоросное
масло и олеум.
Цистерна представляет собой клепаный или сварной ци-
линдр, прикрепленный к раме вагона. ‘На случай увеличения
44
объема кислоты (при повышении температуры) цистерна сверху
снабжается куполом, лазом для проникновения внутрь цистерны.
Цистерна должна быть снабжена воздушными выпускными и
впускными трубами с плотно закрывающимися приспособле-
ниями. Разгружается цистерна большею частью сжатым возду-
хом. Для этого цистерну соединяют через верхний штуцер
с источником сжатого воздуха; кислота выжимается по трубе,
доходящей до дна цистерны.
Употребляющиеся для перевозки серной кислоты цистерны
бывают двухосные грузоподъемностью 15 г и четырехосные
грузоподъемностью 50 т. Выгоднее пользоваться цистернами
большей грузоподъемности; так, например, у 15-тонной ци-
стерны на каждую тонну перевозимой кислоты приходится 0,58 т
тары, а у 50-тонной только 0,47 т.
За границей пользуются ж.-д. цистернами также и для пере-
возки камерной кислоты концентрации 52—55° Вё. Для этого
внутренняя поверхность цистерны освинцовывается. Кислоты
более низкой концентрации, чем камерные, перевозятся в гумми-
рованных цистернах.
Стальные барабаны употребляются для транспортирования
купоросного масла и олеума.
Барабан вмещает 500—600 кг кислоты. За границей внутрен-
няя поверхность барабанов под кислоту 60—65° Вё обрабаты-
вается пассиватором. Для транспортирования кислоты 55 °Вё
стенки барабана освинцовываются или шоопируются, для кис-
лоты 42° Вё — гуммируются.
Кислота заливается в барабан через приваренную железную
втулку с завинчивающейся железной пробкой, которая не
должна выступать выше обручей. При опорожнении барабанов
пробки тщательно завинчиваются для предупреждения разбавле-
ния оставшейся кислоты 'влагой воздуха и, следовательно, трав-
ления металлических стенок. ч Для предохранения наружной по-
верхности от травления барабан окрашивают кислотоупорной
краской. После опорожнения барабан следует промыть сперва
водой, а затем слабым раствором соды.
Перевозка камерной кислоты в бутылях теперь стала очень
редким явлением на сернокислотных заводах. Обычно вся ка-
мерная кислота потребляется на месте производства.
Употребляющиеся для кислоты бутыли должны соответство-
вать стандарту по форме, емкости и качеству стекла.
Литература
1. Thieller Erich, Schwefel, Dresden und Leipzig 1936.
2. Mason D. (История развития мировой серной промышленности), J. Ind.
Eng. Chem. 30, 7, 759 (1938).
3. Ridgwan R. H., Mitchell A. W. (Мировая серная промышлен-
ность 1932—1936), Mineral Heabook, USA, 1301—14 (1937).
4. Duecken W. W. (Новые области промышленного применения серы),
Min. et Met., 473—76 (1938).
4-5
5. Lepsoe Robert, Trail (Brit. Columbia), J. Ind. Eng. Chem. 30, 1,
92—100 (1938).
6. Brutzcus M. (О термохимии соединений серы), Compt. rend. 206, 11,
838 (1938).
7. Preuner, S c h u p p, Ztschr. f. phys. Chem. 68, 157 (1901).
8. T a m m a n G., К r i g e G. (Статика водных растворов SO2), Ztschr.
f. anorg. allg. Chem., 146, 179 (1925).
9. Hudson J. (Растворимость SO2 в воде), J. Chem. Soc. 127, 1732 (1925),
London.
10. M a a s C-, Maas О. (Свойства водных растворов SO»), Chem. Soc. Am.
50, 1352 (1928).
11. Beйцер Ю. И., Путинский Г. П., Химия и физика маскирую-
щих дымов, стр. 49—56, 68—73, Оборонгиз, 1938.
12. Terre s Е., Riihl G. (Изучение бинарной смеси SO2—Н2О), С.
1934, II, 2164.
13. М е s t г е R. (Свойства серной кислоты), С. 1934, 1, 909.
14. К reps Th. (Новые направления в области производства, исходных
продуктов и применения серной кислоты), Chem. Met. Eng. 45,1,22(1938).
15. Дунаевский Е. И., Розенберг Б. С., Экономия серной кислоты
при производстве удобрений, ЖХП № 3, 11 (1939).
16. Мал и и К. М., Производство и потребление сериой кислоты в капита-
листических странах, ЖХП № 1, 52 (1938).
17. Малин К. М., Задачи сернокислотной промышленности в разрезе
решений XVIII съезда ВКП(б), ЖХП № 7, 4 (1939).
18. Waeser В., Handbuch der Schwefelsaurefabrication, ч. I, стр. 1—33,
517—565. Braunschweig 1930.
19. Fairlie, Sulfuric acid Manufacture, стр. 13—64, New York, USA, 1936.
20. Лунге-Берль, Справочник no основной химической промышлен-
ности, Госхимтехиздат, Л. 1933.
21. Кузьминых И. Н., Справочник для инженеров и техников кислотных
заводов, Госхимтехиздат, М.-Л. 1933.
22. Э ф р а и м Ф., Неорганическая химия, ч. II, стр. 46—52, Госхимтехиздат,
Л. 1933.
23. М е н ш у т к и н Б. Н., Курс общей химии, изд. 4, стр. 509—526, 1933,
24. Производство минеральных кислот, сборник статей „Иностранная тех-
ника*, Госхимтехиздат, 1932.
25. Техническая энциклопедия, Справочник физических, химических и техноло-
гических величин, т. V, стр. 467— 469, Москва 1930.
26. Perry J., D a v i s (Номограммы удельных объемов и минеральных
кислот), Chem. Met. Eng. 42, № 2, 87 (1935).
27. Gre епе wait С. (Упругость паров воды над серной кислотой), J. Ind.
Eng. Chem. 17, 522 (1925).
28. Thomas J., Barker W. (Упругость паров H2O и H2SO4 над крепкой
серной кислотой при высоких температурах), J. Chem. Soc., 2820, Lon-
don 1925.
29. Р е г г у J., Davis (Упругость растворов серной и азотной кислот),
Chem. Met. Eng. 41, 4, 188 (1934).
30. Hulsmann О., Bietz (Тепловой анализ систем H2SO4—Н3О),
Ztschr. f. anorg. Chem. 3, 245 (1935).
31. Перри ш, Снеллинг, Концентрирование серной кислоты, стр.
7—19, 1931.
32. Н a u g h t А. (Теплота разбавления серной кислоты), Chem. Met. Eng.
42, 5, 278 (1935).
33. Постников В., Кузьмин Л. (Вязкость олеума, водных раство-
ров сериой кислоты и сернистого натрия), Химстрой № 9, 527
(1934).
34. Lan do It, Phys.-Chem. ТаЫ. 1, 85—86.
35. Бесков С., Слизко некая О. О вязкости сериой кислоты, Хим-
строй № 1, 32 (1935).
36. Foote Н., Leopold G. (Точки замерзания H2SO4 и SO3), С. 1926,
1, 1773.
46
37. Tamm an G. (Физические свойства серной кислоты в зависимости-
от ее концентрации), Ztschr. f. anorg. allg. Chem. 161, 363 (1927).
38. Ф p а й т а г Г., Материалы для изготовления химической аппаратуры.
Госхимтехиздат, 1934.
39. Б о р о д у л и и М. В., Химическое разрушение материалов, ОНТИ,
1935.
40. П о л я к о в К. А., Неметаллические кислотостойкие материалы, ЖХП,.
№ 8, 23 (1939); № 3, 37 (1940).
41. Юшманов Е., Попова А., Коррозия чугуна, железа и свинца
в газовой фазе башениой системы, ЖХП № 4, 13 (1938).
42. Юшманов, Журавлева и Орлова, Коррозия железа, чугуна-
и свинца нитрозами, ЖХП № 7, 2 (1938).
43. В т о р о в М. Н., Стойкость свинца в башенных системах и замена его
другими материалами, ЖХП № 4, 13 (1938).
44. Ю ш м а н о в Е. и Попова А., Замена свинца железом и чугуном,
в башенном сернокислотном производстве, ЖХП № 20, 1387 (1937).
ГЛАВА 2
СЕРНИСТОЕ СЫРЬЕ И ЕГО ПОДГОТОВКА
1. Виды сернистого сырья
Содержание серы в земной коре составляет ~0,1%. Сера
.встречается в виде самородных серных руд, в виде сернистых
металлов (FeSa, ZnS, PbS и мн. др.) и в виде сернокислых солей
(CaSOd, BaSOd, MgSOd, NazSOi и мн. др.). При извержении вул-
канов выделяется сернистый газ, а при гниении животных от-
бросов — сероводород. Сера содержится также в клетках жи-
вотных и растений, в нефти и каменном угле.
Основным сырьем для производства элементарной серы, сер-
нистого ангидрида и серной кислоты являются самородные сер-
ные руды и сульфиды. Некоторые сульфиды, как пирит FeSa,
или специально добываются из-за содержащейся в них серы или
получаются попутно при добыче других сульфидов, используе-
мых для производства цветных металлов.
В некоторых масштабах в народном хозяйстве используется
также сера, попутно добываемая и в других процессах. Так, при
добыче угля попутно добывается и отбирается углистый колче-
дан. В дальнейшем при коксовании угля используется сера, пе-
решедшая в виде сероводорода из угля в коксовый газ. В ста-
дии практического разрешения находится вопрос об использова-
нии серы (превратившейся при горении содержащего ее угля
в SO2) отходящих топочных газов.
Точно так же используется сероводород, получающийся при
очистке нефти.
Производимая промышленностью элементарная сера добы-
вается, главным образом, из самородных серных руд. Значи-
тельно меньшее ее количество получается из сероводорода кок-
совых и других газов, а также из сульфидов и сернистого газа.
Сернистый газ в промышленности получается сжиганием серы
и ее сульфидных руд. Небольшие количества сернистого газа
получаются сжиганием сероводорода, а также восстановлением
сульфатов.
На первый взгляд казалось бы, что серную кислоту проще
всего получать из ее природных солей (гипса, тенардита, мира-
билита и др.). Впервые серная кислота была получена именно
таким образом: алхимик Гебер изготовил серную кислоту сухой
перегонкой железного купороса FeSOd (с тех пор за крепкой
серной кислотой сохранилось название купоросного масла).
-48
Однако залежи сернокислых солей FeSOj, Fe2(SO.j)3, разлагаю-
щихся при относительно низкой температуре, невелики. Боль-
шинство сульфатов разлагается при такой температуре, что в ре-
зультате разложения получается не H2SO4 и не SO3, a SO2.
Кроме того, сульфаты (CaSOi, NaaSCU) начинают плавиться ниже
температуры их разложения, чем затрудняется само разложение.
Экономически же до сих пор более выгодно получать серную
кислоту не разложением сульфатов, а сжиганием самой серы и ее
неокисленных руд (сульфидов). Поэтому промышленное произ-
водство серной кислоты развивается, главным образом, на основе
сернистого газа. Правда, на этой основе долгие годы не могли
получать крепкой серной кислоты. Поэтому до 1875 г. для по-
лучения крепкой серной кислоты пользовались купоросным
сланцем Fe2(SC>4)3. И лишь с открытием контактного способа
стали получать крепкую серную кислоту из сернистого газа.
В ряде промышленных процессов при применении серосодер-
жащего реагента (особенно серной кислоты) сера переходит не
в продукт, а в отход производства, который в свою очередь сам
может стать серосодержащим сырьем. Такого рода отходами
производства являются, например, кислые гудроны, травильные
растворы, фосфогипс и др.
с
1. Самородные серные руды
Залежи самородных серных руд встречаются в трех видах:
1) осадочные месторождения, 2) вулканические месторождения и
3) залежи в шляпах соляных куполов.
Примером залежей первого характера являются месторожде-
ния Италии, главным образом на о. Сицилия. Запасы серы на
о. Сицилия оцениваются в 70 млн. т, содержание серы в руде
колеблется в пределах 15—30%.
Залежи в шляпах соляных куполов находятся в США (в шта-
тах Техас и Луизиана). Это самые мощные месторождения мира,
запасы серы в них оцениваются в 140 млн т. Содержание серы
в руде колеблется в пределах 20—70%.
Крупное месторождение серы в Японии на о. Хоккайдо
представляет собой залежи вулканического характера.
Все другие месторождения самородных серных руд имеют сравнительно
небольшое значение.
В Америке небольшие залежи серы встречаются иа Малых Антильских
островах, в Чили, Перу, Аргентине, Виргинии. В Европе, кроме Италии,
небольшие залежи самородных серных руд имеются во Франции, Испании
и Исландии. В Азии небольшие залежи серы находятся па островах
Киусиу, Кондо и Яве, а также в Турции, Мессопотамии, в Белуджи-
стане; в Африке — в Египте, Тунисе и юго-западной Африке; в Австра-
лии— в Новой Зеландии и на некоторых островах Новой Кале-
донии.
Серных месторождений в СССР чрезвычайно много. Главные
из них следующие:
1. В Поволжье месторождения серы связаны с гипсами и из-
вестняками пермской системы.
Зак. 5019. Технология сериой кислоты.
49
Все они удобно расположены 'Вблизи от путей сообщения и
промышленных районов.
Большое значение приобрели месторождения, расположенные
по плато между реками Волгой, Сок и Самаркой (Алексеевское
и Водинское месторождения).
2. В Крыму в 45 км от г. Керчи находится Чекур-Кояшское
серное месторождение. Сера содержится в отложениях третич-
ного возраста, в глинах и гипсах.
3. В Среднеазиатских республиках насчитывают, примерно,
около 30 месторождений серы. Детально разведана и эксплоати-
руется лишь часть из них.
а) Каракумское в Каракумской пустыне в 226 км от желез-
ной дороги и города Ашхабада в третичных песчаниках с гипсом
в виде цемента.
б) Шор-Су в 38 км от г. Коканда. На границе меловых и тре-
тичных отложений сера находится по трещинам в известняках
и мергелях. . Расположено это месторождение недалеко от
нефтяных источников.
в) Гаурдакское месторождение, открытое в 1926 г., располо-
жено в 50 км от железной дороги, с которой соединяется ко-
лесной дорогой. Сера находится в известняках и плотных гипсах.
г) Чангырташское месторождение расположено в 12 км от
железной дороги на берегу р. Кара-Дарьи и в 18 км от города
Джалалабада. Месторождение находится в освоенном районе.
д) Кенемехское месторождение (Гажды) находится в Узбек-
ской ССР. Разведывалось в 1929 г. Расположено в местности,
не имеющей питьевой воды и удаленной от железной дороги.
Ряд менее значительных серных месторождений находится
также в Уральской области, в Дагестане, в Армянской ССР,
на Камчатке, на Алтае, в Кузбассе, в Карело-Финской ССР,
Северном крае.
2. Рядовой серный колчедан
В большинстве стран основным сырьем для производства сер-
ной кислоты до сих пор служит пирит.
В химически чистом пирите, как это легко высчитать из фор-
мулы, содержится 53,46% серы и 46,54% железа. В природном
колчедане содержится серы 25—52%, а железа 35—44%. Основ-
ные примеси колчедана следующие: сернистые соединения меди,
цинка, свинца, мышьяка, никеля, кобальта, селена, теллура,
углекислые и сернокислые соли кальция, магния и др.; тальк,
кварц и часто в незначительных количествах золото и серебро.
Мышьяк присутствует, главным образом, в виде FeAsS (мышья-
ковый колчедан), медь в виде FeCuS (медный колчедан), CuaS
(медный блеск) и CuS (коввелин).
Серный колчедан FeS2 существует в двух кристаллических
модификациях: 1) пирит — кристаллизуется в правильной си-
стеме, имеет желтоватый и зеленовато-серый цвет, удельный вес
53
Таблица 19
4*
51
его равен 4,95—5,1, твердость 6—6,5 и 2) марказит—встре-
чается гораздо реже, чем пирит; кристаллизуется в ромбической
системе, удельный вес его 4,55. Встречаются также соединения
жедеза с серой более сложного состава, отвечающего формуле
Fe^Sn'+t. Такие соединения называются пирротинами.
Данные о составе некоторых колчеданов приведены в табл. 19.
Уже из этих примеров состава колчеданов ясно, что природ-
ные колчеданы представляют собой комплексные руды.
Серный колчедан — самый распространенный из сернистых
соединений минерал: он встречается во всех зонах земной коры.
Месторождения серного колчедана находятся в Испании, Япо-
нии, Португалии, Канаде, Франции, Норвегии, Италии, Германии,
СССР, Греции и в ряде других стран. Богатейшей страной по
колчедану является Испания, залежи которого там исчисляются
в 485 292 тыс. т.
В СССР залежи сульфидных руд сосредоточены главным об-
разом на Урале. Менее значительные залежи колчедана нахо-
дятся в Закавказье, на Алтае и в Восточной Сибири.
С рудников колчедан поступает в виде кусков размером от
400 до 50 мм в диаметре; в этом случае он называется рядо-
вым или обыкновенным колчеданом. Но в рудниках встре-
чаются и пласты рыхлого колчедана, легко рассыпающегося
в порошок. Этот вид колчедана носит название с ы п у ч к и.
3. Флотационный колчедан
Богатых медью сульфидных руд мало, сжигать же сульфид-
ную руду с содержанием меди 1—3% на медеплавильных заво-
дах нерентабельно. До переработки руды на медеплавильном за-
воде целесообразно обогатить ее в отношении меди. На меде-
плавильных заводах СССР руда обогащается с 3—8% до 15—
25% меди.
Лучшим методом обогащения считается метод флотации, основан-
ный на различном отношении к смачиванию водой разных минералов в спе-
циально создаваемых условиях. Частицы минералов, не смачивающиеся
водой, прилипают к пузырькам воздуха и поднимаются вверх; частицы мине-
ралов, смачивающиеся водой, оседают на дно сосуда. Методом флотации
разделяются различные минералы, входящие в состав одной и той же руды.
Раньше умели проводить только коллективную флотацию, т. е. от-
делять все сульфиды вместе от пустой породы. Теперь разработаны методы
селективной флотации, при которой можно отделить все сульфиды друг
от друга.
Обогащенная (например медью) часть руды называется кон-
центратом, а другая часть руды — флотационными
хвостами или флотационным колчеданом. Флота-
ционный колчедан выходит с обогатительной фабрики очень
влажным (до 80% влаги) и до использования в качестве сырья
для серной кислоты должен подвергнуться фильтрации и сушке.
Состав концентрата и хвостов в первую очередь зависит от со-
става руды. Выход флотационных хвостов составляет 80 —
52
85% от веса руды. Из всей меди, находящейся в руде, перехо-
дит в концентрат 80—82% и остается в хвостах 18—20%. Фло-
тационные пиритные хвосты содержат 32—40% серы. По дан-
ным анализов, проведенных НИУИФ, флотационные хвосты
Красноуральской фабрики характеризуются следующим соста-
вом (в %):
Сера общая..................... 36,70—46,90
Сера сульфатная................0,28— 0,37
Полуторные окисли.............. 50,01—54,40
Мышьяк (считая на As2O3) . . . следы
Селен............................... „
Нерастворимый остаток .... 16,40—23,60
Если хвосты перефлотировать, то содержание серы в полу-
ченном пиритном концентрате можно поднять до 48—50%.
При перефлотации удается извлечь из отбросной пульпы
~92% пирита.
4. Газы металлургических печей
Концентрат, получаемый при обогащении медьсодержащих
сульфидных руд, содержит значительное количество серы. Боль-
шое количество серы содержит также концентрат, получаемый
при обогащении цинковых руд. Так, по данным одного завода
цинковый концентрат имеет следующий состав (в %):
Zu . . .45
РЬ . ... 7
Fe........5
S.........28—30
Си........1,6
СаО .
SiO2
А12О3
Ag •
Аи .
. 1
.6,5
. 1,3
. 100 г/т
.15 .
Концентраты такого состава идут в печи металлургических
заводов. В этих печах основная часть серы концентратов сго-
рает и переходит в сернистый газ. Если взять только те отходя-
щие газы, в которых концентрация SO2 не ниже 3%, то еже-
годно на земном шаре уходит серы с ними около 6 млн. т, при
этом используется пока не больше 3,9 млн. т. В 1938 г. метал-
лургическими заводами Урала в отходящих газах выпущено
490 тыс. т серы.
Использование огромных масс получающегося на медепла-
вильных и цинкоплавильных заводах сернистого газа — чрезвы-
чайно важная задача. Выпускаемый на воздух сернистый газ гу-
бит окрестную растительность, создает антисанитарные условия
для окружающего населения.
Значительная часть металлургических газов (газы обжиговых
печей — см. гл. 3) может быть непосредственно использована
для получения серной кислоты. Эти газы — очень выгодное
сырье для сернокислотного производства. При работе сернокис-
лотного завода на отходящих газах отпадает нужда в рудниках
53
по добыче сырья и сернокислотный завод освобождается от не-
обходимости постройки дробильного и печного отделений.
Часть газов, более бедных по содержанию SO2, может быть
использована с предварительным обогащением (гл. 8) или тоже
непосредственно, но при менее интенсивной работе самой серно-
кислотной установки.
5. Углистый колчедан
Сера в том или ином количестве входит в состав всех иско-
паемых углей. Она присутствует в них в виде органической серы,
сульфатной серы и пиритной серы. По данным Янсея, исследо-
вавшего угли всех стран, среднее содержание различной серы
в них может быть выражено цифрами, приведенными в табл. 20.
Приводим также данные
Таблица 20 о содержании серы в углях
двух рудников Донбасса
(табл. 21).
Потенциальные запасы уг-
ля по СССР расценивают-
ся в 1 654000 млн. т. Если
считать, что можно исполь-
зовать только 1% колчеда-
на от угля, то это составит
~ 16 540 млн. т.
Из этих данных, видно,
что запасы колчедана, со-
путствующего углю, значи-
Форма серы Содержание S в °/0
от ДО среднее
Пиритная . . 0,02 2,61 1,00
Сульфатная . 0,01 0,32 0,05
Органическая 0,23 7,90 1,21
Общая .... 0,44 9,01 2,29
тельно превышают запасы
сплошных колчеданных руд (обыкновенного колчедана). Угли-
стые колчеданы поступают на сернокислотные заводы: 1) как
отход при ручной сортировке угля или 2) как хвосты при обо-
гащении углей мокрым способом и, наконец, 3) как отход от
угля на теплоэлектроцентралях, работающих на пылевидном
топливе.
Таблица 21
Рудоуправле- ние Шахта Сера в угле в %
общее содер- жание пиритная сульфат- ная органи- ческая
Буденновское Смолянинов- № 3/9 2,45 1,46 0,21 0,78
ское .... № 1 1,56 0,89 0,03 0,64
Отборка колчедана от угля — чрезвычайно важная народно-
хозяйственная задача. Колчедан ухудшает качество угля. Ото-
54
бранный углистый колчедан может быть использован как сырье
для производства серной кислоты. При переводе на углистый
колчедан сернокислотных заводов центра и Украины отпадает
необходимость перевозки в эти районы уральского рядового и
флотационного колчедана. Это ощутительно разгружает тран-
спорт.
Отобранный от угля углистый необогащенный колчедан со-
держит 36—42% серы и 6—18% угля.
Особенность углистого колчедана — отсутствие в нем соеди-
нений селена, что особо важно- для сульфитцеллюлозной про-
мышленности.
По данным НИУИФ содержание мышьяка в углистых колче-
данах Подмосковного бассейна незначительно.
6. Отходы углеобогатительных фабрик
При обогащении угля получаются отходы с содержанием
7—8% пиритной серы и 20—25% угля. В 1938 г. по Донбассу
в таких отходах содержалось серы ~ 220 тыс. т.
При вторичном обогащении таких отходов получается, с од-
ной стороны, кондиционное котельное топливо, с другой сто-
роны, отходы вторичного обогащения с содержанием пиритной
серы 12—15% и угля 11—12%.
Во Всесоюзном угольном институте ведутся работы по
обессериванию этих отходов. Обработка этого материала воз-
духом в присутствии влаги дает газ с содержанием 5—6% SO2
при достаточном содержании кислорода. Несмотря на то что
этот газ по качеству (для сернокислотных установок) ниже
газа, получаемого от обжига колчедана, в районах, близких
к этому сырью, может оказаться экономически целесообразным
строить работу сернокислотных заводов на этом сырье.
7. Дымовые газы
При сжигании угля в топках котельных установок органиче-
ская и сульфидная сера угля превращается в сернистый газ, ко-
торый до сих пор вместе с отходящим газом котельных устано-
вок выбрасывается в трубу.
На земном шаре ежегодно сжигается ~ 1050 млн. т угля и
при этом с получающимися газами выбрасывается в атмосферу
не менее 10 млн. г серы.
Помимо того что дымовые газы являются мощным резервом
сырья для получения серной кислоты, вопрос использования SO2
дымовых газов чрезвычайно актуален с санитарно-гигиенической
точки зрения. Очевидно, разрешение проблемы использования
SO2 топочных газов тесно связано с нахождением удачного ме-
тода их концентрирования. Некоторые намечающиеся методы
концентрирования и использования SO2 топочных газов кратко
изложены в гл. 8.
55
8. Сероводород и газоочистительная масса
При коксовании угля ~ 50% всей находящейся в нем
серы переходит в коксовый газ. При этом основная часть серы
(до 95%), перешедшей в газ, находится в виде сероводорода.
Кроме сероводорода в газе находятся органические соединения
серы: сероуглерод, тиофены, меркаптаны и др.
На земном шаре ежегодно коксуется ~ 150 млн. т угля
и при этом в получаемых коксовых газах содержится в виде
сероводорода до 400 тыс. т серы. Сера в виде сероводорода
находится в ряде других промышленных газов: светильном,
генераторном, водяном, а также в газах нефтяных заводов.
Коксовые газы содержат серы в виде H2S 5—25 r/ivft.
В ряде случаев промышленные газы, содержащие сероводо-
род, безусловно подлежат очистке от него: газы, идущие на бы-
товые нужды; газы, употребляемые для синтеза аммиака, и др.
Целесообразно очищать газы от сероводорода почти во всех
случаях. Существуют методы очистки сероводородсодержащих
газов как без всякого использования серы, так и с использова-
нием последней. Методы очистки, использующие серу, фикси-
руют ее либо в виде очень концентрированного сероводорода,
перерабатываемого далее на элементарную серу или серную кис-
лоту, либо непосредственно в виде элементарной серы или эле-
ментарной серы в смеси с применяемой для очистки газа массой.
Наиболее распространен был метод очистки гидратом окиси же-
леза. Так называемая ламингова .масса представляет собой оса-
жденный гидрат окиси железа, получаемый смешением желез-
ного купороса, гашеной извести, глины и воды. Позднее стали
применять естественные гидратированные железные руды, как
бурый железняк и др. Для создания пористости масса переме-
шивается с опилками.
Поглощение сероводорода происходит по реакциям:
2Fe (ОН)8 + 3H2S = Fe2S8 + 6Н2О;
2Fe (ОН)8 3H2S = 2FeS + S + 6Н2О.
В дальнейшем при действии воздуха происходит выделение
серы и регенерация гидрата окиси железа:
w Fe.2S3 + 11 /2О2 + ЗН2О = 2Fe (ОН)8 + 3S;
4FeS-}-3O2 -)-6H2O = 4Fe (OH)3-)-4S.
Обычная газоочистительная установка состоит из четырех
очистительных ящиков. Проходящий газ постепенно переводит
Ре(ОН)з в сернистое железо, и масса все хуже и хуже очищает
газ. Когда предпоследний по ходу газа ящик начинает пропу-
скать сероводород, первый ящик выключается, перезаряжается и
ставится на четвертое место.
Регенерация массы раньше проводилась перелопачиванием ее
56
на воздухе при одновременной поливке водой, теперь добавляют
некоторое количество воздуха в самый очищаемый газ.
Одна и та же масса благодаря производимой регенерации ра-
ботает продолжительное время, постепенно обогащаясь серой.
Когда содержание серы в массе достигнет 40—50%, реакцион-
ная способность ее падает настолько, что ее приходится заме-
нять свежей. Эта окончательно отработанная масса поступает
или на сернокислотные заводы для обжига (в целях получения
SO») или идет на экстракцию серы (гл. 7). В Англии из газоочи-
стительной массы производится до 200 тыс. т серной кислоты
в год.
В США и Германии имеются сернокислотные заводы, исполь-
зующие сероводород. Один сернокислотный контактный завод на
сероводороде, получаемом при очистке нефтяного газа, строится
в СССР.
Методы, по которым при очистке коксового и других газов
от сероводорода получается элементарная сера, описаны
в гл. 7.
9. Доменные шлаки
Доменные шлаки содержат серу, переходящую в них из кокса. П"иводим
содержание серы в шлаках по данным металлургических заводов (в %):
Германские заводы.............................- 2,05
Английские » ............................2,15
Шотландские » .......................... 2,3
Выход шлака на 1 т чугуна в среднем составляет 0,5—0,7 т. Сера
в шлаке содержится главным образом в виде CaS.
Если через шлак пропускать воздух, то происходят реакции:
CaS + i/2O2 + CaSiO3 = Ca2SiO4 + S;
S + O2 = SO2.
Выход SO’ увеличивается, если в шлак добавлять гипс или ангидрит. Гипс
реагирует с сернистым кальцием по реакциям:
3CaSO4 + CaS + 4CaSiO3 = 4Ca2SiO4 + 4SO2;
3CaSO4 + CaS = 4CaO 4-4SO2.
Использование серы доменных шлаков в промышленности еще не осу-
ществлено.
10. Кислые гудроны
Серная- кислота, применяемая для очистки нефтепродуктов,,
переходит в отброс производства нефтеперегонных заводов —
кислые гудроны. Последние весьма обременительны для нефте-
перегонных заводов, так как занимают много места для свалки
и отравляют атмосферу и грунт; кроме того, с гудронами
теряется большое количество серной кислоты.
По составу кислые гудроны — весьма сложная смесь сер-
ной кислоты, различных сульфосоединений, окисленных углево-
дородов и др.
57
Содержание серной кислоты в гудронах колеблется в за-
висимости от сорта их от 20 до 50%.
Кислые гудроны при термическом разложении дают SO2,
воду, кокс, конденсирующиеся и неконденсирующиеся углеводо-
роды. Разложение проводится во вращающихся печах. В каче-
стве теплоносителей применяются топочные газы, нагретый кокс,
песок.
Полученный сернистый газ с успехом может быть направлен
в сернокислотную установку для переработки в H2SO4. В США
имеются сернокислотные установки (по контактному методу),
перерабатывающие газы, получаемые от разложения кислых
гудронов.
В СССР вопрос об использовании кислых гудронов для про-
изводства серной кислоты находится в стадии испытаний на
полузаводских установках.
11. Сульфаты
Залежи сульфатов, особенно гипса, очень многочисленны.
Достаточно сказать, что в Европейской части СССР имеется не
менее 500 месторождений гипса.
Огромные залежи сульфата натрия разбросаны в СССР на
большом пространстве — Кара-Богаз-Гол, Узун-Су, Аральское
море, Кулунда и др., как раз в районах, дефицитных по щелоч-
ным и серным продуктам.
Освоение производства серной кислоты из гипса освободило
бы целый ряд районов от необходимости завоза серной кислоты
или сырья для ее производства. Особенно в этом заинтересованы
Северный край, центральные области, Восточная Сибирь, УССР.
Фосфогипс представляет отход производства концентрирован-
ных фосфорнокислых удобрений (преципитата, двойного супер-
фосфата и аммофоса).
Фосфогипс выгодно отличается от природного гипса тем, что
его не требуется дробить. Если фосфогипс не использовать, то
туковым заводам придется затрачивать много средств, чтобы
убрать его с территории заводов.
Попытки освоения гипса для производства серной кислоты
(или серы)*велись и ведутся в следующих направлениях: 1) вос-
становление сульфата до сульфида,’ вытеснение сероводорода и
дальнейшая переработка последнего на серную кислоту или серу;
2) перевод иона SO4" исходного сульфата в другую промышлен-
но более необходимую соль; 3) разложение сульфата с получе-
нием SO2 и дальнейшая переработка последнего на серную кис-
лоту.
Так как сульфат натрия имеет в качестве катиона основу со-
довых продуктов, то понятно, что при разработке методов ис-
пользования сульфата натрия предусматриваются способы одно-
временного получения серы или серной кислоты, с одной сто-
роны, и соды (кальцинированной или каустической) — с другой.
58
При нагревании с углем при высокой температуре любой
сульфат переходит в сульфид.
Сероводород выделяется тем или иным способом. В промыш-
ленности (в целях использования отходов при производстве
соды по Леблану) были осуществлены способы обработки суль-
фида кальция хлоридом магния по реакции:
CaS + MgCla 4- 2Н2О = H2S + Mg (ОН)2 4- СаС12,
а также углекислотой по реакции:
CaS 4- СО2 4- Н2О = СаСО3 4~ H2S.
В СССР разрабатывается способ получения H2S из гипса
s комплексе с производством соды по Сольвею.
С производством бариевых солей и литопона связано произ-
водство H2S из сульфата бария. Получаемый в качестве отхода
производства BaS обрабатывается углекислотой, при этом осво-
бождается сероводород.
В разрабатываемых способах одновременного получения соды
и сероводорода из сульфата натрия последний или восстанавли-
вается углем до сульфида или приводится в обменное разложе-
ние с сульфидом бария, который получается восстановлением
сульфата бария. В дальнейшем в обоих случаях сульфид натрия
обрабатывается углекислотой с получением соды и сероводо-
рода.
Перевод иона SO/' из состава гипса в состав нового суль-
фата в промышленном масштабе осуществлен для получения
сульфата аммония. Сульфат аммония применяется как удобрение
и производство его на основе гипса освобождает от затраты
соответствующего количества HgSOi. Этот способ впервые был
осуществлен фирмой BASF в Германии.
Получение сульфата аммония из гипса осуществляется:
1) взаимодействием газообразных СОг и NH3 с взмученным
в воде гипсом или 2) взаимодействием раствора карбоната
аммония с гипсом.
Карбонат кальция, получающийся при производстве
(NH4)2SO4 из гипса, может быть использован для получения
цемента.
В СССР разработан промышленный метод одновременного
получения из сульфата натрия соды и сульфата аммония.
Способ разложения гипса с получением сернистого газа ниже
рассмотрен более подробно.
Разрабатываются также способы получения сернистого газа
из сульфата натрия. Так, например, предполагается применить
сульфат натрия для получения каустической соды по Левигу.
При обработке Na2SO4 с Ре2Оз при высокой температуре проис-
ходит реакция:
Na2SO4 4- Fe2O3 -> Na2O • Fe2Os 4- SO2 4- */2Oa.
59
Феррит натрия идет дальше для получения каустической
соды, a SO2 — на производство серной кислоты. По другому ме-
тоду (Академия Наук СССР) из сульфата натрия получается
метасиликат натрия (идущий в стекольную промышленность) и SO2.
12. Травильные растворы
В металлургии и .машиностроении при травлении металла по-
лучаются в отходе так называемые травильные растворы, содер-
жащие '2—4% свободной серной кислоты и до 25% сульфата
железа. Подобные же отходы получаются при производстве ти-
тановых белил. На ряде наших заводов травильные растворы до
последнего времени сбрасывались в реки. Но это совершенно
недопустимо. Загрязненная травильными растворами вода стано-
вится не пригодной для питания котлов, на такой воде устра-
няется возможность гидротехнических сооружений из бетона,
отравляется рыба.
В США имеются установки регенерации серной кислоты из
травильных растворов. Принципиальная схема этих установок
в основном сводится к следующему: травильные растворы, со-
держащие серную кислоту, нейтрализуются во вращающейся
трубчатой печи огарком, получаемым при разложении сульфата
железа, и затем освобождаются от влаги высушиванием за счет
тепла от сжигания топлива. Высушенный сульфат вместе с углем
загружается в печь, где сульфат разлагается на SO2 и огарок.
Таким образом удается регенерировать 90—92% серной кислоты,
затрачиваемой на травление.
Во Франции и Германии имеются установки, в которых из
травильного раствора путем его охлаждения холодильным рас-
твором выделяется весь сульфат в виде FeSOi • 7Н2О. Остав-
шийся маточник после добавления серной кислоты возвращается
обратно в травильную ванну. Железный купорос может быть
использован для получения серной кислоты, а также для других
целей.
13. Соотношение отдельных видов сырья
Соотношение между отдельными видами применяемого сырья
не может оставаться постоянным.
Факторы, влияющие на изменение этого соотношения, в ос-
новном следующие: 1) уровень развития техники — степень
освоенности различных видов сырья; 2) наличие в стране раз-
веданных залежей того или иного вида сырья; 3) развитие при-
мыкающих отраслей (промышленности, поставляющих в виде
своих отходов сырье для промышленности серной кислоты.
В большинстве стран до настоящего времени основным
сырьем для серной кислоты продолжает оставаться серный кол-
чедан. Однако в США большая часть серной кислоты произво-
дится из серы ввиду огромных залежей ее и дешевизны добычи.
В ряде стран значительные количества серной кислоты произво-
60
дятся из газов металлургических печей и из газоочистительной
массы. Соотношение между отдельными видами сырья для про-
изводства серной кислоты по некоторым странам приведено
в табл. 22.
Таблица 22
Виды сырья в % Страны
США (1939 г.) Англия (1938 г.) Канада (1936 г.)
Колчедан .... 25 46,82 13
Отходящие газы 13 11,51 1 74
Сера ...... Газоочиститель- 62 21,38 2 13
ная масса , . . — 20,29 —
Выбор сырья иногда обусловливается особыми обстоятель-
ствами. Так, в Германии во время войны 1914—1918 гг., когда
имморт сырья был затруднен, значительное количество серной
кислоты производилось из гипса.
В СССР раньше основным сырьем для производства серной
кислоты был обыкновенный серный колчедан. В последние годы
этот колчедан заменяется колчеданом флотационным и угли-
стым и газами металлургических печей.
Основными задачами в направлении реконструкции сырьевой
базы сернокислотной промышленности СССР на третью и чет-
вертую пятилетки являются следующие:
1. Богатые газы металлургических заводов (медеплавильных,
цинкоплавильных) должны быть полностью использованы. Не-
обходимо освоить использование слабых и переменных газов
металлургических заводов (конверторов, отражательных печей,
свинцового производства).
2. Должно быть максимально расширено применение угли-
стого колчедана в районах, близких к его добыче.
3. Необходимо полностью регенерировать серную кислоту из
кислых гудронов. Нефтеперегонные заводы в связи с этим
должны потреблять другое сернистое сырье только в количе-
стве, соответствующем производственным потерям.
4. Отходы металлообрабатывающей промышленности — тра-
вильные растворы — должны быть полностью использованы для <.
извлечения железного купороса. При этом за покрытием нужд
народного хозяйства в самом железном купоросе остальное ко-
личество железного купороса должно быть использовано на про-
изводство серной кислоты.
5. Необходимо освоить сжигание элементарной серы.
1 Включая цинковые концентраты.
2 Включая сероводород.
61
6. Необходимо освоить использование серы отходов углеобо-
гащения.
7. Должно быть освоено использование SO-г топочных газов.
8. В остальном потребность сырья будет покрываться колче-
даном, главным образом флотационным. Предназначенный
для сжигания на сернокислотных заводах флотационный кол-
чедан должен подвергаться перефлотации и сушке. Так как по
масштабам развития цветной металлургии флотационного колче-
дана будет получаться значительно больше, чем его требуется
для сернокислотной и бумажной промышленности, необходимо
организовать хранение остатков флотационного колчедана.
9. Необходимо промышленно освоить гипс для получения
серной кислоты и цемента.
II. Дробление колчедана
1. Склад колчедана
Для хранения запаса сырья строится склад колчедана, размер
которого выбирается в зависимости от производительности за-
вода.
Рис. 8. Подвижной ленточный транспортер.
Склад располагается вдоль линии железной дороги. По длине
склада со стороны расположения железнодорожной линии устраи-
вается дамба, к которой вагоны подаются вплотную, образуя так
называемый фронт разгрузки.
Раньше разгрузка вагонов производилась вручную. В послед-
нее время эта работа механизируется: устанавливаются механи-
ческие лопаты, выгружающие колчедан непосредственно из ва-
гона на транспортное оборудование, распределяющее колчедан
по складу; входят в применение саморазгружающиеся вагоны.
62
Распределение колчедана по складу вручную, развозка тач-
ками встречается теперь лишь на старых заводах небольшой
производительности. На новых заводах склады механизированы.
Простейший вид механизации — установка на складе подвижных
наклонных ленточных транспортеров (рис. 8). Загрузка таким
транспортером может быть произведена на высоте до 3 /и и на
участке радиусом от 2 до 6 м в зависимости от длины транспор-
тера (подвижные ленточные транспортеры строятся длиной от 5
до 15 л). Распределение колчедана по складам производится
также при помощи мостового крана с грейфером 2 (рис. 9).
Более усовершенствованная механизация достигается уста-
новкой стационарных транспортеров / и 2 по схеме рис. 10. Ли-
ния железной дороги расположена над бункерами, устроенными
в подземном канале. Колчедан из вагона выгружается непосред-
ственно в бункеры. Под бункерами расположены транспортеры 7,
подающие колчедан к транспортерам 2, а последние к элева-
тору 3. Элеватор поднимает колчедан на центральный транспор-
тер 4, расположенный на верхней площадке склада. Транспортер
снабжен автоматической или ручной сбрасывающей тележкой,
распределяющей колчедан по длине склада.
Разгрузка склада производится транспортером 5, расположен-
ным .в подземном канале склада. В полу склада над транспорте-
ром сделаны отверстия с течками, через которые колчедан за-
брасывается на ленту. Лента эта подает колчедан непосред-
ственно к размольному отделению.
Высота загрузки склада зависит от уровня расположения цен-
трального транспортера. Высота кучи колчедана принимается
в пределах от 3 до 5 л.
63
2. Схема дробильной установки
Для подачи колчедана из склада в дробильное отделение су-
ществуют разные способы.
Рис. 10. Механизация распределения колчедана по складу:
/и2—неподвижные транспортеры; 3 — элеватор; 4— центральный транс-
портер; 5 — транспортер.
Наиболее широко применяется механизированная подача кол-
чедана: транспортерами, проходящими в тоннелях по всей длине
склада, или краном-грейфером в' бункер дробильного отделения.
Применяемый еще на ряде заводов способ подачи колчедана из
немеханизированного склада изображен на рис. 11.
Колчедан вручную тачками или макензеном 7 подается в бун-
кер 2, оттуда пластинчатым транспортером 3 передается в дро-
билку 4. Для равномерной загрузки пластинчатого транспортера
между ним и бункером часто ставят питатель 5 в виде неболь-
шого качающегося лотка. Для этой же цели, а также для отде-
64
ления мелких кусков колчедана, которые нет смысла пускать
в дробилку крупного дробления, перед дробилкой иногда ставят
трясучее сито 6 с отверстиями в днище.
Рис. 11. Схема подачи колчедана:
/—макензен; 2— бункер; 3 — пластинчатый транспортер; 4 — дробилка; 5 — питатель; 6—тря-
сучее сито.
Схема дробильного отделения показана на рис. 12. Руда
с размерами кусков не больше 200 мм подается в дробилку
Блэка 3. Здесь куски руды раздробляются до размера 35—45 мм
Рис. 12. Схема дробильного отделения:
/ — вагонетка; 2— транспортер; 3—дробилка Блэка; 4—элеватор грубого помола; 5—плоский
качающийся грохот; 6—барабанный грохот; 7—вальцовая мельница; Я —бункер; 9—тарель-
чатый питатель; 10— элеватор тонкого помола; 11— транспортер; 12—бункер; 13—кон-
трольная решетка; 14 — течка; /5 —приямок.
в поперечнике. Раздробленный материал поступает в приямок 15,
откуда он захватывается ковшами элеватора грубого помола 4 и
подается в барабанный грохот б. В барабанном грохоте через
отверстия на его поверхности проходят куски размером не более
6—10 мм в поперечнике. Этот готовый материал попадает в бун-
5 Зак. 5019. Технология серной кислоты.
65
кер 8 мелкого помола, из которого через питатель 9 по элева-
тору 10 мелкого помола поступает в печное отделение.
Куски размером 10—45 мт проходят по всей длине барабан-
ного грохота и по течке 14 направляются в вальцовую мель-
ницу 7, где дробятся до размера 6—10 мм. Из вальцовой мель-
ницы материал поступает в приямок грубого помола 15, откуда
элеватором 4 подается в барабанный грохот 6.
3. Щековая дробилка
Для предварительного дробления колчедана ставят почти ис-
ключительно щековые дробилки. Такая дробилка имеет одну не-
подвижную и одну качающуюся щеку; дробление происходит
Рис. 13. Щековая дробилка:
I — рама; 2 — неподвижная щека; 3 — подшипники; 4 — ось; 5 — корпус подвижной щеки;
6— подшипники; 7 — вал; 8—шатун; 9 и 10—клинья; 11—болт; 12— рычажные плиты; 75-
вкладыш; 14— штанга; 15 — пружина; 16—подвижная щека.
раздавливанием при периодическом нажатии. Щековые дробилки
имеются двух типов — Блэка и Доджа. У дробилок Блэка наи-
больший размах качания подвижной щеки осуществлен внизу,
в месте выхода дроблёного колчедана; у дробилки Доджа —
вверху. Для колчедана преимущественно применяют дробилки
Блэка. Дробилка Блэка (рис. 13) состоит из рамы 1, на перед-
ней стенке которой укрепляется щека 2. На подшипниках з, по-
мещенных на корпусе дробилки, установлена ось 4, на которой
шарнирно укреплен корпус 5 подвижной щеки 16- На уста-
новленных на раме дробилки подшипниках 6 расположен эксцен-
триковый вал 7, на который надет шатун 8, делающий при вра-
66
щении вала 7 колебательные движения вверх и вниз. У задней
стенки корпуса машины установлен клин 9, к которому примы-
кает клин 10, могущий передвигаться горизонтально. Клин 9 мо-
жет передвигаться при помощи болта 11.
Рычажными плитами 12, вставленными во вкладыши 13, свя-
заны между собой шатун 8, корпус подвижной щеки 5 и клин 10.
К корпусу подвижной щеки на шарнире прикреплена штанга 14,
оттягивающая при помощи пружины 15 корпус 5.
На валу 7 сидят приводной и холостой шкивы и маховики.
При вращении эксцентрикового вала и при подъеме шатуна 8
плита 12 нажимает на корпус 5 и щеку 16, которая приближается
при этом к щеке 2. При ходе шатуна вниз щека 16 отходит
назад.
Кусок руды, попавший между щеками, дробится при их сбли-
жении. При раздвижении щек колчедан скользит вниз. Для ре-
гулирования величины выходной щели служат клинья 9 и 10.
При подъеме клина 9 вверх клин 10 движется вперед и выход-
ное отверстие суживается. Для регулирования величины щели
можно также прибегать к установке более коротких или более
длинных плит 12. Щеки дробилки подвергаются наибольшему
изнашиванию, поэтому они делаются съемными. Так как наиболь-
шее изнашивание щек происходит внизу, то они могут после не-
которого периода работы переставляться на 180°. Рабочая по-
верхность щек делается зубчатой. Щеки изготовляются из бе-
лого чугуна, марганцовистой стали или кованой хромистой стали.
Для предохранения дробилки от поломок в случае попадания
в нее чрезмерно твердых предметов (куски железа и др.) рычаж-
ные плиты 12 делаются с таким расчетом, чтобы они ломались
Таблица 23
Характеристика дробилок Блэка Заводская марка
Щ-5 Щ-7 Щ-9
Размеры загрузочного отверстия, мм Максимальные размеры загружае- мого материала Диаметр шкивов маховика, мм . Ширина шкива маховика, мм . . Число оборотов шкива в мии. . . Мощность мотора, л. с Ширина выходной щели, мм . . Производительность, м^час . . . Вес дробилки, кг . Габаритные размеры, мм ( длина 1 ширина 1 высота 400X230 350 Х2С0 1 100 170 220 15 35—70 5-10 4 500 2300 1560 1250 600 X 330 550 X 300 1400 200 220 35 45—85 13—25 11000 3050 1790 1680 800X500 750 X 420 1800 240 220 75 70—135 32—53 19300 3700 2330 2150
5*
67
•в первую очередь, предохраняя от поломок более важные части
машины.
Для расчета производительности дробилки Блэка имеется не-
сколько формул. Так, легко вывести следующее выражение:
п K(2C+S)SBn-(
* ~ 2tga • 106 >
где Q — вес измельчае-
мой руды в кг в минуту,
у — вес 1 № материала
в кг, С — ширина выпуск-
ного отверстия дробилки,
aS — размах качания по-
движной щеки в см, а —
угол между щеками, К —
коэфициент разрыхления
руды, величина которого
для колчедана колеблется
в пределах 1/г—*/з, п —
число оборотов вала в
минуту.
Эта формула, как и по-
добные другие, сугубо
ориентировочна. В табл. 23
приведены данные о дро-
билках Блэка, производи-
мых Выксунским заво-
дом дробильно-размольно-
го оборудования.
4. Вальцовая мельница
Т а час ть колчедана .ко-
торая не проходит через
отверстия барабанного гро-
хота, направляется для
окончательного дробления
на вальцовую мельницу.
Здесь кусочки колчеда-
на захватываются двумя
вращающимися навстречу
друг другу горизонталь-
_ ными валками и ими исти-
раются. Для того чтобы кусочки колчедана лучше захватыва-
лись, необходимо, чтобы трение между кусочками руды и по-
верхностью валков было достаточно большим. В связи с этим
поверхность валков делается шероховатой.
Конструкция вальцовой мельницы понятна из рис. 14. В тя-
желой станине 1 находятся неподвижные подшипники 2- В этих
68
подшипниках вращается вал 3, на котором укреплен валок 4. На
валу 3 укреплен также главный приводной шкив 5. На другой
стороне фундаментной плиты расположены подвижные подшип-
ники 6 для валка 4. Они поддерживаются толстыми пружинами 7
с помощью тяги 8, проходящей через всю фундаментную плиту
и имеющую второй упорный подшипник 9. Благодаря этому пе-
редвижной валок имеет некоторый зазор для отклонения в слу-
чае, если на вальцы вместе с рудой попадет кусок металла. Пе-
редвижной валок устанавливается прокладками 10. Этими же
прокладками можно регулировать расстояние между валками,
а следовательно и степень раздробления руды. Давление пру-
жины 7 рассчитывается так, что подвижной валок всегда приле-
гает к прокладкам, и зазор между вальцами имеет определенную
величину. Подвижной валок приводится в действие ременным
шкивом 11. Главная сила привода действует, однако, на шкив 5,
и подвижной валок приходит в движение, главным образом,
вследствие трения.
Теоретическая производительность валков может быть рассчи-
тана следующим образом.
Если окружная скорость вальца и (м/сек), длина вальца b (м),.
расстояние между вальцами 2d (м), то в 1 час проходит через
вальцы лента материала следующего объема (м3):
V = 3600 - и - b - 2d = 7200 • и - b • d.
Если количество оборотов вальца в минуту равно п, а диа-
метр вальца (м) равен D, то:
Таблица 24
Характеристика валковых дробилок Заводская марка
ВМ-2 ВМ-3
Диаметр валка, мм 600 800
Ширина валка, мм Число оборотов валка в мин 300 350
100—180 80—120
Диаметр шкива маховика, мм ........ 1250 1800
Ширина шкива маховика, мм 200 250
Диаметр второго шкива, мм 630 100
Ширина второго шкива, мм • . Максимальный размер входящего материала. . 150 200
ММ 30 40
Ширина щели между валками, мм ...... Производительность максимальной щели, 2—8 2—12
т!час Габаритные размеры, мм 15 30
( длина . . . 2450 2850
| ширина 2195 2535
1 высота • 1340 1620
6»
Производительность в т/час будет:
Q — 377 • п • D • b • d у,
где у — удельный вес руды.
Валковые дробилки изготовляются двух основных типов:
1) тихоходные с числом оборотов до 40 ;в 1 мин. и быстро-
ходные с числом оборотов до 100 и выше.
В тихоходных дробилках валки обычно приводятся в движе-
ние при помощи зубчатой передачи.
В табл. 24 приведена характеристика валковых дробилок,
производимых Выксунским заводом.
5. Барабанный грохот
Барабанный грохот состоит из вращающегося цилиндра, сде-
ланного из дырчатых листов. Листы прикрепляются к металли-
ческой раме грохота, поддерживаемой крестообразно' располо-
женными ручками, надетыми на его ось. Барабан располагается
наклонно под некоторым углом. Колчедан, поступающий в верх-
ний конец грохота, медленно перемещается при вращении гро-
хота к его нижнему концу. При этом происходит просеивание
тех частиц, которые по своим размерам могут пройти через от-
верстия на поверхности грохота. Эта часть материала попадает
в бункер мелкого помола. Непросеявшаяся часть доходит до
нижнего конца грохота и опять возвращается в приямок грубого
помола.
Барабан поддерживается или только фрикционными роликами
или фрикционными роликами на загрузочном конце и короткой
осью на разгрузочном конце; в этом случае на ось надевается
коническое зубчатое колесо.
Снаружи барабанный грохот покрыт железным кожухом для
безопасности работы и лучшего сбора отсеянного материала.
Производительность барабанного грохота может быть опреде-
лена по формуле:
Q = 0,67-ntg2a/^3,
где 7 — объемный вес материала (для колчедана у —2,2) в т,/л3,
л — число оборотов в минуту, а — угол оси барабана с гори-
зонталью, R — радиус барабана в м, h — толщина слоя про-
сеиваемого материала в барабане.
Мощность барабана (в л. с.) определяется по формуле:
лг_/?n(g+25gb)
, 21 500
где R — внутренний радиус барабана в л, п — число оборотов
в минуту, g— вес барабана в кг, go — вес находящегося в бара-
бане колчедана в кг.
'70
6. Подсобное оборудование дробильных отделений
В дробильном отделении обычно имеются два элеватора: пер-
вый (циркуляционный) подает колчедан из приямка грубого по-
мола в барабанный грохот, вторым (продукционным) оконча-
тельно’ размолотый колчедан поднимается в бункер печного отде-
ления. В дробильных отделениях применяются исключительно
цепные элеваторы. Ковши элеватора выполняются из железа тол-
щиной 4 мм.
Производительность элеватора в м3/час равна
Q=3600-ffi ,
о
где р — степень наполнения ковша (0,6—0,7), Е —объем
ковша в м3, v — скорость цепи в м/сек, S — расстояние между
ковшами в м.
Потребная мощность элеватора в л. с. равна
n-S-75 ’
где Р — вес загрузки одного ковша в кг, Н — полная высота
подъема, — коэфициент полезного действия элеватора, рав-
ный 0,75. При учете необходимости преодоления сил инерции
при пуске фактически потребная мощность выше на 100%
определяемой по приведенной формуле.
Как это видно из схемы дробильного отделения, колчедан на
отдельных участках самотеком движется по течкам. Для того
чтобы колчедан в течках не застревал, они должны иметь уклон
не меньше 55° и не должны иметь резких поворотов. Течка де-
лается из 6—7-мм железа, а дно (для большей прочности) выкла-
дывается плитками толщиной 15 мм-
Фактическая производительность дробильного отделения
в первую очередь зависит от соответствия мощности его отдель-
ных аппаратов. Часто, когда мощность дробилки Блэка отстает
от мощности других аппаратов, общая выработка дробильного
отделения может.быть значительно повышена отсеиванием ме-
лочи (уже готовой для обжига) перед дробилкой Блэка (поста-
новка трясучего сита). Часто узким местом отделения является
барабанный грохот; его мощность может быть значительно по-
вышена, если его отсеивающая поверхность выполняется не из
дырчатых листов, а из продольных железных прутьев.
Фактическая производительность дробильного отделения очень
снижается, если циркулирует влажный колчедан: забиваются
течки, отверстия грохота и т. д. Поэтому надо принимать меры
против проникновения воды в приямки и другие места.
Все механизмы дробильного отделения работают в очень тя-
желых условиях, поэтому настоятельно необходимы здесь пра-
вильная постановка планово-предупредительного ремонта, нали-
чие запасных частей и постоянный тщательный уход за аппарату-
рой.
71
Множество движущихся механизмов требует особого внима-
ния к проведению мероприятий по технике безопасности.
Основные правила обслуживания движущихся механизмов
с точки зрения техники безопасности должны сводиться к сле-
дующему: не снимать и не надевать ремней на шкивы на ходу;
не смазывать на ходу движущиеся части аппаратов, а также
подшипники трансмиссий, если вблизи находится ремень или
шкивы; все ремни и движущиеся части аппаратов должны иметь
ограждения; площадки и лестницы должны иметь поручни.
В связи с большим образованием колчеданной пыли дробильное
отделение должно быть оборудовано вытяжной вентиляцией.
Отсос пыли следует производить из мест ее наибольшего обра-
зования — из кожухов элеваторов и барабанного грохота. Во
время работ по очистке элеваторов необходимо пользоваться
противопылевым респиратором.
7. Ситовой анализ
Ни одна машина, дробящая руду, не дает в результате своего дробления
частицы одинакового размера. Для определения, какого размера частицы
составляют наибольший процент раздробленной руды, пробу приходится
рассеивать на фракции с разными размерами частиц.
Ситовой анализ производится с помощью системы сит, в которых вели-
чина отверстий в определенном отношении понижается от сита к ситу.
Сита в их системе принято обозначать числом отверстий на линейный дюйм.
При этом толщину проволоки подбирают так, чтобы величина отверстий
изменялась от сита к ситу согласно принятой шкале. В США принята си-
стема Тайлера, в которой величина отверстий от сита к ситу изменяется
Таблица 25
Нормальные сита системы Тайлера Нормальные сита английской системы
число отверстий в линейном дюйме величина отвер- стий в мм число отверстий в линейном дюйме величина отвер- стий в мм
3 6,68 5 2,54
4 4,70 8 1,57
6 3,33 10 1,27
8 2,36 12 1,06
10 1,65 16 0,79
14 1,17 20 0,64
20 0,83 30 0,42
28 0,59 40 0,32
35 0,42 50 0,25
48 0,295 60 0,21
65 0,208 70 0,18
80 0,175 80 0,16
100 0,147 90 0,14
150 0,104 100 0,13
200 0,074 120 150 . 200 0,11 0,08 0,06
72
в постоянном отношении, равном V '>. В английской нормальной системе
сит толщина проволоки всегда равняется величине отверстия. Это очень
удобно, так как Дает возможность по числу отверстий на 1 дюйм опреде-
лить величину самого отверстия. Так, если на 1 дюйм приходится п отвер-
стий, то величина одного отверстия в мм равна .
В табл. 25 приведены характеристики американских и английских нор-
мальных систем сит.
Сам процесс просеивания производится так.
Взятая проба материала просеивается через сита — от сита к ситу (от
более крупных к более мелким). Просеивание на каждом сите произво-
дится доотказа. Процент просеявшегося материала обозначается знаком (—),
а оставшегося на сите знаком (+). Ситовой анализ считается выполненным
достаточно точно, если в процессе всего анализа потери материала соста-
вляют не больше 1—2% от взятой пробы, а расхождение между повтор-
ными анализами не больше долей процента. Число отверстий сита, прихо-
дящееся на 1 дюйм длины сита, носит наименование меш.
III. Сушка флотационного колчедана
Флотационный колчедан, непосредственно взятый из отвала
медеплавильных заводов (болота), содержит 14—16% влаги.
Транспортировать его в таком виде нерентабельно, во-первых,
из-за загрузки транспорта излишним баластом в виде воды и,
во-вторых, ввиду смерзаемости влажного колчедана в зимнее
время. По опытным данным условия смерзаемости флотацион-
ного колчедана следующие:
Содержание влаги до 3%.............не смерзается
Содержание влаги от 3 до 5%......начинает смерзаться
Содержание влаги выше 5,0%.......смерзается в монолит
Ввиду этого флотационный колчедан подвергается сушке на
месте производства до содержания в нем 4% влаги. Сушильные
установки значительной производительности работают на ряде
Уральских заводов. Продукция этих сушилок направляется на
сернокислотные заводы. Для пылевидного обжига требуется кол-
чедан влажностью 2,0—5%. Фактическая влажность колчедана,
прибывающего* на сернокислотные заводы, бывает в пределах
5—7%, вследствие атмосферных влияний. Поэтому материал до-
полнительно сушится на месте потребления на специальных су-
шильных установках.
Сушка флотационного колчедана производится непосред-
ственно топочными газами. Во избежание возгорания колчедана
сушка ведется параллельным током.
Сушильная установка (рис. 15) состоит из топки 7, откуда
дымовые газы поступают в камеру смешения 2. Смешанный
с воздухом газ поступает в сушильный барабан 3. Сюда же по-
дается влажный колчедан через бункер с питателем 4 и течку 5-
Материал параллельно с газом направляется по барабану (уста-
новленному с наклоном в 4° к горизонту) к разгрузочной ка-
мере б. Сухой колчедан поступает на магнитный сепаратор 7.
Отсепарированные куски железа попадают в бункер 8, а колче-
дан в бункер 9 и на молотковую дробилку 10. Готовый продукт
73
направляется в печное отделение. Газ из разгрузочной камеры
поступает в два последовательно установленных циклона 11 и 12,
обеспыливается и отсасывается вентилятором в выхлопную
трубу 13. На новых сушилках для обеспыливания газа уста-
навливаются электрофильтры.
Объем сушильного барабана определяется напряжением бара-
бана по влаге, т. е. количеством влаги, выпариваемой в час с 1 м3
объема барабана.
Рис ,15. Сушильная установка:
1—топка; 2—камера смешения; 3—сушильный барабан; 4—писатель; 5—течка; 6—разгру-
зочная камера; 7—магнитный сепаратор; 8 и 9—бункер; 10—молотковая дробилка; 11 н 12 —
циклоны; 13—выхлопная труба.
По опытам Всесоюзного теплотехнического' института напря-
жение барабана по влаге равно 58 кг/м3 • час. По заводским дан-
ным напряжение достигает 35—40 кг/м3 • час (при сушке до
0,5% влажности). Напряжение барабана при прочих равных усло-
виях (для одного и того же материала, размеров кусков, темпе-
ратуры газа и т. д.) в значительной степени зависит от конечной
влажности продукта (уменьшается с понижением конечной влаж-
ности).
Диаметр барабана определяется, исхрдя из величины воз-
можно минимального уноса сухого колчедана с отходящими га-
зами.
74
На сернокислотных заводах установлены сушильные барабаны
со следующей характеристикой:
Диаметр барабана D................ 1700 мм
Длина барабана L.................. 9000 »
Число оборотов в мин. п..........2—5 „
Наклон к горизонту а.............4°
Вращение барабана осуществляется посредством зубчатого за-
цепления, состоящего из зубчатого венца, закрепленного на бара-
бане, и малой шестерни, насаженной на вал привода. Вал привода
расположен в подшипниках фундаментной рамы и получает вра-
щение через систему конических зубчатых колес и ременной пере-
дачи от мотора.
Рис. 16. Подъемно-лопастная си-
стема сушильного барабана.
Рис. 17. Ячейковая система сушиль-
ного барабана.
Регулирование работы сушильного барабана сводится к тому,
чтобы обеспечить равномерную подачу колчедана и правильное
соотношение между количествами входящих газов и колчедана
(при учете его влажности).
Температура входящих в барабан газов должна регулиро-
ваться в зависимости от процента влажности поступающего кол-
чедана. Она должна быть такой, чтобы при параллельном токе
колчедана и газов не происходило возгорание колчедана. Как
показывает опыт работающих сушилок, эта температура должна
держаться в пределах 450—650°. Температура уходящих газов
Держится в пределах ПО—115°.
Для получения максимальной производительности сушильного
барабана и наилучшего теплового эффекта необходимо: 1) тесное
соприкосновение материала, подвергающегося сушке, с дымовыми
газами и 2) минимальный унос высушенного материала с выхлоп-
ными газами.
75
Существуют различные системы внутреннего устройства су-
шильного барабана. Наиболее характерные из них: 1) подъемно-
лопастная система и 2) система ячейкового типа.
Подъемно-лопастная система (рис. 16) состоит из ряда лопа-
стей, расположенных по внутренней окружности барабана. При
1—5
Рис. 18. Дроссельный подпорный диск.
вращении барабана материал
поднимается лопастями вверх,
откуда он постепенно ссы-
пается по мере изменения
положения лопаток. При
ссыпании с лопаток по сече-
нию барабана образуется
значительная поверхность
материала, тесно соприка-
сающаяся с перпендикуляр-
но движущимся горячим га-
зом. Недостаток этой систе-
мы — большой унос высу-
шенного материала с вы-
хлопными газами ввиду зна-
чительной высоты падения. Гораздо меньше унос материала
в ячейковой системе (рис. 17) благодаря малому перепаду мате-
риала. В ячейках же достигается более тесное соприкоснове-
ние материала с дымовыми газами. Для увеличения производи-
тельности сушильного барабана рекомендуется установить
Рис. 19. Молотковая дробилка.
в торце .его, в месте выхода сухого материала, специальное под-
порное приспособление, дающее возможность повысить время
пребывания материала в барабане.
Простейшая конструкция подпорного приспособления — это подпорное
кольцо. Кольцо определенной высоты (в зависимости от допускаемого макси-
мального подпора) приваривается к торцу барабана. По поверхности кольца
расположены лючки, служащие для регулирования или уменьшения высоты
подпираемого слоя. Недостаток этой конструкции состоит в том, что кольцо
создает незначительный подпор, так как увеличение ширины кольца умень-
шает сеченне выхода газа. Возможность регулирования подпора весьма огра-
76
•ничеиа. Более совершенным является дроссельный подпорный диск (рис. 18).
Диск в виде сегмента, занимающего */3 сечения барабана, насаживается на
горизонтальную ось, проходящую через стенку неподвижной разгрузочной
камеры. Диск можно передвигать вокруг оси барабана и вдоль нее. Передви-
жением диска в двух направлениях достигается регулирование степени на-
полнения барабана в широких пределах и изменение сечения для прохода
таза.
Для размола комков, образующихся в процессе сушки, после
сушильного барабана устанавливаются молотковые дробилки
(рис. 19).
Молотковые дробилки Союзстроймашины состоят из станины, в боковых
стенках которой проходит вал квадратного сеченни. На вал насажены серьги,
Рис. 20. Электромагнитный сепаратор.
в которых шарнирно укреплены молоты. К низу станины прикреплена ре-
шетка. Измельчение материала, поступающего в приемное отверстие, проис-
ходит от разбивания его иа ходу быстро вращающимиси молотками: комки
разбиваются о стенки станины, на которые они отбрасываются молотками, н
при попадании между молотками и решеткой они окончательно истираются и
выпадают вниз в приемник.
Электромагнитный сепаратор (рнс. 20) представляет собой вращающийся
барабан, внутри которого эксцентрично расположен электромагнит, питаемый
постоянным током. При вращении барабана поверхность его попеременно про-
ходит у полюсов электромагнита, прнтигивает к себе магнитные материалы
и удерживает их, пока находится в зоне магнитного поля. Прн дальнейшем
вращении эта часть барабана выходит нз зоны магнитного поля н куски
железа падают вниз отдельно от потока колчедана.
В отделении сушки должны быть предусмотрены мероприятия
по технике безопасности, в первую очередь связанные с наличием
движущихся частей механизмов и с возможностью большого пы-
левыделения.
77
Все места выделения пыли должны быть закрыты кожухами
с отсосами от вытяжной вентиляции. Помещение, где находится
сушилка, должно иметь приточно-вытяжную вентиляцию с 6—7-
кратным обменом.
Литература
1. Эфраим Ф., Неорганическая химия, ч., II, стр. 3—19, Госхимтехиз-
дат, Л. 1933.
2. Проф. Федоровский Н. М., Курс минералогии, стр. 92—95, Госиз-
дат, М.-Л., 1930.
3. Проф. Самойлов Я- В., Минералогические очерки, Госгорногеоло-
го-нефтеиздат, стр. 64—93, М.-Л. 1934.
4. Л е в и и. А. М., Добыча серного колчедана в капиталистических стра-
нах и мировой рынок серного колчедана, ЖХП, № 11—12, 850 (1937).
5. Die Rohstoffversorgung det deutschen Schwefelsaureindustrie, Berlin 1923.
6. Б ью к e н e н Г. X., Химические явления как орудия флотации, перев.
с аигл., Госхимтехиздат, 1931.
7. Бухгольд К., Флотационный процесс, перев. с нем.. ГНТИ, 1931.
8. Кузьминых И. Н., Флотационный колчедан, Свердловское област-
ное гос. изд., 1935.
9. Фомин, Извлечение серного колчедана нз отходов углемоек, ЖХП
№ 11, 25 (1938).
10. Маркин И. Б., Гольдфлам М. А., Использование углистых пи-
ритов для производства серной кислоты, сборник статей «Сера и сер-
ная кислота”, стр. 149—176, ОНТИ, 1935.
11. Armstrong V., Himus G., Chem. and Ind. 58, № 22, 543—48 (1939).
12. Шнеерсон Б. Л., Получение серной кислоты при обезвреживании
дымовых газов, ЖХП № 9, 24 (1933).
13. Мулерт Ф.. Сера в угле, пер. с нем., Госхимтехиздат, 1932.
14. Ну си нов Г. С., Методы извлечения серы из промышленных газов,
Госхимтехиздат, 1933.
15. С т е п а и о в М. Н., Б р е и е р М. В., Серная кислота из отбросного
гудрона нефтеобрабатывающей промышленности, ЖХП № 2, 47 (1939).
16. Л и х у ш и н К. П., Утилизация кислых гудронов, Азгонти, Баку 1939.
17. Сборник по утилизации отходов и отбросов в нефтяной промышлеииости,
ОНТИ 1934.
18. Труды научного института по удобрениям им. Я- В. Самойлова, Гипс
и фосфогипс, Сборник статей, 1933.
19. Ильинский В. П., Пути получения соды, серы и серной кислоты из
природных сульфатов, Труды солевой лаборатории Академии Наук
СССР, вып. 4, изд. Академии Наук, Л. 1932.
20. Богуславский И. М„ Беньковский С. В., Синчук В. Е.,
Получение едкого натра и серной кислоты из мирабилита, ЖХП № 24,
1467 (1936).
21. Ильинский В. П., Мории Н. В. и др., Новые методы перера-
ботки сульфата натрия на соду и серу, ЖХП № 10, 27 (1934).
22. Тер а ш кевич В. Р., Е зецкий Г. Н., Проблема получения серы
из гипса в сочетании с аммначно-содовым производством, ЖПХ, вып.
9, 12, 2155(1937).
23. К г е р s Th. (Новые направления в области производства исходных
продуктов и применения серной кислоты), Chem. Met. Eng. 41, 1, 22
(1938).
24. Малин. К. М., Производство и потребление серной кислоты в капита-
листических странах, ЖХП № 1, 52 (1938).
25. Малин. К. М., Задачи сернокислотной промышленности в разрезе
решений XVIII Съезда ВКП(б), ЖХП Ns 7, 4 *(1939).
26. Waeser В., Handbuch der Schweielsauerefabrication, I, 34—386, Braun-
schweig 1930.
78
27. T h i е 1 e r Erich, Schwefel, Dresden und Leipzig 1936.
28. Юровский A. 3., Лифшиц M. M. и др., Угли и отходы углеобога-
щения как местное сырье сернокислотной промышленности, ЖХП № 3,
8 (1940).
29. Производство серной кислоты, под ред. Малина К. М., стр. 46—77,
ГОНТИ, 1938.
30. Поляков К. А., Дробление колчедана, ОНТИ, 1936.
31. Бэджер В., Мак-Кэб В., Основные процессы и аппараты химиче-
ских производств, стр. 392—400, 414—426, ОНТИ, 1933.
32. Касаткин А. Г., Основные процессы и аппараты химической техно-
логии, ч. 1. стр. 388—410, 419—428, ОНТИ 1935.
33. К а н т о р о в и ч 3. Б., Размольно-дробильные машины и грохота, стр.
7—73, 208- 293, ОНТИ, 1937.
34. Орт ин М. Ф., Механическое обогащение руд, стр. 13—35, 64—80,
148-153, 159-162, ГНТИ, 1931.
35. Р е з н и к В. Я-, Рациональная схема дробления колчедана, ЖХП № 23,
1613-1618 (1937).
36. Левенсон Л. Б., Прейгерзон Г. И., Дробление и грохочение
полезных ископаемых, Гостоптехиздат, 1940.
ГЛАВА 3
ОБЖИГ СЕРНИСТОГО СЫРЬЯ
И ОЧИСТКА ОБЖИГОВОГО ГАЗА
I. Теоретические основы обжига
1. Химизм горения колчедана и его примесей
Медленное окисление FeSa с выделением SO2 замечается уже
при температуре 170—210°.
Температура воспламенения колчедана зависит от характера
его примесей и от степени его дробления. По данным опытов
Рысс, Журавлевой и Суслова температура воспламенения колче-
дана при изменении размера кусков от 0,130 до 0,485 мм ко-
леблется в пределах 340—406°. По Везеру температура воспла-
менения пирита находится в пределах 400—500°.
Конечными продуктами сгорания колчедана в печах сернокис-
лотных заводов являются SO2 и РегОз. Суммарная реакция об-
жига пирита:
4FeS2 + 11О2 = 2Fe2O3 -f- 8SO2.
На самом деле обжиг колчедана очень сложный процесс, со-
стоящий из нескольких последовательных и целого ряда парал-
лельных реакций. В условиях горения FeS2 в печах сернокислот-
ных заводов одна из промежуточных реакций несомненна: при
простом нагревании FeS2 диссоциирует по реакции:
2FeSa --> 2FeS —S2-
Парциальные давления паров серы над FeS2 в зависимости от
температуры (по Аллену и Ломбарду) приведены в табл. 26.
Таблица 26
Температура в °C Парциальное да- вление паров серы в мм рт. ст. Температура в сС Парциальное да- вление паров серы в мм рт. ст.
575 0,75 645 106,5
595 3,5 655 168
610 13,5 665 251
625 36,3 672 343
635 61,0 680 518
80
Отделяющаяся сера сгорает в газовой фазе по реакции:
S2 + 2О2 = 2SO2.
Горение серы очень легко наблюдать в механических печах.
На верхних этажах печей, где в горящем материале много FeS2,
пламя окрашено в синий цвет, характерный для горения серы. На
следующих этажах, где FeSa в< горящем материале уже мало,
пламя не имеет синего оттенка, однако синие огоньки по-
являются еще вслед за проходящими гребками, которые, перегре-
бая материал, обнажают последние остатки FeSa. Наконец, на бо-
лее нижних этажах синего окрашивания пламени не заметно даже
и около идущих гребков; это значит, что в горящем материале
уже нет FeSa.
При отщеплении серы по 'реакции 2FeSa —- 2FeS + S2 полу-
чается в остатке пористый материал, что облегчает его даль-
нейшее горение. Большое значение для процесса горения пи-
рита имеет также его свойство растрескиваться во время го-
рения.
Суммарной реакцией горения FeS, получающегося при разло-
жении FeSa, является:
4FeS + 7О2 = 2FeaO3 ф- 4SO2.
Поскольку эта многомолекулярная реакция мало вероятна, суще-
ствует целый ряд теорий промежуточных реакций.
По одной из групп этих теорий первой стадией окисления
сульфида являются сульфаты, разложение которых затем дает
уже окислы. По другой группе теорий сульфид непосредс1венно
окисляется до окислов. Поводимому, окисление через сульфаты
идет при низких температурах, при высоких же температурах бо-
лее вероятно прямое образование окислов с возникновением
сульфата через сульфатизацию окислов.
Кроме взаимодействия кислорода с FeS и с переходящей в га-
зовую фазу серой при обжиге происходит целый ряд взаимодей-
ствий между промежуточными продуктами обжига; можно ука-
зать на следующие происходящие реакции:
10Fe2O3 + FeS = 7Fe3O4 ф- SO2,
5FeSO4 ф- FeS = 2Fe3O4 ф- 6SO2;
FeS4-3SO3 = FeO -4-4SO2;
SOa + 1/2O2 = SO3 и др.
В результате этих реакций на верхних этажах печи железо на-
ходится по преимуществу в виде низших окислов, превращаясь
полностью .в FeaO3 только на последних этажах, где и содержа-
ние сульфида в твердой фазе и содержание SO2 в газовой фазе
невелико. Если учесть, что любой вид колчедана содержит раз-
личные примеси, то будет понятно, какое множество реакций
происходит в процессе обжига колчедана.
6 Зак. 5010. Технология сериой кислоты.
81
В конечном итоге большинство сернистых соединений метал-
лов сгорает до окислов металлов и SO2.
Окислы металлов, реагируя с SO3, переходят в сульфаты. Так
как температура разложения ряда сульфатов выше той темпера-
туры, какая развивается в печи, то ряд сернокислых металлов
в этом виде переходит в огарок. Данные о температуре разложе-
ния сернокислых солей по работам Гофмана и Ванюкова приве-
дены в табл. 27.
Т а б л и ц а 27
Сульфаты Температура в °C Продукты распада
начала распада энергичного распада
FeSO4 167 480 FG2O3 • 3SO3
Fe2Oo • 3SO3 492 560 Fe2O3
Al2 (SC4)3 590 639 А12О3
PbSO4 636 705 6РЬО 5SO3
CuSO4 653 670 2СиО • SOs
MnSO4 699 790 Мп3О4
ZnSO4 702 720 3ZnO • 2SO3
2CuOSOs 702 736 CuO
3ZnO • 2SO3 755 767 ZnO
CdSO4 827 846 5CdO • SO3
5CdO SO3 878 890 CdO
MgSO4 890 972 MgO
O) 45 er Oc СЛ О 952 962 2PbO • SO3
CaSO4 1200 — CaO
BaSO4 1510 — BaO
Углекислые соли кальция и магния, если они присутствуют
в колчедане, разлагаются с выделением СОг.
Окислы этих металлов затем переходят в сульфаты.
2. Выход огарка, выгорающий процент серы,
сера в огарке
Основная часть огарка состоит из РегОз. Кроме того, в огарке
могут быть FesOi, FeS, FeSs. В огарок перейдут также и негорю-
чие примеси колчедана, как SiO2, CaSO4 и др., и образовавшиеся
в печи PbSO4, ZnSO4, CuSO4 и т. д.
Для получения практически- пригодной формулы выхода
огарка можно принять, что: 1) колчедан никаких составных ча-
стей (кроме РеЗг), изменяющихся в печи, не содержит и 2) ога-
рок состоит из РегОз и несгоревшей части колчедана (негорючие
примеси плюс невыгоревшая часть Ре8г).
82
Обозначим процент серы в колчедане через PSk, процент серы
в огарке через Psor, количество огарка на 1 кг колчедана че-
рез х.
Сера, процент которой от огарка равен P.sor, составит от кол-
чедана xPsor процентов. Следовательно, при проценте серы
в колчедане PsK, а в огарке Ps(T выгоревшая сера составит от
колчедана (в%):
PS ~ PSK (1)
В чистом пирите сера составляет 53,46%. При выгорающем
проценте серы, равном PSr — xPsOI, из 1 кг колчедана сгорит
FeS2:
У = ~xPs ) = 0,01875(Рч — xPs ),
Оо,4Ък ьк ьог 4 sk sor
а огарка в виде ЕегОз получится:
4FeS2 -> 2Fe2O3
479,84 -> 319,36
0,01875(7% — хРч )->z
v ьк sor
Z = 0,01246 (PSk—xPSor).
Кроме того, в огарок перейдет несгоревшая часть колчедана,
которая на 1 кг загруженного колчедана составит:
1-0,01875 (Р8к-хР8(г),
а всего на 1 кг загруженного колчедана получается огарка:
х = 0,01246 (PSk- xPsJ + [1 - 0,01875 (Ps- Ps<r)],
или по упрощении
160—Ps
Часто в практических расчетах выход огарка без вычислений
и замеров принимают равным 0,7 от колчедана. Фактически при
изменении процента серы в колчедане от 35 до 50, а в огарке
от 5 до 0,8 выход огарка изменяется от 0,805 до 0,685.
Выгорающий процент серы, как мы видели, равен:
₽8 = Ps_-xPEip (1)
Если вместо х подставить его значение из (2), то получим:
160—ps
Р8 = РЧ — Рч -________
S SK sorl60 —Ps
bor
(3)
6*
83
Если процент серы в углистом колчедане равен PsK, процент
угля в нем же Рс, процент серы в огарке Psor. то при помощи
примененного выше метода рассуждений легко вывести сле-
дующее выражение для выхода огарка:
Понятно, что при установлении веса и выхода огарка прямым
замером надо учитывать не только собственно огарок, высыпаю-
щийся из печи, но и пыль, уносимую газами. Отношение количе-
ства пыли к количеству собственно огарка меняется в зависимо-
сти от типа печей, качества колчедана, характера ведения про-
цесса и интенсивности обжига. При нормальной работе механи-
ческих печей на обыкновенном колчедане это отношение при-
мерно равно 0,025. Для горизонтальных вращающихся печей это
отношение составляет 0,04—0,05, а для пылевидных дости-
гает 0,3.
3. Состав обжиговых газов
Обозначим через т отношение между количеством молекул
кислорода, вступающих в реакцию, и количеством молекул SOa,
получающихся по реакции, и через п — процент кислорода в по-
ступающей на горение газовой смеси.
При этих обозначениях на 100 объемов обжигового газа бу-
дет затрачено исходной газовой смеси в объемах:
100--Р^О Н- ^Pso 9
или
100 + (m-l)PSOs,
где Р$о2—объемный процент SO2 в обжиговом газе.
В этом количестве исходной газовой смеси было объемов
кислорода:
^_I100 + (zn-l)PSO!].
При горении израсходовано объемов кислорода mPsos- Следо-
вательно, останется в газах объемов кислорода на 100 объемов
обжигового газа
ТШ ПОО-Нтп— 1) PS0J — mP80i,
или процент О2 в обжиговых газах равен:
'v+Hr1-"‘К- <4)
Это выражение справедливо для расчета состава газов при
горении любого сырья при том условии, что в результате горе-
84
ния кроме SO2 и Н2О никаких газообразных продуктов не полу-
чается. В табл. 28 приведены конкретные формулы для различ-
ных случаев горения, полученные из выражения (4) подстанов-
кой числовых значений тип.
Подставляя в любую формулу табл. 28 вместо Рог нуль, по-
лучим значения теоретических величин процента SO2 в газе, ко-
торые для различных случаев сжигания приведены в последней
графе таблицы. Как видно из формул табл. 28, для всех этих
случаев горения зависимость между
в газах прямолинейная (рис. 21).
Коэфициент избытка воздуха,
т. е. отношение количества факти-
чески затраченного воздуха к тео-
ретически потребному, как это лег-
ко вычислить, равен:
„ — П I О /ЕЛ
— mPs^ ‘ 100m '
Для случая обжига колчедана
в воздухе, например при п = 21 и
m =1,375:
а = ]Д27_|_ 0,057. (6)
<чо2
Из опыта теплотехники наимень-
ший коэфициент избытка воздуха
при обжиге флотационного колче-
дана можно принять равным 1,2. Соответственно этому наивыс-
шая возможная концентрация SO2 будет 13,36%. При обжиге
рядового колчедана наименьший коэфициент избытка воздуха
можно принять равным 1,4.
Практически высшая концентрация SO2 в газах будет 11,37%.
Объем обжигового газа на 1 т сырья равен:
концентрациями ЬО2 и
Рис. 21. Состав газа от обжига
колчедана в зависимости от
процента кислорода в посту-
пающей в печь газовой смеси.
,, 1 000 R,-22,4 100 7007%
= 100-32 * Р^= ~Р^~ ’
или на 1 кг сырья:
v =^
Гв3 /}8О2
Объем потребной на горение 1 т сырья газовой смеси вычис-
ляется равным:
Каз. см = + 7 (m - 1)] Ps М*.
Для случая сжигания колчедана, т. е. при т = 1,375, будем
иметь:
Кюзд ~ (р^ + 2,625^ Ps. (8)
в&
Т Абл ИЦА 28
Теоретиче- ский % SO2 в газе 16,20 15,05 21,00 15,05 37,00
Формула = 21 - 1,296 Р80, (4а) РОа= 21-1,395 Рд0, (46) 1 'аз е» о И Q. 1 см II ©“ и. рОа= 21 -1,395 Ps02 (46) Ро>= 45-1,216 PSOj (4г)
R: сч сч сч СЧ 45
5 1,375 1,5 0‘1 1,5 1,375
Уравнение реакции 4FeS2 +11О2 = 2Fe2O3 + 8SO2 sOS5 + OUZ5 = aOS + SUZ3 . о СЛ II сч о + СЛ сч о со см + о сч X сч II СЧ о со + ол сч X СЧ 6 СЛ СО + О Щ X, сч II О »—1 + сч GO Ф 'Ф
Случай горения Колчедан в воздухе Цинковая обманка в воздухе Сера в воздухе Сероводород в воздухе Колчедан в азото-кислород- ной смеси с 45% кислорода
36
Подобными же рассуждениями выводятся аналогичные фор-
мулы для случая сжигания углистого колчедана.
На рис. 22 показана графически зависимость концентрации О2
и SO2 от коэфициента избытка воздуха для различных значений
Объем обжигового газа, необходимый для производства 1 т
H2SO4, вне зависимости от применяемого сырья равен:
1000 22,4-100 _ 22860 3
£8 ‘ (Р80' + Р80)~Р802+Р80зМ • ( J
В колчеданных печах наряду с SO2 получается некоторое количе-
ство SOs- Обычно в газах механических печей
меньше, чем SO2 по объему.
Относительное количество получаемого
SOs понятно зависит от типа печи и от ре-
жима ведения обжига. В проектных расчетах
количеством SOs задаются и обычно прини-
мают его равным 5% от SOs.
Соотношение между концентрацией ки-
слорода в газах и концентрациями SO2 и SOs
можно установить следующим образом. Поя-
вление SOs в газах можно рассматривать как
результат суммарной реакции:
4FeS2 + 11О2 = 2Fe2O3 -|- 8SO2,
а появление SOs как результат суммарной
реакции
4FeS2 -|- 15О2 = 2Fe2O3 8SO3.
Следовательно, на 1 объем SOs затрачивается
1,375 объема кислорода, а на 1 объем SOs —
1,875 объема кислорода.
Если в газовой смеси объемный процент
SOs равен Pso? а процент SOs равен PSq,
то на 100 объемов обжиговых газов затрачи-
вается воздуха:
100 PSo2 Pso> 1.375Рso2 +
+ 1,875PSO объемов.
Рис. 22. Состав газа, полу-
чаемого при обжиге угли-
стого колчедана.
В затраченном воздухе было кислорода:
21
(100 + 0,375 PSOa 0,875 Р8о) объемов.
Прн горении израсходовано кислорода (1,375 Pso + 1,875 Pso ) объемов.
Следовательно, в 100 объемах обжигового газа осталось кислорода в %:
Ро, = 21 - 1,296 Р80з - 1,692 Pso>. (4д)
В газах колчеданных печей всегда присутствуют также водяные пары.
Во-первых, вода в печь поступает с колчеданом; в разных типах печей
влажность применяемого колчедана колеблется в пределах от 0,5 до 10%.
Во-вторых, влага в печь поступает с воздухом, идущим на горение.
При проектировании влагосодержание воздуха обычно берут равным по-
ловине насыщенного при соответствующей принимаемой в расчет температуре.
87
Вся поступившая в печь влага считается уходящей с газом в виде водяных
паров.
Выражения для величины концентрации отдельных компонентов выше
даны из расчета иа сухой газ.
В газах колчеданных печей в очень небольших количествах всегда при-
сутствует еще целый ряд газообразных соединений, являющихся, с одной
стороны, результатом горения илн разложения примесей колчедана, с другой
стороны, заносимых в печь с поступающим воздухом. К числу этих газовых
примесей относятся: СО2, AsaOs, SeO2 и др.
Все эти примеси не оказывают влияния иа количественные расчеты (за
исключением СОг при обжиге углистого колчедана). Однако некоторые из
этих примесей (см. ниже) могут оказать вредное влияние на ход самого
процесса получения серной кислоты. Поэтому от некоторых из этих при-
месей газы в дальнейшем приходится тщательно очищать.
4. Теплота горения колчедана
Теплота горения химически чистого колчедана легко может
быть вычислена по закону Гесса. Теплота образования моля FeS2
равна 35,5 ккал, FeaOg— 195 ккал, SO2— 70,9 ккал.
Можно написать следующие термохимические уравнения:
1) 4Fe-[- 8S = 4FeS2-|- 4 • 35,5 ккал',
2) 8S~|-8O2 = 8SO.2 + 8-70,9 ккал-,
3) 4Fe-|-3O2 = 2Fe2O3-|-2-195 ккал.
Суммируя эти уравнения, получим:
4FeS2 -|- 1102 = 2Fe.2O3 -f- 8SO2 -f- 815,2 ккал.
Отсюда теплота сгорания 1 кг химически чистого колчедана
равна:
(Ю)
„ 800 • 1000 ,
Q = 479784 ' —1700 ККаЛ‘
Если пренебречь всеми реакциями, которые происходят в печи
с примесями колчедана, то 1 кг любого колчедана при выгораю-
щем проценте серы, равном Ps, выделит тепла при сгорании:
1670 Ps ккал.
53,46
При сжигании углистого колчедана необходимо учесть теп-
лоту, выделяемую при сгорании угля.
Общая теплота горения 1 кг углистого колчедана равна:
Q = 31,8 7>s-j-80,6 Рс ккал. (10а)
При определении теоретической температуры горения колче-
дана надо учитывать следующие статьи прихода тепла в печь:
1) теплоту горения колчедана; 2) теплоту, вносимую воздухом;
3) физическую теплоту, вносимую колчеданом.
Двумя последними статьями привода тепла в интересах упро-
щения расчета можно даже при определении теоретической тем-
88
пературы пренебречь, так как они незначительны по сравнению
с первой.
Если, например, теплотворная способность колчедана равна ,1350 ккал,
а воздуха на горение идет для ,1 кг колчедана 4 л3, то с воздухом при
температуре входа 20° вносится тепла 0,312 - 4.20 = 25 ккал, где 0,312 —
теплоемкость воздуха (средняя между 0 и 20°), соответствующая 1 л3 воз-
духа, при нормальных условиях. Это составляет от теплоты горения:
25-100
“1350---1185 '°-
всего:
Рис. 23. Зависимость теоретиче-
ской температуры горения колче-
дана от концентрации SO2 в обжи-
говом газе.
Средняя теплоемкость колчедана между 0° и 100° равна 0,1301 ккал
на 1 кг; 1 кг колчедана вносит в печь теплоты 0,13 20 — 2,6 ккал- Это
составит от теплоты горения колчедана
= 0,19%.
1350 ,с
Теплота в общем случае рас-
ходуется на нагревание получаю-
щихся газов и огарка. Следова-
тельно, теоретическая температу-
ра горения в общем случае равна:
t= r .vQ+cg-- (п>
где t — теоретическая темпера-
тура горения, Q — теплота горе-
ния, С газ — средняя теплоемкость
1 м3 газовой смеси между 0° и
t°, Vгаз—количество газов в м3,
получившихся от сгорания 1 кг
колчедана, Сог — теплоемкость
огарка, Gor — вес огарка на 1 кг
колчедана.
В печах тех конструкций, где
осуществлен противоток обжи-
гаемого материала (в частности в
пающим воздухом, последним членом в знаменателе можно пре-
небречь. Действительно, здесь огарок выходит из печи не с мак-
симальной температурой горения, а с температурой ~ 500°.
При этой температуре теплоемкость огарка 0,1456 +'
+ 0,000188 • 500 с= 0,23 и с ним уносится тепла из печи (при вы-
ходе огарка, равном 0,7) 0,23 • 0,7 • 500 — 80 ккал. Эта потерн
тепла частично возмещается приходом тепла с поступающим:
в печь воздухом и в виде физической теплоты колчедана.
Следовательно, для таких печей при определении теоретиче-
ской температуры горения колчедана вполне можно пользоваться
формулой:
механических печах) с посту-
_______£_
^'газ ‘ '"'газ
(11а>
8»
На рис. 23 графически изображена зависимость теоретической
температуры горения колчедана в воздухе от процента SO2 в об-
-жиговых газах.
При определении возможной, т. е. практической, температуры
горения колчедана надо учитывать также теплоотдачу вовне.
Для охлаждаемых печей надо учитывать еще теплоту, уносимую
охлаждающим воздухом (или водой).
Теплота, отдаваемая печью вовне, может быть вычислена по
формуле Залесского:
Q = Fz(^(T—t), (12)
где F — поверхность охлаждения печи, г — время, за которое
составляется баланс печи (в часах, Ко — коэфициент теплопровод-
ности стенки печи при 0°, о— толщина футеровки, Т — средняя
температура в печи, t — температура окружающего воздуха,
<р = 0,688 — O,O2678Xo + ^^-|-O,Os151 (Т— t). (13)
Раньше при интенсивности работы механических печей, равной
100 кг 45 %-него колчедана на 1 щ2 пода печи в сутки, теплоот-
дача печи составляла 15—17% от всего прихода тепла. Теперь,
при интенсивности в 200—250 кг, теплоотдача вочвне составляет
25—35%. Теплоотдача, отнесенная к 1 м2 поверхности печи и 1°
средней разности температур, достигает При интенсивной работе
печи 3—4 ккал,/час.
В механических печах теплота расходуется также на подогрев
охлаждающего воздуха или воды. Расходы тепла с охлаждаю-
щей водой в печи Ведже при интенсивности в 205 кг составляют
16% от прихода1 тепла, а в печи ВХЗ (при интенсивности
в 254 кг) с охлаждающим воздухом ~ 20% от прихода тепла.
5. Скорость горения колчедана
, Механизм горения колчедана значительно отличается от меха-
низма горения угля. При горении угля продукт горения (COg)
целиком уходит в газовую фазу. При горении колчедана один
из продуктов горения — FegOs остается на месте, покрывая
оставшуюся несгоревшую часть куска все более и более толстой
твердой коркой. Таким образом горение кускового колчедана
связано с необходимостью диффузии кислорода через корку
огарка к несгоревшей части куска и противоположной диффузии
сернистого газа.
Впервые работа, относящаяся к кинетике обжига сульфидов
(пирротинов), была проведена Мельником с сотрудниками. Наибо-
лее полная работа по кинетике обжига кускового колчедана к на-
стоящему времени сделана Малиным с Лихачевой и Кельман.
Скорость горения колчедана зависит по крайней мере от сле-
дующих факторов: температура, концентрация кислорода в зоне
90
обжига, размер кусков горящего материала, толщина слоя горя-
щего материала, степень перемешивания материала и индиви-
дуальные свойства самого колчедана.
Рис. 24. Зависимость количества выгоревшей серы
от температуры и времени.
Рис. 25. Зависимость количества выгоревшей серы
от концентрации кислорода и времени.
На рис. 24, 25, 26 и 27 представлена зависимость количества
выгоревшей серы от температуры, концентрации кислорода,
крупности кусков и поверхности обжига.
91
Указанными опытами установлено, что скорость горения кол-
чедана при температуре 800° не зависит от концентрации SO2
в газах при постоянной концентрации кислорода в них.
----Минуты
Рис. 26. Зависимость количества выгоревшей серы от крупности
кусков и от времени.
Рис. 27. Зависимость количества выгоревшей серы
от поверхности обжига и времени.
Обработка экспериментальных данных лабораторной работы
привела к следующей формуле для скорости горения одного
куска:
(14>
92
где х — количество сгоревшего колчедана, t — время от начала
горения, F — поверхность обжига, РОг —средняя концентрация
кислорода в зоне обжига, т — показатель степени, R — началь-
ный радиус куска, г — радиус несгоревшей части куска.
В дальнейшем предполагается, что в оболочке (R — г) вся
сера сгорает, а несгоревшая часть сосредоточена в шаре ра-
диуса г. Если обозначить вес куска через g, то можно написать
х
g~~ R3 ’
откуда
х =gRS&r* ’ а dx = —^3rSdr =
где
Л— R3— з./^з — ^>ООЪ
где у — удельный вес колчедана.
Подставляя найденное значение dx в первое выражение и, по-
лагая Pos = const, получим:
dr К
dt R — г ’
откуда
(R — r)dr — Kdt.
Интегрирование последнего выражения в пределах г от R до
rt и t от 0 до t дает:
2Kt = (R—rt)*,
или
R — rt =VKt.
С?--
Из последнего выражения получаем:
rt=R~VKt. (А)
Но
__ $1 __^нач "^выг
Р8 ^нач '^нач
где Sna4 — начальное содержание серы, St — содержание серы че-
рез определенный промежуток времени, 5ВЫг — выгоревшая сера.
Из последнего выражения получается
г. ^выг
Обозначая степень выгорания серы через д, получим:
93
Подставляя в последнее уравнение вместо п его значение из
(А), получим:
»2 г 3 Г~~~ —I2
—j/i-tJ . (15)
Для горения колчедана в слое в последнее уравнение необхо-
димо ввести поправку. В этом случае скорость горения колче-
дана, кроме величин, имеющихся в уравнении (14), зависит еще
от отношения свободного объема между кусками к общей по-
верхности кусков.
Как установлено опытами,
скорость горения колчедана
прямо пропорциональна вели-
Рис. 29. Зависимость константы
скорости горения колчедана от
концентрации кислорода в га-
зах.
Рис. 28. Зависимость константы
скорости горения колчедана от
температуры.
вычислить, что это отношение
окончательным уравнением для
чине этого отношения. Легко
'равно ~ 0,3 R. Таким образом
скорости горения колчедана будет
dx_________________j. F-R
dt К R — г' (14а)
а для времени горения (при введении вместо радиуса также
диаметра куска):
Z = —(15а)
В обоих выражениях концентрация кислорода введена в кон-
станту.
Зависимость константы скорости горения от температуры гра-
фически изображена на рис. 28 в координатах 1g К и Харак-
тер графика — прямая линия показывает, что зависимость кон-
станты скорости горения от температуры выражается уравнением:
94
Зависимость константы скорости горения колчедана [уравне-
ние (15а)] от концентрации кислорода графически изображена
на рис. 29; прямая линия показывает, что эта зависимость,
выражается уравнением
(16)>
Обработка экспериментального материала показала также, что
время горения до определенной степени выгорания прямо про-
порционально толщине слоя материала в некоторой дробной сте-
пени (близкой к 0,5). Таким образом, исключая из К концентра-
цию кислорода и температуру и отражая в формуле найденную
‘зависимость от толщины слоя, получим окончательное уравнение
для времени сгорания колчедана в слое:
п з /--
<17>
где с — величина, обратная толщине слоя.
6. Примерные расчеты
Составить материальный баланс печи ВХЗ за сутки (табл. 29, 30). Дан-
ные: серы в колчедане 41%, в огарке 2%; влаги в колчедане 2%; процент
SO2 в сухих газах 9. Температура воздуха, поступающего в печь, 20э. Кол-
чедана в сутки загружено 30 т.
Тлблицл 29
Материальный баланс печи ВХЗ
Приход
Статья Количество в кг | в ма Расчет
Колчедана . . . Влаги с колче- даном Сухого воздуха . Влаги с воздухом 29 400 600 121 000 990 93500 1230 30000-0,98 = 29400 кг 30000-0,02 = 600 кг ^возД = 29,4 (222.+ 2,625) Л* Ps = 41 — 2 • 0,752 = 39,5 (-^-»«) На 29,4 т колчедана воздуха пой- дет: РЕ03Д = 29,4 (^2 + 2,625) - 39,5 = = 93 500 жЗ При 20° упругость насыщенных па- ров равна 17,54 мм рт. ст. Возь- мем половину от насыщенных во- дяных паров в м3:
И того. . 151 990 1 -
95-
Таблица 30
Материальный баланс печи ВХЗ
Ра сход
Статья Количество Расчет
кг Ж3
•Огарок 22 100 — 29 400 0,0752 = 22 100 кг
Водяные пары . 1590 1940
Сернистый газ . 23260 8140 VSOs = 29,4 • 7 Ps = 29,4 7 • 39,5 = 8 140 ж3
Кислород .... 12064 8 447 Рп = 21,31 — 1,296 9 = 9,34
V . llfO . 9,34 = 8447 ж3 У
78,60 ,,
! Азот 92000 73500 I/ ’ I/ N«~ 100 В03Д
VN> = 93500 = 73500 ж3
Итого . . 151 014 —
Расхождение прихода с расходом составляет:
151 900—151 014 п_.п,
----------------100 = О,64о/0,
-что можно объяснить приближенностью вычислений.
Состав газовой смеси, выходящей из печи, приведен в табл. 31.
Таблица 31
Газ Сухой С влагой
ж3 % ж3 %
SO2 8 140 9,00 8140 8,85
о2 8 447 9,34 8447 9,20
n2 73500 81,66 73500 79,85
Н2О — — 1940 2,10
Итого. . 90 087 100 92027 100
Составить по данным материального баланса суточный тепловой баланс
печи (табл. 32 и 33).
Дополнительные данные следующие: температура поступающего колче-
дана 20°; температура выходящих из печи газов 700°; температура воздуха,
охлаждающего вал печи, при входе 20’, при выходе 200'; температура выхо-
дящего огарка 500°. ,
96
Таблица 32
Приход тепла
Статья ккал % Расчет
Физическая теплота колчедана .... 76440 0,25 Qi = 29 400 • 0,13 • 20 = 76 440 ккал
С воздухом, посту- пающим на горе- ние 556890 Q’ возд ^возд ^возд = = 93 500 (0,308 + 0Д237 20) 20 = = 556 890 ккал
С влагой колчедана 12000 — Q3 = 600 1 20 = 12 000 ккал
С влагой воздуха . Теплота горения колчедана .... 8 700 36 900 000 98,18 Qt = 1230 (0,351 + 0,0д96 - 20) 20 = = 8700 ккал ft = 31,24-39,5-29 400 = = 36 900 000 ккал
Итого. . 37554030 — *11
Таблица 3'3
Расход тепла
Статья ккал % Расчет
С огарком . . . С газами .... Теплоотдача пе- чи вовне . . . 2647 580 22 015 195 9 396000 7,07 58,52 25,00 ft = 22100 (0,1456 4-0,0з188 • 500) 500 Qi = 2 647 580 ккал = <?soa+ Он.0 QSOs = 8140 (0,437-1-0,04847 • 700) 700= = 2 827 860 ккал QOg Na = 81947(0,3084-0,04237 - 700)700 = = 18 619420 ккал fta0 = 1940 (0,351 4- 0,0496 • 700) 7С0 = = 567 915 ккал Q2 = 28278604-186194204-567915= = 22015 195 ккал Считаем равной 25% от прихода тепла Q. = 37 554030 - 0,25=9396000 ккал
Расход с охла- ждающим воз- духом .... 3495255 9,35 Считаем по разности Qt = 37 554 030 — (2 647 580 -J- 4- 22 015 195 4- 9 396 000) = = 3 495 255 ккал
Итого. . 37554030 —
7 Зак. 5019. Технология серной кислоты.
97
Определим количество воздуха, расходуемое в сутки иа охлаждение вала
и гребков печи, если температура воздуха при входе 20°, а при выходе 200":
_ 3 495 255
* ®°зд ~С олл Я . on
°возд. 200° zuu — ьвозд. 20° zu
_______________ 3495 255______________
(0,308 + 0,0*237 • 200) 200 — (0,308 + 0,0*237 • 20) 20 ~
як 60 000 м3/сутки, или 2500 м3/час.
II. Печи для обжига сернистого сырья
1. Механические печи для обжига колчедана
Конструкции механических печей. До изобретения механиче-
ских печей колчедан в виде крупных кусков сжигали в печах-
кильнах и в виде мелочи в ручных печах Малетра и Шаффнера.
В кильнах колчедан обжигался на колосниковой .решетке. Загру-
жался колчедан вручную в форме кусков до 50 мм слоем толщи-
ной до 500—700 мм. Выгоревшие куски удалялись поворотом
колосников, имевших квадратное или овальное сечение. Печи
Малетра и Шаффнера состояли из ряда сводов. Колчедан вруч-
ную (через дверку-печь Малетра или через бункер в печь Шафф-
нера) загружался на верхний свод. Перегребание колчедана
с полки на полку производилось вручную. Вследствие своей ма-
лой производительности и трудоемкости процесса ручного пере-
гребания колчедана эти печи не могли удовлетворить развиваю-
щуюся сернокислотную промышленность и цветную металлургию.
Ручное перегребание материала печей Малетра и Шаффнера за-
менилось механическим.
Были предложены три способа механического перегребания
колчедана: 1) внутри печи вращается вертикальный вал с гори-
зонтальными гребками, на которых насаживаются зубья для пере-
гребания; 2) вал с гребками неподвижен, вращается сама печь,
при этом колчедан передвигается по своду, перегреваясь при его
вращении неподвижными гребками; 3) передвижение колчедана
с полки на полку производится штангами с гребками, устано-
вленными на особой тележке. При посредстве ременной и зуб-
чатой передач гребки получают прямолинейное движение от
трансмиссии и перегребают колчедан с этажа на этаж.
Наиболее удачными и получившими широкое развитие оказа-
лись механические печи с вертикальными вращающимися валами.
Первая печь такой конструкции была предложена в Англии
братьями Мак-Дугаль в 70-х годах XIX столетия. Однако эта
печь имела ряд серьезных конструктивных недостатков. Широкое
внедрение механических печей в сернокислотную промышлен-
ность, а также в цветную металлургию началось в 90-х годах
XIX столетия, после того как Герресгоф (Америка) дал доста-
точно удачную конструкцию механической печи. Все применяе-
мые для обжига колчедана и руд цветных металлов механические
98
печи отличаются от печи Герресгофа не принципиально, а лишь
в конструкциях частей.
В СССР сейчас работают на сернокислотных заводах механи-
ческие печи Герресгофа, Гумбольта, ВХЗ, Лурги, Брак-Моритца
Рис. 30. Вертикальный разрез печи ВХЗ:
1—кожух; 2—вал; 3—гребки; 4—зубья; 5—бункер; 6—питатель; 7 и 8—
течки; 9—дверцы; 10—шкив; 11—редуктор; 12—вал; 13—шестерня; 14—боль-
шая шестерня.
и Г. Наиболее удачными из всех этих типов печей являются печи
ВХЗ, изготовляемые в СССР и принятые для установки при
строительстве и расширении сернокислотных заводов.
Печь ВХЗ (рис. 30) имеет 8 сводов, из них рабочих 7 сводов
для обжига и один — для подсушки колчедана. Железный кожух
7*
99
печи 1 футерован внутри огнеупорным кирпичом. Через центр
печи проходит вал 2, на котором у каждого свода печи посажено
по два гребка 3 с зубьями 4. Колчедан из бункера 5 поступает
на периферию печи и зубьями гребков сушильного свода перегре-
бается к центру свода, где через питатель 6 рабочего свода попа-
дает на / рабочий свод, по которому перегребается от центра
к периферии, где через отверстия просыпается на II рабочий
свод. По II своду колчедан передвигается к центру печи, где
просыпается на /// свод по кольцевому отверстию вокруг вала
и т. д. С последнего свода огарок высыпается или по течке 7
в подставляемую вагонетку, или по течке 8 на транспортер.
Рис. 31. Своды печи ВХЗ (разрез).
Воздух для горения поступает через двенадцать воздушников
на последнем своде. Регистрами воздушников можно регулиро-
вать количество подаваемого воздуха. Дополнительная подача
воздуха в печь может производиться через четыре воздушника
на IV своде. Со свода на свод газы поднимаются навстречу го-
рящему материалу через те же отверстия в сводах, через ко-
торые сыплется и материал. Получившиеся в печи газы уходят
с / рабочего свода через газовую коробку и кривули в общий
газоход.
Для облуживания печи на каждом рабочем своде имеется по
четыре дверцы 9. В каждой дверце прорезано маленькое, прикры-
ваемое шиберком отверстие (глазки) для наблюдения за процес-
сом горения и состоянием рабочего свода.
Вращение вала производится через шкив 10, находящийся на
одном валу с редуктором 11. На валу 12, соединенном с редук-
тором, помещена малая шестерня 13, входящая в зацепление
с большой шестерней 14, насаженной на вал печи. Воздух для
охлаждения вала и гребков поступает в низу вала (во внутренний
его канал), а затем идет в каждый гребок, огибает имеющуюся
в его полбсти и не доходящую до конца гребков перегородку и
100
поступает в вал — в канал между его наружным и внутренним
цилиндрами, откуда иерез трубу выходит в атмосферу или на-
правляется на отопление помещения. На рис. 31 показаны планы
нечетного и четного сводов печи, на которых видны отверстия
для прохода воздуха и колчедана.
Рис. 32. Питатель сушильного свода:
1—тарелка; 2—штанга; 3—стойка; 4—палец питателя; 5—нож.
Для равномерной подачи колчедана из бункера на сушильный
свод служит питатель сушильного свода (рис. 32), Под вырезом
' бункера находится площадка, на которую поступает колчедан из
Рис. 33. Питатель рабочего свода:
Г—тарелка; 2—болты; 3—вал; 4—штанга; 5—рычаг; 6—плита; 7—гайка.
бункера. На гребке сушильного свода помещается тарелка 7,
прикрепленная к штанге 2. Штанги присоединяются к стойкам 3,
на которых установлен основной палец питателя 4; вдигая или
выдвигая этот палец, можно регулировать подачу. Часть колче-
дана, сбрасываемая пальцем 4, попадает на тарелку 7, а осталь-
101
2875
Рис. 34. Вал печи ВХЗ:
1—внутренняя полость; 2—внешняя
«полость; 3—ребра; 4—фланцы; 5—
приливы.
ное попадает на свод. Колчедан,
попавший на тарелку, поворачи-
вается вместе с гребком на 180°.
На противоположной стороне бун-
кера к печи прикреплен неподвиж-
ный нож 5. При прохождении гребка
нож сгребает находящийся на та-
релке колчедан на свод. Таким об-
разом загрузка колчедана на су-
шильный свод происходит в двух
местах, что обеспечивает более
равномерное распределение колче-
дана по сушильному своду.
Питатель рабочего свода (рис. 33)
состоит из чугунной тарелки 7, под-
вешиваемой болтами 2 к лапам,
укрепленным в верхней части цен-
трального вала 3. Колчедан с су-
шильного свода через плиту б про-
сыпается на талеку 7. В кольцевом
зазоре между бортом плиты и плос-
костью тарелки колчедан образует
затвор, препятствующий выбиванию
газа из печи. Над тарелкой 7 с двух
противоположных сторон располо-
жены штанги 4, пропущенные через
трубы, проходящие сквозь футе-
ровку свода. К наружным концам
штанг присоединены рычаги 5. При
вращении вала с тарелкой непод-
вижный конец штанги 4 сгребает
колчедан на первый рабочий свод.
При помощи рычага 5 можно больше
или меньше выдвинуть конец штанги
и тем самым регулировать подачу
колчедана в печь. Подтягиванием
или опусканием гайки 7 на болтах 2
можно регулировать высоту кольце-
вого зазора между тарелкой 7 и
плитой 6.
Вал печи ВХЗ состоит из трех
отдельных частей (по длине печи),
соединенных между собой болтами.
Низ вала заканчивается фланцем 4
(рис. 34), которым он присоединяет-
ся к втулке подшипника. Вал
имеет две полости — внутреннюю 1
внешнюю 2. Внутренняя и внешняя
части вала соединены между собой
102
ребрами 3 и приливами 5, служащими опорами для гребков.
В нижний конец внутренней полости поступает воздух для
охлаждения вала и гребков, верхний конец перекрыт.
Наружная кольцевая полость вала перекрыта внизу и открыта
вверху для выхода нагретого воздуха. Поступающий во вну-
треннюю полость вала воздух распределяется по всем гребкам,
из которых возвращается затем во внешнюю полость вала.
гт гт.'.РПНР Л
г Рис. 35. Гребки рабочих сводов печи ВХЗ:
1—чугунный конус; 2—цилиндр;.?—стейка; 4—отверстие дли входа воздуха; 5—перегородка;
6—отверстие для выхода воздуха; 7—шип; 8—приливы; 9—серьги; 10—зацепка.
Гребок рабочего свода (рис. 35) представляет собой близ-
кую к конической форме чугунную трубу 1. Продолжением боль-
шого основания конуса гребка является цилиндр 2, вставляемый
Рис. 36. Соединение гребка печи ВХЗ
с валом:
1—шип; 2—вырез; 3—кольцевая заточка; 4—
приливы; 5—замок.
Рис. 37. Конструкция зуба:
1—пластинка; 2—прорезь; 3—валик;
4—хвост.
в гнездо вала. Торец цилиндра частично закрыт стенкой 3.
Внутри гребка имеется перегородка 5, не доходящая до конца
гребка. Охлаждающий воздух поступает в отверстие 4, про-
ходит по одной стороне перегородки, огибает ее конец, охла-
ждает вторую половину гребка и выходит из него через отвер-
стие б. Шип 7 служит для крепления гребка в гнезде вала.
Сверху гребка сделаны приливы 8, в которые вставляется замок,
предохраняющий гребок от поворачивания вокруг своей оси во
время вращения вала.
103
При вставке гребка в гнездо вала шип 1 гребка устанавли-
вают против выреза 2 (рис. 36). Цилиндрическая часть гребка
свободно вставляется в гнездо до упора шипа в стенку кольце-
вой заточки 3. После этого гребок поворачивается на 180°, так
чтобы шип 1 заходил в кольцевую заточку; приливы 4 должны
устанавливаться против выреза 2. Между приливами 4 вста-
вляется замок 5, так чтобы заостренный конец его вошел в вы-
рез 2. Ручка замка после этого поворачивается и зацепляется
между приливами 4.
Зуб гребка (рис. 37) представляет собой пластинку 1, снаб-
женную прорезью 2, валиком 3 и хвостом 4. Валик зуба ве-
шается на две смежных сережки гребка. Хвост 4 зуба упи-
рается в нижнюю поверхность гребка и придает зубу устойчи-
вость. Чтобы снять зуб гребка, надо взять его за прорезь 2 и
приподнять вверх. При этом зуб
Рис. 38. Соединение гребков печи Г с ва-
лом:
1—вал; 2—гребки; 3—газовая труба; 4—пробки;
5—болт; 6—гайка.
поворачивается в гнезде и
хвост 4 отходит от по-
верхности гребка.
Другие механические
печи отличаются от пе-
чей ВХЗ конструкцией
вала, гребков и зубьев,
питателя рабочего свода;
способом охлаждения ва-
ла и гребков; соединени-
ем лопаток с гребком и
гребков с валом; устрой-
ством отверстий для про-
хода колчедана и воздуха
через своды; конструкцией передаточного механизма, приводя-
щего во вращение вал печи. Отдельные печи отличаются также
основными своими размерами. В табл. 34 приводятся основные
характеристики механических печей сернокислотных заводов
СССР.
У трехтонной печи Герресгофа охлаждается воздухом только
вал. У печей Ведже, установленных на Невском химическом за-
воде, гребки охлаждаются водой. Вода для охлаждения гребков
поступает из водопровода в коробку, установленную на опоре,
которая прикреплена к верхней части вала и вращается вместе
с ним. Труба, подводящая воду, соединяется с вращающейся
коробкой при помощи сальника. Трубки, подводящий воду
к каждому гребку, присоединены к нижней части коробки. Для
регулирования подачи воды в каждый гребок на подводящих
трубках установлены вентили. Из каждого гребка идет трубка,
отводящая теплую воду. В печах некоторых типов на каждом
своде имеется по четыре гребка.
Способ соединения гребков печи Г и некоторых типов печей
Гумбольдта показан на рис. 38. В конические гнезда вала 7, рас-
положенные с двух диаметрально противоположных сторон,
вставляются своими малыми конусами два гребка 2. Внутри каж-
104
2
£
К я
S
3
о
ecnS
*
3
о
s
х
S х
га s
S
- та
си —।
end
Е
S
га
с[ЙЙ^
Таблица 34
Ведже 8370 1700 1 1 1 1 li 1 1 1 1 1 1 1 1 1 1
Лурги OrF О VO — ос ю оо — — 04 О ЮЮ О Ю тГ 04 СО СО О 00 С4 — — 04 Г'- — Г- о" о^> 2335 0,0066 0,0081 04 СО 00* О 6,8 1 1
Брак- Мо- ритца ООЮССтГ 04 04 СП О °C tF юьо о г-<о <551— со Г-^FCO —<О — iOCOtF ьб ою 1970 0,0027 0,00225 0,36 0,29 3,5 О о о ю tF СО 04
ВХЗ ’ФОС СО СП О О — 00 СП 00 — > о t с г о с *«c<2o2g о -г- 450 2473 0,127 0,016 О — о? — 4,5 оо ОШ ОСО ю
Тип печи Гум- больдта ООО ООО Ю 00 04 ю СО — 1 1 1 1 IS 1 1 1 1 1 11 4,5 5000
ч реконструи- рованная Г ООО 0 — 0 tF о tF ю ь* — Г^Г'’ — COOGOOCtFOcO 0040 — 04 СО О 04 0 — — O^FO СО — о- _Г«= Г4 0,0116 0,0081 00 со сь> — о 3,5 4000—4500
и 5400 7010 1400 b-Ь —сроссо оо О 040— 04 со — О О 04 04 СО — 04 0,0031 0,006 0,29 0,90 4,5 4000 300—350
Геррес- гофа ото ЮО4 о Ю О 04 СО СО — юь ОО , 00 , го 1 1*°$ |й 1 1 1 1 1 1 1 4,5 3600 300-350
со
% S
И t .
о _ X
5=s
Ч
Ч
Ч R
са
х* «
В
5?
Ч
105
дого гребка находится газовая труба 3. Оси газовых труб, при
присоединении гребков с валом, являются продолжением одна
другой. На газовые трубы надеваются пробки 4, через которые
пропускается болт 5; с помощью гаек 6 оба гребка прижимаются
своими конусами к гнездам вала. Газовая труба 3 снабжена
у конца по окружности шестью отверстиями. Воздух из вала
печи входит в газовую трубу и по отверстиям выходит в коль-
цевое пространство, образуемое трубой и полостью гребка. Для
выхода воздуха из гребка конус снабжен тремя отверстиями.
Способ прикрепления гребков к валу, примененный в печи Г,
оказался чрезвычайно неудобным. При поломке гребка и продол-
жающемся вращении вала болт, стягивающий гребки, закручи-
вается вокруг вала. Раскручивание болта и замена гребка явля-
ются весьма затруднительными операциями и вызывают длитель-
ный простой печи. Поэтому в реконструированной печи Г вал
и гребки выполняются по типу печи ВХЗ.
В печах Герресгофа и Брак-Моритца зубья несъемные, гре-
бок отливается вместе с зубьями.
В отличие от печи ВХЗ в некоторых механических печах
газы и материал проходят со свода на свод через разные
•отверстия. Так, в печах Г на нечетных сводах на периферии
имеется по 24 отверстия для прохода газа и по 8 отверстий
для просыпания колчедана. На четных сводах для прохода
газа служит кольцевое отверстие вокруг вала, окруженное бор-
тиком для предупреждения просыпания через него колчедана.
Режим и работа механических печей. Оптимальной эксплоа-
тгацией всякого аппарата считается такая, при .которой аппарат
дает возможно большую производительность, выдает продукцию
(или полупродукт) возможно высокого качества, наиболее полно
использует сырье и долго служит.
Следовательно, нормальными показателями работы печи явля-
ются ее высокая производительность, высокое качество полу-
чаемого обжигового газа, малое содержание невыгоревшей серы
в огарке и температура газов в печи и воздуха из вала, гаранти-
рующая длительную работу деталей печи. Мерой производи-
тельности аппарата может быть пропорциональная ей вели-
чина — интинсивность аппарата.
Под интенсивностью печи понимается количество (в кг) кол-
чедана (в переводе на сухой 45%-ный по содержанию серы),
сжигаемого в сутки на 1 м2 поверхности рабочих сводов печи.
До 1935 г. нормальная интенсивность печи принималась за
100 кг. Стахановское движение и исследовательские работы по-
следних лет довели эту интенсивность до 250 кг и показали
возможность повышения ее до 350 кг.
Содержание серы в огарке должно быть не выше 2%, а кон-
центрация SO2 в газе при обжиге рядового и флотационного кол-
чедана должна быть не ниже 9%. Температура воздуха из вала
печи должна быть не выше 200—250°, а температура газа на
самом горячем своде не выше 850—900.
106
Основные условия, при которых указанные показатели могут
быть достигнуты, следующие:
1) наличие достаточной тяги, открывающей доступ в печь не-
обходимого количества воздуха;
2) равномерное питание педа колчеданом, достаточно измель-
ченным;
3) хорошее охлаждение вала и гребков, предупреждающее
их деформацию;
4) регулярная чистка отверстий и каналов внутри печи от
спекшегося колчедана;
5) регулярное удаление огарка из печи и пыли из штуцеров
печей, газоходов и электрофильтров;
6) систематическое наблюдение за состоянием всей печи
в целом и в особенности гребков и зубьев;
7) выполнение планово-предупредительного ремонта печи и
ее деталей и наличие запасных частей.
Тяга в печи создается, с одной стороны, вследствие разницы
удельных весов столбов воздуха снаружи и газа в печи и, с дру-
гой стороны, благодаря работе вентилятора, оттягивающего газ
из печей. Тяга, получающаяся вследствие разности удельных
весов, приблизительно может быть определена по формуле:
р_ 352(7,—
‘ (273 -J- t2) (273 4- ’
где р — наибольшее разрежение в мм вод. ст., tz — средняя тем-
пература в печи, ti — температура окружающего воздуха, Н —
высота рабочей части в печи в м.
Для того чтобы разрежение на верхнем своде печи было
равно нулю, проталкивание газа через печь должно итти за счет
тяги самой печи. При повышении нагрузки сопротивление печи
возрастает, тяга самой печи становится недостаточной и на
верхнем своде должно получаться разрежение за счет работы
вентилятора. При недостаточной тяге на верхнем своде печи со-
здается давление, превышающее давление наружного воздуха.
Ресурсы самой печи в отношении тяги должны быть исполь-
зованы максимально. Для этого необходимо регулярно чистить
газовые проходы в сводах, не допускать завала воздушников
огарком, подсоса воздуха через питатель, устранять неплотности
в дверцах.
В табл. 35 приведены показатели технологического режима
печей ВХЗ и Ведже при разной интенсивности.
Из табл. 30 видно, что температурный режим печи зависит от
общей интенсивности работы печи и от характера шихты (от со-
держания серы, углерода и влаги в загружаемом колчедане).
Понятно, что при неизменном содержании серы в шихте тем-
пературы в печи повышаются с увеличением содержания угле-
рода и понижаются с увеличением содержания влаги в шихте.
Место наивысшей температуры в печи (температурный пик) за-
висит от тех же факторов.
107
Таблица 35
Печь Ведже ю^о 00 со ь- о —< ССт-^СО О I со' СО CD xF CD Г' —ч СО 1 о ю сч xF СО . 1 О Г-Ь-СО Г-СМ О 1 О СМ —« -f- Nb-Nh-OtOCO CM
со О CD со О СМ О Ю OO^xF о | О xF СМ СО СТ> О *“ СО 1 СО О СМ ОО СО . 1 00 xF CD С" —-> со СО 1 со см —' -|- О Ь b-b-N Ш’Т со
| * Печь ВХЗ Ю Ю ст> Ю Н со СОЮ ’ QO О СМ^ю ОО О Ю со —’ Г" СО см —< Ю о xF СМ о xfX —< C^OlOONDCON СО ОО СО со со со 00 со СО со ю ю •—•
ем со см со —^осо съем съ сс xFlDoT со Ю ОО О ОС ОС О xF СМ —> xF СМ СЪ XF со Г- Ю о О b" xF ОО Ю —* Ю Ь СО ОС ОС Н CD Ю XF —’
xF ое> 00 ОСЪСМ СЪГ-- со xf смсооо о о o’ со ь-со’ ю to’ со см ю сГ Ю xF т— ЮЮЮГ-ЮСО^СЪ СО СЪ СМ Ю Г- 00 СО ОО Г- ь- ю ю
юоосъ^ ст^со °1 S —• О О 00 000 ОООЮОсОоОСОЮ^ч см* Ю XF r-< CM СО XF С" СЬЮ СМ СО Ь" СМ ю ь- со СО 00 г- г- г- ю —<
сою О г- 00<OxF СМ о СО —Г 0*0 t-^O 00 ОсОСМОг-сООЮО Ю XF —~ CM XF • CO-CNLO^CMb-DTFa' СМ Ю Г- СО 00 О Г- СО Ю xF ’
<х> СМ СО ю см см ю 00 СО О 1 xF —- [ со" CM xF »—< xf Ю О СО CM xF СО —" О xF 1 1 ЮОЮСОО СОЮ Ю О СМ СО Г" Г"-СО Г—СО О со XF
ООО»— со lQc2.T:t. со хг со съ ОСО О 1 °° 00XF Ю XF о—. —1 со —< Ю СО СО XF 1 г, ЮОЮСОхМ^—«О XF СО Ю СО СО Ь" ь- со СО xF XF —«
Показатели работы Интенсивность, кг 45 - %-ного колчедана в сутки Шихта, % S С н20 Разрежение, мм вод. ст. в кривуле печи на 1 своде °/0 S02 в газах в кривуле печи . Температура газов, °C в кривуле печи на J своде II » Ш » .IV „ V , VI VII „ Температура огарка, °C Температура воздуха из вала, °C . о/п S в огарке
108
С ростом интенсивности работы печей температуры более
равномерно распределяются по сводам печи — даже самые ниж-
ние своды принимают значительное участие в выжиге серы.
В табл. 36 приведены данные о ходе выгорания серы по от-
дельным сводам печи ВХЗ.
Таблица 36
Свод Интенсивность 203 кг Интенсивность 169 кг Интенсивность 300 кг
сера в % при уходе материала со свода выгорание серы иа своде в % от всей выгоревшей серы сера в % при уходе материала со свода выгорание серы на своде в % от всей выгоревшей серы
0 41,44 41,33
1 29,75 29,56 34,09 23,6
II 23,23 16,56 27,01 25,8
III 19,92 8,36 22,80 7,7
IV 14,92 12,64 18,80 11,3
V 10,02 10,88 13,40 12,6
VI 4,33 15,90 8,48 13,7
VII 1,92 6,10 3,30 5,3
— 100,00 — 100,00
С увеличением интенсивности работы печи и в связи с этим
с повышением средней температуры печи возрастает величина
потерь тепла во-вне как по абсолютной величине, так и в про-
центах от всего прихода тепла (табл. 37)
Таблица 37
Статьи расхода тепла Расход тепла в °/0 от итога при интенсивности в кг
203,7 226 251,5 254 194,1 299,4
С'огарком С газами i С охлаждающим воздухом На удаление влаги иа су- шильном своде .... Потери во-вие 5,90 53,60 22,40 1,50 16,60 5,65 47,10 20,20 2,15 24,90 7,20 37,50 18,25 2,00 35,05 7,15 38,60 20,25 1,85 32,15 5,80 44,30 24,20 2,20 23,50 7,7 42,1 17,1 2,3 30,8
Итого ... 100 100 100 100 100 100
Общий приход тепла в час в тыс. ккал 1662 1922 2049 2097 1564 2340
109
Наиболее часто встречаются следующие отклонения от нор-
мального режима работы механических печей: понижение и по-
вышение концентрации SOa в печном газе, выбивание газа из
печи в атмосферу печного отделения, повышенное содержание
серы в огарке.
Если содержание SO2 в газах низко, то, следовательно, воз-
духа в печь подается больше, чем требуется по нагрузке печи.
Причинами излишнего поступления воздуха в печь могут быть:
слишком большое открытие воздушников, подсосы воздуха че-
рез неплотности печи и очень большое число оборотов вентиля-
тора, протягивающего газ через систему. Мероприятия по повы-
шению концентрации SO2 в газе проводятся в следующем по-
рядке: если все печи установки дают газ низкой концентрации,
то причина этого может лежать и в вентиляторе и в самих
печах; если газ низкой концентрации дает только одна или
только часть печей, то причину надо искать в самих печах.
В этом случае надо сначала проверить, нет ли где-нибудь под-
соса воздуха, вновь промазать все дверцы печи, а затем уже
следует регулировать подачу воздуха в печь регистрами воз-
душников.
Если концентрация печного газа слишком высока, то это
означает, что воздуха в печь подается недостаточно сравни-
тельно с нагрузкой печи. Причинами этого явления могут быть:
недостаточное открытие воздушников; засоренность газоходов
или проходов газа в печи; недостаточное количество оборотов
вентилятора. Если высококонцентрированный газ дают все печи,
то причину надо искать или в работе вентилятора или в засо-
ренности общего газохода.
Причиной выбивания газа из печи в печное отделение яв-
ляется превышение давления газа в печи против давления на-
ружного воздуха. Нормально в печи на нижних этажах должно
быть небольшое разрежение. На I рабочем своде давление
должно быть равно наружному или должно быть очень не-
большое разрежение, не более I мт вод. ст.
Повышенное содержание серы в огарке может зависеть как
от качества самого колчедана, так и от характера работы печи.
Колчедан должен быть раздроблен до величины частиц не боль-
ше 7—10 мм. Повышенное содержание серы в огарке, зависящее
от работы самой печи, получается от несоответствия количества
поступающего воздуха с нагрузкой печи. Если воздуха подается
очень мало, то большая его часть затрачивается на горение на
нижних этажах. На верхние этажи газ приходит с очень малым
содержанием кислорода и в результате сера не успевает выго-
рать. При этом часто вместе с повышением серы в огарке наблю-
дается увеличение концентрации SO2 в газах. Слишком боль-
шое количество подаваемого в печь воздуха гоже часто ведет
к повышению процента серы в огарке. Это объясняется тем, что
с увеличением объема печных газов падает температура во всей
печи и ослабляется интенсивность горения. В этом случае повы-
110
шенное содержание серы в огарке сопровождается уменьшением
концентрации SO2 в газах.
В печных отделениях прежде всего должны быть предусмо-
трены уже упоминавшиеся выше мероприятия против появления
в цехе сернистого газа в больших концентрациях. Необходимо
также проведение мероприятий, направленных против чрезмер-
ного повышения температуры. Кроме правильно организованного
естественного проветривания печного отделения (аэрация) долж-
на быть устроена искусственная подача свежего воздуха как
в целях лучшего проветривания отдельных участков, так и в це-
лях снижения температуры. Для уменьшения выделения SO2
в атмосферу цеха необходима тщательная герметизация огароч-
ных кабинок и отсос газа из них. Для предупреждения несчаст-
ных случаев должны быть запрещены всякие работы в печи,
при вращении ее вала.
Особенности сжигания флотационного и углистого колчедана..
Практикой работы доказана полная возможность обжига в меха-
нических печах флотационного колчедана как в смеси с рядовым,
так и одного. При интенсивности работы механической печи до
200 кг показана возможность сжигания углистого колчедана,
в количестве, соответствующем содержанию 4—6% углерода
в шихте.
Однако- обжиг флотационного и углистого колчедана требует
некоторых особенностей ведения процесса: флотационный (по
причине мелкости дробления) и углистый колчедан (по причине
содержания угля) интенсивнее, чем рядовой колчедан, горят на
верхних сводах. Это вызывает развитие особенно высокой тем-
пературы на одном из верхних сводов, что ведет к размягчению
материала и его спеканию. При сжигании флотационного колче-
дана спеканию благоприятствует и то, что свободные проме-
жутки между его частицами (чрезвычайно мелкими) очень незна-
чительны.
Мероприятия, направленные против спекания, сводятся к тому,,
чтобы относительно уменьшить интенсивность работы верхних
сводов за счет повышения интенсивности работы последующих
сводов. Это достигается или подачей дополнительного воздуха
на более горячие своды (при соответствующем уменьшении по-
дачи воздуха на VII свод) или ускорением прохождения мате-
риала через наиболее горячие своды (путем изменения конструк-
ции или наклона зубьев на этих сводах). Против спекания мате-
риала в печи во всех случаях может быть применено также
примешивание некоторого количества огарка к загружаемой
шихте.
Дальнейшие возможности интенсификации механических пе-
чей. При проектной интенсивности механических печей в 100 кт
рядом заводов уже в длительной работе осйоена интенсивность
в 250 кг. Научно-исследовательской работой НИУИФ в опытах
на обычной заводской печи ВХЗ доказана возможность интен-
сивности в 350 кг. Эти результаты достигнуты: 1) равномерным.
ill
питанием печи колчеданом при наличии достаточной тяги; 2) хо-
рошим обслуживанием печи (своевременная шуровка, регулярная
чистка газовых отверстий в подах, штуцеров и газоходов) и
3) повышением средней температуры в печи и более равномер-
ным распределением температур по сводам печи — интенсифика-
ция работы нижних сводов.
В соответствии с изложенными выше (стр. 90) факторами,
влияющими на скорость горения колчедана, дальнейшие пути ин-
тенсификации лежат: 1) в общем повышении температуры в печи,
что возможно при внедрении жароустойчивых гребков и лопа-
ток, и 2) в применении более мелкого помола колчедана.
В табл. 38 приведены данные о полноте выгорания серы в за-
висимости от степени помола колчедана по печам Ведже и ВХЗ.
Таблица 38
Размер фракций колчедана и огарка в мм Печь ВХЗ при интенсивности в 203 кг Печь Ведже при интенсивности в 205 кг
фракции в шихте В % фракции в огарке В % сера в данной фракции огарка В 7о фракции в шихте в о/о фракции в огарке в «/о сера в данной фракции огарка в %
60,8 63,2 0,9 21,05 66,83 1,96
3—5 13,0 9,72 1,72 6,43 3,93 3,05
5—7 7,0 7,0 4,12 24,76 11,30 4,67’
7—10 6,2 5,0 6,18 31,19 13,27 7,33
10 13,0 15,08 9,70 16,57 4,67 9,67
В табл. 38а показано, какая часть от всей серы, пере-
ходящей в огарок, уносилась отдельными фракциями огарка.
Таблица 38а
Размер фракций в мм Печь ВХЗ при интенсивности в 203 кг Печь Ведже при интенсивности в 205 кг
• фракции в огарке в % сера в данной фракции от всей серы огарка в 7о фракции в огарке в % сера в данной фракции от всей серы огарка В 7о
0—3 63,2 20,5 69:83 42,5
0—5 72,74 26,3 70,76 46,0
0—7 79,94 36,5 82,06 61,0
>7 20,26 63,5 17,94 39,0
112
Таким образом фракции огарка выше 7 мм, составляющие по
печи ВХЗ 20,26% от огарка, уносили в огарок 63,5% от всей
серы огарка. Ясно, что при подаче колчедана в печь кусками не
выше, например, 5 мм работа печи может быть значительно улуч-
шена. Такое требование к помолу не снизит общей производи-
тельности дробильного отделения, если поставить предваритель-
ный отсев мелочи перед дроблением.
Увеличение числа оборотов вала печи в два раза уже пока-
зало, что при этом печь работает лучше. Несомненно, что более
значительное увеличение скорости вращения вала будет интен-
сифицировать процесс горения (в результате более интенсивного
перемешивания материала) и уменьшит опасность спекания мате-
риала.
О конкретных перспективах дальнейшей интенсификации ме-
ханических печей некоторое представление можно составить на
основе учета опыта теплотехники. При интенсивности в 300 кг
на 1 м- пода печи выделяется 17 570 ккал/час. При сжигании раз-
личных топлив в топках котельных выделяется на 1 м2 поверх-
ности решетки 700 000—1 000 000 ккал/час. При интенсивности
печи ВХЗ в 300 кг напряжение ее топочного пространства соста-
вляет 35 000 ккал/м^ в час. Допускаемые тепловые напряжения
для разных топлив колеблются в пределах 200 000—
500 000 ккал/м3 топочного пространства в час. Надо, конечно,
учитывать, что условия обжига колчедана значительно отли-
чаются от условий обжига в топках котельных. Во всяком слу-
чае вполне возможно практически поставить задачу освоения ин-
тенсивности механических печей до 500 кг.
Интенсивная работа печей предъявляет ряд требований к кон-
структивному оформлению отдельных деталей печи.
Так выявилась необходимость изменения конструкции зубьев
сушильного свода в связи с пропуском возросших масс колче-
дана; так как возросшее количество огарка стало заваливать воз-
душники VII свода, встала необходимость поднятия воздушни-
ков. Выявилась необходимость и некоторых конструктивных из-
менений питателя рабочего свода. Повышение средней и макси-
мальной температуры в печи потребовало выполнения зубьев из
более жаростойкого материала. Очень хорошие результаты пока-
зали проведенные испытания зубьев из хромистого чугуна, состав
которого разработан Московским институтом стали (содержание
хрома 15—20%). При параллельной работе зубьев из серого чу-
гуна и из хромистого чугуна на II и III сводах (на одном гребке
зубья из одного материала, на другом из другого) за 44 дня ра-
боты пришлось сменить 126 зубьев из серого чугуна и всего
лишь 2 из хромистого чугуна. Зубья из хромистого чугуна вне-
дряются на всех заводах.
Возможно, что также и для изготовления гребков, а может
быть и вала печи, потребуется применение жароустойчивого ме-
талла. Применение жароустойчивого металла увеличивает началь-
ные затраты на изготовление деталей, но в конечном итоге дает
8 Зак. 5019. Технология серной кислоты.
из
экономию, так как срок службы детали возрастает в большее
число раз, чем растет ее начальная стоимость. Кроме того, при-
менение деталей из жароустойчивого материала (например зубьев
и гребков) сокращает число и продолжительность остановок
аппарата.
Так, конструктивные изменения аппарата (в данном случае
печи), необходимость которых вызвана новыми условиями вы-
сокоинтенсивного режима, будучи осуществлены, в свою оче-
редь открывают новые возможности дальнейшей интенсифика-
ции.
2. Печи пылевидного обжига
Конструкции печей пылевидного обжига. Затруднения при об-
жиге флотационного колчедана в механических печах, связанные
с его склонностью к спеканию, особенно на первых этапах его
Рис. 39. Прямоугольная печь:
/—печь; 2—бункер; 3 шнек; 4—привод; 5—фор-
сунка.
Рис. 40. Цилиндрическая
печь:
1—печь; 2—бункер; 3—шнек;
4—привод; 5—форсунка.
освоения, а также и сам характер флотационного колчедана, его
чрезвычайная мелкость, натолкнули на мысль о применении для
этого случая таких типов печей, где колчедан сгорал бы во взве-
шенном состоянии. Такие печи получили уже некоторое распро-
странение в СССР, США и Канаде — в странах, где имеются
огромные массы флотационного колчедана.
В СССР печи пылевидного обжига для флотационного колче-
дана применяются двух конструкций: прямоугольные (рис. 39) и
круглые (рис. 40).
Прямоугольная печь 7 (рис. 39) представляет собой заключен-
ную в стальной кожух прямоугольной формы шахту, выложен-
114
ную из огнеупорного кирпича. Внутри печь разделена не доходя-
щей до низа перегородкой на две камеры. В передней стенке
бункера расположены несколько наклонно две форсунки 5 для
подачи смеси колчедана и воздуха. Выходя из форсунки, смесь
воспламеняется, образуя факел горящего колчедана, направлен-
ный к потолку печи. Внизу печи расположены два бункера, в ко-
торые собирается падающий огарок.
Рис. 41. Форсунка (Гипро-
хи ма).
Цилиндрическая печь 1 (рис. 40)
представляет высокую круглую
шахту, выложенную из огнеупор-
ного кирпича и заключенную в же-
лезный кожух. В нижней части
печь заканчивается бункером для
огарка.
В прямоугольных печах при
вдувании колчедана снизу его недо-
горевшие частицы долетали до
Рис. 42. Форсунка
(Пермского завода).
верхнего свода печи и налипали на него, образуя так называе-
мые «козлы». Это в значительной степени было устранено тем,
что потолок первой шахты печи стал выполняться из железных
корыт или коробок, охлаждаемых водой.
Еще недавно питание печей пылевидного обжига колчеданом
и воздухом осуществлялось так: из колчеданного бункера 2 кол-
чедан подавался шнековым питателем 3 к форсунке 5, в которую
поступал и воздух для распыления. Так как в связи с измене-
нием качества колчедана (по содержанию серы) приходилось ме-
нять производительность шнекового питателя, для регулирования
числа оборотов последнего между ним и мотором ставился спе-
циальный фрикционный привод 4. Каждый питатель обслужи- •
вался индивидуальным мотором.
Форсунка конструкции Гипрохима (рис. 41) для ввода колче-
дана в круглые печи состоит из трех частей. В верхней части
укреплена труба, по которой из шнекового питателя поступает
флотационный колчедан, в верхнюю же часть подается первич-
ный воздух. Средняя часть снабжена полостью со штуцером,
8»
115
в который подается вторичный воздух. В корпусе средней части
имеются направленные вниз прямоугольные отверстия, через ко-
торые воздух поступает внутрь форсунки. Первичный воздух, по-
ступая в форсунку, содействует падению колчедана вниз, вторич-
ный же, устремляясь через боковые отверстия, завихряет ча-
стицы колчедана и перемешивает их с воздухом. Смесь воздуха
и колчедана поступает в печь через конус в нижней части фор-,
сунки.
Форсунка Пермского завода состоит из двух рукавов, напра-
вленных под углом в 90е друг против друга. При взаимном ударе
двух встречных потоков воздуха, при выходе из форсунки, кол-
чедан распыляется по сечению печи (рис. 42). Эта форсунка со-
Рис. 43. Схема питания печи влажным колчеданом:
1—бункер; 2—питатель; 3—эжектор; 4—съемная плита; 5—сегаратор; 6—бункер; 7—фор-
сунка.
здает лучшее распыление пылевоздушной смеси по всему сече-
нию печи (чем форсунка Гипрохима) и, кроме того, в отличие от
предыдущей имеет только один ввод возд-уха, что упрощает си-
стему воздухопроводов.
Воздух, необходимый для горения колчедана, подается в фор-
сунку и в формы, расположенные над бункером в нижней части
печи. Воздух подается под напором 250—300 мм вод. ст. Распре-
деление воздуха между форсункой и фурмами производится в за-
висимости от степени распыления колчедана, нагрузки печи и вы-
горания серы из колчедана.
Описанная выше схема подачи колчедана имела целый
ряд существенных неудобств. Самое главное неудобство заклю-
чалось в том, что требовалось колчедан сушить до содержания
влаги не больше 0,5%, так как при более высоком содержании
влаги, во-первых, забивались шнековые питатели, а, во-вторых,
влажный колчедан не мог достаточно хорошо распыляться фор-
сунками. Но сушка колчедана вызывала его удорожание, созда-
вала условия чрезвычайно высокой запыленности в сушильном
и печном отделениях (приводившей также к большим потерям
116
колчедана) и вела к образованию катышей, которые не успевали
выгорать за время их пребывания в печи, что повышало потери
серы с огарком.
Указанные недостатки в значительной степени устранены
освоенным сейчас на основе опытов УНИХИМ процессом обжига
влажного флотационного колчедана (до 2—5% НгО). Схемы пи-
тания печи влажным флотационным колчеданом представлены на
рис. 43.
После разрыхления в молотковой мельнице колчедан посту-
пает в бункер 1 и далее через тарельчатый питатель 2 в трубо-
провод, в который нагнетается воздух, предварительно проходя-
Рис. 44. Установка Фримана для сжигания флотационного колчедана:
7—бункер;*2—питатель; 3—шарогая мельница; 4—вентилятор;5—сепаратор; 6—пылеулови-
тель; 7—бункер; 8—питатель; 9~форсунка; 10—вентилятор; 11—печь; 12—пыльная камера;
/3—паровой котел; 14 и 15—промывные башни; 16—эксгаустер.
щий эжектор 3. Эжектор снабжен соплом, которое сообщает
воздушному потоку скорость около 100 м/сек и создает вакуум
перед загрузочным отверстием. Смесь воздуха и колчедана по-
ступает в сепаратор 5. Принцип действия сепаратора состоит
в том, что в вертикальную трубу вдувается пылевоздушная смесь
со скоростью ~ 25 м/сек. Все крупные частицы колчедана вслед-
ствие меньшей скорости в вертикальной трубе падают вниз и
’сталкиваются со встречным потоком вновь поступающих частиц.
Таким образом крупная частица, прежде чем достигнуть низа
трубы и попасть в бункер б, испытывает на пути большое коли-
чество ударов и, постепенно разрыхляясь, превращается в пыль.
При уменьшении скорости пылевоздушной смеси в вертикальной
117
трубе мелкие частицы колчедана ударяются о стенки трубы и
выпадают из воздушного потока. Чтобы избежать этого, в сепа-
ратор вводится дополнительный воздух. Одновременно с разрых-
лением комков и отделением неразрыхленных комков в сепара-
торе происходит добавочное перемешивание колчедана с возду-
хом. Приготовленная пылевоздушная смесь поступает в фор-
сунку 7, которая равномерно распределяет смесь по всему сече-
нию печи.
На рис. 44 представлена установка для обжига флотационного колчедана
системы Фрнмана в США. Колчедан с содержанием влаги не более 1%
поступает в бункер у, оттуда через питатель 2 в шаровую мельницу з.
В мельницу з вентилятором 4 подается воздух. Пылевоздушная смесь идет
через мельницу з в сепаратор 5, из которого более крупные частицы обратно
возвращаются в шаровую мельницу, основная же масса частиц уносится
воздухом в пылеуловитель в. Воздух из пылеуловителя вновь подается
в шаровую мельницу, чем избегается потеря пыли от неполного ее оса-
ждения в пылеуловителе. Из пылеуловителя колчедан поступает в пита-
тельный бункер 7 н далее питателем 6 подается в форсунку .9. Для распы-
ления колчедана подается воздух вентилятором 10. Получающийся в печи и
газ поступает в пыльную камеру j?, а затем в паровой котел уз. После
охлаждения под паровым котлом газ проходит для очистки от пыли через
две промывные башни 14 н уд и эксгаустером 16 подается на производство.
Производительность и технологический режим печей пыле-
видного обжига. Интенсивность работы печей пылевидного об-
жига характеризуется числом килограммов колчедана (считая на
45%-ный по содержанию серы), сжигаемого на 1 м3 свободного
объема печи в сутки. Отраслевая конференция основной химиче-
ской промышленности в 1936 г. приняла нормальную интенсив-
ность печей пылевидного обжига в 500 кг. Дальнейшей работой
стахановцев заводов, а также работами научно-исследователь-
ских институтов, особенно после освоения сжигания влажных
флотационных хвостов, доказана возможность интенсивности
в 700—800 кг.
Тринкс приводит следующие допустимые напряжения топочного про-
странства для различных случаев горения (табл. 39).
При интенсивности печи пылевидного обжига в сутки в 700 кг и при
теплотворной способности колчедана в 1400 ккал напряжение топочного про-
странства составляет
800-1 400
---st---= 40 bob ккал/м1 в час.
24
Из сравнения условий обжига пылевидного колчедана с характеристи-
ками условий, данными Тринксом, нетрудно видеть, что случай обжига рас-
пыляемого колчедана может быть подведен не ниже, чем под третье условие
Гринкса. Существенным различием остается только то, что Тринкс имел
в виду сжигание угля, здесь же мы имеем дело с обжигом колчедана. Но
чем меньше частички сжигаемого колчедана, тем принципиальная разница
между его обжигом и обжигом угля становится меньше. Во всяком случае
нз^приведенного сопоставления ясно, что достигнутая уже интенсивность
работы печей пылевидного обжига может быть повышена в несколько раз.
Нормальная работа печи обеспечивается равномерной подачей
колчедана и тщательным его распылением по сечению печи, точ-
ным соответствием между количеством подаваемого колчедана и
118
Таблица 39
№ пл. Условия горения Напряжение топочного пространства в ккал!м* в час
1 Очень плохое перемешивание топлива с воздухом; несовершенное использование пространства ка- меры горения пламенем или струями газа; круп- 50 000
ные кусочки топлива, холодный воздух
2 Довольно хорошее смешение топлива с воздухом; сравнительно хорошее использование простран- ства камеры горения; крупные куски топлива; холодный воздух или все, что в условии 1, но 144 000
воздух нагрет до 260°
3 Совершенное перемешивание топлива с воздухом; полное использование пространства для горении; очень тонкое распыление топлива; холодный воз- дух или все, как в условии 2, но воздух нагрет 288000
до 260° •
4 Совершенное перемешивание топлива с воздухом; полное использование пространства для горения; очень тонкое распыление топлива или все, как 540 000
в условии 3, но воздух нагрет до 540°
воздуха. Так как колчедан в печи пылевидного обжига пребывает
всего несколько секунд против нескольких часов пребывания
в механических печах, требование равномерной подачи колчедана
и воздуха и соответствие между их количествами особенно
должно полно и точно соблюдаться при эксплоатации этих пе-
чей. Регулирование работы печи заключается в поддержании по-
стоянства концентрации SO2 в обжиговом газе и температуры
в печи. Падение температуры может произойти от того, что
1) стало меньше поступать колчедана и 2) уменьшилось содер-
жание серы в поступающем колчедане.
Конструкция печи пылевидного обжига позволяет получать
более высокую концентрацию SO2 в газах (до 14%) при обжиге
колчедана и более высокую температуру (до 1000° и выше), чем
в механических печах. Это создает благоприятные условия для
использования тепла обжиговых газов, что, как выше упомина-
лось, уже осуществляется в США.
Большим недостатком печей пылевидного обжига остается
еще очень высокая запыленность получаемых газов (до 100 г/да8
нормального газа). При обслуживании этих печей очень затруд-
нителен процесс удаления (имеющего высокую температуру)'
огарка. В целях уменьшения запыленности газов, облегчения ра-
боты по удалению огарка и разрешения проблемы по использо-
ванию его, а также в целях дальнейшей интенсификации печей
119
представляется заманчивым процесс обжига вести так, чтобы
огарок получался в виде агломерата. Этот вопрос несколько по-
дробнее освещен ниже (стр. 145).
3. Горизонтальные вращающиеся печи
Вращающиеся печи обычной конструкции известны по цемент-
ной промышленности. Аналогичные же конструкции применяются
для агломерации железных руд. Применить такого рода печи для
обжига серного колчедана пытались уже около 30 лет -назад. Но
многие попытки приводили к неудачам, так как на некотором
Рис. 45. Питатель печи Лурри.
всей длине печи расположены дюзы
же печи находится питатель для под
расстоянии от ввода в печь
колчедан приобретал такую
высокую температуру, что
спекался и покрывался кор-
кой. Это затруднение в на-
стоящее время преодолено
двумя путями: 1) подача воз-
духа в нескольких местах
печи (печи Лурги) и 2) при-
мешивание огарка к загру-
жаемому в печь колчедану
(печи Кемико).
Остановимся прежде всего на
описании типа горизонтальной
вращающейся печи германской
фирмы Лурги.
Один конец печи оканчи-
вается головкой, через которую
выходят газы, близ другого конца
находится выгрузочное устрой-
ство для отвода огарка. По
для подачи воздуха. В головке
чи колчедана (ри-с. 45). Выходное
отверстие колчеданного бункера оканчивается гладкой чугунной трубкой.
Эта выводящая труба в нижней части закрыта поворачивающимся в обе
стороны цилиндрическим сегментом. Выходное отверстие трубы и цилиндри-
ческий сегмент герметически заключены в коробку, которая на нижнем конце
имеет наставку; через последнюю колчедан при загрузке через шнбер непо-
средственно попадает в печь. Вал внутри кожуха неподвижно соединен
с подвижным телом и имеет рычаг, который при помощи шатуна и эксцен-
трика движется туда и обратно.
На рис. 46 показан продольный разрез самой печи и четыре поперечных
сечения. Выступы обмуровки содействуют продвижению и перемешиванию
горящего материала. В нижней части внутренней поверхности печи имеется
кольцеобразное углубление в виде жолоба. В нем находятся 4—3 отверстия,
оканчивающиеся выгрузочными приспособлениями.
Выгрузочное приспособление (рис. 47) состоит из чугунного кожуха,
в котором находится вращающаяся крестовина. На валу вне чугунного ко-
жуха насажена звезда из полосового железа, концы которой прн вращении
печи бегут мимо упора — под печью на полу помещения. Прн ударе любого
конца об упор барабан поворачивается на оборота и в это время проис-
ходит высыпание огарка.
Дюз для подвода воздуха в печь (рис. 48) работает на всасывание. Часть
дюза, выходящего наружу, представляет собой полый усеченный конус,
120
который закрывается с наружного конца жестяным шибером. Шибер со-
ставлен нз двух круговых секторов — один неподвижный, другой подвижный.
Поворачиванием подвижной части можно увеличивать нлн уменьшать отвер-
стие для прохода воздуха. В печах новой конструкции у шибера имеется
шкала, которая позволяет точно регулировать подачу воздуха.
Рис. 46. Разрез печи Лурги.
Дюзы доводятся до осн печи для того, чтобы предупредить попадание
в них колчедана. По самой 'поверхности печи дюзы расположены по крутой
винтообразной линии. Общее количество дюз 11—13. Есть печи, у которых
дюзы работают под давлением. В этом случае по винтовой линии располо-
жения дюз имеется трубопровод для подвода воздуха.
Наиболее сложная с конструктивной стороны часть печи — головка
(рис. 49).
Рис. 47. Выгрузочное устрой-
ство.
Рис. 48. Дюз.
Чугунное кольцо 1 привинчено к крышке печи н, следовательно, делает
вместе с ней все движения. Внутренняя поверхность этого кольца отделана
как поверхность скольжения. В этом кольце находится второе кольцо
которое не вращается, но может хорошо следовать за движением кольца /
вверх и вниз, вперед и назад и вбок. Чтобы защитить поверхность скольже-
ния от температурных влияний, полое кольцо охлаждается воздухом илн
водой. Охлаждение может быть причиной конденсации SOs, что со временем
121
может вызвать коррозию специального чугуна. Чтобы избежать этого,
кольцо ? выложено высококачественным шамотом. Эта обмуровка одновре-
менно изолирует кольцо 2 от горячих газов. С полым кольцом 2 плотно
соединена коленчатая тру-
Рис. 49. Головка печи:
1 и 2—чугунные кольца; 3—коленчатая труба; 4—гид-
равлический затвор; 5—манжета; 6—кольцо.
Размеры печи этого типа стандартизованы
ба 3 из чугуна, которая
может быть загнута своим
открытым концом кверху
или чаще книзу. Эта труба
не поддерживается коль-
цом, она вместе с кольцом
поддерживается двумя про-
тивовесами, находящимися
слева и справа от ее цен-
тра тяжести. Противовесы
поддерживаются цепями или
канатами на роликах. Гер-
метичность неподвижной га-
зовой вытяжки обеспечи-
вается гидравлическим за-
твором 4. Затвор установлен
так, чтобы в кольцо в не
могла попадать пыль. Удли-
нением штуцера посред-
ством жестяной манжеты 5
совершенно избегается со-
прикосновение гидравличе-
ского затвора и самой жид-
кости с горячими газами.
Все движение печной го-
ловки балансируется в этом
идравлическом затворе.
Соединение между стацио-
нарной засыпной воронкой
и загрузочной трубой не
жесткое, а осуществляется
металлической трубкой или
приспособлением вроде круг-
лой муфты.
таким образом, что отношение
iJt 'WMiHh/ui/
длины к диаметру берется равным 10. Продолжительность одного оборота
печи в минуту равна длине диаметра в метрах. Печи строятся для обжига
от 9 до 50 г колчедана в сутки.
Рис. 50, Разрез печи Кемико.
Содержание SO2 в газе, получаемом из печей, составляет по
данным фирмы Лурги не менее Ю%.
Вращающаяся печь Кемико (рис. 50) представляет собой же-
лезный футерованный шамотным кирпичом цилиндрический бара-
122
бан, установленный наклонно на двух опорах. Внутри барабана
расположен в шахматном порядке ряд лопастей, служащих для
пересыпания колчедана. Обжигаемый материал поступает через
рукав в барабан печи и в виде огарка удаляется через отверстия
в' решетке на винтовой шнек. Печь приводится в движение через
приводный механизм от мотора. Для снижения температуры
в печи в шихте с колчеданом подается огарок (3—6 вес. ч. огарка
на 1 вес. ч. колчедана), температуры в печи порядка 800—850°,
концентрация газа из печи ~ 10%. Печь обжигает ~ 50 т колче-
дана в сутки.
Таблица 40
Об ьект контроля Показатели Место отбора проб или замер Частота анализа или замера
Шихта Вес шихты, за- гружаемой в печь — Каждая загружаемая вагонетка или пока- зание счетчика лен- точных весов
То же Содержание серы, влаги и углерода в % Загружаемая вагонетка Каждая вагонетка Средний анализ - один раз в смену экспрессным методом Унихима
Газ Температура Горловина печи VH свод III свод Каждые 2 часа То же Контрольное опреде- ление 1 раз в сутки
То же W Верх и ииз печи 4 раза в смену; запись производится 1 раз в смену как среднее
То же Разрежение Горловина печи I свод 4 раза в смену (запись) 1 раз в сутки (контрольное опре- деление)
То же Содержание SO2 Горловина печи 1 раз в смену методом Рейха
Воздух или вода, охлаждающая вал и гребки Температура Выход 2 раза в смену
Огарок Содержание серы в % Течка VII свода Отбор каждые два часа; анализ м е то д о м У н и х и м а ил и хроматным методом
123
4. Контроль за работой печного отделения
Правильное регулирование работы печей может быть осуще-
ствлено только при наличии систематического контроля за рабо-
той печей. Основными анализами и замерами, характеризующими
качество работы каждой печи и печного отделения в целом,
являются: анализ газа на содержание SO2 в горловине каждой
печи в общем газоходе перед входом газа в систему и анализ
огарка из каждой печи на содержание серы. Кроме того, в целях
своевременного обнаружения намечающихся отклонений процесса
от нормального режима в печном отделении производится ряд
других анализов и замеров. Перечень их даем в табл. 40.
При обжиге углеродистой шихты газ из горловины каждой
печи и из общего газохода анализируется также на содер-
жание СО2 и Ог. В специальных случаях (обследование работы
печей, освоение нового сырья и др.) производятся и другие за-
меры и анализы (количество воздуха, поступающего в печь, тем-
пература по каждому своду печи, содержание серы в материале
с каждого свода и др.).
5. Печи для сжигания серы.
Сера вступает во взаимодействие с кислородом воздуха при
наличии влаги уже при обыкновенной температуре. Окисление,
которое протекает медленно, приводит к образованию серной
кислоты. Воспламенение серы по одним данным происходит
в воздухе при 360°, а в кислороде при 285°; по другим данным
сера окисляется уже при 214°. При горении серы получается сер-
нистый ангидрид, серный ангидрид может получаться в относи-
тельно незначительных количествах при наличии в сере приме-
сей, контактирующих реакцию окисления SO-2 в SO3.
Горение серы происходит одновременно и в жидкой и в газо-
вой фазах. При этом с обжиговыми газами в зависимости от
условий обжига уносится и некоторое количество паров элемен-
тарной серы. Тот факт, что, несмотря на достаточно высокую
температуру в зоне обжига и избыток кислорода, часть паров
серы не сгорает, объясняется сложностью молекул серы.
Сжигание серы в громадных количествах с получением SO*
для сернокислотной и сульфитцеллюлозной .промышленности ши-
роко применяется в США.
С развитием промышленности элементарной серы применение
ее для сернокислотной и бумажной промышленности становится
возможным и в СССР.
Сера сначала сжигалась в плоских печах. Недостатком этих печей
являются небольшая производительность и громоздкость. Кроме того часть
серы улетучивается, примеси серы собираются на поверхности н на дне, что
требует частой чистки печи.
Дальнейшей конструкцией печи явилась вращающаяся печь (рис. 51).
Такие печи до настоящего времени еще имеются на ряде сернокислотных
заводов. Эта печь состоит нз цилиндрического стального кожуха, к концам
124
которого присоединены на фланцах чугунные конусы. Фланцы обработаны и
являются в то же время бандажами, поверхность которых вращается на
опорных роликах. Один конец печн снабжен воронкой, шнековым питателем
для загрузки твердой серы и цилиндрическим дросселем для регулирования
количества поступающего воздуха. Другой конец имеет такие же регули-
руемые отверстия для ввода воздуха с целью разбавления газа перед его
поступлением в вертикальную стальную, футерованную огнеупорным кирпи-
чом камеру (на рисунке не показана), служащую для догорания сублими-
рованной серы.
Рис. 51. Вращающаяся печь для сжигания серы.
Эта печь имеет крупные недостатки — трудность регулирования равно-
мерной подачн серы, невозможность остановки печи до полного выгорания
серы (а если остановку все же надо сделать, то это связано с потерей серы)
и работа печи на неосушенном воздухе, что усложняет дальнейший процесс,
если газ предназначен для производства контактной серной кислоты.
В установках для сжигания серы в последние годы осуще-
ствляется предварительное расплавление серы и непрерывное пи-
Рис. 52. Схема американской установки для сжигания серы:
1 — приемный сборник; 2—нижннй сборник; 3— насосы; 4—трубопроводы; 5—печь; 6—су-
шильная башня; 7—брызгоуловитель; 8—вентилятор: 9—фильтр; 10—утилизатор-тепла.
тание печи расплавленной серой, обязательная предварительная
осушка воздуха и использование тепла газов для получения пара.
Са.ци печи в этих установках представляют собой или гори-
зонтальные камеры стационарного типа или вертикальные ци-
линдры с внутренними арочными сводами.
Схема американской установки (Chemical Construction Corp.)
Для сжигания серы изображена на рис. 52. Расплавленная сера
125
поступает в верхний приемный сборник 1, змеевики которого обо-
греваются паром под давлением в 4—5 ат. Далее сера идет в ниж-
ний сборник 2, разделенный не доходящей до верха перегород-
кой. В первом отделении находится обогреваемая паром через
змеевики расплавленная сера. В другом отделении установлены
центробежные с паровым приводом насосы 3, подающие серу
через стальные, снабженные рубашкой для обогрева паром (да-
вление 6 ат) и изолированные снаружи трубопроводы 4 к фор-
сункам печи 5. Расплавленная сера распыляется под напором
в 2,5 ат.
Воздух для горения серы вентилятором 8 протягивается че-
рез орошаемую крепкой серной кислотой сушильную башню б и
брызгоуловитель 7. Выходящий из печи газ проходит через
фильтр 9, насаженный кварцем. Назначение фильтра — освобо-
Рнс. 53. Схема американской установки для сжигания серы:
1 и 2—плавильный котел; 3—трубки; 4—насосы; 5—серопровод; 6—печь; 7—сушильная
башня; 8—брызгоуловитель; 9—воздуходувка; 10—паровой koi ел.
дить газ от механических примесей, которые могут оказаться
в газе, несмотря на отстаивание расплавленной серы. Далее газ
проходит через котел-утилизатор тепла 10 и направляется в серно-
кислотную установку- Сама печь представляет собой стальной
цилиндр диаметром в 3,3 м длиной 9 /и, футерованный изнутри
листовым асбестом и огнеупорным кирпичом.
Внутри печи имеются две перегородки. В такой печи, со сво-
бодным объемом 35 /и3, в сутки сжигается 35 т серы, так что
тепловое напряжение печи составляет 78 000—80 000 ккал/м3
в час. На бумажных фабриках применяются аналогичные уста-
новки без предварительной сушки воздуха, поступающего
в печь.
Схема другой американской установки (Monsanto Chemical
Со) понятна из рис. 53. Воздух, идущий в печь, после сушки и
освобождения от брызг подогревается газами, выходящими из
контактного аппарата, до 340—350э. Теплообменник на рисунке
не показан.
Печь представляет собой вертикальный стальной цилиндр диа-
метром 3,9 Л1 и высотой 7,5 /и, выложенный изнутри кислото-
упорным кирпичом. Внутри печи расположены две арки, на кото-
126
рых выложены пирамиды из кислотоупорного кирпича. Распла-
вленная сера поступает в печь — в центр верхней пирамиды, на
поверхности которой и происходит горение. Сера, не сгоревшая
на верхней пирамиде, стекает на нижнюю, где и догорает. Воз-
дух на горение поступает в печь по двум отверстиям, располо-
женным в стенке над верхней аркой. Обжиговые газы уходят из
печи через отверстие в стенке под нижней аркой. Температура
газа, содержащего 8—8,5% SO*, 1000 . В котле-теплоутилиза-
торе температура газов снижается до 450 .
Поскольку большинство сернокислотных заводов СССР обо-
рудовано механическими печами, представляет интерес освоение
обжига серы в этих печах (может быть с небольшой реконструк-
цией этих печей).
Проведенные НИУИФ (Г. Б. Блюм) опыты показали полную
возможность этого. Сера обжигалась в печи ВХЗ в шихте с кол-
чеданом и без него. Серы без добавки колчедана в печи ВХЗ
сжигалось до 23 т в сутки. Сера подавалась на VI свод. Горение
серы в парах (в газовой фазе) происходило до 11 свода. Целе-
сообразно также освоение серы в шихте с другим малосернистым
сырьем (бедный серой колчедан, пирротин и др.), а также
с сырьем, требующим затраты тепла (например железный купо-
рос).
Большой интерес представляет освоение обжига шихты из
цинковой обманки и серы в печах цинкоплавильных заводов. Это
повысило бы концентрацию газов (по SOa) и освободило бы от
необходимости применять другие виды дополнительного топлива
при обжиге цинковой обманки.
6. Металлургические печи
Печи медеплавильных заводов. Получение меди из сернистых
руд проводится по одной из двух схем. Первая схема (старая)
состоит в том, что руда сначала плавится в ватержакетах, где
из нее получается более богатый медью материал, так называе-
мый штейн. Далее штейн подвергается процессу бессемерования
в конверторах, где получается черновая медь.
Во второй схеме (новой) первой стадией обработки концен-
трата является сжигание его в обжиговых печах с целью по-
низить содержание в нем серы. Плавка для получения штейна
проводится в так называемых отражательных печах. Выплавка
черновой меди тоже проводится в конверторах.
Предварительный обжиг. Предварительный обжиг
концентрата производится в механических печах Никкольс-Гер-
ресгофа или Ведже.
Печи Ведже обычно строятся семиэтажными. Гребки охла-
ждаются воздухом или водой, вал — воздухом. Производитель-
ность такой печи до 100 т концентрата в сутки. В обжиговой
печи НиккольсТерресгофа охлаждение вала и гребков воздуш-
127
ное, предусмотрена также подача в печь теплого воздуха из
вала печи.
Если в поступающем на обжиг материале серы содержится
не менее 25, то обжиговая печь работает нормально без доба-
вочного топлива. Степень выжига серы в различных случаях
меняется в пределах от 30 до 75%.
При нормальном концентрате, получаемом из комплексных
сернистых руд, обжиговые газы должны быть нормальной кон-
центрации по SO'2, т. е. как и при обжиге колчедана в механи-
ческих печах.
Получение штейна. По второй (новой) схеме вы-
плавки меди штейн получается в отражательных печах, в кото-
рых обжигаемый материал тонким слоем располагается на длин-
ном горизонтальном поду печи и обогревается сверху горячими
газами, получаемыми от сжигания топлива.
Химизм процесса плавки в отражательных печах заключается в сле-
дующем:
1) Окислы железа и меди шлакуются по реакциям:
2FeO + SiO» = 2FeO • SiO2;
Cu2O + SiO2 = Cu2SiO3.
2) Наряду с этим происходит восстановление окислов меди:
2Ct:0 + Cu,S = 4 Си + SO»;
2CuO CuS = 3Cu + SO».
3) Получившаяся свободная медь превращается обратно в сульфид меди:
2Cu + FeS = Cu»S -|- Fe.
4) Реагируя с сульфидом железа, окислы меди переходят в сульфид
ate ди:
Cu2O + FeS = Cu2S + FeO;
6CuO -f- 4FeS = 4FeO + 3Cu»S + SO».
5) Происходит также реакция:
Fe + Fe2O;i = 3FeO.
6) Действием FeS ошлакованная медь выделяется из шлака:
Cu»SiO.t + FeS = Cu»S + FeSiO3.
В результате отражательной плавки в шлак переходят окнслы железа,
кремнезем н др., а в штейн — сульфиды меди и железа. Но часть сульфида
железа, поступившего с шихтой, в процессе плавки в отражательной печи
окисляется и переходит в шлак. Таким образом по сравнению с шихтой
штейи обогащается медью. Для хорошего отделения шлака от штейна не-
обходимо, чтобы шлак был значительно легче штейна, обладал малой вяз-
костью и легкоплавкостью. Чтобы удовлетворить этим требованиям, к шихте
добавляются флюсы SiO», CaO, Al2O3 и др.
Из всей серы, поступившей в отражательную печь, ~ 25%
сгорает в SO* и уходит с газами. Остальная часть серы выходит
из печи со штейном в виде сернистых соединений меди и же-
128
леза. Состав газов, отходящих
мерно следующий (в %):
СО...0,1
О2.. .0,3—0,5
из отражательном печи, при-
СО2...14—16
SO2... 2—2,5
Сернистый ангидрид этих газов до сих пор не используется.
Путям использования такого рода газов посвящена гл. 8.
Температура газа при выходе из печи должна быть такой,
чтобы шлаки в задней части печи не застывали, т. е. 1100—1300’.
Основной недоста-
ток отражательных пе-
чей —плохое исполь-
зование тепла, так как
газы выходят из печи
при очень высокой тем-
пературе. Поэтому от-
ражательные печи ока-
зались способными кон-
курировать с ватержа-
кетами только после
того, как тепло отхо-
дящих газов стали ис-
пользовать для получе-
ния пара.
По 1-й схеме вы-
плавки меди штейн по-
лучается непосредст-
венно из руды в так на-
зываемых ватержаке-
тах (рис. 54).
Основу ватержакета со-
Рис. 54. Ватержакетная печь:
/, 2, 3—кессоны; 4—колошниковая площадка; 5—окно:
6—распределительная труба; 7—водонапорная линия;
8—лещадь; 9—специальный кессон; 10—жолоб.
ставляют охлаждаемые во-
дой стенки из трех рядов
кессонов 1, 2 н 3- Шихта
поступает в печь с загру-
зочной или колошниковой
площадки 4 через окно 5- Воздух подается снизу по распределительной
трубе 6 к фурмам. Для подачи воды, охлаждающей кессоны, служит водо-
напорная линия 7. Часть шахты, расположенная ниже уровня фурм, назы-
вается внутренним горном и ограничивается снизу лещадью 8. В старых пе-
чах расплавленные материалы выпускались через отверстие в специальном
кессоне 9 по жолобу ю. Позже стали устраивать снаружи передовой горн,
в котором можно было бы накапливать большое количество штейна.
Процесс плавки богатой сернистыми соединениями руды (пиритная
плавка) в ватержакете протекает следующим образом. По мере того как
нижние слои шихты плавятся, верхние опускаются все ниже и ниже и по-
немногу прогреваются. В некоторых местах медный и серный колчедан
разлагаются, выделяя серу и давая сернистое железо и полусерннстую медь.
Пары серы сгорают не здесь, а уже в самом верху печи за счет кислорода
воздуха, засасываемого в загрузочные окна. В этом месте сгорает загружае-
мое с шихтой топливо за счет сернистого газа, пришедшего из нижних
слоев печи. Спускаясь ниже, шихта попадает в область высокой температуры,
называемую фокусом. Здесь происходит окисление сернистых соединений
9 Зак. 5019. Технология серной кислоты.
129
(главным образом FeS) н плавление шихты с образованием SO2 и FeO-
Закись железа соединяется с кремнеземом руды и загружаемого кварца и
в виде силиката окиси железа дает основу шлака. В процессе окисления
части FeS выделяется такое количество тепла, что материал, в том числе
и часть самого FeS, плавится. Расплавленные сернистая медь и сернистое
железо стекают вниз, образуя штейи. Окислившееся железо в виде силиката
закиси железа идет в шлак. В шлак же переходят расплавленные глинозем
н кремнезем руды. Добавленный в руду в виде флюса известняк разлагается
на окись кальция и СО2. СаО идет в шлак, уменьшая его удельный вес
н повышай его легкоплавкость.
В отходящих из ватержакетов газах концентрация SO2 дер-
жится равной 4—6% и может быть доведена до 8%. Такие
Рис. 55. Конвертор:
1—кожух; 2—ролики; 3—ободы; 4—постаменты; 5—фунда-
мент; 6—горловина; 7—привод; 8—воздухораспределительная
трубка; 9—фурмы.
газы могут быть вполне использованы для производства серной
кислоты.
Бессемерование. Штейн состоит, главным образом, из
сернистого железа и полусернистой меди. В конверторы, в ко-
торых происходит бессемерование, штейн поступает из отража-
тельных печей в расплавленном состоянии. В дальнейшем рас-
плавленное состояние материала в конверторе поддерживается
за счет теплоты происходящих в конверторе реакций.
Через расплавленный материал в конверторе продувается воздух. В пер-
вый период процесса горнт, главным образом, сернистое железо:
2FeS 4- ЗОо = 2FeO + 2SO2.
Закись железа переходит затем в силикат закиси железа:
2FeO SiO, = 2FeO • SiO2.
130
Во втором периоде процесса горит полусернистая медь:
2CusS + 3OS = 2Cu2O + 2SO2.
Закись меди далее реагирует с остающейся частью полусернистой меди,
давая металлическую медь:
Cu2S + 2Cu2O = 6Cu + SO:.
Состав отходящих из конвертора газов в среднем следую-
щий (в /<):
SO2...8 02...9,65
S03. ..0,67 Ns...81,7
Содержание SO2 в конверторном газе по причине периодич-
ности работы конвертора колеблется в широких пределах, так
что нормальная работа на конверторном газе возможна лишь
при условии соответствующей организации работы конверторов
(поочередная загрузка и остановка).
Аппарат для бессемерования—конвертор (рис. 55) состоит из горизон-
тального кожуха I с днищами по концам его. Кожух опирается на восемь ро-
ликов 2 посредством ободов з. Ролики стоят на постаментах 4, укреплен-
ных на фундаментах 5. Загрузка штейна и слив шлака меди производятся
через горловину в. Поворот конвертора осуществляется приводом 7. Воздух
подводится по воздухораспределительной трубке 8 н вдувается внутрь кон-
вертора через фурменные рукава 9 и фурменные отверстия.
Количество и качество получаемых газов на 1 т черновой
меди по аппаратам Красноуральского медеплавильного завода
приводятся в табл. 41.
Таблица 41
Газы Общее количе- ство газов в ж3 so2 Сера в т Можно получить H2SO4 в т
в л3 1 в т
Обжиговые Конверторные Отражательных печей . . 25000 5 090 15200 1900 407 213 5,510 1,180 0,618 2,775 0,590 0,309 8,0 1.7 0,9
Всего.. 45 290 2 520 7,308 3,674 10,6
Таким образом из всей серы, поступающей с концентратом
на медеплавильный завод, 75,5% переходит в БОг в обжиговых
печах. Эта часть серы может быть без особых затруднений пе-
реработана в H2SO4. В конверторах от всей серы концентратов
переходит 16%. Эта часть серы может быть переработана
в H2SO4 при соответствующей организации работы конвертор-
ного цеха. В отражательных печах переходит в SO» 8,5% от
всей серы концентратов.
9- . 131
Печи для обжига цинковой обманки. Получение цинка из
его руд происходит или по пирометаллургическому или по ги-
дроэлектрометаллургическому способу. При переработке цин-
ковой обманки, вне зависимости от способа производства цинка,
первой операцией является его обжиг до ZnO.
По пирометаллургическому способу ZnO подвергается затем
восстановлению .углем.
По гидроэлектрометаллургическому способу ZnO обрабаты-
вается серной кислотой и металлический цинк выделяется из
раствора электролитически.
Поступающая на завод цинковая обманка содержит много-
численные примеси, из которых главные: глина, кремнезем, кар-
бонаты кальция и магния, пирит, свинцовый блеск. В небольших
количествах присутствуют также сернистые соединения мышья-
ка, сурьмы, серебра, меди, калия. Часто цинковая обманка со-
держит также плавиковый шпат, барит и изредка киноварь. Хи-
мическое соединение ZnS содержит 67% цинка, и 33% серы.
В природной цинковой обманке содержание цинка колеблется
от 25 до 62%, а серы — от 22 до 30%.
Целью обжига цинковой обманки является максимальное
удаление серы. Однако практически удалить всю серу никогда
не удается. Содержание серы в огарке зависит от характера ве-
дения процесса обжига и, в особенности, от качества самой
РУДЫ.
Цинковая обманка горит по реакции:
2ZnS + 3O2 = 2ZnO + 2SO2.
В дальнейшем ZnO может дать с образующимся в печи SOs
сульфат:
ZnO 4- SO3 = ZnSO4.
Последний полностью разлагается лишь при температуре 900°.
С точки зрения интенсивности обжига и полноты разложе-
ния ZnSOi, а также других сульфатов, обжиг цинковой обманки
желательно вести при возможно более высокой температуре. Но
при очень высокой температуре улетучивается часть ZnO, ZnS,
а также часть присутствующего в руде серебра. Поэтому тем-
пературу обжига держат не выше 900°.
Образование ZnSO< происходит, главным образом, во второй
стадии обжига, когда в материале имеется ZnO. Но в этой ста-
дии обжига за счет горения серы руды не может поддержи-
ваться высокая температура (так как ZnS в руде уже мало). По-
этому, чтобы не допустить образования большого количества
ZnSOi, высокая температура за весь процесс обжига поддержи-
вается за счет постороннего топлива.
Для обжига цинковой обманки применяются печи: отража-
тельные, муфельные и механические. Ведутся также опыты по
сжиганию ZnS в распыленном состоянии.
При обжиге цинковой обманки в отражательных печах по-
132
лучаюгциеся газы не пригодны для производства серной ки-
слоты. Отражательные печи в практике обжига ZnS вытес-
няются муфельными и механическими печами. Основная сущ-
ность муфельных печей состоит в том, что постороннее топливо
сжигается в другой камере, а не в той, где горит обжигаемый
материал. Топочные газы, получаемые от горения постороннего
топлива, не смешиваются с обжиговыми газами. Тепло от горе-
ния постороннего топлива передается обжиговым газам и обжи-
гаемому материалу через стенку.
Хотя муфельные печи имеют небольшую производитель-
ность, все же они еще широко применяются в Западной Европе.
В СССР муфельные печи имеются только на одном заводе. При-
меняемая в цинковой промышленности печь Спирле является
одновременно и муфельной и механической.
Основная масса цинковой обманки обжигается сейчас в ме-
ханических печах, из которых применяются уже упомянутые
выше печи Никкольс-Герресгофа и Ведже, а также Гумбольдта,
Гипроцветмета и др.
Если обжиг в механической печи производится без сжигания
постороннего топлива, то серы в огарке остается 8'—10%. Ога-
рок с таким содержанием дожигается на агломерационных ма-
шинах.
Концентрация SO2 в газах от обжига цинковой обманки ко-
леблется от 5 до 6,5%. Вторичный обжиг на агломерационных
машинах Двайт-Ллойда дает газ различного состава по отдель-
ным секциям машины. Для производства H2SO4 может быть
использовано около 70% серы, выгорающей при вторичном об-
жиге.
По проекту одного завода на 1 т цинка получится 9250 лг
газа следующего состава: 5,7% SO2, 13,03% О2 и 81,27% N2.
На 1 т выплавленного цинка можно получить ~ 2,2 т серной
кислоты, в том числе из газов первичного обжига 1,63 т (74,3%)
и из газов вторичного обжига 0,57 т (25,7%).
На цинковом заводе в Норденгаме (Германия) первичный об-
жиг проводится в печах Гумбольдта. При содержании в руде
31% серы в первичном огарке остается 8% серы. В газах пер-
вичного обжига содержится 4,9—5,6% SO2 и SOg вместе.
При дожиге на двайт-ллойде газы с общим содержанием
SO-2, равным 4,5%, собираемые по двум трубам, направляются
в сернокислотную систему. Газы с содержанием ~ 0,2% SO2, со-
бираемые по третьей трубе двайт-ллойда, выпускаются в: атмо-
сферу.
При желании полностью обжечь цинковую обманку в меха-
нической печи на нижний свод печи подают нефть через фор-
сунку. Продукты горения нефти смешиваются с обжиговыми га-
зами, концентрация SO2 в газах при этом понижается до
3,5—5%.
Полный обжиг цинковой обманки требует сжигания посто-
роннего топлива не потому, как думают некоторые, что нехва-
133
тает теплоты горения самой цинковой обманки, а потому, что
должна быть обеспечена необходимая температура во второй
стадии обжига (стр. 132). Можно доказать тепловым расчетом,
что при соответствующей конструкции печи возможно провести
полный обжиг цинковой обманки и без сжигания постороннего
топлива. Печь подобной конструкции разработана Ингаллсом и
установлена на одном заводе в США.
7. Получение сернистого газа из гипса
Сульфат кальция прямым нагреванием разлагается по урав-
нению;
CaSO4 —► СаО —SO2 —|— i/2O2 — 116,86 ккал.
Полное разложение CaSOi происходит в пределах 1350—
1400°, при этом масса находится уже в расплавленном состоя-
нии. Температура диссоциации CaSO4 значительно понижается,
если процесс идет в присутствии угля. Процесс при этом про-
исходит по реакциям:
CaSO4 + 2С = CaS 4- 2СО2
и
CaS 4- 3CaSO4 = 4СаО 4- 4SO2,
или по реакциям:
CaSO4 4- 4С = CaS 4- 4СО
и
CaS 4- 3CaSO4 = 4СаО 4- 4SO2.
Для более полного использования угля и более полного оки-
сления получающегося в процессе CaS (чему мешает наличие
в газах СО), целесообразнее направлять процесс по первой из
двух вышеприведенных групп реакций. Суммарная реакция про-
цесса, таким образом, выразится уравнением:
2CaSO, —j— С = 2СаО 4~ 2SO2 4~ СО2 — 135,12 ккал.
Температура диссоциации гипса значительно снижается
также при добавках в шихту SiOa, АЬОз и РегОз. Сравнительное
влияние этих добавок на разложение CaSO4 по данным Мар-
шаль представляется цифрами, приведенными в табл. 42.
Таблица 42
Добавки Давление диссоциации CaSO4 в мм рт. ст. прн температуре
940° 1000° 1100° 1170° 1240° 1250° 1280°
SiO2 10,5 20 94 219 575 817
А12О3 .— 9 25 50 .— 133 200
Fe2Os — 12,5 37 90 178 — 273
134
Температура диссоциации CaSO4 еще более понижается,
а упругость диссоциации повышается при совместном присут-
стви этих добавок. Так, по данным Маршаль давление диссо-
циации при добавке каолина выражается следующими цифрами:
При 94(Р . . . 75 мм рт. ст. При 1100°. . . 589 мм рт. ст.
При 1000° . . .173 „ „ . При 1170°. . .1070 . . „
Так как прямое разложение гипса требует большого расхода
топлива, получение SCb этим путем экономически невыгодно.
Получение SO2 из гипса может оказаться экономически целесо-
образным: 1) если будет найден источник отбросного тепла, за
счет которого будет производиться разложение гипса, 2) если
процесс разложения CaSCU объединить с каким-нибудь экзо-
термическим процессом и 3) если производство SO2 из гипса бу-
дет технологически объединено с производством другого полез-
ного продукта.
Промышленное использование гипса для получения SO2
в первую очередь пошло по третьему из указанных путей. Вто-
рым продуктом, получаемым при этом, является портланд-це-
мент. Производство сернистого газа и цемента впервые было
осуществлено фирмой Байера в Леверкузене (Германия) во время
мировой войны 1914—1918 гг. Затем производство SO2 и це-
мента из гипса было организовано также в Биллингеме (Англия).
В СССР подробное изучение условий получения SO2 и це-
мента из гипса проведено Всесоюзным научно-исследовательским
институтом цемента. Полуторные окислы и SiO-j вводились
в шихту добавкой глины. Результаты заводских испытаний
этого института сводятся к следующему:
1) Отходящий из печи газ имел следующий состав: 8,4%
SO2, 1,5% Q>, 21,2% СО2, 68,9% N2. Это, примерно, соответ-
ствует данным о работе завода в Леверкузене.
2) Вредные примеси, как CS2, H2S, СО и COS, в газовой
смеси совершенно отсутствовали.
3) Запыленность газов при выходе из пыльной камеры соста-
вляла 12 г на 1 № газа, приведенного к нормальным условиям,
в пыльной камере пыли оставалось 33 г на 1 л3 газа.
4) Состав и свойства полученного клинкера полностью со-
ответствовали требованиям стандарта.
5) Производительность печи при работе на гипсе уменьшилась
сравнительно с работой на СаСОз на 50%.
Обжигаемая смесь за время пребывания в печи проходит
три зоны: а) зону подогрева, конец которой фиксируется верх-
ним пределом температуры (~ 800°) — температуры начала дис-
социации CaSOi; б) зону диссоциации CaSCh и, наконец, в) зону
спекания клинкера.
Граница между последними двумя зонами фиксируется кон-
цом, диссоциации CaSO4.
Для получения полноценного портланд-цемента необходима
135
температура обжига 1400—1500°. Такая температура должна
достигаться в последней зоне печи — зоне спекания клинкера.
Однако гипсосодержащие шихты практически начинают пла-
виться значительно ниже температуры спекания клинкера, именно
в интервале 1100—1250°, при котором еще не всегда заканчи-
вается процесс диссоциации CaSOi. Это преждевременное пла-
вление шихты нарушает нормальный ход процесса и ухудшает
качество продукта.
Обжиг гипса должен проводиться в слабоокислительной ат-
мосфере — содержание кислорода в отходящем газе должно
быть 1—1,5%. Восстановительная атмосфера содействует по-
явлению большого количества CaS в пеке; сильноокислительная
атмосфера ведет к остатку большого количества CaSO« в мате-
риале.
После разбавления воздухом (для добавки кислорода) кон-
центрация SO2 в газовой смеси получается равной 4—5% SO2.
Таким образом этот газ по концентрации SQ> уступает газу,
получаемому от обжига колчедана. Но преимущество газа, по-
лучаемого от разложения гипса, в том, что он не содержит
мышьяка. В связи с этим значительно упрощается очистка га-
зов при переработке их на серную кислоту методом контактного
окисления. Кроме того, замена привозного колчедана местным
гипсом дает большую экономию на транспортных расходах.
При заводских испытаниях Института цемента на 1 м3 клин-
кера затрачивалось гипса сырого (98% CaSOa • 2НгО) 2 т, глины
(влажность 20%) 0,37 т, реакционного топлива (условного) 0,1 т;
расход форсуночного топлива — 2300 ккйл на 1 кг клинкера.
Результаты опытов Института цемента отнюдь не являются
предельно оптимальными. При повышении коэфициента полез-
ного действия установки (при уменьшении тепловых потерь) по-
высится содержание SO2 в газах и понизится расход форсуноч-
ного топлива.
Постников с сотрудниками провел лабораторную работу по
выяснению возможности добавки колчедана к шихте при полу-
чении SO2 и цемента из гипса. Количество колчедана, вводимого
в смесь, зависит от состава цемента и составляет 6—8% от веса
сырой смеси. Такая добавка колчедана повышает концентрацию
SO2 в получаемом газе на 15—18% и снижает расход угля, не-
обходимого для разложения гипса на 15—16%.
Понятно, что еще больший эффект даст введение в шихту
углистого колчедана.
Будников и Ривлин изучали в лаборатории вопрос о целесо-
образности совмещения процессов обжига углистого колчедана
(экзотермический процесс) и разложения гипса (эндотермический
процесс). Была поставлена задача сбалансирования теплового
расхода без постороннего топлива. Задача использования твер-
дого остатка при этом не ставилась. Опыты показали, что
в смеси, поступающей на обжиг, содержание CaSO^ (предвари-
тельно прокаленного гипса) может быть доведено до 30%. Об-
136
жиг может быть проведен в интервале температуры 1000—1100е.
Процесс разложения CaSCU значительно ускоряется при доба-
влении в шихту SiOa (песка).
III. Подача колчедана к печам и отвоз огарка
1. Подача колчедана к печам
Самая элементарная схема подачи колчедана на печи сле-
дующая.
Колчедан подвозится вагонеткой вручную из склада к печ-
ному отделению. У печного отделения установлена шахта
с подъемником, куда и помещается вагонетка. Подъемником
Рис. 56. Схема подачи колчедана к печам подвесными вагонет-
ками:
1—приемный бункер; 2—весы; 3—подвесная дорога; 4—бункер печи; 5—эле-
ватор.
вагонетка поднимается на уровень площадки, расположенной
над печами, по узкой колее подвозится к печам и разгружается,
на площадке, откуда колчедан подается на печь.
Такой способ применим лишь при малой производительности
печи. При современной интенсивной эксплоатации мощных пе-
чей требуется полная механизация подачи колчедана. На од-
ном из новых подмосковных заводов колчедан из склада по-
дается транспортером к элеватору 5 (рис. 56), установленному
у торцевой стены печного отделения. Элеватором колчедан
поднимается в приемные бункеры 1 печного отделения. Вдоль
бункеров печей расположена линия подвесной дороги з, по ко-
торой водятся вручную подвесные опрокидывающиеся ваго-
нетки. Между бункером и печью расположены весы 2.
Подача колчедана осуществляется также электровагонеткой /
(рис. 57). Преимущество электровагонеток в их быстроходно-
сти (скорость их равна 18 м/сек) и в том, что они автомати-
137
чески разгружаются. При работе на электровагонетках у бун-
керов печей устанавливаются специальные аншлаги 4. Для раз-
грузки вагонетки у данной печи аншлаг поднимают в положе-
Л/
Рис. 57. Подача электровагонетками:
1—вагонетка; 2—ключ; 3—ролик; 4—аншлаг; 5—мотор.
ние И и прикрепляют его шпеньком. Остальные аншлаги оста-
вляются в положении Б. Вагонетка своим роликом 3 упирается
.в выступающий конец аншлага 4, поднимается и приводит в дви-
Рис. 58. Подача колчедана транспортером:
1—элеватор; 2—транспортер; 3—бункер печи; 4—сбрасывающая тележка;
5—питатель.
жение систему рычагов^ открывающих брковые стенки ваго-
нетки. Через образующиеся отверстия колчедан ссыпается в бун-
кер печи.
138
Наиболее спокойная и равномерная работа достигается по-
дачей колчедана на печь транспортером (рис. 58).
Во избежание распыления колчедана скорость ленты транс-
портера не должна превышать 0,5 м/сек для флотационного кол-
чедана и 1 м/сек — для кускового.
Из склада колчедан подается в элеватор /. Последний под-
нимает колчедан и разгружает его в питательный бункерок 5
транспортера 2. Транспортер снабжен подвижной сбрасываю-
щей тележкой 4, которая дает возможность разгрузки колче-
дана в бункер 3 любой печи.
Элеваторы для колчедана работают в весьма неблагоприят-
ных условиях. Вследствие большой твердости колчедана цепи
быстро истираются и часто рвутся. При выборе элеваторов не-
обходимо обратить особое внимание на солидность их конструк-
ции. Толщина железа цепей должна быть минимум 19—20 мм
(%"). Особые требования должны быть предъявлены к элева-
тору при применении его для работы на флотационном колче-
дане или на огарке. Для этих материалов ленточные элеваторы
работают лучше цепных. Во избежание пыления необходимо вы-
бирать элеваторы тихоходные со скоростью 0,6—1,25 м/сек.
В некоторых случаях заменяют элеваторы наклонными транс-
портерами. Угол наклона транспортеров берут от 15 до 18°
к горизонту. Этим достигается более обеспеченная подача кол-
чедана из склада на верхнюю площадку печного отделения. На-
клонный транспортер требует, однако, весьма длинных эстакад:
при высоте течки транспортера над печами, равной 10 м, длина
эстакады от склада должна быть 37,3 м при угле наклона в 15°
и 30 м при угле наклоне в 18°.
2. Выгрузка и удаление огарка
Большинство1 заводов, работающих на механических печах,
до сих пор вывозят огарок вручную. Для этого в кабину под
печью под выгрузочной течкой ставится вагонетка, которая по
наполнении по узкой колее вручную или лошадьми отвозится
на свалку.
Выгрузка огарка вручную — весьма трудоемкая и очень тя-
желая работа. В связи с ростом интенсивности работы печей
ручная отвозка огарка часто не обеспечивает своевременного от-
воза огарка из печей. Уже это одно диктует необходимость
механизации этого процесса.
На ряде заводов, при сохранении приема огарка из печи
в вагонетку осуществлен тот или иной способ механизации даль-
нейшего движения вагонетки к месту свалки. Так, например,
на одних заводах выдвигаемые вручную на продольный путь
наполненные вагонетки сцепляются по 5—6 шт. и отвозятся м о-
товозом на свалку. Там они вручную опрокидываются.
На других заводах устанавливаемые на продольном пути на-
полненные вагонетки захватываются канатом лебедки и затя-
139
гиваются на верхнюю площадку, находящуюся над уровнем
железнодорожной платформы. Там они вручную опрокидываются
и разгружаются в ж.-д. вагоны. По железной дороге огарок
отвозится на свалку или к месту использования.
Более совершенным способом удаления огарка является раз-
грузка вагонеток в бункер (рис. 59). Вагонетки с огарком вруч-
ную подаются к подъемнику-лифту, который поднимает их на
верх бункера, расположенного над железнодорожной линией.
Вагонетки снабжены нижней разгрузкой, что уменьшает пыле-
ние во время спуска материала в бункер. Из бункера материал
спускается в железнодорожную
платформу. Во избежание пыления
во время загрузки ж.-д. вагонов
к течке бункера присоединяется
шнек-увлажнитель. Огарок, проходя
шнек, увлажняется до содержания
3—5% влаги и, попадая в таком
виде на платформу, не пылит. Но
все указанные способы, и даже
последний, еще в значительной части
требуют применения ручного труда..
Полное разрешение вопроса будет
лишь при том условии, если про-
цесс удаления огарка будет пол-
ностью автоматизирован, начиная
с отбора огарка из печи и кончая
его погрузкой в вагон или достав-
кой на место свалки.
Применение ленточных транспор-
теров в данном случае затрудняется
тем, что имеющиеся сейчас транспортерные ленты нормально
допускаются к эксплоатации для материала, температура кото-
рого не превышает 130—150°.
Из полностью автоматизированных способов удаления огарка
применяются или же находятся в процессе освоения гидравли-
ческий и пневматический способы, а также удаление огарка при
помощи трясучего лотка.
Трясучий лоток, или рина (рис. 60), механизирует раз-
грузку огарка внутри печного отделения: он устанавливается
в канале между печами и к нему подводятся течки для огарка
из-под печей. Рина подает огарок к элеватору, установленному
в торец печного отделения. Последний поднимает огарок к бун-
керу, расположенному вне печного отделения. Лоток перекрыт
и из него сделан отсос, вследствие чего во время работы лотка
особого пыления не замечается. При работе лотка без отсоса
он сильно пылит и обслуживание его становится невозможным.
Механизация разгрузки огарка на более далекие расстояния
осуществляется скребками с бесконечным канатом
или скреппером. Этими методами удается механизировать
140
разгрузку огарка от печей до места свалки. Однако большого
распространения эти методы не получили, так как абразивность
материала приводит к весьма быстрому истиранию цепей и ка-
Рис. 60. Трясучий лоток.
Рис. 61. Гидравлическое удале-
ние огарка:
/—течка; 2—труба; 3— сопло; 4—
бункер печи.
Гидравлический способ аналогичен выгрузке золы
из топок котлов1. Огарок из бункера печи 4 ссыпается в течку 1
(рис. 61) и оттуда через решетку (отделяющую крупные куски)
Рис. 62. Пневматическая установка:
1—компрессор; 2—рессивер; 3— воздухопровод; 4—приемный бункер для огарка-5—клапан-
6—течка; 7—трубопровод.
попадает в трубу 2 к устью сопла з. Сильная струя воды из
сопла увлекает с собой огарок, одновременно заливая его. Рас-
ход воды предполагается 7—12 т на 1 т огарка.
141
Этот метод весьма компактный и гигиеничный. Затруднение
вызывает лишь вопрос об удалении смеси (воды с огарком)
ввиду ее больших объемов. Естественные отстойники для оса-
ждения огарка потребуют громадных площадок. В настоящее
время изучается вопрос о принудительном осаждении огарка
в сгустителях Дорра.
Весьма заманчив для разгрузки огарка пневматиче-
ский метод. Пневматические установки для разгрузки мате-
риала распространены в цементной промышленности, для раз-
грузки зерна и т. д.
Для определения основных параметров и работы пневматики
для разгрузки огарка была сделана опытная установка (рис. 62).
Установка состоит из компрессора /, рессивера 2, воздухопро-
вода з, приемного бункера для огарка 4, клапана 5, течки 6 и
трубопровода 7 для аэросмеси.
Установка работает периодически. Огарок из печи поступает
непосредственно в приемный бункер 4. По наполнении бункер
отключается от печи клапаном 5. После этого открывают пу-
сковой клапан и пускают сжатый воздух в низ бункера. При
определенной концентрации смеси и скорости воздуха в трубо-
проводе огарок из бункера подхватывается и транспортируется
во взвешенном состоянии по трубопроводу 7 к месту свалки.
Во избежание пыления в конце трубопровода огарок смачи-
вается водой.
Испытание установки для пневматической разгрузки огарка
дало следующие результаты:
Концентрация смеси при расстоянии от печей до места
свалки 60 л и высоте подъема 10 м............11,1—24,5 кг/кг
Скорость воздуха в начале трубопровода . w . . . 10 —14,8 м1сек
Скорость воздуха в конце трубопровода ск........... 26,7—29,3
при этом давление воздуха в начале трубопровода колебалось,
от 1,3 до 2,2 ати.
Для регулярной работы установки необходимо, чтобы трубо-
провод для аэросмеси был смонтирован из толстостенных труб
(7—9 мм), так как трубы со стенками в 4 мм быстро истираются
и выходят из строя. Особенно подвергаются истиранию колена
и места соединения труб на фланцах. Ввиду этого колена
должны быть сделаны с большим радиусом закругления и из:
твердого материала (чугун, диабаз и т. п.); фланцевые соеди-
нения должны быть сделаны весьма тщательно так, чтобы торцы
соединяемых труб не выступали.
IV. Газоходы печных отделений
Головная часть систем и контактной и башенной работает
под разрежением. Поддержание постоянной величины разреже-
ния (тяги) является условием нормальной работы системы.
Часто величина разрежения уменьшается вследствие засоре-
ния газоходов, идущих от печного отделения к электрофильтрам.
142
При устройстве горизонтальных газоходов между печами и от
печей к электрофильтрам пыль осаждается внутри газоходов,,
часто закладывая сечение газохода на 30% и больше, что уве-
личивает сопротивление системы.
Для удобства очистки газоходов в них устраиваются течки
с затворами. Затворы периодически открываются и накопившая-
ся над ними пыль высыпается. Однако на горизонтальном
участке между бункерами пыль остается лежать под углом есте-
ственного откоса. Для удаления остающегося бугорка огарка
необходимо, чтобы расстояние между течками было возможно
меньше.
Практически течки располагаются через 1,5 м. Для уста-
новки, состоящей из 12 печей, число течек газоходов составляет
Рис. 63. Кривули.
48. Разгрузка огарка из такого большого количества течек —
весьма трудная и негигиеничная работа. В новейших установках
печных отделений горизонтальные газоходы заменены кривулями
(рис. 63). Газоходы от печей делаются под углом 50—60° к го-
ризонту и соединяются с пыльниками. Такой же конструкции
делаются газоходы и между печами, а также и от печей к эле-
ктрофильтрам. При такой конструкции число течек для разгрузки
огарка резко уменьшается, что позволяет чаще очищать газо-
ходы и этим избегать ухудшения работы системы.
V. Использование огарка
1. Извлечение меди
При сжигании колчедана на сернокислотных заводах полу-
чаются огромные количества огарка.
Огарок до настоящего времени является тяжелой обузой хи-
мических заводов, где есть сернокислотные цехи, так как его от-
валы отнимают значительную часть территории заводов. Между
тем, огарок заключает в себе ценные с народнохозяйственной
точки зрения компоненты.
Так, запасы огарка по шести заводам (Щелковскому, Воскре-
сенскому, Войковскому, Невскому, «Красному химику» и Ста-
143
линогорскому) в среднем содержат железа 47,3%, меди 0,59%,
серы 3,45%, серебра и золота небольшие следы. Полное исполь-
зование огарка предполагает его направление на выплавку чу-
гуна. Однако при известной величине содержания в нем цвет-
ных металлов их целесообразно из огарка извлечь до направле-
ния его на металлургическую плавку. Если огарок содержит не
менее 0,75% меди, целесообразно применять сернокислотную экс-
тракцию для извлечения меди. Если наряду с указанным содер-
жанием меди в огарке имеется золота не менее 0,6 г/т, целесооб-
разно применять хлорирующий обжиг огарка, после которого
из огарка выщелачивается не только медь, но также золото и
серебро.
Хлорирующий обжиг огарка заключается в следующем.
Огарок после предварительного измельчения смешивают
с определенным количеством поваренной соли и обжигают
в этажной печи при температуре 450—500°, при этом медь и
благородные металлы переходят в растворимые соединения.
В огарке для обжига должно быть серы не меньше как из
расчета 2—3 атома ее на атом меди, в противном случае к шихте
добавляется соответствующее количество колчедана.
Температура в печи поддерживается генераторным или до-
менным газом.
Выходящие из печи газы содержат НС1, SO3, SO2 и С1г; при
промывке этих газов водой получается слабый раствор смеси
серной и соляной кислот.
Обожженные огарки выщелачиваются в больших железобе-
тонных изнутри покрытых кислотоупорной футеровкой ящиках
•с ложным дном в виде деревянной решетки, на которой находится
слой гравия. Из огарка, подвергнутого хлорирующему обжигу, вы-
щелачивается ~ 95% меди и до 90% золота и серебра. Выще-
лачивание производится смесью серной и соляной кислот, полу-
ченной при улавливании отходящего газа хлорирующего об-
жига. Выщелоченный огарок идет далее на металлургическую
плавку.
Степень извлечения меди из огарка повышается, если выще-
лачивание производится при продувке воздухом.
Степень извлечения меди при выщелачивании достигает 90%, если ога-
рок перед выщелачиванием подвергнуть так называемому сульфатизируюшему
обжигу. Этот процесс состоит в том, что огарок подвергается обжигу
в этажных печах при температуре 550—600°, при этом газы, подаваемые
в печь, содержат SO».
При сульфатнзирующем обжиге происходят следующие реакции:
Cu2S + 202 = 2CuO + SO2;
2SO2 + Оа = 2SOs;
CuO + SOs = CuSOj.
Окисление SOa происходит ври каталитическом содействии окислов же-
леза, присутствующих в шихте.
144
Те огарки, в которых содержание меди меньше 0,75%,
а также и те, из которых медь простым выщелачиванием извле-
чена не полностью, могут быть направлены на выплавку меди-
стого чугуна. Медистые чугуны и стали обладают особыми анти-
коррозийными свойствами и широко применяются в ряде отрас-
лей народного хозяйства.
2. Агломерация огарка
В том состоянии, в каком огарок получается при выходе из
печей, а также после выщ1елачивания меди он не годится для
доменной плавки. Во-первых, огарок часто содержит гораздо
больше серы, чем это допустимо по требованиям доменной
плавки. Во-вторых, по самому физическому состоянию огарка —
мелкости и плотности частиц — он для доменной плавки не
годится.
Подготовка огарка для доменной плавки заключается в его
агломерации. В процессе агломерации из огарка выжигается
сера и огарок превращается в пористые куски.
Для спекания или агломерации огарка применяются, главным
образом, аппараты Двайт-Ллойда. Спекание может произво-
диться и во вращающихся трубчатых печах. В последние годы
ведутся работы по изменению процесса обжига флотационного
колчедана во взвешенном состоянии таким образом, чтобы ога-
рок сразу получался в виде агломерата.
Аппараты Двайт-Ллойда имеются двух типов — ленточные и
круглые.
До поступления на аппарат огарок (или руда) предварительно
смешивается с топливом и смачивается водой приблизительно до
15% влагосодержания. В том случае, когда сама руда содержит
горючее, добавлять топлива не требуется. При спекании огарка
вместо угля можно применять колчедан, смешивая его с огар-
ком. Общий процент серы в смеси в отдельных установках
колеблется от 8 до 15. Понятно, что такого рода материал —
сырье для двайта — можно получить иначе: не дожигая кол-
чедан в тех или иных колчеданных печах.
Как выше уже говорилось, на агломерационной машине на
ряде заводов проводится окончательный дожиг цинковой об-
манки (после механической печи).
По своей конструкции круглый аппарат Двайта представляет собой гори-
зонтально расположенное кольцо 1 (рис. 64), верхняя часть которого состоит
из колосников 2, расположенных по радиусу кольца. Под колосниками про-
ходит кольцевая камера 14, с дверцами но периферии для очистки. Из
этой кольцевой камеры к центру печи ведут горизонтальные трубы 4- Вся
кольцевая камера 14 может вращаться вокруг своего центра, она разделена
вертикальными перегородками з. Ролики ц .в нижней части камеры по-
коятся на кольцевых рельсах 12. Под камерой 14 у ее периферии имеются
зубцы 13, которые находятся в постоянном сцеплении с шестерней.
Для питания печи смесью служит бункер е, оканчивающийся продольной
щелью, через которую смесь поступает на колосники. Топка для разжига-
ния ~ работает иа жидком или газообразном топливе.
10 Зак. 50J9. Технология серной кислоты.
145
При вращении кольцевой части печи под отверстиями бункера 6 смесь
распределяется по решетке ровным слоем ю. При дальнейшем вращении
кольца смесь подходит к месту, где находится топка 7, воспламеняющая
руду. Продукты горения (SO2, N2, О2) вместе с продуктами горения то-
плива просасываются через колосниковую решетку в камеру 14, а из нее по
трубам 4 поступают в центральный сборный коллектор 5, соединенный
с двумя эксгаустерами.
Рис. 64. Конструкция круглого аппарата Двайта:
1—кольцо; 2—колосники; 3—перегородка; 4—горизонтальные
трубы; 5—сборный коллектор; б—бункер; 7—топка; 8—приспо-
собление для удаления огарка; В—вагонетка; 10—слой
огарка; 11—ролики; 12—рельсы; 13—зубцы; 14—кольцевая
камера.
Горение смеси, а затем ее остывание совершается на колосниках, которые
не имеют никакой покрышки, т. е. горение происходит иа виду. Благодаря
работе эксгаустеров газы постепенно просасываются через решетку н не
попадают в атмосферу помещения. Когда кольцо сделает примерно 5/е обо-
рота (считая от места загрузки), вся сера выгорает и образовавшийся огарок
успевает охладиться. Приспособлением 8 огарок постоянно снимается с ко-
лосниковой решетки и попадает в вагонетку 9-
Решетка движется по направлению, указанному стрелками. Если спекается
материал, богатый серой, то богатый SO2 газ отводится с небольшой пло-
146
щадки решетки, а именно от линии 1—I до линии Ц—II (через три трубы).
С площади от линии II—II до линии III—III газ вытягивается в особый кол-
лектор. Этот газ, бедный SO2, для переработки не используется.
Р .зделение газов, богатых и бедных SO2, осуществляется приспособле-
нием, показанным на рис. 65. Цилиндрический коллектор 1 может вращаться
вместе с системой труб 6.
Коллектор 1 покоится на ро-
ликах 2 и опущен в кольцевую
коробку 3, представляющую со-
бой затвор, который наполнен
песком или маслом высокой
температуры кипения. Внутри
коллектора 1 неподвижная часть
4 представляет собой коробку
с горизонтальной щелью 5. Эта
коробка соединяется только с
тремя трубами в. Все же осталь-
ные трубы находятся в постоян-
ном соединении с коллекто-
ром 1.
При вращении колосниковой
решетки всегда три трубы 6 сое-
динены с коробкой 4, а осталь-
ные с коллектором у. Коробка 4
неподвижна и всегда высасывает
газы, образованные на колосни-
ковой решетке между линиями
1—1 и II—II (рис. 64). Из ко-
робки 4 богатые S02 газы всасы-
ваются эксгаустером в сернокис-
лотную систему (рис. 65).
Все трубы 6 вставлены! в
кольцо 7, плотно соприкасаю-
щееся с коробкой 4. Ввиду зна-
чительной скорости газов в тру-
бах пыли осаждается очень мало.
Рис. 65. Детали круглого аппарата
Двайта:
7—коллектор; 2—ролики; 3—кольцевая коробка;
4—неподвижная коробка; 5—щель; 6—трубы;
7 — кольцо..
В наших условиях агло-
мерацию огарка целесообраз-
но производить на крупных
металлургических заводах.
При этом часть огарка
(богатая медью и благород-
ными металлами) должна
предварительно проходить
хлорирующий обжиг и выщелачивание, часть (богатая только
медью) может проходить перед спеканием только выщелачива-
ние (меди), а часть — прямо поступать на спекание.
Широкое использование колчеданного огарка для извлечения
меди, благородных металлов и для металлургической плавки —
задача ближайшего времени.
Большой интерес представляет вопрос о возможности совме-
щения процесса обжига флотационного колчедана с процессом
агломерации получающегося огарка. В установках Сен-Жан для
агломерации пылевидных руд во взвешенном состоянии (рис. 66)
материал вместе с воздухом вдувается в печи 1 через форсунку 2
турбулентного типа. Через фурмы, расположенные на различ-
10»
147
ной высоте печи, вводится дополнительный воздух. Нагрев
печи и поддержание в ней необходимой температуры произво-
дится двумя горелками, расположенными в нижней части печи.
В качестве топлива применяется угольная пыль, жидкое топливо
и генераторный газ. Воздух перед входом в печь подогревается
отходящими газами в воздушном теплообменнике 3 до 350°.
Газ выходит из печи через внутренний корпус 4 форсунки
и, пройдя через теплообменник 3 и циклон 5, выбрасывается
в атмосферу. Готовый агломерат выгружается при помощи вра-
щающегося пода.
Понятно, что при сов-
мещении процессов об-
жига флотационного кол-
чедана и агломерации по-
лучающегося огарка не бу-
дет надобности ввода в
печь дополнительного топ-
лива, так как необходи-
мая для агломерации тем-
пература (1100—1150°)бу-
дет достигнута за счет
горения колчедана и по-
догрева поступающего в
печь воздуха.
3. Другие возможности
использования огарка
Рис. 66. Установка для агломерации руд
во взвешенном состоянии:
7 — агломерационная печь; 2 — форсунка; 3— воз-
душный теплообменник; 4—выход газа; 5 — циклон;
6 — бункер.
Небольшие количества
колчеданного огарка мо-
гут быть использованы
для производства мине-
ральных красок — сурика и
мумии. В этом случае огарок обрабатывается при нагревании
серной кислотой, а затем полученный сульфат окиси железа
с некоторыми добавками (мел, алебастр, глина) обжигается.
В зависимости от температуры обжига и взятой добавки полу-
чается тот или иной оттенок краски.
Как показали опыты НИУИФ, колчеданные огарки, содер-
жащие медь, с успехом могут быть применены в качестве удоб-
рения вместо медного купороса для осушаемых болотистых мест.
Огарок можно применять в качестве полуторного окисла при со-
ставлении шихты в производстве цемента.
VI. Очистка печного газа от пыли
При сжигании колчедана с печными газами уносится часть
огарка в виде мельчайшей пыли. Еще при дроблении колчедана
получается значительное количество мелких частиц, которые при
148
прохождении в печи захватываются потоками газов. Этот про-
цесс особенно усиливается при пересечении газового потока пе-
ресыпающимся с полки на полку колчеданом и при вращении
гребков. Пыль образуется также при распадении крупных ку-
сочков колчедана под влиянием горения и быстрого нагревания.
При конструировании механических печей иногда избегают?
пересечения газового потока пересыпающимся колчеданом. Когда
газы и колчедан проходят через одни и те же междуэтажные
отверстия (печь ВХЗ), степень запыленности газов тем больше,
чем больше скорость газов. При сжигании рядового колчедана
в механических печах печные газы обычно содержат 3—5 г
пыли на 1 норм. л?3. При добавке к обыкновенному колчедану
флотационного содержание пыли в газе увеличивается. Осо-
бенно велико оно при сжигании флотационного колчедана в пы-
левидном состоянии (до 70—100 г на 1 норм. м3).
По химическому составу пыль, уносимая печными газами, не-
сколько отличается от огарка, особенно содержанием мышьяка.
Строение пылинки зависит от природы образования пыли.
При механическом дроблении форма пылинок неправильная,
с острыми углами, почти совершенно отсутствуют шарообразные
пылинки. При сжигании же колчедана в распыленном состоянии
при высокой температуре пылинки приобретают шаровидную
форму вследствие оплавления.
Размер пылинок зависит от качества колчедана и от способа
сжигания. При сжигании рядового колчедана в механических
печах размеры основной массы пылинок 5—20 м, при сжигании
флотационного колчедана пыль получается еще более мелкая.
Освобождение газа от пыли может быть проведено двумя
способами: 1) механическим — под влиянием силы тяжести, цен-
тробежной силы и т. д. и 2) электрическим — под влиянием воз-
действия электрического поля.
1. Механические способы отделения пыли
Осаждение пыли под влиянием силы тяжести. Частицы
пыли под влиянием силы тяжести опускаются вниз. Величина
этой силы, как известно, выражается весом. Вес шаровой ча-
4
стицы равен -у тгг3у. Сопротивление среды при движении лю-
бого тела зависит от поверхности тела (точнее от величины
проекции тела на плоскость, перпендикулярную направлению дви-
жения), от скорости движения тела, от вязкости и плотности
среды. Величина этого сопротивления выражается следующей
общей формулой:
* = (1)
где F —упомянутая выше проекция, у — удельный вес среды,
vvn—относительная скорость тела, g— ускорение силы тяже-
149
сти, <р — коэфициент пропорциональности. Коэфициент пропор-
циональности в свою очередь зависит от ряда факторов — от
вязкости и плотности среды и от размеров и скорости тела;
в общем виде эта зависимость выражается уравнением:
где d — диаметр, — вязкость.
Для шаровых частиц с диаметром 1—100 у формула (2) по
Стоксу может быть выражена так:
। 24
Л —------л
Для мелких частиц формула (1) примет вид:
bFwa\
Q = ----- в
Г
(3)
(4)
Если принять, что частицы имеют форму шара, то F — пг2 и
уравнение (4) примет вид:
о = 6~г®уптг (5)
Из этой формулы видно, что с увеличением скорости дви-
жения тела увеличивается сопротивление среды. Отсюда сле-
дует, что большие скорости осаждения очень трудно дости-
жимы, так как при этом развивается большое сопротивление.
Кроме того, в случае, когда пыль состоит из мелких частиц и,
следовательно, сила притяжения к земле невелика, может быстро
наступить такой момент, при котором сила сопротивления будет
уравновешивать силу притяжения; тогда пылинка будет далее
двигаться с постоянной скоростью.
Определим эту постоянную скорость. Приравняем силу тя-
жести силе сопротивления, получим:
4
— п/-3у = 6-ГГ^пТр
откуда 1
(6)
Из этого уравнения видно, что с уменьшением радиуса ча-
стицы скорость ее падения уменьшается в квадрате. Для того
чтобы частица осела в данном газоходе или в какой-либо отстой-
ной камере, нужно, чтобы она, падая с некоторою скоростью,
определяемой но уравнению (6), успела дойти до дна газохода
за время, пока она в .месте с газами проходит этот газоход.
Обозначим через Л — высоту газохода, b — ширину газохода,
t — время, в которое газ успеет пройти данный газоход, 1 —
длину газохода, V — объем газа, проходящий через газоход за
150
одну секунду, н7 — скорость газа, vvn — скорость падения пы-
линки.
Время падения пылинки, которая при входе в газоход была
на самом верху его, равно . Секундный объем газа равен
V = wbh, но w=-~, следовательно V — Для того чтобы
пылинка успела осесть, время ее падения должно быть равно
или меньше времени пребывания газа в газоходе, т. е. t Э=—, и,
следовательно,
но 1Ь — площадь пола газохода, а V — секундный объем газа
или секундная производительность камеры.
Таким образом производительность камеры или газохода,
т. е. количество газа, которое можно очистить от пылинок круп-
ностью в г, зависит только от площади пола и от скорости па-
дения этих пылинок и не зависит от формы газохода. Стенки
внутри камеры могут способствовать обеспыливанию газа только
в том случае, если газ ударяется о стенку, пылинки тогда те-
ряют свою скорость и выпадают из газового потока. В особен-
ности это явление важно в тех случаях, когда стенка, о кото-
рую ударяется пыль, влажная.
На многих старых сернокислотных заводах употреблялись
камеры Говарда, в которых площадь пола искусственно увели-
чена устройством дополнительных горизонтально расположенных
полок. При этом объем камеры уменьшен очень незначительно,
а площадь пола увеличена в несколько раз.
Пользуясь формулами (6) и (7), можно подсчитать, какой раз-
мер должна иметь камера, чтобы в ней могли осесть все пы-
линки крупнее определенного заданного размера, или, наоборот,
подсчитать, какой крупности пылинки осядут в данной камере-
газоходе, а также процент очистки газа в данной камере.
Разделим газовый поток на несколько слоев и выберем на высоте z от
дна газохода тонкий слой газа. Для того чтобы пылинки из этого слоя
успели осесть в газоходе, необходимо, чтобы время их падения было меньше,
чем время пребывания газа в газоходе, т. е.:
/ — _/= —
«пад— Wn w
где / — длина газохода, a w— скорость газа в газоходе.
Подставим в это неравенство значение wn из формулы (6), получим:
2Т/
(6а)
Все частицы большего размера осядут, а меньшего — будут унесены.
Зная состав пыли по крупности, легко подсчитать, воспользовавшись фор-
151
Рис. 67. Циклон
с улиткой.
мулой (6а), какая часть пыли осядет из каждого слоя, иначе говоря, найти
частные коэфициеиты очистки для каждого слоя.
Для нахождения общего коэфициеита очистки нужно знать среднее зна-
чение частных величин. Наиболее точно эти величины можно найти графи-
ческим методом, для чего по оси абсцисс отложить в выбранном масштабе
общую высоту газохода и выбранные высоты отдельных слоев, а по оси
ординат — частные коэфициенты очистки, соответствующие выбранным слоям.
Через найденные точки провести плавную кривую и подсчитать полученную
площадь. Найденная площадь представляет собой сумму произведений част-
ных коэфициентов очистки на соответствующие высоты слоев. Если эту
площадь разделить на общую высоту газохода, то полученное значение будет
соответствовать среднему коэфициенту очистки газа от пыли.
Циклоны и мультиклоны. В отличие от пыльных камер
в аппаратах, называемых циклонами, для осаждения пыли ис-
пользуется центробежная сила. Простейший аппарат этого типа
состоит из двух цилиндров, вставленных один
в другой; к нижней части большого цилиндра
присоединен конус-бункер. В промежуток между
цилиндрами в тангенциальном направлении вво-
дится газ.
Газ и пылинки в пространстве между цилин-
драми движутся по окружности, а поэтому ис-
пытывают влияние центробежной силы, которая
действует сильнее на пылинки, так как их удель-
ный вес значительно больше удельного веса газа.
Пылинки отбрасываются из газового потока
к стенкам наружного цилиндра, оседают на нем
и затем под влиянием силы тяжести падают
в бункер. Газ же, освобожденный от пыли, вы-
ходит через внутренний цилиндр.
В описанном простейшем типе циклонов про-
исходит столкновение различных струй газов
(вновь входящих и вращающихся внутри аппаратов), а поэтому
образуются вихри, мешающие осаждению пыли и увеличиваю-
щие сопротивление циклонов. Во избежание этих явлений при-
меняют другой вид циклонов, например в виде улитки
(рис. 67).
С целью увеличения поверхности осаждения, что имеет боль-
шое значение, улитку делают с несколькими оборотами.
Выясним, какие факторы влияют на работу циклонов. Обо-
значим через V — секундный объем газа в м^/сек, w — среднюю
скорость в циклоне в м/сек, vvn —среднюю скорость движения
пыли под влиянием центробежной силы по направлению к оса-
дительной поверхности в м/сек, b — ширину входного отверстия
или наибольшее расстояние от внутреннего до наружного цилин-
дров, h — высоту цилиндра, Rt — средний радиус циклона, t —
время пребывания газа в циклоне.
площадь, по которой движется газ со
время пребывания газа в
Произведение bh есть
скоростью w, поэтому
bhv.' V,
(8)
152
но путь газа в циклоне равен 2п/?т, где п — число кругов (обо-
ротов), проходимых газами. Следовательно, скорость газа:
= (9>
Подставляя найденное значение для скорости газа в фор-
мулу (8), получим:
' (10)
Время, потребное для осаждения пылийки, должно быть
равно или меньше времени пребывания газа в циклоне. Время
для осаждения пылинки, находящейся в момент входа в циклон
около внутреннего цилиндра, будет самым большим, так как эта
пылинка более всех удалена от поверхности осаждения, т. е. от
поверхности наружного цилиндра. Эта пылинка должна пройти
путь, равный Ь, в направлении радиуса со скоростью wn за*
время t, т. е. b = wn t или ( = — . Подставив эту величину'
в формулу (10), получим:
V = —? = 2*Rtnh • w„. (11)
Но величина 2-Ri • п • h есть площадь осаждения пыли; если
обозначим ее буквой S, то получим, что:
V=Swn, (12)
т. е. формулу, вполне аналогичную формуле для пыльных ка-
мер. Из этой формулы следует, что производительность цикло-
нов зависит от поверхности улитки и от скорости осаждения
пыли, которую пылинки получают от действия центробежной
силы.
Чтобы найти эту скорость, определим действующие на пы-
линку центробежную силу и силу сопротивления среды. Цен-
- ты2
тробежная сила, как известно, равна —ъ—, где т — масса пы-
линки. Сопротивление среды по закону Стокса равно 6^гщпт(,
где г — радиус пылинки, т; — вязкость газа. Когда пылинки мел-
кие, то скорость их быстро становится настолько большой, что
обе эти силы уравновешиваются и с этого момента пылинки бу-
дут двигаться с постоянной скоростью. Следовательно
mw2
-^- = 6-rWn7],
откуда определим эту скорость:
mW2
Иц = ДЬ---•
153
Масса шаровой пылинки равна у л г5 где у — удельный
вес пылинки, и следовательно:
SgRft
(13)
т. е. скорость осаждения увеличивается с увеличением радиуса
пылинки, удельного веса ее, скорости газа и с уменьшением ра-
диуса циклона. С первого взгляда может показаться, что чем
меньше циклон, тем меньше радиус и больше скорость прохо-
ждения газа в циклоне, т. е. тем больше очистка. Но это не так.
В нашем выводе мы не учитывали того, что с увеличением ско-
рости газа увеличивается унос уже осевшей пыли с поверхно-
сти осаждения. Поэтому существует некоторый оптимум ско-
рости для каждого отдельного случая. Кроме того, с увеличе-
нием скорости сильно возрастает сопротивление циклона. Осо-
бенно сильно возрастает унос в случае мелкой пыли; поэтому
для очистки газа от мелкой пыли (30 и ниже) циклоны упо-
треблять не следует. Нормально скорость газа в циклоне
7—20 м./сек.
Применяя формулы (12) и (13), можно определить необхо-
димую величину аппарата (определить S), если известно, какой
величины пылинки нужно осадить, или, зная размеры аппаратов,
можно определить предельную величину пылинок, которые
осядут (без учета уноса, о котором мы говорили выше).
Для определения процента очистки газа нужно составить формулу для
«аименьшего размера пылинок, которые будут оседать в циклоне, если они
яри входе в аппарат находятся на расстоянии х от внешнего цилиндра.
Из уравнения (13) имеем:
_ 2/-2yw2 dR
W" ~ SgRrt — dt ’
Разделим переменные:
9^i
dt = RvdR,
интегрируем в пределах от 0 до t, заметив, что при t = 0 R — х = рас-
стояние пылинок от центра, а при выходе R = Ri = радиусу внешнего ци-
линдра, тогда:
2r?yw2
/=±(/?;-х2).
Заметив, что t = —-— , получим:
W
rmin
9Дг, — .У') w
4yw’2 • 2r.Rn
(14)
где rnlin — наименьший радиус частиц, при котором еще происходит осажде-
ние. Процент очистки можно определить аналогично вышеописанному на
стр. 151, т. е., разбив входной патрубок на ряд слоев, находящихся иа рав-
ных расстояниях х от центра, найти крупность пылииок, которые осядут из
154
каждого слоя; зная состав пыли по крупности, найти частные коэфициеиты
очистки для каждого слоя и, наконец, графическим методом определить
общий коэфициент очистки.
В последние годы получили большое распространение муль-
тиклоны или циклоны батарейного типа (рис. 68). Эти аппараты,
устроенные на принципе циклонов, обеспе-
чивают относительно высокий процент ула-
вливания пылинок. Они представляют собой
группы-батареи отдельных циклонов ма-
лого диаметра с сильно развитыми удлинен-
ными коническими частями, которые окан-
чиваются общими или отдельными бунке-
рами. Небольшой диаметр, как это видно из
уравнения (13) или (14), способствует хоро-
шему отделению пылинок; удлинение кони-
ческой части дает возможность образовать
устойчивое вращательное движение и уве-
личивает площадь оседания пыли; нали-
чие же отдельного специального бункера
для пыли способствует ее хорошему отде-
лению и затрудняет обратный ее унос.
Газ в мультиклон подается сверху. Для
придания ему вращательного движения
в верхней части устанавливается специаль-
ная розетка, закрепленная между наружным
кожухом и внутренней выхлопной трубой.
Степень очистки в мультиклоне в силь-
ной степени зависит от крупности пыли; по
испытаниям опытных аппаратов на серно-
кислотных заводах она достигает ~ 70%.
Мультиклоны применяются в качестве I сту-
пени очистки, например в контактной си-
Piic. 68. Мультиклон.
стеме Кемико, где остальная пыль улавли-
вается в 1-й безнасадочной промывной башне и в дальнейшем
удаляется из кислоты в отстойнике.
2. Электрическая очистка газа
Теоретические основы. Электрическая очистка газов при-
обрела повсеместное распространение во всех отраслях про-
мышленности и в первую очередь — в химической.
Разберем элементарную теорию этого метода.
Если мы имеем две параллельные металлические пластинки
на расстоянии d друг от друга, противоположно заряженные, то
напряжение поля в любой точке между пластинками будет
равно:
(15)
где V — разности потенциалов между пластинами. »
155
Возьмем теперь цилиндрический конденсатор, т. е. полую ме-
таллическую трубу радиусом R, в центре ее поместим металли-
ческий провод с радиусом г и сообщим заряды -f-Q и —Q на-
ружному цилиндру и проводу.
В этом случае напряжение в любой точке внутри цилиндра
может быть выражено уравнением:
V
хшА’
(16)
где R — радиус цилиндра, г — радиус провода, х — расстояние
данной точки от провода. В отличие от случая плоского конден-
сатора напряжение поля здесь изменяется по сложному закону.
Теперь проведем следующий опыт.
Возьмем плоский конденсатор и
включим его в цепь с источником J
электричества, который может да-
вать различное напряжение конден-
сатору; в ту же цепь включим чув- I
ствительный амперметр (рис. 69) и |
Рнс. 69. Плоский конден-
сатор в цепи с ампер-
метром.
Рис. 70. Ток насыщения
в плоском конденсаторе.
будем постепенно поднимать напряжение, следя за амперметром.
Вначале с повышением напряжения сила тока постепенно увели-
чивается почти пропорционально, затем это увеличение силы
тока замедляется и, наконец, совсем прекращается, несмотря на
увеличение напряжения. В этот момент говорят, что достигнут
ток насыщения. Если напряжение поднять очень сильно,
например до 20 000 в, то между пластинами произойдет пробой
и амперметр покажет очень большую силу тока (рис. 70). Весь
этот процесс можно объяснить следующим образом.
Воздух, находящийся между пластинами конденсатора,
является изолятором, но под влиянием ультрафиолетовых лучей,
радиоактивности и т. д. в этом воздухе все время образуется
некоторое количество ионов. Эти ионы при столкновении друг
с другом могут соединяться вновь в нейтральную молекулу —
происходит так называемая рекомбинация. При-установившемся
процессе за один и тот же промежуток времени образуется и
рекомбинируется одинаковое количество ионов, т. е. число сво-
бодных ионов в некотором 'Объеме постоянно. Под влиянием
поля конденсатора ионы движутся к пластинам и вследствие
156
этого мы наблюдаем по амперметру электрический ток. С увели-
чением напряжения поля ионы начинают двигаться быстрее и
число рекомбинаций становится меньше, т. е. большее количе-
ство ионов проходит к пластинам конденсатора — ток увеличи-
вается. По мере дальнейшего увеличения напряжения число ре-
комбинаций становится незначительным по сравнению с числом
образующихся ионов, т. е. почти все ионы, которые образуются,
участвуют в переносе зарядов в конденсаторе, а поэтому сила
тока становится максимальной и постоянной — наступает момент
насыщения.
Дальнейшее повышение напряжения поля вызывает увеличе-
ние скорости ионов, но так как уже все образовавшиеся ионы
участвуют в образовании тока, то увеличение силы тока не про-
исходит. Если же мы повысим напряжение поля еще больше, то
Рис. 71. Ток насыщения в
цилиндрическом конденса-
торе.
Рис. 72. Поле в цилиндри-
ческом конденсаторе.
может наступить такой момент, когда скорость ионов будет. на-
столько велика, что они, сталкиваясь с молекулами воздуха,
будут эти молекулы разбивать на ионы, которые в свою очередь
также приобретают ббльшую скорость и разбивают следующие
молекулы и т. д., — происходит п р о б о й.
Проведем аналогичный опыт и рассуждения в случае цилин-
дрического конденсатора. Вначале при малом напряжении явле-
ние будет сходно с первым случаем, но период насыщения бу-
дет значительно короче и вслед за ним начнется опять значи-
тельное увеличение силы тока, но не такое резкое, как при пло-
ском конденсаторе. Вначале пробоя не будет, пробой произой-
дет только при дальнейшем повышении напряжения (рис. 71).
Это легко объясняется неравномерностью поля в цилиндриче-
ском конденсаторе (рис. 72). В центре около провода густота
силовых линий, т. е. напряжение, больше, чем на периферии.
Поэтому при значительном повышении напряжения ионы, нахо-
дящиеся в воздухе вблизи центрального провода, под влиянием
большего напряжения будут двигаться с бблыней скоростью и
разбивать встречающиеся на их пути молекулы, ионизируя воз-
дух. Но по мере удаления от центра на ионы будет действовать
меньшее напряжение и их скорость будет уже недостаточна для
157
того, чтобы ионизировать встречающиеся молекулы, т. е. даль-
нейшая ионизация прекратится. Воздух на периферии останется
неионизированным, поэтому пробоя не произойдет. Такое явле-
ние, когда вблизи центрального провода происходит ионизация,
называется коронированием. Внешне корона характери-
зуется свечением и особым звуком стрекотания.
Область короны — область новообразования — становится
проводящей, и можно считать, что в цилиндрическом конденса-
торе при коронировании как бы увеличился диаметр внутреннего
провода. С увеличением напряжения увеличивается пространство,
в котором густота силовых линий еще настолько велика, что
происходит ионизация, т. е. увеличивается размер короны. Это
происходит до тех пор, пока напряжение настолько увеличится,
что произойдет пробой. Определим этот момент. Возьмем урав-
нение:
<»«>
л1п —
Так как корона представляет собой проводящий слой, то,
считая х за радиус короны, можно написать, что х — г, т. е.
Найдем значение Е„ при различных значениях х, выражен-
ных в долях R.
ПРИХ==1^ /? . = 21’7* ’
Too ,n 100
Прих = ^ Ех = 6,7^.
Прих = 4 Ех = 3,1^.
nPHX = W ^ = 2,718Х.
При х = -у £^ = 2,9 — .
Прих=^- Ех = 3,7^.
Из этих данных видно, что до значения х<=-у можно уве-
личивать размер короны, так как до этого значения х в сосед-
них, более удаленных точках Еа меньше, чем в точке х; иначе
в области, где х > Е^я^> Еж, т. е. с этого момента перифе-
рийный слой перестает быть изолятором и наступает пробой.
Следовательно, это является пределом величины короны.
158
Из этих же данных видно, что до некоторого предела с уве-
личением толщины провода падает напряжение, т. е., для того
чтобы вблизи провода получить пробойное напряжение — корону,
нужно с увеличением толщины провода повышать и разность
потенциалов. Поэтому в электрофильтрах ставят возможно более
тонкие провода, так как на них легче получить сильную корону.
Однако чем тоньше провод, тем большее напряжение поля
нужно создать у поверхности проводника для того, чтобы около
него образовалась корона. Это можно объяснить так.
Для образования короны нужно, чтобы ионы приобрели не-
которую скорость, достаточную для разбивания встречающихся
молекул на ионы. Для создания этой скорости необходимо дей-
ствие некоторой наименьшей средней силы поля на определен-
ной, так называемой свободной длине пути.
Рис. 73. Поле близ, тонкого и
толстого провода.
Рис. 74. Поле в цилиндрическом
конденсаторе с короной.
На рис. 73 изображены два провода (тонкий и толстый); на;
поверхности их взято одинаковое напряжение (одинаковая плот-
ность силовых линий) и силовые линии продлены на отрезке /,.
равном длине свободного пути. Из рисунка видно, что около
толстого провода на этом отрезке силового поля средняя плот-
ность силовых линий больше, чем около тонкого; следовательно,
несмотря на одинаковое напряжение поля, у самого провода про-
бивное напряжение у толстого провода будет меньше, чем у тон-
кого.
После того как в цилиндрическом конденсаторе около про-
вода образуется корона, начинается ударная ионизация; вновь
образовавшиеся ионы начинают двигаться под влиянием поля,
при этом в области короны будут ионы обоих знаков, в осталь-
ном же пространстве — только ионы, которые отталкиваются от
коронирующего провода, т. е. заряженные с ним одноименно.
На рис. 74 изображено поле цилиндрического конденсатора,
когда около провода имеется корона (на рисунке область короны
заштрихована). Наличие ионов в области, не занятой короной,
сильно увеличивает число силовых линий в этом пространстве,.
159
т. е. и здесь напряжение поля повышается и может произойти
пробой. Этим объясняется тот факт, что практически размер
короны (диаметр ее) бывает очень невелик, в то время как по
подсчету, приведенному выше, она может занимать довольно
значительное пространство.
Если в цилиндрический конденсатор, в котором образовалась
корона, впустить запыленный газ, то пылинки заряжаются. Под
влиянием электрического поля пылинка испытывает диэлектри-
ческую поляризацию, т. е. отдельные составляющие ее ионы
и электроны несколько сдвигаются, стараясь встать по напра-
влению электрического поля.
Благодаря этому около пы-
линки как бы возникает свое
поле (или искривляется поле
конденсатора). Напряжение
этого поля равно
Рис. 75. Силовые линии при заряже-
нии пыли.
где Ех — напряжение поля кон-
денсатора, вызвавшее поляри-
зацию, е — диэлектрическая
постоянная пылинки. Под влия-
нием этого поля ионы притяги-
ваются к пылинке с силой,
равной:
^(1 + 2Т+г) е>
где е — заряд иона, равный
заряду электрона.
По мере накопления на пылинке зарядов увеличивается куло-
новская сила отталкивания. Заряжение прекращается тогда, ко-
гда сила отталкивания равна силе притяжения, т. е.:
пее____р
rt “--х
ИЛИ
пе = Ехг*(\ 4-2-J-j-^-),
(17)
где п — число ионов, осевших на пылинке, пе — заряд пылинки,
е — заряд иона, г — радиус пылинки.
На рис. 75 изображены силовые линии при процессе заряже-
ния. На пылинку осаждаются те ионы, которые двигаются по
силовым линиям, проходящим через пылинку. По мере заряже-
ния число этих линий уменьшается. При полном заряжении все
силовые линии огибают пылинку — дальнейшего попадания
ионов на пылинку не происходит.
160
Если заряд пылинки пе, то в поле с напряжением Еа пылинка
будет притягиваться к электродам с силой ЕаЛе, под влиянием
этой силы она будет двигаться со скоростью wn. Этому движе-
нию будет препятствовать сопротивление среды 6nrr)vvn (см.
выше).
Выше мы видели, что эти силы быстро уравновесятся, т. е.
пеЕх = 6те/-т]отп,
откуда легко найти
или, заменяя ле по уравнению (17),
Дойдя с этой скоростью до электрода, пылинка отдаст ему
свой заряд и осядет на нем. Из этой формулы мы видим, что
скорость оседания пылинки (т. е. скорость очистки газа) сильно
возрастает с увеличением размера пылинки и еще сильнее с уве-
личением напряжения поля. Последнее обстоятельство необхо-
димо особенно отметить. Электрофильтр работает тем лучше,
чем он выше держит напряжение, а это в значительной степени
зависит от хорошего монтажа и ухода за ним. Внутри электро-
фильтра не должно быть острых углов, заусениц, нецентриро-
ванных деталей, так как в этих местах получается повышение
силовых линий, что может привести к пробою, в то время как
в остальном электрофильтре еще будет очень небольшое напря-
жение; очистка, таким образом, будет неудовлетворительна. По-
этому после всякой остановки, ремонта и чистки нужно прове-
рять пробойное напряжение камеры на воздух; заметив пробои
в каком-либо месте, надо устранить причину их образования.
Такую проверку следует производить до тех пор, пока пробоев
не будет при полностью включенном коммутаторе или когда
пробои все время образуются в разных местах по всей камере.
Подсчитаем скорость осаждения пылинки, диаметр которой
2г = 0,001 лгунью-6 м — 1 р- и е = 4, если она попадет в эле-
ктрофильтр со средним напряжением поля 3000 и на 1 см и если
•Л — 2,5 • 10-6 кг • сек/м2.
Так как величины даны в разных системах измерения (диа-
метр и вязкость в механических единицах, а напряжение поля
в электротехнических), нужно ввести в формулу (18) поправоч-
ный член дор , чтобы ответ сразу получить в см/сек.
Подставляя наши числа, найдем:
3002-0,5-10-6 Л . о3\ о .
Wn ~~ 3002-6п • 2,5-10 6 v + 2 6 ) ~ 2’1 см!сек-
Разделив расстояние от провода до осадительной пластины
на найденную скорость, найдем время, потребное для осаждения
Ц Зак. 5019. Технология серной- кислоты.
161
пылинки, т. е. необходимое время пребывания газа в поле эле-
ктрофильтра (без учета времени заряжения пылинки).
Опыт показал, что очистка лучше происходит при отрица-
тельно заряженном коронирующем проводе.
Наружный цилиндр можно заземлить. Область короны, где
имеются ионы обоих знаков, мала по сравнению с остальной
частью цилиндрического конденсатора — электрофильтра, где
имеются только ионы одноименные с знаком коронирующего
провода (так как ионы этого знака из короны движутся к пери-
ферии), поэтому основная масса пылинок будет заряжаться за-
рядом, одноименным с коронирующим! проводом, а следова-
тельно, притягиваться к наружному цилиндру. В связи с этим
наружный цилиндр называется осадительным электродом.
Электрофильтры. Электрофильтры представляют собой аппа-
раты, построенные на описанном выше принципе и служащие
для улавливания из газов любых примесей, находящихся в твер-
дом (пыль, дым) или жидком (туман) состоянии. Электрофиль-
тры бывают: 1) трубчатые-—из нескольких цилиндрических
конденсаторов и 2) пластинчатые, в которых в качестве осади-
тельных электродов установлены параллельные пластины и ме-
жду ними на некотором расстоянии друг от друга расположены
коронирующие провода.
Трубчатые электрофильтры дают более правильное поле, пол-
ностью охватывающее все пространство, поэтому они лучше ра-
ботают, но они сложны в конструктивном выполнении. У пла-
стинчатых электрофильтров значительно худшее распределение
силовых линий (имеются пустые места, где отсутствует силовое
поле), поэтому они дают худшую очистку, но конструктивно
более просты.
В промышленности применяются оба типа электрофильтров.
На сернокислотных заводах СССР первоначально установлены
были пластинчатые аппараты. Теперь на основе исследований
треста «Газоочистка» будут строиться и трубчатые, причем для
облегчения конструкций трубам придан вид шестигранников —
сот. Нормальный диаметр труб или расстояние между осади-
тельными электродами 200—300 мм. Коронирующие провода из
1—2-мм проволоки устанавливаются точно по центру трубы или
по середине между пластинами на расстоянии в 200—250 мм
друг от друга.
Установка точно по центру и обязательно в натянутом со-
стоянии вызывается тем, что иначе в том месте, где провод
ближе подойдет к осадительной пластинке, может наступить
пробой при значительно пониженном напряжении поля. Хорошая
же очистка происходит только при большой короне, т. е. при
наличии большого количества ионов. Большая корона образуется
при возможно большом напряжении.
Характер влияния напряжения на работу электрофильтров
уже достаточно ясен из вышеизложенного.
Разберем влияние других факторов на работу электрофильтра.
162
Размер пыли и ее свойства. Из уравнения (18)
видно, что скорость осаждения пылинки прямо пропорциональна
ее радиусу, поэтому чем крупнее пылинки, тем меньше времени
нужно на их осаждение. Все же даже для очень мелких пыли-
нок скорость осаждения довольно значительна и для пылинок
в несколько микрон составляет несколько сантиметров в се-
кунду. Диэлектрическая постоянная пылинок не оказывает боль-
шого влияния на осаждение, но некоторые сорта пыли, напри-
мер ZnO- (по неизвестной причине), очень неохотно восприни-
мают заряд.
Работа электрофильтра на проводящей пыли ничем почти не
разнится от работы на непроводящей пыли, за исключением тех
случаев, когда пыль склонна к налипанию, например при осажде-
нии огарковой пыли (в особенности при сжигании флотационных
хвостов). В этом случае пылинки плотно пристают к осадитель-
ным электродам, трудно удаляются с них и создают на их
поверхности слой изолятора, что мешает отдаче зарядов вновь
осевшей пыли. Таким образом на наружном слое прилипшей
пыли будут оставаться заряды, что вызовет резкое уменьшение
напряжения поля и ухудшение очистки. Чтобы этого избежать,
необходимо поддерживать чистоту электродов. Это достигается
сильным и регулярным встряхиванием электродов.
Скорость газа и .время пребывания его
в электрофильтре. Степень очистки газа зависит не
только от времени его пребывания в электрическом поле, но и
от его линейной скорости. При очень больших скоростях газа
происходит унос уже осевшей пыли.
Опыт треста «Газоочистка» показал, что при хорошем встря-
хивании на трубчатом (сотовом) аппарате такого уноса почти
нет. С увеличением скорости процент очистки падает, но не
очень резко даже на больших скоростях.
Особенно интересно то, что основная масса пыли оседает
в самом начале пути газа в поле электрофильтра.
Влажность газа. Если газ насыщен водяными парами
или близок к точке росы, то конденсация паров происходит на
уже имеющихся точках конденсации, т. е. на пылинках, при
этом пылинки укрупняются и их осаждение ускоряется. Такое
сильное увлажнение применяется на сернокислотных заводах,
после 1-й пары мышьяковых электрофильтров. Пылинки AS2O3
очень мелки и их необходимо укрупнить, а для этого газ сильно
увлажняют и охлаждают (гл. 4).
Небольшая влажность может быть также полезной, так как
осевшая в результате увлажнения пыль может стать немного
более электропроводящей, что способствует работе электро-
фильтра.
Запыленность входящего газа. До известного
предела запыленность входящего газа не оказывает влияния на
запыленность выходящего газа.
При большой запыленности, в особенности при большой ско-
П*
163
роста газа, это влияние очень заметно. При входе пылинок
в поле на них скопляется большое число зарядов; заряды вме-
сте с пылинкой двигаются значительно медленнее, чем они дви-
гались бы свободными (из-за сопротивления среды). Это влечет
за собой увеличение числа силовых линий на периферии, т. е.
увеличивает равномерность поля и ускоряет пробой. Пока пыли-
нок мало и число зарядов на них невелико по сравнению с об-
щим числом ионов, влияние первых очень ничтожно, но при
большем числе пылинок, когда сумма зарядов на них становится
значительной по сравнению с суммой зарядов оставшихся сво-
бодных ионов, это влияние будет заметно и придется понизить
напряжение. Такое явление часто наблюдалось на электрофиль-
трах, установленных при концентрационных установках барабан-
ного типа, в которых в электрофильтр поступает большое коли-
чество частиц мелкого кислотного тумана.
Влияние запыленности электродов. Выше мы
уже указывали на некоторое отрицательное влияние налипшей
пыли, но здесь мы еще остановимся на двух специальных слу-
чаях.
Если пыль способна проводить ток и в значительном коли-
честве налипает на коронирующие электроды, то они утол-
щаются, что приводит к снижению напряжения, т. е. к умень-
шению короны и вообще к ухудшению очистки. Кроме того, пыль
может нарастать неравномерно по всей длине провода и образо-
вать в некоторых местах острые углы, а это может вызвать
пробой. Поэтому необходимо очень тщательно производить
встряхивание и следить за хорошей работой.
Выше уже указывалось, что опасно самоналипание непрово-
дящей пыли. Кроме того, если в осевшем слое пыли имеются
трещинки, то силовые линии, стремясь обойти изолирующий
слой, сгустятся около трещин. Может случиться, что плотность
силовых линий будет настолько велика, что в этом месте на-
чнется ионизация. Это явление называется обратным к о р о-
нированием. Образуемые при этом ионы обоих знаков и
ионы одноименные с осадительным электродом будут двигаться
в направлении к центру трубы. Эти ионы могут встретить на
своем пути пылинки, заряженные другим знаком (которые дви-
жутся к осадительным электродам), и нейтрализовать их заряд;
после этого пылинки отделяться от газа больше не будут. Та-
ким образом запыленность осадительных электродов также мо-
жет сильно ухудшить работу электрофильтра.
Огарковый электрофильтр. Электрофильтр для
очистки печного газа от пыли представляет собой вертикальную
камеру-шахту, внутри которой расположены осадительные эле-
ктроды и между ними — коронирующие. Размер камеры зависит
от количества очищаемого газа, а именно: сечение ее должно
быть такое, чтобы скорость газа была около 0,5—0,7 м/сек,
а время пребывания газа в поле 3—5 сек. При таких условиях
можно очистить газ до содержания в нем пыли ~ 0,15 г/м9.
164
Опишем кратко 45-тонный аппарат-электрофильтр (рис. 76),
который состоит из двух параллельных вертикальных шахт, вы-
ложенных из кирпича; сечение камеры 1730 X 2140 мт и высота
около 5,5 м. Внизу каждая камера оканчивается железобетон-
ным бункером 7, выложенным внутри кислотоупорным кирпи-
чом. В каждой камере в качестве осадительных электродов 2
установлено по 9 сеток из З-тим проволоки, натянутой на желез-
ную раму. Расстояние между сетками 250 уйм, ширина каждой
сетки 1,67 м, высота 3 м. Между сетками подвешены рядами
электроды 3 из 2-мм нихромовой проволоки. Расстояние между
коронирующими проводами 200 мм. В электрофильтре все ме-
таллические части, в том числе и осадительные электроды, за-
землены. Коронирующие электроды изолированы и находятся
под напряжением.
Для того чтобы осевшая пыль лучше удалялась с электро-
дов, последние встряхиваются.
Разработано .много различных методов встряхивания. Наи-
более эффективным является для коронирующих электродов
боковой удар молотка 4, направленный снизу вверх. Такой
механизм встряхивания применяется в новых аппаратах.
Опыты показали, что электрофильтр значительно лучше
работает при равномерном распределении газа по всему сече-
нию шахт электрофильтра. С этой целью в современных аппа-
ратах в низу шахты подвешивают железную решетку с отвер-
стиями в 50 мм. Сопротивление этой решетки способствует от-
носительно равномерному распределению газов. Для того чтобы
решетка не забивалась пылью, ее также периодически встря-
хивают.
Пыль, осевшая на электродах, спадает в бункеры, откуда
в каждую смену удаляется через соответствующие затворы
в вагонетку.
Аппарат состоит из двух параллельных камер, для того
чтобы одну из камер можно было выключить, не нарушая ра-
боту всего сернокислотного завода. Перед поступлением
в шахту газ предварительно входит в отделение, отгороженное
кирпичной стенкой от шахты. Газ поступает по'газоходу на вы-
соте 6 м и опускается вниз. В предварительном отделении на
высоте 5 м устроено перекрытие с двумя отверстиями, соеди-
ненными только с одной шахтой. Проходя через одно какое-
либо отверстие, газ далее проходит через соответствующую
шахту-камеру. Для отключения камеры отверстия закрываются
особым приспособлением. Над отверстиями подвешены чугун-
ные колокола, которые могут опускаться и подниматься при по-
мощи штанги и тросов. Подъем и опускание производятся руч-
ным червячным воротом, на который наматывается трос, при
этом колокол поднимается над отверстием и газ свободно про-
ходит в камеру. При опускании колокола он закрывает отвер-
стие и отсоединяет камеру. Аналогичное устройство имеется и
на выходе из камеры. Отключение камер бывает необходимо
166
произвести при ремонте или очистке камеры. В этом случае обя-
зательно нужно тщательно опустить входной и выходной коло-
кола. В случае встряхивания электродов необходимо закрыть
только выходной колокол, для того чтобы газ с большим со-
держанием пыли не прошел в систему.
Электрофильтр должен работать при определенных темпера-
турных условиях. При температурах выше 500° железо дефор-
мируется, что приводит к перекашиванию конструкции камеры.
Кроме того, при таких тем-
пературах может произойти
размягчение и слипание
улавливаемой пыли, что ве-
дет к образованию больших
плотных наростов на эле-
ктродах. Это сильно ухуд-
шает коронирование и может
вызвать даже обрыв прово-
дов. Таким образом темпе-
ратура газов, поступающих
в огарковый электрофильтр,
не должна быть выше
500°.
В газе, идущем от колче-
данных печей, всегда содер-
жится некоторое количество
SO3 и водяных паров, по-
этому при сильном пониже-
нии температуры может про-
изойти конденсация серной
кислоты. Точка росы при
содержании этих компонен-
тов в тех количествах, ка-
кие наиболее часто встреча-
ются при нормальной экспло-
атации печей, находится
около 200—250°. Для пред-
Рис 77. Схема подстанции:
/ — главный рубильник; 2—автотрансформатор;
3— коммутатор; 4 — реостат; 5 — максимально-
нулевой автомат; 6—переключатель; 7 — высоко-
вольтный трансформатор; 8 — механический вы-
прямитель; 9— синхронный мотор.
отвращения конденсации кис-
лоты, а следовательно и кор-
розии камеры, необходимо,
чтобы температура в камере
была выше 250°, а это в свою очередь требует, чтобы темпера-
тура входящего газа была не ниже 275°. Если температура газа
ниже 275°, то следует прекратить подачу тока в электрофильтр,
так как иначе будет происходить осаждение сконденсированного
тумана серной кислоты на электродах. Все же и при отсутствии
тока при низкой температуре частично кислота будет оседать
в камере и сильно разъедать металлические части. Оседание
кислоты на кварцевой трубе вызовет пробои по ней, что повле-
чет за собой снижение напряжения.
167
При нормальной работе электрофильтра газ должен очи-
щаться до содержания пыли в 0,2—0,15 г/м3. В связи с интен-
сификацией сернокислотных установок, даже при таком содер-
жании пыли, количества ее, приходящие в систему, становятся
значительными. Дальнейшее снижение содержания пыли в газе
является поэтому актуальной задачей. При плохой работе
электрофильтра увеличивается не только количество проходя-
щей пыли, но и ее крупность; оба эти фактора сильно влияют
на засорение системы.
Узким местом в электрофильтрах является встряхивание;
механизмы для встряхивания в существующих камерах не
всегда достаточно хорошо и эффективно работают.
Подстанция. Питание электрофильтра требует тока высокого
напряжения постоянного направления. Для получения такого
тока на заводе устраивается повысительно-выпрямительная под-
станция (рис. 77). Трехфазный ток напряжением в 220 или
380 в поступает через главный рубильник 1 в синхронный мо-
тор 9 выпрямителя 8 и автотрансформатор 2, а оттуда через
коммутатор 3 и максимально-нулевой автомат 5 в первичную
обмотку трансформатора высокого напряжения 7. Со вторичной
обмотки трансформированный ток высокого напряжения посту-
пает в механический выпрямитель, с которого положительный
ток направляется в землю, а отрицательный — по кабелю или
по шинам к коронирующим электродам электрофильтра. Опи-
шем кратко устройство и принцип действия основных агрегатов
электроподстанции.
Трансформатор состоит из двух катушек: первичной — из
толстой проволоки с незначительным числом витков и вторич-
ной —- из тонкой проволоки с большим числом витков. Весь
трансформатор погружен в масляный ящик. Масло служит изо-
лятором. Коэфициент трансформации около 350; это значит, что
если на первичной катушке дан ток в 100 в, то на вторичной
получится ток напряжения ~ 35 тыс. вольт. Для регулирова-
ния высокого напряжения служит автотрансформатор; он дает
возможность подавать на первичную обмотку трансформатора
различное напряжение от 50 до 220 в и, следовательно, посте-
пенно менять высокое напряжение от 20 до 75 тыс. вольт.
На железном сердечнике трансформатора (рис. 78) навито
несколько витков провода. От каждого витка отведена клемма;
по клеммам движется контакт. Первичный ток в автотрансфор-
маторе проходит через всю обмотку от точки А до В. Вторич-
ный ток отводится от подвижного контакта \С и до той же
точки В. Двигая контакт по клеммам, можно получить различ-
ное напряжение в зависимости от числа включенных витков
между подвижным контактом и точкой В. Таким образом эта
часть витков играет роль вторичной обмотки трансформатора.
Включая то или иное число витков, мы получим различное на-
пряжение в первичной, а следовательно, и во вторичной об-
мотке высоковольтного трансформатора.
168
Выходные клеммы и подвижной контакт собраны в общий
прибор, называемый коммутатором, который находится за щи-
том. Подвижной контакт 1 вращается от штурвала, который
вынесен на щит (рис. 79). Выводы от автотрансформатора сое-
динены с медными пластинками, расположенными по кругу;
расстояние между пластинками немного больше передвижного
контакта 7. Когда передвигают контакт, то в промежутке ме-
жду пластинками он их не соединяет накоротко. Но при таком
устройстве произойдет другое неприятное явление: при пере-
ходе контакта от промежуточного положения на медную пла-
стинку и от нее дальше на изолированный промежуток будет
сильное искрообразование, а это в свою очередь постепенно
сжигает контакты. Чтобы избежать этого, рядом с подвижным
контактом 7 сделан еще второй подвижный контакт 2, который
Рис. 78. Автотрансформатор.
Рис. 79. Коммутатор:
1 и 2 — подвижные контакты; 3 — сопро-
тивление.
соединен с первым1 через сопротивление 3. Оба контакта так
расположены, что при сдвигании основного контакта с одной
клеммы контакт 2 соединяется со следующей, но при этом ко-
роткого замыкания не происходит, так как контакт 2 соединен
с контактом 7 через сопротивление. Искрообразование также
уменьшается, так как подвижной контакт постоянно остается
под напряжением.
Надолго оставлять контакт 2 и его сопротивление под током
не нужно, так как сопротивление небольшое и может перего-
реть, поэтому к подвижному контакту приделан отщелкиваю-
щий механизм, который облегчает правильный перевод подвиж-
ного контакта от клеммы к клемме.
Коммутатор часто обгорает, поэтому нужно осматривать его
не реже одного раза в месяц. Для удаления нагара можно поль-
зоваться мелкой стеклянной бумагой. При сильном обгорании
лучше обточить коммутатор на станке.
Выпрямитель. Ток высокого напряжения, полученный
в трансформаторе, необходимо выпрямить. Для этой цели слу-
169
жит механический выпрямитель. Он устроен так (рис. 80): на
оси синхронного мотора укреплен крест из четырех изоляцион-
ных лопастей (крыльев). На концах лопастей сделаны метал-
лические наконечники-башмаки; которые попарно соединены
между собой проволокой. По окружности креста также на изо-
ляторах укреплены 4 щетки. К двум противоположным боковым
щеткам 7 и 3 выведены концы вторичной обмотки трансфор-
матора высокого напряжения, к третьей щетке 2 присоединено
заземление и к четвертой щетке 4 — шина, идущая в электро-
фильтр. Мотор, вращающий крест, делает 1500 об/мин или
25 об/сек. Ток же, который подается с электростанции, имеет
50 пер/сек. Таким образом за один период крест повернется на
полоборота.
Рис. 80. Схема работы выпрямителя:
7, 2, 5, 4 — щетки; /, II— проволоки.
Теперь разберем, как работает выпрямитель. Положим, что
мы начинаем с момента, когда на щетку 1 из трансформатора
пришел отрицательный заряд (—), а на щетку з — положитель-
ный (+), и крест находится в положении а, указанном на
рис. 80. Тогда отрицательный ток пойдет с левой щетки 7 по
проволоке 7 на щетку 4 и оттуда в камеру к проводам. С щетки 3
положительный ток пойдет через проволоку II к нижней щетке 2
и оттуда в землю. Через полпериода (0,01 сек.) ток переменит
свое направление, а именно на щетку 7 придет положительный
заряд (-)-), а на щетку 3 — отрицательный (—). Но за это время
крест повернется на четверть оборота и примет положение Ь.
При этом отрицательный ток с правой щетки 3 пойдет по про-
волоке / наверх в камеру, а положительный ток с левой щетки 7
по проволоке // в землю. Через следующие полпериода снова
на левую щетку 7 пойдет отрицательный ток, а на правую 3
положительный, за это время крест повернется еще на полобо-
рота (рис. 80, с). Снова с левой щетки отрицательный ток пой-
дет по проволоке /7 наверх в камеру, а с правой положительный
ток по проводу / — вниз в землю и т. д. Таким образом на
верхнюю щетку всегда подается отрицательный ток, а вниз,
в землю — положительный.
Из описания выпрямителя видно, что щетки должны быть так
установлены, что, когда мимо них проходит лопасть, на
770
1-й щетке, соединенной с трансформатором, должно быть в этот
момент наибольшее напряжение, а на другой — наименьшее. При
этом условии мы и в камеру передадим максимальное напря-
жение. Для того чтобы установить щетки в такое наилучшее
положение, изоляторы, к которым они прикреплены, распола-
гаются на кольце, которое при помощи соответствующего пе-
редаточного механизма может поворачиваться вокруг креста.
В новых аппаратах изоляторы устанавливаются непосредственно
на корпусе трансформатора, мотор — в вертикальном положе-
нии, в центре крышки. В этих аппаратах для правильности уста-
новки креста поворачивают весь мотор.
При первом пуске электрофильтра поворачивают щетки в ту
или другую сторону до тех пор, пока не получится максималь-
ная сила тока при постоянном установленном напряжении. Сила
тока наблюдается по миллиамперметру, который включен в цепь
положительного тока, идущего от выпрямителя в землю. Это
называется регулированием траверсы.
Часто крест выпрямителя повертывается на валу мотора, по-
этому время от времени нужно проверять положение траверсы.
Это необходимо также делать при переключении данной машины
на другую камеру, после ремонта .камеры и т. д.
При обгорании лопастей выпрямителя необходимо удалить
обгоревшую часть острым ножом и покрыть это место шеллач-
ным лаком. Крёст должен равномерно слабо касаться всех ще-
ток и иметь зазор не больше 2 мм. Наконечники креста должны
быть чистые, без нагаров и ржавчины. Для нормальной работы
электрофильтра необходимо к проводам подавать отрицатель-
ный заряд, но при включении тока каждый раз мы не знаем за-
ранее, какой ток ,в этот момент подходит к электрофильтру.
Для того чтобы это узнать, служат синхронизатор, установлен-
ный у вала мотора, и синхронная лампа; в зависимости от ее
показаний включают в ту или иную сторону переключатель.
При правильном включении положительный ток идет в землю
через миллиамперметр, который показывает соответствующее от-
клонение; если же мы неправильно включим и пустим положи-
тельный ток в камеру, а отрицательный — в землю, то милли-
амперметр должен отклоняться в другую сторону, т. е. его
стрелка будет давить в упор. Так как через миллиамперметр
идет ток высокого напряжения и он находится на щите, то при
обрыве провода щит может оказаться под высоким -напряже-
нием; чтобы этого не произошло, за нижней щеткой выпрями-
теля, откуда идет положительный ток в миллиамперметр, уста-
новлен пробойный предохранитель, который замыкает эту щетку
и миллиамперметр на землю сейчас же, как только напряжение
дойдет до ~ 150 в. Пробойный предохранитель представляет
собой обычный патронный предохранитель, между контактами
которого помещен тонкий листок слюды с отверстием. Один
конец пробки соединен с землей, другой — с нижней щеткой вы-
прямителя. Таким образом, как только по какой-либо причине
171
на нижней щетке напряжение поднимается выше 150 в, пробьется
слюда и ток пойдет в землю через предохранитель и вообще
мимо щита, а не через миллиамперметр.
Раньше мы указывали, что для хорошей очистки нужно ра-
ботать на возможно! более высоком напряжении, но при этом
могут происходить под влиянием различных случайных причин
(капли, скопление пыли и т. д.) сильные пробои. При колеба-
ниях напряжения, что бывает часто в первичной цепи, эти про-
бои могут перейти (в дугу, а это повлечет выключение автома-
тов, треншальтеров и даже порчу трансформатора. Для того
чтобы это не произошло, в цепь включают сопротивление, кото-
рое при возрастании силы тока понижает напряжение.
Напряжение трансформатора частично подается к электро-
дам, частично поглощается реостатом. Это напряжение равно,
как известно, произведению сопротивления R на силу тока /.
Если сила тока небольшая, то и RI мало, но если сила тока
возрастает, то сильно возрастает и значение RI, а следовательно
сильно уменьшается напряжение, подаваемое к электродам в ка-
меру, т. е. прекращается искрообразование. Реостат можно
включать и на высоком и на низком напряжении. Обыкновенно
он включен на низком напряжении.
Максимально-нулевой автомат устанавливается как предо-
хранитель: он автоматически выключает ток, подаваемый
в трансформатор (на некоторых схемах в автотрансформатор),
в двух случаях: когда сила тока очень велика — больше уста-
новленной безопасной нормы или когда временно ток в системе
прекратился. Отключение автоматом достигается соответствую-
щими электро- и рычажными механизмами.
Для обеспечения хорошей бесперебойной работы электро-
фильтра и подстанции необходимо кроме тщательного ухода и
ремонта аппаратуры соблюдать абсолютную чистоту. В поме-
щении подстанции часто под влиянием электрических искр об-
разуются вредные газы: окислы азота и озон. Эти газы отра-
вляют персонал и портят аппаратуру (пробои лопастей выпря-
мителя, разъедание и окисление контактов и т. д.). Для устра-
нения этих вредных газов в помещении подстанции должна быть
хорошая, теплая, приточная вентиляция. Под влиянием кислых
газов и грязи изоляторы теряют свои изоляционные свойства, по
ним происходит большая утечка, и обслуживающий персонал
может быть введен в заблуждение, так как при этом на милли-
амперметре наблюдается большая сила тока, но ток идет не через
электрофильтр, а через грязные изоляторы. В связи с этим не-
обходимо изоляторы содержать в полной чистоте и время от
времени проверять состояние изоляторов, наблюдая утечку (силу
тока) при отключенном электрофильтре. Протирку изоляторов
следует проводить в сроки, указанные в инструкции, при обя-
зательном отключении всей линии от высокого напряжения.
Высокое напряжение очень опасно. Прикосновение человека
к высокому напряжению влечет за собой смерть. Поэтому ни при
172
каких обстоятельствах не разрешается: 1) производить какие-
либо работы или прикасаться к приборам, находящимся под на-
пряжением; 2) входить в помещение, где имеется высокое на-
пряжение, не выключив его, а при выключении следует сейчас же
аппарат, бывший под напряжением, заземлить; 3) допускать по-
сторонних лиц в помещение, где имеется высокое напряжение.
Кроме того, необходимо: 1) ®се двери, ведущие в помещения,
где проходит высокое напряжение (дверь для щита, дверь шин-
ного коридора, двери у изолятора и т. д.), запирать на со-
ответствующие замки, а ключи должны находиться только у де-
журного электромонтера.
Перед включением высокого напряжения необходимо убе-
диться в том, что все аппараты закрыты и нигде нет лишних
предметов и посторонних людей.
При ремонте вывешивать соответствующие объявления о за-
прете включения данной линии.
Литература
I. Аллен, Ломбард, Упругость паров серы над пиритом, Ztschr. f.
Elektrochem., 373 (1905).
2. Смирнов В. И., Окислительный обжиг медных руд и концентратов,
ГОНТИ, 1938.
3. Дональд-Лиддель, Справочник металлурга и химика цветной ме-
таллопромышленности, ч. II, стр. 186, Металлургиздат, 1934.
4. Рысс И. Г., Журавлева Г. Г., С у с л о в В. Н., Воспламеняемость
колчеданов и флотационных хвостов, ЖХП № 6, 580 (1935).
5. Залесский И. П., Заводские топки и печи, Госиздат, 1926.
6. М е л ьни к Б. Д., Л е с о х и н И. Г., К о п ы л е в Б. А., Г р а б е р Д. Г.,
Кинетика горения сырья сернокислотного производства, ЖХП № 2.
16 (1938).
7. Мал ин К. М., Кинетика горения колчедана и дальнейшие возмож-
ности интенсификации механических печей, ЖХП Ns 4—5, 14 (1939).
8. Д и е в Н. П., Карякин Ю. В., Окисление сульфидов воздухом,
обогащенным кислородом, ЖПХ, XI, вып. 7—8 и 10—11 (1938).
9. Томсон, Тиллинг, Десульфуризация серного колчедана, J. Soc,
Chem. Ind. 43, 37—46 (1924).
10. W а е s е г В., Handbuch der SchwefelsSurefabrikation, 253—257. Braun-
schweig 1930.
11. К у н и н Т. И., Лапшин Н. С., Сироткин Т. Д., О скорости
горения флотационного колчедана, ЖХП № 1, 49 (1940).
12. П о л я к о в К. А., Печи и аппараты для получения и очистки сер-
нистого газа, ОНТИ, 1938.
13. С о с н о в с к и й Н. П., А р к и н Н. Л., Механические печи для обжига
сериого колчедана, ОНТИ, 1936.
14. Постников Б. Ф., Кунин Г. И., Ваганов А. В., Об увели-
чении производительности механических печей для сжигания колчедана,
ЖХП № 3, 213 (1937).
15. Малин К. М., Смыслов Н. И., Блюм Г. Б., Интенсификация меха-
нических печей ВХЗ и Ведже, ЖХП № 2, 12 (1938).
16. Б л ю м Г. Б., С о к а л ь с к и й А. Г., Интенсификация механических
печей ВХЗ, ЖХП № 4—5, 65 (1939).
17. Аркин Н. Л., Пропускная способность печи ВХЗ и методы ее рацио-
нализации, ЖХП № 9, 30 (1939).
18. Второв М. Н., Смыслов Н. И., Сжигание углистого колчедана
в механических печах, сборник «Сера и серная кислота", Химтеорет
(ЛОГОНТИ), 1935.
173
20. Сосновский Н. П., Вращающиеся печи в производстве серной
кислоты, Химстрой № 4, 2193 (1933).
21. Debuch С. Р., Der Drehrohrofen als Rostofen, Metallgesellschaft, 7 (1932).
22. Debuch С. P., Der Drehrohrofen fiir Piritrostung, Chem. Apparatur 3,
5, 7, 8, 11 (1928).
23. С о с в о в с к и й H. П., Сернокислотная промышленность США, Полу-
чение сернистого газа, ЖХП № 5, 60(1938); Сжигание серы, ЖХП №9,
31 (1938); Утилизация тепла сернистого газа, ЖХП № 1, 47 (1939).
24. С а н д л е р И., Андреев А., Техноэкономическое сравнение печей
для обжига колчедана, ЖХП № 6, 401 (1937).
25. П ос т н и к о в В. Ф., Барс ков Н. М., Опыт расчета основных раз-
меров печи, Химстрой № 1, 29 (1934).
26. Николенко Г. Д.. Пневматическое удаление огарка от печей серно-
кислотных заводов, ЖХП № 4—5, 18 (1939) и № 7, 28 (1939).
27. Эпштейн И. И.. Об использовании тепла сернистых газов, ЖХП
№ 6, 27 (1939).
28. Розен кн о п С. П., Левин А. М., Использование пиритных огар-
ков в третьем пятилетии, ЖХП № 17—18, 1211 (1937).
29, К а т а л ы м о в И. В., К о ш е л ь к о в Г. Н., Об использовании пнрнт-
ных огарков в качестве медных удобрений, ЖХП № 3, 24 (1938).
30. П о м е р а и ц е в И. Н., Сор кии а Д. Э., Переработка колчеданных
огарков сернокислотных производств на медный купорос и ннсекто-
фунгисиды, ЖХП № 1, 47 (1934).
31. С л о б о д с к и й Я. Я., Обработка колчеданных огарков хлорированием,
ЖХП № 4, 44 (1934).
32. С о с н о в с к и й Н. П., Н е м ч и к Г. Л., Сернокислотная промышленность
Германии, ЖХП № 9, 59 (1934).
33. Маковецкий А. Е., Шабалин К. Н., Сульфатизирующий обжиг меди-
стого огарка действием обжигового газа, ЖХП X» 3, 21 (1925).
34. Waeser В. (Серная кислота из сульфатов, доменных шлаков и от-
ходящих газов), Metallborse 817, 875, 930 (1926).
35. М ю л л е р В., Получение серной кислоты из гипса по Байеру, ЖХП
№ 8, 688 (1926).
36. Maqueune (Производство серной кислоты нз гипса в Леверкузене),
Chim. et Ind. № 2, 509 (1930).
37. П о с т н и к о в В. Ф., Кунин Т. И., Бронников А. К., Полу-
чение SO2 и цемента нз гипса с добавкой угля и колчедана, сборник
«Сера и серная кислота", Химтеорет (ЛОГОНТИ), 1935.
38. Рояк С., Г ершманМ., Милославский К., Нагерова Э., Полу-
чение сернистого газа и портланд-цемента из гипса, ЖХП № 7, 35 (1933).
39. Ter res Е. (Гипс как химическое сырье), Ztschr. ang. chem. № 20,
356 (1931).
40. Marschal G., J. de Chim. Phys., 23, 28 (1926).
41. Будников П. П., P и вл ин И. С., Разложение гипса в смеси
с углистым колчеданом, ЖХП № 7—8, 1129 (1938).
42. Славянов Ю. Н., Промышленное получение сернистого газа из
гипса, ЖХП № 4, 22 (1938).
43. Павлов К. Ф„ Л е с о х и н И. Г., Г р а б е р Д. Г., Переработка
гипсов на цемент и серу, ЖХП № 3, 5 (1938).
41. Ляпустина Е. М., Интенсификация и дальнейшее аппаратурное улуч-
шение печей пылевидного обжига, ЖХП № 4—5, 60 (1940).
45. С е н - Ж а и, Обжиг и агломерация пыли и пылевидной руды в турбу-
лентной печи системы Сен-Жан, Revue de metallurgie № 11 (1935).
46. Парфенов, Результаты опытов агломерации железных руд во взве-
шенном состоянии, Советская металлургия № 2 (1938).
47. Шнеерсои, Б. Л„ Электрическая очистка газов в химической про-
мышленности, ОНТИ, 1936.
48. Ш н е е р с о н Б. Л., Е г о р о в Н. Н., Электрическая очистка газов,
ОНТИ, 1933.
* * 4=
174
ГЛАВА 4
ПРОИЗВОДСТВО СЕРНОЙ КИСЛОТЫ МЕТОДОМ
КОНТАКТНОГО ОКИСЛЕНИЯ
I. Основные схемы контактного процесса
Полученный тем или иным способом сернистый газ для пре-
вращения в серную кислоту должен быть окислен до ангидрида
серной кислоты SOs. Однако SO2 с кислородом непосредственно
почти не реагирует, хотя озон довольно легко окисляет SO2.
В промышленности применяются два метода окисления серни-
стого газа: при помощи окислов азота и при помощи твердых
катализаторов.
Высшие окислы азота окисляют SO», сами восстанавливаясь
до NO; окись азота снова окисляется до NO» (или N2O3), соеди-
няясь непосредственно с кислородом. Таким образом в процессе
окисления SO» окислы азота играют роль передатчиков кисло-
рода. На основе окисления SO2 с помощью окислов азота бо-
лее 200 лет назад возник так называемый нитрозный метод про-
изводства серной кислоты. Этот метод производственно оформ-
лен в двух способах: камерном и башенном.
Окисление сернистого газа кислородом в присутствии твер-
дых катализаторов положено в основу контактного метода про-
изводства серной кислоты. Этот метод более молодой, чем ни-
трозный, открыт Филиппсом в 1831 г. и впервые производственно
применен Винклером в 1875 г.
При производстве серной кислоты камерным способом боль-
шая часть продукции получается >в виде 65%-ной H2SO4 и мень-
шая часть в виде 75—76%.-ной H2SO4.
Башенные системы до последнего времени выпускали всю про-
дукцию в виде 75—76%-ной H2SO4. Сочетая башенный процесс
с использованием теплоты обжиговых газов, можно часть или
всю продукцию башенной системы .выдавать :в виде купоросного
масла (90—92% H2SO4). Раньше купоросное масло из башенной
кислоты получали только концентрированием на специальных
концентрационных установках.
Для некоторых потребляющих серную кислоту производств
требуется более крепкая кислота, чем купоросное масло; так,
в целом ряде производств необходим олеум. Из камерной и ба-
шенной кислот концентрированием нельзя получить кислоту с со-
держанием выше 98% H2SO4. В настоящее время нет также спо-
175
соба получения олеума методом окисления SO2 при помощи
•окислов азота. Олеум, или дымящую серную кислоту, вырабаты-
вали в небольших количествах еще в 1526 г. в Пильзене, позже
в Гарце. Это производство велось методом разложения купо-
росного сланца.
Горную породу выветривали, сернокислые соли железа и алю-
миния выщелачивали водой, щелок упаривали; обезвоженный и
подготовленный таким образом «купоросный камень» прокали-
вали в глиняных сосудах, из выделяющегося при этом (наряду
с SO2) ЭОз готовили олеум, с содержанием 10—15% свобод-
ного SO3.
Контактный метод производства серной кислоты не имел ши-
рокого распространения вплоть до 1900 г. Это объясняется сле-
дующим:
Во-первых, Винклер ошибочно предполагал, что для получе-
ния SO3 лучшей является стехиометрическая смесь SO2 и Ог.
Хотя это явно противоречит закону действия масс, но поло-
жение Винклера долгие годы разделялось технологами того вре-
мени. По способу Винклера SO» для производства олеума полу-
чался термическим разложением камерной кислоты. Понятно, та-
кой олеум был дорог и не мог конкурировать с олеумом, выра-
батываемым из купоросного сланца.
Во-вторых, обжиговый газ для контактного производства не
применялся потому, что долгое время точно не знали причин от-
равления катализатора. Эти вопросы были разрешены классиче-
скими работами Книтча.
Недостаточно быстрое решение указанных вопросов связано,
конечно, и с тем, что спрос на олеум был невысок. Он возрос
лишь после того, как стала развиваться (с 1872 г.) промышлен-
ность искусственных красителей. К концу XIX и в начале XX вв.
спрос на олеум возрос также в связи с широкой подготовкой
всех государств к войне.
Впервые обжиговые газы были промышленно применены для
контактного производства серной кислоты Баденской анилиновой
и содовой фабрикой (BASF) ,в 1881 г.
В настоящее время призводство серной кислоты контактным
методом достигло огромных масштабов. Во всех странах доля
контактной кислоты в общей массе сернокислотной продукции из
года в год неуклонно растет. Это объясняется и военным значе-
нием контактной кислоты и ее более высокими качествами по
сравнению с башенной (любая концентрация, более высокая сте-
пень чистоты) и тем, что по последним усовершенствованным
способам контактного метода стоимость контактной кислоты
немногим выше стоимости башенной кислоты.
Так, доля контактной кислоты во всей сернокислотной продук-
ции составляет в Германии: 23—25% в 1928 г., 38% в 1935 г.,
48% в 1936 г.; в Англии 16,4% в 1929 г„ 25% в 1932 г. и 34,5%
в 1938 г. В Японии в 1935 г. мощность контактных заводов со-
ставляла 10% от общей мощности сернокислотных установок,
176
но 38,4% от мощности намеченных к строительству сернокислот-
ных цехов. Особенно велика доля контактной кислоты в общей
продукции серной кислоты в США: с 19,5% в 1913 г. и с 30%
в 1923 г. она к 1939 г. поднялась до 60%.
Особенно быстрое развитие контактного метода производства
серной кислоты в США объясняется тем, что многие заводы там
работают на сере; газы же, получаемые при сжигании серы,
почти не требуют очистки. В связи с этим упрощается вся схема
производства, и кислота получается достаточно дешевой.
Другие методы окисления сернистого газа (озоном — в электрическом
поле, хлором, кислородом — в воде под давлением, кислородом — в растворе
солей железа и марганца и др.) пока не нашли промышленного применения.
Точно так 'же не нашли промышленного применения и методы непосредствен-
ного окисления элементарной серы или серу сульфидной до серной кислоты
(например азотной кислотой), минуя стадию получения SO».
Конкретных технологических схем производства серной кис-
лоты методом контактного окисления много. Однако во всех
этих схемах процесс производства разделяется на следующие
стадии: 1) специальная очистка газа; 2) сушка газа; 3) контакти-
рование и 4) абсорбция серного ангидрида. '
При специальной очистке газы освобождаются от остатков
пыли и от других вредных примесей (соединения мышьяка, ту-
ман серной кислоты и др.). Для лучшей очистки от вредных
примесей газы при промывке слабой кислотой увлажняются па-
рами воды; перед направлением на катализатор газы подвер-
гаются осушке.
Стадия контактирования — центральная операция контактного
процесса: сернистый газ в присутствии катализатора превра-
щается в серный ангидрид.
В стадии абсорбции серный ангидрид, абсорбируясь циркули-
рующей кислотой, превращается в продукцию.
Отдельные конкретные технологические схемы различаются
между собой методами и аппаратурным оформлением процесса
очистки, применяемыми контактными массами, конструкциями
контактных аппаратов, аппаратурным оформлением контактных
отделений в целом и абсорбционных отделений.
В СССР большинство контактных заводов работает по си-
стеме Герресгоф-Байера (рис. 81).
Из печей / через боров 2 и сухой электрофильтр з газы по-
ступают в промывные башни 5 и 6, орошаемые серной кислотой.
Назначение этих башен — окончательное освобождение газов от
пыли, охлаждение и увлажнение их. После промывных башен
газы проходят пару мокрых электрофильтров 7, в которых ула-
вливаются соединения мышьяка.
Пройдя через увлажнительную башню 8, газы поступают во
вторую и третью пару электрофильтров 9 для окончательного
освобождения от мышьяка. В сушильной башне 10 газы осво-
бождаются от паров воды, а в брызгоуловителе 11 (башне, на-
саженной кольцами Рашига, не орошаемой) от брызг кислоты.
12 Зак. 5049. Технология серной кислоты. 177
Рис. 81. Схема контактного завода Герресгоф-Байера.
После компрессора 12 и маслоотделителя 13 газы направляются
в контактный узел. Контактный узел состоит из двух контакт-
ных аппаратов 16 и 17, двух терморегуляторов 14 и 15, пуско-
вого подогревателя 32 и топки 31. При нормальной работе за-
вода газ проходит последовательно оба терморегулятора 14 и 15,
где подогревается теплотой возвращающихся газов. Из второго
терморегулятора газ поступает в первый контактный аппарат 16,
где SO2 частично переходит в SO3; затем горячий газ проходит
через второй терморегулятор и второй контактный аппарат 17,
где заканчивается контактирование. После этого газы, пройдя
через первый терморегулятор 14, где они охлаждаются (нагре-
вая встречные газы), поступают в ангидридный холодильник 18.
В последнее время на заводах СССР в связи с применением но-
вых конструкций контактных аппаратов некоторое изменение
получило все контактное отделение.
Абсорбция SO3 происходит в двух башнях-абсорберах 19 и
20, насаженных кислотоупорными шарами и кольцами и оро-
шаемых серной кислотой. Брызги, увлекаемые газами из абсор-
беров, задерживаются в ловушке 21, после чего хвостовые газы
выбрасываются в атмосферу. Кислота, циркулирующая по про-
мывным башням, охлаждается в холодильниках 26 и подается
насосами 23.
Первый абсорбер 19 орошается олеумом, а второй 20 и су-
шильная башня 10 98%-ной кислотой. Вытекающая из абсор-
беров кислота проходит через сборники 24, насосы 25, холодиль-
ники 26 и идет затем обратно на абсорберы. Кислота в количе-
стве, соответствующем получаемой продукции (за счет абсорб-
ции), отбирается из системы и отправляется на склад или в ци-
стерны. Если кислоту из цеха нужно дать не в виде олеума,
а в виде одного лишь моногидрата или в виде купоросного
масла, то моногидрат или олеум разбавляются в смесителе 33
водой или слабой кислотой. В качестве слабой кислоты иногда
применяется кислота, отбираемая из промывных башен. Из сме-
сителя зз готовая продукция поступает в сборник 34 и насо-
сами 35 перекачивается на склад или в цистерны.
При пуске завода или’ во время расстройства технологиче-
ского режима газы, идущие после маслоотделителя на контак-
тирование, подогреваются в пусковом подогревателе 32 топоч-
ными газами, получаемыми от сжигания топлива в топке 31.
В последние годы в СССР построен ряд контактных устано-
вок по системе американской фирмы Кемико, Схема производ-
ства по системе Кемико показана на рис. 82.
Колчедан обжигается во вращающихся печах 1. Из печей
газ выходит при температуре 550°; содержание SO2 в газе под-
держивается в пределах 10—12%.
Система очистки газа состоит из двух циклонов 2 для каж-
дой печи, двух промывных башен 3 и 4 с холодильниками 15
и 18 и насосами 16 и 17. для циркуляции кислот и электро-
фильтра 5 для осаждения тумана и очистки от мышьяка.
12»
179
Газ поступает в циклоны при температуре 480—550 . Цик-
лоны выделяют из газа от 85 до 95% пыли -— в зависимости от
величины пылинок и способа работы. Из циклонов пыль разгру-
жается в транспортер, которым передается в агломерационную
установку.
Газ охлаждается в две стадии: в первой — с температуры
480 до 60~, во второй — до температуры 33—36 . Газ посту-
пает в первую оросительную башню з (полую) в четырех точках
и пропускается с большой скоростью над кислотной ванной на
дне этой башни (крепость кислоты 10—12 Вё). При этом из
газа вымываются остатки пыли.
Рис. 82. Схема контактного завода Кемико.
После этого газ охлаждается во второй башне 4, наполнен-
ной кольцами Рашига и орошаемой также кислотой (10% H2SO4).
Орошающая башни кислота охлаждается в холодильниках 75 и
18 и подается насосами 76 и 77. После башен влажные газы
поступают в электрофильтр 5, в котором осаждается туман
серной кислоты и улавливается мышьяк.
Сушка газа производится в одной башне 6, дно которой
служит одновременно баком, а верх башни — брызгоуловите-
лем. Башня орошается (насос 20) 93%-ной- кислотой. Кислота
по выходе из сушильной башни охлаждается в холодильнике 19-
Для поддержания оптимальной концентрации SO2 (7—8%) перед
сушильной башней в систему может засасываться воздух (если
концентрация SOs в газах перед сушильной башней больше
180
8%). После сушильной башни устанавливается вентилятор 7,
протягивающий газы через систему.
Контактный узел состоит из топки 8, теплообменника 9 и
контактного аппарата 10. После вентилятора и маслоотдели-
теля 21 газ идет через теплообменник 9 в противотоке с горя-
чими газами из контактного аппарата 10. Из теплообменника
газ поступает в нижнюю часть контактного аппарата, где сна-
чала проходит через слой кварцевой гальки, затем через тепло-
обменные трубки Фильда и, наконец, через слой контактной
массы. Разогрев контактной группы производится топочными
газами из топки 8, Газы после контактного аппарата проходят
теплообменник 9, где охлаждаются с температуры 420—430°
до 210°, и далее проходят через олеумиый 11 и моногидратный
Рис. 83. Схема Теителевского контактного завода.
12 абсорберы. Через олеумный абсорбер может пропускаться по
желанию весь газ или только часть его. Циркулирующая в аб-
сорберах кислота охлаждается в холодильниках 22 и 24 и ка-
чается насосами 23 и 25.
На заводах СССР эта система осуществлена со значитель-
ными изменениями: в большинстве случаев печное отделение
состоит из обычных механических печей, контактные аппараты
Кемико заменены аппаратами советской конструкции и др.
Первой схемой контактного процесса, по которой были по-
строены контактные заводы в России, является схема Тенте-
левского химического завода.
Эта схема была разработана самим Тентелевским заводом..
Способ Теителевского химического завода для своего времени
был настолько совершенным, что он нашел применение в целом
ряде стран. В конструкции отдельных аппаратов! тентелевский
способ не устарел и до сих пор.
На рис. 83 представлена технологическая схема в оформлении Теите-
левского химического завода. Из печей j газы поступают по общей сборной
Р5°е г в пыльную камеру з, оттуда по трубе 4 в холодильник 5, после.
181
чего в форвашер — промыватель в, а затем в промывную башню 7. В фор-
вашере и промывной башне газы освобождаются от последних остатков пыли
и соединений мышьяка. Далее газы последовательно проходят через сушиль-
ные башни 8, из которых первая 1Масажена коксом, ничем не орошается
и служит для улавливания брызг воды, увлекаемых газами из промывной
башии. Остальные три башни орошаются серной кислотой для освобождения
газов от паров воды.
За сушильными башнями газы «дут в компрессор 9, который протяги-
вает газы через систему. После компрессора газы последовательно прохо-
дят два фильтра-маслоотделителя ю. Здесь газы должны освободиться от
масла, захваченного в .компрессоре. Подогревшись в терморегуляторе
газы проходят через контактный аппарат is и затем возвращаются через
терморегулятор н, где отдают свою теплоту идущим в контактный аппарат
газам. Проходя далее через ангидридный холодильник 1:1, газы еше более
охлаждаются и поступают в абсорбер 14. Здесь SO3 абсорбируется оро-
шающей серной кислотой, а остаточный газ через трубу 15 выбрасывается
в атмосферу. Вытекающий из абсорбера 14 олеум частично через монтежю
If: перекачивается на склад. Другая часть олеума идет в смеситель 17, где
разбавляется водой до кислоты крепости 96—98%. Эта кислота проходит
через холодильники 18 и 19 н эмульсером so подается в бак 21, из которого
поступает в абсорбер 14. Во время пуска системы газы нагреваются в по-
догревателе 22.
Из других способов контактного процесса, широко распро-
страненных в недалеком прошлом, следует отметить способы:
Баденской анилиновой и содовой фабрики, Грилло-Шредера и
Союза маннгеймских химических фабрик.
Способ Баденской анилиновой и содовой фабрики мало отличался от
способа Теителевского завода. Основные особенности способа Грилло-Шре-
дера заключались в следующем: очистка газов от мышьяка проводилась
в башнях, насаженных коксом, периодически смачиваемым слабой серной
кислотой; теплота реакции контактирования ие использовалась и газы, иду-
щие на контактирование, подогревались в огневом подогревателе; в каче-
стве катализатора применялась платина, наносимая на сернокислый магний;
абсорбция SO.-, проводилась в горизонтально расположенных цилиндрах по-
верхностным поглощением.
Наибольшее отличие от всех других систем имела система Союза манн-
геймских химиков. Воздух, поступающий на обжиг колчедана, предвари-
тельно осушался в башнях серной кислотой. Газы по выходе из лечи про-
ходили через шахту, наполненную кусковым колчеданным огарком (являю-
щимся катализатором для реакции окисления SOa), где SOa на 40—50%
окислялся до SO.3. После охлаждения газы поступали в абсорберы, а после
абсорбции SOs шли в контактный узел с платиновым катализатором для
окисления остающейся части SOa.
Указанные способы были вытеснены другими, во-первых,
потому, что мощности отдельных установок оказались недоста-
точными в связи с возрастающем спросом «а олеум, а во-вто-
рых, в вытеснении этих схем сыграли роль и недостатки их
оформления. Так, крупным недостатком теителевского способа
является большое сопротивление аппаратуры (барботеры в от-
делениях очистки и абсорбции). Недостаток способа Грилло-
Шредера в самой схеме — неиспользование тепла контактирова-
ния и в связи с этим необходимость большого расхода топлива
на подогрев газов (к этому же трубы подогревателя часто про-
горали). Кроме того, далеко не совершенной была и часть аппа-
ратуры (например абсорбера).
182
Схема контактного процесса очень упрощается, если кон-
тактный завод в качестве сырья потребляет элементарную серу.
Ниже (стр. 304) приведена схема американского контактного за-
вода, работающего на сере.
Значительно изменяется схема контактного завода в случае
применения так называемого мокрого катализа, при котором из
газов, вышедших из контактных аппаратов и содержащих SO3,
серная кислота выделяется охлаждением. Такая схема также
приведена ниже.
II. Специальная очистка и осушка газа
I. Очистка газа от мышьяка н тумана серной кислоты
Теоретические основы очистки. Мышьяк, содержащийся
в колчедане, при сгорании последнего почти полностью пере-
ходит в мышьяковистый ангидрид.
Концентрация AS2O3 в газе зависит от содержания его
в колчедане, от степени выгорания и может колебаться в зна-
чительных пределах от 0 до 40 мг/м3. Так как температура газа
по выходе из печи очень высока, то AS2O3 находится в нец^
в газообразном состоянии.
Применяемые при производстве серной кислоты контактным
способом катализаторы, как мы увидим ниже, отравляются
мышьяком, поэтому необходимо полностью очистить печные
газы от мышьяковых соединений. Очистка газов от мышьяка
производится промывкой их в специальных башнях и в электро-
фильтрах с одновременным охлаждением газа.
В табл. 43 приведены данные по упругости паров AS2O3 при
различной температуре
Таблица 43
Температура в Ч/ Упругость в мм рт. ст. Температура в °C Упругость в мм рт. ст.
60.8 2,4- 10~т 153 0,01
83 2,5 10~г> 165 0,027
103 4,6 10~* 240,8 6,3
119 1,9- 10-з 261,8 18,2
124 2,2-Ю-з 290,9 64,5
142 1,4- IO 2 306,8 110,6
Из этих данных видно, что при охлаждении ниже 60° прак-
тически весь AS2O3 переходит в твердое состояние.
На практике газ охлаждают до еще более низкой темпера-
туры 30—35 . Это делают по следующей причине. Газы после
очистки поступают в сушильную башню, где из них погло-
Шается почти вся влага, которая переходит в абсорбционное от-
183
деление. Чем больше этой влаги, тем больше будет воды в по-
лучаемой кислоте, тем меньше будет получаться олеума.
При охлаждении газов образуется мельчайший туман —
аэрозоль AS2O3 и H2SO4. Частицы -тумана AS2O3 тесно связаны
с частицами H2SO4 и при удалении первого удаляется в такой
же пропорции и последняя. Этот мышьяково-кислотный туман
белого цвета, хорошо виден в охлажденном газе. Точных све-
дений о размере частиц тумана мышьяковистого ангидрида и
серной кислоты у нас нет, но, сопоставляя различные литератур-
ные указания, можно считать размер их ~10-5—Ю-4 см. Такой
туман очень устойчив. Он разрушается или при оседании под
влиянием силы тяжести или при коагуляции, т. е. слипании ка-
пелек при взаимном столкновении под влиянием броуновского
движения.
Определим скорости обоих этих процессов и тем самым докажем устой-
чивость этого тумана. Примем средний радиус частицы тумана 1 • 10-5 см и
температуру газа 100°. Для такой частицы закон Стокса не вполне применим,
так как величина частицы соизмерима с длиной свободного пути молекул
газа, т. е. такая частица будет успевать в некоторые промежутки времени
проходить — падать, не сталкиваясь с молекулами газа, поэтому сопротивле-
ние среды в этом случае несколько меньше. Кеннинг установил, что ско-
рость падения таких частиц можно подсчитать по несколько измененной фор-
муле Стокса, а именно:
где г — радиус частицы; у — удельный вес; — вязкость среды; 1 — длина
свободного пути; А—постоянная, значение которой зависит от рода удара,—
ее можно принять 0,86 при температуре 100°.
В нашем случае
г = 10—3 см; ( = 1,5 ?!см°; ц = 2,10-1 г сек/см'2;
длина свободного пути
где R — радиус молекул ~ 2 • 10—8 с.«; п — число молекул в 1 с.ч3:
6,06 • Ю23-273
" ~ 22 400 • (273 + 100)
2 IO’3;
тг-2-101»-4-10“1«/2 35.5-103 0,355-105’
2 • io-к -1,5 (1 + 0,86
\ 10-° /
и' = ----------92 10-"----------~ ~ 3 см!сек
или ~ 2 с.и/час, т. е. скорость оседания таких частиц очень мала.
Определим теперь скорость коагуляции для этого процесса. В случае
монодисперсного тумана можно применять формулу Смолуховского:
dn
di
/Сп\
(1)
У 2 т:/?2Л ’
184
Гае п — число частиц; R — газовая постоянная, равная 8,48 10* г • с.«/°С • мол;
t — абс. температура, равная 273 + 100 = 373; /V —число Авогадро, равное
6,06 • 1023; S — радиус действия, в большинстве случаев равный 2г;
-г = 2 -10-7 г см!сек-\ I = — 1 == 2,80 • 10- см;
1 0,Зоэ • 105
А = 0,86 (см. выше); г =10- 5 см.
dn 2 • 8,48 • 10* • 2 • 373 Л _ 2,8-10~гА „
dt ~ 3-2-10-7.6,06 • IO-3 V + • ь ) п' ~
= 3,5-10-ю. 3,4 «2= 12-10-м». «2
Предположим, что первоначально концентрация тумана составляла 1 • 10+5
частиц в 1 с.и3. В одну минуту из этого количества коагулирует
12 - 1О-10 ГО10 60 = 720 частиц.
Определим время, за которое половина всех частиц тумана коагулирует,
т. е. когда—— 0,5. Преобразуем уравнение (1) и проинтегрируем его
"о
— Kdt;-------- = Kt
«2 nt w0
или при rtf, = О,5по
1 1 103
t — —— ----------------= — = 8 340 сек. — 140 мин.,
К«о 12 • 1О“10-105 12
т. е. туман скоагулирует на 50% только через 2 часа 20 мин. При большей
концентрации коагуляция будет происходить значительно быстрее, но но-
мере уменьшения числа частиц коагуляция будет замедляться.
Туман не только медленно оседает и плохо коагулирует, но
и медленно абсорбируется. Скорость абсорбции частиц тумана
указанной выше величины примерно в 104 раз меньше скорости
абсорбции газовых молекул.
Все это говорит о большой устойчивости тумана, поэтому и
простая промывка, в частности в башнях с орошаемой насадкой,
для поглощения его недостаточна. Для полного поглощения
необходимо или применять электростатическое осаждение ча-
стиц в электрофильтрах, как это делается на современных заво-
дах, или промывать газы методом барботажа, при котором по-
верхностная жидкая пленка значительно тоньше, чем при
встрече газа с жидкостью в орошаемых насаженных башнях.
На современных сернокислотных заводах в основном приме-
няются три способа очистки.
Способ Тентелевского завода. Газ после очистки от огарко-
вой пыли поступает в газовый пластинчатый холодильник,
представляющий собой полый вертикальный свинцовый ци-
линдр, внутри которого находятся узкие вытянутые трубы-пла-
стины 1 (рис. 84). Снаружи цилиндра устроена водяная рубашка.
Вода находится в кольцевом пространстве между внутренним
цилиндром 2 и наружным кожухом 3, а также в указанных тру-
бах-пластинах. Газ проходит сверху вниз по внутреннему ци-
линдру, соприкасаясь на пути с холодильными пластинами.
Всего имеется четыре ряда пластин; в каждом ряду по три па-
185
раллельных пластины. Холодная вода подается из водопровода
по трубам в пластины, выходит оттуда в кольцевое простран-
ство, подымается и стекает далее в дренаж.
При проходе газа через холодильник часть БОз и Н»О конден-
сируется на холодных
Рис. 84. Холодильник Теи-
телевского завода:
/ — трубы-пластины; 2 — внутрен-
ний цилиндр; 3 — кожух.
поверхностях и в виде кислоты стекает
вниз; газ уходит снизу холодильника.
Крепость стекающей кислоты зависит
от содержания в газе SO'3, HsO и от
температуры воды и колеблется от 30
до 60с; Вё. Кислота эта содержит зна-
чительное количество мышьяка и силь-
но загрязнена пылью. В холодильнике
газ охлаждается до 90—140°. После
холодильника газ по свинцовому газо-
ходу поступает в аппарат предвари-
тельной очистки — форвашер.
Форвашер — аппарат барботажного
типа. Газ пробулькивает в виде пу-
зырьков через слой жидкости и при
этом освобождается от примесей. Фор-
вашер представляет собой свинцовый
вертикальный цилиндр / (рис. 85),
внутри которого опущен колокол 2 с
волнистыми краями 3- В стенке ко-
Рис. 85. Форвашер:
/ — цилиндр; 2 —колокол; 3— края колокола; /—отвер-
стия; 5—входной газоход; 6 — выходной газоход; 7— ко-
жух для воды; 8 — труба для входа воды; 9 — труба для
выхода воды; 10— штуцер для спуска кислоты.
локола, над его краями, сделаны отверстия 4 для образования
.пузырьков. Стенки колокола 2 сделаны из свинца и припаяны
к крышке аппарата. В аппарат заливается вода до такой высоты,
чтобы ее уровень был на 20—25 мм выше отверстий в колоколе.
Газ поступает по газоходу 5, находящемуся в центре крышки,
проходит через отверстия и края колокола в виде пузырьков,
18G
входит в пространство между наружным цилиндром и колоколом
и отсюда направляется по четыре^ выходным газоходам 6, на-
ходящимся по краям крышки аппарата.
В форвашер заливается один раз в сутки около 1—1,3 мя
воды, при этом края колокола опущены в воду на несколько
Рис. 86. Промывная башня Теителевского завода.
1 — газоход; 2 — пространство под колоколом; 3 — воронка; 4 — коло-
кол,- 5 — отверстия в колоколе; 6 — зубчатые края; 7— сифон; 8 — слив-
ная труба; У — труба.
сантиметров. Вода поглощает из промывного газа серный анги-
дрид; при этом образуется серная кислота. По истечении сутоь
в форвашере образуется кислота крепостью 20—30° Вё, ко-
187
торую редко используют, так как она сильно загрязнена пылью
и недостаточно крепкая. Эту кислоту часто выпускали в кана-
лизацию, а иногда употребляли для производства суперфосфата
или солей. Вместе с SO3 в форвашере улавливается и мышьяко-
вистый ангидрид. По исследованиям Пейсахова в форвашере
улавливается около 75,0%, поступающего с газом SO3 и такой
же процент AS2O3.
Для окончательной очистки от мышьяковистого ангидрида и
тумана серной кислоты газ из форвашера поступает в промыв-
ную башню, которая устроена по типу форвашера и представ-
ляет собой как бы пять поставленных друг на друга форваше-
ров (рис. 86). Газ поступает по газоходу / в первый этаж башни
в пространство 2. Центр потолка первого этажа приподнят
в виде опрокинутой воронки 3 с отверстиями. Сверху воронка
накрыта колоколом 4, который зубчатыми краями 6 опирается
на дно второго этажа — потолок первого этажа. В горизонталь-
ной части колокола сделаны отверстия 5. Во втором этаже вода
залита несколько ниже отверстий. Газ из первого этажа прохо-
дит под колокол и через отверстия 5 и края 6 колокола 4 про-
булькивает через слой жидкости. Из второго этажа газ точно
так же поступает в третий этаж и т. д. Всего газ проходит че-
рез шесть этажей, причем 5 раз он пробулькивает через жид-
кость.
Свежая вода, служащая для промывки газа, поступает через
сифон 7 на верхний (шестой) этаж, а избыток жидкости с ше-
стого этажа перетекает на пятый через сливную трубу 8 и по-
ступает в низ четвертого этажа, откуда аналогично на третий,
и т. д. Таким образом осуществляется противоток газа и жид-
кости. Со второго этажа жидкость может быть удалена по
трубе 9 в канализацию. На каждый этаж заливается около
200—250 л жидкости. Ежесуточно опорожняют 2—3 нижних
этажа от жидкости, на опорожненные этажи по очереди сли-
вают жидкость с верхних, а на верхние этажи заливают свежую
воду.
Газ, проходя через промывную башню, обычно полностью
освобождается от тумана серной кислоты и мышьяковистого
ангидрида. По исследованиям Пейсахова на одном заводе на
втором этаже промывной башни улавливалось около 17—
20% SO3 и AS2O3, а суммарный процент улавливания форваше-
ром и вторым этажом промывной башни составлял 94—95. Та-
ким образом на долю остальных четырех этажей оставалось
только 5—6%, в то время как сопротивление их составляло
около 2/з всего сопротивления очистного отделения. Поэтому
такой способ очистки очень дорог в эксплоатации.
Несмотря на то что на долю четырех верхних этажей при-
ходится улавливать всего лишь 5—6% AS2O3, все- же иногда
бывает проскок незначительных количеств АкгОз через про-
мывную башню. Так, в одном случае в газе после промывной
башни содержалось 1—2 .wr/да3, и окончательное улавливание
188
мышьяка происходило в асбестовом фильтре-маслоотделителе.
Вообще асбест очень хорошо отделяет при фильтровании ту-
ман SO3 и AS2O3 и может быть применен наравне с хлопчато-
бумажной ватой для аналитических целей при испытании газа
на содержание As или тумана SO3.
Отработанная жидкость стекает из башни в виде кислоты
в 15—20 Вё. Содержание мышьяка в этой кислоте также ра-
стет параллельно увеличению крепости.
Зависимость растворимости AssOe от концентрации серной
кислоты и температуры графически изображена на рис. 87 (по
Постникову, Кузьмину и Воробьеву).
Е
Рис. 87. Растворимость As3O3 в серной кислоте.
Из рис. 87 видно, что в интервале температуры 10—100°
происходит растворение AS2O3 как твердого мышьяковистого
ангидрида (а не газообразного) и что AS2O3 лучше всего рас-
творяется в кислоте концентрации 76—79% H2SO4.
Тентелевский способ очистки 'газа от мышьяка в настоящее
время редко употребляется из экономических соображений
(большое сопротивление аппаратуры). Значительно большее рас-
пространение получает новый метод с применением мышьяко-
вых электрофильтров — энтарсенирунгов.
Способ Герресгоф-Банера. Газ после огарковых электро-
фильтров последовательно проходит две промывные башни.
Обе башни имеют одинаковое устройство (рис. 88). Свинцо-
вый кожух изнутри футерован кислотоупорным кирпичом.
В низу башни для расположения насадки имеется колосниковая
решетка, устроенная так же, как и в башнях башенных заводов
(см. гл. 5). Снаружи башня окружается железным каркасом и
устанавливается на постаменте высотой около 2,5 м.
189
В башнях в качестве насадки применяются кольца Рашига
с размерами в первой 120 X 120 и 100 X 100, а во второй
100 X' ЮО и 80 X 80 Л1Л1 (см. гл. 5).
Для распределения кислоты по насадке башни применяют
распылительную турбину, работающую от мотора (см. гл. 5).
Первая башня орошается кислотой крепости 55 Вё; вторая —
35 Вё. При прохождении через первую баш-
Рис. 88. Промывная
башня Герресгоф-
Байера:
1 — штуцер для входа га-
за; 2—люк для осмотра;
3 — колосниковая решет-
ка; 4 — трубы каркаса;
5 — кольца из углового
железа; 6—оросительный
прибор.
ню газ охлаждается обычно до 80—90 , во
второй до 30—35'. Одновременно с охла-
ждением газа происходит и частичное вы-
мывание из него тумана серной кислоты и
мышьяковистого ангидрида. В первой башне
вымывается ~ 50%, а во второй ~ 20—25%
тумана SO3 и As2O3.
На орошение башни подается в час 30 и3
кислоты, температура которой ~ 40’ на пер-
вой и 30° на второй башне. При прохожде-
нии через башню кислота нагревается и вы-
текает снизу башни через гидравлический
затвор. Кислота охлаждается в холодиль-
никах с погружными змеевиками из
свинца.
В некоторых случаях для отстоя грязи
кислоту перед холодильником пропускают
через сборник или отстойник. Из холодиль-
ника кислота самотеком поступает в центро-
бежный насос, затем передается на башню.
При остановке насоса вся кислота, находя-
щаяся в башне, сливается в холодильник
или в сборник-отстойник.
Кислота в первой башне при промывке
газа поглощает SO3 и крепость ее постепен-
но повышается, поэтому время от времени
ее разбавляют более слабой кислотой из
второй промывной башни, а во вторую
башню добавляют более слабую кислоту из
бака, в котором собирается кислота из эн-
тарсенирунгов или из увлажнительной баш-
ни (см. ниже). Избыток кислоты из первой
башни выводят, из системы и используют
для приготовления купоросного масла, сме-
шивая кислоту с олеумом, или для произ-
водства суперфосфата или для каких-нибудь других целей.
В промывную кислоту переходит 4—8% от всей сгоревшей
серы. Этот процент зависит от содержания SOs в печном газе,
т. е. от режима работы печей и качества сырья. Из первой про-
мывной башни часть SO3 идет дальше в виде брызг кислоты и
тумана и улавливается в следующих башнях и энтарсенирунгах.
Некоторое количество SO3 остается в газах даже по выходе из
190
всего отделения очистки. Так, по данным одного завода содер-
жание SOs в газах после отделения очистки составляет
0,34 г//и3, по данным другого завода 0,056 г/ти3, по данным
третьего завода—0,20 г/м3.
При нормальной работе сопротивление обеих башен равно
~ 20 мм вод. ст., но часто из-за плохой работы огаркового элек-
трофильтра или из-за разрушения насадки сопротивление сильно
возрастает, и _ в этом случае необходимо производить чистку
башни. Около входного штуцера в'башне вынимается кирпич и
через это отверстие удаляется грязь, осевшая на колосниках
и мешающая проходу газа в башне. Если же эта мера ие дает
необходимого понижения' сопротивления, то приходится промы-
вать башни. Если и это окажется недостаточным, то можно
предположить, что насадка очень сильно разрушена и обва-
лилась.
Плохая очистка газа от пыли в электрофильтрах приводит
также к быстрому загрязнению холодильника первой промыв-
ной башни, что ухудшает охлаждение промывной кислоты. По-
вышение температуры кислоты часто влечет за собой разруше-
ние насадки и футеровки. Кроме того, при повышении темпера-
туры орошающей кислоты в башнях газы будут содержать
больше влаги, что снизит выход олеума в абсорбционном отде-
лении. Таким образом плохая работа огаркового электрофильтра
постепенно разрушает и расстраивает всю контактную систему.
Поэтому необходимо особенно тщательно следить за работой
этого сухого электрофильтра, не допускать его остановки на
продолжительное время. Кроме того, необходимо достаточно
часто чистить холодильники, чтобы не допускать повышения
температуры кислот выше установленной нормы.
В связи с разрушением насадки в первой башне из-за высо-
кой 'температуры на некоторых заводах перед промывной баш-
ней устанавливается газовый холодильник. Последний предста-
вляет собой свинцовый газоход диаметром ~ 1 м и длиной
20 л, уложенный горизонтально в несколько изгибов, на
лотке; сверху газоход обильно орошается холодной водой, сте-
кающей в лоток и далее в дренаж. Газ в таком холодильнике
легко охлаждается до 100—130° и поступает затем в первую
промывную башню. В_ холодильнике частично конденсируется
серная кислота (крепкая) и время от времени спускается по спе-
циальному патрубку. Такой холодильник дал очень хорошие ре-
зультаты: прекратилось разрушение башни, уменьшилось загряз-
нение башен и холодильников и, что особенно валено, улучши-
лась очистка газа от мышьяка и тумана серной кислоты. По-
следнее, очевидно, объясняется тем, что при таком способе
охлаждения газа SO3 и AS2O3 конденсируются в более крупные
частички, которые затем легко улавливаются в электрофильтрах.
После охлаждения газа и отмывки его от значительной ча-
сти мышьяковистого ангидрида газ поступает для окончатель-
ной очистки в мышьяковые или мокрые электрофильтры, назы-
191
ваемые энтарсенирунгами. На каждой контактной системе уста-
новлено последовательно три пары мокрых электрофильтров,
причем в каждой паре газ идет параллельно (рис. 89).'
Такое расположение мокрых электрофильтров дает возмож-
ность, с одной стороны, производить полную очистку, а с дру-
гой стороны, совершенно безболезненно отключать для очистки
и ремонта любой из электрофильтров. Это достигается тем, что
газ после каждой пары энтарсенирунгов выходит через соот-
ветствующие колокольные гидравлические затворы в общий га-
зоход, затем снова через такие же затворы поступает в следую-
щие два парных электрофильтра и т. д.
Опуская колокол в гидравлический затвор на входе и вы-
ходе какого-нибудь энтарсенирунга, заставляют весь газ прохо-
дить через другой парный аппарат.
Рис. 89. Расположение
мокрых электрофильтров.
Для достижения высокой степени очистки после первой
пары электрофильтров, где газ освобождается от основной
массы крупных частиц тумана AS2O3 и HjjSOi, газ дополни-
тельно увлажняют. При этом на мелких частицах тумана (в боль-
шинстве случаев уже заряженных) оседает конденсирующаяся
вода, а это ведет, в свою очередь, к укрупнению частичек и, сле-
довательно, к более легкой очистке. Увлажнение производится
в башне, находящейся после первой пары энтарсенирунгов.
Башня устроена так же, как промывные башни, но имеет не-
сколько иные размеры и наполнена коксом или кольцами. Эта
башня орошается кислотой в 2—3° Вё.
По мере повышения крепости этой кислоты ее разбавляют
водой, а избыток переливают во вторую промывную башню. Газ
проходит ч^рез башню снизу вверх, насыщаясь при этом водя-
ными парами; при дальнейшем проходе по газоходам и электро-
фильтрам газ охлаждается на несколько градусов; часть паров
конденсируется на имеющихся уже центрах конденсации, т. е.
на заряженных частицах тумана AS2O3 и H2SO4.
На полноту улавливания тумана AS2O3 и H2SO4 влияет также
режим работы промывных башен. Выше мы .указывали, что во
второй промывной башне газы охлаждаются до 30—35°, однако
192
дальнейшее понижение температуры может вызвать ухудшение
работы энтарсенирунгов.
Для хорошей очистки желательно также укрупнить частицы
тумана, что достигается увлажнением газа и частичной конден-
сацией этой влаги. При низкой температуре газов влагосодержа-
ние их будет невелико, а поэтому и
щейся влаги мало, т. е. частицы ту-
мана будут мало укрупняться, а это
повлечет за собой ухудшение очист-
ки газа. Такое явление и наблюдает-
ся на некоторых заводах в зимнее
время, когда температура газов
сильно снижается.
Подсчитаем, насколько укрупнят-
ся частицы тумана, если газ, насы-
щенный над 20%-ной кислотой при
20°, охладится на 1° и если он на-
сыщен при 35° и также охлаждает-
ся на 1° при условии, что в газе на-
ходились частицы тумана и концен-
трация его составляла 100 мг/м3. При
20° упругость паров воды равна
15,4 мм, а при 19° — 14,4 мм рт. ст.,
т. е. парциальное давление водяных
паров изменилось на 1 мм, что со-
ответствует, приблизительно, умень-
шению влагосодержания на 1 г/м3.
При 35° упругость паров равна
37,2 мм, а при 34° 35,2 мм рт. ст.,
т. е. парциальное давление измени-
лось на 2 мм, что, приблизительно,
соответствует конденсации 2Н2О.
Если предположить, что все кон-
денсируется на уже имеющихся ча-
стицах тумана и что этот процесс
протекает равномерно на всех части-
цах, то в первом случае вес их уве-
личится со 100 мг до 1,1 г, т. е.
в 11 раз, а во втором до 2,1 г,
Т- е. в 21 раз, и, следовательно, радиус их увеличивается, в пер-
вом случае в 2,2 раза и во втором — 2,8 раза. Так как с уве-
личением размера частиц они сильнее заряжаются и быстрее оса-
ждаются в электрофильтре, т. е. легче улавливаются, то из
этого примера ясно видна роль увлажнения газа перед поступле-
нием его в электрофильтр. Чем больше тумана и чем он мельче, тем
лучше следует его увлажнять перед подачей В' электрофильтр.
Энтарсеиирунг представляет собой пластинчатый вертикаль-
ный электрофильтр. В квадратной свинцовой камере с размерами
основания 1,5 X 1,5 м и высотой 4,5/и подвешены вертикально на
количество конденсирую-
40Ю
JnniU№S//////W, /-У/
Рис. 90. Энтарсеиирунг пластин-
чатый:
1 — гидравлический затвор на входе;
2 — гидравлический затвор на выхо-
де; 3— рама для электродов; 4 — фар-
форовый изолятор; 5 — масляный за-
твор; 6 — решетка.
13 Зак. 5019. Технология серной кислоты.
193
расстоянии 250 мм друг от друга и от двух стенок аппарата пять
свинцовых осадительных электродов (рис. 90) толщиной 3 дли,
длиной 3 м и шириной 1,5 м (по всей ширине камеры). Корони-
рующие электроды из освинцованной медной проволоки подве-
шены рядами между осадительными пластинами на расстоянии
125 ж.м от пластин (посредине) и на расстоянии 200 мм друг от
друга. Снизу коронирующие провода натягиваются свинцовыми
грузами, сверху они все закреплены на медной освинцованной
раме, которая с двух сторон выходит из камеры в боковые ко-
робки, где она подвешена к болтам, также медным освинцован-
ным; последние подвешены к гирляндовым изоляторам. Для того
чтобы газ, находящийся в аппарате, не соприкасался с изолято-
рами, устроены масляные затворы 5. К болту, который висит на
изоляторах, припаян колокол, концы которого опущены в чашу
с трансформаторным маслом. Чаша эта укреплена на боковых
коробках.
Таким образом, с одной стороны, газ отделен от атмосфер-
ного воздуха и изоляторов, с другой — части, находящиеся под
током, изолированы от внешних заземленных частей аппарата
при помощи трансформаторного масла.
Хотя газ, пройдя аппарат, уже значительно очищен от тумана
H2SO4, все же небольшое количество тумана остается в газе и
может осесть на внутренних частях колокола, обращенных к газу.
Эта осевшая кислота будет стекать по стенкам колокола в масло,
но вследствие своего большого удельного веса она не будет
плавать в масле, а сейчас же опустится на дно масляной чаши,
откуда ее время от времени спускают через специальный кран.
Если бы кислота не опускалась на дно, а оставалась на поверх-
ности, то между болтом, находящимся под напряжением,’и стен-
ками чаши образовался бы проводящий слой кислоты, т. е. проис-
ходили бы разряды на поверхности, что повело бы к прекраще-
нию работы электрофильтра.
Колокол устанавливается так, чтобы болт проходил точно по
центру чаши и чтобы края колокола также были на равном рас-
стоянии от дна и стенок чаши. Масло наливается слоем в 500—
600 мм', при скоплении кислоты уровень масла повышается, что
можно заметить по масломерному стеклу. Если накопленная под
маслом кислота не будет во-время удалена, могут произойти
пробои через кислоту под маслом; это сопровождается особым
бурлящим звуком. Допускать такое явление ни в коем случае
нельзя, так как это может повлечь за собой воспламенение масла.
Последнее может также произойти по причине попадания масла
внутрь электрофильтра из-за переполнения чаши маслом и из-за
большого разрежения в системе и при резких изменениях разре-
жения.
Во избежание таких аварий — воспламенения масла, что часто,
влечет за собой пожар, — в новых конструкциях -взамен масля-
ных изоляторных приспособлений устанавливаются обогреваемые
кварцевые изоляторы. Благодаря обогреву влага и туман не кон-
194
денсируются и не осаждаются на кварце и он не теряет изоля-
ционных свойств. Опыт показывает удовлетворительную работу
Рис. 91. Гидравличе-
ский колокольный за-
твор.
таких изоляторов.
Кислота и АзаОз, уловленные в электрофильтре, стекают по
стенкам в нижнюю часть, устроенную в виде бункера; из послед-
него через гидравлический затвор кислота опускается в ящик,
откуда ее насосом перекачивают в бак второй промывной башни.
Так как газ содержит в себе кроме H2SO4 и As2O3 еще ча-
стицы твердого селена, а иногда и небольшие количества остат-
ков огарковой пыли, то в первой паре электрофильтров сильно
загрязняются коронирующие электроды. Поэтому не реже одного
раза в месяц необходимо производить про-
мывку первой пары энтарсенирунгов — луч-
ше всего теплой водой, которая легко смы-
вает все загрязнения с электродов.
Все части аппаратуры, служащие для
крепления свинца, сделаны не из железа, а
из меди. Это вызвано следующими сообра-
жениями. При образовании трещины или не-
плотности в свинце серная кислота, прони- •
кая вглубь и соприкасаясь с железом, будет
выделять водород; последний с мышьяко-
вистым ангидридом дает газообразный AsH3,
который ие осядет в электрофильтре и прой-
дет в контактный аппарат. При соприкосно-
вении же серной кислоты с медью выделе-
ния водорода не будет, так как Си стоит
ниже Н2 в ряде напряжений и не может вытеснить водорода.
Следовательно, образование AsH3 исключено'.
Для выключения каждого энтарсенирунга служат гидравли-
ческие затворы с опускающимися колоколами. Устройство и
принцип их работы ясно видны на прилагаемом схематическом
чертеже (рис. 91). Такие затворы установлены на входе и на
выходе аппарата.
Вторая и третья пара энтарсенирунгов отличается от первой
только тем, что в них установлено не пять, а шесть осадитель-
ных пластин на расстоянии 214 мт друг от друга. Первая пара
энтарсенирунгов питается от одного электрического агрегата, вто-
рая и третья — от другого. Это сделано из следующих сообра-
жений: в первой камере больше оседает грязи, провода как бы
утолщаются, расстояние между проводом и пластиной большее,
поэтому здесь можно держать одно напряжение, во второй и
в третьей камере провода чистые, расстояние меньше, поэтому
здесь при том же самом потенциале градиент значительно
больше. Осаждение конденсата в электрофильтрах следующее
(по работе одного завода):
Электрофильтры Конденсат Концентрация. H2SO4
в м'з в °/0
1-я пара......... 7 19—20
2-я пара.......... 0,15—0,6 4—5
3-я пара.......... 0,0 —0,15 2,5
Приводим часовой баланс АбзОз, составленный бригадой Орг-
хима по работе одного действующего завода.
Поступило АвоОз с колчеданом................... 284,28 г
Осталось в огарке и в пыли форкамеры и сухого
коттреля..................................... 136,99 „
Уловлено в 1-й промывной башне .... ....... 85,90 ,
Уловлено во 2-й промывной башне ................ 25,92 „
Уловлено мокрыми электрофильтрами . ........... 34,47 „
В новых аппаратах, устанавливаемых в настоящее время, в ка-
честве осадительных электродов применяются шестигранные
трубы, а не плоскости. Применение шестигранников дало воз-
можность увеличить скорость газа в аппарате до 0,75—1 м/сек
без ухудшения очистки, что, в свою очередь, повлекло за собой
уменьшение габаритов аппарата и уменьшение расхода свинца.
Наиболее часто устанавливают два последовательных аппарата
с 126 трубами в каждом. Эти аппараты однокамерные, содержат
по одному пакету шестигранников. Внутренний диаметр каждого
шестигранника 250 мм, длина поля 3,5 м. Через аппарат про-
пускают 20 000—25 000 м3/час газа. В качестве коронирующих
электродов употребляется шестигранная звездочка с диаметром
в 9—10 мм.
Для лучшего распределения газа по всему сечению аппарата
в нижней его части установлена деревянная освинцованная ре-
шетка, на которую положен слой кварца или кокса в 500 мм.
Общий вид аппарата показан на рис. 92. Расход энергии около .
9 квт-ч.
Для заводов меньшей мощности устанавливается аппарат, со-
держащий 44 трубы (рис. 93). Этот аппарат двухкамерный, про-
пускает ~ 8000 м3/час газа, расходует энергии 3,5 квт-ч;
в остальном он аналогичен описанному выше.
Для увлажнения газа между 1-й и 2-й ступенью электро-
очистки в газоход (стояк) вбрызгивается форсунками вода. В но-
вых системах увлажнительной башни и 3-й ступени энтарсенирун-
гов не устанавливают, так как описанная двухступенчатая очистка
в трубчатых аппаратах дает достаточно полную очистку газов
от AS2O3, Se и SO3.
При обслуживании электрофильтров необходимо:
1. Следить: а) за чистотой внешних каркасов; б) за чистотой
камеры внутри, регулярно производя промывку камер; в) за уров-
нем масла в затворах, удаляя накопившуюся кислоту; г) за раз-
режением после третьего энтарсенирунга, не допуская сильного
разрежения или толчков, так как при этом может заплеснуть
масло в камеру, в результате чего произойдет пожар; д) за
исправным состоянием колоколов; е) за стеканием кислоты из
электрофильтров, не допуская засорения затворов (в особенности
после промывки камер, когда на дно осело много грязи).
2. Не перекрывать одновременно колоколами обоих электро-
фильтров одной пары (даже на короткое время).
196
3. Поддерживать возможно большее напряжение и силу тока
во всех аппаратах, но не доводить до пробоя.
Признаком сильного загрязнения камеры и необходимости
промывки может служить значительное понижение силы тока
(миллиампераж).
Рис. 92. Сотовый энтарсенирунг 126-трубиый. Рис. 93. Сотовый эитар-
сенирунг 44-трубный.
Общее сопротивление отделения очистки не должно превы-
шать 100 мм вод. ст.
При нормальной работе энтарсенирунгов уже после второй
пары присутствия мышьяка не обнаруживается н третья пара
является контрольной. В настоящее время разрабатывается си-
стема очистки, в которой промывка и очистка производятся в од-
ном аппарате. В этом случае нижняя часть последнего пред-
ставляет собой промывную башню, а верхняя — трубчатый
энтарсенирунг.
197
Система Кемико. Эта система несколько отличается от опи-
санной выше системы Герресгоф-Байера конструкцией и режи-
мом орошения промывной башни.
Горячие газы после циклонов или электрофильтров входят
в первую промывную башню (рис. 94).
Эта башня полая, без насадки, орошается при помощи фор-
сунок кислотой в 50—55° Вё (в некоторых случаях 20—25° Вё).
В нижней части башни 2, отделенной от верхней / кирпичным
сводом, кислотная ванна поддерживается на определенном уровне
при помощи соответствующего положения сливной трубы 6.
Газы поступают в башню по
четырем трубам 3, имеющим
наклон к горизонтальному
положению в 45° и оканчи-
вающимся близко от поверх-
ности кислоты; таким обра-
зом, горячие, содержащие
пыль, газы ударяются о по-
верхность кислоты, при этом
значительно очищаются от
•пыли и поступают в верх-
нюю часть башни, где про-
мываются распыленной кис-
лотой. Для распыла кислоты
установлено 9 или 11 фор-
сунок 9 и 10, приспособлен-
ных к работе на грязных
жидкостях. Башня, имеющая
внутренний диаметр 2,225 м
и высоту (верхней части)
около 3,5 /и, пропускает в
час ~ 15 тыс. мя/час газа и
охлаждает их до 80—90°.
Количество орошающей
кислоты составляет 80 мя/час-
Вся башня стальная, тща-
Рис. 94. Промывная башня системы Ке-
мико:
1 — реакционная часть; 2 — сборник; 3 — вход
газа в башню; 4 — вход газа в верхнюю часть
башни; 5 — выход газа; 6 — кыхък кислоты; 7 —
спуск шлама; 8 — футеровка; 9, 10 — форсунки;
11 — отстойник; 12 — дно; 13 — крышка; 14 —
затвор.
тельно футерованная, причем нижняя часть обложена свинцом.
Для полного спуска кислоты из ванны и для чистки ее слу-
жит специальный спускной клапан.
Нормально кислота стекает по переливной трубе в отстой-
ник 11, установленный под башней. В отстойнике осаждается
уловленная пыль; шЛам из отстойника периодически выводится
через конус (дно) 12 отстойника, а осветленная кислота через
холодильник подается насосом обратно на орошение.
Вторая башня устроена аналогично описанным выше в си-
стеме Герресгоф-Байера, т. е. с насадкой из колец Рашига.
Башня орошается кислотой в 30—35° Вё. Во второй башне газы
охлаждаются до температуры 30—35° и поступают в два после-
довательных мокрых электрофильтра — энтарсенирунга.
198
Для увлажнения газов в вертикальный газоход-стояк, по ко-
торому газы проходят из первого электрофильтра во второй,
вбрызгивается вода.
В остальном система промывки аналогична вышеописанной си-
стеме Герресгоф-Байера.
В этой системе также нужно следить за правильным режимом
кислот, за работой форсунок, за температурами газа, не допу-
ская сильного повышения (выше 36° снижается выход олеума)
и в особенности переохлаждения ниже 25—30° (ухудшение ула-
вливания AS2O3), за работой отстойника, не допуская его пере-
полнения шламом, а также за работой электрофильтров.
2. Осушка газов
В промывных отделениях контактных установок газ в послед-
ней стадии очистки обильно орошается водой или очень слабой
кислотой. В связи с этим выходящий из отделения очистки газ,
освобожденный от различных вредных примесей, содержит боль-
шое количество влаги. Содержание влаги в газе тем больше, чем
выше его температура при выходе из отделения очистки.
До направления газа в контактный узел он освобождается и
от влаги. Влажный газ, содержащий SO2, при низких температу-
рах разъедает железо и сталь, а последующие аппараты (ком-
прессор, маслоотделитель, а также газоходы) выполняются из
этих материалов. Влага (как об этом будет сказано подробно
ниже) является ядом для платиновой контактной массы. Хотя
ванадиевые контактные массы влагой не отравляются, но если
происходит понижение температуры в контактном аппарате, то
образующаяся из НЮ и SO3 и конденсирующаяся на контактной
массе серная кислота разрушает массу. Наконец, при наличии
влаги в газе, в дальнейшем, при охлаждении газа (в отделении
абсорбции) образуется чрезвычайно трудно абсорбируемый серно-
кислотный туман.
Абсорбция паров воды серной кислотой. На всех контактных
установках осушка газов производится высококонцентрирован-
ной серной кислотой. После осушки содержание влаги в газе
должно быть не больше 0,2 г/т9.
Приведем раствор газа в жидкости (с содержанием газа в жидкости,
равным Ci, которому соответствует его упругость над жидкостью р,-) в со-
прикосновение с газовой смесью, в которой фактическое парциальное давле-
ние газа равно рд, причем r>g / P-i- Если при этом рд> Гч< то будет про-
исходить процесс дальнейшего растворения газа в жидкости — абсорбция;
если же pa<Pi, то, наоборот, произойдет процесс перехода части раство-
ренного в жидкости газа обратно в газовую фазу (десорбция, отрицательная
абсорбция, эксорбц.ия).
Абсорбция (процесс растворения) или десорбция (обратный процесс) бу-
дет происходить до тех пор, пока система газ-жидкость не достигнет равно-
весия на новой основе, когда при новом содержании газа в жидкости будет
иметь место равенство рд = pt.
Скорость абсорбции зависит главным образом от процесса диффузии.
Дело в том, что на поверхности раздела газ-жидкость с одной ее стороны
199
имеется тонкий слой газа, в котором концентрация абсорбируемого газа ниже,
чем во всем газовом пространстве над жидкостью. В этом слое газа—i газо-
вой пленке — конвекционные токи газа гораздо слабее, чем в главной массе
газа. С другой стороны плоскости раздела имеется тонкий слой жидкости,
в котором концентрация абсорбируемого газа выше, чем в остальной массе
жидкости. В этом слое жидкости (жидкой пленке) конвекционные токи
получаются гораздо слабее, чем во всей остальной массе жидкости. Эти
пленки н составляют основное сопротивление для перехода вещества из
одной среды в другую.
Обозначим парциальное давление абсорбируемого газа в главной массе
газа через рд: на границе газовой пленки, обращенной к жидкой фазе, пар-
циальное давление будет равно упругости газа, соответствующей дайной кон-
центрации газа в жидкости, т. е. будет равно р{.
Для скорости диффузии через газовую пленку можно написать уравнение:
где Кgg — коэфициент скорости диффузии, отнесенный к единице толщины
газовой пленки, хд— толщина газовой пленки, А — поверхность соприкосно-
вения фаз.
Так как величина хд неизвестна, то отнесем коэфициент скорости диф-
фузии ко всей толщине газовой пленки.
Тогда уравнение (1) примет вид:
^t=Kg(Pg-Pi)A, (2)
где А — площадь соприкосновения, а Кд— коэфициент диффузии через газо-
вую пленку.
Так же рассуждая, получим, что количество прошедшего вещества за
то же время через жидкую пленку будет равно:
dw = Ki (Ct — Сг) Adi, (3)
где Ki — коэфициент диффузии через жидкую пленку. Так как пленки не-
значительны по сравнению с главными массами жидкой и газовой фаз, то
можно считать, что все количество вещества, прошедшее за данный про-
межуток времени через газовую пленку, проходит и через жидкую пленку.
Следовательно, можно написать:
ЙЭ7== К <с‘ -
(4)
Это и есть основное уравнение абсорбции газов жидкостями.
г> dw , ,
В этом уравнении —-----количество абсорбируемого за единицу времени
газа, А — поверхность соприкосновения газ-жидкость, Кд — коэфициент ско-
рости диффузии для газовой пленки, — коэфициент скорости диффузии для
жидкой пленки, рд— парциальное давление абсорбируемого газа в главной
массе газа, р,-— парциальное давление его же в газовой пленке, Ci—концен-
трация абсорбируемого газа в главной массе жидкости, С» —концентрация
газа в жидкой пленке.
Для хорошо растворимых газов сопротивлением жидкой пленки можно
пренебречь, и уравнение абсорбции представится так:
-А) А (2)
200
Разделив обе части уравнения (2) на М (молекулярный вес абсорбируе-
мого газа) и преобразовав его, получим:
^ = %(р0-р<)А<Н. (5)
Пусть общее давление газов в абсорбере Ро и пусть количеству dw
абсорбировавшегося газа соответствует уменьшение его парциального давле-
ния, равное dpа- Возьмем свободный объем абсорбера V, соответствующий
У P«V
поверхности А- В этом объеме будет молей газов: ~2у4-.
Можем написать
М ’ 22,4 — dpH ’ Р°’
откуда
, 22,4 dw
dp° = — ~M-
Умножая обе части уравнения (5) на -р-, получим:
/Г 29 4
dPg = -р~ (Рд—Pi) Adt- (6)
V
Обозначим -—средний радиус свободного объема насадки через р.
Последнее уравнение примет вид:
. Ко 22,4 .
dPg = ~f ~M(Pg—Pi)dt
или, внося 22,4 и М (постоянную для данного газа) в константу, получим:
-JPe-^Udt.
Pg—Pi Р
Интегрируя выражение, получим:
<9
(7)
(8>
f dPg t
J Pg—Pi P
„0
pg
где p°g—парциальное давление газа при входе в абсорбер, а рд—парциаль-
ное давление абсорбируемого газа при выходе из абсорбера.
Для ряда случаев величиной рг- можно пренебречь по сравнению с р„.
Это справедливо, например, при абсорбции SO2 нитрозной кислотой. В дан-
ном случае упругость SO2 над нитрозной кислотой равна нулю, потому что
SOs, попадая в нитрозу, окисляется в ней окислами азота. Это положение
справедливо также для абсорбции N»Os крепкой серной кислотой, а также
Для абсорбции SOs разбавленной серной кислотой. Пренебрегая величиной pit
получим:
(9)
ш4=-
Рв ? "
Время абсорбции от парциального давления р° до парциального давле-
кпя Ру Для этих случаев, следовательно, равно:
^4-
* рд
(Ю)
(11>
201-
Для среднерастворимых газов и для всех газов в случае высокой концен-
трации газа в жидкости величиной pf в уравнении (9) пренебречь нельзя.
Для интегрирования здесь необходимо представить р,- как функцию от pQ.
Время пребывания газа в абсорбере равно
где VCB —свободный объем насаженной части абсорбера, Qra3 — количество
газа, поступающего в абсорбер в одну секунду (расход газа). Подставляя
последнее выражение в (11), получим:
<?газ К Рд'
уабс
а так как р = _— (где А — поверхность насадки абсорбера), то:
А
Л = ^-31п4. (13)
К Рд
Разделив обе части последнего выражения на а — удельную поверхность
-часадки, получим:
(?газ /'о
I/ — га3 |п Л .
Ка n pt
гд
(14)
Из последнего выражения следует, что потребный объем абсорбера (при всех
заданных технологических показателях) прямо .пропорционален расходу газа
« обратно пропорционален величине удельной поверхности иасадки.
В тех случаях когда коэфициент скорости абсорбции повышается с уве-
личением линейной скорости газа, потребный объем абсорбера при заданной
степени абсорбции растет значительно медленнее, чем расход газа, т, е. повы-
диенне нагрузки абсорбера ие вызывает необходимости пропорционального уве-
личения его объема.
Из выражения (13) получаем:
о = ™-
Умножая обе части последнего равенства на выражение
(р°-р')М273
22,4(273 + /°) ’
получим:
Qr^g-P^^m КА (р°д-р*д)М273
22,4 (273 + 7°) 22,4 (273 + 7°) In Ц
Рд
Вся левая часть последнего выражения представляет весовое количество
газа, абсорбировавшегося за ту же единицу времени, к которому отнесена
202
и величина Qra3 . В правой части последнего равенства величины М, 273, 22,4
и (273 + fc) внесем в константу, тогда:
Р°
1п1»
Рд
Р° — р‘
величину в_____я— • представляющую собой среднюю логарифмическую
ш р1
Рд
силу абсорбции, обозначим через др.
Если процесс рассматривать не за единицу времени, а за время z, то
окончательно получим
G=K'ASpz, (15)
где G — весовое количество газа, абсорбировавшееся за время z.
В практических расчетах по абсорбции газов жидкостями применяются
и выражение (11) и выражение (15).
В ряде случаев (растворимость газа не подчиняется закону Генри; про-
цесс абсорбции осложняется одновременно идущим другим процессом, на-
пример абсорбция №Оз окислением NO и др.) процесс нельзя рассчитать
аналитически и приходится применять графические методы расчета.
При осушке газа в контактном производстве серной кислоты
применяется высококонцентрированная серная кислота (близкая
к 98% HsSCU), над которой упругость паров воды практически
равна нулю. Таким образом к этому случаю можно применить
формулу
„ Р°„
(11)
К Рд
Как показал Турхан, для случая десорбции воды из раство-
ров серной кислоты (применительно к процессу концентрирова-
ния серной кислоты см. гл. 6) применима формула
f dp"
₽ -J Pi~Pg
pg
(9a)
Последняя формула, следовательно, применима и для случая
абсорбции паров Н?О раствором серной кислоты, когда величи-
ной р{ нельзя пренебречь по сравнению с величиной pff.
Коэфициент скорости абсорбции паров НгО зависит от кон-
центрации H2SO4 (по опытам Кузнецова и Егоркина)1
% H2SO4 к о/о H,so4 К
57,5 ........... 92 75,8.........322
“ 59,5 ........ 129 97,5.........943
72,6 ........ 242
1 Неопубликованные материалы МХТИ.
203
Коэфициент скорости абсорбции .вычислялся по формуле
где G — количество абсорбированных паров Н2О в г в минуту,
др — средняя движущая сила абсорбции в ат, А — поверхность
орошения в м2.
И опыты Грюневальта и опыты Кузнецова и Егоркина пока-
зали, что коэфициент скорости абсорбции паров НгО серной кис-
лотой не зависит от плотности орошения.
Это же подтвердили и опыты Турхана для коэфициента ско-
рости десорбции Н2О из растворов серной кислоты (см. гл. 6).
Это обстоятельство означает, что в данном случае сопротивле-
ние жидкой пленки ничтожно мало по сравнению с сопротивле-
нием газовой пленки.
Из опытов Грюневальта (а также из опытов Турхана) следует,
что коэфициент скорости абсорбции паров НЮ серной кислотой
в некоторых границах повышается с увеличением линейной ско-
рости газа, чем дополнительно подтверждается, что основным
сопротивлением в этом случае является сопротивление газовой
пленки.
Приведенные выше данные Кузнецова и Егоркина получены
при линейной скорости газа, равной 0,5 м/сек, и при темпера-
туре 15—19°. Как это можно заключить на основе опытов Тур-
хана, коэфициент скорости абсорбции паров НЮ серной кислотой
должен уменьшаться с повышением температуры.
Это вытекает и из теоретических соображений.
Когда основным сопротивлением является газовая пленка,
оптимальным характером контакта газа с жидкостью будет распы-
ление жидкости в газе. В этом случае при пронизывании каплями
жидкости слоя газа газ не имеет сколько-нибудь постоянной по-
верхности раздела. Поэтому газовая пленка не успевает возни-
кать. Наоборот, для тех случаев абсорбции, когда основным
сопротивлением является жидкая пленка, оптимальным характе-
ром контакта газа с жидкостью будет барботаж газа через жид-
кость. Жидкость, пронизываемая пузырьками газа, не имеет
сколько-нибудь постоянной поверхности раздела и жидкая
пленка не успевает возникать.
Сушильное отделение. В тентелевской системе сушильное
отделение состоит из четырех башен и брызгоуловителя, через
которые газ проходит последовательно. По устройству и разме-
рам все башни одинаковы и смонтированы из листового свинца.
Башни загружены кусками кокса размером 15—20 мм, уложен-
ными на колосниковой решетке. В настоящее время все заводы
заменили кокс сушильных башен кольцами Рашига, что умень-
шило сопротивление проходу газа. ,
Для равномерного распределения орошающей кислоты
в крышке башни делается специальная труба с дырками (паук).
Первая башня не орошается и служит для улавливания брызг,
204
Рис. 95. Схема осушки в системе Геррес-
гоф-Банера:
7—сушильная башня; 2 — брызгоуловитель; 3— -
сборник кислоты; 4— насос; 5 — холодильник.
увлекаемых газом из промывного отделения, остальные башни
орошаются серной кислотой различной концентрации: вторая
башня 58—60г Вё (75—78% H2SO4), третья 64—65° Вё (85—
90% H2SO4) и четвертая 65—65,7° Вё (93—94% H2SO4).
Для увеличения концентрации кислот, разбавляющихся
в башнях улавливаемой влагой, часть кислоты из второй су-
шильной башни откачивается в абсорбционное отделение, на ее
место перекачивается кислота из третьей башни. Концентрацию
этой кислоты увеличивают кислотой четвертой сушильной башни,
а последнюю 94—95%-ным купоросным маслом, приготовляемым
в абсорбционном отделении. Установленный за башнями брызго-
уловитель представляет собой свинцовую, насаженную коксом,
башню немного меньших размеров, чем сушильные башни.
В тентелевской систе-
ме специальных холо-
дильников для охлажде-
ния сушильных кислот в
большинстве случаев не
имеется.
Осушка газа в системе
Герресгоф-Байера произ-
водится в одной (а иног-
да в двух) башне, оро-
шаемой крепкой 98%-ной
серной кислотой, которая
лучше, чем любая другая
кислота, поглощает пары
воды. Далее газ проходит
через такую же, но ни-
чем не орошаемую башню
(рис. 95). О регулировании работы сушильной башни подробнее
сказано ниже при рассмотрении режима абсорбционного отде-
ления.
Сушильная башня системы Герресгоф-Байера (рис. 96) пред-
ставляет собой цилиндр 1, сконструированный из отдельных чу-
гунных или железных колец. Башня футерована кислотоупорным
кирпичом 2 и имеет внизу колосниковую решетку 3. Башня на-
саживается кольцами Рашига размером 25 X 25 мм, в самом низу
более крупными. Для распределения кислоты служит располо-
женная горизонтально чугунная плита 4 с прорезами, через кото-
рые кислота и льется в башню. Размеры башни: высота 5510 льи,
внутренний диаметр 1990 мм, наружный 2228 мм-
В системе Кемико осушка производится в одной сушильной
башне, орошаемой серной кислотой крепости 93—94% (купорос-
ным маслом).
Башня (рис. 97) представляет собой железный цилиндр, футе-
рованный изнутри кислотоупорным кирпичом иа кислотоупорном
цементе. Башня разделена сводами на три отделения: кислото-
сборник /, реакционная часть 2 и брызгоуловитель 3. Нижняя
205
часть башни 1 представляет сборник емкостью 15,5 л3, со шту-
цером 4 в самой нижней своей части, по которому кислота вы-
текает в холодильник.
Для определения уровня кислоты в сборнике служат два спе-
Патрубок 5 для входа газа нахо-
дится в верху сборника непо-
средственно под сводом 6, от-
деляющим сборник 7 от сред-
ней части башни 2. Куполооб-
разный свод 6 смонтирован из
циальных водомерных стекла.
Рис. Уб. Сушильная башня
Герресгоф-Байера:
7 — чугунный корпус башни; 2 — футеровка;
3 — колосниковая решетка; 4 — распредели-
тельная плита; 5 — стеклянный колпак для
освещения; 6 — смотровое oki.o.
Рис. 97. Сушильная башня
системы Кемико:
7 — кис лото сборник; 2 — реакционная
часть; 3 — брызгоу ловите ль; 4 — шту-.
иер; 5 — патрубок; 6 — свод; 7 — от-
верстие в своде; 8 — кольца Рашнга^
9 — слой гальки; 10 — чугунные тру-
бы; 77 — крышка; 12 — штуцер.
кислотоупорной керамики и служит одновременно крышкой для
сборника и основанием для реакционной средней части башни 2.
Отверстие 7 в своде предназначается для прохода газа.
На своде лежит насадка из колец Рашига 8 размером
80 X 80 мт, а выше слой гальки или битых колец 9- Над насад-
кой 9 располагаются три чугунных жолоба. В эти желоба по
206
трем чугунным трубам 10 подается орошающая кислота, которая
по ряду трубок входит в насадку 9. Над кислотораспредели-
тельным устройством располагается второй (такой же как ниж-
ний) свод, на котором лежит слой из пяти рядов колец Рашига
80 X 80 мм. Этот слой насадки является брызгоуловителем.
В центре сферической крышки 11 находится штуцер 12, по кото-
рому газ уходит в компрессор. Стекающая из башни кислота
проходит оросительный холодильник и насосом качается обратно
на башни через распределительный бачок.
Для увеличения концентрации сушильной кислоты часть ее
отводится в смеситель при моногидратном абсорбере, смеши-
вается в нем с моногидратом; полученная более концентрирован-
ная кислота качается в тот же распределительный бачок. Этим
путем концентрация кислоты поддерживается постоянной.
В целях возможно большей полноты осушки газа этот про-
цесс должен проводиться при возможно низкой температуре.
Обычно температура орошающей кислоты поддерживается
не выше 45°, а количество ее рассчитывается так, чтобы раз-
бавление кислоты при поглощении влаги было незначительным и,
следовательно, температура кислоты повышалась бы мало.
3. Фильтр-маслоотделитель
После очистки газы поступают в компрессор, а оттуда в кон-
тактный узел, пройдя предварительно через маслоотделитель.
Этот аппарат предназна-
чен для очистки газа от ка-
пель масла, которые могут
быть захвачены им из под-
шипников компрессора.
При применяемых сейчас
конструкциях компрессоров
увлечение капель масла по-
чти невозможно. Так, на од-
ном обследованном заводе
масла в газах после компрес-
сора не обнаружено совсем,
а на другом обнаружены
Рис. 98. Фильтр-маслоотделитель:
1 — стенки фильтра; 2—крышка; 3—днище; 4 —
штуцер для входа газа; 5 — штуцер для выхода
газа; 6 — решетка.
лишь следы его в количестве
миллиграммов на десятки
кубометров газа. Фильтр-
маслоотделитель улавливает
также остатки брызг кислоты, не задержанные брызгоуловите-
лем, и частично туманообразную кислоту, если последняя про-
скакивает из отделения очистки или вновь возникает в сушиль-
ной башне (в случае нарушения в ней нормального технологи-
ческого режима).
^Фильтр-маслоотделитель (рис. 98) представляет собой желез-
ный цилиндрический сосуд 1 с днищем з, снабженный штуце-
207
рами 4 и 5 для входа и выхода газа и крышкой 2. Внутри сосуда
расположены дырчатые диафрагмы 6, между которыми зажат во-
локнистый асбест, служащий фильтрующей массой. Диафрагмы
выполняются из листового железа и соединяются между собой
на болтах с распорами из газовых труб. Собранные болтами
трубы располагаются на кронштейне из углового железа, при-
крепленном к внутренней поверхности сосуда.
4. Перемещение газов
Из контактных систем наибольшее сопротивление прохожде-
нию газа создают аппараты тентелевской системы, у которой при
нормальной работе разрежение на входе в компрессор достигает
100—150 мм рт. ст. и давление на выходе из него 200—250 мм
рт. ст. Это объясняется тем, что во многих аппаратах тенте-
левской системы газ пробулькйвает через слой кислоты.
В системе Герресгоф-Байера, имеющей вдвое большую произ-
водительность, разрежение на входе газа в турбокомпрессор
составляет 300—400 мт вод. ст., а давление на выходе —
2000—2200 Мм вод. ст.
Сопротивление аппаратуры на различных заводах очень
сильно отличается в зависимости от состояния аппаратуры и ко-
личества проходящего газа. Для одного и того же завода сопро-
тивление системы также не остается постоянным, а с течением
времени увеличивается вследствие засоренности аппаратуры,
слеживаемости контактной массы и др.
Для примера приведем сопротивление аппаратуры одного из
заводов системы Герресгоф-Байера:
Разрежение
мм вод. ст
Перед 1-й промывной башней.......................... 34
После 1-й „ башни............................. 62
После 2-й „ „ 90
» 3-й „ „ 125
„ 1-й пары мокрых электрофильтров....................... 141
„ увлажнительной башни.................................. 159
„ 2-й пары мокрых электрофильтров....................... 180
„ 3-й „ „ , .................. 204
Перед турбокомпрессором..................................... 350
Давление
мм рт. ст.
После турбокомпрессора...................................... 154
Перед фильтром.............................................. 145
После фильтра............................................... 140
Перед 1-м теплообменником................................... 130
После 1-го теплообменника................................... 128
2-го „ 125
„ 1-го контактного аппарата ............................. 80
„ 2-го теплообменника (SO3).............................. 77
„ 2-го контактного аппарата.............................. 13
» 1-го теплообменника (SO3)............................... И
208
Перед ангидридным холодильником.......................... 9
> 1-м абсорбером.......................................... 6,6
„ 2-м , 4,0
После 2-го абсорбера.......................................... 0,1
Различные системы отличаются не только сопротивлением
аппаратуры, но и производительностью, поэтому каждая система
оборудуется компрессорами определенной мощности и давления.
Прежде на заводах устанавливались компрессоры различных кон-
струкций (ротационные, поршневые и др.), но в последнее время
почти иа всех заводах они заменены более удобными и надеж-
Рис. 99. Турбокомпрессор:
1 — корпус; 2 — стальной вал; 3 — лопасти; 4 — передний подшипник; 5— задний подшипник;
6 — вход газа; 7 — выход газа; 8—перегородка.
ными в работе турбокомпрессорами. В системе Кемико произво-
дительностью 120 т кислоты в сутки установлен турбовентиля-
тор, который один обеспечивает передвижение по всей системе
необходимого количества газа.
Обычно для одной системы устанавливается один рабочий
турбокомпрессор и к нему один резервный; для двух систем два
турбокомпрессора рабочих и один резервный и т. д.
Турбокомпрессоры устанавливаются в специальном помеще-
нии, изолированном от остальных отделений цеха. Во всех кон-
тактных системах компрессорные отделения никакими характер-
ными для данной системы особенностями не отличаются, поэтому
описание их дается общее для всех систем.
14 Зак. 6019. Технология серной кислоты.
209
Турбокомпрессоры, изготовленные различными заводами, по-
хожи друг на друга устройством и принципом работы, но отли-
чаются некоторыми деталями. На рис. 99 показан в разрезе турбо-
компрессор германской фирмы ККК производительностью 100 м3
газа в минуту.
Турбокомпрессор представляет собой чугунный корпус J,
внутри которого проходит стальной вал 2 с несколькими лопа-
стями 3. Вал укреплен в двух подшипниках: переднем 4 и зад-
нем 5. Сернистый газ поступает через отверстие 6, попадает
в середину первой лопасти 3, которая, вращаясь, сжимает газ,
а имеющиеся в корпусе перегородки 8 направляют его в середину
второй лопасти и т. д. Из компрессора газ выходит под давле-
нием через отверстие 7.
Подшипники смазываются специальным турбинным маслом
периодически вручную или беспрерывно насосом, работающим
от вала компрессора. Для охлаждения нагревающихся от трения
подшипников в их корпус пускается вода. Если же смазка произ-
водится насосом, то охлаждение подшипников производится сма-
зочным маслом, которое, в свою очередь, охлаждается холодной
водой, циркулирующей в змеевиках, помещенных в сборнике
этого масла.
В табл. 44 приводится характеристика турбокомпрессоров,
устанавливаемых для различных систем.
Таблица 44
Характеристика компрессоров Системы
Герресгоф- Байера Тентелев- ская Кемико
Производительность, м31мин . . . 100 50 350
Разрежение на входе, мм вод. ст. 400 2000 600
Давление на выходе, мм вод. ст. 2 200 5000 2000
Число об/мин 2 940 2 930 —
Мощность электромотора, кет . . 70 70 —
Производительность турбокомпрессора может быть значи-
тельно повышена, если уменьшить сопротивление.
Так, если общее сопротивление системы 2600 мм вод. ст.,
производительность турбокомпрессора составит ~ 100 м3/мин. Та-
кое количество газа при содержании в нем 7% SO позволяет
получить 43 т кислоты в сутки. Если же уменьшить сопротивле-
ние аппаратуры до 2250 мм вод. ст., производительность ком-
прессора составит ~ 120 м3/мин, что при тех же условиях
позволит получить 51,6 т кислоты в сутки.
Компрессоры во всех системах ставятся после очистки и
210
осушки газа, т. е. перед контактным узлом. Это место в схеме
создает для’компрессора самые лучшие условия работы (сухие
и чистые газы, низкая температура газов).
5. Контроль работы отделений очистки и осушки газа
Из отделений очистки и осушки газ должен выйти достаточно
сухим, освобожденным от мышьяка и тумана серной кислоты.
Поэтому анализами, показывающими итог работы этих от-
делений, являются: определение содержания мышьяка и тумана
серной кислоты в газах после последнего электрофильтра и со-
держание влаги в газах после сушильных башен. Важнейшим
анализом является также определение содержания SO2 после
отделения очистки.
Кроме того, качество работы отдельных аппаратов, состоя-
ние отдельных аппаратов и намечающиеся отклонения процесса
от нормального режима выявляются рядом других замеров и
анализов. Перечень принятых замеров и анализов по промыв-
ному и сушильному отделениям приводится в табл. 45.
В случае содержания SO2, после прохождения газов через от-
деление промывки, ниже нормы приходится, для выявления места
подсоса воздуха, производить анализ газа на содержание SO2
после каждого аппарата промывного отделения. Эпизодически
необходимо проверять количество орошения по каждой башне.
Таблица 45
Объект контроля Определяется Место отбора проб или замера Частота анализа
Газ Кислота я Газ Кислота Промывное Температура и раз- режение Удельный вес и температура Количество выве- денной из цикла кислоты У в л а ж н и Температура и раз- режение Удельный вес и температура отделение Газоход до и после каждой промыв- ной башни Вход п выход кислоты по каж- дой башне Сборник тельная б а ши я Газоход после башни По выходе из башнн 4 раза в смену Контрольный ана- лиз 1 раз в сутки То же При каждой от- качке 4 раза в смену То же
14*
211
Продолжение табл 45.
Объект контроля Определяется Место отбора проб или замера Частота анализа
Газ Агрегат Газ Кислота » Газ Газ Компрес- сор Маслоот- делитель То же Мокр ые э Температура и раз- режение Содержание мышьяка (по Цинцадзе или Зангер-Блеку) и туманообразной серной кислоты (методом филь- трации газа че- рез гигроскопи- ческую вату или змеевиком Аме- лина) Вольтаж и милли- ампера» С у ш и л ь Содержание SO2 (методом Рейха или газоанализа- тором) Разрежение и тем- пература Температура и со- держание H2SO4 Уровни кислоты Содержание влаги Отделение Разрежение и да- вление Температура Ампераж Давление Конденсат лектрофнльтры После каждой па- ры электрофиль- тров После III пары электрофильтров или перед су- шильной башней На щите ное отделение Перед 1-й сушиль- ной башней В газоходах после сушильных ба- шен и брызго- уловителей По выходе из башен В сборниках После сушильных башен компрессорное До и после ком- прессоров После компрессо- ров После маслоотде- лителя Маслоотделитель То же 1 раз в смену. От- бор газа в тече- ние всей смены 4 раза в смену 2 раза в смену 4 раза в смену 4 раза в смену 1 раз в смену То же 8 раз в смену То же И V Эпизодически
212
III. Теоретические основы
контактного окисления SO2
1. Равновесие реакции 2SO2 4-О2 = 2SO;!
Реакция окисления SO2 протекает с выделением тепла и
с уменьшением числа молекул. Отсюда по ле-Шателье можно
сразу заключить, что на полноту окисления будет благоприятно
влиять понижение температуры и повышение давления^_Для ко-
личественного учёта влияния этих факторов воспользуемся
уравнениями химической термодинамики.
Обозначив парциальные давления компонентов в состоянии
равновесия через pso*, pSQ* и рОг, можем написать:
4^- = ^, О)
РаогРо,
где Ki — константа равновесия. Для не слишком высоких давле-
ний Ki зависит только от температуры. Для расчета равновесных
состояний удобно преобразовать уравнение (1), извлекая из обеих
частей квадратный корень:
= (2)
Зависимость константы К от температуры определяется урав-
нением Вант-Гоффа:
<7 1п /<___Q
dT ~ RF’ '°7
где Q — теплота реакции, отнесенная к 1 молю сернистого газа;
Т — абсолютная температура; R — газовая константа, равная
1,987 кал/град • моль.
Зависимость теплоты реакции от температуры может быть вы-
ражена для ограниченного интервала температур простым уравне-
нием:
+ (4)
где Ьо и bi — постоянные.
Подставляя (4) в (3), интегрируя и переходя к десятичным
логарифмам, находим:
lg 2,303 Rt~Rls
Здесь i — постоянная интегрирования.
Боденштейн и Поль определили экспериментально константы
равновесия в интервале температур 400—600° и рассчитали с их
помощью коэфициенты уравнения (5).
^SO,_
Лзо2 Уро2
213
В результате они получили:
1g К= + 0,611 Ig Т 6,7497.
(6)
Значения К, рассчитанные с помощью этого уравнения, при-
ведены на рис. 100.
Рис. 100. Константы равновесия системы SO3 |-х/2 О., ДД SO ..
Сравнивая (6) и (5), находим
6о = 2372О и ^= — 1,21.
Подставляя эти значения в (4), приходим к следующему выра-
жению для молекулярной теплоты реакции:
Q = 23 720—1,21 Т.
(7)
Вычисленное с помощью этого уравнения значение Q для 293°
(20°) хорошо согласуется с термохимическими данными.
Теоретически возможная степень контактирования. Степенью
контактирования называют отношение парциального давления SO3
в данной смеси к сумме парциальных давлений SO3 и SO2.
Максимальное значение степени контактирования в заданных
условиях достигается, очевидно, в состоянии равновесия. Эта ве-
личина называется теоретически возможной или просто теорети-
214
ческой степенью контактирования и обозначается хт. Согласно
определению:
Pso,
/'so, + /'so, ’
(8)
К
г Ро2
(9)
где п и р п — парциальные давления SO3 и SO2 в состоянии
гSO3 ' SO2
равновесия.
Заменяя величину отношения pSOi к pSOs в уравнении (8) ее
значением из уравнения (2), получим:
х 1_________+_vS_=_
т 1 м. _ >1
«Ур^ УроУ~к кл
Определим теперь р0 — равновесную концентрацию кисло-
рода, как функцию давления и состава газа.
Пусть р — общее давление газа в атмосферах; а — начальный
объемный процент SO2 в газовой смеси (до начала образования
SOs); b — начальный объемный процент кислорода.
При достижении равновесия в 100 объемах исходной газовой
смеси окисляется ахт объемов SO2, что требует 0,5 ахт объемов
кислорода. Остается кислорода b — 0,5 ахт - Но и общий объем
смеси меняется и из 100 объемов остается лишь 100 — 0,5 ахг.
Доля кислорода
разом:
в равновесной смеси составляет таким об-
Ь — 0,5 «хт
100 — 0,5 ах.
и его парциальное давление
b — 0,5 яхт
= Р 100—0,5 ах~ ’
Подставляя это значение в уравнение (9), получим оконча-
тельно для х следующее выражение:
К (Ю)
100 — 0,5 «хт )
р(Ь — 0,5 ллт)
Уравнение (10) определяет хт в неявном виде. Это, однако,
не усложняет сколько-нибудь значительно расчет, так как вели-
чина хт под корнем оказывает малое влияние на значение всего
выражения. Обычно нетрудно грубо оценить ожидаемое значе-
ние теоретически возможной степени контактирования и, под-
ставив его в правую часть уравнения (10), определить хт без
большой ошибки. Если найденная таким путем величина x-t зна-
чительно отличается от предварительно принятой, то расчет
215
повторяют. Второе приближение всегда бывает достаточно
точным.
Влияние температуры. В табл. 46 приведены значения хт при
различных температурах и давлении 1 ат для газа состава:
7% SO2, 11% Оа. 82% N?, Это наиболее распространенный и, как
увидим' далее, наиболее выгодный состав газа перед контактным
аппаратом при работе на колчедане.
Снижение теоретической степени контактирования с повыше-
нием температуры вытекает уже из правила ле-Шателье.
Определяя из уравнения (6) величину константы равновесия-и
подставляя ее в уравнение (10), находим теоретическую степень
контактирования для любой температуры.
Таблица 46
Зависимость хт от температуры
Температура в СС . . . . 400 410 420 430 435 410 445 450 455 460
хт-102 . . . 99,25 99,07 98,8 98,5 98,3 98,1 97,87 97,64 97,4 97,2
Температура в °C ... . 4С5 470 475 480 485 490 495 500 510 520
хт-103 . . . 96,8 96,4 96,07 95,6 95,24 94,77 94,30 93,8 92,5 91,5
Температура в °C ... . 525 530 540 550 560 570 575 580 590 600
х • 102 . . . 91,0 89.7 88,06 86,16 83,9 81,75 80,6 79,3 76,84 73,85
Температура в °C ... . 620 650 675 700 750 800 850 900 1000 —
хт-10а . . . 67,9 59,5 49,9 44,2 31,5 21,6 15,2 10,3 5,0 —
Влияние начального состава газа. Как показывает уравнение
(10), при постоянных температурах и давлении теоретическая сте-
пень контактирования тем выше, чем больше b — начальная кон-
центрация кислорода и меньше а — начальная концентрация 8Ог.„
В производственных условиях невозможно изменять независимо
концентрацию SO2. Так, при работе на колчедане можно лишь
менять избыток воздуха при обжиге. С увеличением избытка воз-
духа Ь возрастает, а падает и теоретически возможная степень
контактирования должна, следовательно, возрастать. В табл. 47
приведены значения хт для газа, получаемого при обжиге кол-
чедана с различным избытком воздуха, для температуры 475° и
давления 1 ат. С уменьшением избытка воздуха хт снижается
вначале медленно, но в дальнейшем, при приближении к стехио-
метрическому соотношению между а и Ь, очень быстро.
При работе на углистом колчедане, вследствие повышенного
расхода кислорода при обжиге, хт при равной концентрации SO2
ниже. При работе на сере, наоборот, выше. Применение для
216
Таблица 47
Изменение хт в зависимости от разбавления воздухом продуктов
обжига колчедана (р = 1 am, t = 475-)
а 2 3 4 5 6 7 8 9 10
ь 18,14 16,72 15,28 13,86 12,43 11,60 9,58 8,15 6,72
97,32 97,16 96,98 96,75 96,47 96,07 95,53 94,61 92,78
обжига воздуха, обогащенного кислородом, также позволит резко
повысить хт при прочих равных условиях.
Влияние давления. С повышением давления второй член зна-
менателя правой части уравнения (10) уменьшается, значит при
прочих равных условиях х возрастает. В табл. 48 приведены
значения хт • Ю2 для различных давлений и температур для газа,
содержащего 7% SOg, 11% Ог и 82% Ng, рассчитанные по
уравнению (10).
Таблица 48
Зависимость теоретически возможной степени контактирования
(в процентах) от давления
Темпера- тура в °C хт при давлении в ата
1 * 5 10 25 50 100
400 99,25 99,67 99,76 . 99,85 99,90 99,93
450 97,61 98,93 99,23 99,52 99,65 99,76
500 93,80 97,15 97,90 98,69 99,06 99,33
550 86,16 93,25 95,0 96,85 97,73 98,36
600 73,85 86,15 89,7 93,3 95,05 96,45
Однако при высоких давлениях уравнение (1), а следовательно
и (10), перестает быть точным вследствие заметных отклонений
от законов идеальных газов. Приведенные значения хт при высо-
ких давлениях надо рассматрйвать как приближенные.
Как показывает табл. 48, применение повышенного давления
значительно расширяет в сторону высоких температур интервал,
в котором могут быть достигнуты высокие степени контактиро-
вания.
2. Скорость окисления SO2
Выше были рассмотрены условия равновесия реакции окисле-
ния SO2.
Как показывает опыт, скорость взаимодействия SO2 с кисло-
родом в отсутствии катализаторов, даже при высоких температу-
рах, очень мала. Это объясняется очень высоким значением энер-
217-
гии активации реакции окисления сернистого газа в отсутствии
катализаторов.
При сближении двух молекул, способных вступить в реакцию,
между ними первоначально возникают силы отталкивания и
только после преодоления этих сил достигается так называе-
мое переходное состояние, при котором возможно образование
новых химических связей. Отсюда следует, что только те из
сталкивающихся молекул смогут вступить в реакцию, энергия ко-
торых больше или равна энергии сил отталкивания. Этот мини-
мально необходимый запас энергии называется энергией актива-
ции (£) и измеряется в кал/моль. Из кинетической теории газов
известно, что доля молекул, обладающих энергией SsE, соста-
_ Е
вляет в первом приближении е БТ, где е — основание натураль-
ных логарифмов.
Поэтому если общее число столкновений между молекулами
реагирующих веществ Z, то число эффективных столкновений,
приводящих к реакции, равно
_ л
Z e БТ. (П)
Общее число столкновений пропорционально произведению
концентраций реагирующих веществ. Так, например, для бимоле-
кулярной реакции:
Z = Л0СлСв.
Отсюда число реагирующих молекул равно: «.
koe~^CACB = kCACB. (12)
а константа скорости реакции
k — koe Ет. (13)
Как следует из уравнения (13), при прочих равных условиях, скорость
’реакции тем меньше, чем больше Е, и при этом, вследствие экспоненциаль-
ной зависимости доли реагирующих молекул от Е, небольшое изменение Е
вызывает резкое изменение скорости реакции.
Если опытным путем определить значение константы скорости реакции
для нескольких, по крайней мере двух, температур, то нетрудно вычислить
значение энергии активации.
Гомогенное окисление сернистого газа даже при высоких
температурах практически не происходит вследствие очень боль-
шой величины энергии активации этой реакции.
Между тем в присутствии многих твердых веществ скорость
взаимодействия SO2 с кислородом становится заметной, а в от-
дельных случаях даже очень большой. Вещества, меняющие ско-
рость химической реакции, >но присутствующие в малых коли-
чествах, не связанных определенными стехиометрическими соот-
218
ношениями с количествами веществ, испытавших превращение,
и не претерпевающие в результате реакции изменения состава,
называются катализаторами.
Если в присутствии катализатора скорость реакции возрастает,
то явление называется положительным катализом, в противном
случае отрицательным. Катализ называется гомогенным,
если реагирующие вещества и катализатор образуют однород-
ную систему, и гетерогенным, если катализатор присут-
ствует в виде отдельной фазы. В контактном сернокислотном про-
изводстве осуществляется положительный, гетерогенный катализ.
Рассмотрим, в чем же заключается причина ускорения реакции
в присутствии катализатора.
Как было сказано выше, скорость химической реакции опре-
деляется произведением числа столкновений на долю молекул,
обладающих необходимым запасом энергии, выражаемой в пер-
___________________________________£
вом приближении множителем е RT, где Е необходимая
энергия активации. При гетерогенном катализе существенны,
очевидно, лишь столкновения, происходящие на поверхности
катализатора, так как только для них возможно взаимодей-
ствие с катализатором. Число таких столкновений, вообще го-
воря, меньше числа столкновений в объеме. Увеличение скоро-
сти реакции при гетерогенном катализе должно быть поэтому
отнесено за счет увеличения второго множителя в уравнении для
скорости реакции, т. е. доли молекул, обладающих необходимой
для возможности реакции энергией. При постоянной температуре
_ Е
увеличение множителя е RT возможно лишь при уменьшении Е —
энергии активации.
Этот вывод подтверждается результатами измерения энергии
активации в присутствии катализаторов. Она всегда меньше энер-
гии активации реакции без катализатора.
Ускорение реакции в присутствии твердого катализатора
сводится к уменьшению необходимой энергии активации.
Труднее объяснить механизм самого снижения энергии' акти-
вации. В этой области господствовало большое число разнооб-
разных теорий. Остановимся кратко лишь) на двух главнейших,
оказавших наибольшее влияние на современные представления.
Это теории — адсорбционная и промежуточных химических сое-
динений. По адсорбционной теории молекулы одного или обоих
реагирующих веществ адсорбируются катализатором. В ре-
зультате адсорбции молекулы деформируются, свойства их ме-
няются.
Для достижения переходного состояния при взаимодействии
адсорбированных на катализаторе молекул или адсорбированной
молекулы со свободной, налетающей на нее из газовой фазы,
может поэтому потребоваться меньшая затрата энергии, т. е.
взаимодействию в присутствии катализатора может отвечать
меньшая энергия активации. Механизм каталитического дей-
219
ствия сводится таким образом к действию специфичных сил на
поверхности раздела фаз. Ряд особенностей действия катализа-
торов, как, например, резкий рост активности с увеличением
дисперсности и вообще нарушением структуры, находятся в хо-
рошем согласии с этой' теорией. Зато другие, в первую очередь
специфичность каталитических реакций, остаются непонятными.
Хорошие адсорбенты способны поглощать вещества с раз-
личными химическими свойствами, в то время как катализатор,
активный в отношении одной реакции, может оказаться совер-
шенно инертным в отношении другой. Поскольку в этой форме
адсорбционная теория не могла объяснить специфичность дей-
ствия катализаторов, постольку она оставалась бесполезной при
поисках ' катализатора для ускорения определенной реакции.
В результате работ последних лет этот недостаток теории уда-
лось значительно смягчить. Было показано, что для гетероген-
ного катализа существенна не всякая адсорбция, а только
так ' называемая активированная адсорбция, протекающая
обычно с заметной скоростью лишь при высоких температурах
и близкая по характеру связи к химическому взаимодействию.
Этот вид адсорбции обладает специфичностью, необходимой
для объяснения особенностей каталитических процессов.
Сущность теории промежуточных соединений сводится
к следующему. Предположим, что реагируют два вещества А и
В с образованием продукта АВ в присутствии катализатора‘К.
Пусть без катализатора реакция
А + В->АВ
требует энергию активации, равную Е. В присутствии катализа-
тора образование продукта АВ может пойти по обходному
пути с образованием промежуточных соединений с катализато-
ром. Например
А+К = АК;
АК + В = АВК-
Если энергии активации этих двух реакций меньше £, то
скорость образования продукта АВ по рассматриваемому обход-
ному пути может значительно превосходить скорость'непосред-
ственного взаимодействия реагирующих веществ. По этой тео-
рии специфичность действия катализаторов вполне понятна, она
связана с общей специфичностью химического взаимодействия.
Но' зато теперь остается неясной зависимость активности ката-
лизаторов от структуры поверхности.
Сторонники теории промежуточных соединений полагали
вначале, что промежуточные соединения реагирующих с ката-
лизатором веществ должны быть идентичными с полученными
иным путем и для доказательства правильности предполагаемой
схемы механизма пытались эти соединения обнаружить и изо-
лировать. В применении к гетерогенному катализу ‘эти попытки
220
были обречены на неудачу, так как промежуточные соединения
не образуют отдельных фаз.
Согласно современным представлениям в промежуточных ре-
акциях участвуют лишь поверхностные атомы кристаллов ката-
лизатора. В соответствии с этим и образующиеся промежуточ-
ные соединения рассматриваются как особые поверхностные сое-
динения, обладающие свойствами, отличными от свойств соот-
ветствующих массивных, кристаллических продуктов. В этой
модификации теория промежуточных соединений способна объ-
яснить влияние состояния поверхности на активность катализа-
тора. 1
Из сказанного видно, что развитие обеих теорий направлено
в сторону их сближения, синтеза. Если в первоначальном своем
виде адсорбционная теория объясняла действие катализаторов
исключительно с помощью поверхностных сил, а теория про-
межуточных соединений только химическим взаимодействием,
то теперь та и другая теории учитывают оба эти фактора. Обе
теории уже не противостоят друг другу и в термины «поверх-
ностное соединение» и «продукт активированной адсорбции»
вкладывается почти одинаковый физический смысл.
3. Катализаторы для окисления SO2
Первым катализатором, предложенным Филиппсом для
ускорения окисления сернистого газа, была металлическая пла-
Рис. 101. Каталитическая активность окислов в отношении
окисления SO2 (по Нейман}).
тина. Этот катализатор сохранял почти монопольное положение
вплоть до 20-х годов нашего столетия. Между тем уже вскоре
после появления патента Филиппса стали появляться указания
на активность большого числа других веществ. В табл. 49 и на
Рис. 101 приведены сравнительные данные по каталитической
активности различных веществ (по Нейману с сотрудниками).
221
Таблица 49
Катали- затор Максималь- ное превра- щение В % Темпера- тура, отве- чающая максимуму превраще- ния, в °C Катали- затор Максималь- ное превра- щение В % Темпера- тура, отве- чающая максимуму превраще- ния, в °C
Fe2O3 69,5 625 wo. 62,5 670
СГ2О3 81,0 580 V2O5 90,0 512
СиО 58,7 700 As2O6 55,0 670
Мп2Оз 22,0 700 Моо3 47,0 700
SnO2 35,0 750 Pt 99,5 425
TiO2 490 700 на асбесте
Данные табл. 49 получены при объемной скорости 15, с га-
зовой смесью, ’ содержащей 7% SO2. Объемной скоростью назы-
вается объем сернистого газа при нормальных условиях, прохо-
дящий через единицу объема катализатора в 1 час. В заводских
контактных аппаратах объемная скорость составляет обычно от
30 до 70. Максимальным превращениям, приведенным в табл. 49,
не следует придавать общего значения, так как каталитическая
активность зависит не только от состава, но и от структуры,
а значит и от способа приготовления катализатора. Как следует
из формы кривых рис. 101, каталитическая активность окислов
начинает заметно проявляться'лишь при высоких температурах,
при которых достижение высоких степеней окисления SO2 ста-
новится невозможным из-за смещения равновесия в сторону
диссоциации SO3. Это можно объяснить повышенным значением
энергии активации окисления SO2 на этих катализаторах по
сравнению с платиной.
Для некоторых катализаторов удалось показать, что резкое
снижение активности (при понижении температуры) связано
с химическим превращением катализатора в неактивное соедине-
ние. Так, активированные ванадиевые катализаторы, а также
окиси железа и меди при низких температурах под действием
сернистого газа переходят в сульфаты, каталитически неактив-
ные. Поэтому лишь при температурах, достаточно высоких для
диссоциации сульфатов, начинает1 проявляться каталитическая
активность этих окислов. Более благоприятные результаты по-
лучаются при применении активаторов.
Активаторами называются вещества, добавка которых
к катализатору повышает его активность. При этом взятый в от-
дельности активатор может вовсе не проявлять каталитической
активности.
В табл. 50 приведены данные о некоторых активированных
катализаторах.
Последняя комбинация — V2O5, П’агСЦКгО), SiO2, начиная
с 20-х годов нашего столетия, используется широко в производ-
222
Таблица 50
Катализатор Макси- мальное превра- щение В % Темпера- тура, отве- чающая максимуму превраще- ния, в °C Катализатор Макси- мальное превра- щение В % Темпера- тура, отве- чающая максимуму превраще- ния, в °C
FeoOa 69,5 625 V2O3 90,0 512
Fe2O3, Bi,O3 72,2 625 V2O5, CuO 91,3 505
Fe»O3, SnO2 76,2 600 V2O3, Ag2O 95,5 475
СГ9ОЗ 80,0 600 V2O3, Na,O
Cr2O3, SnO., 96,0 475 SiO2 98,1 440
стве. Большинство заводов работает на ванадиевых кантактных
массах этого типа. Предложены сотни рецептур их приготовле-
ния, причем во всех без 1 исключения в конечном продукте на-
ряду с другими веществами содержатся пятиокись ванадия,
кремневая кислота и калийная (реже натронная) щелочь.
Заслуживает внимания также комбинация СггОз, БпОг. При-
меняя в качестве добавочных активаторов окиси бария и сурьмы,
Ададуров и сотрудники значительно повысили активность этого
комплекса. Недостаточная термическая устойчивость и высокое
содержание олова препятствуют, однако, практическому исполь-
зованию этого катализатора. Остальные катализаторы, указан-
ные в табл. 49, не позволяют достигать практически полного
окисления SO2. Кроме них в патентной литературе упоминаются
в качестве катализаторов для окисления SO2 окись церия, смесь
церитовых земель, селен, теллур и др.
На большинстве современных контактных заводов приме-
няются ванадиевые катализаторы и лишь на старых заводах
сохранились платиновые контактные массы. Окись железа
в форме колчеданных огарков применялась в качестве катализа-
тора контактными заводами, сооруженными по так называемой
маннгеймской системе.
Платиновые контактные массы. По активности единицы веса
платиновые контактные массы и сейчас превосходят все про-
чие — в том числе и ванадиевые. Особенно резко выступает по-
вышенная активность платиновых катализаторов при темпера-
турах ниже 450°.
Наряду с этим платиновые контактные массы обладают и
двумя серьезными недостатками, а именно: 1) большой чувстви-
тельностью к некоторым примесям газовой смеси, быстро сни-
жающим их активность и 2) высокой стоимостью.
С целью уменьшения затраты платины, для максимального
использования поверхности, ее применяют не в чистом виде,
а нанесенной на различные носители.
Роль носителей не ограничивается, однако, увеличением
активной поверхности.
223
Активность твердых катализаторов очень сильно зависит от
качества поверхности. 'Чем дисперснее вещество катализатора,
чем более нарушена его кристаллическая решетка, тем выше
каталитическая активность.
Способы приготовления активных катализаторов и сводятся
в основном к созданию такой нарушенной структуры. Но с уве-
личением дисперсности вещества растет величина свободной
поверхностной энергии и устойчивость системы уменьшается,
так как равновесию отвечает минимум свободной энергии. Чем
дисперснее, а следовательно, и активнее катализатор, тем ме-
нее он устойчив, тем легче в нем протекают при нагревании про-
цессы рекристаллизации, снижающие активность. Применение
носителей устраняет отчасти этот недостаток, повышая устойчи-
вость активного, тонкодисперсного состояния катализатора, бла-
годаря пространственному разделению отдельных кристалли-
ков и взаимодействию их с носителем.
К носителям катализатора в контактном производстве предъ-
являются следующие требования: 1) устойчивость к химиче-
скому воздействию SO2 и SOs; 2) большое развитие поверхно-
сти на единицу объема; 3) устойчивость к максимальным тем-
пературам, возможным в контактном аппарате (до 650°); 4) ме-
ханическая прочность.
В качестве носителей для платины наибольшее распростра-
нение получили: асбест, сульфат магния и силикагель.
Для нанесения платины обычно употребляется белый длинноволокнистый
асбест—минералы амфибол или хризолит. Эти минералы, особенно первый,
наиболее устойчивы к действию кислот и высоких температур.
Для нанесения платины асбест последовательно обрабатывается раство-
рами платнно-хлористоводородной кислоты и какого-либо восстановителя
(например муравьинокислого натрия).
Содержание платины в полученной контактной массе колеблется от 4
до 10%. Препятствием к снижению содержания платины является высокое
гидравлическое сопротивление носителя. Использование платины сравни-
тельно невелико. На каждую тонну суточной производительности в труб-
чатые контактные аппараты загружается ~ 500 г платины. Для предотвра-
щения увеличения гидравлического сопротивления в результате слеживания,
платинированный асбест помещают в контактных аппаратах на большом
числе решеток, слоем 10—.16 мм на каждой. Срок службы платинированного
асбеста, при условии хорошей очистки газа, ~ 15 лет.
Сульфат магния в качестве носителя платины применялся в системе
Грнлло-Шредера с несовершенной очисткой газа, что приводило к сравни-
тельно быстрому отравлению катализатора. Растворимость носителя в воде
являлась поэтому большим преимуществом, так как позволяла легко извле-
кать платину из отравленной контактной- массы и переносить ее после очистки
на свежий носитель.
Для приготовления носителя кристаллический сульфат магния подвер-
гался двукратному прогреву до температуры темнокрасного каления. Полу-
ченный спек измельчался и фракция крупностью от 3 до 15 мм подвер-
галась платинированию. Для платинирования масса помещалась в керами-
ковые глазурованные противни слоем около 75 мм и равномерно обрызгива-
лась раствором хлористой платины при непрерывном персгребании. В про-
цессе платинирования масса разогревается. По окончании обрызгивания хло-
ристой платиной масса выдерживалась в течение восьми часов, после чего
была пригодна к употреблению.
224
Правильно изготовленная масса должна быть твердой н устойчивой
к истиранию. 1 ,м3 весит около 750 кг. Содержание платины составляет
0,1—0,3%. Активность, отнесенная к единице веса платины, несколько выше,
чем для платинированного асбеста. Количество платины, загружаемое в кон-
тактные аппараты, составляет ~ 300 г на 1 т суточной производительности.
Значительное распространение в качестве носителя платины получил сили-
кагель. Пригодный для нанесения платины силикагель должен быть твер-
дым и устойчивым к нагреванию до 700°. Обычно он представляет собой
прозрачные стекловидные куски неправильной формы или цилиндрики со
средним поперечником 4—5 ,и.и. 1 Л3 силикагеля весит 700—750 кг. Перед
нанесением платины силикагель прогревается при 300—400° и после охла-
ждения насыщается каким-либо восстановителем, лучше всего сернистым
газом. После этого силикагель равномерно обрызгивается водным раствором
какого-либо растворимого платинового соединения, например платинохлори-
стоводородной кислотой. Затем гель высушивается и прогревается в токе
воздуха. Содержание платины составляет 0,07—0,16%, чаще всего 0,125%.
Количество платины, загружаемое в аппараты, составляет 150—250 г на
1 т суточной производительности.
Ванадиевые контактные массы.' Переход на ванадиевые кон-
тактные массы стал осуществляться особенно интенсивно, начи-
ная с 1927 г. после работ Егера и Бертча (США), приготовивших
на основе цеолитов активные и устойчивые ванадиевые контакт-
ные массы, известные под названием массы Монсанто и ванадие-
вой массы Селден К°-
В патентной литературе описано чрезвычайно' большое число
методик приготовления ванадиевых контактных масс. Общим
для всех является содержание в конечном продукте пятиокиси
ванадия, кремневой кислоты и щелочи. В большинстве случаев
содержатся еще и другие добавки.
У нас в СССР широкое использование ванадиевых' контакт-
ных масс началось с 1932 г. на основе исследований, проведен-
ных в Московском химико-технологическом институте им. Мен-
делеева (Шокин'и др.) и Одесском химическом институте (Бо-
ресков с сотрудниками), разработавших ряд оригинальных ре-
цептур приготовления активных ванадиевых контактных масс.
Сырьем для приготовления ванадиевых катализаторов слу-
жит ванадат кальция, получаемый из шлаков 1 металлургической
переработки железных руд, содержащих ванадий. Для перевода
ванадия в растворимую форму ванадат кальция обрабатывается
при кипячении раствором поташа. При этом в осадке образуется
карбонат кальция, ванадий же переходит в раствор в виде ва-
надата калия.
По рецептуре приготовления бариевого алюмованадиевого
катализатора (БАБ) полученный щелочной раствор ванадата
калия смешивается с раствором калийного силиката' и смесь на-
гревается до 70°. В отдельном баке смешиваются растворы хло-
рида алюминия, хлорида бария и соляной кислоты и полученная
смесь медленно 'прибавляется к смеси щелочных компонентов
при непрерывном перемешивании. В месте попадания струй рас-
твора кислых компонентов жидкость окрашивается в интенсив-
ный оранжевый цвет, исчезающий при перемешивании. Вскоре
начинает образовываться белый' хлопьевидный осадок.
15 Зак. 5019. Технология серной кислоты.
225
По окончании добавления кислых компонентов осадок, со-
держащий весь ванадий, отделяется с помощью фильтрпресса.
Продавливанием через отверстия масса формуется в виде ци-
линдриков диаметром‘около 4 мм.
Состав полученного таким путем катализатора:
12SiO2 • О.бАЮз • V2O5. КЮ • ЗВаО.
Окись бария служит в основном для перевода пятиокиси ва-
надия в осадок.1 Добавка окиси алюминия придает катализатору
термическую устойчивость, уменьшает усадку при нагревании.
Рис. 102. Схема установки для испытания активности контактных масс:
7—электрическая печь сопротивления; 2—контактная трубка; 3—термопара никель-нихром;
4—гальванометр; 5—промывалки Дрекселя; 6—смеситель газов; 7—баллон с кислородом
или углекислотой; 8—баллон с сернистым газом; 9, 10— анализаторы для газа высокой и
низкой концентрации; 77 — аспиратор; 7?—реостаты.
В других рецептурах для этой же цели применяются окиси
олова, сурьмы, железа и др.
Конечный продукт представляет собой белые чуть розова-
тые цилиндрики, I м3 которых весит 450—500 кг- При обра-
ботке сернистым газом масса принимает интенсивно желтую
окраску и вес 1 м3 возрастает до 600—650 кг1 вследствие обра-
зования сульфатов.
Этот процесс, называемый предварительным насыщением
сернистым 'газом, сопровождается значительным тепловым эф-
фектом. При действии на свежую массу крепкого сернистого
226
Рис. 103. Разрез контакт-
ной трубки для испыта-
ния активности контакт-
ных масс.
газа температура может повыситься до 900 1000 , что приве-
дет к спеканию контактной массы и полной потере активности.
Операцию 'предварительного насыщения следует проводить по-
этому газовой смесью, содержащей не более 0,5 % SO-2. В про-
цессе насыщения объем контактной массы уменьшается на
10—15%.
Кроме описанной выше гранулированной контактной массы
на наших заводах применяется также и таблетированная ' вана-
диевая контактная масса.
Таблетированная масса особенно пригодна для загрузки в те
части контактных аппаратов, через которые газ движется снизу
вверх, так как благодаря большей крупности и равномерности
кусков таблетированной массы опасность
уноса ее газовым потоком значительно
меньше.
Определение каталитической активности кон-
тактных масс. Активность контактных масс часто
характеризуют степенью контактирования, дости-
гаемой при определенной объемной скорости. При
этом, однако, не учитывается влияние состава
газовой смеси. Более ^строгой характеристикой
активности является величина' константы скоро-
сти реакции, вычисляемая с помощью кинетиче-
ского уравнения. Эта величина зависит только от
рода катализатора и от температуры. Для вы-
числения константы скорости должно быть из-
вестно количество прореагировавшего вещества
(степень контактирования), отвечающее опреде-
ленному времени соприкосновения газа с катали-
затором. Таким образом для полной характери-
стики активности контактной массы должны быть
экспериментально определены степени контакти-
рования при различных температурах и по полу-
ченным значениям вычислены с помощью кинети-
ческого уравнения константы скорости реакции.
Обязательным условием при проведении этих ис-
пытаний является постоянство температуры по
слою контактной массы.
Несоблюдение этого условия может привести
к весьма значительным ошибкам.
На рис. 102 приведена схема стандартной
активности контактных масс. Газовая смесь получается смешением воздуха
с сернистым газом, подаваемым из баллона с жидким SO2- Оба компонента
предварительно высушиваются крепкой серной кислотой. Количества их за-
меряются реометром.
Из смесителя газы идут в контактную трубку (рис. 103). Исследуемая
контактная масса загружается в кольцевое пространство между контактной
трубкой и футляром для термопары. Трубки можно применять кварцевые,
фарфоровые или стеклянные тугоплавкие. Температура замеряется хромель-
алюмелевой или железоконстантановой термопарой, скользящей в футляре,
контактная трубка помещается в электрической печи сопротивления, снабжен-
ои тремя самостоятельно регулируемыми нихромовыми спиралями 1—1,
2, з—з. Это дает возможность отдельно регулировать температуру в раз-
ых частях слоя контактной массы и достигать требуемой изотермичности.
ляш°"УСТИМО коле®ан”е температуры по слою катализатора выше 5°. Выхо-
Щии из контактной трубки газ проходит через поглотитель с серной
установки для испытания
15»
227
кислотой н далее выпускается в тягу или наружу. До и после контактной
трубки определяется концентрация сернистого газа по Рейху.
Для испытания берется 10 мл контактной массы, предварительно
высушенной в токе воздуха при постепенном повышении температуры
до 450°, и смешивается с 20 мл плавленого кварца той же крупности.
Высота слоя смеси в контактной трубке 7,5 см- Температура замеряется
в трех точках: в середине слоя, на уровне 0,5 см выше нижней границы слоя
и на уровне 0,5 см ниже верхней границы слоя массы. Точность опреде-
ления констант скорости указанным способом тем выше, чем дальше отстоит
измеряемая степень контактирования от теоретически возможной в условиях
эксперимента, так как в противном случае небольшие колебания и неточности
замера температуры будут очень резко влиять на значение константы ско-
рости.
Следует поэтому работать с высокими скоростями газового потока. При
испытании активных ванадиевых контактных масс при температурах выше 450°
скорость газовой смеси берется равной 600— 700 мл в минуту при нормальных
условиях, а для более низких температур 200 мл в минуту.
Характерная для данных условий степень контактирования устанавли-
вается обычно не сразу. Необходимо поэтому до начала определения актив-
ности выдерживать установленный температурный режим не менее 2 час. для
температур выше 450° и не менее 4 час. для низших температур.
'Вычисление значения констант скорости из экспериментальных данных
см. стр. 229 («Платиновые катализаторы») и стр. 234 («Ванадиевые контакт-
ные массы»),
В табл. 51 приведены данные о каталитической активности
различных контактных масс. Для' характеристики активности
приведены значения констант скорости реакции, вычисленные по
уравнению Тэйлора-Ленера (стр. 231).
Таблица 51
Сравнительная каталитическая активность различных контактных масс
Температура в °C Платиниро- ванный силикагель (0,2% Pt) Бариевая алю- мованадиевая контактная масса Хромо- ол овянная контактная масса Колчеданный огарок
350 1,2
400 3,3 0,26 0,39 —
450 7,8 8,7 3,4 —
475 11,7 16,2 8,4 —
500 17,1 26,1 20,1 —
525 24,1 41,0 — *
550 33,3 63,0 —
625 — — .—- 0,6
650 — —. — 1.1
675 — — — 2,1
700 — — — 3,9
Кажущаяся 17 000 22 000 39 000 44 000
энергия (при темпера- (при темпера-
активации турах выше турах выше
в кал!моль 450°) 625°)
228
4. Кинетика и механизм окисления SO2
Платиновые контактные массы. Зависимость скорости окисле-
ния SO2 на платине от состава газовой смеси наиболее подробно
изучили Боденштейн и Финк. Эти исследователи' применили ста-
тический метод и следили за скоростью реакции по уменьшению
давления реакционной смеси, находившейся при постоянном
объеме. Большинство экспериментов проведено при 150—250°.
Авторы'нашли, что для смесей, содержащих избыток кислорода
против стехиометрического соотношения, скорость реакции
прямо пропорциональна концентрации SO2, обратно пропорцио-
нальна корню квадратному из концентрации SO3 и не зависит от
концентрации Ог. 1
Эта зависимость выражается уравнением
c>so2
(14)
где т — время и к — константа скорости реакции.
При температурах, применявшихся в' опытах Боденштейна и
Финка, равновесие настолько полно смещено в сторону обра-
зования БОз, что скорость обратной реакции можно было не
учитывать. Между тем в производственных установках' окисле-
ние SO2 ведется при температурах 400—600°, при которых уже
нельзя пренебрегать скоростью обратной реакции. Тейлору и Ле-
неру, изучавшим скорость окисления SO2 по способу нагретой
проволоки при температурах 525—700°, не удалось вывести
самостоятельных уравнений для скоростей прямой и обратной
реакции. Они нашли лишь суммарную формулу, выражающую
скорость приближения к составлению равновесия.
Эта суммарная формула отличается от уравнения Боден-
штейна и Финка заменой концентрации SO2 разностью концен-
траций SO2 в данный момент и в состоянии равновесия:
^SO2^£so3
di di
(15)
здесь C'so2 — концентрация сернистого газа в состоянии равно-
весия при данных температуре и составе исходной газовой
смеси.
Это уравнение учитывает обратимость реакции окисления SO2
и поэтому может быть использовано для расчета скорости про-
цесса в производственных условиях. Для этой цели уравнение
(15) удобно преобразовать, введя в него вместо концентраций
SO2 и SO3 в момент времени т величины, непосредственно заме-
ряемые, а именно, начальную концентрацию сернистого газа а
и степень контактирования в данный момент х.
229
Тогда, если пренебречь изменением объема в результате ре-
акции
с8о,==а(1—х)'> '
CgOj = ах;
£|зо2 = G (1 — хт)-
Подставив эти значения в уравнение (15), получим:
dx
— —k
dt
(16)
Как’уже указывалось, к для данного катализатора зависит
только от температуры, хг — от температуры и начального со-
става газовой смеси. Поэтому при изотермическом процессе оки-
сления SO2 к и хт остаются постоянными и уравнение (16)
можно проинтегрировать в общем виде.
Разделяем переменные
_______ k
хт — х~
Обозначим У х через z, тогда
х = г2; dx — 2zdz
f У* , г— 1/% +г
I —-— dx — Ух In -----------2z + const =
J Хт—х у хт — z
= Vx In —2 Ух 4- const.
У Хт— У х
Уравнение (16) после интегрирования примет вид:
1 В производственных условиях концентрация сернистого газа не пре-
восходит обычно 7% и максимальное уменьшение объема 'меньше 3,5%.
230
Постоянную ' интегрирования можно определить из условия,
что при т=0, х —0. Подставляя это условие в уравнение (17),
получим С = 0. Следовательно, окончательно
к = 25(1^In • (18)
ь \ у ат — Vх. '
С помощью полученного уравнения можно вычислять значе-
ние констант скорости реакции из экспериментальных данных
или, если значение констант скорости известно, вычислять время
соприкосновения т газовой смеси с катализатором, необходимое
для достижения степени контактирования х. При этом необхо-
димо помнить, что уравнение (18) справедливо лишь при условии
протекания реакции при постоянной температуре.
Действительное время соприкосновения газа с катализатором
равно отношению свободного объема катализатора к проходя-
щему через него в 'единицу времени объему газа. При таком
определении т необходимо дополнять характеристику катализа-
тора долей свободного объема.
Чтобы избежать этого, мы будем пользоваться' «фиктивным»
временем соприкосновения Тф, равным отношению всего объема
катализатора к объему проходящего газа при условиях реакции.
При этом различие свободного объема разных' катализаторов
учитывается значениями констант скорости реакции.
Наконец для расчетов протекания неизотермических процес-
сов удобно пользоваться фиктивным временем соприкосновения,
отнесенным к объему газа при нормальных условиях тс.
Очевидно, что
__ t
Ч — 1Г;
273-М_ 273+/
Ч t ₽/ ’ ’
где р — доля свободного объема контактной массы.
Зависимость константы скорости' реакции от температуры вы-
ражается экспоненциальной функцией вида
__Е
k = kQe вт- (13)
Здесь Е — кажущаяся энергия активации. Она представляет со-
бой не просто запас энергии, необходимый для возможности
вступления' в реакцию двух молекул, встретившихся на поверх-
ности катализатора, а включает еще теплоты адсорбции реаги-
рующих веществ и продуктов, определяющих концентрацию их
на поверхности катализатора.
Справедливость уравнения (13) легко проверить.
Прологарифмируем его:
In k =— ^у,-|-1пА0. (21)
(19)
(20)
231
Полученное уравнение показывает, что In к является линей-
ной функцией у. Поэтому, в случае справедливости уравнения
(13)' все экспериментальные точки, откладываемые на графике
•у, 1п к, должны лежать на одной прямой. На рис. 104 прове-
дено соответствующее построение по данным Маклакова и Архи-
повой для платинированного силикагеля в интервале температур
430—500°. 'Полученные точки хорошо ложатся на прямую. По
уравнению (21) тангенс угла наклона полученной прямой равен
Е
— что позволяет вычислить кажущуюся энергию актива-
Рис. 104. Константы скорости окисления SO2 на платине.
ции, равную 16 900 ккал/кг • моль. Для различных платиновых
катализаторов эта величина колеблется от' 14 000 до
17 500 ккал/кг • моль. Для промышленных, платиновых контакт-
ных масс можно принимать £=17 000 ккал/кг • моль. С по-
мощью этой величины можно вычислять по уравнению (13)
значения констант скорости при любых температурах, если
известна активность катализатора при одной какой-либо тем-
пературе.
'Ниже приведены значения констант скорости для платини-
рованного силикагеля, содержащего 0,2% Pt. При расчете этих
констант по уравнению (18) а подставлялось в объемных про-
центах, х и хт в'долях единицы, Тф— фиктивное время сопри-
косновения в секундах.
1° k Е k Е k
400 3,28 485 13,70 550 33,35
425 4,36 500 17,11 575 44,98
450 7,79 525 24,08 600 60,15
475 11,74
232
На рис. 105 приведены для этой контактной массы кривые
зависимости 1 степени контактирования от времени соприкоснове-
ния по уравнению (18) для температур 450—600 . При построе-
нии этих кривых время соприкосновения пересчитано на объем
газа при 0°. Это облегчает расчет неиэотермических процессов,
при котором'приходится пользоваться одновременно несколькими
кривыми.
Детальный механизм окисления SO? в присутствии платины
пока не выяснен.
Рис. 105. Изотермы окисления сернистого газа на платинированном силика-
геле:
Кривые /—400°; 2—425°; 3—450°; 4—475°; 5—500°; 5—525°; 7— 550°; 5—575°; 9—600°
Вероятно процесс слагается из следующих этапов:
1. Активированная адсорбция кислорода платиной.
2. Взаимодействие молекул SOz газовой фазы с адсорбиро-
ванным кислородом с образованием молекул SO3.
3. Десорбция молекул 8Оз с поверхности платины. Скорость
первого этапа сравнительно велика и практически вся свобод-
ная поверхность платины покрыта адсорбированным кислородом.
Этим объясняется независимость скорости реакции 'от концен-
трации кислорода в газовой фазе. Наиболее медленным, а сле-
довательно, и определяющим общую скорость реакции,,
является второй этап, требующий энергию' активации порядка
233
17 000 кал/моль. Уменьшение скорости реакции с увеличением
концентрации SOs связано с тем, что молекулы SOs также
адсорбируются платиной и уменьшают в' результате этого долю
поверхности, занятую кислородом.
В противоположность этим представлениям Боденштейн по-
лагает, что определяющей суммарную скорость процесса
является ' не скорость взаимодействия, а скорость подвода реа-
гирующих веществ в зону реакции.
Для объяснения найденной зависимости скорости реакции от состава газа
Боденштейн предположил, что каталитической активностью в отношении
данной реакции обладают лишь отдельные небольшие участки поверхности
платины, так называемые активные центры. Молекулы SOs и О2, попадающие
на остальную часть поверхности, могут прореагировать, лишь продиффупди-
ровав вдоль поверхности к этим активным местам. Скорость взаимодействия
SO2 с кислородом на активных центрах очень велика и общая скорость
реакции определяется лишь скоростью диффузии компонентов к активным
местам.
Более легкие молекулы кислорода диффундируют скорее и находятся
в избытке на активных центрах. Скорость реакции равйа поэтому числу мо-
лей SO2, диффундирующих в единицу времени к активным центрам. Это
число пропорционально концентрации SO2 и не зависит от концентрации Ог.
Адсорбируемая платиной трехокнсь серы затрудняет диффузию SO2 к актив-
ным местам и тем самым снижает скорость реакции.
Ванадиевые контактные массы. Влияние концентрации SO2 и
Ог на скорость их взаимодействия в присутствии ванадиевого
катализатора'изучали Боресков и Соколова. Они нашли, что ско-
рость реакции в присутствии ванадиевой контактной массы вы-
ражается так:
dt R С°’в
cso3
(22)
Скорость обратной реакции может' быть учтена методом, ис-
пользованным Тейлором и Ленером при выводе кинетического
уравнения окисления SO2 на платине. Для этого предположим,
что скорость уменьшения концентрации SO2 определяется не
всей концентрацией' сернистого газа, а лишь той ее частью, ко-
торая может прореагировать до достижения состояния равно-
весия.
Тогда
^Cso,___, г (CSp2 С5О/8
dt. ~ °«
(23)
где С 'so. — концентрация SO2 в состоянии равновесия при дан-
ной температуре.
Для удобства расчетов' введем а — начальную концентрацию
SO2, b — начальную концентрацию кислорода их — степень пре
вращения в момент времени тф.
231
Тогда1
Cso X)’>
О Wj
си=йО-хт);
CSq3 —ax.
Подставляя в уравнение (23), получаем:
dx
— = k
ох0’8
(24)
Или, если вычислять время
при 0°:
соприкосновения по объему газа
dx 273
— =k--------
dx0 273 -b t
(xT— x)0.8
ex0,8
ах
Ь-----
2
(25)
Разделяя переменные и интегрируя, находим:
(26)
г U a* .
2 ’
С помощью этого уравнения можно определять необходимое
время соприкосновения газа с катализатором, если известны кон-
станты ' скорости, и рассчитывать значения констант из экспери-
ментальных данных. Интеграл в правой части уравнения нельзя
решить в общем виде и в каждом отдельном случае приходится
прибегать к графическому интегрированию.
Влияние температуры на скорость окисления SO2 на ванадие-
вых катализаторах исследовалось Боресковым и Плигуновым.
С целью проверки применимости к 'данному случаю уравнения
Е
Аррениуса к = к<- е RT найденные значения констант скорости
наносились на график In к, \/Т. Оказалось, что эксперименталь-
ные точки не укладываются на одну прямую, а располагаются на
двух прямых, сходящихся вблизи 440°. В дальнейшем при уточ-
нении этих исследований путем увеличения длительности вы-
держки при каждой температуре удалось показать, что эти пря-
мые не пересекаются и между ними располагается область
(425—475°), в которой активность зависит от предыдущего ре-
жима работы катализатора (рис. 106). Наклон прямой,1 * отвечаю-
щей высоким температурам, соответствует величине энергии
активации ~ 22 000 ккал/моль, что близко к энергии активации
на платине. Величина энергии активации, вычисленная'из наклона
нижней прямой, значительно выше, ~ 40 000 ккал/моль.
Найденная закономерность указывает на то, что внутри изу-
ченного интервала температур катализатор испытывает какое-то
изменение. Это подтверждается также изменением состава и
1 Здесь, как и раньше, мы пренебрегаем уменьшением объема в резуль-
тате реакции.
235
цвета катализатора, работавшего при различных температурах.
Так, образцы катализатора, выгруженные после работы в усло-
виях, отвечающих точкам верхней прямой, окрашены в желтый
цвет и содержат ванадий, в' основном, в пятивалентной форме.
Образцы же катализатора, выгруженные после работы при бо-
лее низких температурах, окрашены в зеленоватый цвет и содер-
жат ванадий, в основном, в четырехвалентной форме.
Отсюда можно заключить, что превращение, вызывающее из-
лом на графике Ink, l/T' для ванадиевых катализаторов, неви-
димому, состоит в переходе пятиокиси ванадия, содержащейся
в катализаторе при высоких температурах (возможно в виде ва-
надатов) в сульфат ванадила (VOSO4). Это превращение сопро-
вождается резким снижением активности'катализатора. Поэтому
ванадиевые катализаторы рационально эксплоатировать лишь при
Рис. 106. Константы скорости окисления SO2 на ванадиевом катализаторе.
температурах, отвечающих верхней кривой рис. 106, т. е. выше
440°. Для этого участка зависимость констант скорости от тем-
пературы определяется уравнением
22 000
k — kGe IlT, (27)
где к0— коэфициент, характерный для катализатора и не завися-
щий от температуры.
Подставляя (27) и (25), приходим к следующему уравнению
для расчета скорости окисления SO2:
dx
22 0С0
= "Яг
(хт — X)0-8
(28)
В табл. 52 приведены значения констант скорости реакции
для гранулированных бариево-ванадиевых контактных масс.
В интервале температур 440—460°'даны по два значения кон-
станты скорости реакции. Как уже упоминалось, в этом интер-
вале каталитическая активность зависит от того, как подходить
к исследуемой температуре; со стороны высоких 'или низких
температур, т. е. до или после превращения активного компо-
нента в сульфат ванадила. '
236
Таблица 52
Температура в °C К I Температура в °C К
400 410 420 430 440 после низких температур после высоких температур 450 после низких температур после высоких температур 0,022 0,036 0,053 0,086 0,141 0,87 0,47 1,05 460 после низких температур после высоких температур 475 500 525 550 575 600 1,20 1,32 1,75 2,90 4,58 7,03 10,60 15,68
В производственных условиях низкие температуры обычно
наблюдаются в начале и в конце процесса. Поскольку для ' на-
чальных частей контактной массы опасность сульфатизации осо-
бенно велика для них следует брать при расчетах пониженные
значения'константы скорости.-При определении же объема по-
следних по ходу газа частей контактной массы можно пользо-
ваться повышенными значениями к.
Механизм окисления сернистого газа на! ванадиевых катали-
заторах ’ рассматривается большинством исследователей с точки
зрения промежуточных реакций. Так, Нейман предполагает что
окисление SO2 на чистой пятиокиси ванадия протекает в резуль-
тате следующего цикла последовательных химических реакций:
V2Os + SO2 = V2O4 + SOs
V2O4 + О2 + 2SO2 = 2VOSO4
2VOSO4 = V2O, + SO2 4- SO3
При понижении температуры скорость последней реакции,
диссоциации сульфата ванадила, быстро снижается вследствие
приближения к равновесным условиям.
Нейман полагает, что в присутствии щелочей упругость дис-
социации сульфата ванадила возрастает, и этим он объясняет
повышенную активность катализаторов, активированных ще-
лочью, по сравнению с чистой пятиокисью ванадия.
1 Превращение пятиокиси ванадия в сульфат ванадила осуществляется
по уравнению
VaO3 + SO2 + SO3 = 2VOSO4.
поэтому наиболее благоприятными для этого -перехода будут степени контак-
тирования вблизи 50%.
237
По мнению Борескова активным компонентом промышленных
ванадиевых контактных масс являются щелочные' ванадаты.
В чистом концентрированном виде они малоустойчивы и при
температурах ниже 500° переходят в сульфат ванадила. При на-
личии же в составе катализатора кремневой кислоты активные
щелочные ванадаты распределяются в дисперсном состоянии по
ее поверхности и приобретают вследствие изменения свободной
поверхностной энергии необходимую устойчивость в широком
интервале температур. Точный состав каталитически активных
щелочных ванадатов не удалось установить. Ничего определен-
ного пока нельзя сказать и о деталях механизма их каталитиче-
ского действия. Наиболее вероятным является предположение,
что промежуточные этапы представляют собой поверхностные
реакции, приводящие к образованию поверхностных промежу-
точных соединений.
5. Отравление контактных'масс
Отравлением называется снижение активности катализатора
вследствие наличия в газовой смеси каких-либо примесей. Сни-
жение активности при отравлении твердых катализаторов вызы-
вается выключением 'части активной поверхности в результате
химического взаимодействия яда с катализатором с образова-
нием каталитически неактивного продукта, или химического пре-
вращения яда на поверхности катализатора с образованием неле-
тучих, каталитически неактивных веществ, 'или, наконец, в ре-
зультате активированной адсорбции яда на поверхности катали-
затора.
Если после подачи на отравленный катализатор чистой газо-
вой смеси первоначальная активность восстанавливается, то
отравление называется обратимым или временным. Если же
после прекращения подачи яда активность остается пониженной,
то отравление называется необратимым или ' п о с т о я н-
н ы м.
Мерой отравляемости служит относительное уменьшение кон-
станты скорости реакции в результате подачи на единицу ката-
лизатора весовой единицы яда. Отравляемость обычно не
остается постоянной в процессе отравления. Первые порции яда
вызывают значительно большее снижение активности, чем по-
следующие. В дальнейшем мы будем выражать отравляемость а
относительным уменьшением константы скорости реакции в ре-
зультате подачи 1 г яда на 1 г платины (для платиновых ката-
лизаторов) или на Гл контактной массы (для ванадиевых кон-
тактных масс):
где к — константа скорости реакции, dk — изменение константы
скорости реакции в результате' подачи на катализатор dg г яда.
238
В случае постоянства а уравнение (29) можно проинтегри-
ровать:
г гdk 2,303
= а=—1g-, (30)
11 и
гДе • g__общее количество яда, поданное на катализатор,
k ___ начальное (до отравления) значение константы скорости
реакции, кк — конечное (после подачи g г яда) значение кон-
станты скорости реакции.
Ниже даются некоторые сведения о влиянии возможных при-
месей обжигового газа на платиновые и ванадиевые контактные
массы.
В од яны е пары, по данным некоторых ' исследователей
в небольшой примеси необходимы для возможности каталитиче-
ского окисления SOa в присутствии платиновых катализаторов.
Так, по Кюстеру (Krister) газы, высушенные над пятиокисью фос-
фора, реагируют с меньшей скоростью, чем высушенные над сер-
ной кислотой. Дальнейшее увеличение количества влаги не ока-
зывает заметного'влияния на скорость реакции.
На ванадиевые контактные массы присутствие в газовой
смеси водяных паров также не оказывает вредного действия при
температурах выше температуры конденсации серной кислоты.
При низких температурах в результате‘конденсации серной ки-
слоты контактная масса разрушается с потерей каталитической
активности и механической прочности. Поскольку работавшие
ванадиевые контактные массы содержат значительное количе-
ство SOs, они быстро'разрушаются при соприкосновении с влаж-
ным воздухом. Поэтому перед длительными остановками необ-
ходимо продувать контактные аппараты горячим воздухом для
удаления поглощенного 8Оз и во время остановки не допускать
проникновения ' наружного воздуха в контактный аппарат. Вся-
кого рода перегрузки контактной массы должны производиться
как можно быстрее, после чего содержащие контактную массу
аппараты должны плотно закрываться.
Трехокись мышьяка вызывает очень сильное отра-
вление платиновых контактных масс. Отравление частично
обратимо, особенно при высоких температурах. Причиной отра-
вления является окисление на поверхности платины трехокиси
мышьяка в нелетучую пятиокись, препятствующую проникнове-
нию'реагирующих газов к активным участкам поверхности ката-
лизатора.
Из экспериментальных данных Неймана и Юттнера может
быть вычислена средняя отравляемость при добавлении 100 тиг
мышьяка на 1 г платины. Она равна при 450° — 61 и при
500 —32.
В патентной литературе имеются указания, что платина, на-
несенная на силикагель, менее чувствительна к действию
мышьяка. На ванадиевые контактные массы трехокись
239
мышьяка, вопреки многочисленным патентным указаниям, также
оказывает отравляющее действие, но во много раз меньшее, чем
на платину.
Согласно измерениям Ададурова и Гуминской отравляемость
бариевой оловянно-ванадиевой контактной массы (БОВ) равна
0,01 (относительное уменьшение константы скорости реакции
при подаче 1 г мышьяка на 1 л контактной массы). Приблизи-
тельно того же порядка отравляемость бариево-алюмо-ванадие-
вой контактной массы (БАВ) по данным Гуминской и калиево-
ванадиевого катализатора по данным Зигерта и Ольсена и
Майснера.
В процессе отравления AS2O3 задерживается катализатором
и в отравленных образцах содержится в виде AS2O5. При про-
дувке газовой смесью, свободной от мышьяка, активность отра-
вленного катализатора несколько повышается, но остается зна-
чительно меньше первоначального значения. По отношению
к мышьяку ванадиевые контактные массы в несколько тысяч
раз (в среднем в 5000 раз) устойчивее платиновых. Другими сло-
вами, одинаковое снижение активности произойдет при добавле-
нии к ванадиевой контактной массе в 5000 раз большего коли-
чества мышьяка, чем к платиновой. Отсюда нельзя, однако, де-
лать вывод о том, что при работе с ванадиевыми контактными
массами очистка газа от мышьяка становится излишней.
Действительно, предположим, что сырьем служит колчедан, содержащий
0,1% мышьяка и при обжиге в газовую фазу переходит 76%. В таком слу-
чае в объеме газовой смеси, отвечающем выработке <1 т моногидрата серной
кислоты, будет содержаться 0,7 кг мышьяка. Для выработки 1 т моно-
гидрата серной кислоты в сутки в контактном аппарате должно находиться
200 л ванадиевой контактной массы. Если принять среднюю отравляемость
ванадиевых контактных масс 0,01, то по уравнению (30) нетрудно вычислить,
что для снижения активности в 2 раза на 1 л контактной массы должно быть
подано:
2,3 kK 2,3
G = — 1g — 777ГГ lg 2 = 70 г мышьяка,
а ° kL. 0,01 ь
а на 200 л соответственно 14 кг. Ванадиевая контактная масса снизит, следо-
вательно, в рассматриваемых условиях свою активность в 2 раза за 14:0,7 =
= 20 суток. В действительности нормальная работа контактного узла будет
нарушена еще раньше, так как мышьяк будет задерживаться главным обра-
зом первыми по ходу газа слоями контактной массы и быстро выведет их
из строя.
Мышьяковистый водород, вследствие легкой оки-
сляемости, оказывает такое же действие, как и трехокись
мышьяка. '
Селен и теллур являются очень сильными ядами для
платиновых контактных масс. На ванадиевые контакт-
ные массы селен оказывает вредное действие лишь при темпе-
ратурах ниже 400е. После прогрева при 480—500е восстанавли-
вается первоначальная активность. По действию теллура на ва-
надиевые контактные массы'никаких данных нет.
Углекислота по данным Ризе (Reese), Якимец и Баки-
ной, Борескова и Гуминской, при температурах промышленного
249
проведения контактного процесса не оказывает вредного дей-
ствия на платиновые1 контактные массы даже в количестве
Ю—15% от общего объема газовой смеси. На ванадиевые
контактные массы углекислота также не оказывает вред-
ного действия.
Окись углерода в условиях сернокислотного катализа
чрезвычайно быстро окисляется до углекислоты, не адсорбируе-
мой платиной при высоких температурах,' и поэтому не может
оказывать вредного влияния. Этот вывод подтверждается экспе-
риментальными исследованиями Якимец и Бакиной, Борескова
и Гуминской. В отношении влияния окиси углерода'на ванадие-
вые контактные массы имеются противоречивые указания. Боре-
сков и Соколова, работая' с газовой смесью, содержащей
3—5% СО при 450°, не обнаружили снижения активности вана-
диевой контактной массы в течение 100 час. Приблизительно
в тех же условиях Якимец и' Бакина не обнаружили отравляю-
щего действия окиси углерода на ванадиевую контактную массу
в течение 10 суток. В противоположность этому Зигерт (Н. Sie-
gert) наблюдал,' при работе со смесью 5% SO2, 5% СО и 90%
воздуха при 435° и скорости газа 2 л/мин через 100 мл ванадие-
вой контактной массы, сильное отравление уже через 46 час.
При продувке воздухом активность' медленно восстанавливалась.
При 500° отравление было меньшим и регенерация протекала
быстрее.
Зике указывает, что присутствие больших количеств окиси
углерода может снизить выход SO3, но не в результате отравле-
ния катализатора, а вследствие восстановления SOs по реакции
SOs + CO->SO2 + CO2.
Углеводород ы'также оказывают влияние на активность
контактных масс. Шредеру (Schroeder) неоднократно приходилось
наблюдать пробы платиновых контактных масс, почерневших от
выделившейся на них сажи. Это происходило при применении
руды, подвергавшейся флотации е добавкой органических соеди-
нений. Последние, не полностью сгорая на верхних этажах пе-
чей, могут попадать в обжиговый газ. В контактном аппарате
эти органические соединения 'могут разлагаться с выделением
углерода, поскольку температура недостаточно высока (450—
550°) для полного их сгорания. Выделение углерода сопрово-
ждается резким снижением каталитической активности. Вызы-
вается ли это снижение простым' механическим покрытием по-
верхности платины или образованием неактивного платиноугле-
родистого соединения, точно не установлено.
Возможность выделения углерода из органических соедине-
ний зависит от природы углеводородов, их концентрации, тем-
пературы контактного объема и концентрации кислорода.
В лабораторных условиях, при работе с газовой смесью, со-
держащей-около 3 г/м3 смеси керосина и машинного масла, на-
6 Зак. 5019. Технология серной кислоты.
241
блюдалось небольшое временное снижение активности платино-
вых и ванадиевых контактных масс, исчезавшее после прогрева
при 500°. При добавке к газовой смеси продуктов термического
разложения кислых гудронов в лабораторных условиях наблю-
далось снижение активности, особенно заметное при низких тем-
пературах и полностью необратимое.
Сероводород вызывает отравление платиновых катали-
заторов при окислении аммиака и при многих других каталити-
ческих реакциях. Нет, однако, непосредственных указаний на
вредное влияние сероводорода на платиновые контактные массы
в условиях сернокислотного контактного процесса.
Действие сероводорода на ванадиевые контактные массы про-
веряли в лабораторных условиях Боресков и Соколова при 485°
и 425°. Они нашли, что при содержании в газовой смеси 3%
сероводорода снижение активности не наблюдается в течение
100 час.
По данным Зике на контактных заводах, работающих по ме-
тоду так называемого мокрого катализа, при нарушениях нор-
мального режима на ванадиевую контактную массу попадали
в течение нескольких часов большие количества сероводорода
при недостатке кислорода. После перехода на работу с газом
нормального состава наблюдалось определенное снижение актив-
ности, полностью исчезавшее, однако, после работы в течение
некоторого времени при высокой температуре (500°) с газовой
смесью, содержащей возможно больше кислорода.
Хлор и хлористый водород вызывают сильное отравление пла-
тиновой контактной массы. Так, по наблюдениям Ризе присутствие хлори-
стого водорода в газовой смеси снижает степень контактирования на плати-
нированном сульфате магния до 42%, примесь хлора до 57%.
Шредер (Schroeder) описывает случай, когда не удалось (в 1890 г.)
пустить один завод с платиновой массой, вследствие того что в аппарат для
сушки газа загрузили по ошибке вместо хлористого кальция хлорную известь
и в газ попадало значительное количество хлора. Отравление платиновых
катализаторов хлором и хлористым водородом обратимо. После прекращения
подачи яда первоначальная активность через некоторое время восстанавли-
вается.
На ванадиевые контактные массы хлор и хлористый водород вредного
действия не оказывают.
Фтористый кремний резко снижает активность платиновой массы.
Шредер указывает на факт снижения активности платинированного сульфата
магния иа сернокислотном заводе в Гамборне (Hamborn) в результате работы
иа фторсодержащей руде. При вскрытии аппарата в контактной массе обна-
ружено было значительное количество кремневой кислоты. Вероятно, в про-
цессе обжига образуется SiFn разлагающийся в дальнейшем на контактной
массе с выделением свободной кремневой . кислоты, покрывающей плотным
слоем поверхность катализатора. Отравление (необратимо, но после обрызги-
вания отравленной массы слабой царской водкой первоначальная активность
восстанавливается, вероятно, в результате обновления поверхности платины.
Имеются отдельные указания на вредное действие фтора на ванадиевые
контактные массы, но надежных данных нет.
Сульфат железа вредно действует на платиновые кон-
тактные массы возможно в результате механического покрытия
их поверхности.
242
Для ванадиевых контактных масс, сравнительно устойчивых
к действию рассмотренных выше примесей, сульфат железа
является наиболее вредным загрязнением. Он может образо-
ваться в самом контактном узле в случае недостаточно полной
осушки газа и очистки его от брызг и тумана серной кислоты.
По Зике сульфат железа образует с ванадиевой контактной мас-
сой легкоплавкую эвтектику, в результате чего первый по ходу
газа слой контактной массы может превратиться в спекшийся
ком. Образование твердых корок на первых слоях ванадиевой
контактной массы часто наблюдалось и на наших заводах. Эти
корки слагаются из отдельных гранул контактной массы, склеен-
ных розоватым веществом, состоящим из сульфата железа и
пятиокиси ванадия.
Особенно неприятно это явление в контактных аппаратах
с внутренним теплообменом, где образование корок происходит
вдоль поверхности теплообменных элементов, покрывая их плот-
ным слоем толщиной до 15 мм. Помимо снижения активности
контактной массы накопление сульфата железа приводит к ухуд-
шению теплообмена, росту гидравлического сопротивления и не-
равномерному распределению газа по сечению аппарата. Для
предохранения контактной массы от попадания сульфата железа
Зике рекомендует устанавливать, непосредственно перед кон-
тактным аппаратом, фильтр из керамических фильтровальных
камней. Это приводит, однако, к увеличению гидравлического
сопротивления контактного узла.
Наиболее радикальным мероприятием по борьбе с отравле-
нием сульфатом железа надо признать тщательную осушку газа
и очистку его от тумана серной кислоты.
В производственных условиях отравление платиновых кон-
тактных масс вызывается чаще всего присутствием в газах со-
единений мышьяка.
При небольшом снижении активности частичная регенерация
может быть проведена в самом контактном аппарате путем про-
дувки чистой газовой смесью при возможно более высоких тем-
пературах. При этом происходит частичная диссоциация пяти-
окиси мышьяка и удаление ее из аппарата в виде трехокиси.
Хорошие результаты получаются при добавлении к смеси хлора
или хлористого водорода. В этом случае мышьяк уходит из ап-
парата в виде хлористого мышьяка AsCls. Возможное в этих
условиях снижение активности под влиянием хлора или хлори-
стого водорода несущественно ввиду обратимости отравления
хлором. При более глубоком отравлении контактная масса вы-
гружается из аппарата и регенерируется либо путем промывки
соляной кислотой, либо смачиванием царской водкой. В послед-
нем случае каталитическая активность увеличивается вследствие
обновления поверхности платины. Полностью достигнуть перво-
начальной активности обычно не удается.
Снижение активности ванадиевых контактных масс в произ-
водственных аппаратах обычно происходит в результате терми-
16* _ 243
ческой порчи и накопления сульфата железа. Термическая порча
вызывает настолько глубокое изменение в структуре катализа-
тора, что удовлетворительных методов регенерации пока не най-
дено. Из отработанной массы, однако, легко извлечь соединения
ванадия и использовать их для приготовления свежей контакт-
ной массы.
6. Оптимальный температурный режим процесса окисления SO2
» •
Оптимальной температурой мы называем ту температуру, ко-
торая обеспечивает при других постоянных условиях максималь-
ную скорость образования SO3, т. е. максимальную интенсив-
ность процесса. Ограничимся вначале рассмотрением реакции на
ванадиевых контактных массах. Совершенно аналогично могут
быть определены оптимальные температуры и для окисления
SO2 на платиновых катализаторах.
Для скорости окисления SO2 на ванадиевых контактных мас-
сах выше (стр. 235) было выведено следующее уравнение:
^80, = а dx = Л(хт-х)0.«Л ПЛ \ 273
dtQ dtQ х^*& \ 2 / 273 1
При оптимальной температуре это выражение должно дости-
гать максимума. Из величин, стоящих в правой части уравнения,
от температуры зависят к и хт- Зависимость от температуры
константы скорости реакции определяется уравнением (27). С по-
вышением температуры она быстро растет. Зависимость от тем-
пературы теоретически возможной степени контактирования
определяется уравнением (10). С повышением температуры хт
падает. Нетрудно показать, что скорость образования SO3 с из-
менением температуры, действительно, проходит через максимум.
При низких температурах хт практически постоянно и равно
единице. Скорость образования SO3 растет с повышением темпе-
ратуры вследствие быстрого увеличения к. При высоких темпе-
ратурах хт меньше единицы и падает с ростом температуры. При
некоторой температуре tv, при которой хт становится равным х,
скорость образования SOs делается равной нулю. Значит при
какой-то температуре tx < tp скорость образования SO3 должна
достигать максимального значения. Для определения этой опти-
мальной температуры удобнее всего воспользоваться графиче-
ским методом. Вследствие резкой зависимости скорости реакции
от температуры рациональнее производить построение на лога-
рифмическом графике. Логарифмируя уравнение (31), находим:
lg(«SO = - 2>да+ °’8 <273 + 1g Со> (32)
й027з(*-^)
где Со— ——---08 —-объединяет величины, не зависящие от
температуры.
244
• На рис. 107 отложены значения 1g в зависимости от
температуры для газовой смеси начального состава 7% SO2,
11% Ог и 82% N-2. Из формы полученных кривых следует, что
для каждой степени контактирования скорость реакции дости-
гает резко выраженного максимального значения при определен-
ной температуре. Эта температура и является согласно опреде-
лению оптимальной для данной стадии контактирования. Из гра-
фика следует далее, что чем выше степень контактирования, тем
ниже оптимальная температура. Поэтому нельзя говорить о ка-
кой-то постоянной оптимальной температуре для всего процесса
окисления SO2. Невыгодно поддерживать по всему слою кон-
Chopocmtj реакции
Рис. 107. Зависимость скорости окисления SO2 от температуры
при различных степенях контактирования.
тактной массы одинаковую температуру. Существует определен-
ная оптимальная кривая изменения температуры по слою контакт-
ной массы, отвечающая началу процесса при высоких температу-
рах и постепенному снижению температуры по мере роста степени
контактирования, в соответствии со смещением максимума кри-
вых рис. 107. Проведение процесса, согласно этой оптимальной
температурной кривой, обеспечивает максимальную скорость реак-
ции и, следовательно, наиболее полное использование контактной
массы. Найденные указанным способом значения оптимальных
температур для газа начального состава 7% SO2, 11% Ог и
82% N2 приведены в табл. 53. Значения оптимальных температур
Для ванадиевых и платиновых катализаторов расходятся между
собой незначительно. Изменение начального состава газовой
смеси меняет величину хт и должно, следовательно, сказываться
на значении оптимальной температуры. Данными табл. 53 можно
245
пользоваться и для смесей другого начального состава, но даю-
щих при равных температурах приблизительно те же значения
теоретически возможной степени контактирования, как и смесь
состава 7% SO2, 11 % Ог, 82% N2. Так, например, они применимы
для газа, содержащего 9,1% SO2 и 11,9% О2 (сжигание серы)
или 20% SO2 и 16,8% Ог (смешение чистого SO2 с воздухом),
или 63,5% SO2 и 36,5% О2 (смешение чистого SO2 с кислоро-
дом).
Для смесей, характеризующихся повышенными значениями хт,
оптимальные температуры выше, для смесей с меньшими хт —
ниже, чем указано в табл. 53.
Таблица 53
Оптимальные температуры для газа, содержа-
щего 7% SO2, 11% О2 и 82% N2
Степень контакти- рования х-102 Оптималь- ные тем- пературы ^ОПТ Степень контакти- рования X- 102 Оптималь- иые тем- пературы ^опт
96,4 440 88 503
96 445 85 517
95 455 80 537
94 463 70 572
92 479 60,9 600
90 492
Кривые А и В рис. 107 дают допустимые интервалы колеба-
ний температуры, при которых скорость реакции остается равной
0,9 максимального значения, отвечающего оптимальной темпера-
туре. Полученные значения удобно перенести на график: тем-
пература— степень контактирования (рис. 108). Здесь АВ кривая
теоретически возможных степеней контактирования. Вдоль этой
кривой скорость образования SO3 равна нулю. Точки, лежащие
выше этой кривой, вообще недостижимы для сернистого
газа. Кривая CD соответствует оптимальным температурам.
В точках этой кривой скорость реакции достигает максималь-
ного значения при данной степени контактирования. Кривые KL,
MN, PR и ST отвечают скоростям, пониженным по сравнению
с максимальной, соответственно на 10, 20, 30 и 50% в результате
отклонения температуры от оптимального значения в сторону
высоких и низких температур. Нанося на этот график действи-
тельные кривые изменения температуры со степенью контактиро-
вания, можно судить о правильности температурного режима и,
следовательно, о совершенстве конструкции рассматриваемого
контактного аппарата. Совершенная конструкция контактного
246
аппарата должна, очевидно, обеспечивать соответствие действи-
тельного температурного режима с оптимальной температурной
кривой.
Рис. 108. Диаграмма: температура — степень контактирования:
АВ—равновесная кривая; СО—оптимальная кривая; кривая ST—граница зоны
I кривая PR—граница зоны > 0,7 ; кривая Л1Л7—граница зоны
dx „ /dx\ ,,, dx „ „ (dx\
-г >0,8 (-г) : кривая KL—граница зоны — > 0,9 ( — I
az '«wmax шах
7. Определение объема ванадиевой контактной массы
для заданной степени контактирования
Фиктивное время соприкосновения т0 согласно определению
связано с объемами газа и катализатора соотношением
^о=-у. (33)
Отсюда для объема контактной массы получаем выражение
^=Vt0, (34)
или, вводя коэфициент запаса:
v = cV-0- (35)
247
здесь v — объем контактной массы в ж3, V — объем газа
в м3/сек в пересчете на нормальные условия, т0—фиктивное
время соприкосновения, отнесенное к объему газа при нормаль-
ных условиях, с — коэфициент запаса, вводимый вследствие воз-
можности отклонения действительного режима от предполагае-
мого, а также вследствие снижения активности катализатора
в процессе эксплоатации. Таким образом задача определения не-
обходимого количества контактной массы сводится к расчету
необходимого времени соприкосновения т0.
Интегрирование уравнения (31) дает:
?т^(жЫ“Лг> (Зв)
J К ЛЮ \Хт лJ » __“Л
я?1 2
хг и к зависят от температуры. Подинтегральное выражение
является таким образом функцией степени превращения и темпе-
ратуры. Обозначим:
<3.
Температура в процессе реакции меняется в соответствии с из-
менением степени контактирования. Характер зависимости t от х
может быть различным и определяется условиями теплоотвода
в процессе реакции.
Рассмотрим частные случаи.
1. Изотермический процесс (t = const). Все тепло
реакции отводится. хт и к постоянны. Даже в этом наиболее
простом случае интеграл не удается решить в общем виде и
приходится прибегать к методу графического интегрирования.
На рис. 109 построены кривые изменения f (х, t) в зависимо-
сти от х при изотермическом протекании процесса при темпера-
турах 440, 450, 460, 475, 500, 525, 550, 575, 600° для газа со-
става: 7% SO2, 11% О2 и 82% N2.
Площади, ограниченные этими кривыми, ординатами xi и х-г
и осью абсцисс, равны интегралу уравнения (36), т. е. искомому
времени соприкосновения газа с катализатором, необходимому
для изменения степени контактирования от xi до хг.
Пример. Требуется определить время соприкосновения газа
с катализатором, необходимое для изменения степени контакти-
рования от Х1 = 0,5 до Х2 = 0,75 при 475°.
Решение. Восстанавливаем из точек оси абсцисс, отвечающих
степеням контактирования 0,5 и 0,75, перпендикуляры до пересе-
чения с изотермой 475°. Площадь, ограниченная этими перпенди-
кулярами, изотермой 475° и осью абсцисс, занимает 5,33 клетки.
Как видно из масштабов диаграммы (рис. 109), одна клетка от-
вечает времени соприкосновения, равному 0,1 сек. Искомое время
соприкосновения равно, следовательно, 0,533 сек.
248
Рцс. 109. Диаграмма х,
На рис. ПО приведены вычисленные указанным способом зна-
чения т0, необходимые для достижения различных степеней кон-
тактирования при изотермическом проведении реакции при тем-
пературе 440—600° для газа состава 7% 8Ог, 11% О? и 82% №.
В случае неизотермического процесса необходимое время
можно приближенно определять так же, разбив процесс на ряд
этапов и предположив, что в пределах каждого этапа темпера-
тура остается постоянной. Более точен и удобен, однако, способ,
излагаемый ниже.
Рис. 110. Изотермы окисления SO2 на ванадиевом катализаторе.
2. Оптимальный температурный режим (t =
= <р(х)=^опт.)- Температура меняется со степенью контактиро-
вания согласно оптимальной температурной кривой. В этом слу-
чае по определению оптимальной температуры скорость реакции
на всех стадиях контактирования отвечает максимально возмож-
ному значению, а обратная ей величина f (х, t) — соответ-
ственно — минимальному. Протекание процесса в соответствии
с оптимальной температурной кривой изобразится на диаграмме
кривой, совпадающей для каждого значения х с наиниз-
шей из изотерм, т. е. огибающей семейство изотермических кри-
550
вых. После того как построены изотермы, построение этой оги-
бающей не представит затруднений.
Необходимое время соприкосновения при проведении про-
цесса по оптимальной температурной кривой равно площади,
ограниченной огибающей семейство изотерм, соответствующими
ординатами и отрезком оси абсцисс. Это время будет минимально
возможным, так как огибающая представляет наинизшую из
возможных кривых (рис. 109).
3. Адиабатический процесс. Тепло от контактной
массы не отводится и температура растет за счет тепла, освобо-
ждающегося при реакции.
Введем следующие обозначения:
G—количество газа, проходящее через аппарат, в кг1час;
ав — начальный весовой процент сернистого газа;
ср— средняя теплоемкость газовой смеси в ккал)кг град,
q—теплота окисления 1 кг SO2 в ккал-,
t0 — начальная температура газа;
7газ — вес 1 лг1 газовой смеси при нормальных условиях зкг]м3.
Увеличению степени контактирования на dx соответствует
выделение тепла:
. Gaqdx
(38>
Температура газа возрастает при этом на
dQ a^qdx
dt = GTp=^ <39>
Заменим начальный весовой процент SO2 объемным:
2,857 а
а„ =----->
в у ’
*газ
где 2,857 — вес 1 м3 сернистого газа в кг при нормальных
условиях *.
Тогда
dt = 2;^ gad-. (40)
1®® 7газсГ>
Температура контактной массы в заводских аппаратах ме-
няется в ограниченных пределах (450—600°). Для простоты пре-
небрежем небольшими изменениями q и с9 в зависимости от тем-
пературы и степени контактирования и будем определять обе эти
величины для средних условий (f = 500°; х = 0,5). Согласно
формуле (7):
<75оо°= 22800 ккал/мол или 357 ккал!кг SO2.
1 Молярный объем сернистого газа мы принимаем равным 22,4 л, так '
как в условиях проведения процесса (малое парциальное давление SO?,
высокие температуры) свойства сернистого газа будут мало отклоняться
от свойств идеального газа. Пользование молярным объемом SO± при нор-
мальных условиях привело бы к несравненно большим ошибкам.
251
Подставляя в (40), получим:
2,857 • 357
~~ 100
_^-dx=10,2-V^.
•газ Р Iras Р
Интегрируя в пределах от to до t и от 0 до х, получим:
t — /о=1О,2 —й— х или t-t0~\-Xx, (41)
7газ\р
где.
Х = 10,2—; (42)
гав<'Р
здесь Л — максимальное увеличение температуры газа, отвечаю-
щее изменению степени контактирования от 0 до 1 в адиабатиче-
ских условиях. Найденное уравнение показывает, что на диа-
грамме (t, х) рис. 108 адиабатический процесс изобразится пря-
мой с тангенсом угла наклона, равным Л. Зная начальную кон-
центрацию а и температуру начала реакции t0, эту прямую не-
трудно построить. С помощью этой прямой мы можем опреде-
лить изменение температуры со степенью контактирования и изо-
бразить протекание адиабатического процесса на диаграмме
х, 1 рис. 109. Пусть на этой диаграмме построены изотермы
ti, tz,... С помощью прямой диаграммы (f, х) можно определить
степени контактирования, при которых достигаются указанные
температуры при данном адиабатическом процессе. Пусть они
составляют xi, хг,... Переходя теперь к диаграмме (х, и
восстановив из точек оси абсцисс xi, хг,... перпендикуляры до
пересечения с соответствующими изотермами, мы получим, оче-
видно, точки, отвечающие данному адиабатическому процессу.
Соединив их между собой и началом координат, получим кри-
вую, изображающую протекание данного адиабатического про-
цесса на диаграмме (х, Время соприкосновения, необходи-
мое для изменения степени контактирования от xi и хг при рас-
сматриваемом адиабатическом процессе, равно площади, ограни-
ченной этой кривой, ординатами xi и хг и осью абсцисс. ,
4. Аналогично решается задача и в общем случае.
Обычно, вследствие более или менее интенсивной теплопередачи,
процесс отклоняется от адиабатического. Кривая на рис. 108,
изображающая протекание реакции, пройдет левее адиабаты.
Отклонение ее от адиабаты будет равно на каждой стадии кон-
тактирования величине потерь тепла на пройденных этапах кон-
тактирования, выраженной в градусах изменения температуры
газа. Таким образом, зная потери тепла для отдельных стадий
окисления SO2, пересчитав их в градусы изменения температуры
газа по формуле = - и отложив влево от соответствую-
C/С»,
252
щих точек адиабаты на диаграмме (t, х), мы получим точки, от-
вечающие данному процессу. Соединив найденные таким путем
точки, получим кривую, изображающую рассматриваемый про-
цесс на диаграмме (t, х). С помощью этой кривой можно, так же
как и для адиабатического процесса, построить кривую данного
процесса и на диаграмме (х, Необходимое время соприко-
сновения, как и в предыдущих случаях, равно площади, огра-
ниченной этой кривой, ординатами и осью абсцисс.
Пример. Теплоотвод от контактной массы на отдельных ста-
диях контактирования отвечает данным табл. 54.
Таблица 54
Степень контактирования 0,5 0,6 0,7 0,75 0,8
Общий теплоотвод в градусах изме- нения температуры газа 6 17 35 46 60
Состав газа: 7% SO2, 11% О2, 82% N2. Начальная темпера-
тура 440°. Требуется определить время соприкосновения, необ-
ходимое для достиже-
ния степени контакти-
рования, равной 0,8.
Решение. Прежде все-
го изобразим заданный
процесс на диаграмме
(t, х) рис. 111. Согласно
уравнению (42)
Л =10,2—— =210.
7газсР
Уравнение (41) при-
мет в данном случае
следующий вид:
t = 440 + 210 х.
Изобразив его на диа-
грамме рис. Ill (t, х),
Рис. 111. Диаграмма t, х.
получим прямую ab,
отвечающую адиабатическому протеканию процесса. Для по-
строения кривой, отвечающей заданному процессу, откладываем
от адиабатической прямой влево величины теплоотвода, приве-
денные в таблице. Соединяя найденные таким способом точки,
получаем искомую кривую ас. С помощью этой кривой находим
степени контактирования, отвечающие температурам, для кото-
253
рых построены изотермы на графике (х, — J . Результаты све-
дены в табл. 55.
Таблица 55
Температура в °C 450 460 475 500 525 550 550
Степень контактирования 0,0475 0,095 0,167 0,275 0,44 0,64 0,76
С помощью этих данных строим кривую, изображающую
протекание данного процесса на диаграмме (х, рис. 109.
Площадь, ограниченная этой кривой, осью абсцисс и ордина-
той №0,8 (заштрихована на рис. 109), занимает 4,5 клеток.
Каждая клетка, как показывает масштаб диаграммы, соответ-
ствует времени соприкосновения равному 0,1 сек.
Отсюда искомое время соприкосновения равно:
4,5 • 0,1 = 0,45 сек.
8. Оптимальная концентрация SO2
Рассмотрим вначале вопрос об оптимальной концентрации
газа, получаемого при обжиге рядового безуглеродистого колче-
дана. В этом случае возможность изменения состава газовой
смеси ограничивается ббльшим или меньшим разбавлением из-
быточным воздухом, приводящим к уменьшению концентрации
сернистого газа и к увеличению концентрации кислорода.
Необходимое время соприкосновения газа с ванадиевым ка-
тализатором для достижения степени окисления х определяется
уравнением:
(273-|-Z)x0,8 dx
й(хт-х)0.в(&-^27з'
(36)
Соответствующий объем катализатора, отнесенный к единице
производительности:
^=QT0Ve = С2 = С2 f - (273 + 0x°’8rfx— , (43)
J А(*Т*М*-^)273
л '
где Ve — объем газа в м3/час для единицы производительности.
Ve — обратно пропорционален начальной концентрации SO2.
С увеличением разбавления газа избыточным воздухом концен-
трация кислорода b растет, концентрация сернистого газа а па-
дает, значит растет множитель b —Одновременно повы-
254
шается и теоретически возможная степень контактирования, а,
стало быть, увеличивается и второй множитель в знаменателе
(хт — х) °>8. Таким образом, с увеличением разбавления газа не-
обходимый объем контактной массы снижается. Величина этого
снижения зависит от температурного режима контактного аппа-
рата. Будем полагать в последующем, что процесс протекает
в соответствии с оптимальной температурной кривой. В табл. 56
приведены объемы ванадиевой контактной массы в литрах, необ-
ходимые для достижения различных степеней контактирования
при работе с газом, содержащим от 5 до 9% SO2. При этом коэ-
фициент запаса принят равным 1,5 и количество катализатора от-
несено к производительности 1 т моногидрата в сутки.
Тавлицл 56
Степень контакти- рования Объем ванадиевой контактной массы (в л) при составе газа
5% SO3 13,86% О2 6% so2 12,43% О2 7% SO2 11% о2 8% SO2 9,58% О2 9% О2 8,15% О2
0.94 62,5 81,0 104 149,5 242
0,96 92,5 117 152 220 334
0,97 118 149,5 198 317,7 —
Как видно из данных табл. 56, необходимое количество кон-
тактной массы возрастает с увеличением концентрации SO2 вна-
чале (при малых концентрациях) медленно, а в дальнейшем очень
быстро.
Поэтому с точки зрения расхода контактной массы наиболее-
выгодной надо было бы признать работу на слабом газе. Стои-
мость контактной массы, однако, невелика по сравнению
с остальными капитальными затратами и мало отражается на се-
бестоимости продукции. Поэтому при определении оптимальной
концентрации следует брать за основу не производительность
единицы объема контактной массы, а производительность кон-
тактного аппарата в целом. Производительность контактного ап-
парата можно повысить увеличением сечения или увеличением
высоты слоя массы. Увеличение сечения ограничивается труд-
ностью равномерного распределения газа, увеличение же высоты
слоя — быстрым ростом гидравлического сопротивления.
В современных конструкциях диаметр аппарата ограничи-
вается 3—3,5 м, а высота слоя достижением гидравлического
сопротивления ~ 80 мм рт. ст. Величина гидравлического сопро-
тивления зависит от концентрации SO2. При постоянной произ-
водительности и при постоянном сечении аппарата гидравличе-
ское сопротивление должно резко возрастать и при очень малых
концентрациях сернистого газа (вследствие увеличения объема
перерабатываемого газа, а следовательно и скорости) и при очень
255
больших (вследствие увеличения высоты слоя контактной
массы).
Надо поэтому ожидать, что при определенной промежуточ-
ной концентрации SO2 гидравлическое сопротивление достигает
минимального значения. При постоянной величине гидравличе-
ского сопротивления эта же концентрация сернистого газа бу-
дет, очевидно, отвечать максимальной производительности. Наша
задача и сводится к определению этой оптимальной концентра-
ции, обеспечивающей при постоянном предельном гидравличе-
ском сопротивлении и постоянном сечении максимальную произ-
водительность контактного аппарата.
Гидравлическое сопротивление слоя контактной массы опре-
деляется формулой
Др==С1Г«т"-1Л, (44)
тде Др — перепад давления в мм вод. ст.; W — фиктивная ско-
рость газа в м/сек, т. е. скорость, рассчитанная на все сечение
контактной массы; L — высота слоя в м; у — вес 1 м3 газа в кг,
Сип — постоянные, зависящие от крупности и формы кусков
катализатора.
Для гранулированной контактной массы обычной крупности
С = 675 и п — 1,7. Тогда
Др = 675 W^L. (45)
Величина фиктивной скорости газа, отнесенная к средней тем-
пературе, очевидно, прямо пропорциональна перерабатываемому
объему газа и, следовательно, прямо пропорциональна произво-
дительности Р и обратно пропорциональна концентрации серни-
стого газа:
(46)
Высота слоя контактной массы при постоянном сечении прямо
пропорциональна объему контактной массы и, следовательно,
прямо пропорциональна производительности и необходимому вре-
мени соприкосновения и обратно пропорциональна начальной
концентрации сернистого газа:
L = C2V^C3^^ (47)
Подставляя эти величины в уравнение (44), получаем
/V 1.7 В1,7 0,7^ Рт 1,7/-. 0,7 Р2’7 ,
Др — y С.3 — — ССг С37 т (48)
или
Р2'7 = А Рд2,7 - с
-256
где С.—-----объединяет все величины, не зависящие от
ССУСз-10-7
концентрации (изменением 7 с концентрацией можно прене-
бречь).
Таким образом
Р = а
(49)
Зная зависимость т от концентрации, нетрудно построить
кривые изменения производительности как функции начальной
концентрации. Соответствующее построение выполнено на
рис. 112 для конечных степеней окисле-
ния 0,92, 0,94, 0,96 и 0,97 при течении
реакции по оптимальной тепмературной
кривой. Значения производительности да-
ны в условных единицах. Полученные
кривые показывают максимум производи-
тельности при начальной концентрации
SO-2 около 6,8%. Дальнейшее повыше-
ние концентрации SO2 приводит к сниже-
нию производительности. На практике
отклонения от оптимального температур-
ного режима происходят обычно в сто-
рону перегрева. В этом случае снижение
производительности при переходе концен-
трации SO» за оптимальную величину бу-
дет проявляться еще резче.
Итак, при работе на газе, получаемом
в результате сжигания безуглеродистого
колчедана в воздухе, оптимальная кон-
центрация SO2, т. е. концентрация, обе-
спечивающая максимальную производи-
тельность контактного аппарата, соста-
вляет 6,8%.
При изменении состава сырья меняет-
ся также и оптимальная концентрация.
Так, при работе на углистом колчедане при равной концентра-
ции SO2 кислорода будет меньше. Соответственно снижается и
оптимальная концентрация.
При работе на сере, наоборот, за счет меньшего расхода ки-
слорода при обжиге оптимальная концентрация возрастает и до-
стигает 8,2%. При смешении 100%-ного SO2 с воздухом опти-
мальная концентрация достигает 14,0%. В табл. 57 приведены
значения оптимальных концентраций для газов, получаемых при
обжиге различного сырья в воздухе. При применении воздуха,
обогащенного кислородом, оптимальная концентрация сернистого
газа, естественно, повышается. Нетрудно убедиться, что при ра-
боте с воздухом оптимальная концентрация зависит от числа
объемов кислорода, расходуемых при обжиге на образование
17 Зак. &019. Технология серной кислоты. 257
Рис. 112. Зависимость
производительности кон-
тактного аппарата от кон-
центрации SO2 прн сжи-
гании колчедана в воз-
духе.
одного объема сернистого газа. Это позволяет определять опти-
мальные концентрации для новых видов сырья, не прибегая
к специальным расчетам, путем интерполяции данных табл. 57.
Таблица 57
Оптимальные концентрации SO2 при
сжигании различного сырья в воздухе
Сырье Число объемов О2, расходуе- мое на обра- зование одного объема SO2 прн обжиге Оптималь- ная кон- центрация SO2
Стопроцентный SO2 (при его смешении с воздухом) . . Сера Колчедан . . . Углистый колче- дан : Ро 0,08 С 0,12 0,25 0,36 0,0 1,0 1,375 1,588 1,695 2,042 2,335 14,0 8,2 6,8 6,3 6,0 4,7 4.3
'Так, например, при сжига-
I НИИ сероводорода на один
объем SO2 расходуется
1,5 объема кислорода.
Оптимальная концентра-
ция соответственно этому
равна
с q । а к 0,088 г- го'
6,3 4-0,5 6,5 /о-
Найденные значения опти-
мальных концентраций мо-
гут быть использованы
при решении вопроса о
рациональности примене-
ния того или иного вида
сырья. Так, например, при-
менение углистых колче-
данов требует снижения
концентрации SO2; следо-
вательно, приводит к
уменьшению производи-
тельности установки, уве-
личению амортизационных
расходов и отчасти эксплоатационных. Поэтому работа на углис-
том колчедане может быть выгодной лишь в тех районах, где
разница в стоимости безуглеродистого и углистого колчедана
будет превышать указанное увеличение затрат от снижения про-
изводительности. Аналогично может быть решен вопрос о целе-
сообразности применения кислорода. Последнее будет выгод-
ным только в том случае, если экономия, достигаемая от повы-
шения производительности благодаря увеличению оптимальной
концентрации SO2, будет превышать дополнительные расходы,
связанные с получением кислорода. В настоящее время кисло-
род еще слишком дорог для использования его в контактном
производстве. Стоимость кислорода в ближайшее время должна
резко снизиться благодаря ряду технических усовершенствова-
ний. Дальнейшее развитие контактного производства пойдет,
вероятно, в направлении переработки концентрированных газо-
вых смесей, использования кислорода и предварительного обога-
щения сернистого газа.
9. Ведение контактного процесса при повышенном давлении
Контактный процесс ведется при давлении, близком к атмосферному.
При платиновых и ванадиевых катализаторах в этих условиях может быть
достигнуто практически полное окисление SO2 и, следовательно, в примене-
нии повышенного давления нет необходимости. Однако применение высо-
258
ких давлений в контактном производстве представляет несомненный интерес,
так как позволяет значительно интенсифицировать процесс. Действительно,
время соприкосновения газа с катализатором, необходимое для достижения
заданной степени контактирования, определяется в случае ванадиевых кон-
тактных масс уравнением
Прн увеличении давления в п раз, во столько же раз возрастут концен-
трации SOs и Оа. Если не учитывать изменения с давлением теоретически
возможной степени контактирования, можно заключить, что необходимое
время соприкосновения прн увеличении давления остается постоянным.
Отсюда следует, что объем контактной массы, отнесенный к единице про-
изводительности v = CtV, изменяется обратно пропорционально давлению,
так как при постоянной (производительности обратно пропорционально давле-
нию меняется объем газа.
В действительности эффект применения повышенного давления будет
еще больше, так как с ростом давления увеличивается теоретически воз-
можная степень контактирования, в соответствии с ней растут значения
оптимальных температур на всех стадиях контактирования, а следовательно,
и значение констант скорости реакции в уравнении (36). Коэфициенты тепло-
передачи пропорциональны давлению в степени 0,8. Увеличение давления
позволит поэтому значительно сократить также н размеры теплообменников.
В результате размеры контактных отделений можно будет в несколько раз
уменьшить.* Применение повышенного давления позволит также значительно
интенсифицировать и операцию абсорбции. Высокое гидравлическое сопро-
тивление теперь уже не будет служить препятствием для применения более
компактных и не требующих перекачки кислот, аппаратов барботажного типа.
Станет возможным, кроме того, получать 'непосредственно нз контактных
систем высокопроцентный олеум. Поскольку для сжатия в компрессоре газ
должен быть предварительно охлажден и очищен от тумана сериой кислоты,
отделение очнсткн контактных систем останется неизмененным. Применение
высокого давления рационально ограничить операциями контактирования и
абсорбции.
Для внедрения повышенных давлений в контактное производство необ-
ходимо .преодолеть ряд трудностей. Основная трудность — подбор соответ-
ствующего катализатора. Как уже указывалось, активность ряда контактных
масс ограничена со стороны низких температур условиями сульфатизацни
активного компонента. Так, в частности, для ванадиевых контактных масс
резкое снижение активности, начиная с 440°, связано с образованием суль-
фата ванадила.
Равновесие этого процесса определяется уравнением
Fsoa Pso3 — К-
Увеличение давления в п раз повышает произведение парциальных давлений
SO2 и SOs в п- раз. Так, например, при 20 ат произведение парциальных
давлений возрастет в 400 раз по сравнению с атмосферным давлением.
Константа равновесия во столько же раз увеличится при повышении темпе-
ратуры с 440 до 520°. Снижение активности ванадиевых контактных масс
вследствие сульфатизацни начнется поэтому прн 20 ат не при 440°, а уже
при 520°. В соответствии с зтнм должна быть повышена минимально до-
пустимая температура. Это повышение не скажется резко отрицательно на
возможной конечной степени контактирования, так как одновременно воз-
растает и хт- Так, если прн II ат и 440° хт = 0,981, то при 20 ат та же
теоретически возможная степень контактирования может быть достигнута
при 510°. Однако общее повышение температурного режима нежелательно,
173 259
во-первых, из-за увеличения нагрузки теплообменника, во-вторых, нз-за соот-
ветствующего роста температуры максимального разогрева. В современных
контактных аппаратах при температуре входа в контактную массу, равной
440°, максимальная температура достигает 580—600°, при увеличении темпе-
ратуры входа до 520° она соответственно возрастает до 660—680°, т. е.
достигнет уровня, при котором ванадиевые контактные массы испытывают
быструю термическую порчу.
Таким образом для работы под давлением необходимо подобрать катали-
затор, для которого минимально допустимая температура возрастала бы воз-
можно медленнее с увеличением давления, а область термической устойчи-
вости простиралась бы до 650—700°. К материалу аппаратуры контактного
узла, естественно, будут также предъявлены более высокие требования при
работе под повышенным давлением.
Серьезным недостатком работы под давлением является повышенный рас-
ход энергии. Так, расход энергии для сжатия газа до 20 ат составит около
600 квт-ч на 1 т моногид'рата, в то время как в современных системах весь
расход энергии на (1 т моногидрата не превышает 100 квт-ч. Расход энергии
может быть снижен путем использования в турбине знергнн отходящих газов.
IV. Контактные аппараты и контактные узлы
1. Классификация конструкций контактных аппаратов
Выше была установлена оптимальная кривая изменения тем-
пературы по слою контактной массы, обеспечивающая макси-
мальную интенсивность процесса. Основное требование, предъ-
являемое к конструкции контактных аппаратов, сводится к наи-
более полному осуществлению этого температурного режима.
Одновременно надо стремиться к возможно меньшему гидравли-
ческому сопротивлению, к равномерному распределению газа, про-
стоте конструкции, легкости загрузки и выгрузки массы, доступ-
ности для ремонта и экономии металла.
Рассмотрим наиболее простой возможный тип аппарата. Газ,
предварительно подогретый до температуры, достаточной для
начала реакции, проходит через сплошной слой контактной
массы. Наружные стенки аппарата при этом должны быть доста-
точно изолированы, чтобы можно было пренебречь потерями
тепла через них. Тогда, при прохождении через контактную мас-
су, температура газа будет непрерывно повышаться за счет те-
плоты реакции и максимальная температура будет на выходе
из аппарата. Изменение температуры по слою контактной массы,
таким образом, противоречит требованиям оптимального темпе-
ратурного режима. Вместо начала процесса при высоких темпе-
ратурах и последующего постепенного снижения начало про-
цесса происходит при низких температурах с последующим ро-
стом температуры по мере увеличения степени контактирования.
Степень использования контактной массы оказывается низкой —
вначале из-за недостаточно высоких температур, в конце из-за
перегрева, приближения процесса к равновесной кривой. Избе-
жать этого, перейти от возрастания температуры к ее падению
можно лишь путем отвода тепла в процессе реакции. Поэтому
в совершенных конструкциях контактных аппаратов реагирую-
щие газы обязательно должны охлаждаться. С целью сведения
260
теплового баланса естественно стремление использовать отни-
маемое тепло для предварительного подогрева свежего газа.
Поэтому во всех конструкциях охлаждение в процессе реакции
осуществляется путем теплообмена со свежим газом.
По способу теплообмена контактные аппараты
можно разбить на: 1) аппараты с промежуточным теплообменом
и 2) аппараты с внутренним теплообменом.
В аппаратах первой группы реакция окисления SO2 и тепло-
обмен проводятся раздельно, последовательными этапами. В ап-
паратах с внутренним теплообменом тепло отводится одновре-
менно с реакцией, теплообмен происходит между контактной
массой и холодным газом. В отдельных конструкциях осу-
ществляются обе формы теплообмена, причисление таких аппа-
ратов к той или иной группе определяется преобладающей
формой теплообмена.
В аппаратах с промежуточным теплообменом контактирова-
ние происходит в две или большее число стадий. Наиболее рас-
пространенным представителем контактирования в две стадии
с промежуточным теплообменом является контактный узел си-
стемы Герресгоф-Байера, эксплоатируемый на многих наших за-
водах.
В последнее время разработан ряд конструкций контактных
аппаратов с промежуточным теплообменом и контактированием
в три или четыре стадии.
Аппараты с внутренним теплообменом можно разбить на под-
группы в зависимости от формы теплообменных элементов. Наи-
большим распространением пользуются: 1) аппараты с простыми
трубками, в которых контактная масса помещается внутри трубок,
и 2) аппараты с двойными концентрически расположенными
трубками (так называемыми трубками Фильда) с контактной
массой в междутрубном пространстве.
2. Контактные аппараты с промежуточным теплообменом
На принципе контактирования в две стадии построен кон-
тактный узел системы Герресгоф-Байера (рис. 113).
При нормальной работе свежий газ проходит вначале после-
довательно через меж’дутрубные пространства обоих теплооб-
менников т и IV, где подогревается газом, выходящим из кон-
тактных аппаратов, до 440—450°. Нагретый до этой темпера-
туры газ входит в контактную массу первого контактного аппа-
рата /; степень контактирования здесь достигает 72—77% и теу[-
пература газа повышается за счет тепла реакции до^580—-600°.
После первого контактного аппарата газ проходит по трубкам
второго теплообменника IV и охлаждается, отдавая тепло све-
жему газу в междутрубном пространстве, до 430—450°. Далее,
газ проходит через второй контактный аппарат II. Здесь степень
контактирования возрастает до 93—95% и температура повы-
шается до 470—490°. После этого' газ проходит по трубкам
261
первого теплообменника III, где его тепло используется для по-
догрева свежего газа, и охлажденный до температуры ~ 200°
поступает в абсорбционное отделение.
Контактные аппараты, теплообменники и соединительные га-
зоходы тщательно изолируются диатомовым кирпичом или со-
велитом слоем 250 мм.
Протекание процесса в контактных аппаратах близко к адиа-
батическому, так как теплоотдача через стенки очень невелика
по сравнению с теплотой реакции.
Рис. ИЗ. Схема контактного узла с
промежуточным теплообменом и кон-
тактированием в две стадии:
7—1-й контактный аппарат; II—2-й контактный
аппарат; III—теплообменник; IV—2-й теп-
лообменник; V—огневой подогреватель;
7, 2, 3, 4 и 5, 6—вентили.
Рис. 114. Первый контактный аппа-
рат системы Герресгоф-Байера:
7—крышка; 2—полки для контактной мас-
сы; 3—днище; 4—конус для распределения
газа.
Первый контактный аппарат Герресгоф-Байера (рис. 114)
представляет собой цилиндрический котел со съемной верхней
крышкой. Внутри котла помещены решетчатые полки для кон-
тактной массы. Каждая полка состоит из двух листов толщи-
ной 6 мм с круглыми отверстиями диаметром 3 мм. Листы за-
креплены <на расстоянии 20 мм друг от друга с помощью коль-
цевых прокладок. Решетки скреплены между собой болтами
(22 шт.), из них 10 болтов опираются на днище аппарата. Кожух
аппарата изготовляется из чугуна или железа. Над верхней ре-
шеткой контактного аппарата помещен дырчатый конус для рав-
номерного распределения газа по всему сечению аппарата.
Аналогично устроен и второй аппарат, но в нем не две, а че-
тыре полки и соответственно увеличена высота. Расстояние
между отдельными слоями контактной массы всего около 50 мм.
Катализатор располагается на нескольких полках, а не сплош-
ным слоем, с целью уменьшения давления на нижние слои и воз-
можности промежуточного перемешивания газа. Число .полок
не играет особой роли.
2&
Размеры аппаратов при производительности 36 т моногидрата
в сутки следующие:
1-й аппарат
Внутренний диаметр, мм. 2100
Высота, мм ............ 2150
Число полок............... 2
Расстояние можду полками, мм . . 370
2-й аппарат
2100
3100
4
370
Рис. 115. Теплообменник:
/— ввальцованные трубы; 2—
решетка; 3—промежуточные
решетки.
В первый аппарат загружается 2—
2,5 № ванадиевой контактной массы,
во (второй — 4,5—5 №.
Оба теплообменника устроены оди-
наково и различаются лишь диаметром.
Каждый теплобменник (рис. 115) пред-
Рис. 116. Огневой подогреватель:
1—топка; 2—теплообменник; 3—окна для воз-
духа.
ставянет собой железный вертикальный цилиндр, в котором по-
мещаются трубы, развальцованные в верхней и нижней решет-
ках. Между этими решетками помещаются дополнительные ре-
шетки с отверстиями несколько большими наружного диаметра
труб, служащие для равномерного распределения газа по сече-
нию и для улучшения теплообмена. Горячий газ проходит по
трубкам сверху вниз, а холодный — в междутрубном простраи-
263
стве снизу вверх. Основные размеры теплообменников системы
производительностью 36 т моногидрата в сутки:
1-й тепло- 2-й тепло-
обменник обменник
Высота, мм............................. 5300 5300
Внутренний диаметр, мм................. 1700 1400
Расстояние между решетками, мм . . . 3610 3610
Диаметр труб, мм...................... 46/51 46/51
Число труб . . . •..................... 503 349
Поверхность нагрева, м-................ 282 198
Огневой подогреватель (рис. 116) состоит из топки 7 и теп-
лообменника 2. Нагреваемый газ проходит по междутрубному
пространству теплообменника снизу вверх. Топочные газы раз-
бавляются воздухом, подаваемым через воздушные каналы 3,
для снижения температуры до желаемого уровня и проходят
далее по трубкам теплообменника сверху вниз. Температура то-
почных газов при входе в теплообменник не должна превы-
шать 650°, так как в противном случае возможна деформация
трубной решетки и трубок. При пуске температура топочных га-
зов повышается постепенно, не быстрее чем на 100° в час.
Огневой подогреватель служит для разогрева аппаратов при
пуске, а также для дополнительного подогрева газа при работе
с пониженной концентрацией SO2, когда тепла реакции нехва-
тает для сведения теплового баланса. При пуске задвижки 7,
3 и 5 закрываются, 2, 4 и 6 открываются (рис. 113). В кон-
тактный узел подается сухой воздух в количестве 15—30% от
объема газа при полной нагрузке. Воздух подогревается в огне-
вом подогревателе и затем по обводному газоходу через откры-
тые задвижки 4 и б, минуя теплообменник IJ1, входит в первый
контактный аппарат /. По достижении на входе в первый кон-
тактный аппарат температуры 400—420° к воздуху добавляют
сернистый газ в количестве, отвечающем 2—3% SO2 в смеси *.
После того как в первом контактном аппарате начнется реакция,
что обнаруживается по увеличению температуры на выходе,
концентрацию SO2 повышают до нормальной, открывают за-
движки 3 и 5 и начинают постепенно увеличивать количество
газа, подаваемого в систему, одновременно прикрывая понемногу
задвижки 4 и б и уменьшая нагрузку огневого подогревателя.
Постепенно огневой подогреватель выключают, открывая за-
движку 7 и закрывая задвижку 2. При нормальной работе (ра-
бочая схема) задвижка 2 закрыта, задвижки 1, 3 и 5 — от-
крыты. Задвижки 4 и б частично открыты и служат для регу-
лирования. Открывая задвижку 4, мы увеличиваем количество
холодного газа, проходящего мимо первого теплообменника. Со-
ответственно этому количество тепла, отнимаемого от горячего
1 При наличии в аппаратах ненасыщенной ванадиевой контактной массы
концентрация SO» не должна превышать 0,5% до окончания процесса насы-
щения.
264
газа, уменьшается и температура выхода его из первого тепло-
обменника возрастает. Таким образом, открытие задвижки 4 при-
водит к увеличению потери тепла с выходящим газом и сни-
жению температур в обоих контактных аппаратах. Ее следует
открывать при перегреве обоих аппаратов и закрывать с целью
повышения в них температуры. Открывая задвижку 6, мы уве-
личиваем количество холодного газа, проходящего мимо вто-
рого теплообменника IV- Вслед-
ствие этого подогрев газа в нем
ухудшается и температура входа
в первый контактный аппарат сни-
жается. Одновременно, по той
же причине, уменьшается охла-
ждение горячего газа во втором
теплообменнике и температура
входа во второй контактный ап-
парат II повышается. Задвижка 6
служит, таким образом, для регу-
лирования распределения тепла
между первым и вторым кон-
тактным аппаратом. Открытие ее
увеличивает температуру входа
во второй аппарат, уменьшая
температуру входа в первый; за-
крытие, наоборот, повышает тем-
пературу входа в первый аппарат
и снижает температуру входа во
второй. С помощью обеих за-
движек можно, следовательно,
в любых направлениях менять
температуры в контактных аппа-
ратах. Если при полностью за-
крытой задвижке 4 температуры
в обоих аппаратах низки, следует
включить огневой подогреватель.
Для оценки рациональности
температурного режима в аппара-
Рис. 117. Скорость начальных ста-
дий коитактнровання при различ-
ных температурах.
тах рассматриваемой конструкции
изобразим его на диаграмме (t, х). Минимально допустимая тем-
пература входа газа в контактную массу составляет 440°. При
низших температурах константы скорости быстро падают и пер-
вичный разогрев протекает слишком медленно. Это наглядно
показано на рис. 117.
Зная начальный состав газа, можно рассчитать и построить
на графике t, х (рис. 118) прямую, отвечающую адиабатиче-
скому протеканию процесса в первом аппарате. Возможная сте-
пень контактирования в первом аппарате ограничивается равно-
весной кривой. При работе на 7%-ном газе максимальное пре-
вращение составляет около 75%. При этом приходится значи-
265'
тельно .выходить за пределы оптимальной зоны и заканчивать
процесс вблизи равновесной кривой. Можно было бы, конечно,
ограничиться меньшей степенью контактирования и не выходить
за пределы зоны оптимальных температур, но это еще более
ухудшило бы и без того неблагоприятные условия работы вто-
рого контактного аппарата и привело бы к снижению конечной
степени контактирования. Процесс охлаждения газа на диа-
-----—Температура в °C
Рис. 118. Изменение температуры при двухстадийном контактировании
с промежуточным теплообменом:
/ равновесная кривая; 2—оптимальная кривая; граница зоны 0,9 \4— гра-
«" v «t/max
ница зоны ~ > 0,8 ; 5— граница зоны -£>0,7 ; 6—граница зоны
at '«"/max '«"/max
dx n _ (dx\ . _ x
— > 0,5 I -г I ; /—рабочая линия.
a< '«"/max
грамме t, x изобразится прямой, параллельной оси абсцисс. Охла-
ждение можно доводить также лишь до 440°, так как при более
низких температурах замедляется начало реакции во втором кон-
тактном аппарате. Пренебрегая потерей тепла во втором кон-
тактном аппарате, мы можем изобразить протекание в нем реак-
ции прямой, параллельной адиабатической прямой первого аппа-
рата. Полученный график показывает, что температурные усло-
вия протекания реакции в обоих аппаратах в значительной части
266
отклоняются от оптимальных. Вначале температуры слишком
низки, в конце, наоборот, слишком высоки. Лишь небольшие
участки реакции в обоих аппаратах укладываются внутри опти-
мальной зоны. Процесс в две стадии с трудом умещается между
равновесной кривой и минимально допустимой температурой.
В результате конечная степень контактирова1ния мала и степень
использования контактной массы не превышает 40%.
t>nnpqodnjjMomnoH ujHjnodu
Рнс. 119. Изменение температуры при многоступенчатом контактировании
с промежуточным теплообменом.
Таким образом контактные аппараты с промежуточным тепло-
обменом и контактированием в две стадии не удовлетворяют
основному требованию, предъявляемому к совершенным контакт-
ным аппаратам: не обеспечивают соблюдение оптимального тем-
пературного режима.
Этот недостаток может быть, однако, устранен увеличением
числа стадий контактирования. На рис. 119 изображено проте-
кание процесса с промежуточным теплообменом при пяти и
семи стадиях. В последнем случае удается практически пол-
ностью осуществить оптимальный температурный режим. Уве-
личение числа стадий имеет, однако, и отрицательные стороны.
Оно приводит к увеличению числа аппаратов, усложняет ком-
муникацию и удорожает контактный узел. Одновременно услож-
няется и регулирование, так как число регулирующих задвижек
должно быть равно числу стадий контактирования. Поэтому осо-
267
бенно увлекаться увеличением числа стадий не следует. При
работе на 7 %-ном газе, вероятно, не рационально делать их
больше 3—4.
Для упрощения коммуникации и экономии места обычно
располагают отдельные слои контактной массы и промежуточные
теплообменники непосредственно друг над другом. Теплообмен-
ники конструируются двух типов:
с горизонтальными и вертикальными
трубками. Имеются аппараты с кон-
тактированием в пять стадий (рис. 120)
и в семь стадий (рис. 121).
Расчет аппаратов с промежуточ-
ным теплообменом не представляет
затруднений. Температуры и степень
контактирования в начале и в конце
отдельных стадий определяются с по-
мощью построения на графике (/, х).
Объемы контактной массы в отдель-
Рис. 120. Пятиступенча- Рис. 121. Контактный аппарат Фаузера с кон-
тый контактный аппарат тактированием в семь стадий.
Терентьева.
ных частях аппарата вычисляются по вышеописанному методу
для адиабатического процесса (стр. 251). Размеры промежуточ-
ных теплообменников определяются с помощью известных фор-
мул учения о теплопередаче.
3. Контактные аппараты с внутренним теплообменом
Теоретически необходимый теплоотвод. Предположим, что
процесс окисления SO2 осуществляется в полном соответствии
с оптимальной температурной кривой. Очевидно, что в этом
268
случае на всех стадиях контактирования от контактной массы
необходимо отводить тепло, так как оптимальная кривая отве-
чает снижению температуры с увеличением степени контактиро-
вания. Количества тепла, подлежащие отводу, слагаются из
тепла, выделяющегося при реакции, и уменьшения теплосодер-
жания газа при снижении температуры в соответствии с опти-
мальной температурной кривой. Поскольку нам известно из-,
менение скорости реакции и оптимальной температуры с ростом
степени контактирования, нетрудно рассчитать теплоотвод от
1 ж3 контактной массы в кал/час [(^)опт]> необходимый для
соблюдения оптимального температурного режима на различных
стадиях контактирования.
Соответствующие значения приведены в табл. 58.
Таблица 58
х-10'- в секундах \ dv /опт х-10- ^0 в секундах \ dv /от
50 0,080 1 845000 90 0,818 89 300
60 0,115 1 085000 91 0,924 76 700
70 0.185 560000 92 1,057 67 300
75 0,245 391000 93 1,224 57 000
80 0,340 264000 94 1,440 43 900
83 0,424 202000 95 1,750 32 200
85 0,498 165000 96 2,210 21 700
87 0,597 129000 97 3,000 И 900
89 0,731 104000
На рис. 122 приведены значения интенсивности необходимого
теплоотвода на различных уровнях слоя контактной массы при
коэфициенте запаса, равном 2. Эти данные показывают, что
необходимый теплоотвод резко меняется по высоте слоя катали-
затора. Ог первых по ходу газа частей контактной массы
должно отводиться гораздо больше тепла, чем от последних.
Приведенные в табл. 58 значения меняются в 150 раз.
Если продолжить таблицу в сторону меньших значений времени
соприкосновения, то разница проявится еще резче. К тепло-
обменным элементам, таким образом, предъявляется требование
очень резкого изменения интенсивности теплоотвода по высоте
слоя контактной массы. Резкость этого перепада можно, од-
нако, смягчить путем некоторого отклонения от оптимальной тем-
пературной кривой в начале и конце процесса. Действительно,
совершенно очевидна нерациональность стремления к достиже-
нию оптимальных температур с самого начала реакции. Для
первых стадий контактирования оптимальные температуры очень
высоки, между тем как активность ванадиевых контактных масс
269
начинает снижаться уже три 620°. Но и при этой температуре
нецелесообразно начинать троцесс, так как вследствие очень
большой начальной скорости реакции неизбежен перегрев пер-
вых слоев контактной массы. Высокая начальная скорость реак-
ции приводит к тому, что для достижения первых 60% контак-
тирования требуется ничтожная доля общего потребного коли-
чества контактной массы. Поэтому отклонения в этой части
от оптимального температурного режима не имеют практиче-
ского значения. Ниже приведены значения необходимого вре-
мени соприкосновения для достижения 60%, контактирования
при проведении процесса по оптимальной температурной кривой
и адиабатически при различных
начальных температурах V
Таблица 58а
Количество отводимого тепла 0
ккал/мЗчас при козфициенте запаса Z
Рис. 122. Необходимый отвод теп-
ла для соблюдения оптимального
температурного режима.
Адиабатический процесс при на- чальных темпера- турах в °C Значения т в сек. прн х — 0,6 в различных условиях
430 0,305
440 0,201
450 0,162
460 0,146
475 0,123
Оптимальный тем-
пердтурный режим 0,115
Эти данные пока-зывают, что при адиабатическом проведении
процесса, начиная с 475°, увеличение необходимого времени со-
прикосновения составляет всего 0,008 сек., т. е. ~ 0,3% от всего
т (3 сек. при х = 0,97). Даже в случае начала реакции при 450°
это увеличение составляет всего ~ 1,5%. Таким образом нет
необходимости стремиться к высокой температуре начала про-
цесса и рационально первый этап окисления осуществлять адиа-
батически, начиная с температуры 460—470°.
Начинать теплоотвод с интенсивностью, определяемой дан-
ными табл. 58 а, следует лишь после достижения оптимальной
температуры. Если адиабатический процесс начинается при
460—470°, то оптимальная температура достигается при степени
контактирования 0,57—0,63 и составляет 595—605°. Время со-
прикосновения, необходимое для достижения оптимальной тем-
пературы, составляет 0,13—0,15 сек., что соответствует при-
мерно 5% общего количества контактной массы.
* Здесь и в дальнейшем все числовые значения относятся к газу на-
чального состава: 7% SOa, И % О» и 82%' Na.
270
Начальные участки кривой необходимого теплоотвода, тре-
бующие наибольшей интенсивности теплообмена, на практике,
таким образом, не реализуются.
В последних частях слоя контактной массы реакция проте-
кает медленно, необходимый теплоотвод очень мал. Так, изме-
нение степени контактирования с 93 до 97%, требующее в опти-
мальных условиях 1,75 сек. времени соприкосновения, что отве-
чает более чем половине общего количества контактной массы,
приводит к увеличению температуры газа всего на 8,5 . Этой же
величине отвечает и ширина оптимальной зоны в области высо-
ких степеней контактирования при адиабатическом проведении
реакции. Поэтому можно, не выходя из
зоны оптимальных температур, проводить
последнюю часть процесса без охлажде-
ния, адиабатически.
Таким образом внутренний теплооб-
мен необходимо применять лишь для ча-
сти контактной массы в интервале време-
ни соприкосновения 0,15—1,25 сек., т. е.
примерно лишь для одной трети всего
слоя контактной массы. В этой части
тепло также должно отводиться неравно-
мерно, в первых слоях примерно в 12 раз
интенсивнее, чем от последних. Средняя
интенсивность теплоотвода составляет
~ 200 000 ккал в час. При коэфициенте
запаса 2 это отвечает примерно 100 000
ккал в час от 1 /и3 действительно загру-
женной контактной массы.
Трубчатые аппараты с загрузкой кон-
тактной массы внутри труб. Одна из пер-
вых конструкций трубчатого типа, полу-
Рис. 123. Тентелевскнй
контактный аппарат:
/—железный котел; 2—ре-
шетка; 3—трубка для контакт-
ной массы; 4—решетка; 5 —
штуцер для входа газа; 6—
чившая широкое распространение, — кон-
тактный аппарат Теителевского завода
(рис. 123) — состоит из вертикального цилиндрического кожуха,
в нижней части которого расположена стальная решетка, в ко-
торой развальцовано 200 труб диаметром 127 мм, высотой
2035 мм. В верхней крышке находится штуцер для входа газа.
Под ним располагается распределительная решетка, служащая
лаз; 7-- штуцер для выхода
газа.
для достижения равномерности распределения газа по сечению.
В нижней крышке расположен штуцер для выхода газа и люк
для выгрузки контактной массы. Внутри каждой трубки нахо-
дится по 64 дырчатых диска, на которых располагается плати-
нированный асбест. В аппарат производительностью 12 т моно-
гидрата в сутки загружается 156 кг платинированного асбеста,
содержащего 12 кг платины.
Контактный узел Теителевского завода кроме контактного
аппарата включал еще теплообменник и огневой подогреватель.
Схема расположения аппаратов приведена на рис. 124. При нор-
271
мальной работе задвижки 1 и 4 открыты, 2 и 3 закрыты. Све-
жий газ проходит через междутрубное пространство теплообмен-
ника II, где нагревается за счет тепла газов, выходящих из кон-
тактного аппарата /, до 250° и входит через верхний штуцер
в контактный аппарат. Здесь благодаря конвекционным токам
между трубками газ дополнительно подогревается приблизи-
тельно до 360° и входит в трубы, содержащие платинированный
асбест. Эта контактная масса при 360° обладает еще достаточ-
ной активностью для начала реакции. За счет тепла реакции
температура возрастает вначале до 520°, а затем постепенно
снижается в результате охлаждающего действия свежего газа
до 420—440°. Эта температура обеспечивает достижение вы-
сокой конечной степени контактирования. Тепло газа, выходя-
щего из аппарата и поступающего в трубки теплообменника, ис-
пользуется для подогрева
свежего газа. Задвижка 5
служит для регулирова-
ния. Открывая ее, можно
направить часть горячего
газа мимо теплообменни-
ка и тем самым снизить
температуру газа, входя-
щего в контактный аппа-
рат. Степень контактиро-
вания составляет 95—97%.
При условии хорошей
очистки газа контактный
аппарат может работать
длительный срок (в от-
Рис. 124. Схема контактного узла системы
Теителевского завода:
1—контактный аппарат: /I—теплообменник; Ш—
подогреватель; 7, 2, 3, 4 и 5—задвижки.
дельных случаях 15 лет) без вскрытия. При пуске или расстрой-
стве режима разжигается огневой подогреватель, в котором газ
подогревается, проходя через открываемые задвижки 2 и 3.
При переходе па ванадиевые контактные массы эта конструк-
ция контактного аппарата подверглась изменению, так как по-
догрев газа до 360° недостаточен для начала реакции на ва-
надиевых катализаторах. С целью увеличения интенсивности
теплообмена вход газа был сделан снизу, над трубной решеткой.
Таким образом трубки омывались уже принужденным газовым
потоком. Кроме того, и поверхность внутреннего теплообмена
была увеличена доведением высоты труб до 2,5 м. Для за-
грузки ванадиевой контактной массы в низу труб укреплялись
решетки, на которые засыпался слой кварца высотой 50—100 мм.
а затем сплошным слоем ванадиевая контактная масса в ко-
личестве ~ 25 л. В верхней части труб вначале оставляли не-
сколько дисков с платинированным асбестом для увеличения на-
чальной скорости реакции, в дальнейшем, однако, было найдено,
что в этом нет необходимости.
Видоизмененный указанным образом контактный аппарат
позволял достигать производительности 18 т/сутки и степени
272
контактирования 95—96%. Тем не менее он далек от совер-
шенства. Количество загруженной в него контактной массы со-
ставляет свыше 300 л на 1 т суточной производительности,
т. е. значительно больше, чем в системе Герресгоф-Байера, где
степень использования контактной массы тоже невелика. Это
обусловлено значительным отклонением температуры в средней
части трубок от оптимальной.
Выше было показано, что для достижения оптимальных тем-
ператур интенсивность теплоотвода по слою должна быть не-
равномерной: в начале слоя контактной массы должно отво-
диться тепла во много раз больше, чем в конце. Это условие
в аппарате рассматриваемой конструкции не соблюдается. Дей-
ствительно, количество тепла, отнимаемое от 1 м3 контактной
массы, равно:
Q = (49)
где К — коэфициент теплопередачи в ккал /м2 • час - °C; f —
поверхность труб, приходящаяся на 1 /и3 контактной массы,
в лГ2; tKK — температура контактной массы в данном сечении;
гчг — температура газа в данном сечении межтрубного простран-
ства.
По высоте аппарата f остается постоянным, приблизительно
постоянно также и К, разность же температур пропорциональна
степени контактирования и, следовательно, в нижней части ап-
парата даже несколько больше, чем в верхней.
Таким образом интенсивность теплоотвода от первых слоев
контактной массы не больше, как это требуется для соблюде-
ния оптимального температурного режима, а меньше, чем от по-
следних слоев. В результате (в верхней половине слоя контакт-
ной массы температура значительно превосходит оптимальную и
реакция протекает вблизи равновесной кривой, а следовательно,
с очень малой скоростью. В этой части слоя процесс окисления
SO2 ограничивается не активностью катализатора, а интенсив-
ностью теплоотвода. Степень использования контактной массы
очень мала. Кривая 1 на рис. 125 изображает протекание про-
цесса в аппарате рассматриваемой конструкции. Вторым круп-
ным недостатком аппарата этого типа является высокое гидра-
влическое сопротивление. С целью улучшения подогрева газа
высота труб, а следовательно, и слоя контактной массы увели-
чена до 2,5 м, что, в связи с увеличением нагрузки, приводит
к величине гидравлического сопротивления свыше 100 мм рт. ст.
В ряде случаев столь высокая величина гидравлического сопро-
тивления ограничивает производительность.
Третьим недостатком является малая производительность ап-
парата. Это связано с тем, что контактная масса занимает лишь
небольшую долю всего сечения контактного аппарата (около
30%). Поэтому при равном диаметре аппараты этого типа
должны иметь производительность в три раза меньшую против
аппаратов с промежуточным теплообменом.
18 Зак. 5019. Технология серной кислоты. 273
С целью улучшения температурного режима этих аппаратов
в некоторых конструкциях предусматривается введение холод-
ного газа на различной высоте труб. Это мероприятие без-
условно способствует приближению к оптимальному температур-
ному режиму благодаря увеличению теплопередачи в верхних
частях труб из-за возрастания разности температур, но одновре-
менно снижает общую среднюю разность температур, а следо-
вательно, и производительность аппарата.
Рис. 125. Диаграмма /, х для тентелевского аппарата.
Более рациональным мероприятием для улучшения работы
аппарата является послойная' загрузка в трубки контактной
массы с оставлением промежуточных, охладительных поясов.
Соотношение между высотой слоев контактной массы и охлади-
тельных поясов может быть сделано таким, чтобы достигалось
требуемое для осуществления оптимального температурного ре-
жима соотношение интенсивностей теплоотвода в верхней и ниж-
ней части трубок. Расчет показывает, что для этого достаточно
двух промежуточных охладительных поясов. Протекание про-
цесса в этом случае на графике t, х изображается линией ab
(рис. 125). При этом последний слой, охлаждение которого, как
было показано выше, необязательно, можно не загружать
274
в трубки, а расположить в виде дополнительного слоя в нижней
части аппарата.
На рис. 126 показано наиболее выгодное распределение кон-
тактной массы по трубке, обеспечивающее соблюдение оптималь-
ного температурного режима. Для уменьшения общей высоты
труб целесообразно располагать в полых частях труб какие-либо
завихрители (рис. 127), увеличивающие коэфициенты тепло-
прохождения.
Послойная загрузка позволяет, таким образом, избежать пе-
регревов средней части контактной массы и уложить процесс
внутри оптимальной зоны, а следовательно, повы-
сить степень использования контактной массы.
Рис. 126. Ра-
циональное
распределе-
ние массы в
трубке.
Одновременно этот метод позволяет и резко сокра-
тить величину гидравлического сопро-
тивления (в 2 раза) благодаря умень-
шению высоты слоя массы в трубках.
Сопротивление нижнего слоя массы
мало по сравнению с сопротивлением
трубок, так как скорость газа в нем
примерно в 3 раза меньше.
Из перечисленных выше недостат-
ков трубчатых аппаратов сохраняется
лишь последний — малая производи-
тельность отдельного аппарата. Для
устранения этого недостатка следует
стремиться к возможно более густому
расположению труб. Это выгодно и
с точки зрения увеличения коэфици-
ента теплопередачи благодаря возра-
станию скорости холодного газа
в межтрубном пространстве. Однако
чрезмерное увеличение густоты труб
Рис. 127. За-
вихрители:
1— контактная
масса; 2 — за-
вихритель.
связано с опасностью ослабления трубной решетки. Последнее
можно избежать, если сделать трубы переменного диаметра.
В этом случае трубы можно расположить почти вплотную и
увеличить таким путем долю сечения, занятую контактной мас-
сой, до 55—65%. Благодаря этому при равном диаметре аппа-
рата и величине гидравлического сопротивления производитель-
ность может быть повышена почти в 2 раза.
Контактные аппараты с двойными теплообменными трубками
и расположением контактной массы в междутрубном простран-
стве. Аппараты с двойными трубками были сконструированы
Егером и Бертчем в США в 1928 г. и получили значительное
распространение в американской промышленности.
Аппарат Кемико (рис. 128) предназначен для работы на хо-
лодном газе, предварительно подогреваемом за счет тепла про-
контактированного газа. Аппарат представляет собой вертикаль-
ный цилиндр с тремя горизонтальными решетками. На нижней
решетке расположен слой кварца крупностью 12—15 мм, вы-
18»
275
сотой 250 мм, служащий для выравнивания скоростей по се-
чению аппарата. Во второй решетке развальцованы 733 трубы
диаметром 51 X 2,5 мм, высотой 1700 мм, на которые надеты
закрытые сверху колпаки диаметром 76 X 3 мм, отстающие от
решетки на 70 мм. На эту решетку загружается кварц слоем
150 мм в центре и 200 мм в периферийном кольце шириной
350 мм. На кварц засыпается слой ванадиевой контактной массы
высотой 1400 мм равномерно по всему'сечению аппарата.
На третьей решетке располагается дополнительный слой кон-
тактной массы высотой 500 мм.
Сернистый газ, предварительно подогретый в теплообменнике
до 250°, поступает в нижнюю часть аппарата под первую ре-
шетку, проходит через слой кварца, поднимается по внутрен-
ним трубкам, опускается по кольцевому пространству между
внешними и внутренними трубками, проходит через слой квар-
цевой подстилки и далее поднимается вновь по слою контактной
массы.
При прохождении через трубки газ нагревается за счет
тепла контактной массы до температуры 460—480°, достаточной
для начала реакции, и входит в слой контактной массы. Здесь
температура газа вначале очень быстро повышается за счет теп-
лоты реакции окисления SO2 до 580—590° и затем постепенно
снижается до 480—500°. Степень окисления SO» в нижнем слое
контактной массы достигает 92—93%. Затем газ омывает верхние
части труб, выступающие из слоя контактной массы, охлаждается
до 440—450° и проходит через второй слой контактной массы,
в котором степень контактирования повышается до 96—97% и
температура соответственно на 5—7°.
Кожух и крышки аппарата делаются чугунными или сталь-
ными, теплообменные элементы — из стальных цельнотянутых
труб. Производительность аппарата по американским данным
составляет 40 т моногидрата в сутки.
На рис. 129 изображена схема контактного узла Кемико. Кон-
струкция теплообменника и огневого подогревателя принципи-
ально не отличается от описанных выше. При нормальной работе
Рис. 129. Схема контактного узла системы Кемико:
1— контактный аппарат; II—теплообменник; III—подогреватель; I, 2, 3, 4
и 5—задвижки.
задвижки 3, 4 и 5 закрыты, 1 — открыта, 2—частично открыта
и служит для регулирования температуры входа газа в контакт-
ный аппарат. При пуске задвижки 1, 2 и 4 закрываются, 3 и
5 открываются и аппарат разогревается сухим воздухом, прохо-
дящим через огневой подогреватель. После достижения на
входе в аппарат 400э задвижка 4 частично открывается и к воз-
духу добавляется сернистый газ. При наличии в аппарате ие-
насыщенйой ванадиевой контактной массы концентрация SO2 до
окончания процесса насыщения массы не должна превышать
0,5%. В случае насыщенной контактной массы концентрация
SO2 сразу может быть поднята до 2—3%. После начала реак-
ции, на что указывает быстрое повышение температуры в ниж-
нем слое контактной массы, концентрация SO2 постепенно по-
вышается до нормальной (6,7—7,0%) при одновременном сни-
жении температуры входа газа в аппарат и увеличении нагрузки.
Снижение температуры входа в аппарат достигается понижением
температуры топочных газов огневого подогревателя и пуском
277
части газа через теплообменник путем постепенного открытия
задвижки 7. Наконец, после того как температурный режим
приблизится к нормальному, огневой подогреватель полностью
отключается путем закрытия задвижек 5, 4 и 3 и дальнейшее
регулирование производится с помощью задвижки 2.
На рис. 130 приведена кривая изменения температуры и сте-
пени контактирования в аппарате Кемико. В нижней части ап-
парата наблюдается перегрев, но значительно меньший, чем
в трубчатых аппаратах при сплошной загрузке трубок контакт-
Процент контактирования
-----— Температура в °C
Рис. 130. Кривая изменения температуры в аппарате Кемико.
ной массой. Это связано с тем, что в двойных теплообменных
элементах холодный газ во внутренних трубках движется
в том же направлении, что и в контактной массе. В резуль-
тате от первых по ходу газа частей контактной массы отбирается
больше тепла, чем от последующих, и распределение интенсив-
ности теплоотвода по высоте трубок ближе лежит к теорети-
чески необходимому, чем у простых трубчатых аппаратов.
Это достоинство аппаратов с двойными теплообменными эле-
ментами практически часто сводится на-нет непостоянством тем-
ператур по горизонтальному сечению. Обычно уже вскоре после
278
пуска аппарата, несмотря на правильный температурный режим
средней части аппарата, по периферии нижнего слоя обнаружи-
ваются отдельные участки с значительно пониженной темпера-
турой. Если не обращать внимания на образование этих хо-
лодных участков и допустить в них падение температуры ниже
400°, то контактная масса затухает в этой части сечения по всей
высоте слоя, что приводит к резкому снижению общей степени
контактирования.
Единственным средством, позволяющим поднять температуру
холодных периферийных участков, является повышение темпера-
туры входа газа в аппарат. По мере удлинения срока эксплоа-
тации, для предотвращения затухания периферийных участков,
температуру входа газа в аппарат приходится поднимать на все
более высокий уровень. Так, если при работе на 7%-ном (по
SO2) газе вначале удается держать температуру входа в аппа-
рат не выше 250°, то уже после полугодовой эксплоатации она
возрастает до 270—280°. Возрастание температуры входа газа
в аппарат неизбежно приводит к повышению температуры
в верхнем слое контактной массы и снижению вследствие этого
конечной степени контактирования. Аппарат, изображенный на
рис. 128, дает в первый месяц эксплоатации степень контакти-
рования, равную 96—96,5%, после же полугодовой работы ко-
нечная степень контактирования снижается до 92—94%.
Образование холодных участков представляет сложное явле-
ние, связанное с рядом особенностей работы аппаратов с двой-
ными теплообменными трубками. Наиболее существенны из них
следующие:
1) отсутствие перемешивания газа после подогрева в тепло-
обменных трубках перед вступлением ,в контактную массу;
2) охлаждение подогретого газа в начальных слоях контакт-
ной массы;
3) увеличенная нагрузка периферийных теплообменных труб.
Недостатком аппаратов рассматриваемой конструкции является
также движение газа через контактную массу в направлении
снизу вверх. Это создает опасность выброса массы. Особенно
реальна эта опасность для нижнего слоя контактной массы, в ко-
тором скорость газа повышена из-за наличия теплообменных
труб и высокой температуры. Условие выброса контактной
массы определяется равенством между динамическим напором
газового потока и весом частицы контактной массы, отнесенным
к единице площади проекции на плоскость, перпендикулярную
направлению движения газа. Частицы гранулированной ванадие-
вой контактной массы имеют форму цилиндриков с диаметром
~ 3,5 мп и обладают кажущимся удельным весом около
1,2 г/см3.
Отсюда находим минимальный вес на единицу нлощади
проекции:
2^ 8 ЗЛ4 0 35 12 = 0 33 г}см2 = з,з
279
Выдувание гранулированной контактной массы, следова-
тельно, возможно при динамическом давлении газового потока:
где w — действительная скорость газа в м/сек, рассчитанная на
свободное сечение контактной массы; у — вес 1 /и3 газа в кг',
g — ускорение силы тяжести, равное 9,81 м/сек-.
В действительности, однако, .выдувание контактной массы
происходит при скоростях, отвечающих значительно меньшим
значениям динамического давления. Это связано с некоторой не-
равномерностью распределения газа по сечению, а главным об-
разом с неоднородностью кусков гранулированной контактной
массы. В отдельных участках уже при небольших скоростях
мелкие куски контактной массы уносятся газовым потоком.
В этих местах сечения гидравлическое сопротивление умень-
шается, соответственно, этому возрастает скорость газа и ста-
новится возможным унос и более крупных кусков. Эксперимен-
тальное исследование, проведенное Терентьевым, показало, что
для гранулированной контактной массы уже опасно динамиче-
ское давление газового потока, превышающее 0,5 кг/м2. В ниж-
ней части аппарата Кемико, при производительности 40 т моно-
гидрата в сутки, 7%-ном газе и температуре 500° скорость газа
равна 1 м/сек, а динамическое давление соответственно
0,31 кг/ м2.
Таким образом выдувание не должно было бы иметь места.
Однако в процессе эксплоатации сопротивление контактной
массы растет по сечению неравномерно: в центре быстрее, чем
у периферии. В результате в периферийных участках скорость
газа возрастает и выброс контактной массы становится воз-
можным.
Для предотвращения выдувания следует применять контакт-
ную массу повышенной крупности и возможно более однород-
ную по размерам отдельных кусков. Этим требованиям удовле-
творяет таблетированная контактная масса. Так, для таблетрк
высотой 5 мм минимальный вес на единицу площади проекции
составляет 6 кг/м2 и, следовательно, допускаемое динамическое
давление газового потока возрастает почти вдвое по сравнению
с гранулированной массой. В действительности эффект от при-
менения таблетированной контактной массы еще больше, так
как в ней отсутствуют более мелкие куски, и действительная
скорость газа, вызывающая выдувание, ближе подходит к рас-
четной.
4. Сравнительная оценка различных конструкций. Схема
контактного узла для работы на газе высокой концентрации
Из рассмотренных типов контактных аппаратов лишь две
конструкции позволяют осуществлять процесс окисления SOs
в оптимальных температурных условиях: 1) аппараты с проме-
289
«уточным теплообменом при большом числе стадий контакти-
рования и 2) трубчатые аппараты с послойной загрузкой контакт-
ной массы внутри труб.
И та и другая из этих конструкций имеют свои преимуще-
ства и недостатки и между ними пока не может быть сделан
окончательный выбор.
Преимущество трубча-
тых аппаратов в а) срав-
нительной простоте ком-
муникаций и регулирова-
ния и б) меньшей величи-
не поверхности внутренне-
го теплообмена благодаря
повышенным значениям
коэфициента теплопереда-
чи от трубок, заполнен-
ных контактной массой, по
сравнению с пустыми
трубками. Преимуществом
аппаратов с промежуточ-
Рис. 131. Максимальный разогрев контакт-
ной массы.
ным теплообменом явля-
ется повышенная производи^Вьность (приблизительно на 30%)
при равном диаметре, благодаря тому что контактная масса за-
нимает все сечение аппарата, в то время как в трубчатых аппа-
ратах на долю контактной массы приходится лишь 55—65% от
Рис. 132. Схема контактного узла
при работе на концентрированном
газе:
] и 7/—контактные аппараты; III—теп-
лообменник.
общей площади сечения.
В аппаратах с двойными теп-
лообменными трубками типа Ке-
мико хотя и обеспечивается нуж-
ное распределение теплоотвода
по высоте слоя контактной массы,
оптимальный температурный ре-
жим не может быть осуществлен
вследствие неравномерности тем-
пературы по горизонтальным се-
чениям аппарата. До устранения
этого недостатка от применения
аппаратов с двойными теплооб-
менными трубками следует воз-
держаться.
При переработке газа повышенной концентрации, например полученного
при смешении 100% SOa с воздухом, возникают дополнительные трудности
вследствие опасности перегрева контактной массы. Как видно из построения
на диаграмме t, х_(рис. 131), при температуре входа в контактную массу,
равной 450, для 7%-кого газа максимальная температура разогрева (при
приближении к равновесной кривой) составляет 606°, а для 14%-ного газа
в тех же условиях 660°.
Как уже указывалось, при температурах выше 650° ванадиевые кон-
тактные массы очень быстро портятся. Можно было бы попытаться умень-
шить этот перегрев, обеспечив интенсивный теплоотвод от первых слоев
281
контактной массы. Однако на первых стадиях контактирования скорость
реакции настолько велика, что осуществление нужного теплоотвода оказы-
вается невозможным. Даже расположение контактной массы очень тонкими
слоями с оставлением больших охладительных поясов не приводит к успеху,
так как при загрузке контактной массы с запасом неизбежен перегрев первых
слоев, при загрузке же точно по расчету, после снижения активности при
эксплоатации, первые слои не смогут обеспечить достижения заданной
степени контактирования, процесс сместится к нижним слоям и вызовет в них
опасный перегрев.
Наиболее рационально в этом случае работать по следующей схеме
(рис. 132), предложенной Слинько. Половина 100%-ного SO2, подлежащего
переработке, смешивается с воздухом, взятым в таком количестве, чтобы
конечная с иесь содержала 7—8% SO2. Полученная смесь после подогрева
до 450—-460° в междутрубном пространстве контактного аппарата II и
теплообменнике Ill поступает в контактный аппарат /, где в результате
адиабатической реакции температура повышается до 600—610° и степень
контактирования до 60—70%. По выходе из аппарата 1 газ охлаждается
в трубках теплообменника и к нему добавляется 1100%-ный SO2 в таком
количестве, чтобы суммарная конечная концентрация SO2 и SOs до-
стигла 14%. Полученная смесь поступает далее в трубчатый контактный
аппарат Ц с послойной загрузкой контактной массы. Опасность перегрева
контактной массы при работе по этой схеме полностью исключается. Оба
контактных аппарата п теплообменник могут быть объединены в один
аппарат.
5. Контроль работы контактного узла
Качество работы контактного^^зла зависит от качества по-
ступающего газа (степень его очистки и осушки и содержание
SO2) и температурного режима в контактных аппаратах. Итог
работы контактного узла характеризуется процентом контакти-
рования. О контроле степени очистки газа выше уже говори-
лось в связи с рассмотрением работы отделений промывки и
осушки. Основными определениями,' относящимися непосред-
ственно к контактному узлу, являются анализы газа на SO2 и
замеры температуры.
Перечень принятых анализов и замеров применительно к кон-
тактному узлу нормальной системы Герресгоф-Байера приводим
ниже (табл. 59).
Замер температур производится стационарными термопарами
с выносом показаний на общий щ'ит. Содержание SO2 опреде-
ляется методом Рейха или газоанализаторами.
Процент контактирования по анализу газов на содержание
SO2 высчитывается следующим образом. Если процент SO2 пе-
ред контактными аппаратами равен Pso2, а после контактных
аппаратов P'so., то на 100 объемов газа до контактных аппара-
тов остается после контактных аппаратов в объемах
100-0,5
где у—процент контактирования. На 100 объемов газа до кон-
тактирования остается неокисленногр SO2 в тех же объемах
рк(100-°-5Ш)М >
100 ‘
282
Таблица 59
Объект контроля Определяется Место отбора проб или замера Частота анализа
Газ Газ Топливо Топочные газы Температура Содержание SO2 (методом Рейха или газоанализатором) Количество Температура После подогревателя Вход в 1-й контакт- ный аппарат Вход в массу 1-го контактного аппарата В средине 1-го кон- тактного аппарата Выход из 1-го кон- тактного аппарата Вход во 2-й контакт- ный аппарат Выход из 2-го кон- тактного аппарата Выход из 1-го тепло- обменника Перед 1-м, после 2-го контактного аппарата Вход и выход из по- догревателя 8 раз в смену Каждый час Каждый час
Следовательно, из 100 объемов газов до контактирования окис-
лилось SO2 объемов:
> SO.
S°2 100
Процент контактирования может быть представлен так:
откуда
^so3
100,
10000(Pso3-Ps'o2)
У PSo2(100-0,5Pgoy
Так как при анализе аппаратом Рейха кроме SO2 поглощается
также и SO3, то этим аппаратом определяется после контакти-
рования процент SO2(P'S02) Не ко всей газовой смеси, а к газо-
вой смеси без 5Оз. Поэтому вместо 0,5_yPso., (см. выше) сле-
дует поставить 1,5 уР'^,.
Процент контактирования при анализе газов аппаратом Рейха
вычисляется по формуле:
10000 (PS0a-PsOs)
PSOs(100-1,5Р' )
283
Кроме указанных выше точек контроля в целях выявления
состояния отдельных аппаратов необходимо: 1) не реже одного
раза в десять дней замерять давление после каждого аппарата
и 2) эпизодически проводить анализы газа на содержание
до и после каждого теплообменника и анализ дымовых газов
после подогревателя.
V. Абсорбция серного ангидрида
1. Теоретические основы
После контактирования газы, содержащие SO3 и небольшое
количество SO2 (соответствующее его непроконтактированной
части), направляются в отделение абсорбции, где 5Оз превра-
щается в окончательный продукт. Контактные заводы выпу-
скают продукцию и в виде купоросного масла и в виде олеума.
Товарный олеум выпускается с содержанием 20% свободного
SO3 (или 85,31% общего SO3). Олеум такой концентрации удо-
влетворяет большинство потребителей. Кроме того, олеум этой
концентрации имеет самую низкую температуру замерзания, что
важно с точки зрения условий его хранения и перевозки. В не-
больших количествах для некоторых потребляющих производств
выпускается высококонцентрированный олеум с содержанием
60—65% свободного SO3. Как видно из кривой рис. 2 (стр. 29),
при этой концентрации олеума температура его замерзания опять
имеет минимум.
Возможная концентрация олеума определяется соотношением
между количествами SO3 и влаги, поступающих в отделение аб-
сорбции, а также условиями равновесия между газом и жид-
костью в олеумном абсорбере.
Пусть процент SO2 в газах после очистки равен Pso2, а про-
цент НгО там же — Рн2о» пусть степень контактирования равна
х. Так как влага газов, улавливаемая в сушильной башне, пере-
дается затем в отделение абсорбции, то весовое отношение
между SO3 и Н2О, поступающими в отделение абсорбции, будет:
Рчо -х-во Р„ 0 • 18
сг • сг — _R-2 _ • —Ы
eso8’6H2o 22,4 ' 22,4
или
£so3__4,44 xPSOg
?н2о рв2о
Если вся продукция выпускается в виде олеума и другой
воды в отделение абсорбции, кроме влаги, поступающей с газами
из отделения очистки, не вводится, то максимально возможная
1 концентрацйя олеума (процент общего SO3) выразится следую-
щей формулой:
МО = 444xPSOg
•?so3 + £н,о 4,44 xPSOa + PHj0
284
В табл. 60 приведены вычисленные по этой формуле вели-
чины максимально возможной концентрации олеума в зависимо-
сти от процента SO2 после очистки и температуры в последней
промывной башне (от которой зависит величина Рн.о ) Степень
превращения х принята равной 0,95.
Таблица 60
Температура в последней промывной башне в °C Максимально возможная концентрация олеума (в %) при концентрации SO2 после очистки, равной
3% 4% 57о 6% 7°/о 87о
10 91,10 93,50 94,50 96,00 96,00 96,80
20 84,70 88,00 90,00 92,00 92,50 93,80
30 75,50 80,50 83,30 86,00 87,50 89,00
40 63,30 70,00 74,00 77,80 80,00 82,30
Жирной ломаной линией отграничена верхняя правая область,
заключающая условия, в которых возможно получить всю про-
дукцию в виде 20%-ного олеума (20% свободного SO3 или
85,31% общего St» и выше.
Из формулы (1) легко вывести, что весь олеум может быть
получен с концентрацией не ниже 20% свободного SO3 при усло-
вии, если
хР„(. > 1,31 Рн0.
Если xPso. >1,31 Рщо , то не только всю производимую
системой продукцию можно выпустить в виде 20%-ного олеума,
но еще и ввести в систему со стороны некоторое количество
купоросного масла для превращения его в олеум.
При принятых выше обозначениях на 1 т SO приходится
воды, приносимой газами из отделения очистки:
р. . (2)
4,44 xPSO2
Если в систему вводится купоросное масло и при этом из
системы выводится только 20%-ный олеум, то можно написать
уравнение:
i
Г1 + таА100
85,31 = А___100 ;
, , ^н,о , ’
+ 4.44xPSOj + -V
где у — число тонн купоросного масла, вводимого в систему,
285
считая на 1 т SO3 продукции; z — процент SO3 в купоросном
масле. Из последнего выражения можно получить:
14,69 хР80-19,2 Ря.о
У (85,31-z)x-PSo2 * W
Если в купоросном масле 75,5% SO3 (92,5% H2SO4), то по-
следнее уравнение выразится так:
' 1,96 Р„ 0
j/=l,5----(4>
a?so2
Если приравнять у нулю, то получится, что вводить купорос-
ное масло для превращения в олеум невозможно, если
x^so2 1 >31 •
При проценте SO2 после очистки, равном 7, степени превра-
щения 0,95 и температуре в последней промывной башне 30°
(Рно=4,1%) находим, что на 1 т продукционного SO3 можно
ввести купоросного масла (92,5% по H2SO4) для превращения
в олеум:
1,96-4,1 ЛОП .
>'=1’5-ад5ТТ = 0’29г-
Иначе говоря, на 1 т олеума, получаемого в системе, можно при
этих условиях получить добавочного олеума путем ввода купо-
росного масла со стороны 0,29 • 0,755 — 0,22 т.
Ввод купоросного масла со стороны можно значительно уве-
личить, если разбавляемую в сушильной башне кислоту непре-
рывно выводить из системы, заменяя ее вводимой же со сто-
роны кислотой.
С точки зрения условий равновесия максимально возможная
концентрация олеума определяется тем, что упругость SO3 над
выходящим из абсорбера олеумом должна быть меньше, чем
парциальное давление SO3 во входящем в абсорбер газе. Вели-
чины упругости паров SOs над олеумом в зависимости от его
концентрации и температуры графически изображены на рис. 133.
Рис. 133 иллюстрирует зависимость максимально возможной
концентрации олеума от условий равновесия в абсорбере. Из
этого рисунка видно, например, что если концентрация SOs
в газе составляет 7% (при ..общем давлении газов, равном
760 мм рт. ст.), то максимально возможная концентрация олеума
будет:
Общ. % SO3 Общ. % SO3
При 30°........... 89,0 При 60°... . 87,0
При 40°........... 88,3 При 70° ... . 86,1
При 50°........... 87,5 При 80° .... 85,5
286
Из этого же рисунка видно также, например, что 20%-ный
олеум (85,3% общего 8Оз) может быть получен.
при % SO3 во входящем при температуре
газе, равном олеума не выше
Рис. 133, Упругость SO3 над олеумом.
Если продукцию контактной системы желают получать только
в виде максимально концентрированного олеума, то для опреде-
ления максимально возможной концентрации олеума необходима
28?
провести два не зависимых один от другого расчета: первый по
условиям абсорбции, второй — по соотношению концентрации
SOs и HsO в газах после очистки. Действительной из получен-
ных по этим расчетам величин будет меньшая из них.
От величин Pso2 и х максимально возможная концентрация
олеума зависит во всех случаях. Ограничивается же она или
температурными условиями в отделении очистки (определяю-
щими влагосодержание в газах) или температурой олеума по
выходе из абсорбера (определяющей величину упру-
гости SO3).
Если часть продукции системы выводить в виде купоросного
масла, то максимально возможная концентрация олеума будет
зависеть только от PSo2, х и от температурных условий в олеум-
ном абсорбере. Влагосодержание в газах после очистки в этом
случае не будет оказывать влияния на величину максимально
возможной концентрации олеума, так как избыточная влага (про-
тив соотношения HsO и SOs в олеуме максимально возможной
концентрации) будет выводиться из системы с купоросным мас-
лом. Однако в этом случае величина влагос одержания в газах
после очистки влияет на распределение продукции системы
между олеумом и купоросным маслом.
Раньше считалось, что SOs в газообразном состоянии в при-
сутствии паров воды полимеризуется в SsOe. Этим, в частности,
Сакур (Sackur) объяснял плохую абсорбцию SO3 кислотами кон-
центрацией ниже 98% H2SO4. Теперь после экспериментов Сту-
дека, Истмена и Дулея (Stoodeke, Eastman, Dooley) доказано, что
присутствие водяных паров ведет не к полимеризации SO3,
а к образованию туманообразных частиц, очень плохо абсорби-
рующихся.
Работами Боденштейна, Финка и др. доказано также, что
серный ангидрид в парах состоит
Таблица 61
Общ. % SOs Теплота смешения, счи-
в получаемом тая на 1 кг взятой
олеуме воды в ккал
81,63 1220 4- 2,51 (Р — 18°)
85,00 1263 4-3,16 (Р — 18°)
90,00 1368 4-4.18 (/э— 18°)
95,00 1577 4- 5,42 (Р — 18°)
98,00 1715 4- 6,12 (Р — 18°)
из неполимеризованных моле-
кул SO3.
Как уже указывалось, от
температурного режима в аб-
сорбере зависит максималь-
но возможная концентрация
олеума. Понятно также, что
от температурных условий
в абсорбере зависит и ско-
рость процесса абсорбции.
Температурный режим аб-
сорбера складывается в за-
висимости от температуры
поступающих в абсорбер га-
за и олеума, их концентраций и соотношения между их количе-
ствами. При этом необходимо учитывать теплоту растворения
SO3 в орошающем олеуме.
В табл. 61 приведена теплота смешения воды с жидким SO3
в расчете на 1 лт воды
288
Теплота смешения газообразного SO3 с водой равна:
где Ht — теплота смешения т кг газообразного SO3 с 1 кг воды,
Kt — теплота смешения т кг жидкого SO3 с 1 кг воды и h —
удельная теплота испарения SO3. Величина /* равна 123 ккал при
0°, 116 ккал при 50° и 112 ккал при 100°.
В табл. 62 приведены величины Ht для различных концен-
траций серной кислоты и олеума.
Таблица 62
°/oS03b конечной кислоте Ht в ккал при температуре % SO3 в конечной кислоте Ht в ккал при температуре
0° 50° 100° 0° 50° 100°
72,7 1186 1247 1318 80,80 1631 1720 1823
73,5 1212 1276 1348 81,63 1693 1785 1892
74,3 1251 1317 1393 82,00 1736 1830 2060
75,1 1290 1359 1479 83,00 1783 1886 2133
75,9 1382 1404 1485 84,00 1839 1950 2211
76,7 1375 1450 1535 85,00 1903 2023 2296
78,4 1469 1548 1641 86,00 1982 2106 2395
79,2 1520 1604 1699 87,00 2070 2205 2509
80,0 1573 1659 1758 89,00 90,00 2280 2400 2424 2548 2763 2878
Определим, насколько поднялась бы температура кислоты, если бы
’98%-ная (по H2SO4) кислота абсорбцией газообразного SOs непосредственно
повысила бы свою крепость до 20%-кого олеума (85,31% общего SOs).
В 1 кг 98%-ной H2SO4 содержится 0,816 кг SOs, а воды—>0,184 кг;
для превращения 1 кг этой кислоты в 20%-ный олеум надо поглотить
0,250 кг SOs.
В табл. 62 находим, что теплота смешения газообразного SOs с водой
(на 1 кг воды) при 50° для 98%-ной H2SO4 равна 1659 ккал, а для 20%-ного
олеума (85,31 общего SOs) 2023 ккал.
Разность, считая на 1 кг во^ы, составит 2023 —1659 = 364 ккал, а иа
1,184 кг воды 364 0,1184 = 67 ккал.
Принимая теплоемкость 20%-ного олеума в среднем равной 0,4, получим
повышение температуры кислоты;
т. е. конечная температура олеума будет 50+ 134= 184°. Но при таких
температурных условиях 20%-него олеума не может получиться.
В производственных условиях с той целью, чтобы темпера-
тура абсорбирующего SOs олеума не подымалась выше допусти-
мого предела, применяют один из двух способов: а£ или охла-
ждают кислоту в самом абсорбере (если получают 20%-ный
олеум, производя абсорбцию SOs непосредственно купоросным
маслом, или 98%-ной H2SO4); б) или орошают абсорбер, выпу-
19 Зак. &019. Технология серной кислоты.
289
скающий олеум, большим количеством олеума с тем расчетом,
чтобы олеум при прохождении через абсорбер повышал свою
концентрацию всего на 0,5—1,5% SO3. При этом условии и по-
вышение температуры олеума происходит в допустимых преде-
лах. Понятно, что во втором случае необходимо ставить по край-,
ней мере два абсорбера, так как полностью поглотить SO3 в аб-
сорбере, орошаемом олеумом, невозможно. Второй абсорбер
в этом случае орошают обычно 98%-ной H2SO4 и опять в таком
количестве, чтобы концентрация кислоты при прохождении через
абсорбер увеличивалась немного (примерно на 0,5—1,5% H2SO4).
Наиболее полно вопросы скорости абсорбции SO3 серной
кислотой и олеумом изучены Турханом и Юдиной. Над водной
Рис. 134. Скорость абсорбции SOs в
зависимости от концентрации кис-
лоты.
Рис. 135. Зависимость коэфнциен-
та скорости абсорбции SO3 от тем-
пературы.
серной кислотой упругость SO3 равняется нулю, поэтому для
"расчетов процесса абсорбции, как это следует уже из вышеиз-
ложенного (стр. 201), можно пользоваться формулой
о
Упругость SO3 над олеумом быстро растет с повышением
концентрации олеума и его температуры. Поэтому (см. стр. 201)
при расчетах процесса абсорбции SO3 олеумом нельзя прене-
брегать упругостью ЭОз над олеумом и надо исходить из урав-
нения
f __
J Рд Pi Р
рьв
Еще Книтч установил, что лучшей абсорбционной способно-
стью по отношению к SO3 обладает 98%-ная H2SO4.
290
Зависимость коэфициента скорости абсорбции SO3 от_кон-
центрации серной кислоты и олеума при температурах^ЗО^ГбО®
графически изображена на' рис. 13? (по данным Турхана и Юди-
ной). Уменьшениа-Ка^ф.ициента скорости абсорбции SO3 с пони-
жением1 концентрации JI2SO4 (после 98%-ной ‘ H2SO4) объясняется
Тём что SOs, реагируя с ларами Н2О над водными кислотами^
образует трудно абсорбирующийся сернокислотный туман.
г На рис7135 графически изображен^ 'зависимость коэфици-
ента скорости абсорбции SO3 98%-ной H2SO4 и 20%-ным олеу-
мом от температуры. Из рисунка видно, что в пределах 30—65°
температура мало влияет на коэфициент абсорбции SOs моно-
гидратом (98%-ной H2SO4). Учитывая, что над этой кислотой
упругость SO3 равна нулю, можно сказать, что изменение тем-
пературы в абсорбере, орошаемом моногидратом, в пределе
30—65° не имеет практически большого значения. По тому же
рисунку видно, что коэфициент скорости абсорбции SOs олеу-
мом с повышением температуры уменьшается быстрее, чем коэ-
фициент скорости абсорбции моногидратом.
Так как с повышением температуры олеума, кроме того,
быстро растет упругость SO3 над ним (а движущая сила
абсорбции, следовательно, уменьшается), то понятно, что ско-
рость! абсорбций SOs олеумом быстро уменьшается с повыше-
нием температуры.
Турхан и Юдина показали, что и для моногидрата и для
олеума коэфициент скорости абсорбции SO3 до некоторого пре-
дела линейной скорости газа (в данных опытах до 36 сл/сек)
пропорционален линейной скорости газа в степени 0,5; при более
высоких из изученных ими значениях линейной скорости газа
коэфициент скорости абсорбции SO3 и моногидратом и олеумом
пропорционален линейной скорости газа в степени 0,8.
2. Непосредственная конденсация 5Оз с парами воды
При переработке сероводорода на серную кислоту при сжи-
гании H2S по реакции
2H2S + ЗО2 = 2SO2 + 2Н2О
получается влажный газ. Предварительное выделение влаги, во-
первых, вело бы к потерям некоторого количества SO2, а во-
вторых, требовало бы сильного охлаждения газа после сжига-
ния H2S и обратного нагревания его перед контактированием.
Поэтому была поставлена задача осуществить контактирование
влажного газа. Но это потребовало и нового оформления про-
цесса дальнейшей конденсации SO3. Перед абсорбцией обычным
методом газы охлаждаются. Но при обычном охлаждении газа,
содержащего водяные пары и SO3, получается сернокислотный
туман, который абсорбируется чрезвычайно плохо. Поэтому не-
обходимо процесс охлаждения горячего газа, содержащего SO3 и
НгО, провести так, чтобы сразу конденсировалась крепкая сер-
ная кислота.
19*
291
При переработке на серную кислоту сероводорода (даже без
учета влаги, поступающей с воздухом, идущим на сжигание
H2S) в дальнейшем после контактирования отношение между
SO3 и НгО получается таким, что при полной конденсации того
и другого получается 100%-ная H2SO4. Так как несущие серо-
водород газы содержат некоторое количество влаги, а окисле-
ние SOa в SO3 не проходит полностью, то соотношение между
SO3 и НгО в газах после мокрого катализа обычно таково, что
при конденсации может получиться кислота менее крепкая, че.м
моногидрат. Условия конденсации серной кислоты из горячих,
содержащих SO2 и НгО, газов были изучены в НИУИФ (Аме-
линым).
Реакция SO3 + Н2О—- H2SO4 гомогенна и имеет очень боль-
шую скорость.
Газы, содержащие SO3 и НгО, до температуры выпадения мо-
гут охлаждаться как угодно быстро. В дальнейшем скорость
Таблица 63
Газ перед конденсацией 5,2% SO3; 7,50/0 н2о Газ перед конденсацией 6,58% SO3; 9,95% Н,О
Температура в °C % конден- сации SO3 % H2SO4 в конденсате Температура в °C % конден- сации SO3 °/о H2SO4. в конденсате
260 9,23 97,80 260 29,30 97,70
250 36,60 97,75 250 52.20 97,55
240 56,30 97,65 240 66,60 97,45
230 70,00 97,50 230 77,10 97,25
220 79,30 97,23 220 83,80 96,85
200 90,10 96,50 200 91,70 95,90
180 94,80 95,55 180 96,40 94,65
160 97,40 94,50 160 98,80 93,55
Таблица 64
Температура в °C % конден- сации SO3 Продукционная кислота
паров в верху аппарата газов на выходе из конденсатора температура в °C % H,SO4
283 148 98 04,6
270 144 —. 80 92,7
280 165 99,07 ПО 93,25
286 168 98,43 ПО 92,70
270 150 99,40 105 92,43
— 158 99,31 102 93,80
274 154 99,05 100 93,50
292
охлаждения должна соответствовать скорости конденсации.
Полученные по произведенным расчетам концентрация и выход
кислоты в зависимости от температуры конденсации приведены
в табл. 63.
Процент конденсации серной кислоты, понятно, снижается
при повышении температуры. Концентрация получаемой кислоты
при этом, наоборот, повышается.
В полузаводских опытах (на заводском газе) при конденса-
ции серной кислоты в фарфоровых трубках, наполненных фарфо-
ровыми шариками, получены результаты, приведенные в табл. 64.
3. Схемы и аппаратура абсорбционных отделений
На рис. 136 изображена схема абсорбционного отделения си-
стемы Герресгоф-Байера. Газ, охладившись в ангидридных хо-
Рис. 136. Абсорбционное отделение системы Герресгоф-Байера:
1—олеумный абсорбер; 2— олеумный холодильник; 3— олеумный сборник; 4— насос; 5—
моногидратный абсорбер; 6—моногидратный холодильник; 7—моногидратный сборник; 8 —
насос; Р—ловушка; 10 и 11—кривые; 12—ангидридный холодильник; 13—вентиль.
лодильниках 72, проходит последовательно абсорбер 1, орошае-
(987 '^м^леумом, и абсорбер 5, орошаемый моногидратом
Циркулирующие кислоты охлаждаются в оросительных хо-
лодильниках 2 и б. Схема абсорбционного отделения системы
Кемико отличается от системы Герресгоф-Байера тем, что там
нет ангидридных холодильников. Поддержание необходимой
температуры орошающего олеума достигается увеличенным
охлаждением в олеумном оросительном холодильнике. Таким
образом устранение ангидридных холодильников в системе Ке-
мико вызывает некоторое увеличение оросительных олеумных
293
холодильников. Оросительные холодильники в системе Кемико
поставлены не после насосов, как в системе Герресгоф-Байера,
а до них, и наполнение холодильников происходит вследствие
разностей уровней кислоты в холодильнике и в сборнике кис-
лоты под башней.
По схеме абсорбционного отделения тентелевской системы
(рис. 137) газы после контактного аппарата проходят через ан-
гидридный холодильник V/, а затем последовательно через два
олеумных / и // и один моногидратный /// абсорберы. Кислота
передается из абсорбера в абсорбер навстречу газу. Вода или
слабая кислота (из сушильных башен) добавляется в моногид-
ратный абсорбер. Иногда в тентелевской системе применяется
один абсорбер из нескольких степеней барботажа, аналогичный
описанной выше промывной башне.
4 II—олеумные абсорберы; III—моногидратный абсорбер; IV—чугунная ко-
лонка; V—ловушка; VI— ангидридный холодильник; I—воронка; 2—трубы
для исходящей кислоты.
В ангидридный холодильник системы Герресгоф-Байера
(рис. 138) газ поступает через штуцер 2 в приемную камеру, за-
тем по трубкам 4 спускается вниз в камеру 3 и выходит через
штуцер 1. Охлаждающая вода циркулирует в междутрубном
пространстве. Конденсат, образующийся при плохой осушке
газа перед контактированием, выпускается из камеры 3 через
вентиль 7.
В ангидридном холодильнике системы Грилло-Шредера
(рис. 139) газ проходит через кольцевое пространство, а вода
омывает внутреннюю поверхность внутреннего цилиндра.
В тентелевской системе применялись ангидридные холодиль-
ники, где газы охлаждались наружным воздухом. Такой холо-
дильник отличается от описанного холодильника системы Гер-
ресгоф-Байера тем, что он не имеет наружного кожуха и что
трубы, по которым идет газ, свободно омываются воздухом, ко-
торым и охлаждаются.
Из этих трех типов холодильников, понятно, наиболее эффек-
тивно работает холодильник системы Герресгоф-Байера. Однако
294
недостаток его в том, что трубы его в местах соединения часто
разъедаются конденсирующейся кислотой. Холодильник Грилло-
Шредера обладает .этим недостатком в значительно меньшей
степени. Воздушный же холодиль-
ник опасности разъедания под-
вергается меньше всего, но при
одной и той же производитель-
ности должен иметь значительно
большую поверхность труб, чем
водяной холодильник.
Для абсорбции SO3 в старых
системах Грилло-Шредера при-
менялись горизонтальные цилин-
дрические абсорберы поверхност-
ного поглощения. В тентелевской
системе в качестве абсорберов
применяются барботажные аппа-
раты. В системах Герресгоф-Бай-
ера и Кемико для абсорбции SO3
применяются башни с насадкой.
Олеумный абсорбер системы Гер-
ресгоф-Байера (рис. 140) предста-
вляет железный вертикальный
Рис. 138. Ангидридный холо-
дильник системы Герресгоф-
Байера:
1, 2—газовые штуцеры; 3—нижняя
камера; 4—трубки; 5 и 6—вход и
выход воды; 7—вентиль.
©о©
.©©©©
СЮ
со
©с
ее©
Рис. 139. Ангидридный холо-
дильник системы Грилло-Шре-
дера.
цилиндр /, футерованный внутри кислотоупорным кирпичом 2
и имеющий внизу колосниковую решетку 3 из кислотоупорных
плит. Абсорбер загружен кольцами Рашига. В верхней части
его помещена железная распределительная плита 5, в отверстия
S95
которой вставляются фарфоровые или железные трубки б- Че-
рез эти трубки происходит подача кислоты на насадку абсор-
бера.
В крышке 7 устраиваются четыре люка 8 для осмотра рас-
пределительной плиты.
Газ поступает в нижний штуцер 9, проходит насадку и вы-
ходит сверху через штуцер 10. Олеум подается через штуцер 11
на распределительную плиту, стекает по
Рис. 140. Олеумный абсорбер системы Геррес-
гоф-Байера:
насадке и выходит че-
рез штуцер 12.
Моногидратный аб-
сорбер по конструкции
и размерам почти не
отличается от кон-
струкции и размеров
сушильной башни, так
как эти башни ороша-
ются кислотой одной и
той же концентрации.
У каждого абсорбе-
ра устанавливаются
сборники для орошаю-
щей кислоты. Сборник
олеума делается из же-
леза, а для моногидра-
та — из кислотоупор-
ного чугуна. Сборники
снабжены смотровыми
стеклами и поплавками
для наблюдения за
уровнем кислоты. С бо-
ков сборников устраи-
ваются сифоны, кото-
рые соединяются с на-
сосом. Для заливки си-
7—железный кожух; 2— футеровка; 3—колосниковая
решетка; 4-—угольник; 5—распределительная плита;
6—трубки; 7—крышка; 8—люки; 9—штуцер для входа
газа; 10—штуцер для выхода газа; 11—штуцер для
входа олеума; 12—штуцер для выхода олеума.
фонов в верхней части
их устраиваются па-
трубки.
Конструкция моно-
гидратного абсорбера
системы Кемико аналогична конструкции описанной выше су-
шильной башни этой системы. Олеумный абсорбер отличается
от моногидратного только тем, что не имеет брызгоуловителя.
Олеумный абсорбер тентелевской системы (рис. 141) пред-
ставляет собой железный котел 1 с сферическим днищем 2 и
крышкой 3. Внутри абсорбера помещается железньТй колокол 5
с отверстиями 7, заканчивающийся зубцами 6. Абсорбер напол-
няется олеумом, уровень которого поддерживается на высоте
штуцера 8. Газ поступает по трубе 9, проходит под колокол 5,
зубцы и отверстия которого разбивают его на мелкие струйки,
296
благодаря чему достигается хороший контакт газа и жидкости.
Из колокола газ через штуцер 10 поступает в следующий аб-
сорбер. По трубе 11 в олеумный абсорбер подается купоросное
масло.
Абсорбер снабжен водяной рубашкой 12 для охлаждения
его. Моногидратный абсорбер такой же, как и первые два, но
отливается из чугуна, а внутренняя его поверхность эмали-
руется для предохранения чугуна от коррозии моногидратом.
В хвостовых газах, выходящих из
последнего абсорбера, содержатся брыз-
, и кислоты, захватываемой газом в абсор-
бере. В системе Кемико для улавливания
брызг в сушильной башне и в моногидрат-
ном абсорбере служит верхняя часть
башни (рис. 9). В системе Герресгоф-
Байера для улавливания брызг ставится
или отдельный брызгоуловитель или спе-
циальная ловушка на крышке моно-
гидратного абсорбера. Брызгоуловитель
(рис. 142) представляет собой полую чу-
гунную башню, разделенную на две
Рис. 142. Брызгоулови-
тель:
1 — перегородка, 2, 3t 4f —
штуцеры.
Рис. 141. Олеумный абсорбер тентелевской
системы:
1—железный кэтел; 2 — дннще; 3— крышка; 4 —
фланцы; 5—колокол; б — зубцы; 7—отверстия;
б—штуцер; 9—труба; 10—штуцер; 11—труба; 12—
водяная рубашка.
части вертикальной перегородкой 1, не доходящей до дна. Газ
входит через штуцер 2, опускается вниз, огибает перегородку,,
поднимается вверх и через штуцер 3 поступает в выхлопную
трубу. Благодаря изменению направления и скорости движе-
ния газов (при огибании перегородки) брызги отделяются, па-
дают на дно башни. Кислота стекает из брызгоуловителя через
штуцер 4 в сборник последнего абсорбера. Чтобы кислота
в брызгоуловителе не задерживалась, дно его делается внутри
выпуклым.
297*
Ловушка, непосредственно присоединяемая к крышке моно-
гидратного абсорбера, представляет собой (рис. 143) железный
цилиндр, переходящий в нижней части в конус. В верхней ча-
сти ловушка имеет сферическую крышку 7 с выхлопной трубой.
В корпусе ловушки помещены на расстоянии 250 мм одна от дру-
гой перегородки, идущие попеременно — с отверстиями 2 и без
отверстий з. Газ из абсорбера поступает через штуцер 4, прохо-
дит через отверстия первой перегородки, ударяется о вторую
перегородку, огибает ее и т. д. Вследствие неоднократного из-
менения скорости и направления га-
Рис. 143. Ловушка:
J—крышка; 2—перегородки с отвер-
стиями; 3—перегородки без отвер-
стий; 4—штуцер.
за и вследствие ударов его о пере-
городку брызги кислоты задержива-
ются и стекают обратно в абсорбер.'
Для выделения из газов брызг
кислоты может быть применена
обыкновенная неорошаемая башня с
насадкой.
При применении барботажных
абсорберов циркулирующая кисло-
та охлаждается в процессе самой
абсорбции — абсорберы имеют водя-
ные рубашки. В системах Герресгоф-
Байера и Кемико для охлаждения
циркулирующих кислот применяют-
ся оросительные холодильники.
4. Режим и работа абсорбционного
отделения
Для успешной работы абсорбци-
онного отделения необходимо: хоро-
шее состояние насадки в абсорберах,
равномерное и достаточное ороше-
ние абсорберов, поддержание воз-
можно низкой температуры цирку-
лирующей кислоты.
Газы перед входом в абсорберы
в системе Герресгоф-Байера охла-
ждаются в ангидридном холодиль-
нике до температуры 40—60°. Пер-
вый абсорбер в системе Герресгоф-
Байера орошается 20%-ным олеумом в таком количестве, чтобы
концентрация олеума по свободному SOa повышалась не больше
чем на 0,5—1,5%. На некоторых заводах концентрацию орошаю-
щего олеума поддерживают равной 18% свободного ЗОз, исходя
из того, что олеум такой концентрации замерзает при' темпера-
туре несколько низшей, чем 20%-ный олеум. Но это не следует
рекомендовать, так как олеум такой концентрации оказывает
сильное корродирующее действие на аппаратуру. Концентрация
298
орошающего олеума должна быть не ниже 20% по свободному
SO3, а лучше даже немного выше. Опасности замерзания легко
можно избежать поддержанием определенной температуры охла-
ждающей воды; в самом абсорбере опасности замерзания не
создается.
Обыкновенно считают, что в первом абсорбере абсорбируется
от 30 до 50% от всего поступающего количества SO3. Однако
практически эта степень абсорбции по отдельным системам ко-
леблется в более широких пределах. Она зависит от условий
работы абсорбера: концентрация орошающего олеума, процент
SO3 в поступающем газе, количество орошения, температурный
режим, а также и от аппаратуры: размеры абсорбера, тип на-
садки. По данным бригады Оргхима на одном из обследованных
ею заводов в первом абсорбере абсорбируется 88% от всего ко-
личества поступающего SO3.
Для орошения второго абсорбера в системе Герресгоф-Байера
применяется 98%-пая H2SO4. Насколько важно держать кон-
центрацию орошения второго абсорбера близкой к 98% H2SO4,
показывает следующий пример из практики работы. Нормально
при орошении второго абсорбера 98%-ной H2SO4 содержание
SOs в отходящих газах составляет 0,07%. При обследовании
бригадой Оргхима одного контактного завода содержание SO3
в отходящих газах составляло 0,29%. При этом концентрация
орошающей второй абсорбер кислоты была 94,98% H2SO4, а вы-
текающей 95,08% H2SO4.
Температура кислоты в моногидратном абсорбере без замет-
ного ущерба для процесса абсорбции может доходить до
60—65°.
Регулирование работы абсорбционного отделения в основ-
ном сводится к тому, чтобы держать соответствующие и по-
стоянные концентрации орошающих кислот и поддерживать не-
обходимый температурный режим в абсорберах.
Постоянство концентрации кислоты, орошающей первый
(олеумный) абсорбер, достигается тем, что в его бак-сборник
добавляется кислота из второго абсорбера. Постоянство концен-
трации кислоты, орошающей второй (моногидратный) абсорбер,
достигается добавлением в его бак-сборник кислоты из сушиль-
ной башни и воды. Если необходимо получить максимальный
выход олеума, вместо воды вводят со стороны купоросное
масло. Концентрация орошающего олеума и орошающего моно-
гидрата проверяется анализом. Применяемый метод анализа не-
достаточно точен, поэтому концентрацию орошения второго
абсорбера следует проверять дополнительно наблюдением за
цветом газов. Если концентрация орошающей кислоты ниже
98% H2SO4, то SO3 во втором абсорбере образует с парами воды
туман, который виден в смотровое окно второго абсорбера. Если
концентрация орошения выше 98% H,SO4, то появляется неболь-
шой дымок в верху сушильной башни; это значит, что кислота
концентрации выше 98% H2SO4 уже начинает выделять SOs.
299
Этого никак нельзя допускать, так как образующийся в сушиль-
ной башне сернокислотный туман разрушающе действует и на
компрессор и на контактную массу (при снижении температуры
в контактных аппаратах). Чтобы полностью избежать этой опас-
ности, в последнее время на ряде заводов отказались от парал-
лельного орошения второго абсорбера и сушильной башни и пе-
решли к раздельному циклу орошения.
При нормальном режиме газы и в сушильной башне и во вто-
ром абсорбере будут совершенно прозрачными.
Для того чтобы в ангидридном холодильнике не произошло
замерзания конденсата, он охлаждается темперированной водой,
т. е. такой водой, температура которой поддерживается в пре-
делах 25—30°. Для этой цели используется вода, отходящая из
холодильников при промывных башнях. Эта же вода приме-
няется и для оросительных холодильников при абсорберах.
В отходящих из последнего абсорбера газах содержится
около 0,1% SOa.
Ниже приводится перечень принятых замеров и анализов по
абсорбционному отделению (табл. 65). Кроме указанных анали-
зов >в целях проверки состояния отдельных абсорберов не реже
одного раза в 10 дней должен производиться замер давления
после каждого аппарата. Эпизодически необходимо проверять
количество орошения по каждой башне.
Таблица 65
Объект контроля Определяется Место анализа Частота анализа или замера
Газ Я Кислота Вода Кислота Продукция Температура Содержание SO3 Температура и со- держание H2SO4 Температура Кислотность Уровни Количество, уд. вес, содержание SO3 и H2SO4 Перед олеумным аб- сорбером После каждого абсорбера После моногидратного абсорбера на выхлопе После каждого абсорбера До и после холодильника После холодильника В сборниках Продукционные храни- лища 4 раза в смену 1 раз в смену 4 раза в смену 1 раз в смену 2 раза в смену То же По мере отпуска
VI. Расходные коэфициенты
На контактном заводе сера теряется с огарком, в промывном
отделении, от неполноты контактирования (в виде SO2 с отходя-
щими газами), от неполноты абсорбции (в виде 8Оз и тумана
с отходящими газами), с газами, выходящими через неплотности
аппаратуры и газопроводов в атмосферу при неисправности
ЗСО
аппаратуры. Если всю серу, поступившую с колчеданам, принять
за 100%, то при содержании в колчедане 40% серы, а в огарке
1,5% потеря серы с огарком составит от всей серы, поступившей
в печь,
1^—ТГ1’5’100
------= 2,83%.
В систему поступит от всей серы, загруженной в печь,
100 — 2,83 = 97,17%.
В промывном отделении при абсорбции 8Оз печного газа пе-
рейдет в промывную кислоту ~ 8% от всей серы, пришедшей
с газами, или
97.17 -8_7 7~0;
1С0 ~ ’ /о
от серы, загруженной в печь.
Потери серы в промывном отделении и от неполноты абсорб-
ции трудно подсчитать. Но, исходя из опыта работы лучших
заводов, можно принять, что потери серы в промывном отделе-
нии не должны превышать 1%. Таким образом из всей серы,
поступившей в печи, в контактный узел приходит:
97,17 — 7,77 —1,0 = 88,40.
Нормальный процент контактирования должен быть не
меньше 96. На ряде заводов СССР в последнее время процент
контактирования выше 97. С хвостовыми газами в виде SO2
уйдет 88,40 • 0,04 3,54%.
Учтя потери серы от неполноты абсорбции в размере 1%,
получим общий процент использования серы по системе равным:
97,17 — 1,0 — 3,54 — 1,0 = 91,63.
Отсюда расход 45%-ного колчедана на 1 т продукции дол-
жен быть не больше
100032
98 - 450 • 0,9163
0,790 ш.
Как это понятно из приведенного расчета, такой расходный
коэфициент колчедана выведен в предположении, что вся полу-
чаемая промывная кислота реализуется как продукт.
Фактически на работающих установках расход, колчедана зна-
чительно выше, а следовательно, и коэфициент использования
серы меньше. Перерасход серы (и колчедана) происходит, глав-
ным образом, по .причинам повышенного содержания серы
в огарке и пониженного процента контактирования. Там, где
SO2 хвостовых газов используется (производство бисульфита,
жидкого SO»), потери серы от неполноты контактирования отпа-
дают.
301
На одном заводе при проценте использования серы в преде-
лах 87,1—86,6 потери серы (от всей серы, загружаемой в печь)
распределялись следующим образом (в %):
С огарком........................2,4—3,4
В промывном отделении............1,0—3,6
От неполноты контактирования . . . 6,9—8,0
От неполноты абсорбции н др.....0,3—1,7
Основной расход энергии на контактном заводе падает на
протягивание газа через систему. Расход энергии на протягива-
ние газа через аппаратуру в контактной системе Герресгоф-
Байера достигает примерно 40 квт-ч. Для обычной установки
Герресгоф-Байера считается нормальным расход энергии на 1 т
продукции ~ 80 квт-ч. На тентелевских системах расход энер-
гии значительно больше и достигает 200 квт-ч на 1 т продукции.
Это объясняется тем, что промывные башни и абсорберы пред-
ставляют аппараты барботажного типа, создающие значительно
большее сопротивление газу, чем башни с насадкой.
Вода в контактном цехе расходуется на охлаждение газа и
кислоты в промывном и абсорбционном отделениях. При тем-
пературе поступающей1 воды 15° расход воды для технологи-
ческих целей в системах Герресгоф-Байера и Кемико составляет
50—60 л3 на 1 т продукции.
При 'нормальном состоянии завода расход топлива (подогре-
ватель) совершенно незначителен и составляет не больше 0,01 т
мазута на 1 т продукции.
Борьба за снижение расходных коэфициентов — конкретная
задача всех заводов. Понижение расхода колчедана должно идти
в направлении: уменьшения серы в огарке, повышения процента
контактирования, улучшения работы отделения абсорбции, устра-
нения неплотностей аппаратуры.
Как мы уже показали, процент использования серы, равный
91,63, может быть достигнут на любой установке Герресгоф-
Байера. Уменьшение расхода энергии прежде всего может быть
достигнуто содержанием всей аппаратуры в безупречном состоя-
нии (систематическая чистка от вносимой газами пыли и грязи,
устранение неплотностей аппаратуры, находящейся под разре-
жением). Совершенно несомненно, что считавшийся нормальным
расход энергии в 80 квт-ч на 1 т продукции даже для работаю-
щих установок Герресгоф-Байера не есть минимально предель-
ный. Очень неблагоприятно на расходе сырья и на расходе энер-
гии отражается пониженная концентрация SO2 в газах, посту-
пающих в систему.
Экономия в расходе воды может быть получена, если воду
после холодильников промывных башен использовать для охла-
ждения газа в ангидридном холодильнике (система Герресгоф-
Байера) и для охлаждения олеума.
Значительное увеличение количества охлаждающей воды при
невысокой ее температуре указывает или на загрязнение поверх-
302
ностей холодильника или на неправильный технологический ре-
жим (охлаждение кислот до более низких температур, чем тре-
буется).
VII. Производство высокопроцентного олеума
Некоторые потребители контактной серной кислоты, например
заводы органических красок, требуют олеума очень высокой кон-
Рис. 144. Установка для разложения олеума:
], 2 и 3— реторты; 4—топка; 5—дымовая труба; б, 7 и б—трубы; 9—дефлегматор.
центрации — до 60% свободного SO3. Олеум такой концентра-
ции имеет упругость SOg, равную 185,1 мм рт. ст. при 30°, а при
40°-322,1 мм рт. ст.
Следовательно, чтобы получить 60%- .
ный олеум при 30°, надо поддерживать со-
держание SO3 во входящих на абсорбцию
газах, при нормальном давлении, равным
ЮО = 24,3°/о объемных,
а при 40° — 42%. Поэтому в настоящее
время высокопроцентный олеум произво-
дится на особых установках. Получаемый
на обыкновенных контактных установках
20%-ный олеум разлагается нагрева-
нием в особых ретортах на моногидрат
и почти чистый газообразный SO3. Чис-
тый газообразный SO3 поступает далее
Рис. 145. Реторта уста-
новки для разложения
олеума:
7—труба из реторты.
303
в абсорберы, где
он абсорбируется мо-
ногидратом или 20% -
ным олеумом.
При этом получа-
ется высокопроцент-
ный олеум.
Установка разло-
жения 20%-ного оле-
ума (или олеума дру-
гой концентрации) на
SO3 и моногидрат
состоит из трех тер-
рас ораспол ож е н « ых
чугунных или желез-
ных реторт (рис.
144). 20%-ный олеум
непрерывно поступает
в реторту 3 и затем
перетекает в ретор-
ты 2 и 7. Из ретор-
ты 1 постоянно вы-
водится моногидрат.
Из топки 4, находя-
щейся под ретор-
той 7, топочные газы
проходят под всеми
ретортами (в проти-
вотоке с олеумом)
и затем уходят в ды-
мовую трубу 5. Вы-
деляющийся SO3'
проходит по тру-
бам б, 7 и 8 и ухо-
дит на абсорбцию.
Над ретортой 7 на-
ходится дефлегма-
тор 9, задерживаю-
щий пары серной
кислоты. В дефлегма-
тор заливается 20 % -
ный олеум.
На дне реторты
(рис. 145) устроен
лабиринт перегоро-
док. Олеум входит в
реторту справа, про-
ходит длинный путь
по дну ее и выходит
ЗС4
из нее (из реторты / на рис. 144 выходит моногидрат) по
трубе 7.
Обычно абсорбция полученного почти чистого газообразного
БОз проходит в трех последовательных абсорберах барботаж-
ного типа. Первые два абсорбера выполнены из чугуна, послед-
ний— из железа. Первый абсорбер орошается 20%-ным олеумом;
в нем поглощается до 90% от всего поступившего SOs, и при
этом получается 65%-ный олеум. Последний абсорбер орошается
кислотой концентрации 97—98% H2SO4 и абсорбирует остаток SO3
(2—3% от всего поступившего на абсорбцию).
Абсорберы снабжены мешалками, а также водяными рубаш-
ками для охлаждения.
VIII. Особые формы контактных установок
1. Контактный завод, работающий на сере
Схема производства серной кислоты контактным методом зна-
чительно упрощается, если в качестве сырья применяется сера
(особенно, если она свободна от мышьяка и селена).
На рис. 146 в качестве примера приведена схема системы
Кемико в США. Вместо всего сложного отделения очистки
контактных заводов, работающих на колчедане, при работе на
сере остается только один аппарат — фильтр. Осушка воздуха
перед поступлением в печь осйобождает от необходимости очи-
щать газ от тумана перед контактированием. В связи с этим
газ охлаждается перед контактированием только до темпе-
ратуры начала контактирования.
Аналогичные контактные установки, работающие на сере,
начали строиться также в Италии.
2. Контактная система на принципе мокрого катализа
Схема получения серной кислоты из сероводорода по прин-
ципу мокрого катализа показана на рис. 147. Воздух (через
фильтр 7) и сероводород затягиваются вентилятором 2 и посту-
пают в печь з, где сероводород сгорает до сернистого газа. Да-
лее горячая газовая смесь поступает в контактный аппарат 4, ко-
торый охлаждается воздухом, протягиваемым вентилятором 10
через холодильную рубашку аппарата. Газ, содержащий SO3 и
НгО, из контактного аппарата сразу поступает в конденсатор 5,
где охлаждается водой.
Полученная крепкая кислота через холодильник б идет
в сборник 7 и при помощи монтежю 8 перекачивается на склад
или в цистерны. Получающийся в конденсаторе 5 при кипении
воды пар идет в конденсатор д, из которого конденсат — горя-
чая вода — обратно поступает в конденсатор 5. Остаточный газ,
содержащий избыточные водяные пары и следы SO3, выбрасы-
вается по трубе 77. По такой схеме в Германии работает не-
20 Зак. 5019. Технология серной кислоты.
305
сколько установок, перерабатывающих сереводород — отход кок-
совых заводов.
По данным Зике (Siecke) этим способом получается очень
чистая кислота концентрации 85—90% H2SO4. Расход электро-
энергии составляет 30 квт-ч на 1 т продукции (считая на моно-
гидрат), расход воды 30 м3- Аналогичным способом можно полу-
чать не только водную серную кислоту, но и олеум в том слу-
чае, если процент SO3 в газах больше, чем процент Н-гО.
Рис. 147. Схема контактного завода, работающего на сероводороде:
7—фильтр для воздуха; 2—вентилятор; 3—печь; 4—контактный аппарат; 5—
кислотный конденсатор; 6—кислотный холодильник; 7—сборник кислоты; 8—
монтежю; 9—конденсатор пара; 10—вентилятор; 11—выход остаточного газа;
12—вход холодного воздуха.
По патенту Зике и Водевиль (W. Siecke и М. Wohlewill) газ
после мокрого катализа проходит через аппарат для конденсации
с коррозионноустойчивыми трубами из плохо проводящего тепло
материала. Газ поступает сверху, а в верхней части труб кон-;
денсируется моногидрат в количестве, соответствующем содер-
жанию HgO в газах. Получившийся моногидрат, охлаждаясь
далее, служит для поглощения SO3. В проведенных опытах)
с газами, содержавшими до катализа 9% SOs и 5% НгО, полу-/
ченньщ олеум имел 28% свободного SO3 и температуру 35°.’’
Отходящий из аппарата газ не содержал тумана; SOs в нем
было 1%. Последние остатки SO3 удаляют из газов концентри-
рованной серной кислотой.
ЗС6
IX. Задачи в области интенсификации и дальнейшего
усовершенствования контактного процесса
В последние годы ряд контактных цехов значительно повысил
свою мощность против проектной. Это достигнуто улучше-
нием обслуживания систем, с одной стороны, и конструктивным
улучшением части аппаратуры контактного цеха, — с другой. Со-
вершенно ясно, что в основной аппаратуре контактных цехов
имеются возможности и для дальнейшей интенсификации.
В борьбе за улучшение работы действующих цехов и за даль-
нейший подъем их выработки особенно важно обратить внима-
ние на следующие моменты:
1. Надо всемерно стремиться к максимальному снижению
сопротивления аппаратуры как содержанием ее в чистоте, так
и устранением конструктивных дефектов, вызывающих увеличен-
ное сопротивление. В этих целях целесообразно концентрацию
SO2 в газе до компрессора держать возможно выше, разбавляя
газ до оптимальной концентрации (с точки зрения контактирова-
ния) перед сушильной башней.
2. Особенно строго надо соблюдать технологический режим
отделения мокрой очистки и осушки газа, не допуская проскока
тумана и влаги в контактный узел. При проскоках влаги в кон-
тактные аппараты (во время временных понижений температуры)
происходит порча контактной массы: она слеживается, ее актив-
ность снижается, а сопротивление повышается. Влажный газ,
разрушая аппаратуру, загрязняет также контактную массу про-
дуктами коррозии (соли железа), что тоже приводит и к пониже-
нию активности массы и к повышению ее сопротивления.
3. Необходимо также особенно строго поддерживать постоян-
ство концентрации SO2 в газах перед входом в контактный узел.
В существующих контактных цехах самым узким местом
является сейчас отделение мокрой очистки газов. Поэтому
следует особое внимание сосредоточить на улучшении работы
отделений очистки при одновременном упрощении их оформ-
ления.
Применяемые сейчас в СССР ванадиевые контактные массы
достаточно активны, но еще недостаточно удовлетворительны,
главным образом по причине их быстрой порчи. Необходимо ра-
ботать над дальнейшим усовершенствованием контактных масс
в направлении: 1) повышения их активности при низких темпера-
турах (~ 400 ); 2) повышения их термической стойкости при
температурах около 600—650°; 3) повышения их механической
прочности и 4) удлинения срока их работы в производственных
условиях.
Необходимо также продолжить работы по изысканию более
дешевых, может быть и неванадиевых, контактных масс.
В последнее время в СССР освоены контактные аппараты,
дающие высокую выработку при высоком проценте контакти-
рования (97%). Однако работу по разработке новых еще бо-
20* 307
лее совершенных конструкций контактных аппаратов надо про-
должать. При этом необходимо учитывать следующее:
1. Конструкция контактного аппарата должна быть такой,
чтобы температурная кривая контактирования возможно ближе
подходила к оптимальной температурной кривой.
2. Конструкция контактного аппарата должна обеспечить
равномерное распределение газа и постоянство температуры по
сечению контактного аппарата.
3. Контактный аппарат должен быть не особенно громоздким
и не особенно сложным по конструкции.
4. Мощность одного контактного аппарата должна быть зна-
чительно увеличена против мощности существующих кон-
струкций.
5. Контактный аппарат не должен давать большого сопро-
тивления.
6. Контактный узел должен быть спроектирован так, чтобы
не требовалось специального подогрева газа (кроме момента
пуска системы).
Интенсификация контактного метода может итти в напра-
влении значительных изменений технологической основы произ-
водства: применение концентрированного (по SOa) газа и приме-
нение давления.
Необходимо разработать конструкцию контактного аппарата
для работы на высококонцентрированном газе.
Оформление контактного цеха значительно упрощается при
работе на сере. Точно так же упростится оформление контакт-
ного процесса при применении концентрированного газа. Освое-
ние серы как сернокислотного сырья и разрешение проблемы
концентрирования слабых по SO2 газов являются актуальными
задачами и с точки зрения улучшения и удешевления производ:
ства контактной серной кислоты.
Применение 100%-ного SO2 дает .возможность контактному
цеху работать на 14—16%-ном по SO2 газе, при этом интенсив-
ность контактного цеха повысится больше чем в 2 раза, а расход
энергии понизится в 4 раза.
Как для действующих цехов, так и для вновь проектируемых
актуальной является задача замены свинца черными металлами
и неметаллическими материалами и замена применяемых сейчас
сортов железа и чугуна более коррозионноустойчивыми, но недо-
рогими материалами.
Литература
1. Knietsch R., Вег. 34, 4069 (1901); ЖРХО, 1901.
2 Boden stein М., Pohl, Ztschr. f. Elektrochem. (Равновесие 2SO2-|-
+ O-2 £ 2SO3), 11, 376 (1905).
3. Капуст и некий и Шумовский (Равновесие диссоциации SO3),
Acta Physikochimica URSS, 6, 791 (1936).
4. Grau, Roth, Meichsner (Термохимия серы), Ztsch. f. anorg. allg.
Chem. 193, 161 (1930).
308
5. Раковский А. В., Введение в физическую химию, ОНТИ, 595, 1938.
6. Семенов Н. Н., Успехи химии 1, 1 (1932).
7. Кассель, Кинетика гомогенных газовых реакций, ОНТИ, 1937.
8. Тэйлор, Энергия активации адсорбционных процессов, статья в сбор-
нике „Новые идеи в области катализа", т. I, 161, Госхимтехиздат, 1932.
9. Рогинский С. 3., Адсорбция и катализ, статья в сборнике „Проб-
лемы кинетики и катализа", т. III, 356, ОНТИ, 1937.
10. Шваб Г. М„ Катализ с точки зрения химической кинетики, ОНТИ, 1934.
11. Райд ил и Тэйлор, Катализ в теории и практике, Госхимтехиздат,
Л. 1933.
12. Мэкстед, Катализ и его промышленное применение, ОНТИ, 1936.
13. Phillips Р., Е. Р. 6096 от 21/III 1831 г.
14. Neumann В. и сотр. (Оригинальные работы по изучению механизма
сернокислотного катализа на окисных катализаторах). Ztschr. f. Elek-
trochem. 34, 696, 734 (1928); 35, 42 (1929); 36, 87 (1930); 38, 304 (1932); 41,
589 (1935). См. также: Каталитические процессы в производстве серной
кислоты методом контактного окисления, Сборник переводных статей,
Госхимтехиздат, 1933.
15. Ададуров И. Е. и сотр., ЖПХ VIII, 598, 606, 612 (1935); IX, 603, 1580
(1936). Ряд работ по изучению хромооловянных катализаторов для
окисления сернистого газа.
16. Waeser В., Handbuch der Schwefelsaurefabrikation, т. Ill, 1930.
17. Fairlie A., Sulfuric acid manufacture. New York 1936.
18. Боресков Г. К., Ванадиевые катализаторы для окисления SO2, Сбор-
ник трудов Укр. хим. ин-та, вып. 2 (1935); Доклады VI Менделеевского
съезда, т. II, вып. 2, стр. 159. Сравнение ванадиевых и платиновых ка-
тализаторов.
19. В о d е n ste i n М., Fink С. (Кинетика окисления SO2 на платине),
Ztschr. phys. Chem. 60, 1, 46 (1907).
20. Taylor H., Lenh er S. (Кинетика реакции 2SO2 O2 2SO.. на пла-
тине), Ztschr. phys. Chem., Bodenstein Festband, 30 (1931).
21. В о d e n s t e i n M. (Механизм окисления SO2 на платине), Ztschr. f. phys.
Chem., 2, 345 (1929).
22. Боресков Г. К., Соколова Т. И., Оптимальные концентрации SO2
в контактном сернокислотном процессе, ЖХП, 1241 (1937).
23. Боресков Г. К., П лигу нов В. А., Кинетика окисления SO2 на
ванадиевых катализаторах, ЖПХ VI, 785 (1933).
24. Siege rt Н. (Влияние ядов и активаторов на производительность
ванадиевых катализаторов в контактном сернокислотном процессе),
Ztschr. f. angew. Chem. 50, 319 (1937).
25. Боресков Г. К., Пл игу но в В. К., Механизм окисления SO2 на
активированных ванадиевых катализаторах, ЖПХ, т. XIII, 1910, стр.
329 и 653.
26. Neumann В., Jiittner G. (Отравление сернокислотных катализато-
ров), Ztschr. f. Elektrochem. 36, 87 (1930).
27. А да д у р о в 14. Е., Г ум и некая М. А., Отравляемость ванадиевых
катализаторов As2O3, ЖПХ V, 722 (1932).
38. LJ1 s е n J., М a i s п е г Н. (Отравление ванадиевых катализаторов мышья-
9Q к°М В пР°ИЗводстве серной кислоты), Ind. Eng. Chem., 29, 254—267 (1937).
Боресков Г. К., С о ко л о в а Т. И., Применение углистых колчеда-
нов в контактном производстве серной кислоты с ванадиевыми катали-
заторами, ЖХП № 6, 18 (1934).
’ .J ? ке . V* (Производство серной кислоты контактным способом),
Metall und Erz 20 (1937).
31. Боресков Г. К., Физико-химичесгий метод расчета контактных ап-
паратов, сборник „Реконструкция сернокислотной и серной промышлен-
ности во втором пятилетии", Химтеоретиздат, 1933.
‘ нииР5Оз,бЖХП №П9тп'Ь(1938)ТеМПераТУРа К0нтакт11Р0ВаШ1я п₽н "олуче-
33. Боресков Г. К., Слинько М. Г., Расчет трубчатых контактных
аппаратов для окисления SO,, ЖХП № 4, 221; № 5, 287 (1936).
309
34. Me David (Давление паров дымящей серной кислоты и их приложе-
ние к поглощению серного ангидрида),! Soc. Chem. Ind., 57—61 (1924).
35. Smits A., Schon maker P. (Свойства серного ангидрида), J. Chem.
Soc. 1108, 1603, London (1926).
36. Remy H. (Абсорбция химических туманов), Ztschr. f. ang. Chem., 147 (1926).
37. Remy H., Finnern H. (Улавливание химических туманов жидкими
и твердыми веществами), Ztschr. f. anorg. allg. Chem. 13, 159, 241 (1927).
38. Remy H. (Поглощение тумана SO3), Chem.-Ztg. 677, 698 (1928).
39. Зике, Контактный или камерный способ, ЖХП № 20, 1251 (1936).
40. Л у к ь я н о в П. М., Производство серной кислоты методом мокрого
катализа, ЖХП № 17—18, 1236 (1937).
41. А да Дуров И. Е., Новейший метод производства серной кислоты
контактным способом на влажном газе, ЖХП № 14, 1000 (1937); ЖХП
№ 15, 1083 (1937).
42. А д а д у р о в И. Е., Контактное производство серной кислоты в Союзе
за 20 лет, ЖПХ X, вып. 10—11 (1937).
43. Kroger С. (Новейшие сведения о контактном процессе получения
серной кислоты), Chem.-Ztg., 61, № 86/87, 853 и № 90, 888 (1937).
44. Турхан Э. Я., Юдина В. И., Скорость абсорбции SO3 серной кисло-
той, ЖХП № 12, 12 (1938).
45. С о с и о в с к и й Н. П., Сернокислотная промышленность США. Произ-
водство серной кислоты из серы методом контактного окисления,
ЖХП № 2, 56 (1939).
46. Кузьминых И. Н. и Сигов С. А., К кинетике абсорбции SO3 моно-
гидратом и олеумом, ЖПХ XI, 12 (1938); XII, 9 (1939).
47. А м е л н я А. Г., А а р он о в А. Г., Производство серной кислоты контакт-
ным способом, Госхимиздат, 1939.
48. Сосновский Н. П., Тимко Я. М, Аппаратура производства серной
кислоты контактным способом, ОНТИ, 1934.
49. Амелин А. Г., Конденсация серной кислоты, ЖХП № 10, 14 (1940).
ГЛАВА 5
ПРОИЗВОДСТВО СЕРНОЙ КИСЛОТЫ
ПРИ ПОМОЩИ ОКИСЛОВ АЗОТА
I. Принципиальная схема башенного и камерного
способов
Производство серной кислоты при помощи окислов азота
(нитрозный метод) осуществляется двумя способами: камерным
и башенным.
1. Камерный способ
Принципиальная схема производства серной кислоты камерным
способом изображена на рис. 148. В башне Гловера навстречу
обжиговым газам течет по насадке нитроза — серная кислота,
Рис. 148. Схема производства серной кислоты камерным способом.
в которой растворены окислы азота. Под действием высокой
температуры газов (поступающих в башню при 280—400° и вы-
ходящих из нее при 90—120е) и содержащегося в них SO2
окислы азота выделяются из нитрозы и поступают вместе с га-
зами в камеры. В камерах SO2 окисляется за счет содержаще-
гося в газах кислорода и превращается в серную кислоту. При
этом кислород окисляет SO2 не прямо, а через промежуточные
реакции, в которых участвуют окислы азота. Окислы азота по-
ступают в башню Гловера с орошающей ее нитрозой (идущей из
311
гей-люссаков) в степени окисления, соответствующей N2O13.
К моменту выхода газов из последней камеры не только закан-
чивается окисление SOa, но и окислы азота опять достигают сте-
пени окисления, соответствующей N2O3. В башне Гей-Люссака
окислы азота поглощаются серной кислотой, которая после де-
нитрации в башне Гловера прошла через холодильник. Процесс
окисления SO2, происходящий в основном в камерах, начинается
уже в башне Гловера.
, 2. Башенный способ
В башенных системах (рис. 149) газы последовательно прохо-
дят через ряд башен. Первые по ходу газа башни орошаются
нитрозой, содержащей до 6% окислов азота, считая на HNO?.
Рис. 149. Схема производства серной кислоты башенным способом.
В первых башнях, где содержание SO2 -в газах значительно, ни-
троза теряет окислы азота, которые идут с газами в последую-
щие башни. Прежде чем полностью закончится окисление SO2J
начинается обратное поглощение окислов азота серной кислотой.
В следующих баШнях заканчивается поглощение окислов азота.
Часть денитрированной кислоты по выходе из первых башен пе-
редается на последние башни, часть кислоты из последних ба-
шен (после увеличения содержания окислов азота в них) воз-
вращается в первые башни. Таким образом удается поддержи-
вать постоянную нитрозность кислоты, орошающей каждую
башню.
Все башни, в которых происходит переработка SO2, называ-
ются продукционными. Первые из них по ходу газа,
в которых происходит денитрация нитрозы, называются также
гловерами. Последнюю продукционную башню, в которой
начинается уже обратное поглощение окислов азота, часто назы-
312
вают стабилизатором. Все башни, поглощающие окислы
азота (стоящие после последней продукционной), называются
абсорбционными и г е й-л ю с с а к а м и.
3. Краткие исторические сведения
* Производство серной кислоты при помощи окислов азота является зна-
чительно более старым методом, чем контактный.
Начало нитрозного метода было положено в 1666 г. Николаем Лемери.
который предложил прибавлять селитру к сжигаемой сере. Первый неболь-
шой камерный завод был построен в 1740 г. вблизи Лондона. Основной
аппарат этого завода состоял из стеклянного баллона емкостью в 300 л-
От стеклянного баллона перешли к кубообразным камерам. В 1805 г.
в Эдинбурге работала установка, состоящая из 360 камер емкостью до 6 м3
каждая.
Окислы азота, сыгравшие свою роль в окислении сернистого газа,
в первых установках выбрасывались в атмосферу с хвостовыми газами.
В 1827 г. Гей-Люссак предложил поставить вслед за камерами орошаемую
серной кислотой насаженную (коксом) башню для улавливания окислов
азота. Башни камерных и башенных систем, выполняющие эту роль, носят
и теперь имя этого ученого. В 1859 г. по предложению Гловера начали
устанавливать перед камерами башню, в которой кислота, приходящая из
гей-люссака, освобождалась от окислов азота. Таким образом эта башня,
получившая имя Гловера, стала аппаратом, снабжающим камеры окислами:
азота. -
Этим было закончено создание классической камерной системы. Пре-
вращение камерной системы в башенную шло вытеснением камер насажен-
ными и орошаемыми нитрозой башнями.
Опыт показал, что башня Гловера играет роль не только дснитратора,
но и реакционного аппарата (перерабатывающего SO2) более интенсивного,
чем камеры. Стали увеличивать объем башен Гловера в процентном отно-
шении к объему всей системы. Это в свою очередь вызвало необходимость
увеличивать объем гей-люссаков.
На пути превращения камерной системы в башенную весьма эффектив-
ным оказалось введение Петерсеном в 1905 г. в камерную систему проме-
жуточной башни, так называемой башни-регулятора, для переработки газов
с непостоянной концентрацией SOa. Эта башня была поставлена между
последней камерой и первой башней Гей-Люссака и орошалась крепкой
нитрозой «сама на себя» (т. е. вытекающая из башни кислота опять шла
на эту башню). В зависимости от состава и количества поступающих в си-
стему газов в ней или заканчивалось окисление SOs или происходила
абсорбция окислов азота.
Первая система без камер (собственно башенная система) была по-
строена Оплем в 1907 г. Первая башенная система Петерсена построена
в 1923 г.
Техническое развитие производства серной кислоты шло в связи с воз-
растающим спросом на серную кислоту других отраслей промышленности
и развивающимся сельским хозяйством. Развитие башенных систем стало
возможным лишь • на основе успехов промышленности машиностроения и
строительных материалов. Ярким примером является замена пульзометров
плунжерными и центробежными насосами. Башенные системы при необходи-
мости перекачивать огромные количества кислоты в условиях работы с пуль-
зометрами, коэфициент полезного действия которых составляет ~ 5%, явно
не могли быть рентабельными; они тогда лишь оказались в состоянии кон-
курировать с камерными, когда кислоту стало возможным перекачивать
мощными плунжерными и центробежными насосами с высоким коэфициентом
полезного действия.
В башенных системах, возникших на основе камерных, долгое время,
по примеру камерных систем, применяли свинец. Этот материал, хорошо
работающий в условиях камерных систем (слабая камерная кислота, малан-
313-
ши резкость кислоты), чрезвычайно нестоек в условиях башенных систем
(крепкая кислота, высокая нитрозность). Пользование свинцом как основным
материалом при выполнении аппаратуры башенных систем осложняло и
тормозило работы по их интенсификации.
II. Физико-химические основы нитрозного метода
1. Основные процессы и химические теории нитрозного метода
В камерных и башенных системах происходят следующие
основные процессы:
1) Выделение окислов азота из нитрозы (в гловерах камер-
ных систем и первых продукционных башнях башенных систем).
2) Окисление SO2 до серной кислоты (в гловерах и камерах
камерных систем и продукционных башнях башенных систем).
3) Абсорбция окислов азота серной кислотой (в гей-люссаках
камерных и башенных систем).
4) Обратное окисление окислов азота, восстанавливаемых до
NO при окислении SO2. Этот процесс происходит во всех баш-
нях и камерах, поскольку в газах имеется кислород. Однако ви-
димое окисление NO (т. е. повышение общей степени окисления
окислов азота) начинается только после того, как в основном
заканчивается окисление SO2.
Процесс выделения окислов азота из нитрозы (денитрации
нитрозы) состоит из двух частей. Окислы азота освобождаются
из нитрозы в гловерах и продукционных башнях частично вслед-
ствие увеличения упругости окислов азота при повышении тем-
пературы нитрозы. Это есть процесс, обратный абсорбции —
эксорбция. В результате этого процесса окислы азота перехо-
дят из нитрозы в газы в степени окисления, соответствующей
N2O3.
Вторая часть процесса денитрации связана с кислотообразо-
ванием в нитрозе. Поступающий в нитрозу SO2, окисляясь там
окислами азота, восстанавливает их до NO. При этом вся или
часть NO переходит в газовую фазу.
В тех случаях, когда в первую башню башенной системы по-
дается мало воды (а также в гловере камерной системы), проис-
ходит процесс концентрирования проходящей через башню кис-
лоты за счет теплоты обжиговых газов и за счет образования
H2SO4 при окислении SO2.
Окисление NO происходит в газах по реакции: :
2NO4-O2 = 2NO2.
Повидимому, окисление NO происходит также и в жидкой
фазе (или на границе раздела фаз) абсорбирующимся кислоро-
дом.
Как это будет видно ниже, образование недиссоциирован-
ных N‘_>Ol3 и N2O4 в условиях камерно-башенных систем хотя и
происходит, но в очень незначительных масштабах.
Абсорбция окислов азота серной кислотой, если в газах нет
514
избытка NO2 (против стехиометрической смеси NO + ЫОг),
происходит по реакции:
.ОН
NO2 4- NO + 2H2SO4 2SO2< + Н2О
‘ XONO
Та часть процесса денитрации, которая связана с эксорбцией
окислов азота из нитрозы, происходит по обратной реакции.
На химизм самого процесса кислотообразования среди хими-
ков и технологов-сернокислотчиков нет до сих пор единого
взгляда. Неоднократно проводившимися расчетами установлено,
что в продукционных башнях SO2 перерабатывается значительно
больше, чем могло бы перерабатываться, если бы кислород по-
ступал на окисление SO2 исключительно через стадию окисле-
ния NO в газовой фазе по реакции 2NO + Ог = 2NC>2. В связи
с этим большинство технологов считает, что реакция окисления
SO2 в башнях, орошаемых • нитрозой, в основном протекает
в жидкой фазе, где также происходит и обратное окисление NO.
Теория Лунге-Берля, обновленная за последние годы на осно-
вании работ Берля, сводит процесс кислотообразования к реак-
циям, приведенным в табл. 66.
Таблица 66
Фаза Реакции
В газовой фазе 2NO + О2 = 2NO2
На плоскости разде- ла фаз so2+h2o = h2so8 H2SO3 + no2 = so6nh2 4 SO6NH2 + O2 (2NO2) = 4SO5NH + 2H2O (2NO)
В жидкой фазе SO5NH2£H2SO4+NO
На плоскости раздела фаз 2SO5NH + SO2 + 2 H2O £ 2SO5NH2 + H2SO4
В жидкой фазе 2SO6NH + H2O £ 2H2SO4 + NO + NO2 SO6NH + HNO3 £ H2SO4 4- N2O4
Схема реакций по Мюллеру приведена в табл. 67.
Приведем, наконец; схему реакций, даваемых Чего (табл. 68).
Теории Мюллера и Чего, принимающие передачу кислорода
только в газовой фазе через реакцию окисления NO до NO2,
безусловно неудовлетворительно объясняют процесс кислотооб-
разования.
С этой точки зрения теория Лунге-Берля, допускающая фик-
сацию кислорода нитрозой, имеет преимущества.
315
Таблица 67
Характер реакции и фаза, в которой она происходит Реакции
Газовая реакция 2NO + О, = 2NO-2
Реакция растворения (на поверхности раздела фаз) a) SO2 + Н2О = H,SO:( б) NO + NO2 + 2H-,SO4 = 2SO6NH + Н2О в) NO + NO2 + Н2О = 2HNO2
Реакция гидролиза (жид- кая фаза) SO6NH + H>O = H,SO4 + hno2
Окислительная реакция (жидкая фаза) H2SO3 + 2HNO2 = H2SO4 + 2NO H2O
Таблица 68
Фаза Реакции
Газовая 2NO + О2 = 2NO2
Поверхность раздела фаз (абсорбция) NaO3 + Н2О = 2HNO, so2 + Н2О = H2SO3"
Жидкая фаза 2HNO» + H,SO3 = H,SO4 4- 2NO
Десорбция Офаств.) ^^(ras)
Рядом исследователей было отмечено отставание процесса
десорбции NO от процесса абсорбции ЭО2 нитрозой. Так, боль-
шой экспериментальный материал по этому вопросу дал Лопатто.
В табл. 69 приведены подтверждающие этот же факт дан-
ные Малина, Иткиной и Успенской о количестве выделяющихся
из нитрозы окислов азота при абсорбции ею SOa (из бескисло-
родных газов).
Опыты, результаты которых приведены в табл. 68, проводились в оро-
шаемой трубе при температуре 90°, газа подавалось 1 л/мин, а нитрозы
90 см^мин.
Нитроза, обработанная высококонцентрированным SO2, по
внешнему виду совершенно не похожа на нитрозу, не подверг-
316
Таблица 69
% в нитрозе SO2 в °/0 Перерабо- тано SO2 в % Окислы азота в от- ходящих газах в °/0
H-,SO4 N2O3, считая на HNO3 при входе в колонку при выходе из колонки
76,4 1,0 10,22 7,77 24,0 0,49
83,4 1,0 10,27 8,42 18,1 0,131
93,0 1,03 10,25 8,22 19,8 0,048
76,4 0,98 40,50 30,6 24,5 1,60
нувшуюся обработке SOa. Вместо прозрачной бесцветной жид-
кости получается молочно-фиолетовая жидкость, пронизанная
мельчайшими пузырьками газа. Только через длительное время
нитроза приобретает тот вид, который она имела до обработки
сернистым газом. Указанные же авторы подвергали две одина-
ковые пробы нитрозы после обработки ее SO2 параллельному
(при прочих равных условиях) продуванию — одну кислородом,
а другую азотом. Полученные результаты приведены в табл. 70.
Таблица 70
Проба % окислов азота в пересчете на HNO3 Выделилось см? NO из 1000 сл3 нитрозы
до продувания после продувания
по тит- рованию КМпО4 по нит- рометру по тит- рованию КМпО4 по нит- рометру
Проба, обработанная кис- лородом Проба, обработанная азо- том 0,99 0,99 0,94 0,94 0,92 0,89 0,94 0,87 11,4 17,1
Эти данные говорят о вероятности окисления NO в нитрозе
абсорбирующимся кислородом.
Как сказано было выше, Берль считает, что окисление NO
в нитрозе происходит через предварительное образование проме-
жуточного соединения SO5NH2 — синей кислоты. Манхот
считает, что так называемая синяя кислота не является химиче-
ским соединением, а скорее есть раствор NO в серной кислоте.
Окисляется ли NO в нитрозе (абсорбирующимся кислородом)
как таковая (просто растворенная) или в составе промежуточного
соединения (синей кислоты или другого), окончательно еще не
установлено. Но во всяком случае, повидимому, кислород
317
вводится в процесс окисления SO2 не только через окисление
NO в газовой фазе, но и через окисление NO в жидкой фазе.
Поскольку, как это будет видно ниже, скорость окисления
SO2 повышается вместе с повышением степени гидролиза нитро-
зилсерной кислоты, правильнее считать, что взаимодействие SO*
происходит не с нитрозилсерной кислотой, а с продуктом ее
гидролиза — азотистой кислотой.
В связи со всем вышесказанным наиболее правдоподобными
реакциями в камерных и башенных системах являются реакции,,
приведенные в табл. 71.
Таблица 71
Характер реакции Реакции Место реакции
Десорбция 2SO5NH + Н,О = 2H.,SO4 + N3O3 Гловеры
Газовая 2NO + О2 = 2NO2 Во всей системе
Абсорбция SO2 + Н2О = H3SO3 ’ N9O3 + H.,O = 2HNO3 O2 + 2NO(raCTB.)=2NO2(paCTB.) В гловерах, ка- мерах и реак- ционных башнях
В жидкой фазе H2SO3 + 2HNO, = H0S04 + 2NO + H2O (NO2 + NO)^.) + H2O = 2NO2 То же
Десорбция NO(pacTB.) NO(ras) То же
Абсорбция NO2 + NO(pacTB} + H2O = 2HNO2 То же
Абсорбция N2O3 + 2H2SO4 = 2SO5NH + H2O Г ей-люссаки
В первых башнях происходит также реакция SO3 + НгО==
= H2SO4 — конденсация серного ангидрида, пришедшего с об-
жиговыми газами, а также образующегося при окислении SO2
в газовой фазе.
Разрешение вопроса об окончательном установлении химизма
кислотообразования в нитрозном процессе представляет задачу
для дальнейших исследовательских работ.
2. Окислы азота в газах камерных и башенных систем
В газовой фазе камерных
NO и NO'2, а следовательно,
циированных N2O3 и N2O4.
и башенных систем присутствуют
и некоторые количества недиссо-
318
Окись азота NO — бесцветный газ, под атмосферным давлением, сжи-
жается в бесцветную жидкость при —'150,2°; критическая темпера-
тура —92,9е; критическое давление 64,6 аг, точка плавления —163,7°;
1 л газа при нормальных условиях весит 1,34 г.
Азотистый ангидрид NsOa при обыкновенной температуре — газ стабиль-
ный лишь под высоким давлением. При 25° и давлении 1 ат процент не-
диссоциированного NsOs в равновесии с молекулами NO и NO* равен 10,5.
Двуокись азота NOz— темнобурый газ, NzOi—(бесцветный газ. В за-
висимости от температуры и давления газ представляет смесь NO2 и N*O«
в различных соотношениях.
Окислы азота оказывают раздражающее действие на легкие
и на верхние дыхательные пути. Они вызывают расширение со-
судов, падение кровяного давления, производят наркотизирую-
щее действие на нервную систему и общетоксическое действие
(повреждение сердечной мышцы). По существующему законода-
тельству предельно допустимая концентрация окислов азота в ра-
бочих помещениях 0,005 мг/л. Основными мероприятиями, на-
правленными против выделения окислов азота в атмосферу поме-
щения, являются: герметизация сборников нитроз, холодильни-
ков и кислотопроводов, отвод окислов азота из мест их выделе-
ния в башни, содержание в порядке всей аппаратуры башенного
отделения. Все работающие на случай появления окислов азота
в повышенной концентрации должны иметь противогазы.
На заводах, употребляющих для возмещения потерь окислов
азота азотную кислоту, необходимо соблюдать все меры предосто-
рожности при обращении с ней. Прежде всего надо учесть, что
если прольется азотная кислота на стружки, на дерево и т. п. —
может возникнуть пожар. Азотная кислота при попадании на
кожу вызывает ожоги. Поэтому подача азотной кислоты дол-
жна быть максимально механизирована. В случае, если азот-
ная кислота поступает в бутылях, последние не должны пере-
носиться на спине, а обязательно или на носилках или перево-
зиться на специальных тележках. При необходимости подъема
кверху азотная кислота должна подниматься подъемником.
В башенном отделении должно быть обращено особенно
серьезное внимание на исправность и прочность ограждений всех
площадок и переходов.
Равновесие и скорость реакции 2NO -|- Ог — 2NO2. Уже из
того факта, что реакция идет с уменьшением объема и выделе-
нием тепла (по данным Боденштейна тепловой эффект при 20°
равен 26,92 ккал), следует, что теоретический выход NO2 повы-
шается с увеличением давления и понижением температуры. Бо-
денштейном и Линднером установлена следующая зависимость
константы равновесия от температуры (изохора реакции):
2 п
’g = 1g 2 - = — 4-1,701g Г— 0,0005 Т 4- 2,839. (1)
/’no,
Степень перехода NO в NO2 в момент равновесия можно
вычислить так. Пусть до окисления в одном объеме газа было
319
2 т объемов NO и п объемов Ог. При образовании 2 тх
объемов NO2 израсходовано тх объемов кислорода.
Общий объем газовой смеси в момент равновесия будет ра-
вен 1 — тх объемов, а парциальные давления компонентов
этот же момент (при общем давлении Р) будут:
(2)
р<>=р^ &
р„0 = 2Р^(1~х). (4)
Составляя уравнение изотермы, получим:
дг _ ^no'-Pq, _ Р(1—х)2(ч —тех) s
Р~ Рйо, ~ ^(l-nzx) '
Для определения х равновесной степени окисления NO при
данной температуре и данной исходной газовой смеси необхо-
димо: а) по уравнению (1) определить величину К.р для данной
температуры и б) подставив в уравнение-(5) найденное значе-
ние Кр, определить х.
При температурах ниже 150—200° концентрация NO в равно-
весной смеси (NO2, NO, Ог) практически ничтожна; наоборот,
при температурах выше 700—800° в равновесной смеси могут
присутствовать лишь незначительные количества NO2. Так как
в камерных и башенных системах температура газов ниже 200°,
то там имеются условия для установления равновесия по реакции
2NO + Ог 2NO2. Однако это равновесие в камерных и башен-
ных системах почти никогда не достигается, так как оно сме-
щается другими параллельно идущими процессами (восстановле-
ние NO2 сернистым газом в продукционной зоне, абсорбция
стехиометрической смеси NO' 4- NO2 в абсорбционной зоне).
Так как скорость диссоциации NOa при низких температурах
незначительна, то при выводе формулы скорости окисления NO
скорость обратной реакции можно принять равной нулю.
Пусть начальные концентрации газов в молях на 1 л были:
NO......2а
О3.... b
В момент времени t после достижения степени окисления,
равной х, концентрации компонентов будут:
NO....2а(1—х)
О2... .Ь — ах
NOa ... .Чах
.320
Скорость образования NO2 в момент t будет равна: (6)
= -с at Х NO *'0.
или ^-= К 4аа (1 —л)8 (Ь — ах)
или 2Ка dt = ?. (1 — х)2 (Ь — ах) (7)
Для интегрирования выражения (7) разложим дробь на простые дроби:
1 А , В . D
(1— х)-2(6 — ах) b— ах (I—х)2' I — х '
Получаем:
1 = 4(1 — ху- + В(Ь — ax)+D(b — ах) (1 — х).
После преобразования получим:
(Л 4- Da) хз — (24 + Ba + Db + Da) х + A -f- Bb + Db = 1. (9)
Приравнивая коэфициент в левой и правой частях уравнения (9), полу-
чим систему уравнений:
А + Da = 0;
24 + Ba + Db 4- Da = 0;
4 4- Bb 4- Db = 1.
Решая систему этих уравнений, получим:
4 — °2
(Ь-ау’
а
~(Ь-ау'
Подставляя значения А. В и D в уравнение (7), получим: \
2Kat = f (1—х)2((> —йх) =
= J (b-a)- (b-ах) dx + J (b - а) (1 — х«) dx ~ J (6 —йр (1 — х) dX’
интегрируя, получим:
т - тйт+г-
При г = 0 будем иметь х = 0 и
, 1_________L_.n 1
a(b — а) (b —ay п Ь '
21 Зак. 5019. Технология серной кислоты.
321
Подставляя это значение с в уравнение (10), получим:
о д'/________х________L ' - 1п х
п а(Ь — а)\1 — х)^(Ь — ау- ах
b
(11)
Заменяя натуральный логарифм десятичным для времени
окисления NO до заданной степени, получим следующее окон-
чательное выражение:
2,303
2/С(й —«)2
х(Ь—а) , . 1 —-у
2,303а (1 — х) ‘ J _ ах
(12)
Преобразуем полученное уравнение для случая, когда кон-
станты скорости отнесены к парциальным давлениям в ат. Пусть
в начальный момент доли объема компонентов на 1 объем газо-
вой смеси равны:
NO.....2 m
О2...л
а общее давление газов Р ат. Тогда парциальные давления че-
рез t сек. будут:
PN0 = 2m(l—-х)Р;
Р0=(п—тх)Р;
р^ = 2тхР.
Подставляя в уравнение (12) вместо концентраций в молях
на 1 л парциальные давления, получим:
P22Kt
2,303 Г х (п — т) . . 1 — х
(и— т)2 2,303 т (1—х) ’ wx
п
(13)
Значение К в зависимости от температуры при выражении
концентраций компонентов в ат приводим ниже (по Боден-
штейну).
Температура Температура
в °C К в сС К
0 ............. 34,7 197..............4,24
30 ........... 21,45 241 ............ 3,38
60 ............14,65 291..............2,64
90 ........... 10,50 340.............2,16
141 ............ 6,7 389 ............ 1,83
Как видно из приведенных данных, скорость реакции окисле-
ния NO понижается с повышением температуры. Этим данная
реакция отличается от большинства гомогенных газовых реакций.
Реакция образования Na2Oi. Реакция образования Na^O* идет
по уравнению:
2NO-2 -> N2O4 + 13,6 ккал.
322
Боденштейн получил следующую зависимость константы
равновесия этой реакции от температуры:
(14)
==__2679?_|_1)75 ig 7'4-0,00483 Г—7,144-10-е 7-2^3 062.
Расчет выхода N2O4 в момент равновесия можно произвести так.
Пусть в исходной смеси имеется 1 моль N»O« и вовсе нет NO». Обо-
значим степень диссоциации №04 в момент равновесия через у. Тогда
в равновесной смеси будет:
N2O4 (1 —у’) моля ’NOj 2у моля Всего 1 +_у молей
Обозначим через Р, тогда: парциальное давление NOa и N2O4 в момент равновесия
И ^8o=4tf <15> л«.=т&- <•«>
Подставляя получим: эти значения pNo2 « Pn2o4 в УРавпение изотермы реакции, iz _ КР~ 1—J12’
откуда >= А.Я- <17)
Определим центрацня NO» уравнению (14) концентрацию NsOi в газах при 60°, если исходная кон- равиа 6%, а общее давление газовой смеси равно 1 ат. По определяем Кр равной 1,78; Р — 0,06, следовательно: у = 1 = 0.88; V ^1,78
отсюда парциальное давление N2O4 в момент равновесия равно:
^^8888)-0-0038 ^
I *|-и,оо
а парциальное давление NOs:
2 • 0,88 • 0,06
~ 1+0,88 ==0’0562 ат-
Так как в газах камерных и башенных систем общая концен-
трация окислов азота обычно ниже 6%, а степень окисления NO
редко бывает выше 50%, то возможные концентрации N2O4
21* 323
в аппаратуре камерных и башенных систем чрезвычайно ни-
чтожны.
Реакция образования ЬЬОз. В газах, где имеется NO и NO2,
происходит и реакция образования N2O3:
NO ф- NO2 72 N2O8 ф- 7,925 ккал.
Равновесие этой реакции сдвигается вправо с понижением
температуры и повышением давления и устанавливается очень
быстро. Однако при нормальном давлении и тех температурах,
которые поддерживаются в камерных и башенных системах, при
небольших парциальных давлениях окислов азота нитрозные
газы содержат значительное (по отношению к сумме NO и NO2)
количество недиссоциированной N2O3.
Если нитрозные газы находятся в контакте с раствором, аб-
сорбирующим N2O3, то при идущем в газах процессе окисления
NO все окислы азота могут перейти в раствор в степени окисле-
ния, соответствующей N2O3. Так, если нитрозные газы при
происходящем в них процессе окисления NO находятся в кон-
такте со щелочью, то в результате окислы азота могут целиком
связаться щелочью в форме нитрита. При контакте с концентри-
рованной серной кислотой окислы азота целиком могут перейти
в жидкую фазу с образованием нитрозилсерной кислоты:
/ОН
2H„SO4 ф- N2O8 = 2SO2< 4- НоО
XONO
Но если параллельно с окислением NO не происходит абсорбция
окислов азота, то после достижения максимальной концентрации
недиссоциированной N2O3 в газовой фазе (при стехиометрическом
отношении NO» и NO в газе) дальнейшее окисление NO ведет
к уменьшению концентрации недиссоциированных молекул N2O3
в газе; к концу реакции, если она протекает при температуре и
давлении, когда равновесие 2NO ф- О2 2NO2 далеко сдвинуто
вправо, в смеси практически находятся лишь молекулы NO»,
частично полимеризованные в N2O4. В условиях камерных и ба-
шенных систем степень окисления NO обычно не превышает 0,5
(т. е. степень, соответствующая N2O.4).
3. Окислы азота в кислотах камерных и башенных систем
Растворимость окислов азота в серной кислоте. Раствори-
мость NO в серной кислоте очень незначительна.
Значения для растворимости NO в серной кислоте в зависи-
мости от концентрации кислоты при 15е приведены в табл. 72
(по Тауеру).
Определением растворимости N2O3 в серной кислоте зани-
мались очень многие исследователи — Сорель, Лунге, Матсуи,
Санфурш и Рондье, Берль и Зенгер, Тихонов, Кудрявцев.
Полученные отдельными исследователями результаты значи-
324
Таблица 72
% H2SO4 Коэфициент Бунзена 1 * Ь % H2SO4 Коэфициент Бунзена1
49,4 59,5 71,0 0,0112 0,0110 0,0100 79,6 90,0 0,0109 0,0181
тельно отличаются по абсолютным величинам цифр, но принци-
пиальные выводы всех исследователей совпадают и сводятся
к следующему:
Рис. 150. Зависимость упругости
окислов азота над нитрозой от
содержания N2O3 в нитрозе.
Рис. 151. Зависимость упругости
окислов азота над нитрозой от тем-
пературы.
1) Растворимость N2O3 в растворах серной кислоты подчи-
няется закону Генри, т. е. упругость N2O3 над раствором прямо
пропорциональна концентрации N2O3 в растворе.
1 Коэфициент Бунзена — число см3 газа (приведенного к нормальным
условиям), растворенных в 1 см'" растворителя под давлением этого газа над
раствором, равным 760 мм рт. ст.
325
2) Растворимость N2O3 понижается при повышении темпе-
ратуры.
3) Растворимость N2O3 понижается с уменьшением концен-
трации H2SO4 в растворителе.
Наиболее полные данные по этому вопросу получены
Кудрявцевым.
Зависимость упругости окислов азота над раствором N2O3
в 76,7'4-ной H2SO4 от концентрации N2O3 в растворе при раз-
личных температурах (по данным Кудрявцева) графически изо-
бражена на рис. 150.
Характер графиков (прямые линии) подтверждает примени-
мость к данному случаю закона Генри (в исследованной области
температур). Такой же характер графиков получен и для всех
других исследованных указанными авторами растворов серной
кислоты.
В табл. 73 приведены рассчитанные Малиным по эксперимен-
тальным данным Кудрявцева величины коэфициента Генри для
различных температур и различных растворов H2SO4.
Таблица 73
% H»so4 Коэфициент растворимости (Н) при температуре
30° 40° 50э 60° 70 80° 90° 100° 110 120s 130s 140° 150°
71,6 0,32 0,54 0,91 1,72 2,58 3,76 5,20 7,35
73,4 0,25 0,63 1,01 1,51 2,02 2,65 3,52 4,66 6,04 7,95 9,95 13,10 16,90
76,7 0,18 0,27 0,41 0,68 1,05 1,46 2,09 3,00 3,82 4,82 5,90 7,28 9,90
82 0,048 0,097 0,13 0,16 0,26 0,39 0,60 0,84 1,21 1,71 2,25 2,87 3,52
90 0,045 0,068 0,10 0,17 0,22 0,32 0,45 0,59 0,75 1,01 1,31 1,64 2,05
Приведенные в табл. 73 величины коэфициента растворимо-
сти высчитаны по формуле:
р = Н-С,
$
(18)
где р — упругость окислов азота в мм рт. ст. над раствором
в момент равновесия, С — концентрация N2O3 в растворе в мо-
мент равновесия в весовых процентах, Н — коэфициент раство-
римости, величины которого даны в табл. 73.
Зависимость упругости М»Оз над растворами (в 73,6*Х -ной
H2SO4) от температуры для разных концентраций N2O3 в рас-
творе изображена на рис. 151.
Для других исследованных кислот получается аналогичный
характер графиков.
Если предположить, что зависимость упругости N2O3 от тем-
326
•пературы может быть выражена уравнением Клаузиуса-Клапей-
рона:
lgp = —2_+С, (19)
то график (1g р, у ) должен быть прямой линией.
На рис. 152 изображены такие графики, построенные Мали-
ным по экспериментальным данным Кудрявцева для растворов,
Рис. 152. Зависимость упругости окислов азота над
нитрозой от температуры.
содержащих 4,5$< NasOs и различный процент H2SO4 (по оси
ординат отложены величины Н из табл. 73, пропорциональные
величинам упругостей).
Как видно из рисунка, при температурах выше 60J зависи-
мость упругости окислов азота над растворами N2O3 в серной
кислоте согласуется с уравнением Клаузиуса-Клапейрона.
Вполне возможно, что отступление экспериментальных данных
327
ст этого уравнения при более низких температурах объясняется
значительными (относительными) погрешностями эксперимента.
Если исходить из того, что растворимость N2O3 в растворах
серной кислоты подчиняется законам Генри и Клаузиуса-Клапей-
рона, то можно высчитать упругость окислов азота над серной
кислотой данной концентрации при любой температуре и любой
концентрации N2O3 в растворе. Для этого необходимо иметь
лишь два опытных значения упругости.
По данным Кузнецова и Кудрявцева упругость окислов азота над
73,4%-ной H'sSOi, содержащей 4,5% МгОз, равна при 60° 6,95 мм рт. ст.,
а при 120° — 36,2 .«.« рт. ст.
Пусть требуется найти упругость NaOs над той же кислотой при 90°
и при содержании 3% NSOS в растворе. Уравнение Клаузиуса-Клапейрона
после интегрирования принимает вид:
In ----
Pi R \Tj Тг)
(20>
Подставляя числовые значения, получаем
2,303 lg 6,95 “ 1,987 (зЗЗ 393/’
откуда определяем величину Л равной 7050 кал.
Внося теперь величину К и составляя уравнения Клаузиуса-Клапейрона
для температур 90 и 120°, получаем:
2,3031g —
Р90»
7050
1,987
.363 393)
Из последнего соотношения находим: р90с = 17,5 мм рт. ст.
Для перехода от концентраций NaO3, равной 4,5%, к концентрации, рав-
ной 3%, пользуемся законом Генри и окончательно определяем искомую
упругость окислов азота:
17,5-3
р — —-Гт— = П,6 мм рт. ст.
* 4,3
По опытным данным Кудрявцева и Кузнецова эта величина равна
11,2 мм рт- ст.
Уменьшение растворимости N2O3 в растворах серной кислоты
с понижением концентрации H2SO4 объясняется обратимостью
реакции:
/ОН .ОН
2SO2< 4-N2Os^2SO2< +н2о
ХОН XONO
В 100%-ной H2SO4 нитрозилсерная кислота чрезвычайно
устойчива. С увеличением концентрации воды (не связанной)
в растворе повышается степень гидролиза SO3NH и равновесие
смещается в сторону перехода окислов азота в газовую фазу.
Степень гидролиза нитрозилсерной кислоты увеличивается также
с повышением температуры. В табл. 74 приведена зависимость
между концентрацией H2SO4 и степенью гидролиза нитрозы по
Гантшу при комнатной температуре.
328
Таблица 74
Концентрацию сериой кислоты в % H,SO4 Неразложившийся нитрозилсульфат в молярных процентах Концентрация сериой кислоты в % H2SO4 Неразложившийся нитрозилсульфат в молярных процентах
98,0 98,0 87,0 80,6
95.6 96,6 80,0 72,3
92,0 92.7 70,0 50,2
57,5 0.0
Высшие окислы азота NO2
лоте с концентрацией выше
HNO3.
и NtfOj, растворяясь в серной кис-
67% H2SO4, образуют SO5NH и
При этом в присутствии азот-
ной кислоты в нитрозе упругость
окислов азота над «нитрозой вы-
ше, чем при отсутствии азотной
кислоты (при наличии только ни-
трозилсерной кислоты), но при
неизменной общей нитрозности.
Отсюда следует, что если в ни-
трозных газах наряду с N2O3 име-
ется NO2 (т. е. в газах избыток
NO2 против NO), то раствори-
мость окислов азота ниже, чем
в том случае, когда в газах отно-
шение NO2 к NO равно единице,
так как растворение избыточного
NO2 даст наряду с нитрозилсер-
ной кислотой также и азотную
кислоту в растворе. Следова-
тельно, оптимальным составом
нитрозных газов с точки зрения
их растворимости в концентри-
рованных растворах (выше 67%
H2SO4) серной кислоты является
состав, соответствующий N/Эз.
Рис. 153 иллюстрирует (по
данным Кудрявцева влияние при-
меси HNO3 в нитрозе на упру-
гость окислов азота над ней при исходном содержании H2SO4,
равном 90/с, и при содержании N2O3, равном 4,5%.
Скорость абсорбции окислов азота растворами серной кис
лоты. Коэфициенты скорости абсорбции окислов азота в послед-
ние годы определялись Чего, Малагутти и Ломбарди (в Италии),
Кузьминых с сотрудниками (в УНИХИМ) и Малиным с сотруд-
никами (в НИУИФ).
Зависимость упругости
азота над нитрозой от
Рис. 153.
окислов
содержания HNO3 в нитрозе.
329-
На основе этих работ можно сделать следующие выводы:
1. Для N2O3 коэфициент скорости абсорбции концентрирован-
ными растворами серной кислоты
Таблица 75
0/с H.,SO4 к- юг
NO., N2O3
95 8,6 15,70
80 6,7 14,70
60 5,57 13,00
40 5,39 —
33,6 — 11,12
20 11,27
16,8 — 10,55
Н,0 21,10 13,40
выше, чем для NO2. В табл. 75
приводим данные Чего и др.
Коэфициент вычислялся по
формуле
Х = (21)
Z Ру
2. Коэфициент скорости аб-
сорбции (N2O3) повышается
с ростом концентрации H2SO4
в растворе (доказано опытами
всех вышеупомянутых иссле-
дователей). Приводим данные
Малина, Кельман и Успенской
(табл. 76), рассчитанные по
формуле
g
Hi рА'
(22)
где g — количество абсорбированного N2O3 в / в час, Др — сред-
няя логарифмическая движущая сила скорости абсорбции в мм
рт. ст., А — поверхность контакта фаз в м'2.
Указанные в табл. 76 величины получены .при 30е в опытах с орошаемой
трубкой (внутренний диаметр 2 с.к, длина 20 с.к); подавалось в минуту
“аза 1 л, кислоты—ЛОО с.и3 4).
Таблица 76 * Таблица 77
% H2SO4 К
76,6 18,7
86,05 19,8
88,20 19,8
90.02 20,2
92,70 21,5
94,4 21,9
Линейная ско- рость газа в см!сек 1 р
15 20 40 80 100 1,38 1,79 2,56 2,51 2,56
3. Коэфициент скорости абсорбции N*O« серной кислотой
уменьшается с повышением температуры. Это подтверждается
данными Кузьминых (рис. 154). Коэфициент высчитывался по
только что приведенной формуле.
4. Рис. 155 иллюстрирует (по данным Кузьминых) зависи-.
яость коэфициента скорости абсорбции Ыаз'Оз серной кислоты от
линейной скорости газа. Как показали более поздние опыты Ма-
лина и Добровольской (табл. 77), эта зависимость (которая мо-
330
жет быть выражена формулой К — С • v °’8, где v — линейная
скорость газа) справедлива лишь в том случае, когда основным
сопротивлением диффузии является сопротивление газовой
пленки.
Приведенные в табл. 77 данные получены в опытах с орошаемой труб-
кой (внутренний диаметр 1 с.и, длина 20 с.и, опыты велись с 76%-ной H2SO4
при 30е. орошающей кислоты подавалось 100 с.м3 в минуту).
Последний факт был позднее подтвержден опытами Кузьми-
ных, Юшманова и Апахо-
ва на опытной башне.
Рис. 155. Зависимость коэфициента ско-
рости абсорбции NaO3 серной кислотой
от линейной скорости газа.
Рис. 154. Зависимость коэфи-
циента скорости абсорбции N2O3
серной кислотой (70% H2SO4)
от температуры.
Эти же опыты показали, что коэфициент скорости абсорбции
Рис. 156. Влияние скорости газа иа коэфициент скорости
абсорбции для различных насадок (на 1 м2 поверхности
насадки).
что различные типы насадок создают разную степень турбулент-
ности газового потока и разную степень перемешивания газа
с жидкостью.
331
На рис. 156 представлена по результатам указанных опытов
зависимость коэфициента скорости абсорбции N2O3 от линейной
скорости газа, а на рис. 157 — от плотности орошения для раз-
личных типов насадок.
Рис. 157. Влияние плотности орошения иа коэфц-
циеит скорости абсорбции для различных насадок
(иа 1 м2 поверхности насадки).
Как и следует из только что сказанного, коэфициент абсорб-
ции кварцевой насадки выше, чем кольцевой насадки, однако,
если отнести коэфициент скорости абсорбции к 1 м3 насадки
(т. е. если взять произведение коэфициента скорости абсорбции,
Рис. 158. Влияние линейной скорости газа на коэфициеи! ско-
рости абсорбции для различных насадок (иа 1 м3 насадки).
отнесенного к 1 м'2, на удельную поверхность насадки), то он бу-
дет выше для колец Рашига, чем для кварца. Это видно из
рис. 158.
5. В последние годы в литературе было много споров о том,
каким методом подсчитывать величину движущей силы скорости
абсорбции N2O3. Некоторые предполагали, что величина движу-
332
щей силы скорости абсорбции N2O3 определяется концентрацией
недиссоциированных молекул N2O3; учитывая, что последняя
пропорциональна произведению концентраций NO и NO», они
утверждали, что движущая сила скорости абсорбции окислов
азота серной кислотой также пропорциональна произведению
концентраций NO и NO2 в газовой фазе.
Как показали опыты Кузьминых, а затем Малина, Кельман
и Успенской (табл. 78), движущая сила скорости абсорбции N»O.t
безнитрозной серной кислотой определяется только концентра-
цией стехиометрической смеси NO и NO2.
Таблица 78
°/0 окислов азота в газах перед абсорбцией Коэфициенты
N,O3 (эквивалентная смесь) NO + NO2 Избыточное NO (сверх N3OS) Кт К2
2,19 2,32 0,13 19,20 6,35
2,21 2,87 0,66 18,90 3,79
2,21 2,96 0,75 19,30 3,01
2,39 3,86 1.47 19,10 2,28
2,34 4,35 2,01 19,00 1,72 л
Коэфициент скорости абсорбции высчитывался по формуле
где g —г N2O3 в час, Др — средняя логарифмическая движу-
щая сила скорости абсорбции в мм рт. ст., А — поверхность кон-
такта фаз в м2.
Значения Kt высчитаны, исходя из предположения, что дви-
жущая сила определяется только концентрацией стехиометриче-
ской смеси NO и NO», значения Кг — из предположения, что
движущая сила определяется произведением концентраций NO
и NOo.
Из табл. 78 ясно, что избыточная (против стехиометрической
смеси NO и NO2) NO на движущую силу скорости абсорбции
N2O3 не влияет.
Из всех вышеприведенных данных о растворимости и ско-
рости абсорбции окислов азота серной кислотой следует, что
оптимальным составом нитрозных газов и с точки зрения пол-
ноты и с точки зрения скорости поглощения (коэфициент ско-
рости абсорбции, движущая сила скорости абсорбции) является
стехиометрическая смесь NO и NO2.
„Абсорбция окислов азота серной кислотой состоит по край-
си мере из ^следующих последовательных процессов: диффу-
333
зия N2O3 через газовую пленку, диффузия N2O3 через жидкую
пленку и химическая реакция (образования нитрозилсерной кис-
лоты) в жидкой фазе. В зависимости от конкретных условий
тот или иной из вышеуказанных трех процессов ограничивает
весь процесс абсорбции.
Эксорбция окислов азота из нитрозы. Процесс эксорбции
окислов азота из нитрозы, т. е. та часть процесса денитрации,
которая происходит вследствие превышения упругости окислов
азота над нитрозой против парциального давления окислов азота
в газах, состоит из следующих трех идущих один за другим
процессов: гидролиз нитрозилсерной кислоты, диффузия N2O3
через пограничную жидкую пленку и диффузия N2O3 через по-
граничную газовую пленку.
В зависимости от конкретных условий (температура, содер-
жание H2SO4 и N2O3 в нитрозе) скорость эксорбции зависит
или от скорости диффузии N2O3 через газовую пленку или от
скорости процесса гидролиза. Диффузия N2O3 через погранич-
ную жидкую пленку не оказывает влияния на скорость эксорб-
ции окислов азота.
4. Зависимость скорости кислотообразования
от различных факторов
Так как химизм и механизм кислотообразования в нитрозном
процессе <очно не установлены, то сейчас не представляется
возможным вывести зависимость скорости кислотообразования
от различных факторов на основе данных о кинетике отдельных
слагающих процессов.
Зависимость скорости кислотообразования (как суммарного
процесса) от различных факторов многократно изучалась экспе-
риментально. Из работ этого рода известны работы женевских
исследователей Биттерли и Россиньоля, итальянца Чего, Берля
с сотрудниками в Германии, Уральского научно-исследователь-
ного института химии (УНИХИМ), Института удобрений и ин-
сектофунгицидов (НИУИФ), Московского химико-технологиче-
ского института им. Менделеева (МХТИ).
Выяснению характера влияния отдельных факторов на ско-
рость кислотообразования в башенном процессе способствовали
также проводившиеся в МХТИ (Шульцем) и НИУИФ заводские
работы по интенсификации башенных систем, а также производ-
ственные данные башенных заводов, работающих с высокой ин-
тенсивностью. Особенно в связи с этим необходимо отметить,
что один из наших заводов значительно превысил проектную
интенсивность на 2—3 года раньше других заводов.
По причинам сложности нитрозного процесса, трудности его
экспериментального исследования, различных условий опытов
в работах отдельных исследователей имеются противоречия даже
в этом фактическом экспериментальном материале. Однако ряд
выводов о характере влияния различных факторов на скорость
334
кислотообразования, достаточно надежных в определенных гра-
ницах, на основе имеющихся экспериментальных данных можно
сделать.
Скорость кислотообразования в камерах. Применительно
к условиям камерных систем в лабораторных условиях процесс
кислотообразования наиболее полно был подвергнут изучению
Биттерли, Россиньолем и Кузьминых. Из опытов всех этих ис-
следователей можно сделать заключение, что при полном отсут-
ствии паров воды реакция между SO2 и NO2 или совсем или
почти совсем не происходит.
Скорость окисления SO2 повышается с ростом концентраций
SO2 и окислов азота в газовой смеси.
Скорость кислотообразования по данным Биттерли и Кузьми-
ных пропорциональна первой степени концентрации NO2 в газе,
а по данным Россиньоля пропорциональна концентрации окислов
азота в газах в степени, большей единицы.
В опытах Биттерли газовая смесь пребывала в реакционном
сосуде очень незначительное время. Скорость кислотообразова-
ния оценивалась по количеству образующейся в реакционном со-
суде H2SO4. Таким образом ’ Биттерли фактически изучал ско-
рость взаимодействия между SO2 и NO2 в начальный момент, и
так как начальная концентрация NO2 при этом была больше,
чем SO», то скорость окисления SO2 практически не находилась
в зависимости от скорости обратного окисления NO. Кузьминых
изучал скорость окисления SO2, пропуская через реакционный
сосуд смесь SO2 и NO2, разбавленную азотом. Совершенно по-
нятно, что в этих условиях на скорость окисления SO2 могла
влиять в каждый данный момент только та часть окислов азота,
которая еще оставалась в виде NO2. В опытах Россиньоля на-
чальная концентрация NO2 в газовой смеси, как правило, была
значительно меньше, чем начальная концентрация SO»; таким
образом скорость окисления SO2 находилась в зависимости от
скорости обратного окисления NO. В камерных системах, где
концентрация окислов азота в газах составляет 0,7—1,5^, ско-
рость окисления SO2 в значительной степени зависит от скорости
обратного окисления NO.
В табл. 79 приведены данные Россиньоля о влиянии концен-
трации окислов азота в газовой фазе на скорость кислотообра-
зования в первой камере его установки.
В табл. 80 приведены данные Матсуи (обследовавшего ряд
заводов) о влиянии концентрации окислов азота в газах на ин-
тенсивность работы камер.
Все эти данные позволяют сделать следующее заключение
о характере влияния концентрации окислов азота в газах на ско-
рость кислотообразования в условиях камерных систем.
1. Если концентрация NO» велика по сравнению с концен-
трацией SO2, то скорость процесса переработки SO2 не зависит
°т реакции обратного окисления NO, определяется исключи-
тельно реакцией взаимодействия между SO2 и NO2 и пропор-
335
Таблица 79
№ опытов % окислов азота в газах Отношение концен- трации окислов азота к величине концентрации в первом опыте С мтиишение скорости кислотообразования к скорости кнслото- образования в пер- вом опыте
1 2 3 4 5 6 0,50 1,23 2,46 3,69 4,27 4,92 1 2,46 5,92 7,38 8,54 9,84 1 4,25 9,93 11.4 23,6 47,2
Таблица 80
Количество NaNO3 в циркуляции в °/0 к сере Интенсивность системы в кг]м^ в сутки Рост концентрации окислов азота по отношению к первому примеру Рост интенсивности по отношению к первому примеру
12 13,5 14,5 15,8 18,6 19,7 4 4,5 5,5 6,5 7,2 8,8 1 1,125 1,20 1,31 1,55 1,64 1 1,13 1,37 1,62 1,80 2,20
циональна концентрации NO2. В начальный момент, когда NO
в газах еще очень мало, скорость переработки SO2 практически
пропорциональна общей концентрации окислов азота в газах.
2. В бескислородной газовой смеси скорость переработки
SO2 на всем протяжении процесса прямо пропорциональна налич-
ной концентрации NO2.
3. В газовой смеси, содержащей кислород, скорость окисле-
ния SO2 будет тем сильнее зависеть от реакции обратного окис-
ления NO, чем меньше отношение концентрации окислов азота
к SO2 в газах. В пределе, при весьма малой величине этого
отношения, скорость окисления SO2 целиком определяется ско-
ростью реакции 2NO О» = 2NO2 и прямо пропорциональна
квадрату концентрации окислов азота в газах.
4. В производственных условиях камерных систем скорость
.переработки SO2 пропорциональна концентрации окислов азота
в газах в степени выше, чем первая, но ниже, чем вторая.
По данным Кузьминых скорость кислотообразования в усло-
виях камерных систем пропорциональна также концентрации SO2
в газах.' По данным Биттерли повышение концентрации SOa
только в определенных границах увеличивает скорость перера-
ботки SO2. Однако было бы неправильным полагать, что концен-
трация SO2 имеет какой-то постоянный для различных условий
оптимум. Оптимум концентрации SO2 зависит от всех других
конкретных условий прохождения процесса. С повышением кон-
центрации SO2 в определенных условиях увеличивается ско-
рость переработки SO2 до тех пор, пока скорость общего про-
цесса переработки определяется реакцией взаимодействия между
SO» и NO».
Если в данных конкретных условиях процесс кислотообразо-
вания зависит от реакции обратного окисления NO, то на ско-
336
рость процесса кислотообразования оказывает влияние и концен-
трация кислорода в газах.
По данным Кузьминых скорость взаимодействия между SO2 и
NO2 повышается с ростом температуры (в изученных пределах
от 65 до 150° константа ско-
рости повышается с 0,128 до
0,372). Биттерли пришел к
заключению, что имеется
температурный оптимум, ко-
торый в его опытах оказался
равным 90е. В действитель-
ности очевидно, что темпе-
ратурный оптимум не являет-
ся постоянным и зависит от
всех других конкретных
условий процесса.
Скорость кислотообразо-
вания в условиях башенного
процесса. Вопрос о скорости
кислотообразования приме-
-----* % НЯ03, б нитрозе
Рис. 159. Зависимость коэфициента ско-
рости абсорбции SO2 нитрозой от со-
держания окислов азота в нитрозе.
Условия опыта: скорость газа 1025 см^мин; кис-
лоты 9J смъ'мин; H2SO4 в орошающей кислоте
температура опыта 90°; SOa во входящем
газе 10°/о.
нительно к условиям башенного процесса в последние годы
изучался рядом исследователей. Очень интересные работы в этом
направлении проведены Лопатто, а также Берлем, Гиллебранд-
том и Виннаскером.
Рис. 160. Зависимость коэфициента
скорости абсорбции SO2 нитрозой
от плотности орошения.
Условия опыта: скорость газа 1025 смЧмин-,
H2SO4 в орошающей кислоте 76,5%; HNO
в орошающей кислоте температура
опыта 90°; SO2 во входящей газе 1и»'„.
Рис. 161. Зависимость коэфициента
скорости абсорбции SO2 нитрозой
от скорости газа.
Условия опыта: кислоты 90 слА/мин', H2SO4
в орошающей кислоте 76°JO; температура
опыта 90°; SO2 во входящем газе 10»'о.
Повидимому, процесс окисления SO2 нитрозой слагается из
идущих один за другим процессов: диффузии SO2 через газо-
вую пленку, диффузии SO2 через жидкую пленку и окисления
SO2 в жидкой фазе. Скорость процесса кислотообразования
22 Зак. 5019. Технология серной кислоты. 007
*" % 50? В газе
Рис. 162. Зависимость коэфициента
скорости абсорбции SO2 нитрозой
от концентрации SO2 в газах.
Условия опыта: скорость газа 1000 см3!-
ман; кислоты 90 смг/мин', H2SO4 в оро-
шающей кислоте 76о/о; HNO3 в орошаю-
щей кислоте 1»,0; температура опыта 90°.
в данных конкретных условиях зависит, главным образом, от
факторов, влияющих на тот процесс, который ограничивает про-
цесс кислотообразования в целом.
На рис. 159, 160 и 161 приве-
дены результаты опытов Малина,
Иткиной и Успенской по изуче-
нию зависимости скорости абсорб-
ции SO2 нитрозой от различных
факторов. Все опыты проводи-
лись в орошаемой трубке (вну-
тренний диаметр 2 см, длина
20 см). На рис. 159 и других
нитрозность показана условно
в % HNO3. В применяемой в
опытах нитрозе окислы азота со-
держались в степени окисления,
соответствующей N2O3.
Как показывают эти резуль-
таты, каждый такой фактор, как
нитрозность кислоты, плотность
орошения и скорость газа, изме-
няемый в отдельности при неиз-
менности других факторов, по-
вышает скорость процесса только в определенных границах.
Из рис. 162 видно, что (при достаточном количестве окислов
азота в нитрозе) пропорциональность между скоростью абсорбции
Рис. 163. Зависимость коэфициента скорости абсорбции SO2 нитрозой от со-
держания H2SO4 в нитрозе.
Условия опыта: скорость газа 1000 см’/мин-, кислоты 90 cms/muh; HNOS в орошающей кислоте
1°,ч; температура опыта 90°.
SO2 нитрозой и концентрацией SO2 в газах сохраняется, по край-
ней мере, до 50% SO2 в газах.
Работы всех вышеупомянутых исследователей показывают,
338
4во
440
400
360
ё зго
п0
ЙМ
5 гоо
160
/го
во
40
Рис. 164. Зависимость
скорости кислотооб-
разования от темпе-
ратуры.
башенных систем,
Рис. 165. Влияние концентрации кисло-
рода в газах на количество выдуваемых
из нитрозы окислов азота.
Условия опыта: скорость газа 1000 сзг’/логи; кис-
лоты 90 см^/мпн, H2SO4 в орошающей кислоте
7№jo; HNO, в орошающей кислоте 1о;о; темпера-
тура опыта 90°; SO2 во входящем газе Ю'.'о.
что в некоторых границах скорость кислотообразования повы-
шается с понижением концентрации нитрозы по H2SO4.
Влияние этого фактора наиболее полно изучено Малиным и
Иткиной. На рис. 163 графически изобра-
жена зависимость скорости абсорбции SO2
нитрозой от концентрации H2SO4 в нитрозе.
Повидимому, точка максимума кривой при-
ходится на 57,6%-ную H2SO4, соответствую-
щую гидрату SO3 • 5НгО. Такой характер
кривой можно объяснить следующим:
мере уменьшения концентрации H2SO4 в ни-
трозе повышается доля окислов азота, на-
ходящихся в растворе в форме HNO2, при <S
концентрации H2SO4, равной 57,6%, SO5NH г"
разлагается полностью. При дальнейшем
уменьшении концентрации H2SO4 доля окис-
лов азота, находящихся в HNOa, умень-
шается за счет увеличения части окислов
азота, существующих в виде HNO3.
Как видно из рис. 163, минимум ско-
рости абсорбции SO2 приходится на нитрозу
с содержанием 84% H2SO4, соответствую-
щую гидрату SOs • 2Н2О.
В отношении характера влияния темпе-
ратуры на скорость кислотообразования при
взаимодействии SO2 с нитрозой в условиях
повидимому, наиболее правильны выводы Кузьминых и Лопатто,
по данным которых скорость
кислотообразования возра-
стает с повышением темпе-
ратуры.
На рис. 164 показано
влияние температуры на ско-
рость процесса кислотообра-
зования (по данным Кузьми-
ных и Суркова). На приве-
денных графиках, как по
опытам Кузьминых, так и по
опытам Малина, величины
интенсивности отнесены к
1 /и2 поверхности соприкосно-
вения фаз (в данном случае
орошаемой трубки). Понятно,
что производительность ка-
кого-нибудь аппарата будет
повышаться с развитием
О 50 00
— Температура. °C
удельной поверхности соприкосновения фаз.
Как уже указывалось, вышеописанные опыты Малина и Кузь-
миных проводились в условиях бескислородного газа. К этим
22»
339
условиям башенный процесс фактически приближается по мере
повышения концентрации SO2 в поступающих в систему газах
и по мере повышения отношения между количествами М?Оз и
SO2, поступающими в каждую башню.
В практических условиях производства, когда процесс кисло-
тообразования еще связан с необходимостью обратного окисле-
ния NO в реакционной зоне, вопрос о влиянии отдельных факто-
ров на процесс кислотообразования еще более усложняется.
В этих условиях на скорость процесса кислотообразования влияет
и концентрация кислорода в газовой фазе. Это влияние тем зна-
чительнее, чем меньше отношение между количествами N2O3 и
SO2, поступающими в башню. Как показали опыты Малина и
Иткиной {в условиях орошаемой трубки), а также Кузьминых
(в условиях барботажа), кислород влияет на скорость кислото-
образования даже и в том случае, если в подаваемой нитрозе
окислов азота более чем достаточно для полного окисления SO2.
По данным Малина и Иткиной повышение концентрации кис-
лорода в газах от 0 до 89% увеличило константу скорости аб-
сорбции SO2 с 4,81 до 9,47. С повышением концентрации кисло-
рода в газах уменьшается количество десорбирующейся в газо-
вую фазу NO, несмотря на одновременное увеличение количе-
ства абсорбировавшегося SO». Как видно из рис. 165, при повы-
шении концентрации кислорода в газах в указанных уже преде-
лах концентрация NO в отходящих газах снижалась с 1,1 до
0,4%.
5. Влияние давления на процессы абсорбции и кислотообразования
Скорость диффузии абсорбируемого газа через пограничные газовую и
жидкую пленки можно выразить следующим уравнением:
Adt xgpbm{Ps C*>’
где dW — вес абсорбированного газа за время dt, А — поверхность раздела
фаз, K,ig —коэфициент диффузии через газовую пленку, К(ц —коэфициент
диффузии через жидкую пленку, хд—толщина газовой пленки, х,— тол-
щина жидкой пленки, рЬт —парциальное давление инертного газа, рд—
парциальное давление абсорбируемого газа в основной массе газа, Pi —пар-
циальное давление абсорбируемого газа в газовой пленке (равновесное да-
вление), Ci — концентрация абсорбируемого газа в жидкой пленке, Ci — кон-
центрация абсорбируемого газа в основной массе жидкости.
По данным ряда авторов давление не оказывает влияния на коэфициент
диффузии в жидкости. Наоборот, коэфициент диффузии в газе от давления
зависит.
Зависимость эта по Обермайеру выражается так:
где К°(]д коэфициент диффузии при 0° и 760 лл рт. ст.
Для постоянной температуры:
KlJ9~ р
340
Толщина газовой пленки определяется величиной отношения вязкости и
плотности. Так как вязкость газа не зависит от давления, а плотность про-
порциональна давлению, то можно написать:
В
Х°~Р~'
г от
Учитывая, что коэфициент скорости абсорбции (Кд) для газовой пленки
равен
f( — К rig
хдРЪт
получим окончательно:
Д' — РЪт _ и
РЪш * Р ‘ РЪт Р РЪш
т. е. коэфициент скорости абсорбции для газовой пленки обратно пропор-
ционален давлению инертного газа.
Поскольку абсорбция окислов азота (как хорошо растворимого газа)
серной кислотой определяется скоростью диффузии через газовую пленку,
выведенная зависимость от давления справедлива н для общего коэфициента
абсорбции в выражении:
t = 2.1п^
К
Беря последнее выражение в форме:
Фгаз f Р
=—1ПЙ
можно сделать заключение, что применение повышенного давления не окажет
влияния на потребный объем V абсорбера, так как прн повышеннни давления
в п раз в то же число раз уменьшаются величины 01аз н К.
Однако практически некоторое уменьшение потребного объема гей-люс-
саков с применением повышенного давления все же произойдет по следую-
щим причинам:
Р°
1. Формула t= -yrln—7- выведена в предположении, что упругостью
Рд
газа иад раствором можно пренебречь по сравнению с величиной парциаль-
ного давления абсорбируемого газа.
Практически в условиях гей-люссаков величина р^ является достаточно
большой и ее приходится учитывать.
В формуле
S__G_
&рХ
величину др можно представить как среднюю логарифмическую от величин
(Рд—Р0^ И (Рд— Pi)-
где р°{ и pt— упругости N2O3 над нитрозой в начале и в конце абсорбцион-
ного процесса. Понятно, что величины рд и рд повышаются пропорцио-
нально повышению давления, а величины р® и р[ от давления не зависят.
Отсюда следует, что величины (р°—р?) и (рд — pt), а следовательно
др, растут более чем пропорционально повышению применяемого давления.
Так как величина К, как показано, уменьшается пропорционально повыше-
нию давления, то потребная поверхность абсорбера, а, следовательно, и его
объем (прн заданной насадке) с повышением давления должны снизиться.
341
2. Если одновременно с абсорбцией окислов азота проходит процесс
окисления NO, применение давления ускорит процесс в целом через повы-
шение скорости окисления NO. Если окисление NO (до NaOs) осу-
ществляется (в особом аппарате) до абсорбции NaO-i, то, как это следует
из уравнения, потребное время для окисления NO снизится пропорционально
квадрату давления.
Так как потребный объем окислителя равен V = Qra3t, а величина QrgJ
уменьшается пропорционально увеличению давления, то необходимый объем
для процесса окисления NO до Na03 будет обратно пропорционален кубу
примененного давления.
В продукционной зоне, где на скорость процесса переработки SOs про-
цесс диффузии SOa через жидкую пленку оказывает более значительное
влияние, чем при абсорбции N2O3 нитрозой, повышение давления в мень-
шей степени понижает общий коэфициент скорости абсорбции. Общий
коэфициент скорости абсорбции по концентрациям в газовой фазе можно
выразить так:
г._
Кд + HKl '
где Ki н Кд — частные коэфициенты скорости абсорбции для газовой и
жидкой пленок, а Н—коэфициент Генри.
Из этого выражения видно, что величина К уменьшается менее чем
Таблица 81 прямо пропорционально уменьше- нию и, следовательно, в этом
Давление в ат Темпера- тура в °C Интенсивность продукционной зоны в кг H9SO4 на 1 л3 в сутки случае уменьшается медленнее чем прямо пропорционально по- вышению давления. Величина К тем меньше бу- дет зависеть от давления, чем меньше будет величина К{ отно- сительно Кд (т. е. чем большую относительную роль в процессе абсорбции играет сопротивление жидкой пленки). Если исходить нз предполо-
1,05 6,6 13,1 13,3 13,5 22 21 24 70 24 154 513 3130 2920 2720
жения, что скорость переработки
SOs в продукционной зоне определяется скоростью абсорбции SOa нитрозой,
то, обращаясь к формуле:
'"4.
' Рд
----— ЦоВленсе Ват
Рис. 166. Зависимость коэфициента скорости
абсорбции SO2 нитрозой от давления.
К
приходим к выводу, что
потребной для переработки
'Определенного количества
SOa объем должен пони-
жаться с повышением да-
вления.
Экспериментально вли-
яние повышения давления
на интенсивность работы
продукционной зоны изу-
чалось Берлем, а также
Шустовым (в Московском
химико-технологическом ин-
ституте нм. Менделеева).
Данные Берля о повы-
шении интенсивности про-
дукционной зоны от применения повышенного давления приведены
в табл. 81.
342
На рис. 166 изображена по данным Шустова зависимость коэфициента
.скорости абсорбции SO2 нитрозой (для различных температур) от давления.
Коэфициент скорости аб-
сорбции вычислялся по
формуле:
/<=fin 4-
z рв
Кривые рис. 167 показы-
вают зависимость (для тех
же опытов) количества пе-
реработанного SOa (абсор-
бированного) в реакционном
аппарате от давления. Ха-
рактер приведенных кривых
(опыты Шустова) подтвер-
ждает правильность только
что сформулированной (на
основе теоретических со-
ображений) зависимости ин-
тенсивности продукционно-
го объема от давления.
300
= гоо
Давление В ат
Рис.
167. Зависимость интенсивности продук-
ционной зоны от давления.
6. Окисление аммиака
Для пополнения потерь окислов азота раньше применяли разложение
чилийской селитры. Теперь потери окиСлов азота восполняются вводом
в систему или слабой азотной кислоты, получаемой с азотнокислых заводов,
или газообразных окислов азота, получаемых окислением аммиака на Спе-
циальной небольшой установке в самом' сернокислотном цехе.
Окисление аммиака представляет очень сложный процесс, проте-
кающий в зависимости от условий в различных направлениях. Прежде
всего окисление аммиака может иттн н до NO и до элементарного азота
по реакциям:
4NH3 502 = 4NO + 6Н2О + 216,64 ккал;
4NH3 + 3OS = 2Ns + 6НгО + 303,4 ккал.
Термодинамические Исследования показывают, что обе эти реакция прак-
тически необходимо проходят вправо. Установлено, что в отсутствии катали-
затора реакция окисления аммиака всегда приводит только к молекулярному
азоту. Но и при каталитическом окислении аммиака образование элементар-
ного азота по вышеуказанной реакции возможно — перед катализатором, если
температура газа чрезмерно высока, и после катализатора, еСлн часть ам-
миака пройдет через катализатор неокисленнбй.
Одновременно с окислением аммиак'а может происходить и его диссоциа-
ция: 2NH3 =s Na 4- ЗН2. Однако при атмосферном давлении (окисление
аммиака производится обычно без повышенного давления) и в температур-
ных условиях окисления аммиака (600—900°) скорость диссоциации аммиака
чрезвычайно мала.
Образование элементарного азота может происходить также через распад
окиси азота по уравнению:
2NO ~ N2 4- О2 + 43,2 ккал.
Но константа скорости этой реакции в условиях, в которых проводят
окисление аммиака, примерно в 180 раз меньше константы скорости окисле-
ния NHs в NO. Наконец, если в газовой смеси, прошедшей катализатор,
останется аммиак, то может протекать реакция:
4NHg 4- 6NO = 5N2 -ф 6Н2О 4- 492,6 ккал.
343
Из сказанного понятно, что хотя высокий выход при окислении аммиака
и возможен, но это сопряжено с необходимостью преодолеть целый’ ряд
трудностей.
В отличие от ряда других случаев гетерогенного катализа (например
контактного окисления SOs) роль катализатора здесь сводится не только
к ускорению медленно идущей реакции. Пригодный катализатор в данном
случае из всех термодинамически возможных реакций (окисление аммиака
до NO, окисление аммиака до молекулярного азота н ряда .побочных реак-
ций) должен ускорить реакции, ведущие к образованию окиси азота.
Рис. 168. Оптимальное время и оптимальная температура кон-
тактного окисления NH3.
Катализаторами этой реакции являются окислы железа, марганца и
хрома.
Действие железного катализатора усиливается при добавлении к нему
церия илн висмута. Но самыми лучшими катализаторами в процессе окис-
ления аммиака являются платина н ее юплавы с другими металлами платино-
вой группы. Платиновый катализатор обычно применяется в виде сеток
с диаметром нитей от 0,040 до 0,076 мм с числом петель на 1 с.«2 от 1024
до 3600. Действующая поверхность такой сетки находится в пределах от 1,43
до 2,06 см2 на 1 см2 поверхности сетки (от 27,2 до 35,5 см2 на 1 а металла);
свободная площадь на 1 см2 сетки «Уставляет 0,49—0,62 см2- При темпе-
ратурах около 800° н выше, к которым перешли в последние годы, чистая
платина мало пригодна по причине ее больших потерь. Хорошо при высоких
температурах работают сплавы платины с родием. Обычно применяется сплав
платины с 10% родия.
При большинстве каталитических реакций (окисление SOa, синтез NHs,
конверсия СО и др.) увеличение времени соприкосновения газа с катализа-
тором приводит к непрерывному возрастанию выхода вплоть до достижения
равновесия реакции. В процессе окисления аммиака увеличение времени пре-
бывания газа на катализаторе свёрх необходимого приводит к уменьшению
выхода NO за счет диссоциации NO и взаимодействия NH3 с NO с обра-
зованием молекулярного азота. Оптимальное время контактирования прн ра-
боте с платиновым илн платинородневым катализатором находится в интер-
вале 0,00006 до 0,0003 сек., на железовисмутовом катализаторе ~ 0,01 сек.
344
Очень важен также вопрос об оптимальном температурном режиме про-
цесса окисления 1\Нз- При низких температурах скорость окисления NHs
до NO мала и непрореагировавшнй с образованием окнси азота аммиак раз-
лагается до азота вследствие побочных реакций.
При очень высоких температурах приобретают большое значение реакции
диссоциации NH3 и NO, взаимодействие NHe с NO, а также реакция между
NHs и О» в газовом пространстве до катализатора (с образованием молеку-
лярного азота). Установлено, что оптимальная температурная область про-
цесса окисления аммиака заключается между 800 и 900°. В этом интервале
выход NO может достигать 95—98%. В промышленности процесс ведется
при температуре 650—900°. При 650—680° достигается выход аммиака
в 88—90%.
Оптимальное время контактирования н оптимальная температура взаимно
связаны. С повышением температуры оптимальное время соприкосновения
уменьшается.
Это достаточно ясно иллюстрирусася кривыми рис. 168.
Очень большое влияние на выход NO при окислении NH3 оказывает
отношение кислорода к аммиаку в поступающей на окисление газовой смеси..
Из написанных выше реакций видно, что для окисления 1 моля NH3 до NO
требуется 1,25 моля О2, а для окисления до Ns 0,75 О2. Для хорошего
выхода NO необходимо не менее 1,8—2 объемов кислорода на 1 объем
аммиака. В связи с этим при окислении аммиака с воздухом в поступающей
на катализатор аммиачно-воздушной смеси концентрация аммиака поддержи-
вается равной 9,5%, при этом содержание NO в газах после окисления NHs
получается равным 8,5%.
III. Аппаратура башенных систем
1. Замена свинца
Вся аппаратура камерных систём выполнялась из свинца.
В условиях работы камерных -систем (невысокая концентрация
серной кислоты и небольшая нитрозность газов — до 1,2% и
кислот — до 1,5%) свинец достаточно стоек. Кроме того, он
удобен в изготовлении и монтаже аппаратуры.
В башенных системах до их интенсификации свинец тоже
был основным материалом, хотя он уже и тогда подвергался
коррозии значительно быстрее, чем в камерных системах.
При интенсификации башенных систем в связи с повыше-
нием температуры и нитрозности кислот и газов свинец на-
столько быстро разрушался, что задача замены свинца оказа-
лась неразрывно связанной с проблемой интенсификации.
Проведенные в последние годы лабораторные работы
УНИХИМ (Юшманов) по изучению действия серной кислоты и
нитрозы на металлы, а также работы по изучению стойкости раз-
личных материалов в заводских условиях, проведенные НИУИФ
(Второв), дают теперь возможность всю аппаратуру башенных
систем выполнять без свинца. В разрешении этой задачи боль-
шую роль сыграла также инициатива заводских работников,
установивших опытные аппараты из чугуна и железа (Гри-
горьев, Циреншиков и др.).
Проведенные работы показали, что поведение чугуна и же-
леза в нитрозах прямо противоположно поведению свинца. Кор-
розия свинца резко возрастает с повышением нитрозности и
345
температуры серной кислоты концентраций, обычно применяе-
мых в башенных системах (57—59° Вё), коррозия железа и чу-
гуна в них падает с повышением температуры и нитрозности,
что объясняется образованием пассивной пленки. При этом в пе-
риод образования этой пленки железо подвергается сильной
коррозии нитрозами. Однако при высокой температуре этот
период образования защитной пленки очень короток, и чем выше
температура, тем он короче. Поэтому начавшееся в первый пе-
риод сильное растворение железа быстро падает и железо ста-
новится материалом вполне устойчивым. Чугун ведет себя еще
более надежно, так как он в период образования пленки не
подвергается такому сильному воздействию нитрозы.
Эти свойства черных металлов и дают возможность их при-
менения для постройки почти всей аппаратуры башенных за-
водов.
В горячей денитрированной серной кислоте чугун и железо
ведут себя иначе. Коррозия их при этих условиях настолько
сильна, что применение их для изготовления кислотопроводов
без защитной футеровки для неохлажденной кислоты, выходя-
щей из 1-й башни Гловера, и холодильников для кислоты этой
башни оказалось невозможным. К настоящему времени возмож-
ность замены свинца для змеевиков холодильников 1-й башни
пока практически не выяснена. Лабораторные исследования по-
казали, что единственным материалом, который, невидимому,
возможно будет применить для изготовления змеевиков этих
холодильников, является ферросилиций («термосилид», «си-
лекс» и др.).
Общей аппаратурой для башенных и камерных систем явля-
ются: ' 1) башни Гловера, или продукционные, 2) башни Гей-
Люссака, или абсорбционные, 3) холодильники для кислот,
4) сборники для кислот, 5) насосы для перекачки кислот,
€) приборы для разбрызгивания кислот в башнях, 7) вентиля-
торы для протягивания газа через систему и 8) аппараты для
окисления аммиака.
Аппаратами, имеющимися только в башенных системах,
являются: 1) брызгоуловители (циклоны), 2) окислительные башни.
Аппаратура, присущая только камерным системам: 1) свинцо-
вые камеры, 2) аппараты для фильтрации воды и распыления ее
в камерах.
2. Башни
Газы из пылеочистительных аппаратов (электрофильтров) по
железному цилиндрическому футерованному внутри шамотным
кирпичом газоходу поступают в башни Гловера.
Камерная система имеет обычно одну башню Гловера, иногда
две; в башенной системе продукционных башен, из которых
первые две называются гловерами, обычно имеется три. При
этом последнюю продукционную башню принято называть ста-
билизатором.
346
В камерных системах гловеры включаются в большинстве
случаев параллельно; в башенных — последовательно. Основные
размеры башен Гловера в камерных системах колеблются в сле-
дующих пределах — внешний диаметр 2,8—4 м, высота 8—15 м',
в башенных — диаметр 4—7 м, высота 14—18 м- Башен
Гей-Люссака в камерных системах обычно 2, иногда 3; в башен-
ных системах в большинстве случаев имеются 3 гей-люссака.
Башни устанавливаются на кирпичном или бетонном поста-
менте на высоте, достаточной для осуществления самотека
кислоты, выходящей из башни, в холодильники и далее в сбор-
ники.
В камерных и старых башенных системах оболочки башен выполнялись
нз листсрого свинца толщиной в 5 мм у гловеров ii в 8 .и у гей-люссаков.
Днище башен выполнялось нз lO-.w.w свинца. Оболочка башни выполнялась
из отдельных царг, спаянных вертикальными швами при помощи водородного
пламени. Каждая царга подвешивалась к кольцу из углового железа, при-
крепленному к железным стойкам каркаса башни. Между собой свинцовые
царги соединялись при помощи пайки (сварки) водородным пламенем. В более
старых башнях каркас выполнялся из деревянных брусьев, к которым при-
креплялись металлические междуэтажные кольца, поддерживающие свинец.
В гловерах и продукционных башенях свинец от действия
горячей нитрозы, циркулирующей в башне, защищается толстой
кислотоупорной футеровкой; такой же футеровкой покрывается
и железо.
Толщина футеровки стенок башни Гловера в самой нижней
части делается не менее 450 мм, толщина верхней части
250—300 мм. Футеровка продукционных башен кроме выполне-
ния роли защиты свинца от коррозии служит термоизоляцией
для поддержания в башнях оптимальной температуры. Футеровка
гей-люссаков выполняется меньшей толщины (120—130 мм), чем
у гловеров. Для футеровки башен применяются: 1) кислотоупор-
ный кирпич, 2) керамические фасонные камни, 3) камни, вытесан-
ные из природных кислотоупоров: андезита, бештаунита, фель-
зита и т. д.
Связующим материалом, в большинстве случаев, служит кис-
лотоупорная замазка, сухая часть которой составляется из 960 г
молотого андезита или бештаунита и 40 г кремнефтористого на-
трия. К смеси этих материалов прибавляется 220—250 г жидкого
стекла [уд. вес 1,36 (38° Вё) и отношение SiOz к NazO не ме-
нее 2,8]. В этой замазке в химическое взаимодействие вступают
кремнефтористый натрий и жидкое стекло. Молотый кислото-
упорный порошок (андезит, бештаунит и др.) играет роль на-
полнителя. Эта замазка кроме кислотоупорности обладает свой-
ством быстро схватываться, подобно цементу, что способствует
скорости выполнения работ. Схватившаяся замазка увеличивает
свою прочность при обработке ее серной кислотой.
Оболочки башен теперь выполняются из 6-/ил1 железа. При
этом не требуется каркаса для подвески каждой царги, как это
практиковалось в большинстве случаев при свинцовых оболоч-
347
ках. Листы железа и отдельные царги между собой соеди-
няются электросваркой. Железная продукционная башня пока-
зана на рис. 169.
Рис. 169. Железная башня Гловера (новый тип):
1 — железная оболочка башни; 2~ футеровка; 3—турбинка для разбрызгивания кислоты
с приводом от электромотора; 4 — крышка из кислотоупорного бетона с железной армиров-
кой; 5—колонки колосниковой решетки; б — нижние колосники решетки; 7—верхние
колосники решетки; 8— выстилка дна башни; 9— цоколь фундамента; 10— люк для осмотра;
11 — гидравлический затвор.
Насадка в башне укладывается на колосниковую решетку,
изготовляемую из прямоугольных брусьев, вытесанных из при-
348
родных кислотоупорен — андезита и бештаунита — или выпол-
ненных из кислотоупорной керамики. Материал колосников1
первых башен должен быть не только кислотоупорным, но и
термостойким.
Колосники располагаются на колонках, выложенных из от-
дельных прямоугольных камней. Сверх основных колосников
под насадку укладываются более мелкие колосники в один или
два ряда. Колосниковая решетка располагается на такой высоте
над выстилкой дна башни, чтобы возможно было человеку при
очистке подколосникового пространства или при его ремонтах
поместиться под колосниками в сидячем положении.
В стенках башен устраивают люки для осмотра и очистки
подколосникового пространства.
В верху башни оставляется незаполненное насадкой про-
странство, достаточное для размещения оросительного прибора
таким образом, чтобы брызги кислоты, разбрасываемые этим
Рис. 170. Крышка продукционной башни из армированного железом кислото-
упорного бетона.
прибором, распределялись равномерно по насадке. При устрой-
стве верхнего газовыводного окна в боковой стенке башни учи-
тывается также и возможность его размещения. Обычно высота
этого незаполненного пространства не превышает 1—1,5 м.
Крышки башен, начиная с третьей, выполняются из железа.
Крышки первых башен делаются из кислотоупорного бетона,
армированного железом (рис. 170), так как в связи с высокой
влажностью газов в первых башнях и подачею в них воды на
крышках их может конденсироваться слабая кислота, которая
вызывает коррозию железа.
При выходе кислоты из башни устраивается гидравлический
затвор (рис. 169), который у всех башен, кроме 1-го гловера,
представляет собой чугунную герметически закрытую коробку
с вертикальной перегородкой, не доходящей до дна. Ввиду того
что денитрированная кислота (выходящая из 1-го гловера) при
температуре выше 90° сильно действует на чугун и железо,
кислотовыход и гидравлический затвор этой башни защища-
ются изнутри плотной кислотоупорной футеровкой на кислото-
упорной замазке.
На старых, работающих на заводах, башнях свинец также
заменяется железом, причем эта замена осуществляется без
349
удаления насадки из башни, с оставлением старой футеровки и
с заполнением небольшого концентрического пространства
между футеровкой и железной оболочкой кислотоупорным бе-
тоном.
Железные стенки башен Гей-Люссака в последнее время на-
чинают выполняться уже без футеровки и на некоторых заводах
успешно работают достаточно длительное время (рис. 171).
Рис. 171. Железная башня Гей-Люссака без футеровки с железной решеткой
(низ башни):
1 — желёзиая оболочка башии; 2—железное дно башни; 3—камера под дном башни дла
его осмотра снизу; 4 — железные колонки, поддерживающие решетку башни; 5 — нижиие
колосники решетки в виде железных балок; 6 — верхние колосники решетки (железные
балки).
В этих башнях вместо колосников, вытесанных из андезита,
начинают применяться стальные балки.
Такая конструкция колосниковой решетки изображена на
рис. 171.
Стальные балки расположены на стальных колонках. Под
дном башни имеется камера, из которой можно осматривать это
Дно и в случае нарушения его плотности своевременно произ-
водить ремонт.
350
Возможность постройки железных башен Гей-Люссака без
футеровки вытекает из способности железа пассивироваться
под действием нитрозы. Одно обстоятельство заставляло при
замене свинца железом в башнях Гей-Люссака применять в них
футеровку — это необходимость изредка промывать водой на-
садку этих башен, состоящую из кусков кварца, для устранения
ее загрязнения пылью газов и осадками солей, выделяющихся
из циркулирующих в башнях кислот. При этих промывках обра-
зующаяся слабая серная кислота может вызвать сильную кор-
розию железных стенок башни. Однако опыты неоднократной
промывки водой железной башни на одном из заводов показали,
что в том случае, если эта промывка производится очень быстро
большим количеством воды и затем сейчас же после окончания
промывки башня обильно орошается концентрированной серной
кислотой, стенки башни не подвергаются коррозии. Наоборот,
вполне.основательно опасение, что при наличии футеровки про-
мывка водой может иметь худшие последствия, чем в отсут-
ствие ее, так как при проникании воды или слабой кислоты че-
рез могущие встретиться в футеровке неплотности, слабая кис-
лота при длительном пребывании между футеровкой и железом
должна неминуемо оказать сильное разрушающее действие на
железо. С технологической же точки зрения отсутствие футе-
ровки в башнях Гей-Люссака только желательно. Это создает
условия лучшего охлаждения башни, а происходящие в башнях
Гей-Люссака процессы лучше проходят при низкой температуре
(окисление NO и абсорбция N2O3).
Для подготовки окислов азота к абсорбции (окисление NO
до N2O3) на некоторых башенных заводах между продукцион-
ными и абсорбционными башнями устанавливается так называе-
мая окислительная башня. Это — цилиндрическая башня, вы-
полненная из котельного 6-дни железа, не имеющая ни футе-
ровки, ни насадки. Футеровкой покрывают иногда лишь нижнюю
часть и днище башни. Вследствие наличия в газах, омывающих
стенки этой башни, большого количества окислов азота и' кон-
денсации на них кислоты с высоким содержанием H2SO4 и оки-
слов азота и достаточно высокой температуры газов создаются
условия, благоприятные для образования и поддержания за-
щитной пассивной пленки, предотвращающей коррозию железа.
В окислительную башню газы входят сверху и выходят снизу;
степень окисления NO регулируется обводным газоходом (бай-
пассом), по которому можно из последней продукционной башни
направлять газы непосредственно в 1-ю башню Гей-Люссака.
В байпассе устраивается шибер, при помощи которого большее
или меньшее количество газа может быть направлено помимо
окислительной башни.
Все газоходы между башнями выполняются теперь из листо-
вого железа. Газоходы между 1-й и 2-й башнями (а иногда ме-
жду 2-й и 3-и) должны футероваться внутри кислотоупорными
материалами для защиты от коррозии.
351
3. Насадка башен
Для создания достаточной поверхности соприкосновения
газа с жидкостью все башни (кроме окислительной) заполняются
насадкой. К насадке предъявляются следующие основные тре-
бования: она должна:
1) иметь возможно большую удельную поверхность;
2) в ряде случаев (когда одновременно с абсорбцией в башне
должны происходить процессы в газовой фазе) иметь доста-
точно большой удельный свободный объем;
3) иметь возможно меньший вес на единицу своего объема;
4) быть простой и прочной по своей конструкции и легко
укладываться в башнях;
5) быть кислотоупорной и достаточно термостойкой;
6) не должна давать заметного бокового распора;
7) не должна давать большого сопротивления прохождению
газов;
8) ее форма не должна допускать скопления и застаивания
грязи. °
Насадка, кроме того, должна иметь возможно низкую стои-
мость.
Насадка бывает бесформенная и фасонная.' Из бесформенной
насадки раньше и в башенных и в камерных системах в гей-
люссаках применялся кокс; в настоящее время в башенных си-
стемах для загрузки гей-люссаков применяется кварц. Перед за-
грузкой куски кварца сортируются. Внизу башни загружаются
более крупными кусками с постепенным переходом к более мел-
ким вверху.
Основные достоинства кварцевой насадки — ее низкая стои-
мость и простота укладки; недостатки — легкая забиваемость
грязью, сравнительно большое гидравлическое сопротивление,
малая удельная поверхность, большой боковой распор.
Большинству предъявляемых требований удовлетворяет фа-
сонная насадка. Из фасонной насадки в башенных системах наи-
более широко применяются кольца Рашига — полые цилин-
дрики, изготовляемые из кислотоупорной тщательно обожжен-
ной керамики, иногда из фарфора. Обычно в башенных систе-
352
мах башни насаживаются кольцами Рашига с внешними разме-
рами 80 X 80 и 50 X 50 мм- Лишь нижние ряды, служащие пе-
реходными от колосниковой решетки к главной массе насадки,
состоят из колец Рашига больших размеров, увеличивающихся
от ряда к ряду сверху вниз (рис. 169).
Кольца Рашига устанавливаются вертикально и так, чтобы
стенки цилиндриков каждого вышерасположенного ряда не соз-
давали продолжения стенок цилиндриков нижерасположенных.
В результате такого расположения получается множество
вертикальных канальцев, в которых газ принужден разбиваться
на многочисленные узкие струйки, ударяющиеся о горизОнталь-
Рис. 173. Кладка стабильформатов I и керамиковых призм II.
ные части стенок цилиндриков при переходе в каждый после-
дующий ряд насадки и вследствие этого вновь разбивающиеся
на еще более мелкие струйки.
Применяются и другие формы насадки, особенно в первых
башнях: пятигранные призмы с рифленой поверхностью //, ста-
биль-формат / и др. (рис. 173). Исключительно важен выбор на-
садки для 1-й башни Гловера в том случае, когда башенная си-
стема вырабатывает купоросное масло, так как при этом в ниж-
ней части башни, где быстро и значительно поднимается кон-
центрация кислоты, из нее выделяются кристаллы сернокислых
солей, могущие загрязнить насадку до полной ее закупорки.
В табл. 82 приведены характеристики различных видов на-
садок (стр. 354).
Сопротивление различных насадок изучалось многими ис-
следователями. Применительно к условиям башен сернокислот-
ных заводов недавно была проведена работа Кузьминых и Апа-
23 Зак. 5019. Технология серной кислоты. 353
Таблица 82
Вид насадки Размеры в мм Процент свобод- ного объема Поверхность в м- на 1 ж3 насадки Отношение сво- бодного объма к поверхности (в см)
Обыкновенные камни с высокими ребрами 250 X 150 X 65 37 8,25 4,48
Кварц 150 44 29 1,51
75 46 42,5 1,08
12-25 47 167 028
50 45 63 0,715 !
Кокс 75 58 42,5 1,36
Кольца Рашига . . 15X15X2 82 380 0,216
25 X 25 X 2 89 220 0,40
35 X 35 X3 92 160 0,575
50 X 50 X 4 89 НО 0,81
Гладкие цилиндры 70 X 50 X Ю 61 77,5 0,79
Ю6 Х.80 X 7 56 53 1,05
125 X 125 X Ю 77 46,2 1,66
106 X 80 X 20 73,5 53 1.38
Ромбическая насадка 90 X 300 X 20 45.5 21,8 2,08 :
Х40 53,5 18,6 2,86 :
Х60 58,5 16,6 3,52
70 X 250 X 20 40 27,6 1,45
X 40 50 23 2,17
Х60 57 19,7 2,89
Трехгранные призмы 120X300X20 54 21,6 2,50
X 40 64 18 3,55
Хбо 70 14,8 4,73
90 X 300 X 20 56 27,8 2,01
X 40 63 22,1 2,15
Х60 71 18,4 3,85
Гладкие шары 40 64 90,6 0,71 '
• 50 40 72,2 0,563 :
60 40 59,6 0,67 !
70 4! 50,8 0,81
100 40 36 1,11
30 62 76,2 0,814 1
Пропеллерные кольца Вильсена . . 150 X ’50 80 57,4 1,39
100 X 100 80 73,8 1,08
Спиральные кольца 75X75 72 72 1,0 :
75 X 75 X 63 62,3 105 0,59
Ячейки Гутмана 50 X Ю9 X 150 74 111,5 0.664 j
Круглые диски Кестнера 30X5 77 270 0,285 ’
Туннельные тела 80 X Ю6Х86 58 26 2,33
354
ховым. Результаты этой работы для ряда насадок пр'едставлены
на рис. 174.
Исходя из этих данных, сопротивление башни можно опре-
делить по формуле:
р = К- йттч®9’
где h — высота насаженной
части башни, у — плотность
газа при данных условиях,
w — линейная фиктивная ско-
рость газа в м/сек, р — сопроти-
вление башни в мм вод. ст., К —
коэфициент гидравлического
сопротивления (численно рав-
ный сопротивлению при у =
= 1,13, w = 1 м/сек и й = 1 м).
На величину сопротивления
применяемых насадок (по
Кузьминых и Апахову) изме-
нение плотности орошения в
пределах, имеющих место в
практике башенных систем, не
оказывает значительного влия-
ния. В практике эксплоатации
сопротивление отдельных ба-
шен меняется с течением вре-
мени в очень значительных
пределах. Так, нередки случаи,
когда сопротивление башни с
20—30 мм вод. ст. (в начале
работы башни) возрастает за-
тем по мере загрязнения насад-
---•- иг 6 м/сен
Рис. 174. Сопротивление насадок при
плотности газа 7 = 1,13 и при ороше-
нии серной кислотой~-20 мл1м-1сутки:
1 — кварц 60 мм; 2 — кварц 75 лги; 3 — кварц
100 jwjw; 4 — кольца 50X50 в навалку; 5— пя-
тигранные призмы; 6 — кольца 50 X 50 в
укладку.
башен часто приводит к вы
ки до 100 мм вод. ст. и выше.
Отсюда понятна необходимость
регулярного наблюдения за ве-
личиной сопротивления ка-
ждой башни (которое является
показателем степени ее засо-
рения).
Возрастание сопротивления
нужденному снижению выработки системы и является одной из
основных причин повышенного расхода электроэнергии. Про-
мывкой засорившейся башни слабой кислотой, а также водой
обычно удается значительно отмыть насадку и снизить сопро-
тивление башни. Однако часты случаи, когда по причине, засо-
рения башни ее приходится перенасаживать. Из сказанного
ясно, насколько важно добиваться максимально высокой степени
очистки газа от пыли. Фасонная насадка, особенно кольцевая,
23*
355
подвержена меньшей опасности забивки грязью, чем насадка
бесформенная (кварц).
4. Приборы для разбрызгивания кислоты в башнях
и воды в камерах
Равномерное распределение кислоты, орошающей насадку
башни по всей площади ее поперечного сечения, играет огром-
ную роль в технологическом режиме всякой сернокислотной си-
стемы. При неравномерном распределении кислоты обычно не-
равномерно распределяется и газ в насадке, так как он идет
в этом случае по тем участкам насадки, которые не орошаются
или орошаются в меньшей степени кислотой. Вследствие этого
Рис. 175. Прибор для распределения кислоты по насадке башни
(паук):
1—кран для регулирования количества кислоты; 2—„глушитель" для предот-
вращения разбрызгивания; 3—носики для распределения кислоты по радиаль-
ным отделениям; 4—трубочки с гидравлическими затворами, ведущие кис-
лоту в башню; 5—гидравлические затворы.
соприкосновение газов с кислотой сильно уменьшается, что ве-
дет к расстройству работы башни. Поэтому очень важен выбор
рациональной конструкции оросительного прибора. На старых
камерных сернокислотных заводах, где башни орошались не-
большими количествами кислоты (около 5 м3/час), для этого
применялись так называемые пауки (рис. 175), состоявшие из
небольшого, цилиндрической формы, установленного строго гори-
зонтально свинцового резервуара 2 с впаянными в борта его но-
сиками 3.
Кислота поступала в этот сосуд из напорного бака и стекала
по носикам в радиально расположенные ниже сосуда секции
свинцового противня. В дно каждой такой секции впаяны свин-
цовые трубочки 4, другими своими концами вйаянные в крышку
башни. Торцы трубочек перекрыты свинцовыми колпачками 5,
создающими гидравлические затворы, препятствующие выби-
ванию газа из башни. Эти приборы работали удовлетворительно
356
при незагрязненных орошающих кислотах, в особенности тогда,
когда на сернокислотных заводах сжигался кусковой колчедан
в ручных печах. С появлением на заводах механических печей,
Рис. 176. Стакан Браузе и установка его.
Рис. 177. Брау-
зедержатель:
1—-стакан Браузе;
2—чугунный кор-
пус браузедержа-
теля; 3 — крышка
башни; 4—крышка
браузедержатедя;
5—струбцина.
из которых в систему стали поступать газы, значительно более
загрязненные пылью, работа пауков стала неудовлетворительной
вследствие того, что узкие трубочки, ведущие кислоту из каж-
дой радиальной секции противня к крышке баш-
ни, стали часто забиваться грязью, что вызы-
вало неравномерное распределение кислоты по
насадке башни.
На башенных заводах орошение всех башен,
за исключением гловеров, первоначально осуще-
ствлялось при помощи так называемых стаканов
Браузе. Это — цилиндры, отлитые из гартблея
со множеством мелких отверстий, просверлен-
ных под различными углами в боковых стенках
и в дне (рис. 176). Верхней открытой частью
цилиндр прикрепляется к свинцовой трубе, под-
водящей кислоту из напорного бака. Ввиду не-
удобств, связанных с обслуживанием таким обра-
зом оформленного устройства, в последнее
время введены так называемые браузедержатели,
отлитые из чугуна, в которые вставлены ста-
каны Браузе / (рис. 177). При этом устройстве
смена стакана или выемка его для прочистки мо-
жет быть выполнена быстро после снятия чу-
гунной крышки браузедержатедя 4, прикрепляе-
мой струбциной 5.
Однако несмотря на это усовершенствование, браузе в на-
стоящее время на большинстве заводов заменяются приводя-
щимися в движение от электромоторов турбинками.
Переход от браузе к этим турбинкам вызван следующими
причинами:
357
1) Отверстия стаканов Браузе часто закупориваются грязью
и солями, содержащимися в кислоте.
Рис. 178. Чугунная турбинка для разбрызгивания кислоты:
1—чугунная звездочка; 2—чугунный конус; 3—чугунная крышка конуса; 4 —чугунный
чехол для защиты вала; 5—железная подставка для опоры конуса; 6—железная крышка
башни; 7—чугунный колпачок для защиты вала; Я—стальной вал турбинки; 9—сальниковое
уплотнение; 10—шкив привода; 11—редуктор; 12—электромотор; 13—чугунный кислотопро-
вод; 14—гидравлический затвор.
LtV----------«-*-•-----------OSS
Разрез по /18
Рис. 179. Гидравлическая турбинка (Озаг-Петерсен):
/—воронка; 2—камера турбинки; 3—турбинное колесо; 4—труба для подвода кислоты
в турбинку; 5—звездочка; 6—приемная коробка; 7—переливная воронка; 8—гидравлический
затвор; 9—труба для подвода кислоты помимо турбинки.
2) Эти отверстия теряют данную им первоначально на осно-
вании расчета форму, вследствие чего угол наклона оси отвер-
стия изменяется.
358
Рис. 180. Распылитель Ленерта.
жены на небольшой
Рис. 181. Распылитель,
действующий от удара.
струи о пластинку, и его
установка:
7—дюз; 2—свинцовая трубка;
3—потолок камеры; 4 — гид-
равлический затвор; 5—пла-
стинка; 6—фильтр; 7—водо-
проводная магистраль.
То и другое явления расстраивают правильную работу оро-
сительного прибора, что в сильнейшей степени влияет на техно-
логический режим системы.
3) Браузе могут работать лишь при условии создания до-
статочно высокого напора в подводящей кислоту трубе, для
чего необходимо площадку с
напорными баками поднимать на
большую высоту над крышками
башен (6—9 м). Это требует
большего напора от насосов, что
влияет на набивку сальников и
вызывает часто прорывы кислоты
через неплотности набивки.
Приводные турбинки (рис. 178)
не имеют этих недостатков, ра-
ботают надежно и при правильно
выбранном числе оборотов до-
статочно хорошо распределяют
кислоту по насадке. Для них
не требуется создавать высокого напора в подводящих кислоту
трубах, вследствие чего напорные баки могут быть располо-
дсоте над крышками башен.
На башнях Гловера и на последнем
гей-люссаке, имеющих небольшой диа-
метр, устанавливают одну турбинку в
центре крышки. На остальных башнях
обычно устанавливают по 3 турбинки, рас-
положенные по сторонам вписанного тре-
угольника.
На башнях Гловера раньше применя-
лись гидравлические турбинки, звездочки
которых приводились в движение от на-
пора кислоты (рис. 179). Эти так назы-
ваемые турбинки Озаг-Петерсена в настоя-
щее время почти совсем вытеснены
приводными турбинками. Как у гидравли-
ческой, так и у приводной турбинок кис-
лота разбрызгивается по насадке звездоч-
кой, имеющей различной длины лучи с
целью равномерного распределения кис-
лоты по насадке (рис. 179).
Для разбрызгивания (или вернее —
распыления) воды в камерах применяются так называемые дюзы.
Их действие основано или на ударе двух струй воды друг
о друга (дюзы Ленерта, рис. 180), или на ударе струи воды
о металлическую пластинку (рис. 181), или же на действии цен-
тробежной силы (дюзы Кертинга, рис. 182).
Дюзы устанавливаются в потолке камер (рис. 181). Впуск воды регу-
лируется вентилем на подводящей трубе. Вода, прежде чем поступить
359
в дюзы, сначала пропускается через песчаный фильтр, а затем через фильтр,
наполненный греческими губками, а в последнее время — люфой. Перед
каждой дюзой иногда устанавливается маленький фильтр в (рис. 181), 'пред-
ставляющий (небольшой цилиндрический
сосуд, заключающий одну небольшую гре-
ческую губку. На рис. 183 показана схема
установки для фильтрации воды.
5. Хвостовые вентиляторы
В башенных системах для про-
тягивания газа устанавливаются
почти всегда лишь так называемые
хвостовые вентиляторы между 2-й
и 3-й башнями Гей-Люссака и толь-
ко в виде исключения кроме хво-
стовых устанавливаются перед 1-й
башней Гловера и головные венти-
ляторы, изготовляемые из чугуна.
В настоящее время хвостовые
Рис. 182. Распылитель Кертинга. вентиляторы изготовляются исклю-
чительно из стали, без свинцового
покрытия. Производительность наиболее распространенного на
башенных заводах железного хвостового вентилятора (рис. 184)
50 000 м3/час; напор
920 мм вод. ст., потреб-
ная мощность мотора
300 квт, число оборотов
1475. Колесо такого вен-
тилятора (рис. 185) —
крыльчатка — выпол-
няется из 7-мм же-
леза и приводится в
движение электромото-
ром, сидящим с ним на
одном валу. Корпус
вентилятора построен из
6-лш железа, отдельные
части которого соеди-
нены электросваркой.
Изогнутые лопасти ко-
леса прикреплены к
дискам заклепками.
Вентиляторы эти в
настоящее время уста-
Рнс. 183. Установка для фильтрации воды:
7— напорный бак для воды; 2—труба для подвода
воды к насосу; 3—поршневой насос; 4 — песочный
фильтр; 5 — предохранительный клапан; 6—губочный
фильтр.
навливаются в отдельном помещении, непосредственно прилегаю-
щем к башенному отделению. Выделение вентиляторов в отдель-
ное помещение желательно для предохранения обмотки электро-
моторов высокого напряжения, приводящих в движение крыль-
чатки вентиляторов, от разрушающего действия паров серной
кислоты и окислов азота, иногда выделяющихся в атмосферу
360
помещения башенного отделения через неплотности аппаратуры
и коммуникации. Кроме такой изоляции этих установок на не-
которых заводах применяется предварительная очистка от газов
воздуха, поступающего для охлаждения моторов вентиляторов^
пропусканием его через
особые фильтры.
При введении в практи-
ку хвостовых железных вен-
тиляторов, работающих в
обычных условиях удовле-
творительно, на некоторых
заводах встретились со слу-
чаями сильной коррозии
крыльчаток этих вентилято-
ров. Нормальный срок служ-
бы стальной крыльчатки,
равняющийся одному году,
внезапно сокращался до не-
скольких дней. Кроме этого
на некоторых заводах на-
блюдалась вообще повышен-
ная коррозия вентиляторов.
Причины ненормально бы-
строй коррозии крыльчаток,
обнаруженной на некоторых
заводах, выявлены работами
Второва (НИУИФ).
Внезапные случаи быст-
рой коррозии крыльчаток
после установившегося обыч-
но длительного срока служ-
бы объясняются подсосами
влажного воздуха, прони-
кающего в вентилятор или
в газоход, подводящий к
нему газы, через неплотно-
сти, могущие образоваться
случайно. Увлажнение газа,
происходящее при этом, по-
нижает концентрацию сер-
ной кислоты, осаждающейся
на стенках газохода и попа-
дающей затем в вентилятор,
а также понижает концентрацию серной кислоты, осаждаю-
щейся из тумана на быстро вращающейся крыльчатке.
Чем длиннее газоход, подводящий газы к вентилятору и чем
более он имеет колен, тем более шансов получить в нем кон-
денсат, содержащий слабую серную кислоту, так как в этом
случае из газа, до поступления его в вентилятор, удаляются
261
брызги крепкой кислоты, увлеченные из башни и обычно повы-
шающие концентрацию слабой серной кислоты, осаждающейся
на стенках газохода вентилятора вследствие их охлаждения.
Место установки хвостового вентилятора в системе оказывает
также большое влияние на увеличение или уменьшение его кор-
розии. Чем ближе к голове системы установлен вентилятор, тем
сильнее он подвергается коррозии, так как чем ближе к голове J
системы, тем больше водяных паров содержится в газах, а еле- '
довательно, и осаждается на крыльчатке вентилятора из тумана
кислота более низкой концентрации.
Рис. 185. Колесо (крыльчатка) хвостового вентилятора.
Обычно принятую установку вентилятора между 5-й и 6-й
башнями (при 6-башенной системе) поэтому надо считать удовле-
творяющей условиям меньшей коррозии. При проектировании
установки вентилятора следует выбирать, для него такое место
между этими башнями, чтобы газоход, подводящий к нему газы
из 5-й башни, был возможно более короток и прямолинеен.
6. Брызгоуловители
С газами, уходящими в атмосферу из последней башни си-
стемы, уносится значительное количество сернокислотного ту-
мана, окислов азота, а также брызг серной кислоты.
362
Практическое разрешение вопроса о полном улавливании цен-
ных составных частей выхлопных газов с целью сокращения по-
терь азотной и серной кислот и обезвреживания атмосферы заво-
дов пока не достигнуто. Боль-
Рис. 186. Железный циклон для
улавливания брызг кислоты,
шое количество уносящихся с
газами брызг довольно удачно
Рис. 187. Присоединение циклона к
выхлопной трубе;
1 — выхлопная труба; 2— патрубок, соеди-
няющий выхлопную трубу с циклоном; 3—
дроссель; 4 — регистр для включения цикло-
на; 5 — патрубок для входа газа в циклон;
6 — патрубок для выхода газа нз циклона;
7 — выходная труба; 8—труба для выхода
кислоты.
улавливается циклонами (рис. 186), устанавливаемыми рядом
с выхлопной трубой башенной системы. Газ из заглушенной
дросселем 3 прямой вертикальной выхлопной трубы 1 вводится
по патрубку 5 в верх аппарата, проходит в нем спиральный путь
сверху вниз и выходит по патрубку 6 в выхлопную трубу 7
(рис. 187). Под действием центробежной силы тяжелые капли
363
кислоты, ударяясь о стенки циклона, осаждаются на них и сте-
кают по трубке 8 обратно в башню.
Циклоны эти выполняются из железа толщиной в 6—8 мм.
Нижняя их часть выстилается кислотоупорным кирпичом или
плитками на кислотоупорной замазке.
7. Холодильники
В камерных системах большая часть тепла, приносимого в си-
стему газами и выделяющегося при образовании серной кислоты
и ее разбавлении, отводится через стенки камер. Из башенных
систем тепло, главным образом, отводится через холодильники
кислоты с уходящей из них водой.
Pifc. 188. Цилиндрический холодильник с радиально расположенными
змеевиками.
До сих пор в камерных и башенных системах применялись
исключительно холодильники с погруженными в кислоту змееви-
ками. Кожух резервуаров холодильников и сами змеевики выпол-
нялись ранее из свинца. Теперь на башенных системах свинец
в кожухах резервуаров холодильников всюду заменен железом.
Широкая замена свинцовых змеевиков холодильников на желез-
ные началась лишь недавно.
Резервуары холодильников изготовлялись прямоугольной и
цилиндрической формы. В холодильниках цилиндрической формы
змеевики располагались или концентрически или радиально.
Холодильники с радиально расположенными змеевиками при-
меняются до сих пор на многих заводах (рис. 188). В центре
резервуара холодильника установлен приемный стакан в виде
полого цилиндра из кислотоупорного материала. В нижней ча-
сти стакан имеет четыре выхода для кислоты. Между стаканом
и стенкой резервуара холодильника поставлен второй концентри-
чески расположенный цилиндр из кислотоупорных камней или
железа. Между приемным стаканом и цилиндром расположены
концентрические змеевики. Между вторым цилиндром и стенкой
помещаются радиально расположенные змеевики.
364
Кислота поступает в ста-
кан, выходит из него сни-
зу, поднимается кверху,
переливается через отвер-
стия в верхней части вто-
рого цилиндра, опускает-
ся книзу и затем подни-
мается по вертикальным
трубам, из них собирается
в жолоб, сделанный во-
круг борта холодильника,
из которого и отводится.
Общая поверхность охла-
ждения такого холодиль-
ника ~ 350 щ-.
В настоящее время на
ряде заводов устанавлива-
ются железные змеевики.
После установки в холо-
дильник железные змееви-
ки подвергаются пассива-
ции, которая заключается
в обработкенитрозой(плот-
ностью не ниже 58° Вё
при температуре 90—120е).
Недостатки змеевико-
вых холодильников заклю-
чаются в следующем.
1. Быстрое загрязнение
холодильника сернокис-
лыми солями, приносимы-
ми из башен с кислотой и
осаждающимися в холо-
дильнике вследствие рез-
кого снижения скорости
движения кислоты в нем.
Это явление ведет к бы-
строму снижению коэффи-
циента теплопередачи сте-
нок змеевиков, так как они
снаружи покрываются тол-
стым слоем плохопроводя-
щей тепло грязи (солей
и пр.). Очистка этой грязи
требует выключения холо-
дильников из системы на
продолжительное время,
что неблагоприятно отра-
жается на режиме системы.
Рис. 189. Оросительный холодильник:
водопроводные трубы; 2—железные пластинки; Э—контрольная труба; 4—секции холодильника.
365
2. Стенки труб змеевиков загрязняются не только снаружи,
но и изнутри, и это загрязнение может быть более или менее
быстрым в зависимости от качества воды; его приходится устра-
нять посредством продувки каждого змеевика сжатым воздухом.
Однако это не всегда дает достаточный эффект.
3. Малая скорость движения кислоты через холодильник
является одной из причин невысокого практического коэфи-
циента теплопередачи.
Поэтому при разработке новых типов холодильников из чер-
ных металлов в первую очередь предусматриваются такие кон-
струкции, которые допускают большие скорости кислоты и воды, .
Один из типов холодильников, распространенных на серно-
кислотных заводах, работающих по контактному методу, в на-
стоящее время начинает вводиться и на башенных сернокислот-
ных заводах. Это так называемый оросительный холодильник, -
состоящий из ряда параллельных горизонтальных чугунных труб,
соединенных между собой полукруглыми патрубками (калачами}
на фланцах (рис. 189). В этих холодильниках подлежащая охла-
ждению кислота протекает внутри труб, а вода орошает эти
трубы снаружи.
Коэфициент теплопередачи холодильников этого типа весьма
высок вследствие большой скорости протекания кислоты и мень-
шей возможности загрязнения.
8. Баки и кислотохранилища
Кислота, выходящая из каждой башни, прежде чем поступить
в насосы для перекачки ее вновь на верх башен, направляется
через холодильники или помимо них в нижние сборники (баки).
В этих же баках собирается вся кислота, циркулирующая в баш-
нях, в случае остановки системы. Обычно общий объем таких
баков берут равным 8% о" общего объема всех башен.
Кроме циркуляционных сборников в насосно-холодильном от-
делении устанавливается сборник для денитрированной кислоты,
подготовленной для выпуска в виде продукции. Из этого бака
кислота передается насосами в кислотохранилища на склад. Из
циркуляционных сборников кислота поступает в насосы, кото-
рыми поднимается в напорные баки, расположенные над баш-
нями. При этом раньше, когда применялись для разбрызгивания
кислоты стаканы Браузе, напорные баки помещались на высоте
от 6 до 9 м над крышками башен в особой надстройке над ба-
шенным отделением.
С введением приводных турбинок напорные баки устанавли-
ваются на незначительной высоте над башнями с таким лишь
расчетом, чтобы из них кислота самотеком могла попасть в ма-
ленькие промежуточные бачки каждой отдельной турбинки.
Кожухи баков выполняются теперь исключительно из железа.
Если бак не герметизирован, то его кожух надо изнутри футеро-
вать, так как при колебании уровня кислоты остающаяся на
366
стенках кислота разбавляется влагой воздуха и травит железо.
При переходе на герметически закрытые сборники футеровка
оказалась излишней, так как при этом над кислотой создается
атмосфера, при которой разбавления кислоты на смачиваемых ею
стенках не наблюдается. На рис. 190 изображен нижний цирку-
ляционный сборник. Верхние циркуляционные или напорные баки;
Рис. 190. Циркуляционный сборник (нижний):
1—железное днище; 2—железные стенки; 3—футеровка плитками; 4—чугунное
седло пробки; 5—штурвал пробки; 6—стакан-фильтр.
делаются из железа, причем внутри бака устраивается приспо-
собление для измерения количества кислоты, поступающей
в башню.
9. Насосы
В старых камерных системах для передачи кислоты пользовались чугун-
ными монтежю. Ввиду низкой их производительности и чрезвычайно низкого-
козфяциента полезного действия от этого способа перекачки давно уже
отказались и перешли на насосы. На западноевропейских заводах на камер-
ных системах часто можно встретить поршневые насосы с производитель-
ностью 10,12 м3/час (Форстера, Гайяра и пр.). На башенных системах, где-
приходится перекачивать громадные количества кислоты, эти иасосы не при-
меняются вследствие малой их производительности. Установленные на первых
двух башенных системах в СССР плунжерные насосы Ферраряса с масля-
ной мембраной также быстро вышли нз употребления по той же причине.
В настоящее время на всех башенных, а также и на остав-
шихся в работе камерных системах применяются исключительно
центробежные насосы различной производительности (от 15 до
90 №/час). Схема центробежного насоса изображена на рис. 191.
Принцип действия этих насосов основан на отбрасывании жид-
кости лопастями 3 колеса 2 от центра к периферии корпуса 1
насоса под действием центробежной силы при вращении колеса.
Жидкость, поступающая в турбинку через всасывающую трубу
367
в центре и отброшенная с большей или меньшей силой к стенкам
корпуса, создает давление в корпусе (улите) насоса тем боль-
шее, чем больше число оборотов турбинки и чем больше ее диа-
метр, т. е. окружная скорость концов крыльев колеса, так как
при этом тем больше будет сила отбрасывания кислоты к стен-
кам корпуса. Создающимся при этом давлением жидкость вы-
брасывается через выход 4 из насоса в нагнетательный трубо-
провод.
В насос жидкость подводится непрерывно под действием
атмосферного давления. Вследствие этого высота всасывания
центробежного насоса не может превышать теоретического
столба жидкости, равного по давлению одной атмосфере.. Для
башенной кислоты при удельном весе ее, равном 1,7, эта высота
будет равна не более 10: 1,7 = 6 т.
В практике на эту высоту центробежные насосы не могут
всасывать башенную кислоту вследствие потери давления во
всасывающем трубопроводе, а иногда, кроме
этого, и вследствие подсоса воздуха через
неплотности в сальнике насоса.
Перед пуском в ход насоса необходимо
бывает заполнить всасывающий трубопро-
вод и насос кислотой, что связано с боль-
шими неудобствами. Поэтому на сернокис-
лотных заводах центробежные насосы, слу-
жащие для постоянной перекачки кислоты,
всегда устанавливаются так, что кислота
в них поступает самотеком из
стоящих сборников. И только
бежные насосы, обслуживающие приямок,
всегда помещаемый ниже уровня
Рис. 191. Схема цент-
робежного насоса:
7 — корпус; 2 — колесо;
3—лопасти; 4—выход.
выше их
центро-
пола для
сбора кислоты, попадающей из неплотностей насосов и другой
аппаратуры, ставятся иногда выше этого приямка, что и вызы-
вает необходимость перед каждым пуском насоса заливать его
кислотой. Высота подъема (напора) центробежного насоса зави-
сит от скорости, с которой жидкость покидает колесо. Эта ско-
рость зависит от диаметра колеса и числа оборотов; она не за-
висит от удельного веса жидкости. Поэтому при определенном
числе обортов турбинки и определенном ее диаметре насос мо-
жет поднять жидкости разных удельных весов на одну и ту же
высоту. Разница лишь будет в затрате энергии на работу; по-<
следняя, как известно, равняется произведению веса жидкости
на высоту ее подъема.
Фактическая высота, на которую оказывается поднятой жид-
кость, не соответствует затраченной работе, так как часть ее те-
ряется на преодоление трения в трубопроводах и на преодоле-
ние сопротивления вентилей и кранов. Вследствие этого для
подъема кислоты на башни, высота которых с фундаментом и
высотой верхних баков не превышает 25—30 л, требуются на-
сосы, создающие полный напор в 35 л.
368
Имеется много разновидностей центробежных насосов. На
наших заводах наиболее распространена разновидность насоса,
близкая по конструкции к насосам Амаг-Гильперта с сальниковым
уплотнением. Другая разновидность центробежных насосов хотя
распространена на наших заводах в значительно меньшей сте-
пени, но интересна по своей конструкции — это насосы с бес-
сальниковым уплотнением типа Вейзе.
Ввиду того что в связи с интенсификацией работы башенных
систем потребовалась перекачка кислот высокой температуры,
неблагоприятно отражающейся на сальниковой набивке (что вы-
зывает перебои в работе насосов типа Амаг-Гильперта), невиди-
мому, в 'недалеком будущем заводам придется перейти на на-
сосы с бессальниковым уплотнением.
Рис. 192. Разрез насоса Амаг-Гнльперта:
1—нагнетательный патрубок; 2—всасывающий патрубок; 3 н 4—фланцы;
5 и 8—станина, 6—крышка; 7—защитная втулка, 9—подшипники.
Насосы Амаг-Гильперта. Корпус насоса Амаг-Гильперта
(рис. 192) выполняется обычно из специального чугуна; он за-
жат фланцами 4 и 3, из которых фланец 3 составляет одно це-
лое со станиной. Станина сконструирована так, что на фунда-
менте находится только часть ее 5, другая же часть 8 вместе
с прижатым к ней корпусом насоса выходит за край фундамента
и фундаментной плиты. Такое устройство предусматривает воз-
можность стока вне фундамента кислоты, которая может про-
никнуть через неплотности сальника или других частей корпуса
насоса. Под эту часть насоса устанавливается противень, соеди-
ненный с общим сборным жолобом, уложенным в верхнем строе-
нии пола помещения и ведущим к так называемому аварийному
приямку, в котором собирается кислота от всех насосов. Вал
насоса в станине опирается на два роликовых или шариковых
подшипника 9.
24 Зак. 5019. Технология серной квелоты.
369
Рабочее колесо насоса имеет в своей втулке крыльчатку 4
(рис. 193), которая разгружает сальник, убирая кислоту из ка-
меры 5 в камеру 6, что способствует герметичности сальника.
В случае проникания через неплотности сальника кислоты она
скопляется в камере 7 и вытекает наружу через отверстие 2.
Рис. 193. Разгрузка сальника:
7 —камера; 2—отаерстие; 3—камера; 4—крыльчатка; 5 и 6—камеры.
2.
Станина насоса и электромотор покоятся на одной общей чугун-
ной плите.
В уплотнительное устройство насосов этого типа введено
усовершенствование (Ильченко),
Рис. 194. Бессальниковое уплотнение
Ильченко:
1—упор; 2—станина; 3—пружина.
устранившее недостатки, свя
занные с нестойкостью против
нитрозы применяемых в саль-
никах набивок (рис. 194). Прин-
цип его состоит в следующем.
Подвижная втулка, сидя-
щая поверх защитной втулки
вала, прижимается к гнезду
улитки двумя пружинами, чем
создается уплотнение, и кисло-
та не протекает наружу из на-
соса. При первоначальном пуске
насоса поверхности соприка-
сающихся частей между втул-
кой и улиткой смазываются.
При дальнейшей же работе тре-
ние втулки о гнездо улитки
ослабляется вследствие смазы-
вающего действия кислоты. На-
жатие пружин регулируется
винтами, помещенными на тра-
версе. Зазор между защитной
втулкой вала и уплотнительной втулкой имеет сальниковую
набивку.
Насосы Вейзе. Основное отличие насосов этого типа от
вышеописанного заключается в том, что они имеют дополнитель-
ные детали, заменяющие собой сальник.
370
Главными частями насоса Вейзе являются (рис. 195): корпус,
состоящий из двух частей 1 и 2, рабочее колесо 3, скользящее
колесо 4, разбрызгивающее колесо 5 и особое приспособление
б и 7, которое при пуске насоса сдвигает вал 8 вместе с наса-
женным на него колесом в левую сторону на 0,5 мм.
Передвижение вала в сторону насоса имеет целью отодвинуть
втулку вала 9 от скользящего колеса 4, чтобы дать свободу дви-
жения рабочему колесу насоса. Это передвижение осущест-
вляется при помощи приспособления 10, действующего вследствие
центробежной силы. При остановке насоса это приспособление
Рис. 195. Насос Вейзе (разрез):
£ и 2—корпус; 3— рабочее колесо; 4—скользящее колесо; 5—разбрызгивающее колесо;
6 и 7—особое приспособление для передвигания вала; 8—вал; 9—втулка вала; 70—приспо-
собление для передвижения; 77—пружина.
прекращает свое действие и вал пружиной 11 отодвигается на
свое место. При этом втулка вала 9 вновь тесно прижимается
к колесу 4 и этим предотвращает вытекание кислоты из насоса.
Разбрызгивающее колесо 5 служит во время движения на-
соса для предохранения от попадания в подшипник кислоты, про-
никающей через неплотности. Кислота попадает на колесо 5, от-
брасывается обратно к стенке, стекает по стенке вниз и затем
выходит из камеры.
Корпусы и крыльчатки насосов, служащих для передачи сер-
ной кислоты и нитрозы, отливаются из термосилида — специаль-
ного чугуна, содержащего до 17% кремния (иначе — силекса).
24* 371
Корпусы насосов, служащих для перекачки азотной кислоты,
выполняются из специальной стали типа V2A.
Ввиду того что кремнистый чугун (термосилид, или силекс)
отличается очень высокой твердостью и его обработка связана
с большими затруднениями, а также ввиду большой его хрупко-
сти большинство башенных заводов перешло на применение на-
372
сосов из обыкновенного серого чугуна. Этот чугун подвергается
незначительной коррозии нитрозной башенной серной кислотой и
поэтому вполне удовлетворяет тем требованиям, какие предъ-
являются к насосам на башенных сернокислотных заводах.
10. Коммуникация
Все вышеописанные аппараты — башни, холодильники, сбор-
ники, насосы и прочие — соединены между собой коммуникацией
для циркуляции и транспорта кислот.
Кислотопроводы от башен к холодильникам изготовляются на
всех наших башенных заводах в виде чугунных труб, соединен-
ных между собой при помощи раструбов с законопаткой растру-
бов асбестовым шнуром и
кой его.
Исключение пред-
ставляет кислотопро-
вод, идущий от 1 -й баш-
ни Гловера до холодиль-
ника. В этом кислото-
проводе чугун или же-
лезо не могут быть
применены без тщатель- J
ной футеровки кисло- 1
тоупорными материала-
ми на кислотоупорной
замазке, вследствие то-
го что 75—76%-ная
серная кислота при температуре выше 90° сильно действует на
эти металлы. Поэтому этот кислотопровод выполняется в виде же-
лезного жолоба, обложенного внутри кислото|упорным кирпичом
или плитками на андезитовой или иной кислотоупорной замазке.
Трубопроводы, по которым протекает кислота от холодиль-
ников к сборникам, а также кислотопроводы между башнями
Гей-Люссака и сборниками представляют собой чугунные трубы,
соединенные также на раструбах. Особенность этого участка
коммуникации заключается в том, что для направления кислоты
той или иной башни в любой из сборников в трубопроводах, под-
водящих к сборникам кислоту, имеются распределительные ко-
робки с отверстиями внизу, закрываемыми коническими проб-
ками. Все кислотопроводы располагаются параллельно друг
около друга над сборниками, стоящими в одну линию, причем
над каждым сборником в каждом кислотопроводе имеется рас-
пределительная коробка. Пример такой коммуникации можно ви-
деть на рис. 196. Внутренние диаметры кислотопроводов колеб-
лются от 250 до 350 мм в зависимости от количества кисдоты.
Трубопроводы, транспортирующие кислоты от сборников к на-
сосам, имеют перед насосом запорное приспособление — вентиль
373
(рис. 197), задвижка и т. п., при помощи которых можно сборник
разъединить с насосом, а также регулировать впуск кислоты из
сборника в насос. Нагнетательные трубопроводы ведут кислоту
от насосов (рис. 198) в напорные баки, расположенные над баш-
нями, и далее от этих
баков — к промежу-
точным бакам каж-
дой отдельной тур-
бинки.
Все эти трубопро-
воды выполнены из
чугуна и соединены
при помощи растру-
бов так, как указано
выше.
Размеры нагнета-
тельных трубопрово-
дов в зависимости
от производительно-
сти системы, а следо-
вательно, и от коли-
чества циркулирую-
щих в башнях кис-
лот, колеблются от
450 до 200 тм.
11. Аппаратура для
снабжения башен
и камер окислами
азота
На сернокислот-
ных заводах окислы
азота, теряющиеся
вследствие несовер-
шенства процесса,
пополняются путем
ввода в систему азот-
ной кислоты. Кислоту
эту из бутылей зали-
вают в небольшой
бак, стоящий на зем-
ле и соединенный с
насосом, изготовляе-
мым из стали типа
V2A. Этот насос поднимает азотную кислоту в керамиковый или
изготовленный из стали V2A бачок, стоящий над продукцион-
ными башнями. Из этого бачка кислота спускается непрерывной
струей в продукционные башни. Ток ее регулируется краном.
375
Для транспортирования азотной кислоты в железных цистер-
нах ее смешивают с 10%-ной крепкой серной кислотой. Эта
смесь, называемая меланжем, не действует быстро на железо.
Перевозка излишних 10% серной кислоты окупается устранением
больших расходов на стеклянные бутыли и всех неудобств, свя-
занных с громоздкой упаковкой бутылей, требующих значительно
ббльшего количества вагонов.
Для приема и хранения меланжа на сернокислотных заводах
применяются железные герметически закрытые резервуары, в ко-
торые меланж из ж.-д. цистерны перекачивается насосами. Над
продукционными башнями устанавливаются для меланжа желез-
ные, футерованные кислотоупорными плитками, небольшие сбор-
ники.
На некоторых башенных и камерных сернокислотных заводах
питание башен осуществляют вместо азотной кислоты и меланжа
окислами азота, полученными на самом заводе окислением амми-
ака, который транспортируется с аммиачных заводов или в виде
жидкого аммиака или чаще в виде аммиачной воды.
Аммиачная вода с содержанием 20% МНз из ж.-д. цистерны 12
(рис. 199) перекачивается насосами 11 в хранилища 13, откуда
теми же насосами подается в напорный бак 1‘, из него аммиачная
вода поступает в колонку 7 через уравнитель 2, регулирующий
поток воды, и через теплообменник 3, нагревающий ее те-
плом выходящей из колонны жидкости, имеющей температуру
100°.
Разделительная колонна состоит из трех частей: 1) нижней,
в которую подается пар, 2) средней, куда вводится воздух, и
3) верхней, служащей для улавливания брызг и смешения ам-
миака с воздухом.
Подогретая в теплообменнике аммиачная вода поступает в ко-
лонну через разбрызгиватель; навстречу ей поступают пар и воз-
дух, подводимый воздуходувками 5.
Выделенный из аммиачной воды аммиак смешивается с воз-
духом. Для поддержания правильной концентрации аммиака
в аммиачно-воздушной смеси в верхнюю часть колонны подво-
дится дополнительный воздух. Правильная и равномерная кон-
центрация аммиачно-воздушной смеси необходима во избежание
образования хлопков, связанных с большим, чем 10—12%, со-
держанием аммиака в смеси.
Температура выходящей из колонны газовой смеси равна
30—35°.
Из колонны газовая смесь поступает в трубчатый дефлегма-
тор 4, где температура его падает до 25° вследствие охлаждения
водой, циркулирующей в пространстве между стальными труб-
ками, по которым проходят газы. При этом происходит конден-
сация водяных паров и полученная вода стекает из дефлегматора
обратно в колонку.
Далее газовая смесь проходит отсекатель 8, назначение кото-
рого — прекращать (отсекать) доступ газа из колонны в контакт-
376
ные аппараты 9. Отсекатель имеет клапан с электромагнитом.
Последний сблокирован с моторами воздуходувок и потенцио-
метром контактных аппаратов.
При остановке по какой-либо причине мотора вентилятора
размыкается электромагнит и в отсекателе закрывается клапан.
То же происходит при увеличении температуры в контактном
аппарате выше 800°.
Из дефлегматора газы поступают в контактные аппараты 9,
в которых катализатором служат платинородиевые сетки (90%
Pt и 10% Rh). До поступления
на сетку аммиачно-воздушная
смесь подогревается в тепло-
обменнике '10 выходящими
нитрозными газами. Послед-
ние из контактных аппаратов
поступают по трубопроводу в
1-ю башню Гловера с темпера-
турой 600°.
Подаваемый в разделитель-
ную колонну воздух до посту-
пления в воздуходувки 5 под-
вергается очистке в фильтре 6.
Рнс. 200. Гидравлический
затвор в камере.
Рис. 201. Камеры Морица с же-
лезным каркасом.
12. Камеры
Свинцовые камеры в старых камерных системах устраивались следующим
образом.
На деревянном каркасе, расположенном на деревянном помосте (пол) и
состоящем из нижней и верхней обвязок и вертикальных стоек с промежу-
точными деревянными брусками, навешивался рольяый свинец в виде верти-
кальных стенок; пол выстилался тоже рольным свинцом; края его загибались
кверху в виде бортов, образуя противень, в который спускались нижние края
377
стенок. Крышка камеры состояла из рольного свинца, подвешенного к гори-
зонтально лежащим на верхней обвязке деревянным балкам. Противни (под-
доны) камер заливались серной кислотой, которая создавала гидравлический
Рис. 202. Система Мильс-Паккара (схема):
1—вход газа; 2—басня Гловера; 3—камеры; 4—башни Гей-Люссака; 5—выход газа.
стеклянные колпаки (фонари), соединенные с камерным пространством и да-
вавшие возможность по изменению цвета газа своевременно замечать начи-
нающееся отклонение от
Рис. 203. Турбодисперсер
Мартена:
1—электромотор; 2—турбин-
ка; 3—вход кислоты.
нормы в процессе. На стенках камер также име-
лись термометры и так называемые капельницы,
т. е. узкие стеклянные цилиндрические стакан-
чики, в которые непрерывно из свинцовых тру-
бочек с гидравлическими затворами поступала
с внутренней поверхности свинцовых стенок камер
та кислота, которая только что образовалась на
стенках (капельная кислота). При помощи по-
стоянно находившегося в капельнице ареометра
определялась крепость этой кислоты, что необхо-
димо было для контроля за впуском распылен-
ной воды в камеры. Колебания температуры в ка-
мерах сигнализировали также об уклонениях хода
процесса от нормы. Разрез конструкции нижней
части камер с гидравлическим затвором и кон-
струкции ее потолка показан иа рис. 200.
Современные камерные системы имеют иную
конструкцию. В связи с тенденцией придавать им
форму очень узких и высоких камер для улучше-
ния охлаждения газа (камеры Морица, рис. 201)
каркасы их выполняются из железа, что одно-
временно придает им большую противопожарную
безопасность.
В противоположность старому типу камер
с деревянным каркасом, высота которых обычно
не превышала 8 м, современные камеры со сталь-
ным каркасов имеют высоту .18—ЙО .и и незначи-
тельную ширину 5 м-
На некоторых иностранных заводах приме-
няются свинцовые камеры с наклонными стенками, орошаемыми снаружи во-
дой, так называемые камеры Милльс-Паккара (рис. 202). Такие камеры обес-
печивают более усиленное охлаждение газов, возможность вести процесс
с повышенным содержанием окислов азота в газах и уменьшение коррозии
378
свинца стенок. Для отвода излишнего тепла из обычных прямоугольных
ка ..ер на иностранных заводах применяют распыление внутри камер вместо
воды слабой серной кислоты, которая по выводе ее из камер охлаждается
в змеевиковых холодильниках и затем насосами вновь поднимается в резер-
вуары, находящиеся над камерами, для впуска ее в камеры.
Распыление кислот осуществляется диоперсерами (рис. 203, система Мар-
тена). Работа дисперсеров всех систем основана на быстром вращении
диска или турбинки, приводящихся в движение моторчиком с вертикальным
валом: на диск непрерывной струей притекает кислота. Дисперсеры уста-
навливаются на потолке камер и снабжены гидравлическими затворами. Для
того чтобы в камерах с внутренним орошением распыленной кислотой равно-
мернее распределялась эта кислота, Гайяр предложил тип цилиндрических
камер.
В настоящее, время в камерах Милльс-Паккара кроме наружного охла-
ждения применяют- также внутреннее распыление холодной слабой серной
кислоты, что дает возможность довести интенсивность работы этих камер
до 20—25 «г/.м3.
Для увеличения поверхности соприкосновении газов с кислотой в США
применяют свинцовые камеры с насадкой из кислотоупорного материала (на-
пример из кирпичей). Свинцовые стенки камер изнутри футеруются кислото-
упорным кирпичом. Камеры имеют вертикальные кирпичные перегородки и
образуют, таким образом, ряд последовательно соединенных камер, орошаемых
охлажденной серной кислотой.
Эта система камер, так называемая система Лариссона, является, по
существу, переходом от камерных к башенным системам.
IV. Технологический режим башенных
и камерных систем
1. Характер встречи газа и кислоты в башенных системах
На отдельных башенных заводах устанавливается от 5 до
7 башен. Большей же частью башенная система состоит из шести
башен.
В 1-й башне для более полной денитрации кислоты (а при по-
лучении продукции в виде купоросного масла и для более значи-
тельного ее упаривания) необходим противоток газа и жидкости.
В абсорбционных башнях для полноты улавливания окислов
азота противоток газа и жйдкости более эффективен, чем прямо-
ток. Предпочтение противотоку надо отдать и в отношении по-
следней продукционной башни: лучше охлаждается газ, что
важно для последующих процессов окисления NO и абсорбции
TWa.
При оформлении системы наряду с технологическими сообра-
жениями приходится учитывать и аппаратурно-конструктивные.
Так, чередование прямотока с противотоком дает возможность
сократить протяженность газоходов. Практически без ущерба
процессу прямоток может быть осуществлен во 2-й башне (если
3-я башня является последней продукционной) и в 4-й башне
(1-й абсорбционной). Однако практически целесообразнее вклю-
чать 2-ю башню также противотоком, так как при этом, в случае
необходимости выключить 1-ю башню (серьезный ремонт, пере-
насадка и др.), 2-я башня легко может выполнять роль
1-й башни.
379
2. Орошение башен
Схема орошения. Схема орошения (т. е. порядок перетока ки-
слоты с башни на башню) неодинакова на разных заводах. На
рис. 149 (стр. 312) показана четырехнитрозная схема орошения,
на рис. 204 — пятинитрозная.
Четырехнитрозная схема орошения означает, что в системе
вращается четыре сорта нитроз, различающихся концентрацией
окислов азота в них. Наиболее нитрозная кислота подается на'
продукционные башни. Самую нитрозную кислоту в системе
в практике называют крепкой нитрозой. Наименее нитрозная ки-
слота подается на последнюю башню в системе (последний гей-
люссак). Эту кислоту называют слабой нитрозой. Другие кислоты
Рис. 204. Схема орошения башенной системы
Винницкого завода.
называют средними
нитрозами.
В последние годы
с целью наиболее
обезопасить башни
от засорения пылью,
приносимой газами,
1-я башня или оро-
шается в цикле с по-
следней или «сама
на себя» — на другие башни вытекающая из 1 -й башни кислота
не передается. В первом случае пыль может попасть только
в 1-ю и в последнюю башни, а во втором — только в 1-ю баш-
ню. Эти башни для предупреждения от засорения насаживаются
обязательно кольцевой насадкой.
О количестве пыли, приносимой в башни с газами, дают пред-
ставление следующие цифры.
После электрофильтров, очищающих газы, получаемые от об-
жига колчедана в механических печах, содержание пыли соста-
вляет 0,15—0,25 г на 1 м3 нормального газа. Если содержание
SO2 в газе равно 9%, то, следовательно, на 1 т продукции при-
ходит пыли в систему:
• 0,15 = 381 г = 0,382 кг.
В систему, выпускающую в сутки 250 т H2SO4, в течение ме-
сяца, таким образом, будет принесено пыли с газами:
0,382 • 250 • 30 с= 2865 кг.
При работе на печах пылевидного обжига запыленность газа
перед башнями часто составляет 0,5 г на 1 м3 нормального газа,
а иногда доходит до нескольких граммов.
’ Количество приносимой в систему пыли во всех (при неизмен-
ности общей выработки системы) случаях повышается с пониже-
нием концентрации SO2 в печном газе. Из приведенных фактов
понятна актуальность всех мероприятий, направленных против
3S0
засорения башен грязью (дальнейшее повышение степени очистки
газов в электрофильтрах и, в> частности, установка при печах
пылевидного обжига двух последовательных ступеней электро-
очистки; применение типов насадок, минимально задерживающих
грязь в башнях; регулярная очистка сборников и холодильников
кислоты от грязи; своевременная промывка засорившейся башни;
применение отстойника грязи для кислоты, вытекающей из пер-
вой башни).
Засорение башен грязью повышает сопротивление башен, что
в практике работы заводов часто приводит к вынужденному зна-
чительному снижению выработки, а иногда и к остановке си-
стемы.
Количество орошения. Общее количество орошения башен
принято оценивать величиной отношения количества поступающей
во все башни кислоты к количеству кислоты, производимой си-
стемой за то же время.
Это отношение принято называть кратностью орошения. Крат-
ность орошения в практике отдельных заводов колеблется в пре-
делах от 25 до 120.
Приводим данные о количестве орошения по отдельным заво-
дам (табл. 83)..
Табл ц а 83
Институты, проводившие заводские опыты Интенсив- ность в кг'м3'сутк > Орошение в jw3 на 1 т продукции в моногид- рате
МХТИ 1930 г 15,8 43,5
МХТИ 1931 г 26,8 28
МХТИ 1936 г 104 67,5
НИУИФ 1937 г 124,9 52,5
НИУИФ 1937 г 61,9 60,50
Распределение общего количества орошения по отдельным
башням зависит от диаметра башен, от их относительной вели-
чины и от ряда других факторов?
Так как из 1-й башни необходимо получать максимально де-
нитрированную кислоту (продукция, кислота, направляемая на по-
следнюю башню для возможно полной абсорбции окислов азота),
то орошение этой башни не может быть большим. Так как сма-
чивание всей насадки башни обязательно, то 1-я башня должна
иметь относительно небольшой диаметр.
Исходя из того, что последняя башня должна орошаться по
возможности малонитрозной кислотой, а количество получаемой
в системе (из 1-й башни сверх продукции) денитрированной ки-
слоты невелико, последнюю башню тоже следует ставить о воз-
можно малым диаметром.
381
На многих системах денитрированной кислоты (сверх продук-
ции) нехватает для полного орошения последней башни (доста-
точного смачивания всей насадки) и приходится последнюю
башню частично орошать «саму на себя».
Концентрация орошающих кислот. Как это видно из рассмо-
трения процессов кислотообразования и абсорбции окислов азота,
продукционные башни целесообразнее орошать мало концентри-
рованной, а абсорбционные башни возможно более концентриро-
ванной (по % HaSO4) нитрозой. При этом приходится принимать
во внимание переток кислоты из продукционных башен в абсорб-
ционные и обратно, и в связи с этим затруднительность полной
диференциации орошения (продукционных башен, с одной сто-
роны, и абсорбционных — с другой) по концентрации H2SO4. Так
как в нитрозе концентрации 75—78% H2SO4 сернистый газ оки- -
сляется достаточно хорошо и достаточно хорошо растворяются
окислы азота, то концентрации циркулирующих во всей системе
кислот обычно поддерживаются в этих пределах. Поднимая кон-
центрацию нитроз (по % H2SO4) в интересах повышения скоро-
сти и увеличения полноты абсорбции окислов азота в гей-люсса-
ках, наносимый этим ущерб процессу в продукционных башнях
можно устранить в некотором пределе соответствующим повы-
шением температуры и нитрозности нитроз, орошающих продук-
ционные башни.
Стремление провести возможно полную диференциацию кон-
центрации (по H2SO4) орошающих нитроз в последнее время
привело к схеме орошения в три цикла: внешний цикл (1-я и по-
следняя башни) с концентрированной нитрозой (для улучшения
условий процесса абсорбции N2O3 в последнем гей-люссаке),
внутренний цикл (3-я и 4-я башни в 6-башенной системе, или
3-я башня орошается «сама на себя» в 5-башенной системе) с са-
мой мало концентрированной нитрозой (по % H2SO4) в интере-
сах интенсификации процесса кислотообразования в 3-й башне,
устраняющей проскок сернистого газа в гей-люссаки. В среднем
цикле орошения (охватывающем 2-ю и предпоследнюю башни)
циркулирует средняя (по концентрации H2SO4) нитроза.
Петерсен в последних своих статьях рекомендует 4-башенную
систему с двумя циклами орошения: внешний цикл (1-я и 4-я
башни) с крепкой кислотой и внутренний цикл (2-я и 3-я башни)
со слабой кислотой. Для внешнего цикла Петерсен рекомендует
кислоту концентрации 75—84% H2SO4, а для внутреннего
цикла — концентрации 70—75% H2SO4. По мнению Петерсена
при такой схеме 80% всей продукции будет получаться во 2-й
башне, а 90% улавливаемых окислов азота будет абсорбиро-
ваться в 3-й башне.
3. Температура кислот и газа
Температура орошающих кислот выбирается из тех сообра-
жений, что скорости процессов кислотообразования и денитра-
ции увеличиваются с повышением температуры, а условия аб-
382
сорбции окислов азота улучшаются с понижением температуры.
Раньше, в связи с тем, что повышение температуры нитрозы вы-
зывало быстрое усиление коррозии свинца, а также приводило
к разрушению набивки сальников насосов, температура кислоты,
орошающей первые башни, поддерживалась ниже 50е. В послед-
нее время температура кислоты, орошающей первые башни, по
отдельным системам поддерживается в пределах 50—75 .
С техническим разрешением вопроса о перекачке горячей ки-
слоты температуру кислоты, орошающей продукционные башни,
целесообразно держать выше. Температура кислоты, выходящей
из 1-й башни как готовая продукция, обычно колеблется в пре-
делах 100—130 , а при выпуске купоросного масла достигает
200 . Температура кислот, орошающих гей-люссаки, выбирается
в зависимости от конкретных условий (температура охлаждаю-
щей воды, схема орошения). Надо стремиться к тому, чтобы тем-
пература кислоты, орошающей последний гей-люссак, была не
выше 35, а предпоследний — не выше 45е.
И с точки зрения полноты денитрации нитрозы в 1-й башне,
и с точки зрения процесса концентрирования серной кислоты
(если система выпускает купоросное масло), и с точки зрения
интенсивности кислотообразования желательно, чтобы темпера-
тура обжиговых газов, входящих в 1-ю башню, была возможно
выше. Верхний предел температуры здесь обусловливается тем,
что темдература газов, входящих в применяемые сейчас электро-
фильтры, не должна превышать 500 . Обычно температура газа
перед 1-й'башней поддерживается в пределах 300—400°. Для
того чтобы всю продукцию башенной системы получать в виде
купоросного масла, необходимо, чтобы температура газа перед
1-й башней (концентратором) была выше 375°.
Для поднятия температуры газов по выходе из печного от-
деления и для поддержания этой температуры постоянной, вне
зависимости от нагрузки печного отделения, есть предложение
осуществить регулируемое возвращение в печь горячего воздуха,
выходящего из вала печи.
Температура газов в дальнейших точках башенного отделения зависит ог
целого ряда причин: интенсивность работы башни, плотность орошения, ха-
рактер встречи газа с кислотой (противоток или прямоток) и др. Обычно
температура газа, выходящего из il-й башни, выше средней арифметической
(между входом и выходом) температуры кислоты в башне на 2—15°. Темпе-
ратура газа, выходящего из последней башни, обычно превышает среднюю
температуру кислоты в этой башне меньше чем на 10°.
I
4. Концентрация SO± при входе в систему
Чем меньше процесс окисления БСД в продукционных башнях
зависит от обратного окисления NO, тем справедливее прямо
пропорциональная зависимость интенсивности работы продук-
ционной зоны от концентрации SO» в газах. Этот характер зави-
симости приобретает все большую устойчивость в производствен-
ной практике, где одним из основных методов интенсификации
3С3‘
является неуклонное повышение отношения между количествами
Ы2О3 и SO2, подаваемыми в продукционные башни.
В производственных условиях башенных систем, работающих
на газах, получаемых от обжига колчедана в воздухе (даже в том
случае, когда NaOb подается в продукционные башни значи-
тельно меньше, чем в эквивалентном отношении к SOs), прямая
пропорциональность между концентрацией SO2 и интенсивностью
работы башен сохраняется по крайней мере до содержания
9—9,5% SO2 в газах.
Петерсен дает следующую зависимость (из опыта работы его
установок) производительности системы (постоянного объема) от
процентного содержания SO2 в обжиговых газах (табл. 84).
Таблица 84
% SO2 Производитель- ность системы в mj су тки Рост концентра- ции SO2 против первого примера Рост ности производите.! ь- против первого примера
2,5 15 1 1
5,0 30 2 2
7,5 40 3 2,66
2-я башня одного из заводов во время опытов в 1932 г. при
прочих равных показателях режима произвела кислоты в сутки 114491 кг
при средней концентрации SO2 в башне <1Д8% и 26803 кг при средней
концентрации SO2 в башне 2,21%. Таким образом увеличение концентра-
2 21
дни SO2 в ’ = 1,87 раза повысило интенсивность работы башни
1,10
26 803 , ос
® 14491 =1’86 Ра3а-
Хотя в газах, поступающих в гей-люссаки, SO2 быть не
должно, концентрация SO2 в начале системы имеет определенное
и значительное влияние на процесс в гей-люссаках.
Если считать что на 1 т образующейся H2SO« выделяется
определенное количество окислов азота из нитрозы, то концен-
трация NO в газах при входе в гей-люссаки будет обратно про-
порциональна величине объема газов, приходящих в гей-люссаки,
падающей на 1 т продукции.
Как показано выше, объем обжигового газа, приходящегося
, 22860 „ „
на 1 т продукции, равен —р—. Так как при образовании 1 т
H2SO4 расходуется 228 /и3 SO2 и 114 /и2 Ог, то на 1 т H2SO4, если
нет подсоса воздуха и без учета окислов азота, в гей-люссаки
приходит газов:
ив-
газ
/22860
\ Р8Ог
342) ж8.
384
Если задаваться равными потерями окислов азота с хвосто-
1 рд
выми газами на 1 т продукции во всех случаях, то величина ~
рд
{р°д — парциальное давление окислов азота перед входом в гей-
люссаки, а р* — парциальное давление окислов азота при выходе
из системы) будет неизменной и, следовательно, неизменной бу-
дет и величина:
Так как величина абсорбера пропорциональна произведению
Qra3 t (где Qra3 —секундный расход газа), а величина Огаз про-
порциональна Vra®’ , то объем гей-люссака имеет такой же ха-
рактер зависимости от концентрации SO2 в обжиговых газах, как
и величина V*®’ . Таким образом потребный объем абсорберов
(гей-люссаков) уменьшается более чем прямо пропорционально
повышению концентрации SO2 в газах.
Если учесть еще, что с повышением концентрации окислов
азота в газах перед гей-люссаком (благодаря повышению концен-
трации SO2 в обж.иговых газах) повысится движущая сила аб-
сорбции окислов азота, то сокращение потребной кубатуры гей-
люссаков (на 1 т суточной продукции) от повышения процента
SO2 в газах, поступающих в систему, должно быть еще больше.
В общем получается, что потребный объем всей башенной си-
стемы уменьшается (а интенсивность системы увеличивается) не
менее чем пропорционально концентрации SO2 в газах, посту-
пающих в систему.
Кроме того, как это ниже подробно обосновывается, расход
азотной кислоты на 1 т продукции H2SO4 понижается более чем
прямо пропорционально содержанию SO2 в поступающих в си-
стему газах.
5. Нитрозность кислот и денитрация нитроз
Нитрозность орошения по отдельным башням устанавливается
на основании следующих соображений: 1) повышение нитрозно-
сти кислот, поступающих на Продукционные башни, в значитель-
ных пределах ускоряет процесс кислотообразования; 2) нитроз-
ность кислоты, орошающей 1-ю башню, должна быть такой,
чтобы при учете всех других факторов происходила денитрация
продукции до стандарта.
Приводим данные по некоторым заводам о выделении окис-
лов азота из нитроз в первых башнях (табл. 85).
Коэфициеиты денитрации высчитаны по формуле:
j,-.- °
А PHN03 РИО2 ’
где q — количество десорбированных окислов азота в г HNOa в час,
А__поверхность насадки башни в м2,' PHNOj — средняя нитрозность кислоты
25 Зак. 50И. Технология серной кислоты.
385
Таблица 85
Режим в 1-й башне | линейная скорость газа в м/сек ю Ю Оз СО Tf CN СО СМ СО СМ СМ 10 О
средняя концентра- ция кислоты в 0 Вё ** ’“1 ° со со со' со со сз Ю 1Q Ю Ю ю 1О
нитрозность кислоты в °/0 HNO3 I выход со со ю ю о со 03 О О С СМ О о о о о о о
| вход rf UOb-ЮГ-- ю со °°- со со со со CM Tf“
°/о SO2 в газе |выход СМ ю о о со СО со со тН со ио"
I вход СО со LQ г- °Ч. °° оз* оо оз* оГ
темпера- тура кислот выход оо со с? см о со —I —' О СМ СМ —<
вход | СО О оэ со со ю со ю
темпера- тура газа вход |выход со СО Ю 03 05 оз СО СО 03 СО 03 03
О оз о о о О ' Ь- Сз СО ю со СО см см см со
Коэфи- циеит денитра- ции Ю Ю Г- Ю Tf СО 03^ Оз со Сз ьС со ю см
Институты, проводившие заводские опыты • • • . • со u г • е* с2 СО °? СО со со со о> о со со со _ „ О О> С5 g ” ~ "I s е & S S S х s S Н Н f-1 S >> >> X X X X S S g S S >> х х
386
в башне в % HNCh; Pso2 —средний процент SOs в газах в башне. Коли-
чество окислов азота, десорбированных в первых башнях, по приведенным
заводам находилось в пределах 0,094—0,4)10 молей HNO3 на 11 моль SOs,
поступившего* в систему.
Нитрозность кислот, орошающих гей-люссаки, в интересах
полноты и скорости улавливания окислов азота должна быть
возможно низкой.
В башенных системах современного аппаратурного оформле-
ния нитрозность крепкой нитрозы колеблется в пределах 3—6%,
а слабой нитрозы — в пределах 0,4—‘1,5%, считая на HNO3.
В табл. 86 приведены данные о количестве подаваемых окис-
лов азота по отношению к SO2 по отдельным башням.
Таблица 86
Институты, про- водившие завод- ские опыты Общая интенсив- ность системы 5 кг/м*/ сутки Подано в башни
1-я 2-я 3-я
молей n2o3 на моль посту- пившего so2 молей N2O3 на моль перера- ботанно- го в баш- не SO2: молей n2o3 на моль посту- пившего so2 молей N2O3 на моль пе- рерабо- танного в башне . so2 молей n2o3 на моль посту- пивше- го в башню so2
МХТИ, 1930 г. 15,8 0,058 0,48 . 0,153 0,324 0,485
МХТИ, 1931 г. 26,8 0,20 0,50 0,53 0,86 5,36
МХТИ, 1936 г. 77,5 0,055 0,304 0,425 0,547 4,75
УНИХИМ, 1936 г 76,3 0,032 0,28 0,48 0,63 4,10
НИУИФ,1937 г. 61,9 0,023 0,725 0,84 1,79 2,32
МХТИ, 1936 г. 104 0,039 0,284 0,66 0,767 4,85
Из приведенных цифр видно, что в 1-й и 2-й башнях окислы
азота совершали по нескольку циклов обращения (окисления и
восстановления).
Так в 1-й башне поданные окислы азота делали от 1,51 обо-
рота (опыт НИУИФ 1937 г.) до 3,58 оборота (опыты УНИХИМ
1936 г.).
25*
387
Таблица 87
Институты, проводившие заводские опыты Азот*- оборот в кг HNOg на 1 т H2SO4 Кислотообразова- иие по башням в % от общей вы- работки Выделение окислов азота в газ в % от азотооборота
1-я 2-я 3-я 1-я 2-я 3-я
МХТИ, 1931 г 261 42,1 40,2 17,7 67,42 32,581 —
МХТИ, 1932 г. . .’ . . . 167 52,7 20,5 25,3 1 2 1001 — —
НИУИФ, 1937 г 272 31,4 32,4 ^,4 2 60,2 39,81 —
МХТИ, 1936 г 246 18,23 .63,72 15,302 22,9 77,11 —
МХТИ, 1937 г 335 18,32 26,9 34,4 2 30,7 69,71 —
НИУИФ, 1937 г 530 89,5 9,5 2 32,7 67,3 1 —
УНИХИМ, 1936 г. . . . 210 11,4 67,5 18,60- 12,8 87,21 —
Таблица 88
Институты, проводившие заводские опыты Азото- оборот в кг HNO3 на 1 т H2SO4 Абсорбция по башням в % от общего количества абсорбированных окислов азота
Башни
2-я 3-я 4-я 5-я 6-я 7-я
МХТИ, 1931 г 261 45,33 44,07 10,6
МХТИ, 1932 г 167 21,44 27,75 29,59 21,22
НИУИФ, 1937 г 272 1,5 57,5 19,9 14,9
МХТИ, 1936 г 246 60,7 29,3 5,1 4,1 0,8
МХТИ, 1937 г 335 18,40 16,87 42,10 18,45 4,18
НИУИФ, 1937 г 500 52,4 41,4 6,2
УНИХИМ, 1936 г. . . . 210 14,9 81,6 2,0 1,3 0,2
1 В следующей башне начинается абсорбция окислов азота.
2 Остатки SO2 окисляются в последующей башне.
388
6. Переработка SO2,
эксорбции и абсорб-
ция окислов азота по
отдельным башням
Общее количество
окислов азота, выде-
ляемое в1 продукцион-
ной зоне в газовую
фазу в кг HNOs в
пересчете на 1 тпро-
дукции H^SCU, носит
название а з о то-
обор о т а.
Как величина азо-
тооборота, так и рас-
пределение общего
выделения окислов
азота по отдельным
башням зависят от
всех конкретных ап-
паратурных' и тех- с
нологических уело- 00
вий.
В табл. 87 приве- я
дены данные о рас- щ
пределении денитра-
ции и кислотообра-
зования по отдель-
ным башням по ряду
заводов.
Абсорбция окис-
лов азота (как видно
из приведенных дан-
ных и как об этом
уже; говорилось вы-
ше) начинается зна-
чительно раньше, чем
окончательно закон-
чится окисление
сернистого ангид-
рида.
Нетрудно вынести
следующее прибли-
женное соотношение
между процентом
SO2 в газах, посту-
пающих в систему,
Расход HNO3 в кг на 1 ш D л С-1 Е Ю сх со N СО Ю ОО т— —< СМ
Общая интенсив- ность в кг HoSCLc 1 53 м и оэ 3 Ю со Ю СП N СО О N Ф О СО
Абсорбционная зона свобод- ный объ- ем башен на 1 m суточной придук- ЦИИ < О СО со CM iO
поверх- ность на- садки в м2 на 1m суточ- ной про- дукции СО СО ьГ со со СП ОО СП ю см со со см
объем абсорб- ционной части на 1 m суточной продукции’ без футе- ровки СО ОО о см 1О со О СО СО •-«' Г-—М
общий N CM N GO <О N СО ОС О? Ю
Продукционная зона I интенсивность в кг H2SO4 в сутки на 1 л3 свобод- ного объема башен то Ю О О ,-7 см ОО ОО С5 см СМ тг
на 1 л2 поверх- ности насадки со со - ю ’ СП СО^ см см со
объем продукци- онной части си- стемы на 1 m су- точной продукции без футе- ровки 00 Ю со 05 ОО СП QO См rf rj7 см со
общий г-. Xf ь- О со р,- ь
Институты, проводившие заводские опыты | МХТИ, 1936 г УНИХИМ,1936г НИУИФ, 1937 г Опыты одного из заводов 1937 г
389
Таблица 90
№ башен по ходу газа Плотность кислот в сВё Нитрозность кислот в % HNO3 Температура кислот в °C Темпе- ратура газа в °C
вход ВЫХОД вход ВЫХОД вход ВЫХОД
1 58,4 58,4 1,70 0,20 46 124 348
2 59,5 59,2 5,00 3,22 76 94 104
3 59,5 59,3 5,17 4,76 76 89 102
4 59,3 59,4 4,50 5,17 52 78 62
5 59,2 59,3 3,43 4.50 46 51 55
6 58,4 58,4 1,27 1,77 42 47 40
Рис. 205. Кривые выделения и абсорбции окислов азота по башням
одного завода.
390
величиной азотооборота и наивысшей концентрацией окислов
азота в- газах:
р О, J04 • GHNOs • Pg02
N0“ 66,7-PS02 ’
где Ghno3—азотооборот в кг, PNo — наивысший процент оки-
слов азота (считая на NO) в газовой фазе.
В табл. 88 приведены данные р распределении абсорбции
окислов азота по отдельным башням. ‘
В табл. 89 приведены данные об интенсивности работы от-
дельных частей некоторых башенных систем.
Приведем подробные данные о режиме башенной системы
одного из заводов^ в сентябре 1938 г., работавшей в это время
с интенсивностью 90 кг H2SO4 на 1 лг3 объема башен в сутки
с расходом азотной кислоты 22 кг HNO3 на 1 т H2SO4 (табл. 90).
Процент SO2 перед входом в 1-ю башню был равен 9,6; про-
цент кислорода в хвостовых газах — 7,1; окислов азота в выхо-
дящих из системы газах — NOa 0,05%, NO 0,12%. На рис. 205
представлен процесс выделения и абсорбции окислов азота, а
также переработка SO» по отдельным башням.
7. Тепловой баланс башенной системы
и схема охлаждения кислот
Приход теплоты в башенную систему составляется из: 1) фи-
зической теплоты газов, 2) теплоты реакции образования моно-
гидрата, 3) теплоты разбавления моногидрата до концентрации
продукционной кислоты и 4) теплоты конденсации водяных па-
ров, приходящих в систему с печными газами.
Расход тепла составляется из: 1) теплоты, уносимой уходя-
щими из системы газами, 2) теплоты, уносимой охлаждающей
водой, и 3) теплоты, теряемой аппаратурой системы в окружаю-
щее пространство.
В табл. 91 приведены тепловые балансы башенной системы
для случаев концентрации газа, поступающего в систему, в 7 и
9,5% SO2 при 250 и 350°. Температура газа, уходящего из си-
стемы, для всех случаев принята равной 35°.
Влажность поступающих в систему газов принята в соответ-
ствии с 5%-ным содержанием НгО в колчедане. Концентрация
продукционной кислоты принята равной 76% H2SO4. Теплота раз-
бавления моногидрата до 76%-ной H2SO4 высчитывалась по фор-
муле Портера (стр. 418).
Так как потери тепла в окружающую среду обычно не боль-
ше 10% от общего прихода, то целесообразно (также и в целях
большей надежности результатов расчета потребной поверхности
охлаждения холодильников) относить весь расход тепла в си-
стеме (кроме тепла, уносимого хвостовыми газами) на охла-
ждающую воду. Весь расход тепла, указанный в табл. 91,
также отнесен на охлаждающую воду.
391
Раньше считалось, что на 1 т H2SO4, производимую сиотемой
в сутки, требуется от 8 до 12 № поверхности охлаждения холо-
дильников. Как показали заводские обследования и опыты
НИУИФ (Малин, Андронов, Кириченко и Отвагин), действи-
тельно потребная поверхность охлаждения составляет значи-
тельно меньшую величину. Обследование холодильников на
одном из заводов показало, что коэфициент теплопередачи по
отдельным холодильникам и змеевикам колебался в пределах
74—250 ккал/м2 • час. При должном уходе за. холодильниками
(регулярная очистка холодильников от грязи и продувка змееви-
ков) коэфициент теплопередачи по всем холодильникам и змее-
викам может быть не меньше 200.
Другой резерв интенсификации работы холодильников — по-
вышение средней температурной разности между охлаждаемой
кислотой и охлаждающей водой 'путем рационализации самой
схемы охлаждения. Раньше охлаждались кислоты, вытекающие
из всех башен. Попятно, что холодильники, охлаждавшие кис-
лоты из последних башен, работали в условиях незначительной
температурной разности. Рациональная схема охлаждения сво-
дится к. тому, что вся отводимая из системы теплота отбирается
охлаждением кислот, вытекающих или только из первых двух
или (максимум) из первых трех башен. При этом холодильники
работают в условиях более высокой разности температур ки-
слоты и воды.
НИУИФ составлена следующая схема охлаждения кислот
(расчет на 1 т H2SO4 продукции) применительно к летним усло-
виям химического завода (табл. 92).
Таблица 92
Башни
Предел ох-
лаждения
кислоты
в °C
119—30
111-54
111—35
Предел
нагрева
воды в ° С
Отнимается
тепла
в ккал
20—40
20-35
20—35
33
50
37,5
194000
86000
620 000
900000
Средневзвешенная разность температур при этом получается
равной 37,6°. При коэфициенте теплопередачи 200 потребная по-
верхность охлаждения равна:
900001 , „
37,6-200^24 ~ 4,95 М •
Те же авторы применительно к заводским условиям соста-
вили схему охлаждения, приведенную в табл 92а
Средняя разность температуры при этом получилась равной
48 . При коэфициенте теплопередачи 200 и количестве подлежа-
392
393-
щего отъему на 1 т H2SO4 тепла, равном 900 000 ккал, потреб-
ная поверхность охлаждения составляет
900000
48 • 200 • 24
= 3,9 №.
В табл. 92 и 92а количества теплоты отнесены к 1 т продук-
ции в моногидрате, считая выпуск продукции в виде 76%-ной
H2SO4.
Если система выпускает продукцию в виде! купоросного ма-
сла, то 'потребная поверхность охлаждения может быть еще
Т а в л и ц а 92а
Башни Предел ох- лаждения кислоты в °C Предел на- грева воды в °C Д/
1-я 130-37 20—40 43
2-я 105-55 20-35 50
меньше, так как: 1) умень-
шается теплота разбавления
моногидрата и 2) в связи со
значительным повышением
температуры кислоты, выте-
кающей из 1-й башни, еще
больше повышается средняя
разность между температу-
рой кислоты и температурой
воды. При работе системы
на выпуск купоросного масла
и при рациональной схеме охлаждения потребная поверхность
«охлаждения на 1 т суточной продукции составит не больше 3 м~.
Сверх приведенных цифр минимально необходимой поверхности
охлаждения в практике надо иметь резервную поверхность
в размере ~ 25% от минимально необходимой.
8. Подготовка окислов азота к абсорбции
Неоднократно проводившимися расчетами было показано, что
если окисление NO и абсорбция N2O3 протекают одновременно,
то течение обоих этих процессов значительно задерживается.
Этот факт объясняется тем, что часть окислов азота, перешед-
шая в NO2 вместе о эквивалентной концентрацией NO, быстро
абсорбируется кислотой, в связи с этим понижается концентра-
ция NO в газе, чем уменьшается скорость дальнейшего окисле-
ния оставшейся в газах NO. Гораздо скорее процесс проходит,
если сначала (до абсорбции) провести окисление NO до степени,
соответствующей N2Q3, а затем проводить процесс абсорбции
N2O3. Учитывая, что (как это раньше было показано) N2O3
в крепких кислотах растворяется лучше, чем другие окислы
азота (NO, NO2 N2O4), и что коэфициент скорости абсорбции
N2O3 крепкими растворами выше, чем коэфициент скорости аб-
сорбции NO2 (N2O4), приходим к заключению о необходимости
доводить окислы азота перед абсорбцией до степени окисления,
соответствующей N2O3.
Раньше, когда процессы окисления NO и абсорбции N2O3
проводились одновременно, около 0,75 всего объема башен-
ной системы приходилось на гей-люссаки. Только разделение
394
этих процессов открыло настоящие возможности для интенси-
фикации гей-люссаков. В условиях одновременного протекания
указанных процессов нельзя было полностью использовать, на-
пример, такой метод интенсификации работы гей-люссака, как
применение насадки с более развитой поверхностью (без одновре-
менного увеличения свободного объема), так как это мероприя-
тие, интенсифицируя процесс абсорбции, <не могло интенсифици-
ровать протекающий параллельно с абсорбцией процесс окисле-
ния NO. К сожалению, некоторые заводы до сих пор недоста-
точно оценили необходимость предварительного (до абсорбции)
окисления NO до N2O3.
Из мероприятий, которыми пользуются на практике для под-
готовки окислен азота к абсорбции, известны два — применение
полого окислительного объема и орошение последней продукци-
онной башни нитрозой с пониженной концентрацией H2SO4.
395
Полый окислительный объем пока чаще всего осуществляется
путем выемки части насадки из башни. Обычно некоторое коли-
чество насадки вынимается сверху последней продукционной и
первой абсорбционной башен, если эти башни соединяются газо-
ходом поверху.
Более прогрессивным выполнением окислительного объема
является установка в шунт между последней продукционной и
первой абсорбционной башнями специальной полой башни. Регу-
лируя количество газа, пропускаемое через окислительную башню,
легко поддерживать окислы азота перед первым гей-люссаком"
в степени окисления, соответствующей N2O3 вне зависимости от
состава газа (и других условий) при его выходе из последней
продукционной башни.
Регулируя концентрацию H2SO4 в кислоте, орошающей по-
следнюю продукционную башню (чем изменяется концентрация
окислов азота в газах), можно также поддерживать окислы
азота при входе газов в первый гей-люссак в степени окисле-
ния, соответствующей N2O3.
На рис. 206 графически показана (для случаев азотооборота
в 200, 300, 400 и 500 кг) зависимость времени окисления NO до
N^Os от концентрации SO2 в обжиговом газе для температуры
60° и в предположении, что все окислы азота выходят из про-
дукционной зоны в виде NO, a SO2 целиком окислено в продук-
ционной зоне. На рисунке кроме кривых для времени окисления
NO показаны кривые концентраций Ог (нисходящие прямые) и
NO (восходящие прямые) в газах при входе в окислительный
объем.
Необходимый окислительный объем на 11 т суточной продукции может
быть рассчитан так:
^к. — Qras • Z
14,65
К
где Qra3—секундный расход газа, a z— время окисления NO до N2O3 (обо-
значенное на рис. 206), К — коэфициент скорости окисления NO при данной
температуре, 14,65 — коэфициент скорости окисления при 60°.
п _ Ггаз-100 (273 + /)
^газ (100 Рок ). 24 -3600 -273’
где Vra3—объем газов на 1 т H2SO4, равный Г?? — 342 Y t — средняя тем-
\ 'so2 /
пература газов в окислительном объеме, Р ок— процент окислов азота в газах.
Окончательное выражение для величины окислительного объема в расчете
на 1 т суточной продукции примет вид:
V г <22860 ?49У273+0
59 (100 - Рок.) к ( PSOa 273 •
С достаточной точностью величину Qra3 можно принять равной
О - 22860 - /273 + А
Л м Лзо, • 24 ’ 3600 \ 273 ) ’
396
Потребный объем окисления при этом (с небольшим запасом) равен:
= 3,87 z /2734-/\
ок- 273 J'
9. Технологический режим камерной системы
Требования к технологическому режиму башни Гловера камерной си-
стемы такие же, как и к 1-й башне башеииой системы. Башня Гей-Люссака
орошается кислотой из башни Гловера. До поступления на гей-люссак эта
кислота должна охлаждаться до 30—35°. '
Для лучшего распределения в объеме камер газ лучше вводить с верху
камеры, ic этой же целью газ следует вводить в камеру в нескольких местах.
Лучшее перемешивание газа получается, если часть газа из гловера вво-
дить не в первую, а в одну из последующих камер.
Работа камер зависит от следующих факторов: а) концентрация SOs и
окислов азота в газах, б) температура газов, в) концентрация кислот, находя-
щихся в камерах, и г) условия перемешивания газов в камерах.
В разных камерных системах концентрация окислов азота в газах ко-
леблется в пределах 0,5—1,5%. Концентрация окислов азота в газах дан-
ной системы на всем протяжении камер остается приблизительно постоянной.
Несмотря почти на постоянную концентрацию окислов азота, интенсив-
ность камер по ходу газов падает по причине снижения процента SO2 (и Ог)
в газах.
В камерах, особенно в первых по ходу газов, задерживающей реакцией
во всем процессе образования серной кислоты, повидимому, является реакция
окисления NO. • Эта реакция идет лучше при более низких температурах.
При образовании (серной кислоты, а затем и ее разбавлении до концентрации
камерной кислоты, выделяется много теплоты. Чтобы температура в каме-
рах была нормальной и не повышалась до нежелательного предела, необхо-
димо интенсивно отводить теплоту из камер. В камерах, особенно в тех,
в которые вода поступает в виде пара, главная статья расхода теплоты —
отдача теплоты в окружающую среду. Матсуи приводит тепловой баланс
по камерам одной обследованной им системы:
Приход тепла (в %) Расход тепла в (%)
1. С газами.........21,35 1. С газами.........5,92
2. Образование H2SO4 . 76,85 2. С отходящей кисло-
той ................................................ 5,79
3. Образование нитро- 3. Теплоотдача в атмо-
зилсериой кислоты . 0,01 сферу............. 88,29'
4. Вносится с водой . . 1,79
Всего. .100,00 Всего. .100,00
По данным одной камерной системы из общего расхода тепла по всем
камерам приходится на теплоотдачу в окружающую среду 82,7% и на теп-
лоту, уносимую кислотой, 7,1%.
Вот почему одним из условий интенсификации камерных систем были и
остаются мероприятия по усилению охлаждения камер. Это же обстоятель-
ство определило и развитие самой формы камер. Форма камеры с точки
зрения процесса тем лучше, чем больше ее удельная поверхность, т. е. отно-
шение -р, где S — поверхность камеры в .и2, а V — объем камеры в м3.
С точки зрения теплового баланса камер понятно, что камеры лучше орошать
водой, чем паром, так как в том и в другом случае весовое количество НгО
подается одно и то же, но приход теплоты с паром несравненно больше,
чем с тем же весовым количеством воды.
397
Матсуи сообщает, что одной только заменой питания камер водой вместо
пара ему удалось повысить интенсивность одной системы почти на 50%.
Температура в камерах по ходу газов понижается от камеры к камере
в соответствии с понижением интенсивности работы камер. В первой камере
температура держится обычно в пределах 100—80°, а в последней она па-
дает до 50—30°.
Концентрация кислот поддерживается в камерах, во-первых, такой»
чтобы в камерной кислоте возможно меньше растворялись окислы азота.
Если концентрация кислоты в камерах будет высокой, то окислы азота
будут растворяться в ней с образованием нитрозил серной кислоты. Если
концентрация камерной кислоты будет очень низкой, окислы азота будут
растворяться в ней с образованием азотной кислоты. И то и другое поведет
к уводу окислов азота из сферы кислотообразования и к повышенному рас-
ходу азотной кислоты. Кроме того, повышенная нитрозность кислот усилит
травление свинца камер, особенно, когда окислы азота растворяются с обра-
зованием азотной кислоты.
Мы видели выше, что в 57 %-ной HaSOa и ниже нитрозилеерная кислота
разлагается полностью. Разложение нитрозилсерной кислоты начинается,
примерно, с 73%-ной концентрации и особенно интенсивно происходит, начи-
ная с 67 %-ной H2SO4. В более слабых кислотах образуется уже азотная кис-
лота. Эти цифры дают указание о тех пределах, в которых должна держаться
концентр|ация кислоты в условиях камер. Можно заранее сказать, что кон-
центрация кислоты должна падать от камеры к камере по ходу газов —
в первых камерах (здесь больше SO2 и выще температура) условия для рас-
творения окислов азота хуже, чем в последних. Кроме того, в первых ка-
мерах интенсивнее идет образование новой H2SO4. В трехкамерной системе
концентрация донной кислоты от камеры к камере может колебаться, при-
мерно, в следующих пределах:
° Вё
1-я камера.........53—55
2-я камера.........50—52
3-я камера ..... 47—49
Концентрация капельной кислоты, текущей по стенкам камер, несколько
ниже, чем концентрация дюнной. Для тех же трех камер концентрация ка-
• пельной кислоты от камеры к камере будет изменяться примерно так:
° Вё
1-я камера • . . . . 50—53
2-я камера.........48—50
3-я камера.........45—47
Концентрация кйслоты в камере может чрезмерно' повышаться, если в ка-
меру недостаточно вводится воды; наоборот, если в камеру воды вводится
слишком много, то кислота будет чрезмерно разбавляться. Воду требуется
подавать не только в достаточном количестве, яо и хорошо распыленной.
Если вода распылена плохо, то она быстро падает на дно камеры, в газовой
фазе воды будет недостаточно и процесс образования серной кислоты пойдет
плохо. Как продукция камерная серная кислота отбирается не из всех камер,
а обыкновенно из одной.
Мы уже видели, что интенсивность образования серной кислоты падает
от камеры к камере по ходу газов. Но даже и в одной камере не во всех
ее частях образование серной кислоты происходит одинаково интенсивно.
Одной нз причин является образование мертвых пространств — пло-
хое перемешивание газов. Замечено, что в первой части последующей камеры
образование серной кислоты идет интенсивнее, чем в последней части пред-
шествующей камеры. Это объясняется тем, что газы, пройдя через газоход,
соединяющий камеры, успевают хорошо перемешиваться.
398
10. Контроль производства башенных и камерных систем
Для любой сернокислотной установки основным условием ее
нормальной бесперебойной работы является постоянство коли-
чества и концентрации газа, поступающего в систему. Концен-
трация БОг^еред входом в систему должна непрерывно опреде-
ляться автоматическим газоанализатором. На передовых заводах
показания концентрации газа перед входом в систему пере-
даются, с одной стороны, в печное отделение (для принятия мер
в случае понижения или чрезмерного повышения содержания SO2
против нормального), а с другой стороны, в башенное отделение
(для того чтобы башенщики своевременно могли регулировать-
процесс в соответствии с изменением концентрации газа).
Замер количества проходящего через систему газа удобно
производить посредством калибрированных диафрагм.
Сравнением вычисленного (на основе величины выработки
системы и концентрации газа при входе в систему) и замерен-
ного диафрагмой количества газа можно определить величину
подсоса воздуха в системе. Однако эта величина скорее может
быть вычислена на основе анализа хвостовых газов на содержа-
ние кислорода. Величина подсоса воздуха является одним из.
важных показателей состояния системы, поэтому она должна
определяться систематически и, следовательно, систематически
должен делаться и анализ хвостовых газов на содержание кисло-
рода.
При данной нагрузке системы и качестве поступающего в нее
газа характер работы самой системы определяется величиной
потерь азотной кислоты. Величину потерь азотной кислоты в ка-
ждый данный момент характеризует содержание окислов азота
в хвостовых газах. Если при этом дается не только общее со-
держание окислов азота, но раздельное их содержание (т. е. от-
дельно NO и NO2), то можно судить не только о величине по-
терь азотной кислоты в данный момент, но и об основных при-
чинах этих потерь.
Быстрое и достаточно точное (для целей регулирования про-
цесса) определение общего и раздельного содержания окислов
азота может производиться при помощи колориметров, скон-
струированных УНИХИМ (Кузьминых и Турхан).
Как выше рассмотрено, хорошая работа абсорбционной части
системы требует предварительного окисления окислов азота до
N2O3. Поэтому частое определение степени окисления окислов
азота перед абсорбционной зоной (выполняемое также колори-
метрами) входит в обязательный объем контроля производства.
Перечисленных выше газовых анализов вполне достаточно
для регулирования процесса при более или менее нормальном его
ходе.
При обследовании системы, а также при глубоком расстрой-
стве технологического режима необходимо бывает установить
степень переработки SO2, денитрации и абсорбции окислов азота
39»
Таблица 93
Объект кон- троля 1 । Определяется Место анализа или замера Частота анализа или замера
Башенное отделение
Сопротивле- ние каждой башни Разрежение мм Газоход перед 1-й башней и у вентиля- тора После каждой башни 4 раза в смену 1 раз в смену
Газ Температура Содержание SO2 (аппарат Рейха или Газоход перед 1-й башней и после каждой башни 4 раза в смену
газоанализатор) Перед 1-й башней 8 раз в смену
Содержание SO2 и NO + NO2 (ме- тодом эвакуиро- ванных колб) После последней продукционной башни 1 раз в сутки
Содержание NO? и NO+NO2 Перед 1-й абсорбци- онной башней 2 раза в смену
(колориметром) После последней башни 4 раза в смену
Содержание О2 (аппаратом Орса) Перед 1-й башней После последней башни При наличии угле- рода в шихте 1 раз в смену 1 раз в смену
Подсос воздуха (определяется анализом газа на содержание кислорода) После каждой башни По мере надобности, но не реже 1 раза в 5 дней
Кислота Содержание брызг После последней 1 раз в 5 дней при
н тумана (фильтра- цией газа через гигроскопическую вату) башни отсутствии брызго- уловителя и при на- личии брызгоулови- теля 1 раз в 10 дней
Количество ороше- ния каждой башни (кислотомером Шишелова или методом водо- слива) Перед поступлением в башню или при выходе из нее 4 раза в смену
Нитрозность (ти- трованием перман- ганатом), плот- ность по Вё и температура Орошающие и выте- кающие нитрозы 4 раза в смену
400
Продолжение табл. 93
Объект контроля Определяется Место анализа или замера Частота анализа или замера
Конденсат Е Плотность по Вё ашенное отдел Трубка для стока е н и е 1 раз в смену
вентилятора Содержание H2SO4 конденсата 1 раз в сутки
Продукция и окислов азота (титрование пер- манганатом) Плотность по Вё Коробка или жолоб Каждый час
Количество Содержание H8SO4 и окислов азота В “/о Замер уровней до холодильника П редукционный бак Циркуляционн ые Каждый ба к *перед выдачей кислоты 1 раз в конце смены.
КИСЛОТЫ в сборниках и вырабо- танная про- дукция в сборниках сборники Сборник продукцион- ный До и после дачи кислоты
Производст- Кислотность отра- Колодцы 1 раз в смену
венный и дренажный колодцы Кислота ботаииой воды (качественный анализ) Хо Температура лодильное отде После каждого холо- л е н и е 2 раза в смену
Охлаждаю- * Плотность по Вё Температура дильиика До и после каждого холодильника До н после охлажде- По мере надобности но ие реже 1 раза в 5 дней 1 раз в смену
гцая вода Кислотность (ка- чественная реакция) 1 ння Выход воды из каж- дого холодильника 2 раза в смену
по отдельным башням. В этих случаях приходится определять
содержание SO2 и окислов азота (общее или раздельное), а ино-
гда брызг и тумана кислоты после ряда (а иногда и после всех)
башен. Содержание окислов азота и SO2 при совместном их при-
сутствии определяется при помощи эвакуированных колб.
Бесперебойная работа системы требует не только равномер-
ной подачи газа в систему, но и равномерного орошения башен.
Поэтому замер количества орошения по всем башням необходим,
особенно в условиях интенсивной работы. Количество орошения
замеряется непрерывно показывающими кислотомерами тех или
иных конструкций.
26 Зак. 5019. Технология серной кислоты.
401
Для полной характеристики хода процесса во всех частях си-
стемы, кроме рассмотренных выше систематически (или непре-
рывно) выполняемых газовых анализов, требуется и наблюдение
за жидкой фазой (нитрозами). В обязательный объем контроля
производства входит замер температур и плотности, а также
определение нитрозности орошающих и вытекающих нитроз. Эти
определения дают возможность судить о качестве работы ка-
ждой башни.
Для наблюдения за состоянием насадки башен систематиче-
ски определяется сопротивление каждой башни установленными
между башнями манометрами.
Состояние холодильников и качество их работы определяется
анализом кислотности отходящей воды и периодической провер-
кой величины коэфициента теплопередачи по отдельным змееви-
кам. Перечень принятых анализов и замеров по башенному и
холодильному отделению приведен в табл. 93.
11. Расходные коэфициенты в башенных и камерных системах
Как сказано выше (гл. 3), потери серы с огарком в печном
отделении не должны превышать 2,83% от всей загруженной
в печи серы. С отходящими из системы газами некоторое коли-
чество серы теряется в виде тумана и брызг кислоты.
Концентрация тумана и брызг кислоты в отходящих газах
зависит от интенсивности работы системы, технологического ре-
жима работы и аппаратурного оформления системы (количество
башен, их размер, тип насадки и др.). После 6-й башни (по-
следней) на одном из заводов! при интенсивности 79 кг/м8/сутяя
содержание брызг кислоты в газах составляло 13,38 г/м3, а ту-
мана 1,38 г/м3. После циклона (поставленного здесь в хвосте
системы) в газах оставалось: брызг кислоты 1,361 г/м3, а ту-
мана 0,357 г,/мл.
Очистка хвостовых газов башенной системы при интенсив-
ной работе (установкой циклона или другим способом) безу-
словно обязательна. В газах, выбрасываемых в атмосферу, со-
держание брызг и тумана, считая на SOs, не должно быть больше
1 г/ м3.
При 9 %-ном газе потери H2SO4 на 1 т продукции (без учета
подсоса воздуха в системе) составят, таким образом:
(22J*6?— 343^ = 2197 г, или 0,22% от продукции.
Если учесть и некоторые другие неизбежные потери серы
(потеря печного газа, разлив кислоты и др.), общий процент
использования серы в башенной системе должен быть не ниже
95%. Отсюда расходный коэфициент по колчедану (считая на
45%-ный по сере колчедан) не должен быть выше:
1000-32
98 • 450 - 0,95
= 764 кг.
402
На некоторых башенных заводах расход колчедана значи-
тельно выше, в отдельных случаях* он достигает даже
850—900 кг. Это объясняется несоблюдением правил техниче-
ской эксплоатации и нарушением •нормального технологического
режима на седельных заводах.
Повышенный расход сернистого сырья обычно происходит
по причинам — ненормально высокого содержания серы
в огарке, выпускания больших масс печного газа в атмосферу
цеха и большого разлива кислоты в цехе.
Потери азотной кислоты происходят — с отбираемой продук-
цией, с хвостовыми газами, при выделении окислов ^азота в атмо-
сферу цеха и с разливаемой нитрозной кислотой.
На всех башенных заводах основная потеря азотной кислоты
приходится на окислы азота, теряемые с уходящими газами.
Величина расхода азотной кислоты с продукцией может быть
определена так:
Щ = кг
^H2SO4
где Ph2so. —.процент H2SO4 в продукции, a PHno3 — процент
окислов азота (считая на HNO3) в продукции. По стандарту
на башенную кислоту содержание H2SO4 в продукции должно
быть 75—76,5%, а содержание N2O3 не должно быть выше
0,02%, таким образом потеря HNO3 с продукцией на 1 т H2SO4
не должна быть выше:
1000 • 100 • 0,02 • 126
75 • 100 76
= 0,44 кг.
Потеря азотной кислоты с уходящими из системы газами
равна:
1/хв . /?Ок-. 6?
П* газ г g п ЛЛО*7 1 7х »• ок.
2 = 'тжкг = °>0037 ре
где — объем хвостовых- газов на 1 т H2SO4 (при нормаль-
ных условиях), р°“ — парциальное давление окислов азота
в хвостовых газах в мм рт. ст. •
Парциальное давление окислов азота в хвостовых газах
можно, во-первых, представить как сумму двух величин:
где р1 парциальное давление эквивалентной смеси NO и
6 NO
NO2, а р g — парциальное давление избыточной (против NaOs)
NO. Наличие величины является результатом недостаточ-
ной подготовки окислов азота перед абсорбционной зоной. По-
нятно, что при окислении окислов азота перед абсорбционной
26* 403
зоной до степени окисления, соответствующей N2O3, величина
р^° обращается в нуль. В свою очередь величину p‘g можно
представить как сумму двух величин:
Pg~Pt+Pg •
В последнем выражении р, — равновесная упругость окислов
азота над нитрозой, орошающей последнюю башню, /?“6с —пре-
вышение парциального давления N2O3 над равновесной упру-
гостью его над нитрозои. Ясно, что наличие величины
является результатом неполноты процесса абсорбции окислов
азота, подготовленных к абсорбции (окисленных до степени
окисления, соответствующей N2O3).
Анализируя состав величины p°g, можно установить главные
причины потерь азотной кислоты с хвостовыми газами.
Величины р™ и р*° определяются анализом отходящих из
системы газов. Величину р: можно определить или по данным
табл. 73 (составленной на основании исследований Кудрявцева)
или по приближенной формуле Беньковского:
p=(f. JOO,2(71.8 - П)+ 0,023 («-85)
где р — упругость окислов азота над нитрозой в мм рт. ст., d —
процент HNO3 в нитрозе, В — содержание H2SO4 в нитрозе в %,
t — температура орошающей нитрозы. Формула Беньковского
выведена на основании экспериментальных работ Матсуи, Берля
и Зенгера и Тихонова, охватывающих изменение различных фа-
кторов в следующих пределах: % HNO3 от 0 до 3,5, концентра-
ция кислоты от 57 до 60° Вё, температура нитрозы от 30 до 90°.
Определим величину упругости окислов азота над нитрозой,
содержащей 76,7% H2SO4, 1% HNO3 при температуре 40°. По
табл. 73 эта упругость 0,325 мм рт. ст. По формуле Беньков-
ского эта величина получается равной 0,266. Если процент SO2
в газах 9, а подсоса воздуха в системе нет, то V"‘s —2197 /и3.
Потери азотной кислоты, обусловленные упругостью окислов
азота над нитрозой, будут равны:
V™'- Рок -63 2197 • 0,325 • 63 „
,аз гок —______________________9 КД л-2
760 • 22,4 760 • 22,4 ~ ’
В производстве технологические условия в последнем гей-
люссаке (% HNO3 и H2SO4 в орошающей нитрозе, температура
нитрозы) могут быть не только не хуже (с точки зрения до-
стижения возможно минимальной упругости окислов азота над
нитрозой), но и лучше условий, приведенных в разобранном
выше примере. Из этого следует, что величина потерь HNOs,
обусловленная упругостью окислов азота над нитрозой, вполне
может быть не выше 3—5 кг на 1 т H2SO4.
404
Потерь HNO3 в виде избыточного NO в отходящих газах
можно избежать совершенно. Улучшением процесса абсорбции
(полнота смачивания насадки, достаточная поверхность соприкос-
новения газа iv кислоты и др.) можно практически величину
р^ сделать как угодно малой. Отсюда следует, что при ра-
боте на газах от обжига колчедана расход азотной кислоты
может быть не выше ~ 5 кг- Учитывая неизбежность некото-
рых других потерь азотной кислоты (в атмосферу цеха из-за
неполной герметичности аппаратуры), а также нецелесообраз-
ность увеличения абсорбционной аппаратуры для улавливания
небольшой последней части величины р“бс (необходимость со-
хранения достаточной движущей силы абсорбции), можно при-
нять нормальной величиной расхода азотной кислоты 8—10 кг
на 1 т H2SO4.
В практике: многих действующих заводов расход азотной
кислоты значительно выше указанной цифры. Как это вытекает
из только что проведенного анализа, основными причинами по-
вышенного расхода азотной кислоты могут быть:
1) Низкая концентрация SO2 в поступающем в систему газе
или большой подсос воздуха в системе;
2) Недоокисление окислов азота перед абсорбцией (наличие
избыточного NO сверх стехиометрической смеси NO и NOa
в газах).
3) Недостаточная полнота абсорбции подготовленных к аб-
сорбции окислов азота (плохое распределение газа и кислоты
в башне, недостаточная величина поверхности соприкосновения
газа и кислоты и др.).
4) Повышенная упругость окислов азота над нитрозой (по-
вышенное содержание HNO.3 в нитрозе, пониженное содержание
HaSOi в нитрозе, высокая температура нитрозы, наличие в ни-
трозе свободной HNO3. Последнее является результатом нали-
чия избыточной NO2 против стехиометрической смеси NO 4- NO2
в газе — результатом переокисления окислов азота).
Приводим в табл. 94 пример распределения потерь азотной
кислоты на башенной системе одного из заводов по данным
обследования Блюма 1
Таблица 94
Общие потерн азотной кислоты в кг на 1 т Потери с продукцией Потери с хвостовыми газами
в кг в % в кг в %
21,7 18,3 0,52 0,52 2,38 2,84 21.18 17,78 97,62 97,16
> Материалы фонда НИУИФ.
405
Распределение потерь с хвостовыми газами приводим в табл. 95.
Таблица 95
Общие потери HNO3 с хвостовыми газами в кг В том числе
потери статиче- ские (определяе- мые величиной pi) потери от непол- ноты абсорбции (определяемые ве- личиной р*бс’) потери от непол- ноты окисления (определяемые ве- личиной Pg°)
в кг в % в кг в % в кг в %
21,18 17,78 4,18 5,27 20,2 29,6 8,52 6,60 40,2 37,2 8,32 5,91 39,6 33,2
На образование продукции, в том случае если башенная кис-
лота получается в виде 76%-ной, на 1 т H2SO4 требуется воды:
1^ + (1роо 1оо 1ООО)= 499>8 кг
Значительно большее количество воды требуется для охла-
ждения кислот.
Величина этого расхода зависит от целого ряда условий:
схема охлаждения кислот, технологический режим системы, тем-
пература охлаждающей воды (местные климатические условия),
конструкция и состояние холодильников и др. В зависимости
от конкретных местных соображений при неизменности всех
других условий может оказаться экономически целесообразным
пойти или на сокращение величины потребной поверхности охла-
ждения за счет некоторого повышения расхода охлаждающей
воды, или, наоборот.
Можно принять, что в среднем, считая на 1 т продукции (при
получении башенной 76%-ной кислоты), отнимается охлаждаю-
щей водой 900 000 ккал тепла. Таким образом расход воды в /и3
на 1 т башенной кислоты равен:
где t-2 — средневзвешенная температура воды по выходе из хо-
лодильников, a ti — температура приходящей на охлаждение
воды.
В большинстве случаев расход воды на 1 т башенной кис-
лоты составляет ~ 40—50 /и3- Расход воды по отдельным ба-
шенным системам в зависимости от конкретных местных усло-
вий и времени года колеблется от 15 до 75 ж3 на 1 т H2SO4.
406
Расход энергии на 1 т H2SO4 по печному отделению (на вра-
щение печей и на проталкивание воздуха через вал и гребки
печи) составляет 10—±2 квт-ч. В башенном отделении энергия
затрачивается на перекачку орошающих кислот и на протягива-
ние газа через систему.
Пусть в час производится 1 т H2SO4. Пусть орошающая кислота под-
нимается на высоту Н м, а количество орошения в м3/час на 1 т H2SO4
продукции составляет А м3.„ Тогда энергия подъема орошающей кислоты
будет равна:
_ Л ТУ -0,736
^подъем — 36С0-75 ’
где d — удельный вес кислоты, 0,736 кет = 1 л. с. = 75 кг-м/сек,
Л
- —количество орошения в м^/сек- Можно принять, что потеря энергии
3600
на преодоление сопротивлений и иа создание живой силы составляет 25%
от энергии подъема.
Если высота подъема кислоты 25 м, количество орошения в час 30 л’,
вес I м3 орошающей кислоты 1700 кг, а суммарный коэфициент полезного
действия насоса 0,4, то расход энергии иа перекачку кислоты иа 1 т H2SO4
будет:
1700-30-25-0.736-1,25
3600 75 • 0,4
= 10,9 квт-ч.
Потребную мощность на продвижение газа через систему можно опреде-
лить по формуле:
Qra, • И' 0,736
дг —------------------
75>)
кет,
где Q газ — объем газов в м3/сек, Н'— сопротивление системы в .и.м вод. ст.
(кг/л3), ч — коэфициент полезного действия вентилятора, который можно
принять равным 0,4.
При концентрации SO2 в обжиговом газе, равной 9%, температуре газов
перед вентилятором, равной 50°, секундный объем газа равен:
22 860 • 323
9 • 273 • 3600
= 0,832 м3/сек.
Если общее сопротивление системы 200 мм вод. ст., то расход энергии
на продвижение газа через систему иа 4 т H2SO4 будет равен:
0,832-200.0,736
-----75^4-----=41 Квт'4-
Общий расход энергии на 1 т H2SO4 при указанных условиях таким обра-
зом будет ~52—54 квт-ч.
Из сказанного следует, что величина расхода энергии для
данной системы по башенному отделению определяется: крат-
ностью орошения, концентрацией газа при входе в систему
(а также величиной подсоса воздуха в системе) и сопротивле-
нием системы.
407
12. Задачи и пути дальнейшей интенсификации
и усовершенствования нитрозного метода
Башенные системы раньше работали с интенсивностью
18—22 кг H2SO4 на 1 м3 общего объема башен в сутки. Отдель-
ные заводы уже несколько лет назад значительно превысили
эту интенсивность, считавшуюся тогда нормальной. Так, коллек-
тив одного из заводов! (Б. Д. Мельник и др.), начиная
с 1931 г. непрерывно повышал интенсивность башенной системы.
Большую роль в подъеме интенсивности сыграли проводившиеся
в течение ряда лет проф. В. Н. Шульцем заводские опыты по
интенсификации башенных систем.
С развитием стахановского движения интенсивность на ряде
заводов сразу дала большой скачок вверх.
Освоение интенсивности 60—100 кг при небольшом расходе
азотной кислоты (8—12 кг HNO3 на 1 т H2SO4) на всех ба-
шенных заводах является ближайшей задачей в области произ-
водства серной кислоты нитрозным методом. Такая интенсив-
ность при указанной величине расхода азотной кислоты обеспе-
чивается следующими условиями: 1) концентрация SO2 в газе
при входе в систему 9—9,5$; 2) минимальный подсос воздуха
в системе; 3) нитрозность крепкой нитрозы 4,5—6$ HNO3;
4) нитрозность слабой нитрозы не выше 0,8% HNO3; 5) воз-
можно низкая температура кислоты, орошающей последний гей-
люссак (при орошении башенной кислотой не выше 40 ); 6) бе-
зусловное проведение мероприятий для подготовки окислов
азота к абсорбции (доведение до степени окисления, соответ-
ствующей N2O3).
При 'высокоинтенсивном режиме требуется особо строгое со-
блюдение правил эксплоатации: высококачественное проведение
капитального ремонта, серьезная постановка планово-предупре-
дительного ремонта и точное выполнение его графика; равномер-
ная нагрузка системы; возможно полная очистка газа от пыли;
регулярная промывка засоряющихся башен и чистка холодиль-
ников; систематическая проверка работы аппаратов (турбинок,
насосов, холодильников и др); точное соблюдение утвержден-
ных норм технологического режима. Необходимо также закон-
чить полную замену свинца железом и чугуном в аппаратуре ба-
шенных систем. В условиях интенсивной работы (высокая ни-
трозность и температура кислот) свинец настолько быстро тра-
вится, что расходы на ремонт свинцовой аппаратуры поглощают
значительную долю выгод, получаемых от интенсификации
системы.
Надо также провести герметизацию аппаратуры (холодиль-
ники, сборники).
На ряде заводов, если получаемая кислота подлежит отправке
или если при заводе имеются цеха, требующие серной кислоты
в виде купоросного масла, необходимо освоить процесс постоян-
ного выпуска продукции с концентрацией 92—92,5% H2SO4.
408
Выпуск всей продукции в виде купоросного масла вполне воз-
можен, если температура газов, поступающих в систему, не
ниже 375е.
Наметились две схемы орошения при работе системы с вы-
пуском купоросного масла. По первой схеме на 1-ю башню
дается кислота в количестве, соответствующем продукции. Же-
лательно, чтобы при этом на 1-ю башню давалась такая же
высоконитрозная кислота, которая подается и на 2-ю башню.
1-я башня в этом случае не только играет роль концентратора
продукции, но и принимает значительное участие в перерабеггке
SO?. Если в 1-й башне высоконитрозная кислота не денитри-
руется до стандарта, то приходится подавать менее нитрозную
кислоту.
По второй схеме 1-я башня орошается в цикле с последней,
при этом количество орошающей кислоты больше, чем количе-
ство, соответствующее продукции (избыток вытекающей кис-
лоты против продукции направляется на орошение последней
башни).
Последняя башня частично орошается «сама на себя».
Часть кислоты, вытекающей из последней башни, передается на
1-ю башню (количество этой кислоты определяется тем, что
с ней должны быть возвращены в голову системы все окислы
азота, улавливаемые в последней башне).
Работа по второй схеме предпочтительнее, если 1-я башня
имеет сравнительно большой диаметр и количества кислоты, со-
ответствующего продукции, недостаточно для смачивания всей
насадки башни. Для осуществления второй схемы требуется не-
сколько более высокая температура входящих газов, так как
кроме концентрирования продукции в этом случае некоторое ко-
личество теплоты тратится на концентрирование кислоты, при-
ходящей с последней башни (орошаюшая последнюю башню
кислота в этой башне несколько разбавляется поглощенными во-
дяными парами из газов).
Работа системы с выпуском продукции в виде купоросного*
масла предъявляет новые требования к материалу 1-й башни: ма-
териал колосников башни и насадки башни должен противостоять
разрушающему действию высококонцентрированной и горячей
кислоты (до 200°).
Перевод работы башенной системы на выпуск купоросного
масла особо настоятельно требует также окончательного вытес-
нения свинца из аппаратуры башенных систем.
Рассмотренные выше зависимости интенсивности процессов
кислотообразования и абсорбции от технологических факторов
таят в себе дальнейшие возможности интенсификации ннтроз-
ного процесса, как современного (башенные системы), так и
особенно нового аппаратурного оформления.
О возможностях нитрозного процесса прежде всего говорят
имеющиеся в нем внутренние противоречия: 1) между концен-
трациями сернистого газа и кислорода в обжиговых газах;
409
2) между концентрацией кислоты (процент H2SO4) в продук-’
ционной и абсорбционной зонах; 3) между процессами окисле-
ния NO и абсорбции окислов азота при их одновременном про-
текании и др.
Возможность, и при том не однозначного разрешения ка-
ждого из этих противоречий, открывает множество путей ин-
тенсификации башенного процесса и дальнейшего усовершен-
ствования нитрозного метода.
Так, частичное разрешение первого из вышеуказанных про-
тиворечий может быть проведено следующим образом.
В печном газе поддерживается максимально возможная кон-
центрация SO2, после продукционной зоны для повышения кон-
центрации кислорода подсасывается некоторое количество воз-
духа.
Более радикальным путем разрешения первого из указанных
противоречий является применение обогащенного по кислороду
газа для обжига сернистого сырья. В этом случае высокая кон-
центрация SO-2 в обжиговом газе (не только без ущерба для
процесса обжига, но даже при интенсификации его) достигается
при одновременном повышении и концентрации кислорода. По-
вышение концентрации SO2 в обжиговом газе в п раз дает уве-
личение интенсивности всей системы в п раз, сокращение расхода
азотной кислоты минимум в п раз и уменьшение расхода энергии
на протягивание газа через систему в п- раз.
В связи с намечающимся разрешением проблемы концентри-
рования слабых по SO2 металлургических газов может оказаться
возможным снабжать некоторые сернокислотные заводы жидким
100%-ным сернистым ангидридом. Зависимость между концен-
трациями SO2 и Ог в смесях, получаемых смешением 100%-ного
SO2 с воздухом, имеет вид: Ро., =21—0,21 Psos-
Если в смеси содержание SO2 равно 24%, то кислорода в ней
будет 16%.
Содержание Ог в хвостовых газах при этом будет:
™ (ро — °.5 Рщ) 100
Рхв- '__у8 b,)g С ОСО/
°- ~ (100-1,5Р302)
Применение такой газовой смеси даст возможность повысить
интенсивность системы и сократить расход азотной кислоты
(против работы на нормальных обжиговых газах: SO2 — 9%) по
24
крайней мере в j = 2,67 раза.
Переработку 100%-ного SO2 на серную кислоту нитрозным
методом можно организовать совершенно по новой схеме:
100%-ный SO2 окисляется нитрозой, полученная газообразная
100%-ная NO разбавляется воздухом (получается смесь с со-
держанием ~ 40% NO) и после окисления до N2O3 улавли-
вается серной кислотой с образованием очень крепкой нитрозы,
идущей обратно на окисление SO». Возможна также организа-
ция процесса взаимодействия жидкого SO2 с нитрозой.
410
Применение 1009? -ного SO2 избавит сернокислотный цех от
дробильного и печного отделений — источников пыли и грязи.
Возможность точного дозирования подаваемых в систему чистого
SO2 и воздуха и точного регулирования состава получаемой
смеси создаст условия для полной автоматизации сернокислот-
ного завода.
Выше уже упоминался ряд новых схем башенного процесса,
направленных к разрешению противоречия между концентра-
циями (по H2SO4) нитроз, орошающих продукционную й абсорб-
ционную зоны. Есть предположение о целесообразности (в этих
же целях) орошения последней продукционной башни совсем
слабой (~ 60% H2SO4) кислотой. Есть также предположение
о возможности орошения всех башен купоросным маслом. При
этом для возможности переработки SO2 в продукционных
башнях предполагается значительно повысить в них темпе-
ратуру.
О ряде методов разрешения противоречия между процессами
окисления NO и абсорбции N2O3 (при их одновременном проте-
кании) уже говорилось выше.
В последние годы сделано не мало попыток применения в ни-
трозном процессе вместо орошаемых башен с насадкой (скруб-
беров) аппаратов другого типа: барботеров, полых башен,
в которых разбрызгивается нитроза, и др. Несомненным пре-
имуществом барботеров и полых башен перед скрубберами
является*значительно меньшая опасность их забивания приноси-
мой газами грязью. Полые башни, кроме того, обладают совер-
шенно незначительным гидравлическим сопротивлением. Наобо-
рот, барботеры создают значительно большее сопротивление, чем
скрубберы. Английской фирмой Ватсон (Watson) сконструиро-
вана камерная система, где роль башен Гловера и Гей-Люссака
также играют пустые камеры, в которых нитроза распыляется
посредством особых разбрызгивателей.
Коэфициент скорости абсорбции N2O3 серной кислотой (слу-
чай хорошей растворимости), повидимому, имеет наибольшее
значение для случая разбрызгивания жидкости в газе и наимень-
шее значение для случая барботажа газа через жидкость.
Однако из сказанного не следует, что аппараты барботаж-
ного типа не могут иметь перспективы применения. Вопрос ре-
шается не только величиной коэфициента абсорбции, но и вели-
чиной удельной (считая на единицу объема аппарата) поверх-
ности соприкосновения фаз и рядом других соображений (ги-
дравлическое сопротивление аппарата, условия для уноса брызг
кислоты из аппарата и др.).
Интенсивность работы башенных систем до сих пор характе-
ризуется числом кг H2SO4, получаемой в сутки с 1 ai3 общего
объема системы. Однако оценка работы системы не может быть
дана только на основании этого показателя. Может быть такой
случай, что повышение съема кислоты с 1 м3 системы сопрово-
ждается ростом себестоимости кислоты (увеличение расхода
4П
азотной кислоты, увеличение расхода на ремонты и др.). По-
длинная интенсификация должна обязательно давать понижение
правильно скалькулированной себестоимости продукции, что бу-
дет характеризовать повышение производительности обществен-
ного труда на данном участке. Только такая интенсификация,
которая дает повышение производительности общественного
труда, является действенным методом разрешения задачи догнать
и перегнать капиталистические страны Европы и США в эко-
номическом отношении.
Рядом вполне конкретных мероприятий (большая дифе-
ренциация концентрации орошающих кислот по отдельным баш-
ням, более низкие температуры в гей-люссаках, применение бо-
лее совершенных типов насадок, применение более совершенных
конструкций распыливающих кислоту аппаратов и др.) вполне
возможно величину интенсивности повысить в 2—3 раза. Меро-
приятиями более глубокого характера, из которых ряд приме-
ров приведен выше, представляется возможным получить ин-
тенсивность и значительно более высокого порядка.
Однако надо иметь в виду, что прогрессивность того или
иного мероприятия не может быть оценена только величиной
съема кислоты с 1 м3 объема системы в сутки.
Литература
1. Lunge G., Handbuch der Schwefelsaurefabrikation, Braunschweig 1916
2. Bitterli E., These 587, Paris 1925.
3. Кириченко H. E., Беньковский С. В., Кислород в башенном
и камерном производстве, ЖХП № 4, 27 (1934).
4. L u п g е О., В е г 1 Е. (Теория камерного процесса), Ztschr. f. angew. Chi tn.,
1713 (1907).
5. Brin er E., Rossignol M-, Chem. Metall. Eng., 885 (1923).
6. Raschig F. (Химизм камерного процесса), Ztschr. f. angew. Chem. 1001
(1925).
7. Waeser В. (Теория камерного процесса), Metallborse, 1786 (1926).
8. Berl E., Saenger H. (Синяя кислота), Ztschr. f. angew.Chem. 291
(1931); Ztschr. f. anorg. allg. Chem. 208, 113 (1932).
9. Muller W., Foerber D., Fort R. (Гидролиз ннтрсзилсериой ки-
слоты), Ztschr. f. angew. Chem. 1, 782 (1932).
10. Мюллер В., К теории камерного процесса, Хим. реф. журн. № 1. 7
(1932).
11. Абель, Кинетика образования серной кислоты, ЖХП №1, 90 (1934).
12. Berl Е., Winn а скег К., Saenger Н. (О синей кислоте), Ztschr. f.
anorg. allg. Chem. 211, 373 (1933).
13. Berl E., Hildebrand H., Winn acker К- (О механизме переноса
кислорода к SO2 нитрозилсерной кислотой), Ztschr. f. anorg. allg. Chem.
214, 13, 369 (1933).
14. M а п c h о t W. (О синей кислоте), Ztschr. 1. anorg. allg. Chem. 210, 135
(1933).
15. Szego L., Исследование камерного процесса, ЖХП № 5, 63 (1933).
16. VI о 11 е г W. (К теории камерного процесса), Ztschr. f. anorg. allg. Chem.,
218, 13, 307 (1934).
17. Ma nc hot W., Hansen H., Синяя кислота в камерном процессе
ЖХП № 6, 631 (1935).
412
18. Лопатто Э. К., Окисление сернистого газа в жидкой фазе, изд.
Одесского индустриального института, 1939.
19. ЭпштеКн Д. А., Химия и технология связанного азота, ОНТИ, 1934.
20. Bodenstein, Ztschr. Phys. Chem. 100. 66 (1922).
21. Sanfourche A., Rondier L. (Свойства смесей H2SO4 с азотис ей
и азотиой кислотами), Compt. Rend. 187, 291 (1928); С. 1928, II, 1704.
22. Sanfourche A., Rondier L. (Упругости окислов азота иад интроз-
иой серной кислотой), Bull. Soc. Chim. France. 815 (1928); С. 1929, I, 623.
>3 Матсуи, Упругость окислов азота над нитрозой, ЖХП № 28- 30,
1837 (1930).
24. Hants ch A., Rose А. (О нитрозилсерной кислоте и цветных про-
дуктах ее восстановления), Ztschr. f. anorg. allg. Chem. 190, 321, (1930).
25. Berl E., S a e n g e г H. (Система: нитрозилсериая кислота, серная ки-
слота, вода), Ztschr. f. anorg. allg. Chem. 13, 202, 1’3 (1931).
26. Тихонов А., Парциальное давление паров и воды н окнелов азота .
иад нитрозой. ЖХП № 8, 58 (1938).
27 Ада Дуров И., Цейтлин А’., Гидролиз нит; озы. Ж. физ. хим. 8, 1096
(1935)
28. Беньковский С. В., Потери азотиой кислоты в башенном и камерном
процессах, ЖХП № 6, 330 (1933).
29. К у д р я в ц е в А. А., Упругссть окнелов азота над нитрозами, ЖХП (1941).
30. Lewis W., Whitman W. (Принципы абсорбции газов жидкостями),
J. Ind. Eng. Chem. 1215 (1925).
31. Чего, Малагути и Ломбарди, О скорости поглощения окис-
лов азота серной кислотой, ЖХП № 2, 53 (1933).
32. Кузьминых И., Яхонтова Е„ Сурков Е., Кинетика поглощения
окислов азота серной кислотой, ЖХП № 11, 38 (1934).
33. К у з ь м и н ы х И., Ю д ина В., Влияние скорости газа на скорость
поглощения окислов азота серной кислотой, Химстрой № 6, 315 (1934).
34. Мал'ин К., Кельман Ф., Успенская М., О движущей силе аб-
сорбции окислов азота серной кислотой, ЖХП № 4, 25 (1938).
35. Малнн К., Кельман Ф., Успенская М., Милованова М., Эк-
сорбция окислов азота из нитрозы, ЖХП № 6, 24 (1938).
36. Кузьминых И., Т у р х а н Э-, Архипова М., Механизм и кинетика
окисления SOo двуокисью азота в газовой фазе. Zischr. f. anorg. allg.
Chem. 226, 31o“(I936); ЖХП № 10 615 (1936).
37. Матсу н, Равновесие реакции между NO2 и SO2, ЖХП № 7, 502 (1930).
38. M a t s u i M. (Изучение камерного процесса), С. 1928, 1, 2642.
39. К у з ь м и н ы х И., Сурков Е., Интенсивность кислотообразования
при взаимодействии SO2 и ннтрозы, Химстрой № 8, 473 (1935).
40. Тихонов А., Изучение кнтрозиого процесса, ЖХП № 3, 147 (1936'.
41. Petersen Н. (Башенный сернокислотный процесс в свете новейших
исследований), Chem.-Ztg. 63, № 69, 585 (1939).
42. Ч е р е п к о в И. Ф., Туманообразная кислота в нитрозном сернокислотном
процессе, ЖХП № 3, 19 (1940).
43. Безбашенный завод серной кислоты фирмы Ватсон, J. Ind. Eng. Chem.
XV, № 171, 159 (1939).
44. К у з ь м и н ы х И., Яхонтова Е., Температуры замерзания нитроз,
ЖХП № 6, 25 (1939).
45. М а л^и^ К., О расчетных формулах башенного процесса, ЖХП № 3,
46. Berl Е. (Получение сериой кислоты под давлением), Chem. Met. Е’g.
41, № И. 1577 (1934).
47. Stlecke W. (Контактный или камерный способ), Zischr. f. angew. Chem.
49, 29, 475 (1936).
48. Пашевский Г. Д.. Савинкова Е. И., Состав грязи в циркуля-
ционных кислотах башенных систем, ЖХП № 16, 1153 (1937).
49. Поляков К., Васильев А., Бессвинцовые башенные системы, ЖХП
№ 4, 284 (1937).
50. Юшманов Е„ Бессвинцовые башенные системы, ЖХП № 14, 1019
(1937).
413
51. Юшманов Е„ Попова А., Замена свинца железом и чугуном в ба-|
шенном сернокислотном производстве, ЖХП № 20, 1387 (1937).
52. Второв М. Н., Стойкость свинца в башенных системах и его замена
другими материалами, ЖХП № 4, 13 (1938); ЖХП № 5, 3 (1938).
53. Юшманов Е, Попова А., Коррозия чугуна, железа и свинца в газо-
вой фазе башенной системы, ЖХП № 4, 13 (1938).
54. Юшманов, Журавлева и Орлова, Коррозия железа, чугуна
и свинца нитрозами, ЖХП № 7, 2 (1938).
55. Григорьев Г. С., О заменителях свинца в железных холодильниках,
ЖХП № 9, 44 (1939).
56. Малин К. М., Кириченко Н. Е., Андронов В. П„ От ва-
гин Н. Н., О рационализации холодильного хозяйства на сернокислот-
ных башенных заводах, ЖХП № 3, 10 (1938).
57. Шульц В. Н., Регенерация окислов азота в башенном сернокислотном
производстве, ЖХП № 1, 18 (1937).
58. Кузьминых И. Н„ Об окислительном объеме и других вопросах башен-
ного процесса, ЖХП № 14, 1007 (1937).
59. Шульц В. Н., Окисление окислов азота и окислительная башкя в ба-
шенном сернокислотном процессе, ЖХП № 23, 1605 (1937).
60. Шульц В. Н., Интенсификация башенного сернокислотного процесса,
ЖХП № 24, 1673 (1937).
61. Кузьминых и А п а х ов. Сопротивление насадок, ЖОХ № 4—5 (1940).
62. П а ш е в с к и й Г. Д., П о и о м а р е в В. Д., С а в и н к о в а Е. И., Сни-
жение расхода азотной кислоты на Кировоградской башенной системе,
ЖХП № 1, 20 (1938).
63. Пащевский Г. Д., Савинкова Е. И., Анализ потерь азотиой
кислоты на Кировоградской башенной системе, ЖХП № 14, 1014 (1937).
64. Малин К. М., Пути снижения расхода колчедана и азотной кислоты
в башенных системах, ЖХП № 8, 13 (1940).
65. Поляков К. А., Свинцовая аппаратура, ОНТИ, 1935.
66. П о л я к о в К. А., Аппараты для охлаждения н подъема серной
и азотной кислот, ОНТИ, 1934.
67. Б е и ь к о в с к и й С. Б., С м ы с л о в Н. И„ Р а й н о в К. К., Производ-
ство серной кислоты башенным способом, Госхимиздат, 1940.
68. W а е s е г В., Handbuch der SchwefelsMurefabrikation, Braunschweig 1930.
69. Вебб Г. Окислы азота, их поглощение и переработка, Техническое
издательство, Харьков 1931.
70. F a i г И е, Sulfuric acid manufacture, New York 1936.
ГЛАВА 6
КОНЦЕНТРИРОВАНИЕ СЕРНОЙ КИСЛОТЫ
I. Теоретические основы
Многие производства потребляют серную кислоту в виде ку-
поросного масла концентрации 92—95% H2SO4: производство
соляной кислоты, концентрирование азотной кислоты, производ-
ство взрывчатых веществ и многие другие. Кислоту такой кон-
центрации можно получить разбавлением олеума или моно-
гидрата. Однако до настоящего
времени купоросное масло в
значительных количествах про-
изводится концентрированием
более слабых кислот — камер-
ной и башенной и отбросной
кислоты различных произ-
водств. В ряде производств ку-
поросное масло расходуется в
незначительном количестве;
оно лишь разбавляется водой.
Так, например, при концентри-
ровании азотной кислоты сер-
ная кислота выходит из про-
цесса производства с концен-
трацией 68% H2SO4. Вышед-
шая из процесса кислота опять
концентрируется до купорос-
ного масла и возвращается в
Рис. 207. Зависимость температуры
кипения и % H2SO4 в парах от содер-
жания H2SO4 в растворе.
процесс производства.
Процесс концентрирования серной кислоты заключается в уда-
лении из нее части воды. Если из кислоты концентрации а про-
центов H2SO4 надо получить В тонн кислоты концентрации b
процентов H2SO4, то количество воды, подлежащее удалению,
определяется следующим выражением:
В применяемых сейчас в промышленности установках кон-
центрирования серной кислоты вода удаляется исключительно
термическим методом, т. е. нагреванием.
415
Диаграмма рис. 207 показывает зависимость температуры ки-
пения серной кислоты и концентрации H2SO4 в парах, при тем-
пературе кипения, от концентрации H2SO4 в растворе. Из диа-
граммы видно, что для всех кислот концентрации ниже 98%
H2SO4 процент H2SO4 в парах меньше, чем процент H-2SO4 в рас-
творе. Для кислот концентраций ниже 70% H2SO4 серная кис-
лота в парах отсутствует совсем. Отсюда ясно, что при кипя-
чении серной кислоты концентрации ниже 98%, H2SO4 происхо-
дит постепенное повышение ее концентрации до тех пор, пока
она не достигнет 98*4 H2SO4. Так как 98*/ -ная H2SO4 перего-
няется полностью, то получить кипячением серную кислоту кон-
центрации выше 98'/ невозможно.
Из приведенных выше (см. гл. 1, стр. 29) данных видно, что
уже при температурах, лежащих значительно ниже точки кипе-
ния, водные растворы серной кислоты имеют значительные
упругости. При этом для всех концентраций ниже 98% H2SO4
отношение H2SO4 к НгО в парах меньше, чем в жидкости.
В большинстве применяемых сейчас в промышленности концен-
трационных установок серную кислоту концентрируют при тем-
пературах, лежащих ниже точки ее кипения. Так делается из
следующих соображений. При кипении кислоты значительные ее
количества увлекались бы в виде капель (брызг). Кроме того, и
упругости паров H2SO4 над кипящей серной кислотой конечной
концентрации значительно выше, чем упругость этих паров над
той же кислотой при той температуре, при которой обычно про-
водится концентрирование.
Полезный расход теплоты, потребной для концентрирования
серной кислоты, слагается из следующих статей:
1) теплота нагревания, т. е. та теплота, которая требуется для
повышения температуры кислоты от исходной до температуры
концентрирования;
2) теплота дегидратации — обратная теплоте разбавления; эта
теплота носит также название диференциальной теплоты раз-
ведения;
3) скрытая теплота испарения удаляемой воды.
Каждая из этих статей расхода высчитывается отдельно. Те-
плоту, потребную для нагревания кислоты, нельзя вычислять по
формуле
Q = GC(f2 — /Д
так как теплоемкость кислоты изменяется прн повышении тем-
пературы. Эту теплоту можно вычислить по формуле
Q1 = gSoA
где Gso,—вес SO3 в нагреваемой кислоте в кг, а /,2 и М— ко-
личества теплоты в больших калориях (теплосодержание), ко-
торое требуется для нагревания данной кислоты от 0° и соот-
ветственно до tz° и ti° (считая на 1 кг SOs). Величины тепло-
содержания даются в таблицах. Это же количество теплоты, по-
416
требное для нагревания кислоты, можно определить по графику
рис. 208.
Чтобы найти количество теплоты, потребное для нагревания с 20 до 220°
кислоты, содержащей, например, 55% SOs, берут ординату точки пересечения
—»- Температура в °C
Рис. 208. График для определения количества тепла,
потребного на нагревание кислоты.
наклонной, идущей от цифры 55, с вертикалью, идущей от цифры 20. Эта
ордината по шкале калорий равна 20. Далее берут ординату пересечения
той же наклонной с вертикалью,
идущей от цифры 220. Эта орди-
ната по шкале калорий равна 207.
Разность этих ординат
207 — 20= 187
дает число больших калорий, по-
требных для нагревания данной
кислоты с 20 до 220° в количе-
стве, соответствующем 1 кг SOs- ।
Наконец,теплоту, потреб-
ную для нагревания кислоты, |
можно вычислить по диа-
грамме Цейсберга (рис. 209).
Правда, эта диаграмма дает
возможность найти теплоту, |
потребную для нагревания *
данной кислоты не до любой |
температуры а лишь до тем-
пературы кипения.
Рис. 209. Диаграмма Цейсберга.
Определение по этому графи-
ку производится следующим об-
разом. Пусть концентрация кисло-
ты 70% H2SO4 и температура 40°.
Берем точку пересечения верти-
кали, соответствующей цифре 70, с кривой, соответствующей температуре 40°.
Ордината этой точки по шкале калорий дает 93, т. е. число больших кало
рий, которое требуется для нагревания данной кислоты с 40° до темпера-
туры кипения в количестве, соответствующем 1 кг моногидрата H2SOs.
27 Зак. 501Я. Технология серной кислоты.
417
Теплота, потребная для выделения воды из кислоты, пред-
ставляет величину, обратную теплоте разведения, численно ей
равна и может быть вычислена по той же формуле Томсева
(см. также стр. 32):
,, т, ____ «2-17 870 «1-17 860
2 1 “ л2 + 1,7983 — И! 1,7983 ’
где п-г — количество молей НЮ на моль H2SO4 в начальной
(слабой) кислоте, m — количество молей НЮ на моль H2SO4
в конечной (крепкой) кислоте. Количество теплоты получается
по этой формуле в больших калориях на килограмм-моль H2SO4.
Из этой формулы легко вывести следующее соотношение:
Г100 — а" 100 — а'1
? 1150—а!' 150—в]
а
где а" — процент H2SO4 в начальной кислоте, а а' — процент
H2SO4 в конечной кислоте, q — число больших калорий, потреб-
ных для удаления воды, считая на 1 кг H2SO4.
Теплоту, необходимую для выделения воды из кислоты,
можно вычислить по формуле Портера, учитывающей также тем-
пературу. Портер для теплоты разведения дал следующую фор-
мулу:
,, 504.2 М . 0,714(7—15)
П ~М± 0,2013 ** М + 0,062 ’
где Н — число больших калорий на количество кислоты, соот-
ветствующее 1 кг SO3; М — отношение веса всей НЮ' к весу
SO3 в данной кислоте; t — температура.
Диференциальная теплота разведения или, что то же, теплота,
потребная для выделения воды (считая на 1 кг SO3), по Портеру
равна:
w „ Г 504,2 412 ( 0,714 442(7 — 15)1
— [Ж2_|_ о,2013 “Г 442 + 0,062 J
Г 504,2 441 . 0,714 441(4—15)]
[ 471+0,2013 441 + 0,062 J’
где M-г относится к начальной, т. е. слабой кислоте, a Mi — к ко-
нечной (крепкой) кислоте.
Скрыта^ теплота испарения воды может быть взята из
табл. 17 (стр. 31) или вычислена по формуле
£ = 83,9(365 —/°)Ч
где L — число больших калорий на 1 кг испаряемой воды, а (° —
температура, при которой происходит испарение воды, т. е. тем-
пература концентрирования.
Общее количество теплоты, потребное для концентрирования,
может быть определено графическим способом (рис. 210). Пунк-
тир на рис. 210 поясняет, как это количество вычисляется: от на-
чальной концентрации кислоты (76% H2SO4) ведется нисходящая
параллель до пересечения с? ординатой конечной концентрации
кислоты 92% H3SO4 (верхняя левая шкала). Далее ведется гори-
зонталь до пересечения с ординатой температуры концентриро-
вания (220—верхняя правая шкала), наконец, ведется восхо-
дящая параллель, точка пересечения которой с правой шкалой и
покажет потребное количество теплоты на 1 т продукции. Этот
график дает лишь приблизительные значения, однако достаточ-
но точные для заводских
целей.
Рассмотренными стать-
ями не исчерпывается весь
расход теплоты при кон-
центрировании. При про-
ектировании ' концентра-
ционной установки дол-
жны быть также учтены
потери тепла аппаратурой
в окружающую среду и!
с отходящими из уста-
новки газами.
При концентрировании
серной кислоты происхо-
дят два физических про-
цесса: испарение воды и
переход теплоты от на-
Конечная концентрация Нг504 Т-рс
/5/200
/76400
20/600 6
226800 §
252000 ч
277200 *
302600 'ё
327600 ?
352800 §
37В 000 §
603200 °
628600 е
653600 Q
678BOO '
506000 §
529200^
556 600
579600
606 BOO
гревающих газов к кис-
лоте. Общая скорость
концентрирования опреде-
ляется скоростью наи-
более медленно идущего
Рис. 210. График для определения полного
количества тепла, потребного для концен-
трирования кислоты.
процесса, каковым в зависимости от конкретных условий будет
или процесс теплопередачи или процесс эксорбции водяных
паров из раствора серной кислоты.
Единственное более или менее полное исследование скорости
процесса эксорбции водяных паров из растворов серной кислоты
было проведено Турханом и Андреевой. Этими исследованиями
установлено, что в изученном пределе скорости проходящего газа
коэфициент скорости эксорбции водяных паров пропорционален
линейной скорости газа в степени 0,8. Увеличение плотности
орошения не оказывает влияния на коэфициент скорости эксорб-
ции водяных паров. Значительно повышается коэфициент ско-
рости эксорбции паров Н2О с увеличением концентрации упари-
ваемой кислоты. Так, при 137° и при линейной скорости газа
50 см/сек коэфициенты скорости эксорбции К имеют такие зна-
чения для разных концентраций H2SO4 (в см/сек):
о/о H2SO4 к %H2SO4 К
65 .... 0,81 80.......1,23
70........0.92 85 ..... 1,39
75........1,11
27*
419
С повышением температуры коэфициент скорости эксорбции па-
ров НгО из растворов серной кислоты уменьшается. Значения
коэфициента скорости эксорбции паров воды в зависимости от
температуры для 75%-иой кислоты при линейной скорости газа
50 см/сек приводим ниже:
Р Р К
97,0 .......... 1,65 127,0 ........ 1,22
107 ........... 1,50 137 .......... 1,11
117 ........... 1,34 147 .......... 1,С0
Все эти данные приводят к заключению, что при эксорбции
паров НгО из растворов серной кислоты основное сопротивление
оказывает газовая пленка. Для расчета процесса испарения воды
из растворов серной кислоты, если он не тормозится процессом
теплопередачи, можно пользоваться уравнением
f dPB
J Pi Pg P
где pt — упругость паров НгО над кислотой; р„ — парциальное
давление паров НгО в газах; р — средний радиус свободного
объема аппарата (отношение свободного объема аппарата к его
орошаемой поверхности) и t — время пребывания газа в аппа-
рате.
Для концентрирования серной кислоты применяются два
основных типа установок.
Первый тип — концентрирование серной кислоты непосред-
ственным соприкосновением горячих газов с упариваемой кис-
лотой. Этот тип установок в настоящее время доминирует. Сюда
относятся установки Кесслера, Гайяра и Кемико.
Второй тип — концентрирование серной кислоты путем ее на-
гревания через стенку. Сюда относятся установки Паулинга, Бю-
шинга и Фришера. К этому же типу установок можно отнести
и вакуум-концентраторы (ом. ниже).
Аппараты для концентрирования серной кислоты стали применяться в на-
чале прошлого столетия. Первые аппараты (в Англии) делались из стекла и
работали по принципу наружного обогрева кислоты (т. е. через стенки).
К тому же периоду относится появление платиновых чаш и свинцовых со-
судов. Но в свинцовых сосудах кислота концентрируется лишь до 80%,
так как более крепкая горячая кислота разрушает свинец.
В конце прошлого столетия появились чугунные аппараты. Позднее,
в начале XX в., стали применять аппараты из свинца и фарфора. Подобные
аппараты из кислотоупорного чугуна, работающие по принципу наружного
обогрева, применяются и в настоящее время для концентрирования серной
кислоты до высокой крепости (95—98% H2SO4). Аппарат Кесслера впервые
был установлен в 4891 г. в Шотландии. В 1906 г. во Франции была по-
строена первая установка Гайяра, а в 1925 г. в Америке — новая установка
фирмы Кемико. В аппаратах Кесслера, Гайяра и Кемико практически полу-
чают кислоту концентрации 92—93% H2SO-1.
В последние годы начали также строиться появившиеся впервые в США
аппараты для концентрирования, работающие под вакуумом. Аппараты такого
типа изготовляются в США фирмами Свенсон, Симонсон-Мантиуса и др.
На башенных системах в ряде случаев для концентрирования продукции
теплотой обжиговых газов используется первая башня системы.
420
II. Установки" для концентрирования
с непосредственным обогревом кислоты
1. Схемы
производства
Концентрирование
кислоты непосред-
ственным ее обогре-
вом производится в
установках Кессле-
ра, Гайяра и Кемико,
а также в насажен-
ной башне башен-
ной системы, когда
для концентрирова-
ния продукции си-
стемы используется
теплота обжиговых
газов.
В принципе уста-
новок Кесслера, Гай-
яра и Кемико лежит
одна схема.
По технологиче-
ской схеме Кесслера
(рис. 211) топочные
газы из топки 2 по-
ступают в концен-
тратор 7, который
соединяет в себе са-
туратор и рекупера-
тор. Выйдя из кон-
центратора, газы про-
ходят через подогре-
ватель 4, где отдают
свое тепло напра-
вляемой в концен-
тратор кислоте. Да-
лее газы проходят
через холодильник 8,
где осаждается часть
брызг кислоты и кон-
денсируется часть
водяных паров. Для
более полного осво-
бождения газов! от
увлекаемой ими из
концентратора сер-
ной кислоты, ОСО-
421
бенно в виде тумана, они проходят далее через коксовый
фильтр 9 и, наконец, вентилятором 10 выбрасываются в атмо-
сферу. Кислота, направляемая на концентрирование, из напор-
ного бака 7 поступает в подогреватель 4, затем идет в. концен-
тратор 1. Готовая кислота из концентратора проходит холодиль-
ник 3, поступает в сборник 5 и насосами 13 перекачивается в бак
(большой сборник) 14 или в другое место. Конденсат серной
кислоты, уловленный из газов в подогревателе 4, холодильнике 8 *
и фильтре 9, а также осаждающийся и в вентиляторе 10, соби-
рается в сборнике 11. Из сборника 11 конденсат насосом 12 по-
дается в напорный бак 7, т. е. вводится обратно в цикл произ-
Рис. 212. Схема концентрационной установки Ганяра:
7—генератор; 2—сатуратор; 3—холодильник; 4—рекуператор; 5—же-
лоб; 6—-коксовый фильтр; 7—вентилятор; 8~холодильник; 9—сборник
для конденсата; /О—сборник кислоты; 77—монтежю.
водства, или (как обычно делается) передается в сборник кон-
денсата 1/5 для использования его на другие цели. В последние
годы на ряде установок Кесслера вместо коксовых фильтров
поставлены электрофильтры.
В установке Гайяра (рис. 21(2) генераторный газ из генератора 1 сгорает
в канале, соединяющем генератор с башней-сатуратором г. Газы—.продукты
горения — поступают в башню 2 снизу и проходят через нее навстречу раз-
брызгиваемой распылителями серной кислоте. Башня 2 полая, она выполнена
из вольвикской лавы или андезита. Из нее газы еще горячие поступают
снизу в такую же другую башню-рекуператор 4, где они также встречаются
с кислотой, которую предварительно подогревают и концентрируют. Для
освобождения от увлекаемой из башен серной кислоты газы потом напра-
вляются в коксовый фильтр-конденсатор в и, наконец, выбрасываются венти-
лятором 7 в атмосферу.
Кислота, подлежащая концентрированию, подается в сборник 10, из ко-
торого при помощи монтежю н перекачивается к пульверизаторам башен 2
и 4- Кислота, вышедшая из башни 4 и предварительно немного сконцентри-
рованная и подогретая, через жолоб 5 идет в холодильник 8, а из него
в сборник ю, куда подается и свежая, подлежащая концентрированию кис-
лота. Готовая кислота (купоросное масло) по выходе из башни 2 охлаждается
в холодильнике з н затем перекачивается на склад.
422
В установке Кемико (рис. 213) ^ефть или мазут сжигаются
в топке 7, распыляясь там при помощи форсунки. Для горения
топлива и для разбавления продуктов горения в топку вентиля-
тором подается под давлением воздух. Горячие газы — про-
дукты горения — по газоходу 2 поступают в первую камеру кон-
центратора 3 — камеру окончательного упаривания (сатуратор).
Затем газы по внешней трубе переходят во вторую камеру кон-
центратора — камеру подогрева и предварительного концентриро-
вания. После этого по трубе 5 газы идут в электрофильтр б, где
улавливается увлекаемая из концентратора туманообразная сер-
ная кислота. Хвостовые газы через выхлопную трубу поступают
/to обратное упарийоние
Рис. 213. Схема концентрационной установки Кемико:
1—топка; 2—газоход; 3—концентратор; 4—выхлопная труба; 5—шлемовая труба; 6—эле-
ктрофильтр; 7—напорный бак; 8—контрольная коробка; 9—выходная труба для кислоты;
10—холодильник; 11—сборник; 12—сборник; 13 и 14—насосы.
в атмосферу. Подлежащая концентрированию слабая серная кис-
лота из напорного бака 7 через контрольную коробку 8 посту-
пает в камеру предварительного концентрирования, а из нее пе-
ретекает в камеру окончательного концентрирования через на-
ружную трубу 9. В нижнюю часть этой трубы для облегче-
ния перетока кислоты, а также для продувки оседающего шлама,
подается сжатый воздух.
Готовая кислота по выходе из концентратора стекает по трубе
в холодильник 10, затем в сборник 77, из которого насосом 13
перекачивается на склад или в цех^потребитель.
Улавливаемый в электрофильтре б конденсат стекает по трубе
или жолобу через кислотный затвор в сборник 72, из которого
насосом 14 перекачивается обратно в напорный бак 7, т. е. воз-
вращается в производство. ч
423
2. Аппаратура
Единая схема установок Кесслера, Кемико и Гайяра кон-
кретно оформлена следующей основной аппаратурой (табл. 96).
Таблица 96
Аппараты Установка
Кесслера Кемико Гайяра
Топка Топка Топка Топка
Сатуратор Сатуратор—часть концентратора Сатуратор (ка- мера окончатель- ного упарива- ния)— часть кон- центратора Башня-сатуратор
Рекуператор Рекуператор (часть концентра- тора); подогрева- тель кислоты Камера подогрева и предваритель- ного концентри- рования — часть концентратора Башня-рекупе- ратор
Конденсатор Подогреватель кислоты; газовый холодильник, коксовый фильтр (или электро- фильтр) Электрофильтр Коксовый фильтр
Кроме того, установки снабжены холодильниками, насосами,
вентиляторами, сборниками и другим вспомогательным оборудо-
ванием.
Остановимся на описании конкретной аппаратуры примени-
тельно к установкам Кесслера и Кемико.
Топки. Полугенераторная топка установки Кесслера (рис. 214)
сложена из красного кирпича и облицована изнутри огнеупорным
кирпичом. Топливо — антрацит, освобожденный от мелочи, загру-
жается в воронку 1, снабженную двойным затвором с верхней
крышкой и конусом и рычагом, служащим для поднятия конуса.
Горение антрацита происходит на чугунной колосниковой ре-
шетке 2, состоящей из отдельных стержней овальной или ква-
дратной формы. Каждый колосник на одном торце имеет выступ
квадратного сечения. На этот выступ надевают ключ 6, кото-
рым повертывают колосники таким образом, чтобы большие оси
их овального сечения приняли вертикальное положение. Прн
этом положении колосников зола проваливается в зазоры между
ними и падает в воду, налитую в углублении пода поддувала 9-
Шуровка топлива производится через боковое окно 4 в кладке
топки. Для предохранения колосников от перегорания на ко-
424
лосниковой решетке всегда поддерживается небольшой слой
шлака. Воздух подается в зазоры между колосниками в таком
количестве, чтобы в топке образовался полугенераторный газ.
Сгорание этого газа происходит if камере догорания 7. Воздух
в камеру догорания засасывается через воздушную прослойку
между огнеупорным и красным кирпичом кладки. Количество
воздуха, подаваемого под колосники и
в камеру догорания, регулируется за-
слонками 5. Горячие газы выходят из
камеры догорания через газоходы. Во
время разогрева топки газы выпускаются
в атмосферу через растопочную трубу,
которая при нормальной работе топки
перекрывается шибером 3.
В установках Кесслера применяются
также и топки, работающие на мазуте.
Топка установки Кемико, работающая
на мазуте, представляет собой железный
барабан 1 (рис. 215), насухо выложенный
листовым асбестом и футерованный тре-
мя слоями кирпича: изоляционный — диа-
томитовый или трепельный (только в
верхней части топки), красный и, нако-
нец, огнеупорный. Внутри топки прохо-
дят две перегородки: первая 1'3 по всему
сечению топки в виде мелкой решетки,
вторая 8 — сплошная с окном в нижней
части для выхода газа в камеру смеше-
-ния. Первая перегородка предохраняет
вторую от действия голого огня и способ-
ствует перемешиванию выходящих из то-
почного пространства газов, равномер-
ному горению мазута, аккумулируя тепло.
Воздух подается вентилятором в же-
лезный короб 12, из которого идет, во-
первых, в топочное пространство 4 через
отверстие в центре передней стенки
топки и, во-вторых, по двум боковым и
нижнему каналам 7 в кладке топки —
в камеру смешения 5. Общее количество
Рис. 214. Полугенератор-
ная топка установки Кес-
слера:
7—воронка; 2— колосниковая
решетка; 3—шибер; 4—шу-
ровочное окно; 5—заслонка;
б—ключ для поворота колос-
ников; 7—камера догорания;
8—каналы дчя воздуха; 9—
поддувало.
подаваемого воздуха регулируется за-
движкой на всасывающем патрубке вентилятора, а распределе-
ние этого общего количества между главным входом в топку и
каналами регулируется дополнительными задвижками и реги-
страми на нагнетательных воздухопроводах. Нефть или мазут
подается под давлением через фильтр в форсунку, проходящую
через короб печи. Газы из топочного пространства через решетку
первой перегородки поступают в камеру смешения, где они сме-
шиваются с дополнительным воздухом, поступающим через каналы.
425
Из камеры смешения газы по штуцерам и чугунным газохо-
дам идут или в концентратор или в атмосферу (при растопке).
Ввнтилипюю
Рис. 215. Нефтяная топка аппарата Кемико:
/—железный цилиндр; 2—красный кирпич; 3— огнеупорный кирпич; 4—камера горения;
.5—камера смешения; б—передняя горловина; 7—каналы; в—перегородка; 9—смотровое
•окно; 70—термопара; 11—штуцер для выхода газа; 12—короб; 13—решетчатая перегородка.
Рис. 216. Концентратор Кесслера:
1—рекуператор; 2—сатуратор; 3—каллоты; 4—полки рекуператора;
5—крышка-шлем; 6—отверстие для чистки.
Концентраторы. Концентратор Кесслера объединяет в одном
аппарате и сатуратор 2 и рекуператор 1. Ннжняя часть концен-
•426
Рис. 217. Концентратор Кемико:
1—перегородка; 2—камера предварительного и 3—камера
окончательного упарнва шя; 4—отверстия для входа и вы-
хода кислоты.
тратора (рис. 216) — сатуратор — представляет собой прямоуголь-
ную коробку, на дне которой всегда держится некоторый слой
кислоты, упариваемой здесь до купоросного масла. Уровень
кислоты в сатураторе может изменяться при помощи фарфо-
ровой керамиковой перегородки, вставляемой в трубу, выводя-
щую кислоту из аппарата. Перегородка закрывает нижнюю
часть сечения спускной трубы, поднимая тем самым уровень кис-
лоты в сатураторе. Для более тесного соприкосновения газов
с кислотой сатуратор разделен рядом перегородок (колосников),
немного не доходящих до уровня кислоты. Для чистки сатура-
тора от грязи и шлама с каждой его стороны сделано по три
отверстия, которые во время нормальной работы аппарата за-
крыты особыми камнями.
Рекуператор представляет собой квадратную семиполочную
башню, поставленную на сатуратор и составляющую с ним, та-
ким образом, один
аппарат. Полку 4 ре-
куператора образует
андезитовая плита с
бортами; в этой пол-
ке имеются оваль-
ные отверстия с вы-
тесанными в камне
бортами; на борта от-
верстий устанавли-
ваются фарфоровые
колпачки 3 (каллоты) с зазубренными краями. Благодаря этим
колпачкам достигается тесное соприкосновение кислоты с га-
зами, проходящими через отверстия. Колпачки на разных полках
путем наклейки кислотоупорных пластинок на борта каллот уста-
навливаются на разной высоте над отверстиями. Боковые отвер-
стия каждой полки рекуператора предназначены для чистки
ее от грязи м шлама и для смены каллот. Во время нормальной
работы эти отверстия заделаны особыми камнями.
Сверху рекуператор покрыт свинцовой конической крыш-
кой — шлемом 5- Он состоит из спаянных между собой свин-
цовых листов, поддерживаемых железным каркасом, тоже опаян-
ным сверху свинцом. К шлему на фланцах присоединен свин-
цовый газоход, по которому газы уходят из концентратора.
Стенки сатуратора и рекуператора выложены на кислотоупор-
ной замазке из андезитовых камней и обтянуты листовым свин-
цом. На ряде заводов рекуператор выполнен в виде башни с на-
садкой (кварцем).
В последние годы свинец в аппаратуре установок Кесслера
успешно заменяется железом: на стенках, днище и крышке са-
туратора, на стенках рекуператора и на стенках и днище холо-
дильников купоросного масла.
Концентратор Кемико (рис. 217) представляет собой гори-
зонтальный сварной цилиндр из листового железа. Барабан
427
внутри выкладывается слоем асбестового картона, смоченного
жидким стеклом. По асбесту кладется слой листового свинца,
затем еще один слой асбестового картона и, наконец, футеровка
из кислотоупорного кирпича или андезита. Днище барабана —
сферическое. Пространство между стенкой днища и футеровкой
засыпается щебнем из андезита и заливается андезитобетоном.
Барабан разделен свинцовой перегородкой 1 на две камеры:
подогрева и предваритель-
ного упаривания — реку-
ператор 2 и окончательно-
го упаривания — сатура-
тор 3. Перегородка с обе-
их сторон футеруется.
Размеры барабана (с про-
ектной произвЬдитель-
ностью 60 т, в сутки) и его
камер следующие
Длина барабана . . . 7830
Диаметр.............2824
Внутренний диаметр 2170
Длина камеры окон-,
нательного упари-
ваиия ............. 3200
Соусх конденсата Длина камеры пред-
мм
Рис. 218. Подогреватель слабой кислоты.
верительного упа-
ривания ......... 2815 ,
Барабан сверху снабжен штуцерами для присоединения га-
зоходов и люками для обслуживания его во время ремонта и
чистки. Для входа и выхода кислоты в каждой камере концен-
тратора имеются по два отверстия, расположенные в сатура-
торе на 50 мм ниже, а в рекуператоре на 300 мм выше горизон-
тальной оси барабана. Высота уровня кислоты в каждой камере
регулируется кольцами-перегородками, вставляемыми в соеди-
нения кислотных труб у выхода кислоты.
Слабая кислота поступает в рекуператор через штуцер из
кислотоупорного металла, вставленного в отверстие в стенке
барабана. Присоединением к штуцеру свинцовой трубы с ко-
леном создается гидравлический затвор из кислоты, препят-
ствующий выбиванию газа из концентратора. Переход кислоты
из рекуператора в сатуратор происходит по внешнему сифону,
выполненному из дюрайрона' или другого кислотоупорного
сплава. В нижнюю часть сифона по свинцовой трубе подво-
дится сжатый воздух.
Купоросное масло вытекает из сатуратора в холодильник
также по трубе из дюрайрона, состоящей из патрубка, встав-
ленного в аппарат, колена и патрубка, опускаемого в кислоту
в холодильнике. Погружение последнего патрубка в кислоту
создает гидравлический затвор, исключающий выбивание газа
428
из аппарата. Для спуска кислоты во время чистки или ремонта
аппарата каждая камера имеет клапан.
Газоходы от топки к камерам выполнены из чугуна, а от
камеры в камеру и от концентратора к электрофильтру отлиты
из кислотоупорного сплава. Газоход из рекуператора в электро-
фильтр сделан из листового железа, выложенного внутри ли-
стовым асбестом, затем освинцован и, наконец, футерован кис-
лотоупорным кирпичом.
Аппараты для улавливания тумана и брызг серной кислоты.
В установке Кесслера конденсат осаждается в подогревателе
кислоты, газовом холодильнике, коксовом фильтре и вентиляторе.
Основное назначение подогревателя кислоты — лучшее исполь-
зование теплоты газов, т. е. этот аппарат продолжает выполнение
•функций рекуператора. Газовый холодильник служит для охла-
ждения газов, что создает лучшие условия работы для газового
вентилятора, который в этой установке стоит в хвосте системы.
Рис. 219. Газовый холодильник:
1—деревянный корпус; 2—свинцовая оболочка; 3—свинцовые трубки; 4—люк.
Подогреватель слабой кислоты представляет собой цилиндр
(рис. 218) с крышкой, вокруг которой устроен гидравлический
затвор. Внутри цилиндра помещены свинцовые змеевики, отде-
ленные один от другого перегородками, которые газ принужден
огибать. Кислота из общей трубы течет во все три змеевика
и выходит из них сверху, подогреваясь до 40—50°.
Газовый холодильник установки Кесслера (рис. 219) пред-
ставляет собой свинцовый ящик, разделенный по длине свинцо-
выми перегородками на три части. В перегородки своими откры-
тыми концами впаяны свинцовые трубы. Газы входят в первую
камеру ящика, проходят далее через свинцовые трубы и уходят
из последней камеры ящика. В средней части ящика свинцовые
трубы охлаждаются водой. Размеры такого холодильника для
установки с проектной производительностью 12 т в сутки сле-
дующие:
Длина ящика..........................5900 мм
Ширина ящика......................... 2200 ,
Высота . •........................... 1500 ,
Длина свинцовых труб................. 2725 .
Диаметр свинцовых труб...............50/60 „
Число свинцовых труб . •.............67 шт.
Поверхность охлаждения...............32 м2
429
Коксовый фильтр установки Кесслера представляет co6oir
ящик, на дне которого поставлено на ребра несколько кислото-
упорных кирпичей, на них в виде решетки укладываются освин-
цованные железные прутья, на которые загружается кокс, сна-
чала более крупный, а затем более мелкий. Между коксовой
Рис. 220. Электрофильтр.
насадкой и крышкой
ящика остается за-
зор в 20—30 срт). Газ
поступает в ящик
сверху, проходит че-
рез кокс и выходит
через газоход в низу
ящика.
Благодаря тесно-
му соприкосновению
газов с поверхно-
стью кокса частицы
тумана осаждаются
на коксе и собира-
ются в капли, кото-
рые стекают в низ
ящика и удаляются
оттуда через гидрав-
лический затвор в
сборник.
В установке Ке-
мико осаждение ту-
мана и брызг кисло-
ты из газа происхо-
дит в электрофильтре. Принцип работы электрофильтров под-
робно описан выше. Там были рассмотрены принципы осаждения
кислотного тумана электрофильтрами и дано описание работы
электрофильтров, применяемых на контактных сернокислотных
заводах. Здесь мы коротко остановимся
лишь на конструкции электрофильтров, при-
меняемых на концентрационных установ-
ках.
Электрофильтр на установках Кемико в
СССР представляет собой квадратную баш-
ню из андезита или андезитобетона разме-
ром 4 X 4 м и высотой 9 м (рис. 220).
На высоте 6,5 м в башне устроены сво-
ды в виде колосниковой решетки. Между
графитовые трубы, подвешенные на сводах
на равном расстоянии друг от друга. В верхнем конце каждой
трубы снаружи имеется углубление, в этом месте трубу охваты-
вает графитовая пластинка с круговым вырезом, разделенная на
две части. Пластинки с зажатой в них трубой опираются на
своды.
2мм
Ф=10мм
Ф-8мм
Рис. 221. Сечение
короиирующего про-
вода.
сводами находятся
430
Внутри каждой трубы проходит провод, состоящий из 1,5—
2-Л7И жилы и свинцовой оболочки (рис. 221). Снаружи освинцо-
ванный провод имеет вид пятигранника. Провода подвешены
к общей раме, которая находится над верхними концами труб.
Снизу на каждом проводе подвешен груз для натягивания; все
грузы соединены между собой, для того чтобы отдельные про-
вода " не раскачивались и чтобы их можно было точно устано-
вить в центрах труб. Верхняя рама подвешена к балке, которая
обоими концами выходит из камеры в боковые коробки, где
балка сама подвешена при помощи хомутов к болтам. Другой ко-
нец болтов соединен с гирляндовыми изоляторами в коробках.
К болту, висящему на изоляторах, припаян колокол, опущенный
в чашу с трансформаторным маслом (масляный затвор).
Газ через трубу поступает в междутрубное пространство-
(немного ниже сводов), опускается вниз и входит в трубы; здесь
газ очищается от тумана серной кислоты и выходит вверх через
трубу в атмосферу. Кислота оседает на трубы, стекает вниз и
через карман самотеком поступает в бак для конденсата, откуда
время от времени подается в напорный бак слабой кислоты,
т. е. возвращается в цикл производства.
В электрофильтрах, устанавливаемых в последние годы
(рис. 220), газ вводится снизу под кварцевую насадку. Кварц
улучшает распределение газа по сечению аппарата и предвари-
тельно несколько очищает газ, чем облегчает работу электро-
фильтра.
Для перекачки кислот в установках Кесслера и Кемико упо-
требляются горизонтальные центробежные насосы, для охлажде-
ния кислот — цилиндрические (свинцовые) змеевиковые холо-
дильники.
3. Режим и работа концентрационных установок
Кесслера и Кемико
Основными условиями нормальной работы установок Кесслера
и Кемико являются: соответствие между количеством газов и
количеством кислоты, поступающих в концентратор; достаточ-
ная чистота газов; достаточно высокая температура поступаю-
щего газа; отсутствие в кислоте вредных примесей; поддержание
аппаратуры в порядке.
Температура газов, поступающих в аппарат Кесслера, под-
держивается в пределах 900—950°. Чем выше температура по-
ступающих газов, тем это лучше с точки зрения более интен-
сивной теплопередачи от газа к кислоте.
Температура газа, однако, не должна быть настолько высо-
кой, чтобы началось разложение H2SO4 с образованием SO2.
В газах, поступающих в концентратор, должны отсутствовать
и вредные газообразные компоненты — продукты неполного сго-
рания топлива. Эти вещества восстановляюще действуют на
серную кислоту, увеличивая ее потери. Кроме того, неполное
431
сгорание топлива означает недостаточно эффективное его исполь-
зование и, следовательно, удорожает стоимость "продукции. Из
этих соображений вытекает основное требование: отношение ко-
личеств воздуха и топлива, подаваемых в топку, должно быть
таким, чтобы, во-первых, сгорание топлива было полным и, во-
вторых, чтобы температура отходящих газов была достаточно
высокой. Кроме того, для полноты сгорания требуется равномер-
ная подача топлива и воздуха в топку, а также (в установке
Кемико) хорошее распыление горючего и его перемешивание
с воздухом.
В установке Кемико температура топки поддерживается
в пределах 1000—1100°. Однако в камере смешения темпера-
тура газов понижается (раньше до 650°, теперь до 800—900°)
при разбавлении воздухом (поступающим по каналам). В отли-
чие от концентратора Кесслера, где горячие газы.лишь поверх-
ностно соприкасаются с кислотой, в концентраторе Кемико го-
рячие, газы пробулькивают через кислоту, благодаря этому даже
и при более низкой температуре входящих газов достигаются
хорошие условия теплопередачи. Для максимального использо-
вания теплоты газов следовало бы газы выпускать из концен-
тратора с возможно более низкой температурой. Но при умень-
шении разности температур между газами и кислотой требуется
все большая и большая поверхность соприкосновения для пере-
дачи определенного количества тепла, а это связано с увеличе-
нием размеров аппаратуры. Кроме того, из несущих большое ко-
личество влаги газов водяные пары обратно конденсируются
в холодной кислоте и в определенной части аппарата получается
круговая циркуляция некоторого количества водяных паров.
Отсюда ясно, что должна существовать оптимальная темпера-
тура выпуска газов из концентратора. Нормальной температурой
выпуска газов из концентратора Кесслера (температура по вы-
ходе из рекуператора) и концентратора Кемико (по выходе из
камеры предварительного упаривания) считается 125—150°.
Правда, теплота газов, вышедших из рекуператора, в установке
Кесслера еще частично используется — в подогревателе для
предварительного подогрева кислоты; газы прн этом охла-
ждаются до 90—95°. Но это связано с усложнением установки.
Нормально на концентрирование поступает кислота концен-
трации 68—76% H2SO4. В поступающей кислоте не должно
быть большого количества вредных примесей, например азот-
ной кислоты и окислов азота, солей и органических веществ.
В поступающей кислоте не должно быть больше 0,1% окислов
азота и азотной кислоты, считая все на HNO3. Выделяющиеся
при нагревании кислоты окислы азота сильно разъедают аппа-
ратуру, главным образом электрофильтры (свинец). Присут-
ствующие в кислоте соли и взвешенные примеси осаждаются
в виде шлама, загрязняя аппараты и кислотопроводы. Органи-
ческие вещества, присутствующие в кислоте, при нагревании
взаимодействуют с ней, восстанавливая часть ее до SO». По аме-
432
риканским нормам в примесях кислоты, поступающей на концен-
трирование, не должно быть более 3% углерода.
В установках Кемико и Кесслера концентрирование кислоты
обычно ведут до 92—93% H2SO4.
При дальнейшем повышении концентрации кислоты она
сильно разрушает футеровку. Для большинства потребляющих
производств концентрация купоросного масла в 92—93% H2SO4
вполне достаточна.
Готовое купоросное масло вытекает из концентратора с тем-
пературой 220—250° и охлаждается в холодильнике. В уста-
новке Кесслера газы после подогревателя охлаждаются в газо-
вом холодильнике с тем расчетом, чтобы температура их при
входе в вентилятор была не выше 60°. В установке Кемико газы
выпускаются в атмосферу при температуре ~ 120°. Как уже ука-
зывалось, брызги и туман серной кислоты в установке Кесслера
улавливаются в подогревателе, газовом холодильнике, коксовом
фильтре и вентиляторе.
Концентрация получаемого конденсата примерно следующая
(в % H2SO4):
Из газового холодильника.............. 21—22
Из коксового фильтра..................... 45—48
Из вентилятора........................ 39—40
Чем ниже температура газов при конденсации, тем слабее
получается конденсат, так как тем больше вместе с туманохм
H9SO4 конденсируется водяных паров. Поэтому температура га-
зов в электрофильтре установки Кемико и поддерживается
выше 100°.
В установке Кесслера всего конденсата получается около
15% от продукции (считая и продукцию и конденсат в моноги-
драте), в установке Кемико в электрофильтре улавливается
6—13% от продукции. Потери в установке Кесслера составляют
3—5% от продукции, в установке Кемико — 2%. Большая часть
потерь приходится на неуловленную туманообразную кислоту,
а меньшая, очень незначительная часть, получается от разложе-
ния H2SO4 до SO2.
Производительность концентратора выражается обычно в тон-
нах готовой продукции в сутки. Понятно, что производительность
одной и той же установки тем больше, чем выше концентрация
начальной кислоты и чем ниже концентрация продукции. Уста-
новки Кесслера раньше ставились производительностью от 6 до
30 т купоросного масла в сутки, в последнее время они проекти-
руются производительностью до 60 т в сутки.
Проектная производительность описанного аппарата Кемико
составляет 60 т в сутки 93%-ной кислоты при исходной концен-
трации 68—70% H2SO4. Опыт стахановцев показал, что аппа-
раты Кесслера и аппараты Кемико имеют большие резервы для
интенсификации. Основным условием осуществления интенсифи-
кации аппарата Кемико является наличие достаточного напора и
28 Зак. 5019. Технология серной кислоты.
433
производительности вентилятора, а основным методом интенси-
фикации— повышение температуры входящих газов. При работе
концентратора Кемико на горячей кислоте (до 120°) производи-
тельность достигала 86 т в сутки. Как показали заводские опыты,
повышением температуры газов на входе в первую камеру
можно увеличить суточную производительность аппарата Кемико
минимум до 90 т в сутки.
Понятно, что связанные с повышенной интенсивностью неко-
торые изменения в режиме работы аппарата Кемико вызывают
необходимость некоторых аппаратурных изменений (увеличение
диаметра труб подводящих газ в аппарат, выполнение барботаж-
ных труб из материала, устойчивого в более тяжелых условиях
работы и др.).
Величины расходных коэфициентов зависят от большинства
тех же факторов, от которых зависит и производительность аппа-
рата и в первую очередь от начальной концентрации кислоты.
Расходные коэфициенты на 1 т продукции в установке Кес-
слера при упаривании кислоты до 92—93% H2SO4 примерно сле-
дующие.
При начальной концентрации
68% H2SO4 75% H2SO4
Мазут (10 тыс. ккал), кг........ 60 53
Электроэнергия, квт-ч........... 13,5 11
Вода (начальная температура
20°), л3......................... 10 10
Пар (давление 2 ат), кг .... . 20 20
Расходные коэфициенты на 1 т продукции купоросного масла
(в пересчете на моногидрат), при питании концентратора холод-
ной кислотой (температуры 20—25°) с начальной концентрацией
68% H2SO4, по установке Кемико примерно следующие:
Топливо (мазут 10 тыс. ккал), кг............ 57
Пар (давление 2 ат), кг..................... 25
Электроэнергия, квт-ч...................... 25
Вода (начальная температура 2(Р), мЛ ... . 6
Если концентратор питается горячей кислотой, то расход то-
плива, энергии и пара несколько снижается.
4. Контроль производства
Контроль производства на установках Кемико и Кесслера
сводится к анализу: 1) слабой кислоты на H2SO4 и на примеси
(азотную кислоту и окислы азота, твердый остаток), 2) продук-
ции на H2SO4 и на твердый остаток, 3) топочных газов на СО_>,
4) отходящих газов на туман H2SO4 и 5) конденсата на H2SO4;
к замеру температуры газов и разрежения (давления — установка
Кемико) по точкам и температуры кислоты по выходе из концен-
тратора (а также из подогревателя кислоты — установка Кес-
слера).
431
Таблица 97
Место контроля Объект контроля Определяется Частота анализа или замера
Вентилятор Топка Концентра- ционный аппа- рат Холодильник Электрофильтр Мотор венти- лятора Воздух Газ Нефть Жиклеры Газ Кислота Купоросное масло Вода Электроток Ампера ж Давление после венти- лятора Температура Процент СО2 в газах (аппаратом Орса) Температура в °C Давление Расход по уровню замера Температура: на входе в 1-ю камеру на входе во 2-ю Камеру- на шлеме Давление: на входе в 1-ю Камеру- на входе во 2-ю Камеру- на шлеме Поступающая на пере- работку: °Вё при 15° температура процент HNO3 процент H.2SOj Вытекающее купоросное масло: температура до холо- дильника °Вё при 15° после холо- дильника и температура до холодильника Температура после холодильника А. V, mA Через 2 часа Через час Через час Через 2 часа То же Через час, запись один раз в смену По мере необ- ходимости Через час » » » » Через час. я » » » Я я » н 1 раз в смену То же Через час Через час 1 раз в смену То же Через час
28'»
435
Продолжение табл. 97
Метод контроля Объект контроля Определяется Частота анализа или замера
Электрофильтр Продукция Газ Конденсат Купоросное масло Содержание H2SO4 в г/ж3 иа выходе и входе Температура и °Вё при 15° °Вё при 15°, темпера- тура и процент H2SO4. Количество выработан- ного масла по уровням бачков 1 раз в смену То же По мере на- полнения бачков
Работу установки в основном характеризуют следующие по-
казатели: температура газов при входе и выходе из концентра-
тора, температура (и концентрация) кислоты по выходе из кон-
центратора. Если, например, при нормальной температуре входа
газов температура их при выходе и температура выхода кислоты
(и ее концентрация) понизились, то это означает недостаточную
подачу газов или перегрузку аппарата кислотой. Если при нор-
мальной температуре входа газов температура их при выходе по-
высилась при одновременном понижении температуры и концен-
трации выходящей кислоты, то это означает неисправность ап-
парата: загрязнение или разрушение полок рекуператора (уста-
новка Кесслера), прохудение газоподводящих труб внутри камер
барабана (установка Кемико и др.).
Найти неисправное место в аппаратуре часто можно по изме-
нению показаний манометров.
Перечень принятых анализов по концентрационной установке
Кемико приведен в табл. 97. Аналогичный объем контроля про-
изводства принят также по установке Кесслера.
5. Концентрирование башенной кислоты
теплотой обжиговых газов
Найдем температуру обжиговых газов при входе в башню концентра-
тора, достаточную для того, чтобы теплотой этих газов можно было про-
дукцию— башенную кислоту — сконцентрировать до купоросного масла. Если
концентрация башенной кислоты 76% HsSO«, а купоросного масла 92,5%
HsSOn, то на 1 т продукции в моногидрате надо выпарить воды:
1000-100 1000-100 оос
~76----------923- = 235 кг’
На выпаривание воды при 200° потребуется теплоты
(?! = 463 • 235 = 109 000 ккал.
436
Для нагревания кислоты от температуры ее входа в концентратор (при-
мерно 60°) до 200° потребуется теплоты:
@2 = 815 (150 — 45) = 85 000 ккал,
где 815 — кг SOs в 1 т моногидрата; 150 и 45 — теплосодержание в ккал
76%-ной кислоты, при 200 и 60°, считая иа 1 кг SOs.
На выделение воды из кислоты потребуется теплоты (теплота, обратная
теплоте разведения):
Г100 - 76 100 — 92,51
Q' ~ 2,2' [150-'76 150 — 92,5] 1000
<2з = 53 000 ккал.
Всего потребуется теплоты для концентрирования, считая на 1 т продукции
в моногидрате:
Q = 109 000 + 85 000 + 53 000 = 247 000 ккал.
Определим теперь температуру входящих в концентратор газов, принимая
температуру выходящих газов равной 120°, а концентрацию SO2 во входящем
газе равной 9%. Теплоемкости SO2, О2 и Na в зависимости от темпера-
туры (считая на количество газа, соответствующее 1 .и3 при 0°) имеют сле-
дующие выражения:
Cs02 = 0,437 4-0,04847/,
со2 = cn2 = 0.308 + 0,04237.
Из этих выражений для теплоемкости обжигового газа можно составить
формулу:
<?га8 = 0,308 4- 0.0Д29 PSOa 4- (0,0,237 4- 0Д61 PS02) /,
где Ps0 —.объемный процент SOa в обжиговом газе. При Pso, равном 9,
объем обжиговых газов, считая на 1 т продукции, составит:
10009^-9-0-° = 2530 ^
При температурном перепаде от tBV до 120° газ отдаст тепла
<2 = 2530 (С сгаз, -120).
' гав£вх. газ120 ’
Подставляя в последнее выражение значения* Сгаз для температуры
входа и 420° по выведенному выше выражению теплоемкости и 247 000,
вместо Qi получим уравнение:
/^4-10900 — 4700000 = 0,
откуда /вх = 450°.
Так как при подаче на J-ю башню неденитрированной кислоты в пей
будет окисляться некоторое количество SO2, практически можно получить
всю продукцию башенной системы с концентрацией до купоросного масла и
при более низкой температуре входящих газов (~ 376—400°).
Для те^ случаев, когда теплота обжиговых газов недостаточна, было
предложено подогревать поступающую в концентратор кислоту горячей кис-
лотой, выходящей из концентратора (теплообмен). Однако это мероприятие
не может улучшить тепловой баланс концентратора. При повышении темпе-
ратуры входящей в концентратор кислоты иа такое же количество градусов
повышается и температура уходящих из концентратора газов. При повыше-
нии температуры входящей кислоты на 1° приход тепла (считая на 1 т
437
продукции в моногидрате) возрастет на 0,5-1316 = 658 ккал- При повы-
шении же температуры уходящих газов на Iе расход тепла (считая опять
на 1 т- продукции) возрастет на 0,33 2530 — 840 щц.
Отсюда следует, что предварительный подогрев поступающей в концен-
тратор кислоты не улучшает тепловой баланс концентратора. Это мероприя-
тие можно рекомендовать только при условии наличия избыточной теплоты
газов против требуемой для концентрирования продукции до купоросного
масла. Уменьшение количества теплоты, которое должно быть передано от
газа к кислоте, снижает потребную поверхность теплообмена, а следова-
тельно, и потребный объем концентратора. Повышение температуры газов,
выходящих из концентратора, ведет также к улучшению условий работы
первой продукционной башни.
III. Установки с внешним подогревом
1. Установки, работающие по принципу дефлегматорных колонн
К этой группе установок относятся аппараты Бюшинга, Пау-
линга и Фришера.
Общий принцип их работы заключается в следующем. 4 В обогреваемом
через стенку сосуде выпаривается 98%-ная кислота; пары этой кислоты та-
кого же состава, как и упариваемая жидкость,
Рис. 222. Схема концентрационной установки
Бюшинга:
7—реторта; 2—топка; 3—дефлегматор; 4—ловушка;
5—конденсатор; 6—холодильник купоросного масла;
7—мешалка.
поступают иа первую тарелку
дефлегматорной колонки. На
этой тарелке кипит не-
сколько более слабая кис-
лота, чем в выпарном со-
, суде; следовательно, здесь
содержание H2SO« в парах,
находящихся в равновесии
с кипящей жидкостью,
меньше, чем в парах, при-
шедших из выпарного со-
суда. Поэтому на первой
тарелке часть H2SOa перей-
дет в жидкость и в выпар-
ной сосуд отойдет более
концентрированная по H»SO«
кислота, чем кислота, при-
шедшая на первую тарелку.
Наоборот, в парах, выходя-
щих с первой тарелки на
вторую, содержание H2SO«
будет меньше, чем в парах,
поступивших на первую та-
релку снизу. По такому же
принципу пойдет процесс и
на следующих тарелках.
Если на какой-нибудь бо-
лее верхней тарелке нахо-
дится кислота концентра-
ции ниже 70% H2SO1, то
в парах, находящихся с ней
в равновесии, даже при
температуре кипения будет
кислота находится на сле-
содержаться только Н2О- Чем более слабая
дующих тарелках, тем больше воды будет переходить в пары. В отходящих
из колонки парах, таким образом, теоретически совсем не должно быть HsSOa.
Практически в них могут присутствовать лишь в незначительных количе-
ствах увлеченные брызги кислоты.
438
Принципиальная схема установок Бюшинга, Паулинга й Фри-
шера одна и та же и установки эти отличаются лишь деталями
аппаратурного оформления.
В установке Бюшинга (рис. 222) реторта (котел) из кислото-
упорного чугуна вмазана в топку и обогревается снаружи горя-
чими газами, получаемыми в топке. В реторту заливается
98' < -ная кислота, пары которой поднимаются в колонку-дефлег-
матор, где им навстречу течет с тарелки на тарелку более слабая
кислота. Всего в колонке 9—10 тарелок. На каждой тарелке
происходит конденсация части паров H2SO4, пришедших с пред- <
шествующей (более нижней) тарелки. С последней (верхней) та-
релки, на которую поступает 67—70%-ная кислота, уходят уже
только пары воды и капли серной кислоты, увлекаемые механи-
чески и задерживаемые в ловушке. Пары воды из ловушки по-
ступают в конденсатор, где они конденсируются холодной водой
и вместе с ней уходят в канализацию. Действием конденсатора
в установке создается некоторый вакуум, чем достигается кипе-
ние кислоты при несколько пониженной температуре и устра-
няется разложение кислоты, начинающееся при более высокой
температуре. Готовая продукция из котла непрерывно стекает
в холодильник, а в дефлегматор поступает слабая кислота из
бака. Котел служит, главным образом, для получения паров сер-
ной кислоты, необходимых для концентрирования в дефлегма-
торе. Мешалка внутри котла препятствует образованию осадка
на его стенках, благодаря чему в аппарате возможно упаривать
и грязную кислоту.
Установки Паулинга, Бюшинга и Фришера практически дают
более концентрированную кислоту (до 98% H2SO4), чем уста-
новки Кесслера, Гайяра и Кемико, с незначительными потерями
кислоты при концентрировании (1—2% от продукции). Однако
расход топлива в этих установках значительно больше, чем
в установках с непосредственным обогревом. Большим недостат-
ком этих установок является также их незначительная произво-
дительность (10—20 т в сутки), а также частое прогорание ре-
торт.
21 Вакуум-концентратор
Под вакуумом кислота (как и всякая жидкость) кипит при
более низкой температуре. По кривым рис. 223 видно, например,
что 95%-ная H2SO4, кипящая под атмосферным давлением при
295 , под вакуумом в 756 мм рт. ст. кипит при 130°. Такое
понижение температуры кипения позволяет сохранять большую
разность температур между упариваемой кислотой и нагреваю-
щим телом. Кроме того, упариваемая кислота и ее пары при низ-
кой температуре гораздо меньше разъедают аппаратуру. Наконец,
значительно уменьшаются потери H2SO4, а разложения H2SO4 до
SO2 не происходит вовсе.
Концентрирование под вакуумом особенно имеет смысл для
слабых кислот, содержащих органические примеси (например
439
кислота, сепарированная из кислого гудрона), тдк как под ваку-
умом значительно уменьшается восстановление кислоты.
В вакуум-выпарной установке Симонсон-Мантиуса (рис. 224)
слабая серная кислота из сборника 7 заливается в концентра-
тор / в первое время самотеком, а затем подсасывается вслед-
ствие вакуума, создающегося в концентраторе. Обогрев кисло-
ты производится при помощи нагревательных труб насыщенным
паром. Выделяющиеся из кислоты
рез брызгоуловитель 2 в конден-
сатор 4, где и конденсируются.
Воздух из конденсатора, который
впускается в аппарат для пере-
мешивания кислоты, а также воз-
дух, который выделяется из во-
ды, поступающей на конденса-
цию паров, отсасывается двух-
пары воды отсасываются че-
295
280
160
140
120
760
200
180
240
220
so
60
40
20
235’
280
268
240
220
200
ISO’
160^.
I
140 &
120 &
too
80
60
40
JO 31 3? 33 34 35 3637 38 39 404! 4243 44 45 48 47 48 43 50 51 52 53 54 55 56 57 58 59 60 6! 62 63 64 65 66
------------------------------*-°Вё
Рис. 223. Кривые температур кипения растворов серной кислоты под
вакуумом.
ступенчатым эжектором 5. Конденсирующийся пар вместе с во-
дой выходит из конденсатора в виде подкисленной воды, кото-
рая проходит через нейтрализатор в канализацию. Греющий пар
конденсируется в трубах нагревательной поверхности концен-
тратора и конденсат выходит в конденсационные горшки, от-
куда откачивается на обратное питание паровых котлов. Скон-
центрированная серная кислота стекает из аппарата в холодиль-
ник 6, в котором охлаждается до температуры 50°, позволяю-
щий перекачку ее насосами 9 в склад.
Установка Симонсон-Мантиуса работает периодически, выда-
вая за каждую операцию 50 т купоросного масла в пересчете на
моногидрат при упаривании 68%-ной H2SO4. Одна операция упа-
ривания 50 т кислоты продолжается 12 час. Суточная произво-
дительность аппарата составляет 100 т готовой продукции.
Вакуум-концентратор установлен на фундаменте высотой 4 м
от уровня пола. Такое расположение дает возможность кислоте
410
поступать самотеком из концентратора в холодильник и, кроме
того, обеспечивает барометрический затвор конденсатора, распо-
ложенного на высоте 11 м от уровня пола. Брызгоуловитель
находится непосредственно на концентраторе и представляет
с ним как бы одно целое. Брызгоуловитель соединен с конден-
сатором при помощи вакуум-усилителя или бустера. Рядом с кон-
центратором на полу установлен холодильник и насосы для пе-
рекачивания .серной кислоты.
Рис. 224. Схема выпарной установки Симонсоп-Ман-
тиуса:
1—вакуум-концентратор; 2—брызгоуловитель; 3—вакуум-усили-
тель; 4—конденсатор; 5—двухступенчатый эжектор; 6—холодиль-
ник; 7—сборник для слабой кислоты; 8—сборник для купоросного
масла; 9—насосы для купоросного масла; 10—сточная канава;
11—редуцирующие вентили для пара.
В а к у у м-к онцентратор (рис. 225) представляет собой
цилиндр диаметром 4870 мм и высотой 6080 мм. Аппарат внутри
футерован кислотоупорным материалом (андезит, керамика и др.).
Керамическая футеровка обтянута свинцом. После свинцовой
оболочки аппарат покрыт железным кожухом. В центре концен-
тратора имеется керамическая полая колонка 3, служащая для
создания жесткости аппарата. Кроме того, колонка поддержи-
вает брызгоуловитель 6, установленный на верхней крышке кон-
центратора.
Поверхность нагрева концентратора состоит из 128 труб 4,
изготовляемых из железа с содержанием 14,5% кремния. Все
трубы вставлены через штуцера, укрепленные на кожухе концен-
тратора.
Поверхность нагревательных труб составляет 101 №. Трубы
должны выдерживать рабочее давление в 8 ат.
441
Концентратор имеет лаз 10 для осмотра аппарата, люк для
наблюдения за работой концентратора 13, а также штуцера 5
для входа и выхода кислоты и присоединения контрольных изме-
рительных приборов.
Брызгоуловитель (рис. 226) состоит из двух керами-
ковых цилиндров, вставленных один в другой, что создает лаби-
Рнс. 225. Вакуу.м-концентратор:
1—кожух; 2—крршка; 3—колонка; 4—наг-
ревательные трубы; 5—кран для впуска и
выпуска кислоты; 6—брызгоуловитель; 7—
паровой эжектор; 8—конденсатор смешения;
9—барометрическая труба; 10—лаз; 11—
кран для выпуска пара; 12—термометры:
13— смотровое стекло.
Рис. 226. Брызгоуловитель:
1—внутренний керамиковый цилиндр, 2—
футеровка.
ринтный ход для паров, выходящих из концентратора. Эти ци-
линдры взяты в керамиковую футеровку (андезит, диабаз), сна-
ружи обтянутую листовым свинцом. Брызги серной кислоты,
увлекаемые парами воды, улавливаются в брызгоуловителе и
стекают обратно в концентратор.
442
Конденсатор противоточного типа (рис. 227) выпол-
няется из гартблея или из кислотоупорной меди. Внутри конден-
сатора имеются дырчатые плиты 3 для равномерного распреде-
ления воды; сверху конденсатор по-
крыт сферической крышкой, через
которую в центре проходит дырча-
тая труба 2 для подачи воды, в
низу конденсатрра — крышка с от-
верстием для выхода воды. К шту-
церу 4 присоединяется двухступен-
чатый паровой эжектор для отсоса
воздуха из аппарата.
Температура воды, подаваемой в
конденсатор, не должна быть вы-
ше 27°, так как при более высокой
температуре трудно получить высо-
кий вакуум (99'/). Температура от-
ходящей воды держится обычно не
выше 35—36'.
Вакуум- усилитель, б у с-
т е р, для отсоса водяных паров из
концентратора сделан из кислото;
упорного металла (ферросилиций) и
рассчитан на создание остаточного
абсолютного давления паров воды
в концентраторе в 7,5 мм рт. ст. Он
представляет собой (рис. 228) ком-
бинацию из двух конических труб,
соединенных» узкими концами в цен-
тре, и колена, в центре которого
вставлено сопло для подачи пара
в бустер.
Двухступенчатый паро-
вой эжектор с промежуточным
конденсатором (рис. 229) изготовлен
Рис. 227. Конденсатор:
1—корпус конденсатора; 2—распре-
делительная дырчатая труба; 3—рас-
пределительная плита для воды; 4—
штуцер для отсоса воздуха; 5—дыр-
чатые плиты.
из кислоупорного металла.
Пар подводится от коллектора к фильтру, пройдя который
он направляется в два диффузора 2 и 3 первой и второй паро-
Рис. 228. Паровой эжектор (бустер).
вой ступени эжектора. В промежуточный барометрический кон-
денсатор подается холодная вода для конденсации пара через
443
распылитель 4, вставленный в верхнюю крышку эжектора. Ме-
таллические ребра внутри конденсатора способствуют лучшей
отдаче тепла корпусом эжектора, что сокращает расход воды,
потребной для конденсации пара. Эжектор работает при давле-
нии пара в 7 ат.
При начальной
бота аппарата проходит в
Bioinapa
food ши
Bui Ыаалп-
саанСаемко ю
KCHuempamp^y
fyiiobCntoia
ВытОМы
Рис. 229. Двухступенчатый паро-
вой эжектор:
1—корпус; 2—первая ступень эжектора;
3—вторая ступень эжектора; 4—распы-
литель воды; 5—конусная часть конден-
сатора; 6—фильтр; 7—штуцер дчя воды.
концентрации кислоты в 68% H2SO4 ра-
две стадии: в первой — концентрация
кислоты с 68% H2SC>4 доводится
до 81 %, во второй — кислота упа-
ривается до купоросного масла
(93% Н.ДО4). Вначале пускают
воду в конденсатор и после со-~
здания в нем вакуума в ~ 75 мм
выдувают воздух из концентра-
тора паром из парового коллек-
тора. В это же время пускаются
пар и вода в двухступенчатый эже-
ктор, а затем пар в нагреватель-
ные трубы. До впуска кислоты в
аппарат в нем создается вакуум
до 685 мм рт. ст. После этого в
аппарат впускается 50 т кислоты,
температура которой ~ 75°, со
скоростью примерно в 1,5 м3/мин.
Если кислота холодная, ее надо
впускать очень медленно, чтобы
предотвратить внезапное охлаж-
дение и вследствие этого растре-
скивание труб. Первце 30 м3 кис-
лоты заливаются в течение 20 мин.
Последние 16,5 м3 кислоты впу-
скаются более медленно — при-
мерно в течение 1’/2 час. после на-
чала работы. Давление пара уве-
личивается постепенно, примерно
на 0,33 ат в минуту, до тех пор,
пока оно не достигнет 10 ат.
часов по мере упаривания кис-
постепенно возрастает до 724 мм
В течение первых четырех
лоты, вакуум в концентраторе
рт. ст. В это время происходит кипение кислоты (и ее упарка),
причем температура кипения постепенно повышается с НО до
127°. К концу этого периода абсолютное давление в концентра-
торе должно быть около 40 мм рт. ст. Кислота при этих усло-
виях достигает концентрации в 81% H2SO4. После того как до-
стигнутая концентрация кислоты проверена (взятием пробы), аб-
солютное давление в концентраторе снижают в течение часа до
20 мм рт. ст., а затем до 6—7 мм в течение следующих 6 час.
Это осуществляется пуском в ход парового инжектора при по-
степенном открывании парового вентиля, пока манометр не пока-
444
жет давления ;в 5,5 ат. Когда концентрация кислоты достигнет
86% H2SO4 (в это время кислота кипит при 130°), в концентра-
тор впускают через воздушные вентили воздух со скоростью
15 м^/час. Воздух усиливает циркуляцию кислоты вокруг нагре-
вающих труб. Когда температура кипения кислоты поднимается
до 149° (в это время давление в концентраторе составляет 7—
7,5 мм рт. ст.), концентрация кислоты достигает 93% H2SO4,
т. е. процесс концентрирования заканчивается.
В это время в змеевики холодильника пускают воду, а в меж-
змеевиковое пространство выпускают кислоту из концентратора
(примерно в течение 15 мин.). Концентратор может начать новый
цикл работы. Один цикл продолжается 12 час.
Установка описанного концентратора Симонсон-Мантиуса
рассчитана на получение 100 т кислоты в сутки (в пересчете
на моногидрат) при питании аппарата кислотой 68% H2SO4 и упа-
ривании ее до купоросного масла 93%. При этом температура
кислоты, поступающей на упаривание, достигает ~ 75°. При упа-
ривании 68%-ной кислоты той же температуры (75°) до купорос-
ного масла расходные коэфициенты на 1 т кислоты в моноги-
драте следующие:
Пар (насыщенный при давлении 8 am и температуре 170°). . 1150 кг
Вода (при температуре 18°)...................... 50 мл
Вода (при температуре 20э)......................56,2 „
Электроэнергия расходуется в очень незначительном коли-
честве лишь на перекачивание кислоты (~ 0,2 квт-ч).
Потери .кислоты при уносе ее парами воды в конденсатор
составляют по данным практики действующих за границей уста-
новок 0,5—2% от производительности аппарата, причем вся эта
кислота уходит из конденсатора, растворенная в большом коли-
честве воды.
IV. Дальнейшие задачи в области концентрирования
серной кислоты
Темпы роста химической промышленности, а в частности
большие масштабы производств, связанных с установками по кон-
центрированию серной кислоты, предъявляют требования на бо-
лее мощные аппараты-концентраторы, так как даже мощных ап-
паратов Кемико при некоторых производствах приходится ста-
вить много.
Неустраненным недостатком аппаратов (особенно установки
Кесслера и Кемико), работающих по методу непосредственного
обогрева кислоты, является недостаточно полное улавливание
брызг и тумана кислоты из хвостовых газов, что важно не
столько с точки зрения уменьшения потерь кислоты, сколько
с точки зрения санитарно-гигиенической и устранения коррозии
крыш зданий, водосточных труб и т. п. от конденсирующейся на
них серной кислоты. Улучшение очистки отходящих газов кон-
445
центрационных установок продолжает оставаться, таким обра-
зом, актуальнейшей задачей.
Установки Кемико работают на дефицитном привозном жид-
ком топливе. Разрешение задачи перевода этих установок на
твердое и газообразное топливо даст возможность (применять на
них местное топливо.
Для тех химических комбинатов, где имеется и цех концентрирования
азотной кислоты и цех башенной серной кислоты, представляет интерес сле-
дующая схема: 68%-иая H2SO4, выходящая из цеха концентрирования азот-
ной кислоты, направляется в башенную систему, где концентрация ее увели-.
чпвается примерно до 76% H2SO4 и уже с такой концентрацией она идет
в цех концентрирования серной кислоты, где упаривается до купоросного
масла. Таким путем значительно может быть интенсифицирована работа цеха
концентрирования серной кислоты.
Представляет интерес вопрос об использовании для концентрирования
серией кислоты абсолютно сухих хвостовых газов контактных сернокислотных
цехов.
Литература
]. Производство серной кислоты, Коллектив авторов под редакцией Малина
К. М., стр. 265-325, ГОНТИ, 1938.
2. Кирш А. И., С о с и о в с к и й Н. П-, Концентрирование серной кислоты,
Госхимиздат, 1940.
3. Перри ш, Снеллинг, Концентрирование серной кислоты, 1931.
4. Ш н е е р с о и Б. Л. и Егоров Н. Н., Электрическая очистка газов,
ОНТИ, 1933.
5. F a i г I i е, Sulfuric acid Manufacture, стр. 285— 329, New York, U.< S. A., 1936.
6. Waeser B., Handbuch der Schwefelsaurefabrikation, B. 2, Braunschweig 1930.
7. Турхан Э. Я-, Андреева E. А., Скорость концентрирования серной
кислоты, ЖХП № 6, 18 (1939).
ГЛАВА 7
ПРОИЗВОДСТВО ЭЛЕМЕНТАРНОЙ СЕРЫ
1. Извлечение серы из самородных серных руд
Главнейшими производителями элементарной серы из ее са-
мородных руд (и в то же время главнейшими производителями
элементарной серы вообще) являются США, Италия и Япония.
В 1936 г. было добыто серы в США 2048 тыс. т, в Италии —
350 тыс. т, в Японии 175 тыс. т. Максимальная добыча серы
из самородных серных руд во всем мире была достигнута
в 1931 г. и составляла 3070 тыс. т.
Элементарная сера получается еще из коксовых и других про-
мышленных газов, в которых сера находится в виде сероводо-
рода, а также из пиритных руд и отходящих газов металлурги-
ческих печей, содержащих сернистый газ. Неоднократно дела-
лись и продолжают делаться попытки получения серы и из
сульфатов.
В объеме мирового производства элементарной серы все дру-
гие источники, кроме самородных серных руд, занимают незна-
чительное место. Однако в отдельных странах сера произво-
дится исключительно из других источников. Так, в Германии
сера, производимая из сероводорода (коксовых и других газов),
покрывает 65—70% всей потребности страны в сере. Норвегия,
добывающая серу из пиритов, является экспортером серы.
В промышленности применяются следующие методы извлече-
ния серы из ее самородных руд: 1) термический — выплавка
серы за счет теплоты сгорания части ее самой, за счет теплоты
обогревающего инертного газа или пара и т. п.; 2) физико-терми-
ческий — выплавка серы из руд в воде под давлением или в вы-
сококипящих жидкостях и 3) извлечение серы из ее руд при
помощи растворителей (экстракцией). Кроме того, применяются
еще методы предварительного обогащения руд: механический,
электростатический и флотационный.
Метод механического обогащения основывается на том, что при дро-
блении сера размельчается легче, чем содержащая ее порода. В результате
этого при рассеивании раздробленной руды по фракциям содержание серы
в мелких фракциях получается значительно выше, чем в исходной руде.
Электростатический метод обогащения состоит в том, что раздробленная
руда проходит в электростатическом поле между двумя проводниками (пло-
ским заземленным и цилиндрическим положительным). При этом частицы
серы и породы, имеющие разные проводимости, отталкиваются с различной
447
-силой и падают па различных расстояниях от оси цилиндрического провод-
ника (иа горизонтальном проводнике).
На ряде серных заводов СССР серная руда перед плавкой
в автоклавах подвергается флотационному обогащению.
1. Термический метод
Термический метод извлечения серы из ее самородных руд
в промышленности осуществлен в виде калькарон, печей Жилля
и паровой выплавки в тоннельных печах.
Почти исключительно этим методом добывается вся сера,
производимая Италией. Термический метод (калькарона, печи
Рис. 230. Калькарона.
Жилля) получил также
применение в СССР.
В калькаронах и пе-
чах Жилля нагревание
всей руды и расплавле-
ние серы происходят
за счет сгорания части
самой серы.
Калькароны. Калька-
рона (рис. 230)предста-
вляет собой открытую
сверху камеру цилинд-
рической формы. В за-
висимости от удобства
расположения калька-
роны ставятся на усту-
пах скал (причем зад-
няя стенка прилегает к скале), или углубляются в землю, или
выкладываются на специальных, поднятых над уровнем мест-
ности площадках. Задняя стенка делается вровень с подом или
несколько выше него. Отверстие для выпуска расплавленной
серы, расположенное в низу передней стенки, заделывают тон-
кой перегородкой из гипса, оставляя в перегородке маленькие
отверстия, через которые можно видеть нижнюю часть внутрен-
ности калькароны. При кладке перегородки камни связываются
гипсовым тестом. Загрузка калькароны производится вручную
или механическим путем.
На под калькароны сначала кладут самые большие куски
руды, оставляя между ними промежутки. Второй ряд составляют
менее крупные куски руды, и дальше при кладке оставляют во
всей массе руды проходы по всем направлениям. Вертикальные
ходы для тяги обкладывают более крупными кусками руды. Чем
ближе к вершине калькароны, тем куски руды должны быть
мельче. В результате над стенками калькароны образуется конус
из руды со скатами около 30°. Сверху калькарона покрывается
выжженной мелочью от предыдущих плавок и промазывается
гипсом, глиной и т. д.
448
Воздух поступает в массу через нижнюю часть покровного
слоя, а отходящие газы, содержащие SO2 и пары серы, выходят
через верхнюю часть этого слоя. Жидкая сера стекает вниз, по-
переменно затвердевая и снова расплавляясь по мере распро-
странения огня, и доходит до наклонного пода, по которому те-
чет к выпускному отверстию.
Благодаря медленности горения стекающая сера почти совер-
шенно не подвергается действию огня. Из серы же, впиты-
ваемой пустой породой, часть сгорает, а часть отгоняется. Пары
серы частично уносятся газами и, соприкасаясь по выходе на-
ружу с воздухом, горят. Другая часть паров диффундирует
в массу руды и конденсируется в ее низших зонах.
До известной степени работа калькароны может регулиро-
ваться толщиной покровного слоя. Оптимальный температурный
режим обжига серы в калькаронах, как и в нижеописываемых
печах Жилля, определяется рассмотренными уже в первой главе
физическими свойствами серы: сера плавится при 112—114°, при
214 в присутствии воздуха горит с образованием SO2; жид-
кая сера при нагревании до 160° густеет и темнеет, при 220°
принимает красно-бурую окраску и становится очень вязкой.
При 300° она снова становится подвижной. Отсюда следует, что
отделять жидкую серу от породы надо при возможно низкой
температуре. При этом надо учесть, что отделение никогда не
бывает полным, так как часть серы смачивает пустую породу и
удерживается ею в силу явлений капиллярности. Практикой
установлено, что эта часть серы составляет 7—22% от веса пу-
стой породы, в среднем 13%. Таким образом руда, содержащая
24% серы (76% пустой породы), удерживает (по отношению
к весу руды) в среднем 9,88% серы, которую можно выделять
лишь дестилляциёй. Однако и отгонкой не удается выделить
всю серу и уменьшения ее потери ниже 5% от веса руды трудно
добиться.
Способ выплавки серы в калькаронах связан с очень большим
расходом серы: сера затрачивается не только на получение
тепла, необходимого для выплавки остальной серы, но большое
количество тепла теряется также с отходящими горячими газами
и выгружаемым горячим огарком.
Печи Жилля. Печи Жилля выгодно отличаются от калькарон
тем, что используют значительно большую часть тепла сго-
рающей части серы. Предложенная в 1881 г. печь Жилля со-
стояла из двух камер. Воздух, идущий на горение в камеру вы-
плавки серы, подогревался теплом горячих огарков от предыду-
щей плавки, находящихся в другой камере. В дальнейшем печь
Жилля непрерывно совершенствовалась путем введения допол-
нительных камер. В настоящее время применяются четы
рех-, пяти- и шестикамерные печи. В многокамерных печах ис-
пользуется тепло и горячих огарков и отходящих горячих газов.
Двухкамерная печь Жилля (рис. 231) составляется из двух
отделений 7 в каменной кладке. Под камеры наклонен к перед-
29 Зак. 5019. Технология серной кислоты.
449
ней части и построен из обожженной рудной мелочи или какого-
либо другого материала, например из кирпича, и сверху обмазан
гипсовым тестом. Отверстие для выпуска серы делается в виде
арки 1—2 №. высотой и 0,1 м в диаметре; арка после нагрузки
отделения закладывается кирпичом или гипсовым камнем и шту-
катурится. Вверху оба отделения сообщаются горизонтальной
трубой 2 с квадратным сечением, на половине которой имеется
шибер для разобщения камер. Отверстия 4 в низу каждой камеры
сообщаются каждое со своей дымовой трубой. Дымовые трубы
с квадратным сечением возвышаются над печью на 1—Р/г м.
Газоходы, ведущие к трубам, имеют окна, которые во время
работы данного отделения замуровываются. Эти окна необхо-
димы для очистки газоходов, так как наблюдается довольно ча-
стое засорение боровов серным цветом.
Разрез по А-В
Рис. 231. Двухкамерная печь Жилля:
/—камеры, 2—горизонтальная труба, 5—отверстие в своде печи, 4— отвер-
стие в низу^камеры.
Загрузка начинается через нижние отверстия, а заканчи-
вается через верхние отверстия 3 в своде камеры. При загрузке
руды в камеру у ее основания помещают более крупные куски,.
250—300 мм в поперечнике, а затем более мелкие. В эксплоати-
руемых у нас печах руда сортируется по крупности кусков на
следующие классы:
Низ печи..............выше 150 мм
Середина печи........ 150—60 „
Верх печи............60—10 »
Более мелкие куски можно загружать в печи после предва-
рительного брикетирования с неорганическими связующими ма-
териалами (гипс, известь и др.).
У нижнего отверстия ставят железные решетки или делают
арку из крупных кусков руды (последние употребительнее).
Кладку руды стараются вести так, чтобы к нижнему отверстию’
шли как бы трубы, образованные рудой.
При нормальной работе двухкамерной печи одна пусковая
камера наполнена теплыми осколками выплавленной и выжжен-
ной руды от предшествующей плавки, а другая загружена об-
рабатываемой рудой. Когда в этой последней камере процесс
выплавки заканчивается, вторая камера посредством шибера
изолируется от первой. Первая камера освобождается от находя-
щихся в ней остатков от выплавки и загружается свежей ру-
450
дой, после чего шибер открывается и процесс начинается вновь
только в обратном порядке.
При пуске двухкамерной печи роль пусковой камеры выпол-
няет одна из камер, загруженная наполовину рудой, которую
разжигают сверху и которая также дает жидкую серу. Таким
образом при первоначальном пуске выплавка идет одновременно
в обеих камерах.
Воздух, потребный для горения, входит через нижнее от-
верстие в камеру с раскаленными от предшествовавшей плавки
огарками, нагревается, протекая между ними, и через верхнюю
горизонтальную трубу входит в камеру, наполненную свежей
рудой, пронизывает ее и через отверстие у основания камеры
уходит в дымовую трубу вместе с выделяющимся сернистым
газом. Описанная циркуляция газов происходит исключительно
благодаря тяге через дымовую трубу.
В начале выплавки теплый воздух высушивает руду; когда
руда высыхает, сера воспламеняется сама собой (температура
горячего воздуха, поступающего в камеру плавления, около
200°) сначала в верхней части камеры; затем огонь медленно
распространяется сверху вниз и начинается выплавка серы.
Печь регулируется притоком воздуха.
В начале операции, когда руда холодна и сыра, дают интен-
сивный воздушный поток, когда же начинается выплавка, газо-
вый поток умеряют и потом процесс ведут таким образом,
чтобы температура вытекающей серы была не больше 150°.
За работой печи следят по консистенции серы, вытекающей
из выпускного отверстия. Если температура серы близка к точке
плавления, она легко затвердевает даже внутри выпускного ка-
нала. Это называется «холодной плавкой» и в этом случае не-
обходимо усилить циркуляцию воздуха, для чего расширяют от-
верстие, через которое поступает воздух. Если температура вы-
сока, то сера становится вязкой и темнеет. В этом случае (слу-
чае «горячей плавки») приходится ослаблять поток воздуха
уменьшением отверстия для его впуска.
По окончании плавки закрывают шиберами газоход между ка-
мерами, а также и дымоход и приступают к разгрузке камеры,
бывшей пусковой, чтобы загрузить ее свежей рудой. После за-
грузки и приведения в порядок этой камеры открывают шибер
в газоходе между камерами и пускают в ход парную печь, но
в противоположном направлении. Продолжительность отдельных
операций следующая;
Разгрузка огарка и загрузка руды........10— 15 час.
Нагревание (подготовка к плавке)........ 25— 30 „
Плавка.................................. 25— 50 „
Ожидание перед загрузкой другой камеры 10— 15 „
Охлаждение камеры, работавшей в каче-
стве пусковой........................ 50—100 »
Всего. . 120—210 час.
В среднем двухкамерная печь дает ~ 100 плавок в год.
29*
451
В печах Жилля с двумя камерами на ожидание наполнения
рудой одной камеры, когда еще идет плавка в другой, и на
охлаждение разгружаемой камеры теряется более 50% времени
всего цикла процессов в печи и, кроме того, теряется много
тепла. В четырехкамерной печи используется не только теплота
камеры, содержащей раскален-
ные остатки предшествовавшей
выплавки, но и теплота газов,
идущих из камеры плавления.
Кроме того, улавливается зна-
чительная часть серы, выходя-
щей в парообразном состоянии
f из камеры плавления. Дости-
гается это тем, что газы, вы-
ходящие из камеры плавления,
D проходят через камеру, загру-
женную свежей рудой. Таким
образом в цикле процессов
оборота печи одна камера раз-
гружается или нагружается,
Рис. 232. Горизонтальный разрез че- вторая С раскаленными огар-
тырехкамерной печи Жилля. ками от предыдущей плавки
служит для нагрева воздуха,
идущего в камеру плавления; в третьей камере идет процесс
плавления, а в четвертой — предварительный перед плавлением
прогрев. Таким образом все четыре камеры находятся в дей-
ствии одновременно.
а б
Рис. 233. Вертикальный разрез печи Жилля (по каналам и по камерам):
а — разрез по камерам (СО), б — разрез по каналам (ЕР).
Четырехкамерные печи разжигаются различно в зависимо-
сти от их конструкции. Иногда около вертикальных ходов
устраивают небольшие подтопки. В этом случае через нагревание
подтопки зажигается руда верхней части данной камеры, дальше
газы перебрасываются во вторую камеру, и таким образом начи-
нается операция. При отсутствии специальных подтопок розжиг
452
производится так: одна камера нагружается наполовину и под-
жигается, дальше продукты горения идут уже в другую камеру,
нагруженную нацело, и таким образом печь начинает действо-
вать. Первый цикл печь делает лишь на двух камерах, как
в двухкамерной печи, и только когда печь разгорается, вклю-
чается третья и потом четвертая камера.
На рис. 232 изображен горизонтальный разрез четырехкамер-
ной печи, на рис. 233 — вертикальный разрез по каналам и ка-
мерам.
Вертикальные каналы устраиваются вблизи выпускных отвер-
стий таким образом, чтобы в них можно было проникнуть для
чистки. На рис. 232 каналы для теплого воздуха обозначены
цифрами 7, каналы для отходящих газов цифрами 3, а цифрами
2 — каналы, непосредственно соединяющие камеры. Воздух вхо-
дит через отверстие 4 (рис. 234) в низ камеры, загруженной го-
рячими огарками, и пронизывает последние. Через верхние отвер-
стия 7—7 образовавшиеся газы выходят в канал и далее
направляются в камеру плавления, пересекают ее сверху вниз,
выходят в канал 5, по которому поднимаются и входят (сверху)
в камеру нагревания. Из камеры нагревания газы выходят
через нижнее отверстие б в каналы дымовых газов 3—3 и дальше
в дымовую трубу. Как и в двухкамерной печи, во время нагре-
вания и просушки руды дается сильная тяга, а во время плавле-
ния — слабая. Регулирование производится, главным образом,
через отверстия для подвода воздуха.
Едва прекращается процесс плавления, как камера плавления
должна превратиться в нагревательную. В этот момент печь
имеет в серии только две камеры — нагрева газа и прогрева
руды. Камера нагрева газа продолжает оставаться включенной
через газоход, пока в камере нагрева руды не начинается пла-
вление, тогда включается новая камера нагрева газа, бывшая
перед этим камерой плавления. Вследствие постепенного охла-
ждения уходящих газов при большом сопротивлении кругового
потока происходят значительные изменения в скорости газов,
а потому в это время нужно особенно внимательно наблюдать за
тягой.
453
Продолжительность каждого цикла для одной камеры
четырехкамерной печи примерно следующая:
Разгрузка огарка и загрузка руды........10— 15 час.
Очередное ожидание........................ 30— 65 ,
Прогрев................................... 50— 90 ,
Плавка.................................... 30— 70 „
Работа камеры в качестве пусковой .... 40— 80 „
Всего. . 160—320 час.
В среднем в год проводится ~ 150 плавок.
Рис. 235. Ход воздуха и газов
в шестикамерной печи Жнлля:
7—камеры; 2—газоходы; 3—загру-
зочная воронка; 4—рукоятки шибе-
ров; 5—выгрузочные отверстия; 6—
отверстия для выпуска серы; 7—фор-
мы для серы.
Последовательно улучшая работу
печи Жилля, стали строить пятика-
мерные и шестикамерные печи
(рис. 235).
В шестикамерной печи (рис. 235)
в каждый момент одна камера яв-
ляется пусковой, в двух камерах
идет процесс плавки, в двух — про-
грев руды и одна камера находится
в процессе загрузки и разгрузки.
В шестикамерной печи продолжи-
тельность отдельных процессов в
каждой камере примерно следующая:
Разгрузка и загрузка ....
Очередное ожидание н дру-
гие потери времени . . .
Прогрев руды..............
Плавка ...................
Работа в качестве пусковой
камеры...............• . .
15— 25 час.
5-25 „
10— 50 „
50— 90 „
25—50 .
Всего. . 105—240 час.
В среднем в год производится
по 250 плавок.
При содержании в руде 24%
серы количество воздуха, подавае-
мое в печь на 1 т руды (по данным
М. Гатто), следующее:
Для четырехкамерной печи .... 580 м3
Для пятикамерной , .... 370 „
Для шестикамерной , . ... 312 „
Ниже приводим выхода серы и
потери ее в % от веса руды для раз-
личных печей (для руды среднего
качества, содержащей 24% серы) по
данным М. Гатто и на основе итальянской практики:
451
Выход в % Потери в %
Калькарона............... 15 9
Двух- и трехкамерные печи. 16 8
Четырехкамерная печь.... 17 7
Шестикамерная печь.......... 19 5
Потери тепла на 1 т руды (с учетом потерь на излучение,
с огарками и отходящими газами и т. п.) составляют
100—108 тыс. ккал. При теплотворной способности серы, рав-
ной 2400 ккал.,/кг, теоретический расход серы составляет. таким
образом 41—45 кг, или 4,1—4,5$ от веса руды.
Плавка серы из руд как в калькаронах, так и в печах Жилля
предъявляет особые требования к руде, а именно:
1) руда должна быть достаточно плотной, чтобы обеспечить
сохранение минерального костяка после выплавки (песчанистая,
рассыпающаяся руда для выплавки в калькаронах и печах Жилля
не годится);
2) руда должна быть сухая, иначе будут большие потери
тепла на испарение влаги;
3) гипсовые руды, содержащие кристаллизационную воду,
не годятся и могут лишь примешиваться в небольшом количестве
к мергелитовым и известковым рудам (20% от шихты).
При управлении технологическим режимом печей Жилля ру-
ководствуются следующим: 1) температура массы руды по ходу
вниз должна снижаться, чтобы расплавляемая сера, стекая вниз,
становилась более жидкой и текучей; кроме того, более низкая
температура дает возможность конденсироваться парам серы,
идущим в газовом потоке; 2) горение серы должно быть очень
медленное, чтобы: а) дать возможность стечь жидкой сере;
б) способствовать испарению большей части серы, первоначально
впитавшейся в руду; с) • поддерживать температуру ниже 214°
с целью воспрепятствовать воспламенению большого количества
серы.
Значительная часть серы руды уходит из печи Жилля с га-
зами, главным образом в виде SOs, который отравляет воздух
и губит растительность. В Сицилии обезвреживают выпускае-
мые газы: а) предварительным разбавлением газов воздухом;
б) поглощением SOs твердыми адсорбентами в специальных ка-
мерах; в) поглощением SOa водой в скрубберах. Есть сведения,
что в Сицилии на отходящих газах одной установки печей
Жилля работает камерный сернокислотный завод. Национальный
конгресс чистой и прикладной химии, собиравшийся в Палермо
в 1926 г., дал заключение, что лучше всего утилизировать сер-
нистый ангидрид из печей Жилля на контактных сернокислот-
ных установках.
Паровая выплавка серы. Для богатых серой серных руд, осо-
бенно руд гипсовых, применяется выплавка перегретым паром.
Установка для паровой выплавки серы из руды (рис. 236) со-
стоит из железного или чугунного цилиндрического котла, поме-
щающегося слегка наклонно (10—12 ). Руда загружается в ва-
455
гонетки, которые вставляются в котел. Форма вагонетки макси-
мально подгоняется под форму котла, с. тем чтобы избежать
мертвого пространства. На дне вагонетки имеются отверстия
для стока серы. Очень мелкая руда для выплавки этим методом
не годится. Обычно в котел помещается четыре вагонетки
с 1000 кг руды в каждой. После ввода вагонеток в котел по-
дают пар при температуре ~ 140°. Сера собирается у низ-
кого конца котла и выпускается из него через спускное отвер-
стие. В одном котле проводится 16 плавок в сутки. При вы-
плавке паром в остатке содержание серы (13—15%) значительно
Рис. 236. Паровая выплавка серы.
выше, чем в огарке из печей Жилля. Понятно, что для паровой
плавки могут применяться только богатые серой руды. Хвосты
паровой плавки часто передаются на доработку в печи Жилля.
Паровая выплавка требует значительно больших эксплоата-
ционных затрат, чем выплавка в печах Жилля и калькаронах.
Однако капитальные затраты на сооружение установок паровой
'плавки значительно ниже капитальных затрат на сооружение
печей Жилля.
Некоторые сравнительные показатели выплавки серы различ-
ными способами по итальянским данным приведены в табл. 98.
Таблица 98
Показатели Выплавка паром Выплавка в калькаро- нах Выплавка в шестика- мерных печах
Содержание серы в руде, °/0 . . . Извлечено, % к руде Потери, % к руде Выход серы, % 23,14 12,00 11,14 52,00 23,14 13,20 9,94 57,00 23,14 17,16 5,98 77,40
Основные показатели работы калькарон и печи Жилля в Чан-
гырташе за 1938 г. приведены в табл. 99.
456
Таблица 99
Показатели Калькарона Печь Жилля
Извлечение серы, % Сгорело серы на 1 т руды, кг Содержание серы в огарке, °/0 Содержание серы в продукте, % 57,3 130 1,01—3,27 97,63- 68,0 85 0,14—2,98 -99,38
В отношении других способов термической выплавки серы де-
лались и делаются попытки нагрева руды инертными (топочными)
газами, а также нагрева через стенку. Однако эти способы пока
промышленного применения не получили. Много делается попы-
ток осуществить непрерывный процесс термического извлечения
серы из руд (вращающиеся печи и др.). Эти способы также
не получили еще прочного промышленного применения.
2. Физико-термический метод
Способ Фраша. Способ Фраша, состоящий
перегретой водой на месте залегания серной
в свое время переворот в серной промышлен-
ности. Этим способом работает почти вся сер-
ная промышленность США (около 2/з всей ми-
ровой продукции серы).
Способ Фраша, однако, применим не ко
всякому месторождению серы. Для того чтобы
применить способ Фраша, необходимы сле-
дующие условия: 1) достаточная глубина за-
легания серных пластов, 2) достаточная плот-
ность покрывающих пород, 3) руда должна
быть богатой по содержанию серы и 4), так
как при применении способа Фраша расхо-
дуется большое количество воды и топлива,
то вблизи месторождения должны быть доста-
точные запасы того и другого.
Из всех известных сейчас месторождений
в выплавке серы
руды, произвел
самородных серных руд указанным условиям
удовлетворяют только мощные месторождения Рис. 237. Способ
США. Сущность способа Фраша понятна из Фраша.
рис. 237. В скважину, доходящую до основа-
ния сероносного пласта, вставлены одна в другую четыре кон-
центрические трубы. Внешняя труба 4 проходит только через
крышу серного пласта 8 и служит термоизолятором. Через
трубу 7 в кольцевое пространство между трубами 3 и 2 по-
дается перегретая вода, по трубе 5 и 1 подается сжатый воздух.
Перегретая до 150—160° вода выходит в сероносный пористый
известковый пласт 10 и расплавляет серу. Расплавленная сера
457
собирается в низу скважины и входит через отверстия в конец
трубы, изолированный от отверстий, через которые накачивается
вода. Давлением сжатого воздуха, входящего по трубе 5—7,
сера выдавливается на поверхность земли по кольцевому про-
странству между трубами 2 и 1 и по трубе б.
Жидкая сера, выходящая из скважины, поступает в большие
деревянные резервуары емкостью до 25 000 т серы, где она осе-
дает на дно и затвердевает. По застывании серы формы разби-
раются, а образующуюся глыбу серы разбивают с помощью
сверл или взрывают и собирают куски, удобные для погрузки.
Для приема серы применяют также конвейерные стальные ленты,
на которых сера застывает в виде чешуи. Сера принимается
Рис. 238. Автоклав
для выплавки серы:
7—крь!шка; 2—труба
для впуска пара и во-
ды; 3—патрубок для
выпуска серы.
также и в разборные железные резервуары.
Для правильного ведения процесса надо
располагать громадными количествами воды.
Возвращаемую частично на поверхность воду
можно употреблять вновь после декантации,
но большая часть воды теряется в пластах.
В среднем расход воды составляет 9 /и3 на
1 т серы, расход топлива (нефти на 1 т серы)
330 ДЛ
По качеству сера, выработанная по Фрашу,
получается исключительно чистой: 99,8—99,9%.
Способ Фраша экономически дает самую де-
шевую серу, но технологически рациональным
его назвать нельзя, так как он позволяет из-
влечь из месторождения не больше 259< имею-
щейся там серы.
Выплавка серы в автоклавах. На ряде сер-
ных заводов СССР плавка в перегретой воде
измельченной руды производится в автокла-
вах. Автоклав (рис. 238) выполняется из ко-
тельного железа, он выдерживает давление до
6 ат. Через крышку автоклава 7 загружается
руда; через эту же крышку подается вода
в таком количестве, чтобы она смочила руду (приблизительно
15% от веса руды). Люк герметически закрывается и в авто-
клав пускается через вентиль трубы 2 острый пар, прогревающий
всю массу до 130—140е. Выплавляющаяся сера собирается
в нижней части автоклава, заполняя пустоты между кусками
руды. По окончании стекания на дно автоклава жидкая сера вы-
пускается под давлением через патрубок 3 в нижней части авто-
клава, пар выпускается в атмосферу, давление снижается до
атмосферного, и через люк в низу автоклава выбрасывается от-
работанная порода, смешанная с водой.
Руда перед загрузкой в автоклавы подвергается дроблению
до кусков размером 5—80 мп- Общая продолжительность одного
цикла 3—4 часа, причем прогрев руды одного автоклава зани-
мает приблизительно 1 час, а собственно плавка 20 мин.
458
Выход серы из руд колеблется в зависимости от начального
содержания серы в руде приблизительно так:
Начальное содержание
серы в руде
в °/0
40-60
30—40
15—20
Выход серы
из руды
в %
60-70
50-60
30—50
В СССР автоклавный метод применяется также в сочетании
с предварительным флотационным обогащением серной руды.
На плавку в этом случае поступает концентрат с содержанием
серы 60—80%.
Извлечение серы при этом составляет 65—75%, а при введе-
нии некоторых добавок в шихту может быть повышено
до 85—90%.
В воде выплавлять серу из руд можно только с применением
повышенного давления; в жидкостях, кипящих выше точки
плавления серы, можно проводить выплавку и без применения
повышенного' давления.
В Италии ряд лет работало предприятие, которое для вы-
плавки серы применяло концентрированный раствор хлористого
кальция. Предприятие было закрыто по причине больших по-
терь хлористого кальция, что привело к нерентабельности спо-
соба.
В СССР имеется установка, работающая по этому методу на
богатой рудной мелочи. Извлечение серы составляет 70—73%.
3. Экстракция серы растворителями
Одним из методов извлечения серы из самородных серных
руд, в направлении разработки которого не прекращаются ра-
боты, является экстракция серы растворителями.
Изучению в этом отношении подвергнуты растворители: серо-
углерод, керосин, соляровые масла, дихлорэтан, хлористый наф-
талин, сернистый аммоний и др.
Принцип технологической схемы извлечения серы из руд тем
или иным растворителем состоит в следующем: после обработки
руды растворитель, несущий в себе растворенную серу, напра-
вляется на дестилляцию, где испаряется, а сера выделяется
в жидком виде или в виде крйсталлов. Пары растворителя за-
тем конденсируются и опять возвращаются в процесс.
Несмотря на кажущуюся простоту технологической схемы,
этот метод не получил значительного промышленного приме-
нения.
Широкому внедрению методов извлечения серы из руд
экстракцией препятствуют: легкая воспламеняемость (а следо-
вательно и огнеопасность) ряда растворителей, ядовитость и
большие потери их, приводящие к малой рентабельности про-
459
цесса. Однако все вышеуказанные обстоятельства не закрывают
возможности разработки более оптимального процесса по этому
методу в дальнейшем.
4. Размол комовой серы
Комовая сера после дробилки Блека элеватором подается
для тонкого размола в мельницу Раймонда. Размолотая сера
увлекается из мельницы инертным газом, из которого она выде-
ляется в циклон-аппарате, а оттуда собирается в бункере и да-
лее засыпается в бумажные пакеты весом по 40 кг. Каждые два
пакета упаковываются в мешок и в этом виде отправляются
потребителям. Продувание мельницы инертным газом необхо-
димо потому, что смесь серы с воздухом взрыво- и огнеопасна.
Инертный газ получается в специальной топке при сжигании
мазута. Получаемые обжиговые газы подвергаются охлаждению
и очистке в двух скрубберах, насаженных коксом и орошаемых
водой.
II. Получение серы из сероводородсодержащих газов
1. Метод сухой очистки
Сера может быть получена из газоочистительной массы (при
очистке сероводородсодержащих газов) одним из тех методов,
которые применяются для извлечения серы из самородных руд.
Обычно это метод экстракции тем или иным растворителем.
Наиболее подходящими растворителями для этой цели являются
сероуглерод и сернистый аммоний. Недостаток извлечения серы
из газоочистительной массы заключается в том, что вместе с се-
рой переходят в раствор и смолистые вещества, абсорбирован-
ные из газа газоочистительной массой. Эти вещества загрязняют
получаемую серу.
Самый способ очистки газов от сероводорода при помощи
газоочистительной массы не может полностью удовлетворить
промышленность, так как: 1) такого рода установки, при большом
количестве подлежащего очистке газа, очень громоздки, тре-
буют больших капитальных затрат и 2) сера переходит в сравни-
тельно малоценный продукт — газоочистительную массу.
Было предложено многочисленное количество способов
очистки газов от сероводорода, из которых целый ряд получил
промышленное применение.
Все способы очистки газов от сероводорода можно разде-
лить на две основные группы: 1) очистка при помощи адсорбен-
тов или, как принято называть, сухая очистка; 2) очистка при
помощи абсорбционных растворов или, как принято называть,
мокрая очистка.
При этом по некоторым способам H2S подвергается окисле-
нию в газовой фазе, а газ затем очищается уже от продуктов,
окисления.
460
Рис. 239. Схема очистки газа активирован-
ным углем:
7 и 2—фильтры; 3—баки; 4—сосуд; 5—кипятиль-
ник; 6—центрофуга; 7—конденсатор; 8—бак; 9 и
10— насосы.
Очистка активированным углем. Кроме очистки болотной ру-
дой из способов сухой очистки применяется очистка активиро-
ванным углем. Этот способ широко применяется в Германии
фирмой И. Г. (в Оппау очищается водяной газ, в Леуна — гене-
раторный газ).
Хороший активированный уголь на 1 т3 (400 кг) адсорбирует
400—500 кг серы. Наиболее пригодным для этой цели является
уголь, активированный с помощью ZnCL. Окисление H2S до серы
на активированном угле
можно проводить как при
комнатной, так и при по-
вышенной температуре.
При температуре 120—
170 сера выделяется в
жидком виде и часть се-
ры переходит в газы в ви-
де паров. При обыкновен-
ной температуре сера от-
лагается на катализаторе в
твердом виде и в дальней-
шем извлекается из него
методом экстрагирования
растворителем—чаще все-
го сернистым аммонием.
Есть указания на то,
что окисление H2S на ак-
тивированном угле воз-
можно проводить посред-
ством SO2 по реакции
2H2S-j- SO2 = 2Н2О -f- 3S.
Коксовый газ, смешанный с 3—4% воздуха и с примесью
небольшого количества аммиака 0,3 г NH3 на 1 м3 газа), про-
пускается (рис. 239) через фильтр 7, наполненный активирован-
ным углем. Здесь сероводород окисляется до серы. После того
как уголь фильтра 7 насытится серой, ток газа переключается
в фильтр 2- В это время уголь фильтра 7 обрабатывается раство-
ром (N144)28, который растворяет серу, выделившуюся на угле,
образуя многосернистый аммоний.
Фильтр последовательно промывается растворами: сначала из
бака За, затем из бака ЗЬ, Зс и, наконец, из бака 3d. Фильтр 7
после промывки выпаривается, пары поступают в конденсатор 7,
откуда конденсат стекает в бак 8 для чистого (NPta^S. Из бака 8
он по мере надобности насосом 9 перекачивается в бак 3d-
Раствор, насыщенный многосернистым аммонием, из бака За
насосом 10 перекачивается в сосуд 4, откуда стекает в кипя-
тильник 5- Здесь многосернистый аммоний разлагается. При
этом сернистый аммоний (вернее NH3 и H2S) переходит в пары,
а сера выпадает. Пары сернистого аммония поступают в кон-
461
денсатор 7, где конденсируются. Выпавшая из раствора в ки-
пятильнике 5 сера образует вместе с оставшейся частью раствора
так называемую серную кашу. Эта серная каша направляется
далее в центрофугу 6, где сера отделяется от маточного рас-
твора. Сера получается в виде зернистого продукта с влажно-
стью I—2%. Если хотят получать комовую серу, то кипячение
раствора полисульфида проводится при давлении 2,5—3 ат при
температуре выше точки плавления серы. В этом случае осво-
бодившаяся в кипятильнике сера осаждается в расплавленном
состоянии и стекает в сборник. Маточный раствор тоже посту-
пает в конденсатор 7. Здесь он служит абсорбционной жидко-
стью для паров сернистого аммония, поступающих из кипятиль-
ника 5 и из фильтра при его выпаривании. Из конденсатора 7
сернистый аммоний обратно передается в производство. Активи-
рованный уголь, загружаемый в фильтры, является одновре-
менно и катализатором для окисления H2S и адсорбентом для
получающейся при окислении серы. Насыщенный раствор сер-
нистого аммония, передаваемый на выделение серы в кипятиль-
ник, содержит 200—300 г серы на 1 л раствора. Получаемая по
этому методу сера представляет довольно чистый продукт
(99% серы).
К недостаткам метода относятся: большие потери активиро-
ванного угля при его регенерации (5—15%) и некомпактность
установки (занимает большую площадь).
2. Методы мокрой очистки
Способы очистки посредством абсорбционных растворов осо-
бенно многочисленны. Однако все эти способы можно разде-
лить на две группы:
1. Нейтрализационный метод, где сероводород обрабаты-
вается как кислота:
H2S + 2ОН' X S" + 2Н2О
2H2S+ ОН' 72 2HS' 2Н2О
2. Окислительный метод:
H2S */2О2 — Н2О 4- S 4- 53 ккал
H2S 4- 2О2 = H2SO4 4-169 ккал
По первой группе в результате получается концентрирован-
ный сероводородный газ. Процесс протекает таким образом,
что при низкой температуре происходит абсорбция сероводо-
рода, а при повышении температуры — выделение сероводо-
рода. Конечный продукт по окислительному методу получается
или в виде элементарной серы или в виде серной кислоты. В по-
следнем случае, связывая серную кислоту аммиаком, получают
сульфат аммония. Необходимый для окисления кислород вво-
дится с воздухом.
462
Нейтрализационный метод. При этом методе применяются
аммиачный раствор, известковое молоко, карбонат калия, фос-
форнокислые соли калия и натрия, моно- и триэтаноламины, соли
слабых органических кислот и сильных оснований, феноляты ка-
лия и натрия’ и некоторые другие. Наиболее простым, на пер-
вый взгляд, кажется поглощение H2S аммиачным раствором.
Особенно он представляется целесообразным по отношению
к коксовому газу, содержащему наряду с H2S и аммиак. Однако
зтот способ имеет ряд крупных недостатков: 1) сероводород хо-
рошо поглощается только концентрированным раствором ам-
миака, для получения которого требуется специальная концентра-
ционная установка; 2) аммиак поглощает не только H2S, но и все
другие кислые компоненты газа (HCN, СО2 и др.), что приводит
к большому расходу поглотителя; 3) приходится улавливать
аммиак (серной кислотой) как после поглощения H2S (из отходя-
щих из поглотительной колонки газов), так и после отгонки H2S
из раствора.
При применении карбоната калия процесс абсорбции проте-
кает по реакциям
К2СО3 ф- H2S -> KHS -f-КНСО3
и
К2СО3 + СО2 4- Н2О 2КНСО8
Для регенерации H2S через раствор пропускают СО2. При
этом происходит реакция
КНСОз 4- KHS 4- СО2 4- Н2О 2КНСО3 + H2S
Вместе с H2S уходит и значительное количество СО2. В газах,
отходящих из регенератора, содержание H2S составляет 40—50%,
а СО2 50—60%.
Поглощение раствором фосфорнокислого калия основано на
обратимости реакции
К3РО4 4- H2S К2НРО4 4- KHS
Применяется 50%-ный раствор К3РО4. Отличительной осо-
бенностью этого раствора является то, что при разбавлении его
водой резко падает упругость H2S над ним. Поэтому поглоти-
тель вводят в абсорбер в двух точках: крепкий регенерирован-
ный раствор в середину колонны, а разбавленный для поглоще-
ния остатка H2S — сверху.
Известное применение нашли амины в способе Гирботол (Gir-
botol) в США. Применяемый в качестве поглотителя триэтанол-
амин К(СН2СН2ОН)з смешивается во всех отношениях с водой.
Наилучшая поглощающая смесь содержит 50% триэтаноламина.
В Германии разрабатываются способы, основанные на приме-
нении солей слабых органических кислот. При абсорбции проис-
ходят реакции
2RCOONa 4- H2S = 2RCOOH ф- Na2S
RCOONa 4- H2S = RCOOH 4- NaHS
463
При нагревании . слабая органическая кислота, взаимодействуя
с сульфидом и гидросульфидом, образует опять соответствую-
щие соли и сероводород. При применении фенолятов калия или
натрия процесс основывается на обратимой реакции
NaO С6Нб + H2S NaHS Н- С6Н5ОН
В стадии абсорбции реакция идет слева направо. В процессе
регенерации, при нагревании раствора до кипения, реакция идет
в обратном направлении. Фенол в этом процессе играет балан-
сирующую и регулирующую роль: без него легко идет только
прямая реакция, регенерация же затрудняется.
На рис. 240 изображена схема извлечения H2S из газа крекинга при
нефтеочистительном заводе в Эль Сегундо (США). Газы с крекинг-завода
поступают в два газгольдера: высокого (.111,6 ат) и низкого (1,5 от) давле-
Рис. 240. Схема очистки газа крекинга от сероводо-
рода фенолятным методом:
1—абсорбер высокого давления; 2—абсорбер низкого давле-
ния; 3—-теплообменник; 4—дегазатор; 5—холодильник; 6—деф-
легматор; 7—активатор.
ния, из которых идут в соответствующие абсорберы 1 и 2. Последние пред-
ставляют собой работающие в противотоке колпачковые колонны с 12 та-
релками; на девятую снизу тарелку подается активированный раствор цир-
кулирующего в системе фенолята натрия, на верхнюю же тарелку — не-
большое количество свежей воды для отмывки уходящего газа от реагентов,
увлекаемых из раствора. Вытекающая из абсорбера жидкость проходит через
теплообменник, охлаждая поступающий в абсорбер активированный раствор, и
идет далее в расширительную камеру — дегазатор 4. В последнем улетучи-
ваются растворившиеся в абсорбере углеводороды. Освобожденные здесь
газы и пары через регулятор давления пропускаются вновь через абсорбер
низкого давления для улавливания выделившегося в дегазаторе HaS.
Из дегазатора раствор перекачивается на верхнюю тарелку 12-тарельчатой
колпачковой колонны — регенератора, где -поступающим водяным паром рас-
твор регенерируется, выделяя газообразный HaS-
Регенерированный раствор проходит теплообменник з, затем холодиль-
ник д и опять поступает в абсорбер. Сероводородный газ, смешанный с водя-
ными парами и реагентами, очищается в дефлегматоре в и направляется
464
х месту потребления. На заводе в Эль Сегундо сероводород передается на
сернокислотный завод, где сжигается в специальных печах.
Окислительный метод. Как уже было сказано, во всех спо-
собах этой группы H2S окисляется или до серы или до H2SO4
кислородом воздуха. В качестве катализаторов применяются или
нестойкие соединения серы (сернистая кислота, тиосерная кис-
лота, тионовые кислоты) или другие соединения (железа, мышь-
яка, меди и др.). Из способов, дающих элементарную серу, из-
вестны: железо-содовой и железо-аммиачный (Феррокс), никеле"
вый способ, мышьяково-содовый и мышьяково-аммиачный (Тай-
локе) и электролитиче-
ский способ Фишера.
Из способов, дающих
более полное окисление
H2S, известны: политио-
натовый метод Фельда,
тиосульфатный способ,
катасульфпроцесс и дру-
гие.
Очистка железо-содо-
вым и железо-аммиачным
растворами (способ Фер-
рокс). Способ очистки, из-
вестный под названием
Феррокс, состоит в том,
что H2S абсорбируется из
газа при помощи суспен-
зии гидрата окиси железа
в содовом растроре. По-
сле очистки от аммиака
и смолы газ поступает
в абсорбер (рис. 241),
где сероводород газа
вступает в следующие
реакции:
Рис. 241. Схема сероуловительной
установки Феррокс:
1 — абсорбер; 2— центробежный насос; 3 —
башия-регенератор; 4 — флотационный апп -
рат; 5—труба; 6—сероотстойник; 7—пере-
ливной бачок; 8 н 9—баки; 10 — иасос; 11—
подогреватель.
Na2CO3 + H2S = NaHS-|-NaHCO3 (I)
Далее сульфгидрат натрия реагирует с гидратом окиси же-
леза:
3NaHS + 2Fe (OH)S = 2FeS S + 3NaOH -J- 3H2O (2)
Образовавшаяся по второй реакции щелочь разлагает бикарбо-
нат с обратным образованием соды:
NaOH + NaHCO3 = Na2CO3H-H2O (3)
По выходе из абсорбера жидкость центробежным насосом 2 по-
дается в нижнюю часть второй башни-регенератора 3. Сюда же
30 Зак. 5019. Технология серной кислоты.
465
нагнетается воздух. Образовавшийся в абсорбере FeS окисляется
здесь кислородом воздуха с выделением серы
4FeS + 6Н2О +ЗО2 = 4Fe(OH)3-j- 4S (4>
Выделяющаяся под действием воздуха сера собирается
в виде пены в верхней части регенератора. Отсюда вся масса
поступает во флотационное устройство 4, находящееся в верх-
ней части абсорбера; здесь серная пена отделяется от циркули-
рующей жидкости. Посредством перемещения трубы 5 можно
менять количество серной пены, стекающей в сероотстойник 6,
и получать серную пену более или менее густой консистенции.
Отделенная от серной пены циркулирующая жидкость поступает
обратно в абсорбер через переливной бачок 7. Для возмещения
потери соды и гидрата окиси железа новые растворы их приго-
товляются в баках 8 и 9, откуда они добавляются в абсорбер
насосом 10.
Если просуммировать реакции (1), (2), (3) и (4), то получим,
что весь процесс абсорбции и регенерации сводится к одной
реакции:
H2S4- О = Н2О -f- S
Из этого получается, что теоретически расхода NazCOg и
Fe(OH)g в процессе производства не должно было бы быть.
Однако фактически это далеко не так. Наряду с основными
реакциями протекает ряд побочных реакций как в абсорбере,
так и особенно в регенераторе. В коксовом газе, поступающем
в абсорбер, наряду с H2S имеется синильная кислота HCN, кото-
рая, реагируя с содой и гидратом окиси железа, дает нерегенери-
руемые соединения — роданистый и железосинеродистый натрий
по реакциям:
Na2COs + HCN = NaCN + NaHCO3
NaCN4-S = NaCNS
6NaCN + Fe (OH)3 = Na3Fe (CN)6 -J- 3NaOH
В регенераторе происходит частичное окисление серы с после-
дующим образованием тиосульфата натрия NazSzQg по реакциям:
2NaOH + S + О2 = Na2SO3 -J- Н2О
Na2SOg S = Na2S2O3
В результате теряется часть соды и гидрата окиси железа.
Гидрат окиси железа, кроме того, теряется с удаляемой из про-
цесса серой, так как его не удается полностью отделить от
образующейся серной пены. После некоторого периода работы
установки циркулирующий раствор настолько загрязняется про-
дуктами побочных реакций, что в дальнейшем он становится
неспособным выполнять свою основную роль. Поэтому время от
времени циркулирующий раствор приходится спускать в канали-
зацию, заменяя его свежим раствором.
466
Для хорошей работы абсорбера требуется его хорошее оро-
шение, своевременная добавка свежих порций растворов соды и
гидроокиси железа, замену циркулирующего раствора новым по
мере его загрязнения продуктами побочных реакций. Работа ре-
генератора в сильной степени зависит от температурного режима.
Оптимальная температура 35—40°. Для поддержания этой тем-
пературы раствор приходится подогревать в специальном подо-
гревателе 11 (рис. 241) или пропусканием пара через змеевик,
находящийся в регенераторе.
На работу всей установки очень сильное влияние оказывает
щелочность циркулирующего раствора. Повышение щелочности
ускоряет абсорбцию сероводорода, но и увеличивает роль по-
бочных реакций. Поэтому держать концентрацию NagCOs в рас-
творе выше 2—3% не рекомендуется.
Расходные коэфициенты и другие показатели работы этого
способа примерно следующие:
Степень очистки газа..........................99,65%
Процент использования серы....................73,6 %
Расход соды на 1 кг выделевного H-S............0,6 кг
Расход гидроокиси железа (считая на чистое железо)
на 1 кг H2S.................................0,11 »
Расход всздуха иа 1 кг H<,S..................... 7 .и3
Как уже указано, одним из крупных недостатков способа
Феррокс является большой расход соды. В связи с этим было
предложено заменить содовый раствор аммиачным. Газ по спо-
собу Феррокс очищается от H2S после освобождения от смолы и
аммиака, по железо-аммиачному способу очистка от H2S ставится
сразу после смолоотделения.
В технологической схеме очистки аммиачно-содовым раство-
ром в отличие от схемы Феррокс есть дополнительный аппарат —
кислотный промыватель, в котором улавливается (серной кисло-
той) выдуваемый воздухом из регенератора аммиак. Полученный
при этом сульфат аммония после достижения определенной кон-
центрации передается в сульфатное отделение завода.
Очистка мышьяково-содовым и мышьяково-аммиачным раство-
рами (способ Тайлоке). Один из крупных недостатков, свой-
ственный и способу Феррокс и железо-аммиачному, состоит
в том, что активный реагент — гидроокись железа, находящаяся
в циркулирующем растворе, взвешена в растворе в виде суспен-
зии. По этой причине чрезвычайно трудно отделить достаточно
полно образующуюся серную пену от гидроокиси железа, .в ре-
зультате чего сера выходит сильно загрязненной соединениями
железа. Понятно поэтому, что возник вопрос о замене гидро-
окиси железа другими реагентами, устраняющими этот недостаток.
Применение с этой целью сернокислого никеля, хорошо растворимого
в абсорбционной жидкости, в значительной степени устраняет указанный не-
достаток. Получаемый продукт содержит около 97% серы, считая на сухое
вещество. Из уловленного HeS удается регенерировать до 85% серы в виде
элементарной. Остальная часть серы переходит в гипосульфит и сульфоцио-
иат натрия.
30*
467
Применять никелевый способ для очистки коксового газа нецеле-
сообразно, так как никелевый катализатор быстро отравляется синильной
кислотой коксового газа с образованием неактивного и химически весьма
устойчивого соединения Na2[Ni(CN4)].
Для очистки водяного, генераторного и других газов, в которых содер-
жание HCN незначительно, этот способ с успехом может быть применен.
Широкому внедрению способа препятствует дороговизна катализатора и
сравнительно высокие расходные коэфициенты.
Наиболее удачным из этой группы считается способ Тайлоке.
В принципе установка для очистки коксового газа по спо-
собу Тайлоке (рис. 242) не отличается от установок по способу
Феррокс и железо-аммиачному способу.
В качестве поглощающего раствора здесь служит раствор
соды и соединений мышьяка. Химизм процессов, происходящих
в абсорбере и регенераторе, окончательно не установлен. Пови-
Рис. 242. Схема установки Тайлоке:
7—абсорбер; 2—приемник для раствора; 3— регенераторы; 4 — приемник для серы; 5—-при-
емник для серного шлама; 6—вакуум-приемник; 7 —сосуд для расплавления; 8 — формы для
серы; 9 — циркуляционный иасос; 10 — воздушный компрессор; 11 — вакуум-насос; 12 —
фильтр-насос; 13 — засасывающий насос.
димому, он заключается в том, что введенный в раствор AS2O3
образует сульфосоли и оксисульфосоли мышьяка; в дальнейшем
в абсорбере кислород этих соединений частично замещается се-
рой, наоборот — в регенераторе при продувке воздухом сера
вновь вытесняется кислородом. Все соединения мышьяка, полу-
чающиеся по ходу процесса, легко растворимы, поэтому сера,
выделяющаяся в виде пены в регенераторе, легко отделяется от
циркулирующего раствора.
После введения А$аОз в раствор образуются сульфомышьяко-
вистые соли:
As2O3 + 2Na2CO3 -|- Н2О = 2Na2HAsO3 4- 2СО2
Na2HAsO8 + 2H2S = Na2HAsSaO + 2Н2О
При продувке в регенераторе сульфомышьяковистые соли пре-
вращаются в сульфомышьяковые соли:
Na2HAsS2O 4- ’/гО2 = Na2HAsS2O2
468
Затем наступает нормальный ход процесса, при котором один
атом кислорода сульфомышьяковой соли замещается в абсорбере
серой по реакции
Na2HAsS2O2 Д- H2S = Na2HAsS3O + Н2О
а в регенераторе один атом серы обратно вытесняется кисло-
родом:
Na2HAsSgО 4- »/гО2 = Na2HAsS2O2 S
Циркулирующий раствор поступает из абсорбера в приемник, где
подогревается до 25°. Регенерация производится продувкой воз-
духом в количестве, значительно превышающем теоретическое.
Серная пена после ее отделения от раствора флотацией соби-
рается в приемнике для серного шлама, из которого передается
в вакуум-фильтр, где теряет 40—50$ своей влажности.
На степень очистки большое .влияние оказывает плотность
орошения. Степень очистки повышается с увеличением концен-
трации №гСОз и AS2O3 в растворе. На ход процесса регенера-
ции прежде всего влияет величина отношения концентрации ще-
лочи к концентрации соединений мышьяка. Повышение содер-
жания щелочи против оптимального отношения усиливает побоч-
ные реакции и понижает выход серы. Изменение температуры
в пределах 30—50° заметного влияния и на ход процесса абсорб-
ции и на ход процесса регенерации не оказывает. Содержание
мышьяка в растворе, считая на AS2O3, поддерживается в пре-
деле 5%.
При очистке мышьяково-содовым раствором, кроме поглоще-
ния H2S, происходит также ряд побочных реакций. В скруббере
происходит 100%-ное поглощение цианистых соединений с обра-
зованием роданистых соединений. В регенераторе образуется
гипосульфит. Роданистые соединения и гипосульфит связывают
15—20% серы, поглощенной из газа. На образование роданистых
соединений и гипосульфита, кроме того, расходуется часть ще-
лочи. Поэтому в раствор приходится добавлять щелочь. По
мере накопления роданистых солей и гипосульфита удельный
вес циркулирующего раствора повышается. Поэтому его перио-
дически приходится разбавлять свежим, а в связи с этим часть
раствора из процесса выводить. Из сбрасываемой части раствора
предварительно выделяют мышьяк. Для этого в раствор до-
бавляют серную кислоту. Выпадающий при этом AS2O5 затем
растворяют в свежем содовом растворе, с тем чтобы опять вер-
нуть в процесс. Неизбежные в процессе небольшие потери AS2O3
компенсируются растворением новых порций AS2O3.
Сера, получаемая по этому способу, гораздо чище, чем по
способу Фёррокс и железо-аммиачному: зольность ее не превы-
шает 2%. После расплавления паром отлитая в формы сера до-
стигает степени чистоты в 99,5—99,9%.
Хотя при способе Тайлоке также протекают побочные реак-
ции, но в гораздо меньшей степени, чем в способе Феррокс.
469
В связи с этим потери соды и серы в способе Тайлоке значи-
тельно меньше, чем в способе Феррокс и железо-аммиачном.
При проценте извлечения серы из газа, равном 98, удается пе-
ревести до 80% серы, содержавшейся в газе, в продукт. Расход
соды 150—250 кг; расход мышьяка ~ 20 кг на 1 т получаемой
серы. В последнее время вместо содового раствора в процессе
Тайлоке стали применять аммиачный раствор. Так как аммиач-
ный раствор получается здесь же из аммиака коксового газа, то
этим самым уже удешевляется процесс очистки. Кроме того,
при применении аммиачно-мышьякового раствора меньше обра-
зуется гипосульфита и достигается более высокая степень
очистки. Получаемая после фильтрации (в процессе Тайлоке)
пастообразная сера чрезвычайно дисперсна: до 95% ее принадле-
жит к классу частиц с диаметром не более 3 у. В связи с этим
эта сера с успехом применяется в качестве фунгисида (в виде
мытой пасты — суспензии в виде тончайшей серной пыли после
осторожного просушивания). Сера, полученная по методу Тай-
локе, может также перерабатываться переплавкой или возгонкой.
Освобожденный от мышьяка выводимый из процесса раствор,
содержащий роданистые соединения, может быть использован
как средство для уничтожения сорняков. Способ Тайлоке ши-
роко распространен в США, Японии, Германии. Развитие этого
способа в США и Японии, добывающих достаточные количества
элементарной серы из самородных серных руд, является нагляд-
ным доказательством удачности этого способа. Несколько уста-
новок по этому же способу работает и в СССР.
Голландский способ Пти. Способ Пти представляет собой комбинацию
улавливания сероводорода раствором карбоната калия с последующим
окислением сероводорода гидроокисью железа. Как уже выше указано, после
улавливания HsS карбонатом калия, если HaS регенерируется пропусканием
через раствор СО2, получающийся газ состоит примерно наполовину
из HaS и наполовину из СОа.
В способе Пти этот газ направляется в очистной ящик с гидроокисью
железа. Сероводород при этом полностью поглощается, а СОа возвращается
в процесс. Прн продувании очистной массы воздухом выделяется элемен-
тарная сера, которая затем отделяется от массы экстракцией сероуглеродом
илн трихлорэтиленом. Сера получается очень чистая, так как в этом про-
цессе в ящик, наполненный гидроокисью железа, не могут попасть ни ча-
стицы смолы, ни цианистые соединения.
Электрический способ Фишера. В этом способе в качестве поглощающей
жидкости применяется щелочной раствор железосинеродистого калия. Для
ослабления хода побочных реакций в абсорбционной жидкости наряду с со-
дой или поташом целесообразно иметь также соответствующий бикарбонат.
При поглощении сероводорода происходит следующая реакция:
2КзРе (CN)6 + 2К2СО34- H2S = 2K4Fe (CN)6 + 2КНСО3 + S
Вытекающая из абсорбера жидкость отфильтровывается от серы и поступает
на регенерацию, осуществляемую путем анодного окисления.
В электрорегенераторе протекает реакция
2K*Fe (CN)6 + 2КНСО3 = 2KFe (CN)6 + 2К2СО3 + Н2
Выделяющийся водород отводится для использования или поступает
в общую газовую сеть
470
Получаемая по этому способу сера достигает высокой степени чистоты —
после промывки и сушки продукт содержит до 99% серы. Степень улавли-
вания сероводорода очень высока—'дополнительной очистки газа не тре-
буется. Способ заслуживает внимания при наличии дешевой электроэнергии.
Способ Фе льда. Идея окисления H2S непосредственно до H2SOi возникла
из следующих соображений. Коксовые заводы для использования получае-
мого из коксовых газов аммиака, для перевода последнего в сульфат аммо-
ния потребляют большие количества привозной серной кислоты. Между тем
большое количество серы в виде HaS имеется в самих же газах. Предста-
вилось заманчивым построить такой процесс, чтобы из NHs и HaS, содер-
жащихся в газах, получать сульфат аммония, минуя стадии сложного’ про-
изводства серной кислоты. Первой попыткой в этом направлении является
политионатовый способ Фельда. В качестве поглотительной жидкости при-
меняется раствор политионатов (три- и тетра-) аммония. Эта жидкость гото-
вится из аммиачной воды, серы и сернистой кислоты. В абсорбере происхо-
дят реакции:
(NH4)2 S3Oe + 2NH3 + H2S = 2 (NH4)2 S2O3
(NH^ S4O6 + 2NH3 + H2S = 2 (NH4)2 S2O:! + S
Раствор, вытекающий из абсорбера, отделяется от серы и обрабатывается
сернистым газом от сжигания части серы, полученной в самом процессе, или
от сжигания другого сернистого сырья. При этом тиосульфатный раствор
обратно переводится в политионатный:
2 (NH4)2 S2O3 + 3SO2 = (NH4)2 S3O6 4- (NH4)2 S4O6
Из приведенных реакций видно, что за цикл обращения количество
политионатов удваивается за счет улавливаемых NH3 и H2S и вводимого SO2-
Половина всего количества жидкости поэтому обратно направляется на
абсорбцию, а другая половина выводится из цикла. Эта часть жидкости под-
вергается обработке для получения продуктов.
При нагревании жидкости политионаты разлагаются с образованием суль-
фата аммония по реакциям
(NH4)2 S3O6 = (NH4)2 SO4 + SO2 + S
(NH4)2 S4Oe = (NH4)2 SO4 + SO2 + 2S
жидкость отстаивается; сульфат отделяется от серы и упаривается, сернистый
газ возвращается в процесс. Сера частично используется для получения не-
обходимой для процесса SO2, а частично выдается в виде продукта. Основ-
ной недостаток способа Фельда в том, что, вследствие 'непостоянного отно-
шения NHs и HsS в коксовом газе все время приходится менять количество
добавляемого в процесс того или иного недостающего компонента, что за-
трудняет управление процессом. Вредит процессу повышенное содержание
СОг в газе, так как это дает попутное образование (NHahCOs, приводящее
к нарушению нормального хода процесса. Кроме того, политионатовые рас-
творы сильно корродируют все металлы, что вызывает высокие амортиза-
ционные расходы.
Хотя способ Фельда в описанном виде не нашел применения в промыш-
ленности, он дал толчок и основу для дальнейших изысканий в этом на-
правлении. Известен очень похожий иа способ Фельда способ CaS Cop-
pers Со, по которому работает несколько промышленных установок в Гер-
мании. В этом способе в растворе циркулируют тиосульфат и политионаты
железа (наряду с политионатами и тиосульфатом аммония).
3. Катасульфпроцесс
В последние годы в Германии особенно настойчиво разраба-
тывается и уже получает промышленное применение так назы-
ваемый катасульфпроцесс.
471
Катасульфпроцесс заключается в следующем. К очищаемому
коксовому газу добавляется необходимое количество воздуха
(8—10%) и полученная газовая смесь пропускается через кон-
тактный аппарат (при температуре ~ 300°). Благодаря специфич-
ности катализатора и большой скорости прохождения газа че-
рез контактную массу протекает только реакция
H2S -f-11/2O2 = SO2 + H2O4-123,9 ккал-,
ни окисления водорода, ни обратного восстановления SO2 не про-
исходит. На каждый грамм серы в 1 м3 газа температура газа
повышается на 12—14° и для поддержания температуры в кон-
тактном аппарате хватает теплоты реакции, если серы в виде
H2S в поступающем газе не меньше 5 г/лт3.
Катализатор состоит из металлов или их окислов. Невиди-
мому составными частями катализатора являются соединения ме-
таллов группы железа и щелочные соединения.
После контактного аппарата газы, содержащие МНз и SO2,
проходят через промывную башню, где происходят реакции
2NH3 + SO2 + Н2О = (NH4)2 SO3
и
2NH3 4- 2SO2 + 2H2O = 2NH4HSO3
Чем больше отношение содержания H2S к МНз в газах, тем
больше получается бисульфита. Так как с повышением кон-
центрации бисульфита в растворе растет упругость SO2 над ним,
что означает унос SO2 из промывной башни, то в случае, если
указанное отношение концентрации H2S к концентрации МНз
больше 1,5, часть H2S отбирается из газов в неизмененном со-
стоянии до контактного аппарата. Отбор может производиться
следующими способами: 1) часть H2S вымывается из газа перед
контактным аппаратом раствором сульфита-бисульфита из про-
мывной башни и 2) газ делится на два потока, из которых один
проходит через контактный аппарат, и другой мимо него прямо
в промывную башню, 3) можно также в газовую смесь давать
недостаточное количество воздуха с тем расчетом, чтобы часть
H2S проходила через контактный аппарат без изменения.
В том случае, когда содержание H2S в газе меньше, чем
необходимо для связывания всего МНз, в процесс вводится до-
бавочное количество SO2, получаемого из другого источника.
Поглощение H2S раствором сульфита-бисульфита происходит по
реакции
2 (NH4)2 SO3 4- 2NH4HSO3 4- 2H2S = 3 (NH4)2 S2O3 4- 3H2O
Полученный щелок подвергается далее переработке на суль-
фат аммония и серу.
Щелок может быть переработан несколькими способами. Так.,
при давлении в 50—80 аг и при 250° все аммонийные соедине-
472
ния раствора превращаются в сульфат аммония, сернистый ам-
моний и серу. При уменьшении давления сероводород и аммиак
выделяются и снова возвращаются в процесс (вводятся в газы
перед контактным аппаратом).
Для переработки щелока, при более низких давлении и тем-
пературе, известны способы двухступенчатый и одноступенча-
тый. При двухступенчатом способе при варке раствора с серой
получается тиосульфат. Дальнейшая обработка раствора серни-
стым газом дает политионаты по реакции
6 (NH4)2 S2O8 + 3SO2 = 4 (NH4)2 +
4- (NH4)2 S4O6 + (NH4)2 S3O6
Тиосульфат и политионаты при температуре 120—150° (что
соответствует давлению 1—5 ат) превращаются по реакции
4 (NH4)2 S2O3 + (NH4)2 S4O6 4- (NH4)2 S3O6 =
= 6(NH4)2SO44-9S
Сера после сожжения до SO2 используется для образования
политионатов.
В одноступенчатом способе при температуре 140—150° в рас-
творе происходит реакция
(NH4)2 SOs + 2NH4HSO3 = 2 (NH4)2SO4 4- S 4- H2O
Тиосульфат взаимодействует с бисульфитом по реакции
(NH4)2 S2O8 + 2NH4HSOs = 2 (NH4)2 SO4 4- 2S -f- H2O
Для полного превращения сульфита и тиосульфата отноше-
ние концентрации бисульфита к концентрациям сульфита и тио-
сульфата в автоклаве должно быть больше, чем оно может быть
получено в растворе, отбираемом из промывной башни. С этой
целью отобранный раствор после промывной башни предвари-
тельно обрабатывается сернистым газом от обжига серы, полу-
чаемой в самом процессе. Для обработки раствора вместо сер-
нистого газа (вводимого для образования бисульфита) можно
применить любую минеральную кислоту, под действием которой
сульфит и тиосульфат также превращаются в сульфат и серу.
На рис. 243 изображена технологическая схема очистки
коксового газа от сероводорода по катасульфметоду односту-
пенчатым способом. Назначение отдельных аппаратов понятно
из рисунка. При данной схеме превращение сульфита и тио-
сульфата совершается с помощью вводимой H2SO4. Если на за-
воде отбросной H2SO4 (которую в этих целях можно использо-
вать) нет, то предусматривается печь для обжига серы, полу-
чаемой в самом процессе.
Обычно наряду с H2S в коксовых газах содержатся также
HCN, CS2 и COS. При применении катасульфметода синильная
473-
кислота на том же катализаторе под действием водяных паров
‘полностью разрушается по реакции
HCN + Н2О = NH3 4- СО
На этом же катализаторе происходит и частичное окисление
CS2 и COS до SO2. Если перед указанным контактным аппа-
ратом поставить другой контактный аппарат (форконтакт)
с гидрирующим катализатором (олово, свинец, медь, цинк или
их соединения), то в температурном интервале 300—600е эти
Газотистно Переработка ‘щелонпб
Рис. 243. Одноступенчатый способ очистки газа методом катасульф:
Т — электрофильтр; 2—форвашер (частичная отмывка HaS); 3 — теплообменник; 4 — контакт-
•ный аппарат; 5, 6 — промыватели газов; 7, 8—холодильники растворов; 9 — автоклав: J0 —
оросительная колонка для улавливания газэз, выделяющихся в автоклаве.
сернистые соединения при взаимодействии с водородом газа
также превращаются в HaS:
CS2-|-4H2 —CHt4~2H2S
cos+4н2=сн, 4- h2s 4- н2о
Как выше было сказано, для окисления H2S к газу доба-
вляется воздух,что приводит к некоторому разбавлению получае-
мого газа азотом, а следовательно, и к некоторому снижению его
калорийности. Так, калорийность газа при исходной в 5000 ккая/нР
понижается примерно до 4700 ккая/м3.
III. Получение серы из сероводорода
Концентрированный сероводородный газ идет в производство
элементарной серы или непосредственно на производство сер-
ной кислоты.
Сера из сероводорода может быть получена или сжиганием
(по реакции 2Нг§ Ог ;=? 2НЮ + 2S), или взаимодействием
HaS с сернистым газом (по реакции SO2 + 2H->S — 2НаО + 3S),
474
или взаимодействием с сульфатами (по реакции MeSCh -|-
+ 4H2S = MeS + 4НЮ -f-4S).
В промышленности реализован способ сжигания сероводорода
до элементарной серы.
Сероводород без содействия катализатора плохо горит, если
его сжигают с теоретическим количеством воздуха. Если же
сжигание производить с избытком воздуха, то в результате по-
лучается главным образом SO2, а не сера.
Как показал Клаус, реакция 2H2S + О2 = 2НгО + 2S идет
хорошо в присутствии катализатора (окись железа, бурый же-
лезняк, боксит и др.). Выход серы не бывает, однако, полным
Рис. 244. Печь Клауса:
J—колосники; 2 — кирпичные стойки; 3—водяной затвор; — выходная труба.
и в отходящих газах остается 19—22 г серы на 1 лг'! нормального
газа в виде H2S и SO2, которые улавливаются путем промывки
водой или известковым молоком. Добавкой SO2 к газу, посту-
пающему на катализатор, можно увеличить полноту вступления
сероводорода в реакцию.
Печь Клауса (рис. 244), применяемая для сжигания сероводо-
рода до серы, состоит из толстого железного кожуха, футеро-
ванного изнутри огнеупорным кирпичом. Печь разделена пере-
городками на четыре камеры. Контактная масса помещается на
колосниках /, положенных на кирпичные стойки 2. Смесь серо-
водорода с воздухом через водяной затвор 3 поступает в печь
и пронизывает контактную массу сверху вниз. При этом проис-
ходит беспламенное горение сероводорода. Образовавшаяся
жидкая сера собирается на поду печи и стекает через трубу 4
в приемник. Через эту же трубу удаляются отработанные газы.
475
IV. Получение серы из сульфидных руд
и сернистого ангидрида
Сера из сульфидов, в частности из пирита, может быть по-
лучена или непосредственно или предварительным обжигом
сульфида с получением SO2 и дальнейшим восстановлением
последнего до элементарной серы.
Для непосредственного получения серы из FeS2 было сделано
много попыток провести процесс обжига пирита по реакции
4FeS2 + ЗО2 = 2Fe2O3 + 8S
Однако этот процесс неизменно не удавался и в результате об-
жига FeS2 при недостатке кислорода всегда получалось большое
содержание серы в огарке, а в газовой фазе получался SO2.
Много попыток было сделано также в направлении получения
элементарной серы путем обжига пирита с применением водяного
пара. Однако в результате взаимодействия Нг'О с FeS2 (и то при
большом избытке пара) основная часть серы получается в виде
H2S и меньшая в виде SO2 и S2.
1. Метод Шабалина
Возможность термической отгонки первого атома серы из
FeS2 известна давно. Нагревание с этой целью FeS2 через стенку
требует большой затраты тепла. Понятно, что для отщепления
первого атома серы по реакции
Fe^-^FeS + S
можно использовать тепло реакции
4FeS-J-7O2 = 2Fe2O3-|-4SO2-|-574 ккал
На этом принципе основан способ получения элементарной серы,
разработанный Шабалиным и осуществленный в производствен-
ных условиях.
В основе этого способа лежат следующие данные, которые
получаются элементарными материальными и тепловыми расче-
тами (все величины отнесены к 1 кг исходного FeSe):
Теплота, потребляемая для диссоциации FeS2 и для
превращения ромбической серы в газообраз-
ную ......................................... 2С0 ккал
Теплота, выделяющаяся при сгорании FeS........ 1212 ,
Парциальное давление паров двухатомной серы в гагах 49,6 мм рт. ст.
Из этих данных видно, что теплота сгорания FeS более чем
достаточна для разложения FeS2 по реакции
2FeS2-» 2FeS-J-S2
Упругость паров серы над FeSe при 635° достигает 61 мл
рт. ст. (табл. 26, стр. 80). Следовательно, при этой температуре
476
и температурах более высоких возможно полное разложение FeS2,
так как упругость паров серы значительно выше ее парциального
давления в газах (49,6 мм рт. ст.). Нетрудно рассчитать также,
что при обжиге FeS без избытка воздуха теоретическая темпера-
тура получается выше 1200е. Отсюда понятно, что в зоне раз-
ложения FeSa практически достижимы температуры, обеспечи-
вающие возможность полного разложения FeS2.
По технологической схеме Шабалина производства серы из
колчедана (рис. 245) кусковой колчедан (или специально приго-
товленные брикеты из флотационного колчедана) шахтным подъ-
емником 7 подается на верхнюю площадку печи 2. В верху печи
имеется загрузочный колокол.
Рис. 245. Схема серного завода по методу Шабалина:
/—подъемник; 2—печь; 3—пыльник; 4 — паровой котел; 5—кривули; 6 — вентилятор;
7 —кварцевый фильтр; 8 —вентилятор; 9 —серная камера; /0— вентилятор; 11— холодилъ-
ник-кривули; 12—кварцевый фильтр.
В верхней зоне печи происходит подсушка колчедана, затем
идет зона диссоциации и, наконец, зона сгорания FeS. Вывод
газа из печи производится в двух местах: основной газ, который
больше не возвращается в печь, выводится в верху печи. Не-
сколько ниже выводится другая часть газа (оборотный газ), ко-
торая после выделения из нее серы опять подается в печь
(вместе с воздухом, подаваемым на обжиг FeS) с целью сни-
жения температуры в печи; без подачи оборотного газа в печи
развивалась бы такая температура, при которой неизбежно проис-
ходило бы спекание материала.
Вывод из печи оборотного газа сделан несколько ниже вы-
хода основного газа с целью устранения паров воды в оборот-
ном газе (колчедан подсушивается основным газом выше вывода
оборотного газа). Присутствие водяных паров в печи в зоне
диссоциации осложняло бы процесс образованием сероводорода.
Оборотный газ, выйдя из печи при температуре 450—480е, про-
ходит в паровой котел 4 через пыльник 3. Далее газ в воздуш-
ном холодильнике-кривулях 5 охлаждается до 180? и чугунным
477
вентилятором 6 подается в кварцевые фильтры 7. Газы имеют
при выходе из фильтров температуру 130—135°. В кривулях и
фильтрах улавливается около 97% всей серы оборотного газа.
После этого оборотный газ вентилятором 8 обратно заворачи-
вается в печь. Подача воздуха вместе с оборотным газом регу-
лируется шиберами иа газоходах перед вентилятором.
Температура выходящего из печи прямого газа 270—300°;
вентилятором 10 газ проталкивается через холодильник-кривули
11, в кварцевый фильтр 12 и серную камеру 9, где конденси-
руется сера.
Основные недостатки этого метода заключаются в следу-
ющем:
1. Низкий процент выхода серы: даже теоретически можно
получить в виде элементарной не более 50% от всей серы кол-
чедана; практически же выход серы на действующем заводе
составляет 35—37%.
2. Большие потери серы: а) с огарком, так как FeS обжи-
гается без избытка воздуха, и б) с отходящими газами в виде SO2.
На металлургических заводах в атмосферу выбрасываются
огромные массы SO2, использование которого представляет инте-
рес не только с точки зрения возможности получения серы, но
и настоятельно диктуется санитарно-гигиеническими соображе-
ниями, а в методе Шабалина обработке подвергается колчедан
и при этом опять получается SO-л в отходящих газах. Если все
это учесть, то будет понятно, что этот метод не является пер-
спективным.
2. Получение серы восстановлением SO2
Сернистый ангидрид может быть восстановлен: углеродом,
окисью углерода, водородом, сероводородом, метаном и др.
Практически сернистый ангидрид восстанавливают либо про-
пусканием через раскаленный уголь или кокс, либо смешением
содержащих SO2 газов с воздушно-генераторным газом. При
восстановлении SO2 воздушно-генераторным газом в реакцион-
ную аппаратуру вводится больше инертного газа (с генератор-
ным газом и с содержащими SO2 газами), чем при восстановле-
нии SO2 углем (только с содержащими SO2 газами). Увеличе-
ние объема инертных газов (считая на единицу SO2 или серы)
вызывает увеличение реакционной аппаратуры и потерь серы
с отходящими газами. Поэтому процесс восстановления SO»
углем является более совершенным, чем процесс восстановления
генераторным газом.
Основная реакция при восстановлении SO2 воздушно-генера-
торным газом:
1) 2СО -J- SO2 2С О2 -I- »/2S2
Кроме того, происходят побочные реакции:
2) СО + >/2S2 Z2 COS
478
и
3) 2COS Z2 СО2CS2
Зависимость константы равновесия этих реакций от темпера-
туры выражается так:
, РрП • pc2 11 300
1g ** = ’8 * “7-----1 >375 ’S
Рсо Pso2 ‘
4- 0Д67—ОД 772—1,77;
. ,, Ггоч 49,20
igtf =lg-i££^= _4,6
• А Pco-Ps’
и
Т
1g К
[pcosp
790
-у- -|-0,33.
В равновесной газовой смеси сероуглерод в интервале
350—1000° присутствует в незначительных концентрациях
(в ат от 3 • 10~6 до 2 • 10-5). Таким образом наличием CS2 в равно-
весной смеси и реакцией 2COS ;=» СО2 4 CS2 при расчете равно-
весного состава смеси можно пренебречь.
Если исходный состав газовой смеси был 66,7% СО и
33,3% SO2 (при общем давлении в 1 ат), то состав равновесной
смеси в зависимости от температуры будет следующий
(табл. 100).
Таблица 100
Темпера- тура в СС Состав газа в % Процент восстанов- ления серы
со2 СО COS SOa
1200 75,2 16,2 0,33 8,27 90,5
1000 87,0 7,9 0,80 4,30 94,5
800 < 95,6 2,0 1,00 1,40 97,5
700 97,8 0,7 0,80 0,70 98,5
600 98,5 0,2 0,80 0,50 98,8
500 99,4 0,02 0,40 0,20 99.4
300 99,94 <0,01 0,04 0.02
При восстановлении SO2 углем процесс идет таким образом,
что образующаяся СО2 снова восстанавливается углем до СО,
т. е. в этом случае, кроме уже вышеуказанных, происходит реак-
ция
2СО СО2 + С
Зависимость константы равновесия этой реакции от темпе-
ратуры следующая:
]g Д'= ^2 — 2,451 1g 74- 0,00 17— ОД 1 Г2—2,76.
479
Газовая смесь, получаемая при действии газообразного SO2 на
уголь и находящаяся в равновесии с углем, имеет следующий
состав (табл. 101):
Таблица 101
Темпера- тура в °C Состав смеси в % Процент восстанов- ления серы
СО2 СО COS CS2
1000 од 82,4 3,5 13,7 26,5
800 7,2 66,8 13,3 12,7 17,7
700 21,3 44,4 21,8 12,5 15,4
600 34,8 17,5 37,5 10,2 5,7
500 49,3 3,9 37,2 9,6 3,0
350 84,2 0,2 14,9 ‘ 0,7 82,5
При низких температурах ~ 350° скорость процесса совер-
шенно недостаточна для технических целей. При более высо-
ких температурах SO2 полностью вступает в реакцию, но при
этом лишь незначительная часть серы получается в виде эле-
ментарной. Исходя из сказанного, процесс восстановления SO»
углем ведут следующим образом.
Газы, содержащие SO-2, пропускаются через уголь с такой
скоростью, чтобы в газах, уходящих из сферы взаимодействия
с углем, оставалось SO2 в количестве, достаточном для даль-
нейшего взаимодействия с CS2, COS и СО с образованием серы
и СОг. В этих же целях можно также газовую смесь, достиг-
шую равновесия в сфере взаимодействия с углем, смешать да-
лее с новой порцией газов, содержащих SO2.
В отсутствии угля, как выше уже было показано, происходя-
щие между компонентами этой газовой смеси реакции в очень
большом интервале температуры практически полностью про-
текают в направлении образования элементарной серы.
В присутствии влаги, которая заносится в сферу реакции
или с углем или с газами, содержащими SO2, происходят также
следующие реакции:
1) 2H2O + 3/2S2-2H2S + SO2
2) h2+‘/2S2z:h2s
3) SO2 + 2Н2 72 2Н2О + ‘/2S2
Эти реакции, понятно, протекают и в том случае, когда водо-
род вносится в сферу реакции как компонент восстановительного
газа.
480
Зависимость константы равновесия этих реакций от темпера-
туры следующая:
1g К, = 1g -1,381g Т +
Alois' т
4- 0,0006 Т— 0,0;7 Г 4- 0,79.
рнч 4180
1g к2 = 1g - н* , =-----0,47 lg Т + 0,0336 Т 4- 0,0.8 У2 — 0,36.
/’h/J’s” Т
р„ л • p'i* 6900
1g = 1g-а—./ ---------Ь 0,4351g Т—0,0013 Л 4-
Pso//’h2 т
4-0,0е237-! —1,5.
По вычислениям, произведенным Лепсе в газовой системе
окись углерода — сернистый газ, потери превращения серы, обу-
словленные присутствием паров воды в количестве 1%, дости-
гают 0,6%. -
Содержание сероводорода в конечных газах снижается при
наличии избытка SO2. Так, если конечный газ содержит 1% SO2,
то на каждый процент воды в нем получается 0,5% сероводо-
рода.
Таким образом присутствие влаги в газовой смеси: окись
углерода — сернистый газ нежелательно.
Как уже было сказано, в системе окись углерода — серни-
стый газ все реакции достаточно полно протекают в направле-
нии образования элементарной серы. Однако опыты показали,
что без содействия катализатора все эти реакции идут с очень
малой, неприемлемой для промышленных целей скоростью.
В качестве катализаторов для рассмотренных реакций могут
быть применены AI2O3, смесь AI2O3 и БегОз и природный боксит,
а также катализаторы, приготовленные из шлакоглиноземистого
цемента и активированные некоторыми промоторами.
Процесс восстановления SO2 углем был осуществлен на уста-
новке «малой серы» у нас bi Союзе. Шведская фирма Болиден
применила этот метод на металлургическом заводе в Рен-
шере. Схема технологического процесса на заводе в Реншере
состоит в следующем. Часть исходного сернистого газа, пред-
варительно подогреваемого в подогревателе до 250—300° тепло-
той газов, идущих из катализаторной камеры, разбавляется воз-
духом. Далее эта газовая смесь реагирует с раскаленным коксом
в печи, напоминающей по своей конструкции обыкновенный газо-
генератор; кокс подается в печь непрерывно. Процесс в печи
ведется при 1100—1200е. Предварительный подогрев содержа-
щих SO2 газов и их разбавление воздухом (для повышения
в смеси концентрации кислорода) производятся для создания
достаточно высокой температуры в печи. Выходящая из печи
газовая смесь смешивается с другой частью исходного серни-
31 Зак. 5019. Технология серной кислоты.
481
стого газа и получаемая смесь проходит последовательно две
катализаторные камеры, наполненные контактной массой из
активированных окисей железа и алюминия. Взаимодействие
SO2 с СО, CS2 и COS происходит в температурном интервале
400—600 . После второй камеры газы проходят через холодиль-
ник, механический пылеуловитель, подогреватель (где отдают
тепло сернистому газу, идущему в печь-генератор) и через вто-
рой холодильник. Во втором холодильнике газы охлаждаются до
140е, при этом выделяется около 30% от всей содержавшейся
в них серы. Остальная часть серы выделяется в двух последо-
вательно соединенных серных электрофильтрах. Жидкая сера
подвергается очистке от угольной пыли и перекачивается в де-
ревянные бункера, где и застывает. Годовая производительность
установки — 25 тыс. т серы. Сернистые газы здесь получаются
при обжиге мышьяковой руды с очень незначительным избытком
воздуха, при этом обжиговые газы сод^эжат 11—12% SO2 и
1—2% О2.
Непосредственное восстановление углем сернистого газа при
его низком содержании в несущих его газах невыгодно, так как
очень много восстановителя расходуется на связывание кисло-
рода. Так, при газе, содержащем 3% SO2 и 12% Ог, требуется
для восстановления SO2 (считая на 1 т серы) 500 кг кокса,
а для связывания кислорода — 2000 кг кокса. Кроме того, при
бедном (по содержанию SO2) газе требуются очень большие
объемы аппаратуры (считая на 1 г суточной продукции).
В Биллингеме (Англия) фирмой ICI осуществлена установка,
в которой газы, содержащие SO2, предварительно обогащаются
при помощи раствора основного сульфата алюминия. В даль-
нейшем концентрированный газ перерабатывается на серу по
той же схеме, какая применяется и фирмой Болиден.
3. Метод Оркла
Из методов получения серы из сульфидов в промышленности
наиболее широко применяется метод Оркла. Этим методом из
медистого колчедана в ватержакете получается, с одной сто-
роны, медный штейн, а с другой стороны, газообразная сера и
ее газообразные соединения. По этому методу шихта, соста-
вленная из медистого колчедана, кокса, кварца и известняка,
поступает через двойной затвор в совершенно закрытую печь
(ватержакет) 1 для плавки (рис. 246). Воздух подается через
нижние фурмы и количество его регулируется так, чтобы вы-
ходящие из ватержакета газы совершенно не содержали кисло-
рода.
По происходящим в печи процессам ее можно разделить на
следующие основные зоны (считая сверху): 1) зона подогрева
и подсушки, 2) зона возгонки первого атома серы, 3) восстано-
вительная зона и 4) зона окислительной плавки и шлакообразо-
вания.
4?2 *
В первой зоне шихта нагревается до 550°, во второй зоне
(температура 500—800е) отгоняется один атом серы из желез-
ного колчедана. В третьей зоне идущий снизу сернистый газ
встречается с раскален-
ным коксом, в резуль-
тате чего получаются
элементарная сера, серо-
углерод и сероокись
углерода. В нижней зо-
не (окисления и шлако-
образования) сернистое
железо окисляется по
реакции
2FeS + 3O2 =
= 2FeO + 2SOa
Часть сульфида же-
леза, не окисленная воз-
душным дутьем, соеди-
няется с сульфидом ме-
ди и образует штейн,
окисленная же часть
(ЕеО) соединяется с
флюсом шихты и обра-
зует шлак. Вследствие
присутствия влаги в
шихте и в поступающем
в печь воздухе всегда
образуется некоторое
количество сероводоро-
да. Образовавшиеся в
печи серосодержащие
соединения в дальней-
шем разрушаются под
действием SO2 в кон-
тактных камерах с вы-
делением серы. В этих
целях процесс регули-
руют так, чтобы в вы-
ходящих из ватержа-
кета газах был некото-
рый избыток SO2. По-
лучающийся в первой
ватержакетной плавке
штейн перерабатывает-
ся в другом ватержакете 2 (сократительная плавка). При этой
переработке штейна выгорает еще некоторое количество серы и
достигается дальнейшая концентрация меди (до 35—40%).
В выходящем из печи газе из всей находящейся в нем серы
31»
483
до 80% приходится на элементарную, а остальные 20% на серу,
связанную в SO2, H2S, 'COS и CSa.
В смесителе-пыльнике 3 соединяются газы из ватержакетов
первичной и вторичной (сократительной) плавки. Здесь из газа
выделяется крупная пыль. Полная очистка газа от пыли про-
исходит в электрофильтре 4. Очищенный от пыли газ поступает
в реакционную камеру 5, где при содействии катализатора SO2
реагирует с COS и CS2 с образованием серы по приведенным
выше реакциям. Процесс происходит при температуре 400—450°.
В качестве катализатора применяется смесь окисей алюминия и
железа или боксит. После первой реакционной камеры газы
проходят паровой котел б, где конденсируется основное коли-
чество серй. Из парового котла отбирается пар (образующийся
за счет теплоты газов) двух давлений: в 5 и 1,5 ат. Газ (тем-
пература его 130°) выходит из котла и поступает в электро-
фильтры 7, где улавливается остальное количество серы.
В первой ступени катализа находящийся в газах сероводо-
род практически не реагирует. В связи с этим после фильтра 7
газ подогревается в подогревателе до 250° и затем проходит
через вторую контактную камеру, где при содействии того же
катализатора реагируют сернистый газ и сероводород. Реакция
происходит при температуре 250—275°. Для улавливания серы
газы проходят через вторые серные холодильники-конденсаторы,
затем через вентилятор 10 и серный фильтр и направляются на
выхлоп. Отходящие газы содержат еще серы в различных со-
единениях ~ 20 г на 1 /и3 нормального газа. Для обезврежива-
ния выхлопных газов на заводе в Тамсгавне (Норвегия) они про-
пускаются через несколько железобетонных башен 9, насаженных
известняком и орошаемых водой. Вода спускается в море.
Сера, поступающая в печи с пиритом, распределяется затем
примерно так (в %):
Извлекается в элементарном виде (уходит в продукцию) . . 80
Остается в штейне.....................................10
Остается в шлаке......................................4,7
Уходит с отбросными газами............................5,3
Получаемая по этому методу в Норвегии сера экспортируется
и конкурирует на европейском рынке с элементарной серой, по-
лучаемой из самородных серных руд. По этому же методу по-
строены медносерные заводы в Испании, Португалии и Канаде.
Один мощный медносернистый завод пущен в СССР.
Метод Оркла имеет и ряд недостатков. Во-первых, оконча-
тельно не разработан вопрос об использовании той части же-
леза, которая переходит в шлак. Во-вторых, метод предъявляет
ряд ограничений к руде: а) максимально допустимое содержа-
ние цинка в руде 2,2—2,3%; при большем содержании цинка
он конденсируется в верхней части печи, накапливается в ней,
образует настыли и останавливает процесс плавки; б) в мате-
риалах шихты совершенно не должно быть хлористых соеди-
нений, которые тоже могут вызвать забивку конденсационной
484
аппаратуры, качество и физические свойства кокса должны
быть строго определенны; в) поступающая в ватержакет руда
не должна иметь мелочи, она должна быть, кроме того, доста-
точно твердой. В-третьих, получаемая по этому методу сера бы-
вает загрязнена мышьяком и селеном, если соединения их при-
сутствуют в руде; это затрудняет использование серы в ряде
производств или же требует специальной очистки серы.
Наконец при производстве серы по методу Оркла (как оче-
видно и при всяком другом методе, где применяется восстано-
вление SO2 углем) получаются хвостовые газы, которые необ-
ходимо обезвреживать. Таким образом метод Оркла, решая одну
^гасть использования медносернистых руд: комплексное исполь-
зование серы и меди, другую часть — ликвидация выпуска вред-
ных газов в атмосферу в самом процессе производства —
разрешает не полностью.
V. Очистка и рафииировка серы
Сера, добываемая по методу Фраша в США, обычно на-
столько чиста, что она не требует дальнейшей очистки. Итальян-
ская же, а также сера, получаемая из самородных руд в СССР,
содержит примеси пустой породы, песка и т. п. Получаемая
из некоторых месторождений сера загрязнена битумами. Сера,
полученная из сульфидных руд, содержит примеси пыли,
мышьяка, селена и др. Сера, полученная при очистке коксовых
и других газов от сероводорода, всегда загрязнена смолистыми
веществами.
Способы очистки серы многообразны и .выбор их в каждом
случае зависит от характера загрязнений серы и от требований
потребителя к качеству серы. Для удаления механических при-
месей (пустая порода, песок и др.) может оказаться достаточ-
ной простая переплавка с последующей фильтрацией плава.
Мышьяк успешно удаляется обработкой расплавленной серы рас-
твором щелочи, селен обработкой азотной кислотой в растворе
хлорида — лучше всего в растворе хлорида магния. Значительно
труднее очистить серу от примесей смол, масел и битумов, так
как во всех растворителях серы обычно растворяются и эти
вредные примеси.
Известны многочисленные способы очистки серы, основанные
на обработке расплавленной серы окислителями и адсорбентами.
В качестве окислителей применяются: серная кислота, азотная
кислота, хлорсульфоновая кислота и др. При этом битумы и
смолистые вещества окисляются, а сера окисляется очень не-
значительно. Недостаток этих способов в том, что, освобождаясь
от битумов или смолистых веществ, сера в то же время загряз-
няется реагентами, применяющимися для обработки.
В качестве адсорбентов применяются активированный уголь,
силикагель, инфузорная земля и т. п. Эти способы дают не-
достаточную степень очистки, громоздки и ведут к большим по-
терям серы.
485
В тех случаях, когда сера получается экстракцией (напри-
мер экстракцией сероуглеродом из газоочистительной массы,
а также при очистке коксового газа активированным углем —
экстракция сернистым аммонием), очистка от смолистых веществ
проводится до выделения серы из раствора (окислителями или
Рис. 247. Установка для рафини-
ровки серы:
1 — котел; 2 — конденсационная камера;
3 — перегонная реторта; 4 — топка; 5 —
отверстие для выпуска серы; 6 — прием-
ник серы.
адсорбентами).
Старейшим способом очистки
серы, и пожалуй еще самым рас-
пространенным, является рафини-
ровка. Сырая сера расплавляется
в котле 7 (рис. 247), откуда
по боковой трубе направляется
в чугунную реторту з, обогре-
ваемую топкой 4. Реторта 3
соединена с кирпичной кон-
денсационной камерой 2. В за-
висимости от температурного ре-
жима процесса сера или оседает
на стенках камеры в виде серно-
го цвета (если температура не
выше 114°) или (если процесс ве-
дется при более высокой темпе-
ратуре) она собирается на дне ка-
меры в жидком виде, откуда че-
рез отверстие 5 поступает в приемник б. Сера, получаемая в жид-
ком виде, отливается в формы и носит название черенковой
или литрованной серы. Производительность рафинировочного
аппарата при работе с выпуском серного цвета значительно ниже,
чем при работе с выпуском черенковой серы.
Литература
k Gatto М., Trattomento Mineralurgico dei minerali di Solfo, Torino 1928.
2. Армстронг Б. и Химус В., Сера в угле, Chem. and Ind. 58, № 23,
543—48 (1939).
3. Пауэлл А., Извлечение серы из заводских газов, J. Ind. Eng. Chem.
31, № 7, 789—95.
4. Krczil (Каталитическое окисление сероводорода в присутствии акти-
вированного угля), Chem.-Ztg. 61, № 23, 247; № 25, 267 (1937).
5. Bahr Н. (Катасульф—новый каталитический процесс получения серы
и сульфата аммония в Германии),!. Ind. Chemist. 14, №157, 82—83(1938).
6. Mui 1 е n J. (Извлечение сероводорода посредством нормального фосфор-
нокислого калия), Oil, Gas J. 37, № 48, 37 (1939).
7. Получение серы из сероводородсодержащих газов по методу Тайлокса,
ЖХП № 7, 60 (1939).
8. Извлечение серы из нефтяных газов, Chem. and Met. Eng. 45, № 8, 416
(1938).
9. Wood W., Storss В. (Удаление сероводорода из газов очистительных
заводов посредством метода Girbotol), Oil, Gas J. 37, № 2, 47 (1938).
10. в a h г H. (Способ очистки газа .Катасульф*), Gliickauf 73, № 40,901 (1937).
11. Новый германский процесс очистки серы из коксовых газов, Chem.
Trade J. 101, № 2618, 62 (1937).
486
12. Craise F., Brown (Сероводород нефтеочистительных заводов как
экономически выгодный источник серной кислоты), Chem. and Met. Eng.
44, 376 (1937).
13. Ну с и но в Г. О., Андрианов А. П., Мышьяковый процесс газо-
очистки, ОНТИ, 1937,
14. Му л ер т Ф., Сера в угле, Госхимтехиздат, 1932.
15. Нусинов Г. О., Абсорбция H2S растворами тиоарсенатов, ЖХП № 23,
1420 (1936).
16. Thiele г Erich, Schwefel, Dresden und Leipzig 1936.
17. Lepsoe R. (Термодинамика химического восстановления сернистого
газа), J. Ind. Eng. Chem. 30, 1, 92-100 (1938).
18. Получение элементарной серы из пирита в Канаде, Canad. Min. № 3.
123—24 (1938).
19. De mag пу (Получение серы из бедных сернистых газов), LTnd. Chim.
№ 299, 801—02 (1938).
20. А р р 1 е b а у М. (Добывание серы из газов плавильных заводов), J. Soc.
Chem. Ind. 56, 5, 139, Т (1937).
21. Сера из обжиговых газов. Работа Биллингемской установки, J. Chem.
Trade 100, 2601, 280 (1937).
22. Roesner О. (Регенерация серы из отходящих газов. Физико-хими-
ческие основы и производственный способ восстановления сернистого
. ангидрида), Chem.-Fabr. 10, 9/10, 101 (1937).
23, Авдеева А. В., Термодинамические данные для газообразных серни-
стых соединений, ЖХП № 24, 1688 (1937).
24. Авдеева А. В., Взаимодействие сернистого ангидрида с сероуглеро-
дом и сероокисью углерода, ЖХП № 15, 1077 (1937).
25. Ам. пат. 1904481, 1904482, 1904483.
26. Шабалин К. Н., Переработка флотационных хвостов на серу, сернистый
газ и железорудные брикеты, Сборник Техническая реконструкция
сернокислотной и серной промышленности во втором пятилетии,
ОНТИ, 1934.
27. С. Г. Аронов, Сера. Извлечение из промышленных и отбросных газов,
Металлургиздат, Харьков-Москва 1940.
ГЛАВА 8
КОНЦЕНТРИРОВАНИЕ СЕРНИСТОГО ГАЗА
Жидкий сернистый ангидрид, широко применяемый в промыш-
ленности — в холодильном деле, для консервирования, для оку-
ривания вредителей сельского хозяйства и т. д., — получается из
обжиговых газов методом водной абсорбции. В Канаде в по-
следние годы применяется также метод вымораживания. Мас-
штабы производства 100%-ного SO2 из обжиговых газов срав-
нительно невелики. Исключительное значение приобретает про-
блема .получения 100%-ного SO2 (или во всяком случае кон-
центрированного газа) из бедных по содержанию SO2 газов
(проблема обогащения SO2) в связи с задачей использования
огромных масс сернистых газов.
Выше (стр. 53) были приведены цифры огромных потерь
серы в виде SO2 с отходящими газами заводов цветной метал-
лургии и с отходящими топочными газами электро- и теплоцен-
тралей. Важность использования этого сернистого газа диктуется
соображениями экономного расходования природных ресурсов
серы, а также санитарно-гигиеническими. В социалистическом
хозяйстве эти соображения являются обязательными. Для ка-
питалистического хозяйства, где стимулом хозяйственной дея-
тельности является частная прибыль, хищническая эксплоатация
природного сырья представляет нормальное явление, для социа-
листического же хозяйства комплексность использования сырья
есть одна из основ техно-экономической политики.
Все отходящие сернистые газы по величине их концентрации
по SO2 можно разделить на следующие основные группы:
1) Сравнительно богатые газы, содержащие 4—7% SOs
(газы ватержакетов и обжиговых печей медеплавильных заво-
дов, газы обжиговых печей цинкоплавильных заводов и др.).
2) Бедные газы, содержащие от 0,5 до 4,0% SO2, и газы пе-
ременной концентрации по SO2. Сюда относятся: а) газы ватер-
жакетов никелеплавильных (а иногда и медеплавильных) заво-
дов; б) газы отражательных печей медеплавильных заводов
(0,5—2,5%' SO2); в) меняющиеся по концентрации и количеству
газы конверторов медеплавильных заводов; г) газы агломери-
рующего обжига цинкоплавильных заводов (1,5—2,5% SO2);
д) отходящие газы сернокислотных контактных заводов, содер-
жащие при недостаточно хорошем контактировании 0,5—0,7%
SO2.
488
3) К третьей группе относятся огромные массы отходящих
топочных газов, содержащих менее 0,5% SO2, и отходящие газы
контактных сернокислотных цехов при хорошем контактирова-
нии.
4) Все другие отходящие газы с концентрацией SO* менее
0,1%.
Пути целесообразного использования тех или иных отходя-
щих газов зависят прежде всего от концентрации SO2 в них,
а затем и от ряда других конкретных условий.
Совершенно понятно, что газы первой группы целесообразнее
всего перерабатывать непосредственно «а серную кислоту. Од-
нако в ряде случаев, если серная кислота не может быть по-
треблена на месте или в достаточной близости, приходится изы-
скивать другие способы использования и этой группы газов.
Газы второй группы непосредственно могут применяться
с успехом лишь на приготовление сульфитных солей. Однако
масштаб потребности сульфитных солей ничтожно мал в срав-
нении с наличными массами этих газов. Поэтому этот путь ис-
пользования нельзя рассматривать как решение в какой-либо сте-
пени проблемы использования этой группы газов. Эта проблема
может практически быть разрешена только при условии, если эти
газы могут быть применены для производства элементарной серы
или серной кислоты.
Так как прямое получение серной кислоты или серы из этой
группы газов (по, причине их малой или непостоянной концен-
трации) технологически затруднительно и экономически нерен-
табельно, то необходимо предварительное обогащение этих газов.
Основные методы концентрирования сернистого газа следую-
щие: 1) выделение SO2 из газовых смесей вымораживанием;
2) циклические методы обогащения сернистого газа посредством
воды и различных абсорбционных растворов; 3) метод физиче-
ской адсорбции; 4) метод химической адсорбции.
I. Метод вымораживания
На рис. 248 изображена схема одной канадской установки
производства жидкого SO2 методом вымораживания (произво-
дительностью 1350 кг в сутки).
Обжиговые газы после такой же промывки и сушки, какая
применяется на контактных заводах, поступают в компрессор 1.
Осушка газа в данном производстве необходима, так как иначе
влага газов, образуя сернистую кислоту, разрушала бы аппара-
туру — в первую очередь компрессор. Кроме того, влага газов,
замерзая в дальнейшем процессе производства, нарушала бы его
нормальный ход.
В этой установке, работающей на 6% -ном обжиговом газе,
поставлен компрессор на 20 ат с водяным охлаждением. Газ
выходит из компрессора при температуре 25°. При этой темпе-
ратуре и данном давлении (20 ат) понятно не может произойти
конденсации (преждевременной) SO2. Действительно, парциаль-
ное давление SO2 в газах после компрессора будет:
20-6 , о
Тоо-^1’2 ат-
Как видно из табл. 7 (стр. 19), парциальное давление SO?
над жидким SO2 при 25° равно 3,97 ат- Из этой же таблицы
видно, что парциальному давлению SO2 в 1,2 ат соответствует
температура начала конденсации, лежащая в интервале между
—5° и —10° (точнее —6,75°). Конденсация SO2 происходит
Рис. 248. Схема производства жидкого SO2 методом вымораживания:
J — компрессор: 2 — конденсатор; 3 — экспансионная машина; 4 — цистерна; 5 — генератор;
6 — выхлопная труба.
в конденсаторе 2. Конденсатор представляет собой вертикальный
стальной цилиндр (рис. 249), разделенный по высоте плитами на
четыре камеры. Нижняя камера служит сборником для жидкого
SO2. Она смонтирована из двух половинок стальной литой трубы,
скрепленных то всей длине болтами. Вторая снизу камера
является собственно конденсатором. В этой камере проходят
16 полудюймовых труб, развальцованных своими концами в ее
верхней и нижней плитах. В каждой из этих полудюймовых труб
проходят ‘/в-Дюймовые медные трубки. Последние своими верх-
ними концами примыкают к плите, отделяющей третью камеру ог
четвертой (верхней). Нижние концы медных трубок проходят
в первую камеру (нижнюю); они собраны в медном кольце, ко-
торое соединено с конусообразным медным диском, обращен-
ным расширением книзу. Диаметр второй снизу камеры 400 м,
высота 9150 мм, диаметр третьей и четвертой камеры 250 мм,
высота 500 мм.
490
Сжатый до 20 ат газ поступает по трубе в камеру, попадает
в верхние открытые концы полудюймовых труб и идет по ним
через вторую камеру, где и конденсируется SO2 (см. ниже). Жид-
кий SO2 вместе с газами входит в нижнюю ка-
меру, выпадает на дно этой камеры, а газы
через диск поступают в Ув-дюймовые медные
трубки. Конусообразный медный диск предот-
вращает унос капель жидкого SO2 газами. Про-
ходй по медным трубкам, отходящие газы
охлаждают газы, идущие на конденсацию по
полудюймовым трубам. Выйдя из медных тру-
бок в верхнюю камеру, отходящие газы далее
при температуре 15° и давлении 20 ат посту-
пают в1 экспансионную машину 3 (рис. 248).
Здесь газы расширяются до давления, равного
1 ат, при этом они охлаждаются и производят
работу. Температура, до которой при этом газ
охлаждается, может быть вычислена по фор-
муле адиабатического расширения:
12
где Ti и Pt — начальные температура и да-
вление, Тг и Рг — конечные температура и да-
вление, у — отношение удельных теплот (при
постоянном давлении и объеме), равное в дан-
ном случае 1,4.
Газ из экспансионной машины при темпера-
туре — 80° и давлении 1 ат поступает в кон-
денсатор (рис. 249) в низ второй камеры, омы-
вает снаружи ‘Л-дюймовые трубы и отни-
мает дополнительно теплоту от конденсирую-
щего внутри этих труб газа. По выходе из вто-
рой камеры газы выхлопываются в атмосферу.
По расчету конденсация SO2 происходит
при температуре — 65°. Парциальное давле-
ние SO2 при этой температуре в газах над
жидким SO2 равно 0,04 ^г. Следовательно, со-
держание SO2 в несконденсировавшихся, а
также и в выхлопных газах равно:
°’0 20^ ~ = в»2% объемных-
Потери SO2 с выхлопными газами соста-
вляют от всего SO2, поступившего в установку
(при концентрации SO2 в обжиговых газах
в 6%),
0,2 100_о оо/
g — /0.
Рис. 249. Конден-
сатор:
1,2, 3 и 4— камеры;
5 — трубы; 6 —*
медные трубы; 7 — ко-
нусообразный диск;
8 — вход газа в кон-
денсатор; 9 — выход в
экспансионную маши-
ну; 10 — вход газов
после экспансионной
машины; 11 — выход
газов в выхлопную
трубу.
491
Экспансионная машина может вращать генератор (мотор) s
(рис. 248). Получаемая энергия компенсирует 10% энергии, за-
трачиваемой на сжатие .поступающих в установку газов.
Жидкий SO2 перепускается из нижней камеры конденсатора
в цистерну, из которой разливается в баллоны. При большей
концентрации SO2 в исходных газах относительные потери SO2
с выхлопными газами уменьшаются.
Метод вымораживания может быть применен для получения
жидкого сернистого ангидрида только из богатых сернистых
газов. Применение этого метода для концентрирования слабых
газов (и даже нормальных по содержанию SO2 газов — 6—8%)
для дальнейшей переработки концентрированного SO2 на серную
кислоту и серу, при современном состоянии техники, нерента-
бельно. Этот метод может быть применен в небольших масшта-
бах для получения жидкого SO2, непосредственно потребляемого
в таком виде для некоторых целей. Метод вымораживания по-
этому ни в какой мере не может разрешить проблему использо-
вания слабых газов.
II. Циклические методы обогащения SO2
Принципиальная схема циклического метода обогащения с по-
лучением 100%-ного сернистого газа изображена на рис. 250. Га-
зовая смесь, содержащая SO2, проходит через башню-абсорбер 7,
Рис. 250. Принципиальная схема цик-
лического метода обогащения SO2:
/ — абсорбер; 2—теплообменник; 3—десор-
бер.
где SO2, абсорбируясь орошаю-
щим башню раствором, более
или менее полно извлекается
из газовой смеси. Газ, осво-
божденный от SO2, выбрасы-
вается в атмосферу.
Насыщенный SO. раствор
проходит через теплообмен-
ник 2, где он подогревается
теплом освобожденного от SO?
раствора, идущего из башни-
десорбера 3. Подогретый рас-
твор поступает на орошение
башни-десорбера, в низ кото-
рой для нагревания раствора
подается пар. Выделяющийся
в десорбере из раствора при
его нагревании SO2 вместе с некоторым количеством водяных
паров идет на осушку и на компрессию или же на непосред-
ственную переработку в газообразном виде. Освобожденный от
SO2 и охлажденный раствор вновь поступает на орошение абсор-
бера.
Если необходимо получить не 100%-ный SO2, а лишь газ,
значительно более концентрированный (по SO2), чем исходный,
то отгонку SO2 из раствора можно производить током воздуха
492
или другого инертного газа при более низкой температуре, чем
в первом случае, при этом разница между температурами раство-
ров, выходящих из абсорбера и десорбера, значительно меньше.
Поэтому во втором случае теплообмен между растворами
устанавливать не следует.
Указанный способ частичного обогащения SO» промышлен-
ного применения пока не получил.
При применении в качестве абсорбирующей жидкости воды
принцип цикличности нарушается: после десорбции вода с не-
большими остатками SO2 спускается в канализацию, а абсорбер
непрерывно орошается свежей водой, так как выгоднее расхо-
довать большие массы воды, чем охлаждать циркулирующую
воду до надлежащей температуры. Несмотря на указанное об-
стоятельство метод водной абсорбции в принципе мало отли-
чается от циклических методов обогащения другими поглоти-
телями (можно представить, что поступающая на абсорбер вода
есть охлажденная после десорбции вода).
Первое и главное требование к абсорбционным растворам за-
ключается в том, чтобы они обладали достаточно большим тем-
пературным коэфициентом растворимости по отношению к SO2,
т. е. чтобы растворимость SO2 в применяемом абсорбционном
растворе резко падала с повышением температуры.
При растворении SO2 в воде устанавливаются следующие
равновесия:
SO2 (газ)^28О2 (раствор) -ф- Н2О X
X HSO8' + Н’ SOs" + 2Н’
Способность водного раствора к поглощению SO2 при дан-
ном давлении газа обратно пропорциональна концентрации водо-
родных ионов в растворе. Растворение двуокиси серы в воде
вызывает быстрое повышение концентрации ионов водорода и
поэтому растворение скоро прекращается. Если в растворе при-
сутствуют щелочи, то повышение концентрации ионов водорода
предотвращается и могут быть растворены большие количества
SO2. Но в этом последнем случае очень малы возможности для
последующего извлечения SO2 при повышении температуры рас-
твора.
В связи с этим хороший поглотитель, дающий возможность
обратимости процесса растворения SO2, должен обладать низ-
кой кислотностью (или высоким pH) на холоду и высокой кис-
лотностью (или низким pH) при нагревании.
Из сказанного следует, что более или менее успешного при-
менения в циклическом методе обогащения SO2 можно ожидать,
во-первых, от растворов слабых оснований или их сульфитов, и
во-вторых, от растворов солей сильных оснований и слабых не-
летучих кислот, константа диссоциации которых возрастает с по-
вышением температуры.
Испытаны следующие растворы в качестве абсорбентов-обо-
гатителей для SO2: сульфитбисульфит аммония, основная соль
493
сернокислого алюминия, ароматические основания — амины (на-
пример ксилидин С«Нз(СН4)гКН2, диэтилентриамин, триэтилен-
тетраамин, моноэтаноламин и др.), натриевая соль фосфорной
кислоты.
Зависимость растворимости газа в жидкости от температуры
можно выразить уравнением Клаузиуса-Клапейрона:
^Р — ^т+С, (2>
где X — скрытая молярная теплота абсорбции абсорбируемого
газа, R — газовая постоянная, Т — абсолютная температура.
Рис. 251. Сравнительная характеристика температурных коэфицпентов раз-
личных абсорбционных растворов.
Отсюда следует, что 1g Р является линейной функцией от у .
Угол наклона прямой 1g Р — у + С характеризует величину тем-
пературного коэфициента растворимости.
На рис. 251 показана в координатах lg Р Иу зависимость
растворимости SO2 от температуры в ряде уже упомянутых
абсорбционных растворов (по Шокину). Как видно из наклона
полученных прямых, величина температурного коэфициента рас-
полагается в убывающей степени в следующем порядке: 1) кси-
лидин, 2) основной сульфат алюминия, 3) сульфитбисульфит
аммония, 4) фосфат натрия, 5) вода и 6) сульфитбисульфит
магния.
Второе важное требование к абсорбционным растворам заклю-
чается в том, что раствор не должен химически изменяться ни
под действием SO2, ни под действием примесей, могущих быть
в поступающих на абсорбцию газах (сера, селен, серный анги-
491
дрид и др.). В частности, не должно быть значительного окис-
ления SO? в 8Оз (с образованием сульфатов в растворе), так как
это требует расходов по регенерации растворителя. Особенно
важна степень сульфатизации раствора при применении относи-
тельно дорогих органических оснований. Наконец, давление па-
ров применяемых растворов не должно быть значительным, так
как в противном случае будут большие потери абсорбентов с от-
ходящими газами или придется предусматривать специальную
аппаратуру для улавливания паров растворителя.
Основным условием работы установки обогащения SO2 ци-
клическим методом при любом абсорбционном растворе является
предварительная тщательная очистка несущего SO-2 газа от пыли
и туманообразующих примесей. Некоторые примеси газа (сера,
селен, SO3 и др.) вызывают необратимые химические измене-
ния растворов (образование сульфатов, .политионатов и т. п.),
а следовательно их порчу. При водном способе обогащения
(при непрерывном сбросе воды в канализацию) особо тщательной
очистки газа не требуется.
При данной концентрации SO2 в исходной газовой смеси и
при заданной степени извлечения SO2 необходимое количество
циркулирующего раствора, расход пара и величина поверхности
теплообмена определяются характером зависимости парциаль-
ного давления SO2 над данным раствором от концентрации
SO2 в растворе и от температуры. Размеры потребной аб-
сорбционной и десорбционной аппаратуры (считая на 1 т
SO2, получаемого в час) зависят, кроме того, от величины
коэфициента скорости абсорбции SO2 данным раствором и от ряда
других технологических и аппаратурных факторов (тип насадки,
характер встречи газа с жидкостью и т. п.).
Величина необратимых потерь абсорбционного раствора опре-
деляется степенью его химической порчи, зависящей от природы
растворов и от степени предварительной очистки газа.
Краткое описание отдельных способов обогащения SO2 ци-
клическим методом приводим ниже.
1. Способ водного обогащения
Подробные данные об упругости SO2 и Н2О над растворами
SO2 в воде приведены в гл. I (табл. 9).
Если температура орошающей воды 20°, процент SO2 в по-
ступающем газе равен 7,75, то теоретический расход воды на 1 т
извлекаемого из газов при абсорбции SO2 будет следующий.
При равновесии водного раствора с 7,75%-ным газом (пар-
циальное давление SO2 при общем давлении газов 760 мм рт. ст.
составит 59 мм рт. ст.) содержание SOs в растворе равно 1 кг
на 100 кг воды или 10 кг на 1 т воды, следовательно, на аб-
сорбцию 1 т SO2 потребуется 100 м3 воды. Расход воды будет
тем меньше, чем ниже ее температура и чем выше содержание
SO2 в поступающем газе.
495
В табл. 102 приведен теоретический расход воды на погло-
щение 1 т SO2 из газа в зависимости от концентрации SO2 в по-
ступающем газе и температуры орошающей воды 1.
Таблица 102
u/o SO2 в поступаю- щем газе Теоретический расход воды в мл на 1 m поглощаемого SO2 при раз- ной температуре воды
10’ 15° 20’
4 140 165 200
6 92 106 123
8 70 81 96
10 57 67 78
12 48 55 66
Практически, конечно, равновесие между раствором и газом
не достигается и практический расход воды всегда несколько
больше теоретического. Понятно, что с уменьшением практиче-
ского расхода воды система газ-раствор больше приближается
к равновесию, уменьшается движущая сила абсорбции и увели-
чивается потребный объем абсорбционной аппаратуры.
В случае применения для отгона SO2 из водного раствора
только пара (когда надо получить 100%-ный ЭО2), для более
или менее полной отгонки SO2 из раствора необходим нагрев
раствора до температуры ~ 100е. При этом с отходящим SO2
удаляется некоторое количество и пара, после конденсации ко-
торого получается чистый SO2. Количество пара, уносимого
с десорбированным SO2, в значительной степени зависит от кон-
кретного оформления процесса отгонки.
При достаточной высоте и соответствующей конструкции ко-
лонки можно достигнуть того, что температура уходящего из ко-
лонки газа будет выше температуры поступающего в нее из аб-
сорбера раствора всего на 10—20е. В этом случае унос водяного,
пара с SO2 будет незначителен (20—30 кг на 1 т SO2). При
этих условиях можно учитывать только расход пара, потребный
на подогрев раствора до 100°. В табл. 103 приведены данные
о расходе пара для нагрева на 1 т отгоняемого SO2 в зависимо-
сти от начального содержания SO2 в поступающем на абсорбцию
газе и температуры абсорбирующей воды.
Расход пара может быть значительно сокращен, если осуще-
ствить теплообмен между растворами: идущим из абсорбера
и уходящим после отдувки SO2. Но тогда вместо полученной
экономии на сокращении расхода пара возникают расходы на со-
оружение теплообменников. Если принять коэфициент тепло- * 3
1 Эти данные, а также цифры следующих двух таблиц взяты из работы
3. П. Розенкноп, Обогащение SO2 циклическим методом (фонд НИУИФ).
496
передачи теплообменника равным 150 ккал/м2 • час, то при ис-
ходной концентрации SO-» в газе, равной 6%, происходит сле-
дующее сокращение расхода пара (табл. 104).
Таблица! 03 Таблица! 04
°/п SO2 в поступаю- щем на абсорбцию газе Теоретический расход пара на 1 т SO2 при разной температуре абсор- бирующей воды
10’ 15° 20’
4 21 23,4 26,7
6 13,8 15 16,4
8 10,5 11,5 12,8
10 8,5 9,5 10,4
12 7,2 7,8 8,7
Степень подогрева раствора в теплооб- меннике Расход пара при отгонке SO2 Потребная поверхность теплообмен- ника в м^т SO2 в час
Не подо- гревается До 50° До 70’ До 90° 13,8 7,65 4,60 1,50 487 1410 2900
До какой величины будет целесообразно сокращать расход
пара за счет увеличения поверхности теплообмена — это зави-
сит от целого ряда конкретных условий. Если SOs отгоняется
не одним паром, а смесью инертного газа с паром (при этом
получается не 100%-ный, а лишь обогащенный против исход-
ного сернистый газ), раствор требуется подогревать до более
низкой температуры. Пусть нам из раствора, содержащего на
1 м3 воды 10 кг SO2, при отгонке паром и воздухом надо по-
лучить смесь воздуха с SOa, концентрация которого равна 16%.
В газе такой концентрации парциальное давление SOa равно
121 мм рт. ст. Как видно из табл. 9 (гл. 1), для создания такой
величины упругости газа над раствором необходимо подогреть
раствор до 40е. Теоретический расход пара при этом при ис-
ходной температуре раствора 10° (полученного абсорбцией SO2
нз 6%-ного газа), считая на 1 т SO2, составит 4,6 т вместо
13,8 т, требующихся при отгонке только одним паром (с полу-
чением 100%-ного SO2). Расход пара, конечно, еще может быть
понижен, если применяемый для отгонки инертный газ сам го-
рячий.
Для расчета процесса абсорбции SOa водой вполне может
быть применена формула:
t==K^pA
Константа скорости абсорбции SO-» водой определялась ря-
дом исследователей (Haslam, Ryan и Weber, Haslam, Herschey и
Kean, W. Adams и др.).
Этими авторами выведен ряд формул, выражающих зависи-
мость константы скорости абсорбции SO2 от ряда технологиче-
ских величин (скорость газа, плотность орошения, температура).
32 Зак. 5019. Технология серной кислоты.
497
Однако все эти формулы применены лишь в ограниченных пре-
делах. В табл. 105 и 105 а приведены величины константы скоро-
сти абсорбции SO2 водой, рассчитанные по вышеприведенной
формуле (в г SO2 на 1 м2/час на мм рт. ст.), полученные в ла-
боратории серной кислоты НИУИФ в опытах по методу орошае-
мой трубки.
Таблица 105 Таблица 105а
Скорость газа в см/сек (считая объем газа при 0°) Константа скоро- сти абсорбции 8О2 при температуре
30° 50° 6Z
1,59 15,95 12,25 7,93
2,65 17,55 14,35 11,60
5,30 23,50 20,95 16,85
6,60 24,35 22,55 19,00
7,95 26,70 24,30 19,60
Орошение в см2/мин Константа скоро- сти абсорбции 8О2 при температуре
30’ 50’ 62’
160 13,85 11,55 7,25
200 19,30 16,60 —
300 23,35 20,95 16,85
350 23,30 22,70 17,2
400 23,30 21,25 16,80
Орошаемая трубка имела внутренний диаметр 2 см и длину 20 см.
Процент SO2 в поступающих газах колебался в пределах 4,30—1.1,22.
В опытах, отражеиных в табл. 105, количество орошения было 300 см3/мин.
В опытах, отраженных в табл. 105а, скорость газа была 5,30 см/сек (считая
объем газа при 0°).
В этой же лаборатории проведены опыты по изучению кон-
станты скорости десорбции SO2 из его водного раствора. Эти
опыты показали, что константа скорости десорбции SO2 имеет
(в аналогичных условиях) ту же величину, что и константа ско-
рости абсорбции.
Как видно из табл. 105 а, константа скорости абсорбции SO2-
водой возрастает с увеличением плотности орошения лишь
в ограниченной области. Невидимому, в ограниченной области
возрастает константа скорости абсорбции SO2 водой и от увели-
чения линейной скорости газа.
По методу водного обогащения SO2 (методу Гениш-Шредера)
горячие обжиговые газы поступают под свинцовую сковородку 1
(рис. 252), где они отдают свою теплоту сернистокислому рас-
твору в целях освобождения его от SO2. Далее газы поступают
в высокую башню 2, орошаемую большим количеством воды.
Вода в противотоке с газами насыщается сернистым газом ДО'
содержания 10 кг на 1 м3 раствора, а уходящие из башни в атмо-
сферу газы при хорошей ее работе содержат всего 0,05% (объ-
емных) SO2.
Насыщенный сернистым газом раствор из башни 2 течет
в ящик 3, где он подогревается протекающей по змеевикам теп-
лой водой. Предварительно подогретый в ящике раствор течет
далее по длинной свинцовой сковороде /, к концу которой он
498
в противотоке с горячими обжиговыми газами, идущими по ка-
налу 4, подогревается гГочти до температуры кипения. При этом
из раствора выделяется SO2, который вместе с водяными па-
рами по трубе 5 идет далее в охлаждаемый водой спиральный
змеевик 6. Часть водяных паров, сопутствующих SO2, в змее-
виках конденсируется. Для окончательного освобождения от
водяных паров газ по трубе 7 поступает в сушильную башню 8-
Осушка газов в башне производится помощью сухого СаОг или,
что лучше, посредством орошения насадки башни крепкой сер-
ной кислотой. Совершенно сухой газ идет через трубу 9 дальше
в компрессор. Для возможности последующего сжижения SO2
приходится в зависимости от времени года применять давление
от 2 до 3,5 ат. Для смягчения колебаний давления служит ре-
гулятор 10.
Рис. 252. Схема производства 100%-ного SO2 методом водной абсорбции:
1—свинцовая сковорода; 2—башня; 3 — ящик; 4—каналы; 5—труба; б —змеевик; 7 —труба;
8 — сушильная башня; 9—труба; 70— регулятор давления; 77—змеевик; 72—котел; 73, 14—
трубы; 75 — колонка; 76 — кран; 77 — труба.
Сжатый газ поступает в змеевик 77, там охлаждается, сжи-
жается и течет в толстостенный железный кованный котел 72,
из которого и разливается в железные баллоны или в железно-
дорожные цистерны. Поступающие в котел 72 вместе с SO2
в небольших количествах кислород и азот отводятся при помощи
вентиля через трубу 13 обратно в абсорбционную башню 2.
Вытекающая со сковороды 7 жидкость еще содержит неко-
торое, хотя и небольшое, количество SO2. Для полного осво-
бождения от SO2 эта жидкость через кран 76 направляется в ко-
лонку 75. Сюда же стекает и конденсат, образовавшийся в змее-
вике 6. Последние остатки SO2 из жидкости, поступившей со
сковороды 7, и конденсата из змеевика 6 выделяются действием
подводимого по трубе 14 пара. Собирающаяся на дне колонки 75
свободная от SO2 горячая вода используется для предваритель-
ного подогрева сернистокислого раствора в ящике 3, куда она
по трубе 17 передается помощью ротационного насоса.
Как видно из приведенных выше данных, по причине не-
большой растворимости SO2 в воде, расход воды и пара при вод-
ном обогащении SO2 велик и быстро растет с понижением кон-
32* 499
центрации SO2 в исходном газе. По этим причинам способ вод-
ной абсорбции не может быть применеи для обогащения отхо-
дящих слабых газов и, следовательно, не может решить проблему
использования слабых газов. Способ водной абсорбции может
применяться для получения 100%-ного SO2 из нормальных обжи-
говых газов, особенно там, где в избытке имеется холодная и
дешевая вода. При этом условии может оказаться также вы-
годным применение способа водной абсорбции для относитель-
ного обогащения (например до 20% SO2) нормального обжи-
гового газа в целях интенсификации производств, потребляющих
сернистый ангидрид (производство серной кислоты, сульфитцел-
люлозы и др.).
2. Обогащение посредством аммиачного раствора
Растворы сульфитбисульфит аммония обладают по отноше-
нию к SO2 большей молярной растворимостью, чем растворы
всех других сульфитных солей.
При абсорбции SO2 раствором сульфитбисульфит аммония
особенно большое значение имеет величина отношения сульфита
к бисульфиту в растворе. Понятно, что упругость SO2 над рас-
твором (при данной концентрации аммиака в растворе) будет
тем больше, чем больше отношение бисульфита к сульфиту.
Упругость аммиака от изменения этого отношения изменяется
в обратном направлении.
Джонстон вывел следующие формулы для определения упру-
гости SO2 и NHs над растворами сульфитбисульфит аммония:
и
Af(2S—С)"
^soa— с-S
_NC(C—S)
^nh3— 2S — С ’
(4)
(5)
где pso2 и Pnh, — упругости SO2 и NHs в момент равновесия
в мм рт. ст., S и С — концентрации SO2 и МНз в растворе в мо-
лях на 100 молей воды; М и N — множители, следующим об-
разом зависящие от температуры:
1g/И = 5,865 —(6)
и
lg/V = 13,680 — (7)
Как показал Джонстон, упругость паров воды над раство-
рами сульфитбисульфит аммония подчиняется закону Рауля и,
следовательно, выражается следующим уравнением:
1 ° /ел
—AcH.o-j-S-)- ’ W
где pw — упругость паров воды над водой при той же темпера-
туре.
500
Зависимость упругости SOs и NH3 от концентрации SO2
в растворе графически изображена иа рис. 253; концентрация
NH3 в растворе при этом неизменна и составляет 22,5 моля NH>
на 100 молей воды. Эта концентрация по данным Джонстона
представляет тот максимум концентрации аммиака, который мо-
жет применяться в циклическом процессе без опасности кри-
сталлизации соли, если исходный газ содержит 0,3% SO2 и
процент извлечения SO2 из газа равен 90. При исходном 0,3%-ном
(по SO2) газе насыщенный рас-
твор (упругость SO2 над рас-
твором при равновесии и при
температуре 35° равна 2,28 мм
рт. ст.) содержит 20,45 моля SO»
на 100 молей воды (рис. 253).
Пределом отгона SO2 является
та точка, в которой при данной
температуре упругость аммиака
равна упругости SO2. Вести
дальнейший отгон нецелесообраз-
но, так как газовая фаза будет
обогащаться не сернистым газом,
а аммиаком.
Как видно из рис. 253, если
отгонка происходит при 90°, то
концентрация SO2 в регенериро-
ванном растворе будет равна
17,7 моля на 100 молей воды. Та-
ким образом емкость этого рас-
твора при разобранных условиях:
20,45—17,7 = 2,75 моля SO»
на 100 молей воды
Рис. 253. Упругость SO2 н NH3
над растворами сульфит-бисуль-
фит аммония.
На 100 кг циркулирующего раствора его емкость будет:
64-2,75-100
18 -100+ 17-22,5-1-64-17,7 ’ '
Понятно, что емкость раствора повышается при повышении
концентрации SO2 в исходных газах.
При исходной концентрации газа, равной 3% SO», емкость
раствора сульфитбисульфит аммония в 12 раз больше емкости
воды. Если учесть также, что и нагрев сульфитбисульфитного
раствора аммония требуется проводить в меньшем интервале тем-
пературы (от 35 до 90°), чем нагрев водного раствора SO2 (от
10—20 до 100°), то понятно, что расход пара при применении
этого раствора в несколько раз меньше, чем при водном обога-
щении SO-2.
Расход энергии на перекачку жидкостей при различных аб-
сорбентах обратно пропорционален отношениям емкостей рас-
501
творов. С этой точки зрения раствор сульфитбисульфит аммония
вполне может применяться для концентрирования слабых газов.
Серьезный недостаток этого раствора заключается в образо-
вании в нем сульфата (из SO3 как содержащегося в поступаю-
щих газах, так и образующегося вследствие окисления части SO2
содержащимся в газе кислородом) и саморазложения растворов
сульфитбисульфита на сульфат и серу.
Образование сульфата из SO» входящих газов может быть
сведено к достаточному минимуму предварительной очисткой
газа от сернокислотного тумана. Гораздо труднее избежать об-
разования сульфата в результате окисления раствора. Кроме
того, раствор подвержен саморазложению. Процессу самораз-
ложения подвержен не только раствор сульфитбисульфит ам-
мония, ио и растворы других солей сернистой кислоты и водный
раствор SO2. Разложение идет через промежуточное образова-
ние гипосульфита, при этом с увеличением содержания гипосуль-
фита в растворе возрастает скорость его разложения. Устано-
влено, что разложение раствора ускоряется в присутствии серы
или селена. Повышение температуры также ускоряет процесс
разложения раствора.
В связи с подверженностью этого раствора окислению и само-
разложению на нем пока не удалось осуществить обогащения SO2
чисто циклическим методом. Задача применения раствора суль-
фитбисульфит аммония значительно упрощается, если сразу же
предусматривается вывод части SO2 из цикла в виде (NH^zSCh.
3. Основной сульфат алюминия как абсорбционный раствор
при циклическом методе
Основной сульфат алюминия имеет избыток AI2O3 против со-
отношения AI2O3 и SO3 в соединении АЦЗО^з. Отношение из-
бытка AI2O3 к общему количеству его в растворе, выраженное
в процентах, носит название основности раствора.
Поглотительная способность раствора по отношению к SOa
повышается с повышением основности раствора. Но при этом
повышается также наклонность раствора к выделению осадков.
Практически употребляют раствор с основностью в 40% с об-
щим содержанием AI2O3 в растворе в 10%.
На рис. 254 изображены кривые растворимости SO2
в различных растворах основного сульфата алюминия, на
рис. 255 — кривые температур кипения растворов, содержащих
различные количества SO2. Из этих данных видно, что SO2 при
известных условиях может отгоняться при достаточно низких
температурах.
Применение этого реагента в промышленном масштабе в ци-
клическом методе обогащения SO2 затрудняется выделением из
раствора твердых фаз. Апплебей сообщает, что из раствора
основного сульфата алюминия могут выпадать осадки, указан-
ные в табл. 106.
502
В настоящее время найдены условия, до минимума устраняю-
щие возможность образования всех этих осадков. Образующийся
в растворе избыточный SO/' время от времени выводится путем
добавления известкового молока к части регенерированного
Рис. 254. Растворимость SO2 в рас-
творах основного сульфата алюминия:
Рис. 255. Точки кипения растворов
SO2 в основном сульфате алюминия:
Кривые А1 в растворе
сво- бод- ный свя- зан- ный всего %
ZZ, Па, ПЬ 3,44 4,00 6,61 5,82 10,05 9,82 34,2 40,8
III 4,65 5,32 9,87 46,1
Кривые Al в растворе
сво- бод- ный свя- зан- ный всего °/о
/ 3,44 6,61 10,05 34,2
II 4,00 5,82 9,82 40,8
III 4,55 5,32 9,87 46,1
раствора. После осаждения гипса отфильтрованный раствор воз-
вращается в поглотительную систему. Саморазложения раствора
основного сульфата алюминия почти не происходит.
Таблица 106
Наименование осадка Состав осадка Условия образования
Г идролизный 4А12О3 • 3SO3 • 24Н2О При разбавлении раствора водой
Растворимый 3A12O3-4SOs-31H2O Медленно образуется при поддерживании темпе- ратуры ниже 50°
Нерастворимый .... А1,О3 • SO3 - 3,5Н2О Осаждается при продол- жительном кипячении раствора
Щелочный ЗА12О3 • 5SO3 • R2O Медленно отлагается в виде накипи при кипячении в присутствии солей натрия или калия
503
Этот метод разработан английской фирмой 1CI. На заводе
в Биллингеме (Англия) была построена опытная установка про-
изводительностью 20 т SO2 в сутки, проработавшая несколько
лет. С 1936 г. крупная установка (производительностью 52 т
в сутки) работает в Финляндии близ Иматры. На этой установке
обогащению подвергается конверторный газ с начальной концен-
трацией SO» от 2,5 до 10%.
4. Сульфидиновый способ
Фирмой Лурги (Германия) разработан способ циклического
обогащения SO» смесью ксилидина СбНз(СНз)2МН2 с водой. Этот
способ известен под названием сульфидинового.
Применяется сырой ксилидин, представляющий собой смесь
из шести изомеров. Сам ксилидин очень мало растворим в воде,
но получающийся при абсорбции SOs ксилидинсульфат образует
с водой гомогенный раствор. При десорбции SO2 раствор опять
разделяется на два несмешивающиеся раствора — ксилидин и
воду.
Растворимость SO» в смеси ксилидина с водой и эффективная
емкость этой смеси очень высоки и приводятся в табл. 107.
Таблица 107
% SO2 в газе г SO, на 1 л раство- рителя % so, в газе г SO2 на 1 л раство- рителя
0,05 И 1.2 150
0,1 80 2,5 175
0,3 70 5 200
0,7 120 8 220
Поглощенный SO-2 легко отгоняется при температуре
80—100°. Образовавшийся ксилидинсульфат также легко разла-
гается содой с образованием свободного основания и воднорас-
творимого сульфата натрия, переходящего в водный слой, из
которого он может быть затем выделен.
Оказалось возможным производить абсорбцию SO2 при не-
котором постоянном содержании сульфата натрия в водной ча-
сти циркулирующего раствора. Чтобы избежать накопления
сульфата натрия, все время выводят часть его водного раствора
из цикла, добавляя вместо этого в цикл соответствующее коли-
чество свежей воды или раствора соды. С отходящими из аб-
сорбера газами увлекается ксилидина до 2,5 г на 1 и3 газа. Для
улавливания этого ксилидина отходящие газы промываются сер-
ной кислотой, которая при этом почти полностью нейтрализуется
504
с образованием ксилидинсульфата. Регенерация уловленного та-
ким образом ксилидина происходит во время отгонки SO2, как
уже описано выше.
Сульфидиновый метод был впервые применен на опытной
установке в Гамбурге: получалось 5—6 т SO2 в сутки из нор-
мального обжигового газа и 3—4 т SO2 в сутки из конвертор-
ного газа (со средним содержанием SO», равным 3%). Есть со-
общения о строительстве по этому способу установки с суточ-
ной производительностью в 30 т SO2 на газах свинцовоплавиль-
ного завода (исходная концентрация 4% SO2).
Расход пара при применении ксилидина меньше, чем при при-
менении любого другого известного раствора.
Основным недостатком этого раствора является его дорого-
визна и необходимость постановки добавочной аппаратуры для
улавливания паров ксилидина из отходящих после абсорбции
газов.
Ш. Концентрирование SO2 методом адсорбции
Попытки обогащения сернистого газа при помощи твердых
адсорбентов (силикагеля, активированного угля) делались уже
давно. По данным Чуфарова активированный уголь и силика-
гель могут поглощать следующие количества SO2 (табл. 108):
Таблица 108
Адсорбент % so2 в газе Поглощается SO2 в г на 100 г адсорбента прн тем- пературе
0° 10э 20°
Силикагель 2 9,5 8,5 7,0
Активированный уголь 2 19 16 12,5
Активированный уголь 4 24 20 17,5
Для удаления SO2 требуется нагрев активированного угля до
250°, а силикагеля до 125—130°. Для хорошей работы адсор-
бента требуется предварительная особо тщательная очистка газов
от пыли н осушка от влаги. Этим обстоятельством, а также и
тем, что после некоторого времени работы адсорбент теряет свою
активность и, кроме того, отличается громоздкостью потребной
установки, объясняется тот факт, что концентрирование SO2 при
помощи активированного угля и силикагеля не получило приме-
нения в промышленности.
Повидимому, большую перспективу имеет метод концентри-
рования SO2 твердыми химическими адсорбентами. Так, тре-
стом «Газоочистка» разработан для очистки от SO2 отходящих
505
топочных газов так называемый магниевый метод. По этому ме-
тоду SO2 улавливается пульпой окиси магния, крепко связы-
ваясь в сульфит магния. Регенерация окиси магния с освобожде-
нием концентрированного SO2 проводится прокалкой MgSOs.
Ведется также работа по изучению концентрирования SO2
окисью цинка.
IV. Другие методы использования
и концентрирования слабого SO2
1. Аммиачный метод
Трудности процесса обогащения SO2 при помощи раствора
сульфитбисульфит аммония, о которых говорилось выше, отпа-
дают, если насыщенный SO2 раствор не регенерируется, а обра-
батывается серной кислотой.
При этом происходят следующие реакции:
2NH4HSO3 + H2SO4 = (NH4)2 SO4 + 2SO2 + 2H2O
(NH4)2 SO3 -f- H2SO4 = (NH4)2 SO4 + SO2 + H2O
Таким образом весь извлеченный из газов SO2 получается
концентрированным, но вместе с этим (как бы попутно с кон-
центрированием) производится сульфат аммония. Циркулирую-
щий раствор освежается вводом в него свежего аммиака и вы-
водом из процесса сульфата аммония.
Установка по этому способу работает на хвостовых газах од-
ного контактного завода СССР. Хвостовые газы контактного
завода поступают частично непосредственно в абсорбционную
башню 2 (рис. 256), частично предварительно пройдя кристал-.
лизатор 3. В абсорбционной башне 2, орошаемой раствором
аммиачных солей (МН^гБОз, NH4HSO3 и (NH^zSOi, происходит
абсорбция SO2. Газ, почти освобожденный от SO2, пройдя через
ловушку 14, выбрасывается в атмосферу. В ловушке серной кис-
лотой улавливаются следы NH3, могущего быть увлеченным из
орошающего раствора проходящим газом. Образовавшийся рас-
твор (NH4)2SO-4 отводится из ловушки в реакционную колонну 7-
Из абсорбционной башни 2 орошающий раствор поступает
в сборный бак б- В этот же бак поступает маточный раствор
(NFUJaSCU из кристаллизатора 3 и центрофуги 5 и добавляется
аммиачная вода через бачок 75 и мерник 76 из сборника 4. Ам-
миачная вода добавляется со следующими целями: в маточном
;растворе, приходящем из центрофуги и кристаллизатора, может
быть свободная серная кислота, которая при действии на суль-
фит и бисульфит аммония уже в баке могла бы выделять SO«;
добавленная же аммиачная вода нейтрализует свободную серную
кислоту. Кроме того, добавлением в бак 6 соответствующего
количества аммиачной воды поддерживается постоянная кон
506
центрация аммиачных солей в растворе, орошающем башню 2,.
а также постоянное количество циркулирующего раствора.
Через змеевик бака б пропускается пар, в случае если про-
исходит кристаллизация солей. Из бака 6 раствор насосом 7
частью подается обратно на орошение башни 2, а частью на-
правляется в сборник 72, из которого через мерник 73 поступает
в реакционную колонну 7. В эту же реакционную колонну из.
бака 9 через мерник 70 поступает серная кислота. В реакцион-
ной колонне происходят вышеприведенные реакции взаимодей-
ствия серной кислоты с сульфитом и бисульфитом аммония.
Реакционная колонна разделена двумя плитами на три отде-
ления. Серная кислота вводится в реакционную колонну не-
сколько выше, чем раствор аммиачных солей, в целях некото-
рого подсушивания отходящего SO2. Верхняя часть реакционной
колонны ничем не орошается и является брызгоуловителем. Для
подогрева раствора аммиачных солей в нижней части реакцион-
ной колонны имеется змеевик, через который может подаваться
глухой пар. Выделившийся в реакционной колонне SOs, пройдя
холодильник 7 7, уходит в сушильное отделение. Раствор суль-
фата аммония из реакционной колонны при температуре 100—120°
поступает в кристаллизатор Цана 3, где навстречу горячему рас-
твору идут сухие хвостовые газы контактного завода. Газы на-
сыщаются парами воды, а раствор сульфата аммония упаривается^
при этом часть (NH^aSOi выпадает в виде кристаллов. Насы-
щенные водяными парами газы с SO2 идут в абсорбционную
башню 2. Маточный раствор вместе с кристаллическим
(NH4)2SO4 поступает в центрофугу, 5, где кристаллы освобо-
ждаются от маточного раствора; последний направляется в бак б-
Сернистый газ идет из холодильника 7 7 через две сушиль-
ные башни 77 и 20. Для освобождения от брызг кислоты, увле-
каемых из сушильных башен, SO2 проходит через брызгоулови-
тель 24, далее сжимается компрессором 25 в зависимости от
времени года (т. е. температуры окружающей среды) до 2—4 ат-
После этого газ Проходит через маслоотделитель 26.
Для охлаждения и конденсации SO2 поставлено два конден-
сатора 27. Температура газа, входящего в конденсаторы, 40—50°.
Конденсаторы представляют собой деревянные чаны, внутри ко-
торых помещены железные змеевики с циркулирующей охла-
ждающей водой. SO2 входит в змеевик, в них конденсируется
и стекает через измерительные бачки 28 в цистерны 23.
Между маслоотделителем и компрессором поставлен газголь-
дер 8 для регулирования давления в системе. Он представляет
собой колпак, опущенный в чан, в который налито минеральное
масло. От газопровода между брызгоуловителем и компрессором
сделан отвод, который идет под колпак газгольдера. Колпак
этот опускается или поднимается в зависимости от выделяюще-
гося количества SO2, тем самым поддерживается постоянное
давление в системе, что необходимо для устойчивости режима:
производства.
508
В описанной схеме обогащения SO* аммиачным методом цик-
личность устранена полностью. Можно, однако, на кислотную об-
работку выводить раствор лишь в таком количестве, которое
обеспечивает его обновление в степени, достаточной для поддер-
жания нормального процесса.
В этом случае большая часть концентрированного SO* полу-
чается не в процессе обработки раствора серной кислотой, а от-
гоном SO2 паром. Цикличность метода, таким образом, сохра-
няется.
Большая установка обогащения SO2 аммиачным методом
с одновременным получением сульфата аммония имеется в Ка-
наде (в Трейле).
По данным Лепсе и Киркпатрика газы от обжига цинковых и
свинцовых концентратов, с содержанием от 0,8 до 6—8% SO2,
после очистки в электрофильтрах и промывки водой проходят
через четыре наполненные хордовой насадкой абсорбционные
башня, в противотоке орошаемые раствором. В отходящих газах
содержание SO2 доводится до 0,1%. Насыщенный раствор обра-
батывается серной кислотой. Концентрированный SO* напра-
вляется в установку получения элементарной серы.
2. Кислотный и известковокислотный методы использования
SO2 топочных газов
Джонстоном, а затем Научно-исследовательским институтом
газоочистки изучался метод прямого получения серной кислоты
из SO2 топочных газов. Схема этого метода состоит в следую-
щем.
Газы после тщательной очистки от золы проходят через
скрубберы, орошаемые растворами солей железа- или марганца.
При взаимодействии с солями железа окисление SO2 суммарно,
очевидно, происходит по такой схеме:
SO2 -|- 2FeSO4 4- О2 = Fe2 (SO4)3
Fe2 (SO4)8 + SO2 + 2H2O = 2FeSO4 -f- 2H2SO4
В результате получается серная кислота концентрации
35—40%.
Эта кислота может несколько упариваться теплотой самих
отходящих газов, если они не используются для подсушки угля,
или другим путем.
Основное затруднение этого метода в том, что катализатор
(раствор соли) временами отравляется. Опыты Института газо-
очистки выявили ряд условий работы по этому методу: оптималь-
ную концентрацию раствора соли, максимальную концентрацию
H2SO4 в растворе, при которой очистка и окисление SO2 проте-
кают хорошо.
Если в указанных скрубберах уловить половину содержавше-
гося в газах SOa, а другую уловить далее известковым молоком,
509
то в результате половина SO2 будет превращена в слабую сер-
ную кислоту, а другая будет получена в виде CaSOs. При взаи-
модействии слабой серной кислоты с сульфитом кальция полу-
чится 100%-ный SO2 и гипс.
Можно конечно весь SO2 уловить известковым молоком,,
а полученный шлам сульфита кальция (и сульфата кальция, ча-
стично получающегося вследствие окисления сульфита) перера-
ботать на цемент и SO2.
V. Выбор метода использования
и концентрирования слабого SO2
Из данного выше обзора видно, что существует много мето-
дов концентрирования и использования слабых сернистых газов.
Несомненно также, что разработанными и разрабатываемыми ме-
тодами имеющиеся в этой области возможности далеко не исчер-
паны.
Выбор метода для промышленного применения должен
определяться характером подлежащих использованию газов и ря-
дом других конкретных условий.
Как правило, газы с концентрацией выше 4% SO2 целесооб-
разно непосредственно перерабатывать на серную кислоту. Од-
нако в особых условиях, когда для серной кислоты нельзя найти
достаточно близко потребителя, может оказаться целесообраз-
ным обогащать и такие газы в целях выпуска жидкого SO2 или
переработки SO2 на элементарную серу.
Циклические методы, характеризующиеся тем, что поглоти-
тель связывает SO2 непрочно, и требующие предварительной тща-
тельной очистки газов, применимы для концентрирования газов
с исходной концентрацией не ниже 0,7—0,5% SO2.
Для очистки огромных количеств отходящих газов тепло- и
электроцентралей с концентрацией SO2, равной 0,1—0,5%
(очистка которых с особой настоятельностью требуется и по са-
нитарно-гигиеническим соображениям), требуется применение реа-
гентов, прочно связывающих SO2. Здесь наиболее применимы
такие методы, как известковый, магниевый и кислотноизвест-
ковый.
Во многих случаях (для разных концентраций SO2 в исходном
газе) целесообразно применять аммиачный метод с выдачей суль-
фата аммония или сульфата аммония и элементарной серы. По-
нятно, что этот метод применим там, где имеется аммиак и где
есть потребность в сульфате аммония.
При современном состоянии техники использование SO2 из
газов с исходной концентрацией ниже 0,1% SO2, повидимому,
нецелесообразно. Но так как с санитарно-гигиенической точки
зрения может требоваться очистка и таких газов, то целесооб-
разна разработка методов одновременной очистки этих газов и
от SO2 и от летучей золы.
510
Литература
1. Thieler Erich, Schwefel, Dresden und Leipzig 1936.
2. H.Johnstone (Извлечение SO2 нз дымовых газов. Равновесие парциаль-
ных давлений паров над раствором в системе аммиак — сернистый ангид-
рид— вода), J. Ind. Eng. Chem. 27,587—593 (1935).
3. Н. Johnstone, D. Keyes (Извлечение SO2 из дымовых газов. Пе-
регонка трехкомпонентной системы аммиак—сернистый ангидрид —
вода), J. Ind. Eng. Chem. 27, 659- 665 (1935).
4. Н. Johnstone (Извлечение SO2 из дымовых газов. Расчет скрубберов
для большого количества газов), J. Ind. Eng. Chem. 20, 286—297 (1937).
5. Н. Johnstone (Извлечение SO2 из дымовых газов. Влияние концен-
трации растворителя на производительность и расход пара при работе
с растворами сульфита и бисульфита аммония), J. Ind. Eng. Chem. 29
1396—1398 (1937).
6. Н. Johnstone, Н. Read, Н. Blankmeyer (Извлечение SO2 из-
дымовых газов. Равновесие давлений паров иад раствором сульфитов
и бисульфитов), J. Ind. Eng. Chem. 30, 101—109 (1938).
7. Edeleanu (Сернистый ангидрид, его поглощение и рекуперация
J. Ind. Eng. Chem. 19, 222, 5С4 (1932).
8. Адамс Ф., Абсорбция сернистого ангидрида водой. Сборник Серная.
кислота и сера, Химтеорет (ЛОГОНТИ), Л. 1935.
9. Haslam, Ryan, Weber, Trans. Amer. Inst. Chem. Eng. 15, 177 (1923)
10. Haslam, Herschey, Kean, J. Ind. Eng. Chem. 16, 1224 (1924).
11. Applebey, Chem. et Ind. 53, 1097 (1934); J. Soc. Chem. Ind. № 56
139 T (1937).
12. G. Roesner, Metall u. Erz № 1, 5 (1937).
13. Lepsoe R., Kirkpatrick W., The Canadian Min. & Metallurg., Bu
№ 304, 399 (1937).
14. Kirkpatrick, Chem. and Met. Eng. 45, 483 (1938).
15. Шоки н, Егорки и, Хайтов и ч, Использование отбросных газов
низкой концентрации, ЖХП № 8, 10 (1938).
16. Постников, Асташева, Абсорбция сернистого газа ксилидином
ЖХП № 3, 14 (1940).
17. Авербух, Кооперация химии и цветной металлургии, ЖХП № 4—5
22 (1940).
18. Авербух, Стрежнев, Извлечение сернистого ангидрида из отброс-
ных газов, ЖХП № 6, 13 (1940).
19. Чуфаров, Адсорбционное обогащение бедных сернистых газов
Труды съезда основной химической промышленности, Госхимтехиздат
1932.
20. Пей с ахов И. Л., Чертков Б. А., Очистка дымовых газов от.
сернистого ангидрида, ЖХП № 10, 6 (1940).
предметный указатель
-Абсорбер 207, 295, 296, 297
Абсорбционное отделение
Герресгоф-Байера 293 сл.
контроль работы 300
режим работы 298, 299
Теителевского завода 294
Абсорбционные башни 313, 350, 351
-Абсорбция
окислов азота серной кислотой
314, 315, 329, 330, 331, 394, 395
ларов воды серной кислотой
199 сл.
сернистого газа
влияние давления на процесс
340, 341
водой 496, 497, 498
нитрозой 337, 338, 342
серного ангидрида 284 сл.
скорость 290, 291
теоретические основы 284 сл.
Автоклав 458
Агломерация
огарка 145 сл.
руд во взвешенном состоя-
нии 147, 148
Адиабатический процесс 251, 265,
268, 270
Адсорбент 505
Адсорбция 505
активированная 220
кислорода платиной 233
Азота окись см. также Окислы
азота
время окисления 395, 396
свойства 319
Азота двуокись см. также Окислы
азота
свойства 319
скорость диссоциации 320
Азота окислы см. Окислы азота
Азотистый ангидрид
растворимость в серной кислоте
328
реакция образования 324
свойства 319
Азотная кислота
потери 403 404 40'
расход 385, 403, 404
техника безопасно'"” 319
Азотооборот 3S8, 389 Зг’
Аккумуляторная серная кислота 36
Активаторы 222, 223
Активированный уголь 461, 505
Активность
катализатора 223, 243
контактной массы 226, 227
Алюминий 41
Аммиак
окисление 343, 344
установки для окисления 375,
376
Анализ ситовой 72
Андезит G6
Аппаратура
башенной системы 345 сл.
дробильного отделения 64 сл.
контактного процесса 260 сл.
концентрирования серной кисло-
ты 420 сл.
материалы для нее 35 сл.
расположение в башенной систе-
ме 372, 374
сопротивление прохождению га-
за 208, 209
сушильная установка 74, 75, 76
Асбест 36
Базальт 38
Бак (и) 366, 367
Баланс тепловой
башенной системы 391, 392
камерной системы 397
Барабанный грохот 70
Бариевая алюмованадиевая кон-
тактная масса 228
Бариево-ваиадмевая контактная мас-
са 236, 237
Бариево-оловянно-ванадиевая кон-
тактная масса 240
Башенная серная кислота, стандарт
33
Башенные системы
контроль производства 399, 400
производительность 384
расходные коэфициенты 402 сл.
тепловой баланс 391, 392
технологический режим 379 сл.
Башня (и) 346 сл.
абсорбционные (гей-люссаци) 313,
350, 351 J
»>
4л
512
Башня (и)
концентрация орошающих кис-
лот в них 382
орошение 380, 381
охлаждение кислот 393
продукционные (гловеры) 312,
348, 349
промывная
Герресгоф-Байера 190
Кемико 198
Теителевского завода 187
распределение кислоты в них
356, 357
режим в них 382, 383, 386
сушильная
Герресгоф-Байера 205, 206
Кемико 130
Бессемерование 130
Бисульфит натрия 21
Боденштейн и Катаяма, константа
диссоциации H2SO« 30
Боденштейн и Финк
равновесие реакции окисления
SO2 229
скорость процесса окисления
SO2 234
Браузе стакан 357
Браузедержатель 357
.Брызгоуловитель 297, 362, 363, 441,
442
Бунзена коэфициент 325
Бустер 443
Бюнтинга концентрационная уста-
новка 438, 439
Вакуум-концентратор 439 сл.
Вакуум-усилитель 443
Вал печи
ВХЗ 100, 102
Герресгофа 104
Вальцовая мельница 68, 69
Ванадиевые катализаторы 223 сл.
Ванадиевые контактные массы 225
сл., 234 сл.
объем 247, 248, 255
отравление 240, 241
скорость реакции 234 сл.
Вант-Гоффа уравнение 213
Ведже печи 104, 127
Вентили 373
Вентиляторы 360, 361
Водяные пары 239
Воздух, расчет объема для сжига-
ния колчедана 87
Газоочистительная масса 56
Газоходы печных отделений 142,
143
Газ (ы)
дымовые 55
медеплавильных заводов 131
Газ (ы)
обжиговые (печные) 96 см. так-
же Обжиговые газы
осушка 199, 211, 212
очистка 148 сл. см. Очистка га-
зов
перемешивание (мертвое про-
странство) 398
перемещение в системе 208, 209
сопротивление прохождению 208
характер встречи с кислотой 379
Гайяра концентрационная установ-
ка 4'22
Гей-Люссака башни 313, 350, 351
Г ерресгоф-Байера
контактная система 177, 178
промывная башня 190
расходные коэфициенты 300, 301,
302
сушильная башня 205, 206
схема процесса 177, 178
холодильник 244, 295
Герресгофа печи 98
Гидравлический затвор
камеры 377
энтарсенирунга 195
Гипосульфит натрия 21
Гипс 58, 134, 135
Гаварда камера 151
Гребки 103, 104
Гудроны кислые 57
Двайт-Ллойда аппарат 133, 145
Двуокись азота 319, 320 см. также
Окислы азота
Денитрация нитроз 386, 387, 388
Десорбция молекул SOs 233
Диабаз 36
Диференциальная теплота разве-
дения 416
Доменные шлаки 57
Донная кислота 398
Дробилка
Блэка 66
молотковая 76
Дымовые газы 55
Дюзы 120, 121
Жилля печи 448 сл.
Завихрители 275
Залесского формула 90
Изотермы
окисления SOs 250, 252
реакции окисления NO 320
Изохора реакции окисления NO
319
Интенсивность башенной системы
387
38*
513
Интенсификация
башенного процесса 408 сл.
контактного процесса 307, 308
печей 111 сл.
Калькароны 448
Каменный товар 37
Камера (ы>
гидравлический затвор 377
Мильс-Паккара 378
Морица 377
Камерная серная кислота, стандарт
33
Камерная система
контроль производства 399, 400
расходные коэфициенты 402 сл.
технологический режим 397, 398
Капельная кислота 378, 398
Катализ 219, 220
Катализатор (ы)
алюмованадиевый 225
активность 221, 222, 223, 243
бариево-алюмованадиевый 228
бариево-ванадиевый 236, 237
ванадиевый 225
определение 219
отравление 242, 243
платиновый 229, 234, 344
синтеза аммиака 344
способы приготовления 224
хромооловянный 228
Каталитическая активность 224, 234
Ква.рц 36
Кемико
концентратор 427
концентрационная установка 423
печи горизонтальные вращаю-
щиеся 120, 122, 123
промывная башня 198
сушильная башня 130
схема контактного завода 180
Керамика 37
Кернинга распылители (дюзы) 359,
360
Кесслера
концентратор 426, 427
концентрационная установка 421
Кислород, концентрация .в печных
газах 87
Кислота
концентрация в башнях 382
характер встречи с газом 379
Кислотообразоваине
в условиях башенного процесса
337
интенсивность 409
скорость в камерах 335
факторы 334 сл.
Кислотоупорные бронзы 40
Кислотоупорные цементы 37
Кислотохранилища 367
Клаузиус-Клапейрон, уравнение рас-
творимости газа в жидкости 494
Клауса печь 475
Колчедан серный
дробление 62 сл.
обжиг 80, 129 см. также Обжиг
колчедана
подача
в дробильное отделение 64, 65
к печам 137 сл.
рядовой 50, 51, 52
склад 62, 63
сыпучка 52
углистый 54, 111
флотационный 52, 117, 118
Коммуникация аппаратов 373
Конвертор 130, 131
Конденсатор 424, 490, 491
плоский 156
устройство 443
цилиндрический 157
Конденсация серного ангидрида,
условия 293
Константа
диссоциации HsSOt 30
скорости окисления SOa 228, 229
зависимость от температуры
231
на ванадиевом катализаторе
236
на платине 232
Контактирование
влияние температуры 253, 254
время соприкосновения 233, 249,
252, 253
двухстадийное 266
изотермический процесс 248, 249
многоступенчатое 267
определение объема ванадиевой
массы 247, 248
скорость начальных стадий 265
степень 214. 215, 217, 246, 247,
279
температурный режим 250, 269,
270, 271
теоретические основы 213 сл.
Контактные массы
активность 226, 227, 228
бармево-ванадиевые 236, 237
ванадиевые 225, 226. 227
определение объема 247, 248
влияние примесей 239 сл.
высота слоя 256
максимальный разогрев 281
отравление 238 сл. см. также
Контактные яды
распределение 274, 275
завихрители 275
свойства 224, 225
сопротивление 256, 280
температура 251
теплоотвод 269 сл.
514
Контактные трубки для испытания
активности контактных масс 227
Контактные яды
окись углерода 241
сероводород 242
сульфат железа 242
углеводы 241
фтористый кремний 242
хлор и хлористый водород
242
Контактный аппарат (ы)
Герресгоф-Байера 262
Кемико 276, 276.
конструкции 260, 280, 281
производительность 255, 257, 273
с двойным теплообменом 275,
276, 281
с промежуточным теплообменом
261 с л.
Тентелевский 271
Терентьева пятиступенчатый 268
Фаузера семиступенчатый 268
Контактный завод
работающий на сере 304, 305
работающий на сероводороде
306
Контактный метод окисления SOs
175, 176
Контактный процесс
на' принципе мокрого катализа
305
при повышенном давлении 258,
259
расходные коэфициенты 300,
301, 302
системы
Герресгоф-Байера 177, 178
Кемико 179, 180
Тентелевская 181
способы 182
схемы 175 с л.
Контактный узел
Герресгоф-Байера 261 сл.
Кемико 277, 278
контроль работы 282, 283
работа на газе высокой концен-
трации 280, 281
температурный режим 281
тентелевской системы 271, 272
Концентратор
Кемико 427
Кесслера 426
Симонсон-Мантиуса 445
Концентрационные установки
Бюшинга 438, 439
Кемико 423
Кесслера 421
Паулинга 439
режим работы 431, 432, 433
Фришера 439
33 Зак. 5019. Технология серной кислоты.
Концентрация
орошающих кислот 382
сернистого газа см. Сернистый
газ
Концентрирование серной кислоты
415 с л.
обжиговыми газами 436. 437
аппаратура 420 сл.
диаграмма Цейсберга 417
контроль производства 434, 435,
4.36
определение количества тепла
417, 419
теоретические основы 415 сл.
технологическая схема Кесслера
421
установки с внешним подогре-
вом 438 сл.
установки с непосредственным
обогревом кислоты 421 сл.
Концентрирование сернистого газа
488 сл.
метод
абсорбции аммиачными рас-
творами 500, 501, 506, 507
абсорбции водой 495 сл.
адсорбции 505, 506
вымораживания 489, 490, 491
кислотный 509
сульфидиновый 504
циклический 492 сл.
Корона 158, 159
К'Оронирование 158
обратное 164
Коэфициент (ы)
Бунзена 325
избытка воздуха при сжигании
колчедана 85
растворимости окислов азота
326
расходные башенных и камер-
ных систем 402 сл.
расходные контактного произ-
водства 300, 301, 302
скорости абсорбции
окислов азота 329, 333
SO2 нитрозой 338
SOs 291
теплопередачи 273
Кремнистые чугуны 39
Купоросное масло, стандарт 33
Ленер и Тейлор формула 229
Ленерта распылитель (дюз) 359
Ловушка 297, 298
Лурги печи горизонтальные вра-
щающиеся 120 сл.
Мак-Дугаля печи 98
Малетра печи 98
515
Материалы для аппаратуры ' I
природные 35, 36
искусственные 36, 37, 38
Металлы 38 ел.
Механические печи 98 сл.
Ведже К)4, 127
ВХЗ 99 сл. I
Герресгофа 98, 104, 106
интенсивность 109
интенсификация 111 сл.
конструкции 98 сл.
Мак-Дугаля 98
материальный баланс 95, 96
(работа и режим 108 сл.
тепловой баланс 109
теплоотдача 90
характеристика 105
Моногидрат 27, 291, 391
Моногидратный абсорбер 207
Мультиклоны 155
Мышьяк
очистка газов от него 183 сл.
трехокись 239
Мышьяковистый ангидрид
баланс при очистке 196
растворимость 189
упругость паров 183
Мышьяковистый водород 240
Насадка башеи 352, 353, 354
влияние на скорость абсорбции
окислов азота 331, 332
сопротивление 355
Насосы 367 сл..
Амаг-Гильперта 369
Вейзе 370, 371
центробежные, схема 368
Никкольс-Герресгофа'печи 127, 128
Нитроза
абсорбция SO2 337
свойства 316, 317
Нитрозность кислот 385, 386, 387
Нитрозный метод 313 сл.
интенсификация 408, 409
основные реакции 315, 316, 318
Обжиг колчедана 80, 129
константа скорости горения 94,
95
механизм горения 90
особенности сжигания флота-
ционного колчедана 117, 118
промежуточные реакции 81
скорость горения 90, 92
температура горения теоретиче-
ская 89
теоретические основы 80, 81, 82
теплота горения 88, 89, 90
теплотворная способность 89
факторы горения 91, 92
химизм горения 80
Обжиговые газы 84 сл., 13В
влажность 87
коицеи! рация SOs в них 133,
136, 254, 258
примеси 88
расчет объема 87
состав 84 сл., 133, 135
Огарок
агломерация 145 сл.
выгрузка 139 с л.
выход 82, 83, 84
гидравлическое удаление 141
извлечение меди 143 сл.
использование 143 сл., 148
каталитическая активность 228
пневматический способ удаления
142
теплоемкость 89
удаление 139 сл.
хлорирующий обжиг 147
Огневой подогреватель 263
Окисление NO 394, 395 см. также
Окислы азота
Окисление сернистого газа
влияние давления 217
влияние начального состава газа
и температуры 216
гомогенное 218
кинетика 229 сл.
константа равновесия реакции
213, 214
константы скорости 218, 232
контактным методом 175, 176
214, 215
механизм 229 сл., 233, 237
нитрозой 337
скорость 247 сл., 229, 232, 245,
318
температурный режим 244, 245,
250
условия 229
Окислительный объем 396
Окислы азота 311 сл.
абсорбция серной кислотой 314,
315
в камерных и 'башенных систе-
мах 318 сл.
действие на организм 319
десорбция из нитрозы 339
концентрация в газах 335, 336
коэфициент растворимости 326
окисление NO 394, 395
окисление NO в нитрозе 317
парциальное давление 403 J
подготовка к абсорбции 395
растворимость в серной кислоте
324 сл., 333
реакции образования 319, 322,
324
реакция окисления 314, 319
516
Окислы азота
содержание в газах перед аб-
сорбцией 333
упругость над нитрозой 325 сл.,
404
эксорбции из нитрозы 334
Окись азота см. также Окислы
азота
время окисления 395, 396
свойства 319
Олеум
высокопроцентный 303, 304
концентрация максимально воз-
можная 284, 285, 286
коэфициент скорости абсорбции
SOs 291
получение 176
стандарт 33
установка для разложения 303
Оркла метод 482, 483
Осушка газов 205, 211, 212
Отравление контактных масс
298 сл. см. также Контактные
яды
Охлаждение кислот 396
Очистка г^зов 148 сл., 154, 183
активированным углем 461, 462
контроль 211, 219
способы
механический 149 сл.
Герресгоф-'Байера 189 сл.
Кемико 198, 199
Тентелевский 185 сл.
электрический 155 сл.
Паулинга концентрационная уста-
новка 439
Печи
ватержакетные 129
вращающиеся для обжига серы
125
горизонтальные ’ вращающиеся
120 сл.
головка 122
для обжига цинковой обманки
132 с л.
для сжигания серы 124 сл.
Жилля 448 сл.
кильиы 98
Клауса 475
контроль работы 123, 124
металлургические 127 сл.
механические 98 сл. см. Механи-
ческие печи
муфельные 133
отражательные 132, 133
ручные Малетра и Шаффнера
98
тепловой баланс 97, 109
Печи пылевидного обжига 114 сл.
конструкции 114
Печи пылевидного обжига напря-
жение топочного пространства
119
питание 115, 116
производительность 118 сл.
режим 118 сл.
условия горения 119
Пирит 83
Пиритная плавка 129
Питатель печи 101, 102
Платина как катализатор 221 сл.
Платиновые контактные массы
223 сл., 229 сл.
отравление 242
Подогреватель
олеума 43
слабой серной кислоты 428, 429
Подстанция 167, 168
Портера формула 391, 418
Пробой 157
Продукционный бак 42
Промывная башня
Герресгоф-Байера 190
Кемико 198
Теителевского завода 187
Пыль, способы осаждения 149 сл.
Пыльная камера 151
Распылитель Ленерта 359
Расходные коэфициенты
башенных и камерных систем
402 сл.
контактного производства 300,
301, 302
Реакции горения
колчедана 81
серы 81
Рейха метод 123, 212,227,282, 283,400
Рекуператор 424, 427
Рина (трясучий лоток) 140
Сатуратор 424, 427
Сборник циркуляционный 367
Свинец 40, 345, 349
Селен 240
Сера
выгорающий процент 82, 83, 84
выплавка
в автоклавах 458, 459
паровая 455 сл.
выхода из руды 455, 457, 459
газообразная 15
горение 81
дисперсная 17, 18
комовая 17
размол 460
метод обогащения
механический 447
флотационный 447, 448
электростатический 447. 448
методы получения
Фраша 457, 458
517
Сера
методы получения
физико-термический 447. 457 сл.
экстракцией растворителями
447, 459, 460
модификации 14
очистка и рафинировка 485, 486
парциальное давление паров 80
печи для сжигания 124 сл.
получение см. также Сера, спо-
собы получения
из самородных руд 447 сл.
из сернистого газа 478 сл.
из сероводорода 174, 175
из сероводородсодержащих
газов 460 сл.
из сульфидных руд 476, 477
из сульфидов 482 сл.
применение 13, 16
производство 447 сл.
растворимость 15
реакция горения 81
свойства 14, 15, 16
содержание в огарке 82, 83, 84
сорта 17
сырье для получения 447
температура воспламенения 124
теплоемкость 14
теплота испарения 14
упругость паров 15, 80, 476
установки (американские) для
сжигания 125
элементарная 14 сл.
Сера, способы получения
Пти (голландский) 470, 471
железо-аммиачный 465 сл.
железо-содовый 465 сл.
катасульф 465, 471, 472, 473
мокрой очистки 462 сл.
мышьяково-аммиачный 465, 467 сл.
мышьяково-содовый 465, 467 сл.
нейтрализационный 463 сл.
никелевый 465
окислительный 465
Оркла 482, 483, 484
сухой очистки 460 сл.
Тайлоке 465, 467 сл.
тиосульфатный 465
Фельда 471
фенолятный 464
Феррокс 465 сл.
Шабалина 476. 477
Серная кислота
башенный способ производства
312
вязкость растворов 32
Диференциальная теплота разве-
дения 31
камерный способ производства
311
Серная кислота
константа диссоциации 30
контактный способ производства
175 с л.
на сере 304, 305
концентрация в орошающих
кислотах 396
концентрирование 415 сл.
мировое производство 26
нитрозиый способ
исторические сведения 313
теория 314 сл.
плотность растворов 27
получение из топочных газов
509
применение 13, 23, 24, 25
свойства
физические 27
химические 23, 24, 25
стандарты 33
температуры замерзания и кипе-
ния 28, 29
теплоемкость 28
теплота
парообразования 31
разведения 31, 391, 416, 418
растворения 31
смешения 31
упругость паров 29, 30
Сернистое сырье 48 сл.
газоочистительная масса 56
газы металлургических печей 53
доменные шлаки 57
дымовые газы 55 '
кислые гудроны 57
колчедан см. Колчедан серный
обжиг 80 сл. см. Обжиг колче-
дана
отходы углеобогатительных фаб-
рик 55
реакции горения S6
самородные серные руды 49
сероводород 56
соотношение отдельных видов
60, 61
сульфаты 58, 59
травильные растворы 60
Сернистый газ (ангидрид) 18 с л.
абсорбция
водой 496, 497, 498
нитрозой 337, 338. 342, 343
влияние на организм 21
восстановление до серы 478, 479
изотермы окисления 233
катализаторы для окисления
221 сл.
конденсация 490, 491
концентрация
в башнях 384
в газовой смеси 338, 410
в конверторных газах 130
518
Сернистый газ
•концентрация
(В обжиговых газах 85, 90, 254
при входе в систему 383, 384
концентрирование 488 сл., 505
аммиачный способ 500, 501,
506, 507
водный способ 495 сл.
сульфидиновый способ 504
циклический способ 492, 493,
494
критическая температура 18
окисление см. Окисление сернис-
того газа
получение 18
из гипса 134 сл.
реакции 86
растворимость 19
в основном сульфате алюми-
ния 503
свойства
физические 18, 19, 20
химические 19, 21
скорость абсорбции нитрозой
338, 342, 343
стопроцентный, получение 499
теплоемкость 18
упругость паров 19
«ад растворами сульфит-би-
сульфит аммония 501
Серный ангидрид 28, 29
абсорбция 284 сл.
скорость 290, 291
теоретические основы 284 сл.
в печных газах 87
газообразный, теплота смешения
289
конденсация с парами воды 291,
292
полимеризация 288
получение 124, 173
свойства 22
соединения с водой 27
теплота смешения с водой 288
упругость паров над олеумом
287, 290
Сероводород 56, 174, 175, 469 сл.
Силикагель 225, 228, 232, 233, 239,
505
Силовое поле 159, 160, 161
Симонсон-Мант.иуса выпарная уста-
новка 440 сл.
Смеситель для кислот 43
Стокса формула 150
Сульфат алюминия 502, 503
Сульфат ванадила 236
Сульфат кальция, разложение 134
Сульфат магния как носитель ката-
лизатора 224
Сульфаты, температура разложения
82 *
Сульфит-бисульфиг аммония 501
Сульфит натрия 21
Сушильная установка для флота-
ционного колчедана 73, 74
Сушильное отделение 204 сл.
Сушка флотационного колчедана
73 сл.
Тайлоке, способ получения серы
467, 468, 469
Тейлор и Ленер, формулы 229, 234
Теллур 240
Температура горения колчедана 88,
89
Температурный режим (оптималь-
ный) контактного узла 281
Тентелевская схема контактного
завода 181
Теплоемкость
сернистого газа 18
серной кислоты 28
серы 14
Теплообменник 263, 265
Теплоотвод, интенсивность 269, 270,
271, 273
Теплоотдача механических печей
90
Топка
Кемико нефтяная 425, 426
Кесслера 424, 425
Трясучий лоток (рина) 140, 141
Турбинка для разбрызгивания кис-
лоты 358
Турбокомпрессор 209, 210
Углистый колчедан 64
теплота горения 88
особенности сжигания 111
Фарфор 37
Феррокс, способ получения серы
465, 466
Фильтр-маслоотделитель 207, 208
Флотационный колчедан 52
обжиг 117, 118
особенности горения 117, 118
сушка 73 сл.
Флотация, метод 52
Форвашер 186
Форсунка 115, 116
Фосфогипс 58
Фраша способ получения серы 457.
458
Фримана установка для сжигания
флотационного колчедана 117
Фришера концентрационная уста-
новка 439
Холодильник (и) 364, 365
ангидридный
Герресгоф-Байера 294, 295
519
Холодильники
ангидридный
Грилло-Шредера 294, 295
газовый 191, 429
интенсификация 393
оросительный 365
поверхность охлаждения 394
Теителевского завода 186
цилиндрический 364
Хромистые стали 39
Цейсберга диаграмма 417
Циклоны 152 с л.
Циклоны (брыэгоуловители) 362,
363
Цинк, получение 132
Цинковая обманка 132 сл.
Черные металлы 38, 39
Шамот 36
Шаффнера печи 98
Эжектор двухступенчатый паровой
443, 444
Эксорбции
водяных паров из серной кис-
лоты 419
окислов азота 334, 3S9, 390
Элеватор 71
Электрическая очистка газов 155 сл.
Электровагонетка 137, 138
Электромагнитный сепаратор 77
Электрофильтр (ы) 162 сл., 424, 430
мокрые 192
огарковые 164 с л.
степень очистки газа 163
трубчатые 162
условия работы 167
факторы, влияющие на работу
163
Эмаль 37
Энергия активации для катализа-
торов 231 сл.
Энтарсенирунг (и) 191
пластинчатый 193
сотовый 197
Отв. редактор М. А. Григорук.
Тираж 3000 экз. Подписано к печати 2?/П 1941 г. Л 13875.
Печатных листов 32,5. Учетно-издат. листов 36,96. Тип. зн. в 1 печ. листе 43.120.
Цена в переплете 14 р. 25 к. Без переплета 12 р. 25 к. Зак. № 5019.
4-я типография ОГИЗа РСФСР треста «Поаяграфкнига» вмени Евгении Соколовой.
Ленинград, пр. Кр. Командиров, 29.