






















































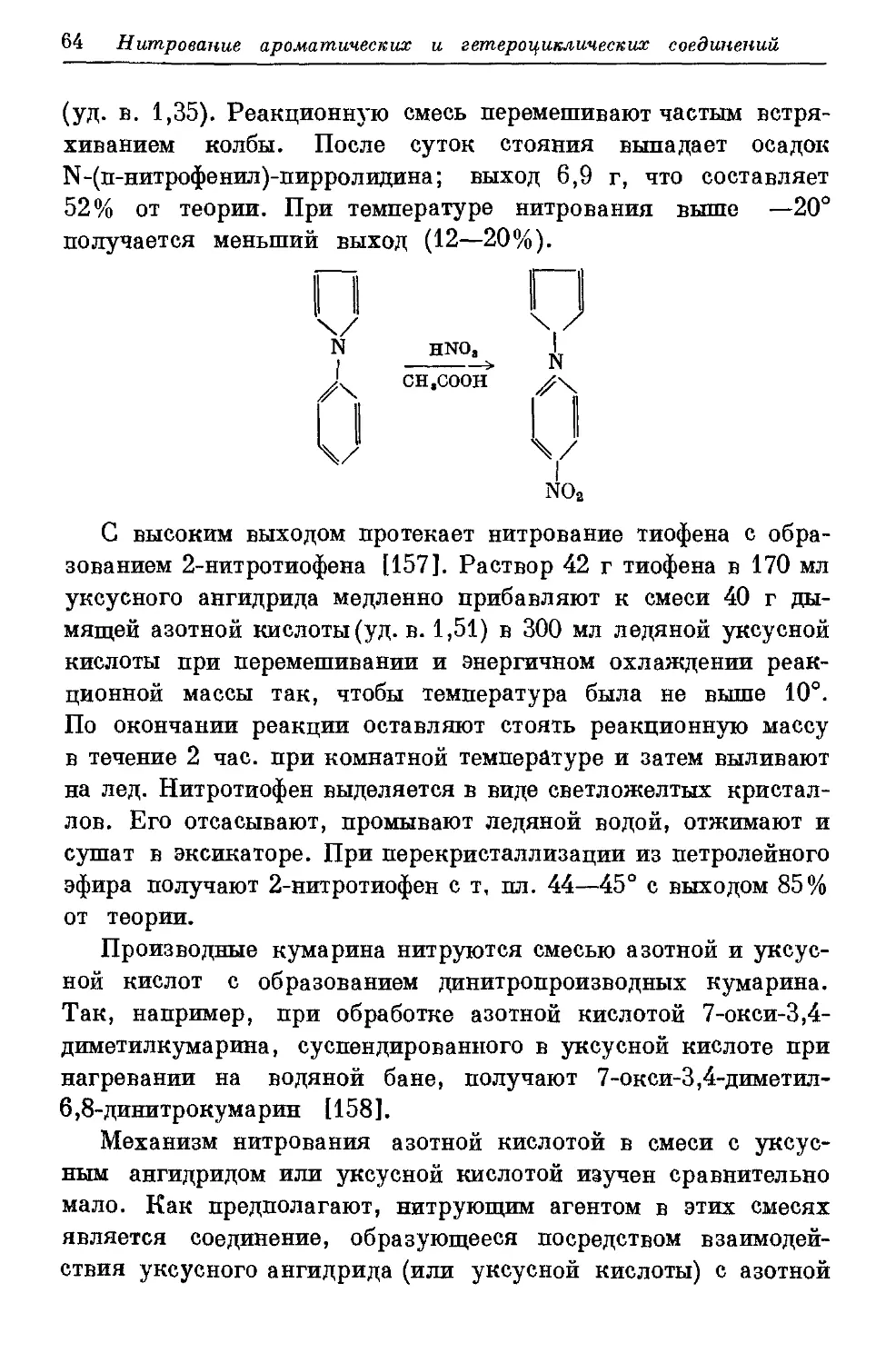




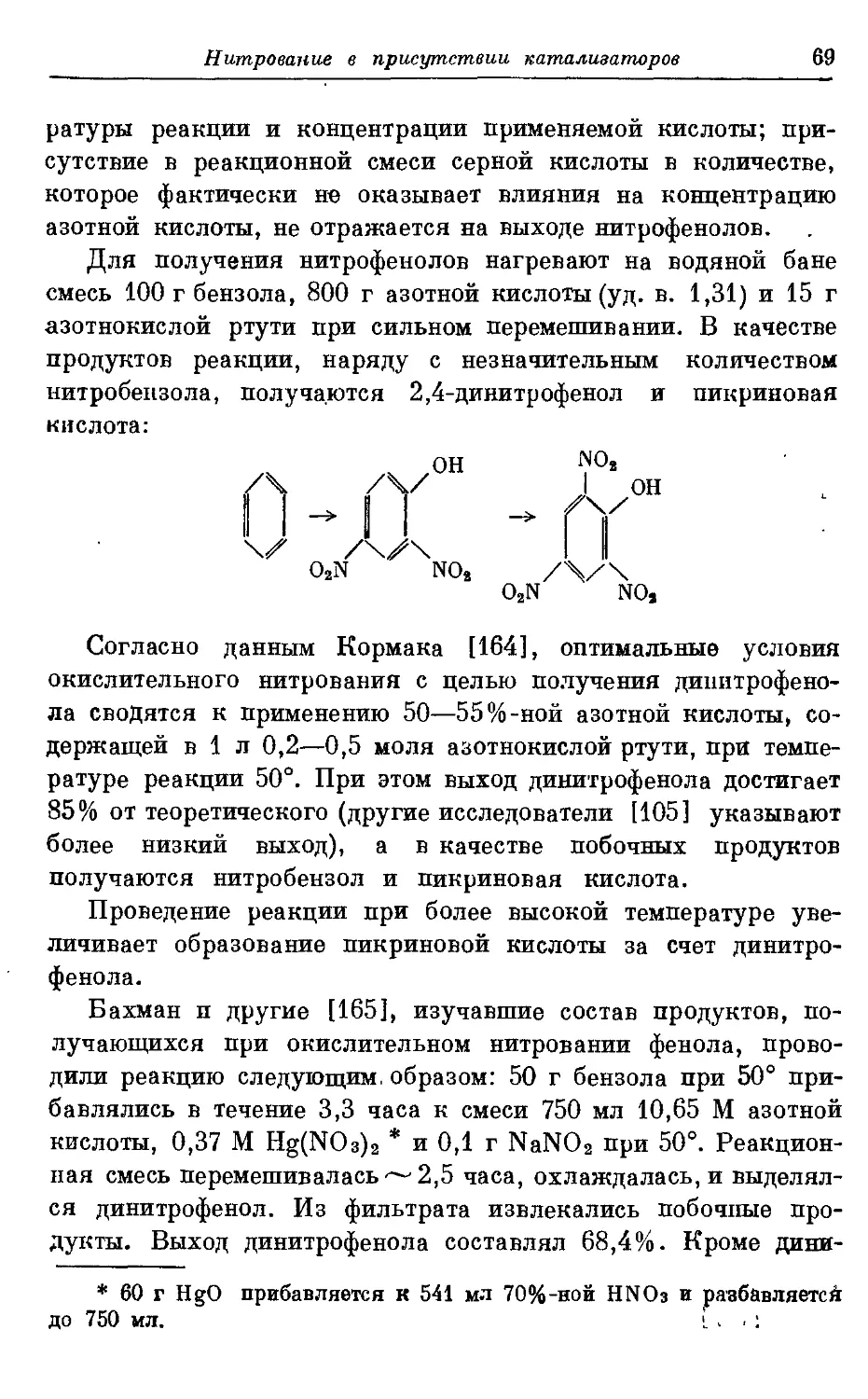


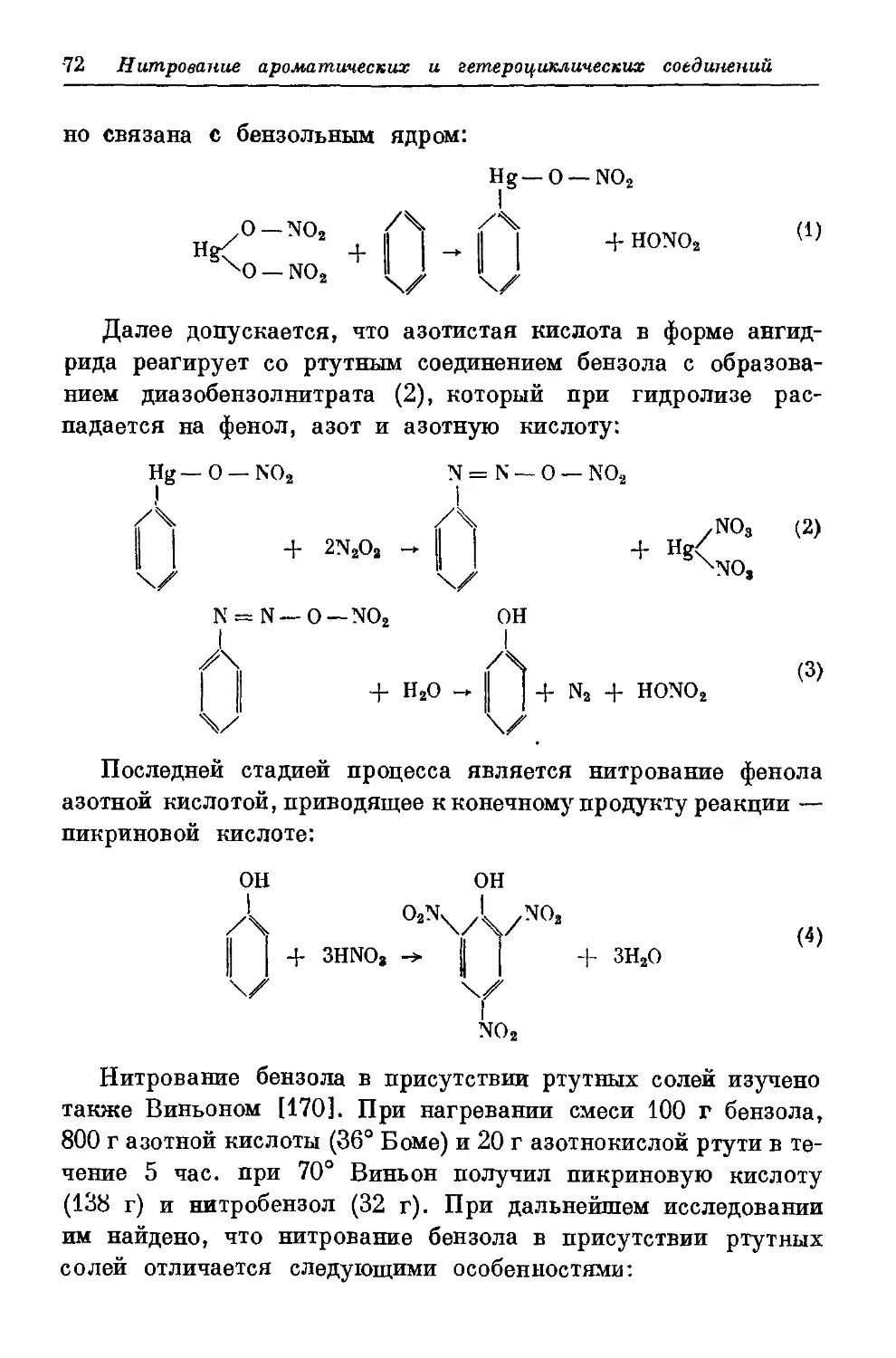

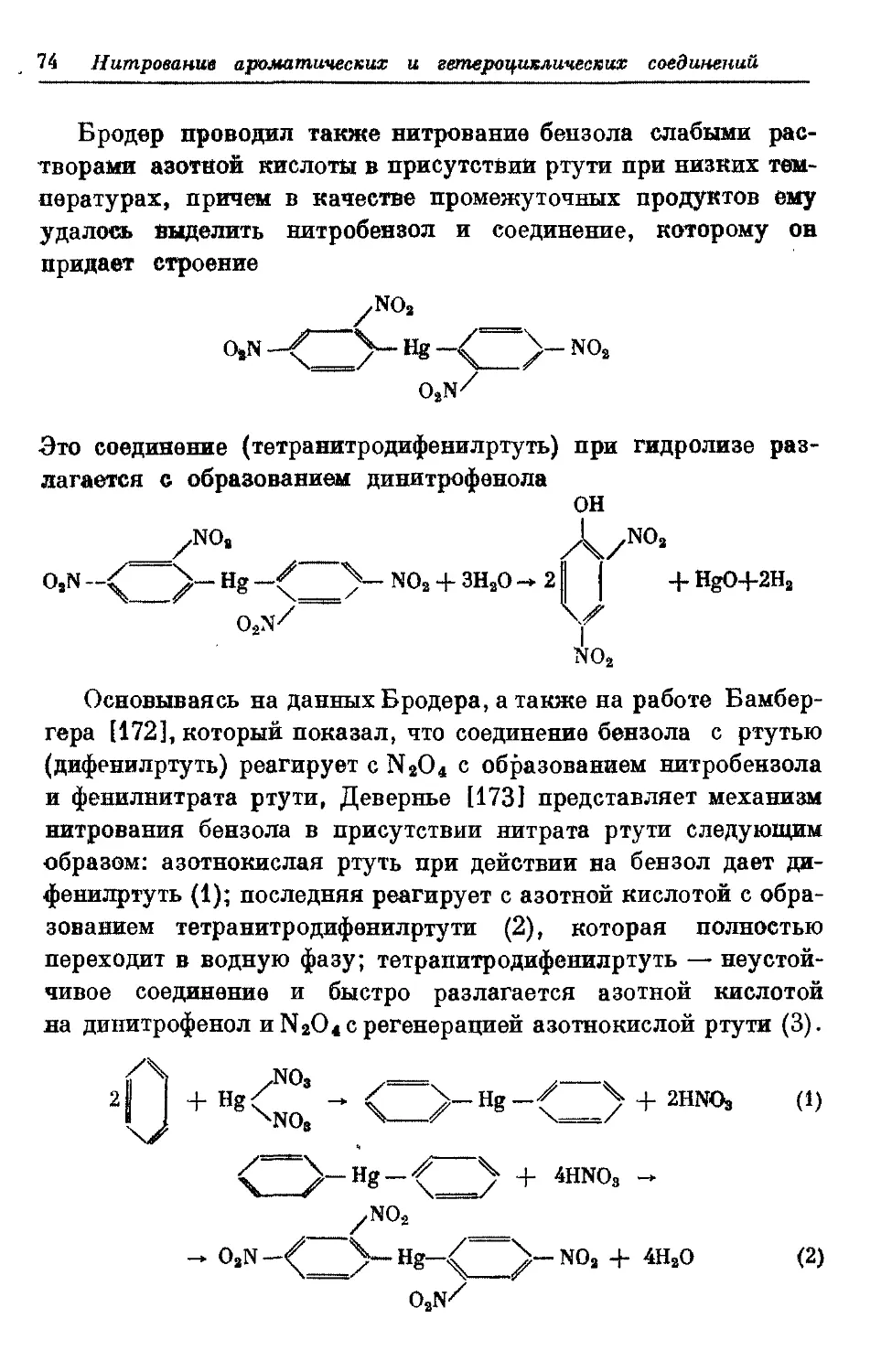


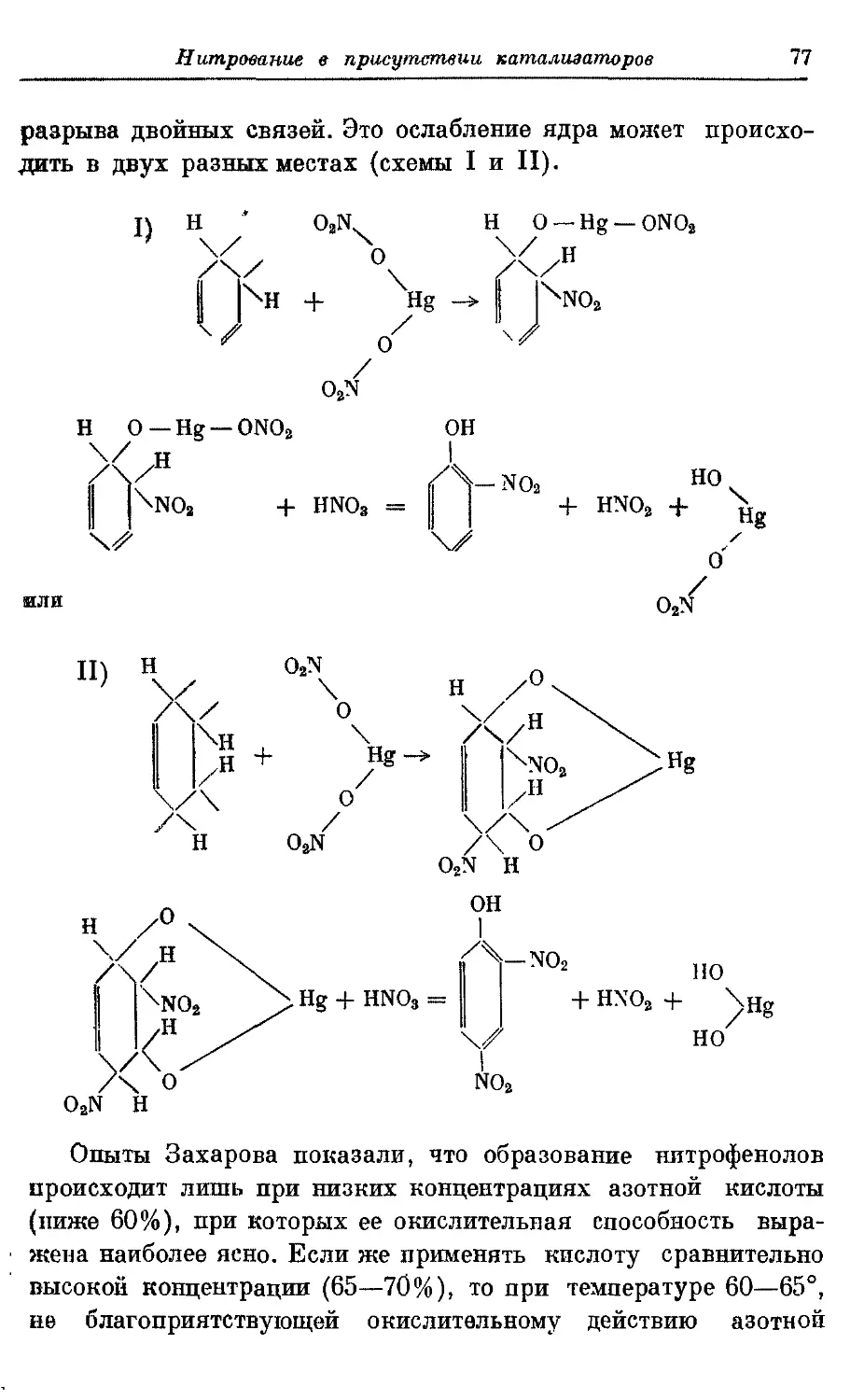











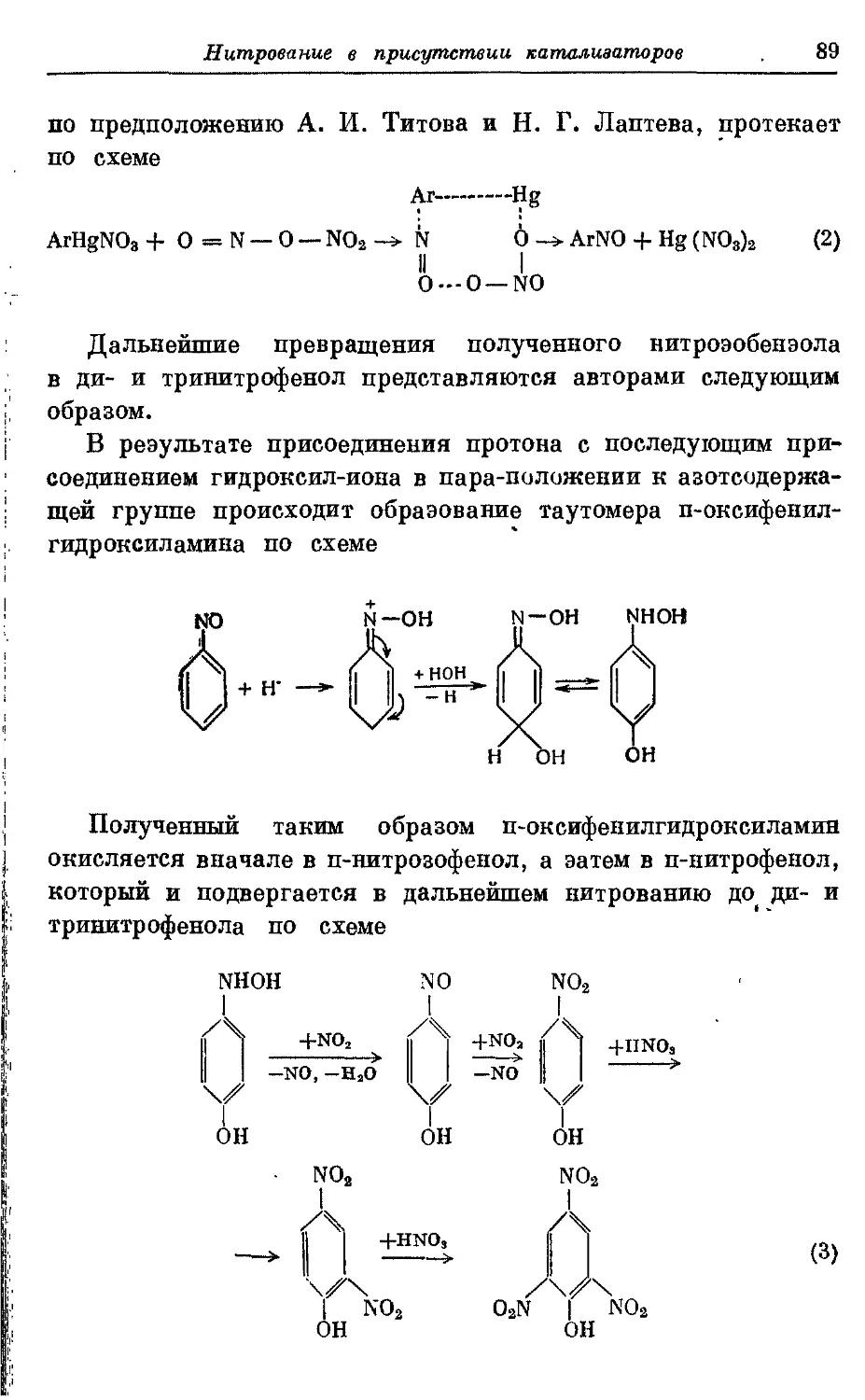

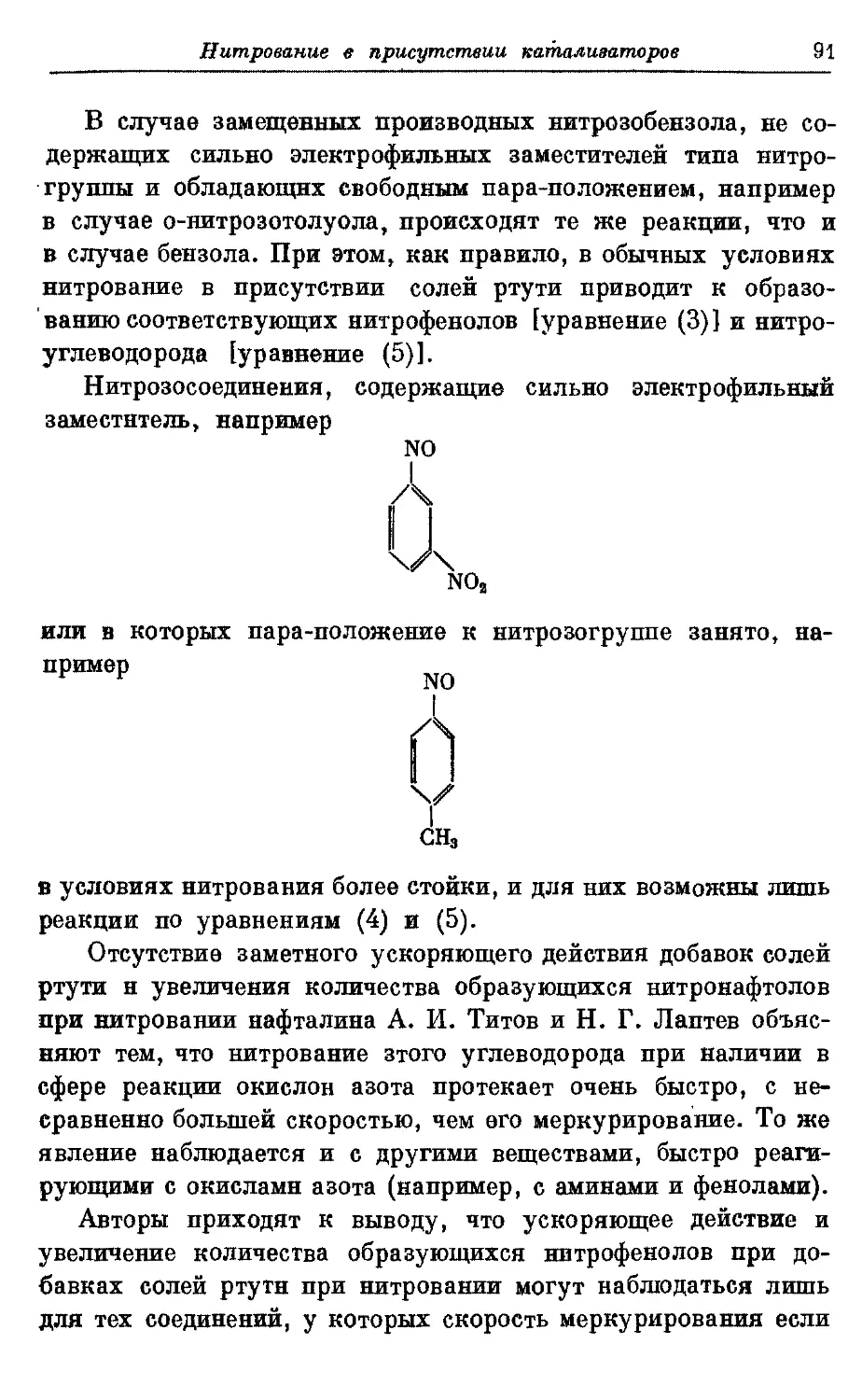
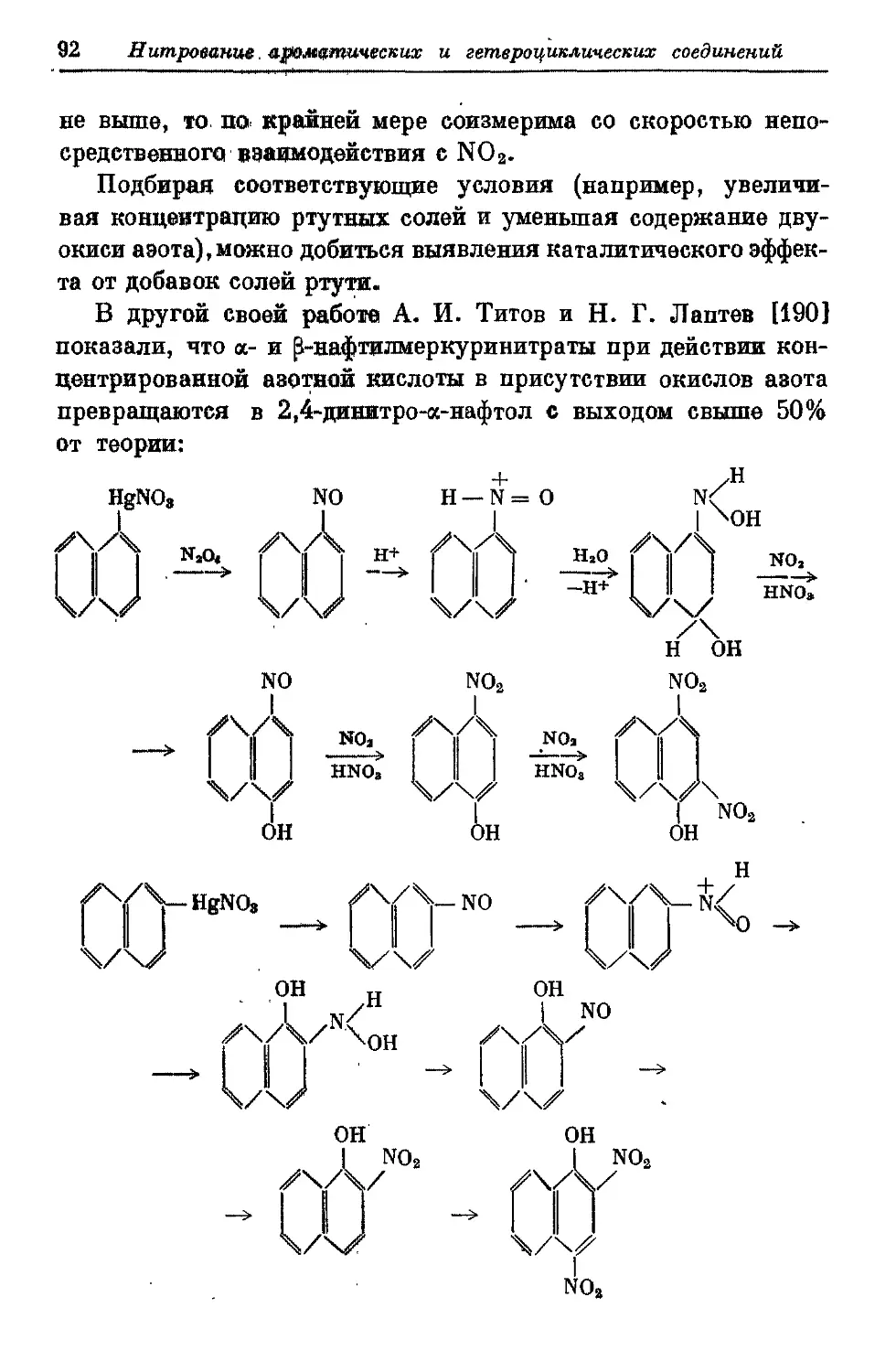




























































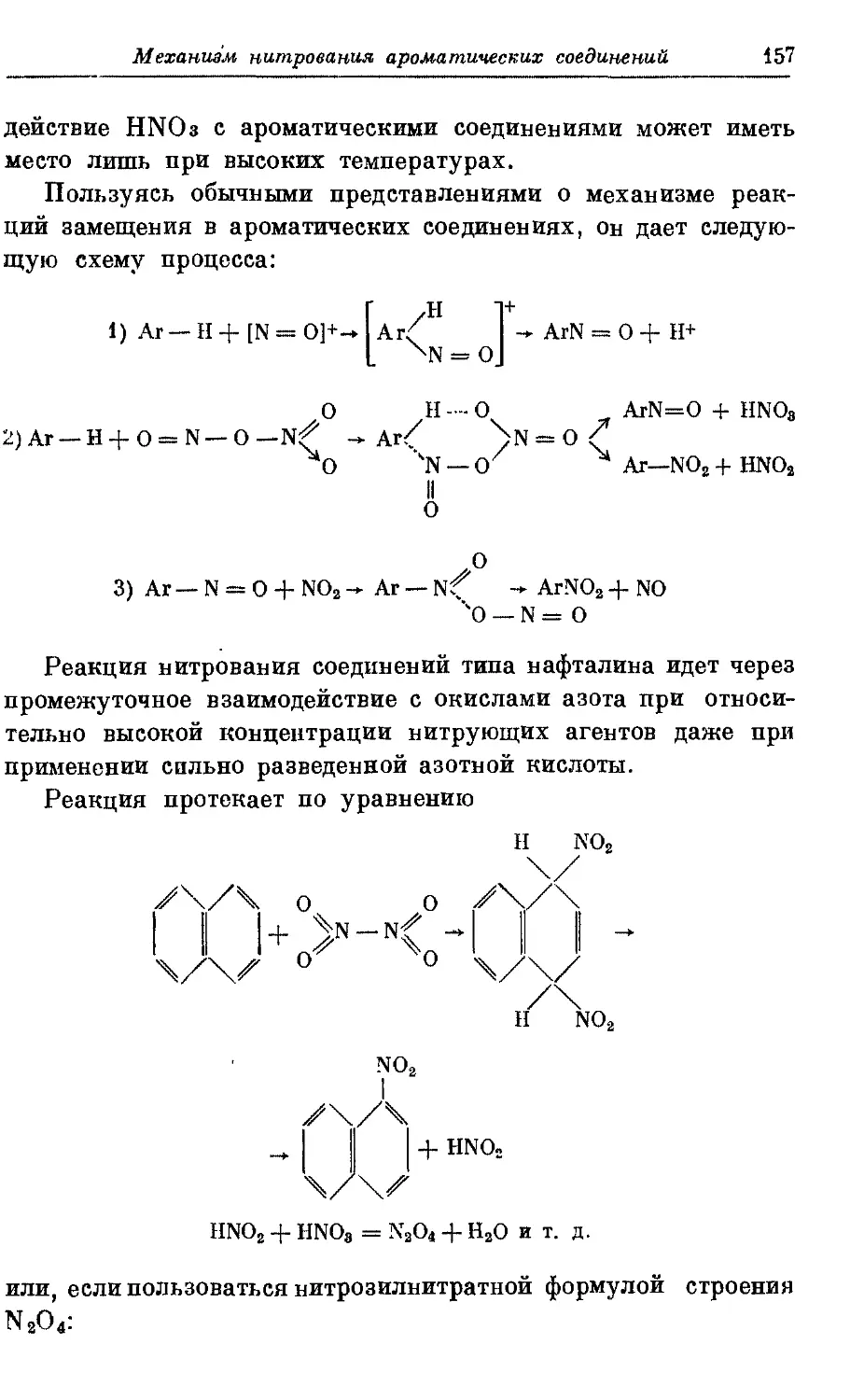
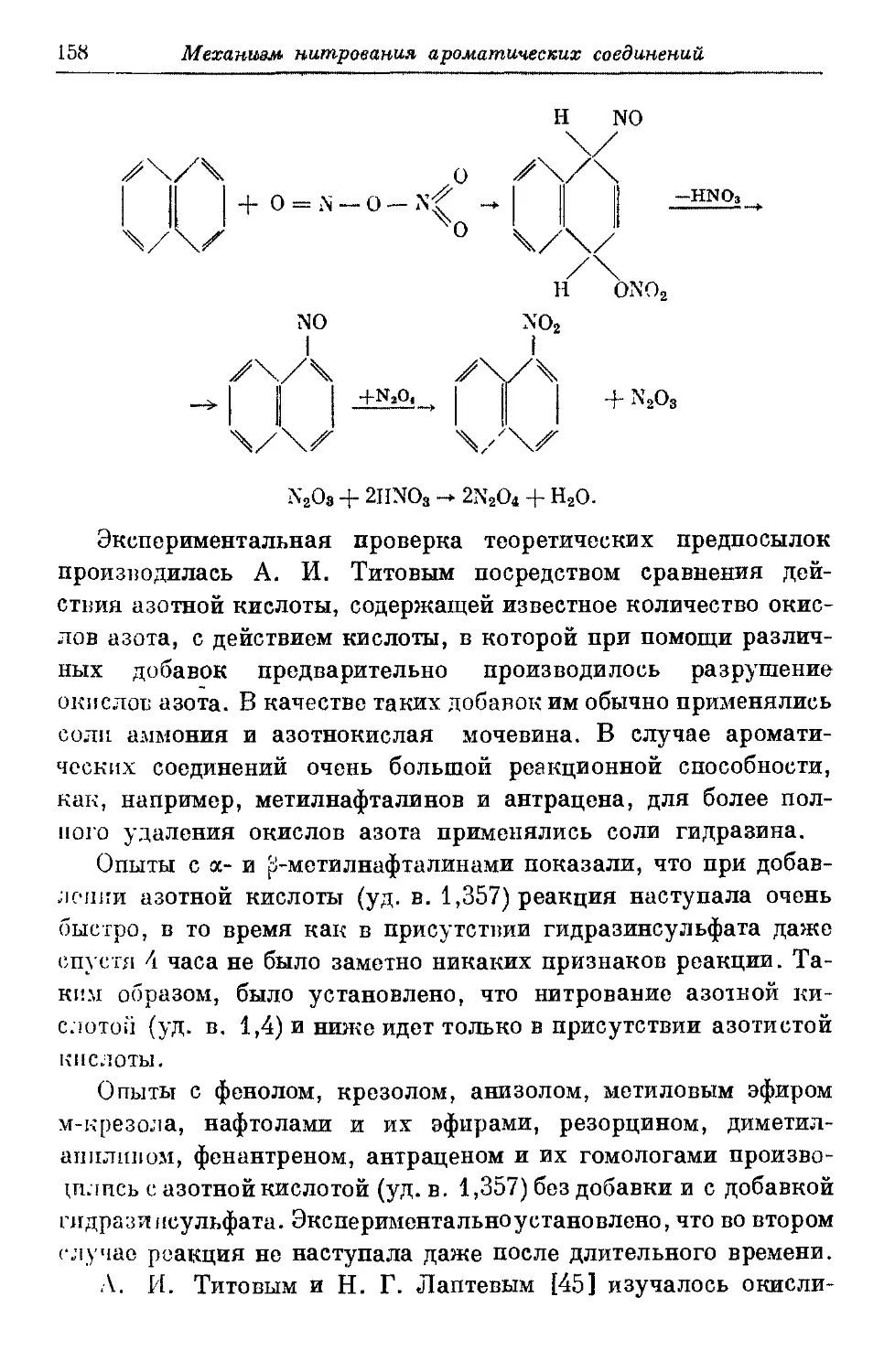

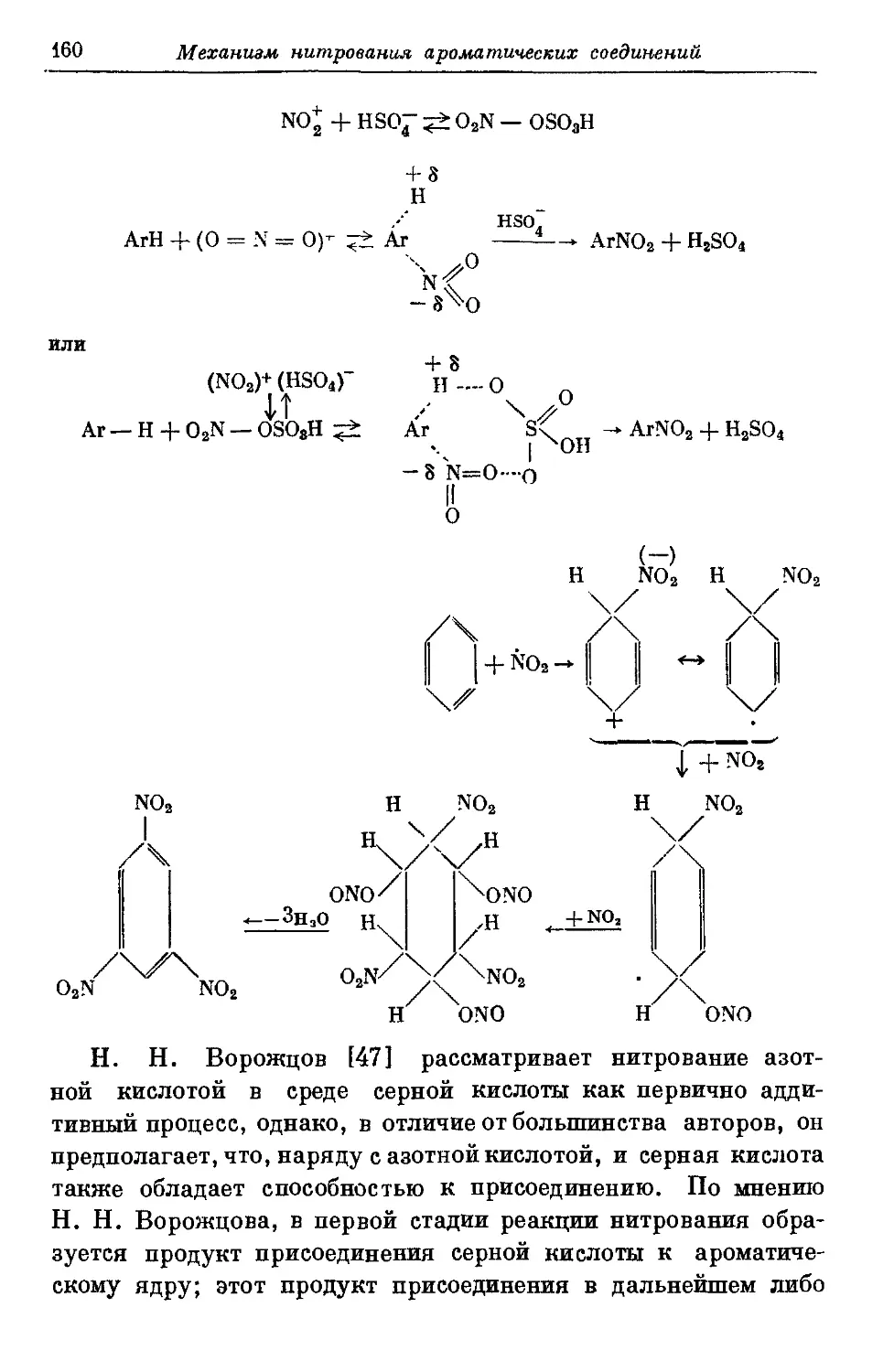
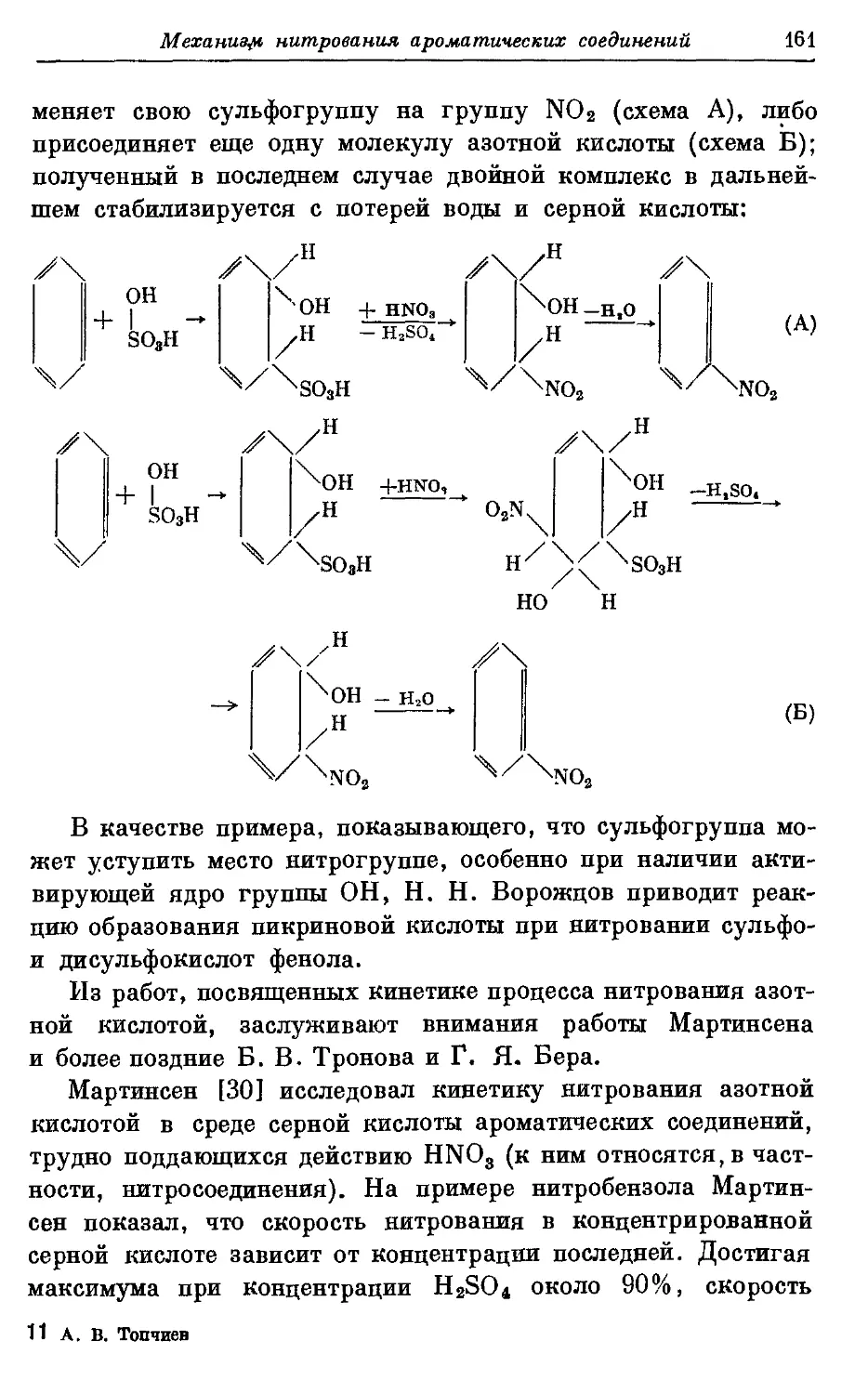








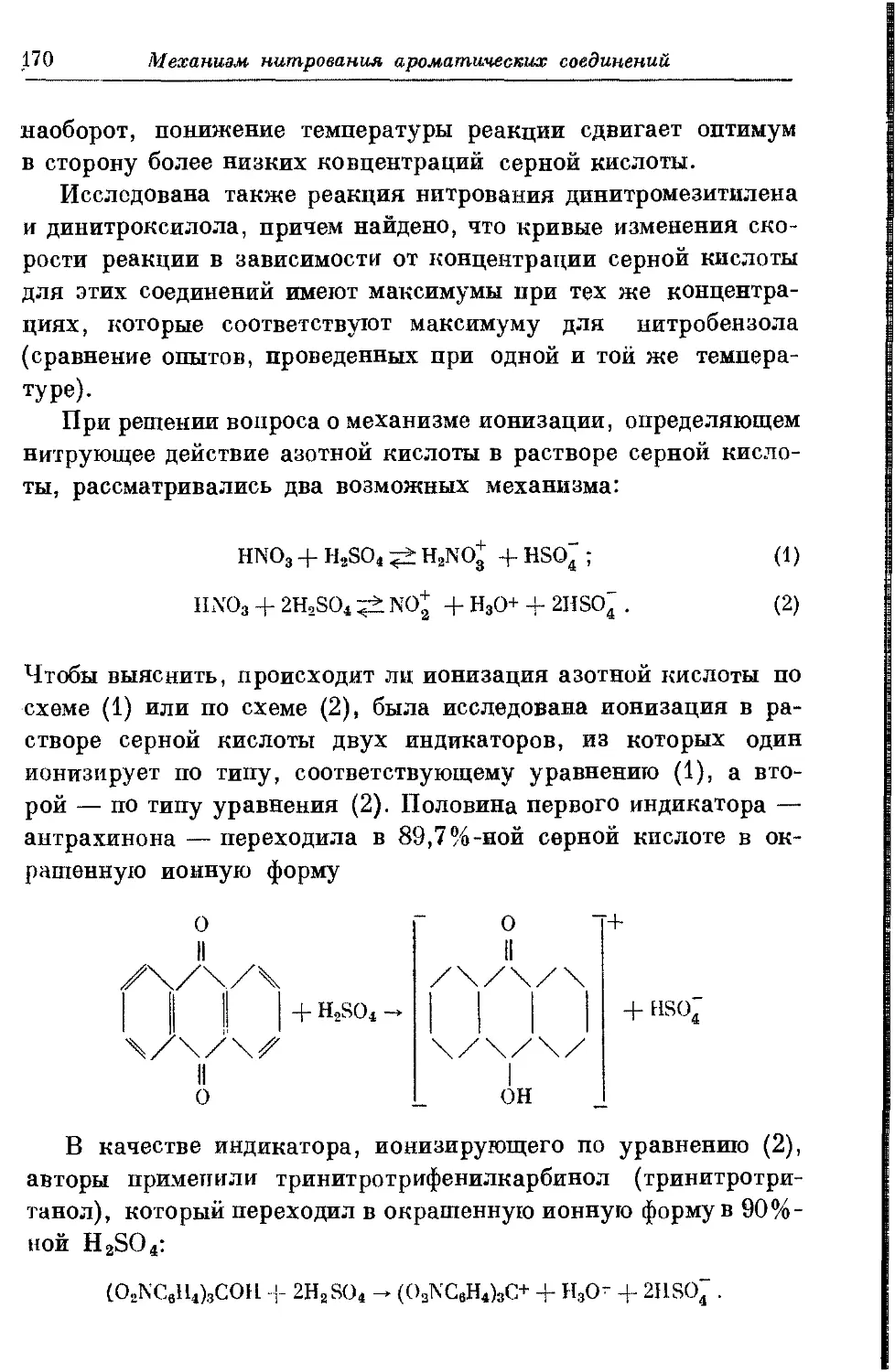




























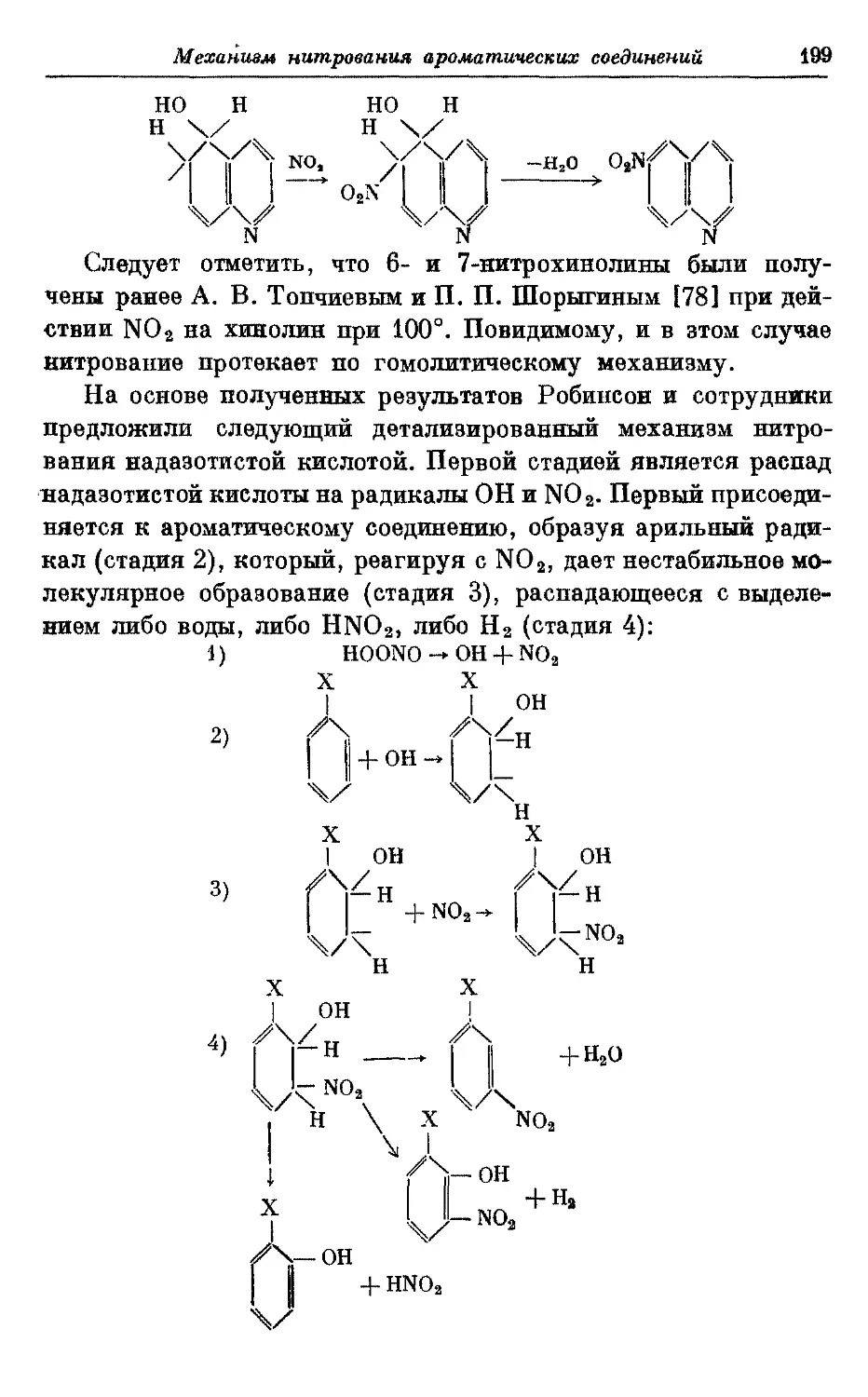






























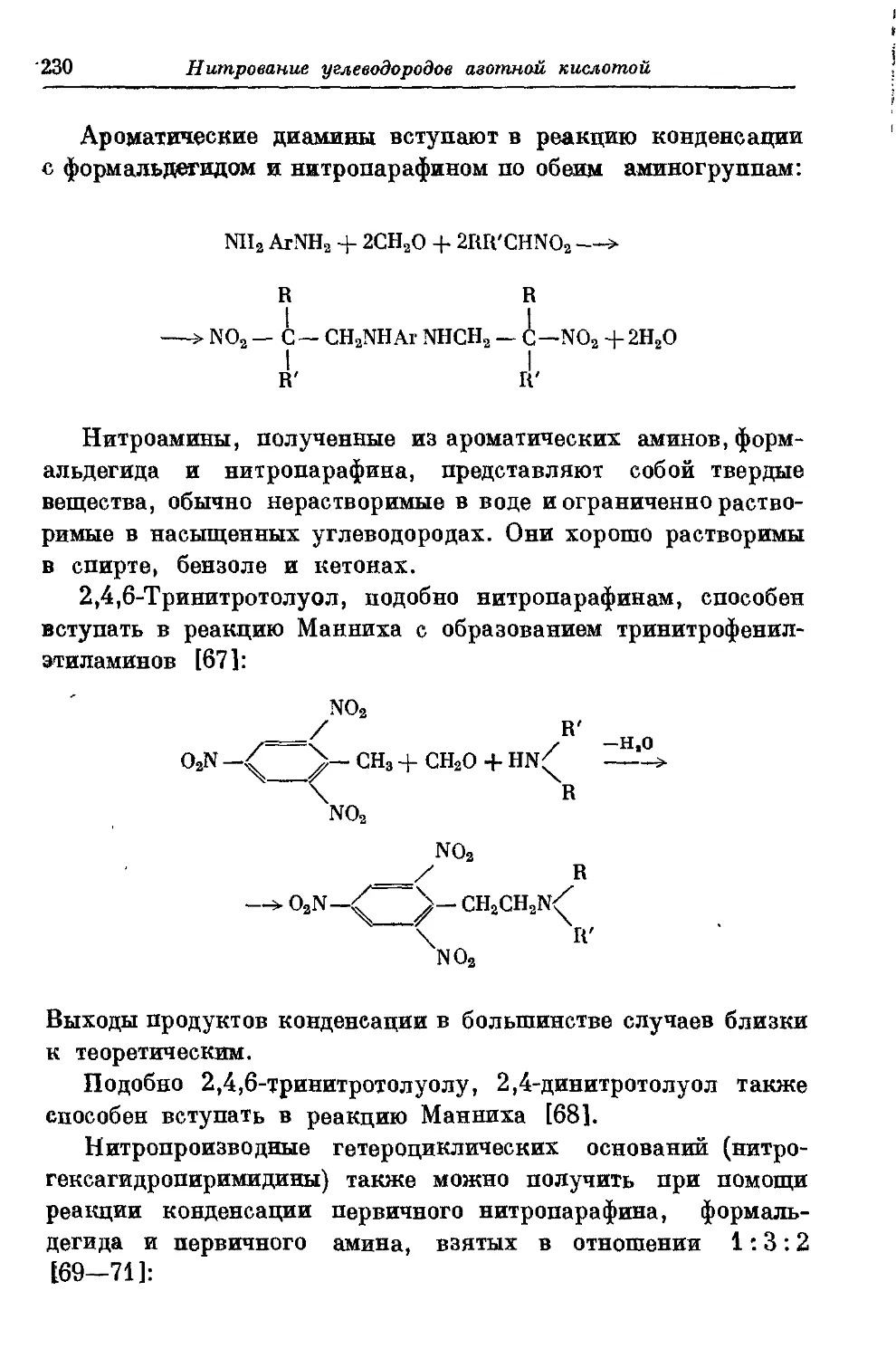



















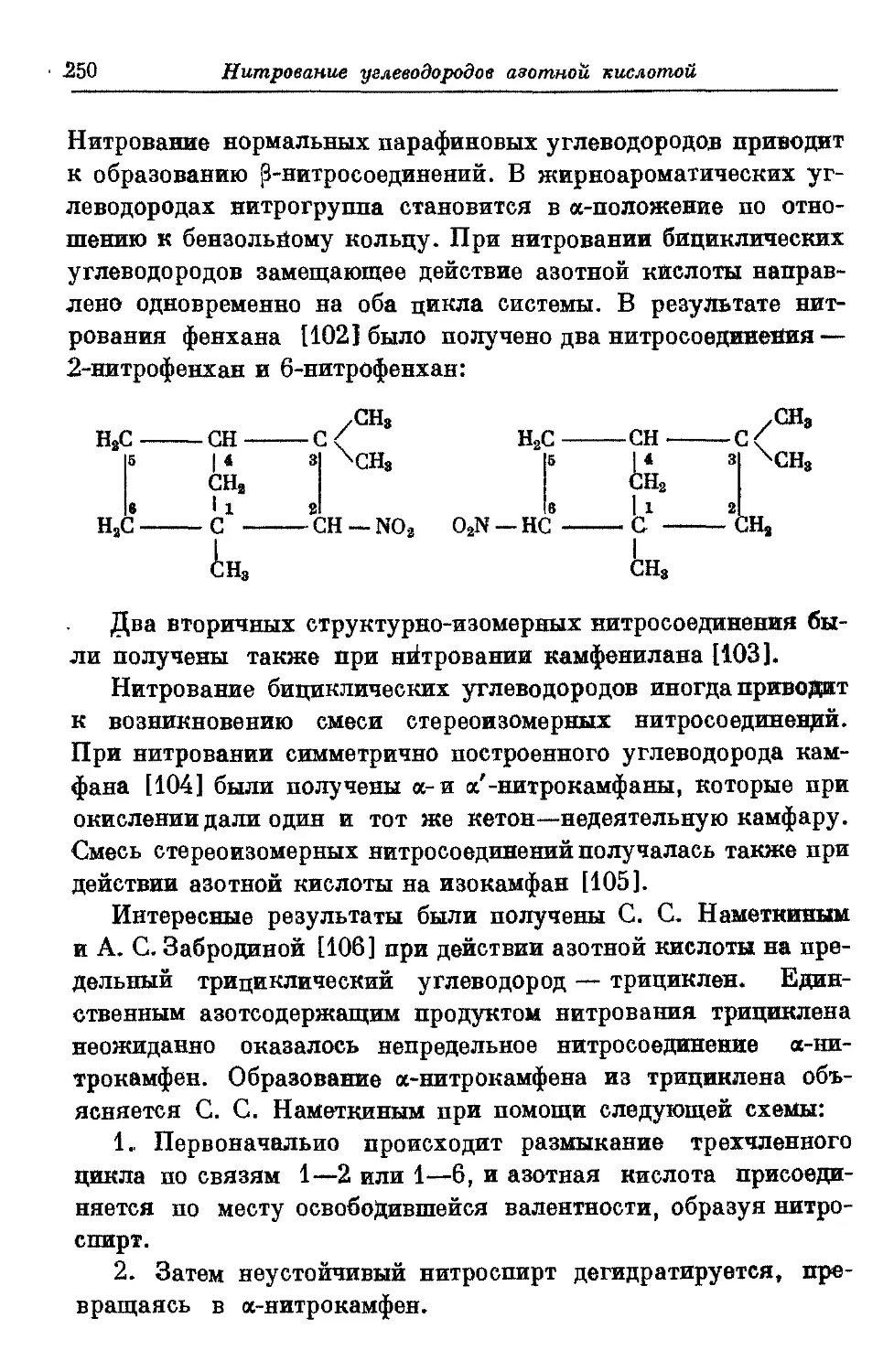
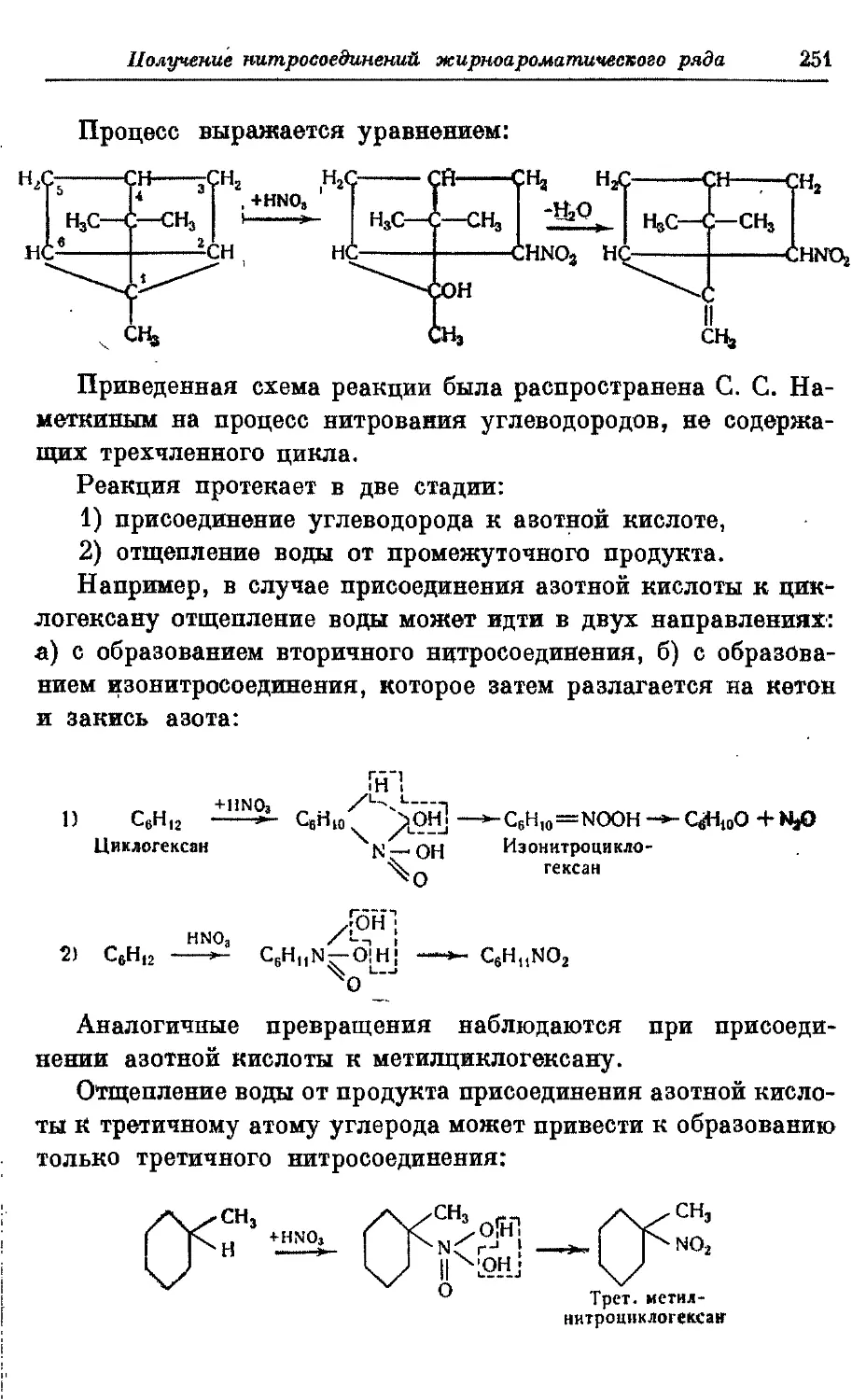















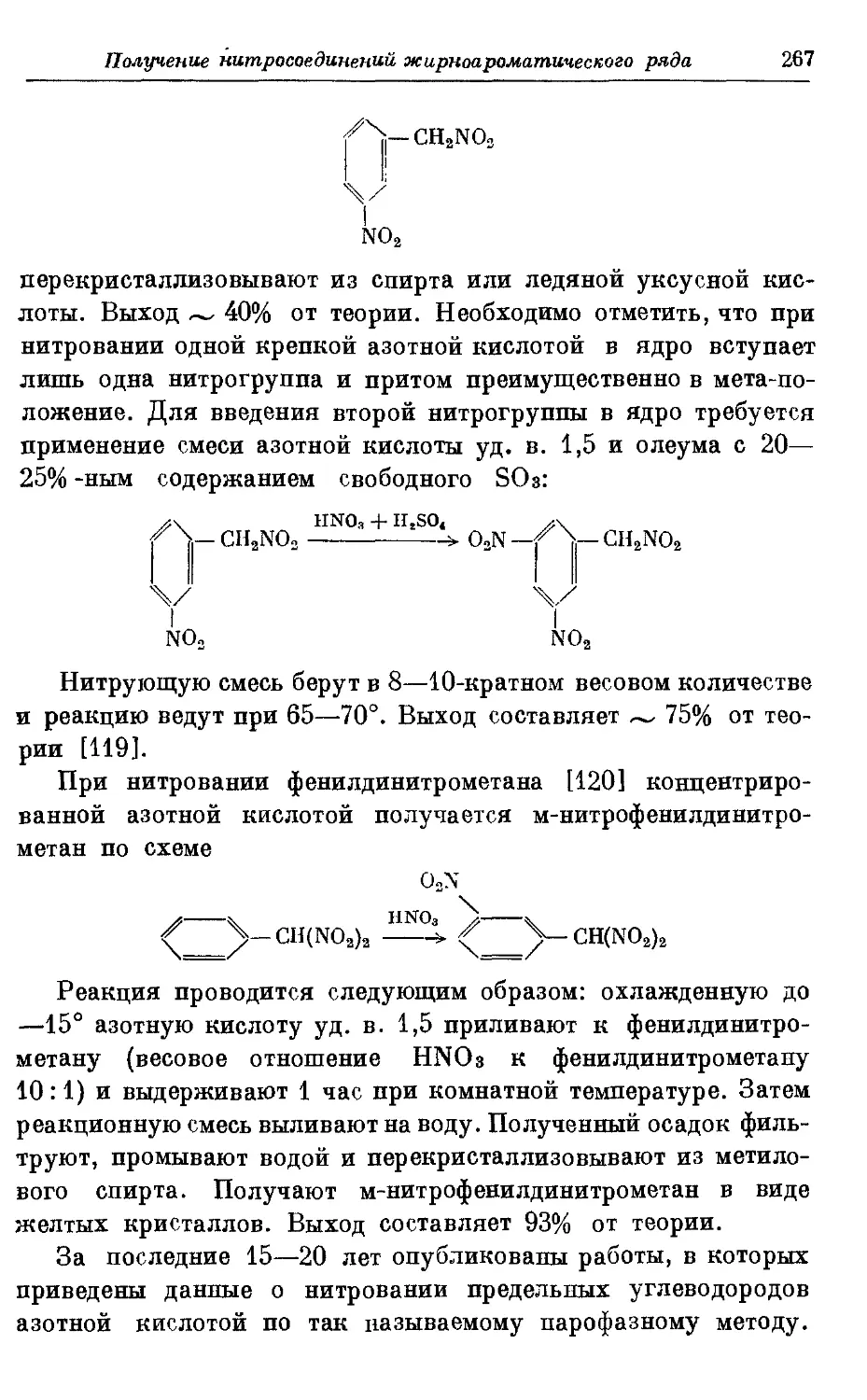


















































































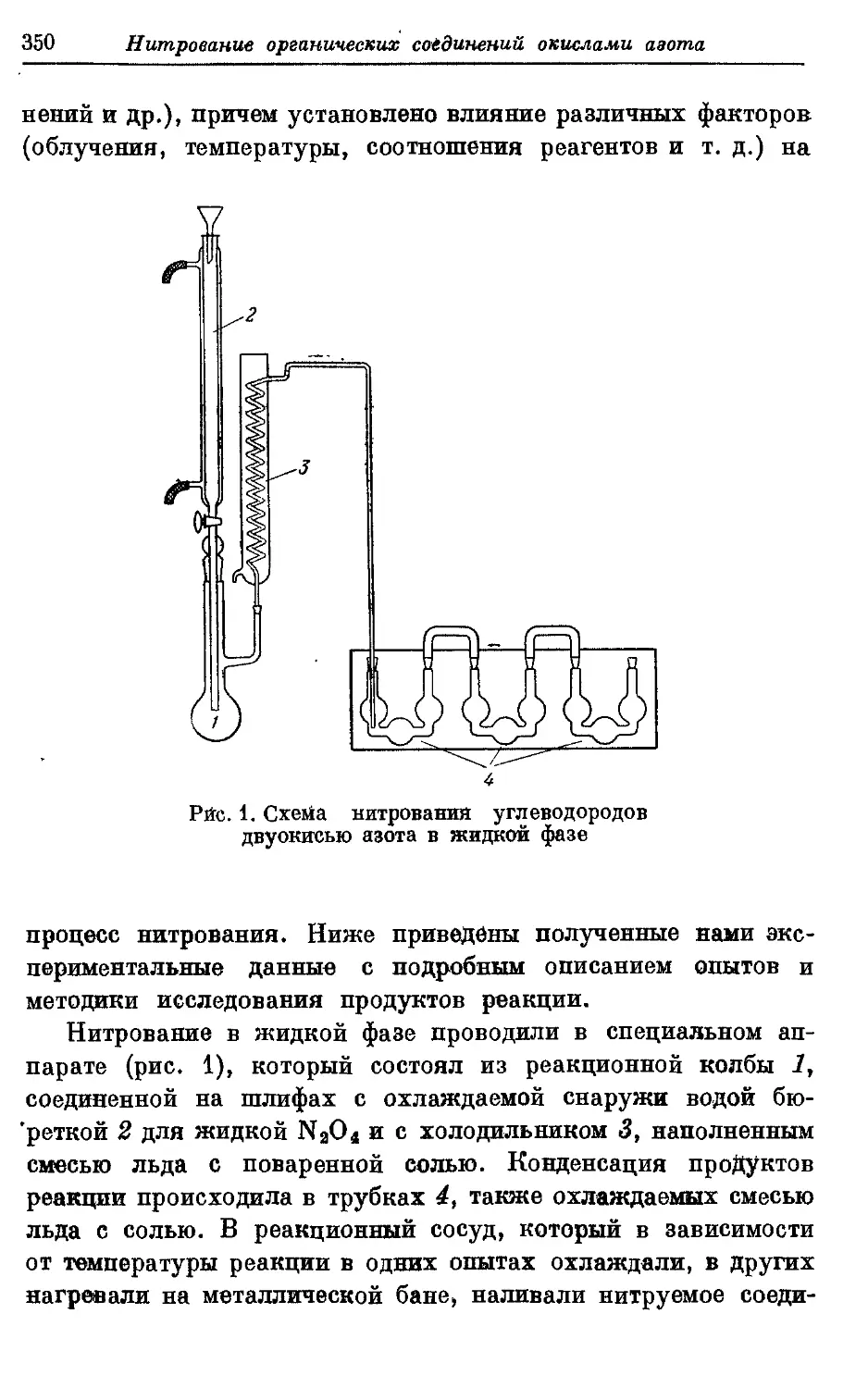

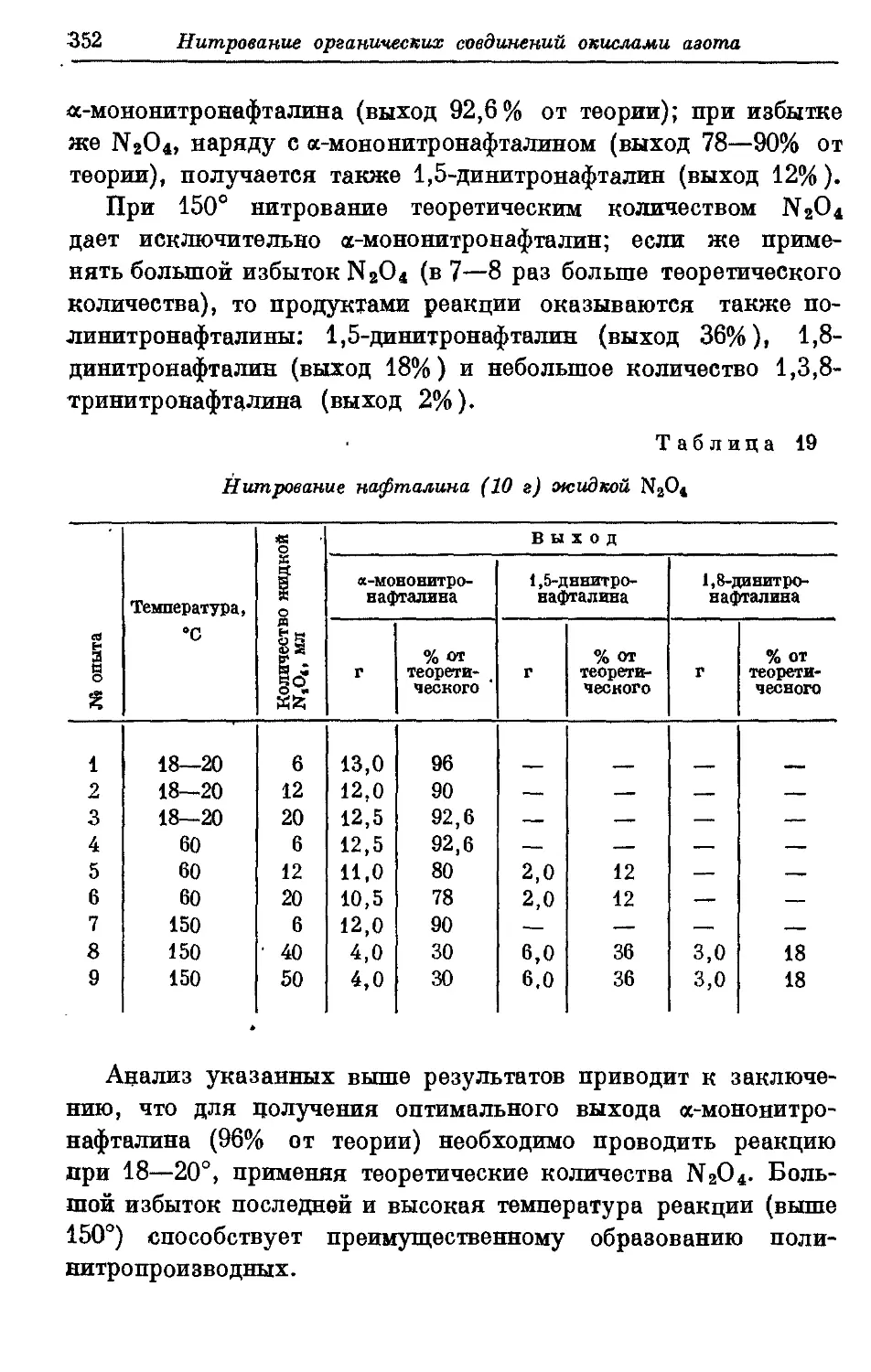









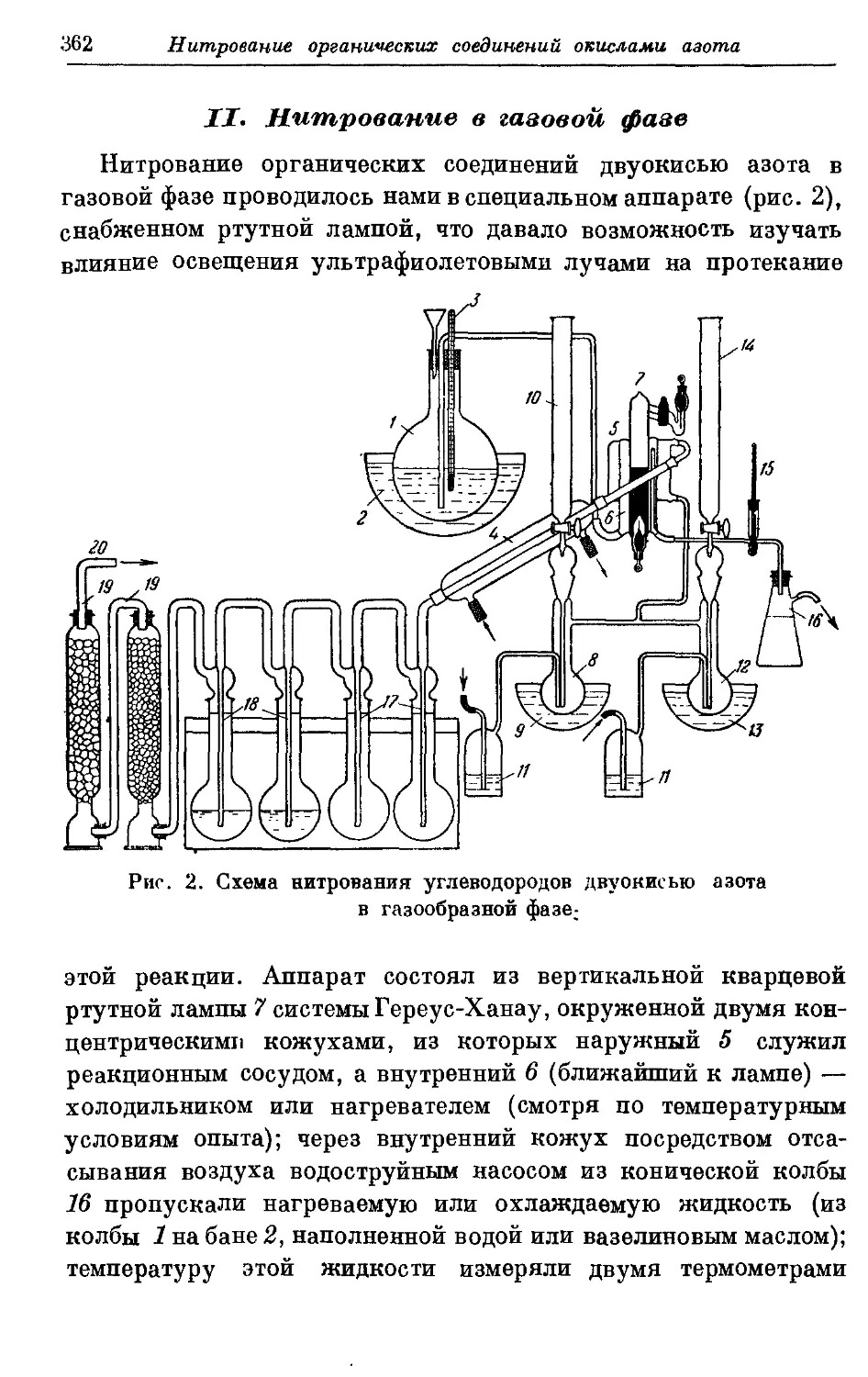





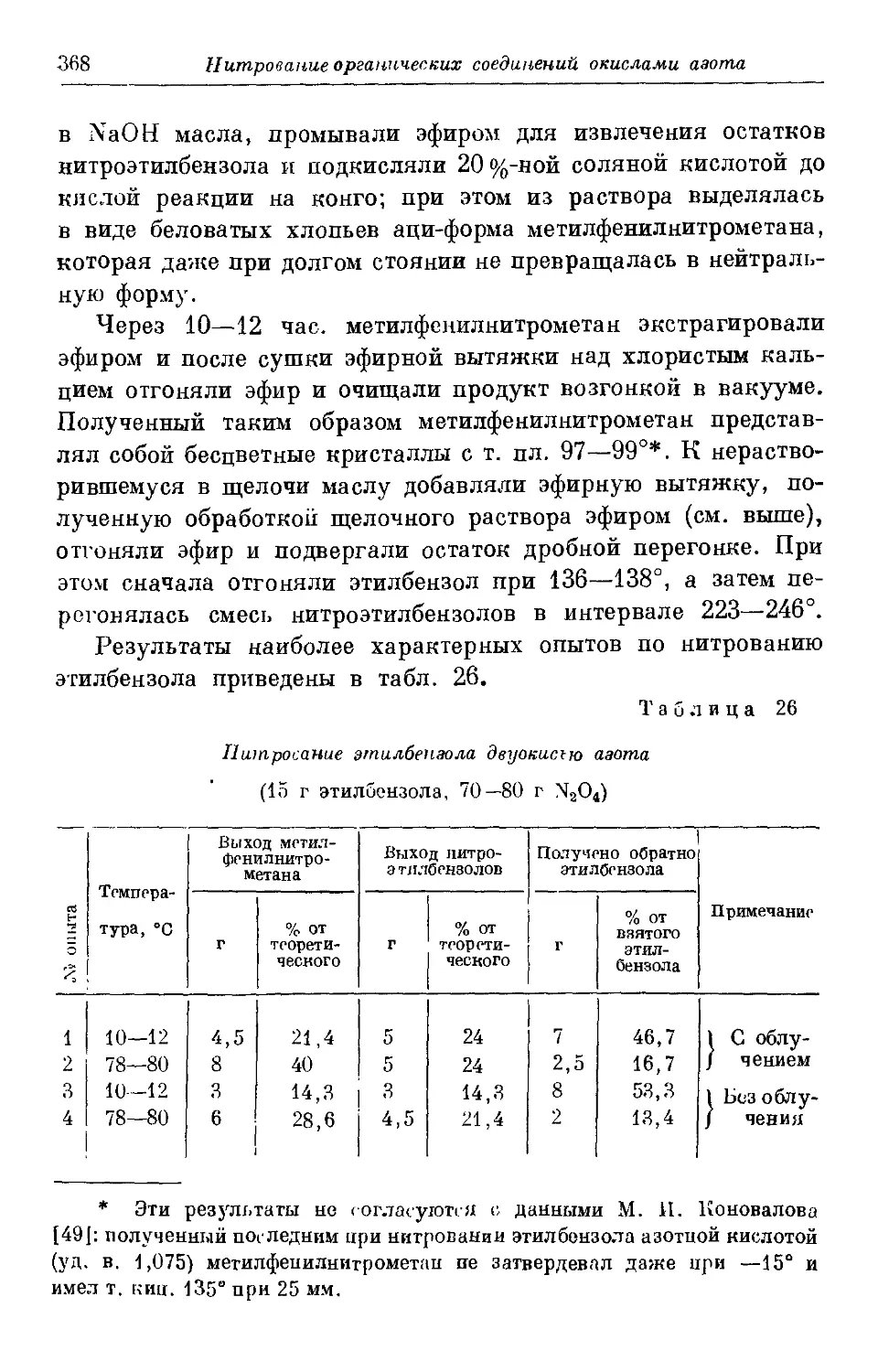





















































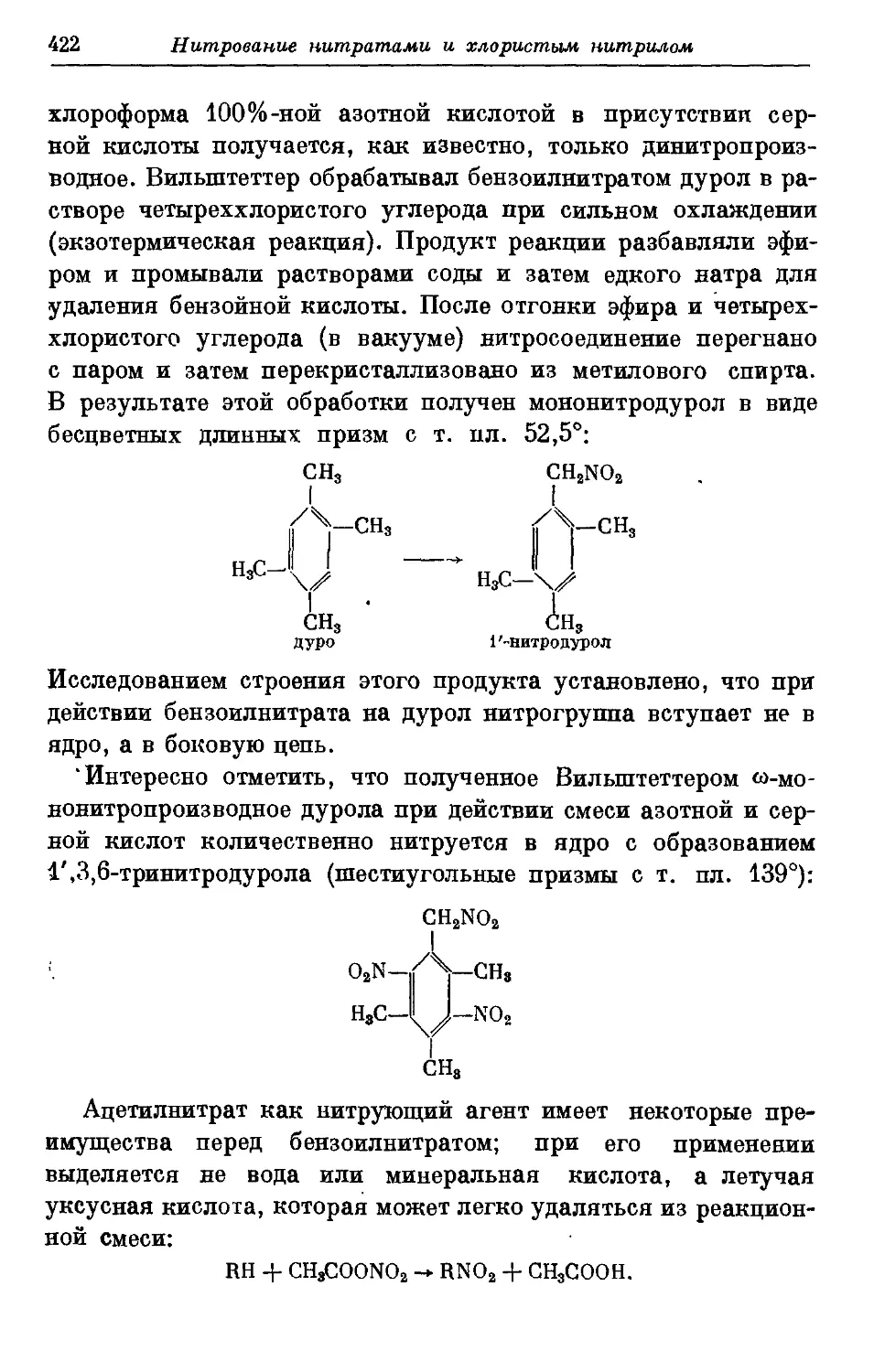

















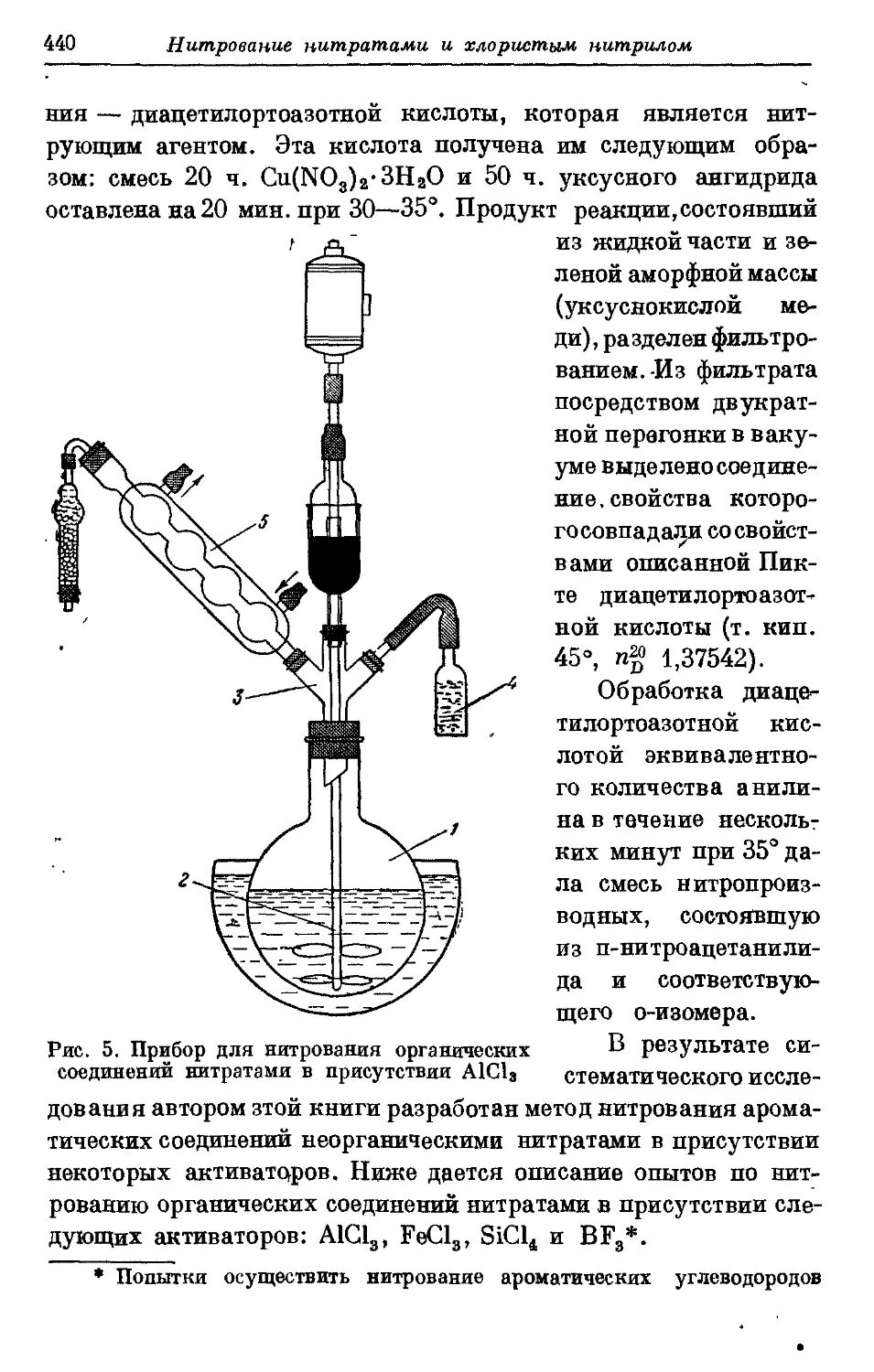



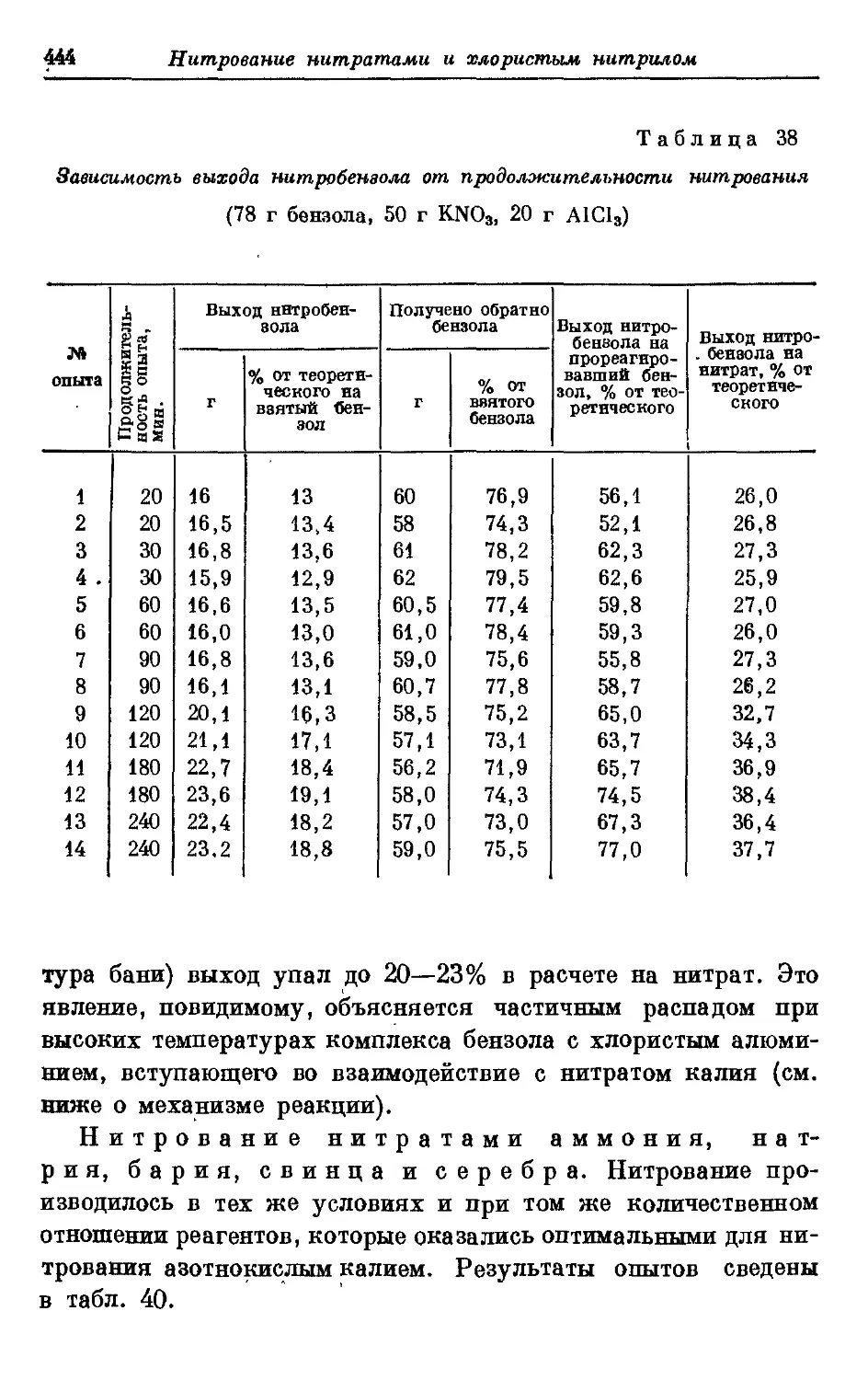

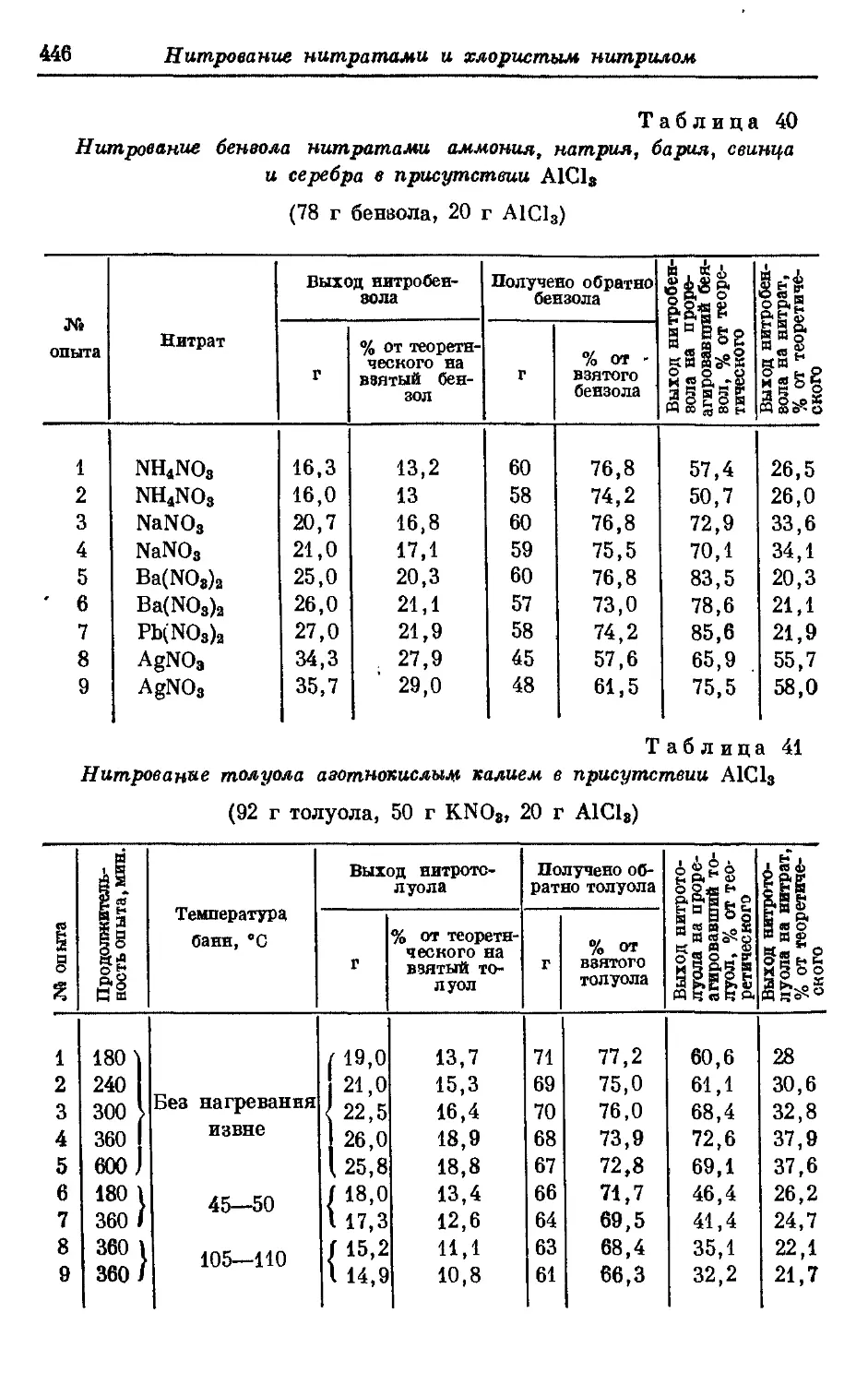



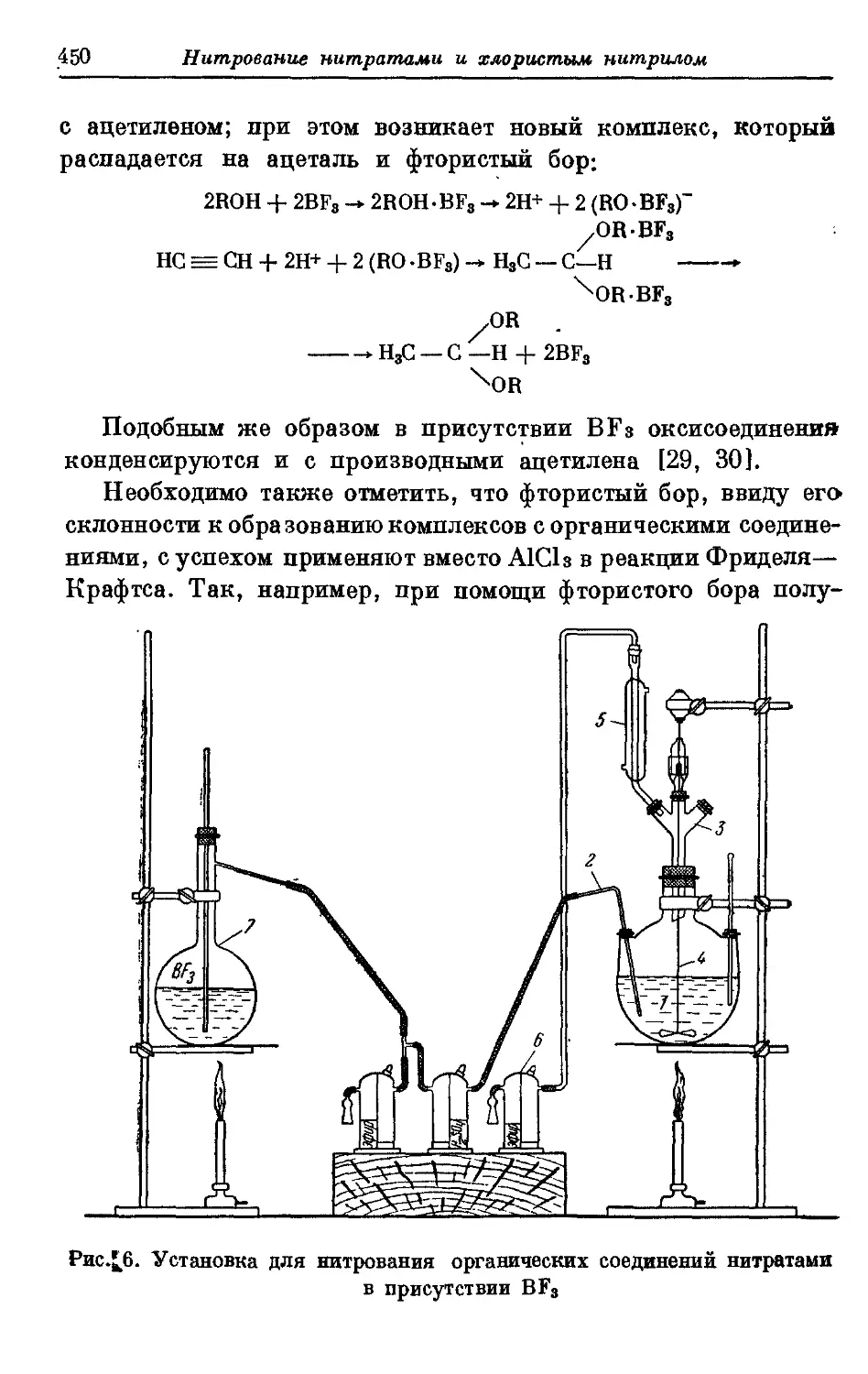


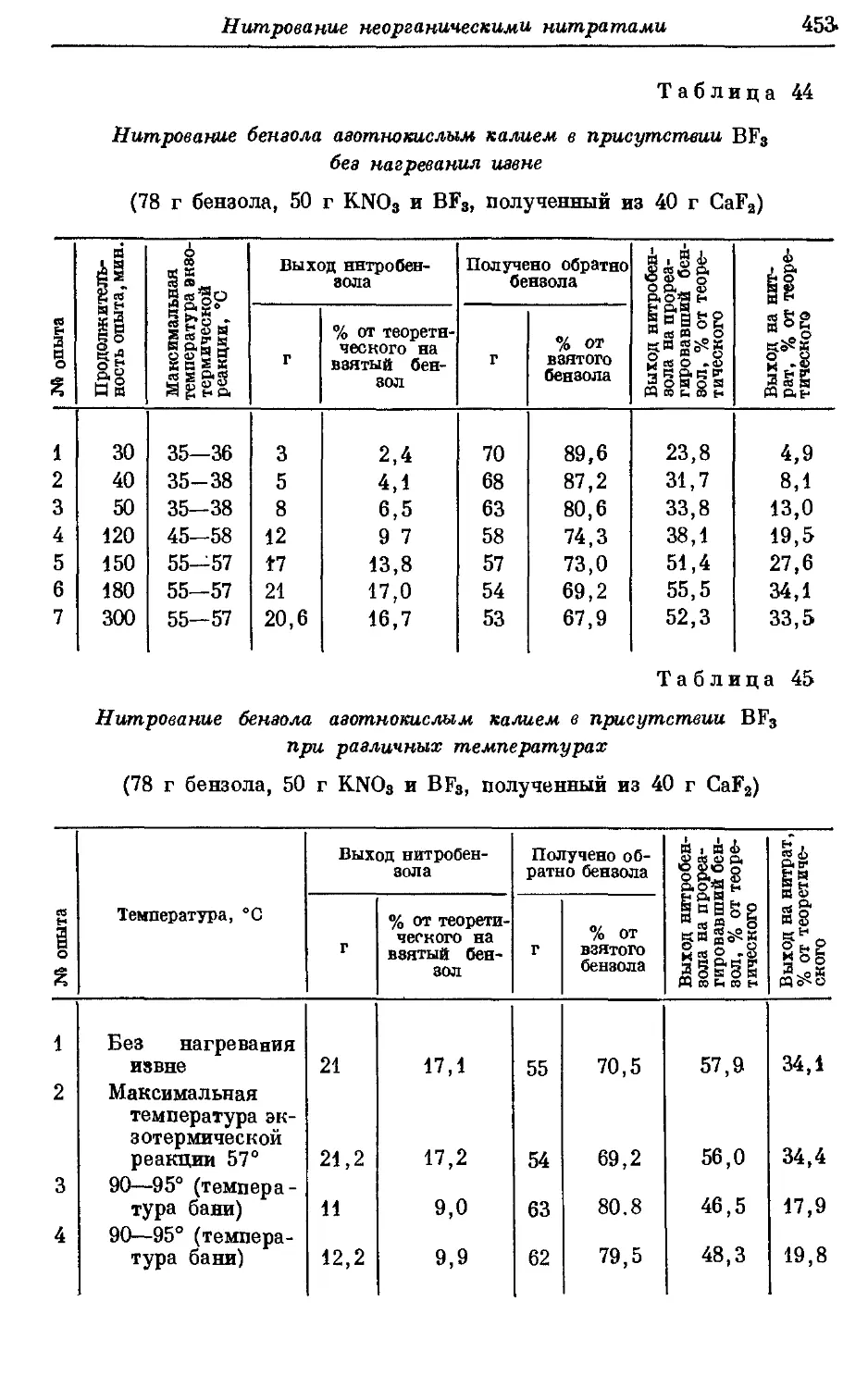







































Текст
АКАДЕМИЯ НАУК СССР
Академик
А. В. ТОПЧИЕВ
НИТРОВАНИЕ
УГЛЕВОДОРОДОВ
И ДРУГИХ
ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Дополненное
ИЗДАТЕЛЬСТВО АКАДЕМИИ НАУК СССР
М осжед— J966
ВВЕДЕНИЕ
Реакция нитрования органических соединений, открытая
в 1834 г. (Митчерлих, синтез нитробензола), является одной
яз важнейших и наиболее распространенных реакций в
органической химии.
Цель нитрования — замена одного или нескольких атомов
водорода ароматического или гетероциклического ядра, либо
предельного углеводорода одной или несколькими нитрогруп-
яами.
Нитросоединения содержат одновалентную нитрогруппу
NOg (одну или несколько), у которой атом азота
непосредственно связан с атомом углерода органического радикала.
Нитросоединения изомерны эфирам азотистой кислоты RONO, в
которых азот связан с углеродом не непосредственно, а через
кислород; например
f Н,С —О —N = 0
нитрометан метиловый эфир
азотистой кислоты
В настоящее время нитрогруппе приписывается строение
Таким образом, принимается существование в нитрогруяде
одной семиполярной связи.
Следует отметить, что такая формула не отражает
экспериментально доказанной симметричности нитрогруппы.
Равноценность обоих атомов кислорода в нитросоединениях
лучше отражает следующее строение:
Названяя нитросоединений производятся от названий
соответствующих углеводородов добавлением приставки «нитро».
Например: нитрометан, нитропропан, нитробензол, нитротолуол
и т. д. (но женевской номенклатуре положение нитрогрупцы
обозначается не буквой, а цифрой).
В зависимости от характера углеводородного радикала,
с которым связана нитрогруппа, различают первичные,
вторичные и третичные нитросоединения.
Нитросоединения играют очень важную роль в качестве
исходных и промежуточных продуктов химической
промышленности, в качестве взрывчатых веществ (например, ди-
и тринитробензол, тринитротолуол, тетранитротолуол,
пикриновая кислота и др.), в качестве душистых веществ
(например, нитробензол, тринитро-трет. бутилтолуол). Нитропроиз-
водные — необходимые промежуточные продукты в
производстве анилиновых красителей и различных синтетических
препаратов.
Нитросоединения оказывают сильное действие на
микроорганизмы (нитрофенолы [1], хлорпикрин). Хлорпикрин
применялся в качестве отравляющего вещества во время первой
мировой войны. Он является очень ценным инсектисидом и
фунгисидом и применяется для обработки зернохранилищ и
посевных материалов.
Большое количество различных нитросоединений, в том
числе нитрокрасителей, подвергалось
химико-терапевтическому испытанию при ряде трипанозомных инфекций
(возвратный тиф и др.) и в качестве дезинфекционных средств на
стафилококках и бактериях кишечной группы [2].
Некоторые ароматические оксинитросоединения могут
применяться для борьбы с вредителями сельского хозяйства.
Высокой активностью против яиц некоторых насекомых — сель-
скохозяйственных вредителей —обладает раствор дднитро-о-
циклогексилфенола в керосине [3].
Низшие нитропарафины, особенно в смеси со спиртами,
являются хорошими растворителями для эфиров целлюлозы
и винилитовых смол.
Полинитропарафины находят применение в качестве
добавок к различным видам жидкого топлива [4].
Некоторые нитроолефины являются активными фунгисидами.
Широкое применение нитросоединений для получения
различных органических веществ стало возможным лишь после
того, как выдающийся русский химик Н. Н. Зинин в 1842 г.
сделал замечательное открытие, значение которого в истории
органической химии вряд ли можно переоценить. Им было
показано, что при восстановлении сернистым аммонием
нитробензол превращается в анилин. Благодаря найденному Зини-
аым переходу от нитросоединений к первичным аминам химики
получили возможность применить нитросоединения для
синтеза самых разнообразных органических соединений.
Ароматические амины являются в настоящее время важнейшими
полупродуктами анилокрасочной, фармацевтической и многих
других отраслей промышленности органической химии, а исходя
из первичных аминов, через диазосоединения можно получить
фенолы, простые эфиры, галоидопроизводные, нитрилы и т. д.,
которые в свою очередь находят применение в разных
областях промышленности органической химии.
Для введения нитрогруппы в органические соединения
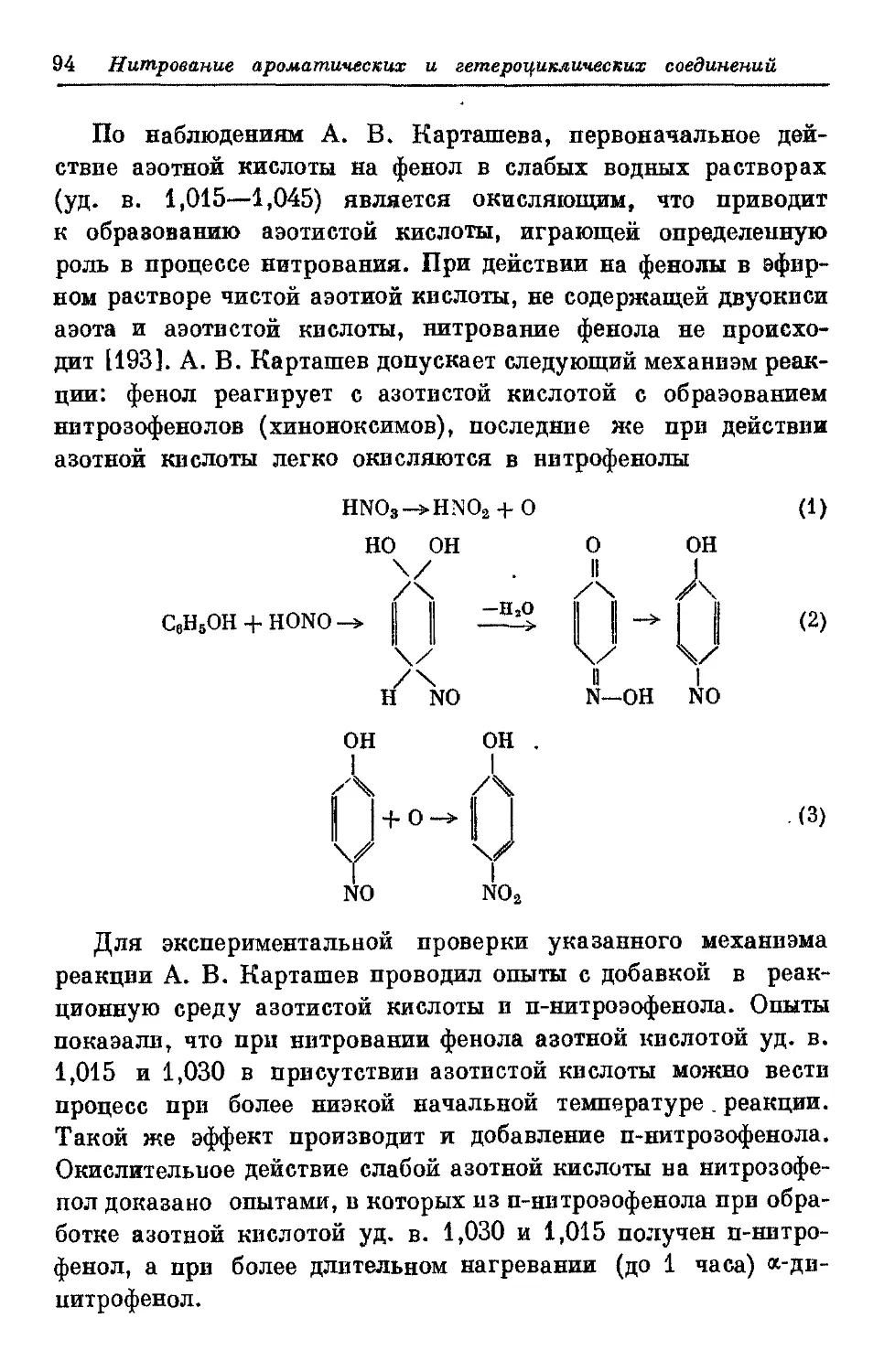
применяют главным образом азотную кислоту.
Схематически реакцию нитрования можно изобразить
следующим образом:
RH + MONO, -> RNO* +Н*О,
где RH — предельный или ароматический углеводород,
гетероциклическое соединение, либо их производные,
обменивающие свой атом водорода на нитрогрупну.
Необходимо отметить, что указанная схема не отображает
течения реакции, а дает лишь общее представление о ее
результате. В действительности реакция протекает через ряд проме-
6
жуточных стадий, при этом в реакцию с органической
молекулой нередко вступает не сама азотная кислота, а продукты
ее превращения.
Число вступающих в органическую молекулу нитрогрупп
зависит от концентрации применяемой кислоты, от
температуры и продолжительности процесса, от природы нитруемого
вещества и от других факторов. В процессе нитрования, наряду
с основной реакцией, протекают также и побочные процессы,
которые приводят к уменьшению выхода основного продукта —
нитросоединения. К числу таких нежелательных побочных
процессов относится окислительное действие азотной кислоты
на органическую молекулу. Эта окислительная способность
увеличивается при повышении температуры реакции и при
увеличении содержания воды в реакционной смеси и окислов
азота в азотной кислоте. При очень жестких условиях нитрования
может произойти даже распад органической молекулы,
например: при получении тринитробензола в качестве побочного
продукта образуется тетранитрометан.
Из приведенной выше схемы реакции нитрования видно,
что вступление нитрогруппы в органическую молекулу
сопровождается выделением молекулы воды. Накопление воды в
реакционной смеси понижает концентрацию азотной кислоты.
В случае применения в качестве нитрующего агента одной
азотной кислоты при снижении концентрации ее ниже
определенной величины реакция практически прекращается, и, таким
образом, полное использование азотной кислоты не
представляется возможным. Поэтому на практике для нитрования
применяют смесь азотной и серной кислот или так называемую
нитрующую смесь. Однако, как показано ниже, реакция
нитрования протекает с наибольшей скоростью при применении
не 100% -ной, а приблизительно 90 % -ной серной кислоты.
Это свидетельствует о том, что в процессе нитрования серная
кислота служит не только дегидратирующим агентом.
Применение вместо одной азотной кислоты нитрующей
смеси имеет большое техническое преимущество. Нитрующая
смесь не разъедает железа и ее можно длительно хранить в
железной аппаратуре и передавать по железным трубам. Необ-
ходимо отметить, что присутствие воды оказывает
значительное влияние и на самый характер действия азотной кислоты.
Как правило, чем концентрированнее применяемая азотная
кислота, тем выше ее нитрующая способность и тем в меньшей
степени проявляется ее окислительное действие. В некоторых
же случаях наблюдается обратное явление, т. е. ослабление
окислительных свойств азотной кислоты в присутствии воды.
Так, например: при действии раствора азотной кислоты в
уксусной кислоте на антрацен, наряду с 2,7-динитроантраценом,
получается и антрахинон. При этом присутствие воды
способствует образованию динитроантраценапри одновременном
уменьшении количества образующегося антрахинона. Удаление
воды из реакционной смеси (применением смеси азотной кислоты
с ледяной уксусной кислотой и уксусным ангидридом)
приводит к полному подавлению реакции нитрования и к
количественному образованию антрахинона [5].
Образование в процессе нитрования, наряду с нитросоеди-
неииями, ряда побочных продуктов отмечалось уже давно.
Обычно в результате побочных процессов при нитровании
получаются оксинитросоединения, что указывает на
одновременное вхождение в органическую молекулу окси- и нитро-
групны. Так, например, при нитровании нитробензола в
качестве побочного продукта образуется 2,4,6-тринитрорезор-
цин [6], а при нитровании нафталина в качестве примеси
образуется 2,4-динитро-*-нафтол [7 ]. При нитровании бензола
наблюдалось образование в качестве побочного продукта ди-
нитрофенола и пикриновой кислоты [8]. Беннет [6]
предполагает, что яри нитровании нитробензола сначала образуется
нитрофенол в результате взаимодействия с ароматическим
соединением не азота нитрующего агента, а его кислорода,
имеющего положительный заряд. В результате последующего
нитрования нитрофенола получается 2,3,4,6-тетранитрофенол,
в котором, как известно, нитрогруппа в положении 3
отличается большой подвижностью, и при замене ее на оксигруппу
образуется 2,4,6-тринитрорезорцин. Однако, исходя из
изложенного выше, нельзя объяснить образования м-хлорфенола
при нитровании хлорбензола и ряд других реакций.
8
Получение побочных оксисоединений (например, при
нитровании бензола), по мнению А. И. Титова [9], объясняется
первоначальным взаимодействием бензола с нитрозиясерной
кислотой с образованием нитрозобеязола. Полученный нитрозо-
бензол присоединяет протон, затем присоединяет в
пара-положение к азотсодержащей группе ион гидроксила, при этом
образуется п-оксиарилгидроксиламин. Последний окисляется
в п-нитрозофенол, а затем в п-нитрофенол, который в процессе
нитрования превращается в динитро- и тринитрофенол. Однако
этот механизм не пригоден для объяснения образования 2,4,6-
тринитрорезорципа из нитробензола. Установление механизма
образования побочных продуктов в процессе нитрования
нельзя считать окончательно установленным, и этот вопрос должен
быть предметом дальнейшего исследования.
В качестве нитрующего агента, кроме азотной и серной
кислот, в некоторых случаях применяются и другие реагенты.
Так, например, смесью азотной и уксусной кислот удобно
пользоваться в тех случаях, когда необходимо получить те или иные
мононитропроизводные, а применение концентрированной
азотной кислоты в смеси с серной кислотой приводит к более
глубокой степени нитрования, т. е. дает ди- или полинитро-
производные.
При смешении азотного и уксусного ангидрида образуется
ацетилнитрат — эфир строения CHgCOONOg, который также
применяется в качестве нитрующего агента [10]. В литературе
имеется много работ, в которых описывается применение @тил-
нитрата и бензоилнитрата в качестве нитрующих агентов. Так,
например: при действии этилнитрата в присутствии этилата
натрия на циклопентадиен получается нитроциклопентадиея[11].
Бензол энергично реагирует с этилнитратом в присутствии
хлористого алюминия. При нитровании в этих же условиях
толуола получается преимущественно о-нитротолуол
(отношение в смеси орто-изомер: пара-изомер — 5 : 1) [12].
При помощи этилнитрата в присутствии этилата натрия
можно синтезировать фенилнитрометан из цианистого
бензила [13]. В лабораторной практике в качестве нитрующего
средства находит применение бензоилнитрат. Посредством это-
9
го реагента удается получить о-нитропроизводные втех случаях,
когда другими способами получаются преимущественно
соответствующие п-производные. Так, при взаимодействии бензоил-
нитрата с анизолом и фенетолом получается с теоретическим
выходом о-нитроанизол или,соответственно, о-нитрофенетол [14].
Интересно отметить, что полиалкилзамещенные бензолы
при взаимодействии с бензоилнитратом образуют с
хорошим выходом соответствующие нитропроизводные, в которых
нитрогруппа содержится не в ядре, а в боковой цепи;
например: дурол превращается в 1'-витродурол [15].
В последнее время появились работы [15а] по
нитрованию некоторых органических соединений ацетоциангидрин-
нитратом в щелочной среде.
Вместо обычной нитрующей смеси (смесь азотной и серной
кислот) для нитрования может быть применена смесь серной
кислоты с солью азотной кислоты (обычно с натриевой
селитрой). Процесс проводится при нагревании; при этом серная
кислота выделяет из селитры свободную азотную кислоту.
Неудобство применения такой смеси состоит в том, что при этом
в качестве отхода образуется значительное количество
бисульфата, который не находит широкого применения.
При замене натриевой селитры на аммиачную в качестве
отхода получается сульфат аммония, который может быть
использован как удобрение. Однако, благодаря высокой
стоимости аммиачной селитры, применение ее в качестве
нитрующего агента весьма ограничено.
Автором настоящей книги исследовалось применение
фтористого бора в качестве катализатора при нитровании
органических соединений солями азотной кислоты. На основе этих
исследований выдвинута теория механизма нитрования
ароматических соединений неорганическими нитратами в
присутствии AlClg и BFg.
Давно известно каталитическое влияние ртути или ее солей
при сульфировании ароматических соединений. Однако первые
опыты по изучению влияния ртутных солей при нитровании
не дали положительных результатов. Но уже в 1913 г. Воль-
фенштейн и Бетерс [16] нитрованием бензола разбавленной
10
азотной кислотой в присутствии солей ртути получили
нитробензол и нитрофенол. При этой реакции одновременно идут
процессы нитрования и окисления. Течение реакции зависит
от концентрации азотной кислоты.
Катализаторами при реакции нитрования могут служить
также соединения хрома, вольфрама, молибдена, тантала,
ниобия, ванадия, галлия и индия [17]. Особо следует
отметить применение фтористого бора как катализатора при
реакции нитрования ароматических соединений [18].
В некоторых случаях нитрования азотистая кислота также
может обладать каталитическим действием. Так, например,
при нитровании фенола [19], алифатических кетонов [20],
нафталина [21 ] азотистая кислота ускоряет реакцию.
Иногда нитрование приходится проводить в среде
растворителя. В технике в качестве растворителей при нитровании
применяется, в частности, о-дихлорбензол или хлорбензол.
В качестве примера можно привести 3-нитроализарин, который
получают нитрованием ализарина азотной кислотой в среде
о-дихлорбензола. В качестве растворителей в процессе яитро-
вания могут применяться и хлорпроизводные этана. При ни-
троваяии фенола в пикриновую кислоту может быть применен
этиловый спирт [22] или, еще лучше, метиловый спирт [23],
который значительно устойчивее к действию азотной кислоты.
На примере нитрования фенола в среде различных
растворителей было показано влияние растворителей на ход
процесса [24].
Температура нитрования — также весьма важный фактор,
влияющий на ход процесса. Нитрование является реакцией
экзотермической, а в случае применения смеси азотной и
серной кислот (при нитровании ароматических соединений)
большое количество тепла выделяется дополнительно в результате
разбавления серной кислоты водой, образующейся в процессе
реакции. Поэтому, как правило, при аитрованип
ароматических соединений приходится прибегать к внешнему
охлаждению и постепенному смешиванию реагентов. Изменение
температуры нитрования оказывает влияние как на количество
вступающих в молекулу нитрогрупп, так и на место вступления
нитрогрупны. Само собой понятно, что для каждой отдельной
реакции нитрования существует своя оптимальная
температура.
В то время как химия нитросоединений ароматического
ряда успешно развивалась и нашла широкое
промышленное применение уже много лет назад, удобные методы
получения нитросоединений жирного ряда разработаны лишь в
последние десятилетия. -
Отсутствие до недавнего времени удобных и дешевых
методов получения нитросоединений жирного ряда было
основным препятствием широкого внедрения этого класса
соединений в практику. Причиной этих трудностей является
большая инертность парафиновых углеводородов по сравнению
с ароматическими углеводородами к действию азотной кислоты.
В течение многих лет попытки ввести нитрогруппу в
ациклические углеводороды прямым действием азотной кислоты не
давали положительных результатов (это также относится к
нитрованию боковой цепи ароматических углеводородов). Однако
широкая доступность парафиновых углеводородов (особенно
СССР богат естественными газами, которые и представляют
источники низших парафиновых углеводородов) заставила
многих химиков обратиться к изучению вопроса переработки
предельных углеводородов в нитропарафины. Этот класс
соединений может быть использован в различных областях
химической промышленности. Кроме того, нитропарафины
являются весьма реакционноспособными веществами, и на
их основе можно синтезировать многие новые, весьма ценные
химические продукты, из которых некоторые уже нашли себе
применение.
Нитроспирты — продукты конденсации нитропарафинов
с альдегидами — имеют большое практическое применение. Их
производные и продукты взаимодействия с различными
реагентами применяются как пластификаторы для эфиров
целлюлозы и могут употребляться в качестве взрывчатых веществ [25]
и др.
Галоидопроизводные нитропарафинов применяются как
инсектисиды и как растворители для очистки смазочных масел.
12
Для осуществления синтеза яитропарафинов приходилось
прибегать к обходным путям, иногда довольно сложным.
В 1872 г. В. Мейер впервые осуществил синтез нитропара-
финов действием солей азотистой кислоты на галоидные ал-
килы
CgHsONO Ч- AgJ
C*H,J + AgNO*/
C±H*NO% 4- AgJ
Одновременно с нитропарафином получается эфир
азотистой кислоты.
В конце прошлого столетия крупному русскому химику
М. И. Коновалову [26] впервые удалось показать возможность
введения нитрогрупны в углеводороды жирного ряда и в
боковую цепь жирноароматических углеводородов прямым
нитрованием азотной кислотой. На основе этих работ и возникли
современные методы промышленного нитрования парафиновых
углеводородов, которые привели к созданию новой отрасли
промышленности органической химии — производству нитро-
парафинов и их многочисленных производных.
М. И. Коновалов установил ряд закономерностей хода
реакции нитрования парафинов. Он указал на значение для
процесса нитрования концентрации азотной кислоты,
температуры реакции, давления. Работами Коновалова было
установлено, что изопарафины нитруются легче, чем парафины яор-
мального строения, что увеличение давления способствует
повышению выхода нитропродуктов, а повышение температуры
увеличивает выходы первичных нитропродуктов по отношению
ко вторичным. При нитровании пропана, бутанов, пентанов
получаются смеси изомерных нитросоединений со
значительным (иногда больше 50%) содержанием вторичных
нитросоединений.
Работы М. И. Коновалова послужили стимулом к изучению
парафинов, олефияов и нафтенов. Реакцией Коновалова часто
пользуются для характеристики отдельных продуктов,
входящих в состав нефти при процессах ее переработки.
Метод М. И. Коновалова был в дальнейшем подробно изучен
другим русским ученым В. В. Марковниковым [27], который
13
установил зависимость реакции нитрования от концентрации
азотной кислоты и других условий. Марковдиков использовал
реакцию нитрования предельных углеводородов для
определения состава нефти но характеру нитропроизводных.
При действии азотцой кислоты на предельные
углеводороды, как показал С. С. Наметкин с сотрудниками [28], в
первоначальной стадии получается изонитросоединение, которое,
в зависимости от условий реакции, может либо превращаться
в устойчивую форму нитросоединений, либо разлагаться
с образованием альдегида, кетона, карбоновых кислот:
ОН О
2ВСН = л/ -»2RC^ +H*O + N*O
О Н
Исследование С. С. Наметкиным и его сотрудниками
действия азотной кислоты на октадекан С^Щ* и гексатриаконтан
CggH*, подтвердило выводы, сделанные Коноваловым.
Единственным продуктом реакции нитрования октадекана оказался
вторичный нитрооктадекан СНзССНаХбСЩМОаУЗНз, а при
нитровании гексатриаконтана была получена смесь
вторичного мононитропродукта со вторичным р,р'-динитропродук-
том [29].
При нитровании азотной кислотой бициклических
углеводородов предельного характера получаются структурно
изомерные нитросоединения со вхождением нитрогруппы в
различные циклы системы. Например, при нитровании фенхааа
получаются 2-нитрофенхан и 6-нитрофенхан [30].
Вопрос о механизме реакции нитрования уже давно является
предметом многочисленных исследований.
Интерес различных исследователей к вопросу о механизме
нитрования несомненно связан с огромным значением
нитросоединений в различных отраслях промышленности и со
стремлением при помощи теории процесса способствовать
разрешению практических вопросов. В случае нитрования
ароматических углеводородов теория, объясняющая процесс
нитрования, тесно связана со строением бензольного ядра и
механизмом его реагирования.
14
Многочисленные (подчас и противоречивые) работы по
вопросу о механизме реакции нитрования разбросаны по
страницам советской и иностранной журнальной литературы, что
в значительной мере затрудняет ознакомление с ними.
Поэтому ниже, в отдельной главе, нами систематизированы основные
работы по механизму нитрования ароматических соединений.
Хотя реакция азотной кислоты с олефимами изучена
значительно менее подробно, чем реакция между азотной
кислотой и предельными углеводородами, все же имеется ряд работ
и в этом направлении.
Ненасыщенные соединения алифатического ряда легче
подвергаются нитрованию, чем предельные углеводороды.
Сравнительно легко нитруется изобутилен [31 ]. Ненасыщенные
боковые цепи ароматических соединений нитруются легче,
чем сами ароматические ядра. Также легко нитруются
ацетиленовые углеводороды. Из ацетилена при действии
концентрированной азотной кислоты получается, наряду с
другими продуктами, тринитрометан [32].
При нитровании концентрированной азотной кислотой
изомерных октенов [33] получаются непредельные нитросоедине-
ния первичного характера. Вступление нитрогруппы в
молекулу олефина сопровождается перемещением кратной связи
в ^-положение.
Большое значение приобрел способ нитрования
углеводородов и их производных в паровой фазе. Особенно зто
относится к алифатическим углеводородам. Некоторые
парафины лучше нитруются в паровой фазе, чем в жидкой.
Нитрование в газовой фазе было осуществлено X эссом,
Ходжеми Вандербильтом [34], Хзссом и Александером [35],
Данцигом и Хзссом [36], Буллоком и Митчелом [37] и др.
Этот способ нашел большое практическое применение, так как
дал возможность работать без применения давления. При
повышенных температурах нитрования (до 400—500°)
наблюдается деструктивный распад исходных углеводородов на
свободные радикалы, которые присоединяют нитрогруииы и дают
нитропарафины с меньшим числом углеродных атомов, чем
в исходном углеводороде. Это наблюдение не распространяется
15
на ряд нафтенов. В процессе нитрования полинитросоединения
не образуются.
Особенный интерес представляют работы последних двух
десятилетий по нитрованию окислами азота.
Нитрование органических соединений окислами азота
исследовалось многими химиками, как, например, Виландом,
Н. Я. Демьяновым, П. П. Шорыгиным, А. В. Топчиевым,
A. И. Титовым и др. При изучении механизма реакции
нитрования непредельных соединений окислами азота было
установлено, что двуокись азота присоединяется, подобно галоидам,
по двойной связи, давая динитропродукты.
Последние яри действии щелочей с отщеплением азотистой
кислоты (в виде соли) переходят в мононитрозамещениые зти-
леновые углеводороды:
— НС —СН— — С = СН —
I I - I + HNO,
NO,NO, NO,
При действии двуокиси азота на ароматические соединения
также получаются нитропродукты. Так, например, бензол,
толуол, нафталин, фенантрен, нафтол, хлорбензол, фенол,
крезол образуют нитросоединения. Хорошие результаты
получаются при применении вспомогательных реагентов, таких,
как концентрированная серная кислота, хлористый алюминий,
хлористое железо. Низшие нормальные парафины от метана до
пентана нитруются в газообразной фазе двуокисью азота с
образованием моно- и динитропроизводяых, причем получаются
исключительно первичные нитросоединения. Однако наряду
с нитрованием парафинов происходит их расщепление. Из
пропана получаются: нитрометан, нитрозтан, 1-нитропропан и
2-нитропропанол [38 ].
Нитрование окислами азота было осуществлено П. П. Шоры-
гиным и А. В. Топчиевым [39], А. В. Топчиевым и В. П.
Алания [40], Мак-Ки и Вильгельмом [41], Н. А. Баляшко,
B. И. Близнюковым и А. Е. Луцким [42] и др.
Большое практическое применение имела работа П. П. Шо
рыгина и А. В. Топчиева (1935) по нитрованию углеводородов
окислами азота при низких температурах. В дальнейшем зта
работа дала возможность автору настоящей книги разработать
оригинальный метод нитрования предельных углеводородов
в газовой фазе посредством одновременного воздействия хлора
и окислов азота [40].
Для нитрования органических соединений может примеяять-
ся также пятиокись азота [43].
При нитровании нарафинов азотным ангидридом при
температуре ниже 0* образуются алкилнитраты (основной
продукт), нитроалканы и карбояовые кислоты [44].
Использование окислов азота для нитрования имеет
большое промышленное значение, так как этот способ дает
возможность применять газовую смесь, получающуюся при окислении
азота воздуха иля аммиака. По этому вопросу имеется ряд
патентов [45, 46].
Дальнейшее развитие, усовершенствование и техническое
освоение методов нитрования предельных углеводородов,
а также нитрование в газовой фазе относятся к актуальным
вопросам современной прикладной химии.
ЛИТЕРАТУРА
1. С, III, 252, 288 (1922).
2. С, II, 800 (1934); С, II, 79, 2234, 2539, 2545 (1935).
3. С, II, 831 (1934); С, II, 3348 (1936).
4. Амер. патент 2590009 (1952); С. А., 46, 5823а (1952); амер. патент
2542193 (1951); С. А., 45, 10545а (1950).
5. G. Odd а. С, I, 1671 (1901).
6. G. М. В enn e t, Р. V. J о u I e. J. Chem. Soc, 1816 (1938); G. М.
В е п net, J. F. Grove. J. Chem. Soc, 378 (1945).
7. H. E. Fierz-David, R. Sponagel. Helv. Chim. Acts,
26, 98 (1943).
8. А. И. Титов. Журя. общ. хямин, 17, 382 (1947).
9. А. И. Титов. Н. Г. Лаптев. Журн. общ. химии, 18, 741
. (1948); 19, 267 (1949).
10. A. Picket, Е. Khotinsky. Вег., 40, 1163(1907).
11. J. Tiele. Вег., 33, 760 (1900).
12. Boedtker. Bull. Soc. Chim. France, [4], 3, 726 (1908).
17
13. J. Meieenheimer. Ann., 355, 284(1908).
14. F. Francis. Ber., 39, 3801 (1906).
15. R. Willstatter, H. Kubil. Ber., 42, 4152(1909).
15a. W. D. Em mo us, J. R. Freeman. J. Am. Chem. Soc.; 77, 4387,
4391, 4673 (1955).
16. R. Wolf ens tein, 0. Boters. Ber., 46, 586 (1913); C, I,
1106 (1913).
17. Франц. патент 821767 (1937).
18. R. J. Thomas, W. F. Anzilotti, С F. Hennion.
Ind. Eng. Chem., 32, 408 (1940).
19. H. Martin веп. Z. phys. Chem., 59, 605 (1909).
20. R. В eh rend, J. Schmitz. Ann., 277, 313(1893).
21. А. И. Титов. Жури. общ. химии, 11, 1125 (1941).
22. Франц. патент 345441; Frdl., 8, 131 (1908).
23. Е. Plazek. С, 1,1427(1931).
24. F. Arhall. J. Chem. Soc, 125, 811 (1924).
25. F. Hof wimmer. Schiess- und Sprengstoffwesen, 7, 43 (1912);
J. Soc. Chem. Ind., 31, 204 (1912).
26. M. И. Коновалов. ЯСРХО, 25, 472 (1893); 31, 57 (1898).
7. В. В. Марковников. Ann., 302, 15(1898); Ber., 32, 1444
(1899); ЖРХО, 31, 47, 530 (1899); 32, 1441 (1900); 35, 1033 (1803).
28. С. С. НаметкипиА. С. Забродина. Докл. АН СССР,
75, № 5, 701 (1950).
29. С. С. Наметкин, С. С. Нифонтова, Р. Я. Сущид.
Докл. АН СССР, 70, № 2, 241 (1950).
30. С. С. Наметкин. ЖРХО, 47, 1596 (1915).
31. L. Haitinger. Ann., 193, 366 (1878).
32. G. T e s t о n i, L. M a s с a r e 1 1 i. C, II, 177 (1901).
33. А. Д, Петров, М. А. Б у л ы г и н а. Докл. АН СССР, 77,
1031 (1954).
34. Н. В. Hass, E. В. Hodge, В. Vanderbilt. Ind. Eng.
Cbem., 28, 339 (1936).
35. H. В. Hass, L. G. Alexander. Ind. Eng. Chem., 41, 2266 (1949).
36. M. H. Danzig, H. B. Hass. J. Am. Chem. Soc, 66, 2017
(1944).
37. J. L. Bullock, E. T. Mitchell. J. Am. Chem. Soc, 65,
2426 (1943).
38. С Gabriel. Ind. Eng. Cbem., 32, 887 (1940).
39. П. П. Ш о р ы г и н, А. В. Топчиев. Журя. общ. химии,
№ 5, 549 (1935); № 7, 193 (1937); № 8, 981 (1938).
2 А. В. Топчиев
18
40. А, В. Топчи* в, В. П. Алания. Докл. АН СССР, 67,297 ($949).
41. R. Н. М с К ее, R. Н. Wilhelm. Ind. Eng. Chem., 28, 662 (1936).
42. H. А. Валяшко, В. И. Близяюков, А, Е. Луцкий,
Тр. Харьковского технология, вп-та, вьш. 4, стр. 48 (1948).
43. Герм, патент 211198.
44. А. И. Титов, Н. В. Щитоъ. Докл. АН СССР, 81, № 6, 1085(1951).
45. Англ. патент 262097 (1926).
46. Патент СССР 9294 (1929).
НИТРИРОВАНИЕ АРОМАТИЧЕСКИХ
И ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ
АЗОТНОЙ КИСЛОТОЙ И НИТРУЮЩЕЙ СМЕСЬЮ
Введение нитрогруппы в ароматические и
гетероциклические соединения, обладающие ароматическим характером,
обычно осуществляется действием азотной кислоты или
нитрующих смесей.
При нитровании одной азотной кислотой удалось получить
ряд нитросоединений, хотя в большинстве случаев нитрующая
смесь и другие нитрующие агенты дают лучшие результаты.
Даже в случае применения для нитрования ароматических й
гетероциклических соединений безводной азотной кислоты из-
за разбавления ее водой, образующейся в процессе реакции,
повышается окислительная способность азотной кислоты и
соответственно понижается ее нитрующее действие. Тем не
менее для получения некоторых нитросоединений применение
азотной Кислоты имеет преимущество перед другими
нитрующими агентами. При нитровании одной азотной кислотой
приходится употреблять ее в большом избытке против теории (обычно
избыток азотной кислоты составляет 50%). После отделения ни-
тропродукта отработанную азотную кислоту подкрепляют
концентрированной HNOs и используют для повторного
нитрования.
В отдельных случаях нитрования легко нитрующихся
соединений (например, фенолов, эфиров фенолов, ализарина и
применяют разбавленную азотную кислоту; при этом
20
и
нитрование иногда проводят в растворителе, как, например,
в хлорбензоле, полихлорбензолах и др.
Ниже мы даем обзор описанных в литературе важнейших
экспериментальных данных, относящихся к нитрованию
азотной кислотой ароматических и гетероциклических соединений
как в отсутствие, так и в присутствии активаторов.
§ 1. Нитрование азотной кислотой
Нитрование бензола, хлорбензола, бромбензола и толуола
азотной кислотой различной концентрации исследовано Шпинд-
лером [1]. Нитрование производилось посредством медленного
приливания яитруемого соединения к азотвой кислоте, взятой
в избытке (на 1 r-моль нитруемого соединения взято грамм-
молей азотной кислоты: для бензола 3,8, толуола 4,6,
хлорбензола 4;4, бромбензола 4,6). Опыты по нитрованию
углеводородов азотной кислотой различной концентрации (одно и то
же количество кислоты разбавляли различными количествами
воды) показали, что с увеличением разбавления выход нитро-
соединений падает сначала очень быстро, затем медленно;
начиная с концентрации, соответствующей объемному
отношению азотной кислоты (уд. в, 1,5) и воды 10 : 6, выход нитро-
производных опять быстро падает с дальнейшим разбавлением,
и при объемном отношении 2 : 1 нитрование полностью
прекращается.
При нитровании разбавленной азотной кислотой (5—10
объемов воды на 1 объем азотной кислоты) в течение 120 час.
при нагревании на водяной бане не обнаружено образования
нитройроизводных; анализ полученных продуктов показал, что
При этих условиях реакция направляется в сторону окисления.
Сравнивая экспериментальные данные по нитрованию бензола,
толуола, хлорбензола и бромбензола кислотой одной я той же
концентрации, Шпиндлер приходит к выводу, что присутствие
галоида и метильяой группы в ароматическом ядре при
нитровании концентрированной и мало разбавленной азотной
кислотой повышает выход нитропродуктов. При нитровании же
разбавленной кислотой средних концентраций влияние
заместителей в ядре на нитрование менее заметно, так как оно отчасти
21
компенсируется влияяием разбавления (с увеличением разбавт
ления азотной кислоты выход нжтропроизводных толуола, хлор-
бензола и бромбенаола понижается в большей степени, чем это
наблюдается у самого бензола).
При нитровании бензола концентрированной азотной
кислотой при комнатной температуре основным продуктом реакции
является мононитробензол; при длительном нагревании с
кипящей азотной кислотой образуется м-динитробензол [2 ].
Предварительное введение алкильвыас групп в
ароматическое ядро способствует более легкому вступлеаию нитрогруп-
пы. Так, например: толуол нитруется азотной кислотой даже
при комнатной температуре, причем основными продуктами
реакции являются моновитротолуоды, а именно: о- и п-нитро-
толуолы (м-нитротолуол образуется лишь в небольшом
количестве) [2—4J. Если нитрование производить дымящей
азотной кислотой при высоких температурах, то основным
продуктом реакции оказывается 2,4-динитр|отолуол [2].
Голлеман [5] исследовал нитрование толуоЛа азотной
кислотой уд. в. 1,475 при различных температурах. Опыты
показали, что, несмотря на варьирование температуры в широких
пределах (от —30 до 60*), отношение количеств получению*
изомеров нитропроизводных изменяется не8начите#нЬ(табл.1Х
Как видно из приведенных в мой таблице данных, с BOB*-?
шежием температуры выходы о- и м-витротолуояов повышаются*
а выход п-нитротолуола понижается.
При нитровании п-хлортолуола при 0° азотной кислотой
уд. в. 1,48, взятой в отношении к углеводороду 4:1,
получена смесь нитропродуктов, которая содержала 58% 4-хлор-З-
нитротолуола и 42 % 4-хлор-2-нитротолуОла.
Нитрование при более высоких температурах (30 и 60*)
приводит к образованию, кроме указанных выше нитропроиз*
водных, еще полинитро-4-хлортолуолов.
Ксилолы также легко нитруются концентрированной
азотной кислотой уже при комнатной температуре. При действии
дымящей азотной кислоты на м-ксилол образуются 4-иитро-
м-ксилол и 4,6-Дивитро-м-ксилол щ ?]; если же нитрование
м-ксилола производить избытком концентрированной HNO @
22
к
яри нагревании на водяной бане, то продуктами реакции
являются 4,6-динитро-мчксилол и 2,4-Динитро-м-ксилол [8 ].
Й-Ксилоя дает при нитровании дымящей НМОзпри охлаждении
льдом 2-нитро-п-ксилол [9]; если же нитровать п-ксилол при
нагревании, то продуктами реакции являются 2,3-дввжтро-
а-ксилол и 2,6-динитро-п-ксилол [10].
Таблица 1
толуола HNOg *
от
Температура
реакции, *G
—30
0
30
.60
Содержание в продуктах реакции (в А*
количеству ннтросоединеинй)
о-витротолуола
55,6
56,0
56,9
57,5
п-нитротоауола
41,7
40,9
39,9
38,5
к общему
м-ннтротолуола
Г
2,7
3,1
3,2
4,0
При нитровании о-ксилола дымящей азотной кислотой при
комнатной температуре получены 4-нитро-о-ксилол и 3-питро-
0-ксилол [11]. о-Ксилол (50 г) нитровали [12] азотной кислотой
(300 г), охлажденной до —15°, причем углеводород приливали
по каплям в азотную кислоту; во время реакции поддерживали
температуру —4°. Продуктами реакции оказались: 4-нитро-о-
ксилой, 3-аитро-о-ксилол, 3,4-динитро-о-ксилол, 4,6-диаитро-
о-ксилол и 4,5-динитро-о-ксилол. При нитровании о-ксилола
(25 г) ббльшим избытком дымящей азотной кислоты (250 г)
при температуре 20—25° получается смесь 3,4-динитрочмкси-
дола, 4,6-динитро-о-ксилола и 4,5-динитро-о-ксилола с общим
выходом динитроксилолов, близким, к теоретическому.
Мезитилен при нитровании азотной кислотой (уд. в. 1,38)
при нагревании дает мононитромезитилены; если же
нитрование производить избытком азотной кислоты (уд. в.
1,5) при нагревании на водяной бане в течение 2 час,
то получается смесь ди- и тримитромезитилевов, которые можно
выделить посредством разбавления продукта реакции водой*
жмслотой 23
Для разделения этих нитропроизводных смесь полинитромези-
тилежов обрабатывают спиртом, в котором растворяется лишь
2,4-динитромезитилен (т* ил. 87,5*) [81.
При титровании фенола разбавленной азотной кислотой
образуется смесь о- и п-нитрофецола. Изучение вопроса об
относительном содержании изомерных нитрофенолов и нитро-
крезолов в продуктах нитрования нашло отражение в работе
Вайбеля [131. Лучшие результаты при нитровании фенолов
были получены [141 при применении азотной кислоты средней
концентрации. Процесс проводился следующим образом:
к охлажденной до 8—9* азотной кислоте (уд. в. 1,35) при
энергичном перемешивании прибавляют по каплям фенол
(предварительно превращенный в жидкость добавлением небольшого
количества воды). Реакционная масса тотчас окрашивается,
и через 10—20 мин. начинается выделение желтого продукта
реакции. По окончании прилжвания фенола реакционную
массу перемешивают еще 30 миж., поддерживая ту же температуру,
и смесь охлаждают до —10*. Полученную смесь о~ л п-витро-
феяола отсасывают досуха при 0* и при помощи водяаога пара
отгоняют о-нитрофенол. Выход составляет приблизительно
40% от теории. Остаток от перегонки с водяным паром
несколько раэ экстрагируют на кипящей водяной бане 2%+#ой соляной
кислотой, слегка охлаждают и фильтруют. При охлаждении
фильтрата выпадают пластинчатые кристаллы п-шитрофеяола.
Выход —40% от теории. Из фильтрата можно выделить
небольшое количество 2,4-динитрофевола. П ри ки тровании
п-оксибензойной кислоты 8 н.НМОз была, получена пикриновая
кислота с выходом 95% от теории [15].
Динитро-о-крезол может быть получен непрерывным
способом по следующей методике [16]: 12,9 кг о-крезола я 34 кг
70%-ной азотной кислоты пропускают в течение 1 часа через
калиброванные сопла в охлаждаемую водой металлическую
трубу длиной 1 м и диаметром 38 мм. Выход дшнитрокревола
80% от теории.
Что касается эфиров фенола (например, анизол, фенетол),
то эти соединения легче нитруются при применении
концентрированной азотной кислоты.
24
м-Нитроащ##шоя можно волучтъ о хорошим выходом
следующим обедом [17]. В ожджжденяую авотвую кислоту
(уд. в. 1,51) ифвливааУг &о каплям ацетофенон. Процесс
проводят яри ожттждежжж (0—8*) ж иеремешивавии. Продукт
реакции яылжюают ва лед, фильтруют и яерекристаллиэовы-
ватт из метилового спирта. Выход м-нитрокрезода
составляет ^ 90% о* теории.
Нитрование^ ирониофевоиа дымящей азотной кислотой
при охлаждевиж получева смесь о- и м-вя?роаровиофеноиа,
которая разделяется 95%-ним этиловым спиртом [18].
При обработке феаилукеусной кислоты избытком дымящей
азотвой кйслтш образуется 2,4-Дииитрофбиилуксусиая
кислота [19].
Гаясбергер [20] примевяд витроваиие азотжоё кислотой для
аитропронзводных дифеиялуксуежой жиелоты* При
дифенилуксусжой кислоты к избытку ааотвой
кислоты (уд. в. 1,5) при Ж,вылжвааииреаЩиотноймасоы иа воду
ж кршоталливащии иа уксусной каолоты автор получил с ве-
@на9ителъвш« ммодом (7,4% от теории) ЗД'-дияитродифеаил*
ужуовую кислоту.
При вжтро%жид1 ашкилированямж жромводжвнс бензола
раэбавленяой Паствой кислотой в зажаяижоё трубке витрогруп-
яа вступает ж$|А ядро^ ж в баковую цепь. Тек, яапример^ жри
вагревааии этщ^бекаола с 5^6-кратяым количеством
разбавленной азотной кислоты (уд. в, 1,075) в дждаянвой трубке при
105* образуется %-нитроэтилбензол С^(Ш(МО^)СН, с
выходом 44% or теории. Ивопродилбежзол ж втж% же условиях
дает феаиляитроизодропан С^Н#С(МО,)(СН$)$ [21].
Легге [22] юучал действие азотной кислоты яа 1,4"Д*ифет.
бутжлбенаол. При постепеаиом прибавлении 100 г 1,4-ди-
трет. бутилбежэола при 15—Л0* к 300 мл 96%-ной азотной
кислоты и последующем нагревании реакционной смеем в
течение 5 чао. при 60* им было подучено 2,6-данштропроиавод-
ное с выходом 53% от теории. При охлаждении фильтрата
удается выделить некоторое количество 2,5-дяжятропрожввод-
ного. При нятрованвя 1,4<щ~трет.бутяабев8ола азотной
кислотой в смеси с уксусным ангидридом а уксусной кислотой
же авторы получала же 2,6-дтштро-» а 2-витродройзвод-
вое с выходом 80% от теории.
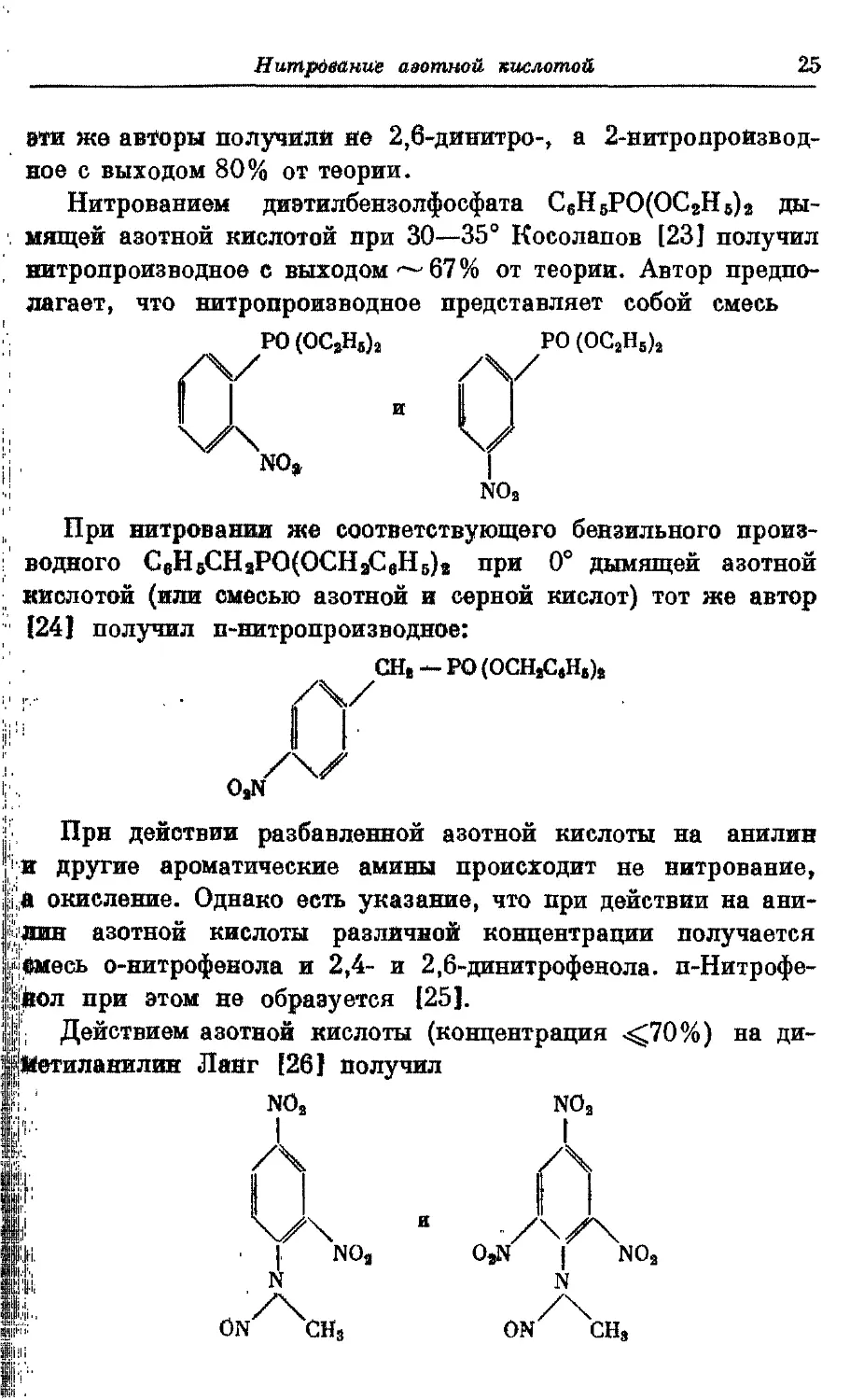
Нитрованием диэтилбензолфосфата С,НвРО(ОС*Н,)*
дымящей азотной кислотой при 30—35° Косолапов [23] получил
витропроизводное с выходом ^-67% от теории. Автор
предполагает, что нитропроизводное представляет собой смесь
РО (ОСА)з РО
У /\У
NO,
При нитровании же соответствующего бензильного прошв-
водного С,НдСН^О(ОСН/],Нб), при 0* дымящей азотной
кислотой (или смесью азотной и серной кислот) тот же автор
(24] получил п-нитропроизводное:
РО(ОСНаС*Н&)*
iii'i
Прн действии разбавленной азотной кислоты на анилин
я другие ароматические амины происходит не нитрование,
ia окисление. Однако есть указание, что при действии на ани-
[лин азотной кислоты различной концентрации получается
IgjWecb о-нитрофенола и 2,4- и 2,6-динитрофенола. п-Нитрофе-
при этом же образуется (25].
Действием азотной кислоты (концентрация ^70%) на ди-
Лажг [26] получил
NO. NO,
Р
\У\
МО,
N0%
|
N
ON CH,
СН,
Если же применить азотную кислоту концентрадии выше
70%, то получается
N0.
O.N
Далее Ланг установил, что эти же соединения получаются
при действии азотной кислоты концентрации выше 70% на
2,4-Динитрометиланилин и 2,4,6-тринитрометилавилин.
В случае ацилированных ароматических аминов
наблюдается нитрование ароматического ядра с одновременным
омылением ацильной группы 127].
Арилсулъфонвлъйые производные ароматических аминов
легко нитруются разбавленной азотной, кислотой. Так,
например, при действии на п-толуолсулъфанилид разбавленной
азотной кислоты образуется мононитропроизводное [28].
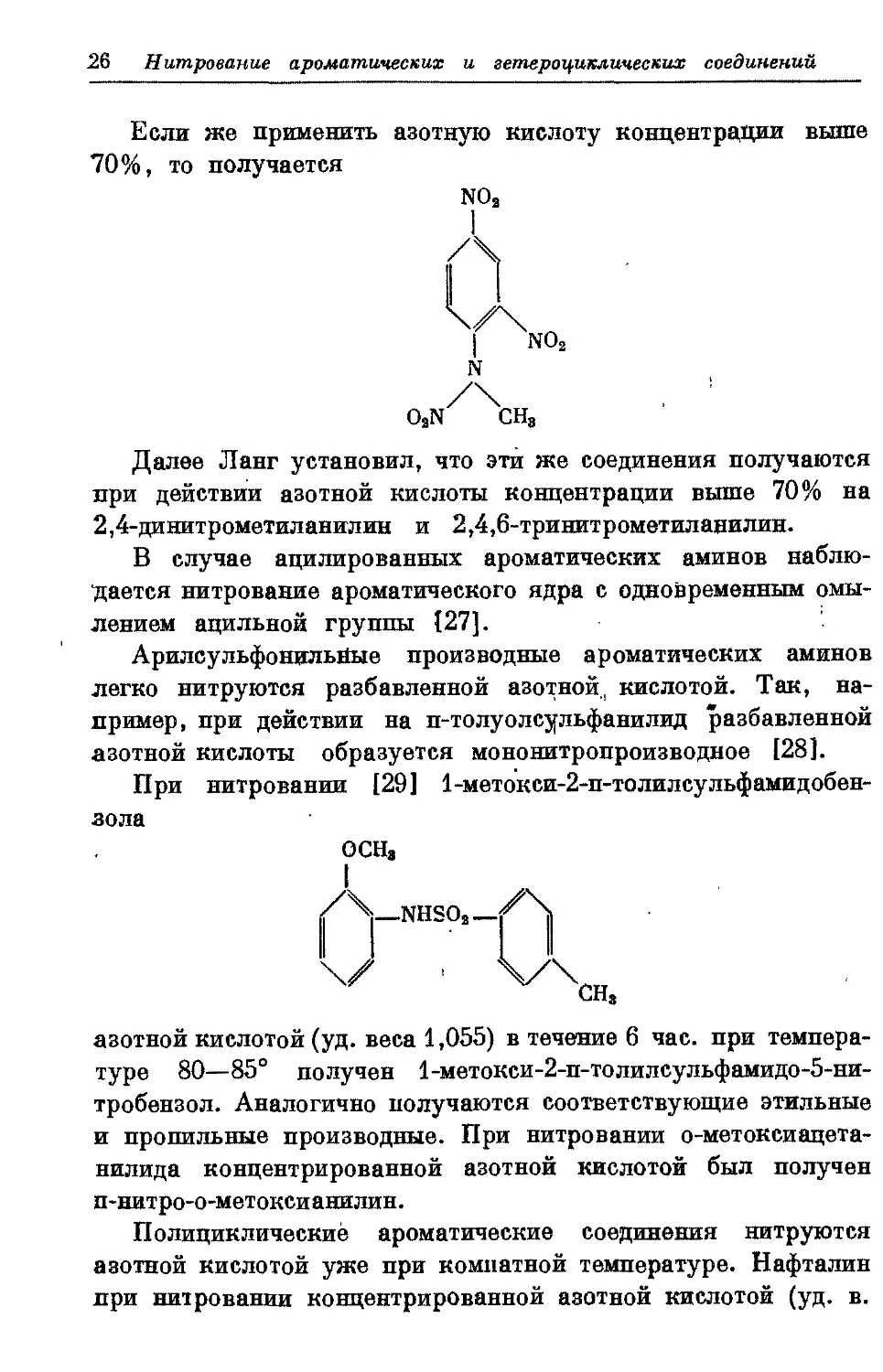
При нитровании [29] 1-метокси-2-п-толилсульфамидобен-
зола
ОСИ,
/\_NHSO,
U ;
азотной кислотой (уд. веса 1,055) в течение 6 час. при
температуре 80—85° получен 1-метоксн-2-п-толилсулъфамидо-5-ни-
тробензол. Аналогично получаются соответствующие этильные
и пропильные производные. При нитровании о-метоксиацета-
нилида концентрированной азотной кислотой был получен
п-нитро-о-метоксианилин.
Полициклическиё ароматические соединения нитруются
азотной кислотой уже при комнатной температуре. Нафталин
дри нитровании концентрированной азотной кислотой (уд. в.
27
1,33) жри 20° дает %-нитронафталин [30]. При действии на
нафталин дымящей азотной кислоты при температуре кипения
последней образуются 1,5-динитронафталин и 1,8-динитро-
нафталин [31].
Кроме %-нитронафталина при нитровании нафталина в
качестве побочных продуктов удалось выделить 4,5% (3-нитро-
нафталина и 0,5—3,5% 2,4-двнитро-1-нафтола [32].
Я. Я. Макаров-Землянский и В. П. Бибишев [33] показали,
что при нитровании тетралина азотной кислотой получаются
1,2- и 1,3-динитротетралин, в то время как при применении
нитрующей смеси продукт реакции не удается выделить ввиду
значительного осмоления.
Нитрование алкилпроазводныж нафталина изучали С. И.
Сергиевская и Г. Я. У редкая [34]. При нитровании 1-про-
яилнафталижа азотной кислотой при 40° авторы получили
4-нитро-1-пропилнафталин с выходом 35% от теории.
Получение нитроиройзводных 1-хлор-2-метоксииафталина
описано в английском патенте [35].
1-Хлор-2-метоксинафталин медленно прибавляется к 2,5-
кратвому против теории количеству 62%-ной азотной кислоты
при 40* в растворе хлорбензола. При этом получают смесь
изомеров 4-, 6- и 8-нитро-1-хлор-2-метоксинафталина.
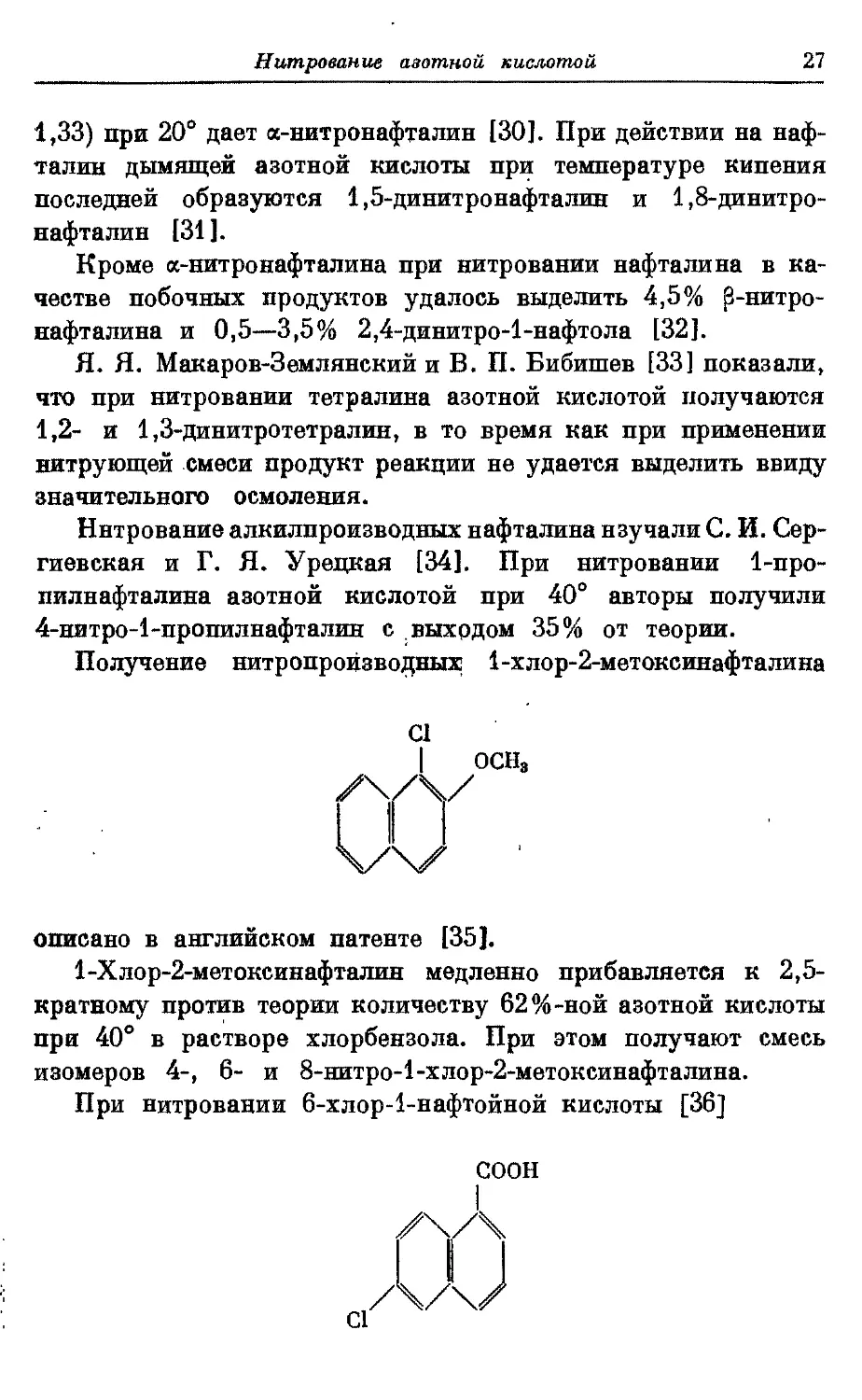
При нитровании 6-хлор-1-нафтойной кислоты [36]
28 Дытроммые д^хмюмгыческыа и
избытком азотной кислоты (уд. в, 1,49) в течение 5—10 миа. на
водяной бане получена б-нитро-б-хлор-^аафтойная кислота
соон
0
с выходом 27,5% от теории (после очистки). Есж исходить
из метилового эфира &-хлор-1-нафтойной кислоты, то в этих
же условиях получают с выходом 30% от теории 5-н%тро-6-
хлор-1~м&гиловый эфир нафтойяой кислоты:
СООСНд
I
При нитровании дифенила дымяЩей азотной кислотой
образуются 4,4'-динитродифенил ш 2,4'-динитродифенил [37, 38].
Белл и Кеаьон [39] при нитровании дифеаила дымящей
азотной кислотой получили 4,4'-динитродифенил, 4,2'~динит-
родифеаил (т. пл. 70—80**) и 2,2'-динитродифенил (т. ял. 127—
128°). Они синтезировали также 2-нитродифенил и 4-яитро-
дифенил по методу Гюбнера и Людденса [40] (обработка
дифенила дымящей азотной кислотой в растворе уксусной
кислоты при 60*). При нитровании 20 г 2-нитродифевнла 14 мл
дымящей азотной кислоты, разбавленной 2 мл воды, в течение
получаса продуктами реакции являются 4,2'-диннтродафенил
и 2,2'-диннтродифенил; 4-нитродифенил в этих же условиях
дает 4,2'-дннитродифенил.
При действии азотной кислоты на антрацен происходит
окисление его в антрахинон. Концентрированная азотная
кислота уже на холоду превращает диацетилализарин в нитропро-
ивводное
жысдолюД 29
/\/\/\У
О ОСОСНд О ОСОСН*
_^ Я L ,осос% _ II 1 ,ососн,
HNO:
О О N0%
Довольно хорошо изучено нитрование фенантрена. Еще
в 1879 г. Г. А. Шмидт [41], действуя на фенантрен большим
избытком дымящей азотной кислоты, получил смесь *-, (3-
и %-нитрофенантренов; из этой смеси ему удалось затем
выделить отдельные изомерные нитрофенантрены. Впоследствии
Ю. Шмидт [42], нитруя фенантрен при комнатной температуре
азотной кислотой (уд. в. 1,56), синтезировал 3-аитрофенантрен
с т. пл. 170—171°, идентичный %-нитрофенантрену,
описанному Г. А. Шмидтом.
Ю. Шмидт [43] нитровал азотной кислотой также феная-
тренхинон но следующему методу: смесь 30 г чистого фе-
нантренхивона и 900 мл концентрированной азотной кислоты
(уд. в. 1,4) нагревали до полного растворения феаантренхивона,
после чего дицячеяие смеси продолжали еще в течение 20 мин.
При вливании продукта реакции в 2 л воды нитропроизвод-
ные выделялись в виде желтых хлопьев, осадок
отфильтровывали от жидкой части, промывали водой и обрабатывали
кипящим спиртом; при этом в раствор переходил 4-нитрофе-
нантренхинон, который выкристаллизовывался из спирта при
охлаждении. После перекристаллизации из ледяной уксусной
кислоты получен чистый 4-нитрофенантренхинон с т. дл. 176—
177°. Нерастворившийся в спирту остаток после извлечения
4-нитрофенантренхинона растворяли в ледяной уксусной
кислоте, из которой при охлаждении выделен 2-нитрофенантрен-
хинон в виде желтых игольчатых кристаллов с т. пл. 256—260°.
Из новых нвтросоединений, нашедших значительное
применение для борьбы с вредителями виноградников и
фруктовых садов, следует отметить 1,3,6,8-тетранитрокарбаэол [44].
Нитрованием флуоренона дымящей азотной кислотой при
кипячении в течение 3 час. получается 2,7-динитрофлуоренож [45].
30
Взаимодействием дибензантрона с 70%-ной азотной
кислотой в присутствии о-нитротолуола при нагревании получается
мононитропроизводное [46].
И. С. Иоффе и А. С. Эфрос [47] описывают нитрование
дибензпирена азотной кислотой. Нитрование ведется в среде
нитробензола при 25—30° азотной кислотой (уд. в. 1,52). Для
получения 5,10-динитродибензпирена приходится применять
значительно больший избыток азотной кислоты. При нитровании
дибензпирена и его монозамещенных производных натрогруп-
пы вступают в наиболее реакционноспособные положения
(5 и 10), независимо от характера заместителя, содержащегося
в молекуле. :
Бруннер и Витт [481 применяли нитрование азотной кисло-
той для получения динитробензидина, исходя из диацетобен-
зидина: 10 г диацетобепзидина вносили в 100 г дымящей
азотной кислоты при комнатной температуре; полученное динитро-
соединение осаждали ледяной водой, отфильтровывали и
промывали горячей водой. Выход динитродиацетобензидина со-
ставлял!2,5 г (93% от теоретического). При умеренном
нагревании динитродиацетобензидина с 1,5-кратным количеством
концентрированного раствора едкого кали в присутствия
небольшого количества спирта получен с количественным
выходом динитробензидин
N0%
ОО
Динитробензидин нерастворим в воде, растворим в феноле и
мало растворим в спирту; при восстановлении дает тетраами-
нодифенил
При растворении динитробензидина в серной кислоте и
последующей обработке нитритом получается диазосоединение;
кипячение последнего в растворе абсолютного спирта приводит
смесью мотммоД и ссрмой жмслот 31
к разрушению диазогрупп с выделением азота, вследствие
чего образуется 3,3'-динитродифенил (т. пл. 197—198°)
OaN NO,
Гетероциклические соединения, обладающие ароматическим
характером, при действии азотной кислоты могут быть
превращены в соответствующие нитроироизводные. Мейер и Штад-
лер [49] получили нитротиофен посредством пропускания
воздуха, насыщенного парами тиофена, через дымящую азотную
кислоту, причем на 1 объем тиофена затрачивалось 4 объема
азотной кислоты. По окончании опыта реакционную смесь
разбавляли водой и собравшееся над водным слоем масло
отделяли и перегоняли с водяным паром. Продуктами реакции
оказались мононитротиофен (т. пл. 44°) и два изомерных ди-
нитротжофена, из которых один (получающийся с больший
выходом) плавился при 52°, а другой — при 75—76°.
Нитрование хиволина азотной кислотой при нагревании
дает 5- и 8-нитрохинолиа с почти одинаковыми выходами
наряду с незначительным количеством 6,8- я 5,7-диаитрохино-
лина [50], Значительно труднее нитруется азотной кислотой
ииридия; по данным Фридля [51], при кипячении пиридина
с концентрированной азотной кислотой в течение 24 час.
образуются лишь незначительные количества нитропиридина.
§ 2. Нитрование смесью азотной и серной кислот
Наиболее распространенным методой нитрования
ароматических соединений является нитрование смесью азотной
кислоты с ковцентрированной серной кислотой («нитрующей
смесью»). Нитрующая смесь — более энергичный нитрующий
агент, чем сама азотная кислота.
Помимо водуотнимающих свойств серной кислоты (точао
так же, как и смеси ледяной уксусной кислоты с уксусным
ангидридом), присутствие серной кислот* способствует
усилению нитрующего действия азотной кислоты при
одновременном уменьшении ее окислительных свойств. Кроме того, сер-
82
ы
ная кислот* <№УШ# аорошж растроритежем дд* многих ни-
ю также является благоприятммм факт*
ром для процеоса^ритрования.
Процесс иитрошния бензола на мононитропроизводное
освоеа уже много! десятков лет назад и осуществляется
в производстве. Дж того чтобы избежать образования дввитро-
произ#дного, аа^фЩая кислота берется с некоторым (5%)
недоот&тком прот#и Чгеории [44], Льюис и другие {521 показа-
перемаиивание реагентов достаточно для
высоких выходов. При нитровании бен-
условиях (температура 75—80", концен-
что даже ел
достижения наи
аола в более
тращня Ьтработант
дтшЪробенвола.
1—3% пара-
Коб и Миле |
хождевию оптима
Авторы изучали
количества азотя
концентрации. О
следующих у слоя
СИЛИКДТыОГО vTG
и 120 г серной к
серйоё кислоты и
теория) постепен
Температура нит
кислоты 86%) получается смесь изомеров
Йеь содержит 90% мета-, 8—9% орто- и
(58].
провели детальное исследование по на*
х условии получения моаоаитробеязода.
ияниё на выход продукта температуры,
ислоты, количества серной кислоты и ее
альные результаты были подучены при
проведения реакция: # реактор изборо-
Загружают при охлбжден&и 250 г бензола
юты (84%-вой). Нитрующая смесь (180 г
тная кислота с избытком 1,5—1,8% от
подается в реактор яри размешивании,
ния равна 60* при продолжительности
процесса 40 мин. Ш окончании нитрования реакционная
масса обрабатывается $ Делительной воронке 400 г льда.
Полученный нитробензол роняется с паром и сушился. Выход 98—
99% от теории. '44 \
Мак-Кормак [5$*^ осуществил синтез м-дюштробензола
непосредственно из #ё#ола. К нагретой до 70° смеси, состоящей
из 617 г азотной к%#оты (59,9%-вой) и 836 г дымящей серной
кислоты (олеум с #%-ным содержанием SO»), медленно
приливают из капельш№ воронки 69 г бензола; через 2 часа после
начала приливанйА' бензола температура реакционной смеси
(без Шедшего названия) достигает 109*. По окончании
выделения тепла смеЫь при сильном размешивании нагревают
жы&мл* 33
еще в течение 2 час, поддерживая температуру 109*. Выход
динитробеазола составляет 79% от теории. Можно получить
156] м-динитробеваол нитроважи^м беизола при более низкой
температуре, если вести реакции сначала при 30—60*, а потом
заканчивать при 80*. Соотношение реагентов:
«3-*-б/1; HNCWC,H,='2,1/1.
Прямое нитрование бензола до тринитробензола протекает
значительно труднее, чем нитрование его гомологов,
например толуола. При нитрования дявитробензола даже в особо
жестких условиях удается получить лишь очень низкие
выходы тринцтробензола. Поэтому этот процесс не получил
практического применения.
Наличие метильной группы в толуоле значительно
облегчает его нитрование. i
Нитрование толуола на мощонитротолуол протекает гладко
я в тех же условиях, что и нитрование бензола. При этом
получается смесь изомеров следующего состава:
орто-изомер 66%
дара-жзоывр 39%
метачкаомер 5%
С повышением температуры нитрования увеличивается
содержание в смеси мета-изомера [57]. Нитрование толуола на
мононитропроизводное можно осуществить и непрерывным
способом. Смесь нитротолуолов при дальнейшем нитровании
превращается в динитротолуол. Нитрование ведется при тем-
«пературе 75* и концентрации отработанной кислоты —80%;
при этом получается смесь динитротодуолов, в которой
содержится —75% 1,2,4-изомера, —21% 1,2,6-изрмера, наряду с не-
; большими примесями других изомеров [58]. Тринитротолуол
I получается нитрованием нитротолуола в одну или более
стадий, в зависимости от того, что является исходным продуктом
Гмоно- или динитротолуол. Из трех изомерных китротолуолов
о- и п-нитротолуол превращают только в 2,4,6-1риниг*ротО'*
дуол. Превращение динитротолуола в тринитротолуол произ*
Годится безводной смесью азотной и рериой кислот с
содержанием азотной кислоты 20—25% прщ отношении азотная
18 А. В. Топчиев
34
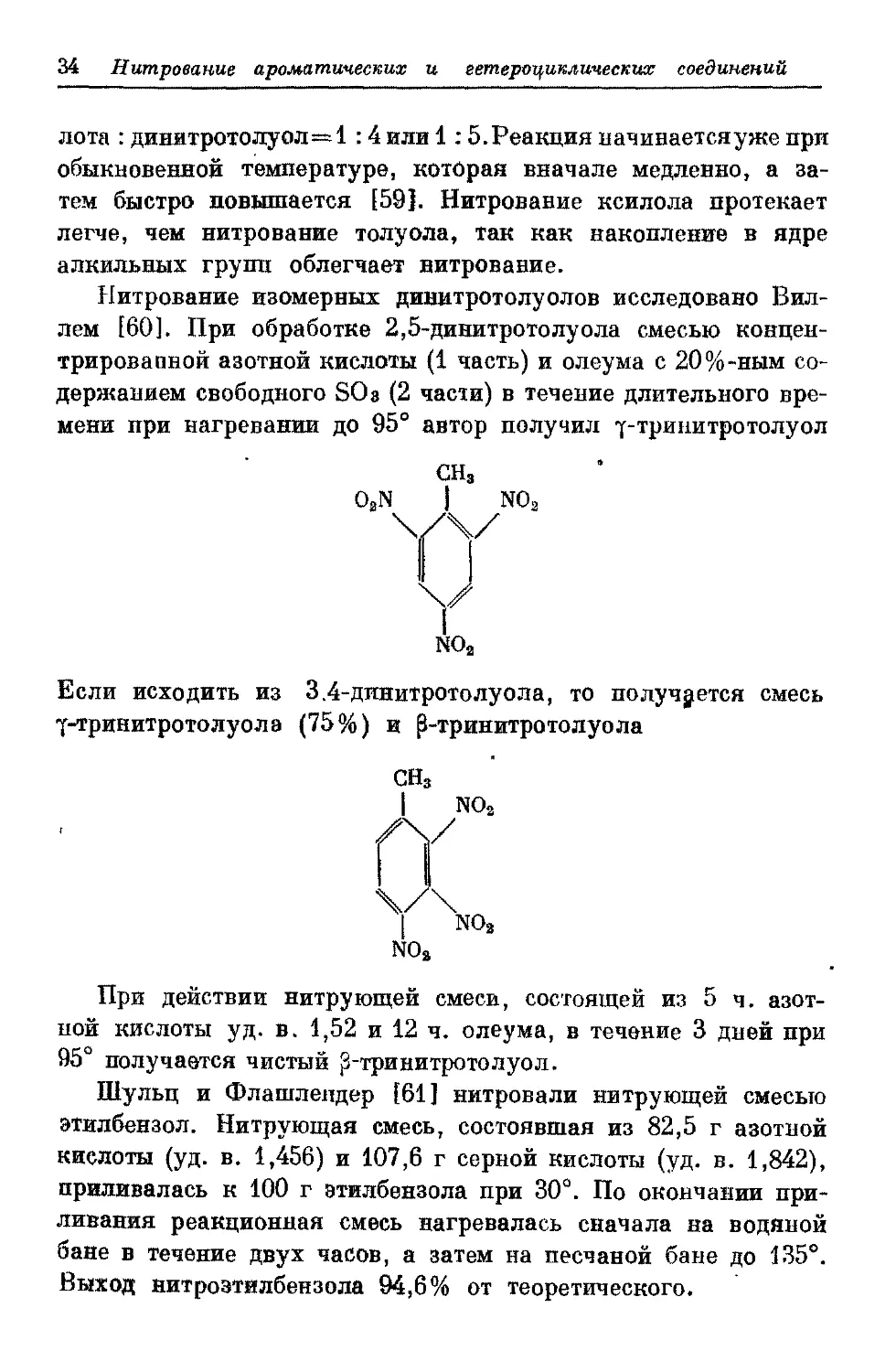
лота : динжтротолуол=1:4 или 1:5.Реакция начинаетсяуже при
обыкновенной температуре, которая вначале медленно, а
затем быстро повышается [59]. Нитрование ксилола протекает
легче, чем нитрование толуола, так как накопление в ядре
алкильпых групп облегчает нитрование.
Нитрование изомерных дивмтротолуолов исследовано
Биллем [60]. При обработке 2,5-динитротолуола смесью
концентрированной азотной кислоты (1 часть) и олеума с 20%-ным
содержанием свободного 80s (2 часки) в течение длительного
времени при нагревании до 95° автор получил т-трииитротолуол
Если исходить из 3.4-длпитротолуола, то получается смесь
у-тринитротолуола (75%) и ^-тринитротолуола
При действии нитрующей смеси, состоящей из 5 ч.
азотной кислоты уд. в. 1,52 и 12 ч. олеума, в течение 3 дней при
95° получается чистый р-тринитротолуол.
Шульц и Флашлелдер [61] нитровали нитрующей смесью
мжл бензол. Нитрующая смесь, состоявшая из 82,5 г азотной
кислоты (уд. в. 1,456) и 107,6 г серной кислоты (уд. в. 1,842),
приливалась к 100 г втжлбенвола при 30°. По окончании
приливают реакционная смесь нагревалась сначала на водяной
бане в течение двух часов, а затем на песчаной бане до 135*.
Выход нитрозтилбевзопа 94,6% от теоретического.
кислот 35
2,4-Динитроэтилбенэол был получен Вейсвейлером [62]
при медленном приливании 30 г этилбензола к смеси 2 ч.
концентрированной серной кислоты (уд. в. 1,88) и 1 ч.
концентрированной азотной кислоты (уд. в. 1,43). Затем смесь
нагревалась в течение 10 мин. до 125—130°. Строение 2,4-динитроэтил-
бензола было доказано его окислением азотной кислотой
в 2,4- динитробензойную кислоту.
Вейсвейлер получил симметричный тринптроэтилбензол
с 70%-ным выходом при прибавлении 10 г отилбензола в смесь
40 г азотной кислоты (уд. в. 1,525) и четырехкратного объема
дымящей серной кислоты (SO*"2H*SO*), охлаждаемую льдом,
с последующим нагреванием реакционной смеси до 100°.
Продукты реакции состоят почти исключительно из тринитроэтия-
бензола, содержащего следы дияитропроизводного [62].
Нитрование отилбензола нитрующей смесью изучали
Б. И. Долгов и Н. А. Кучумова [63]. Прибавлением 25 г этилбен-
зола к смеси 20,5 г азотной кислоты (уд. в. 1,5) и 27 г серной
кислоты (уд. в. 1,86) на холоду и последующим нагреванием до 135*
авторы получили главным образом 2-нитроэтилбеизол. Подобным
же образом м-дяэтилбензол дает 2,4,6-тринитровроиаводное.
Нитрование изопропилбензола детально изучено Хауном
и Кобом [64]. При обработке 250 г кумола, эмульгированного
в 300 г 97%-ной серной кислоты и 34,2 г воды, нитрующей
смесью, состоящей из 215,5 г 97%-ной серной кислоты и 225 г
70%-вой азотной кислоты, было получено 323,7 г нятрокумола.
I Выход нитрокумола 94,5% от теоретического. Авторы установи-
н; ли, что нитрокумол, полученный при температуре нитрования
от 0 до 45°, на 24% состоит из орто-и на 76% из параизомера»
Г Ханш и Хельмкамп [65] получили 2,4-динитроизопропил*
' бензол с выходом 96% при нитровании 253 г и-нитрокумола ни-
чттрующей смесью, состоящей из 542 г концентрированной азо*-
:;& мой кислоты и 800 г концентрированной серной кислоты, при
^температуре 45—60°.
|#t При нитровании трет, бутилбенэола нитрующей смесью (со*
^гава 22,3% HNO#, 65,6% H%SO& и 12,1% Н%О) обраауется
пы о 96%-ным выходом смесь о-, м и п-нитро-трет.
Глов [66].
36
Браун и Боннер [67] изучали распределение изомеров в
смесях монояитропроизводпых, полученных при нитровании зтил-
и изопропилбензола. Углеводороды нитровались нитрующей
смесью состава: 22,3% HNO,, 65,6% H,SO. и 12,1% Н%0.
Продукты реакции перегонялись на высокоэффективной
ректификационной колонке. Состав мононитропроизводных зтилбен-
зола и изопропилбензола, вместе с составом
мононитропроизводных толуола и трет.бутилбензола [66], приведен в табл. 2.
Таблица 2
Углеводород
Толуол
Этилбеязол . . .
Изопро пил бензол
Трет, бутилбензол
Изомер
орто-
58,45
45,4
30,0
15,8
мета-
пара-
4,1
6,5
7,7
11,5
37,15
48,2
62,3
&72,7
Из таблицы видно, что количество орто-изомера заметно
снижается с увеличением углеводородного радикала за счет
значительного увеличения содержания napa-изОмера.
Количество мета-изомера с увеличением боковой цепи заметно
увеличивается. Такое изменение содержания орто-изомера
связано, невидимому, с сильным стерическим влиянием алкиль-
ной группы на замещение в орто-положение.
Нитрование о-ксилола нитрующей смесью изучалось Кросс-
леем и Ренуфом [68]. Нитрующая смесь, состоящая из 25 г
азотной кислоты и 100 г концентрированной серной кислоты,
медленно приливается к охлажденному до 0* о-ксилолу. В
качестве основных продуктов реакции получаются 3-нитро-о-
ксилол и 4-нитро-о-ксилол наряду с небольшим количеством
динитро-о-ксилолов. Если же нитровать о-ксилол (30 г)
избытком азотной кислоты, приливая углеводород к смеси,
состоящей из 50 г азотной кислоты и 100 г серной кислоты, т, е,
в обратном, чем в предыдущем синтезе, порядке, то образуются
динитро-о-ксилолы с теоретическим выходом.
смесью даотной и серной жислот 37
МаркеролиЛориет [69] нитровали о-ксилол (35 г) в
растворе серной кислоты (65 г 7096-ной серной кислоты), постепенно
приливая нитрующую смесь (85 г), содержащую 56% H*SO«,
28% НМОз и 16% Н,О. Нитрование проводилось при
перемешивании реакционной массы, при этом температура
поддерживалась ниже 30*. Выход мононитроксилола составил 45,2 г.
При нитровании м-ксилола смесью азотной и серной кислот
яри температуре ниже 0° образуется 4-нитро-м-ксилол и 2-
нитро-м-ксилол [70].
Нитрование п-ксилола описано в работе Боузна [71].
К 1 молю п-ксилола при 25—30° прибавляется по каплям
смесь 133 г концентрированной азотной и 212 г
концентрированной серной кислот. После отделения органического слоя и
его нейтрализации продукт реакции перегоняют с паром,
экстрагируют бензолом и после отгонки бензола получают нитро-п-
ксилол с выходом 78—87% от теории.
Максимальный выход (90%) мононитро-п-ксилола
получили Коб и Левин [72] нри применении избытка азотной кислоты
11% (при обезвоживающем значении серной кислоты —3,0),
температуре 30* и продолжительности реакции 30 мин. В
качестве примесей обнаружены 2,3- и 2,6-динитро-п-ксилол и
п-толуиловая кислота.
Из трех изомеров ксилола наиболее легко нитруется м-кси-
лол [73]. Максимальный выход мононитро-м-ксилола (98%)
получается при нитровании в следующих условиях; к 1 молю
м-ксилола при 30° прибавляется 1,08 моля H,SO* (81%-ной)
и 10%-ный избыток НМОз. Реакция продолжается 1 час.
Для нитрования м-ксилола применяется вдвое меньшее
количество серной кислоты, чем при нитровании орто- и пара-
изомеров. Данные по мононитрованию изомерных ксилолов
сведены в табл. 3.
Полинитроксилолы получаются при обработке ксилолов
нитрующей смесью. Нельтинг и Гейсман [74] при нитровании
п-ксилола смесью, состоящей из 80 г дымящей азотной
кислоты и 40 г серной кислоты, в течение 24 час. при нагревании
на водяной бане получили смесь двух динитро-п-ксилолов:
2,3-динитро-п-ксилола (т. пл. 24*) и 2,6-динитро-п-ксилола
38
драмамычбсжыа и
(т. пл. 124°). Смесь удается разделить дробной
кристаллизацией из толуола. При нагревании 20 г п-ксилола на водяной
бане в течение 16 час. с 80 г дымящей азотной кислоты и 120 г
серной кислоты авторы получили 2,3,5-тринитро-п-ксилол
(т. пл. 139-140°).
Таблица 3
Молярное отношение
HgSO*: ксилол
о-Ксилол, 2,22 . . .
п Ксилол, 2,2 ...
м-Ксилол, 1,08 . . .
Температура, "С
о m
KB
6-35
20—40
5—55
i
ее
§«
25
30
30
о
И
3
со
15,8
5,0
10
Концентрация
H.SO., %
§s
80
85
81
сти-
&=
о ^
KS
78—82
77—85
77—85
а"
к
3
Е ш
60
30
60
3
а;
аз
90
92
98
При нитровании п-ксилола при 80° избытком (10%) азотной
кислоты (при обезвоживающем значении серной кислоты 8,0)
и продолжительности реакции 15 мин. Коб и Левин [72]
получили с выходом 95% смесь динитро-пчкеилолов, состоящую
из 60—80%, 2,3-динитро-п-:ксилола и 40—20% 2,б-динитро-п-
ксилола.
В работе Лекорша и Аубертейна [75] описано получение
тринитро-м-ксилола. 53 г м-ксилола медленно прибавляют
к 365 г смеси серной и азотной кислот (62,5% HsSO*, 14,5%
HNOs, 23% HgO) при 50°. Продолжительность реакции 1,5—
2 часа. В результате реакции получают 85 г смеси равных
частой моно- и динитроксилола. Полученвую смесь в растворе
320 г серной кислоты (92%-ной) приливают к 65 г смеси азот-
вой и серной кислот (86,7% HNOs, 8,4% H*SO«, 4,9% Н,О)
при 70°. В дальнейшем постепенно повышают температуру до
100°, а затем быстро до 120°. Получают 100—108 г тринитро-
ксилола хорошего качества.
Большой практический интерес представляют нитропроиз-
водные хлорбензола. При нитровании хлорбензола получается
смесь п- и о-нитрохлорбензола. Мак-Кормак [55] изучал нитро-
ддомной и серной *%сл@л& 39
вание хлорбензола смесью азотной и серной кислот различшях
концентраций. Предварительно охлажденный до —5°
хлорбензол частями добавлялся в нитрующую смесь при сильном
охлаждении. Оптимальные результаты получены при
поддержании температуры реакционной смеси 10°. Продолжительность
нитрования около 4 час. Для повышения производительности
процесса Мак-Кормак проводил нитрование хлорбензола
также при нагревании в интервале температур 80—95°. Общая
продолжительность нитрования 2,5 часа. В последнем случае
он применял нитрующую смесь, состоящую иа 106,5 ч. азотной
кислоты (65,5%-пой) и 107,9 ч. серной кислоты (93,6%-яой)
на 100 ч. хлорбензола. В обоих случаях выход нитрохлорбен-
зола составлял 98% от теоретического, при этом 7% азотной
кислоты оставалось без изменения. Продукт реакции
представлял собой смесь, состоящую из 66% п-нитрохлорбензола и
34% о-нитрохлорбонзола. При дальнейшем нитровании смеси
изомеров нитрохлорбензола получается смесь динитрохлор-
беызолов, в которой преобладает 2,4-динитрохлорбензол
наряду с заметным количеством 2,6-динитрохлорбензола. В
качестве небольших примесей смесь содержит и другие изомеры.
При нитровании п-дихлорбензола получается главным
образом 2,6-динитро-1,4-дихлорбензол наряду с двумя другими
возможными изомерами [76 ].
Нитрованием о-дихлорбензола получают 3,5-динитро-1,2-
дяхлорбензол [77], в случае же м-дихлорбензола образуется
преимущественно 4,6-динитро-1,3-дихлорбензол наряду с
некоторым количеством 2,4-динитро-1,3-дихлорбензола [78].
При взаимодействии 1,2,4-трихлорбензола со 100 мл смеси
серной кислоты (уд. в. 1,82) и азотной кислоты (уд. в. 1,52) в
отношении 3 : 1, в течение 24 час. при комнатной температуре
и 8 час. при 50—60° получено 11 г 1,2,4-трихлор-3,5-динитро-
бензола с т. пл. 101—103° [79].
Получение динитрофторбензола описано в работе Цана
и Вюрца [80]. 40 г фторбензола приливают по каплям к смеси
120 г азотной кислоты (уд.в. 1,52) и 280 г серной кислоты (уд. в.
,1,84) при температуре 0—20°. По окончании приливания смесь
в течение 2 чцс. нагревают л а водяной бане, охлаждают,
40 #wmpoe&w%e дрождтмчрскш; % гемгбро^мклм^скмаг соединений
выливают на лед. Кристаллический продукт содержит некоторое
количество кислоты, для удаления которой его расплавляют
в теплой воде, затем фильтруют, повторяя эту операцию до
тех пор, пока промывные воды не станут совершенно
нейтральными. Продукт окончательно очищают перегонкой в
глубоком вакууме (при этом происходит частичная полимеризация).
Выход 65,8 г, т. пл. 25,8*.
Фингер и другие [81 ] рекомендуют при нитровании бензотри-
фторида применять следующие условия: избыток
концентрированной азотной кислоты 10% против теории; начальная
температура реакции 30—35°, а к концу 60°. Выход 90% от теории.
Ходжсон и Бэрд [82] пользовались нитрующей смесью для
получения нитропроизводных ароматических альдегидов. Ни-
тробензалЪдегид был ими получен следующим образом. К смеси
160 мл азотной кислоты (уд. в. 1,4) и 700 мл концентрированной
серной кислоты добавляют в течение 2,5 часов 180 г
бензальдегида, при этом температура реакции не должна превышать 10°.
После внесения бензальдегида перемешивание продолжают еще
в течение 45 мин. Выделившийся продукт реакции — м-нитро-
бензальдегид отфильтровывают от жидкой части (масла). Для
удаления остатков маслянистого .продукта промывают
последовательно водой, а затем водным раствором соды, снова
водой и, наконец, сушат. Выход сырого м-нитробензальдегида
составляет 152 г. Маслянистый фильтрат экстрагируют эфиром,
вытяжку отфильтровывают, сушат над хлористым кальцием и
эфир отгоняют. При этом в остатке получается смесь о- и п-
нитробензальдегидов. Общий выход технических нитробензаль-
дегидов составляет 215 г. Из содовой вытяжки получают 10 г
смеси бензойной кислоты и о- и п-нитробензойных кислот.
С. Н. Ушаков и Е. Н. Фрейдберг [83] получили из толуола
смесь нитробензальдегидов. Реакция проводилась следующим
образом: 106 г толуола в течение 4—5 час. приливались к
раствору 110 г азотнокислого калия в 325 мл концентрированной
серной кислоты при 5*. Был получен м-нитробензальдегид
с выходом 46—61% от теории. Из маточного раствора
выделяли 25% орто-изомера. Если вести реакцию при 25—35°, то
выход мета-изомера повышается до 75—78%.
амесью дэотной w серной жыслот 41
При взаимодействии 25 мл толуола со смесью 18 мл дымящей
азотной кислоты и 63 мл серной кислоты в течение 3 час. при
температуре 40°, а затем 90° (при температуре выше 100*
реакция протекает со взрывом) Ниши и Токи [84] после очистки
сырого продукта получили 18 г м-нитробензойной кислоты.
С высокими выходами получаются нитробензальде-
гиды из коричной кислоты [85]. 30 г коричной кислоты в
250 мл концентрированной серной кислоты обрабатывается
при температуре ниже 20° 11 мл азотной кислоты (уд. в. 1,52).
После соответствующей обработки получают о-нитробензальде-
гид с выходом 50% от теории и 39% п-иитробензальдегида.
Нитрованием фталевого ангидрида А. А. Пономаренко [86]
получил 3-нитрофталевую кислоту с выходом 25% от теории.
Нитрующую смесь применяют для получения динитрофе-
нилуксусной кислоты [87] с высоким выходом. К 163 г фенил-
уксусной кислоты яри 0° постепенно приливают смесь 260 мл
дымящей азотной кислоты и 600 мл концентрированной серной
кислоты с такой скоростью, чтобы температура реакции была
60°. Получают 2,4-динитрофенилуксусную кислоту с выходом
95% от теории.
Нитропроизводные диарилсульфата [88] получают при
обработке 16 г его смесью азотной кислоты уд. в. 1,48 (11 г) и
серной кислоты уд. в. 1,84 (14,3 г). После перекристаллизации
полученного сырого продукта (21 г) получают чистый п-нитро-
фенилсульфат (7 г). При обработке последнего смесью азотной
кислоты и олеума (20% 8Оз) получают 2,4-динитрофенилсуль-
фат, который после гидролиза дает 2,4-динитрофенол.
Постепенным прибавлением 1 мл п-бромтолуола к смеси
4 мл серной кислоты (98 % -ной) и 2 мл азотной кислоты (70 % -ной)
при 90° получают 2,3-динитро-4-бромтолуол [89].
Нитрование 2,3,5-трибромтолуола смесью дымящей азотной и
концентрированной серной кислот дает 3,5,6-трибром-2,4-дини-
тротолуол [90].
Для получения нитроаминов обычно предварительно
защищают аминогруппу, замещая атом водорода в аминогруппе
на ацильную группу. При постепенном внесении раствора
100 ч. ацотанилида в 400 ч. серной кислоты в смесь из 76 ч.
42 JEfumpogaKug адхьмдлымесжы* u
серной кислоты (91%-ной) и 73 ч. азотной кислоты (61,3%-вой)
Мак-Кормак [55] получил п-нитроацетааидид с выходом 83,5%
от теории.
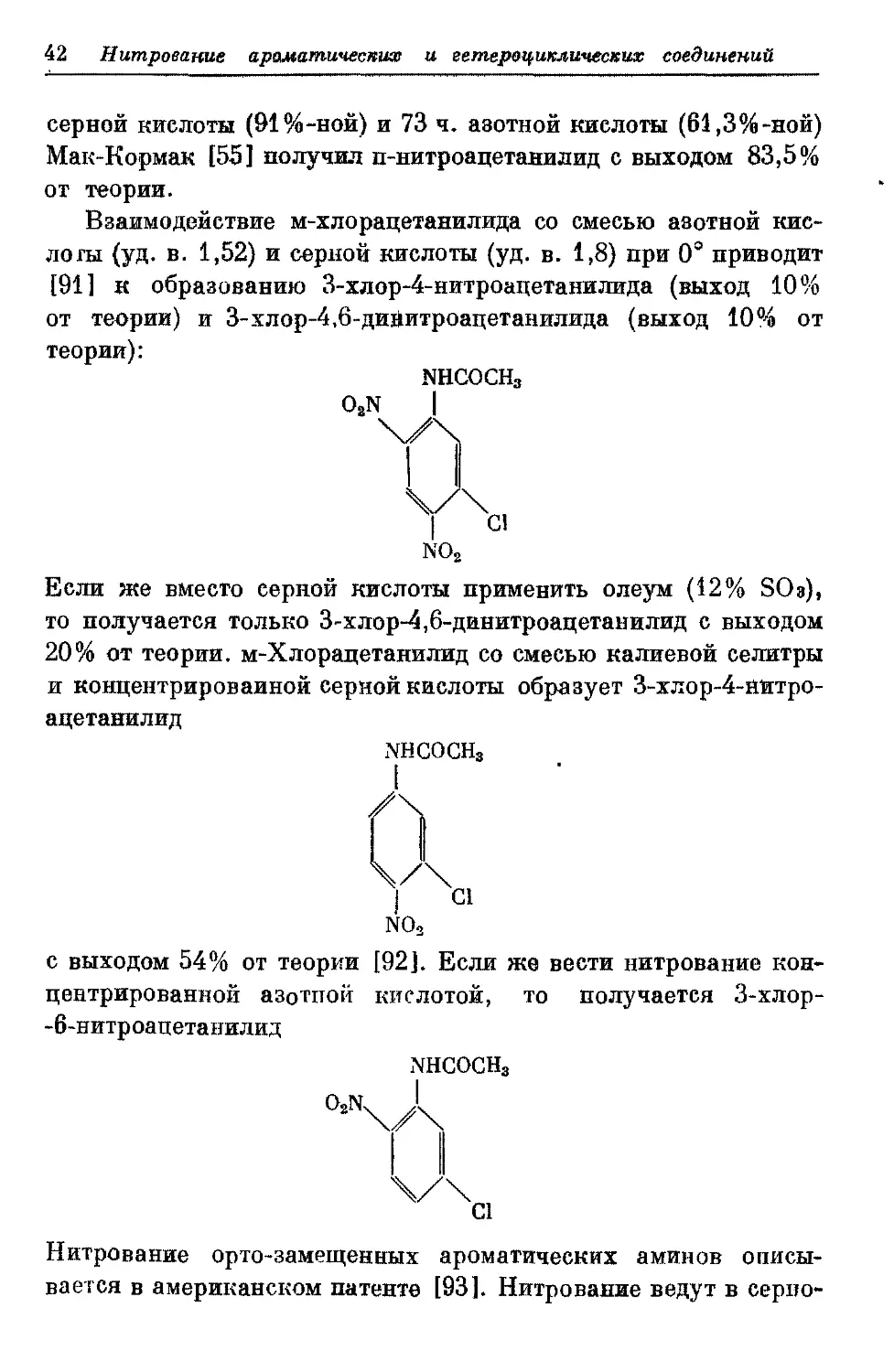
Взаимодействие м-хлорацетанилида со смесью азотной
кислоты (уд. в. 1,52) и серной кислоты (уд. в. 1,8) при 0" приводит
[91] к образованию 3-%лор-4-нитроацетанилпда (выход 10%
от теории) и 3-хлор-4,6-диаитроацетавилида (выход 10% от
теория):
NHCOCH,
N0*
Если же вместо серяой кислоты применить олеум (12% SO в),
то получается только 3-хлор-4,6-динитроацетанилид с выходом
20% от теории. м-Хлорацетаяилид со смесью калиевой селитры
и концентрированной серной кислоты образует З-хлор-4-ажтро-
ацетаяилид
NHCOCH,
с выходом 54% от теории [92]. Если же вести нитрование кон-
центрированной азотной кислотой, то получается 3-хлор
-6-нитроацетаниляд
NHCOCH,
Нитрование орто-замещевных ароматических аминов описы*
вается в американском патенте [93]. Нитрование ведут в серно-
43
кислом растворе амина при 0—5°. В качестве примера
приводится нитрование о-анизидина, который дает 5-нитро-о-аии-
зидип.
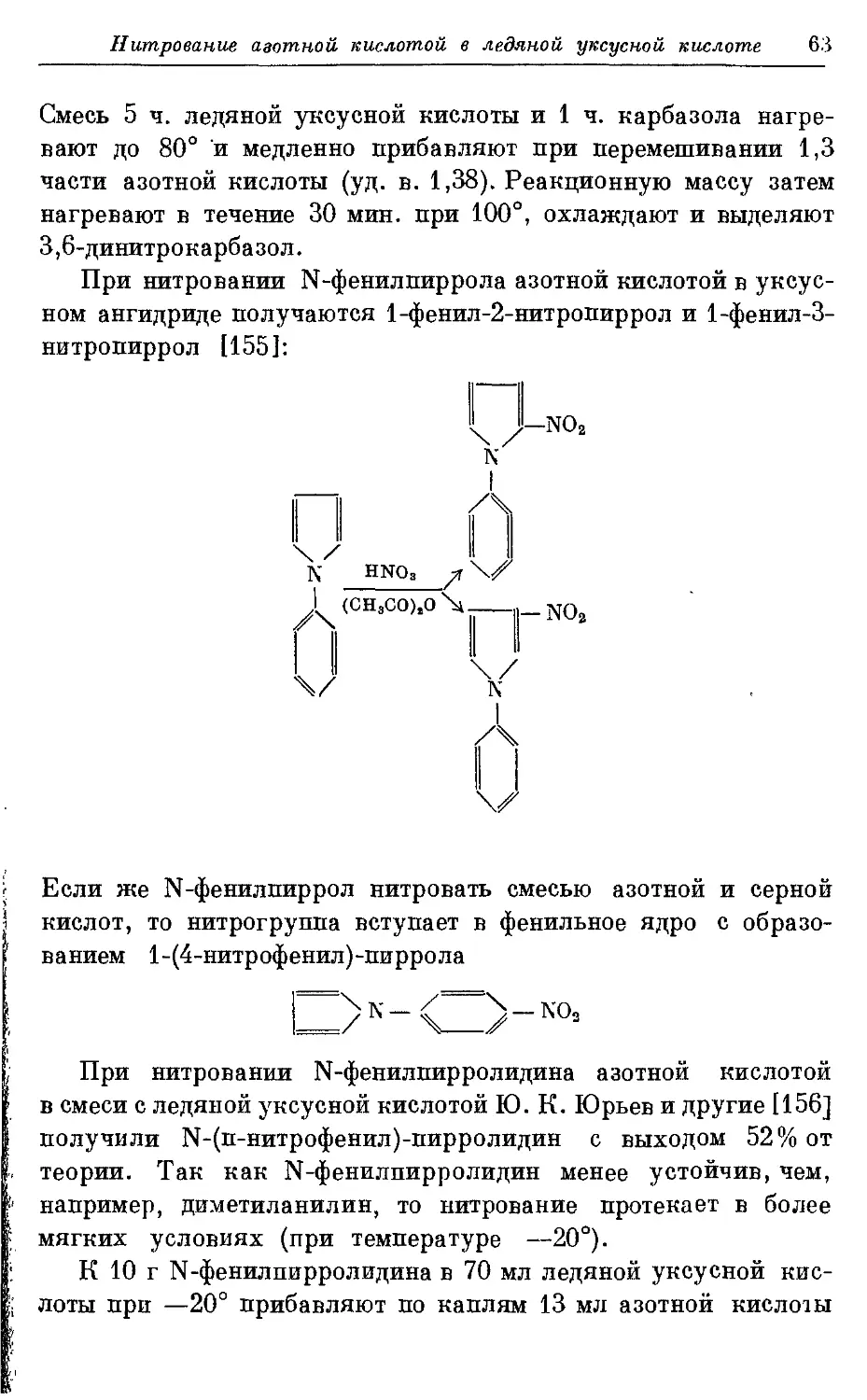
Весьма гладко протекает нитрование ацяльных
производных дифениламина нитрующей смесью. При этом получают 2,4-
динитро дифениламин [941 с теоретическим выходом. Если же
вести нитрование смесью дымящей азотной кислоты и
концентрированной серной, то получается 2,4,6-тринитро дифениламин.
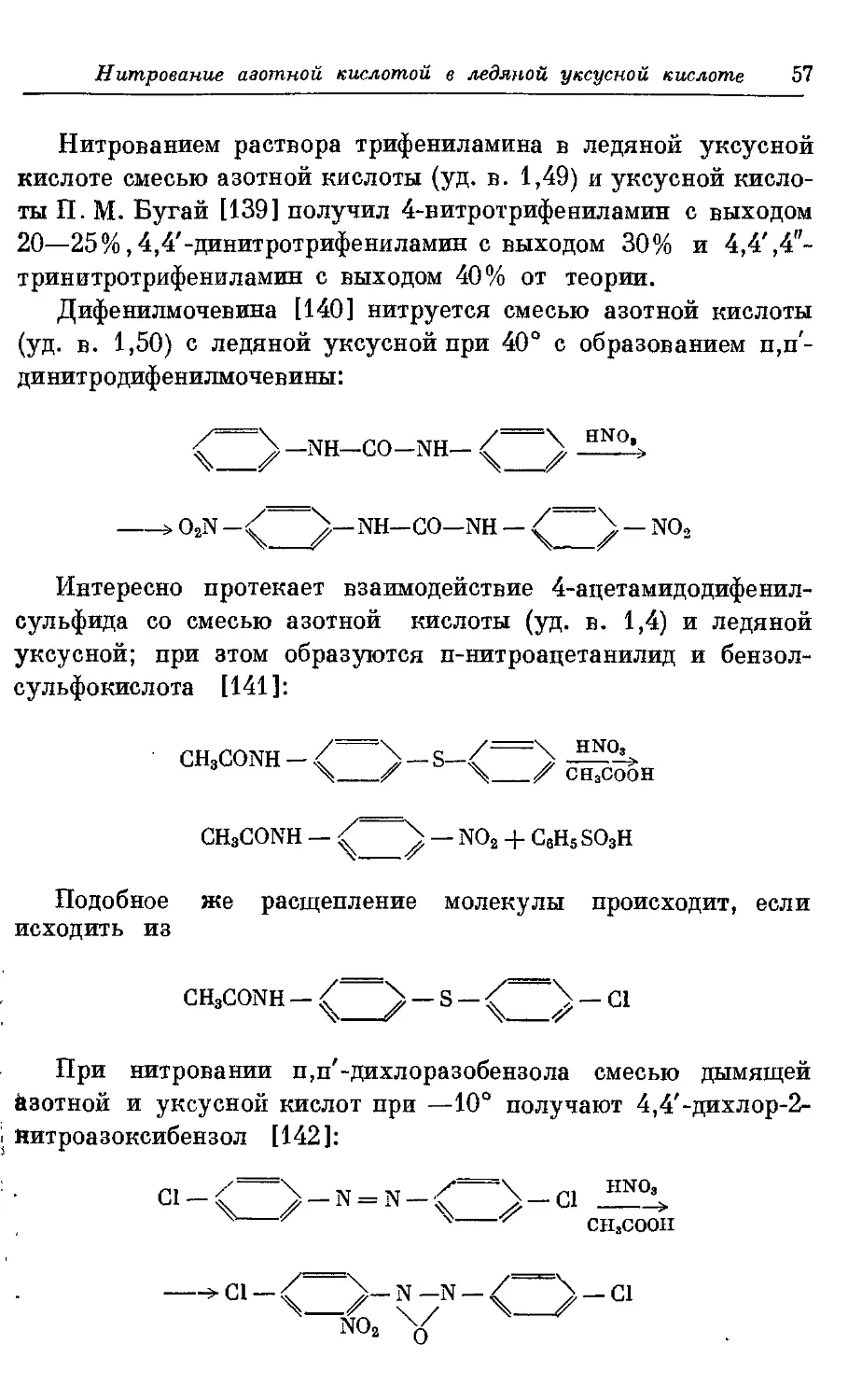
Получение нитропроизводных дифенилмочевины было
изучено И. М. Коганом и Д. Ф. Кутеповым [95]. При нитровании
дифенилмочевины смесью 68,3%-ной азотной кислоты и
концентрированной серной кислоты при 0° получается 4,4'-ди-
иитродифенилмочевина
Тетранитропроизводное при этом обнаружить не удалось.
Изучение влияния концентрации азотной кислоты показало,
что наилучшие выгоды получаются при использовании 48—
67%-ной HNOg. Оптимальная температура реакции —15*.
Интересно отметить, что концентрация серной кислоты в ряде
случаев оказывает большое влияние и на положение
вступающей в ядро нитрогруппы. Так, например, при нитровании ацет-
аиплида нитрующей смесью замена моногидрата 78,8%-ной
серной кислотой приводит к увеличению выхода орто-изомера
ia счет пара-изомера [96].
Большой практический интерес представляют нитропроиз-
водные нафталинового ряда. Нитрование нафталина при 25°
смесью 83 ч. азотной кислоты (61,3%-ной) и 233 ч. серной
кислоты (91%-ной) приводит к образованию %-нитронафталина
с выходом 85% от теории.
В. А. Ленхольд (лаборатория Кинешемского ззвода)
доказал образование в этих условиях очень незначительной при-
lii меси р-нитрояафталина.
: При действии нитрующей смеси на нафталин (в более жест-
i кмх условиях) получается смесь 1,5- и 1,8-дпнитронафталина.
44
Получение динитронафталина обычно проводят в две
стадии: вначале одной азотной кислотой (^62%-ной) при
охлаждении превращают нафталин в %-мононитронафталин, а
затем к полученной реакционной массе медленно при
размешивании и охлаждения приливают смесь 3 ч. серной кислоты
(92%-ной), 1 ч. олеума (60% 80s) и 1,5 ч. азотной кислоты
(;—62%-вой). После соответствующей обработки получаются
1,5-динитронафталин с выходом 27% от теории и 1,8-динитро-
нафталин с выходом 67% [97]. При последующем нитровании
смеси динитронафталинов получается смесь три- и тетранитро-
нафталинов [98], которые находят применение в качестве
взрывчатого вещества.
Если нитровать 2-метилнафталин [99], то получается смесь
изомеров, причем основным продуктом реакции является
1-нитро-2-метилнафталин. Наибольший выход (57%) получается
при применении 70%-вой HNOs с избытком 70% при
температуре от 0 до 30*. Если применять 15%-ный избыток смеси,
состоящей из 25% HNOs, 55% H^SO* и 20% ЩО, то в качестве
основного продукта реакции также получается 1-нитро-2-ме-
тилнафталин. Если же применять при нитровании уксусный
ангидрид, то 2-метилнафталин дает динитропроизводное.
Нитрование %-нафтола протекает гладко: при зтом
получается 2,4-динитро-%-нафтол. А, А. Пономаренко и Т. Е. Ма-
циевич [100] нитрованием 1 моля тетрагидронафталина смесью
170 г азотной кислоты (уд. в. 1,48) и серной кислоты (уд. в. 1,83)
получили 1,2,3,4-тетрагидро-6,8-дииитронафталин с выходом
33% от теории.
При нитровании 5-нитро-1-нафтойной кислоты нитрующей
смесью реакция идет уже при 0°, при зтом получается 4,5-
динитро-1-нафтонная кислота с выходом 52% от теории [101].
Отмечается, что если вместо серной кислоты применять
ледяную уксусную кислоту, то нитрование не идет даже при
нагревании.
Из других нитропродуктов полициклических соединений
следует отметить динитроантрахинон, который получается
нитрованием растворенного в крепкой серной кислоте антрахи-
пона нитрующей смесью при 70—80°. При зтом образуется
слисмо бдопъноД и серной кысдот 45
смесь, содержащая главным образом 1,5- и 1,8-динитроантра-
хиной наряду с другими изомерами [102]. При нитровании
фенантрона получается смесь 9-, 2-, 4- и 3-мононитрофенаитре-
на, и в небольшом количестве (1%) образуется 1-нятрофеваа-
трен [103].
Смесь азотной и серной кислот применяется также для
нитрования труднонитрующихся соединений, например,
пиридина. По мнению ряда исследователей, содержащийся в
пиридине третичный атом азота оказывает тормозящее действие
на нитрование; при нейтрализации отрицательного влияния
третичного азота введением амино- или гидроксильной группы
нитрование пиридина протекает удовлетворительно.
Фридль [104] осуществил нитрование пиридина, действуя
на него смесью серной и азотной кислот (последняя получилась
ги cWw иаасегмй при взаимодействия азотнокислого калия
с серной кислотой). В раствор 20 г пиридина в 120 г олеума
(18% 80s), который нагревали на песчаной бане, вносили
частями 110 г азотнокислого калия и одновременно через
реакционный сосуд и приемник пропускали сильный ток воздуха.
Нитрование производили в течение 1 часа при 330*. Дестиллят,
состоящий из азотной кислоты, пиридина и нитропиридина,
выпаривали на водяной бане для освобождения от большей
части азотной кислоты и после нейтрализации содой
обрабатывали эфиром (в эфир переходили нитропириджн и избыток
пиридина). После отгонки зфира и пиридина остаток
растворяли в азотной кислоте. Из раствора при стоянии выделялись
кристаллы нитропиридин-нитрата; последний растворяли в
разбавленной азотной кислоте и, добавляя соду, выделяли нитро-
пиридин. Очистку сырого продукта производили
растворением его в бензоле или спирте и осаждением из этих растворов
лигроином или водой. Нитруя пиридин указанным методом,
Фридль получил нитропиридип(т.пл. 41°) с выходом 15% от
теоретического.
Восстановлением в аминопиридин, который был
идентифицирован как ^-аминопиридин, Фридль доказал, что
продуктом нитрования является 3-нитропиридкн. В данном случае
подтвердилось правило, что присутствие третичного атома
46 Д^шм^обдуюб apwwimww^CJM&p и
азота ориентирует заместители отрицательного характера
прежде всего в р-положеяпе.
При нитровании 1-оксипиридина смесью
концентрированной серной и азотной кислот при 100° получается 4-нитро-1-
оксипиридив [105]. Выход продукта увеличивается при
применении дымящей серной кислоты и при температуре 130\
Дальнейшее увеличение температуры до 140° приводит я
уменьшению выхода.
Нитрованием 1-оксипиридина смесью дымящей серной
кислоты и калиевой селитры получают 4-нитро-1-оксипиридин с
выходом 52,4% от теории [106].
При прибавлении 4,5 г 3,5-диэтоксипирижна в 10 мл
концентрированной серной кислоты к смеси 11 мл крепкой
азотной и 2 мл дымящей HNO* в течение 30 мин. при 70° получают
3,5-димокси-2-вжтрошфидин с выходом 45% от теории [107 L
Если же вести нитрование при более высокой температуре (80°)
и с большим избытком дымящей азотной кислоты, получают
3,5-д*этокси-2,6-динятро1шридиа с выходом 40% от теории.
4-Нитропвридин-М-оксид [108] с выходом 85—90% от теории
получается при прибавлении по каплям 1,5 г пирждвн-N-
оксида в 3 мл концентрированной серной кислоты к смеси
5 мл дымящей азотной (уд. в. 1,52) ж 3 мл концентрированной
серной кислоты. По окончании приливания реакционную
массу нагревают 1,5 часа при 90*, выливают на лед, подщелачивают,
экстрагируют эфиром и после удаления эфира выделяют
готовый продукт.
Главным продуктом реакции при действии нитрующей
смеси на хиволин [109] являются два изомера мононитропродукта»
которые образуются в равных количествах, а именно:
5-нитрохинолин 8-нитрохиаолин
\/
ел*а?ь# даотноб и серкоё жыслол* 47
Введение в хинолиновое ядро второй нитрогруппы с
помощью нитрующей смеси представляет значительные
трудности и не всегда возможно.
Кауфман и Деккер [НО] разработали метод получения ди-
иитропроизводньп хинолина из соотАетствующих мононитро-
производных. 6-Нитрохинолин Же нитруется смесью азотной
и серной кислот даже при кипячении в течение многих дней.
Нитрование же 6-нитрохинолина рассчитанным количеством
KNO@ и H*SO# в запаянной трубке в течение 10 час. при 130—
140* дает два динитропропзводных: 6,8-Динитрохияолин
с т. пл. 154° (идентичный полученному синтезом из 2,4-дини-
троанилипа) и 5,6-джнитрохинолин с т. пл. 185°. Значительно
легче протекает нитрование 7-нитрохинолина. При
нагревании 5 г последнего смесью 100 мл азотной кислоты (уд. в. 1,5)
и 150 мл концентрированной сержой кислоты в течение 6 час.
с обратным холодильником Кауфман и Деккер получили два
нитропроизводных: одно ст. пл. 225° и другое с т. пя. 175*(по
форме кристаллов и другим физическим свойствам последнее
соединение представляло собой, невидимому, 5,7-дивжтрохинолин).
8-Нитрохинолин при действии нитрующей смеси в течение
20 час. при нагревании дал с дочти количественным выходом
6,&-дйнитрохинолин ст. пл. 154°, идентичный продуктам,
получающимся при нитрования 6-нитрохинолина (см. выше),
а также синтезом из 2,4-дишюроапилииа. 6,8-Динитрохинолин
может быть получен в чистом виде (без примеси 5,6-дюштрохи-
аолииа), если 6-нитрожинолин (1 ч.) нитровать смесью Г ч.
SO* с 2 ч. концентрированной азотной кислоты при
перемешивании и постепенном повышении температуры до 100° в
течение 2 час. (6-нитрожиполин вносится в Нитрующую смесь).
При нитровании 5-нитрохинолина по этому же методу получеа
5,8-динитрохинолин [111]. 1-Оксихинолин при действии смеси
H2SO4+ KNO* при 70" дает 1-окси-4-нитрохинолин с хорошим
выходом [105].
При прибавления раствора 10 г 4-оксихинолина в 45 мл конц.
H3SO4 при 0—5** к смеси 10 мл азотной кислоты (уд. в. 1,5) и
10 мл концентрированной серной кислоты Адаме [112] получил
9,9 г 6-нитро-4-оксихинолина.
48
При нитровании 2,4-диметилхинолина получается, наряду
с 8-нитро-2,4-диметилхинолином, также 6-нитро-2,4-диметил-
хннолин [ИЗ], разделение которых производится при помощи
разбавленной горячей серной кислоты. 4,6-Диметилхинолин
с нитрующей смесью при температуре ниже 0* дает с очень
хорошим выходом 5-нитро-4,6-диметилхинолин [114].
По вопросу нитрования 4-аминохинолнна см.
оригинальную литературу [106, 115, 116].
С очеяь хорошим выходом и в качестве единственного
продукта реакции получается 4-хлор-6-метокси-8-нитрохинальдин
[117] при обработке 4-хлор-6-метоксихинальдина смесью
серной кислоты и калиевой селитры.
При нитровании бензотиазола смесью азотной кислоты
(уд. в. 1,5) и концентрированной серной кислоты при 0°
получают смесь 6-нитробензотиазола и мононитропроизводного
бензотиазола, в котором положение нитрогруппы точно не
установлено [118].
Предполагают, что это нитропроизводное является
молекулярным соединением 6-нитро- и 7-нитропроизводного
бензотиазола.
Нитрование 5-метил-2-тиофенкарбонобой кислоты
нитрующей смесью при —5* приводит к образованию 4-нитро-5-ме-
тил-2-тиофенкарбоновой кислоты [119] с хорошим выходом
наряду с образованием в небольшом количестве динитро-2-
метилтиофена. Если проводить реакцию при более высокой
температуре, то выход мононитропроизводного тиофенкарбо-
новой кислоты падает и увеличивается количество динитропро-
изводного тиофена.
Клоза [120] осуществил нитрование 4-оксикумарина смесью
серной и азотной кислот. При этом получается нитрооксику-
марин с плохим выходом. Нитрование 4-оксикумарина в среде
хлороформа протекает с лучшим выходом по схеме:
ОН ОН
о
ж%*слотой @ лбблной ужсусмой кис^оттге 49
Азотсодержащие производные кумарина представляют
интерес в физиологическом отношении (связь с антибиотиками).
§ 3. Нитрование азотной кислотой в ледяной уксусной кислоте
или в уксусном ангидриде
Довольно распространенным методом нитрования
ароматических соединений является нитрование смесью азотной
кислоты с уксусной кислотой, а также с уксусным
ангидридом.
Систематическое исследование процесса нитрования
ароматических углеводородов смесью азотной и уксусной кислот
проведено М. И. Коноваловым и К. Гуревичем [121]. Авторы
изучали нитрование толуола, ксилолов (орто-, мета-и
пара-изомера), этилбензола, дизтилбензола и других, применяя
азотную кислоту уд. в. 1,495 и ледяную уксусную кислоту. Во
всех случаях, за исключением этилбевзола и диэтилбензола,
реакция проводилась при кипячении с обратным
холодильником в течение 1,5—2 час.
В случае же этилбензолов реакция осуществлялась при
легком нагревании. По окончании нагревания реакционную массу
выливали на воду, промывали содой и обрабатывали водно-
спиртовым раствором щелочи для удаления продуктов
нитрования боковой цепи. Соединения с нитрогруппой в боковой
цепи выделялись пропусканием углекислоты и
экстрагированием эфиром. Продукты нитрования в ядро извлекались
эфиром, а затем фракционировались.
На основе своих исследований авторы приходят к
следующим выводам.
1. Уксусная кислота при разбавлении ею азотной кислоты
играет ту же роль, что и вода. Она ослабляет действие
азотной кислоты на ароматическое ядро и направляет это действие
(при нагревании) на боковую цепь.
2. Способность реагировать с азотной кислотой,
смешанной с уксусной, для различных углеводородов в общем
аналогична способности реагировать с разбавленной азотной
кислотой.
4 А. В. Топчиев
50
u
3. Одновременно с нитрованием в боковую цепь происходит
и окисление ее (особенно при проведении реакции в жестких
условиях) с образованием соответствующих карбоновых
кислот.
Благодаря тому, что ледяная уксусная кислота является
растворителем, можно избежать применения избытка азотной
кислоты. Кроме того, ледяная уксусная кислота, а еще в
большей степени уксусный ангидрид при нитровании служат для
связывания образующейся в процессе реакции воды. При
нитровании смесью азотной и ледяной уксусной кислот
наблюдаются те же закономерности, что и при нитровании водной
азотной кислотой [122]. Замена при нитровании
концентрированной серной кислоты уксусным ангидридом представляет
интерес в том отношении, что при этом исчезает свойственная
серной кислоте способность направлять нитрогрунну в
определенное положение.
Основными условиями, определяющими течение реакции
при нитровании ароматических углеводородов смесью азотной
кислоты с уксусным ангидридом, являются отношения
количества азотной кислоты к количествам воды и нитруемого
соединения.
Нитрование толуола азотной кислотой, взятой в избытке
по отношению к углеводороду (3—4 моля НМОз на 1 моль
толуола), в отсутствие уксусного ангидрида приводит лишь
к небольшому (17—18%) выходу нитропроизводных (отношение
азотной кислоты к воде, имеющейся в растворе и
образующейся при нитровании, составляет 2,66 : 1). При проведения
реакции в присутствии уксусного ангидрида получается
количественный выход нитропроизводных (смесь о- и п-нитротолуолов)
(123].
Смесь азотной и уксусной кислот с успехом применяется
для нитрования мезитилена.
Нитромезитилен получают с выходом 50% при нагревании
до кипения в течение 1,5 часа 1 ч. мезитилена,
растворенного в 4 ч. ледяной уксусной кислоты, с 1 ч. дымящей азотной
кислоты. Продукт реакции осаждают водой и, после отделения
от последней, подщелачивают едким натром и перегоняют
даотноЯ ммлотой б лбЭяной ужсусной кислоте 51
с водяным паром. Из водного дестиллята отгоняют мезитилен
и оставшийся нитромезитилен перекристаллизовывают из
спирта; при охлаждении нитромезитилен выделяется из спиртового
раствора в виде призматических кристаллов [124]. .
Бамбергер и Ризинг [125] для получения мононитромези-
тилена применяли следующий метод нитрования: 100 г
концентрированной азотной кислоты (уд. в. 1,51) вносят в смесь
100 г мезитилена и 400 мл ледяной уксусной кислоты, при
этом происходит энергичная реакция с выделением тепла, и
жидкость начинает кипеть; кипение жидкости поддерживают
еще в течение 50 мин. По окончании нитрования охлажденную
реакционную массу вливают в 1 л воды со льдом; выделившееся
при этом тяжелое масло отфильтровывают от взвешенных
кристаллов мезитиленовой кислоты и обрабатывают эфиром;
эфирную вытяжку несколько раз встряхивают с 20%-ным
раствором поташа и затем с 15%-ным раствором едкого кали для
извлечения мезитиленовой кислоты и ксилилнитрометана (последний
из щелочных растворов выделяется при насыщении СО*). Из
эфирной вытяжки отгоняют эфир и остаток подвергают
фракционированной перегонке (с паром); сначала отгоняется мези-
тилен, а затем нитромезитилен; последний при охлаждении
выделяется в виде кристаллов с т. пл. 125°. Основным продуктом
реакции при этом способе нитрования служит
нитромезитилен в отличие от нитрования по М. И. Коновалову [126], когда
преобладающим продуктом является ксилилнитрометан,
т. е. замещение группой NO* происходит преимущественно
в боковой цепи.
Нитрование мезитилена может быть проведено смесью
азотной и ледяной уксусной кислот и уксусного
ангидрида [127]. Смесь 31,5 г дымящей азотной кислоты (уд. в. 1,51),
20 г ледяной уксусной кислоты и 20 г уксусного ангидрида
постепенно приливают к охлажденной до 10° смеси 40 г
мезитилена и 60 г уксусного ангидрида. Скорость приливания и
охлаждение регулируют таким образом, чтобы температура
была не выше 20°. По окончании прилив ания реакционную
массу оставляют при комнатной температуре в течение 2 час,
нагревают до 50°, а затем по охлаждении дылипают при переме-
52
шнвании в 800 мл ледяной воды. Затем прибавляют 40 г
хлористого натрия, продукт отделяют, извлекают эфиром водный
слой и вытяжку присоединяют к основной массе продукта.
Далее эфирную вытяжку промывают 10—15%-ным раствором
едкого натра (до явно щелочной реакции), эфир отгоняют,
прибавляют к остатку 150 мл 10%-ного раствора едкого натра и
отгоняют нитромезитилен с водяным паром. В полученном
дестилляте отделяют маслянистый слой, который растворяют
.в эфире. После отгонки на водяной бане эфира перегоняют
остаток при температуре 243—250°. Полученный сырой продукт
очищают перекристаллизацией из метилового спирта и
получают нитромезитилен с выходом 75—76% от теории.
Нитрование о-ксилола постепенным прибавлением его к
смеси дымящей азотной кислоты с половинным по объему
количеством ледяной уксусной кислоты при 20—25* после
соответствующей обработки реакционной массы приводит к
образованию 4-нитро-о-ксилола [128] в виде желтых призм с т. пл. 30°.
Из фильтрата можно выделить 3-нитро-о-ксилол. о-Ди-
этилбензол нитруется смесью азотной и уксусной кислот при
температуре 10—20* с образованием 1,2-дизтил-4-нитробензола.
Выход 41% от теории [129].
Синтез нитрофенолов также осуществляется обработкой
фенолов смесью азотной и уксусной кислот. Например,
нитрование м-крезола можно проводить следующим образом: смесь
140 г м-крезола и 140 г ледяной уксусной кислоты охлаждают
до 5° и медленно вливают в охлажденную до —15° смесь,
состоящую из 200 г азотной кислоты (уд. в. 1,5) и 400 г ледяной
уксусной кислоты. После добавления всего количества
м-крезола реакционную смесь оставляют еще на 1,5 часа
(температура не должна опускаться ниже —1°). Для выделения
продуктов реакции массу выливают в воду со льдом; при этом
отделяется вязкая жидкость, которая через некоторое время
закристаллизовывается; при перегонке с паром получаются
4-нитро-м-крезол (т. пл. 56°) и 6-нитро-м-крезол (т. пл. 129°)
[130].
При прибавлении о-пропилфенола (20,4 г) в уксусной
кислоте (20,4 г) к смеси 40,8 г азотной кислоты (уд. в. 1,42) и 61,2 г
53
уксусной при температуре от —4 до —6* получается 2-нптро-6-
иропилфенол с выходом 39% от теории [131].
2,6-Ди-трет. бутилфенол нитруется смесью
концентрированной азотной и уксусной кислот (1 : 1) с образованием 4,6-
динитро-2-трет. бутилфенола [132].
Хлористый бензил нитруется смесью азотной кислоты и
уксусного ангидрида при 25° с образованием 32% орто-, 15,5%
мета- и 52,5% пара-нитробензилхлорида [133].
5-Нитропроизводное о-метоксифенола (гваякола)
получается [134] прибавлением раствора 135 г ацетильного
производного гваякола в 120 мл уксусной кислоты к раствору 115 мл
азотной кислоты (уд. в. 1,5) в 180 мл уксусной кислоты. При
этом температура поднимается до кипения. Полученный
продукт гидролизу ют 50 мл 10%-ного едкого натра. Выход 5-нитро-
производного, считая на исходное ацетильное производное,
составляет 42% от теории.
Базани и Пианка [135] предлагают улучшенный метод
нитрования ароматических о-оксикарбоновых кислот и их зфи-
ров, который состоит в следующем: 1 моль соответствующего
соединения в небольшом количестве уксусной кислоты быстро
обрабатывается смесью 1 моля азотной кислоты с 9 объемами
уксусной при комнатной температуре и затем нагревается до
тех пор, пока раствор станет коричневым. Полученное нитро*
соединение высаживают водой. Салициловая кислота дает при
этом 5-витропроизводное
ОН
СООН
с выходом 41% or теории. Метиловый и этиловый эфир
салициловой кислоты дают 5-нитропроизводное с выходом
соответственно 51 и 64% от теории.
Нитрование п-оксибензальдегида [136] смесью азотной и
ледяной уксусной кислот приводит к образованию 2-нитро-
54
4-оксибензальдегида:
ОН
У\
Нитрованиерастворап-оксибензалъдегидавледянойуксусной
кислоте ведется при слабом нагревании с азотной кислотой уд. в. 1,4.
При нитровании бензойной кислоты смесью азотной
кислоты и уксусного ангидрида получается с хорошим выходом
м-нитробензойная кислота.
Смесь азотной и уксусной кислот с успехом применяют для
превращения анилина и его производных в соответствующие
нитросоединения. Как известно, при нитровании азотной
кислотой обычно обрабатывают ею не анилин, а ацетанилид. В этом
случае происходит образование преимущественно п-нитроаце-
танилида и лишь в незначительных количествах о-нитроаце-
танилида. Витт и Утерман [137] показали, что применение
вместо азотной кислоты смеси ее с уксусной кислотой
существенно изменяет отношения образующихся изомеров.
По методу Витта и Утермана, в раствор 45 г ацетанилида
в 22 г ледяной уксусной кислоты при охлаждении вливают
раствор 23 г азотной кислоты (уд. в. 1,5) и 1 г мочевины в 23 г
ледяной уксусной кислоты; смесь оставляют на 24 часа.
Выделение сырого продукта реакции и разделение изомеров
производят следующим образом: к реакционной смеси добавляют
360 г льда и выпавший осадок, состоящий из смеси о- и п-ни-
троацетанилидов, обрабатывают спиртовым раствором едкого
кали; при этом омылению подвергается лишь о-нитроацетани-
лид, а п-изомер остается без изменения. Основным продуктом
реакции (в отличие от нитрования азотной кислотой в отсутствие
уксусной кислоты) является о-нитроацетанилид(отношениеколи-
честв о- и п-изомера равно 3:1). Общий выход нитропроизвод-
ных (в виде сырого продукта) составляет 87% от теоретического.
Нитрование производных анилина для получения нитро-
аминосоединений осуществляется также с хорошими
результатами при применении смеси азотной кислоты с уксусным
даотм*о& к^слотом. g лдАямой уксусной лсаслоте 55
ангидридом; так, например, триброманилин этим путем может
быть количественно превращен в трибромнитроаминобензол
(2,4,6-трибромфенилнитроамин C@HgBrgNHNOa) [123]. Метод
заключается в следующем. Нитруемое производное анилина
растворяют в 98—99%-ной ледяной уксусной кислоте и к
раствору при охлаждении приливают азотную кислоту в
отношении 2—3 моля на 1 моль нитруемого соединения. К получен-
вой смеси, также при охлаждении, медленно добавляют
уксусный ангидрид, следя за тем, чтобы температура не превышала
25°. Для получения хорошего выхода нитроаминов необходимо
применять азотную кислоту, не содержащую HNOg, так как
последняя может диазотировать нитроамин
Освобождение азотной кислоты от азотистой достигается
перегонкой в вакууме при 20—30 мм. Лучшие результаты дает
обработка азотной кислоты нитратом мочевины.
Нитрование аминосоединении, которые содержат мало
заместителей и поэтому легче могут подвергаться ацетилированию
(например, 2,4-дихлоранилин), можно производить,
растворяя нитруемое соединение в ледяной уксусной кислоте и
медленно приливая раствор к смеси, состоящей из азотной кислоты,
уксусной кислоты и уксусного ангидрида; нитрование
осуществляется в этом случае при низких температурах
(реакционный сосуд охлаждают водой со льдом). Этот метод дает
превосходные выходы нитросоединений при нитровании производных
анилина, у которых не замещены одно или оба о-положения по
отношению к аминогруппе. В качестве побочных продуктов
при нитровании этих соединений образуются в
незначительных количествах изомерные нитроаминам нитроанилины.
При нитровании диалкиланилинов по этому методу
обнаружено, что азотистая кислота активирует процесс, т. е.
оказывает действие, противоположное тому, которое она проявляет
при нитровании других производных анилина. Смесь уксусного
ангидрида с азотной кислотой, предварительно освобожденной
от азотистой кислоты, не реагирует с диалкиланилинами.
Если нитрование последних производить смесью уксусного
u
ангидрида с обычной азотной кислотой, содержащей
азотистую кислоту, то реакция протекает с выделением тепла, и
получаются динитропроизводные. Из N-диметиланилина,
например, образуется 2,4-динитро-К-диметиланилия.
Ацетанилид и п-хлорацетанилид нитруются смесью
азотной кислоты с уксусным ангидридом в соответствующие нитро-
производные с количественными выходами (оптимальное
отношение реагентов: 4 моля азотной кислоты, не содержащей
HNOg, на 1 моль нитруемого соединения). С другой стороны,
2,4-дихлорацетанилид при обработке смесью азотной кислота
и уксусного ангидрида не подвергается нитрованию.
Бамбергер [138], изучавший реакцию нитрования
анилинов смесью азотной кислоты с уксусным ангидридом, высказал
предположение о механизме этой реакции. По его
представлениям, анилин при действия азотной кислоты образует ани-
линнитрат, который затем превращается в фенилнитроамин
с потерей одной молекулы воды
Н Н Н
С,Н#( ->С,НбМ <^—Н -+ СЛ,Г?/ + Н,0 (1)
Н ^ ONOg . NO,
Уксусный ангидрид при этом играет роль дегидратирующего
агента. Дальнейшие исследования показали, что у
ароматических нитроаминов нитрогруппа, соединенная с атомом азота,
переходит в ядро в присутствии минеральных кислот,
являющихся катализаторами этой перегруппировки. Например, из
фенилнитроамина при каталитическом действии
минеральных кислот образуются о- и п-нитроанилины
NH,
СА-т-гю,-т>с,нУ (2)
N0,
Таким образом, применяя две указанные выше реакции (1)
и (2), можно осуществить нитрование ароматических оснований
в две стадии, из которых первая состоит во вступлении нитро-
группы в боковую цепь (получение ароматических
нитроаминов), а вторая — в переходе нитрогруппы в ядро (получение
нитроанилипов).
даотной кхклотлоД б лд&жноД уксусной кислоте 57
Нитрованием раствора трифениламина в ледяной уксусной
кислоте смесью азотной кислоты (уд. в. 1,49) и уксусной
кислоты П. М. Бугай [139] получил 4-витротрифениламин с выходом
20—25%,4,4'-йинитротрифениламинсвыходом 30% и 4,4',4"-
тринитротрифениламин с выходом 40% от теории.
Дифенилмочевина [140] нитруется смесью азотной кислоты
(уд. в. 1,50) с ледяной уксусной при 40° с образованием п,п'-
динитродифенилмочевины:
Ъ— NH—CO—NH —
Интересно протекает взаимодействие 4-ацетамидодифенил-
сульфида со смесью азотной кислоты (уд. в. 1,4) и ледяной
уксусной; при этом образуются п-нитроацетанилид и бензол-
сульфокислота [141]:
Подобное же расщепление молекулы происходит, если
исходить из
— С1
При нитровании п,п'-дихлоразобензола смесью дымящей
Азотной и уксусной кислот при —10° получают 4,4'-дихлор-2-
: йитроазоксибензол [142]:
-Cl ^
СИзСООИ
0
58
Это же соединение можно получить нитрованием п,п'-дихлор-
азоксибеазола.
Синтез нитродифенилов проводят также в среде уксусной
кислоты. Например, нитруют 1 моль дифенила 2 молями азот-
нон кислоты (уд. в. 1,5) в присутствии уксусной кислоты при
нагревании; при этом образуются п-нитродифенил (бесцветные
иглы, т. пл. 113°), о-нитродифенил (т. пл. 200—201°) и 3,5,4'-
тринитро-4-оксидифенил (т. пл. 202°) [143].
При нитровании окиси дифенилена смесью азотной и
уксусной кислот получается окись 2-нитродифевилена с выходом
56% от теории [144].
Томсон и другие [145] описывают получение с высокими
выходами моно- и динитропроизводных 1,5-диметоксинафталина.
10 г 1,5-диметоксинафталина в 400 мл уксусной кислоты
медленно нагревают со смесью 4 мл азотной (уд. в. 1,42) и 4 мл
уксусной кислот. Реакционную массу выдерживают 30 мин.
при 80°. При этом получается 4-нитро-1,5-диметоксинафталин
в виде желтых кристаллов с т. пл. 167° и выходом 89% от
теории. Если взять больший избыток азотной кислоты (8 мл вместо
4 мл) и кипятить реакционную массу,, то получается 4,8-ди-
нитропроизводное 1,5-диметоксинафталина с выходом 84%
от теории. Это соединение (2 г) в 150 мл уксусной кислоты яри
нагревании его с обратным холодильником в течение 1 часа
с 15 мл азотной кислоты дает 2,4,8-тринитро-1,5-диметокси-
нафталин с выходом 43% от теоретического. Этот продукт при
прибавлении его малыми порциями в азотную кислоту (уд. в.
1,5) при 0° дает2,4,6,8-тетранитропроизводноесвыходом 23%
от теории.
С хорошим выходом (70—77% теоретического) протекает
нитрование 1-окси-2-нафтойной кислоты и ее метилового эфира
смесью азотной и уксусной кислот. При этом получается
4-нитро-1-окси-2-нафтойная кислота [135].
Галоидные производные нафталина также нитруются
азотной кислотой в смеси с ледяной уксусной. Так, например,
нитрованием 2 г 1,5-дифторнафталина (в 8 мл ледяной
уксусной кислоты) 8 г азотной кислоты (уд. в. 1,52) при нагревании
на водяной бане в течение 4 час, получают 1,5-дифтор-4-нитро-
ддотноД кислотой б ^б^яной уксусном кислоте 59
нафталин с выходом 82%. Более энергичное нитрование
приводит к смеси трудноразделимых изомеров [146]. Нитрованием
1'-хлор-1-метилнафталина В. А. Измаильский и А. И.
Козин [147] получили его нитропроизводное (8-нитро-1-хлор-
метилнафталин).
Нитрование ацетил-^-нафтиламина приводит к образованию
1-нитро-2-ацетилнафтиламина [148]. 200 г азотной кислоты
(уд. в. 1,4) медленно и при постоянном перемешивании
добавляют к смеси 300 г ацетил-^-нафтиламина в 500 мл ледяной
уксусной кислоты. При помощи охладительной смеси
поддерживают температуру реакционной массы не выше 40°. После
прибавления всей азотной кислоты перемешивают еще в
течение 10 мин. и затем реакционную массу оставляют в ледяной
воде в течение 3 час. Полученные кристаллы отсасывают,
промывают 50%-ной уксусной кислотой и эфиром и перекристал-
лизовывают сначала из бензола, а затем из 95%-ного спирта.
Полученный нитропродукт имеет т. пл. 123—124°. Выход
f—50% от теории.
Смесь азотной и уксусной кислот может быть применена
также для нитрования антрацена. При действии 63%-ной
азотной кислоты на антрацен, суспендированный в ледяной
уксусной кислоте, при температуре не выше 30—35* получается
антрацен-нитроацетат
/\
Н ОСОСНз
При добавлении воды к этому соединению выделяется вязкая
масса, которая при нагревании с разбавленным раствором
едкого натра постепенно переходит в нитроантрацен. Иитро-
антрацен может быть получен также обработкой реакционной
смеси, содержащей антрацен-нитроацетат, концентрированной
серной кислотой при комнатной температуре [149].
Мейзенгеймери Коннераде [150] исследовали действие
азотной кислоты на антрацен в присутствии уксусной кислоты и
60
w
уксусного ангидрида. К взвеси 50 г антрацена в 200 мл
ледяной уксусной кислоты медленно приливали 20 мл азотной
кислоты (уд. в. 1,4); реакцию проводили при охлаждении, причем
температура поддерживалась в интервале 30—35*. По
истечении 15 мин. в реакционной колбе образовался прозрачный
раствор, который выливали тонкой струей в холодную воду;
выделившуюся при этом желтую массу промывали водой и
после сушки в вакууме растворяли в эфире. Из раствора после
долгого стояния выделялись кристаллы антрацен-нитроаце-
тата
Н ОСОСНД
Выход составлял 45% от теоретического.
При применении избытка азотной кислоты Мейзенгепмером
и Коннераде были получены другие продукты реакции. И 20 г
смеси антрацена с 80 мл ледяной уксусной кислоты медленно
приливали при охлаждении 20 мл концентрированной азотной
кислоты (уд. в. 1,4), разбавленной 40 мл уксусной кислоты.
Температура реакции поддерживалась в интервале 30—35°.
По истечении двух часов из реакционной смеси начали
выделяться кристаллы. После двухдневного стояния выделившиеся
кристаллы были отфильтрованы от жидкой части, промыты
уксусной кислотой и высушены. Из этих кристаллов исследователи
выделили следующие продукты:
динитроантрацен (т. пл. 294°)
NO,
NO,
даотмной кислотой g лбДяной уксусной кыслолм 61
тринитродигидроантрацен (т. пл. 139—140")
Н
нитроантрон (т. пл. 148°)
О
Нитро- и динитроантрацены могут быть получены и прямым
нитрованием антрацена, если в качестве нитрующего агента
применять смесь азотной кислоты с уксусным ангидридом и
уксусной кислотой. Метод нитрования состоит в следующем:
10 г тонко измельченного антрацена вносят в 40 мл ледяной
уксусной кислоты и к этой смеси медленно приливают из
капельной воронки при сильном охлаждении смесь 10 мл не-
содержащей окислов азота азотной кислоты (уд. в. 1,5), 6 мл
уксусного ангидрида и 15 мл ледяной уксусной кислоты.
Температуру реакции поддерживают в интервале 15—20°. Через
некоторое время из реакционной смеси начинают выделяться
кристаллы, которые отфильтровывают через 24 часа по
окончании реакции, промывают спиртом и обрабатывают кипящим
спиртом; при этом в раствор переходит нитроантрацен, который
выкристаллизовывается при охлаждении (т. пл. 145—146°).
Нерастворившиеся в спирту кристаллы представляют собой
динитроантрацен, который очищают перекристаллизацией из
ксилола.
Нитрование фенантрена смесью азотной кислоты и
уксусного ангидрида изучено Шмидтом иГейнле [151]. 80 г чистогс
фенантрена растворяли в 160 мл горячей уксусной кислоты и
к раствору при нагревании на водяной бане медленно добавляли
смесь 120 мл уксусного ангидрида и 60 мл концентрировав-
ной азотной кислоты (уд. в. 1,45). После добавления смеси
62
азотной кислоты и уксусного ангидрида реакционную жидкость
еще некоторое время нагревали на водяной бане до полного
прекращения выделения нитрозных газов. Продукт реакции
выливали в холодную воду, при этом выделялся осадок нитро-
фенантренов; последний отделяли от жидкой части и промывали
водой до полного удаления азотной кислоты и уксусного
ангидрида. Для очистки сырого продукта от смолистых примесей
его растворяли в спирте. Из этого раствора посредством
дробной кристаллизации выделено четы; е мононитрофенантрена:
2-нитрофепантрен с т. пл. 99°, 3-нитрофенантрен с т. пл. 170—
171°, 4-иитрофенантрен с т. пл. 80—82° и 9-нитрофенантрен
с т. пл. 116—117°. Основным продуктом реакции оказался 9-
нитрофепантрен (60% общего количества мононитрофенантре-
нов, содержащихся в сыром продукте). Общий выход моно-
нитрофенантренов составляет 60—61% от теории.
Для превращения галоидных производных фепантренов
в соответствующие нитрогалоидопроизводпые можно также
пользоваться смесью азотной кислоты с уксусным ангидридом.
Например, к смеси 9-бромфенантрена (20 г), уксусной кислоты
(20 мл) и уксусного ангидрида (8 мл) медленно приливают 5 мл
азотной кислоты (уд. в. 1,45) при нагревании на водяной бане
в течение 20 мин. При охлаждении реакционной смеси
выделяется твердый продукт, который перекристаллизовывают из
толуола и затем из ацетона. Выход нитропроизводного (с т. пл.
175—190°) составляет 8 г [1521.
С более высоким выходом (79% от теория) удается провести
нитрование флуорена [153] азотной кислотой (уд. в. 1,42)
в ледяной уксусной кислоте. Реакция ведется при
перемешивании и температуре 50°, которая затем поднимается до 65°,
а под конец до 80°. Получается 2-нитрофлуорен в виде желтых
игл, который плавится при 156°.
При нагревании 4-оксипиридин-М-оксида в ледяной
уксусной кислоте с азотной кислотой (уд. в. 1,36) на водяной бане
в течение короткого времени получается 3,5-динитропроизвод-
ное с выходом 80% от теоретического.
Карбазол нитруется азотной кислотой в смеси с ледяной
уксусной кислотой с образованием динитрокарбазола [154].
азотной кислотой б
уксусной кислоте
63
Смесь 5 ч. ледяной уксусной кислоты и 1 ч. карбазола
нагревают до 80° и медленно прибавляют при перемешивании 1,3
части азотной кислоты (уд. в. 1,38). Реакционную массу затем
нагревают в течение 30 мин. при 100°, охлаждают и выделяют
3,6-динитрокарбазол.
При нитровании N-фенилпиррола азотной кислотой в
уксусном ангидриде получаются 1-фенил-2-нитропиррол и 1-фенил-З-
нитропиррол [155]:
—NO*
/\
I
— NO,
Если же N-фенилпиррол нитровать смесью азотной и серной
кислот, то нитрогруппа вступает в фенильное ядро с
образованием 1-(4-нитрофенил)-пиррола
ко,
При нитровании N-фенилпирролидина азотной кислотой
в смеси с ледяной уксусной кислотой Ю. К. Юрьев и другие [156]
получили М-(п-нитрофепил)-пирролидип с выходом 52% от
теории. Так как N-фенилпирролидип менее устойчив, чем,
например, диметиланилин, то нитрование протекает в более
мягких условиях (при температуре —20°).
К 10 г N-фенилпирролидина в 70 мл ледяной уксусной
кислоты при —20° прибавляют по каплям 13 мл азотной кислоты
64
(уд. в. 1,35). Реакционную смесь перемешивают частым
встряхиванием колбы. После суток стояния выпадает осадок
М-(п-нитрофенил)-пирролидина; выход 6,9 г, что составляет
52% от теории. При температуре нитрования выше —20°
получается меньший выход (12—20%).
HNO,
сн.соон
I
С высоким выходом протекает нитрование тиофена с
образованием 2-нитротиофена [157]. Раствор 42 г тиофена в 170 мл
уксусного ангидрида медленно прибавляют к смеси 40 г
дымящей азотной кислоты (уд. в. 1,51) в 300 мл ледяной уксусной
кислоты при перемешивании и энергичном охлаждении
реакционной массы так, чтобы температура была не выше 10°.
По окончании реакции оставляют стоять реакционную массу
в течение 2 час. при комнатной температуре и затем выливают
на лед. Нитротиофен выделяется в виде светложелтых
кристаллов. Его отсасывают, промывают ледяной водой, отжимают и
сушат в эксикаторе. При перекристаллизации из петролейного
эфира получают 2-нитротиофен с т, пл. 44—45° с выходом 85%
от теории.
Производные кумарина нитруются смесью азотной и
уксусной кислот с образованием динитроироизводных кумарина.
Так, например, при обработке азотной кислотой 7-окси-3,4-
диметилкумарина, суспендированного в уксусной кислоте при
нагревании на водяной бане, получают 7-окси-3,4-диметил-
6,8-динитрокумарин [158].
Механизм нитрования азотной кислотой в смеси с
уксусным ангидридом или уксусной кислотой изучен сравнительно
мало. Как предполагают, нитрующим агентом в этих смесях
является соединение, образующееся посредством
взаимодействия уксусного ангидрида (или уксусной кислоты) с азотной
даопшоД кислотой е лб^яной уксусной жысдотд 65
кислотой (диацетилортоазотная кислота, или, по другому
представлению, ацетилнитрат).
Пикте и Женекан [159] нашли, что при смешении азотной
кислоты с уксусным ангидридом или уксусной кислотой
образуется смешанный ангидрид уксусной и ортоазотной кислот —
диацетилортоазотная кислота, имеющая строение
сн,соо /ОН
/ N — ОН
СН,СОО ^ду
Если азотную кислоту (уд. в. 1,4) добавлять к равному объему
уксусного ангидрида, то происходит энергичная реакция с
выделением тепла, приводящая к кипению жидкости. При
фракционированной перегонке продукта реакции удается выделить
основную фракцию, которая перегоняется при 127,7° (при
730 мм). Тот же продукт образуется при смешении ледяной
уксусной кислоты с дымящей азотной кислотой (уд. в. 1,52),
но в этом случае реакция протекает менее энергично. Если на
1 моль азотной кислоты брать 2 моля уксусной, то продукт
реакции почти полностью перегоняется при 127—128°.
Элементарный анализ показал, что полученный продукт
соответствует формуле C^HgNOg, т. е. содержит элементы одной
молекулы азотной кислоты и двух молекул уксусной кислоты.
Выделение тепла во время реакции, а также тот факт, что
температура кипения получаемого продукта выше температур
кипения исходных веществ, свидетельствуют об образовании
в данном случае нового соединения, а не смеси компонентов.
Диацетилортоазотная кислота представляет собой
дымящую на воздухе бесцветную жидкость со следующими
константами: уд. в. 1,197 (при 15°), Пд= 1,38432, т. кип. 127,7° (яри
730 мм) и 45° (при 17 мм). В водных растворах
диацетилортоазотная кислота полностью диссоциирует на две молекулы
уксусной кислоты и одну молекулу азотной кислоты. На
органические соединения диацетилортоазотная кислота действует
очень энергично ацетилирующим, окисляющим или нитрующим
образом в зависимости от условий проведения реакции и при*
роды вещества, подвергающегося ее действию.
б А. В. Топчиев
66 #%7Mj?o#ZH%e ддхшдлшческыа.
Пикте и Женекан полагают, что в смесях азотной кислоты
с уксусным ангидридом или уксусной кислотой нитрующим
агентом является диацетилортоазотная кислота. Ова была
применена Пикте [160] для нитрования ряда ароматических
соединений. При этом выяснилось, что как нитрующий агежт
диацетилортоазотная кислота приближается к разбавленной
азотной кислоте, так как она проявляет слабое нитрующее и
довольно сильное окисляющее действие. Анилин, фенол,
нитробензол диацетилортоазотной кислотой не нитруются.
Коген и Вибо [161] изучили кинетику нитрования
ароматических соединений в присутствии уксусного ангидрида,
причем предварительно ими исследована реакция взаимодействия
азотной кислоты с уксусным ангидридом в отсутствие
нитруемого соединения. Скорость последней реакции измерялась по
изменению титра азотной кислоты (в молях на 1000 мл
раствора). Найдено, что при небольших начальных концентрациях
азотной кислоты скорость реакции несоразмерно меньше по
сравнению с опытами с более высокими начальными
концентрациями.
Результаты кинетических измерений показали также, что
в первые 75—100 мин. в реакционной смеси еще имеется
свободная азотная кислота, которая в дальнейшем вступает в
реакцию с уксусным ангидридом с образованием ацетилжитрата
HNO, + (СНзСО)аО ^± CH,CONO, + СН,СООН.
Через 150 мин. после начала реакции становится заметной
убыль ацетилнитрата, который вступает во взаимодействие
с уксусным апгидридом с образованием нитропроизводных
последнего (нитроацетатов).
Для изучения кинетики взаимодействия бензола с
азотной кислотой в присутствии уксусного ангидрида ставились
опыты, которые отличались друг от Друга различными
начальными концентрациями азотной кислоты, причем варьировались
также и количественные отношения компонентов. При
небольших концентрациях азотной кислоты и бензола нитрования
практически не происходит. Реакция протекает с заметной
скоростью лишь при следующих начальных количествах pea-
ддотно& кислотой g лд8яно& уксусгбой жыслотб 67
гентов: азотной кислоты 0,8 мол. и бензола 1,2 мол. Для
получения более ясной картины течения процесса произведены
одновременные измерения концентраций азотной кислоты и
нитробензола. Оказалось, что нитрование бензола вначале
протекает быстро, но с течением времени замедляется, причем
общий расход азотной кислоты и ацетилнитрата превышает то
количество этих реагентов, которое затрачивается, на
образование нитробензола. Полученные результаты указывают, что
процесс нитрования ароматического соединения сопровождается
побочной реакцией взаимодействия между ацетилнитратом и
уксусным ангидридом (см. выше).
Сравнение обеих исследованных реакций дает следующее
представление о реакции нитрования бензола азотной кислотой
в присутствии уксусного ангидрида: в начале процесса
единственным нитрующим агентом является азотная кислота, и
лишь в дальнейшем происходит образование другого
нитрующего агента — ацетилнитрата. Последний не только нитрует
ароматическое соединение, но и вступает в реакцию с
уксусным ангидридом, образуя нитроацетаты.
Изучено также влияние на реакцию нитрования азотистой
кислоты; опыты с добавлением мочевины, разрушающей
азотистую кислоту, показали заметное снижение скорости реакции
по сравнению с опытами в присутствии HNO%, что указывает
на каталитическое действие азотистой кислоты. В опытах без
добавок мочевины обнаружено, что концентрация азотистой
кислоты возрастает со значительной скоростью по мере
течения реакции, невидимому, в связи с окислительным действием
азотной кислоты на бензол, приводящим к образованию новых
количеств HNO%. С повышением концентрации азотистой
кислоты повышается ее каталитическое действие. Однако это
происходит лишь до известного предела, после которого даль-
вейшее образование азотистой кислоты уже не отражается за*
метным образом на скорости реакции.
Кинетика нитрования ароматических углеводородов и их
галоидных производных азотной кислотой в уксусном
ангидриде исследована также Б. В. Троновым, Г. X. Камай и А. Г.
Коваленко [162].
б*
Смесь эквимолекулярных количеств азотной кислоты,
уксусного ангидрида и испытуемого вещества нагревали при
температуре от 0 до 20°. Через определенные промежутки времени
смесь оттитровывали раствором едкого натра. Относительную
скорость нитрования различных соединений вычисляли по
времени, в течение которого происходило одинаковое
уменьшение кислотности, т. е. реагировало одно и то же количество
азотной кислоты (при этом принимали в расчет и количество
щелочи, пошедшее на титрование уксусного ангидрида). Опыты
показали, что скорость нитрования углеводородов повышается
в следующем порядке (скорость нитрования бензола принята
за единицу): бензол (1)<См-ксилол (7)<Смезитилен (25)<Спсевдо-
кумол (28). Галоидные соединения могут быть расположены
в следующий ряд по возрастанию относительной скорости их
нитрования:
С,Н,С1 (0,15) < СбНвВг (0,25) < С,Н,СН,С1 (0,4) <
<м-СЩСАС! (0,5)<п-СН,С,Н,С1 (1,15)<
<о-СН,С,Н,С1(1,2).
§ 4. Нитрование в присутствии катализаторов
Еще в 1906 г. обнаружено каталитическое действие
азотнокислой ртути (закисной) при нитровании антрахинона и метил-
антрахинона [163]. Несколько позднее найдено своеобразное
каталитическое действие солей ртути при нитровании бензола.
Установлено, что реакция нитрования бензола в присутствии
Hg(NO@)a, при определенной концентрации азотной кислоты,
направляется в сторону образования нитрофенолов. При
действии на бензол концентрированной азотной кислоты или
смеси азотной и серной кислот в присутствии ртути получают
только нитробензол; для образования нитрофенолов
необходимо соответствующее разбавление азотной кислоты, при
котором она может проявить свое окислительное действие.
Образование нитрофенолов при нитровании ароматических
угяеводородов разбавленной азотной кислотой в присутствии
азотнокислой ртути в значительной степени зависит от темпе-
69
ратуры реакции и концентрации применяемой кислоты;
присутствие в реакционной смеси серной кислоты в количестве,
которое фактически не оказывает влияния на концентрацию
азотной кислоты, не отражается на выходе нитрофенолов.
Для получения нитрофенолов нагревают на водяной бане
смесь 100 г бензола, 800 г азотной кислоты (уд. в. 1,31) и 15 г
азотнокислой ртути яри сильном перемешивании. В качестве
продуктов реакции, наряду с незначительным количеством
нитробензола, получаются 2,4-динитрофенол и пикриновая
кислота:
O,N NO, /\/\
O,N NO,
Согласно данным Кормака [164], оптимальные условия
окислительного нитрования с целью получения
динитрофенола сводятся к применению 50—55%-ной азотной кислоты»
содержащей в 1 л 0,2—0,5 моля азотнокислой ртути, при
температуре реакции 50°. При этом выход динитрофенола достигает
85% от теоретического (другие исследователи [105] указывают
более низкий выход), а в качестве побочных продуктов
получаются нитробензол и пикриновая кислота.
Проведение реакции при более высокой температуре
увеличивает образование пикриновой кислоты за счет
динитрофенола.
Бахман и другие [165], изучавшие состав продуктов,
получающихся при окислительном нитровании фенола,
проводили реакцию следующим, образом: 50 г бензола при 50°
прибавлялись в течение 3,3 часа к смеси 750 мл 10,65 М азотной
кислоты, 0,37 М HgfNOa)* * и 0,1 г NaNO* при 50°.
Реакционная смесь перемешивалась ^2,5 часа, охлаждалась, и
выделялся динитрофенол. Из фильтрата извлекались побочные
продукты. Выход динитрофенола составлял 68,4%. Кроме дини-
* 60 г HgO прибавляется к 541 мл 70%-ной НМОз и разбавляется
до 750 мл. L i '1
70
трофенола выделены
о
Пикриновая кисяота 4
Нитробензол 7,8
СО. 9,7
Щавелевая кислота 2,5
Нитрофенолы образуются в результате комбинирования
двух процессов: окисления и иитрования, причем окисление,
повидимому, предшествует нитрованию. Последнее допущение
подтверждается тем, что при действии азотной кислоты на
нитробензол он остается без изменения или же переходит в ди-
нитробензол, но не в нитрофенол. Реакция образования
пикриновой кислоты из бензола протекает по следующему
уравнению, выражающему конечный результат нитрования:
С.Н, + 4HNO, = СА (ОН) (N0,), + ЗН.,0 + HNOg.
Предполагается, что вначале в качестве промежуточного
продукта образуется комплекс бензола со ртутью, который
распадается под влиянием избытка азотной кислоты [166].
Показано также, что азотнокислая ртуть оказывает
каталитическое действие и на процесс нитрования бензойной кислоты
азотной кислотой, направляя реакцию в сторону образования
тринитрооксибензойной кислоты (в обычных условиях
нитрования, т. е. без катализатора, бензойная кислота дает, как
известно, нитробензойные кислоты). Образующаяся тринитро-
оксибензойная кислота, переходящая при нагревании с
отщеплением СО* в пикриновую кислоту, представляет собой 2,4,6-
тривитро-3-оксибензойную кислоту
соон
ОЛ I N0
\/\/
Таким образом, и в данном случае присутствие ртути
способствует вхождению гидроксильной группы в ароматическое
ядро [167].
71
Действие азотной кислоты в присутствии ртути на
ароматические соединения и их производные исследовали также
Блехта и Патек [1681. При нитровании толуола 50%-ной
азотной кислотой, в которой растворено 2% ртути, образуются
нитротолуолы и п-нитробензойная кислота с выходом 10% от
теории (т. нл. п-нитробензойной кислоты 237—238°). В
продуктах реакции обнаружен также тринитро-м-крезол. Эти
авторы не подтверждают образования нитросалициловой кислоты,
наблюдавшегося другими исследователями [167].
При нитровании бензола смесью азотной и сераой кислот
в присутствии 2% ртути получены нитробензол и тринитрофе-
нол. Нитрование этой же смесью толуола дает нитротолуолы,
нитрокрезол и п-нитробензойную кислоту, т. е. те же
продукты, которые образуются и при нитровании азотной кислотой.
Эти результаты указывают, что добавление серной кислоты не
влияет на нитрование ароматических соединений азотной кисло-
той в присутствии ртути. Далее показано, что нитрование
нитробензола 80—85%-ной азотной кислотой в присутствии ртути
приводит к большим выходам динитробензола, чем
нитрование без активатора.
Механизм нитрования ароматических соединений в
присутствии ртути объяснен Блехтой и Патеком следующим
образом: ртутная соль образует промежуточный ртутьоргаиический
комплекс с бензолом C@HgHgNO3, который разлагается при
действии азотной кислоты, выделяя углеводород m aW%
лдзсежК; активированный таким образом углеводород легко
нитруется азотной кислотой. Авторы изображают
схематическое течение реакции следующим образом:
2С*Н, — Hg — NO, + 4HNO, -» 2QH.OH + 2Hg (NO,), + N,O, + И.О.
Образующийся при этом фенол тотчас же вступает в реакцию
с азотной кислотой с образованием нитрофенола.
Для объяснения образования пикриновой кислоты из бен-
^ %ола при нитровании его в присутствии ртутных солей
предложен следующий механизм реакции [169].
^ В первой стадии процесса предполагается образование
ртутного соединения бензола, в котором ртуть непосредствен-
72
но связана с бензольным ядром:
О —N0,
HONO,
(1)
Далее допускается, что азотистая кислота в форме
ангидрида реагирует со ртутным соединением бензола с
образованием диазобензолнитрата (2), который при гидролизе
распадается на фенол, азот и азотную кислоту:
Hg-0
= N — О — NO,
А
+ 2N.0:
N —О
NO,
+
(2)
А
+ н,о
HONO*
Последней стадией процесса является нитрование фенола
азотной кислотой, приводящее к конечному продукту реакции —
пикриновой кислоте:
ОН ОН
+ ЗШО, -* | | + 3H.0
Нитрование бензола в присутствии ртутных солей изучено
также Винъоном [170]. При нагревании смеси 100 г бензола,
800 г азотной кислоты (36° Б оме) и 20 г азотнокислой ртути в
течение 5 час. при 70° Виньон получил пикриновую кислоту
(138 г) и нитробензол (32 г). При дальнейшем исследовании
им найдено, что нитрование бензола в присутствии ртутных
солей отличается следующими особенностями:
73
1) При концентрации выше определенного предела
пикриновая кислота, по мере своего образования, полностью окие-
дяется азотной кислотой до двуокиси углерода.
' 2) Избыток бензола по отношению к азотной кислоте,
создавая благоприятные условия для образования дивитрофено-
лов, за счет частичного уменьшения выхода пикриновой
кислоты, повышает общий выход оксинитропроизводных. Так,
например, применяя 5 ч. бензола на 10 ч. азотной кислоты, можно
получить выход нитрофенолов около 85% от теории, в то время
как при отношении реагентов 1 ч. бензола на 10 ч. азотном
кислоты выход составляет только 34%.
Варьируя условия процесса нитрования бензола в
присутствии HgfNOa)*, Виньон получил оптимальные результаты при
следующем способе проведения реакции: смесь 100 г азотной
кислоты (36° Боме), 50 г бензола и 2,5 г азотнокислой ртути
нагревают (с обратным холодильником) в течение 6 час. при
75—80°. Избыток бензола облегчает поддержание температуры
смеси на этом уровне и растворяет нитрофенолы, которые,
таким образом, меньше подвергаются окислительному
действию азотной кислоты. По окончании нитрования отделяют
бензол, содержащий в растворе нитрофенолы, от водного слоя;
последний промывают бензолом, после чего оба бензольных
раствора соединяют и отгоняют сначала бензол и затем
нитробензол (с водяным паром). Остаток представляет собой смесь
нитрофенолов (52% динитрофенола и 48% пикриновой кислоты).
В случае необходимости обрабатывают смесь
нитрофенолов азотной кислотой или NaNOg и серной кислотой, в
результате чего динитрофенол переходит в пикриновую кислоту.
Бродером [171] проведено тщательное исследование всех
газообразных продуктов, образующихся при реакции
нитрования бензола азотной кислотой в присутствии нитрата
ртути. Ему не удалось обнаружить среди них азота, а найдены
лишь окислы азота (NOа и N*()*). Далее Бродер показал, что
при нитровании бензола образуется некоторое количество о- в
п-динитробензолов, появление которых может быть объяснено
Окислением азотной кислотой соответствующих
промежуточных продуктов — нитронитрозобензолов.
74 //штроеднке дролюткчбскмл: w
Бродер проводил также нитрование бензола слабыми
растворами азотной кислоты в присутствий ртути при низких
температурах, причем в качестве промежуточных продуктов ему
удалось выделить нитробензол и соединение, которому ов
придает строение
NO,
Это соединение (тетранитродифенилртуть) при гидролизе
разлагается с образованием динитрофенола
ОН
/NO,
O,N __/"""%- Hg -/ У- NO, + 3H,0
Основываясь на данных Бродера, а также на работе Бамбер-
гера [172], который показал, что соединение бензола с ртутью
(дифенилртуть) реагирует с N,0* с образованием нитробензола
и фенилнитрата ртути, Девернье [173] представляет механизм
нитрования бензола в присутствии нитрата ртути следующим
образом: азотнокислая ртуть при действии на бензол дает #*-
фенилртуть (1); последняя реагирует с азотной кислотой с
образованием тетранитродифенилртути (2), которая полностью
переходит в водную фазу; тетранитродифенилртуть —
неустойчивое соединение и быстро разлагается азотной кислотой
на динитрофенол и N%0* с регенерацией азотнокислой ртути (3).
^ + 4HN0,
у" NO, + 4Н,0 (2)
75
O.N
4HNO
он
NO,
(3)
Появление в продуктах реакции нитробензола Девернье
объясняет действием N#0*, образующегося по уравнению (3),
на дифенилртуть с последующим окислением азотной кислотой
нитрозобензола в нитробензол (4):
(4)
Девернье также полагает, что азотная кислота может
действовать и нитрующим образом на нитрозобензол, превращая
его в витронитрозобензол (5); при дальнейшем окислении
последнего образуется п-динитробензол (6), также найденный
в продуктах реакции.
N0 N0
Н.0
(5)
(6)
76
Для выяснения оптимальных условии реакции нитрования
бензола в присутствии Hg(NOg)a Девернье поставил ряд
опытов, в которых он варьировал концентрацию азотной кислоты,
отношения реагентов, температуру и продолжительность
нитрования, В некоторых опытах нитрование проводилось
посредством пропускания паров бензола через реактор с азотной
кислотой и катализатором. Эти опыты дали менее
благоприятные результаты по сравнению с опытами, в которых
нитрованию подвергался жидкий бензол. Оптимальные результаты
(выход нитрофенолов около 70% теории) получены при
следующих условиях: отношение реагентов — 1,4 ч. бензола, 3 ч.
азотной кислоты (61%-ной), 0,06 ч. Hg(NOg)a;
температура реакции 75—80°; продолжительность титрования
6 час.
Девернье показал также, что при нитровании бензола в
присутствии нитратов марганца, алюминия, меди и цинка
получается небольшой выход нитросоединений.
А. И. Захаров [174] приводит следующие возражения
против предложенных Девернье схем образования динитрофенола
и нитробензола: 1) превращение бензола в динитрофенол, по
Деверпье, сопровождается выделением водорода; между тем
при исследовании продуктов реакции Захарову не удалось
обнаружить образования водорода даже при нитровании больших
количеств бензола; 2) образование нитробензола посредством
взаимодействия дифенилртути с NaO* с последующим
окислением нитрозобензола (см. выше) маловероятно. По мере
течения реакции количество окислов азота должно постепенно
возрастать, достигая наибольшего накопления к концу
процесса. Если принять схему Девернье, следовало бы ожидать к
концу нитрования преимущественного образования нитробензола
за счет понижения выхода нитрофенолов, чего в
действительности не наблюдается; даже при дополнительном насыщении
реакционной массы NaO* выход нитробензола заметно не
повышается.
А. И. Захаров выдвигает другую схему нитрования, исходя
из допущения, что основная роль катализатора сводится
к ослаблению общей стойкости бензольного ядра посредством
e
77
разрыва двойных связей. Это ослабление ядра может
происходить в двух разных местах (схемы I и II).
I)
\/
\
\
н +
О
\
О
Н О —Hg
Хл
\/
0N0
Н О—Hg
Хл
или
ОН
N0,
+ HNO,
^ ^
О
O.N
Опыты Захарова показали, что образование тштрофенолов
происходит лишь при низких концентрациях азотной кислоты
(лише 60%), при которых ее окислительная способность
выражена наиболее ясно. Если же применять кислоту сравнительно
высокой концентрации (65—70%), то при температуре 60—65°,
ве благоприятствующей окислительному действию азотной
/8 Яылукюдны*
w
кислоты, в начале реакции будет получаться только
нитробензол; появление нитрофенолов наблюдается лишь через
некоторое время после начала процесса вследствие понижения
концентрации азотной кислоты в реакционной смеси.
Таким образом Захаров приходит к выводу, что введение
гидроксильной группы при этой реакции зависит не от
присутствия катализатора, а только от концентрации
применяемой азотной кислоты и температуры реакции.
Далее Захаров считает, что образование нитробензола
происходит по следующей схеме:
\
NO,
"ЪН,
При повторном использовании отработанной кислоты, как
показал Захаров, увеличиваются и скорость реакции и выход
динитрофенола. Так, например, при нитровании свежепри-
готовленяым раствором, содержащим 59—60% HNO,, при
30%-ном содержании катализатора и температуре 35—40°,
выход динитрофенола составляет —40%. При вторичном
использовании отработанной кислоты (при сохранении
остальных условий) выход двяятрофееола увеличивается до 50—55%.
При последующем использовании отработанной кислоты
в 3, 4 и 5-й раз выход динитрофенола соответственно
достигает 65, 75 и 80%. Автор объясняет это тем, что в отработанной
кислоте катализатор находится в более активном состоянии.
А. И. Захаров исследовал также каталитическое нитрование
производных бензола в присутствии Hg(NOs),. Оказалось,
что эти соединения дают только нитропроизводные, т. е. у них
отсутствует способность подвергаться одновременному
окислению, как это наблюдается у бевзола. Например, фенол при
нитровавии его в присутствии Hg(NO#), разбавленной азот-
щой кислотой (3—4%) при низких температурах (до —5
дщет почти чистый п-ивомер витрофенола с выходом 60%.
Оптимальные условия яитровая^я фенола: загрузка
небольшими порциями фенола (50 г) в разбавленную азотную
кислоту (из расчета 36 г моногидрата), содержащую катализатор
(5 г). При нитровании салициловой кислоты 10—12%-ной
азотной кислотой в присутствии Н§(МОв)% получена смесь изомер-
жых мононитропроизводных с общим выходом 92—93%;
преобладающий продукт нитрования п-нитросалициловая
кислота. Реакцию необходимо проводить при сильном
охлаждении (оптимальная температура 44—47°).
Ускоряющее действие азотнокислой ртути на процесс
нитрования нафталина отмечали Энц и Пфистер [175]. При
этом, по утверждению авторов, увеличивается выход 2,4-ди-
иитро-а-нафто ла.
В противоположность этому Захаров утверждает, что
полициклические ароматические углеводороды (нафталин, антрацен)
н их производные при нитровавши азотной кислотой в
присутствии Hg(NOa)* дают только житропродукты (а ие окси-
нитропроизводные) и притом мононитропровзводные.
Нитрование ^-нафтола приводит исключительно к а-вятро-р-вафтоду.
Захаров рекомендует следующий метод нитрования р-нафтола:
50 г р-пафтола загружают в 2—4%-вую азотную кислоту (из
расчета 44 г моногидрата), содержащую катализатор; реакцию
ведут при 30—35* с перемешиванием. Выход %-нитро-{3-нафтола
достигает при этих условиях 84—85% от теоретического.
Е. И. Орлов [176] исследовал нитрование м-ксилола
разбавленной азотной кислотой в присутствии ртути. Смесь
м-ксилола с 45—50%-вой азотной кислотой кипятили в
присутствии 2% ртути с обратным холодильником в течение 6 час.
Продукт нитрования подвергался перегонке с водяным паром,
при этом отогналось небольшое количество масла с запахом
нитробензола. Остаток состоял из смеси двух веществ, из
которых одно представляло собой желтые кристаллы,
растворимые при нагревании в воде, эфире, спирте, бензоле и ксилоле,
а второе имело вид оранжевых комков. Исследование
показало, что второе вещество является таутомерной формой первого.
#0
ajMwwzпимбскиа
Для определения содержания нитрогрупп в желтых
кристаллах последние восстанавливались SnCl* в солянокислой среде;
после удаления НС1 образовавшийся амин разрушался серной
кислотой и азот определялся по методу Кьельдаля.
Данные анализов на содержание карбоксильных групп и
азота показали, что продукт нитрования представляет собой
оксинитробензолдикарбоновую кислоту.
Нитрование ксилола азотной кислотой в присутствии ртути
изучено также Н. А. Холево и И. И. Эйтингтоном [177]. В круг-
лодонную колбу, снабженную обратным холодильником и
мешалкой, загружали 800 г азотной кислоты (50,2% НКОз)
и 16 г металлической ртути (или соответствующее количество
азотнокислой соли); содержимое колбы нагревали при
перемешивании до 75* и постепенно приливали (по каплям) 100 г
м-ксилола; после введения всего ксилола нагревание при
температуре (75—80°) продолжали еще в течение 5 час. при
перемешивании. Во всех опытах продуктами реакции оказались
4-нитро-м-ксилол и 4-нитро-З-метилбензойная кислота. Окси-
нитросоединений получено не было.
При проведении нитрования м-ксилола в указанных выше
условиях, но без катализатора, Холево и Эйтингтон получили те
же соединения и с теми же выходами; например, выход моно-
нитроксилола из 100 г м-ксилола составлял 35 г, независимо
от того, проводилась ли реакция с катализатором или без
него.
Холево и Эйтингтон проводили также нитрование
м-ксилола в условиях, указанных в работе Е. И. Орлова [176].
Полученные результаты не подтвердили, однако, данных
последнего об образовании оксинитробензолдикарбоновой
кислоты в качестве основного продукта реакции. Холево и Эйтинг-
тоиом выделены при этом мононитроксилол и, в преобладающем
количестве, 4-нитро-З-метилбензойная кислота.
На основании экспериментальных данных Холево и
Эйтингтон приходят к выводу, что ртуть не оказывает
каталитического действия на реакцию нитрования м-ксилола в отличие от
нитрования бензола. Это объясняется, по их мнению,
способностью ксилола к легкому образованию мононитрокарбоновых
кдлимиадлкумю 81
кислот, которые уже в дальнейшем не дают возможности
ртути проявить каталитическое действие. В этом случае
карбоксильная группа как бы противодействует вступлению в ядро
оксигруппы, подобно тому как это наблюдается в случае
присутствия в ядре некоторых других остатков, например
груППЫ N0;.
Реакция нитрования ароматических соединений азотной
кислотой детально исследовалась также Дэвисом с
сотрудниками [178, 179]; в этих работах выяснялись механизм
реакции и роль добавок азотнокислой ртути.
Опыты с бензолом производились этими авторами в круг-
лодонной колбе из стекла пирекс с пришлифованным
холодильником. В колбу загружали сначала азотную кислоту,
в которой растворялась окись ртути или Hg(NO%)a, затем
приливали бензол и смесь нагревали на песчаной бане до
умеренного кипения и легкого выделения окислов азота. По
окончании реакции добавляли равный объем воды и смесь
подвергали перегонке с водяным паром. При этом в дестиллят в первую
очередь переходили непрореагировавший бензол и
нитробензол; последний содержал довольно значительное количество
двнитрофенола, от которого его освобождали взбалтыванием
с раствором соды. Во вторую очередь отгоняли динитробензолы
и 2,4-динитрофенол. Было выделено два динитробензола,
которые отделялись один от другого фракционированной
кристаллизацией. Остаток после перегонки с паром, содержавший
в осадке соль ртути, подвергали горячему фильтрованию. Из
фильтрата при охлаждении выделялись кристаллы пикриновой
кислоты. Найдено, что для получения хороших результатов
необходимо применять относительно большее количество
азотной кислоты, чем зто указано в сообщении Вольфенштейна и
Бетерса [166].
При нитровании в течение 7 час. 200 г бензола 600 мл
азотной кислоты уд. в. 1,42, в которой растворено 10 г окиси ртути,
получено: 41 г исходного бензола, 61,3 г нитробензола, 105 г
пикриновой кислоты и 4 г смеси, состоящей из равных частей
м-динитробензола и 2,4-динитрофенола и некоторого
количества н-динитробеизола.
6 А. В. Топчиев
82
Далее показано, что в зависимости от концентрации азотной
кислоты (от 100 до 20,6%) изменяются количественные
отношения и характер получаемых продуктов реакции. При
концентрации азотной кислоты 70% и выше получаются
удовлетворительные выходы моно- и ди нитробензолов, во пикриновая
кислота образуется лишь в незначительных количествах.
Нитрование толуола азотной кислотой в присутствии ртутж
протекает значительно более энергично, чем нитрование бензола.
Реакцию проводили в течение длительного периода нрж
постепенном повышении температуры (12,5 час. при 65—85°,
2 часа при 85—95°, 1 час при 95° и, наконец, 7 час. при
нагревании на песчаной бане). При нитровании 200 г толуола 600 мл
азотной кислоты (уд. в. 1,4), в которой растворено 10 г HgO,
получены следующие продукты: 48 г яепрореагировавшего
толуола, 15 г о-нитротолуола, 12 г п-нитротолуола, 8,8 г п-нитро-
бензойяой кислоты и 6 г трияитро-м-крезола. Образование ббль-
швх количеств п-нитробензойной кислоты, невидимому,
объясняется окислением азотной кислотой п-нитротолуола.
Влияние различных окислов металлов и их нитратов на
соотношение изомерных яитротолуолов, получающихся
яри нитровании толуола, изучали Дзудзуми и др. 11801. При
нитровании (без добавок) 60 г толуола 66 г HNO* (65%-ной)
в течение 8 час. при 30° соотношение о-, м- и п-нитротолуола
составляет: 63,1 : 3,7 : 33,2. При проведении нитрования
в этих же условиях, во с добавкой одного из окислов (AgO,
PbO,CuO,WO,, PtO,) или нитратов (Pd, Cu,Ba, Cd, Co.Mg,
Zn, Cr, Fe, Mn, Al или Bi) соотношение изомеров почти не
меняется. Если же вести нитрование с добавкой HgO (2 г),
Hg(NOa), или Ni(NO*), (в количестве, эквивалентном 2 г
соответствующей окиси металла), то получается следующее
соотношение изомеров:
HgO ИДНО,), N1(NO,).
Орто 50,5 40,5 58,i
Мета 3,7 3,4 3,9
Пара 45,8 56,1 38,0
Таким образом, применение этих добавок сказывается лишь
на изменении соотношений орто- и пара-изомера. Что
касается м-нитротолуола, то его содержание почти не меняется.
83
Эти же авторы [181] изучали влияние добавок различных
жвтратов на соотношение изомеров, получающихся при
нитровании хлористого бензила. Нитрование проводилось имя в
уксусном ангидриде азотной кислотой (82—94%-ной). При
нитровании без добавок отношение орто- и пара-изомера
составляет 0,55—0,6 (при содержании мета-изомера 3,9—4,1%).
В присутствии нитратов ртути, серебра, меди, бария или свид-
ца отношение орто- к пара-изомеру достигает —0,92 при
сохранении того же содержания мета-пзомера.
В отличие от толуола хлорбензол в присутствии ртути
реагирует с азотной кислотой значительно медленнее, чем бензол.
Нитрование хлорбензола (100 г) в течение 10 час. азотной
кислотой уд. в. 1,4 (300 мл), в которой растворено 5 г HgO,
дало следующие продукты реакции; 57,6 г вепрореагировав-
шего хлорбензола, 7,6 г п-нитрохлорбензола и 1,5 г тринитро-
м-хлорфенола.
Дэвис [178, 179] проводил также нитрование нафталина,
в условиях, близких к тем, которые оказались оптимальными
для бензола. 5 г HgO растворяли в 300 мл азотной кислоты
(уд. в. 1,42) и раствор небольшими порциями приливали к 100 г
нафталина. По окончании первоначальной бурной реакции
реакционную смесь нагревали на кипящей водяной бане дрн
достоянном перемешивании. Продукт нитрования (в виде
масла) отделяли от азотной кислоты, промывали горячей водой
и обрабатывали горячим раствором соды; щелочной раствор
подкисляли соляной кислотой и взбалтывали с эфиром. Из
эфирного раствора выделен нитронафтол в количестве 1,5% от
общего веса иитродроизводных. Из остатка после экстракции
эфиром получены фракционированной кристаллизацией из
опирта, этилацетата и ацетона: <%-нитронафталин (20%),
$,3-дишггронафталин (т. пл. 144°; 35%) и 1,5-динитропафталип
|(т. пл. 213—213,5°; 40%). В отработанной азотной кислоте
!жайден еще один кристаллический продукт, который оказался
Житронафтойяой кислотой (т. пл. 207—208°).
к Дэвис проводил нитрование нафталина и в более мягких
, а именно: разбавленной азотной кислотой при ниа-
температурах. Опыты показали, что в этих условиях
84
образуются преимущественно нитронафтолы, т. е. реакция
направляется в сторону образования гидроксилированных
ни троярои зводвых.
Например, смесь 1 моля нафталина с 1 л раствора,
содержавшего 600 мл азотной кислоты (уд. в. 1,35) и 1 моль
азотнокислой ртути, нагревали до 35° яри перемешивании в течение
37 час. Основным продуктом реакции оказался 2,4-динитро-%-
нафтол (т. ял. 138°); кроме того, получено небольшое
количество 2-ыитро-ос-нафтола (т. ял. 128°). Общий выход нитронафто-
лов составлял 55 г.
, Для выяснения роли азотнокислой ртути Дэвис и
сотрудники кипятили нитробензол с азотной кислотой в присутствии
Hg(NOJ: в течение 4 час. При этом образования пикриновой
кислоты не обнаружено. Единственным продуктом реакции
оказался динитробевзол в небольшом количестве. При
кипячении с азотной кислотой и Hg(NOg)a м-дпнитробензола 82% его
осталось без изменения, а остальная часть окислилась до
щавелевой кислоты; нэ найдена также пикриновая кислота при
кипячении с азотной кислотой 1,3,5-тринитробензола, который
при действии других окислителей дает пикриновую кислоту.
Из этих опытов сделан вывод, что ртуть ие играет роли
катализатора, способствующего окислению атомов водорода
ароматического ядра, содержащего группы N0%, т. е. образование
пикриновой кислоты из бензола не проходит через
предшествующую стадию нитрования бензола.
Дэвис предполагает, что роль ртутной соли заключается в
образовании промежуточного комплекса с бензолом, который в
дальнейшем окисляется, невидимому, в ртутную соль динитро-
фенола; последняя яри действии азотной кислоты
превращается в пикриновую кислоту с регенерацией ^азотнокислой ртути.
Присоединение Hg(NOg)@ к ароматическому ядру в первой
стадии реакции происходит, невидимому, таким образом, что
группа —N0% присоединяемся к одному из атомов углерода,
а остаток —О—Hg—ONO, к другому.
При нитровании толуола и хлорбензола в присутствии
Hg(NO@)a образуются фенолы с гидроксильной группой,
находящейся в м-положении по отношению к метальной группе
85
или к атому хлора, которые обычно ориентируют в о- и п-по-
ложения. Этот неожиданный результат может быть
удовлетворительно объяснен, если предположить, что группа —N0,
нитрата ртути присоединяется в о- или я-яоложеяии по
отношению к метильной группе или к атому хлора, а остаток
—О—Hg—0N0, занимает соседнее, т. е. мета-положение
по отношению к метильной груяпе ил», соответственно,
к атому хлора. Таким образом, благодаря образованию
комплекса с нитратом ртути нитрование и гидроксилирование
ароматического ядра происходят одновременно.
Мак-Ки [182] исследовал действие Hg(NO,)& при
нитровании азотной кислотой или нитрующей смесью ряда
ароматических соединений: ксилола, нафталина, бромбензола, фенан-
трена, фенола, хлорфенола, %-нафтола, бензальдегида и др.
Оныты Мак-Ки показали, что в большинстве случаев
азотнокислая ртуть оказывает благоприятное действие на нитрование,
повышая общий выход нитронроизводных, в том числе гидро-
ксилиропанных нитросоединений. При нитровании фенантре-
на азотной кислотой уд. в. 1,5 в присутствии Hg(NO*)& подучен
выход нитрофенантрена на 13% больше, чем при нитровании
без катализатора; выход нитрофенола при нитровании фенола
разбавленной азотной кислотой повышается от добавления
ртутной соли на 22—25%; %-нафтол при нитровании смесью
азотной и серной кислот в присутствии ртути дает выход
%-нитронафтола нэ 10% больше, чем при нитровании без
катализатора.
Основные причины, затрудняющие использование солей
ртути в качестве катализатора при нитровании, следующие:
1) необходимость значительного расхода дефицитной ртути;
2) трудность регенерации ртути;
3) нежелательность загрязнения нитросоединений солями
ртути.
При нитровании фенола в присутствии азотнокислого
свинца при 20* наблюдается увеличение количества п-нитрофенола
за счет орто-изомера. Если же вести нитрование при более
высокой температуре, то соотношение обоих образующихся
изомеров становится обычным [183].
#ыгмроа%ныб
Как показали А. М. Володарский и В. С. Калинина [184],
в присутствии сернокислой меди нитрование ос-нафталянсуль-
фововой кислоты в 1,8-нитронафталинсульфоновую кислоту
протекает с более высоким выходом, чем без добавления
катализатора.
Согласно французскому патенту [185], в качестве
катализаторов при нитровании могут быть применены соединения
хрома, вольфрама, молибдена, тантала, ниобия, ванадия, галлия
'ж индия. По утверждению патента, при применении одного из
этих катализаторов можно легко получить с малым расходом
крепках кислот тетранитронафталин и другие полиннтропрояз-
водшае.
Как катализатор [186] применяется также трехфтористый
бор. Схематично реакцию нитрования ароматических
соединений в присутствии ВРз можно представить так:
RH 4- HNO, + BF, ^ ПЖ)* + BF, Н@О.
Нитрование нитробензола в присутствии ВРз проводилось
следующим образом: к 31 г (0,25 М) нитробензола прибавляют
,19 г (0,3 М) дымящей HNO* (уд. в. 1,5) и пропускают при
перемешивании 17 г BF*. Температура'реакции не должна быть
выше 80*. Затем ведут нагревание при 100* в течение 30 мин.
,Продукт реакции выливают в 500 мл горячей воды и
перемешивают до охлаждения. Полученный динитробензол фильтруют
и перекристаллизовывают из спирта. Выход 36,5 г (87% от
теории). Аналогично ведут нитрование бензойной кислоты,
фталевого ангидрида и других соединений (табл. 4).
Фтористый бор можно регенерировать добавлением, по
окончании реакции, достаточного количества воды для
превращения BFg"H*O в BFg-2H,O. Последний выделяют отгонкой
в вакууме в виде тяжелой прозрачной жидкости. Обработкой
фтористым кальцием удается регенерировать ВРз по схеме
2BF,.2H,O 4- СдР, -> Са (BF,), + 4Н,О;
Ca(BF*),-»2BF,+ CaF,.
По вопросу о механизме каталитического нитрования
ароматических соединений в присутствии солей ртути особого
внимания заслуживают работы А. И. Титова,
87
Таблица 4
Иозодное
соединение
Нитробензол
Беааойвая
кислота
п-Толуолсулъ-
фокислота
Метиловый
эфир
бензойной КИСЛОТЫ
Фталевыи ад-
гидрид
0,25
0,25
0,10
0,10
0,10
о
К
моли
0,30
0,30
0,15
0,15
0,15
0,25
0,37
0,12
0,12
0,28
&
с»
РОИ
0,5
0,5
0,5
1,0
13,0
1"
80-100
80
100
0-10
100
Полученный продукт
м-Динитробеазол
м-Нитробеазойная
кислота
2-Нитро-4-толуолсулъ-
фокислота
Метиловый эфир м-ни-
тробеазойнон к ис-
лоты
3-нитрофталевая
кислота
*
а
87
82
92
77
81
В своей работе совместно с А. М. Барышниковой [187] и
несколько позднее с Н. Г. Лаптевым [1881 А. И. Титов
показал, что при взаимодействии ароматического соединения с азот-
жокислой ртутью в среде азотной кислоты вначале происходит
меркурирование ароматической молекулы с образованием
ртутьорганического соединения, причем эта реакция является
обратимой:
Аг — Н + Hg (NO,)% ^ Ar — HgNO, + HNO,.
В отсутствие окислов азота реакция исчерпывается этой
схемой. Из бензола, таким образом, получается февилмеркури-
нитрат, который и был выделен из реакционной массы.
В связи С — HgNOs углерод поляризован несколько более
отрицательно, чем в обычной связи С — Н, и с точки зрения
общей теории активности ароматических соединений в
реакциях электрофильного замещения он может быть в известной
мере уподоблен орто- и пара-атомам углерода таких реакцион-
воспособных соединений, как фенолы и амины, остаток же
HgNOs соответствует так называемому подвижному водороду.
-88 #%гмробдмиб др&мдпшчбсжыа: и
Вместе с тем несомненно, что в превращениях групп С — HgNOa
будет проявляться известная специфичность химического
поведения ртути. Установлено, что с увеличением концентрации
аэотной кислоты глубина меркурирования уменьшается.
Аэотная кислота до известной степени ее концентрации
при отсутствии окислов азота действует на арилртутное
соединение только расщепляющим образом, приводя к
регенерированию исходного углеводорода. Авторы изображают механизм
обратимой реакции меркурирования следующей схемой:
Аг
(i)
Расщепление 40—50 % -ной аэотной кислотой идет очень
медленно при обыкновенной температуре и быстрее при 100°.
Совершенно по-другому относится ртутноароматическое
соединение к азотной кислоте, содержащей окислы аэота, или
к одной двуокиси азота. Фенилмеркуртштрат или ацетат
взаимодействует с 50—60%-ной аэотной кислотой, содержащей
окислы аэота, очень быстро с разогреванием даже при
пониженной температуре.
При медленном прибавлении арилртутного соединения и
механическом перемешивании реакция протекает довольно
гладко и приводит к образованию той же смеси продуктов, что
и при нитровании бензола в присутствии солей ртути, т. е. ни-
трофенолов, нитробензола и п-динитробенэола.
На основе своих экспериментальных данных авторы
приходят к выводу, что каталитическое действие солей на
реакцию окислительного нитрования ароматических углеводородов
проявляется лишь в присутствии окислов аэота. В азотной
кислоте, содержащей окислы азота, арилмеркуринитрат
быстро реагирует с образованием нитроэосоединения, что было
доказано выделением его из реакционной массы.
Образование нитрозосоединеней иэ ртутных производных
при взаимодействии с N,0* в ее нитроэилнитратной форме,
89
до предположению А. И. Титова и Н. Г. Лаптева, протекает
по схеме
Ar Hg
. *
ArHgNO* +O = N — О — NOa-^N 6-» ArNO + Hg (NO,)* (2)
O — O —NO
Дальнейшие превращения полученного нитроэобенэола
в ди- и тринитрофенол представляются авторами следующим
образом.
В результате присоединения протона с последующим
присоединением гидроксил-иона в пара-положении к
азотсодержащей группе происходит образование таутомера п-оксифенил-
гидроксиламина по схеме
Пояучеиный таким образом поксифенилгидроксиламит
окисляется вначале в п-нитрозофенол, а эатем в п-нитрофенол,
который и подвергается в дальнейшем нитрованию до ди- и
тринитрофеиола по схеме
NHOH
N0
+N0,
N0,
ОН
NO,
+HN0,
он
N0,
(3)
90
При этом группа ОН оказывается в пара-положении к месту,
которое занимала нитрозогрунна.
Следует отметить, что реакция образования бензолмер-
куринитрата является наиболее медленной, определяющей
скорость всего процесса превращения бензола в ди- и трннитро»
фенол, и зависит от концентрации азотной кислоты (с
уменьшением концентрации HNO* скорость меркурирования резко
падает). Кроме приведенного главного направления реакции
превращения иитрозобензола в нитрофенолы,. возможна и
другая схема реакции, которая состоит в первоначальном
взаимодействии нжтрозобензола с окисью азота с образованием
нитрата диазония [1891 по схеме
CoH*NO + 2NO -> ArN = N — NO*.
Полученное дкгазонкгевое соединение разлагается с
образованием фенола, который легко нитруется разбавленной
азотной кислотой до ди- и тринитрофенола с оксигрупной в том
месте, где была группа —N0:
Аг — N = N — N0, ->
N0*
(4)
Образование при окислительном нитровании бензола в
присутствия Hg(NOg)@ в качестве побочных продуктов жщтробен-
зола и п-динитробензода также объясняется соответствующим
превращением нитрозобензола по схеме
NO NO,
N,0,
-3N0
(5)
91
В случае замещевыых производных ижтрозобензола, не
содержащих сильно электрофильных заместителей типа нитро-
группы и обладающих свободным пара-положением, например
в случае о-нитрозотолуола, происходят те же реакции, что и
в случае бензола. При этом, как правило, в обычных условиях
нитрование в присутствии солен ртути приводит к
образованию соответствующих нитрофенолов [уравнение (3)] и нитро-
углеводорода [уравнение (5)].
Нитрозосоединеиия, содержащие сильно электрофильный
заместитель, например
N0
или в которых пара-положение к нитрозогрупне занято,
например
в условиях нитрования более стойки, и для них возможны лишь
реакции по уравнениям (4) и (5).
Отсутствие заметного ускоряющего действия добавок солей
ртути н увеличения количества образующихся нитронафтолов
при нитровании нафталина А. И. Титов и Н. Г. Лаптев
объясняют тем, что нитрование этого углеводорода при наличии в
сфере реакции окислов азота протекает очень быстро, с
несравненно большей скоростью, чем его меркурирование. То же
явление наблюдается и с другими веществами, быстро
реагирующими с окислами азота (например, с аминами и фенолами).
Авторы приходят к выводу, что ускоряющее действие и
увеличение количества образующихся пвтрофенолов при
добавках солей ртутн при нитровании могут наблюдаться лишь
для тех соединений, у которых скорость меркурирования если
92
и
не выше, то по крайней мере соизмерима со скоростью непо-
средственжом вэашыкщействия с N0,.
Подбирая соответствующие условия (например,
увеличивая концентрацию ртутных солей и уменьшая содержание
двуокиси азота),можно добиться выявления каталитического
эффекта от добавок солей ртути.
В другой своей работе А. И. Титов и Н. Г. Лаптев [1901
показали, что ос- и р-жафтилмеркуринитраты при действии кон-
центрировавной азотной кислоты в присутствии окислов азота
превращаются в 2,4-динитро-а-нафтол с выходом свыше 50%
от теории:
HgNO
А
\-HgNO
он
/\/\/
мшт&роданыд 93
§ 5. Каталитическое действие азотистой кислоты
при нитровании
Фенол принадлежит к числу наиболее легко нитруемых
веществ. При действии на фенол сильно разбавленной азотной
кислоты получаются монопитропроизводные.
Рядом исследований было установлено, что нитрование
фенола представляет собой сложную автокаталитическую
реакцию.
Мартинсен [8] при изучении нитрования фенола азотной
кислотой различных концентраций нашел, что присутствие
азотистой кислоты каталитически действует на процесс
нитрования. Дальнейшее исследование показало, что концентрация
азотистой кислоты в растворе повышается по мере течения
реакции вследствие частичного восстановления азотной кислоты
при побочных окислительных процессах. Скорость
превращения азотной кислоты в азотистую значительно уменьшается
в присутствии ртути, серебра или их солей. Применением этих
веществ удается регулировать процесс нитрования [191).
Таким образом, нитрование фенола является автокаталитиче-
скпм процессом, протекающим без внесения катализатора
извне. Мартинсен установил, что нитрование ускоряется с
повышением концентрации азотной кислоты и замедляется с
повышением концентрации фенола.
А. В. Карташев [1921 при исследовании нитрования фенола
разбавленной азотной кислотой (уд. в. 1,010—1,060) нашел, что
нитрование протекает с удовлетворительными результатами
лишь при избытке азотной кислоты. Варьируя концентрацию
азотной кислоты, ее избыток и температуру, А. В. Карташев
установил, что влияние этих факторов на нитрование
заключается в ^следующем: 1) избыток азотной кислоты понижает
начальную температуру реакции значительно более резко, чем
увеличение ее концентрации; 2) понижение температуры
реакции уменьшает окислительные процессы; 3) азотная кислота
действует на фенол при концентрации не ниже 2,8% (уд. в.
1,015); 4) высокая температура вызывает сильные
окислительные процессы даже при концентрации азотной кислоты
около 2%.
94
По наблюдениям А. В. Карташева, первоначальное
действие азотной кислоты на фенол в слабых водных растворах
(уд. в. 1,015—1,045) является окисляющим, что приводит
к образованию азотистой кислоты, играющей определенную
роль в процессе нитрования. При действии на фенолы в
эфирном растворе чистой азотной кислоты, не содержащей двуокиси
азота и азотистой кислоты, нитрование фенола не
происходит [193]. А. В. Карташев допускает следующий механизм
реакции: фенол реагирует с азотистой кислотой с образованием
нитрозофенолов (хиноноксимов), последние же при действии
азотной кислоты легко окисляются в нитрофенолы
+ 0 (1)
QH.OH + HONO
НО ОН
\/
Н N0
а,о
(2)
/
ОН
+ О-»
N0
Для экспериментальной проверки указанного механизма
реакции А. В. Карташев проводил опыты с добавкой в
реакционную среду азотистой кислоты в п-нитроэофенола. Опыты
показали, что при нитровании фенола азотной кислотой уд. в.
1,015 и 1,030 в присутствии азотистой кислоты можно вести
процесс при более низкой начальной температуре _ реакции.
Такой же эффект производит и добавление п-нитрозофенола.
Окислительное действие слабой азотной кислоты на нитрозофе-
пол доказано опытами, в которых из п-нитрозофенола при
обработке азотной кислотой уд. в. 1,030 и 1,015 получен п-нитро-
фенол, а при более длительном нагревании (до 1 часа) %-ди-
нитрофенол.
95
А. В. Карташев исследовал также процесс нитрования
фенола в растворе уксусноэтилового эфира. Произведенные им
наблюдения показали, что в ходе процесса можно выделить
два периода: начальный период, проходящий без изменения
температуры реагирующей смеси, и второй период
экзотермической реакции, причем подъем температуры происходит
вначале медленно, а эатем более быстро. Такое течение реакции
указывает, что при взаимодействии между фенолом и аэотнои
кислотой имеет место автокаталитический процесс, развитие
и протекание которого происходят за счет некоторой
промежуточной реакции, являющейся источником образования
катализатора.
Влияние азотистой кислоты на процесс нитрования фенола
показано опытами с добавкой мочевины, а именно найдено,
что, несмотря на незначительную растворимость в уксусво-
этиловом эфире, мочевина вызывает заметное замедление
процесса.
При проведении нитрования в среде СО,, т. е. при отсутствии
окислительного действия кислорода воздуха на HNO%,
которое наблюдается в обычных условиях нитрованжя, реакция
протекала с большей скоростью.
Другие исследователи [194] поддерживают точку зрения
А. В. Карташева о механизме каталитического действия
азотистой кислоты при нитровании фенола. Однако это
объяснение нельзя считать достаточно обоснованным. Относительное
значение реакции образования нитрозофенола (при обычно
малой концентрации азотистой кислоты в реакционной массе)
:' вряд ли может быть большим. Кроме того, необходимо отметить,
: что при нитровании фенола аэотнои кислотой получается смесь
|: почти равных количеств о- и п-нитрофенола. Если бы механизм
I реакции нитрования сводился лишь к окислению нитроэофе-
i: нола азотистой кислотой до нитрофенола, то следовало
ожидать образования не'смеси нитрофенолов, а лишь одного пара-
^ изомера (как известно, при взаимодействии азотистой кислоты
: и фенола получается лишь п-нитрозофенол). При нитровании
! фенола в присутствии больших концентраций азотистой кис-
: доты образование нитрофенолов может протекать через нитро-
96
зофенол. Однако, как было показано в этом случае (например,
при нитровании фенола в присутствии 2 молей азотистой
кислоты на 1 моль азотной кислоты), вместо обычного соотношения
о-нитрофенол : п-натрофснол=1 : 1 получается соотношение
1 : 9. Как и следовало ожидать, количество пара-изомера
резко увеличивается [195].
Кинетика нитрования фенола в растворе абсолютного
спирта исследована Арналлем [196]. Он проводил нитрование
эквивалентным количеством азотной кислоты при 25,5°, Скорость
реакции измерялась по количеству свободной кислоты,
найденной в растворе через определенные промежутки времени; при
этом установлено, что реакция ускоряется автокаталитически,
а это объясняется, по мнению Арналля, влиянием азотистой
кислоты, образующейся при побочных реакциях. Для
выяснения правильности этого допущения Арналль добавлял к
нитрующей смеси небольшие количества азотистой кислоты
(0,02%); опыты показали значительное повышение скорости
реакции по сравнению с контрольными, особенно заметное в том
случае, когда HNO% добавляли с самого начала реакции.
В другой серии опытов Арналль применял избыток азотной
кислоты как в отсутствие азотистой кислоты, так и в
присутствии последней; оказалось, что 100%-вый избыток азотной
кислоты повышает скорость реакции, но в начальном периоде
реакция протекает относительно медленно, ускоряясь лишь
позднее; при нитровании же 50%-ным избытком азотной
кислоты, но в присутствии азотистой кислоты, скорость реакции
быстро возрастает уже в начальном периоде, причем повышение
скорости происходит более равномерно, чем в первом случае.
Полученные результаты подтвердили, таким образом,
допущение, что образование нитрофенолов ускоряется автокаталити-
чески по мере течения реакции под влиянием азотистой
кислоты; каталитическое действие последней доказано также
опытами с добавкой мочевины (0,5%), гидразинсульфата и
солянокислого фенилгидрааина; в зтих опытах наблюдались
значительное замедление нитрования в присутствии мочевины и
полвое прекращение реакции в присутствии двух последних
агентов.
Дебетам* даотмспгой жыаюты лрц милгробдм%% 97
Далее Арналль нашел, что добавление и-нитрозофенола
не влияет на реакцию нитрования фенола. Он приходит к
выводу, что при нитровании фенола реакция протекает не через
. стадию образования нитрозофенола, а другим путем. По его
мнению, возможен следующий механизм нитрования фенола:
I HNO, Ч- HNO, -^ NA + НдО;
Н 4-
Опыты Арналля по нитрованию фенола в растворе уксусной
, кислоты показали, что применение этого растворителя
приводит к почти количественным выходам нитрофенола, причем
реакция протекает гладко и быстро, не сопровождаясь
образованием смол; при 45* нитрование 20%-ной азотной кислотой
дает выход мононитрофенолов 98,7%; реакция может быть
осуществлена и при комнатной температуре.
Анализ продуктов реакции производился Арналлем
следующим образом: сначала определяли общее количество м-,
о- и п-нитрофенолов посредством титрования хлористым
титаном, затем отгоняли с водяным паром о-нитрофенол и в
остатке по хлористому титану определяли общее содержание м- и
п-нитрофенолов; посредством обработки бензилхлоридом
последние переводились в соответствующие м- и п-нитрофенил-
бензиловые эфиры; состав смеси зфаров устанавливали
определением понижения точки замерзания при добавлении
взвешенного количества этой смеси к определенному количеству
!, чистого п-производного.
: При нитровании фенола в растворе уксусной кислоты Арналль
обнаружил некоторые изменения в отношениях полученных
^ нитрофенолов в зависимости от температуры проведения
реакции и концентрации азотной кислоты, как это видно из табл. 5.
|1 Наилучшие выходы нитрофенолов получены при натрова-
i вии 10%-ной азотной кислотой при 25°; Арналль применял
следующие весовые отношения реагентов: 23,5 г (0,25 моля) фе-
вола, 15,75 г(0,26 моля) азотной кислоты (уд. в. i,42) и 157,5 г
]%гксусной кислоты. ,
р Кроме уксусной кислоты, исследовано также влияние
1|Мекоторых других растворителей на нитрование фенола.
.7 А. В. Топчиев
Таблжца 5
нш%ро0ено*д а
5
10
20
0-
57,
57,
58,
6
8
9
iC
ц-
40
39
38
,0
,0
,3
м-
3,4
3,1
2,8
0-
58,
59,
80,
1
2
3
п-
39,0
38,1
37,3
м
2,
2,
2,
9
7
4
0-
54,
60,
61,
9
6
5
45*
п-
38,
37,
36,
2
2
4
м-
2,
2,
2,
4
2
1
При применении 10%-ний азотной кислоты получены следующие
результаты: в уксусном ангидриде реакция протекает почти
мгновенно; в ацетоне нитрование в течение 3 час. дает выход
нитрофенолов 80%; в эфире через 12 час. выход составляет
68%; в смеси спирта (2 ч.) и уксусной кислоты (1 ч.) получен
выход 75% через 48 час, а в абсолютном спирте — выход
55% через 48 час. Отношения количеств изомерных
нитрофенолов изменяются в зависимости от примененного растворите-
ля, как это видно из табл. 6 (концентрация азотной кислоты
10%, температура реакции 10° за исключением первых двух
опытов, где нитрование проводилось при 25°).
Нитрование в уксусной кислоте в присутствии
азотнокислой меди и иода не изменило обычных результатов, но в
присутствии азотнокислой ртути получено следующее
отношение изомеров: 53,5% о нитрофенола, 42,7% п-нитрофенода и
3,8% м-яитрофенола.
Вайбель [13, 197] для выяснения механизма нитрования
фенола в присутствии азотистой кислоты исследовал
зависимость направления реакции, т. е. отношения образующихся
о- и п-нитрофенолов, от концентрации азотной кислоты.
Нитрование проводили при 25° азотной кислотой в виде
разбавленных растворов (1; 1,5; 2 и 3 н.), причем в каждом опыте
применяли значительный избыток азотной кислоты по
отношению к фенолу (20 : 1), т. е. практически количество азотной
кислоты оставалось во время опыта постоянным.
Таблица 6
дэотмой жмслотой б
Растворитель
Нитрофенол, %
0-
П-
Уксусный ангидрид
Уксусная кислота
То же
Ацетон
Эфир
Спирт (2 ч.) и уксусный ангидрид
(1ч.)
Спирт
59,6
59,2
57,8
57,4
57,8
57,6
57,7
37,8
38,!
39,1
39,6
39,2
39,3
39,2
2,6
2,7
3,1
3,0
3,0
3,1
3,1
В кислоту вносили определенвое количество раствора
NaNO%, которое соответствовало в одних опытах 0,001 н.,
в других 0,003 н. HNO%; затем приливали до каплям раствор
фенола в количестве 1/20 моля на 1 моль азотной кислоты.
После прибавления всего фенола реакционную смесь оставляли
на некоторое время до полного окончания реакции. Разделение
образовавшихся продуктов реакции производили следующим
образом: избыток азотной кислоты нейтрализовали едким
натром, раствор подкисляли уксусной кислотой до слабокислой
реакции, после чего из него отгоняли с водяным паром о-нитро-
фенол.
Определение о-нитрофенола в дестилляте производили
титрованием хлористым титаном. В остатке от перегонки с во-
дяиым паром определяли п-нитрофенол и побочные продукты
нитрования; при выщелачивании остатка водой в раствор
переходил и-нитрофенол, не растворимый в воде побочный
продукт нитрования отделяли от п-нитрофенола фильтрованием.
Этот побочный продукт при исследовании оказался продуктом
окисления индофенола. Как предполагает Байбель, индофенол
мог образоваться в результате конденсации одной молекулы
100 Лытроедмиб ду%).л*ат#%ескшс и
фенола с одной молекулой нитрофенола или же с
промежуточным продуктом реакции.
Опыты Вайбеля показали, что яри яитроваиии фенола
азотной кислотой концентрации в пределах от 1 до 3 я. образуются
почти одинаковые количества о- и н-нитрофевола. Отношение
о-нитрофенол: п-нитрофенол повышается с увеличением
концентрации азотной кислоты, но в незначительной степени.
Выход продукта конденсации понижается от 12 до 3% при
увеличении концентрации азотной кислоты от i до 3 н.
Вайбелем изучено также нитрование о- и м-крезолов.
Нитрование м-крезола протекает медленнее, чем нитрование
фенола и о-крезола. Скорость образования продукта
конденсации такая же, как у фенола и о-крезола; таким образом, при
нитровании м-крезола получается больше продуктов
конденсации по сравнению с другими фенолами. Нитрование
м-крезола приводит к образованию 6-нитро- и 4-витро-м-крезолов
в весовом отношении 2:3, причем это отношение не зависит
от концентрации азотистой и азотной кислот. При
титровании о-крезола продуктами реакции являются 6-нитро- и
4-нитро-о-крезолы в отношении 1:1.. Это отношение остается
постоянным, независимо от начальной концентрации азотистой
кислоты; с другой стороны, оно незначительно возрастает при
повышении концентрации азотной кислоты. При дальнейшем
нитровании 6-нитро- и 4-нитро-о-креголы дают динитропроиз-
водные в равных отношениях. При нитровании 6-нитро- и
4-нитро-м-крезолов образуются динитропродукты с
преобладанием производных 6-нитро-м-крезола.
Вайбель исследовал также реакцию нитрозирования
фенола, причем установил, что отношение о-нитрозофенол:
n-нитрозофенол не соответствует отношению о-нитрофенол:
n-нитрофенол, полученному при нитровании фенола.
На основании экспериментальных данных Вайбель
допускает следующий механизм нитрования фенола: фенол
образует с азотистой кислотой комплекс, который при действии
азотной кислоты дает нитрофенол (схема 1). Наряду с этой
основной реакцией нитрования происходит побочная реакция
превращения комплекса в о-нитрозофенол, окисляющийся
101
далее азотной кислотой в нитрофенол (схема 2). Другая
побочная реакция — это образование продукта конденсации
комплекса с фенолом, который при действии азотной кислоты
дает окисленный продукт конденсации (схема 3).
он
ОН
он
/\н.нж),
+ HNO, =±
\
ОН
+ HNO,
/\__NO,
о-питрофепол
ОН
HNO,-»
ОН
А
HNO, ^HNO,
он
NO,
п-нитрофепол
витровавие
+ Н,0
(1)
ОН
ОН ОН
V_NO /\
о-иитровофепол
ОН
н-шо,
п-пнтроэофевол
житровироваиио
ОН
ОН
HNO,->
N0.
о-аитрофеиол
ОН
4- НЛО,
N0
N0
п-шгррофенол
омислеине
HNO
UNO
(2)
102
ОН
он
о-продунт
конденсации
о-продукт
конденсации
окисленный
продукт
конденсации
+HNO:
ОН
п-продукт . п-продукт
конденсации' конденсации
окисленный
продукт
конденсации
+ HNO,
(3)
конденсация
о кисление
Этот механизм удовлетворительно объясняет: 1)
установленное рядом исследователей каталитическое действие
азотистой кислоты при нитровании фенола, 2) несоответствие
между отношением о-нитрозофенол: п-нитрозофенол и о-нитро-
фенол: п-нитрофенол. Образование нитрофенолов только
частично происходит по схеме (2) (нитрозирование и окисление);
основная часть о- и п-нитрофенолов образуется по схеме (1),
т. е. по реакции нитрования.
Сравнение схем (1), (2) и (3) показывает, что только
скорость реакции нитрования (1) находится в прямой
зависимости от концентрации азотной кислоты, что подтверждается
экспериментально; эта зависимость не одинакова для о- и п-
нитрофенолов, поэтому отношение орто : пара варьирует с
изменением концентрации азотной кислоты. Реакция
образования нитрофенолов из яитрозофенолов (2) не находится в яря-
мой зависимости от концентрации азотной кислоты; в
количественном отношении выход нитрофенолов по этой
реакции, однако, значительно меньше, чем по реакции (1),
вследствие чего указанная зависимость отношения орто : пара от
концентрации азотной кислоты не подвергается значительным
изменениям.
Скорость реакции образования окисленных продуктов
конденсации по схеме (3) также не находится в зависимости от
концентрации азотной кислоты, что согласуется с
экспериментальными данными (при концентрации HNOs, равной 3 н.,
деомшстоД жысжотм nj?w ншпдхюамш* 103
образуется меньше продуктов конденсации, чем при
концентрации 1 и., в то время как константы скорости других
реакций возрастают с повышением концентрации кислоты).
Дальнейшие работы Вайбеля имели задачей изучить в
отдельности механизм основной реакции нитрования и процесса
образования нитрофенолов из нитрозофенолов.
Произведенные Вайбелем кинетические измерения скорости реакции ни-
трозирования фенола показали, что при варьировании
концентрации фенола и азотной кислоты в одном направлении
изменение констант скорости было неодинаковым. Вайбель
объясняет зто явление тем, что при избытке азотной кислоты,
наряду с нитрозированием, происходит динитрозирование, что
приводит к более высоким константам скорости. При избытке
фенола образуется больше продуктов конденсации, и
поэтому наблюдается уменьшение констант скорости.
При изучении кинетики реакции окисления п-нитрозофе-
нола в п-нитрофенол Вайбель нашел, что скорость реакции
сильно возрастает с увеличением концентрации азотной
кислоты, но, с другой стороны, не зависит от концентраций нитро-
зофенола и азотистой кислоты. Опыты показали, что
окисление протекает нормально лишь в том случае, если имеется
минимальная концентрация азотистой кислоты. Окисление
о-нитрозофенола протекает значительно быстрее, чем окисление
и-нитрозофенола (в 25 раз).
Измерения скорости реакции нитрования фенола при
применении азотной кислоты концентраций 1, 1,5 и 2 и.
показали, что для концентрации 1 н. конставта скорости оставалась
постоянной в течение опыта, при концентрациях же 1,5 и 2 и.
в начале опыта величина константы возрастала и только спустя
определенное время становилась постоянной. Зто явление
объясняется недостатком азотистой кислоты; при увеличении
вдвое начальной концентрации последней нитрование
протекало нормально уже с самого начала реакции, т. е. константа
скорости реакции оставалась постоянной в течение всего
опыта.
Скорость реакции нитрования фенола также зависит от
концентрации последнего; повышение концентрации фенола
104
вдвое (от 0,2 до 0,4 н.) при постоянных концентрациях азот-
вой и азотистой кислот приводит к резкому падению скорости
реакции. Это объясняется особенностью реакции нитрования*
которая состоит из двух стадий; 1) образования комплекса
с азотистой кислотой и 2) собственно нитрования. При двойной
по отношению к фенолу начальной концентрации азотистой
кислоты относительно большее количество фенола вступает
в реакцию с азотистой кислотой, образуя комплекс,
участвующий в дальнейшем процессе.
В отсутствие окислов азота при комнатной температуре
нафталин не реагирует с азотной кислотой уд. в. 1,17 и 1,36,
а с концентрированной азотной кислотой реакция протекает
очень медленно. При внесении в реакционную смесь
небольшого количества азотнокислого калия upon ходят бурная
реакция с образованием %-нитронафталина [19 ].
Таким образом, изучение реакций нитрования фенола и
других ароматических соединений азотной кислотой
показывает, что эти реакции безусловно катализируются азотистой
кислотой. При определенной концентрации азотной кислоты
скорость реакции прямо пропорциональна концентрации
азотистой кислоты.
Предположение Н. Н. Ворожцова (младшего), что
каталитическое влияние нитрозофеяола при нитровании фенола
сводится к образованию из азотной кислоты двуокиси азота (за
счет окисления нитрозофенола в нитрофенол), представляется
вполне вероятным, так как N0% катализирует нитрование
фенола гораздо сильнее, чем азотистая кислота [199].
ЛИТЕРАТУРА
1. P. Spindler. Ana., 224, 283 (1884).
2. D e v i 11 е. Ann. cbim. pbys., 3, 187.
3. Ф. Ф. Бейльштейн, А. А. Курбатов. Ann., 155, i
(1870).
4.0. Witt, E. No el ting. Ber., 18, 1337(1885).
5. A. Holleman. Chem. Rev., 1, 187 (1925).
6. W. H a r m s e n. Ber., 13, 1558 (1880).
7. Ф. Ф. Бейл ьшт**н. Ann., 144, 274 (1867),
8. H, Martinson. Z. phys. Chem., 50, 425 (1905); 59, 605 (1907).
10&
9. P. Jannasb. Ann., 176, 5 (1875).
10. R. Fittig, Geiazer. Ann., 144, 17 (1867).
11. 0. Jacob son. Ber., 17, 160 (1884).
12. A. W. С г о s s 1 e y, N. R e n о u f f. J. Chem. Soc, 95, 215
(1909).
13. S. Veibel. Bar., 63, 1582, 2074 (1930).
14. К. В e a u с о u r t, E. H a m m e r 1 6. J. prakt. Chem. [2], 120,
185 (1928).
15. С A., 4471b (1947).
16. Герм, патент 805645 (1951); С, А., 1041с (1952).
17. N. I. Leonard, S. N. Boyd. J.Org. Chem., 11, 405(1946);
A. Spada, Ezio Casiai. Gazz. chim. iW., 60, 642 (1950).
18* B. L. Z enitz, W. H. H ar f uag. J.Org. Chem., 11, 444 (1946).
19. G. M а с h e к, РЖХим., № 3, 83, реферат 14477 (1954);
J. Blanch ard, Goer ing. J. Chem. Soc., № 8, 2977 (1954).
20.1. M. Hunaberger. J. Am. Chem. Soc., 71, 2635 (1949).
21. M. И. Коновалов. Ber., 28, 1856 (1895).
22. D. I. L egge. J. Am. Cbem. Soc., 69, 2079 (1947).
23. G. M. К о 8 о 1 a p о f f. J. Am. Cham. Soc., 7i, 4021 (1949).
24. G. M. Koaolapoff. J.Am. Chem. Soc, 71, 1876(1949).
25. E. M а с с i о 11 a. C, I, 3054 (1932).
26. Fr. M. L an g. Compt. rend., 226, 1381 (1948).
27. F* Re wer di n. Helv. Chim. Acta, 12, 1053 (1929).
28. И. Г у б en. Методы органической химии, т. IV, кв. 2. М., Гос-
химиздат, 1949, стр. 1094.
29. С. Bosma, W. Meerburg, P. E. Verkade. Rec. trav.
chim., 67, 235 (1948).
30. G. Staedeler. Ann., 78, 32 (1851).
31. F. В ei Is t ein, A. Kiihlberg. Ann., 169, 85 (1874).
32. H. E. F i e r z-D a v i d. Helv. Chim. Acta, 26, 98 (1943).
33. Я. Я. M а к a p о В-Э e м л я н с к и и, В. П. Б и б и ш е в.
Журн. общ. химии, № 7, 1280 (1937); ср. также A. Schroeter.
Ann., 426, 1, 17, 83 (1922).
34. С. И. Сергиевская, Г. Я. У р е ц к а я. Журн. общ. химии,
23, 1522 (1953).
35. Англ. патент 635635; С. A., 6438g (1950); Швейк, патент 265838 и
271563; С. А., 183 (1951), 1045 (1952).
36. С. С. Price, Т. I. В ard os. J. Am. Chem. Soc., 70, 1978
(1948).
37. R. Fit tig. Ann., 124, 276 (1862).
38. R. Wills tatter. Ber., 39, 3478 (1906).
39. F. Bell, I. Ken у on. J. Chem. Soc., 2705 (1926).
40. H. Hubner, H. Ltd d ens. Ber., 8, 871(1875).
41. G. A. Schmidt. Ben, 12, 1153 (1879).
106
42. J Schmidt. Ben, 34, 3531 (1901).
43. J. Schmidt, 0. Spoua. Ber., 55, 1194(1922).
44. H. H. Вороясцов (младш.). Журн. хим. пром., № 6, 21 (1947).
45. J. Schmidt, К. Bauer. Ber., 38, 3758(1905).
46. Амер. патент 2052614; С, I, 438 (1937).
47. И. С. Иоффе, А. С. Эфрос. Журн. общ. химии, 46, 111
(1946).
48. Р. В г u n a e г, О. Witt. Ber., 20, 1023 (1887).
49. V. Meyer, О. Stadler, Ber., 17, 2648 (1884).
50. A. Clans, H a r t m a a n. J. prakt. Chem., 53, 198 (1851).
51. Fr. Fried 1. Moaalsb, Chem., 34, 766 (1913).
52. W. K. Lew) в, Т. Y. Suea. Ind. Eng. Chem., 32, 1095 (1940).
53. Fr. E. Pounder. J. Mas son. J.Cbem. Sou, 1352(1934).
54. A. Kobe, Mills. Ind. Eng. Chem., 45, 287 (1953).
55. H. McCormack. Ind. Eng. Cbem., 29,1933 (1937); C. A., 153
(1947).
56. G. Taylor, L. Russe. Амер. патент 2643271 (4953).
57. W. W. Jones, M. Basse!. J. Chem. Soc, 921 (1947).
58. W. H. Gibso n. J. Chem. Soc, 121, 270 (1922).
59. А. Ш т e т б a x e p. Пороха и взрывчатые вещества. М., ОНТИ,
1936, стр. 379.
60. W. Will. Ber., 47, 704 (1914).
61. G. Schultz, J. Flasch lander. J. prakt. Chem., 66,
153 (1902).
62. G. W eis wei 11 er. Monatsh. Chem., 21, 39 (1900).
63. Б. И. Долгов, Н. А. Кучумова. Журн. общ. химии,
20, 445 (1950).
64. J. W. Haua, A. Kobe. Ind. Eng. Chem. 43, 2355 (1951),
65. G. Hansh, G. H elm к am p. J. Am. Chem. Soc., 73, 3080
(1951).
66. K. L. Nelson, H. G. Brown. J. Am. Chem. Soc, 73, 5605
(1951).
67. H. С Brown, W. H. Bonner. J. Am. Cbem. Soc., 76, 605
(1954).
08. A. W. Cross ley, N. Renouff. J. Chem. Soc., 95, 215
(1909).
69. Marqueroll, Loriette. Bull. Soc. chim. France, 27, 424
(1920).
70. E. Noeltiag, S. Fore]. Ber., 18, 2674 (1885).
71. D. M. Bowen, R. W. В e 1 f i t, R. A. Walser. J. Am.
Chem. Soc., 75, 4307 (1953).
72. A. Kobe, H. Levin. Ind. Eng. Chem., 42, 352 (1950).
73. A. Kobe, В г е а а е с к e. Ind. Eng. Chem., 46, 728 (1954).
74. E. Noeltiag, C. G eis man. Ber., 19, 144 (1886).
107
75. Н. Lee о re h e, P. A u b er t ei n. C. A., 919a (1952).
76. A. Hoi la n d e r, E. H a e f f en. C, III, 338 (1920).
77. R. Ni et z к i, А. К on wal dt. Ber., 37, 3892 (1904).
78. H. S u b b а и др. С, I, 2866 (1931).
79. J. Kl о sa. РЖХим., № 16, реферат 38639 (1954).
80. H. Zah n, A. Wiir z. Angew. Chem., 63, 147 (1951).
81. G. G. Fi n ger, N. H. N a cht ri e b, E. H. Ree d.
C, I, 1009 (1940).
82. H. H. H о d gso a, H. Cr. Bear d. J. Soc. Chem. lud., 45, 91
(1926).
83. С. Н. У ша к о в, 1С. Н. Ф р ей дбе р г. Изв. АН СССР, ОХН,
268 (1950).
84. Т. Nishi, Н. Toki. С. А., 11099, (1950); J. Soc. Cbem. Ind.,
Japan, 45, 37 (1942).
85. W. 10 av e y, J. R. Gwi It. J. Chem. Soc, 204 (1950).
86. А. А. П он о марен ко. Журн. общ. химии, 20, 469 (1950).
87. I. F. Me Gh i е, С. М о rt о а, В. L. Re у п о Id s,
J. W. Sp enc e. J. Soc. Chem. Ind., 68, 328 (1949).
88. L. J. В ol linger. C. A., 173(1949).
89. R. К 1 e e n e. J. Am. Chem. Soc, 7i, 2259 (1949).
90. Walter Q v 1 s t. С A., 2397 (1947).
91. B. B. Dey, R. K. Mailer, B. Pai. С A., 5755(1949).
92. B. B. Dey, R. K. Mailer, B. Pai. С A., 4668(1951).
93. Амер. патент 2525508; С. А., 2504 (1951).
94. I. Forrest, D. Lid d ell, S. H. Tucker. J. Chem.
Soc, 454 (1946).
95. И. М. Коган, Д. Ф. Кутепов. Журн. общ. химии, 21,1297
(1951).
96. А. П. Терентьев, Б. М. Кедров. Уч. зап. МГУ, 6, 213
(1936).
97. И. Г у бен. Методы органической химии, т. IV, вып. 1. М.—Л.,
Гослитиздат, 1949, стр. 260.
98. Н. Н. Hodgson, E. R. Ward. J. Chem. Soc, 533(1946).
99. J. Brink, S. N orris. Ind. Eng. Chem., 46, 694(1954).
100. А. А. Пономаренко и Т. Е. Мациевич. Журн. общ.
химии, 20, 1845 (1950).
101. С. F. Koelsch, D. О. Hoffmann. J. Am. Cbem. Soc,
65, 989 (1943).
102. M. А. Ильинский. Полупродукты и красители
антраценового ряда. М.— Л., Госхимтехиздат, 1932, стр. 68.
103. P. M. Bavin, M. J. Dewar. Chem. and Ind., 53, № 22, 543
(1953). РЖХим., № 3, реферат 14492 (1954).
104. F. Fried 1. Ber., 45, 428 (1912).
Ю5. E. Ochiai, M. Katada. C. A., 9541(1951).
108 Лит^оеднме драмалгычасжыз
108
109
НО
106. Е. Ochiai, M. К at ad а. С. А., 9538 (1951).
107. Н ertog van W е е г е п. Вес. trav. cbim., 67, 984 (1948).
.1. О b er h о f f, H. Y. Her tog. Rec trav. chim., 69, 468
(1950); С. А., 6418 (1950).
.A. Glaus, Th. Kramer. Ber., 18, 1243(1885).
. A. Kaufman n, H. Decker. Ber., 39, 3648(1906).
Д1..А. Kaufman n, Н. Hussy. Ber., 41, 1735 (4906).
112. A. Adams, D. H. Hey. J. Chem. Soc, 255 (1949).
113. С. С Price, B. H. Welz en, D. B. Gutrie. J. Org.
Chem., 12, 203 (1947); Och ia*, Sch imizu. С A., 3502 (1950).
114. J. L. C. Marais, O. G. Backeberg. J. Chem. Soc, 2207 (1950).
115. H. Jen s eh. Ann., 568, 73(1950).
116. Simpson, G. F. Wright. J.Chem. Soc, 2023(1948).
117. В a zu Chandra п. С A., 7573 (1951).
118. H. N i s h i z a w a. C. A., 609 (1951).
119. E. E. Campaign @, H. G. Grose. J. Am. Chem. Soc,
73, 3812 (1951).
120. J. Klosa. Pharmazie, 8, № 3 (1953).
121. M. И. Коновалов, К. Гурепич. ЖРХО, 37, 537(1905).
122. М. И. Коновалов. ЖРХО,25, 389, 472 (1893).
123. К. J. Or ton. Ber., 40, 370(1907).
124. G. Schultz. Ber., 17, 477 (1884).
125. E. Bamberger, A. Rising. Ber., 33, 3625(1900).
126. M. И. Коновалов. Ber., 29,2201(1896).
127. Синтезы органических препаратов, т. IV. М,—Л., ОНТИ, 1936,
стр. 68.
128. A. W. Cross ley, N.Renouff. J.Chem. Soc, 95, 202(1909).
129. J. P. Lamboy. J. Am. Chem. Soc, 71, 3756 (1949).
130. W. Staedel, A. Kolb. Ann., 259, 208(1890).
131. B. D. Tiffany. J. Am. Chem. Soc, 70, 592(1948).
132. J. Am. Chem. Soc, 73, 3779 (1951); С A., 930d (1952).
133. С. К. In go Id, F. R. Shaw. J.Chem. Soc, 575 (1949); С A.,
7454f (1949).
134. N. L. Drake, H. C. Harris, С B. Jaeger. J. Am.
Chem. Soc, 70, 168 (1948).
135. H. С. В a zany, M Pianka. J.Ghem. Soc, 956(1946).
136. И. Г у б е н. Методы органической химии, т. IV, вып. 1. М.—Л.,
Госхимиздат, 1949, стр. 277.
137.0. Witt, A. Utermann. Ber., 39, 3901 (1906).
138. Е. Bamberger. Ber., 27, 585 (1894); 28, 399 (1895).
139. П. М. Бугаи. Жура. общ. химии, 4, 605 (1953).
140. К. G s u d a, S. S a k a m a t о. С. А., 2887 (1951).
141. R. Passerini. С. А., 7976с (1951).
142. С. D. Houghton, A. Waters. J.Chem. Soc, 1018(1950).
108
143. Van Hove. Bull. Acad. Belg., 8, 505 (1922).
144. W. Borsch e, W. Bothe. Ber., 41, 1940 (1908); W.
Borsch e, Schaeke. Ber., 56, 2498(1923).
145. R. H. Thomson, E. Race, F. M. R о we. J. Chem. Soc,
350 (1947).
446. РШХим., стр. 134, реферат 10558 (1954).
147. В. А. Измаильский и А. И. Козий. Докл. АН СССР*
28, 621 (1940). '
148. Синтезы органических препаратов, т. III. M.— Л., ОНТИ, 1935, стр. 58.
149. О. Dimroth. Ber., 34, 219 (1901).
150. J. Meisenheimer, E. Connerade. Ann., 330, 133 (1904).
151. J. Schmidt, E. Heinle. Ber., 44, 1488 (1911).
152. R. K. Callow, J. M. G u 11 a n d. J. Chem. Soc, 2424 (1929)'.
153. Синтезы органических препаратов, т. III. М.—Л.,ОНТИ, 1935, стр. 64.
454. Е. Tauber. Вег., 25, 128(1892).
155. Dhont, I. P. Wibaut. Rec.trav.chim., 62, 177(1943).
156. Ю. К. Юрьев, М. С. Корсакова, А. В.
Арбатский. Изв. АН СССР, ОХН, № 2, 166 (1951).
157. Синтезы органических препаратов, т. IV. М.— Л., ОНТИ, 1936, стр. 69.
158. N a i k, J a d n a v. С. А., 2519 (1950).
159. A. Pictet, P. Genequand. Ber., 35, 2526(1902).
160. A. Pictet. Ar. Science Phys. et Nature. Geneve, (4), 16, 191.
161. F. H. Cohen, J. P. W i b a u t. Rec. trav. chim., 54, 409 (1935).
162. Б. В. Тронов, Г. X. Намай, А. Г. Коваленко.
ЖРХО, 60, 1013 (1928).
163. H. Holdermann. Ber., 39, 1250 (1906).
164. M. С а г m а с k. J. Am. Chem. Soc, 69, 785 (1947).
165. W. E. Bachman, J. M. Chemefda, N. C. Deno,
E. С Horning. J. Org. Chem. 13, 390 (1948); см. также D о*
n aid S 1m ma. С A., 10213c, (1951).
166. R. Wolffenstein, O. Boeters. Ber., 46, 586 (1913).
167. R. Wolffenstein, W. P a a r. Ber., 46, 589 (1913).
168. F. В 1 e с h t a, K. P a t e k. Schiess- und Sprengstoffwesen, 22
(1927); C, I, 780 (1928).
169. G. D a r z e a s. Nitration du benzene en presence de mercure. Com-
mim. priv., Paris, 1914.
170. L. Vignon. Bull. Soc. Chim., 27, 547 (1920).
171. Broders. Trav, effect an laborat. de recherches de la Paudreriede
Saint-Fous, 1919.
172. E. Bamberger. Ber., 30, 506(1897).
173. L. Desvergens. Chim. et Ind., 22, 451 (1929).
174. А. И. Захаров. Журн. хим. пром., 8, 31 (1931).
175. W. E n z, К. Р f i s t e r. Helv. Cbim. Acta, 13, 194 (1930).
176. E. И. Орлов. Укр. хим. журя., 2, 370 (1926).
НО
177. Н. А. Холево, И. И. Эйтингтон. Журя, прикл. химии,
5, 612 (1932).
178. Т. L. Davis, D. E. Worral, N. L. Drake. J. Am.
Chem. Soc, 43, 594 (1921).
179. T. L. Davis. 'J. Am. Chem. Soc, 44, 1588 (1922).
180. S. Tsutsumi, E. Iwata. С A., 6604a (1952).
181. S. Tsutsumi, Ё. Twata. С A., 11126c (1951).
182. R. V. Me Ke e. J. Soc. Chem. Ind., 46, 261T'(1927).
183. В. В. Dey, Т. R. Govindachari, M. T. Govind-
r a Jan. С. А., 38, 3264 (1944).
184. A. M. Володарский, В. С. Калинина. Патент
СССР 55967.
185. Франц. патент 821627 (1937).
186. R. I. Thomas, W. F. Anzilotti, G. F. H e nn i о п.
Ind. Eng. Chem., 32, 3, 408 (1940).
187. А. И. Титов, А. М. Барышникова. Журн. общ. химии,
17, 829 (1947).
188. А. И. Титов, Н. Г. Лаптев. Журн. общ. химии, 19, 267
(1949).
189. Е. Bamberger. Ber., 30, 512 (1897); 51, 634 (1918); см.
также А. Н. Несмеянов, С. Т. Иоффе. Журн. общ.
химии, 11, 392 (1941).
190. А. И. Титов, Н. Г. Лаптев. Доил. АН СССР, 66, 1101
(1949).
191. A. Klemeaz. С, II, 442 (1922).
192. А. В. К а р т а ш е в. ЖРХО, 59, 819, 833 (1927); 62, 2129 (1930).
193. A. Klemeaz, Monatsch., 39, 641 (1918).
194. R. M. S с h r a m m, F. H. Westheimer. J. Am. Chem.
Soc, 70, 1782 (1948).
195. C. A. Bunt on, E. D. Hughes, G. J. Mink off,
Y. R eed. Nature, 158, 514 (1946).
196. F. A mall. J. Chem. Soc, 123, 311 (1923); 125, 811 (1924).
197. S. V eib el. Z. phys. Chem. 13, 10, 22 (1930).
198. А. И. Титов. Журн. общ. химии, 11, 1125 (1941).
199. Н. Н. Ворожцов (старщ.). Основы синтеза промежуточвнх
продуктов и красителей. М.—Л., Госхимяздат, 1950, стр. 135.
МЕХАНИЗМ НИТРОВАНИЯ
АРОМАТИЧЕСКИХ СОЕДИНЕНИИ АЗОТНОЙ КИСЛОТОЙ
II НИТРУЮЩЕЙ СМЕСЬЮ
Настоящая глава посвящена теоретическим работам,
освещающим кинетику и механизм реакции нитрования азотной
кислотой и нитрующей смесью (HNO3+ H*SO*).
Следует отметить, что этому вопросу посвящено большое
число работ как отечественных, так и иностранных авторов.
Мы остановимся лишь на важнейших из этих работ.
В 20-х годах текущего столетия Виландом [1, 2] была
предложена теория, которая рассматривает нитрование как
реакцию присоединения нитрующего агента (в данном
случае азотной кислоты) по двойной связи нитруемого
соединения.
Виланд исходит при этом из выдвинутого еще Тиле [3] и
Голлеманом [4] положения, что всякое замещение в
ароматическом ряду есть результат первичного присоединения.
Например, реакция бромирования ароматических соединений,
приводящая к замене атома водорода ароматического
соединения бромом, протекает, согласно такому представлению,
через первичное присоединение по схеме
/\
+ Вг,
\/
н
н
У\
\/
сн
+ НВг
СВг
112
При реакции нитрования по его воззрениям происходит
присоединение азотной кислоты по двойной связи
ароматического ядра с последующим отщеплением воды:
У\
У\
\/
Изучение нитрования непредельных соединений привело
Виланда к допущению, что первичным продуктом нитрования
является нитрогидрин (нитроспирт), образующийся в
результате присоединения элементов азотной кислоты к двойной
связи нитруемого непредельного соединения
СН,: СН, + HONO, -> HOCH,CH,NO,.
Нитрование первого члена ряда олефинов — этилена
осуществлено еще в 1869 г. Некуле [5]: при обработке этилена
смесью азотной и серной кислот ои получил масло, которое,
по его мнению, представляло собой нитрит-нитрат этиленгли-
коля
CH,(ONO)CH*(ONO,).
Вяланд, повторяв этот синтез, применил более
совершенный метод очистки (перегонка с паром и последующая
многократная перегонка в вакууме). Он нашел, что масло,
получающееся при нитровании этилена, состоит из смеси двух
продуктов, из которых один (с более низкой точкой кипения)
идентичен динитрату гликоля CH:(0N02)CH:(0N02), а другой,
образующийся в меньшем количестве, представляет собой эфир
азотной кяслоты и (3-нитроэтилового спирта CH*(ONOa)CH*(NO*).
По мнению Виланда, последнее соединение образуется в
результате этерификации азотной кислотой первичного
продукта реакцяи р-нятроэтилового спирта ф-нитрогядряна)
СН*(МОд)СНг(ОН). Для проверки этого предположения
Виланд подвергал чистый нитроэтиловый спирт действию
нитрующей смеси; продукт этой обработки оказался
идентичным нитроэтилнятрату, полученному при нитровании этилена.
митроеамил
113
i Строение р-вжтроэтилового спирта доказано Виландом сдн-
:' ^езом его по В. Мейеру из этилевиодгидрива и азотястокислого
серебра:
AgJ
|
СН&ОН
Н,ОН
При перегонке нитроэтялового спирта над фосфорным ан
гядридом или безводным бисульфатом Вяланд получил с хо
рошям выходом нитроэтилен
СН,ОН
CIIXO,
сн,
+Н.0
Этот продукт может быть получен также из нлтроэтялнятрата
СН,(ЖО,
HNO
СН,
На основаняя приведенных выше экспериментальных данных
Вяланд допускает следующий механизм реакция нитрования
этилена азотном кяслотон:
сы,
C1L ОН
СН;\О;
(^-интроэтадовый спирт)
NO
ОН CH;ONO%
нитроотнлнитрат
Образование второго продукта реакции — динятрата гликоля—
Внланд объясняет следующим образом: остаткя азотной кисло*
ты (N0% и ОН) присоединяются по месту двойной связи
нитруемого олефяна, причем группа N0, связывается с олефйаем
8 А. В. Топчиев
114
мылцкюдныл
не через атом азота, а через атом кислорода; образовавшийся
таким образом мононитрит гликоля при дальнейшем действии
нитрующей смеси переходит в двнптрат:
СН,
ОН СН,ОН
присоедиветхе i
,
2H,ONO
CH*ONO
этилеигликоля
CH,ONO,
NO
CH,ONO
нитрит-нитрат
этилеигликоля
CH,ONO,
окисление
CH,ONO
дннитрат гликоля
При нитровании %,ос-дифевилэталена азотной кислотой
в растворе СС14 получен «,%-дифенил-^яитроэтиловый спирт [61
(т. пл. 106°):
(С$Н@), С = СН% Н^. (СЛв)% С (ОН) CH
При этом осуществляется лишь первая фаза реакции, а
именно процесс присоединения.
При действии раствора безводной азотной кислоты в че-
тыреххлористом углероде на изоамилен Виланд получил,
помимо других продуктов, питроизоамилев, который является,
таким образом, продуктом замещения
(СН»), С « СН — СН,
ЦСН@), С (ОН) — СН (КО.) — СН,]
нитроизоамилен
Колер и Дрэйк [7] показали, что нитрование %,%-дифенил-
этилеяа осуществляется с хорошими выходами, если к
углеводороду постепенно добавлять азотную кислоту при низкой
температуре и хорошем перемешивании. В качестве продукта
реакции в этом случае получаются %,%-дифениляптровтилев и
а,а-джфеаиляитроэтиловый спирт.
145
Из ароматических соединений Виланд исследовал
нитрование фенантрена, который легко присоединяет азотную
кислоту, причем происходит одновременное отщепление элементов
воды от яитроспирта с образованием соответствующего
простого эфира. Нитрование осуществлялось им следующим образом:
в раствор 20 г фенантрева в 100 мл СС1*, охлажденный до —15°,
медленно приливали по каплям 10 мл концентрированной азот-
пой кислоты при перемешивании. Продукт реакции, имевший
вид смолистого осадка, перекристаллизовался из бензола.
После вторичной перекристаллизации из ацетона получен,
по мнению автора, эфир, образовавшийся из двух молекул
8-окси-9-нитрофенантрена (т. пл. 167*). Виланд предполагает,
что при нитровании ароматических соединений, как и при
взаимодействии этиленовых соединений с концентрированной
азотной кислотой, первоначально происходит присоединение
молекулы азотной кислоты к двойной связи ароматического ядра
с образованием неустойчивого нитрооксипроизводного,
которое разлагается на соответствующее ароматическое нитро-
соединение и воду. Например, при нитровании бензола в
качестве первичного продукта реакции образуется нитрояси-
циклогексадиен — весьма неустойчивое соединение, которое
затем распадается на нитробензол и воду
N0,
+ HONO,
\/
\/\
+Н,0
н
\/
Признак существования промежуточного продукта Виланд
видит в коричневой, быстро исчезающей окраске,
появляющейся при смешении азотной кислоты с бензолом. Однако
Виланд ве приводит никакого прямого доказательства
существования промежуточного продукта присоединения азотной
кислоты к бензолу. ~
В связи с работами Виланда по нитрованию непредельных
соединений, на основе которых этот исследователь построил
свою теорию нитрования ароматических соединений, упомя-
Жем также о работах Аяшютда и Гильберж* [8].
116 М«я*?*%*л*
Эти авторы нитровали &,*-дифеяилэтилен р растворе
уксусной кислоты равномолекуляряым количеством азотной
кислоты сначала при обыкновенной температуре, а затем при 100*
до тех пор, пока в пробе раствора при разбавлении водой
выделялся не маслянистый, а кристаллический продукт; по
окончании нитрования добавляли воду, причем получался осадок.
Посредством дробной кристаллизации последнего из спирта
авторы выделили три продукта: %,%-дифелтл-р-питроэтиловый
спирт (т. пл. 106—107°), ос,%-дифешт ^ннтроэтилел (т. пл.
87—88°) и а,%-дифенпл-р,Э-динитроэтилен (т. пл. 146—147°).
Образование этих трех продуктов реакции объяснено
следующим образом:
КО,
(СдНя)ч С ;= СН? -г
а, %-дифенил-^-нитро -
й сппрт
С (ОН) CH,NO, =2^ (С^@Х С = CHNO*
этилен
ОН , .
ло
-дишггроэтилсн
Образование нитроспирта наблюдалось также нрп действии
азотной кислоты в& ос,ос-ди-п-толилэтплеи.
В литературе встречаются указания о получении
некоторыми авторами продуктов присоединения при нитровании
ароматических соединений. Так, например, Одда [9] при нитровании
нафталина азотном кислотой получил маслянистый продукт,
который, по мнению автора, представлял собой молекулярный
комплекс Вафталипа с азотной кислотой. Этот комплекс
оказался весьма неустойчивым соединением и очень быстро коагу-
с отщеплением воды и образованием остыононитроваф-
вЁ. Для объясйенЬя механизма нитрования и образований
присоединения Одда принимает, что бЫаольнае
молекулы нафталина не обладают
ф,.
117
способностью: одно из них имеет ароматический, другое
алифатический характер (алицикдическое ядро); последнее более
реакционноспособно, содержит две конъюгированные двойные
связи и обладает наибольшей реакционной способностью
в %,%'-положении. В этом положения я происходит
присоединение HOMO* при смешении нафталппа с азотной кислотой
с образованием комплекса, который отщепляет воду я дает
%-мононятронафталия. При дальнейшем нитровании
нафталина, т. е. при введении второй нитрогруппы, Одда допускает
предварительйук) перегруппировку ядер с переходом
алифатических свойств к другому ядру, причем присоединение HNOg
происходит в положениях 5,8. Образовавшиеся комплексы
далее отщепляют воду и дают динитросоединения (1,5- и
1,8-динитронафталины).
Помимо работ Одда, имеются указания на присоединение
азотной кислоты к бензальфталимидину я к бензальдифепил-
малеиду [10]. Кроме того, показано, что при действии смеси
азотной и ледяной уксусной кислот на N-ацилпроизводяые ку-
мароно-(2,3,2',3')~индола происходит присоединение групп
—N0* и — ОСОСНз к двойной связи и образование
соответствующих Битроацетатов [11]
О
ОСОСИз
N ОСОСНз
ОСОСП,
Мейзеягёймер [12] получил продукт присоединения а8от
ной кислоты к антрацену — дигидронитроантранол C^H
Н ОН
У\/\/\
При обработке этого продукта щелочью выделено соответствую
щее нитросоединение — мезонитроантрацен
строение которого доказано восстановлением цинком и НС1
в мезоантрамин C^HgNHg; последний оказался идентичным
продукту, получающемуся действием водного аммиака на ан-
транол.
Предложенная Виландом схема образования ароматических
нитросоединений отщеплением воды от первично
возникающего продукта присоединения молекулы HNO, к двойной
связи ароматического ядра встретила ряд возражений, из которых
заслуживают внимания следующие:
1) образующиеся после присоединения азотной кислоты
производные циклогексадиена должны быть крайне нестойкими,
и последующее превращение их только в направлении
отщепления воды является мало вероятным;
2) против присоединения молекулы азотной кислоты к
двойным связям ароматического ядра говорят несоответствие
скоростей различных реакции замещения, например,
чрезвычайная трудность бронирования бензола (без катализаторов), и
легкость нитрования.
Считая на этом основании концепцию Виланда
неудовлетворительной, Б. В. Тронов [13, 14], а также С. С. Наметкин
и А. С. Забродина [15] выдвинули другую схему нитрования
ароматического ядра, согласно которой ароматическое ядро
присоединяется и атому азота азотной кислоты путем
образования связи с С — N и одновременной миграции атома
водорода, до того соединенного с атомом углерода, к одному из
кислородных атомов молекулы азотной кислоты.
О О
+ Н,О. (1)
\
он
R-
\
\
О
он!
н I
119
При таком допущении промежуточный продукт реакции
сохраняет свой ароматический характер, что делает понятным
отсутствие у него склонности к различным превращениям,
свойственным производным циклогексадиена.
Образование азотнокислых зфиров нитроспиртов при
действии азотной кислоты на непредельные алифатические
соединения объясняется, по мнению этих авторов, предварительным
нитрованием:
О О
, + , —N— ОН -» СН, = , + %
ОН ОН
Вторая молекула азотной кислоты присоединяется к двойной
связи с образованием азотнокислого эфира нитроснирта
сн, = си N0,+изо, -> о,:ч — о — сы,—сн,—:чо*.
В противоположность мнению Виланда, согласно которому
азотная кислота может присоединяться к двойной связи
только с разрывом по связи НО — N0%, Михаэль и Карлсон [16]
предполагают, что азотная кислота при действии на
непредельные соединения при низкой температуре может
присоединяться по схеме Б. В. Тронова и С. С. Наметкина с разрывом по
связи Н — 0N0*. Образование нитросоединений есть
результат вторичной реакции. Таким образом, процесс нитрования
ароматических соединений должен протекать по схеме
О О
С*Н& __ Н + N = О -» С*Нв — % -ОН -> C#H,NO,+H:O,
ОН ОН
а при нитровании циклогексана реакция должна идти так:
О О
%, + N = 0-» С,Ыц __N —0H-> Qhio = NOOH + Н%0
ОН ОН
изонитроциклогенсан
Согласно этой схеме можно предположить, что нитрование
ароматических углеводородов должно идти так же, если
120 Меаити&м мылухмдныя
вместо азотной кислоты взять эфиры азотной кислоты. В этом
случае следовало бы ожидать следующей реакции:
О О
= О -» С#Н* - Г< — ОН ^ СоН#)40д + ROH,
OR OR
но в действительности нитросоединсние не образуется.
В отношении нитрования непредельных соединений эти же
авторы считают, что при действии азотной кислоты происходит
присоединение по двойной связи согласно схеме
II И
\
с
\
X
с —с
X
Таким образом, схема Б. В. Тронова, С. С. Наметкина и
А. С. Забродиной ве могла объяснить Отсутствия нитрующих
свойств по отношению к ароматическим соединениям у эфиров
азоФвой кислоты. При смешении бензола и его гомологов
(ксилола й псевдокумола), а также легко нитрующегося ани»
Аола с этилнитратом и изоамилнитратом не наблюдалось
никаких изменений даже при стоянии этих смесей в течение 4—
5 лет. Трудно допустить, чтобы замена водорода в азотной
кислоте на углеводородный остаток сама по себе могла оказать
влияние на распад молекул HNO, на N0% и ОН или на разрыв
двойной связи азота с кислородом. Скорее всего можно было
прийти к выводу, что в реакции нитрования принимает
участие атом водорода азотной кислоты.
Исходя из указанных выше фактов, а также данных Гирш-
баха я Кесслера [17], которые при изучении действия
азотной кислоты на бензол в нитробензольном растворе нашли,
что эта реакция по отношению к HNO* является
бимолекулярной, Б. В. Тронов допускает, что в реакции нитрования
ароматических соединений азотной кислотой принимают участие
две молекулы последней, причем одна из них является
катализатором я действует, невидимому, своим подвижным атомом
водорода. Механизм реакции состоит а том, что HNO,
присоединяется к атому углерода ароматического ядра, образуя комплекс
(А). Этим расшатывается связь между углеродом и связанным
с ним водородом; последний становится подвижным и легко
отщепляется, вследствие чего создаются условия для
присоединения комплекса (А) к новой молекуле кислоты с образованием
комплекса (Б); далее происходит распад комплекса (В) с
отщеплением одной молекулы азотной кислоты и молекулы воды,
что и приводит к образованию нитробензола:
Y"
сн Ън
CH N—ОМ CH
НС" С' "[ОН , НС ^С—N
нс^^сн ' нс^сн
СН (6) СН
Отсутствие у эфиров азотной кислоты нитрующей способвостиг
по отношению к ароматическим соединениям Б. В. Троаов
объясняет тем, что реакция может протекать только в
присутствии катализатора, каковым в случае нитрования азотжой
кислотой является она сама.
Основываясь на данных Бедткера [18], который показал,
что этиляитрат энергично нитрует бевэол в нрисутотвяи
Б. В. Тронов исследовал влияжие ряда хлоридов (BClg,
, PCI,, FeClg, Aid*, SiCI^, SbCl@) на реакцию
нитрования бензола зтилнятратом. К эквимолекулярным количествам
компонентов добавляли 0,5 моля испытуемого в качестве
катализатора вещества (опыты с А1С1* производили и с
варьированием количества катализатора). Смеси выдерживали при
комнатной температуре в течение 48 час, затем разбавляли
водой и эфиром. Эфирный раствор сушили хлористым кальцием
и .вываривали в тщчение 1 часа при 105—110° для удаления
этилнитрата и бензола (нитробензол определяли в остатке).
Опыты показали, что из исследованных веществ лишь А1С1,,
8дС1д, SbCl& и FeClg обнаружили каталитическую активность
и притом в различной степени, как это видно из табл. 7.
Таблица 7
и ГсС1з
Катализатор
А1С1,
AICI3
S11CI4
SbCl*
Количество
катализатора,
моли
од
0,3
0,5
0,5
0,5
0,5
Выход
нитробензола, % от
теории
12,0
39,5
50,8
35,0
7,1
9,8
Исходя из данных Вернера [19] и Пфейфера [20], которые
показали, что эти четыре хлорида обладают способностью
к образованию разнообразных комплексов, Б. В. Тронов
объясняет их каталитическое действие при нитровании этил-
нитратом присущими им комплексообразующими свойствами
(в отличие от ВО,,, SiCl* и PCIJ. Так как известно, что А1СЦ
и ЗпСЦ, которые оказались наиболее активными
катализаторами (см. табл. 7), энергично реагируют с этилнитратом,
Б. В. Тронов допускает, что А1С1з(8пСЦ) образует комплекс
с этилнитратом, который затем вступает в реакцию с бензолом Л
С,НвО —N = O4-AlCl,-*CgHaO--N==O ..AlCl, (1)
О
О. ...'
о
N = О ... А1С1,
ii
О
с,н,он + о
но с,
о + Aici;
(2)
* Данная схема построена Б. В. Триповым по типу реакции
образования кетонов при взаимодействии хлораыгидридов с ароматияе-
123
По аналогии с указанной выше схемой нитрования этилпитра-
том в присутствии cAlClg Б. В. Тронов предполагает, что
нитрование ароматических соединении азотной кислотой протекает
ве по схеме (1), как он допускал ранее (см. выше, стр. 118),
а по схеме (3) (нитрование ароматического соединения
осуществляется комплексом из двух молекул HNO,, из которых одна
является нитрующим агентом, а другая комплексообразова-
телем)
НО О... HONO,
НО—N = 0 HONO, + C*H, -* N -*
О О Call*
НО — N = О ШЖО,-» Н,0 + О =N = О HONO,-»
НО СдН* СдН*
+ HNO,. (3)
Как указано выше, нитрование, по представлению Вилан-
да, протекает через первичную стадию присоединения HNOa,
в результате чего образуются насыщенные ннтрогидрины.
У алифатических соединений иитрогидрины разлагаются далее
на нитроолефины и воду или, подвергаясь этерификации, дают
соответствующие эфиры азотной кисло™ (оба эти процесса
могут происходить также и одновременно). Нитрование
ароматического ядра Виланд представляет себе как присоединение
скими углеводородами в присутствии AlClg; как известно, для этой
реакции обычно принимается первичное образование комплекса из 1
моля хлорангидрида и 1 моля А1СЦ:
C=0
ci a
—С —О&Л Aicu
124
остатков НО ж N0* к атомам углерода ядра то двойной связж
с дальнейшим разложением подученного танам образом
комплекса ла нитропроизводное и воду.
Михаэль ж Карлсон [21] считают эту схему № доказанной
экспериментально; в тех условиях, в которых Виландом про»
изводилось нитрование непредельных соединений ааотяой
кислотой и нитрующей смесью, процесс нитрования значительно
усложняется побочными реакциями, приводящими к
образованию соединений, которые могут играть роль нитрующих
агентов. При нитровании этилена нитрующей смесью Виланд,
наряду с продуктами окисления, подучил азотный эфир нитро-
спирта (нитронитрат) CH*(ONO*)CH*NO*, образование
которого он объясняет этерифжкацией азотной кислотой нитро-
спирта — первичного продукта прямого присоединения
молекулы HNOs к этилену.
По мнению Михаэля и Карлсона, в данном случае более
вероятен другой механизм реакции, так как в смеси серной
я азотной кислот нитрующим агентом является не HNOs,
а смешанный аягвдрид H0SO,ON0,, который обладает, как
все смешаяяые ангидриды, большой способностью к
присоединению. При присоединении этого ангидрида к этилену
образуется промежуточный продукт реакция СНа(ОЗОзН)СН%МОд,
в котором остаток SO,H может быть легко замещен болеь
отрицательным радикалом N0%, что приводит к ийтронитрату
L-H,
пжросеряая
кислота
Далее, с появлением в реакционной смеси в результате
окислительных процессов окислов азота, создается
возможность для вступления в реакцию NsO* и N±0*. Взаимодействие
125
окислов о этжлевом (присоединение) также приводит
к образованию нитрожитрата.
Как указывалось типе, при нитровании изоамилена в
растворе ССЦ Виланд в качестве основного продукта реакции
лолучил ос-нитроизоамилен наряду с некоторым количеством
вйтронитрата
Исходя из правила положительно-отрицательного
присоединения, Михаэль и Карлсон полагают, что если бы реакция
протекала по схеме Впланда, то в качестве первичного продукта
должен был бы получиться главным образом
я Лишь небольшое количество
Этерификация первого продукта азотной кислотой
должна была бы привести к конечному продукту
<CH,)^(NO*)CH(ONO,)CH*, а не к тому изомеру, который
Виланд получил в действительности. Если же допустить, что
реакция происходила посредством присоединения N*O*, может
наблюдаться образование полученных Виландом продуктов:
Е,С
Н,С
СН —СН,
N0%
с—си.
нитровзоамилен
СИ,
ОН
СЫ
СН
СН - СН, + HOMO, ->
ОН N0%
СИ — СН,
ONO
426 Джамны*** тлллуюддмма
Анализируя экспериментальные данные Виланда по
нитрованию азотной кислотой а,*-дифеяилэтилеяа, Михаэль и
Карлсон приходят к выводу, что в данном случае наблюдалось
участие в процессе нитрования окислов азота, возникающих
в результате побочных окислительных реакции (как известно,
с N,0, %,%-дифенилэтилен дает маслообразный продукт,
который затем легко разлагается на нитрогидрин и производные
нитроэтилена; с N^O*, в присутствии воды, дифенилэтшюн
образует %,0-нитрогидрин).
Для экспериментальной проверки высказанных ими
взглядов о действии азотной кислоты и нитрующей смеси на
непредельные углеводороды Михаэль и Карлсон проводил*
титрование олефинов в условиях, при которых устранялись
окислительные процессы, приводящие к образованию окислов
азота, а именно в присутствии растворителей (хлороформа, че-
тыреххлористого углерода и хлористого метилена) и при
низкой температуре и сильном перемешивании.
Нитрование этилена азотной кислотой (98,6 % -ной) они
осуществляли или посредством барботирования этилена через
азотную кислоту в растворе хлористого метилена при —25°,
или же добавляя HNO% в растворитель, через который при —28*
пропускали этилен при сильном перемешивании; в обоих
случаях получен маслянистый продукт реакции, который
разлагался даже яри умеренном нагревании. Отходящие газы
состояли из окислов азота.
Нитрование бутилена, который обладает большей
способностью к присоединению НВг и воды, чем этилен, привело лишь
к образованию продуктов окисления. Из последних выделен
метилэтилкетон, который мог бы образоваться при окислении
азотнокислого эфира вторичного бутилового спирта.
При нитровании изоамилена и изобутилена получены
более удовлетворительные результаты, что объясняется большей
способностью этих углеводородов к присоединению кислот
по сравнению с этиленом и бутиленом, а также большей
устойчивостью эфиров с третичным радикалом по отношению к оэкис-
лению сравнительно с соответствующими вторичными
производными.
мытроддныя ap&wamwxfCKiw; бм^ынднмД 127
В противоположность указа шло Виланда и Рана [2], при
нитровании ивоаиилева в качестве основного продукта реакции
Михаэль и Карлсон получили азотнокислый эфир третичного
амилового спирта. Нитрование проводили добавлением 98,6%-
ной аэотвой кислоты в раствор изоаыилеяа при —20°. При
таких же условиях авторы нитровали изобутилен, причем по-
лучей азотнокислый эфир третичного бутилового спирта.
Авторы объясняют образование нитратов присоединением
молекулы азотной кислоты к молекуле олефина по схеме
СН,
ONO,
При нитровании ос,ос-дифенилэтилена, которое Михаэль и
Карлсон проводили в растворе хлороформа или ССЦ при 0*,
обнаружено, что углеводород не реагирует с азотной кислотой
с образованием вптропроивводного, но частично превращается
в полимер. В растворе ледяной уксусной кислоты полимер не
реагировал с азотной кислотой при комнатной температуре; при
проведении же реакции при нагревании из реакционной смеси
удалось выделить %,*-дифенил-(3-нитроэтиловый спирт. Эти
данные подтверждают, что полученяыеВиландом соединения
могли образоваться в результате взаимодействия углеводорода с
окислами азота, а не прямым присоединением азотной кислоты.
Переходя далее к титрованию ароматических
углеводородов, Михаэль и Карлсон указывают на неправильность
предложенной Виландом схемы процесса нитрования бензола по
следующим причинам: в обычных условиях бензол обладает
слабой способностью к реакциям присоединения (по
отношению, например, к галоиду), по благодаря
внутримолекулярным энергетическим изменениям присоединение к одной паре
атомов углерода влечет за собой передачу способности к
присоединению двум оставшимся парам атомов углерода.
Аналогичные отношения должны наблюдаться и при нитровании
азотной кислотой. Отсюда можно сделать вывод, что если бы ни-
арование бензола азотной кислотой происходило по схеме
128 ЛЛмгдниал* ми/мротзммл
Влланда, оно дйлжно было бы привести к образованию
исключительно триаитробензола.
При житровавии фенантреяа азотной кислотой Виланд
получил эфир 8-окси-9-нитрофенаятрена, образовавшегося, по
ого мнению, в результате конденсации двух молекул 8-окси-
9-нитропроизводного с выделением одной частицы воды. 8-окси-
9-нитропроизводноо фввбятрена является первичным
продуктом прямого присоединения групп НО— и N0%— к атомам
углерода в положениях 8 и 9 (по Виланду, только эти атомы
углерода обнаруживают способность к присоединению,
аналогичную этиленовым углеводородам). Более "ранние
литературные данные [22] не подтверждают этих положении Вилан-
да, так как при нитровании фенантрена в растворе уксусной
км слоты получены не только 9-нитро-, но также 3-, 2- и 4-ни-
грофенантрены. Эфир, выделенный Виландом, повидимоыу,
образовался при действии яитрозиых газов на фенадтрен (по
данным Шмидта [23], нитрование феваатрена житрозными
газами приводит к тому же эфиру, который долучен Виланд ok).
Михаэль н Карлсон при обработке фенант^ейА 98,6%-ной HNO@
в растворе хлороформа при —20" получили продукт реакция,
представлявший собой смесь нитрофенантренов с выходом 25%.
Основываясь на экспериментальных данных, полученных
ими при изучении нитрования олефинов и ароматических со-
едияений, Мжхаэль й Карлсон выдвигают теорию <альдолизав
Пии», которая исходит из следующих принципов: кислород
азотной кислоты, связанный двумя валентностями с азотом,
должен обладать значительно большей химической энергией,
чем частично гидрогенизированный кислород группы ОН, и;
следовательно, большей способностью «Притягивать» водород
бензольного кольца, отделяв его тем самым от связанного
с ним углерода. Основные силы, которые определяют
нитрующую способность азотной кислоты, это химические потенциалы
кислорода по отношению к водороду ароматического ядра и
азота по отношению к фенильному радикалу (арилу). С этой
точки зрения нитрование бензола может быть представлено
следующей схемой: ' '.,,,.,,.'
С@Нд + НСЖО* -^ Сд
оод^ммемый 129
Как эта, так и другие ранее изложенные теории
нитрования ароматических соединении исходят из допущения, что
нитрующим агентом является молекула азотной кислоты,
соединяющаяся с ароматическим соединением в первичный
комплекс, в дальнейшем распадающийся на нитросоединение и
воду. При этом предполагается, что присоединение HNOs может
происходить или к двойным связям ароматического ядра (Ви-
яанд), или же, наоборот, ароматическое ядро присоединяется
к молекуле азотной кислоты (Б. В. Тронов, С. С. Наметкин и
А. С. Забродина, Михаэль и Карлсон).
Оригинальный взгляд на механизм процесса нитрования
ароматических углеводородов и на природу нитросоединений
выдвинут А. М. Беркенгеймом [24].
По мнению автора, в случае нитрования, например, бензола
имеют место слабо выраженная диссоциация молекулы бензола
на ионы Н' и С*Н/ и амфотерная диссоциация молекулы
азотной кислоты, с одной стороны, на Н'+ МОз' (1)и, с другой
стороны, на NOg + ОН' (И). Диссоциация азотной кислоты
в первом направлении происходит в значительно большей
степени. Однако ббльшая концентрация ионов Н*,
препятствующая слабо выраженной диссоциации молекулы бензола,
приводит к тому, что диссоциация азотной кислоты в
направлении (I) не может вызвать образование нитросоединений.
Диссоциация азотной кислоты в направлении (II), вследствие
образования Н*О за счет ионов Н" и ОН', усиливает диссоциацию
молекулы бензола и приводит к образованию плохо
ионизированного нитросоединения.
В своих взглядах на природу нитросоединения А. М. Бер-
кенгейм исходит из предположения, что молекула бензола
состоит из попеременно заряженных четырьмя отрицательными
и тремя положительными с одним отрицательным зарядами
атомов углерода, согласно следующей схеме:
////
с
С""'
с
////
9 А. В. Топчиев
130 Af&cdwu&M Nwm^xwiHUA дрожатмцчбсжаа:
При нитровании бензола атом азота азотной кислоты,
имеющий пять положительных зарядов, связан с атомом углерода,
имеющим четыре отрицательных заряда, что приводит к
образованию нормального нитросоединения.
Ниже мы остановимся на теориях нитрования, в основу
которых положено допущение, что в системе ароматическое
соединение — азотная кислота нитрующими агентами
являются несколько форм азотной кислоты в ионизированном и не-
иониэированном состояниях.
Ганч [25, 26] при изучении физических и химических свойств
растворов азотной кислоты в воде, серной кислоте и
органических растворителях пришел к выводу о существовании в
растворах различных форм азотной кислоты, количественные
отношения которых друг к другу меняются в зависимости от
концентрации растворителя.
До появления работ Вернера все кислородные кислоты
рассматривались как гидрокеилыше соединения, а соли кислот
и эфиры — как производные гидроксильного соединения
O.N. ОН; О„М. ОМе; O*N
Эти структурные формулы не могли объяснить основных
различий между электролитами и неэлектролитами и между
солями и сложными эфирами кислот. Вернер, исходя из принципов
своего учения о координационных связях и о строении
комплексных соединений, пришел к другой концепции, согласно
которой лишь сложные эфиры имеют строение производных
гидроксильного соединения; соли же и кислоты, являющиеся
электролитами, представляют собой комплексы с комплексным
анионом и ионогенпо связанным катионом металла или
водорода, находящимися во второй сфере притяжения центрального
атома, в частности азота
N —О —С*Н*. ON CH, ON H
ON
0"
0
сн,
ON
0
0
ншт/юадныл дрождти^^сжшг соебкненый 131
Исследования Шеффера [271, который определял спектры
поглощения гомогенной азотной кислоты и ее растворов в не-
ионизирующих средах, а также соответствующих солей и
эфиров, показали, однако, что спектры поглощения азотной
кислоты и ее эфиров имеют определенное сходство; лишь в водных
растворах при сильном разбавлении спектры поглощения
азотной кислоты аналогичны спектрам ее солей.
Ганч при исследовании спектров поглощения различных
кислот, их солей и сложных эфиров обнаружил, что соли и
сложные эфиры одной и той же кислоты отличаются друг от
друга по своим оптическим свойствам, кислоты же занимают
промежуточное положение и, в зависимости от их природы
и растворителя, являются оптически идентичными либо солям,
либо сложным эфирам. Ганч также установил, что ионизация
не изменяет оптических свойств. Действительно кислоты,
дающие такой же спектр поглощения, что и их соли, идентичны
последним не только в неднссоциированном состоянии, но и
в виде ионов.
Для объяснения полученных результатов Ганч допускает,
что оптические свойства исследованных соединений
находятся в зависимости от их строения. Например, те кислоты,
которые оптически идентичны сложным эфирам, построены
аналогично последним, т. е. содержат гидроксил, соответствую*
щий алкоксилыюй группе сложных эфиров
Эти кислоты, не являющиеся электролитами, представляют
собой, по Ганчу, шеевдокл слоты». К истинным кислотам
относятся кислоты, идентичные солям, т. е. содержащие водород,
соответствующий металлам солей; в отличие от псевдокислот
истинные кислоты обладают свойствами электролитов, так
как атомы водорода, образующие катионы, связаны ионо-
геино (во второй сфере притяжения центрального атома
азота) с несколькими (по крайней мере с двумя) атомами
кислорода.
9*
132 Мммжныал*
выше отношения в общем виде могут быть пред
ставлены следующим образом (НХО*— кислота):
^0 О
I. Сложные эфиры Х\ Псевдокислоты Х%
неэлектролиты гидроксильные формы
электролиты комплексные формы
Соединения, входящие в каждую из этих групп, имеют
сходные структуры.
Для изучения строения кислот Ганч пользовался также
химическим методом, основанным на различном характере
взаимодействия истинных кислот и псевдокислот с диазоуксусным
эфиром.
Как известно, диазоуксусный эфир реагирует с кислотами
с выделением азота по следующему уравнению:
(СООС*Н,) CHN* + НХ -+ (COOCgH.) CH%X + Ng.
Кислородные кислоты, содержащие лишь один атом
кислорода, инертны по отношению к диазоуксусному эфиру или
реагируют с ним лишь очень медленно. Кислоты же, содержащие
более одного атома кислорода (ХО*Н, ХОвН и т. д.), даже в не-
диссоциированном состоянии легко вступают в реакцию с
диазоуксусным эфиром с выделением азота, но лишь в том
случае, если они содержат ионогенно связанный водород, т. е.
ведут себя в растворе, как истинные кислоты. Если же они
представляют собой псевдокислоты, т. е. являются гидроксиль-
яыми соединениями ОХ-ОН, они не реагируют с
диазоуксусным эфиром или реагируют очень слабо, подобно другим ^идро-
ксильным соединениям.
Пользуясь реакцией с диазоуксусным эфиром, Ганч
установил, что азотная кислота в эфирном растворе легко
переходит из формы истинной кислоты (NOg)'H' в псевдокислоту
О
II
О = N — ОН. Эти результаты совпадают с данными Шеф-
133
фера [27], который показал, что в эфирном растворе азотная
кислота оптически идентична ее сложным эфирам.
При изучении водных растворов азотной кислоты
оптическим методом Гаич подтвердил данные Шеффера,
указывающие на присутствие в этих растворах истинной кислотной
формы H(NOs), и показал, что по мере увеличения концентрации
азотной кислоты спектры ее водных растворов все более и
более приближаются к спектрам сложных эфиров, что служит
доказательством постепенного перехода истинной формы
H(NOs) в гидроксильную форму псевдокислоты O*N-OH.
Дальнейшие исследования Ганча привели его к выводу,
что в водных растворах азотная кислота находится не в
свободном состоянии, а в виде соли гидроксония. Вода
представляет собой очень слабое оснований, которое вступает в
реакцию с азотной кислотой с образованием соли. Переход
псевдоформы азотной кислоты в соль гидроксония при разбавлении
азотной кислоты водой может быть представлен следующим
уравнением:
или
азотнокислая соль = (МО,У Щ") (H.OL.
гидроксония * / \ 4 ,а
Сравнительное изучение поглощения ультрафиолетовых
лучей гомогенной (абсолютной) азотной кислотой и ее
сложными эфирами показало, что азотная кислота поглощает заметно
слабее эфиров (это явление не может быть приписано столь
незначительной разнице в строении, которая возникает
вследствие замены гидроксила псевдоазотной кислоты алкокспль-
ной группой). Для объяснения этой оптической аномалии,
которая вызвала резкую критику теории Ганча со стороны Га; ь-
бана [28], Ганч допускает существование в гомогенной
азотной кислоте, наряду с псевдокислотой O*NOH, еще и другой
формы азотной кислоты, которая поглощает
ультрафиолетовые лучи в меньшей степени по сравнению с
псевдокислотой и является электролитом (последнее допущение
объясняет сравнительно высокую электропроводность
гомогенной азотной кислоты, несмотря на то, что псевдокислота не
является электролитом). Ганч считает, что эта форма азот-
134 Масдниам
ной кислоты образуется посредством ассоциации трех
молекул псевдоазотной кислоты с внутримолекулярным
перемещением атомов водорода:
О
Ъ
; — ОН
О N0,
-но
LHO
он
Полученный комплекс, имеющий солеобразный характер,
который Ганч назвал нитронийдинитратом, диссоциирует на
отрицательные нитрат-ионы и положительный ион нитрония *:
(N (ОН),] Z 2N0 ^
Если исходить из позднейших работ Ганча, его теория
строения гомогенной азотной кислоты и ее водных растворов
сводится к следующему.
Абсолютная (гомогенная) азотная кислота представляет
собой раствор обладающего электропроводностью аитрацидий-
динитрата (^Оз)а[(НО)зМ] в нсевдокислоте, не обладающей
электропроводностью. Эти две формы азотной кислоты
находятся между собой в равновесии
%,Х.ОН^(Ж)з):! 1(110), N].
В 98,6%-ной азотной кислоте концентрация нсевдоазотной
кислоты составляет 80%, остальная часть кислоты находится
в форме нитрацидийдинитрата. При разбавлении азотной
кислоты водой происходит перегруппировка нсевдокислоты в соль
гндроксония и истинной кислоты, т. е. наступает новое
равновесие
O.NOH + л Н,0 Z (NO,)' 1H".(ОН,)*].
Нитрацидийдинитрат при этом исчезает. Начиная с
концентрации HNOs 77% в растворе содержатся нсевдокислота и
истинная кислота (в недиссоциированном состоянии в виде
соли гидроксония и в виде ионов). При дальнейшем разбавле-
* ГанчназываетмошдН^^Оа и H3NO3 ионами нитрония. Позднее
эти иопы получили названии аитрацндин-иопов, а нитроннем стали
называть ион NOg . И дальнейшем мы будем придерживать н этой
терминологии (позднейшей).
135
нии содержание псевдокислоты уменьшается, а содержание
истинной кислоты повышается; при концентрации 31,6 %
HNOs, т. е. после добавления 8 молей воды, перегруппировка
псевдокислоты в форму истинной кислоты полностью
заканчивается, причем около 40% истинной кислоты находится
в диссоциированном состоянии, а остальная часть — в виде
недиссоциироваяной соли гидроксояия.
Ганч дает для водных растворов азотной кислоты таблицу,
показывающую взаимоотношения между псевдоформой и формой
истинной кислоты в зависимости от содержания воды (табл. 8).
Таблица 8
псееёожыслоты ы ысл&ынкоД кислоты #
HNO@ е ддеысылюсты от со^е^кмсднш* д
трация
HNO,, %
77,3
.48,3
31,6
Жалей Н,0
та 1 моль
HNO,
1,0
7^5
Псевдо-
н не лота
70
50
2
Недиссоции-
роваыная
истинная
кислота
%
25
32
60
Ионизированная истинная
кислота
(NOg' + Н')
5
18
38
Ганч исследовал также систему азотная кислота — серная
кислота, причем установил, что эта система отличается
значительно более высокой электропроводностью, чем азотная
кислота, а также аномально большим понижением точки замерзания*
Для объяснения этих явлений Ганч допускает существование
в смеси HgSO^-f HNOs электролитов, образование которых
происходит в результате взаимодействия молекулы азотной
кислоты с одной или с двумя молекулами серной кислоты (так как
псевдоазотная кислота имеет два ненасыщенных атома
кислорода, возможно возникновение двух солеобразных комплексов):
O*NOH + HSO4H -+ [NO (ОН)*] [SO*H];
иитрацидийсульфат
(*)
[NO (ОН),] [SO.H] Z H,NO+ + HSO";
O&NOH + 2HSO4H Z (N(OH),][SO4Hk;
нитрацнднйдиоульфат
(2)
136 Месдмы&м мытдкхюнмл
[N (ОН),] [SO&H], ^ Н^)О++ + 2HSO"
Подобным же образом, как показал Ганч, происходит и
взаимодействие азотной кислоты с хлорной кислотой с образованием
солей (нитрацидийперхлоратов)
OgNOH +HCIO4-+ [NO(OH)a] CIO,;
+ 2НС1О. -^ [N (ОН),] (СЮ*),.
Эти соединения, являющиеся очень стойкими, выделены Ган-
чем при смешении азотной кислоты с безводной хлорной
кислотой в виде бесцветных твердых продуктов. Реакция
проводилась при сильном охлаждении и перемешивании. После
перекристаллизации моноперхлората (из азотной кислоты)
и диперхлората (из хлорной кислоты) Ганч получил эти
продукты в чистом виде в форме игольчатых кристаллов.
Используя данные Ганча и Шеффера о строении азотной
кислоты Фармер [29] приписывает этерифицирующую и
нитрующую функцию азотной кислоты ее псевдоформе. Азотная
кислота легко переходит в псевдоформу. Это свойство резко
отличает ее от других сильных кислот, например от серной
кислоты, так как, согласно Ганчу, способность перехода
в псевдоформу у сильных кислот, в противоположность
слабым кислотам, выражена менее заметно. Из всех состояний
азотной кислоты нитрующая функция присуща только
псевдоформе.
Максимальное значение константы скорости нитрования
при определенном содержании воды в серной кислоте Фармер
объясняет следующим образом: при увеличении относительного
содержания серной кислоты в нитрующей смеси (H2SO4+
+ HNOs) содержание псевдоформы вначале возрастает, но
начинает уменьшаться при значительном избытке серной
кислоты вследствие образования за счет этой псевдоформы
некоторого количества нитрацидийсульфата. Поэтому нитрующая
активность смеси по мере возрастания содержания в ней
серной кислоты вначале увеличивается, но, достигнув известного
максимума, начинает уменьшаться.
137
Кинетика реакции нитрования изучалась рядом
исследователей. Мартинсен считает, что реакция нитрования
ароматических соединений эквимолекулярным количеством азотной
кислоты в избытке серной кислоты является бимолекулярной
и что скорость ее прямо пропорциональна концентраций как
азотной кислоты, так и нитруемого соединения [30].
Лауэр и Одда [31] при изучении кинетики реакции
нитрования антрахинона азотной кислотой в водных растворах
серной кислоты в пределах концентраций последней от 87 до
95,6% нашли, что максимальная скорость реакции
соответствует концентрации 89%. Это связано с наличием в
нитрующей смеси различных форм азотной кислоты в зависимости
от концентрации серной кислоты. Как мы увидим дальше,
концентрация серной кислоты и воды не является единственным
решающим фактором для скорости нитрования.
Исходя из полученных результатов, Лауэр и Одда допу-
скаю^г, что в смесях азотной кислоты с серной в присутствии
воды нитрующим агентом является форма азотной кислоты
иная, чем в смесях азотной кислоты с безводной серной
кислотой.
Анализируя полученные результаты, Лауэр и Одда
приходят к следующим выводам:
1) при концентрациях серной кислоты ниже 89% азотная
кислота состоит из двух модификаций: истинной (Н—NO@)
и псевдоазотной (НО — N0%) кислот, причем нитрующим
агентом является только псевдоазотная кислота, содержание
которой уменьшается с разбавлением водой;
2) при концентрациях серной кислоты от 89 до 98%
азотная кислота состоит в основном из псевдокислоты наряду с
некоторым количеством нитрацидийсульфата [N0(0H)2](S0*H),
содержание которого повышается с увеличением концентрации
3) при повышении концентрации серной кислоты от 98
до 100% содержание нитрацидийсульфата в смеси сильно
возрастает;
4) в смеси азотной и дымящей серной кислот нитрующим
агентом является исключительно нитрацидийсульфат.
138 Л/асямиаж мктроддмшк
Тот факт, что реакция нитрования в безводной серной
кислоте протекает с меньшей скоростью, чем в водных растворах
этой кислоты, авторы объясняют тем, что азот в ионе нитра-
цидия, являющемся в данном случае нитрующим агентом,
более «защищен», чем в псевдоазотной кислоте, а именно: тремя
гидроксильными группами.
В водных растворах с концентрацией серной кислоты ниже
89% понижение скорости нитрования по мере дальнейшего
разбавления кислоты водой объясняется уменьшением
содержания псевдоазотной кислоты, являющейся нитрующим
агентом. Понижение скорости нитрования при переходе к более
высоким концентрациям серной кислоты объясняется
уменьшением содержания псевдоазотной кислоты вследствие
частичной замены последней нитрацидийсульфатом.
Таким образом, интенсивность нитрования в водных
растворах серной кислоты определяется содержанием псездо-
азотной кислоты, которое максимально при концентрации,
равной 89%.
Лауэр и Одда изучили также реакцию нитрования
нитробензола азотной кислотой в 90 и 100%-ной серной кислоте.
Опыты показали, что основным продуктом реакции является
м-динитробензол; при увеличении концентрации серной
кислоты выход о-дшштробензола повышается, а выход м- и
п-изомеров понижается.
Механизм реакции нитрования ароматических соединений
азотной кислотой в водных растворах серной кислоты, по
мнению авторов, состоит в том, что молекула псевдоазотной
кислоты, предварительно распадающаяся на N0% и ОН,
присоединяется к двойной связи ароматического ядра (причем группа
NOg присоединяется к анионоидному атому углерода). Нитро-
производное образуется во второй стадии нитрования
посредством отщепления воды от комплекса HNOs с ароматическим
соединением.
При нитровании в безводных растворах серной кислоты
механизм реакции состоит в присоединении нитрацидийсуль-
фата по двойной связи с последующим отщеплением воды и
серной кислоты:
нытмроадмыд
О
(HSO4J
О N0%
4- Н,0 4-
Оригинальную точку зрения на механизм реакции
нитрования ароматических соединений выдвигает в своих работах
французский исследователь Ланц [32]. Он рассматривает нитрование
как процесс, связанный с кислотно-основным равновесием
в системе, исходя при этом из представлений Бренстедта.
Бренстедт [33] определяет «кислоту» как вещество,
способное дать протоны другому веществу, которое он называет
«основанием». Обратимая реакция, которая при этом
возникает, дает начало новому «основанию» и новой «кислоте».
Кислота (1) + основание (1) ±^ основание (2) + кислота (2).
По Бренстедту, свойства «кислоты» и «основания»
проявляются только в том случае, если кислота и основание
находятся вместе. Некоторые вещества в присутствии некоторых
кислот имеют свойства оснований, а в присутствии некоторых
оснований обнаруживают свойства кислот. Например,
органические карбоновые кислоты, фосфорная и азотная, которые
являются кислотами по отношению к обычным основаниям,
становятся уже «основаниями» в присутствии более
энергичных кислот — серной, пиросерной и хлорной.
При действии серной кислоты на азотную возникает
равновесие, которое, принимая концепцию Ганча, можно
изобразить следующим образом:
+ HNO, % 2HS0"
Иногда вещества могут дать равновесие кислота —
основание без участия других соединений, т. е. в гомогенном
140 М&сдны&м
состоянии или в индифферентном растворителе: вещество
одновременно может быть и акцептором и донором протонов.
Хорошо известным примером таких веществ является вода,
которая может дать равновесную систему с участием двух ее
молекул
Такими же свойствами обладает и аммиак
Установленные Ганчеи равновесия в гомогенной серной
кислоте, определяющие ее химические свойства, можно также
рассматривать с точки зрения Бренстедта— Ланца,
предполагая, что они возникли в результате взаимодействия двух
молекул («кислоты» и «основания»); при этом образуются
отрицательный ион HSO&" и положительный ион H*
+ H,SO, z HBO" + H.SO+
Взаимодействие молекул может происходить и по другому
уравнению
H2SO4 + 2H2SO4 Z 2HSQ-' 4- H^SO^+
2HSO4H
Влияние добавления воды на реакции, происходящие в
среде серной кислоты, Ланц объясняет тем, что вода образует
гидратированные ионы, например ион (SOiHs"H*O)* или
S(OH)s\ Посредством потери протона этот ион превращается
в кислоту HgSO^-HgO, существование которой доказывается
кривыми температур плавления для системы HaSO*—HgO.
Образование гидратировашшх ионов вызывает уменьшение
количества ионов H,SO/".
Переходя к реакции нитрования азотной кислотой в среде
серной кислоты, Ланц предполагает следующий механизм
этой реакции в зависимости от изменений концентраций воды
и серной кислоты.
В моногидрате азотная кислота находится полностью в фор-
141
ые соли нитрацидия, что может быть изображено уравнением,
которое мы уже приводили выше:
2H,SO4 + HNO, % 2 (HSO4)" + (OH)gN++. (1)
При добавлении воды образуется гидратированная серная
кислота HgSO&mHgO (вероятно, л = 1); последняя, являясь
основанием по отношению к серной кислоте, приводит к воз-
рикновеншо нового равновесия основание — кислота, которое
может быть выражено уравнением
HBO" + пН^ОдНзО+. (2)
В результате последней реакции происходит образование
нового количества ионов HSO/\ что вызывает сдвиг
равновесия в уравнении (1) слева направо и, соответственно,
увеличение количества ионов (OH)@N** и уменьшение количества
свободной (недиссоциированной) кислоты. При введении
нового количества воды может наступить момент, когда среда
будет содержать сильно гидратированную серную кислоту или
даже свободную воду, являющиеся веществами, относительно
сильно основными. Последние вызывают новое равновесие,
в котором свободная (недиссоциированная) азотная кислота
действует, как кислота, образуя ионы NOs~. Например, в
присутствии свободной воды имеет место следующий процесс:
+ %О Z NO" + НзО+, (3)
который приводит к уменьшению количества неионизирован-
ной свободной кислоты.
Как показывают уравнения (1)—(3), недиссоциированная
кислота не может существовать в среде как слишком «кислой»,
т. е. содержащей мало воды [уравнение (1)], так и слишком
«щелочной», т. е. с содержанием большого количества воды
[уравнение (3)].
Исследуя реакции нитрования нафталинсульфоновых
кислот нитрующей смесью при различных концентрациях серной
кислоты, Ланц установил, что при концентрации HaSO* 90%
и достаточном избытке НМОз у нафталиндисульфоновых
кислот вступают в ядро две нитрогруппы, у нафталинтрисульфо-
142 Л/еа%т%&м нытроеднил д^хымлгц^сжыа; соединений
новых — только одна нитрогруппа, а нафталинтетрасульфо-
новые кислоты в этих условиях не нитруются. Скорость
реакции нитрования при вступлении последней нитрогруппы
максимальна при концентрации серной кислоты от 85 до 90%
в зависимости от природы нитруемого соединения и условий
проведения нитрования. При повышении содержания HgSO*
скорость реакции очень быстро убывает и при концентрации
100% практически равна нулю (при нормальной температуре).
Анализируя результаты своих опытов и применяя к ним
представления Бреистедта, Ланц приходит к следующим
выводам .
1. Нитруемые соединения не пассивны и участвуют в
равновесии кислота — основание посредством присоединения
протона или положительного иона. Скорость реакции нитрования,
по этой причине, зависит не только от активности нитрующего
агента, но и от строения нитруемого соединения: чем больше
последнее содержит групп, сообщающих ему основной
характер, тем оно более реакционноспособно по отношению к
«кислоте» (в данном случае нитрующему агенту).
2. При нитровании азотной кислотой в водном растворе
серной кислоты нитрующим агентом является недиссоцииро-
ванная молекула HNO@; при нитровании в безводной серной
кислоте нитрующим агентом является ион нитрацидия.
Гезерингтон и Мессон [34] на основании кинетических
данных также пришли к выводу, что в нитрующей смеси
активным нитрующим агентом является недиссоциированная
молекула азотной кислоты, однако, в отличие от Ланца, они
полагают, что азотная кислота вступает в реакцию не с
ионизированным ароматическим соединением, а с комплексным
катионом, образуемым из ароматического соединения и
азотной кислоты. Изучая реакцию нитрования нитробензола
смесью серной и азотной кислот при различных концентрациях
компонентов этой смеси (HgSO*, HNO@ и Н%0), Гезерингтон
и Мессон установили, что реакция прекращается еще до
момента полного израсходования нитробензола и азотной
кислоты в реакционной смеси, если количество находящейся в
последней серной кислоты недостаточно для образования с водой,
143
присутствующей в начале реакции и возникающей в
результате нитрования, гидрата состава Н *SO&' Н gO. Критической
точкой нитрующей смеси является содержание серной кислоты
по отношению к смеси кислот (в молях), равное 0,5. При
содержании HgSO*, меньшем этого количества, азотная кислота
участвует в нитровании только частично; при более высокой
концентрации H%SOa азотная кислота полностью нитрует
нитробензол. Нитрование нитротолуола, которое в общем
протекает со значительно большей скоростью, чем нитрование
нитробензола, характеризуется аналогичной зависимостью
скорости нитрования от концентрации HgSO*.
Полученные Гезерингтоном и Мессоном результаты
привели их к выводу, что первое условие, необходимое для того,
чтобы реакция приобрела заметную скорость,— это сдвиг
равновесия между компонентами нитрующей смеси,
определяемого уравнением (1), вправо
H*SO, + (H,O)+4-NO" Z (НзО)+-НН8ОаГ + HNO,. (1)
Основываясь далее на опубликованных ранее работах
одного из них [35], Гезерингтон и Мессон полагают, что
нитробензол может образовать комплексы с HaSO* и HNO@ [см.
уравнения (2) и (3)]; дальнейший механизм реакции состоит
в том, что катион этих комплексов (CgHgNOgH)^ вступает
во взаимодействие с HNO@, образуя динитробензол
[уравнение (5)]:
H*SO, + C,H,NO, Z (C,H,NO,'H)" (HSO*)'; (2)
HNO, + C,H,NO, ^ (C*H&NO%. H)" (NO,)'; (3)
(C,H,NO,.H)" + H,0 Z C,H,MO, + (H,O)"; (4)
H)" + HNO, - C@Hi (N0^ + (H3O)". (5)
При израсходовании серной кислоты равновесие,
выражаемое уравнением (1), сдвигается влево, что приводит к
понижению концентрации безводной азотной кислоты. Сильно
замедляющее действие воды, обнаруживаемое
экспериментально, объясняется тем, что вода не только гидратирует
кислоты, но и реагирует с органическим катионом комплексов
нитробензола с безводными кислотами (CgHgNO^H)' [урав-
144
нение (4)], удаляя этот нов из сферы реакции и тем самым
препятствуя образованию динитробензола по уравнению (5).
Таким образом, но нредставлению Гезерингтона и Мессона,
нитрующим агентом является недиссоциированная молекула
азотной кислоты, которая не только вступает в реакцию с
образованием комплекса с нитробензолом, но и реагирует с
активным ионом этого комплекса (CgHgNOgH)', давая
соответствующее динитросоединенае. Роль серной кислоты заключается
в том, что она препятствует диссоциации азотной кислоты и
участвует, наравне с азотной кислотой, в возникновении
активного иона (C*H*NO*H)".
М. Усанович с сотрудниками [36], исследуя
электропроводность двойных систем, одним из компонентов которых является
HNOs, нашел, что действие этих смесей определяется амфо-
терной природой азотной кислоты и кислотно-освоввыми
функциями компонентов. Так, например, по отношению q воде
НМОз проявляет свойства сильной кислоты, по отношению
же к H*SO* она ведет себя, как вещество с резко выраженными
основными функциями. Опыты показали, что HNO? образует
двойное соединение с СНзСООН и СС1#СООН, представляя,
невидимому, кислоту по отношению к СНзСООН и основание
по отношению к ССЬСООН; с СН&С1СООН и СНСЦСООН
азотная кислота совсем не реагирует, что указывает на
отсутствие у этих моно- и дихлорзамещенных уксусных
кислот кислотно-основных функций по отношению к НМОэ.
При рассмотрении действия нитрующих смесей на
ароматические углеводороды обнаруживается интересный факт, что
нитрованию этих соединений способствует прибавление к HNOg
таких веществ, как H^SO^ или H^PO*, т. е. веществ, по
отношению к которым HNOg является основанием; наоборот,
прибавление к HNOg веществ, по отношению к которым она являет*
ся кислотой, как, например, воды, нитробензола и других,
препятствует нитрованию ароматических соединений.
Так как HNOg при взаимодействии с кислотами образует
катионы нитрацидия [NO(OH)%]' и Щ0Н)з1", а с
основаниями — нитрат-анион, указанный выше факт свидетельствует
о том, что нитрующим агентом при нитровании ароматических
соединений 145
соединений служат катионы житрацидия, яитрат-аяиоп же
является инертным. Обратные отношения наблюдаются нри
нитровании алифатических соединений, а именно:
разбавление водой или уксусной кислотой, т. е. веществами, для
которых НМОз является кислотой, способствует нитрованию
этих углеводородов. Это указывает, что нитрующим агентом
по отношению к алифатическим соединениям является анион
No;.
Для проверки высказанного им взгляда относительно
действия нитрующих смесей М. Усанович исследовал в трех
сериях опытов нитрование ароматических углеводородов (на
примере толуола) в присутствии растворителей трех родов, из
которых одни — основания, другие — кислоты, третьи
—индифферентны по отношению к HNO*.
В первой серии опытов нитрование толуола производилось
в присутствии уксусной кислоты и нитробензола, причем
варьировались отношения компонентов нитрующей смеси.
Опыты, проводившиеся на водяной бане яри 90—92° в течение
3,5 часа, показали, что яри действии HNOa в присутствии
СНзСООН и C,H*NO* нитруется не только ароматическое
ядро, но и боковая цепь (ср. с данными М. И. Коновалова и
П. П. Шорыгияа, гл. III, § 2). Образование фенилнитро-
метана увеличивается (ж, соответственно, уменьшается выход
нитрояродуктов, замещенных в ядре) с увеличением
концентрации растворителей. В растворе уксусной кислоты
наибольший выход фенилаитрометана получен с нитрующей смесью,
состоящей иэ 20% HNO* и 80% СНзСООН, а в растворе
нитробензола максимальный выход фениляитрометана дает смесь
30% HNO* и 70% CeHsNO*. Одновременно с фенилнитроме-
таном получаются также ди- и полинитросоединения. При
нитровании толуола смесями, содержащими 80—90% СНзСООН
и 5—10% азотной кислоты, образуются почти исключительно
нитропродукты, замещенные группой N0% в ядре. Это
объясняется тем, что в первом случае нитрование концентрированной
азотной кислотой ядра идет со значительно большей скоростью,
чем боковой цепи; во втором случае, при наличии 5—10%
НМОз, относительное количество азотной кислоты недостаточно
10 А. В. Топчиев
146 Mgaawu&w нилгроеднил дрождти^сскиа; соединений
для нитрования боковой цепщ. При действии на толуол
азотной кислоты в присутствии уксусной кислоты или
нитробензола получается, кроме нитропродуктов, также и
бензойная кислота; это свидетельствует о том, что азотная кислота
при этих условиях является также окислителем, особенно при
большом избытке растворителя.
Во второй серии опытов нитрование толуола проводилось
в присутствии серной и трихлоруксусной кислот (при
нагревании на водяной бане до 90—95° в течение 3,5 часа). Эти
опыты показали, что в присутствии указанных выше
растворителей, играющих роль кислот но отношению к HNO@,
происходит главным образом нитрование ядра, причем выходы
нитротолуола и динитротолуола (последний образуется только
при нитровании смесью HNOs и HgSO*) повышаются с
увеличением относительного количества растворителя.
При нитровании смесью HNO@— H*SO* максимальный
выход нитротолуола получается при содержании в смеси от 60
до 40% HNOs. При дальнейшем понижении концентрации
азотной кислоты выход нитротолуола резко падает, и при
концентрации 15% и ниже единственным продуктом реакции
является динйтротолуол. При нитровании смесью HNO@—
CClsCOOH динйтротолуол не образуется (выход нитротолуола
цри этом выше, чем при нитровании смесью HNOs и H*SOJ.
Влияние растворителей на процесс нитрования очень
значительно, и сопоставление опытов первой и второй серий,
таким образом, приводит к выводу, что растворители, являющиеся
кислотами по отношению к HNOs, направляют реакцию в
бензольное ядро (нитрование катионами нитрацидия), а
растворители, имеющие основные свойства по отношению к HNOs,
способствуют нитрованию боковой цепи (нитрование
анионом Щ).
Опыты третьей серии по нитрованию толуола в присутствии
индифферентных растворителей — монохлоруксусной
кислоты и этилнитрата — показали, что при этих условиях
нитруется не только ядро, но и боковая цепь. При увеличении
относительного количества растворителя нитрующая
способность азотной кислоты падает и притом в большей степени.
147
чем это соответствует уменьшеяяю концентрации HNO@ в
растворе. Это указывает на то, что нитрование идет за счет ионов
N0^, IIgNO+и H3NO++, концентрация которых с разведением
быстро убывает, а не за счет недиссоциированной молекулы
азотной кислоты (в последнем случае абсолютные выходы ни-
тропродуктов должны были быть прямо пропорциональны
концентрации азотной кислоты).
М. Усанович объясняет механизм реакции нитрования
ароматических соединений следующим образом. Ароматические
соединения, вследствие координационной ненасыщенности
атомов углерода, могут присоединять HNOg, причем
координационная связь осуществляется за счет пары электронов
координационно ненасыщенных атомов углерода. Ион NOg
не будет присоединяться к бензолу вследствие своего
отрицательного заряда. Молекулы азотной кислоты и катионы
нитрацидия могут присоединяться к ароматическому ядру,
причем с увеличением положительного заряда при переходе
от HNOg к N(OH)++эта способность должна возрастать.
Присоединение HNO*, NO(OH)^ и N(OH)j-+ к одному из атомов
углерода бензольного кольца должно увеличить подвижность
водорода, стоящего при этом атоме углерода. Этот водород
отщепляется вместе с одним из гидроксилов, входящих в
состав иона нитрацидия, и образует молекулу воды, например:
СН СН
СН СН
Второй продукт этой реакции [уравнение (1)] — комплексный
катион С(,НэМ(ОН)^+ при разбавлении реакционной смеси водой
дает нитробензол [уравнение (2)]:
C,H,N (OH)g+ -f- 2H,() -+ C,H*NO, -|- 2 (Н&Ор. (2)
При нитровании же алифатических углеводородов, не
имеющих координационно ненасыщенных атомов углерода, ион
N0*" может присоединиться к RCHg, no при этом он должен.
10*
148
предварительно вытеснить водород и занять его место
RCH, + NO" Z RCH*NO*(OH)-.
Ввиду большой затраты энергии на эту перегруппировку
образовавшееся соединение неустойчиво и распадается в обратном
направлении. Но в кислой среде реакция может стать
необратимой, так как образующиеся комплексные анионы могут
отщеплять гидроксил, который связывается ионом гидроксония
RCH*NO, (ОН)" + Н,0+ -+ RCHJfO, + 2Н*0.
Предлагаемая М. Усановичем трактовка реакции нитрования
разъясняет известный факт, что водные растворы нитратов,
несмотря на содержащиеся в них ионы N0@", не способны
вступать с углеводородами в реакцию Коновалова.
В связи с представлениями о роли нитрат-иона в реакции
нитрования следует отметить также работу Гальбана и Эйзен-
бранда [28], исследовавших спектры поглощения нитратов и
растворов азотной кислоты различных концентраций в воде и
других растворителях (серной, хлорной, фосфорной, уксусной
кислотах).
По данным этих авторов, спектры сильно разбавленных
водных растворов HNO@ обнаруживают большое сходство со
спектрами нитратов, т. е. светопоглощение разбавленных
растворов обусловливается присутствием ионов NOs .
При дальнейшем повышении концентрации HNO@ спектры
сохраняют свое сходство со спектрами нитратов до
концентрации 10 н. (60% HNO@); при зтой концентрации происходит
существенное изменение характера спектра, что указывает
на переход нитратной (ионной) формы азотной кислоты в
другую форму. Изучение парциального давления паров
азотной кислоты над водными растворами последней
показывает, что при концентрациях около 10 н. содержание
недиссоциированных молекул азотной кислоты еще очень
незначительно. Гальбан и Эйзенбранд поэтому предполагают,
что изменение спектра при концентрации Юн. обусловливается
образованием ассоциированных или же комплексных ионов.
149
Если принять первое допущение, которое более вероятно, то
в водных растворах HNO@ средней концентрации должно
существовать равновесие, которое выражается уравнением
Ы,О" + N0^ ;: Н,О'М(% % HNO, + Н*0
ассоциированная
ионная пара
Изучение спектров растворов HNOs в эфире и уксусной
кислоте привело авторов к выводу, что в зтих растворах азотная
кислота состоит главным образом (на 60%) из оптически
однородных ассоциированных ионов (HgO^NOs") или комплексных
ионов (HgNOs ); остальная часть азотной кислоты находится
в недиссоциированной форме («псевдокислота»).
При исследовании спектров растворов HNOa в серной
кислоте Гальбан и Эйзенбраыд обнаружили, что если исходить из
водных растворов серной кислоты и постепенно повышать
концентрацию последней, то наблюдаются такие же
закономерности, как и в водных растворах азотной кислоты. При
приближении к более высоким концентрациям серной кисло гы
спектры становятся аналогичными спектрам эфиров. В момент
перехода к безводной серной кислоте (если, например,
повышать концентрацию Н *SO4 постепенным добавлением SO 3)
происходит внезапное падение поглощения. В этом
случае спектр, по мнению Гальбана и Эйзенбранда,
соответствует не нитрацидийсульфату, а смешанному ангидриду
серной и азотной кислот. Следует отметить, что В. В. Марков-
нмков [37] значительно ранее высказывал соображения о роли
в процессе нитрования смешанного ангидрида серной и азотной
кислот (раствор нитрооерной кислоты в избытке серной
кислоты), образующегося при нитровании в нитрующей смеси:
HgSO* + ШО, -> HOSO,ONO, + Н*0.
Вследствие высокой реакционной способности остатка азотной
кислоты, содержащегося в нитросерной кислоте, нитрующая
смесь является сильнодействующим реагентом нитрования.
Образование смешанного ангидрида доказывается выделением
тепла при смешении кислот и действием нитрующей смеси на
150 Afea%ZHW&w
органические соединения. В то время как азотная кислота
реагирует с третичными углеводородами жирного ряда уже яри
0°, нитрующая смесь действует на последние лишь нри
нагревании. С другой стороны, ароматические соединения
значительно легче нитруются смесью кислот, чем азотной кислотой.
В. В. Марковников объясняет это большей но сравнению
с азотной кислотой способностью нитрогрунны смешанного
ангидрида вступать в реакции двойного обмена.
Нитрование яитросерной кислотой но В. В. Марковжикову
протекает согласно схеме
ОН
8ОУ + АН -+ RNO, + HsSO,
ОНО,
Однако образование промежуточного продукта им не было
доказано.
Совершенно своеобразно подошли к вопросу о механизме
витровавия ароматических соединений азотной кислотой Кде*
менц и Шеллер [38], которые рассматривают нитрование как
окислительно-восстановительиьш процесс (этой же точки
зрения придерживается Фергюсон [39]). При этом они исходят
жз сравнения двух следующих процессов:
HNO, + 2H-» HNOg + Н*О;
HNO, + RH -. RNOg f H*O.
При изучении кинетики нитрования фенола азотной
кислотой в водных растворах, а также в среде эфира они аашли, что
реакция каталитически ускоряется не только азотистой
кислотой, как показали Мартинсев [30] и другие исследователи, но
и окислами азота, и притом последними в значительно
большей степени. На основании изучения электропроводности
нитрующих растворов Клеменц и Шеллер пришли к выводу,
что каталитическое действие окислов азота объясняется их
участием в образовании промежуточного соединения (s-кисло-
ты), которое и является нитрующим агентом (при этом реакция
протекает с регенерацией N0*):
2HNO* + NOs % H,N*O, (е-кислота); (1)
2RH + ЩМзОб -, ZRNOg + NO* + 2Н*О. (2)
По мнению авторов, этот же промежуточный продукт,
играющий роль активного агента, образуется и при участии
азотистой кислоты, которая является источником NOg.
Таким образом, с точки зрения Клеменца и Шеллера
нитрование фенола в водном и эфирном растворах представляет
собой ав токаталитический процесс *: наряду с нитрованием
происходят окислительные реакции, которые приводят к
возникновению окислов азота и азотистой кислоты; последние
участвуют в образовании собственно нитрующего агента —
--кислоты. Этот автокаталитический процесс аналогичен реакции
окисления органических соединений перманганатом в
присутствии H*SO#.
Клеменц и Шеллер решили экспериментально проверить
допущение некоторых исследователей о том, что роль
азотистой кислоты заключается в нитрозировании фенола; по этому
представлению, образующийся таким образом нитрозофенол
окисляется далее в нитрофенол, причем одновременно
происходит регенерация HNO&
С,Н*ОН + HNO* -+ СА (N0) ОН + Н,0;
нмтровофеяол
СА (N0) ОН + TWO* - С,Н* (N0,) ОН 4*
Клеменца й Шеллера по окислению нятроэофенола
азотной кислотой показали, однако, что эта реакция носит
автокаталитический характер, а именнЬ: процесс вначале
протекает медленно, затем скоробть реакции быстро возрастает,
после чего опять наступает замедление. С другой стороны,
при нитровании фенола в присутствии нитрозофенола они не
могли обнаружить положительного влияния последнего За
реакцию нитрования. На основании этих результатов Кле-
менц и Шеллер пришли к выводу, что нитрование фенола не
проходит через стадию витрозофенола.
При исследовании нитрования фенола азотной кислотой
в растворе концентрированной серной кислоты эти авторы
нашли, что реакция не ускоряется каталитически при добав-
* См. выше гл. I о нитровавши фенолов.
лении к нитрующей смеси азотистой кислоты и окислов азота;
это указывало, что нитрование в данном случае осуществляется
не е-кислотой, а каким-то другим нитрующим агентом.
Клеменц и Шеллер установили, что константы скорости
реакции одинаковы при нитровании в сорной кислоте как
азотной кислотой, так и азотным ангидридом, если в последнем
случае расчет вести на N^O^/Z. В обоих случаях реакции
являются бимолекулярными. Дальнейшие исследования привели этих
химиков к выводу, что нитрование в концентрированной
серной кислоте осуществляется нитрующим агентом X, который
находится в равновесии с HNOg и N2O5:
HNO,
Основываясь на данных Вибо [40], Клемеац и Шеллер
принимают такой же механизм реакции для нитрования и в среде
уксусной кислоты и в среде уксусяого ангидрида *.
В связи с выдвинутым Клеменцом и Шеллером
механизмом нитрования азотной кислотой в растворе серной кислоты
отметим более ранние работы А. В. Сапожникова [41], который
объясняет нитрующее действие смеси HNO3+H2SO4 при
содержании HgSO* от 70 до 90% присутствием в реакционной
среде азотного ангидрида, образующегося в результате
дегидратации азотной кислоты.
При исследовании упругости паров смесей серной и азотной
кислот с различными концентрациями компонентов (HgSO*,
HNOs и HgO) А, В. Сапожников нашел, что упругость паров
азотной кислоты постепенно повышается при увеличении
концентрации H^SOa в нитрующей смеси, достигая своего
максимального значения при содержании H^SO* около 60%. А. В. Са-
яожников объясняет это повышение упругости паров азотной
кислоты увеличением числа частиц свободной НМОз,
образующейся в результате дегидратирующего действия серной кисло-
См. выше, гл. I, § 3.
153
ты на гидраты азотной кислоты
HNO,-mH*O + H2SO4 -+ HNO,(m —я) Н,0 +
При дальнейшем повьппении содержания серной кислоты
происходит понижение упругости паров азотной кислоты,
особенно заметное при концентрации HaSO* около 90%, что
вызывается, по мнению А. В. Сапожникова, более глубоким
дегидратирующим действием серной кислоты на молекулу азотной
кислоты, приводящим уже к превращению последней в
азотный ангидрид. На примере нитрования целлюлозы А. В.
Сапожников установил, что нитрующая смесь, соответствующая
максимальной упругости паров азотной кислоты, обладает
максимальной нитрующей способностью.
По Шааршмидту [42 ], механизм нитрования смесью
HNO3+ Н&ЗОа заключается в дегидратации азотной кислоты
до азотного ангидрида, который затем присоединяется к
бензольному ядру. Образующееся при этом дигидропроизводвое
бензола
\
н
н
ONO,
представляет собой неустойчивое соединение и распадается на
нитробензол и азотную кислоту. Последняя снова подвергается
действию серной кислоты с образованием ангидрида. Шаар-
шмидт дает следующую схему нитрования азотной кислотой
ароматических соединений в растворе серной кислоты:
2HNQ
H,SO*.H,0
С.Н
.Н.
I1NO,
У
C,H*NO,
Допущение, что азотная кислота, а не N,O&, присоединяется
154 Л/баганма* нклфоаднкя &ро.м&лгич@с*ц#
к бензольному ядру, является, по мнеято ШааршАшдта,
вероятным, так как оно не объясняет роли в этом процессе
серной кислоты, которая не участвует, но его мнению, в
образовании комплекса с бензолом. Для доказательства влияния
окислов азота на процесс титрования Шааршмидт в качестве
примера приводит нитрование феиола, которое ускоряется
окислами азота и, наоборот, замедляется азотнОкйслбЙ ртутью.
Тормозящее действие азотнокислой ртути вызывается, по его
мнению, тем, что она препятствует образованию окиблов аЗО-
та. При нитровании же в водных растворах и в эфире
необходимо добавлять извне окислы азота или HNOa, так как в этих
условиях они, в отличие от нитрования в концентрированной
серной кислоте, не могут образоваться и& НМОз ввиду
отсутствия дегидратирующего агента *.'
А. И. Титов [43], на основании многочисленных
исследований нитрования ароматических соединений азотной
кислотой, разработал интересную гипотезу о механизме этого
нитрования. Согласно этой гипотезе, нитрование ароматических
углеводородов протекает через стадию первичного
взаимодействия ароматического ядра с различными кислородными
соединениями азота, находящимися в сфере реакции и
возникающими из них катионами. В обычной азотной кислоте имеются,
по его мнению, следующие частицы, обладающие электро-
фйльйобтью:
ВО —Nt,0 O = N=iO 0=N — O--N = 0
О О
О О
* Позднейшие исследования Бенпета, Бранда и Вильямса [44] же
подтвердили данных Клеменца й Шеллера о существовании в витрующей
смеси N*O* в виде нсдисооциировагшой молекулы (что соответствует также,
как мы видели выше, взглядам А. В. Сапожникова и Шааршмпдта). При
изучении электролиза раствора N#0* в олеуме Беннет и Бранд установили,
что NaOs находится в этом растворе в ионизированном состоянии.
Результаты опытов Клеменца и Шеллера могут быть объяснены тем, что
в среде H:SO* диссоциация азотного ангидрида приводит к образованию
тех же ионов, производящих нитрующее действие, что и диссоциация
азотной кислоты.
О
НО —N [Х = О1 [О = N *- ОН)
Движущей силой реакций нитрозирования ж нитрования,
способствующей образованию промежуточных комплексов,
является, согласно представлению А. И. Титова, энергетический
выигрыш при переходе электронов от ароматического
соединения к атомам азота яитрозирующих и нитрующих агевтов
через соответствующие атомы углерода ядра.
Необходимое для образования промежуточного комплекса
различие в сродстве к электрону зависит от пространственной
доступности я координационной ненасыщенности ижтфужмцжж
я нитрозирующих агентов»
Так как по опытным данным координационное чжсло азота
в его кислороджых соединениях не превышает 3, атом ааота
в молекуле азотной кислоты является координационно
насыщенным, обладающим лишь слабой способностью к
проявлению электрофвльиости, что, по мнению А. И. Титова,
затрудняет образование комплексов азотной кислоты с ароматжчесй*-
ми соединениями.
С другой стороны, атомы азота в указанных выше
соединениях, обладающие координационными числами 1 и 2,#М"ут
действенно проявлять свой электрофильный характер и вступать
во взаимодействие с соответствующими нуклеофильными
атомами углерода ароматических ядер. На этом основ&зжи
особенно высокой активностью должны обладать нитрозил-катион
|N = О] и нитрозилнитратная форма димера двуокиси азота
.0
Высокой реакционной способностью должен также обладать
мономер двуокиси аэота О = N -» О (при образовании
первичных комплексов этого мономера с ароматическими
соединениями получается дополнительный ваюгрыш энергии ва счет
обменного взаимодействия электрода . N0& с
электронами ^-связей ядра).
15G
На основании указанного выше теоретического анализа
А. И. Титов пришел к выводу, что при нитровании азотной
кислотой реакция протекает через стадию взаимодействия
ароматического соединения с нитрозилнитратной или мономерной
формой двуокиси азота или с возникающими из них катионами.
Азотная кислота служит лишь источником двуокиси азота,
а также регенерирует последнюю из низших окислов азота,
образующихся во время реакции.
Такое течение реакции нитрования через образование
промежуточных комплексов с двуокисью азота, по мнению
А. И. Титова, наблюдается лишь до известной концентрации
азотной кислоты. При дальнейшем повышении концентрации
азотной кислоты процесс нитрования происходит при участии
других агентов, возникающих из азотной кислоты и
обладающих большей активностью, чем N0%. По представлению автора,
к таким энергично нитрующим агентам принадлежит «сухой»
нитроний-катион [О = N — О]", обладающий высокоэлек-
трофильным и координационно ненасыщенным атомом азота,
а также соединения, содержащие в своих молекулах группу
NOg, которая легко отщепляется (например, 0*N—OSOaH,
0*N—0N0, ит. д.).
Наиболее энергично нитрующие агенты (N0%, ON—ONO2»
[О = NT) будут находиться в равновесии
О
; O^O = N —О ,q[ Г^
A О CD
В
Повышение температуры и разбавление во всех случаях будут
сдвигать равновесие в пользу мономера А.
Увеличение полярности среды будет также способствовать
диссоциации димера В, переходу его в полярную нитрозилни-
тратную форму С, возникновению нитрозил-катиона D.
Учитывая, что азотная кислота значительно менее реакцион-
неспособна, чем указанные выше кислородные соединения
азота, А. И. Титов предполагает, что непосредственное взаимо-
драма лшчбскыа
157
действие HNOs с ароматическими соединениями может иметь
место лишь при высоких температурах.
Пользуясь обычными представлениями о механизме
реакций замещения в ароматических соединениях, он дает
следующую схему процесса:
Ar
ArN = О + Н+
О
(
Н -О
"N —О
II
О
\
С
ArN=O + HNO,
Ar—NO, + ШО,
3)
= О
Реакция нитрования соединений типа нафталина идет через
промежуточное взаимодействие с окислами азота при
относительно высокой концентрации нитрующих агентов даже при
применении сально разведенной азотной кислоты.
Реакция протекает по уравнению
Н N0*
У\/\ О
О
О
О
Н N0,
N0
HNO.
н т. д.
или, если пользоваться нитрозилнитратнои формулой строения
N,0,:
158
= N —О
и
(Г
н
\
/\/
\/\
N0
/
<
/
ONO.
-HN0,
N0
хо,
\±0# + 2IIN0, -ч. 2N%0* + H%0.
Экспериментальная проверка теоретических предпосылок
производилась А. И. Титовым посредством сравнения
действия азотной кислоты, содержащей известное количество
окислов азота, с действием кислоты, в которой при помощи
различных добавок предварительно производилось разрушение
окислов азота. В качестве таких добавок им обычно применялись
соли аммония и азотнокислая мочевина, В случае
ароматических соединений очень большой реакционной способности,
как, например, метилнафталинов и антрацена, для более
полною удаления окислов азота применялись соли гидразина.
Опыты с ос- и ^-метилнафталинами показали, что при
добавлении азотной кислоты (уд. в. 1,357) реакция наступала очень
быстро, в то время как в присутствии гидразинсульфата даже
спустя 4 часа не было заметно никаких признаков реакции.
Таким образом, было установлено, что нитрование азотной
кислотой (уд. в. 1,4) и ниже идет только в присутствии азотистой
кислоты.
Опыты с фенолом, крезолом, анизолом, метиловым эфиром
м-крезола, нафтолами и их эфирами, резорцином, диметил-
атшлшюм, фенантреном, антраценом и их гомологами
производились с азотной кислотой (уд. в. 1,357) без добавки и с добавкой
гидразинсульфата. Эксперименталъноустановлено, что во втором
случае реакция не наступала даже после длительного времени.
А. И. Титовым и Н. Г. Лаптевым [45] изучалось окисли-
тельное нитрование ароматических нитрозосоединений и арил-
гидроксиламинов (ем. гл. I; стр. 87—92).
А. И. Титов [46] установил ряд закономерностей процесса
нитрования ароматических соединений азотной кислотой и
нитрующей смесью, которые заключаются в следующем.
Бензол и его гомологи медленно реагируют с NOg и со слабой
азотной кислотой, но легко нитруются крепкой азотной
кислотой или нитрующей смесью, причем присутствие в нитрующей
смеси N0* не играет существенной роли.
При проведении реакции с азотной кислотой нитрование
ароматических соединений протекает не в органическом слое,
а в нитрующей смеси. Значительно ускоряет реакцию
нитрования ароматических соединений добавление к азотной
кислоте сильных протонных и апротонных кислот (А1С1з, TiCl*,
BFg, H*SO*). Положительная роль активаторов процесса
нитрования — сильных протонных и апротонных кислот —
заключается в том, что они способствуют усилению координационной
неиасыщенноетт и электрофильности атома азота нитрующих
средств. Эти соединения легко отщепляют нитроний-катион при
химическом взаимодействии;
н
NO*.. .OSOgH NO,.. .о/
N0*.. .0([ N0%.. .0X0.. .А1С1* 0N0.. .A1CI,
AIC1,
По наблюдениям А. И. Титова, скорость нитрования в парах
сильно уменьшается по сравнению с реакцией в жидкой фазе.
Нитрование ароматических соединений азотной кислотой
умеренной концентрации ускоряется в присутствии солей
ртути.
А. И. Титов рассматривает реакцию нитрования
ароматических соединений как ионно-комплексную реакцию.
Для реакции нитрования ароматических соединений
нитрующей смесью им предложена следующая схема:
НОЖ), + 2Н,9О. 2> (О = N = 0)+ + Н,0+ + 2HS0
160
нилгроедншс
NO:+HSOr<±O,N —
АгН + (0 = N
Н
± Аг
HSO
X ^0
ArNOg
или
Ar—
,N —0SO.H
4-8
N0
N0
— О
Аг
X
-8 N=0-0
II
Н
H N0,
ON0
Н
NO* H
N0,
f N0%
H N0,
0N0
Н. Н. Вороящов [47 ] рассматривает нитрование
азотной кислотой в среде серной кислоты как первично
аддитивный процесс, однако, в отличие от большинства авторов, он
предполагает, что, наряду с азотной кислотой, и серная кислота
также обладает способностью к присоединению. По мнению
Н. Н. Ворожцова, в первой стадии реакции нитрования
образуется продукт присоединения серной кислоты к
ароматическому ядру; этот продукт присоединения в дальнейшем либо
соединений
161
меняет свою сульфогруппу на группу N0% (схема А), либо
присоединяет еще одну молекулу азотной кислоты (схема Б);
полученный в последнем случае двойной комплекс в
дальнейшем стабилизируется с потерей воды и серной кислоты:
н
он
+ I
зол
\
HN0
У\/
ОН
+ I
н
н
ОН -fHNO,
н
SO.H
,н
\
ОН _н,0
н
N0,
(Б)
В качестве примера, показывающего, что сульфогруппа
может уступить место яитрогруппе, особенно при наличии
активирующей ядро группы ОН, Н. Н. Ворожцов приводит
реакцию образования пикриновой кислоты при титровании сульфо-
и дисульфокислот фенола.
Из работ, посвященных кинетике процесса нитрования
азотной кислотой, заслуживают внимания работы Мартинсена
и более поздние Б. В. Тронова и Г, Я. Бера.
Мартинсен [30] исследовал кинетику титрования азотной
кислотой в среде серной кислоты ароматических соединений,
трудно поддающихся действию HNOg (к ним относятся, в
частности, нитросоединения). На примере нитробензола
Мартинсен показал, что скорость титрования в концентрированной
серной кислоте зависит от концентрации последней. Достигая
максимума при концентрации Н #80 4 около 90 %, скорость
11 А. В. Топчиев
162
реакции затем быстро падает при переходе к безводной серной
кислоте (скорость реакции в 95%-ной H^SO* в три раза больше,
чем в 100%-ной).
Скорость нитрования повышается при замене водорода в
молекуле нитробензола метильной группой и понижается, если
заместителем является карбоксильная группа; при этерифи-
кации карбоновой кислоты замедляющее действие, ранее
вызываемое карбоксильной группой, уменьшается. Нитрогруппа
понижает скорость реакции еще более значительно, чем
карбоксильная.
п-Нитроанилин нитруется смесью HNOg + H^SO^ с
одновременным вступлением двух нитрогрупп, причем образуется пик-
рами д. ос-Нитронафталин нитруется леМе нитробензола.
Нитрование фенола в водном растворе HNOg представляет собой
сложную автокаталитическую реакцию; скорость реакции
повышается с повышением концентрации азотной кислоты и
понижается с повышением концентрации фенола. При
добавлении веществ, содержащих ион N0^" (нитратов), скорость
нитрования повышается.
Азотистая кислота является положительным катализатором
реакции нитрования фенола. Применением колориметрического
метода Мартинсену удалось доказать образование азотистой
кислоты во время нитрования фенола, чем и объясняется, по
его мнению, автокаталитический характер этой реакции.
Нитрование п-крезола — также автокаталитическая
реакция, протекающая с образованием нитрита.
При дальнейших исследованиях реакций нитрования
различных производных бензола в присутствии H^SO* найдено,
что наблюдаемое повышение скорости реакции при переходе от
моногидрата к серной кислоте уд. в. 1,839 зависит от природы
нитруемого вещества.
Мартинсен, исследуя влияние концентрации серной кислоты
на скорость нитрования, при температуре 25° для различных
соединений получил следующие данные (табл. 9).
Указанные в таблице вещества могут быть распределены на
две группы.
Первая группа — это те соединения, для которых скорость
163
Таблица 9
ныт/хмдния от
Наименование вещества
4,6-Д инитро-Л1-ксил ол
п-Хлорнитробснзол .
м-Хлорнитробснзол .
о-Хлорнитробснзол .
Нитробензол
2,4-Динитроанизол . .
2,4-Динитрофснол . .
Бензолсульфокислота
Бензойная кислота .
К,(для
H.SO. .
. 0,3 If ,0)
0,0040
0,18
0,39
7,15
1,50
0,17
0,85
- 26
-100
к,
0
0
0
2
0
0
0
2
-5
(для
,80.)
,0014
,050
,14
,18
,37
,053
,39
Л
2,86
3,60
2,78
3,28
4,05
3,21
2,18
11,30
18,50
реакции увеличивается приблизительно втрое при переходе от
моногидрата (100%-ная H@SO*) к серной кислоте состава
Н*5О*- 0,3HgO (95% -ная Н *SO*). Вторая группа веществ
характеризуется тем, что для них скорость реакции при этих
условиях значительно больше, чем скорость реакции
соединений первой группы. Первая группа — нитропроизводные
и хлорнитропроизводные ароматических углеводородов;
вторая группа — карбоксильные и сульфопроизводные
ароматических углеводородов.
Данные табл. 9 подтверждают положение, что в
определенных пределах концентрации серной кислоты скорость
нитрования резко уменьшается с повышением этой концентрации.
Величина константы скорости реакции нитрования
нитробензола уменьшается при переходе от среды НгбО^О^НдО
к среде Н*5О* 0,4 8Оз пропорционально уменьшению
количества воды.
Различные заместители по характеру и степени их влияния
на скорость нитрования могут быть расположены в
следующий ряд:
> SQ,H > СООН > С1 < СН, < ОСИ, < ОС;]** < ОН.
11*
164
Хлор занимает серединное положение, так как он действует
иногда замедляющим, а иногда ускоряющим образом. Справа
от хлора расположены заместители, повышающие скорость
реакции, слева — уменьшающие ее. При этом оказалось, что
заместители, замедляющие реакцию, ориентируют нитрогруп-
пу в м-положение, заместители же, повышающие скорость
реакции, а также хлор, ориентируют группу N0% в о- или
л-положение.
На основании кинетических данных установлено, что
нитрование азотной кислотой нитробензола и 2,4-динитротолу-
ола в сернокислой среде представляет собой реакцию второго
порядка.
Для проверки высказываемого некоторыми авторами (Шаар-
шмидтом, А. В. Сапожниковым и др.) взгляда, что серная
кислота играет в реакции нитрования нитрующей смесью роль
дегидратирующего агента, Мартинсен проводил ряд опытов в
присутствии фосфорного ангидрида. Опыты показали, что
последний не влияет на скорость реакции, из чего можно заключить,
что дегидратирующее действие серной кислоты не является
фактором, определяющим скорость реакции.
Боннер и другие [48], изучая нитрование нитробензола
и п-хлорнитробензола, нашли, что относительные скорости
нитрования этих соединений мало меняются с изменением
концентрации серной кислоты. Оптимальная концентрация
Ы&ЗОд при нитровании этих соединений равна 90,4%.
Изучением реакционноспособности различных
ароматических соединений при нитровании занимался Огата [49]. Им
установлено, что при нитровании 2-фенилантрахинона, 1-ан-
трахинонкарбоновой кислоты и нафтантрахинона нитрогруп-
па направляется в бензольное или нафталиновое ядро.
Некоторые из исследованных соединений по своей
реакционноспособности располагаются в следующий ряд:
Б. В. Тронов и Г. Я. Бер [50] исследовали кинетику
нитрования ароматических соединений в растворе нитробензола при
молярном отношении HNOg : C@HgNOg = 1:2.
драмдтгш^есжкг боб^ынемий 165
Опыт проводился следующим образом: вещество
растворялось при 0° в смеси 2 молей нитробензола и 1 моля азотной
кислоты, после чего раствор оставлялся на некоторое время
при 16—18°. Через определенные промежутки времени
отбирались пробы жидкости и титрованием устанавливалось
количество непрореагировавшей азотной кислоты.
Относительная скорость нитрования различных
соединений вычислялась по промежутку времени, в течение которого
затрачивается одинаковый процент HNOs, причем скорость
реакции измерялась трижды, а именно после вступления
в реакцию 5, 15 и 25% азотной кислоты. Опыты Б. В. Тронова
и Г. Я. Бера показали, что заместители влияют на скорость
реакции ускоряющим или замедляющим образом, причем на
разных стадиях нитрования ускоряющее (или замедляющее)
действие заместителя неодинаково.
При расположении заместителей в ряд по их влиянию на
реакцию нитрования Б. В. Тронов и Г. Я. Бер получили три еле-
дующих ряда, соответствующих моментам вступления в
реакцию 5, 15 и 25% азотной кислоты (справа от водорода,
соответствующего незамещенному бензолу, находятся замедляющие»
слева—ускоряющие заместители):
1) для момента вступления в реакцию 5% HNOg
СН,С1 >Вг> СА > Н <
2) для момента вступления в реакцию 15%
CHgCO > CHaCl > Br > Н < СА < СН, < С^
3) для момента вступления в реакцию 25% HNOs
СН,СО>...>СА>Н<СН,<Вг<С1.
Опыты Б. В. Тронова и Г. Я. Бера показали отсутствие
простой зависимости между скоростью реакции и
ориентирующим влиянием заместителей, как это обычно принималось
ранее. Например, метильная группа, ориентирующая в о- и
п-положение, замедляет нитрование; замена в метильной группе
166
водорода на хлор, который вызывает увеличение выхода
м-проязводного, ускоряет реакцию. Дифенилметан дает почти
исялючщтельжо о- и н-нитросоединения, причем реагирует
медленнее, чем толуол. Галоиды вначале ускоряют нитрование,
но с течением времени их влияние переходит в
противоположное.
Только группы, сильно ориентирующие в м-положение,
вполне подчиняются правилу и очень сильно замедляют
реакцию (NO*,CN, CHgCN, C*H*CO). Ацетильная группа,
ориентирующая менее определенно, оказалась сильным ускорителем.
Вторая и особенно третья жетильная группы влияют
противоположно первой, заметно ускоряя нитрование: о*, м- и п-кси-
лолы нитруются соответственно в 1,6—1,9; 4,5—4,9; 5,7—
10,5 раз быстрее толуола. Значительно скорее нитруются мези-
тилен и псевдокумол.
При нитровании бром- и нитропроизводных бифеиилоксида
(51} введение нитрогруппм в положение 4 протекает легче,
чем а положение 1.
Нитрование фенил тиазолов азотной и серной кислотами
цра 0° дало исключительно н-нитропроизводное, тогда как
нитрование бензилового спирта дает только м-иитропроизвод-
пое [52].
Концентрация применяющейся при нитровании серной
кислоты, по исследованию А. П. Терентьева и Б. М. Кедрова
[53], играет большую роль в смысле направления вступающей
в ядро нитрогруппы. Так, при нитровании ацетанилида
нитрующей смесью они нашли, что если применять не 100%-ную
серную кислоту, а 78,8%-ную, то выход п-нитроацетанилида
падает при одновременном увеличении выхода орто-изомера.
Изучением влияния фтористого бора как катализатора
реакции нитрования фенола и анилина занимались А. В. Топчиев,
В. П. Алания, Г. С. Шнайдер [54].
Исследование проводилось следующим образом: фенол
предварительно насыщался фтористым бором до постоянного веса,
после чего комплекс фенола с BFs загружался в круглодонную
колбу, куда прибавлялась азотная кислота при температуре
от 5 до 20* при постоянном перемешивании. По окончании ре-
167
акция полученная смесь нитрофенол ов обрабатывалась 20%-ным
раствором щелочи с целью разрушения комплекса.
Нитрование фенола в присутствии BFg проводилось азотной
кислотой безводной, 20, 10 и 5%-ной в растворителе и без
растворителя.
В результате проведенных опытов выяснилось, что в
присутствии BFg образуется до 9,4% м-нитрофенола, в то время
как нитрование фенола разбавленной HNOg без BFg приводит
к получению лишь о- и п-изомеров.
Таким образом фтористый бор оказывает влияние как на
место вступления группы N0% в ядро, так и на увеличение
выхода нитропродуктов.
Отступление от правила ориентации в данном случае
авторы объясняют следующим образом: гидроксильная группа
в бензольном кольце увеличивает плотность электронного
облака о- и п-атомов углерода бензольного кольца. При действии
на однозамещенный бензол электрофильным реагентом электро-
фильная группа реагента (в случае нитрования фенола это
будет группа N0*) при реакциях замещения направляется к
тому атому бензольного кольца, у которого имеется повышенная
электронная плотность. Образование комплекса BFg с
фенолом будет сказываться на распределении электронной
плотности в бензольном кольце. Плотность электронного облака
у о- и п-атомов углерода должна быть уменьшена, а
следовательно, и орто- и пара-положения должны быть
пассивированы в связи с образованием трехвалентного положительно
заряженного атома кислорода в комплексе с BFg no схеме
Н—О—ВК
Так как положение, занимаемое вновь вступающим
заместителем, определяется относительными скоростями трех
одновременно конкурирующих между собой реакций, а орто- и пара-
положения пассивированы, то вероятность вступления нового
168
заместителя в м-положение должна увеличиваться, что и было
подтверждено опытными данными как для фенола, так я для
анилина. При нитровании анилина нитрующей смесью в
присутствии BF@ было получено 98% теоретического выхода моно-
нитроавилижов, причем 67,2% приходится на долю
мета-изомера, а 12,8 и 20,0%, соответственно,— на долю орто- ж пара-
изомеров.
Модик [55] изучал вопрос о соотношении изомеров,
получающихся при титровании 2,5-дихлорнитробензола и 2,5-ди-
бромнитробензола в серной кислоте различной концентрации
при различных температурах. Данные этих исследований
(основанных на определения инфракрасных спектров
реакционных смесей) сведены в табл. 10.
Вестгеймер и Караш [56], исследуя кинетику нитрования
ароматических соединений в серной кислоте, нашли, что
основным фактором, влияющим на скорость реакции, является ки-
Таблжца
жолычесл**
и@ол*ерое при
Концевтрация H.
Дымящая
96,2
96,0
89,0
96,2
Дымящая
96,8
90,0
96,8
96,2 (+KHSO*) . . . .
96,2 (Ч-КС1О*)
Дымящая НРЮз+20%
НС1О.
Температура, ?С
100
100
100
100
140
140
100
100
120
100
100
100
мета/орто
3,7
2,4
2,0
1,6
*,?
4,7
2,0
1,9
1,4
орто/пара
0,45
0,77
0,95
1,2
0,91
0,33
0,89
0,98
1,6
мета/орто
18,6
3,6
1,7
2,6
1,6
opio/naga
0,07
0,38
1,4
0,67
1,4
169
слотность среды. Последняя в жж опытах оказалась
оптимальной при концентрации Н*5О*, равной 90%, что согласуется
с более ранвшш данными Мартжвсена.
Определяя константы скорости иитроваиия
нитробензола в серной кислоте умеренных концентрации при 25*, Вест-
геймер и Караш нашли, что эти константы возрастают при
падении концентрации H*SO* от 95,6 до 90%; при дальнейшем
уменьшении концентрации HgSO* наступает резкое падение
константы скорости, которая при нитровании в 80%-ной серной
кислоте уменьшается в 3000 раз по сравнению с 90%-ной
серной кислотой.
Вестгеймер ж Караш испытали далее влияние различных
добавок на скорость нитрования. При этом оказалось, что
вещества, не изменяющие кислотности среды, как, например,
динитробензол и фосфорный ангидрид, не влияют на скорость
реакции.
Добавление к реакционной смеси бисульфата калия,
который по отношению к Я*8О< является основанием (увеличивает
концентрацию ионов HSO^), оказывает различное
влияние в зависимости от кислотности среды, в которой проводится
нитрование. В растворах, кислотность которых выше
оптимальной (яри концентрации HaSO* больше 90%), добавление
бисульфата понижает кислотность, приближая ее к оптимуму,
т. е. благоприятно влияет на скорость реакции; в растворах
с кислотностью среды жиже оптимальной добавление
бисульфата, который еще более уменьшает кислотность среды, влечет
за собой понижение скорости реакции.
Аналогичным образом влияет добавление азотной кислоты,
которая является основанием по отношению к серной кислоте:
если растворителем служи* 90%-ная HgSO*, т. е. кислотность
среды оптимальна, добавление азотной кислоты понижает
скорость реакции; в растворах же, более кислых по сравнению
с оптимумом, добавление HNOs понижает кислотность, т. е.
приближает ее к оптимуму, что приводит к повышению
скорости реакции.
При повышении температуры реакции от 25 до 40°
оптимальная концентрация серной кислоты повышается до 91%;
170
дрол*дтыч*ежыар
наоборот, понижение температуры реакции сдвигает оптимум
в сторону более низких концентраций серной кислоты.
Исследована также реакция нитрования дияитромезитилена
и динитроксилола, причем найдено, что кривые изменения
скорости реакции в зависимости от концентрации серной кислоты
для этих соединений имеют максимумы при тех же
концентрациях, которые соответствуют максимуму для нитробензола
(сравнение опытов, проведенных при одной и той же
температуре).
При решении вопроса о механизме ионизации, определяющем
нитрующее действие азотной кислоты в растворе серной
кислоты, рассматривались два возможных механизма:
HMO, + H,SO,
No:
, +HSO,;
H,O+ + 2H5O7 .
(1)
(2)
Чтобы выяснить, происходит ли ионизация азотной кислоты по
схеме (1) или по схеме (2), была исследована ионизация в
растворе серной кислоты двух индикаторов, из которых один
ионизирует по типу, соответствующему уравнению (1), а
второй — по типу уравнения (2). Половина первого индикатора —
антрахинона — переходила в 89,7%-вой серной кислоте в
окрашенную ионную форму
о
_ ОН _
В качестве индикатора, ионизирующего по уравнению (2),
авторы применили тринитротрифенилкарбинол (тринитротри-
танол), который переходил в окрашенную ионную форму в 90%-
ной Н
2Н,5О,
2I1SO, .
нытроадлол ajwuwamwvc*war co^^wwewww 17 i
Исследование ионизации этих двух индикаторов
производилось при различных концентрациях серной кислоты, причем
степень превращения в окрашенную форму определялась
колориметрически.
Сопоставление кривых, представляющих ионизацию антра-
хинона и тринитротританола в растворах серной кислоты в
пределах концентраций ее от 80 до 90%, с кривой зависимости
константы скорости реакции нитрования нитробензола в растворах
серной кислоты от ее концентрации показало полное
совпадение последней кривой с кривой ионизации трняитротрнтанола.
Авторами сделан вывод*, что ионизация при нитровании
нитробензола происходит по уравнению (2), т. е. нитрующим агентом
в этой реакции является ион N0**.
Согласно взглядам Беннета [57] и ряда других
исследователей, роль серной кислоты заключается в том, что она:
1) необходима для образования нитрующего агента (N0**)
из азотной кислоты, при этом предполагается, что образование
иона ыитрония протекает в две стадии:
_ +Н„0;
2) является наиболее сильным донором протона;
3) служит растворителем для углеводорода.
Кинетику нитрования азотным ангидридом исследовали
Голд, Хьюзидр. [58]. Нитрование азотным ангидридом
изучалось в апротонных растворителях (СС1*), причем авторы
считают, что, с одной стороны, идет нитрование ароматической
молекулы азотным ангидридом, а с другой стороны, благодаря
ионизации NgOs по схеме
2HN0, -+ N,0, -{ HgO; Х.О, - NOg + \(\ ,
нитрующим агентом является нитроний-ион.
Вильяме и Лоуэн [59] исследовали реакцию нитрования
* Данные авторов не дают основания для этого сделанного имя
заключения.
172
соединений, содержащих активное ароматическое ядро; в
частности, oak измерили скорость нитрования иона триметил-п-
толияамяЫ)Ёиж в серной кислоте (при концентрациях 75—82%)
в пределах температур 17,5—45*. Они нашли, что при 25°
скорость нитрования быстро растет, если концентрация H%SO*
больше 80%, и приближается к нулю при концентрации H#SOa,
равной 75%. Нитронрй-ион образуется в этих условиях в коли-
честве, достаточном, чтобы действовать как нитрующий агент,
хотя этого количества недостаточно, чтобы он был открыт до
раман-сджтру. Беннет, Бранд и другие [44] установили, что
при элежролязе раствора азотной кислоты в дымящей серной
кислоте Жзотаая кислота находится в растворе в виде катиона
(это обнаруживается ее движением к катоду). В дымящей
серной кислоте азотная кислота диссоциирует, давая ион NOg,
который является активным нитрующим агентом. Отсутствие
свободно!* азотной кислоты в нитрующей смеси подтверждается
ни&вжм Давлением паров азотной кислоты, умеЛьшающимсж
с умеаыШяиеы содержания воды. При прибавлении воды в смесь
новая активная форма переходит в обычную с уменьшением
жжтружя#й способности. Диссоциацию азотной кислоты
в H%SOa можно представить уравнением
HNO, + 2H%SO# -^ NOg + Н*О^ + 2HSO[.
Реакция образования нитронии-иона [60], повидимому,
протекает через присоединение протона к HNO* по уравнению
НО.
HONO% + № Z N+ = О Z Н%0 Ч- N0,
Растворы азотной кислоты в серной кислоте подвергались
также спектральному анализу [61].
Вильяме и Симкинс [62] исследовали кинетику обратимого
превращения нитрата гуанидина в нитрогуанидин в 71,5—
83%-ной серной кислоте при температуре 14,8—35°. Общее
кинетифское уравнение реакции имеет вид:
мытреаднил ajpouwamzt^c^wa; смАжбмыб 173
^ - Z: [CH+] X [HNO,] - Z
где СН+ и РН+ — ионы гуанндопия и нитрогуанидония.
Первый член уравнения соответствует прямой, а
второй—обратной реакции. Константы j^g и А\ увеличиваются с
повышением кислотности, причем ЛГ* зависит от кислотности в
значительно большей степени, чем ЛТ%. Определяемая зависимость
скорости нитрования приводит к следующему: для 78,3%-ной
H,SO, Я, = 1,6*10?ехр(-1323О/ДГ), для 74,5%-пойН,8О4
X, = 7,7110'ехр(— 13790/ДГ), для 81,1%-яой Н^О^ ЛГ% =
= 8,15-10* ехр (— 8940/ДГ). Изменение концентрации ШЩ
почти не влияет на величину ЛГ*. При повышении кояцецтра-
ции гуанидина константа J^g несколько падает. Это влияние»
как и влияние добавок неорганических солей, также
понижающих величину ЛТ#, невидимому, связано с изменением
кислотности среды,
Бешет, Бранд и другие [63] исследовали кинетику
нитрования 2,4-жвжтротолуола азотной кислотой в среде серной
кислоты различных концентраций, начиная от водных
растворов с содержанием 87,4% серной кислоты и до олеума о
содержанием 29,1% свободного SO*. Температура реадщщ
нитрования варьировалась от 60 до 120*. Нитрование проводилось
в гомогенных средах. При проведении реакции в водных
растворах серной кислоты брали различные количества серной
кислоты и воды, в которых были растворены в небольших
концентрациях динитротолуол и азотная кислота. Из компонентов
нитрующей смеси серная кислота и вода были взяты в
большом избытке, вследствие чего создавалась постоянная среда
для нитрования. Например, в опытах, где нитрование
проводилось с 95,7%-ной серной кислотой, исходный состав системы
был следующим: 0,04 моля двнвтротолуола, 1,8 моля H*SO*,
0,02 моля HNOa (0,7% всей нитрующей смеси), 0,44 моля воды.
Таким образом, количества образовавшихся во время реакции
воды и азотистой кислоты (последняя является продуктом
побочной реакции окисления) были настолько незначительны,
что они не могли существенно влиять на состав системы и ско-
174 Л/е%лн%ак нилуюаянмя
рость реакции. Была исследована зависимость скорости
реакции от следующих факторов: 1) состава среды, в которой
происходило нитрование; 2) концентраций реагентов (азотном
кислоты и динитротолуола); 3) добавки различных количеств
бисульфата калия и нитрозилбисульфата.
При постоянных начальных концентрациях нитруемого
вещества и азотной кислоты (0,4 моля динитротолуола и 0,2 моля
HNOg) и изменении состава среды от 87%-пой HgSO* до олеума
с содержанием 29% 80% авторы нашли максимум скорости
реакции в среде, состоявшей из 92% серной кислоты и 8% воды
(оптимальная среда). При изучении кинетики реакции в
гетерогенной среде установлено, что практически реакция идет
только в кислотном слое, в органическом же слое скорость реакции
мала. .
Опыты показали, что влияние концентрации нитруемого
вещества и азотной кислоты и добавки бисульфата на скорость
реакции различны в зависимости от того, содержала ли среда
серную кислоту в количестве, большем или меньшем
оптимальной концентрации. В средах с содержанием серной кислоты
больше оптимального бимолекулярная константа скорости
реакции возрастала: 1) с повышением начальной концентрации
азотной кислоты (при постоянной начальной концентрации
динитротолуола); 2) с повышением начальной концентрации
динитротолуола (при постоянной начальной концентрации
азотной кислоты); 3) с повышением концентрации бисульфата,
который добавлялся или в виде бисульфата калия, или в виде
нитрозилбисульфата (при постоянных концентрациях азотной
кислоты и динитротолуола). Отношения, указанные в пп. 1 и 2,
наблюдались также в среде олеума.
В средах с содержанием серной кислоты меньше оптимального
бимолекулярная константа скорости убывала: 1) с повышенном
начальной концентрации азотной кислоты; 2) с повышением
начальной концентрации динитротолуола; 3) с повышением
концентрации добавляемого бисульфата.
Для объяснения этих результатов сделаны допущения, что
нитрующим агентом во всех исследованных сернокислотных
средах, от олеума до эквимолекулярных смесей сорной кисло-
ншн/юсдныя духкмд/мичсскыл соеJwffMWft 17
ты и воды, является ион N07. В среде сорная кислота — вода
концентрация иона N07 находится в зависимости от
равновесий (1) л (2)*:
НОЖ), -г 2Н„5О, Z NOg + Hg(f + 21150^ ; (1)
, -г 2Н„5О,
Далее было сделано допущение, что полнота нитрования
достигается лишь в том случае, если, наряду с присоединением
иона NOg к ароматическому ядру, происходит и отдача
протона от ядра специальному акцептору, т. о. среда проявляет
и кислотные, я основные функции. Таким образом скорость
нитрования определяется частотой столкновений между
молекулой динитротолуола, ионом NOJ и молекулой акцептора
протона.
Наиболее активным акцептором в среде сорная кислота —
вода может быть бисульфат-ион. Однако он не единственный
акцептор, так как константа скорости нитрования в 100%-нон
серной кислоте только в три раза меньше ее максимального
значения для 9296-ной серной кислоты. Наиболее вероятным
акцептором протона считается молекула сорной кислоты, которая,
как показал Ганч, способна к автоиоштзации; возможный
механизм участия сорной кислоты в нитровании следующий:
Исходя из указанного выше, нитрование динитротолуола мо
жет быть представлено следующими уравнениями:
,Н, (КО,): + NO; -4- HSO, - CHgCgllg (ХО,)з Ч-11,50,;
Дз (N0^)2 + NOg -4-11^04 -' CHgCH, (NO:), 4" 1^50;
В смесях серной кислоты с водой с молярным отношением
30,= 1 концентрация ионов N0^ падает до нуля и нитрование
прекращается (нитрование имеет место лишь нри молярных отношениях
176
Причина уменьшения скорости реакции с увеличением
кислотности среды выше оптимальной может быть объяснена
с этой точки зрения падением концентрации бисульфат-иона
за счет увеличения концентрации серной кислоты. Поэтому
добавление бисульфата в реакционную смесь при кислотности
среды выше оптимальной дает положительный эффект, так как
оно восстанавливает необходимую для реакции концентрацию
активного акцептора протона (бисульфат-иона).
Падение скорости реакции при понижении кислотности
среды ниже оптимальной, согласно этим представлениям,
вызывается понижением концентрации ионов N0^, образующихся
по уравнению (1) (см. выше). Добавление бисульфат-ионов
должно приводить к падению скорости реакции, так как оно
способствует сдвигу равновесия в уравнении (1) справа налево,
т. е. еще большему понижению концентрации ионов N0^".
Влияяие начальной концентрации азотной кислоты на скорость
реакции определяется концентрацией в системе бисульфат-
иона, так как повышение начальной концентрации HNO@
влечет за собой, как это видно из уравнения (1), повышение
концентрации бисульфат-иона. Поэтому в тех случаях, когда
повышение концентрации бисульфат-иона оказывает
положительное действие на скорость реакции, повышение начальной
концентрации HNOs приводит к такому же результату, а в тех
случаях, где влияние бисульфат-иона оказывается
отрицательным, скорость реакции с повышением начальной концентрации
HNOg понижается.
При действии азотной кислоты на ароматическое соединение
в среде олеума основными акцепторами протона являются
молекула серной кислоты и пиросульфат-ион. Нитрование
в этом случае может быть представлено следующими
уравнениями:
A (NO,), + NOg + H,SO. -+ СН,СА (NO,), + H,SO%
%
CH,C,H, (NO,), + N0% + HS,O^ Z CH,C,H; (NO,), + H,S,O,.
В отсутствие серной кислоты ион N0% может образовываться
согласно ур авнению
3HN0, 3 N0% + Н,0+ +2 N0;.
,
нылцммяныл д/Юыид/т*4А;жиа; см^ынбмыД 177
Другое объяснение максимума скорости реакции при
определенной концентрации серной кислоты дает А. И. Титов [46].
Он указывает, что катионы, образующиеся при соединении
ароматических веществ с протоном, например CeHgNOgH^,
должны вступать в реакцию замещения значительно труднее,
чем соответствующие нейтральные молекулы.
Уравнение скорости реакции было дано Н. Н. Воронцовым
[64]. Суммарная скорость реакции соответствует уравнению
х [NO; ] х [HSO; ] + ^ [ДНТ] х [NO; ] X [H.SO.I,
где [ТНТ] и [ДНТ] — концентрации тринитротолуола ж ди-
нитротолуола, J^ и ЛГ' — константы скорости, величины
которых не зависят от среды (концентрации серной кислоты).
Так как концентрация NOg может быть найдена из выражения
для константы равновесия
HNO, + 2H,S0, "
причем [HN0@] = [N0%4+ [HONOg] (концентрация
азотной кислоты равна сумме концентраций нитроний-катиона и
оставшейся недиссоциированной HONOg), то доля () азотной
кислоты, превращенной в N0^, равна
Откуда (NO,] = Q
и уравнение скорости нитрования может быть написано в виде
[ДНТ]
Уравнение бимолекулярной реакции соответствует скорости
реакции при постоянной концентрации серной кислоты:
(f [ТНТ]
12 л. в. Топчиев
178 Меагднм&м милгроеднил
Следовательно, константа скорости бимолекулярной реакции
равна
При нитровании в серной кислоте и в олеуме акцепторами
протона могут являться HaSO* и ион HS%0; . Поэтому уравнение
скорости реакции нитрования в общем виде (как в разведенной
а, так и в олеуме) может быть изображено так:
= (f Щ8О; ] + Г [Н^О*] + f [HSgO; ]) ЩИТ] [UNO,] Q.
Концентрация Н8%0? вычислялась исходя из допущения, что
константа равновесия реакции
11,50. + Н^шО? ^ ^ ^
равна 1.
Большое влияние на процесс нитрования оказывает
растворитель, о чем мы вкратце упоминали и ранее. Из работ,
заслуживающих внимания в этом отношении, отметим исследование
Холстеда и Ламбертона [65]. Они изучали реакцию нитрования
в различных растворителях без серной кислоты. Ими показано,
что реакция нитрования уретана, метилуретана и мочевины
необратима в среде уксусная кислота — уксусный ангидрид.
При нитровании уретана, метилуретана и мочевины в среде
с отношением (СН*СО)*О : СНзСООН = 24 : 1 при 25° в
течение 1 часа выходы нитросоединений достигают 60—80%. При
нитровании уретана при 25° в течение 20 час. в средах с
отношением (CHgCO)gO : CHgCOOH = 1:1 и 1 : 19 выходы
нитросоединений составляли, соответственно, 80 и 40%. В системе
уксусная кислота — вода с отношением СНзСООН : Н%О =
= 199 : 1 нитрование ве происходит. При растворении
соответствующих нитрамидов в указанных выше растворителях
свободной азотной кислоты не обнаружено. Реакция нитрования
уретана в 60%-пой НС1О* обратима. Положение равновесия
для уретана при 70° достигается через 20 мин. Выход нитро-
уретана 53%.
В приведенных выше работах Беннета и др. [63]
предполагается, что реакция нитрования протекает как тримолеку-
ляряый процесс:
- N0% + АгН + HSO[ -*- ArNO, + H
В таком процессе удаление протона из ароматического ядра;
должно иметь существенное значение для кинетики реакции,
поскольку оно тробует для своего осуществления наличия
основания.
С целью проверки этого предположения Меландер [66]
исследовал нитрование толуолов, содержащих тритий в
различных положениях, и на основе полученных результатов пришел
к заключению о несостоятельности основного предположения
Беинета и соавторов.
При динитровании 2-Т-толуола Меландером был получен
2,6-динитротолуол, причем количества активного и
неактивного производных оказались одинаковыми. Подобный факт
можно объяснить лишь тем, что в общем процессе нитрования
I стадия отщепления протона является очень быстрой и потому
1 не определяет общую скорость реакции. В случае же течения
^реакции как одностадийного тримолекулярного процесса из-за
н наличия изотопного эффекта количества активного и неактив-
; ного дияитротолуолов оказались бы различными,
! Этими опытами Меландер подтвердил, что процесс нитро-
| вания протекает не как тримолекулярная реакция.
'|| Обширное исследование кинетики и механизма нитрования
;| ароматических соединений было проведено Ингольдом и со-
{трудниками.
I, В работах этих авторов [67, 68] было изучено нитрование
| нитрующей смесью (HNOg+ HgSO*), одной азотной кислотой,
а также азотной кислотой в органических растворителях (нит-
рометане и уксусной кислоте). Основные экспериментальные
факты, найденные при этом, сводятся к следующему.
В случае нитрования ароматических соединений нитрующей
смесью реакция протекает по второму порядку
Скорость в H,SO« = ЛГ [НАг] [НГЩ] (1)
12*
180 ЛзГездниал*
Это установлено при нитровании нитрозамещенных бензолов,
бензойной и бензолсульфоновой кислот и антрахияонов.
При нитровании одной азотной кислотой имеет место
реакция первого порядка:
Скорость в HNO, = Ai [HAr] ([HNO»] = const) (2)
(установлено при нитровании нитробензола, п-хлорвнтро-
бензола и 1-нитроантрахинона).
В случае применения в качестве растворителей нитрометана
шш уксусной кислоты нитрование достаточно реакционно-
способных в влектрофильным замещениям ароматических со-
эдинений протекает по нулевому порядку, а мало реакционно-
способных к таким замещениям — по первому порядку:
Скорость в CH3NO3 = #о ([HNO,] = const) (3)
(установлено при нитровании бензола, толуола в этил-
бензола).
Скорость в CHgNO» == #i [HAr] ((HNO,| «* const) (4)
(установлено при нитровании п-дихлорбензола и 1,2,4-
трихлорбензо ла).
При нитровании в органических растворителях
ароматических соединений с промежуточной реакционяоспособ-
ностью реакция протекает по дробному порядку (от 0 до 1):
Скорость в CH&NO*: средняя между скоростями, приведенными в уравве-
вжях (3) и (4) (5)
(установлено для галоидзамещенных бензола).
Далее было показано, что нитрование ароматических
соединений азотной кислотой и особенно в органических
растворителях ускоряется добавками серной кислоты и замедляется
добавками диссоциирующих нитратов металлов. При этом
наблюдается линейная зависимость как между добавками серной
жислоты и скоростью реакции нитрования, так и между
добавками нитрата и обратной величиной скорости реакции.
Следует подчеркнуть, что влияние добавок серной кислоты и
нитратов сказывается только на скорости реакции, но меняя се
181
(первого яри нитровании азотной кислотой и нуле-
#@го иди первого яра нитровании в растворителях CH,NO& или
и Было изучено также влияние воды. Оказалось, что добавка
незначительного количества воды уменьшает скорость
нитрования в растворителях также без изменения самого порядка
реакции. При этом, как и в случае добавок нитрата, обратная
величина скорости изменяется линейно с концентрацией воды*
В общем, по сравнению с эффектом, оказываемым серной
кислотой и нитратом, влияние добавки относительно небольших
количеств воды очень невелико. Необходимо отметить, что
сохранение порядка реакции наблюдается только при небольшие
добавках воды. Прибавление же больших количеств воды ядре-
водит реакцию нулевого порядка (например, нитрование без*
зола азотной кислотой в органическом растворителе) в pew?
цжю первого порядка.
При исследовании того, как влияет изменение состава ере-
дм на скорость нитрования в органических растворителях* было
найдено, что увеличение содержания азотной кислоты резко
увеличивает вонот*нту скорости как нулевого, так и первого
порядка ж что константы скорости нитрования обоих этих поряд?
ков при употреблении в качестве растворителя нитромет#щ#
значительно выше, чем при употреблении уксусной кислотц.
Далее было показано влияние изменения состава среды и на
порядок .реакции. Уменьшение концентрации азотной кислоты
приводит к переходу от кинетики реакции нулевого к первому
порядку. При сохранении постоянной концентрации азотной
кислоты и замене растворителя нвтрометана уксусной
кислотой порядок реакции изменяется от нулевого к первому.
На основании полученных экспериментальных данных Ин-
гольд с сотрудниками пришли к ряду заключений относительно
механизма нитрования ароматических соединений азотной
кислотой как в присутствии органических растворителей, так
и без них.
Первое заключение авторы делают в связи с нулевым
порядком нитрования в органических растворителях аромати*
ческих соединений, достаточно способных к электрофиль-
182
ным замещениям. Такая кинетика нитрования в данщом
случае означает, что скорость реакции зависит от некоторого
предварительного процесса, происходящего без участия
нитруемого соединения и могущего из-за наличия растворителя
протекать достаточно медленно, чтобы явиться той стадией,
которая определит скорость общей реакции. Этот
предварительный процесс не включает в себя и самого растворителя, так как
различные по своей химической природе растворители
оказывают одинаковое действие. Таким образом, этот
предварительный процесс должен происходить только с самой азотной
кислотой. Изменение, которое она при этом претерпевает,
происходит с измеримой скоростью, определяющей общую скорость
нитрования нулевого порядка. Такое изменение не может
поэтому заключаться только в перемещении протона, которое в
кислородных кислотах происходит мгновенно. Авторы приходит,
таким образом, к выводу, что предварительный процесс,
протекающий с известной энергией активации, должен включать
в себя разрыв связи. Исходя далее иэ того, что устаяовлевшае
при нитровании ароматических соединений законы
ориентации и влияния заместителей на реакционную способность явно
подтверждают электрофильный характер этой реакции, авторы
заключают, что разрыв связи в молекуле азотной кислоты
должен иметь гетеролитическую природу. Только в этом случае
возникнет электрофильный нитрующий агент, т. е. атомная
группировка с атомом азота, несущим неполное число
электронов. Наиболее простое осуществление этого можно было бы
видеть в следующей реакция:
O,N
ОН -» O*N+ + H(T. (1)
Однако тот факт, что добавки кислот более сильных, чем
азотная, значительно увеличивают скорость нитрования,
привел авторов к предположению, что гетеролитическому
распаду предшествует захват молекулой азотной кислоты одного
протона с превращением ее в ион Н%МОз\ Последующий
распад этого иона произойдет по уравнению
183
O,N | OHg ^ 0^ + OH, (2)
и приведет к возникновению яитроний-иона
Таким образом, протон, присоединяющийся к молекуле
азотной кислоты, поставляется, очевидно, наиболее сильной
кислотой, имеющейся в системе, и весь механизм первого
этапа реакции нитрования — образование нитрошш-иопа — может
быть изображен уравнениями (3):
а) HNO, + Н, SO*
б) H^OZNO
в) Н%0 + HgSO* Z H3O""
(3)
При отсутствуй кислоты более сильной, чем азотная,
последняя сама является датчиком протонов, при этом
образуется нитрат-ион:
а) HNO, + HNO,
б) H,NO,ZNO%
в) Н,0 + HNO, ^
Такая схема образования ндтроний-иона, предложенная Ин-
гольдом с сотрудниками, дает возможность объяснить
установленную этими авторами кинетику нитрования ароматических
соединений.
Второй порядок нитрования в серной кислоте является
следствием того, что в ее присутствии нитрошш-иои
образуется быстро, т. е. равновесие стадии б) процесса (3) сильно
сдвинуто слева направо.
В случае, если нитрование приводят одной азотной кислотой
(т. е. более сильной кислоты не имеется), то аитроний-ион
хотя тоже образуется с большой скоростью, но уже только в
небольшой равновесной концентрации. Эта концентрация будет
сохраняться в процессе нитрования достоянной, что и
приведет к первому порядку реакции по нитруемому соединению.
По той же самой причине при титровании в органических
растворителях мало реакционноспособных ароматических
184 Л/еаиш«ал* нылцхюдмыд
соединений кинетика реакции будет также первого порядка по
этим соединениям. Действительно, даже если в этих условиях
малая равновесная концентрация нитроний-ионов создается
медленно, все же можно думать, что скорость этого процесса
гораздо больше скорости их вхождения в реакцию с
нитруемыми ароматическими соединениями. В результате
концентрация нитроний-иона будет стационарной и близкой к
равновесной, а это и обозначает кинетику первого порядка по
нитруемому соединению.
Особенное значение имеет возможность нахождения иа
основе предлсЬкенной схемы причины протекания в органических
растворителях нитрования достаточно рсакцноаноспособных
ароматических соединений по кинетике нулевого порядка.
Такая кинетика будет определяться тем фактом, что
относительно медленно образующийся в органическом растворителе
аитроний-ион будет немедленно реагировать с высокореакцвон-
ным ароматическим соединением. Таким образом,скорость
нитрования будет равна скорости образования нитроний-иона.
Эта же последняя постоянна, если азотная кислота
присутствует в постоянном избытке. Нитрование, следовательно,
будет реакцией нулевого порядка.
Дальнейшим серьезным экспериментальным
подтверждением предложенной схемы образования нитроиий-иона является,
по мнению авторов, уменьшение скорости нитрования и нулевого
и первого порядков, происходящее при добавлении нитратов
металлов. Замедление нитрования при добавке нитратов
указывает, что реакция, приводящая к образованию нитроний-жона,
включает в себя образование также и нитрат-иона и что эта
реакция может быть сдвинута в обратную сторону
добавленными извне нитрат-ионами.* Действительно, предложенная
схема образования нитроний-иона вполне удовлетворяет этому
требованию, поскольку обратимая стадия а) процесса (4)
включает в себя возникновение нитрат-иона.
Особенно существенным представляется авторам факт
уменьшения нитратами металлов скорости нитрования
нулевого порядка, без изменения при этом самого порядка реакции.
Нулевой порядок нитрования означает, что весь образующийся
мылумадмш* а/юлмпш^сдеаа; соб^цмениД 185
житроний-иож расходуется жа иитрование ароматического со-
едижежия. Согласовать это с фактом уменьшения скорости
нитрования при добавке нитрата металла можно лишь в
предположении, что процесс образования житроюш-иона,
являющийся стадией, определяющей скорость всей реакции нитрования
нулевого порядка, в свою очередь сам состоит из двух стадий.
При этом та его стадия, на которой происходит образование
иитрат-ионов, должна быть, как мы выше видели, обратимой;
вторая же стадия, являющаяся конечной стадией
образования нитроннй-иожа, должна быть необратимой. Только в этом
случае добавка нитрат-ионов сможет уменьшить скорость
нитрования нулевого порядка, не меняя яри этом самого
порядка.
Таким образом, специально для случаев нитрований
нулевого порядка в схемы (3) и (4) достаточно внести лишь то из*
менение, что стадии б) в них протекают необратимым путам.
Образование нитроний-иона будет происходить теперь по
уравнениям (3*) и (4°);
ж) HNOl + H,SO« Z ;
в) Н,0 + HaSO^ Z HaO+ 4- HSO^
а)
в) Н,0 + HNO* Z HNOgHgO.
Из этих уравнений непосредственно следует
пропорциональность между концентрацией нитроний-иона и равновесной
концентрацией иона H*NOg. Таким образом, предложенная
авторами схема объясняет наблюденный на опыте для нитрования
нулевого порядка линейный прирост скорости при добавках
серной кислоты и линейный прирост обратной величины
скорости при добавках нитратов металлов. Следует отметить, что
нахождение линейного, а не квадратичного эффекта обозначает*
что на стадии а) процесса (3°) молекулой азотной кислоты
захватывается только один протон, а на стадии а) процесса (4*)
возникает только один нитрат-ион. Иначе говоря, в реакции
нитрования возникает ион HgNOg, а не Н
186 Мйсамыал*
Согласно схемам (3*) и (4*), вода появляется только на не-
обратимте стадиях, и уже до одному этому ее действие при
нитровании нулевого порядка не может быть значительным. Для
объяснения наблюденного на опыте незначительного
тормозящего влияния добавок воды авторы формулируют наличие еще
одного процесса распада иона H*NOg в обратимой реакции
+
а) HNO, + Н,О+ Z H,NOg + Н.О; |
б) 1уЮ НО NO j
При этом предполагается, что равновесие стадии а) процесса (5*)
столь сильно сдвинуто слева направо, что относительно
небольшие добавки воды способны только незначительно
сдвинуть это равновесие влево и, следовательно, только очень
не намного уменьшить стационарную концентрацию иона
HgNOg. Следствием этого явится лишь незжачщтелыюе
уменьшение скорости образования нитроний-иояа и отсюда
слабое тормозящее действие добавок води, Линейшый же
эффект добавок воды вполне согласуется с уравнениями (5*).
Переход к нитрованию, протекающему по первому порядку,
связан, по мнению авторов, с тем, что в последнем случае
обратное взаимодействие нитроний-иона с водой [т. е. обратная
реакция стадии б) в процессах (3*) и (4*)] протекает значительно
быстрее, чем взаимодействие этого иона с нитруемым
ароматическим соединением. В этом случае, следовательно, обе стадии
а) и б) процесса образования нитроний-иона должны
рассматриваться как обратимые, и мы приходим к уравнениям (3) и (4).
Нитрование первого порядка определяется тем, что жа всем
его протяжении поддерживается стационарная концентрация
нитроний-иона, которой и пропорциональна константа
скорости первого порядка. Как ясно из уравнений (3) и (4), эта
стационарная концентрация нитроний-иона пропорциональна
добавкам серной кислоты и обратно пропорциональна добавкам
нитратов металлов. Отсюда и следует наблюдаемая на опыте
линейная зависимость скорости нитрования первого порядка
от этих добавок.
Вторым этапом реакции нитрования как одной азотной
кислотой, так и в присутствии органических растворителей явля-
187
по мнению авторов, взаимодействие иоиа нитрония с нит-
ароматическим соединением. Для этого взаимодействия
овж дают следующую схему:
медлевиая Н
j. стадия /
а) N0» + АгН > Аг+^
NOg
Н быстрая
/ стадия
б) А^
N0,
I
Такой механизм предполагает, что очень скоро после
соединения нитроний-иона с ароматическим атомом углерода
протон, соединенный с этим атомом, отщепляется. Таким
образом этим механизмом исключается тримолекулярная реакций.
Подтверждение этому автора видят в том, что, исходя из всей
предложенной ими схемы нитрования, удается вывести
кинетические эффекты, которые были экспериментально найдены при
изменении растворителей. Вывод этот сводится к следующему.
Первый этап реакции нитрования — образование ндтро-
ний-иона, например, ио схеме (4)— состоит из прямых и
обратных процессов.
В результате прямых процессов три нейтральные молекулы
превращаются в одну нейтральную и два иона. Обратные
процессы приводят к противоположному результату.
Следовательно , такой процесс образования нитрожий-иожа должен
значительно ускоряться при переходе к нитрованию в более
полярной среде, так как увеличение сольватирующей
способности среды будет ускорять прямые процессы и замедлять
обратные.
Второй этан реакции нитрования, протекающий по схеме
(6), не включает в себя ни образования, ни исчезновения ион-
кых зарядов. Поэтому его скорость ве будет зависеть от по-
кярности среды.
Теперь можно предсказать, как скажется на скорости и
порядке реакции нитрования изменение растворителя.
Выше было показано, что скорость нитрования нулевого
порядка зависит только от прямых процессов образования пит-
188 Мезоны**
ронии-иона, а последние сильно зависят от полярности средни
Поэтому увеличение содержания азотной кислоты или ваыен*
уксусной кислоты на нитрометан (что увеличивает полярность
среды) должны ускорить нитрование, что и было установлено
на опыте.
Скорость нитрования первого порядка зависит от двух
факторов: стационарной концентрации иитроний-иона и
константы скорости взаимодействия нитроний-иона с нитруемым
соединением. Первый фактор — стационарная концентрация
NOg соответствует равновесной концентрации, в свою
очередь определяемой обратимым превращением трех нейтральных
молекул в два иона и одну молекулу. ОтсЭюда ясно, что
стационарная концентрация нитроний-иона будет расти с
увеличением полярности среды. Второй фактор не чувствителен к
изменению полярности среды, так как взаимодействие N0**
с нитруемым соединением не сопровождается возникновением
тли исчезновением ионных зарядов. В итоге нитрование пер*
вого порядка должно существенно ускоряться при увеличении
полярности среды. Это также было констатировано на опыте.
Что касается порядка реакции, то, как ясно иэ предшествую*
щего, он будет определяться тем, по какому пути — в обрати*
мых ли стадиях процесса образования NOg или при
взаимодействии NOg с нитруемым соединением — будет
расходоваться подавляющая часть нитроний-иона. Расход по первому
пути приведет к кинетике первого порядка, расход по
второму — к кинетике нулевого порядка. Как было показано
выше, скорость процессов, осуществляющихся на первом пути,
мсзает быть уменьшена ростом полярности среды; на скорости
процессов второго пути изменение полярности среды влияния
не окажет. Отсюда результатом увеличения полярности среды
должно быть установленное на опыте уменьшение порядка ре-:
акции от первого к нулевому.
Распространение выдвинутого Ингольдом с сотрудниками
механизма С-нитровавшя реакционноспособных
ароматических соединении в органических растворителях на N- и 0-нит-
ровапае было произведено Блэкколлом и Хьюзом [69].
Основным предположением этих авторов было то, что в случае, еслк
189
N- и Очштрование происходят также при помощи нитроний-
иона, то нитрование нулевого порядка должно протекать с
одинаковой скоростью независимо от природы нитруемого
соединения. Скорость нитрования будет определяться только
скоростью образования нитроний-иона, которая при прочих
равных условиях не будет зависеть от того, происходит ли N-,
С- или О-нитрование.
Экспериментальные результаты Блэкколла и Хьюза по
проверке этого предположения приведены в табл, 11,
Таблица 11
р
(4 М
Нитруемое
соединение
Толуол
Метилпинрамид .
Метиловый спирт
Начальная
концентрация.
г-моль/л
0,099
0,119
0,491
Скорость,
г-моль/л-сен.
10,3x10-*
10,4x10"»
10,2x10-"
Как ясно из данных таблицы, скорости С-, N- и О-нитрований
нулевого порядка действительно оказались одинаковыми, и,
следовательно, предположение авторов об одинаковом
механизме С-, N- и О-нитрований при помощи нитроний-иона
получило некоторое экспериментальное обоснование.
Дальнейшим развитием работ Ингольда с сотрудниками
было изучение нитрования ароматических соединений водной
азотной кислотой с добавкой более сильной кислоты (серной
или хлорной) [70]. В этих условиях нитрованию были
подвергнуты 2-фенилэтилсульфоновая и бензилсульфоновая кислоты.
Авторы считают наиболее вероятным, что в водных растворах
титрованию подвергаются анионы этих сульфоновых кислот,
причем их реакционная способность (к нитрованию) сравнима
с реакционной способностью ароматических соединений,
нитрующихся в органических растворителях по кинетике
нулевого порядка.
190
Было найдено, что нитрование указанных сульфововых
кислот водной азотной кислотой (с примесью хлоршой кислоты)
является реакцией первого порядка по ароматическому
соединению. Кроме того, было установлено очень резкое влияние
изменения состава среды на скорость реакции. Действительно,
уже небольшое изменение в определенных узких пределах
концентрации либо азотной, либо хлорной кислот, либо воды
сразу переводит реакцию от очень малой к очень большой
скорости. Существуют, таким образом, пороги концентраций
компонентов среды, при переходе через которые медленная
реакция становится очень быстрой. Вблизи таких порогов
добавка NaClO* ускоряет, а добавка NaNOg замедляет
нитрование.
Переходя к установлению механизма нитрования водными
растворами азотной кислоты, авторы в первую очередь
подвергают обсуждению возможные изменения при
образовании нитроний-иона [по схеме (4°), относящейся к кинетике
нулевого порядка], при переходе от нитрования в органических
растворителях к нитрованию в водных растворах.
Согласно этой схеме, вода образуется одновременно с
нитроний-ионом. Это означает, что при добавлении
достаточного количества воды эта стадия должна стать
обратимой с сильным сдвигом равновесия влево. В этом случае,
следовательно, подавляющая часть образующегося нитроний-
иоиа снова превратится в ион H*NO,, и лишь незначительная
часть пойдет на нитрование ароматического соединения. Та
ким образом переход к водным растворам должен быть связан
с переходом кинетики реакции от нулевого к первому порядку,
что и наблюдается на опыте.
Возможно, однако, и другое объяснение перехода
нитрования под действием воды от нулевого к первому порядку.
Можно предположить, что наличие больших количеств воды
полностью подавляет образование нйтроняй-иоиа, в
результате чего нитрование будет уже осуществляться меяее
активным ионом НаМОз\ Чтобы такое нитрование протекало по
первому порядку, достаточно, чтобы из двух возможных реакций
расходования иона H*NOa+ (отрыва протона и взаимодействия
кылухюдмад дрОАЮмгм^ескшс соб^цнемый 191
с ароматическим соединением) первая происходила значительно
быстрее второй.
Дополнительные данные, необходимые для установления
механизма нитрования водной азотной кислотой, были
получены при исследовании раман-спектров нитрующей смеси,
употреблявшейся авторами в этих опытах. Состав смеси был
следующий: Н,0 72 мол.%, НМОз 17 мол.%, СЩ 11 мол.%.
Исследование раман-спектров показало, что практически
вся хлорная кислота присутствует в виде ионов Н* и СЮ%,
азотная же кислота в подавляющей своей части (до 96%) не-
нонизнрована, и лишь 4% ее (т. е. 0,68% от всей смеси)
находятся в виде житрат-ионов. Необходимо, однако, учитывать, что
исследование раман-спектров не может дать ответа ни на
вопрос о том, в какой мере молекулы азотной кислоты, проявляя
основную функцию, присоединяют к себе протоны
(отщепленные от НС1О*) и существуют в виде ионов Н*МОз\ ни на
вопрос о том, в какой мере происходит присоединение протонов
к молекулам воды с образованием гидроксониевых ионов*. Тем
не менее тот факт, что только 4% имеющейся в рассматриваемом
случае азотной кислоты находятся в ионизированном
состоянии, Приводит к заключению, что значительная часть ее
присутствует в виде молекул HNO*. Действительно, если даже
принйть, что все 11% протонов, образующихся при
ионизации хлорной кислоты, присоединяются к молекулам азотной
кислоты, давая ионы H*NO,, то и тогда все же сохранится
свыше 5% (в расчете на всю смесь) молекулярной азотной
кислоты. Это является важным фактом, если принять во внимание,
что даже небольшие изменения состава нитрующей смеси,
которые не могут сколько-вибудь существенно повлиять на
концентрацию молекулярной азотной кислоты, вызывают очень
значительное изменение скорости нитрования. Из этого
несомненно следует заключение о том, что в опытах с водной азотной
кислотой нитрование молекулами НМОз не происходит.
* Это объясняют обычво тем, что такие ионы (H*NO, , HgO+) обме-
вивают свои протонЪг со столь большой частотой, которая препятствует
созданию точно определенного вибрационного уровня энергии.
192 Afaawwww* %имгро*днил
Второе заключение, которое можно вывести из спектральных
данных, сводится к тому, что если нитрующим агентом
является ион H*NO*% то он должен присутствовать лишь в
относительно малых концентрациях: иначе говоря, не может
происходить преимущественное присоединение протонов,
получающихся из хлорной кислоты, к молекулам азотной кислоты,
а не к молекулам воды. Действительно, если бы это
происходило на самом деле, то концентрация иона Н*МОз не могла
бы быть столь чувствительной функцией состава нитрующей
смеси: она должна была бы, например, меняться
пропорционально введенному количеству хлорной кислоты, в то время
как даже незначительное увеличение концентрации этой
кислоты сверх некоторого значения вызывает очень резкое
увеличение скорости нитрования.
Указанное выше наличие порогов концентраций, при
переходе через которые скорость резко возрастает, авторы, приняв
ион H*NO#f в качестве нитрующего агента в водных
растворах азотной кислоты, объясняют следующим образом,
рассматривая три возможных случая.
1. Концентрации азотной кислоты и воды сохраняются
неизменными, содержание же хлорной кислоты увеличивается.
Образующиеся яря диссоциации хлорной кислоты протоны
взаимодействуют главным образом с водой с образованием гидро-
ксониевых ионов НдО*. К молекулам азотной кислоты в
присутствии воды протоны присоединяются лишь в слабой
степени, так как вода обладает ббльшими основными свойствами по
сравнению с азотной кислотой. Следует учитывать, что вода,
помимо соединения с протонами, гидратирует еще и молекулы
азотной кислоты, присоединяясь к положительно заряженному
центру последних. Авторы отражают это следующей формулой
гидрата азотной кислоты: Н%0... HONO*. Следует думать, что
н протон, и молекула азотной кислоты, невидимому, способны
давать не только моно-, но и более высокие гидраты. В
результате постепенного увеличения концентрации хлорной кислоты
наступит момент, когда вся имеющаяся в системе вода окажется
связанной в виде гидратов. Начиная с этого момента, все
добавляемое количество протонов (из НС1О*) будет уже реагиро-
193
вать с азотной кислотой, образуя ион H3NO3. Поскольку этот
ион является нитрующим агентом, постольку скорость
нитрования с этого момента начнет резко возрастать*
2, Концентрации воды и хлорной кислоты постоянны,
увеличивается содержание азотной кислоты. Как и в предыдущем
случае, вода связывает протоны и гидратирует молекулы
азотной кислоты. Постепенное увеличение содержания
азотной кислоты приведет в некоторый момент к связыванию
подавляющей части воды. Начиная с этого момента, прибавляемая
азотная кислота будет взаимодействовать с протонами,
образуя ионы H3NO3, т. е. резко увеличивая скорость
нитрования.
3. Концентрации хлорной и азотной кислот постоянны,
увеличивается содержание воды. В этом случае в самом начале
реакции (в отсутствие воды) скорость нитрования максимальна,
так как все образующиеся протоны соединяются с азотной
кислотой, давая ионы H%NOg. Постепенное прибавление
воды приводит ко все более увеличивающемуся расходованию
протонов на образование ионов Н,О*. и, следовательно,
к постепенному уменьшению скорости реакции. При
достижении некоторого содержания воды количество
протонов, присоединяющихся к азотной кислоте, резко
уменьшается, и, начиная с этого момента, скорость нитрования резко
упадет.
При дальнейшем изучении нитрования различных
ароматических соединений азотной кислотой в органических
растворителях Ингольд с сотрудниками [71, 72] показали, что
нитрование фенолов, ароматических аминов и их алкилированных
производных отличается от нитрования других ароматических
соединений. Это отличие проявляется в различном влиянии
азотистой кислоты на скорость нитрования. Как мы видели,
нитрование в органических растворителях, достаточно ре-
акционноспособных к электрофильным замещениям
ароматических соединений (бензола, толуола и др.), протекает по кине-
тике нулевого порядка, причем добавление азотистой кислоты
несколько снижает скорость реакции. Выражение для скорости
в этом случае имеет следующий вид:
13 А. В. Топчиев
194 Afaamzwwaw нытроддмыл
Иначе действует азотистая кислота при нитровании фенолов,
ароматических аминов и их алкилированных производных.
Прибавление азотистой кислоты в этом случае увеличивает
скорость реакции, причем порядок реакции из нулевого
становится вторым
Если при нитровании этих соединений азотистая кислота
образуется при сопутствующем нитрованию окислении, реакция
носит автокаталитический характер.
Таким образом при нитровании фенолов, ароматических
аминов и их алкилированных производных мы встречаемся
с иным механизмом реакции, названным Иигольдом «саеци-
альшдм» механизмом. При этом оказалось, что для нитрования
этих высоко реакционноспособных соединений возможен
переход от обычного нитрования нитроний-ионом к этому
специальному механизму. Такой переход можно осуществить
изменением концентраций азотной и азотистой кислот. При низких
концентрациях азотной кислоты реакция сильно
катализируется присутствующей азотистой кислотой и протекает по
кинетике второго порядка (см. выражения скорости).
Постепенное повышение концентрации азотной кислоты приводит
сначала к уменьшению каталитического действия азотистой
кислоты, затем к полному его исчезновению, когда порядок
реакции становится нулевым, и, наконец, к появлению
незначительного тормозящего действия азотистой кислоты.
При установлении механизма действия азотистой кислоты
Ингольд с сотрудниками в первую очередь исходят из того,
что нитрование фенолов и ароматических аминов, подчиняясь
обычным законам ориентации, является электрофальнш*
замещением. Следовательно, механизм его должен включать
электрофильный реагент. Учитывая, что скорость реакции при
нитровании в присутствии азотистой кислоты зависит от
концентрации последней (и не зависит от концентрации азотной
195
кислоты), авторы делают естественный вывод, что веществом,
поставляющим этот электрофильный реагент, должна быть
сама азотистая кислота.
В цитируемых работах Ингольда азотистая кислота для
большинства опытов приготавливалась растворением чистого N^O*
в азотной кислоте. При этом было показано, что растворенный
в азотной кислоте N^O* является сильным электролитом.
Природа образующихся при этом ионов была установлена опытами
Гульдена и Миллена [73], изучавших раман-спектры
растворов NaO* в азотной кислоте. Оказалось, что N*O* диссоциирует
не только гомолитически, но и гетеролитически по уравнению
N,O* Z N0+ + Ж\ .
В результате авторы приходят к заключению, что электро-
фильным реагентом при нитровании в присутствии азотистой
кислоты является нитрозоний-ион N0+.
Принятие нитрозоний-иона в качестве нитрующего агента
сразу же дало возможность Ингольду сформулировать
механизм нитрования, ускоряемого азотистой кислотой, по
аналогии с ранее выдвинутым им механизмом нитрования при
помощи нитроний-иоиа. Первым этапом нитрования,
ускоряемого азотистой кислотой, является образование нитрозоний-
иона N0+:
HNO, + НЖ)
NO
Второй этап этой реакции — присоединение нитрозоний-
^ дона к нитруемому соединению (что представляет собой
медленную стадию реакции), последующее очень быстрое выделение
протона с образованием нитрозосоединения, которое далее
Зугаюне быстро окисляется в нитросоедннение:
ArH +N0+
медленная
стадия
Аг/
N0+
13*
196
Н быстрая
/ стадия
Аг ^ ^ ArNO + Н+
быстрая
стадия
ArNO + HNO, ^ ArNO, + HNO,
Школой Ингольда был также рассмотрен давно известный
экспериментальный факт влияния на скорость нитрования
концентрации присутствующей серной кислоты.
Как уже указывалось выше, различными исследователями
было констатировано, что скорость нитрования максимальна
при концентрации серной кислоты около 90%. Снижение
скорости нитрования при уменьшении концентрации серной
кислоты ниже 90% объясняется в настоящее время сопутствующим
уменьшением концентрации питронни-иона. Причина же
падения скорости нитрования при увеличении концентрации
серной кислоты сверх 90% до сих пор остается неясной.
Выдвинутое Беннетом и другими [63] объяснение, основанное на
принятии тримолекулярной реакции в качестве определяющей
стадии процесса нитрования, оказалось, как установил Меландер
Д66], несостоятельным. Выше было показано, что наличие
тримолекулярной реакции при нитровании ароматических
соединений отрицает и Ингольд. В литературе, однако, имеется
и другое предположение о причине замедления скорости
нитрования с ростом концентрации серной кислоты свыше 90%,
которое связывает этот факт с основным характером
нитруемого соединения.
С целью проверки этого предположения и установления
истинной причины рассматриваемого явления Гиллеспи и
Нортон [74] провели изучение влияния состава нитрующей смеси
на скорость нитрования при 25° триметилфениламмоний-иона
и я-хлорфенилтриметиламмопий-иона (обоих в виде нитратов)
и п-хлорнитробензола, обладающего основным характером.
Нитрование проводилось в сернокислой среде как водной (до
12% НаО), так и содержащей свободный серный ангидрид (до
13% SO*). Было найдено, что скорость нитрования всех трех
изученных соединений уменьшается с увеличением концентра-
митроедмыл дрождти^ескщ; собЭымбмг&Й 497
ции серной кислоты от 90 до 100%. Из этого следует, что в ос-
яовности нитруемого соединения нельзя видеть главную при-
чину уменьшения скорости реакции.
При переходе к олеуму оказалось, что с увеличением
содержания в нем серного ангидрида скорость нитрования
йодов растет, а скорость нитрования п-хлоряитробензола
продолжает падать. Последнее, по мнению авторов, вызывается
основным характером п-хлорнитробензола, т. е. способностью
его нитрогруппы присоединять протон. Такая протонирован-
ная иитрогруппа оказывает более сильное дезактивирующее
действие, чем обычная нитрогруппа.
Основной же факт падения скорости нитрования всех трех
изученных соединений при изменении концентрации серяой
кислоты от 90 до 100% и последующий рост скорости при
переходе к олеуму авторы объясняют изменением при этом
диэлектрической постоянной растворителя и связанным с этим
изменением его сольватирующеи способности. Авторы считают,
что при нитровании в момент переходного состояния
происходит распространение заряда, сконцентрированного в ни-
троний-ионе, на ароматическое ядро. Поэтому уменьшение
диэлектрической постояваой растворителя, которое, по
предположению авторов, происходит из-за образования иояов
при добавке воды или серного ангидрида к 100%-ной серной
кислоте, действительно должно увеличить скорость нитрования
при переходе как от 100 к 90%-ной серной кислоте, так и при
увеличении содержания серного ангидрида в олеуме.
Примером гемолитического, а не гетеролитического
механизма нитрования является открытая Робинсоном [75, 76]
реакция нитрования и гидроксилирования надазотистой
кислотой. Робинсоя с сотрудниками показал, что при
взаимодействии азотистой кислоты с перекисью водорода образуется над-*
азотистая кислота, которая способна нитровать и гидроксилиро-
вать ароматические соединения. По мнению авторов, надазо-
тистая кислота распадается гомолитически аа два
свободных радикала:
HOONO->HO"
198 Л/#вдми&м мшироедныл
Налжчре свободных радикалов доказывается тем, что над-
азотистая кислота способна инициировать процесс полимери*
зации метилакрилата. Кроме того, при действии надазотистой
кислоты на бензол образуется фенол.
Реакция нитрования надазотистой кислотой проводится
в следующих условиях: ароматическое соединение в, количестве
100 г смешивается со 100 мл 5%-ной НаОз, 5 мл 3 н. НС1 и 250 мл
Н%О; в течение 30 мин. при комнатной температуре
прибавляется яри постоянном перемешивании 150 мл 5%-ного
NaNO,.
Робинсон с сотрудниками изучил нитрование надазотистой
кислотой ряда ароматических соединений; бензола, толуола,
хлорбензола, нитробензола, фенола, диметиланилина, фене-
тола, фенилпропилового эфира и ацетофенона. Выход нитро-
и гидроксилированяых производных невелик (порядка 10%).
Обсуждая возможный механизм нитрования надазотистой
кислотой, авторы приходят к выводу, что гетеролитический
механизм в этом случае должен быть отвергнут. Действительно,
в таких слабокислых растворах, в которых проводится это
нитрование, маловероятно образование иитроний-нояа N0%.
Кроме того, образующиеся продукты отличаются по своему
отроению от продуктов, получающихся при нитровании тех
же веществ нитрующей смесью. Так, при электрофильном
замещении группа N0% вступает обычно в орто- или
пара-положение при наличии в молекуле соответствующих ориентантов
(например^!, ОН, СНзи др.). При нитровании надазотистой
кислотой нитрогруппа в присутствии тех же ориентантов встает в ме-
ra-положение (например, хлорбензол превращается в м-нитро-
хлорбензол и т. п.),
К идентичным выводам пришли и Уотерс с сотрудниками
[77], показавшие, что при нитрованиихинолина надазотистой
кислотой получаются 6- и 7-нитрохинолины. Обычно при гете-
ролитическом нитровании (смесью HNO3+ H*SO*) образуются
5- и 8-яитрохинолины. Такое различие авторы объясняют гомо-
литическим механизмом нитрования надазотистой кислотой:
радикал ОН присоединяется в «положение, открывая при
этом ^-положение для взаимодействия с N0$:
190
НО Н
но н
н \/
\
NO,
"Н.0
Следует отметить, что 6- и 7-нитрохинолины были
получены ранее А. В. Топчиевым и П. П. Шорыгиным [78] при
действии N0% на хинолин при 100°. Невидимому, и в этом случае
нитрование протекает по гемолитическому механизму.
На основе полученных результатов Робинсон и сотрудники
предложили следующий детализированный механизм
нитрования надазотистой кислотой. Первой стадией является распад
жадазотистой кислоты на радикалы ОН и N0*. Первый
присоединяется к ароматическому соединению, образуя арильный
радикал (стадия 2), который, реагируя с N0%, дает нестабильное
молекулярное образование (стадия 3), распадающееся с выделе-
вием либо воды, либо HNOg, либо Нз (стадия 4):
]) HOONO-»OH + NO,
X X
I I ОН
+ ОН
\/
X
I ОН
н
X
он
NO,
X
н
X N0,
он
N0,
+ HNO,
300
Для выяснения вопроса, является ли потеря протона в
процессе нитрования стадией, определяющей скорость реакции,
Лауэр и соавторы [79] изучали нитрование монодейтеробензола.
При нитровании монодейтеробензола, содержащего 91,5 4%
+ 1$7 мол.% C@HgD, образуются нитробензол, содержащий
74,9 ± 0,7 мол.% монодейтеронитробензола, им-динитробензол,
содержащий 60,9 + 2,0 мол.% монодейтеро-м-динитробензола.
Уменьшение содержания дейтерия не является, как
показывают опыты, следствием реакции дейтерообмена, так как при
обработке обычных С*Н#, СбНдМО*, и м-динитробензола дей-
теросерной кислотой происходит лишь ничтожное введение
D в эти молекулы. При обработке С^НД) концентрированной
НзбОд при 50—60° менее чем за 1 час происходит уменьшение
содержания C^HsD до 76,7 мол.%. Если бы реакция изотопного
обмена дошла до равновесия, содержание C*H#D должно было
уменьшиться до 41 мол.%. Следовательно, реакция детейро-
обмена проходит с меньшей скоростью, чем реакция
нитровавия. В результате проведенных опытов авторы пришли к
заключению, что отщепление протона от реакционного комплекса не
является стадией, определяющей скорость реакции нитрования.
Из работ по механизму нитрования ароматических
соединений заслуживает внимания также работа Джонса, Торна, Лайна
и Тейлора [80]. Эти авторы при исследовании спектров
поглощения азотной кислоты, начиная с 80%-ного водного раствора
до раствора, содержащего 24% избыточного N*O*, нашли,
что максимум поглощения находится вблизи 2650 А и что
интенсивность этого максимума изменяется в зависимости от
состава раствора.
Спектроскопические измерения показали, что интенсивность
поглощения повышается с увеличением концентрации
кислоты, достигая максимального значения при содержании воды,
равном 7% (для водных растворов); при последующем
увеличении концентрации азотной кислоты поглощение резко
падает (минимум при содержании воды 5%). При дальнейшем
уменьшении содержания воды интенсивность поглощения снова
возрастает, однако при переходе к безводной азотной кислоте
максимум поглощения становится размытым.
Добавление азотного ангидрида приводит к возрастанию
интенсивности поглощения, но максимум становится все ме
нее и менее заметным (при содержании 12% N#0* максимум
исчезает).
Эти результаты были объяснены наличием в системе
следующих равновесий:
+Н,0 +2Н.0
Растворы с содержанием 7% воды, дающие наибольшую
интенсивность поглощения, соответствуют, но мнению этих
исследователей, наибольшей концентрации недиссоциирован-
ной азотной кислоты. При дальнейшем уменьшении содержания
воды, которое приводит к размыванию максимума, азотная
кислота переходит в N#0*, причем создается равновесие
ротный ангидрид частично ионизирует по схеме
Н. С. Спасокукоцкий [81] дает обнаруженным
указанными выше исследователями закономерностям другое толкова-
I ние. Он исходит из предположения, что в водном растворе,
кроме обычной ионизации по схеме
HNQa + Н,0 Z Щ0+ 4 N0",
^одновременно происходит в некоторой степени и автоионизация
схеме
3HN0, Z N0+ + Н,0+ Г
^Максимум поглощения 2650 А, по мнению Спасокукоцкого,
кможно приписать присутствию иона NOg. При дальнейшем
^увеличении концентрации кислоты процесс автоионизации
Ыелается все более и более заметным. Усиление автоиониза-
р щии идет, однако, медленнее, чем уменьшение но рмальной
Ионизации с водой, вследствие чего количество недиссоцииро-
важной азотной кислоты увеличивается.
202 Меамнмам ммтроедмкя ярол*лтичеся;%*
По достижении концентрации, отвечающей 7%-ному
содержанию воды, начинается образование азотного ангидрида
по схеме
Так как этот процесс протекает с выделением воды, то
пополнение количества недиссоциированной кислоты происходит
за счет равновесия автоионизации, что сопровождается
исчезновением из системы некоторого количества ионов N0%*.
По этой причине наблюдается падение интенсивности
поглощения. Когда содержание воды достигает 5%, наступает
ионизация азотного ангидрида, в связи с чем начинает
увеличиваться поглощение для длив волн около 2650 А- Таким
образом, общая схема протекающих процессов выражается, по
Спасокукоцкому, следующей системой уравнений:
ч-ан.о
4- 2
it # —ЗН,0 (нормальная ионизации)
N0" + N0% 2NQ- + Н.0+ + N0%
автоиовявация
В настоящей главе был приведен обзор механизмов,
предложенных за последние три десятилетия для реакции нитрования
ароматических соединений азотной кислотой или нитрирующей
смесью. Из обзора видно, что господствовавшее вначале
представление о протекании этой реакции путем последовательных
процессов присоединения и отщепления сменилось в дальней-
шем признанием того, что истинным содержанием нитрования
является процесс замещения.
Нитрование, как процесс присоединения-отщепления,
в трактовке Виланда (1920—1921 гг.) заключался в
присоединении элементов азотной кислоты по двойной связи
ароматического ядра и в последующем отщеплении воды с
восстановлением двойной связи, а в трактовке Б. В. Тронова (1924—
1929 гг.), С. С. Наметкина и А. С. Забродиной (1925 г.), а также
Михаэля и Карлсона (1935 г.) — в присоединении молекулы
азотной кислоты целиком к одному из атомов углерода
ароматического ядра с одновременным переходом атома водорода от этого
атома углерода к одному из атомов кислорода присоединяющей-
203
ся азотной кислоты и в последующем отщеплении воды. В
дальнейшем представление о титровании как о процессе
присоединения-отщепления, длительное время (до конца 40-х годов)
было общепринятым — менялись только взгляды на природу
нитрующего агента, причем всегда принималось отщепление
многоатомного соединения (воды, серной кислоты и т. п.),
а не одного лишь атома водорода, на место которого в
результате нитрования должна встать нитрогруппа.
Развитие представлений о природе нитрующего агента,
отличной от природы не диссоциированной молекулы азотной
кислоты, ведет свое начало от работ Ганча (1906—1909 и 1917—
1926 гг.). Как указывалось, Ганч на основании криоскопиче-
ских измерений и измерений электропроводности пришел к
выводу, что в сильных кислотах в отсутствие воды происходит
переход протона от одной молекулы кислоты к другой. В
результате молекула, явившаяся донором протона,
превращается в отрицательный ион, а молекула, играющая роль
акцептора протона,— в положительный ион. В азотной кислоте
такой процесс приведет к образованию нитрацидий-иона
(HaNOg, HaNO^) и нитрат-иона (NOa~). В случае смеси двух
кислот, например азотной и серной, донором протона явится
более сильная кислота, и, следовательно, возникнут ионы
H*NOa* (или HsNOg*) и HSO*~. Серьезным подтверждением
зтих заключений явилось получение Ганчем в
кристаллическом виде перхлоратов нитрацидия [HsNOsMClOJ и
Сам Ганч не изучал нитрования ароматических соединений,
однако его теория строения гомогенной азотной кислоты и ее
смесей с серной кислотой оказала, как мы видели, сильное
'Влияние на все последующее развитие исследований этого
;1 ^процесса. Теперь при вскрытии механизма нитрования арома-
i: этических соединений уже нельзя было игнорировать наличия
ж нитрующей смеси ионных образований и сводить реакцию
'К воздействию только нейтральных молекул азотной кислоты
|';Жа нитруемое соединение. Большая реакционная способность
I Ыонных образований заставляла предполагать их несомненное
|'участие в реакции, и не учитывать их стало невозможным.
204
Одним из первых отражений этого были взгляды Геверинг-
тона и Мессона, которые, изучая нитрование нитробензола^
пришли в 1933 г. к заключению о первоначальном образовании
в этой реакции комплекса [C#H&NO,- H Г [МОзГ, катион
которого при взаимодействии с недиссоциированной
молекулой азотной кислоты превращается в СдН/МО*)* и [НзО]\
Таким образом, эти авторы хотя и рассматривают
нитрование как иояно-комплексную реакцию, однако принимают, что
ионизированным образованием является само нитруемое
соединение, превращающееся в катион в результате
присоединения протона, а нитрующим агентом — яедиссоциировавная
молекула азотной кислоты.
Такой же точки зрения придерживался Ланц (1939 г.).
Он также приходит к выводу, что при нитровании в водном
растворе серной кислоты нитруемое ароматическое соединение
путем присоединения протона превращается в катион, который
в дальнейшем взаимодействует с недиссоциированной
молекулой азотной кислоты.
Двойственную позицию заняли Лауэр и Одда. В работе
1936 г., посвященной нитрованию нитробензола и антра-
хинона, они проводят резкое различие между механизмами
реакции в зависимости от того, протекает ли она в водных или
безводных средах. В первом случае нитрующим агентом
является, по мнению авторов, недиссоциированная молекула азот-
вой кислоты, элементы которой присоединяются по двойной
связи с последующим отщеплением воды. Следовательно, для
этого случая полностью принимается механизм Виланда.
Во втором случае, т. е. при нитровании в безводных
растворах серной кислоты, авторы считают уже, что нитрующим
агентом является нитрацидий-сульфат [H*NOa]+ Ш8О*Г,
который, присоединяясь по двойной связи ароматического ядра,
дает промежуточное соединение. Последнее» отщепляя воду
я серную кислоту, переходит в нитросоединение. Такой
механизм уже полностью отвечает представлению Ганча о строении
азотной кислоты.
Ионный механизм нитрования ароматических соединений
выдвигал в 1940 г. и М. И. Усанович. Принимая по Гаячу на-
205
Жичие в нитрующей смеси нятрацкдки-жона, М. И. Усановяч
Считает, что первым актом нитрования является присоедине-
вие этого иона к одному из атомов углерода ароматического
ядра. Образовавшееся промежуточное соединение, отщепляя
молекулу воды и протон, дающие Н,О\ превращается в нит-
росоединение.
Помимо Лауэра и Одда и Усановича к началу 40-х годов
другие авторы также присоединились к точке зрения,
согласно которой нитрующим агентом при нитровании
ароматических соединений является нитрацидий-ион. Следует при
этом отметить, что такая «замена» нитрующего агента
(нитрацидий-ион вместо недиссоциированной молекулы азотной
кислоты) вполне укладывалась в представление о нитровании
как о процессе присоединения —отщепления.
Однако уже в середине 40-х годов предположение, что нит*
рующим агентом является нитрацидий-ион, не могло считаться
общепринятым. Более того, в работах второй половины 40-х
годов были получены объективные доказательства того, что
истинным нитрующим ароматические соединения агентом
служат не нитрацидий-ион HgNOg или H,NO^% а нитроний-
ион N0/.
' Впервые мысль о существовании иона N0% еще в 1903 г.
высказал Эйлер [82]. В работе 1922 г. [83] он снова
возвращается к ней. Этот ион упоминался в литературе и в
последующие годы [84—86], однако вплоть до работ Ингольда и
сотрудников [87] в 1946 г., в которых были изучены влияние азотной
кислоты на точку замерзания серной кислоты, а также спектры
Комбинационного рассеяния смесей азотной кислоты с другими
/кислотами, объективные подтверждения действительного
существования нитроний-иона отсутствовали.
В обзоре механизмов нитрования ароматических
соединений, данном в этой главе, из соображений цельности
изложения не рассматривались эта работа Ингольда, а также другие
:j работы, хотя и надежно подтверждавшие существование иит-
1роний-иона, но не путем изучения процесса нитрования,
ГЁ на основе криоскопических и спектроскопических измерений
^смесей азотной кислоты с другими кислотами. В заключение
206 AfgaMZHUfjw нот/юбдныя д/юлмтцчесжиа; соединены &
обзора целесообразно, однако, привести и эти доказательства
наличия нитронмй-монов в нитрующих смесях.
В 1946 г. Ингольд с сотрудниками [87], применив
улучшенную по сравнению с использовавшейся Ганчем криоскопическую
технику, смогли показать, что понижение точки замерзания
серной кислоты, вызываемое растворением в ней азотной
кислоты, в четыре раза больше понижения в случае идеального
раствора. Ганч, наблюдавший в 1908 г. трехкратное по
величине понижение точки замерзания серной кислоты при
растворении в ней азотной, объяснил его превращением азотной
кислоты в нитрацидий-ион:
Протонярованме азотной кислоты, предположенное Ганчем,
полностью принимается и Ингольдом. Объяснение же четырех-,
а не трехкратного понижения точки замерзания Ингольд
находит в дальнейшем распаде нитрацидий-иона на ионы нит-
рония NOg и гидроксония HgO\ Суммарное уравнение,
следовательно, приобретает следующий вид:
Ингольд считает, что нельзя предложить никакого другого
объяснения четырехкратного понижения точки замерзания для
системы HNO* — HaSO*. С таким выводом, действительно,
трудно не согласиться.
Второе объективное доказательство реального
существования нитроний-иона было получено при изучении спектров
комбинационного рассеяния смесей азотной с другими,
главным образом с серной, кислотами. В спектрах этих смесей
всегда наблюдаются две линии с частотами 1400 и 1050 см~\
причем их интенсивность исключает предположение о том, что
источником этих линий является неизмененная молекула
азотной кислоты. Шедэн [88], однако, смог показать присутствие
этих частот в спектре комбинационного рассеяния гомогенной
азотяой кислоты. Следовательно, их источником является
какой-то продукт, образующийся из азотной кислоты. Шедэну
удалось далее открыть, что такой продукт получается в резу ль-
207
тате дегидратирования азотной кислоты и что он разрушается
при добавлении воды, приводящем к регенерации молекулярной
азотной кислоты. В то же время Сусц и Брияер [89] нашли
частоты 1400 и 1050 см"^ в спектре комбинационного
рассеяния раствора азотного ангидрида NaO* в азотной кислоте, после
чего стало несомненным, что их источником является особая
форма азотного ангидрида. В цитированной выше работе Беннет,
Бранд и Вильяме [44] предположили, что такая форма
возникает при ионизации азотного ангидрида; частота 1400 см"^
отвечает нитроний-иону, а 1050 см"* — нитрат-иону.
Решающее подтверждение этого взгляда было получено Ингольдом
с сотрудниками [87] в работе 1946 г. В ней были изучены спектры
комбинационного рассеяния смесей азотной и хлорной, а
также азотной и селеновой кислот. В обоих случаях была
обнаружена частота 1400 см~\ частота же 1050 см"^ отсутствовала,
а вместо нее появлялись частоты, отвечающие анионам хлорной
и селеновой кислот. Дальнейшее исследование этих авторов
установило, что появление линии с частотой 1400 см ^
вызвано источником, который кроме нее не дает никаких других.
Известно, что одну основную линию в спектре
комбинационного рассеяния могут давать только двухатомные и линейные
трехатомные частицы с одинаковыми конечными атомами. В
рассматриваемом случае такой частицей может быть только нитро-
ний-ион NOg.
Можно, наконец, привести еще одно доказательство
существования нитроний-иона. Ингольд с сотрудниками [90], ис-
яользуя улучшенную методику, повторили эксперимент Ганча
по получению твердых продуктов взаимодействия азотной
и хлорной кислот. Как уже выше сказано, Ганч приписал этим
продуктам состав и строение перхлоратов витрацидия,
Ингольд же показал, что на самом деле они представляют собой
смесь перхлоратов нитрония и гидроксония (N0%)^ (С1О*)~
и (H,O)+(C1OJ-.
Подводя итог приведенным результатам криоскопических
ж спектроскопических измерений самой азотной кислоты и
главным образом ее смесей с другими кислотами, можно
констатировать, что в конце 40-х годов реальность существования
208 Масонкам
в этих средах нитроний-иона уже ие могла подвергаться
сомнению. Это естественно сказалось на предложенных в даль*
нейшем механизмах нитрования ароматических соединений.
Действительно, все механизмы этого процесса, появившиеся
в литературе, начиная с конца 40-х годов, принимают нитро-
ний-ион в качестве нитрующего агента.
Характерной и положительной чертой исследований,
проведенных в эти годы (Титова, Вестгеиыера и Караша, Вилльям-
са и Лоуэна, Беняета, Бранда и Вилльямса, Томменсона и
Гроггияса и особенно Ингольда с сотрудниками), является то,
что в них, наряду с изучением химизма реакций, все ббльшую
роль начинает играть установление ее кинетических
особенностей. При этрм, как следует из приведенного выше обзора этих
работ, оказалось, что ббльшая часть кинетических данных о
нитрования ароматических соединений получила
удовлетворительное объяснение при принятии нитроиий-иона в качестве
нитрующего агента. Это в первую очередь относится к такой
важной кинетической характеристике, как изменение порядка
реакции в зависимости от состава нитрующей смеси и
растворителя. Следует признать, что Ингольд и его школа блестяще
разрешили эти казавшиеся мало понятными кинетические
особенности реакции.
Таким образом можно считать, что в конце 40-х и в начале
50-х годов было не только констатировано реальное
существование нитроний-иона в нитрующих средах, но и осуществлено
посредством его нитрование ароматических соединений*. Та-
кая замена в механизме реакции нитрацидий-иона на нитро-
пий-ион означает переход к представлению о нитровании как
о процессе замещения. Действительно, теперь реакцию
нитрования представляют как присоединение нитроний-иона к атому
* Следует напомнить, что, по Ингольду, в определенных условиях
нитрующим агентом не всегда является ион нитрония. Так, при нптрова-
аии разбавленной азотной кислотой нитрующим агентом будет нитра-
цидий-ион (см. стр. 190), а при нитровании фенолов, аминов и алкилиро-
важных производных в присутствии азотистой кислоты возможен переход
от обычного нитрования витроний-ионом к нитрованию ионом нитрово-
авя (см. стр. 194).
209
углерода ароматического ядра с последующим или
одновременным отрывом от этого атома углерода связанного с жим атома
водорода в виде протона. Здесь нет, следовательно, образования
нового вещества соединением отщепляющегося протона с
какой-то частью присоединившееся нитрующего агента.
Определение такого механизма как процесса замещения еще
не устанавливает того, осуществляется ли он в один акт или
в стадийной последовательности двух актов. Беннет, Бранд
и Вилльямс предположили первое, Ингольд с сотрудниками —
второе. Как указывалось, вопрос был решен опытами Мелан-
дера, воспользовавшегося методом меченых атомов и
показавшего, что процесс нитрования протекает в две стадии, из
которых первая — присоединение нитроний-иона к ароматической
молекуле — происходит с относительно небольшой скоростью,
а вторая — отщепление протона — с очень большой скоростью.
В результате скорость первой стадии определяет скорость всего
процесса нитрования.
Как ясно иа всего материала этой главы, основным выводом
длительного исследования реакции нитрования ароматических
соединений следует считать установление для нее ионного
механизма. Такое нитрование, следовательно, является гетероли-
тической реакцией. Как будет показано в следующей, главе,
имеется много оснований предполагать совершенно иной,
свободно радикальный механизм для нитрования алифатических
соединений, которое поэтому представляет собой
гемолитическую реакцию. До недавнего времени можно было считать,
что нитрование ароматических и алифатических соединений
принципиально различается именно в этом. В последние годы,
однако, появились первые работы, которые заставляют
предполагать возможность пересмотра такого резкого и
принципиального разграничения процессов нитрования этих двух
классов соединений. Эти изложенные выше работы Робинсона
с сотрудниками и Уотерса с сотрудниками свидетельствуют о
том, что в известных условиях нитрование ароматических
соединений может осуществляться по свободно радикальному
механизму. Авторам удалось показать, что при нитровании
ряда ароматических соединений азотистой кислотой в
14 А. в. Топчиев
210 .М&гдныам
присутствии перекиси водорода процесс протекает
гомологически. Никакого механизма такого нитрования ароматических
соединении яри этом выдвинуто не было, за исключением
предположения, что образующаяся надазотистая кислота
распадается на радикалы ОН и N0%, которые и «ведут* процесс:
HOONO-+OH + NO,.
Можно, однако, предположить, что в данном случае
образование свободных радикалов происходит тая же, как и в
реакциях с участием перекиси водорода и ионов переменной
валентности. Известно, что Н. Н. Семенов [91], воспользовавшись
данными Ури [92] об изменении свободной энергии в
следующих двух процессах:
Fe#+ + Н,О, ^ Fe*+ + Н+ + НО,
и
Н,О, % Cn+ +H+ + НО,,
рассчитал равновесные концентрации радикала НО*,
возникающего в указанных системах. Эти реакции оказались вполне
достаточными для инициирования цепной реакции.
Аналогично образование свободных радикалов ОН и N0,
в системе азотистая кислота — перекись водорода можно
представить следующим образом:
1) HONOZOH- + NO+;
2) N0+ + Н,0, Z NO, + H+ + ОН;
3) 0Н+ HONO %Г Н,0 + N0%;
4) N0* + Н^)* Z N0^ + Н+ + ОН;
5) 0H + N0.ZHN0,.
В основе такой схемы лежит предположение о возможной
диссоциации азотистой кислоты на ионы ОН" и N0* (реакция 1).
Нитрозояий-ион N0% реагируя далее с перекисью водорода
по типу приведенных выше реакций ионов Fe** и Си^
с Н#0,, превращается в свободный радикал N0%, причем
одновременно возникает радикал ОН (реакция 2), В отсутствие
ароматического соединения в такой системе будут протекать
211
реакции 3, 4 и 5, и суммарное уравнение всего процесса будет
'иметь следующий вид:
1, 2HNO± + 211,0, -^ 2HN0: + 211,0.
I Если же к такой системе добавить ароматическое соедине*
Г'вне, то свободные радикалы ОН и N0%, помимо взаимодействия
I до реакциям 3, 4 и 5, частично будут вступать в реакции
I'
I 6) АгН + ОН->Аг+Н,О,
| 7) Ar+NOa-^ArNO,,
I'
[цриводящие к образованию нитропроизводшк.
;| Таким образом, в тех случаях, когда в системе создается
I возможность образования свободных радикалов, нитрование
' ароматических соединений может протекать и по радикальному
'; механизму.
i
I ЛИТЕРАТУРА
1. Н. Wieland, E. Sakellarios. Ber., 52, 898 (1919); 53,
201 (1920).
2. Н. Wieland, F. Rahn. Ber., 54, 1771(1921).
3: J. Tiehle. Ann., 306, 128 (1899).
4. A. H о 11 em a n n. Die direkte Einfiibrung von Subslituenten m
den Benzolkem. Leipzig, 1910.
5. А. К 6 k n 1 e. Ber., 2, 329 (1869).
6. Сравни С, III, 618 (1923).
7. E. P. К о h 1 er, N. L. Drake. J. Am. Cbem. Soc,, 45, 1281
(1923); C, HI, 618 (1923).
8. R. AnBBhAtz, A. Gilbert. Ber., 54, 1854 (1921); 57, 1697
(1924).
0. G. Odd о. С, Н, 4598 (1925).
O. S. Gabriel. Ber., 18, 2442 (1885).
l.S. R. Cawley, S. G. Plant. J.Chem. Soc, 12H (1938).
. J. Meisenbeimer, Ber., 33, 3547 (1900).
|*3. Б. В. Тронов. Изв. Томск, технол. нж-та, 45, № 3 (1924). Труды
Ь IV Менделеевского съезда, 157 (1925); ЖРХО, 61, 2388 (1929); Изв.
jli Сжб. химиколгехжол. нн-та, № 2 (1931); Б. В. Тронов,
II Н.Х. Сибгатулл ин. ЖРХО, 62, 2267 (1930).
1*4, Б. В. Тронов, Л. В. Ладыгина. Укр. хнм. журв., 7,
:| 55 (1932).
14*
212
15. С. С* Наметкжя, А. С. Заброджя#. ЖРХО, 57, 87
(1*5),
16. A. Michael, G. H. Carlson. J.Am.Chem.Soc.,57,1268(1935).
17. J. Glerschb ach, A. Kessler. Z. phy5.Ghem.,2, 676 (1888).
18. Boedtker. Bull. Soc. Chim. France, 726 (1908).
19. A. Werner. Neue Anachanngen auf dem Geblete der anorgaai-
Bche Chemie.
20. P. P f e i f er. Ofganmche №Wkulv@rbiadimgea. Stwttg#Af 1922.
21. A. Mich a el, С H.Carlson. Bcr.,29,1795(1896); J.Am.Chem.
Soc., 57, 1268(1935).
22. L Schmidt, E. Heinle. Ber., 44, 1488 (1911).
23. I. S с b m t d t. Bar., 33, 3251 (1900).
24. A. M Вержеаге#м- Журя. общ. жшшяж, 3, 381 (1033).
%5L A, BantKach. 2, phy,. (&#m., 61, %7 (1907); W, *7@, 626 (1907);
&5, 41 (1906); 08, Ж (19Р9).
26. A. H a n t z s с h. %. Elektrochem., 24, 201 (i918); 29, 221 (1923);
&), 194, 397 (1924); 31, 167, 455 (1925); B*r., SO, 1413, 1422 (1917);
52, 1&44 (1919); 58, 612, 941 (1925); 59, 1096 (1926).
27. E. Schaeffer. Z. Wias* Phot., 8, 312 (1910); 17, 193 (1913);
Z. angew. Ghem., 97,285 (1916); 98, 70 (1916).
28. H. H alb аи, J.Eisenbrand. t.phys. (hem., 132, 433(1928).
29. E. Farmer. J. Soc. Chem. Ind., 50, 73 (1934),
30. H. M@r$t6s*#. 2* phy». Qhom., Щ 385 (19Q4); 99, 605 (1907).
31. K. Laaer, R. Odd a. J. prakt. Chem., 144, 17g Ц936).
32. R. Cant z, Boll. Soc. Chip*, Praace, [5], g, 280 (1939).
33. I. N. Breasted, Rec. trov. chim., 4&, 718 (1923); Ber., 61, 2p49
(1928).
34. J. A, Hetheriagton, J.M*B8O%_ J. Chem, Soc, 105 (1933).
35. f. M ass on. J. Gbem. Soc., 3200 (1931). Ср. Д. И. Титов.
Шура. общ. хжишж, 18, 140, 733 (1948).
30. М. И. Ус аяовж ч. Журв* общ. zmorn, 219 (1940); М. g. У с а-
вович, Ш. А б п д о в. Жури. общ. химярк, iq, 224 (1940);
М. И. Ус ажовж^И.Глуж о в. Жури. общ. worn, 1ф, #7(1940);
М. М. Усановжж, Т. Сушкевич. Шура. общ. химии,
Ю, 230 (1940).
37. В. В. Маряовяяко*. ЖРХО, 3!, 48 (1%9).
38. А. К I em en t, R, 6 с h о 11 e г. Z. anorg, aUg, Chem., 141, 231
(1924).
,39. L. N. Ferg*s$Qo. J. Cbem., Educ., 23, 550 (1940).
40. J. P. Wiha*t. R#e. tmv. chim., 94, 241 (1915).
41. А. В. Сапожя**+в, ШРХ0, *, 10Ю (1903); 37, 374 (1905);
38, 1192 (1906).
42. A. SchaarschmWL Z. angew, Cbem., *, 1457 (1926).
43. А. И. Титов. Журя. общ. химии, 17, 382 (1947).
I . J7wm%%p*ny&pa 213
1Щ G, At. В e n a et, 1, C. I). В r a n d, G. W i 11 i a m s. J. Chem.
I'll Soc, 869 (1946).
l#. A. И. Т ж т о а, Н. Г. Л а п т е в. Журя. общ. аимиж, 18, JN& 4, 741
% (1948).
j,46. А. И. Т и т о в. Журж. общ. химия, 18, № 2, 190 (1948); 18, 733
I! (1948).
|:47. Н. Н. В о р аж щ о в. Остовы стте&а промежуточных продуктов
I,,, ж яр&сителей, гл. Ill, М., Госхвмиадат, 1940.
Iv#8. Т. О. В о а а ег, М» Е. J a m ев, А. М. L о w e n, G. W i 1-
i I i a m 8 Р?attire, 463, 955 (1949).
149. О g a t а. С. А., 4557@ (1947).
|50. Б, В. Трожов, Г. Я. Б ер. ЖРХО, 62, 2357(1930).
| 51. С. A., 4487d(1947).
:@2. Н. Е г 1 0 n m ey e r. Helv. Chim. Acta, 30, 2058 (1947).
|:,53. А. П. Т @ р 0 п т ь е в, Б. М. Кедров. Уч. вап. МГУ,
} 213 (1936); С, II, 1993 (1937).
154, А. В. Топчиев, В. П. Алания, Г. С. Шшайдер.
- Докл. АН ОССР, #95, W 1, 89 (1954).
L 55 F. L Modi с, Jowa State Coll. J. Sci., 27, No 2, 219 (1953).
! 56. P. U. W eathelmer, M. S. Kharagch. J. Am. Chem. Soc.^
': 68, 1871 (1946),
157. G. M. В en n 0L Ch#m. and Ind., 235 (1949).
"58. V. Gold, E. D. H»gh ев. J. Cbem. Soc, 2452 (1950).
! E. W. Dnnnine, C. W. Nutt. Trans. FaradaySoc, 47, 15
Hi (1951); E, S. H a 1 b А г s t a d t. Nature, !58, 448 (J946).
I 59. G. William*, A. M. Low en. J. Chem. Soc, 3812 (1950).
I 00. M. Tomlin&on, P. H. Groggins. Chem. Eng., 57, №12,
131 (1950).
:61. С K. In gold, D, Mlllen, H. G. Poll 6. Nature, 158,
|| 480 (1B46).
!|62. G. Willlems, R. SimkinB. J. Chem. Soc, 1386(1953).
g| 63. G. M. В en net, J. C. D. Brand, D. M. James,
G.Williams, T. G. Saunders. J. Chem. Soc, 464 (1947).
Г 64. H. H. В о р о ж д о в. Основы ситтеза промежуточных продуктов
jij и «расителей. М., Госхимиздат, 1950, стр. 151.
i 65. С. Related, A. H. Lamhertom. J Chem. Soc, 3349 (1953).
; 66. L. Mel and »r Nature, 163, 599 (1949).
i 67. G. А. В en ford, С. К. In gold. J. Chem. Soc., 929 (1938);
. E. D. Hughes, С. К. In gold, R. Reed. Nature, 158,
I , 448 (1946),
68. E. D. Hughea, С R. Ingold, R. Reed. J. Chem. Soc,
I 2400 (1950).
! 69. E. Black all, E. D. Hughes. Nature, 170, 972 (1952).
214
70. Е. S. Hal b erst ad t, E. D. H ugh ев, С. К. I n go Id.
J. Chem. Soc, 2441 (1950).
71. С. А. В an ton, E. D. H ugh ев, С. К. Ingo Id,
M. B. J acobs, R. N. J ones, C. J. Ail nkoff, R. Reed.
J. Chem. Soc, 2628 (1950).
72. J. Glaz er, E. D. H ugh es, C. K. Ingo Id, R. N. J ones,
D. M. J am ев, E. R obe rls. J. Chem. Soc, 2657 (1950).
73. L. D. S. Go ul den, D. J. M i lien. J. Chem. Soc, 2620 (1950).
74. R. J. G i 11 e s p i e, L. Norton. J. Chem. Soc, 971 (1953).
75. E. Halfpenny, P. L. R ob inson. J. Chem. Soc, 939 (1952).
76. W. Heslop, P. L. Robinson. J. Chem. Soc, 1271 (1954).
77. W. A. Waters, J. R. Laville, J. Chem. Soc, 413
(1954).
78. А. В. Топчиев, П. П. Шорыгин. Вег., 69, 1874 (1936).
79. W. M. La ue r, W. E. N ol and, J. Am. Chem. Soc, № 15,
3689 (1954).
80. R. N. Jones, С D. Thorn, M. Lyn e, E. C. Taylor.
Nature, 159, 163 (1947).
81. H. С. Спасокукоцкий. Усп. химии, 17, № 1 (1948).
82. H. Е n 1 ег. Ann., 330, 280 (1903).
83. Н. Е и 1 е г. Z. angew. Chem., 35, 580 (1922),
84. P. W aid en. Z. angew. Chem., 37, 390 (1924).
85. T. Ri, H. Eyrin g. J. Chem. Phys., 8, 433 (1940).
86. С. С. Price. Chem. Rev., 29, 51 (1941).
87. E. D. Hughes, С, К. Ingo Id, R. I. Reed. Nature, 158,
488 (1946).
88. J. Ch edin. Compt. rend., 200, 1397 (1935); 201, 552, 714 (1935);
202, 220, 1067 (1936); 203, 772, 1509 (1936); Ann. Chim., 8, 243
(1937).
89. B. Snsz, E. В riner, Helv. Chim. Acta, 18, 378(1935).
90. D. R. Goddard, E. D. Hnghes, С. К. Ingold.
Nature, 158, 480 (1946); J. Chem. Soc., 2559 (1950).
91. H. Н.Семенов. О некоторых проблемах химической кинетики
и реакционной способности. М., Изд-во АН СССР, 1954.
92. N. Uri. Chem. Rev., 50, 375 (1952).
I
НИТРОВАНИЕ ПРЕДЕЛЬНЫХ, ЖИРНОАРОМАТИЧЕСКИХ
И НЕНАСЫЩЕННЫХ УГЛЕВОДОРОДОВ АЗОТНОЙ
КИСЛОТОЙ
В связи с успехами, достигнутыми при синтезе нитропара-
финов, появился ряд работ яо изучению свойств нитрояарафи-
нов для изыскания различных путей их применения в
органическом синтезе.
Ввиду того что материалы о свойствах нитроиарафинов
разбросаны по страницам советской и иностранной журнальной
литературы, мы считаем целесообразным дать краткий обзор их
в настоящей главе*.
§ 1. Свойства нитропарафннов
' Почти все мононитронарафшш представляют собой
бесцветные жидкости с т. кип. от 101,7° и выше. Нитрометан раство-
i ряется в воде в объемном отношении 1:10 при 20*; растворимость
^остальных иитропарафинов уменьшается с повышением моле-
Окулярного веса (табл. 12).
I * По нитропарафияам смотри:
il. А. И. Якубович. Усп. химии, 15, вьш. 5, 577 (1946).
! 2. Р. Гольдштейн. Химическая переработка нефти, стр. 74—81,
. М , 1952.
УЗ. G. Eglof f, M. Aleiand er. Oila. Gas J.,4d, 23, 39, 49 (1942).
ijji*. О. Schickh. Angew. Chem., 62, 527—556 (1950).
|l$. W. Craber. Ind. Eng. Chem., 40, 1631 (1948); 43, 1989 (1951); 44,
"" 204! (1952); 46, 1098 (1953).
По витроолефинам смотри:
.В. Перекали в, А. С. Со п ов *. Усп. химия, 24, выя. 5 (1955).
216
азотной
Таблица 12
Нятросоедииенир
,20
"20
о
D
§
Нитрометан .
Нитроэтан . .
1-Нитропропан
2-Нитропропан
61,04
75,07
89,09
89.09
1,139
1,052
1,003
0,992
29
- 90
108
93
101,2
114,0
131,6
120,3
27,8
15,6
7,5
12,9
1,3818
1,3916
1,4015
1,3941
9,5
4.5
1,4
Большинство органических растворителей (ароматические
углеводороды, спирты, сложные эфиры, кетоны, эфиры, кар-
боновые кислоты) смешиваются с нитропарафинами.
Нитрометав — прекрасный растворитель для эфиров
целлюлозы; такими же свойствами обладает, 2-метил-2-нитропропан.
Смеси натропарафинов со спиртами являются хорошими
растворителями не только для сложных эфиров целлюлозы, но
и для винияитовых смол, простых эфиров целлюлозы и
смешанных сложных эфиров целлюлозы, как,например, ацетата-бутира-
та и ацетата-дропионата целлюлозы.
Нитропарафины представляют собой соединения, довольно
своеобразные с химической стороны. Переходя в таутомерную
форму «нитроновой кислоты», они обладают способностью
к различным превращениям и реакциям конденсации. При
конденсации с альдегидами они дают нитроспирты, которые
в свою очередь могут служить источником получения сложных
и простых эфиров, аминоспиртов и др.
Из продуктов превращения нитропарафинов заслуживают
также внимания нитроолефины, которые, обладая активной
двойной связью, могут давать различные продукты
конденсации.
217
Восстановление нятрояарафияов
Восстановление нитропарафинов до аминов осуществляется
яри действии различных реагентов: цинка, свинца или железа
в кислой среде [1—5], амальгамы алюминия [6] и т. д.; нитро-
яарафины гидрируются в паровой фазе в присутствии
катализатора Адамса [7], а также никеля, меди и платины [8];
хорошие результаты получены при гидрировании с никелевым
катализатором Ренея.
Нитропарафины восстанавливаются до алкилгидроксил-
аминов при действии металлов, например 8я и Za, в
присутствии кислот [9, 10] или в нейтральной среде [11] при действии
SaClg [12], а также при каталитическом гидрировании.
Нитропарафины удается гидрировать [13, 14] до алкилгидроксил-
аминов в присутствии палладированного BaSO*. Алкилгид-
роксиламины могут быть получены посредством восстановления
нитропарафинов электрохимическим путем [15].
Превращение нитропарафинов в гидроксиламины может
производиться алкилметаллами и алкилметаллгалогеяидами [16].
Из нитропарафинов могут быть получены и оксимы. Так,
например, оксимы получены [17] при обработке
нитропарафинов цинковой пылью в присутствии ледяной уксусной кислоты.
Первичные и вторичные нитропарафины могут быть
превращены в оксимы посредством пиролиза продукта алкилирования
их до нитроновых сложных эфиров:
R,C = NO,Ka + CH:J ->R,C = N — OGH, + NmJ;
О
B,C = N — ОСН. f^E^l R,C = NOH + HCHO.
Действие минеральных кислот
на нитропарафины
При действии концентрированных сильных минеральных
кислот нитропарафины дают в качестве продуктов реакции
!йарбоновые кислоты и соли гидроксиламииа с хорошими
выходами. Так, например, этим путем синтезирована уксусная
218
кислота из нитроэтана [18] с выходом 90%, пронионовая из
1-нитропронана с выходом 96% и изомасляная из 1-нитроизо-
бутана с выходом 90%. Одновременно яри этой же реакции
получены соли гидроксиламина с выходом 86—90%.
Для осуществления этого процесса смесь 1 моля нитро-
иарафива и 1 моля 85%-ной серной кислоты нагревали с
обратным холодильником нри перемешивании до наступления
экзотермической реакции, но окончании которой нагревание
продолжали еще в течение 8 час.
Как предполагают, механизм указанной выше реакции
состоит в предварительной таутомеризациинитрояарафиновв аци-
форму (нитроновую кислоту), которая присоединяет по
двойной связи минеральную кислоту; полученное соединение
переходит, теряя воду, в нитрозонроизводное; нитрозопроизводное
перегруппировывается в смешанный ангидрид серной и гидро-
ксамовой кислот, который, подвергаясь гидролизу, дает гицро-
ксамовую (алкилгидроксамовую) кислоту:
О
/
НСН = NO,H + HgSO* ^ RCH — N — ОН -> RGH — N = О -^
Н OS%H
нптрозопронзводвое
g ОН
ангидрид гидронсамовая
серной и гидроксамо- кислота
вой кислот
Гидроксамовая кислота в свою очередь подвергается гидро
лизу с образованием конечных продуктов реакции — карбо
новой кислоты и соли гидроксиламина:
RC =
Некоторым авторам при действии минеральных кислот на
нитропарафижы удалось выделить алкилгидроксамовую
кислоту, которая, согласно указанной схеме, является промежуток
219
аым продуктом реакции*. Бамбергер и Рюст [18] получили
соответствующую алкилгидроксамовую кислоту из феяиляитро-
метана с выходом 2%. Лишшякот и Хэсс [19] синтезировали
пронангидроксамовую кислоту из 1-нитроиропана с
выходом 44%.
Действие разбавленных сильных минеральных кислот на
нитропарафины приводит к синтезу альдегидов и кетонов [20]:
2ДСН = NO,Na + 2Н^О# -^ 2RCHO + 2NaHSO< + N,0 +11,0;
2R.C = NO.Na + 2H,S0, -^ 2R.C0 + 2NaHS0& + N,0 + И.О.
Кальциевые соли аци-нитрокарафинов реагируют с
разбавленными кислотами с лучшими выходами, чем натриевые
соли. Применяя кальциевые соли, Джонсон и Дегеринг 121]
осуществили превращение нитронарафинов в альдегиды и ке-
тоны с выходом 80—85%.
Действие оснований на нитропарафины
При действии оснований на нитронарафивы получаются
триалкилизоксазолы [22]; например, при действии щелочи на
нитроэтан получается триметилизоксазол:
—С С —СЩ
сн —с
о
В отличие от других нитродарафияов нитрометан дает при
действии основании метазояовую кислоту. Последняя реакция,
как предполагают, протекает по следующей схеме:
+Н,0
CHg СН
II + 1
%о,к и
/\
НО ОК
снсн
-> II
кон
* Гидронсамовые кислоты применяются в качестве флотадиоввого
агента для некоторых медных руд.
220
С алкоксвдаый натрия житропарафжны реагируют с
образованием продукта прясоедияевжж основания к н&трогрупие
R' О R' О
R__C — N + R*O№-^R—С —N—О —№
НО Н OR*
Получение галоидояроизводных
нитронарафинов
Галоиднитросоедивения содержат галоид при том же
углеродном атоме, что и нитрогрупна. Они могут быть
использованы в качестве окислителей при различных реакциях.
Некоторые вещества такого типа могут быть использованы как
фумиганты для борьбы с различными вредными насекомыми.
Для получения хлорпроизводных нятропарафяиов
применяются весьма разнообразные методы. При действии хлорной
воды на натриевую соль натромегава получен моножлорнитро-
метан [23]. Хлорированием нитрометана в присутствии водной
суспензии СаСО&, которую, добавляли для поддерживания
слабокислой реакции, получен с количественным выходом
хлорпикрин [24]* Последний продукт может быть также получен из
нитрометана при действии гипохлоритов щелочных и
щелочноземельных металлов [25].
Монохлорпроизводные яитроэтана, 1-нвтропропана, 2-ни-
тропропана и 1-нитро-2-метилярояана получают посредством
пропускания газообразного хлора в водный раствор натриевой
соли аци-нитропарафижа [26, 27]. Хлорнитропарафины
получаются при действии хлористого нитрозила NO^Cl на
ненасыщенные углеводороды [28]. Хлорнитропарафины могут быть
получены и другими, весьма разнообразными путями,
например нитрованием хлорпарафинов азотной кислотой [29, 30],
хлорированием ннтроолефинов [31, 32], действием хлористого
яитроаила или гипохлоритов на оксимы [33], действием РС1*
на нитроспирты [26], m т. д. Разработан метод получения
дихлорнитропарафияов посредством хлорирования нитропа-
рафинов или же их монохлорпроизводнмх [34].
221
При фотохимическом хлорировании иитроалканов в без-
нодвых средах хлор замещает водород, стоящий у атома
углерода, не содержащего N0$. Так натроэтан дает почти
исключительно 2-хлор-1-витроэтан [35].
Изхлорнитропроизводных 1,1-дихлор-1-нитроэтан является
инсектисидом; другие хлорнатропарафины применяются в
качестве селективных растворителей для очистки смазочных
масел [36].
Бромароваже нитропарафинов хорошо изучено; эта
реакция протекает настолько быстро, что ее применяют как
способ определения превращения ннтропарафинов в таутомерную
нитроновую кислоту.
Иодпроизводвые также легко получаются прямым
иодированием жжтропарафивов, но эти соединения
неустойчивы [37].
Механизм реакции галоидироваяия нвтропарафивов
(в присутствии основания) заключается, как предполагают,
в присоединении гаяожда X к нитроновой кислоте или к соли
последней с последующей потерей НХ или МеХ:
R,C = NOgH + Х,-» R,C — NOgH ^ R,CNO, + НХ
R,C = NOgMe + X, -»R*C — NO#e -^ R,CNO% -f MeX
XX X
Конденсация нитропарафинов с
альдегидами (образование нитроспиртов)
Реакция конденсации иитропарафинов с альдегидами,
которая впервые была открыта Анри [38], приводит к
образованию весьма интересных соединений — яитроспжртов, имеющих
большое практическое применение. Эта реакция в общем виде
может быть представлена следующей схемой:
N0, N0%
*R —С —(Ш(0Н)- В"
R'
R'
222
или для случая конденсации нитропарафинов с формальдеги
дом
N0,
RCH,NO, + 2НСН0 -^ R — С — СН*ОН
Конденсация нитропарафинов с альдегидами относится
к альдольному типу и состоит в том, что один, два или три
атома водорода, связанных с атомом углерода, при котором
находится нитрогрунпа, присоединяются к кислороду
альдегида с образованием оксиалкилзамещенных
нитропарафинов. Необходимое условие для осуществления этой реакции
заключается в предварительной таутомеризации нитроиара-
фина в нитроновую кислоту (аци-форму). Значение оснований,
применяющихся в очень слабых концентрациях в качестве
катализаторов этой реакции, заключается в том, что они
повышают концентрацию нитроновой кислоты, сдвигая
равновесие между нитрояарафином и его аця-формой (нитроновой
кислотой) в сторону образования последней по мере того, как
имеющаяся в наличии нитроновая кислота прореагирует с
альдегидом. Эта реакция может быть осуществлена с большей
эффективностью [39], если альдегид заменить его бисульфитным
соединением, а нитропарафин — соответствующей натриевой
солью: бисульфитное соединение освобождает нитроновую
кислоту без повышения концентрации водородных ионов, которые
действуют отрицательно, превращая аци-форму нитронарафина
в его нейтральную форму.
В качестве катализаторов при указанном выше синтезе
нитроспиртов, по данным Вандербильта и Хэсса [40], могут
быть применены основания, карбонаты, метилат и этилат
натрия, сульфит натрия, ацетат натрия и бура. Исследования этих
авторов показали также, что активность щелочного катализа-
тора в большей мере зависит от его рН, чем от концентрации его
в реакционной смеси. Так, например, 0,9%-ный NaOH более
активен, чем Na,CO, более высокой концентрации.
При конденсации нитропарафинов с альдегидами основной
процесс образования нитроспиртов сопровождается следующими
кытро дд;рдд9ыное 223
побочными реакциями; 1) альдольной конденсацией; 2)
полимеризацией реагирующего альдегида; 3) превращением нитро-
парафияа в изоксазолили,в случае витрометана, в метазоновую
кислоту (см. выше, стр. 219); 4) образованием
соответствующего яитроолефива и продукта его полимеризации. Для
устранения первых трех побочных реакций необходимо применять
минимальное количество щелочного катализатора. Последняя
реакция доводится до минимума посредством применения
низких температур, которые ве благоприятствуют возникновению
нитроолефинов (по данным Вандербильта и Хэсса, оптимальной
температурой при синтезе нитроспиртов является 30—35°).
Кроме того, для уменьшения полимеризации поддерживают
низкую концентрацию альдегида, добавляя его к нитропара-
фину постепенно, что одновременно устраняет и возможность
образования оксиальдегидов.
При конденсации формальдегида с нитропарафинами
реакционная способность последних уменьшается в
гомологическом ряду с повышением молекулярного веса. Нитрометан
может легко конденсироваться с тремя молекулами
формальдегида с образованием (HOCHa)gCNOs. При конденсации нитро-
метана с другими альдегидами реакция протекает труднее,
особенно если альдегид содержит более пяти атомов углерода;
|в последнем случае реакция часто останавливается после при-
| соединения одной или двух молекул альдегида.
При конденсации гомологов нитрометана с уксусным аль-
; Дегидом и следующими по гомологическому ряду альдегидами
№олько одна молекула альдегида присоединяется сравнительно
пяегко, другие молекулы присоединяются значительно труднее.
Вторичные нитропарафивы реагируют намного медленней,
|Р*ем первичные, а третичные, которые не имеют атомов
водорода в ос-положении, не реагируют с альдегидами.
! Очистка нитроспиртов от обычной примеси нитроолефинов
Же представляет никаких трудностей, так как нитроолефины
Щегко полимеризуются и могут быть удалены в виде
Щмол [40].
J; Другой интересный метод получения нитроспиртов — зто
Ыяденсация окисей олефинов с нитритами [41—43].
224
Алифатические полинитроооединения такше легко вступают
в реакцию конденсации с альдегидами с образованием поли-
нитроопиртов. Так, при добавлении калиевой соли динитро-
метана к раствору формалина образуется калиевая соль
2,2-дянитропропандиола-1,3, из которой при яодкислении
уксусной кислотой получается с выходом 66% 2,2-динитро-
пропандиол-1,3 [44].
При конденсации 1,4-динитробутана с формальдегидом в
присутствии Са(ОН)% образуется 2,5-бис-(оксиметил)-2,5-дини-
трогександиол-1,6 (1) с выходом 40%. При конденсации тем
же методом 1,5-динитропентана с формальдегидом образуется
с выходом 50% 2,6-бис-(оксиметил)-2,6-динитрогептандиол-1,7
(II). Наконец, при взаимодействии 1,6-динитрогексана
с формальдегидом образуется 2,7-бис-(оксиметил)-2,7-динитроок-
тандиол (III) [45].Реакция протекает по следующему уравнению:
OaNCH* (CHa^CHgNOm + 4СЩ0 -^ С —(CH,)* __ С
HOCHg СЩОН
I. л = 2; II. л = 3; III. л = 4.
Тринитроспирты можно получить взаимодействием витро-
формн CH(NOg)3 с альдегидами или кетонами. Например:
тринитроэтиловый спирт (МО^дССНаОН получается при
взаимодействии нитроформа с формальдегидом [46] или с ц*ра-
формальдегидом [47]. При конденсации уксусного альдегида
с нитроформом образуется вторичный тринитропропиловый
спирт CH,CH(OH)C(NO,), [46].
Тетранитрогликоли получаются при взаимодействии
калиевой соли динитрометана с диальдегидами [48]. Так, при
конденсации калиевой соли динитрометана с глиоксалем
образуется дикалиевая соль 1,1,4,4-тетранитробутандиола-2,3,
которая при действии разбавленной серной кислоты дает 1,1,4,4-
тетранитробутандвол-2,3:
NO, NO*
КНС (NO^b + СНО — СНО ->КС —СН (ОН) СН (ОН) СН
NO,
22b
NO: NO:
НС —СН(ОН)СН(ОН)—CH
Подобным же образом из соли динитрометана и диальдегида
янтарной кислоты получается 1,1,6,6-тетранитрогександиол-2,5.
Из нитросяиртов промышленное применение получили
(HOCH,),CNO:;|
С(СН)т и
Среди соединений, получаемых из нитроспиртов, наибольшее
значение имеют их сложные эфиры и аминосяирты. Сложные
эфиры азотной кислоты могут быть получены при действии на
нитрояарафины охлажденной (от 0 до —20°)
концентрированной азотной кислоты в присутствии серной кислоты, например:
+ 3HNO, —2f (O*NOCH,),
Эфир азотной кислоты и нитроспирта (HOCH^CNOg обладает
большей взрывчатой способностью (на 7% больше
нитроглицерина) [49].
Нитросяирты реагируют с хлорокисью фосфора с
образованием соответствующих сложных эфиров фосфорной кислоты
[501. Сложные эфиры фосфорной кислоты и нитроспиртов
находят применение в качестве пластификаторов для эфиров
целлюлозы. Эфиры органических кислот и нитросяиртов
получаются при действии на последние кислот, ангидридов кислот
1ж хлорангидридов в присутствии серной кислоты. Эти эфиры
применяются в качестве пластификаторов.
Аминоспирты могут быть получены из нитроспиртов
посредством восстановления водородом m *Wx* лдмегий яри действии
юловэ и соляной кислоты или же железа и уксусной кислоты,
также посредством гидрирования. Амйноспирты были
приготовлены гидрированием нитроспиртов при применении в
качестве катализатора палладия в растворе щавелевой кислоты
il]; они могут быть также яолучены в присутствии яикелевого
1тализатора Ренея [52 Ь
16 А. В. Топчиев
226 #мгмробднме уелебо&цю^об дэотной
Из аминоспиртов применяются
NHa; (HOCH*)C(CH,)*NH,;
и (ОН
Эти соединения дают с высшими жирными кислотами мыла,
обладающие высокой эмульгирующей способностью, что
делает их особенно ценными.
Можно предполагать, что алифатические нитроспирты, так
же как и их сложные и простые эфиры, в дальнейшем
приобретут большое техническое значение.
Как указано выше, ароматические альдегиды в присутствии
неорганических оснований конденсируются с нитропарафи-
нами с образованием нитроспиртов. Если же конденсацию
проводить, применяя в качестве катализатора амины [53],
то продуктом реакции будут нитроолефины:
АгСНО + RNH* -> АгСН = Ж + Н*О;
АгСН = NR + RCHgNOg -^ ШЩ + АгСН =
Этим путем можно, например, из бензальдегида и нитроме-
тана [54% легко получить о-нитростирол:
С#НдСНО + CH3NH2 -^ СЛбСН = NCH@ + Н*0;
NCH,
Если конденсация проводится в присутствии алкоголята,
то в первую стадию реакции образуется щелочная соль аци-
формы нттроспирта, при подкислении которой слабой
кислотой выделяется нитроспирт:
АгСНО + СЫ»МО, + КОН ^ ArCH(OH)CH = NO&K + И.О.
При действии на соль нитроспирта минеральной кислоты
образуется непредельное нитросоединение [55, 56]:
АгСН(ОН)СН = NOgK + HC1 -» АтСН = СНМОд + НдО;
ArCH(OH)CH = NO&K + СНзСООН ^ АгСН(ОН)СНдМОд + СН,СООК.
Нитроолефины могут быть получены также из ацетатов
нитроспиртов при нагревании этих соединений в присутствии
227
карбонатов [57]
Н ОСОСН,
I I
2RC — CR
2RC = CR*
СО, + Н*О
Действие на нитроспирты водуотнимающих средств также при
водит к нитроолефинам [58]:
СН& = CHNO*
В качестве водуотнимающего средства рекомендуется
применять фталевый ангидрид, а также сложный катализатор,
состоящий из орто-фосфатов Са и Mg (10%) [59].
Нитроолефины могут быть получены из нитроспиртов при
действии щелочи на нитронитраты, содержащие группы N0%
и ONOg у соседних атомов углерода [60].
Нитроолефины в свою очередь могут подвергаться различным
превращениям; так, например, они могут быть восстановлены
в соответствующие амины. 1-фенил-2-нитропропен-1 при
гидрировании в присутствии никелевого катализатора Ревея дает
соответствующий амин с выходом 40% [61]. Из о-нитростирола
посредством гидрирования в присутствии окиси платины может
быть получен р-фенетиламин с выходом 93%, а 3,4-метвдеядиок-
си-о-нитростирол и З-нитро-ы-нитростирол в присутствии этого
же катализатора гидрируются в соответствующие амины 17],
Каталитическим гидрированием нитроолефинов в
присутствии окиси платины можно получить также предельные
нитросоединения [62].
Действием на нитроолефины железных стружек и соляной
кислоты [31] получены кетоны (при избытке НС1) и кетоксимы
(при минимальном количестве НС1). Оксимы были получены
также при восстановлении нитроолефинов цинковой пылью
и уксусной кислотой [63].
Подробнее о свойствах нитроолефинов см. ниже: гл. Ill,
4
15*
228
Конденсация нитропарафинцв с
альдегидами и аминами (получение нитроаминов)
Реакция конденсации нитроиарафинов с альдегидами
и аминами, которая представляет собой частный случай реакции
Манннха, приводит к образованию нитроаминов — весьма
интересных органических соединений, содержащих
одновременно а амино- и нитрогруппу. Эту реакцию в общем виде
можно представить следующей схемой:
R В
CHNO, + СН$О + R'NHg %5^ С — CH^NH — R*
R' R' NO,
При конденсации первичного нитропарафина, обладающего
двумя подвижными атомами водорода, реакция протекает
о.образованием или нитроамина, или нитродиамина:
RCHaNOs + СНдО + R'NHg -> RCH(NO^CH: NHR' + ЩО;
RCH*NO, + 2СН:О + 2R*NH% -^ R'NHCHa — CR — CH% NHR' + 2H,O.
. , NO.
Третичные нитропарафины или третичные амины, не
имеющие подвижного атома водорода, в реакцию Манниха не всту-
Конденсацию первичных или вторичных нитропарафинов
4; формальдегидом и первичными или вторичными аминами мож-
во проводить двумя способами [64, 65].
.1 . 1^ Формальдегид вначале смешивается с амином, затем к
образующемуся N-метилольному производному амина
добавляется нитропарафин:
СН%0 + RNHs -^ RNH — СНдОЫ
R , R
CHNO: + В*МНСЩОН —!2 С ^
Л 1Е.,Вдачалб при конденсаций нитросарафина с
формальдегидом получается нитроспирт в случае вторичного нитропарафана
229
или нитрогликоль в случае первичного нитропарафина. Полу
ченнын нитроспирт или нитрогликоль вводится затем в
конденсацию с амином:
R
CHNO, + СНдО
R'
С — СНдОН + R'NH, =5
R' NO*
RCH3NO3 + 2СН:0
СН*ОН — CR — CH3OH + Ж
N0%
R
С — СНдОН
R' N0%
R
С — СЩ — NHR"
R' N0*
— CR—СЩОН
NO,
— CR —
NO,
Ароматические амины также вступают в реакцию
конденсации с формальдегидом и первичными или вторичными нитро-
парафинами. Реакция проводится в присутствии сильно
основного катализатора [66].
В случае вторичного нитропарафина, обладающего одним
подвижным атомом водорода, реакция протекает с
образованием ароматического нитроамива, тогда как в случае первич-
k ного витропарафина, обладающего двумя подвижными
атомами водорода, в реакцию с нитропарафином вступают по две
молекулы ароматического амина и формальдегида с
образованием ароматического нитродиамина:
R
+ СН*О + RR'CHNOg
— C —NO: + H3O
R'
2ArNH% + 2СН3О + RCH3NO3
R
C —
NO,
2Н,0
230 #м#цэобд%ме уедеаоДоро&м
Ароматические диамины вступают в реакцию конденсации
с формальдегидом и нитропарафином по обеим аминогруппам:
R R
—С— CHsNHAr NHCH& — С—NO&+2H%O
Нитроамины, полученные из ароматических аминов,
формальдегида и нитропарафина, представляют собой твердые
вещества, обычно нерастворимые в воде и ограниченно
растворимые в насыщенных углеводородах. Они хорошо растворимы
в спирте, бензоле и кетонах.
2,4,6-Тринитротолуол, подобно нитропарафинам, способен
вступать в реакцию Манниха с образованием тринитрофенил-
этиламинов [67]:
N0:
-н,о
R
Выходы продуктов конденсации в большинстве случаев близки
к теоретическим.
Подобно 2,4,6-тринитротолуолу, 2,4-динитротолуол также
способен вступать в реакцию Манниха [68].
Ыитропроизводные гетероциклических основании (нитро-
гексагидропиримидины) также можно получить при помощи
реакции конденсации первичного нитропарафина,
формальдегида и первичного амина, взятых в отношении 1:3:2
№-71]:
231
RCH2NO3 + ЗСНдО ^4
Реакция конденсации нитронарафинов с аминами и
формальдегидом открыла возможность синтеза ряда нитроаминов,
получение которых другими методами связано с огромными
экспериментальными трудностями, а в отдельных случаях едва
ли вообще возможно.
Восстановление полученных по реакции Манниха нитро-
аминов приводит к образованию нолиаминов.
При пиролизе солянокислых солей нитроаминов, полу;
ченных конденсацией первичных нитронарафинов с
формальдегидом и аминами, образуются соответствующие нитроолефи-
ны [72]:
RCH — CHsNR'R'.HCl -^ RC === СН& + R'R'NHHCl
Выход нитроолефинов но этой реакции можно увеличить до
80—90%, заменив хлористоводородные соли нитроаминов
молекулярными соединениями нитроаминов с фтористым бором
173].
При нагревании 1- и 2-нитроиропана с 1-(диметиламино)-
2-нитробутаном (I) образуются 3,5-динитрогеитан (II) и,
соответственно, 2,4-динитро-2-метилгексан (III) с 34%-и22%-ным
выходом. Образование указанных динитропарафинов Снай-
дер и Хамлин [74] объясняют деааминированием нитроамина
с последующим присоединением к получающемуся при этом
2-нитроолефину 1- или 2-нитропропана:
-HN(CH,).
L С,Н,СН — CHN(CH)
NO
2
232
RR'CHNO,
СЩ1 > QHgCH — СН@ — CRR'
N0, ' N0, N0
2 nwg
II. R«=CA
III. В=а' з
Предложенный Снайдером и Хамлином механизм реакция
подтверждается тем, что 1-(диметиламино)-2-нитро-2~метилг
пропан
СН,
N0*
йе содержащий атома водорода у |3-атома углерода, не способен
вследствие этого к дезаминированию и не алкилируется нитро-
парафивом.
§ 2. Получение монопитропарафидов и нитросоединенйй
жирноароматического ряда
Нитрование парафинов и других алифатических соединений
обычно производится не концентрированной, а разбавленной
азотной кислотой. Это объясняется тем, что с
концентрированной азотной кислотой нитрование парафиновых углеводородов
происходит менее гладко, а в основном протекают окислительные
процессы. Смесь серной и азотной кислот, которая применяется
для нитрования ароматических соединений, непригодна при
нитровании парафинов* так как первичные нитропарафиша
быстро гидролизуются горячей серной кислотой, а вторичные
# третичные изомеры осмоляются.
В 1888 г. выдающемуся русскому химику М. И. Коновалову
впервые удалось успешно осуществить нитрование предельных
углеводородов разбавленной азотной кислотой на примере
нонанафтена —одного из циклических углеводородов,
входящих в состав кавказской нефти. Нонавафтен имеет следующую»
структурную формулу:
23&
СЕ —СН,
Н.С
/\
СН
СН - СН,
СИ
До М. И, Коновалова изучением реакции нитрования
циклических предельных углеводородов зажимался ряд
исследователей (Байер, Вреден, В. В. Марковников, Ф. Ф. Бейль-
штейн, А. А. Курбатов и др.). Применяя в качестве
нитрующего агента концентрированную азотную кислоту
(удельный вес не ниже 1,38), эти исследователи получали при
нитровании главным образом продукты окисления углеводородов
при небольших выходах нитросоединений.
В отличие от этих авторов М. И. Коновалов нитровал нона-
нафтен разбавленной азотной кислотой различных концентра*
ций(уд. в. 1,155; 1,075 и 1,036) в запаянных трубках при
120—130°. Оптимальный выход нитропроизводных (51 %) получен
;:М. И. Коноваловым [75] при нагревании до 120—130° вованаф-
II тена с азотной кислотой уд. в. 1,075, взятой в объемном отноше-
1|#ии к углеводороду 4:1. Преобладающими продуктами
редакции нитрования нонанафтена по методу Коновалова оказались
третичные нитропроизводные, образовавшиеся посредством
вамещеняя нитрогруппой атомов водорода в группщ ]>СН;
#*роме третичных в небольшом количестве обнаружены ж
вторичные нитросоедииения.
Опыты по нитрованию нона&афтена, доказавшие возможность
едежя яитрогрушш в предельные циклические угдеводоро-
[, навели М. И. Коновалова на мысль исследовать действие
збавленной азотной кислоты и на предельные углеводороды,
ирного ряда (синтез нитропарафипов прямым нитрованием
леводородов азотной кислотой считался в то время неосу-
ествимым).
В 1893 г. М. И. Коновалов сообщил о результатах своих
щытов по нитрованию алифатических углеводородов разбав-
ой азотной кислотой, которые дали вполне удовлетвори-
%сьные выходы нитропарафинов. Там, при нитровании я* гек-
ва азотной кислотой уд. в. 1,075 при 140° и продолжитель-
234
ности нагревания 4—6 час. получен нитрогексан с выходом
около 40%, а н. гептан и н. октан при действии азотной
кислоты той же концентрации в течение 5—9 час. при 125—130°
дала соответствующие нитросоединения с выходом 47% (для
гептана) и 49—52% (для октана) [76].
При нагревании диизобутила с азотной кислотой уд. в.
1,075 в запаянной трубке при 105—110* М. И. Коновалов [77],
наряду с другими продуктами, получил мононитродиизобутил
При проведении этой реакции при более высокой температуре
(120—125°) им был получен кристаллический динитродиизобу-
ТЙЛ
Аналогично протекает образование нитроирои&водвых при
нитрования диизопропила и диизоамила, причем в случае ди-
изоамила реакция протекает при более высокой температуре.
Имеется указание, что нитронроизводные н. гексана и н.
гептана я наиболее легко н. октана получаются при нитровании
и более концентрированной азотной кислотой (уд. в. 1,42).
Выход моно- и динитрооктана составляет 60—70% от
теории [78].
2-Нитронроизводные высших парафиновых углеводородов
легко получаются при пропускании через нагретый
углеводород паров азотной кислоты [79].
В дальнейшем М. И. Коновалов показал, что разработан-
ный им метод нитрования разбавленной азотной кислотой
может служить ж для введения нитрогруппы в жирную цепь
ароматических углеводородов, вопреки установившемуся взгляду,
что слабая азотная кислота при нагревании действует на жирно-
ароматические соединения слишком энергично, окисляя
боковую цепь, но не нитруя ее.
М. И. Коновалов [80] не только блестяще доказал
возможность нитрования жирной цепи ароматических
углеводородов слабой азотной кислотой, но, варьируя основные
факторы, влияющие на течение реакции нитрования (концентра-
235
цию азотной кислоты, температуру и продолжительность
процесса), выяснил также оптимальные условия этой реакции
для ряда жирноароматических углеводородов. Им было
установлено, что при нагревании алкилированных производных
бензола с разбавленной азотной кислотой в запаянной трубке
нитрогруппа вступает не в ядро, а в боковую цепь. Так,
например, этилбензол при нитровании 12,5%-ной азотной
кислотой (уд. в. 1,075) в течение 9^-11 час. при 105—108° дает выход
фенилнитроэтана 44%. Уменьшение концентрации азотной
кислоты до 6,5% (уд. в. 1,036) приводит к снижению выхода
фенилнитроэтана до 26%. Опыты Коновалова также показали,
что азотная кислота очень низких концентраций (0,25—0,14%)
еще обладает способностью нитровать этилбензол, тогда как
при применении концентрированной азотной кислоты
нитрование происходит лишь при стоянии смеси кислоты с этил-
бензолом при комнатной температуре в запаянной трубке в
течение 45 час. Более эффективно по сравнению с этилбензолом
протекает нитрование пропилбензола: при действии азотной
кислоты уд. в. 1,075 при 105* и продолжительности реакции
4—5 час. этот углеводород дает выход фенилнитропропана 74%.
Пропилбеязол нитруется даже 1%-ной азотной кислотой при
100—105° и продолжительности нагревания 90 час. При
нитровании исевдокумола азотной кислотой уд. в. 1,075 при 110°
и продолжительности реакции 5 час. получена смесь ксилил-
витрометанов с общим выходом нитропроизводных 33%.
Интересные результаты дали опыты М. И. Коновалова [80]
шо нитрованию алкилбензолов (толуола, м- и п-ксилолов, ме-
&итилена и трет, бутилтолуола) слабой азотной кислотой уд.
в. 1,075 (при объемном отношении азотной кислоты к
углеводороду 4:1). Для уменьшения окислительного действия азот-
#ой кислоты М. И. Коновалов проводил нитрование алкилбен-
' золов при температурах около 100°.
I Из указанных выше углеводородов наиболее высокий выюд
1иитроироизводных (около 74%) дал трет, бутилтолуол при
Проведении реакции в течение 4—5 час. при 105°. При нитро-
ёании м-ксилола получен выход нитросоединения 51% (сред-
|Еяя продолжительность реакции 16 час). п-Ксилол, мезити-
236
лен и толуол нитруются в указанных условиях с меньшими
выходами щитропроизводнмх (около 30%). М. И. Ков овалов
в дальже&пем щитровал толуол азотной кислотой уд. в. 1,2
нри продолжительности реакции 48 час. Выход февилнитроме-
тана при этих условиях достигал 43%,
Несколько более высокий выход февилвитрометана
получается нри применении большого избытка разбавленной
азотной кислоты. Процесс проводится следующим образом.
Нагревают толуол с шестикратным количеством разбавленной
азотной кислоты уд. в. 1,075 в запаянвой трубке при 105° в
течение 5 час. Для выделения фенилнитрометана реакционную
смесь обрабатывают раствором едкого кали и экстрагируют
эфиром. При обработке щелочного раствора соли фенилнитро-
метана током углекислоты образуется желтое масло фенилг
нитрометана. Полученный продукт извлекают и после
отгонки растворителя перегоняют в вакууме,
М. И, Коноваловым проводились также опыты по изучению
скорости нитрования алкилбензолов азотной кислотой уд. в.
1,075 при осуществлении реакции в открытых сосудах с
обратным холодильником (отношение HNOa к углеводороду 4 : 1).
Оказалось, что быстрее других углеводородов при кипячении
с азотной кислотой нитруется мезитилен, за ним следует
и-ксилол; сравнительно медленно реагируют бутилтолуол и
м-ксилол и очень медленно толуол.
При нитровании жирной цепи ароматических углеводородов
во всех указанных случаях образуются преимущественно моно-
нитроироизводные. Для получения полинитроироизводных
необходимо применять концентрированную азотную кислоту
и проводить реакцию при охлаждении. М. И. Коновалов
рекомендует, например, следующий способ проведения реакции
для получения динитромезитилена с хорошим выходом; моно-
нитромезитилен (1 ч.) постепенно вносят в охлажденную до
—10** азотную кислоту уд, в. 1,48 (5 ч.); по окончании реакции
продукт вливают в ледяную воду. Для синтеза тринитро-
мевитжлена обрабатывают динитромезитилен (или мононитро-
мезитилен) большим избытком концентрированной азотной
кислоты уд. в. 1,51.
237
Проведением параллельных опытов также было показано,
что нитрование жирноароматических и алифатических
углеводородов в открытых сосудах дает меньшие выходы питропро-
ивводжых, чем нитрование под давлением (в запаянных трубках),
причем в наибольшей степени это различие заметно у
алифатических углеводородов. С азотной кислотой уд. в. 1,2
нитрование парафинов в открытых сосудах протекает очень слабо;
с азотной кислотой уд. в. 1,42 реакция идет более энергично,
но приводит лишь к небольшим выходам нитропроизводных
(смеси моно- и динитросоединений)*.
Основные выводы, вытекающие из классических работ
М. И. Коновалова до нитрованию предельных углеводородов
и жирной цепи ароматических углеводородов, сводятся к
следующему.
1. При действии разбавленной азотной кислоты можно
вводить витрогруппу как в насыщенные алифатические и
циклические углеводороды, так и в жирную цепь ароматических
углеводородов.
2. Основными факторами, влияющими на скорость реакции
нитрования и выход нитросоединений, являются концентрация
азотной кислоты, температура реакционной смеси, а также
продолжительность процесса, причем степень влияния этих
факторов зависит ет природы и строения нитруемого
углеводорода. Опыты по нитрованию предельных углеводородов и
жирной цепи ароматических углеводородов в общем показали,
что чем слабее азотная кислота, тем труднее протекает
образование нитросоединений, и для осуществления реакции
требуются более высокая температура и ббльшая продолжительность
Процесса.
3. Нитрование предельных и жирноароматических углево-
I дородов наиболее благоприятно протекает при повышенном
Давлении. Нитрование в открытых сосудах приводит к сравни-
* Эти результаты не совпадают с опубликовавшими ранее данными
нУорстолла [811, который при проведении реакции в открытых сосудах
Ьолучил сравнителъво высокие выходы нитропарафинов, а именно:
Ы 70% ив нонаяа, 55% из октана и 40% из гептана (ошибочвость этих
результатов признал впоследствии и саЫ Уорстолл).
238
ддотлмой
тельно более низким выходам нитросоедииений (жирноарома-
тические соединения), или же реакция протекает в очень слабой
степени (алифатические углеводороды). С другой стороны, при
взаимодействии углеводородов с азотной кислотой в запаянных,
трубках в течение продолжительного времени натросоединения
образуются даже в том случае, если процесс проводится при
комнатной температуре.
4. При нитровании углеводородов разбавленной азотной
кислотой наблюдаются следующие закономерности в зависи*
мости от строения нитруемого углеводорода:
а) Нитрование ароматических углеводородов в ядро
протекает значительно труднее, чем нитрование предельных
углеводородов. Так, например, бензол не нитруется азотной
кислотой уд. в. 1,075 при 130°, т. е. в условиях, при которых
происходит нитрование парафинов.
б) Циклические предельные углеводороды, содержащие
в молекуле третичные атомы водорода, относительно легко
нитруются разбавленной азотной кислотой. Так, например,,
нонанафтен нитруется при 120—130° азотной кислотой уд. в.
1,075 с выходом 51%; незамещенные иолиметилены, у которых
отсутствует группа Z> CH, по позднейшим данным В. В, Мар-
ковникова, нитруются значительно труднее.
в) Жирная цепь ароматических углеводородов нитруется
легче предельных углеводородов как при проведении реакции
в открытых сосудах, так и под давлением (в запаянных трубках),
так как соседство ароматического ядра активирует, повиди-
мому, реакцию нитрования жирной цепи.
5. Оптимальные результаты как по выходу, так и по чистоте
продуктов получаются при нитровании углеводородов азотной
кислотой уд. в. 1,075 в запаянных трубках при температурах
в интервале 110—140°.
6. Легкость замещения нитрогруппой повышается при
переходе от первичного водорода (в группе — СН,) ко вторич-
повку (в группе ]> СНа); еще легче происходит замещение
третичного водорода (в группе-^>СН). С повышением
концентрации азотной кислоты наблюдается увеличение образования
вторичных и первичных нитросоединений.
239
7. У алифатических углеводородов нормального строения
замещение нитрогрупиой при нитровании азотной кислотой
происходит у второго атома углерода.
8. Нитрование разбавленной азотной кислотой
сопровождается побочными реакциями окисления. М. И. Коновалов
рассматривает эти реакции как процессы вторичного характера
(действие азотной кислоты на первично образовавшиеся ни-
тросоединения). В подтверждение своего взгляда М. И.
Коновалов приводит следующие факты:
а) при перегонке ос-фенилнитрозтанаС@НдСНСНз наблюдается
N0,
превращение его в бензойную кислоту;
б) при уменьшении выхода нитросоединений с
повышением температуры реакции, продолжительности нитрования
или концентрации азотной кислоты соответственно
увеличивается выход тех кислот, которые могут возникнуть из
первоначально образующихся нитросоединений. Так, например,
при нитровании мезитилена азотной кислотой уд. в. 1,155 при
100° образуется ксилилиитрометан (СН„)%СаН^СН,N0,; при
продолжительном кипячении мезитилена с азотной кислотой
получается главным образом мезитиленовая кислота.
Последняя может быть также получена из ксилилнитрометана при дли-
уельном нагревании этого соединения с азотной кислотой
уд. в. 1,155. Другим примером может служить образование трет,
бутилтолуиловой кислоты при нитровании трет, бутилксилола
[82]; эта же кислота получается и при непосредственном
действии окислителей на калиевую соль первичного нитросо-
едищения трет, бутилксилола.
М. И. Коноваловым [83] исследовано также нитрование
жирноароматических соединений азотной кислотой в растворе
уксусной кислоты, причем реакция велась при кипячении
с обратным холодильником в течение 1,5—2 час. При
нитровании этил бензола реакция протекает очень бурно, поэтому
реакционную смесь лишь умеренно нагревают до легкого
выделения окислов азота. Опыты М. И. Коновалова показали, что
240 JTwmpoazwwe у*л**о&уюДо* азотной
исследованные им алкилбензолы обладают различной
реакционной способностью, причем по легкости вступления в реакцию
они располагаются в том .же порядке, как и при нитровании их
водными растворами азотной кислоты. Труднее всего реагирует
трет, бутилбензол, за которым следуют, в порядке повышения
легкости нитрования, о-ксилол, и-ксилол, псевдокумол, этил-
бензол и диэтилбензол. По мнению М. И. Коновалова,
уксусная кислота ослабляет действие азотной кислоты на
ароматическое ядро, направляя это действие на боковую цепь.
Нитрование м-ксилола и мезитилена показало, что чем сильнее
разбавлена азотная кислота уксусной, тем больше получается
первичных и других нитросоединений, замещенных группой N0%
в боковой цепи. При слабом разведении реакция нитрования
направляется главным образом в ядро. Также установлено,
что и в присутствии уксусной кислоты (как и в водном
растворе) реакция нитрования сопровождается окислительными
процессами. Так, например, при нитровании п-ксилола и п-диэтил-
бензола в продуктах реакции обнаружена терефталевая
кислота.
Наличие в изобутане нескольких первичных атомов
водорода и лишь одного третичного давало основание надеяться
получить мононатроизобутан с хорошим выходом, так как, по
М. И. Коновалову, наиболее легко замещается на нитрогруппу
третичный водородный атом, в то время как первичные менее
склонны к этому. Опыт подтвердил ожидания, и, таким образом,
нитроиаобутан впервые был получен нитрованием изобутана
азотной кислотой в запаянной трубке при 150° по методу
М. И* Коновалова [84].
Нитрование предельных циклических и алифатических
углеводородов изучалось также В. В. МарковникОвым [85]. На
основе своих многочисленных исследований он показал, что
нормальные парафиновые углеводороды и особенно парафины,
содержащие четвертичные атомы углерода, лишь с трудом
подвергаются нитрованию ири взаимодействии с разбавленной азот-
цой кислотой. Так, например [86], для превращения изогек-
сааа <СН,),ССН,СН, в нитроизогексан (CHa),CCH(NO,)CH,
и изогептана (СНд^ССНаСНаСН., . в нитроизогептан
241
p.
CHgCHg приходится в течение длительного
времени (27 час.) нагревать углеводород с азотной кислотой
уд. в. 1,235 при 110—115°.
В 1926 г. был опубликован патент [87], согласно которому
моно- или динитросоедвненвя алифатических углеводородов
получаются из соответствующих углеводородов при
нагревании с азотной кислотой при температуре ^ 115° в присутствии
азотнокислого алюминия. Однако, как было установлено
впоследствии [88], азотнокислый алюминий при этом не является
катализатором, и роль его сводится лишь к повышению
температуры кипения азотной кислоты.
При нитровании циклических предельных углеводородов
В. В. Марковниковым показано, что слабой азотной кислотой
нитруются не только соединения, у которых имеются алкилза-
мещенные метиленовые группы, но и незамещенные полимети-
лены. Однако в последнем случае нитрование протекает
значительно труднее, вследствие чего требуется применение более
концентрированной азотной кислоты. При нитровании
азотной кислотой уд. в. 1,235 гексанафтена по методу М. И.
Коновалова в течение 9,5 час. при 100° получено около 18% нитро-
гексанафтена С@НцМО*, считая на взятый в реакцию
углеводород. Одновременно с нитрованием наблюдается реакция
окисления (в продуктах реакции обнаружено большое
количество адяпиновой кислоты).
Изучая действие дымящей азотной кислоты уд. в. 1,52 на
парафины и нафтены, В. В. Марковников [85] установил, что
% азотная кислота этой концентрации действует медленно на
нормальные парафины и очень энергично на парафины,
содержащие группу-^> СИ. Азотная кислота уд. в. 1,52
реагирует относительно быстро с цента- и гексаметиленом и очень
^быстро — с замещенными циклическими углеводородами,
например с гомологами циклопентана. При обработке азотной
кислотой этой концентрации циклических и алифатических
редельных углеводородов получены лишь незначительнее
количества нолинитросоединении; основными же продуктами
реакции оказались продукты окисления: СОз, летучие жирные
кислоты (при нитровании парафинов), двухосновные кислоты
В. Топчиев
242 #wmpoea»w€ уелб*о&ум>#о* д@отмо& кислотой
(при нитровании парафинов и в большем количестве при
нитровании полиметиленов).
В. В. Марковвиков показал также, что нитрующая смесь
(НМОз-Ь HaSO*) не действует на предельные углеводороды
(парафины и вафтенм) при обыкновенвой температуре и лишь
очень медленно реагирует при нагревании.
В. В. Марковников [89] посвятил ряд работ изучению
углеводородов, входящих в состав нефти; в этих работах ов
пользовался реакцией Коновалова как методом исследования,
который дает возможность легко идентифицировать отдельные
углеводороды, руководствуясь при этом в качестве критерия
отмеченной выше различной реакционной способностью этих
соединений к замещению нитрогруппой в зависимости от строения.
Изучение реакции нитрования н. гептана и 2,7-диметилок-
тана сравнительно недавно проводилось А. И, Титовым [90].
Автором было показано, что даже азотная кислота уд. в. 1,42
сама по себе не реагирует с парафинами. Нитрование азотной
кислотой происходило лишь при наличии окислов азота. Были
поставлены параллельные опыты нитрования н. гептана в
трубках как в присутствии, так и в отсутствие окислов азота при
нагревавии в течение 3 час. В первом случае было получено
(из 5,8 г гептана, 10 мл азотной кислоты уд. в. 1,2 и 1 г N,0*)
2,52 г сырого 2-нитрогеятава (после очистки 1,37 г). Во втором
случае при такой же загрузке, но без добавления N,0^ не
наблюдалось избыточного давления, и получены лишь следы
нитросоединения.
На примере, нитрования 2,7-диметилоктава автор показал,
что азотная кислота уд. в. 1,42 не оказывает ускоряющего
влияния на взаимодействие NaO* с этим углеводородом.
Новые стороны реакции Коновалова были выявлены в
результате глубоких исследований С. С. Наметкина.
С. С. Наметкин [91] при исследовании нитрования
предельных углеводородов слабой азотной кислотой обратил внимание
на значение в этом процессе относительного количества азотной
кислоты. Этот факт в отличие от концентрации азотной
кислоты и температуры, влияющих на скорость реакции, определяет
главным образом направление реакции, т.е. характер обра-
243
вующихся продуктов. С. С. Наметкин показал, что подбором
количества азотной кислоты можяо значительно снизить эф-
побочвых реакций окисления и повысить выход нитро-
дннений.
Нитруя гексагидропсевдокумол азотной кислотой уд. в.
^1,3 в открытых сосудах при различных отношениях кислоты
: углеводороду, С. С. Наметкин нашел оптимальные условия
еакции для получения нитропроизводных гексагидропсевдо-
умола с хорошим выходом.
Нитрование производилось в соединенной с холодильником
рленмейеровской колбе, которую нагревали на песчаной бане
о слабого кипения (для устранения толчков и перегревов
жидкость вносили алюминиевую проволочку). По оконча-
реакции, которая обычно продолжалась 6 час, верхний
ой, содержавший смесь углеводорода и яитропродукта, отде-
яли от кислотного слоя, промывали последовательно водой,
одой и затем снова водой и после сушки над хлористым каль-
ием подвергали перегонке с дефлегматором.
Опыты по нитрованию гексагидропсевдокумола дали сле-
ющие результаты: при отношении углеводорода к азотной
слоте 1 : 0,75 выход нитропродукта составляет 65%; прв
еличении относительного количества азотной кислоты вдвое
ход падает до 58—59% ; дальнейшее повышеяие относительно-
количества азотной кислоты (отношение углеводорода
HNOs = 1 : 2,5) приводит к понижению выхода до 45—46%.
Для определения относительных количеств образовавшихся
и нитровании моно- и полинитросоединений С. С. Наметкин,
еле отгонки не вошедшего в реакцию углеводорода, подвер-
л продукт реакции фракционированной перегонке в вакууме
и 40 мм; мононитросоединения при этом давлении перегоня*
сь в интервале 140—145*, поливитропроизводаые определяв
сь в остатке.
Сравнение опытов с различными относительными количе-
ами азотной кислоты показало, что с увеличением отноше-
азотной кислоты к углеводороду количество полинитро-
изводных повышается и, соответственно, понижается вы-
мононитросоединений.
16»
244 #ипфое&н%€ ya/tgeo&yWoa деотной кислотой
Сопоставление полученных результатов с литературными
данными привело С. С. Наметкина к следующим выводам.
1. Действие азотной кислоты является смешанным , т, е.
наряду с продуктами нитрования образуются продукты
окисления.
2. При одной и той же действующей массе азотной кислоты
эффективность нитрования не зависит от ее концентрации
(при соответствующем подборе температуры и давления).
Так, например, с кислотой уд. в. 1,3 (47,5%) в опытах
С. С. Наметкина получен такой же выход мононитросоедяне-
най, как и в опытах М. И. Коновалова в запаянных трубках
с кислотой уд. в. 1,075 (13,5%).
3. Скорость реакции нитрования зависит от трех факторов:
температуры, давления и концентрации кислоты, которые в из*
вестных пределах не влияют на направление реакции. С по-
вышением температуры и давления реакция ускоряется и тем
сильнее, чем концентрированнее кислота. С азотной кислотой
уд. в. 1,3 реакция протекает при атмосферном давлении о
практически приемлемой скоростью; при меньшей концентрации
(уд. в. 1,075—1,055) реакция идет медленно, вследствие чего
требуется применение давления.
4. Относительное количество азотной кислоты и
продолжительность процесса влияют главным образом на направление
реакции, т, е. на характер получающихся продуктов. При
продолжительном кипячении с большим избытком азотной
кислоты реаццщя приводит к преобладанию продуктов окисления.
С уменьшением относительного количества азотной кислоты
и продолжительности нагревания выход нитропродуктов
повышается.
Исходя из найденной им зависимости течения реакции
нитрования предельных углеводородов от указанных факторов,
С. С. Наметкин в дальнейшем успешно осуществил нитрование
гексаметилена(гексанафтена)*. При проведении реакции под
давлением посредством кипячения в грушевидной запаянной
колбе равных объемов углеводорода и азотной кислоты уд, в. 1.3
* Гексанафтен представляет собой основную часть фракции
кавказской нефти с т. кип. 80—82".
24$
С. С. Наметкин получил выход нитрогексаметилена 58,6% от
Теоретического.
! С. С. Наметкин проводил также нитрование синтетического
гексаметилена, приготовленного гидрированием из бензола»
рагревая его с азотной кислотой уд. в. 1,2 при 105—110° под
Давлением (в запаянных трубках). Опыты показали, что при
увеличении относительного количества азотной кислоты в 4 ра-
ва (в первой серии опытов отношение количеств азотной кислоты
ж углеводорода составляло 1,5 : 1, во второй 6 : 1) выход ни-
^ропроизводных падает с 49 до 36%, причем соответственно
повышается выход продуктов окисления (органических кислот).
Для выяснения правильности положения, высказанного
|'еще М. И. Коноваловым, что окисление является процессом
Вторичного характера, С. С. Наметкин изучил действие окисли*
телей, в частности азотной кислоты, на продукт описанной выше
реакции — нитрогексаметилен. Оказалось, что при действии
азотной кислоты уд. в. 1,3 на нитрогексаметилен реакция
протекает довольно медленно, причем выход адипиновой кислоты
получается в 2 раза меньше, чем при непосредственном
действии азотной кислоты на гексаметилен. С другой стороны,
;если вносить в кипящую азотную кислоту нитрогексаметилен
|р слабощелочном растворе, в котором это соединение находится
|В виде соли соответствующей аци-формы (изонитросоединеная),
реакция протекает очень быстро, приводя к выходу адипино-
вой кислоты 83,3%. Из этих фактов С. С. Наметки* делает
вывод, что окислению подвергается не нитрогексаметилен,
'а продукт его превращения — изонитрогексаметилен, который
(Является очень нестойким соединением.
Окисление нитропроизводного в виде аци-формы (изонитро-
;-,еоединения) в органическую кислоту происходит, невидимому,
керез промежуточное образование альдегида (если лабильная
форма соответствует первичному нитропроизводному) или ке-
|*она (в случае вторичного нитросоединения):
2RCH = N — ОН -> 2RC + Н,0 + N„0 (1)
246 #илфобяние удлббО&уюДое азотной
2R.C = N —ОН ^ 2ВзС = O + N,O + H.0 (2)
II
По представлениям С. С. Наметкина, при действии азотной
кислоты на предельные углеводороды в первой фазе
образуются изонитросоединения, являющиеся нестойкими в кислой среде
и при высоких температурах. Эти изонитросоединения либо
переходят в стабильную форму нитрососдинепий, либо через
альдегиды и кетоны, образующиеся по приведенному выше
уравнению, окисляются в соответствующие карбоповые кислоты.
Образование промежуточных продуктов окисления
наблюдалось М. И. Коноваловым [92], который выделил альдегид
1,6,6-триметилгептиловой кислоты при нитровании диизо-
амила крепкой азотной кислотоц, а также С. С. Наметкиным
при нитровании кавказской нефти (фракция с т. кип.
142-144°).
Промежуточные изонитросоедипения образуются лишь при
нитровании углеводородов, содержащих водород, связанный
6 первичным или вторичным атомом углерода:
+ O = N=O->RCH = N = 0 + H,0 -> RCH,NO, fl)
ОН ОН
(лабильное ичо-
нитрососдинсние,
или аци-форма)
R,CH, + О = N = O^R^C = N = O -> R.CHNO, (2)
I I
он он
I I (стабильная
он он ^LS
Углеводороды, содержащие водород, связанный с
третичным атомом углерода, при нитровании образуют сразу
соответствующее стабильное нитросоединение:
RgCH + HOMO, ^ R,CNO, 4- Н,0.
С. С. Наметкин и А. С. Забродина [93] изучали нитрование
изооктана [2,2,4-триметилпентана (СН^ССНзСЩСН^а] по
методу Коновалова в запаянных трубках. В каждую трубку
загружалось 5 г углеводорода, 35 мл азотной кислоты уд. в.
1,075 и нагревалось при температуре 143—147° в течение
20 час. с перерывом через каждые 5 час. для спуска давления.
247
Верхний масляный слой промывался раствором бикарбоната.
Из 315 г 2,2,4-триметилпентана До лучено:
нитрометан и изооктан, не вошедший в реакцию. 138 г
2.2-диметилпентанон-4 (СН,),ССН,СОСН, 1,7»
2,2,4гтримстилпентаяон-3 (СН,),ССОСН(СН,), 5»
(образование этого кетона рассматривается как результат
распада, получающегося в качестве промежуточного продукта,
вторичного изонитрооктана).
ацетон небольшое количество
вторичный нитроизожтан (СН,)@ССН(МО#)СН(СН,)д. , 2,4 г
третичный нитроизооктая (CH@)gCCH^C(NOa)(CHg)%. * . . 60 ж
дшштроивооктап (CH,)@CCH(NO#)C(NO,)(CH*)9,. . 5,1 ж
Ранее уже упоминалось, что при нитровании нормальных
парафинов слабой азотной кислотой (13,15%) на примерах ге-
ксана, гептана и октана М. И, Коновалов показал, что едвж-
ственными продуктами этих реакций являются вторичные
Р-нитросоединения. Изучая действие слабой азотвой кислоты яа
октадекан (С^Ндд) при иагреважии в запаянвых трубках (по
Коновалову) С. С. Наметкин, С. С. Нифонтова и Р. Я. Сущик
[94] установили, что единственным продуктом реакции
оказалось вторичное Р-нитросоединенже СН,(СНж)1бСН(МО*)СН,,
т. е. образуется соединение, аналогичное соединению, получеж-
ному М. И. Коноваловым из гексана. При действии рбвбавлек-
ной азотной кислоты на гексатриаконтаж (C@,H,J в
аналогичных условиях главным продуктом реакции оказалось двувто-
ричное р,В'-динитросоединение СН,СН(МО*)(СН))мСН(МО,)СНэ,
т.е. нитрование идет с обоих концов молекулы, причем обе ни-
трогруппы становятся в ^-положение.
Выход динитропроизводного в два раза больше выхода моно-
нитропроизводного. На основании своей работы указанные
авторы высказывают предположение, что до известного предела
молекулярного веса единственным продуктом реакции
нитрования нормальных парафиновых углеводородов является
соответствующее вторичное (3-нитросоединение. В случае
превышения некоторого предельного молекулярного веса главным
продуктом реакции нитрования no M. И. Коновалову становится
348
,р'-динитропроизводяое, являющееся, повядимому, продуктом
дальнейшего нитрования мояонитропроизводного.
Однако, как было показано Пони и Костаческу [95], ивопен-
тан яри нитровании HNOa (уд. в. 1,38—1,42) в запаянной трубке
при 140* дает, наряду с 2-нитро-2-метилбутаном, 2,3-динитро-
2-метилбутан (CHg)^C(NOi)CH(NO,)CH@, а также с
незначительным выходом 1,2,3-тринитро~3-метилбутан (CH*)/Z(NO,)
CH(NO#)CH^O. (с т. ш%. 189-190*).
При нитровании изооктана разбавленной азотной кислотой
уд. в. 1,075 яо методу Коновалова (в запаянных трубках)
С С. Наметкин и А. С. Забродина [96] выделили ив кислотного
сдоя: уксусную, изомасляную, триметилуксусную, трет, бутил-
ужсусяую, *,%-диметилянтарную кислоты и следы щавелевой.
Эти же авторы [97, 98] на примере изооктана изучали
взаимосвязь между реакцией нитрования и окислением азотной
кислотой углеводородов предельного характера.
Основываясь на том, что предельные нитросоединеяия
весьма устойчивы к азотной кислоте даже при кипячении и ожщслв-
ются значительно медленнее соответствующих предельных
углеводородов, авторы приходят к выводу, что промежуточными
продуктами при окислении предельных углеводородов азотной
кислотой являются же нитро-, а изояитросоединения.
Последние очень неустойчивы в кислой среде. Образуясь в качестве
начальных продуктов реакции, изонитросоединения либо
тотчас же изомеризуются в соответствующие витросоедижевия»
либо разлагаются с образованием закиси азота я альдегидов,
которые в свою очередь окисляются в карбоновые кислоты.
В случае изооктана образуется изонитроиаооктан, затем
вторичный яитроизооктан (2,2,4-триметил-З-яитрояевтан),
кетон — 2,2,4-триметиляентанон-З и продукты его окисления.
Если же действие азотной кислоты направлено ра третичный
водород, то сразу образуется третичное нитросоедияение.
Третичные алифатические нитросоедижения могут являться
промежуточными продуктами при переходе к продуктам окжс-
леяшя.
С. С. Наметкиным была впервые детально исследована
реакция нитрования азотной кислотой предельных бицжклических
249
углеводородов. Вследствие трудной доступности эти соединения
оставались долгое время совершенно не изученными.
С. С. Наметкин установил, что бициклические углеводороды
обладают некоторыми особенностями, отличающими их от
парафинов и моноциклических нафтенов.
Как уже упоминалось выше, при нитровании парафинов
и нафтенов, содержащих третичный атом водорода, основным
продуктом реакции является третичное нитросоединение.
Большинство бициклических углеводородов содержит третичный
атом водорода, стоящий у атома углерода, общего обоим циклам
системы. Исследования С. С. Наметкина показали, что при
нитровании слабой азотной кислотой бициклических углево*
дородов, состоящих ив двух сочлененных пятичленных циклов»
эти атомы водорода никогда не замещаются на иитрогруппу.
Третичные витросоединения могут быть получены только и&
углеводорода, содержащего группу > СН — СН@. Например»
при нитровании изоборнилана [99] был получен 2-нитроизобор-
нияая следующего строения:
Н^С — СН —- ' CHg
С —СН,
Ж),
Интересно отметить, что при нитровании бициклических
нафтежов, состоящих из двух шестичленных циклов или из^
одного шестжчленного и одного пятичлеиного, образуются
третичные нитросоединения с нитрогруппой у атома углерода,,
общего обоим циклам. В результате нитрования декалина
С. С. Наметкин [100] получил третичный нитродекалин, а прв
нитровании гидриядана [101 ] — третичный нитрогидриндав.
Другая характерная особенность бициклических
углеводородов, установленная С. С. Наметкиным,заключается в их
способности к образованию структуряо-иаомерных нитросо-
единений. При нитровании парафинов и моноциклических
нафтенов замещающее действие азотной кислоты имеет строга
определенное направление, так что из числа возможных
вторичных нитросоедииений образуется всегда только одно.
250
уалб*оЯоро#о* даотном
Нитрование нормальных парафиновых углеводородов приводит
к образованию р-нитросоединений. В жирноароматических
углеводородах нитрогруппа становится в %-пояожевие по
отношению к бензольному кольцу. При нитровании бициклических
углеводородов замещающее действие азотной кислоты
направлено одновременно на оба цикла системы. В результате
нитрования фенхана [102] было получено два нитросоедижеаия—
2-иитрофенхан и 6-нитрофенхан:
НаС
СН
I *
СН,
1%
СН
СНд
СН,
НС
СН
4
н,
/
СН,
Два вторичных структурно-изомерных вятросоедивенжа
были подучены также при нитровании камфевилажа [103].
Нитрование бициклических углеводородов иногда приводит
к возникновению смеси стереоизомерных нитросоединещй.
При нитровании симметрично построенного углеводорода кам-
фана [104] были получены ос-и %'-нитрокамфаны, которые при
окислении дали один и тот же кетон—недеятельную камфару.
Смесь стереоизомерных нитросоедижений получалась также при
действии азотной кислоты на изокамфаа [105].
Интересные результаты были получены С. С. Наметкиным
и А. С. Забродиной [106] при действии азотной кислоты на
предельный трициклический углеводород — трициклен.
Единственным азотсодержащим продуктом нитрования трицивлена
неожиданно оказалось непредельное нитросоедивеяже а-ни-
трокамфен. Образование %-нитрокамфена из трицинлена
объясняется С. С. Наметкиным при помощи следующей схемы:
1. Первоначально происходит размыкание трехчленного
цикла по связям 1—2 или 1—6, и азотная кислота
присоединяется по месту освободившейся валентности, образуя нитро-
спирт.
2. Затем неустойчивый нитроспирт дегидратируется,
превращаясь в %-нитрокамфен.
Процесс выражается уравнением:
Приведенная схема реакции была распространена С. С.
Наметкиным на процесс нитрования углеводородов, не
содержащих трехчленного цикла.
Реакция протекает в две стадии:
1) присоединение углеводорода к азотной кислоте,
2) отщепление воды от промежуточного продукта.
Например, в случае присоединения азотной кислоты к цик-
логексану отщепление воды может идти в двух направления*:
а) с образованием вторичного ндтросоединения, б) с
образованием изонитросоединения, которое затем разлагается на кетой
и закись азота:
+IINO,
Циклогексан
—
Иэоммтроцикло-
гексан
СлН,
lI
Аналогичные превращения наблюдаются при
присоединении азотной кислоты к метилцжклогексану.
Отщепление воды от продукта присоединения азотной
кислоты к третичному атому углерода может привести к образованию
только третичного нитросоединения:
+HNO,
а
252
С. С. Наметкин высказал предположение, что нитрование
ароматических углеводородов также проходит через стадию
присоединения углеводорода к азотной кислоте. Ароматические
нитросоединения образуются при отщеплении воды от
промежуточного продукта
Л Л -н.о
СД, + N-O-» CAN — ОН >
Изучением нитрования жирноароматических соединений
занимались также П. П. Шорыгин и А. М. Соколова [107]^
которые в качестве нитрующего агента применяли азотную
кжолоту в растворе уксусной кислоты.
Работа этих авторов имела своей задачей найти наиболее
благоприятные условия нитрования жирной цепи толуола для
получения фенилнитрометаиа. Опыты производились как
в запаянных трубках, так и в открытых сосудах. В первом
случае смесь 10 мл толуола, 10,7 мл азотной кислоты уд. в.
1,5 и 29,3 мл ледяной уксусной кислоты нагревали в течение
24 час, причем был получен фенилнитрометан с выходом 14,5%.
Опыты при атмосферном давлении проводили следующим
образом: смесь 150 мл (130 г) толуола, 120 мл азотной кислоты
уд. в. 1,5 и 480 мл ледяной уксусной кислоты нагревали в колбе
с обратным холодильником на масляной бане до 110° в течение
3,5 часа. При этих условиях, которые на основании
предварительных исследований оказались оптимальными, выход фе*
яилнитрометана составлял 14%. Одновременно происходят
также нитрование ядра (выход нитротолуолов 24%) и
образование продуктов окисления (выход бензойной кислоты 31,5%).
Обработка продуктов реакции производилась П. П. Шоры-
гиным и А. М. Соколовой следующим способом. Выделившийся
при вливании реакционной смеси в воду маслянистый слой
взбалтывали с 5%-ным раствором соды для извлечения
бензойной кислоты. Оставшееся масло, содержавшее
фенилнитрометан и нитротолуолы, перегоняли с водяным паром.
Перегнанное масло обрабатывали 20%-ным раствором NaOH для
извлечения фенилнитрометаиа, после чего оно подвергалось пе-
HwmjDocoe#wN<?HwzZ жырнодролмтычбсжодо jpw^a 253
регонке, причем сначала переходил толуол (109—111°), а за-
^гем о- и п-нитротолуолы (217—238°). Из щелочного раствора
'фенилнитрометан выделяли при подкислении 20%-ным раство-
фом соляной кислоты.
Подробное исследование и раскрытие механизма нитрова-
жия предельных и жирноароматических углеводородов
принадлежит А. И. Титову.
Рассматривая механизм нитрования предельных углеводоро-
щов и их производных, А. И. Титов [108] приходит к следующим
выводам.
1. При нитровании парафинов азотная кислота не проявляет
самостоятельного химического действия, но служит лишь
источником возникновения и регенерации окислов азота.
2. Активным химическим агентом в этой реакции служит
исключительно лшпь радикалоподобная молекула мономерной
формы двуокиси азота N0%.
В противоположность бесцветной и диамагнитной N %О&
мономерная форма N0% парамагнитна и окрашена, что и
доказывает радикалоподобный характер структуры ее молекулы.
3. Начальным элементарным химическим процессом
нитрования парафиновой цепи является возникновение
углеводородного радикала при взаимодействии мономерной формы
двуокиси азота с исходным органическим соединением по схеме
На основе экспериментальных данных А. И. Титов считает,
что предельные углеводороды и их остатки в других
соединениях, как правило, реагируют со свободными радикалами (А)
с незначительной энергией активации (не больше 10 ккал/моль)
с отрывом атома водорода и образованием углеводородного
радикала по схеме
Тот факт, что скорость реакции нитрования возрастает
параллельно увеличению относительной стабильности ради-
кала R и параллельно уменьшению энергии активации отрыва
254
атома водорода, является подтверждением начального
возникновения радикалов,
4. Образовавшиеся в первой стадии реакции алкильные
радикалы быстро реагируют с радикалоподобпыми молекулами
окиси и двуокиси азота с образованием нитрозо- и нитросо-
единений и алкилнитритов ио схеме
R"+NO-*RNO; (1}
Наиболее вероятными превращениями и взаимодействиями
свободных радикалов с другими компонентами реакционной
системы, по мнению Титова, являются:
а) R Ч- N3O4 -» RONO + N0%;
б) R' + N,O,-*RNO + NO,;
в) R" + N,0, -^ RONO + NO;
г) R" f N*O,^RNO% + NO;
д) R" + HONO2 -^ ROH + NO,.
Однако осуществление таких превращений
углеводородных радикалов маловероятно и приобретает значение лишь при
очень значительных концентрациях N&0*, N#0*, HNO^ или при
высокой темпе рату р е.
Автор утверждает, что все разнообразие конечных
продуктов взаимодействия окислов азота и азотной кислоты с
предельными углеводородами и их производными есть результат
дальнейшего превращения в условиях нитрования нитрозосоедине-
ний, эфиров азотистой кислоты, спиртов и в меньшей степени
нитросоединений.
Особенности нитрования азотной кислотой в первую
очередь определяются состоянием равновесия, которое выражается
схемой 2НМ0в + NO ^± 3N0* + Н%0. и факторами, от
которых зависит установление этого равновесия.
На основе экспериментальных данных А. И. Титов доказал,
что азотная кислота в отсутствие окислов азота практически не
реагирует ни с нормальными парафинами, ни с изопарафинами,
ни с циклопарафинами, ни с боковой цепью алкилбензолов.
ныщросоеДыненмД жмр моа^олтмгцчбсжого jwA% 255
В присутствии окислов азота химическое действие возрастала
параллельно увеличению содержания двуокиси арота.
Изучением действия разнообразных смесей окислов азота (N0,
N0%, N%Os, N,Oi), азотистой и азотной кислот автор установил,
что химически активную роль при нитровании продельных
углеводородов играет только мономер двуокиси азота N0%,
Насыщение реакционной смеси окисью азота уменьшает
скорость реакции в соответствии со сдвигом равновесия
МО*+МОз±^*О, слева направо.Если же вводить в реакциоявук»
смесь кислород с целью удаления окиси азота из сферы
реакции, то это приводит к ускорению реакции.
Скорость нитрования парафиновых углеводородов
определяется скоростью начальной стадии образования
углеводородного радикала. Эта реакция относится к молекулярно-ради-
кальному типу, что доказывается тем, что увеличение объема
кислотной фазы не сказывается на течении реакции; при
увеличении объема углеводородной фазы степень прохождения
реакции увеличивается. Отсюда следует, что нитрование
парафинов азотной кислотой протекает не в кислотной фазе, а в
углеводородной. Это дает объяснение известным фактам того, чта
а) скорость реакции заметно не меняется в зависимости от
степени полярности среды, б) присутствие сильных протонных
и апротонных кислот (H*SO*, А1С1з,Т1С14)не ускоряет
нитрования парафиновой цепи.
А. И. Титов [109, НО] предполагает, что аналогично с
предельными углеводородами основными агентами, титрующими
жирную цепь ароматических углеводородов при действии азот-
дой кислоты, являются окислы азота (N0* и N0),
находящиеся в сфере реакции. Роль азотной кислоты сводится
лишь к регенерации двуокиси азота и повышению ее
концентрации посредством окисления низших окислов азота,
образующихся во время реакции.
В начальной фазе нитрования под влиянием нитрующего
агента происходит первичный распад углеводородов с
образованием свободных радикалов:
N0% -» САСН", -f- HNO,.
256
Правильность предположения о возникновении свободных
радикалов при действии N0% на жирноароматические
углеводороды подтверждается наблюдением автора [111] о сильном
возрастании скорости реакции в ряду: толуол, дифенилметан,
трифеннлметан — параллельно легкости образования
соответствующих радикалов. Свободные радикалы далее реагируют
с радикалоподобными молекулами окиси азота и двуокиси
азота
CACH'+NO -*С,Н,СЩМО; (I)
+ N0* -^ С@Н@С%01Ч О.
(III)
Образующиеся по схеме (I) нитрозосоединения дают в
дальнейшем динитропроизводные, причем реакция проходит через
промежуточную стадию превращения нитрозосоединений в оксимы
СаН@СН = NOH;
= NOH
(1)
(2)
[ Изучая продукты реакции, полученные нитрованием
толуола только N0%, с одной стороны, и азотной кислотой
(содержащей N0%), с другой, А. И. Титов получил следующие
данные (табл. 13).
Таблица 13
NOg u азотной
Продукт реакции
Фенилнитрометан . . .
Фенялдинитрометан . .
Бензойная кислота . .
Остаточное масло . . .
Содержание, мол. %
Нитрование
N0,
52,5
2,7
11,2
33,6
Нитрование
HN0,
55,2
3,7
11,9
29 2
257
Таким образом, в обоих случаях получается смесь
практически одного ж того же состава.
Опыты А. И. Титова показали, что при нитровании жирной
цепи ароматических углеводородов в зависимости от условий
образуются преимущественно либо w-мононитросоединения,
либо w-динитросоединения. Образование обоих соединений
протекает по двум совершенно различным путям, и, таким образом,
мононитросоедивения не являются промежуточной ступенью
образования гем. динитросоединений. Это доказывается тем,
что при действии двуокиси азота на фенилнитрометан он
остается без изменения, действие разбавленной азотной кислоты
приводит к образованию бензилового спирта и бензойной
кислоты, а в результате действия крепкой азотной кислоты про-
исходит нитрование в ядро. При низких температурах
получаются главным образом динитропроизводные. Например,
смесь 100 мл азотной кислоты (уд. в. 1,42), содержавшей 0,3 г
двуокиси азота, и 1 л толуола оставляли стоять в неплотно
закрытой колбе при температуре около 25* на 20 дней. При
этих условиях основным продуктом реакции оказался февил-
динжтрометан. Повышение температуры до 50° приводит к
увеличению выхода феиилнитрометааа и к понижению выхода фе-
вилдинатрометава. При температуре около 100° из продуктов
нитрования боковой цепи образуется лишь фенилнитрометав.
Например, к 500 мл толуола при нагревании в колбе с
обратным холодильником на кипящей водяной бане в течение 2,5
часа прибавляли 300 мл азотной кислоты (уд. в. 1,38). В
результате реакции получен фенилнитрометав с выходом 52% наряду
с продуктами окисления: бензойной кислотой (выход 24,5%)
и беязальдегидом (выход 3%).
Понижение выхода фенилдинитрометана с повышением
температуры объясняется, по А. И. Титову, понижением
растворимости окиси азота, необходимой для образования нитрозо-
соединевий, и повышением диссоциации NgO*, превращающей
оксимы в динитропроизводные (см. выше схему образования
последних).
Другим фактором, отрицательно влияющим на выход
фенилдинитрометана, является высокая концентрация N0%, которая
17 А. В. Топчиев
258 #итроеян%г удлеео&цю&ю
приводит к уменьшению возможности столкновения свободных
радикалов с N0 и к понижению концентрации последней
вследствие сдвига равновесия слева направо в уравнении
С другой стороны, повышение концентрации окиси азота,
например, при специальном насыщении ею реакционной смеси
приводит к повышению выхода фенилдинитрометана.
Образование w-мононитросоединений происходит при
действии мономерной молекулы N0% (при применении большого
избытка нитруемого углеводорода N0% растворяется в
последнем). В начальной стадии нитрования присутствуют лишь
незначительные количества двуокиси азота, растворенные
в азотной кислоте (для повышения этой начальной
концентрации А. И. Титов обычно добавлял к реакционной смеси триокси-
метилен, который при взаимодействии с азотной кислотой
легко образует N0%),
Приводим описание некоторых опытов А. И. Титова по
нитрованию толуола на фенилнитрометан, давших хорошие
результаты.
В реакционную колбу загружали толуол, 0,5 г триоксимети-
лена и буферную смесь из 8 г азотной кислоты уд. в. 1,38 и 40 г
50%-ной серной кислоты. Затем при нагревании на водяной
бане в течение 3,5 часа в нитрующую смесь приливали из
капельной воронки 50 мл азотной кислоты уд. в. 1,5. Таким
образом, крепкая азотная кислота не соприкасалась с толуолом,
чем устранялась возможность нитрования ядра.
В этих опытах при варьировании количественного
отношения толуола к азотной кислоте оптимальные результаты
(выход 54,6%) получены при употреблении 1750 мл толуола на
50 мл азотной кислоты.
Обработка продуктов реакции по окончании опыта
производилась следующим образом. После промывки водой
углеводородный слой энергично перемешивали 4 часа с 200 мл
20% -ного раствора бикарбоната калия, причем в течение второго
и третьего часа дополнительно было прибавлено 50 мл 50%-ного
раствора поташа. Выпавшую при этой операции калиевую соль
259
фенилдинитрометана отсасывала и затем промывали водой,
спиртом и эфиром. Из раствора оставшийся фенилдинитроме-
таи выделяли бромом в виде фенилдинитробромметана. После
отделения последнего выделяли из маточного раствора
бензойную кислоту.
Для извлечения фенилнитрометана реакционную массу
перемешивали 4 часа со 100 мл 10%-ной щелочи.
А. И. Титов приходит к выводу, что для успешного
нитрования толуола азотной кислотой в боковую цепь требуются:
1) начальное присутствие в сфере реакции окислов азота,
2) накопление их до известного предела при помощи азотной
кислоты,
3) создание условий, препятствующих нитрованию азотной
кислотой ароматического ядра и деструктивному действию
на образующийся фенвлнитрометан.
Окисление парафиновой цепи окислами азота и азотной
кислотой при нитровании предельных углеводородов нередко
является одной из основных реакций. Механизм этих реакций
А. И. Титов представляет следующим образом:
RONO + HgO ^ ROH + HNO,;
ROH
алиилввтрат
Создавая благоприятные условия для сохранения спиртов,
их азотисто- и азотнокислых эфиров, можно направить
реакцию в сторону получения этих продуктов в значительном
количестве. Так, например, при применении большого избытка
углеводорода, воду отнимающих средств и при введении
кислорода для устранения окиси азота из сферы реакции можно
направить взаимодействие между толуолом и N0% при обычной
температуре в сторону преимущественного получения бензил-
нитрата, бензилового спирта, а из него кислоты (схема,
стр. 260).
Свою теорию нитрования парафиновых и жирноароматиче-
ских углеводородов А. И. Титов [112] подтвердил и на опыте
нитрованием дифенилметана и циклогексана.
17*
260
RNO »- оксниы
NO, ,^NOi, N,O,
RONO,
ROM »" кетоны и альдегиды
КИСЛОТЫ
В зависимости от применяющихся условии реакции глав-
ним продуктом нитрования дифенилметана является дифенил-
мононитрометан или дифенилдинжтрометан. Основные пути
образования этих продуктов автор представляет следующим
образом. Вначале из дифенилметана и N0% образуется бензгидрил:
(CAhCHH + NOg ^ (СбН&)зСН' + HN0,.
далее (СеН*),СН' + NO.^(CA),CHNO%,
или
+ NO ^ (C@H,),CHNO -», (СН,)*С = NOH -^ (С A
Наряду с нитропродуктами автор доказал образование
продуктов окислительного направления реакции, как то: бенз-
гидрилнжтрата, бензгидрола, дибензгидрилового эфира и бен-
зофенона, по схемам
ONO, N,Oa, N,0.
а) (СН,ЬС1Г ^ (СбНв^СН — 0N0 ^
+н,о што,
^ (С$Н»кСНОН 7—^ (СаНбЬСНОМОж;
-нмо, н.о
б) %С,Н,),СНОН ^=± (СбНжкСН - О - СН (С,Н,),;
N0,
в) (С@Нб),СНОН > (СаН»),СО + N0 + Н/).
В одной из работ последнего времени ва :примере
нитрования циклогексана азотной кислотой и окислами азота
261
А. И. Титов [113] показывает, что при увеличении отношения
NaO* : NO» все ббльшую роль при реакции гексильного
радикала с димером двуокиси азота играет образование циклогек-
силнитрита
СбН^-|- О = N "N = O^CgHnONO + NO. (1)
Образовавшийся циклогексилнитрит в кислой среде
реагирует с водой с образованием циклогексанола, который легко
окисляется в циклогексавон и затем при более глубоком
окислении дает адипиновую кислоту. При применении большого
избытка циклогексана и продолжительности процесса 1 час автор
получил смесь следующего состава:
Мол. %
яятрациклогексан 50
цжнлогексанол 8
циклогексилнитрит 8
другие эфиры 14
адипивовая кислота 20
С увеличением общей концентрации димера двуокиси азота
выход продуктов окисления в соответствии с возрастанием роли
реакции циклогексила с N*O* по уравнению (1) повышается
значительно больше, чем выход нитроциклогексана.
Положительная роль повышенного давления при нитровании
предельных и жирноароматических углеводородов наблюдалась
М. И. Коноваловым, который отмечал резкое падение выхода
нитросоединений при проведении процесса в открытых сосу-
. даж. А. И. Титов провел интересную работу по изучению
роли герметизации реакторов и процессов диффузии при
нитровании предельных углеводородов азотной кислотой в жидкой
фазе. Активно нитрующий агент — двуокись азота при
нитровании углеводородов по методу Коновалова остается
преимущественно в углеводородном слое, где и протекает реакция.
Скорость реакции, следовательно, определяется содержанием
N0% в этом слое. Газообразная окись азота, которая также
остается в сфере реакции, способствует поддержанию необходимой
262
концентрации двуокиси азота благодаря ее участию в
равновесии
2HNO, + NO=± 3NO, Ч- Н,О. (I)
В начале реакции содержание двуокиси азота в
углеводородном слое, особенно при нитровании разбавленной азотной
кислотой, очень незначительно. По мере течения реакции
RH + NO*-*R" 4- HNO, (II)
содержание N0% в углеводородном слое падает, и для
поддержания реакции необходимо постоянное пополнение расхода
N0%. Таким источником пополнения N0% является кислотный
слой, куда проникает N0 из углеводородного слоя.
Регенерация двуокиси азота в прилегающем к углеводороду
верхнем кислотном слое связана с расходом азотной кислоты и,
следовательно, требует ее поступления из более глубоких
слоев. Из-за отставания процессов диффузии при отсутствии
перемешивания концентрация N0% в углеводородном слое
становится ниже, чем требуется согласно уравнению (I).
Недостаточность концентрации N0, сказывается тем больше, чем больше
скорость реакции углеводорода с N0%. Отсутствие достаточного
пополнения углеводородного слоя двуокисью азота ведет к
замедлению скорости нитрования. В конце реакции нитрования
по М. И. Коновалову, когда велика концентрация N0
вследствие побочных окислительных процессов и сильно понижена
концентрация азотной кислоты, роль N0 как регулятора
содержания N0, особенно велика.
При нитровании по Коновалову малоактивных
углеводородов (например, диизоамила, циклогексана) при обыкновенной
температуре отставания диффузионных процессов от реакции
нитрования практически не наблюдается. В случае нитрования
таких углеводородов, как, например, дифенилметан, это
отставание становится весьма значительным, так что реакция
происходит только в тонком слое на поверхности раздела слоя
углеводорода и кислоты.
При повышении температуры скорость диффузионных
процессов несколько увеличивается (при повышении температуры
263
на 1° коэффициент диффузии увеличивается на 2—3%), но при
этом скорость реакции увеличивается в гораздо большей
степени. Таким образом, нитрование при повышенной
температуре влечет за собой еще большее отставание диффузии от
скорости реакции.
Влияние диффузии на скорость реакции изучалось
А. И. Титовым при нитровании м-ксилолав реакторах различного
сечения. При этом оказалось, что степень прохождения реакции
в сосуде сечения 100 см% в 2,6 раза больше, чем в сосуде сечеяия
20 см%. Точно так же в горизонтальных трубках выход в 2—3
раза выше, чем в вертикальных трубках.
При изучении влияния перемешивания оказалось, что во
вращающемся автоклаве выход продуктов реакции в 1,5 раза
выше, чем без перемешивания.
На основе своих опытов А. И. Титов приходит к следующим
выводам.
1. При титровании парафиновой цепи по методу Коновалова
роль герметизации реактора (применение запаянных трубок)
сводится к обеспечению сохранения двуокиси и окиси азота
в сфере реакции.
2. При нитровании парафинов азотной кислотой в жидкой
фазе наблюдается отставание диффузионных процессов от хода
реакции, что приводит к замедлению реакции и к изменению
соотношения продуктов реакции в сторону увеличения выхода
моно- и динитросоединения и уменьшения выхода продуктов
окисления.
3. Отставание диффузионных процессов и связанные с ним
явления становятся более заметными с увеличением химической
активности углеводорода и повышением температуры, а также
зависят от аппаратурного оформления процесса.
Рассматривая различия нитрования ароматических и
предельных углеводородов, А. И. Титов [114] приходит к
следующим заключениям:
1. Ароматические соединения легко нитруются
концентрированной азотной кислотой или нитрующей смесью, причем
присутствие в них N0, не имеет значения, в то же время N0%
является активным нитрующим агентом при нитровании пре-
даотной
дельных углеводородов. При их нитровании азотная кислота
сама по себе не оказывает заметного влияния.
2. При гетерогенном проведении реакции нитрование
ароматических соединений протекает в кислотном слое; нитрование
предельных углеводородов протекает в углеводородном слое.
3. Добавка к реакционной смеси сильных протонных и анро-
тонных кислот (AlClg, TiCl*, BFg) сильно ускоряет титрование
ароматических соединений и не влияет заметным образом в
случае парафиновых углеводородов.
4. Скорость нитрования ароматических углеводородов
в парах значительно меньше, чем в жидкой фазе; в случае
предельных углеводородов скорость нитрования в парах и жидкой
фазе почти одинакова.
5. Добавка ртутных солей при нитровании ароматических
углеводородов азотной кислотой умеренной концентрации
ускоряет реакцию, а при нитровании предельных углеводородов
не влияет на ход процесса.
6. При нитровании ароматических соединений (азотной
кислотой, нитрующей смесью) выход иитропроизводных, как
правило, близок к теоретическому, в то время как при
нитровании предельных углеводородов выход соответствующих
моионитропроизводных составляет не более 60% от теории
наряду со значительным количеством образовавшихся продуктов
окисления.
7. Благодаря координационной ненасыщенности и наличию
кратных связей в ароматических ядрах реакция их нитрования
начинается со взаимодействия нитрующего агента с атомом
углерода; в случае предельных углеводородов действие
нитрующего агента (N0%) направлено на атом водорода.
8. Нитроваиие ароматических соединений в ядро
представляет ионно-комплексяую реакцию, а нитрование предельных
углеводородов — радикально-молекулярную реакцию.
В своей работе А. И. Титов и Н. В. Щитов [115] показали»
что в ряде случаев нитрования парафинов азотной кислотой
и N0%, особенно при высокой температуре, необходимо
допустить участие в реакции радикала N0^, который образуется
из азотного ангидрида по схеме
265
O,N — С) — NO, 3±NO; + ONO,.
Авторы исследовали взаимодействие нормальных парафинов
и циклопарафинов с азотным ангидридом. Реакция идет с
достаточной скоростью даже при 0° в среде инертных
растворителей. При смешении реагентов взаимодействие происходит
быстро и с разогреванием.
Авторы считают, что N0' , образовавшийся из азотного
ангидрида, реагирует с углеводородом с образованием
углеводородного радикала по схеме
Авторы предполагают, что образование весьма активного
радикалоподобного остатка N0 и объясняет наблюдаемую в
некоторых случаях более высокую, чем с N0%, скорость
нитрования азотной кислотой.
Фенилтринитрометан был впервые получен в 1952 г. А. И.
Титовым и В. В. Смирновым [116] действием N,0* на калиевую
соль фенилдинитрометана по схеме
"*' „.о,
NOOK
К суспензии калиевой соли фенилдинитрометана в четырех-
хлористом углероде при—12* приливают раствор NgO* в том
же растворителе из расчета 2,4 моля N#0* на 1 моль соли.
Реакция протекает почти мгновенно. Реакционную смесь
обрабатывают водой и раствором соды и после отгонки
растворителя в вакууме очищают сырой фенилтринитрометан
концентрированной серной кислотой. Выход 70% от теории.
Попытка получить фенилтринитрометан действием N,0*
не на соль, а на свободный фенилдинитрометан не увенчалась
успехом. Реакция идет лишь при прибавлении основания (даже
такого, как пиридин). Это указывает, что фенилдинитрометан
реагирует с N^O* только в виде аци-формы.
При действии азотной кислоты уд. в. 1,4, содержащей окислы
266
азота, на фенилдинитрометан также получается фенилтринитро-
метан с выходом 50% от теории. Кроме того, при этом
образуется п-нитробензойная кислота с выходом 35% от теории.
В отсутствие окислов азота эта реакция не идет.
Интересный метод введения двух нитрогрупп в боковую цепь
ароматического ядра был предложен итальянским химиком
Пончио [117]. Метод основан на действии N,0* на бензальдок-
по схеме
N.O.
= NOH >Сб
При этой реакции существенное значение имеет избыток N^O*.
Если вести процесс с эквимолекулярным количеством NgO^, то
развиваются побочные реакции. Пончио предложил следующую
методику проведения реакции. К эфирному раствору бензаль-
доксима добавляют N^O* в количестве 2 молей на 1 моль ок-
сима. Процесс ведут вначале при охлаждении, а затем при
комнатной температуре. Полученный фенилдинитрометан
извлекают 5%-ным водным раствором щелочи, а затем выделяют его
подкислением щелочного раствора 10% -ной серной кислотой.
Выход фенилдинитрометаиа 75% от теории.
Исследовалось превращение окснмов в нитросоединения
[118] посредством окисления гипобромитом с последующим
восстановительным дебромированием. С альдоксимами и кето-
ксимами алифатических и ароматических кетонов реакция
совершенно не удается.
Лишь алициклические кетоксимы дают соответствующие
нитросоединения. В качестве бронирующего реагента
рекомендуется N-бромимид янтарной кислоты. В качестве
восстановителей наилучшие результаты дает борогидрид натрия
NaBH..
Уместно будет отметить действие крепкой азотной кислоты
ва фенилнитрометан и фенилдииитрометан.
При нитровании фенилнитрометана его медленно
прибавляют к 7—10-кратному весовому количеству азотной кислоты
уд. в. 1,49—1,53 при температуре 10—15° (реакционную массу
приходится охлаждать). Реакционную массу выливают на
лед и выпавший осадок м-нитрофенилиитрометана
267
CH.NO?
N0,
дерекристаллизовывают из спирта или ледяной уксусной
кислоты. Выход ^х 40% от теории. Необходимо отметить, что при
нитровании одной крепкой азотной кислотой в ядро вступает
лишь одна нитрогруппа и притом преимущественно в
мета-положение. Для введения второй нитрогруппы в ядро требуется
применение смеси азотной кислоты уд. в. 1,5 и олеума с 20—
25% -ным содержанием свободного 80s:
^ O
N0%
Нитрующую смесь берут в 8—10-кратном весовом количестве
а реакцию ведут при 65—70°. Выход составляет ^, 75% от
теории [119].
При нитровании фенилдинитрометана [120]
концентрированной азотной кислотой получается м-нитрофенилдинитро-
метан по схеме
Реакция проводится следующим образом: охлажденную до
—15° азотную кислоту уд. в. 1,5 приливают к фенилдинитро-
метану (весовое отношение НМОз к фенилдинитрометаяу
10:1) и выдерживают 1 час при комнатной температуре. Затем
реакционную смесь выливают на воду. Полученный осадок
фильтруют, промывают водой и перекристаллизовывают из
метилового спирта. Получают м-нитрофенилдинитрометан в виде
желтых кристаллов. Выход составляет 93% от теории.
За последние 15—20 лет опубликованы работы, в которых
приведены данные о нитровании предельных углеводородов
азотной кислотой по так называемому парофазному методу.
268
Особенностью этого метода, который осуществляется при
атмосферном давлении, являются высокие температуры реакции,
значительно превышающие температуры нитрования по методу
Коновалова.
Эта особенность парофазного метода придает ему
своеобразный характер, так как в этих условиях, наряду с нитрованием
парафинов, происходит пиролиз последних (или свободных
радикалов, см. ниже) с образованием соединений (или радикалов)
меньшего молекулярного веса, которые, подвергаясь
нитрованию, дают нитропродукты с меньшим числом атомов углерода,
чем в исходном парафине.
Промышленное производство нитропарафинов нитрованием
парафинов непрерывным методом в газовой фазе, начатое
в 1940 г., открыло новую область органической химии.
Для увеличения выхода нитропарафинов и во избежание
опасности взрыва весьма важно регулировать соотношения
реагентов таким образом, чтобы соотношение углеводорода к
азотной кислоте равнялось по крайней мере двум.
Кроме смеси нитропроизводных с различным числом атомов
углерода, при нитровании по парофазному методу образуются
также продукты окисления.
В работе Хэсса, Ходжа и Ваыдербильта [121] имеются
данные по нитрованию парафинов: этана, пропана, н. бутана и изо-
бутана. Синтез соответствующих нитропарафинов осуществлялся
этими авторами посредством пропускания паров
углеводорода через азотную кислоту со скоростью 150 л/час (молярное
отношение углеводорода к азотной кислоте 2 : 1);
образовавшаяся таким образом смесь поступала далее в реактор, который
нагревали на нитрит-нитратвой бане до 420° (реактор
представлял собой стеклянную трубку диаметром 10 мм).
При нитровании этана получены в качестве продуктов реакции
витрометан и нитроэтан; нитрование пропана дало уже четыре
продукта: нитрометан, нитроэтан и в преобладающем
количестве (65% общего выхода нитропарафинов) 1- и 2-нитропро-
паяы. При нитровании бутана основными продуктами реакции
оказались 1- и 2-нитробутаны (77% общего выхода
нитропарафинов); кроме того, получены 1-нптропропан, нитроэтан и
269
нитрометан, образовавшиеся в результате нитрования
продуктов пиролиза.
Нитрование изобутана привело к следующим результатам:
из двух образовавшихся яитропроизводных, соответствующих
по числу атомов углерода нитруемому парафину, первичный
1-нитроизобутан получен с выходом 65% общего выхода нитро-
производных, третичный же 2-нитроизобутан — с выходом
всего 7% ; кроме того, в продуктах реакции обнаружен 2-нитро-
пропан (выход 20%) и нитрометан. Результаты этих опытов
сведены в табл. 14.
Таблица 14
Углеводород
Продукт реакции,
&
I
a
CO
эпро-
a w
@5
a w.
?5
1
(NO
#
Этан . .
Пропан .
Бутан . .
Изобутан
10—20
9
6
а
80—90
2G(?)
12
32
5
50
20
65
9
21
28
25
При нитровании изобутана по методу Коновалова при 150*
эти же исследователи получили в качестве единственного
продукта реакции кристаллический 2-нитроизобутан, который
при нитровании по парофазному методу, как мы видели выше,
образуется с выходом всего 7% . Сравнение этих двух методов
наглядно указывает на особенности парофазного нитрования:
благодаря высоким температурам реакция становится менее
селективной и дает изомеры, из которых первичный,
полученный со значительно ббльшим выходом, чем третичный,
является, невидимому, более устойчивым в условиях реакции. Как
уже было отмечено выше, высокие температуры приводят
также к образованию, вследствие пиролитических реакций,
нитросоединений с меньшим числом атомов углерода, чем в
нитруемом парафине.
270
ддотмой
Обстоятельное исследование парофазного нитрования
пропана и бутана азотной кислотой провел в 1952 г. Бахман с
сотрудниками [122—125], установивший, что в результате реакции
нитрования, наряду с нитросоединениями, получаются и
кислородсодержащие соединения и пепредельные углеводороды. При
температуре 425* (оптимальная температура) и времени
контакта 1,6 сек. конверсия достигает 36% . Этими авторами было
изучено влияние добавок кислорода и галоидов на выход и
состав продуктов нитрования бутана [123] и пропана [124, 125].
Было найдено, что добавка двух молей 0% на моль HNOg не
меняет оптимальной температуры (425°) нитрования бутана,
увеличивает выход нитропродуктов в расчете на пропущенную
HNOg, но понижает выход в расчете на превращенный бутан.
Было также найдено, что при последовательном введении 1,
2 и 3 молей кислорода содержание карбонильных соединений,
олефинов и СО в общем увеличивается, количество же СО%
достигает минимума при максимальном количестве дитропара-
финов, полученном при содержании 1—2 молей 0% на моль
НМОз.
Бахман следующим образом истолковывает результаты, по-
лученяые с добавками кислорода. Добавки кислорода
увеличивают концентрацию свободных радикалов, которые,
соединяясь с двуокисью азота, дают нитропарафин. В нитропарафины
всегда превращается только часть алкильных радикалов,
а остальные алкильные радикалы взаимодействуют с
кислородом. С увеличением количества кислорода выше оптимального
скорость окисления растет, и в эту реакцию вовлекается
дополнительная часть радикалов, превращавшихся при
меньшем количестве кислорода в нитропарафины. Наличие
олефинов в опытах, проведенных без кислорода, подтверждает
участие свободных радикалов в нитровании.
Добавки 0,015 моля брома на моль HNOg увеличивали
выход нитропарафинов от 23 до 28% в расчете на пропущенную
HNOg и от 23 до 50% в расчете на превращенный бутан [124].
Добавки кислорода повышали положительное влияние брома.
Так, выход нитропарафинов, рассчитанный на пропущенную
HNOg, достигает максимальной величины 48% при содержании
271
2 молей 0% на моль HNOg, а выход нитропарафинов,
рассчитанный на прореагировавший бутан, достигает максимальной
величины 57% при наличии 1 моля О* на моль HNOs.
По мнению авторов, положительное влияние брома связано
с его способностью генерировать радикалы по схеме
1) Вг,^
2) С.Н*^, + Вг -> C*n;*+i + НВг;
+ С*Н^+, + НВг + Вг";
5) НВг+[О]-»НОЧ-Вг'.
Преимущество брома по сравнению с кислородом Бахмав
объясняет легкостью распада галоидных алкилов с
регенерацией атомов брома. Положительное влияние добавок кислорода
при нитровании в присутствии брома объясняется, по мнению
Бахмана, окислением НВг до Вг".
По данным Бахмана, для достижения максимального
выхода нитропродукта (в присутствии 0,1 моля 0% на моль пропана)
хлора необходимо в два раза больше(0,05 моля С1% на мольНМОэ),
чем брома (0,025 моля Вг* на моль HNOs). Этот
результат объясняется Бахманом следующим образом. Хлор и бром,
реагируют по схеме
1) Х,-»2Х";
2)
3)
4)
5) НХ + NO'-^Xa + HgO-L низшие окислы азота;
6) RX + NOg-*X# + продукты окисления R + низшие окислы азота.
Бромистый водород и бромистый алкил окисляются быстрее,
чем соответствующие производные хлора. Поэтому количества
молекул галоида, способное превратиться в свободные радикалы,
больше в случае брома, чем для хлора. Падение выхода нитро-
продуктов с повышением концентрации галоида выше
оптимальной объясняется расходованием N0% по уравнениям 5 и 6.
272
В работах Бахмана быяо также исследовано влияние
параметра Д/У (отношение величины поверхности реактора к его
объему) на выход нитропарафинов при нитровании бутана в
присутствии кислорода [123]. Кривая зависимости выхода RNO&,
рассчитанного на пропущенную НМОз, от количества
кислорода, проходила через максимум при всех
исследованных величинах 6"/F (20, 28 и 300). Максимальный
выход (43% RNOg) оставался постоянным при всех трех
значениях 6"/У, но в зависимости от параметра 6"/У достигался при
различном содержании кислорода, причем при максимальном
выходе наибольшему значению Д/У соответствовало
наименьшее количество кислорода (1 моль 0% на моль НМОз).
Увеличение параметра .9/V вызывало понижение отношения
прореагировавший бутан—пропущенный бутан. Бауман приводит
следующее объяснение найденным фактам. Понижение
количества прореагировавшего бутана с увеличением 6"/У
объясняется исчезновением части радикалов на поверхности. Однако,
несмотря на исчезновение части радикалов, количество
нитропарафинов, получающихся из радикалов, не падает (сохраняется
высота максимума). Следовательно, увеличение J/У позволяет
получить большее количество натропарафинов за счет обрыва
цепей окислительной цепной реакции, т. е. благодаря
подавлению окислительного направления.
Бахман и Поллак [126] изучали парофазное нитрование
пропана в присутствии хлора в качестве катализатора и
сопоставляли полученные результаты с опубликованными в 1952 г.
данными по парофазному нитрованию в присутствии кислорода
и хлора.
Как указывают эти авторы, при обычном парофазном
нитровании пропана получается смесь нитропарафинов,
состоящая из 25% 1-нитропропана, 40% 2-нитропропана, 10% нитро-
этана и 25% нитрометана.
Если при нитровании применять в качестве катализатора
смесь кислорода и хлора, то превращение в нитропарафины
увеличивается на 32% , а при применении одного хлора
происходит увеличение на 46% . Бром действует так же, как и хлор,
но недостаток его применения состоит в том, что образующаяся
/Толченые нылуюсоеЭыненцД акы^рмоа^олмтм^сжоео ря^д 273
НВг восстанавливает нитросоединеншя с образованием
аммониевых и гидроксиламмониевых солен.
При парофазном нитровавши природного газа состава:
75,56% СН.,6,36% С,Нв,3,45% СдНв,13,65% N,.0,69%
СаНю и при отношении природный газ: HNO^: Н&О: С1, =
=13,6 : 1 : 1,6 :0,4, времени контакта 1,7—1,8 сек. и
температуре 450° оптимальное превращение составляло 13%.
Такое же превращение получается при молярном отношении
СдНа : HNOs : Н,0 : СН^ = 0,483 ; 1 ; 1,6 : 14,0 при
нитровании без хлора. Таким образом, если в смеси преобладает
метан, то хлор не оказывает каталитического влияция.
Согласно американскому патенту [127], пропан нитруется
40—70 %-ной азотной кислотой при температуре 190° и
давлении ^9 атм. На 2 моля пропана берут 1 моль кислоты и
получают смесь нитропропанов [(СНз)%СНМО% и (CH,),C(NO,)*
с выходом 8—10%, считая на азотную кислоту].
Нитрованию пропана азотной кислотой уделялось
большое внимание как в отношении аппаратурного оформления
процесса (создание условий изотермического протекания
реакции), так и в отношении применения катализаторов
(кислород, воздух) для интенсификации процесса [128].
Хэсс и Александер [129] изучали влияние кислорода при
нитровании пропана азотной кислотой при температуре 410°.
При этом им удалось увеличить превращение пропана от 28
до 62%.
При температуре 395° и введении 3,8 моля кислорода на
1 моль HNOg превращение пропана в нитропродукты
увеличивается от 20 до 76%. =
В недавно опубликованном патенте- [130] указывается на
значительное увеличение выходов нитроалканов и ценных
побочных продуктов окисления, получающихся при
парофазном нитровании алифатических углеводородов СН&, С,Н$,
QHg, QHiQ (нормальный и изо-) в присутствии
кислорода.
Добавление кислорода незначительно влияет на
нитрование метана.
При температуре 435* изменение молярного отношения
18 л. В. Топчиев
274
O&/HNO; от 0 до 5 увеличивает конверсию азотной кислоты,
в нитрометан с 17,6 до 24,2% [129].
При проведении процесса под давлением и в присутствии
кислорода конверсия HNO@ в CH,NO, резко падает по мере
увеличения концентрации кислорода. Это видно из
приводимых ниже данных табл. 15 [131], полученных при температуре
444° и давлении 7,8 атм.
Таблица 15
л HNO@ б CHgNOg пой Аделемы&м б дрысутсямыы
Ковцеитрация О*
Конверсия, . . .
Выход
11
0
0
21
21
,8
,0
,0
,1
,3
12
0
2
18
21
,0
,38
Л
Л
12
0
2
20
23
,3
,37
Л
Л
,6
11,9
0,60
3,9
19,3
22,4
10,5
4,30
8,8
0,0
0,0
При высоком содержании кислорода нитрометан не
получается; образуется главным образом фррмал&дегид.
Изучением реакции нитрования жидких дестиллатов вефяи
в паровой фазе занимались Б. Н. Тютюнняков, Н. П. Мань-
ковская и Т. Д. Явлинский [132]. Смесь углеводородов
состояла из 24,2 % нафтеновых и 72,6 % парафиновых углеводородов.
Выход нвтропродуктов аа один проход составлял 60—65%
от смеси углеводородов. Полученные нитропродукты
содержали: 90% мононитросоединений, состоящих из 20% первичных
нитросоединений, 40% вторичных и 40% третичных.
Применялась 67%-ная азотная кислота в отношении к лигроину
1:1, продолжительность контакта 4— 5 сек., температура
нитрования 330—350*. Как указывают авторы, при просасы-
вании паров углеводородов при помощи тока воздуха выход
нитропродуктов повышается на 90%, что, повидимому, связано
с каталитическим действием кислорода воздуха.
Хэсс и Паттерсон [133] исследовали реакцию нитрования
пентана. Пары пентана поступали со скоростью 260 мл/час
в реактор, где их перемешивали с парами азотной кислоты,
которую пропускали со скоростью 70 мл/час (молярное
отношение пентана к азотной кислоте 2,3:1). Нитрование производили
275
при 400°. В результате нитрования получены следующие
продукты: I-, 2*, 3-витропентанм (с выходом 65% от
общего количества житропарафинов), 1-нитробутая, 1-иитро-
пропан, нитроэтан и нитрометан. Общий выход нитртшрафи-
нов иа азотную кислоту, вступившую в реакцию, составлял 31% .
Сейгль я Хэсс [134] разработали метод иарофааного
нитрования изоаентана. При молярном отношении у г леводородов
к кислоте 1,7 : 1 и температуре нитрования 420 авторы полу*
чили 9 нитропарафинов: З-метил-1-нитробутан, 2-метвл-1-яя-
тробутан, З-метил-2-нитробутан, 2-метил-2-нитробутан, 2-ни-
тробутан, 2-метил-1-нитропропан, 2-нитропропаи, нитроэтан
и нитрометан. При 380° получены те же продукты, во о
меньшими выходами.
На парофазпый метод нитрования 1,1,1-трифторпроиана
указывают Хэсс и Робинсон [135], которые вводили в реактор
трифторпропан и азотную кислоту при температуре 395° со
скоростью 0,42 и 0,52 мол/час ври продолжительности контакта
2,5—3,5 сек. Получается 39% смеси 1,1,1-трифторнитроэтана
и 1,1,1-трифтор-З-нитропропаяа в отношении 2 : 3. В нитропро-
дукты превращается 11,9% исходного продукта.
Если действовать азотной кислотой на хлорпроизводные
углеводородов в газовой фазе, то получаются хлорнитро-
углеводороды, применяющиеся как растворители иясектисидов,
а также как промежуточные продукты [136],
Иа работ, относящихся к нитрованию парафинов в газовой
фазе, отметим еще работу Данцига и Хэсса [137] по жзучеяию
реакции нитрования 2,3-диметилбутана, Эти адторы поставили
задачу выяснить на примере 2,3-диметилбутава, происходит
ли в условиях иарофазного нитрования образование динитро-
парафинов (наряду с мононитронарафинамн), как это
наблюдается при проведении реакции по методу Коновалова.
При нитровании 2,3-диметилбутана в запаянной трубке при
125° М. И. Коновалов [138] получил, кроме 2,3-диметил-2-
нитробутана, также 2,3-диметил-2,8-динитробутан,
образование которого может Чэшъ объяснено замещением нитрогруяпами
третичных атомов водорода при втором и третьем атомах
углерода.
18»
276
2/глеао<%ух>()оа даолгной
Опыты Данцига и Хэсса проводились при температуре
400—430°и молярном отношении углеводорода к азотной кислоте,
равном 1,58 : 1. Найдено, что оптимальная конверсия 2,3-
диметилбутана в нитропарафины происходит при 410° и
продолжительности контакта 1—2 сек. При дальнейшем
повышении температуры конверсия уменьшается, причем
наблюдается образование побочных продуктов окисления, выход
которых достигает наибольшей величины при 430°. При 408°
конверсия по азотной кислоте, вступившей в реакцию,
составляет 17,5% .
В отличие от нитрования но методу Коновалова динит-
ропарафины в опытах Данцига и Хэсса не обнаружены,
Получены лишь мононитроиарафины (2,3-диметил-1-нитробутан,
2,3-диметил-2-нитробутан, З-метил-2-нитробутан, 2-нитропро-
пан и иитрометан) наряду с продуктами окисления (кетоны)
и крекинга (2,3-диметил-1-бутен и 2,3-диметил-2-бутен).
На примере нитрования бутана Мак-Клирли [139] показал
влияние температуры на состав продуктов реакции (табл. 16).
Таблица 16
Температура, ^С
395
393
393
445
450
Ницрометав
2,1
6,1
5,0
5,9
9,0
Нитроэтая
12,7
19,0
11,0
18,2
25,0
1-Нитропро-
яаи
4,9
7,0
5,0
6,5
7,0
2-НитроОутав
49,0
41,0
46,0
37,0
28,0
i-Яитробугаи
30,5
27,0
32,0
31,8
31,0
ридно из этой таблицы, увеличение температуры ре-
акции ведет к уменьшению образования иитробутанов и
соответствующему увеличению количества везших нитродара-
финов, в том числе нитрометана.
277
Следует отметить, что получение нитрометана прямым
нитрованием метана — довольно трудная проблема из-за
небольшой конверсии и трудности: регенерации непрореаги-
ровавшего углеводорода. Имеется патент, согласно которому
рекомендуется нитровать метан, разбавленный инертным
газом, например азотом, с рециркуляцией. При этом выход
яитрометана увеличивается по мере разбавления его ааотом.
Так, конверсия 100%-ного метана составляет 2,2%, в случае
же нитрования 10% -ного метана конверсия увеличивается
почти в 3 раза [140].
В связи с большими трудностями нитрования метана, витро-
метан в основном получают при нитровании пропана азотной
кислотой.
В зависимости от условий проведения реакции содержание
нитрометана в продуктах реакции может колебаться от 9 До
49% [141]. Для удаления нитрометана из смеси продуктов
реакции рекомендуется применять перегонку с н. гексаном.
Последний дает азеотропную смесь с CHgNOg, низшими
алифатическими спиртами и водой.
Алифатические эфиры, спирты, кетояыи карбоновые кислоты
при парофазном нитровании азотной кислотой дают выходы
нитропроизводных не выше 20% (от 5 до 20%); теаЖпература
реакции 400° [142].
Парофазному нитрованию подвергались и жирноаромати-
ческие углеводороды. Так Булок и Митчелл [143] при
нитровании толуола азотной кислотой в паровой фазе получили смесь
изомерных нитропроизводяых, которая содержала 55% о-нит-
ротолуола, 5% м-нитротолуола и 40% п-нитротолуола. В
продуктах реакции обнаружены незначительные количества поли-
нитропродуктов и продукты окисления. Соотношение изомеров
примерно такое же, которое получается при обычном
нитровании.
Парофазное нитрование бициклических нафтенов
исследовано на примере норкамфана. Действуя яа норкамфан азотной
кислотой при температуре 400—410*, Хэсо и Бликенстафф
[144] получили третичный нитроноркамфан
278
-сн — сн%
сн.
н,с—с—сщ
Образование этого продукта показало, что при парофазном
нитровании исчезает инертность третичного водорода, стоящего
у атома углерода, общего обоим циклам.
Обзор опубликованных экспериментальных данных по
нитрованию парафинов в газовой фазе приводит к следующим
заключениям об особенностях парофазного метода.
1. Нитрование в газовой фазе осуществляется при
обыкновенном давлении и высоких температурах от 250 до 600°;
в большинстве случаев синтез нитропарафинов проводят при
температурах 400—450°, которые являются, повидимому,
оптимальными.
2. В отличие от нитрования по методу Коновалова, которое
дает один или два продукта реакции, нитрование по парофаз-
ному методу приводит к образованию ряда изомерных нитро-
соединений (при высоких температурах реакция становится
менее селективной).
3. Нитрование в газовой фазе, наряду с нитропроиЗвод-
ными, соответствующими нитруемому углеводороду, дает в
качестве продуктов реакции ряд нитропарафинов с меньшим
числом атомов углерода, чем в исходном парафине.
Для объяснения этого факта допускают образование из
парафинов свободных радикалов, которые, подвергаясь
дальнейшему пиролизу, дают радикалы с меньшим молекулярным
весом; продукты пиролиза, присоединяя группу NO,, образуют
нитропарафины с различным числом атомов углерода. При
повышении температуры относительный выход
нитропарафинов, появляющихся в результате нитрования продуктов пиро-
лжза, повышается.
4. При парофазном нитровании образуются исключительно
моновитропарафины, причем преобладают первичные нитро-
производные. С повышением температуры выход первичных
279
нитросоединений повышается, а вторичных и третичных —
понижается. Из этих данных можно сделать следующие выводы:
а) полинитропарафины легко подвергаются пиролизу, вследствие
чего они не могут существовать при высоких темвературах
парофазного нитрования; б) из мононитропарафинов наиболее
устойчивыми являются первичные нитропроизводные.
5. Конверсия (количество образовавшихся нитропарафинов
но отношению к азотной кислоте, пропущенной через реактор)
является постоянной в довольно широких температурных
пределах при соответствующем подборе времени контакта. При
одинаковом времени контакта кривая зависимости конверсии
от температуры проходит через максимум; при температурах
ниже оптимальной нитрование является неполным, а при более
высоких температурах происходит пиролиз нитропарафинов.
6. Реакция нитрования сопровождается процессами
окисления парафинов и их аитропроизводных, в результате которых
получаются спирты, альдегиды, кетоны, карбоновые кислоты,
а также СО и СО,. Оптимальное использование азотной кислоты
при синтезе нитропарафинов достигает 40%. Посредством
регенерации окислов азота, образующихся при реакциях
окисления, выход нитропарафинов по азотной кислоте может
быть повышен (на больших установках промышленного синтеза
нитропарафинов, где производится регенерация окислов азота
до азотной кислоты, выход нитропарафинов составляет
90 молей на 100 молей азотной кислоты).
7. Из парафинов наиболее трудно нитруется метан, который
при более высоких температурах и большем времени контакта
дает меньшую конверсию в нитропроизводные сравнительно
с другими гомологами (при 450* и времени контакта 0,42 сек.
выход нитрометана составляет 15%). Далее, до пропана
включительно, наблюдается последовательное повышение легкости
нитрования. Однако углеводороды, следующие за пропаном,
не обнаруживают при нитровании заметных различий в
реакционной способности.
8. Механизм реакции нитрования в газовой фазе недостаточно
изучен. По представлениям Мак-Клирли и Дегеринга [139],
сначала происходит расщепление азотной кислоты на двуокись
280
азота и радикал ОН [уравнение (1)]; при действии последнего
ва углеводороды образуются свободные радикалы [уравнение
(2)], которые реагируют с N0% или с азотной кислотой [уравнения
(3) и (4)]:
(1)
(2)
R
R'
R"
R
\
R' _ CH 4
/
R"
R
R' —
/
R*
\
— С _|_
/
c-
. HO" -
bNO,
HONOg -»
R
\
_ R/ _ С + I
/
R'
R
\
-+R' —CNO,
/
R"
R
\ _
R' _ CNO, 4
/
R'
(3)
НО' (4)
Таким путем возникают нитросоединения с числом атомов
углерода, соответствующим нитруемому парафину. Для
объяснения образования нитросоединений с меньшим числом
атомов углерода Мак-Клирли и Дегеринг допускают
дальнейшее расщепление радикалов под влиянием высоких температур
и последующее нитрование продуктов пиролиза двуокисью
азота или азотной кислотой.
Другая теория,автором которой являетсяЭвелл[145],основана
на предположении, что углеводороды, соединяясь с азотной
кислотой, образуют промежуточные комплексы двух видов*
Последующее расщепление комплексов первого вида дает
в качестве продуктов реакции спирты инитропарафины с
меньшим числом атомов углерода, чем в нитруемом соединении;
расщепление комплексов второго вида приводит к образованию
воды и нитропарафинов, соответствующих исходному
углеводороду. Например, нитрование этана проходит, согласно взгляду
Эвелла, через образование следующих промежуточных
комплексов:
НО
H\l
6
0
N = 0
i
!
С—11
\н
но
j
[
1
н
н
0
/
—\ = 0
1
1
]
1 ^
\
— с —н
н
II
При дальнейшем течении реакции первый комплекс
распадается на нитрометан и метанол, а второй — на яитроэтая и воду.
Подтверждение теории комплексов мы находим в работе
Бертона [146], который показал, что при нитровании
оптически активного 3-метилгексана образуется также оптически
активный 2-нитробутан.
Нитрование в газовой фазе, как мы указывали, обычно
осуществляется при атмосферном давлении. В некоторых
случаях, однако, для повышения эффективности реакции
применяется нитрование под давлением. Хэсс, Гибсман и Пирсон
[147] при нитровании этана под давлением 7 атм. получили
нитропарафины с выходом 33% при одном проходе через
реакционную камеру (при атмосферном давлении конверсия
составляет 9%).
В патентной литературе последнего времени имеются
сообщения о проведении нитрования азотной кислотой в газовой
фазе в присутствии катализаторов. Норман Леви [148]
пропускал смесь парафинов с азотной кислотой при 300—450°через
реакционную камеру с катализатором, состоявшим из соединений
мышьяка или сурьмы, смешанных с силикатами. Оптимальная
конверсия получена при отношении парафинов к азотной
кислоте 0,5 : 1 и времени контакта 1—5 сек.
Имеются также предложения [149] проводить реакцию
парофазного нитрования алифатических, а также и
ароматических углеводородов в реакторе с пылевидным контактом
(размер частиц 5—600 р), находящимся во взвешенном состоянии
в струе реагентов.
282
Опубликованы данные [150] по исследованию
непрерывного процесса нитрования циклогексана азотной кислотой.
Применяется стеклянная аппаратура, через которую жидкость
проходит с определенной, регулируемой в широких пределах
объемной скоростью. Рабочее давление равно 7 атм. Как
исходные вещества, так и продукты реакции вигде не
соприкасаются с металлическими частями и подаются под давлением .
азота. Оптимальными условиями процесса нитрования
оказались: температура 120° и отношение HNOg/CgHia = 0,91.
Выход нитроциклогексана 13,9%.
Необходимо отметить, что синтез ряда низших нитропара^
финов парофазным методом в настоящее время осуществлён
в промышленном масштабе.
§ 3. Получение полинитропарафинов
В отличие от синтеза мононитропарафинов получение поли-
нитропарафинов прямым нитрованием азотной кислотой
не так распространено. В настоящем разделе мы даем
краткий обзор различных методов получения полинитро-
парафинов.
Олефины при действии N*O* присоединяют группы N0,
с образованием полинитропарафинов. Например, 2,3-диметил-
2-бутен, реагируя с N*O*, дает 2,3-диметил-2,&-дивитробутан
C = C —CH,+ N*0* -^ СНз—С — С — СН;
Н, СНз
По разработанному Сейглем и Хэссом [151] методу, динит-
ропарафины могут быть получены из вторичвых иитропарафн-
нов при последовательной обработке их основанием и галоидом.
Сущность метода состоит в конденсации натриевых солей
аци-формы нвтропарафинов с продуктом взаимодействия этих
солей с галоидом, причем выделяется натриевая соль
последнего:
NaOH -* R,C = NO$Na + H:O; (1)
283
+ R,C = NO^Na -к R^ __ С — С — R.+NaCl
Например, натриевая соль аци-формы нитропропана
(СН,)уС = NOgNa ^ яри нагревании в течение 7 час. с
(CH,)*CClNOa дает 2,3-диметил-2,3-динитробутан (выход 9%):
СЩСНз
С
(СН,):С = NO%Na |- (CH,),CC1NO*
Реакция протекает более эффективно, если вместо хлор-
производного действовать на натриевую соль аци-формы бром-
производным (выход динитронарафияа 29%); еще лучшие
результаты получаются при взаимодействии (CHg)*C = NO%Na
с иодопроизводным (CH,)*CJNO* (выход 43%). Подобным
же образом из натриевой соли аци-формы нитробутана
(СаНд)(СНд)С = NO^Ta и (C*H,)(CH,)CBrNO, получен
3,4-диметил-3,4-динитрогексан с выходом 16%:
СН/Ш, — С = NOgNa + CH^DH. — CBrNO
СН
СН, СН,
С —С— СНаСЩ + №Вг
N0% N0$
Натриевая соль нитроциклогексана при нагревании (с
обратным холодильником) в течение 3 час. в растворе 80% -ного
спирта с (CHg)aCBrNOg дает 1-нитро-1-(2-нитроизопропил)-
циклогексан с выходом 19%.
Первичные нитросоединения не дают этой реакции.
Анджели и Алессандри [152] нашли, что серебряная соль
нитрозтана способна самопроизвольно разлагаться с
последующей конденсацией продуктов распада в 2,3-динитро-
бутан:
284
CHgCH=NO#Ag СН, —СН —СН —СН,
^ NO, NO,
Этот синтез, повидимому, имеет общий характер а может
слушать для перехода от моно- к полинитропарафинам. Например^
пз серебряных солей нитропентана а фенилнитрометана
получаются , соответственно, 5,6-динитродекан и 1,2-динитро-
1,2-дифенилэтан.
Динитропарафины могут быть синтезированы а
электрохимическим путем, а именно: электролизом солей аци-витро-
парафинов [153, 154]
электролиз
2R,C = NOgMc > R:C — CR%
| | + 2Me
Электролиз проводится в ячейке с катодом из меди и анодом
из полированной платины при напряжении 6—10 в и
температуре 23—27°. В этих условиях из 2-нитробутана образуется
в основном 3,4-динитро-3,4-диметилгексав и в небольшом
количестве 2,2-динитробутан [155].
Динитросоединения могут быть также получены действием
различных окислителей на соли аци-форм вторичных витро-
парафивов. Наилучшие результаты дает применение
персульфатов. Были получены: из 2-нитропропана 2,3-диметил-2,3-
динитробутан (выход 51%), из 2-нитробутава 3,4-двметил-3,4-
динитрогексан (37%), из витроциклогексава 1,1-динитро-
бициклогексил (30%). В то же время неудачными оказались
попытки превратить методом окислительной димеризации 1,1-
динитроэтан в тетравитробутав и тривитрометав в гексавитро-
этан. В отличие от вторичных первичные нитроалкавы
реагируют с персульфатом лишь с образованием производных изо-
ксазола [156].
Синтез динитропарафинов, у которых обе витрогруппы
находятся при одвом и том же атоме углерода, может быть
осуществлев по методу, который заключается в окислевии псев-
донитролов R*C(NO)(NO%) хромовой кислотой; псевдонитролы
[157] получают действием N^O* на оксимы:
285
'2*""\
(1)
N0,
R,C
у Н,СгО*
N0,
ХО,
/
(2)
N0,
По патентным данным [158], при обработке псевдонитро-
лов в растворе CCl* окислами азота или 70%-вой азотной
кислотой получается 2,2-динитропропан с выходом 27—36%.
При нитровании парафинов при температуре 180* удается
получить смесь соответствующих моно- и динитропроизвод-
ных наряду с продуктами окисления и непрореагировавшим
углеводородом. Соотношения продуктов реакции зависят от
соотношения количеств углеводорода и азотной кислоты.
Так, например, нитрование додекана [159] при 180—190° азот-
яой кислотой уд. в. 1,4 дает следующие продукты (табл. 17).
Таблица 17
Отношение доде-
наи/HNO,
1 :4
1 :2
1 :1
2 :1
Продукт реакции, %
додекан
24
33
43
58
Битродеканы
4
25
10
36
полинитро-
деканы
47
38
15
5
жирные
кислоты
25
4
2
1
Как видно из таблицы, при большом избытке азотной
кислоты (углеводород: HNOs = 1:4) получаются главным
образом динитропроизводные и жирные кислоты, при недостатке
азотной кислоты (углеводород: HNOg = 2:1) только 42%
додекана вступает в реакцию, при этом основным продуктом
реакции являются мононитропроизводные.
Парафины, имеющие более 8 атомов углерода, хорошо
нитруются в токе N0%, воздуха и СО а при температуре 160—
286
190°. В 300 г додекана пропускают N0%, СО% и воздух при
175—180° в течение 110 мин. Продукт реакции фракционируют
и получают 130 г мононитродекана, 40 г высших нитропара-
финов, 20 г жирных кислот и 130 г непрореагировавшего
додекана.
Так же нитруют углеводороды, содержащие 12—20
атомов углерода, и получают соответствующие нитропара-
фины [160].
При использовании в качестве нитрующего агента двуокиси
азота, большом времени реакции и температуре 200—250*
наблюдается образование полинитросоединений с меньшим
числом атомов углерода. Так, при реакции пзобутана с N0%
при 200—250*, наряду с 2-метил-2-нитропропаиом и 2-метил-
1,2-динитропронаном, получается 2,2-динитропропан [161].
Нитропарафины конденсируются с кетонами с образованием
динйтропарафинов. Так, например, реакция между нитроме-
таном и ацетоном протекает по схеме
сн* сн, с н,
I I GH.NO, J
С = О + H,CNO, -к G = CHNO: ^ O.NCH: — С — СН^О.
СН, СН, СНз
Реакция проводится в присутствии основных катализаторов,
предпочтительно вторичных аминов. Б продуктах реакции,
наряду с динитроалканами, содержатся витроолефины.
Понижение температуры реакции и увеличение концентрации ке-
тона благоприятствуют образованию динитроалканов [162].
Конденсация калиевой соли динитрометана с диальдеги-
дами приводит, по патентным данным [163], к получению
тетранитродиолов общей формулы:
(NO^CHCHfOHKCH,),, (OH)CHCH(NO,),, где а = 1-2.
Тетранитродиолы могут быть исходными веществами для
синтеза различных полинитросоединений.
Полинитропарафины общей формулы
N0% Н N0.
Hi — Li — Li — U f!*,
Н R, R,
287
где Ri, Rg, Rg = Н или алкия, a R^ = алкил, нитроалкил,
алкоксил, получаются [164] взаимодействием щелочных солей
нитропарафияов общей формулы
Вз О
В^ ОМе
гдеМе=]\а, К, с нитроолефинами общей формулы OgNCH=CHR
с последующим разложением полученного продукта конденсации
слабой кислотой, например, уксусной. Так, из К-соли 2-нитро-
пропана и 1-нитро-1-бутена получается 1,3-динитро-2-зтил-
3-метилбутан. Взаимодействием К-соли 2-нитропропаиа с 2-
нитробутеном-1 получен [165] 3,5-динитро-З-метилгексан с
выходом около 90% .
Щелочные соли полинитропарафинов также способны
реагировать с нитроолефинами с образованием полияитросо-
единений. По литературным данным[166], 1,1,1,3-тетранитро-
пропан получается конденсацией нитроформа с нитрозтиленом.
Взаимодействием К-соли 1,1-динитрозтана с 2-нитро-
зропиленом был получен [163 и 167] 2,2,4-тринитропентан с
выходом 93% .
Другой метод получения полинитросоединений заключается
в обменной реакции Ag-соли тринитрометаиа с галоидалкилами
а галоиднитроалкилами. Реакцию в основном удается провести
з галоидметильнымипроизводными ароматического ряда [168]:
О
<Г ^— СЩС (N0g)3 + AgX т. пл. 135°
Аналогично получается 2,4,6-тринитро-2,2,2-тринитрозтил-
эензол CgH^OigNg (т. пл. 153°). Из алифатических полияитро-
зоединений в литературе описано получение 1,1,1,2- и
1,1,1,3-тетранитропропава [169 ].
Взаимодействием 1-бром-1-нитрозтана или 1-бром-2-нитро-
этана с AgC(NO*)s в среде эфира получается 1,1,1,2-тетра-
288
нитропропан—жидкость с уд. весом около 1,6 и т. кип. ^235°.
Наконец, полинитропарафины в отдельных случаях могут
быть получены нитрованием солей нитропарафинов. Так,
дикалиевая соль тетранитрозтана при действии смеси
концентрированных серной и азотной кислот превращается в гекса-
иитрозтан с т. пл. 142°.
Согласно американскому патенту [170], мононитропара~
фины с 3—5 атомами углерода с нитрогруппой, стоящей у
вторичного углерода, при обработке азотной кислотой при
температуре 150—250° и давлении приблизительно 10 атм. дают
динитропарафины с группой N0, у вторичного атома
углерода. Так, например, (СНз)зСН(МОз) в этих условиях образует
(СНз)^2(МО*)з, в то время как первичный нитропропан
O2NCH2CH2CH3 не образует динитросоединения.
По данным других патентов [171] нитрованию подвергается
смесь мононитропарафина и исходного углеводорода.
Концентрация применяемой HNOg составляет 40—70%. Реакция
проводится под давлением 56—85 атм. отношение HNOs :
{RNO3 + RH) = 0,5-+-1,0, Для лучшего контроля процесса
рекомендуется применение разбавителей N*, НдО, СО@.
§ 4, Свойства витроолефинов*
Известны два типа нитроолефинов: ос-нитроолефины,
содержащие нитрогруппу у непредельного атома углерода, и
нитроолефины с цитрогруппой в аллильном положении.
Нитроолефины представляют собой слегка желтоватые
прозрачные жидкости с резким раздражающим запахом. При
хранении нитроолефины полимеризуются, превращаясь в
темные вязкие смолы. Склонность к полимеризации уменьшается
с увеличением молекулярного веса. В присутствии щелочей
скорость полимеризации резко увеличивается.
%-Нитроолефины присоединяют спирты, амины, нитро-
ларафины и другие вещества. Эта способность обусловлена
наличием в молекуле нитроолефина сопряженной системы двой-
* Некоторые свойства нитроолефинов см. также § 1 данной главы
289
4 2 3 4
ных связей Z>C = С — N = О. Вещества, содержащие подвиж-
I
ный атом водорода, присоединяются к нитроолефинам в
положение 1,4. Возникающая при этом псевдокислота изомери-
зуется в соответствующее нитропроизводное:
О
Ас--с = м -+\с-
R Н ОН R
Способность нитроолефинов к реакциям присоедияения
позволяет использовать их в качестве исходных веществ для
синтеза нитро- и аминокетонов, динитропарафинов, эфиров
нитроспиртов и других соединений.
Восстановлением нитроолефинов можно получить амины,
оксимы, кетоны и нитропарафины. Характер продуктов
восстановления определяется условиями реакции (см. § 1 данной
главы).
Действие минеральных кислот
на нитроолефины
При действии концентрированной соляной кислоты на
нитроолефины образуются солянокислый гидроксиламин и
эс-окси-илиос-хлоркарбоновая кислота [172]. Эти же продукты
можно получить при обработке нитроолефинов эфирным
раствором хлористого водорода. Взаимодействие низших
нитроолефинов с эфирным и водным растворами НС1 Гыло изучено
Хисом и Розе [173]. Авторы предполагают, что в первую
стадию реакции хлористый водород присоединяется к
нитроолефинам в положение!—4. Образовавшаяся при этом аци-
форма хлорнитропарафина превращается под действием
соляной кислоты в солянокислую соль гидроксиламина и ос-хлор-
или ос-оксикарбоновую кислоту. Реакция протекает по схеме,
аналогичной схеме превращения первичного нитропарафина
в карбоновую кислоту:
19 А. В. Топчиев
290
О О
4-BCI ' Л 4-HCI /*
RsC = CHNO: > R2CCICH = N » R.CCICHONНОН -+
\
ОН
CCCHClNO , R3CCICCI = NOH , R.CCICOOH 4- NH.OHHCI
Такой механизм реакции авторы подтверждают следующими
фактами.
При действии эфирного раствора НС1 на 1-нитропропеи-1
и 1-нитро-2-метилпропен-1 в качестве промежуточных
продуктов были выделены соответствующие 1,2-дихлороксимы.
2-Хлор-1-нитропропан (продукт присоединения НС1 к
1-иитропропену в положение 1,2) не взаимодействует с эфирным
раствором НС1, тогда как 1-нитропропен в тех же условиях
превращается в ос-хлорпропионовую кислоту и гидрохлорид гид-
роксиламина. Это обстоятельство исключает возможность
присоединения НС1 к нитроолефину в положение 1,2 в первую
стадию реакции.
При действии соляной кислоты или эфирного раствора
НС1 на нитроолефины, не содержащие атома водорода у
углерода, несущего нитрогруппу, авторы получили темносиние
жидкости, обладающие свойствами лакриматоров;
предполагается, что это дихлорнитрозосоединения, образующиеся по схеме
О
4-HCI У 4-HG1
RR'C = CR^NOg > [RR'CCICR' = NOH] »
О
- tRR'CClCClR"NНОН] , RR'CCICCIR'NO
Нитроолефины легко растворяются в серной кислоте
(3 объема HsSO* на 1 объем Н%0). Растворение сопровождается
разложением, в результате которого образуются нитропара-
фин и альдегид. Нитропарафин под действием серной кислоты
превращается в карбоновую кислоту и сернокислую соль
гидроксиламияа [174 ]:
Нж80.
RCH = С (NO*) R' + Н.0 , RCH (ОН) CHNO,R' -^ RCHO + R'CH*NO%;
+ %0 + H,SO* -^ R'COOH
291
Взаимодействие нитроолефинов
со спиртами
Присоединение к нитроолефину молекулы спирта приводит
к образованию простого эфира нитроспирта:
\C = CHNO:+ROH-+ )
Реакция протекает при комнатной температуре в присутствий
щелочного катализатора.
Впервые конденсация спиртов с нитроолефинами была
осуществлена Мейзенгеймером и Геймом [175], Авторы
получили метиловый и этиловый эфиры ос-фенил-р-нитроэтиловога
спирта, действуя спиртовым раствором алкоголята на
о)-нитростирол и насыщая затем реакционную смесь
углекислотой. В данном случае реакция идет в две стадий:
сначала образуется натриевое производное аци-формы эфира
%-фенил-|3-нитроэтилового спирта, из которого углекислота
выделяет свободный эфир:
О
=^ CHNO, + CH,ONa - C^Hs
ОСН3 ОЩ
Н
Присоединение алкоголята к арилзамещенным гомологам
нитроэтилена описано в некоторых работах [176].
Конденсация алкоголятов с низшими нитроолефинами была
проведена Ламбертом и другими [177]. При действии на нитро-
этилен, нитропропилен и нитроизобутилен спиртового раствора
метилата и бутилата натрия и подкислении реакционной смеси
уксусной кислотой были получены соответствующие эфиры
нитросииртов. Взаимодействие 2-нитропропеиа-1 с метилатом
на*грия, наряду с метиловым эфиром 2-витропропилового спирта,
19*
приводит к образованию метилового эфира 2,4-диаитро-2-ме-
тиламилового спирта. Последний образуется в результате
присоединения нитропропена к первоначальному продукту
лонденсации — эфиру мононитросяирта:
jZHgC (NO.) = СН, + СН.ОН - СН.СН (N0.) СЩОСН, -^
^СН/Ш (NO,) CR,C (CH,) (NO,) СН.ОСН,
Взаимодействие АЖОГОЛЯтов с нитрооктеном описано в
работе Банера [178].
Взаимодействие нитроолефинов
с нитропарафинами
Нитроолефины присоединяют первичные и вторичные нитро-
парафины с образованием 1,3-динитропарафинов. Реакция
протекает по уравнению
ВВ'СШО, + СНВ' = CR"?fO* -^ ВВС (NO*) CHR"CHR"NO,.
Несмотря на то, что эта реакция представляет собой
удобный метод синтеза динитропарафинов, ей посвящено лишь
незначительное число работ,
Ламберт и Пиггот [179] исследовали реакцию
присоединения нитроэтана и 2-нитропропана к 2-нитропропену-1, 2-нитро-
бутену-2 и нитроциклогексену. Авторы пришли к выводу, что
реакция имеет общий характер, но выходы сильно колеблются
при работе с различными нитропроизводными. Так, при
взаимодействии 2-нитробутена-2 с 2-нитропропаном в присутствии
этилата натрия образуется 2,4-динитро-2,3-диметилпентан с
выходом 47% , а при взаимодействии 2-нитропропана с 2-нитропро-
иеном-1 выход продукта присоединения составляет 26%.
Наряду с дивитропарафинами часто получаются высоко-
полимерные побочные продукты, так как нитроолефины легко
аопимеризуются под действием щелочных агентов.
Продукт присоединения нитрометана к 1-нитро-2-метил-
арожвву-1 описан в работе Хэсса [180].
В: литературе имеются также сведения о црисоединении
феящшитрометааа к нитростильбену [181].
293
Банер и Кайт [182] нашли, что при конденсации щелочных
солей нитропарафинов с нитроолефинами диви^росоединения
получаются со значительно более высоким выходом, чем яри
конденсации нитропарафинов с нитроолефинами в присутствии
щелочного катализатора.
Взаимодействие нитроолефинов с
аминами и другими органическими
основаниями
Присоединение первичных и вторичных аминов к нитро-
олефинам приводит к образованию нитроаминов. Впервые
эта реакция была проведена Виландом и Саккеларнусом [183],
которые действием анилина на эфирный раствор нитроэтилена
получили кристаллический продукт присоединения —
М-(*-яитроэтил)-анилин:
Подробное исследование реакций взаимодействия о-нитро-
стирола с органическими основаниями было проведено Уорре-
лом [184]. В работе этого автора было испытано действие на
ю-нитростзфол 40 различных органических оснований. В
результате исследования было установлено, что нитростирол
взаимодействует со следующими основаниями: анилином,
п-толуидином, фенилгидразином, дифенилгидразином, р-яафтил-
гидразином, п-толилгидразином, семикарбазидом и
пиперидином. Согласно данным Уоррела, наиболее активно
взаимодействуют с нитростиролом производные гидразина. Введеаие
в молекулу ароматического амина или арилзамещенного
гидразина отрицательного заместителя понижает реакционную
способность основания. Так, например, анилин и фепилгид-
разин энергично взаимодействуют с нитростиролом, а п-алор*
анилин, п-нитроанилин и п-бромфеяилгидразин не образуют
продуктов присоединения.
Продукты конденсации аминов с ос-нитростильбеном б*ли
получены Дорноу и Робергом [1851.
294
Присоединение низших нитроолефинов к алифатическим
и ароматическим аминам описано в работе Ламберта и других
[177].
Получены также продукты присоединения нитроцикло-
гексена к фенилгидразину, семикарбазиду [186], тиосемикар-
базиду и п-толуидину.
Взаимодействие нитроолефинов с
соединениями, содержащими метиленовые
группы, активированные карбонильными
и карбоксильными группами
Нитроолефины конденсируются с веществами, содержащими
подвижные атомы водорода в метиленовых и метильшдх группах.
Так, например, при конденсации w-нвтростирола с
натриевым производным малонового эфира был получен
этиловый эфир 1-нитро-2-фенил-3-карбэтилмасляной кислоты [187]:
СН (СООС,Н,),
/ нвслота
- CHNO, + NaCH (COOC$H@)% - СА — СН »
СН =
СН
.^ С*Н,СН
Продукты конденсации w-иитростирола с ацетоуксусным
эфиром, бензоилацетоном и ацетилацетояом были получены
В. В. Перкалиным и А. С. Соповой [188]. Реакция
проводилась при комнатной температуре в бензольном растворе. В
качестве катализатора применялся триэтиламин. Выход продуктов
конденсации 77—98 %.
Авторы отмечают, что <*-нитростирол не взаимодействует
с метилэтилкетоном, в котором метиленовая группа
активирована только одной карбонильной группой.
В обзорной статье Хэсса [189] имеется указание, что при
конденсации ацетона с 1-нитро-2-метилпропеном-1 был
получен 5-нитро-4,4-диметилбентанон-2:
295
СН,
+ (СН,),С = CHNO* -^ O*NCH,C — СН,СОСН,
Условия реакции в работе не приводятся.
Дорноу и Роберг [190] осуществили присоединение ос-нитро-
стильбена к малоновому эфиру, ацетоуксусному эфиру, ацетил-
ацетону, бензоилацетону и фенилацетону. Малоновый эфир
вводился в реакцию в виде натриевого производного. Другие
кетоныприсоединялись к ос-нитростильбену только в присутствии
этилата натрия. Выход продуктов конденсации был невысоким
(не более 29%).
Присоединение нитроолефинов к диенам
Так как нитроолефины содержат активную двойную связь,
они могут конденсироваться по реакции Дильса — Альдера.
При обработке нитроэтилена, 1-нитропропена-1 и 1-нитропен-
тена-1 циклопеатадиеном получаются продукты конденсации
по следующей схеме [191]:
НС
НС
СН СН
/\ ~ CHR /
НС
CHR
СН,
НС I CHNO;
1-Нитропентен-1 конденсируется также с бутадиеном, метил-
бутадиеном и 2,3-диметилбутадиеном.
Моно- и бициклические нитросоединения были получены
при конденсации 2-нитробутена-2 и 2-нитропропена-2 с
бутадиеном и циклопентадиеном [192].
Фенилзамещенные нитропронзводиые циклогексена были
получены Вилдманом при конденсации w-нитростирола с
бутадиеном и метилбутадиеном [193].
Полимеризация нитроолефинов
Нитроолефины обладают большой склонностью к реакции
полимеризации [183, 194]. Так, нитроэтилен СНз = CHNO*,
296 ffw%/?ogaHwe з/гледо&фоЭое дэотной
полученный дегидратацией янтроэтилового спирта над H
полимеризуется при комнатной температуре в течение 2—3
дней в прозрачную вязкую жидкость. В то же время продукт,
полученный дегидратацией нитроэтилового спирта надКНЗО,,
не полимеризуется [195].
В присутствии водного раствора бикарбоната калия 2-нитро^
пропен полимеризуется [196 ] с образованием растворимого
в органических растворителях высокомолекулярного продукта
СН*
N0.
Этот полимер при гидрировании в присутствии катализатора
Ренея дает соответствующий поливиниламия
который хорошо растворим в воде, но в отличие от полимера
2-нитропропена нерастворим в большинстве органических
растворителей.
§ 5. Нитрование ненасыщенных углеводородов *
Ненасыщенные соединения нитруются довольно легко,
причем в иитропродуктах преобладают мононитросоединения.
Нитрование непредельаых углеводородов слабой азотной
кислотой было впервые исследовано М. И. Коноваловым.
Реакция проводилась в условиях, выработанных для
нитрования парафинов. Объектами исследования служили: мен-
тен, октен-1, триметилэтилен [197], боряилен, пинен [1981
и камфен" [199].
* Некоторые способы получения нмтроолефинов см. также в § 1
данной главы.
297
При нитровании ментена азотной кислотой (уд. в. 1,075)
в запаянных трубках было получено два изомерных мононитро-
соединения. Нитроментены растворялись в щелочи.
Щелочной раствор яри добавлении хлорного железа давал красные,
растворимые в эфире осадки (реакция, характерная для
первичных и вторичных нитрогрупи).
При нитровании октена-1 в условиях, аналогичных условиям
нитрования ментена, было получено нитросоединение состава
CgHigNOa Нвтросоединение растворялось в щелочи и давало
с хлорным железом резкую реакцию на первичное или
вторичное нвтросоединеяие.
Триметилэтилен нитруется слабой азотной кислотой
значительно труднее ментена и октена. Образовавшиеся нитропро-
дукты растворялись в щелочи.
Растворимые в щелочи нитросоединения были получены
также при нитровании непредельных бициклических
углеводородов — борнилена, камфена и пилена.
На основании полученных результатов М. И. Коновалов
пришел к выводу, что нитрующее действие азотной кислоты
направлено не на углеводородные группы, связанные
двойной связью, а на соседние с ними.
Работы М. И. Коновалова остались незаконченными. Не
все нитросоединения были выделены в частом состоянии, не
было окончательно установлено положение нитрогруппы в
молекуле углеводорода.
Позднее было показано, что при нитровании камфена [2001
азотной кислотой (уд. в. 1,075) образуются два структурно-
изомерных нитросоединения: вторичный ос-нитрокамфен и
(о-нитрокамфен с расположением витрогруппы у атома
углерода, связанного двойной связью.
При нитровании двизобутвлена [201] получается смесь
изомерных мононитросоединенвй. Нитрование проводилось
крепкой азотной кислотой при температуре немного выше 60°.
Нитрование двизобутилена 70-% ной азотной кислотой
проводится при температуре 70—90°. Это же нитрование может быть
осуществлено и разведенной азотной кислотой (20%) в случае
применения избытка азотной кислоты, добавления в нее окислов
азота и проведения реакции при температуре 70*. Последнее
указывает на то, что в приведенных условиях реакции
олефинов с HNO, они нитруются окислами азота, генерируемыми
азотной кислотой, т. е., вероятно, реакция идет по тому же
механизму, что и нитрование предельных углеводородов.
2-Этилгексен-1 нитруется так же легко, как диизобутилен.
При нитровании 2-этилгексена-1 80%-ной азотной кислотой
при температуре 70—75* получается 1-нитро-2-этилгексен-2
с выходом около 85,9%:
СН,= ССНвСЩСЩСНз NO,CH: — С = СНСН,СН,СН,
I -^ !
СЩСНз СН,СН,
Для нитрования октена-1 в 1-нитрооктен-2 требуются
более жесткие условия (температура 90—100*).
Нитрование олефинов в этих условиях, как мы видим,
аналогично хлорированию олефинов, т. е. введение в молекулу
аитрогруппы сопровождается передвижением кратной связи
в р-положение.
Мак-Ки [202] исследовал реакцию взаимодействия
этилена с концентрированной азотной кислотой. Работа была
предпринята с целью найти источник получения тетранитро-
метана. В результате исследования было установлено, что
основным азотсодержащим продуктом реакции этилена с
концентрированной азотной кислотой является тринитрометан. Наряду
с тринитрометаном были получены также р-нитроэтиловый
спирт, щавелевая кислота и двуокись углерода. Тринитрометан
не выделялся в чистом состоянии, а подвергался дальнейшему
нитрованию в тетранитрометан (при нагревании в присутствии
серной кислоты).
В работе Мак-Ки ток сухого этилена пропускался через
склянку Дрекселя с концентрированной азотной кислотой.
В различных опытах концентрация азотной кислоты
изменялась в интервале 95—100%. При пропускании этилена через
азотную кислоту наблюдались незначительное разогревание
и обильное выделение двуокиси углерода. Реакционная масса
выливалась в воду, нейтрализовалась и извлекалась эфиром.
ненасыщенные уелмо&^^о&м 299
При разгонке продуктов нитрования в вакууме был выделен
|3-нитроэтиловый спирт.
Р-Нитроэтиловый спирт и тринитрометан получались в
отношении 1 : 2,4. При добавлении к реакционной смеси
нитрата ртути выход нитросоединения повышался почти в полтора
раза. По мнению авторов, нитрат ртути подавляет
конкурирующую реакцию окисления этилена.
Образование тринитрометана из этилена и азотной кислоты
Мак-Ки объясняет при помощи следующей схемы:
UNO, ^
СН, = СН, ^ СЩ (ОН) — CHgNOg ^ СН*(NO.) Cf -
н
-СО, HN0,
^ СН. (N0.) СООН ^ CHgNOg ^ СН (N0.) = NOH -^
HN0,
-^ СН, (N0*). , (N0^ С = NOH ^ СН
При действии концентрированной азотной кислоты на изо-
бутилен Гайтингер [203] получил небольшое количество
непредельного нитросоединения состава C^H^NOg.
Нитросоединение растворялось в щелочи, а при нагревании
с водой в запаянной трубке распадалось на ацетон и нитрометан.
При восстановлении нитроизобутилена были получены
смесь нейтральных соединений, небольшое количество амина
и аммиак.
Гутри [204] пропускал амилен через нагретую до кипения
дымящую азотную кислоту. Основным продуктом реакции
являлось кристаллическое вещество состава С*Ню(Ж)*)*, природа
которого установлена не была. Это же соединение
образовывалось при действии на амилен двуокиси азота [205].
Невидимому, это было динитросоединение.
В работе Д. П. Коновалова [206] исследовалось
взаимодействие с азотной кислотой диизобутилена и триизобутилена.
Диизобутилен реагировал с азотной кислотой (уд. в. 1,4) очень
бурно, и опыт неизменно заканчивался взрывом.
Нитрование в среде уксусной кислоты также не дало
положительного результата, так как углеводород полимеризовался.
При нагревании диизобутилена с азотной кислотой уд. в.
300
1,28 на водяной баше образовалась зеленая жидкость, частично
растворимая в щелочи. При восстановлении ее железом и
уксусной кислотой была получена смесь аминов, из которой удалось
выделить амия состава CgHisNHa. Наряду с аминами было
получено вещество котонного характера. Моно- и диамины
образовывались также яри восстановлении продукта нитрования
триизобу тил еяа.
Буис [207] действовал концентрированной азотной кислотой
на каприлеж. Реакция протекала очень энергично, поэтому
реакционную смесь приходилось охлаждать или же сначала
проливать разбавленную азотную кислоту. В результате
реакции образовался жидкий продукт, содержание азота в котором
было средним между отвечающим моно- и динитрокап-
рилену.
При нитровании циклогексена [186] азотной кислотой
уд. в. 1,2 при температуре 60—65° в течение 3 час. были
получены 1-нитроциклогексен-1, вторичный яитроциклогексен
и исевдоиитрознт циклогексена. В кислотном слое
присутствовала адипиповая кислота. Средний выход смеси изомерных
нитроциклогенсенов, считая на вошедший в реакцию цикло-
гекбев, равен 16%. При взаимодействии этилена [208]
с#6,6%-ной азотной кислотой (без окислов азота) при
температуре 0—25° можно было предположить образование тринитро-
метана, р-нитроэтилового спирта или его эфира. Однако эти
соединения ив были обнаружены, а полученные продукты
реакции из-за неустойчивости не могли быть определены.
При пропускании ацетилена [209] в концентрированную
азотную кислоту можно получить тринвтрометан, из
которого дальнейшим нитрованием получается тетранвтрометан.
При нитровании бутадиена [210] азотной кислотой при 30°
(1 ч. азотной кислоты уд. в. 1,49—1,50 медленно приливается
при перемешивании к 3,5 ч. бутадиена) получается нитробута-
джев (лакриматор), содержащий в качестве примеси димер.
Нитрованием ретева Ci@Hi@ можно получить смесь моно-
штроретенов с выходом до 75% от теории [211]. Процесс осу-
щестжлялея следующим образом: в концентрированную азот-
яую кислоту (75 мл) прибавляли 20 г ретепа в 200 мл ледяной
301
уксусной кислоты при температуре не ниже 6—7°. Полученное
масло выливается в 1,5 л воды и экстрагируется петролейным
эфиром.
Продукты реакции при нитровании ретена можно разделить
%роматографированием на А1*О@. Главные продукты реакции:
9-нитроретен (28—31%), 3-нитроретен (17—18%), 4-нитро-
ретен (11—13%).
Получаются также с небольшим выходом 5(?)- или 8(?)-
нитроретен. Такие ненасыщенные соединения, как, например,
стирол, коричная кислота и жх производные, нитруются
так же, как и чисто ароматические соединения.
При нитровании стирола концентрированной азотной
кислотой Симон [212] получил смолистое вещество, при перегонке
которого с водяным паром был выделен кристаллический
w-нитростирол.
Влит ж Гофман [213] получили нитростирол с более
высоким выходом, вводя стирол в охлажденную льдом дымящую
азотную кислоту.
Механизм нитрования ненасыщенных соединений изучался
А. И. Титовым [214], который считает, что в зависимости от
условий реакции нитрование олефинов, как и ароматических
соединений, протекает по типам ионной или радикальной
реакций. Взаимодействие в обоих случаях начинается с атаки
электрофильных ; нитрующих агентов на подвижные и
пространственно доступные тг-электроны. Подвижность т-злек-
тронов обусловлена их сравнительно малой энергией свяви
с ядрами атомов углерода, а доступность—периферическим
расположением их орбит относительно связей между атомами
углерода.
При действии окислов азота на олефины образуются
свободные радикалы, что было доказаноА. И. Титовым опытными
данными.
* * *
В заключение настоящей главы, посвященной нитрованию
азотной кислотой парафинов и жирной цепи жирноаромати-
ческих углеводородов, необходимо дать оценку достоверности
предложенных для атой реакции схем механизма.
#%лгро«дние у&маоАоробо* дао/мной
Все описанные в литературе современные схемы такого
нитроваиия (А. И. Титова, Мак-Клирли и Дегеринга, Бахмана
с сотрудниками) совпадают в том, что оно представляет собой,
по терминологии Титова, молекулярно-радикальный процесс.
В этот термин Титов вкладывает представление о реакции,
происходящей путем образования из исходных веществ
свободных радикалов (R и N0%) и последующей рекомбинацией
последних. Процесс, являясь радикальным, не приводит
к возникновению цепной реакции, поскольку рекомбинация
алкильного радикала с двуокисью азота, как и всякая
рекомбинация, не сопровождается появлением новых, свободных
радикалов и, следовательно, представляет собой обрыв цепи.
Таким образом согласно современным взглядам, существует
принципиальное различие механизмов нитрования азотной
кислотой ароматических и жирных (в том числе и жирной
цепижирноароматических) углеводородов: нитрование первых,
как ясно из обзора главы II, представляет собой ионную ре~
акцию, нитрование же вторых — молекулярно-радикальную.
Столь резкое отличие механизмов, несомненно, должно
отражаться в значительных экспериментальных различиях. И
действительно, выше было показано, что, во-первых, двуокись
азота, являясь активным нитрующим агентом для жирных
углеводородов, не оказывает влияния на процесс нитрования
ароматических углеводородов азотной кислотой или
нитрующей смесью; во-вторых, добавка сильных кислот
значительно увеличивает скорость нитрования ароматических соедине-
.ний и мало сказывается на нитровании жирных углеводородов
и, в-третьих, скорость парофазного нитрования
ароматических соединений значительно меньше скорости жидкофаз-
ного, в то время как в случае жирных углеводородов
скорости нитрования в нарах и в жидкой фазе почти одинаковы.
К этому можно прибавить еще одно различие, наблюденное
различными авторами и заключающееся в положительном
каталитическом воздействии некоторых окислов металлов и
солей на нитрование ароматических соединений и
практически полное отсутствие влияния этих же веществ на паро*
фазное нитрование жирных углеводородов.
Ионный механизм нитрования ароматических соединений
может считаться общепринятым и обладающим значительной
степенью достоверности. Что касается нитрования жирных
углеводородов, то приведенные выше экспериментальные
отличия свидетельствуют лишь о неприменимости ионного
механизма течения процесса. Нужно, однако, подчеркнуть, что,
опровергая ионный механизм, эти экспериментальные отличия
еще не определяют, является ли титрование жирных
углеводородов молекулярной или молекулярно-радикальной или
цепной реакцией.
Однозначно решить, протекает ли этот процесс по чиста
молекулярному механизму или же по одному из двух свободна
радикальных (молекулярно-радикальному или цепному),
можно было бы, если бы имелись прямые, объективные методы
констатирования (и идентификации) свободных радикалов
в ходе такой сложной химической реакции. К сожалению,
слабое еще развитие химии свободных радикалов исключает
в настоящее время решение этого вопроса таким путем.
Другая, правда более косвенная возможность для суждения
о природе (молекулярной или свободно-радикальной) реакции
могла бы возникнуть при рассмотрении кинетических
закономерностей нитрования жирных углеводородов. Сюда
относятся такие экспериментально определяемые характеристики
реакции, как ее порядок по каждому из исходных веществ ^
энергия активации, изменение скорости в зависимости от
времени, температуры и давления, действие поверхности ^
добавок и т. д.
Приходится, однако, констатировать, что исследование нит
рования жирных углеводородов до настоящего времени
развивалось главным образом по линии синтетической, т. е. в
направлении изыскания методов получения ценных
азотсодержащих и других промежуточных и конечных продуктов,
возникающих в ходе этой реакции. Изучение же ее формально-
кинетических характеристик сильно отстало от этих
синтетических исследований и находится лишь на самой начальной
стадии, что исключает возможность установления таким путем
природы процесса с достаточной степенью достоверности,
304 #%гмроадн%е у&дббо&уюДое даотной
В результате, принятые в литературе представления о мо-
лекулярно-радикальной, а не чисто молекулярной природе
реакции нитрования жирных углеводородов не могут считаться
однозначно доказанными. Наглядной иллюстрацией этого
может, например, служить следующее. Бахман с сотрудниками
[215], подыскивая подтверждения свободно радикальной
природы парофазного нитрования жирных углеводородов как
азотной кислотой, так ж двуокисью азота, вынуждены признать,
что до сих пор наиболее убедительным доказательством такого
механизма является самый факт протекания этого процесса
в газовой фазе. Понятно, что доказательная сила такого
аргумента не может считаться значительной.
Несмотря на все это, автор настоящей книги также
склоняется к принятию свободно радикальной природы нитрования
жирных углеводородов. При проводившемся им изучении этой
реакции среди прочих был получен один экспериментальный
факт, наиболее естественное объяснение которого
предполагает, как считает автор, участие свободных радикалов в ходе
превращения. Автор имеет в виду открытое в работе совместно
с В. П. Алания [216] быстрое протекание термической реакции
одновременного нитрования и хлорирования метана в его
смесях с двуокисью азота и хлором в условиях, когда в отсутствие
либо двуокиси азота, либо хлора реакция практически не идет.
Это может быть объяснено образованием при совместном
присутствии двуокиси азота и хлора нитрозилхлорида
NOC1.Последний, вследствие малой анергии связи N—С1 (^38ккал/моль),
может относительно легко распадаться с образованием
окиси азота и атома хлора, дающего при взаимодействии с
исходным углеводородом алкильные радикалы (RH -}- С1 -*
->В"+НС1)лри более низкой температуре, чем двуокись азота.
Таким образом, процесс нитрования должен включать в себя
образование ж последующее превращение алкильных радикалов.
Важными и также утверждающими свободно радикальную
природу нитрования жирных углеводородов представляются
автору книги данные Хэсса и Рилея [217], показавших, что при
аарофазном нитровании тетразтилсвинца азотной кислотой
получаются азотнокислый свинец, нитроэтан и зтилнитрат.
305
Термический распад тетравтилсвинца с возникновением втиль-
ных радикалов сейчас хорошо изучен и, как известно,
представляет собой один из широко распространенных методов
получения свободных алкилъвых радикалов. Поэтому вряд ли можно
сомневаться в том, что в опытах Хэсса и Рилея нитрозтан и
зтилнитрат образовались в результате взаимодействия
двуокиси азота с зтильными радикалами. Образование при этом
нитрозтана не требует объяснения, образование же зтилнитрата
можно представить себе в результате первичного
возникновения этилнитрита по схеме CgHg-j- ONO-^CgHgONO с
последующим окислением в этилвитрат.
В общем, цитируемые работы свидетельствуют о
несомненной реальности образования нитропродуктов при
взаимодействии алкильных радикалов с нитрующим агентом.
Таким образом несмотря на то, что до настоящего времени
не удалось прямым и объективным методом обнаружить
свободные радикалы при нитровании жирных углеводородов,
их действительное участие в ходе этой реакции все же может
быть принято на основании некоторых косвенных доказательств.
Более детальное рассмотрение двух основных схем
нитрования жирных углеводородов (Титова и Бахмана) позволяет
сформулировать следующие замечания.
1. Доказательства, приведенные авторами этихмолекулярно-
радикальных схем в подтверждении их достоверности,
сводятся к наличию именно тех стабильных промежуточных
и конечных веществ, которые действительно найдены в
качестве продуктов нитрования жирных углеводородов.
Методологически такое подтверждение схемы является,
конечно, обязательным, но не однозначным. Действительно,
при той степени проникновения в истинный механизм
нитрования жирных углеводородов, которая существует в настоящее
время, образование одних и тех же стабильных продуктов
реакции может быть объяснено не одной, а несколькими
схемами с различными свободными радикалами.
2. Необходимо подчеркнуть, что образование основных,
включенных в схемы промежуточных веществ — нитрозосое-
20 А. В. Топчиев
306
динений и алкилнитритов, остается в известной мере
гипотетическим. Нитрозосоединения в числе продуктов нитрования
обнаружены не были, и потому образование из них через
стадию оксимов динитросоединений является лишь
предположением; что же касается алкилнитритов, то они, вероятно,
образуются, но их промежуточная роль никем объективно
доказана не была.
Здесь стоит отметить, что, согласно рассматриваемым
схемам Титова (главным образом для жидкофазного) и Бахмана
(для ларофазного нитрования), алкилнитрит представляет
собой тот промежуточный продукт, дальнейшее превращение
которого приводит к образованию продуктов окисления —
спиртов, карбонильных соединений. При этом, как мы видели,
Титов предполагает, что в рамках жидкофазвой реакции,
протекающей при низкой температуре, начальнаястадия превращения
алкилнитрита представляет собой его гидролиз; для
высокотемпературной же ларофазной реакции этой стадией, по Бах-
ману, является мономолекулярный распад с образованием
алкоксильного радикала, инициирующего цель с небольшим
числом звеньев. Такое предположение было сделано
Бауманом на основе имеющихся в литературе работ по изучению
термического распада алкилнитритов, происходящего в
отсутствие воды,— условия, которое никак нельзя считать
осуществляющимся при парофазном нитровании азотной кислотой.
Таким образом, если принять предположение Титова о
гидролизе алкилнитрита в ходе жидкофазного нитрования,
то становится сомнительным отсутствие такого гидролиза
в условиях ларофазной реакции. Возникает, следовательно,
вопрос, можно ли, принимая промежуточную роль
алкилнитритов, считать естественным такое совершенно различное
(в зависимости от температурных условий) их дальнейшее
превращение, постулируемое обеими схемами.
3. Схема Бахмана не расшифровывает яутей образования
олефинов, получающихся при ларофазном нитровании жирных
углеводородов; путь же, предлагаемый для этого схемой
Титова, не может, как будет сейчас показано, считаться
удовлетворительным.
307
Для вскрытия истинного механизма реакции нитрования
вопрос об образовании олефинов значительно более серьезен,
чем это может показаться с первого взгляда. Проще всего
предположить образование непредельных углеводородов
путем распада алкильвых радикалов с разрывом либо связей
С—С, либо связей С—Н:
Щ - СН', + <%
Известно, однако, что такой крекинг предельных
углеводородов происходит с измеримыми скоростями лишь при
температурах, значительно превышающих температуры ларофаз-
ного нитрования, и, следовательно, не может служить
источником олефинов в этом случае. Отсюда следует, что в ходе
нитрования создаются условия, благоприятствующие
образованию олефинов.
Можно предположить, что таким благоприятным условием
могло бы явиться возникновение олефинов яе из алкильного
радикала, а на одной из стадии его дальнейшего превращения.
При этом, разумеется, каждая из стадий такого превращения,
включая и окончательное возникновение олефина, должна
протекать с большей легкостью, чем распад алкильного
радикала.
Именно это, невидимому, предполагает Титов, приводя
следующую последовательность стадий:
N0, Н,0 HN0,
В » RONO ;zt ROH —Zt RONO, ^ олефииы + HNO,.
HNO,
Здесь алкильный радикал после ряда превращений переходит
в алкилнитрат, последний же, отщепляя азотную кислоту,
дает соответствующий олефин.
Трудно оценить, является ли такой путь более вероятным,
|! чем образование олефинов непосредственно из алкильных
радикалов, тем более, что ни в одной из опубликованных
I работ по исследованию термического распада алкилнитратов
J!j [218] ни разу не констатировалось образование олефинов.
20*
308 #цлухниу»ыб углееоДоробое азотной
Не предрешая вопроса о том, правильно ли предположение
об образовании олефинов не непосредственно из алкильного
радикала, а на одной из стадий его дальнейшего превращения,
и возможно ли найти более удачную, чем это сделал Титов,
химическую конкретизацию такого предположения, автор
настоящей книги хочет обратить внимание читателя на хорошо
известный факт значительного понижения температуры
крекинга жирных углеводородов (с образованием олефинов) в
присутствии кислорода. Механизм такого действия кислорода до
сих пор неясен, однако самый факт не подлежит сомнению и
представляется автору возможной причиной образования
олефинов с измеримыми скоростями уже при температурах
парофазного нитрования.
Если бы такое объяснение оказалось верным, это означало
бы наличие свободного кислорода в зоне реакции парофазного
нитрования. Трудно переоценить значение такого
обстоятельства для протекания всего процесса, особенно ввиду
многочисленных экспериментальных подтверждений положительного
влияния добавок кислорода на нитрование жирных
углеводородов. В частности, несколько по-иному, чем это сделано
в рассматриваемых схемах, можно представить образование
алкильного радикала, инициирующего нитрование (см.
ниже, л. 5).
4. Схемы ларофазного нитрования Бахмана и Титова не
предусматривают также путей образования получающихся при
этом процессе СО и СО а.
В рамках этих схем образование продуктов глубокого
окисления и деструкции парафиновой цели, какими являются
окисл# углерода, естественнее всего рассматривать как
результат дальнейшего воздействия нитрующего агента на
получающиеся в ходе нитрования альдегиды и олефины. Можно
думать, что оба эти класса соединений легче подвергаются
воздействию со стороны, например, двуокиси азота, чем
жирные углеводороды.
Из современных работ по исследованию взаимодействия
двуокиси азота с альдегидами можно назвать работу Мак-
Доуэлла и Томаса [219], которые изучали реакцию двуокиси
309
азота с ацетальдегидом (окислы углерода найдены не были),
и работы Полларда и Виатта [220], исследовавших
взаимодействие двуокиси азота с формальдегидом при температурах
118—184°. При этом констатировано образование окиси азота,
воды и окислов углерода (СО и СО*). Авторы предлагают
следующий механизм реакции, включающий возникновение
промежуточного бинарного комплекса между формальдегидом и
двуокисью азота:
1) НСНО Ч- NO* -»(HCHO-NO,);
2) (HCHO.NO,) + NO, -^ СО + Н,0 + NO + N0,;
3) (HCHO.NO,) Ч- NOg -^ СО, + Н,0 + 2NO.
Хотя авторы считают, что экспериментальные результаты
объясняются предложенной ими схемой, следует, однако,
признать, что она носит скорее характер стехиометрическпх
уравнений, чем совокупности истинных элементарных
процессов. Действительно, осуществление, например, стадии 3) в один
элементарный акт включает разрыв шести и образование четырех
валентных связей. Трудно предположить наличие столь
сложного элементарного акта.
Исследованию взаимодействия двуокиси азота с
непредельными углеводородами посвящены появившиеся в последние
годы две работы — Котрела и Грехема [221] и Томаса [222].
В обеих работах (в первой исследовалась реакция двуокиси
азота с этиленом, а во второй с ацетиленом) в числе продуктов
превращения были обнаружены окислы углерода. Механизм
их образования в первой работе не предложен, а во второй
сводится к следующему:
СН = СН 1 СН—СН
2N0;
1)
2)
3)
4)
5)
СА-
СО-
со-
нсо
f 2N0,-
-СНО +
-СНО-+
+ HNO,
сн =
.оно
-NO,-
N0,-»
С0 +
—»
НаО
+ С0
со.
= сн -
0N0_
— СНО
+ НСО
+ CO + NC
).
сн—
II
HNO,;
N0;
СН
II
310
Предложенный механизм сводится к первоначальному
образованию альдегида (глиоксаля) и последующему его окислению
кислородом двуокиси азота, превращающейся при этом в
азотистую кислоту и окись азота. Схема этого окисления довольно
сложна и мало убедительна. Если еще стадия 2) отщепления
атома водорода из карбонильной группы с образованием
свободного радикала СО — СНО и HNO% может считаться
вероятной, то предположение о стадиях 3) и 5) представляется
пока мало обоснованным.
Таким образом в современных механизмах, предложенных
для реакции двуокиси азота с альдегидами и непредельными
углеводородами и предполагающих прямое и непосредственное
воздействие двуокиси азота как на исходные вещества, так
и на все промежуточные образования, мы не находим серьезно
обоснованных путей образования окислов углерода. Такое
положение этого вопроса в настоящее время может, конечно,
иметь временный характер. Не исключена возможность
выдвижения в дальнейшем других схем, хотя и основанных на том
же принципе непосредственного воздействия двуокиси азота,
но с другими промежуточными лабильными образованиями
и с более вероятными при этом путями образования окислов
углерода.
Автор настоящей книги, однако, не считает исключенным
образование в условиях парофазного нитрования окислов
углерода путем окисления альдегидов не двуокисью аэота, а
свободным кислородом. Хотя такой механизм возникновения
окислов углерода пока является лишь предположительным,
все же необходимо отметить, что, предположив наличие в зоне
реакции ларофазного нитрования свободного кислорода (см.
л. 3), трудно оправдать дальнейшую окислительную
деградацию альдегидов взаимодействием с двуокисью азота,
а не с кислородом. Легкая окисляемость альдегидов кислородом
хорошо известна, а предложенные для этого
радикально-цепные схемы вполне логичны. Поэтому автор считает
необходимым при вскрытии механизма нитрования жирных
углеводородов иметь в виду и такую возможность образования окислов
углерода.
311
5. Отсутствие цепного механизма реакции, утверждаемое
схемами Титова и Бахмана, не может считаться окончательно
установленным. Действительно, как уже было сказано,
кинетические характеристики нитрования лона еще мало изучены,
а ведь именно они являются основным материалом для решения
вопроса о цепной природе химической реакции.
К этому можно добавить, что предположение о наличии в
зоне реакции нитрования свободного кислорода заставляет
с особой осторожностью отнестись к отрицанию цепной
природы этого процесса. Возможно, разумеется, что цепная природа
присуща не стадиям получения азотсодержащих продуктов,
поскольку образование последних скорее всего является, как
справедливо принимают обе схемы, следствием реакции
рекомбинации радикалов (R и N0,), а окислительному направлению,
приводящему к дальнейшему превращению уже с помощью
кислорода таких промежуточных продуктов, как альдегиды,
спирты и др. В этом случае в рамках общей реакции нитрования
мы имели бы интересный случай одновременного протекания
как чисто радикальной, так и цепной реакции.
Не следует думать, что оба эти направления реакции
должны развиваться независимым образом,не влияя друг на друга.
Свободные радикалы, ведущие, например, цепное окисление
альдегидов, несомненно способны взаимодействовать и с
исходным углеводородом, хотя, возможно, и с меньшей
эффективностью. В результате такого взаимодействия естественно
предположить образование алкильного радикала (RH + А->
R'+АН, где А'—свободный радикал), который путем
рекомбинации с двуокисью азота будет поддерживать чисто радикальное
направление реакции.
Таким образом совсем не обязательно представлять себе,
как это делает Титов, первичное образование алкильных
радикалов только в результате реакции исходного углеводорода
с двуокисью азота:
RH + N0% -* R + HNO,.
Необходимо, кстати, отметить, что такой элементарный
процесс вообще еще никем не был констатирован.: По крайней мере
312
на него нет ни одной ссылки в книге Стиси «Реакции атомов
и свободных радикалов» (2-е издание, 1954 г.).
Предположение о наличии кислорода в зоне реакции
нитрования дает еще и другую возможность возникновения
алкильного радикала
RH + O,-* R' + HO,.
Такой элементарный акт предполагается в ряде современных
схем окисления углеводородов; его принимает также и
Н. Н. Семенов [223].
Вся совокупность развитых в этом заключении соображений
о возможном протекании нитрования жирных углеводородов,
включая и критические замечания в отношении некоторых
частей схем Титова и Бахмана, привели автора настоящей
книги к постановке в его лаборатории соответствующего
исследования, имеющего целью получение дополнительного
экспериментального материала, необходимого для более
детального и оправданного раскрытия истинного механизма
этой реакции. Это исследование включает в себя: 1) выяснение
всей формально кинетической картины протекания нитрования
жирных углеводородов, 2) установление кинетики по
израсходованию исходных и накоплению промежуточных и
конечных продуктов по всему ходу превращения и, наконец,
3) изучение в чистых условиях некоторых из основных
элементарных процессов, осуществление которых предполагается
в реакции.
ЛИТЕРАТУРА
1. I. I. -Be ? ad. J. prakt. Chem., 48,345 (1893); 63, 193 (1901).
2. H. Prins. Rec. tray, chlm., 44, 1051 (1925).
3. V. M ey er. Ber., 5, 203 (1872).
4. H. Кг а 08 e. Chem. Ztg., 40, 810 (1916).
5f R.Lyons, L. Smith. Вел, 60, 13, 173 (1927).
6. M. Montmollin, А с h e rm a n n. Helv. Cbim. Acta, 12,
873 (1929).
7. O. Shales. Ber., 68, 1579 (1935).
8. P. S a b a t ie r, J. Send erena. Compt. rend., 135, 225 (1902);
Ann. Cbim. et Phys., 4, 414 (1905).
9. J. Lub 1 eln. Ber., 10, 2083 (1877); 34, 284 (1901).
313
10. V. Meyer, Hoffmann. Ber., 27 1347 (1894).
11. E. Bamberger. Ber., 27, 1347 (1894).
12. A. Kir pa 11. Ber., 25, 1714 (1892).
13. W. Tranbe, A. Schulz. Ber., 56, 1856 (1923).
14. E. S с hmid t, A. As с b er 1, L. Meyer. Ber., 58,2430(1925).
15. P. Pi err on. J. Chem. Soc, 76, 844 (1899).
16. И. Б ев а д. ЖРХО, 39, 947 (1907).
17. К. J ohnson, E. D egering. J.Am.Gbem. Soc.,61,3194(1939).
18. E. Bamberger, Rest. Ber., 35, 45 (1902).
19. S. B. L i p p i n с о t, H. В. Н a s a. Ind. Eng. Chem., 31, 119
(1939).
20. J. Nef., Ann., 280, 263 (1894).
21. K. Johnson, E. D eg e г in g. J. Org. Chem., 8, 7 (1943).
22. S. B. L i p p i n с о L J. Am. Cbem. Soc., 62, 2604 (1940).
23. J. Tsch erniak. Ber., 8, 609 (1875).
24. W. D. R amag e. Амер. патент 1996388 (2/IV 1935).
25* В. Vanderbilt. Амер. патент 2181411 (28/XI 1939).
26. L. Henry. Bull. Acad. Belg., 34, 547 (1898).
27. F. R. S ch aw. Bull. Aead. Belg., 34, 1019 (1898).
28. W. S t eink op f, M. К oh n el. Ber., 75, 1323 (1942).
29. M. И. Коновалов. ЖРХ0, 38, 607, (1906).
30. E. Mills. Ann., 160, 117 (1871).
31. A. S oaie. Thesis, Purdue Univ. (1939); H. B. H ass, A. G. S тз-
sie, С W. Scaife. J. Org. Cbem., 15, 8(1950).
32. P on z i o. Gaz. Chim. Ital., 36, 2, 100, 338 (1905).
33. Rein bold. Ann., 451, 161 (1927).
34. W. S t ein к op f. Ber., 42, 3925 (1927).
35. H. H a s s, E. R i 1 e y. Chem. Rey., 32, 373 (1943).
36. Амер. патент 2138166 (29/XI 1938).
37. V i 11 i e r s. Bull. Soc. Chim., 43, 323 (1928).
38. L. Henry. Compt. rend., 120, 1265 (1895).
39. К am 1 e t. Амер. патент 2151517 (21/111 1939).
40. В. Vanderbilt, H. В. Haas. Ind. Eng. Cbem., 32, 34 (1940).
41. Японск. патент 156256 (1949); С. А., 48, 2008 d (1950).
42. Японск. патент 6910 (1951); С. А., 48, 1412 d (1950).
43. D a r z e n s. Compt. rend, 229, 1148 (1949).
44. H. F e u e r, G. В. В а с h m a n, J. P. Kiaperaky.
J. Am. Qnem. Soc, 73, 1360 (1951).
45. Амер. патент 2387019; С А., 40, 1171 (1946).
46. Шведск. патент 195832 (27/V 1952); С. А., 6974 (1953).
47. N. S. M a ran a, R. R. Z е 1 i n s k i. J. Am, Chem. Soc., 72,
5329 (1950).
48. Амер. патент 2544103 (1951); С. А., 49, 7587f (1951).
49. F. Hofwimmer. Schiess- und Sprengatoffweaen, 7, 43 (1912);
314
J. Soc Chem. Ind., 31, 204 (1912).
50. В. Vanderbilt. Амер. патент 2177757 (31/Х 1939).
53. Е. Schmidt, R. W ilk end or f. Ber., 52, 391 (1919).
52. H. В. Haas, E. В. Hodge, В. Vanderbilt. Ind. Eng.
Chem., 28, 339 (1936).
53. E. Knoevenagel, Walter. Ber., 37, 4507 (1904); Grand,
H a p p i n. J. Org. Chem., 48, 1 (1953).
54. D. E. W or all. Organie Synthesis, New York, vol. 1,405(1932);
vol. IX, 662 (1929).
55. K. W. Rosenmnnd. Ber., 46, 1034 (1913).
56. H. B. H ass, E. F. Riley. Chem. Re?., 32, 410 (1943).
57. E. Schmidt, G. Rutz, M. T теп el. Ber., 61, 472(1928).
58. H. Wieland, E. Sakkelarioa. Ber., 52, 898 (1919).
59. G. Buckley, C. Skaife. J. Chem. Soc. 1471 (1947); С A.,
44, 653e (1950).
GO. С S cd i f e. Амер. патент 2460243 (1945); С, I, 1910 (1950).
61. J. T i n d a 11. Private communication; H. B. H a s s, E. F.
Riley. Chem. Rev., 32, 410 (1943).
62. Cerf d e Manny. Bull. Soc. Chim., [5], 7, 133 (1940).
63. В о live a ult, Wahl. Bull. Soc. Chim., [3], 29, 517 (1903);
D. N a i g h t i и g a 1 e, J.R.Janes. J. Am. Chem. Soc., 66,
352 (1944).
64. M. Senkna. J. Am. Chem. Soc, 68, 10 (4946).
65. H. Johnson. J. Am. Chem. Soc., 68, 42 (1946).
66. H. Johnson. J. Am. Chem. Soc., 68, 14 (1946).
67. H. В r us on, G. Butler. J. Am. Ghem. Soc., 68, 2348 (1946).
68. W. Kermack, W. Muir. J. Chem. Soc., 300 (1933).
69. Амер. патент 2465958 (29/111 1949); С. А., 5424 (1949).
70. Амер. патент 2391847 (25/ХН 1945).
.71. М. Sen k us. J. Am. Chem. Soc, 68, 1611 (1946).
72. А. В 1 о m q u i s t, J. Shelley. J. Am. Chem. Soc, 70, 147
(1948).
73. W. E m m о n d s, W. С a n а о u, J. D a w s о n, R. Ross.
J. Am. Chem. Soc, 75, 1993 (1953).
74. H. Snider, W. Hamlin. J. Am. Chem. Soc, 72, 5082 (1950).
75. M. И. Коновалов. ЖРХ0, 25, 389 (1893).
76. M. И. Коновалов. ЖРХ0, 25, 472 (1893); Ср. также
В. В. Марковников. Ber., 33, 1906 (1900).
77. М. И. Коновалов. Вег., 28, 1855 (1895).
78. R. A. Worst all. С, I, 926 (1898).
79. С. G r u n d m a n a. Ber., 77, 82 (1944).
80. М. И. Коновалов. ЖРХО, 31, 255 (1899).
81. R. A. Worst а 11. J. A m. С h e т., 202, 664 (1898).
82. М. И. К он ов а лов. ЖРХО, 36, 232 (1904).
315
83. М. И. Конов а л о в, X. Гтрев и ч. ЖРХО, 37, 537 (1905).
84. Н о р к ins, В ас. Амер. патент 1694097 (1928).
85. В. В. Марковников. Ann., 302, 15 (4898); В. В. М а р к о в-
нпков. ЖРХО, 31, 47, 530 (1899); 32, 1441 (1900); 35, 1033 (1903).
36. В. В. М а р к овников. Бег., 33, 1906 (1900).
87. Амер. патент 1588027; С, Н, 1189(1926).
88. Н. В. Н ass, Е. Т. R i 1 е у. Ghem. Rev., 32, 377 (1943).
89. В. В. Марковников. ЖРХО, 20, 118 (1888); 27, 174 (1895);
28, 125 (1896); 30, 151 (1898); 31, 215 (4899); 32, 302 (1900).
90. А. И. Т и т о в. Журя. общ. химии, 19, № 8, 1464 (4949).
91. С. С. Н а м е т к п я. ЖРХО, 40, 184, 1570 (1908).
92. М. И. Коновалов. ЖРХО, 38, 134 (1906).
93. С. С. Наметкин, А. С. Забродина. Докл. АН СССР, 75,
№ 3, 395 (1950).
94. С. С. Наметкин, С. С. Н и ф о в т о в а, Р. Я. Сущик.
Докл. АН СССР, 70, № 2, 241 (1950).
95. P. P on I, N, Cos t ас hes с о. С, I, 624 (1903).
96. С. С. Наметкин, А. С. Забродина. Докл. АН СССР,
75, # 4, 543 (1950).
97. С. С. Наметкин, А.С.Забродина. Докл. АН СССР, 75,
№ 5, 701 (1950).
98. С. С. Наметкин, А. С. Забродина. Докл. АН СССР,
81, № 1, 55 (1951).
99. С. С. Н аметкин. ЖРХО, 47, 405 (1945); 47, 1609 (1915).
100. С. С. Наметкин, О. С Мадаева-Сычева. ЖРХО,
57, 382 (1925).
101. С. С. Наметкин, М. Г. Р у д е н к о, В. Н. Громова.
Иов. АН СССР, серия хим., 61 (1941).
102. С. С. Н а м е т к и н. ЖРХО, 47, 1596 (1915).
103. С. С. Наметкин, А. М. Хухрикова. ЖРХО, 47, 425 (1915).
104. С. С. Н а м е т к и н. ЖРХО, 47, 409 (1915).
105. С. С. Наметкин, Л. Н. Абакумовская. ЖРХО,
47, 414 (1945).
106. С. С. Наметки в, А. С. Забродина. ЖРХО, 57, 87 (1925).
107. П. П. Шорыгип, А. М Соколова. ЖРХО, 62, 673
(1930).
108. А. И. Т и т о в. Жури. общ. химии, 16, № 11, 1897 (1946).
109. А. И. Т и т о в. Журн. общ. химии, 7, 1695 (1937).
110. А. И. Титов. Журн. общ. химии, 8, 465, 473, 534 (1938).
111. А. И. Титов. Усп. хим., 21, 881—914 (1952).
112. А. И. Т и т о в. Журн. общ. химии, 18, 1312 (1948).
113. А. И. Титов, М. К. Матвеева. Сборник статей по общей
химии, I, 247 (1953), Изд-во АН СССР.
114. А. И. Т иго в. Журн. общ. химии, 18, № 2, 190 (1948).
316
115. А. И. Титов, Н. В. Щитов. Докл. АН СССР, 81, 1085 (1951).
116. А. И. Титов, В. В.Смирнов. Докл. АН СССР, 83, 243 (1952).
117. Р о n z i о. Gazz. Chim. Ital., 36, 588 (1906).
118. D. Iffland, G. Griner. J. Am. Ghem. Soc., 75, 4074 (1953).
119. A. F. H ol lemann. Rec tray, chim., 14, 121 (1895); W. В aker
C. K. In go Id. J. Ghem. Soc., 2462 (1926); C. U rbanski
и др. С. А., 33, 1528 (1939).
120. L. Fieser, Doer ing. J. Am. Ghem. Soc., 68, 2252 (1946);
M. Mil one, A. Mass а. С. А., 34, 4571 (1940).
121. H. B. H ass, E. B. Hodge, B. Vand erbilt. Ind. Eng.
Cbem., 28, 341 (1936).
122. G. B. Bachmann, L. M. Add is on, J. V. Hewett,
L. Kohn, A. M i 1 1 i k a n. J. Org. Ghem., 17, 906 (1952).
123. G. B. Bachmann, H. B. H ass, L. M. Add is on. Ibid.,
17, 914 (1952).
124. G. B. Bachmann, J. V. Hewett, A. G. Millikan.
Ibid., 17, 933 (1952).
125. G. B. Bachmann, L. Kohn. Ibid., 17, 942 (1952).
126. G. Bachmann, M. Pollack. Ind. Eng. Ghem., 46, № 4,
713 (1954).
127. Амер. патент 2489320; С. А., 44, 2545в (1950).
128. L. Stengel. Амер. патент 2575855; С. А., 46, 1308 (1952); Амер.
патент 2512587; С. А., 44, 8942 d (1950).
129. Н. В. Hass, L. G. Alexander. Ind. Eng. Cbem., 41, 2266
(1949).
130. Амер. патент 2609401; С. А., 48, 2758 (1954).
131. R. Kirk, D. О th mer. Encyclop. Ghem. Technol., 9, 441 (1952).
132. Б. Н. Тютюнников, Н. П. Маньковская, Т. Д.
Явлинский. Укр. хим. жури., 8, № 1, 87 (1954).
133. Н. В. Hass, Patterson. Ind. Eng. Cbem., 30, 67 (1938).
134. Seigle, H. B. Hass. Ind. Eng. Chem., 31, 648 (1939).
135. H. B. Hass, Robinson. J. Am. Chem. Soc., 72, 3579 (1950).
136. H. H epf t, O. Schickk. C, II, 5105 (1953).
137. M. H. Danzig, H. B. Hass. J. Am. Ghem. Soc., 66, 2017
(1944).
138. M. И. Коновалов. ЖРХ0, 37, 1119 (1905).
139. M с Clearly, Degering. Ind. Eng. Chem., 30, 64 (1938).
140. Амер. патент 2291345 (1942); С. А., 47, 2766c (1953).
141. G. Bachmann, M. Atwood, M. Pollack. J. Org.
Chem., 19, 3 (1954); G. Tin da 11. Амер. патент 2465959 (1946).
142. H. В. H ass, H nd gin. J. Am. Chem. Soc., 76, №10, 2692(1954).
443. J. L. Bullock, E. T. Mitchell. J. Am. Cbem. Soc., 63,
3230 (1941); 65, 2426 (1943).
317
144. R. Т. Blickenstaff, II. В. 13 ass. J. Am. Chem. Soc, 08, 1431
(1946).
145. E well. Ber., 35, 3554 (1902); 41, 2219 (1908).
146. Burton. Thesis, Purdue University.
147. H. Hass, H. Hihsmann, E. Pierson. 1Ы. Eng. Chem.,
32, 427 (1940).
148. Norman Levy. Амер. патент 2394315; С. А., 40, 2454 (1946).
149. W. Marschall. Амер. патент 2654788 (1953); С. А., 48, 114791
(1954).
150. О. Wichterle, M. Kolinsky, S. Svastal. РЖХим.
№ 17, реферат 40439 (1954).
151. Seigle, H. Hass. J.Org. Chem., 5, 100(1940).
152. A. An g el i, Z. Alessandri. С. А., 2634 (1910).
153. Амер. патент 2181531 (1939).
154. H. Д. Зелинский. ЖРХО, 26, 610 (1894).
155. С. Р a h n е г. Ind. Eng. Chem., 44, 317 (1952); С. А., 44, 2876 (1950).
156 Н. Sh eohte r, R. К а р 1 а п. J. Am. Chem. Soc, 75, 3980](1953).
157. G. Born. Ber., 29, 9 (1896).
158. Амер. патент 2469396 (1949); С. А., 43, 6646 (1949); амер. патент
2489122 (1949); С. А., 44, 1526 (1950).
159. P. H. Groggins. Unit processes in organic synthesis. New York,
стр. 6 (1947).
160. Герм, патент 956547 (1950); С. А., 1024е (1952).
161. М. L ет у, J. R os e. Quart. Re?., 2, 358 (1948).
162. M.L arris on, H. Hass. С A., 40, 3475 (1946); H. H ass,
J. В our land. С. А., 44, 2969 (1950).
163. H. Plan t. Амер. патент 2544103 (1951); С. А., 45, 7587 (1951).
164. С. В ah пег, Н. Kite. Амер. патент 2477162 (1948); С, I, 2031
(1950).
165. С. Banner, H. Kite. J. Am. Cnem. Soc., 71, 3597 (1949).
166. R. Kirk, D. Othmer. Encyclop. Chem. Technology 9, 451
(1952).
167. H. Shech ter, L. Z eldin. J. Am. Chem. Soc., 73, 1276 (1951).
168. W. R eich, G. Rose, W. Wilson. J. Chem. Soc., 1235 (1947).
169. J. H ann urn. Амер. патент 2583048; С. А., 46, 3735 (1952).
170. Амер. патент 2425367; С. А., 7409 (1947).
171. W. Den ton. Амер. патент 2538298; С. А., 45, 3862 (1951);
герм, патент903453; С, 5170 (1954); С, 2026 (1953).
172. Priebs. Ann., 225, 319(1884); Н. Н. Haitinger. Monatsh.
Chem., 2, 287 (1881).
173. R. H. Heath, J.D.Rose. J. Cnem. Soc., 1485 (1947).
174. H. Hass, A. Susie, H eider. J. Org. Cnem., 15, 8 (1950).
175. J. Meisenheimer, F. Helm. Ber., 38, 466 (1905).
176. J. Meisenheimer, H. Johonson. Ann., 355, 293 (1907).
318
A. D о г п о w, F. Roberg. Bar., 83, 261 (1950); амер. патент
2562151; С, А., 46, 3565 (1952).
177. A. Lambert, С. W. Scaife, A. E. Wilder-Smith.
J. Chem. Soc, 1474 (1947).
178. С. Bahaer. J. Tenn. Acad. Sci., 23, 281 (1948); С. А., 43, 17116
(1949).
179. A. Lambert, H. A. P iggo tt. J. Chem. Soc., 1489(1947).
180. H. В. На 98.' Tnd. Eng. Chem., 35, 1151 (1943).
181. F. Heim. Ber., 44, 2019 (1911); D. Worral. J. Am. Chem.
Soc, 57, 2299 (1935).
182. C. T. Banner, H. T. Kite. J. Am. Chem. Soc., 71, 3597(1949).
183. H. Wiel and, E. Sakkelarious. Ber., 52, 898 (1919>.
184. D. Worral. J. Am. Chem. Soc, 49, 1598 (1927).
185. A. Dornow, F. Roberg. Ann., 578, 95 (1952).
186. А. В. Топчиев, E. Л. Ф а н т а л о в а. Докл. АН СССР, 88,
№ 1, 83 (1953).
187. Е. Р. К о Ы е г, Н. Engelbrecht. J. Am. Chem. Soc, 41,
366 (1919).
188. В. В. Перекалин, А. С. Сопова. Журн. общ. %имвж, 24,
513 (1954).
189. Н. В. На as, E. Т. Riley. Chem. Нет,, 32, 414 (1943).
190. A. Dornow, F. Roberg. Ann., 578, 101(1952).
191. К. A d ler, H. F. R i с k or t, E. Windemnth. Ber., 71,
2451 (1938).
192. D. V. Naightingal, M. Maintal, A. J, Gallher.
J. Am. Chem. Soc., 73, 4852 (1953).
193. W. С Wild man, R. B. Wild man. J. Org. Chem., 17,
581 (1952).
194. Schmidt, R u tz. Ber., 61, 2142(1928).
195. Kaishi Norn a. Chem. High. Polymers, 5, 99—103 (1948); С. А.,
46, 4471 (1952).
196. Blomquist, Topp, Johnson. J. Am. Cbem. Soc, 67,
1519 (1945).
197. M. И. Коновалов. ЖРХ0, 26, 380 (1894).
198. M. И. Коновалов. ЖРХ0, 31, 57 (1899).
199. M. И. Коновалов. ЖРХ0, 34, 121, 43 (1902).
200. С. С. Наметкин, Е. Л. Фанталова. Докл. АН СССР, 87,
№ 6, 979 (1952).
201. А. Д. Петров, М. А. Б у л ы г и и а. Докл. АН СССР, 77, № 6»
1031 (1951).
202. М с К ее. J. Chem. Soc, 962 (1927); Or ton, Me К ее.
J. Chem. Soc, Щ 283 (1920).
203. H. H aitinger. Ann., 193, 366 (1878); Monetsh., 2, 286 (1881).
204. F. G u th ri e. Ann., 116, 248 (1860).
319
205. F. Guthrie. Ann., 119, 83(1861).
206. Д. П. Коновалов. Bull. Acad. Sci. St. Petersb., 27, 38 (1888).
207. J. В о u i s. Ann. Ghim. phys., 44, 118 (1855).
208. Michael, К aria on. J. Am, Chem. Soc, 57, 1268 (1935).
209. K. F. Eager. Ind. Eng. Chem., 41, 2168 (1948).
210. С 1 e arena, Сок, Doumani. Амер. патент 2478243 (1950).
211. Karrman, Berqkwist. С A., 5447b (1948).
212. E. Simon. Ann., 31, 269 (1839).
213. J. Blyth, A. H of man. Ann., 53, 289 (1845).
214. А. И. Титов, А. М. Барышникова. Докл. АН СССР, 91,
№ 5, 1099 (1953).
215. Bachman, Addison, Hewett, Kohn, Millikan.
J. Org. Chem., 17, 906 (1952).
216. А. В. Т о п ч и е в, В. П. А л а н и я. Докл.АН СССР, 67,297(1949).
217. Haas, Riley. Chem. Revs., 32, 383 (1943).
218. S t e а с i e. Atomic and radical reactions. 2d edition, New York, 1954.
219. McDowell, Thomas. Trans. Farad. Soc, 46, 1030 (1950).
220. Pollard, W у a 11. Trans. Farad., Soc, 45, 760 (1949).
221. Cottrell, Graham. J. Chem. Soc, 556 (1953).
222. Thomas. Trans. Farad. Soc, 48, 1142 (1952).
223. H. H. С е м е и о в. Некоторые проблемы химической кинетики в
реакционной способности. М.» Изд-во АН СССР, 1954.
НИТРОВАНИЕ АМИНОВ
В течение последнего десятилетия большое значение
получили нитролроизводные первичных и вторичных аминов —
N-нитроамины, которые в небольших количествах были
получены еще в прошлом столетии. Однако методы нитрования
аминов детально разработаны лишь недавно. Разработка
новых методов нитрования аминов сделала N-нитроамины вполне
доступными соединениями и некоторые из них производятся
в промышленном масштабе (динитрооксидиэтилнитроамин,
этилендинитроамип и др.).
Третичные амины, не имеющие водородного атома,
связанного с азотом аминогруппы, N-нитросоединений,
естественно, не образуют.
В отличие от аминов, обладающих основными свойствами,
первичные нитроамины имеют характер кислот, причем их
кислотность выше, чему соответствующих карбоновых кислот.
Способность N-нитроаминов к солеобразованию объясняется
тем, что они изомеризуются аналогично нитропарафинам.
Например, строение калиевой соли фенилнитроамина можно
выразить формулой C@HsN = NOOK.
Нитроамины по внешнему виду представляют собой
белые довольно стабильные кристаллические вещества. Некоторые
арилнитроамины, лронитрованные в ядро, окрашены в
слабожелтый цвет. Однако интенсивность их окраски заметно ниже
интенсивности окраски соответствующих нитролроизводных
анилина. Некоторые нитроамины разлагаются со взрывом при
нагревании. Вместе с тем многие вторичные алифатические
нитроамины перегоняются в вакууме [1].
321
Алифатические первичные нитроамины быстро разлагаются
концентрированными кислотами и вследствие этого не могут
быть получены прямым нитрованием соответствующих аминов.
Общий препаративный метод получения таких N-нятроаминов
заключается в ацетилировании амина с последующим
нитрованием полученного вторичного амида в нитроамид и
разложением последнего щелочами с образованием соли нитроамина.
Нитроамины получаются при осторожном лодкаслении их
солей [1].
Часто в качестве промежуточных продуктов получения
китроаминов применяются уретаны. При обработке эфирного
раствора сырого алкилнитроуретана газообразным аммиаком
(или алифатическим амином) получаются соль нитроамина
и растворимый в эфире уретан. Так,метилнитроамин можно
получить по следующей схеме [2]:
С1СООС.Н, HNO
СНЛНССАС% — N ^
N0,
!^ CH^N — NO, + ^
Недавно Смартом и Райтом [3] был разработан новый
метод нитрования первичных аминов. По этому методу
первичный амин при взаимодействии с хлором превращается в
N-дихлорамин,который в дальнейшем подвергается нитрованию
нитрующей смесью, состоящей из 98—99%-ной азотной кислоты
и уксусного ангидрида. В результате такого нитрования
образуется М-хлор-М-яитроамин — очень нестабильное соединение,
которое при гидролизе в восстановительной среде (например,
раствором бисульфита натрия) дает нитроамин.
Типичный пример получения первичных нитроаминов —
нитрование этилендиамина. В первой стадии реакции при
взаимодействии хлора и этилендиамина получают N-тетрахлор-
1,2-диаминозтан (I), при нитровании которого азотной
кислотой и уксусным ангидридом образуется М,М'-дихлор-]\,]Х'-
динитро-1,2-диаминоэтан (III). Последнее соединение при
21 А. В. Топчиев
322
обработке бисульфитом натрия превращается с хорошим выходом
в этилен-М,М'-динитроамин (IV).
CH%—NCI; 2HN0,
сн,
:а:о:н
:о:о:
:а:о:н
н
СИ,—X
—N
о:
о:
:а:о:н
CH2—NH—NO
2HO + ZNaHSO,
CHg—NH—NO»
2CIOH
По мнению авторов, при нитровании К-тетрахлор-1,2-
диаминоэтана азотной кислотой первоначально образуется
N, ^'-координационное соединение (II), которое можно рас
сматривать как соль слабого донора электронов. Это
соединение самопроизвольно теряет хлорноватистую кислоту с
образованием М,М'-дихлор-М,М'-дииитро-1,2-диаминоэтана (III).
который, невидимому, является слишком слабым донором
электронов и вообще не может образовать соли. Это
подтверждается тем, что тстранитроамин совершенно не образуется.
Приведенная схема не отражает роли уксусного ангидрида
при нитровании. Присутствие уксусного ангидрида необ
ходимо для поддержания продуктов реакции в безводном
состоянии. Действительно, при применении большого избытка
азотной кислоты в отсутствие уксусного ангидрида
реакционная смесь получалась очень нестабильной.
При гидролизе N, М'-дихлор-N, К'-динитро-1,2-диамино-
этана (III) водой вместе с нитроамином образуется
хлорноватистая кислота. Это разложение водой представляет собой.
невидимому, обратимую реакцию. Поэтому для получении
323
хорошего выхода этилендинитроамияа авторы предложили
проводить разложение N, М'-дихлор-М,М'-динитро-1,2-диами*
яоэтаяа в этилен-N, N'-динитроамия восстанавливающими
реагентами (например, бисульфитом натрия), которые разлагают
хлорноватистую кислоту при проведении гидролиза.
Описанным выше методом Смартом и Райтом были
получены и охарактеризованы следующие N-нитроамины: этилен-
динитроамин, втор, бутилнитроамия, н. бутилнитроамия,
я. октилнитроамин, изопропилнитроамин и циклогексилнит-
р о амин.
В промышленном масштабе этилеядинитроамия получают
нитрованием этиленмочевпны (имидазолидона-2) с
последующим гидролизом этилендинитромочевины кипячением с во-
дой [4]:
N0*
СН, —МЩ HNO3CH2 —К п,0 CH.NHNO,
) | )СО-^ | +СО
)СО^ | )СО |
g — NR/ СН, — N/ CHgNHNO,
N0%
Нитрование этилеямочевииы производят нитрующей смесью,
состоящей из 74% серной кислоты, 15,4% азотной кислоты
и 10,6% воды. К 10 вес. ч. нитрующей смеси, охлажденной
ниже 10°, небольшими порциями прибавляют 1 вес. ч. эти-
ленмочевины с такой скоростью, чтобы температура не подви-
малась выше 10°. По окончании реакции смесь выливают На
лед, и выпавшую этилендинитромочевину отфильтровывают
и промывают водой. Полученная этилендинитромочевина
кипятится с водой до прекращения выделения газа. При
охлаждении раствора до комнатной температуры этилеядини-
гроамин выделяется в виде блестящих кристаллов, которые
отфильтровывают, промывают водой и высушивают.
Этиленмочевина в свою очередь получается многими
методами: конденсацией двуокиси углерода с этилендиамином,
взаимодействием этилеядиамина с мочевиной, конденсацией
диэтилкарбоната или фосгена с этилеадиамином, и др. [5].
Разработаны и другие методы синтеза этилендинитроамйна,
например: нитрованием и последующим гидролизом этиЯен-
21*
324
бис-уретана (I) и М,М'-этиленоксамида (II) [6]:
NO,
СН, - А - СО.СА
aHg ^ СН„ — N— CO%C*H, Н.О
NO
,
-NH-CO__ CH:-N-CO^ CH.-NHNO:
CH: —NH —CO ' CH, —N —CO H,O,NH, )
II I CH,-
NO,
Однако эти методы не имеют промышленного значения.
Из нитропроизводных первичных аминов отметим еще
получение весьма интересного соединения — метилендинит-
роамина, который первоначально был выделен из продуктов
нитрования уротропина [7].
Метилендинитроамин получается при нитровании в
уксусном ангидриде и последующем гидролизе метилен-бис-N-
ацетамвда, который в свою очередь образуется при конденсации
ацетамида с параформальдегидом. Реакция проводится по
следующей схеме:
4CH,CONH: + НСНО -+ 2CH,CONHCH:NHCOCH,
метилен бис-М-ацетамид
CHgCONHCHgNHCOCHg > CH3CON— CHg— NCOCHg + 2Н%0
NO, NO:
метилен-бис-М-(М-нитро)-ацетамид
гидролиз
CH,CON - СН: — N — СОСН, -——-^ O.NNHCH.NHNO* + CH3COONH4
I НОМН
метилендииитроамин
Метилендинитроамин выделяется из реакционной смеси
осаждением его в виде бариевой соли с последующим
разложением соли соляной кислотой.
325
Нитрование вторичных аминов — диметиламина и
пиперидина — было изучено еще в прошлом столетии Бамбергером
[8, 9]. N-нитродиметиламин и N-нитропиперидин получались
действием уксусного ангидрида на азотнокислые соли
диметиламина и пиперидина. Выходы нитропроизводных авторы не
сообщают.
Райт с сотрудниками [10] повторил синтез Бамбергера
и получил N-нитропиперидин с выходом 22% и
N-нитродиметиламин с выходом лишь 5%.
С другой стороны, было установлено, что ряд вторичных
алифатических аминов легко нитруется безводной азотной
кислотой с образованием N-нитроаминов [11]. Этим способом
были получены N-нитропроизводные имино-бис-ацетонитрила
(I), имино-бис-уксусной кислоты (II), диамида имино-бис-
уксусной кислоты (III) и 2,6-диоксопиридазина (IV)
О О
N==C — CH, НООС —СН, Н.К — С—СН, С—СИ,
NH NH NH HN NH
% НООС —СИ, H%N—С —СН, С—СН%
II II
О О
I II HI IV
По сравнению с диметиламином и пиперидином указанные
соединения представляют собой очень слабые основания.
Нитрование по этому методу очень невыгодно, так как
требует применения довольно большого избытка безводной
азотной кислоты. Кроме того, метод нитрования безводной
азотной кислотой не является общим, и многие вторичные
амины в этих условиях N-нитросоединений не образуют.
Райт с сотрудниками [10] разработали новый метод
каталитического нитрования вторичных аминов азотной кислотой
в среде уксусного ангидрида. При нитровании диэтаноламина
им было установлено, что хлористый водород, введенный в
реакционную смесь в небольшом количестве, чрезвычайно
активный катализатор нитрования вторичных аминов азотной
326
кислотой в среде уксусного ангидрида и резко увеличивает
выход р,р'-двнитрооксидиэтилнитроамина.
Первоначально р,р'-динитрооксидиатилнитроамин с
выходом 98 % авторы получали взаимодействием дитрата динитро-
оксидиэтиламмония с уксусным ангидридом в присутсгвии
хлористого цинка, хлористого ацетила или хлорида динитро-
оксидиэтиламмония как катализаторов. Однако этот метод
оказался неудобным вследствие необходимости получения и
выделения нитрата динитрооксидизтиламмояия.
В связи с этим был разработан метод нитрования диэтанол-
амина в одну стадию. В качестве катализатора в этом процессе
применялась солянокислая соль диэтаноламина.
Для получения хорошего выхода {%,}%'-динитрооксидиэтил-
нитроамина диэтаноламцц, азотную кислоту, уксусный
ангидрид и солянокислый диэтаноламин брали в молярном отношении
1 : 3,2 : 3,4 : 0,05. Азотную кислоту и диэтаноламин
добавляли к перемешиваемому уксусному ангидриду в течение
45 мин. при 5—15° с такой скоростью, чтобы предотвратить
накопление в реакционной смеси свободного диэтаноламина.
По окончании добавления реагентов реакционную смесь
нагревали до 40° в течение 40 мин. Затем продукты реакции
выливали в холодную воду. Осадок ^,^-двнитрооксидиэтил-
нитроамина отфильтровывали и промывали разбавленным
раствором аммиака. Выход сырого р,р'-динитрооксидиэтил-
нитроамина был равен 90% , т. пл. 49,5—51,5°.
Для очистки р,р'-динитрооксидиэтилнитроамина его
суспензию в воде обрабатывали паром (для удаления }%,8'-динитро-
оксидиэтилнитрозоамина), и продукт перекристаллизовывали
из ацетона.
Каталитический метод был успешно применен и для
нитрования других вторичных аминов. Нитраты вторичных аминов
обрабатывали уксусным ангидридом, содержащим хлористый
цинк. Превращение нитратов диметиламина, диэтиламииа,
пиперидина и морфолина в нитроамины было, соответственно,
равно 65, 60, 58 и 65% от теоретического. Солянокислый
ди-н. бутиламин также нитровался азотной кислотой в среде
уксусного ангидрида [10, 12].
327
с сотрудниками установил [13], чго легкость
нитрования вторичных аминов зависит от основности аминогруппы.
Было найдено, что основные свойства аминов, которые
нитруются в отсутствие катализатора с хорошим выходом, значительно
слабое, чем у морфолина. Для очень сильных аминов (например,
дин. бутиламина) катализатор лучше всего применять в
эквимолекулярном количестве.
Относительная основность некоторых вторичных аминов
определялась колориметрическим титрованием. В качестве
индикатора авторы применяли о-яитроанилин, который в
ледяной уксусной кислоте не диссоциирует в значительной
степени и имеет оранжево-желтую окраску. В серной и хлорной
кислотах при их концентрации, достаточной для превращения
индикатора в ион аммония, о-нитроанилин почти бесцветен. Если
добавить один эквивалент вторичного амина к одному
эквиваленту раствора серной или хлорной кислоты в уксусной
кислоте, содержащей индикатор, то отношение недиссоцииро-
ванных (окрашенных) молекул индикатора ко всему
количеству индикатора будет наибольшим, когда исследуемый амин
будет иметь максимальную способность к присоединению
протона.
Авторы не пытались оценить степень ассоциации
индикатора в уксусной кислоте в виде нщррфеяиламмоний-
ацетата и поэтому выражали результаты титрования только
в виде отношения интенсивности окраски индикатора в
присутствии определенного количества амина и серной (или хлор-
яой) кислоты к интенсивности окраски того же количества
индикатора в уксусной кислоте.
В табл. 18 дана относительная способность присоединения
протона некоторыми аминами по отношению к серной и
хлорной кислотам (см. стр. 328). Эта таблица показывает, что
все амины (№ 14—17 включительно), которые нитруются ь
хорошим выходом в отсутствие хлористоводородного
катализатора [11 ], являются наиболее плохими акцепторами
электронов. Напротив, диметиламин и пиперидин (сильно
основные амины) в отсутствие катализатора дают крайне низкий
выход нитросоединений (соответственно, 6 и 22%) [10].
328
и
и"
%
Относительная склонность л/жсое&бнемыя рфотонд
Амии
Таблица 18
по отношению
Интенсивность
окраски индикатора
вСН.СООН
серная
кислота
хлорная
кислота
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
Дииаопропиламив
Диизобутиламин
Дициклогексиламин
Ди-н. бутиламин
Диэтиламин
Ди-н. октиламин
Диметиламин
Лизидин
Метилэтаноламин
Диизоамиламин
Морфолин
Имино-бис-продионитрил
Диэтаноламин
Имино-бис-ацетонитрил
Имино-бис-метилацетонитрил (твердый диасте-
реомер)
Имиво-бис-метилацетонитрил (жидкий диасте-
реомер)
Имино-био-диметилэцетонитрил
1
1
1
1
1
1
1
0
0
0
0
0
0
,06
,06
,05
,04
,02
,02
,01
,99
,98
,945
,915
,85
,77
0,675*
0,536*
0,531
0,527
0,516*
0,516
0,511*
0,500
0,495*
0,478
0,463*
0,430*
0,389*
0,310
— 0,182
0,084
0,0825
* Звездочкой отмечены амивы, которые не титровались хлорной
кислотой, и цифры пересчитаны по данным, полученным с сераой
кислотой. Склонность присоединения протона пиперидином почти такая же,
как и у метилэтаноламина.
Таким образом, амины, которые являются плохими акцепторами
электронов, нитруются наиболее легко.
При изучении реакционной способности вторичных
алифатических аминов в реакции нитрования Райтом и его
сотрудниками были установлены следующие закономерности [12, 13]:
1) реакционная способность вторичных аминов
уменьшается с увеличением склонности амина к присоединению нротона;
329
2) слабо основные амины (№ 14—17 включительно)
нитруются в отсутствие катализатора с хорошими выходами N-витро-
соединений;
3) для получения хороших выходов N-нитроаминов из
более основных аминов (№ 1—13 включительно) процесс
витрования необходимо проводить в присутствии катализатора
(хлористого водорода, хлористого цинка и др.). Количество
катализатора необходимо увеличивать по мере увеличения
способности амина к присоединению протона. Для нитрования
наиболее основного амина — диизопропиламина уже
требуется эквимолекулярное количество хлористого водорода.
При нитровании вторичных аминов в присутствии
хлористого водорода или хлористого цинка в реакционной смеси
был обнаружен электроположительный хлор, например,
в виде хлорноватистой кислоты или хлористого ацетила [12].
Эти соединения с положительно заряженным хлором могут
вызвать превращение вторичного амина в соответствующий
менее основной хлорамин. Если свежеприготовленный раствор
азотной кислоты и хлористого водорода в уксусном ангидриде
будет давать хлорамин из амина, то хлористый ацетил также
должен находиться в реакционной смеси при нитровании
вторичных аминов в присутствии хлористого водорода, и он будет
действовать как хлорирующий реагент.
Это предположение проверялось при нитровании диэта-
яоламина и его производных. При нитровании р,р'-диокси-
диэтиламмояийхлорида и р,р'-динитрооксидиэтиламмоний-
хлорида выход р, р'-динитрооксидиэтилнитроамина был выше
(94 и 97%, соответственно), чем выход (92%), полученный из
диэтаволамина и небольшого количества хлористого водорода
[10].
При нитровании динитрооксидиэтилхлорамина азотной
кислотой был получен 78%-ный выход ^,^-динитрооксидиэтил-
яитроамина, но реакционная смесь была нестабильной, повиди-
мому, вследствие выделения хлорноватистой кислоты. С другой
стороны, при нитровании этого хлорамина азотной
кислотой в уксусном ангидриде выход р,р'-дииитрооксидиэтилни-
троамина (96 %) практически совпадал с выходом, полученным
330
яри иснользованищ динитродиэтиламмоний%лорида, и
реакционная смесь была совершенно стабильна. Это увеличение
выхода и стабильности реакционной смеси, яовидимому,
связано с тем, что уксусный ангидрид превращает хлорноватистую
кцелоту в хлористый ацетил. Довольно высокий вауод р,^-
дицдтрооксадиэтилнитроамана, яолученный при щщювании
хлорамина азотной кислотой, наяоминает нитрование сдабых
амцнов типа имино-бис-ацетонитрила [12, 14]. Следовательно,
унеуевцй ангидрид при каталитическом нитровании является
стабилизирующим реагентом.
Присутствие хлорамина в реакционной смеси, однако, не было
обнаружено, хотя наличие электроположительного хлора и
было установлено. Райт с сотрудниками [15] определили
количество электроположительного хлора, который выделяется из
хлористого цинка и азотной кислоты в уксусном ангидриде,
и нашли, что его образуется 39% от количества, ожидаемого
согласно нижеследующему уравнению:
L ZaCI,+ 2HNO.+ 3 (CH,C0),0 Z Za (OCOCH,), + N,0, +
Ч- ZClOCOCHg + 2Н0С0СН*.
На основании изложенного можно полагать, что хлорамин
может быть выделен из нитрационной смеси, которая,
согласно уравнению I, будет содержать количество азотной кислоты,
достаточное для образования хлорацетата, но недостаточное,
чтобы вызвать последующее нитрование. Такая смесь была
приготовлена согласно уравнениям II и III и дала 30%-ный
выход предполагаемого хлорамина.
П. 2НС1 + ZHNO, + 3 (СНзСО)дО -» 2СЮСОСН, + N,0, + 4H0G0CH*.
III. (МОз — СНд — CH^NH + С10С0СН, - (NOg — СН, — СНа)ЛС1 +
+НОСОСНз.
Наряду с хлорамином был получен р,р'-динитрооксдди*
этилнитрозоамин с выходом 36% от теории. Невидимому,
реакция протекала по уравнениям
длимо* 331
+ %Оз + 2НС1 - 2NOC1 + 2НОСОСН,
V. N0C1 + (NO, — CH, — CH,)*NB ^ (NO,- CH, — CH,), N — NO + HCL
Следовательно, хлорамин может превращаться в нитро-
амин согласно уравнению VI:
VI. (NO* —CH%^CH%):NCI+HNOa^(NO$"CHr"CH:):N -NOg+HOCl.
Тая как ни азотистая кислота, ни азотистый ангидрид по
реакции VI не образуются, можно полагать, что эти соединения
не будут присутствовать в продуктах реакции. Действительно,
ни азотистая кислота, ни азотистый ангщдрвд в продуктах
реакции не обяаружены.
Согласно уравнению VI, при нитровании хлорамина
образуется хлорноватистая кислота. Если хлорноватистая
кислота стабилизируется превращением в хлорацетат, то она
может использоваться для получения большего количества
хлорамина. Таким образом, каталитическое нитрование
вторичных аминов можно рассматривать как цепную реакцию,
так как электроположительный хлор регенерируется в
процессе реакции.
Хотя нитрование вторичных аминов можно проводить в
отсутствие уксусного ангидрида, выход N-нитросоединений
заметно увеличивается в присутствии этого реагента.
Применение уксусного ангидрида особенно необходимо при
каталитическом нитровании сильно основных аминов. Это
объясняется тем, что хлорноватистад кислота быстро разрушается
азотной кислотой и, следовательно, де успевает прореагировать
с амином в том случае, когда скорость реакции хлораминиро-
вания будет относительно низкой.
Райт и сотрудники считают, что титрование вторичных
аминов происходит через образована комплекса амияа и
азотной кислоты. Они полагают, что такие комплексы не образуются
с сильно основными аминами, которые дают нормальные
электровалентные соли. Поэтому при каталитическом нитровании
сильно основные свойства амина ослабляются превращением
его в соответствующей хлорамин.
332
Первая стадия реакции каталитического нитрования
вторичных аминов заключается, следовательно, в образовании
хлорамина. Присоединение хлорацетата в активном состоянии
к амину происходит согласно такой схеме:
Cl
I
н о
R:N: + c=o
R см.
н:о:
R:N:c:o:
R сн]"
R
1,3
—*
С1
: N: с: о: н
R СМ,
R:ы: ч- с:о:н
R
В продукте присоединения хлорацетата к амину
может затем произойти 1,3-перегруппировка водорода и 1,3-
перегруяпировка хлора. При разложении комплекса, после
перегруппировок водорода и хлора, образуются хлорамин и
уксусная кислота.
Во второй стадии реакции происходит образование кова-
лентной связи между атомами азота хлорамина и азотной
кислоты. Это соединение самопроизвольно выделяет
хлорноватистую кислоту с образованием нитроамина.
Хлорноватистая кислота, таким образом, выделяется в свободном
состоянии и стабилизируется ацетилированием
Р BONO. ?\9\. -НОС1 ^"9" ..
R:N: > R:N:N:O: ^ R:N:N::O
:С1: :С1:О:"
H
Исследование побочных продуктов реакции, полученных яри
нитровании хлораминов, показало, что нитрование хлорамина
протекает медленнее образования его в реакционной смеси.
ЛИТЕРАТУРА
1. А. Н. L amb ert on. Quart. Rev., 5, № 1, 75 (1951).
2. P. N. Franchimont. Rec.trav.chim., 13, 1308(1894).
333
3. G. N. R. Smart, G. F. W right. Canad. J. Res., 26 B, 284(1948).
4. Encyclop. Chem. Techno]. (New York), 6, 23 (1952).
5. J. F. M u 1 v a n e y, R. L. Evans. Ind. Eng. Chem., 40, 393 (1948).
6. W. F. Bachmann, W. J. Morton, E. L. Jenner,
N.W. Mac Naughton, E. E. Maxwell. J. Am. Chem.
Soc. ,72, 3132 (1950).
7. F. Charm an. J. Chem. Soc, 1633 (1949).
8. E. Bamberger. В or., 28, 399 (1895).
9. E. Bamberger, A. Kir pal. Ber., 28, 553(1895).
10. W. J. Chute, K. G. Herring, L. E. Tombs,
G. F.Wright. Canad. J. Res., 26B, 89(1948).
11. A. P. N. F r a n с h i n m о n t, J. V. D a b sk y. Rec. trav.
chim., 36, 70 (1916).
12. W. J. Chute, G. E. Dunn, J. E. Mackenzie, G. S.
Myers, G. N. R. Smart, J. W. Suggit, G. Wrigbt.
Canad. J. Res. 26B, 114 (1948).
13. G. R. Dunn, J. С MacKenzie, G. F. Wright. Canad.
J. Res. 26B, 104 (1948).
14. J. V. Dabsky. Ber., 49, 1045(1916).
15. G. S. Myers, G. F. Wrigh t. Canad. J. Res., 26B, 257 (1948).
Ллдед пятмдд
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ОКИСЛАМИ АЗОТА
А. НИТРОВАНИЕ ОКИСЛАМИ АЗОТА БЕЗ КАТАЛИЗАТОРОВ
Метод нитрования окислами азота, который начал
разрабатываться еще в 70-х годах прошлого столетия, приобрел
актуальное значение лишь с 1910—1915 гг. в связи с
освоением химической промышленностью синтетических методов
получения азотной кислоты из атмосферного азота через
жислы азота. Начиная с этого периода, проблема
использования окислов азота (нитрозных газов) для нитрования орга-
зических соединений привлекает усиленное внимание
исследователей, которые посвящают ей значительное число работ.
Это объясняется главным образом тем, что метод нитрования
жислами азота обладает определенным техническим
преимуществом перед обычно принятыми методами нитрования
&зотной кислотой и нитрующими смесями, так как при его
1рименении устраняется необходимость в переработке окислов
кзота в азотную кислоту (как известно, синтез азотной кислоты
13 окислов азота представляет собой довольно сложный
процесс и состоит в окислении кислородом низших окислов азота
%о азотного ангидрида в присутствии воды и получении, таким
эбразом,слабой азотной кислоты,которая затем концентрируется
%ри помощи HgSO*).
Обычно применяемую для нитрования N^O* (температура
(япения 21,3° при 760 мм) рассматривают как равновесную
месь согласно уравнению
335
При низких температурах преобладает димерная форма
двуокиси азота. Измерение плотности пара показывает, что при
температуре выше 140* наблюдается полная диссоциация
N*O*, а при 27° около 20% N#0* находится в форме мономера.
Внешний вид подменяется в зависимости от температуры и,
следовательно, состояния равновесия от бесцветных
кристаллов при температуре—50° до черных паров при 183°.
Структуру NgO* нельзя считать точно установленной.
Из всех предложенных формул наибольшего внимания
заслуживают
О О
O = N —O — Nf иО = М —О
О 0-
В пользу такой формулы говорит ряд химических реакций
NgO*, в частности, реакция с водой по уравнению
\:О* + Н,0 -» UNO, + HNO*.
Клузиус и Векчи [1] на основании исследования
взаимодействия меченого тетраметиламмонийнитрата (СНз)&М^М0 у
с жидкой NzO*, считают, что N^0* частично существует в форме
нитрозилнитрата NONOa, способного в свою очередь к
ионизации
+
\:О* Z NO-NO» ^ NO + NO".
Образование трехокиси азота N0@ допускается некоторыми
авторами. Так, Уэлш [2], объясняя механизм окисления
различных соединений двуокисью азота, считает, что кислород
образуется в результате диссоциации N0@ по следующей схеме:
Л:0* -+ N0, + N0;
N0@ -+ N0 + О*.
А. А. Баландин [3], Т. В. Заболоцкий [4], Блэсет [5]
рассматривают N0» в качестве первичного продукта окисления окиси
азота озонированным кислородом.
336 /fwmjDogaNi^ ореднычеслсмс
§ 1. Нитрование непредельных соединений
Из работ,посвященных нитрованию органических соединении
окислами азота, необходимо прежде всего отметить работы
по получению нитросоединений из непредельных соединений
жирного ряда. Обычно продукт реакции представляет собой
нестойкое твердое вещество или масло, из которого подчас
бывает очень трудно выделить чистое вещество.
Виланд [6, 7] исследовал реакцию взаимодействия
алифатических непредельных соединений с N*O* и яитрозными
газами *. Им найдено, что продуктами этой реакции являются
дияитросоединения («динитрюры»), которые образуются по
схеме
Д__СН = СЕ —R' + NgO* -+R — CHNOg — CHNO, — R%
где R и R^ — насыщенные углеводородные радикалы.
Подобные динитросоединеяия получаются и при нитровании NgO*
смешанных жирноароматических непредельных углеводородов,
имеющих общую формулу С*Нб — СН = СН — R, а также
соединений общей формулы Аг — СН = СН — Аг.
Так, например, стильбен при действии N,0^ даетдифенил-
цияитроэтан.
= СНСаНж + \:0, - QH* — СН — СН — С^Щ
Строение этого динитросоединения доказано Виландом
синтезом этого соединения из фенилбромнитрометана при
взаимодействии последнего с серебром:
/Н
^ 4. CgHg — СН — СН — С^Щ + 2AgBr
| |
* Вилояд рассматривает нитрозяые газы как смесь NaOa и
активную роль в этой смеси играют, по его мнению, свободные радикалы
N0" и N0;.
337
Продукт взаимодействия стильбена с N,0^ оказался
идентичном дифенилдижитроэтапу, полученному из фенилбромнитро-
метана. Виланд исследовал ташке реакцию нитрования NgO*
непредельных углеводородов с сопряженными двойными
связями следующего строения:
При обработке суспензии 1,4-Дифенилбутадиена в абсолютном
эфире раствором NsO* в смеси эфира с газолином при
охлаждении Виланд получил 1,4-Дифенил-1,4-динитробутен-2
СИ = СН — СН (NOg
Таким образом, у 1,4-Дифенилбутадиеяа присоединение нитро-
групп происходит не к двум соседним атомам углерода (в
положениях 1,2), а к двум крайним атомам системы сопряженных
связей (1,4), что согласуется с правилом Тиле.
При действии спиртовой щелочи дифенилдинитробутеа
отщепляет молекулу HNO% и дает 1,4-дифенил-1-нитробутадиен
С,Н,СН(Юд) — СН = СВ — CH(NO,)C.H, -»
= СН — СН = СНС.Н, Ч-
Далее Виланд показал, что при действии иитрозных газов
на непредельные соединения, содержащие карбонильную группу
и имеющие общую формулу
Р *
С„Н, —С = С —С —R
н н о
получаются следующие три типа соединений.
1) Нитрогруппа присоединяется к %-атому углерода,
а витрозогруппа — к р-атому углерода (1); при дальнейшем
течении реакции в результате внутримолекулярных дере-
22 А В. Топчиев
338 Дцт/юедмиб ореднмуедсыа? соб^ымемцц ожисллмц
группировок соединение (I) переходит в оксим (II), последний
легко теряет воду и дает азоксазол (III)
Н Н NO, NO,
II I I
С,Нв—С — С--С —R-Xyi, —С— С = С —Н^СдНв —С—С = С —R
I N0 N0, О Н NOH ОН IH N О
2) Нитрогруппы присоединяются по месту двойной связи
к атомам углерода * и (3, образуя динитросоединение:
Н Н
СдНв —С —С—С —R
N0, N0, О
3) Происходит прямое нитрование в радикале, связанном
с кетогруппой (R), которое приводит к ненасыщенному нитро-
кетону:
С#Нв — СН = СН^СО —'ЩН + N,0* -^ СдНд— СН=СН —СО — Ri+ HNOg
N0%
Взаимодействие NgO^ с тетраметилэтиленом изучалось,
некоторыми исследователями. Шмидт [8] утверждает, что при
применении эфира в качестве растворителя основным
продуктом реакции является динитрит строения:
сн, сн,
HgC — С — С — GHg
ONO 0N0
Это утверждение было в дальнейшем опровергнуто Н. Я.
Демьяновым и К. В. Сидоренко [9, 10], которые также занимались
изучением реакции нитрования непредельных соединений
окислами азота. При нитровании тетраметилэтилена до остающегося
веболыиого избытка углеводорода эфирным раотвОром окислов.
азо*а, долучеайых при действии азотной кислоты на Аа&Оз,.
эти исследователи получили динитросоединение состава
бед кдлмлыадтйо/ю* 339
I
и продукт с эмпирической формулой CgHigNgOs (II), имевший
т. пл. 101—104°. Строение динитросоединения (I) доказано
восстановлением его в диамин; второй продукт реакции (II)
давал при восстановлении смесь диамина и оксамина, на
основании чего Н. Я. Демьянов и К. В. Сидоренко приписывают
ему следующее строение:
н,с . сн,
НзсЪю—с/
1 I
II
Этими же исследователями, при применении в качестве
нитрующих агентов N^O*, N,0^ и М%Оз, получены продукты
присоединения N0% к изобутилену, гексилену, диаллилу
и другим непредельным углеводородам жирного ряда.
Взаимодействие тетраметилэтилена с N#0* подробно
изучали Михаэль и Карлсон [11], При проведении реакции в
эфирном растворе авторы получали до 22% 2,3-динитро-2,3-диме-
тилбутана, в отсутствие же растворителя или в растворе пет-
ролейного эфира динитропродукт получается лишь с небольшим
выходом. Во всех своих опытах авторы отмечают получение
нитронитрата, которому Н. Я. Демьянов и К. В. Сидоренко
приписывали строение II.
По данным этих авторов, этот продукт получается в
результате присоединения NgO^ и последующего окисления. Михаэль
и Карлсон приходят к выводу, что динитрит не образуется.
Эти же авторы изучали взаимодействие NgO* с изобутиленок
в различных условиях. При проведений процесса в эфирном
растворе продукты реакции восстанавливались, и по составу
полученных продуктов восстановления можно было судить
о первоначальном составе продукта взаимодействия с N*O*.
Таким образом, было определено, что в растворе серного
22*
340
эфира получаются соединения
СН, Н
HgC —С —СН с выходом 16—23% от теории;
ONO N0*
сн,
HgC — С = СН с выходом 5—12% от теории;
N0,
СП,
НзС — С — СН% с выходом 12% от теории.
O.NO N0%
При проведении реакции в отсутствие растворителя или
в петролейном эфире, получается нитрозонитрат с выходом
—13% от теории
СН,
НдС С СН%
0,N0 NO
Если вместо изобутилена исходить из триметилэтилена [12],
то в эфирном растворе получается
СН,
Н,С — С--СН —СН,
ONO
с выходом 35% от теории.
Шаарщмидти Гофмейер [13, 14] нашли, что при действии
жидкой NgOa щ& непредельные углеводороды получается смесь
продуктов различного характера, причем состав этой смеси
я ^аждом отдельном случае зависит от природы данного оле-
и уемпературнцх условий проведения реакции. По
ию этих .^имикрв, в результате присоединения
рдефинам возможно образование следующих четырех
бед лсдлгдлмадлгорое 341
типов соединений:
— СН —NO —CH — NO, — СН —N0& _СН —0N0
I I I I
— СН — 0N0, — СН — 0N0 — СН — N0* — СН — 0N0
I. Нитроваты II. Авотистые III. Динитро- IV. Азотистые
(нитровони- эфиры нитро- соединения эфиры глинолей
траты) спиртов (нитро- («динитрюры») (динитриты)
нитриты)
Из этих соединений устойчив лишь динитрюр (III), у которого
оба атома азота непосредственно связаны с атомами углерода;
соединения остальных типов (I), (II) и (IV) легко подвергаются
превращениям и дают другие продукты реакции. '
Соединение типа I (нитрозонитрат), аналогично
отщеплению НС1 у продуктов присоединения хлора, отщепляет
HNO@; последняя окисляет образовавшийся нитрозоолефин.
Соединение типа II (нитронитрит) может распасться на нитро-
олефин и азотистую кислоту, которая далее разлагается по
уравнению 3HN0% = HNO3+ HgO^ 2N0. Азотистая кислота
и, вероятнее, азотная, полученная по указанному выше
уравнению, действуют на нитроолефин, окисляя последний. Ди-
нитрит (IV) легко отщепляет азотистую кислоту с одновременным
образованием весьма нестойкого олефин-нитрита.
При пропускании чистого этилена в очищенную жидкую
двуокись азота при 0° Смит [15] получил с хорошим выходом
%, р-динитроэтан. Согласно этому же автору, при пропускании
N^O* в эфирный раствор тетраметилэтилена получается 2,3-
динитро-2,3-диметилбутан с выходом ^22%. Аналогичным
образом получаются 1,2-динитропропан и 1,2-динитроизо-
бутан [16].
Описан процесс получения 1,2-динитроциклогексана из
N:0* и циклогексена при 300* и давлениях до 100 атм. [17].
Леви [18] получал динитроэтан одновременным
пропусканием этилена (со скоростью 30 л/час) и кислорода (со скоростью
8 л/час) в жидкую N^O* (3 л) при 0° в течение 10,5 часа. За
время реакции адсорбировалось 228,5 л этилена и 24,4 л
кислорода. После удаления сухим воздухом избыточной NgO^ из
полученного масла при сильном охлаждении (твердая
342 /fwnzpoeawwe о/%а%%чбск%а; соб&шбмый ожысд&иы
OH) выделяют—400 г 1,2-динитроэтана,что составляет
^33% от теории, считая на этилен, и ^40%, считая на NsO*.
Леви с сотрудниками [19] исследовали также реакцию
взаимодействия пропилена, бутилена, циклогексена, 2,4,4-
триметилпентона-1 и 2,4,4-триметилпентена-2 с N%Og. При
работе с газообразными олефинами ток углеводорода
пропускался через раствор N*O* в эфире. Жидкие олефйны
приливались к N*O*, или же, наоборот, эфирный раствор N^0^
добавлялся к углеводороду. Реакция проводилась при
невысоких температурах (от —5 до 0°). Во избежание образования
азотистого ангидрида в некоторых опытах через реакционную
смесь пропускался слабый ток кислорода.
В результате исследования было установлено, что
взаимодействие N*O* с олефинами приводит к образованию двух
веществ: динитросоединения и нитронитрита. В случае
несимметричной молекулы олефина группа ONO всегда
присоединяется к атому углерода, связанному с меньшим числом атомов
водорода. Так, например, при присоединении четырехокиси азота
к пропилену образуются динитропропан
и %,р-нитроизопропилнитрит CH
Ввиду неустойчивости нитронитритов они не выделялись в
чистом виде, а подвергались гидролизу с образованием нитро-
спиртов. В некоторых опытах в смеси продуктов реакции были
обнаружены нитронитраты, которые возникли в результате
окисления нитронитритов.
Согласно наблюдениям Леви и Скейфа, при взаимодействии
олефинов с N*O*B отсутствие растворителя, наряду с реакцией
присоединения, происходит также окисление олефвна. В
качестве растворителей использовались простые и сложные
эфиры: диэтиловый эфир, диоксан, этилацетат и др. Авторы
полагают, что эфиры образуют комплекс с молекулой N^O*, что
подавляет окислительное действие последней [20].
Действием двуокиси азота (N0%) на окись этилена [21]
получается 2-нитроэтиловый спирт, если реакцию вести
следующим образом: к 92 г NO* и 1000 г СНС1з при 0° медленно
прибавляют 50,г окиси этилена и 250 г СНС1з, охлажденных
до 0°. Оставляют стоять на 12—15 час. при комнатной темпера-
6*9 ждтдлиедторое 343
туре, после чего отгоняют СНС1з при 50° и получают 135 г
нитроэтилнитрата, который после омыления 10% -ным раствором
Na%COs дает 2-нитроэтиловый спирт.
^-Нитроэтиловый спирт получается также при обработке
окиси этилена нитритом натрия [22] при одновременном цро-
иускании углекислого газа: в раствор 200 г NaNO* в 1 л воды
пропускаются СО* и окись этилена при 25—30*. Выпадающий
NaHCOs отфильтровывается. Фильтрат перегоняется, давая
235 г O*N — СН, — СН* — ОН (85%) с т. кип. 107—110°
при 20 мм.
Кроме того, р-нитроэтиловый спирт получается при
пропускании окиси этилена в раствор нитрита бария. 130 г окиси
этилена барботируют через раствор 4<J0 г Ba(NO*)z в 100 мл
ЩО; 2-нитроэтанол получается с выходом 93%.
Нагреванием тетрахлорэтилена (или тетрабромэтилона) с
NgO* в запаянной трубке при 100° в точение 3 час. Бильц
[23] получил симм. динитротетрахлорэтан с хорошим
выходом. В этих же условиях ос,ос-дифенил-^,^-дихлорэтан
(СдН*)аС = CClg образует дифенилдинитродихлорэтан
(CaH,)%C(NO,) - C(NO,)C1,.
При взаимодействии этилена с двуокисью азота(N0%),
содержащей примесь NO, при температуре 20—60° и давлении
выше 50 атм.получаются неустойчивый продукт присоединения,
легко разлагающийся при 50—100° в присутствии пара, и
2-нитроэтанол, который выделяется обычным сдособом
[24].
При взаимодействии N*O* с углеводородами, содержащими
систему сопряженных двойных связей, происходит
присоединение в положение 1,4. Так, например, при проведении реакции
с 1,3-бутадиеном как в различных растворителях» так и в
паровой фазе образуется 1,4-диннтро-2-бутен [25].
Как указывалось выше, аналогичное течение реакции
отмечалось Виландом [6, 7].
В некоторых случаях наблюдается присоединение N0%
в положение 1,2 конъюгированной системы. Так, например,
при действии NaO* на 1-циано-1,4-дифенил-1,3-бутадиен
получается 1-циано-1,4-дифенил-3,4-динитро-1-бутен [26 ].
344
ааолгд
еЩ СбНа-^СаНв — С = СН—СН —СН —CgHg
CN , CN NO, NO*
Наряду с этим Аллен [27] отмечает присоединение как в
положение 1,2 так и в положение 1,4 сопряженной системы.
Так, например, действие NgO* на 2,3-дифенил-1,3-бутадиен
выражается уравнением:
^ Н%С=С
§ 2. Нитрование ароматических и гетероциклических
соединении
Нитрование ароматических соединений окислами азота
изучается сравнительно давно. Еще в 1871 г. Газенбах [28]
нашел, что при взаимодействии N*O* с бензолом в течение 7
дней при комнатной температуре образуется нитробензол
наряду со щавелевой кислотой.
Азотистый ангидрид, легко присоединяясь к непредельным
соединениям, вступает в реакцию с ароматическими
углеводородами лишь в результате разложения на окись и двуокись
азота но уравнению [29]:
Лидс [30] насыщал двуокисью азота бензол в течение
многих дней на холоду; при этом получены нитробензол,
щавелевая и пикриновая кислоты. Невидимому, при этом бблыпая
часть бензола^ остается без изменения. При действии же N0%
на кидящий бензол, кроме указанных выше продуктов,
образуются еще два соединения» из которых Лидсом идентифици-
роваао только одно (монооксинроизводное с т. нл. 205°). Этот
же автор нитровал двуокисью азота толуол, ксилол, цимол,
беа каталымлкую* 345-
нафталин, антрацен и фенол. При насыщении толуола
двуокисью азота на холоду в течение 3 мес. получены о-нитро-
толуол и другие продукты окисления: метилдинитродиоксибен-
зол СбН(СНз)(МОа)з(ОН)з (игольчатые кристаллы с т. ял.
110°), щавелевая и диоксибензойная кислоты. При проведении
реакции аналогично нитрованию толуола ксилол дает о-нитро-
ксилол и продукты окисления: щавелевую, п-толуиловую
и фталевую кислоты, а цимол дает ос-нитроцимол, щавелевую
и п-толуиловую кислоты. При нитровании нафталина
двуокисью азота получены: мононитронафталин, динитронафталин,
тетраоксинафталин и нафтодихинон. Фенол, который нитруется
очень легко, дает пикриновую кислоту. При насыщении
двуокисью азота раствора антрацена в ледяной уксусной
кислоте в качестве единственного продукта реакции образуется
антрахинон с выходом 58 г при загрузке 50 г антрацеяа.
Фридбург [31] видоизменщл метод Лидса, проводя
нитрование бензола при повышенной температуре. Окислы азота
предварительно растворялись в сероуглероде и получеявый
раствор смешивался с бензолом; при этом была получена смесь
нитро- и п-динитробензола.
Нитрование антрацена двуокисью азота исследовано также
Либерманом и Линдеманом [32] и Мейзенгеймером [33],
которые, в отличие от Лидса, получили при этой реакции нитро-
соединения. ЛиберманиЛиндеман пропускали двуокись азота
медленной струей в антрацен, смешанный с 4 ч. ледяной
уксусной кислоты, причем температура реакции поддерживалась
не выше 10—15°. Полученное нитросоединение отделялось
от избыточного антрацена посредством обработки кипящим
бензолом, в котором это соединение нерастворимо (в раствор
переходил только антрацен). Мейзенгеймер получил 9,10-
динитро-9,10-дигидроантрацен при действии жидкой N^O*
на антрацен, смешанный с хлороформом. В то же время при
действии NgO* на антрацен, суспендированный в бензоле иди
растворенный в нитробензоле, образование нитропроизводных
антрацена не наблюдается [34]. В первом случае образуются
лишь следы антрахинона,а во втором случае получается высокий
выход антрахинона.
346
На этих примерах ясно видна зависимость получающихся
продуктов реакции цри нитровании N#0* от характера
растворителя. 1-Хлорантрацен [35] с сухой N^O* в среде четырех-
хлорвстого углерода образует продукт присоединения, который
при нагревании с пиридином превращается в 1-#лор-9(или
10)-#итроантрацен.
1,5-Дихлорантрацен дает 1,5-дихлор-9,10-дивитро-9,10-
дигидроантрацен, который при нагревании с пиридином
превращается в 1,5-дихлор-9-ндтроантрацен.
Виланд [36 ] нитровал двуокисью азота ароматические
углеводороды, фенолы и амины. Нитрование бензола, которое
проводилось в запаянных трубках при 80°, дало лишь
небольшой выход нитробензола. При применении эквимолекулярных
количеств бензола и N0% получены, наряду с продуктами
окисления (СО,, щавелевая кислота и др.), главным образом
1,3,5-тринитробензол и пикриновая кислота, причем часть
бензола осталась без изменения. Опыты по нитроваяию
нитробензола показали, что последний не нитруется цри проведении
реакции аналогично нитрованию бензола. Анализируя эти
результаты, Виланд приходит к выводу, что нитрование
бензола не является ступенчатой реакцией. Автор полагает, что
бензол сразу присоединяет 6 частиц N0% (аналогично
присоединению к бензолу хлора и брома на солнечном свету) с
образованием гексанитроциклогексана; последний расцадается
далее на тринитробензол и 3 молекулы азотистой кислоты:
СН CHNO*
НС
СН O.NHC
+ 3N.0*
CHNO*
CHNO*
HCL /СН O*NHC
СН CHNO,
CHNO
O&NHC
CHNO
O*NHC
CHNO
Нитрование нафталина в растворе эфира на холоду приводит
лишь к небольшому выходу нитросоедине^ий, в то время как
бед кдлмлыадлгорое 347
применения растворителя реакция протекает энергично
с образованием %-нитронафталина.
Фенолы легко реагируют с N0% со значительно лучшими
выходами, чем при нитровании их азотной кислотой. Для
нитрования этих соединений Виданд применял растворы
двуокиси азота в смесях бензола с цетролейным зфиром или
газолином и проводил реакцию при хорошем охлаждении и
сильном перемешивании. Им получены следующие результаты:
фенол дает в качестве продуктов реакции п- и о-нитрофенолы
с преобладанием первого, причем общий выход нитропродуктов
ло отношению к фенолу составляет 125% от веса взятого
фенола. При нитровании о-, а- и м-крезолов получается смесь
изомерных нитросоединений. При действии N0% на 1, 3,4-кси-
ленол образуется с хорошим выходом 5-нитропроизводное
(т. пл. 72°), а ос-нафтол дает при нитровании смесь 2-нитро»
нафтола и 2,4-динитронафтола.
При нитровании ацетанилида NgO* в среде эфжра
на холоду Виланд получил диазобензоилнитрат и
уксусную кислоту. Следует отметить, что при действии N%Oa
на анилин в бензольном растворе Витт [37] также получил
бензолдиазонитрат с количественным выходом (ср. с
работами автора настоящей книги по изучению взаимодействия
анилина и ацетанилида с NaO* в хлороформе, см. стр. 357).
Нитрование дифениламина проводилось Виландом в
растворе абсолютного эфира или же смеси бензола с петролей-
ным эфиром в отношении 4 : 1. В первом случае в качестве
основного продукта реакции получен дифенилнитрозоамин
(кристаллическое соединение с т. пл. 66*), во втором случае —
п-нитродифенилнитрозоамин. Для объяснения образования
дифенилнитрозоамина Виланд допускает, что у N2O4, наряду
с молекулами со структурой 0*N — N0%, существуют молекулы,
имеющие строение О = N — О — N0%, с которыми
дифениламин реагирует по следующему уравнению:
(СА), NH + О = N — О — NOs -* (CA),N-- NO + HONO,,
Взаимодействие ди-п-толилгидроксиламина с N^O* в
холодном эфирном или бензольном растворе приводит к образо-
348
ванию о,о'-дияитро-п-толиламина [38]. В этой же работе
Виланд указывает, что дифениламиноксид с NgO^ образует
п, п' -динитродифени л аминоксид.
Относительное количество применяемой для нитрования
N3O4 оказывает значительное влияние на течение реакции.
С большой убедительностью это было показано в работе
Баттегей и Керна [39], которые исследовали реакции нитрования
NgO* монометиланилина и диметиланилина. При
взаимодействии монометилаиилина с эквимолекулярным количеством
NsO* в бензольном растворе в течение 5 дней получается с
выходом 55,4% п-нитро-М-нитрозомонометиланилин (т. пл. 100°).
Если же при указанных условиях применять избыток N^O*,
то основным продуктом реакции будет 2,4-дижитромонометил-
анилин с т. пл. 175° (выход 50—60%). При нитровании
диметиланилина, которое проводилось N^O*, растворенной в бен-
эоле, при температуре реакции не выше 10° (молярное
отношение реагентов 1 :1) получены п-нитродиметиланилиж и
п-витрозодиметиланилин (в виде нитрата). Образование этих
продуктов Баттегей и Керн объясняют следующими реакциями:
CAN (CR,), + N,0, -* O.NCAN (СН,), + HNO,;
д + HNO* -^ ONCAN (СН*)* + Н*О;
N,0. + Н%0 -^ HNO, + HNO,;
+ HNO,-*HON = С@Щ = N
При увеличении относительного количества N%0* до 2
молей на 1 моль диметиланилина образуется главным образом
2,4-динитродиметиланилин наряду с небольшим количеством
п-нитродиметил анилина.
Шааршмидт и Смолла [40] нитровали некоторые
ароматические соединения жидкой NsO* на холоду при соотношении:
1 ч. NgO* на 3 ч. углеводорода. В этих условиях бензол дает
нитробензол, пикриновую кислоту и в небольшом количестве
динитробензол; при действии N%O* на толуол продуктами
реакции оказались нитротолуол, динитрокрезол, нитрофенол,
бензальдегид, щавелевая и бензойная кислоты. С увеличеяием
продолжительности реакции значительно повышается отно-
349
сительный выход бензойной кислоты, а выход нитропроизвод-
ных несколько уменьшается.
Ксилол и мезитилен реагируют с NgO* весьма энергично
с выделением тепла. При этом вместо нитросоединеяий
получаются смолистые массы, которые, по данным анализа,
содержат азот и значительные количества кислорода. Эти результаты
указывают, что при действии NgO* на ксилол и мезитилен
преобладают реакции окисления.
Шааршмидт, Бальцеркевич и Ганте [41] нитровали ди-
метиланилин раствором NgO* в четыреххлористом углероде.
Раствор 9,3 г NgO* (0,1 моля) в 20 г четыреххлористого
углерода вливали постепенно в раствор 12,3 г диметиланилина
(0,1 моля) в 100 г четыреххлористого углерода при
перемешивании и наружном охлаждении смесью льда с поваренной
солью. После прибавления всего количества NgO*
реакционную смесь оставляли в охлаждающей смеси еще в течение
3 час. По окончании нитрования реакционную массу
размешивали с водой и извлекали органическую часть эфиром. После
отгонки растворителей и перекристаллизации остатка из
спирта получен с выходом 86% от теории п-нитро-М-диметил-
анилин (желтые иглы с т, пл. 162°).
Нитрование галоидированных ароматических соединений
в отсутствие серной кислоты, А1С1@ и других катализаторов
протекает недостаточно удовлетворительно. Фторбензол не
реагирует с NgO* в растворе четыреххлористого углерода
даже после длительного (72 часа) взаимодействия. Иодбензол
в этих же условиях дает смесь о- и п-иоднитробензола с
выходом 45% от теории. Хлорбензол и бромбензол образуют
соответствующие хлорнитро- и бромнитропроизводные,нос
меньшим выходом.
Обширнф систематическое исследование реакции
нитрования органических соединений окислами азота проводилось
с 1934 г. автором настоящей книги (до 1939 г. совместно
с П. П. Шорзагиным[42]). Эта реакция была изучена как в
жидкой, так и в газообразной фазах на многочисленных
примерах разнообразных органических соединений (ароматических
углеводородов, фенолов, аминов, гетероциклических соеди-
350
нений и др.), причем установлено влияние различных факторов
(облучения, температуры, соотношения реагентов и т. д.) на
Р*с. 1. СхеМа нитрования углеводородов
двуокисью азота в жидкой фазе
процесс нитрования. Ниже приведены полученные нами
экспериментальные данные с подробным описанием опытов и
методики исследования продуктов реакции.
Нитрование в жидкой фазе проводили в специальном
аппарате (рис. 1), который состоял из реакционной коябы 7,
соединенной на шлифах с охлаждаемой снаружи водой
бюреткой 2 для жидкой N,0* и с холодильником 5, наполненным
смесью льда с поваренной солью. Конденсация продуктов
реакции происходила в трубках 4, также охлаждаемых смесью
льда с солью. В реакционный сосуд, который в зависимости
от температуры реакции в одних опытах охлаждали, в других
нагревали на металлической бане» наливали нитруемое соеди-
бед лдлгалиадмго/юе 351
нениё и затем из бюретки постепенно, по каплям, прибавляли
Нитрование нафталина
Опыты проводили при различных температурах и
различных соотношениях реагентов» N%0* прибавляли по каплям
из бюретки в продолжение 30-^-40 мин. По окончании реакции
содержимое колбы и приемников переносили в колбу Вюрца
и при 22—40° отгоняли непрореагиройавшую N%0*. Остаток
выливали в насыщенный раствор соды, причем выпадал гряз-
ножелтый осадок; последний отфильтровывали, промывали
водой и обрабатывали холодным бензолом (или сероуглеродом),
который растворял <%-мононитронафталин. После отгонки
растворителя и перекристаллизации остатка из горячего спирта
получен <х-мононитронафталин (удлиненные желтые иглы с
т. пл. 57—58,5°). Полинитронафталины, нерастворившиеся
в. бензоле, экстрагировали кипящим ацетоном, в который
переходили 1,8-динитронафталин и 1,3,8-тринитронафталин,
(1,5-динитронафталин в ацетоне не растворяется).
Ацетонную вытяжку после отгонки ацетона обрабатывали
горячим спиртом, из которого при охлаждении
выкристаллизовывался 1,8-динитронафталин; последний очищался повторной
перекристаллизацией из спирта (т. пл. 172°). Нерастворив-
шийся в спирте 1,3,8-тринитронафталин растворяли в горячем
хлороформе, из которого выделяли при охлаждении в виде
ромбических табличек желтого цвета продукт с т. пл. 218°.
1,5-Динитронафталин (см. выше) после перекристаллизации и&
бензола и ледяной уксусной кислоты выделен в чистом вид&
(желтые кристаллы с т. пл. 215°).
Приводим результаты наиболее характерных из
проведенных опытов по нитрованию нафталина (табл. 19).
Как видно из данных табл. 19, при 18—20° нафталин дает
как с теоретическим, так и с избыточным количеством NgO*
<%-мононитронафтаЛин с выходом 90—96% от теории но вая-
тому в реакции аафталину. При 60° нафталин нитруется
теоретическим количеством N%O* с образованием исключительно
352
<%-мононитронафталина (выход 92,6% от теории); яри избытке
же NgO^, наряду с <%-мононитронафталином (выход 78—90% от
теории), получается также 1,5-динитронафталин (выход 12%).
При 150° нитрование теоретическим количеством NgO*
дает исключительно %-мононитронафталин; если же
применять большой избыток NgO^ (в 7—8 раз больше теоретического
количества), то продуктами реакции оказываются также по-
линитронафталины: 1,5-динитронафталин (выход 36%), 1,8-
динитронафталин (выход 18%) и небольшое количество 1,3,8-
тринитронафтщлипа (выход 2%).
Таблица 19
№ опыта
1
2
3
4
5
6
7
8
9
Температура,
*С
18—20
18—20
18—20
60
60
60
150
150
150
о
2
m
II
6
12
20
6
12
20
6
40
50
Выход
а-М0Е0ЕИТР0-
нафталина
г
13,0
12,0
12,5
12,5
11,0
10,5
12,0
4,0
4,0
% от
теоретического
96
90
92,6
92,6
80
78
90
30
30
1,5-ДЕЕИТрО-
нафталина
г
—
—
2,0
2,0
.
6,0
6,0
% ОТ
теоретического
___
___
___
12
12
___
36
36
1,8-ДИЕитро-
нафталина
г
—
—
—
—
—
—
3,0
3,0
% от
теорети-
чесного
—-
—
—
—-
—
—-
18
18
Адализ указанных выше результатов приводит к
заключению, что для получения оптимального выхода <%-мононитро-
нафталина (96% от теории) необходимо проводить реакцию
дри 18—20°, применяя теоретические количества N2O4.
Большой избыток последней и высокая температура реакции (выше
150*) способствует преимущественному образованию поли-
витропроизводных.
бее ждтдлмадто^хю 353
Нитрование дифенила
Нитрование проводили при комнатной температуре в
течение 1—2 час. избытком N^O*. По окончании опыта, после
удаления NgO*, не вошедший в реакцию дифенил отгонялся с
водяным паром. Остаток растворяли в ледяной уксусной кислоте,
из которой дробной кристаллизацией выделены о- и п-нитро-
дифенил: при охлаждении раствора выкристаллизовывался
п-изомер, при разбавлении маточного раствора водой
выделялся о-изомер. Для очистки этих продуктов они несколько
раз были перекристаллизованы из спирта, причем получены
п-нитродифенил с т. пл. 113—114° и о-изомер с т. пл. 37°.
Нитрование антрацена
Нитрование антрацена проводили в растворе
хлороформа (в отсутствие растворителя реакция протекает
очень энергично, сопровождаясь осмолением). К 5 г антрацена
в 100 мл хлороформа прибавляли по каплям в течение 45—
50 мин. 30 мл N„0* при температуре около 0° (охлаждение
льдом). В результате нитрования получен 9,10-динитроантра-
цен, который очищен многократной перекристаллизацией сы
рого продукта из уксусной кислоты и спирта. Выход 9,10-
динитроантрацена составлял 6 г, что соответствует 80% от
теории.
Нитрование фенантрена
5 г фенантрена в 100 мл хлороформа обрабатывали 30 мл
жидкой NgO* в течение 45 мин. при 0°. Избыток N^O* удаляли
из реакционной смеси продуванием сухой СО%. После отгонки
хлороформа остаток выливали в холодную воду и
выделившееся при этом масло растворяли в спирту при нагревании.
Для разделения продуктов реакции (мононитрофенантренов)
применен метод дробной кристаллизации. Спиртовый раствор
масла охлаждали и выделившиеся на стенках стакана
кристаллы отделяли от масла. Масло растворяли в спирту, охлаждали,
причем выделялась вторичная фракция кристаллов; оставшееся
23 А В. Топчиев
354 #%лфоеам%г <фаан%чбски# соб^мненмй окцслдлФМ ядол&д
масло после отделения от кристаллов опять растворяли в
спирту и т. д. В результате такой обработки получены 2-монони-
трофенантрен в виде светложелтых кристаллов с т. пл. 99° (выход
1,7 г, что соответствует 32% от теории), 9-мононитрофенантрен
с выходом 0,7 г (11% от теории) в виде оранжево-желтых
кристаллов с т. пл. 116—117°, 4-мононитрофенантрен в виде
желтых игл с т. пл. 80—82° (выход 3% ) и в незначительном
количестве 3-мононитрофенантрен (т. пл. 170—171°).
Необходимо отметить, что метод дробной кристаллизации,
примененный для разделения нитрофенантренов, приводил к
значительным потерям исходного продукта; при выходе сырого
продукта?—7,5 г общий выход перекристаллизованных
продуктов составлял 2,6 г. (Следует отметить, что Шмидт и Гейнле
[43], нитровавшие фенантрен смесью азотной кислоты и
уксусного ангидрида, получили выход сырого нитропродукта в
2—2,5 раза меньше.)
Нитрование фенола
Для выяснения оптимальных условий реакции опыты при-
водили при различных температурах. 5 г фенола
обрабатывали 20—30 мл жидкой N%O* в течение 30 мин. По окончании
нитрования отгоняли не вошедшую в реакцию NgO* и
реакционную смесь подвергали перегонке с водяным паром; при
охлаждении из водного дестиллята выделялись кристаллы
в виде желтоватых листочков. Остаток обрабатывали эфиром;
экстрагированный продукт, после отгонки эфира, извлекали
спиртом и спиртовым раствор обесцвечивали животным углем
при кипячении; при прибавлении воды к спиртовому раствору
выделен 2,4-динитрофенол, который при возгонке дал
кристаллы в виде золотистых игл с т. пл. 113—114°.
Кристаллы, выделившиеся из водного дестиллята,
идентифицированы как 2,4-динитрофенол (после возгонки они
плавились при 113—114°). Мононитрофенолы в продуктах реакции
не обнаружены.
Необходимо отметить, что нитрование фенола NsO* яв-
355
ляется экзотермической реакцией: при отсутствии охлаждения
температура поднимается до 100—105°.
Результаты опытов по нитрованию фенола приведены в
табл. 20.
Таблица 20
(5 г фенола, 25 мл N
№ опыта
1
2
3
4
Температура,
"С
0
5&-60
98—100
150—154
Выход 2,4-динйтрофенола
г
7,5
7.0
6,0
4 0
% от
теоретического
76
71
61
41
К&к видно из данных табл. 20, оптимальной является
температура реакции 0° (наибольший выход 2,4-динитрофенола
76%).
Нитрование м-крезола
Нитрование проводили аналогично обработке фенола. Из
водного дестиллята, полученного при перегонке продукта
реакции с водяным паром, выделен 4,6-динитро-м-крезол (т. пл.
63—65°).
Остаток после отгонки 4,6-динитрокрезола с водяным паром
экстрагировали эфиром, который затем отгоняли; полученный
таким образом 2,4,6-тринитро-м-крезол очищали от примеси
смолистых веществ многократной перекристаллизацией из горячего
лигроина (т. пл. 107—109°). Реакция нитрования м-крезола
сопровождается значительным выделением тепла (при
отсутствии охлаждения температура в реакционной колбе
поднимается до 115°).
Варьирование температур реакции дало следующие
результаты (табл. 21).
23*
356
Таблица 21
ж-креаола
(10 г м-крезола, 40—50 мл N,0^, продолжительаость реакции
50—55 мин.)
№
параллельных
ОПЫТОВ
1
2
3
Температура,
"С
0
98—100
Около 115
Выход 4
1
1
1
г
,5-2
,0-1
,0-1
,6-дипитрокре8ола
,0
,5
,5
% от
теоретического
8—10
5,5-8
5,5-8
Выход
г
6
7,0-
7
2,4,6-тринитронрезола
,0
-7,4
,0
% от
теоретического
26,7
31,1-33
31
Основной продукт нитрования — это 2,4,6-тринитрокре-
эол, наибольший выход которого (31—33%) получен при
температуре реакции 98—100°; при понижении температуры до
0° (наружное охлаждение) выход тринитрокрезола несколько
падает, причем повышается выход другого продукта реакции —
4,6-динитрокрезола.
Нитрование ^-нафтола
Нитрование проводили в растворе хлороформа (1 : 20),
так как в отсутствие растворителя происходит значительное
осмоление продукта. К 5 г р-вафтола в 100 мл хлороформа
постепенно приливали 25—30 мл жидкой NgOa при
продолжительности нитрования 55—60 мин. По окончании нитрования
отгоняли избыток NgOa и реакционную смесь обрабатывали
раствором соды; водао-щелочной слой отделяли от хлороформен-
ного и подкисляли соляной кислотой; при этом выпадал осадок,
который отфильтровывали, высушивали и растворяли в спирту;
к спиртовому раствору, обесцвеченному кипячением с
животным углем, добавляли воду, причем выделялся 1,6-динитро-р-
нафтол (т. пл. 195—196°). Небольшое количество 1,6-динитро-
р-нафтола получено также из хлороформенного раствора.
Результаты наиболее характерных опытов приведены в
табл. 22.
бед
357
Таблица 22
(5 г р-нафтола, 25—30 мл N,0*, продолжительность
нитрования 55—60 мин.)
№ опыта
1
2
3
4
Температура, °С
0
0
55—60
55—СП
Вызод 1,6-дт
6,5
5,5
5,0
4,5
% от
теоретического
81
69
62,5
56,2
Как видна из табл. 22, оптимальной температурой
нитрования {3-нафтола является 0° (выход 1,6-динитро-|3-нафтола до
81% от теории). При повышении температуры до 55—60*
значительно увеличивается количество образующихся смолистых
продуктов, и выход 1,6-Динитронафтола соответственно
понижается.
Нитрование анилина
К 10 г анилина в 100 мл хлороформа приливали 30 мл NgO*
в течение 40—45 мин. при наружном охлаждении реакционной
смеси снегом с солью. По окончании нитрования отгоняли
N, О* (при 40°) и хлороформ (при 80*); остаток перегоняли с
водяным паром. Выделившиеся из водного дестиллята желтоватые
кристаллы (листочки) 2,4-динитрофенола очищали возгонкой,
причем получены кристаллы в виде золотистых игл с т. пл.
113—114°. Этот продукт идентифицирован по отсутствию
понижения точки плавления смешанной пробы его с 2,4-дини-
трофенолом. Остаток в колбе после отгонки с паром
2,4-динитрофенола подщелачивали и затем экстрагировали эфиром. Из
эфирной вытяжки отгояяли эфир, и полученный в остатке
нвтроанилин перекристаллизовывался из спирта и воды
(игольчатые кристаллы с т. пл. 145—147°). Опыты по нитрованию ани-
358
лина яри указанных выше условиях проведения реакции дали
5 г 2,4-динитрофенола, что соответствует 25% от теории, и
около 1 г п-нитроанилина, что соответствует 7% от теории.
При нитровании анилина без охлаждения выходы обоих
продуктов понижаются приблизительно в 2 раза.
Полученные результаты совпадают с данными Ринкса [44].
Однако необходимо отметить, что этот автор не обнаружил
образования п-нитроанилина при нитровании анилина N#0*.
Варма и Кришамурти [45] при изучении действия окислов
азота (из смеси HNO@ и As*O@) на ароматические амины нашли,
что последние подвергаются диазотированию с последующим
нитрованием, в результате чего происходит образование нитро-
фенолов. При нитровании хлористоводородного анилина в
водном растворе при 0° они получили 2,5-динитрофенол.
Ринке, однако, показал, что при нитровании анилина
двуокисью азота получается не 2,5-, а 2,4-динитрофенол.
Нитрование диметиланилина
К 5 г диметиланилина в 50 мл хлороформа приливали 30 мл
N*O* в течение 45—50 мин. По окончании реакции отгоняли
N*O* и хлороформ, а остаток растворяли в ледяной уксусной
кислоте;, раствор выливали в холодную воду, причем выпадал
светложелтый осадок п-нитродиметиланилина; после
перекристаллизации из спирта выделен чистый п-нитродиметил-
анилин в виде желтых игл с т. пл. 164—166°. Кроме
п-нитродиметиланилина получен в незначительных количествах м-изо-
мер.
Варьирование температуры реакции показало, что
оптимальной температурой является 0° (выход
п-нитродиметиланилина 4,7 г, что соответствует — 70% от теории). При
повышении температуры до 55—60° выход п-нитродиметиланилина
снижается до — 55%.
По литературным данным [46], при действии N0% на диме-
тиланилин в четыреххлористом углероде (молярное отношение
диметиланилина к N0% = 1,6) наблюдается преимущественное
замещение в ядро с образованием п-нитродиметиланилина.
359
Условия реакции были следующие: к взвеси 0,05 моля диметил-
анилина в 50 мл четыреххлористого углерода прибавляют по
каплям раствор N0% в СС1*. Начальная температура реакции 15",
продолжительность опыта 2,5 часа, конечная температура 5*.
Выход п-нитродиметиланилина не указан. Реагируя с
избытком N0%, п-нитродиметиланилин превращается в 2,4-динитро-
диметиланилин и далее в 2,4-динитро-М-нитрозомонометил-
анилин. Выход последнего продукта 62% от теории. 2,4,6-
тринитродиметиланилин не реагирует с N0^. Из 2,4,6-три-
хлордиметиланилина и N0% в описанных условиях реакции
получен 2,4,6-трихлор-М-нитрозомонометиланилин с
выходом 86%. Из М,М-диметил-п-толуидина и N0^ образуется
продукт одновременного замещения в ядро и в боковую цепь, а
именно: 4-метил-2,6-динитро-М-нитрозомонометиланилин с
выходом 44% *
Нитрование ацетанилида
Раствор 5 г ацетанилида в 150 мл хлороформа нитровали
30—40 мл N*O* в течение 35—40 мин. при 60—70°. По
окончании реакции отгоняли избыток N^O* и хлороформ, и остаток
подвергали перегонке с водяным паром. Водный дестиллят
экстрагировали эфиром. После отгонки эфира получен о-нитро-
ацетанилид, который был перекристаллизован из горячей
воды и спирта (т. пл. 91—92°).
Остаток в колбе после отгонки с паром о-нитроацетанилида
экстрагировали эфиром; нерастворившийся в эфире продукт
промывали холодной водой и раствором двууглекислой соды,
а затем растворяли в горячей воде: из горячего водного
раствора выделен п-нитроацетанилид, который был перекристал-
лпзован из спирта (т. пл. 204—206°).
Из эфирной вытяжки получено еще некоторое
дополнительное количество о-нитроацетанилида с т. пл. 92—93°. _
Ацетанилид яри нитровании в указанных выше условиях
дал выход п-нитроацетанилида 3,5—4 г, что соответствует
53—60% от теории; о-нитроацетанилид получен с выходом 2 г
(30% от теории).
360
ожыслджы
Опыты по нитрованию аминов N^O^ дали, таким образом,
следующие результаты: анилин, кроме небольшого количества
п-нитроанилина, дает в качестве основного продукта реакции
2,4-дивитрофенол (предполагаемый механизм реакции — ди-
азотирование аминогруппы с последующим нитрованием ядра);
амины же с защищенной аминогруппой (диметиланилин и ацет-
авилид) образуют лишь замещенные в ядре нитросоединения,
т. е. ацил- и диалкиламиногруппа, как и следовало ожидать,
остается без изменения.
Нитрование хинолина
Из гетероциклических соединений изучено нитрование хи-
нолина в жидкой фазе. Опыты проводили при весовом
отношении N2O4 к хинолину 5:1, причем варьировалась температура
реакции.
По окончании реакции содержимое колбы выливали в воду
(туда же прибавляли остаток из конденсационных трубок
после отгонки из них N*O*); выделившийся при этом осадок
отфильтровывали от кислого фильтрата, в котором находились
растворенные мононитрохинолин и хинолин (в виде солей), и
обрабатывали 40%-ным раствором КОН; из щелочной
жидкости динитрохинолин экстрагировали бензолом. Бензольный
раствор, после сушки над твердым КОН, кипятили в течение
нескольких часов с животным углем для осветления
первоначально темной жидкости, затем из него отгоняли
растворитель и продукт очищали многократным осаждением водой из
спиртового раствора и лигроином из бензольного. В результате
указанной обработки выделен динитрохинолин в виде
желтоватых игл с т. пл. 170—180°. Невидимому, этот продукт
представлял собой смесь 5,7-динитрохинолина с другими нитро-
производными (по Кауфману и Гюсси [47] чистый 5,7-дини-
трохинолин плавится при 180°). К кислому фильтрату
добавляли NaOH и щелочной раствор экстрагировали смесью эфира
с бензолом. После сушки эфирно-бензольной вытяжки над
КОН отгоняли эфир в бензол и затем в вакууме — хинолин.
Остаток — сырой 7-мононитрохинолин был очищен двукрат-
361
ной перегоякой с водяным паром (т. пл, очищенного 7-моноии-
трохинолина 130—131*); смешанная проба последнего продукта
с 7-мононитрохинолином, приготовленным по методу Скрауна
из м-нитроанилина, глицерина, серной и мышьяковой кислот,
плавилась при 130—131°.
Результаты опытов по нитрованию хинолина N*O* при
различных температурах приведены в табл. 23.
Таблица 23
eg
С
О
1
2
3
4
5
6
7
8
Температура,
14—16
95—100
100—105
150—155
155—160
150—160
155—160
150
(20
1!
II
55
60
60
65
60
55
90
95
г хинолина, 100 мл )
ВЫХОД
мононитрох ино-
лина
г
(
3
3
1
0,5
1
0,5
1
% от
ретического по
взятому
хинолину
2леды
11
11
3,7
1,85
3,7
1,85
3,7
динитрохинолина
г
—
—
3,5
3,5
4
3
2
% от
ретического по
взятому
хинолину
_
—
10,3
10,3
11,8
9
5,9
Получено обрати
хинолина
г
18
15
14
13
14
12
12
12,5
% от
В8ЯТ0Г0
хинолин %
90
75
70
65
70
60
60
62,5
Как видно из этих данных, нитрование хинолина N%O*
протекает с небольшим выходом нитропроизводных (по взятому
хинолину); при температуре реакции около 100° образуется
только мононитрохинолин (выход 11%); при более высоких
температурах (150—160**) основной продукт реакции — динитро-
хинолин (выход 10—12%), выход же мононитрохинолина
значительно уменьшается. Несмотря на применение большого
избытка N2O4 (пятикратное количество по отношению к
хинолину), основная часть взятого хинолина не вступает в реакцию
(при 155—160° до 60—70%). При комнатной температуре
нитрование хинолина не происходит.
362
e гдао@о&
Нитрование органических соединений двуокисью азота в
газовой фазе проводилось нами в специальном аппарате (рис. 2),
снабженном ртутной лампой, что давало возможность изучать
влияние освещения ультрафиолетовыми лучами на протекание
Рис. 2. Схема нитрования углеводородов двуокисью азота
в газообразной фазе;
этой реакции. Аппарат состоял из вертикальной кварцевой
ртутной ламды 7 системы Гереус-Ханау, окруженной двумя
концентрическими кожухами, из которых наружный 5 служил
реакционным сосудом, а внутренний 6 (ближайший к лампе) —
холодильником или нагревателем (смотря по температурным
условиям опыта); через внутренний кожух посредством
отсасывания воздуха водоструйным насосом из конической колбы
76 пропускали нагреваемую или охлаждаемую жидкость (из
колбы 7 на бане 2, наполненной водой или вазелиновым маслом);
температуру этой жидкости измеряли двумя термометрами
бе* ждлмлыдд/лодх)* 363
Зи 7J. Испарение углеводорода и N2O4, которые приливали по
каплям из бюреток 70 и 74, происходило в колбах # и 72,
нагревавшихся в банях Р и 7J до необходимой температуры
(бюретка 7^cNgO4 окружена водяной рубашкой для предотвращения
испарения N^O^).
Для регулирования скорости поступления в реакционную
камеру паров углеводорода и N^0^ через колбы # и 72
пропускали ток СО*, которую предварительно обезвоживали
пропусканием через скляяки 77 с серной кислотой. Пары продуктов
реакции конденсировали сначала в холодильнике 4, затем в
охлаждаемых снаружи конденсационных склянках 7Z и
поглотительных склянках 7Д с раствором соды и, наконец, остатки
улавливали активированным углем в турмах 79. Для
облегчения прохождения газов и паров через конденсационно-по-
глотительную систему их отсасывали насосом 20, помещенным
в конце системы. Все части аппаратуры соединены шлифами
или спаями.
Нитрование бензола
40 г бензола, 180 г NgO^ и 40—50 л СО а пропускали в
течение 2 час. через реакционную камеру. По окончании опыта
отгоняли при 25—40° невошедшую в реакцию N#0*, а остаток
подвергали перегонке для отделения нитробензола от непрореа-
гировавшего бензола. Как и при нитровании толуола, динитро-
производных бензола в продуктах реакции не обнаружено.
Результаты наиболее характерных опытов приведены в табл. 24.
Из данных табл. 24 видно, что облучение
ультрафиолетовым светом не улучшает выхода нитробензола по сравнению
с опытами без облучения. Это указывает на то, что
ультрафиолетовые лучи влияют на нитрование положительным образом
лишь в тех случаях, когда полная симметрия молекулы
нарушена наличием боковой цепи, благодаря которой возможно
таутомерное превращение с образованием таутомернои системы
конъюгированных двойных связей (как это имеет место в
случае толуола, см. ниже).
Оптимальные результаты (выход нитробензола около 32%
от теории) получены при 55—60
°
364
Таблица 24
б^ндолд ^т/окисью
(40 г бензолы, 180 г К%0«)
(б
%
1
2
3
4
5
6
7
8
Температура, *с
10-12
25—30
55—60
80—85
10-12
25—30
55—60
80-85
Выход
нитробензола
Г
8
18
20
8
8
17
20
8
% от
ретического
12 7
28,6
31,7
12,7
12,7
27,0
31,7
12,7
Получено обратно
бензола
г
33
25
20
22
32
25
22
20
% от
ретического
82,5
62,5
50,0
55,0
80,0
62,5
55,0
50,0
Примечание
С облучением
Без облучения
В другой серии опытов но нитрованию бензола
варьировалось отношение N%O& к бензолу. Эти опыты показали, что при
применении 100 г NgO* на 40 г бензола (что соответствует
молярному отношению 2 : 1), выход нитробензола остается без
изменения, по сравнению с выходами в предыдущей серии
опытов (см. табл. 24), где это отношение равнялось 4:1. При
уменьшении же количества N#0* до 50 г (отношение 1 : 1)
выход нитробензола значительно понижается (до 6,5%). Таким
образом для получения оптимальных результатов необходимо
применять избыток N*O* (2 моля NgO* на 1 моль бензола).
Нитрование толуола
Опыты по нитрованию толуола двуокисью азота
производились нами как с облучением ультрафиолетовым светом, так
и без облучения.
Нитрование толуола двуокисью азота проводилось
следующим образом. Через реакционную камеру в течение 2 час.
пропускали 40 г толуола, 200 г N*O* и 40—45 л СО*. По окончании
365
опыта содержимое конденсационных склянок 17 (рис. 2)
переливали в колбу, соединенную (на шлифах) с холодильником,
через который пропускали охлажденную до 0° воду, и при
25—40° отгоняли непрореагировавший избыток N%0& для
использования в дальнейших опытах. Остаток выливали в
большое количество воды, куда добавляли промывные воды от спо-
ласкивания всех частей аппарата, содержимое поглотительных
склянок 7Д и конденсат, полученный при отгонке с перегретым
паром продуктов, адсорбированных в турмах 79
активированным углем. После стояния продукты реакции разделялись на
два слоя: масляный и водный. Масляный слои отделяли,
водный слои обрабатывали эфиром и эфирную вытяжку
присоединяли к масляному слою. Масло с эфирной вытяжкой
промывали раствором соды и водой и после отгонки растворителя
перегоняли с водяным паром. Масло перегналось полностью, что
указывало на отсутствие динитротолуолов. Выделившееся в
водном дестилляте масло отделяли от воды и обрабатывали 20% -ным
раствором NaOH. После стояния в течение ночи отделяли
щелочной раствор от нерастворившегося в NaOH масла и об-
рабатывали последнее эфиром (эфирный раствор А). При
нейтрализации щелочной вытяжки соляной кислотой выделялась
кристаллическая аци-форма фенилнитрометана, которая
постепенно приобретала маслообразную консистенцию вследствие
перехода в нейтральную форму. Последнюю экстрагировали
эфиром (эфирный раствор Б). После отгонки эфира из раствора Б
получен чистый фенилнитрометан с т. кип. 140—142* при
35 мм.
Нерастворившееся в NaOH масло и эфирный раствор А
дали смесь изомерных мононмтротолуоловст. кип. 217—238°,
Результаты наиболее характерных опытов по нитрованию
толуола двуокисью азота приведены в табл. 25.
Данные табл. 25 показывают, что при нитровании толуола
двуокисью азота образуются главным образом мононитро-
толуолы наряду с фенилнитрометаном. Оптимальные
результаты при освещении ультрафиолетовыми лучами получены при
14—15° (выход мононитротолуолов 46% , фенилнитрометана9% ).
С повышением температуры до 58—60° выход фенилни-
366
Таблица 25
(40 г толуола, 200 г N%0^)
(б
§
1
2
3
4
5
6
7
Температура,
*С
14—15
58—60
105—110
14-15
58—60
105—110
140—145
Выход
фенил-
нитрометана
г
5
5,5
3
4
5
3
2,5
&:
8,5
9,4
5
7
8,5
5
4,2
Выход
яитро-
толуолов
г
27
15
10
19
11
11
10
S|
46,2
25,5
17
32,5
18,8
18,8
17
Получено
обратно
толуола
г
16
23
15
22
27
20
16
о
40
57,5
37,5
55
67,5
50
40
Примечание
L С облучением
!- Без облучения
трометана остается почти без изменения, а выход нитротолуолов
значительно уменьшается (до 17%).
Без облучения нитрование протекает менее эффективно.
Необходимо отметить, что относительные количества фенилнит-
рометана и нитротолуолов в опытах с освещением остаются
без изменения по сравнению с опытами без освещения, несмотря
на увеличение выходов.
Н. А. Валяшко, В. И. Близнюков и А. Е. Луцкий [48]
нитровали толуол двуокисью азота следующим способом:
реакция NgO* (3 моля) с толуолом (1 моль) проводилась в
запаянной стеклянной трубке при температуре до 70°. Уже при
комнатной температуре реакция шла очень энергично. При
температуре 70° наблюдались взрывы. Основным направлением
реакции было вступление N0% в метильную группу, побочное
направление реакции — вступление в ядро с образованием
мононитротолуолов. В процессе нитрования образуются
продукты окисления — бензойная и щавелевая кислоты.
Катализаторы, обычно направляющие нитрогруппу в ядро, в этих ус-
бед ждтндлыддлкфОб 367
ловиях оказались недеятельными. Замедление реакции
NgO^ с толуолом было достигнуто заполнением трубок
стружками из нержавеющей стали, разведением реакционной смеси
ССЦ и уменьшением количества NaO* до 1,7—1 моля на 1 моль
толуола. Ускорение реакции нитрования в ядро достигнуто
прибавлением к реакционной смеси 0,27—0,4 моля уксусного
ангидрида на 1 моль толуола, причем было получено 2,5—3%
2,4,6-тринитротолуола и до 30% нитротолуолов.
Образовавшиеся мононитротолуолы дальше не нитруются. При
нитровании толуола в ядро получаются только моно- и тринитрото-
луолы без промежуточного образования динитротолуола.
Замена ангидрида на уксусную кислоту не вызывает ускорения
реакции.
Нитрование этилбензола
15 г этилбензола (т. кип. 136—138°), 70—80 г N,0* и
15—20 л СО* пропускали через реакционную камеру в течение
50—55 мин. По окончании нитрования отгоняли NgO*H3
конденсационных склянок 77 (рис. 2) и смешивали их содержимое
с содержимым поглотительных склянок 7# и с конденсатом,
полученным при отгонке с перегретым паром продуктов,
адсорбированных активированным углем. Смесь при стоянии
разделялась на два слоя: масляный и водный. Масляный слой
отделяли и к нему присоединяли эфирную вытяжку,
полученную посредством обработки эфиром водного слоя. После
промывания 5%-ным раствором соды, а затем водой отгоняли эфир
и масло подвергали перегонке с водяным паром; при этом в
водный дестиллят переходили метилфенилнитрометан и моно-
нитроэтилбензол. В жидкости, оставшейся после отгонки
указанных выше нитросоедииений, динитроэтилбензола ие
обнаружено.
Для отделения метилфенилнитрометана от мононитроэтил-
бензола масло, находящееся в водном дестилляте,
обрабатывали 20%-ным раствором едкого натра при энергичном
встряхивании в течение 20 мин. и оставляли для отстаивания на ночь.
Щелочной раствор отделяли после этого от нерастворившегося
368
в NaOH масла, промывали эфиром для извлечения остатков
нитроэтилбензола и подкисляли 20 %-ной соляной кислотой до
кислой реакции на конго; при этом из раствора выделялась
в виде беловатых хлопьев аци-форма метилфенилнитрометана,
которая даже при долгом стоянии не превращалась в
нейтральную форму.
Через 10—12 час. метилфснилнитрометан экстрагировали
эфиром и после сушки эфирной вытяжки над хлористым
кальцием отгоняли эфир и очищали продукт возгонкой в вакууме.
Полученный таким образом метилфенилнитрометан
представлял собой бесцветные кристаллы с т. пл. 97—99°*. К нераство-
рившемуся в щелочи маслу добавляли эфирную вытяжку,
полученную обработкой щелочного раствора эфиром (см. выше),
отгоняли эфир и подвергали остаток дробной перегонке. При
этом сначала отгоняли этилбензол при 136—138°, а затем
перегонялась смесь нитроэтилбензолов в интервале 223—246°.
Результаты наиболее характерных опытов по нитрованию
этилбензола приведены в табл. 26.
Таблица 26
(15 г этилбсызола, 70—80 г Х^
о
1
2
3
4
Температура, °о
10—12
78—80
10-12
78-30
Выход
фенилнитрометан а
г
4,5
8
3
6
% от
теоретического
21,4
40
14,3
28,6
Выход нитро-
этллбсвволов
г
5
5
4,5
% от
теоретического
24
24
14,3
21,4
Получено обратно
этилбензола
г
Т
2,5
8
2
% от
взятого
этял-
бензола
46,7
16,7
53,3
13,4
Примечание
1 С облу-
j чением
1 Ьсз облу-
; чения
* Эти результаты ыо согласуются с данными М. И. Коновалова
[49|: полученный последним цри нитровании этилбензола азотной кислотой
(уд. в. 1,075) метилфенилнитрометан кге затвердевал даже при —15* и
имел т. кии. 135° при 25 мм.
369
Из данных этой таблицы видно, что нитрование этилбен-
зола двуокисью азота приводит к образованию мононитроэтил-
бензолов наряду с продуктом замещения группой N0% в
боковой цепи — метилфенилнитрометаном. При 10—12° и
проведении реакции при освещении ультрафиолетовыми лучами
выход нитроэтилбензолов несколько превышает выход метилфе-
нилнитрометана (общий выход нитросоединений 45%); при
повышении температуры до 78—80° общий выход нитросоеди-
нений достигает 64% , причем выход метилфенилнитрометана
повышается почти вдвое (до 40%), выход же
нитроэтилбензолов остается без изменения. Такая же закономерность
(увеличение выхода с повышением температуры главным образом
нитросоединений, замещенных в боковой цепи) наблюдается
и в опытах без облучения, которые в общем дали более низкие
выходы нитропроизводных сравнительно с опытами,
проводившимися при облучении.
Нитрование м-ксилола
Нитрование м-ксилола двуокисью азота и разделение
продуктов реакции проводили аналогично нитрованию толуола и
этилбензола.
20 г м-ксилола с т. кип. 139—140°, 140—150 г N,0. и 20—
25 л СО g пропускали через реакционную камеру в течение 50—
60 мин. при 12—14°. В результате нитрования выделены: толил-
нитрометан (желтоватое масло с т. кип. 128—132° при 19 мм),
смесь моноыитроксилолов, которые перегонялись при 225—
245°, и небольшое количество динитро-м-ксилолов.
Опыты с освещением ультрафиолетовыми лучами дали 21 г
мононитрокенлолов (73,9% от теории по взятому м-ксилолу),
7 г толилнитрометана (24,6% от теории); кроме того, получено
незначительное количество (0,5 г) динитро-м-ксилолов. Опыты
без облучения дали более низкие выходы мононитропроизвод-
ных: 16 г мононитроксилолов, что соответствует выходу 56,3%
от теории, и 6 г толилнитрометаыа (21% от теории); выход
24 \. В.
370 #%лгро*дн%е ореднхмдсжыа; сое^онений ожыдлдл*;*
динитро-м-ксилолов оказался несколько больше, чем в опытах
с облучением (3,5% от теории).
Эти результаты указывают, что нитрование м-ксилола
двуокисью азота в газовой фазе протекает вполне
удовлетворительно уже при 12—14°, причем основным продуктом реакции
являются мононитроксилолы. При применении освещения
ультрафиолетовыми лучами получен почти количественный общий
выход нитропроизводных м-ксилола, при нитровании же без
освещения общий выход этих соединений составляет 81% .
Нитрование пиридина
Нами изучено также нитрование двуокисью азота пиридина,
который, по литературным данным, нитруется с большим
трудом. Так, например, Фридль [50] действовал на пиридин
смесью 18% -ной дымящей серной кислоты с нитратом калия при
330°, пропуская одновременно через реакционную смесь
сильную струю воздуха. В результате нитрования им получен
3-нитропиридин(т. пл. 41°) с выходом 15% от теоретического.
Шааршмидт, Бальцсрксвич и Ганте [41] нитровали
пиридин N*O* как без катализаторов, так и в присутствии А1С1%,
но не получили положительных результатов.
Нитрование пиридина осуществлялось нами следующим
образом: 20 г пиридина (предварительно высушенного и
перегнанного над ВаО), 100—150 г NaO* (т. кип. 22—25°) и 15—20 л
СО & пропускали в течение 1 часа через реакционную камеру.
По окончании нитрования все части аппарата промывали
чистой N*O*, промывную жидкость смешивали с содержимым
конденсационных склянок и выливали в колбу с
пришлифованным термометром и с пришлифованным либиховским
холодильником, через муфту которого пропускалась ледяная вода. При
слабом нагревании при температуре 25—40° отгоняли непрореа-
гировавшую NaO*. Остаток в колбе обрабатывали раствором
соды и экстрагировали эфиром; из эфирного раствора отгоняли
эфир, а затем не вошедший в реакцию пиридин; оставшуюся
бурую массу растворяли в азотной кислоте (уд. в. 1,4). После
долгого стояния в этом растворе образовались кристаллы азот-
371
нокислой соли р-нитропиридина. При добавлении соды к
водному раствору этой соли выделялось свободное основание,
которое перекристаллизовали из смеси бензола с лигроином; по-
лученяый таким образом jj-нитропиридин представлял собой
бесцветные иглы с т. пл. 41—42°.
Опыты при более высоких температурах (200—300°)
проводили в тугоплавкой стеклянной трубке (трубке для сожжения),
наполненной стеклянными бусами [51].
Попытки осуществить нитрование пиридина N*O* в
газовой фазе при температурах выше 300° привели к сильному
взрыву, разрушившему аппаратуру. Очень сильные взрывы
наблюдались также при смешивании пиридина с жидкой NaO*
при комнатной температуре.
В табл. 27 приведены результаты опытов по нитрованию
пиридина двуокисью азота при различных температурах.
Таблица 27
(20 г пиридина, 100—150 г N„0*)
Температура,
14—15
58—60
115—120
14—15
58—60
115—120
220—230
Выход В-нитро-
пиридина
г
1,0
1,5
2,0
1,0
1,5
2,0
2,0
% от
теории
3,2
4,8
6,4
3,2
4,8
6,4
6,4
Получено обратно
пиридина
г
13
12
12
11
13
12
10
% от
ВВЯТОГО
пиридина
65
60
60
55
65
60
50
Примечание
С облучением
Без облучения
Из указанных опытов по нитрованию пиридина можно
сделать следующие выводы: 1) освещевие ультрафиолетовым
светом не оказывает влияния на выход нитросоединений, что
372
объясняется симметричным строением молекулы пиридина
(аналогия с бензолом и нафталином); 2) оптимальной температурой
нитрования является 115—120°; повышение температуры до
220—230° не изменяет выхода ыитропиридина; 3) при вит-
ровании большая ч%сть пиридина не вступает в реакцию
(60—65%).
При нитровании пиридина двуокисью азота в продуктах
реакции обнаружена также азотнокислая соль пиридина
C^HgNHNOg, образование которой может быть объяснено
действием на пиридин азотной кислоты, получающейся по
уравнению
N,0, -!- Н„О -» НКО„ + Н\О,
(пиридин, благодаря свойственной ему гигроскопичности,
содержит воду, которая и реагирует с NgO*).
Азотнокислый пиридин образуется также при действии
паров NgO* на жидкий пиридин при комнатной температуре или
при приливании по каплям жидкой NgO* к хорошо
охлажденному (смесью льда с поваренной солью) пиридину, выделяясь
в обоих случаях в виде красного кристаллического осадка;
при перекристаллизации этого осадка из горячего спирта
нитрат пиридина получается в чистом виде (бесцветные
кристаллы с т. пл. 117—118°).
Для выяснения возможности использования для
нитрования ароматических соединений окислов азота (нитрозных
газов), получающихся в больших масштабах при производстве
азотной кислоты из атмосферного азота, проведены опыты с
искусственной газовой смесью, которая по своему составу
соответствовала промышленным нитрозпым газам (10% М%О*, 80%
N,, 5% О, и 5% Н,О).
Эта смесь приготовлялась следующим образом: из
бюретка, охлаждаемой водяной рубашкой, приливали жидкую
NgO* в колбу Вюрца, которую нагревали на водяной бане;
образовавшиеся пары NgO* переходили во вторую колбу, где
ждтдлыддпмроа 373
происходило смешение их с газовой смесью, поступавшей из
газометра и составленной в определенных объемных отношениях
из Ng, О2, Н%0; из смесителя газы направляли в реакционную
камеру-нитратор, куда поступали пары нитруемого соединения
(из испарителя).
Нитрование бензола
В служившую испарителем колбу Вюрца, которую
нагревали до температуры кипения бензола, приливали по каплям
бензол из пришлифованной к колбе бюретки, охлаждаемой
водяной рубашкой; пары бензола через отводную трубку
направляли в колбу-нитратор емкостью 500 мл, где они встречались
с нитрозными газами, поступавшими из смесителя. Продукту
реакции вместе с непрореагировавшим бензолом и избытком
N*O* конденсировали сначала в холодильнике, затем в системе
соединенных друг с другом (на шлифах) приемников, которые
охлаждали сухим льдом до —80°. По окончании опыта
содержимое приемников сливали вместе и отгоняли при 25—40*
избыток N#0*; остаток подвергали перегонке для разделения нс-
прореагировавшего бензола и нитробензола.
Результаты наиболее характерных опытов по нитрованию
бензола нитрозными газами, проводившихся при различных
температурах (в каждом опыте смесь 40 г бензола и нитроз-
ных газов в количестве, содержащем 47 г N,0*, пропускали
через реакционную камеру в течение 2 час), приведены
в табл. 28.
Как видно из этих данных, оптимальные результаты
(выход нитробензола по взятому бензолу около 13% ) получаются
при 55—60°, причем значительная часть (80% ) бензола остается
без изменения (выход до бензолу, вступившему в реакцию,
65%).
Нитрование нафталина
10 г нафталина и нитрозные газы в количестве, отвечающем
8 г NgO&, пропускали через реакционную камеру в течение
374
Таблица 28
(40 г бензола, 47 г N,0*)
№ опыта
'1
2
Температура,
15—18
55—60
90—100
Выход нитробензола
г
5
8
4,5
% от
теоретического
8
12,9
7,25
Получено обратно бензола
г
35
32
35
% от взятого
бензола
87,5
80,0
87,5
2 час. при 18—20°. В результате нитрования получено 13 г
сс-мононитронафталина, что соответствует выходу 96% от
теории. Динитросоединенмй в продуктах реакции не оказалось.
ВЫВОДЫ
Анализируя результаты наших исследований по
нитрованию окислами азота ряда ароматических и гетероциклических
соединений, можно прийти к следующим основным выводам.
1) При нитровании бензола, нафталина и фенантрена
получаются главным образом мононитропроизводные; наиболее
легко нитруется нафталин; выход ос-мононитронафталина при
температуре реакции 18—20° составляет 96% (без примеси
полинитропроизводаых). При температурах выше 60°
образуются также полинитропроизводяые. Нитрование
фенантрена при 0° приводит к смеси мононитрофенантренов с общим
выходом 48% . Бензол нитруется труднее, чем предыдущие
соединения; выход нитробензола составляет 32% при температуре
реакции 55—60°.
2) В отличие от указанных углеводородов, при нитровании
антрацена основным продуктом реакции является 9,10-динит-
роантрацен с выходом 80% от теории (оптимальная
температура 0°).
3) Освещение ультрафиолетовыми лучами не оказывает
никакого влияния на реакцию нитрования ароматических угле-*
беа «атдлиэатло^об 375
водородов с симметричным строением молекулы (бензол,
нафталин и др.).
4) Нитрование ароматических соединений с боковой цепью
(жирноароматические соединения) приводит к одновременному
образованию мононитропроизводных, замещенных как в
боковой цепи, так и в ядре, с преобладанием последних.
Например, при освещении ультрафиолетовыми лучами толуол
дает 46% мононитротолуолов и 9% фенилнитрометана (при
температуре реакции 14—15°), а этилбензол — 40% нитроэтил-
бензолов и 24% метилфенилнитрометана (при 78—80°).
5) Освещение ультрафиолетовыми лучами при нитровании
ароматических соединений несимметричного строения приводит
к повышению выхода нитропроизводных сравнительно с
опытами без освещения (например, м-ксилол нитруется с общим
выходом нитропроизводных 99% при освещении и 81% без
освещения).
6) Нитрование фенолов протекает сравнительно легко,
причем образуются динитро- и тринитропроизводные. Фенол
дает 2,4-динитрофенол с выходом 76% , м-крезол—смесь 2,4,6-
тринитро- и 4,6-динитро-м-крезолов с общим выходом 41%,
а ^-нафтол — 1,6-динитро-^-нафтол с выходом 81% .
Оптимальные температуры нитрования: для фенола 0°, м-крезола 98—
100°, р-пафтола 0°.
7) Нитрование аминов протекает различно в зависимости
от того, защищена аминогруппа или нет. Нитрование анилина
при незначительном выходе п-нитроанилина приводит к
преимущественному образованию 2,4-динитрофенола, что указывает
в качестве возможного механизма реакции на диазотирование
аминогруппы с последующим нитрованием ядра; нитрование
аминов с защищенной аминогруппой— диметиланилина и ацет-
анилида — протекает значительно легче, причем диметил-
анялин нитруется с образованием п-нитродиметиланилина с
выходом 72% при температуре реакции 0*, а ацетанилид дает
смесь п-ио-нитроацетанилида; выход п-изомера 60% и о-изо-
мера 30% при температуре 60—70°.
8) Нитрование гетероциклических соединений протекает
значительно труднее соединений ароматического ряда;
376
хинолин при 95—100° нитруется с образованием мононитрохино-
лина с выходом 11% ; при 155—160° образуется главным
образом динитрохинолин с выходом 10—12% ; нитрование
пиридина приводит к небольшому выходу (6%) 8-нитропиридина
(оптимальная температура 115—120°). При нитровании как
хинолина, так и пиридина, значительная часть нитруемого
соединения (65—70%) не вступает в реакцию и может быть
регенерирована. Необходимо отметить, что пиридин нитруется
двуокисью азота в газовой фазе уже при комнатной
температуре, тогда как по методу Фридля нитрованием HNOs в смеси
с дымящей серной кислотой реакция осуществляется лишь
при 330°. .
9) Опыты по нитрованию газовой смесью, соответствующей
по составу промышленным нитрозным газам, показали, что
последние могут быть использованы для нитрования
ароматических соединений; например, нафталин нитруется нитрозными
газами при температуре 18—20° аналогично нитрованию
чистой NgO* с выходом 96% от теории.
А. И. Титов [52], посвятивший ряд работ нитрованию
двуокисью азота органических соединений, установил, что при
соответствующих условиях нитрование жирноароматических
соединений может привести к вполне удовлетворительным
выходам w-нитропроизводных, причем отношение выходов нитро-
и динитропроизводных находится в зависимости от
концентрации NO и NOg в реакционной смеси и температуры нитрования.
Влияние первого фактора доказано им экспериментально
следующим образом: он смешивал одни и те же количества
двуокиси азота (25 мл) и безводной CuSOa (20 г) с различными
количествами толуола и выдерживал эту смесь при 20° в течение
30 дней. Результаты оказались следующими:
Дед ждтмдлыддтмороб 377
Количество толуола, мл
1000 250 50
Выход фенилдинитромстана, г .... 23,5 7,3 2,6
Выход фенилиитромстана, г 5,0 3,9 2,1
Эти данные показывают, что с увеличением разбавления,
т. е. с уменьшением концентрации N0% (при низких
температурах), выход динитропроизводных, в данном случае фенил-
динитрометана, повышается, что согласуется с выводами*
А. И. Титова. Действительно, понижение концентрации N0%
создает более благоприятные условия для соединения
радикалов с NO, т. е. для образования нитрозосоединений, которые,
по мнению Титова, являются источником возникновения дини-
тропроизводных:
NO, . NO
С,Н,СН, —> С,Н,СН, -> С,Н,СН,КО ^
Влияние температуры установлено А. И. Титовым опытами^
в которых последняя варьировалась от 20 до 95°.
Температура реакции
20* 40* 70* 95'
Выход фенилдинитрометана, г ... 23,5 18,4 6,8 1,4
Выход фенилнитрометана, г .... 5,0 7,2 15,9 13,4
Увеличение выхода фенилдинитрометана с понижением
температуры объясняется А. И. Титовым увеличением
растворимости окиси азота и уменьшением диссоциации на NO* димер-
ной формы NsO*, присутствие которой необходимо для
превращения нитрозосоединения в фенилдинитрометан (см. выше
схему реакции).
Выход фенилдинитрометана по отношению к фенилнитроме-
тану может быть повышен посредством насыщения
реакционной смеси окисью азота. Например, смесь 18,4 г двуокиси азота
с 2 л толуола была предварительно насыщена окисью азота в
затем оставлена при комнатной температуре на 6 дней (опыт
№2). В сравнении с контрольным опытом (№ 1) без насыщения
окисью азота получены следующие результаты:
378
Опыт Опыт
Выход фенилнитрометана, г 3,7 2,3
Выход фенилдинитрометана, г 14,8 11,7
Отношение количеств динитро- и иоио-
нитросоединения 4:1 5:1
Приводим описание одного из опытов А. И. Титова по
нитрованию толуола двуокисью азота на фенилдинитрометан,
давшего хорошие результаты: смесь 1500 мл толуола, 10 г
двуокиси азота и 30 г безводной CuSO*, предварительно
насыщенная при охлаждении окисью азота до привеса 1 г, была
оставлена на 15 дней при температуре около 25—30°, причем в
течение первых 9 дней через каждые 3 дня прибавлялось по 10 г
двуокиси азота. В результате опыта получены следующие
выходы продуктов реакции:
г %
Фени лнлтрометан 4,6 10,3
Фенилдинитрометан 28,1 46,7
Бензойная кислота 4,3 10,6
В противоположность &)-динитросоединениям, образование
которых происходит благодаря взаимодействию с NO, w-моно-
нитросоединения возникают при действии на боковую цепь
жирноароматического углеводорода мономерной формы
двуокиси азота (применяющаяся обычно для нитрования жидкая
N2O4 представляет собой равновесную смесь мономерной N0%
и димерной NaO* форм двуокиси азота и, кроме того,
некоторого количества NO).
Важнейшим фактором, определяющим направление
реакции в сторону образования ы-мононитропроизводных, является
достаточно высокая температура реакции (оптимальная
температура 100°), способствующая сдвигу равновесия
слева направо, что создает высокую концентрацию N0% в
реакционной смеси.
Образован ию &)-мононитросоединений благоприятс твует
введение в сферу реакции кислорода, переводящего окись азота
беа кдлмлиддтдро» 379
в двуокись. Например, смесь 1750 мл толуола с 20 г безводной
CuSO& насыщали в течение 4 час. двуокисью азота при
нагревании на кипящей водяной бане. Одновременно через смесь
пропускали кислород (опыт № 2). В сравнении с контрольным
опытом № 1, без введения кислорода, получены следующие
результаты:
Выход в % от теоретического
на прореагировавший толуол
Опыт № 1 Опыт № 2
Фенилнитрометан 51 52,5
Фенилдинитрометан 2,7 2.7
Бензойная кислота 7,1 11,2
Выход по связанному азоту с учетом регенерированной NgO*
составлял в опыте с введением кислорода (№ 2) 93%, в опыте
без применения кислорода (№ 1) 63% от теоретического.
Другим фактором, благоприятствующим выходу &)-моно-
нитропроизводных (в данном случае фенилнщтрометана),
является уменьшение концентрации раствора (при высоких
температурах). При прочих равных условиях (температура 100°,
количество N0% 43 г, продолжительность насыщения 2 часа,
расход кислорода 8 л) опыт, где было взято 1750 мл толуола,
дал абсолютный выход фенилнитрометана в 3 раза выше, чем
при использовании 175 мл толуола.
Нитрование бензола двуокисью азота А. И. Титовым также
рассматривается как свободно-радикальный процесс. Роль
первичного радикала, по мнению А. И. Титова, выполняет ради-
калоподобный комплекс — продукт присоединения электро-
фильной и радикалоподобной молекулы NO* к бензольному
ядру
н
При этом по мере увеличения разведения бензолом одного
и того же количества окислов азота (NgO*) выход нитропроиз-
380 Иитдроашые о^юдничесляи; сое&шенЫк ожислдлш
водных возрастает. Из продуктов реакции А. И. Титовым
выделены нитробензол, м-и п-динитробензолы, симм. тринитро-
бензол, 2,4-динитрофенол, 2,4,6-тринитрофенол и щавелевая
кислота*
После опубликования работ П. П. Шорыгина и А. В.
Топчиева по нитрованию ароматических соединений двуокисью
азота Урбанский и Слон [53, 54] исследовали реакцию
нитрования алифатических предельных углеводородов двуокисью
азота в газовой фазе. Смесь паров NaO* и углеводорода
пропускали через стеклянную трубку, наполненную стеклянными
кольцами, которую нагревали в электропечи до температуры
около 200°. Летучие продукты реакции конденсировались.
Избыток NaO* удаляли на холоду током воздуха и остаток,
состоявший из нитропродуктов и непрореагировавшего
углеводорода, промывали водой. После сушки над Na*SO* из этой
смеси отгоняли углеводород. Оставшуюся смесь
нитропродуктов фракционировали в вакууме.
Экспериментальные данные показали, что выходы
нитропродуктов варьируют от 30 до 80% в зависимости от условий
нитрования и размеров реактора. Отношение между моно- и
динитропроизводными обычно составляет 60 : 40.
При нитровании пропана .получены нитропропан (т. кип.
31—34°) и %,%'-динмтропропан (т. кип. 42°) с общим выходом
имтропроизводных до 70% от теории; нитрование пентана дало
смесь 60% моно- и 40% динитропентана, из которой выделены
нитропентан (т. кип. 164—165° при 750 мм) и динитропентан
(в остатке). Гексан и гептан нитруются с образованием
соответственно мононитрогексана (т. кип. 66—72° при 14 мм) и моно-
нитрогептана (т. кип. 199—200° при 750 мм) и динитросоедине-
ний. При нитровании метана в качестве продуктов реакции
получены нитрометан (с небольшим выходом) и полинитросо-
единения до тетранитрометана включительно.
Показано также, что при нитровании предельных
углеводородов в указанных выше условиях основными продуктами
реакции являются первичные нитросоединения, содержащие
группу СН%]\О2(эти продукты идентифицировались по
образованию нитроловых кислот и восстановлению последних дс
бб* ждлгддиадлм^об 381
альдегидов при действии цинка и уксусной кислоты). Наряду
о мононитропродуктамн, отвечающими общей формуле
OgNCHg — (GHg)^— CHg, нитрование дает также динитропроиз-
водные общей формулы 0%NCH* — (СН*)* — CH^NOg.
Исходя из аналяза жидких и газообразных продуктов
реакции, Урбанский и Слон дают следующую схему нитрования
алифатических углеводородов:
2RCH,
Кроме нитропроизв^одных в продуктах реакции найдены
альдегиды и жирные кислоты, образующиеся в результате
окислительного действия окислов азота.
Нитрование алифатических углеводородов двуокисью азота,
до данным авторов, представляет собой взрывоопасный
процесс (при проведении опытов в некоторых случаях
наблюдались взрывы, сопровождавшиеся разрушением аппаратуры).
Хэсс и другие [55] при нитровании пропана окислами азота
KN2O4) при температуре 790—795° получили примерно равные
количества нитроэтана, 1- и 2-нитропропана. Более низкая
температура благоприятствует образованию 2-нитропропана.
Кроме нитропарафинов образуются различные продукты
окисления: альдегиды, кислоты. Значительная часть исходного
вещества остается без изменений.
А. И. Титов [56] при нитровании н. пентана двуокисью
.азота при температуре 260—270° получил почти
исключительно вторичный нитропентан и следы первичного.
Нитрование парафиновых углеводородов, согласно
патентным данным [57], можно осуществить, так же как и нитрование
ароматических углеводородов, нитрозными газами,
получающимися при каталитическом окислении аммиака. Продукты
реакции окисляются кислородом или воздухом с образованием
литропарафинов (предполагается, что первичная стадия
процесса состоит в образовании нитрозосоединений).
П. П. Шорыгин и А. В. Топчиев [58] установили, что при
взаимодействии н. гексана с N^O^, разбавленной углекислотой,
получается небольшой выход 2-нитрогексана. Реакция
382
проводилась при температуре 10—80^ в течение 1 часа. Цикло-
гексанв этих условиях дает около 15% мононитроциклогексана.
Эти же авторы [42] еще в 1935 г. показали возможность
нитрования парафиновых углеводородов окислами азота при
низких температурах. В последнее время автором настоящей книги
разработан оригинальный метод нитрования предельных
углеводородов в газовой фазе посредством одновременного
воздействия хлора и окислов азота. Пользуясь этим методом, удалось
осуществить с хорошими результатами нитрование метана,
который, как известно, нитруется азотной кислотой и окислами
азота лишь при довольно высоких температурах с небольшим
выходом нитропроизводных.
Работа проводилась с природным саратовским газом,
содержащим 92—93% метана, 2,8% этана, 1,49% пропана, 0,56%
бутана и др. Для очистки от примеси высших гомологов метана
газ перед поступлением в реактор пропускали через колонку,
наполненную активированным углем. Опыты по изучению
реакции взаимодействия двуокиси азота и хлора на метан [59]
проводились в газообразной фазе в интервале температур от
комнатной (25—30°) до ^ 300° при отношении метан ( окислы
азота = 4:1 и метан :хлор=1 : 3. Жидкие продукты реакции
растворяли в эфире, эфир промывали водой, затем 2%-ным
раствором двууглекислой соды и сушили над хлористым кальцием.
После отгонки эфира на водяной бане масло переносили
в колбу с дефлегматором и подвергали фракционированной
перегонке, в результате которой выделены фракции с
температурой кипения 100—103° (45—49° при 22 мм), 120—124°, 128—132°
и 133—137°. Полученные фракции очищали вторичной
перегонкой. Опыты показали, что при 150—230° и отношениях
хлор : метан=1 : 3 и метан : окислы азота=4 : 1 получаются нит-
рометан, нитроформ, хлорнитрометан и хлортринитрометан с
общим выходом 77—93% по отношению к окислам азота,
вошедшим в реакцию. При 230°, кроме перечисленных продуктов,
образуются также хлороформ, метиловый спирт и муравьиная
кислота.
Полученный нитрометан имел т. пл. —29°. Взаимодействие
метана и хлора в этих условиях (без N,0*) не наблюдалось;
беа ждтдлиддтороб 383
также не наблюдалось нитрование метана в отсутствие хлора.
При температуре 150—230° без повышенного давления
реакция идет главным образом в сторону окисления.
На основании другой работы, проведенной в лаборатории
автора настоящей книги [60], было установлено, что при
нитровании циклогексана двуокисью азота в паровой фазе в
присутствии галоидов при низких температурах (140—200°) реакция
идет значительно быстрее, чем без галоидов. Роль галоида
заключается^ одной стороны (при малых концентрациях), в
каталитическом ускорении реакции циклогексана с N0% по
уравнению
C@Hia + 1,5 К()„ - СиНцЖ)з + 0,5 NO + 0,5 Н„О,
и, с другой стороны, в активном участии в реакции
С,Н,а + N0* + 1,5 Хз ^» С^Ж)* + НХ + Хз.
В присутствии больших концентраций галоида увеличиваются
выходы по двуокиси азота. Термодинамические расчеты
указывают на крайне малую вероятность возникновения
свободного радикала циклогексила в качестве промежуточного
продукта при реакции циклогексана с N0%. Очевидно, роль
активных центров в изучаемой цепной реакции должны выполнять
не радикалы, а сложные радикалоподобные промежуточные
комплексы [61]. Небольшой выход нитроциклогексана
объясняется тем, что наряду с основной реакцией протекают
побочные процессы, при которых происходит разложение
нитроциклогексана (при более высоких температурах). Продукты
разложения вступают в реакцию с NO* и галоидом.
Бахман и сотрудники [62] исследовали влияние хлора,
брома и иода на нитрование пропана окислами азота и азотной
кислотой.
При нитровании пропана двуокисью азота в присутствии
брома при температуре 325°, времени контакта 2 мин. и составе
смеси QHg : NO* : О* : Вг% = 4,2 : 1,0 : 0,5 : 0,031 выход ни-
тросоединений возрастает с 41,7 до 50,2% . Такое же
возрастание выхода наблюдается и в присутствии иода (NO, : Ja =
=1 : 0,0085).
384
Концентрация галоида не должна быть чрезмерно большой,
так как в противном случае выход и конверсия уменьшаются.
С другой стороны, по литературным данным [63], для
активирования реакции фотохимического нитрования насыщенных
углеводородов рекомендуется содержание хлора в смеси,
равное 33—50%.
В лаборатории автора настоящей книги было проведено
Рис. 3. Схема нитрования углеводородов двуокисью%азот&
. паровой фазе
изучение нитрования н. гептана, изооктана (2,2,4-триметил-
пентана) и изопентана двуокисью азота в паровой фазе [64].
Реакция проводилась при разбавлении реакционной
смеси азотом, воздухом и кислородом при 300, 325, 350 и 375°
в приборе, изображенном на рис. 3. Смесь углеводорода с
двуокисью азота загружалась в капельную воронку 7, откуда она по
каплям подавалась в испаритель 2. Температура в испарителе
поддерживалась электрообогревом несколько выше температуры
кипения углеводорода. В испаритель 2 из газометра 3 подавались
азот, воздух или кислород. Смесь паров углеводорода,
двуокиси азота и газа-разбавителя поступала в реактор 4,
погруженный в расплавленную смесь нитрита натрия с нитратом
калия. Из реактора 4 реакционная смесь быстро 'выводилась в
холодильник 5, а затем в дополнительный холодильник 6,
соединенный с приемником 7. Жидкие продукты реакции
собирались в приемнике Z, а газообразные продукты через отводную
бе*
385
трубку б поступали в систему поглотительных склянок Р,
охлаждаемую смесью льда и соли. В первых двух склянках
собирались унесенные с газом жидкие продукты реакции, а в двух
последующих, заполненных водой, поглощалась не вошедшая
в реакцию двуокись азота. Газы, не вступившие в реакцию
с водой» собирались в газометре. В газах определялось
содержание двуокиси углерода и окиси азота.
Жидкие продукты реакции разделялись в делительной воронке
на углеводородный слой с растворенными в нем продуктами
реакции и на водно-кислотный слой. Углеводородный слой
промывался водой, обрабатывался 5%-вым раствором бикарбоната
натрия и вновь промывался водой. Промытые продукты реакции
высушивались над безводным сульфатом натрия и затем
отгонялись с дефлегматором не вошедшие в реакцию
углеводороды. Остатки не вошедшего в реакцию углеводорода
отгонялись под вакуумом.
Результаты опытов по нитрованию гептана и изооктанд
двуокисью азота в паровой фазе приведены в табл. 29, а при
разбавлении — в табл. 30.
Таблица 29
идоожл&дмд
Температура реакции, *С
Время контакта, сек. . . .
Взято:
углеводорода, г . . . .
двуокиси азота, г . . .
азота, л
Получено:
углеводорода, г ....
водного слоя, г
газа, л
витросоединений, г ...
Конверсия на NO@, % . .
25 А. В. Топчиев
300
3,7
170
40
13
146
10
28
20
19,8
Гептан
325
1
170
40
7
148
8
24
21
20
,5
,5
,5
350
1
170
40
10
142
7
26
21
20
,2
,5
,5
375
0,
170
40
15
143
9,
33
21
20,
7
4
5
Ивоонтав
300
6,0
170
40
10
146
12
23
22
21,5
325
5
170
40
15
144
10
27
22
21,
1
5
350
2,8
170
40
11
142
10
27
21
20,8
375
1,8
170
40
11
10
32
18
17,i
386
Таблица 30
сидсы азотов %
Температура реакции, *С .
Время контакта, сек. . . .
В зато:
углеводорода, г ....
двуокиси азота, г ...
азота, л
воздуха, л
Получено:
витросоединений, г ...
Конверсия на NOa, % . .
Гептан
325
3,9
170
40
7,5
23
20,5
325
2,5
170
40
10
23
20,5
Иэооктан
325
170
40
15
22
23,5
325
5,9
170
40
16
22
21,5
Изолентам
325
1,7
140
40
9
15
17,2
325
2,5
140
40
9
16
18,1
325
2,4
140
40
15
17,2
Полученные нитросоединения разгонялись на
ректификационной колонке в вакууме.
Из табл. 29 и 30 видно, что характер разбавителя (азот,
воздух), а также его количество при нитровании
парафиновых углеводородов в даровой фазе не оказывают
существенного влияния на выход нитросоединений. Во всех
случаях выход нитросоединений зависел только от характера
нитруемого углеводорода.
При изменении температуры реакции от 300 до 375°
конверсия двуокиси азота в нитросоединения существенным образом
не изменялась, яри условии если с увеличением температуры
реакции уменьшалось время пребывания смеси в реакторе. Во
всех случаях конверсия двуокиси азота в нитросоединения была
около 20% для гептана и изооктана и около 18% для изопен-
тана.
При нитровании гептана состав продуктов реакции с
изменением температуры от 300 до 375° существенно не
изменялся, однако при нитровании изооктана с изменением
387
температуры опыта происходило довольно заметное изменение
и фракционного состава.
При нитровании гептана основная масса продуктов реакции
перегонялась в пределах 57—63° (до 70% по весу от общего
количества образовавшихся нитросоединений) и представляла
собой мононитрогептан, в основном, вторичного характера.
Содержание в продуктах нитрования мононитрогеятанов при
изменении температуры от 300 до 375° заметно не изменялось.
Характер зтих мононитрогептанов с изменением температуры
опыта также существенно не изменялся. С повышением
температуры реакции основная масса мононитрогептанов попреж-
нему состояла из вторичных нитрогептанов (в основном из
2-нитрогептана), хотя при этом, невидимому, несколько
увеличилось количество первичного нитрогептана, так как реакция
с образованием нитроловых кислот стала более ярко
выраженной.
При нитровании изооктана основная масса (55%)
полученных нитросоединений перегонялась в пределах 60—69° при
5 мм. Повышение температуры реакции от 325 до 350° привело
к заметному уменьшению выхода третичного нитроизооктана.
Мононитросоединения, полученные при 325°, состояли на 25%
из третичного нитроизооктана, тогда как в опыте,
проведенном при 350°, содержание третичного нитроизооктана
снизилось до 20% . Основная масса нитросоединений, полученных
при нитровании изооктана в паровой фазе, состояла из
первичных нитроизооктанов (до 48 % по весу от общего 'количества
нитросоединений).
Нитрование изопентана приводит к образованию довольно
сложной смеси мононитросоединений. Наряду с ожидаемыми
2-метил-2-нитробутаном, 2-метил-З-нитробутаном и 2-метил-4-
нитробутаном образуется довольно значительное количество
(до 16%) нитросоединений с меньшим числом атомов углерода,
чем в изопентане. Нитросоединения меньшего молекулярного
веса давали реакции как на образование нитроловых кислот,
так и на образование псевдонитрола. Невидимому,
нитросоединения меньшего молекулярного веса в основном состояли из
2-нитробутана и 2-метил-2-нитропропана.
25*
388
При нитровании окислами азота в паровой фазе гептана,
изооктана и изопентана, наряду с мононитросоединениями,
образуются динитросоединения указанных углеводородов.
При нитровании гептана в паровой фазе динитросоединений
было получено около 25% от общего веса нитросоединений.
Полученные при нитровании гептана динитросоединения
обладали свойствами первичных нитросоединений.
Выход динитросоединений при нитровании изооктана был
равен 40% от общего веса полученных нитросоединений.
Динитросоединения изооктана также обладали первичным
характером.
Наконец, яри нитровании изопентана выход
динитросоединений составлял 35% от общего веса нитросоединений.
Как видно из приведенных результатов, при нитровании
углеводородов в газовой фазе выход диыитросоединений
зависит от строения нитруемого углеводорода. Выход
динитросоединений для углеводородов изостроения значительно выше,
чем для углеводородов нормального строения.
Нитрование углеводородов изостроения в паровой фазе
протекает со значительно меньшей скоростью, чем нитрование
нормальных углеводородов. При нитровании в присутствии
воздуха требуется приблизительно в полтора раза большее
время контакта, чем при нитровании в присутствии азота.
При нитровании в паровой фазе лишь около 20% двуокиси
азота идет на образование нитросоединений, остальная часть
действует, как окислитель. Окисляя углеводород, двуокись
азота в основном восстанавливается до окиси азота. При
нитровании с введением в зону реакции кислорода или воздуха
количество окиси азота в газообразных продуктах реакции
резко уменьшается. Одновременно с уменьшением количества
окиси азота в газообразных продуктах реакции увеличивается
содержание высших окислов азота, улавливаемых в
поглотительной системе.
При нитровании в паровой фазе около 50% углеводород;
расходуется на образование нитросоединений, остальная част!
углеводорода окисляется. Процессы окисления при нитрова
нии в паровой фазе идут значительно глубже, чем при нитро
de* ждгшиммдто/х)* 389
вании в жидкой фазе. При нитровании в паровой фазе
получается очень мало кислот, плохо растворимых в воде. В
продуктах окисления в основном присутствуют низшие карбоно-
вые кислоты, дикарбоновые кислоты и вода.
Наряду с этим образуется небольшое количество
карбонильных соединений. Детально продукты окисления не
исследовались.
При нитровании с введением в зону реакции кислорода или
воздуха получается несколько больше продуктов окисления,
чем при нитровании с введением в зону реакции азота. Повиди-
мому, часть кислорода расходуется на окисление углеводорода.
Нитрование парафиновых углеводородов двуокисью азота
в паровой фазе с введением в зону реакции азота или воздуха
протекает спокойно. Напротив, нитрование с введением в зону
реакции кислорода представляет довольно опасный, в
отношении образования взрывчатых смесей, процесс.
В той же работе [64] было изучено нитрование н. гептана
и изооктана (2,2,4-триметилпентана) окислами азота в жидкой
фазе. Реакция проводилась как при атмосферном, так и при
повышенном давлениях. Реакция при атмосферном давлении
проводилась в приборе, изображенном на рис. 4.
Кислород из газометра 7 подавался в склянку 2 с окислами
азота. Смесь кислорода с окислами азота из склянки 2
поступала в подогреватель J. Подогретая до температуры 250—400°
смесь окислов азота с кислородом поступала в реактор 6 через
капилляр 4. В реакторе находился кипящий углеводород. Пары
углеводорода конденсировались в холодильнике 6, и
конденсат стекал обратно в реактор. Газообразные продукты реакции
отводились в поглотительную систему Z. В склянках с водой
поглощалась двуокись азота. Непоглотившиеся водой газы
собирались в газометр. В них определялось содержание
двуокиси углерода и окиси азота. В поглотительных склянках
титрованием щелочью определялось количество не вошедпшх-
в реакцию окислов азота.
Результаты опытов по нитрованию н. гептана и изооктана
двуокисью азота при атмосферном давлении в присутствии
кислорода приведены в табл. 31.
390
н
it мзооктдкд
Таблица 31
дзота три
Температура нагрева N0%, *С . . .
Продолжительность опыта, часы . .
Б з я т о:
углеводорода, г
двуокиси азота, г
кислорода, л
Получено:
углеводорода, г
водного слоя, г
газа, л
нитросоединоний, г
двуокиси азота, г
Мононитросоодинений, %
Динитросоединений, %
Конверсия на N0%, % . ,
Выход на прореагировавшую N0%, %
Гептан
300
12
170
60
7,5
147
4
20
50
66
34
12,5
75
И8ОС
250
12
170
40
5
149
6
7
21
30
27
73
21,5
86
400
12
170
40
5
145
7
7
23
28
35
65
22,8
76
Нитрование изооктана и н. гептана при повышенном
давлении проводилось в запаянных трубках. В каждую трубку
помещалось 4 г двуокиси азота и 15 мл углеводорода.
Запаянные трубки помещались в металлические карманы, которые
затем погружались в баню с кипящей водой. Нагревание трубок
продолжалось до тех пор, пока вся двуокись азота не вступала
в реакцию. Как при нитровании н. гептана, так и при
нитровании изооктана этот момент наступал через 90 мин. после
погружения запаянных трубок в кипящую воду. По окончании
реакции трубки охлаждались в ледяной воде и вскрывались.
В газообразных продуктах реакции определялось содержание
двуокиси углерода и окиси азота. При нитровании и и. гептана
и изооктана окислы азота использовались практически на-
391
Рис. 4. Схема нитрования углеводородов двуокисью азота
в жидкой фазе
цело. Количество двуокиси углерода достигало 10% от
газообразных продуктов реакции (по объему).
Результаты опытов по нитрованию н. гептана и изооктана
двуокисью азота при повышенном давлении приведены в
табл. 32.
Из таблиц 31 и 32 видно, что выход нитросоединений,
считая на окислы азота, взятые в реакцию при
нитровании под давлением, значительно выше, чем при
нитровании без давления. Это различие в выходе можно объяснить
значительно большей концентрацией реагирующих веществ
при нитровании под давлением. Выход жо нитросоедине-
ний, считая на окислы азота, вступившие в реакцию, был
выше при нитровании без давления в присутствии
кислорода. Это можно объяснить тем, что окись азота,
образующаяся в процессе реакции, окислялась кислородом до
двуокиси азота, а не вступала в реакцию окисления
углеводорода.
Жидкие продукты реакции, полученные при нитровании
п. гептана и изооктана, переносились в делительную воронку,
392
органически*
дао/мл
одьщгоа %о
Таблица 32
Гелтая
160
64
1,15
105
20
17
60
50
50
37
66
Изоо
189
58
3,3
140
18
15
57
50
50
39
70
Взято:
углеводорода, г
двуокиси азота, г
Молярное отношение углеводород : N0% . .
Получено:
углеводорода, г
водного слоя, г
потери с газом, г
нитрососдинений, г
мононитросоединений, %
дивитросоединенпй, %
Выход нитросоединений на окислы
азота, %
Выход нитросоединений на углеводород, %
222
92
0,975
140
22
10
112
32
68
50
82
где водный слой отделялся от углеводородного слоя,
содержавшего нитросоединения и часть продуктов окисления.
Углеводородный слой затем промывался водой, 5% -ным раствором
бикарбоната натрия и вновь водой и высушивался над
безводным сульфатом натрия. Невошедший в реакцию углеводород
отгонялся с дефлегматором. Полученные после отгонки
углеводорода нитросоединения разгонялись на ректификационной
колонке в вакууме.
При нитровании н. гептана введение кислорода в зону
реакции привело к значительному уменьшению выхода динитро-
соединений. Однако введение кислорода в зону реакции при
нитровании изооктана существенно не влияло на выход ди-
нитросоединений. Выход динитросоединений во всех случаях
яри нитровании изооктана оставался высоким (около 70%).
Выход нитросоединений, считая как на окислы азота, так
без ждл&алыадпыумю
и на углеводород, вступивший в реакцию, достаточно высок
и мало отличается от выхода, который получается при
нитровании азотной кислотой.
Основная масса нитросоединений, полученных при
нитровании изооктана двуокисью азота в жидкой фазе, состояла из
динитросоединений. При разгонке продуктов реакции на
ректификационной колонке в вакууме динитросоединения
оставались в перегонной колбе и затем перегонялись с небольшим
дефлегматором. Полученный динитроизооктан представлял собой
тяжелое масло светложелтого цвета с т. кип. 117—123° при
3 мм, d*o 1,117 и %^ 1,4744. Определение азота по Дюма
дало значения, соответствующие вычисленным для динитроизо-
октана.
Полученные при нитровании изооктаиа
динитросоединения обладают свойствами первичных нитросоединений.
Около 25% по весу от общего количества нитросоединений,
полученных при нитровании изооктана, выкипало в пределах
63—69° при 5 мм рт. ст. Эта фракция нитросоединений
представляла собой масло слегка желтого цвета с d^ 0,9584 и
показателем преломления %^ 1,4360. Оно не растворялось в
щелочи. Определение для этого масла азота по Дюма дало
значения, соответствующие мононитроизооктану. Это нитросоеди-
яение может быть лишь третичным нитроизооктаном (2,2,4-три-
метил-4-нитропентаном).
При нитровании гептана двуокисью азота в жидкой фазе под
давлением около 50% нитросоединений выкипало в пределах
64—66° при 6 мм. При нитровании н. гептана без давления в
присутствии кислорода в этих пределах выкипало около 66%
нитросоединений.
Определение азота, восстановление в амин и
положительная реакция на образование псевдонитрола свидетельствуют
о том, что это нитросоединение является 2-нитрогептаном.
При нитровании гептана двуокисью азота в жидкой фазе
образуется довольно значительное количество
динитросоединений, выкипающих в пределах 85—90° при 5 мм.
Определение азота в этих соединениях дало значения, соответствующие
вычисленным для динитрогептана.
394 ffwmpogaHwg рреднычбскшс соб^инбный ожыслдлм*
Динитросоединения гептана на ^/* растворялись в растворе
щелочи. Нерастворимая в щелочи часть динитросоединений
гептана может быть лишь динитросоединением с нитрогруп-
пами у одного вторичного атома углерода. Поскольку при
нитровании углеводородов нормального строения в основном
образуются нитросоединения с нитрогруппой у второго атома
углерода, можно полагать, что нерастворимая часть
динитросоединений является 2,2-динитрогептаном. Строение
растворимых в щелочи динитросоединений не установлено.
При нитровании н. гептана и изооктана двуокисью азота
в жидкой фазе происходило образование значительного
количества продуктов окисления. В продуктах окисления гептана
были найдены двуокись углерода, вода, уксусная,
валериановая, капроновая и щавелевая кислоты, а также некоторое
количество карбонильных соединений. В продуктах окисления
изооктана обнаружены двуокись углерода, вода, триметилуксус-
ная, трет.-бутилуксусная и уксусная кислоты, дикарбоновая
кислота неустановленного строения и некоторое количество
карбонильных соединений.
Процесс нитрования циклогексана окислами азота
изучался А. И. Титовым [61]. На основании своих опытов
(взаимодействие циклогексана с двуокисью азота при температурах
от 20 до 330° в запаянных трубках или открытых сосудах, в
жидкой или газовой фазе) А. И. Титов пришел к следующим
выводам.
1. Взаимодействие циклогексана с двуокисью азота в
начальной стадии окисления и нитрования протекает
сравнительно быстро и дает те же продукты нитрования, что и при
действии азотной кислоты (по методу Коновалова — Намет-
аина):
CgHia + NO, -» C,Hu + HNO,
СбНп + NO, -> C,HuNO,
О
О
бе* ждп*длы#дтороб 395
2. Отсутствие заметного ускорения реакции между N0%
и циклогексаном при добавлении сильных протонных и апро-
тонных кислот (HgSO^, TiCl^, А1С1з) указывает на радикальный
характер первичной стадии реакции и свидетельствует о малой
роли ионного характера этой стадии.
3. При высокой концентрации димера двуокиси азота
возможно развитие достаточно длинных цепей по схеме
R + N,0, -> RONO
ж т. д.
4. Нитрование и окисление циклогексана протекают по
схеме, предложенной ранее для нитрования парафинов.
Согласно литературным данным [65], осуществлено
нитрование насыщенных углеводородов четырехокисью азота в
жидкой фазе в проточной системе.
Реакция проводится при температуре 150—200° и давлении
выше 20 атм. в аппаратуре из нержавеющей стали. Основная
часть аппаратуры реактор — стальная трубка длиной 1 м и
внутренним диаметром 4 мм, обогреваемая водой (под
давлением).
Получены следующие результаты.
1. Нитрование н. додекана:
1000 г додекана и 300 г N^O* пропускают при 190°, 40 атм.
и объемной скорости 204 объема реакционной смеси на единицу
объема реактора в час. После обработки продуктов реакции
получено 560 г нитродекаиа и 12 г жирных кислот.
2. Нитрование и. гептана:
1000 г С,Ни и 380 г Ng()а,взаимодействуя при 170° и 40 атм.
(объемная скорость 155), далн 630 г сырого нитрогептана и 8 г
лис лот.
3. Нитрование 2,2,4-триметилпентана:
1000 г CgHig и 400 г N,0, при 170° и 40 атм. дали 645 г сы-
jporo нитроизооктана.
4. Нитронание циклогексана:
396 Дит/юедние органические соеЭш*бныД ожыслдл#ы
1500 г C*Hi% и 500 N*O* при 150° и 40 атм. (время реакции
30 сек.) дали 570 г сырого нитроциклогексана. В свою очередь
продукты нитрования могут быть вновь аналогичным образом
подвергнуты нитрованию с получением ди- и полинмтропро-
дуктов.
Из опытных данных получены значения энергии активации
процесса. Они составляют для н. гептана 5850 кал/моль и для
изооктана 7500 кал/моль.
Нитрованием циклопропана Хэсс и другие [66] получили
нитроциклопропан по следующему методу: избыток
циклопропана (2396 г) и 316 г N2O4 пропускали через трубку яри 420—
455°. Время контакта было равно 2,36—2,44 сек. Из
конденсатов, собранных при 0—80° (после удаления окислов азота и
циклопропана), обработанных водой, нейтрализованных и
экстрагированных эфиром, фракционированием выделяют 45 г
витроциклопропана. Выход 15% , считая на М*О*.
Б. НИТРОВАНИЕ ОКИСЛАМИ АЗОТА В ПРИСУТСТВИИ
КАТАЛИЗАТОРОВ
В предыдущем разделе было показан^, что двуокись азота
(в мономерной и димерной формах) легко присоединяется к
непредельным алифатическим соединениям с образованием
соответствующих нмтропродуктов, но, с другой сторояы,
реагирует довольно медленно с ароматическими соединениями.
Причина такой инертности последних заключается, повиди-
мому, в их относительно большей насыщенности по сравнению
с непредельными соединениями жирного ряда. Для
повышения реакционной способности ароматических соединений к
присоединению группы N0% обычно ведут нитрование в
присутствии катализаторов, активирующих ароматическое ядро.
Например, при пропускании смеси паров бензола, толуола,
нафталина или хлорбензола с нмтрозными газами,
содержащими окислы азота, через серную кислоту (катализатор) при
повышенной температуре получаются нмтропроизводные [67].
Пинк [68] предложил при нитровании ароматических
соединений окислами азота добавлять серную кислоту, которая.
397
по его мнению, не только играет роль дегидратирующего
агента, но и активирует процесс нитрования, выделяя HNOs:
ОН
N.Oi + Н*504 Z So/ + HNOg
ONO
При применении избытка серной кислоты реакция
практически протекает слева направо, так как азотная кислота, вступая
во взаимодействие с углеводородом с образованием нитросо-
единений, постоянно удаляется из сферы реакции.Нитрозилсер-
ная кислота и избыток серной кислоты, не вступивший в
реакцию с N*0*, являются дегидратирующими агентами.
По представлению Пинка, реакция взаимодействия
ароматических углеводородов с N*0* в присутствии серной кислоты
протекает по следующему уравнению (на примере бензола):
ОН
С@Не + N,04 + Н,50,—> САМО, + SO* (^ + Н*0
0N0
Как видно из уравнения, только 50% окислов азота
используются в этом процессе для превращения углеводородов в ни-
тросоединения, остальные 50% вступают в реакцию с серной
кислотой, образуя нитрозилсерную кислоту.
Практическое применение двуокиси азота в присутствии
серной кислоты для нитрования ароматических соединений
зависит от того, насколько полно возможно регенерировать
двуокись азота, связанную в нитрозилсерной кислоте.
В одном из американских патентов [69] предложено
анодное окисление нитрозилсерной кислоты с применением
пористой диафрагмы для регенерации окислов азота в процессе
нитрования. В результате реакции образуются азотная и
серная кислоты. Выход на азотную кислоту 92%.
Окисление нитрозилсерной кислоты протекает по
уравнению
2 ONOSO@H + 0 + H,0-> HgSOi + HNO,. (1)
Отработанную витрозилсерную кислоту подвергают действию
сернистого газа по уравнению
2 ONOSO@H + 50а + 2Н,0 -> 3*1*50* + 2N0. (2)
398 ЛЪпфоедние оредническшс
Выделившуюся окись азота окисляют воздухом в двуокись
азота и применяют для нитрования. Получается серная
кислота более мысок ой концентрации, чем в отработанной
кислоте, за счет моды, вступающей м реакцию [уравнение (2)].
Нитрование бензола проводилось Пинком следующим
образом: к раствору 1,05 моля N^O* м 1,25—1,75 моля серной
кислоты прилипали из капельной воронки при постоянном
перемешивании бензол (1 моль), причем температуру реакции
поддерживали в интервале 5—15^; по окончании добавления бен-
зола температуру повышали до 40—60°. Общая продолжи-
тельность нитрования 5 час.
Варьируя температуру от 40 до 60^, количество серной
кислоты от 1,25 до 1,75 моля на 1 моль бензола и концентрацию ее
от 85 до 95% при постоянном молярном отношении М%О*:бен-
зол =1,05 : 1, Пюшпоказал, что оптимальный выход
нитробензола (94,4%) достигается при температуре 55—60°, применении
большого избытка HgSO* (1,75 моля) и концентрации ее
около 95%.
Основным фактором, способствующим получению высоких
выходом нитропроизводных при нитровании по методу Пинка,
является большой избыток серной кислоты, который не
благоприятствует побочным окислительным процессам; при этом
условии нитрование может быть осуществлено при более
высоких температурах (до 60°), что приводит к повышению
скорости этой реакции.
Для синтеза нитротолуола реакционную смесь,
полученную посредством приливания 1 моля толуола к раствору 1,05
моля N%O* в 1,6 моля 95%-ной серной кислоты, перемешивают
в течение 3,5 часов при 50—55° и по окончании реакции избыток
толуола отгоняют в вакууме (выход нитротолуола 87,5%
от теории).
При нитровании нафталина N^O*, которое проводилось
посредством добавления 1 моля нафталина к раствору 1,1 моля
NsO* в 1,5 молях серной кислоты получен выход чистого ос-ни-
тронафталина88,4% от теоретического. Пинк применил
разработанный им метод и для синтеза динитросоединений из моно-
нитропроизводных. Так, например, при медленном прилива.
399*
нии 1 моля нитробензола к раствору 1,05 моля N^O* в 2,8 моля
дымящей серной кислоты с содержанием 4,28% свободного
серного ангидрида получен динитробензол с выходом 93,4%
от теоретического.
При нитровании антрахинона окислами азота в
присутствии олеума, содержащего 5% 80s, Шааршмидт, Бальцер-
кевич, Ганте получили нитропроизводное антрахинона [41].
Баттегей [70], исследовавший реакцию нитрования
ароматических соединений N^O* в присутствии серной кислоты,
пришел к заключению о существовании известного параллелизма
между указанной выше реакцией и реакцией Фриделя —
Крафтса. Активирование ароматического ядра в первой
реакции достигается, по его представлению, присоединением
HgSOaK ароматическому ядру с образованием неустойчивого
комплекса, аналогичного комплексу с А1С1з, который, как
полагают, является промежуточным продуктом реакции Фриделя —
Крафтса. Для доказательства этого параллелизма Баттегей
хлорировал (и бронировал) бензол в присутствии относительно
небольших количеств 90%-ной серной кислоты, причем
реакция протекала так же легко, как и при применении в качестве
активаторов А1С1з и FeCls; даже при низких температурах
хлорирование в присутствии серной кислоты приводит к
количественным выходам хлоропроизводных (при температурах ниже
0° основным продуктом реакции является С@Н@С1б). В отличие
от Пинка (см. выше), который добавлял углеводород к раствору
N*O* в серной кислоте, Баттегей приливал жидкую N*O* к
нитруемому соединению или же насыщал углеводород N0 а в
присутствии концентрированной серной кислоты. Поглощение
окислов азота происходило быстро, сопровождаясь некоторым
повышением температуры. Температура реакции
поддерживалась в пределах 15—20° посредством наружного охлаждения.
Для нитрования ароматических соединений окислами азота
Баттегей применял предпочтительно жидкую N*O*, которую
приливали из сосуда, охлаждаемого льдом, в реакционный
аппарат, где находилась смесь нитруемого соединения с серной
кислотой. Во время нитрования реакционную смесь хорошо
перемешивали.
400
Опыты Баттегея привели к выводу, что оптимальная
концентрация применяемой серной кислоты зависит от природы
нитруемого соединения, например, для бензола, толуола и
ксилолов она составляет 78—80%, при нитровании же нитро-
соединений для превращения их в полинитропроизводные
требуется концентрация кислоты около 100%.
Показано также, что конечная концентрация серной
кислоты не должна быть ниже определенной величины (например,
для бензола 61%). Так как во время реакции происходит раз-
банление серной кислоты вследствие образования нитрозил-
серной кислоты и воды, необходимо применять такое
количество серной кислоты, чтобы концентрация ее во время
нитрования всегда оставалась больше некоторой предельной
величины, ниже которой происходит неполное поглощение
окислов азота.
В табл. 33 приведены результаты опытов Баттегея для ряда
органических соединений.
Таблица 33
дрол*длгическ%# соединений #еуо*иеья> даопгд а
Нитруемое соединение
Нзято для реакции
нитруемого
вещества
H,SO,
Концентрация
Выход нитро-
продунтов ва
взятую в
реакцию NgO*.
Бензол . . .
Толуол. . .
о-Ксилол . .
м-Ксилол . .
и-Ксилол . .
Хлорбензол
Нитробензол
Антрахинон
50
50
50
50
50
150
61
20
50
50
44
44
44
52
47
9,5
350
609
400
600
400
370
250
100
80,2
77,6
78,8
77,6
78,8
84,5
100,0
Олеум
(5% SO,)
95
94
77
89
95
95
94
84,5
Прв нитровании ароматических соединений N*O* в
присутствии серной кислоты Баттегей обратил внимание на окраску
отработанных кислот, которая очень устойчива. Интенсивность
401
этой окраски зависит от концентрации кислоты; например,
отработанная кислота, остающаяся после нитрования бензола,
имеет оранжево-красную окраску в присутствии 73—74%
HgSOa и тёмнокрасную—яри концентрации серной кислоты 82%
и выше. Если отработанную кислоту после нитрования бензола
N*O* промыть бензолом и, таким образом, освободить ее от
следов нитробензола и затем остатки бензола отогнать в
вакууме, то получается окрашенный продукт, который
обесцвечивается при добавлении холодной воды; при этом выделяется
бензол наряду с продуктами разложения нитрозилсерной
кислоты. Обесцвечивание происходит также при добавлении
избытка азотной кислоты с выделением нитробензола.
По представлению Баттегея, в отработанных кислотах
содержится окрашенный комплекс, состоящий из трех
компонентов: CgHg, HNSO5 и H2SO4, для образования которого
необходима определенная концентрация, серной кислоты (например,
в случае бензола не ниже 74%). Механизм образования
комплекса состоит в том, что серная кислота своими остаточными
валентностями присоединяется к бензолу, повышая степень
активности бензольного ядра, вследствие чего последнее
приобретает способность к присоединению молекулы
нитрозилсерной кислоты:
№50, . . , уС,Н,] +
Исходя из способности H2SO4 активировать бензольное ядро,
что показано на примере образования окрашенных комплексов
в отработанных кислотах, а также из структурной аналогии
между нитрозилсерной кислотой и N^O**, Баттегей допускает
следующий механизм реакции яри взаимодействии N^O* с
ароматическими соединениями: образующиеся в первой стадии
молекулярные комплексы, состоящие из нитруемого
углеводорода! NgO* и HgSOa, быстро превращаются в динитросоеди-
* По Баттегею, нитрующим агентом при нитровании ароматических
соединений NaO* является форма двуокиси NOgONO («нитрозилнитрат»).
26 А. в. Топчиев
402 #итроед;б%е оргдмическшс coe^wtgftzwl ожислдлм*
нения или в нитроаитриты, которые, будучи весьма
неустойчивыми производными дигидробензола, распадаются далее с
образованием соответствующего нитропроизводного и нитро-
зилсерной кислоты:
СоЩ + N,0* + Н%80* -^ СбНб.. . N,0*. . . H*SO&;
N0%
H.SO* + С$Н@ (^ -4. CaH#NO% + HNSO# 4- Н,0.
0N0
А. И. Титов и А. Н. Барышникова [71] полагают, что
нитрование ароматических соединений двуокисью азота в
присутствии HsSO* протекает по следующей схеме:
RH + N,0.-> RNO, + HNO%; (1)
^:0i + HgSO^ Z HNOa + HKSO,; (2)
2HNS06 + H,0 ^ N%0* + 2H%S0i; (3)
2HN0, + N.O, ^ 2^0^ 4- H^O; (4)
H:SO*.gHgO + Щ0 ^ H,S04(g + 1) H,0. (3)
Общая схема:
RH + \,0* -! /%HgS04 mHgO -> RNO% + ONOSO,H +
+ (n —l)UaS0&.(m + i)H%0. (6)
Нитрование идет за счет азотной кислоты, образующейся при
взаимодействии двуокиси азота с серной кислотой. Главную
роль в процессе активирования реакции играет огромная
способность концентрированной серной кислоты передавать
протон бензольному кольцу и активировать его. Выдвигая такой
механизм реакции, указанные выше авторы исходят из
представления, что нитрование ароматических соединений связано
с насыщением силового поля сорной кислоты элементами
азотистой кислоты и воды. Для достижения наиболее полного
использования NsO* отношение между количествами свободной серной
кислоты и воды Цл—!)/(%&+!) по уравнению (6)] к концу
нитрования не должно быть ниже некоторого минимального
значения, характерного для каждого соединения (как показали
экспериментальные данные, для бензола это отношение равно
приблизительно 4:1, для хлорбензола 5:1, для толуола 1,8 : 1).
Степень использования серной кислоты, как это следует из
уравнений (2) — (6), может быть повышена при увеличении
концентрации NOg или удалении из сферы реакции N*0:*.
403
Нитрование ароматических соединений NsO* в присутствии
серной кислоты осуществлялось А. И. Титовым и А. Н.
Барышниковой посредством добавления раствора N2O4 в серной
кислоте к углеводороду, взятому в избытке, что давало
возможность автоматически поддерживать необходимую
концентрацию серной кислоты без применения большого избытка
последней. Нитрование бензола N^O* проводили следующим образом:
к 29,2 г углеводорода приливали при перемешивании
пропеллерной мешалкой раствор 13,8 г N^O* в39,2г94%-нойсервойкис-
лоты при температуре около 40° в течение 20 мин. По
добавлении всего количества N^O* нагревание продолжали еще в
течение 1 часа при 50°, после чего нитробензольяый слой сливали
и фракционировали. Оптимальные результаты (выход
нитробензола 98,4% от теории) получаются при температуре
нитрования 40—50° и концентрации серной кислоты к концу
нитрования 80,6%. При повышении остаточной концентрации H^SO*
до 89,5% выход нитробензола понижается вследствие усиления
побочных реакций.
Скорость нитрования хлорбензола раствором N*O* в серной
кислоте значительно меньше по сравнению со скоростью
нитрования бензола особенно при низких температурах; при 0°,
продолжительности процесса 5 час. и остаточной
концентрации серной кислоты 69% выход нитрохлорбензола составляет
72% (при этих же условиях бензол дает выход нитробензола
90%). Опыты показали, что для повышения скорости
нитрования хлорбензола необходимо после проведения первой фазы
реакции с раствором N^O* в 95%-ной серной кислоте
постепенно добавлять концентрированную серную кислоту, чтобы
к концу нитрования концентрация свободной HgSO* была не
ниже 82—84% (общий расход серной кислоты составляет 2,5
моля на 1 моль нитропродукта). При этом условии выход нитрс-
хлорбензола может быть повышен до 98—99% при
проведении реакции в температурном интервале 40—50*.
Нитрование толуола раствором N^O* в серной кислоте
протекает очень энергично даже при 10° и невысоких
концентрациях серной кислоты. Хорошие результаты получаются при
нитровании толуола 36,3%-ным раствором N^O* в 94%-ной
26*
404 #%гуфодан%е (фганичесяш: соединений ожмсдал*ц
серной кислоте при 0—15° и остаточной концентрации серной
кислоты 64,5%; при этих условиях выход чистого нитропродук-
та составляет 98% от теоретического.
При нитровании толуола, кроме нитротолуолов, обнаружен
еще один продукт реакции, который, при исследовании, ока-
зался 2,6-двнитрокрезолом. Образование последнего А. И.
Титов объясняет следующими реакциями:
сн, сн, сн, сн,
+HNO, /\ /^
N = NOH ОН
В подтверждение этого механизма А. И. Титов ссылается на
имеющиеся в литературе данные [72, 73] о получении диазопро-
изводных при действии азотистой кислоты на ароматические
соединения; кроме того, установлено, что при нитровании
толуола в больших количествах происходит образование диазо-
соединений, которые даже удалось выделить из реакционной
смеси.
А. И. Титов [74] также показал, что при нитровании
раствором N*O* в серной кислоте мононитропроизводных
ароматических соединений они могут быть превращены в
соответствующие полинитросоединения. Например, к 12,3 г нитробензола
при интенсивном перемешивании в течение 3 час. при 5—7°
приливали из капельной воронки раствор 10,1 г N2O4 в 25 г
45%-ного олеума. По окончании добавления всего количества
нитрующей смеси реакционную массу, после выдержки в
течение 1 часа при той же температуре, подвергали нагреванию,
причем температуру постепенно повышали до 100° в
продолжение 1 часа. После этого температуру снижали до 40° и к
реакционной смеси приливали 100 мл воды. Не охлаждая,
переводили содержимое реакционной колбы в стакан и
отфильтровывали динитробензол после его затвердевания. Сырой динитро-
бензол очищали обработкой водой и затем слабым раствором
соды при температуре около 80°. Оптимальные результаты (вы-
405
ход динитробензола 97—98%) получены при температуре
реакции 5—15° и соотношении реагентов 0,14—0,15 моля Н*$О*
и 0,11 моля N*O* на ОД моля СбН*МО*.
При нитровании нитротолуолов, которое производилось
аналогично нитрованию нитробензола, оптимальные
результаты (выход 2,4-Динитротолуола 98% от теории) достигаются
при применении олеума из расчета 1 моль серного ангидрида на
1 моль N2O4. При нитровании 2,4-динятротолуола получен
85%-ный выход 2,4,6-тринитротолуола. Опыты по нитрованию
нитрохлорбензолов показали, что для получения оптимальных
результатов (выход динитропродукта 98,5% от теории)
необходимо применять некоторый избыток N2O4, равный около
10%.
Шааршмидт [75] для активирования бензольного ядра при
нитровании ароматических соединений N2O4 применял в
качестве катализатора хлориды металлов AlClg и FeCI*.
Например, к смеси 3 молей углеводорода и */,- моля AlClg добавляли
жидкую N*O* при охлаждении и сильном перемешивании или
же насыщали эту смесь газом, содержащим NO& при
обыкновенной температуре. При этом, после постепенного
растворения AlClg, получался интенсивно окрашенный в красный цвет
раствор комплекса, состоящего из AlClg, С^Н* и М%Од в избытке
углеводорода. В отличие от непредельных соединений,
которые образуют весьма неустойчивые продукты присоединения
(см. выше), продукт взаимодействия N*O* с ароматическим угле-
водородом состоит в основном из весьма устойчивого нитронит-
рита. При нагревании этого продукта в бензольном растворе
на водяной бане не наблюдается никаких заметных признаков
разложения. При дальнейшем исследовании Шааршмидт
нашел, что комплекс, состоящий из хлористого алюминия,
углеводорода и N*O*, при действии воды разлагается очень легко
уже на холоду с образованием небольшого количества хлор-
замещенного нитруемого углеводорода (например,
хлорбензола). Образование последнего Шааршмидт объясняет
следующим образом: вода разлагает незначительное количество
хлористого алюминия, что вызывает отщепление HNO%; последняя
далее гидролизует еще некоторую часть А1С1з и окисляет соля-
406
пую кислоту, являющуюся продуктом частичного гидролиза
AlClg, до хлора, который и хлорирует углеводород.
Шааршмидт на примере бензола доказал, что комплекс из
C*Hj, AlClg и N2O4 является аасыщенныл:: NgO^, хлор и бром
не реагируют с этим комплексом. Экспериментальные данные
показали, что при нитровании 1 моля бензола 1 молем NaO*
получают 1 моль нитробензола, если применяют не менее */*
моля AlClg. При нитровании хлорбензола (1 моль) также
можно получить выход нитрохлорбензола, близкий к
теоретическому, если молярное отношение AlCl^ к N^O* составляет
% : 1. На основании этих результатов Шааршмидт принимает,
что комплексы, образующиеся при нитровании, имеют состав
2A1C1,"3C,H,.3N*O, для бензола и 2A1CI, ЗСбН,С1 3N,O, для
хлорбензола.
Влияние AlClg на реакцию нитрования N^O* зависит от
природы нитруемого соединения. Реакция нитрования
хлорбензола протекает при обыкновенной температуре и может
быть осуществлена и при более низких температурах. Реакция
нитрования бензола состоит из двух фаз: образование
продукта присоединения N*O* происходит при температурах ниже
10*, для разложения же комплекса, т. е. получения нитро-
соединения, необходимо умеренное нагревание выше 10°. FeClg
действует менее энергично, но дает более благоприятные
результаты. При реакции с толуолом наряду с нитрованием
наблюдаются и окислительные процессы, в результате
которых образуются нитрокрезолы (в виде эфиров азотной и
азотистой кислот).
По Шааршмидту, механизм реакции нитрования
ароматических соединений N2O4 в присутствии А1С1з состоит в
следующем: AlClg присоединяется к ароматическому соединению,
активируя бензольное ядро, которое далее присоединяет NsO*
с образованием тройного комплекса; при разложении
последнего водой AlClg отщепляется; оставшийся продукт
присоединения N2O4 к бензолу, являющийся весьма неустойчивым
производным дигидробензола, распадается на нитросоединение,
которое остается растворенным в избытке углеводорода, и
азотистую кислоту, переходящую в водную фазу; азотистая кис-
ждлмлиддлмро? 407
лота разлагается далее по уравнению 3HNO,.—HNO3+H%O
+ 2NO.
Указанный выше процесс может быть представлен в виде
следующей схемы (на примере нитрования хлорбензола):
2 AICI3 + ЗС*Н,С1 -^
(неустойчивое
производное дигидробензола)
N0,
зс А( + зномо
1_ Н,0 + 2N0
Нитрование бензола NgO^ в присутствии А1С1, проводилось
Шааршмидтом следующим образом. В суспензию 140 г
сублимированного AlCIg в 200 г бензола медленно приливали тонкой
струей раствор 92 г NaO* в 100 г бензола при температуре 10—
15°. После внесения всего количества нитрующего агента
реакционную смесь нагревали до 30—35* и медленно выливали
струей на мелкораздробленный лед. При этом получены
нитробензол (в бензольном растворе вместе с AIC1*). Бензольный
раствор отделяли от водной фазы, промывали водой и затем
разбавленным раствором NaOH и подвергали перегонке для
отделения нитробензола от бензола. Выход сырого нитробензола
(с примесью смолистых продуктов) составлял 118 г.
Галоидозамещенные производные бензола [76] дают при
нитровании окислами азота в присутствии катализаторов
или PeClg) следующие количества о-и п-изомеров (в %):
и о
С*НЛОгР 91 9
C,H*NO*Cl 78 22
95 5
95 5
При нитровании хлорбензола двуокисью азота в присутствии
концентрированной серной кислоты главным продуктом
реакции является п-нитрохлорбензол.
408
ожыслдлм*
А. И. Титов [74] при изучении реакции нитрования
ароматических соединений двуокисью азота в присутствии А1С1%
пришел к следующим выводам:
1) AlClg переходит в раствор в 3 раза меньшем количестве,
чем это требуется для образования комплекса Шааршмидта;
2) при дальнейшем прибавлении NgO* реакция протекает
менее энергично;
3) действие воды на комплекс, которое Шааршмидт считает
необходимым для образования нитросоединений, не
обязательно.
По наблюдениям А. И. Титова, при нагревании в отсутствие
воды реакция протекает по уравнению
2А1С1, + ЗИН + 3N,O* - 3RNO, + 3NOC1 + А1,С1, (ОН),.
А. И, Титов выдвигает механизм постадийного протекания
реакции яитровашая двуокисью азота в присутствии A1CI,,
дричем первая фаза, окончание которой совпадает с моментом
полного перехода хлористого алюминия в раствор,
соответствует наиболее интенсивному координационному насыщению
силового поля атома алюминия в
RH + N,O*+ 2А1С1г» RNOz-А1СЦ+ A1C1% (OH).NOCl Z RNO,AlCl% (OH) +
(1)
На следующих стадиях насыщение силового поля атомов
алюминия происходит менее энергично:
(2)
Aid*(OH)-NOCl + N2O4 + RH ^ A1CI (OH)*'RNO,.2NOC1. (3)
При нагревании комплекс диссоциирует на компоненты.
Механизм активирующего действия хлористого алюминия А. И.
Титов представляет следующим образом. Алюминий, благодаря
своей резко выраженной координационной ненасыщеяяости
в галоидных солях, производит глубокое поляризующее
действие на молекулы, способные подвергаться деформации, и
может даже вызвать их ионизацию
А1С1, + N,O*-> CI3AI - NOz — NO
oeawwe g
409
(0.NA1C1,)
В образовавшемся соединении водород, связанный с тем же
углеродом, что и нитрогруппа, обладает большой
подвижностью и легко отщепляется в виде иона, что и приводит к
образованию нитробензола.
Хорошие результаты получены при нитровании
хлорбензола, бензола и толуола М*Од в присутствии AlClg.
Смесь 55 г хлорбензола (0,35 моля) и 10,7 г AICI, (0,08 моля)
при постоянном встряхивании насыщали двуокисью азота, что
сопровождалось энергичной экзотермической реакцией. После
полного растворения AlClg реакция протекала с меньшей
интенсивностью. Насыщение окислами азота продолжали до
поглощения 10,5 г М*Од (0,114 моля), что соответствует 95%
теоретического количества. Реакционную смесь, превратившуюся
в студнеобразную массу, постепенно нагревали до кипения,
одновременно пропуская через нее СО*. После кипячения
отделяли жидкую фазу от осадка основных солей алюминия и
подвергали ее фракционированной разгонке, причем было
выделено 17,2 г смеси нитрохлорбензолов, что соответствует 96%
от теории. Аналогичные результаты получены при нитровании
бензола.
Нитрование толуола, которое в опытах Шааршмидта
проходило неудовлетворительно и с образованием продуктов
окисления, А. И, Титов осуществил с выходом нитротолуолов 88%
от теории, изменив порядок смешивания компонентов:
AlClg прибавляли к смеси МгО^и толуола, что уменьшало
вероятность возникновения побочных реакций с азотистой
кислотой.
М. И. Богданов [77], исследовавший реакцию нитрование
ароматических соединений N^O* в присутствии FeClg, пришел
к выводу, что одновременное пропускание хлора активирует
реакцию нитрования. По его мнению, хлор при действии на
тройной комплекс, состоящий из FeClg, C,H, и N2O4, лишает
410
хлорное железо его дополнительных валентностей и переводит
в соединение с более высоким содержанием хлора. Благодаря
этому происходит разрушение тройного комплекса с
образованием неустойчивого производного дигндробензолаСбНб'МаОа,
в дальнейшем разлагающегося на нитробензол и азотистую
кислоту. Образовавшийся из FeClg продукт с высоким
содержанием хлора также неустойчив и распадается на хлор и
хлорное железо, которое затем снова вступает в реакцию с бензолом
и N*O*.
По представлению М. И. Богданова, при нитровании
бензола в присутствии FeClg в начале реакции образуется комплекс
состава
\
FeCI, ...
Н
ONO
который в дальнейшем распадается на хлорное железо,
нитробензол и азотистую кислоту.
При; нитровании и одновременном хлорировании 53,32 г
бензола в течение 5 час. при 20° в присутствии 5,6 г железа
М. И. Богданов получил выход нитробензола на взятый бензол
35,&% от теоретического.
Дальнейшим развитием работ по нитрованию органических
соединений двуокисью азота в присутствии катализаторов были
работы А. В. Топчиева, который использовал в качестве
катализатора трехфтористый бор.
Необходимая для опытов чистая N^O* получалась
взаимодействием нитрозилсерной кислоты с азотнокислым калием.
Кристаллическую нитрозилсерную кислоту, синтез которой
осуществлялся насыщением 80% раствора крепкой азотной кислоты
в дихлорэтане при 25—30°, перемешивали с порошком нитрата
калия в фарфоровой чашке и смесь быстро переносили в широ-
когорлую круглодонную колбу, соединенную с конденсацион-
но-поглотительной системой, состоявшей из холодильника Ли-
g /gpuci/mcmeww ждтдлмадторм 411
биха и двух широких U-образных трубок, помещенных в
охладительную смесь. Колбу подвергали нагреванию ва
водяной бане до 50—60°, причем происходило образование N2O4
по уравнению
N0,80, Н + KNO,.» М%Од + KHSO4.
Реакция протекала равномерно и достаточно быстро и
заканчивалась обычно через 1,5—2 часа. При избытке нитрата
{2 моля на 1 моль нитрозилсерной кислоты) выход N2O4
достигает 85%.
Нитрование бензола N^O* в присутствии BFg
осуществлялось приливаннем NgO^ по каплям из бюретки в реакционную
колбу с бензолом, через который одновременно пропускали
BF,*.
Нитрование производили без наружного нагревания;
ввиду экзотермичности реакции температуру в начале нитрования
повышали до 55—60° и затем поддерживали в этих пределах
в течение 1,5—2 час.
Через 10—15 мин. после начала опыта реакционная масса
приобретала темножелтый цвет, который через некоторое время
переходил в темнобурый, причем на стенках колбы выделялся
осадок.
По окончании опыта непрореагировавшие N^O* и бензол
отговяли на водяной бане. Остаток перегоняли с водяным
паром и перешедший в дестиллят нитробензол экстрагировали
эфиром, из которого он выделен фракционированной
перегонкой (после отгонки растворителя).
При отношении реагентов 0,5 моля N2O4 на 1 моль бензола
получено 22 г нитробензола, что соответствует 18% от
теоретического выхода на взятый бензол.
Мак-Ки и Вильгельм [78] при нитровании ароматических
соединений двуокисью азота применяли в качестве
катализатора силикагель. Нитрование бензола проводилось
следующим образом. Окислы азота, полученные при взаимодействии
Og с HNOg, проходили через реометр, а из последнего
* Метод получения фтористого бора описан ниже в гл. VI.
412
в испаритель, где происходило испарение бензола, поступавшего
с определенной скоростью из сборника. Образовавшуюся в
испарителе смесь паров бензола и двуокиси азота направляли в
реакционную камеру (стеклянную трубку со слоем
катализатора длиной 110 см), которую нагревали в длинной
электропечи. Жидкие продукты реакции и непрореагировавший
бензол собирали в приемнике, соединенном с обратным
холодильником и охлаждаемом льдом, а окислы азота, образовавшиеся
во время реакции, вместе с избыточной двуокисью азота
проходили через поглотительную систему с водой, далее через
растворы окислителей, где окись азота N0 окислялась до NO2,
которая затем улавливалась 10%-ным раствором NaOH.
Опыты, в которых варьировались объемная скорость,
отношения реагентов и температура реакции, показали, что
конверсия бензола в нитробензол возрастает с повышением
температуры (но не выше 310°), уменьшением объемной скорости и
увеличением отношения NO2 к бензолу. При повышении
температуры выше 310° конверсия уменьшается вследствие
усиления побочных реакций окисления, которые становятся
особенно заметными при 380°. Падение выходов при температурах
выше 310° объясняется также понижением активности
катализатора.
Найдено, что при 310° и скорости поступления бензола
0,5 мл/мин конверсия в нитробензол составляет 30,1% (на
взятый в реакцию бензол), если в течение 1 часа пропускать чере&
реакционную камеру 31,1 г NO» и 26,4 г С@Не (молярное
отношение двуокиси азота к бензолу 2 : 1). При увеличении
молярного отношения NO* : С@Нб до 7 : 1 (54,5 г NO» на 13,2 г
бензола) конверсия при той же температуре достигает 83,6%.
Интересно отметить, что при действии окислов азота на
бензол, наряду с нитробензолом, образуется также нитрофенол,
который при температурах выше 330° представляет собой
преобладающий продукт реакции. Исходя из данных анализа
жидких и газообразных продуктов, получающихся при
нитровании бензола двуокисью азота, Мак-Ки и Вильгельм
предполагают, что эта реакция протекает по следующему уравнению:
б + 3NO, -> 2C#H,NO, + NO + Н,О.
413
По мнению этих исследователей, действие силикагеля
объясняется отчасти его дегидратирующими свойствами (как
известно, силикагель успешно применяют в качестве
катализатора при синтезах сложных эфиров, алкилировании в паровой
фазе аммиака и анилина действием спиртов и других реакциях,
при которых, как и в данном случае, происходит выделение
воды).Несомненно, однако, что силикагель обладает также
специфичностью действия при реакции нитрования N0%
ароматических соединений, так как другие дегидратирующие
катализаторы, как, например, окись магния и другие, не оказывают
на эту реакцию никакого каталитического действия.
Опыты по нитрованию толуола дали, сравнительно с
бензолом, менее благоприятные результаты. При пропускании
через каталитическую камеру при 275° в течение 1 часа 31 г
NO2 и 31 г толуола, что соответствует молярному отношению
первой ко второму 2:1, Мак-Ки и Вильгельм получили
нитротолуол с выходом всего 6,8% от теоретического.
Леви [79] разработал метод нитрования парафинов сухой
NO2 в присутствии катализатора, содержащего соединения
мышьяка или сурьмы. Например, смесь 30% изобутана и 70%
Путана пропускали в количестве 5 л в течение 1 часа через
смесительную камеру, в которую поступала N0% со скоростью
10 л/час. Смесь реагентов проходила через сушильную колонку
о PgOg в конвертор, в котором находился катализатор,
состоявший из мышьяковистокислого натрия, смешанного со стеклом.
Нитрование проводили при 200° и времени контакта 120 сек.
После удаления избыточной NO» выделен продукт, который
состоял из смеси триметилнитрометана (CHg),CNOa (60%),
2-нитробутана (20%), 2,3-динитробутана (20%).
Общий выход нитросоединений составлял 45% на
пропущенный или 90% на вошедший в реакцию бутан. По этому же
способу Леви нитровал нормальный бутан (без примеси
изобутана) и пропан.
При пропускании метана а окислов азота (NgO^) через
трубку из стекла яирекс при температуре 440—680° в
присутствии различных катализаторов (платина, пятиокись ванадия,
пемза и никель) Фрелих [80] получил формальдегид с выходом
414
25%, считая на использоваяяый метан. Среди продуктов
реакции метанол не обнаружен.
Согласно американскому патенту [81], смесь равных
объемов пропана и N^O* пропускали при температуре 270° в
течение 80 сек. (время контакта) через катализатор, состоящий
из А1 с 1,7% Си и 1,3% Fe. Получаются нитропарафияы и
продукты окисления.
Другой американский патент [82] указывает, что при
нитровании ароматических углеводородов окислами азота при
температуре 130—430* в присутствии таких катализаторов, как
метафосфат металла, фосфорная кислота, фтористый бор,
получаются мононитропроизводные ароматических
углеводородов.
Следует отметить, что в отличие от двуокиси азота ни
закись, ни окись азота не реагируют с ароматическими
углеводородами.
ЛИТЕРАТУРА
1. К. Clusias, M. Veechi. Helv. Cbim. Acts, 36, 930 (1953).
2. A. D. Walsh. Fuel, 33, 243 (1954).
3. А. А. Баландив, Я. Т. Э и д у с, Н. Заломив. Журн_
физ. химии, 6, 389 (1935).
4. Т. В. Заболоцкий. Журн. общ. химии, 20, 1992 (1950).
5. F. Blacet. Ind. Eng. Chem., 44, 1341 (1952).
6. H. Wieland. Ann., 328, 154 (1903); 329, 225 (1903); 340, 63
(1903); 360, 299 (1908); 424, 71 (1921).
Т. H. Wioland, H. S teazel. Ber., 40, 4823 (1907).
8. J. Schmidt. Ber., 36, 1773 (1903).
9. Н.Я. Демьянов, К. В. Сидоренко. ЖРХО, 4i, 832
(1900).
10. Н. Я. Демьянов. ЖРХО, 36, 13 (1904); Анило-красочн. пром.,
4, 132 (1934).
11. A. Michael, G. H. Carlson. J.Org. Cbem., 5, 1—23 (1940).
12. A. Michael, G. H. Carlson. J. Am. Chem. Soc, 59, 843
(1937).
13. A. Sen a arse hm id t, H о f f m ei e r. Ber., 58, 1047 (1937).
14. A. S rh aarschmid t. Z. angew. Chem., 36, 335, 565 (1923).
15. A. S m i t h. Амер. патент 2384087; С. А., 40, 347 (1046).
16. II. В a] dock, A. Smith. Ind. Eng. Chem., 44, 2041 (1952).
17. T. Doumanl. Амер. патент 2621203; С. А., 47, 10532b (1953).
415
18. N. Levy, С W. Scaif e, A. E. Wilder. J. Chem. Soc., 1093
(1946).
19. N. Levy, С W. 5c aif e, Л. E. W ild er. J. Chem. Soc, 52
(1948).
20. H. В a Id о ck, X. Levy, C. W. Scaife. J. Chem. Soc,
2627 (1949).
21. G, Darzens. Compt. rend., 229, 1148 (1949).
22. S. M in га. С. А., 48, 1412d (1954); C. A., 2208d (1950).
23. H. Bill %. Ber., 35, 1528 (1902); Bur roe, W unt or. J. Chem.
Scx\, 134, 1357 (1932).
24. Патент СССР 66299 (1946).
25. Frank I in, Wi Ik ins. Англ. патент 532686 (1940).
26. P. W. N eb e r, S. Paeschke. Ber., 59, 2440 (1926).
27. C. F. Allen, C. G. Eliot, A. Bell. С. А., 33, 6284
(1939).
28. Hasenbach. J. prakt. Cbom., 4, 17 (1871).
29. И. Г у б е н. Методы органической химии, т. IV, кн. 1. М., Госхимид-
дат, 1949, стр. 303.
Ж Л. Leeds. Her., 13, 1093 (1880); И, 482 (1881).
31. Fried bnrg, M and el. J. Am. Chem. Son., 12, 7 (1890).
32. С Liebermann, L. Lindenmann. Ber., 13, 1584 (1880).
33. I. Meisenheimer. Ann., 330, 149 (1904); В a rn e t I.
J. Chem. Soc, 127, 2040 (1925).
34. L. W. Bass, M. B. Johnson. J. Am. Chem. Soc, 46, 456
(1924); M. А. Ильинский, Журн. общ. химии, 5, 469 (1928).
35. В а г n e 11. J. Chem. Soc, 127, 2040 (1925).
36. Н. Wi el an d. Ber., 54, 1776 (1921).
37. O. Witt. C, II, 226 (1880).
38. H. Wiel and. Bor., 53, 210 (1920).
39. M. Battegay, Kern. Bull. See. Cbim., 43, 114 (1928).
40. A. Schaarschmidt, E. S m о I I a. Ber., 57, 32 (1924).
41. A. Schaarschmidt, H. Balzerkewicr, J. Gante.
Ber., 58, 499 (1925).
42. П. П. Ш о р и г и в, А. В. Топчиев. Журя. общ. химии,
5, 549 (1935).
43. J. Schmidt, E. Heinle. Ber., 44, 1488(1911).
44. I. J. R inke s. Rec. trav. chim., 46, 506 (4937).
45. V arm a, Kr'isb am ur ty. J.Indian Qiem. Soc, 3, 323 (1926).
46. L. Homer, F. Hubenett. Ann., 579, 193 (i953).
47. A. Kaufmann, H. Hussy. Ber., 44, 1736 (1908).
48. H. А В а л я ш к о, В. И. Б л и з н ю к о в, А. Е. Л у ц к и м.
Тр. Харьковского технологич, ин-та, вьш. 4, 48 (1944).
41*. М. Н. Коновалов. ЖРХО, 25, 514 (1893).
50. Fr. Fried I. Ber., 45, 428(1912).
416 #%троедн%е (федничбсжыа; coe^wHgftww окисла л*и
5J.H. П. Шорыгин, А. В. Топчиев. Бег., 69, 1874(1936).
52. А. И. Титов. Журн. общ. химии, 18, 465, 473 (1948).
53. М. U г b a a s к у et al. Compt. rena., 203, 620 (1936).
54. М. U f bansky et al. Compt. rend., 204, 870 (1937).
55. H. B. H a ss, J. D orsk y, E. B. Hodge. Ind. Eng. Chem.,
33, 1138 (1941).
56. А. И. Титов. Журя. общ. химии, 19, № 8, 1471 (1949).
.57. Амер. патент 2597027(1952); С. А., 47, 1726f (1953).
58. П. П. Шорыгин, А. Б. Топчиев. Вег., 67, 1362 (1934).
59. А. В. Топчиев, В. П. Алания. Докл. АН СССР, 67,
297 (1949).
60. Н. Капцов. Диссертация. Москва, 1949.
61. А. И. Т и т о в, М. К. Матвеева. Журн. общ. химии, № 2,
238 (1953).
62. G. Bachmann, L. К о h n. J. Org. Chem., 17, 942—54 (1952);
G. Bachmann, J. H e w e t t, A. M i 1 1 i k a n. J. Org.Chem.,
17, 935 (1952).
63. H. Wegofer. Герм, патент 841287 (1952); С, I, 7570 (1952).
"64. Л. К. Мухин. Диссертация. Москва. 1954.
€5. G. G e i s е 1 е г. Angew. Chem., 67, № 9/10, 270 (1955).
66. Н. В. Н a ss et el. J. Am. Chem. Soc, 75, № 6, 1382 (1953).
67. Фокин. Патент СССР, 9294 (1929).
68. L. A. Pine k. J. Am. Chem. Soc, 49, 2536 (1927).
69. Амер. патент 1640737 (1927).
70. M. Battegay. Bull. Soc. Chim., 43, 109 (1928).
71. А. И. Титов, А. Н. Барышникова. Журн. общ. химии,
6, 1800 (1936).
72.. В. М. Родионов, В. К. Матвеев. Вег., 57, 1714 (1924).
73. W e s e ] s k у. Вег., 8, 98 (1875).
74. А. И. Титов. Журн. общ. химия, 7, 667 (1937).
75. A. S ch a ar schmidt. Вег., 57, 2065(1924).
76. I. Riebsomer. Chem. Rev., 36, 178 (1945).
77. М. И. Богдане в. Аяило-крагочн. пром., 4, 133 (1933).
78. R. H. McKee, R. H. Wilhelm. Ind. Eng. Chem., 28, 662
(1936).
79. N. Levy. Амер. патент 2382241 (1945).
80. F г б I i с h. J. Am. Chem. Soc, 50, 3216 (1928).
81. Амер. патент 2464572 (1949); С. А., 43, 4285e (1949).
82. Амер. патент 222994 (1947); С. А., 1605 (1948).
НИТРОВАНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
ОРГАНИЧЕСКИМИ И НЕОРГАНИЧЕСКИМИ
НИТРАТАМИ И ХЛОРИСТЫМ НИТРИЛОМ
J.
О применении органических нитратов в качестве нитрую-
щих агентов в литературе имеется сравнительно мало данных.
Наибольшее число работ но применению органических
нитратов для нитрования относится к использованию этилнитрата,
бензоилнитрата и ацетилнитрата.
Этилнитрат иля азотноэтиловый эфир C2H5ONO2 представ-
ляет собой жидкость уд. в. 1,105 (при 20°), растворимую в
воде, спирте и эфире. Температура кипения этилнитрата 87,5°.
Этилнитрат получается из азотной кислоты и этилового спирта
в присутствии азотнокислой мочевины. При перегреве
этилнитрат разлагается со взрывом.
Подобно многим эфирам органических кислот, этилнитрат
конденсируется в присутствии натрия или этилата натрия с
соединениями, содержащими активную метиленовую группу.
При этом образуется натриевая соль изонитросоединения.
Например [1]:
+ CAONO, + СН,О\а -> С@Н#С = NOOXa + 2С,Н*ОН
С = NOONa ^ QH, СН = NOOH ^ Сб,%*
СХ СООХа фенилиаонитрометан фенилиитрометан
27 А. В. Топчиев
418
При действии этилнитрата на циклонентадиен Тиле
получил нитроциклопентадиен.В25 г абсолютного спирта
растворяют 2,3 г натрия и к раствору прибавляют 1,1 г
этилнитрата. По охлаждении до комнатной температуры в смесь
вносят 6,6 г циклопентадиена и образовавшуюся прозрачную
бурую жидкость оставляют стоять несколько часов.
Выделившуюся в виде мелких красновато-бурых пластинок натриевую
соль отсасывают и промывают эфиром. Свободный нитроцикло-
пентадиен выделяется при подкислении из не слишком
разбавленного раствора соли в виде почти бесцветных быстро
затвердевающих капелек масла. Вещество, однако, очень легко
разлагается.
Пиррол и его производные [3], а также индол при
нитровании этилнитратом дают соли соответствующих ни^роловых
кислот, например:
СН СИ с,н,охо, НС С = ХООХа
СИ СН ** НС СН
натриевая соль лиррол-
&-нитроловой кислоты
пиррол
При обработке бензотиазола этилнитратом в
концентрированной серной кислоте при 0—5° получается 6-нитробензотиа-
зол в виде желтых игл с т. пл. 173—174°. 2-Аминобензотиазол
при обработке в концентрированной серной кислоте
этилнитратом дает 2-амино-6-нитробензотиазол [4].
Мононитротолуолы можно получить нитрованием
этилнитратом [5] по следующему методу: к перемешиваемой смеси
толуола и этилнитрата при температуре 40° постепенно
приливают 98%-ную серную кислоту. Температура реакции
поддерживается не выше 60°, после чего прибавляют еще этилнитрат
и перемешивают 10 мин. После охлаждения разбавляют водой,
отделяют водный слой, верхний слой промывают водой,
растворяют в растворе углекислого натрия и перегоняют. Получают
мононитротолуолы с выходом 94,5% от теории. При
фракционированном разделении можно выделить п-нитротолуол. В при-
419
сутствии хлористого алюминия этилнитрат энергично
реагирует с бензолом с образованием нитробензола (подробнее см.
гл. I).
Бензоилнитрат представляет собой смешанный ангидрид
бензойной и азотной кислот. Бензоилнитрат получается по
методу Френсиса действием хлористого бензоила на
азотнокислое серебро при температуре 15°:
САС0С1 + AgONO, -+ AgCl + С
Полученный этим методом бензоилнитрат всегда содержит
примесь 15—20% бензойного альдегида и представляет собой
маслянистую жидкость уд. в. 1,3° (при 0°). При действии воды
бензоилнитрат легко разлагается на бензойную и азотную кислоты:
СоН,СООЖ), + Н,0 -» СбЩСООН 4- HNO,.
Подобно другим смешанным ангидридам, бензоилнитрат
при перегонке распадается на соответствующие простые
ангидриды:
2C,H,COO\O: -»(СЛбСОкО + \%Об -» (СоЩСО^О + \%Од + О.
Исследования Френсиса [6] показали, что нитрование бен-
зоилнитратом дает хорошие результаты, если его добавлять
при охлаждении к избытку нитруемого соединения. В тех
случаях, когда реакция протекает слишком энергично, нитруемое
соединение растворяют в четыреххлористом углероде. Иногда
растворяют в этом растворителе также и бензоилнитрат, так
как при этом условии удается поддерживать достаточно
высокую температуру реакционной смеси, необходимую для
получения оптимальных выходов нитросоединений. Например, тио-
фен при проведении нитрования в растворе ССЦ дает
соответствующее мононитропроизводное с теоретическим выходом.
О нитрующей способности бензоилнитрата свидетельствует
его отношение к спиртам и ароматическим аминам. Так,
например: бензоилнитрат легко превращает этиловый спирт в
этилнитрат, а при действии на вторичные ароматические
амины дает с прекрасными выходами соответствующие нитроамины.
Бензоилнитрат энергично реагирует и с ароматическими угле-
28 А. В. Топчиев
420 ДЪтродднме нитратами и
водородами. Бензол, толуол и особенно легко м-ксилол дают
соответствующие мояонитрояроизводные. При нитровании
толуола, ксилола и мезитялена нитрогруппа вступает в ядро,
а не в боковую день. Мезитилен в растворе четыреххлористого
углерода уже при низких температурах нитруется бензодлни-
тратом в мононитромезитилен с теоретическим выходом. С
другой стороны, бромбензол и бензоилхлорид реагируют с бензо-
илнитратом с большим трудом и дают лишь следы
соответствующих п-нитропроизводных.
При действии бензоилнитрата на фенол образуется смесь
о- и п-нитрофенолов с преобладанием о-изомера. Еще легче
протекает нитрование яри взаимодействии бензоилнитрата с
эфирами фенола — анизолом и феяетолом, причем яолучаются
соответствующие о-нитропроизводные с выходами, близкими
к теоретическим. Следует отметить, что яри нитровании фенола
и его эфиров другими, обычно применяющимися нитрующими
агентами в преобладающем количестве образуются п-нитро-
яроизводпые.
В отличие от фенола нитрование бензоилнитратом ос- и р-
нафтолов приводит лишь к незначительным выходам 2,4-ди-
нитро-ос-нафтола (т. ял. 138—139") и 1,6-динитро-р-нафтола
(т. ял. 195°). Эфиры же *- и р-яафтолов в растворе четырех-
хлористого углерода легко нитруются бензоилнитратом; яри
действии последнего на этиловый эфир ос-нафтола образуется
4-нитропроизводное этого эфира; метиловый и этиловый эфиры
^-нафтола нитруются в положение 1, т. е. дают
соответствующие эфиры 1-нитро-2-яафтола. По отношению к большинству
ароматических альдегидов (бензальдегиду, анисовому
альдегиду, салициловому альдегиду и др.) бензоилнитрат действует
главным образом как окислитель; нитросоединеяия образуются
в этих реакциях лишь в ничтожных количествах. С другой
стороны, яри обработке бензоилнитратом р-иафтальдегид
количественно превращается в соответствующее нитропроизводное
(т. пл. 194—195°), а ванилин также с количественным выходом
дает 3-нитровавилин (т. пл. 178°). Кумарин нитруется
бензоилнитратом с образованием 5-нитрокумарина (т. пл. 185°).
Полученные Френсисом экспериментальные данные показы-
421
вают, таким образом, что бензоидпитрат как нитрующий
агент обладает следующими особенностями: 1) реакция
протекает при полном отсутствии воды, что особенно важно при
нитровании тиофена; 2) в некоторых случаях нитрование бензоил-
нитратом приводит, в отличие от обычно применяющихся
методов нитрования, к исключительному образованию о-нитро-
производных или к преобладанию последних (анизол, фенетол»
фенол).
Действие бензоилнитрата на амины исследовано Батлерои
[7]. С первичными аминами бензоиляитрат реагирует, давая
бензоильные производные:
CACOONO, + 2RNH, -> CACONHR + RNH.-HNO*.
При действии бензоилнитрата на м-хлоранилин образуется
м-хлоранилид бензойной кислоты (т. ял. 120°); аналогичным
образом в-хлоранилин дает в-хлоранилид бензойной кислоты
(т. вл. 190°). При обработке бензбилнитратом Вганизидина,
о-толуидинаи ксилидинов получаются соответствующие анизи-
диды, толуидиды и ксилидиды бензойной кислоты.
Вторичные амивы ври действии бензоилнитрата дают также
соответствующие бензоильные производные:
R. R,
QHsCOONOa + 2 )NH-+ C@H,CONRR'+ )NHHNO*.
R'/ R'/
В некоторых случаях, однако, бензоилнитрат реагирует со
вторичными аминами с образованием соответствующих нитро-
аминов. Так, например, N-метил-н-толуидин дает ври
действии бензоилнитрата N-метил-в-толилнитроамин (т. вл. 74,5°)
с теоретическим выходом:
Н,СХН Н3СЖО2
Вилыптеттер [8], применив метод Френсиса, получил
(о-мононитродурол из дурола.При нитровании дурола в растворе
ае
422 jifwmj9O6awwe wwmpayyta^tw % амодмютыл*
хлороформа 100%-ной азотной кислотой в присутствия сер-
вой кислоты получается, как известно, только динитропроиз-
водвое. Вильштеттер обрабатывал бензоилиитратом дурол в
растворе четыреххлористого углерода при сильном охлаждении
(экзотермическая реакция). Продукт реакции разбавляли
эфиром и промывали растворами соды и затем едкого ватра для
удаления бензойной кислоты. После отговки эфира и четырех-
хлористого углерода (в вакууме) витросоединение перегнано
с паром и затем перекристаллизовано из метилового спирта.
В результате этой обработки получев мовонитродурол в виде
бесцветных длинных призм с т. ил. 52,5*:
СН, CH,NO,
/\
_СН
С
Щ Н,
дуро 1 '-нитродурол
Исследованием строения этого продукта уставовлево, что при
действии бензоилвитрата на дурол нитрогруппа вступает ве в
ядро, а в боковую цепь.
'Интересно отметить, что полученное Вилыптеттером &>-мо-
вовитропроизводное дурола при действии смеси азотвой и сер-
вой кислот количествевно нитруется в ядро с образованием
1',3,6-тринитродурола (шестиугольные призмы с т. пл. 139°):
CH,NO,
Ацетилнитрат как нитрующий агент имеет некоторые
преимущества перед бензоилвитратом; при его примевении
выделяется ве вода или минеральная кислота, а летучая
уксусная кислота, которая может легко удаляться из
реакционной смеси:
RH + CHfOONO, -» RNO, + СЩСООН.
423
Ацетилнитрат получают из азотного ангидрида
(приготовленного перегонкой азотной кислоты с фосфорвым авгидри-
дом) и уксусного ангидрида
CHgCO. ,
)О + О(
СН3СО/ ^
Ацетилинитрат представляет собой очень гигроскопичную бес-
цветвую жидкость, дымящую ва воздухе; уд. в. 1,24 (15**).
Сивтез ацетилнитрата осуществлен Пикте и Хотинским
следующим образом [9]: азотный авгидрид, полученный
посредством перегонки концевтрировавной азотной кислоты вад
РгО@, вносили равными частями в уксусный авгидрид, причем
ов растворялся в последнем без заметного повышения
температуры реакционной смеси. Для отделения ацетилнитрата от
избытка уксусного ангидрида продукт реакции перегоняли
в вакууме при 70 мм и 22* (если перегонку производить при
обыкновенном давлении, то ацетилвитрат взаимодействует с
уксусвым авгидридом с образовавием тетрапитрометана).
При действии воды ацетилвитрат быстро разлагается на
уксусную и азотвую кислоты с выделением тепла.
По своему действию ва органические соединения ацетил-
нитрат обнаруживает большое сходство с бензоилвитратом.
Ов реагирует со спиртами с. образованием соответствующих
сложных эфиров азотвой кислоты.
По отношению к ароматическим соединениям ацетилпит-
рат — очень энергичный витрующий агент (его обычво
применяют при низких температурах и в виде сильно разбавлен-
вых растворов в четыреххлористом углероде или уксусном
ангидриде). Высокая нитрующая активность ацетилнитрата
объясвяется тем, что витрование протекает без выделения воды
и образования сильных минеральных кислот.
Исследоваво действие ацетилнитрата ва векоторые
ароматические соедивения. Бензол, толуол, бензилхлорид, бевзои-
вая кислота, фенол, анизол, ацетанилид, вафталип, хиволип
дают при нитровании соответствующие моновитросоедивения
с теоретическими или почти теоретическими выходами. При
действии ацетилвитрата на производные бензола получаются,
424
как и при нитровании бензоилнитратом (см. выше), главным
образом о-нитропроизводные. Так, например, нитрование
толуола, фенола и бензилхлорида приводит к смеси о- и п-нитро-
производных.
В табл. 34 дано содержание о- и п-нитропроизводных в
продуктах реакции при нитровании ацетилнитратом.
Таблица 34
Нитруемое
вещество
Толуол ....
Февол ....
Бензилхлорид
Ацетанилид .
Содержание
о-изомера
Содержание П-И80-
мера
%
88
52
60
100
12
48
40
0
Как видно из таблицы, ацетанилид дает в качестве продукта
реакции исключительно о-нитропроизводное. На амины аце-
тилнитрат также действует ацетилирующим образом. При
взаимодействии ацетилнитрата с анилином в растворе четыреххло-
ристого углерода при сильном охлаждении (смесью льда с
поваренной солью) получаются с теоретическим выходом
эквимолекулярные количества ацетанилида и анилин-нитрата.
6-нитро-о-толуидин получен нитрованием ацетилаитратом
ацетил-о-толуидинапо следующему методу: к смеси 66 г ацетил-
нитрата и 150 г ледяной уксусной кислоты при —10°
прибавляют 85 г ацетил-о-толуидина и оставляют на ночь. По
прибавлении раствора поваренной соли из реакционной смеси
выпадает нитропродукт (92 г), который омыляют соляной кислотой
и подвергают перегонке с водяным паром. Выход
6-нитро-о-толуидина 53%.
Тиофен реагирует с ацетилнитратом без применения
растворителей с образованием нитротиофена (т. пл. 44°); реакцию
необходимо проводить при сильном охлаждении. Антрацен с
425
ацетилнитратом также дает нитропроизводное. При действии
ацетилнитрата на хлороформ получается хлорпикрин. При
действии уксусного ангидрида и уксусной кислоты на ацетил-
нитрат получается тетранитрометан.
Следует, однако, отметить, что трудно нитрующиеся
соединения, как, например, нитробензол, хинон и пиридин, не
нитруются ацетилнитратом даже при высоких температурах.
Вместо готового ацетилнитрата можно с успехом применять смесь
концентрированной азотной кислоты с уксусным ангидридом
[10]. В. А. Измаильский и А. И. Козин [11] получили таким
путем из 1'-хлор-1-метилнафталина его 8-нитропроизводное.
Введение одного атома хлора в метильную группу %-метил-
нафталина уже настолько снижает активирующее действие ме-
тильной группы, что реакция направляется главным образом
на незамещенное второе кольцо нафталина. Нитрованием
1'-хлор-1-метилнафталина смесью уксусного ангидрида и
азотной кислоты (уд. в. 1,52) при температуре 5—10° получают
8-нитро-1'-хлор-1-метилнафталин.
Ацетдднитрат рекомендуется применять для нитрования
легкоомыляющихся сложных эфироя, когда необходимо
избежать действия воды, например для получения метилового
эфира нитроопиановой кислоты и других эфиров.
Ингольд и другие [12] проводили нитрование бензола и
толуола в различных условиях (различные температуры и
концентрация уксусного ангидрида), а также изучали кинетику
нитрования ацетилнитратом галоидбензолов совместно с
бензолом при применении различных растворителей и различных
температур нитрования.
Зммонз и Фриман [12а] применяли в качестве нитрующего
агента нитрат ацетонциангидрина.
Авторы установили, что нитрат ацетонциангидрина является
единственным в своем роде нитрующим агентом аминов в
щелочной среде.
Нитрат ацетонциангидрина легко получается нитрованием
ацетонциангидрина дымящей азотной кислотой в уксусном
ангидриде. Этим методом нитрозфир ацетонциапгидрина был
получен с выходом 65—67%:
426
ОН ONO%
I (СН.С0),0 |
з — С — СНз + HNO, > СНз—С — д
CN CN
Нитрат ацетонциангидрина—подвижная бесцветная жидкость,
легко перегоняющаяся в вакууме.
Нитрат ацетонциангидрина в щелочной среде легко
вступает в реакцию с первичными и вторичными аминами, образуя
нитроамины.
Первичные алифатические и алициклические нитроамины
были получены с 50—60 %-ным выходом, а вторичные
нитроамины с 55—80%-ным выходом. Реакция протекает по схеме:
ONO,
R,NH + СНз — С — СНз -> R2NNO3 + (СН,), СО + HCN.
CN
Эммонз и Фриман показали также, что цианистый водород
и ацетон, образующиеся в продуктах реакции, в свою очередь
реагируют с избытком амина с образованием соответствующего
ос-аминонитрила:
R,NH 4- HCN + (СНд): CO-^R^f—С— CN
СНз
Авторы исследовали влияние различных растворителей
на реакцию нитрования аминов нитратом ацетонциангидрина.
Вторичные амины сами по себе являются хорошими
растворителями, хотя реакция может также производиться в растворе
ацетонитрила и тетрагидрофурана. Амин применялся в
5-кратном избытке и нагревался в течении 5 час. с нитратом
ацетонциангидрина.
При применении растворителя с низкой диэлектрической
постоянной или избытка амина выход нитроамина был лишь
20—25 %. При нагревании 3-кратного избытка первичного амина
с нитратом ацетонциангидрина в течение 6 час. в растворе
ацетонитрила или тетрагидрофурана получался хороший выход
нитроаминов.
427
Для нитрования аминов авторы применяли также нитроэфи-
ры циангидринов циклопентанона и циклогексанона:
CN
В другой работе [126] Эммонз иФриман изучали реакцию
нитрования малоноцого, ацетоуксусного эфиров и их алкильных
производных нитратом ацетонциангидрина в щелочной среде.
В качестве основания авторы применяли гидрид натрия.
Малоновый эфир первоначально превращался в его натриевое
производное и затем к полученному соединению добавлялся
нитрат ацетонциангидрина. Реакция протекает по уравнению:
NaH
—> Na[CH(CO,QH,):]
Na [СН (СОаСаНвЫ + (СН,), CONO: -^ (СН,), СО + NaCN + СН (СО,С*Н,),.
CN @
Реакция обычно проводилась в избытке натриевого
производного малонового эфира в растворе тетрагидрофурана. При
применении 3-кратного избытка натриевого производного
малонового эфира выход нитропроизводного диэтилмалонового эфира
был равен 45 %.
При проведении реакции нитрования натриевых
производных малонового и ацетоуксусного эфиров и их алкильных
производных в избытке гидрида натрия образующиеся нитро-
производные тут же в реакционной массе подвергались
расщеплению с образованием %-нитроэфиров:
RCH (C<% QH,), ^ ((^ CONO, !^2 R СН СО,С
нснсо,с,н. ^ ^
С
ОСНз
Выход %-нитроэфироы, в зависимости от применяемого
производного малонового или ацетоуксусного эфира, изменялся
в пределах 42—70%.
Авторы считают, что разработанный ими метод нитрования
малонового и ацетоуксусного эфиров и их алкильных
производных с одновременным щелочным расщеплением полученных
428
нитропроизодных является наиболее общим методом синтеза
%-нитроэфиров и протекает в одну стадию. Этот метод может
применяться для нитрования производных малонового и ацето-
уксусногоэфиров, содержащих группы (например, ароматические
кольца), которые взаимодействуют с обычными нитрующими
смесями.
Арилацетонитрилы также нитруются нитратом ацетонциан-
гидрина с образованием соответствующих нитроацетонитриль-
ных производных.
Нитроацетонитрильные производные авторы не выделяли
в чистом виде, а превращали в арилнитрометаны обычным
способом:
NaH NaOH
ArCH*CN + (СНз)г CONO%—> АгС = NOgNa-—% ArCH,NO,.
i : 2 HCI
CN CN
Этим способом фенилацетонитрил превращался в фенилнит-
рометан с 70%-ным выходом, а о-хлорфенилнитрометан был
получен из о-хлорфенилацетонитрилас 42 %-ным выходом.
Эммонз и Фриман изучали также реакцию нитрования
нитратом ацетонциангидрина ряда алкоголятов спиртов [12в].
Ими установлено, что эта реакция приводит к образованию двух
соединений—нитроэфира и соответствующего эфира %-оксиизомас-
ляной кислоты:
NaOR + (СНз)г CONO, —-» RONO* + (CH,), CCOOR.
OH
Нитрование ароматических углеводородов в боковую цепь,
а также нитрование нафтеновых и парафиновых
углеводородов неорганическими нитратами в литературе освещено очень
слабо. Имеется всего лишь несколько работ об этой реакции.
В 1894 г. М. И. Коновалов [13] показал, что азотнокислая
медь (без прибавления воды) энергично взаимодействует с изо-
октанафтеном в запаянных трубках яри 160—170° или яри
кипячении в открытом сосуде. При этом получаются нитрояро-
дукт и кислоты.
429
В 1901 г. М. И. Коноваловым [14] опубликована работа по
титрованию в боковую цепь дифенилметава и этилбевзола
различными неорганическими нитратами. Опыты проводились в
запаянных трубках.
Для каждого опыта бралось 5 мл углеводорода (этилбен-
зола или дифенилметана), 40 мл воды и к смеси прибавлялась
азотнокислая соль в таком количестве, чтобы при полном
гидролизе соли получался 12%-ный раствор азотвой кислоты.
В некоторых опытах М. И. Коновалов применял 10%-ный
раствор соли.
Нитрат висмута Bi(NO@)a, а также его основная соль
Bi(NO,)(OH)%, нитрат серебра AgNO,, нитрат ртути Hg(NO,)*,
смесь алюминиевых квасцов и нитрата алюминия витруют ди-
фенилметан при нагревании (с водой) до 110—125* в течевие
6 час.
Нитрат висмута и смесь алюминиевых квасцов с нитратом
алюминия нитруют дифенилметан даже при нагревании (с
водой) до 100—106° в течение 6 час. Нитрат серебра и нитрат
ртути при этой температуре витросоединений с дифенилметаном
не образуют. Нитрат свинца не образует нитросоединений с
дифенилметавом при нагревании до 125°.
При нитровании дифенилметава неорганическими
нитратами ни в одном опыте не наблюдалось выделения
газообразных продуктов. Это указывает на отсутствие заметвого окис-
левия углеводородов в процессе витрования.
Нитраты ватрия и калия (которые применялись в виде
10%-ных растворов) не образуют витросоединений с этилбев-
золом при нагревании реакциоввой смеси до 125° в течение
7 час.
Как показали опыты М. И. Коновалова, на витрующую
способность витрата калия оказывает большое влияние
добавление в реакционную смесь небольшого количества витрита
калия. Так, например, взаимодействие этилбензола с 10%-вым
раствором нитрата калия с небольшой примесью нитрита
калия приводит к образованию нитроэтилбензола.
На основании проведенных исследований М. И. Коновалов
пришел к следующим выводам.
430
1. Нитраты щелочных металлов и аммония при
температурах до 125° или совсем не гидролизуются водой или гидроли-
зуются очень слабо, так что концентрация получающейся
азотной кислоты будет очень низкой, вследствие чего эти соли не
дают нитросоединений при нагревании их растворов с
углеводородами.
2. Нитраты висмута, алюминия, ртути, серебра при
температуре 125° и ниже значительно гидролизуются, вследствие
чего эти азотнокислые соли (особенно висмута и алюминия)
можно применять для нитрования углеводородов при
нагревании.
3. При нитровании углеводородов водными растворами
нитратов металлов не наблюдается заметного окисления
углеводородов, что имеет определенные преимущества перед
нитрованием в тех же условиях разбавленной азотной кислотой.
С. С. Наметкин [15] применял азотнокислый алюминий для
нитрования нафтеновых углеводородов.
Смесь 78 г циклогексана и 210 г азотнокислого алюминия
нагревалась в запаянных трубках яри температуре НО—115°.
Б -трубках развивалось сильное давление. После обычной
обработки продуктов реакции было получено 27 г мононитрояро-
дукта, т. е. 56,7% от теоретического выхода. При нитровании
азотной кислотой такого высокого выхода нитроциклогексаяа
получить не удается, и этот спосиб нитрования С. С. Наметкин
считает наилучшим для получения нитроцикяогексана.
Метилциклогексан в запаянных трубках нитруется легко
как азотной кислотой уд. в. 1,2, так и сухим азотнокислым
алюминием. Основным продуктом реакции в обоих случаях
является третичное нитросоединение. При нитровании азотаой
кислотой в получающемся небольшом количестве смеси
первичного и вторичного нитросоединений преобладает вторичное
нитросоединение, тогда как при нитровании азотнокислым
алюминием преобладает первичное. Выход сырого нитропро-
дукта достигает при нитровании азотной кислотой 58%, при
нитровании нитратом алюминия — 72%, считая на
углеводород, вошедший в реакцию.
Циклопентан нитровался нитратом алюминия в запаянных
431
трубках яри 110—115°. В каждую трубку загружалось 10 мл
углеводорода и 20 г соли. В результате было получено 2 г
чистого нитроциклопентана (из трех трубок). В продуктах
реакции обнаружена глутаровая кислота.
Как и М. И. Коновалов, С. С. Наметкин объясняет
нитрующее действие нитрата алюминия гидролизом при температуре
реакции нитрата алюминия с образованием азотной кислоты.
Азотнокислый алюминий А1(МОз)з'9НвО плавится, не
разлагаясь, при 73°. При более высокой температуре начинается
гидролиз. При 140° происходит полный распад нитрата
алюминия на гидроокись алюминия и азотную кислоту. При
температурах выше 73° и ниже 140°, очевидно, будет наблюдаться
некоторое равновесие между солью и продуктами ее гидролиза.
В присутствии углеводорода это равновесие, вследствие
вступления HNOg в реакцию, будет постепенно нарушаться, и для
восстановления его гидролиз должен будет идти все дальше
и дальше. Таким образом, при нитровании азотнокислым
алюминием кислота действует не сразу всем своим количеством,
а постепенно. Поэтому выходы на нитросоединения в этом
случае получаются лучше, чем при нитровании азотной кислотой,
если последнюю взять с тдким расчетом, чтобы относительное
количество ее было равно количеству кислоты, которое может
образоваться при полном гидролизе нитрата алюминия,
взятого для реакции.
Из парафиновых углеводородов М. И. Коновалов [16]
изучал нитрование неорганическими нитратами диизоамила
Нитрование диизоамила нагреванием углеводорода до
температуры кипения в течение 8 час. с 32%-ным раствором
нитрата ртути дало отрицательные результаты.
При кипячении диизоамила с 13%-ным раствором нитрата
алюминия в течение 10 час. в продуктах реакции обнаружены
нитросоединения, растворимые в щелочи.
При нагревании 60 г диизоамила до 110° с 60 г сухого
нитрата висмута в течение 8 час. получается 63 г масла, из
которого около 40 г растворяется в щелочи. В этом растворе
обнаружены первичные и вторичные нитросоединения. Нераство-
432
римая в щелочи часть состоит главным образом издиизоамила.
Из нее было выделено около 1 г третичных нитросоединений.
М. И. Коновалов изучал также нитрование диизоамила
нитратом алюминия в запаянных трубках. Нитрование
производилось как с раствором нитрата алюминия, так и с сухой солью.
Диизоамил нагревался с 13%-ным раствором нитрата
алюминия при 130—150° в течение 8 час. В результате реакции
получалось небольшое количество смеси первичного, вторичного
я третичного нитросоединений. Из 10 г углеводорода 7 г не
вступило в реакцию.
При нитровании диизоамила сухим азотнокислым
алюминием в течение 8 час. при 130° получалась в основном смесь
первичного и вторичного нитросоединений. Из 10 г
углеводорода 5 г в реакцию не вступало. Третичного нитропродукта было
получено всего лишь 0,5 г.
Возможность применения неорганических нитратов для до-
лучения нитросоединений как жирного, так и ароматического
ряда изучалась автором настоящей книги. Так, была
исследована реакция нитрования п.гептана я изооктана (2,2,4-три-
метилпентана) солями азотной кислоты*. Нитрование
указанных углеводородов производилось азотнокислым алюминием
A1(NO,),.9H*O, азотнокислым висмутом Bi(NO,),.5H,0,
азотнокислой медью Си(МОз)з' 6Н&О, азотнокислым железом
Fe(NOg)g - 9Н%О и азотнокислым цинком ZH(NOg)2.6H&O. Реакция
проводилась в запаянных трубках ирв температуре 140° и
лишь в опытах по нитрованию азотнокислым цинком — при
температуре 160°.
При нитровании изооктана и п. гептана азотнокислыми
солями висмута, меди и железа продолжительность опытов
равнялась 8 час, а с солями алюминия и цинка — 20—40 час.
При проведении опытов с солями висмута, меди и железа
наблюдалось выделение окислов азота.
По окончании опыта ампулы охлаждались, после чего
вскрывались. Углеводородный слой, в котором содержались
растворенные нитросоединеяия, отделялся от водного слоя, содержа*
* Неопубликованные данные.
433
щего не вошедшую в реакцию соль. Углеводородный слой
промывался водой, 5%-ным раствором бикарбоната натрия, вновь
водой и высушивался над безводным сульфатом натрия. Не
вошедший в реакцию углеводород отгонялся при атмосферном
давлении, а остатки отгонялись в вакууме.
Продукт, полученный после отгонки углеводорода,
представлял собой нитросоединения с небольшой примесью
карбонильных соединений.
Результаты опытов по нитрованию н.гептана и изооктана
солями азотной кислоты приведены в табл. 35.
Таблица 35
Углеводород
Соль
Взято:
углеводорода,г. .
СОЛИ, Г
Молярное отношение
углеводород: N0, .
Температура опыта,
*С
Продолжительность
опыта, часы . . .
Получено нитросое-
двнений, г ....
Выход на N0%, % .
Иаоонтан
АГ"
124
117
1,2:1
140
20
32
25
* и.Гептаи
АГ"
109
104
1,34:1
140
20
22
20
Bi"
109
104
1,7:1
140
8
24
28
С*'"
109
104
1,58:1
140
8
20
36
Fe"
109
104
1,02:1
140
8
45
45
Zn"
109
104
1,58:1
160
40
5
Полученные нитросоединения разгонялись на
ректификационной колонке в вакууме. Основная масса продуктов
нитрования п.гептана (85—90%) перегонялась в пределах 59—62*
при 6 мм. Относительное количество этой фракции не зависело
от характера применяемой для нитрования соли. Продукты
нитрования изооктана на 75% перегонялись в пределах 60—
63° при 5 мм. Перегнанные продукты реакции исследовались
обычными методами.
При изучении реакции нитрования н.гептана и изооктана
различными солями азотной кислоты было установлено следующее.
434
1. Нитрование изооктана азотнокислым алюминием
происходит с несколько большей скоростью, чем нитровапие
н.гептана. Например, при витровании изооктана азотнокислым
алюминием выход нитросоединевий составлял 25%, считая на
азотнокислый алюминий, тогда как при нитровании н.геп-
тана выход был равен 20%.
2. Скорость реакции в большей степени зависит от
характера применяемой соли, точнее от характера катиона, чем от
характера нитруемого углеводорода. Из изученных
азотнокислых солей наибольшей активностью в реакции нитрования
н.гептана обладает азотнокислое железо. Азотнокислый цинк
давал наименьший выход нитросоединений.
3. При нитровании различными солями азотной кислоты
получаются одни и те же продукты реакции. При нитровании
н.гептана основная масса продуктов реакции состояла из
вторичного нитрогептана, а при нитровании изооктана — из
третичного нитроизооктана. Наряду с этим образуется небольшое
количество первичных нитросоединений.
4. При нитровании н.гептана и изооктана солями
азотной кислоты, помимо мононитросоединейий, образуется
небольшое количество дивитросоединений. При нитровании
н.гептана образуется значительно меньше динитросоединений (около
10% по весу), чем при нитровании изооктана (около 18% по
весу). Как в случае нитрования н.гептана, так и в случае вит-
рования изооктана образуются дивитросоединения
первичного характера. При нитровании н.гептана получается также
небольшое количество вторичных динитросоединений.
5. Нитрование н.гептана и изооктана солями азотной
кислоты сопровождалось окислением нитруемого углеводорода.
Продукты окисления подробно не исследованы.
Нитрование ароматических соединений азотнокислыми
солями металлов обычно осуществляется в смеси с другими
компонентами, которые, как предполагают, играют роль активаторов.
Менке [17], анализируя действие на ароматические
соединения нитрующих смесей азотной кислоты с уксусным
ангидридом, пришел к выводу, что последний не только служит во-
дуотнимающим средством, но и каталитически ускоряет процесс.
435
Предполагая, что при замене азотной кислоты ее солями
нитрующая смесь все же будет достаточно активной ввиду
каталитических свойств уксусного ангидрида, Менке изучил
нитрование ароматических соединений смесями уксусного
ангидрида с нитратами, имеющими достаточно низкие
температуры разложения (к числу этих нитратов относятся нитраты
железа, меди, никеля, кобальта, алюминия, церия и ряд
нитратов других металлов I, II, III, IV и VIII групп периодической
системы). Опыты Менке показали, что из этих соединений
нитраты щелочных металлов не обладают нитрующим действием
или же реагируют лишь в слабой степени.
Нитрование анилина проводилось Менке следующим
образом^ смеси Ювес.ч.СъЦМО^з.ЗНзО и 30ч.уксусного ангидрида
добавляли по каплям 8 ч. анилина при перемешивании и
охлаждении льдом (температура реакции не должна превышать 30°).
Через полчаса, считая от начала приливания анилина,
реакционную смесь вливали в 150 ч. воды. Образовавшуюся при этом
кристаллическую массу отделяли от воды, измельчали в
порошок и промывали водой для освобождения от включенной
азотнокислой меди. После кристаллизации сырого продукта
из 50%-ного спирта получено 11ч. чистого о-нитроацетанилида
с т. пл. 92°.
Так как уксусный ангидрид с анилином уже при
обыкновенной температуре образует ацетанилид, то реакцию
нитрования анилина смесью Cu(NOg)% с уксусным ангидридом можно
рассматривать как нитрование ацетанилида. Полученный
результат указывает на различие между нитрованием
ацетанилида смесью уксусного ангидрида и нитрата и смесью азотной
и серной кислот (в последнем случае образуются одновременно
о- и п-нитроацетанилиды).
Для нитрования фенола его растворяют в уксусном
ангидриде в количестве 5 г и раствор вливают по каплям в смесь
15 г азотнокислого железа и 25 г уксусного ангидрида при
70—85°. Продукт реакции — пикриновая кислота.
Подобным же образом при нитровании бензола при 80°
получается нитробензол, а из хлорбензола при 40—45° п-ни-
трохлорбензол. Толуол дает при действии смеси Fe(NO»)»
436 ZTwmpoaawwe н%лгрдлг&л(% % злорыслгыа
с уксусным ангидридом о-нитротолуол (при температуре
реакции 40°).
Далее показано, что если нитрат применять в смеси не с
уксусным ангидридом, а с ацетилхлоридом (или ацетилброми-
дом), то, наряду с нитрованием, происходит я галоидирование
ароматического соединения. Так, например, при действии на
фенол Си(МОз)з и ацетилхлорида (реакция проводится при
охлаждении смесью льда с солью) получается 4,6-д&хлор-2-
нитрофенол.
Смесь азотнокислой меди и уксусного ангидрида может
также служить для получения нитроацетоуксусного эфира из
ацетоуксусного эфира.
Ароматические соединения нитруются также смесью
нитрата с уксусной кислотой. Следует, однако, отметить, что эта
смесь является менее активным нитрующим средством, чем
смесь нитрата с уксусным ангидридом. Для объяснения этого
различия предполагают, что в первой смеся в качестве
промежуточного продукта, играющего роль нитрующего агента,
образуется не ацетилнитрат, как в смеси с уксусным ангидридом,
а ацетилортоазотная кислота (Менке).
Некоторые соединения (фенол, о-нитрофеяол, диметилани-
лин) нитруются смесью нитрата с уксусной кислотой даже при
комнатной температуре. С другой стороны, нафталин нитруется
азотнокислой медью или азотнокислым железом в смеси с
уксусной кислотой лишь при 105°, а ацетанилид — при 75* (в смеси
с уксусным ангидридом нитраты реагируют с двумя
последними соединениями уже при 30°).
В качестве примера нитрования ароматических соединений
смесью нитрата с уксусной кислотой укажем на нитрование
фенола. Раствор фенола (5 г) в ледяной уксусной кислоте (10 г)
добавляют по каплям при сильном перемешивании к смеси 8 г
Cu(NOa)* и 25 г ледяной уксусной кислоты (реакцию
необходимо проводить с охлаждением, поддерживая температуру 26°).
Через 20 мин. после первого приливания раствора фенола
реакция заканчивается. При вливании реакционной смеси в 10 мл
воды выделяется продукт реакции в виде кристаллической
массы, которую отфильтровывают и промывают холодной во-
милую пиши 437
дой. В результате этой обработки получается чистый о-нитро-
фенол с т. пл. 44—45° (как указано выше, при нитровании
фенола смесью нитрата с уксусным ангидридом образуется не
о-нитрофенол, а пикриновая кислота). Таким образом
нитрование фенола нитратом металла в смеси с уксусным
ангидридом или уксусной кислотой дает в том или другом случае
различные продукты реакции в зависимости от второго компонента
нитрующей смеси.
Реакция нитрования ароматических соединений нитратами
металлов в присутствии уксусного ангидрида обстоятельно
исследована Бахарашем [18—20]. Он поставил своей задачей
выяснить экспериментально: 1) является ли этот метод нитро*
вания общим для всех ароматических соединений или же он
применим только к определенной группе этих соединений и
2) существует ли известная зависимость между природой
нитрата и строением полученного нитросоединения, т. е.
ориентируется ли нитрогруииа в то или иное положение в
зависимости от применяемого нитрующего агента.
Для разрешения первого вопроса исследовано действие
азотнокислой меди и азотнокислого железа в присутствии
уксусного ангидрида на бензальдегид, бензойную кислоту,
нитробензол и п-толуидин. Опыты с бензальдегидом,
бензойной кислотой и нитробензолом, которые проводились при
различных температурах, дали отрицательный результат. С
другой стороны, п-толуидив нитровался чрезвычайно легко.
Исходя из полученных им результатов и данных других
исследователей*, Бахараш приходит к выводу, что
нитрование ароматических соединений неорганическими нитратами
дает положительный результат лишь в том случае, если нитруе*
мые соединения содержат о- и п-ориентирующие заместители;
соединения же, у которых заместители ориентируют в м-поло-
жение, не нитруются неорганическими нитратами в смеси с
уксусным ангидридом.
* Менке [17], Шпигеля и Гаимана [2t]; последние два автора при
нитровании анилина, фенола и анизола азотнокислым висмутом в
присутствии уксусного ангидрида получили во всех случаях смеси о- и п-нитро-
производных.
29 А. в. Тгпчиеы
438 #%ттфобоние ммтр&тдлФм ы агдормстьм*
Для выяснения зависимости строения образующегося нит-
росоедииения от ярироды применяемого нитрата Бахараш
нитровал я-толуидин азотнокислыми солями железа, марганца^
кобальта, никеля, меди, ртути, серебра, натрия и лития в
присутствии как уксусной кислоты, так и уксусного ангидрида.
Опыты показали, что я-толуждин сначала реагирует с
уксусным ангидридом или уксусной кислотой с образованием ацето-
толуидида, после чего наступает нитрование. Все указанные
выше азотнокислые соли дали в качестве продукта реакции
3-нитро-п-ацетотолуидвд
сн,
А
У
HNCOCH,
за исключением нитратов натрия и серебра, которые оказались
неактивными.
Показано также, что наибольшая скорость реакциинаблюдает-
ся при применении нитрата меди (продолжительность процесса
10 мин. при 30°). Продолжительность нитрования п-толуидина
нитратами металлов группы железа составляет 15—20 мин. в
интервале температур 65—85° (аналогичные результаты получены
и при нитровании азотнокислой ртутью). Наименее активным
оказался нитрат лития; нитрующее действие его обнаружено
лишь при кипячении реакционной смеси с обратным
холодильником в течение 2 час.
При нитровании анилина нитратом лития продуктом
реакции оказался п-нитроацетанилид (а не о-изомер, который, по
данным Менке, получается при нитровании анилина нитратами
тяжелых металлов). Сопоставляя эти результаты, Бахараш
приходит к заключению, что нитраты, которые реагируют яри
относительно низких температурах (например, нитраты
тяжелых металлов), приводят к образованию о-нитросоединений,
в то время как нитраты, действующие лишь при высоких
температурах, например нитрат лития, дают п-нитропроизводные.
Однако эта закономерность наблюдается не во всех случаях;
439
например, п-толуидия со всеми нитратами дает одно и то же ни-
тропроизводиое.
Бахараш изучил также действие нитратов на некоторые
двуядерные соединения. При нитровании ОДМОз^-ЗНаО этот
нитрат сначала обрабатывали уксусным ангидридом до
получения зеленой аморфной массы, после чего к последней
добавляли при перемешивании раствор нитруемого соединения в
уксусном ангидриде. Нитрование азотнокислым литием
производили при кипячении в течевие нескольких часов, причем
для повышения активности добавляли 4% нитрата меди.
При действия азотнокислой меди на дифенил получены
п-нитро- ип,п'-динитродифенил;дибензилдал п,п'-динитроди-
бензил. Нитрат лития не реагировал ни с дифенилом, ни с ди»-
бензилом. Бензил и стильбен Ее нитровались нитратами лития
и меди, а бензоив лишь окислялся ими в бензил. Опыты с
ядерными соединениями подтвердили, таким образом,
указанные ныше выводы, что смесью нитрата с уксусным ангидридом
нитруются TO,j?Ko те ароматические ядра, которые содержат
заместители, ориентирующие в о- и п-положения, но не в
м-положение.
Дальнейшие исследования Бахараша, однако, показали,
что в некоторый случаях соединения, содержащие заместители,
ориентирующие в м-положение, также могут подвергаться
нитрованию смесью нитрата с уксусным ангидридом. Так,
например, хинолин, который при действии азотной кислоты или
смеси азотной и серной кислот дает только 5- и 8-иитропроиз-
водные, нитруется азотнокислым литием с образованием
7-нитрохинолина.При нитровании фенилуксусной кислоты
азотнокислой медью в уксусном ангидриде получены м-иитрофенил-
уксусная кислота с выходом 50% от теоретического и о-яитро-
фенилуксусная с выходом 10% (как известно, при действии
азотной кислоты фенилуксусная кислота дает смесь м-нитро-
фенилуксусной кислоты с выходом 14% и о- и п-нитропроизвод*
них фенилуксусжой кислоты с выходом 80%).
Механизм реакции нитрования нитратами металлов в
присутствии уксусного ангидрида и уксусной кислоты состоит,
по мнению Бажараша, в образовании промежуточного соедине-
30 А. В. Топчиев
440
ния — диацетияортоазотной кислоты, которая является
нитрующим агентом. Эта кислота получена им следующим
образом: смесь 20 ч. Cu(NOg)*"3H*O и 50 ч. уксусного ангидрида
оставлена на 20 мин. при 30—35*. Продукт реанции,состоявший
из жидкой части и
зеленой аморфной массы
(уксуснокислой
меди), разделен
фильтрованием. Из фильтрата
посредством
двукратной перегонки в
вакууме выделено
соединение, свойства которо-
госовпадали со
свойствами описанной
Пикте диацетилортоазот-
ной кислоты (т. кип.
45°, ng 1,37542).
Обработка диаце-
тилортоазотной
кислотой
эквивалентного количества
анилина в течение несколь?
ких минут при 35*
дала смесь нитронроиз-
водных, состоявшую
из п-нитроацетанили-
да и
соответствующего о-изомера.
В результате
систематического
исследования автором зтой книги разработан метод нитрования
ароматических соединений неорганическими нитратами в присутствии
некоторых активаторов. Ниже дается описание опытов по
нитрованию органических соединении нитратами в присутствии
следующих активаторов: AlClg, FeCl,, SiCl* и BF,*.
* Попытки осуществить нитрование ароматических углеводородов
Рис. 5. Прибор для нитрования органических
соединений нитратами в присутствии AlCl
4
§ 1. Нитрование в присутствии А1С1@
Нитрование проводили (рис. 5) в круглодонной колбе jT
емкостью 0,5 я, снабженной трехгорлым форштоссом 3, через
центральный канал которого проходила ось мешалки 2;
боковые трубки форштосса соединялись с обратным
холодильником бис мерным сосудиком ^, из которого производили
засыпку активатора.
При проведении опыта в колбу загружали безводный
нитрат в виде порошка, затем нитруемый углеводород и после
пуска мешалки через боковую трубку форштосса постепенно
вносили активатор (из мерного сосудика). Опыты
производили как на холоду, так и при нагревании.
Изучено действие на ароматические - углеводороды в
присутствии хлористого алюминия следующих нитратов: KNO@,
NaNO,, NH4NO3, Ba(NO,),, Pb(NO,), и AgNO,.
Нитрование бензола
Для нитрования применяли бензол, предварительно
очищенный от тиофена и других примесей тщательной обработкой
концентрированной серной кислотой. Нитраты перед
употреблением обезвоживали, расплавляли и полученный плав
подвергали измельчению.
Нитрование азотнокислым калием. В
первоначальных опытах молярное отношение нитрата к бензолу
составляло 1:1; эти опыты, однако, показали, что яри
указанном отношении реагентов происходит загустевание
реакционной массы, что затрудняет перемешивание. Поэтому в
дальнейшем нитрование проводили в присутствии избытка бензола,
который, таким образом, играл роль растворителя. Обработку
продуктов реакции, полученных после нитрования бензола
азотнокислым калием, производили так же, как и при
нитровании бензола окислами азота (см. гл. V).
Первая серия опытов имела своей задачей выяснение
зависимости процесса нитрования от количества активатора и дро-
(бензола, толуола, ксилола и мезитилеяа) в отсутствие активаторов дали
отрицательный результат: даже при продолдснтельностщ нитрования 3 ча^
са при различных температурах исходные углеводороды не подверглись
никаким изменениям.
80*
442
должительности его загрузки (табл. 36 и 37); В реакционный
сосуд загружали 1 моль бензола, 0,5 моля азотнокислого
калия я реакцию проводили в течение 3 час, причем в одних
опытах варьировали количества хлористого алюминия, который
Вносили в течение 30 мин. (табл. 36), в других — изменяли про-
Таблица 36
А1С1з
(78 г бензола, 50 г KNO,)
опыта
Выюд
нитробензола
% от
теоретического яа
бен-
воя
обратно
бензола
% от
взятого
бензола
Выход
нитробензола на
ярореагиро-
вавпшй
бензол, % от
теоретического
Выход
нитробензола на
нитрат, %
от
теоретического
3
4
5
6
7
8
9
63
34
20
20
10
10
3
3
7
23
23,1
23,3
22,7
12,5
11
5
4,2
2,2
18,7
18,8
18,9
18,4
10,1
8,9
4,1
3,4
4,8
53
55
58
57
62
65
70
72
73
67,9
70,5
74,3
73,0
79,5
83,5
89,7
92,3
93,7
58,3
63,6
73,6
68,5
49,5
53,6
39,6
44,3
27,9
37,3
37,6
37,9
37,0
20,3
17,9
8,2
6,8
3.6
должительность загрузки активатора при постоянном его
количестве (табл. 37). Опыты проводили без нагревания извне,
но ввиду экзотермичности реакции температура реакционной
смеси во время нитрования повышалась до 30—40°.
Данные табл. 36 и 37 показывают, что оптимальные
результаты получаются при загрузке 20 г (0,15 моля) активатора в
течение 30 мин.
В дальнейшем изучена также зависимость выхода нитро-
беазола от продолжительности процесса нитрования при про-
ведении реакции без нагревания. Оказалось, что при
443
увеличении продолжительности нитрования с 20 до 90 мин.
выходы нитробензола остаются без изменения; при повышении
продолжительности реакции с 90 мин. до 3 час. выходы
нитробензола постепенно повышаются; дальнейшее увеличение
продолжительности нитрования приводит уже к падению выхода
нитробензола, т. е. трехчасовой период нитрования является
оптимальным (табл. 38).
Таблица 37
(78 г бензола, 50 г RNO,, 20 г А1СУ
опыта
1
2
3
4
5
6
Продолжительность загрузни
AlClg, мин.
5
30
60
60
120
120
Выход
нитробензола
г
16,0
23,3
21,5
22,0
20,5
19,8
% от
теоретического на
взятый
бензол
13,0
19,9
17,5
17,9
16,7
16,1
Получено обратно
бензола
г
61
58
56
54
57
58
% от
взятого
бензола
78,2
74,3
71,8
69,3
73.0
74,3
Выход
нитробензола на
прореагировавший
бензол, % от
теоретического
59,6
73,8
61,9
58,1
61,8
62,7
Выход на
нитрат, % от
теоретического
26,0
37,9
34,9
35,8
33,4
32,2
Опыты по нитрованию бензола нитратом калия в
присутствии AlCls показали, таким образом, что добавление
активатора в количестве 0,15 моля при молярном отношении бензола
к KNOs 2 : 1 приводит к выходу нитробензола 38,5% на нитрат
и 74,5% на прореагировавший бензол, если реакция
осуществляется в течение 3 час. без нагревания извне.
Совершенно неожиданный результат получен при попытке
активирования процесса повышением температуры реакции
(табл. 39).
Опыты с нагреванием извне на бане привели ж
уменьшению выхода нитробензола; например, при 90—100° (темпера-
444
Таблица 38
от дро&ик%смпге.&ьмосг%м
(78 г бензола, 50 г KNO& 20 г А1С1,)
опыта
1
2
3
4 .
5
6
7
8
9
10
ii
12
13
14
g ^
gg
А
20
20
30
30
60
60
90
90
120
120
180
180
240
240
Выход
нитробензола
г
16
16,5
16,8
15,9
16,6
16,0
16,8
16,1
20,1
21,1
22,7
23,6
22,4
23.2
% от
теоретического на
взятый
бензол
13
13.4
13,6
12,9
13,5
13,0
13,6
13,1
16,3
17,1
18,4
19,1
18,2
18,8
Получено обратно
бензола
г
60
58
61
62
60,5
61,0
59.0
60,7
58,5
57,1
56,2
58,0
57,0
59,0
% от
В8ЯТ0Г0
бензола
76,9
74,3
78,2
79,5
77,4
78,4
75,6
77,8
75,2
73,1
71,9
74,3
73,0
75,5
Выход нитро-
бенвола на
прореагировавший
бензол, % от
теоретического
56,1
52,1
62,3
62,6
59,8
59,3
55,8
58,7
65,0
63,7
65,7
74,5
67,3
77,0
Выход нитро-
. бензола на
нитрат, % от
теоретического
26,0
26,8
27,3
25,9
27,0
26,0
27,3
26,2
32,7
34,3
36,9
38,4
36,4
37,7
тура бани) выход упал до 20—23% в расчете на нитрат. Это
явление, невидимому, объясняется частичным распадом при
высоких температурах комплекса бензола с хлористым алюми-
яием, вступающего во взаимодействие с нитратом калия (см.
ниже о механизме реакции).
Нитрование нитратами аммония,
натрия, бария, свинца и серебра. Нитрование
производилось в тех же условиях и при том же количественном
отношевии реагентов, которые оказались оптимальными для
нитрования азотнокислым калием. Результаты опытов сведены
в табл. 40.
445
Таблица 39
ом:
(78 г бензола, 50 г KNO,, 20 г А1С1,)
опыта
Температура
бани, "С
Выход
нитробензола
% от
теоретического
на взятый
бензол
Получено обратно
бензола
% от
взятого
беязола
з&Е
#
Д
1
2
3
4
Без нагрева-
вания извне
45—50 . . ,
90—100 . .
23,1
21,2
18,7
17,9
,14,2
112,8
18,9
17,2
15,2
14,5
11,5
10,4
57
56
58
59
57
60
73,0
71,0
74,3
75,6
73,0
76,8
69,7
60,9
59,2
59,7
42,9
45,1
37,6
34,5
30,4
29,1
23,1
20,8
Сравнение данных табл. 39 и 40 показывает, что из всех
исследованных нитратов наиболее активным нитрующим
агентом в присутствии AlCls является AgNOs; остальные нитраты
осуществляют нитрование в меньшей степени, причем наименее
активны нитраты бария и свинца* Если расположить все
исследованные нитраты в порядке убывания активности
нитрования по отношению к бензолу, получим следующий ряд:
AgNO,>KNO,> NaNO, ,> NH.NO, > Pb (NO,), > Ba(NO,),.
Нитрование толуола
Из данных табл.41 видно, что при нитровании толуола без
нагревания извне в течение 6 час. выход нитротолуола достигает
38% по нитрату и 72,6% по толуолу, вступившему в реакцию.
Повышение температуры посредством внешнего обогрева и
понижение продолжительности процесса оказывают
неблагоприятный эффект на выход нитротолуола.
О Ф*
№ опыта
Продолжительность опыта, мин
О
н
m
мл
*
g
II
Выход
нитротолуола на
прореагировавший
толуол, % от
теоретического
Выход
нитротолуола на нитрат,
% от
теоретического
р
§
g
к
8 #
о g
8 2
G Л
3 go
со
N0,
СЛ
СО
о
со
о
*-*
СЛ
СЛ
СЛ
К
о
06
>
N0,
К
СО
*&»
СЛ
СЛ
-ч
G5
СЛ
СО
СЛ
Z
!g
о
на.
СО
СЛ
ио
д*~
по
СЛ
О)
(35
Со
(N0,
К
о
IV)
Сл
СО
о
со
СЛ
РЭ
(N0,
СЛ
о
о
.
о
о
со
по
СЛ
о
РЭ
о
СЛ
со
СЛ
СЛ
о
на.
4^
на.
в»
N0,
со
сэ
с
о
со
со
о
сэ
о
СЛ
ио
СЛ
о
^]
сэ
о
Р
оэ
ее
ее
s>
о
сэ
со
СП
*&~
сэ
СЛ
!%!з
я о о
1
ом
8 3»*
бен-
Н
м
о
Выход нитробен-
вола на
прореагировавший бея-
вол, % от
теоретического
Выход нитробен-
вола на нитрат,
% о/г
теоретического
со
447
Нитрование м-ксилола
При нитровании м-ксилола применяли следуювдие
количества реагентов: 0,2 моля м-ксилола, 0,1 г-моля KNO* и 0,03"
г-моля А1С1@; отделение продуктов реакции от непрореагировав-
шего м-ксилола и разделение мононитроксилолов от динитро-
м-ксилола производили таким же путем, как и при нитровании
окислами азота (см. гл. V).
Опыты по нитрованию ксилола (табл. 42) показали, что
оптимальный выход изомерных мононитроксилолов (29%, считая
на нитрат) получается при проведении реакции без
нагревания извне. При повышении температуры выход
мононитроксилолов понижается, выход динитро-м-ксилола, наоборот,,
повышается, достигая максимальной величины (10—11%) при
105—110°.
Таблица 42
А1С1з
(21 г м-ксилола, 10 г KNO,, 4 г А1С1,)
опыта
3
Температура
бани, "С
Без нагревания
извне
45—50
105—110
Выход мовояатронсилола
г
f 4,1
14,2
3,4
I 2^6
% от
теоретического на
ввятый
ксилол
13,6
13,9
11,3
7,9
8,6
% от
теоретического на
ввятый
нитрат
27,0
27,8
22,5
15,9
17,2
Выход дижитроксилола
г
1,5
1,2
1,8
2,0
2,1
% от
теоретического на
ввятый
ксилол
3,8
3,1
4,6
5,1
5,3
% от
теоретического на
взятый
нитрат
7,65
6,2
9,2
10,2
10,6
§ 2. Нитрование в присутствии FeCl@ и
Нитрование бензола в присутствии SiCl* производили при
тех же количественных отношениях реагентов, как и в
присутствии AlClg; при нитровании в присутствии FeCls количе-
448
ства реагентов были уменьшены в 4 раза против обычных
(0,25 моля бензола, 0,125 моля KNOs и 0,037 моля FeCls).
Результаты опытов приведены в табл. 43.
бензола KNO
Таблица 43
опыта
Актвват ор
Выход
нитробензола
% от теорети
ческого на
взятый
бензол
Получено обратно
бензола
% от
взятого
бензола
Н%
ш
хз
1
2
3
4
FeCl,
SiCl*
3,1
3,3
5,2
4.8
10,3
11,0
3,3
3,8
16
15
69
72
84
80
88,4
92,2
65,3
52,5
36,6,
50,7
20,2
21,4
8,45
7,8
Сравнивая эти результаты с данными, полученными при
нитровании в присутствии AlCls (см. табл. 37), можно прийти
к выводу, что FeCls менее активный катализатор, чем AlCls,
который дает выход нитробензола около 38% на нитрат и 70%
на прореагировавший бензол. Еще менее активный
катализатор SiCl* (выход на нитрат в 5 раз меньше, чем в присутствии
AlCls).
§ 3. Нитрование в присутствии
Как известно, фтористый бор широко применяется в
качестве катализатора в органическом синтезе, что объясняется его
способностью к образованию промежуточных комплексов с
ароматическими соединениями, обладающих активными
свойствами. Так, например, Ньюланд и сотрудники [22]
пользовались BFs как катализатором при конденсации пропилена с
органическими кислотами (уксусной, хлоруксусной, дихлоруксус-
ной, бензойной и др.) в соответствующие эфиры. Реакцию
проводили при 70° и давлении на 25 мм выше атмосферного (без
катализатора эта реакция протекает лишь при высоких
температурах и давлениях).
449
' По данным того же исследователя [23], олефины в
присутствии BFs конденсируются также с оксикислотами
(салициловой, п-оксибензойной, м-оксибенэойной и др.). Например,
пропилен конденсируется с салициловой кислотой с образованием
изопропилсалицилата и алкилированных в ядре сложных эфи-
ров салициловой кислоты. Процесс протекает таким образом,
что пропилен вначале конденсируется с салициловой кислотой
с образованием изопропилсалицилата, который затем
подвергается изомеризации, причем изопропильная группа вступает
в бензольное ядро. Механизм действия фтористого бора в
данном случае, вероятно, состоит в том, что он образует комплекс
:С салициловой кислотой, присоединяясь к атому кислорода
гидроксила группы СООН; водород карбоксильной группы
делается при этом более подвижным, чем и обусловливается
.возможность конденсации кислоты с олефивом.
Образовавшийся сложный эфир соединяется далее с BFg. Это ослабляет
связь между кислородом и углеводородным радикалом, что
облегчает изомеризацию.
При помощи фтористого бора может быть осуществлена
конденсация олефинов с ароматическими углеводородами [24].
Например, бензол конденсируется с пропиленом в сернокислой
среде с образованием моно- и диизопропилбевзола; при этом,
в отличие от реакция в присутствии А1С1з, которая дает
преимущественно м-диизопропилбензол, конденсация в присутствии
BFg приводит к п-диизопропилбензолу. Бензол конденсируется
также с этиленом и бутиленом.
Фтористый бор применяется в качестве катализатора также
при конденсации олефинов с фенолами, причем, как и в случае
салициловой кислоты, при взаимодействии олефина с фенолом
происходит конденсация с последующей изомеризацией [25].
В присутствии фтористого бора ацетилен коденсируется с
одноатомными спиртами [26], гликолями и оксикислотами [27],
а также с карбоновыми кислотами [28].
Механизм конденсации спиртов с ацетиленом, приводящий
к синтезу ацеталей, заключается, как полагают, в следующем:
образовавшийся в результате соединения спирта с BFg
молекулярный комплекс вступает в реакцию в ионизированной форма
450
с ацетиленом; при этом возникает новый комплекс, который
распадается на ацеталь и фтористый бор:
2ROH + 2BF, -» 2ROHBF* -#2Н+ + 2 (RO-BF,)"
НС = СН + 2Н+ + 2 (RO
Н,С —С—Н
\
/OR .
С—Н + 2BF*
OR-BF,
Подобным же образом в присутствии BFs оксисоединения
конденсируются и с производными ацетилена [29, 30].
Необходимо также отметить, что фтористый бор, ввиду его
склонности к образованию комплексов с органическими
соединениями, с успехом применяют вместо А1С1з в реакции Фриделя—
Крафтса. Так, например, при помощи фтористого бора полу-
&б. Установка для нитрования органических соединений нитратами
в присутствии
неорганическими нит^рду^д^и 451
чают гомологи бензола, конденсируя его с алкилгалогенидами;
из бензола, толуола и анизола при взаимодействии с уксусным
ангидридом в присутствии BFg синтезируются
соответствующие кетоны [31].
Основываясь на указанных выше каталитических свойствах
фтористого бора и его способности заменить А1С1з в реакции
Фриделя — Крафтса, автором этой книги сделана попытка
применить фтористый бор в качестве катализатора при нитровании
органических соединений солями азотной кислоты. Ниже
приводятся результаты некоторых из проведенных в этом
направлении опытов.
В качестве нитратора применяли круглодонную трехгорлую
колбу 7 (рис. 6), снабженную трехрогим форштоссом 3,
центральный канал которого служил для прохождения оси
мешалки 4; одна из боковых трубок форштосса соединялась с конден-
сационно-поглотитёльной системой, состоявшей из
холодильника J и склянки Тищеико 6 с эфиром, в котором происходило
поглощение не вошедшего в реакцию фтористого бора.
Фтористый бор поступал в реакционную колбу из аппарата 7, где
производили его синтез, но трубке 2, проходившей через
левое горло колбы 7. Необходимый для опытов фтористый бор
получался смешением 20 г В%Оз, 40 г CaF% и 200 г HgSO^ уд. в.
1,84. Реакция происходит но следующему уравнению:
3CaF, + 3H,SO, + B,O, -* 3CaSO. + 3H,0 + 2BF,.
В качестве исходных веществ для синтеза фтористого бора
применяли чистый кристаллический CaF% (флюорит) и борный
ангидрид, полученный прокаливанием технической борной
кислоты при 800—1000* (в муфельной печи) до прекращения
выделения пузырьков Н%0. Флюорит и борный ангидрид
измельчали в порошок и затем хорошо перемешивали друг с другом.
Смесь вносили в колбу Вюрца емкостью 500 мл и заливали сер-
иой кислотой. Во время реакции колбу Вюрца время от
времени встряхивали, чтобы облегчить прохождение фтористого
бора через реакционную кашицеобразную массу.
Образовавшийся фтористый бор перед направлением в нитратор
проходил предварительно через склянку Тищенко с серной
кислотой.
452
Нитрованиебензола
Опыты по нитрованию бензола азотнокислым калием в
присутствии BFs проводились следующим образом: после
загрузки реагентов (0,5 моля K.NO* и 1 моль бензола) смесь при
перемешивании насыщали BFs, который пропускали из колбц
Вюрца, где происходил его синтез. Опыты велись как без на-г
гревания извне, так и при нагревании (да водяной или
вазелиновой бане). При нитровании без нагревания извне темпера*
тура реакционной смеси, вследствие экзотермичности процесса,
быстро повышалась до 34—38°, а затем более медленно до 55—
57°. По окончании насыщения фтористым бором реакционная
масса, имевшая темнобурый цвет, разделялась на жидкую часть
и твердый осадок на дне колбы. Жидкую часть отделяли от
осадка, промывали водой, а затем раствором соды и после
сушки над хлористым кальцием подвергали перегонке
(вначале отгонялся бензол, а затем при 207—210* нитробензол).
Твердый осадок Для извлечения адсорбированного им
нитробензола перегоняли с водяным паром.
В табл. 44 и 45 приведены результаты опытов по нитрованию
бензола азотнокислым калием в присутствии BFs,
проводившихся как с различной продолжительностью (табл. 44), так и
яри различной температуре (табл. 45).
Данные табл. 44 указывают, что при нитровании бензола
азотнокислым калием в присутствии BFs получаются лучшие
выходы нитробензола, если длительность процесса нитрования
увеличивается от 30 минут до 3 часов. Наилучшие результаты
получаются при трехчасовой продолжительности нитрования.
Что касается влияния температуры реакции на выход
нитробензола при нитровании азотнокислым калием в присутствии BFs,.
то из данных табл. 45 видво, что наилучшие результаты полу*
чаются при отсутствии нагревания извне.
Нитрование толуола и м-ксилола
В табл. 46 и 47 приведены результаты опытов по
нитрованию толуола и м-ксилола.
РЭ
Cl
РЭ
5
со
со
оь
со
ел
»t-
со
ее
н^
СО
Ъо
со
со
ел
^^
g
н
<D
-о
РЭ
i
РЭ
РЭ
со
^)
сэ
ее
00
00
ьС-
сэ
ел
со
ее
I
со
ел
CD
К
н
CD
"О
рэ
i
CD
ел
о
\э
ел
*~
со
ел
сэ
1^)
W
03
о
ф
В
CD
О
Я
о
3
<D
РЭ
*<3
13
рэ
Я
1
К
о
а
РЭ
а
а
м
1^
ел
ел
о
ел
ел
1о
ее
<D
Ш
CD
И
рэ
Я
а
№ опыта
§
4
О
g 2 §
2 Д (В
Я а) 4
i
ggS
В
и
о
«а
gm
*ь
о
к
Выход
нитробензола на
прореагировавший
бензол, % от
теоретического
Выход на нитрат,
% от
теоретического
00
<D
S
р
м
.^3
сэ
в
о
ш
3
к
н
3
к
3
*%
А
с*»
6
р
"8
:
А
S
2
РЭ
РЭ
^
f
8
сэ ел
$88
ее
её
ел ел ел &- ее ее се
ел ел ел ел ее ее се
-J -J ~Д 00 СО СО О
20,6
со ел се
сэ ^ ее со сэ
-л О со ^ ел
ьэ
ел ел ел ел сэ сэ ^
ее ^ ^з оо ее со о
сэ сэ ^з ^ оо со со
-л со ее ^ о -л со
со ьэ о ее сэ ьо оэ
ел ел ел ее ее ее ьо
N> ел »-»- оо се »->. се
се ел
со
со
W се
се ^
-л со се со ^
сэ ел О *^- со
№ опыта
Продолжительность опыта, мин
Максимальная
температура
экзотермической
реакции, *С
Н:
взятого
бензола
1{
I!
Выход
нитробензола на
прореагировавший
бензол, % от
теоретического
Выход на
нитрат, % от
теоретического
со
I
8
м
о
а
5*
I
А
S %
454
Анализируя результаты опытов по нитрованию
азотнокислым калием бензола, толуола и м-ксилола в присутствии BF@,
можно прийти к выводу, что процесс протекает аналогично
нитрованию нитратами в присутствии А1С1з; оптимальные
условия достигаются при проведении реакции без нагревания извне
в течениеЗ часов. Повышение температуры приводит к
понижению выхода нитропродуктов, невидимому, вследствие того,
что при высоких температурах происходит распад комплекса,
образуемого BFa с ароматическим соединением.
Таблица 46
ffumpoemtwe люл&олд KNO* б %j%/ci/r%cr%*uu BF3
(92,1 г толуола, 50 г KhOg и BF@, полученный из 40 г CaF,)
опыта
1
2
Максимальная
температура
экзотермической
реакции, *С
40
40
Выход
нитротолуола
г
23,8
24,3
% от
теоретического на
взятый
толуол
17,4
17,7
Получено обратно
толуола
г
71
69
% от
взятого
толуола
77,1
75,0
Выход
нитротолуола на
прореагировавший
толуол, % от
теоретического
76,2
70,9
Выход на нитрат,
% от
теоретического
34,7
35,5
KNO
Таблица 47
(11 г м-ксилола, 10 г KNO, и BFa, получеавый из 40 г
опьпа
1
2
Максимальная
температура
экзотермической
реакции, °С
35
35
Выход моноявтроксилола
г
3,6
3,4
% от
теоретического на взятый
нитрат
23,8
22,6
Выход динитроксилола
г
i,i
1,3
% от
теоретического на взятый
нитрат
5,6
6,6
455
Получение динитропроизводных
Для выяснения возможности введения в ароматическое
ядро нескольких нитрогрупп действием нитратов в присутствии
BFg проведены также опыты по нитрованию азотнокислым
калием нитробензола.
В смесь 0,8 моля нитробензола и 0,25 г КЖ)в при
перемешивании в течение 3 час. пропускали фтористый бор,
полученный из 20 г CaFg и 10 г В*О*. По окончании опыта жидкий слой
сливали, осадок в колбе промывали эфиром и эфирный раствор
присоединяли к жидкому слою. После сушки над хлористым
кальцием из жидкости отгоняли эфир, а затем непрореагиро-
вавший бензол; остаток охлаждали, и полученная при этом
твердая масса перекристаллизовывалась несколько раз из
спирта. В результате этой обработки получен динитробензол в
виде кристаллов с т. пл. 87—88°.
Таблица 48
3
1
2
3
(яитроваяие нитробензола KNO# в
Температура банн,
"С
Без нагревания .
95—100
95—100
Получено дянит-
робензола
г
0,5
3,2
3,8
% от
ретического на
взятый бензол
0,36
2,3
2,8
присутствии ВРз)
Получено обратно
нитробензола
г
98
94
93
% от
взятого
бензола
98
94
93
Выход динитро-
бензола на
прореагировавший
нитробензол, %
от теоретического
18,4
39,2
39,9
Ь в у
g Е-
Ж!
1,2
7.6
9,1
Опыты показали (табл. 48), что при проведении нитрования
без нагревания извне, т, е. при температуре реакции
25—30°, получаются лишь незначительные выходы динитро-
бензола (1,2% на нитрат и 18% на прореагировавший
бензол). При применении внешнего обогрева (температура бани
95—100°) выход повышается (максимальный выход 9% на
иитрат и 40% на прореагировавший бензол).
Полученные результаты указывают, что при нитровании
KNO* в присутствии BF@ введение второй нитрогруппы в аро-
456
матическое ядро протекает значительно труднее, чем введение
первой.
§ 4. Механизм нитрования ароматических соединений
неорганическими нитратами в присутствии активаторов
(А1С1, и ВРз)
Автором настоящей книги выдвинута следующая трактовка
механизма реакции нитрования ароматических соединений
неорганическими нитратами в присутствии А1С1з, сближающая
эту реакцию с реакцией Фриделя — Крафтса. Принимая
представление Ганча о структуре азотной кислоты, можно
предположить существование равновесия между псевдоформой и
истинной формой соли азотной кислоты, причем равновесие сдвинуто
Б сторону истинной формы, т. е. последняя находится в
преобладающем количестве
О
О N
О
NaO
Проведение реакции в присутствии А1С1* изменяет условия
равновесия, сдвигая его в сторону образования псевдоформы
благодаря тому, что эта форма способна реагировать с
активированной хлористым алюминием молекулой бензола (с ком-
шлексомСбНбА1С1зН). При взаимодействии псевдоформы нитрата
с этим комплексом водород из внешней сферы комплекса
переходит к кислороду нитрата, связанному с азотом семиполярной
связью [уравнение (2)]; возникающий при этом продукт
присоединения в дальнейшем распадается по уравнению (3) с
образованием нитропроизводного
Na
О
О N
О
№0 —N
CgHg + A1C1, -> (QHtAlCl,) H (1)
NaO — N<(* + (СбН#А1С1,)Н -^ (С,ЩА1СЦ)— З—ОН (2)
- N—OH -^ C,H,NO, 4- A1C1%OH+ NaCl (3)
457
Нами также экспериментально проверена возможность
протекания реакции и по другой схеме, в основу которой положено
допущение об образовании в системе свободной азотной кислоты
вследствие гидролиза AlClg под действием влаги, проникающей
в реакционный сосуд извне, и последующего разложения
нитрата действием НС1. В этом случае нитрующим агентом могла
бы быть азотная кислота
А1С1, + ЗНОН -* А1(ОН)з + 3HCI; \
НС1 + КЛО, -» HNO, + KC1; ) II
Для выяснения правильности схембс (II) проведены опыты
по нитрованию ароматических соединений нитратами в
присутствии НС1. Реакционную смесь (1 моль бензола+ 0,5 моля KNOa)
насыщали сухим хлористым водородом при 40—45°, т. е. при
той температуре, которая создается в реакционной смеси при
проведении реакции в присутствии А1С1з (без нагрева извне).
Опыты показали, что в присутствии НС1 образуются лишь
незначительные количества нитробензола.
В другой серии опытов реакцию проводили с применением
AlClg в качестве активатора и с одновременным насыщением
реакционной смеси хлористым водородом. Оказалось, что
введение хлористого водорода значительно понижает выход
нитробензола. Таким образом, опыты с насыщением реакционной
смеси хлористым водородом показали, что принятие в данной
реакции в качестве нитрующего агента свободной азотной
кислоты, образующейся из нитрата, не соответствует
действительному течению реакции.
Механизм нитрования азотнокислыми солями в присутствии
BF@ протекает, по нашему мнению, согласно схеме,
предложенной нами для нитрования в присутствии AlClg. Например,
для случая нитрования бензола при помощи K.NO* реакцию
можно представить следующим образом.
Как показывают уравнения I и III, механизм реакции в
присутствии активаторов (BFs и AlClg) состоит из следующих
стадий:
31 А. В. Тодчиев
458
1) переход истинной формы азотнокислой соли в
псевдоформу;
2) образование комплексного соединения нитруемого
вещества с активатором;
3) образование тройного комплекса ароматическое
соединение — активатор — нитрат;
4) разложение тройного комплекса, приводящее к
образованию нитробензола и регенерации активатора (при этом в
качестве одного из продуктов реакции выделяется гидрат окиси
металла, нитрат которого является нитрующим агентом).
к
С*Н
О
ON
О
КО
о
*Н,
КО
BF,
о
(2)
(3)
QH5NO2 + BF, + КОН (4)
На основании полученного нами экспериментального
материала можно прийти к следующим выводам об особенностях
метода нитрования неорганическими нитратами.
1. Нитрование азотнокислыми солями в присутствии
активаторов (А1С1зи ВРз) протекает с достаточно хорошими
выходами, причем непрореагировавшее ароматическое соединение
может быть легко регенерировано посредством отгонки из
реакционной массы.
2. Из нитратов наиболее активен AgNOa; менее активные
нитраты щелочные металлов (KNOs и NaNOs) дают, однако,
удовлетворительные результаты. Так, например, при
нитровании бензола KNOs в присутствии AlCls получен выход иа
прореагировавший бензол 70% (38% на нитрат); с NaNOs
выход составляет 73% (34% на нитрат). Менее активны нитраты
аммония, бария и свинца.
влорыстьы* нитрх^ол* 450
3. Из катализаторов наиболее активны AlClg и BF,
следний несколько уступает А1С1* в активности), менее актируя
FeClg и мало активен как катализатор SiCl*.
4. Реакция нитрования азотнокислыми солями протекает
более удовлетворительно при нормальной температуре и избыт»
ке ароматического соединения, которое играет роль
растворителя; нитрат применяется в твердом виде, поэтому при
проведении реакции необходимо перемешивание.
5. Нитрование неорганическими нитратами не
сопровождается реакцией окисления.
Ни в одном опыте по нитрованию ароматических соединении
неорганическими нитратами (но нашему методу) образования
продуктов окисления не обнаружено. Этот метод более удобен,
чем нитрование органическими нитратами, при котором обра*-
зуются побочные продукты вследствие разложения нитрата
при взаимодействии с нитруемым соединением.
Вопрос практического применения нитрования
неорганическими нитратами требует дальнейшей разработки.
Хлористый нитрил ClNOg можно рассматривать как хлоран-
гидрид азотной кислоты.
Хлористый нитрил получают следующим образом [32]:
к 400 г дымящей азотной кислоты при 0* добавляют по каплям
644 г 30%-ного олеума, а затем 370 г хлорсульфоновой кислоты
в течение 12 час; выдерживают 1 час при 20°. В приемнике
(охлажденном сухим льдом) собирают хлористый нитрил с
т. кип. 15—16*. Выход продукта 91% от теории.
Хлористый нитрил можно применить для нитровавия
ароматических соединений средней активности, обычно в присутствии
катализаторов (HF, A1C1@, BFa). Высокоактивные
ароматические соединения сильно окисляются хлористым нитрилом;
так, фенол дает следы о- и п-нитрофенола, анизол — следы
о-нитрофенола, дифениловый эфир— 7% 4гнитродифенилового
эфира, N-диметиланилин образует смолу, нафталин дает
1-яитронафталинс выходом 26—31% от теории. Прв нитровании
31*
460 Дигрфоедние нитрдмм%л<м и
нафталина получается смесь ос-нитронафталина и ос-хлорвитро-
иафталина [32].
Бензойная кислота, хлорбензойная кислота, п-толуиловая
кислота, нитробензол, п-нитротолуол, ацетофенон и бензаль-
дегид с хлористым нитрилом не реагируют.
Нитрование хлористым нитрилом Прайс и сотрудники [33]
осуществляли следующим образом. К 0,2 моля ароматического
соединения в 100 мл CS* прибавляют 0,25 моля А1С1з при 0°
и 0,25 мола хлористого нитрила. Перемешивают 1—2 часа при
0° и выдерживают при 20° до исчезновения хлористого нитрила.
Выливают на лед и получают из бензола нитробензол с выходом
27—35% (без катализатора; после смешения всех компонентов
кипятят 12 час). При действии катализатора HF или AlClg
и соотношении реагентов 0,5 моля бензола + 0,55 моля
хлористого нитрила + 250 мл жидкого HF выход нитробензола
достигает 70—89%.
Из толуола получают о- и п-нитротолуол (47 и 24%), из бром-
толуола—2- и З-нитро-4-бромтолуол (23 и 16%), из бромбензо-
ла — п- и о-нитробромбензол (67 и 8%), из дихлорбензола —
2,4-дихлорнитробензол (31%) и из салициловой кислоты —
5-нитросалициловую кислоту (58%).
ЛИТЕРАТУРА
1. J. Meisenheimer. Ann., 355, 284 (1908).
2. J. T h i e 1 е. Бег., 33, 670 (1900).
3. A. An g el i, Z. Alessandri. C, I, 420 (1911).
4. M. Col on n a. C. A., 754(1947).
5. Амер, патент 2416974; С. А., 3485а (1947).
6. F. Francis. Ber., 39, 3798 (1906).
7. Т. Н. В и 111 e r. Ber., 39, 3804 (1906).
8. R. WilsttSter. Ber., 42, 4151 (1909).
9. A. Picket, E. Kb о tin sky. Ber., 40, 1163 (1907).
10. O. Witt, A. U term a nn. Ber., 39, 3901 (1906).
11. В. А. Измаильский, А. И. Козин. Докл. АН СССР,
28, 621 (1940).
12. С. Ing old, A. Lap wort, G. В en ford. J. Cbem. Soc,
1959 (1931); 918 (1938).
12a, W. D. Emm one, I. R. Freeman. J. Am. Cbem. Soc., 77,
4387(1055).
461
126. W. D. Emmons, I. R. Freema'n. J. Am. Chem. Soc., 77, 4391,
(1955).
12в. W. D. Em mo ng, I. R. Free ma n. 1 Am. Chem. Soc, "77,
4673 (1955).
13. M. И. К о н о в а л о в. ЖРХО, 26 (2), 61 (1894).
14. М. И. К он о в а л о в. ЖРХО, 33, 393 (1901).
15. С. С. Н а м е т к и н. Избранные труды. М., Изд-во АН СССР, 1949.
16. М. И. Коновалов. ЖРХО, 39, 109—41(1906).
17. J. В. М en k e. Rec. trav. chim., 44, 141, 269 (1925).
18. G. В а с b а г а с h. J. Am. Chem. Soc, 44,1522 (1922); Chem. News,
142, 305 (1931).
19. G. В а с h а г а с h. Rec tray, chim., 50, 732 (1931); 52, 413 (1933).
20. G. В а с h а г а с h. Ber., 64, 2136 (1931).
21. L. Spiegel, H. Haymann. Ber., 59, 202(1926).
22. J. A. N i en w 1 a n d, D о r r i s, T. J. S о w a. J. Am. Chem.
Soc, 56, 2698 (1934).
23. J. A. Nienwland, С r ox all, T. J. Sowa. J. Am. Chem.
Soc, 56, 2054 (1934).
24. J. A. Nienwland, SI an on a, T. J. Sow a. J. Am. Chem.
Soc, 57, 1547 (1935).
25. T. J. Sow a, H. D. H in ton, J. A. Nienwland. J.Am.
Chem. Soc, 54, 3694 (1932).
26. H. D. H i n t о n, J. A, Nienwland. J. Am. Chem. Soc, 52,
2893 (1930).
27. J. A. Nienwland. J.Am. Chem Soc, 52, 1018(1930).
28. Ю. C. 3 а л ь к и н д и др. Тр. Ленингр. хим.*технол. ин-та, II,
21 (1935).
29. J. A. Nienwland. J. Am. Chem. Soc, 56, ИЗО, 1786 (1934).
30. И. А. Ротенберг, М. А. Фаворская. Журн. общ.
химии, 4, 185 (1936).
31.J. Meerwein, Vossen. J. pr. Chem., 14i, 149 (1934).
32. W. S t e i n k о p f, M. К n h n e 1. Ber., 75, 1323 (1942).
33. С. С Price. J. Am. Chem. Soc, 75, № 13, 3277 (1953).
ИМЕННОЙ УКАЗАТЕЛЬ
Адаме (Adams A.) 47
Алания В. П. 15, 166, 304
Александер (Alexander L. G.) 14,
273
Алессандри (Alesaaadri Z.) 283
Аллен (Allen G. F.) 344
Анджели (Angeli A.) 283
Анри (Henry L.) 221
Аншютц (Anaobutz R.) 115
Арналль (Amall F.) 96 ел.
Аубертейя (Aubertein P.) 38
Базани (Bazaay H. С.) 53
Байер (Ваеуег А.) 233
Баландин А. А. 335
Бальцеркевич (Balzerkewiez H.)
349, 370, 399
Бамбергер(ВашЬег&ег Е.) 51,56,
74, 219, 325
Банер (Bahner С. Т.) 292 ел.
Барышникова А. Н. 87, 402 ел.
Баттегей (Battegay M.) 348, 399,
401
Бахараш (Bacharach С.) 437
Бахман (Bachmann G. В.) 270 ел.,
302, 311
Бахман (Bachman W. Е.) 69
Бедткер (Boedtker) 121
Бейлыптейп Ф. Ф. 233
Белл (Bell F.) 28
Беннет (Bennet G. М.) 7, 154,
171 ел., 179, 196, 207 ел.
Вер Г. Я. 161, 164 ел.
Беркенгейм А. М. 129
BeptOH (Burton) 281
Ветерс (Boters О.) 9, 81
Бибишев В. П. 27
Бильц (Biltz H,) 343
Блехта (Blechta F.) 71
Близнюков В. И. 15, 366
Влит (Blyth J.) 301
Блэккол (Blaekall E.) 188
Блэсет (Blacet F.) 335
Богданов М. И. 409 ел.
Боннер (Bonner W. Н.) 36
Бойлер (Bonner Т. С.) 164
Боуэн (Bowen D. М.) 37
Бранд (Brand G. D.) 154, 172
ел., 207, 208
Браун (Brown H. С.) 36
Бренстедт (Bronsted I. N.) 139 ел.
Бликенстафф (Blickenstaff R.T.)277
Бриаер (Briner E.) 207
Бродер (Broders) 73, 74
Бруннер (Впшпег Р.) 30
Бугай П. М. 57
Буис (Buois J.) 300
Буллок (Bollock J. L.) 14, 277
Бутлер (Buttler Т. Н.) 421
Бэрд (Beard H. Сг.) 40
Вайбель (Veibel S.) 23, 98 гл.
Валяшко Н. А. 15, 366
Вандербильт (Vanderbilt В.) 14,
222 ел., 268
Дленной
Варма (Уагша) 358
Вейсвейлер (Veisweiller G.) 35
Векчи (Veeehi M.) 335
Вернер (Werner A.) 122, 130
Вестгеймер (Westheimer F. U.)
168 ел., 208
Виатт (Wyatt) 309
Внбо (Wibaut J. Р.) 66, 152
Виланд (Wieland Н.) 15, 111 ОД.,
118, 123, 125 ел., 202, 293,
336, 346 ел.
Вилл (Will W.) 34
Вильгельм (Wilhelm R. Н.) 15, 411
Вильдман (Wildman W. С.) 295
Билыптеттер (WilstatterR.) 421,422
Вилльямс (Williams G.) 154, 171,
172, 207, 208
Виньон (Vignon L.) 72, 73
Внтт (Witt О.) 30, 54, 347
Володарский А. М. 86
Вольфенштейя (Wolfenstein R.) 9,
81
Ворожцов Н. Н. 160 ел.
Ворожцрв Н. Н. (младший) 104
Вреден 233
Вюрц (Wiirz A.) 39
Газенбах (Hasenbach) 344
Гайман (Haymann H.) 437
Гайтингер (Haitlnger H.) 299
Гальбан (Halban Н.) 133, 148 ел.
Гаясбергер (Hunsberger I. M.) 24
Ганте (Gante J.) 349, 398
Ганч (Hantzsch A.) 130—136, 140,
203, 204, 206, 207
Гезернягтон (Hetherington I. A.)
142 ел., 204
Гейм (Heim F.) 291
Гейяле (Heinle E.) 61, 354
Гейсман (Geisman С.) 37
Гвбсмая (Hibsmann H.) 281
Гядлесяж (Gillespie R. J.) 196
Гильберт (Gilbert A.) 115
Гиршбах (Gierschbach J.) 120
Голд (Gold V.) 171
Голлеман (НоНешадп А.) 21, 111
Гофман (Hofman Д.) 301
Гофмейер (Hoffmeier) 340
Грехем (Graham) 309
Гроггинс (Grogging P. H.) 208
Гульден (Goulden L. D. S.) 195
Гуревич К. 49
Гутри (Guthrie F.) 299
Гюбнер (Hiibner H.) 28
Гюсси (Hussy H.) 360
Данциг (Danzig Н. М. ) 14, 275 ел.
Деверяье (Desvergens L.) 74 сд.
Дегеринг(Degering Е.) 219,279ел.,
302
Деккер (Decker H.) 47
Демьянов Н. Я. 15, 338 ел.
Джонс (Jones R. N.) 200
Джонсон (Johnson К.) 219
Дзудзуми (Tsutsumi S.) 82
Долгов Б. И. 35
Дорноу (Dornow A.) 293, 295
Дрэйк (Drake N. L.) 114
Дэвис (Davis Т. L.) 81, 83 ел.
Женекая (Genequand P.) 65, 66
Заболоцкий Т. В. 335
Забродина А. С.118,120,129,202,
246, 248, 25
Захаров А. И. 76 мл.
Зияия Н. Н. 5
Измаильский В. А. 59, 425
Ингольд (Ingold С. К.) 170, 183,
188, 189, 194ел.,205 ел., 425
Иоффе И. С. 30
Кайт (Kite H. Т.) 293
Калинина В. С. 86
Кама* Г. X. 67
Караш (Kharasch M. S.) 168 ел., 208
Карлсон (CarlsonG.H.) 119,124ел.,
202, 339
Карташев А. В. 93 ел.
Кауфман (Kaufmann A.) 47, 361
464
Кедров Б. М.
Кекуле (Keknle A.) 112
Кеньон (Кепуоп I.) 28
Керн (Kern) 348
Кесслер (Kessler A.) 120
Клеменц (Klemenz A.) 150 ел., 154
К лоза (Klosa I.) 48
Клузнус (Clusius К.) 335
Коб (Kobe A.) 32, 35 ел., 37
Коваленко А. Г. 67
Коган И. М. 43
Коген (Cohen F. Н.) 66
Козин А. И. 59, 425
Колер (Kohler E. Р.) 114
Коновалов Д. П. 299
Коновалов М. И. 12, 13, 49, 51,
232 ел., 239 ел., 245 ел.,
248, 261 ел., 275 ел., 278,
296 ел., 368, 394
Конрад (Connerade E.) 59, 60
Кормак (Karmack M.) 69
Косолапов (Kosolapoff G. М.) 25
Костаческу (Costachescu N.) 248
Котрел (KottreH) 309
Кришамурти (Krishanmrty) 358
Кроссле (Crossley A. W.) 36
Курбатов А. А. 233
Кутепов Д. Ф. 43
Кучумова Н. А. 35
Лайв (Lyne M.) 200
Ламбертон (Lamberton А. Н.)178,
291 ел.
Ланг (Lang Fr. M.) 25, 26
Ланц (Lanbz R.) 139 ел., 204
Лаптев Н. Г. 89, 91 ел., 158
Лауэр (Lauer W. М.) 200
Лауэр (Lauer К.) 137 ел., 204, 205
Левн (Levy Norman) 28f, 341 ел.,
413
Левин (Levin Н.) 37 ел.
Легге (Legge D. I.) 24
Лекорш (Lecorche В.) 38
Ленхольд В. А. 43
Либерман (Liebermann С.) 345
Лидс (Leeds A.) 344 ел.
Линденман (Lindenmann L.) 345
Липпинкот (Lippiucot S. В.) 219
Лорнет (Loriette) 37
Лоуэн (Lowen A. M.) 171, 208
Луцкин А. Е. 15, 366
Льюис (Lewis W. К.) 32
Людденс (Liiddens HJ 28
Мак Доуэлл (Me Dowell) 308
Мак-Ки (Me Kee R. Н.) 15, 85,298,
411 ел.
Мак-Нлирли (МсС1еаг1у)276,279ел.,
302
Ман-Кормак (Me Cormack H.) 32,
38, 39, 42
Макаров-Землянский Я. Я. 27
Маяажх 228
Маньковская Н. П. 274
Маркерол (Marqueroll) 37
Марковников В. В. 12, 13, 149 ел.,
233, 238, 240 ел.
Мартинсен (Martinson H). 93, 150,
161 ел.
Мациевич Т. Е. 44
Меиер (Meyer V.) 12, 31, 112
Мейзевгеймер (Meisenheimer J.) 59,.
60, 117, 291, 345
Меландер (Melander L.) 179,
196, 209
Менке (Menke J. В.) 432 ел.
Мессон (Masson J.) 142 ел., 204
Миле (Mills) 32
Миллен (Milien D. J.) 195
Щитчел (Mitchell E. Т.) 14, 277
Мятчерлих 4
Михаэль (Michael А.) 119,124,202,
339
Модик (Modic F. I.) 168
Наметкин С. С. 13, 118 ел., 129,
202, 242 ел., 246, 248 ел.,
394, 427 ел.
Нельтинг (Noelting E.) 37
Дженмо&
465
Нифонтова С. С. 245
Ниши (Niscbi) 41
Нортон (Norton L.) 196
Ньюланд (Nieuwland J. А.) 448
Огата (Ogata) 164
Одда (Oddo G.) 116
Одда (Odda R.) 137 ел., 204 ел.
Орлов Е. И. 79 ел.
Натек (Patek К.) 71
Паттерсон (Patterson) 274
Перекалин В. В. 215, 294
Пиаяка (Pianka M.) 53
Пиггот (Piggott H. А.) 292
Пикте (Pictet A.) 65, 66, 423
Пинк (Pink L. А.) 396
Пирсон (Pierson E.) 281
Поддай (Pollack М.) 272
Поллард (Pollard) 309
Пони (Poni P.) 248
Пономаренко А. А. 41, 44
Пончио (Ponzio) 266
Прайс (Price С. С.) 460
Пфейфер (Pfeifer P.) 122
Пфистер (Pflater К.) 79
Райт 321, 323, 325
Рая (Rahn F.) 127
Ревуф (Renonff N.) 36
Ризинг (Rising A.) 51
Рилей (Riley) 304 ел.
Ринке (Rinkes I. J.) 358
Роберг (Roberg F.) 293, 295
Робинсон (Robinson P. L.) 197,
199, 209
Розе (Rose J. D.) 289
Рюст (Riist) 219
Саккелариус (Sakkelarious E.) 293
Сапожников А. В. 152 ел., 164
Сейгль (Seigle) 275, 282
Семенов Н. Н. 210, 312
Сергиевская С. И. 27
Сидоренко К. В. 338 ел.
Симкинс (Simkins R.) 172
Симой (Simon E.) 301
Скейф (Scaife С. W.) 342
Скрауп 360
Слон (Slon) 380 сл^
Сыарг (Smart G. N. R.) 321, 323
Смирнов В, В, 265
Смит (Smith A.) 341
Смолла (Smolla E.) 348
Снайдер (Snider Н.) 231 ел.
Соколова А. М. 252
Сопова А. С. 294
Спасокукоцкий Н. С. 201
Сусц (Snsz В.) 207
Сущик Р. Я. 247
Тейлор (Taylor E. С.) 200
Терентьев А. П. 166
Тиле (Tiehle J.) Ill, 337,*418
Титов А. И. 8, 15, 86 ел., 91 ел.,
154 ел., 177, 208, 242,253сл.,
261, 301 ел., 305 ел., 311 ел.,
376 ел., 394, 402, 40»
Токи (Toki H.) 41
Томас (Thomas) 308 ел.
Томливсон (Tomlinson) 208
Томсон (Thomson R. Н.) 58
Топчиев А. В. 15, 166, 199, 380,
381, 410
Торн (Thorn С." D.) 200
Тропов Б. В. 67, 118 ел., 129, 161
Тютюнников Б. Н. 274
Уоррел (Worral D.) 293
Уорстолл (Worstall R. А.) 237
Уотерс (Waters W. А.) 198, 209
Урбанский (UrbanskyM.) 380 ел.
Урецкая Г. Я. 27
Ури (Uri N.) 210
Усанович М. И. 144 ел., 204 ел.
Утермав (Utennann A.) 54
Ушаков С. Н. 40
Уэлш (Walsh A. D.) 335
Фармер (Farmer E.) 136
Фергюсон (Pergnsson L. N.) 150
468
указатель
Фингер (Finger G. G.) 40
Флашлендер (Flaschlander J.) 34
Фрейдберг Е. Н. 40
Фрелих (Frolich) 413
Френсис (Francis F.) 419 ел.
Фрид бур г (Friedburg) 345
Фридль (Friedl F.) 31,45,370, 376
399
Фриман (Freeman I. R.) 427, 428
Ханш (Hansh G.) 35
Хаылш (Hamlin W.) 231 ел.
Хаун (Haim J. W.) 35
Хельмкамп (Helmkamp G.) 35
Хис (Heath R. Н.) 289
Ходж (Hodge Е. В.) 14, 268,
Ходжсон (Hodgson Н. Н.) 40
Холево Н. А. 80
Холстед (Hoisted С.) 178
Хотжвский (Khotinsky Е.) 432
Хьюз (Hughes E. D.) 171, 188
Хэсс (Haas H. В.) 14, 219, 222,
273 ел., 268, 275, 281, 282,
292, 294, 304 ся., 381
Цан (Zahn H.) 39
Шааршмидт (Schaarschmidt A.) 153
ел., 164, 340, 348, 349, 370,
399, 405 ел.
Шедэн (Ghedin J.) 206
Шеллер (SchoIIer R.) 150 ел., 154
Шеффег (Shaeffer Е.) 130,132ел.,
136
Шмидт Г. A. (Schmidt G. А.) 29
Шмидт Ю. (Schmidt J.) 29, 61,
128, 338, 354
Шнайдер Г. С. 166
Шорыгин П. П. 15, 199, 252, 349,
380 ел.
Шпигель (Spiegel L.) 437
Шшшдлер (Spindler P.) 20
Штадлер (Stadler О.) 31
Шульц (Schnltz G.) 34
Щитов Н. В. 264
Эвелл (Ewell) 280
Эйзенбранд (EisenbrandJ.) 148 ел.
Эйлер (Euler H.) 205
Эйтингтон И. И. 80
Эммонс (Emmons W. D.) 427, 428
Эиц (Enz W.) 79
Эфрос А. С. 29
Юрьев Ю. К. 63
Явлинский Т. Д. 274
Якубович А. И. 215
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адипиновая кислота, получение
245, 261, 300
Азота двуокись
взаимодействие с альдегидами
308 ел.
взаимодействие с непредельными
углеводородами 309
нитрующий агент 15, 261 ел.,
286, 334 ел.
Азота окислы как катализаторы
150
Азота пятиокись (азотный
ангидрид),нитрующий агент 16,171
Азотистая кислота как катализатор
10, 67, 93, 162
Азотная кислота, нитрующий агент
170, 179
Ализарин, нитрование 10, 19
Алифатические углеводороды,
нитрование 376
Алкилнитраты, получение 16
Алюминиевые квасцы в смеси с нн-
Фратом алюминия, нитрующий
агент 429
Амилен, нитрование 299
2-Амннобензотиазо л, нитрование
418
2-Амино-8-нитробензотиазол,
получение 418
Аминоспирты, получение и при-
меиеиие 225, 226
4-Аминохинол ин, нитрование 48
Амины, нитрование 25, 26, 320 ел.
326 ел.
Аммиачная селитра, нитрующий
агент 9
Аиизидиды бензойной кислоты,
получение 421
о-Анизидин, нитрование 43
п-Анизндин, нитрование 421
Анизол, нитрование 9, 23, 158,
420, 421, 423, 435, 459
Анилин, нитрование 25, 166,
168, 347, 357 ел., 375, 432,436
Анилин, производные, нитрование
54 ел.
Анисовый альдегид, нитрование 420
Антрахинон, нитрование 44, 45,68,
137, 180, 304, 399, 400
Антрахннон, получение 7, 28, 345
Антрацен, нитрование 7, 28,59—61,
79, 117, 158, 345, 353, 374,
424 ел.
Антрацен-ннтроацетат, получение
59
Арилгидроксиламияы, нитрование
159
Ароматические соединения,
нитрование 14, 26
азотной кяслотой 19
в жидкой фазе 334 ел.
Ароматические соединения,
нитрование
кинетика 66—68
468
механизм 71, Ш ел.
нитратами металлов428 ел.,437
нитрующей смесью 19
скорость нитрования галоидо-
производных 68
хлористым нитрилом 459 ел.
Ароматические углеводороды,
конденсация с олефинами 447
Ароматические углеводороды,
нитрование разбавленной азотной
кислотой 238
Ацетальдегид, нитрование
двуокисью азота 308 ел.
4-Ацетамидодифенилсульфид,
нитрование 57
Ацетанилид, нитрование 41—43,54,
56, 166, 347, 359 ел., 375, 423
Ацетилен, нитрование 14, 300
двуокисью азота 309
Ацетиленовые углеводороды,
нитрование 14
Ацетил-Р-нафтиламия, нитрование
59
Ацетофенон, нитрование 24
надазотистой кислотой 198
Ацетилнитрат, нитрующий агент
8, 347, 422 ел.
Ацетил-о-толуидин, нитрование 424
Ацетоцианг идриннитр ат,
нитрующий агент 9
Бензальдегид, нитрование 40, 85,
420, 435
Бензальдифенилмалеид,
нитрование 117
Бензальдоксим, нитрование 266
Бензальфталимидин, нитрование
117
Бензиловыйспирт,нитрование
азотной и серной кислотами 166
Бензилхлорид, нитрование 423,424
Бензоилнитрат, нитрующий агент
8, 9, 419—422
Бензоилхлорид, нитрование 420
Б ензойная кислота
нитрование 54, 70,86, 87, 180,.
423, 435
образование 40, 146, 239,
252, 256, 259, 348, 366, 37»
Бензол, нитрование 7, 9, 10,.
15, 20, 21, 31 ел., 66—72, 76,.
81, 90, 115, 118, 120 ел., 159»
180, 181, 198, 346, 348, 363 ел.,
373, 374, 379,396,397,400,405,
407 ел., 419, 423, 425, 433, 439,
445, 449, 455, 458
механизм 74
окислами азота 344
при повышенной температуре*
345
схема 76
Бензолдиазонитрат, получение 347
Бензол сульфокислота, нитрова-
н:э 163
Бензолсульфоновая кислота,
нитрование 180
Бензотиазол, нитрование 48, 418
Бензотрифторид, нитрование 40
Б ензофенон, получение 260
Бифенилоксид
бромпроизводные, нитрование 166
нитропроизводные,нитрование!66
Борнилен, нитрование 296, 297
Бром как катализатор 272
Бромбензол, нитрование 20, 85,.
349, 420, 458
Бромтолуолы, нитрование 41* 458:
9-Бромфенантрен, нитрование 62
Бутадиен, нитрование 300, 343
Бутан, нитрование 12, 268, 270,
272, 276, 413
трет. Бутилбензол,нитровавие 35,
36
Бутилен, нитрование 126, 342
трет. Бутилксилол, нитрование 239
Бутилнитроамины, получение 323
трет. Бутилтолуол, нитрование 235
Вавилип, нитрование 420
469
Гваякол, нитрование 53
Гексагидропсевдокумол,
нитрование 243
Гексаметилен (гексанафтен), нитро-
ваше 241, 244, 245
н. Гексан, нитрование 233, 234
Гексанафтен, нитрование 241
Гексанитроциклогексан, получение
346
Гексанитроэтан, получение 288
Гексатриаконтан,нитрование 13,247
Гексилен, нитрование 339
а. Гептан, нитрование 24, 234, 242,
284, 385—389, 395, 432—434
Гидриндан, нитрование 249
Гетероциклические
соединения,нитрование 31
азотной кислотой 19
в жидкой фазе 344
нитрующей смесью 19
Гликоля динитрат, получение 112
Дейтеробензол,нитросоединения200
Декалин, нитрование 249
Диазобензоилнитрат,получение 347
Диалкиланилины, нитрование 55
Диаллил, нитрование 339
Диамид имино-бис-уксуснон
кислоты, М-нитропроизводные325
Диарилсульфат, нитрование 41
Диапетилализарин, нитрование 28,
29
Диапетилортоазотная кислота
(ацетилнитрат), нитрующий
агент 64—66, 437
Диапетобензидин, нитрование 30
Дибензантроа, нитрование 30
Дибензил, нитрование 436
Днбензпирен, нитрование 30
2,5-Дибромнитробензол,
нитрование 168
Ди-п. бутиламин, нитрование 326
1,4-Ди-трет. бутилбензол,
нитрование 24
2,6-Ди-трет. бутилфенол,
нитрование 53
Дигидронитроантранол,
получение 117
Диены, присоединение нитрооле-
финов 295
Диизоамил, нитрование 234, 262,
431, 432
Диизобутил, нитрование 234
Диизобутилен,нитрование 297—300
Диизопропил, нитрование 234
Диметиламин, нитрование 325, 326
Диметиланилин, нитрование 25,26,
158, 348, 349, 358 ел., 375,434
надазотистой кислотой 198
N-Диметиланилин, нитрование 56,
459
2,3-Диметилбутан, нитрование 275
2,3-Д иметил-2-бутен, нитрование
282
2,3-Диметил-2,3-динитробутан,
получение 275, 282—284
3,4-Диметил-3,4-динитрогексан,
получение 283, 284
2,3-Диметил-1-нитробутан,
получение 276
2,3-Диметил-2-нитробутан,
получение 275, 276
2,7-Диметилоктан, нитрование 242
N ,М-диметил-п-толуидин,
нитрование 359
2,4-Диметилхинолин, нитрование 48
4,6-Диметилхинолин,нитрование 48
1,5-Диметоксинафталин,
нитрование 58
2,4-Динитроанизол, нитрование 163
1,5-Динитроантрахинон, получение
45
1,8-Динитроантрахинон, получение
45
Динитроантрацен, получение 60,61
2,7-Динитроантрацен, получение 7
9, 10-Динитроантрацен, получение
353, 374
470
Динитробеизидин, получение 30
2,4-Динитробензойная кислота,
получение 35
м-Динитробензол, получение 21,
32, 81, 87, 138, 200
о-и п-Дивитробевзолы, получение
73, 75, 81, 138, 345
Динитробевзолы, получение 32,33,
84, 86, 143, 348, 404,453
1,1-Динитробициялогексил, полу-
чевие 284
2,3-Дияитро-4-бромтолуол,
получение 41
1,4-Динитробутан, конденсация с
формальдегидом 224
2,3-Динитробутан, получение 283,
413
1,4-Динитро-2-бутен, получение
343
4,6-Динитро-2-трет. бутилфенол,
получевие 53
1,6-Динитрогексан, кондевсацвя с
формальдегидом 224
2,2-Дивлтрогептаи, получевие 394
5,6-Джиитродекаи, получение 284
Днвитродиацетобензидин, полу-
чевве 30
п ,п'-Двнитроднбензил, получение
436
5,10-Динитродибензпирен,
получение 30
9,10-Дивитро - 9-10-дигидроантра-
цен, получение 345
Динцтродиизобутил, получение
234
2,4-Дввитро-М-диметиланилин,
получевие 56
2,4-Двнитродиметиланилив, но-
лучевве 348, 359
2,3-Дввитро-2,3-диметилбутаа,
получевие 339, 341
2,4-Д нвитро-2,3-димети лпеитаи,
получение 292
2,2'-Динитродифенил, получение 28
2,4'-Динитродифенил, получение 28
4,2'-Динитродвфенил, получение 28
4,4'-Динитродифеннл, получение28
м-Динитродифенил, получение 30
п,п'-Динитродифенил, получение 28
2,4-Дннитродифениламин,
получение 43
п,п' - Динитродифениламиноксид,
получение 348
4,4^Динитродифенилмочевива, по*
лучение 43
п,п'-Динитродифенилмочевина,
получение 57
3,4^-Динитродифевилуксусная
кислота, получение 24
1,2-Динитро-1,2-днфеннлэтан,получение 284
2,4'Динитро-1,3-дихлорбензол,
получение 39
2,6-Динитро-1,4-Днхлорбензол, gq_
лучение 39
3,5-Д инитро-1,2-дихл ор бензол, по -
лучение 39
4,6-Динитро-1,3-дихлорбензол,
получение 39
1,2-Д инитроизобутан, получение
341
Динитроизооктан, получение и
свойства 393
2,4-Д ивитроизопропидбевзо л
получение 35
Динитрокапрвлев, получевве 300
3,6-Динитрокарбазол, получение 6&
Динитрокарбазолы,получевие 62,63
2,6-Динитрокрезол, получение 404
4,6-Динитро-м-крезол, получение
355, 375
Диннтро-о-крезол, получение 23
Диннтрокрезолы, получение 23,
348, 355, 375, 403
Динитро-м-ксилол, получение 369,
444
2,4-Диннтро-м-ксилол, получение
22
471
4,6-Д ииитро-м-кснлол
нитрование 163
получение 21, 22
3,4-Дипитро-о-ксилол,получеаие 22
4,5-Динитро-о-ксилол, получение
22, 36
4,6-Динитро-о-ксилол,получение 22
36
2,3-Диннтро-п-ксилол, аолучение
22, 37 ел.
2,6-Динитро-п-ксило л, получение
22, 37 ел.
Динитроксилолы, нитрование 170
Дипитромезитилен
нитрование 170
получение 22, 236
2,4-Динитрометиланилин,
нитрование 26
2,3-Динитро-2-метилбутан,
получение 248
3,5-Дннитро-З-метилгексаи,
получение 287
Динитро-2-метилтиофен, получение
48
2,4-Динитромонометиланилин,
получение 348
1,3-Диннтронафталин, получение 83
1,5-Динитронафталин, получение
27, 43, 44, 83, 117, 351
1,8-Динитронафталин, получение
27, 43, 44, 117, 351
Динитронафталнны, получение 345
4,5-Динитро-1-нафтонная кислота,
получение 44
2,4-Д инитр 0-%-н афто л, по л у чение
7, 27, 44, 79, 84, 92, 347, 420
1,6-Динитро-Р-нафтол, получение
356 ел., 375, 420
2,4-Д инитро-N - нитрозомономети ла-
нилин, получение 359
Р ,Р^Динитрооксндиэтил аммоний-
хлорид, нитрование 329
р,Р\Цииитроокснднэтилнитроамин,
получение 326, 329
амин получение 330
Динитрооксидиэтилхлорамищ,
[нитрование 329
Динитропарафипы, получение 231
1,5-Динитропентан, конденсация с
фор мал ьдегидом 224
1,2-Дииитропропаа, получение 341
2,2-Динитропропан, получение 286
Динитропропаны, получение 342;4
2,2-Динитропропандиол-1,3,
получение 224
1,2-Динитротетралин, получение 27
1,3-Динитротетралии, получение 27
симм. Динитротетрахлорэтан,
получение 343
ДинитротиофенЫизомерные,
получение 31
о ,о'-Динитро-п-голиламин,
получение 348
2,4-Динитротолуол,нитрование 405
азотной кислотой 164, 173 са,
2,4-Динитротолуол, получение 21,
405
2,5-Динитротолуол, нитрование 34
2,6-Дннитротолуол, получение 179
3,4-Динитротолуоя, нитровааве 34
Динитротолуолы, получение 146
Динитротолуолы изомерные,
нитрование 34
4,4'-Д инитротрифениламии,
получение 57
2,4-Динитрофенилсульфат,
получение 41
2,4-Диннтрофенилуксусная
кислота, получение 24, 41
2,4-Динитрофенол, получение 23,
25, 41, 69, 81, 163, 354, 357,
358, 360, 375
2,5-Динитрофенол, получение 358
2,6-Дипнтрофенол, получение 258,
*-Динитрофенол, получение 94
Динитрофенолы, получение 7, 74,
78
472
указатель
2,7-Динитрофлуоренон, получение
29
Дипитрофторбензол, получение 39
5,6-Динитрохинолин, получение 47
5,7-Динитрохинолин, получение 31,
47, 360
5,8-Динитрохинолин, получение 47
6,8-Динитрохинолин,получение 31,
47
Динитрохинолины, получение 360,
376
1,2,4-Динитрохлорбеизол,
получение 39
^,6,1-Динитрохлорбензол, получе-
вше 39)
1,2-Динитроцикло1 ексан,получение
341
Динитроэтан, получение 341
-(%,Р-Дипитроэтап, получение 341
2,4-Динитроэтилбензол, получение
35
1,3-Динитро-2-этил-3-метилбутан,
получение 287
«Динитрюры», получение 336, 341
"Р,Р'-Диоксидиэтил аммоний-хлорид,
нитрование 329
2,6-Диоксопиридазин, N-нитроиро-
изводные 325
Ди-п-толилгидроксиламин,
нитрование 347
<%,<%-Ди-п-толилэтнлен, нитрование
116
Дифенил, нитрование 28, 58, 353,
436
Дифениламин, нитрование 347
Дифениламин, апильные
производные, нитрование 43
Дифениламиноксид, нитрование 348
1,4-Дифенилбутадиен, нитрование
337
2,3-Дифенил-1,3-бутадиен,
нитрование 344
;1,4-Дифенил-1,4-динитробутен-2,
получение 337
Д ифени лдинитро д их л ор этан, по -
лучение 343
Д ифенилдинитроэтан ,получение 336
<%,<%-Дифенил-Р,р-динитроэтилен,
получение i!6
«,<%-Д ифенил-р, 0-дихлор этан,
нитрование 343
Дифенилметан,нитрование 166,259,
260, 262, 426
Д ифонилмононитрометан,
получение 260
Дифенилмочевина, нитрование 43,
57
1,4-Дифенил-1-иитробутадиен,
получение 337
Д ифенилнитрозоамин, получение
347
Дифенилнитрометан, получение 260
<%,(%-Дифенилнитроэтилен,
получение 114, 116
<%,(%-Дифенилнитроэтиловый спирт,
получение 114, 116, 127
Дифениловвш эфир,нитрование459
Дифенилуксусная кислота,
нитрование 24
<%,(%-Дифенилэтилен, нитрование
114, 126, 127
1,5-Дифторнафталин, нитрование
58
1,5-Дифтор-4-нитронафталин,
получение 58
п,п' -Дихлоразобензол,
нитрование 57
2,4-Дихлоранилин, нитрование 55
1,5-Дихлорантрапен, нитрование
346
м-Дихлорбензол, нитрование 39
о-Дихлорбензол, нитрование 39
о-Дихлорбензол как растворитель
при нитровании 10
п-Дихлорбензол, нитрование 39,
180
Дихлорбензолы, нитрование 458
N ,М'-Дихлор-М ,М'-динитро-1,2-
473
дяамивоэтав, получение 321
N ,N '-Д ихлор-N ,N '-динитро-1,2-
дидминоэтаи, гидролиз 322
1,5-Дихлор-9,10-динитро4),10-диги-
дроантрацен, получение 346
4,4'-Дихлор-2 -нитроазоксибензол,
получение 57
1,5-Дихлор-9-нитроаитрапон,
получение 346
2,4-Дихлорнитробензол, получение
458
2,5-Дихлорнитробепзол,
нитрование в серной кислоте 168
Дихлорнитропарафины, получение
220
4,6-Дихлор-2-нитрофенол,
получение 433
Диэтаноламии,нитро*авие 325,326,
329
Диэтаволамин, солянокислая соль,
как катализатор 325
м-Диэтилбенэол, нитрование 35
о-Диэтилбеизоя, нитрование 52
Диэтилбензолы, нитрование 49,240
Диэтилбензолфосфат, нитровавие25
1,2-Диэтил-4-нитробешзол,
получение 52
3,5-Д иэтокси-2,6-динитропиридин,
получение 46
3,5-Диэтоксипирндин, нитрование
46
3,5-Диэтокси-2-нитрониридин,
получение 46
Додекан, нитрование 285, 395
Дурол, нитрование 9, 421, 422
Жирноароматические соедцнеаия,
нитрование в жидкой фазе 376
Заместители, изменяющие
скорость иитровадая 163 ел.
Изоамиден, нитрование 114,
Изоборнилан, нитровавие 249
Э2 А. В. Топчиев
Изобутан, нитровадие240, 268,286
Изобутилен, нитрование 1Д6, 299,
339
Изогексая, нитрование 240
Изогептан, нитрование 240
Изокамфаи, яитрование 250
Изонитроизооктан, получение 248
Изонитроциклогексаи, получение
119, 251
Изооктан (2,2,4-триметилпеитав),
нитрование 248, 384 — 386,
432 ел.
Изооктаяафтен, нитрование 428
Изопевтан, нитрование 248, 275,
384, 386 ел.
Изопропилбензол,нитрование 24,35
Изопропилнитроамип, получение
323
Имино-бис-ацетонитрид, N-нитро-
производиые 325
Имино-бис-уксусная кислота,М-ни-
тропроизводные 325
Индол, нитрование 418
Иодбензол, нитрование 349 о-и
н-Йоднитробензолы, получение 349
Камфан, нитрование 250
Камфеш, нитрование 296 ел.
Камфенилан, нитрование 250
Каприлен, нитрование 300
Нарбазол, нитрование 62 ел.
Катализаторы при нитровавии 9,
68 ел., 121 ел., 281, 396 ел., 45б
оежовные 286
хлористоводородный 327
Кетоксимы, нитрование 266
Кетоны, нитрование 10, 277
е-Кислота, нитрующий агент 150
Кислоты карбоновые, нитрование
277
Коричная кислота, нитрование 41,
301
м-Крезол, нитрование 52, 355 сд.,
375
474
указатель
м-Крезода метиловый эфир,
нитрование 100, 158
о-Крезол, нитрование 23, 100
п-Крезол, нитрование 162
Крезолы, нитрование 158, 347
1,3,4-Ксиленол, нитрование 347
Ксилидины, нитрование 421
Ксилилнитрометан, получение 51,
235, 239
м-Ксилол, нитрование 21, 37, 38,
79, 80, 235 ел., 369 ел., 37,5
400, 420, 444, 451
скорость нитрования 68
о-Ксилол, нитрование 22, 36, 37,
52, 240, 400
n-Ксилол, нитрование 22, 37, 38,
235 ел., 240, 400 *щ,
Ксилолы, нитрование 33,49,85, 349
двуокисью азота 344
Кумарин, нитрование 420
Кумарина производные,
нитрование 64
Кумол, нитрование 35
Мезитилен, нитрование 22, 50, 166,
235 ел., 239, 349, 420
Мезонитроантрацен, получение 118
Ментен, нитрование 296, 297
Метан, нитрование 273, 277, 279,
304,382
Метилантрахинон, нитрование 68
З-Метилгексан, нитрование 281
Метилдинитродиоксибензол,
получение 345
4-Метил - 2,6-динитро-№нитро-зомо-
нометиламинанилин,
получение 359
2-Метил-1 ;2-динитропропан,
получение 286
3,4-Метилендиокси-*>_нитро-стирол,
гидрирование 227
Метилендииитроамин, получение
324
Метилен-бис-М-(М-нитро)-ацетаашд,
получение 324
(К- и Р-Метилнафталины,
нитрование 44, 158
Метилнитроамин, получение 321
2-Метил-1-нитробутан, получение
275
2-Метил-2-нитробутан, получение
275, 387
2-Метил-З-нитробутан, получение
387
2-Метил-4-нитробутан, получение
387
З-Метил-1-нитробутан, получение
275
З-Метил-2-нитробутан, получение
275, 276
2-Метил-2-нитропропан, получение
286, 387
трет. Метилнитропиклогексан,
получение 251
Метиловый спирт, нитрование 189'
Метиловый эфир бензойной
кислоты, нитрование 87
Метиловый эфир м-нитробензойной
кислоты, получение 87
Метилпикрамид, нитрование 189
5-М етил-2-тиофенкарбонован
кислота, нитрование 48
N-Метил-п-толилиитроамин,
получение 421
N-Метил-п-толуидин, нитрование
421
Метилуретан, нитрование 178
Метилфенилнитрометан, получение
367, 375
Метилпиклогексан,нитрование 251,
427
О'Метоксиацетанилид, нитрование
26
1-Метокси-2-п-толилсульфамидо-5-
нитробензол, получение 26
1тМетокси-2-п-толилсульфамидо-
бензол, нитрование 26
о^Метоксифенол (гваякол), нитро-
ванже 5S
указатель
4/0
Механизм реакции нитрования см.
Нитрование, механизм реакции
Монодейтеробевзол, нитрование 200
Монодейтеро-м-динитробензол,
получение 200
Монодейтеровитробевзол, получе-
ние 200
Мовометиланилин, нитрование 348
Мововитрокаприлен, получение 300
Моноаитродиизобутил, получение
234
Мовохлоруксусвая кислота как
растворитель при нитровании 146
Мочевина, нитрование 178
Надазотистая кислота, нитрующий
агент 197 ел.
Нафталин, нитрование 7, 10, 15,
26, 27,43, 44, 79,83, 85, 91,
116, 157, 346, 351 ел., 374,
376, 396, 398, 423, 434, 457
двуокисью азота 345
Нафталиндисульфоновые кислоты,
нитрование 141
*-Н афталинсульфоновая кислота,
нитрование 86
Нафталинсульфоновые
кислоты,нитрование 141
Нафталинтрисульфововые кислоты,
нитрование 141
Нафталивтетрасульфововые
кислоты, витровавие 142
Р-Нафтальдегид, витров&ние 420
*-Нафтилмеркуринитрат,
витровавие 92
Р-Нафтилмеркуривитрат,
нитрование 92
Нафтавтрахивон, иитровавие 164
«-Нафтол, витрование 44, 85, 347,
420
Р-Нафтол, витровавие 74, 356 ел.,
375, 420
Нафтолы, эфиры, витровавие 158,
420
Нафтолы, яитровавие 15, 158
Непредельвые соедивения, витро*
вавие 14, 296 ел., 301
азотной кислотой 215 ел.
в жидкой фазе 336 ел.
Нитраты металлов (нитрующие
агенты) 347, 425 ел., 437
алюминия 427 ел.
аммония 442
бария 442
висмута 426, 429
железа 429
калия 439
лития 436, 437
меди 429
натрия 422
ртути 426
свинца 442
серебра 426, 442
цинка 429
Нитраты органические как
нитрующие агенты 347
Нитрацидийдисульфат 134, 135
Нитрацидийперхлораты 136
Н итр ацидийсул ьфат, нитрующий
агент 137
З-Нитроализарин, получение 10
Нитроалкавы, получение 16
Нитроалкоголь циклогексадиена,
получение 115
Нитроамивы, получевие и свойства
228 ел., 320, 321
5-Нитро-о-авизидив, получение 43
о-Нитроавизол, получение 9
п-Нитроааилин
нитрование 162
получение 358, 375
1-Нитроантрахивов,витрование 180
Нитроавтрацен, получевие 59, 61
Нитроавтрон, получение 61
о-Нитроацетавилид, получение 54,
359, 375, 432, 435
n-Нитроацетанилид, получение 42,
54. 359, 375, 436—437
Нитроацетать!, получение 66, 67
32*
р^ф,
получение 5б
м-Нитрофцетрфенрн, получение 24
Н итробензальде! иды, получеаже ^0,
41
II итробензидрлориды иаомерные,
получение 53
м-Нитробеязойная кислота,
получение 41, 54, 87
о-Нитробеязойная кислота,
получение 40
п-Нитробеязойная кислота,
получение 40, 71, 82, 266
Нитробеазойные кислоты,
получение 70
Нитробензол, нитрование 7, 84,
86, 87, 138, 142, 161 гл., 163,
180, 204, 400, 404,425, 452 ел.
азотной кислотой 164
иоиязацяя дрв 171
константа скорости 169
надазотистой кжелотой 198
Нитробензолы, получение 10,
21, 32, 68—72, 75, 78, 115,
121, 147, 198, 344—346, 348,
363, 373, 398, 400 ел., 403,
405, 407, 408, 411, 419, 433,
. 445, 450, 458
6-Нитробензотиазол, получение 48,
418
2-Н итро-4-бромтолуо л, получение
, 458
З-Нитро-4-бромто луол, получение
458
Нитробутадиен, получение 30Q
1-Нитробутан, получение 268 ел.,
275
WWpogyraa, нитрование 284
2-Нитро(%у?аж, получение 268 ел.,
275, %&1, 4*3
Нитробутаны, получение 268 ел.,
27Д
2-Нитробут^н, конденсация с цик-
лодещтадиеяощ 295
2-Нитро0утен-1, нитрование 287
2-Нитробутен-2, конденсация с
бутадиеном 295 '
Цитро-трет. бутилбеязолов
изомеры, получение 35
Нитрование
в газовой фазе 14—15,281, 362
в жидкой фазе 336 ел.
влияние добавок на скорость
реакции 169
кинетика 67, 68, 111 ел., 137,
164 ел., 179
механизм реакции 13 ел., 56,
111 ел., 129, 138 ел. 147, 179,
190, 200 ел., 279, 301, 305 ел.,
406, 453 ел.
нитратами металлов см.
Нитраты металлов
окислами азота 334 ел.
парофазный метод 278
скорость реакции 102 ел., 169
смесью азотной кислоты с
уксусным ангидридом 49
смесью азотной и уксусной
кислот 49
температура 10
З-Нитрованилин, получение 420
Нитрогексаметилея, нитрование
и получение 245
Нитрогексан, получение 234
Н итрогексанафтен, получение 241
2-Дитрогептан, получение 242, 387,
393
Нитрогептаны, получение 387, 395
<%,Р-Нитрогидрин, получение 126
трет. Нитрогидриндан ,получение249
Нитрогидрины насыщенные,
получение 123
трет. Нитродекалин, получение 249
Нитродеканы, получение 285, 286,
395
Нвтродиамияы, получение 228,
Нитродигидроантраценацетат
получение 60
477
Нитродиизобутил, получение 234
N-Нитродиметиламин, получение
825
п-Нитродиметиланилин, получение
348, 349, 358, 375
5-Нитро-4,4-диметилпентанон-2,
получение 294
5-Нитро-4,6-диметилхинолин,
получение 48
6-Нитро-2,4-Диметилхиаолин,
получение 48
8-Нитро-2,4-диметилхиволин,
получение 48
4-Нитро-1,5-диметоксинафталин,по-
лучение 58
2-Нитродифенил, нитрование и
получение 28
4-Нитродифенил, нитрование и
получение 28
о-Нитродифенил, получение 58, 353
п-Нитродифенил,получение 58,353,
436
2-Нитродифенилена окись,
получение 58
п-Н итродифеии лиитрозоамин,
получение 347
4-Н итродифениловый эфир,
получение 459
Нитродифенилы, получение 58
l'-Нитродурол, получение 9, 422
«а-Нитродурол, получение 421
Нитрозилхлорид, образование 304
Нитрозилсерная кислота,
образование 397
Нитрозные газы, нитрующий агент
128, 372 ел.
Нитробензол, нитрование 75
п-Нитрозодиметиланилин,
получение 348
Нйтрозосоединения,нитрование 159
0-НЯтрозотолуол, нитрование 91
о-Нитрозофенол, получение 100 ел.
п-Нитрозофенол, получение 100 ел.
Нитрозофенол как катализатор 104
Нитрозофенолы, получение 94 151
Нитроизоамилен, получение 114,125
2-Нитроизоборнилан, получение 249
Нитроизогептан, получеаие 240,241
Нитроизобутаны, получение 240,269
Нитроизогексан, получение 240
Н итроизооктаны, получение 38?, 395
<%, Р-Н итроизопропилнитрит,
получение 342
<х-и*'-Нитрокамфаны,получение 250
ос-Нитрокамфен, получение 250
втор.к-Нитрокамфен, получение 297
<а-Нитрокамфен, получение 297
Нитрокаприлен, получение 300
4-Нитро-м-крезол, нитрование и
получение 52, 100
6-Нитро-м-крезол, нитрование и
получение 52, 100
Нитрокрезолы, получеаие 71
2-Нитро-м-ксилол, получение 37
4-Нитро-м-ксилол, получение 21,
37, 80
З-Нитро-о-ксилол, получение 22,
36, 52
4-Нитро-о-ксилол, получение 22,
36, 52,
о-Нитроксилолы, получение 345
2-Нитро-п-ксилол, получение 22
Нитро-п-ксилолы, получение 37
Нитроксилолы, получение 37; 369,
452
5-Нитрокумарин, получение 420
Нитрокумол, нитрование и
получение 35
Нитромезитилен
нитрование 236
получение 22, 50—52, 420
Нитроментены, получевие 297
Нитрометан
взаимодействие с основаниями
219
' как растворитель 180
конденсация с альдегидами 223,
226
478
получение 15, 268, 272,274—276,
299, 382
хлорирование 220
4-Н итро-3-метил бензойная
кислота, получение 80
2-Нитро-2-метилбутан, получение
248
1-Нитро-2-метилнафталин,
получение 44
4-Н итро-5-метил-2-тиофенкар боно-
ваа кислота, получение 48
п-Нитро -о-метоксианилин,
получение 26
<%-Н итронафталин
нитрование 162
получение 27, 43, 44, 83, 104,
116,347,351,352,374,398,457
Р-Нитронафталин, получение 43
Н итронафталины, получение 345
1,8-Нитронафталинсульфоноваа
кислота, получение 86
8-Нитро-<х-нафтметилхлорид,
получение 59
Нитронафтойнаа кислота,
получение 83
5-Нитро-1-нафтойнаа кислота,
нитрование 44
%-Нитронафтол, получение 85
2-Нитронафтол, получение 347
2-Нитро-*_нафтол, получение 84
<х-Нитро-Р-нафтол, получение 79
1-Нитро-2-нафтола эфиры,
получение 420
Нитронафтолы, получение 83, 84
п-Н итро-М-нитрозомонометилани-
лин, получение 348
1-Нитро-1-(2-нитроиаопропил)- ци-
клогексан, получение 283
Нитронийдинитрат, получение 134
З-Нитро-ы-нитростирол,
гидрирование 227
трет.Нитроноркамфан,получение277
2-Нитро-4-оксибензальдегид,
получение 53 ел.
Нитро-3-оксибензолдикарбоноваа
кислота, получение 80
Нитрооксикумарин, получение 48
4-Н итро-1 -окси-2-нафтойная
кислота, получение 58
4-Нитро-1-оксипиридин, получение
46
6-Нитро-4-оксихинолин, получение
47
втор. Нитрооктадекав,получение 13
1-Нитрооктен-2, получение 298
Нитроолефины, взаимодействие о
р азличными соединениями
294 ел.
с аминами 293
с нитропарафинами 292 ел.
с органическими основаниами
293
с щелочными солями полинит-
ропарафинов 287
со спиртами 291
восстановление 227
кояденсапиа с алкоголятами 291
полимеризация 295 ел.
получение 226 ел., 231
присоединение к диенам 295
свойства 288 ел.
Нитропарафины
бромирование 221
взаимодействие с нитроолефипа-
ми 292
восстановление 217
гал оидопроизводные, получение
220 ел.
действие на них минер альвых
кислот 217 ел.
действие па них оснований 219
иодирование 221
кояденсапиа
с альдегидами 11, 221—223
с альдегидами и аминами 228
с кетонами 286
получение и применение 11, 12,.
232 ел., 272
479
свойства 215 ел.
4-,"*"2-,*3-Нитропентаны, получение
275
1-Нитропентен-1,конденсациас
диенами 295
N-Нитропиперидин, получение 325
4-Н итропиридин-N -оксид,
получение 46
Р-Н итр ояиридия, получение 4 5,
370 ел.
Литропиридины, получение 31, 45
N-Нитропроизводные
диамида имино-бис-уксусной
кислоты 325
2,6-диоксоииридазина 325
нмино-бис-ацетонитрила 325
1-Нитропропан, получение 15, 268,
272, 275
2-Нитропроиан
нитрование 284
получение 268, 272, 275, 276
2-Нитропропанол, получение 15
2-Нитропропен, полимеризация 296
2-Нитропропен-2, конденсация с
бутадиеном 295
с пиклопентадиеном 295
4-Нитро-1-пропилнафталин, полу-
чевие 27
2-Нитро-6-пропилфенол, получение
53
о- и м-Нитропропнофеноны,
получение 24
З-Нитроретен, получение 301
4-Нитроретен, получение 301
9-Нитроретен, получение 301
Нитроретены, получение 300
5-Н итросалициловая кислота,
получение 460
л-Н итросалицил овая кислота,
получение 79
Нитросерная кислота, нитрующий
агент 150
Н итросоединениа
жирноароматического рада 232 ел.
изомерные 13
нитрование 161
общаа характеристика 3,4
применение 4 ел.
Нитроспирты
гидрирование 225
их аминоспирты, получение 225
и хсложныеэфиры, получение 225
получение 11, 221 ел.
применение 11
Н итростильбен, присоединение к
фенилнитрометану 292
<а-Нитростирол
гидрирование 227
конденсациа с бутадиеном 295
конденсация с метилбутадиеном
295
получение 226, 301
6-Нитро-о-толуидин, получение 424
о-Нитротолуол, получение 8, 82,
345, 433, 460
п-Нитротолуол, получение 82, 418,
460
2-Нитро-4-толуолсульфокислота,
получение 87
Нитротолуолы
нитрование 143, 404
получение 21, 22, 33, 50, 71, 146,
348, 365 ел., 375, 398, 403,
413, 443, 452 ел.
Нитротиофен, получение 31, 64, 424
4-Н итротрифенил амин, получение
57
Нитроуретан, получение 178
2-Нптрофенантрен, получение 62,
128, 354
З-Нитрофенантрен, получение 29,
62, 128, 354
4-Нитрофенантрен, получение 62,
128, 354
9-Нитрофенантрен, получение 62,
128, 354
2-Н итрофенаятренхинон,
получение 29
480
указатель
4-Нитрофенантренхинон, получение
29
Н итрофенантрены, получение 29,
85, 128
о-Нитрофенетол, получение 9
м-Н итрофенилдивитрометан,
получение 267
1 -Н итр о-2-феа ил-3-кар бэтил мае ля-
ной кислоты этиловый эфир,
получение 294
м-Нитрофшилнитрометав,
получение 266
1-(4-Нитрофенил)-пиррол,
получение 63
М-(п-Нитрофенил)-пирролидин,
получение 63, 64
п-Н итрофенилсульфат, получение
41
м-НитрофенияуксУсная кислота,
получение 437
о-Нитрофенилуксусная кислота, по-
лучеаие 437
м-Нитрофенол, получение 167
о-Нитрофенол, нитрование 23, 25,
347, 434, 459
п-Н итр офенол, получение 23, 94,
347, 459
Ыитрофенолы
нитрование 7
получение 7, 10, 68 ел., 70, 71,
85, 91, 94, 97, 100, 151, 277,
348, 400, 412
2-Нитрофенхан, получение 13, 250
6-Нитрофенхан, получение 13, 250
2-Нитрофлуорен, получение 62
Нитроформ
взаимодействие с альдегидами
или кетонами 224
получение 382
З-Нитрофталевая кислота,
получение 41, 87
5-Нитрохинолин
нитрование 47
получение 31, 46, 198
6-Н итрохинол ин
нитрование 47
получение 198
7-Н итрохкнолин
нитрование 47
получение 198, 360, ел., 437
8-Нитрохинолин
нитрование 47
получение 31, 46, 198
Нитрохинолины,получение 360,376
м-Нитрохлорбеазол, получение 1#8
о-Н итрохлорбензол ,получение38,39
п-Нитрохлорбензол, получение 38,
39, 83, 407, 433
Нитрохлорбензолы, получение 403,
425
8-Нитро-1 '-хлор»4-метилнафталин,
получение 425
5-Н итр о-6-хл ор-1 -метиловый эфир
нафтойной кислоты, получение
28
4-, 6- и 8-Нитро-1-хлор-2-метокси-
нафталивы, получение 27
5-Нитро-6-хлор-1-нафтойная
кислота, получение 27
Н итроциклогексаи
нитрование 284
получение 261, 282, 383, 396, 427
втор. Нитроциклогексен,
получение 300
1-Нитроциклогексен-1, получение
300
Нитроппклопентадиен, получение
8, 418
Нитроциклопентан, получение 426
Нитропикловропан, получение 396,
428
ос-Нитроцимол, получение 345
Нитроэтан
взаимодействие со щелочью 219
получение 15, 268, 269, 272, 275,
276, 304 ел., 342
М-(<%-Нитроэтил)-анилин, получение
293
481
2^НЖ^ро@тилбенэол, получение 35
о^-Нитроэтилбензол, получение 24
НйАгроэтилбензолы, получение 34,
367, 375, 426
1-Нитро-2-этилгексен-2, получение
298
Цитроэти лен, полимеризация 295с л.
Нитроэтвлеи, получение 113
Нитроэтилнитрат, получение 343
Нитроэтиловые спирты,
нитрование 112
Р-Нитроэтиловый спирт, получение
112, Ж, 342, 343
Нитрующая смесь 31, 179
Вдтрующяе агенты 8 ел., 19,156
Нонаиафтен, нитрование 232 ел.,
238
Норкамфан, нитрование 277
Окислы азота, нитрующий агент
15, 16, 334 ел.
Окись дифенилена, нитрование 58
п-О%сибен8альдегид,нитрование 54
п-Оксибензойная кислота,
нитрование 23
7-Окси-3,4-диметил-6,8-динитроку-
ясариА, получение 64
7-Окси-3,4-Диметилкумарин,
нитрование 64
о-Оксикарбоновые кислоты
ароматические, нитрование 53
4-Оксикумарин, нитрование 48
2,5-бис-(Оксиметил)-2,5-динитр оге-
ксандиол-1,6, получение 224
2,6-бис-(Оксиметил\-2,&-динитрогеп-
т^вдиол-1,7, получение 224
2,7-бис-(Оксиметил)-2,7- динитроок-
таидиол, получение 224
1-Окси-2-нафтойная кислота,
нитрование 58
Оксинитросоединония, получение 7
8-Окиси-9-нитрофенантрена эфир,
получение 115, 128
1-Окси-4-нитрохинолин, получение
47
1-Оксш1иридин, нитрование 46
4-Оксипиридин-М-оксид,
нитрование 62
1-Оксихинолин, нитрование 47
4-Оксихинолип, нитрование 47
Октадекан, нитрование 13, 247
Октаа, нитрование 234, 247
Октен-1, нитрование 296 ел.
Октены изомерные, нитрование 14
н. Октилнитроамин, получение 32&
Олефины
конденсация
с ароматическими углевоД&рода-
ми 447
с оксикислотами 446
с фенолами 447
нитрование 126, 282
Парафины, нитрование 12, 15, 16,
241, 285, 425
в газовой фазе 278 ел.
Парофазноб нитрование 277 али-
фатичеёких углеводородов^ 273
ел., 277
Пентаметилен, нитрование 241
Пентаны, нитрование 12, 274
Пикрамид, получение 162
Пикриновая кислота, получение 7,
10, 23, 69, 70, 81, 84, №,
344—346, 348, 433
Пикриновая кислота, механизм
получения 71 ел.
Пинен, нитрование 296 ел.
Пиперидин, нитрование 325, 327
Пиридин, нитрование 31,45, 370 ел.,
376, 425
Пиридин азотнокислый, получение
372
Пиридин-М-оксид, нитрование 46*
Пиросерная кислота, получение 124
Пиррол, нитрование 418
Платины окись как катализатор 227
Полиамины, получение 231
Поливиниламин, получение 296
482
указатель
Полишитрододекавы, получение 285
Полишитропарафины, получение
282 ел.
Полинитропарафины, щелочные
соли, взаимодействие с нитро-
олефинами 287
Полинитроепирты, получение 224
Полинитро-4-хлортолуолы,
получение 21
Полихлорбензолы как
растворители при нитровании 20
Предельные углеводороды,
нитрование 13, 14, 237, 253 ел.
в газовой фазе 16
механизм 253 ел.
Предельные бициклические
углеводороды, нитрование 248 ел.
Пр сдельные жирно ар оматические
соединения, нитрование
азотной кислотой 215 ел.
Пропан, нитрование 12, 15, 268,
273, 277, 383, 413
парофазное 270, 272
Пропилбензол, нитрование 235
Пропилен
конденсация с органическими
кислотами 446
нитрование 342
1-Пропилнафталин, нитрование 27
о-Пропилфенол, нитрование 52 ел.
Пропиофенон, нитрование 24
Псевдоазотная кислота,нитрующий
агент 137
Псевдокумол
нитрование 166, 235, 240
скорость нитрования 68
Разбавление, влияние на
нитрование 20
Растворители при нитровании 10,
20, 97 ел., 126, 145 ел., 342
Резорцин, нитрование 158
Ретен, нитрование 300
Ртуть и ее соли как катализаторы
9, 10, 68, 70, 78 ел.
Салициловая кислота, нитрование
53, 79, 460
Салициловый альдегид,
нитрование 420
Серная кислота
влияние ее концентр ацин ша
скорость нитрования 162 ел.
как катализатор 396 ел.
как растворитель 31
Силикагель как катализатор 411
Спирты, нитрование 277
Стильбен, нитрование 336 ел.
Стирол, нитрование 301
Тетрааминодифенил, получение 30
Тетрабромэтилен, нитрование 343
1,2,3,4-Тетр агидро-6,8-динитроиаф-
талин, получение 44
Тетрагидронафталин, титрование 44
Тетралин, нитрование 27
Тетраметилэтилея, нитрование 338,.
339, 341
1,1,4,4-Тетранитробутандиол-2,3,
получение 224
1,1,6,6-Тетранитрогександиол-2, 5,
получение 225
Тетранитродиолы, получение 286
Тетранитродифенилртуть,
получение 74
1,3,6,8-Тетравитрокарбазол,
применение 29
Тетрапитрометан, получение 298,300
Тетранитронафталин, получение 86
Тетрашитронафталины, получение
и применение 44
1,1,1,2-Тетранитропропан,
получение 287, 288
1,1,1,3-Тетранитропропан,
получение 287
Тетранитроэтан, дикалиевая соль,
нитрование 288
483
Тетраоксинафталин, получение 345
К-Тетрахлор-1,2-диаминоэтан,
получение и нитрование 321
Тетрахлорэтилен, нитрование 343
Тетраэтилсвинец, нитрование 304
Тиофен, нитрование 31, 64, 419,
421, 424
Толилнитрометан, получение 369
п-Толуидин, нитрование 435, 436
п-Толуиловая кислота, получение
37, 345
Толуол, нитрование 8, 15, 20—22,
33, 36, 40, 44, 49, 50, 71, 82,
84, 145 ел., 166, 180, 189,
236 ел., 252, 256—258, 277,
348,364сл.,396,398, 400, 403,
408,409,413,418,420,423—425
433, 443, 451, 458
азотной кислотой 258, 259
двуокисью азота 344
надазотистой кислотой 198
п-Толуолсульфанилид, нитрование
26
п-То луолсульфокислота,
нитрование 87
Триалкилизоксазолы,получение 219
Триброманилин, нитрование 55
3,5,6-Трибром-2,4-ДИнитротолуол,
получение 41
Трибромнитроаминобензол (2,4,6*
трибромфенилнитрометан),
получение 55
2,3,5-Трибромтолуол, нитрование
41
Триизобутилен, нитрование 299
Триметилнитрометан,получение 413
2,2,4-Триметил-З-нитр опентан
(втор, нитроизоонтан),
получение 248
2,2,4-Триметил-4-иитропентан, по -
лучеаие 393
2,2,4-Триметилпентан, нитровапие
395
2,4,4-Триметилпевтен-2,
титрование 342
2,2,4-Триметилпентанон-З,
получение 248
Триметилэтилен, нитрование 296,
297, 340
1,3,5-Тринитробензол, получение
33, 84, 346
Тринитродигидроантрацен,
получение 61
2,4,6-Тринитродиметиланилин,
нитрование 359
2,4,-8-Тринитро-1,5-диметоксинаф-
талин, получение 58
2,4,6-Тринитродифениламин,
получение 43
1' ,3,6-Тринитродурол,получение422
2,4,6-Трииитро-м-крезол,
получение 355 ел., 375
Тринитро-м-ксилол, получение 38,
71, 92
2,3,5-Трииитро-и-ксилол,
получение 38
Трияитромезитилев, получение 22,
236
Тринитрометаа, получение 14,298-—
300
2,4,6-Тринитрометиланилин,
нитрование 26
1,2,3-Тринитро-3-метилбутан,получение 248
1,3,8-Тринитронафталин,
получение 351
Тринитронафталины, получение и
применение 44
Тринитрооксибензойная кислота
(2,4,6-тринитро-З-оксибензой-
ная кислота), получение 70
2,2,4-Тринитропентан, получение
287
втор. Тринитропропиловьш спирт,
получение 224
484
2,4,6-Тринитрорезорпин,
получение 7, 8
Трияитроепирты, получение 224
Р-Трияитротолуол, получение 34
Т-Трияитротолуол, получение 34
2,4,6-Трииитротолуол, получение
33, 405
Тринитротолуолы, получение 33
2,4,6-Тринитро-2,2, 2-тринитро-
эти л бензол, получение 287
4,4%4^Тринжтротрифениламин, по-
лучение 57
Тривитрофеяийэтиламииы,
получение 230
Трижитрофенол, получение 71
Тринитро-м-хлорфенол, получение
83
сими. Тринитроэтилбензол,
получение 35
Тринжтроэтиловый спирт,
получение 224
Трифеанламин, нитрование 57
1,1,1-Трифтор-З-нитропропан,
получение 275
1Д ,1-Трифторнйтроэтан,
получение 275
1,1,1-Трифторпропан, нитрование
275
1,2,4-Трихлорбензод, нитровавие
39, 180
2,4,6-Трихлордиметиланилин,
нитрование 359
1,2,4-Трихлор-3,5-диаитробензол,
получение 39
2,4,6-Трихлор- N-нитрозомономети-
ланилин, получение 359
Трициклен, нитрование 250
Триэтиламин как катализатор 294
Уксусная кислота
как растворитель при
нитрований 178, 180
образование 248
Уксусный ангидрид как
растворитель при нитровании 178
Уретан, витрование 178
Хинодив, нитрование 31, 46, 360
ел., 376, 423, 437
надазотистой кислотой 198
Хинон, нитрование 425
Хлор как катализатор 272 ел.
Хлорамин, получение и нитрование
330 ел.
м-Хлоранилия, нитрование 421
п-Хлоранилин, нитрование 421
1-Хлорантрацен, нитрование 346
м-Хлорацетанилид, нитрование 42
п-ХлорацетанилиД, нитрование 56
Хлорбензол
как растворитель при
нитровании 10, 20
нитрование 15, 20, 38 ел., 83,
84, 396, 400, 403, 405—408,
433
надазотистой кислотой 198
З-Хлор-4-динитроацетанилиД,
получение 42
3-Хлор-4,6-динитроацетанилия;,
получение 42
Хлориды металлов как
катализаторы 405
Хлористый бензил, нит^бвание 53„
83
Хлористый водород как
катализатор 325
Хлористый нитрид, нитрующий
агент 447, 459
Хлористый цинк как катализатор
329
%-Хлоркарбоновая кислота,
получение 289
1 '-Хлор-1-метилнафталин,
нитрование 59, 425
6-Хлор-1-нафтойной кислоты:
метиловый эфир, нитрование 28
1-Хлор-2-метоксиаафталин, нитро-
производные 27
4-Хлрр-6-метокси-8-нитрохиналь-
дин, получение 48
4-Х лор-6-метоксихинальдиа,
витрование 48
6-Хлор-1-нафтойная кислота, нит-
рование 27
JV -X лор-М-нитроамин, получение
321
1-Хлор-9(или 10)-нитроантрацен,
получение 346
З-Хлор-4-нитроацетанилид,
получение 42
З-Хлор-6-нитроацетанилид, получе*
ние 42
п-Хлорнитробензол, нитрование
164, 180
Хлорнитробензольг, нитрование
1@3, 164
Хлорнитрометан, получение 382
<^-Хлорнитронафталин, получение
460
Хлорнитропарафияы, получение
220
4-Хлор-2-нитротолуол, получение 21
4-Хлор-З-нитротолуол, получение
21
Хлорпикрин, получение 220, 425
п-Хлортолуол, нитрование 21
Хлортривитрометая, получение 382
Хлорфевод, витровавие 85
Фенантрен, вжтрощание 15, 29, 45,
61, 85, 114 ел., 128, 158, 353,
374
Фенантренхинон, нитрование 29
&-Фенетиламив, получение 227
Фенетол,нитрование 9, 23, 198, 421
2-Фенилантрахинон, нитрование 164
Фенилдинитрометан, получение
256—258, 266, 377
витрование 265—267
Феаилизонитрометан,получение 347
Феяилнитроизопропан, получение
24
Феиилнитрометан
витрование 266 ел.
получение 9, 145, 236, 252, 258,
347, 365, 375, 377
Фенилнитрометан
присоединение к яитростиль-
бену 292
1-Фенил-2-нитропиррол, получение
63
1 -Фенил-3-нитропирро л ,нитрование
63
Феиилнитропропаа, получеяие %
1-Фенил-2-нитропропен-1,
гидрирование 227
<%-Ф енил-Р-нитро этиловый спирт,
эфиры, получение 291
Фенилпролиловыи эфир,
витрование надазотистой кислотой 198
Фенилнитроэтан, получеяие 235
N-Фенилдирролидин, нитрование
63
Фенилтиазолы, нитровавие 166
Фенилтринитрометан, получение
265
Феяилуксусная кислота,
нитрование 24, 41, 437
2-Фенилэтилсульфоновая кислота,
нитрование 1р9
Фенол
конденсация с олефинами 447
нитрование 10, 19, 23, 69, 78 ел.,
85, 93 ел., 150, 154, 158, 162,
166, 347, 354 ел., 375, 420,
421, 423, 424, 433, 459
двуокисью азота 345
надазотистой кислотой 198
нитрозирование 100 ел.
Фенола сульфокислоты,
нитрование 161
Фенхан, нитрование 13, 250
Флуорен, нитрование 62
Флуоренон, нитрование 29
Фталевый ангидрид, нитрование
41, 86, 87
Фторбензол, нитрование 39, 349
486
Фтористый бор как катализатор 9,
10, 86, 166 ел., 410, 446 ел.
1-Циано-1,4-дифенил-1,3-бутадиен,
нитрование 343
1-Циано-1,4-дифенил-3,4-динитро-
1-бутен, получение 343
Цианистый бензил, нитрование 8
Циклические предельные
углеводороды, нитрование 238, 241
Циклогексан, нитрование 119, 251,
259, 260, 262, 341, 342, 383,
394 ел., 430
азотной кислотой 260 ел., 282
окислами азота 260 ел.
Циклогексанрл, получение 261
Циклогексанон, получение 261
Цпклогексен, нитрование 300
Циклогексена псевдонитрозит,
образование 300
Циклогексилнитрит, получение 261
Циклогексилнитроамин, получение
323
Циклопентадиен, нитрование 8, 418
Циклопентан, нитрование 430
Циклопропан, нитрование 396
Цимол, нитрование двуокисью
азота 344
Этан, нитрование 268
Этилбензол, нитрование 24, 34—36,
49, 180, 235, 239, 240, 367 ел.»
375, 429
2-Этилгексеш-1, нитрование 298
Этилен, нитрование 112, 124, 126,
298, 300, 341, 34%
двуокисью азота 309
механизм реакции 113
Этилена окись, нитрование 342
Этилендиамин, нитрование 321
Этилен-N ,N '-динитроампн,
получение 322, 323
Этилендинитромочевина,
получение и гидролиз 323
Этиленгликоля нитрит-нитрат, по*
лучение 112
Этиленмочевина (имидазолидон-2),
нитрование 323
N ,М'-Этиленоксамид, нитрование
324
Этилнитрат
как нитрующий агент 8, 121»
347, 417, 418
как растворитель при
титровании 146
получение 304
Этилен-бис-уретан, нитрование 324
Эфиры алифатические, нитрование
277
Эфиры ароматических о-оксикар-
боновых кислот, нитрование 53,
Эфиры фенолов, нитрование 19
ОГЛАВЛЕНИЕ
Введение 3
Литература 16
Л&дед лередл. Нитрование ароматических и гетероциклических
соединении азотной кислотой и нитрующей смесью 19
§ 1. Нитрование азотной кислотой 20
§ 2. Нитрование смесью азотной и серной кислот 31
§ 3. Нитрование азотной кислотой в ледяной уксусной
кислоте или в уксусном ангидриде 49
§ 4. Нитрование в присутствии катализаторов 68
§ 5. Каталитическое действие азотистой кислоты при
нитровании 93
Литература 104
Глдед *тордл. Механизм нитрования ароматических соединений
азотной кислотой и нитрующей смесью 111
Литература 211
Нитрование предельных, жирноароматических и
ненасыщенных углеводородов азотной кислотой 215
§ 1. Свойства нитропарафинов 215
§ 2. Получение мононитропарафинов и нитросоединенийжир-
ноароматического ряда 232
§ 3. Получение полинитропарафинов 282
§ 4. Свойства нитроолефинов 288
§ 5. Нитрование ненасыщенных углеводородов... . 296
Литература 312
Нитрование аминов 320
Литература 332
Нитрование органических соединений окислами
азота 334
А, Нитрованке окислами азота без катализаторов 334
I. Нитрование в жидкой фазе 336
# *, Нитрование непредельных соединении 336
§ 2. Нитрование ароматических и гетероциклических
соединении 344
II. Нитрование в газовой фазе 362
III. Нитрование ароматических соединений нжтрозныыи
газами 372
IV. Нитрование окислами азота жирноароматических и
алифатических углеводородов . 376
Б. Нитрование окислами азота в присутствии
катализаторов 396
Литература 414
Улд*д шастая. Нитрование органических соединений органическими
и неорганическими нитратами и хлористым нитридом . . 417
I. Нитрование органическими нитратами 417
И. Нитрование неорганическими нитратами 428
§ 1. Нитрование в присутствии AlClg 441
§ 2. Нитрование в присутствии FeClg и SiCl* 447
§ 3. Нитрование в присутствии В?з 446
§ 4. Механизм нитрования ароматических соединений
неорганическими нитратами в присутствии активаторов
(А1С1, и BF,) #6
III. Нитрование хлористым нитрилом 459
Литература 460
Именной указатель - ...... 462
Предметный указатель 467
Нитрование углеводородов
и других органических соединений
у СССР
Редактор издательства Я). Т. Струим* Текиический редактор Е. Н.
РИСО АН СССР № 10—6В. Сдано в набор 7/1 1956 г. Подп. в печать 13/iy &
Формат бум. 60x92'/». Печ. л. 30'/,. Уч.-ивд. л. 25,3. Тираж 3000. Ивд. JA
T-02G54 Дено ^ р. У# к. *Жп.
Ивдательство Академия наук СССР. Москва, Подсосеиомвй пер., д. 2*
2-я типография Издательства АН СССР. Москва, Шу6жао«ш# дер., д. *0
Опечатки и исправления
Строка
Яопечотдко
Долэюно
19
180
208
214
218
219
224
229
2 св.
2 св.'
9 св.
ссылка
79
18 св.
7 сн.
4 ся.
5 св.
318
326
329
,,
330
466
468
А. В.
ссылка
192
6 св.
45 св.
20 св.
5 св.
1 сн.
21—22 ев
Топчиев
НИТРИРОВАНИЕ
и бензолсульфоновой
кислот
Томменсона
(1954)
СН —С N
О
N0,
I
нислота
N0%
С
R' N0,
J. Am. Cbem. Soc, 73
хлористого ацетила
хлористого ацетила
хлористый ацетил
хлористый ацетил
Якубович А. И.
Беязолсульфоновая
кислота, нитрование 180
НИТРОВАНИЕ
кислоты и бензолсульфо-
«ислот
Томлинсона
(1953)
СИ, —С N
О
N0,
i
нислота .
N0,
R
С
R'
J. Am. Chem. Soc., 75
хлорацетата
хлорацетата
хлорацетат
хлорацетат
Якубович А. Я.
исключить