








































































































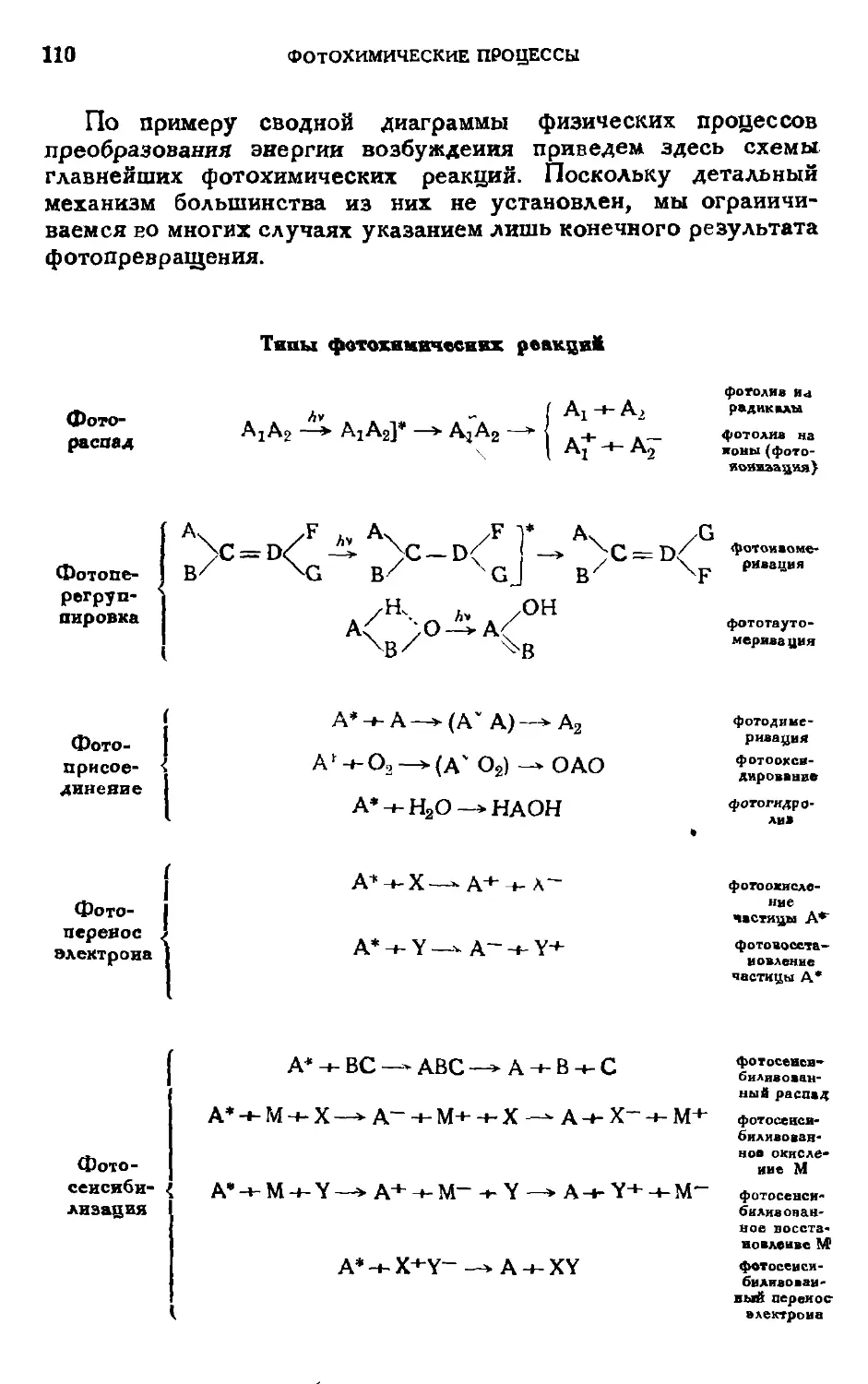

























































































































































































































































Текст
55
Т55
АКАДЕМИЯ НАУК СССР
НАУЧНО-ПОПУЛЯРНАЯ СЕРИЯ
А na.beмин
А.Н.ТЕРЕНИН
Т 3
ФОТОХИМИЯ
КРАСИТЕЛЕЙ
И РОДСТВЕННЫХ
ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
ЦЕЯТГАЛЬЧДЯ Е
ИЗДАТЕЛЬСТВО АКАДЕМИИ НАУК СССР
МОСКВА
1947
ЛЕНИНГРАД
ПРЕДИСЛОВИЕ
Настоящая книга возникла из потребности в современном
обзоре и критическом пересмотре накопившегося, трудно
обозримого фактического материала по фотохимическим реакциям
красителей, рассеянного по многочисленным журнальным
статьям.
Выцветание красителей, фотосинтез в растениях и
сенсибилизация фотопленки к инфракрасным лучам — общеизвестны.
Между тем мы еще далеки от понимания этих явлений, а
следовательно лишены возможности сознательно управлять ими.
В соответствии с поставленной задачей при составлении
книги были, главным образом, использованы работы
теоретического направления, имевшие целью выяснить механизм
данного явления. Необозримо большой опытный материал,
имеющийся в технической литературе, смог быть использован
только в той мере, в какой он отвечал этой цели. Многие
интересные статьи в специальных технических журналах,
касающиеся узко практической темы, не смогли быть включены
в книгу, посвященную основам фотохимии красителей, тем
более что автор значительно превысил намеченный ранее
объем.
Автор надеется, что эта книга послужит в некоторой мере
установлению общего языка между представителями отраслей
промышленности, которые синтезируют и используют
красители, и представителями химической физики, посвятившими
себя разработке принципиальной стороны вопроса.
Сотрудничество специалистов различных областей для
совместного решения проблем, выдвигаемых грандиозным
Сталинским планом развития нашей Родины, должно
обеспечить новый подъем нашей науки и техники в послевоенную
пятилетку.
Автор не скрывает от себя, что физические разделы книги,
изложенные по необходимости весьма сжато, могут оказаться
трудными для неподготовленного читателя. Поэтому рекомен-
4 ПРЕДИСЛОВИЕ
дуется предварительно ознакомиться с основами квантовой
теория в объеме курса строения вещества, излагаемого в
программах химических вузов. Из числа книг, могущих
способствовать предварительной подготовке читателя, можно указать
книгу М. В. Волькенштейна „Строение молекулы", выходящую
в Научно-популярной серии Академия Наук СССР, а также
обзорные статьи на соответствующие темы, печатающиеся
в журнале „Успехи химии". Ссылки на некоторые из этих
статей даны в указателе литературы, в конце книги.
Необходимо также подчеркнуть, что некоторые из
затрагиваемых в книге вопросов являются новыми и излагаются
здесь впервые, чем объясняется трудность их изложения
достаточно понятным языком для неспециалистов.
Работа автора над книгой, прерываемая неоднократно,
продолжалась с 1944 по 1946 г. Несомненно, что в книге есть
упущения и даже ошибки, за указания на которые автор будет
признателен.
В заключение автор должен выразить свою глубокую
благодарность А. Т, Вартаняну, которому он обязан за помощь
при составлении литературного обзора и за многочисленныэ
критические замечания при просмотре рукописи.
Академик А. Е. Порай-Кошиц и проф. В. А. Измаильский
любезно согласились просмотреть главу вторую и предложили
ряд изменений и исправлений ее текста.
Л. Теренин
Сулв-Ярви, август 1946
ВВЕДЕНИЕ
Среди миогообразия соединений органической химии
красители выделяются интенсивным поглощением видимого света.
Последнее сообщает им те яркие цвета, которые послужили
причиной их широкого использования с давних времен для
крашения. Однако значение красителей не исчерпывается
нх эстетическим применением. Свет есть энергия, а,
следовательно, красители представляют собой мощные приемники
радиации, превращающие ее в иные формы.
На продуктивном использовании природными красителями
энергии света основана возможность жизни на земле.
Действительно, от него зависит ассимиляция растениями углерода из *
углекислоты атмосферы и чувствительность глаза к свету.
Активное поведение красителя на свету по отношению к
окрашенному объекту проявляется в ряде явлений. С ним связано
очувствление фотографической эмульсии к излучению,
нормально на нее не действующему. Оно вызывает механическое
разрушение окрашенного объекта и приводит к разложению
живой клетки.
Краситель сам испытывает разрушающее действие света,
что приводит к его выцветанию. Техника использовала это
явление для цветной фотографии, но в больших масштабах
изыскивает меры к его предотвращению.
Внимание физиков привлекали в красителях оптические
явления, не связанные с их химическим изменением, как
например испускание света флуоресценции, избирательное
поглощение и отражение, аномальная дисперсия. Эпизодически
красители применялись также в опытах по фотоэлектричеству.
За последнее десятилетие большие успехи сделала теория
цветности окрашенных соединений и красителей.
Несмотря на разнообразие отраслей знания и техники,
занимающихся красителями и использующих их на практике,
мы не имеем до настоящего времени достаточно полной
глубокой теоретической трактовки физических и химических процес-
6 ВВЕДЕНИЕ
сов, идущих в ннх под действием света. Причиной такого
состояния этой важной области являются: сложность
молекулярного строения красителя, разнообразие его
физико-химических состояний, усложняющее участие среды в фотореакциях
и, наконец, многообразие путей превращения красителя,
создающее запутанное протекание фотореакции в реальных
условиях.
Совершенно ясно, что проблема фотохимии красителей
вступила в настоящее время в такую стадию, когда требуется
систематизация громадного и разнообразного фактического
материала и его обобщение с точки зрения современных
представлений. Такое подведение итогов послужит отправным
моментом для постановки методически новых изысканий
принципиального характера, важность которых для практики не
требует дополнительного обоснования.
Данная книга является попыткой дать обзор
теоретических основ фотохимии красителей1,1)
Для понимания фотореакций в красителях необходимо также
рассмотреть действие света на более простые окрашенные
соединения и даже некоторые бесцветные циклические
соединения, поглощающие ультрафиолетовую радиацию. Поэтому
термин молекула красителя используется нами иногда
в расширенном смысле, включающем цветные органические
соединения, а не только такие из них, которые применяются для
крашения, т. е. способны фиксироваться на волокне, и кото-
р ым собственно присвоено наименование „краситель".
Последний признак для фотохимии красителей является важным,
но все же побочным фактором. Существенное значение имеет
то обстоятельство, что большинство красителей представляет
собой молекулярные ионы. С этим связано не только резкое
возрастание поглощения света красителем, по сравнению
с цветными соединениями, но и его физико-химическое
поведение в растворах, своеобразие которого следует учитывать
при рассмотрении фотопроцессов.
Мы намеренно не затрагиваем в этой книге два обширных
раздела фотохимии красителей, относящихся к фотобиологии,
а именно: фотосинтез в растениях и
фотодинамическое действие на живую клетку. Включение их значительно
увеличило бы и без того возросший против первоначального
намерения объем книги. Кроме того, по этим разделам имеются
современные монографии.2
1) Цифры петитом без скобки относятся к литературе в конце книги.
ЧАСТЬ ПЕРВАЯ
•ФОТОФИЗИЧЕСКИЕ ПРОЦЕССЫ
В КРАСИТЕЛЯХ
ГЛАВА ПЕРВАЯ
ПОГЛОЩЕНИЕ СВЕТА
1» Цвет и спектр поглощения
Воспринимаемый нами цвет окрашенных объектов есть
результат избирательного поглощения красителем
определенных участков в непрерывном спектре падающего белого
света. Только в редких случаях наблюдаемый цвет обусловлен
избирательным отражением от поверхности красителя,
присутствующего в большой концентрации, например в виде
кристалликов.
Поглощение света лучше наблюдать после его прохождения
через однородные окрашенные среды — растворы и пленки,
где оно не осложнено многократными внутренними
преломлениями и отражениями, присущими объектам с дисперсной
структурой, например тканям.
В зависимости от положения в спектре участка
избирательного поглощения, глаз ощущает тот или иной цвет как
результат совместного действия всех остальных, пропущенных
красителем областей видимого спектра.
На фиг. 1 заштрихованы различные участки спектра,
не пропускаемые каким-либо красителем; воспринимаемые при
этом глазом цвета указаны на фигуре для каждого случая
справа. Они являются дополнительными к
поглощенному цвету в том смысле, что при совместном их действии
глаз испытывает ощущение белого света. Из сказанного
янствует, что цвет, возникающий после прохождения белого
света через окрашенное тело, как правило, не есть чистый
спектральный, а смешанный.
Ощущение одного и того же смешанного цвета можно
создать самым разнообразным сочетанием поглощаемых и
пропускаемых участков спектра. Поэтому для однозначной
характеристики поглощения света красителем нельзя ограничиться
10
ПОГЛОЩЕНИЕ СВЕТА
указанием на цвет последнего, а необходимо привести положение,
а также вид его полосы поглощения в спектре.
Полосы поглощения красителей не имеют столь резко
очерченных границ, какие изображены на фиг. 1. В
действительности имеется преобладающий максимум поглощения,
спадающего в обе стороны, т. е. некоторое распределение
поглощения по длинам волн, характерное для каждого краси-
k
Длина
домны ■ А= 4QQ
Сини 0 \3еленый\^\ Красный
500 800 700 т и.
Желтый
Фиг. 1. Цвет я спектр поглощения.
теля. Для локализации максимума поглощения в
„односторонних полосах, лежащих на краях видимого спектра (фиг. 1)
необходимо распространение наблюдений на смежные невидимые
области — ультрафиолетовую для красителей желтого цвета,
инфракрасную — для сине-зеленых красителей. Красители
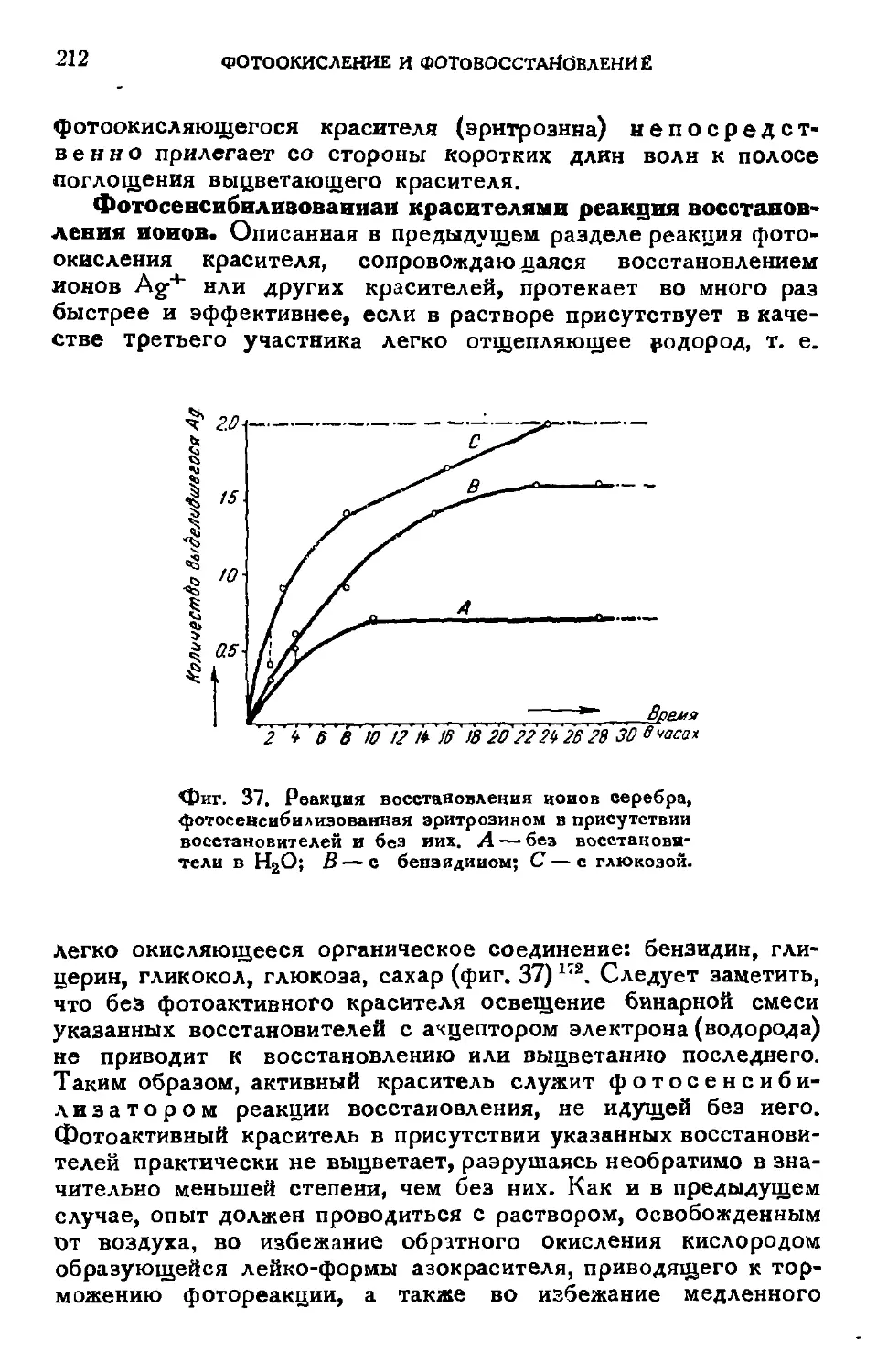
коричневого и черного оттенка отличаются нерезкой полосой
поглощения, захватывающей широкое протяжение длин волн.3
Как следует из фиг. 1, последовательное смещение полосы
поглощения от фиолетового конца видимого спектра к красному
вызывает следующую последовательность воспринимаемых
нами цветов:
1. ЦВЕТ И СПЕКТР ПОГЛОЩЕНИЯ 11
желтый -»■ оранжевый —> красный -> пурпурный —> синий -»■ сине-
зеленый.
Такое чередование цветов получило в химии красителей
укоренившееся название „углубления" цвета, а обратная
последовательность — „повышения" цвета. Структурные изменения
или действия среды, вызывающие „углубление" цвета
красителя! называют батохромными причинами, а приводящие
к повышению первоначального цвета — гипсохромными
(по-гречески „батос"— глубина, „гйпеос" — высота, „хрома" —
цвет).
Из предыдущего явствует, что батохромный эффект
вызывается смещением полосы поглощения в сторону длинных волн,
а гипсохромный — в сторону коротких.
Кроме смещения, полоса поглощения красителя может
испытать усиление (гиперхромный эффект) или ослабление (гипо-
хромный эффект), с сопутствующими изменениями
насыщенности или даже оттенка цвета (по-гречески „гипёр" — над;
„гиаб" — под).
Сопоставим здесь обозначения и единицы измерения
величин, которые характеризуют положение в спектре.
^(т[/.): длина волны монохроматического света,
выраженная в миллимикронах (1 т£*. = 10~7см = 10А); в видимом
спектре ^ меняется от 400 до 760 /п(л (фиг. 1).
v (сек."1 или ): частота, колебания света в секунду,
\ сек* /
связанная с длиной волны соотношением v^ = с, где с — скорость
света = 3*1010 , если 1 выражена в тру это соотношение
CM
сек.'
,17
сек
принимает вид: v\^ = 3 • Ю
Частоты колебания имеют для видимого света порядок
величины ДО15 сек.""1. Если за единицу частоты колебания принять
1 френель (1 /= 1012 сек. "*), то в этих единицах частоты
колебания выражаются трехзначными числами.
v [см~г или —): число волн X в 1 см или „волновая"
\ см /
частота (волновое число), определяемая соотношением
vXCM=l или, в миллимикронах:
Величины v выражаются пятизначными числами (фиг. 1), В
старых работах встречается v, выраженное в мм"1, которое
в 10 раз меньше v, выраженного в см"1.
12
ПОГЛОЩЕНИЕ СВЕТА
2. Законы поглощения света
Пусть ввесьма тонкий слой однородной окрашенной среды
вступает поток монохроматической световой энергии с длиной
волны \ поглощаемой красителем (фиг. 2), Если через каждый
см2 поверхности слоя вступает ежесекундно / эрг, то
световой поток равен /
эрг
см2 сок.
. После прохождения слоя,
имеющего толщину Ьх см, поток уменьшается в результате
поглощения на положительную величину ] §/|, представляющую
собой энергию, поглощаемую ежесекундно в этом слое.
Допуская, что ослабление потока определяется толь.;о
числом поглощающих частиц — молекул, находящихся на его
пути, и не зависит от
абсолютной величины потока, а также
от взаимного влияния молекул,
можно получить два основных
/-16/1 закона поглощения,
подтверждаемых опытом:
1) Каждый тонкий слой
постоянной толщины
внутри однородной окрашенной
среды поглощает
определенную долю входящего в него
/см* /
Фиг. 2. Поглощение светового
потока в тонком слое.
потока радиации: закон Буге
(1729) —Ламберта (1760).
2) Поглощение данным тонким слоем
пропорционально числу окрашенных молекул, в нем содержащихся,
а следовательно, также числу их в единице объема среды,
т. е. их концентрации: закон Бэра (1852).
Первую закономерность можно сформулировать как
независимость поглощательной способности молекул от энергии
светового потока /. Строгое соблюдение этого закона было
показано Вавиловым (1920) посредством опыта, при котором
поток изменялся от крайне малого значения до очень большого,
а именно с 10 u до 108
эрг
, т. е. при изменении потока
см- сек.
в 1019 раз14
Напротив, закон Бэра, утверждающий независимость
поглощательной способности молекул от их сближения при
увеличении концентрации, для красителей обычно нарушается
и может рассматриваться лишь как правило, справедливое
только при достаточном разведении раствора.
Выразим концентрацию красителя числом п его молекул
в 1 см3. В таком случае, в заштрихованном на фиг. 2 объеме
2. ЗАКОНЫ ПОГЛОЩЕНИЯ СВЕТА 13
слоя, равном Ьх см3, находится п Ьх молекул красителя. Погло-
щаемая в этом объеме доля светового потока есть —-±-=:——^
где | Ь/\ есть абсолютное значение величины, на которую
уменьшается поток, а $Г—сама, отрицательная, величина
изменения потока (&/<0). Согласно приведенным двум законам
поглощения, эта доля пропорциональна числу молекул,
находящихся в объеме, т. е.
kb закон Ламберта-Бэра, (1)
где коэффициент пропорциональности к называется
молекулярным коэффициентом поглощения. Ои зависит,
разумеется, от примененной длины волны и представляет собой
константу, характеризующую поглощательную способность
молекулы для данной длины волны.
В формуле (1) слева от знака равенства стоит отвлеченное
число. Поэтому к измеряется единицей, размерность которой
обратна размерности произведения пЬх или —<р см, т. е. к
измеряется в см2.
Обратимся к вопросу измерения молекулярной константы
к, важность которой явствует из последующего изложения.
Выше мы намеренно рассматривали поглощение в очень
тонком слое для выяснения сущности явления. В
действительности всегда имеют дело с окрашенным слоем конечной
толщины / см, поглощение в котором настолько велико, что
для него выражение (1) непосредственно неприменимо. Однако
такой слой может быть мысленно разложен на ряд тонких
слоев, последовательно проходимых светом, для каждого
из которых справедлива формула (1). Перепишем последнюю
в виде Slogf/ = — КЬх и проинтегрируем это выражение
от начальной величины светового потока /0, вошедшего в слой
при значении координаты лг=О, до конечного его значения 1г
после прохождения слоя, когда координата .г — /. Получаем
— log-/0 = — КЫли logy-= — Л7,
откуда следует обычное выражение закона Буге-Ламберта для
слоя конечной толщины /:
-; =е~*1, (2)
где е — основание натуральных логарифмов (log).
14 ПОГЛОЩЕНИЕ СВЕТА
По закону Бэра константа К пропорциональна концентрации
красителя; К = кп.
Отношение величины потока, вышедшего из слоя 1г к
вошедшему /0, носит название „пропускания" слоя Т\
Г*1 —hnl /о\
~~^ ',' ,7* g I J I
Энергия светового потока, поглощенная слоем А$ равная /0 —119
получается из выражения (3) вычитанием обеих его половин из 1:
— У /— т (л А~кп1\ (д\
Концентрацию растворенного вещества принято в химии
выражать числом с грамм-молей в 1 литре раствора; при этом,
вместо молекулярного коэффициента поглощения к, необходимо
соответственно пользоваться новым молярным
коэффициентом поглощения е, который удовлетворяет условию:
ее = к п = К. (4')
Отсюда получаем обычное выражение закона Ламберта-Бэра:
Численное соотношение между обоими коэффициентами
получается равным:
_е^ _п 6.06-10*3 с ^ fi
к ~ с 1000 с ^
или
На практике, для большего удобства, пользуются десятичными
логарифмами и закон Ламберта-Бэра представляют в
следующей форме:
или
li = £ю c/i О)
где е10—десятичный „молярный" коэффициент поглощения
принято называть коэффициентом погашения (экстинк-
ц и и). Он связан с предыдущим соотношением: е10 = 0.43 е
и следовательно:
к = 1.6 • КГ81 в»?- 10"22 е10. (8)
2. ЗАКОНЫ ПОГЛОЩЕНИЯ СВЕТА
1
В работах по флуоресценции красителей часто можно
встретить концентрацию т красителя, выраженную в г на 1 см3
раствора. Численное значение соответствующего коэффициента
поглощения а связано с предыдущим соотношением am —ее
и, следовательно, а — ~Xf~s> где М — молекулярный вес
красителя, j
Измерения пропускания -4- однородной окрашенной среды,
производимые с помощью спектрофотометра, дают возможность
определить по формулам (2) и (5) коэффициенты поглощеиия
е для каждой длины волны, поглощаемой красителем.
Полученные данные наносят на график в виде спектральной
Ктох
Фиг. 3. Типичная спектральная кривая полосы
поглощения красителя (сафранин)3.
кривой поглощения, характерной для каждого красителя,.
а также условий, в которых он находится. На фиг. 3
изображена такая кривая, отложенная в шкале \ и на фиг. 4 —
в шкале v.
На фигуре видно, что поглощателъная способность
молекулы имеет максимальную величину егаа1 для определенного
значения 1 = Х и что эта способность падает в обе стороны
от максимума, притом заметно более круто в сторону
больших длин волн (меньших частот).
Ввиду большого диапазона изменения коэффициента
поглощения е, обычно по вертикальной оси откладывают значения
его десятичного логарифма log"10e.
Для окрашенных соединений молярный коэффициент
поглощения е имеет в максимуме порядок величины 10*, для
истинных красителей *гаахда105 —105.
Необходимо отметить, что спектральные кривые
поглощения, приводимые в старых работах, изображены большей
16 ПОГЛОЩЕНИЕ СВЕТА
частью иначе, чем принято в настоящее время, а именно:
по вертикальной оси в них отложен логарифм такой толщины
/ слоя разбавленного раствора данного соединения, при котором
происходит „полное" поглощение данной длины волиы 1.
Другими словами, наносят границы поглощения данной
полосы. Получающийся график качественно воспроизводит
спектральную кривую logs, но в-перевернутом виде, таким
образом, что максимум поглощения изображается здесь как
минимум кривой. Для сопоставления с современными кривыми
такой график следует рассматривать в перевернутом положении.
Необходимо также учесть, что „числа колебаний",
отложенные в старых графиках по горизонтальной оси, представляют
собой значения -*-, выраженные в —, а не , как принято
А * г мм ' см ' г
теперь, т. е. они в 10 раз меньше v cm~j. 3
Закон Ламберта-Бэра (2) или (5) не может быть
непосредственно использован для практически важного случая
прохождения света через неоднородную окрашенную среду, имеющую
дисперсное строение. Расчет светового потока, испытавшего
поглощение внутри такой среды и вышедшего обратно в виде
рассеянного по всем направлениям света, представляет собою
сложную математическую задачу, требующую учета деталей
внутренней структуры среды. Для таквх объектов
ограничиваются обычно приведением спектральной кривой диффузно
отраженного (рассеянного) белого света. Максимум поглощения
красителей проявляется на таких кривых в виде минимума
интенсивности рассеяния в соответствующем участке спектра.
3. Эффективность или выход процесса поглощения
Перепишем закон Буге-Ламберта-Бэра (1) в виде: т==
= кЬп$ где Ьп есть число молекул, заключенных в тонком
слое Ъх на фиг. 2. Этот вакон сводится, таким образом,
к утверждению, что каждая молекула поглощает определенную
долю, равную kt падающего на слой светового потока. Из
порядаа величины молярного коэффициента поглощения в
максимуме спектральной кривой, £mai я& Ю5 для красителей,
применяя формулу (6): к =1.6 • 10~"21е, получаем, что поглощаемая
каждой молекулой световая энергия составляет 10~16-ую
часть от падающей на 1 см2 поверхности слоя. Следовательно,
каждая молекула красителя уподобляется непрозрачной
площадке в 10~16 см2, поглощающей целиком падающую на нее
часть светового потока. В предыдущем параграфе мы показали,
что молекулярный коэффициент поглощения к имеет, действи-
3. ЭФФЕКТИВНОСТЬ ИЛИ ВЫХОД ПРОЦЕССА ПОГЛОЩЕНИЯ 17
тельно, размерность см2. Мы видим здесь, что к дает
величину непроницаемой для света площади, окружающей молекулу.
Молекула красителя имеет линейные размеры порядка
10 А = 10~7 см, а следовательно, ориентировочно, занимает
площадь 100 А2 = 10~14 см2. Из предыдущего следует, что
эффективная или „непроницаемая" для света площадь в 100 раз
меньше площади молекулы5.
Основной особенностью поглощения света материей является
его прерывистость во времени. Молекула поглощает
энергию светового потока не непрерывно, а с перерывами,
извлекая каждый раз из потока сразу целую порцию или
квант энергии- Иначе говоря, световой поток дискретен и
состоит из отдельных порций энергии — фотонов, величина
которых пропорциональна частоте света v сек."1, а именно
равна, как известно, Av, va^ h есть универсальная постоянная
Планка: А = 6.57 • 10~27 эрг X сек.
Способность молекулы воспринимать энергию только
квантами определенной величины есть существенная черта явлений
атомарного масштаба. Внутренняя энергия молекулы не может
иметь произвольную величину, а способна принимать только
вполне определенные значения или, образно выражаясь,
подниматься только до вполне определенных дискретных
уровней, специфичных для данного химического индивидуума —
атома или молекулы.
При восприятии энергии любого рода внутренняя энергия
молекулы скачкообразно повышается с нормального или
основного, свойственного данной молекуле, уровня на
более высокий. Обозначим энергию основного состояния
молекулы через Е°, а более высокий уровень энергии того
состояния возбуждения, в которое молекула может
переходить поглощением света, через Е*. Величина фотона Av,
который молекула способна поглотить, равна разности энергий
Е* — Е° этих состояний:
hv — E* F°
Энергия фотона в видимой области имеет порядок
величины Av==6 • 10~27 • 5 • 1014« 10~12 эрг. Однако энергию при
молекулярных процессах удобнее измерять не этими малыми
количествами, а относить энергию к граммолекуле вещества,
умножая на 6.06 • 1023, т. е. на число молекул в моле. Для
удобства сопоставления с теплотами химических реакций эту
„молярную" величину поглощенного кванта выражают в кгкал
путем деления на механический эквивалент большой калории
2 А. Н. Теркина Л •>* Jf^/
I 'л\ К
18 ПОГЛОЩЕНИЕ СВЕТА
в эргах: 1 кгкал = 4.2 • 1010 эрг. Отсюда получается следую
щая переводная формула:
, кгкал 6.06 • 102J Av эрг. оо 1000
/jV == _—__ == 2ХЬ -
моль 4.2 • 1010
Последнее численное соотношение непосредственно следует
из формул, приведенных в конце параграфа 1. На фиг. 1
внизу даны значения энергии фотонов для видимого спектра,
выраженные в этих единицах.
При изучении процессов возбуждения в системах
атомарного размера в физике принято пользоваться иной единицей
энергии, а именно кинетической энергией электрона (заряд
электрона е —4.77-Ю~10 CGSE), сообщаемой ему при
прохождении разности потенциалов в 1 вольт. Эта единица энергии
л т/ 4.77 - 10-ю
носит название электрон-вольт и равна: 1 ev = ■
= 1.6'10~12 врг. Значения квантов энергии, выраженные
в таких единицах, имеют более удобный для прикидочных
вычислений вид двузначных чисел (фиг. 1).
Приведем дополнительно следующее полезное соотношение
между химической и физической единицей:
1 е К= 1 электрон-вольт = 23 ?
г моль
а также соотношение:
В соответствии с прерывчатым характером процесса
поглощения света, последний может быть формально описан как
попадание отдельных фотонов в „непроницаемую" площадь
величиной к см2, которая, по сказанному выше, окружает
молекулу. Если фотон „удачно" попадает в эту миниатюрную
мишень, то происходит его поглощение и изъятие из
светового потока. Если монохроматический световой поток равен
I —^— * то число содержащихся^ нем фотонов есть /Vv = т- •
Число фотонов, исчезнувших в результате поглощения в
элементарном слое Ьх (фиг. 2), будет й/У¥ = т^-> где S/, по
предыдущему, есть величина уменьшения энергии
монохроматического светового потока после прохождения этого слоя.
Подставляя выражения для /"и о/ в формулу (1), имеем:
3. ЭФФЕКТИВНОСТЬ ИЛИ ВЫХОД ПРОЦЕССА ПОГЛОЩЕНИЯ 19
где Ьп — число поглощающих молекул в слое, а кЪп— общая
непроницаемая площадь, покрываемая молекулярными
„мишенями" в пределах 1 см2, предполагая, что они не перекрывают
друг друга, т. е., что концентрация поглощающих молекул
невелика. Перепишем (10) в виде:
— W,=kNM (11)
где справа стоит произведение числа пролетающих через слой
за 1 сек. фотонов иа число молекул в слое, а слева число
исчезнувших фотонов. Это выражение может быть уподоблено
уравнению кинетики химической реакции с тем отличием, что
здесь происходят соударения или встречи не между
материальными частицами, а между фотонами и молекулами. В таком
толковании коэффициент k представляет собой значение
„выхода" или эффективности рассматриваемой реакции
поглощения фотонов.
Если световой поток имеет величину 1 —£—j то из за-
J см15 сек.
кона (1) величина поглощенной энергии в слое получается
при /= 1 равной: — Ъ/= кпЪх=кЪп. Пусть концентрация
., л—р. МОЛЬ
поглощающих молекул равна 10 ° 1 тогда число молекул
л
/г iП23
п в см3 равно 10~6 • 1Qqq «101Б. Пусть толщина слоя Ьх =
= 10~1 мм = 1(Г2 см, тогда &п = 1013 и —Ы=к> 1013. Отсюда
для красителей в максимуме спектра поглощения, где кя& 10~16,
^ 7 1 П— Ч ЭРГ 5/ 10"~3 g
получаем:—о/да 10 6 > что дает для — -г- « 1П_12 = 10у
фотонов, поглощаемых за секунду данной тонкой окрашенной
пленкой площадью в 1 см2 под действием светового потока
1 —г Такой световой поток осуществляется, например,
см с^к*
при обычной фотографической экспозиции. На ярком
солнечном свете, где поток по порядку величины в 100 000 раз
больше, т. е. /да Ю5—~ , поглощаемая энергия —оТ,
согласно закону поглощения (1), а следовательно и число
поглощаемых фотонов —u7Vv возрастут пропорционально в 105 раз.
В этиз^ условиях ДО14 фотонов будут поглощаться ежесекундно
1013 молекулами красителя в рассматриваемом окрашенном
тонком слое.
4. Испускание света красителем
Молекулу красителя в нормальном или основном
состоянии с энергией Е° мы будем обозначать в дальнейшем через А,
2*
20 ПОГЛОЩЕНИЕ СВЕТА
а молекулу, переведенную поглощением фотона Av на более
высокий уровень энергии Е* мы будем называть
возбужденной молекулой и обозначать ее через А*.
В отличие от основного состояния, возбужденное состояние
молекулы неустойчиво и имеет весьма краткую
продолжительность существования т. По истечении, в среднем, 10~8 с к,
большинство из числа одновременно возбужденных молекул А*
возвращается в исходное основное состояние А и вновь может
поглотить квант света.
Многие красители, растворенные в жидких или твердых
средах, поглощая свет, дают интенсивное свечение, не
испытывая при этом никаких химических изменений. На\ичие
такого явления флуоресценции или
фото-люминесценции означает, что энергия, освобождаемая при
возвращении возбужденной молекулы в основное состояние,излучается
в виде фотона6. Рассматриваемый элементарный процесс
поглощения и испускания световой энергии может быть записан
следующей схемой:
поглощение: Ava-*-A->A*
испускание: А* -> А -+- Av/e
Спектр испускания красителей представляет собой
широкую полосу, занимающую в спектре, как и полоса поглощения,
протяжение порядка 100 т\)*ь но всегда сдвинутую по
отношению к последней в сторону длинных волн. Такова первая
закономерность явления флуоресценции, которая была давно
обнаружена и сформулирована под названием правила
С т о к с а, согласно которому частота излучаемого света v -
должна быть всегда меньше частоты va света поглощенного,
а следовательно,
Av/<Ava: правило Стокса.
Этот факт означает, что в виде фотона всегда испускается
только часть поглощенного кванта. Остаток Ava — Av.
задерживается в молекуле в виде избыточной термической энергии,
быстро отдаваемой окружению.
В полосе флуоресценции также имеется максимум у
определенной длины волны со спаданием интенсивности испускания
в обе стороны. На фиг. 4 сопоставлены спектральные кривые
полос поглощения и испускания. Для полосы поглощения по
оси ординат отложены значения коэффициента поглощения к
в условных единицах, а для полосы флуоресценции — числа
испускаемых фотонов данной частоты v. Последние числа
получаются путем деления относительных значений интенсив-
4. ИСПУСКАНИЕ СВЕТА КРАСИТЕЛЕМ
'21
полоса
полоса
поглощения
ности испускания F4 иа Av. Для удобства сравнения обеих
спектральных кривых единицы интенсивности подобраны таким
образом, чтобы максимумы кривых были бы на одной высоте.
Характерной особенностью спектральной кривой испускания,
хорошо заметной на фигуре, является ее несимметричный
вид с более крутым спаданием в сторону больших частот от
максимума, обратно тому, что имеет место для кривой
поглощения. Спектральные кривые поглощения и испускания
являются взаимным зеркальным отражением по
отношению к пунктирной вертикали на фиг. 4.
Эта вторая закономерность спектра флуоресценции была
количественно
установлена Левшиным (1931)7.
Третья
закономерность, наблюдающаяся
при флуоресценции
красителей и родственных
им более простых
ароматических соединений,
заключается в том, что
прн возбуждении
монохроматическим светом
(спектральной линией)
в любом месте полосы
поглощения
испускается вся полоса
флуоресценции целиком с
неизменным
распределением в ней интенсив- .—
ности. Разумеется, при возбуждении длинами волн не в
максимуме полосы поглощения, общая интенсивность полосы
флуоресценции соответственно падает.
На фиг. 4 обращает на себя внимание частичное
перекрывание полос поглощения и испускания. При возбуждении
спектральными линиями на участке полосы поглощения,
лежащем влево от пунктирной вертикали, также испускается
вся полоса флуоресценция, включая участок, лежащий вправо
от пунктирной вертикали. При этом, следовательно, нарушается
правило Стокса, так как испускаются, хотя и со слабой
интенсивностью, фотоны, по величине превышающие
поглощенные: Av^ > Ava. Такие частоты спектра флуоресценции
носят название ант и ст о к с о вы х. Дополнительная энергия
k*f— h\9 прибавляющаяся к поглощенному фотону Ava,
черпается из запаса тепловой энергии молекулы.
Факт постоянства спектра испускания при изменении час-
Фиг. 4. Зеркальная симметрия полос
поглощения и флуоресценции родамина 6 G в
ацетоне7.
22 ПОГЛОЩЕНИЕ СВЕТА
тоты возбуждающего монохроматического света в широких
пределах приводит к важному заключению о механизме этого
внутримолекулярного процесса.3) Действительно, поглощение
фотонов различной величины hva несомненно переводит
молекулу из основного состояния на различные уровни энергии.
При монохроматическом возбуждении достигается только
один определенный из этих возможных уровней. Если при
эголг испускается целый набор фотонов hv/9 ипритом всегда
с неизменным распределением интенсивности, то это значит,
что между актом поглощения и актом испускания происходит
какая-то единообразная перестройка возбужденной молекулы,
приводящая к одинаковому состоянию, исходному для
испускания. Такая нивелировка различных состояний
возбуждения является внутренним свойством молекулы, а не вызвана
взаимодействием молекулы с окружающей средой, так как
неизменность спектра флуоресценции наблюдается и в
простейших ароматических соединениях в состоянии разреженного
пара в вакууме, как это было установлено в лаборатории
автора книги8? 21.
Способность молекул отдавать поглощенную энергию в виде
световой энергии количественно характеризует величиной
выхода флуоресценции.
Под квантовым выходом <р флуоресценции понимают
отношение числа фотонов Av/f излученных на всем протяжении
полосы испускания, к числу поглощенных за то же время
фотонов при монохроматическом возбуждении светом данной
частоты va:
число излученных фотонов число молекул излучивших
ф —. .—. с= —_— ■
1 число поглощенных фотонов число молекул поглотившах
Вавилов (1924) показал, что квантовый выход флуоресцен*
ции остается постоянным при возбуждении различными длинами
волн на всем протяжении полосы поглощения9. Для типичных
флуоресцирующих красителей, как например флуоресцеина-Na
(уранина) <р — 0.80, т. е. 80°/0 актов поглощения квантов
света сопровождается излучением энергии в виде фотонов.2)
!) Детальное рассмотрение вопроса о внутренней энергии молекулы см.
в главе 3.
*г) Кроме квантового выхода, можно пользоваться энергетическим
выходом флуоресценции, понимая под ним отношение суммарной
испускаемой энергии к поглощенной монохроматической эяергии. Если для
упрощении принять одну среднюю частоту v>. для всей полосы флуоресценции,
то между квантовым и энергетическим выходом получается очевидное
^v/ л
соотношение: эиергвг, выход = ср ■=—• = Ф<^-» При постоянстве ф и Aj-
энергетический выход пропорционален поглощаемой длине волиы
5. СТРУКТУРНЫЕ СКЕЛЕТЫ И ТИПЫ КРАСИТЕЛЕЙ
23
Выход флуоресценции зависит от структуры соединения,
природы среды и температуры, значительно повышаясь,
например, с понижением температуры10. Ничтожный выход или
полное отсутствие флуоресценции у большинства красителей
в обычных условиях указывает на наличие в молекуле иных
путей превращения энергии поглощенного кванта,рассмотрению
которых посвящается глава 3. К закономерностям
флуоресценции мы возвратимся в параграфе 11 этой главы.
ГЛАЩ ВТОРАЯ
СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
5. Структурные скелеты и типы красителей
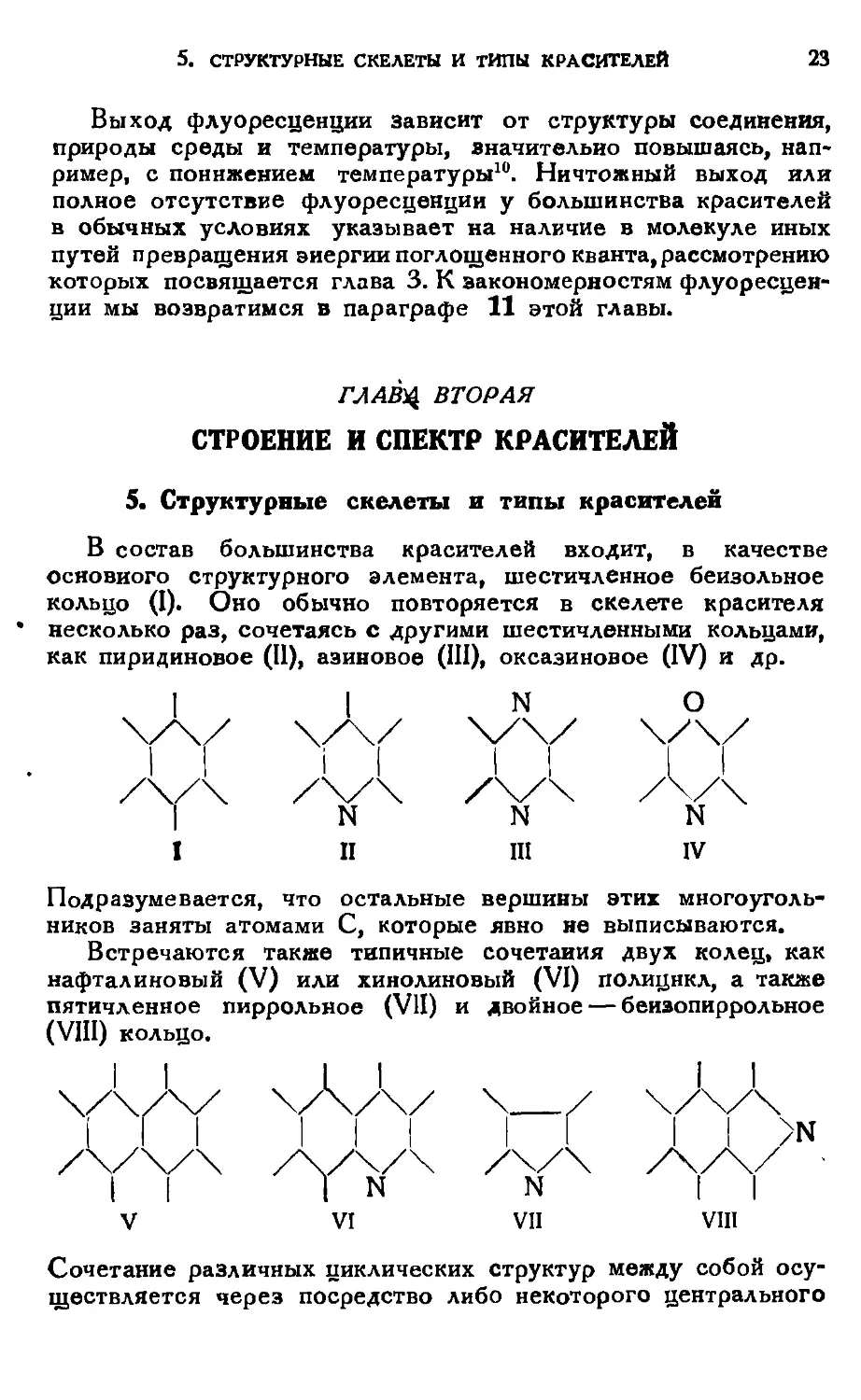
В состав большинства красителей входит, в качестве
основного структурного элемента, шестичленное бензольное
кольцо (I). Оно обычно повторяется в скелете красителя
несколько раз, сочетаясь с другими шестичленными кольцами,
как пиридиновое (11), аэиновое (III), оксазиновое (IV) и др.
I
N
\У\/
N
III
О
N
IV
Подразумевается, что остальные вершины этих
многоугольников заняты атомами С, которые явно не выписываются.
Встречаются также типичные сочетания двух колец, как
нафталиновый (V) или хинолиновый (VI) полицнкл, а также
пятичленное пиррольное (VII) и двойное — беизопиррольное
(VIII) кольцо.
\
N
VII
VIII
Сочетание различных циклических структур между собой
осуществляется через посредство либо некоторого центрального
24 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
атома, либо цепочки атомов, например, азогруппы —N = N—,
полиметиновоЧ цепи —СН —СН— СН-—, азометиновой цепи
— CH = NH—CH=NH— и других звеньев.
В диаграмме 1 (на отдельном листе) приведены
схематические структуры почти всех классов красителей, а также
формулы строения конкретного представителя каждого класса.
Внутри циклон нами совсем не обозначено положение
двойных валентных связей, фигурирующих при обычном способе
написания этих формул. Это сделано намеренно по той
причине, что вторая черта валентности имеет существенно иную
природу, чем первая, и не может быть приписана определен^
ному месту в молекуле (см. следующий параграф). Агомы Н
при циклах как обычно не выписаны. Приведенные структуры
не являются собственно формулами строения красителей,
а воспроизводят, в основном, только геометрическую
конфи гурацию атомов в молекуле, т. е. дают ее скелет.
Диаграмма содержит только наиболее простые в
структурном отношении красители. Замещение атомов Н при кольцах
радикалами СН3, С2Н5 или галоидными атомами;
использование, вместо бензольного кольца, полициклон; введение суль-
фогруппы SO3H и тому подобные структурные усложнения —
создают богатое разнообразие красителей, используемых на
практике.
Все приведенные структурные скелеты являются по
существу плоскими. Когда водородные атомы при кольцах
замещаются объемистыми группами, может произойти нарушение
плоскостного расположения и деформация молекулы, так как
замещенные кольца вынуждены выйтн из общей плоскости.
Это обычно сопровождается отчетливым гипсохромным
эффектом, т. е. сдвигом полосы поглощения в сторону коротких
длин волн.
При составлении диаграммы структурных скелетов
красителей мы не имели намерения дать классификацию
красителей, что представляет собой трудную и поныне еще
окончательно не решенную задачу.
Геометрическое расположение атомов, разумеется,
недостаточно для характеристики более тонких деталей строения
красителя, определяющих его химическое я оптическое
поведение. К этим особенностям структуры мы и переходим
в дальнейшем изложении.
Красители, имеющие наибольшее значение» являются
солеобразными соединениями, т. е. представляют собой
положительные или отрицательные молекулярные ионы, как
явствует из примеров диаграммы 1. Соответственно имеются
„катионные" и „анионные" красители, хотя такое название на
5. СТРУКТУРНЫЕ СКЕЛЕТЫ И ТИПЫ К->АСМТ£\Е Й 25
практике не привилось, а пользуются иными терминами,
приводимыми ниже.
Существенным в структурной формуле красителя является
присутствие концевых групп —NR2, —OR (R_=H или ра-
дилал), которые получили в химии красителей название ауксо-
хромов (греч. „ауксо" — помогаю). Такое название
указывает, что эти группы выявляют или усиливают цвет исходного
слабоокрашенного соединения. Оптическое действие
заместителей типа NH2 или ОН заметно в простейших производных
бензола, спектр поглощения которых остается в пределах
ультрафиолетовой области. Так, например, спектр анилина
CtH5-NH2 не только сместился в сторону длинных волн, по
сравнению с бензолом, но и поглощение в нем увеличилось,
примерно, в 10 раз. Аналогичные батохромный и гиперхромный
эффекты вызывают введение подобных групп а более сложные
соединения, являющиеся исходными для получения красителей.1)
Однако следует подчеркнуть, чго типичный для красителя
резкий сдвиг полосы поглощения в видимую область с
одновременным сильным возрастанием поглощения не обусловлен
одним только присутствием указанных характерных
заместителей — ауксохромов. Более существенно то обстоятельство,
что ауксохромы способствуют приобретению нсей окрашенной
молекулой заряда, положительного или отрицательного,
либо создают условия для появления противоположных
зарядов внутри нейтральной молекулы. Другими словами, особое
значение имеет нонообразующее нли ионогенное действие
ауксохромов (см. 8). -
Причина появления избыточного заряда в молекуле
красителя заключается в том, что ауксохромы или иные полярные
группы исходного соединения обладают отчетливо выраженной
основной или кислотной функцией, т. е. обладают
способностью присоединять или отдавать протон Н+. Первая
способность характерна для трехвалентного атома азота,
например в группах =NH или —NR2, вторая—для фенольной
группы —ОН.
Они сообщают исходному, слабо окрашенному, соединению
свойства основания или кислоты. Взаимодействие с сильной
кислотой в первом случае, со щелочью — во втором и
приводит к солеобразным сильно окрашенным красителям
согласно реакциям: i_i+
Chr = NH +- Н+, СГ -> Chr = NH, СГ -> [Chr — NH2]+ СГ
Chr-OH + Na+,OH'-> [Chr — ОГ-*-H+ -4- Na+, OH"-*
->[Chr —
Четкое определение понятия „ауксохром" было дано Измаильским
15
26 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
В этих формулах и в дальнейших Chr условно обозначает не
рассматриваемую более детально основную часть структуры
красителя (хромоген).
Красители, представляющие собой катионы [А]"1",
образовались, таким образом, в результате реакции слабого
органического основания Chr = NH с сильной кислотой НХ.
Кати о иные красители называются также основными.
Наряду с указанным выше, так называемым хиноидиым
основанием Chr = NH, для красителей с центральным атомом С
(ди- и трифенилметановых) имеется продукт присоединения
к основанию молекулы воды H2N—Chr — ОН, который
содержит группу ОН, связанную с центральным атомом С,
подобно спиртам или карбинолам. По этой причине такое
соединение называют карбииольным основанием.
Превращение в солеобразный краситель под действием кислоты
протекает здесь иначе, а именно:
— Ph)3C — ОН ч- Н+, СГ -> [(R2N — Ph)3 С]+СГ -+- Н2О,
бесцветный краситель
где для сокращения ароматическое кольцо обозначено
знаком Ph (фенил). Под действием щелочи на краситель реакция
протекает в обратном направлении (см. 18). Замена группы
ОН атомом водорода превращает карбинольное основание
в бесцветное же гидрированное производное красителя
(R2N — Ph)3C — Н, которое носнт название лейкоосно-
вания красителя (греч. „лаукос"— белый).
Группы — NR2» а также > NR и ^N, присутствующие в кати-
онном красителе, могут также присоединять протон Н+, т. е.
продолжают проявлять в ослабленной степени свойства
основания. При этом цвет красителя изменяется (см. 14).
Присоединение к красителю, наряду с протонами Н4" эквивалентного
числа электронов, нейтрализующих заряд, т. е. процесс
гидрирования красителя двумя атомами водорода 2Н уничтожает
«го цвет, превращая в бесцветную лейко-форму (28).
Красители с цветным анионом [А]~ или [А]
представляют собой, наоборот, соль исходной слабой органической
кислоты Chr — ОН, Chr — СООН или Chr — SO:jH, обычно
натриевую, получившуюся при реакции этой кислоты со
щелочью (см. схему реакции, приведенную выше). Анионные
красители принято называть кислотными.
Ионизации фенольной группы ОН в исходном соединении
Chr — ОН с отщеплением протона благоприятствует
присутствие таких заместителей, как — NO2, — NO. Однако часто
5. СТРУКТУРНЫЕ СКЕЛЕТЫ И ТИПЫ КРАСИТЕЛЕЙ
27
образование аниона красителя обеспечивается наличием, в
качестве заместителей, заведомо кислых групп:
или
о
и придающих молекуле
отно но о
карбоксильная сульфогруппа
легко отщепляющих протоны Н"
рицательный заряд.
Кислотные и основные группы не только делают краситель
растворимым в воде, но также служат для непосредственного
фиксирования красителя на белковом волокне животного
происхождения (шерсть, шелк).
Примером основного красителя может служить желтый азо-
краситель хризоидин (1), а кислотного — краситель той же
группы оранж II (II).
СГ
Na
НО
II
Простейшим кислотным красителем является нитрокраситель —
пикриновая кислота (диаграмма 1).
Существуют, однако, многочисленные красители с
электрически нейтральными молекулами (диаграмма 1). В иих
имеется возможность внутренней ионизации путем
пространственного разделения противоположных зарядов (стр. 35).
Необходимо считаться с тем обстоятельством, что при
изменении концентрации водородных ионов среды краситель
может изменить число атомов водорода, заряд и спектр
поглощения, присоединяя новые протоны или отдавая их среде.
На этом, как известно, основано применение некоторых
красителей в качестве индикаторов рН среды. В диаграмме 1
даны структурные формулы, свойственные нормальным
условиям применения данного красителя.
Наряду с разделением красителей на основные и
кислотные, своеобразие технологических приемов крашения тканей
28 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
привело к выделению некоторых красителей в особые группы
по признаку метода фиксации на волокне.
Красители, окрашивающие непосредственно волокна путем
адсорбции из раствора, назынаются субстантивными.
Однако в узком смысле субстантивными называют группу
азокрасителей, способную прямо окрашивать целлулозные
волокна (хлопок) путем адсорбции коллоидных частиц крася-
теля из водного раствора. Их называют также прямыми.
Наиболее важный технологический метод, приводящий
к прочной фиксации красителя, есть синтез красителя
непосредственно на волокне с помощью той или иной химической
реакции. В соответствии с этим различают следующие главные
группы:
_К_у б овые красители, которые образуются путем отнятия
кислородом воздуха водорода от бесцветной лейко-формы
красителя, раствором которой пропитывается ткань. К ним
принадлежат индигоиды и полициклокетоновые красители
(индантрены).1) Для кубовых красителей типична обратимость
реакции окисления, т. е. присоединение двух атомов водорода
восстанавливает исходную леЙко-форму.
Ледяные красители „проявляются" на ткани путем реакции
слабо окрашенных или бесцветных соединений непосредственно
на волокне. Например, путем сочетаний соли диазония (I)
с натриевой солью (II) ^-нафтола на целлулозном волокне
образуется азокраситель (III):
Na"
II
OaN -/ V- N = N —<^ \-ь Na-% СГ
нет
III
пара-красный
Протравные красители фиксируются на тканях,
предварительно обработанных растворами солей металлов Al, Fe,
г) Следует отметить, что гидрированная, т. е. лейко-форма последних
красителей отнюдь не бесцветна, но интенсивно окрашена.
6. ВАЛЕНТНЫЕ СТРУКТУРЫ 29
Cr, Sn. К ним принадлежат некоторые антрахиноновые
красители, например ализарин (диаграмма 1).
Краситель образует с металлом прочное комплексное
соединение, в котором ион металла, замещая водород гидро-
ксильной группы, входит в скелет молекулы:
Органические протравы с кислотными свойствами
применяются для крашения хлопчатобумажных тканей основными
красителями.
6. Валентные структуры
Атомы С, N, О, S, входящие в скелет красителя, соединены
между собой, прежде всего, одиночными валентными
связями С — С, С — N, С— О, N — N, С — S, имеющимися в
предельных соединениях. Черта валентности здесь условно
изображает два „поделенных" электрона, связывающих только
оба этих атома.
Элзктроны одиночной связи не удаляются сколько-нибудь
значительно за пределы междуатомного промежутка, создавая
в последнем некоторую избыточную плотность отрицательного
заряда, как бы стягивающую положительно заряженные остовы
атомов.
Иное имеет место для второй пары электронов в
двойной снязи, валентное насыщение которых принято изображать
в классических структурных формулах с помощью второй
черты валентности, тождественной первой. Одинаковому
с простой связью способу начертания противоречит, однако,
большая реакционноспособность и подвижность этих
электронов „второго рода" в непредельных и ароматических
соединениях, несмотря на то что прочность молекулы в целом
и увеличивается.
Локализац/ я или приурочивание двойных связей к
определенным местам в цепочке сопряженных связей или в аро-
30
СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
матическом кольце не передает химических свойств
соединения. Бензол, например, одинаково описывается как формулой 1>
так и формулой II, в которой двойные связи передвинуты
на одно звено:
Н Н Н
А с — Г Г — R ш
н н н
А=С—С=С—В iv
Полиметиновая цепочка, связывающая два ауксохрома в
красителе, также допускает два равноправных способа
написания (III) и (IV), так как ауксохромные группы А и В способны
изменять свою валентность на единицу.
Подвижность второй черты валентности в подобных
соединениях пытались, по предложению английских химиков
(Робинсон, Ингольд), изображать изогнутыми стрелками или дугами:
Стрелки указывают на способность электронов смещаться из
одного междуатомного промежутка в другой. Это смещение
неизбежно влечет за собой передвижение электронов по всему
скелету, причем, как указано, в этом перемещении участвуют
также свободные электроны ауксохромных групп.
Делокализацию таких электронов „второго рода"
правильнее изобразить пунктиром, охватывающим все
участвующие атомы, например:
НИН
Необходимо, таким образом, допустить, что электроны
второй черты валентности принадлежат в некоторой мере
всей молекуле как целому, а не одной лишь паре атомов.
Электроны, осуществляющие одиночную связь и
„поделенные" только между соседними атомами, получили
спектроскопическое наименование с-электронов. Обобществленным же
6. ЗАЛЕНТНЫЕ СТРУКТУРЫ
электронам присвоено название подвижных электронов, или
электронов второго рода, или те-электронов.
В современной теории электронной структуры органических
соединений принят следующий несовершенный способ
изображения валентных структур, вызванных подвижностью тс-элек-
тронов11. Выписывают все возможные, для данной
конфигурации атомов, распределения вторых черт валентности или,,
другими словами, все электромеры молекулы. Ни одна из
этих структур не существует в отдельности. Истинное
распределение тс-электронов в реальной молекуле может быть
передано, как сосуществование всех этих структур
в одной суммарной результирующей или „гибридной"
структуре. Каждая отдельная валентная структура входит в нее со
своим относительным или „удельным* весом. Например,
электронное строение молекулы бензола передается в основных
чертах двумя структурами Кекуле (I) и тремя структурами
Дьюара (И). Пропорция, в какой эти структуры представлены
в бензоле, выписана под ними:
Необходимо помнить, что разложение реальной молекулы
на составляющие „электромерные" структуры является данью
ограниченности наших средств изображения. Сложность
соотношений, имеющих место в электронной оболочке молекулы,
недоступна наглядному представлению с помощью
упрощенных валентных структур, но может быть выражена
математическим языком квантовой механики.
Сосуществование в одной молекуле различных валентных
структур было впервые осознано самими химиками, которые
дали этому явлению название мезомерии (греч. „мёзос" —
средний). Физики обосновали этот эффект теоретически н
называют его квантово-механическим резонансом структур.
К этому вопросу мы возвращаемся в следующем параграфе.
Принцип Паули» Ионизованные структуры. Прн
построении электронных формул строения оказалось необходимым
учитывать и такие валентные состояния атома, когда в нем
создается избыточный положительный или отрицательный
заряд. Такая внутримолекулярная ионизация атома возникает
32 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
в результате утраты или приобретения им электрона и
существенно меняет его привычную классическую валентность.
Рассмотрим подробно этот важный пункт п.
Спектроскопия позволяет провести тонкое разграничение
между различными видами электронов, которые в химических
структурных формулах фигурируют как совершенно
тождественные точки или валентные черты.
В атоме или молекуле электроны могут занимать только
совершенно определенные квантовые состояния, или орбиты
по старой терминологии, которым присвоена нумерация с
помощью определенных квантовых чисел, некоторые из
которых для удобства обозначения заменяются буквами. Так,
например, в атоме имеются электроны на орбитах Is, 2s, 2р0,
2р+1, 2p_j, а в молекуле—электроны та, шг,
Квантовый принцип, лежащий в основе периодической
системы элементов Менделеева и носящий название принципа
исключения или принципа Паули, гласит, что в каждой
орбите, занумерованной определенными квантовыми числами,
может поместиться не более пары электронов. Электроны
должны при этом быть „спарены", т. е. взаимно насыщены
s валентном смысле.
Последнее выражение может быть сформулировано более
точно на основании того факта, ч/го электрон ведет себя как
заряд, обладающий вращением вокруг собственной оси или,
по-английски, — спином (spin), наподобие волчка. Электрой
проявляет себя поэтому не только как элементарный
отрицательный заряд, но и как элементарный магнитик,
обозначаемый стрелкой f . Последний символ дает ось и
направление вращения, а следовательно, расположение магнитных
полюсов, создаваемых вращением.
При валентном насыщении илн „спаривании" двух
электронов, их магнитные стрелки должны иметь противоположные
Направления f[ или, как говорят, спины должны быть
антипараллельными.
На квантовой орбите атома может присутствовать
одинокий электрон. Тогда он свободен для валентного
взаимодействия с другим атомом, т. е. представляет собой свободную
валентность. В ту же квантовую орбиту может вступить и
второй электрон, но только при условии насыщения или
антипараллельности спинов. Свободные валентности этих электронов
как бы. смыкаются, не проявляясь во вне.
Если второй электрон имеет спин, совпадающий по
направлению или параллельный спину первого \\, то такие два
электрона по принципу исключения не могут находиться на
<одной квантовой орбите, но должны разместиться иа разных
b. ВАЛЕНТНЫЕ СТРУКТУРЫ
33
орбитах в качестве одиноких, валентно свободных
электронов.
Внешняя оболочка атомов С, N, О, имеющая главное
квантовое число 2 (оболочка L по терминологии
рентгеновских спектров), содержит 4 орбиты, которые символически
могут быть представлены в виде четырех квантовых „ячеек
заполняемых электронами:
I I I
Если все эти ячейки
содержат по паре электронов, то мы получаем завершенный
октет электронов химически инертного атома неона.
Предшествующие стадии заполнения этих ячеек реализуются
в оболочках предшествующих неону атомов С, N, О, приводя
к нормальным валентностям этих атомов, а именно: IVC,
mN, UO, как показано в крайнем левом столбце диаграммы 2-
Однако в условиях взаимодействия с другими атомами
внутри молекулы появляется тенденция к утрате или
приобретению электрона. В следующих столбцах диаграммы 2
показано, что удаление электрона из квантовой ячейки илн
вступление в нее нового электрона не только создает у атома
избыточный заряд соответствующего знака -ь или—, но
существенно меняет его нормальную валентность, т. е. число
валентных электронов, которыми он располагает. Валентные
связи атомов С, N, О с соседними атомами в структурном
скелете молекулы показаны в диаграмме в виде различных
комбинаций черт валевтности, исходящих из дайного атома.
Спаренные электроны, не занятые в связях, обозначены парой
точек; свободный непарный электрон — одной точкой. Знак
заряд атома указан справа сверху, валентность — римской
цифрой слева сверху.
t
t
г*
IIIN
н
t
t
> N- >v Ы
"0
Н
n
t
t
t
t
с
t
t
>c< >c=
t
t
♦
(Аммониевый)
'o-
H
u
t
III.
♦
♦
(Карбетеббш)
H
t
t
>N~
• •
(AasHvamoBbiu)
'"o+
M
t
t
t
w
t
t
t
>c
(НарбениатоВый)
I t t t
(АммениеВый)
>0 =0 -0:"t " ' =O-
Диаграмма 2. Валентные состоянья атомов.
3 А. Н. Твревив
34 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
Из диаграммы 2 следует, что четырехвалентный атом
углерода IVC, заряжаясь положительно или отрицательно,
неизбежно утрачивает или приобретает один валентный электрон,
а следовательно, теряет одну свободную валентность, делаясь
трехвалентным ШС+, ШС~. Атом кислорода может
принимать валентные состояния Ю~ и П1О+. Большее
разнообразие мы встречаем у атома азота, а именно JIN~, IVN"*",
mN+ i)
Изображенные валентные состояния атомов энергетически
неравноценны, т. е. их осуществление путем приема илн
отдачи электрона требует затраты энергии различной величины.
По этой причине состояния ШС~, nN~, ШО"*" осуществляются
реже и имеют меньший удельный вес, a IVC превалирует над
111 С"1". Наибольший вес в красителях имеют структурные
формулы с IVN"*" и Ю~~. Следует, однако, оговориться, что все
возможные валентные состояния должны быть учтены при
расчете структуры соединения.
Преобладание внутриионизованных структур в
нейтральных молекулах органических соединений непосредственна
сказывается в аномальной большой величине их дипольиого
момента11.
В молекулярных ионах красителей, несущих избыточный
заряд, вес ионизованных структур должен быть значительным
по той причине, что этот заряд в некоторой доле должен
обязательно осуществляться какими-либо атомами.
В свете приведенных разъяснений структурные скелеты
диаграммы 1 теряют свою необычность и могут служить
канвой для начертания всех валентных структур данного красителя.
Например, электронная оболочка иона аурамина может быть
представлена совокупностью следующих электромериых
структур (см. след. стр.).
Числа в скобках обозначают число вариантов данной струк ■
туры, если принять во внимание две различных формулы
Кекуле бензольного кольца. В дальнейшем, для упрощения,
зеркально симметричные структуры, заключенные в фигурных
скобках, будут выписываться только в виде одной формулы
и снабжаются сверху двойной стрелкой вида: -с—> • Наличие
двух структур Кекуле будет всегда подразумеваться.
Структуры (III) и (IV) аурамина с энергетической точки
зрения менее выгодны, чем предыдущие, так как содержат на
1) Наличие этих состояний валентности в солеобрпзитях соединениях
давно известно химикам, которые да\и им следующие наименования: ионы
карбения Н1С+, карбениат* ШС", аммония IVN ' р пвеииати UN"™, аяения HN*
аммония HIN4
6. ВАЛЕНТНЫЕ СТРУКТУРЫ
35
R2N
I
(г)
NR.
X\tO
'NH.
1
I]
III
I +■
IV
Аурамнн
одну связь меньше. По этой причине структуры с IVN+
будут всегда преобладать над структурами с П1С+.
При написании структурных формул красителей обычно мы
встречаемся с характерным сочетанием ароматического (бен-
зоидного) кольца с хиноновым или хиноидным, как это имеет,
например, место в формулах (I) аурамина. Возможность хнно-
идной формулировки структуры красителей принималась одно
время за причину цветности. Она служит до сих пор основой
классификации красителей.
Приведем в заключение в качестве дополнительного
примера главные внутриионизованные структуры красителя с
нейтральной молекулой — индиго11а:
0
и
H
H
I
c&-<fc>
A
н
6
(4)
36 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
Из предыдущего следует, что атомы в скелете красителя
скреплены не только одиночными валентными связями,
имеющимися у предельных соединений, но также тг-электронами
вторых связей, распределенными по всему скелету.
В результате междуатомные связи приобретают прочность
промежуточной величины между одиночной и двойной связью.
Междуатомные расстояния соответственно также сокращаются
по сравнению с чисто одиночной связью.
Нивелировка одиночных и двойных связей в молекуле
красителя убедительно показана на примере фталоцианииа,
в котором с помощью рентгеновских лучей было установлено
полное равенство междуятомных расстояний в цепочке сопря-
I I I
женных одиночных и двойных связей: = С—N = C — N = ...
1 34 1.34 1.34
внутреннего цикла этой молекулы (диаграмма 1 и фиг. 7)12«
7# Строение электронной оболочки
Движение электронов в молекуле не подчиняется обычным
механическим законам, справедливым для макроскопических
тел с большой массой, а управляется совсем иной —
квантовой механикой. Природа электрона в атоме н молекуле такова,
что мы не в состоянии точно нарисовать его траекторию,
а можем лишь приблизительно дать как бы
статистическую картину его пребывания в различных точках
пространства, окружающего положительные ядра атомов.
Другими словами, в явлениях атомарного масштаба электрон
проявляет себя не как точечный заряд, а как расплывчатое
облако с общим отрицательным зарядом, равным заряду
электрона е. Можно себе представить это явление так, как
если бы электрон за краткое мгновение был способен обежать
всё предоставленное ему в атоме или молекуле пространство.
Тогда плотность отрицательного заряда около каждой точки
пространства, окружающего атом или молекулу, будет вызвана
большей или меньшей длительностью пребывания
электрона около этой точки. Заряд в электронном облаке
распределен отнюдь не равномерно, и строение облака рассчитывается
квантовой механикой.
Два спаренных электрона, вступивших в одиночную или,
иначе, (г-связь, образуют единое Еытянутое облако отрицттель-
ного заряда, симметричное вокруг прямой, соединяющей атомы.
Большая часть заряда, равного 2е, сосредоточена и интервале
между атомами. Группы атомов, сочетаемые одиночной связью,
обладают свободой вращения покруг нее по причине осевой
симметрии электронного облака.
7. СТРОЕНИЕ ЭЛЕКТРОННОЙ ОБОЛОЧКИ
37
Прочно связанные электроны одиночной связи не участвуют
в поглощении света красителем в видимой области, а потому
нас интересовать в дальнейшем не будут.
Облака 7г-электронов, о которых была речь в предыдущем
параграфе, имеют совсем иную ориентацию по отношению
к прямой, соединяющей атомы. А именно: каждый тс-электрон
дает двойное облако, показанное на фиг. 5* Заряд электрона
в таком двойном облаке распределен симметрично вокруг оси,
перпендикулярной направлению одиночной связи, а еле-
7Z- электрон
7Г электрон
Фиг, 5. Двойные облака двух разделенных 7г-элек-
тронов.
Контуры соединяют места с одинаковой
плотностью электронного заряда, подобно
геодезическим линиям на карте. Плотность заряда плавно
убывает от центра облака к его периферии, что
показано убывающей толщиной линии.
Распределение заряда в облаке симметрично вокруг
пунктирной вертикальной оси.
довательно, перпендикулярной плоскости ароматического
кольца. Большая часть заряда тг-электрона сосредоточена
вблизи двух „фокусов", расположенных по обе стороны
плоскости молекулы, на расстояниях порядка 1 А от нее.1) При
приближении к плоскости кольца плотность электронного заряда
быстро падает и делается в ней равной нулю. Это означает,
что 7г-электрон все время пребывает вне плоскости молекулы,
что делает понятным большую его реакционную способвость
по сравнению с <г-электронами одиночной свяэи.
В своеобразной форме облака электрона, изображенной
на фиг. 5, непосредственно проявляются волновые свойства
-1)
Междуатомное расстояние С — С в ароматическом кольце составляет
38
СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
квантовой механики, управляющей его движением.
Возможность пребывания электрона с двух сторон плоскости при
отсутствии связующего пути, пересекающего последнюю, есть
следствие хорошо известной в оптике и акустике способности
волн к интерференции. Действительно, пространство,
окружающее положительные ядра атомов, может быть
уподоблено картине стоячих волн, образующихся в результате
пробегания волной объема, ограниченного отражающими
стенками. Плотность электронного облака минимальна или даже
равна нулю в тех местах, где волны взаимно гасятся из-за
противоположности фаз и вычитания амплитуд
перекрещивающихся колебаний. Наоборот, там, где колебания складываются,
сосредоточивается максимальная доля заряда электрона.
Амплитуда стоячей
волны в каждой точке
пространства
обозначается в квантовой механике
буквой ^. Плотность
заряда в каждой точке
электронного облака
пропорциональна
интенсивности колебания, т. е.
квадрату его амплитуды ф2*
л - л , В ароматическом кольце
Фяг. 5а. Срединонные облака тг-алек- имеется Щесть ^дектро-
тронов, осуществляющих 7Г-связь. г
нов, доставляемых шестью
атомами С, а следовательно, шесть облаков изображенного
на фиг. 5 вида, накладывающихся друг на друга.
Они образуют два общих кольцеобразных облака,
расположенные по обеим сторонам плоскости кольца (фиг. 5а). Каждому
тс-электрону предоставляется, таким образом, возможность
выходить за пределы своего атома и находиться в любом
месте обобществленного электронного облака, расположенного
с двух сторон скелета. Свободные пары электронов у атомов —
N <С и — О — также принадлежат к разряду тс-электронов
и вливаются в общую электронную оболочку молекулы.
Распространение заряда одного тг-электрона по всему
скелету молекулы иллюстрируется в фнг. б, основанной на кван-
тово-механическом расчете. В ней изображен костяк радикала
трифенилметила (ССН5)3С-, обладающего одним стюбодным
непарным электроном, причем числа на фигуре означают долю
заряда этого электрона, приходящуюся на различные места
молекулы. Только около трети заряда электрона
сосредоточено у центрального атома; остальная часть распределена
7. СТРОЕНИЕ ЭЛЕКТРОННОЙ ОБОЛОЧКИ
О
00/в
Ю07В
р
бодного ЭА«ктРона по
дикала трифеиилметила
равномерно между атомами С в орто- и параположениях.
Пунктирные окружности соединяют все точки, где амплитуда
стоячей волны ф, а
следовательно, и плотность электрона
(пропорциональная ф2) равна
нулю. В промежуточных зонах
амплитуда и величина заряда
достигают максимального
значения. Детальное рассмотрение
поведения тг-электронов самих
бензольных колец показывает,
что они перераспределяются
талим образом, что
неравномерность заряда в кольцах, созда- "'
ваемая свободным электроном, Фиг. 6. Распределение заряда
своими компенсируется к ни один
агол* С колец не выделяется
по величине электронной плотности от другого.
Наличие в ароматических циклах общего электронного
облака, охватывающего
все атомы С, а также
подвижность -^-электронов
непосредственно
доказывается измерениями
магнитной восприимчивости
производных бензола- А
именно» помещение этих
соединений в магнитное
поле вызывает
противодействие, эквивалентное
индукции в молекуле
электронного тока, текущего
по замкнутому контуру.
Из измерения величины
этого диамагнитного
Фиг. 7, Кольцевые токи в молекуле фта- эффекта получается, что
лоцианина1*. Контуры токов обведены ПОПервЧНИК
жирной чертой. Разделенные облака
^-электронов показаны в плане в виде
концентрических окружностей.
кольцевого
тока, индуктированного в
молекуле, сравним с
размерами бензольного
шестиугольника. В красителе фталоцианине, наряду с кольцевыми
токами бензольных ядер, наблюдается также кольцевой
электронный ток, охватывающий внутренний цикл сопряженных связей
— C = N — C = N — ♦••• (фиг. 7)с площадью порядка 50 А2 и.
40
СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
Картина распределения общей плотности электронного
заряда («у-и ти-электронов совместно) может быть получена для
молелул с помощью рентгеновских лучей. На фиг. 7а
приведена такая „топографическая карта" молекулы фталоцианина.
Квантово-механическая интерференция волн ф,
принадлежащих всем ти-электронам, не только приводит к
обобществлению облаков и опре-
f деленному
распределению плотности
электр онного заряда
1 в пространстве,
окружающем скелет
молекулы, но
одновременно вызывает и
снижение энергии
результирующего
состояния по сравнению
с суммой энергии
тс-электронов,
раздельно взятых (фиг. 8,
на стр. 45). Разность
энергий А/?
представляет собой
отдаваемую энергию, т. е.
экзоэнерге тический
эффект образования
молекулы из атомов,
"' вследствие
взаимодействия их
валентных электронов.
Такова
энергетическая сущность всякой
валентной связи, бу-
Фнг, 7а. Распределение суммарной плотмости Дет ЛИ она Простой
электронного заряда в молекуле фталоцианина
О
2
3
\
5А
по рентгеновским данным12.
ОДИНОЧНОЙ СВЯЗЬЮ
(У-электронов или де-
локализованной
связью тс-электронов. Выигрыш энергии А/? и измеряет
прочность связи.
Действительно, как показывают термохимические данные для
Теплот сгорания и гидрогенизации, энергия бензола на34.4кгкал
ниже, чем сумма энергий, выделяющихся при образовании
шести связей С — Н, трех связей С — С и трех связей С = С
по формуле Кекуле. Та^м образом, благодаря
обобществлению 7>электронов, бензол на 34.4 кгкал стабильнее или
7. СТРОЕНИЕ ЭЛЕКТРОННОЙ ОБОЛОЧКИ 41
прочнее, чем одна структура Кекуле. Изобразим это
соотношение с помощью термохимического уравнения:
+ 3 *384кгкал = g jl ч- 344 какал
Н Н^^|^ЧН энергия образования Н"^Ч^^Н энергия
трех ддоиных связей 1 обобществления
" (резонанса)
В более сложных ароматических полициклах такая
стабилизация молекулы в результате обобществления те-электронов
доходит до величин свыше 100 кгкал (фиг. 9 иа стр. 47).
Указанный здесь экзоэнергетичегкий эффект является
непосредственным следствием суперпозиции или, как говорят,
„резонанса" различных возможных валентных структур
данного соединения, которые были рассмотрены в предыдущем
параграфе. По этой причине энергию дополнительной
стабилизации молекулы (34.4 кгкал, в случае бензола) называют
энергией „резонанса" или „резонансной" энергией11.
Необходимо указать еще на одно следствие обобществления
7г-элелтронов, а именно: увеличение жесткости плоскостной
конфигурации соединения. Действительно, всякая деформация
молекулярного скелета, выводящая атомы из общей плоскости,
либо при поперечном сгибе, либо при кручении одной части
молекулы относительно другой, неизбежно нарушает
параллельное расположение и максимальное перекрывание двойных
облаков re-электронов в фиг. 5. Полное их разобщение
наступает при повороте оси симметрии одного из них вокруг
одиночной связи на 90° по отношению к оси другого. Это
произойдет, например, если 7г-облако концевой группы ауксохрома
— N <С путем вращения выводится из совпадения с 7г-облаками
ароматического кольца, с которым она связана. Но для такого
разобщения спаренных между собой обобществленных
электронных облаков необходимо затратить работу. Это равносильно
поднятию энергетического уровня молекулы с самого низкого
его нормального положения, обусловленного „резонансом",
до более высокого, когда „резонанса" практически нет.
По этой причине связи, соединяющие ауксохромные группы
с ароматическим кольцом в красителе, отличаются от
одиночной ^вязи. В частности, они не только обладают несколько
повышенной прочностью по сравнению с последней, но
обладают сопротивляемостью кручению, т. е. утрачивают в
некоторой мере свободу вращения, возможную для нормальной-
одиночной связи.
42 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
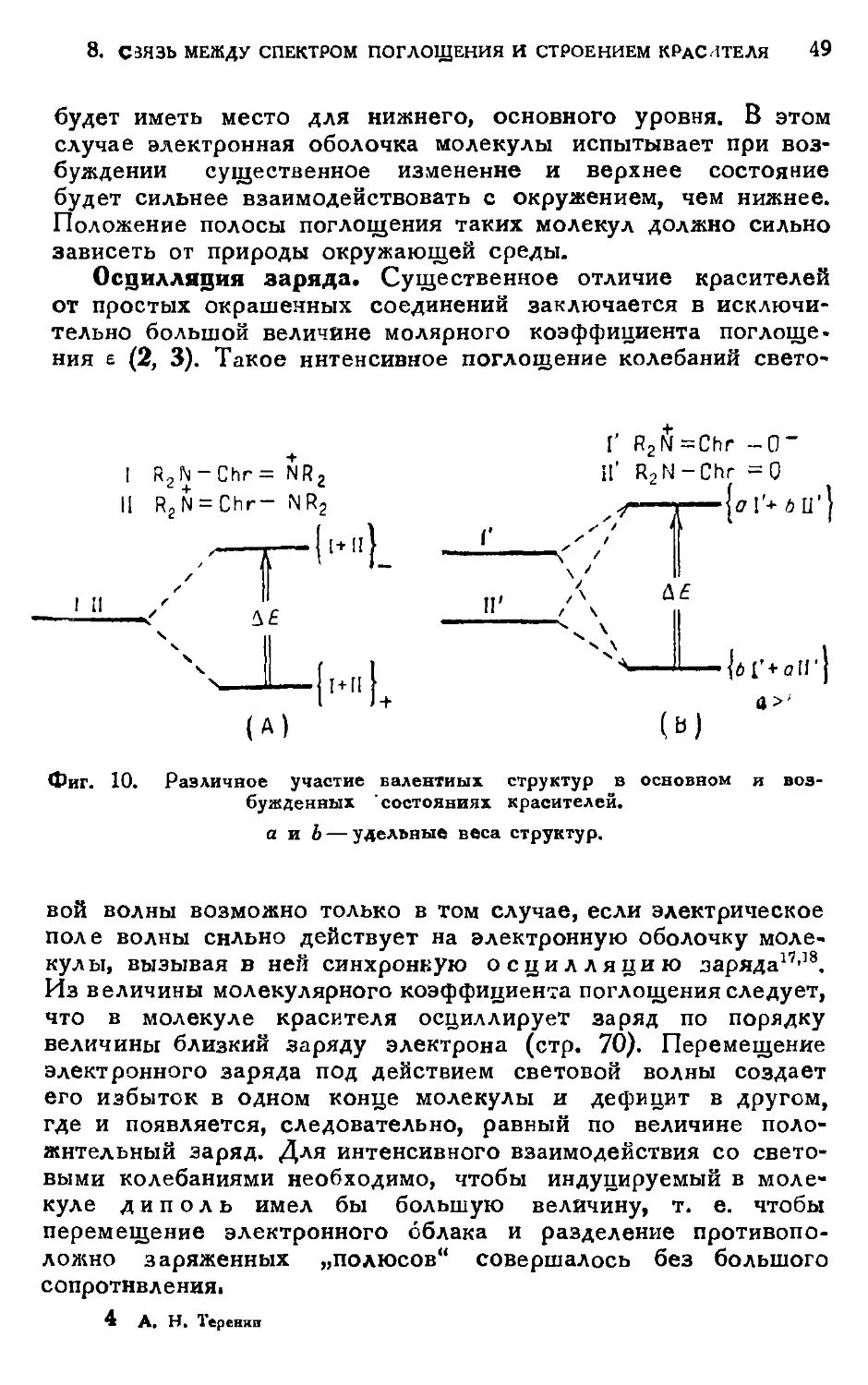
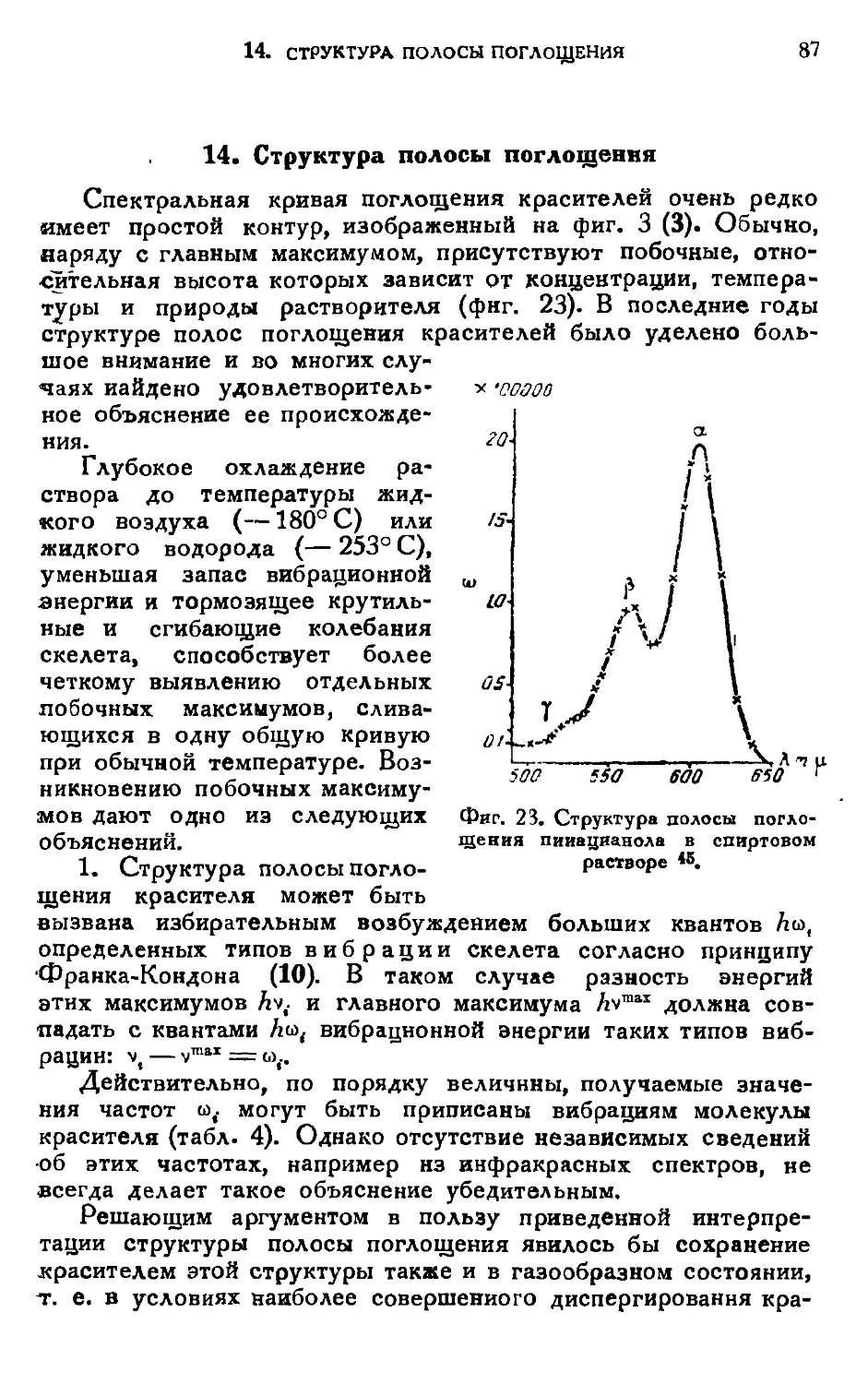
Sm Связь между спектром поглощения и строением красителя
Проблема происхождения цветности органических
соединений и ее зависимости от структуры молекулы уже давно
привлекает внимание химиков. Начиная с середины прошлого
века предлагались различные теории цветности, в
которых цвет, т. е. появление и передвижение полосы
поглощения в видимом спектрэ (1), приписывался той илн иной
особенности структурной формулы соединения.
Гребэ и Либерман (1868) впервые указали на
ненасыщенный характер окрашенных соединений. Витт (1876) при^
писал цветность присутствию определенных валентно ненасы-
I I
щенных групп, так называемых хромофоров: — С=С —
— N = N — ,]>С = О, — NO2 и др. Кроме того, он
подметил существенную роль других групп ауксохромов:
— NH2, — NR2, — ОН, — OR и. др., углубляющих цвет (1) и
придающих соединению красящие свойства, т. е. способность
фиксироваться на тканях. Нецкий и Армстронг выдвинули
в 80-х годах сохранившуюся до сих пор в кла^ификации
красителей точку зрения о хиноидной структуре
окрашенных соединений (стр. 35).
Знаменательным результатом попыток создать теорию
цветности, исходя только из химических данных, было
признание неудовлетворительности структурных формул
классического учения о валентности. Неспособность этих формул
передать такое основное свойство органического соединения,
как цвет, дало позод к изображению окрашенных соединений
с помощью „осциллирующих" валентных структур (Байер,
1907; Порай-Кошиц, 1910). Позднейшие химические теории
цветности еще больше приближаются к современной
концепции омезомерии молекулы (Измаильский, 1915—1939; Арндт,
1924; Кёниг, 1925; Ингольд, 1930; Эйо-терт, 1935 и др.).
Измаильский впервые ввел понятие о „мезозтроении" или „мезо-
состоянии" красителей, отличая его от предельных идеальных
структур молекулы. Он же впервые изобразил строение
красителей в виде молекулярных ионов, поместив знак заряда
за скобку и не фиксируя его на определенном атоме К таким
формулам пришли затем Фнрц (1918), Гантш (1921) и другие.
Развитию вопроса о цветности органических соединений
посвящена богатая литература, рассмотрение которой выходит
за рамки настоящей книги15. Можно упомянуть книгу Вицин-
гера (1933), последовательно прозодящую точку зрения,
высказанную ранее Дильтеем (1920), согласно которой цветность
•обусловлена координативио ненасыщенными атомами и и оно-
8. СВЯЗЬ МЕЖДУ СПЕКТРОМ ПОГЛОЩЕНИЯ И СТРОЕНИЕМ КРАСИТЕЛИ 43
идным состоянием молекулы красителя. Теория Дилътея-
Внцингера в своих качественных формулировках удачно под-
м^гила некоторые особенности строения, ответственные
за появление интенсивного поглощения в видимой области
спектра. Недостатком ее является произвольный выбор
хромофорных центров и ауксохромов, а также отсутствие учета
электронных формул строения. Развиваемые химиками теории
охватывали большей частью ограниченный круг красителей
и требовали нередко натянутых предположений или даже
неверных утверждений, как, например, пятивалентности
азота.
Химические теории цветности сводились обычно к чисто
внешнему, феноменологическому описанию качественных
соотношений между строением и цветностью. Действие света
на молекулу в них сформулировано нечетко.
Общий их недостаток состоит, прежде всего, в том
обстоятельстве, что они ограничивались большей частью
рассмотрением различных структур нормального состояния молекулы,
проячляющегося в химическом опыте. Между тем акт,
поглощения кваита света вызывает переход молекулы из
нормального ее состояния в энергетически более высокое состояние.
Положение полосы поглощения в спектре, а следовательно
и цветность, определяется не одним состоянием молекулы,
а комбинацией или разностью энергий двух состояний
А* и А (3). Заранее нельзя сказать, имеет ли состояние
А* то же электронное строение, как нормальное состояние
молекулы А, к которому относится структурная формула,
получаемая из химических даииых. Состояние А*, достигаемое
при поглощении кванта света, может вообще не быть
осуществимо химическими средствами не только потому, что оно
требует затраты энергии порядка 40—70 кгкал/моль, т. е. его
осуществление представляет собой сильно эндотермический
процесс, но такжз ввиду специфичности действия света.
Следовательно, структурная формула молекулы красителя в
энергетически верхнем из двух состояний остается нам
неизвестной. Однако, положение дела не столь безнадежно, как может
показаться на основании приведенного соображения. Самый
факт существования и качественной применимости химических
теорий цветности, исходящих из нормальной структурной
формулы соединения, показывает, что поглощение кванта
видимого света не изменяет существенно скелета и
электронного строения молекулы красителя. Тем не м^нее, для
создания подлинной теории цветности принципиально необходимо
учитывать оба электронных состояния молекулы А* и А, между
которыми совершается переход при поглощении фотона.
44 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
Квантово-механическая теория цветности. Разработка
проблемы цветности методом квантовой механики, начатая
физиками сравнительно недавно (Склар, 1937; Фёрстер, 1939),
положила начало количественному этапу ее развития16. Для
бензола, полицчклических углеводородов и некоторых других
простейших соединений оказалось возможным рассчитать длину
волны поглощаемого им света, исходя только из геометрической
конфигурации скелета и взаимодействия тг-электронов.
Подобное количественное предсказание спектра для типичных
красителей затрудняется исключительной сложностью
математического расчета и громоздкостью вычислений, которые пытаются
в настоящее время обойти полуэмпирическим путем. Не
производя полного расчета, оказывается тем не менее возможным
предсказать направление смещения спектра красителя при
изменении его структуры.
В основе квантово-механической теории цветности лежат
те же волновые свойства электронов, которые обуслоэливают
валентность, мезомерию и химическую связь.
В предыдущем параграфе было указано, что делокализация
те-электронов, несовершенно изображаемая посредством
суперпозиции валентных структур, приводит к понижению
энергетического уровня молекулы по сравнению с энергией
изолированной структурной формулы. Такая т стабилизация соединения
не имеет прямого отношения к величине поглощаемого им кванта
света. Она говорит лишь о повышении его химической
стойкости, например, к гидролизу. Однако интерференция стоячих
волн ф, эквивалевтная суперпозиции различных валентных
структур давного соединения, имеет своим следствием не только
понижение его уровня энергии, но также появление второго,
более высокого уровня. Поглощаемый квант света, а
следовательно длина волны полосы поглощения, определяется
интервалом между этими уровнями (3).
Рассмотрим происхождение второго уровня иа простейшем
примере бензола.
Пусть строение последнего описывается суперпозицией
только двух структур Кекуле Кг и К2 (6). Эти структуры,
если они могли бы существовать изолированно, имели бы
одинаковую энергию, которую можно изобразить в виде двух
уровней Кг и KVf находящихся по шкале энергии на одной
высоте (фиг, 8). Каждая структура Кекуле соответствует
в квантовой механике своя стоячая волна ф, с частотой
колебания и. Энергия каждой отдельной структуры Кекуле
определяете частотой и колебания, согласно знакомому иам
универсальному соотношению Ек =£к =EK=hv. Следует помнить,
8. СВЯЗЬ МЕЖДУ СПЕКТРОМ ПОГЛОЩЕНИЯ И СТРОЕНИЕМ КРАСИТЕЛЯ 45
что и не есть частота световой волны, а стоячей волны
с амплитудой ф, определяющей распределение электронного
заряда в молекуле.
Суперпозиции двух структур Кекуле в бензоле отвечает
в квантовой механике интерференция двух стоячих волн
tyK и ф^. Это явление отдаленно напоминает картину
сложения двух связанных между собой колеблющихся систем,
например двух взаимно „связанных" электрических колебательных
контуров. По аналогии с ними две стоячие волны ф^ и ф^а,
взаимодействуя друг с другом, либо складываются в каждой
точке пространства: tyK -+- tyK (симметричное колебание), либо
I
S
I
Ed'
>3 /
/
/
/
/
/
LR
Е-
Фнг. 8. Два электронных уровня бензола, как
результат резонанса валентных структур.
вычитаются: tyK—tyK (антисимметричное колебание). Другими
словами, в результате „связи" колеблющихся систем
появляются два новых типа колебания.
В первом типе результирующая стоячая волна имеет
частоту более низкую, чем чаотота исходного невозмущеиного
колебания и: и+ <и; во втором результирующем колебании
частота и_>и. Частоты и_^ и и_ будут тем больше отличаться
от первоначальной частоты и изолированных друг от друга
систем, чем сильнее их взаимодействие или „связь". Стоячие
волны структур Кекуле Кх и ЛТ2 сильно „связаны" или
возмущают друг друга, а потому частоты и+ и и_ весьма
различны.
Каждому результирующему стационарному колебанию <|>
соответствует определенная энергия молекулы бензола, а именно:
£.=Ли_- и ZT_ = Au_.
46 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
Получается, что суперпозиция двух структур Кекуле,
обладавших одинаковой энергией, приводит к расщеплению
первоначального общего энергетического уровня Ек на два
новых (фиг. 8).
Нижний уровень Е+ соответствует нормальному или
основному состоянию бензола. О нем была речь выше и А/?
иа фиг. 8 означает энергию стабилизации или „резонанса".
Верхний энергетический уровень, возможный для молекулы
бензола, соответствует возбужденному состоянию А*, о
котором была речь в параграфах 3 и 4.
Величина кванта света, поглощаемого бензолом, а
следовательно положение полосы поглощения в спектре,
определяется разностью энергий Е_ и E4_\hv=E_— Е+=^&Е. Как
указывалось выше, сильное взаимодействие структур Кекуле
сказывается в большой величине расщепления Л£, равной
у бензола 110 кгкал/моль. Поэтому, согласно соотношению,
приведенному на стр. 18, спектр поглощения бензола
оказывается расположенным вблизи 260 тр, т. е. в
ультрафиолетовой области.
Необходимо указать на характерную особенность
происхождения спектра поглощения бензола, типичную для
соединений подобного рода. В верхнем состоянии молекулы А*,
достигаемом поглощением света, протяженность электронной
оболочки остается такой же, как в ниж'нем состоянии А. Оба
состояния отличаются только способом сочетания стоячих
волн $к и ijv, а именно с одинаковыми нли
противоположными фазами колебания. Отсюда следует, что верхнее
возбужденное состояние бензола столь же мало подвержено
влияниям среды, как и его основное состояние. В этом заключается
существенное отличие ароматических соединений от более
простых молекул и атомов, возбуждение которых приводит
к увеличению протяженности электронного облака, а также
частичному разрыву валентных связей, т. е. создает
предпосылку для большей чувствительности возбужденного состояния
к внешним воздействиям.
Выше мы оставили без внимания три структуры Дьюара
(6). Их учет приводит к увеличению расщепления &Е только
на 8°/о и к появлению новых уровней, расположенных над £_.
Если пренебречь растянутой валентной связью
противоположных (пара) атомов С, то каждая структура Дьюара примерно
на одну 7г-связь, т. е. примерно на 38.4 кгкал (ср. стр. 41)
менее прочна, чем структура Кекуле, и, следоиательно,
расположена на более высоком уровне энергий Ео (фиг. 8).
Стоячие волны ф структур Дьюара интерферируют со стоячими
8. СВЯЗЬ МЕЖДУ СПЕКТРОМ ПОГЛОЩЕНИЯ И СТРОЕНИЕМ КРАСИТЕЛЯ 47
волнами структур Кекуле, несмотря на такое значительное
различие энергий, а следовательно, частот колебания и.
Интерференция волн квантовой механики тем самым принципиальна
отличается от явлений механического и оптического резонанса,
требующих совпадения энергий и частот колебания
взаимодействующих систем. Укоренившееся наименование эффекта
квантово-механического взаимодействия структур термином
„резонанс" структур следует поэтому признать неудачным.
На фиг. 9 воспроизведено относительное расположение
нижнего и верхнего энергетических уровней для ряда а рома-
хгхал
50-
л
и
50-
/00-
150-
>с=с<
1
Л/9
"о
—
со
—
—
—
ссо -
т ОХО -
—
6
VI
—
/У rYVVY4! VII
сохо
Фиг. 9, Электронные уровня основного а возбужденного состояния для ряда
ароматических углеводородов.
тических углеводородов. В качестве нулевого уровня отсчета
принята здесь энергия двойной связи этилена. В результате
резонанса валентных структур нижний — основной энергетический
уровень углеводородов — испытывает смещение внизу на
величину энергии резонанса. Мы видим, однако, что верхний, эоз-
бужденный, уровень смещен вниз на еще большую величину.
В результате, при переходе от бензола к пентацену (V)
интервал между уровнями, а следовательно поглощаемый молекулой
фотон, прогрессивно уменьшается и спектр поглощения
сдвигается в сторону длинных волн (батохромный эффект). У фуль-
вена (VI), имеющего одинаковое число двойных связей с
бензолом, но иное строение, энергия резонаиса в нижнем состоянии
составляет, примерно, половину энергии резонанса бензола,
но верхний уровень смещен вниз значительно больше, чем
48 СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
у бензола. Поэтому спектр фульвена сильно смещен в сторону
длинных волн и фульвен окрашен. С другой стороны, у
антрацена (III) н фенантреиа (VII), имеющих почти одинаковую
энергию резонанса в нижнем состоянии, возбужденнее верхние
уровни заметно отличаются по высоте расположения, вызывая
несовпадение спектров.
Отличие верхнего состояния от нижнего, ясно показанное
на фиг, 9, вызвано тем, что, хотя оба состояния
осуществляются одними и теми же валентными структурами,
относительные удельные веса структур в верхнем состоянии могут
быть иными, чем в нижнем. Расчеты показывают, что в
верхнем состоянии ароматических углеводородов усиливаются
такие структуры, которые слабо представлены в нормальном
нижнем состоянии соединения. А именно, преобладает обмен
и взаимодействие далеко отстоящих тг-электронов, как это
имеет место для растянутой валентной сбязи между
противоположными атомами в структурах Дьюара (стр. 31). Такие
структуры, равносильные увеличению делокалиаации
электронов, способствуют увеличению энергии резонанса в верхнем
состоянии и большему смещению верхнего состояния по
сравнению с нижним. Однако в бензоле в верхнем состоянии
превалируют именно структуры Кекуле, а не Дьюара.1*
Данное выше описание сущности эффекта сохраняется
и для более сложных соединений типа красителей. Число
сочетаемых различных валентных структур в них значительно
больше; энергетические уровни структур сближаются, так как
взаимное возмущение их стоячих волн i|/t- падает, а
следовательно, уменьшается и величина энергетического
расщепления Д£ между основным и ближайшим верхним уровнем.
В результате уменьшается величина кванта поглощаемого
света и полоса поглощения передвигается в область видимого
спектра.
Если нон красителя построен симметрично (фиг. 10,А),
то „резонирующие" структуры I и II входят в равной мере
в основное и возбужденное состояние. Если же структура
красителя несимметрична (фиг. 10, В), то составляющие
структуры I' и II' различны. Энергия рнутриионизованной структуры
Г выше нейтральной II' из-за работы, требующейся для
разделения противоположных зарядов в первой. По этой причине
в результирующем верхнем уровне внутриионизованиая
структура I' будет присутствовать с большим весом, „а", чем
классическая структурная формула Ц' (вес „Ь"). Обратное
:
2) Изложенные здесь сведения любеяио сообщил мие до их опубляко*
ваиия Я. К, Сыркии.
8. СЗЯЗЬ МЕЖДУ СПЕКТРОМ ПОГЛОЩЕНИЯ И СТРОЕНИЕМ КРАСИТЕЛЯ 49
будет иметь место для нижнего, основного уровня. В этом
случае электронная оболочка молекулы испытывает при
возбуждении существенное изменение и верхнее состояние
будет сильнее взаимодействовать с окружением, чем нижнее.
Положение полосы поглощения таких молекул должно сильно
зависеть от природы окружающей среды.
Осцилляция заряда. Существенное отличие красителей
от простых окрашенных соединений заключается в
исключительно большой величине молярного коэффициента
поглощения е (2, 3). Такое интенсивное поглощение колебаний свето-
[' R2Й =Chr -
R?N-Chr= NR2 II* R2N-Chr =0
R2N = Chr- NR2 r
/
! il
/
f
4
^ /
\
4
ч
4
4
N \
^
(A)
Фиг. 10. Различное участие валентных структур в основном и
возбужденных состояниях красителей.
а и Ь — удельные веса структур.
вой волны возможно только в том случае, если электрическое
поле волны сильно действует на электронную оболочку
молекулы, вызывая в ней синхронную осцилляцию заряда17'18.
Из величины молекулярного коэффициента поглощения следует,
что в молекуле красителя осциллирует заряд по порядку
величины близкий заряду электрона (стр. 70). Перемещение
электронного заряда под действием световой волны создает
его избыток в одном конце молекулы и дефицит в другом,
где и появляется, следовательно, равный по величине
положительный заряд. Для интенсивного взаимодействия со
световыми колебаниями необходимо, чтобы индуцируемый в
молекуле диполь имел бы большую величину, т. е. чтобы
перемещение электронного облака и разделение
противоположно заряженных „полюсов" совершалось без большого
сопротивления!
4 А. Н. Теренин
50
СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
Подвижность электронов в молекуле зависит от
способности входящих в ее состав атомов принимать н отдавать
электроны. Таким образом ова должна быть связана с
легкостью осуществления внутриионизованных структур. Не следует,
однако, думать, что световая волна вызывает осцилляцию
самих валентных структур с переходом одной в другую.
Как было сказано выше, осциллирует в молекуле под
действием электрического поля световой волны только ничтожная
доля общего заряда электронной оболочки.
Представление об осцилляции заряда в молекуле под
действием световой волны подтверждается следующими
фактами17. Длина волны поглощаемого света непосредственно
зависит от протяжения цепи сопряженных связей, соединяющей
ауксохромы, на которых появляется заряд при „резонансе"
структур.
Известно, что чем длиннее растянутый проводник, тем
больше длина волны собственных электрических колебаний,
которые в нем могут быть возбуждены, а следовательно, тем
больше длина волны, на которую он лучШе всего отвечает.
Если эту аналогию распространить на молекулу красители,
то следует ожидать, что длина волны поглощаемого света
для гомологического ряда красителей будет пропорциональна
длине молекулы. Фактически это и наблюдается для ряда
карбоциакинов типа:
R -
о
- (СН= СН)Я - СН =
— R
и
С —(СН = СН)„ —СН =
для которых найдена линейная зависимость между длиной
волны максимума поглощения Хша* и числом п метановых
групп (фиг. 11).
Если заряд имеет возможность появиться на каком-либо
промежуточном атоме длинной цепочки, то такое „укорочение"
размаха его перемещения сказывается также в виде уменьше-
8. СВЯЗЬ МЕЖДУ СПЕКТРОМ ПОГЛОЩЕНИЯ И СТРОЕНИЕМ КРАСИТЕЛЯ 51
ния длины волны поглощаемого света (гипсохромный эффект).г)
Так, желтый краситель аурамин (I) имеет максимум
поглощения у более коротких длин волн и, следовательно, более
„высокий" цвет, чем сходный по строению гидрол Михлера
(II) или зеленый Биндшедлера (III) с более длинным путем
заряда:
R2N
К vox
£20 my
111
720my,
Повышение цвета или гипсохромный эффект введения атомов
N, О, S в качестве мостиков между кольцами делается также
понятным с этой точки зрения, так как создает более короткий
путь для перемещения заряда. Это следует, например, из
сопоставления „резонирующих" структур малахитового зеленого
(IV) и акридинового оранжевого (V):
„Рассеивание" мезомерного эффекта по терминологии Измаильского16
52
СТРОЕНИЕ И СПЕКТР КРАСИТЕЛЕЙ
W00 Л
800
1-6GG
Введение третьего ауксохрома—NR2, имеющее место в
кристаллическом фиолетовом (VI), хотя и увеличивает число
„резонирующих" структур по сравнению с (IV), тем не менее
перемещает поглощение в сторону коротких длин волн, т. е;
производит действие, равносильное укорочению пути заряда.
Последний парадоксальный факт выводится теоретически
при квантово-механическом рассмотрении соо гветствующих
структур16- Наглядно
объяснить его можно тем,
что, при перемещении
заряда между двумя аук-
сохромами — NR2, третий
ауксохром в (VI) „оттяги*
вает" часть заряда на себя,
укорачивая его путь.
Подобные
поверхностные аналогии с
электрическими колебаниями в
проводниках помогают
только при качественном
рассмотрении
соотношений между спектром и
структурой красителя.
Более углубленная трактовка
требует применения
квантовой механики.
Особенно рельефно
факт осцилляции
электронного заряда в молекуле
под действием световой
волны, ею поглощаемой,
проявляется при поглощении поляризованного света,
в котором колебания электрического поля совершаются
перпендикулярно лучу в одной плоскости. При падении такого
луча на молекулы, ориентированные определенном образом
по отношению к плоскости его колебании, величина поглоще-
ния, а также поглощаемая длина волны зависит от ориентации .
Коэффициент поглощения наибольший, когда световые
колебания в падающем луче совершаются вдоль плоскости
молекулы; другими словами, когда затрагивается наибольшее
протяжение электронной оболочки. Поглощение имеет
значительно меньшую величину и происходит и другом участке
спектра, когда колебания света совершаются перпендикулярно
плоскости молекулы. Этот эффект, давно установленный
на кристаллических окрашенных соединениях (например азо-
400
POQ
О
о
} 2 3 4 # 5 6 ;
Число двойных связей
Фиг. 11. Пропорциональность
поглощаемой длины волны света
геометрической длине молекулы красителя17.
8. СВЯЗЬ МЕЖДУ СПЕКТРОМ ПОГЛОЩЕНИЯ И СТРОЕНИЕМ КРАСИТЕЛЯ 53
бензоле), носит название дихроизма. Действительно, бывают
случаи, что пропускаемый свет дважды меняет цвет при
повороте кристалла по отношению к плоскости
поляризации падающего света, но большей частью различия в
полосах поглощения обнаруживаются только спектральным
путем.
Как неоднократно указывалось выше, интенсивная полоса
поглощения в видимой области может появиться в слабо
окрашенных соединениях в результате перехода нейтральной
молекулы в ионное состояние. Это имеет место при соле-
образовании, когда от молекулы отрывается или к ней
присоединяется протон Н~*" (5). Прд этом все тс-электроны образуют
пары и число их остается четным.
Но есть другой путь ионизации молекулы, который можно
назвать окислительным (стр. 185), заключающийся в том,
что из молекулы удаляется один нз спаренных электронов,
например один электрон из свободной пары атома N в аук-
сохроме — NR2,
В результате молекула приобретает не только
положительный заряд, но и валентную ненасыщенность, превращаясь
в весьма реакционно-способный свободный радикал с одним
непарным электроном. Опыт показывает, что все такие
неустойчивые формы соединения сильно окрашены. К ним
относятся, например, красители типа Вурстера, окраска которых
вызвана „резонансом" валентных структур с непарным
электроном: ,
/—\ ь
t I )
Глубокая и интенсивная окраска подобных „окисленных"
форм соединения, по сравнению с исходной, нередко
бесцветной молекулой, есть результат повышенной подвижности
электрона из-за появления пустой квантовой „ячейки" в атоме
N~*~. Катионы, содержащие атом азота в таком валентном
состоянии ^ N+f носят название аммениевых (ср. 6).
Радикалы подобного типа получаются на промежуточной
стадии „окисления" соединения, когда нейтральная молекула
лишается одного электрона (28). Они носят название полу-
или семихинонов, в связи с тем, что такая неустойчивая
54 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
форма возникает на промежуточной стадии „окисления*
гидрохинона
б—С >—О" или б—< >—О:".
ГЛАВА ТРЕТЬЯ
ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
9. Вибрационная энергия
Внутренний запас энергии молекулы, т. е. высота ее
энергетического уровня, определяется, прежде всего, состоянием
ее электронной оболочки, как это было описано выше. Этот
основной вид молекулярной энергии, который носит название
электронной энергии, в дальнейшем будет обозначатьси
Ее. Скачкообразное изменение электронной энергии в акте
поглощения фотона (3, 4) приводит к повышению
энергетического уровня молекулы от первоначального нормального
значения Ее° до значения Е* верхнего возбужденного
состояния электронной оболочки (фиг. 14 а на стр. 57).
Однако внутренняя энергия молекулы обусловлена не
только состоянием ее электронной оболочки- Другим
существенным видом энергии, которым она обладает, является
энергия вибрационного движения, совершаемрго_около поло-
жевий равновесия ядрами атомов, входящих в ее состав.
Главную долю * теплоемкости сложной молекулы составляв*
именно запас вибрационной энергии, возрастающий
пропорционально температуре. При комнатной температуре для
красителей он достигает величины порядка 100 кгкал/моль.
Вращательное движение молекулы красителя, как целого,
совершается весьма медленно, и его кинетическая энергия
вносит лишь ничтожную долю в общий энергетический баланс;
поэтому мы ею пренебрегаем.
Траектории вибрационного движения атомов в скелете
молекулы чрезвычайно сложны, но, в отличие от электрона,
могут быть при малых амплитудах вибрации достаточно точно
воспроизведены простой механической моделью масс,
соединенных друг с другом упругими связями, наподобие пружин,
сопротивляющихся также и изгибу. В механике доказывается,
что сложное движенае такой системы можег быть разложено
на простые составляющие, а именно: на дискретный ряд линей-
9. ВИБРАЦИОННАЯ ЭНЕРГИЯ
55
ных колебаний с различными частотами ы19 ыл, ы3 f
тп (сек."1). Таким приемом сложное колебательное движение,
фактически имеющее место в молекуле, разлагается на своего
рода механический спектр частот ^(/=1, 2, 3, л),
содержащихся в суммарном движении. Каждая из них
соответствует совершенно определенному типу вибрации скелета
молекулы.
На фиг. 12 изображены 5 типов вибрации бензольного
кольца из 30 для него возможных. Через полупериод
направление движения ядер атомов, указанное стрелками, меняется
на обратное. Чем сложнее соединение, тем разнообразнее
формы его вибраций. Число различных типов вибраций,
совершаемых скелетом красителя, достигает 100—200.
Ш
(с-с)
608 см-1
IV
-Цс-с)
О
V
Фиг. 12. Некоторые типы вибрации скелета бензола и нх частоты (в см""1).
Частоты вибраций молекулярного скелета по порядку1
величины в 10—50 раз меньше частот v видимого спектра,
а следовательно, соответствующие длины волн ^ в 10—50
раз больше видимых и лежат в инфракрасной области.
Действительно, ряд частот вибрации молекулярного скелета
обнаруживается непосредственно в виде отдельных узких
полос поглощения в этой области. Это означает, что свет
подходящей частоты 6>t способен возбудить соответствующий
тип вибрации молекулярного скелета. Обычно же
вибрационному движению энергия доставляется междумолекулярными
столкновениями. Но особенно большие порции энергии
подводятся вибрациям одновременно с актами поглощения
и испускания фотона Av, о чем подробно говорится ниже.
Всякое увеличение или уменьшение вибрационной энергии
подчиняется основному квантовому закону скачкообразного
изменения, т.' е. перехода с одного уровня вибрации на
другой.
Таким образом, для каждого вибратора, колеблющегося
с частотой «t>o существует последовательность дискретных
56
ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
уровней его энергии, и энергетический интервал между
уровнями определяет величину кванта Ло\ вибрационной энергии,
воспринимаемого или отдаваемого нм (фиг. 13).
По мере увеличения амплитуды и энергии данного типа
вибрации, связи, удерживающие атомы в положениях
равновесия, ослабевают, следовательно частота о>( вибратора
уменьшается. Это сказывается на фигуре тем, что интервалы между
соседними вибрационными уровнями, т. е. Лсо(, делаются
меньше по мере роста вибрационной энергии-
В условиях теплового равновесия при данной температуре
Г(абс.) на каждый независимый вибратор или, как говорят,
вибрационную „степень свободы" молекулы приходится в
средою г» /о кгкал \
нем энергия К/, где R — газовая постоянная \&I МЛ^1,ГЯ7Г у
моль rf ад
ЙТ
Фиг. 13. Уровни энергии отдельных типов вибрации.
При комнатной температуре (293° абс.) это составляет
около 0.6 кгкал/моль. Эта энергия, как показано на фигуре,
поддерживает вибратор с малой частотой «3 на высоких
уровнях его энергии, которые образуют нерасчлененную,
фактически сплошную, полосу. Но эта энергия недостаточна для
возбуждения даже одного кванта вибрации высокой частоты
Wj. Однако фактически вибраторы взаимодействуют между
собой при больших амплитудах колебаний, и средняя энергия
каждого из ннх повышается за счет взаимной связи их
движения.
Общая вибрационная энергия Ev молекулы, т. е.
вибрационная часть ее теплоемкости, равна сумме энергий всех ее
вибраторов. Суммарная энергия, очевидно, также изменяется
скачкообразно с одного результирующею уровня Еп на
другой EJ (фиг. 14 а).
В нижнем своем электронном состоянии Е^ молекула уже
обладает некоторым запасом вибрационной эйергии Evt соз-
9. ВИБРАЦИОННАЯ ЭНЕРГИЯ
57
даваемым тепловым движением. В результате статистического
выравнивания (см. 10) подавляющее большинство молекул
будет находиться на уровне £е = .св -
Допустим, что, при поглощении фотона hv и переходе на
верхний электронный уровень Ее*у запас вибрационной
энергии, сосредоточенный в молекуле, не изменяется. В таком
случае энергия поглощенного кванта hva равна в точности
интервалу Av0 между уровнями электронной элергии Е* и Ев°
(фиг. 14 а). Но такой случай является исключением. В дей-
Л £t
■дг.
Фиг. 14. (а и Ь). Электронные Ее и вибрационные Е9
уровни энергии молекулы и переходы между ними при
поглощении и испускании света.
ствительности всегда существует сильное взаимодействие
между состояннем электроиной оболочки и внбрацией атомов
по той причине, что упругие силы, удерживающие атомные
ядра в положении равновесия, обусловлены именно
электронами.
При электронном возбуждении с уровня Е6° на уровень
Е*, одновременно, как правило, происходит и изменение
вибрационной энергии. Пусть при акте электронного
возбуждения вибрационная энергия повысилась с уровня EV=EV до
уровня EJ, т. е. испытала прирост EJ — EvT=kEv (фиг. 14 а).
Б таком случае, по закону сохранения энергии, произошло
поглощение не Av0, д большего кванта Ava, равного сумме
58 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
изменений электронной и вибрационной энергий, вместе
взятых:
hva = hv0+bEe. (1)
Наглядно это совместное изменение электронной и
вибрационной энергий изображается обычно таким способом, что
система уровней вибрационной энергии £* вычерчивается не
только при нижнем электронном уровне 2ГД но и при
верхнем Е* (фиг. 14 Ь). В таком случае легко видеть, что
поглощаемый квант fo*a изобразится стрелкой, начинающейся на
уровне Е9 и оканчивающейся на уровне EJf так как его
величина дается суммой (1).
Мы молчаливо допустили здесь, что интервалы между
уровнями Еь1 т. е. вибрационные частоты tot, актом
электронного возбуждения не изменяются, т. е., что верхняя
система вибрационных уровней тождественна с нижней. Это
условие для красителей большей частью приближенно
соблюдается.
Обилие различных вибраций со( в сложных молекулах
типа красителей, взаимное возмущение вибраторов при
больших амплитудах, а также беспорядочное влияние силового
поля среды создают практически сплошное заполнение
интервалов уровней суммарной вибрационной энергии Ev (ср. фиг. 13).
В результате, поглощаемые громадным числом молекул
различные частоты va представляют собой совокупность
нерасчлененных, тесно примыкающих друг к другу
спектральных линий, т. е. практически непрерывную полосу
поглощения, как это и наблюдается на опыте (фиг. 3 и фиг. 4).
Постепенное спадание поглощения в полосе в сторону
больших частот указывает на то, что увеличение
вибрационной энергии при акте поглощения фотона не может
происходить беспредельно, но делается все менее вероятным по мере
того, как сообщаемая порция вибрационной энергии &Е9
увеличивается. Максимальное протяжение полосы в сторону
больших частот дает представление о максимальной порции
вибрационной энергии, доставляемой фотоном. Она составляет
по порядку величины 10—20 кгкал/моль.
Закономерности флуоресценции. Какова дальнейшая
судьба избытка вибрационной энергии &E9f сообщенного
молекуле при поглощении света и повышающего вибрационную
анергию молекулы выше среднего равновесного уровня Е/ при
температуре опыта? За краткое время т«10~8 сек.
возбужденного состояния молекулы эта избыточная энергия успевает
распределиться между многочисленными вибраторами моле-
9. ВИБРАЦИОННАЯ ЭНЕРГИЯ 59
кулы или передаться окружающей среде, восстанавливая
первоначальное равновесное распределение, соответствующее
уровню Еп , На фиг. 14 6 этот процесс утечки вибрационной
энергии показан волнистой стрелкой, обращенной вниз.1)
Если молекула принадлежит к разряду флуоресцирующих,
то для большинства молекул испускание света происходит
именно с равновесного верхнего уровня Ер после
завершения, процесса рассеяния избытка Д£„ (ср. 4).
Величина испускаемого при флуоресценции фотона hif
определится тем нижним уровнем Ev", на котором
вибрационная энергия молекулы окажется в результате испускания.
Явление, имевшее место при поглощении света, сохраняется
и здесь: акт испускания света снова приводит к
возрастанию вибрационной энергии молекулы против равновесного
значения на некоторую величину Д£/>0 и, следовательно:
f0/, (V)
где
Из фиг. 14 Ъ непосредственно следует, что испускаемый
кнант fay будет всегда меньше кванта поглощенного Avfl
на величину суммы избытков энергии, сообщаемых
вибрационным „степеням свободы" молекулы при поглощении Д£в
и испускании &Е/:
hsa-h,f = LE9 + LE/. (2)
Первая порция Д£в утрачена молекулой к моменту
испускания, вторая же Д£/ остается в ней после испускания.
Правило Стокса, требующее соотношения Ava > Av^ (4),
находит, таким образом, углубленную интерпретацию в
терминах внутримолекулярной энергетики. Его соблюдение
связано с тем фундаментальным фактом, что вибрационная
энергия молекулы при восприятии или отдаче фотона, как правило,
всегда повышается.
Зеркальная симметрия спектральных кривых поглощения
и флуоресценции, изображенная на фиг. 4 (4), приводит к
важному выводу. Пунктирная вертикаль на фигуре, которая служит
— ■■ •
*) Необходимо подчеркнуть, что в актах поглощения и испускания
света участвует сравнительно небольшая доля всех вибраторов молекулы.
Избыток Д^Гр сообщается, в основном, только определенвым типам
вибрации. Остальные „неактивные" вибраторы служат своего рода емким
резервуаром, в Который утекает избыток вибрационной энергии, создаваемый
светом, или из которого черпается энергия (см. ниже).
60 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
осью симметрии для обеих кривых, находится по шкале энергий
у значения кванта Av0 чисто электронного перехода Е*— Ее°
(фиг. 146). Кванты Ave и Av^ с одинаковой относительной
гврэятностью (3, 4) поглощения и испускания расположены,
как это следует из фиг. 4, на одинаковом расстоянии справа
и слева от Av0. Но это расстояние в энергетической шкале,
согласно (1) и (1'), есть не что иное, как повышение \ЕЩ
вибрационной энергии, сопровождающее акты поглощения
н испускания. Подавляющее большинство* поглощающих
и испускающих молекул красителя находится в исходном
в з
соответствующем тепловому равновесию молекулы со средой. Таким
образом, из свойства зэркальной симметрии следует вывод,
что акты поглощения и испускания, исходицие с одинакового
нижнего и верхнего вибрационного уровня (фиг. 146), имеют
одинаковую относительную вероятность, если
они сопровождаются одинаковым возрастанием &EV
вибрационной энергии молекулы.
Максимумы на спектральных кривых поглощения ииспускания
находятся у таких значений Ava и hv ., которые соответствуют
наиболее вероятной величине повышения вибрационной
энергии Д2Гв. Вероятность восприятия молекулой порций
вибрационной энергии больших или меньших этой оптимальной падает,
причем особенно быстро в сторону меньших порций, как
это следует из более крутого спадания кривых в сторону оси
симметрии, т. е. с приближением "к Л^о.
Для поглощенных квантов hvay лежащих слева от
пунктирной вертикали: Avff <[ Av0. Из (1) мы видим, что для них кЕь9
т. е. изменение вибрационной энергии, должно быть
отрицательным: &EV <C 0, а следовательно, при акте поглощения
происходит не возрастание, а, наоборот, убывание
вибрационной энергии по сравнению с исходным равновесным зна-
чением Ev . Аналогичное имеет место для квантов
испускаv
ния, лежащих справа от оси симметрии Av, ^> Av0) для которых
также Д^^О: согласно (1') испускание фотона,
превышающего Av0> происходит также за счет уменьшения равновесного
запаса вибрационной энергии молекулы Е9 . Малая
термодинамическая вероятность процесса уменьшения равновесного
запаса термической энергии сказывается спектрально в виде
более крутого падения кривых поглощения и испускания
в том участке^ где они перекрываются.1)
1) По вопросу о соблюдении второго начала термодинамики при ф>
оресц нцни см. Вавилов10*1 и дискуссию в Journal of Physics USSR 10 (1946)
495—506.
9. ВИБРАЦИОННАЯ ЭНЕРГИЯ
^
Г1
Увеличение испускаемого кванта по сравнению с
поглощенным за счет термической энергии молекулы, т. е.
нарушение правила Стокса (4), представляет большой
принципиальный интерес.
На фиг. 15 показан стрелками механизм, каким
излучаемый фотон hvj может оказаться больше поглощенного Ava.
Поглощение исходит из нижнего уровня Ev f на котором
находится ^максимальное число молекул при данной температуре
и заканчивается на верхнем уровне EJy лежащем ниже равно-
весного Еь . За время т существования возбужденного
состояния происходит восстановление равновесного теплового запаса
вибрационной энергии, что по-
казано волнистой короткой =
стрелкой, имеющей длину &Еьй,
Испускание фотона
происходит, следовательно, с верхнего
равновесного уровня
вибрационной энергии Ev . Если акт
испускания, как показано на
фиг. 15, также сопровождается
падением запаса вибрационной
энергии на величину AEV , то
испускаемый фотон будет
превышать поглощенный на сумму
двух порций энергии Д2Гед -н^Д/
черпаемых из теплового
движения.
Нарушение правила Стокса
делав!ся очень большим, когда
поглощение фотона Ava исходит нэ с равновесного уровня
Ео , а более высокого. Поглощение этих фотонов имеет место
в области частот близких максимуму полосы флуоресценции
и ничтожно по своей величине. Чем ниже по сравнению с Е/
расположен уровень, достигаемый при поглощении такого
фотона, тем затруднительнее молекуле за краткое время
возбужденного состояния успеть восстановить равновесное
распределение и подняться до уровня Е/. В этом случае полоса
флуоресценции красителя утратит свое неизменное распределение
интенсивности, обусловленное, как мы видели выше,
достижением одного и того же исходного верхнего уровня (ср. 4).
Этот эффект сделается особенно резким при понижении
температуры, т. е. понижении запаса вибрационной энергии
молекулы и сопутствующего замедления ее перераспределения
между вибраторами. Такое нарушение неизменного вида
Фи1. 15. Переходы между
энергетическими уровнями прв
антистоксовом испускании света.
62 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
полосы испускания действительно наблюдается на опыте 20.
Оно более значительно для соединений с малым числом
атомов, а следовательно, малым запасом энергии в вибрациях,
как например эскулина. Перераспределения интенсивности,
наблюдаемые в спектре флуоресценции прн монохроматическом
возбуждении, служат, таким образом, плодотворным методом
изучения внутримолекулярной энергетики21'22.
Количества энергии, черпаемые при антистоксовой
флуоресценции из запаса термической энергии молекулы,
сравнительно невелики, порядка 10 кгкал. Аномально большое
превышение испускаемого фотона над поглощенным, равное
около 20 кгкал, было установлено для такой простой
молекулы, как анилии в газообразном состоянии при температуре
200° С23.
10. Основной принцип внутримолекулярной энергетики
Максимумы спектральных криных указывают на тот факт»
что поглощение и испускание фотонов определенной
величины имеет наибольшую вероятность, т. е.
происходит наиболее часто. При этом, согласно разъяснениям
предыдущего параграфа, молекулой воспринимается заметный
избыток вибрационной энергии, порядка 2—5 кгкал. Какова
причина такой избирательности процесса?
Объяснение этого явления дает очень важный принцип,
являющийся основой молекулярной энергетики и носящий
название принципа Франка-Кондона.
Для его изложения плоскостной чертеж уровней энергии,
приведенный на фиг» 14, недостаточен. Необходимо его
существенно дополнить картиной изменения запаса энергии моле*
кулы при деформации ее скелета. Другими словами,
требуется изобразить зависимость энергии молекулы от смещения
ядер атомов из положений равновесия. Из фиг. 12 и
сопутствующего текста следует, что виды деформации скелета могут
быть самыми разнообразными, и возрастание энергии
молекулы при одинаковых смещениях атомов будет весьма
различным в зависимости от того, в каких направлениях она
деформируется Если, например, растягивать шестиугольник
бензола, сохраняя его форму (фиг. 12, II), то этому
растяжению будут сопротивляться все шесть достаточно прочных
связей кольца. Напротив, сгибание шестиугольника
перпендикулярно его плоскости, придающее ему седловидную или
ваннообразиую форму (фиг. 12, V), встречает меньшее
сопротивление, так как в этом случае происходит только некоторое
перераспределение плотности тг-электронов в его оболочке
10. основной принцип внутримолекулярной энергетики 63
(7), Различная податливость скелета может быть
непосредственно выведена из величины частот со,- рассматриваемых
типов вибрации: 993 и 400 см"1.1)
Чем больше „сопротивление" fif препятствующее данному
виду искажения скелета, тем больше будет частота mt
вибрации, которую скелет будет совершать при этом типе
смещения ядер. Соотношение здесь такое: квадрат частоты о\.2
пропорционален „сопротивлению" /„ если участвующие
в вибрации массы одинаковы. Предполагая соблюдение
последнего условия, можно было бы, на основании численных
значений частот «о, сказать, что сопротивление ароматического
/ 993 \2
р
/ 993
кольца симметричному растяжению примерно в ("лап/ да" Раз
больше, чем сопротивление поперечному сгибанию (при
неизменности длины связей С — Н). Еще меньшее усилие,
чем для сгибания кольца, требуется для кручения частей
скелета красителя Друг относительно друга вокруг
соединяющей их одиночной, точнее квазидвойной, валентной связи,
как оси (ср. стр. 41).
Упругая сила, сопротивляющаяся деформации, возрастает
сначала пропорционально величине смещения. Если последнее
обозначить через £, то упругая сила в конце смещения будет
/Д. Работа, затрачиваемая на преодоление этой упругой
возвращающей силы на пути £, будет "у/^Х?, т. е.
пропорциональна /,£2. Такова величина повышения энергии молекулы
при данном типе искажения. Отсюда следует, что искомая
зависимость запаса энергии молекулы от деформации ее
скелета может быть графически изображена на небольшом участке
координаты £ в виде параболической кривой fj?. Чем больше
сопротивляемость /, скелета данному типу искажении, тем
круче поднимаются ветви параболы; другими словами, тем
меньше будет величина деформации Д£ при данной затрате
энергии &Е (фиг. 16).
Энергия, накопленная в молекуле при данной деформации
ее скелета, и есть потенциальная энергия
соответствующего типа вибрации, равная Еш (фиг. 16). Каждому
дискретному уровню вибрационной энергии соответствует
определенная дискретная амплитуда ^ максимального смещения атомов
в обе стороны от положения равновесия.
Положение равновесия £ = 0 находится в минимуме
параболической кривой потенциальной энергии и проходится ато-
*) Как н в случав частот V света, здесь удобнее пользоваться
значениями оз не в сек.""1, а в см""1 v2).
64
ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
мами при колебаниях, аналогично примеру маятника с
максимальной скоростью и кинетической энергией, равной исходной
потенциальной энергии Ev. Наоборот, в точках поворота
5=±се, где совершается изменение направления колебания
на обратное и энергия имеет всецело потенциальную форму,
атомы проводят наибольшее время.
Поглощение и испускание снета молекулой может
происходить на всем протяжении изменения £ при вибрации. Однако
эти акты застают скелет молекулы большей частью именно
в поворотных точках, где он задерживается более длительное
время. Акты поглощения и испускания фотона совершаются
настолько "Моментально, что медленно движущиеся ядра атомов
Фиг. 16. Потенциальная энергия двух типов (/ и //)
деформации скелета молекулы, как функция смещения атомов из
положений равновесия (v =1, 2 , ... суть квантовые числа уровней).
не успевают изменить своего положения. Следовательно
сразу после этих актов атомы находятся в исходных
положениях, а именно: в положениях максимального смещения
5в. В этом приблизительно и заключается сущность принципа
Франка-Кондона. Фундаментальное значение этого простого
соображения для молекулярной энергетики делается понятным
если изобразить графически акты поглощения и испускания
фотонов в диаграмме не просто уровней EvJ как на фиг. 14
а с помощью кривых потенциальной энергии Ес(%)9
приближенно изображенных на фиг. 17 в виде парабол»
Предположим сначала, что увеличение электронной
энергии в возбужденном состоянии А* молекулы совершенно не
сказывается на величине и характере взаимной связи
вибрирующих атомов. В таком случае для верхнего электронного
состояния должна быть нарисована такая же параболическая
кривая, как и для нижнего (фиг. 17), причем минимумы
кривых будут находиться на одной вертикали, поскольку пред-
И), основной принцип внутримолекулярной энергетики
65
полагается, что для возбужденного состояния сохраняется
прежнее положение равновесия атомов.
Поглощение фотона изобразится согласно принципу
Франка-Кондона вертикальной стрелкой Av(r, исходящей из
поворотной точки вибрации с энергией Ех (?).
Вертикальность перехода между потенциальными кривыми и есть
следствие этого принципа, требующего сохранения
положения ядер (£ = const) в акте поглощения света. Длина
стрелки Avrt между исходной и конечной потенциальными
кривыми и дает величину поглощаемого молекулой фотона.
Как мы видим, при полном тождестве верхней и нижней
Фиг. 17. Принцип Фран-
ка-Кондона
вертикальности переходов.
Фяг. 18. Изменения энергии молекулы
при актах поглощения и нспуска иия
света.
парабол стрелка заканчивается вверху на том же уровне
вибрационной энергии, из какого она исходила, и,
следовательно, никакого возрастания запаса вибрационной энергии
±EV, наблюдаемого на опыте (9), при таком предположении
ие будет,
Д\я согласия с наблюдениями необходимо, чтобы ветвь
верхней потенциальной кривой имела большую крутизну, чем
нижняя. Только при этом условии поглощение фотона
приведет к приросту &ЕЬ вибрационной энергии, как показано на
фиг. 18 а. За время т возбужденного состояния происходит
утечка избытка вибрационной энергии &ЕО как было описано
в предыдущем параграфе и изображено волнистой стрелкой
на фиг. 14. Таким образом, к моменту испускания молекула
да
будет находиться на верхнем уровне Ev . Испускание фотона
снова подчиняется принципу сохранения ядрами атомов своих
положений при этом акте. Оно также происходит из положе-
Ъ А. Н. Терендц
66 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
ния максимального смещения в верхнем состоянии, возмож-
it
ного для уровня Еь , и изобразится стрелкой Av^ спускающейся
из этого положения вертикально вниз на нижнюю кривую.
Зеркальная симметрия полос поглощения и испускания,
подробно рассмотренная выше (9), требует, как мы видели,
равенства избытков вибрационной энергии AEV, сообщаемых
молекулой при равновероятных актах поглощения и
испускания. Это накладывает на крутизну ветвей потенциальных
кривых, между которыми происходит акт испускания,
соотношение, обратное предыдущему. Ветвь нижней потенциальной
кривой должна быть более крутой, чем ветвь верхней кривой,
с которой происходит испускание. Нижние и верхние кривые
в половинах аи Ь фиг. 18как бы перемевяются местами. Только
при таком виде и относительном расположении потенциальных
кривых может осуществиться зеркальная симметрия
распределения интенсивности в полосах поглощения и испускания.
В том случае, когда она нарушается, очевидно, имеют место
иные соотношения; например, происходит сдвиг положения
равновесия верхней потенциальной кривой по отношению
к нижней, как это указано на фиг, 17 пунктиром.
Переходы, изображенные на половинах а и Ъ фиг. 18
вертикальными стрелками, означают, что при поглощении,
как и при испускании, исходным состоянием является
состояние максимального смещения атомов при данном запасе
термической энергии Ev . Это смещение происходит в
направлениях легкой деформируемости молекулы и соответствует
медленным вибрациям с малыми частотами tot (параболы
плоские, сопротивляемость деформации /t. мала). Наоборот,
в конечном состоянии обоих актов поглощения и
испускания избыток энергии AEV подводится в иной тип упругой
деформации скелета, встречающий большее сопротивление
и соответствующий вибрациям больших частот о\. (параболы
крутые, /,- велики).
Необходимо оговориться, что приведенная качественная
картина упрощает имеющие фактически место более сложные
взаимоотношения, поскольку в молекулах типа бензола
возможны несколько десятков различных типов деформации,
а, кроме того, при больших амплитудах смещения
потенциальные кривые будут сильно отличаться от параболической
формы.
Значение рассмотренного здесь принципа выходит далеко
за рамки процессов поглощения и испускания света. Им
управляются буквально все явления внутри- и
междумолекулярных превращений электронной энергии (см. 12).
П. СТАТИСГИЧЕСКИЕ ЗАКОНЫ ПОГЛОЩЕНИЯ И ИСПУСКАНИЯ 67
11. Статистические законы поглощения и испускания
Мы рассмотрели выше акты поглощения и испускания,
как они протекают, в основном, для отдельной молекулы.
В действительности мы всегда имеем дело с громадным числом
Т» I r\—ft МОЛЬ
молекул, 1ак, например, при концентрации с = 10 ь число
молекул в слое толщиной ^ мм и 1 см2 понерхности будет
1013 (3). В каждый данный момент какая-либо из них
поглощает фотон в пределах полосы поглощения.
Спектральная кривая кч или sv (фиг. 4) дает, как мы видели,
значения „выхода" или эффективности поглощения фотонов
различной величины (3). Если бы все молекулы находились
точно на одном и том же нижнем уровне вибрационной
энергии Е% (фиг. 14), то эта спектральная кривая передавала бы
наглядно, как зависит от величины фотона вероятность
молекулы его воспринять. А так как, согласно формуле (1)
предыдущего параграфа, поглощаемый квант Avfl есть сумма
постоянного члена чисто электронного перехода Av0 и
изменения вибрационной энергии A/iB> то спектральная кривая
воспроивводила бы способность молекулы или вероятность
воспринять избыток вибрационной энергии &ЕГ, в зависимости
от величины последнего- Принцип Фраика-Кондона (10)
в изложенной выше форме описывает только наиболее
вероятный процесс, т. е. наиболее часто происходящие акты
поглощения. Но менее часто могут происходить и такие переходы
между потенциальными кривыми, которые изображаются
стрелками, отклоняющимися от вертикали.
Дело в том, что хотя атомные ядра как частицы более
массивные, чем электроны, и подчиняются в своих: движениях
обычной механике, но отнюдь не строго. Фактически
колебательное движение атомных ядер в молекуле также
подлежит трактовке с помощью квантовой механики, из которой
следует, что точка поворота колебаний представляет собой
на самом деле не точку, а некоторый расплывчатый участок
смещений, в котором невозможно точно фиксировать
положение ядра (ср. сказанное в 7 об облаке электрона). Таким
образом, на фнг. 18 „точки", из которых исходят стрелки Ava
и Av,, представляют собой фактически некоторые „зоны"
изменения амплитуды, переходы из которых ограничены
пунктирными вертикалями.
Высказанное здесь предположение, что все молекулы
находятся на одном и том же исходном уровне Et , приближенно
оправдывается только для очень низких температур, например
5*
68
ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
температуры жидкого водорода (20° абс), когда большинство
молекул действительно сосредоточивается на узком интервале
уровней вблизи одного уровня.
При обычной температуре такое упрощение условий не
имеет места. В результате теплового движэния молекулы
приобретают и теряют самые различные пррции
вибрационной энергии и, следовательно, распределяются по разно-
t>
11
<20eat>s
?ООО
4000
6000
8000 ЮОООса"
Фиг. 19. Населенность молекулами различных вибрационных уровней
в зависимости от температуры (для молекулярного скелета с 12 типами
вибрации).
На второй кривой ординаты увеличены в два раза.
образным уровням, значительно отстоящим от того уровня
Ev , на котором сосредоточивается большинство молекул при
температуре опыта.
„Населенность" уровней молекулами представляет собой
картину, графически изображенную на фиг. 19. В ней
приведена доля Sn общего числа молекул п, приходящаяся на узкий
участок вибрационных уровней, заключенных между
заданным значением Ev и Е9 -+- ЬЕ„. При обилии вибрационных
уровней в молекуле красителя можно считать, что уровни
практически непрерывно заполняют весь этот узкий интервал.
значений.1)
г) Фигуру 19 любезно предоставил П. В. Мейкляр.
11. СТАТИСТИЧЕСКИЕ ЗАКОНЫ ПОГЛОЩЕНИЯ И ИСПУСКАНИЯ 69
Максимально населены уровни вблизи значения энергии
Ещ , которое близко по величине средней вибрационной
энергии молекул при данной температуре Т. Доля молекул,
обладающих вибрационной энергией, большей и меньшей, быстро
падает, причем обращает на себя внимание более крутое
спадание в сторону значений EV<^EV . Чем выше температура,
тем к более высоким значениям вибрационной энергии Еь
передвигается максимум кривой и тем более он расширяется.
С каждого из вибрационных уровней нижнего состояния
молекулы А возможен переход в верхнее состояние А* с
поглощением фотона. Допустим, что с каждого нижнего уровня
поглощается только один фотон с соблюдением принципа
вертикального перехода между потенциальными кривыми (10).
Предположим также, что вероятность поглощения одинакова
для всех этих уровней. Тогда поглощение света на участке
спектральных частот от v до vh-Sv в полосе поглощения
будет зависеть только от населенности того участка
исходных уровней, с которого происходит поглощение данных
фотонов. Следовательно, в этом предположении спектральное
распределение в полосе поглощения будет воспроизводить
статистическую картину населенности исходных нижних
уровней.
В действительности же, как было изложено в начале
параграфа, согласно расширенному принципу Франка-Кондона,
возможен не один, а целый ряд переходов с каждого
вибрационного уровня в неопределенной зоне поворота
колебаний атомов. В результате образуется целый участок
поглощаемых фотонов, группирующихся вокруг фотона, обладающего
максимальной вероятностью поглощения, согласно строгому
принципу Франка-Кондона. В итоге спектральная кривая
поглощения, изображенная на фиг. 19*а, представит собой
наложение двух статистических распределений: 1) вероятности
поглощения фотонов различной величины с данного
исходного уровня; 2) населенности исходных уровней
вибрационной энергии, даваемой фиг. 19.
Пусть Kw есть получаемый из опыта, согласно формуле
(2) в параграфе 2, монохроматический коэффициент
поглощения на узком участке спектральной кривой поглощения для
частот, заключенных в интервале между v и v^-Bv (фиг. 19 а).
В таком случае произведение K$v, изображенное
заштрихованной полоской на этой фигуре и дающее величину
поглощения на рассматриваемом „монохроматическом" участке,
определяется совместным действием двух перечисленных
факторов 1) и 2). Нам требуется, однако, молекулярная посто-
70
ЭНЕРГЕТИК\ МОЛЕКУЛЫ КРАСИТЕЛЯ
янная, характеризующая суммарный акт электронного
возбуждения молекулы вне зависимости от детальной
картины распределения поглощения на фотоны различной
величины, которая вызвана возможностью различных
вибрационных состояний молекулы.
Математическая обработка этой задачи приводит к
следующему результату. Искомая суммарная вероятность или
эффективность поглощения света, которая была уже приведена
в параграфе 3 и обозначена там через к, будучи умножена
на число молекул п в см3, равна площади контура
спектральной кривой поглощения,
изображенной на фигуре, т. е. равна
Другими словами, имеем со-
отлошение:
= Г K.h
(3)
или
Фиг. 19а. Площадь контура
полосы поглощения красителя.
где Лч =
Площадь контура полосы поглощения красителя дает еще
одну важную постоянную» характеризующую процесс
поглощения света. Как мы указывали (8), поглощающая свет
молекула красителя может быть уподоблена электрическому
заряду, осциллирующему под действием переменного
электрического поля световой волны. Относительная величина этого
заряда, выраженная по отношению к заряду электрона е,
получила неудачноз название „силы" молекулярного
осциллятора 1е:
- осцилл. заряд в молекуле /л\
е заряд электрона "
Теория приводит к следующему соотношению между
площадью спектральной кривой поглощения и f,:
(5)
где m — масса ие — заряд электрона, ev— молярный
монохроматический коэффициент поглощения для частоты v в см""1 (2).
11. СТАТИСТИЧЕСКИЕ ЗАКОНЫ ПОГЛОЩЕНИЯ И ИСПУСКАНИЯ
71
В отличие от окрашенных органических соединений, для
которых fe^0.01, красители отличаются в десятки раз
большей величиной осциллирующего заряда, близкой к величине
заряда электрона.
В табл. 1 даны значения ie для некоторых красителей*
Таблица 1
Величина осциллирующего заряда fe при поглощении света красителями1
Краситель
Фуксин
Пикахром ....
Эритрозин ....
Родамин 6G . .
Флуоресцеин-Ыа .
Метиленовый го-
Тионин
Феносафраиин S .
Индиго красный .
Индиго синий . .
Хлорофилл . . .
Растворитель
С2Н5ОН
II
9}
п
(СН3)2СО
н2о
)>
w
п
»
пары
II
твердая пленка
549
526
597
578
54Э
530
585
/ 660
\ 290
288
525
500
546
546
Г 1.16
\ 0.48
0.87
0.90
0.99
ок. 0.63
0.72
0.41
0.60
ок. 1.38
ок. 1.05
0.82
0.51
0.34
0.16
0.2
Ссылки
на литературу
25,26
(твердая пленка)
25
25
25
28
28,30
25а
5° (0.86а9)
29
29
25а
\
11
V о
4 29а
Длительность возбужденного состояния. Сказанное выше
о распределении интенсивности в полосе поглощения
сохраняет значение и для полосы испускания с тем отличием, что
исходные вибрационные уровни находятся вверху и переходы
совершаются вниз.
В явлении испускания света необходимо, однако, принять
во внимание также статистику иного рода. Как указывалось
в 4, верхнее электронное состояние молекулы неустойчиво
и через весьма краткий промежуток времени самопроизвольно
переходит в нижнее нормальное состояние с испусканием
фотона, если молекула принадлежит к разряду
флуоресцирующих.
Длительность пребывания в возбужденном состоянии при
отсутствии внешних возмущающих факторов или так
называемое естественное время жизни возбужденной моле-
Таблицу любев.чо составила М. В. Савостьянова.
72 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
кулы определяется некоторыми внутренними динамическими
свойствами электронной оболочки и может для отдельной
молекулы варьировать в широких пределах. Однако, если
провести наблюдение над громадным числом молекул, как
это фактически имеет место на опыте, то среднее
статистическое время существования возбужденного состояния имеет
вполне определенное значение, характерное для данной
молекулы и обозначаемое т.
При описании процесса поглощения света (3) было
введено понятие о статистической вероятности совершиться акту
поглощения фотона молекулами, помещенными в световом
потоке, т. е, о „выходе" или эффективности процесса
поглощения к» Аналогично тому, испускание фотонов множеством
возбужденных молекул характеризуется вероятностью
испускания или численным коэффициентом, который означает,
сколько рассматриваемых актов происходит в среднем за
1 сек. с каждой возбужденной молекулой. Численные
значения таких „вероятностей", полученные из опыта, являются
статистическими постоянными молекулярных процессов,
знание которых позволяет описать поведение отдельной
молекулы.
Пусть £уП* есть число актов испускания фотона,
наблюденных за секунду для большого числа п* возбужденных
молекул А*. Эмпирическая постоянная kf и есть вероятность
испускания.
Если возбужденные молекулы создаются поглощением
фотонов п нормальными молекулами, помещенными в
световом потоке /, то в стационарных условиях для идеальных1)
флуоресцирующих молекул число испусканий за секунду
kjTi* должно равняться числу поглощений kn ~j— (формула (11)
в 3, в которой вместо §п написано здесь л). Следовательно,
^ = к^\ (6)
Постоянная вероятность испускания kf дает, таким
образом, число испускания за секунду, отнесенное в среднем
к единичной возбужденной молекуле из данного множества.
При такой интерпретации коэффициента kj процесс,
фактически идущий со множеством возбужденных молекул,
переносится на единичный средний статистический его
представитель—молекулу А*. Подразумевается, что послед-
Т. е. с квантовым выходом испускания — 1.
П. СТАТИСТИЧЕСКИЕ ЗАКОНЫ ПОГЛОЩЕНИЯ И ИСПУСКАНИЯ 73
няя, после акта испускания, немедленно восстанавливает свое
возбужденное состояние. Отсюда следует, что среднее время
т существования молекул в возбужденном состоянии равно
интервалу времени между последовательными испусканиями
молекулы А* за 1 сек., т. е. т = -т- или kf = —.
Как упоминалось в 4, по порядку величины т^10~~8—10~~9 сек.,
отсюда вероятность испускания к*ж\0Р—Юэ сек"1.
Величина т настолько мала, что доступна
непосредственному измерению только с помощью сверхбыстрых оптических
„затворов", открывающихся на время порядка 10~~9 сек.
Последняя конструкция такого „флуорометра" и новые
результаты, полученные для красителей, описаны в статье Тумер-
мана31. Для флуоресцеина-Na (уранина) т = 4.8*10~9 сек.; для
эозина т = 5.7-10~~9 сек.; для родамина В т=2.1-10~8 сек.1)
Время жизни возбужденного состояния молекулы может
быть, однако, приблизительно оценено с помощью величины
молекулярного коэффициента поглощения. Действительно,
уравнение (6) непосредственно связывает к^ = -— с
вероятностью поглощения к. Приближенно мы можем из него сразу
сделать вывод, что, при прочих равных условиях, чем больше
вероятность поглощения к, тем больше будет и вероятность
испускания kfy а, следовательно, тем меньше т.
Точный теоретический расчет приводит к следующей
зависимости между kf илв т и площадью &/К заключенной
под контуром спектральной кривой поглощения:
£,=--- = const-*2 1 *А (7)
где 1—некоторая средняя длина волны полосы
поглощения, выраженная в см.
Уравнение (6) соответствует идеальному случаю, когда
каждый акт поглощения сопровождается актом испускания;
другими словами, когда квантовый выход флуоресценции
9 (4) равен единице. В действительности он, как правило,
меньше единицы (табл. 2) и большинство красителей в
обычных условиях неспособно испускать свет.
Причина отсутствия флуоресценции заключается в том,
что электронная энергия возбуждения тратится до акта
испускания на иные процессы, которые подробно
рассматриваются в следующем параграфе. Тем не менее, соотношение
]) В ацетоне, уксусной кислоте, глицерине.
74 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
Таблица 2
Квантовый выход ? флуоресценции
в годном растворе при концентрации ок. 10~*
л
/
Флуоресцени-Na 0.8
Родами:! 5G 0.5
В 0.25
Эовин • 0.2 1.0 (в адетоне)3*
Антрацен 0.2 (в бензоле)54
Порфирины ок. 0.1
Этил-хлорофиллид 0.08
Акридии-оранж 0.03
Эрятроэин 0.05 1.0 (в ацетоне)^
Бенгальская роза 0.02
Нейтр. красный ]
Феио сафранин > '<0.01
Метилеаовый голубой . . - J
(6) сохраняет свое значение и для случая <f<Clt если под
т понимать „естественную" длительность возбужденного
состояния молекулы, какой она обладала бы при отсутствии
возмущений.
12# Превращения поглощенной световой энергии
Отсутствие флуоресценции у большинства красителей
^несмотря на интенсивное поглощение ими света, указывает
на то, что энергия возбуждения преобразуется в иную форму.
К рассмотрению соответствующих элементарных процессов
мы и переходим ниже.
Дезактивация, Если длительное освещение не приводит
к химическому изменению красителя, то единственный путь,
каким могла исчезнуть электронная энергия возбуждения»
это — переход ее в термическую энергию. Действительно,
окрашенные объекты всегда испытывают заметное нагревание
под действием тех длин волн, которые поглощает краситель.
Несмотря на кажущуюся простоту этого явления, его
механизм далеко не выяснен. Рассеяние электронной энергии
возбуждения в тепло может осуществиться двумя путями:
1) внутримолекулярным и 2) междумолекулярным.
Процесс внутримолекулярной дезактивации преобладает
в красителях и соединениях с малым выходом флуоресценции.
Он осуществляется в том случае, если электронная энергия
возбуждения способна непосредственно переключагься на
вибрацию скелета, другими словами, если существует
возможность всей электронной энергии возбужденного состояния
12. ПРЕВРАЩЕНИЯ ПОГЛОЩЕННОЙ СВЕТОВОЙ ЭНЕРГИИ 75
целиком превратиться в потенциальную энергию сильно
деформированного скелеча.
Такая возможность имеет место в случае, изображенном
на фиг. 186, когда кривая потенциальной энергии нижнего
электронного состояния пересекает кривую верхнего состояния.
В точке или, точнее, „зоне", где обе кривые сближаются,
и делается возможным переход возбужденной молекулы в
состояние высокой потенциальной энергии определенного вида
деформации нижнего состояния молекулы. Превратившись
в вибрационную форму, эта избыточная энергия уже легко
перераспределяется между многочисленными типами вибрации
и отдается окружающей среде, как термическая энергия. В ре-
зультате возбужденная молекула плавно переходит на нижний
электронный уровень без испускания фотона34.
Элементарная физическая „реакция" для такого процесса
внутреннего превращения или конверсии электронной энергии
в вибрационную может быть кратко записана следующим
образом:
А^ -> А, (I)
где звездочка обозначает наличие у молекулы электронной,
а значок ~~ — вибрационной энергии. Описанный процесс
удовлетворяет требованию „неподвижности" ядер в момент
перехода, т. е. принципу Франка-Кондона (10), так как
происходит в точке пересечения кривых.
Преобразование всей электронной энергии возбужде шя
в потенциальную энергию движения атомных ядер типично
для многоатомных молекул даже более простого строения.
В них оно сопровождается разрывом наиболее слабой
валентной связи, и известно под названием предиссоциа-
ции21'22. При возбуждении молекул красителей малыми
фотонами видимого света, энергия возбуждения недостаточна для
отщепления атомов или атомных групп, но хватает для
приведения их в энергичные колебания продольного или
крутильного вида. Для рассеяния электронной энергии последние
имеют особое значение. Вызываемое ими нарушение
плоскостной конфигурации атомов, разобщая облака тс-электронов,
уменьшает „резонансное" расщепление уровней (стр. 46),
а следовательно, благоприятствует сближению их
потенциальных кривых при больших амплитудах вибрации z4.
Действительно, флуоресцирующие красители обладают
„жестко*— построенным скелетом, не допускающим свободного
кручения концевых групп35. Весьма знаменательно, что если
в нефлуоресцирующий ион фенолфталеина ввести
промежуточный адостик — О—, жестко связывающий два ароматических
76 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
ядра, то получается типичный флуоресцирующий ион флуорес
ценна (см. диаграмму на стр. 24)'*' *5.
Междумолекулярный процесс дезактивации состоит
в отводе энергии возбужденной молекулы при приближении,
к ней другой молекулы. Сохранение ароматическими
соединениями способности флуоресцировать в растворенном состоянии
показывает, что молекулы инертных растворителей неспособны
отводить электронную энергию возбуждения этих соединений.
Но флуоресценция „тушится", т. е. выход флуоресценции
падает, если в раствор ввести определенные посторонние
вещестна или же повысить концентрацию флуоресцирующего
соединения (21, 35, 36). Не следует думать, что квант
электронной энергии возбуждения может целиком, в одном
первичном акте, превратиться в поступательную энергию
встретившихся молекул согласно элементарной реакции:
*
М -» А -н М, (II)
где ш есть молекула „тушителя , а стрелки указывают на
избыток кинетической энергии партнеров, получившийся
за счет электронной энергии и рассеиваемый затем в тепло»
Такой процесс имеет исключительно малую вероятность.
Прибавление к флуоресцирующим парам ароматических
соединений одноатомных газов (Не, Ne, Ar) давлевием до 1
атмосферы не тушит заметно их испускания, хотя за время
возбужденного состояния эти соединения испытывают от 100 до 1000
столкновений с атомами указанных газов21> 22. Между тем,
единственная возможность отвода электронной энергии
возбуждения здесь заключается в переходе ее в поступательную
энергию согласно (II).
Малую вероятность имеет также переход энергии
возбуждения целиком в вибрационную энергию партнера, согласно
схеме:
А*н-М->Ан-М (III)
с переводом молекулы М на соответствующий высокий уровень
вибрации.
На возможность такого процесса указывает наблюденная
при флуоресценции газообразных производных бензола
некоторая избирательность тушения узких участков в сплошной полосе
флуоресценции, которые совпадают по своим частотам с
последовательностью высоких вибрационных уровней молекул
прибавляемого двуатомного газа (Н2, N2, СО):0.
Однако в атмосфере многоатомных газов (NH3l C2H4, эфир,
пентан), могущих воспринимать большие порции энергии
12. ПРЕВРАЩЕНИЯ ПОГЛОЩЕННОЙ СВЕТОВОЙ ЭНЕРГИИ 77
в виде вибрационной, никакого тушения флуоресценция,
свидетельствующего о механизме (III), не наблюдается22.
В резком отличии от электронной энергии избыток
вибрационной энергии Д£в, сообщаемый молекуле при акте
поглощения фотона (9,10), легко передается посторонним молекулам,
в особенности многоатомным 22. Другими словами, значительно
большую вероятность, чем предыдущие процессы, имеет для
ароматических соединений в газообразном и растворенном
состояниях процесс:
| А*-*-М-»А*ч~М. (IV)
По существу тот же процесс чисто вибрационной дезактивации,
не затрагивающий электронную энергию возбуждения, описан
был нами при рассмотрении Стоксова смещения и неизменности
вида полосы флуоресценции красителей (9).
Электронная энергия возбуждения_^легко передается по сто-
ронней частице М _трлькр_ в, том случае, если последняя
способна ее воспринять почти целиком в виде электронной
же энергии. сЗто будет иметь место, когда молекула М имеет
уровен^_электронной энергии, расположенный на одной высоте
или ниже уровня А*. "Если „тушитель" М способен испускать
•свет, то наблюдается испускание его спектра флуоресценции
за счет энергии света, первично поглощаемой молекулами А,
т. е. явление так называемой сенсибилизованной
флуоресценции ~
.
(V)
Такое явление установлено в бинарных смесях паров
простейших ароматических соединений, а именно: бензол* -+- анилин37,
анилин*-ь индиго23, бензол* н- нафтиламин22, где * отмечает
возбуждаемый светом компонент. Для бинарных смесей
красителей и более простых соединений в растворах сенсибилизо-
ванная флуоресценция не была обнаружена, повидимому, из-за
полной деградац и передаваемой электронной энергии в тепло
по механизму (I), где под А* следует теперь понимать
бинарный комплекс А*М двух встретившихся частиц В
жидкой среде время пребывания двух частиц в сфере их
взаимного действия более длительно, чем при
кратковременных столкновениях молекул в газообразном состоянии.
Весьма интересно, что, подобно антистоксовой
флуоресценции (4, 9), при которой происходит испускание молекулой
фотона Av большей величины, чем поглощенный Avfl> наблю-
73 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
даются случаи антистоксовой сенсибилизованной
флуоресценции23. Явление состоит в розбуждении флуоресценции
частицы М в процессе (V), когда ее уровень электронной
энергии расположен выше уровня возбужденной молекулы А*.
В этом случае недостаток энергии покрывается из общего
теплового запаса вибрационной энергии двух частиц А*М,
рассматриваемых как одно целое8.
Конверсия в метастабильное состояние. Наряду с
флуоресценцией, немедленно1) прекращающейся после выключения
возбуждающего света, красители, а также более простые
родственные им ароматические соединения обнаруживают
в определенных условиях длительное послесвечение —
фосфоресценцию, продолжающуюся секундами. Благоприятствует
этому явлению „жесткое" закрепление молекулы в
стеклообразных органических или неорганических средах, как
например частично обезвоженной борной кислоте или сахарном
леденце, или же фиксация молекул на поверхности адсорбентов,
как силикагель, целлулоза, желатина и т. п. Особо длительная
и интенсивная фосфоресценция наблюдается при охлаждении
таких объектов до низкой температуры жидким воздухом
(—180° С). В последнем случае могут быть использованы
в качестве растворителя и такие жидкости (спирт), которые
способны замерзать при этой температуре в виде
стеклообразной среды, не кристаллизуясь. В этих условиях способность
фосфоресцировать обнаруживается у громадного числа оргави-
ческих соединений38.
Обращает на себя внимание тот факт, что при комнатной
и не очень низкой температуре спектр послесвечения в
жесткой среде мало отличается по положению и виду от полосы
обычной флуоресценции, наблюдаемой в парах данного
соединения или его жядком растворе. Отличием является лишь
аномально большая длительность послесвечения спектра, которая
в 108 раз превышает нормальную длительность возбужденного
состояния (4, II)39. Это послесвечение с неизмененным
спектром правильнее поэтому называть „замедленной
флуоресценцией". Наряду с ним в послесвечении присутствует новая
полоса испускания, которая делается преобладающей при
низкой температуре, заменяя обычную полосу флуоресценции
Красителя (фиг. 20). Новая полоса сдвинута в сторону меньших
частот по отношению к последней. Тонкая структура спектра
флуоресценции, наблюдаемая для простейших ароматических
соединений, сохраняется практически неизменной в таком
„сдвинутом" спектре фосфоресценции38. Примечательно^
]) Т. е, зч время порядка 10 8
12. ПРЕВРАЩЕНИЯ ПОГЛОЩЕННОЙ СВЕТОВОЙ ЭНЕРГИИ
79
что как замедленная флуоресценция, так и новый спектр
фосфоресценции возбуждаются поглощением света в одной
и той же, т. е. нормальной полосе поглощения красителя,
которая обычно лишь немного изменяет свое положение и
протяжение при понижении температуры. Спадание обоих видов
послесвечения во времени t подчиняется показательному за-
i
кону е т, справедливому для нормального возбужденного
состояния А*, с тем отличием, что постоянная т, т. е. время
жизни увеличивается с 10~8 до 1 сек.39.
?во
cu
too
520
440 А п
Фиг. 20. 7 — Спектр послесвечения
кристаллического фиолетового в глицерине при —195°С (а —
полоса замедленной флуоресценции, 6—"полоса
фосфоресценции); 2— полоса обычной
флуоресценции в тех же условиях; 3— полоса поглоще-
.41
Это замечательное явление послесвечения органических
соединений в жестких средах, открытое в 1896 г, Видеманом
и Шмидтом и подробно изученное многими исследователями,
получило исчерпывающее объяснение только в самое
последнее время. Оно имеет первостепенное значение для понимания
некоторых особенностей фотохимических реакций
красителей 24.
Первая интерпретация наблюдаемых фактов была дана
Яблонским (1933)40, который объяснил взаимную связь и сдвиг
спектров замедленной флуоресценции и фосфоресценции
простым допущением существования промежуточного
электронного уровня „mete", расположенного ниже обычного
возбужденного уровня £* (фиг. 21). Переход с него на нижний —
основной уровень Е° „запрещен" в том смысле, что он обла-
so
ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
дает исключительно малой вероятностью испускайия kJ9
а следовательно, аномально большим временем жизни т (11).
Такие уровни или состояния, установленные для более
простых электронных систем — атомов и двуатомных молекул,
получили название метастабильных, отмечающее их
повышенную устойчивость по сравнению с обычным
возбужденным состоянием. Нами принято для таких уровней
обозначение 2Г, а для соот етствующего этому уровню состояния
молекулы обозначение A v. На фиг. 21 изображены
дополнительные возможности
электронных переходов,
возникающие из-за наличия метаста-
бильного уровня.
_^__^ ,, Возбужденная квантом Ava
\ Г^ и ° до уровня Е* молекула
может, вместо испускания
фотона Av -f перейти на уровень
/Г , растрачивая только часть
энергии возбуждения в
вибрацию, как показано
волнистой стрелкой. Очевидно, на
метастабильный уровень ЕУ
попадает некоторая доля тех
£0 __.. J молекул А*, которая испыты-
Л вает процесс внутренней де-
Фиг. 21. Метустабильиый уровень зактивапии, согласно меха-
(meta) электронной энергии в краси- низму (I), Т. е. такая КОНВвр-
сия идет по схеме:
h-i
}ph
^^
телятс.
А;' -* А -* А \
(VI)
Если термическая (вибрационная) энергия молекулы А* и ее
окружения достаточны для перекрывания интервала Е*—Е
между уровнями, то будут происходить обратные переходы
А ->А* (фиг. 21, волнистая стрелка, обращенная вверх).
Эти переходы будут происходить и после выключения
действующего света и продолжаться, пока имеются метастабильные
молекулы А\ При этом с уровня Е* будет испускаться
обычный спектр флуоресценции hv/f но с аномально большой
длительностью.
Глубокое охлаждение понижает запас тепловой энергии
и исключает, таким образом, возвращение метастабильной
молекулы А* на первоначальный уровень возбуждения А*.
В этом случае остается только путь испускания фотона
12. ПРЕВРАЩЕНИЯ ПОГЛОЩЕННОЙ СВЕТОВОЙ ЭНЕРГИИ 81
фосфоресценции Av л, совершающегося весьма редко, а потому
растягивающегося на время порядка секунды.
Положение уровня 2Г ниже Е* обусловливает неравенство
hvph <[ fay и объясняет, тем самым, сдвиг полосы
фосфоресценции в сторону меньших частот.
При интенсивном непрерывном облучении такого
органического „фосфора" при низкой температуре, накопление
молекул в метастабильном состоянии Av может быть
обнаружено появлением новой полосы поглощения Av*,
соответствующей переходу на более высокий электронный уровень Ег
метастабильной молекулы (фиг. 21)41»42.
Интервал Е*—Е" между уровнями может быть определен
из разности fay—hvph или вычислен из температурного
коэффициента поглощения h\ • Для красителей он имеет величину
2—10 кгкал, для простейших ароматических соединений он
больше (табл. 3).
Измерения различными методами приводят к выводу, что
от 5 до 10°/0 всех возбужденных молекул красителя переходят
в метастабильное состояние в таких фосфорах при низкой
температуре42'43.
Та б л ица 3
Разность энергий возбужденного А* и метастабилъного Av состояний
в в. вольтах
Бензол, иафгалии ок. 1 \
Анилин 0.6 \ и
Антрацен 0.5 J
Флуоресцеии 0.4 1 41
Кристаллический фиолетовый 0.25 /
Родулнн оранж NO 0.3 \
Бенэофлавнн 0.2 > *2
Трипафлавшг 0.1 J
О природе метастабильного состояния и причине его
аномальной длительности высказывались самые разнообразные
предположения. Автором настоящей книги эти предположения
были подвергнуты критическому рассмотрению и была
обоснована в 1943 г.2* следующая точка зрения, независимо
выдвинутая позднее Дж. Льюисом с сотрудниками (1945)*4.
Прд_ОеРех°Де с нижнего электронного уровня ^^на
возбужденный Е* спаренность вхед электронов не "Меняется и нх
спины сохраняют антипараллельное расположение 1(6), чт0
можно записать схематически следующим образом:
Ш, (V)H
6 А. Н, Терении
82 ЭНЕРГЕТИКА МОЛЕКУЛЫ КРАСИТЕЛЯ
где скобки обозначают отдельные квантовые ячейки (стр. 33}„
Метастабильное состояние А" отличается от возбужденного
А* тем, что один из электронов изменяет направление своего
спина на противоположное, т. е, обращает вращение:
A*[f] Ш^А'М [f]. (VIII)
В результате появляются два неспаренных электрона* т. е
свободных валентности, и молекула приобретает свойства
двувалентного бирадикала. Обращение направления спина,
т. е. размыкание спаривания электронов, требует затраты
энергии за счет энергии возбужденного состояния А*. В
результате электронный уровень бирадикала будет ниже уровня
возбужденного состояния на величину затрачиваемой энергии.
Из табл. 3 следует, что эта энергия уменьшается с
усложнением соединения, а также сопутствующим ему уменьшением
энергии возбуждения.
Самопроизвольный переход молекулы из состояния
бирадикала А* в основное, нижнее состояние А с испусканием
фотона hvph затруднен, так как он требует обращения спина
электрона. В этом смысле он „запрещен", т. е. имеет
исключительно малую вероятность, а состояние Av аномально
большое время жизни. Однако при междумолекулярных
соударениях и сопровождающих их сильных возмущениях
бирадикал может превратиться в нормальную молекулу путем
вибрационного механизма деградации электронной энергий
в термическую, описанным выше (I). Бирадикальное
электронное состояние дезактивируется особенно легко в том случае,
если соударение происходит с атомами или свободными
радикалами, также обладающими свободными электронами с неспа-
ренными спинами (правило Вигнера сохранения суммарного
спина в элементарной физической реакции обмена энергии):
Av [f] Ш + М[Ц-*А [U] + M[f]. (IX)
Несмотря на то, что бирадикальное состояние молекулы
проявляется спектрально только в особых условиях, оно имеет
существенное значение для ее фотохимического поведения,
так как представляет собой не только длительное состояние
повышенной электронной энергии, но также состояние
валентной ненасыщенности. На значение этого факта было впервые
обращено внимание автором настоящей книги2*. Отсутствие
флуоресценции, свидетельствующее о внутренней
дезактивации по механизму (I), отнюдь не лишает молекулы
фотохимической активности, так как при вибрационной деградации элек-
12. ПРЕВРАЩЕНИЯ ПОГЛОЩЕННОЙ СВЕТОВОЙ ЭНЕРГИИ 83
тронной энергии может произойти также описанная выше (VI)
конверсия молекулы в бирадикальное состояние
повышенной реакционной способности.
Концентрация возбужденных молекул, создаваемая
поглощением света в обычных условиях, может быть подсчитана
на основании уравнения (6) предыдущего параграфа. Ранее (3,
стр. 19) мы приводили число актов поглощения за секунду,
даваемое левой половиной этого уравнения. Для конкретного
случая окрашенной пленки оно было равно 10й; стационарная
концентрация п* возбужденных молекул, создаваемых
поглощением света в этих условиях, получается согласно
уравнению (6), равной п* = т X число актов поглощения в сек,
—10~8- 10u= 106 молекул. По сравнению с общим числом
п = 1013 молекул, концентрация возбужденных молекул, как мы
106
видим, ничтожна: T77jg=lU '.
Предположим теперь, что 1°/0 возбужденных молекул
переходит в бирадикальное состояние со времевем жизни т в 108 раз
большим. В таком случае стационарная концентрация мета-
стабильных молекул будет в 108 • 10~2 раз, т. е. в миллион раз
больше концентрации возбужденных.
В дальнейшем изложении, при рассмотрении механизма
разнообразных фотохимических реакций, нам неоднократно
придется для объяснения наблюдений предполагать участие
метастабильного состояния красителя.
В заключение этой главы приведем в виде сводной схемы
основные элементарные процессы внутримолекулярного
и междумолекулярного превращения энергии, которые можно
условно назвать физическими реакциями.
Типы физических фотореакцин
(* или v обозначает избыток электронной внергнв, "* обовначает избыток нибрационной
анергии)
Первичный акт поглощения
фотона uva-*-A-*A*
Флуоресценция А* —> А* -*• Ач-Л^(т^ 10-8 ceiu)
Конверсия в метастабиль-
ное состояние А* * А—>AV
А
Замедленная флуоресценция Av —> А* —> А
Фосфоресценция Av —*■ А **- Лу ,
Дезактивация анутримоле- ^ ""*" А
кулярная (конверсия в вн- J, + и
брации) А + М
6
*
8 \ СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕНИЯ КРАСИТЕЛЯ
Дезактивация мождумоле- ,
куляриая (электроннойэнергии) А*-*-М—> AM—*А-4- М
Дезактивация
междумолекулярная (вибрационной впер"
гии). . . . . А" -*-м -* А -*" м
Сенсибилнзованная
флуоресценция А*ч-М—>А-*-М*
ГЛАВА ЧЕТВЕРТАЯ
СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕЙИЯ
КРАСИТЕЛЯ
13. Влияние среды на положение полосы поглощения
Растворитель или иная среда, в которой находятся молекулы
(ионы) красителя, воздействуют в большей или меньшей
степени на их электронную оболочку, а следовательно,
оказывают влияние на положение максимума (Хта* или vffiait)
и величину коэффициента поглощения (&тах или J кы ovj*
Различные действия среды ва спектр могут быть разбиты
на две главных группы: а) слабые влияния силового поля
молекул среды; б) сильные воздействия на „резонансную"
электронную структуру красителя.
а) К первой группе относится действие электрического
молекулярного поля растворителя на осцилляцию заряда
в красителе (8). По законам электростатики среда с
диэлектрической постоянной D > 1 уменьшает работу, требующуюся
для смещения электрона, а следовательно, и частоту v его
осцилляции. Смещение Av максимума поглощения в
растворителе, по сравнению с его положением в вакууме, определяется
выражением Av = vBaK# — vcpefla tt 1 — -^- Наблюдаемые
фактически смещения Av возрастают с увеличением D обычно
быстрее, чем требует это выражение45. Это несоответствие
может быть приписано тому обстоятельств}^ что для быстрых
осцилляции электронного заряда с частотой v ж 1015 сек."1
не имеет смысла пользоваться диэлектрической постоянной
растворителя D, измеряемой при гораздо более низких
частотах электрического поля. Для частот инфракрасного света,
как показынает теория, роль диэлектрической постоянной
*) Правильнее смещение Av относить к положению максимума
поглощения ие для газообразного состояния в вакууме, что, вообще говоря, для
большинства красителей неосуществимо, а к его положению в инертном
неполирном растворителе, каким является, например, гексан.
13. ВЛИЯНИЕ СРЕДЫ НА ПОЛОЖЕНИЕ ПОЛОСЫ ПОГЛОЩЕНИЯ 85
играет квадрат показателя преломления /iM2fc. Очевидно, для
частот в области видимого спектра должно существовать
некоторое -соотношение между положением полосы поглощения
и показателем преломления среды, если имеет место
предполагаемая электростатическая картина.
Действительно, небольшое смещение максимума
поглощения красителей различных классов в неполярных
растворителях плавно возрастает с увеличением показателя
преломления пЛа для желтой линии Na или с величиной дисперсии
nF — пс, где nF и пс — показатели преломления синей (F)
и красной (С) линий водорода, как это было установлено
правилом смещения Кундта (1878).
В полярных неассоциированных жидкостях смещение
Av имеет большую величину, чем для растворителей со
сходным строением молекул, но не содержащих дипольных
заместителей. Для электрически нейтральной молекулы красителя —
мероцианина Av возрастает пропорционально дипольному
моменту молекулы растворителя и остается постоянным, когда
дипольный момент постоянен, например, в гомологическом
ряду спиртов. Это также подтверждает электростатическую
природу сил, воздействующих на электронную оболочку
красителя45* ^.
б) Резкое изменение окраски, вызванное смещением полосы
поглощения, испытывают в полярных растворителях такие
окрашенные ароматические соединения, в валентную структуру
которых, в особенности возбужденного состояния, входят
внутриионизованные структуры со значительным
пространственным разделением противоположных зарядов, как
например в случае паранитроанилина (фиг. 22).
© е
Аналогичный эффект наблюдается также при адсорбции
бесцветных или слабоокрашенных соединений на твердых
адсорбентах (37).
Предполагается, что большая диэлектрическая постоянная
или большой дипольный момент окружения ослабляет силу
взаимного притяжения зарядов ф и © *) в молекуле,
облегчая их разделение, что приводит к преобладанию
ионизованной структуры, написанной J
J) Т. е. мест недостаточной и избыточной плотности электронного
облака* (8).
86 СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕНИЯ КРАСИТЕЛЯ
Для красителей аномально большие эффекты смещения
спектра поглощения в сторону длинных волн производят
жидкие ароматические амины и гетероциклические основания
(пиридины), а также растворители, молекулы которых
способны вступать в „водородную" связь (Н2О, спирты, жирные
кислоты и т. п.)50. Несомненно, здесь имеет место более
тесная, квазивалентная связь
красителя с растворителем, а
не чисто электростатическое
воздействие молекулярного
поля среды 45.
Молекула растворителя,
в особенности если она также
способна принимать
различные ионизованные структуры,
образует с молекулой
красителя своеобразную един/ю
резонансную систему, в
которой появляются
дополнительные возможности резонанса
з-
го
30
rt
Фиг. 22. Положение полосы
поглощения пара-нитроаиилияа в различных „ / \
F F r при наличии мостиков (....)
растворителях17, /—в гексане; 2 — в
диоксаие? 3—в воде.
„водородных связей, как
например в структурах:
N<
Если один из участников обладает сильно выраженной
основной или кислотной функцией, то протон Н"*" перемещается
к соответствующему" партнеру и спектр поглощения красителя
испытывает изменения, аналогичные тем, которые наступают
при избытке или недостатке водородных ионов в водном
растворе (инднкато^Ыд см^ 5),
Сильная пертурбация электронной структуры наступает не
только при частичном переносе протона, но и при переносе
электрона. Подобный механизм образования
междумолекулярной связи типичен для соединения хинона с гидрохиноном
и подобных ему мерохиноидных междумолекулярных
соединений, обладающих интенсивной окраской, в резком отличии
от почти неокрашенных компонентов.1)
J) Вопросу о сильных смещениях полос поглощения при междумолеку-
ляриых взаимодействиях ароматических соединений друг с другом и с
неорганическими веществами („галохрэмия") посвящена особая работа аатора
книги.
14. СТРУКТУРА ПОЛОСЫ ПОГЛОЩЕНИЯ
87
'QOQQ0
го-
/5-
10
OS-
14. Структура полосы поглощения
Спектральная кривая поглощения красителей очень редко
имеет простой контур, изображенный на фиг, 3 (3). Обычно,
наряду с главным максимумом, присутствуют побочные,
относительная высота которых зависит от концентрации,
температуры и природы растворителя (фнг. 23). В последние годы
структуре полос поглощения красителей было уделено
большое внимание и во многих
случаях найдено
удовлетворительное объяснение ее
происхождения.
Глубокое охлаждение
раствора до температуры
жидкого воздуха (—180° С) или
жидкого водорода (— 253° С),
уменьшая запас вибрационной
анергии и тормозящее
крутильные и сгибающие колебания
скелета, способствует более
четкому выявлению отдельных
побочных максимумов,
сливающихся в одну общую кривую
при обычной температуре.
Возникновению побочных
максимумов дают одно из следующих
объяснений.
1. Структура полосы
поглощения красителя может быть
вызвана избирательным возбуждением больших квантов Ль>(
определенных типов вибрации скелета согласно принципу
Франка-Кондона (10). В таком случае разность энергий
этих максимумов Avf. и главного максимума Avmax должна
совпадать с квантами кы{ вибрационной энергии таких типов виб-
рацин: vf — vra" = wtf.
Действительно, по порядку величины, получаемые
значения частот o)t. могут быть приписаны вибрациям молекулы
красителя (табл. 4). Однако отсутствие независимых сведений
•об этих частотах, например нз инфракрасных спектров, не
всегда делает такое объяснение убедительным.
Решающим аргументом в пользу приведенной
интерпретации структуры полосы поглощения явилось бы сохранение
красителем этой структуры также и в газообразном состоянии,
т. е. в условиях наиболее совершенного диспергирования кра-
ог-
500
550
6OQ
650
Фиг. 23. Структура полосы
поглощения пииацианола в спиртовом
растворе *Б.
88 СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕНИЯ КРАСИТЕЛЯ
сителя на отдельные молекулы. Такой опыт с положительным
результатом был проведен с мероцианином45.
Таблица 4
Рааности частот между главным и побочным максимумам в полосах погло-
щения некоторых красителей 46
vj — v ЙЖ(см-1)
Изоциаяины • • от 860 до 1420
Карбоциаинны » 1200 „ 1650
Ин докарбоцианины 1520
Оксикарбэцнанины 1600
Днфенилп элиены 1593
Другим доводом в пользу вибрационного происхождения
структуры служит присутствие побочных максимумов с т е м и же
значениями интервалов от главного также и в полосе
флуоресценции, зеркально отображающей структуру полосы
поглощения (4, 11). Подобное тождество, установленное для
радикала трифенилметана при глубоком охлаждении раствора, не
оставляет сомнения в вибрационной природе данной
структуры47. Были наблюдены побочные максимумы со следующими,
разностями частот по отношению к главному:
в поглощении: 190, 600, 820, 970, 1090, 1520 см -1:
в испускании: [230, 670, 970, 1080, 1550 см ~\
2. Побочный максимум поглощения может принадлежать-
геометрическому изомеру или таутомеру
красителя. Различная пространственная конфигурация атомных
групп в стереоизомерах сказывается на величине
взаимодействия тс-электронов (8), а следовательно, на интервале между
возбужденным и основным уровнями энергии, определяющим
спектральное положение полосы поглощения, а также на
величине коэффициента поглощения. Наиболее длинноволновое
положение полосы и наиболее сильное поглощение дает
плоскостное расположение атомов, участвующих в резонансе
(глава 2), Чрезмерное сближение некоторых атомов при
наличии в молекуле объемистых групп, например при цисконфи-
гурации, неизбежно приводит к выходу циклов из общей
плоскости скелета и появлению спектра разобщенных
циклов. Вследствие этого максимум полосы поглощения цис-
формы будет лежать у более коротких длин волн и будет
ниже, чем для трансформы как это наблюдается для стиль-
бена и азобензола. Сходные системы полос поглощения,
сдвинутые как целое по шкале частот, наблюдаемые в азо-
красителях, могут быть также приписаны цис- и трансиэо-
формам "• ^ 49,
14. СТРУКТУРА ПОЛОСЫ ПОГЛОЩЕНИЯ 89
Другим примером служит побочный максимум,
расположенный вблизи главного в полосе поглощения иона
кристаллического фиолетового, с разностью частот, эквивалентной
0.5 кгкал. Льюис с сотр.Б0 приписывают этот побочный
максимум изомеру красителя, в котором одно из фенильных колец
нарушает правильное лепесткообразное расположение колец,
имеющее место для главного изомера. *>
Таутомерные формы красителя, вызванные, например,
перемещением атома водорода (кето-энольная, имино-имидоль-
ная н т. п. формы), будут давать, как правило, далеко
отстоящие спектры и не могут объяснить близко лежащие побочные
максимумы. Необходимо, однако, учесть, что ионы красителей
могут вступать в тесное взаимодействие с молекулами
растворителя, образуя междумолекуляриые соединения или соль-
ваты определенного стехиометрического состава, каждый из
которых может давать спектр, смещенный относительно
главного на определенный дискретный интервал. Известно,
например, что многие красители при высушивании прочно
удерживают воду, которая, очевидно, входит в качество структурного
элемента в кристаллическую или аггрегироваииую фазу
красителя51. Подобно тому как было изображено выше на стр. 86,
молекулы растворителей с группой ОН (спирты, кислоты)
способны вступать в водородную связь с концевыми амино-,
сульфо- и карбонильными группами красителя, образуя также
различные по составу и ориентации междумолекулярные
соединения со смещенными полосами поглощения52,
3. Иной причиной сложности контура спектральной кривой
поглощения является присутствие в растворе различных
ионов красителя, образующихся из исходного путем
присоединения или отдачи одного-двух и т. д. протонов Н~*\ Как
было сказано в 5, все красители обладают более или менее
отчетливо выраженной либо основной, либо кислотной
функцией, а следовательно, в зависимости от концентрации ионов
водорода в растворе, будет преобладать тот или иной ион при
]) Данная интерпретация не представляется нам убедительной. Побочный
максимум воспроизводится в зеркально симметричной кривой испускания,
а следовательно, по сказанному выше, должен иметь вибрационное
происхождение. Для изомера же побочный максимум в полосе испускания должен
был бы, наоборот, сместиться в сторону больших частот, следуя смещению
побочного максимума поглощения, н нарушить тем самым зеркальную
симметрию. Фё'рстер 16 (8) приписывает побочный максимум трифеннлме-
тановых красителей, наблюдаемый также в катионе трнфенилметана,
расщеплению верхнего электронного уровня на два, однако этому
объяснению противоречит исчезновение побочного максимума прн понижении
температуры.
"90 СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕНИЯ КРАСИТЕЛЯ
наличии меньших концентраций других ионов. На фиг. 24
приведены спектральные кривые различных катионов
кристаллического фиолетового 17*53. Присоединение протона к одной
из концевых (ауксохромных) аминогрупп нормального катиона /
как бы фиксирует свободную пару электронов атома N,
выключая ее из резонанса с ти-электронами остального скелета. В
результате максимум поглощения такого дважды положительно
заряженного иона (кривая 2) занимает приблизительно то же
положение, как в однократно
заряженном катионе малахитового
зеленого, содержащем только
две аминогруппы.
Присоединение еще одного протона
выключает обе аминогруппы из
резонанса; в результате спектр
поглощения перемещается в
ультрафиолетовую область (кривая
3). Для красителей типа
индикаторов кислотности условия
для преобладания ионов
определенного вида наиболее
благоприятны. В общем случае
это не имеет места, и сложная
структура спектра поглощения,
а также флуоресценции иногда
о
22* W00 см
-t
''Фиг. 24. Изменения спектре
поглощения катиона кристаллического
фиолетового при различной
концентрации ионов водорода в
водном растворе17
/ — (MegN-Phb C]+; Me = СН3;
Ph = С Н
2—Ме2 HN^h-CfPh-NMeg
J (MHP C-Ph-NMeaJ
вызвана присутствием
различных типов ионов данного
красителя, не учитываемых
авторами исследований.
4. Спектральная кривая
поглощения красителей нередко
обнаруживает такие максимумы,
относительная высота которых
в сильной степени зависит от концентрации красителя,
температуры и диспергирующей способности растнорителя. Эти
максимумы вызваны наличием в растворе различных степеней
молекулярной аггрегации красителя.
В большинстве растворителей, особенно в воде, ионы
- л—3 моль
•красителей, начиная с концентрации порядка W ~^~> ПРИ"
сутствуют в виде димеров, т. е. двойных ионов, несмотря
на одноименный заряд, который должен был вызвать их
взаимное отталкивание 5*. Это явление наблюдается для красителей
самых различных классов: трифенилметановых, ксантеновых
(эозин), тиазиновых, а в особенности полиметйновых (цианины).
14. СТРУКТУРА ПОЛОСЫ ПОГЛОЩЕНИЯ
91
Максимум поглощения димерного иоиа смещен на величину
от 1 до 50 ту* в сторону коротких длин волн по отношению
к максимуму мономерного иона (фиг. 25), см. табл. 5.
Та блица 5
Длины волн Q^m\>) максимумов поглощения моиомерных А
и димерных А24"4" катионов красителей
597
656 5
575
583
528
557
600
540
540
500
и А
55
55,56
56
50
33
24"4" СИЛЬНО
/о}-с
Тионин
Метиленовый голубой ....
Оксоиип
Кристаллический фиолетовый .
Родамин 6G
Относительная высота максимумов А"
зависит от температуры (фиг. 25), указывая иа обратимую
термическую диссоциацию
димерного иона. Из
температурного
коэффициента этого явления следует,
что прочность связи ионов
в димере тионина около 7
кгкал. Димерные ионы не
флуоресцируют (36), ио
Ьохраняют способность
сенсибилиэовать
фотографическую пластинку (стр.
272). В абсолютном
этиловом спирте димеризация
ионов практически отсут-
стнует.
Молярный
коэффициент поглощения е (2) ди-
мера приблизительно в 2
раза больше такового для
мономера.
С повышением концентрации в некоторых растворителях
(вода) происходит дальнейшая ассоциация молекул красителей
в тримеры и аггрегаты более высокого порядка, вплоть до
коллоидных частиц. Силы, удерживающие ионы в этих аггрегатах,
не представляют собой валентных связей, а
неориентированные аддитивные силы молекулярного сцепления, имеющие
для красителей значительную величину, ввиду большого раз-
700 А т
Фиг. 25. Максимумы поглощения
мономера и днмера катяоиа тионниа
(с = 2.5 • 10"~* М)^5. При повышении
температуры димеры частично диссоциируют
иа мономеры.
92 СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕНИЯ КРАСИТЕЛЯ
Х/000
(50
мера электронной оболочки и большой подвижности в ней
электронов54 ,х)
Существование димеров, как особой разновидности
междумолекулярного соединения, на которой останавливается
ассоциация на своем первом этапе, даже в случае легко аггреги-
рующих цианинов, указывает на специфичность сил,
связывающих два иона и исключающих присоединенве других. Таким
свойством насыщения — силы молекулярного сцепления не
обладают. Необходимо
поэтому предположить
существование
„резонансной" связи между
плоскими структурами
красителей,
ориентированными параллельно друг
другу 56.
Беспорядочная аг-
грегация молекул
красителей с образованием
коллоидных частиц,
неудачно называемая „по-
лимериза цией"
красителя, не приводит
обычно к появлению
новых побочных
максимумов на кривой
поглощения, а только к расши-
100
60
18
20
22XW00LM'1
Фиг. 26. Переход псевдо-иэоцнанина в
состояние упорядоченной аггрегации,
обладающее узким максимумом поглощения.
/ — с = 1.05- Ю-2 в Н2О
2—с = 1.20- Ю-2 в С2Н5ОН
рению и смещению
максимума димера. Иногда
это вызывает кажущееся
расширение и сдвиг в
красную сторону главного максимума поглощения мономера
красителя.2) Усиление в спектре поглощения
флуоресцирующего красителя максимума, принадлежащего не
флуоресцирующим аггрегированным формам, которое происходит
при увеличении концентрации красителя или введении в
раствор ионов солей, вытесняющих красители из раствора, может
привести к кажущемуся падению выхода флуоресценции, т. е.
тушению (36).
^Величина подобной „дисперсионной" связи двух электронных систем
пропорциональна fe2^3, где {е есть „сила" электронного осциллятора,
определяемая из величины площади кривой поглощения (стр. 70), а Л—длина
волны максимума поглощения.
2) В пленках твердого красители, полученных осаждением из раствора,
спектр красителя испытывает аналогичное смещение и расширение 57.
14. СТРУКТУРА ПОЛОСЫ ПОГЛОЩЕНИЯ 93
Дая некоторых полиметиновых красителей (псевдоизоциа-
кинов) была открыта, наряду с указанной, иная, а именно
обратимая упорядоченная аггрегация ионов с включением
молекул воды и галоидных анионов 58>59. В результате такой
аггрегации получаются громадные макромолекулы,
содержащие от Ю3 до 106 отдельных молекул и обнаруживающие
своеобразные оптические свойства 60f 6I.
При образовании такого упорядоченного аггрегата
обычный широкий максимум поглощения красителя исчезает,
заменяясь исключительно узким и нысоким максимумом,
смещенным в сторону длинных волн (фиг. 26). Ширина этой новой
полосы, обозначаемой в иностранной литературе / или Р/,
примерно в 10 раз меньше ширины нормальной полосы
поглощения красителя, составляющей от 800 до 1700 см"*1;
молярный коэффициент поглощения в максимуме доходит до значения
2
£тах =430000 у вместо обычного значения 50000—100000.
моль
Площадь, заключенная внутри контура этой тонкой полосы,
однако, примерно, равна исчезнувшей площади нормальной
полосы красителя. Поразительным отличием от поведения всех
сложных молекул является тот факт, что спектр флуоресценции
этой новой формы красителя состоит из такого же узкого
максимума, почти точно совпадающего с узким максимумом
поглощения.1)
Обращает на себя внимание, что мономерные молекулярные
ионы, на которые распадается такая аггрегнрованная форма
красителя в спиртовом растворе, неспособны к
флуоресценции.
Исключительно высокая чувствительность испускания к
внешним возмущениям, выражающаяся в полном тушении
флуоресценции в присутствии 1 молекулы тушителя на 103 —10°
молекул, входящих в состав макромолекулы, заставляет думать,
что энергия возбуждения в последней беспрепятственно
распространяется по всему ее протяжению, передаваясь от одного
составляющего иона красителя к другому без деградации
электронной энергии в тепло. Очевидно, мы имеем здесь дело с новой
системой обобществленных 7г-электронов, сходной по своему
поведению с кристаллом (ср. 35 и 39).
1) Другими словами, имеет место ивление. резонансного испускания
падающей частоты света, наблюдаемое при оптическом возбуждении
атомарных паров металлов. Квантовый выход флуоресценции, однако, невелик:
Ф = 0.10—0.15. Испускание узкого У-макевмума может быть вызвано также
поглощением света более коротких длни волн но второй, более широкой,
полосе, сохраняющейся у упорядоченного „полимера".
94 СТРУКТУРА И ИЗМЕНЧИВОСТЬ ПОЛОСЫ ПОГЛОЩЕНИЯ КРАСИТЕЛЯ
Плоские молекулярные ионы красителя, составляющие
макромолекулу, ориентированы, как и в димерах, своими
плоскостями параллельно друг другу. Присутствие молекул
воды в структуре такой аггрегированной формы существенно
для ее образования 45. Повидимому, молекулы воды
осуществляют промежуточные мостикн водородной связи,
необходимые для обобществления электронов (ср. стр. 86).
Упорядоченная аггрегированная форма красителей
обратимо переходит в состояние беспорядочной аггрегации или
распадается на отдельные ионы красителя при изменении
условий (растворителя, температуры и т. п.).
ЧАСТЬ ВТОРА
ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
КРАСИТЕЛЕЙ
ГЛАВА ПЯТАЯ
ФОТОХИМИЧЕСКИЕ ПРОЦЕССЫ
15« Формальные законы фотохимии
Необходимый предварительный этап всякой фотохимической
.реакции состоит в поглощении света. Это условие было
отчетливо сформулировано в начале XIX века Гротгусом
{1817) и Дрэпером (1830) в виде положения, что только
поглощаемый Свет может произвести химическое превращение.
Гротгус, рассматривая общий вопрос о связи цветности и
фотохимического действия света, пришел к заключению, что
действует только тот свет, цвет которого является
дополнительным к окраске тела (ср. 1). Подтверждением закона Гротгуса
•было наблюдение Гершеля (1842), что выцветание окрашенных
веществ, содержащихся в цветах растений, происходит наиболее
сильно под действием света дополнительного цвета.
Однако поглощение света есть только необходимое,
но недостаточное условие дая протекания фотохимической
реакции. Поглощая свет, окрашенные вещества отнюдь не всегда
претерпевают химические изменения. В предыдущем параграфе
мы описали целый- ряд физических процессов, идущих под
действием света в красителе и обратимо восстанавливающих
его первоначальное состояние.
Закон Гротгуса-Дрэпера содержит высказывание только
качественного характера. Первое количественное
фотохимическое исследование было проведено Буизеиом и Роско (1855)
для реакции соединения хлора с водородом. Закон Бунзена-
Роско гласит, что количество продукта фотохимической
реакции (названное ими фотохимическим „эффектом") однозначно
определяется произведением интенсивности падающего света /
на время освещения t. При условии постоянства произведения
It получается одинаковое количество продукта Q, независимо
от численных значений, принимаемых / и i в отдельности.
7 А. Н. Теревва
98 ФОТОХИМИЧЕСКИЕ ПРОЦЕССЫ
Этот закон называется также заковом взаимозаменамо~
сти интенсивности и времени освещения; следовательно, если.
/1*1 = /г*2» тО Qi==Q2- закон Бунзена-Роско. (1)
Для светочувствительных систем более сложных, чем смеси
простых газов, закон взаимной заменяемости /и t нередко
нарушается, В законе Бунзена-Роско по сути дела содержится
утверждение, что существует прямая зависимость между
фотохимическим действием и энергией падающего света, которая
пропорциональна произведению IL Но только Вант Гофф (1904)
впервые отчетливо высказал мысль о
пропорциональности количества фотохимически измененного вещества
поглощенной энергии света. Отсюда непосредственно
получается закон Бунзена-Роско, если принять, что за все
время освещения система поглощает некоторую постоянную
долю падающей энергии It.
Закон Вант Гоффа лежит в основе формальной кинетики
фотореакций и может быть представлен в следующей
дифференциальной форме (Лазарев, 1911)62.
Пусть за малый интервал времени bt ковцентрация с
светочувствительного вещества изменилась (уменьшилась) на велв-
чину Ьс (<0), например, в результате реакции выцветания.
При этом с обозначает значение концентрации в любой
выбранный момент протекания реакции. С другой стороны, если
светочувствительная система представляет собой окрашенный
слой толщиной /, то, согласно формуле (4) в 2, световая
энергия, поглощаемая ежесекундно при значении концентрации
с, равна Л = /0 (1 — е~ш), где /0 есть энергия падающего
потока монохроматического света на каждый см2 в сек., а е —
молярный коэффициент поглощения- В этих обозначениях
закон Вант Гоффа может быть записан следующим образом:
— Ьс= const X A bt= const X /0 (1 — * 6C) °t
или: (2)(
— g^= const X/o (1—e~tcl): закон Вант Гоффа.
Другими словами, скорость реакции — g- пропорциональна
„скорости" А поглощения световой энергии.
Закон Вант Гоффа охватывает, таким образом, в четкой^
математической формулировке предыдущие законы Гротгуса-
Дрэпера и Бунзена-Роско.
Для тонкого слоя толщиной Ьх, A = lQzcoxr согласно
формулам 1 и 4'(2) и, следовательно, скорость реакции пропорцио-
Is. ФОРМАЛЬНЫЕ ЗАКОНЫ ФОТОХИМИИ 99
малмни просто имеющейся в данный момент концентрации
г поглощающего свет и исчезающего компонента. Наоборот,
при полном поглощении света, когда ее или / велики, вторым
членом в скобке выражения для Ал приведенном выше, можно
пренебречь и скорость реакции — gr не будет зависеть от
концентрации до той поры, пока последняя не снизится настолько,
что сделанное в формуле пренебрежение будет неприменимо.
Из выражения (2) для закона Вант Гоффа путем
интегрирования по времени t может быть получен ход падения
концентрации (выцветания) во времени, а также распределение
продукта реакции по толщине слоя (J переменное) при
отсутствии диффузии.
Закон Вант Гоффа, связывающий кинетику фотохимической
реакции с поглощением световой энергии, является
формальной закономерностью в том смысле, что не затрагивает
существа вопроса о механизме действия света.
Основным законом фотохимии следует считать закон
квантовой эквивалентности Эйнштейна (1912),
согласно которому каждый поглощенный фотон Av вызывает
„изменение" одной молекулы1. Это изменение может быть
физическим или химическим. В первоначальной формулировке
закон эквивалентности предполагал, что каждый поглощенный
фотои неизбежно вызывает химическое превращение молекулы.
Как мы видели, это не так: существуют разнообразные
физические процессы деградации поглощенного молекулой фотона
в тепло, без химического изменения молекулы. Если
поглощенная за некоторое время энергия монохроматического света
есть Л, то число поглощенных фотонов Л^ равно г-. По закону
эквивалентности оно равно числу „измененных" молекул Nm:
= ^ = УУт: закон квантовой эквивалентности. (3)
Число фотонов» содержащихся в 1 эрге поглощенной энергии,
будет зависеть от величины фотона, т. е. частоты или длины
волны света. Поэтому, при рассмотрении механизма
фотохимической реакции, вызываемой различными длинами волн,
принципиально неправильно относить количества
прореагировавших молекул к одинаковым значениям поглощенной
энергии. Необходимо вводить поправку на различные числа
фотонов, содержащихся в одинаковых значениях энергии. В
пределах сравнительно узкой полосы поглощения красителя эта
поправка не столь существенна (ср. стр. 160).
100 ФОТОХИМИЧЕСКИЕ ПРОЦЕССЫ
В соответствии с законом эквивалентности была предложена
единица Эйнштейн, равная 6.06*10а8*Лл т, е, световой энергии,
вызывающей изменение или фотохимическое превращение
одного моля вещества.
В большинстве фотохимических реакций с участием
возбужденных молекул красителя последние утрачивают свою
энергию и превращаются в нормальные молекулы до момента
приближения второго реагента и вступления с ним в реакцию.
Поэтому, по аналогии с введенным выше (4, 11) понятием
квантового выхода флуоресценции <р, первой количественной
характеристикой фотохимической реакции является измерение
ее квантового вы]ход}а у, под которым понимают
отношение;
число прореагировавших молекул - « /Vv
- - = v = квантовый выход реакции) (4)
число мэлекул, поглотивших фон тоны '
Определяемый на опыте суммарный квантовый выход
реакции зависит от конкуренции с химической реакцией тех
рассмотренных выше процессов дезактивации возбужденной
молекулы, которые непроизводительно растрачивают энергию
возбуждения в тепло. Наряду с вероятностью дезактивации
путем испускания фотона к,% подробно рассмотренной выше
(11), каждый элементарный процесс вывода возбужденной
молекулы из строя, либо путем внутреннего преобразования
энергии, либо путем химической реакции, может быть
охарактеризован соответствующей вероятностью кгу имеющей смысл
эффективности или выхода данного процесса.
Суммарный квантовый выход у складывается из значений этих
элементарных выходов, умноженных на концентрации
участвующих в данном процессе частиц. Задаваясь определенной
схемой протекания данного фотохимического превращения,
можно составить определенное выражение для у, как функции
всех этих величин. Экспериментально полученная
зависимость у от концентрации реагентов открывает, следовательно,
возможность проверить правильность предполагаемой схемы
и даже, в отдельных случаях, определить численные значения
элементарных выходов к,. "R,главе 9 мы встретимся с
простейшим примером такой процедуры.
Приведем теперь основное выражение фотохимической
кинетики, как оно следует из закона эквивалентности и
сравним его с законом Вант Гоффа. Пусть число
светочувствительных молекул, исчезнувших за малый интервал времени oty будет
, а число фотонов, поглощенных за это время, будет
/а. Тогда по определению квантового выхода^
15. ФОРМАЛЬНЫЕ ЗАКОНЫ ФОТОХИМИИ 101
Рмделив обе половины на §/, т. е- отнеся все величины
к 1 сек., имеем для скорости изменения числа молекул
кинетическое соотношение:
где выражение для А было приведено выше. Это уравнение
дает определенное толкование коэффициенту
пропорциональности const в кинетическом уравнении Вант Гоффа (2).
Для тонкого слоя §х поглощаемая ежесекундно энергия А,
как мы видели (2, формула 1), равна A = I0 knbx = IokNm,
где /0 обозначает интенсивность падающего света. Подставляя
это выражение для А в уравнение (5), имеем:
Это соотношение представляет собой наиболее простую
формулировку основного закона фотохимической кинетики.
Мы видим, что при заданной интенсивности /0 падающего
света величина относительного фотохимического изменения
вещества за единицу времени —лГ~л7 зависит от произведе-
* МП "'
иия двух величин к и у, из которых первая есть вероятность
поглощения фотона или вероятность фотону эффективно
„попасть в молекулу" (3), а вторая — вероятность молекуле,
поглотившей фотон, вступить в реакцию.
Произведение hff от которого зависит величина
фотохимического действия, можно назвать
фоточувствительностью данной системы63.
Для яркого солнечного света -г- составляет, примерно,
-mi* ФОТОНОВ ' „
lUia 2~ в фиолетовом участке спектра, о случае
фотохимической реакции С12-1-Н2, молекулярный коэффициент
поглощения светочувствительного компонента — хлора имеет
весьма малую величину &даЮ*~21, но квантовый выход этой
цепной реакции очень велик у= 105. Отсюда &у»10~1в
и ^•^^^10~16-1015^=10"1, т. е. падение концентрации
хлора в результате реакции за секунду составляет около 1О°/о-
Примером другой высокочувствительной реакции является
выцветаине зрительного пурпура (родопсина), для которого
квантовый выход у равен единице, но к% как для всех
красителей, чрезвычайно велико &^10~16. Отсюда h( имеет также
102 ФОТОХИМИЧЕСКИЕ ПРОЦЕССЫ
порядок величины 10~16 и результат получается такой же, как
и в предыдущем случае.
Противоположным примером слабосветочувствительной
реакции является выцветание обычных красителей в
результате реакции с кислородом воздуха. Хотя поглощение света
красителем велико: £»10~16, квантовый выход исключительно
низок у«10~4, а потому krffi&lQT20 и> следовательно,
выцветание красителей происходит в 10000 раз медленнее
выцветания зрительного пурпура.
16. Основные типы фотохимических реакций в красителях
Перечислим предварительно основные фотохимические
реакции, возможные для красителей, а также родственных
им соединений.
Из общего вида спектра поглощения следует, что
необходимо различать 3 области фотохимической чувствительности
красителя: а) видимый или близкий ультрафиолетовый участок
спектра, где расположена интенсивная, характерная для
красителя, полоса поглощения, свойственная электронному резонансу
валентных структур всей молекулы в целом; б) область
ультрафиолетовых длин волн короче 300 пць9 где расположена
примерно в 10 раз менее интенсивная полоса поглощения
ароматического кольца в различных состояниях замещения,
свойственная, наряду с красителями, всем простейшим ароматическим
соединениям, а также лейко-форыам красителя; в) область
значительного поглощения в коротком ультрафиолете.
Практический интерес представляет первая спектральная
область, так как соответствующие длины волн представлены
в солнечной радиации с достаточной интенсивностью*
Фотохимические реакции красителя, вызываемые полным
светом кварцевой ртутной лампы, богатой ультрафиолетовым
излучением, протекают, как правило, в результате активации
красителя во втором участке фоточувствительности, а не
первом, что объясняет, в частности, установленное практикой
несоответствие между порядком выцветания выкрасок под
действием кварцевой лампы и солнечного света.
Фотораспад. В отличие от простейших многоатомных
молекул и производных бензола, спектр поглощения которых
расположен в ультрафиолетовой области, поглощение кванта
видимого света молеку\ой красителя, как правило, недостаточно
для разрыва какой-либо из ее валентных связей. По этой
причине явление фотораспада или предиссоциации,
характерное для первых соединений21'22, для красителей не имеет
места в видимой полосе поглощения, но может осуществиться
16. ОСНОВНОЕ ТИПЫ ФОТОХИМИЧЕСКИХ РЕАКЦИЙ В КРАСИТЕЛЯХ 103
т ультрафиолетовых полосах поглощения, имеющихся у
красителей или их лейкосоединений. Своеобразие сложных орга-
нгическнх соединений заключается, однако, в том, что разрыв
некоторых одиночных валентных связей, соединяющих две
части скелета молекулы, требует энергии пониженной величины,
по сравнению с нормальной прочностью связи, наблюдаемой
в предельных соединениях. Это имеет место, когда
получающиеся в результате распада атомные группы испытывают
перегруппировку валентной структуры, сопровождающейся
выделением энергии. Если перегруппировка валентных
электронов происходит одновременно с поглощением фотона
в одном акте, то выделяющаяся энергия входит в общий
энергетический баланс, снижая величину кванта, потребного
для распада молекулы. Примером такой энергетической
компенсации является фотораспад или фотолиз диаэосоедине-
ния, например:
Аг — N = N]"" СГ—*-> Аг — С1 -+- К
с выделением молекулы азота N273. Эта фотореакция
происходит в само1 первой полосе поглощения, расположенной в
фиолетовой и близкой ультрафиолетовой области спектра, т. е. под
действием фотонов, вызывающих возбуждение молекулы (17).
Совершенно аналогичное снижение прочности связи имеет
место, когда в распавшихся частях молекулы появляются новые
возможности структурного резонанса, а следовательно,
происходит их стабилизация за счет снижения электронного уровня
(7). Примером может служить гексафенилэтан Ph3C — CPh3
(Ph = СбН5), энергия разрыва связи С — С, в котором составляет
только 12 кгкал по сравнению с 72 кгкал в этане Н3С — СН3.
Причиной является выигрыш энергии резонанса в 60 кгкал
у двух свободных радикалов 2Ph3C' по сравнению с недиссоции-
рованньш соединением (точка обозначает непарный: электрон,
см. 7). В соответствии с этой стабилизацией продуктов распада,
гексафенилэтан распадается на свободные радикалы под
действием длин волн близкого ультрафиолета (300—250 т^),
r то время как этан требует длин волн короче 180 т\ь.
Подобное же поведение обнаруживают создинения, содержащие
в своем составе радикал дифенилазот Ph2N-, например тетра-
фенилгидразин Ph2N—NPh2. Под действием 11 300— 250 т\ь
происходит распад последнего соединения с появлением
голубой окраски, обусловленной полосой поглощения указанного
свободного радикала (максимум у 730 т(л). В жидких
растворах концентрация свободных радикалов, создаваемая
освещением, незначительна из-за быстрой рекомбинации. Замораживая
растворы жидким воздухом, Льюису с сотрудниками удалось
104 фотохимические процессы
проследить фотолиз разнообразных соединений по появлении»
характерных видимых полсс поглощения, присущих свободным
радикалам ■ NPh2, • CPh3, • SPh, например:
рь3с - NPh2 -^|Ph3q. и-;- NPh2-
Наряду с распадом на свободные радикалы был также
установлен распад нейтральной молекулы на противоположно
заряженные ионы (ср.^18):
Ph2N — NH2—v-
Очевидно, сходные с указанными процессы могут иметь
место и в красителях, но, повидимому, только при
поглощении в ультрафиолетовых полосах.
Фотоперегруппировка* Перечисленные выше реакции могут
быть объединены в одну группу реакций первичного
фотораспада, нарушающих целостность молекулы. Иной тип первичной
фотореакции, наблюдаемой в сложных соединениях, состоит
в перегруппировке атомов в скелете под действием света~
Если в результате поглощения фотона происходит поворот
одной группы атомов по отношению к другой или иное
изменение геометрической конфигурации атомов, то такой тип
реакции следует назвать фотонзомериаацией (точнее
фото-стереоизомеризацией)65. В качестве примера можно
привести переход под действием ультрафиолетового света
трансформы азобензола и стильбена в цисформы:
Ph
Ph
■\
Ph
\
1
траве
с=с<
/
транс
4Phj
Н
/
xph
Av
Ph
\
Ph
., v
i
sN.= N/
ЦВС
Ph
1 хн
ЦИС
В этих соединениях стереоизомеризация имеет место
непосредственно после акта поглощения фотона и представляет собой
один из разнообразных путей превращения, открывающихся
для возбужденной молекулы А*, наряду с другими,
перечисленными в конце параграфа 12. Вместо возвращения в
основное свое состояние, возбужденная молекула затрачивает часть
энергии на работу поворота одной группы по отношению^
16. ОСНОВНЫЕ ТИПЫ ФОТОИМИЧЕСКИХ РЕАКЦИЙ В КРАСИТЕЛЯХ 10S
к остальным вокруг соединяющей их связи, как оси вращения.
В результате конечное состояние молекулы представляет
собой стереоизомер исходного с повышенным содержанием
электронной энергии, достигающим 15—20 кгкал. Совершение
подобного поворота возможно при условии, если возбуждение
1>лектронной оболочки настолько ослабляет взаимодействие
тт-электронов, что двойная связь между атомами N —N н С=С
теряет свою обычную жесткость, допуская выход
соединяемых ею групп из общей плоскости и их вращение (ср. стр. 41)65.
Переход из общего для двух стереоизомеров возбужденного
состояния А* в устойчивый стереоизомер исходного
соединения может происходить также путем испускания фотона
флуоресценции Av,b6, но чаще всего путем указанной выше
конверсии электронной энергии возбуждения в потенциальную
энергию крутильных колебаний большой амплитуды вокруг
ослабленной двойной связи, по аналогии с вибрационной
конверсией типа I (12). Такие крутильные или вращательные
колебания весьма быстро теряют свою избыточную энергию и
молекула стабилизуется либо в исходной^ [форме, либо в внде
стереоизомера.
Процесс, который можно назвать незавершившейся стерео-
изомеризацией, с конверсией электронной энергии возбуждения
в крутильные колебания большого размаха возможен и для
красителей, где он создает предпосылку внутренней
дезактивации24.
Разумеется, при рекомбинации свободных радикалов,
получившихся при фотолизе, также может образоваться
стереоизомер.
Другой тип внутренней перегруппировки под действием
света есть фототаутомеризация. Он состоит в
перемещении отдельного атома из одной части скелета в другую.
Обычным примером этой реакции служит превращение орто-
нитробензальдегида (а) в ортоннтрозобензойную кислоту
(Ь) под действием Хк < 436 тр в области первого максимума
поглощения исходного соединения67:
106 фотохимические процессы
В газообразной фазе квантовый выход этой реакции близок
единице: у = 0.75, но падает до 0.5 при прибавлении 500 мм
азота. В растворах и твердом состоянии также у = 0.5 Этот
результат объясняют тем, что, хотя обе связи N — О в нитро-
группе способны активироваться, только та связь, которая
оказывается вблизи альдегидного атома Н приводит к
внутримолекулярной реакции между О и Н с образованием гидро-
ксильиой группы кислоты67. Свободное вращение нитрогруппы,
имеющее место в газообразной фазе, благоприятствует
сближению любого из двух атомов О этой группы с атомом Н
за время возбужденного состояния. Таким образом водородная
связь, намеченная пунктиром в альдегиде (а), существующая
для растворенного и твердого его состояния, будет снижать
квантовый выход по сравнению с газообразным состоянием.
Наряду с указанным, имеется ряд других примеров
легкости фотоперемещеиия атома О, сопровождающегося его
включением между соседними атомами Н и С ароматического
кольца, например в азоксисоедннениях6В:
\/
г/
N—N—/Ч JU —N=N—
О W НСК
Обычные реакции таутомеризяцни молекул в растворах,
заключающиеся в перемещении атома водорода на большие
расстояния в молекуле (прототропия), вызваны участием
посторонних доноров и акцепторов протона Н**, а не фактическим
передвижением этого атома.1)
Фотоприсоединение* Процесс, противоположный
фотораспаду, заключается в присоединении к возбужденной светом
молекуле другой молекулы того же или иного рода. Сюда
относятся реакции фотодимеризации и
фотоконденсации, а также фотохимического присоединения кислорода.
Типичным примером реакции первого типа является
образование в растворе антрацена двойных молекул диантрацена,
наблюдаемое при больших его концентрациях под действием
длин волн, возбуждающих его флуоресценцию. В темноте
молекулы диантрацена обратимо распадаются вновь на
одиночные молекулы (19). Фотоаггрегацией ионов красителя в димеры
и более сложные образования мы объясняем происхождение
неизвестных окрашенных фотопродуктэв, наблюдаемых в вод-
С этой точки зрения д^лущенивуО фотоииграция атома Н или даже
радекалл СН3 в изолированной мо\екуле 69 неправдоподобно*гК
16. ОСНОВНЫЕ ТИПЫ ФОТОХИМИЧЕСКИХ РЕАКЦИЙ В КРАСИТЕЛЯХ 107
еных растворах некоторых красителей (20). Ряд фактов
указывает также на возможность присоединении молекулы
растворителя к возбужденной молекуле красителя силами более
прочными, чем обычные силы сцепления Ван дер Ваальса.
В пределе возбужденная молекула может прореагировать
с молекулой растворителя, испытывая, например в воде,
реакцию фотогядролиза (26).
Реакциям фотопрясоедииения благоприятствует глубокое
охлаждение, например до температуры жидкого воздуха .
Способность присоединять иные молекулы сильно
возрастает, если возбужденная молекула переходит в метастабиль-
ное состояние бирадикала с двумя свободными валентными
электронами и большим запасом электронной энергии24 (12).
Возбужденные и метастабильные молекулы красителя
способны связывать молекулу кислорода О2 с образованием
на первом этапе более или менее устойчивой перекиси или
пероксида типа АО2. Эгому важному типу фотореакций
посвящена целая глава 7.
Присоединение кислорода к органическим соединениям при
обычной темпгратуре носит в химии название реакции ауто-
или самоокисления. Для обозначения аналогичного
фотохимического связывания молекулы О2 был предложен
Рабиновичем2 громоздкий термин „фотоаутоксидация". По аналогии
с общепринятым термином „гидрирование" для обозначения
реакция присоединения молекулы Н2, мы предлагаем и будем
в этой книге иногда пользоваться термином „оксидирование"
и „фотооксидирование" для обозначения реакции
присоединения молекулы кислорода.1)
Необходимость установления четкой терминологии следует
из того факта, что под словом „окисление", широко
применяемом в химии, понимают, наряду с указанной выше реакцией,
также и такие реакции, которые идут по принципиально иному
механизму, даже без всякого участия кислорода. А именно:
а) Окислением называют реакцию отщепления водорода,
т. е. дегидрирование по суммарной схеме:
А<н~*-М -» A-f-H>M,
н ъ 9
н
где М может, в частности, быть молекулой О2 (см. стр. 169)»
Эта реакция протекает обычно в два этапа, на каждом из
которых отщепляется толькэ один атом водорода:
А-Н-н-М -» Aj-f-H-M
Ч Для иностранной терминологии предлагается термин „фотооксигени-
■зация*' (pboto-oxygfenation) по аналогии с „гидрогенизацией* (hydrogfenation)*
108 ФОТОХИМИЧЕСКИЕ ПРОЦЕССЫ
Такая элементарная реакция может явиться промежуточным
этапом в развитии реакции присоединения кислорода, а именно:
А-Н'-ьО2-* А-О-О-Н-* A-f-НОг.
б) Термином окисление широко пользуются для обозначения
обратимых реакций переноса электрона в растворах между
нонами и молекулами, идущих без участия кислорода, например
,Fe+-" ^=* Fe-»-++ -+- e.
восстановлевн е
Весьма распространенные фотореакции, идущие по такому
электронному механизму, называются в этой книге реакциями
фотоокисления и фотовосстановления; они рассмотрены
в главе 8. Ниже дается их краткая характеристика.
Фотопереиос влектроиа. Под действием ультрафиолетового
света некоторые ароматические соединения, а также красители
отщепляют электроны. Такая фотореакция может быть
обнаружена по появлению проводимости или окраски, свойственной
образующемуся при этом положительному иону, например71
Ph2NH -2
дифениламин его иов
ь бесцветный синий J
Такой фотопроцесс, идущий под действием сравнительно-
коротких ультрафиолетовых длин волн, мы называем
элементарным фотоокислением.
Образовавшийся молекулярный остаток с избыточным
положительным зарядом обычно неустойчив и распадается
затем с выделением протона по схеме71:
В итоге происходит кажущееся отщепление от молекулы
нейтрального атома Н, т. е. реакция фотораспада:
Ph2NH ^ Ph2N н- Н.
Подобная реакция квазидегидрирования типична для
бесцветных лейко-форм красителей, которые под действием
ультрафиолетового света в первой полосе поглощения отдают два
атома водорода акцептору водорода X и приобретают, окраску*
16. ОСНОВНОЕ, ТДЛЫ ФОГОХЛМЛЧЕСЛЛХ РЕАКЦИЙ В КРАСИТЕЛЯХ 104
свойственную красителю (глава 8). Эту суммарную реакцию
•фотоокисления можно записать следующей схемой:
JM И
Обратная реакция фотовосстановления происходит
с красителями некоторых классов (например с метиленовым
голубым) при действии света в их видимое полосе поглощения
в присутствии веществ, легко отдающих атомы^ Н (точнее,
доноров е + Н+):
А-4-и>У-А<н . Y.
н н
Фотосенсибилизация. Примером физической реакции фото-
сенснбилизации является сенсибилнзованная флуоресценция
(12), заключающаяся в передаче электронной энергии
возбуждения от одного компонента бинарной смеси другому. Если
передаваемая энергия приводит к распаду второй молекулы,
то мы имеем дело с простейшим случаем фотохимической
сенсибилизации. Примером реакции такого типа может служить
диссоциация йодистого метила в бензольном растворе в
присутствии возбужденных светом молекул нафталина (Napht*)"2:
Napht*-b'CH3J -> СН3-ь J
Элементарный механизм преобразования энергии
возбуждения молекулы А* в энергию разрыва связи второй молекулы,
очевидно, аналогичен процессу конверсии электронной энергии
в потенциальную энергию скелета (I), рассмотренному в 12*
Более сложные и до сих пор невыясненные процессы
происходят при очувствлении бромистого серебра красителями
к длинноволновому свету, поглощаемому последним (глава 10).
Реакции окисления и восстановления, протекающие в
сложных системах и требующие больших квантов энергии, могут
быть также очувствлены красителями к действию малых
фотонов видимого света. Типичные примеры такой фото-
сенсибилизации, имеющей громадное значение для фотосинтеза
(В растениях, будут рассмотрены в главах 7 и 8/'
110
ФОТОХИМИЧЕСКИЕ ПРОЦЕССЫ
По примеру сводной диаграммы физических процессов
преобразования энергии возбуждения приведем здесь схемы
главнейших фотохимических реакций. Поскольку детальный
механизм большинства из них не установлен, мы
ограничиваемся ео многих случаях указанием лишь конечного результата
фотопревращения.
Фото-
распад
Типы фотокнмкчгснвх реакций
фотолив иа
радикалы
фот олив на
жоыы (фото-
Фотопс-
рвгруп-
пировка
>
C-D
Gj
Av
В
/G
к/ фотоиаоме-
\г ривзция
фототауто-
меривзция
Фото-
прнсое-
динеяие
O2)
А*-нН2О->НАОН
фотодиме-
ривация
фотоохсв-
дировваве
фотогидр о-
ли«
Фото-
перенос
электрона
А* -н Y —^ а- -+- Y+
фото
окисление
частицы А*"
фотовосста-
иожление
частицы А*
Фото-
сеисиби-
лизация
ВС — ABC
В
Y+-I-M-
фотосенеа*
бнлнвоаан-
ыый распад
фотосенси-
биливошзн'
нов окисле-
иие М
фотосеиси-
билнвопав-
вое восста-
новлеиже №
фотосеиси-
биливоваи-
вый перенос
влектроиа
17 ФОТОЛИЗ ДИАЗОСОЕДИНЕНИЙ ПИ
ГЛАВА ШЕСТАЯ
ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
17. Фотолиз дназосоединений
Диазосоедйиения распадаются с выделением азота под
действием фиолетовых и близких ультрафиолетовых длин волн
в области первой полосы поглощения. Фотолиз идет по схеме:
Ar —N = N —X ^Ar — X-bN
или
где Ar обозначает ароматический остаток молекулы,
реагирующий далее с образованием устойчивых соединений Аг — X
или Аг— С1.
Такая фотореакция имеет место для твердых диазосоединений'
и их растворов в жидкостях и органических пленках73.
Простейшие диазосоединения испытывают фотораспад и в
газообразном состоянии (диазометан, диазоэтан).
Повышенная светочувствительность диазосоединений,
сравнимая, в отдельных случаях, с таковой для хлористого
серебра, привела к их применению для получения фотокопий
(диазотипия). „Проявление" полученного невидимого
изображения производится растворами аминов или фенолов,
реагирующих с неразложенным диазосоединением с образованием
светопрочных красителей. Введение в ди аз о пленку аминов
и фенолов, а также солей тяжелых металлов, образующих
с красителями глубоко окрашенные комплексы, позволяет
получить позитивное изображение и непосредственно, без
дополнительного проявления.
Светочувствительность диазосоединений сильно зависит
от их строения, повышаясь с введением определенных
заместителей в ароматическое ядро.
Квантовый выход наименее светочувствительных из них
составляет для ^^366/и(л от 0.01 до 0.03, а для наиболее
светочувствительных (напр, для 2-диазоднфениламинсульфата)
доходят до 0.52. Квантовый выход остается приблизительно
постоянным на большом протяжении спектра и не зависит,
в некоторых пределах, от температуры и концентрации. В
спиртовом растворе квантовый выход повышается, повндимому,
вследствие устранения ассоциации диазомолекул и связанного
с ней концентрационного торможения фотореакции (ср. 36),
112 ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕк
Концентрация ионов водорода оказывает большое влияние
на квантовый выход фотореакцин диазосоедииений основного
и кислотного характера, очевидно, из-за структурных изменений
молекулы (5, 13, 14).
Большинство диазосоедннений в сильно кислой среде под
„действием света количественно разлагается с выделением
азота и образованием соответствующего фенола по схеме
реакции
ArNJ+ Hal" H2O -^ Na -+- HHal -+- ArOH
Вместо иона Hal~ здесь может фигурировать иной остаток
кислоты.
В щелочной, нейтральной и слабо кислой средах имеют
место побочные процессы, главным образом, сочетание
образующегося фенола с неизмененным диазосоединением.
Наибольшее практическое применение получили диаэосоеди-
нения бензольного и нафталинового рядов, содержащие в пара-
или ортоположениях к диазогруппе заместители типа NHPh,
NPh2 и ОН, например:
N—/
\
Cl
\ /
H
Литература по светочувствительным соединениям, ввиду
их практического значения, обширна. Более новые работы
приведены в указателе литературы в конце книги74.
Фотораспад диазосоедннений представляет значительный
интерес и с принципиальной стороны в связи с эффектом
энергетической компенсации, упомянутым на стр. 103.
18. Фотолиз на ионы (фотононизация) лейкокрасителей
Бесцветные исходные соединения, из которых получаются
трифенилметановые красители, могут быть представлены
формулой Ar3C — R, где Аг обозначает простое или замещенное
бензольное кольцо, a R —валентно связанную с центральным
атомом С группу ОН, CN, SO3H и т. п. Эти соединения
представляют собой, таким образом, лейкокарбинолы, лейко-
цианиды, лейкосульфиты (сульфоновые кислоты)
соответствующих красителей.
Под действием ультрафиолетового света с длиной волны
<С330/п[/. (ртутно-кварцевая лампа или дуга с электродами
Pe-Ni) бесцветные спиртовые растворы этих соединений
18. ФОТОЛИЗ НА ИОНЫ (ФОТОИОНИЗАЦИЯ) ЛЕЙКО КРАСИТЕЛЕЙ 113
окрашиваются в характерный цвет иона красителя Аг3С"*.
В темноте раствор с течением рремени вновь обесцвечивается
(Лифшиц, 1919)76. Наблюдаемая фотореакция может б|лть
записана следующим образом:
бесцветное
Ar3C — R
не электролит
окрашенное
Аг С**~ -4- R
электролит
Особо высокую светочувствительность обнаружили лейко
цианиды, которые окрашиваются после минутной экспозиции.
Квантовый выход фотореакции для лейкоцианида
кристаллического фиолетового равен единице, т. е. каждый
поглощенный фотон вызывает появление одного иона красителя.
Концентрация образующихся иоиов красятеля не превышает
5°/о исходного лейкоцианида; тем не менее, из-за большого
коэффициента поглощения ионов вызываемый ими оптический
эффект значителен.1)
Итак, поглощение фотона рассматриваемыми здесь лейко
формами трифенилметановых красителей приводит к явлению
распада нейтральной молекулы на сольватированные ионы.
Принципиальная возможность превращения валентной связи
при поглощении фотона в ионную и распад молекулы на ионы
был обнаружен автором этой книги на простых двуатомных
молекулах TlHal, AgHal в газообразном состоянии7ва.
Рассматриваемая здесь фотореакция протекает в более сложных
условиях, не позволяющих решить, состоит ли механизм
явления из прямой фотодиссоциации лейкосоединения на ионы,
как это имеет место для упомянутых двуатомных молекул, или
же из возбуждения лейкосоедииения с последующей
ионизацией растворителем возбужденной молекулы. Высокий
максимум поглощения лейкоцианидов (log-1Q е ^ 5) занимает
участок от 300 до 250 т\ь\ в сторону длинных волн от него
имеется второй, более плоский и низкий максимум (log10 e « 4.5)
в виде ступени на общей кривой поглощения, простирающейся
до резкой длинноволновой границы спектра, т. е. 330/п[л.
Из того факта, что квантовый выход равен единице для
ртутных линий 313 и 254 т^, попадающих в различные
максимумы, повидимому, следует, что спектр фотохимической
чувствительности совпадает со спектром поглощения. ]Уе*кду
тем, наличие в последнем сравнительно узких максимумов,
:) Такие благоприятные особенности рассматриваемой фотореакцш:
побудили воспользоваться ею длн акаинометрин, т. е, измерения и дозировки
ультрафиолетовой радиации 7<\ Природа растворителя имеет существенное
значение: реакция идет значительно медленнее в эфире и бензоле; спирт
должен быть безводным.
8 А. Н. Тереннн
114 ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
расположенных в области поглощения бензольного кольца,
достаточно определенно свидетельствует о том, что первичный
процесс заключается в переходе молекулы в возбужденное
состояние:
Ar3CR -^ Ar3CR*, (1)
а распад на ионы представляет собой вторичный процесс:
Ar3 CR* ^ (Аг3С-)С0ЛЬ.
ИЛИ
Ar3CR*^Ar3C-R 5=
З^14 —'^*3^ 1Ч ' V"X3~ /солы. ' V1^ /соль».
При втором написании реакции распад на ионы
осуществляется после конверсии электронной энергии в
потенциальную энергию растянутой связи С —R.1) Энергия сольватации
ионов при механизме (II) не входит в баланс первичного акта
поглощения фотона. Разрыв валентной связи С — CN с
превращением ее в ионную связь С"*" CN~ требует меньшей энергии,
чем в молекулах более простого строения из-за выигрыша
значительной энергии резонанса в получающемся ионе красителя.
Электростатическое же притяжение противоположно заряженных
ионов незначительно вследствие большого размера катиона
красителя.
Как было упомянуто в 16, Дж. Н. Льюис с сотрудниками
обнаружил реакцию фотоионизации на более простом
соединении Ph2N — NH2 по появлению в замороженном растворе
полосы поглощения, принадлежащей иону Ph2N**~. Представило
бы значительный интерес определение спектральной кривой
фотохимической чувствительности и сравнение ее с кривой
поглощения.
Возвращаясь к описанной выше фотореакции лейко-форм
красителей, необходимо отметить, что ее протекание
осложняется целым рядом обстоятельств.
Обратная, темаовая реакция обесцвечивания идет весьма
медленно, продолжаясь сутками; в случае кристаллического
фиолетового обесцвечивается за секунду только 10~6 часть
образовавшихся при освещении ионов. Медленно исчезает
в темноте и электропроводность раствора, вызванная
освещением. Дифенилметановые красители, которым также свой-
*) Некоторые авторы пишут лейкоцианид в виде нитрила АГ3С — NC,
который только при поглощении фатонз, переходит в тэутомериый цианид
Аг3С — CN. Для такого допущении достаточных оснований нет, но, возможно,
что фотохимически чувствительной является именно форма нитрила, обла-
Д1ющая менее прочной связью С — NC, а не С—-CN.
18. ФОТОЛИЗ НА ИОНЫ (ФОТОИОНИЭАЦИЯ) ЛЕЙКОКРАСИТЕЛЕЙ 115
ствеина рассматриваемая фотореакция, обесцвечиваются
быстрее.
Детальное исследование темяовой реакции обесцвечивания,
проведенное в последнее время77, установило, что в присутствии
воды обесцвеченный раствор содержит не исходный лейко-
цианид, а неизвестный, довольно стойкий фотопродукт с
характерным максимумом поглощения у 360 /п[л. Это соединение
также испытывает фотораспад под действием ультрафиолетового
света (или НС1), регенерируя ион красителя. Предположение,
что этот фотопродукт есть лейкокарбинол Аг3С — ОН,
образовавшийся в результате гидролиза, встретило возражение,
и это неизвестное вещество было приписано продукту
присоединения спирта к иону красителя, а именно эфиру типа
Аг3С — OR. Аналогично лейкоцианидам ведут себя и лейко-^
карбинолы трнфенилметановых красителей.
Фотолиз на ноны наблюдается и для кислотных красителей:
обесцвеченный избытком щелочи, вследствие образования
карбинола строения Аг3С — ОН] , бензаурин н фенолфталеин
вновь приобретают окраску первоначального аниона красителя
Аг3С~ под действием ультрафиолетового света, который,
очевидно, отщепляет ион ОН~75.
Фотореакция трифенилметана Аг3С — Н с его хлорпроиз-
водным Аг3С — С1, приводящая к образованию гексафенил-
этана по схеме:
Аг3СС1-ь Аг3СН %> Аг3С —САг3-нНС1, (III)
возможно, также проходит через промежуточную стадию
образования ионов Аг3С+ и С1 и заслуживает более
подробного изучения.
Уместно здесь упомянуть н о следующих фотореакциях
лейкокарбинолов трифенилметановых красителей, изученных
Лифшицем (1930)75, которые могут быть представлены
следующими суммарными схемами:
/2Аг3СОН -*- ОН — ("\— ОН1 % Аг3С - О -/—ч>— О -
I \—/ icaH5oH \—/
— CAr3-i-2H2O
{Ar3COH + С2НБОН} СбНб -^ Ага СН н- СН3СНО +- Н2О. (V)
В последней реакции происходит необратимое дегидрирование
спирта в альдегид под д йствием света, поглощаемого лейко-
карбинолом.
Кислые растворы лейко-форм Аг3СН трифенилметановых
красителей под действием поглощаемого ими ультрафиоле-
8*
116 ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
тового света также дают окрашенные ионы красителей Аг3С+.
По Лифшицу75 реакция идет через промежуточное образование
карбинола, реагирующего с кислотой по схеме:
Аг3СН + {оЛ Аг3СОН
3
(VI)
Аг3СОН —> Аг3С+, СГ -ь Н2О.
Присутствие следов красителей, даже другого класса, очувст-
вляет реакцию к видимому свету, превращая ее в
самоускоряющееся фотосенснбилнзованное окисление лейко-формы
кислородом (см. 34).
Однако сам трифенилметан Ph3CH в спиртовом растворе
испытывает под действием ультрафиолетового света в
присутствии О2 иное, более глубокое превращение с появлением
продукта желто-зеленого цвета и интенсивной флуоресценцией.
Аналогичного типа фотопродукты рассматриваются в
следующем параграфе (ср. эскулин).
19. ФотодимеризацЕЯ полициклических углеводородов
Под действием света в первой длинноволновой области
спектра поглощения на растаоры антрацена в различных
растворителях происходит, наряду с флуоресценцией, фото-
химическая реакция образования двойных молекул или
дим еров антрацена78.
Допустим, что для реакции требуется встреча возбужденной
молекулы антрацена А* с нормальной молекулой А по схеме:
А'-ьА^А,, (I)
где ку обозначает постоянную скорости, т, е. „выход" этой
реакции (1).
Обозначим концентрацию возбужденных молекул антрауена
через [А*], концентрацию нормальных молекул и молекул
диантрацена, на данном этапе реакции, соответственно через
[А] и [А2]. Эти концентрации означают числа молекул в единице
объема. Из принятой выше схемы реакции (1) следует, что
скорость образования диантрацена, т. е. возрастание его
8 ГАо]
концентрации за секунду—^~—> должна быть пропорциональна
числу встреч А* с А за то же время, а следовательно,
произведению концентраций [А*] и [А]:
(1)
19. ФОТОДИМЕРИЗАЦИЯ ПОЛИЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ Ш
Определим концентрацию [А*] возбужденных молекул.
Последние создаются поглощением /Vv фотонов в секунду,
т. е. скорость образования молекул А* есть:
АГ£!=а дг = COMt#
При таком написания предполагается, что все вступающие
в единицу объема фотоны /Vv поглощаются. Это условие
обеспечивается при достаточно большой концентрации
молекул А.
Коэффициент пропорциональности а<1отмечает тот факт,
что только некоторая постоянная доля поглотивших фотоны
молекул оказывается способной вступать в реакцию. Другими
словами, здесь учитывается возможность внутренней
дезактивации, немедленно переводящей остальную часть молекул
(1 — а) с возбужденного уровня на основной (12).
Возбужденные молекулы А* исчезают двумя путями; либо
вступая в реакцию димеризации (I), либо испуская энергию
возбуждения в виде фотона (флуоресценция):
Скорость первого процесса была дана выше (1); скорость
второго, согласно формуле (6) в 11, равна &ДА*], где
к^=— есть вероятность испускания, а т—время
„естественного" пребывания молекулы в возбужденном состоянии.
Необходимо, однако, для полноты картины учесть также
возможность процесса дезактивации возбужденных молекул
при столкновении с нормальными (12), т. е. предположить, что
встреча молекул А* с А не обязательно сопровождается
реакцией димеризации (1), но может ограничиться физическим
процессом междумолекулярного отвода и рассеяния энергии
возбуждения:
А*-+-А -* АА -* А-4-А. (Ш)
Если „выход" последнего процесса обозначить через kdt
то исчезновение возбужденных молекул будет происходить
В результате трех процессов (I), (II), (Ш) с суммарной скоростью*.
- -Ц£^ = (к, ч- kd)JА*] [А] -н к, [А*]. (3)
118
ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
Концентрация возбужденных молекул [А*] создается в
результате компенсации скорости их образования скоростью
их исчезновения, ^следовательно равенством выражений (2)и(3):
Откуда*
= (*, + kd) [А*] [А] -ь к, [А*].
j к;-*г
(4)
(5)
Подставляя это выражение в уравнение скорости реакции
фотоднмернзацин (1), имеем:
В|А21
(6)
02-
at-
JL
.a
Фиг- 27. Зависимость квантовых выходов фотореакции у и флуоресценции 9
от концентрации [А] поглощающего сает соединения.
Справа—зависимость обратной величины кзантового выхода флуоресценции
— от концеитрацвя [А].
Отсюда, согласно 15, квантовый выход у рассматриваемой
фотохимической реакции будет равен:
Т
■■«.-,
ткг[А]
[А]
1 -+- т {kr -+- kd) [А]
(7)
т. е. будет представлять собой следующую функцию
концентрации исходных молекул [А]:
(8)
где а, £, С—постоянные. Графически эта зависимость
представлена на фиг. 27 в виде кривой. При увеличении концентра-
19, ФОТОДИМЕРИЗАЦИЯ ПОЛИЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ 119
ции [А], когда С[А] делается значительно больше 1, кривая
асимптотически приближается к предельному значению
Исследования кинетики фотодимеризации антрацена
действительно привели к функциональной зависимости от
концентрация (8), выведенной нами из предположения, что в реакции
участвуют возбужденные светом молекулы А*.
Экспериментальное предельное значение у0 оказалось равным 0.25.
С другой стороны, квантовый выход флуоресценции (4) дается
выражением:
kf [A*] ab a
kf [A] ab
или:
которое также графически изображено на фнг. 27. При понижении
концентрации [А], когда С[А]<^1, кривая приближается
к значению <Ро = а- Для удобства сравнения с опытными данными
зависимость (10) представляют в линейной форме:
которая изображается прямой (фиг. 27). Пересечение этой
прямой с осью ординат дает величину —; угловой коэф-
с
фициент — постоянную — •
Исследование падения квантового выхода флуоресценции
с повышением концентрации, т. е. так называемого
„концентрационного тушения" флуоресценции (36) действительно приводит
для антрацена к линейной зависимости — от [А].
Эмпирическая постоянная С оказалась при этом, как того требует наш
вывод, совпадающей с эмпирической постоянной С для
квантового выхода фотодимеризации у в формуле (8).
Предельное значение квантового выхода флуоресценции,
достигаемое при малой концентрации антрацена, равно
<р0 —ос = О24 (11, табл. 2). Полученное численное значение
для а приводит к следующему важному заключению. Поскольку,
120 ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
как указано выше, предельный выход фотореакции уо = О.25,
что практически совпадает с а = 0.24, то из (9) следует, что
—г—^-т—«1. Другими словами, kd*&0, а следовательно, про-
цессом дезактивации возбужденных молекул при их встречах
■г нормальными молекулами (III) можно пренебречь:
возбужденные молекулы, кроме испускания, исчезают только путем
димеризации. Из четырех молекул, поглотивших фотон, при
малой концентрации испускает только одна [<?о^—г-}] при
большой концентрации флуоресценция тушится потому, что,
вместо испускания, возбужденные молекулы вступают в реакцию
(Yo = O.25).
Реакция фотодимеризацнн не объясняет явления
концентрационного тушения флуоресценции в других случаях (36),
но несомненно должна быть учтена при пользовании
концентрированными растворами красителей и родственных им
соединений.
Зная величину т из опытов по тушению флуоресценции
посторонними вещестаами (35), можно из опытного значения
постоянной С=^хкг получить величину выхода кт рзакции
димеризации (I).
Таким образом, на конкретном примерз антрацена здесь
обрисован тот путь, каким изучение кинетяки фотохимической
реакции, т. е. измерение зависимости суммарного выхода
у от различных факторов позволяет проверить предполагаемый
механизм реакции и даже определить выход или эффективность
элементарных процессов, лежащих в его основе.
20* Фотохимические реакции ксантеновых красителей
в растворах
Окрашенные фотопродукты. Под интенсивным облучением,
в особенности ультрафиолетовым светом, испускание света
растворами флуоресцирующих красителей ксантеновой группы
(флуоресцеин-Na, эозин, родамин)1) исчезает, причем растворы
либо обесцвечиваются, либо в них появляются новые
окрашенные фотопродукты79.
Исчезновение флуоресценции под действием мощного
облучения дало повод Ж. Перрену (1919) предложить объяснение
флуоресценции красителей, в корне отличающееся от
общепринятого. Он предположил, что флуоресценция красителей
представляет собой свечение, сопровождающее разрушение моле-
1) См. диаграмму иа стр. 24.
20. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ КСАНТЕНОВЫХ КРАСИТЕЛЕЙ 121
кулы светом. Испускает свет продукт реакции в виде фотона,
представляющего собой лишь часть энергии, выделившейся
при химической реакции. Этим, по Перрену, и объясняется
стоксово смещение спектра флуоресценции (4). Гипотеза Пер-
рена находится в резком противоречии с закономерностями,
наблюдаемыми при флуоресценции красителей, и вызвала сразу
заслуженную критику. Однако произведенные для
опровержения этой теории эксперименты представляют
непосредственный интерес для фотохимии красителей, а потому обсуждаются
ниже.
Интерпретация опытов затрудняется тем, что большинство
авторов не обращало внимание иа тщательное исключение
кислорода воздуха и совсем не учитывало изменения состояния
аггрегацин красителя при переходе от одного растворителя
к другому или при нагревании раствора. Исчерпывающее
описание полученных, иногда противоречивых результатов
заняло бы много места. Ограничимся приведением только
общей картины явления и попыткой ее интерпретации. Особо
подробному исследованию подверглись анионные красители:
флуоресцеин-Na (уранин) и его галоидозамещенные: эритрозин
(тетра-бром уранин) и эозин (тетра-иод уранин).
В неводных растворителях (э гиловый спирт, пиридин,
ацетон), в которых преобладает мономолекулярная дисперсность
красителя, флуоресценция интенсивна1) и фотохимическая
реакция состоит в медленном выцветании как в присутствии
кислорода, так и без него; окрашенные фЪтопродукты при этом не
образуются.2) Без кислорода краситель может быть тем или
иным методом регенерирован из бесцветного продукта.
Очевидно, имеет место обратимая фотореакция красителя с
неводным растворителем, повидимому, типа фотовосстановления (32).
Действительно, аля эозина установлено, что при
непродолжительном облучении в присутствии восстановителей происходит
обесцвечивание красителя в результате фотогидрирования80.
В темноте окраска вновь появляется, если облучение не было
слишком длительным. Так как в этом опыте кислород не
исключался, то необратимый процесс связан, очевидно, с
медленной реакцией оксидирования (глава 7).
Выцветание трифенилметановых красителей под действием
ультрафиолетового света в спиртовых растворах,
освобожденных от воздуха, отчасти также принадлежит описанному типу
1)Для эозина в ацетоне квантовый выход флуоресценции равен единице.
2) По Прингсхсйму (1922) в этиловой, амиловом спиртах и глицерине
получаются окрашенные фЪтопродукты, зависящие от природы
растворителя и отличные от получающихся в воде. Возможно, что примененный
свет содержал более короткие длины волн.
122 ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
реакций. Для малахитового зеленого обесцвечивание
наблюдалось Гебгардом; для бриллиантового зеленого—Лифшицем
{1927); розанилин оказался гораздо более светоустойчивым75.
Последний автор приписывал такое выцветание гидрированию
иона красителя за счет спирта с образованием лейкосоедине-
ния красителя по схеме:
Аг3С*--*-С2Н5ОН --^ Аг3СН.
Подобная реакция фотовосстановления идет трудно с три-
фенилметановыми красителями, склонными к фотоокислению,
а потому происходит только под действием больших квантов
ультрафиолетового света. Для ксантеновых красителей можно
предположить, что описанное выше обесцвечивание в неводных
растворах есть результат реакции такого же
восстановительного типа, но идущей под действием видимого света.
В водном, нейтральном, лишенном кислорода, растворе
реакция обесцвечивания идет очень медленно. В таком
растворе следует предполагать значительную степень ассоциации
красителя81. В воде квантовый выход флуоресценции эозина
(9 = 0.18) и эритроэина (<р = 0.05) весьма низок, по сравнению
с таковым в органическом растворителе — ацетоне (см. 11,
табл. 2). Причина, повидимому, заключается именно в
ассоциации в воде ионов красителя в димеры и более высокие аг-
грегаты. Замедление реакции обесцвечивания указывает, таким
образом, на большую фотохимическую устойчивость последних
по сравнению с мономерными ионами.
Прибавление спирта к водному раствору прежде
всего значительно повышает скорость фотохимического
процесса, очевидно вследствие участия мономерной формы. Самое
интересное заключается, однако, в том, что в водиоспиртовом
растворе уранин, эозин и эритрозин через несколько минут
интенсивного облучения-видимым светом дают нефлуоресци-
рующие, но окрашенные фотопродукты неизвестного со-
стаза (так называемые фотоуранин, фотоэозин и т. п.). В
случае уранина максимум полосы поглощения фотопродукта
смещен в длинноволновую сторону от первоначального
положения; для остальных красителей существенного изменения
положения не происходит. Эти фотопродукты получаются также
в растворе, освобожденном от воздуха откачкой, и,
следовательно, не обусловлены реакцией с кислородом (Натансон79).г)
Следует попутно отметить, что флуоресценция рассматриваемых
здесь красителей не тушится кислородом (стр. 129, 170).
1) В противоположность утверждению Блума и Шпилмана*
20. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ КС4НГЕНОВЫХ КРАСИТЕЛЕЙ 123
Если сопоставить с описанными здесь фактами приведенные
выше данные об изменении спектра красителя при ассоциации
его ионов (14,4), то напрашивается вывод, что окрашенные,
но не флуоресцирующие фотопродукты в водном растворе
являются результатом фотоаггрегации молекулярных
ионоа красителя, вплоть до образования коллоидных частиц. *)
Скорость образования окрашенных фотопродуктов сильно
увеличивается в щелочном растворе (Прингсхейм79). Добавка
NaOH(10~3N) к раствору эозина (10~5М) вызывает резкое
увеличение скорости фотореакции, примерно в 10 раз (Натансон).
Наличие спирта, даже в количестве 0.5°|0, сильно ускоряет
превращение, что, повидимому, указывает на активное участие
мономерной формы аниона красителя в фотореакцни. При этом
на начальной стадии образуется иной фотопродукт, чем в
щелочном, чисто водном растворе (Прингсхейм), который обладает
интенсивным максимумом поглощения, сдвинутым ккоротким
длинам воли, и интенсивно флуоресцирует цветом,
отличающимся от цвета флуоресценции исходного красителя. Продукт
сравнительно устойчив, так как его флуоресценция сохраняется
при дальнейшем длительном и интенсивном облучении, причем
параллельно происходит потребление щелочи (ионов ОН~).
Повышение концентрации щелочи до 0.1 N вызывает
дополнительное изменение спектра поглощения в
коротковолновой части, что указывает на одновременное образование еще
нового фотопродукта.
Аналогичные эффекты вызывает по Натансону добавление,
вместо ОН", ионов SO3 (0.3 N). Явление имеет также место
и в ииелочных растворах метилового, амиловэго спиртов и
глицерина.
Отсутствие окрашенного фотопродукта в щелочном водно-
*) С этим выводом согласуется тот факт, что Вуд наблюдал образование
таких фотопродуктов при очень высокой концентрации красителя, а в случае
нафталинового красного наблюдал в горячей виде выделение под действием
света суспензии синеватого цвета. Натаисон упоминает, что при
длительном стоянии после фотореакцни из раст> ора выпадает осадок.
Натансон, повторивший опыты Вуда и Прингсхейма в более тщательных
условиях, установил для галоидозамещевных красителей (эозин, эритрозни)
появление в растворе ионов галоида после фотореакции. На Этом
основании, а также из даичых микроанализа на элементы небольшого количества
выделенного фотоэозина, ои предположил, что рассматриваемая реакция
есть фотогидролиз, приводящий к зимен2 атомов галоида на гидроксилы.
Такое существенное структурное изменение краси1бля должно было бы
вызвать сдвиг максимума поглощения в сторону коротких длин волн, что
противоречит наблюдениям д\я нейтрального водного раствора красителя.
Кроме того, Вуд получал окрашен дый фотопролукт и д\я уранива, в котором
нет атомов галоида. Объяснение фотогидр лизом более применимо для
фотопродукгов, получающихся в присутствии щелочи (см. инже).
124
ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
5Ю
450
-Кту.
спиртовом растворе у уранина, дающего только выцветание,
и сходство спектров поглощения и флуоресценции фотоэозина
и фотоэритрозина со спектрами аниона ураиина, не
содержащего атомы галоида (фиг. 28), побудило Натансона высказать
предположение, что окрашенные фотопродукты эозина и эри-
трозина образуются в результате замены атомов галоида
в скелете этих красителей на ОН (в случае щелочи) или Н
(в случае сульфита). Это объясняет возникновение
флуоресцирующих фотопродуктов, получающихся в щелочном или
сульфитном растворах, но не происхождение нефлуоресцирую-
щих фотопродуктов,
образующихся в нейтральном водном
растворе и отличающихся
положением полосы поглощения.
Дальнейшее длительное
освещение щелочного и
сульфитного растворов приводит к их
полному обесцвечиванию с
утратой флуоресценции.
Поскольку эта реакция идет также в
отсутствии кислорода, то ее
причиной, действительно, следует
считать необратимый, более
Фиг. 23. Полоса поглощения фо- глубокий ГИДрОЛИЗ фОТОПрО-
тоэозина79. ДУКТа
Характерно что кислород
подавляет образование
окрашенного фотопродукта в
щелочном растворе и вызывает
лишь обычную медленную
реакцию окислительного разрушения и выцветания красителя
без появления новых максимумов поглощения (глава 7).1)
Торможение кислородом установлено и для других
фотохимических реакций, однако здесь оно не может быть вызвано
реакцией О2 с возбужденными молекулами или тушением их
флуоресценции, так как кислород не оказывает влияния иа
выход испускания рассматриваемых флуоресцеииовых
красителей (стр. 170). Сильный тушитель флуоресценции красителей
ион J~ совершенно приостанавливает образование фотоэозина;
однако выход флуоресценции эозина уменьшается при этом
только в 2.3 раза (Натансон). Напрашивается вывод, с которым
3) Наблюденное Вудом и Приигсхеймом образование окрашенных фото-
продуктов в водноспи^товэм растворе на воздухе не противоречит этому,
та:< как ими применялась значительная концентрация красителя и щелочи,
.понижавшие растворимость кислорода.
2 — тот же раствор через 15 мии.
освещения;
3—раствор уранина (10~5 М)
cNa2SO3(0.3M).
20- ФОТОХИМИЧЕСКИЕ РЕАКЦИИ КСАНТЕНОВЫХ КРАСИТЕЛЕЙ 125
мы неоднократно встретимся в дальнейшем, что в описываемой
реакции образования фото продуктов участвуют, главным
образом, не возбужденные молекулы А*, способные к испусканию
света, а те поглотившие фотоны молекулы,которые не
испускают света флуоресценции, но испытывают внутреннюю
конверсию в бирадикальное, метастабильное состояние Av (12).
Явление торможения реакции кислородом сводится, повидимому,
к облегчению возвращения длительно живущего бирадикаль-
ного состояния красителя в основное состояние красителя А.
Торможение, с нашей точки зрения, аналогично сильному
тушению кислородом фосфоресценции красителей (21).
Описанные выше факты относились к анионным красителям
флуоресцеиновой группы.
Из катионных (основных) красителей акридиновый
оранжевый ведет себя приблизительно как уранин, в то время как
флоксин, резоруфин, нафталиновый красный только выцветают
{Вуд79). Опыты были произведены в растворах на воздухе,
а потому толкование их не может быть одночначным. Родамин,
принадлежащий также к катионным красителям (5, диаграмма 1),
отличается от флуоресцеиновых красителей тем, что образует
окрашенный, сильно флуоресцирующий фотопродукт только
в неводных растворителях-—этаноле и метаноле. В спектре
поглощения фотородамина присутствует с большой
интенсивностью коротковолновой из двух максимумов полосы поглощения
родамина. Такой побочный максимум в катионных красителях
принадлежит, как мы Еидели, димерному иону (14,4). Однако
димерный ион, как правило, теряет способность
флуоресцировать, а, кроме того, неводные растворители мало способствуют
ассоциации ионов.
Мы предполагаем поэтому, что фотородамин представляет
собой продукт ассоциации иона родамина с молекулами
растворителя, предположительно обусловленной водородными
связями гидроксильной группы спирта с атомами N родамина.
Ассоциации такого типа приписывают коротковолновый
побочный максимум кристаллического фиолетового, наблюдаемый
в неводных растворителях50, что подтверждает наше
предположение.
Под действием концентрированного
ультрафиолетового свэта глицериновые растворы красителей флуоресцейно-
вой группы на воздухе весьма быстро теряют флуоресценцию
и выцветают с образованием иных фотопродуктов, чем
описанные выше, полученных при облучении видимым светом (Астер-
блум)79. Наиболее светочувствительным оказался эритрозин
(т^тра-иод-уранин), наименее — родамин. Эозин иуранин
занимают промежуточное положение, как это имеет место и при
126 ФОТОРАСПАД И ФОТОПРИСОЕДИНЕНИЕ
действии видимого света. При облучении длинами волн < 254
зеленая флуоресценция ураннна пропадает, но заменяется
голубой, которая при дальнейшей экспозиции исчезает. Ввиду
присутствия кислорода трудно решить, является ли этот
фотопродукт результатом фотогидрирования, Т. е. лейко-формой
красителя (5,16). Но в случае родамина обесцвеченный раствор
вновь приобретает окраску при соприкосновении с воздухом,
что является доводом в пользу втого толкования.
Образование промежуточных фотопродуктов при действии
света на водные растворы флуоресцирующих соединений в о т-
сутствии кислорода свойственно не только ионам
красителей. Эскулин — бесцветное соединение со структурой:
не
обладает интенсивными максимумами поглощения в
ультрафиолетовой области, расположенными у 340 тр. и 270 mfi и яркой
синей флуоресценцией.
При облучении освобожденного от воздуха водного
раствора мощным ультрафиолетовым светом наблюдается па-
деиие интенсивности флуоресценции до некоторого
постоянного значения, сохраняющегося длительное время (2 часа)
неизменным (Мак Леннан и Кэйл, Кирни79). Как и для красителей,
при дальнейшем увеличении экспозиции флуоресценция
постепенно пропадает из-за более глубоких процессов разрушения
молекулы. В неводных растворителях — спирте и глицерине —
начальная скорость падения флуоресценции больше, но
промежуточный фотопродукт, обнаруживаемый по постоянству
интенсивности испускания, более устойчив. В бензиловом спирте
скорость фотореакции особенно велика.
Фотохимически активным является поглощение света
только во втором коротковолновом максимуме.
Если повторить опыт в присутствии кислорода воздуха, та
происходит монотонное падение интенсивности флуоресценции,
вызванное, очевидно, фотореакцией с О2, и раствор
приобретает желтоватую окраску. Фотохимически активным является
в этом случае поглощение и в первом максимуме (340 тр).
Параллельно с падением флуоресценции этот длинноволновой
20. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ КСАНТЕНОВЫХ КРАСИТЕЛЕЙ 127
максимум исчезает. Любопытные явления последействия
рассматриваются в главе 11 (стр. 316).
В опытах Вуда79 при освещении растворов красителей
мощным потоком света большой интенсивности было обнаружено на-
рушение законов Бунзена-Роско и Вант Гоффа (15), согласно
которым количество разложившегося красителя должно было
быть пропорциональным поглощенной энергии, независимо
от ее концентрации во времени или в объеме.
Помещая раствор в пучке света, концентрируемом вогнутым
зеркалом, сначала вне фокуса, а затем в самом фокусе лучей,
Вуд, а за ним ПрингсхеЙм показали, что во втором положении
количество исчезнувшего красителя несравнимо больше.
Количество энергии в световом потоке было в обоих случаях
совершенно одинаково. Одинаковым было и поглощение света
в очень широких пределах. Различие было лишь в
плотности энергии, т.е. количестве энергии или числе фотонов на
единицу объема.
Было сделано несколько попыток объяснить это явление,
но без успеха. Вейгерт, открывший торможение реакций
фотохимического оксидирования избыточным давлением кислорода83
(глава 7,24), полагал, что наблюдавшаяся Вудом фотореакция
представляла собой реакцию с кислородом воздуха,
замедленную избыточным содержанием последнего в растворе. В фокусе
пучка света имело место по его мнению быстрое потребление
кислорода, а следовательно, при медленности диффузии
понижение его концентрации, что должно было вызвать ускорение
реакции. Толкование Вейгерта несомненно применимо для
других случаев, а именно, нэредко наблюдаемого интенсивного
возгорания флуоресценции красителей в пленках при
локальном интенсивном облучении ультрафиолетовым светом. В этом
случае действительно происходит связывание растворенного
кислорода, оказывающего тушащее действие на флуоресценцию
красителя, присутствующего в избытке. Однако для
рассматриваемой здесь фотореакции такое объяснение неприменимо, так
как она, по сказанному выше, не требует присутствия кислорода.
ПрингсхеЙм высказал гипотезу, что рассматриваемая
фотореакция требует либо последовательного поглощения одной
молекулой двух фотонов, либо сближения двух возбужденных
молекул. Такие процессы, пропорциональные второй степени
интенсивности падающего света, весьма редки. Сомнительно,
чтобы они имели место для обычных возбужденных молекул
из-за кратковременности их существования. Однако при
наличии длительного метастабяльного состояния (12) вполне
возможны двукратное поглощение фотонов той же молекулой
(ср. фиг. 21) или же встреча двух метастабильных молекул.
128 ФОТОРЕАКЦИЯ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
с использованием их совместной энергии. Такое суммирование
разновременно подведенных порций энергии приводит к
аномальной пропорциональности скорости фотохимической
реакции квадрату интенсивности света вместо обычной
линейной зависимости, что и обнаружено в условиях заведомо
благоприятных для возникновения метастабильных молекул44.
Местное нагревание раствора, выззанное концентрацией
света, ие могло вызвать обсуждаемого здесь эффекта, так как
опыт по нагреванию раствора до 100° С не привел к заметному
увеличению скорости фотореакции. Примечательно, что легко
выцветающие, но не флуоресцирующие красители не дают
нарушения основных законов фотохимической кинетики. Вопрос
заслуживает детальной экспериментальной проверки.
ГЛАВА СЕДЬМАЯ
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО
КИСЛОРОДА
21. Тушение флуоресценции кислородом
Из всех постоянных газов только кислород вызывает
исключительно сильное тушение флуоресценции ароматических
соединений и, в частности, многих красителей, при
неизменности поглощения возбуждающего света. Явление наблюдается
для растворенного8*1 85, газообразного88и адсорбированного87*81
состояния.
Тушение обратимо, т е. после удаления кислорода
откачкой или вытеснения его азотом, первоначальная
интенсивность флуоресценции полностью восстанавливается.
Из других газов также N0 оказывает тушащее действие,
сравнимое с кислородом88. В качестве примера на табл. 6
приведена величина ослабления интенсивности флуоресценции
ряда соединений в спиртовом растворе, вызванного присутствием
кислорода, давлением в 1 атмосферу над раствором.
Концентрация растворенного кислорода в этих
условиях для различных растворителей составляет: в воде около
1 • 10~*, в спирте и бензоле около 7 • 10~3, в ксилоле 7.5 • 10~3,
в гексане 15.6* 10~3 моль/л 8Э.
В табл. 7 приведены дополнительные данные для
растворимости кислорода в различных растворителях.
В табл, 8 даны значения квантового выхода флуоресценции
антрацена в различных растворителях на воздухе.
21 ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КИСЛОРОДОМ 129
Сравнение ее с табл. 7 для растворимости кислорода
в тех же растворителях обнаруживает только в единичных
случаях обратное соотношение между растворимостью О2
и квантовым выходом флуоресценции. В общем случае эта
зависимость не имеет местами, кроме концентрации
растворенного кислорода, на квантовый выход флуоресценции влияют
также специфические свойства растворителей. Аналогичное
несоответствие с растворимостью О2 наблюдалось для другого
полициклического углеводорода — бензпирена: несмотря на
одинаковое содержание кислорода в диэтиловом эфире и петро-
Таблица б1)
Величина тушения флуоресценции (в процентах) при наличии 1 атм
кислорода над спиртовым раствором вещества с концентрацией ок. 10~5М
(Вейсс, Вейл-Малерб)85.
З-4-Бензпирен 91.7 Этил хлорофиллид .... 32.1
Пнрен 87.7 Сернокислый хинин в мета-
Хрнзен 8S.7 ноле 17.5
Рубрен 68.0 Эозин 0
Антрацея 60.6 Акридин ....-•.. 0
Таблица 7 э°
Растворимость кислорода, выраженная в см3 газа (при 0° 760 мм) в 1 см3
растворителя при указанных температурах (° С)
Диэтиловый эфир . . 0.41 20° Бензол
Петролейный эфир . 0.41—0'2918° Этнлацетат
Четыреххлористый Изоамил. спирт . . .
углерод 0.22 25° Этиловый спирт, абз.
Ацетон 0.21 19° Этилов. спирт 95°/0 .
Хлороформ 0.20 16° Пиридин 0.12—0.10 28Р
Метанол 0.17 19° Диоксан 0.12 28е
Ксилол 0.17 16° Нитробензол .... 0.07 18°
Толуол 0.17 18°
Таблица 8
Квантовый выход флуоресценции 9 антрацена в различных растворителях
в присутствии О^ (Боуен, Нортон) 85
Парафин 0.28 Ацетон 0.18
Уксусная кислота 0.24 Гексан 0.15
Этиловый спирт 0.22 Пиридин 0.13
Этилэцетат 0.19 Хлорбензол 0.11
Тетралин 0.19 Хлороформ 0.10
Бензол 0.19 Тетрахлорэтилен .... 0.08
Ксилолы 0.19 Четыреххлористый углерод 0.02
Диэтнловый эфир .... 0.18 Бромбензол 0.01
Твердый антрацен .... 0.8—1.0
0.16
016
0.16
0.14
0.12
19°
20°
17°
20°
28°
См. также интересную таблицу такого же рода в
9 А. Н. Теренин
130 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
лейном эфире, интенсивность флуоресценции бензпирена в пер-
иом в 2 раза больше, чем во втором. То же имеет место
в случае изоамилового спирта и этилацетата: при одинаковой
растворимости О2 интенсивность флуоресценции в первом
растворителе в 2 раза больше. В нитробензоле, несмотря на
минимальную растворимость кислорода, флуоресценция
бензпирена полностью отсутствует, но этот растворитель сам
поглощает возбуждающий ультрафиолетовый свет, а следовательно,
играет роль светофильтра.:)
Хотя тушение, как сказано выше, полностью обратимо,,
но при достаточно длительном или интенсивном освещений
раствора в нем наблюдается поглощение кислорода с
образованием продуктов необратимого окисления, которые для
антраценовых углеводородов и рубрена были выделены и
отождествлены с перекиси ым продуктом присоединения
молекулярного кислорода. Эти перекисные соединения, получающиеся
на свету, получили название фотооксидов (см. след.
параграф).
Измерение скорости реакции по поглощению О2 или
исчезновению углеводорода и сравнение ее с величиной тушения
флуоресценции кислородом, взятой, например, из табл. 6, позволяют
качественно судить, имеет ли место параллелизм между обоими
явлениями. При таком качественном сопоставлении оказывается,
что только для бензола, его метилированных производных
и рубрена можно говорить о параллелизме обоих явлений;
флуоресценция нафталина, антрацена, аценафтена 3—4-бензпи-
рена, дифенила и трифенилметана тушится значительно сильнее,
чем это можно было бы объяснить необратимой фотореакцией
с кислородом (фиг. 29). Бензпирен, испытывающий согласна
табл. 6 исключительно сильное тушение флуоресценции
кислородом, вообще не образует стойких фотооксидов.2)
С другой стороны, можно усмотреть прямую связь между
тушением и фотореакцией в том, что акридин, не
испытывающий тушения кислородом, не образует также устойчивого
фотооксида.
1) Интенсивность флуоресценции не является надежным критерием для
суждения об изменении выхода испускания, так как при переходе от одного
растворителя к другому происходит смещение узких полос поглощения
рассматриваемого полициклического углеводорода по отношению к
возбуждающим линиям ртутной лампы (глава 4), что нарушает постоинство
поглощения света.
2) Факт отсутствии фотооксндов не является, однако, всегда
убедительным. Так, например, авторы, пытавшиеся подучить ф это оксиды нафталина
и его производных, пользовались солнечным светом, не содержащим
коротковолновой ультрафиолетовой радиации, необходимой для фотоактивации
этих соединений.
21 ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КИСЛОРОДОМ
131
Для решающих выводов необходимы количественные данные
о квантовом иыходе у фотореакции с кислородом и о квантовом
выходе <р флуоресценции для ряда соединений. Действительно у
дает долю от всех поглотивших фотоны молекул, которая
вступает в реакцию (15); <р— долю от всех поглотивших фотоны
молекул, которая испускает свет флуоресценции (4). Если
тушение флуоресценции при увеличения концентрации
кислорода вызвано реакцией возбужденной молекулы А* с моле-
Антрацен в бензоле Нафталин в гексане Бензол в гексане
ОП/К
ПООЙ
0008
Фиг, 29. Зависимость для ряда ароматических углеводородов кэантовых
выходов флуоресценции 9 н фотореакции поглощения кислорода f
93ft \
от давления последнего (по Боуену >'
кулой О2, то имеет место конкуренция между двумя путями
исчезновения возбужденных молекул, а именно:
О
АО,
i0X
(D
(И)
Увеличивая давление кислорода, мы увеличиваем число
процессов (I) за счет процессов (11). В таком случае выход
фотореакции: будет возрастать за счет уменьшения выхода
флуоресценции, а их сумма будет оставаться постоянной
и равной предельному выходу флуоресценции <ро> когда
кислорода иет и реакция (I) не происходит:
у-ь <р = const = ф0. (1)
Такое соотношение приблизительно соблюдается для
тушения флуоресценции бензола, его метилзамещенных (кроме
СвМев) и рубрена. В этих случаях можно считать, что тушение
кислородом вызвано реакцией возбужденной молекулы
углеводорода с О2. Для большинства остальных флуоресцирующих
132 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
улгеводородов в растворах выход реакции фотооксидирово-
ния у(1) значительно ниже, чем величина тушения кислора-
дом о0—9, что указывает на легкость распада комплекса АО2,
образовавшегося в первичной реакции (I). Для этих
углеводородов необходимо принять независимый от (I) третий
преобладающий путь исчезновения возбужденных молекул, а именно,
в результате процесса дезактивации:
А*-*-О2^А*О2-*А+О2, (III)
при котором электронная энергия возбужденной молекулы
рассеивается в тепло (12).
Кинетика тушения кислородом. По аналогии с реакцией
димеризации антрацеиа (19), рассмотрим кинетику тушения
флуоресценции полициклическнх ароматических углеводородов,
оставляя открытым вопрос о механизме тушения. Другими
словами, мы не будем различать процессы (I) и (III), объединяя
их в один процесс исчезновения возбужденных молекул в
результате сближения А* с О2, имеющий кинетическую
постоянную kq = кох ч- кл\
'ЧАО, (kj (IV)
Увеличение собственной концентрации растворенного
углеводорода вызывает также тушение его флуоресценции,
независимо от присутствия кислорода, т. е. явление
концентрационного тушения, о котором речь была выше (19). Природу
последнего мы также оставляем здесь без уточнения, изображая
схемой:
(V)
Необходимо отметить, что повышение собственной
концентрации углеводорода в растворе вызывает снижение величины
тушения, производимого кислородом. Это значит, что наличие
концентрационного тушения (V) сокращает время т
пребывания молекулы А в возбужденном состоянии, а следовательно,
и возможность ей встретиться с молекулой О2. Итак, мы имеем
дело с тремя независимыми процессами (II), (IV), (V),
вызывающими совместно уменьшение концентрации [А*]
возбужденных светом молекул со скоростью:
8 ГА*] . ^
^__^_ *- _^* | Иш I /\ 1* I 1 Шш I /\ Ф I I /\ I ■ / _ ■ /\ r^ llflll fill
5, /L J ffL JLJ QV JL2J' V/
'*
21 ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КИСЛОРОДОМ 133
Скорость появления возбужденных молекул н §—
определяется постоянной скоростью поглощения Wv фотонов в сек.,
из которых только некоторая доля а < 1 реализует
возбужденное состояние А* (19). Необходимость введения а вызвана
наличием процесса внутренней дезактивации молекулы,
совершающегося в значительно более короткий промежуток времейи,
чем длительность возбужденного состояния (12)»
Численное значение постоянной ос можно получить в
предельном случае разбавленного раствора, без кислорода, когда
процессами тушения (IV) и (V) можно пренебречь и все
молекулы <xjVv, достигающие возбужденного состояния, испускают
свет согласно (II). Если значение предельного выхода
флуоресценции при таких условиях обозначить <ро> То а^ = ¥о^»
а следовательно а = <ро (ср. 19)-
Таким образом постоянная скорость появления
возбужденных молекул равна:
^ ЛГ (3)
Приравнивая выражения (2) и (3) для скоростей
противоположных процессов исчезновения и появления возбужденных
молекул, определяем из получившегося уравнения концентрацию
[А*] этих молекул в установившихся условиях при заданных
значениях [А] и [О2]:
г д *1 ^
Квантовый выход флуоресценции в этих условиях 9 выразится
следующей зависимостью от [А] и [О2]:
9о
или:
^ (5)
где То=1Г" ©CTa «естественное" время жизни невоьмущенной
молекулы А* (11). Отношение <ро/? должно представлять собой,
таким образом, линейную функцию переменных [А] и [О2].
Такая зависимость действительно находится в
удовлетворительном согласии с опытными данными. Опыт дает,
следовательно, возможность определить кинетические константы к^к^
соответственно, концентрационного и кислородного тушения
134
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
флуоресценции для различных ароматических полициклических
углеводородов. В табл. 9 приведены полученные значения для
растворов этих соединений в гексане,1)
При сопоставлении констант тушения кислородом k2 с
выходами реакции фотооксидирования соответствующих
углеводородов (определенных по поглощению Оа) вырисовывается,
в большинстве случаев, отсутствие параллелизма
между тушением и фотореакцией. Так, хотя константы тушения
кислородом к2 нафталина и аценафтена очень высоки (табл. 9),
выход фотореакции, напротив, весьма низок. Для антрацена
и его производных константа к2 имеет наинизшее значение,
между тем оии как раз легко образуют фотооксиды. Только
для бензола и его производных тушение и фотореакция,
повидимому, тождественны (фиг. 29).
Та блнца 9
Постоянные ki концентрационного и к2 кислородного тушения
флуоресценции ряда ароматических углеводородов в гексане по формуле (5) 2)
Углеводород фо &1 к2 Углеводород Фо кг k2
Бензол. . . .
Толуол. • . . ,
о-Ксилол. . . ,
м- „ . . . .
П~ н • • ■ ,
1-3-5 триметил-
беизол . . . .
Гекеаметнлбенэ.
0.11 <0.5 600 Нафталин. . . 0'38 < 0.5 2400
0.23 <0.5 1100 Аценафтен . . 0.70 2.6 2200
0.29 < 0.5 1060 Антрацен . . . 0.46 178
0.30 <0.5 800 Фенантрен . . 0.27 < 0.5 960
0.41 < 0.5 1050 Флуорен ... 1.0' 2.5 530
Днфеннл . . . 0.23 < 0.8 440
0.16 < 0.5 850 Трифоннлметан 0.23 < 0.5 580
0.04 < 0.5
Из изложенных данных по ароматическим полициклическим
углеводородам следует, что наблюдаемое тушение
флуоресценции кислородом отнюдь не сопровождается образованием
стабильного продукта присоединения—фотооксида, а
ограничивается преходящим взаимодействием возбужденной молекулы
с О2 согласно физической реакции (III). На это указывает
также легкая восстанавливаемость первоначальной
интенсивности испускания, после удаления кислорода, а также отсут-
1) Кинетика фотооксидирования рубрена не укладывается в
приведенную простую формулу, предполагающую независимое тушащее действие
молекул рубрена и О,, а требует для своего объяснения допущение
длительной ассоциации Рубрен-О2 (22). Антрацен, растворенный в бензоле,
также дает кинетическое выражение длн тушения флуоресценции более
сложное, чем (5).
2) Концентрации [А] и [О2] выражены в моль/л, 9о есть квантовый
выход флуоресценции в отсутствии тушителей при возбуждении X254
(Боуен» Унлнамс)85.
21. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КИСЛОРОДОМ 135
-ствие каких-либо новых полос в спектре поглощения раствора
после длительного освещения.
Переходим к вопросу о тушении кислородом флуоресценции
красителей.
Тушение флуоресценции красителей кислородом. Особый
интерес в этом отношении представляют работы Каутского,
в которых красители (трипафлавин, уранин, хлорофилл, пор-
фирины и др.) фиксировались на поверхности прозрачных
адсорбентов (силикагель, алюмогель, органические
адсорбенты)87. В вакууме такие адсорбаты не только интенсивно
-флуоресцируют, но дают явление замедленной флуоресценции,
описанное выше (12, стр. 78). Впуск кислорода при ничтожном
давлении порядка 10~3 мм достаточен для исчезновения этого
аномального послесвечения флуоресценции. Дальнейшее
тушение нормальной флуоресценции адсорбата требует уже
значительно больших давлений кислорода (100 мм)88. Этот
факт находит свое естественное объяснение в различной
длительности возбужденного и меТастабильиого состоянии
молекулы красителя. Аномально большое время жизни
в 105—-109 раз превышающее нормальное время жични
{тдаЮ"8 сек-), делает метастабильные молекулы
чувствительными к значительно меньшим концентрациям кислорода.
Сказанное служит примером повышения реакционной способности
.метастабильного состояния А* молекулы красителя,
обусловленного одним только фактом его большей длительности.
Наряду с этим результатом, Каутский обнаружил, что
явление замедленной флуоресценция красителя (с
длительностью порядка 10~2 сек.) можно наблюдать и в жидком
,растворе при обычной температуре, если тщательно удалить
растворенный кислород. Эффект слаб в воде, отчетлив в этаноле
и особенно значителен в ацетоне8i.
Однако самое замечательное явление, открытое Kayv-
ским (1933), есть переход кислорода в активную форму при
тушении им замедленной флуоресценции красителей87'91. Этот
факт был установлен следующим остроумным экспериментом.
В эвакуированный сосуд была помещена смесь двух родов
зерен силикагеля: на одних зернах был адсорбирован
трипафлавин, на других— бесцветная лейко-форма малахитового
зеленого, способная при дегидрировании принимать зеленую
окраску вследствие образования красителя. Заметим, что
лроцесс дегидрирования данной лейко-формы и появления
красителя может быть осуществлен только освещением
ультрафиолетовым светом, поглощаемым лейко-формой, в
присутствии большого давления кислорода (18). При малом давлении
кислорода (10~4 мм) освещение смеси зерен видимым светом,
136
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Св_ет
поглощаемый
который действует только на трипафлавин, не вызывает
никаких изменении. Однако при некотором оптимальном давлении
кислорода в 1.8 • 10~3 мм, когда имеет место тушение
послесвечения трипафлавина, на первоначально бесцветных зернах,
несущих лейкосоединение, появляется зеленая окраска,
вызванная дегидрированием последнего. При давлении кислорода
4 • 10~3 мм, несколько большем оптимального, явление не имеет
места. На фиг. 29а для наглядности представлены
схематически два соседних зерна силикагеля, из которых одно имеет
на поверхности молекулы
трипафлавина, а второе сплошь
покрыто адсорбированным лей-
косоединением. Таким образом
происходит своеобразное
явление сенсибилизованного трипа-
флавином окисления
кислородом другого вещества,
пространственно отделенного
от места поглощения света.
Этот многократно
обсуждавшийся опыт допускает только
следующее толкование:
молекулы кислорода, приходя в
соприкосновение с поглотившим
фотоны видимого света
молекулами трипафлавина, переходят
в некоторую длительно
живущую активную модификацию,
отлетающую в объем и
способную дегидрировать лейко-
соединение (фиг. 29а). Существование оптимального и
притом низкого давления кислорода объясняется, очевидно,
тем, что при очень малом давлении метастабильные, а тем
более возбужденные молекулы трипафлавина успевают отдать
свою энергию до столкновения с молекулой О2. При большом
давлении кислорода активная модификация О2 не доходит до
лейкосоединения, но тем или иным путем дезактивируется или
реагирует с самим красителем.
Вопрос о природе активной модификации кислорода вызвал
оживленную дискуссию и до сих пор остается нерешенным Jl.
Каутский предположил, что молекула О2 воспринимает энергию
возбуждения красителя и переходит с основного своего
электронного уровня (32) на один из ближайших, выше
расположенных уровней (:2 или :A), требующих подведения энергии
от 22.5 до 37.8 кгкал. Эти уровни молекулы О2 метаста-
Фиг. 29а. Схгма опыта Каутского
по фотосеисибилизованному
окислению на расстоянии.
Заштрихованные ззрна окрашены
трипафлаэином; белые — несут лей-
косоедияение малахитового
зеленого.
21. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КИСЛОРОДОМ 137
бильны, т. е. возвращение с них на основной уровень мало
вероятно, а потому молекула кислорода в таком состоянии
повышенной энергии может существовать длительное время.
Теренин (1943)24 привел дополнительное обоснование
предположения Каутского, указав на сохранение суммарного
спина электронов при таком процессе (ср. стр. 82), а именно:
он представил процесс передачи энергии следующей схемой:
.meta
1+оГЧПП ], (VI)
где квадратные скобки обозначают не концентрации, а
квантовые ячейки; спаренные электроны изображены
антипараллельными, а непарные электроны — параллельными
стрелками— спинами (6). Молекула О2 в основном, нормальном,
состоянии имеет два непарных электрона, в соответствии
со спектральным символом этого состояния. (32), и ведет себя
как своеобразный бирадикал. В метастабильной же модифи-
Oraeta gr-.
2 электроны молекулы кислорода спарены.
Обратное имеет место для метастабильного А* в основного А
состояния красителя. Молекулы Av и О2 как бы обмениваются
электронами. Избыток внергии рассеивается в вибрационной
форме, что указано знаками—. Молеку\а Оа"в * обладает
возможностью принять дополнительный электрон в
освободившуюся квантовую ячейку, т. е. действовать как окислитель
по отношению к лейкосоединению малахитового зеленого НМ,
производя реакцию по схеме:
НМ -* О," н- НМ+ -> НО2 -+- М.
Противники Каутского считают опровержением его
предположения наблюдение Гаффрона (1935)92. Последний установил
в растворе наличие реакции окисления (дегидрирования)
аминов кислородом, когда присутствующий сенсибилизирующий
краситель поглощает инфракрасный свет (с Х = 800 mjx),
т. е. при поглощении энергии 35 ккал, недостаточной для
перевода молекулы О2 на метастабильный уровень :2 (37.3 ккал).
Но Оо имеет также более низкий метастабильный уровень
аА(22.5 ккал). Это опровержение нельзя считать поэтому
убедительным* Мы полагаем, кроме того, что в растворе
реакция фотосенсибилизованного окисления кислородом может
идти по иному механизму, чем в вакуумных условиях,
осуществленных Каутским.
Вейсс предположил, что активная модификация молекулы
кислорода в опытах Каутского есть иои О2~, образовавшийся
138 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
путем захвата электрона от возбужденной молекулы
трипафлавина А*93. Между тем, в состоянии иона О2"~ молекула
кислорода по энергетическим данным должна утратить
окислительную способность по отношению к нейтральному лейко-
соединению НМ, как указано было автором этой книги. *)
Позднее Вейсс присоединился к точке зрения Франка и Леви
(1935)94 и Франке и Ливингстона (1941)ti9, согласно которой
активной модификацией кислорода в опыте Каутского является
радикал НО2. образовавшийся в результате
дегидрирования самого сенсибилизирующего красителя нА кислородом
по схеме:
нАтеЬа -I- О2 -* hA+<V -* А-О-О-Н -> А ■ -ь Н0'2, (VII)
где буквой н нами обозначен структурный атом водорода
в скелете молекулы красителя А. Знак meta означает, что
молекула красителя в результате поглощения света перешла
в длительное метастабильиое состояние повышенной энергии.2)
Радикал НО'2, являющийся незавершенной перекисью
водорода, должен обладать дегидрирующими свойствами и
окислять лейкосоединение НМ, что также согласуется с
обнаруженным Каутским свойством активной модификации молекулы
кислорода.
Приведенная концепция Франка-Ливингстона-Венсса
встречает, однако, то возражение, что сам краситель нА претерпевает
при этом процесс необратимогр дегидрирования, превращаясь
в форму, существенно отличную по структуре от исходной.
Действительно, при отрыве атома Н должно произойти
изменение строения, затрагивающее резонанс валентных структур,
а следовательно и спектр поглощения. Иными словами,
краситель-сенсибилизатор испытывает сам химическое изменение
в этом фотопроцессе и не может функционировать второй раз,
как это принимается для процесса сенсибилизации. Между
тем, в опыте Каутского нет никаких указаний на одновременное
разрушение трипафлавина. Подробное выяснение этого обстоя-
) Но Ofc~ может отщепить протон от катиона красителя
]*, превращаясь при этом в активный окислитель НОг-
2) По Франку н Ливннгстону метастабильная форма красителя есть
„таутомер" молекулы красителя, вызванный перемещ нием „лабильного"
атома водорода в скелете красителя. На необоснованность такого пред-
лоложения автор книги указал в двух статьяхljl*. Поэтому в приведенной
схеме вместо принятого нами обозначения А поставлено обозначение Ametft,
не прецизирующее природу метастабильного состояния. Теперь, когда
известно, что „фосфоресцирующее" состояние красителя есть б и
радикальная (триплетная) его форма, схема Франка-Ливингстояа должна быть
применена именно к ней.
21. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КИСЛОРОДОМ 139
тельства явилось бы решающим для принятия данной точки
зрения. Ей противоречит легкость отрыва активной модификации
молекулы кислорода от красителя, установленная в опыте
Каутского, и которая больше согласуется с физическим
процессом простой передачи энергии, чем с химической реакцией
перегруппировки и последующего отрыва радикала НО2.
Мало вероятно, чтобы энергия, требующаяся для различных
этапов схемы (VII), могла быть покрыта электронной энергией
метастабильного уровня и притом в одном акте. Заметим
попутно, что фотооксиды ароматических углеводородов при
термическом распаде выделяют молекулу О2 в нормальном,
а не активном виде95.
Эксперимент Каутского ввиду его фундаментального
значения несомненно заслуживает повторения и более детального
изучения.
Переходим к механизму тушения кислородом обычной
флуоресценции красителей в растворах и на адсорбентах,
требующего, как мы упоминали, значительно большего давления этого
газа из-за кратковременности возбужденного состояния А*.
Указаний на появление модификации молекулы О2 в этом
процессе, подобной описанной 4ыще, не имеется. Дезактивация
молекукы А* при встрече с молекулой Оа может заключаться,
согласно схеме III (21), в превращении электронной энергии
возбуждения в вибрационную энергию и рассеянии последней
в тепло. Для О2, в качестве партнера столкновения, такой
процесс имеет повышенную вероятность ввиду большей
химической активности кислорода по сравнению с другими газами.
Последняя обеспечивает более тесную ассоциацию молекулы
кислорода с возбужденной молекулой А*, а следовательно!
более длительное взаимодействие. В комплексе А*О2
создаются благоприятные условия для вибрацининой конверсии
электронной энергии возбуждения в вибрации комплекса, как
целого с последующим распадом на А-+-О2.г)
Однако данные по сенсибилизованной флуоресценции (12)
показывают на большую вероятность передачи части
электронной энергии, как таковой, если партнер способен ее
воспринять, т. е. имеет соответствующий высокий уровень. Как
было указано выше, молекула О2 может воспринять энергию
порядка 1 э. вольта с переходом на уровень метастабильного
состояния Ог"*- Поэтому есть основание ожидать появления
этой модификации молекулы кислорода и при тушении флуо-
г) Ср. схему между молекулярной дезактивации электронной энергии
■на стр. 84, где М = О2, нлн = NO.
140 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
ресценции, притом не только красителей, но и более простых
ароматических соединений.
Теренин24 высказал предположение, что рассматриваемый
здесь процесс тушения, как передача энергия, должен
подчиняться правилу неизменности суммарного электронного спина
партнеров, установленного для передачи электронной энергии
между более простыми частицами (правило Вигнера). Это
дополнительное требование приводит здесь к выводу, что при
передаче энергии возбуждения молекулам с непарными
электронами, как О2 или N0, возбужденная молекула А* должна
переходить на метастабильный, а не на основной уровень,
как это явствует из следующей развернутой схемы:
A* [f] [J] + 02 [f] [f] —* Av [f] [f] -к ОГ1
A* [f] [|] -+- NO [f] —> A" [f] [f] -*- NO
(обозначения были приведены выше на стр. 137). Действительно,
одновременно с передачей энергии, здесь соблюдается
сохранение суммарного спина системы двух частиц, не имеющее
места, если А* переходит при этом процессе в основное
свое состоявие A[fy]. Между партнерами столкновения здесь
происходит как бы обмен электронами с сохранением
направления их спинов без необходимости поворота спина в обратном
направлении, который требует затраты энергии. В этом
автор видит причину исключительно сильного тушения*
производимого О2 и N0, выделяющее их из ряда других газов,
для которых доступен только вибрационный механизм
дезактивации.
Действительно, возбужденные молекулы простейших
ароматических соединений в газообразной фазе, как например анилина,
нафтиламина, антрацена, „тушатся" при первом же столкновении
с молекулой О2> тогда как выход „тушащнх" столкновений
всеми другими газами (H2f N2, СО, СО2> меньше 1:100021'22.
Молекуле О2 по этой концепции передается разность
энергий возбужденного и метастабильиого уровней
флуоресцирующей молекулы (А* и Av, см. 12). Если для данного
соединения эти уровни расположены настолько близко, что
передаваемая энергия недостаточна для осуществления пере-
2 ->vJ2 « то тушение кислородом не будет иметь
места. Отсутствие тушения флуоресценции кислородом у ряда
ксантеновых красителей 96 (уранин, эозин В, родамин G)
приписано автором книги именно такой причине, хотя несомненно
играют роль н особенности структуры красителя, благоприят-
22. ФОТОРЕАКЦИИ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ С КИСЛОРОДОМ 141
ствующие взаимодействию с О2, как, например, наличие легко
отщепляемого атома Н (см. ниже)-1)
Встреча Av со второй молекулой О2 сопровождается
передачей последней электронной энергии согласно (VI) и
завершает, тем самым, возвращение первоначально возбужденной
светом молекулы в основное состояние. В конденсированной
фазе процесс дезактивации А* -» А может осуществляться
и средой путем рассеяния электронной энергии в тепло (12).
Механизм сенсибилизованного красителями фотоокисления
кислородом посторонних органических соединений,
присутствующих в растворе, рассматривается в главе 8 (34).
22. Фотореакции ароматических углеводородов
с кислородом
Мы рассмотрим здесь подробнее фотохимическую сторону
действия кислорода на возбужденные светом молекулы
ароматических углеводородов, о тушении флуоресценции которых
говорилось выше.
Простейшие производные бензола (толуол, ксилолы)
в жидком и растворенном состояниях при освещении в
присутствии кислорода ультрафиолетовым светом, попадающим
в первую область поглощения (300—240/п^), связывают
газообразный кислород07. Наряду с кон°чным продуктом реакции —
бензойной кислотой (с), образуются также фотопродукты
присоединения кислорода, имеющие свойства перекисей.
Охлаждение или прибавление нитроароматических соединений
стабилизует промежуточные продукты реакции и позволяет
выделить бензальдегид (а), гидроперекись бензола, иначе —
пербензойную кислоту (Ь), задерживая их дальнейшую реакцию
с кислородом, которая идет самопроизвольно и в темноте
!) Тереинв выдвинул также гипотезу, что молекулы О2 и NO могут
■облегчать „запрещенные" электронные переходы А*->А% и AN->A(12)
на основании лишь факта наличия у вих иескомпенсироваиного
электронного спина, а следовательно парамагне тиама'Ч Облегчение поворота
спина в присутствии парамагнитных атомов или радикалов установлено
в ряде явлении. Электронная энергия, выделяющаяся при этих переходах,
должна рассеиваться в виде вибрации столкнувшихся частиц. Возможность
такого парамагнитного механизма тушения флуоресценции (А*—> А') и
фосфоресценции (А' —> А) требует постановки i роверочных опытов, дли
которых парамагнитные ноны в растворах непригодны из-за больших размеров
сольватиой оболочки, препятствующей достаточному сближеаию с
возбужденной молекулой. Автором такие опыты в настоящее время проводятся.
142 .ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА'
(реакция самоокисления), но ускоряется ультрафиолетовым:
светом, также поглощаемым этими продуктами.
/ \—сн3*-+-о2—
н
1
fN r\
н-о-с/ но
(Ь) (с)
Присутствие воды сильно ускоряет реакцию.
Первичный фотопродукт присоединения О2 к молекуле
толуола не может быть выделен, так как он очень неустойчив
и быстро разлагается при вынимании из охлажденной смеси,
давая бензойную кислоту. Предполагали, что первичный
фотопродукт имеет строение (е) и образуется при реакции О2
с гипотетической таутомерной формой (d) толуола по схеме::
н хн
(d)
СН2 ( >-С-СН
0-0-Н 6-0-Н
(О (?)
Следующий этап, согласно этому предположению,
заключается в перегруппировке первичного пероксида (е) в
устойчивую форму надкислоты (f), которая и обнаруживается
аналитическими реагентами на перекиси.1) Но можно с
уверенностью сказать, что возбужденная молекула толуола не
представляет собой таутомера (d), существенно отличающегося
z) Органические перекиси обнаруживаются, например, по выделению
молекулярного иода из подкисленного раствора KJ, т. е. реакцией раэрндкк
анионов иода.
22. ФОТОРЕАКЦИИ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ С КИСЛОРОДОМ 143
от нормальной молекулы, и что предполагаемое строение (е)
первичного пероксида мало вероятно из-за пространственной
близости скопившихся атомов, неизбежно вызывающей
их взаимное отталкивание. Таким образом указанное
предположение не имеет под собой почвы. При самоокислении этил-
бензола образуется надкислота (g1), которую естественно
приписать перемещению лабильного атома водорода группы — СН2 —
(у— положение по отношению к ароматическому ядру)
(ср, стр. 223).
Исключительно большое значение для выяснения протекания
реакции молекулярного окисления ароматических соединений
имеют работы Бэкстрёма по кинетике фотооксидирования
бензальдегида в растворах98. Обнаруживаемая аналитически
перекись в этом случае есть пербензойная кислота, иначе —
гидроперекись бензола (Ь). Квантовый выход реакции
образования бензойной кислоты (с) составляет 10000 молекул на один
фотон ультрафиолетового света (к 254 т(л), поглощенный бен-
зальдегидом. Такой громадный выход указывает на цепное
протекание реакции, заключающееся в том, что образующаяся
первоначально под действием света по нижеуказанной схеме IX
реакционноспособная молекула бензальдегида (V)
регенерирует вновь одновременно с появлением конечного продукта.
Присоединяя О2, реакционноспособная молекула
бензальдегида образует сначала активный промежуточный продукт
(А О2), который превращается в конечный продукт (пербензой-
ную кислоту) при встрече с нормальной молекулой бензаль^
дегида. Последняя при этом активируется или теряет атом Н
и начинает новое звено темновой реакции:
Ph
\ СО -^ \rrv Ph = C6H5 (IX)
Н
Ph Ph
\со
Н—0-0 Н
НО
144
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Активными продуктами являются первичные
лабильные пероксиды с непарными электронами.1) Они служат
переносчиками кислорода на другие соединения,
присутствующие в зоне реакции". При этом обычно развитие реакции
прекращается или, как говорят, такие соединения „обрывают"
цепи. Таким „ингибитором" является, например, антрацен,
который, будучи введен в раствор, оксидируется одновременно
с бензальдегндом, причем последний дает бензойную кислоту
/О
(см. схему IX). В стабильных же пероксидах типа А\ | или
А—О—О—Н, обнаруживаемых аналитически и выделяемых
химическим путем, молекула О2 связана настолько прочно,
что они, как правило, неспособны продолжать реакцию при
обычной температуре.
Многочисленные полициклические ароматические
углеводороды, как упоминалось выше, обладают способностью
образовывать более или менее устойчивые фотооксиды, химически
изолируемые в некоторых случаях в виде бесцветных
кристаллических продуктов. Последние при нагреиании выделяют
кислород полностью или частично, а иногда при этом взрывают.
Такая способность характерна для антрацена (а) и его
производных, образующих легко диссоциирующие фотооксиды 10°.
Углеводороды с фенантреновой (Ь), а также нафталиновой
структурой не дают выделяемых фотооксидов, хотя и
подвергаются окислению кислородом101. Замена одного из —СН =
мостиков в антрацене на —/N= приводит к акридину (с),
не образующему стойкого фотооксида102. Нафтацен (d) также
дает продукт присоединения и выделяет кислород обратно
при нагревании300.
Н
/\у Ч/ч
N
(с)
(а)
*) Мы намеренно обозначаем здесь активные формы молекул значком
бирадикала , в отличие от всех авторов, предполагающих наличие только
некоторого состояния возбуждения (см. ниже}.
22. ФОТОРЕАКЦИИ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ С КИСЛОРОДОМ
Ph Ph
(d)
Получаемые препаративно фотооксиды обладают
ультрафиолетовым спектром поглощения, расположенным в области
более коротких длин волн, чем исходный углеводород. Под
действием поглощаемого им света они могут распадаться
с выделением кислорода и первоначального углеводорода,
обнаруживаемого спектрально 1Oi.
Получению и свойствам разнообразных фотооксидов
посвящены многочисленные работы Дюфресса с сотрудниками,
содержащие громадный фактический материал, требующий
самостоятельного обзора 100~103.
Исключительно большое внимание было посвящено
изучению фотооксидирования углеводорода рубрена (тетра-фенил-
^нафтацена), имеющего строение (е)105. Он представляет собой
красное вещество с характерным спектром поглощения,
интенсивно флуоресцирующее в растворах желтым светом, способное
действовать как „переносчик" кислорода иа другие,
присутствующие в растворе, органические соединения, т. е. быть
фотосенсибилизатором оксидирования.
При освещении бензольного раствора рубрена (конц.
> 10~2 М) каждая молекула рубрена присоединяет 1 молекулу О2,
которую при нагревании отдает обратно, регенерируя полностью
в первоначальном состоянии. Это сходство рубрена с
гемоглобином объясняет повышенный интерес к нему биохимиков.
Негенерация рубрена при нагревании сопровождается
испусканием света. Квантовый выход образования фотооксида
рубрена. зависит от природы растворителя. Присутствие двух
атомов Нв положениях 1, 1' значительно снижает обратимость
выделения О2106.
я объяснения сложной кинетики фотореакцин рубрена
с кислородом, сильно зависящей от концентрации
углеводорода, оказалось необходимым допустить следующую картину
процесса (см. схему X ниже).
При малой концентрации рубрена (<Ю~2М) н достаточной
концентрации растворенного кислорода, возбужденные светом
молекулы Ru* прн встрече с молекулой О2 образуют
неустойчивый легко распадающийся продукт присоединения Ru O2,
10 А. Н. Тереняи
146 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
который стабилизуется только путем столкновения с другой
молекулой Ru, преобразуясь сам в устойчивый пероксид Ru 02
или превращая в таковой столкнувшуюся молекулу Ru.
При концентрациях рубрена ^> 3 • 10~2 М, т. е. в диапазоне
концентраций, типичных для наступления концентрационного
тушения флуоресценции (19, 36), путь реакции изменяется.
Возбужденная светом молекула Ru* сталкивается сначала
с другой молекулой Ru, преобразуясь в модификацию Ru,
способную реагировать с кислородом, давая устойчивый
пероксид Ru O2.
Сказанное можно изобразить следующей схемой, где hvf и Av r
обозначают фотоны испускаемого света, Ru О3—неустойчивый,
a RuO2—устойчивый пероксид.
[ О
[ 2 -> R^O2
Ru--* Ru* -ь ) RuO2 (X)
Ru - "У
Ru
В отличие от антрацена и красителей, рубрен не
испытывает столь значительного концентрационного тушения
флуоресценции и не обнаруживает также концентрационного падения
квантового выхода фотореакции молекулярного окисления.
При увеличении концентрации рубрена квантовый выход
поглощения им кислорода непрерывно возрастает.
О природе модификация рубрена Ru авторы не делают
никаких предположений. Между тем напрашивается вывод,
что мы имеем здесь пример фотохимического проявления
метастабильного бирадикального состояния (12, стр, 80)
молекулы рубрена. В согласии с этим концентрированные растворы
рубрена обнаруживают послесвечение (замедленную
флуоресценцию) длительностью 10~3 сек.107.1)
1) Дан объяснении высокого квантового выхода фотореакции при малой
концентрации кислорода, Гаффрон 105 еще ранее допустил существование,
варнду с обычным возбужденным состоянием рубрена, также длительно
жинущей (10~2 —10 1 сек.) активной формы рубрена. Однако Шумахер
опровергает экспериментальные данные Гаффрона и находит, что обе
модификации рубрена Ru* и Ru должны обладать нормальной
длительностью существования порядка 10 8 секр Следует отметить, что оппоненты
не обращают внимания на существенную зависимость фотореакции от
природы растворители. Вопрос заслуживает более внимательного изучения.
23. МЕХАНИЗМ ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ КИСЛОРОДА 147
Изолируемый химическим путем фотооксид, как и пероксид
темновой реакции самоокисления, представляет собой
сравнительно стабильный промежуточный продукт. Он должен
отличаться н по структуре и по реакционноспособности от исходного
неустойчивого продукта присоединения молекулы О2 к
активированной светом илн теплом молекуле окисляемого
соединения.
Следуя предложению Энглера и Штаудиигераш,
первичный лабильный пероксид неизвестного строения мы будем
называть мольоксидом и будем обозначать АО2, в отличие
от изолируемых химически устойчивых фотооксидов или пере-
кисных соединений реакции самоокисления, которые мы
обозначаем АО о.
23. Механизм ф этореакции присоединения молекулярного
кислорода
Способность присоединять молекулярный кислород, т. е.
испытывать реакцию так называемого самоокисления,
присуща большому числу органических соединений и без
участия света. Кинетике и механизму этой реакции, идущей
путем термической активации, посвящена громадная
литература108. Поглощение света увеличивает способность веществ
присоединять кислород.
Рассмотрим последовательные этапы такой реакции
фотооксидирования (по нашей терминологии), используя
картину, полученную при изучении термической реакции
самоокисления, дополнив ее новым представлением о бирадикальном
состоянии ароматических соединений (12).1)
Исключительно сильное тушащее действие кислорода
на флуоресценцию, по сравнению со всеми другими
постоянными газами, указывает на сильную пертурбацию, испытываемую
возбужденными молекулами ароматических соединений при
их сближении с молекулой О2.
Обратимое и притом немедленное восстановление
первоначальной интенсивности флуоресценции при удалении кислорода
откачкой указывает, однако, на то, что это взаимодействие
является преходящим и что оно не сопровождается, как
правило, химической реакцией.
*) В отличие от простейших органических молекул,
ультрафиолетовые фотоны, реакция кислорода с соединениями типа
красителей, идущая под действием видимого света, не имеет цепного характера
(стр. 167) и укладывается в схемы, приводимые ниже.
10*
148 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Суммарное протгканиэ такой физической реакции может
быть представлено схемой:
А*
\+о2
У А*,О2 (I)
А*
Конверсия энергии возбуждения, обозначенной звездочкой,
в интенсивные вибрации, повидимому, и расщепляет слабую
междумолекулярную ассоциацию А*,О2 на исходные
составляющие. Согласно предыдущему параграфу, часть энергии
возбуждения может при этом остаться в молекуле О2)
превращая ее в реакционно способную модификацию Ог , причем
молекула А* также переходит в бирадикальное состояние Av.
При ближайшем столкновении с нормальными молекулами А
и О2 или под дейстзием окружающей среды такие богатые
энергией частицы возвращаются в итоге в свое основное
состояние, что и обозначено в схеме (I). Вспомним, что
активная модификация кислорода в опыте Каутского исчезает уже
при незначительном давлении кислорода и что столь же малые
его давления достаточны для устранения молекул А ,
вызывающих послесвечение. Отсутствие параллелизма между
тушением флуоресценции кислородом и фотореакцией его
присоединения для большинства полициклических углеводородов (21)
свидетельствует о том, что междумолекулярная ассоциация,
обозначенная в схеме (I) A*fO2f осуществляется слабыми силами.
Следовательно, такой вид взаимодействия между А* и О2
не приводит, как правило, к образованию того активного
продукта присоединения кислорода, с которого начинается
реакция, т. е. мольоксида, о котором была речь в предыдущем
параграфе.
О строении мольоксидов было высказано предположение,
согласно которому молекула кислорода присоединяется
по месту двойной связи окисляемого соединения, размыкая
одну из своих связей и образуя мостик О — О109. Например,
в случае бензальдегнда и дифенилэтилена мольоксид
изображался структурами (а) и соответственно (Ь):
\r П
н >с~° Ph 1 |
(a/ 4b) '
23. МЕХАНИЗМ ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ КИСЛОРОДА 149
Для фотооксидов антрацена и его производных
предполагают строение (с), в котором кислородный мостик соединяет
противоположные атомы С среднего кольца и лежит вне
плоскости молекулы (трансаннулярные перекиси)300.
Однако Рихе110 привел ряд доказательств того, что
и реакциях, идущих через свободные органические радикалы,
например РЬ3С" илн R— С = О, молекула О2 способна
присоединяться к ним одним концом, образуя пероксидный
радикал типа РЬ3С— О — О* или R— С = О. При даль-
•о—о
нейшем развитии реакции последние, присоединяя атом Н или
второй радикал, дают стабильные гидропероксиды или
пероксиды типа:
R —О —О —Н или Rj — О — О — R_. (d)
Например, гидропероксид тетралина имеет по Рихе структуру:
(е)
>
н—о—о н
Вейсс держится взгляда, что органические перекиси имеют
ионное строение А+О2~ и состоят из потерявшего электрон,
т. е. „окисленного" органического соединения А"*" и
отрицательного молекулярного нона кислорода О2> аналогично
неорганическим перекисям типа К^О^111.
Приведенные структурные формулы (а—е) не
воспроизводят свойств первичного мольоксида, который, согласно
данным кинетики реакции самоокисления, должен представлять
собой быстро реагирующий активный продукт
присоединения кислорода, обладающий оксидирующей способностью,
превосходящей таковую молекулы О2 в нормальном ее
состоянии.
В фотореакциях первичный активный мольоксид может
иметь строение, аналогичное формуле пероксидного радикала
Рихе, но с двумя непарными электронами, обозначенными
ниже точками, т. е. представлять собой бирадикал типа:
150
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
R
с—о-
I
о—о-
н н
н о—о-
л /\
нон н
.-н
н
о
(а')
(с')
(е')
Такой лабильный активный мольоксид мы обозначили выше
АО2, в отличие от формы междумолекулярной ассоциации
А,О2 и стабильного пероксида или фотооксида АО2,
доступного химическому выделению.
• • • •
Лабильность мольоксида АО2 или А-О-О проявляется
в том, что он легко перегруппируется, а его чрезвычайная
реакционная способность в том, что он реагирует по схеме:
АО,-*-А —> АО-ьОА,
(И)
где АО условно обозначают конечные продукты
оксидирования (альдегиды и кислоты).
Превращение мольоксида в стабильный пероксид
совершается по Дюфрессу лишь при встрече его с частицей
„положительного катализатора" В, которым, в частности, может
служить и молекула исходного соединения А. При этом
образуется, очевидно, временный комплекс трех частиц А, О2
и В, распадающийся на устойчивый пероксид АО2 и
неизмененную частицу В; в наших обозначениях:
—> А-СЬВ
АО,-*-В
(Ш)
Не исключена возможность, что сравнительно устойчивый
пероксид АО2 может также преобразоваться в конечные
продукты реакции оксидирования вследствие расщепления
присоединенной молекулы О2 с закреплением атома О в
структуре получившегося соединения АО:
АО.
АО-*-О А,
(IV)
В соответствии со сказанным, изобразим графически на
фиг. 30 в виде энергетических уровней промежуточные
стадии реакции фотооксидирования, т. е. фотосамоокисления.
23. МЕХАНИЗМ ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ КИСЛОРОДА
151
Взаимодействие между возбужденной светом молекулой А*
и молекулой О2, при тушении флуоресценции, согласно преды*
дущему состоит из слабой ассоциации молекул А*,О2, которая
сопровождается выделением незначительной энергии qf
обозначенной короткой стрелкой, обращенной вниз. Комплекс А*,О2
нскоре распадается на части с рассеянием в тепло избыточ*
ной электронной энергин, что обозначено волнистой стрелкой»
Однако, при благоприятных структурных свойствах
соединения А, этот комплекс преобразуется в активный моль-
оксид АО2 с выделением энергии Q. Подобный энергетический
уровень может быть достигнут, исходя из нижнего состояния
О,
а
1
АО:
в
• AD + OA
Фиг. 40. Разложение фотореакции молекулы А с кислородом на ряд
энергетических этапов.
А -I- О2 путем подведения некоторой термической энергии
активации, как это и осуществляется в темновой реакции
самоокисления. *)
Присоединение молекулы кислорода к такой термически
актипированной молекуле дает также пероксидный радикал АО2.
Мольоксид АО2 лабилен потому, что находится на энерге-
гичгском уровне, расположенном выше исходного уровня
A i i\. Поэтому, подобно комплексу А?О2, он стремится
распасться на состапные части, что показано на фиг. 30
волнистой стрелкой, обращенной вниз. Встреча с молекулой „поло-
1) Мы предполагаем, что анергия вктитции необходима, главным обра-
вом, для преобразования валентной структуры частицы А в форму б и
ради кала А\ что требует размыкания в ней спаренных электронов.
В термической рзакцчи самоокисления могут принимать участие иные
бирадик&льные еоетояния молеиулы, энергетически боле« низкие, чем
метастабнльный уровень ароматического соединении, достигаемый
оптическим путем, о котором была речь выше (12). Существование такого низко
расположенного бираднкальчого уровни на выоте около 1 ». вольта для
полицнклическнк углеводородов выведено недавно теоретически
112
152 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
жительного катализатора" В (в частности молекулой А),
способной воспринять часть избыточной электронной энергии
АО2» стабилизирует его, переводя на уровень устойчивого
фотооксида АО2. При этом выделяется энергия, так как
свободные валентности пероксидного радикала смыкаются (фиг. 30).
Специфическая роль катализатора В состоит, повидимому,
не столько в отводе избытка энергии, как в облегчении
„запрещенного" перехода пероксидного бирадикала, обладающего
неспаренными спинами электронов, в состояние фотооксида,
у которого эти спины спарены. *)
Для стабильного фотооксида уровень АО2 лежит
несколько ниже исходного уровня А + О2 и при подведении
термической энергии он более или менее обратимо выделяет
кислород, притом в нормальном состоянии90. Для легко
распадающегося фотооксида он лежит выше этого уровня,
В дальнейшие стадии химической реакции оксидирования
вступает именно пероксидный бирадикал ДОг по
приведенной выше схеме (II).
Разумеется, приведенная на фиг. 30 последовательность
этапов передает только общую картину процесса. В частности,
на пути от пероксидного радикала АО2 до стабильного
фотооксида или конечного продукта АО существует ряд
промежуточных ступеней. Протекание химического процесса в
несколько этапов, на каждом из которых выделяется или
поглощается сравнительно небольшая энергия, есть явление,
характерное для превращения сложных органических соединений.
Оно является следствием трудности деградации больших
порций электронной энергии прямо в тепловое движение.
Электронная энергия стремится преобразоваться целиком
в электронную с минимальным рассеянием в тепло, что и
осуществляется путем перегруппировки в промежуточные
валентные структуры.
Может оказаться, что соединение В само перейдет в бира-
дикальное состояние за счет части воспринимаемой энергии
и захватит молекулу кислорода от АО2, преобразуясь сначала
в ВО2, а затем в ВО2. Если уровень пероксида ВО2 или ВО2
окажется выше уровня В + О3 (фиг. 30), то произойдет обра
тимая отдача им кислорода, что показано на фигуре волни-
*) Специфическая роль среды сказывается в наблюденном Дюфресом
факте108, что антрацен дает фотооксиды в тех растворителях, в которых
оя ие флуоресцирует (например CS2). В бевэоле, в котором антрацен
флуоресцирует, образование фотоокснда, напротив, незначительно. Отсюда
напрашивается вывод, что присоединяют кяслород именно те молекулы,
которые лишаются способности флуоресцировать, но остаются достаточно
активными, очевидно, вследствие превращения в бирадикальное состояние»
23. МЕХАНИЗМ ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ КИСЛОРОДА 15$
стой стрелкой. В таком случае В будет тормозить
оксидирование соединения А, т. е. функционирует как ингибитор илн
„отрицательный катализатор", не изменяясь сам.
Если уровень пероксида ВО2 лежит ниже уровня В -*- О2,
то он преобразуется дальше до получения конечного продукта
ВО деструктивного окисления вещества В. В этом случае мы
будем иметь реакцию переноса кислорода от А к В.
Изложенное можно изобразить следующей схемой
последовательных реакций:
А62-ьВ >А-ьВб2
\пл (Перенос кисло- fu\
yfDKJ рода на В) *
вол
\а« /~v (Торможение
О ~+~ vJ2 оксидиро- (VI)
вания А)
В том случае, когда активный мольоксид АО2 образуется
при поглощении света молекулами А, присутствующими в
системе А-+-В-ьО2, то тнп реакции V получает название
фотосенсибнлизованного веществом А переноса кислорода
иа акцептор В, являющийся нормально стойким к реакции
самоокисления.а)
1) В реакции присоединения О2 к равличиым органическим акцепторам
(алифатические амины, аллнлтиомочевина), фотосенсибилнзованной
флуоресцирующими, красителями, Гаффрон92 обнаружил» что кислород на90°/д
связывается в виде перокснда акцептора, но, против ожидании, не
пероксида красителя-сенсибилизатора (эритрозина, рубрена, этилхлорофил-
лида, гематопорфнрина). Отсюда им был сделен вывод, что переносчиками
кислорода здесь не могут являться временно образующиеся перокснды
красителя и что фотосенсибилизатор активирует не молекулу кислорода,
а молекулу акцептора, которан, воспринимая энергию, переходнт в длительно
живущее состояние, способное присоединять кислород. Такой механизм нм
прннимаетсн н в тех случаях, когда наблюдается попутное фотооксидированне
самого сенсибилизатора.
В отличие от оксидирования аминов перекисью водорода, в
рассматриваемой здесь фотореакцни кислород затрагивает первично аминогруппу.
Выход реакции сильно зависит от растворителя, возрастая в 100 pas при
замене воды ацетоном или пирндяном, что, повндимому, указывает, как
и в других аналогичных случаях (стр. 122), иа участие мономерной формы
красителя.
Квантовый выход фоторез.кции непрерывно возрастает с увеличением
концентрации акцептора, приближаясь в пределе к единице на всем
протяжении спектра поглощенин сенснбнлнватора (см. фиг. 50 в 34.)
Нам представляется маловероитным допущение длительного метаста-
бильного активного состояния у молекул предельных алифатических ами-
154 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
24. Деструктивное окислительное выцветание красителей
на волокне под действием света
Для всех красителей, независимо от их строения и свойств,
при достаточно длительном или мощном освещении, в
особенности ультрафиолетовым светом, в присутствии кислорода
имеет место фотореакция необратимого деструктивного
окисления. Этот медленный процесс накладывается на
разнообразные типы фотохимических превращений красителя, если
опыт производится иа воздухе (20). Даже те красители,
выцветание которых нормально обусловлено реакцией фотовосста-
иовления, например, метиленовой голубой (32), при
длительном освещении на воздухе испытывают необратимое
деструктивное окисление. В технической литературе этому
вопросу уделяется большое внимание, поскольку с этой
реакцией связано выцветание окрашенных тканей. Мы ограничимся
здесь рассмотрением работ, стремившихся выяснить механизм
явления.
Выцветание красителей под действием солнечного света
давно приписывалось их реакции с кислородом воздуха110.
Было показано, что в атмосфере инертного газа или при
удалении кислорода из воздуха выкраски на тканях более
светостойки и что влажность благоприятствует выцветанию114,
Гебгард115 рядом классических опытов с красителями
различных классов в водных растворах и на тканях установил,
что выцветание представляет собой фотохимическое окисление
кислородом воздуха, сопряженное с гидролизом. Он показал,
что иет сходства между окислением красителя перекисью
водорода или персульфатом и реакцией оксидирования, идущей
на свету. Кроме того, он установил различные действия света
в видимой и ультрафиолетовой полосах поглощения красителя:
малахитовый зеленый при выцветании в первом случае дает,
главным образом, карбинол (5), а во втором случае в нем
затрагиваются аминогруппы.
Специальным опытом Гебгарду удалось выделить на первой
стадии выцветания перекисное соединение красителя
(фотооксид). Галлер н Циршш значительно позднее повторнли ука-
занвые Гебгардом аналитические реакции на перекисное
соединение красителя с положительным результатом почти для
всех испытанных субстантивных, основных, индигоидных
и сульфокрасителей иа хлопке. Однако, ими также было
нев. Наблюдении Гаффрона совместимы и со схемой переноса кислорода,
данной выше, если принять, что активный пероксндный радикал красителя
AOg но образует устойчивого пероксида АОг» нэ либо распадается на
.А -+- Оз, либо реагирует с акцептором.
24. деструктивное*окислительное;выцветание красителей 155
установлено, что образование пероксида красителя не стоит
в прямой связи ни с потерей прочности волокна (27), ии со
степенью видимого выцветания, но связано со строением
красителя.
Конечные продукты фотореакции могли быть отождествлены
в ряде случаев. Уже давно Геффтер 115 показал, что при
длительном освещении эозина в водном растворе получается НВг,
щавэлевая и фталевая кислоты.
Шарвин и Пакшвер117 изучали выцветание ряда красителей
(метиленовый голубой, кристаллический фиолетовый, конго
красный, эозин) на целлулозном волокне под действием
солнечного света и близкой ультрафиолетовой радиации ртутной
лампы. Они нашли, что выцветание наблюдается в
присутствии кислорода, а также окиси и закиси азота. В качестве
газообразного продукта выцветания была обнаружена СО2.
Мунье ш исследовал выцветание азокрасителей (хризоиднн,
диаминовый голубой 2В и росселнн) на целлулозе, шерсти
и стекле при освещении кварцевой ртутной лампой. Путем
капельных реакций ему удалось показать, что выцветание
представляет собой реакцию окисления, но ему, как и другим
авторам, не удалось отождествить продуктов, как это удается
делать для нитрокрасителейш. Продукты, образовавшиеся
в поверхностном слое, стабилизируют краситель к свету:
вторичная выкраска по выцветшему красителю обнаружинает
большую светостойкость.
Трифенилметановые красители в порошке при экспозиции
на солнечном свете на воздухе переходят в соответствующие
кетоны:
Например при освещении малахитоного зеленого был выделен
пара-диметиламинобензофенон (а); при освещении
кристаллического фиолетового — кетой Михлера (Ь)ш:
(CH3)2N (CHS)2N
V V\
(a) \-/ (b)
(CH8),N
156 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
В отсутствии воздуха реакция идет по схеме:
Аг3С+ ~* Аг3СН,
т. в. краситель восстанавливается с образованием трифенил-
метана за счет присутствующего аниона щавелевой кислоты.1)
Необходимо здесь повторить сказанное выше (стр. 102),
что фотохимическое поведение красителя под действием
ультрафиолетового света может быть совсем иным, чем под
действием видимого света, поглощаемого в первой полосе спектра.
Беленький120 установил повышение кислотности водной
среды при выцветании ряда выкрасок в видимом свете и,
наоборот, повышение щелочности при освещении
ультрафиолетовым светом. При длительном облучении на воздухе краситель
фактически „выгорает" с выделением СО2 и Н2О.
Бесцветные лейко-формы трифенилметановых красителей
(5) под действием поглощаемого ими ультрафиолетового
света в присутствии О2 легко фотоокисляются
(дегидрируются) до соответствующего красителя, как это подробнее
рассматривается ниже. Эта реакция, повидимому, идет через
стадию карбинола (стр. 116). Деструктивное
фотооксидирование самих трифенилметановых красителей в тех же условиях
ультрафиолетового освещения идет значительно медленнее.
Еще медленнее оно идет в видимом свете. Замечательно, что
краситель действует в качестве фотосвнсибилизатора
окисления собственной лейко-формы кислородом воздуха. А именно,
краситель, поглощая видимый свет, вызывает реакцию
окисления кислородом (дегидрирование) лейко-формы, требующую
нормально поглощения ультрафиолетовых фотонов.2)
Впервые это явление было установлено в классической
работе Гроса, который, изучая реакцию лейко-форм
красителей флуоресцеиновой и розанилиновой групп с кислородом
в водном растворе, измерял манометрически потребление
кислорода при освещении раствора солнечным светом.
Незначительная исходная концентрация красителя (Ю~ВМ) была достаточна,
чтобы вызвать быстрое появлевие окраски в результате
дегидрирования лейко-формы красителя кислородом под действием
поглощаемого красителем видимого света. Реакция испытывала
своего рода фотоавтокаталитическое ускорение190.
Красители способны также быть фотосенсибнлизаторами
реакции деструктивного оксидирования другого красителя.
*) Красители представляли собой солн щавелевой кислоты.
2) Механизм подобной фотосенсибнлизации рассматривается н 34.
24. ДЕСТРУКТИВНОЕ ОКИСЛИТЕЛЬНОЕ ВЫЦВЕТАНИЕ КРАСИТЕЛЕЙ 157
Так, прибавление эозина (2.5*10"БМ) к водному раствору
уранина (5 • 10~3М) вызывает увеличение скорости поглощения
кислорода в 2.7 раза.
Фотооксидирование хинина в водном растворе, изученное
Вейгертом12, имеет некоторые черты, общие с
фотооксидированием красителей. Прн реакции образуются сравнительно
устойчивые промежуточные продукты перекисного характера^
так как после освещения раствор хинина способен окислять
ионы иода. Эти продукты ие представляют собой перекиси
водорода и являются существенной стадией реакции, так как
до 42.5 °/о кислорода связывается в такой форме. Особенно
интересным является торможение фотореакции хинина,
а также флуоресцеиновых красителей избыточным давлением
кислорода. После некоторого оптимального значения
дальнейшее увеличение давления приводит к падению скорости
реакции, которая изменяется обратно пропорциональво давлению
кислорода (см. стр. 124). Реакция тормозится повышением
кислотности раствора, очевидно, вследствие изменения
структуры молекулы хинина, содержащей атом N в гатероцикле
и обнаруживающей поэтому основные свойства.
Светостойкость.1) С точки зрения технологии
светостойкость выкрасок красителями различных классов может быть
качественно охарактеризована следующей таблицей.
Таблица 10
Красители Светостойкость
Основные (трифенилметановые) Исключительно низкая
Субстантивные Низкая
Сернистые Посредственная
Ледяные Хорошая
Протравные (ализариновые) . Высокая
Кубовые (иядигоиды, индантреновые) Очень высокая
Стойкость текстильных выкрасок к свету условно
оценивается пятибальной шкалой путем визуального срайне-
ния с некоторыми выбранвыми стандартными выкрасками
(эталонами), выцветающими совместно с данной. Наинизший
балл присвоен наиболее легко выцветающим выкраскамш.
Описание методики проведения испытаний на
светостойкость и обширной технической литературы по этому вопросу
*) Мы предпочитаем пользоваться термином „светостойкость" вместо
общеупотребляемого „светопрочность", твк как последний термин уместнее
употреблять в свяэи с разрушающим действием красителя на м о х а н и*
ческую прочность ткани.
158 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
выходит за рамки настоящей книги. Эта проблема безусловна
заслуживает радикального пересмотра, имеющего целью
введение более объективных приемов сравнения и оценки123.
Светостойкость выкрасок зависит от многих факторов,
из которых главными являются: (а) молекулярное строение
красителя; (б) способ его фиксации на носителе, т. е. метод
крашения, и природа окрашиваемого объекта; (в) состав
окружающей среды.
Одновременное действие стольких переменных создает
весьма сложное протекание реакция в реальных условиях,
а минимальные количества получающихся продуктов
выцветания не дают возможности провести детальный анализ процесса.
По пункту (а) следует сказать, что классическая структурная
формула красителя с равноценными чертами валентности
и локализованными двойными связями неприменима, как мы
видели в главе 2. Она не отражает имеющего место гораздо
более товкого явления обобществления электронов, некоторое
отдаленное представление, о котором дает понятие „резонанса"
валевтных структур. Поэтому попытки объяснить
наблюдаемые различия в светостойкости красителей структурными
деталями обычной формулы их строения (см., например, Геб-
гард115, 1909) заранее обречены на неудачу.
Из структурных свойств, благоприятствующих
окислению кислородом, как при реакции отнятия их водорода, так
и при его первичном присоединении с образованием пероксида
(ср. стр. 143), следует отметить присутствие слабо связанных
и реакционноспособных атомов водорода в структуре
соединения. Еще Гебгард указал на понижение светостойкости при
наличии групп—NH2, — ■ ОН и ее поиышение введением
карбоксильной или сульфогруппы, которые в кислотных красителях
ионизованы (5), т. е# не содержат атома водорода.
Образование в красителе внутримолекулярной водородной
связи, возможной в антрахиноновых, ализариновых н индан-
треновых красителях, согласно схемам:
сильно снижает способность к окислению и деструктивному
выцветанию красителя, так как клещеобразно удерживает
атомы Н, склонные к отщеплению при реакции окисления24*124.
24. ДЕСТРУКТИВНОЕ ОКИСЛИТЕЛЬНОЕ ВЫЦВЕТАНИЕ КРАСИТЕЛЕЙ 159
Аналогичная замкнутая валентная структура молекулы
создается при комплексообразовании красителей с солями
металлов, когда атом Н водородной связи в приведенных
схемах замещается атомом или катионом металла, например
А1 в случае ализаринового „лака", Си в случае фталоцианина,
Mgf в случае порфирнна (ср. стр. 29). Исключение реакционно-
способного атома водорода в таких комплексах сильно
повышает их стойкость к фотоокислеиию с сохранением высокого
коэффициента поглощения, свойственного красителю. Введение
ионов металлов есть распространенный прием для уменьшения
выцветания вы красок.
Обстоятельства, упомянутые в пунктах (б) и (в), имеют
не меньшее значение. Краситель в виде мономера обладает,
как правило, большей способностью к реакции, чем в аггреги-
рованной форме, например в виде коллоидных частиц (см.
концентрационное самотушение флуоресценции и самоторможение
фотохимических реакций в 36). Сам носитель (волокно)
может оказаться стабилизующим агентом, если обладает
способностью быстро отводить избыточную энергию,
доставляемую молекуле красителя поглощением света (12). Для
такой дезактивации необходимо более тесное и
специфическое взаимодействие адсорбированного красителя с
подкладкой, чем это осуществляется обычными силами
междумолекулярного сцепления (ван-дер-Ваальса). Повндимому,
благоприятные для дезактивации красителя условия осуществляются
при создании водородных связей между красителем и
структурными звеньями волокна. Известный факт, что краситель
не светостойкий на хлопке, делается светостойким на шерсти,
можно объяснить созданием связующих водородных
„мостиков" между красителем и амфотерным белковым волокном
шерсти, способным адсорбировать как основные, так и
кислотные красители.
Об ингибиторах — веществах, тормозящих выцветание,—
будет речь ниже, при обсуждении его механизма (26).
Концентрация водородных ионов водной среды, в которой
идет деструктивное выцветание, имеет исключительно большое
влияние на его скорость. В кислом растворе светостойкость
красителей повышается; в щелочном — катастрофически падает.
Это значит, что излишние ионы водорода стабилизируют, а ионы
гидроксила способствуют деструктивному фотоокислению
красителя.
Концентрация красителя имеет также существенное
значение для светостойкости выкрасок, причем в литературе есть
противоречивые указания, с одной стороны, на то, что более
слабые выкраски более светостойки, чем насыщенные, а с дру-
160 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
гой — что в определенном диапазоне концентраций красители
обладают наименьшей светостойкостью. Следует ожидать,
что увеличение концентрации должно повести к торможению
фотореакции вследствие сильного дезактивирующего
действия тождественных молекул (см. 36).
25. Кинетика деструктивного выцветания красителей
при фотореакции с кислородом
Кинетика деструктивного выцветания нестойких к свету
цианиновых красителей (пинавердол, лепидинцианин, цианин,
пинахром, хинальдинцианин и др.) в коллодионной пленке
была изучена в классических работах Лазарева (1907—192l)liJ,
применившего спектрофотометр для измерения выцветания.
Скорость выцветания, т. е. уменьшения поглощения для
данной длины волны, при освещении окрашенной пленка
монохроматическим светом той же длины волны служила, согласно
закону Ламберта-Бэра (2)t мерой скорости уменьшения
концентрации с красителя в результате его разрушения.1) Лазарев
нашел подтверждение фотохимического закона Вант Гоффа,
требующего пропорциональности между скоростью уменьше-
ния концентрации поглощающих свет молекул *- и
количеством поглощенной световой энергии данной волны А
^)х= у. А, = аЛ«(1 - е-1'). (1)
Зная коэффициент поглощения Кх1 для различных длин
волн lf можно определить спектральную кривую поглощенной
энергии Av С другой стороны, можно построить спектральную
кривую скорости выцветания—\~Tf) B различных длинах волн.
В соответствии с требованием закона Вант Гоффа, по Лазареву
обе спектральные кривые оказались в определенном масштабе
практически совпадающими в пределах одной простой полосы
поглощения. Это значит, что одинаковые количества
поглощенной энергии оказывают одинаковое фотохимическое
действие, независимо от длины волны. Такой результат,
как мы упоминали ранее, противоречит основному
фотохимическому закону квантовой эквивалентности (стр. 99).
Действительно, число фотонов, содержащихся в одинаковых значениях
энергии, падает с уменьшением длины волны, а потому»
2) Опыту благоприятствовало то обстоятельство, что дианиновые
красители, выцветая, дают бесцветные продукты.
25 КИНЕТИКА ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛЕЙ 161
принимая постоянство квантового выхода у в пределах полосы,
скорость реакции, измеряемая числом исчезающих молекул за
сек., должна была бы падать в сторону коротких длин волн
быстрее, чем поглощаемая энергия А\. Между тем, по Лазареву
постоянная а в законе Вант Гоффа (J), как сказано выше,
оказалась независящей от длины волны и даже, наоборот, имеет
тенденцию возрастать в сторону коротких длин воли, что
совместимо только с возрастанием каактового выхода на
протяжении полосы в коротковолновую сторону.
Вейгерт121 нашел, однако, что светочувствительность
(скорость выцветания) цианина падает в сторону коротких длин
волн быстрее, чем поглощаемая энергия Ах что совместимо
с постоянством квантового выхода. Квантовый выход
флуоресценции 9 Аля флуоресцирующих красителей (иных
классов, чем циаииновые) по Вавилову (4) остается также постоянным
на всем протяжении полосы (даже при налички побочных
максимумов).х)
Измерения Лазаревым (1921) зависимости скорости
выцветания тех же окрашенных пленок от давления кислорода
показали, что в интервале давлений меньших 1 атм., скорость
для данное длины волны пропорциональна давлению кислорода
Ро2) при повышении давления сверх этого значения скорость
растет медленнее и, наконец, в интервале давлений от 40
до 120 атм. скорость выцветания практически не зависит
от давления.
Лазарев выразил наблюдаемую зависимость эмпирической
формулой:
-! = ^А(*,-Ы-е^), (2)
в которой Лх обозначает поглощенную энергию для данной
длины волны, ки £2, къ — постоянные. Предельное постоянное
значение скорости реакции при больших значениях Ро2 равно
kj Ax если пренебречь небольшим постоянным членом кг
по сравнению с единицей.
В более поздней работе Гамбурцев]2Б показал, что скорость
выцветания коллодионноЙ плейки, окрашенной цианином, начи-
1) Выбор Лазаревым коллодиоиной плеики, т. е. нитроцеллюлозы,
нельзя признать удачным» так как последняя иа свету оказывает
самостоятельное выцветающее действие на краситель и без кислорода [см. ниже
постоянный член к% в кинетическом уравнении (2)]. Возможно, что
описываемое здесь монотонное возрастание квантового выхода реакции
выцветания в сторону коротких длин волн вызвано фотоактивацией
нитроцеллулозы, имеющей ультрафиолетовую полосу поглощения, простирающуюся
с постепенным спаданием в видимую область.
11 А. Н, Теренмн
162 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
иает замедляться при давлениях даже меньших 1 атм. (фиг. 31),
удовлетворительно укладываясь в теоретическую зависимость:
и ~-^^«... (з)
выведенную из предположения, что реакция происходит при
встрече молекулы О2 с возбужденной молекулой красителя А*
за время т возбужденного состояния последней. Вывод
совершенно аналогичен приведенному выше выводу формул для
тушения флуоресценции и реакции фотоприсоединения
кислорода к полициклическим углеводородам (21), а также выводу
формулы (8) для реакции фотодшлеризаций антрацена (19),
что объясняет их внешнее сходство. Зависимости такого рода
дают возможность вычислить время т, если предположить,
что первая же встреча возбужденной молекулы А* с О2
приводит к ее устранению (см. также главу 9). Гамбурцев
вычислил из данных опыта с помощью формулы (3), что т
имеет обычное значение порядка 10~8—Ю~° сек.,
наблюдаемое н при флуоресценции красителей.1)
Для твердых пленок самих цианиновых красителей,
осажденных на стекло без коллодия, Лазарев и Гамбурцев получили
1) При выводе формулы Гамбурцев делает следующие два
предположения: а) давление кислорода внутри пленки пропорционально его внешнему
давлению Po2f т. е. соблюдается закон Генри для растворенного газа;
б) число встреч молекул О2 с молекулой красителя за секунду
определяется из выражения кинетической теории газов, причем площадь,
занимаемая молекулой красителя, а также поперечное сечение молекулы
кислорода оцениваются весьма неточно. (См. аналогичные расчеты в работеt8
по тушению флуоресценции адсорбироаавных красителей кислородом.)
Между тем, кроме газообразного кислорода, в пленке имеется в
значительно большем количестве кислород, адсорбированный на внешней
поверхности мицелл нитроклетчатки, на которой закреплены ионы
красителя. С этим кислородом, мигрирующим по поверхности со скоростью,
отнюдь не даваемой выражением кинетической теории газов, также
реагирует возбужденный краситель. Содержание адсо рбир ованного
кислорода в пленке с увеличением внешнего давления этого газа стремится
к насыщению. По замечанию Гамбурцева не исключена возможность, что
форма кривой на фиг. 31 обусловлена н этим эффектом и что вследствие
этого вычисленное по формуле (3) значение т имеет преувеличенную
величину. Действительно» время жизни возбужденной молекулы, как и выход
флуоресценции, должны испытать резкое сокращение из-за
дезактивирующего действии носителя. С другой стороны, последний может
способствовать переходу возбужденной молекулы в длительное реакционноспособное
Эираднкальное состояние (12). Кроме того, использованная Гамбурцевым
картина явления совсем не учитывает тормозящего действия на
ротореакцию деструктивного окисления избыточного давления кислорода,
обнаруженного Вейгертом (24). Эти критические замечания указывают на
лалую надежность значения т, вычисленного по формуле (3). Вопрос
заслуживает тщательной экспериментальной проверки иа других объектах.
25. КИНЕТИКА ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛЕЙ
163
строгую пропорциональность скорости выцветания давлению
кислорода на том интервале давлений, где окрашенные кол-
лодионные пленки обнаруживают отступление от этой
зависимости (фиг. 31).1) Этот результат может быть приписан
сокращению времени т вследствие возросшей дезактивации
возбужденного светом красителя тождественными
окружающими молекулами (36), В пользу такой интерпретации говорит
факт расширения и изменения высоты максимума поглощения
в пленках твердого красителя по сравнению с его
раствором (Н).2)
В высоком вакууме _ Sc
осажденные пленки циа- 6/
ниновых красителей по
Лазареву и Гамбурцеву
при освещении видимым
светом не выцветают.
Тот же результат был
получен для флуоресцеи-
новых красителей
Натансоном79. Введение в
вакуум паров воды
(упругостью 14 мм ртутного
столба) также не
приводит к фотореакции флу-
оресцеиновых красителей.
Те же слои выцветают
полностью в тщательно
высушенном воздухе за
10 часов и за 1.5 часа в атмосферном, т. е. влажном, воздухе.
Последний результат указывает, что деструктивное фото-
окислеиие красителя кислородом может происходить и без
участия воды, но присутствие воды его сильно ускоряет.
Однако осажденные пленки трифенилметановых
красителей выцветают под действием видимого света и в высоком
О гОО 400 600 Ро? мм
Фиг. 31. Зависимость скорости
выцветания цианина от давления кислорода125.
О — в коллоднонной плеике
# — в твердом слое.
*) В отличие от коллодионных пленок прямая здесь проходит через
начало координат, что снова указывает на фотохимическую активность
самого коллодия.
2) Табл. 1 (11) указывает на заметное уменьшение площади
спектральной кривой поглощения (пропорциональной fe) дли красителей
фуксина н синего инднго в твердой пленке, по сравнению с их состои-
нием в растворе или парах. См. также результат Диксона57 длн
родамина В. Площадь спектральной кривой поглощения согласно
формуле (7) в 11 должна находитьси в обратной зависимости от
„естественного" времени существования т0 возбужденного состояния. Таким образом т^
в твердой плеике примерно ш 2 рага больше и молекула более подвержена
дезактивации.
И*
164 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
вакууме, что, повидимому, объясняется взаимным
восстановлением и окислением молекул красителя (гл. 8)-
Температурный коэффициент скорости фотохимического
выцветания незначителен, составляя 1.04—1.08 на 10° для
цианиновых красителей в коллодиониой пленке126 (в интервале
от ч- 60° до 120° С) н 1.03—1.05 для кристаллического
фиолетового в целлофане123 (в интервале от —40° до-4-60° С).
Повышение температуры увеличизает коэффициент
поглощения красителей в коллодионной пленке (Лазарев62, 1922),
особенно длинноволнового края полосы. Глубокое охлаждение
вызывает сужение максимумов, поглощения, а также более
отчетливое разделение побочных максимумов, как установлено
измерениями Левшина7 над замороженными ра-творами (ср. 11).
Температурный коэффициент скорости темнового
выцветания коллодионных пленок, окрашенных цианиновыми
красителями при нагревании их в интервале 60°—120°> имеет, наоборот,
большую величину, а именно 2.0 на 10°, типичную ^ля
обычной химической реакции127. Реакция, в основном, происходит
за счет нитроцеллулозы, так как осажденные пленки красителя
на стекле термически стойки в этом интервале температур
на воздухе.
В случае пленок, окрашенных цианиновыми красителями,
для каждой длины волны в пределах полосы поглощения
константа закона поглощения К\у пропорциональная
концентрации красителя (2), уменьшается по мере выцветания
пропорционально времени t.
Проверка Лазаревым закона Бунзена-Роско (15),
требующего постоянства количества выцветшего красителя при
постоянстве произведения Itf т« е. интенсивности падающего
света на время, показало его применимость в данном случав.
Однако такой монотонный ход выцветания имеет место
только для слабо окрашенных пленок. В толстых пленках
или большой концентрации красителя в ходе начального
выцветания при наблюдении в белом свете имеют место
периодические потемнения и просвэтления слоя со временем,
которые могут иметь различную причину.
Одна из них, чисто оптическая, состоит в том, что
вследствие падения интенсивности поглощаемого света по
показательному закону по мере проникновения в пленку (2) быстрее
всего выцветает самый поверхностный слой, обращенный
к падающему свету. Если в начальной стадии выцветания
образуется продукт, обладающий хотя бы слабым
поглощением, но поглощением» перекрывающим спектральную область
пропускания глубинными слоями пленки, где краситель еще
не затронут, то этот продукт будет действовать как свето-
25. КИНЕТИКА ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛЕЙ 165
фильтр дополнительного цвета, поглощающий свет,
пропускаемый незатронутым красителем. В результате, при
наблюдении пленки на прозрачность в белом свете, участок пленки,
подвергнутый освещению, будет делаться темнее, а не светлее,
пока не произойдет дальнейшее разрушение этого первичного
продукта и процесс выцветания не распространится на
большую глубину.
Действительно, спектрофотометрические промеры
спектральной кривой поглощения в процессе выцветания выкрасок
на бумаге установили, наряду с уменьшением полосы
поглощения красителя, появление новой слабой полосы в области
прозрачности данного красителя120'128. В этом может состоять
объяснение некоторых случаев потемнения окрашенных тканей
под действием света.1)
Другая причина наблюдаемых аномалий временного хода
выцветания может иметь чисто химическое происхождение.
А именно: внутренние слои пленки обедняются кислородом,
потребляемым на фотореакцию, если диффузия его
совершается медленно. Аналогичное действие будет производить
накопляющийся продукт, если он способен тормозить
фотореакцию. Быстро начавшаяся реакция будет в таких
случаях постепенно замедляться.
Этой причине, повидимому, следует приписать замедление
скорости фотореакции по мере уменьшения концентрации
красителя в ходе реакции, наблюденное Фонда в тонкой
цленке ацетилцеллулозы, окрашенной родамином В130. Автор
измерял исчезновение красителя по падению интенсивности его
флуоресценции и установил показательный закон падения
концентрации с красителя во времени с = сое~а', откуда скорость
реакции — ^г^=ас. Кроме того, он обнаружил, что скорость
фотореакции пропорциональна не интенсивности падающего
света, а только логарифму интенсивности, в нарушение
основной фотохимической закономерности (15). Такое отступление
от пропорциональности интенсивности могло быть вызвано
медленностью диффузии кислорода ■ пленку, снижающее
скорость реакции при больших значениях интенсивности.
Естественно, что кинетика выцветания таких сложных
по своей текстуре окрашенных объектои, как выкрасок
х) Потемнение выкрасок под действием интенсивного теплового
излучения в область 670—30J0 т\к 128 может иметь такое же происхождение,
причем реакция оксидирования здесь вызывается, повиднмому,
нагреванием.
166 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
на тканях, не укладывается в какую-либо простую
зависимость.1)
Периодическое увеличение и уменьшение скорости
выцветании тонких, окрашенных цианиновыми красителями коллодион-
ныч пленок с увеличением концентрации красителя вызвано
явлением интерференции света, так как пропускание света
испытывает такие же периодические изменения при
увеличении концентрации13>.
После выцветания окрашенных пленок под действием
поляризованного света Вейгерт133 открыл, что пленка
делается дихроичной, т. е. обнаруживает различную
величину поглощения, в зависимости от направления
электрических колебаний в падающем поляризованном свете. Это
явление объясняется тем упомянутым иа стр, 52 фактом, что
молекулы красителей поглощают особо сильно свет, если
его колебания совершаются вдоль наибольшой длины молекулы.
При действии поляризованного света исчезают, таким образом^
в первую очередь те из беспорядочно ориентированных
молекул пленки, которые удовлетворяют этому условию. Поэтому
после выцветания слой будет более прозрачен для колебаний
световой волны, совершающихся в этом направлении, и менее
прозрачен для колебаний перпендикулярных ему.
26. Механизм деструктивного выцветания красителей
Квантовый выход у фотореакции разрушения красителей
кислородом был подсчитан, по данным Лазарева, Винтером
и Боденштейном134, которые нашли, что примерно из 500—1000
поглотивших фотон молекул только одна вступает в реакцию,
а именно: у = 2-10"~3 для цианина и у = 1-10~3 для пинавер-
дола в коллодионной пленке.
Для фотореакции родамина В в ацетилцеллулозной пленке
с кислородом, скорость крторой измерялась по падению
флуоресценции красителя, Фонда130 нашел исключительно
низкое значение у = 3*10~7. Этот результат объясняется, пови-
димому, медленностью диффузии кислорода в пленку. Поэтому
более надежные данные следует ожидать от опытов в
растворах, в которых обеспечивалось насыщение кислородом. Так,
длч фотореакции хинина, уранина и эритрозина в водных
растворах с кислородом, из данных Вейгерта121, измерявшего
1) Предлагавшиеся по этому поводу формулы имеют лишь
эмпирическое значение, например: етепень выцветания выкрасок пропорциональна
п \Ji где t — время освещения, п —число, характеризующее светостойкость
красителя по отношению к стандартам, а степень выцветании условно
оценивается по Циршу 1з1,
26. МЕХАНИЗМ ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛКЙ Ih/
скорость поглощения кислорода, Боденштейн ш получил
значение квантового выхода у = 5-10~3.1)
Для квантового выхода фотовыцветания эозина (10~5 М)
вводном растворе в присутствии щелочи (10~3N) Натансон711
из спектрофотометрических измерений получил у = 1.5' 10~4.2)
Другими словами, из 10000 молекул красителя, поглотивших
фотои, реагирует только одна. Приведенные данные указывают
на существенное отличие деструктивной фотореакции
красителей с кислородом от цепных термических н
фотохимических реакций присоединения кислорода, в которых имеется
обратное соотношение, а именно: на одну поглотившую фотон
молекулу реагирует 100000 молекул в результате регенерации
активной реакционноспособной формы — пероксидного радикала
(стр. 143). Торможение деструктивного фотовыцветания, т. е.
повышение светостойкости красителей посторонними
примесями, не может быть поэтому приписано обрыву цепи.
Действительно, стабилизирующие вещества прибавляются здесь в
значительных количествах, иногда превышающих концентрацию
красителя.
Переходим к рассмотрению механизма деструктивного
фотовыцветания, вызванного кислородом. Ряд авторов
приходит к выводу, что этот процесс заключается в
фотохимическом присоединении кислорода, сопровождающемся
гидролизом. Присутствие воды, как было сказано выше, существенно
для повышения скорости реакции. Несомненно, что процесс
идет через ряд стадий, из которых только некоторые
обнаруживаются по неравномерности временного хода изменения
поглощения света, а также изменения электропроводности
и концентрации ионов водорода120'123. О природе этих этапов
высказывались разнообразные предположения, которые
сгруппированы нами под тремя рубриками.
а) Первая теория процесса выцветания состоит в том, что
принимается на начальном этапе образование перекиси
водорода в результате предполагаемого сенсибилизующего
действия поглотившего свет красителя на кислород и воду 82t 1J5.
Такую реакцию можно представить следующим стехиометриче-
ским уравнением:
* -+- Н2О -*- -| О2 -> А ч- Н2О2. (1а)
1) Для фотореакции хинина с кислородом в присутствии хромовой
кислоты квантовый выход значительно выше (f — 0.7).
2) В присутствии щелочи кислород особенно сильно тормозит образова
ние окрашенного фотопродукта, затрудвяющего намерении процесса
деструктивного фотовыцветакня, т. е. реакции с кислородом (см. 20).
168
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Перекись водорода, согласно этой теории, и окисляет
краситель А, а также действует разрушающе на волокно (27).
При этой реакции предполагается промежуточное образование
лабильного пергидроксида илн гидроперекиси Н-А-ООН:
Н.О.
\оон
(На)
распадающегося далее по различным путям:
= О-4-Н,О
н
ООН
ОН
он
он
н
(1)
(2)
(3)
(Ilia)
Все эти реакции обычно сопровождаются расщеплением
скелета красителя А на более простые структурные элементы.
В реакциях (1—4) образуются вещества кетоиного, карбиноль-
ного, альдегидного и кислотного (фенольного) характера,
появление которых при выцветании обнаружено на опыте.
Выше они обозначались просто как АО (23). При реакциях
(3) и (4) появляются активные частицы О и ОН, которые
могут вступить в реакиию с другой молекулой красителя или
с носителем (волокном).
В старых работах135 на первом этапе предполагалось
образование при реакции кислорода с „ионизованной" водой
не перекиси водорода, а гипотетической перекиси воды
НО-О-О-Н, распадающейся в водной среде на ионы ОН"|"(?)
и НО2~» которые и должны были присоединяться к красителю
с образованием лабильного пероксид-гидрата НО-А-ООН,
обладающего повышенной реакционной способностью. Другими
словами, предполагались следующие этапы процесса
выцветания:
А*н~Н9О-*-О9 -^ А-ь НО-О-О-Н
НО-О-О-Н -+- А
Л)Н
А\оон
(ia')
(ИаО
26. МЕХАНИЗМ ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛЕЙ
169
ОН
= О + Н2О2
/ОН
< +0
\он
/ОН
0
\о
он
(2')
(3')
(4')
(Ша1)
В этой группе теорий принимается также, что две
гидроперекиси типа Н-А-ООН или два гидрата перекиси типа
НО-А-ООН отщепляют Н2О (ангидрофикация), образуя более
стойкие перекисные формы:
2А
\оон~*
/ОН
А
А—О—О—А-ь2Н2О
/О
-»А/ |-ьН20
(IVa')
В изложенной трактовке процесса выцветания
сомнительным является именно первый этап (1а) или (1а'), построенный
по аналогии с теорией Траубе самоокисления органических
соединений кислородом137. Предположение, что возбужденная
видимым светом молекула красителя сенсибилизует
присоединение кислорода к воде с образованием Н2О2 или даже
гипотетической перекиси воды НО-О-О-Н, не имеет под собой
оснований.
б) Более правдоподобным объяснением начального этапа
выцветания было бы распространение на фотореакцию
основного представления теории Виланда о механизме самоокисления
органических веществ137. По этой теории реакции с кислородом
предшествует присоединение к органической молекуле
составные частей воды, т.е. гидратация. Окисление кислородом,
по Виланду, состоит в отнятии молекулой О2 двух атомов
водорода от этого гидрата с получением в качестве
конечного, а ие началоного продукта — перекиси водорода.
Распространим эту точху зрения на процесс фотовыцветания;
тогда получается следующая последовательность этапов:
/ОН
Н,0 -> А/ (16)
А*
ОН
н
\н
(Пб)
170 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Для начала реакции выцветания необходимо, таким
образом, по теориям (а) и (б), чтобы было обеспечено присутствие
молекулы воды около возбуждаемой светом молекулы
красителя с тем, чтобы за краткое время возбужденного состояния т
последняя смогла вступить в реакцию фото гидратации (1б).
Гигроскопические свойства волокна, как правило,
обеспечивают это условие. Однако такой механизм не дает объяснения
фотовыцветанию, наблюдаемому в тщательно высушенном
воздухе (стр. 163).
в) Вместо малоправдоподобных начальных этапов,
допускаемых описанными выше теориями, уместнее предположить,
-что сама возбужденная молекула красителя скорее способна
присоединить химически активный кислород, чем инертную
молекулу воды или сенсибилизовать непонятным образом
реакцию присоединения О2 к Н2О.
Образование перекисных соединений красителя при
выцветании действительно было установлено на опыте136 (24),
но оказалось, что оно не стоит в связи ии с видимым
выцветанием красителя, ни с потерей прочности волокна116 (27).
Однако, как указано было выше, выделяемые химически
стабильные фотооксиды отличаются от тех промежуточных
лабильных мольоксидов, через которые развивается реакция
оксидирования (23). Целесообразнее поэтому построить картину
процесса деструктивного выцветания красителей светом
по аналогии с механизмом термических реакций
самоокисления, предложенным Бахом и Энглером137.
Началом процесса мы считаем лабильное присоединение
О2 не к возбужденной молекуле красителя, а к молекуле^
находящейся в процессе внутренней дезактивации (10, 12)
по схеме:
(5)
Основанием для такого допущения является тот факт,
что флуоресцирующие красители ксантеновой группы (уранин,
эозин, родамин) испытывают описываемую здесь
деструктивную фотореакцию с кислородом, но никакого тушения
флуоресценции кислородом не обнаруживают134. Поскольку квантовый
выход флуоресценции этих красителей заметно меньше единицы
(табл. 2 в 11), иными словами, большая часть поглотивших
26. МЕХАНИЗМ ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛЕЙ 171
фотоны молекул не испускает, то такой факт означает, что
в фотореакции принимают участие именно те молекулы, которые
испытывают внутреннюю дезактивацию согласно
приведенной схеме (5). Весьма важно, что при этом краситель способен
перейти в длительное бирадикальное состояние А4', как
отмечено в этой схеме.1)
Независимость фотореакции деструктивного выцветания
от флуоресценции, а следовательно возбужденного состояния
красителя, проявляется также в неодинаковом действии на них
ионов иода. Концентрация ионов иода (0.3 N KJ), достаточная
д\я полного прекращения выцветания эозина в водном
растворе, тушит его флуоресценцию только в 2.3 раза 135.2*
Эти факты л дают нам основание считать активной формой
красителя не возбужденную А*, а бирадикальную форму А",
в которую она переходит при вибрационной дезактивации (12).
К аналогичному выводу мы приходим при рассмотрении
реакции кислорода с посторонними соединениями, в которой
краситель участвует в роли фотосенсибилизатора (33, 34).
Таким образом, первым этапом фотореакции
деструктивного выцветания мы считаем встречу молекулы О2 с
красителем в бирадикальном состоянии, приводящую к их временной
лабильной ассоциации:3)
\
Химическая реакция делается возможной, если лабильная
ассоциация двух молекул с избытком электронной энергии,
обозначенная Av, O2> перестраивается в активный моль-
оксид, т. е. в рассмотренный выше пероксигдный бирадикал
АО2 (23). Последний реагирует с ионами вод^г, приводя
к образованию гидрата перекиси НО-А-О-О-Н
предыдущих теорий или, всего вероятнее, к гидролизу моль-
оксида с расщеплением структуры красителя на более
простые части:
*) См. процесс конверсии (VI) в 12.
2) О механизме фотореакции эозина с нонами иода см. 33.
3) Согласно разобранному в 21 опыту Каутского, молекула кислорода
при втом получается в активной форме, способной дегидрировать лейко-
соедннении. По соображениям, изложенным на стр. 148, сомнительно, чтобы
эта модификация кислорода имела бы значение при обычном давлеиии
кислорода.
172 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
/ОН
А-,0, - А&+Н---НОН--» А^оон (Пв)
АО2 или 'А-О-О-Н или "А
ООН
Гипотетический гидрат перекиси красителя НО—А—ООН
или продукты расщепления красителя могут эволюционировать
далее согласно вариантам (1'—4') в этапе (Шд') первой теории.
При этом образуются активные продукты О, ОН, Н2О2,
способные разрушать другие молекулы красителя. Активный
пероксндный бираднкал АО2 до реакции гидролиза может,
однако, преобразоваться в стабильный фотооксид красителя
АО2, доступный химическому обнаружению, аналогично
поведению полициклических ароматических углеводородов (23).
Тем самым развитие реакции будет задерживаться уже в самом
начале (см. схему III в). Этот устойчивый фотооксид имеет, пред-
/О
положительно, строение АС | или 'А—ООН (23), где штрих
ХО
обозначает, что атом Н заимствован из структуры красителя.
Перекисная форма 'А—ООН без воды устойчива, но под
действием света способна распадаться дальше.1) Если в ней
явно выписать второй атом водорода Н, как это сделано
в конце схемы Шв» то такой пергидроксид может испытывать
все реакции (Ша), предполагаемые в первой теории
выцветания. Однако более вероятно, что он испытывает в
присутствии воды фотогидролиз с выделением активных
продуктов (О, ОН, Н2О2) согласно реакциям, изображенным
под (Ша'), и при этом краситель А расщепится на составные
части. •
Для возможности начала реакции необходимо, таким
образом, чтобы было обеспечено присутствие молекулы О2 вблизи
возбужденной молекулы за краткое время ее возбужденного
состояния. Это достигается, как показали опыты Лазарева
и Гамбурцева, увеличением даэления кислорода (25). Однако
и в этих условиях предельный квантовый выход фотореакции
остается ничтожным. Последнее обстоятельство делается
понятным, если учесть следующие неблагоприятные факторы:
1) лабильность ассоциации возбужденной молекулы красителя
с молекулой кислорода в комплексе Avf O2, обратимо
распадающемся на первом же этапе (1в); 2) редкое преобразова-
!) См. Гебгард 135.
26. МЕХАНИЗМ ДЕСТРУКТИВНОГО ВЫЦВЕТАНИЯ КРАСИТЕЛЕЙ 173
ние этой ассоциации в реакционноспособный бирадикал АО2
и 3) возможность боковой реакции преобразования бирадикала
АО2 в стабильный фотооксид, как изображено в схеме (Шв).
Гидролиз иа втором этапе (Нв) есгь ионная реакция, воз-
можвая только в присутствии жидкой воды, что объясняет
понижение светостойкости выкрасок во влажной атмосфере,
когда в порах тканей обеспечена капиллярная конденсация
воды, а также сильное ускоряющее действие повышения
концентрации ионов ОН~ иа выцветание.
Перекись водорода по этой трактовке если и образуется,
то только на конечном этапе (реакция 4 в Ша и реакция
1' в Ша'). Для протекания процесса выцветания она не имеет
существенного значения.
Опыты Гебгарда135 относятся именно к стабильному
фотооксиду красителя АО2. Он показал, что такие фотооксиды
хорошо обрадуются под действием длин волн в пределах
видимой полосы красителя. Наблюдение Гебгарда, что
фотооксиды имеют более темную окраску, чем краситель, почти
черную, объясняется тем оптическим эффектом, что они
поглощают свет на спектральном участке прозрачности
красителя, оставшегося незатронутым в глубине слоя (см. стр. 165).
В некоторых случаях освещение вызывает регенерацию
красителя. Однако большей частью фиолетовый и улыра-
фиолетовый свет разрушают фотооксид АО2 с переносом
активного кислорода на незатронутый краситель, вызывая
его выцветание. Такая фотореакция идет только в присутствии
воды; в сухом воздухе фотооксид фотохимически стабилен.
Так, например, длительно освещенная в сухом воздухе выкраска
на ткани в опыте Гебгарда не и меняет своего цвета; однако,
если освещение продолжать затем во влажном воздухе, то
участок выкраски, подвергнутый предварительной засветке
в сухом воздухе, выцветает хаметно быстрее, чем окружающие
участки, не подвергнутые этой обработке.
Собственная фоточувствительность подобных
промежуточных продуктов выцветания^ естественно, должна вызвать
неравном рный временный ход реакции выцветания,
отмеченный выше (25). Такая многостадийность процесса и ничтожные
количества образующихся продуктов делают детальный анализ
его механизма в реальных условиях на тканях чрезвычайно
затруднительным. Поэтому исследователи предпочитают брать
более простые объекты, как, например, жидкие растворы
красителей или окрашенные пленки, хотя результаты таких
исследований и не могут быть непосредственно применены к
окрашенным тканям.
174 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Присутствие веществ, способных отнимать кислород от
пероксидного бирадикала АО2 и выделять его обратно, будет
тормозить реакцию выцветания, играя роль ингибитора
или отрицательного катализатора (23)ш. Так, прибавление
гидрохинона в небольшой концентрации полностью
останавливает выцветание уранина в водном растворе. Подобные
гидрохинону вещества, тормозящие также термические реакции
самоокисления, получили название „антиоксидантов". Их
действие может быть представлено следующей суммарной схемой>
не предрешающей истинного механизма (ср. стр. 237):
AO2-hZ -+ А-н ZO2
Действие легко дегидрируемых веществ — антиоксндантов —
на флуоресценцию красителей рассмотрено подробно в 35»
Для красителей с большим квантовым выходом
флуоресценции и обнаруживающих тушение ее кислородом начальный
этап реакции деструктивного выцветания будет подобен
начальному этапу рассмотренной выше фотореакции
присоединения кислорода к ароматическим углеводородам (23),
В заключение следует отметить, что не всякое выцветание
красителей под действием видимого света связано с
рассмотренной здесь фотореакцией деструктивного оксидирования.
Определенные красители выцветают в результате присоединения
водорода, т. е. путем фотогидрирования, или иначе —
фотовосстановления (32). Но всегда в присутствии кислорода
(н воды) достаточно длительное освещение, в особенности
короткими длинами волн, приводит к деструктивному
фотооксидированию.
27. Фотосенснбилнзованное красителем раврушенне волокна
В рассмотренной выше картине выцветания роль носителя —
волокна — была лишь побочной и сводилась к подведению
других участников реакции — кислорода и воды — к
возбуждаемому светом красителю. Между тем, природа носителя
оказывает специфическое влияние на скорость выцветания, а кроме
того, носитель сам может участвовать в фотохимическом
процессе 139.
Короткая ультрафиолетовая радиация затрагивает носитель
непосредственно. Так, в вакууме при освещении кварцевой
ртутной лампой целлулоза обесцвечивает флавантрен и другие
прямые красители путем восстановления (гидрирования);
27. ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЕМ РАЗРУШЕНИЕ ВОЛОКНА 175
испытывая при этом окисление140. В отсутствии целлулозы
прямые красители, осажденные на стекло, не выцветают в тех
же условиях. На солнечном свете, не содержащем столь
коротковолновой радиации, а потому ие затрагивающем
целлулозу непосредственно, этот процесс не имеет Места,
но краситель в вакууме может медленно разрушаться сам
в результате фотореакции взаимного восстановления-окисления
(ср. стр. 164).
На белковом волокне (шерсти), обладающем меньшей
восстанавливающей способностью, чем целлулоза, выцветание
в коротком ультрафиолетовом свете в вакууме идет более
медленно.
Отбеленные целлулозные и белковые волокна (хлопок,
шерсть, шелк) постепенно разрушаются при освещении
на воздухе под действием ультрафиолетовой радиации с
длинами волны короче 400 ш[х. Наибольшее повреждение
оказывают >Х<350 m/л. В результате волокно теряет свою
первоначальную механическую прочность. Это явление объясняется
реакцией фотооксидирования с образованием оксицеллулозы
(фотоцеллулозы) и выделением СО2, так как в вакууме или
атмосфере индифферентного газа разрушение волокна при
освещении останавливается.
На солнечном свете, не содержащем короткой
ультрафиолетовой радиации, поглощаемой неокрашенной
целлулозой, разрушение последней пренебрежимо. Иное имеет
место для окрашенных целлулозных волокон.
Хлопок, окрашенный светостойкими красителями, в
особенности желтыми и оранжевыми кубовыми красителями,
испытывает значительное повреждение волокон. Синие и зеленые
красители того же типа действуют заметно слабее. Все эти
красители являются светостойкими и химически
устойчивыми по отношению к кислороду и другим окисляющим
агентам.
Желтые субстантивные и нафтоловые красители не только
не производят этого явления, но некоторые из них проявляют
даже защитное действие, в особенности голубые красители.
Однако из основных красителей некоторые бывают активными.
Прямая зависимость разрушения целлулозы от цвета, т. е.
спектра поглощения красителя, указывает на то, что имеет
место фотосеисибилизованиая красителем реакция целлулозы
с кислородом н что активной является фиолетовая и
ультрафиолетовая радиация, поглощаемая желтыми и оранжевыми
красителями.
Интересно, что светостойкие и неактивные красители синего
цвета, присутствующие на волокне вместе с активными жел-
176 ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
тыми красителями, выцветают быстрее, чем когда они
присутствуют одни. При этом волокно разрушается не столь
сильно, как без синего красителя141. Очевидно, последний
принимает на себя воздействие активного красителя и тем
самым предохраняет волокно от повреждения. Подобному
сенсибилизованному выцветанию подвергаются синие красители
не только кубовые, но также и субстантивные, сернистые
и другие.1)
Фотосенсибилизованное разрушение белкового волокна
(шерсть, шелк) в общих чертах аналогично целлулозному,
но шерсть устойчивее целлулозы, в то время как шелк
разрушается легче. При этом красные и фиолетовые красители
оказываются более активными разрушителями для
натурального шелка, чем для целлулозы.
Между способностью красителей к флуоресценции н их
разрушающим действием непосредственной связи не обнару-
Т4.9
живается ♦
Явление разрушения волокна представляет собой фотосен-
сибилизованную красителем реакцию окисления целлулозы
за счет кислорода воздуха143. Активные красители, поглощая
фотон фиолетового или ультрафиолетового света, действуют
как своеобразные „передатчики" кислорода на инертные
к нему субстраты (целлулоза, синие красители). Однако
фотореакция не состоит в простом переносе активного
кислорода от фотооксида красителя на субстрат, поскольку кубовые
красители инертны по отношению к окислителям. Кроме
того, фотореакция может протекать и без кислорода, в
присутствии совсем иных окислительных агентов. Сенсибилизация
не может быть также объяснена передачей энергии
возбуждения красителя субстрату с активацией последнего по
отношению к кислороду, поскольку не намечается какой-либо
связи с флуоресценцией.
Сколфилд, Гудийр, Тернерш и Ландольтm связывают
описываемое здесь фотокаталитическое действие кубовых
красителей на целлулозу со способностью этих красителей
восстанавливаться (гидрироваться), с образованием леЙко-формы,
под действием сильных восстановителей (стр. 28). Было
показано145, что разрушающее действие активных красителей
делается исключительно сильвым тогда, когда краситель
присутствует на волокне в виде лейко-формы и имеет место
1)ФотосенсибиАизапию выцветания синего красителя на волокне могут
вызвать и бесцветные вещества, интенсивно поглощающие
ультрафиолетовую радиацию, как например Р-нафтол-сульфокислый натрий.
27. ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЕМ РАЗРУШЕНИЕ ВОЛОКНА 177
реакция фотоокисления последней кислородом воздуха.1) Было
также установлено, что выкраски активными желтыми и
оранжевыми кубовыми красителями под длительным освещением
изменяют опенок, приближаясь к цвету своих лейко-форм-2)
Далее был поставлен опыт с заменой целлулозы этиловым
спиртом. Суспензии активных желтых и оранжевых
красителей 3) в освобожденном от воздуха этиловом саирте,
заключенные в запаянные стеклянные трубки, при освещении изменяли
цвет в сторону цвета своих лейко-форм. При этом спирт
дегидрировался до альдегида, количество которого изменялось
параллельно разрушающему действию тех же красителей
на целлулозу. При взбалтывании изменившейся суспензии
с воздухом она вновь приобретает первоначальный цвет.
Аналогичную фотореакцию дает исходное для этих красителей
более простое соединение — антрахинон в спиртовом растворе
при освещении ультрафиолетовым светом.
Поскольку лейко-формы кубовых красителей имеют феноль-
ный характер, другой способ их обнаружения заключался
в обработке инсолированной выкраски диазооснованиями,
приводящей к типичной реакции сочетания с образованием азо-
производных, обладающих характерным цветом.
Совокупность изложенных данных привела Сколфилда,
а также Ландольта к следующему пониманию механизма
разрушающего действия кубовых красителей. На первом этапе
краситель фотовосстанавливается (гидрируется) в лейко-
краснтель Н2А за счет двух атомов водорода, отнимаемых
от целлулозы:
/Н
А* -+- целлулоза -»• А<^ -ьоксицеллулоза. (I)
Лейкокраситель, содержащий легко отщепляемые атомы
водорода, способен вступить в реакцию с кислородом воздуха
и образует гидропероксид Н-А-ООН, с которым мы
встретились в предыдущем параграфе.
А/ +О2 -* A<f (II)
\н 2 \оон
г)Эта реакции сенсибилнзуется к более длинноволновой радиации
следами красителя, получающимися в результате первоначального окислении,
и развивается самоускоряясь (см. cip. 156).
2) Как было упомянуто на стр. 28, лейко-формы антрахинодовых н др.
кубовых красителей окрашены.
■*) Цибанои оранжевый R, антра желтый GC, дурантрен
золотисто-оранжевый т, индантрен желтый 3R.
\Z A. H, Теренин
178
ФОТОРЕАКЦИИ ПРИСОЕДИНЕНИЯ МОЛЕКУЛЯРНОГО КИСЛОРОДА
Последний представляет собой лабильный промежуточный
продукт, доставляющий активный кислород, разрушающий
целлулозу или любой другой субстрат S, например, краситель
синего цвета, с регенерацией исходного красителя А:
н
ООН
н2о2
н2о
OS
(III)
Хотя перекись водорода иногда и образуется, но она
не является необходимым звеном этой реакции. Так было
показано, что в кислой среде1) или в отсутствии влаги Н2О2
не образуется, но, тем не менее, реакция фотосенсибилизо-
ваниого разрушения целлулозы идет. Однако влага все же
ускоряет последнюю, а также упомянутое выше фотосенсиби-
лизованиое выцветание синих красителей в присутствии
желтых, очевидно вследствие образования более лабильного
пероксидгидрата, рассмотренного в предыдущем параграфе.
Не все желтые и оранжевые красители оказывают
фотодеструктивное действие на волокно. Очевидно, легкость
образования и распада гидропероксида Н-А-СОН, являющегося
переносчиком активного кислорода, зависит от специфических
свойств строения красителя. Выделяются в этом отношении
антрахиноновые структуры. А именно, было установлено, что
антрахинон (а) и его простейшие производные способны фото-
сеисибилизовать реакцию присоединения кислорода к другим,
веществам, причем было установлено, что они под действием
света претерпевают превращение в соответствующие антра-
гидрохиноны (Ь) и обратно в хиноны (а), не расходуясь
в реакции
146'
О
2Н
О
(а)
*) В щелочной ередэ окислителем целлулозы является молекулярный
кислород, а не Н2О2, которая в такой среде иониэуетси и теряет
активность.
28. ПЕРЕНОС ЭЛЕКТРОНА 179
Наряду с антрахиноновыми красителями фотосенсибилизация
оксидирующего действия была найдена у некоторых инди-
гоидных, сернистых, акридиновых, флуоресцеиновых и ряда
основных красителей, а также исходных, более простых
соединений.
Наличие аминогруппы в молекуле, а особенно циклически
связанного азота (в производных пиридина), снижает
фотодеструктивное действие красителя на волокно. Ослабление
основных свойств атома азота в цикле соответствующими
заместителями неизменно повышает активность красителя147.
Итак, фотодеструктивное действие кубовых красителей
на волокно связано со следующими их свойствами: а)
повышенной стойкостью этих красителей к окислительным агентам,
обеспечивающее их самих от разрушения; б) способносттю
поглощать большие кванты энергии, доставляемые
ультрафиолетовым светом; в) способностью обратимо и легко гидрирв-
ваться (восстанавливаться) и дегидрироваться (окисляться).
К рассмотрению явления обратимого
окисления-восстановления под действием света мы переходим в следующей
главе.
ГЛАВА ВОСЬМАЯ
ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
28. Перенос электрона
Некоторые красители принадлежат к разряду соединений,
способных обратимо обмениваться электронами с другими
частицами, присутствующими в растворе. Сущность этого
важного явления, которому в химии присвоено название
окисление-восстановление, заключается в
следующем148. От частицы А, изображающей атомарный или
молекулярный ион любого знака или же нейтральную молекулу,
другая частица X отнимает электрон, что можно записать
таким образом:
А -+- X -> °А -ь *Х: окисление частицы А, (I)
где значок * обозначает переданный электрон, а значок ° —
его отсутствие в структуре частицы А. Например, в
простейшем случае катионов металлов имеем;
и*
180 ФОТООКЧСЛЕН4Е И ФОГОЗОССТАНОВЛЕНИЕ
где фигурные скобки с обозначением aq указывают на то, что
процесс происходит в водном растворе, В дальнейшем это
обстоятельство будет подразумеваться и приведенное
обозначение опускаться.
По укоренившейся в химия терминологии говорят при таком
процессе, что ^онор электрона А претерпевает „окисление",
а захвативший электрон акцептор X—„восстановление", хотя
процесс вообще не требует участия кислорода. Это
наименование возникло по той причине, что в водном растворе
происходят, наряду с реакцией (I), также побочные реакции
обмена электронами с ионами воды Н+ нли ОН~. Разрядка же
последних вызывает появление частиц Н и ОН и связанное
с их рекомбинацией вторичное участие газообразного
водорода или кислорода в процессе.
Выражая реакцию (I) в активной словесной форме, говорят,
что окислитель X „окисляет" частицу А. Другие же
вещества Y, которые, наоборот, стремятся отдать электрон частице А,
способной его воспринять, называются восстановителями,
и элементарная реакция восстановления может быть изображена
следующим образом:
А-ь Y->*A-h°Y: восстановление частицы А. (II)
Каждая из реакций (I) или (II) является двусторонним
процессом окисления-восстановления: если один из
участников взаимодействия „окисляется", то другой
„восстанавливается",
Перенос электрона от А к X или от Y к А делается
возможным, если, во-первых, электронная оболочка второго
участника X или, соответственно, А имеет свободное место
для электрона и может его воспринять и, во-вторых, когда
такой переход энергетически выгоден, т. е. сопровождается
понижением или постоянством общей энергии системы
двух частиц.
Изобразим графически на фнг. 32 начальное состояние
системы (I) двух рассматриваемых частиц А и X в виде уровня
электронной энергии, находящегося на высоте Еи При
переносе электрона система опускается на более низкий уровень
энергии Ег> выделяя энергию Ег — E2 — kEf рассеиваемую
окружающей средой Этот выигрыш энергии означает, что
перемещенный электрон был в А менее прочно связан, чем в X.
Аналогичное имеет место для реакции восстановления (II), где
теперь А играет роль X, a Y—роль окисляемой частяцьг.
Мерой прочности связывания электрона соответствующими
частицами является та энергия /, которая требуется для пол-
28. ПЕРЕНОС ЭЛЕКТРОНА 181
ного его отрыва от носителя с удалением электрона в вакуум
за пределы силового поля, притягивающего электрон к
лишившемуся его носителю. Такое состояние отсутствия притяжения
между электроном, помещенном в вакууме, и лишившейся его
частицей служит условным универсальным нулевым
уровнем для отсчета энергии электрона в любых частицах.
Он приведен вверху фиг, 32 под знаком Ео и принимается
за нуль, от которого отсчитываются вняз значения энергии .
При рекомбинации удаленного электрона с
возможными его акцепторами X н °А, находящимися в растворе,
выделяются соответственно энергии /х и JA, которые
тождественны энергиям отрыва электрона от частиц *Х и А. Эти
t ■
Фиг. 32. Энергетические уровни реакции переноса электрона (окислении-
восстановления).
X—окислитель, Y — восстановитель по отношению к частице А.
значения энергии и отложены на фиг. 32 вниз от условного
нулевого уровня EQ. Из фигуры следует, что Jx > JA и JeA >/F,
Другими словами, вступая в оболочку X, электрон прочнее
в ней связывается, чем в оболочке А. Наоборот, в А он связан
прочнее, чем в Y, Энергетический эффект переноса, как видно
из фигуры, представляет собой разность Jx—JA и JeA—JF.
Мысленно мы можем перенос электрона при реакции
окисления разложить на следующие два этапа:
A >cA + e (затрата энергии JA)
е-ьХ-—>*X (выделение энергии Ух)
Аналогично при реакции восстановления:
Y-—>°Y~he (затрата энергии А)
(И')
еч-А—>еА (выделение энергии JeA)
182 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Энергию отрыва собственного структурного электрона
от какой-либо частицы принято в физике называть энергией
ионизации, а энергию, выделяющуюся при присоединении
к такой частице избыточного электрона, — энергией
сродства к электрону.
Из предыдущего следует, что реакция переноса электрона
идет в том направлении, в котором выделяющаяся энергия
с избытком комаенсирует затраченную. Который из участников
реакции будет играть роль донора электрона (восстановителя)
и который акцептора (окислителя) — будет зависеть от
относительного расположения энергетических уровней электрона
в различных частицах.
В роли окислителя X, т. е. акцептора электрона,
фигурируют частицы, которые способны в данной ситуации прочно
связать электрон Так, например, катион Со1"*"1" в щелочном
растворе быстро разряжает ион гидроксила ОН~, вызывая
выделение кислорода в результате следующей
последовательности реакций:
Со ь+ч- -ь ОН" -> Со++-+- ОН
Это значит, что Jq0*++^>JOh-9 t* е* уровень ОН~ расположен
выше уровня Со4"1"**. Но катион Со4"1"*" не может присоединить
к себе электрон из анионов NO3~ и SO4 , так как их уровни
лежат ниже уровня Со4""1"1" и электрон связан в них прочнее,
чем он будет связан, когда перейдет к катиону Со"1"1"4".
Типичными окислителями, т. е. акцепторами электрона для
органических соединений являются катионы металлов, анионы
МпО4" и СгО4~~, выделяющие атомарный кислород, а также
другие анионы, содержащие определенные сочетания
электроотрицательных атомов с большим сродством к электрону.
Молекула О2 также является слабым окислителем; при
присоединении к ней электрона в газообразной фазе выделяется
энергия сродства к электрону, равная около 1 э, вольта150.
Окислительной функцией обладают также кислородные
соединения азота.
В роли госстановителей, т. е. доноров электрона, могут
выступать атомарные ионы обоих знаков, молекулярные анионы
и электронейтральные органические соэдинения.
Восстанавливающая способность многозарядных катионэв, например Fe+"*~,
Sn4"4", Ti*"*"*", объясняется тем, что в условиях водной среды
энергия отрыва от них электрона сильно понижена, а поэтому
их энергетические уровни расположены сравнительно высоко.
28. ПЕРЕНОС ЭЛЕКТРОН \ 183
В случае слабых, двуосновных органических кислот типа
R(COOH)2 донорами электронов являются анионы с двойным
отрицательным зарядом, появлению которых благоприятствует
щелочная среда.
Элементарная реакция окисления-восстановления. Мы
рассмотрели выше первичный элементарный акт, имеющий
место при реакции окисления-восстановления. При протекании
окисления-восстановления с участием органических
соединений этот первичный акт немедленно сопровождается
последующими их структурными изменениями. А именно, если
донор электрона Y представляет собой электронейтральную
органическую молекулу, то положительный заряд,
возникающий после удаления электрона, сосредоточивается на том
лабильном атоме водорода, который склонен отщепляться
в виде иона водорода, С другой стороны, электрон,
перенесенный на восстановленное соединение еА, концентрируется в нем
преимущественно на том атоме, например атоме О, который
способен его воспринять согласно внутриионизованной
валентной структуре получающегося ионв (6).
Система двух молекул eAY~*" достигает нового минимума
энергии тем, что от Y4" отделяется протон Н+ и переносится,
вслед за электроном, к молекуле dA, нейтрализуя заряд
перенесенного электрона. Весь процесс восстановления можно
представить следующей схемой, в которой значок н
обозначает „лабильный" атом водорода в структуре
восстанавливающего соединения:
A-hhY -> «AHY+ -> eAH+Y -> HA-h'Y. (Щ)
Аналогично можно изобразить обратный процесс окисления
соединения А каким-нибудь окислителем X:
Х-ьнА -> *ХЯА+ -> £Х,- А -» НХ + 'А (IV)
Значок ' обозначает утрату атома водорода соответствующей
частицей.
Когда роль окислителя выполняет молекулярный кислород,
т. е. когда Х = О2, такой перенос атома водорода
сопровождается образованием перекисного радикала Н-О-О • или НО2-
Электрон и протон в обычных условиях следуют друг
за другом настолько быстро и неразрывно, что весь процесс
производит впечатление единого процесса переноса
нейтрального атома водорода.
Применение глубокого охлаждения позволило отделить акт
переноса электрона от акта переноса протона в процессе
184 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
элементарного фотоокислення (стр, 108). Очевидно,
перемещение протона, как частицы большей массы, требует
дополнительной затраты некоторой термической энергии активации,
кбторая не требуется для электрона.
Необходимо эдесь подчеркнуть, что, хотя итог процесса (III)
состоит в разрыве валентной связи Н — Y и образовании
валентной связи Н — А,1) фактический элементарный механизм
реакции окисления-восстановления органических соединений
заключается в переносе не атома Н, а электрона и протона
раздельно, т. е. имеет существенно ионную природу.
Вводной или ионной среде такое ступенчатое прохождение реакции
требует на промежуточных его этапах значительно меньшей
энергии активации, чем непосредственный отрыв нейтрального
атома водорода, хотя суммарный энергетический эффект
реакции согласно закону сохранения энергии и не зависит от его
расчленения на отдельные этапы. Облегчение реакции имеет
место также потому, что обмен электроном может происходить
и без необходимости тесного сближения частиц, как этого
требует обычное химическое превращение.
Концентрация ионов Н4" и ОН" среды имеет исключительно
большое значение для протекания реакции
окисления-восстановления, изменяя относительную высоту рассматриваемых
здесь уровней анергии.
Если соединение HY представляет собой слабую кислоту,
то в щелочной среде происходит электролитическая
диссоциация на Н+ и Y~~, Донором электрона по отношению к А
является в таком случае анион кислоты Y~~ и элементарная
реакция восстановления завершается последующим
присоединением протона Н+ к молекуле вА, воспринявшей
электрон.
Если А обладает основными свойствами, то
последовательность присоединения электрона и протона в кислой среде
будет обратной схеме (III). А именно, сначала А присоединяет
протон от иона гидроксония Н+, Н2О, образуя положитель*
ный ион промежуточного соединения аммониевого или оксо-
ниевого типа. Реакция восстановления тогда завершается
нейтрализацией заряда такого иона электроном. Таков, повидимому,
механизм восстановления в кислой среде карбонильной группы
в антрахиноновых красителях:
>с = о —* [>с = о — н «—> >с —о —hJ —* ^с — он,
где в квадратных скобках изображены возможные
„резонирующие" валентные структуры получившегося иона (6).
1)Иаи, соответственно, Н — А и Н — X в (IV).
28. ПЕРЕНОС ЭЛЕКТРОНА 18S-
Однако в большинстве случаев отрыв или присоединение
только одного электрона, вместе с протоном, создает
незамкнутую валентную структуру соединения, обладающую
непарным электроном. Мы встретились с этим выше в случае
реакции окисления молекулярным кислородом (стр. 138). То же
имеет место для органических носстановителей, как например
аниона гидрохинона (а), который, утратив только одни
электрон, преобразуется в преходящую форму полу- или семи-
хинона (Ь) (см. стр. 53), стремящегося отдать и второй
избыточный электрон с превращением в незаряженное стабильное
соединение — хинон (с), в котором все электроны спарены:
Значок ■*—^, согласно стр. 34, означает сосуществование двух
зеркальных валентных структур, придающее некоторую
преходящую стабильность промежуточной форме (Ь),
предотвращающую, например, димериэацию с другой такой же валентно
ненасыщенной частицей.
Аналогичное имеет место, когда восстановителем служит
электронейтральная молекула, как например
пара-феиилендиамин, окисление которого происходит, предположительно, через
следующие этапы:
X=/=NH.-.
Таким образом, при реакции окисления-восстановления,
исходящей ив валентно-насыщенных соединений и приводящей
к конечным стабильным соединениям с парным числом
электронов, необходимо должны переноситься два электрона
и два протона совместно. Какова последовательность
промежуточных этапов, в большинстве случаев неизвестно. Схе-
^) Возможен также следующий вариант развитии реакции на средней
стадии:
186 ФОТООКИСЛЕЛИЕ И ФОТОВОССТАНОВЛЕИИЕ
матически изобразим процесс восстаноаления А соединением Y
следующим образом:
л -+- HY -> €A HY+ -> eA H+Y -» НА -+- 'Y
НА-+- .'Y -» еНА H+'Y -> H2A+''Y. l '
Промежуточные этапы протекают настолько быстро, что
итог окислительно-восстановительной реакции внешне
представляется как реакция присоединения молекулы водорода,
т. е. гидрирования соединения путем
дегидрирования восстановителя 1IY, где значки « обозначают атомы
водорода в структуре участвующих соединений:
Неразделимость промежуточных этапов оправдывает
привычную терминологию, отождествляющую окисление с отдачей,
а восстановление с присоединением водорода. В этом смысле
можно говорить о донорах и акцепторах не электрона, а
водорода.
В случае восстановления катионами металлов,
присоединяемый водород доставляется ионами водорода среды.
Например, восстановление азокраснтелей хлорным оловом SnCl2
в кислой среде, сопровождающееся исчезновением их окраски,
может быть представлено таким образом:
Sn^-bRj — N = N— R2 ^Sn^^-Ri — N — N — R2.
H H
Для красителей, формулы которых с двойными связями
представляют собой часть резонирующих валентных структур,
присоединение атомов Н, устраняя резонанс, приводит тем
самым к исчезновению окраски. Бесцветный гидрированный
продукт Н2А реакции восстановления красителя по этой
причине называется его лей ко, т. е. бесцветной формой
(стр. 26). Для азокрасителей эта реакция необратима. Но для
красителей иных типов (кубовые) атомы водорода в лейко-
форме связаны непрочно и могут быть отщеплены путем
реакции с кислородом воздуха по схеме (IV), сопровождаясь
обратимой регенерацией красителя (ср. стр. 177).
Семихинон типа HY, получившийся в результате отдачи
только одного атома Н, например частично дегидрированный
гидрохинон (а) стремится отдать протон и превратиться
28. ПЕРЕНОС ЭЛЕКТРОНА
147
в радикал (Ь), так как резонанс
непарного электрона в последнем
сопровождается понижением
энергии на величину около 1.5 э.
вольт
-ОН
— 35 ккал
(а)
6<>о-
(Ь)
н+.
Однако последний радикал, как мы
видели выше, имеет
кратковременное существование я быстро
отдает свой электрон подходящему
акцептору, превращаясь в хинон.
Исчезновение семихинонов может
происходить также в результате
реакции окисления-восстановления
между ними:
НА-ьНА -* Н2А-ьА, (VII)
(реакция ди с мутации),
сопровождающейся полным
восстановлением одной из частиц А и
регенерацией другой93.
Приводим ниже
предположительную картину
последовательных ступеней восстановления двух
основных красителей — оксонина
и метиленового голубого,
являющихся типичными красителями,
выцветающими в результате
восстановления, но обратимо
регенерируемых при окислении нх
лейко-формы148'ш.
Резонанс валентных структур,
приведенных в средних
квадратных скобках, стабилизует семихи-
нон, позволяя его обнаружить^
даже спектрально, в особых
условиях (сильно щелочная среда).
188 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Окислительно-восстановительный потенциал. Когда в
растворе присутствует одновременно с данной частицей также
ее окисленная или восстановленная форма, то между ними
обмен электронов и протонов при встрече совершается весьма
легко, так как энергия, затрачиваемая при отрыве, точно
компенсируется энергией присоединения и энергетический эффект
такой реакции равен нулю. Например, если в растворе имеются
одновременно ионы Fe4"4" и ге++ f то между ними
совершаются процессы обмена электроном в прямом и обратном
направлениях согласно схеме:
Fe"1"1"!-*- Fe1 ' ' ^± Fe~*"*~+ -t- Fe4"1" (VIII)
12 12
Аналогичное имеет место для окисленной или
восстановленной формы органических соединений, например:
\ Q— | Q У N=0 ~*" О / \: Q |
Т 7 Т
~°-\_У-°~ (ix)
2
= NH
Встретившиеся частицы как бы обмениваются своими
структурами с сохранением неизменности энергетического
уровня системы двух таких частиц, Обратимся к вопросу
о положении этого уровня по шкале энергии, изображенной
на фиг. 32. Энергия, затрачиваемая на перенос электрона
от Fe"*"1" к Fe++H" при реакции (VIII) может быть определена
путем разложения этого процесса на два этапа: 1) отрыв
электрона от Fe"*""4" в растворе и перемещение электрона
на бесконечно большое расстояние в вакуум, на что требуется
энергия /л++> которая определяет удаление исходного уровня
энергии системы Fe^^-KFe"1"*"*" от нулевого уровня Ео на фиг. 32;
2) обратный процесс присоединения электрона с этого
нулевого уровня к Fe' ' ', при котором выделяется энергия,
28. ПЕРЕНОС ЭЛЕКТРОНА 189
в точности равная затраченной в предыдущем случае, так как
Fe"*""*"*" превращается в Fe++. В итоге, система двух частиц (VIII)
при переносе электрона не меняет своего энергетического
уровня и &Е=0.
Совершенно аналогичное положение дела мы имеем для
реакций (IX), при которых между участниками переносятся
два электрона. Для характеристики усредненной энергии
отрыва одного электрона в таких случаях откладывают
половину суммарной энергии отрыва двух электронов
на шкале энергии в фиг. 32.
Между двумя формами данного соединения в растворе
устанавливается согласно обратимым реакциям (VIII, IX)
состояние подвижного равновесия, соответствующее
некоторым равновесным значениям концентраций участников.
Погруженная в раствор пластинка инертного металла
участвует в этом равновесии, его не нарушая, так как металл
способен как принимать электроны от участников реакции,
так и отдавать им электроны. В зависимости от того, какой
из этих процессов превалирует, металл заряжается
отрицательно или положительно, с установлением некоторого
равновесного потенциала. Численное значение последнего при
стандартных условиях однозначно характеризует уровень
электронной энергии системы двух частиц, обменивающихся
электронами (см. стр. 314), и носит название стандартного
окислительно-восстановительного, или сокращенно р е д о к с -
потенциала системы .
Значение редокс-потенциала для рассматриваемого здесь
процесса переноса электрона состоит в том, что его величина
непосредственно зависит от положения исходного
энергетического уровня, отщепляемого электрона или, что то же, от
конечного уровня присоединяемого электрона в реакциях (VIII, IX).
Последовательность численных значений редокс-потенциалов
воспроизводит, таким образом, относительное расположение
электронных уровней для различных частиц, а следовательно,
дает возможность сказать, в каком направлении совершится
перенос электрона между заданными участниками или,
другими словами, какой из них будет функционировать
окислителем, а какой—восстановителем ш.
Опытные значения окислительно-восстановительных
потенциалов в электрохимии отсчитывают не от предельного
уровня Е^9 принятого в теоретической физике, а от
некоторого промежуточного уровня, условно считаемого за нуль
редокс-потенциала. В качестве последнего принят потенциал
платинового электрода в стандартных условиях при насыщенин
19Э ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
водного раствора газообразным водородом и обусловленный
равновесием обратимой реакции по суммарной схеме:
_l * 7
И —*• —Н
Значок aq лишний раз подчеркивает протекание
рассматриваемых здесь реакций в водной среде.
Опытные значения редокс-потенцналсв, расположенные
выше этого условного „водородного уровня", приобретают,
тем самым, согласно фиг. 33 отрицательный знак; значения же
потенциалов, находящиеся ниже, — положительный знак.
Численные значения тех и других возрастают по мере
удаления от „водородного уровня" в обе стороны.
Для перехода от редокс-потенциалов к численным
значениям / энергии отрыва электрона, относимым к предельному
теоретическому уровню Ео, необходимо, очевидно» знать
положение „водородного Уровня" по отношению к последнему,
T^fe. величину /н. Из косвенных данных приближенно следует,
что_7нда 3.8 э.вольт149' 149\
Поэтому электрохимические значения стандартных редокс-
потенциалов S0 преобразуются в энергии отщепления
электрона / от соответствующих частиц с помощью соотношения:
У=^°-н3.8 э. вольт. (1)
При переносе двух электронов, как это обычно имеет место
в суммарной окислительно-восстановительной реакции между
органическими соединениями, из значения ^° по формуле (1)
получается усредненная энергия отрыва только одного
электрона.
На фиг. 33 приведено примерное расположение
электронных уровней, т. е. значения / для различных неорганических
ионов, органических соединений и красителей, вычисленные
по формуле (1). С левой стороны шкалы приведены частицы,
отрыв электрона от которых требует затраты энергии,
задаваемой глубиной расположения уровня. С правой стороны
приведены частицы, при воссоединении с которыми электрона,
наоборот, выделяется энергия, определяемая глубиной
расположения уровня под предельным теоретическим нулевым
уровнем Ей. Чем выше по шкале находятся частицы левой
половины, тем легче от них оторвать электрон, а следовательно
в эту сторону возрастает восстанавливающая
способность частиц, расположенных на этой половине. Чем ниже
по шкале находятся частицы правой половины, тем большая
28. ПЕРЕНОС ЭЛЕКТРОНА
191
энергия выделяется при восприятии ими электрона и тем
больше их способность окислять другие частицы.
В окислительно-восстановительной реакции между части-
цами различного рода электрон с занятого им уровня в какой-
Зонтые
уровни
Н
fudpL хинон
ОН'
Со'
20
40
урсбм
153
Сг
Виолоеены
Нейтральный кросныд
Феносафранин
■ Н +— Метилвновый голубой
- Тиянин
Фенолы, индофенолы
~ Хинон
Tif++ -
| J2— Дифениламин
о - Фенантролин - F в
5
1
I
■Co
OH
Фиг. 33. Шкала абсолютных значений '^окяслятельно-вос-
становнтельных потенциалов.
либо из частиц левой половины переходит на одий из
вакантных уровней правой половины, расположенный ниже его или
на одной высоте. В последнем случае мы имеем
рассмотренную выше обратимую окислительно-восстановительную реак-
192 ФОТООК-илЕНИЕ И ФОГОВОССТАНОВЛЕНИЕ
переноса электрона между двумя формами той же частицы,
приводящую к установлению равновесия между ними с
определенным значением редокс-потенцнала. Для большинства
органических соединений, в том числе и красителей, такой
обратимый перенос электронов (совместно с протонами)
не имеет места, и редокс-потенциал таких систем
непосредственно не может быть получен на опыте.1)
29. Фотоокисление неорганических ионов
Акт переноса электрона между частицами происходит
самопроизвольно, когда соблюдено относительное положение
уровней, изображенное на фиг. 32 предыдущего параграфа.
Обратное перемещение электрона от акцептора к лишившемуся
его донору есть эндоэнергетический процесс, требующий
восполнения разности энергий &Е. В настоящем параграфе мы
рассмотрим простейшую реакцию такого рода, когда
необходимая энергия доставляется поглощением фотона Av ^ &E.
Мы опишем сначала элементарный акт отрыва светом
электрона от растворенного иона или нейтральной молекулы,
который назовем элементарным фотоокислением, в от*
личие от более сложных типов реакции фотоокисления, о
которых речь ниже.
Ряд неорганических анионов и катионов, как например
Hal", OH~, SO3 , Fe"1""*", а также некоторые органические
анионы, как RCOO~, обнаруживают в водном растворе
интенсивные ультрафиолетовые полосы поглощения (ешах«104),
.которые приписываются именно этому элементарному
фотоэлектрическому эффекту разрядки иона в результате отрыва
электрона светом. Спектральное положение наблюдаемых
максимумов поглощения приведено в табл. 11 совместно
с некоторыми энергетическими данными, используемыми
ниже154»158.
Существует несколько попыток обосновать указанную
интерпретацию этих полос путем подсчета энергии, требующейся
для такого процесса, и сравнения ее с величиной поглощаемого
фотона.2)
Франк и Шейбе165, предложившие впервые такое
объяснение спектра, исходили из картины, по которой электрон, отры-
1) Его значение может быть, однако, вычислено из величины
энергетического эффекта при реакции переноса двух атомов водорода, т. е.
реакции гидрирования или дегидрирования, получаемой из термохимических
данных 153.
-) Д\я однозариженных анионов спектр, связанный с отрывом электрона,
получил название „спектра сродства" (к электрону) нейтральной частицы,
получающейся в результате отрыва.
29. ФОТООКИСЛЕНИЕ НЕОРГАНИЧЕСКИХ ИОНОВ
193
Таблица 11
и Avmax
Максимумы полос поглощения гидратированных ионов (Хт
энергии их гидратации (W) и энергии отрыва электрона от ннх в газо
образном состоянии в вакууме (!)
Ионы
ci-
г
он-
Ag+
TI+
Fe++
Fe
181
/ «к. 190
\ 195
f 194
1 226
186
Г 210.5
I 193
214
180
225
(граница 275)
230
(граница 340)
1
Av
(eV)
6.84
6.50
6.34
6.35
5.46
6.65
5.86
6.40
5.77
6.86
5.48
5.36
W
(eV)
2.82
2.52
2.08
2.48
7.05
4.65
20.9
20.9
ск. 51
/
(eV)
3.75
3.53
3.22
3.7—4.8
22.5
20
(36)
ваемый от гидратированного иона галоида Hal aq, удаляется
от него в окружающую водную среду и „растворяется" в ней.
Другими словами, подобно гидратированньш ионам, он
оказывается окруженным оболочкой молекул воды, диполи которых
ориентированы в создаваемом нм электрическом поле.
Процесс изобразится, следовательно, схемой:
Hal
aq
HaL
(0
Энергия, требуемая для его осуществления» может быть
подсчитана, если расчленить весь процесс на ряд этапов,
энергия которых известна из опыта или доступна вычислению.
На фиг. 34 изображена такая последовательность процессов,
приводящая в итоге к тому же результату, что и поглощение
фотона. Направление каждого отдельного процесса дано
стрелкой; затрата энергии дана без знака плюс; выделение
обозначено знаком минус впереди. Суммарная энергия,
затрачиваемая на таком обходной пути, равна энергии этапа,
замыкающего цикл, т. е. искомой энергии фотона /iv.
Поглощение фотона с отрывом электрона есть настолько
мгновенный акт, что тепловое движение не успевает дезорнен-
13 А. Н« Терсвпн
194 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
тировать диполи молекул воды, которые в первый момент
остаются в первоначальной ориентации вокруг разряженного аниона.
Между тем, теплота гидратации атома галоида Wa в фиг. 34
как величина, получаемая в условиях теплового равновесия
с окружением, при котором диполи расположены беспорядочно
около незаряженного атома, не содержит этой принудительной
ориентации диполей Н2О. Поэтому необходимо затратить на
такую ориентацию дополнительную энергию Р9 чтобы получить
конечное состояние водной среды вокруг атома галоида
в первый момент после акта поглощения. Отсюда получается
0^- л /-> Фиг. 34. Цикл Франка-Шейбе
^ для вычисления фотона Av, по-
Вокуу
JU
'WQ
'/;
для вычисления фотона Av,
поглощаемого гидратированны м
анионом галоида.
w_ —
_ — энергия гидратации ани-
W
л - .//,,/// она: Wa — энергия гидратации
vOPO атома; We — энергия гидрата-
■/->-' Цми электрона; /__ — энергия
W_ ^~ ч сродства атома к электрону
t ^ * 1 ^ в 1азообразном состоянии в
вакууме; Р— энергия перевода
конечное диполей (ч—>) молекул еоды из
Состояние беспорядочного в
ориентированное состояние. Величины
энергии, приведенные без знака, означают, что осуществление процесса в
направлении, указанном стрелкой, требует затраты энергии; величины
энергии с отрицательным знаком впереди означают, что при процессе,,
указанном стрелкой, выделяется энергия.
теоретическое уравнение, составляющие члены которого
приведены в фигуре:
hs=z W -+-I W» W -+- Р (1\
Сдедует сразу отметить, что энергия гидратации Wa
незаряженной частицы пренебрежима по сравнению с энергиями
гидратации ионов, так как имеет "Величину порядка только
1 кгкал/моль-
Франк и Шейбе получили согласие вычисленной таким путем
величины фотона с наблюдаемыми максимумами поглощения
ионов галоидов в водном растворе. Однако это совпадение
оказалось совершенно случайным, когда Паулинг166
теоретическим подсчетом показал, что энергия гидратации электрона
We равна 88кгкал, а не 18.5 кгкал, как принимали Франк
и Шейбе. Отсюда следует, что результат действия света на
гидратированный ион неправильно воспроизводится схемой (I)
и картиной, изображенной на фиг. 34. Электрон не перехо-
29. ФОТООКИСЛЕНИЕ НЕОРГАНИЧЕСКИХ ИОНОВ 195
дит в раствор, а следовательно учет энергии для создания
орийнт^цинГ'^диполей в гидратной оболочке атома, а также
вычисление величины We излишни15*.
Между TdM, наблюдаемая величина hi не передается
остающимся в (1) выражением W_ -+-/_, которое согласно фиг. 34а
есть энергия отрыва электрона от гидратированного аниона
с удалением электрона в вакуум, т. е. величина У предыдущего
параграфа. Действительно AvHa6A. <С J-, где 7_ есть неличина
редокс-потенциала аниона галоида, отнесенная, согласно
формуле (1) предыдущего параграфа, к теоретическому нулевому
уровню Ео (ср. фиг. 32).
О
Ш у////////////,
Вода
0
7-
/-+W-
вачалоное Конечное
состояние состояние
Фиг. 34а. Энергия ,/_■=/_-*- М^__, требующаяся
длн отрыва электрона от гидратированного аниона
с переводом электрона в вакуум. Энергия
гидратации атома Wa положена равной нулю.
Наиболее простое объяснение такого неравенства
заключается в том, что одна из молекул воды, находящаяся в^
непосредственной близости к иону, воспринимает электрон с
выделением некоторой энергии „сродства" JHQ = J_ — А^дабд>
Подобная трактовка спектров поглощения гидратирован-
ных анионов галоидов была выдвинута братьями Фаркас157,
которые для различных анионов получили приблизительно одно
и то же значение разности /_—AvHa6A. ^18кгкал и сделали
отсюда вывод, что эта величина и дает сродство молекулы
воды к электрону (ок. 0.8 э. вольта).
Разложение перемещения электрона на отдельные этапы
изображено на фиг. 35. Энергия гидратации частицы Н2О~
при переходе из газообразного ее состояния в раствор,
'но—, положена равной энергии испарения молекулы воды
о, поскольку гидратная оболочка вокруг иона остается
практически неизменной в конце акта поглощения фотона,
а следовательно никакой гидратации иона Н2О~, собственно,
не имеет места.
196 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Рассматриваемый перенос электрона от донора На1~к
акцептору Н2О укладывается в схему уровней элементарной реакции
окисления-восстановления, приведенной на фиг. 32 предыдущего
параграфа с той разницей, что реакция протекает здесь в
сторону увеличения энергии, т. е. с уровня Е2 на уровень Еи за
счет поглощения фотона /iv = A£.
Пребывание электрона в молекуле Н2О не может быть
длительным именно из-за более высокого расположения
энергетического уровня Еи а потому рассматриваемая система
частиц возвращается вскоре на исходный уровень Е29 растрачивая
избыток энергии &Е н виде тепла, рассеиваемого средой. Весь
процесс может быть записан в виде схемы:
НаГ\Ч2О aq |* Hal,H2CTaq (II)
fa = W_ + /_ — /н,о (+ xV) (2)
Если в непосредственном соседствз с „возбужденной"
светом системой HaljH^O" появляется катион или другая частица,
обладающая большим сродством к электрону, чем молекула
Н2О157> например ион водорода Н^ или Fea^++f то электрон
переходит к последнему. Вакантный электронный уровень
в последних частицах расположен настолько высоко» что они
неспособны „окислять" ион галоида са юпроизвольно, без
участия света (фиг. 33).
Таким путем, действуя ультрафиолетовым светом, погло-
щазмым гидратированным ионом нода, можно вызвать разрядку,
г. е. восстановление ионов водорода в растворе с выделением
молекул J2 и Н2, которые получаются в результате рекомби-
*) Концепция Фаркасов нуждается в некоторых коррективах. Прежде
всего, при перемещении электрона от иона галоида к одной из молекул
воды гидратной оболочки конфигурация зарядов, ^ следовательно, потен-
циальнан энергия электрического поля изменяется одновременно
с актом переноса на некоторую величину ^К. Далее, /_= W—-+- /_t п^рлучеч-
ное из величины электрохимического редокс-потенциала, есть по своей
природе значение энергии отрыва электрона в условиях достижении
потерявшей электрон частицей теплового равновесия с окружением, т. е. полной
беспорядочной ориентации окружающих диполей воды. Для получения
конечного состояния, осуществляемого мгновенным поглощением света,
необходимо, следовательно, затратить кроме/— еще энергию
принудительной ориентации Р, упоминавшуюся выше. Как Д Vt так и Р не могут быть
подсчитаны сколько-нибудь точно, составляй вместе некоторую
неопределенную поправку х в теоретическом выражении для Av. По этой причине
величина /u q, подсчитанная Фаркасами, нг равна значению сродства и
электрону газообразной молекулы воды, а содержит укаваяные неопред
ленные члены. См. также 159.
29. ФОТООКИСЛЕНИЕ НЕОРГАНИЧЕСКИХ ИОНОВ
197
нации первично образующихся атомов иода и водорода157,
Реакция протекает по следующей схеме:
J,H2O
aq
н
aq
J
aq
н
aq
I"
H2
н
->
Квантовый выход у фотореакции для Н^невелик: у =0.1—0.3,
что вызвано, вероятно, быстро идущей обратной реакцией
в схеме (II). Для галоидных
солей щелочных металлов выход н 0
еще меньше н выделение
водорода не наблюдается.
По схеме (И) может протекать
- Iн,о
н?о
элементарное фотоокисление
также многократно заряженных
анионов, а также катионов, на-
7.
Н20
-И/ .-
0
1'
О
Начальное
гостсяние
состояние
Фиг. 35, Цикл промежуточных Э1а-
пон по б р. Фаркас.
W-q 0— энергия гидратацин^отдель-
ной молекулы НгО гидратной
оболочки; W-Q q — энергии гидратации
газообразной молекулы НоО,
воспринявшей электрон и
перенесенной н гидратную оболочку;
/д о — энергия сродства.
пример Fe"*""1". В уравнениях,
построенных подобно (2),
должна при этом фигурировать не
энергия гидратации исходного
иона, а разность энергий
гидратации иона в двух состояниях
зарядки. Это обстоятельство
делается ясным из фиг. 36, в
которой изображены циклы,
позволяющие вычислить энергию /
отрыва электрона от
соответствующего гидратиро-
ванного иона на основании
величины энергии / отрыва
электрона от того же иона в газообразном состоянии в
вакууме и теплот гидратации W иона в исходном и конечном
состоянии. Обозначения аналогичны тем, которыми мы
пользовались в фиг. 34. Энергия / есть не что иное, как
абсолютная величина редокс-потенциала соответствующего иона
(стр. 190).
Правый цикл применим и для того случая, когда исходной
частицей является нейтральный атом, теплота гидратации
которого Wa да 0. Тогда Ja=Ia — W+, где Н^+ теплота гидратации
образовавшегося катиона. Особенно разительно действие
гидратации на положение уровня электрона в атоме водорода.
На шкале фиг. 33 этот уровень отстоит всего только на 1.53
э. вольт от предельного нулевого уровня по сравнению-
193
ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
с /н =13.53 э. вольт — энергией отрыва электрона от атома Н
в газообразном состоянии. Отсюда делается понятной
легкая ионизация гидратированного атома водорода частицами,
обладающими низко расположенным вакантным электронным
уровнем.
Энергии J и /+ в фиг. 36 соответствуют такому
конечному состоянию, когда гидратная оболочка полностью
приспособилась к изменившемуся заряду и объему иона. Иное
имеет место при расчете величины поглощаемого фотона."
Акт поглощения фотона и отрыва электрона»
совершающийся мгновенно, сопровождаются столь же мгновенным из-
штж
Фиг. 36. Цикл промежуточных этапов для вычислении энергии отрыва
электрона от гидратнрованиых ионов в условиях термического равновесия.
мененнем потенциальной энергии электростатического поля
в гидратной оболочке. Диполи молекул воды, как было сказано
выше, не успевают занять равновесную конфигурацию около
получившегося нового иона в соответствии с новым минимумом
энергии. Поэтому при отрыве электрона светом в отличие от
процессов, изображенных на фиг. 36, разность теплот
гидратации W__— W_ или Н^2+—W+ сказывается не в полной мере.
При расчете величины поглощаемого фотона это^обстоятельство
заставляет ввести некоторую неопределенную поправку в
величину разности теплот гидратации. Кроме того, для
многозаряженных ионов требуемые для расчета величины W и /
часто неизвестны. Поэтому бр.Фаркас367 предложили для ионов
железа в уравнении для Av, составленному по образцу уравнения
(2), вместо совокупности неизвестных членов /++—(Н^3+ —
— Н^2+)> использовать равную ей, согласно фиг. 36, величину
редокс-потенциала /++ реакции Fe++ -* Fe+++ в абсолютной
шкале. Вместо (2) отсюда получается уравнение:
= /+-+ — 7н2о -+- х,
(3)
29. ФОТООКИСЛЕНИЕ НЕОРГАНИЧЕСКИХ ИОНОВ 199
где /++= 0*°-»-+ 4-3.8 э. вольт, если <?0+-ь есть
электрохимический стандартный редокс-потенциал. Для энергии /но, т.е.
электронного „сродства" молекулы воды в газообразном
состоянии, они полагают, как сказано выше, значение 18 кгкал =
= 0.8 э. вольт. Неопределенная величина х9 согласно
предыдущему, означает некоторую поправку к равновесному
термодинамическому значению /++. Она представляет собой по сути дела
избыточную энергию гидратной оболочки иона Fe+++ в первый
момент его получения действием света по сравнению с
конечным, более низким уровнем энергии, когда гидратная оболочка
приспособилась к изменившимся условиям.
Бр. Фаркас показали, что уравнение (3) дает для гидрати-
рованного катиона Fe++ величину поглощаемого фотона Av,
согласную с опытом, если положить лт^1э. вольт. При
неточности значения /н о можно считать, что х — ГцО, и уравнение
(2) преобразуется в форму (4), приданную ему Данном159:
= 7R^dox= <£°-ь 3.8 э. вольт. (4)
Действительно, этому соотношению удовлетворяет
длинноволновая граница полосы поглощения не только гидратированного
катиона Fe++ 157, но и Сг++ 15°, а также СгО4 159\
Под действием света в ультрафиолетовой полосе
поглощения гидратированного катиона Fe++, так же как в случае
иона ]~у происходит выделение газообразного водорода из
раствора100. Механизм этой фотореакции должен отличаться,
однако, от (II'), так как ее весьма низкий квантовый выход
у =0.02—0.05 не находится в прямой зависимости от
концентрации ионов водорода ЬГ*~. Бр. Фаркас157 предполагают, что ионы
Н+не могут подойти к „возбужденной" системе Fe~*~++, H2O~
ввиду сильного электростатического отталкивания и принимают,
что в этих случаях водород выделяется изводы путем
реакции распада:
\
н,
при которой выделяется значительная электростатическая
энергия притяжения между ионами Fe++*~ и ОН~, способствующая
расщеплению молекулы воды. Подобный распад молекулы Н2О,
выдвинутый Франком и Хабером в качестве второго варианта
200 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
объяснения полосы поглощения гидратированных ионов Halaq" ?
подробно рассматривается ниже.1)
Обратная реакция Н с Fe+++, выписанная в (III), является
также возможной причиной низкого квантового выхода
выделения водорода. Для иона Сг++ фотолиз воды с выделением
водорода имеет больший квантовый выходу = 0.2 — 0.4159.
Ввиду несостоятельности предложенного Франком и Шейбе
механизма (I) Франк и Хабер162 выдвинули иную трактовку
происхождения спектра поглощения гидратированных анионов,,
отличающуюся от разобранной выше схемы (II) бр. Фаркас.
К этой трактовке присоединился затем Вейсс и другие.16*
Франк и Хабер предполагают, что переход электрона к
молекуле Н2О сразу вызывает ее диссоциацию на Н-кОН~.
Электрон как бы нейтрализует заряд протона в молекуле воды
Н+ОН~, вследствие чего электростатическая связь в последней
исчезает. Эта точка зрения имеет обоснование в опытах по
прохождению медленных электронов в вакуумных условиях через
пары воды, в которых установлен такой тип распада при
достижении электронами некоторого критического значения
кинетической энергии15^:
^нН2О -* Н-ьОН".
По Франку-Хаберу поглощение света гидратированным
ионом галоида вызвано следующим процессом фотолиза
молекулы воды:
Hal", H2O ^ Hal -н Н -ь ОН", (V)
*) Совсем иной тип расяада молекулы воды имеет место при освещении,
щелочного раствора ионов Се^~*" в области ультрафиолетовой полосы
поглощении последнего, так как в отличие от предыдущей реакции при этом
выделяется газообразной кислород с квантовым выходом у =0.1 161.
Предполагают следующий механизм втой реакции:
Се4+, Н2О -^ С 2
Uoh- UV>
Н2О -*- ОН" <- СеЗ ь Н2О -I- ОН
\-hCH
V2O2+H2O.
Первичный процесс здесь состоит не в присоединении электрона
к молекуле Н2О (восстановление воды), как раньше, а наоборот — ее
окислении с переходом электрона на вакантный уровень катиона Се^+ 158.
Другими словами, происходит фотовосстановление катноиа, прл котором
молекула Н2О играет роль донора электрона (ср 32). Ультрафиолетовые
полосы поглощения гидратированиых катионов Си++, Zn-*-**, Cd-*~*", Hg+-*-r
Sn-*-*~, Pb++ по Рабиновичу158 могут быть связаны с такой же реакцией,.
Вейсс и Поррет допускают даже распад Н2О *"-»• Н++ ОН в первичном акте 161.
29. ФОТООКИСЛЕНИЕ НЕОРГАНИЧЕСКИХ ИОНОВ 201
а величина фотона, требующаяся для этой реакции, должна
вычисляться согласно этим авторам по уравнению (5),
заменяющему первоначально предложенное Франком-Шейбе
уравнение (1):
iIbI HnO Oil ^ *
в котором DH o есть энергия диссоциации газообразной
молекулы Н2О на Н-ьОН:
н,о^Ь2н +
Обозначения остальных величин были приведены ранее
при фиг. 34. Неопределенная энергия х, как и раньше,
представляет собой часть энергии поглощенного фотона,
удерживаемую в виде потенциальной энергии системой, не успевшей
придти в тепловое равновесие с окружением. Она не может
быть предвычислена, а следовательно лишает выражение (5)
возможности проверки.
V В уравнении (5) явно отсутствует энергия гидратации W_
иона галоида, которая, повидимому, учитывается членом л:.
Энергия гидратации газообразного аниона гидроксила ОН~
справедливо не принимается во внимание, поскольку
мгновенность акта поглощения не успевает нарушить исходное
состояние гидратной оболочки. Весь процесс как бы происходит
в вакууме, что объясняет присутствие в (5) величин,
относящихся только к газообразным частицам.
В кислом растворе образовавшийся в (V) ион ОН~,
соединяясь с ионом водорода, дает молекулу воды.
По механизму (V), как и по механизму (II'), выделяется
водород. Различие заключается в том, что по (II') атом появляйся
в результате восстановления постороннего иона водорода,
тогда "как в (V) он доставляется распадающейся молекулой
воды/ В случае катиона Fe++, как мы видели (ср. III), обе
трактовки совпадают.
Однозначный выбор между механизмами (II) и (V) для
анионов галоидов затруднителен. По второму механизму разрядка
аниона неизбежно сопровождается появлением иона гидроксила,
т. е. увеличением рН. Действительно, при освещении
нейтральных водных растворов иодидов щелочных металлов
ультрафиолетовым светом в области полосы поглощения нона J~
Бутков установил повышение щелочности раствора и выделение
молекулярного иода (в виде иона J3~)m« Однако выход был
незначителен и реакция могла идти по механизму (II'), так как
выход увеличивается с подкислением раствора157.
202 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
f Необходимо отметить, что тот же спектр поглощения ио-
(нов галоида (спектр „сродства") наблюдается без
существенных изменений и в спиртовом растворе ш. Сродство молекулы
С2Н5ОН к электрону неизвестно, но, вероятно, близко к
значению для молекулы воды. Между тем схема (V) делается здесь
неприменимой.
Частицы Н и ОН~, образовавшиеся по схеме (V), не
успевают, как правило, разойтись, ввиду препятствия со стороны
окружающих молекул воды, и должны немедленно рекомбини-
ровать с восстановлением заряда иона галоида. Таким образом
атом Н в этой схеме доступен для дальнейших реакций только
[за короткое время своего свободного существования в то время,
'как по схеме (II') ему предоставлена возможность диффузии
от места образования.
В кислом растворе для анионов галоидов безусловно должна
иметь место фотореакция (II') переноса электрона к иону Н+
через гидратные оболочки. Для катионов же справедлив
механизм (III).
Выделение газообразного водорода при освещении
ультрафиолетовым светом в области спектра „сродства" было
установлено для водных растворов, содержащих анионы SO3 ,
НСОСГ, RCOCT, С2О4—165а'159. Первичная фотореакция
приписывается процессу (V) с разложением молекулы воды.
Раствор должен быть тщательно освобожден от
растворенного кислорода, В присутствии последнего атом Н
захватывается молекулой О2,образуя реакциониоспособный радикал
НО2, с которым мы встретились ранее (стр.137.)1)
Концентрация растворенного кислорода в воде при
атмосферном давлении воздуха составляет, как мы упоминали (21v,
величину порядка 10~4 моль/л, что эквивалентно парциальному
давлению 2 мм кислорода в растворах. Такая концентрация
молекул кислорода достаточна, чтобы был обеспечен захват
ими атомов Н, образующихся при фотоокислении гидратирован-
ных ионов в растворах. В нейтральном или щелочном растворе
1) Существование радикала НО2 постулируется при объяснении
кинетики реакций окисления как кислородом в газообразной фазе, так и в
растворах1Ь5. Прочность связи атома водорода в НО2 оценивается в 45—50
кгкал/моль. Присоединение второго атома Н с образованием перекиси во-
дорэда выделяет значительную энергию 94 кгкал:
Этн данные относятся к газообразному состоянию. Таким образом
.обладает значительной дегидрирующей (окисляющей) способностью.
29. ФОТООКИСЛЕНИЕ НЕОРГАНИЧЕСКИХ ИОНОВ 203
растворенный кислород выступает в роли акцептора
электрона. Действительно, если численное значение /н о«
^1 э. вольт, полученное бр. Фаркас для электронного сродства
молекулы воды, справедливо, то оно, примерно, совпадает
с величиной электронного сродства молекулы О2 по расчетам
Казарновского: /о ^ 0.9 в. вольт150*
Из совпадения этих величин следует, что молекула О2,
находящаяся вблизи поглотившего фотон гидратированного
иона На1,Н2О~или Fe"1""1""1", H2O~, способна захватить электрон
от заряженной молекулы воды, превращаясь в молекулярный
ион О2~. В кислой среде последний присоединяет Н+ и
превращается в радикал НО2, который дает в конечном итоге
перекись водорода. Процесс протекает по схеме16D:
Ion,H2O"4-O2-^Ion,H2O-+- О2"
|-*-Н+ (VI)
НО2
(Н+-> Н О„+ ~> Н2О9
2 2 "
Перекись водорода является, таким образом, конечным
продуктом восстановления кислорода, подобно тому как
конечным продуктом восстановления органического соединения
является присоединение двух атомов Н.
Итак, в отсутствии кислорода элементарная реакция
фотоокисления ионов, рассмотренная в эгом параграфе, приводит
к выделению из водного растзора водорода. В присутствии
растворенного кислорода образуэтся вместо этого перекись
водорода. Активные длины волн для этой реакции лежат
в пределах полос поглощения гидратированных ионов,
приведенных в табл. 11.
30. Фотоокисление органических соединений
s В отличие от атомов, для которых из спектральных данных
| хорошо известны энергии отрыва электрона в газообразном
\ состоянии, такие сведения для молекул почти полностью
? отсутствуют. Особый интерес эти сведения представляют для
f органических соединений в связи с вопросом об окислительно-
восстановительных процессах, в которых последние участвуют.
Факт отщепления электрона, т. е. фотоэлектрического эффекта
для органических молекул, в твердом и растворенном
состоянии был установлен давно (42). Было также давно показано,
пары антрацена, дифенилметана, дифениламина и а-наф-
204 ФОТСОККСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
тяламииа обнаруживают проводимость под действием
короткого ультрафиолетового света кварцевой ртутной лампы166.
Эти старые работы имели целью выяснить связь между
отщеплением электрона и флуоресценцией органических
соединений. Никакой связи установлено не было, так как оба эффекта
вызываются светом различной длины волны. Серков167 показал
наличие заметной проводимости в разреженных парах анилина
при освещении ртутной кварцевой лампой. В лаборатории
автора настоящей книги этот опыт был повторен и установлено,
что освещение паров «анилина упругостью 0.1 мм искрой цинка
вызывало значительный фототок порядка 10~9А. Активны
короткие ультрафиолетовые длины волн с 7Л <С 220 /л;л,
В новую плодотворную фазу развития вопрос об
отщеплении электрона светом от органических соединений вступил
в самое последнее время благодаря работам Льюиса с
сотрудниками ' . Эти исследователи изучали спектры окрашенных
продуктов, образующихся под действием близкого
ультрафиолетового излучения на замороженные (температура жидкого
воздуха) растворы ряда бесцветных ароматических соединений,
в том числе и лейко-форм красителей. Подходящим
растворителем, не кристаллизующимся при замораживании, служила смесь
эфира-изопентаиа-этанола (в пропорции 5:5:2 по объему),
остающаяся прозрачной при температуре жидкого воздуха
и не выделявшая растворенного вещества.
В этих условиях под действием света из исходного
бесцветного ароматического соединения возникает вследствие
отрыва электрона окрашенный положительный молекулярный
ион с непарным числом электронов (стр. 103), который в
некоторых случаях может быть получен и чисто химическим
действием слабого окислителя. Спектры таких ионов были изучены
ранее Михаэлисом с сотрудниками. Следовательно, мы имеем
здесь в чистом виде явление элементарного
фотоокисления нейтральной молекулы. Так, например, при освещении
раствора трифениламина наблюдается появление видимой
полосы поглощения его положительного иона согласно механизму:
PhsN X?hzN++e. (0
Аналогичная фотореакция имеет место для дифениламина,
парафенилендиамина и его производных, а также производных
дифенила и для аниона дифенилазота:
R^N— <^ V- NR2 —^R2N—/ \—NR2+-be
30. ФОТООКИСЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
205
он
Av
_/
-OH+
но
Явление наблюдается только для ароматических
соединений, содержащих атомы N,O,S, имеющие основные
свойства, г. ^е. обладающие свободной парой электронов и могущие
присоединить протон Н~*\ Очевидно, свет отщепляет одни
злектрбнГ'йЗ этой пары, как представлено на схемах.
Огделнвшнйся электрон остается прикрепленным при низкой
температуре к молекулам растворителя, но достаточно
небольшого повышения температуры для немедленной его
рекомбинации с ионом.
Принципиально важным для понимания механизма
окислительно-восстановительных реакций является наблюдение, что
получившиеся положительные ионы неустойчивы и через
некоторое время, в условиях низкой температуры,
самопроизвольно отщепляют протон Н+. Это наблюдение
рассматривается нами как первое экспериментальное доказательство
раздельности актов переноса электрона и протона (28).
Так, например, после первичного получения указанных ниже
молекулярных ионов происходит медленный темновой процесс
распада с образованием новых окрашенных форм:
—> Ph2N : -
НО -
-ОН+ —* НО -
- 6: -ь Н+
-- 6: -ь Н (II)
Вся фотореакция может быть записана следующим образом,,
«ели обозначить исходную молекулу через НА:
НА
(III)
Так ведут себя оксазин (а) и тиазин (Ь), дающие при
освещении форму семихинона HA+, превращающуюся затем в дру»
*гую форму семихинона А168.
О S H2N S NH2
206
ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Me2N
NMe2 Me2N
\/\
Ме = СН3
NMe2
Полоса поглощения иона НА+ смещена в сторону более
коротких длин волн, чем полоса радикала А, и часто имеет
вибрационную структуру, отчетливо выраженную из-за низкой
температуры.
Фотореакция окисления лейко-форм красителей: лейкотио-
нина (с), лейкометиленового голубого (d), лейкокристалличе-
ского фиолетового (е), лейкомалахитового зеленого, гидрола
Михлера (стр. 51) и других, "протекает более сложным путем.
А именно, она идет только при значительной концентрации
лейко-формы НА, когда большинство молекул лейкокрасите-
лей присутствуют в виде димеров (НА)2. Фотоокислению
подвергается, главным образом, димер, причем первым этапом
является отщепление двух электронов при поглощении только
одного фотона. Мы полагаем, что первичный акт состоит
все же в отрыве одного электрона, но что получившаяся
полуокисленная форма димера НААН+ настолько неустойчнва,
что сразу отщепляет еще один электрон и протон с
образованием неустойчивой формы димера НАА+. Последняя
самопроизвольно распадается далее по одному из трех путей,
указанных в следующей схеме (IV), причем для различных
лейкокрасителей преобладают различные пути распада. Все
эти стадии были прослежены спектральным путем по
появлению промежуточных окрашенных продуктов в первоначально
бесцветном растворе
168.
hi
НААН --*НААН
НАА
(IV)
АА-ьН+ А + А + Н
для лейкометиленового
голубого
для леикотионнна
где А"*" обозначает катион красителя.
30. ФОТООКИСЛЕНИЕ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ 207
Для понимания механизма окислительно-восстановительных
процессов представляет большой интерес тот факт, что
поглощение только одного фотона является уже достаточным
для полного двухстадийного окисления с отдачей 2е, а в
случае тионина также и 2Н+. Однако эти наблюдения относятся
к димерам лейкокрасителей. В какой мере этот результат
воспроизводит поведение мономера при фотоокислении —
не выяснено. Лейкокрасители с центральным атомом С
(например, е) требуют для фотоокисления в этих условиях длин волн
короче 270 т\ь9 в то врэмя как лейкокрасители с
центральным атомом N (с, d) испытывают фотоокисление светом
с П<400 т|л.
Начальный квантовый выход у фотореакции, определяемый
как отношение числа образовавшихся катионов красителя
А+ к числу фотонов, поглощенных лейко-формой НА, равен
0.04 для метиленового голубого и 0.10 для синего Вурстера168^
Однако поглощающими свет частицами были не только димеры,
но и мономеры лейкокрасителей, интенсивно
флуоресцирующие в этих условиях, т. е. отдающие обратно поглощенную
энергию в излучение без превращения в ион. Отсюда следует,
что полученные значения квантового выхода фотореакции
преуменьшены.
В заключение этого параграфа рассмотрим вопрос об
энергии, требующейся для отрыва электрона ог растворенной
молекулы или иона органического соединения, и о том, в какой
мере эта энергия доставляется поглощаемым фотонам. Общая
схема вычисления величины этой энергии должна быть построена
аналогично рассмотренной выше (29) схеме процесса
фотоокисления растворенных неорганических ионов и атомов.
Однако, в отличие от небольших по своим размерам
неорганических ионов, теплота гидратации которых значительна,
энергия гидратации исходной молекулы или иона
органического соединения не превышает 0.5 э. вольта и обусловлена,
повиднмому, образованием водородных связей с
определенными атомными группами соединения170. Теплота гидратации
больших молекулярных нонов красителей также должна быть
невелика, поскольку „резонанс" валентных структур
распределяет заряд иона на большой объем, а следовательно, диполи
воды находятся в слабом электростатическом поле.1)
Отсюда следует, что, в отличие от атомов и
неорганических катионов, теплота гидратации ие может значительно
1) Ослабление электростатического поля ионов красителей явствует
из того факта, что они легко димеризуются, а также присоединяют
протоны, несмотря иа отталкивание одноименных зарядов (ср. стр. 229).
208 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
снизить энергию отрыва электрона от нейтральных и
заряженных молекул органических соединений.
Незнание теплот гидратации можно обойти,
воспользовавшись, по примеру иона Fe~*~+f значением стандартного окисли-
тельио-восстановительного потенциала <£°, если лейко-форма
красителя способна к обратимой реакции:
Допустим, что механизм фотореакции первичного отрыва
электрона здесь аналогичен механизму (II) предыдущего
параграфа и что энергия фотона, требующегося для этого
процесса, может быть вычислена по приведенному там
уравнению (2), в котором опускаем величину х в предположении,
что перенос электрона к молекуле Н2О не сказывается на
потенциальной энергии гидратной оболочки, имеющей равновесное
значение (ср. стр» 199). Уравнение принимает вид:
Ь = /ВА —/1ЬО = £°ЛЛ -4-3.8 — llh0 э. вольт.
Поскольку величина /я 0> по оценке бр. Фаркас, составляет
около 0.8 э. вольта (18 ккал), то энергия отрыва электрона
-от молекулы НА в водной среде будет, примерно, на 1 э. вольт
меньше стандартного редокс-потенциала /НА данного
соединения в абсолютной шкале. Для дифениламина /НА=4.5 э. вольт.
В таком случае в среде, обладающей сродством к электрону,
близким /„ 0, Av»3.5 э. вольт, что по формулам пересчета,
приведенным в 2, соответствует 350 та в согласии с опытом.1)
Значительный интерес представило бы определение
спектральной кривой скорости фотоокисления под действием
различных длин волн и сравнение ее со спектром поглощения
исходного лейкосоединения.
31. Фотоокисление красителей в присутствии акцепторов
электрона
Красители некоторых классов, в частности красители,
вызывающие так называемый фотодина to ический эффект
{34), поглощая видимый свет, действуют как восстановители,
т. е. доноры электрона по отношению к присутствующим
в растворе неорганическим катионам или другим окислителям.
Эта фотореакция наблюдается, например, в водном растворе
1) Роль акцептора электрона в смеси растворителей, цримеаяемой
Льюисом, играл, вероятно, эфирный атом кислорода 1(*8.
31. ФОТООКИСЛЕНИЕ КРАСИТЕЛЕЙ АКЦЕПТОРАМИ ЭЛЕКТРОНА 209
красителя в присутствии гидратированных ионов серебра
А£+ (из Ag- NO3)171.
Первичный механизм реакции состоит в переносе
электрона от фотоактивированной молекулы красителя А* или А"
к иону — акцептору электрона с выделением металлического
серебра:
A*v А+ —> °А -+- А£. (I)
Двойной значок** имеет тот смысл, что реакция
приписывается нами не только возбужденной, но также бирадикальной
форме Av молекулы красителя, в которую она переходит
путем внутренней конверсии (стр. 80). Основания для этого
допущения приводятся ниже (стр. 223). Значок ° обозначает
отсутствие электрона (28).
К красителям, обнаруживающим эту фотореакцию,
принадлежат: эозин, флуоресцеин, сафранин G, хромотроп 2R,
метиловый фиолетовый и ряд других. Малахитовый зеленый
действует очень слабо.
Большинство этих красителей в результате отдачи электрона
необратимо выцветает даже в отсутствии кислорода. Родамин
В и феносафранин, выделяя серебро, тем не менее сохраняют
первоначальную окраску. Для остальных красителей первичный
акт развивается дальше, приводя к структурному изменению
красителя.
При длительном освещении (свыше 10 часов) количество
выделившегося серебра перестает возрастать, стремясь к
пределу, что указывает на существование обратной
компенсирующей темновоЙ реакции, состоящей, повидимому, в процессе:
Ag- -+- ° А —> А%+ -к А. (II)
Присутствие кислорода воздуха оказывает
замедляющее действие на фотореакцию выцветания и параллельно
на выделение серебра. Особо значителен этот эффект для
сафранина и хромотропа. Флуоресценция сафранина, а также
некоторых других фотоактивных красителей (эозин), не тушится
кислородом. Следовательно, тормозящее действие кислорода
не может быть приписано дезактивации возбужденных
молекул красителя А* (ср. стр. 124). Торможение кислородом
установлено и для других типов окислительных фотореакций
(157). Как и там, в рассматриваемой здесь фотореакции в
присутствии кислорода появляются перекисные соединения
красителя, обнаруживаемые аналитически. Естественно
предположить, что эти стабильные фотоокснды являются побочными
продуктами дезактивации кислородом фотоактивированной
молекулы, как это изображено на схеме (III) и фиг. 30 в 23.
14 А. Н. Тиренни
2Ю ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Фотореакция выцветания хлорофилла в присутствии таких
окислителей как ион Fe+++, обратимая в темноте176, во всех
отношениях аналогична описанной выше и может быть
представлена следующей схемой69:
> или (III)
СЫ к Fe^+ ^ I 'Chi + FT- -+- Fe++.
Полуокисленная форма хлорофилла, обозначенная здесь
°СЫ или 'СЫ, имеет желтый цвет и в свою очередь
обнаруживает фоточувствительность69.
Хлорофилл испытывает также медленное окисление ионами
ре+++ в темноте| очевидно за счет термической активации
бирадикальной формы Chi .
Быстрое обратимое фотовыцветание раствора хлорофилла
в отсутствии окислителей Франк и Ливингстон69 приписывают
окисляющему действию нормальной молекулы на
фотоактивированную форму СЬГ по схеме:
СЬГ-+-СЫЧ
5 'СЫ -н НСЫ, (IV)
СЫ -чChK
причем получающийся здесь однократно дегидрированный
хлорофилл 'СЫ отличается от семихинона 'СЫ в схеме (III).
Торможение реакции кислородом этой, как вообще и других
окислительных реакций, эти авторы приписывают иной причине,
чем предполагаемая нами. А именно, торможение кислородом
объясняется ими побочной реакцией дегидрирования
семихинона НСЫ, регенерирующей хлорофилл (или краситель)
согласно механизму:
НСЫ -+- О2 > СЫ -н НО2; (V)
НО2 -+- 'СЫ —> СЫ -+- О2 (VI)
Второй этап в данной схеме, допускающий у радикала
НО2 наличие восстанавливающей (гидрирующей) способности,
мало правдоподобен, поскольку этот радикал обладает высокой
окисляющей способностью (стр. 202). Образование же
перекиси водорода не наблюдается, а кроме того, оно
привело бы к деструктивному выцветанию хлорофилла.
Реакция фотоокисления активного красителя с
восстановлением присутствующего в растворе окислителя имеет также.
31. ФОТООКИСЛЕНИЕ КРАСИТЕЛЕЙ АКЦЕПТОРАМИ ЭЛЕКТРОНА 211
место, когда акцептором электрона (с протоном) является
другой краситель172. Так светостойкие красители различных
классов (азокрасители, антрахиноновые трифенилметановые
и др.) тем не менее выцветают путем восстановления
(гидрирования), когда в освещаемом водном растворе,
освобожденном от воздуха, добавляется эритрозин или флуоресцеин.
Последние принадлежат к „фотодинамически" активным
красителям (34)« При впуске воздуха первоначальная окраска
выцветшего красителя обычно вновь обратимо появляется,
что и указывает на то, что продукт выцветания представляет
собой лейко-форму исходного красителя.1) Сам эритрозин или
флуоресцеин при этом необратимо окисляется, утрачивая
флуоресценцию.
Фотореакцию можно, таким образом, представить
следующей условной схемой:
Э^ + Азо +--1 О0Э -I- Н2Азо, (VII)
в которой Э обозначает фотоактивированный анион эритро-
зина, Азо — катион азокрасителя; в результате реакции
образуется потерявший два электрона анион эритрозина и
гидрированная, т. е. лейко-форма азокрасителя.
При длительном освещении наступает, как и в случае
восстановления ионов серебра, явление насыщения, связанное,
повидимому, с торможением продуктами реакции.2)
Однако из испробованных многочисленных комбинаций
пар красителей реакция фотогидрирования типа (VII)
наблюдается лишь у немногих172. Так оказались неактивными эозин,
родамин В, цианин В, желтые красители (аурамин), а также
соединения, поглощающие в ультрафиолетовой области (хинин,
аскулин). С другой стороны легко и обрнтимо
восстанавливаемые (гидрируемые) кубовые красители, метиленовый голубой
и сафранич не фотогидрируются аритроэином и не выцветают
в его присутствии. Трудно сказать, в какой мере имеет
значение относительное положение уровней редокс-потенциала
этих сочетаний красителей, а в какой мере близость спектров
поглощения, на которую обратил внимание автор описываемого
исследования. А именно, оказывается фотореакция типа (VII)
наблюдается почти всегда, когда полоса поглощения активного
2) Азокрасители, например фиолетовый Виктории 4BS, необратимо
распадающиеся иа амины, при восстановлении не регенерируют при впуске
воздуха.
2) Следует отметить, что поскольку смесь красители подвергалась
освещению нераэложенным светом на широком интервале, то активации
подвергался одновременно и акцептор электрона (водорода) (ср. 32).
14*
212
ФОТООКИСЛЕНИЕ И ФОТОВОССТАЙОВЛЕНЙЁ
фотоокисляющегося красителя (эрнтрознна)
непосредственно прилегает со стороны коротких длин волн к полосе
поглощения выцветающего красителя.
Фотосенсибилизоваииаи красителями реакция
восстановления иоиов. Описанная в предыдущем разделе реакция
фотоокисления красителя, сопровождаю ^цаяся восстановлением
ионов Ag-+ нли других красителей, протекает во много раз
быстрее и эффективнее, если в растворе присутствует в
качестве третьего участника легко отщепляющее родород, т. е.
Время
2 * 6 д Ю 12 !*> )В 18 20 222*26 28 30 в часа*
Фиг. 37. Реакция восстановления ионов серебра,
фотосененбнлиэованная эритрозином в присутствии
восстановителей и без иих. Л — без
восстановители в Н^О; В — с бензидииоы; С — с глюкозой.
Легко окисляющееся органическое соединение: бензидин,
глицерин, гликокол, глюкоза, сахар (фиг. 37)172. Следует заметить,
что без фотоактивного красителя освещение бинарной смеси
указанных восстановителей с акцептором электрона (водорода)
не приводит к восстановлению или выцветанию последнего.
Таким образом, активный краситель служит
фотосенсибилизатором реакции восстановления, не идущей без него.
Фотоактивный краситель в присутствии указанных
восстановителей практически не выцветает, разрушаясь необратимо в
значительно меньшей степени, чем без них. Как и в предыдущем
случае, опыт должен проводиться с раствором, освобожденным
От воздуха, во избежание обратного Окисления кислородом
образующейся лейко-формы азокрасителя, приводящего к
торможению фотореакции, а также во избежание медленного
31. ФОТООКИСЛЕНИЕ КРАСИТЕЛЕЙ АКЦЕПТОРАМИ ЭЛЕКТРОНА 213
побочного процесса деструктивного фотооксидирования
(глава 7). *)
В предыдущем разделе было показано, что реакция
восстановления акцептора имеет место и без постороннего
органического соединения восстановителя и состоит в
фотоокислении активного красителя. Упомянутое выше относительное
расположение полос поглощения активного и гидрируемого
красителя, благоприятствующее фотореакции, сохраняет свое
значение и здесь. Очевидно, ускорение фотореакции,
наблюдаемое в присутствии постороннего восстановителя „Y, имеет
свою причину в том, что потерявший электрон фотоокислен-
ный красвтель немедленно обратимо регенерируется
восстановителем HY и вновь вступает в фотореакцию. Внешне
же получается впечатление, что фотосенснбилизатор облегчает
перенос электрона или атома водорода от восстановителя HY
к акцепторам: иону серебра, красителю или молекуле киело-
рода. Такая концепция легла в основу теории сенсибилизации
красителями фотолиза бромистого серебра, предложенной
Кбгелем под названием теории дегидрирования-гидрирования
(стр. 294). По аналогии с теорией окисления Виланда (стр. 238)
Когель предполагает, что первичный процесс состоит
не в фотоокисления красителя, как было сказано выше,
а в его фотовосстановлении веществом nY.
Предполагается при этом, что поглотивший свет сенсибилизатор
„активирует" лабильный атом водорода в соединении „Y путем
передачи энергии.2)
*) Отметим, что тормозящее действие кислорода при трехкомпонентной
системе меньше сказывается на фотореакции с участием иона серебра.
2) Когель и Штейгчан17"3 обнаружили явление, названное ими „эффект
метнленового голубого", которое заключается в том, что экспонированные
места желатиновой пленки, окрашенной этим красителем, получают
способность выделить серебро нэ раствора AgNC>3, т. е. восстанавливать ионы
Ag~*~. По общепринятым представлениям метилеиовы i голубой при
поглощении света испытывает реакцию фотовосстаноэления самыми
разнообразными восстановителями, в данном случае — желатиной, превращаясь в свою
лейко-форму (32). В данном опыте, когда выцветания не было заметно,
„скрытое" изображение состояло, повидимому, из полувосстановленной
формы красителя, т. е. семнхинона, обладающего, как всикий такой
радикал, свойствами восстановителя. Аналогично метиленовому голубому ведут
себн многие красители: трипафлавин, тиазины, оксазины, аэины н даже
тряфеиилметановые (кристаллический фиолетовый, этилфиолетовый, метил-
фиолетовый, роэоловая кислота), а также хиион-диааиды—
светочувствительные соединения, использованные КЬгелем для получения
фотографических отпечатков.
На основании этих опытов Когель и предложил свою теорию гидри-
роваиия-дегидрирования. Однако подобного эффекта не дают
„фотодинамические" красители: эозин, эритрозин, феносафрании, родамин В, а также
пинахром, пннафлавол и хлорофилл, к числу которых принадлежат тем
214 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Однако, поскольку та же реакция восстановления иона
Ag1"1" или другого красителя имеет место и без постороннего
восстановителя за счет необратимого разрушения
фотоактивного красителя, то последовательность этапов правильнее,
повидимому, передается следующей условной схемой:
Азо —% оо Э -+- Н2Азо
Второй этап, как всякий процесс восстановления, протекает
в несколько ступеней переноса электрона и протона, быстро
следующих одна за другой (см. 28).1)
Для некоторых из перечисленных красителей-сенсиб :ли-
заторов все же не исключена возможность, что на первом
этапе имеет место не фотоокисление сенсибилизатора
акцептором, а фотогидрирование сенсибилизатора А
восстановителем с образованием семихинона НА, переносящего затем
атом водорода акцептору (см. э2—34). Вопрос требует более
тщательного изучения.
Интересно, что ту же реакцию восстановления ионов серебра
или красителей весьма эффективно сенсибилизует также
кристаллическое твердое тело — окись цинка под действием
поглощаемой ею близкой ультрафиолетовой радиации172 (41).
Подобие фотохимического поведения столь различных по своей
природе сенсибилизаторов, как кристалл — полупроводник
и ион растворенного красителя, наводит на мысль об
общности первичного механизма в обоих случаях, состоящего,
очевидно, в переносе электрона.
В 1918 г. Баурm выдвинул своеобразную
электрохимическую теорию фотосенсибилизации. Он предположил, что погло-
ие менее фотоСенеибилизаторы рассматриваемой в настоящем параграфе
реакции.
Следует, однако, заметить, что „эффект метиленового голубого"
по Шмидту 1?3 в более или менее выраженной степени дают все красители,
как кислотные, так н основные, фотографические сенсибилизаторы или
десенсибилиэаторы. Это означает, что красители всех классов обладают
способностью фотовосстанавлнваться в присутствии соответствующего
восстановители (32).
г) Аналогичная трактовка фотосеисибилиэованиого красителем
восстановления катионов серебра в растворе осложняется тем обстоятельством,
что краситель — сенсибилизатор часто не индифферентен по отношению
к ним: кислотные красители, как эритроэин, образуют с Ag"*" нерастворимые
з воде соли, а основные красители, в частности хииолиновые, образуют
с иовами серебра растворимые комплексы, постепенно распадающиеся
-г выделением серебра, без действия света.
31. ФОТООКИСЛЕНИЕ КРАСИТЕЛЕЙ АКЦЕПТОРАМИ ЭЛЕКТРОНА 215
тившая свет молекула сенсибилизатора А (краситель, ZnO)
испытывает пространственное разделение заряда с
образованием двух полюсов © и © согласно схеме:
или А{||. (I)
Такое состояние молекулы А Баур назвал фототроп-
ным и рассматривал его как своего рода электрохимическую
ячейку молекулярных размеров. Сходство продуктов фото-
сенсибилизованных реакций окисления-восстановления с
продуктами электролиза побудило Баура предположить, что
процессы обмена электронами, происходящие на полюсах фото-
тропной молекулы, тождественны с теми, которые имеют место
на электродах. Катодные деполяризаторы, т. е. соединен
ния с окисляющей функцией X, как и при электролизе,
воспринимают электрон от отрицательного полюса фототроп-
ной молекулы. Легко окисляемые соединения Y, служащие
анодными деполяризаторами и являющиеся по своей природе
восстановителями, по Бауру отдают электроны положительному
полюсу фототропной молекулы.
Весь процесс фогосенсибилизации изобразится в наших
обозначениях схемой:
/\
(9-ьХ->вХ-
А Л \ <">
(
Роль фотосенсибилизатора заключается, следовательно,
в облегчении переноса электрона от Y к X, совершающегося
путем двух актов обмена. По Бауру последние происходят
одновременно. Но одновременная встреча трех частиц за время
фототропного, т. е. активированного состояния молекулы мала,
а кроме того, пространственно они будут мешать друг другу.
Достаточно принять, что два этапа переноса электрона
совершаются последовательно во времени, причем на первом
этапе образуется преходящая полу окисленная или
полувосстановленная форма молекулы сенсибилизатора А. Процесс
завершается переносом протона от Y+ к восстановленному
окислителю Х~, как изображено в (II). В итоге от Y к X
переносится атом Н через посредство фотоакти^ ированного
сенсибилизатора. Порядок двух этапов безразличен.
В основе теории Баура лежит правильная мысль, что
поглотивший фотон краситель способен либо отдать, либо принять
216 ФОТООКИСЛЕНИЕ И ФОТОВО ССТАНОВЛЕНИЕ
электрон, в зависимости от партнера. Протекание реакции
в том или другом направлении, как мы видели выше,
определяется относительным положением уровней электрона
в участниках, которое может быть в некоторых случаях
получено из значений редокс-потенциалов соответствующих
систем.
Теория Баура в свое время не встретила широкого
признания, так как казалась примитивной и не укладывалась в
общепринятое представление о возбужденном состоянии атома
по теории Бора. Однако через 15—20 лет, совсем недавно^
она возродилась в модернизованной форме в концепции
Вебера (1935) и Вэйсса (1936) об электронном механизме
фотореакций красителей в растворах, о чем речь ниже.
32. Фотовосстановление красителей в присутствии
доноров электронов
Реакция обратимого переноса электронов и протонов между
лейко-формой и красителем по суммарной схеме:
А-нН2А ^Н2
характеризуется, подобно окислительно-восстановительной
реакции (IX) параграфа 28, определенным значением редокс-
потенциала, значения которого в абсолютной шкале приведены
в фиг. 32. Чем ниже расположен этот энергетический уровень,
тем больше энергия отрыва электрона от лейко-формы
Н2А и, следовательно, тождественная ей энергия
присоединения электрона к молекуле А (иону) красителя.
Действительно, азины, оксазины, тиазины, индофенолы, индамины,
антрахиноны и некоторые другие кубовые красители легко
ипритом обратимо восстанавливаются различными
химическими агентами в том случае, когда уровень электрона в
последних расположен выше вакантного уровня электрона в
молекуле красителя. Положение уровней и дается абсолютными
значениями редокс-потеициала.1)
В том случае, когда электронный уровень восстановителя
лежит ниже вакантного уровня красителя, реакция
восстановления итти не может, так как электрон прочнее связан
в первом, чем он будет связан в красителе. Опыт показывает^
1) Определение значений редокс-потенциалов для кубовых красителей
дало возможность получить некоторые сведения об нх структуре и езди-
1 v«Br
ствах
32. ФОТОВОССТАНОВЛЕНИЕ КРАСИТЕЛЕЙ ДОНОРАМИ ЭЛЕКТРОНОВ 217
что поглощение фотона видимого света указанными выше
красителями, достаточного для восполнения дефицита энергии
А£* между этими уровнями (ср. фиг. 32), делает возможным
восприятие электрона красителем и реакцию его
восстановления.
Действительно, Гебгард (1909) уже давно установил, что
метиленовый голубой выцветает в вакуумных условиях при
освещении видимым светом в результате реакции
восстановления и обратимо принимает первоначальную окраску в темноте.
Лазарев (1912), позднее, изучил кинетику обратимого выцвета-
ння коллодионной пленки, окрашенной метиленовым голубым,1)
hv
У
Фиг. 38. Образование вакантного электронного
уровни при поглощении красителем фотона.
—О— пустующие уровни; —#— заполненные
уровни. Левый рисунок относится к состоянию
до поглощения фотона, правый — сразу после его
поглощения; прерывистая стрелка обозначает
переход электрона на освободившееся место I
Такую же реакцию в слабой степени обнаруживают
малахитовый зеленый и ксантеновые красители. Возвращение
первоначальной окраски требует в некоторых случаях присутствия
кислорода, регенерирующего краситель путем окисления
образовавшейся его бесцветной лейко-формы.
В результате акта поглощения света, очевидно, сродство
красителя А к электрону увеличивается, т. е. вакантный
уровень красителя опускается вниз на величину анергии
фотона и оказывается тем самым ниже заполненного
электроном уровня восстановителя, как показано на фиг. 38.
Сущность этого необычного действия света2) заключается
в том, что фотон выводит электрон из прочно связанной
*) Необратимое изменение цвета метиленового голубого,
наблюдавшееся в этих опытах, повидимому, выввано химической активностью
нитроклетчатки.
2) Мы привыкли к картине повышения энергетического уровня при
поглощении фотона, но следует помнить, что речь здесь идет о
вакантном уровне, а не о том уровне, на который переходит электрон.
218 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
квантовой орбиты оболочки красителя, освобождая тем самым
вакантное место для постороннего электрона.
Поведение кристалла во всех отношениях аналогично
поведению кристалла-полупроводника, который в
современной физике рассматривается как макромолекула огромных
размеров с обобществленными электронами.
Полупроводники разделяются на два типа по механизму
возникновения в них электропроводности под действием света
или тепловой энергии. В первом типе, обладающем, как
говорят, электронной проводимостью, поглощение фотона
переводит электрон на более высокий пустой уровень в „зоне"
проводимости, находясь в которой электрон свободно
перемещается по кристаллу, как если бы последний представлял
собой металл. С этого уровня электрон может быть совсем
удален из полупроводвика и притом более легко чем с
исходного уровня. Такому типу полупроводников соответствуют
фотоокисляющиеся красители, отдающие электрон
акцептору при освещении длинами волн в пределах видимой полосы
поглощения* По аналогии с электронными полупроводниками
можно допустить, что в фотоактивированном состоянии
электрон менее прочно связан в молекуле и имеет возможность
перемещения до ее „периферии". Незатронутые светом
остальные электроны красителя остаются прочно связанными и не
мигрируют в указанном смысле.
Во втором типе полупроводников поглощение фотона
также переводит электрон на более высокий энергетический
уровень, но электрон в этом состоянии продолжает быть
прочно закрепленным, освобождая вакантное место иа
исходном, более низком уровне. Способностью мигрировать в
кристалле обладает теперь не перемещенный светом электрон,
а его пустое место или „дырка" в электронной оболочке,
имеющая избыточный положительны й заряд. Механизм
проводимости в таком, как говорят, „дырочном" или
дефектном полупроводнике состоит в том, что вакантная орбита
заполняется электроном из другого близлежащего нижнего
уровня (или орбиты), а получившееся новое пустое место
заполняется в свою очередь с соседнего уровня и так далее,
вызывая сравнительно свободное перемещение электронного
дефекта по кристаллу. Этому типу полупроводников,
несомненно, аналогичны фотовосстанавливающиеся
красители, рассматриваемые в этом параграфе. Подвижностью в них
обладает не электрон, а электронный „дефект", достигающий
за *время фотоактивированного состояния периферии
электронной оболочки красителя и нейтрализуемый посторонним
электроном. В шкале вакантных уровней, изображенных справа
32. ФОТОВОССТАНОВЛЕНИЕ КРАСИТЕЛЕЙ ДОНОРАМИ ЭЛЕКТРОНОВ 219
на фиг. 33, поглощение фотона равносильно увеличению
„сродства" красителя к электрону, т. е. снижению вакантного
уровня, как и изображено здесь на фиг. 38.1) ^
Простейшим примером реакции фотовосстановления является
обратимое выцветание водного подкисленного раствора тионина
или метиленового голубого в присутствии ионов железа под
действием поглощаемого красителем видимого света17Ь.
Суммарное протекание процесса изобразится механизмом:
A*v -ь Fe-*-* > А~ -+- Fe
H-jj-H- (I)
H A -t- H A ^z:± H2 A +- A
семихинон лейко-форма
Фактическое протекание реакции значительно сложнее, как
показал Рабинович 177.
Из фиг. 38 следует, что для возможности реакции
восстановления красителя А, обладающего пустующим уровнем,
расположенным выше заполненного уровня восстановителя Y,
<= <? 178,179.
должно быть соблюдено условие
^>JY-lA, , (1)
где Jr и Je дают глубину расположения энергетического
уровня электрона соответственно в частицах Y и €А.2)
Перенос электрона должен сопровождаться мгновенным
изменением исходного состояния участников и взаимодействия
их с растворителем. Следовательно, в первый момент система
двух обменявшихся электроном растворенных частиц обладает
энергией, отличающейся от той, какую она принимает,
позднее, при новой равновесной конфигурации всех
участников. Поэтому здесь, как и в случае фотоокисления
неорганических ионов (29), в балансе энергии мгновенного акта пере-
1) Остается открытым вопрос, происходит ли смещение вакантного
уровня вниз иа полную Энергию поглощенного красителем фотона, как
допускается Вебером178 н Вейссом1™, илн только на часть его.
Все красители, способные восстанавливаться в темноте, обладают,
очевидно, свободной „ячейкой" (6) для электрона уже в нормальном состоянии
молекулы Поглощение фотона облегчает реакцию восстановления тем, что
уводит од ш из электронов, мешающих приему нового.
2) См. правую часть фиг. 32 с тем отличием, что здесь уровень A + Y
расположен ниже уровня йА-н°У и разность энергий компенсируется
фотоном.
220 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
носа будет фигурировать неопределенная величина х удаления
энергии системы от равновесного значения н (1) перепишется
в виде:
J J . (2)
Энергии ]Y н/в , как мы знаем, представляют собой редокс-
потенциалы обратимых окислительно-восстановительных
процессов:
Ач-'А ^> <Ан-~А, (И)
12 12
которые либо наблюдаются на опыте, либо могут быть
подсчитаны нэ косвенных энергетических данных.
Следовательно, чем ближе редокс-потенциалы
восстановителя "Y и красителя А, тем более вероятно, что энергия
поглощаемого красителем фотона будет достаточна для
осуществления реакции восстановления согласно фиг. 38179.
Если восстановитель представляет собой органическое
соединение, то вместе с электроном, как мы знаем,
переносится от полуокисленного восстановителя °Y также протон
и происходит в целом присоединение атома водорода
к красителю, с превращением его в преходящую форму
семихинона НА (стр. 185). Завершение реакции
восстановления осуществляется переносом второго атома Н от
восстановителя или его полуокисленной формы, или от другой
молекулы семихинона красителя. Реакция обратимого
фотовосстановления протекает, следовательно, по суммарной схеме69.
Н2А -+-н Y (
VlA-+-"Y (Щ
"а' I I . НД
Н2А-нА
Обесцвеченный краситель Н2А вновь принимает
первоначальную окраску в темноте. Поэтому в схеме (III) фигурируют
обратные реакции, приводящие к исходным соединениям.
Быстрая реакция выцветания и медленное возвращение окраски
в темноте, наблюдаемые для метиленового голубого в
присутствии восстановителя тиозинамина (аллилтиомочевины),
указывают на быстрое протекание прямых реакций а и Ь, и
медленное— реакций b и а'в обратном направлении69.
32. ФОТОВОССТАНОВЛЕНИЕ КРАСИТЕЛЕЙ ДОНОРАМИ ЭЛЕКТРОНОВ 221
Реакцию обратимого фотовосстановления обнаруживают
не только катионные красители, указанные в начале параграфа,
но, в слабой степени, и анионные красители, как эозин, эритро-
зин, ураиин и другие ксантеновые красители, даже в
присутствии такого слабого восстановителя, как ион J~~.
Одноименный знак заряда ионов не существенен для возможности
протекания реакции (ср. стр. 229). Полагают, что слабости
эффекта с ксантеновыми красителями вызвана быстрой
обратной реакцией а'.1) -
Реакция (с) диспропорционирования или дисмутации семи-
хинонов НА требует большой их концентрации, а потому
ею можно практически пренебречь. Только очень слабое
и притом необратимое фотовосстановление хлорофилла может
быть приписано этой реакции.
В отсутствии кислорода фотовоссгановление делается
необратимым, если окисленный восстановитель (HY или "Y
в схеме III) по своей структуре неспособен к обратному
присоединению водорода. К таким веществам относятся
многоатомные спирты (маннит, гликоль, глицерин), амнны,
органические кислоты и такие вещества, как тиомочевина и ее
производные. Без ноздуха получившаяся в результате освещения
таких систем бесцветная лейко-форма красителя сохраняется
неопределенно долгое время. При допуске воздуха
первоначальная окраска более или менее быстро регенерирует, так как
кислород дегидрирует лейко-форму, возрождая краситель,
который вновь вступает в фотореакцию с восстановителем.
Эта реакция рассматривается ниже (34) под названием фото-
сенсибилизованного окисления в присутствии кислорода
(фотодинамическое действие красителя).
Большинство восстанавливающих агентов способно
действовать на краситель и в темноте, но некоторые органические
соединения, как тиозинамин (аллилтиомочевина), вступают
в реакцию только при освещении.2)
Без доступа воздуха метиле новый голубой испытывает
реакцию фотовосстановления также под действием ультра-
*) Ливингстон 189 показал, что для обратимости реакции эозина с ионом
иода необходимо присутствие кислорода (см. 33).
2) Веществам, приводящим к быстрому выцветавию красителей в
результате фотовосстановления, в фотографической литературе иногда
присваивают неудачное название „сенсибилизаторов" или „акцепторов"
выцветания 18°. Требующаяся для ускорении выцветании концентрация этих веществ
иногда в несколько десятков раз превышает концентрацию красителя
и фактически они являются средой, непосредственно участвующей
в фотореакции. Выцветание красителей путем фотовосстановления есть
один из приемов цветной фотографии, не нашедший широкого практического
применении иэ-ва малой светочувствительности 18°.
222 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
фиолетового света, поглощаемого во второй полосе
поглощения красителя Х^=290 m\L18lu Реакция наблюдается в
подкисленном спиртовом растворе и имеет ничтожный квантовый выход
у да 1:6000, что объясняется отчасти тем, что
ультрафиолетовый свет действует также на лейко-форму, регенерируя
краситель (30). В видимом свете квантовый выход
фотовосстановления метиленового голубого в тех же условиях
значительно больше у = 1:35. Спиртовый раствор способствует,,
очевидно, преобладанию мономерной формы красителя (14).
В присутстаии катионов Fe"1"*" фотореакция обратимого
выцветания происходит значительно быстрее, причем устанавливается
равновесие между различными формами участников.
При наличии кислорода освещение раствора метиленовога
голубого ультрафиолетовым светом XX < 270 т}). приводит
к необратимому выцветанию182, очевидно, вследствие
фотооксидирования.
Нитрокраснтели (моно-, ди-, тринитрофенолы и нитроамины),.
а также бесцветные нитросоединения испытывают под
действием поглощаемого ими света также реакцию восстановления,
но иного — необратимого типа118. Продукты имеют
коричневую окраску и представляют собой конечный результат
фотореакции отнятия кислорода от нитрогрупп,
сопровождающегося сочетанием двух получившихся остатков в азокси-
соединение, переходящее затем в окси-азоформу. Так,
например, для пикриновой кислоты имеют место следующие стадии,
каждая из которых также обладает светочувствительностью:
ОН
ОН—>
Реакцию фотовосстановления испытывают не только
красители, но также более простые соединения под действием
поглощаемого ими ультрафиолетового света. Так,
ароматические кетоны (ацетофенон, антрахинон), а также хинон,
фотс?гидрируются в присутствии спиртов183. „Лабильным",,
т. е. гидрирующим атомом водорода в спиртах оказался
32. ФОТОВОССТАНОВЛЕНИЕ КРАСИТЕЛЕЙ ДОНОРАМИ ЭЛЕКТРОНОВ 223
не гидроксильный водород, а атом Н в группе —СН2
—ближайшей к карбонильной группе (так называемое а-положение).
Световое окислительно-восстановительное равновесие.
Большой интерес представляют такие фотореакции, когда
восстановитель H2Y, потерявший атомы водорода при
фотовосстановлении красителя А, способен в свою очередь под
действием света в собственной полосе поглощения фотовос-
станавливаться лейко-формой красителя согласно обратимой
световой реакции:
А
А + H2Y ^г± Н2А н-Y .
2
В действительности реакция протекает, конечно, через
промежуточные стадии семихинонов, как было разъяснено выше.
Под действием света, поглощаемого красителем, происходит
его обесцвечивание с появлением окисленной
(дегидрированной) формы Y соединения H2Y. Если теперь систему освещать
светом, поглощаемым Y, то реакция течет в обратном
направлении и краситель регенерирует. Приведем пример такого
смещения равновесия действием света, тщательно изученный
Холстом ш.
В качестве красителя А был взят метиленовый голубой,
катион которого мы обозначим Mb"*", а лейко-форму— Н2МЬ+.
Восстановителем H2Y служил анион сульфофеннлгидраэина:
Ph—NH—NH — SO3~i представляющий собой лейко-форму
окрашен ног о аннона сульфодиазобензола: Ph — N = N — SO3~,
имеющего желтый цвет. Для краткости последние ио^ы
обозначим -N2H2 -]~~ и, соответственно, -N2 -]~.
При освещений ультрафиолетовым светом в области
максимума поглощения, лежащего у 280—285 ту* (край полосы —
330 та), лейко-форма -N2H2-]~ выделяет газообразный
водород из кислого водного раствора (рН^=2 — 3), превращаясь
В отсутствии кислорода в окрашенный анион -N2-]~\
Механизм такой реакции фотодегидрирования состоит,
очевидно, в процессе фотоокисления аниона -N2H2-]~ с разрядкой
ионов водорода, наподобие схемы (II') фотоокисления
неорганических анионов в 29, с тем отличием, что после отрыва
электрона анион здесь самопроизвольно освобождается также
от протона и второго атома водорода, как это имеет обычно
место для лейко-форм красителей (ср. 30).
Аналогичная реакция фотоокисления имеет место для
лейко-формы метиленового голубого Н2МЬ"+", которая фото-
дегидрируется (с регенерацией красителя) под действием
ультрафиолетового света на кислый водный раствор лейко-
224
ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
формы (рН = 1). Квантовый выход этой реакции у ==0.2—0.3
постоянен на широком протяжении полосы поглощения
(405—313 ту-)9 немного повышаясь в сторону коротких длин
волн. Спектры поглощения метиленового голубого н его
лейко-формы даны на фиг. 39.
Освобожденный от кислорода воздуха, слегка
подкисленный водный раствор, содержащий окрашенные ионы Mb"1",
вместе с бесцветными ионами -N2H2-]~~ при освещении
видимым светом в области 700—500 ту., поглощаемым
только Mb"*", обесцвечи-
667500400333 250 200 А ту влется> приобретая слабую
40 ■
/7 -
25 -
го
45 л желтую окраску,
принадлежащую ионам -N2-]—.
Очевидно, произошел процесс
фотовосстановления Mb"1" в
лейко-форму Н2МЬ+ за счет
нонов -NJ-I2-]~~, не
подвергавшихся действию света.
Квантовый выход этой
реакции у&ОЛ невелик. Если
выцветший раствор теперь
осветить ультрафиолетовым
светом в области длин волн
440—400 m\f.9 поглощаемых
ионами "N2-]~~, то
фотореакция идет в обратном
направлении в результате
фотовосстановления этих ионов
лейко-формой Н2МЬ"*~,
присутствующей в избытке.
Квантовый выход последней
реакции, измеренный при
избытке ионов -N2-]~~ над HgMb"*", равен у ^0.2. Появлению
окраски Mb"*" благоприятствует также фотоокислеяие его лейко-
формы, вызываемое тем же ультрафиолетовым светом.
Итоговый фэтопроцесс гидрирования-дегидрирования может
'быть записан следующей схемой:
ЮООО 20000 За000 4000D 50000ся
•Фиг. 39. Спектры поглощения
метиленового голубого (/) н его лейко-
формы (II) в растворе 0.1 НС1184.
Mb
-N2H2-]
i Н?МЬ-
-N.-I
На самом деле, еще до освещения, в растворе
устанавливается некоторое равновесие между 4 участвующими ионами
и свет лишь меняет состояние равновесия в ту или иную
33. ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 225
сторону. Если выключить свет после достижения стационарного
состояния, то система медленно возвращается в исходное
равновесное состояние.
Весьма интересно наблюдение Холста, что если выключить
свет до установления стационарного состояния, то реакция
продолжается в темноте в течение нескольких минут. Этот
факт непосредственно указывает на участие в реакции
длительно живущих активных форм, какими могут быть только
радикалы — семихиноны.
Кислород тормозит реакцию тем, что является более
сильным, чем Mb"*", акцептором водорода от аниона -N2Ha-]~~
(ср. следующий параграф).
Дальнейшее детальное исследование подобных обратимых
фотохимических систем представляет большой интерес
не только теоретического характера.
33. Фотосенсибилизованное красителями окисление
неорганических ионов
Рассмотренная в предыдущем параграфе реакция
обратимого фотовосстановлення красителя сопровождается,
естественно, окислением восстановителя. Однако в отсутствии
кислорода после прекращения освещения процесс идет нацело
в обратном направлении и система обратимо возвращается
в исходное состояние, как было описано выше. Иное
положение создается в присутствии растворенного кислорода. В этом
случае с восстановленной формой красителя вА или НА
реагирует не лишившийся электрона окисленный
восстановитель °Y, а молекула О2, являющаяся более сильным акцептором
электрона, а также атома водорода. Образующийся на первом
этапе ион О2~9г) присоединяя протон, превращается в НО^,
а затем—в перекись водорода185. Регенерированный в
результате этих реакций с кислородом краситель А вновь фотовос-
станавливается, окисляя тем самым новую молекулу
восстановителя Y и процесс развивается в том же направлении
дальше. Кислород прн этом непрерывно поглощается187. Таким
способом, действуя видимым светом на краситель, можно,
не изменяя его концентрации, фотохимически окислить
неопределенно большое количество неорганических
ионов-восстановителей, требующих для непосредственного
фотоокисления, как мы видели (29), больших фотонов ультрафиолетового
*) Существование свободного Газообрвэного иоиа О2 показано
опытами по прохождению пучка Электронов через разреженный кислород
в вакуумных условиях1^6.
15 А. Н. Т еренкя
226 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
света. Так, например, ионы J~~, требующие для
фотоокисления поглощения фотонов в области длин волн <С! 250 m\L
(\тах — 226 тир-), легко окисляются с выделением
молекулярного иода j2 в присутствии эозина при освещении раствора
иодида видимым светом, т. е. длинами волн на участке
полосы поглощения эозина (Хтм =518 т}*.)188. Поскольку
реакция окисления иона-восстановителя, требующая
большого фотона ультрафиолетового света, „очувствляется" в при-
сутстнии красителя к малому фотону видимого света, то весь
указанный процесс называют фотосенсибилизованным
окислением субстрата Y, в данном случае аниона иода.
Без кислорода выделения молекулярного иода не наблюдается,
так как атомарный иод успевает претерпеть обратную реакцию,
присоединяя электрон и регенерируя краситель. Схема
процесса выглядит следующим образом189:
А*'
/°' J, (IV)
Д | ■ О — Вторичные
2 реакции
но2
н2о2
В такую схему укладывается фотореакция окисления
ионов: Fe"*"1", J~, SO3 , AsO3 , оксалат , фотосенснби-
лизованная красителями: флуоресцеин (уранин), эозин, рода-
мины, бенгальская роза, нейтральный красный, феносафранин,
сафранин, метиленовый голубой и др.1) Детальному
кинетическому исследованию подвергалась реакция окисления ионов
иода, фотосенсибилизованная эозином189, н ионов
сульфита SO3 и арсенита AsO3 , сенсибилизованная эозином,
уранином, родамином190. Было давно замечено, что наиболее
значительный эффект фотосенсибилнзации дают, как правило,
флуоресцирующие красители, В табл.12 значения
квантового выхода флуоресценции <р сопоставлены с значениями
квантового выхода у фотосенсибилизованной реакции окисле-
J) Неорганический ион уранила UO2"**"*" также фотосенсибилизует
окисление указанных анионов. Фотохимическое поведение этого
флуоресцирующего иона во многих отношениях сходно с поведением флуоресцирующих
красителей 1В1.
33. ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ
227
ния иона иода, иона оксалата и тиозянамина (аллнлтиомоче-
вины) для различных красителей.
Т а бл н ца 12
(по Ливингетону, Хёрду, 189 1940)
Краситель
Квантовый
выход
флуоресценции 9
Квантовый выход фотосенси-
бнлизованного окксленвя y
оксалат"
тиозн-
намни
Флуоресцеии-Na (уранин)
Эовин
Бенгальская роза . . .
Нейтральный красный .
Феиосафранин
Сафранин
Метиленовый голубой .
Порфирнны ......
Эгил хлорофнллид . . .
0.8
0.2
0.02
^0.01
^0.01
<0.01
<0.01
о к* 0.1
0.03
1.0
1.0
0.14
0.13
0.18
0.15
З-10-з
<5-10-*
Из таблицы следует, что типичные флуоресцирующие
красители, действительно, дают наибольший выход фотореакции
окисления иона иода. Однако для слабо флуоресцирующих
красителей нет прямого соотношения между способностью
к флуоресценции и сенсибилизирующим действиям. Далее,
хотя окисляемый в реакции субстрат — восстановитель и тушит
флуоресценцию красителя-сенсибилизатора, никакой
непосредственной связи между реакцией и тушением не имеется, подобно
фотореакции присоединения кислорода (21, 22).
Данные, приведенные в таблице, показывают, что, как
правило, квантовый выход окисления значительно превосходит
выход флуоресценции ф (наблюдаемый в отсутствии реагентов),
например, в 5 раз в случае эозина и иона иода. Для этилхло-
рофиллида выход реакции окисления тиозинамина равен
единице, в то время как тушения флуоресценции тиозннамином
нет вовсе, и квантовый выход испускания сохраняет в
присутствии этого восстановителя свою первоначальную небольшую
величину в 8°/0 (ср. наблюдение Каутского на стр. 242).1)
г) Аналогичное несоответствие между тушением флуоресценции и
фотосенсибилизацией наблюдается также для иона уран ила в качестве фого-
сенсибилизатора окисления авиона оксалата. Так, добавление к этому
раствору ионов J~~ с концентрацией ок. 10~3 N полностью тушит
флуоресценцию иона уран ила? между тем скорость фотосенснбнлизованного
окисления иг изменяется.
15*
228 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
Кислород, как было упомянуто выше, вообще не тушит
флуоресценцию большинства указанных
красителей-сенсибилизаторов190 (стр. 170).
Из этого несоответствия следует, что на начальном этапе
фотореакции, изображенной выше (схема IV), используются
отнюдь не только возбужденные светом молекулы
красителя А*, способные к испусканию, но, главным образом,
те не испускающие, но активные молекулы А\ в которые они
преобразуются. Такую длительно живущую модификацию
молекулы мы отождествили с метастабильной бирадикальной формой
красителя (стр. 81). Для эозина, например, квантовый выход
флуоресценции в отсутствии ионов иода равен только 20°/о« Тем
не менее остальные ь0% поглощающих фотоны молекул
доступны для реакции, как показывает 1000/0-й квантовый
выход реакции окисления иона иода (см. табл. 12). В случае
флуоресцеина в реакции должны участвовать и возбужденные
молекулы, поскольку квантовый выход флуоресценции близок
единице (<р—0.8). Однако только аномально большая
длительность существования метастабильного состояния А ,
превосходящая в 108 раз длительность возбужденного
состояния, может обеспечить эффективную встречу с
окисляемым ионом иода и наблюдаемый высокий выход
фотореакции.
Тот факт, что только флуоресцирующие красители являются
наилучшими сенсибилизаторами, не противоречит этой
концепции. Как было указано на стр. 75, характерным для
флуоресцирующих красителей является жестко построенный скелет,
не допускающий непроизводительного растрачивания избытка
электронной энергии в деформацию и вибрации составляющих
атомных групп. В красителях с такой структурой имеются,
следовательно, налицо благоприятные условия для сохранения
значительной доли энергии поглощенного фотона также
и на уровне метастабильного состояния молекулы.
Обратимся снова к первому этапу всего процесса
(см. схему IV), заключающемуся в восстановлении
фотоактивированной молекулы Av или А* гидратированным
анионом Y = J~~ (Или катионом Fe"*"*"). Без
красителя-сенсибилизатора отрыв электрона от последнего, т. е. реакция прямого
фотоокисления аниона, требует, как мы видели (29), фотока
ультрафиолетового света в 1.5—2 раза большей величины,
чем подводимый здесь красителю фотон видимого света.
Возникает вопрос, за счет какого источника восполняется
здесь столь большой дефицит энергии, достигающий в случае
нона иода и эозина 70 кгкал. Следует при этом отметить,
что квантовый выход фотосенсибилизованного окисления даже
33. ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 229
в несколько раз больше прямого фотоокислйшя
ультрафиолетовым светом.
Допустим, по аналогии с рассмотренной в предыдущем
параграфе реакцией фотовосстановления, что и здесь
поглощение фотона видимого света, создавая вакантную орбиту,
увеличивает сродство красителя-сенсибилизатора к электрону
настолько, что энергетически делается возможным перенос
электрона на краситель от окисляемого иона. Однако
положение дела здесь не столь просто, так как краснтели-сенси-
бнлиэаторы не создают обратимого
окислительно-восстановительного равновесия со своими лейко-формами и не дают
определенного значения редокс-потенциала, по которому
можно было бы оценить величину исходного сродства этьх
красителей к электрону (32). В отличие от типичных легко
восстанавливающихся на свету и в темноте красителей,
фотосенсибилизаторы окисления представляют собой, большей
частью, ксаитеновые красители с двукратно заряженным
анионом, типичной реакцией которого является, наоборот,
фотоокисление (31, 32).1)
Приходятся здесь сослаться на выявленную Когелем также
и у ксантеновых красителей некоторую способность к реакции
фотовосстановления (см. примечание на стр. 213).
Возможны следующие экзоэнер! етические эффекты,
освобождающие энергию, способную восполни!ъ недостаток энергии
в красителе до значения, которое требуется для отрыва
электрона от окисляемого им аниона. Прием электрона анионом
ксантенового красителя сопровождается, во-первых, усилением
электростатического взаимодействия с гидратной оболочкой,
окружающей краситель, во-вторых, — резким изменением
электронного состояния молекулы красителя, всегда вызывающим,
как мы знаем, резкое изменение запаса вибрационной энергии
молекулы (10), Отличие заключается здесь в том, что
изменение электронного уровня происходит не путем поглощения
илн испускания фотона, а вследствие приема лишнего
электрона в электронную структуру фотоактивированной
молекулы.2) Вполне мыслимо, что при рассматриваемом акте
переноса происходит по этой причине повышение уровня
электронной энергии красителя за счет термической энергии
вибрация красителя, как это имеет место в явлении антистоксовой
флуоресценции (стр. 61). Как мы видели, этим путем воспол-
г) Одноименный заряд ие препятствует сближению ионов в растворе,
что следует, например, из того факта, что флуоресценция аниона эозина
сильно тушится анионами же, как например: J~, салицилат~ и фенолят".
2) Такой же эффект будет иметь место и при отдаче электрона
красителем, т. е. при элементарном окислении фотоактивированной молекулы
230 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
няется, как правило, недостаток энергии порядка только Юкгкал
(ок. 0.5 э. вольт). Более существенный источник энергии
появляется в том случае, если в результате приема
красителем нового электрона или его отдачи создается возможность
дополнительных валентных структур, „резонанс" которых
сопровождается выделением значительной энергии.
В результате всех этих эффектов принявшая электрон
восстановленная молекула красителя СА* или СА' 2) оказывается
как бы располагающей энергией, превосходящей в 1.5—2 раза
энергию поглощенного фотона видимого света, т. е. делается
способной осуществить реакцию фотоокисления постороннего
аниона, требующую без красителя фотоиа ультрафиолетового
света.
Приведенная здесь картина переноса электрона от
окисляемого аниона к фотоактивированному красителю не
предполагает иного участия окружающих молекул воды, как только
промежуточных пассивных передатчиков электрона.
Полувосстановленная форма красителя еА, возникшая на первом этапе
реакции, превращается в полугидрирэванную форму семихи-
нонж НА в результате вторичной реакции присоединения
протона от иона водорода, содержащегося в растворе.
Франк, Леви192 и Вейсс187 предложили иной механизм
появления семихинона НА. Оии предположили, что схема
Хабера-Франка для прямого фотоокисления гидратнроваиного
аниона Y~ (29, схема V) сохраняется и здесь для его фото-
сенсибилизованного окисления. А именно, по их мнению,
электрон, перенесенный на молекулу воды гидратной оболочки,
вызывает диссоциацию этой молекулы на Нч-ОН~, причем
атом водорода немедленно гидрирует краситель в семихинон
НА. Первый этап сенсибилизованной красителем фотореакции
выглядит согласно такому предположению следующим
образом:
A* -hH2O,Y" -^НА-ьУч-ОН-. (V)
Следовательно, в отличие от фотоокнсления аниона Y"
прямым действием ультрафиолетового фотона, величина которого
вычисляется по уравнению (5) в 29, теперь в балансе членов,
составляющих это уравнение, необходимо учитывать
выделение энергии связи Н — А, которая, по мнению авторов,
частично компенсирует недостающую энергию. Однако,
поскольку эта величина невелика и не может покрыть наблю-
*) Признание главной роли метастабильной формы молекулы красителя
увеличивает дефицит энергии, так как метастабильный уровень лежит ниже
возбужденного на 0,1—0.5 э. вольт (табл. 3 на стр 81),
33. ФОТОСШСИБИЛИЗОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 231
даемого дефицита, авторы полагают, что основным
источником компенсации недостающей энергии служит
одновременное выделение большей части энергии гидратации иона ОН",
а также энергии гидратации получившейся частицы Y, если
она заряжена (Fe"4"4""*", SO3~).
При акте прямого фотоокисления (29) изменение энергии
гидратации, как мы видели, в схеме Хабера-Франка не
учитывается вовсе на основании мгновенности акта поглощения
фотона. Здесь же, в фотосенсибилизованной реакции, авторы
допускают, что перенос электрона к активированному светом
красителю совершается постепенно, за достаточно длительное
время возбужденного состояния красителя, в плавном
(адиабатическом) процессе перестройки системы. В коице этого
процесса получившиеся частицы успевают разойтись и занять
новые положения, отвечающие новому минимуму энергии
системы, т. е, состоянию полной гидратации. Заметим, что
теплота гидратации иона ОН", равная W =57 кгкал
(стр. 193), может покрыть упомянутый выше дефицит в 60 кгкал
только на пределе. Подтверждением гипотезы Франка-Вейсса
было бы отсутствие реакции фотосенсибилизованного окисления
иона иода в абсолютном спирте, в котором сольватация иона
гидроксила освобождает существенно меньшую энергию,
а спектр „сродства" иона иода сохраняет свое положение.
Последовательность этапов фотосенсибилизованного
окисления неорганических ионов в присутствии кислорода
согласно гипотезе Франка-Вейсса изобразится следующей схемой
(в наших обозначениях)189:
А-4-Н2А
A*v -ь H2O,Y-
+НА
НА -+- НО,
\
ОН-ч-Y
й,/ н,о, + о, * |+v
/U Uho, Y2 (VI)
А -+- Н2О, Y" |
А-*-НО2
Y-*-OH~-+-H2O2
I
Эта схема удовлетворительно воспроизводит кинетические
закономерности, наблюдающиеся при окислении иона J" (Y")
232 ФОТО ОКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИ^
в присутствии эозина А в качестве фотосенсибилизатора.
В частности, воспроизводится пропорциональность квантового
выхода реакции у квадратному корню концентрации О2 и обрат-
з
ная пропорциональность у/, где /—интенсивность света. Как
было сказано вначале, первый этап (а) состоит в обратимом
восстановлении фотоактивированного красителя А* или А .
Кислород действует только вторично, дегидрируя семихинон
НА до наступления обратной реакции а! илн дегидрируя
лейко-форму красителя Н2А; если она успевает образоваться
при встрече двух семихинонов согласно обратимой реакции Ь.
В результате получается окисленная частица Y, которая
в случае нода рекомбинирует в молекулу Y2, а
регенерированный краситель вновь вступает в фотореакцню. Кислород
связывается первично в виде радикала НО2, обладающего
повышенными окислительными свойствами (стр. 202), который
эволюционирует далее, образуя перекись водорода,
распадающуюся на Н2О и О2, или же окисляет ион H2O,Y~.
Действительно, в таких опытах удавалось обнаружить образование
незначительного количества перекиси водорода, но перекиси
красителя-сенсибилизатора обнаружено не было. Последнее
обстоятельство, а также отсутствие тушения флуоресценции
красителя-сенсибилизатора кислородом рассматривается как
веский довод против участия кислорода в первом этапе
реакции.
Подтверждением приведенной выше схемы является
ускорение фотосенсибилизованной реакции (VI) ионами Fe~*~~*~
и Мп"1""*" (переносчиками кислорода), которые ускоряют тем-
новое окисление кислородом лейко-форм красителей.
Наблюдаемые на опыте в рассматриваемой здесь реакции
явления светового насыщения и темнового продолжения
реакции после прекращения освещения указывают иа длительное
существование промежуточных реакционноспособных форм,
которыми могут быть как,семихиноны, так и радикал НО2.
Выход реакции для уранина и эозина увеличивается
в щелочной среде и тормозится повышением концентрации
ионов водорода, что указывает на то, что
фотосенсибилизатором является именно анион этих красителей.
По нашему мнению, не исключена, в особенности для
ксантеновых красителей, склонных к окислению, возможность
того, что кислород участвует именно на первом этапе
реакции, который состоит не в фотовосстановлении, а в
фотоокислении кислородом. Полуокисленная форма красителя, т. е.
его семихинон, и является акцептором электрона от
неорганического аниона (субстрата). Начальные стадии в таком
33» ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 233
случае изобразятся следующей последовательностью
реакции:
2
\
У °А -+- О2" или 'А -ь НО2 (VII)
где °А обозначает краситель, лишенный электрона, а 'А —
лишенный атома Н. В остальном реакция развивается по
приведенной выше схеме (VI). Другими словами, мы
допускаем, что процесс протекает по типу фотосенсибилиэованной
красителем реакции восстановления, рассмотренной
в 31 (стр. 212), свойственной именно те\х же красителям,
какие активны и здесь. Роль восстанавливаемой частицы, т. е.
акцептора электрона (и протона) играет здесь молекула О2|
вместо иона Ag1**" в приведенном там примере.
Многозаряженным анионам ксантеновых красителей более естественно
отдать электрон, чем приобрести лишний. Потерявший
электрон краситель °А, приобретая его от постороннего аниона Y~,
выделяет значительную энергию „резонанса", так как
переходит в более стабильное состояние и может этим путем
компенсировать недостающую энергию, о чем была речь выше.
Действительно, в последней работе по окислению иона
J", фотосенсибилизованному эозином, Ливингстон (1942)18Э
для исчерпывающей трактовки кинетики реакции вынужден
допустить возможность и прямой реакции
фотоактивированного эозина с кислородом, сопровождающейся
дегидрированием красителя, и образованием НО2, как показано в нашей
схеме. Семихинон красителя 'А восстанавливается в
краситель А путем принятия атома Н от семихинона НА,
образующегося первично при реакции эозина с гидратированным
ионом J~ согласно первоначальной схеме (стр. 231), которая
Ливингстоном также сохраняется.
Вопрос заслуживает тщательного пересмотра в свете новых
данных, с использованием и неводных растворителей.
Исследователи этого вопроса уделяли недостаточное
внимание изменению состояния аггрегации красителя при изменении
растворителя (например, прибавлением спирта), а также не
учитывали одновременно идущие фотореакции иного типа,
которые для тех же красителей-сенсибилизаторов были
описаны в 20*
234 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
34. Фотосенсибилизованное красителями окисление
органических соединений в присутствии кислорода
(фотодииамическое действие)
Фотохимический процесс с участием красителя, в качестве
сенсибилизатора, во всех отношениях аналогичный
описанному в предыдущем параграфе, имеет место, когда окисляемый
субстрат представляет собой не анион, а органическое
соединение, которое мы обозначим "Y, где н указывает на
присутствие „лабильных" атомов водорода в структуре
соединения (ср. 28).
При освещении видимым светом раствора, содержащего
наряду с красителем данное соединение, происходит
поглощение газообразного кислорода, а органическое соединение
непрерывно необратимо окисляется путем дегидрирования
или присоединения кислорода (оксидирования)103. В отсутствии
кислорода происходит только реакция дегидрирования,
органического соединения, сопровождающаяся фотовосстановле-
иием красителя по типу процесса, описанного в 32. Эта
фотохимическая реакция в литературе рассматривается поэтому
заодно с предыдущей и обычно укладывается в приведенную
иа стр. 231 схему (VI).
Атом водорода, присоединяемый красителем, здесь
доставляется непосредственно органическим соединением
(восстановителем), без необходимости допущения распада молекулы
воды гидратиой оболочки по схеме (V), Развитие вторичных
реакций в основном считается тождественным с предыдущим.
Имеем, следовательно, такую общую картину процесса е9;
Л*' . HV YO ИЛИ Ч
Ан-НО
2
н2о2 -*- о
2о2 -*- о2
Некоторое отличие заключается в том, что, наряду с
образованием Н2О2, кислород может связываться с HY или Y,
триводя к продуктам необратимого окисления (оксидирования)
условно обозначенным YO (глава 7). Дегидрируемое исход-
toe органическое соединение HY носит в биохимии название
шбо „субстрата" окисления, либо „акцептора" кислорода.
34. ФОТОСЕНСИБИЛИЗЭВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 235
Акцепторами кислорода служат разнообразные органические
соединения, обладающие некоторой восстановительной
активностью, но не способные ее проявить без, подведения
дополнительной энергии, как, например, полиспирты, сахара,
органические кислоты, фенолы, алифатические амины, бензидин,
анетол, тиозинамин и ряд биологически важных объектов
(эргостерол, тирозин, гуанин), откуда делается понятным
интерес к этой реакции в биохимииш. В частвости,
освещаемые видимым светом красители-сенсибилизаторы
разрушающе действуют на жнвые клетки по тому же механизму
фотосенсибилизованного окисления, которое в биологии
получило название фотодинамического действия
красителя. Белки в результате фотореакции окисляются до
СО, и NH3.
К фотодинамически активным красителям принадлежат
метиленовый голубой, бенгальская роза, феносафранин, эозин,
эрнтрозин, хлорофилл, гематопорфнрин и др. Установлено, что
способность красителя к флуоресценции существенна для его
активности.
Так, например, фотодинамически неактивны следующие
красители, не флуоресцирующие в обычных условиях: три-
и дифенилметановые (малахитовый зеленый, аурамин), азо-
красители, пикриновая кислота, нафталиновый зеленый В,
индиго кармин, некоторые ксантеновые красители. Интересно,
что замещение магния в хлорофилле медью приводит к не-
флуоресцирующему красителю, которое лишено способности
фогосенсибилизовать окисление бензндина. Аналогично
введение в гематопорфирин медн или серебра дает нефлуоресци-
рующие соединения, которые неактивны фотодинамически,
тогда как соединение с цинком флуоресцирует и
одновременно активно. Хотя флуоресценция и является необходимым
условием рассматриваемой здесь фотосенсибилизации,
непосредственной связи между тушением флуоресценции и
фотохимическим действием, как мы видели выше (34), не имеется
(стр. 228). Концентрация окисляемого субстрата
(восстановителя) "Y для получения квантового выхода фотореакцни у
порядка единицы должна быть значительной. Так, при
окислении тиозинамина сенсибилизованном хлорофиллом (этилхлоро-
филлидом) в ацетоновом растворе на 1 поглощенный фотон
приходится 1 связанная молекула тиозияамина когда концентрация
этого субстрата в 100 раз превышает концентрацию красителя;
при меньшем избытке субстрата у быстро падает (фиг. 40).
Гематопорфирин и флуоресцирующие красители ведут себя
аналогично. Квантовый выход не зависит от применяемой
длины волны на широком интервале спектра поглощения сен-
236
фотоокисление и фотовосстановление
as-
0А-
о.з-
02-
0.1
о
сибнлизатора. Растворитель имеет существенное значение:
в воде фотореакция окисления амиламина кислородом, сенси-
билизованная хлорофиллом, имеет в 100 раз меньшая выход,
чем в ацетоне и пирнднне ш.
Примером рассматриваемого здесь фотохимического
процесса может также служить окисление лейко-формы красителя
Н2А кислородом, фотосеисибилизованное к видимому свету
самим красителем А или другим красителем. Это явление
уже давно установил Грос (1901)195 в классическом
исследовании, в котором он изучал
_. реакцию фотоокисления в
присутствии кислорода как красителей
флуоресцеиновой группы (урани-
на, эозина, эритрозина), так и их
лейко-форм в водном растпоре.
Измерялось поглощение кисло-
* рода растворами в стеклянных
| сосудах под действием солнеч-
: ноге? света. Таким образом,
действующей была только видимая
1о 20 30 40 50 60 70 8090 юо радиация, поглощаемая в случае
лейко-формы на начальной
стадии только краситалем,
присутствующим в ничтожной
концентрации. Краситель, образующийся
из бесцветной лейко-формы, в
результате реакции сильно
повышает скорость поглощения
кислорода, причем Грос доказал, что
активными являются длины волн,
поглощаемые красителем. Сенсибилизуют реакцию все
красители данной группы, если их ввести в раствор какой-
либо лейко-формы. Например, скорость окисления лейко-
ураиииа увеличивается в 1.5 раза при добавлении уранина,
в 3.3 раза при добавлении эозина и в 4.4 раза — эритрозина.
Другие галоидозамещенные флуоресцеины увеличивают
скорость реакции даже в 6 раз. Нитрозамещенные флуоресцеины
менее активны. Концентрация добавляемого сенсибилизатора
должна быть малой (10~6—10~3М); иначе наступает
торможение фотореакции (ср. 36). Фотореакция необратимого
(деструктивного) окисления кислородом самого красителя (25)
протекает значительно медленнее, чем фотосенсибилизованное
окисление его лейко-форм,
Грос показал, что прибавление к водному раствору лейко-
уранйиа солей металлов Си, Fe, Ni, Co, U, Ag и др., так
Ссы
Фиг. 40. Квантовый выход у
окисления тиозинамина, фото-
сеисибилизованного этилхлоро-
филлидом, как функция
концентрации тиоэинамяна
и сенсибилизатора ^
Асе
34. ФОТОСЕНСИБИЛИЗОБАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 237
называемых переносчиков кислорода, не оказывает
существенного влияния на ^скорость поглощения кислорода.
Аналогичную фотосенсибилизованную реакцию окисления
следами красителя обнаруживает лейко-форма (лейкоосиова-
иие) малахитового зеленого и его гомологов в коллодионных
пленках196. Ускоряют реакцию Основания, кислоты ее
задерживают. Гетероциклические и алифатические амины действуют
сильнее ароматических. Следует отметить, что только лейко-
основания ксантеновых и трифеиилметановых красителей
принадлежат к числу стойких в темноте; лейкоиндигоиды
окисляются кислородом уже в темноте; лейкоиндамины вообще
не обнаруживают фотосенсибилизованного окисления. Как
всякий процесс окисления молекулярным кислородом,
рассматриваемая здесь реакция тормозится легко окисляемыми
соединениями „антиоксидантами" или „ингибиторами" окис-
левия, если их ввести в раствор. Таковыми здесь служат фенол,
гидрохинон, пирогаллол, а также неорганические анионы J~~,
CNS~~ и др. Для заметного торможения реакции концентрация
ингибитора должна быть значительной; часто она превосходит
концентрацию красителя-сенсибилизатора более, чем в 10 раз.
Предполагается, что действие ингибитора заключается здесь
в том, что ингибитор hZ (а не субстрат «Y) вступает в
реакцию фотовосстановления, испытывая дегидрирование.
Кислород реагирует преимущественно с полуокисленной формой
ингибитора hZ, образуя НО2. Именно последний радикал,
а не О2 дегидрирует теперь семихннон красителя НА,
регенерируя краситель. Радикал НО2 обладает большим
сродством к электрону, чем О2| судя по некоторым косвенным
данным ш.
Итак, с изложенной выше точки зрения механизм
фотосенсибилизованного красителем окисления субстрата
кислородом состоит не в том, что краситель „переносит" кислород
на субстрат, а в том, что краситель дегидрирует субстрат,
как он делает и без кислорода, выцветая сам. Действительно,
было показано, что эозин, эритрозин, феносафранин н е
выцветают под действием света в присугствии тиомоче-
вины или тиозинамина, если „лабильные" атомы водорода
последних замещены алкильными радикалами (этил или аллил).
Действие кислорода заключается только в регенерации
красителя путем дегидрирова! ия полувосстановленной НА,
а также лейко-формы красителя Н2А.
Трактовка процессов окисления органических соединений
кислородом, как реакции дегидрирования, была впервые
выдвинута Виландом (стр. 238) и связана с его именемlti7.
Изложенная выше точка зрения Вейсса187, развитая Франком
238 ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
и Ливингстоном69, представляет собой, в сущности,
модернизацию концепции Виланда и перенесение ее на
фотохимические реакции.
Биохимические теории окисления кислородом. Ввиду
большого значения окислительных процессов с участием
кислорода в биологии (дыхание), этот вопрос разрабатывался
главным образом биохимиками, выдвинувшими несколько
теорий о механизме реакции. Противопоставляются две
основные точки зрения, которые можно кратко сформулировать,
как теория мобилизации водорода (Виланд) и теория
мобилизации кислорода (Варбург, Бах и Энглер). Согласно
первой предполагается, что при реакции окисления на первом
этапе активируются структурные атомы водорода
окисляемого соединения; согласно второй — молекула кислорода.137
По Виланду главным и первичным актом является
отщепление атомов Н от окисляемой молекулы, которые
временно связываются каким-либо акцептором водорода
(катализатором реакции) и только затем передаются кислороду.
В рассматриваемой нами фотохимической реакции таким
переносчиком водорода служит активированный светом
краситель. Если окисление органического соединения состоит
не в дегидрировании, а в необратимом присоединении
молекулярного кислорода (оксидировании), то Виланд полагает,
что реакция происходит без прямого участия молекулы О2,
а за счет кислорода воды. А именно предполагается, что
в присутствии последней образуется гидрат органического
соединения Y типа HYOH, который н отщепляет атомы
водорода в первичной реакции дегидрирования с образованием
устойчивого кислородосодержащего продукта YO:
Н
ОН
Так, например, окисление ацетальдегида в уксусную кислоту
трактуется Виландом так:
c-o =
н ннб ь но
1) В приведенной структурной формуле гидрата альдегида составные
части молекулы воды присоединены обычными валеятнымн связями по
месту двойной связи карбонильной группы, т. в. предполагается образова-
34. ФОТОСЕНСИБИЛИЗОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ 239
Кислород, по Виланду, действует только на
промежуточный носитель водорода, т. е. на гидрат альдегида, его
дегидрируя-
Если бы окисление всегда состояло только в
дегидрировании субстрата (или его гидрата), как это принимает Виланд,
то эта реакция всегда приводила бы к одинаковому продукту,
независимо от присутствия кислорода, при замене его
окислителями другого рода. Между тем, ряд фактов не согласуется
с таким предположением и показывает, что молекула
кислорода способна присоединяться сама к субстрату, притом
в неводной среде, т. е. субстрат выступает как акцептор
кислорода.
По Варбургу, первый этап кислородного окисления
органических соединений заключается именно в активировании
молекулы О2, а не атомов Н субстрата, как это принимает
■Виланд Согласно теории Баха и Эиглера, активирование
кислорода и реакция самоокисления непредельного или
ароматического соединения происходит путем присоединения О2
по месту двойной связи с образованием неустойчивого
соединения перекисного типа, которое схематически изображают
.о r °-9
как Y<f j , например \ Л г\ (ср. стр. 148). Некоторые
X) /^ U
Н
вие нового устойчивого соединения с соответствующей перегруппировкой
валентных электронов. С нашей точки зрения ввиду инертности молекулы
воды это вряд ли имеет место. Гидрат может представлять собой три типа
взаимодействия (а), (Ь) и (с), из которых первый есть простая
междумолекулярная ассоциация »лектрост«1ическими силами притяжения взаимно
противоположных диполей, направление которых показано стрелками.
R R R
>с=о с=о >с—о-
Н^О-Н / ; н О-Н+
/ <+■ н-о—н /
н / н
н
(а) (Ъ) (с)
Второй тнп (Ь) есть „водородная связь" между молекулой воды с
кислородом карбонильной группы, а также, возможно, между альдегидным
атомом Н и кислородом воды. Схема (с) соответствует резонансной
структуре ассоциированных молекул, частично осуществляемой при водородной
связи. В полной мере такая валентная структура и изображенный в (с) тип
взаимодействии имеет место, когда к альдегиду присоединяются готовые
иоиы H~*~ и ОН~. Таким образом, изменение ультрафиолетового спектра
поглощения альдегида, наблюдаемое при его растворении в воде, имеет
своей причиной совместное действие указанных трех типов взаимодействия,
а не образование обычных валентных связей, указанных в структурных
формулах Виланда,
24Э ФОТООКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
вещества выполняют, по Мурё и Дюфрессу, роль
„переносчиков кислорода" (прооксидантов) тем, что в их присутствии
ускоряется термическая реакция окисления кислородом более
стойких субстратов. Этот вопрос подробно рассмотрен нами
в главе 7, посвященной фотореакциям присоединения
кислорода к молекулам, активированным светом.
Для фотосенгибилизованной красителями реакции
окисления, описываемой здесь, можно было бы попытаться
нарисовать механизм, использующий представление о первичном
образовании лабильного пероксида красителя АО2(23), который
и служил бы „переносчиком" активной формы кислорода на
окисляемый субстрат HY, осуществляя либо дегидрирование
последнего, либо присоединение к нему кислорода по схеме:
Варианты:
f A-»-НО2 ~t-„Y
О2 -* AO. + IY -* нА-нО2+ „Y (1Х)
В первом варианте развития реакции, как и в схеме (VIII)
на стр. 234, образуется радикал НО2, но сразу в результате
дегидрирования субстрата, а не через семихинон НА
красителя. В последнем варианте гидропероксид HOOYH
эволюционирует дальше по аналогии со схемами, приведенными на
стр. 168, с выделением перекиси водорода.
Действительно, образование перекиси субстрата hY
было обнаружено Гаффроном191 при фотосенсибилизован-
лом хлорофиллом окислении аминов, притом с большим
квантовым выходом. В неводном растворителе (ацетоне, пиридине)
количество перекиси было в сотни раз больше, чем в воде.
Напротив, никаких устойчивых перекисей красителя-
сенсибилизатора найдено не было. Порфирины под действием
света могут образовывать перекисные соединения, но при
этом происходит расщепление их кольцевой структуры,
а следовательно, необратимое изменение молекулы, которая
не может таким образом функционировать в качестве
сенсибилизатора реакции окисления. Этот факт устраняет
возможность второго варианта схемы (IX), но не опровергает
остальные. Однако гипотеза о первичном образовании
лабильного пероксида (мольоксида) красителя АО2, неуловимого
обычными аналитическими приемами, требует дополнительного
обоснования. Между тем семихиноны типа НА, как
промежуточные, легко окисляемые формы красителей, хорошо известны.
34. ФОТОСЕНСИБИЛИЭОВАННОЕ КРАСИТЕЛЯМИ ОКИСЛЕНИЕ
Это обстоятельство вместе с тем фактом, что в отсутствии
кислорода сенсибилизатор, поглощая свет, дегидрирует
субстрат, а также тем наблюдением, что флуоресценция с, н-
сибилизатора не тушится кислородом, послужили в
совокупности к установлению той точки зрения на механизм
процесса, которая изложена в начале этого параграфа и
суммирована в схеме (VIII). Согласно этому механизму, как мы
видели, активированный светом сенсибилизатор служит
переносчиком водорода от окисляемого субстрата к молекуле
кислорода- Такой механизм фотохимического окисления,
в развитие идеи Виланда, был впервые предложен Когелемт
(см. стр. 213, 294). В последнее время та же концепция
возродилась в трактовке Вейсса-Франка-Ливингстона, которая
и была дана в начале этого параграфа (схема V1I1).
При первоначальной дискуссии возможного механизма
фотосенсибилизованного окисления была выдвинута несколько
иная точка зрения. А именно предполагалось, что
возбужденный светом краситель передает энергию возбуждения
молекуле суГстрата, „мобилизуя" или активируя тем самым
атомы водорода последнего к реакции с кислородом. Молекула
О2 дегидрирует такой активированный субстрат:
A*-4-HY-> А-ьН — Y
Y-*-HO2
Эта реакция представляет собой вариант первого
механизма в предыдущей схеме (IX), но без активации кислорода.1)
Впервые предположение о возможной активации
субстрата, а не кислорода, высказал Гаффрон (1933)194, который,
между прочим, рассматривал активированную форму
субстрата Н - Y как метастабильное его состояние, сущестау-
ющее 1(Г"2 - 1(Г"г сек., что встретило возражения со стороны
Шумахера (стр. 146). К точке зрения Гаффрона
присоединились затем Франк и Леви (1934) ^ которые выступили
против первичной активации молекулы О2 на том основании,
что субстрат (бензидин) производит в растворе более сильное
тушение флуоресценции возбужденной молекулы красителя А*,
чем кислород, который вообще не тушит флуоресценцию таких
сенсибилизаторов, как эозин, эритрозии. В свете позднейших
фактов, установивших отсутствие непосредственной связи
1) Когель и Штейгманн173 высказали эту точку зрения уже давно,
но в отношении к фотографической сеисибялчзации, где ояа неприменима
вовсе (стр. '294).
16 А. Н. Tt-ренин
242 ФОТО ОКИСЛЕНИЕ И ФОТОВОССТАНОВЛЕНИЕ
между тушением флуоресценции и фотохимическими реакциями,
этот довод не является в настоящее время решающим. Кроме
того, Каутский показал как раз обратный эффект для
хлорофилла и гематопорфирина, флуоресценция которых н е
тушится окисляемыми субстратами, как тиозинамин или
изоамиламин, но тушится кислородом91.1) Каутский является
сторонником точки зрения о первичной активации молекулы
кислорода, выдвинутой им на основании проведенного им
остроумного эксперимента в вакуумных условиях, подробно
описанного выше (стр. 135). Он полагает, что при фотосенси-
билизованном окислении активна именно длительно живущая
метастабильная форма красителя, особо чувствительная к
присутствию кислорода, которая и реагирует с субстратом, его
дегидрируя по механизму:
А*
I
При достаточно большой концентрации субстрата HY
(„акцептора кислорода"), когда обеспечена его реакция
с каждой метастабильной молекулой кислорода O3mPta кваи*
товый выход реакции у будет равен единице и не будет
зависеть от концентрации кислорода, как это действительно была
установлено Гаффроном 91.
Несомненно, что открытые Каутским факты несколько
нарушают стройность механизма фотосенсибилизованного
окисления, предлагаемого Вейссом, а также Франком
и Ливингстоном.
Из изложенного можно сделать вывод, что
дегидрирование субстрата является существенным этапом реакции, но
что детальный механизм ее протекания не может быть
однозначно установлен на основании изучения только кинетики.
Настоятельно необходима постановка опытов, использующих
новые методические приемы для кардинального решения
затронутых вопросов.
1) Тушится как обыкновенная, так и „замедленная" флуоресценция,
которая обязана метастабильному состоянию (стр. 78), По Каутскому,
последнее представляет собой димер красителя, что мало вероятно307.
С нашей точки врения, увеличение длительности послесвечения при
большой концентрации, возможно, вызвано стабилизацией бирадикальных форм
молекул, вступивших во временную ассоциацию с другими молекулами того.
же рода.
35. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ 243
ГЛАВА ДЕВЯТАЯ
ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
35. Тушение флуоресценции красителей посторонними
веществами
Если в раствор флуоресцирующего красителя ввести
определенные посторонние вещества, не поглощающие
возбуждающего света, то интенсивность и выход испускаемого света
падает или, как говорят, происходит тушение
флуоресценции. Наиболее типичными тушителями являются различные
анионы, в особенности ион J~, но тушат также катионы Ag%
Cu++, Fe"*"*" и нейтральные молекулы (анилин, гидрохинон
и др.).
Анионы по своему тушащему действию на флуоресценцию
обширной группы красителей и других флуоресцирующих
веществ могут быть расположены приближенно в следующий
ряд спадающего вффекта1Э8.
J-> CNS~> Вт~> CI- >• (СОО)2— > СНаCOO- > SO4— > NO3~ > F~.
Такая последовательность тушащего действия наблюдается
для флуоресценции уранина, эозина, родаминов, сернокислого
хивина, акридона, а-и Р-антраценсульфоновой кислоты, иона
уранила и некоторых других флуоресцирующих красителей
и соединений199. .
Однако другая группа флуоресцирующих соединений,
обладающая интенсивной полосой поглощения в близком
ультрафиолете („ультрафиолетовые красители"), тушится
анионами в иной последовательности, а именно199:
J(V > BrO3- > NOr > S А~ > AsO4— > С1О
К этой второй группе принадлежат следующее соединения,
флуоресцирующие видимым светом: антраниловая кислота,
сульфоанилат натрия, нафтионат натрия, 7-нафтол, 1-4-на-
фтолсульфоновая кислота и др.
Обе груты тушатся теми катионами, которые являются
окислителями (акцепторами электронов), как:
Явление тушения не может быть, в общем случае,
приписано темновой реакции с тушителем, сопровождающейся
16*
244 ТОРМОЖЕНИЕ ФОТО РЕАКЦИЙ КРАСИТЕЛЕЙ
образованием комплексов, лишенных способности
флуоресцировать. Действительно, сильное тушение часто наблюдается
при столь малой концентрации тушителя, когда нет никаких
признаков такого комплекс^бразовакия. Далее, во многих
случаях спектр поглощения красителя нэ испытывает
изменений даже при значительной концентрации тушителя,
превышающей в тысячи раз концентрацию красителя. Следует
оговориться, что указа* ио^ постоянство спектра поглощения имеет
место только при достаточно низкой концентрации самого
красителя «10~4М). Иначе введение посторонних солей
в раствор приводит к кажущемуся тушению в результате
„высаливания" красителя и * раствора, т. е. аггрегации и даже
коагуляции его в нефлуоресцирующие частицы202. Характерным
признаком изменения состояния дисперсности красителя в ра-
стзоре является искажение его спектра, о чем было сказано
в главе 4 (14)203.
В большинстве случаев тушение не может быть также
приписано исчезновению возбужденных светом молекул в
результате необратимой ф отохи мическо й реакции с тушителем,
так как после удаления последнего, например осаждением,
первоначальная интенсивность флуоресценции
восстанавливается. Далее, при разбавлении потушенного раствора
растворителем, которое приводит к уменьшению концентрации
тушителя, выход флуоресценции ер увеличивается. Кроме
того, величина тушения не зависит от времени освещения.
Всестороннему исследованию явления тушения
флуоресценции красителей в нашей стране посвящены систематические
работы Вавилова и его школы.200 Этими работами было на
многих типичных примерах показано, что действию тушителя
подвергаются только возбужденные светом молекулы
красителя за короткое время этого состояния (т^10~8 сек.).
А именно, тонкими измерениями степени поляризации
испускаемого света при тушении было показано, что оно приводит
к сокращению времени т и что выход флуоресценции
9 падает пропорционально т, т. е.:
.1- = JL, (1)
Фо Ч
где <р0 и т0—значения выхода и времени вэзбужденного
состояния в отсутствии тушителя, а <р и т— значения этих величин
при определенной концентрации последнего.
Сокращение времени т для флуоресцирующего красителя
действием тушителя было также показано непосредственным
измерением его длительности31.
3% ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ
245
При небольшой концентрации флуоресцирующего красителя
кинетика тушения удовлетворительно воспроизводится следу-
о о/5 аз
Флуоссгцеун
л Анилин
• мооистый
копий
Jo
т
6
5
3
2
1
0.1 02
Антрацен
т Нитробензол
о ГЗаякол
D Гидрохинон
О 015
Родулин kp
03
3
2
О QJQ5 015 0J5
Родам ин В
о
Нснртионот No
Фнг. 41. Зависимость величины тушения флуоресценции от концентрации
тушителя с
201
Наклоны прямых дают коьстанты тушения kg.
ющей зависимостью выхода флуоресценции
сивности F от концентрации тушителя с:
или ев
интен(2)
где с — концентрация тушителя, а к — постоянная тушения
(фиг. 41).
246 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
С выраженном такого вида мы уже встретились ранее при
рассмотрении тушения флуоресценции ароматических
углеводородом кислородом (формула 6 и 21, для случая £1[А] = 0).
Оно шнподится здесь, как и там, из кинетической картины
бимолекулярных „встреч" между возбужденными молекулами
Л* и молекулами (ионами) тушителя. Для сталкивающихся
газообразных частиц формула (2) была выведена Штерном
и Фольмером (1920)204.
Постоянная kq представляет собой произведение следующих
величин:
*в=К*. (3)
где z есть число встреч с молекулами (ионами) тушителя,
испытываемых за секунду любой молекулой красителя, a toz —
число встреч, испытываемых возбужденной молекулой
за ее естественное время жизни т0. Коэффициент Р^ 1 здесь
представляет собой величину „выхода" тушащих встреч, т. е.
эффективности встречи в смысле тушения. Число
столкновений z для газообразных молекул дается выражением
кинетической теории газов:
771] 7712
/Tli -Ь- 1712 /
где гх и r2 обозначают радиусы сталкивающихся молекул;
т1 и т2— их массы; Т—абсолютная температура; к— газовая
постоянная Больтцманна.
Расчет числа встреч или соударений, испытываемых
молекулой (ионом) красителя в растворе со стороны других ионов,
представляет собой трудную задачу из-за многочисленности
и сложности факторов, определяющих подвижность частиц
в этих условиях. В жидкостях соударения молекул имеют
совсем иной характер, чем в газах. Две встретившиеся прн
диффузии молекулы долго находятся друг около друга,
испытывая ряд повторных соударений.
Исходя из картины диффузии и встреч растворенных частиц
при совершении ими броуновского движения, Вавилов (1928)200
вывел для z следующее выражение:
•i
где f(rlf r2) есть некоторое выражение, содержащее радиусы
частиц, а тз—коэффициент вязкости растворителя.
35. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ 247
Другими словами, тушащее действие данного вещества
должно падать при увеличении вязкости среды,
затрудняющей диффузию и сближение частиц,
1]ри выводе формулы (4) учитываются лишь первые
соударения, что равносильно предположению, что вероятность
тушения £J равна единице.
Формула (2) дает зависимость величины -- или w от
концентрации с в виде прямой, из наклона которой легко
определить к % а зная т0, и $z. Для тушения флуоресценции в
газообразной фазе такая зависимость неизменно наблюдается
на опыте. Для растворов в формулу (2) необходимо
подставить вместо z выражение (4), что дает линейную зависимость
как от концентрации тушителя, так и от „текучести" —
растворителя. Однако в растворах, в особенности ионных, имеют
место значительные отступления от простой линейной формулы
(2), которые побудили Вавилова и Франка200 ввести для этих
случаев представление о „сфере действия" возбужденной
молекулы красителя с радиусом, значительно превышающим
нормальный ее радиус, известный из опытов по диффузии.
В результате формула (2) усложняется путем введения в пра-
вую часть общего множителя е , зависящего от концентрации,
в котором v обозначает объем „сферы действия".
Между тем, оказалось возможным Объяснить отступления
от линейной зависимости, не прибегая к представлению об
аномально большой сфере действия (Вавилов, 1935200 и
Свешников, 1935 201). При учете того обстоятельства, что ва
короткое время т0 возбужденного состояния красителя диффузию
нельзя рассматривать как установившийся процесс и что имеют
значение повторные соударения возбужденной молекулы
с тушителем, удается получить удовлетворительное согласие
с наблюдениями. Последнее обстоятельство означает, что
эффективность тушения (3 всегда меньше единицы даже для
сильного тушителя. Так, ншример, для объяснения хода
тушения флуоресценции уранииа ионом иода необходимо принять,
что вероятность тушения при встрече возбужденной молекулы
с этим ионом составляет 0.25201<Ш7*.
Трудности интерпретации кинетики тушения флуоресценции
красителей обусловлены в значительной мера их ионной
природой, так как необходимо учитывать специфику ионного
равновесия и ионные „силы" вводимых посторонних ионов,
не говоря об изменении структуры красителя в результате
изменения концентрации водородных ионов. Эти обстоятельства
получили должное внимание только в самое последнее время205'206.
'-48 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
О механизме действия тушителя существуют различные
предположения. Сторонники чисто физической трактовки
явления (Вавилов, Перрен, Свешников и др.) рассматривают
тушение, как акт передачи энергии возбуждения от красителя
тушителю. Такой процесс, известный для газовых молекул,
происходит с большим выходом только тогда, когда тушащая
частица способна воспринять передаваемую электронную
энергию целиком или большую ее часть, т. е. когда верхние
электронные уровни обеих частиц близки, а следовательно, и полосы
поглощения примерно совпадают (ср. стр. 211).2) 1Уежду тем,
для сильных тушителей такое соотношение между
энергетическими уровнями красителя и тушителя никогда не
наблюдается. Гидратироваиный анион иода J~~, являющийся одним
из самых сильных тушителей, обладает, как мы видели выше
(29), ультрафиолетовой полосой поглощения, а следовательно,
ближайшим электронным уровнем, весьма далеко отстающим
от таковых для красителя. С другой стороны, ион иода
совершенно не тушит ультрафиолетовую флуоресценцию нафталина»
спектрально более близко расположенную. Анион NO3~,
имеющий полосу поглощения у 300 mj/., т. е. в близком
соседстве с видимой полосой красителей, является тем не менее
значительно более слабым тушителем. Катионы редких земель
с узкими полосами поглощения в видимой области,
совпадающими с полосами поглощения и испускания красителей,
практически не тушат их флуоресценцию.2)
Передача энергии от возбужденного светом органического
соединения неорганическому катиону Ей"*"*"*" была доказана
по появлению дискретного спектра флуоресценции катиона
при поглощении ультрафиолетового света органической
молекулой, комплексно связанной с ним208. Однако это имеет место
!} Передача энергии возбуждения может осуществить и такой
электронный переход в воспринявшей эту энергию частице, который неосуществим
светом, как, например, перевод молекулы кислорода в метастаэильное
состояние О2Ш а» подробно рассмотренный иа стр. 137. В этом случае
тушитель может и ив иметь полосы поглощения в спектральной области, где
расположен спектр'красителя.
2) Последний факт не может, однако, рассматриваться как решающий
довод против возможности передачи энергии, так как гидратняя оболочка,
прочно связанная с высоко заряженными катионами, несомненно должна
препятствовать достаточвому нх приближению к красителю. Приведенный
выше ряд анионов от J~ до F~, расположенный в порядке уменьшения
тушащего действия, почти в точности соответствует их порядку в смысле
увеличения энергии гидратации.
Объемистая гидратная оболочка мешает сближению частиц до такого
расстоянии, когда возможен акт тушения путем передачи энергии. Эта
препятствие отпадает для нейтральных, слабо гидратированных молекул
органических соединений и растворенного кислорода (ср. стр. 141).
35. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ 249'
только при наличии прямой валентной связи между
участниками, и простого их соседства недо .таточно.
Единственный факт, который можно трактовать как
передачу внергии возбуждения,— это падение выхода
флуоресценции красителя при увеличении собственной концентрации,
т, е. явление самотушения или концентрационного
тушения, о котором речь ниже. В этом случае совпадение
электронных уровней полное- Однако известны флуоресцирующие
красители и соединения, которые вообще не обнаруживают
самотушения, как например акридон, хинин, пинакриптол
желтый и др.
Резонансная передача энергии возбуждения, если она имеет
место, должна сопровождаться полной деградацией переданной
энергии в тепло, так как явление сенсибилизованнон
флуоресценции акцептора энергии, столь распространенное в
газообразных системах, в растворах еще никогда не наблюдалось.1)
Отсутствие ярко выраженной зависимости тушения от
температуры показывает, что оно не требует дополнительной
термической энергии активации199.
Принципиально важен при выяснении механизма тушения
вопрос о природе сил взаимодействия между возбужденной
молекулой красителя и тушителем, так как установлено, что
они взаимодействуют на расстоянии, заметно превышающем
размеры частиц, даже е^лн внести поправку на неточность
измерения последних.
Были попытки приписать это взаимодействие силам
электростатического происхождения, т. е. величине заряда иона или
дипольного момента тушащей нейтральной молекулы. Никакой
отчетливой зависимости от этих величин, а также от
диэлектрической постоянной растворителя, существенной для
проявления таких сил (ср. 13), получено не было198. Вещества без
дипольного момента, как нафталин, являются сильными
тушителями, многие же вещества с дипольным моментом не тушат
вовсе210. Как неоднократно отмечалось выше, кулоновское
отталкивание одноименно заряженных участников не играет
никакой роли в смысле затруднения их сближения; это
следует, например, из сильного тушения флуоресценции аниона
эозина анионом же иода211. Но испускание катиона хинина
все же сильнее тушнтся анионами, а аниона нафтионата
наоборот,— катионами.
Приведенное в начале этого параграфа расположение
анионов от J~~ до F~~ в порядке уменьшения тушащего действия
!) Свечение за счет передача красителю выделяю щей си химической
энергии реакции, т. е. явление хемилюмииесценции красители тем не менее
в растворах хорошо известно (стр. 307, см. также2^9)
250 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
•совпадает в общих чертах с их расположением в порядке
уменьшения величины электрической поляризуемости их
электронной оболочки. Это указывало бы на зависимость
тушения от смещаемости электрона тушителя в электрическом
поле, а следовательно, от величины „податлмвости" электрона
при взаимодействии с возбужденной молекулой.
Среди органических веществ повышенной электрической
поляризуемостью или смещаемостью электронов обладают
непредельные и ароматические соединения. Кроме того, такие
соединения обнаруживают так называемую „экзальтацию"
рефракции, т. е. превышение над суммой рефракций состав-
Таблица 132Ю
Тушение флуоресценции хииииа
Константа тушения
Тушитель
Бе и вол
о-ксилол *
М-КС И АО А
П-КСИЛОА
Нафталин •«....
ляющих атомных групп. Такой эффект обусловлен наличием
подвижных тс-электронов в этих соединениях (7, 8). Эйзенбрандт
(1933)2i0 показал, что, действительно, имеется прямая
зависимость между величиной экзальтации и тушащим действием
данного соединения иа флуоресценцию хинина (табл. 13).
Однако некоторые сильно тушащие соединения не дают
экзальтации. Существенным является присутствие
сопряженных двойных валентных связей, которое, по данному выше
разъяснению, есть признак наличия обобществленных
электронов (6).
Необходимо отметить, что, в отличие от неорганических
ионов, органические тушители не могут быть расположены
в единообразный ряд для любого флуоресцирующего
соединения, как это, например, следует из табл. 14 и фиг. 41,
приведенной раньше. Таким образом, тушение проявляет
специфичность бимолекулярной химической реакции, зависящей от
природы обоих партнеров.
1) См. объяснение обозначения с1 Ниже.
с
1
11
20
50
125
1
1 ■■
2
*_/fbacuvDi ли
— 0.15
-+-0.19
-+-037
-+-0.39
н-1.91
35. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛГ.Й
•гы
В табл. 14 приведены некоторые численные значения,
характеризующие тушащее действие различных органических
веществ, а именно даны численные значения концентрации
-с1 тушителя (в молях/л), соответствующие падению исходной
интенсивности флуоресценции FQ на половину. Из формулы
(2) на стр. 245 следует, что когда F=-^ FQi то
соответствующая концентрация ci=--r. Иными словами, значения с\ об-
Т ** Т
ратно пропорциональны тушащему действию прибавляемого
вещества.
Таблица 14
Значения концентрации с г (моль/л) „половинного тушении"
т
Cj = _—, где k —постоянная тушения в формуле (2),
Тушитель
Флуоресцирующяе соединения212
нафтио-
нат-Na
в Н2О
векулин
в С2Н5ОН
флуореиои
в CaHfl
Фенол . . .
Гидрохинон •
Резорцин . *
Бренцкатехии
Пирогаллол .
а-нафтол . .
Анилин • .
0.04
2.9
0.2
0.08
0.13
0.S0
0.19
0.11
0.08
0.22
10-10-»
18-Ю-з
14.10-3
Тушители флуоресценции хинина в кислом СН3ОН210
'Стильбен 4.4-10—ъ Фу ран
Диметнловын эфяр ги- Тиофен
14-Ю-3
18-10-3
дрохинона
Нафталин
Диметиловый эфвр
бренцкатехина ....
Диметиловый эфир
резорцина
Дигидроиафталин . .
ок.
4.6-10—3 Пара-кснлол ок. 2010—3
8.3-10—3 Тетрагидронафталин . . 33-10 3
Циклопентадиен ....
8.6-10 3 мета-ксилол
Орто-ксилол
12-10—3
50-10--1
50-Ю-3
90-10-3
252 ТОРМОЖЕНИЕ ФОТОРЕ^КЦИЙ КРАСИТЕЛЕЙ
Наиболее сильными тушителями являются ароматические
соединения» содержащие группы ОН, NH2 и NO2, а именно:
фенолы (фенол, резорцин, пирогаллол), амины (анилин,
дифениламин, аминофенол); кроме того, тушат: тиомочевина, фта-
лимид, мочевая кислота, гидроксиламин, фенилгидраэин,
нитробензол. Алифатические амины оказывают значительно меньшее
действие. Пиридин тушит очень слабо, даже в
концентрированном растворе.
Все указанные вещества принадлежит к типу легко
окисляющихся соединений — восстановителей. Зто обстоятельство
наводят на мысль, что тушение объясняется переносом
электрона, т. е. состоит в реакции окисления-восстановления.
Родоначальником этой концепции, в применении как к
явлению тушения флуоресценции, так и фотосенсибилизованным
реакциям, является Баур174»213. Идеи Баура, высказанные в
несовершенной и примитивной форме, были возрождены в
последнее время Вебером138'178'214 и Вейссом179*187, исходившими
из современных представлений.
Схема реакции „молекулярного электролиза",
происходящего на полюсах поглотившей свет „фототропной" молекулы
красителя, была изображена по Бауру на стр. 215. Тушение
флуоресценции красителя А* трактуется по тому же механизму,
как двукратный перенос электрона с дополнительным
предположением, что энергия возбуждения красителя при этом
растрачивается в виде тепловой. Например, тушение ионами железа
изображается схемой:
а тушение анионом гидрохинона схемой:
-ь О = / \=О -» -О-
- "°-С>-°^ -
Другими словами, заряд „анода" вовбужденной молекулы
красителя нейтрализуется и имеет место ее восстановление
анионом гидрохинона. Электроны с „катода" молекулы переносятся
на нейтральную молекулу хинона, присутствующую также
в растворе (вследствие темнового окислительно-восстановитель-
35. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ 253
ного равновесия гидрохинон*—>хинон). Действительно,
типичные органические тушители принадлежат к классу анодных
деполяризаторов, т. е. являются по своей природе легко
окисляющимися соединениями или „антиоксидантами".
Вебер т впервые попытался связать эффективность и
постоянную тушения флуоресценции красителей с редокс-потенциа-
лом тушителя. Для флуоресценции хинина (сернокислого)
и нафтионата-Na тушение гидрохиноном, пирогаллолом
и бренцкатехином возрастает с перемещением редокс-потен-
циала тушителя в сторону отрицательных значений, т.е.
смещением занятого электроном уровня вверх (фиг. 33 в 28).
Веберу удалось в некоторых случаях даже установить
прямолинейную зависимость между логарифмом постоянной тушения
к из уравнения (2) и величиной редокс-потенциала указанных
соединений, а также ряда анионов галоидов. Такая же
зависимость была выведена теоретически Вейссом и Фишгольдом179.
Однако для катионов нет никакого соотношения между
величиной тушения и редокс-потенциалом. Специфические
^особенности участников играют настолько большую роль, что
отчетливую связь с потенциалом обратимого
окислительно-восстановительного равновесия установить не удается.1)
Веберlj8, а также Шнейдер188 отождествили тушение
флуоресценции красителей анионами галоидов с первичным
актом фотосенсибилизованного окисления этих анионов (33).
Тушение не сопровождается химическим изменением красителя,
так как предполагается, что краситель регенерируется быстро
гекУЩеЙ обратной реакцией. Придер живая с ь схемы Франка-
Леви (стр. 230), предполагающей первичное гидрирование
возбужденного красителя атомом водорода, доставляемого
молекулой воды из гидратной оболочки,' Вебер объясняет
тушение ионами галоида следующей обратимой реакцией:
А*-ъН,О,На1-
} Н А -+- Hal -f- ОН". (II)
А ~ь Н2О^
В кислом растворе, содержащем повышенную
концентрацию ионов водорода, скопляющихся в виде „роя" около
аниона галоида, гидрирующий атом водорода доставляется именно
ими:
А* -I- Н2О,НаГ,Н+
>НА-*-Н,О,НаГ. (IF)
А -ь Н2О,НаГ,Н-^
!) По Веберу линейная зависимость между log kq и величиной редокс-
стотенииала галоидных авиовов лучше всего получаете и в кислых
водных растворах хинина и уранина2l4.
254 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
Действительно, Вебер нашел, что повышение концентраций
ионов подорода усиливает тушение флуоресценции хинин-
сульфата, эскулина и уранина галоидными ионами.1)
Следует, однако, отметить, что такое действие водородных
ионов на тушение не всегда наблюдается, а кроме того,
нельзя вообще ожидать однозначной интерпретации эффекта
по той причине, что изменение рН затрагивает также
структуру носителя флуоресценции, а в случае органического
тушителя — и структуру последнего. Так, например, анилин
тушит флуоресценцию акридина в кислом растворе слабее^
чем в нейтральном, но в кислом растворе флуоресцируют
не нейтральные молекулы акридина, а положительные его
ионы, образовавшиеся вследствие присоединения протона,
притом с иным цветом свечения. Точно так же тушителем здесь
являются уже положительные ионы анилина, а не нейтральные
молекулы.
Вейсс187 отождествил тушение с реакцией элементарного
фотовосстановления возбужденного красителя путем
восприятия электрона от тушителя, например:
А* -+- Fe^ -> А~ -н Fe4"*^. (Ill)
По Вейссу возбуждение повышает сродство красителя
к электрону и делает энергетически возможным перенос
электрона от тушителя (32). Вейссу и Фишгольду также удалось
сопоставить логарифм постоянной тушения с величиной редокс-
потенциала анионов галоидов и дать объяснение этой
зависимости, основываясь на теории электродных процессов,
принадлежащей Гэрни179.
Против универсальности механизма тушения Баура-Вебера-
Вейсса говорят, однако, следующие факты.
Флуоресценция пинакриптола желтого в кислом растворе
тушится гидрохиноном сильнее, чем в нейтральном, хотя
в первом растворе носители свободных электронов — анионы
гидрохинона — заменены недиссоциированными молекулами
гидрохинона (Вебер214, 1932).
Далее, многие сильные органические тушителн, как анилин,
бенэиловый спирт, совсем не способны давать обратимую
окислительно-восстановительную систему, т. е. не способны
обратимо отдавать и принимать электрон, как того требует
обратимость явления тушения. Более того, Эйэенбрандт210
показал, что если в гидрохиноне и аналогичных ему дифено-
лах заместить „лабильные" атомы Н в гидроксильных группах
1) Катионы Fe"1"*" также тушат флуоресценцию метиленового голубого-
наиболее сильно в кислом растворе при рН = 3 (Хеллстрём*81).
35. ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ 255'
ОН на алкильные радикалы как СН3, то получающиеся эфиры,
утрачивающие восстанавливающую способность, тушат тем
не менее флуоресценцию хинин-сульфата так же хорошо, как
дифенолы (см. табл. 14).
Кроме того, сильное тушение производят не только
восстановители, но также окислители, как например Ag4', Си4"1".
С другой стороны, сильно тушатся ионом иода даже такие
красители (ксантеновые, хлорофилл), которые по своим
свойствам легче испытывают реакцию фотоокисления, чем фото-
восстановлеиия138 (1936>'192.
Отсутствие даже следов продукта предполагаемой
фотохимической реакции, несмотря на большое количество фотонов,
поглощаемых потушенным раствором, неизменность спектра
поглощения, а также остающейся флуоресценции, при
длительном освещении приводят к выводу об исключительно быстрой
регенерации первоначальной формы краснтеля, не совместимой
с протеканием фотохимического процесса. Мы указывали,
кроме того, неоднократно выше (стр. 228) на отсутствие
прямой связи между тушением флуоресценции и фотореакцией
и подчеркивали, что в последнюю вступает именно та часть
поглотивших свет молекул, которая по внутренним
причинам неспособна отдавать энергию в виде испускания света.
Совокупность изложенных данных приводит к выводу
о ничтожной химической реакционной способности красителя
в возбужденном состоянии А*, в отличие от метастабиль-
ной бирадикальной формы А*, и склоняет к заключению
о физической природе тушения. Однако против гипотезы
простой передачи энергии имеются, как мы видели, достаточно
веские аргументы. Нам представляется поэтому, что тушение
обусловлено временным о бм еном электронами между
встретившимися частицами. Перенос электрона, действительно,
имеет место, но он немедленно компенсируется переносом
другого электрона в обратном направлении до того, как частицы
успевают разойтись. В растворах последнее условие
удовлетворяется в полной мере.1) Направление переноса первого
электрона от тушителя к возбужденной молекуле или от
возбужденной молекулы к тушителю зависит от относительного
расположения занятых и вакантных электронных уровней
и принадлежности красителя к типу электронных или
дефектных систем, т. е. к типу фотоокисляемых или фотовосстанав-
ливаемых соединений (32). Изложенное можно изобразить
в виде следующей схемы (фиг. 42), в которой горизонтальные
]) С этой точки зрения представляет интерес изучение тушения
аналогичных систем в газообразном состоянии и сравнение с тушением в
растворе.
256 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
черты обозначают уровни энергии, жирные точки — занятость
уровня электроном, а кружки — пустой, вакантный уровень.
Слева изображено возбужденное состояние А* молекулы,
причем пунктирные горизонтальные стрелки указывают на
„мобильность" электрона или дефекта в смысле его миграции
к „периферии" молекулы красителя (32), где происходит
взаимодействие с тушителем. Справа изображен второй этап.
Тип I соответствует фотовосстанавливаемому, иначе
„фотодефектному" красителю, а тип II—фотоокисляющемуся, иначе
д*
А*+
U А*
Фиг. 42, Схема процессов переноса электрона прн тушении
флуоресценции.
„фотоэлектронному" красителю. Наклонные стрелки
показывают направление переноса электрона с заполненного уровня
на пустой. Взаимный обмен электронами между возбужденной
молекулой А* и тушителем Q происходит в два
последовательных акта, изображенных в левой и правой половинах
фигуры. Волнистые стрелки обозначают деградацию
выделяющейся электронной энергии в тепло путем перехода
в вибрационную энергию частиц, находящихся в переходном
состоянии электронного обмена и взаимодействия. Таким
образом, возбужденная молекула дезактивируется, в сущности,
в два раздельных этапа, на каждом из которых в тепло
превращается только часть энергии возбуждения. Как мы
указывали уже выше (32), акт принятия или отдачи электрона
молекулой красителя должен неизбежно сопровождаться
изменением запаса ее вибрационной энергии, совершенно аналогично
тому, как это происходит в результате поглощения фотона,
"В обоих случаях распределение валентных электронов, которое
35* ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ КРАСИТЕЛЕЙ 257
обусловливает междуатомную связь и жесткость
молекулярного скелета, перестраивается внезапно.
Однако, как мы неоднократно убеждались, четкого
разграничения красителей двух типов провести нельзя, и один и тот
же краситель может вести себя как донор или как акцептор
электрона, в зависимости от природы второго участника.
Сильное тушение флуоресценции красителей, производимое
ароматическими и непредельными углеводородами, обладающими
легко смешаемыми электронами (ср. табл. 13), возникает по
той же причине. Оно свидетельствует о сильном
взаимодействии электронных оболочек возбужденного красителя и
тушителя, которое, согласно концепции квантовой механики,
обусловлено „обменом" электронами между этими частицами.за
краткое время их встречи, пока они образуют
ассоциированный комплекс.
Такова предположительная трактовка механизма тушения,
основная сущность которого была дана в несовершенной форме
еще Бауром.
В заключение необходимо отметить, что тушение
флуоресценции зависит от природы растворителя. В спиртовом и аце-
тонэвом растворах ион иода значительно слабее тушит
флуоресценцию эозина 7(1935) и флуоресцеина-Na.1)
Тушение флуоресценции самим растворителем (спиртами),
если оно вообще имеет место, происходит с ничтожной
эффективностью порядка одной тушащей „встречи" на 10 000215.
Значительная вариация в величине квантового выхода
флуоресценции, наблюдаемая для различных растворителей (см.
табл. 2 на стр. 74), не может быть объяснена этой
причиной, а вызнана двумя факторами: 1) изменением состояния
дисперсности красителя (14); 2) изменением условий
внутренней дезактивации возбужденной молекулы, т. е. конверсии
в вибрационную энергию или в метастабильное, бирадикальное
состояние (12).
Повышение температуры раствора, наряду с уменьшением
его вязкости и условий диффузии тушащих частиц, сказывается
одновременно на этих двух факгорах. Кроме того,
усиливающееся тепловое движение вызывает такие деформации скелета
молекулы,_которые нарушают ее плоскостную конфигурацию
*) Если механизм тушения ионом иода приписать реакции гидрирования
красителя атомом Н из молекулы воды, как это принимает Вебер
(стр. i.53), придерживающийся схекы реакции Фраика-Леви, то ослабление
тушащего действия в неводном растворителе служило бы аргументом
в пользу этой сжемы (см. стр. 231). Однако, как теперь выяснено, тушение
флуоресценции и фотореакция не связаны друг с другом (33).
17 А. Н. Тер с нин
258 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
я затрагивают самый спектр поглощения красителя (стр. 89),
Наблюдающееся при нагревании уменьшение коэффициента
поглощения и площади спектральной кривой 7(Ш5) связаны,
главным образом, с этим эффектом. Но следует учесть также
возможность аггрегирования красителя в более крупные частицы
при нагревании, как это имеет, например, место для родамина
(20).1)
Параллелизм между тушащим действием на флуоресценцию
и торможением фотохимической реакции красителя будет
иметь место только в тех случаях, когда в реакцию вступает
именно возбужденная молекула красителя А*, а роль
ингибитора состоит в простом отводе энергии возбуждения. Как
неоднократно указывалось выше, подобное соответствие между
тушением и фотореакцией в большинстве случаев не
обнаруживается. Вебер138 установил параллелизм между тушением
флуоресценции различными веществами и торможением ими
же реакции фотовосстановления красителей. В ряду
органических тушителей: гидрохинон—пирогаллол—фенол,
наибольшее торможение этой реакции производит первый, в
соответствии с тушащим действием. Однако полной убедительности
это заключение не имеет, поскольку торможение реакции
сравнивается с тушением флуоресценции не тех же
красителей, а других объектов, как хиниисульфат, нафтионат-Na,
эскулин.
Торможение гидрохиноном фотореакции деструктивного
присоединения кислорода к флуоресцирующим красителям210
не может также рассматриваться, как довод в пользу точки
зрения дезактивации возбужденной молекулы этим тушителем,
поскольку гидрохинои является ингибитором также и темновых
реакций самоокисления органических соединений и его
действие, как „антиоксиданта", укладывается в иную схему (26).
Факты торможения фотореакций флуоресцирующих
соединений ионами иода, как например деструктивного
фотооксидирования хинин-сульфата и эозина, а также образования
фотоэознна в отсутствии кислорода, не могут быть
истолкованы как результат тушения флуоресценции. Обе фотореакции
полностью приостанавливаются при такой концентрации ионов
иода, когда флуоресценция еще сохраняется (стр. 124). В
присутствии О2, кроме того, наблюдается появление молекул j2
в результате фотосенсябилизованного окисления иона J~,
1) Проводимые в лаборатории автора этой книги исследовании тушеиня
флуоресценции ароматических соединений в газообразном состоянии
допускают более однозначное решение затрагиваемых здесь вопросов о
механизме процесса, чем это могут сделать опыты в растворах (см., напри»
мер,21'22).
36. КОНЦЕНТРАЦИОННОЕ ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ И ТОРМОЖЕНИЕ 259
свидетельствующего, как мы видели, об активности
поглотивших свет, но не испускающих его обратно метастабиль-
н ы х молекул красителя.
36. Концентрационное тушение флуоресценцин и
торможение фотохимических реакций
Увеличение собственной концентрации красителя сверх
определенного весьма низкого значения (порядка 10~3М для
спиртового и 10~4 М для водного) вызывает явление
самотушения флуоресценции, а именно, начиная с некоторого
порога концентрации выход флуоресценции 9 падает по
показательному закону (Ф. Перрен, 1924217; Вавилов, 192521*)*:
- f (5)
<р
где <р0 есть квантовый выход флуоресценции до порога, а сш
1 1
то значение концентрации, когда выход снижается до —
С
С а. I
своей первоначальной величины (фиг, 43).
Среднее расстояние между молекулами красителя, когда
наступает самотушеиие, равно 50—100 А и примерно в 5 раз
больше радиуса молекул, получаемого из диффузии и других
кинетических явлений. Отсюда следует повышенная
чувствительность возбужденной молекулы красителя к соседству
второй, невозбужденной молекулы, причем такое
„дальнодействие" неизменно сопровождается растрачиванием энергии
возбуждения. По Ф. Перрену вероятность такого
взаимодействия уменьшается с удалением молекул друг от друга, как
шестая степень их расстояния.
Сущность явления была приписана „резонансному"
переносу энергии между двумя частицами, обладающими
тождественными энергетическими уровнями („резонансная индукция"
по Ж. Перрену216).1)
]) Ж. Перрен описывает также опыты, в которых нефлуоресцирующнй
краситель — метнленовый голубой, с полосой поглощения, точно
совпадающей с полосой и с п у с к а н и я другого красителя (флуоресцентного
голубого^, сильно тушит флуоресценцию последнего и притом более эффективно,
чем ето делает эквивалентное повйшение собственной концентрации
флуоресцирующего красители. Тот же метиленоЕЫЙ голубой слабо тушит
флуоресценцию эозина, ураннна и еще слабее — хинин-сульфата, полосы
испускания которых не дают столь полного совпадения. Аналогичяо полоса
флуоресценции уранина сильнее тушится эозином с совпадающей полосой
поглощения, чем при повышении собственной концентрации илн при при-
6авЛ1 нии эскулина с ультрафиолетовой полосой поглощения.
г)ти наблюдения нуждаются в количественной проверке, в особенности же
принимай ко внимание сопутствующие явления ассоциации красителей,
искажения нх спектров поглощения и возможность фотореакций.
17*
260 ТОРМОЖЕНИЕ ФОТОР£А.КЦИЙ КРАСИТЕЛЕЙ
Однако картина явления, как вызванного сближением
молекул красителя в результате диффузии, не воспроизводит
того основного факта, что концентрационное тушение не
находится в прямой зависимости от вязкости растворителя
и мало изменяется при переходе даже к твердым растворам,
в которых молекулы красителя лишены подвижности.
Например, самотушение уранина имгет примзрно одинаковую
величину как в воде, так и в растворе глюкозы с вязкостью
в 2000 раз большей, и даже в твердом леденцеш. В
глицерина же, обладающем промежуточной величиной вязкости,
самотушение, напротив, значительно менее выражено:
при той же концентрации
уранина (10~2 М) глицериновый
раствор светится в 10 раз ярче
917
водного .
Поэтому первоначально
была дана статическая или чисто
конфигурационная картина
явлений самотушения как выззан-
ная случайным
распределением неподвижных молекул
в „сфере действия",
окружающей возбужденную молекулу
(ср. стр. 247). Такая трактовка
Фиг. 43. Падение выхода флуо- позволяет вывести показатель-
ресцен^ни родамина В с увели- ную зависимость от концентра-
^внием собственной ко дентрацнй цИИ (5?, НО не ВОСПРОИЗВОДИТ
н метиловом спирте^. другие особенности явлений, о
которых речь ниже.
Естественно напрашивается объяснение падения выхода
флуоресценции тзм, что при повышении концентрации
красителя, особенно в водном растворе, происходит обратимая
ассоциация и аггрегацкя нормальных молекул красителя
в нефлуоресцирующие димеры, полимеры и даже
коллоидные частицы (Левшин203). Действительно, в диапазоне
значений концентрации, когда наблюдается сильное
самотушение, имеет M.eCiO и отчетливая деформация спектральной
кривой поглощения родамина, родулина и уранина, вызванная
усилением побочного максимума димера или полимера (14).
Вид спектра флуоресценции, напротив, осгается без
изменения. Далее, в спиртовом и ацетоновом растворах, в которых
преобладает мономерный ион красителя, выход флуоресценции
родамина не падает на широком интервале увеличения
концентрации (вплоть до 10~3 W»), в то время как в водном растворе,
благоприятствующем аггрегации, он сильно уменьшается уже
36. КОНЦЕНТРАЦИОННОЕ ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ И ТОРМОЖЕНИЕ 261
при 2*10~4 М, а при 3'10~3 М выход флуоресценции
составляет только 30 выхода при концентрации 10~~e M.1)
Повышение температуры увеличивает выход
флуоресценции в концентрированном водном растворе, в соответствии
с представлением о распаде димеров и других аггрегатов
(стр. 91). Наоборот, в разведенном водном и спиртовом ра-
9(17
створе нагревание вызывает понижение выхода .
Однако объяснение самотушения аггрегацией нормальных
молекул красителя, а также картина конфигурационного
(статического) тушения, находятся в противоречии с другим
основным фактом, а именно тем, что при этом сокращается
длительность т возбужденного состояния молекулы, измеряемая
косуенно или непосредственно флуорометром (стр. 73). На
опыте для самэтушения установлена прямая
пропорциональность выхода у времени т, как это имеет место для
постороннего тушителя (формула 1 на стр. 244).
Это доказывает, что воздействию при концентрационном
тушении подвергаются именно флуоресцирующие
молекулы за время т0 их естественной жизни, а не нормальные
молекулы красителя 2001201) 220.
Для полициклических углеводородов и уранина в водном
растворе (при соблюдении постоянства термодинамической
„активности" или „силы ионов") формула (2) предыдущего
параграфа удовлетворительно воспроизводит также и
концентрационное тушение219. В тех случаях, когда имеет место
статическое тушение с аномально большой сферой действия,
в эту формулу вводится показательная функция от
концентрации200 (стр. 247):
Несмотря на приведенный аргумент против трактовки
концентрационного тушения, как ассоциации нормальных молекул
красителя, тем не менее обойти последний факт не
представляется возможным. В связи с этим была выдвинута компромис-
*) Банов202 приписал тушение флуоресценции красителей посторонними
ионами эффекту „высаливания", т. е. вытеснению красителя и» раствора
вследствие еильнон гидратации ионов соли и вызванного этим связывания
ими части растворителя. Это равносильно повышению концентрации
красителя и должно повести к его ассоциации. Таким образом, по Баноиу,
тушение посторонними неществамн тождественно с концентрационным.
Хотя эффект „высаливания" несомненно должен учитываться в йодном
растворе при больших концентрациях красителя и тушителя, для
разбавленных растворов, применяемых обычно в опытах по тушению, объяснение
Б а нова неприменимо.
262 ТОРМОЖЕНИЕ ФОТО РЕ АКЦИЙ КРАСИТЕЛЕЙ
сная точка зрения, обыиняющая концентрационное тушение
переносом энергии возбуждения не одиночной молекуле
красителя, а нефлуоресцирующему димеру или полимеру,
находящемуся в непосредственной близости к возбужденной
молекуле221. Предполагается, что небольшое различие
энергетических уровней димера и мономера не уменьшает эффективность
такого переноса. Ассоциированные молекулы красителя
по этому предположению производят тушение двояким
способом: во-первых, поглощая свет в области спектра поглощения
красителя, но не излучая его обратно; во-вторых, и главным
образом, дезактивируя возбужденные молекулы путем
восприятия от них энергии
В последйее время Вавилов218 дал исчерпывающую
интерпретацию наблюдаемых при концентрационном тушении
кинетических закономерностей, исходя из представления о квантово-
механическом резонансе тождественных молехул,
сопровождающемся переяосом энергии возбуждения между ними на большом
расстоянии, причем оказалось необходимым допустить
зависимость вероятности переноса от времени.
Следует отметить, что явление концентрационного тушения
не менее специфично, чем тушение посторонними веществами.
Целый ряд флуоресцирующих соединений его не обнаруживают,
как например акридон219, пирен222, пинакриптол желтый214;
перилен в бензольном растворе и хининсульфат проявляют
слабое самотушение, хотя последний обнаруживает большую
„сферу действия" при тушении органическими соединениями
(стр. 251). Наоборот, нафтионат-Na, обнаруживающий
сильное самотушение, слабо тушится посторонними веществами214.
Для полициклических ароматических углеводородов, как
например антрацена, было показано выше (19), что тушение
флуоресценции собственными молекулами имеет химическую,
а не физическую природу и состоит в реакции дчмеризации
при встречах возбужденных молекул с нормальными.1)
Для фотохимии красителей особый интерес представляет
падение выхода фотореакции с увеличением собственной
концентрации красителя, аналогичное тушению флуоресценции.
В некоторых случаях удается установить параллелизм между
концентрационным тушением и торможением фотореакции, что
1) В последних исследованиях было обращено внимание на
необходимость исключения растворенного кислорода, так как последний является
сильным посторонним туши гелем н может запутать кинетические
закономерности 2а2. В частности, нагревание раствора, понижающее
растворимость воздуха, может вызвать усиление флуоресценции, помимо других
возможных интерпретаций температурного эффекта*
36. КОНЦЕНТРАЦИОННОЕ ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ И ТОРМОЖЕНИЕ 263
служит доказательством участия именно возбужденных
молекул, вступающих в реакции за время жизни т0.
Было, например, установлено, что скорость
фотовосстановления метиленового голубого в освобожденном от воздуха
глицериновом растворе падает с увеличением концентрации
красителя параллельно тому, как уменьшается интенсивность
флуоресценции223. Так, при возрастании концентрации
с 0.7-10~3М до 2'10~3М интенсивность флуоресценции упала
в 10.5 раз, а скорость фотореакции в 11 раз. Раствор
содержал для поддержания постоянства рН ацетат натрия в качестве
буфера, так как иначе получаются ненадежные результаты.
С эозином также наблюдается резкое торможение выцветания
в глицериновом растворе при увеличении концентрации.
Глицерин (полиспирт) дегидрируется при этом до альдегида или
кетона ш.
Торможение фотосенсибилизоваяного окисления
увеличением концентрации красителя-сенсибилизатора наблюдалось
неоднократно и получило соответствующее выражение в
кинетическом уравнении этих реакций. Связь этого явления с
концентрационным тушением, однако, не всегда сознавалась.
Интересный пример представляет поведение хлорофилла
(этилхлорофиллида), обладающего громадной константой
самотушения kqy в 2000 раз превышающей таковую для
полициклических углеводородов и красителей. Это обстоятельство
делает понятным, почему при обычных разведениях (10~J—
10~4 М) выход флуоресценции раствора этилхлорофиллнда
очень низок (9 = 0.08) и мало подвержен действию
посторонних тушителей222.
Если механизм фотосенсибилиэованного окисления
хлорофилла состоит на первом этапе в реакции возбужденной
молекулы СЫ * с органическим субстратом Y, то тушащие
встречи ее с собственными молекулами и должны, главным
образом, определять кинетику реакции.
Выражения для скорости и квантового выхода фотореакции
выводятся здесь совершенно так же, как это было сделано
в 22 при рассмотрении кинетики тушения кислородом с
заменой О2 на Y.
Пренебрегая в данном случае константой испускания kf
по сравнению с константами кд и kit доминирующих процессов
СЫ * -*- СЫ и СЫ*-нУ, мы получаем для стационарной
концентрации возбужденных светом молекул выражение:
где Nv есть число поглощаемых за секунду фотонов, a f0 —
264 ТОРМОЖЕНИЕ ФОТОРЕАКЦИЙ КРАСИТЕЛЕЙ
предельный квантовый выход флуоресценции в отсутствии
каких-либо тушителей- Это выражение, будучи подставлено
в уравнение для скорости исчезновения Y в результате
реакции, дает:
ГГЫ*1ГУ1- WVJY]
LChn[Y]-*.[Chl]H_Myj-
Для квантового выхода фотореакции, обусловленной только
возбужденными молекулами СЫ*( способными к флуоресцен-
ции, отсюда имеем:
t , k
j [Chi]
Это выражение совпадает с эмпирической зависимостью,
найденной Гаффроном для фотосенсибетлизованного
хлорофиллом (этилхлорофиллидом) окисления тиэзинамина (=Y)b
присутствии кислорода, которая приведена на фиг. 40 (стр. 236).
Из значения постоянной £=0.2, наблюдаемой на опыте,
следует, что kq = 5 kr9 т. е., что при (предполагаемых) равных
условиях диффузии возбужденные молекулы хлорофилла
взаимодействуют с собственными молекулами в 5 раз эффективнее,
чем с молекулами субстрата (тиозинамина). В основу втого
вывода положено предположение, что в реакцию вступают
только возбужденные молекулы за время их пребывания
в этом состоянии, сокращенное в результате собственного
тушения хлорофилла. Для проверки этого допущения
необходимы независимые измерения изменения выхода флуоресценции
и т, что при слабой интенсивности флуоресценции хлорофилла
затруднительно сделать.
Вейсс и Вейль-Малерб222, которым принадлежит этот вывод,
принимают для фотосенсибилизации хлорофиллом реакций
окисления кислородом иной первичный механизм, чем
дегидрирование субстрата Y (или "Y) по схеме (VIII) на стр. 234,
предложенной Франком и Ливингстоном. В соответствии с тем
фактом, что хлорофилл более склонен подвергаться
фотоокислению в присутствии акцепторов электрона, например Fe4"*""*"
(см. стр. 210), предполагается, что первичный этап состоит
в переносе электрона от возбужденной молекулы хлорофилла
субстрату, являющемуся акцептором электрона:
Для тиозинамина, являющегося типичным донором водорода
при реакциях фотовосстановления метиленового голубого
37. СПЕКТР ПОГЛОЩЕНИЯ АДСОРБИРОВАННОГО КРАСИТЕЛЯ 265
и др. красителей, такое допущение сомнительно. Мы полагаем,
что более применима здесь схема, где первичная реакция СЫ*
имеет место с кислородом, а не молекулой субстрата (стр. 240),
тем более что квантовый выход реакции Гаффрон измерял
по отношению к поглощаемому кислороду» а не по
исчезновению субстрата и нарочито указывал на отсутствие
ассоциации между молекулами хлорофилла и субстрата.
Остается неясным, заключается ли тушащее взаимодействие
СЫ*-нСЫ в простой дезактивации или фотодимеризации
с обратной темновой реакцией, как это имеет место в случае
антрацена (19).
ГЛАВА ДЕСЯТАЯ
ФОТОРЕАКЦИЯ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ
КРИСТАЛЛИЧЕСКИХ ТЕЛ
37. Спектр поглощения красителя, адсорбированного
кристаллическим телом
Изложенный в 24—27, а также других параграфах материал
дает некоторое представление о фотохимическом поведении
красителей, фиксированных на носителе органического
происхождения. Неорганические кристаллические тела в виде
порошка или коллоидного золя также способны адсорбировать
красители, более или менее прочно их удерживая.
Ионы красителей подчиняются общим закономерностям для
адсорбции ионов из растворов с тем отличием, что, обладая
значительно большими размерами, они слабее удерживаются
чисто электростатическими силами, исходящими из поверхности
кристалла225.
Катионы основных красителей адсорбируются
преимущественно „кислыми" адсорбентами с поверхностными отрица-
тельно заряженными группами, например /S*\ _>
образовавшимися в результате отделения протонов в окружающую
водную среду, или при наличии избытка отрицательных ионов
на поверхности кристалла, например ионов Вг~ на AgBr.
Анионы кислотных красителей, наоборот, адсорбируются,
главным образом, „основными" адсорбентами, поверхность
которых несет избыточный положительный заряд в результате
отдачи ионов ОН~ в раствор или предварительной адсорбции
катионов, например Agf"*" на AgBr.
"266 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
Однако для больших ионов красителя, для которых силы
сцепления ван-дер-Ваальса и другие типы взаимодействия
^превалируют над электростатическим притяжением или
отталкиванием, адсорбция может происходить и на одноименно
заряженной поверхности.
Установлено интересное явление преимущественной
адсорбции красителей на совершенно определенных
кристаллографических гранях226.
Хедвалл271 наблюдал усиление адсорбции красителя из
раствора, вызванное освещением твердого тела, К этому
явлению мы вернемся ниже (стр. 304).
Фотохимическое поведение адсорбированного красителя
осложняется его взаимодействием с кристаллическим носителем,
приводящим к новым видам элементарных физических и
химических реакций. В рамках этой книги мы остановимся только
на наиболее разработанных вопросах. Обратимся прежде всего
к влиянию адсорбции на спектр поглощения красителей й
родственных им соединений.
Вид и положение полос поглощения красителя испытывают
большие изменения только на таких адсорбентах, которые
способны существенно изменить структуру красителя путем
приема или отдачи протона (кислотно-основной тип
взаимодействия), электрона (окислительно-восстаиовительныйтип
взаимодействия) или же путем изменения состояния дисперсности
красителя, в частности, с образованием упорядоченной аггреги-
рованной формы красителя (14).
В большинстве случаев происходит сравнительно
небольшое смещение главного максимума полосы поглощения с
некоторым изменением величины коэффициента поглощения,
вызванное действием сильного электростатического подя,
исходящего от ионов на поверхности кристаллической решетки.
Каргин и Фодиман227 показали, что при адсорбции красителей
(метилеиовый голубой, тионин, метиловый голубой, эритрозин)
в растворе на золях SiO2, TiO2f As2O3 спектр поглощения
испытывает наиболее значительное изменение, когда
поверхность золя имеет знак, противоположный знаку иона
адсорбированного красителя. Однако характер изменения спектра
указывает на то, что эффект не обязан действию
электрического поля, а аггрегации ионов красителя на поверхности
в димерыиболее крупные частицы,1) На одноименно заряженных
1) Так, в случае адсорбции катионов метиленового голубого и тионина
на отрицательно заряженном воле TiO2 в водной среде превалирует именно
коротковолновой побочный максимум димерного иона (ср. стр. 91).
Концентрация красителя в растворе при описываемых опытах не была
значительной (около 4-10~6М).
37. СПЕКТР ПОГЛОЩЕНИЯ АДСОРБИРОВАННОГО КРАСИТЕЛЯ 267
золях изменение полосы поглощения адсорбированного
красителя было незначительным- Следует отметить, что коллоидные
частицы золя окружены плотной гидратной оболочкой,
препятствующей тесному взаимодействию красителя с адсорбентом.
В этом отношении опыты, проводимые в вакуумных условиях
(см. ниже), интерпретируются более однозначно.
Однако адсорбент может действовать и более активно,
вызывая распад бесцветной валентно связанной молекулы на
ионы с появлением окраски, присущей иону228""230. Так,
например, лейко-трифенилгалогениды PhgC-—Hal в бензольном или
хлороформенном растворе при адсорбции на тщательно
высушенном силикагеле, введенном в раствор, приобретают окраску,
характерную для иона Ph3C+ (ср. 18). При добавлении спирта
или ацетона, вытесняющего адсорбированные молекулы с
поверхности силикагеля, окраска исчезает. Явление протекает
обратимым образом230.
При адсорбции может произойти и отщепление протона.
Если адсорбировать амидофталевокислый гидр азид (а) на
катионах Th+"4"H, покрывающих отрицательно заряженную
поверхностьсиликагеля,то его нормальная голубая
флуоресценция, наблюдаемая в нейтральном растворе или при адсорбции
на отрицательном силикагеле, меняет цвет, превращаясь в
желтую, свойственную этому соединению вщелочном растворе
и принадлежащую структуре (Ь)231:
NH.HO
В чистых условиях спектры адсорбированных молекул были
получены в ряде исследований Де-Бура с сотрудниками232,
использовавшего в качестве адсорбента тонкие прозрачные
пористые слои CaF2, BaF2f ВаС12ит- п., полученные
сублимацией этих солей в вакууме. Большая внутренняя поверхность
слоев смогла быть измерена особыми остроумными приемами.
Главный интерес для нашего изложения представляют
результаты, полученные с фенольными индикаторами
кислотности: пара- и ортонитрофенолом, фенолфталеином, тимол-
268 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
фталеином, а также ализарином, Эги соединения адсорбируются
дипольной группой ОН на галоидных анионах F~, СГ~,
образующих, как было установлено, внешнюю поверхность слоистой
микроструктуры адсорбента. Как показали измере шя количества
адсорбированных молекул, при полном заполнении поверхности
кольца молекул паранитрофенола лежат плоско на поверхности
адсорбента даже при большой поверхностной концентрации.
Это объясняется сильным электростатическим взаимодействием
большого дипольного момента нитрогруппы с ионами решетки.
Когда нитрогруппа находится в молекуле в непосредственном
соседстве с группой ОН, как в о-нитрофеиоле, ароматическое
кольцо при большом заполнении поверхности отходит от нее
и располагается перпендикулярно к ней, ориентируясь
параллельно кольцам других молекул в результате их взаимного
притяжения.
Бесцветные фенолы при адсорбции приобретают окраску,
аналогично тому, как это имеет место в их водном растворе при
добавлении щелочи. Это значит, что адсорбция производит действие,
аналогичное электролитической ионизации молекулы, когда
отщепляется протон и образуется окрашенный в желтый цвет
фенолят-ион. Однако максимумы полос поглощения
адсорбированных фенолов не совпадают в точности с таковыми для
фенолят-иона и обнаруживают характерную зависимость ог
электростатического поля на поверхности кристаллической
решетки, которое для BaF2, например, сильнее, чем для CaF2
(ср. табл. 15).1)
Таблица 15
Положение максимума поглощения фенолов прн адсорбции на солях
(в тир.) (по Де-Буру232)
Наранитрофяиол . . .
Тимолфталеин
Анион ализарина,
адсорбированный на
катионах решетки М++ . .
CaF2
365
475
482
SrF2
495
BaF2
413
536
520
В гексане, нейтральном
водном растворе или
большом заполнении
поверхности СОЛН: 3i6 77IJA
В щелочном водной
растворе: 400 т}Х
(фенолят-анион)
*) Ввиду большего рздчуса иона Ва++ по сравчению с Са++, он в
меньшей степени нейтрализует поле противоположно заряженных поверхностных
ионов Hal", под которыми эти катионы находится. Когда галоидные анноны
37. СПЕКТР ПОГЛОЩЕНИЯ АДСОРБИРОВАННОГО КРАСИТЕЛЯ
269
Из факта отличия полосы адсорбированною паранитро-
фенола по положению от полосы для фенолят-аниона можно
сделать вывод, что полного отщепления протона в
адсорбированном состоянии не имеет места, а происходит только
усиление соответствующей внутриионизованной валентной
структуры нейтральной молекулы (6).
При повышении поверхностной концентрации
мономолекулярного слоя образуется, как показал Де-Бур, второй слой
молекул, который уже не взаимодействует непосредственно
с поверхностью адсорбента, а только с ^
покрывающими ее молекулами пер- ^
вого слоя. В соответствии с этими, "§
более слабыми, обычными силами §
сцепления, максимум поглощения *>
второго слоя практически сонпадает |
■с нормальным спектром, наблюдае- §
мым в инертном растворителе.
Из спектральных данных Де-Бур
выводит следствие, что второй слой |
невозмущенных молекул начинает %
образовываться до полного покры- ^
тия поверхности монослоем. Оче- о.
видно, образуются димерные моле- Щ
кулы, в которых только один
компонент находится непосредственно
на поверхности.
Cah
3
§/754
600 500 400
Длина волны в т^
Фиг. 44. Смещение полосы
поглощения аниона ализари-
Спектр фотографической сеНСИ- на, адсорбированного на
билизацаи. В связи с практическим фторидах * вакууме 231.
значением особенно подробному
исследованию в последнее время был подвергнут вопрос о
спектре поглощения красителей-сенсибилизаторов,
адсорбированных на бромистом серебре239.
Фогель, открывший в 1873 г. фотосенсибилизаияю
фотографической эмульсии красителями, установил также тот
факт, что максимум сенсибилизирующего действия смещен
на несколько десятков mj/. в красную сторону по сравнению
с максимумом полосы поглощения красителя в растворе.
Это отличие следовало, очевидно, приписать изменению
спектра поглощения красителя, наступающему в результате
удалены с поверхности путем реакции фенола с протоном и образованием
летучих соединении HHal, то остающийся фенолят-аннон (ализарина)
оказывается уже под непосредственным действием катионов Са~*~~*~, Sr**-"*~F
Ва~*~"*~. Тогда смещение полосы поглощения аииоиа ализарина,
происходящее в сторону коротких длин волн, убывает в этой
последовательности, как видно из фнг. 44 (ср. табл. 15).
270 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
адсорбции. Действительно, позднее удалось показать, что если
адсорбировать некоторые красители из парообразного их
состояния на дисперсном слое AgBr, полученном возгонкой
в вакууме, то спектр поглощения адсорбированного в этих
условиях красителя смещеи в красную сторону и совпадает
со спектров вызываемой им сенсибилизации в эмульсии.
Этот результат был получен для эритрозина, иод-эозина,
флоксина и родамина В23а. Совпадение спектров поглощения
и сенсибилизация было показано и непосредственно для
эмульсии бромистого серебра, окрашенного типичными
сенсибилизаторами— цианиновьши красителями2154. Интересно, что
на спектральной кривой, воспроизводящей сенсибилизирующее
действие красителей, воспроизводится (со сдвигом) не только
главный максимум поглощения (а на фиг. 23 в 14), но и
побочный максимум (|3), приписываемый димеру красителя235.
Интервал (в частотах v) между максимумами спектра поглощения
сохраняет свою величину и в спектре сенсибилизации.
Смещение полосы поглощения красителя, как целого,
в сторону меньших частот в результате адсорбции объясняется
взаимодействием электронной оболочки красителя с
кристаллом. Полагают, что она является непосредственной мерой
теплоты адсорбции2^5. В таком случае из наблюдаемой величины
смещения в 20 — 60 /пи. следует, что теплота адсорбции циани-
новых красителей составляет от 5 до 16 кгкал/моль, что
правдоподобно*
Большой сдвиг порядка 120 /nj/. и деформация спектра
сенсибилизации по сравнению со спектром поглощения
разбавленного раствора красителя свидетельствуют об аггрегации
красителя при адсорбции на бромистом серебре. В этих
условиях влияние молекул красителя друг на друга превалирует
над их взаимодействием с адсорбентом238t 2J8it.
Максимум поглощения аниона эритрозина,
адсорбированного на золях AgBr и AgJ в воде, отличается по положению
от такового для иона в растворе, но очень близок максимуму
того же аниона в кристалле эритрозината серебра229»2-16.
Так, для эритрозина, адсорбированного на AgJ, lma* = 547 m\t<
(фиг. 45), а для эритрозината серебра V"ax = 552 тр..
Это показывает, что анионы этого кислотного красителя
связываются на поверхности кристаллов галогенидов серебра
именно катионами Ag+ и испытывают при этом столь же
сильную деформацию, как в кристаллической решетке эри-
трозяната серебра. По всем признакам здесь имеется не столько
электростатическая поляризация аниона эритрозина в сильном
электрическом поле катиона Ag4", как наличие частичного,
обобществления электронов.
37. СПЕКТР ПОГЛОЩЕНИЯ АДСОРБИРОВАННОГО КРАСИТЕЛЯ
271
Малое значение электростатических сил для адсорбции
больших ионов красителей явствует из того факта, что нет
существенного различия в величине адсорбции и заполнения
поверхности AgBr между катионами основных и анионами
кислотных красителей (карбоцианинами и флуоресцеинами)237.
С увеличением концентрации красителя в растворе сначала
поверхность покрывается мономолекулярным слоем, в котором
каждая молекула (ион) красителя приходится на 2—6 ионов
Вг~~ бромистого серебра. После заЕершения монослоя
происходит новый подъем количества адсорбированного красителя
до второго состояния насыщения,
соответствующего наращиванию
сверх монослоя нескольких слоев
молекул красителя или изменению
состояния аггрегации в монослое.
Катионы цианиновых
красителей — типичных
фотографических сенсибилизаторов—
представляют собой длинные цепочки из
полимэтиновых сопряженных
связей, на концах которых
находятся гетероциклы, содержащие
атомы N (см. диаграмму 1 в 5).
Из валентной структурной фор- фиг- 45- Полоса ™гл°Щения
г* J г г аниона эритрознна в водном
мулы красителя следует, что по- растворе \7) и в адСОрбирован-
ложительный заряд его катиона ном состоянии на золе AgJ "
Л
550 600 ту.
будет осуществляться, главным
образом, этими атомами при
резонансе структур. В таком случае атомы азота будут
взаимодействовать с отрицательно заряженными ионами брома
на поверхности кристалла. С другой стороны тот же атом N,
обладающий в неионизованном трехвалентном состоянии
свободной парой электронов, способен взаимодействовать
с катионами Ag1"1", обобществляя с ним электроны наподобие
аммиакатов.1)
Поэтому несомненно, что имеет место частичный взаимный
обмен электронами между красителем и поверхностными
1) Действительно, многие основные красители легко обравуют в водном
растворе комплексы с нонамн серебра. Эти комплексы разлагаются уже
в темноте с выделением металлического серебра, чем обусловлено
вуалирующее дейстзие этих красителей на фотографическую эмульсию. Но это
явление ие связано непосредственно с сенсибилизацией254-1 (см. С7р. 288).
272 ФОТОРБАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
ионами Ag4" и Вг~, создающий новые возможности для
„резонанса" структур с выделением соответствующей энергии.
Последняя и обусловливает, главным образом, величину теплоты
адсорбции красителя и одновременно смещение спектра
поглощения.
Значение галоидных анионов для возможности адсорбции
красителя следует из того факта, что на карбонатах, оксалатах
и фосфатах серебра, а такжэ на сульфате и оксалате свинца
адсорбция сенсибилизаторов не имеет места.
Вытянутая плоская конфигурация скелета цианиторого
красителя удовлетворяет условию наибольшего резонанса
и обобществления электронов в молекуле (глава 2). Было
показано, что в самом первом адсорбироваииом монослое
молекулы красителя располагаются своей плоскостью почти
перпендикулярно поверхности кристалла бромистого серебра,
прилегая к ней длинными ребрами. Соседние молекулы
ориентируются своими плоскостями параллельно друг другу
наподобие колоды карт23'. Кислотный краситель — эритрозин—
также адсорбируется ребром к грани октаэдра кристалла,
взаимодействуя главным образом, с ионами Ag"~*~.
Как было описано в 14 и неоднократно отмечалось выше,
склонность к ассоциации свойственна ионам красителей
различных классов, независимо от структуры и заряда. Подобная
же аггрегация наблюдается при большой концентрации и для
адсорбции красителя на бромистом серебре. Образование
на поверхности димерных и полимерных ионов и других
аггрегированных* форм сказывается спектрально появлением
новых максимумов как в спектре поглощения раствора, так
и в спектре сенсибилизации. В зависимости от природы
растворителя (водного или спиртового) на поверхности AgBr
может преобладать мономерное, полимерное или коллоидное
состояние дисперсности красителя238.
Различным состоянием аггрегации красителя, создающим
различные максимумы спектра поглощения, свойственна также
фотосенсибилизующая способность, как это выяснилось путем
сравнения спектра сенсибилизации со спектром поглощения
красителя в растворе при различной концентрации2383.
Спектр поглощения коупакт^ых твердых пленок красителей,
получаемых осаждением из спиртового раствора на стекло,
обнаруживает по сравнению со спектром раствора также
сдвиг максимума в красную стороьу и значительное его
расширение57. Это значит, что взаимодействие тождественных
молекул красителя приводит к спектральным эффектам,
аналогичным тем, которые производит адсорбция на бромистом
серебре.
38. СЕНСИБИЛИЗОВАННЫЙ ФОТОЛИЗ БРОМИСТОГО СЕРЕБРА 273
38. Сенсибилизованный красителями фотолиз бромистого
серебра
Адсорбированные на галоидных солях серебра красители-
сопоибилизаторы, поглощая свет, производят выделение серебра
и, тем самым, дают начало „скрытому" изображению, про-
ипляемому в дальнейшем химическими или физическими
средствами. Процесс схематически можно записать следующим
образом:
Разложение галоидного серебра производится фотонами
в пределах полосы поглощения красителя А, далеко отстоящей
от спектральной области собственной фотохимической
чувствительности кристалла. Так, например, пентакарбоцианин,
содержащий две концевые гетероциклические группы,
соединенные длинной полиметиновой цепочкой:
Не^ — СН=СН — СН = СН— СН = СН — СН = СН —
— СН = СН — СН = Het2]+
придает бромистому серебру чувствительность к
инфракрасным длинам волн вплоть до ^ = 1350 т(л (Av = 21 кгкал/моль),
в то время как нормальная фоточувствительность AgBr
не простирается далее 7, = 480 ту* (Av = 60 кгкал/моль).
Сенсибилизирующей способностью, кроме цианиновых
(полиметиновых) красителей, обладают в меньшей степени
также многие другие красители, принадлежащие к ксантеновой,
фталеиновой и даже трифенилметановой группам.
Проблеме фотографической сенсибилизации посвящена
громадная литература239. В рамках настоящей книги ниже
рассматривается только вопрос о механизме этой фотореакции.
Энергетика процесса. Основной вопрос, возникающий
в связи с механизмом сенсибилизации, касается
минимальной величины энергии q, требующейся для разложения
кристаллического бромистого серебра без света. Энергия
фотона для максимума или длинноволнового края полосы
поглощения, как правило, всегда превышает ту работу,
которую требуется затратить для перевода системы из одного
равновесного состояния в другое также равновесное
состояние. Мы встретились с этим уже при рассмотрении
акта поглощения фотона отдельной молекулой (10), при
котором последней всегда сообщается избыток нибрационной
13 А. Н. Терснмн
274 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
энергии над равновесным его значением. Аналогичное имеет
место при фотораспаде кристаллов: акт диссоциации при
поглощении фотона происходит настолько мгновенно, что
образующиеся частицы сохраняют в первый момент свое
исходное положение, а окружающая их кристаллическая
решетка — свое состояние. Только в дальнейшем происходит
перестройка частиц в новую равновесную конфигурацию
с выделением энергии. С таким эффектом мы встретились
при расчете энергии фотона, вызывающего отрыв электрона
от гидратированного аниона галоида (29).
Таким образом, энергия фотона длинноволнового края
спектра поглощения бромистого серебра, находящегося,
примерно, у 480 т\ь (60 кгкал), должна явно превышать ту
минимальную работу q, которая требуется для плавного,
достаточно медленного (адиабатического) перевода системы в новое
состояние равновесия.
Попытки вычислить q9 исходя из различных вариантов
процесса разложения, привели к противоречивым
результатам239"241.
Из термохимических данных можно вычислить, что энергия,
требующаяся для разложения твердого бромистого серебра
[Ag-^Br"] на твердое металлическое серебро [Ag] и
газообразный атомарный (Вг), составляет 48 кгкал/моль:
-f-48 кгкал
Эту энергию, эквивалентную фотону с Х = 600 т[л,
отождествляют с величиной q. Действительно, собственная
чувствительность бромистого серебра может быть прослежена до
Х = 625 /п[л, где она в 100 000 раз ниже, чем в максимуме
чувствительности, расположенном у 450 mj/.. Ничтожная
чувствительность может быть обнаружена даже вплоть до 750/nj/.242.
Однако столь далекое продолжение в красную сторону
собственной чувствительности наблюдается только в эмульсии
и не имеет места аля чистого бромистого серебра без
связующего. Следовательно, вто продолжение вызвано
химическим изменением поверхности зерен AgBr в процессе
изготовления фотографического слоя, а не свойственно самому
бромистому серебру.
Затем, по существу неправильно отождествлять искомую
энергию q с энергией процесса (1), в котором образуются
раздельные фазы твердого [Ag] и газообразного (Вг).
В элементарном процессе разложения [Ag^Br"] получающиеся
первично атомы Ag1 и Вг остаются в окружении
кристаллической решетки и продолжают взаимодействовать с нею.
38. СЕНСИБИЛИЗОВАННЫЙ ФОТОЛИЗ БРОМИСТОГО СЕРЕБРА 275
Еще более ошибочно минимальную энергию, требующуюся
для фотолиза бромистого серебра, приравнивать
термохимическому значению энергии образования бромистого сереб ра
из элементов, т. е. суммарной теплоте реакции:
В самом деле, при фотолизе первично выделяются
пространственно разделенные атомы брома, которые рекомбинируют
в молекулы только впоследствии путем вторичного
процесса, а поэтому выделяющаяся при их рекомбинации энергия
23 кгкал (на один атом) никак не может быть включена
в энергетический баланс первичного акта, как это сделано
в (II) и как вто допускают многие авторы для самой фото-
реакции .
Правильнее энергию q сопоставить с значением
термической энергии активации темнового разложения бромистого
серебра. Для чистого AgBr эта величина еще не измерена.
Однако из температурного коэффициента скорости темнового
образования центров проявления в фотографической э м у л ь-
сии при нагревании следует, что энергия активации этого
процесса равна 15 кгкал/моль2*4. Это соответствует фотону
в инфракрасной области с длиной волны 1900 т[л.
Следовательно, если фотон способен произвести действие,
эквивалентное термической активации распада
бромистого серебра в эмульсии, то собственная фоточувствительность
последней может простираться в инфракрасную область еще
дальше, чем найденная на опыте граница у 750 тп{ь9 и даже
заходить за ту длину волны (1350 т[л), до которой удается
сенсибилизовать эмульсию красителями в настоящее время.
В связи с этим интересно, что Эбней в 1880 г. осуществил
случайно фотографическую эмульсию, чувствительную до
~к = 2000 тр., воспроизвести которую позднейшим
исследователям не удалось2-19» 266.
Таким образом, краситель-сенсибилизатор очувствляет
фотографическую эмульсию в пределах спектральной области,
где она еще сохраняет ничтожную, но все же
собственную фоточувствительность. Фотолиз бромистого серебра под
действием инфракрасного фотона, в 4 раза меньшего, чем
фотон синего света, поглощаемый кристаллом в максимуме
чувствительности, не представляется с этой точки зрения
удивительным и не противоречит энергетической возможности
процесса. Действие сенсибилизатора по втой концепции
заключается в том, что краситель создает интенсивное
276 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОЗЕрХНоСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
поглощение световой энергии в такой спектральной области,
где поглощение эмульсией настолько ничтожно, что оно
не обнаруживается прямым путем.
Одинаковое использование световой энергии, незазисимо
от того, поглощается ли она самим AgBr в области
собственной (максимальной чувствительности) или же поглощается
красителем-сенсибилизатором, следует из ряда фактов,
приводимых ниже.
Остается открытым главный вопрос о механизме
использования поглощенной красителем энергии.
Квантовый выход и выход сенсибилизации. Некоторым
шагом на пути решения поднятого вопроса является
измерение квантового выхода у фотореакции сенсибилизованного
разложения галоидного серебра, т. е. числа выделяющихся
атомов Ag9 приходящихся на один поглощенный красителем
фотон. Определение этой величины для высокодисперсного
объекта, каким является эмульсия или золь галоидного серебра,
представляет большие затруднения, что объясняет
расхождение данных, полученных различными методами. Интенсивность
прошедшего через такой объект светового потока зависит
не столько от поглощения света, сколько от его рассеяния
во все стороны; по этой причине расчет энергия,
поглощенной красителем, как и самим галоидным серебром, не может
быть произведен с помощью простых формул,
приведенных в 2.
Для прямого фотолиза AgBr и AgCl в состоянии
фотографической эмульсии или водного золя квантовый выход
близок единице, когда путем добавления акцепторов брома
_, 242
подавляют обратную реакцию воссоединения
Применяя типичные сенсибилизаторы из группы тиокарбо-
цнанинов, Шеппард с сотрудниками2^ нашел для квантового
выхода фотосенсибилизованного разложения эмульсии
бромистого серебра значение также близкое единице: у = 0.78 —1.28.
Выделившееся серебро при этом измерялось непосредственно
оптическим методом.
Сопоставление этих результатов приводит к выводу, что
фотон, поглощенный красителем, вызывает такое же
фотохимическое действие, как значительно больший фотон,
поглощаемый непосредственно бромистым серебром. Важность
этого факта для теории фотографической сенсибилизации
будет пояснена ниже.
Для эритроэина квантовый выход сенсибилизации оказался
определенно меньше единицы: у = 0.1—0.3. Другими словами,
из 3—10 поглотивших фотоны молекул эритрозина только
одна способна произвести выделение атома серебра.
38. СЕНСИБИЛИЗОВАННЫЙ ФОТОЛИЗ БРОМИСТОГО СЕРЕБРА
277
Сводка всех полученных численных данных приведена
в табл. 16.
Т аб л иц а 16
Квантовый выход у сенсибилизованного красителями фотолиза бромистого
серебра, ивмеренный по видимому почернению
(По Зохеру)247
Краситель
ствующая
X тпц,
Акцептор
брома
Литературная
с.ылка
Эритрозян
Пиронии G
Эритрознн .
Меэо-метил-тиазоловый пурпурный .
Фотослой, сенснбилизоваиный Орто-
хромом Т
С проявлением
579
579
579
546
546
590
0.06
0.1
0.06
03
1.04
1
не было
нитрит-Na
»
ацетонсемн-
карбазои
\ 246
245
0.004
0.08
желатина
242
Описанные определения квантового выхода основаны
на измерении выделившегося серебра или брома в условиях
большой интенсивности света, приводящей уже к видимому
почернению без дополнительного проявления. Эти условия
существенно отличаются от нормального использования
фотографической эмульсии при слабой засветке, когда образуется
только „скрытое" изображение. При малых ныдержках можно
подсчитать, что в этом случае примерно из 150 фотонов,
вступающих в фотослой, только один выделяет атом
серебра. Поскольку поглощается только часть их, то
квантовый выход еще ниже (ср. табл. 16, низ).
Расчет показывает, что для образования, после проявления
почернения с плотностью 0.1, достаточно в среднем
поглощения фотона одной молекулой красителя из 700 000 молекул,
покрывающих отдельное зерно AgBr в фотографической
эмульсии *42.1)
С другой стороны, для создания такого же почернения
без проявления, а только интенсивным 'освещением
3) На самых чувствительных дернах
по фотону 3—4 молекулы красителя.
в этих условиях поглощают
278 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
эмульсии, требуется поглощение каждой из 700 000 молекул
красителя, покрывающих зерно громадного числа 520
фотонов, причем каждая молекула выделяет только 2 атома
серебра. Если квантовый выход сенсибилизации в первом
случае составляет, как показывают измерения, 0.08, то во
втором случае ои равен только ^п== 0.004.
В гомологическом ряду полиметиновых сенсибилизаторов
удлинение цепочк^ сопряженных связей приводит не только
к последовательному смещению максимума полосы поглощения
в инфракрасную сторону (фиг. 11 в 8), но также к
прогрессирующему увеличению коэффициента поглощения красителя
(в растворенном состоянии). Между тем, эффективность
фотосенсибилизации по мере передвижения полосы в
инфракрасную область падает. Это должно указывать на
исключительно быстрое падение квантового выхода сенсибилизации
по мере поглощения красителями все меньших фотонов.
Для выяснения механизма фотореакции еще большее
значение, чем квантовый выход, имеет измерение так
называемого выхода сенсибилизации, т. е. числа
выделившихся атомов серебра, приходящихся за время экспозиции
на одну молекулу красителя.
Если в элементарной реакции фотосенсибилизации
молекула красителя разрушается, то она сможет вызвать выделение
только одного-двух атомов серебра. Если же в акте
сенсибилизации молекула красителя испытывает лишь обратимое
изменение, немедленно регенерируя в исходном состоянии,
то на одну молекулу адсорбированного красителя должно
приходиться большое число атомов серебра при достаточно
длительной экспозиции.
Трудности измерения этой величины, которую мы
обозначим с, не привели к однозначному ответу на этот вопрос.
С одной стороны, ряд исследователей218 получил для а
значения в несколько десятков атомов Ag, приходящихся на
молекулу сенсибилизатора, каковым служил эритрозин245' 246. Это
указывало бы на способность красителя многократно
поглощать фотоны и производить фотолиз AgBr. В более
совершенных условиях проведения опытов численное значение снизилось
до нескольких атомов серебра: ада 4—5. При общей
неточности этих измерений полученный результат не позволяет
однозначно выбрать между двумя приведенными выше
альтернативами.
При количественных исследованиях сенсибилизации
выяснилось новое обстоятельство, осложняющее интерпретацию
измерений. А именно было обнаружено, что бром, выделяю-
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 279
щийся при фотолизе бромистого серебра, реагирует
с красителем, приводя к его выцветанию. Последнее имеет
место как при освещении синим светом, поглощаемым только
AgBr, так и при освещении в полосе поглощения
адсорбированного краситзля. Наблюдается это явление как для эритрозина,
так и для основных красителей цианинового типа. На каждую
выцветшую молекулу эритрозииа приходится 1—2 атома
выделившегося серебра. Эритрозин, адсорбированный на BaSO4)
не выцветает245.
В присутствии акцепторов брома (нитрит-Na, фенол,
желатина и др.) сенсибилизатор не выцветает. Выяснилось, что
краситель функционирует, как своеобразный переносчик брома,
а именно способен обратимо его присоединять, легко отдавая
затем акцептору. Пииацианол, например, может присоединить
до 6 атомов брома. Если к выцветшему бромированному
отдельно раствору красителя прибавить золь серебра, то
образуется AgfBr, а краситель регенерирует246.
Последнее наблюдение Шеппард связывает с тем фактом,
что большая поверхностная концентрация сенсибилизатора
понижает собственную фоточувствительность бромистого
серебра, т. е. тормозит реакцию прямого фотолиза синим
светом. А именно он полагает, что в этих условиях
выделяющийся бром связывается, главным образом, красителем,
который легко его отдает, вызывая обратную реакцию с
выделившимся серебром. Такое объяснение не является, однако,
убедительным, так как возможны и другие эффекты.
39. Механизм фотосенсибилизации бромистого серебра
красителями
Прямой фотолиз бромистого серебра, с максимумом
спектральной чувствительности у 450 m^f не может быть
приписан переносу электрона с аниона Вг" к катиону Agf+,
как это происходит в кристаллах галоидных солей
щелочных металлов, например Na~43r~.
Величина поглощаемого фотона для бромистого натрия
может быть подсчитана в удовлетворительном согласии с
опытом, если разложить акт переноса электрона от Вг~ к
соседнему иону Na~*~ на ряд мыслимых этапов, энергия каждого
из которых известна, в полной аналогии с тэм, как это было
сделано в 29 по другому поводу. Если такой обходный путь
применить для Ag-Br, то для величины фотона, требующегося
на фотолиз по схеме
-U А*, Вг, (III)
280 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
получается 114 кгкал/моль, что соответствует ^ = 255 р,
вместо фактически наблюдаемого максимума поглощения
у 450 щ*. (60 кгкал)210.
Далее, в отличие от щелочио-галоидных солей, поглощение
света в кристаллах галоидного серебра вызывает не только
фотолиз, но и фотоэлектропроводность. Это явление
обусловлено передвижением по кристаллу в приложенном извне
электрическом поле тех электронов, которые вырваны светом
с анионов Вг~. Фотопроводимость обнаруживается в пределах
спектральной области поглощения А^Вг и может быть
прослежена даже до Ъ = 546 т\ъ.
Г
1
I
Пустая зона уровней
проводимости
AgBr
т
Jl
Ьг
лок
зона энергетических уровней
Фиг. 46. Зоны уровней энергии электрона в кристалле бромистого
серебра и механизм фотолиза.
По современным взглядам, ионный кристалл, каким является
Ag-Br, представляет собой подобие гигантской молекулы
с еднной электронной системой обобществленных электронов,
обладающей собственными уровнями энергии, отличными
от уровней энергии составляющих ионов Вг™ и Ag*~*~.
Энергетические уровни кристалла тесно группируются в непрерывные
полосы или зоны возможных значений энергии, разделенные
пустыми интервалами совершенно так же, как в атоме или
молекуле отдельные дискретные уровни „квантованной" элек-
тронной энергии разделены пустыми интервалами » .
Уровни самой нижней зоны заполнены электронами
аналогично тому, как заполнен в атоме или молекуле основной
уровень, а также более глубокие уровни, затрагиваемые
только большими фотонами рентгеновского излучения. На фиг. 46
эта нижняя зона уровней заштрихована. Она отвечает
нормальному устойчивому состоянию электронов в кристалле*
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 281*
Электроны, заполняющие уровни этой зоны, доставляются
нонами Вг~. Но наряду с зоной за нятых уровней, кристалл
имеет зону свободных пустых уровней, расположенную
на некоторой высоте над первой (фиг. 46), аналогично тому,
как у атома или молекулы имеется свободный от электрона
уровень возбужденного состояния. Величина поглощаемого
кристаллом фотона Л^а3Вг определяется разностью энергий
верхней и нижней зоны, а действие света состоит в переносе
электрона из заполненной зоны в пустующую.
Свободные уровни последней не являются вакантными
уровнями ионов Ag*4", а принадлежат всей кристаллической
решетке как целому. Поэтому, когда поглощение фотона
/*VAgBr переводит электрон с нижней зоны на верхнюю,
электрон ие принадлежит отдельному иону Ag1"*", а всему кристаллу,
и может передвигаться по кристаллической решетке, удаляясь
от места отрыва (фиг. 46). В этом заключается причина
возникновения электропроводности бромистого серебра при
освещении.
Поскольку исходный и конечный уровни акта поглощения
принадлежат непрерывным последовательностям уровней,
сгруппированным в виде зон, то совокупность поглощаемых
кристаллом фотонов образует непрерывную полосу,
занимающую широкое протяжение спектра. Минимальное
расстояние между границами зон определяет величину наименьшего
поглощаемого фотона, т. е. длинноволновую границу спектра
поглощения. Как мы видели, для чистого кристалла AgBr эта
граница находится приблизительно около 500 тр. и только
после химической обработки кристаллических зерен, имеющей
место при приготовлении эмульсии, она может быть
прослежена значительно далее в красную сторону.
Остановка в миграции электрона происходит тогда, когда
он достигает местного нарушения правильного строения
кристаллической решетки. Такие нарушения создаются уже
одним фактом наличия внешней поверхности, ограничивающей
кристаллическое зерно, а также наличием внутренних
поверхностей в микротрещинах и иных дефектах реального
кристалла. В этих местах имеются аналогичные вакантные уровни
для электрона, но в отличие от ненарушенной
монокристаллической решетки они не лежат в пустой зоне электронной
проводимости, а ниже ее (фиг. 46). Попадающий на такое место
электрон, естественно стремящийся занять более низкий
уровень энергии, заполняет „локальный" уровень и теряет тем
самым подвижность, свойственную нахождению в зоне
проводимости. Однако локальные уровни нарушений подобного
рода расположены настолько близко к зоне проводимости,
282 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
что теплового движения кристалла обычно достаточно, чтобы
снова вывести электрон в ^ону проводимости. Более
существенное значение для фотографической эмульсии имеют
иные нарушения кристалла, называемые „зародышами"
чувствительности, создаваемые иа поверхности зерна
технологическим процессом „созревания" эмульсии. Онн состоят
из ничтожных вкраплений металлического или сернистого
серебра и способны более прочно удержать электрон, чем
предыдущие дефекты.
Прикреплением электрона к такому центру и заканчивается
первый этап процесса, вызванный поглощением фотона.
В реальном зерне AgBr с решеткой, сильно нарушенной
в процессе кристаллизации, всегда имеется значительное
число катионов Agf'*", высвобожденных из своих нормальных
положений и имеющих возможность, благодаря малому раз-
меРУ* диффундировать через решетку, просачиваясь между
ее ионами. Этим объясняется, в частности, проникновение
я перемещение посторонних катионов через нагретый кристалл
Ag-Br в приложенном электрическом поле.
Появление при освещении отрицательного заряда на
центрах чувствительности решетки, вызванное прикреплением
к ним электрона, создает электрическое поле, которое
притягивает диффундирующие катионы Ag"*" к этим центрам.
Катион Ag""4", нейтрализуя заряд электрона в центре
чувствительности, выделяется там же в качестве атома, и процесс
захвата электрона центром и нейтрализации его заряда может
повториться.
Это объясняет тот факт, что, хотя акты поглощения
фотонов распределяются статистически по всему объему
кристаллического зерна, получающиеся атомы серебра
концентрируются только в определенных местах на поверхности, наращивая
при продолжающемся освещении коллоидную частицу
металлического серебра, служащую в дальнейшем центром
конденсации серебра при проявлении. „Скрытое" изображение и состоит
из таких частиц.1)
1) Визможно, что электроны нейтрализуют катионы серебра,
прилегающие к центру чувствительности, без необходимости допускать
диффузию ионов Agf*\ По мнению Боденштейяаа41, в баланс* энергии
должна участвовать теплота конденсации атома Ag на частице иегал-
лического серебра, равная теплоте испарения, т. с 66 кгкал. При учете
последней в первоначальном балансе энергии» составляемом для
вычисления величины поглощаемого фотоиа (III), теоретическая граница поглощения
сдвигаетси от 2S5 тц (114 кгкал) до 450 т(х (60 кгкал), в соответствии
с наблюдаемым ее положением. Однако сомнительно, чтобы миграция
електрона в зоне проводимости н выделение атома серебра происходили
настолько быстро, что можно было бы говорить об едином элементарном
акте изменении энергии всей системы как целого.
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 283
Как видим, реальный процесс фотолиза зерна
бромистого серебра в эмульсии гораздо сложнее, чем простая
картина переноса электрона между соседними ионами,
изображаемая схемой (Ш).
Разложение бромистого серебра при
фотосенсибилизации его красителем происходит, как указывают опытные
данные, по тому же механизму, какой был описан выше для
прямого фотолиза, с тем отличием, что энергия доставляется
теперь красителем. К такому заключению приводит факт, что
спектральная кривая фоточувствительности сенсибилизованной
эмульсии в точности воспроизводит ее спектральную кривую
поглощения, если последнюю относить к одинаковым
значениям поглощенной энергии. Различия между эффективностью
действия разных количеств энергии в спектральной области
собственной чувствительности эмульсии и области
сенсибилизации не наблюдается247. Это показывает
эквивалентность поглощенной энергии, независимо от того, поглощается
ли она кристаллом непосредственно нли красителем.1) Такой
результат получен для панхроматической эмульсии; для
ортохроматической эмульсии поглощение света красителем
менее эффективно, чем поглощение непосредственно
бромистым серебром.
Одинаковое использование световой энергии, независимо
от того, поглощается ли она самим AgBr, в области собстиэн-
ной чувствительности или же сенсибилизатором, следует
также из фактов одинакового нарушения фотохимического
закона взаимозаменимогти (закона Бунзена-Роско, см. 15),
а также одинакового действия прерывчатого освещения в обеих
спектральных областях, как это установлено для эмульсий250.
Наконец, наиболее веский аргумент состоит в том, что
отложение коллоидного металлического серебра в виде центров
проявления происходит в объеме кристаллического зерна
как при прямом, так и сенсибилиэованном фотолизе, несмотря
на то, что в последнем случае поглощающий свет краситель
локализован на внешней поверхности зерна.
Главная проблема теории фотографической сенсибилизации
заключается в вопросе: каким путем поглощение красителем
фотона в 3—4 раза меньшего, чем фотон, требующийся для
прямого действия, способно привести к фотолизу кристалла.
Разнообразные точки зрения на механизм действия сенси-
]) Разумеется, это не затрагивает вопроса о квантовом выходе фото-
реакцин, т. е. фоточувстителъности, отнесенной к одинаковому числу
поглощенных фотонов, которая для далеко отстоящих длин воли в этих
областях будет различной.
284 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
билизатора могут быть сведены к двум основными представле*
ниям:
1) краситель передает поглощенную им энергию
кристаллу;
2) краситель передает электрон кристаллу.
Передача энергии* Согласно первой теории,
адсорбированный краситель-сенсибилизатор передает бромистому серебру
энергию поглощенного им фотона по аналогии с процессами
переноса энергии возбуждения, наблюдаемого для частиц
в газообразной фазе239 (12). При такой природе
сенсибилизации краситель заведомо не испытывает необратимых
изменений структуры и может повторить тот же акт передачи
энергии боль/шое число раз через весьма короткие интервалы
времени. Большое численное значение выхода сенсибилизации с
(стр. 278) послужило бы решающим доводом в пользу этой
концепции. Между тем, измерения приводят к значениям
а = 1—5 (атомов серебра на одну молекулу красителя), что
при их неточности не дает возможности сделать
определенный вывод.
Главный вопрос теории сенсибилизации о несоответствии
поглощенного красителем фотона Ava зиачительво большему
интервалу между зонами энергии AgBr (фиг. 46) с точки
зрения передачи энергии трактуется следующим образом.
В главе 3 (9—11) мы рассмотрели подробно, как изменяется
запас вибрационной энергии молекулы при актах поглощения
и испускания фотона. Мы объяснили механизм антистоксового
испускания, прн котором испускаемый фотон превосходит
по величине поглощаемый путем заимствования из запаса
тепловой энергии вибрационного движения молекулы (стр. 61).
Этот запас (вибрационная молекулярная теплоемкость)
достигает значительной величины ввиду сложности структуры
молекулы и многочисленности различных типов вибраций,
совершаемых ее скелетом.
В нашей лаборатории было обнаружено Прилежаевой
явление антистоксовой сенсибилизованной
флуоресценции, объясняемое тем, что увеличенный за счет тепловой
энергии квант передается второй молекуле другого рода
и возбуждает испускание в последней фотона,
превосходящего по величине фотон, поглощенный первой молекулой
(стр. 78). Явление наблюдалось в газообразной фазе.
Количество энергии, черпаемое при этом из запаса термической
энергии молекулы, обычно невелико, составляя около 10 кгкал.
Это наблюдение согласуется с ориентировочным расчетом
Франка-Герцфельда251 той дополнительной энергии, которая
может быть доставлена из запаса вибрационной энергии при
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 285
-фотореакциях с квантовыми выходами в пределах от у =0.1
у = 1. Из подсчета следует возможность получения
дополу у
нительной энергии в 5—10 кгкал.
Участие термической энергии в реакции фотографической
сенсибилизации явственно сказывается в том важном факте,
что с понижением температуры чувствительность эмульсии
падает в спектральной области сенсибилизации
значительно быстрее, чем в области собственной чувствительности
бромистого серебра252.
Выше было нами отмечено быстрое падение эффективности
сенсибилизации при перемещении полосы поглощения красителя
в инфракрасную область, т. е. при увеличении дефицита
энергии по сравнению с фотолизом в области собственной
чувствительности эмульсии. В гомологическом ряду цианино-
вых красителей эффект сенсибилизации1) падает в 70 раз при
переходе от сенсибилизатора с максимумом чувствительности
1 = 590 т\ъ (48 кгкал) к сенсибилизатору с максимумом
Х = 780 т\>* (36 кгкал). С другой стороны, температурный
коэффициент сенсибилизовавной чувствительности для
последнего сенсибилизатора в 30 раз больше, чем для первого:
0.03 вместо 0.001. Эти данные были использованы Мейкляром
и Степановым200 для расчета числа типов (степеней свободы)
вибрации красителя, тепловая энергия которых может
восполнить поглощаемый фотон до фотона границы собственной
чувствительности бромистого серебра (1 = 480 т\ьр Av = 6lKncaA).
Оказалось, что, независимо от длины полиметиновой цепочки
сенсибилизатора, вполне достаточно термической энергии
около 30 типов вибрации молекулы красителя из всех 150—
200 типов для покрытия недостающей энергии в каждом
случае. Другими словами, согласно этой концепции, для
покрытия дефицита, изменяющегося от 13 кгкал (для X 590 т\ь)
АО 25 кгкал (для > 780 т\ь), всегда используется
определенная часть (примерно 20 %) тепловой энергии молекулы
сенсибилизатора.
По мысли авторов, поглотившая фотон молекула красителя
в результате дополнительного участия тепловой энергии
оказывается фактически на высоком уровне энергии,
совпадающем с верхней зоной кристалла. Осуществленное таким
способом совпадение нижнего и верхнего уровней
благоприятствует квантово-механнческому резонансу красителя и
кристалла, приводя к весьма эффективному переносу энергии от
первого к последнему. Падение эффекта сенсибилизации по мере
1) Не величина квантового выхода, которая дли указанных здесь сенси-
билиэованных эмульсия: пока ие определилась.
286 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
уменьшения величины поглощаемого сенсибилизатором фотона
вызвано с этой точки зрения исключительно все
уменьшающейся вероятностью сенсибилизатору восполнить возрастающий
дефицит энергии из своего теплового запаса.
Факт снижения эффективности сенсибилизации при
повышении поверхностной концентрации красителя с изложенной
выше точки зрения объясняется дезактивацией собственными
молекулами, аналогичной концентрационному тушению
флуоресценции (36),
Наличие положительного температурного коэфицнента в
эффекте сенсибилизации с несомненностью указывает на необ*
ходимость дополнительной термической активации
фотореакции. Однако картина переноса суммарной энергии от
красителя кристаллу допускает далеко идущую аналогию
с реакциями обмена энергией возбуждения между
газообразными молекулами при их столкновении. Не учитывается
непродуктивная деградация поглощенного красителем фотона
в тепло, которая вызывает отсутствие флуоресценции у
большинства сенсибилизаторов в жидких и твердых растворах при
обычной температуре. Способность красителя к флуоресценции
и его эффективность в качестве сенсибилизатора скорее
взаимно противоположны. Так, например, в ряду уранин-эозин-
эритрозин выход флуоресценции уменьшается, а сенсибили-
зующая способность возрастает в этой последовательности.1)
Принимая использование всей энергии поглощенного
сенсибилизатором фотона, необходимо допустить одновременно,
что обычная дезактивация возбужденной молекулы с
деградацией поглощенного фотона в тепло полностью устраняется
при адсорбции красителя, поскольку квантовый выход
сенсибилизации для типичных сенсибилизаторов близок единице^
а следовательно каждый поглощенный ими фотон ведет
к реакции.
Концепция передачи энергии не- требует, однако, чтобы
доставляемый красителем квант был обязательно равен
интервалу между зонами, составляющему 60 кгкал, как это следует
из границы фоточувствительности (фиг. 46). Было указано
выше, что собственная чувствительность бромистого серебра
в эмульсии продолжается до 625 тр и можег быть про-
1) Это наводит на мысль, что при фотографической сенсибилизации,
как н при других рассмотренных выше реакциях, главную роль играет
не возбуждевиое, а мвтастабильное состояние красителя. Проверке этого
теаисв посвящена одна из работ лабораторяи автора книги. Следует,
однако, отметить, что квантовый выход флуоресценции эритрозина»
а также эозина завнсвт от растворителя и в ацетоне равен единице-
(стр. 74).
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 287
слежена до 750 т\ь (38 кгкал) и что минимальная
термическая энергия q= 15 кгкал, требующаяся для выделения
центров проявления без освещения, эквивалентна границе
собственной чувствительности, лежащей у Ъ = 1900 тр*
Краситель-сенсибилизатор всегда поглощает фотон заведомо
большей величины, чем q> и располагает, следовательно,
достаточной энергией, в особенности при дополнительном
участии термической энергии вибраций.
Возникает теперь вопрос, какова причина продолжения
собственной чувствительности бромистого серебра, хотя
и сильно ослабленной в сторону столь длинных волн. Это
объясняется тем, что граница спектра поглощения и
фоточувствительности AgfBr у 480 т[л принадлежит его
неискаженной кристаллической решетке и наблюдается в
монокристалле. В реальном микрокристаллическом бромистом
серебре, особенно в эмульсии, существуют многочисленные дефекты
и нарушения правильной структуры решетки, локализованные
преимущественно на внешней поверхности кристаллического
зерна. В местах таких нарушений уровень, занятый
электроном иона Вг~, представляет собой о б о с о б ленный уровень,
не связанный с нижней зоной кристалла и расположенный
над нею (фиг. 46). Это значит, что требуется меньший
квант энергии для отрыва электрона от деформированного
места в решетке и перевода его в зону проводимости.
Непрерывная градация степени нарушения решетки и создает
непрерывный набор таких обособленных электронных уровней,,
а следовательно, вызывает продолжение фоточувствительности
за четкую границу, характерную для преобладающего
ненарушенного в объеме кристалла.1)
Адсорбция красителя происходит на поверхности зерна
и имеются все основания предполагать, что в первую очередь
краситель адсорбируется именно на дефектах поверхности
кристалла, как местах повышенного силового поля.
Из предыдущего следует, что адсорбированные в местах
нарушения молекулы красителя находятся в особо
благоприятных условиях для осуществления обязательного этапа
фотолиза, а именно перевода электрона в зону проводимости,
так как на это здесь требуется меньшая энергия. С этой
точки зрения не удивляет, что оптимум сенсибилизации
достигается прн такой поверхностной концентрации красителя,
когда полное покрытие монослоем еще не достигнуто.
*) Как было сказано выше, нарушения того же или иного типа создают
те обособленные вакантные уровни, расположенные ниже верхней зоны
проводимости, попадай на которые, мигрирующий электрон закрепляется
в месте нарушения.
288 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
Однако сомнительно, чтобы наблюдаемый большой
квантовый выход сенсибилизации мог быть обеспечен
поглощением света только теми, сравнительно немногочисленными
молекулами красителя, которые адсорбированы на местах
нарушения поверхности кристаллического зерна. Поэтому
вопрос об источнике восполнения дефицита энергии для
большинства молекул красителя, адсорбированных не на дефектах
поверхности, остается открытым.
Передача электрона. Вторая группа теорий
фотографической сенсибилизации предполагает, что электрон доставляется
в верхнюю зону проводимости (в конечном счете — иону Ag4")
не от аниона Вг~, как при прямом фотолизе, а от поглотившей
фотон молекулы красителя254. По химической терминологии
последняя испытывает реакцию фотоокисления в присутствии
акцептора электрона (31), которым здесь является бромистое
серебро (или Ар;-*-).
Действительно, как показали Мекке и Земерано254, циани-
новые инфракрасные сенсибилизаторы выделяют из раствора
нитрата серебра металлическое серебро, т. е. передают
электроны ионам серебра даже без освещения. В нейтральном
растворе на одну прореагировавшую молекулу красителя
выделяется один атом серебра. Но краситель при этом
необратимо разрушается. Тот же краситель, адсорбированный на
водном золе бромистого серебра в темноте, устойчив, а на
свету выделяет серебро из золя, но одновременно также
разрушается.1) Отсюда был сделан вывод, что рассматриваемые
инфракрасные сенсибилизаторы обладают по отношению к
бромистому серебру под действием света явно выраженной
восстанавливающей способностью и что в этом явлении состоит
сущность фотографической сенсибилизации. Однако
одновременное разрушение красителя служит доводом против такой
чисто химической концепции, в особенности, когда было
установлено, что выход сенсибилизации с больше единицы.
Между тем, наблюдаемое фотохимическое разрушение
нестабильного инфракрасного сенсибилизатора, вуалирующего
фотографическую эмульсию даже при хранении в темноте,
является побочным обстоятельством, вызванным вторичной
реакцией, потерявшей электрон молекулы красителя с
кислородом воздуха. Вполне мыслимо, что в условиях адсорбции
первичный акт отдачи электрона красителем сопровождается
приемом им же другого электрона от иона брома или нижней
\) Для дианнновых сенсибилизаторов к видимому спектру (например
пинавердола) также наблюдается химическое взаимодействие с ионами
серебра, но это явление скорее характеризует вх вуалирующую» а не сен-
■снбнлизующую способность, как покавалн Гороховский и Натансон 254а.
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 289
заполненной электронами зоны кристалла бромистого
серебра.
Действительно, сенсибилизаторы принадлежат к красителям,
обладающим способностью легко отдавать электрон, т. е.
окисляться в присутствии акцепторов электрона.
В случае цианиновых сенсибилизаторов такое свойство
присуще концевым хииолиновым кольцам, содержащим азот
с парой свободных электронов. При резонансе валентных
-структур тот же атом азота может использовать эту
свободную пару электронов иным путем, делаясь четырехвалентным
и заряжаясь положительно (6). Отсюда следует, как было
упомянуто выше, что атом азота в молекуле сенсибилизатора
способен взаимодействовать как с катионом Ag"+, так и с
анионом Вг~. Таким образом, возможно, что при поглощении
сенсибилизатором фотона происходит перенос электрона от
последнего к первому через посредство электронной
структуры красителя. Такой механизм давно допускал Баур в своем
объяснении фотосенсибилизации, включающем сенсибилизацию
бромистого серебра (см. стр. 215), и изображал схемой;
Л i © -+- Вг~ -> Вг
По существу тот же механизм сенсибилизации выдвинули
позднее Мекке и Земерано.1)
1) Эти авторы приводят следующую конкретную картину переноса
электрона:
п
Предполагается, что под действием электростатического поля
левого аниона Вг~„ принадлежащего поверхности кристалла, находящийся
над ним атом N красители эарижастси положительно, принимая валентную
структуру —N^ с перемещением электрона к атому С ближайшей мети-
новой группы. В результате последний заряжается отрицательно,
приобретая валентную структуру — СН—, н отдает электрон катиону
поверхности. Первоначальный заряд красителя восстанавливается путем отдачи
электрона от правого аниона Вг~~ соседнему атому N"*~. Такая конкретизация
механизма сенсибилизации, оперирующая только одной валентной структурой
красителя, весьма спорна. Однако вполне мыслимо, что воздействие ионов
поверхности решетки на возбужденную молекулу красителя может повести
к усилению удельного веса структуры, благоприятствующей обмену
-электронами между сенсибилизатором и кристаллом.
1У А. Н. Тс рении
290 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
Гэрни и Мотт254 предложили аналогичную теорию
сенсибилизации, по которой исходный (основной) уровень электрона
в красителе-сенсибилизаторе расположен в промежутке
между зонами кристалла (фиг. 47) на такой высоте, что
энергия поглощаемого красителя фотона AvA вполне достаточна
для переноса электрона от красителя в зону проводимости.
Возможность отрыва электрона от сенсибилизатора
действием света в пределах его видимой полосы поглощения
установлена опытами Щодро над фото-
электропроводностью коллодионных пленок,
окрашенных цианином, пинавердолом, пина-
хромом, и опытами Вартаняна над
фотоэлектропроводимостью осажденных слоев
красителей. К этим работам мы возвращаемся
в следующей главе (42).
Было показано далее, что бромистое
серебро, окрашенное эрнтрозином и цианяно-
вымн красителями, делается проводящим при
освещении не только в спектральной
области собственной чувствительности, но также
бОНгкач
-Т
Г255
Фиг. 47. Механизм
сенснбилизоваино-
го красителем фо-
голиза бромистого
серебра по
гипотезе передачи
электрона.
НИИ
в пределах полосы поглощения красителя-
Шеппардом25а было в последнее время
обнаружено, что слой бромистого серебра на
серебряном электроде в растворе влектро-
лита, окрашенный одним из тиокарбоциа-
нинов, дает фотовольтаический эффект
(явление Беккереля, см. 44) при освещении
длинами волн, поглощаемыми красителем.
Этот факт также свидетельствует о появле-
электронов в зоне проводимости кристалла при действии
света на к р а с и т ел ь.
Главная трудность; в концепции переноса электрона от
сенсибилизатора есть объяснение механизма, каким красителю
доставляется второй электрон, необходимый для его
регенерации (см. фиг. 47).
Гэрни и Мотт полагают, что избыточный положительный
заряд, возникающий на молекуле красителя после утраты
электрона, достаточен для отталкивания соседних с ней
катионов Ag"**, способных диффундировать по решетке согласно
описанной выше картине образования скрытого изображения
(стр. 282). По этой причине в непосредственной близости
к красителю в решетке создается пустое место, не занятое
катионом Ag+. He испытывая притяжения последнего,
электрон ближайшего аниона Вг" оказывается более слабо
связанным, а, следовательно, занятый им уровень энергии поднимется
39.( МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 291
выше заполненной нижней зоны кристалла. Есля этот уровень
поднимется выше опустевшего основного уровня
сенсибилизатора, произойдет переход электрона на последний и
регенерация красителя за счет соседнего с ним аниона Вг~.
Наличие тесного взаимодействия между электронной
оболочкой адсорбированного красителя и электроном иона
брома принимается всеми последними теориями
сенсибилизации. Присутствие в окружении красителя нонов брома имеет
решающее значение не только для адсорбции (стр. 272),
ио и для возможности сенсибилизации. Так, хотя эритрозинат
серебра и эрнтрозин, адсорбированный на AgBr, имеют
практически тождественный спектр поглощения (стр. 270)
при экспозиции, вызывающей значительное выделение серебра
в случае адсорбированного эритрозина, эритрозинат серебра
заметно не разлагается236.
Шеппард235 придает также большое значение ориентации
плоской молекулы сенсибилизатора своей длиной параллельно
направлению кристаллографической плоскости AgBr,
содержащей ионы брома, допуская взаимодействие осциллирующего
в молекуле заряда (8) с этими нонами.
В связи с изложенным следует упомянуть гипотезу Франка
и Теллера об источнике энергии, покрывающей дефицит при
сенсибилизации257. Они полагают, что в результате разрядки
аниона брома выделяется значительная потенциальная энергия
поляризации кристаллической решетки, вызванная действием
электростатического поля иона брома на окружающие ионы»
Эта энергия не входит в баланс прямого фотолиза
бромистого серебра, так как он происходит в виде мгновенного акта,
по завершении которого и независимо от которого наступает
затем более медленная перегруппировка решетки вокруг
потерявшего заряд атома брома. При поглощении света
красителем предполагается, что возбужденное состояние
последнего достаточно длительно, чтобы изменение
конфигурации решетки успело за это время завершиться. В таком
случае выделяющаяся энергия должна быть учтена в общем
энергетическом балансе процесса наряду с поглощенным
фотоном. В результате поглотивший фотон краситель и
непрерывно взаимодействующий с ним ион Вг~ оказываются
располагающими суммарной энергией, достаточной для
перекрывания интервала от нижней до верхней зоны кристалла.
Роль сенсибилизатора, как донора электрона» заставляет
помещать его исходный основной электронный уровень на
определенной высоте по отношению к зонам
кристаллической решетки. А именно, он должен быть помещен в
соответствии с абсолютными значениями энергий отрыва электрона
19*
292 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
От красителя и от AjjBr, выводимыми из величины
соответствующих редокс-потенциалов (28). Если исходный занятый уровень
красителя помещен настолько низко, что поглощаемый им
фотон недостаточен для перевода электрона в зону
проводимости, то такой краситель будет не способен к сенсибилизации.
Это может сразу объяснить, почему при тождественном
положении полос поглощения и одинаковой адсорбции
только некоторые красители являются сенсибилизаторами
в противовес концепции простой передачи энергии, в которой
такой дискриминации нет и требуется дополнительное
допущение.
Против картины процесса, даваемой теорией Гэрни и Мотт,
следует, однако, выдвинуть то замечание, что нахождение
верхнего уровня возбужденного состояния красителя в преде*
лах вакантных электронных уровней зоны проводимости
(фиг. 47) по кваитово-механическим представлениям должно
было бы вызвать аномально большое расширение и искажение
формы спектральной кривой поглощения адсорбированного
красителя, электрону которого при возбуждении был бы
предоставлен тем самым непрерывный и широкий диапазон
возможных для него уровней. Между тем, кроме небольшого
сдвига, вид полосы поглощения или полосы сенсибилизации
адсорбированного красителя мало отличается от вида полосы
для того же красителя в растворе. Этот факт, а также
необходимость дополнительной термической энергии активации при
сенсибилизации (стр. 285) заставляют думать, что верхний —
возбужденный—уровень электрона в красителе всегда
расположен ниже зоны проводимости и достигается только путем
затраты дополнительной энергии, доставляемой тепловым
движением.
Некоторую аналогию можно видеть в факте наличия
положительного температурного коэффициента для фотоэлектро-
проводиости у твердых слоев красителей, обнаруженном Варта-
нявом (42). Из его величины можно сделать заключение, что
необходима дополнительная затрата термической энергии
активации порядка 10 кгкал, для того чтобы электрон от
возбужденной светом молекулы красителя мог перейти в зону
проводимости кристаллической решетки самого красителя.
Повидимому, такое же явление происходит, когда молекула
красителя адсорбиронана на кристалле бромистого серебра.
Из изложенного выше можно сделать вывод, что если
возмещение электрона сенсибилизатору происходит вскоре же
после его отдачи, то механизм сенсибилизации путем переноса
электрона фактически будет неотличим от механизма
передачи энергии.
39. МЕХАНИЗМ ФОТОСЕНСИБИЛИЗАЦИИ БРОМИСТОГО СЕРЕБРА 293
Остановимся на одном структурном факторе, существенном
для сенсибилизации. Выше было отмечено растянутое
расположение адсорбированной молекулы, прикасающейся своим
длинным ребром к поверхности кристалла (стр. 272). Бруке-
ром с сотрудниками258 было открыто, что изомеры цианиновых
сенсибилизаторов, имеющие не плоский скелет, полностью
лишены способности сенсибилизации. Так, красители (а) и(Ь)
имеют изомеры (а') и (Ь'), которые одинаково хорошо
адсорбируются бромистым серебром.
/V
/\/\
Et
Et
(b')
Красители (а) и (Ь) имеют плоский скелет и являются
сенсибилизаторами. Но в изомерах (а') и (Ь'), из-за близости
водородных атомов, размещение всех атомов молекулы в одной
плоскости невозможно и эти изомеры неспособны к
сенсибилизации.
В связи со склонностью некоторых цианиновых
красителей к упорядоченной аггрегации в громадные макромолекулы
(14, п. 4), Шейбе259 выдвинул гипотезу о возможности
аккумуляции в последней энергии нескольких разновременно
поглощенных фотонов, с отдачей всей накопленной энергии
в виде единого кванта при сенсибилизации бромистого серебра.
Это объяснение несостоятельно потому, что если бы для
выделения одного атома серебра требовалось поглощение 2—3
фотонов, то в кинетическом уравнении следовало бы ввести
произведение „концентраций" фотонов и
фотографическое действие сенсибилизатора было бы пропорциональным
не первой, а второй или третьей степени интенсивности света,
что противоречит наблюдениям.
294 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТ*ЦКРИСТАЛЛЧЧЕСКИХ ТЕЛ
Однако эффект сенсибилизации, наблюдающийся в
спектральных полосах, заведомо принадлежащих агтрегатам и поли-
иоиам, а также коллоидным частицам красителя, заставляет
поставить самостоятельный вопрос о механизме такого
явления. Поскольку поглощают свет любые молекулы, входящие
в состав такого аггрегата, а сенсибилизируют только те,
которые расположены непосредственно на поверхности
бромистого серебра, то, очевидно, существует беспрепятственный
перенос энергии или влектрона по такому аггрегату даже
в том случае, когда он не имеет кристаллического строения.
Измерения квантового выхода сенсибилизации для таких
аггрегатов представляет с этой точки зрения значительный
интерес.
Сказанное выше относительно бромистого серебра может
быть применено и к другим галогенидам серебра, а также
ионным кристаллам аналогичного типа, ио ввиду его
практического значения подробные данные относятся» главным
образом, к бромистому серебру.
40. Вуалирующее действие красителей на бромистое
серебро и десенсибилизация
В главе 8 (34) нами упоминалась теория фотографической
сенсибилизации Кбгеля-Штейгмана, по которой источником
электрона как при прямом, так и сенсибилизованиом фотолизе
AgfBr является желатина эмульсии или иной органический
восстановитель. Вместе с электроном переносится и протон,
т. е. в целом атом водорода, который вновь гидрирует
сенсибилизатор, дегидрированный бромистым серебром в акте
сенсибилизации. В духе концепции Виланда об окислении,
как дегидрировании, указанные авторы изображают прямой
фотолиз следующей схемой 173«260:
а -*.г» — AgBr _
А§г"*"ог -+-желатина > Ag Br +H + окисленная желатина
Н ч- Ag*Br- > А^-+- НВг,
где поглотившее фотон бромистое серебро путем передачи
энергии „мобилизует" атом водорода восстановителя
(желатины). Роль красителя-сенсибилизатора, по мысли авторов,
состоит в том, что он, поглощая фотон, передает эту энергию
желатине, „мобилизуя" и передавая атом водорода
бромистому серебру, как в предыдущем случае. Краситель при этом
испытывает лишь обратимое изменение (ср. стр. 241). Этой
схеме противоречит возможность прямого и сенсибилизован-
40. ВУАЛИРУЮЩЕЕ ДЕЙСТВИЕ КРАСИТЕЛЕЙ НА БРОМИСТОЕ СЕРЕБРО 295
ного фотолиза бромистого серебра в отсутствии
восстановителей, а также тот факт, что фотографические
сенсибилизаторы не принадлежат к обратимо гидрирующимся красителям.
В 31 на стр. 213 был описан „эффект метиленового
голубого", который рассматривается Кбгелем и Штейгманом, как
подтверждение выдвинутой ими концепции. Он состоит в том,
что желатиновая пленка, окрашенная указанным красителем,
после освещения оказывается способной выделять серебро
из раствора нитрата, т. е. быть донором электрона. Однако
Шмидт показал, что тот же эффект в большей или меньшей
степени воспроизводят все красители, как сенсибилизаторы,
так и десенсибилизаторы 173. Активность окрашенной пленки
была приписана нами образованию при освещении
полувосстановленной формы, т. е. семихинона красителя.
Способность красителей осуществлять обратимым образом
перенос электрона от постороннего восстановителя акцептору,
в данном случае иону Agf *", заслуживает внимания.
Известно, что метиленовый голубой, а также зеленый
Биндшедлера и другие (кубовые) красители вуалируют
эмульсию бромистого серебра в желатине без с в е та, т. е. создают
в темноте центры скрытого изображения 2б1. Явление
наблюдается только при большой поверхностной концентрации
красителя. Каждая адсорбированная молекула метиленового
голубого вызывает при этом появление одного центра
скрытого изображения, который образуется сразу при адсорбции
молекулы. Площадь последней способна покрыть иа
поверхности кристалла 40 ионов Ag"** и Вг~, если адсорбция
происходит плоскостью молекулы. Атом серы в структуре
красителя имеет существенное значение, так как аналогичные
по структуре красители без этого атома не адсорбируются
и не вуалируют.
Такое явление стоит, повидимому, в связи с редокс-потен-
циалом красителей, определяющим положение уровня
электрона в красителе по отношению к вакантной зоне
проводимости в акцепторе (AgfBr) и к занятому уровню в доноре
электрона (восстановитель — желатина). Вуалирующие
красители принадлежат к классу красителей, способных к
обратимой реакции окисления-восстановления.
Интересен тот факт, что вуалирующее действие тем больше,
чем более положителен редокс-потенциал красителя 2б2,
т. е. чем ниже расположен его уровень в абсолютной шкале
энергий отрыва электрона (28).
Рассмотрим предположительный механизм вуалирующего
действия с помощью схемы уровней электрона (фиг. 48).
Поскольку желатина сама не вуалирует бромистое серебро,
296 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
ее занятый электроном уровень Red находится ниже зоны
проводимости кристалла. С другой стороны, легко
восстанавливающийся краситель типа метиленового голубого в нормальном
своем состоянии обладает, наряду с занятым уровнем (О),
вакантным уровнем (V), определяющим сродство
нормальной молекулы к электрону (ср. 32). Перемещение
электрона с уровня О на уровень V может быть осуществлено
поглощением фотона или за счет термической энергии. *) Тогда
в метиленовом голубом появляется вакантный уровень
электрона V, лежащий ниже уровня Red восстановителя—жела-
_____^ тины (фиг. 48, i), и может
произойти перенос электрона от
последнего к красителю (ср. стр. 217)^
Располагающий теперь
избыточным электроном иа уровне О'
краситель легко отдает его в
свободную зону проводимости,
возможно при некоторой
дополнительной термической активации»
Таким путем краситель выполняет
роль переносчика электро-
_ ■ ~ ■ на.2)
, , (ь) ^ Описанный выше „эффект
метиленового голубого", имеющий
Фиг. 43. Механизм вуалирую- место в результате освещения ок-
щего действия красителей. рашенной желатиновой пленки,.
имеет тот же механизм, с тем
отличием, что электрон перемещается с уровня О на уровень V
при поглощении фотона, а образующаяся нестабильная форма
красителя, когда оба уровня О' и V заняты (семихинов),
отдает свой избыточный электрон катиону Agf+ в растворе.
1 Электрон с уровня Red может при Затрате некоторой энергии за*
полнить вакантный уровень V , приводя к тому же результату. Уровень
Red произвольно помещен в промежутке между О и V; он может быть
расположен и ниже V, увеличивая общую энергию требующейся термической
активации.
2) Необходимо здесь обратить внимание на упрощенный характер рас-
суждения, оперирующего с фиксированным положением уровней электрона,,
отвечающих стандартным редокс-потенциалам. При адсорбции красителя
на кристалле фактическое расположение уровней должно несомненно
отличаться от того, какое устанавливается в водной среде в условиях
измерения стаядартного потенциала. Более того, не для веяного участника
переноса электрона такой потенциал может быть определен. Поэтому
сопоставление с значениями редокс-потеицналов не всегда оправдывается* By а лиг
рующан способность красителей, как упомянуто выше (стр. 271), тесно
свявана с их склонностью образовывать комплексы с ионами серебра»
в водном растворе25*11.
40» ВУАЛИРУЮЩЕЕ ДЕЙСТВИЕ КРАСИТЕЛЕЙ НА БРОМИСТОЕ СЕРЕБРО 297
Чем положительнее редокс-потенциал, тем ниже
располагается уровень О и тем более будет обеспечено
благоприятное положение вакантного уровня V7 илн V по отношению
к уровню Red желатины.
Десенсибилизация. Среди красителей, а также
родственных им слабо окрашенных или даже бесцветных соединений,
имеются такие, которые при адсорбции на галоидном серебре
понижают собственную чувствительность последнего
к свету, но не затрагивают центры скрытого изображения,
образовавшиеся до их адсорбции 263. Следует подчеркнуть,
что это явление не вызвано поглощением света десеисибили-
затором, а представляет собой свойство нормальной молекулы
последнего. Типичным красителем такого рода является,
например, феносафранин. Предполагают, что действие десенсибили-
затора А состоит в реакции окисления, т. е. удалении
электрона с атомов серебра, образующихся в бромистом серебре
при освещении, причем электрон нозвращается красителем
атому брома решетки по схеме260:
Вг —-i Ag Br = *
Десенсибилизаторы находятся в близком структурном
родстве с сенсибилизаторами. Более того, изменением условий,
например, концентрации ионов брома в растворе можно
обнаружить у сенсибилизатора способность к десенсибилизации263*.
А также при повышении концентрации
красителя-сенсибилизатора сверх оптимального значения наблюдается
понижение собственной чувствительности бромистого серебра, т. е.
десевсибилизующее действие 263.
Пользуясь схемой уровней фиг. 48, можно объяснить
явление десенсибилизации тем, что электроны, попавшие в зону
проводимости вследствие поглощения света бромистым
серебром, захватываются вакантным уровнем V красителя,
расположенным ниже зоны проводимости, но выше уровня
металлического серебра в центрах скрытого изображения.
Избыточный электрон с уровня О передается тогда красителем
в нижнюю зону кристалла, возмещая утраченный.
Выделяющаяся электронная энергия, как обычно, рассеивается в
тепловую.
Таким образом, десенсибилизатор функциоиирует как
переносчик электрона из верхней зоны в нижнюю,
аналогично его обратному действию при сенсибилизации согласно
393 Ф0ТОРЕАКЦ4И КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
второй теории, изложенной в предыдущем параграфе
(стр. 288).х)
Метиленовый голубой, который при большой
поверхностной концентрации вуалирует эмульсию бромистого серебра,
как было описано выше, действует, наоборот, как
эффективный дэсенсибилизатор при ничтожном (0.5 °/0) покрытии
поверхности кристалла. Этот факт йе только подтверждает
расположение его вакантного уровня ниже зоны проводимости, как
изображено на фиг. 48. Он показывает, что электроны,
попадающие в зону проводимости при освещении бромистого серебра,
мигрируют из объема зерна, где происходит поглощение
света к его поверхности, где находится адсорбированный
краситель.
Механизм десенсибилизации, однако, более сложен, чем
приведенная простая схема. Было обнаружено, что для
десенсибилизирующего действия необходимо присутствие
кислорода 26*. В вакууме или атмосфере азота десенсибилизация
не имеет места. Повидимому, молекула кислорода
необходима как переносчик избыточного электрона от красителя
в нижнюю зону кристалла, т. е. на втором, а не на первом
этапе реакции. Но десенсибилизация, производимая
красителями-сенсибилизаторами, не требует присутствия кислорода.
С другой стороны, посторонний восстановитель (желатина,
сахар) также участвует в процессе, возможно отдавая атом
водорода на некоторой стадии процесса.
Весьма знаменательно в связи с описанной здесь
электронной трактовкой механизма явлений, что присутствие десенси-
билизатора повышает светостойкость сенсибилизатора, но
одновременно уменьшают его сенсибилизирующее действие.
Этот факт не вызван вытеснением сенсибилизатора с
поверхности бромистого серебра 2&. Так, например, десенсибилиза-
тор пинакриптол желтый устраняет выцветание пинахрома,
но вместе с тем понижает сенсибилизацию фотографической
эмульсии последним. Этот эффект естественно объяснить
обратимым захватом электрона десенсибилизатором от
возбужденной молекулы сенсибилизатора наподобие тушения
флуоресценции по электронному механизму (стр. 256).
В заключение упомянем о действии красителей на
фотографический эффект Гершеля. Последний заключается в
рассасывании центров скрытого изображения при интенсивном
облучении эмульсии красным и инфракрасным светом. Автором
■книги было обнаружено ускоряющее действие на это явле-
!) См. в работе Сивонона^бл првмененив^кЗдесенсибялнэации
концепции Баура о переносе елекгронов.
41. ФОГОСЕНСИБИЛИЗ\ЦЧЯ ТВЕРДЫМ^ГЕЛОМ ВЫЦВЕТАНИЯ 299
яие некоторых красителей, в особенности малахитового
и бриллиантового зеленого, обладающих интенсивной полосой
поглощения в красной области спектра, простирающейся при
адсорбции в инфракрасную сторону26G.
Формально это действие можно приписать реакции
фотовосстановления красителя за счет электронов атомов серебра,
входящих в состав коллоидной частицы металлического серебра,
т. е. центра скрытого изображения. Приобретающая
избыточный положительный заряд коллоидная частица рассасывается
по кристаллу в виде ионов серебра в процессе, обратном ее
образованию в результате диффузии этих ионов к центру
чувствительности. Красители-десенсибилизаторы могут
ускорять рассасывание коллоидного серебра и без необходимости
предварительного поглощения ими фотона красного или
инфракрасного света. По своей природе они должны
способствовать переносу электронов с атомов серебра, образующихся
при распаде коллоидной частицы, в нижнюю зону уровней
кристалла. Явление заслуживает более подробного изучения.
41. Фотосенсибилизация твердым телом выцветания
красителей
Выцветание красителей-сенсибилизаторов,
адсорбированных на бромистом серебре, под действием света,
поглощаемого бромистым серебром, объясняется реакцией красителя
с выделяющимся бромом (стр. 279). Обратный эффект
появления окраски красителя (малахитового, зеленого), вызванный
фотоокислением его лейко-формы в присутствии кислорода
ускоряется при адсорбции, в особенности на солях серебра,
как это обнаружил Грос195. В теории сенсибилизации,
исходящей из представления о передаче энергии от красителя
кристаллу, принимается принципиальная возможность и
обратного процесса переноса энергии от освещаемого кристалла
красителю с активацией последнего.
В частности, такому переносу энергии приписывается
понижение собственной чувствительности бромистого серебра при
повышении поверхностной концентрации сенсибилизатора253.
Факт фотосенсибилизации твердым телом наиболее
убедительно установлен для реакций окисления-восстановления
в присутствии окиси цинка и окиси титана267. В частности,
установлено, что эти окислы, поглощая свэт, ускоряют
реакцию выцветания красителей, которая без них не идет вовсе
или идет чрезвычайно медленно. Твердое тело при этом
не испытывает явных химических изменений. Такое фотосен-
снбилнзирующее действие на красители, наряду с ZnO, TiO2
300 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
и отчасти Sb2O3, свойственно, по Гудиву268, в меньшей
степени всем белым окислам металлов, обладающим сильным
поглощением в близкой ультрафиолетовой области. Карбонаты
н сульфаты, как MgCO3f ZnCO3, BaCOs BaSO4, прозрачные
в ультрафиолете до "к <240т[/-, не вызывают выцветания
адсорбированных на них красителей. Сульфиды (ZnS, CdS),
поглощающие в близкой ультрафиолетовой области, также
способны вызывать при освещении выцветание красителей
269
в растворе269.
В табл. 17 даны границы спектра поглощения некоторых
порошкообразных веществ (белых пигментов).
Т абл иц а 17
Длинноволновые границы поглощения (Aq) белых
кристаллических порошков
(по Гуднву 268)
400
385
274
| <240
Av0(кгкал)
71
74
104
тю2 .
ZnO .
PbCOg
BaSO4> ff3
BaCO3> ZnCO3
Особо подробному исследованию подверглась окись цинка,
обладающая сильным фотосенсибилизующим действием н
способностью флуоресцировать, что наводило исследователей
на мысль о полной аналогии ZnO с фотодинамическими
красителями (34).
Окись цинка при освещении ультрафиолетовым светом
в области собственного поглощения (^<380mf*) в отсутствии
кислорода действует как восстановитель на катион серебра
или метилеиовый голубой в водном растворе, выделяя
металлическое серебро в первом случае и обесцвечивая краситель
во втором172 (ср. стр. 208 и 214). Обесцвеченный продукт
состоит на 80°/0 из лейко-формы красителя, который
регенерирует с появлением первоначальной окраски при
соприкосновении лейко-формы с кислородом. Но 20 °/о представляют собой
необратимо измененный продукт окисления красителя.1)
г) Такой большой процент разрушенного красителя, возможно, был
вызван недостаточным удалением воздуха ив раствора, поскольку оно про-
нвводилоеь только водяным иасосом (остаточное давление 11 мм), а кроме
того, ие была исключена возможность действии света непосредственно
в ультрафиолетовых полосах краеителя.^
41. ФОТОСЕНСИБИЛИЗАЦИЯ ТВЕРДЫМ ТЕЛОМ ВЫЦВЕТАНИЯ 301
Если в освещаемый раствор ввести дополнительно легко
окисляющиеся органические соединения (восстановители),
такие как глицерин, глюкоза, бензидян, то метилеиовый
голубой почти целиком превращается в лейко-форму. Явление
совершенно аналогично тому, какое наблюдается при
фотореакции сенсибилизованного восстановления, когда поглощает
свет, вместо ZnO, другой краситель-сенсибилизатор (ср. фиг.
37 на стр 212).
Школа Баура трактует эти явления с его точки зрения,
применяя концепцию переноса электрона через посредство
сенсибилизатора, которым здесь является ZnO172'267,
Фотосенсибилизованное окисью циика обратимое
выцветание красителей наблюдается для многих красителей
различных классов172: для кубовых (тиоиндиго, индигокармин), ази-
новых (сафранин, феносафранин, пинакриптол желтый, инду-
лин), оксазиновых (нильский голубой), тиазиновых
(метиленовый голубой, циба), ксантеновых (церулеин), антрахиноновых
(псевдопурпурин), трифенилметановых (малахитовый зеленый).
Азокрасители восстанавливаются (гидрируются) необратимо.
Желтые красители, например аурамнн, не подвергаются
сенсибилизирующему влиянию ZnO и TiO2l повидимому,
действуя как экран, не допускающий ультрафиолетовый свет
до кристалла (ср. сказанное в параграфе 1 о связи цвета со
спектром).
Двуокись титана (модификация анатаза) также является
фотосенсибилизатором выцветания красителей в растворе,
а также адсорбированных красителей в сухом состоянии,
хотя она и не флуоресцирует в обычных условиях (Краснов-
ский267). Так, в растворе в присутствии TiO2 значительно
ускоряется выцветание следующих красителей под действием
линий ртути 436, 406 и 365 ту* в области поглощения
двуокиси титана: прямой голубой (кислотный), метиленовый голубой
{основной), нафтол оранжевый (кислотный), хризоидин
(основной). Эта последовательность соответствует снижению эффекта.
В отсутствии воздуха (неполном) для метилевового голубого
получается 40% лейко-формы и 6О°/о продуктов необратимого
окисления.
Наблюдается некоторая противоположность действия TiO2
и ZnO, заключающаяся в том, что красители, быстро
выцветающие на одном, светостойки на другом, хотя оба окисла
поглощают ультрафиолетовый свет примерно в одной
области. Было показано Красновским267, что эта
противоположность действия не может быть приписана различной адсорби-
руемости данного красителя, вызванной тем, что ZnO
обладает в присутствии воды основными свойствами, TiO2 —
302 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
/
кислыми, а следовательно, эти окислы должны адсорбировать
различные красители.
Далее, сенсибилизирующая активность ZnO — наибольшая
в присутствии воды; при полном осушении окись цинка
неактивна. Двуокись титана, наоборот, наиболее фотоактивна1
в отсутствии воды, сохраняя способность фотосенсибилнзации
даже в органических растворителях. Это наводит на мысль
о различном механизме процесса в обоих случаях.
Для трактовки механизма фотосенсибилизации твердым
телом можно воспользоваться наглядной схемой переноса
электрона, изображенной на фиг. 42 (стр. 256) для тушения
флуоресценции различных типов красителей. Здесь, вместо
флуоресцирующих молекул, мы рассматриваем кристаллы —
полупроводники двух типов, к которым, собственно, схема
фигуры и относится. Окись цинка принадлежит к типу И,
„электронных" полупроводников, а следовательно при
поглощении света электрон попадает в зону проводимости, по
которой он мигрирует до поверхности кристалла. Там он
захватывается адсорбированным красителем, обладающим
вакантным уровнем и способным к восстановлению (метиленовый
голубой). Посторонний восстановитель (глицерин и т. п.)*
возмещает утраченный электрон кристаллу, и процесс может
* повториться снова. Протон отделяется от восстановителя,
потерявшего электрон (ср. 28), и идет на образование обычной
полувосстановленной формы (семихннона) красителя. Весь
процесс может быть изображен следующей схемой:
A/ZnO -Д. A/'ZnO > А" / ZnO+ > НА/ ZnO,
где А изображает молекулу красителя, адсорбированную
на ZnO, * есть условное обозначение подвижного электрона
в ZnO и „лабильного"электрона восстановителя HY; НА есть,
как обычно, семихинон красителя. Образование лейко-формы
красителя Н2А может произойти при повторном протекании
того же процесса,*) но более вероятно через реакцию „дис-
мутации":
НА-+-НА >Н2А-4-А.
Весь процесс фотосенсибилизованного окисью цинка
восстановления красителя А за счет восстановителя HY проте-
г) Участие при образовании одной молекулы левюкрасителя двуж
фотонов должно было бы повести к зависимости скорости реакции от-
квадрата иитеисивиости света, что не наблюдалось.
41 ФОТОСЕНСИБИЛИЗАЦИЯ ТВЕРДЫМ ТЕЛОМ ВЫЦВЕТАНИЯ 303
кает аналогично схеме (И) на стр. 215, с тем отличием, что
сенсибилизатором служит ZnO, а окислителем X — краситель А.
В присутствии кислорода может происходить его конкуренция
с адсорбированным красителем А в смысле захвата электрона
с поверхности ZnO, Тушение флуоресценции окиси цинка
кислородом, обнаруженное Гачковским и Терениным270,
вызвано, вероятно, таким захватом. Кроме того, кислород может
дегидрировать семихинон и тормозить реакцию, регенерируя
краситель. Однако более вероятно, что кислород реагирует
с семихиноиом необратимо по типу реакций оксидирования
(глава 7).
Необратимое изменение красителя, имеющее место при
отсутствии постороннего восстановителя HY, вызвано,
очевидно, тем, что другая молекула красителя А выполняет его
роль, т. е. возмещает электрон окиси цинка, окисляясь
(и испытывая затем необратимую реакцию в присутствии
кислорода).
Поведение второго типа полупроводников, а именно
„дефектного", иллюстрирует верхняя половина фиг. 42 (иа стр. 256).
После поглощения фотона кристаллом к его поверхности
мигрирует не электрон, а электронный дефект нижней зоны, имеющий
положительный заряд. Адсорбированный на поверхности
краситель на первом этапе отдает электрон кристаллу, т. е.
окисляется, превращаясь далее в необратимый продукт
окисления в том случае, когда присутствующий восстановитель
HY неспособен его регенерировать или когда присутствует
кислород.
Процесс изобразится схемой, в которой П обозначает
полупроводник дефектного типа:
i
Продукт Д—
окиеления
где ° обозначает электронный дефект, появившийся в П
в результате поглощения фотона. Свой избыточный электрон
П~ передает другой адсорбированной молекуле красителя,
производя такое же действие, как ZnO. Это приведет к
получению лейко-формы красителя одновременно с необратимо
окисленным красителем.г)
*) Для выяснения механизма весьма желательно определение
квантового выхода реакции и его зависимости от интенсивности свет а итаких
системах.
"304 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОВЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
Роль постороннего восстановителя здесь может привести
к тому, что он, а не молекула красителя, будет терять
электрон на первом этапе, передавая его °П, и тем самым
увеличивать образование лейко-формы красителя А~ за счет
уменьшения выхода окисленного продукта (ср. стр. 215).
Весьма слабая флуоресценция двуокиси титана,
наблюдаемая при низкой температуре, н е тушится кислородом в
отличие от ZnO, Молекула О2 должна быть, действительно, инертна
по отношению к положительно заряженному электронному
дефекту, мигрирующему в кристалле.
Однако, согласно физическим измерениям, TiO2 не
принадлежит к типу дефектных полупроводников и приведенная
здесь схема явления к ней, повидимому, неприменима. Таким
образом вопрос о причине различного поведения ZnO и TiO2
остаетоя открытым.
Весьма интересно и важно в практическом отношении
установленное Красновским2в7 торможение выцветания
красителей, фотосенсибилизованного двуокисью титана. Торможение
вызывают адсорбированные на TiO2 анионы J~, CNS~, Br~~,
С1~, а также катионы Со"*"*", Мп4"*", Сг4"*"4". Последовательность
расположения анионов соответствует снижению их
тормозящего действия и обнаруживает полный параллелизм с
тушащим действием этих анионов на флуоресценцию (35). Такое
далеко идущее подобие не случайно и, очевидно, первичный
механизм обоих явлений имеет тождественную природу, т. е.
состоит в переносе электрона от тушителя, как это
разъяснено выше.
В связи с изложенным представлением о миграции
положительного или отрицательного заряда к поверхности
полупроводника при поглощении им света, значительный интерес
имеют наблюдения Хедвалла, обнаружившего увеличение
адсорбции фталеиновых красителей из раствора на
сульфидах ртути и кадмия при освещении последних271.
Такое явление „фотосорбции" следует по всей
вероятности приписать повышенной адсорбируемости молекул на
центрах избыточного заряда, возникающих вследствие
внутреннего фотоэффекта на поверхности кристалла. Аналогичные
явления повышенной сорбции молекул из газовой фазы
при освещении адсорбента изучаются в лаборатории автора
книги .
Миграции энергии. Измерение квантового выхода
фотохимических реакций, в которых участвует порошкообразные
твердые тела, представляет значительные затруднения. Тем
не менее, Гудиву268 удалось измерить относительные значения
квантового выхода выцветания красителя, адсорбированного
41. ФОТОСЕНСИЗИЛИЗАЦИЯ ТВЕРДЫМ ТЕЛОМ ВЫЦВЕТАНИЯ 315
на ТЮ^ в двух спектральных областях: в видимом свете, где
расположена полоса поглощения красителя, и на
ультрафиолетовом участке спектра, поглощаемом кристаллом. Эти
измерения были проведены с двуокисью титана, окрашенной азо-
красителем — хлоразол небесно голубой FF,- в сухом состоянии
на воздухе. Измерения показали, что поглощение фотона
самим красителем примерно в 100 раз менее эффективно
для выцветания, чем поглощение фотона ультрафиолетового
света адсорбентом.
Попытка измерения абсолютной величины квантового выхода
выцветания красителя при фотоактивации адсорбента —
двуокиси титана в этих условиях привела к тому результату,
что одна выцветшая молекула красителя приходится на 240
частиц TiO2, поглотивших по фотону, т. е. что суммарный
квантовый выход реакции Т = 240~" ^3 сРеднег0 размера
^отдельных кристалликов порошка ТЮ2, употреблявшегося в опыте
(диаметр ок. 0.3 м), из концентрации красителя и
приблизительной величины площади, занимаемой молекулой красителя,
следует, что поверхность кристалликов была целиком заполнена
адсорбированным мономолекулярным слоем красителя. В таком
кристаллике на каждые 150 частиц TiO2 объема приходится
одна, лежащая на его поверхности в непосредственном
соседстве с красителем. Если поглощение света имеет место
во всем объеме кристаллика, а фотохимически активно
поглощение света только поверхностными частицами TiO2, то
максимальная величина суммарного квантового выхода будет
Y—Тсп~* При неточности измерении можно считать это зна
чение совпадающим с наблюдаемым у^-^-д * В этом объяснении
сделано, однако, допущение, что почти каждое поглощение
фотона поверхностными частицами TiO2 приводит к реакции
адсорбированной молекулы красителя, т. е. что истинный
квантовый выход реакции фотосенсибилизации близок к единице,
что мало вероятно.
Гудив и Китченер пришли к иному выводу268. В опытах
применялась концентрация красителей, при которой одна
молекула красителя приходилась на 3000 частиц TiO2. Из приве-
1
денного выше значения суммарного квантового выхода Y=~240~
следует, что 240 из этих частиц, т. е. -г* часть поглощает
по фотону, приводя к выцветанию одной молекулы красителя.
20 А. Н. Теренжн
306 ФОТОРЕАКЦИИ КРАСИТЕЛЕЙ НА ПОЗЕРХНОСТИ КРИСТАЛЛИЧЕСКИХ ТЕЛ
Авторы полагают, что такая большая эффективность света*
поглощаемого кристаллом, возможна только в том случае,
если действуют фотохимически на краситель, главным образом,
такие поглотившие фотоны частицы TiO , которые
расположены в глубине кристалла.
Передача действия из глубины на поверхность кристалла
может совершаться путем миграции либо электрона в зоне
проводимости или электронного дефекта в нижней зоне, как
описывалось выше.
Однако в ненарушенном кристалле принципиально возможно
также явление миграции энергии.
Представление о миграции энергии в виде кванта
электронной энергии возбуждения в идеальном кристалле было
впервые выдвинуто Френкелем, который назвал этот
мигрирующий квант „экситоном"27 3. Эту концепцию применял»
затем неоднократно различные авторы для объяснения
флуоресценции ничтожных примесей в кристалле под влиянием
длин волн, поглощаемых кристаллом, а не примесью274.
Длительное сохранение электронной энергии кванта при его
переносе по кристаллу без деградации в тепло возможно в том
случае, когда передача кванта от одного центра
кристаллической решетки другому совершается достаточно быстро,
не порождая вибраций решетки. Монокристалл при низкой
температуре является именно такой средой, для которой
можно ожидать беспрепятственвого распространения экситова
от места поглощения фотона.
Наиболее благоприятны условия для миграции „экситона"
по кристаллу, когда он построен из отдельных молекул
органического соединения, т. е. для так называемых
молекулярных кристаллов, таких, как нафталин, антрацеи и др„
Для неорганических кристаллов типа полупроводников, таких
как ZnO и TiO2, более вероятен механизм миграции
электрона нли его дефекта по решетке, чем перенос экситона*
Миграцию экситона можно рассматривать как перемещение
электронного дефекта по кристаллу вместе с электроном,
который все время остается на локальном уровне,
расположенном над основным уровнем кристалла и принадлежащем
дефекту.
Мигрирующий экситон, подобно электрону, достигает места-
нарушения или поверхности кристалла и растрачивает там
свою энергию в тепло или передает ее на возбуждение
постороннего включения, способного ее воспринять.
Последнее либо испускает полученную энергию в виде фотона
флуоресценции, либо активируется для реакции с окружающей*
средой.
41. ФОТОСЕНСИБИЛИЭАЦИЯ ТВЕРДЫМ ТЕЛОМ ВЫЦВЕТАНИЯ 307
Красители, адсорбированные на силикагеле и некоторых
других адсорбентах, сохраняют способность к флуоресценции
и даже фосфоресценции с большим выходом, несмотря на
адсорбцию (21)87t8tt. Эгот факт указывает на слабое
взаимодействие адсорбированного красителя с этими адсорбентами,
так как иначе энергия возбуждения была бы рассеяна путем
превращения в многочисленные вибрации кристаллической
решетки. В соответствии со слабостью взаимодействия с кри-
сталлом, полоса поглощения красителя сравнительно мало
смещена и искажена на этих адсорбентах, в отличие от
красителей-сенсибилизаторов, адсорбированных на галоидных солях
серебра.
С другой стороны, сильное взаимодействие красителя с
кристаллом способствует деградации энергии возбуждения в тепло.
Это соображение делает понятным отсутствие достоверно
установленного факта переноса энергии от кристалла
адсорбированному на нем красителю с испусканием света последним,
когда освещению подвергается кристалл в области
собственного поглощения.1)
Перенос энергии, вызывающий свечение адсорбированного
красителя, наблюдается, однако, в условиях» когда энергия
подводится не сьетом, а сильно экзоэнергетической
реакцией с кислородом, идущей на поверхности твердого тела.
Это явление можно назвать сенснбилизованной оксилюми-
несценцией. Так, если окислять действием Мп О4 в
растворе производное кремнеорганического соединения— сило-
ксена, обладающего слоистой структурой и чрезвычайно
большой реакционной способностью, на котором
предварительно был адсорбирован способный к флуоресценции
краситель (родамин В, родамин сульфонат, изохинолиновый
красный), то появляется яркая люминесценция275. Спектр
этого свечения тождественен спектру флуоресценции
адсорбированного красителя, если возбуждать последний
непосредственно светом, им поглощаемым. Свечение не вызвано
реакцией окисления красителя, а только окислением адсорбента.
Отсюда следует вывод, что большая порция первично
выделяющейся энергии реакции способна сохраниться и дойти
в виде кванта до адсорбированной молекулы красителя,
приводя к ее возбуждению.2)
*) Некоторые, относящиеся сюда наблюдения автора кяиги описаны в70»
2) Аналогичное явление было наблюдено дли гетерогенной реакции.
окисления паров спирта воздухом иа аэрогелях силикагеля и окиси магния
в лаборатории автора2"^.
20*
308 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
ГЛАВА ОДИННАДЦАТАЯ
ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
42. Внешний фотоэлектрический эффект н
фотопроводимость твердых пленок красителей
Освещение твердой пленки красителя, полученной
осаждением из раствора, в вакуумных условиях приводит к двум
типам фотоэлектрических явлений: I) внешний или
поверхностный фотоэффект, состоящий в вырывании электронов
с поверхности красителя и перенесении их в окружающее
эвакуированное пространство; 2) внутренний фотовффект
или фотопроводимость, вызванный отрывом электронов
и их переносом внутри слоя.
На красителях первое явление было обнаружено вскоре
после открытия внешнего фотоэффекта на металлах277.
Внешний фототок наблюдался при этом даже без
эвакуации, на воздухе, при освещении ультрафиолетовым светом
электрода, покрытого пленкой твердого красителя или жидкой
пленкой концентрированного водного раствора,х)
/ Наряду с красителями (малахитовый зеленый, метиловый
фиолетовый, розанилин и др.) внешний фотоэффект способны
давать и их лейкооснования (Кноблаух 277).
Длины волн, вызывающие заметный внешний фотоэффект
в красителях, находятся, как и в случае более простых
органических соединений (30), в короткой ультрафиолетовой области
^<^25Om[/-. Величина фототока меньше, чем наблюдаемая для
металлов, но всё же значительнее, чем для остальных твердых
органических соединений.
Спектральная кривая фотоэлектрической чувствительности
имеет продолжение в сторону длинных волн, обнаруживая
максимумы вблизи полос поглощения слоя красителя (фиг. 49)279.
Около 220/п[л начинается крутой подъем
фоточувствительности, подобный наблюдаемому у металлов.
Длинноволновые максимумы кривой для шести красителей
различных классов (фуксин, эозин G» метиловый фиолетовый,
хинолиновый голубой, бриллиантовый зеленый, патентованный
1) Отсутствие эффекта у спиртового раствора, возрастание фототока
по мере старения и механического упрочнения жидкой пленки и другие
наблюдения — с несомненностью показали, чт^ испускание электронов
и в этом случае происходит с пленки твердого красителя, а ие из раствора
{Роде277). См. также исследование фотоэффекта с пленок анилина, ди- и три-
•фениламина, нанесенных на раствор электролита278.
42, ВНЕШНИЙ ФОТОЭЛЕКТРИЧЕСКИЙ ЭФФЕКТ И ФОТОПРОВОДИМОСТЬ 309
голубой) оказываются, примерно, на одних и тех же местах
(270 и 240 лп[л). Электроны отщепляются, повидимому, от
определенных структурных элементов молекулы красителя по
механизму внутренней конверсии энергии, описанному на стр. 103.
Фотоэффект наблюдается, примерно, до А — 360 /П[л, как
границы, но обращает на себя внимание наличие небольшой
фотоэмлссии электронов в области, близкой к видимой полосе
поглощения красителя (фиг. 49), т. е. в длинах волн,
вызывающих внутренний фотоэффект красителя.1)
Внутренний фотоэффект обнаруживается по падению
сопротивления слоя
красителя при освещении, т. е.
по появлению
фотопроводимости281.2)
Вартанян в
лаборатории автора показал
существование внутреннего
фотоэффекта для
многочисленных красителей
самых разнообразных
классов (ди- и трифенилмета-
НОВЫХ, ЦИанинОВЫХ, КСан- Фиг. 4°. Спектральная кривая фото-
тенОВЫх) И установил ПОЛ- электрической чувствительности для вне-
с:
ч
слоя
шиего фотоэффекта с твердого
красителя (фуксияв) в вакууме; Хэ, X2,
Х3 — селективные максимумы
фотоэффекта 279.
ное подобие явления
поведению неорганических
типичных
полупроводников'281. В отличие от
внешнего фотоэффекта, фотопроводимость наблюдается под
действием видимого света и полосе поглощения красителя. *
При освещении слоя красителя в вакуумных условиях
проводимость медленно нарастает в течение 10—30 мин.
до стационарного значения. После выключения света
проводимость быстро спадает, но в некоторых, особо
фоточувствительных красителях, таких, как кристаллический фиолетовый,
она сохраняется длительное время (несколько суток).
Фотопроводимость значительно увеличивается с повышением
температуры от — 50° до -+-100° Сш. Из величины темпера-
х) Предводителей и Нечаева обнаружили периодическое увеличение
и уменьшение фототока по мере увеличения толщины осужденного слоя
красителя (в диапазоне толщин от 0.2 до 1 р.) 280. Это явление
исчерпывающе объясняется интерференцией света в слое, создающей периодическое
увеличение и уменьшение плотности световой энергии на поверхности
слоя (Глучка) 28°.
2) Фотопроводимость сопровождается изменением диэлектрической
постоиииой слоя, которое было обнаружено уже в старых работах под
названием „актиио-диэлектрического" эффекта *^2.
310 ФОТОЭЛЕКГРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
турного коэффициента следует, что для освобождения
электронов внутри слоя поглощения фотона видимого света
недостаточно, а необходима дополнительная термическая
энергия порядка 10—15 кгкал/моль. Отсюда можно заключить,
что для красителя в твердом кристаллическом состоянии
существует также зона проводимости, подобно кристаллу или
другим полупроводникам (фиг. 46), которая в данном случае
лежит на 10—15 кгкал выше уровня возбужденного состояния
молекулярного иона красителя, входящего в состав его
кристаллической решетки.
Величина квантового выхода внутреннего фотоэффекта еще
не измерялась, но можно ожидать, что она будет невелика
вследствие значительной дезактивации возбужденной молекулы
окружающими тождественными молекулами, как это следует
из явления концентрационного тушения флуоресценции (36),
Значительный интерес представляет действие газов на
проводимость красителя24'281. Теиновая проводимость слоя,
медленно спадающая в вакууме после прекращения освещения,
падает очень резко при впуске в эвакуированный сосуд
кислорода давлением а несколько мм или паров иода давлением
в 0.001 мм.
Явление можно объяснить тем, что сорбированные молекулы
О2 (а также J2) захватывают электроны, обусловливающие
проводимость. *)
В результате описанного выше напускания кислорода слой
красителя утрачивает свою первоначальную
фоточувствительность, если даже восстановить вакуум, г, е. слой оказывается
„отравленным". Фоточувствигельность слоя может быть
регенерирована только длительной откачкой и прогревом до 70° С
в вакууме, удаляющими кислород, сорбированный слоем.
Если освещение слоя производить в присутствии кислорода,
то после нарастания фотопроводимости во времени до
некоторого максимума происходит в дальнейшем монотонное ее
спадание вследствие необратимой реакции красителя с кислородом.
Такой чувствительный электрический метод дает возможность
следить н измерять кинетику процесса окислительного
выцветания твердого слоя красителя до того, как эта фотореакция
обнаруживается по выцветанию.
Фотопроводимость растворов красителей. В разбавленном
растворе антрацена в гексаие (2.5'10~4М) было давно
обнаружено появление проводимости при освещении ультрафиолето-
1) Увеличение фотопроводимости при напускаиии водорода, иаблюдеи-
иое в аналогичных опытах Петрикальном, вызвано влажностью вводимого
газа, как доказал Вартаиян.
42. ВНЕШНИЙ ФОТОЭЛЕКТРИЧЕСКИЙ ЭФФЕКТ И ФОТОПРОВОДИМОСТЬ 311
вым светом с ^ <С 225 rrajx282. Аналогичный объемный или
внутренний фотоэффект дают гексановые растворы
дифениламина, (i-нафтола и, в особенности, а-нафтиламииа и дифенил-
метана.
Насыщенный раствор антрацена в гексане обнаруживает
фотопроводимость при более длинных волнах вплоть до 400 m\j.t
но это продолжение фоточувствительности принадлежит
внутреннему фотоэффекту кристаллического антрацена,
отлагающегося на электродах (44).
Отрыв электрона с поверхности твердого антрацена
и упомянутых выше ароматических соединений в
кристаллическом состоянии, т. е. и с внешний фотоэффект в вакууме требует
* < 250 т[х (ср. 30)да.
В более недавнее время применение весьма
чувствительного электрического метода (импульсного счетчика
вылетающих электронов) позволило обнаружить внешний фотоэффект
с твердого бензофенонаш. Длинноволновая граница лежит
в области 7. <d 315 лп^ и, предположительно, совпадает с
границей полосы поглощения бензофенояа с максимумом у 260 mjx. х)
Щодро в ряде классических исследований обнаружил
и изучил фотопроводимость цнаниновых красителей (цианин,
галлоцианин, пинавердол, пинахром) не только в коллодионной
пленке (глава 7), но и в бензольном растворе под действием
видимого света .
При кратковременном освещении раствора (около 10 мин.)
появляется проводимость, медленно исчезающая после
прекращения освещения. При длительном освещении (до 8 часов)
раствора или окрашенной пленки происходит непрерывное
увеличение проводимости, идущее параллельно с выцветанием
красителя. Последний эффект вызван, очевидно, фотореакцией
красителя с кислородом воздуха, которая приводит, как мы
знаем, к появлению продуктов феиольного и кислотного
характера (глава 7), способных отщепить ионы водорода.
Больший интерес представляет первая — обратимая —
фотопроводимость, которая может быть объяснена двояким
образом: 1) краситель в слабо диспергирующем растворителе,
^Слабая фотопроводимость жидких предельных углеводородов под
действием л .< 25Э тр., обнаруженная в ряде работ28*, несомненно
принадлежит продуктам их окисления кислородом воздуха (т. е. альдегидам
и кислотам), на что убедитель to указывает спектральная область фого-
активных длин воли, совпадающая с областью поглощения карбонильной
группы8. Сами углеводороды в чистом виде начинают поглощать только
в короткой ультрафиолетовой области (X <; 190 тр.).
Все описанные выше результаты были получены давно и, несомненно»
требуют воспроизведения и развития современными экспериментальными
«средствами, чго делается в лаборатории автора книги.
312 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
каким являв!ся бензол, присутствует в виде коллоидных частиц»
наблюдаемая в видимом свете фотопроводимость есть
результат частичного перехода в раствор электронов внутреннего'
фотоэффекта этих частиц; 2) краситель в неполярном
растворителе, как бензол, присутствует в виде аггрегатов — ионных
пар A"*" Hal", т. е. в виде ионного недиссоцнированного
соединения, которое распадается на отдельные ионы только под
действием света.
Трудно без дополнительных опытов решить в пользу
первого или второго объяснения. Интересно, что нагревание
раствора уменьшает фотопроводимость, что может быть
приписано как дезаггрегации коллоидных частиц, так и
уменьшению сольватации, а следовательно, возможности диссоциации
ионных пар.
Весьма знаменателен неоднократно подчеркиваемый нами
факт отщепления электронов от красителей и более простых
ароматических соединений под действием света в первой же
полосе поглощения, соответствующей возбуждению
молекулы (ср. 30). Если этот еще недостаточно четко
установленный результат подтвердится в дальнейшем, то это означает,
что возможна внутренняя конверсия электронной энергии
с передачей ее одному из слабо связанных электронов,
43. Фотогальванический аффект
В водном растворе, содержащем обратьмо
восстанавливающийся краситель А, например метиленовый голубой, вместе
с его лейко-формой Н2А, устанавливается подвижное
равновесие в результате реакции переноса атомов водорода от одьой
молекулы другой, которая была подробно рассмотрена выше (28)
и может быть изображена суммарной схемой:
Как было там сказано, реакция (1) в действительности состоит
из нескольких быстро следующих один за другим этапов, при
которых частицы обмениваются не нейтральными атомами
водорода, а электрическими зарядами: электронами е
и протонами Н4". Опуская для простоты изложения
промежуточное образование полуокисленной или
полувосстановленной формы семмхинона НА, истинный механизм написанной
выше обратимой окислительно-восстановительной реакции
можно представить следующим образом:
А -+- Н2А ^± А" ~н 2HV- А ^ АН2 -ь А. (2)
1 '21 21 2
43. ФОТОГААЬВАНИЧЕСКИЙ ЭФФЕКТ; 313
Например:
"]<^ у~-он ^ "О—\ ~~ /—°~ ■+■
н- О=/ \=О ^ НО-/ ^>—ОН -#- O=/S=O.
Первичный акт состоит в обмене электронами; протоны,
как менее подвижные, отщепляются и присоединяются вслед
за электронами (стр. 183). Реакция (2) на самом деле, как
показал Михаэлис, состоит из двух ступеней, на каждой
из которых переносится по одному электрону и протону (28)148.
Быстрое и практически нераздельное следование этих этапов
один за другим позволяет оперировать в дальнейшем суммар~
ной картиной (2).
Наличие в этом процессе промежуточной стадии, на
которой появляются заряды, дает возможность обнаружить и
измерить концентрацию заряженных частиц путем введения
в раствор химически инертного зонда, способного вступить
в обратимое равновесие с зарядами. Таким зондом и является
металлический (платиновый) электрод, который заряжается
до определенного потенциала в результате непрерывно идущих
противоположных процессов захвата электронов от частиц А
и отдачи электронов непосредственно частицам А или ионам
водорода.
Отсюда следует, что наряду с предыдущим подвижным
равновесием (1) или (2), на платиновом электроде происходят
элементарные процессы, которые условно могут быть
записаны таким образом:
Ptl ^ A-f-IPt
[PtJ—н-2Н+ ^ [Pt]-+-2H
Стационарный потенциал, принимаемый плат?новым
электродом | Pt | в результате этих противоположных действий,
измеренный по отношению к какому-либо стандартному
электроду, зависит от специфических свойств и относительной
концентрации участвующих в равновесии частиц А ~, Н~*~, А.
При обычных концентрациях внесение электрода не
сказывается на первоначальном равновесии (1) или (2), и потенциал
электрода служит электрическим выражением состояния
раствора.
Электрохимический закон Нернста устанавливает для таких
обратимых систем следующую | линейную зависимость
314 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
потенциала электрода S от логарифма отношения
концентраций участвующих частиц, а именно:
S = S° ■+- W-ln [I--] -*- 2 2F ln
или для 20° С: (4)
где In есть знак натурального логарифма, F ==96500 кулон
есть заряд, переносимый одним граммэквивалентом ионов,
R — газовая постоянная, Т— абсолютная температура, а
<§°—постоянная. Легко видеть, что последняя равна тому
потенциалу, который установится при равенстве концентраций
[А] = [А ] и когда концентрация водородных ионов [Н+] = 1,
т. е., когда логарифм в выражении (4) обращается в нуль.
Константа <£°, характеризующая свойство данной системы
частиц независимо от концентраций, представляет собой
стандартный (нормальный) редокс-потенциал, о котором
была речь в 28. Умноженный на 2е, он дает величину энергии,
требующуюся для совместного переноса двух электронов
от А~7 к платиновому электроду и от него к 2Н+, согласно
схеме процесса (3).
Но перенос электронов от | Pt к 2Н и обратно, т. е.
вторая половина процесса (3), осуществляется на так
называемом нормальном водородном электроде, представляющем
собою платиновый электрод, покрытый платиновой чернью,
погруженный в водный раствор с концентрацией ионов
водорода [Н+] = 1 при насыщении газообразным водородом.
Потенциал, принимаемый таким электродом, условно
считается нулем отсчета редокс-потенциалов. Если его
абсолютная величина была известна, то, учитывая ее, можно
было бы получить из измеряемой величины энергии 2е£°
абсолютную энергию совместного отрыва двух электронов
от граммолекулы частиц А , иначе (2е£°)абс
Половина этого значения представляет сэбой среднюю
-абсолютную энергию / отрыва электрона от частицы А ,
о которой была речь в 28. Следует помнить, что, выражая
энергию в 3AeKTpoH-BOAbTax> приводят только численное зна-
чзние потенциала <£°абс в вольтах (2).
Как было сказано в 28, абсолютное значение потенциала
водородного электрода из косвенных данных принимается
1)Все концентрации выражены в молях или граммионах на л.
43. ФОТОГАЛЬВАНИЧЕСКИЙ ЭФФЕКТ
315
равным 3.8 вольт, а следовательно, если стандартный
потенциал рассматриваемой системы есть S0 вольт, то энергия
отрыва одного электрона от частицы А будет: <^oa6C::=:^==
= ё° -4-3.8 (Э. ВОЛЬТ),
Совершенно ясно, что изменение концентрации и природы
частиц в окружающем электрод растворе, возникающее
в результате фотохимической реакции при освещении раствора,
должно сказаться иа равновесии частиц, а следовательно,
и на потенциале электрода.
Действительно, уже давно было обнаружено, что при
освещении раствора красителя (в частности — флуоресцирующего)
в зоне, непосредственно
прилегающей к одному из
двух одинаковых электродов,
погруженных в раствор,
между электродами возникает
разность потенциалов *DX>*-°\
Этот эффект, обнаруженный
в чистом виде Свенсоном,
мы в дальнейшем называем
фотогальваническим
эффектом, в отличие от
эффекта Беккереля,
возникающего только при
освещении самого электрода (44)
(фиг. 50).
Сдет
Фиг. 50. Различные виды фотопо-
ЕСЛИ К указанным выше тенциалов, „аЬлюда.мые при освеще-
J нии растворов красителей. /—фото-
двум Электродам была При- гальванический эффект (Свенсона-Бек-
керели)? 2—фотовольтаический
эффект (Беккереля).
ложена внешняя
электродвижущая сила, то
возникновение дополнительного
фотопотенциала проявляется в виде кажущегося изменения
сопротивления раствора. Фотопотенциалы достигают величины
милливольта и выше.
Несомненно, что фактически фотогальванический эффект,
в особенности растворов флуоресцирующих красителей,
осложняется наложением эффекта Беккереля, так как близость
зоны освещения раствора к электроду, необходимая для
получения максимального фотопотенциала, неизбежно связана
с освещением самого электрода, который всегда несет на себе
поверхностную пленку, обладающую некоторой
фотоэлектрической чувствительностью. На это указывает зависимость
фотопотенцнала от природы электрода2 \ Такое наложение
двух эффектов тем более имеет место, когда освещается сам
электрод и притом через раствор. В частности, на электродах
316 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
может присутствовать слой адсорбированного или аггрегиро-
ванного красителя. Это следует, между прочим, из того факта,
что спектральная область длин волн, вызывающая фотопотен»
циал, простирается в красную сторону з а границу длин волн,,
возбуждающих флуоресценцию раствора красителя, хотя
освещение раствора производилось сквозь полупрозрачный
электрод, нанесенный непосредственно на стенку сосуда во
избежание поглощения света раствором290. Как указывалось ранее,
полоса поглощения адсорбированного красителя смещена
в сторону красных длин воли по сравнению с полосой
поглощения для красителя в растворе (37).
Решающим доказательством в пользу объяснения
фотопотенциала, как вызванного ф отохимическими
изменениями красителя в растворе, служат опыты, в которых было
установлено, что такой же по величине и знаку потенциал
может быть вызван погружением электрода в
предварительно освещенный раствор флуоресцирующего красителя
(Грумбах 289).
Возникновение потенциала в результате фотохимической
реакции объясняется тем, что освещение раствора вблизи
одного из двух электродов изменяет здесь концентрацию
исходных частиц290. Если выравнить концентрацию
перемешиванием раствора илн заставить раствор быстро циркулировать
мимо электрода, то величина фотопотенциала сильно
снижается 291.
Наоборот, если затруднить диффузию от одного электрода
к другому использованием тонкого капилляра или повышением
вязкости растворителя, например заменой воды глицерином,
то фотопотеициал увеличивается292*293.
Было показано, что диффузия к электроду продуктов,
образовавшихся в месте действия света, является
определяющим фактором как для величины фотопотенциала, так и для
его временного хода при освещении и после прекращения
последнего292. Исходя из этой предпосылки, оказалось
возможным составить уравнение, связывающее величину
фотопотенциала с интенсивностью света, временем освещения
и временем затемнения, которое удовлетворительно
согласуется с опытом.
Описанные эксперименты производились с уранином,
эозином, родамином в водных и спиртовых растворах, т. е. как раз
с теми красителями, которые дают сравнительно устойчивые
окрашенные фотопродукты (20). Поэтому было высказана
предположение, что именно последние, достигая электрода
и, возможно, адсорбируясь на нем, вызывают наблюдаемый
фотопотенциал29О> 292'293.
43. ФОТОГАЛЬВАНИЧЕСКИЙ ЭФФЕКТ 317
Однако, если из раствора уранина удалить воздух откачкой,
то фотопотенцнал исчезает293. Такое наблюдение указывает
на главное значение для эффекта наличия продуктов
фотохимического окисления кислородом, а не окрашенных
фотопродуктов, образующихся и в отсутствии кислорода (20).
Интересно, что фотопотенциал не зависит в некоторых пределах
от концентрации ионов водорода288.
Наблюдаемая зависимость фотопотенциала от концентрации
красителя и от корня квадратного интенсивности света
приписывалась тому, что фотопотенциал вызывается
неустойчивым фотопродуктом, который образуется при встрече
возбужденной, способной флуоресцировать молекулы
красителя А* со второй — нормальной молекулой A29i.
Предполагалось, что распад или потеря активности фотопродукта имеет
место при встрече двух молекул фотопродукта. Заманчиво
отождествить этот неустойчивый фотопродукт с семихиноном
красителя, но экспериментальные данные для такого
заключения недостаточны.
Было показано далее, что образующийся в растворе уранина
фотопродукт, приходя в соприкосновение с электродом,
заряжает его отрицательно290. Это наблюдается при освещечии
слоя раствора на расстоянии в 15 мм от электрода.1)
Природа красителя имеет значение для знака заряда288'291'293.
Так, например, хризоидин дает положительный потенциал. Но,
возможно, что это изменение знака следует приписать все же
поверхностному слою красителя, осаждающем ся на
электроде (44).
Постепенное повышение концентрации красителя в водном
растворе сначала увеличивает, а затем уменьшает
фотопотенциал. Причина последнего явления может заключаться либо
в дезактивации возбужденных молекул красителя, либо
в аггрегации молекул красителя в растворе в коллоидные
частицы, т. е. в концентрационном торможении фотохимической
реакции (36).
Обратимое окислительно-восстановительное световое
равновесие (32), типичное для тиазиновых и оксазиаовых красителей
(метиленоваш голубой, тионин и др.)» также сопровождается
изменением потенциала электрода, погруженного в освещаемую
1) Если же освещать сам электрод с прилегающим слоем раствора
то знак фотопотенциала — положительный. Это и означает, что иа электроде
имеется фотоэлектрически чувствительный слой, дающий эффект Беккереля
знака, обратного знаку фотогальванического эффекта, вызванного
фотохимической реакцией в среде раствора.
Для родамина в условиях циркуляции раствора этот „поверхностный"
положительный потенциал исчезает, что также указывает на то, что он
вызван адсорбцией или осаждением красителя на электроде 291,292.
318 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
электролитическую полуячейку296. Новое стационарное
состояние равновесия под освещением устанавливается исключительно
быстро для растворов, содержащих, кроме красителей А н его
лейко-формы Н2А, также неорганические катионы, например
Fe+4~*" и Fe+~*~, способные быстро и обратимо обмениваться
электронами с красителем и его лейко-формой. Такие катионы,
носящие название „медиаторов" или „посредников",
способствуют быстрому установлению и темнового редокс потенциала
различных органических систем.
Весьма высокой фотохимической и одновременно
фотогальванической чувствительностью обладает система:
Av
тионин -b2Fe~*"l~ ^± лейко-тиотинн- 2Fe+++, (5)
детально изученная Рабиновичем'96.
В темноте равновесие смещено целиком в левую сторону.
При освещении видимым светом, поглощаемым тионнном,
раствор полностью выцветает за несколько секунд и столь же
быстро принимает первоначальную окраску, если кислород
из раствора был удален. Кислород, вызывая частично
окисление лейко-тионина и ионов Fe++, постепенно уничтожает
обратимость эффекта.
Фотопотенциал значителен (—250 mV), имеет
отрицательный знак и следует без задержки за изменением интенсивности
освещения.
Квантовый выход фотовыцветания по порядку неличины
составляет 0.25. Для представления о чувствительности
фотореакции можно привести следующие численные данные. При
концентрации тионина 5-10~5 М и поглощении раствором 1019
фотонов в сек. выцветает 50% исходной концентрации
красителя (ср. 3).
В условиях столь интенсивного освещения (применялась
лампа в 1000 ватт) каждая молекула красителя в растворе
должна была поглощать по 1 фотону два раза в секунду
и, следовательно, она находилась в среднем г12 секунды
в обесцвеченном состоянии лейко-формы.
Схема (2) реакции (43), применимая и здесь, изображает
как обычно лишь суммарный механизм, который фактически
протекает через стадию переноса только одного электрона
и протона, т. е. форму семихннона красителя (28).
В действительности, кинетика процесса (5) значительно
осложняется из-за присутствия семихинона и лейко-формы
не только обычного однозаряженного катиона тионина, но также
семихинонов и лейко-форм всевозможных других модификаций
^4. ФОТОВОЛЬТАИЧЕСКИЙ ЭФФЕКТ (ЭФФЕКТ БЕКККРЕЛЯ)
молекулы красителя, образующихся в растворе в результате
присоединения или отдачи протонов, поскольку наблюдения
относятся к кислому раствору, благоприятствующему
выцветанию. Со всеми этими формами реагируют катионы железа,
создавая весьма запутанную картину явления. Подробный
анализ различных кинетических уравнений в общем случае
затруднителен, но для большой величины выцветания можно
сделать вывод, что потенциал электрода вызывается семихино-
ном обычного однозарядного катиона тионина, т. е. формой
Н-тионин"1" и продуктом присоединения к нему иона водорода:
Н-тиоНин*" Н~*\ Эти ионы отдают электроны платиновому
электроду, заряжая его отрицательно. Электроны от
платинового электрода забираются катионами Fe"1""1""1". Иовы Fe++~,
а также форма лейко-тионина Н2-тионив+Н'*" не участвуют
в электронном обмене с электродом.
Отрицательный знак фотопотенциала наблюдается также
для разнообразных ароматических и магний-органических
соединений при освещении их неводиых растворов в
непосредственной близости к электроду286. Характерно, что безинер-
циоиные фотопотенциалы дают аминопроизводные, обладающие,
как мы подчеркивали (30), склонностью легко отщеплять
электрон при освещении, т. е. обнаруживающие эффект
элементарного фотоокисления297.
44. Фотовольтаический аффект (эффект Беккереля)1)
В 1839 г. Беккерель обнаружил появление разности
потенциалов между двумя одинаковыми электродами, погруженными
в раствор прозрачного электролита (например КС1), если они
покрыты слоем фоточувствительного вещества (например Ag-Br)
и один из электродов подвергается освещению (фиг. 50). Это
явление подверглось в дальнейшем всестороннему изучению 2S6.
Д,\я фотохимии красителей представляют интерес
исследования Стора, в которых освещаемый электрод (обычно
платиновый) нес на себе краситель либо в виде окрашенной
пленки (коллодионной), либо твердого осажденного слоя298.
Второй электрод помещался в стандартную электролитическую
полуячейку, соединенную с освещаемой полуячейкой проме-
1) В литературе иет согласованности в терминологии при наименовании1
разнообразных фотоэффектов, наблюдаемых на электродах, погруженных
в растворы электролитов. Фотогальванический эффект, описанный выше,
называют таиже эффектом Беккереля II рода Нередко его объединяют
С собственно эффектом Беккереля под общим названием „фоговольтаи-
ческий" эффект (см., например, обзор Копеланда, Блэка, Гаррета286).
"320 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
жуточной трубкой, наполненной электролитом (КС1) для
создания общей цепи.
Было установлено, что фотоэффект локализован на границе
раздела электрода и нанесенной иа него окрашенной плевки.
Знак потенциала, принимаемого электродом, соответствует
тому заряду, который должна принимать молекула красителя
•на первичном этапе фотореакции со средой. Красители хинон-
имидного строения (метиленовый голубой), которые испытывают
реакцию фотовосстановления, т. е. воспринимают электроны,
дают отрицательный фотопотенциал. Напротив, легко
фотоокисляющиеся флуоресцеиновые, ди- и трифенилметановые
красители, отдавая электроны среде, заряжают электрод
положительно. В безводном глицерине эти красители
испытывают реакцию необратимого фотовосстановления;
в соответствии с обратным направлением переноса Электрона
и знак фотопотенциала для тех же красителей в глицериновой
среде, вместо водной, делается отрицательным.
Явление происходит так, как если бы знак потенциала
электрода определялся последующим обменом электронами
ляэжду ним и красителем, испытавшим первичную фотореакцию
отрыва или принятия электрона.
Освещение короткими ультрафиолетовыми длинами волн
(^ <^*300 т\к) в присутствии кислорода вызывает, как мы
указывали выше, для красителей всех классов необратимую фото-
реакцию деструктивного окисления (глава 7). В соответствии
с этим поведением красителей фотопотенциал всех красителей
при освещении коротким ультрафиолетовым светом неизменно
положителен. Это указывает на то, что первичный акт
фотореакции в этом случае состоит в отщеплении электрона
от молекулы красителя.
Введение в раствор легко окисляющихся веществ
(ароматические амины, полифенолы и т. п.), тормозящих реакцию
окисления кислородом (антиоксиданты), вызывает уменьшение
и даже исчезновение положительного фотопотенциала,
обусловленного фотоокислением красителя при освещений как
видимыми, так и короткими ультрафиолетовыми длинами волн.
Наоборот, при повышении щелочности среды, которая ускоряет
окислительное выцветание, положительный фотопотенциал зна-
чительно^растет. *)
1) В случае трифенилметаноаых красителей повышение щелочности
раствора свыше определенного значения (рН > 10) резко меняет знак фото-
пот^нциала иа положительного на отрицательный. Очевидно, это обращение
знака с вяза к о с превращением красителя в карбинол путем присоединения
ионов ОН~~ (стр. 159).
44. ФОТОВОЛЬТАИЧЕСКИЙ ЭФФЕКТ (ЭФФЕКТ..БЕККЕРЕЛЯ)
321
Итак, обнаруживается далеко идущий параллелизм между
знаком и даже величиной фотопотенциала и природой
фотохимической реакции, нанесенного на электрод красителя.
Особый интерес для понимания механизма возникновения
фотопотенциала представляют опыты с оксазиновыми и тиази-
новыми красителями (нильский голубой, метиленовый голубой),
способными в отсутствии кислорода, как мы знаем, испытывать
обратимую фотореакцню окисления-восстановления (32).
В темноте для этих
красителей устанавливайся
определенный по знаку и
величине потенциал электрода,
в соответствии с отношением
концентраций катиона
красителя — метиленового
голубого Mbь и его
лейко-формы Н2МЬ+ на электроде (43). § *200'
Для см мщения равновесия '§
в сторону красителя или же *§
его лейко-формы в раствор ^
прибавлялся в возрастающей ^
концентрации неорганический
окислитель (хлорная вода)
или восстановитель (TiCl3).
При этом темновой
потенциал изменяется как
п ,
+6QQ-
+400-
-200-
Фиг. 51. Зависимость темиового по»-
тенциала и фотопотенциала эффекта
разной Кривой (фИГ. 51), Беикерели от возрастания концентра-
лейко-формы метиленового голу-
бого В коллодионной пленке на пла-
тиновом электроде
обычно по типичной S-об-
разной кривой (фиг. 51),
средняя точка перегиба
которой соответствует 50°/0
содержания восстановленной
формы HglV.b"1". С двух сторон
от перегиба находятся участки особо быстрого изменения
темнового потенциала, сопровождающиеся переменой знака,
как это следует из шкалы, изображенной на фиг. 51 слева.
Если параллельно этому изменению содержания Н2МЬ+ измерять
фотопотенциал, то оказывается, что он принимает
значительную величину именно на этих участках быстрого
изменения темнового потенциала (см. фигуру). В отсутствии
кислорода фотопотенциал положителен на всем протяжении
изменения концентрации лейко-формы I-^Mb"1" и связан,
очевидно, с ее фотоокислением. Высокий максимум фотопотенциала
на фиг. 51 наблюдается на том участке концентраций
восстановителя, который соответствует большой скорости
образования лейко-формы. Когда весь краситель превращен в лейко-
л1 А, Н. Теренин
322 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
форму, фотопотенциал падает до нуля. Это наводит на
предположение о связи потенциала с присутствием лабильной
полувосстаиовленной формы, т. е. семихинэна НМЬ"1".
Наибольшее значение этого положительного
фотопотенциала получается при освещении длинами волн близкой
ультрафиолетовой области спектра, поглощаемыми лейко-формой
красителя. В отсутствии восстановителя, когда преобладает
краситель, фотопотенциал имеет небольшую величину и
отрицательный знак, что свидетельствует о наличии слабой
реакции фотовосстановления красителя iVib"1" в рассматриваемых
условиях. Действительно, наибольшее значение этого
отрицательного фотопотенциала имеет место при освещении красным
светом в максимуме полосы поглощения красителя (метилено-
вый или нильский голубой).1)
С противоположностью действия на
окислительно-восстановительную систему радиации различной длины волны
мы ознакомились ранее в 32.2)
Изучением влияния рН на положительный
фотопотенциал, вызванный фотоокислением лейко-формы красителя
(нильский голубой), было установлено в ки:лой области
= 2.0—5.8), что фотопотенциал мал, т. е. фотоокисляемость
1
Н2№1+ невелика.
В этой области концентраций ионов водорода лейко-форма
красителя, обладающая основными свойствами из-за
присутствия свободных аминогрупп (ср. 5), присоединяет
дополнительный протон и превращается в форму HgNil^H"1". Если
концентрацию водородных ионов уменьшать, то при рН = 6.5,
когда не только последняя, но и сама лейко-форма b^Nil"1"
освобождается от структурного протона Н~*~, превращаясь
в слабоокрашенное свободное основание красителя (5),
фотопотенциал резко увеличивается. Этот важный факт указывает
на то, что присоединение протонов, связывающее свободные
пары электронов трехвалентного азота в аминогруппах,
препятствует фотоокислению, а следовательно, что последнее
*) Для получения стационарного отрицательного фотопотенциала ока-
залось необходимым присутс:вие кислорода, окисляющего восстановленную
формулу красителя Н2МЬ"Ь обратно в краситель, — иначе изменяется
концентрация последнего.
2) Интересно, что поглощение света в ультрафиолетовых полосах
красители практически не вызывает какого-либо фотопотенциала, в отличие
от лейко-формы, хотя положение полос для МЬ"*~и Н2МЬ+ почти одинаковое.
Если придерживаться установленного выше параллелизм1» между
фотохимической реакцией и фотопотенциалом, то следует заключить о слабой
фотохимической активности поглощения ультрафиолетовых фотонов
красителем (в отсутствии кислорода).
44. ФОТОВОЛЪТАИЧЕСКИЙ ЭФФЕКТ (ЭФФЕКТ БЕККЕРЕЛЯ) 323
состоит именно в отщеплении электрона и притом от атомов N
(ср. 30).
Большой интерес представляют наблюдающиеся
соотношения между фотопотенцналом и строением красителя.
Противоположность фотохимического поведения фотоокисляющихся
красителей с центральным атомом С и фотовосстанавливаю-
щихся краснтелей с центральным атомом N (см. диаграмму 1 в 5)
сказывается и в знаке фоювольтаического эффекта.
Красители второй группы, обладающие выраженной
способностью к обратимой реакции окисления-восстановления, для
которых типична в присутствии восстановителя реакция
фотовосстановления (32), дают отрицательный фотопотенциал
в соответствии с приемом ими электрона, переносимого затем
на электрод. К этой группе красителей принадлежат и
фотографические десенсибилизаторы (40).
Поведение красителей с центральным атомом С более
сложное. Химическая реакция восстановления соответствующими
реагентами протекает для них с трудом и полученные лейко-
красители сравнительно устойчивы по отношению к
кислороду, который, напротив, быстро окисляет лейко-формы
красителей предыдущей группы. Таким образом, они неспособны
быстро приходить в окислительно-восстановительное равновесие
с окружающей средой. Несмотря на интенсивное поглощение
света фотоэлектрохимически, они мало активны. Типичной для
этих красителей является реакция необратимого окисления
кислородом, приводящая к разрушению молекулы (глава 7).
В соответствии с этим поведением знак их фотопотенциала
либо равен нулю (трифенилметановые), либо имеют
значительную положительную величину в присутствии кислорода
(аурамин, флуоресцеины). Следовательно, они склонны к
отдаче электрона, заимствуя его затем от электрода. К ним
принадлежат и некоторые фотографические сенсибилизаторы.
Под действием ультрафиолетового света обе группы
красителей дают значительный положительный фотопотенциал,
связанный с необратимым выцветанием в результате окисления
кислородом воздуха.
Таким образом, эффект Ееккереля с красителями,
нанесенными на электрод, подтверждает электронную природу
первичных фотохимических реакций красителей и дает некоторую
возможность проследить за направлением переноса электрона.
Необходимо, однако, считаться с рядом обстоятельств,
осложняющих интерпретацию явления, таких как участие колло-
дионной пленки в рассматриваемых фотореакциях, медленность
установления фотопотенциала, не достигающего стационарного
значения, наконец, изменение темнового потенциала в резуль-
21*
324 ФОТОЭЛЕКТРОХИМИЧЕСКИЕ ЭФФЕКТЫ КРАСИТЕЛЕЙ
тате накопления продуктов фотореакции в непосредственном
соседстве с электродом (см. предыдущий параграф).
Однако, несмотря на эти недостатки, применение
электрических измерений в фотохимии красителей открывает
определенные перспективы научной и технической значимости
и следует интенсивно разрабатывать эту область.
Произведенная в этой книге попытка систематического
изложения многообразного фактического материала по
фотохимическим реакциям красителей позволила выявить некоторые
типачные особенности поведения красителей различных классов.
Последовательная электронная трактовка первичного
фотохимического акта и учет метастабильного (бирадикального)
состояния красителя дали возможность объединить разнородные
явления в одну общую картину. Разумеется, еще много
необходимо потратить труда, чтобы доработать поставленные вопросы
экспериментально и теоретически. Но уже одно сопоставление
полученных до сих пор результатов и точек зрения на механизм
различных фотореакций, несомненно, послужит более глубокой
теоретической трактовке фотохимического поведения красителей
в тех гораздо более сложных конкретных условяях, с
которыми приходится иметь дело при техническом их применении.
ЛИТЕРАТУРА
1. По фотохиыии красителей специальной монографии нет; отдельные
реакции затронуты в ел'дующих книгах: Лазарев, Основы учения
о химическом действии света, /, //, ///(1919—1920); Kistiakowsky,
Photochemical Processes (1928); Griffith, McKeown,
Photo-Processes in Gaseous and Liquid Systems (1929); P 1 о t n i k о w, Allg-emeine
Photochemie (19j6); Ellis, Wells, Chemical Action of Ultraviolet
Light (1941).
2. Rabinowitch, Photosynthesis and Related Processes (1945); Blum,
Photodynamic Action and Diseases, caused by Light (1941); по
фотосинтезу см. сбзор: Краен овский, Успехи современн. биол., 21
(1946) 155.
3. О спектрах поглощения красителей сведения можно получить из
следующих источников: Ley, Handb. d. Physik, XXI (1929) 1, 57;
Forma n e k, Untersuchung und Nachweis organicher Farbstoffe (1908);
Formanek, Knop, Untersuchung" und Nachweis organischer
Farbstoffe (1926); Кравец, Абсорбция света в растворах окрашенных
веществ, М. (1912); L a z a r e w, Nedopekin, Atlas des spectres des
substances colorantes, Ac. Sc. URSS (19-7); Uhler, Wood, Atlas of
Absorption Spectra, Carneg-ie Inst. (1907); Colour Index, Soc. Dyers
a. Col.; S с hu 1 t z, Farbstofftabellen, Akad. Verl. (1931); Tables annuel-
les de Gonstantes et Donnees nuraeriques VII (1930) 1.
4. Вавилов, Изв. Физ. инст. и нист. биол. физ. 1(1920, вып. 3) 92, 96.
5. Вавилов, Там же, 1 (1920, вып. 2) 77.
6. По флуоресценции красителей специальной монографии нет. История
изложена в книге: К ays e r, Handb. Spe^troskopie, I*/ (1908);
состояние вопроса даио в несколько устаревшей книге: Pringsheim,
Fluoreszenz und Phosphoreszenz (1928) и Handb. Exp. Physik, XXIII. 2,
Teil (1928) 956.
7. Леашин, Ж. фив. хим. 2 (1931) 641; 6 (1935) 991; 9 (1937)1; Acta
phys. chim. URSS 2 (1935) 221; 6 (1937) 213; Z. Phys. 72 (1931) 368,
382.
8. Terenin, Acta phys. polon., 5 (1936) 229.
9. Vavilov, Phil. Mag. 43 (1922) 307; Z. Phys. 22 (1924) 266; 42 (1927)
311.
10* См. многочисленные статьи Вавилова и его сотрудников. Некоторая
сводка результатов дана в статье: Вавилов, О фотолюминесценции
растворов, Изв. АН СССР, физ., 9 (1945) 283.
Юа. Vaviiov, J. phys. USSR 9 (1945) 68.
11. Паулинг, Усп. хим. 7 (1938) 1312; Сыркнн, Дяткина, Жую-
в и цк и ж, Усп. хим. 10 (1941) 121; Сыркии и Дяткина,
Химическая связь и строение молекул (1946); Paulingf, Nature of the
Chemical Bond, Cornell Un. (1944).
326 ЛИТЕРАТУРА
11а. VanAlphen, Rec. Trav. Chim. 60 (1931) 138? Arndt, Ber. 72 (1939)
860; Докунихин, Левин, ДАН 35 (1942) 1/0, см. также*5,124.
12- Robertson, J. Chem. Soc. London (1^35) 615; (1936) 1195.
13. H iicke I, Z. Electrochem. 43 (1937) 75 J, 8/7.
14. Lonsdale, Proc. Roy. Soc. 159 (1937) 149.
15. Вицингер, Органические красители, ОНТИ (1936); Wizingfer,
Org-anische Farbstoffen (1933); Измаильский, Тр. IV Совещ.
анилокрас. химии и техи. АН СССР, Отд. хим. и. (1940)41; Eistert,
Tautomerie und Mesomene, Gleichjrewicht und Resonanz (1938);
Измаильский и сотр. ДАН 26(19*0) 9, 101, 906, 912; Ж. Общ.
Хим. 11 (1941) 650, 691, 6^7; 13 (194°*) 6^5. 8И, 348.
16. Pauling, Proc. Nat. Acad. 25 (1939) 577; Bury, JACS 57 (1935) 2115;
Forster, Z. Electrochem. 45 (1939) 548; Уегг. хим. 9 (1940) 71;
Infold, Proc. Roy. Soc. 169(1^39)149; Nature 141(1938) 314; Воль-
кенштейн, Природа (I9i3, Xs 5) 18.
17. Lewis, Calvin, Chem. Rev. 25 (1939) 2731; Усп. хнм. 10 (1941) 32.
Lewis, JACS 67 (1945) 770
18. Henrici, Z. phys. Chem. (B) 47 (1940) 93.
19. Феофилов ЖЭТф 12 (1942) 328; J. Phys. URSS 7 (1943) 68;
Изв. Ак. Наук СССР, физ. 9 (1945) 283; Lewis, Bigreleisen,
JACS 65 (1943) 2102, 2107; Ku hn, D iihr kop, Martin, Z. phys.
Chem. (B) 45 '1939) 21.
20. Тумермаи, Тр. ФИАН. 1 (1913, вып. 4), 77.
21. Тереиии, В а р т а и я и, Непорвит, Trans Farnd Soc, 35 (1939)
39; Вартаи я и, Изв. Ак. Наук СССР, физ. (1938, №4)3 1; Ж. фиа.
хяи., 12 (1938) 308; J. Phys USSR, I (1939) 213; 3 (1940) 467.
22. Непоре ит, Ж. физ. хим.. 13 (1919) 916; дисс ГОИ (1945).
23. Прилежаева, Acta phys. chim, URSS, 1 (1935) 785.
24. Тереиии, Acta phys. chim. URSS. 18 (L943) 210; Ж. физ. хим., 18
{ (1944) 1; Изв. АН СССР, фиа., 9 (1945) 305.
25. К р а в е и, Абсорбция света в растворах окрашенных веществ (1912).
25а. Auschka~p, Acta Univ. Latv., 1 (1930, № 12) 279.
26. Konigsberger, Ki 1 с h li n g, Ann. Physik, 28 (1909) 889.
27. Konigsber^er, Kiipf erer, ib., 37 (191J^ 601.
28. Аевшин, Acta phys. chim. URSS, 1 (1935) 685; 2 (1935) 221.
29. M i с h a e 1 i s, G r a n i с k, JACS, 67 (1945) 1215.
29a. Prins, Nature, 134 (1934) 457.
30. Rabino witch, Epstein, ib., 63 (1941) 69.
31. Тумерман, ЖЭТФ, 11 (1941) 515 [J. Phys. USSR, 4 (1941) 151].
См. также G о 111 i n g-, Phys. Rev., 22 (1924) 566; Gaviola,
Z. Physik, 35 (1926) 708.
32. Hurd, Livingston,.]. Phys. Chem., 44 (1940) 565.
33. Л е в ш ни, Ж. физ. хим. 6 (1935) 991; Acta phys. chim., URSS, 2 (1935)
221; Ж. физ. хим., 2 (1931) 641.
34. В о w е П> N о г t о п. Trans. Farad. Soc., 35 (1939) 44.
35. Феофилов, ДАН, 45 (1944) 387.
36. Прилежаева, Ж. физ. хим., 10 (1937) 342 [Acta phys. chim. URSS.
Z. (1937) 149].
37. Прилежаева, Климова, Ж. фиа. хим., 10 (1931) 353; Acta phys.
chim. URSS, 7 (1937) 163.
38. Handb,Experim.Phy iktXXIII. I Teil(19^8) 55 6; Борисов, ЖРФХО,
37 (1905) 249; Gold stein, Verb. Deutsch. Phys. Ges., 6 (1S04) 156,
185; Phys. Z., 11 (1910) 430; 12 (1911) 614; 13 (19b) 188, 577; Phil.
Mag., 2Э (1910) 619; F i s с h e r, Z. wiss. Phot, 6 (19o8) iO5; Dickson,
ib., 10 (1912) J68; Marsh, J. Chem. Soc, London (19/7) 1/5;
Kowalski, C. R., 144 (1907) 836? 145 (1907) 391, 1270; 148 (1909)
ЛИТЕРАТУРА 327
280; 151 (1910) 810, 943; 152 (1911) 83; Phys. Z., 12 (1911) 956» 964;
15 (1914) 322. См. интерпретацию спектров в статьях: Тег en in,
Acta phys. chim. URSS, 12 (1 40) 617; 13 (1940), 1; 14 (1941) 566;
Изв. Ак. Наук СССР, хим. (1940) 59. См. также 2*.
3% Вавилов, Левшин. Z. Physik, 35 (19^6) 920; Wawilow,
Р г i n g s h e i m, ib., 37 (1926) 705; G о 1 о ц Ь, Sow. Phys., 7 (1935) 49;
Лев шин, Винокуров, Sow. Phys., 10(1936) 10; ДАН, 2 (1936)
133; Левшин, Повунова, Тр. ФИАН, 1 (19*8) 38; Kaut-
s k у 87.
40. Jablonski, Nature, 131 (1933) 839; Z. Physik, 94 (1935) 38; Acta
phys. polon, 5 (1936) 271.
41. Lewis, Lipkin, Magel, JACS, 63 (1911) 3005; 64 (1042) 1774.
42. I Чердынцев И Чердынцев иВассерман, (1940).
43. Вавилов, Шишловскии, Sow. Phys., 5 (1934) 379.
44. Lewis, Kasha, JACS, 66 (19*4) 2100; Lewis, Calvin, ibid., 67
(1945) 1232.
45. Sheppard, Rev. Mod. Phys., 14(1942) 303; Shepp a r d, Brig-ham,
JACSP 66 (194*) 380.
46. Sheppard, Lambert, Walker, J. Chem. Phys., 9 (1941) 96.
47. Lewis, Lipkin, Magel, JACS, 66 (1944) 1579.
48. Smakula, Wassermann. Z. Phya. Chem., A, 155 (1931) 353
Z. Phys. Chem. (B) 25 (1934) 90; Lewis, Magel, Lipkin, JACS,
62 (1940) 2973.
49. Hartley, J. Chem. Soc, London (1938), 633.
50. Lewis, Magel, Lipkin, JACS, 64(194.) 1774.
51. Holmes, Scanlan, JACS, 49(1927) 159.;51 (1929) 1204; см.также&7.
5 A Holmes, Ind. Eng. Chem., 16 (19^4) 35; Lewis, Mage I, Lipkin60.
53. Adams, R о s e n s t ei n, JACS, 36 (1914) 1452.
54. Kortum,Z. Phys. Chem. (B), 33 (1936) 1.
55. Rabinowitch, Epstein, JACS, 65(1941)69; Lewis, Gold-
schmidt, Magel, Bigeleisen, ibid., 65 (1943) 1150; Vicker-
stoff, Lemin, Nature 157 (1946) 373.
56. Michaelis, Granick, JACS, 63(1941)1636; 65(1943) 1747.
57. D i x о n, J. Opt. Soc, 21 (1931) 250; Holmes, Peterson, J. Phys.
Chsm., 36 (1932) 1243; Sheppard, Rev. Mod. Phys., 14 (194*) 303;
Соловьев, Ж. тех. фиэ. 15 (1945) 765; Acta phys. chim. URSS
19 (1944) 448. См. т кже^в.
5S. J el ley, Nature 138 (19%) 1009; 139 (1937) 631.
59. S с h e i b e, Z. an<rew, Chem., 50 (1937) 212.
60. Socher, Fortchritte d. Photographie, Bd. Ill (1944).
61. Scheibe, Schontag.Katheder, Naturw. 21 (1939) 499; Kathe-
der, Koll., Z. 92 (1940) 292; 93 (1940) 28.
62. Л a r a p e в, Выцветание красок и пигментов в видимом спектре,
М. (1911); Ann. Physik. 24 (,Р07) 661; 37 (1912) 812; Z. Phys. Chem.
78 (1911)657; 98 (13j1) 94; 100 (19j2) 266.
63. Goodeve. Nature 143 (1939) 10J7.
64. Lewis, Lipkin, JACS 64 (l9-±2) 2801; Lewis, Bigeleisen,
ib. 65 (1943) 5Л).
65. Olson, T-ans. Farad. Soc. 27 (191?) 69; J. Chem. Phys. 1 (1933) 418;
Olson, Hudson, JACS, 55 (1933) 1410; О 1 s on, M aun e y, ib.. 56,
(1934) П20; Mul liken, Phys. Rev. 41 (193*) 751; Теренин, Изв.
АН СССР, фнз. 9 (1945) 305; Феофилов, ДАН 45 (1944) 387.
66. Lewis, Magel, L i p ki n. JACS 62 (19 0) 2973,
67. Kiichler, Pa tat, Z. Electrochem. 42 (1936) 529; Leigh ton,
Lucy, J. Chem. Phys. 2 (1934) 756, 760.
323 ЛИТЕРАТУРА
68. K6ge), Phot. Korr. 63 (1927) 106, Seyewetz, Mounier, CR 18S
(1927) 1279.
69. Franck, Livingston, j. Chem. Phys., 9 (1941) 184; Franck,
Pringsheim, ib., 11 (1943) 21.
70. Terenin, Acta phys. chim. URSS 12 (1940) 617; 13 (1940) 1; Изв.
Ar. Наук СССР, хим. (1940) 59.
71. Lewis, Lipkin, JACS, 64(1942) 2801; Lewis, Big- eleis en, ib „
65 (1943) 5/0, 1144, 2419, 2424.
72. West,J. Chem.Phys. 8 (i940)849; Ann. New-York Ac Sc. 41 (1941)203.
73. Kogel, Z. wiss. Phot. 24 (19-7) ^16; К 6 g- e 1, Steigman, ib., 24
(1927) 18; E g s e r t, S с h г о t e r, Z. Electroch., 34 (1928) 602? S с h г Й-
ter, ib. 28(1930); Ramsper^er, JACS 50(1928) ЬЗ; 49(1927)
912; Proc. Nat. Ac. 13 (1927) 549; Seyewetz, M о u n i e r, CR. 186
(1923) 953; Feldmann, Z. ph. Chem. 12 (1931) 449; Wolf, ib., 17
(193£) ^6; Eder, Z. wiss. Phot. 33 (193 ) 1 (история;; S a under*,
The Aromatic Diaso- Compounds (1936), Ch. IX [Cay и дере,
Аромат, диазосоедииеиия, М. (1938)].
74. Левкоев, Петров и Дурмашкина, Фотохии. пром. 4 (1936,
вып. 1) ^3; Сов. кииофото (1936, вып. Ю) 40 См. также1: Р I о t n i-
kow, All^em. Photochem. (1936) 781; Ellis, Chem. Act. UV Light
(L941) 624.
75. Lifschitz, Ber. 52 (1919) 1919; 58 (1925) 2444; Lifschitz,
Girbes, ib.f 61 (1928) 146"*; Lif schitx, Joffe, Z. phys. Chem. 97
(19-1) 4/6; Z. wiss. Phot. 29(1930^ 91; Holmes, JACS 44 (192/) 1002;
Ariga, Bull. Chem. Soc. Japan 2 (1927) 65.
76. Weyde, Frankenburger, Zimmerman n, Naturw. 18 (1930)
'/06; Z. phys. Chem. (B), 17 (1932) /76; Trans. Farad. Soc 27 (1931) 561.
76a. Те ренин, Фотохимия паров солей, ГТТИ (1934).
77. Harris, Kaminsky, S i m a r d, JACS 57 (1935) 1151; Germann,
Le Roy, Gibson, ib, 62 (1940) 110.
78. Weigert, Naturw. 15 (19/7) U4; Bowen, Trans. Farad. Soc. 35
'1919) 20; Bowen, Norton, ib.f 35 (1939) 44; Weiss, W e i 1-
Malherbe, J. Chem. Sjc. London (I94i) 544.
79. J. Perrin, Ann. physique 10 ^1*П9) 33; Wood. Phil. Maff. 43 (1922)
757; Prin^sheim, Z. Physik 10(1922) 176; 16 (19/3, 71; Weigert,
ib., 10 (1922) 349; McLennan, С a 1 e, Proc. Roy. Soc. 102(19.3)
256; Kearney, Phil. Mag. 47(1924)648; Asterblum, Bull Ac.
Polon., № 7—8A (19.4) 297; Натансон, Ж. физ. хим. 14 (19-iO) 16.
80. Heffter, Вгг., 18 (1905)3643.
81 Лев шин, Ж. физ. хим. 2 (1931) 641 [Acta phys. chim. URSS 2
(1935) 221].
82. Blum, SpealmanJ. Pbys. Chem. 37 (1938) 1123.
83. Wei^ert, Z. Physik 5 (1921) 410; Nat irw. 9(1921) 53^10(1922)349.
84. Kautsky, Ber. 64 (1931) 2677; 65 (1932) 1762; 68 (1935) 152.
85. Bowen, Nor t он, Trans. Farad. Soc 35 (1939) 44? В о w e n, W i I-
liams, ib., 35 (1939) 765; Weil-Malherbe, Weiss, Nature 149
(1942) 471, 612; 153 (1913) 449; J. Chem. Soc. London (1944) 541;
Weigert, ib.,149 (1942) 56; Miller, Baumann, JACS 65 (1943)
1540.
85a. С he с ha n, CR 222 (1946) 80.
86. Шишлове кий, ЖЭТФ 7 (1947) 1252; Вартаиян^; Рыскииа,
дипл. раб. ГОИ (1939); НепоренТ22.
87. Kautskv, Ber. 64 (1931) 1610, 2053, 2446; 65 (1932) 401; 66 (19Щ
1583; Biochem. Z. 291 (1937) 271; Trans. Faraday Soc. 35 (1939) 216;
PringsKeim, Vogels, Nafurw. 19 (1931) 964; Voxels, Paris,
(брошюра) (1935).
ЛИТЕРАТУРА 329'
88. Кондратьев, 3 и с к и н, Sow. Phys. 6 (1934) 293.
89. Euler, Brunius, Z. phys. Chem. (A) 139 (Haber-Festbd) 615.
90 Critical tables. свлш Miller, Baumann^'.
91. Kautsky, Trans. Farad. Soc. 35 (1939) 216.
92. Gaffron, Ber. 68 (1935) 1409; Biochem. Z. 264 (1933) 251; 287
(1936) 130.
93. Weiss, Trans. Farad. Soc. 35 (19^9) 48, 224.
93a. Weiss, Trans. Farad. Soc. 42 (1946) 116, 133; Bowen, ib., 138.
94. Fran ck, Lev i, Naturw. 23 (1935)229.
95. Audubert, Racz, Chem. Abstr. 38 (1944) 270.
96. Шпольский, Шереметьев, Ж. физ. хим. 8 (1936) 640 [Act»
phys. chim. URSS S (1936) 575].
97. Suida, Monatsh. 33 (1912) 1255,1268;Ber. 47 (1914) 467, 471; Ciami-
cian. Silber, Ber. 45(1912)40; 58(1925) 313 см. Красновский,
Ж. общ. хим. 10 (1940) 1094. (1940); Hi bbert, J. Soc. Dyers Col. 44
(1928)377, Sudborouffh, Watson, Narayanan, J. Ind. Inst.
Sc. 8A (1925) \; John, Ber. 58 В (1925) 1563; Eckert, ib., 58-
В (1925) 313.
93. Backstrom Beatty, J. Phys. Chem. 35(1931) 2530; Z. ph. Chem.
(B) 25 (1934) 99.
99. Bod en stein, Ber. (1931) 73; Z. phys. Chem. (B) 12 (1931) 141.
100. Dufraisse, Gerard, CR 201 (1935) 423; 202 (П31) 1859; Bull, soc
chim. 4 (1947)21)52; Dufraisse, Veluz, CR 203 (19%) 327.
101. Dufraisse, Prfou, CR 204 (1937) 127; Bull. Soc chim 5(19%)
611, Dufraisse, Le Bras, Velluz, Velluz, Bull. Soc. Chim. 4
(1937) 349, 12o0; 5 (1938)192,606, 1073; W i 1 le m ar t, CR 201 (1935)
1201; 202 (1936) 140; 203 (1936) 1372; 205 (1937) 866, 993; Bull. soc.
chim. 4 (1934) 357, 510, 1447; 5 (1938) 556.
102. Dufraisse, HoupilU t, Bull. soc. chim. 5 (1938) 626,
103. Dufraisse, Hor do is, Bull soc, chim. 3 (1936) 187 „ 1380; D uf-
raisse, Momer, CR. 196 (1933) 13/7; Dufraisse, Chovin,
CR. 197 (1933) 1127; Dufraisse, Loury, CR. 199 (1934) 957; Ba-
doche, CR. 198 (1934) 662, 1515; 200 (1935) 750; Bull. Soc. Chim. 3
(1936) 2Э40; Dufraisse, Veluz, ib., 3 (1936) 254.
104. Cook. Martin, Roe, Nature 143 (1939) 102.
105. Moureu, Dufraisse, Dean, Lotte Butler. CR 182(1926)
1440; 183 (1926) 101; 190 (19^0) 143; Dufraisse, Badoche 200
(1935) 1103; Schoenber?, Ber. 67 (1933) 633, 1404; Ann. Chem.
518 (L935) 20; Mfiller, Miiller-RodI of f, ib., 517 (19'5) 134;
Gaffron, Biochem. Z. 264 (1933) 251; Z. phys. Chem. (B) 37 (1937)
437, <53; Bowen, Steadman, J. Chem. Soc. (193 ) 1098; Kautsky,
Biochem. Z. 284 (1936) 412; Schumacher, Z. Electrochem. 42 (I93o)
522; Z. phys. Chem. (B) 37 (1937) 462; Kohlitz, Schumacher,
ib., 35 (19^7) 11; Petrikalis, Balodis, Acta Univ. Latviensis (Kim.
Fak. Ser.) 5 (1940, № 4) 75 [Phys. Ber. 24 (1943) 55].
106. Dufraisse, Loury. С R.200 (1935) 1673; D uf r a i s s e, L e Br a s.
Bull. Soc. chim. 4 (1937) 319.
107. Kautsky, Merkel, Naturwiss. 27 (1939) 195; Kautsky, Miiller
ib., 29 (1941) 150.
108. Dufraisse, Traite de Chmie Org-anique (Grigfnard) //(1936) 1147r
Dufraisse, Chovin, Hdb. der Katalyse (Schwab), //(1940) 339.
109. Bodenstein, Z. phys. Chem. (B) 12 (1930) 151; Бах, Усп. хим. 3
(1934) 177; Staudin?er, 2-е Conseil Chim. Solvay (1925).
110. Rieche, Angew. Ch. 50 (1937) 2o [Усп. хим. 6(1937) 1350];
Oxidation (Gen. Discussion) Trans. Farad. Soc. 42 (1946) 99.
111. Weiss, Nature 145 (1939) 744.
330 ЛИТЕРАТУРА
112. Сыркин иДятхина, Изв. АН СССР, хим. (1945) 543; (1946) 153;
Усп. хим. 16 (1*47) 29.
113. Chevreuil (1837), Abney. Rus s e 1 (1839), S ch u nk (1879).
114. Joffre, Fisher's Jahr. ber. (1839) 1157; Оглоблин, ЖРФХО 26
(189 ) 107; 27 (1895) 8J; Z. anorg. Chem. 8 (1895) 415; 12 (1896) 230,
Watson, Proc. Chem. Soc. (London) 25 (1909) *2i, 290; Worel,
Eder's Jahrb.Phot. 18(193±)42; 19 (1905) 7; Heffter, Ber. 18 (1S05)
3633.
115. Gebhard, Z. angew. Chem. 22 (19Э9) 82,433. 1390, 1890,1963,
1966, 2484; 23 (19Ю) 820; 24 (П11) <i74, 1291, 1616, 1990. ^<u6; 26
(1913) 79; 27 (1914) 4; J. Chem. Soc. (London) 98 (1910) ^05; Ber. 3
(1910; 751; Phot. Korr. (1909) 457; Chem. Z. (1913) 601, 622, 638, 662,
717, 755, 879; J. prakt. Chem. 84 (1911) 561.
116. Haller, Ziersch., Z. angew. Chem. 43 (1930) 209; MelL Tertber. Ю
(1929) 951.
117. Curtis, JACS 42 (1920) 7.0; Ш а рв ин, Пак ш вер, ЖФРХО 59
(19/7) 459; Z. angew. Chem. 40 (1927J 1003; Holmes, JACS 44
(193,!) 100-; Mounier, Tiba (1931) (см. Журн. ЗОТ, сер. Текстиль
1933, № 1); Де Шалыт, Ломанова, Изв. хлопч.-бум. пром.
(1932, № 2); Те ренин, Тр. IV Совещ. анил. кр. АН СССР (1940) 63.
118. Sey ewetz, Mounier, CR 185 (1927) 1279; Шар вин,
Шатрова. Ж. прикл. хнм. 2 (19/5), 109; Schultz, Ganguly
Ber. 58 (1925) 702; Kogel, Phot. Korr. 63 (1927) 107; Ворожцов
с сотр. Ж. общ. хим. 7 (19V7) 1610.
119. Iwamoto, Bull. Chem. Soc. Japan 10 (1935) 420.
120. Беленький, За реконстр. текст, пром. 12 (1933, № 6) 38.
121. Weig-ert, Z. phys. Chem., Nernst Festbd. (19Ь) 464; W i n the r,
_ Ann. Phys. 37 (1912) 8b; Bodenstein, Z. phys. Chem. 85 (1913)
357, 361
122. ОСТ 10085—39; см. также: Textile Colorist 62 (1940) 538.
123. Вартанян, Ж. техн. физ. 14 (1^44) 703; Вартанян, Адрианов,
Отчет Гос. Опт. инст. Ш— 2 (1940).
124. G i I Ь, S t о п е h i 11, J. Soc. Dy. Col. 60 (1944) 183 См. также "а .
125. Гамбурцев, Ж. прякл. физ. 6 (1929) 53.
126. Schwetzow, Z. wiss. Phot. 9 (191J) 65
127. Wawilow, 2, phys Chem. 88 (1914) 35.
128. Lei ber, Koll, Z. 50 (1930) 68, 117; Alberts, Z. wiss. Phot. 33
(1935) 234, 249
m Rein, Z. ang-ew Chem 47 (193') 157.
130. Fonda, J, Opt. Soc. 26 (1936) 316, 132.
131. Sommer, Z. an^ew. Chem. 44 (1931) 61.
132. Предводителе в, Нечаева, Z. Physik, 32 (1925) 226; 29 (1924)
332; Предводителе в, Блинов, ib, 35 (1925) Ч8.
133. Weigert, Z. Physik 5 (1921) 410, 423; 10 (1922) 349. Koll. Z. 28
(1921) 115; Naturw."9 (1921) 583.
134. W i n t h e r, Z. wiss. Phot. 11 (1911) 92; В о d e n s t e i n, Z. phys. Chem.
85 (1913) 357, 368; Натансон, Ж. физ. хнм. 14 (1940) 16.
135. Gebhard, Z. angew. Chem. 22 (1909) 437; 26 (1913) 79, Phot. Korr.;
Beyer, Le papier (mars 1925) 310. См. также110.
136. Gebhard, Z. an<rew. Chem. 22 (1909) 1890, 2484; 23 (1910) 820; 26
(1913)79; J. Soc. D. Col. (1910) 1/2.
137. Вюрмэер, Биологическое окисление и восстановление (19 5); Бах,
Усп. хим. 3 (1934) 177; В ил а ид, Усп. хнм. 8 (19*9) 561-
13а Weber, Z. Phys, Chem. (В) 15 (1931) 18; Ber. 69 (1936) 1026;
Z. Electrochem 43 (1937) 633; Radiolo ;ica 1 (1937) 233 [Chem. Zbl.
1939 11 825]; Weber Inhibitorwirkungen (1938).
ЛИТЕРАТУРА 331
139. Обзор литературы вопроса дан в книгах: Map к узе, Влияние света
на прочность текстильные подокон, Новости те*н. текст, пром., Гия-
легпром (1940); Садов, Действие света и атмосф. условий иа хлопч.-
бум. ткани (1946). Валько, Коллоидно-химические основы
текстильной технологви, //, гл. XIII Гизлегпром (194э).
140. Cunliffe, Ь Text. Inst 15 (1924) А 173; 19 (1928) 169; Ellis,
Chem. Ag-e 13 (1925) 470; Harrison, J Soc. Dy. Col. 28 (1912. 225.
141. Scholefi eld, Goodyear, Mell. Textber. 10 (1929) 867.
142. Haller, Wyszewiansky, Mell. Textber. 17(193o>45, 138,217,325.
143. Scholefield. Turner, J Text. Instr. 24 (1933) P. 130, 330; Mell.
Textber. 10 (1929)867; см. также: Baur, Perret, Helv. chim. Acta
7 (19^4) 910.
144. Land о It, Mell. Textber. 14 (1933) 32; S с h e II e r, ib , 16,(1935) 787.
145. Scholefield, Patel, ]. Soc. D. Col. 44 (1928) 268; 45 (192Э) 175.
146. Benrath, Meyer, Ber. 45 (1912) 2707; M e у e г, Е с k e r t u. Eckert
ib., 58 (1925) 303, 313; 63 (1927) 1693.
147. Jones. J. Soc. D. Col. 52 (1936, №8) 285; Henk, Mell. Textber.,
19 (1938, № 10) 730; Kunz, Z angew. Chem 52 (19*9) 269.
148. Michaelis, Schnbert, Chem. Rev. 22 (1938) 437; Michaelis,
Schubert, Granick, J^CS 61 (1939) 1981; 62 (1940) 1822.
149. Gurney, Ions in Solutbns (1936); Гэрни, Ионы в растворах (1933).
149а. Г an он, Ж. физ. хим. 20 (1946) 1023
150. Казарновский, Ж. физ. хим. 14 (1940) 320; Касаточкин, ДАН
47 (1945) 199.
151. Lewis, Bigeleisen, JACS 65 (1944) 1144.
152. Michaelis, Granick, JACS 63 (1941) 1636.
153. Michaelis, Oxidation-Reduction Potentials (1929) [Михаэли с.
Окислителъно-Босстанонительный потенциал (1929,]; Вюрмэер,
Биологическое окисление и восстановление, ГТТИ (1935); Некрасов,
Окислительно-воссгановительный иотенцяал в биологии, Усп. биол.
хим. 10 (19 Ч) 12; S tone hi 11, J. Soc. Py. Col. 60 (1944) 176.
1154. Те ренин, Усп. фнз н. 17 (1^37)1.
155. Franck, Scheibe, Z. phys. Chem. A 139 (1928) 22.
156. Pauling, Phys. Rev., 34 0929) 828.
157. A. Farkas, L. Farkas, Trans. Farad. Soc. 34 (1938) 1113, 1120.
158. Rabinowitch, R-v. Mod. Phys. 14 (1942) 112.
159. Дайн, Дисс. Инт. Физ. Хим УССР (194^); Dain, L i b e r s on, Acta
phys. chim. URSS 19 (1944)410.
159a. Вейденбах, ДАН (1947).
160. Potteril, Walker, Weiss, Proc. Roy. Soc. 156 (1936)338,561;
Weiss, Nature 136 (1^35) 794
161. Weiss, Porret, Nature 139 (1937) 1019.
162. Franck, Haber, Stzb. Preuss. Akad. Wiss. 13 (1931) 250.
163. W e i s , Trans. Farac'. Soc. 31 (1935) 668, 966. Bray, JACS 60 (1938) 82.
164. Б утков, Z. Physik, 62 (1930) 71.
165. Weiss, Naturw. 20 (1935) 64? Nature 133 (1934) 643.
165a. Haber, Wansbroug-h Jones, Z. phys. Chem. (B) 18 (1932) 103;
Farkas, Wansbrouo-h Jon a, ib., 18 (1932> 124.
166. Schmidt, Ann. 64(1898) 7 8; Stark, Phys. Z. 10(1939) 614; Pauli,
Ann. Phys. 40 (1913) 677; Hughes, Hhil. Mag. 24 (1912) 380.
167. Серков, ЖРФХО, фич ч. 44 (1912) 293.
168. Lewis Lipkin, JACS 64 (i 942) 2801.
169. Volmer, Ann. Physik 40 (1913) 775
170. Butler, Trans. Farad. Soc. 31 (19 >7) 229.
171. Burgherr, Z. wiss. Phot. 24 (1927; 393; Banckroft, Proc. Nat
Ac. 17 (1931) 407.
332 ЛИТЕРАТУРА
172. Neuweiler, Z. wiss. Phot. 25 (1927) 211.
173. KSffel, Steigmann, Z. wiss. Phot. 24 (1926)-18, 171; Schmidt,
ib., 24 (1927) 221; Steigmann, ib., 27 (1929) 24; Kogel, Ber. V1IL
Kongr. Photogr. (1931) 363; KolL Z. 39 (1940) 52.
174. Baur, Helv. Chim. Acta 1 (1918) 186; Z. phys. Chem. (B) 16 (1932) 465.
175. Geake, Lemon, Trans. Farad. Soc. 34(1938) 1395, 1409; Apple-
ton, Geake, ib., 37 (1941) 45, 60; Geake, ib., 37 (1941) 88; Clib-
bens, J. Soc. Dy. Col. 59 (1943) 275; S to n e h i 11, ib., 60 (1944) 176.
176. Rabinowitch, Weiss, Nature 136 (1936) 10^8; Porret, Rabi-
nowitch. ittd, 140 (1937) 321; Weiss, ib., 141 (1938) 248;
Rabinowitch, Weiss. Proc Roy. Soc 162 (1937) 251.
177. Rabinowitch, J. Chem Phys. 8 (1940) 551, 560.
173. Weber, Naturw. 23 (1935) 849; Nature 137 (1936) 570.
179. Weiss, Nature, 138 (1936) 80; Trans. Farad, Soc. 32 (1936)1331;
Weiss, Fish gold, Z. phys. Chem. (B) 32 (i936) 135.
180. Mudrovcic, Z. wiss. Phot. 26 (1928/9) 171. См. также: Хюбль,
Гребе, Уолл, Цветная фотография, М- (1933) § 14; Friedman,
History of Color Photography (1945) Ch. 28.
131. Euler, Hells tr 6 m, Brandt, Naturw, 23(1935)486; Hellstroni
ib., 24 (1936) 76.
182. Niirnberger, Arnow, J. Phjs. Chem. 38 (1934) 71.
183. Weizmann, Hirschberff, Bergmann, Nature 141 (1938) 1012;
JACS 60 (1938) 1530.
184. Hoist, Z. Phys. Chem. (A) 169 (1934) 1; 175 (1935) 117; 183 (1938)
423; Acta phys. Chim. URSS 6 0937) 137; Radiologica 2 (1938) 151
[Chem. ZbU 1939, II, 1833]; Hoist, Zur Photochemie d. reversiblen
Redoxprocesse, Diss. Lund. (193_>).
185. W*eiss, Naturw. 23 (1935) 610; Nature 136 (193!)) 794.
186 T u x e n. Z. Pbys. 103 (1936) 463; Nilsen, Bradbnry, Phys. Rev. 51
(1937) 69.
187. Weiss, Trans. Farad. Soc. 35 (1939) 48.
188. Schneider, Z. phys. Chem. (B) 23 (1935)311.
189. Living-ston, Hurd, J. Phys. Chem. 44 (1940) 865; 45 (1941) 547;
Livingston ib,, 46 (1942)233.
190. Шпоаьскнй, Колесникова, Ж. физ. хим. 5 (1<П4) 1197;
Шпольскнй, Ильина, Acta phys. chim. URSS 3 (1945) 269;
Шпольский, Шереметьев, ib., 5 (3936) 576; Гришку н,
Шп ольский, Ж. фиэ. хнм. 19(1945)97, 107; Гришкун, Гуретич,
там же (1946), Гришку и, Ракииова, там же (1946).
191. Weiss, Tra-s. Farad. Soc. 34 (19->8) s51.
192. Franck Levi, Z. phys. Chem. (B) 27 (1935) 409.
193. Blum, Photodynamic Action (19 1), см.2.
194. Gaffron, Ber 60 (1927) 75S, 2229; Bioch. Z. 264 (1933) 251.
195. Gros, Z. phys. Chem 37 (1901) 157.
196. Car о 11, J. phys. Chem. 30 (Ю26) 130.
197. Gorin, Chem. Abstr. 35 (1941) 2778.
198. West, Mull er, J e 11 et Proc. Roy. Soc 121 /1928) 234, 299, 313.
199. Rollers on, Stouahton, JACS 63 (1941) 1517.
200. Вавилов, Z. Physik 50 (1928) 52; Acta phys. polon. 5 (1936) 417;
Вавилов, Франк, Z. Physik 69 (1931) 100. См. также10.
201. Свешников, Тр. Гос. Опт. инст 12 (1948J вып. 108; Acta phys.
chim. URSS 3 (1935) 257; 4 (1936) 354; 7 (1937) 755; ДАН 3 (1936) Ы.
202. Банов, Z. Phys. 58 (19^9) 811; 64 (1930) 121; Z. phys. Chem. (A) 163
(1933; 172; ЖРФХО (физ.) 68 (1931) 465; Ghosh, Sen Gupta,
Z. ph,s. Chem. (B) 41 (1933) 117.
203. Лев тин, Ж. физ. хим. 6 (1935) 1; Z. Pbysik. 43 (1927) 260.
ЛИТЕРАТУРА 333
204. Stern, Volmer. Z. wiss. Phot. 19 (1920) 275.
205. Relief son, JACS 61 (1949) 2634; 62 (1940) 2264.
206. Umberger, La Mer, JACS 67 (19.5) 1099; Montroll. ib.t 14
(1946) .02.
207. West, Ann. New-York Acad. Sc. 41 (1941) 203.
208. Weissmann, J. Chem. Phys. ll) (1942) 214.
209. Свешников, Природа П941, № 1)\ Изш. АН СССР, физ., 9
П 945) 341.
210. Eisenbrandt, Z. Phys. Chcm. (В) 22 (1933) 145.
211. Kortum, Z. phys. Chen. (B) 40 (P33) ^34.
212. Weber Savic, Z. Phys. Chem. (B) 24 (1934) 68.
213. Baur, Helv. Chim. Acta 12 (1929) 793; Z. phys. Chem. (B) 16 (1932) 465.
214. Weber, Z. phys. Chem. (B) 19 (1932) 22; 30 (1935) 69; Z. Elektroch.
43 (1937) 633
215. Вавилов, ДАН 3 (1936) 271; Вавилов, Севченко, там же 3
(1936) J77; Севченко, Тр. ГОИ (1949).
216. F. P err in, CR 184 (19 7) 1121; Р n vaul t, ib., 184 (19J7) 1120;
J. Perrin. CR 184 (1927) 1097; Acta phys. polon. 5 (П36) 319.
217. F. Per r in, CR 178 (1924) 1978, /25-; 192 (1931) 1727; Ann. phys. 17
(1942) .83.
218. Вавилов, Z. Physik 31 (1925) 750; ЖЭТФ 13 (1943) 1; ДАН 35
(194 ) ПО; J. Phys. USSR 7 (1944) 1 L
219. Rollefson, Dodger, J. Chem. Phys. 12 (1914) 107.
2*0. Пекерман, ДАН 52 (19 6) 09,771
2Л. F. Perrin CR 192 (194) 17 7; Д у тине кий, ДАН 14 (1947) 73
222. Weiss, Weil-Malherbe.J. Chem. Soc (London) (19^4) 544.
22S. J. Perrin, Choucroun, CR 183(1926) 329, 357.
224. Levaillant, CR 177 (1924) 398; J. Perrin, Choucroun CR
178 (1924, 1401; 177 (1924) 612. 665
225. Freundlich Kapillarchemie, / (1ГЧ)); //(1942).
2„6. Davies, France, J. phys. Chrm. 40 (1946) 81, 177.
227. Фодиман, Каргин, Acta phys. chim URSS 1 (1934) 220.
228. Deutsch, Z. phys. Chem. (А), Ш (1928) ^54.
229. Fajans, Adsorption Indicators [Newer Methods ot Volumetric
Chemical Analysis (Bottger) (1938) 226J.
230. Weitz, Schmidt, Ber. 72 (1939) 1740, 2099; Z. Electrochem. 46
(1940) 222; 47 (1941) 65.
231. Kautsky, Roll. Z. 75 (1936) 164.
232. DeBoer, aha, Z. phys. chem (B), 13 (1942) 18; 16 (1932) 397, 403;
17 (1932) 161; 18 (1942) 49; 25 (1934) 225, 233, 408; Physica 3 (1946)
407; D e Boer, Electron Emission and Adsorption Phenomena, Cambr.
Univ. (1945) Ch. VII, § 64.
233. С. Натансои, Ж. фна. хим. 11 (1948) 157; Nature 140 (1937) 197.
244. Leermakers, Carroll, Staud, J. Chem. Phys. 5(1937)878,889.
235. SHeppard, Lambert, W-aIker, ibid., 9 (1941) 96.
236. С. Натансон, Ж. фиа. хим. 14 (1940) 989; Тр. Совещ. научн. фот.
ГОИ (194)
237. Соловьев, Труды Совещ. научн. фот. ГОИ (1941); Sheppard,
Lambert, Walker, J. Chem. Phys. 7 (1939, 265.
238. Schontag.Z. wiss. Phot. 39 (1940) 18, I Никифоров I,Тр. Совещ.
науч. фот. ГОИ (1941).
238а. Scheibe Schontag, Kopske, Henle. Z. wiss. Phot 38 (1939) 1;
Davey, Trans. Fared. Soc 36 (1940) 323; Гороховский, ДАН
39(1943)300; Левкоев, С. Натансон, Acta phys. chim. URSS
21 (1946) 437, С. Натансон, там же 21 (1946) 451.
334 ЛИТЕРАТУРА
239. Б о к и ник, Оптическая сенсибилизация фотографических слоев ()j
Багдасарьян, Уси, хим. 8 '19^9) 1617; А» Рабинович, Новые
работы по теории оптической сенсибилизации [Тр. Совещ. научо.
фот. ГОИ, 1941], М е е s, Theory of the Photographic Process (1944),
Part VI; Socher, Fortschntte der Photogfraphie, Bd. /// (1943),
Кар. III.
240. Meidinger, Fortschntte d. Photographic, ///(1943) Кзр. I.
241. Bodenstein, Naturw. 28 (1940) 14S; H i p p e 1, Z. Physik 93 (1934)
86; 101 (1936) 680.
242. Egbert, Biltz, Trans. Farad. Soc. 34 (1938) 892, Z. wUs. Phot. 39
(1940) 340.
243. Sheppard, Vanselow, J. Phys. Chem. 33 (1929) ?50; 34 (1930)
2719; А. Рабинович, Ж. фи», хим. 7 (1936) 276; Acta phys. chim.
URSS 3 (1935) 368; IX« Congres Intern. Phot. Scl. Appl. Paris (1938)
316; Fran с k. Teller, J. Chem. Phys. 6 (1938) 8Ы; Bo d e nst e in2*1.
244. Blair, Leigh ton, J. Opt. Soc. Am. 24 (1934) 185
245. Sheppard, Lambert, Walker, J. Chem. Phys. 7 (1939) 426.
246. Багдасарьянй39; Aeta phys. chim. URSS 9 (1938) 205.
247. Socher, Fortschritte d. Photographic, ///(1944) 168.
248. Leszinski, Z. wiss. Phot. 24 (1925) 261; Tollert, Z. phys. Chem.
(A) 140 (1929) 355; Б о к ин и к, Ильина, Ж. физ хим. 7 (1936) 237;
Acta phys. Chim. URSS 3 (1935) 383; Sheppard, Lambert,
Walker **&.
249. S e i t z, J о h n s о n, J. Appl. Phys. 8 (1937) 84, ]86,246;[Усп.физ.н. 23
(1940; 89, 293]} Mott, Gurney, Electronic Processes in Ionic
Crystals (1940).
250. Webb, J. Opt. Soc. Am. 23 (1933) 3167.
251. franck, Herzfeld, J. Phys. Chem. 41 (1934) 97.^
252. Evans, J. Opt. Soc. 32 (1942) 214; Бартенева, Гороховский,
Ж. xeiH. физ. 14 (1944) Ш
25S. Мейкляр, Степанов, ^АН 54(1946)799;Ж.физ.хнм. 20(1947)17.
254. Sheppard, Chem. Rev. 4 (1927) 340; Mecke, Semerano,
Z. wiss. Phot. 36 (1937)25: Gurney, Mott, Proc. Roy. Soc. 164
(1938) 151. [Уси. хим. 7 (1938) 1755].
254a. Гороховским, Крюков, Федотова, Ж. физ. хим 14 (1940)
180? С. На та не о и, Acta phys. chim. URSS 21 (1946) 430.
255. Kameyama, Kikuchl, J. Soc. Chem. Ind. (Japan) 40 (1937) 396r
К a m e у am a, Fukumoto, ib., 42 (1939) 244 B; Kameyama,
Mizuta, ib., 42 (1939) 6 В 489.
256. Sheppard, Vanselow, Happ, J. Phys. Chem. 44 (1940) 411;
Nature 145 (1940) 386.
257. Franck, Teller, J. Chem. Phys. 6 (1938) 861.
/58. Brooker, Keyes. JACS 58 (1936) 659. См. также*5.
259. Scheibe, Naturw. 25 (1937) 474.
260. Gr I tanner, Helv. Chim. Acta 24 (1941) 725.
261. Meidinger, Handb. wiss. Phot. V (\9%j) 258.
262. Steigrnann, Z. wiss. Phot. 30 (1942) 69.
263. E d e r, Liip p o-Cr a m e r, Sensibilisierung u. Desensibilisierung
(1932/ Ed^r's Hdb. Phot. Ill, Teil 3; Бокиник**», гл. V, § 11;
Mees'^9, Ch. XXIV, p. 1040; Seyeweh, Bull. Soc. Chim. 4 (1937)
1 [Уса. хич. 8 (1939) 546]; Sihvonen, Z. wUs. Phot. 25 (19277 1;
Eg-gert, Reitstotter, Z. vviss. Phot. 24(1927) 450; Kornf eld, J.
Phys Chem. 42 (1938) 795; Багдасарьян, Acta phys. cbm. URSS
19 (1944) 436.
263a. Bokinik, Jljina, Z. wiss. Phot. 31 (1932) 210; Ca rr о 1, Kr
etch man, Bur. Stand. J. Res. 10 (1933) 449.
ЛИТЕРАТУРА 435
264. Blau, Wambacher, Z. wiss. Phot. 33 (1934) 191; 34 (1935) 253.
265. Hiibl. Z. wiss. Phot. 24 (1927) 143.
266. Те ренин, Z. Physik 23 (19J^ 294; Тр. Гос. Опт. инст. 4 (1928) № 47.
Carrol, J. Phys Chem. 29(1925) 683; Blau. Phot. Korr 71(1935)21;
Weber, ib.. 71 (1935) 107; Мейкляр, ?K. физ. хим. 19 (1945) 441.
267. Winther, Z. wiss. Phof. 21 (1921) 45, 141, 168, 175; Baur, Perret,
Helv. Chim. Acta 7 (1924) 910; Perret, J. Chim. Phys. 23 (1926) 97
(обзор); Trans. Farad. Soc. 21 (1925) 6^7; Baur, Neuweiler, Helv.
Chim. Acta 10 (1°27) 901; Neuweiler, Z. wiss. Phot. 25 (1028) 187;
Arro [Chem. Abstr. 26 (19*2) 4255; Chem. Zbl. 1931 / 18831;
Краснове кий, Лисе. МХТИ Л941).
268. Good eve, Trans. Farad. Soc. 33 (1937) 340, Goodeve,
Kitchener, ibid., 34 09*8) 570, 902.
269. Gloor, Helv- Chim. Acta 20 (J937) 854.
270. Гачковский, Теренин, Изв. Ак. Наук СССР (1936) 805; Acta
phys. Chim. URSS 7 (1937) 521.
271. Hedvall, Borgfltrom, С о h n, Koil, Z. 94 (1941) 57; Hedvall,
Cohn. Nature, 143 (1939) 330; Hedvall, Nord, Arkiv Kemi,
Min. Geol. 17 А (Ш4, № 11), Hedvall, Wallffren, Manssen,
Trans. Farad. Soc. 36 (1940) 697.
272. Теренин, Ж. фиэ. хим. 6 (1935) 189; Курбатов, там же, 14
(1940) 1049; Тибнлов, там же 14 (1940) 9S3.
273. Френкель, ЖЭТФ 6 (1936) 647; Sow. Phys. 9 (1936) 158.
27l. Mosrlich, Schon, Naturwiss. 27 (1949).
275. Kautsky, al. Z. Electrochem 29 (1923) 308; Z. Physik 31 (1925) 60;
Trans. Farad. Soc. 21 (1926) 591.
276. Тереиин, Качур, Иав. Ак. Наук СССР, хим. (1945)271;
Курбатов, ДАН (1947).
277. Hug-hes, UchtelektrizlfcSt (1915), Кар. VIII; Handb. Radiologie, ///
(1916); Hall wachs, Die Lichtelelektrizitat. §4, 36; Hallwachs,
Ann. Physik 37 '1889) 666; Phys. Z. 5 (1904) 489; Столетов, Собр.
соч. 1 (1939) 217; ЖРФХО (физ.; 21 (1839) 159; CR 106 (1888; 1593;
Phys. Rev. (1892) 1; Knoblauch, Z. Phys. Chem. 29 (1899) 527;
Rhode, Ann. Physik 19 Г1906) 935; см. также: Лазарев, Родзе-
вич, ДАН (1929) 464; (19Ч>) 10S.
278. С ass el, Tohmfor, Z. Physik 90 (1934) 427.
279. HI иска, Z. Physik 81 (1933) 66, 521; 95 (1935) 486.
280. Предводителев, Н е ч а е в a, Z. Physik 29 (1924) 332; HI uc к а,
ib., 38 (1926) 589; 81 (1933) 76, 516; 84 (1933) 364; 89 (1934) 497; 103
(1935) 246; 104 1937) 654.
281. Hetrikiln, Z. Phys. Chem. (B) 10 (1930) 9; Vartanian, Tere-
nin, J. Phys. 4 (1941) 173; Вартанян, Ж. физ. хнм. 20 (19 6) 1065.
282. Pauli, Ann. Physik 40 (1913) 677- Volmer, ib. 40 V191S) 775; Byk,
Borck, Verh. Deutsch. Phys. Ges. 12 (1910) 621; см. тлкже^.
283. Visser, Rec. Trav. Chim. 47"(1928) 1037.
284. Huahes, DuBridg-e, Photoelectric Phenomena (1932) Ch. VIII, § 26
285. Щодро, Изв. Росс. Ак. Наук 6 (1919) 1 Л; Изв. Физ. Ин. 1 (1920).
571, 132; J. Chimie Phys. 26 (1929) 59, 117, 178; Soul an, CR 172
(1921) 581.
286. Ries, Das Licht (1909); Hughes, DuBri d ?e, Photoelectric
Phenomena (1942) Ch. IX; Cop eland, Black, Garrett, The Photovoltaic
Effect, Chem. Rev. 31 (1912) 177. См. также Веселовский, Ж.
физ. хим. 13 ^939) 1544; 20 (1946) 269, 1493; Ada phys. chim. URSS
14 (1941) 483; 21 (19 6) 803.
287. Riffollot, J. phys. 6(1897) 520; Camichel, ib., 4 (1905) 873;
Cunningham, Proc. Camb. Phil. Soc. 11(1902); Regner, Phys.
336 ЛИТЕРАТУРА
Z. 4 (1903) 862; Stoletow, CR, 106 (1888)1149, 1593; Nichols,
Merritt, Phys. Rev. 19 (1901) 18, 396; Goldmann, Ann. Phys. 27
(1908) <49; Hodge, Phys. Rev 26 (1908) 540; 28 (1909) -5;
Thompson, ihid., 5 (1915)47; Cope land, Black, Garrett2^, part. Ц.
288. Lifschitz, Hooghoudt, Z. Phys. Chem. 128 (19j7) 87; 133
(1928)43; 141(1929) э2; 146 (1930) 145; LIfschitz, Reffianni,
ib., 155 (1931) 431,
289. Grumbach, CR 176 (1923) 88; ib., 177 (1923) 395.
290. Grumbach, CR 177 (1928) 395; 180 (192э) 1102; Phil. Mag. 2 (1926)
313; Grumbach, Taboury, ibid., 1>3 (19 VL) 1178; 194 (1932; 84;
Mnrdock, Proc. Nat. Ac. U. S. 12 (19 6) 504; Trans. Farad. Soc. 23
(1927) 593; Rule, Phil. Mag. 1 (1926) 532; Proc. Nat Ac. U. S. 14
(1928) 272.
291. Jenkins, Phys. Rev. 18(1921) 402; Murdock, ibid.r 17 (1921) 626;
Lowry, Phys. Rev. 33 (1929) 1081; 35 (1930) 1270; Rao, J. Phys.
Chem 38 (19*4) 693.
292. Rule, Ph.l. Maff. 1 (1926) 532; Proc. Nat. Ac. U. S. 14 (1928) 272;
Russell, Phys. Rev. 32(1928) 667.
293. Grumbach, CR 184 (1927) 1169; Grumbach, Schlivitch,
ib., 189 (1929) 753; Schlivitch, ib., 190 (1930) 302.
294. Ghosh, Z. phys. Chem. (B) 3 (.929) 419.
295. Grumbach, CR 177 (1923) 395.
296. Rabinovit-h, J. Chem. Phys. 8 (1940) 551, 560.
.297. N<ra,CR 202 (19^6) 10 9 204 (1937) 763; J. Chim. phys. 32 (1935) 731,
298. Stora, CR 198 (194) 1763; 200 '1935) 552, 1034, 1191; 202 (1936)
48, 408, 1666, 2152; J. Chim. phys. 29 (1932) 168; 34 (1937) 536; Nga,
ibM 32(1935) 564, 72j.
299. Тартаковскнй, Внутренний фотоэффект в диэлектриках, ГТТИ
„, (1940); Волькенштейи Ф„ Электронные процессы в реальных
кристаллах, Уса. физ. н. 28 (1946) 389.
300. Clar, Aromatische Kohlenwasserstoffe (1941).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аггрегация красителей 90, 122, 123
Адсорбции красителей 265
Азобензол 104
Акридин 129, 144
Акридиновый оранжевый 51, 74, 125
Акридон 243, 262
Акцептор кислорода 153, 235, 242
„ электрона 209
„ выцветания 221
Аллнлтиомочевина 227, 242
Ализарин 24, 29, 268
Анилин 25, 62, 77, 81, 140, 204, 308
Аитиоксиданты, см. Ингибиторы
Антистоксова флуоресценция, см.
Правило Стокса
Антрахинон 178 222
Антрацен 47, 74, 81, 116, 129, 131, 134,
140, 144, 203, 245, 262, 306, 310, 311
Антрацевсульфоиовая кислота 243
Ауксохромиые группы 25, 41, 42, 52
Аурамин 24, 35, 51, 211, 235f 301, 323
Аутоохисление, см. Самоокисление
Аценафтеи 130, 134
Батохромныи эффект 11, 25, 47
Бенгальская рова 74, 226, 235
Беиэаурин 115
Бензол 30, 31, 41, 44, 47, 55, 62, 77,
81, 131, 250
Бензофенон 311
Беизофлавин 81
Бензпиреи 129
Бирадикальное (метастабнльиое, три-
плетное) состояние 78, 125, 127,
171, 209
Бриллиантовый зеленый 122, 299, 308
Валентные состояния атома 33, 34
Валентные (резонансные)структуры 31,
34, 49, 50, 53, 85, 187
Вероятность, см. Выход
22 А. Н. Теренин
Вибрации молекулы
„ энергия 54, 63, 75
типы 55, 62, 285
,, частоты 55, 63, 87
Водородная связь 86, 106, 158
Возбужденное состояние 17, 20, 43, 47,
57, 246, 255
Время жизни (длительность) 20, 71
83, 241, 244, 261
Вуалирующее действие 295
Выход (эффективность, вероятность),
см. также Квантовый выход
Выход процесса поглощении света 19,
83, 246
Выход элементарный 100, 116, 264
„ процесса испускания
(флуоресценции) 22, 72, 131
Выход тушения 257
„ сенсибилизация бромистого
серебра 278
Выцветание
„ деструктивное
окислительное 154
Выцветание кинетика 160
„ механизм 166
н адсорбированных молекул
299, 304
V
Галлоцианин 311
Гематопорфирин 235, 242
Гидрирование и дегидрирование 26,
107, 186, 213, 237, 241, 294
Гидраэнд амидофталевокислый 267
Гид рол Михлера 51
Гидроперекись бенэоила 143
Гидропероксиды, гидроиерекиси, см.
Перекиси
Гидрохинон 54, 86
Гипохромный эффект 11
риперхромиый „ 11, 25
Гипсохромный „ 11, 24, 51
338
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Двойная свявь 24, 29
Дезактивация 74, 83, 117, 132
Дегидрирование, см. Гидрирование
Долокалиэация электронов 30
Деполяризаторы (анодные, катодные)
215
Десенсибилизация 297
Диаэосоедивения 111
Диазотипия 111
Димеры 90, 116, 206, 260
Диполь, ем. Осцилляция заряда
Дииольный момент 34
Дисмутация (диспропорционированяе)
187, 221
Дифеннл 130, 134, 205
Дифеннлазот 205
Дифениламин 108, 203, 204, 208, 308,
311
Дифенилметан 203, 311
Дихронэм 52, 166
Закон
„ поглощения света Буге-Лам-
берта 12
„ поглощения света Бэра 12
„ Буязена-Роско 97, 164, 283
„ Вант-Гоффа 98, 160
„ Гротгуса-Дрэпера 97
„ Нернста 313
„ v Эйнштейна (квантовой эквя-
валеитности) 99
Зеленый Биндшедлера 24, 51
Зеркальная симметрия спектров 21,
59, 66
Зеркалъиосимметричные структуры 34
Зоны энергии, см. Энергетические
уровня
Изомеры красителей 88, 293
Иэохинолиновый красный 307
Ингибяторы 144, 153, 237, 258, 320;
см. также Торможение
Индиго 24, 35, 71, 77
Индвго кармии 235, 301
Иидякаторы кислотности 24, 27
Индулии 301
Интерференция стоячих волн, см.
Квантовая механика
Ион иода 226, 227, 233, 248, 257
Ион (молекулярный) кислорода 225
Ионы иеоргаиические 192, 209, 226,
243, 248, 304
Ионы серебра 193, 209, 271
Ионизация внутренняя 27, 33, 35
Ионизации красителей 25
Испускание света 19, 58, 65, 71, 78.
83
Кйрбинольное основание (карбинол,
лейко-карбииол) 26, И2, 115, 116
К»ант 17, 56
Квантовая механика 36
Квантовые числа 32, 64
Квантовые ячейки 33, 82, 137
Квантовый выход
„ „ флуоресценции 22f
74, 118, 122, 129, 131, 133, 227, 263
Квантовый выход фотореакцнй 100,
118, 131, 143, 166, 199, 207, 222,
224, 227, 242, 263, 305, 318
Квантовый выход фотолиза
бромистого серебра 276
Кинетика
„ поглощения фотонов 18, 83,
101
Кинетика процесса испускания 72
„ ф ото днме риза ции 116
„ тушения кислородом 132
„ выцветания 160
„ тутевня флуоресценции 245
„ концентрационного
торможения 263
Комплексы с ионами металлов 159
Коэфициент
„ поглощения молекулярный
13
Ковфициент поглощения молврный 14
„ погашения (экетинкцни) 14
Красители
„ азиновзде 24, 213, 216
азо 211, 223, 235, 301,30$
„ акридиновые 24, 179
„ ализариновые 24, 157, 158
„ анионные 26, 214
„ антрахиноновые 24, 158,
179, 211, 216, 301
Красители Вюрстера 53
я днфенилметановые 24, 114,
235, 309, 320
Красители индаминовые 24
„ индоанилиновые 24
н индофеноловые 24, 216
„ индантреновые 24, 28, 158
„ икдигоидные 24, 28, 157,
179
Красители карбоцианияовые, циани-
новые 24, 32, 50, 52, 88, 211, 270,
273, 309, 311
Красители катионные 24
„ кислотные 26, 214
„ ксантеновые (фталеиновые)
24, 120, 217, 235, 301, 304, 309
Красители кубовые 28, 157, 158, 175,
177, 211, 216, 301
Красители ледяные 28, 157
ПРЕДМЕТНЫЙ УКАЗАТЕЛ Ь
339
Красители иитро 24, 222
„ нитрозо 24
„ нафтохиноновые 24
„ оксазнновые 24, 301, 317,
321
Красителя основные 26, 125, 157, 179,
214
Красители полициклокетонные 24
„ порфярнны 24
„ протравные 28, 157
и прямые 28, 174
я сернистые 157, 176, 179
„ солеобразные 25
„ структурные смелеты 23
,, субстантивные 28,157, 175,
176
Красители тиазиновые 24,301, 317,321
„ трнфенилметановые24, ИЗ,
115, 121, 157, 211, 235,301, 309, 320,
323
Красители флуоресцемноиые 179, 320,
323
Красителя фталоцианиновые 24
я хинолнновые 24
Кристаллический фиолетовый 52, 81,
91, 125, 213, 309
Лабильный атом водорода 183, 213,
222, 234, 254
Лабильный перокснд, ом. Перекиси
Лейкосульфнты 112
Лейко-форма (лейкокрасителн) 26, 28,
112, 186, 224
Лейко-форма дегидрирование 156
„ фото реакции 177, 206,
211, 236, 299, 318
Лейко-форма малахитового Беленого
135
Лейкоцианнд 112
Малахитовый зеленый 24, 51, 122, 135,
200, 217, 235, 299, 301, 308
Меэомерия, см. Резонанс
Мерохиноидные соединения 86
Мероцианин 88
Метастабильное состояние или уровень
Бяергнн, см. Бирадикальное
состояние
Метиленовый голубой 24, 71, 74, 91.
109, 211, 213, 217, 220, 222, 223,
224,226, 235, 263 266, 295, 298, 301,
302, 312, 317, 320, 321, 322
Метиловый фиолетовый 209, 213,
308
Миграция электрона 218
Мольокснд 147, 150; см. также
Перекиси
Нафталин 47, 81, 109, 130, 134, 140,
248, 250, 306
Нафталиновый зеленый 235
п красный 125
Нафтацен 47, 145
Нафтнламин 77, 204, 311
Нафтмонат натрия 251, 262
Нафтол, 243, 301 311
Нафтосульфоновая кислота 243
Нейтральный красный 74, 226
Ннтроааилин 85
Нитробеивалъдегнд 105
Нитробенэойная кислота 105
Нильский голубой 301, 321, 322
Нитрофенолы 268
Облако электрона 36
Одиночная или простая связь 29, 36
Окисление-восстановление, как
перенос электрона 179
Окисление-восстановление,как
элементарная реакция 183
Окисление-восстановление при
фотолизе бромистого серебра 288—292
Окислительно-восстановительный
потенциал, см. Редокс-потенцнал
Окисление фотосенсибиливованиое,
см. Фотосенсибнлиаованиое
окисление
Оксазнн 205, 213, 216
Оксидирование (присоединение
кислорода), см. Фотооксидированне
Оксоннн 91, 187
Ортохром 277
Основное (нормальное) состояние 17»
19, 43, 46
Основание хниоидное 26
„ карбинольное 26
Осцилляция заряда 49, 70, 84
„ структур 42, 50
Отражение света 9
Патентованный голубой 308
Парафенилен-днамнн 204
Перекиси или пероксяды 107, 142, 154
163, 239
Перенос электрона, см. Окисление-
восстановление
Перилен 262
Пер оке иды, см. Перекиси
Пикриновая кислота 24, 27, 222, 2q5
Пинавердол 311
Пивакриптол 262, 298, 301
Пинафлавон 213
Пинахром 71, 213, 298, 311
Пнрон 129, 262
Пироннн 277
340
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Повышение цвета, си. Гиспохромный
эффект
Полицнклические углеводороды, см.
Углеводороды вроматическяе
Поглощение света 9—13. 42, 5$, 57—
62, 64—71, в4—94
Полоса испускания, см. Флуоресценция
Полоса поглощения, см. Поглощение
света
Полупроводники 218, 302, 303, 306
Полухиноны, сы. Семихиноиы
Полярнзоваияого света поглощение,
см. Дихроизм
Порфирины 74, 135, 227
Поел свечение, см. Фосфоресценция
Потенциальные кривые 63
Правило Вигнера 82
„ Кувдта 85
Стокса 20, 59, 61
Предиссоциагдии, см. Фотораспад
Принцип Паулн (принцип исключения)
31
Прнжцип Франка-Кондона 62, 67, 69,
75, 87
Пропускание 14
Прочность связи, см. Энергия связи
Прямой голубой 301
Псевдоизоцианины 93
Псевдопурпурии 301
Пурпур зрительный (родопсин) 101
Радикалы свободные 38, ЮЗ, 105, 115
Редокс-потенциал 188, 208, 216. 253,
254, 312, 314
Резонанс (квантовомеханический). 31,
41, 44, 50
Резонансная энергия, сы. Энергия
резонанса
Резонансные структуры, см. Валентные
структуры
Резонансный перенос энергии 248,249.
2S9, 262, 285
t Резоруфин 125
V Роааиилин 122, 308
-i Родамин 71, 73, 74. 91, 120, 125, 126,
340, 209, 211, 213, 226> 243, 245,
270, 307, 317
Родулнны 81, 245
Рубрен 129, 131, 145
Самоокисление 107, 147
Самотушеняе, см. Тушение
концентрационное
Сафранин 209, 211, 226, 301
Светостойкость (светопрочность) вы-
красок 157
Связи <г и тс 30, 36, 40
Семихиноны 58. 185, 187, 302, 318,322
Сенсибилизованная флуоресценция 77,
84, 249, 284
Сенсибилизаторы выцветаиня 221
Сенсибилизаторы фотографические,
см. Фотосенсибилизация
бромистого серебра
Силв осциллятора 70, 92
Силы сцепления Ван-дер-Ваальса 91
Скрытое изображение 273, 277
Спектр поглощения (спектральные
кривые) 15, 21, 70, 79, 86, 87, 90—
92, 124, J24, 269, 271
Спектр (полоса) испускания 20, см.
также Флуоресценция,
Фосфоресценция
Спектр сродства, см. Сродство
Спектр фотографической сенсибнлива-
ции, см. Фотосенсибилнзация
бромистого серебра
Спектральная кривая фотоэффекта 309
Спин электрона 32, 81, 137
Сродство к электрону (энергия
сродства, спектр сродства) 182, 194, 197,
199, 202, 203; см. также Энергия
отрыва влехтрона. Энергия нони-
зацни
Статистические законы (поглощения,
испускания) 67—73
Стереоизомерия 104
Стнльбен 104
Структуры бензола (Кекуле и Дьюара)
31, 34, 41, 44, 48
Субстрат окисления 226, 232, 237, 240,
264
Сульфоанилат натрия 243
Суперпозиция валентных структур,
см. Кваитовая механика, Резонанс
Таутомерия 89, см. Фототаутомерия
Температурный коэффициент, си.
также Энергия активации
Температурный коэффициент
выцветаиня 164
Температурный коэффициент распада
(темнового) бролшетого серебра 275
Температурный коэффициент сеиси-
бнлнзации бромистого серебра 285
Тетралин 129
Тетрафенилгндразин 103
Тимолфталеин 268
Тиозинамии, см. Аллилтиомочевниа
Тиазииы 205, 213, 216
Тионндиго 301
Тионин 71, 91, 266, 317, 318
Титана двуокись 299, 301
Толуол и метилированные бензолы 134
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
341
Торможение фотореакций 258, см.
также Ингибиторы, Антиоксиданты
Торможение кислородом 127, 157, 209,
225
Торможение концентрационное 263
Торможение выцветания 1Ь8, 304
Трппэфлавин 81, 135, 136, 138, 213
Триплетное состояние, см. Бирадикаль-
ное состояние
Трифениламин 204, 308
Трифенилметан 115, 130, 134, 203
Тушение флуорееценции 76, 77, 124,
227, 241, 243—258
Тушение флуоресценция кислородом
128—141
Тушение флуоресценции
концентрационное 119, 146, 259—265
Тушители 243, 250—252
Углеводороды ароматические 47, 144
Углубление цвета, см. Батохромный
эффект
Уранила ион 226, 243
Урании, см. Флуоресцеии-натрин
Уровни электрона (при окислении-
восстановление) 181, 190
Феиантрен 48, 134, 144
Феносафраиин 24, 71, 74, 209, 213,226,
235, 297, 299, 301
Фенолфталеин 115, 267
Флуорее 134
Флуоренон 251
Флоксин 125, 270
Флуоресценн-натрия (ураиии) 22, 24,
71, 73, 74, «1, 120, 121, 124, 125,
126, 135, 140, 209, 211, 226, 236,243,
245 254 257
Флуоресценция 20, 58, 73, 83, 88, 117,
135; см. также Тушение
флуоресценции
Флуоресценция фотопродуктов 123,
125
Флуоресценция замедленная 78
Фосфоресценция 77, 81, 83, 135, 139
Фотовосстановлеиие 109, ПО, 121, 122,
211, 216—225, см. также
фотогидрирование
Фотоокнсление (фотодегидрирование)
109, 110, 223
Фотоокисление элементарное 108, 204
Фотоокислеиие акцепторами электрона
208
Фотогальваиический эффект 315, 319
Фотовольтанчесхий эффект 319
Фотогидратация (фотогидролив) 110,
Фотогидрированне 109, 110, 121, 126;
см. также Фотовосстановлеиие
Фотографическая сенсибилизация, см.
Фотосенсибилизация бромистого
серебра
Фотодимеризации, см.
Фотоприсоединение
Фотодинамический эффект 208, 211,
213, 235
Фотоивомеривацня 104, 110
Фотоионияация, см. Фотораспад на
ионы
Фотоконденсацин, см. Фотоприсоеди-
ненне
Фотолиз, см. Фотораспад
Фотолиз молекулы воды 200, 23Q
„ бромистого серебра 273
„ бромистого серебра сенсиби-
лиэоваиный, см. Фотосенснбилнза-
цня бромистого серебра
Фотолив на ионы, см. Фотораспад
Фотолюминесценция, см.
Флуоресценция
Фотон 17
Фотоокисление красителей нонами
в растворах 209
Фотоокислеаие элементарное
Фотоокислеиие неорганических иоиов
192
Фотоокмслеиие органических
соединений /03
Фотооксидирование 107—110, 124,
12з 179
Фотоохснды 130, 134, 139, 144
Фотоперенос электрона 108, 110
Фотопроводимость 280, 309, 310
Фотоприсоединение 106, 110 116, 123
Фотореакция с кислородом, см.
Фотооксидирование
Фотораспад 75, 102, 110—112
Фотораспад на ионы 104, ПО, 112
Фотосеасибилнзация 109, ПО
„ переноса
кислорода 153
Фотосенсибилизация бромистого
серебра 109, 269—294
Фотосенсибилиэация реакции
восстановления 215
Фотосенсибилизация окисления
неорганических ионов 225
Фотосенсибилиэация красителем
разрушении волокна 174
Фотосинтез 6, 109
Фотосорбция 304
Фототаутомеризация 105, Ц0
Фоточувствительность 101
Фотоэффект внешний 303
342
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Фотоэффект внутренний, см.
Фотопроводимость
Фталоцнанин 36, 39, 40
Фуксин 71, 308
Фульвен 47
Хемилюминесцевция (оксилюминес-
ценция) 307
Хинин сернокислый(хинмясульфат) 129,
151, 211, 245,250, 251, 254
Хиноидные структуры 35, 42
Хииолиновый голубой 308
Хиион-диазиды 86, 213, 222
Хлоразол голубой 305
Хлорофилл 71, 74, 129, 135, 210, 213,
227, 235, 242, 243, 263
Хриэен 129
Хризоиднн 301, 317
Хромоген 26
Хромотроп 209
Хромофорные группы 42
Цвет дополнительный 9
[ветность 9
[ветностн теория 42
[ерулеин 301
Цианян 71, 90, 211, 311
Циба 301 _
Цинка окись (как фотосеясябиАиаатор)
214, 300
Частота колебаний света 11
„ вибраций 55
„ стоячей волны в кваитово й
механике 37—44
Частота осцилляции наряда 49
Эйнштейн (единица) 100
Экситон 306
Электромерные структуры 31
Электроны ff и я 21, 30, 37, 40
Электроннаи оболочка 36
Энергии активация (термическая) 275,
285, 286, 292, 310; см. также
Температурный коэффициент
Энергия
гидратации 193—198, 201
„ ионизации 182
„ отрыва электрона 193, 197,
199
Энергия резонанса 40, 46
связи 40, 103
электронная (возбуждения)
44, 54, 57, 75; см. также
Энергетические уровня
Энергетические уровни 17, 45. 49, 56,
151, 181, 218, 256, 280, 290, 296
Энергетические уровни кристалла
бромистого серебра 280
Эозин (тетраиод-ураиин) 71, 73, 74,
90, 120, 121, U% 123, 124, 125, 229,
140, 209, 211, 213, 226, 235, 236,
243, 257, 270, 308
Эритрозин (тетрабром-уранин) 71, 74,
121, 122, 124, 125. 211, 212, 213,
235, 236, 266, 270, 277
Эскулин 62, 211, 251, 254
Этил фиолетовый 213
Этилхлорофиллид 74, 129; см. также
Хлорофилл
Эффект Гершелн 298
Эффективность, см. Выход
УКАЗАТЕЛЬ АВТОРОВ
Цифры обозначают номера страниц, где упоминается работа данного
автора; цифры петитом, вынесенные наверх, укаэывают номер в указателе
литературы, где находится упоминаемая работа. Тире, поставленные после
или перед запятой, обозначают фамилию автора, стоящую вначале.
Адрианов, см. Вартанян, —
Армстронг 42
Арндт 42
Вагдасарьян 269, 273,274, 275, 28423В;
277, 278?48; 297 283
Байер 42
Баиов 244, 261 а02
Бартенева, см. Гороховский, —
Баур, см. Ваиг
Бах 1ч810в; 238, 170137
Беккерель 315, 319
Беленький 156, 165, 167120
Блинов» см. Предводителев,—
Бокииик 269, 273, 274, 275, 28428в;
297 «»
—, Ильина 3. 278248; 29728ба
Борисов 78 36
Боуен, см. Bowen
Буге 12, 16
Бунзен, Роско 97, 127, 283
Бутков 2011в4
Бэр 12, 13, 14, 16
Вавилов 124; 17е; 22 в; 23"; 601Оа? 161,
24 *, 246, 247 *>°; 257 21*; 259, 262318
248 :
—, Левшин 78, 7939
— , Prin"sheiin78, 79 зв
—, Севченко 257 215
—, Фганк И. 247 200
— . Шишловский 81 43
Валько 1741з8
Ван-дер-Ваальс, см. Силы В. д. В.
в предм. указателе.
Ваит-Гофф 98, 99, 100, 101, 127
Варбург 238
Вартанян 22, 62, 75, 76, 102»; 128м;
158, 164, 1671аэ; 290, 292, 309,
ЗЮ281
— f Адрианов 158, 164, 167123
Вассерман, см. Чердынцов, —
Вебер, см. Weber
Вейдеибах 19916вО<
Вейгерт, см. Weigert
Вейсс, см. Weiss
Веселовский 315 286
Вигнер 140
Видеман, Шмидт 79
Виланд, см. Wieland
Винокуров, см. Левшин,—
Вицингер, см. Wlzingfer
Волькенштейн М. 44, 5216
Волькенштейн Ф. 2802ВВ
Ворожцов 155, 222 »•
Вюрмэер (Wurmser) 169, 189, 192,
238137, 153
Гамбурцеа 161, 163, 172125
Гантш (Hantsch} 42
Гач^овский, Теренин 303 2?0
Гебгард, см. Gebhard
Гершель 97, 298
Гороховский 270, 27223sa
— , Бартенева 285 252
—, Крюков, Федотова 271, 288264R
Гребэ 42
Гришкун, Гуренич 226, 2281в0
— , Ракинова 226, 228180
—, Шпольский 226, 2281В0
Грос, см. Gros
Гротгус (Grothus) 97, 98
Гудив, см. Goodeve
Гудийр, см. Goodyear
344
УКАЗАТЕЛЬ АВТОРОВ
Гуревнч, СМ. Грншкун,—
Гэри , см. Gnrney
Дайн 199, 200, 2021Б9
Де Бур, см. De Boer
Де-Шалыт, Ломаиова 155117
Дильтей (Dilthey) 42
Докувнхин Левин 35,llft
Дурмашкниа, см. Левкоев,—
Душинский 262 221
Дрэпер 97, 98
Дюфресс, см. Dufraisse
Дяткина, см. Сыркин, —
Дьюар 31
Жуховицкин, см. Сыркив, —
Зискчн, см. Кондратьев, —
Измаильский 25, 42, 51 16
Ильина А., см. Шпольский,—
Ильина 3., см. Боки ни к,—
Казарновский 182, 203. 225
Каргии, Фодиман 266 227
Касаточкин 182. 203, 22516°
Каутский, см. Kautsky
Качур, см. Теренин, —
Кекуле 31, 34, 40, 41
Кёниг (фп*8г) 42
Климова, см. Прилежаева,—
Колесникова, см. Ш польский,—
Кондон, см. Принцип Франка-Кондоиа
Кондратьев, Зискин 128, 135, 162,
307 68
Кравец 10, 15, 16»; 71"
Красновский 6, 1342; 141 В7; 299, 301,
Кундт 85
Курбатов 304 272; 307 276
Лазарев б1; 98, 160, 164, 166, 172,
21762
—, Родэевич 303277
—, Недопекин 10, 15, 163
Ламберт 12. 13, 16
Левин, см. Докунихнн, —
Левкоев, Петрпв, Дурмашкииа 1127*
—, Натансон С 27Oa3Sft
Левшни 21,164. 257, 2587; 71 28; 7433;
122"; 244,260, 261303
—, Винокуров 78, 79 зв
—, Повунова 78, 79 зв
Либерзон см. Дайн, —
Лнберман 42
Ломанева, см. Де-Шалыт,—
Льюис, см. Lewis
Маркузе 174139
Мейкляр, Степанов 285 263
Менделеев 32
Натансон Г. 120, 122, 123, 124, 163,
167, 171, 258 7в, 134
Натансон С. 270233.23Sa; 291236; 271,
288, 2961М« см. Левкоев,—
Некрасов 189, 192 153
Непореит 75, 76, 77, 102, 140, 258 2аг
1288в? см. также Тереиин, Варта-
нян, —
Нерист 313
Нчкифоров 272 238
Нецкий (Nietzki) 42
Нечаева, см. Предводнтелев, —
ОглобАни 154114
Пакшвер, см. Шарвин,—
Паулн 31
Пекерман 261 220
Перрен, см. Perrin
Петров, см. Левкоев, —
Плаик 17
Порай-Кошип 42
Предводителев, Блинов 166132
—, Нечаева 166 132; 309 ™*
Прилежаева 62, 77, 78, 28423; 76^
—, Климова 77 з7
Рабинович А. 269, 273ззв; 275243
Рабинович £., см. Rabinowitch
Ракниова, см. Гриш кун, —
Робиисон 30
Родзевич, см. Лазарев,—
Роско, см. Бунзен,—
Рыскина 128 е6
Савостьянова 71
Садов 17413*
Саундерс 11173
Свешников 245, 247, 248 *«; 24920»
Севченко, см, Вавилов, —
Серков 204167
Склар4416
Сколфилд, см. Scholefield
Соловьев 89, 9267; 271 237
Степанов, см. Мейкляр, —
Стоке 20
Столетов 308 277; 315 287
Сыркин 48
—, Дяткина 31, 32, 34, 41"; 151112
■—, —, Жуховнцкий 31, 411Х
Тартаковский 280 29в
Тереннн 228; 75, 76, 79, 81, 82, 106,
УКАЗАТЕЛЬ АВТОРОВ
345*
107, 137, 140, 158, 310 24; 7838;
105вБ; 107, 30770; 1137ба; 170117;
192, 195, 200, 2021В4; 275, 299 266;
304272; см. также Гачковский,—
Теренни, Вартанян, Непореит 22, 62#
75, 76, 102, НО. 25821
—, Качу о 307 278
Тибилов 304 272
Траубе 169
Тумерман 6220; 73, 244 31
Уолл, см. Хюбль, Гребе, —
Феофилов 521в; 75, 76 Зб; 10566
Фирц 42
Фодиман, см. Каргив,—
Франк И., см. Вавилов,—■
Франк, см. Franck J.
Френкель 306 273
Хюбль, Гребе, Уолл 2211в0
Чердынцев, Вассерман 81 42
Шар вин, Пакшвер 155117
—, Шатрова 155, 222U8
Шатрова, см. Шарвин, —
Швецов 164128
Шереметьев, см. Шпольский,—
Шишловский 12886. См. также
Вавилов,—
Шмндт, ей. Вндеман,—
Шпольский, см. Гришкун,—
—, Ильина А. 2 6. z28180
—, Колесникова 226, 228m
—, Шереметьев 140"; ^26, 228 1В0
Штаудингер 147
Штерн 246
Щодро 290; 311 2S5
Эбвен, см. АЬпеу
Эйнштейн 99
Эвстерт 42
Увглер 147, 170, 238
Яблонский, см. Jablonski
Abney 275 ■"
Abney, Rugsel 154ll3
Adams, Roaenstein 90 и
Alberts 165 m
Appleton, Geake 216 "*
Ariga. 115 7б
Avndt 42; 35Uft
Amow, см. Nurnbergfer,—
Arro 299287
Asterblum 120» 12578
Audnbert, Racz 139, 1529B
Auschkap 71 25ft
Backstrom, Beatty 14398
Badoche 144103, см. Dufraisse,—
Balodis, см. Petrikalis, —
Banckroft 299m
Baumann, см. Miller, —
Baur 214, 252, 289 *74; 252, 254, 257 «3
—, Perret 176143; 299, 301 a"
— , Neuweiler 299, 301 *«7
Beatty, см. Backstrom, —
>Benrath, Meyer 17814e
Bergmann, en* Weizmann,—
JBeyer 167, 168, 171IM
BijfeJeisen, см. Lewis, —
Biltz, см. Eggfert, —
Black, см. Copeland,—, Garret
Blair, Leig-hton 275244
Blau 299 266
—, Wambacher 298 2в4
Blnm 6B; 234, 2471вз
—, Speaiman 122, 167 м
Bodenitein 144"; 148loe; 157 *«; 166,
167, 1701S4; 274, 282241, 275243
Borck, см* Byk,—
Borgrstrom, см. Hedvall, —
Bowen 116 78; 131 »»
—, Norton 74 84; 116 78; 128, 129, 134 8«
— , Steadman 14510Б
— , Williams 128,129, 134 e6
Bradbury, см. Nilsen,—
Brandt, см. Euler, —
jBray 200"»
Brijrham, см. Sheppard, —
Brooker, Keyes 293 f6S
Brunius, см. Euler, —
Burg-herr 209 m
Bury 44 52ie
Butler 207 17°
Byk, Borck 309, 311 262
Cale, см. McLennan,—
Calvin, ем. Lewis, •—
Camichel 315 aS7
CaroiJ 2371B0; 299 »e; см. также Leer-
makers, —, Staud
—, Kretchman 2972e3a
Casael, Tobmfor 308 a?e
ChecKan 12У 86a
Chevreuil 154113
Choucroun, см. Perrin ].t —■
Chovin, см. Dufraisse, —
Ciamiclan 141 B7
Clar 144 30°
CJibbens 216 "5
Coha» см. Hedvall,—
Condon, см. Franck, —
Cook, Martin, Roe 145104
Copeland, Black, Garret 315, 31928e;
315 887
Cunliffe 175 uo
Cunniffham 315 2e7
Curtis 155117
Davies, France 266 22<J
Dean, см. Mouren, —
De Boer 267, 268. 269 *32
Deutsch26722e
Dickson 78"; 89, 92, 272"
Dodger, см, Rollefson,—
Du Bridge, см. Hugfhes, —
Dufraisse 147, 152loe; см. таило Mou-
reu,—
—, Chovin 1451OS; 152108
— , Badoche 14510«
—, Gerard 144, 145 10°
— f Horclois 144, 145103
УКАЗАТЕЛЬ АВТОРОВ
347
Houpillart 144, 145102
Le Bras, Vellnz, Velluz 144, 145 101
Le Bras 145 106
Loury 145 30B; 145106
Momer 145 1W
Priou 144, 145 101
Veluz 144, 145 10°; 145103
Duhrkop, см. Kuhn, —
Eckert 141 в7( см. также Meyer,—
Eder 11173; 297263
— , Livpo-Cramer 297 263
Egbert, Biltz 274. 276, 277 2«
— , Reitstotter 297 283
—, Shroter 111"
Eisenbrandt 249, 250, 251 254210
Eistert 42 15
Ellis 175 140
Ellis, Wells 61; 1127*
Epstein, см. Rabinowitch,—
Euler, Brunnina 12868
—, Helistrom, Brandt 222161
Evans 285 262
Farkas A., Farkas L. 195, 196, 197,
198, 199, 201, 208l67
Farkas A.. Wansbrou?h Jones 202 165a
Fajans 267, 270, 271 22e
Feldmann 11173
Fisher 782S
Fisbgfold. см. Weiss, —
Fonda 165, 166130
Formanek 10, 15, 16 3
— , Knop 10, 15, 16 8
F6rster44, 52, 8816
France, см. Davies, —
Franck JM Condon 62, 75
—, Haber 201)16Я
—, Herzfeld z84251
—, Lev 138, 235, 24184; 230, 255192
—, Livingston 106, 138, 210, 220, 234
238, 2*1 6e
—, Pringsheim 106, 138, 210, 220, 234,
23ee
— , Scheibe 192, 194, ЛЮ1"
—, Teller 275, 291 2"
Frankenburg-er, см, Weyde,—
Freundlich 26b 226
Friedman 221ieo
Fukumoto см Kame^ama, —
Gaffron 137, 15392; 145, 146105; 236,
240,241,242,264,265 194
Ganguli, см. ShuLtz, —
Garrett, см. Copeland, Black, —
Gaviola 73, 244 3l
Geake, см« Apploton, —
— , Lemon 21617«
Gebhard 122, 154, 158»«; 170l36; 167
168, 171, 172, 173125
Gerard, см. Dufraisse, —
Germann, Le Roy, Gibson 115 77
Ghosh 244 202; 317 2M
Gibson, см. Germann, Le Roy, —
Gilb, Stonehill 35, 158l24
Girbes, см. Lifschitz, —
Gioor 300 2&g
Goldmann 3152e7
Goldschmidt, см. Lewis,—
Goldstein 78 3S
Goloub 78, 79 39
Goodeve 101 63; 300, 304 8se
— , Kitchener 300, 304 268
Goodyear, см. S^holefield, —
Gorin 237187
Gottlinff 73, 24431
Granick, см. Michaelis, —
Griffith, McKeown 61
Gritanner 294, 297 260
Gros 156, 236, 2991Вб
Grumbach 3162SB; 316290; 316, 3172M;
317 285
317
—, Schlivitch 316.317293
—, Tab^nry 316 2eo
Gurney 181, 189, 19014e; 28024B
—, Mott 290254
Haber, см. Franck, —
— , Wansbrou^h Jones 20216fia
Haller, Wyszewiansky 176 142
— , Ziersch 154, 170116
Hallwachs 308 277
Happ, см. Sheppard, —
Harris, Kaminsky, Simard 11577
Harrison 175 140
Hartley 89 40
Hedvall, Borgstrom, Cohn 266, 304 271
—, Cohn 266, 30-*71
—, Nord, 266, 304 27i
—, Wallgren, Manssen 266, 304 271
Heffter 121so; 155114
Helistrom 222, 25416\ см. также Euler,
Brandt, —
Henk 179147
Henle, см. Scheibe, Schontagf, Kopske, —
Henrici 4918
Herzfeld, см. Franck, —
Hibbert 141 ft7
Hippel 274241
H rschbergf, см, Weizmenn, —
Hlucka 30827fl; 309 260
Hodge 315 267
Holmes 89 62; 115", 155 "7
— , Peterson 89, 92, 272 B7
348
указатель авторов
— , Scanlan 89 **
Hoist 223184
Hoodhoudt, см. Lificbitz, —
Horclojs, см. Dufraisce, —
HoupiLlart, см. Dufraisse, —
Hiibl 298265
Hiickel 3913
Hudson, см. Olson, —
Hughes 308277; 204166
—, Dn Brudge 311 2e4; 315, 319286
Infold 30, 42, 4416
lwamoto 155119
Jablonski 79 40
Jelley 93 68
Jenkins 316, 3172»1
Jette, см. West, —
Joffe, см. Lifschitz, —
Joffre 154 1U
John 141 "
Jones 179 U7
Kameyama, Fukumoto 290 255
—, Kikuchi 290 255
— , Mizuta 290 266
Kaminsky, см. Harris, —
Kasha, см.^-ewis, —
Katheder 93 6\ см. Scheibe, —
Kautsky 78, 79 s9-, 128, 135 s4-, 128, 135.
30767; 135,148,242B1; 145105; 26723a;
307 276
— , Merkel 146, 242107
— , MuHer 146 1Q7
Kayser 20 s
Kearney 120, 126 7e
Keyes, см. Brooker, —
Kikuchi, см. Kazneyama, —
Kilchling, см. Konigsberger, —
Kistiakowsky 6 г
Kitchener, см. Goodeve, —
Knoblauch 308 277
Knop, см. Formanek, —
Koblite, Schumacher 145 l05
Kogel 106 88; 11173; 155 m
— t Steigmann 11173; 213,229, 241, 294,
295173
Konigsberger, Kilchling 71 2S
—, Kupferer 71 27
Kopske, см Scheibe, Schontag, Henle, —
Kornfeld 297 263
Kortum 90, 925i; 249 *u
Kowalski 78aft
Kretchman, см. Car oil, —
Kuchler, Pstat 105, 106 67
Kuhn, Duhrkop. Martin 52le
Kunz 179147
Kupferer, см. Konijrsberger, —
Lambtrt, см. Sheppard, —, Walker
La Мег, см. Umberger, —
Landolt 176144
Le Bras, см. Dufraisse, —
Leermakers, Caroll, Staud 270, 27223*
Leiber 165 m
Leighton, см. Blair, —
—, Lucy 105, 10667
Lemin, см. Vickerstoff, —
Lemon, см. Geake, —
Le Roy, см. Germann, —
Leszinski 278 2i*
Levaillant 263 224
Levi, см. Franck, —
Lewis 49r 50, 52, 85, 86, 89, 9017
—, Bigeleisen 52"; 103 е4; 204, 108";
187 161
—, Calvin 49, 50, 52, 85, 86, 89, 9017;
81 44
—, Goldshmidt, Magfel, Bigeleisen 91 55
—, Kasha SI, 128"
—, Lipkin 103, 11464; 108, 204, 205,
206, 20771- 16e
— , —, Magel 79, 81«; 88 47; 89 4»; 86,
89, 125 60; 89 52
— , Maffel, Lipkin 10S в6
Ley 10, 15. 163
Lifschitz 113, 115, 116, 122 ™
—, Girbes m, 115, 11675
—, Hooghoudt 315, 3172S8
—, Joffe 113, 115, 116 75
— , Regianni 315, 317 28S
Lipkin, см. Lewis, —
Livingston 221, 226, 227, 233, 237l8e;
см. также Franck, —
—, Hurd 74 32; 226, 227, 231188
Lonsdale 39 14
Lotte, см. Moureu, —
Loury, см. Dufraisse, —
Lowry 316, 317 281
Lucy, см. Leighton, —
Luppo-Cramer, см. Eder, —
Magel, см. Lewis, —
Manssen, см. Hedvall, —
Marsh 78 3e
Martin, см. Cook, —, Roe
Martin, см. Kuhn, Diihrkop, —
Mauney, см. Olson, —
McKeown, см. Griffith, —
McLennan, Cale 120, 126 79
Mecke, Semerano 288 264
Mees 269, 273,274. 275, 284238; 297 2«
Meidinger 274, 280 24°; 295 261
УКАЗАТЕЛЬ АВТОРОВ
349
Merkel, см. Kautsky, —
Merrit, см. Nichols, —
Meyer, см. Benrafch, —
— , Eckert 178 14e
Michaelis 71 2B; 189, 1921БЗ
— , Granick 92 Бв. 152
—, Schubert 179, lS7t 204, 31314e
—, —, Granick 179, 187, 313148
Miller, Baumann 1.8 8б; 129 eo
Mizuta, см. Kameyama, —
Moglich, Schon 306 274
Momer, см. Dufraisse, —
Mott, Gurney 280 24e
Mounier 155 117, см. также Seyewetz,—
Moureu, Dufraisse, Dean, Lotte, Butler
14510Б
Montroll 247 20G
Mudrovcic 221180
Muller, Miiller-Rodloff 145 10Б
Miiller, см. Kautsky, —
Muller, см. "West, — , Jette
Mulliken 104, 105 65
Murdock 316 2B0; 316, 317 2ei
Narayanan, см, Sudborougfh, —
Neuweiler 211, 212, Л4, 300, 301172;
299, 301 267, см. также Baur, —
Nga 319 2B7; 319, 321208
Nichols, Merritt 315287
Niisen, Bradbury 225 15°. 186
Nord, см. Hedvall, —
Norton, см. Bowen, —
Nurnberger, Arnow 222182
Olson 104, 105 e5
—, Hudgon 104, 105 G5
— , Manney 104, 105 6б
Patat, см. Kuchler, —
Patel, см. Scholefield, —
Pauli 309, 311 lee. 282
Pauling: 31,32, 34, 41 "• 44, 52"; 19415e
Perret 299, 301 2в7, см, Baur, —
Perrin F. 2S921*; 259, 260217; 262221
Perrin J. 120 7e; J59216
— , Choucroun 263 223; 263 224
Peterson, см. Holmes, —
Petrikaln 309, 310 281
Petrikalis, Balodis 145105
Porret, см. Weiss, —
—, Rabinowitch Л0, 219176
Potteril, Walker, Weiss 199160
Priou, см, Dufraisse, —
Privau t 253, 259 216
Prinssheim 20<>; 120, 123, 127 78f см.
Franck, —, см. Вавилов, —,
Voxels 128, 307 87
Prlns 71 2ea
Rabinowitch E. 6f 107,134 2; 71 3e; 318 20e;
см. также Porret, —
— , Epstein 91ББ; 196, 20Э168
— , Weiss 210, 21917e; 219177
Racz, см. Audubert, —
Ramsper^er 111 73
Rao 316, 317 2ei
Regianni cm. Lifschitz, —
Reguer 315 287
Rein 165 12e
Reitstotter, см. Egbert, —
Rhode 308 377
Rieche 149 uo
Ries 315, 319 28e
Rigollot 315287
Robertson 36, 4012
Roe, см. Cook, Martm, —
Rolbfson 243, 249 205
—, Dodder 261, 2622ie
—, Stoughton 243, 249lee
Rosenstein, см. Adams, —
Rule 316 2B0; 31o, 317 202
Russell 316, 317 2B2
Saunden 11173
Savic, см. Weber, —
Scanlan, см. Holmes, —
Scheibe 935B; 2932Бв; см. также
Franck, —
— , Schontag, Kantheder 93 61
_ t _, Kop ke, Henle 270288-
Scheller 176114
Schlivitch 316, 3172ВЗ; см. также Grum-
bach, —
Schmidt, см. Weitz, —
Schmidt 214, 294 173
Schmidt 204lee
Schneider 226, 253 188
Schoenberjr U5 105
Scholefield, Goodyear 17614t
— , Patel 17614Б
— , Turner 17614S
Schon, см. Moglich, —
Schubert, см. Michaelis, —
Schultz 10, 15, 162
— , Ganguli 155, 222 118
Schumacher 145, 241105; см. также
Koblitz, —
Seitz, Johnson 280 2"
Semerano, см. Mecke,—
Seyewetz 297 2e3
—, Mounier 106 68; 11173; 155, 222 ш
Sheppard 33, 84, 85, 86, 87, 88, 944Б;
9z 272b7t 288254
— , Brigham'35,84, 85, 86, 87, 88, 9415
350
УКАЗАТЕЛЬ АВТОРОВ
— .Lambert, Walker 85, 88"; 270,
272, 390, 291235; 271*"; '276, 277,
278, 279 245f а48
— , Vanselow 275 243
—, —, Нарр 29025в
Schontag 272 ав8; см. также Scheibe, —,
Kopske, Henle
Shroter, см. Egbert, —
Shunk 154Ila
Sihvonen 297, 298268
Silber» см. Ciamictan, —
Simard, см. Harris, —
Sklar 44
Smakula, Wasserman 89 48
Socher 93e0; 269, 273, 274, 275, 28423e;
277, 283a47
Sommer 166lsl
Soulafl 3112a^
Spealman, см. Blum, —
Stark 204lee
Staud, см. Leermakers, —
Staudinger 148 loe
Steadman, см. Bowen, —
Stei^mann 241, 294, 29517з; 295 262; см.
также Kogel, —
Stern, Volmer 246 204
Stonehill Л6175; 189, 1921"; см. также
Gilb. -
Stora 3ft, 3212es
Stoughton, см. Rollefson, —
Sudborough, Watson, Narayanan 141 e7
Suida 141 87
Taboury, см. Grumbach, •—
Teller, см. Franck, —
Thompson 315287
Tohmfor, см Cassel, —
Tollert 278 248
Turner, см. Scholefield, —-
Tuxen22516°, 18e
Uhler, см. Wood, —
Umbergrer, La Mer 24720e
Van Alphen 35 lia
Vanselow, см. Sheppard, —
Velluz, см. Dufraisse, —
Vickerstoff, Lemin 91 55
Visser 311 283
Voxels 128, 307", см. Prinffsheim, —
Volmer 204, 309, 311 U9> 282
Walker, см. Sheppard, Lambert, — ; см-
тахж.е PotteriJ, — , Weiss
Wallgren, см. Hedvall, —, Manssen
Wambacher, см. Blau, —
Wansbrough Jones, см. Haber, —, см-
Farkas A., —
Wassermann, см. Smakula, —
Watson 154114
Watson, см. Sudborough, —
Webb 283 2Б0
Weber 174, 252, 253, 255, 258 138; 219,
^52178; 216, 252, 253, 254, 262214;
299 ase
—, Savic 251212
Weig-ert 116 78; 120 7°; 127 83; 12885^
157, 161, 166121; 1661M
Weil-Malherbe, см. Weiss, —
—, Weiss Ь8, К9"
Weiss 136, 138, 187 вз; 136 83aJ 149 ш;
299UO. д)01вз; 202, 2031вБ; 21017e;
216, 219, '220, 241, 252"» 22518R;
230,237, 225, 254, 252le7; 2261ВЧ
см. также Potteril, Walker; — , Ra~
binowitch, —
—, Fishgold 219, 253, 25417B
— , Porret 200 161
— , Weil-Malherbe 116 78; 129 85; 262,
263 264222
Weisemann 248 208
Weizmann, Hirschberg, Bergmann222 18S
Weitz, Schmidt 267 a30
Wells, см. Ellis,—
West 109 72; 248207
— , Miiller, Jette 243, 249108
Wevde, Frankenburorer, Zimmerman^
113 w
WieJand 169. 237, 238, 241137
Willemart 144101
Williams, см. Bowen —
Winther 157iai; 166 134; 299, 301 a67
Wizin^er 4215
Wolf 11173
Wood 120, 123, 125, 1277e
-, Uhler 10, 15, 16 3
Worel 154 m
Wyszewiansky, см. Haller, —
Ziersch, см. Haller, —
Zimmermann, см. Weyde, —
ОГЛАВЛЕНИЕ
От pi
Введение 5
Часть первая. Фотофизические процесоы в красителях
Глава 1. Поглощение света 9
1. Цвет и спектр поглощения —
2. Законы поглощения света • 12
3. Эффективность или выход процесса поглощения 16
4. Испускание света красителем 19
Глава 2. Строение и спектр красителей 23
5. Структурные скелеты и типы красителей —
6. Валентные структуры • . . . . 29
Принцип Паулн. Ионизованные структуры 31
7. Строение электронной оболочки 36
8. Связь между спектром поглощения и строением красителя. 42
Квантово-механнческая теория цветности • • • • • « . 44
Осциллнцня зарнда • . . 49
Глава 3* Энергетика молекулы красителя 54
9. Вибрационная энергия —
Закономерности флуоресценции 58
10. Основной принцип внутримолекулярной Энергетики • . 62
11. Статистические законы поглощения в испускания . . . . 67
Длительность ноабужденного состояния 71
12. Превращении поглощенной световой энергии ...... 74
Дезактивация —
Конверсия в метастабильное оостонние 78
Типы физических фотореакций • . . • • 83
Г л а н а 4. Структура и изменчивость полосы поглощения красителя 84
13. Влннние среды на положенно полосы поглощенвя . . . . —
14. Структура полосы поглощения 87
Часть вторая. Фотохимические реакции красителей
Глава 5. Фотохимические процессы 97
15. Формальные ваконы фотохимии —
16. Основные типы фотохимических реакций в красителях • 102
352 ОГЛАВЛЕНИЕ
Фотораспад • .... 102
Фотоперегруппнровка . • • - 104
Фотоприсоединенне • 106
Фотоперенос электрона «... * 108
Фотосенсибнлизация 109
Типы фотохимических реакций 110
Глава 6. Фотораспад и фотоприсоединение 111
17. Фотолнэ диазосоединений —
18. Фотолнв на ноны (фотононнэацвя) лейкокраснтелей . . • 112
19. Фотодимернзацин полнциклнческнх углеводородов .... 116
20. Фотохимические реакции ксантеноаых красителей в
растворах. Окрашенные фотопродукты 120
Глава 7. Фотореакуии присоединения молекулярного кислорода. 128
21. Тушение флуоресценции кислородом —
Кинетика тушения кислородом 132
Тушение флуоресценции красителей кислородом . . . 135
22. Фотореакцнн ароматических углеводородов с кислородом. 141
23. Механизм фотореакцин присоединения молекулярного
кислорода • 147
24. Деструктивное окислительное выцветаине красителей иа
волокне под действием света 154
Светостойкость 157
25. Кинетика деструктивного ныцзетания красителей при фото-
реакции с кислородом 160
26. Механизм деструктивного выцветания красителей . • • . 166
27. Фотосеисибнлнзоваяное красителем разрушение волокна. 174
«лава 8. Фотоокисление и фотовосстановление * «•••*••• 179
28* Перенос электрона —
Элементарная реакция окисления восстановления • > 183
Окислительно-восстановительный потенциал . . . • • 188
29. Фотоокисленне неорганических ионов . . • • • 192
30. фотоокисленне органических соединений 203
31. Фотоокисленне красителей в присутствии акцепторов
электрона 208
Фотосенснбилнзованнан красителями реакции
восстановления ионов ...*.• 212
32. Фотовосстановленне красителей в присутствии доноров
электронов •••• 216
Световое окислительно-восстановительное равновесие. 223
33. Фотосенснбилизованное красителями окисление
неорганических ионов • •• 225
34. Фотосенсибилнэованное краентелнмн окисление
органических соединений в присутствия кислорода (фотодннамиче-
ское действие) • 234
Биохимические теории окисления кислородом 238
а л а в а 9» Торможение фотореакуий красителей 243
35* Тушение флуоресценции красителей посторонними
веществами <—
ОГЛАВЛЕНИЕ 353
36. Концентрационное тушение флуоресценции и торможение
фотохимических реакций 259
Глава 10. Фотореащии красителей на поверхности
кристаллических тел 265
37. Спектр поглощения красятеля, адсорбированного
кристаллическим телом —
Спектр фотографической сенсибилизации 269
38. Сенснбнлизонанный красителями фотолнв бромистого
серебра 273
Энергетика процесса —
Квантовый выход и выход сенсибилизации 276
39. Механизм фотосенснбилизацни бромистого серебра
красителями • 279
Передача энергии 284
Передача электрона 288
40. Вуалирующее действие красителей на бромисто» серебро
и десенсибилизация 294
41. Фотосенсибилизация твердым телом выцветания
красителей 299
Миграция энергии 304
Глава 11. Фотоэлектрохимические аффекты красителей 308
42. Внешний фотоэлектрический эффект и фотопроводимость
твердых пленок красятелей —
Фотопроводимость растворов красителей 310
43. Фотогальв'ннческий эффект 312
44. Фотовольтаический эффект (эффект Беккереля) . . . 319
Литература 325
Предметный указатель 337
Указатель авторов . . » 343
Под общей редакцией Комиссии АН СССР
по изданию иаучно-популяриой литературы
Председатель комиссии
Президент АН СССР академик С. И. ВАВИЛОВ
Зам. председатели
Члеи-корреспондевт АН СССР П. Ф. ЮДИН
Печатается по постановлению
Редакуионко-иэдательскою совета
Академии Наук СССР
*
Технический редактор Р. С. Певзнер
Корректор Р. С. Ролубовская
#
РИСО АН СССР J4 2543. М-04791. Подписано к печати
12/VII 1947 г. Формат бумаги 60 X MVto-
Печ. а. 221/8 + 1 »**• Учетн.-иадат. л. 24. Тираж 5000
1-я Типография Издательства Академия Наук СССР.
Ленинград, В- О., 9 лиц., д. 12.
Нитрозо-краситеми
no
Ншйро-красители
N0*
Ни/прозо ^-нафтол
в щелотопсреде
Хинолинобь/е
Полиуиклокетомобые
НафтохиноноВые Антрахинонобые
нч нх
о 9 fl о
-Он
N0;
Пикриновая
кислота
ЦианинШФшметтбш}
ц/Шфтазорин о
Индигоидные ^
Н
ФлабаИилин
М ифенилметамодые
I
мн?
Аурами*
с?н5 с
Пинацианол
Трифенилметонобые
Индиго
Индатретдый
синий
Индомины
Малахитбыйзелешй
Зеленый Биндшедлера
Индоанилины
Акридино8ь№
н
Трипотодм
он
он
дщелочсреде
Аррин
Индофенолы
^ндофенолб
ырмшивйсреде
Окъазинобые
Фосфин
(Хризаиилин)
но
Родамин В
он
+ (CH3)N
Напри синий
Тиазинобые
О
а
N]
Метиленобыи голубой
Азокрасители
д кислой' среде б щелочной среде
Флуоресцеия (урамн)
Фталоциаяины
Хризоидин
Сафранин Т
Порфирины
N
П N
Метилоранж 3J
б щелочной среде
Фталацияям
СН
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ
Страница
Строка
Нап
ечатано
Следует читать
14 j 1 снизу
I
55 i 13 сзерху
81 13 снизу
99 '11 и 12 снизу
131 'i3h14chh35
137 i 9 сверху
186 5 сверху
(конец
формулы)
212 2 снизу
292 18 снизу
Теренич, „Фотохимия красителей
7- 10-22.
10
^ . кг кал
моль град
й"
азэкрасытеля
■ . и достигается
а-<. № 70.
Av[f][f]
Г К1Л
ноль град
1
Л',,,'
А*ч-О2
"
акцептора
, которая достигается